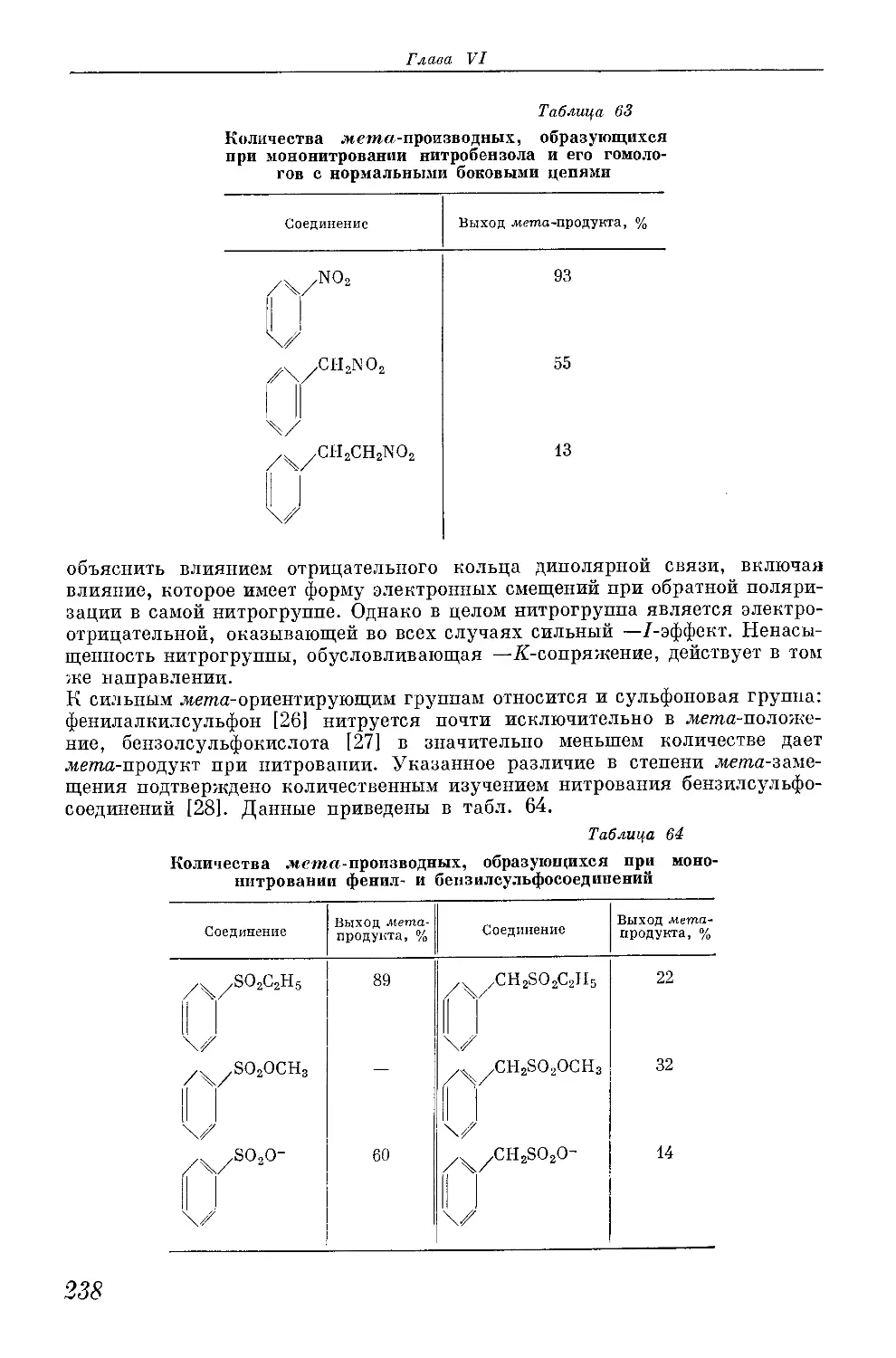
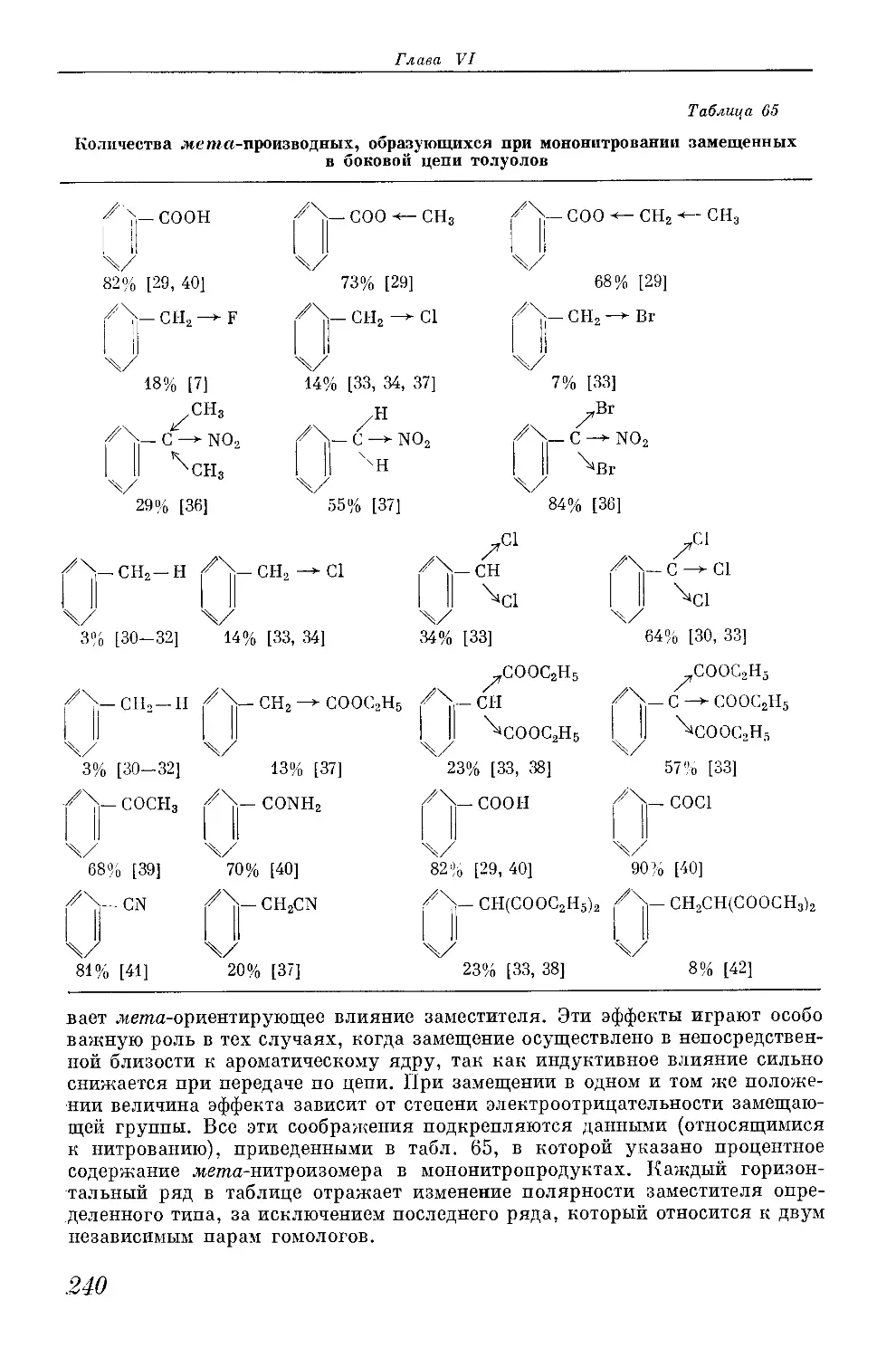
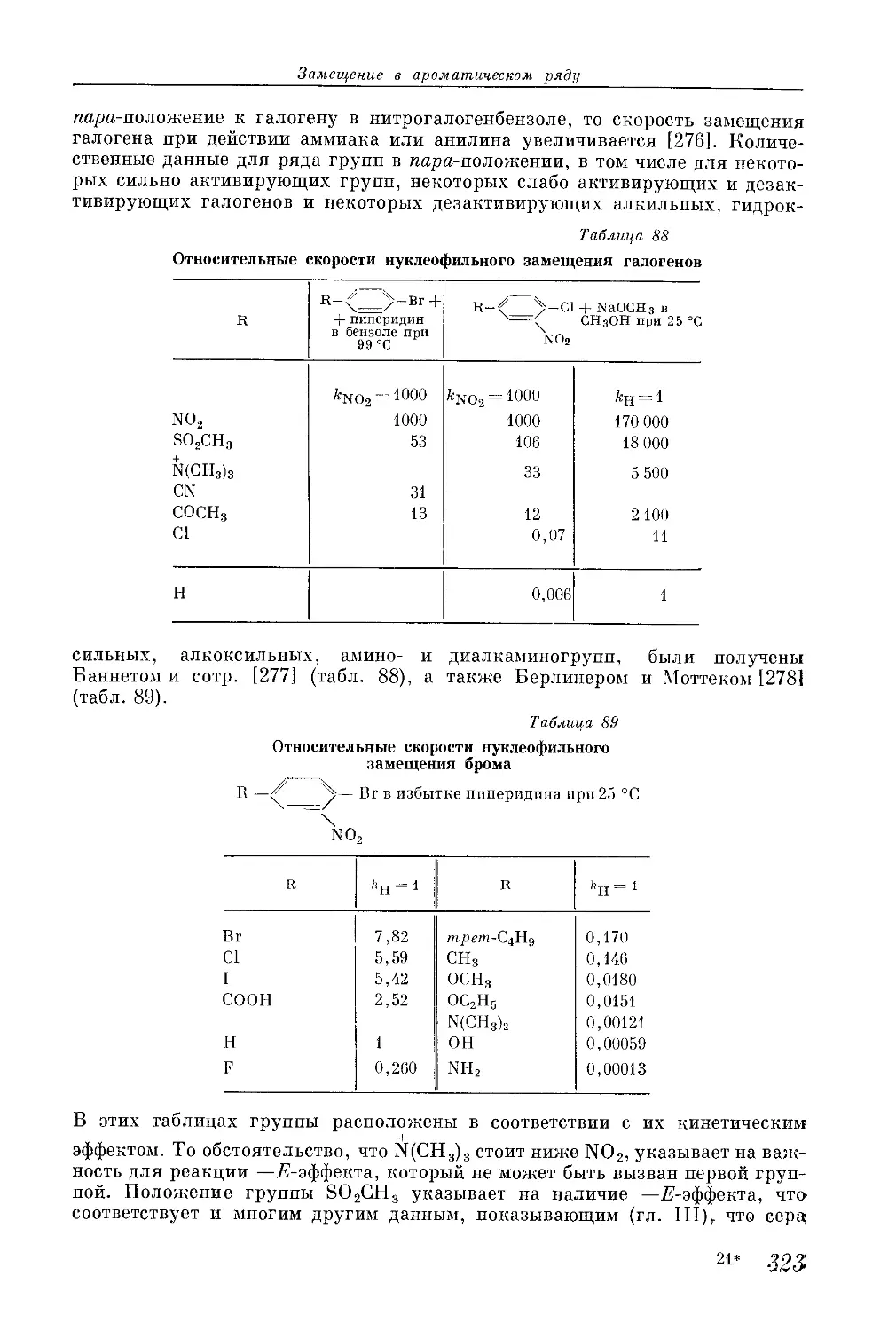
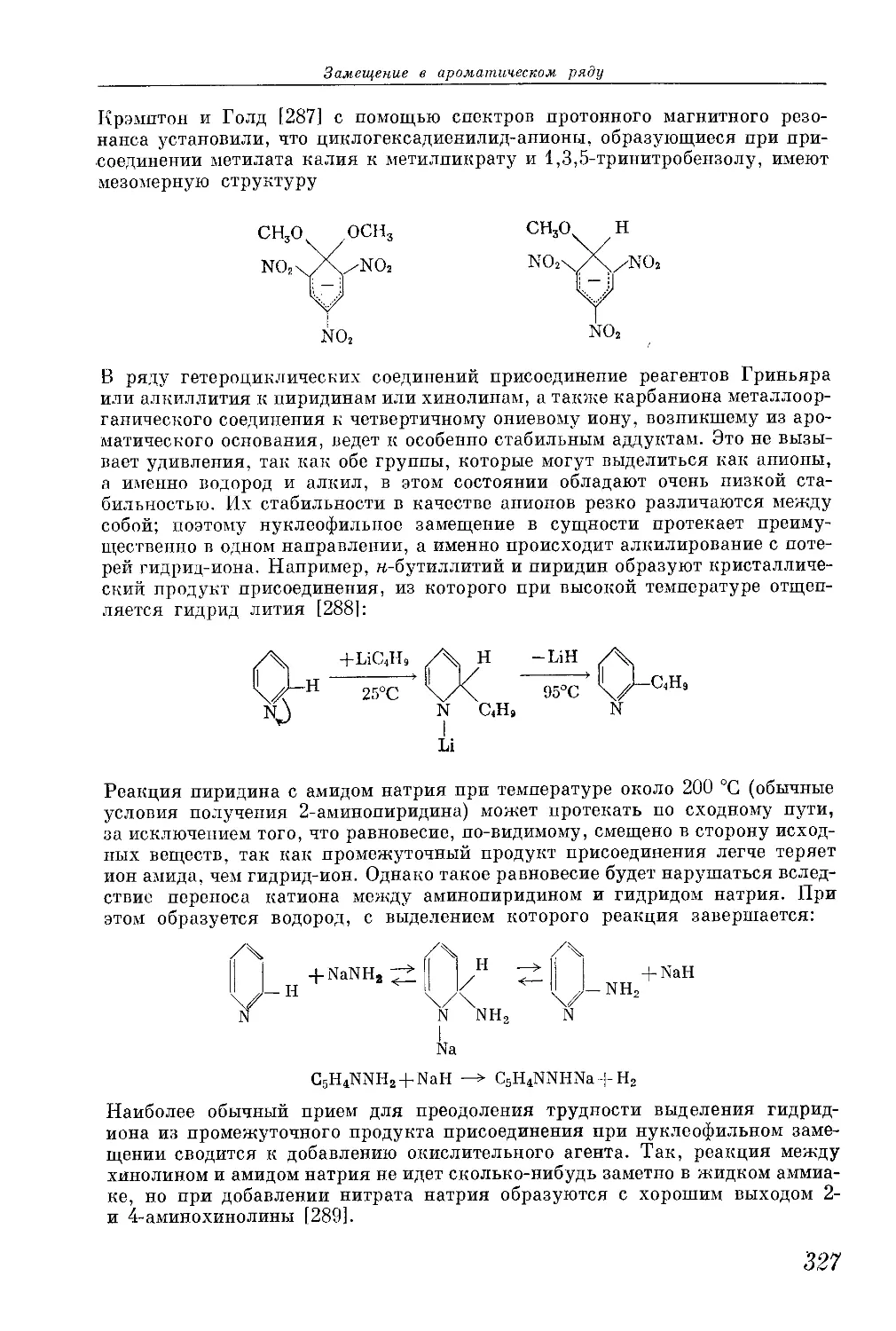
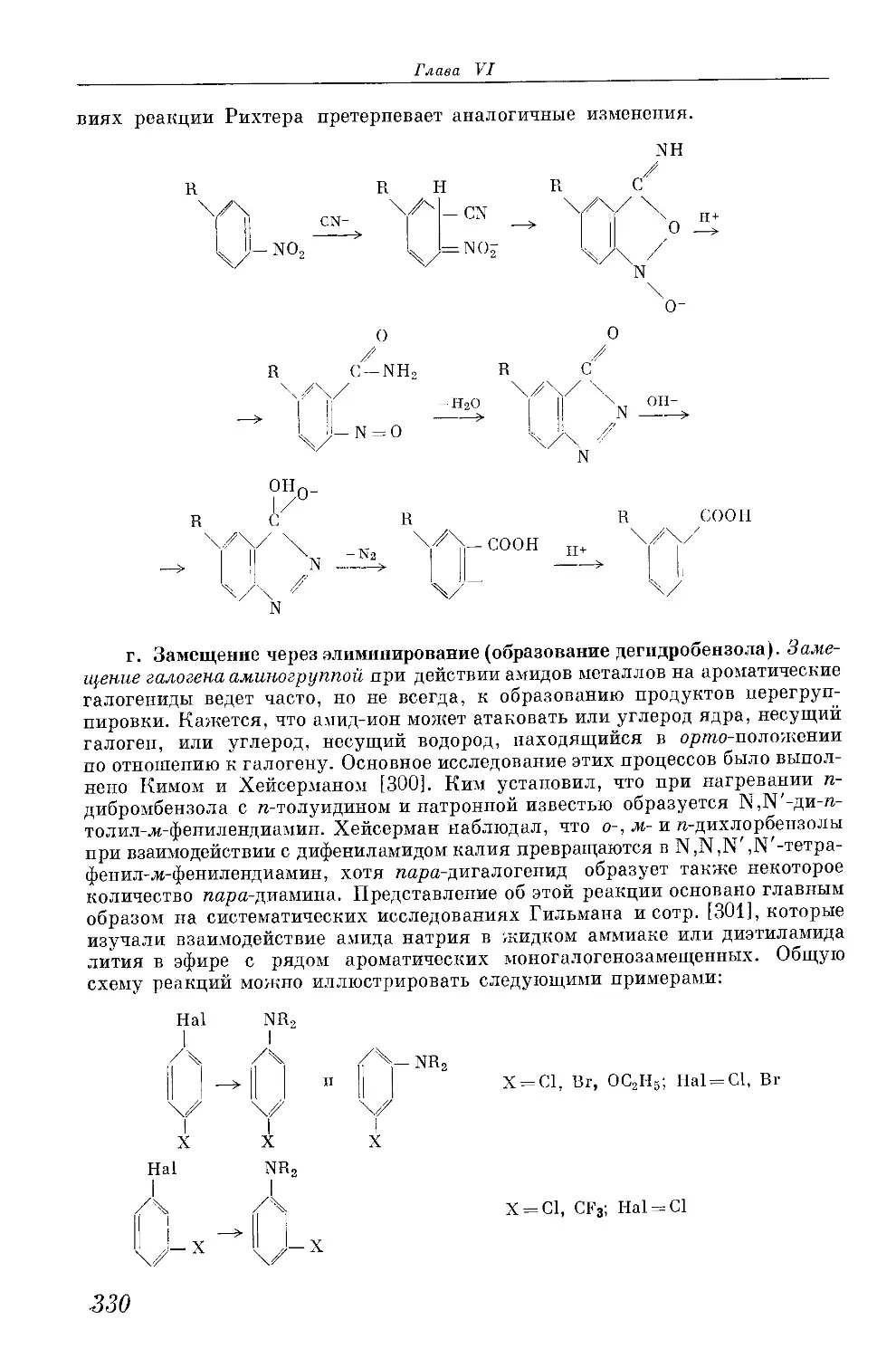
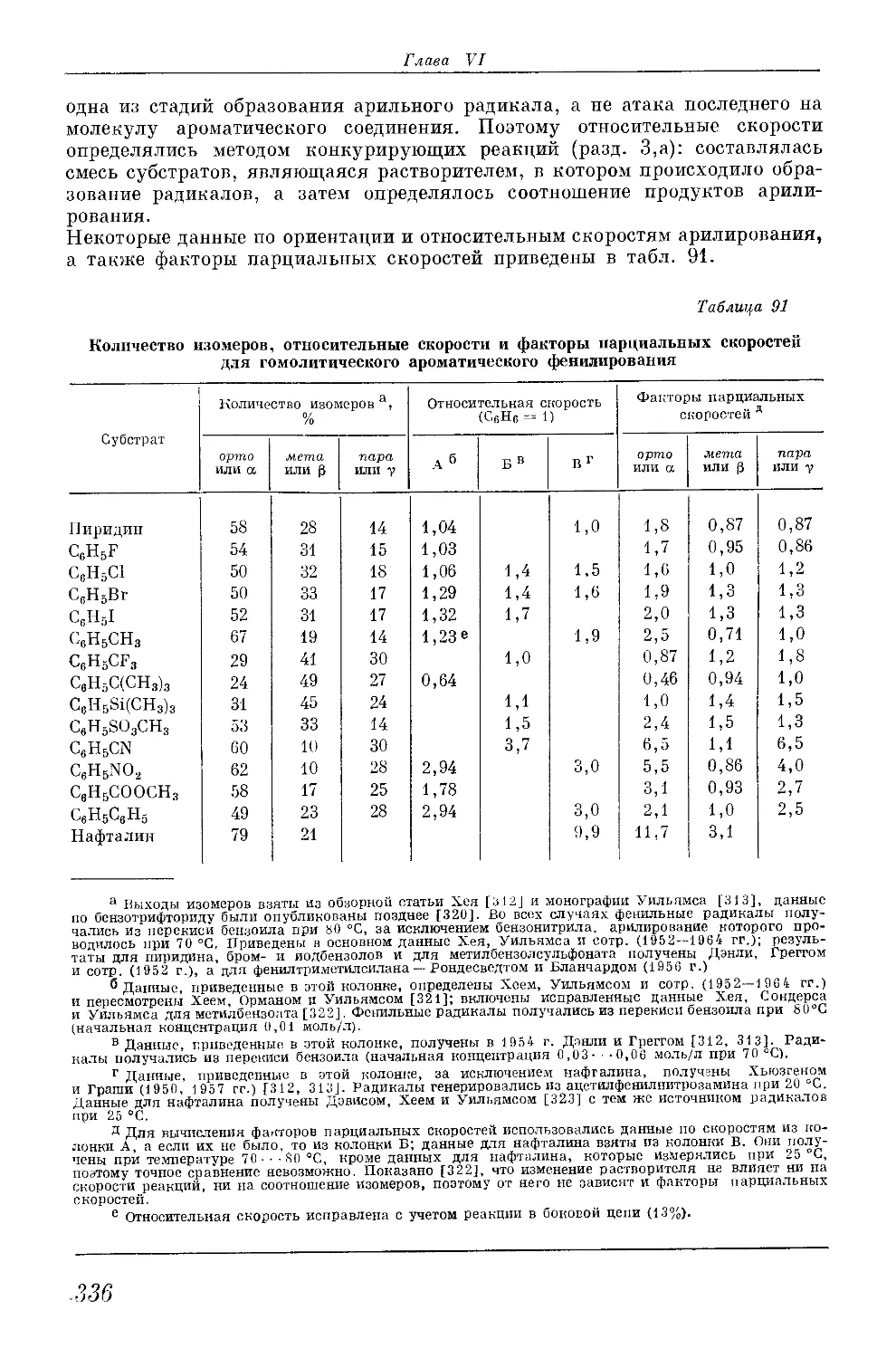
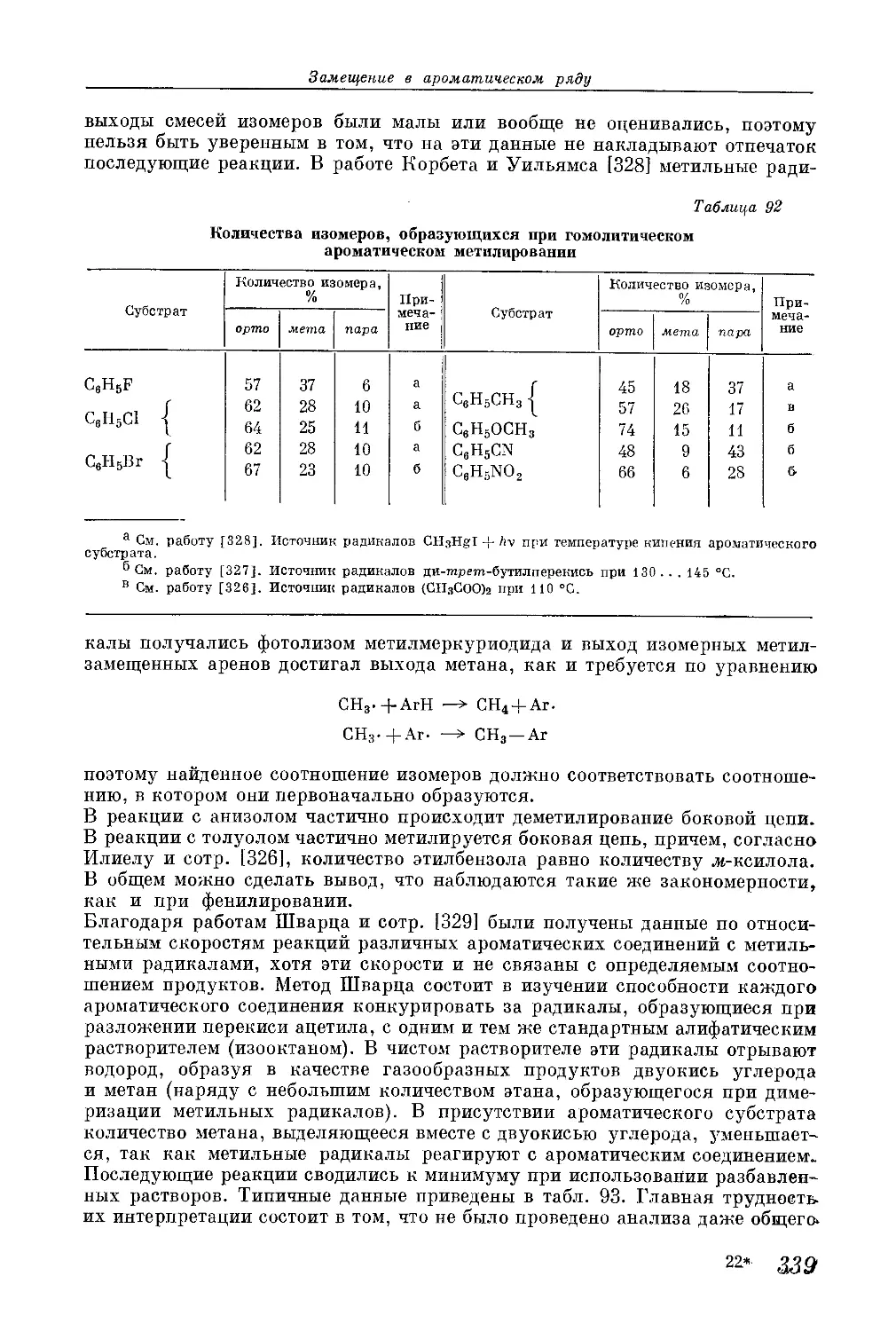
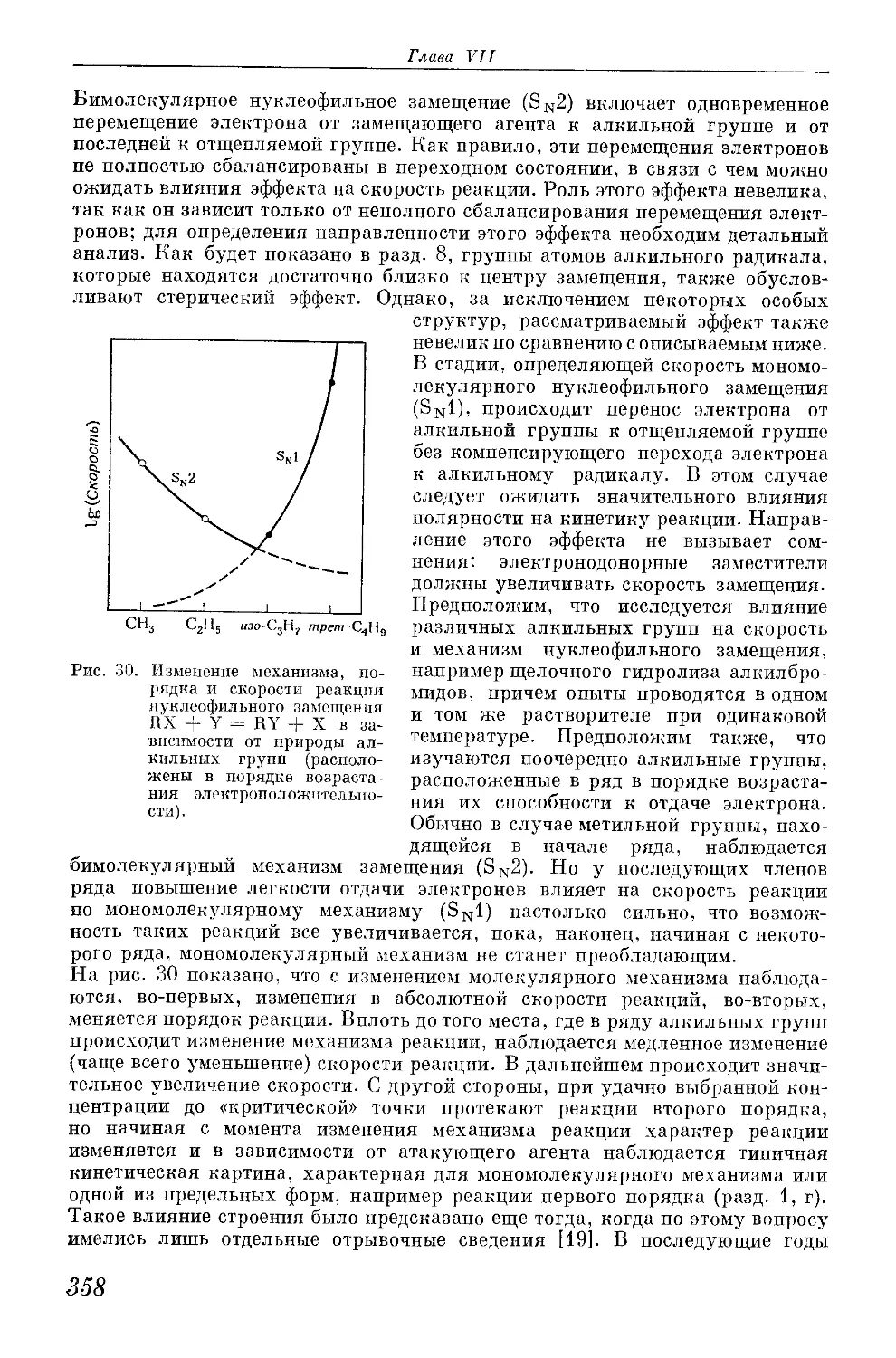
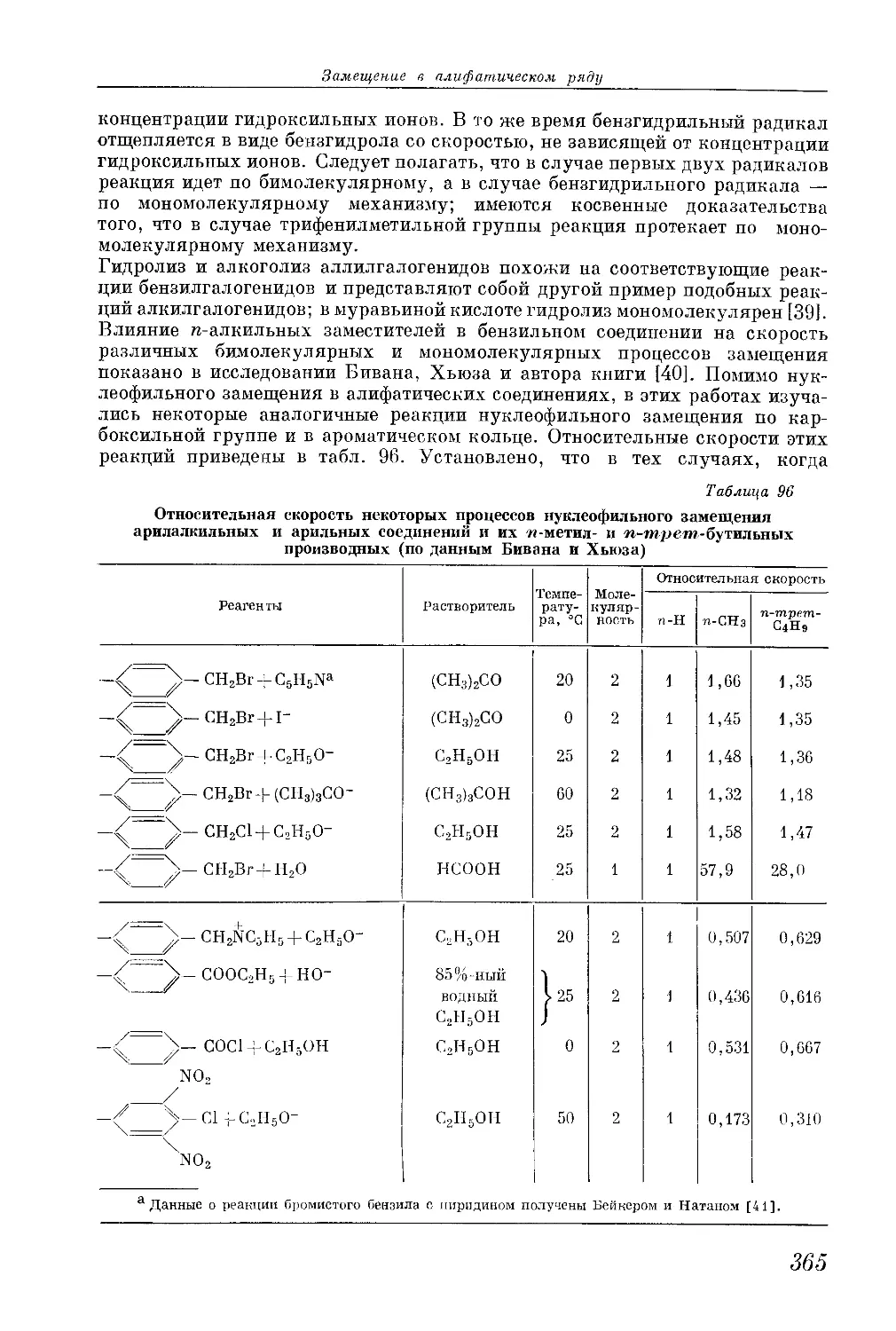
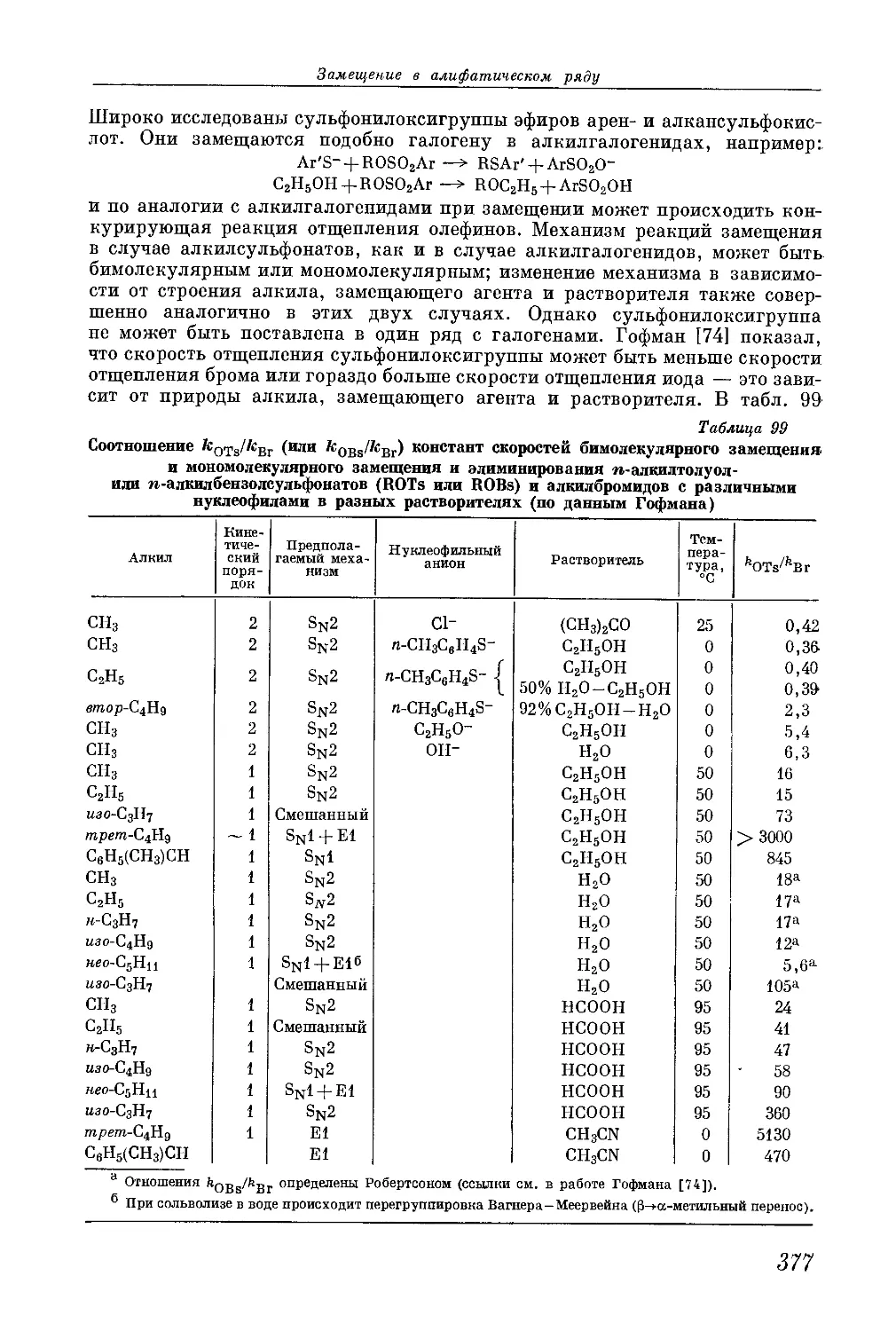
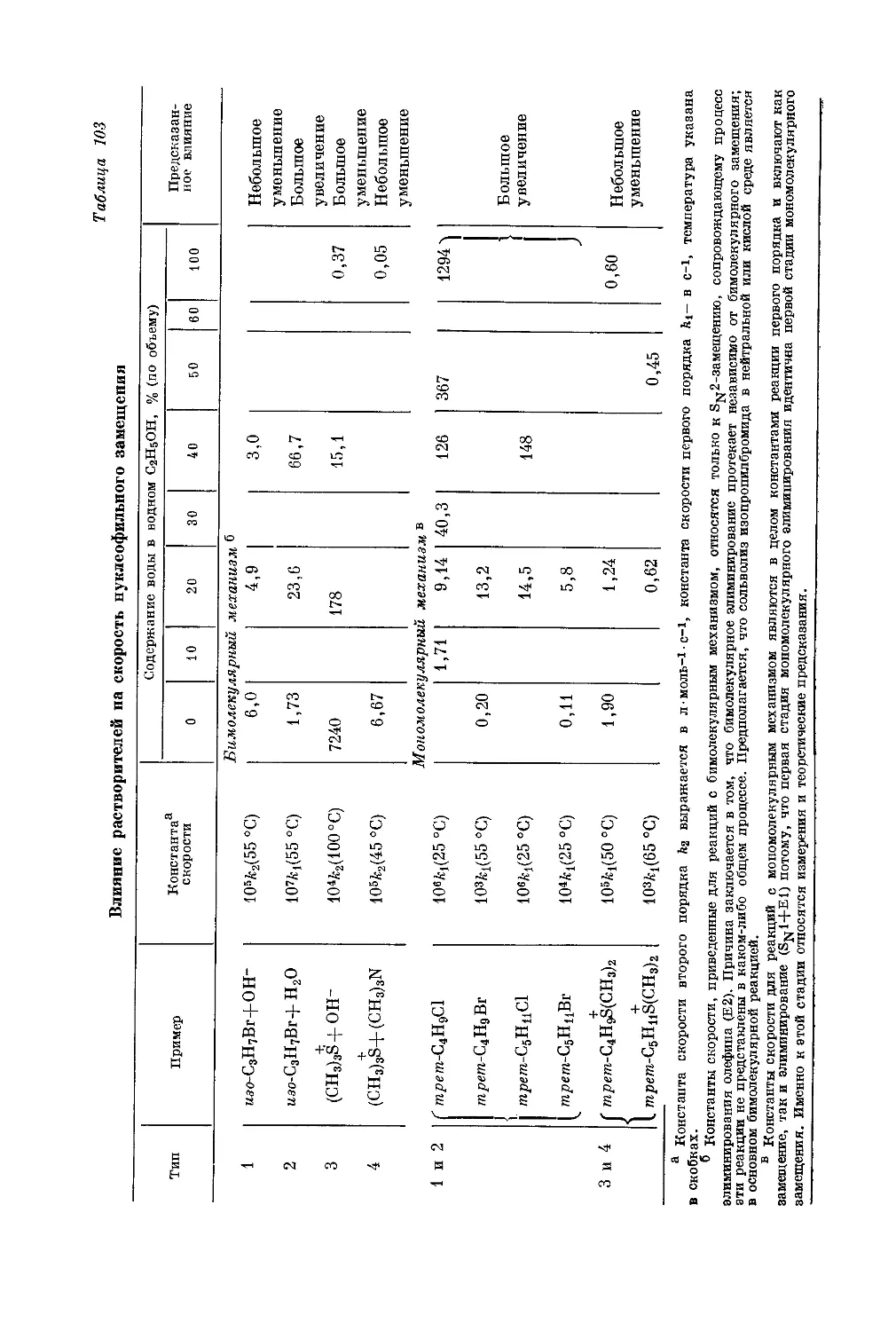
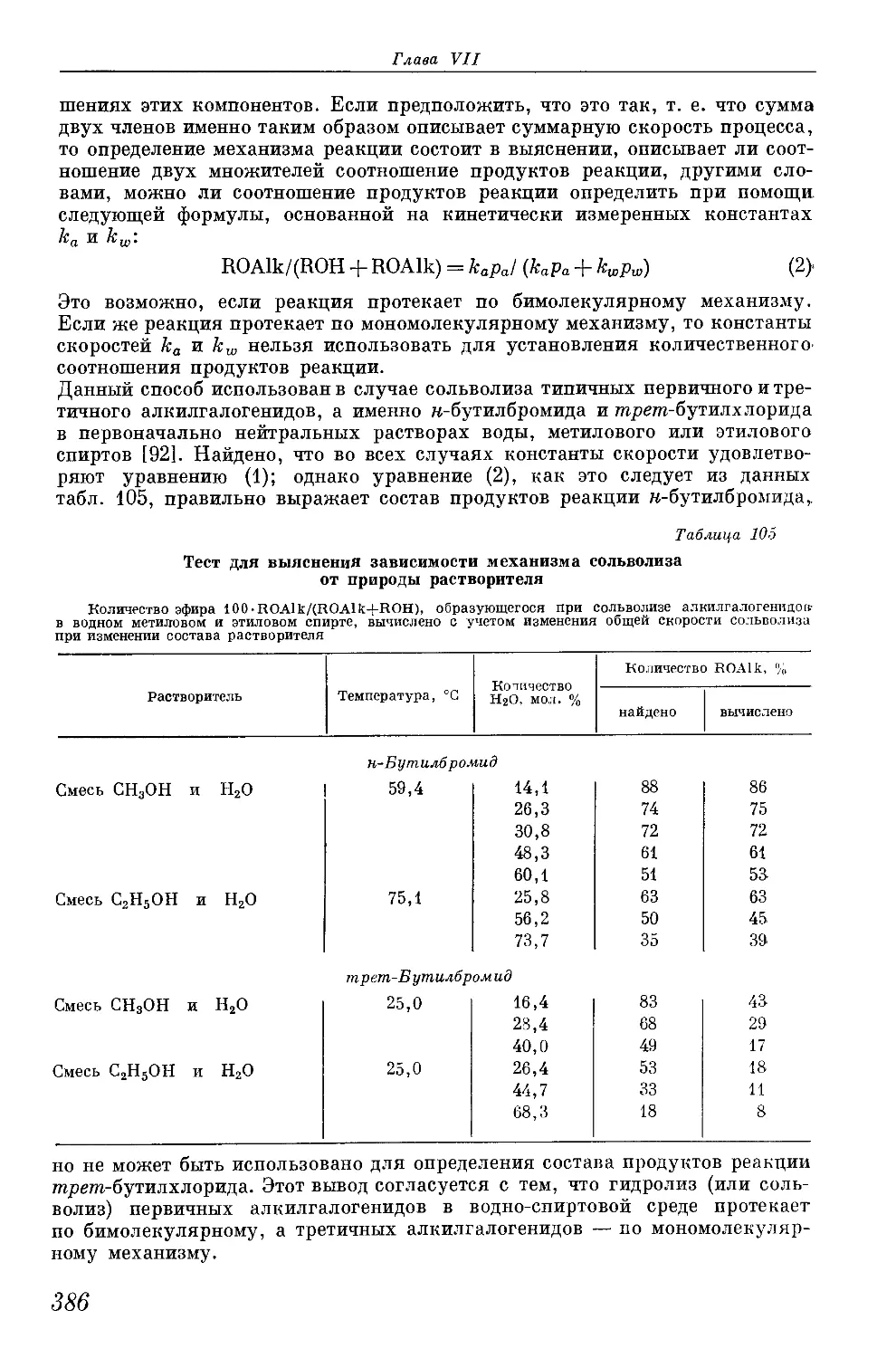
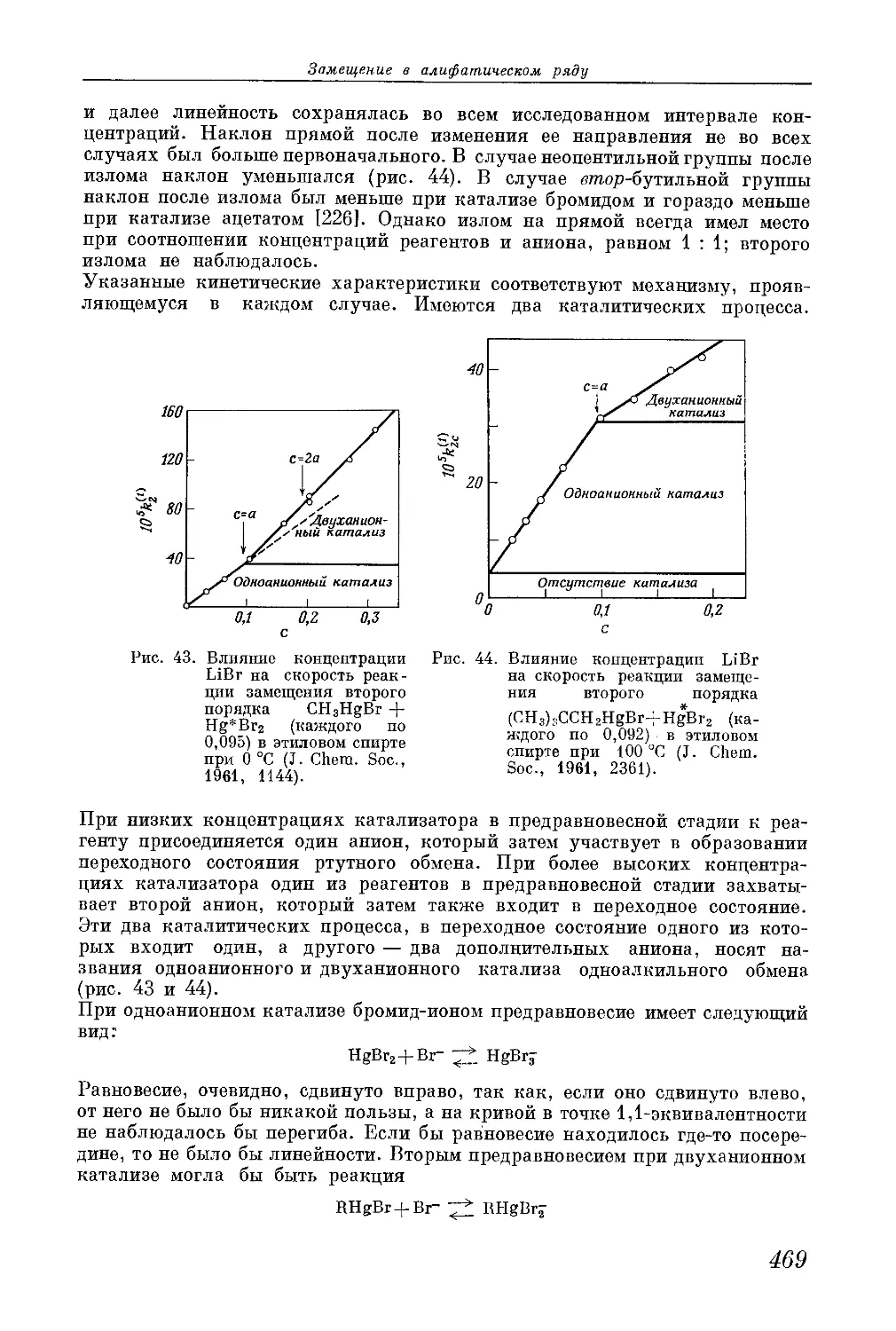
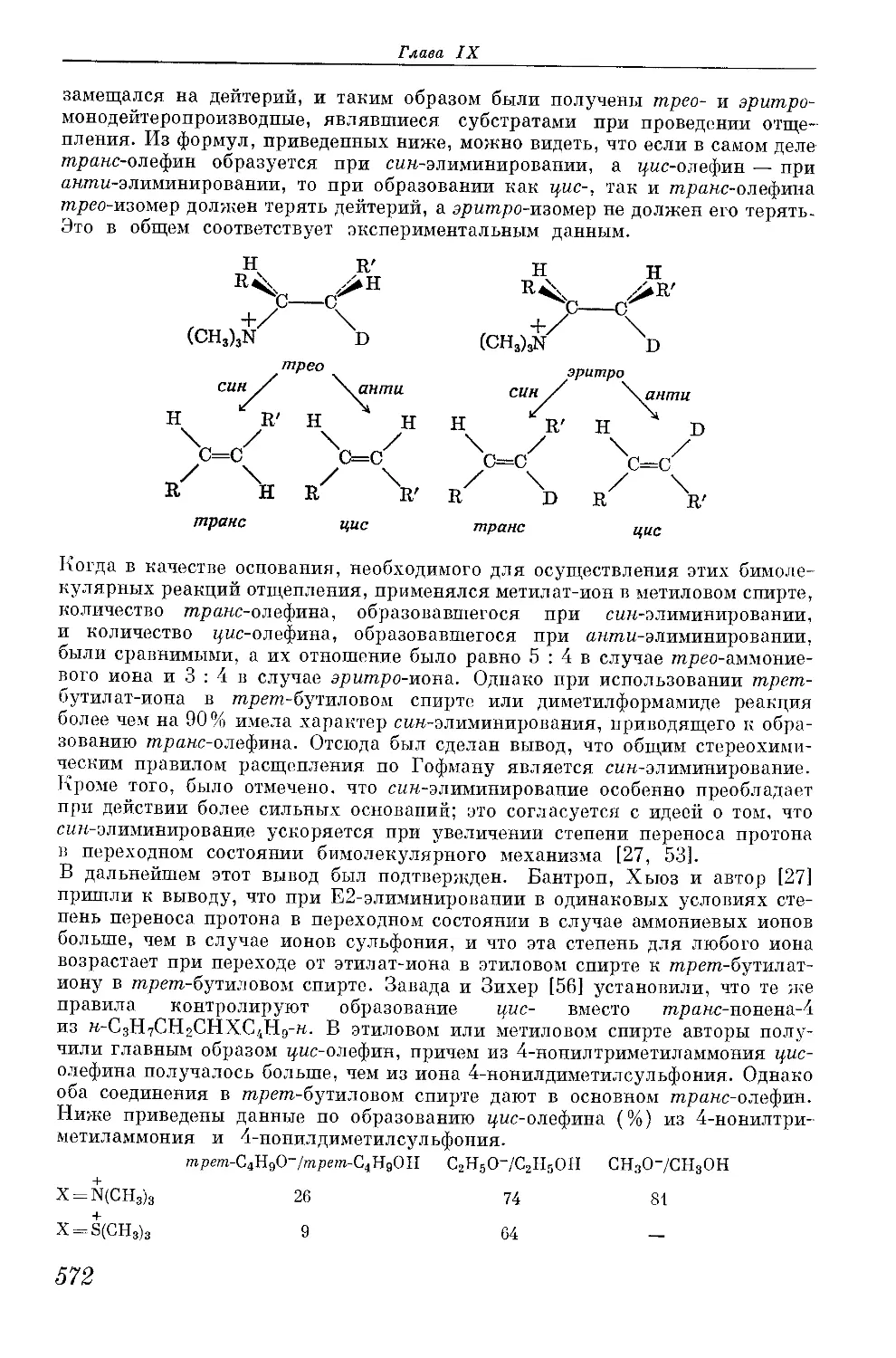
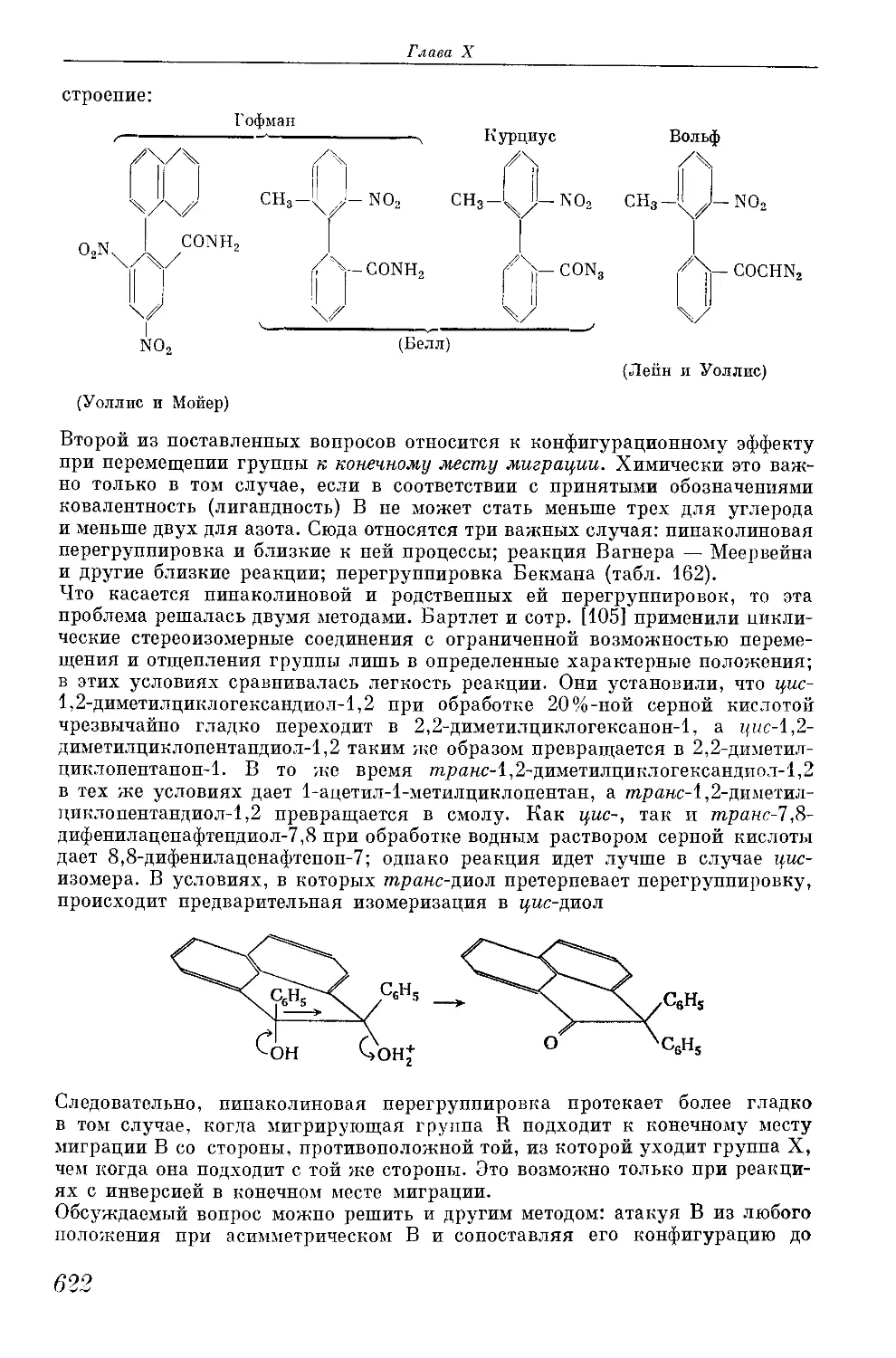
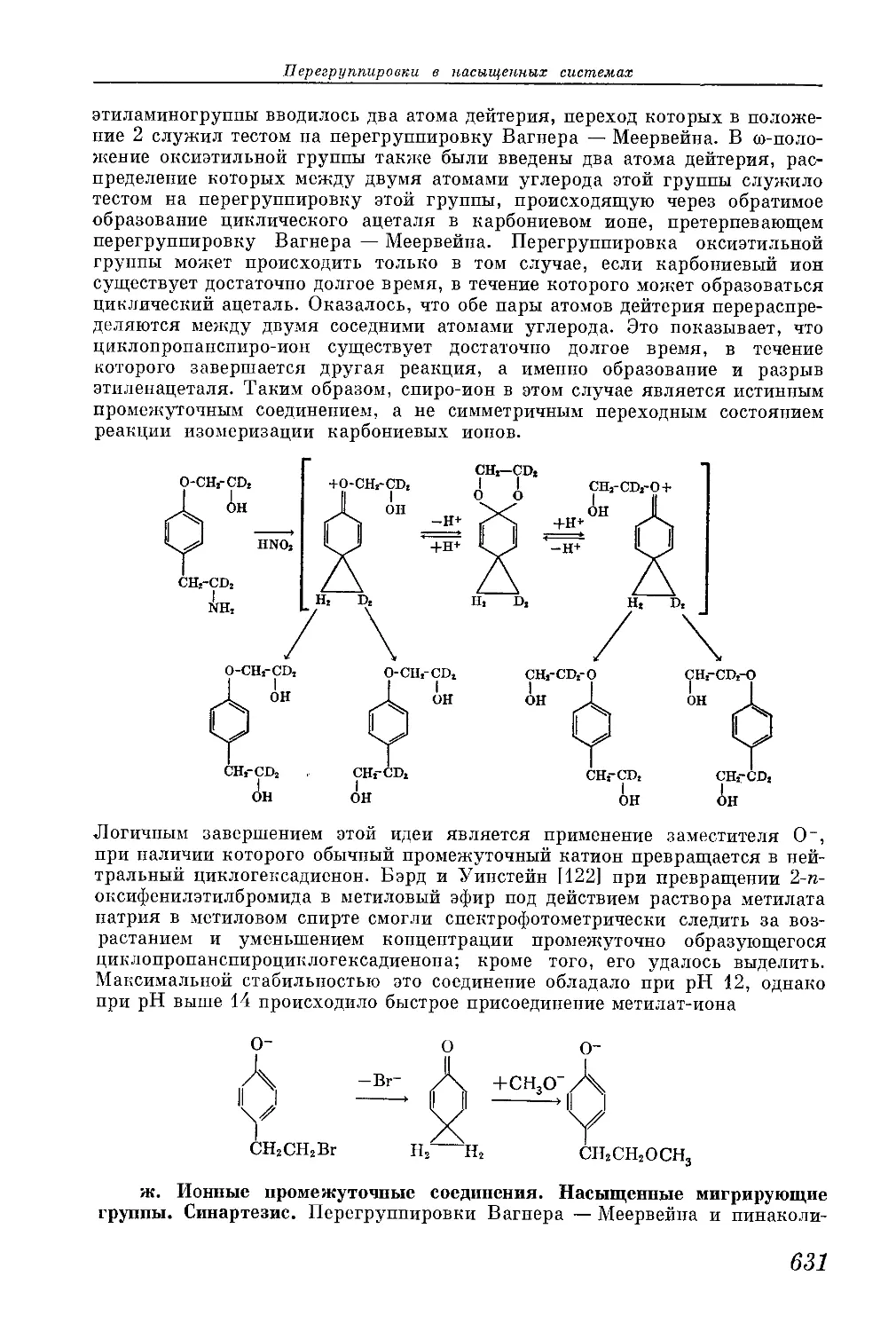
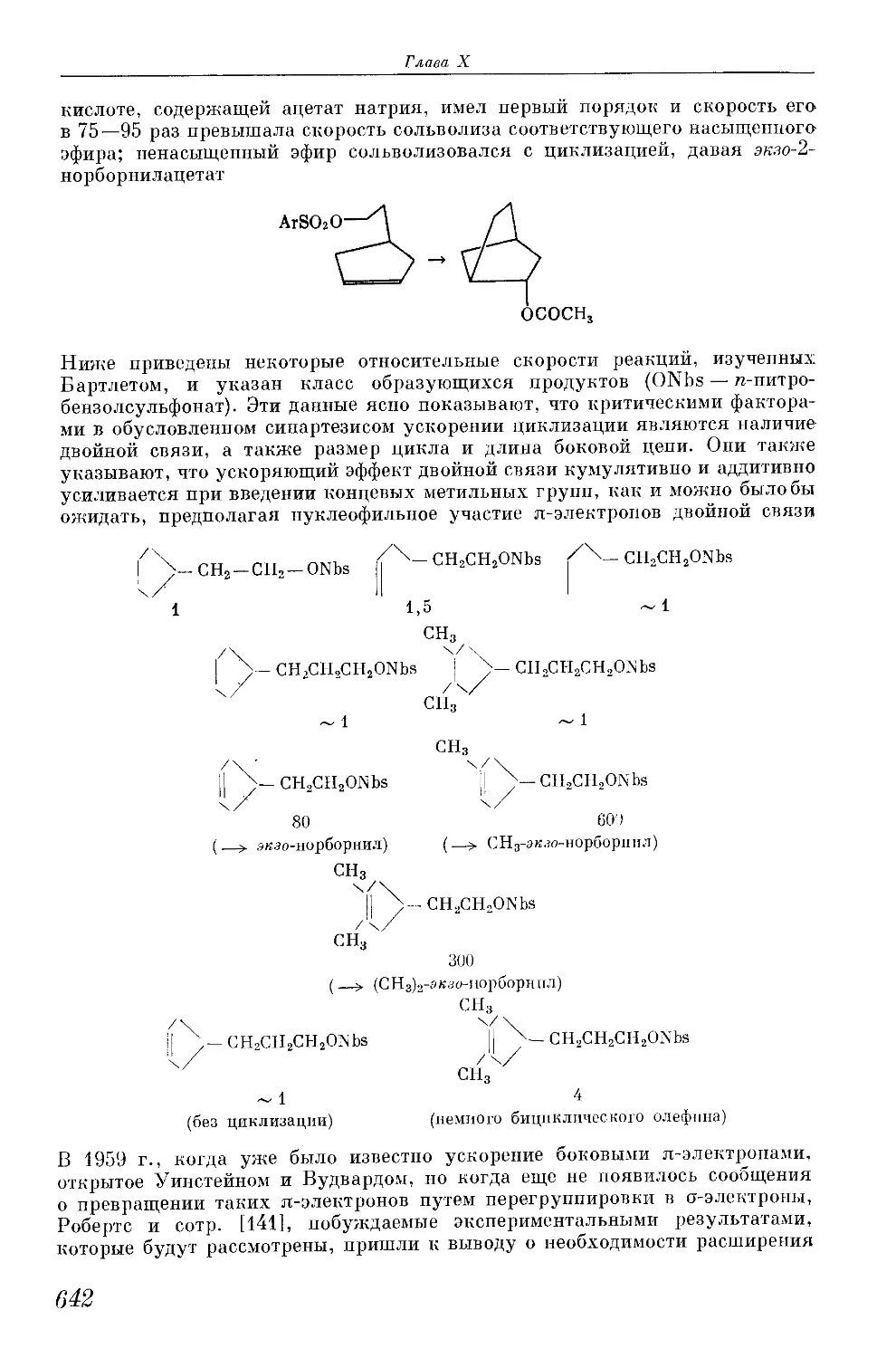
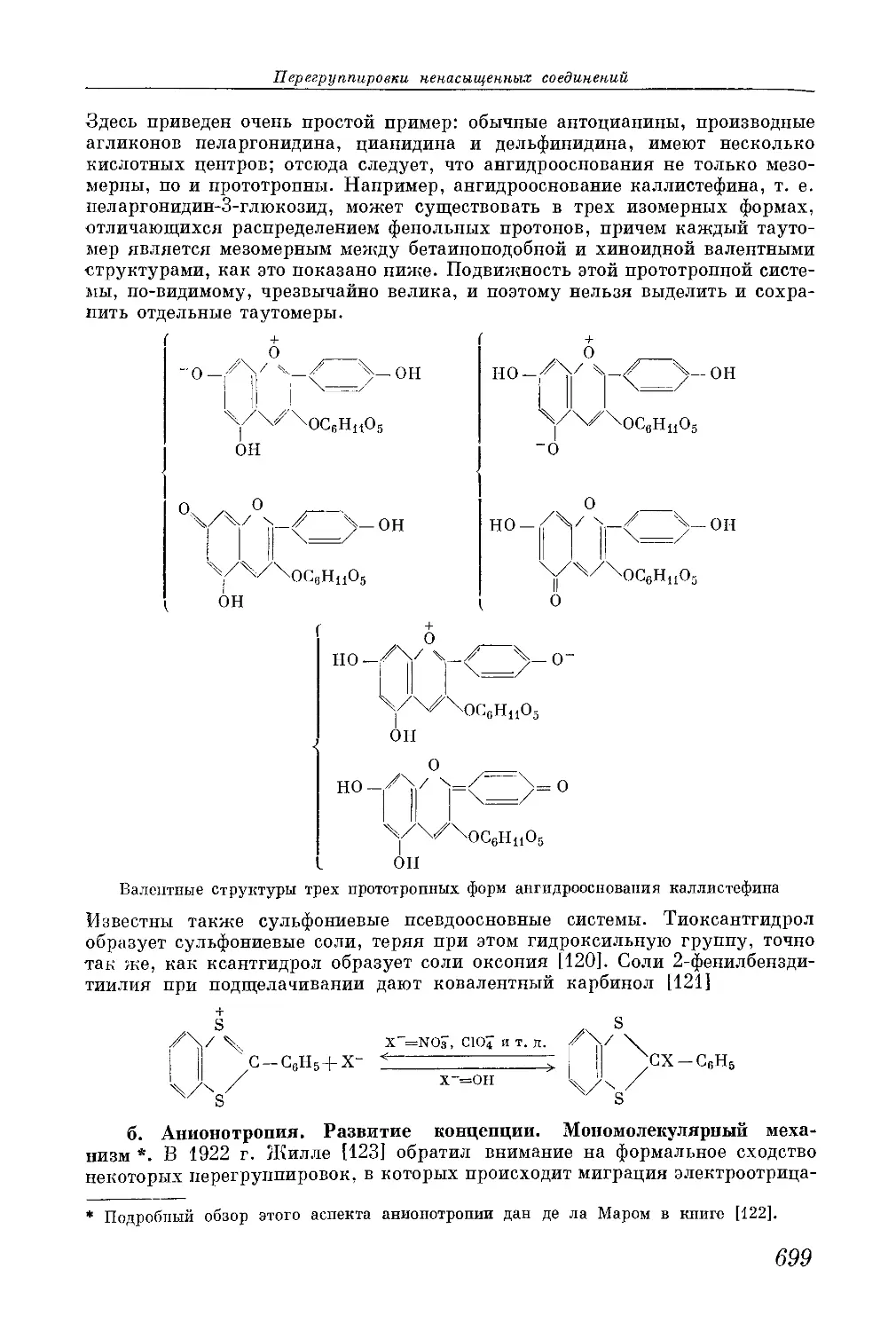
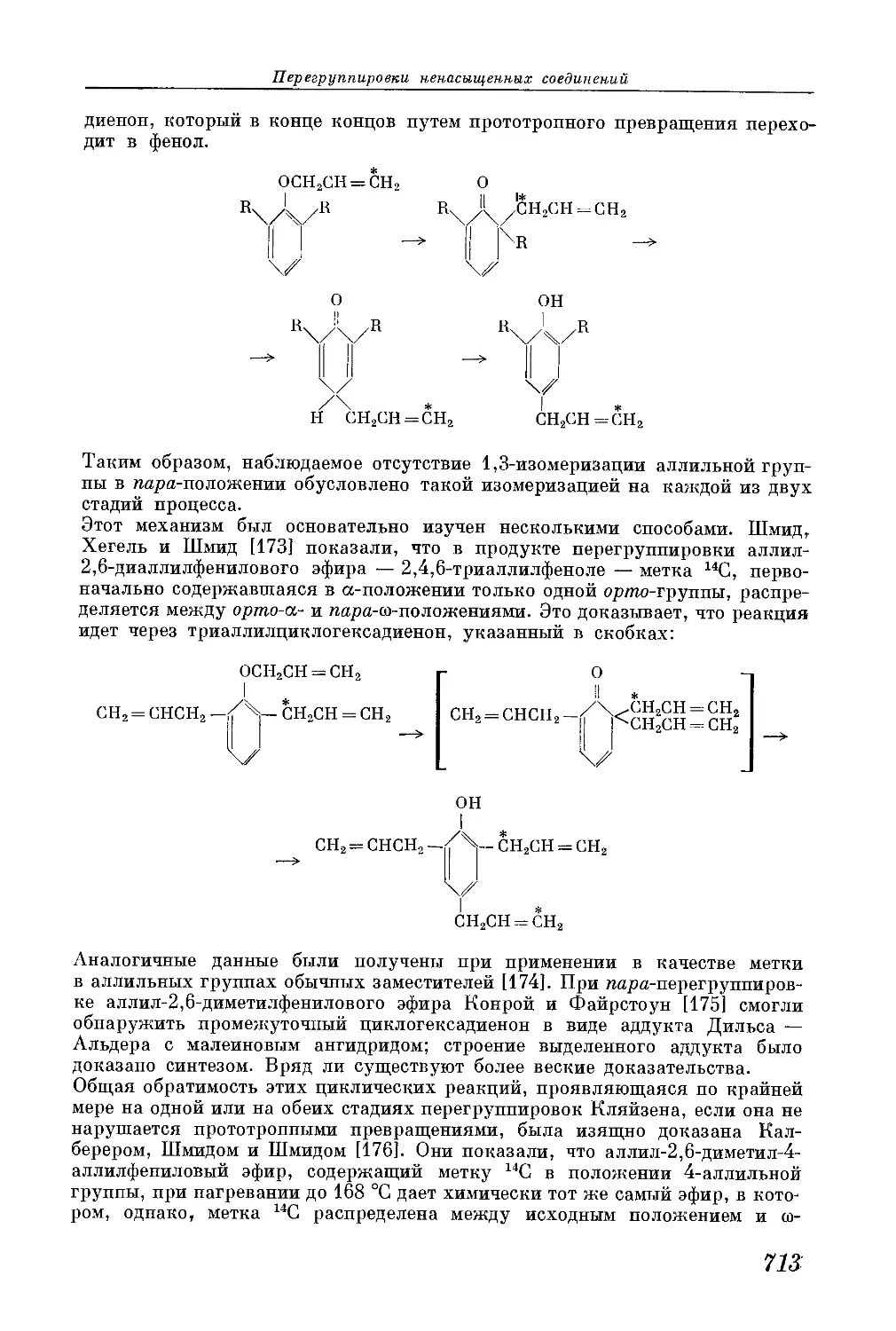
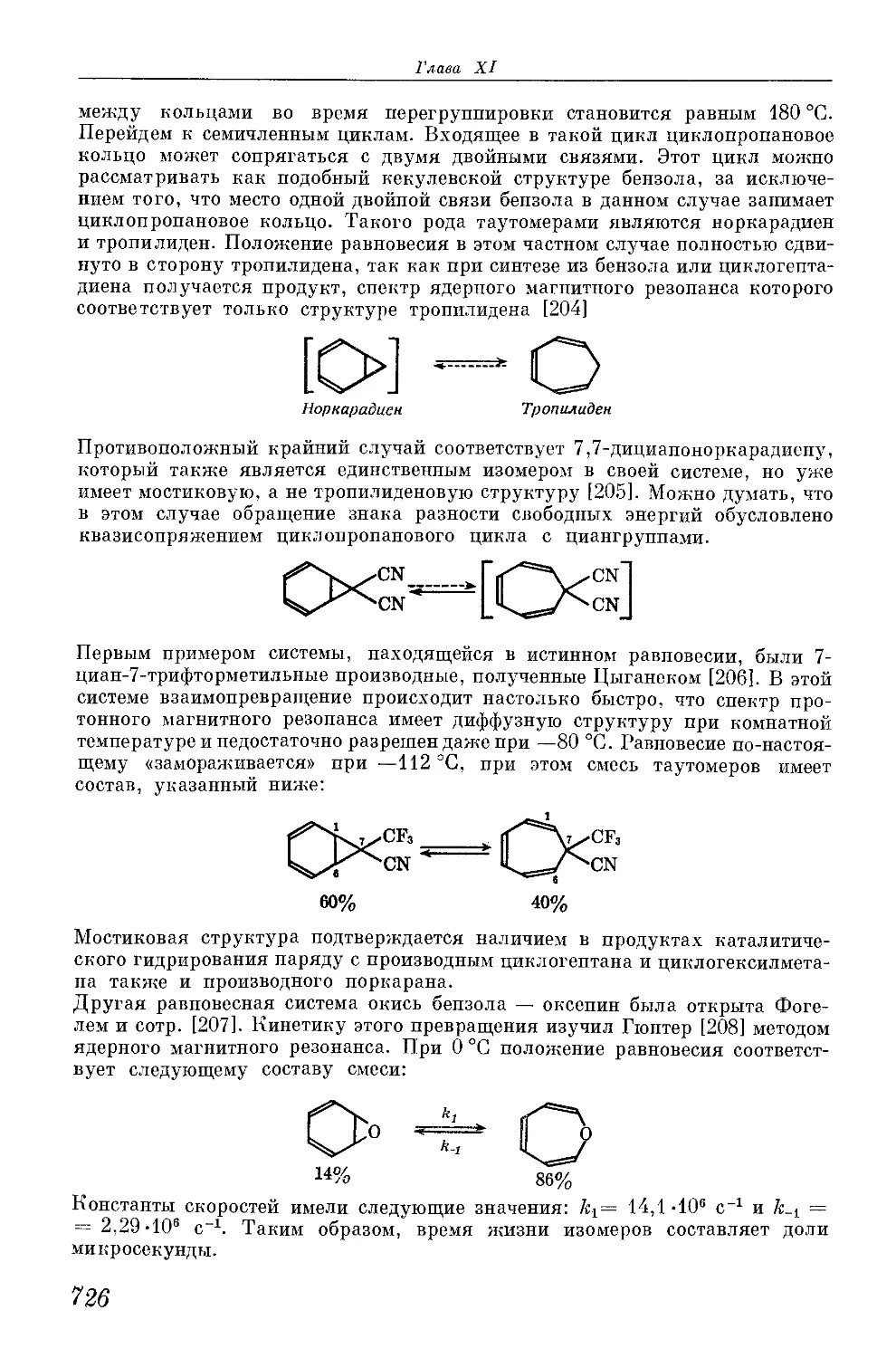
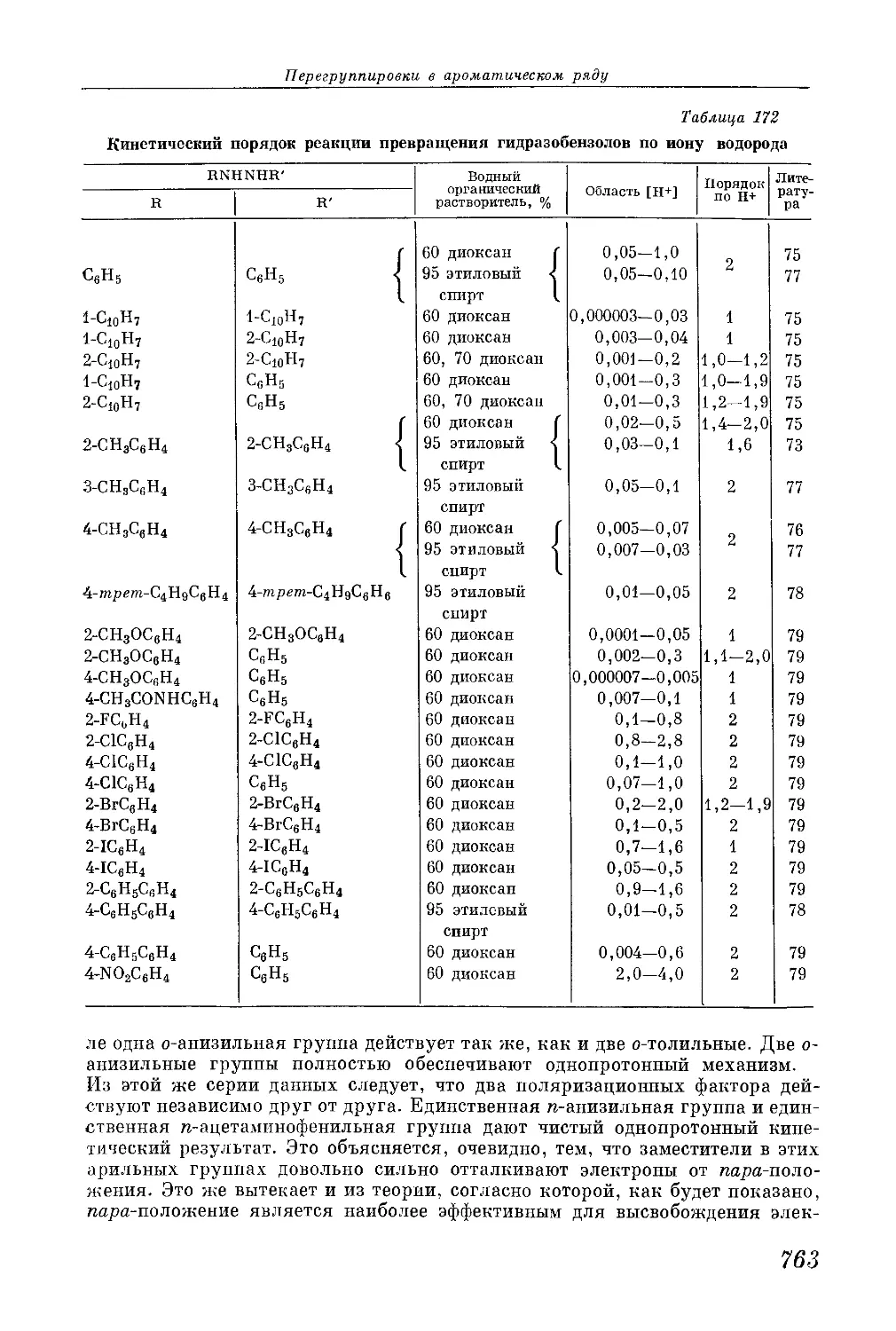
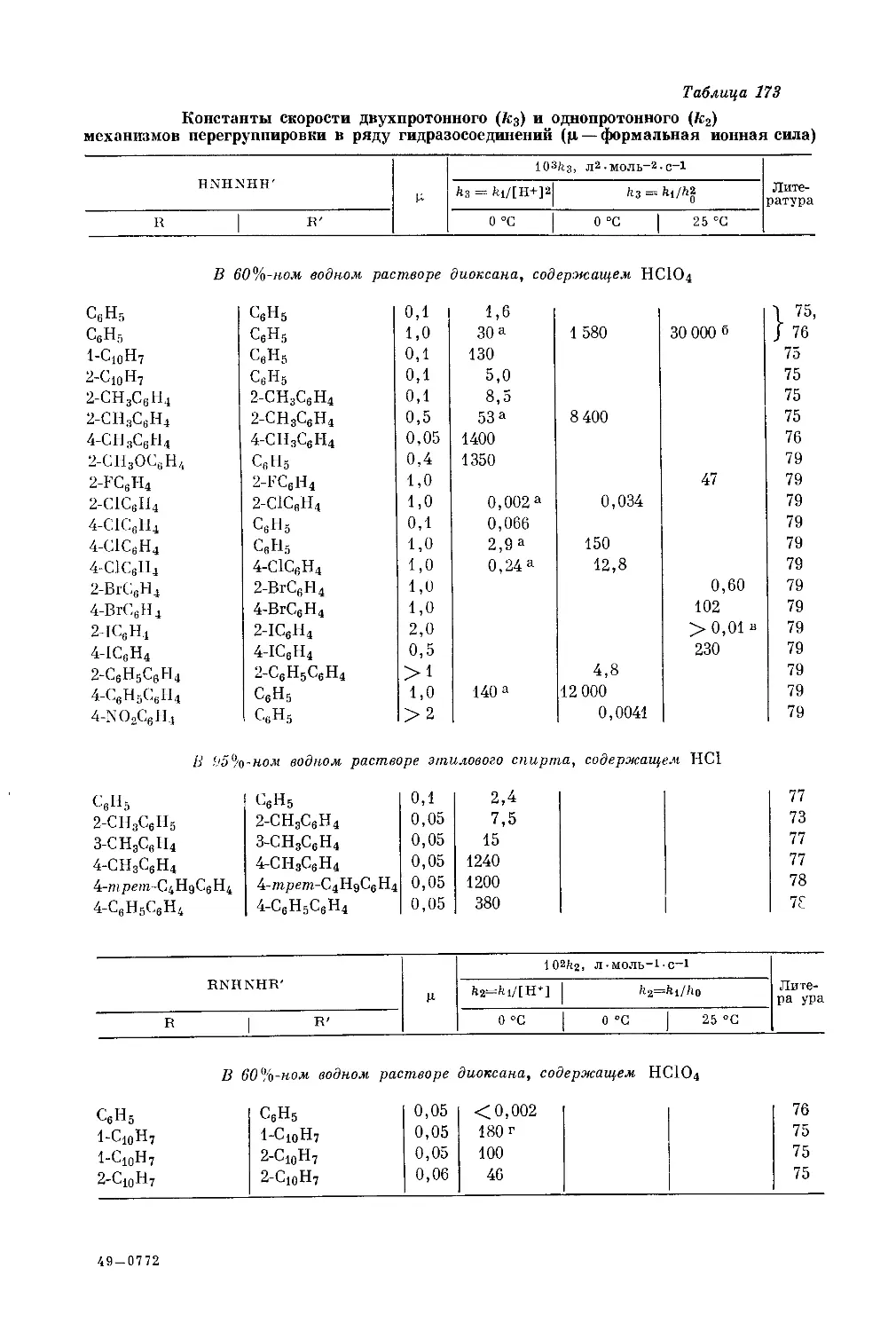
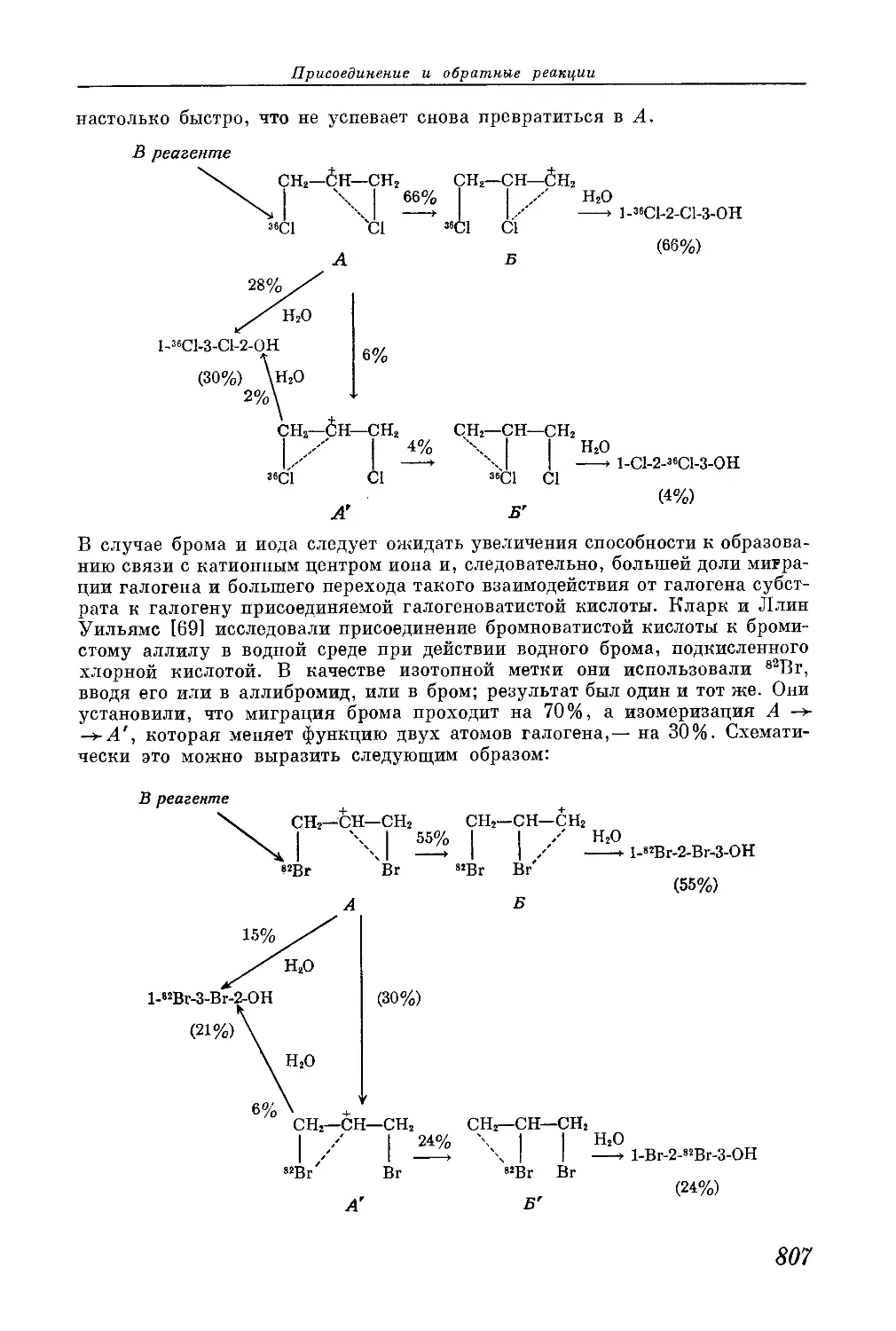
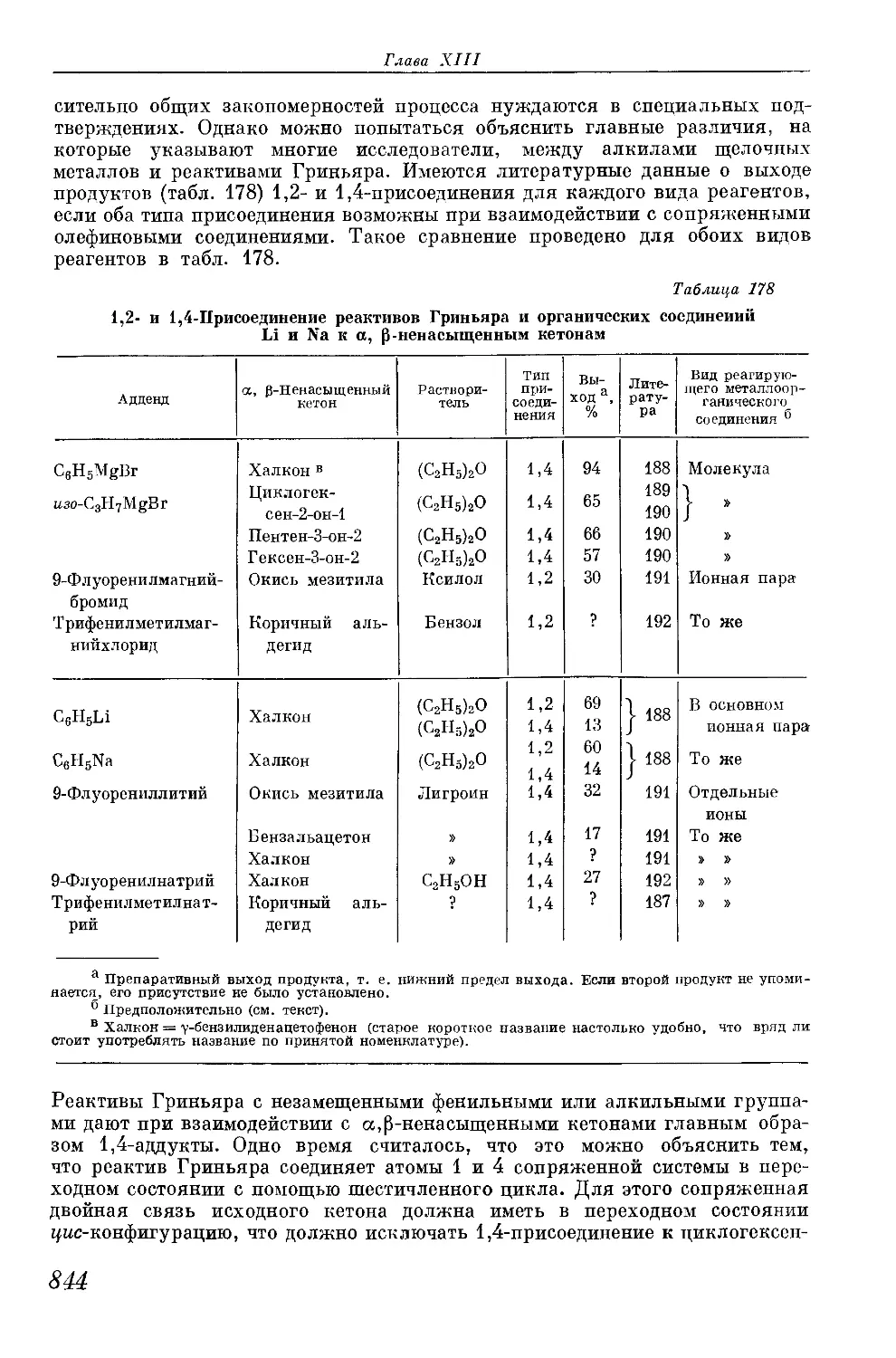
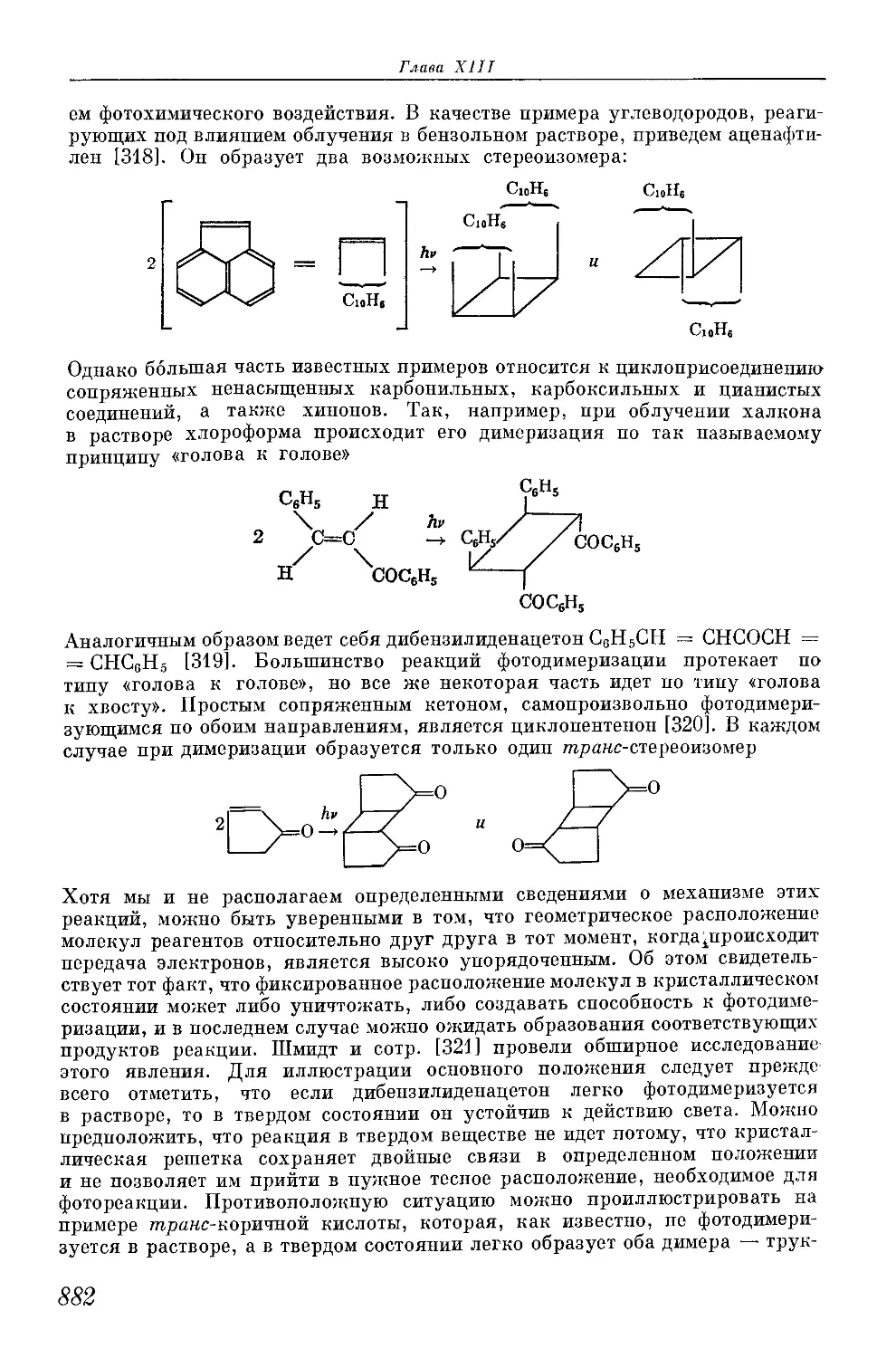
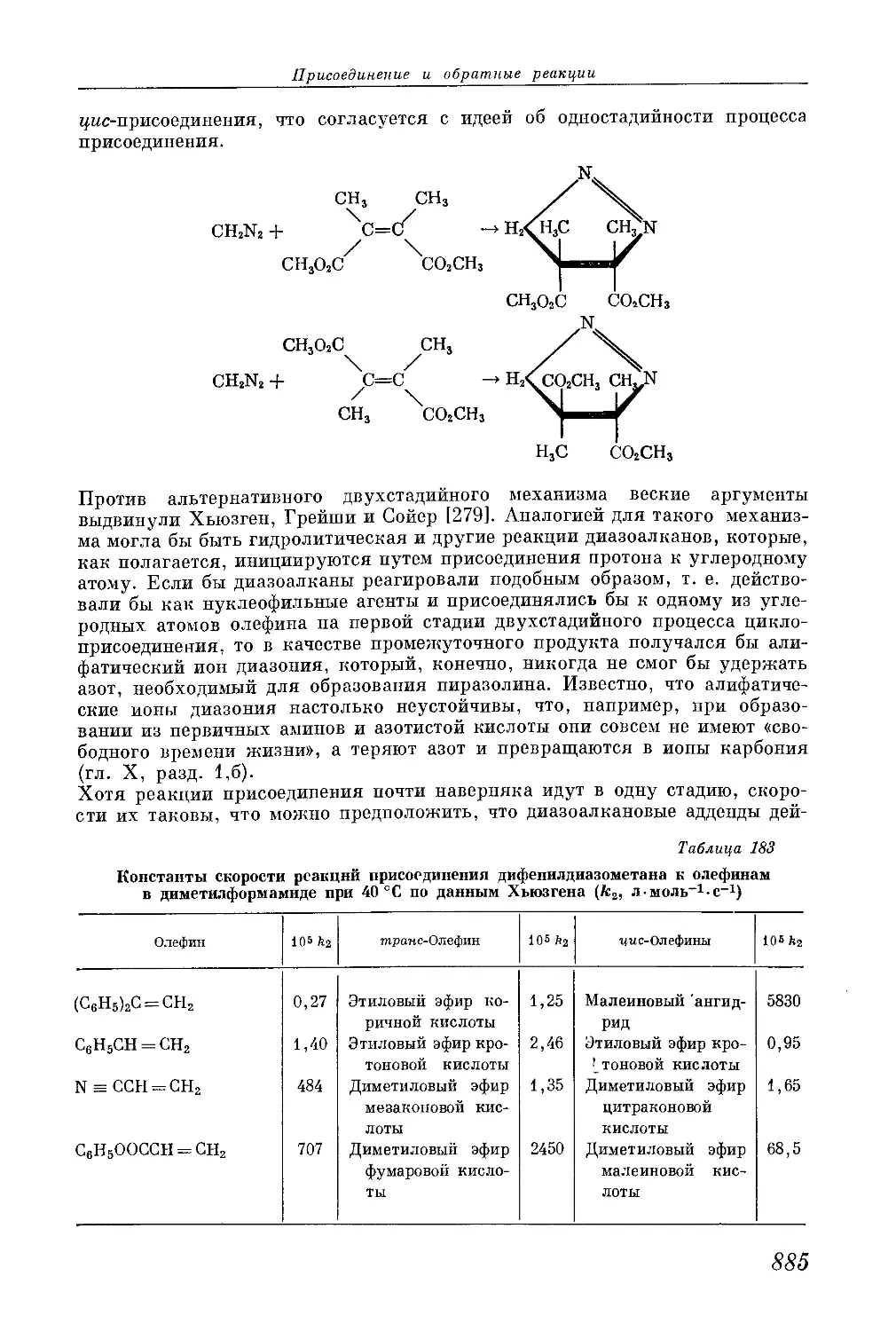
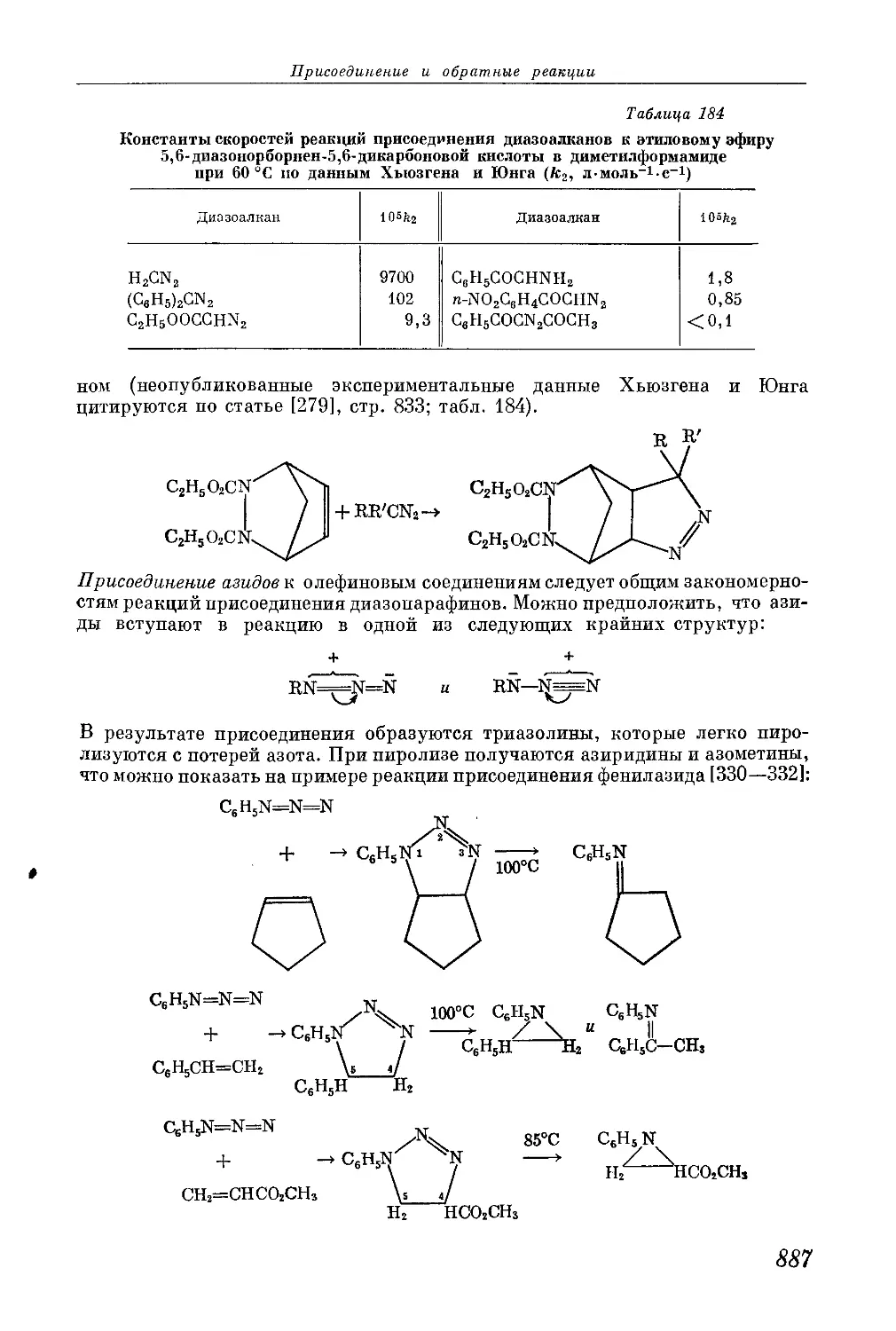
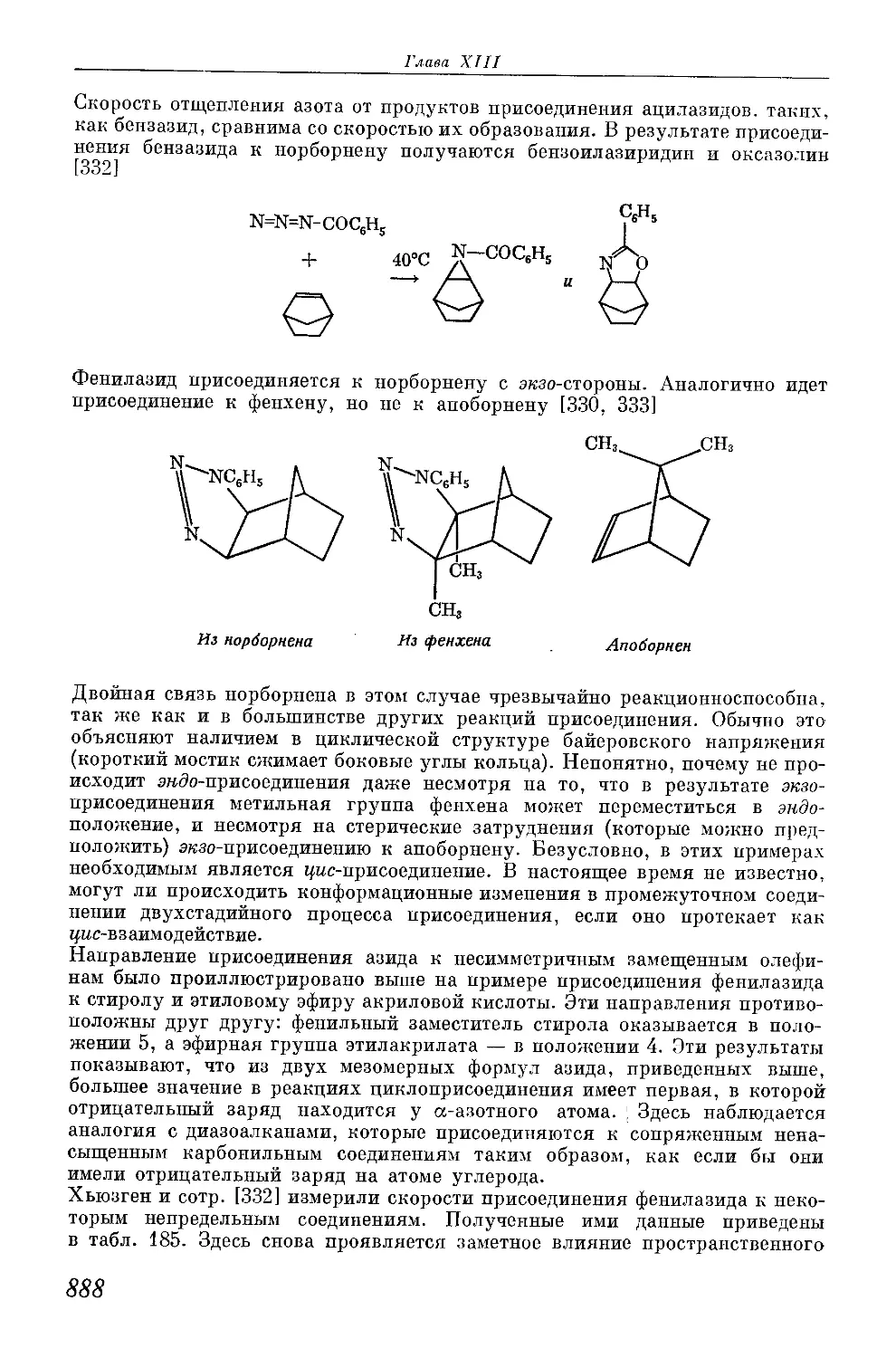
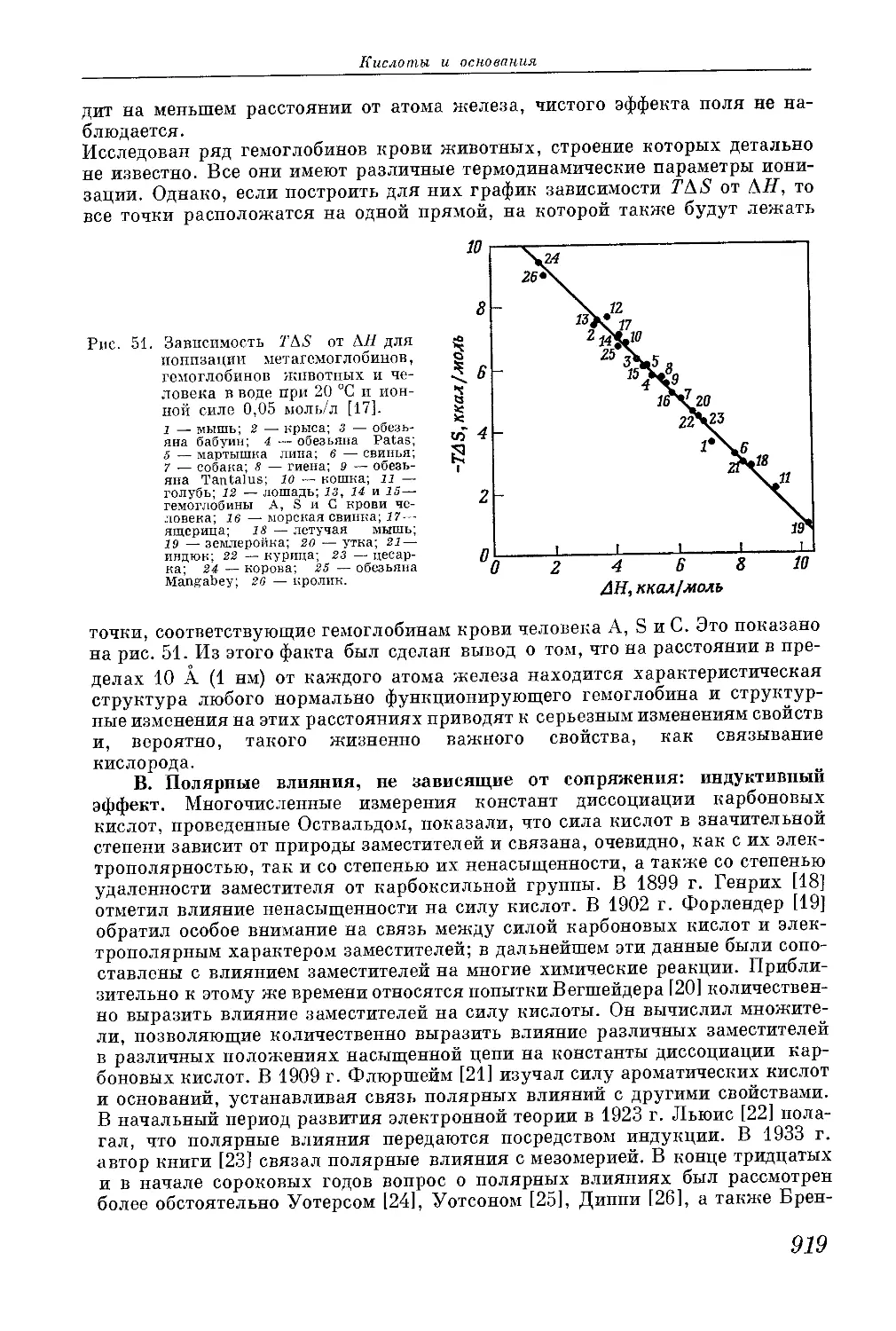
/






































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Author: Ингольд К.
Tags: классификация органических соединений элементоорганические соединения органическая химия
Year: 1973




Text
XUM'UU
STRUCTURE AND MECHANISM
IN ORGANIC CHEMISTRY
SECON D EDITION
By C.K.INGOLD
Professor of Chemistry
University College
University of London
CORNELL UNIVERSITY PRESS
ITHACA AND LONDON
19 6 9
к. ингольд
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
ОРГАНИЧЕСКОЙ ХИМИИ
перевод с английского
{андид ат а
химических наук
КП. БУ ТИНА
под редакцией
пр о ф ессора
И. П.БЕАЕЦКОЙ
ИЗДАТЕЛЬСТВО • МИР • МОСКВА -19 7 3
УДК 547.1
Автор книги К. Ингольд — ученый с мировым
именем, основатель физической органической хи-
мии — хорошо известен советским химикам. Издан-
ная в русском переводе его книга «Механизм реакций
и строение органических соединений» (ИЛ, 1959)
давно стала библиографической редкостью. Объем
второго издания этой фундаментальной и широко
известной монографии значительно расширен за счет
включения результатов исследований за последние
15 лет. В книге рассмотрены механизмы по существу
всех основных типов органических реакций, вклю-
чая радикальные, которые не были достаточно осве-
щены в первом издании.
РЕДАКЦИЯ ЛИТЕРАТУРЫ ПО ХИМИИ
тт В253 347
и 041(01)-7з" 73-72
ПРЕДИСЛОВИЕ
Среди многих книг по органической химии монография выдающегося
английского химика профессора Кристофера Ингольда занимает особое
место. В этой книге собраны и обобщены все достижения органической
химии в области изучения механизмов органических реакций и установления
связи между структурой органических соединений и их реакционной спо-
собностью, т. е. все то, что составляет теоретический фундамент современной
органической химии. Энциклопедичность и фундаментальность книги,
несомненно, связаны с тем, что ее автор был одним из главных создателей
той области науки, которая составляет предмет книги и которая получила
название «физическая органическая химия».
Характерной особенностью книги является глубокий анализ исторических
аспектов возникновения существующих взглядов на механизмы органиче-
ских реакций. При этом отдается должное всем исследователям, внесшим
существенный вклад в развитие теории органической химии.
Первое издание монографии Ингольда, написанное в 1953 г. и вышедшее
в русском переводе в 1959 г., сейчас является библиографической редкостью.
Между тем, по этой книге учились многие химики.
С момента выхода в свет первого издания в работу по выяснению механизмов
органических реакций включилось большое число химиков во всем мире,
что привело к накоплению огромного количества новых фундаментальных
данных, касающихся механизмов органических реакций. За это время были
развиты представления об участии ионных пар в реакциях замещения
и отщепления, был открыт ферроцен, что способствовало углублению взгля-
дов на природу ароматичности, были вскрыты закономерности термических
и фотохимических реакций электроциклизации (правила Вудварда — Гоф-
мана), был развит корреляционный анализ. В последние 10—15 лет большие
успехи были достигнуты в исследовании механизмов свободнорадикальных
реакций в растворе, начато изучение механизма электрофильного замещения
у насыщенного атома углерода и нуклеофильного замещения в ароматиче-
ском ряду. Наконец, значительный прогресс был достигнут в теории влияния
растворителя на скорость реакций, и диполярные апротонные растворители
стали широко применяться в химических лабораториях и в производствен-
ной практике. Кроме перечисленных важнейших достижений и открытий,
было решено множество других более частных, но трудных проблем, напри-
мер установлен механизм бензидиновой перегруппировки. Выросли в само-
стоятельные области химия карбониевых ионов и карбанионов, развита
химия карбенов, большое внимание в изучении механизмов реакций стало
уделяться промежуточно образующимся нестабильным частицам. Все эти
вопросы нашли отражение в книге Ингольда, поэтому по сравнению с первым
5
Предисловие
изданием объем книги значительно увеличился- Следует отметить, что автор
и его школа принимали участие в разработке многих из перечисленных
проблем, поэтому изложение характеризуется глубоким знанием насущных
задач органической химии и заботой о дальнейшем совершенствовании
теории механизмов органических реакций.
Первые главы книги, в которых излагаются основы теории строения молекул,
природа химической связи, электронные эффекты, физические свойства
молекул, представления об ароматичности и классификация реагентов
и реакций, принципиально не отличаются от первого издания. Последующие
главы, связанные с механизмами органических реакций, существенно изме-
нены и дополнены. Так, сильно расширена глава, посвященная замещению
в ароматическом ряду, в результате включения в нее реакций нуклеофиль-
ного и радикального замещения в бензольном ядре. Естественно, что основ-
ная часть этой главы посвящена электрофильному замещению в бензольном
кольце. Этот раздел также существенно расширен за счет новых данных,
полученных в 1953—1969 гг. В первом издании основные закономерности
в ароматическом ряду (природа электрофильного агента, механизм реакции,
правила ориентации) разбирались на примере реакции нитрования. Во вто-
ром издании эти вопросы оказалось более удобным разбирать на примере
галогенирования, поскольку большинство имеющихся в настоящее время
данных получено именно для этой реакции. Кроме классических реакций
электрофильного ароматического замещения, где уходящей группой является
протон, рассмотрена большая группа реакций протодеметаллирования
ароматических производных элементов IV группы АгЭА1к3 (Э = Si, Ge,
Sn, Pb).
При рассмотрении нуклеофильного замещения в ароматическом ряду глав-
ное внимание уделено различию в механизмах замещения (SN1, SN2, заме-
щение через дегидробензол). Что касается радикального замещения в аро-
матических соединениях, то такого раздела в первом издании вообще не было,
поскольку эта интереснейшая область химии бензола получила развитие
лишь в последние годы.
Столь же значительно изменено содержание главы, посвященной замещению
в алифатическом ряду. Эта глава, особенно первые два ее раздела, относя-
щиеся к реакциям нуклеофильного и электрофильного замещения, интересны
еще и тем, что существенный вклад в понимание природы указанных про-
цессов внес сам автор. Поэтому обзор по механизмам замещения у насыщен-
ного атома углерода читается с особым интересом. По сравнению с первым
изданием значительно расширены разделы, посвященные сольволитическим
реакциям и отличию между SN1- и 8к2-сольволизом, на более высоком
уровне обсуждаются температурные зависимости при нуклеофильном заме-
щении, энтропия и теплоемкость и их значение для идентификации моно-
и бимолекулярных механизмов сольволиза. Выделены в самостоятельные
разделы каталитические эффекты при нуклеофильном замещении (катализ
растворителем, кислотный катализ, катализ солями металлов) и влияние
6
Предисловие
солей с общим и необщим ионами на скорость SN1- и 8к2-реакций. В обсуж-
дение стереохимии нуклеофильного замещения включены алициклические
системы. Появился важный раздел по нуклеофильному замещению у оле-
финового атома углерода, где рассматриваются механизмы присоедине-
ния — отщепления и прямого замещения, а также пространственная направ-
ленность этих реакций.
Вторая часть главы посвящена электрофильному замещению у насыщенного
атома углерода — новому разделу химии, возникшему в последние 15 лет.
На примере реакций изотопного обмена ртути рассмотрены механизмы
SE2. SEi, SE1 и каталитический механизм SE1-2X~, где X- — анион гало-
гена.
Столь же новой является часть, относящаяся к реакциям гомолитического
замещения водорода в алканах. Газофазные реакции здесь сравниваются
с реакциями в растворах и приводятся доказательства цепного
механизма.
Новой в настоящем издании является глава «Замещение у гетероатомов»,
в которой рассмотрены механизмы электрофильного замещения у азота
и кислорода, а также механизмы нуклеофильного замещения у атомов крем-
ния, фосфора и серы. Особенно интересен раздел, посвященный диазоти-
рованию и связанным с ним процессам, написанный ярко, в великолепной
запоминающейся критической манере.
Глава «Отщепление с образованием олефинов, ацетиленов и карбенов»
значительно расширена по сравнению с первым изданием за счет новых
данных, касающихся 1,2-элиминировапия в газовой фазе и фрагментации
Гроба, а также важных с практической точки зрения реакций 1,1-элимини-
ровапия с образованием карбенов.
Существенно расширены главы, посвященные перегруппировкам, появились
отдельные разделы, связанные с реакциями электроциклизации и с валент-
ной таутомерией. В главе «Перегруппировки в ароматическом ряду» особый
интерес представляет раздел по механизму бензидиновой перегруппировки,
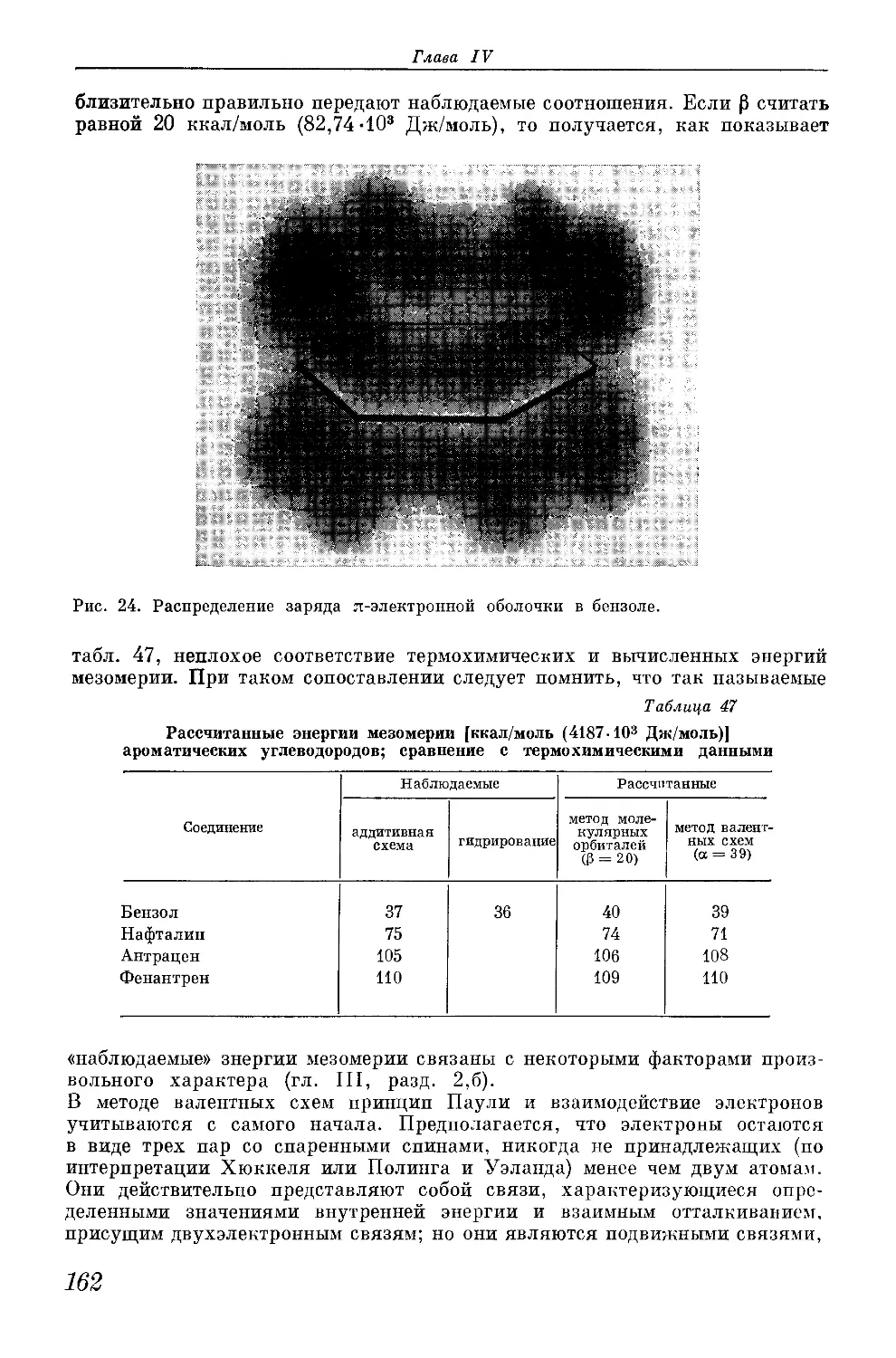
который был выяснен в последние годы главным образом благодаря работам
автора книги и его сотрудников. Автор убедительно доказывает внутримо-
лекулярный характер этой сложной перегруппировки, которая в зависимости
от природы заместителя в гидразобензоле, кислотности среды (однопротон-
ный и двупротонный катализ) и других условий реакции может проходить
через различные переходные состояния и в связи с этим давать смеси продук-
тов различного состава.
В главе «Реакции присоединения» существенно расширен раздел, посвященный
механизму реакции Дильса—Альдера, а в главу «Реакции карбоксилатной
и фосфатной групп» включен раздел по реакциям производных фосфорной
кислоты. Наконец, две последние главы —«Полярные влияния» и «Ста-
бильные радикалы»— вообще отсутствовали в первом издании. В первой
из них разобран принцип линейности свободной энергии и применение
pff-анализа для выяснения механизмов органических реакций, а во второй
1
Предисловие
дан обзор свойств стабильных органических радикалов и обсужден механизм
их стабилизации.
Следует отметить, что личность автора и его вкусы чувствуются на протя-
жении всей книги и это придает ей особый колорит; впрочем, иногда это
имеет и теневую сторону. Так, благодаря приверженности автора старым
представлениям при рассмотрении природы химической связи явное пред-
почтение отдается методу валентных схем и в тени остается метод молекуляр-
ных орбиталей, хотя заслуги этого метода несомненны, и в настоящее время
именно он является основным оружием квантовой химии.
Прогресс в органической химии в последние годы, связанный с широким
развитием физико-химических методов, столь велик, что практически невоз-
можно написать книгу, в которой весь материал сохранял бы новизну до
момента ее выхода в свет. Представление о механизмах реакции непрерывно
меняется и углубляется. Примером этого могут служить представления
об участии стадий одноэлектронного переноса во многих реакциях, которые
с классических позиций относятся к электрофильному и нуклеофильному
замещению- Огромную роль в развитии этих представлений сыграл метод
химически индуцируемой динамической поляризации ядер. Однако все то,
что сейчас волнует многих исследователей, занимающихся механизмами
органических реакций, появилось уже после написания и издания книги.
Тем не менее, эта книга, как ни одна другая монография по механизмами
органических реакций, является энциклопедией современных знаний
и, несомненно, по этой книге будут учиться многие поколения химиков.
И. Белецкая
ОГЛАВЛЕНИЕ
Предисловие автора ко второму изданию................ 10
I. Валентность и строение молекул..................... 11
II. Внутримолекулярное и межмолекулярное взаимодействия 37
III. Физические свойства молекул .............................101
IV. Ароматичность и связанные с ней физические свойства . . 155
V. Классификация реагентов и реакций..................201
VI. Замещение в ароматическом ряду.....................227
VII. Замещение в алифатическом ряду.....................351
VIII. Замещение у гетероатомов...........................507
IX. Отщепление с образованием олефинов, ацетиленов и кар-
бенов .........................................................537
X. Перегруппировки в насыщенных системах....................599
XI. Перегруппировки ненасыщенных соединений..................657
XII. Перегруппировки в ароматическом ряду.....................737
XIII. Присоединение и обратные реакции.........................783
XIV. Кислоты и основания......................................909
XV. Реакции карбоксильной и фосфатной групп..................935
XVI. Полярные эффекты ........................................987
XVII. Стабильные радикалы ....................................1017
Предметный указатель.............................. 1040
ПРЕДИСЛОВИЕ АВТОРА КО ВТОРОМУ ИЗДАНИЮ
Книга «Механизм реакций и строение органических соединений» была
подготовлена для первого издания в 1950—1951 гг., когда рассматриваемая
область химии уже сформировалась, а ее развитие только еще начиналось.
В настоящее время в этом направлении достигнуты большие успехи и поэто-
му назрела необходимость рассказать о них. В новом издании сохранено
примерно две трети первоначального текста, в который внесены измене-
ния, а остальное составляет новый материал.
В моей жизни было только три периода, когда я был относительно свободен
от повседневных обязанностей и мог интенсивно писать — писать с целью
помочь развитию органической химии. Первый период относится к 1932 г.,
когда, работая в Стэнфордском университете, я написал обзорную статью,
опубликованную в 1934 г. Вторая благоприятная возможность представилась
в 1950 г., когда я читал лекции в Корнелльском университете и подготовил
первое издание этой книги. В 1964 г. я получил приглашение стать членом
Национального научного общества Вандербилдского университета; это
позволило мне приступить к подготовке второго издания книги, работе над
которым способствовала спокойная обстановка и контакт с близкими по духу
людьми. Таким образом, тремя перерывами в работе, когда я мог спокойно
размышлять, я обязан Американской академии.
Я очень признателен за помощь, оказанную мне в этой работе. Несколько
очень полезных уроков по гомолитическим органическим реакциям пре-
подал мне мой сын; он дал также сотни ссылок на оригинальные источники
и проверил, а в некоторых местах сильно изменил мой первоначальный
вариант обзора по этой проблеме. Эти части книги могли бы выйти в соавтор-
стве с сыном. При написании других частей книги неоценимую помощь
оказали мне мои коллеги, особенно д-р Банторп, д-р Гофман, д-р Джонсон,
проф. Макколл, д-р Ридд, проф. Вернон и д-р Вассерман. Я особенно бла-
годарен моей жене, которая проверила многие ссылки, исправила множе-
ство мелких ошибок и прочитала большую часть корректуры. Она также
перепечатала на машинке мою неразборчивую рукопись.
К. К. Инголъд
ГЛАВА I
ВАЛЕНТНОСТЬ И СТРОЕНИЕ МОЛЕКУЛ
1. Развитие теории химического строения....................... 12
2. Электронная природа ковалентной связи ..................... 14
3. Физическая природа ковалентной связи......................... 20
4. Стереохимические представления ............................ 28
Список литературы ......................................... 36
1. Развитие теории химического строения
В 1808 г. Дальтон опубликовал свою атомистическую теорию, а в 1812 г-
Берцелиус предложил первую теорию химической связи. Эта так назы-
ваемая дуалистическая теория основывалась на изучении неорганических
веществ и рассматривала химическую связь как электростатическое притя-
жение противоположно заряженных атомов. Результаты работ Дюма и дру-
гих исследователей показали несовместимость дуалистической теории с на-
копившимся фактическим материалом органической химии, и к 1840 г.
от этой теории отказались. Значительно позже, в связи с выдвинутой Арре-
ниусом в 1887 г. теорией электролитической диссоциации, идея об электри-
ческой природе связи между атомами была возрождена, но уже только для
соединений, способных к ионизации.
В период главным образом между 1840 и 1860 гг. в работах Жерара, Лорана,
Канниццаро, Франкланда, Вильямсона, Кекуле и многих других исследо-
вателей был выяснен вопрос о численной характеристике валентности.
В значительной мере благодаря этим работам унитарная теория, пройдя
через промежуточные стадии — теорию радикалов и теорию типов, разви-
лась в структурную теорию. Авторы структурной теории не предпринимали
никаких попыток установить физическую природу сил, связывающих атомы.
Предполагалось, что эти силы являются неотъемлемым свойством самих
атомов и что число связей, образуемых атомом, т. е. его валентность, харак-
теризует данный вид атомов.
Первоначально возникшее представление о том, что химическая связь также
должна иметь и определенную пространственную направленность, не сразу
было принято как аксиома. Это представление получило признание благо-
даря работам Вант-Гоффа и Ле Беля, которые в 1874 г. установили опре-
деленное пространственное расположение связей атома углерода.
В конце прошлого столетия один и тот же термин «валентность» применяли
для обозначения как заряда элемента в ионной форме, так и числа связей,
которые атом образует с другими атомами в молекуле. То обстоятельство,
что эти числа часто совпадают, способствовало такому двоякому употребле-
нию термина «валентность». Возникла значительная путаница; наличие
структурных связей часто постулировали там, где их не было. В этот период
Вернер впервые указал на четкое различие между электрически нейтраль-
ными совокупностями кинетически разделимых ионов и кинетически инди-
видуальными молекулярными или ионными единицами и, следовательно,
между числом, выражающим заряд иона, и координационным числом атома
(по терминологии Вернера), т. е. числом атомов, структурно связанных
с данным атомом в молекуле.
Уже давно внимание исследователей привлекали закономерности в значе-
ниях валентных чисел элементов. Менделеев, сформулировавший в 1869 г.
свой периодический закон, показал, что валентность атома тесно связана
с номером его группы в периодической системе и обычно изменяется на еди-
ницу при переходе от одной группы к следующей. После того как Томсоном
и Вихертом в 1897 г. был открыт электрон, делались неоднократные попытки
выразить с помощью электронных представлений связь между валентностью
атома и группой периодической системы, к которой он принадлежит. Так,
Абегг приписал каждому элементу положительную валентность (равную
номеру группы) и отрицательную, причем сумма обеих (без учета знаков)
всегда равна восьми; наименьшее значение валентности независимо от знака
он назвал нормальной валентностью элемента. Абегг [1] считал, что любой
атом имеет восемь мест для электронов, а значение положительной валент-
ности, соответствующее номеру группы периодической системы, указывает
на число таких мест, действительно занятых в нейтральном атоме. По мнению
12
Валентность и строение молекул
Друде [2], нормальная положительная валентность по Абеггу соответствует
числу электронов, легко отдаваемых атомом, а отрицательная валентность —
числу легко присоединяемых электронов. В более ясной форме октетная
теория едва ли могла появиться ранее 1913 г., т. е. до того, как благодаря
работам Фаянса, Содди и особенно Мозли стало известно количество элек-
тронов в атомах.
Уже в 1911 г. Резерфорд предложил ядерную теорию атома. В 1916 г. были
опубликованы знаменитые статьи Косселя [3] и Льюиса [4]. По представле-
ниям этих авторов, электроны в атомах образуют концентрические оболочки:
первая из них содержит два электрона (дублет), вторая и третья — по
восьми. Количество электронов в более высоких оболочках не столь постоян-
но. однако последняя оболочка в атомах инертных газов всегда содержит
восемь электронов (октет). Впоследствии было доказано, что эти положения
Косселя и Льюиса являются верными, хотя они были высказаны еще до
открытия правил квантования. Предположения, выдвигаемые теорией Кос-
селя о пространственном расположении электронов, отличаются от представ-
лений Льюиса, но это различие несущественно. Общим для них, что весьма
важно, является утверждение о наибольшей степени устойчивости и запол-
ненности электронных оболочек в атомах инертных газов, а именно: для
гелия — двухэлектронная оболочка, для неона — двух- и восьмиэлектрон-
ная оболочка и т. д. Для атомов других элементов, имеющих больше или
меньше электронов, чем атом инертных газов, характерно стремление
к отдаче или присоединению электронов с образованием электронной струк-
туры инертного газа. Таким образом, можно было объяснить образование
многих устойчивых ионов, например ионов калия, кальция, сульфид- и хло-
рид-ионов и др.
Льюис сделал принципиально важный шаг вперед, указав на обобществле-
ние электронов как на второй способ образования устойчивых электронных
группировок. Таким образом, Льюис ввел в химию электронную интерпре-
тацию структурной связи. Льюис предположил, что химическая связь осу-
ществляется за счет электронной пары, принадлежащей одновременно
двум атомам и дополняющей электронную оболочку каждого из них. Бла-
годаря обобществлению электроны используются более экономно, так что
свободные атомы с недостаточным количеством электронов могут заполнить
свои оболочки, образуя связь. Связь насыщает единицы сродства обоих
атомов (каждой единице сродства соответствует один электрон). Таким обра-
зом, связь осуществляется двумя электронами, которые образуют стабиль-
ную группировку. Льюис считал такую электронную пару, или дублет,
наиболее важной электронной группировкой и полагал, что октеты валент-
ных электронов состоят из четырех дублетов независимо от того, все ли
электроны обобществлены или нет. Эта идея также оказалась правильной,
хотя опа постулирована за десять лет до открытия спина электрона и прин-
ципа Паули.
Связь, образованная двумя обобществленными электронами, представляет
собой наиболее важный тип связи; она определяет строение молекулы
и является обычной связью классической структурной химии. Ленгмюр
назвал ее ковалентной связью. Известна также более слабая связь, осуще-
ствляемая при обобществлении только одного электрона. Процесс обобще-
ствления электронов обычно называют ковалентным связыванием. Если
степень обобществления электронов двумя связанными атомами одинакова,
то такую связь называют гомеополярной связью.
Электростатическое притяжение между ионами было названо Ленгмюром
электровалентностью. Хотя такое взаимодействие весьма сильно, его очень
13
Глава I
часто бывает недостаточно для связывания разноименных ионов в растворе
вследствие существования столь же сильного электростатического притяже-
ния между ионами и растворителем. Как правило, электростатическое
притяжение может возникать между противоположно заряженными ионами,
между ионами и постоянными или наведенными диполями и между двумя
диполями. Электростатическое взаимодействие является одним из главных
факторов, определяющих строение молекулы. Ковалентная связь часто
включает в себя некоторую долю такой электростатической связи.
2. Электронная природа ковалентной связи [5]
а. Связь между атомами. Льюис предположил, что при образовании
молекул из двух атомов водорода, из атома водорода и атома фтора или
из двух атомов фтора каждый атом, стремясь дополнить свою валентную
оболочку, приобретает по одному электрону в результате обобществления
электронной пары, осуществляющей связь; при этом у атома водорода будет
заполняться электронная оболочка до дублета, а у атома фтора — до октета
электронов. Другие атомы, по предположению Льюиса, могут также соеди-
няться аналогичным образом с заполнением их устойчивых валентных
оболочек; так, например, углерод может образовывать связи с четырьмя
атомами водорода или фтора. Свои представления Льюис выражал форму-
лами, в которых валентные электроны изображались точками, а общепри-
нятые символы элементов были использованы для обозначения не нейтраль-
ных атомов, а положительно заряженных остатков атомов, т. е. ядер вместе-
со всеми заполненными внутренними электронными оболочками.
Н- + Н- -> Н:Н
:F- + :F- -> :F:F:
Н- + :F- -» H:F:
H
•С- + 4Н- -» Н:С:Н
Н
:F:
•С- + 4:F- -> :F:C:F:
:F:
Процесс образования ковалентной связи путем обобществления двух элек-
тронов по одному от каждого атома мы будем называть коллигацией. Колли-
гацию следует отличать от другого способа образования двухэлектронной
связи, называемого координацией, который будет рассмотрен в следующем
разделе. Необходимо подчеркнуть, что различие состоит лишь в путях
образования связей, а не в природе самих связей.
В слове «коллигация» приставка «ко», как и в слове «ковалентность», озна-
чает качественное сходство в поведении соединяющихся друг с другом атомов
в процессе образования связи. Иначе говоря, термин «коллигация» отно-
и
Валентность и строение молекул
сится к такому взаимодействию между атомами, когда каждый из атомов
выступает в роли «лиганда» по отношению к другому атому *.
Читая справа налево уравнения, аналогичные вышеприведенным, можно
составить представление об одном из способов разрыва ковалентной связи.
При этом способе каждый из атомов удерживает по одному электрону из ранее
обобщенной пары. Такой тип разрыва связей называется гомолитическим.
б. Координация. Приведенные выше формулы по числу и характеру
связей вполне соответствуют формулам классической структурной теории.
Однако, как подчеркивал Лыоис, это соответствие не носит общего характера;
теория двухэлектронной связи и валентного октета приводит к ряду струк-
тур, в значительной степени отличающихся от ранее принятых. Более того,
новые формулы не содержат некоторых недостатков прежних формул, в том
числе и тех, на которые указал в свое время Вернер [6]. Электронная теория
включает теорию Вернера, объясняет ее и в то же время идет дальше, отка-
зываясь от оставшихся в теории Вернера нереальных различий в некоторых
типах связей. Так, Вернер, рассматривая образование и строение аммоние-
вых солей и борфторидов, считал, что атом азота в аммиаке использует свои
три главные валентности, а для удержания четвертого атома водорода в соли
аммония — побочную валентность, отличающуюся от главной; аналогично
описывал Вернер и образование борфторидов. Согласно Льюису, не суще-
ствует никаких различий между связями, удерживающими четыре атома
водорода или фтора в указанных соединениях. Образование ионов аммония
и борфторида Льюис изображает следующим образом:
Н
Н++
ii
:F:
:F:B+ :F:-->
":F:
H:N:H
ii
:F:
:F:B:F:
":F:”
Процесс образования связи с использованием побочной валентности Вернер
называл координацией. Согласно электронным представлениям, этот про-
цесс, очевидно, является другим общим способом образования двухэлек-
тронной связи, а именно таким, при котором один из атомов, имеющий в своей
валентной оболочке место для двух дополнительных электронов, присоеди-
няет веподеленную электронную пару другого атома. Все реакции, которыми
Вернер иллюстрировал связывание с помощью побочных валентностей,
укладываются в рамки электронных представлений. В настоящее время
не усматривают никакой разницы между двумя различными (по Вернеру)
видами валентности; даже связи, которые, как считал Вернер, образованы
за счет главных валентностей, можно получить координацией. Так, молекула
водорода может образоваться не только из двух атомов водорода, но и из про-
тона и гидрид-иона; аналогичным образом молекула фтористого водорода
* Термины «лиганд», «лигапдность», «четырехлигандный» и т. д. в настоящее время
часто используются вместо более старого слова «валентность», «четырехвалептный» и др.
Эти термины отражают только то, какие атомы и группы связаны друг с другом, но не дают
сведений о природе рассматриваемых связей.
15
Глава I
может получаться (и обычно получается) из протона и фторид-иона
Н+ + Н:-->Н:Н
Н++ :F:~-»H:F:
*• *•
По Сиджвику, все такие процессы образования связи обычно объединяются
под названием «координация». Несмотря на то что с точки зрения этимологии
это слово не совсем точно отражает существо предмета, по исторически
сложившимся обстоятельствам термин Вернера, вероятно, не следует
заменять.
Коллигация и координация представляют собой два различных пути обра-
зования одной и той же связи, структура которой не зависит от механизма
образования. Некоторые авторы для описания связей используют различные
обозначения в зависимости от того, каким образом они понимают механизм
образования этих связей; однако это может привести к неправильному пред-
ставлению о строении продукта реакции, которое не зависит от пути проте-
кания реакции.
Читая справа налево последние четыре уравнения, можно представить
себе второй способ разрыва ковалентных связей; при этом оба электрона,
образующих связь, удерживаются у одного из разделяющихся атомов.
Такой тип разрыва связей называется гетер олитическим.
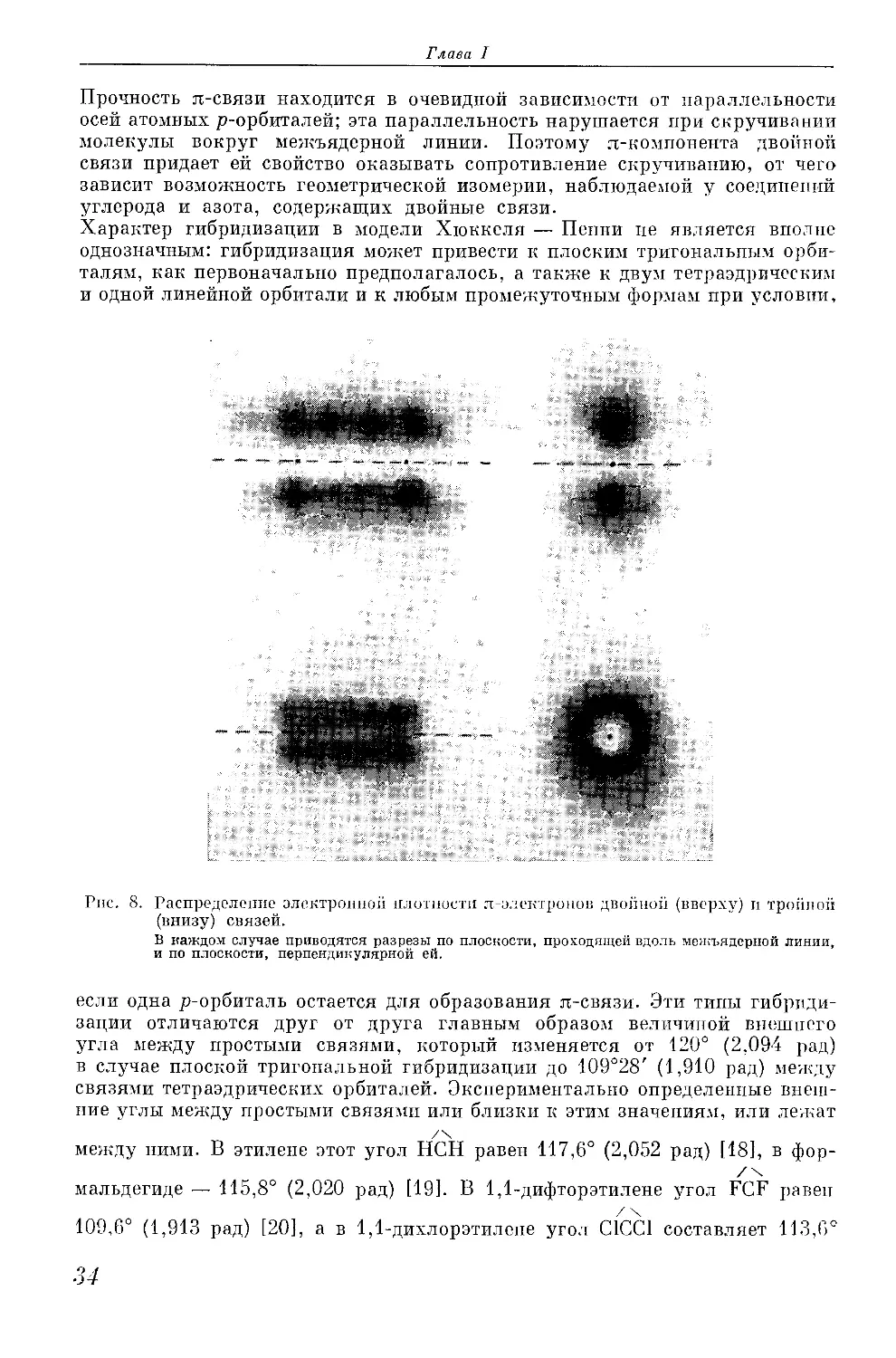
в. Кратные связи. Электронная интерпретация некоторых двойных
и тройных связей классических структурных формул очень проста. Двойная
связь в этилене и карбонильных соединениях представляется в виде двух,
а тройная связь в ацетилене и цианидах — в виде трех обобществленных
электронных пар
Н. ,Н Н. .
.С::С.‘ :С::О; Н:С:::С:Н H:C:::N:
н- -н И- •
г. Диполярные связи. Два примера могут иллюстрировать характер
этих связей. Рассмотрим сначала соединение аммиака с триметилбором,
ранее считавшееся «молекулярным» соединением. Вернер считал, что атомы
азота и бора соединены в этом случае побочной валентностью, и приписывал
этому соединению формулу H3N • • •В(СН3)3. Согласно электронной теории,
эта связь является ковалентной, а процесс образования соединений —
процессом координации
HR HR
H:N: + B:R-»H:N:B:R
HR HR
Однако электронная формула показывает, что атом азота находится в четы-
рехковалентном состоянии, характерном для иона аммония; таким образом,
по отношению к атому азота это соединение является замещенным ионом
аммония. С другой стороны, атом бора также находится в четырехковалент-
ном состоянии, как, например, в ионе борфторида; это соединение можно
рассматривать как замещенный борат-ион. Для того чтобы подчеркнуть оба
эти обстоятельства, можно написать в структурной формуле над символами
азота и бора знаки соответствующих зарядов и рассматривать это соединение
+ —
как биполярный ион H3NBR3.
16
Валентность и строение молекул
В качестве другого примера можно привести окись триметиламина, формулу
которой ранее изображали с двойной связью между атомами азота и кисло-
рода (CH3)3N = О. Однако электронная теория рассматривает эту связь
как простую ковалентную связь. Она может образоваться посредством коор-
динации между молекулой триметиламина и атомом кислорода
R R
R:N: + О: -+R:N:O:
R R '
В этом соединении атом азота связан так же, как в ионе аммония. Атом
кислорода находится в одноковалентном состоянии, типичном для иона гид-
роксила, фенолят-иона и других алкоголят-ионов. В этом случае также
можно написать знаки зарядов над атомами азота и кислорода и рассматри-
+ —
вать окись амина как биполярный ион R3NO.
Связь такого типа, т. е. простая ковалентная связь между атомами, обра-
зующими формальный диполь, может быть названа диполярной связью.
Согласно теории обобщенного дублета и валентного октета, такие связи
имеются во многих простых неорганических соединениях, главным образом
в кислородных кислотах, их галогепангидридах, ангидридах и других
производных. По одной такой связи содержится в нитрогруппе, азокси-
группе, сульфоксидах и по две связи — в сульфонах. Существуют так назы-
ваемые диполярные двойные связи, т. е. ковалентные двойные связи между
атомами, образующими формальный диполь, например в диазогруппе
и в азидной группе *.
+ + ••- -о-
R:N:O: R:N:O: p/atn?
.... R:S:O:-
:O: :N:R R ++j^ "
^C::N::N: R:N::N::N:
R-
По мере развития физической теории химической связи менялись и пред-
ставления, лежащие в основе приведенных структур; однако совершенство-
вание теории протекало главным образом в направлении нового толкования
структур, которые остаются правильным исходным пунктом для дальней-
шего развития теории.
д. Полярность связей. Полярность характеризует строение молекул
более точно, чем это вытекало бы только из наличия формальных зарядов.
Рассмотрим снова ковалентную связь между формально не заряженными
атомами. Как предусмотрительно подчеркивал Лыоис, гипотеза обобще-
ствления электронных пар не предполагает, что обобществленные электроны
совершенно в равной мере принадлежат обоим связанным атомам. Очевидно,
что в молекулах водорода или фтора обобществленная электронная пара
в равной степени принадлежит обоим атомам. Но в молекуле фтористого
водорода обобществленные электроны принадлежат атому фтора в большей
* Связь, названная диполярной связью, раньше обозначалась как семиполярная двойная
пли координационная связь. Термин «биполярная связь» является предпочтительным,
так как этот тип связи в некоторых соединениях никогда не рассматривался как двойная
связь, а координация далеко не во всех случаях сопряжена с возникновением зарядов.
По мнению Льюиса, пет необходимости вводить особый термин для рассматриваемого
типа связей; однако, с пашей точки зрения, трудно обойтись без специального термина.
2-0772
17
Глава I
степени, чем атому водорода. Следовательно, молекула фтористого водорода
должна обладать дипольным моментом (с водородом в качестве положи-
тельного и фтором в качестве отрицательного концов диполя). Такая элек-
трическая асимметрия должна снижать величину энергии, необходимую
для расщепления этой молекулы на ионы водорода и фтора.
Более подробно рассмотрим этот вопрос на примере следующего изоэлек-
тронного ряда'.
Н И И
Н:С:Н :N:H :О:Н :F:H :Ne:
Н Й
Молекулы этого ряда характеризуются одинаковым суммарным ядерным
зарядом, равным десяти, в поле которого находятся все электроны каждой
молекулы. Однако если рассматривать этот ряд молекул слева направо,
то можно видеть, что положительные заряды последовательно перемещаются
от периферии молекулы к ее центру, откуда они должны сильнее воздейство-
вать на октет валентных электронов. Следовательно, отдельная обобще-
ствленная электронная пара, например связывающая водород справа от
центрального атома в первых четырех формулах, будет в возрастающей
степени больше принадлежать центральному атому и меньше атому водорода
в ряду CH4, NH3, Н2О, FH. При этом атом водорода становится все более
положительным, что должно отразиться на таких физических свойствах,
как дипольные моменты, и на таких химических реакциях, как, например,
диссоциация на ионы кислот. Если рассматриваемая электронная пара свя-
зывает не атом водорода, а другой, общий для данного ряда соединений
атом, например углерод, то в таком ряду она должна постепенно удаляться
от атома углерода. В этом случае также должны наблюдаться изменения
физических и химических свойств, которые будут рассмотрены ниже.
С помощью вышеприведенного изоэлектронного ряда можно также показать,
что неподеленная электронная пара, изображенная левее символов централь-
ных атомов в последних четырех формулах, должна все более прочно удер-
живаться центральным атомом в ряду NH3, Н2О, HF, Ne. Этот эффект также
скажется на физических свойствах, например на поляризуемости, и хими-
ческих свойствах, например на основности.
На протяжении всей истории развития химической теории строения молекул
термины «электроотрицательный» и «электроположительный» применялись
по отношению к атомам и атомным группам. Согласно взглядам, ведущим
начало от Берцелиуса, атомы и группы атомов считались электроотрица-
тельными, если введение их в какое-либо соединение придавало ему кислот-
ные свойства или усиливало эти свойства, и электроположительными, если
их действие оказывалось в противоположном направлении. Например, атом-
ные группы —СН3, —NH2, —ОН, —F или, что то же, центральные атомы
этих групп расположены в порядке возрастания электроотрицательности.
Из предыдущего ясно, что электроотрицательность характеризует спо-
собность атома притягивать электроны от связанных с ним атомов, т. е.
характеризует напряженность положительного электрического поля данного
атома. Наоборот, электроположительность отражает способность атома
отталкивать электроны и свидетельствует о преобладании отрицательного
электрического поля.
Следует ожидать, что электроотрицательность в общем случае будет увели-
чиваться по мере перемещения положения атома (или центрального атома
группы) вправо в таблице Менделеева, т. е. при увеличении атомного номера.
18
Валентность и строение молекул
Это положение соблюдается только при сравнении атомов, являющихся
формально нейтральными, или по крайней мере атомов, обладающих одина-
ковыми формальными зарядами. Следует подчеркнуть, что атомы и группы,
обладающие положительным ионным зарядом, уже благодаря этому факту
должны быть электроотрицательными. Они принадлежат к числу наиболее
электроотрицательных групп. Так, группа —NH* является значительно
более электроотрицательной, чем — NH2 или даже чем любая из групп ряда
— СН3, —NH2, — ОН, —F. Аналогично атомы и группы, несущие отрица-
тельный ионный заряд, являются электроположительными. Так, группа
—О- значительно более электроположительна, чем —ОН или любая ней-
тральная группа.
е. Водородная связь. Атом водорода имеет значительно меньшие раз-
меры, чем другие атомы, и поэтому его ядро может особенно близко под-
ходить к неподеленным электронным парам других атомов. Вследствие этого
свойства связанного водорода в значительной степени определяются элек-
тростатическим притяжением. Этим обусловлены два очень важных
эффекта.
Первый заключается в большой протонной подвижности водорода, свя-
занного с электроотрицательными атомами, имеющими неподеленные элек-
тронные пары, особенно с атомами, принадлежащими к группам азота, кис-
лорода и фтора. Связанный с такими атомами протон очень мало экраниро-
ван, благодаря чему к нему может близко подойти другой атом, обладающий
активной неподеленной электронной парой; энергия возникающего при
этом электростатического притяжения оказывается соизмеримой с энергией
прежней связи. Таким образом, лишь очень небольшой энергетический
барьер препятствует переходу протона от одного электроотрицательного
атома к другому, например, в следующей реакции:
:F:H+ :F:-+ H:F:
Именно вследствие этого не наблюдается изомерии, связанной с положением
протона относительно электроотрицательных атомов, например, для неорга-
нических кислородных кислот.
Второй эффект состоит в том, что при сближении двух молекул до расстоя-
ния, допускающего легкий переход протона, последний притягивается
к обоим атомам силами той или иной природы; при этом связь может пере-
мещаться и даже неоднократно от одного атома к другому. Однако силы,
действующие в обоих направлениях, остаются, в результате чего все три
атома будут удерживаться вместе. Такая форма связывания двух атомов
«через водород» называется водородной связью. Можно упомянуть три типа
явлений, обусловленных водородными связями: 1. Водородная связь может
оказаться достаточно прочной, чтобы обеспечить длительное существование
комплексов с водородной связью как кинетически независимых частиц
в растворе. Таким кинетически устойчивым образованием является, напри-
мер, ион дифторида водорода (FHF)~. 2. Более слабые водородные связи
отчетливо проявляются в ассоциированных системах, где они могут образо-
вываться с такой частотой, что, несмотря на непродолжительность их суще-
ствования, общее количество таких связей всегда велико. Этим объясняется
ассоциация молекул, вызывающая уменьшение летучести, увеличение вяз-
кости и изменение других физических свойств, которое наблюдается во мно-
гих чистых жидкостях, например в аммиаке, воде, фтористом водороде,
первичных и вторичных аминах, спиртах, фенолах, минеральных и орга-
2* 19
Глава I
нических кислотах. 3. Известны случаи устойчивых внутримолекулярных
водородных связей, если структура самой молекулы способствует образо-
ванию таких связей. Внутримолекулярные водородные связи также оказы-
вают влияние на физические свойства, подавляя образование межмолеку-
лярных водородных связей. Вследствие этого увеличивается летучесть,
а вязкость уменьшается. Они оказывают влияние также и на химические
свойства веществ, изменяя обычные функциональные свойства групп, напри-
мер способность к кислотной ионизации. Одним из многих известных при-
меров является значительно большая летучесть салицилового альдегида
по сравнению с «-оксибензальдегидом.
3. Физическая природа ковалентной связи
Льюис установил материальную основу ковалентной связи. Однако он не мог
вскрыть характер действующих сил, так как их природа не описывается
классической физикой — открытие этих сил явилось одним из достижений
квантовой механики.
а. Квантовая механика [7]. Понятие кванта действия h было введено
в 1900 г. Планком. В 1905 г. Эйнштейн высказал предположение о двой-
ственной корпускулярно-волновой природе излучения. В 1913 г. Бор успеш-
но использовал законы квантования для создания первой теории строения
атома. В 1923 г. де Бройль применил идею корпускулярно-волнового дуа-
лизма к электрону. В 1925 г. Гейзенберг и Шредингер одновременно, но
в различной форме сформулировали основы квантовой механики. Метод
Шредингера более непосредственно отражает двойственную природу
частиц.
В этом методе поведение элементарной частицы, в частности электрона,
описывается функцией пространственных координат и времени (обычно
обозначаемой Ч7), подвергнутой действию оператора, связанного с кинети-
ческой и потенциальной энергиями. Оператор придает Ч7 свойства волны
с частотой v, которая связана с энергией Е уравнением Планка Е = hv;
в то же время | Ч7 |2 является вероятностной функцией распределения части-
цы. Если рассматривается несколько электронов, то волновая функция Ч7
является функцией координат всех этих электронов и времени.
С помощью квантовой механики нельзя рассчитать точных траекторий частиц,
но можно вычислить вероятности их положений и импульсов. Это обстоя-
тельство имеет фундаментальное значение, потому что, как показал Гей-
зенберг, невозможно точно определить и положение и импульс частицы
одновременно. Выдвинутый Гейзенбергом принцип неопределенности мате-
матически выражается соотношением, согласно которому произведение
пределов неопределенности координат и соответствующих импульсов не
может быть меньше постоянной Планка h. Этим свойством обладает любая
пара динамических переменных, произведение которых имеет размерность
действия (энергия и время образуют другую такую пару): чем точнее опре-
деляется один из множителей, тем более неопределенным становится другой.
Именно в этом и заключается сущность кванта действия.
б. Атомные состояния и орбитали [8]. При возбуждении механиче-
ского колебательного движения в какой-либо замкнутой системе возникают
стоячие волны, имеющие строго определенные формы и частоты, зависящие
от характеристики системы. Аналогично этому Ч7-функция электронов,
ограниченных потенциалом притяжения атомных ядер, может принять
форму стоячих волн с определенными частотами. Именно этими волновыми
20
Валентность и строение молекул
свойствами и пользовался Шредингер при описании стационарных состоя-
ний атомов.
При рассмотрении атомных состояний время считается неопределенной
величиной, и поэтому величины энергии могут быть точно вычислены. Таким
образом, концепция состояния предполагает, что электроны движутся
вокруг ядра по неопределенным траекториям, но с вычисляемой вероятно-
стью распределения их в пространстве. Для многих целей это распределение
можно считать распределением усредненной плотности электронного заряда.
Поэтому его называют распределением электронной плотности.
Основной проблемой квантовой механики атомных состояний является
проблема стационарных состояний атома водорода. Опа была полностью
разрешена. Набор стационарных состояний определяется орбитальными
квантовыми числами п, I и тр закономерности в численных значениях
последних видны из данных табл. 1. От различных значений этих чисел
Таблица 1
Закономерность изменений квантовых чисел
(приводится для значений п, равных 1, 2 п 3)
п 1 2 3
1 0 0 1 0 1 2
mi 0 0 —1 0 4-1 0 — 1 0 4-1 -2 —1 0 +1 42
Символ 1s 2s 2р 3s 3/? 3d
зависят симметрия и ориентация волновых функций V и функций элек-
тронной плотности | Y | 2, а также формы узловых поверхностей * этих
функций.
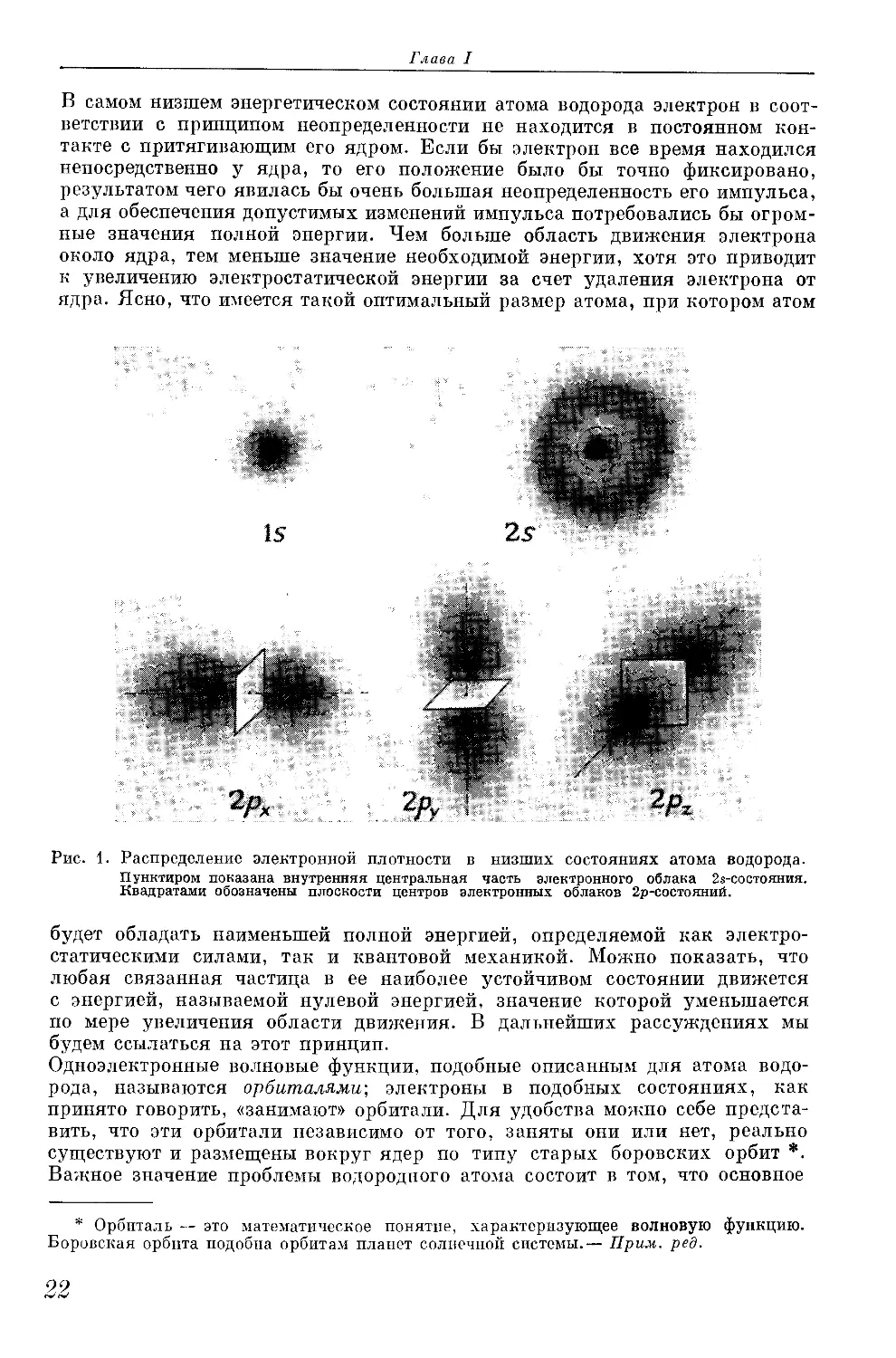
На рис. 1 показано распределение электронной плотности для первого
квантового состояния атома водорода (и = 1) и формы четырех функций
для второго квантового состояния (п = 2). Is-Состояние, т. е. нормальное
состояние атома водорода, характеризуется сферической симметрией рас-
пределения плотности заряда, которая уменьшается в радиальном направ-
лении по экспоненциальному закону. Каждое из квантовых состояний
с п = 2, являющихся первично возбужденными состояниями с равными
величинами энергии, характеризуется наличием одной узловой поверхности.
25-Состояние характеризуется сферической симметрией и узловой поверх-
ностью сферической формы, в то время как три 2р-состояния, отличающиеся
друг от друга лишь ориентацией во взаимно перпендикулярных направле-
ниях, характеризуются аксиальной симметрией и плоскими узловыми
поверхностями. Девять квантовых состояний с п = 3 являются вторично
возбужденными состояниями с равными величинами энергии; каждое из них
имеет две узловые поверхности. Одно 3s- и три Зр-состояния аналогичны
2s- и 2р-состояниям, но каждое обладает дополнительной сферической узло-
вой поверхностью. Пять Зс?-состояний имеют несколько более сложные фор-
мы электронных облаков.
* Узловая поверхность — это поверхность, образованная точками пространства,
в которых значение функции равно нулю.— Прим. ред. первого издания.
21
Глава I
В самом низшем энергетическом состоянии атома водорода электрон в соот-
ветствии с принципом неопределенности не находится в постоянном кон-
такте с притягивающим его ядром. Если бы электрон все время находился
непосредственно у ядра, то его положение было бы точно фиксировано,
результатом чего явилась бы очень большая неопределенность его импульса,
а для обеспечения допустимых изменений импульса потребовались бы огром-
ные значения полной энергии. Чем больше область движения электрона
около ядра, тем меньше значение необходимой энергии, хотя это приводит
к увеличению электростатической энергии за счет удаления электрона от
ядра. Ясно, что имеется такой оптимальный размер атома, при котором атом
Рис. 1. Распределение электронной плотности в низших состояниях атома водорода.
Пунктиром показана внутренняя центральная часть электронного облака 2$-состояния.
Квадратами обозначены плоскости центров электронных облаков 2р-состояний.
будет обладать наименьшей полной энергией, определяемой как электро-
статическими силами, так и квантовой механикой. Можно показать, что
любая связанная частица в ее наиболее устойчивом состоянии движется
с энергией, называемой нулевой энергией, значение которой уменьшается
по мере увеличения области движения. В дальнейших рассуждениях мы
будем ссылаться на этот принцип.
Одноэлектронные волновые функции, подобные описанным для атома водо-
рода, называются орбиталями-, электроны в подобных состояниях, как
принято говорить, «занимают» орбитали. Для удобства можно себе предста-
вить, что эти орбитали независимо от того, заняты они или нет, реально
существуют и размещены вокруг ядер по типу старых боровских орбит *.
Важное значение проблемы водородного атома состоит в том, что основное
* Орбиталь — это математическое понятие, характеризующее волновую функцию.
Боровская орбита подобна орбитам планет солнечной системы.— Прим, ред.
22
Валентность и строение молекул
и возбужденные состояния атома водорода послужили моделью для при-
блаженного расчета основных состояний всех атомов. Аналогично атому
водорода ядро с данным зарядом имеет серию орбиталей (квантовые числа),
а следовательно, симметрию и узловые поверхности. Электроны в количе-
стве, соответствующем заряду ядра, располагаются на энергетически наи-
низших орбиталях в соответствии с «электронной емкостью» последних.
На каждой орбитали может находиться не более двух электронов. В 1925 г.
Уленбек и Гаудсмит обнаружили, что электрон обладает свойством, назван-
ным спином, определяющим его собственный угловой и магнитный моменты;
оказалось, что в магнитном поле этот
момент может быть ориентирован толь-
ко двумя способами — либо по направ-
лению поля, либо против него. Поэтому
говорят, что, кроме трех пространст-
венных квантовых чисел и, I и mi, элек-
трон имеет спиновое квантовое число
ms, равное -Н/г или —Нг-
В том же году Паули сформулировал
принцип запрета, согласно которому
на одной устойчивой орбитали может
находиться не более двух электронов,
причем В ЭТОМ случае спины электро- рИс. 2. Приблизительное соотношение
нов, а следовательно, их угловой и маг- энергетических уровней атом-
нитный моменты должны иметь про- них орбиталей.
тивоположные направления или, как
иногда говорят, должны быть спарены. Согласно принципу запрета, два
электрона в атоме не могут иметь одинаковые квантовые числа. Например,
так называемые эквивалентные электроны с одинаковыми пи/ должны
иметь разные mt или ms- Таким образом, если электроны занимают одну
и ту же орбиталь тг, они должны иметь разные ms, т. е. должны быть спа-
ренными.
Энергетическая последовательность орбиталей в многоэлектронных атомах
качественно отличается от последовательности для водорода. В многоэлек-
тронных атомах источник потенциального поля, действующего на каждый
отдельный электрон, не локализован в ядре, а распределен по всему атому.
Поэтому энергия орбитали в таких атомах зависит не только от главного
квантового числа п, но также от I (однако не от тг). Таким образом, группы
орбиталей с одинаковым значением п энергетически разделяются на следую-
щие подгруппы: р-орбитали лежат выше, чем s-орбитали; d-орбитали —
выше, чем р-орбитали, и т. д. Последовательность устойчивости орбиталей
основных состояний атомов элементов, находящихся в начале периодической
системы, качественно представлена [9] на рис. 2.
В основных состояниях атомов порядок заполнения электронами эквива-
лентных орбиталей, т. е. орбиталей с одинаковыми пи/ (например, трех
2р-орбиталей), определяется правилом Гунда; сначала каждая из р-орби-
талей принимает по одному электрону, спины которых параллельны, а затем
происходит присоединение вторых электронов с противоположными спи-
нами. Это правило максимальной мулътиплетности выводится из общего
принципа запрета. Электроны с одинаковыми спинами не только занимают
разные орбитали, но также коррелируют движение друг друга таким обра-
зом, чтобы быть максимально удаленными в пространстве. Благодаря этому
достигается минимизация электростатической энергии отталкивания. Мно-
гоэлектронная система с параллельными спинами более стабильна, чем
23
Глава I
система с антипараллельными спинами электронов на разных орбиталях,
так как в последнем случае не происходит пространственной корреляции
движения в соответствии с принципом запрета. Таким образом, для нор-
мальных и возбужденных атомов и даже для неэквивалентных орбиталей
состояние с параллельными спинами неспаренных электронов па разных
орбиталях будет иметь более низкую энергию, чем состояние с тем же самым
распределением электронов по орбиталям, т. е. с той же самой электрон-
ной конфигурацией, но с антипараллельными спинами неспаренных
электронов.
Примером может служить распределение электронов по орбиталям, т. е.
электронная конфигурация, атомов элементов первого периода. В табл. 2
показан порядок заполнения К- и Т-оболочек, т. е. дублета и первого октета
в атомах в их основных состояниях; электроны изображены стрелками,
пары противоположно направленных стрелок обозначают спаренные
спины.
Таблица 2
Электронная конфигурация атомов в их основных состояниях
Атом Л’-Оболочка
U 2s
н t
Не п
Li н t
Be п н
В н U
С н W
N н U
О н н
F н
Ne н п
Дублет
L-Оболочка
t
t
t
н
n
u
Октет
2pz
Симме г-
рия
s
s
s
s
p
p
s
p
p
s
t
u
По принятой терминологии считают, что атом со всеми спаренными спинами
находится в синглетном состоянии. В основном состоянии атомы гелия
и неона — синглеты. Если атом имеет один неспаренный спин, то говорят,
что он находится в дублетном состоянии. Атомы водорода, а также атомы
бора и фтора в основном состоянии представляют собой дублеты. Атомы
с двумя параллельными спинами находятся в триплетном состоянии.
В согласии с правилом Гунда нормальные (свободные) атомы углерода и кис-
лорода— триплеты. По той же причине нормальный атом азота находится
в квартетном состоянии. Атом с п параллельными спинами находится
в (ге+ 1)-летном состоянии, а величина (и + 1) называется мулътиплет-
ностъю. Такая же терминология применима и к молекулам. Молекулы
с нечетным числом электронов, например окись азота, обычно находятся
в дублетном состоянии. Почти во всех молекулах с четным числом электронов
все спины спарены, и поэтому в основном состоянии эти молекулы синг-
летны. Исключение, однако, составляет обычная молекула кислорода; она
24
Валентность и строение молекул
имеет два параллельных спина и, таким образом, находится в триплетном
состоянии.
Интересным геометрическим свойством эквивалентных орбиталей является
то, что при одинаковом заполнении их электронами, как, например, 2р-орби-
талей атомов азота и неона в основном состоянии, распределение электрон-
ной плотности, определяемое У1, | V | 2, имеет сферическую симметрию.
Следовательно, когда одна из эквивалентных орбиталей оказывается запол-
ненной не так, как другие, распределение электронной плотности имеет
аксиальную симметрию, соответствующую одной из орбиталей этой под-
группы. В табл. 2 буквой S (см. правую колонку) обозначены атомы, обла-
дающие сферической симметрией, и буквой Р — атомы, имеющие лишь
аксиальную симметрию сфероида.
в. Орбитали связей. Если при рассмотрении атомных состояний суще-
ственным было обращение к свойствам стоячих волн, то при обсуждении
вопроса о связывании атомов столь же важно обратиться к другому общему
свойству волн, называемому резонансом. В механике резонанс наблюдается
в тех случаях, когда две системы, в которых могут возникнуть идентичные
стоячие волны, связаны или способны до известной степени взаимодейство-
вать между собой. При этом стоячая волна одной из систем возмущает другую
систему, в результате чего сама перестает быть стоячей. Таким образом,
устанавливается единая система двух новых стоячих волн, одна из которых
будет иметь уменьшенную, а другая — увеличенную частоту. В 1926 г.
Гейзенберг открыл аналогичное свойство волн, описываемых ^-функцией,
и установил влияние этого свойства на энергии состояний. Последнее обстоя-
тельство, как показали в 1927 г. Гейтлер и Лондон, и является основной
причиной прочности ковалентной связи.
Сначала лучше рассмотреть простейший случай связи при помощи одного
обобщенного электрона, например связь в молекулярном ионе водорода Щ.
Предположим, что два протона А та Б находятся на различных фиксируемых
расстояниях друг от друга и что у протона А находится электрон, образую-
щий вместе с протоном обычный атом водорода. Пусть сначала расстояние
между протонами достаточно велико, чтобы исключить взаимодействие.
Затем это расстояние уменьшается настолько, что занятая орбиталь А
слегка перекрывается с незаполненной орбиталью Б. В этом случае электрон
способен переходить с одной эквивалентной орбитали на другую и поэтому
последние перестанут быть стационарными: в результате резонансного
взаимодействия эти две атомные орбитали превратятся в две симметричные
по отношению к обоим протонам молекулярные орбитали, которые описывают
движение электрона в поле обоих протонов. Одна из этих орбиталей обла-
дает пониженной, а другая — повышенной энергией; первая, обозначаемая
по соображениям симметрии символом Iso, является связывающей орбиталью
(не имеет узловой поверхности), а вторая, обозначаемая 2рсг,— разрыхляю-
щей орбиталью с узловой поверхностью, которая перпендикулярна линии,
соединяющей ядра, и проходит через ее середину. Пас будет интересовать
главным образом более стабильное состояние системы, в котором электрон
занимает низшую орбиталь. При дальнейшем уменьшении межъядерного
расстояния резонанс увеличивается. При этом энергия более устойчивой
орбитали будет уменьшаться, а менее устойчивой — увеличиваться. Однако
наступает момент, когда дальнейшее уменьшение межъядерного расстояния
не приведет к уменьшению энергии более устойчивой орбитали, так как
при достаточно малом расстоянии сильно возрастает энергия отталкивания
между ядрами. Поэтому должно существовать оптимальное расстояние
между ядрами с минимальным значением энергии; это расстояние и являет-
25
Глава I
ся равновесной длиной связи невозбужденного молекулярного иона
водорода.
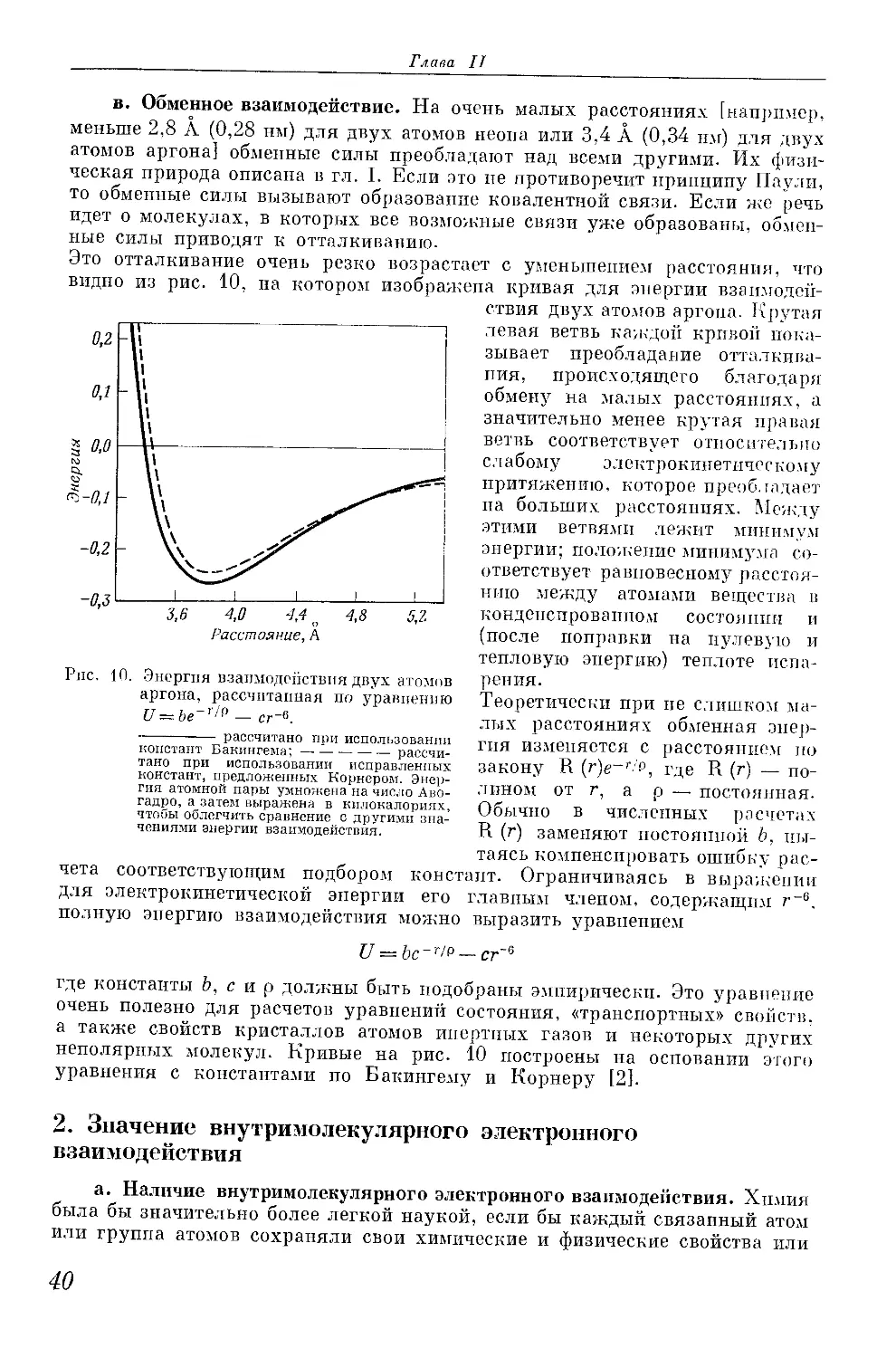
Расчеты, подтвержденные спектроскопическими данными, показали, что
длина связи в этом ионе равна 1,06 А. (10,6-IO-2 нм), а энергия связи —
61 ккал/моль (255,39 -103 Дж/моль) [10]. Резонанс обусловливает примерно
80 % этой энергии. Остальные 20% возникают вследствие так называемой
деформации: протон и атом водорода взаимно притягиваются даже в отсут-
ствие резонанса вследствие способности атома поляризоваться под влиянием
протона. На рис. 3 представлено вычисленное распределение электронной
плотности. Диаграмма показывает, что электрон находится большей частью
в межъядерном пространстве вокруг линии, соединяющей ядра, и значи-
тельно меньше с противоположной стороны обоих ядер. Величина и ширина
Рис. 3. Кривые равной электронной плотно-
сти в молекулярном ионе водорода (по
Барроу).
Вверху показан разрез по линии, соеди-
няющей ядра; в центре — план на плоско-
сти, проходящей через ядра; внизу — разрез
по плоскостям, перпендикулярным линии,
соединяющей ядра: слева — в плоскости,
проходящей через ядро, справа — через
середину линии, соединяющей ядра.
этой функции в плоскости, перпендикулярной межъядерной линии в середине
последней, показывает, что электрон находится в этом положении значи-
тельно чаще, чем в параллельных плоскостях, проходящих через ядра.
Такое распределение электронной плотности указывает на то, что резонанс
заключается не в чередующемся обладании электроном каждым из двух
атомов, а является формой молекулярного движения электрона, возни-
кающей в результате наложения прежних движений электрона вокруг
отдельных атомов. Так как заполнение этой молекулярной орбитали озна-
чает образование химической связи, то такая орбиталь может быть названа
орбиталью связи. Такие орбитали обладают аксиальной симметрией сфероида
относительно линии связи и обозначаются сг-орбиталями; занимающие их
электроны часто называются cr-электронами, а сами связи — сг-связями.
г. Ковалентные связи. Образование связи между двумя атомами водо-
рода рассматривается Гейтлером и Лондоном примерно так же, как и в слу-
чае молекулярного иона водорода. Два протона А и Б помещаются на опре-
деленном расстоянии друг от друга; вокруг протона А вращается электрон 1,
вокруг протона Б — электрон 2, как в двух обычных атомах водорода.
Допустим, что сначала расстояние между А и Б столь велико, что всякое
взаимодействие между атомами исключено. Тем не менее, чтобы было воз-
можно последующее обсуждение взаимодействия, оба атома с самого начала
следует считать единой системой, состояние которой описывается двухэлек-
тронной функцией Y, так что | V |2 будет характеризовать распределение
двух электронов по всем возможным парам положений в пространстве. Если
электроны поменять местами, то состояние системы будет выражаться дру-
гой, но энергетически эквивалентной двухэлектронной функцией 4f; однако
при первоначальном способе размещения электронов вторая функция будет
незаполненной, а первая — заполненной. Допустим, что расстояние между
26
Валентность и строение молекул
Рис. 4. Зависимость энергии двух вза-
имодействующих нормальных
атомов водорода от расстояния
между ядрами (по Сегиура).
За единицу энергии принята величи-
на 13,53 эВ = 312 ккал/моль, за едини-
цу длины 0,528 А (5,28-10-2 нм).
1 — устойчивое состояние молекулы;
2 — неустойчивое состояние моле-
кулы.
атомами уменьшено настолько, что каждый из электронов может оказаться
в поле действия протона другого атома. Тогда будет иметь место резонансное
взаимодействие: электроны могут в этом случае самопроизвольно переходить
от одного атома к другому, а распределение электронов, которое раньше
описывалось только заполненной ^-функцией, может теперь описываться
и первоначально не заполненной ^-функцией. Таким образом, вследствие
возможности электронного обмена ни одна из этих функций не будет ото-
бражать стационарного состояния системы. Эти функции заменятся моле-
кулярными двухэлектронными ^-функциями, в каждую из которых будут
симметрично входить оба альтернативных распределения электронов, бла-
годаря чему и будет учтен обратимый электронный обмен. Таких двухэлек-
тронных функций будет две: одна, занятая, выражает состояние с умень-
шенной энергией, другая — с увеличенной энергией. При уменьшении
расстояния между протонами величина
энергии более устойчивого состояния
уменьшается вследствие увеличения
резонанса, затем достигает минималь-
ного значения и, наконец, увеличива-
ется, так как возрастает отталкивание
между ядрами. Энергия менее устойчи-
вого состояния при этом постепенно
увеличивается. Первое состояние от-
вечает образованию устойчивой моле-
кулы, второе — отталкиванию между
атомами. Соответствующие кривые за-
висимости энергии от межъядерного рас-
стояния приведены на рис. 4.
Спектроскопические измерения пока-
зали, что длина связи в молекуле водо-
рода равна 0,740 А. (7,4-10-2 нм),
а энергия связи — 102,6 ккал/моль
(429,57 Дж/моль). Большая часть
(80%) этой энергии, как показали вы-
числения Гейтлера и Лондона, расши-
ренные и уточненные Сегиура и Вангом,
обусловлена энергией резонанса электронного обмена. По Вейнбауму, 5%
энергии можно получить, если отказаться от допущения, согласно которому
оба электрона никогда не бывают у одного и того же ядра. Оставшиеся 15%
приходятся на долю различных типов взаимодействия, объединяемых под
названием деформации. Эти взаимодействия были учтены в расчетах Джеймса
и Кулиджа [11], которые получили точное значение энергии связи, а также
и других физических констант молекулы.
Мпогоэлектронные системы в случае как атомов, так и молекул прибли-
женно можно описывать с помощью одноэлектронных волновых функций,
т. е. орбиталей. Считается, что электроны, занимающие каждую из таких
орбиталей, движутся в поле ядер и однородном поле одного или нескольких
других электронов. Так, двухэлектронная волновая функция, соответствую-
щая низшему из двух гейтлер-лондоновских состояний молекулы водорода
(рис. 4), может быть аппроксимирована произведением двух совпадающих
идентичных одноэлектронных функций, или орбиталей, и в соответствии
с этим описана как «дважды занятая» орбиталь. Эта орбиталь не имеет узлов
и является связывающей (Iso; приведена на рис. 3 для молекулярного иона
водорода); соответствующая орбиталь молекулы водорода в общем имеет
27
Глава I
точно такую же форму, хотя и несколько иные размеры. Если такая орби-
таль занята двумя электронами, как, например, в низшем энергетическом
состоянии молекулы водорода, то в соответствии с принципом Паули эти
электроны должны иметь спаренные спины.
Аналогично высшее энергетическое гейтлер-лондоповское состояние (рис. 4)
может быть аппроксимировано произведением двух орбиталей, каждая
из которых занята одним электроном, но одна относится к типу Iscr, а дру-
гая — к энергетически менее выгодному типу 2рсг. Последняя имеет узловую
плоскость, проходящую через середину линии, соединяющей ядра атомов.
В результате образуется общая разрыхляющая орбиталь с высокой энер-
гией (рис. 4). Два электрона на разных орбиталях в соответствии с принци-
пом запрета будут иметь параллельные спины, их движение будет согласо-
ванным, и они будут удалены друг от друга в пространстве, что приведет
к более стабильной электронной конфигурации, чем конфигурация со спа-
ренными спинами.
Принцип Паули требует не только того, чтобы в устойчивом состоянии моле-
кулы (кривая 1 па рис. 4) спины были антипараллельны, но и чтобы в не-
устойчивом состоянии (кривая 2) спины были параллельны. Основываясь
па этом, Гейтлер и Лондон показали, что их теория взаимодействия двух
атомов водорода является в принципе общей теорией образования ковалент-
ной связи, а также теорией отталкивания несвязывающихся атомов. Два
атома водорода вследствие электронного обмена могут либо соединяться,
либо отталкиваться друг от друга, а атом водорода и атом гелия или два
атома гелия могут только отталкиваться. Принцип Паули разрешает обмен
лишь электронов с параллельными спинами, а это в случае атомов гелия
приводит к состоянию отталкивания, описываемому кривой 2 на рис. 4.
Поэтому при межатомных расстояниях, достаточно коротких для того, чтобы
стал возможен обмен электронами, неспаренные электроны при обмене
отталкивают электронные дублеты со спаренными спинами, а все такие
дублеты отталкиваются друг от друга. В то же время два неспаренных элек-
трона могут образовывать ковалентную связь. Вообще две атомные орбитали,
па каждой из которых имеется по одному электрону, могут образовывать
орбиталь связи, которая при занятии ее двумя электронами становится
ковалентной связью.
Образование связи путем электронного обмена находится в соответствии
с принципом неопределенности, согласно которому связанные частицы в наи-
более устойчивых состояниях обладают нулевой энергией, величина которой
зависит от области пространства, в которой происходит движение. Обмен
увеличивает возможности движения электронов, уменьшая тем самым
нулевую энергию системы. В результате образуется более устойчивая систе-
ма, если только состояние с нулевой энергией разрешено принципом
Паули.
4. Стереохимические представления
а. Направленные простые связи. Развитие взглядов Гейтлера и Лондона
на ковалентную связь применительно к молекулам, образующимся из мпо-
гоэлектронных атомов, и в особенности при рассмотрении направленности
связей дано главным образом в работах Полинга и Слейтера [12]. На осно-
вании формальных теоретических представлений Полинг вывел определен-
ные общие правила, касающиеся образования связи. Он указал, что атомные
орбитали двух электронов, участвующих в образовании связи, стремятся
к взаимному перекрыванию и избегают перекрывания с орбиталями других
28
Валентность и строение молекул
электронов; это обусловлено тем, что перекрывание облегчает электронный
обмен, который является главной причиной устойчивости связей и неустой-
чивости перекрывающихся несвязывающих электронных пар. Отсюда сле-
дует, что несферическая орбиталь стремится к образованию связи в направ-
лении наибольшей электронной плотности. Хотя, как правило, более устой-
чивые орбитали образуют более устойчивые связи, однако для орбитали,
электронная плотность которой сконцентрирована в одном направлении,
характерно образование более прочных связей, чем для орбитали эквива-
лентной устойчивости, но с менее
концентрированной в определен-
ном направлении электронной плот-
ностью.
Сравнительные плотности и про-
странственные ориентации атомных
орбиталей с одним и тем же глав-
ным квантовым числом можно схе-
матически изобразить с помощью
полярных графиков их угловых
компонентов. Зависящий от угла
множитель, который содержится в
выражении для V, служит ради-
усом-вектором для описания неко-
торой поверхности; расстояние
любой точки этой поверхности от
центра ее дает величину Ч*1 и ее
зависимость от направления. По-
Рпс. 5. Полярные графики s- и р-орбиталеп
(приведены угловые величины).
лярным графиком s-орбитали является
сферическая поверхность,
а р-
орбитали — две равные сферические поверхности, соприкасающиеся в
точке, где расположено ядро (рис. 5). Если зависящая от угла вели-
чина V для s-орбитали принимается равной единице во всех направлениях,
то для р-орбитали эта величина по направлению максимума равна ]/ 3 =
= 1,73. Учитывая принцип перекрывания, Полинг предположил, что эти
числа отображают прочности связей, образованных орбиталями, и ввел для
них название «прочности» орбиталей; по изложенным ниже причинам мы
будем называть их просто угловыми величинами, избегая утверждения, что
эти числа являются точной мерой прочности связи. Полинг полагает, что
р-орбиталь должна образовывать более прочную связь, чем s-орбиталь с тем
же главным квантовым числом, а также что две р-орбитали одного атома
должны стремиться образовать связи во взаимно перпендикулярных направ-
лениях.
Эти заключения в известной мере подтверждаются опытом. В атоме кисло-
рода в невозбуждешюм состоянии имеются две р-орбитали, каждая из кото-
рых занята только одним электроном, и никакое перераспределение элек-
тронов в Л-оболочке не может привести к увеличению числа таких орби-
талей (табл. 2). Следовательно, атом кислорода и другие атомы с аналогичной
электронной конфигурацией (например, сера) могут образовывать две кова-
лентные связи с атомами типа водорода или фтора, каждый из которых
имеет одну орбиталь, запятую одним электроном. Известно, что связи в моле-
кулах, образующихся из таких атомов, расположены всегда под некоторым
углом друг к другу, и эти углы почти всегда несколько больше прямого.
Так, в воде угол составляет 105,0° (1.832 рад), в окиси фтора 101,5° (1,771 рад)
и в сероводороде 92,3° (1,611 рад). Имеется несколько причин, обусловли-
вающих образование валентных углов, превышающих 90° (1,571 рад). Одна
29
Глава I
из них заключается в электростатическом отталкивании между связями
одинаковой полярности. Другой причиной является стерическое отталкива-
ние между связями, если предположить, что заполненные орбитали связи
концентрируются между атомами, а по другую сторону двухвалентного
атома имеется лишь очень незначительная электронная плотность. Третья
причина состоит в стерическом отталкивании между связанными атомами,
если они достаточно велики. Четвертая возможная причина заключается
в том, что увеличение угла вследствие гибридизации (обсуждаемой ниже)
приводит к более стабильным связям, но к менее стабильным неподеленным
дублетам; действие этих двух противоположных эффектов может благо-
приятствовать некоторому увеличению угла.
В атоме азота в невозбужденном состоянии каждая из трех р-орбиталей
занята одним электроном (табл. 2), и, следовательно, атом азота, как и дру-
гие атомы с подобной электронной конфигурацией, может образовать три
ковалентные связи с одновалентными атомами. Известно, что молекулы,
образованные таким образом, всегда имеют пирамидальную структуру;
в этом случае углы между связями также больше прямых: например, в аммиа-
ке 106,8° (1,864 рад), в трехфтористом азоте 102,5° (1,789 рад) и в тригало-
генидах фосфора, мышьяка и сурьмы 96 . . . 104° (1,675 . . . 1,815 рад).
Атомы бериллия, бора и углерода с одновалентными атомами типа водорода
или фтора могут образовывать больше связей, чем это можно предположить,
исходя из их обычной электронной конфигурации (табл. 2). Причиной этого
является наличие у этих атомов в Л-оболочке как незанятых орбиталей,
так и орбиталей, занятых двумя электронами. Последнее обстоятельство
определяет возможность перераспределения Л-электронов, что приводит
к возбужденным атомным состояниям с электронной конфигурацией, пока-
занной в табл. 3. Можно считать, что такое возбуждение возникает при обра-
зовании связей в тех случаях, когда энергия возбуждения меньше допол-
нительной энергии, выделяющейся за счет возникновения большего числа
связей. В этих примерах энергия, освобождающаяся при связывании, допол-
нительно увеличивается за счет процесса гибридизации, который и следует
рассмотреть.
Таблица 3
Электронная конфигурация атомов, находящихся
в возбужденных состояниях
Атом К-Обо- лочка Ь-Оболочка Симмет- рия
Is 2s 2px 2py 2Рг
Ве * и t f p
В * н t t p
С * п s
По аналогии с вырожденными колебаниями и волнами в механических систе-
мах число орбиталей с точно равными энергиями всегда равно числу их неза-
висимых суперпозиций. Так, если три эквивалентные р-орбитали вступают
в суперпозицию и образуются три новые независимые орбитали, то эти
последние совершенно одинаковы; наложение приводит лишь к повороту
осей координат. Расширяя этот принцип, можно утверждать, что в любом
энергетическом процессе с участием орбиталей в суперпозицию могут всту-
30
Валентность и строение молекул
и р-орбиталей) по сравнению
s-
L — линейная (угловая величина
1,93); Р — плоская тригональная
(угловая величина 1,99); Т — тетра-
эдрическая (угловая величина 2,00).
пать лишь такие орбитали, разница энергий которых не превышает энергии
процесса. Суперпозиция приводит к образованию максимально неустой-
чивых орбиталей.
Например, возбужденный атом углерода (С* в табл. 3) способен образовать
четыре связи. Однако для этой цели непосредственно не используются
электроны его s- и р-орбиталей, так как суперпозиция этих орбиталей при-
ведет к возникновению гибридных орбиталей, электронная плотность кото-
рых концентрируется более локализованно и которые, следовательно, могут
образовывать более прочные связи. Если s- и р-орбитали вступают в супер-
позицию так, что образуют хотя бы одну гибридную орбиталь с максимальной
угловой величиной (эта величина равна 2,00 в тех же единицах, которые
уже использовались для характеристики
со всеми орбиталями, которые могут
возникнуть при их суперпозиции, то
при этом обязательно образуются че-
тыре эквивалентные орбитали с такой
же угловой величиной; связи, образу-
емые этими орбиталями, направлены
одна по отношению к другой под тетра-
эдрическими углами, равными 109°28'
(1,910 рад). Полярный график такой
тетраэдрической орбитали показан на
рис. 6.
Это положение лежит в основе стерео-
химии углерода. Аналогичные выводы
приложимы к атому кремния и к дру-
гим атомам с подобными связываю-
щими орбиталями, в том числе к ато-
мам азота, фосфора и другим, с одной
стороны, и к атомам бора, алюминия
и т. п., с другой, в том случае, когда
они вследствие координации образуют четыре ковалентные связи. Эти рас-
суждения применимы к четырехковалентности с участием только s- и р-орби-
талей.
Атом бора и другие атомы с такой же, как у бора, внешней электронной
конфигурацией могут образовывать три эквивалентные гибридные орбитали.
Направления, в которых образуются связи, лежат в одной плоскости под
углами 120° (2,094 рад), угловая величина такой плоской тригональной
орбитали равна 1,99, т. е. практически такая же, как и в случае тетраэдри-
ческой орбитали (рис. 6). Показано, что ряд соединений трехвалентного
бора [BF3, ВС13, ВВг3, В(СН3)3] имеет плоскую тригональную конфигу-
рацию.
Атом бериллия и атомы, имеющие аналогичную валентную оболочку, могут
образовывать две эквивалентные гибридные орбитали. Направления, в кото-
рых образуются связи, находятся под углом 180° (3,141 рад), что определяет
в этих случаях образование линейных структур. Угловая величина такой
линейной орбитали, равная 1,93 и являющаяся почти одинаковой с угло-
выми величинами других гибридных орбиталей (рис. 6), указывает на то.
что эта орбиталь сходна с другими гибридными орбиталями по своей способ-
ности образовывать прочные связи, в частности более прочные, чем связи,
образующиеся за счет р-орбиталей. Доказано, что линейная ковалентная
связь имеется в соединениях тяжелых элементов, таких, например, как
ртуть (HgCl2, HgBr2, Hgl2, Hg2Cl2 и т. д.).
31
Глава I
Тетраэдрическая орбиталь, являющаяся «смесью» 1/48-орбитали и 3/4р-орби-
тали, часто обозначается символом spa; тригональная орбиталь, содержащая
1/3s- и 2/3р-орбиталей, называется 8р2-орбиталыо, а линейная орбиталь
(1/2s + х/2р) — sp-орбиталыо.
Наиболее поразительны успехи орбитальной теории связи, развитой Полин-
гом, в приложении к соединениям переходных металлов. Последние спо-
собны к образованию связей не только за счет s- и р-орбиталей с высшими
главными квантовыми числами, но также и за счет ci-орбиталей с меньшими
главными квантовыми числами (энергии таких с/-, s- и р-орбиталей имеют
близкие значения), которые не заняты пеподеленными электронными пара-
ми. Это значительно повышает возможности гибридизации. Полинг [13]
показал, что если наряду cs-и р-орбиталями имеется ci-орбиталь, то в резуль-
тате гибридизации могут образоваться четыре эквивалентные орбитали
с угловыми величинами, равными 2.69. т. е. значительно большими, чем
в случае тетраэдрических орбиталей; связи, которые образуются за счет
этих орбиталей, направлены к вершинам квадрата. По Полингу, в известных
квадратных комплексах переходных металлов доступность орбиталей для
образования связей точно совпадает с требованиями теории; он предсказал
наличие квадратных конфигураций, в частности для четырехковалентных
комплексов двухвалентного никеля, что в дальнейшем было подтверждено.
Более того, Полинг показал, что, когда в образовании связей наряду с s-
и р-орбиталями могут принимать участие две ci-орбитали, гибридизация
всех шести орбиталей может привести к шести эквивалентным орбиталям,
имеющим значительную угловую величину, равную 2,92; связи, образован-
ные за счет этих гибридных орбиталей, направлены к вершинам октаэдра.
Наконец, Полинг установил, что способность орбиталей образовывать
связи в известных октаэдрических комплексах переходных металлов согла-
суется с теорией. Если для образования связи используются четыре или
пять ci-орбиталей, возможны и другие геометрические конфигурации связей
[14], причем некоторые из них известны [15].
Полинг предположил [12], что угловые величины и, следовательно, связы-
вающие свойства sp-гибридпых орбиталей существенно улучшаются, если
в гибридизации частично участвуют d- и /-орбитали. Эти дополнительные
орбитальные компоненты в большинстве случаев имеют высокие главные
квантовые числа и, будучи занятыми,— относительно высокую энергию.
Это обстоятельство приводит к тому, что доля их участия в гибридизации
невелика. Однако, как указал Малликен, небольшая доля гибридизации
может привести к большим последствиям: включение дополнительных орби-
талей улучшает связывание. Полинг вычислил, что максимальное улучшение
связеобразующпх свойств тетраэдрической орбитали, приводящее к увели-
чению угловой величины от 2,00 до 2,76, происходит при включении в гибри-
дизацию 4% d- и 20% /-орбиталей. Это, конечно, максимальная величина,
одпако определенная доля (трудно сказать какая) d- и /-орбиталей всегда
участвует в образовании связывающих орбиталей, поскольку такие связи
энергетически выгодны. Этот эффект не проявляется в случае орбиталей,
занятых пеподеленными электронами.
б. Двойные п тройные связи. Для двойных связей, как, например,
в этилене, были предложены две модели. В одной модели, первоначально
предложенной Полингом и Слейтером [12] и недавно далее развитой Полин-
гом [15], оба обобществленных дублета считаются одинаковыми, а именно
изогнутыми простыми связями, образованными из тетраэдрических орби-
талей и направленными под значительными углами к направлению макси-
мального перекрывания. В другой модели, предложенной Хюккелем и под-
32
Валентность и строение молекул
держанной Пенни [16], один обобществленный дублет образует обычную
простую сг-связь, а второй дублет — химически реакционноспособную
часть двойной связи, которая возникает за счет бокового перекрывания
атомных уз-орбиталей. Эти две модели не так сильно отличаются одна от
другой, как могло бы показаться. При применении приближенного метода
расчета молекул, известного под названием метода молекулярных орбита-
лей. использование этих двух моделей приводит к одинаковой картине
общего распределения электронов [17]. В этом методе можно условно про-
водить подразделение на дважды занятые орбитали связей. В других при-
ближениях это не так, и вопрос о том, какая модель будет наилучшей, зави-
сит от рассматриваемой конкретной задачи. Тем не менее в общем модель
Рис. 7. Взаимодействие р-орбиталей (изображенных в
виде полярных графиков), приводящее к образо-
ванию л-орбптали, которая при занятии двумя
электронами становится л-компонептой двойной
связи в этилене.
п-Электронные пары изображены только их осями симмет-
рии, которыми являются межъядерные линии.
Хюккеля — Пении имеет большое преимущество, так как она с почти авто-
матической легкостью объясняет «сопряжение»— явление, с которым мы
столкнемся в гл. II. Метод Хюккеля применим не только к полипенасы-
щеппым системам с открытой цепью, например бутадиену-1,3 (для которого,
конечно, можно переоценить важность сопряжения), но также (и совер-
шенно точно) к ароматическим системам, например бензолу.
Согласно этой модели (которую можно рассмотреть на примере этилена),
в результате гибридизации s-орбитали и двух р-орбиталей каждого атома
углерода образуются три плоские тригональные орбитали, две из которых
участвуют в образовании связей с водородами, а третья — в образовании
обычной о-связи между углеродными атомами. Все эти связи лежат в одной
плоскости. У каждого атома углерода, кроме того, имеется занятая одним
электроном р-орбиталь. ось симметрии которой перпендикулярна плоскости
атомов; в результате «бокового» взаимодействия между этими атомными
р,-орбиталями возникает вторая компонента двойной связи (рис. 7). Если
в каждой компоненте двойной связи спины электронов спарены, то принцип
Паули удовлетворяется.
Как для исходных р2-орбиталей, так и для орбитали связи, образовавшейся
при их «боковом» взаимодействии, плоскость ядер является узловой пло-
скостью. Такие орбитали связи называются л-орбиталями, электроны, зани-
мающие эти орбитали,— л-электронами, а соответствующая компонента
двойной связи — л-связью. Распределение электронной плотности в л-свя-
зях приводит к образованию двух слоев, по одному с каждой стороны пло-
скости молекулы (рис. 8). Характерно, что электронная плотность л-связи
равна пулю на линии, соединяющей ядра, и очень мала около нее, в то время
как плотность сг-связи концентрируется непосредственно вокруг межъядер-
пой линии. Такое разделение электронных пар, несомненно, является фак-
тором, определяющим устойчивость двойной связи. Однако с точки зрения
возможности возмущения эти пары резко отличаются друг от друга: сг-элек-
троны размещены глубоко внутри, а л-электроны — снаружи, и, следова-
тельно, именно последние легче отрываются или фотолитически возбуж-
даются и наиболее легко вовлекаются в химические реакции.
3 — 0772
33
Глава I
Прочность л-связи находится в очевидной зависимости от параллельности
осей атомных р-орбиталей; эта параллельность нарушается при скручивании
молекулы вокруг межъядерной линии. Поэтому л-компонента двойной
связи придает ей свойство оказывать сопротивление скручиванию, от чего
зависит возможность геометрической изомерии, наблюдаемой у соединений
углерода и азота, содержащих двойные связи.
Характер гибридизации в модели Хюккеля — Пенни не является вполне
однозначным: гибридизация может привести к плоским тригональным орби-
талям, как первоначально предполагалось, а также к двум тетраэдрическим
и одной линейной орбитали и к любым промежуточным формам при условии,
Рис. 8. Распределение электронной плотности л-электропов двойной (вверху) и тройной
(внизу) связей.
В каждом случае приводятся разрезы по плоскости, проходящей вдоль межъядсрпой линии,
и по плоскости, перпендикулярной ей.
если одна р-орбиталь остается для образования л-связи. Эти типы гибриди-
зации отличаются друг от друга главным образом величиной внешнего
угла между простыми связями, который изменяется от 120° (2,094 рад)
в случае плоской тригональной гибридизации до 109°28' (1,910 рад) между
связями тетраэдрических орбиталей. Экспериментально определенные внеш-
ние углы между простыми связями или близки к этим значениям, или лежат
между ними. В этилене этот угол НСН равен 117,6° (2,052 рад) [18], в фор-
/х
мальдегиде — 115,8° (2,020 рад) [19]. В 1,1-дифторэтилене угол FCF равен
109,6° (1,913 рад) [20], а в 1,1-дихлорэтилепе угол C1GC1 составляет 113,6°
34
Валентность и строение молекул
(1,982 рад) [21]. В изобутилене и тетраметилэтилене внешний угол ССС
равен 111° (1,937 рад) [22].
Из аналогичного теоретического рассмотрения тройной связи (например,
в ацетилене) следует, что из s-орбитали и одной р-орбитали каждого атома
углерода получаются две гибридные линейные орбитали, одна из которых
образует связь с водородом, а другая — сг-связь между атомами углерода.
В данном случае все атомы лежат на одной прямой линии. При этом у каж-
дого атома углерода имеются еще две р-орбитали, оси симметрии которых
перпендикулярны друг другу и межъядерной линии. При «боковом» взаимо-
действии р-орбиталей каждого из атомов углерода с р-орбиталями другого
Рис. 9. Взаимодействие л-орбиталей (изобра-
женных в виде полярных графиков) с
образованием л-орбиталей тройной свя-
зи в ацетилене.
атома образуются две л-связи. Каждая из л-орбиталей занята двумя элек-
тронами со спаренными спинами. Можно сказать, что эти четыре электрона
образуют л-оболочку тройной связи (рис. 9).
Вследствие ортогонального расположения атомных р-орбиталей узловые
плоскости образовавшихся л-орбиталей вдоль межъядерной линии должны
пересекаться под прямыми углами. Это приводит к тому, что максимум
электронной плотности каждой электронной пары лежит в плоскости нулевой
электронной плотности другой пары. Кроме того, на основании рассмот-
ренных геометрических представлений (см. разд. 3,6), согласно которым
в случае двух одинаково заполненных р-орбиталей суммарное распределение
электронной плотности характеризуется круговой симметрией относительно
оси третьей р-орбитали, следует считать, что две одинаково заполненные
л-орбитали будут приводить к электронному распределению с круговой
симметрией относительно межъядерной линии. Из этого следует, что л-обо-
лочка тройной связи имеет форму цилиндрического слоя, причем внутреннее
пространство вдоль межъядерной линии имеет очень незначительную плот-
ность л-электронного облака (см. рис. 8). В то же время вдоль этой оси элек-
тронная плотность сг-компоненты связи будет наибольшей.
Так же как и в случае двойной связи, л-электроны тройной связи, располо-
женные с внешней стороны, наиболее легко подвижны и л-связь легко рас-
щепляется или возмущается фотолитическим или химическим путем. В отли-
чие от л-компоненты двойной связи, легко разрушаемой при скручивании
молекулы, аналогичное разрушение л-облака тройной связи, очевидно,
невозможно; это обстоятельство объясняет относительно высокую терми-
ческую устойчивость некоторых простейших соединений, содержащих трой-
ную связь.
Известно, что в ряду метан, этилен и ацетилен связь между водородом и угле-
родом становится все более полярной, а водород — все более положитель-
ным; лучшим доказательством этого является способность ацетилена давать
металлические производные. По Уолшу [23], это увеличение полярности
СН-связей объясняется увеличением относительного содержания (1/4, V3
и х/2) s-компоненты 5,р-гибридных орбиталей углерода в рассматриваемом
3* 35
Глава I
ряду соединений. В соответствии со свойствами s- и р-орбиталей s-компо-
нента гибридной орбитали в большей степени, чем p-компонента, способна
к удерживанию электронов у ядра углерода и соответственно дальше от
протонов *•
СПИСОК ЛИТЕРАТУРЫ
1. Abegg R., Z. anorg. Chem., 39, 330 (1904).
2. Drude P., Ann. Physik, 14, 722 (1904).
3. Kossel W., Ann. Physik, 49, 229 (1916).
4. Lewis G. N., J. Am. Chem. Soc., 38, 762 (1916).
5. Lewis G. N., Valence and the Structure of Atoms and Molecules, Chemical Catalog Co..
New York, 1923.
6. Werner A., Neuere Anschauungen auf den Gebiete der Anorganischen Chemie, Vieweg,
Braunschweig, 1905.
7. Rojansky V., Introductory Quantum Mechanics, Prentice-Hall, New York, 1946.
8. Herzberg G., Atomic Spectra and Atomic Structure, Blackie, London and Glasgow,
1937.
9. Pauling L., Nature of the Chemical Bond, Cornell University Press, Ithaca, New York,
3rd Edn., 1960, p. 49.
10. Burrau O., Kgl. Danske Videnskab. Sclskab, 7, 1 (1927); Pauling L., Chem. Revs., 5,
173 (1928); Finkelstein B. N., Horowitz G. E., Z. Physik, 48, 118 (1928); Hylleraas E. A.,
ibid., 71, 739 (1931); Dickinson B. N., J. Chem. Phys., 1, 317 (1933); Jaffe G., Z. Physik,
87, 535 (1934).
11. Heitler W., London F., Z. Physik, 44, 455 (1927); Segiura T., ibid., 45, 484 (1927);
Wang S. C., Phys. Rev., 31, 579 (1928); Rosen N., ibid., 38, 2099 (1931); Weinbaum S.,
J. Chem. Phys., 1, 593 (1933); James H. M., Coolidge A. S., ibid., 1, 825 (1933).
12. Pauling L., Proc. Nat. Acad. Sci. (U.S.), 14, 359 (1928); J. Am. Chem. Soc., 53, 1367
(1931); Nature of the Chemical Bond, 3rd Edn., 1960, p. 108; Slater J. C., Phys. Rev.,
37, 481 (1931).
13. Pauling L., J. Am. Chem. Soc., 53, 1367 (1931).
14. Hultgren R., Phys. Rev., 40, 891 (1932).
15. Pauling L., Nature of the Chemical Bond, 3rd Edn., 1960, Chap. V, p. 145.
16. Hiickel E., Z. Physik, 60, 423 (1930); Penney W. G., Proc. Roy. Soc. (London), A144,
166 (1934); A146, 223 (1936).
17. Hall G. G., Lennard-Jones J. E., Proc. Roy. Soc. (London), A205, 357 (1951).
18. Из инфракрасных спектров: Allen H. C., Plyler Е. К., J. Am. Chem. Soc., 80, 2673
(1958). Из спектров комбинационного рассеяния: Dowlong J. М., Stoicheff В. Р.,
Canadian J. Phys., 37, 703 (1959).
19. Из микроволновых спектров: Ока Т., J. Phys. Soc. Japan, 12, 2274 (1960).
20. Из микроволновых спектров: Edgell И7. F., Kinsley Р. A., Amy J. W., J. Am. Chem.
Soc., 79, 2691 (1957).
21. Из микроволновых спектров: Sekino S., Nishikawa Т., J. Phys. Soc. Japan, 12, 43
(1957).
22. Дифракцией электронов: Pauling L., Brockway L. O., J. Am. Chem. Soc., 59, 1223
(1937).
23. Walsh A. D., Disc. Faraday Soc., 2, 18 (1947).
* Такой вывод можно сделать на основании рис. 2, пз которого видно, что s-уровни
расположены ниже соответствующих р-уровнеп. Это, конечно, согласуется с рис. 11,
из которого видно, что у ядра s-орбиталь имеет максимальную плотность, а р-орбпталь —
нулевую плотность.
ГЛАВА II*
ВНУТРИМОЛЕКУЛЯРНОЕ И МЕЖМОЛЕКУЛЯРНОЕ
ВЗАИМОДЕЙСТВИЯ
1. Электронные взаимодействия между нереагирующими молеку-
лами .............................................................. 38
2. Значение внутримолекулярного электронного взаимодействия 40
3. Передача внутримолекулярного взаимодействия............... 64
Список литературы......................................... 98
* Эта и три последующие главы написаны с широким использованием работы автора
«Принципы электронной теории органических реакций», опубликованной в журнале
Chem. Revs., 15, 225 (1934) (см. сборник «Электронная теория в органической химии»,
ОНТИ, 1936).
1. Электронные взаимодействия между нереагирующими
молекулами
Прежде чем рассмотреть электронное взаимодействие между частями моле-
кулы, полезно классифицировать все известные типы взаимодействия между
отдельными нереагирующими молекулами. Можно ожидать, что различные
типы взаимодействия, которые возникают между молекулами, должны
проявляться и во внутримолекулярном взаимодействии между соответственно
расположенными частями одной молекулы. Впрочем, как будет видно из
дальнейшего изложения, имеются также определенные эффекты, характер-
ные только для взаимодействия внутри молекулы, которые в основном
зависят от типов связей. Сначала удобнее рассмотреть межмолекулярные
взаимодействия. Их можно разделить на три основных вида: электростати-
ческое, электрокинетическое и обменное.
а. Электростатическое взаимодействие. Почти все молекулы обладают
рядом электростатических характеристик, которые объединяются общим
термином «поляризация». Это означает, что молекула создает внешнее элек-
тростатическое поле. Как правило, такие поля проще всего рассматривать
как совокупности накладывающихся полей, каждое из которых характе-
ризуется своим пространственным распределением. Поля сил дальнего
действия возникают только в тех случаях, когда молекула представляет
собой ион *.
Электростатический потенциал результирующего заряда иона ±Ze про-
порционален заряду, обратно пропорционален расстоянию г и не зависит
от направления. Если молекула не имеет результирующего заряда, но
обладает дипольным моментом, она образует поле сил среднего действия.
Потенциал этого поля пропорционален дипольному моменту ц, меняется
с расстоянием обратно пропорционально г2 и зависит от направления,
поскольку ц — вектор. Так, например, в молекуле фтористого водорода
потенциал вблизи атома водорода будет положительным, а вблизи атома
фтора — отрицательным. Если молекула не обладает ни дипольным момен-
том, ни высокой симметрией, она может иметь квадрупольный момент,
который приводит к возникновению поля сил близкого действия. Потенциал
этого поля пропорционален квадрупольному моменту 0, изменяется с рас-
стоянием обратно пропорционально г3 и зависит от направления, поскольку
0 — тензор. Так, например, в молекуле водорода потенциал будет положи-
тельным вблизи ее концов и отрицательным по обе стороны от линии
связи.
Разумеется, на любой электрический заряд или систему зарядов в пределах
эффективного радиуса какого-либо из этих потенциальных полей будет дей-
ствовать сила. В любом потенциальном поле сила, действующая на одиноч-
ный электрический заряд или, как говорят, на «ноль», пропорциональ-
на градиенту потенциала; сила, действующая на диполь, пропорцио-
нальна скорости изменения градиента по данному направлению простран-
ства и т. д.
Все молекулы обладают электростатическим свойством поляризуемости.
Она зависит от легкости, с которой смещаются в противоположных направ-
лениях ядерные и электронные заряды в электростатическом поле. При
этол: возникает индуцированный дипольный момент. Индуцированный момент
пропорционален поляризуемости а поляризуемой молекулы и градиенту
потенциала индуцирующего электрического поля; этот момент зависит
* Под термином «молекула» мы понимаем любую кинетически независимую частицу,
будь то атом, нейтральная молекула, одноатомный или многоатомный ион.
38
Внутримолекулярное и межмолекулярное взаимодействия
от ориентации (а представляет собой тензор). Если источником электриче-
ского поля является другая молекула, то между последней и индуциро-
ванным дипольным моментом возникает сила. Подобно силе, действующей
на постоянный диполь, сила, действующая на индуцированный диполь,
пропорциональна скорости изменения градиента потенциала по данному
направлению пространства; она также пропорциональна индуцированному
моменту.
Все вышеупомянутые силы независимо от того, возникают ли они вследствие
только поляризации молекул или вследствие поляризации и поляризуемости,
вносят значительный вклад в электростатическое взаимодействие, которое
обусловливает когезию молекул.
б. Электрокинетическое взаимодействие. В предыдущем разделе было
принято, что электроны в молекуле достаточно хорошо описываются стати-
ческим распределением электронного заряда, и не учитывалось действи-
тельное движение электронов. Поэтому не был рассмотрен другой тип сил
близкого действия, а именно силы электрокинетического взаимодействия.
Электрокинетические силы существенным образом зависят от движения
электронов, они обязательно приводят к притяжению. Природа этих сил
была рассмотрена Лондоном [1]. Как показал Лондон, эти межмолекулярные
силы возникают во всех без исключения случаях и часто представляют собой
сильнейшие из сил притяжения, известных под собирательным названием
сил Ван-дер-Ваальса: эти силы определяют агрегатное состояние веще-
ства.
Природу электрокинетические сил * можно понять из рассмотрения двух
атомов гелия, расположенных достаточно далеко друг от друга, чтобы мож-
но было не учитывать электронный обмен. Одни атомы притягивают другие,
хотя не имеют постоянного дипольного или квадрупольпого моментов. Ядро
и каждый электрон одного атома взаимодействуют отдельно с ядром и каж-
дым электроном другого; при этом движение электронов изменяется так,
что суммарная энергия взаимодействия становится минимальной. Так, если
представить, что атомы расположены справа и слева один от другого, то,
когда электроны одного атома будут двигаться к правой стороне данного
атома, электроны другого атома будут, вероятнее всего, находиться в правой
стороне, а не в левой; когда электроны одного атома будут двигаться к левой
стороне, электроны другого атома будут стремиться влево. В результате
этого наблюдается притяжение. По существу, это представляет собой при-
тяжение флуктуирующих взаимно индуцированных диполей, энергия взаи-
модействия которых меняется с расстоянием обратно пропорционально г6.
Это только главный член ряда электрокипетических взаимодействий: должны
существовать силы притяжения еще более близкого действия, энергии
взаимодействия которых меняются обратно пропорционально г8, г10
и т. д.
Как указывал Лондон, электрокинетические силы не вызывают насыщения;
другими словами, притяжение между двумя атомами не мешает заметным
образом притяжению каждого из этих двух атомов к третьему. Ясно, что
это свойство необходимо учитывать при объяснении конденсированного
состояния вещества. Оно резко отличает электрокинетические силы от сил
электронного обмена, которые ведут к образованию ковалентных связей.
* Оти силы обычно называются дисперсионными; Лондой показал, что их можно
приблизительно вычислить из данных по оптической дисперсии. Они также называются
силами Лондона, и такое название, вероятно, было бы общераспространенным, если бы
Лондоном не были открыты и более важные обменные силы.
39
Глава II
в. Обменное взаимодействие. На очень малых расстояниях [например,
меньше 2,8 А (0,28 нм) для двух атомов неона или 3,4 А (0,34 нм) для двух
атомов аргона] обменные силы преобладают над всеми другими. Их физи-
ческая природа описана в гл. I. Если это не противоречит принципу Паули,
то обменные силы вызывают образование ковалентной связи. Если же речь
идет о молекулах, в которых все возможные связи уже образованы, обмен-
ные силы приводят к отталкиванию.
Это отталкивание очень резко возрастает с уменьшением расстояния, что
видно из рис. 10, па котором изображена кривая для энергии взаимодей-
Рпс.
ствия двух атомов аргона. Крутая
левая ветвь каждой кривом пока-
зывает преобладание отталкива-
ния, происходящего благодаря
обмену на малых расстояниях, а
значительно менее крутая правая
ветвь соответствует относительно
слабому электрокинетическому
притяжению, которое преобладает
па больших расстояниях. Между
этими ветвями лежит минимум
энергии; положение минимума со-
ответствует равновесному расстоя-
нию между атомами вещества в
конденсированном состоянии и
(после поправки на пулевую и
тепловую энергию) теплоте испа-
10. Энергия взаимодействия двух атомов
аргона, рассчитанная по уравнению
U — be~r,p — сг~в.
---------- рассчитано при использовании
констант Бакингема;------------рассчи-
тано при использовании исправленных
констант, предложенных Корнером. Энер-
гия атомной пары умножена на число Аво-
гадро, а затем выражена в килокалориях,
чтобы облегчить сравнение с другими зна-
чениями энергии взаимодействия.
рения.
Теоретически при не слишком ма-
лых расстояниях обменная энер-
гия изменяется с расстоянием по
закону R (г)е-г,'Р1 где R (г) — по-
лином от г, а р — постоянная.
Обычно в численных расчетах
R (г) заменяют постоянной Ь, пы-
таясь компенсировать ошибку рас-
соответствующим подбором констант. Ограничиваясь в выражении
электрокинетической энергии его главным членом, содержащим г~в.
чета
Для
полную энергию взаимодействия можно выразить уравнением
U — Ъс~т^р — ст 6
где константы Ь, с и р должны быть подобраны эмпирически. Это уравнение
очень полезно для расчетов уравнений состояния, «транспортных» свойств,
а также свойств кристаллов атомов инертных газов и некоторых других
неполярных молекул. Кривые на рис. 10 построены на основании этого
уравнения с константами по Бакингему и Корнеру [2].
2. Значение внутримолекулярного электронного
взаимодействия
а. Наличие внутримолекулярного электронного взаимодействия. Химия
была бы значительно более легкой наукой, если бы каждый связанный атом
или группа атомов сохраняли свои химические и физические свойства или
40
Внутримолекулярное и межмолекулярное взаимодействия
их влияние на соответствующие свойства молекул оставалось неизменным
во всех молекулах, в состав которых они входят. На самом деле это не так;
свойства, которые особенно характерны для данного атома или группы ато-
мов, обычно заметно изменяются в зависимости от свойств других групп,
имеющихся в молекуле, и от того, как эти другие группы связаны и рас-
положены по отношению к рассматриваемому атому или группе. Поэтому
электронная теория атома и теория валентной связи атомов в молекулах
сами по себе не являются достаточной теоретической основой для интер-
претации физических и химических свойств. Тем не менее они представляют
собой фундамент, на котором необходимо возвести надстройку, а именно
теорию внутримолекулярного электронного взаимодействия, чтобы учесть
очевидное влияние, которое атомы или группы атомов в молекуле оказы-
вают друг на друга. Ниже будет показано, что эта теория весьма похожа
на теорию валентной связи и характеризуется, подобно последней, наличием
электростатического и резонансного аспектов; опа. разумеется, может счи-
таться развитием теории валентности, если принять во внимание, что теория
электронного взаимодействия рассматривает изменения в характере связен
вследствие взаимодействия между частями молекулы.
б. Внутримолекулярное взаимодействие и физические свойства. Наи-
более наглядное представление о внутримолекулярном взаимодействии
можно получить, изучая физические свойства молекул. Например, как уже
отмечалось, молекулы обладают комплексом электростатических свойств,
объединяемых термином «поляризация», наиболее важной количественной
характеристикой которой в случае нейтральных молекул служит их диполь-
ный момент. Следует рассмотреть, в какой мере понятию «поляриза-
ция» может быть придан строгий смысл не только по отношению к целым
молекулам, но также и к отдельным комбинациям атомов и атомных групп
и, в частности, в какой мере дипольные моменты молекул можно разложить
на составные части, характеризующие отдельные атомы или группы атомов.
Этот вопрос имеет свою историю. После того как принципы, лежащие в основе
определения молекулярных дипольных моментов, были уже даны Дебаем
[3], по еще до того, как многие дипольные моменты были измерены, Томсон
[4] высказал предположение, что дипольные моменты можно вычислять как
векторные суммы характеристических моментов атомов и групп. Вскоре
после этого были измерены более точно дипольные моменты очень многих
соединений и стало ясно, что предположение Томсона приближенно верно.
Затем последовал ряд попыток па основе принципа аддитивности построить
таблицы со значениями моментов групп, на основании которых по правилам
векторного сложения можно вычислить моменты молекул. Естественно, что
эти попытки не могли быть успешными, во-первых, потому, что внутренняя
индукция должна вызывать новые моменты, поскольку молекулы характе-
ризуются не только поляризацией, но и поляризуемостью; во-вторых, пото-
му, что точная аддитивность предполагает отсутствие внутримолекулярного
электрического взаимодействия, которое па самом деле является основой
для объяснения общих химических свойств молекул [5]. По мере того как
измерения делались более многочисленными и более точными, становилось
ясно, что отклонения от аддитивности являются правилом и они слишком
велики, чтобы ими можно было пренебречь. Следующая фаза в развитии
рассматриваемой проблемы, а именно установление соотношения между
отклонениями в дипольных моментах и структурой, а также связи этих
отклонений с другими явлениями, зависящими от внутримолекулярного
взаимодействия, рассмотрена в гл. III. Итак, хотя точная аддитивность
п не может быть установлена, сам принцип аддитивности в первом прибли-
41
Глава II
жении сохраняется; качественно можно считать обоснованным, что группы
обладают определенной поляризацией, хотя соответствующие численные
значения не являются строго постоянными и характеристичными.
Примерно сходное положение создается при попытке разложить на атомные
и групповые составляющие поляризуемость молекул. Среднее значение
поляризуемости молекулы (среднее значение тензора), если не считать
постоянного численного множителя, определяется молекулярной рефрак-
цией газа или жидкости; уже давно известно, что молекулярная рефракция
приближенно подчиняется правилу аддитивности. При этом существуют
отклонения; однако большие отклонения встречаются реже, чем в случае
дипольных моментов (гл. III), и наблюдаются главным образом при наличии
высокой поляризации и ненасыщенности любого типа, включая неподелен-
пые валентные электроны, незаполненные валентные электронные оболочки
и кратные связи. Поэтому в раннем периоде изучения молекулярных рефрак-
ций большие отклонения приписывали особенностям строения, и малые
отклонения сглаживались в процессе усреднения при вычислении атомных
или групповых значений. При этом также наблюдаются отклонения от адди-
тивности, свидетельствующие о явлениях внутримолекулярного электрон-
ного взаимодействия; все же остается приближенно верным, что связанные
атомы и группы имеют собственную поляризуемость, хотя измеряемые вели-
чины нельзя считать строго постоянными и характеристичными.
в. Внутримолекулярное взаимодействие и равновесия химических
реакций. В последующих главах будет рассмотрен вопрос о влиянии вну-
тримолекулярного электрического взаимодействия на различные химиче-
ские реакции. При таких исследованиях необходимо строго различать тер-
модинамический и кинетический аспекты.
Начнем рассмотрение с термодинамического аспекта. Следует отметить, что
при таком подходе не рассматривается скорость реакции, которая зависит
от механизма процесса. Термодинамика реакций не зависит от механизма.
При таком подходе имеет значение лишь степень превращения исходных
веществ по любому механизму в конечные продукты без всяких ограничений
во времени. Равновесная степень превращения не зависит от механизма
реакции, а зависит только от исходных веществ и конечных продуктов
и от таких физических условий, как давление и температура.
Равновесная степень превращения для реакции
А+В+... X+Y+...
может быть выражена константой равновесия К
к ;[X] [Y]...
IА] [В] ...
Обычно принимают, что К равняется нулю, когда нет превращения, и ста-
новится бесконечной, когда равновесие достигается при полном превраще-
нии. В выражении для К величины [X]... являются, строго говоря, актив-
ностями; однако активности часто заменяют концентрациями в тех или иных
единицах (парциальные давления газов, мольные доли, моляльность, моляр-
ность и т. д.) в том случае, когда концентрации можно считать приблизи-
тельно пропорциональными активностям. Константа равновесия * зависит
* Нет необходимости рассматривать подробнее способы определения К пли вопрос
о том, каким образом соответствующие термодинамические функции исходных и конеч-
ных продуктов определяются для того, чтобы отвечать избранным условиям (условие
«стандартного состояния»). По этому вопросу см. работу [6].
Внутримолекулярное и межмолекулярное взаимодействия
от разностей термодинамических функций для исходных веществ, рассматри-
ваемых как одно целое, и конечных продуктов реакции, также рассма-
триваемых как целое. Во-первых, она зависит от разности (А//) энтальпий
исходных и конечных продуктов; эта разность обычно равна или очень
близка к разности (ДЕ1) их внутренних энергий, причем эти величины совпа-
дают с точностью до слагаемого (рАк), представляющего собой внешнюю
работу, которая выполняется, если реакция не проводится при постоянном
объеме. Во-вторых, константа равновесия зависит от разности (ЛА) энтро-
пий исходных и конечных продуктов; энтропия является термодинамической
мерой статистической вероятности системы. Обе рассмотренные величины
часто выражаются в виде одной разности (AG) свободной энергии, и связь
с константой равновесия может быть выражена поэтому в нескольких альтер-
нативных формах:
AG = АЯ - T\S = \Е + рАу - T\S = - RT In К
Если переписать это уравнение в форме
__ e\S/Re-MI/RT
то можно сразу увидеть, что конечные продукты будут образовываться тем
в большем количестве, чем больше их энтропия и чем меньше их энтальпия
по сравнению с соответствующими свойствами исходных продуктов.
Чтобы понять, как, например, внутримолекулярные электрические эффекты
могут различно влиять на энтальпию и свободную энергию исходных и конеч-
ных продуктов реакции, рассмотрим исходную молекулу А — Ви конечную
молекулу А — В' при реакции
А-В+... А-В'+...
Допускается, что В' является более электроположительным, чем В, т. е.
В' отталкивает электроны сильнее, чем В, по направлению к общему моле-
кулярному остатку А. Наряду с этой системой рассмотрим вторую сходную
систему, в которой А заменено на Ар
At — В+... Д± Ai —В'+...
причем At электроотрицательное А, т. е. At имеет большее сродство к элект-
ронам, чем А. В этом случае дополнительная поляризация В' по сравнению
с В и дополнительная поляризация At по сравнению с А будут в молекуле
Aj — В' действовать в одном направлении, что приведет к отрицательной
энергии взаимодействия. Другими словами, образующаяся молекула Ai — В'
будет более устойчива по сравнению с исходной А — В, нежели молекула
А — В' по сравнению с исходной А — В. Следовательно, при переходе
от первой реакции ко второй положение равновесия должно сместиться
вправо. Такой вывод основывается на рассмотрении ожидаемого различия
в разностях энергий, если, как часто бывает, аналогичной разностью энтро-
пий можно пренебречь.
Характер последнего допущения может быть выяснен на следующем при-
мере. Предположим, что А — С6Н5, В = СООН, В' = ССН и Aj =
= и-ХО2С6Н4. причем первая реакция—ионизация бензойной кислоты, а вто-
рая — ионизация и-нитробензойной кислоты в воде:
СвН5СООН ~> СвН5СОО- + Н+ (в воде)
NO2CeH4COOH 7» NO2C6II4COO-- Н+ в (воде)
43
Глава II
Тогда дополнительная поляризация В' по сравнению с В по существу заклю-
чается в отрицательном заряде карбоксилат-иопа. Это сильно сказывается
на общей энергии, во-первых, вследствие ориентации постоянных диполей
воды и их притяжения и, во-вторых, благодаря индуцированию дополни-
тельных диполей в органических остатках и в ближайших молекулах воды.
При этом возникает также большой энтропийный эффект главным образом
благодаря тому, что положения ближайших молекул воды становятся более
фиксированными, что уменьшает статистическую вероятность нахождения
иона в сольватированном состоянии. Предположим, что величины этих
энергетических и энтропийных эффектов для первой и второй реакции
не сильно отличаются друг от друга. Можно допустить, например, что поле
диполя нитрогруппы мало влияет на силу притяжения или устойчивость
положения тех молекул воды, которые находятся ближе всего к карбокси-
лат-иону и поэтому оказывают определяющее влияние на энергию и энтро-
пию сольватированного иона. Это допущение можно также легко подверг-
нуть критике для энергетических и энтропийных эффектов, возникающих
при взаимодействии питрогруппы с ее ближайшим окружением. В этом
случае высказанным предположениям можно придать другую эквивалентную
форму, а именно: влияние заместителей в исходных соединениях сохраняется
неизменным в конечных продуктах реакции. Но. помимо всех этих эффектов,
которые по предположению взаимно уничтожаются в разностях разностей
энергий и в разностях разностей энтропий начальных и конечных продук-
тов с заместителем и без него, остается некомпенсированное внутреннее
взаимодействие двух дополнительных полярных связей, совместно присут-
ствующих в ионе ге-питробензойпой кислоты. Это должно вызвать остаточ-
ный энергетический эффект. В такой молекуле, где взаимодействующие
центры отделены друг от друга и имеется лишь небольшая возможность
стереохимической деформации, взаимодействие пе может вызвать большого
остаточного энтропийного эффекта.
По этим причинам следует ожидать большего значения для равновесной
степени ионизации и-нитробензойпой кислоты по сравнению с бензойной
кислотой: это и подтверждается экспериментом. Обычно (гл. XIV) степень
равновесной ионизации выражают через константу кислотности.
xa=[ii3o+][B-7iHB]
называемую также силой кислоты НВ в воде; уравнению часто придают
логарифмическую форму
РХ„ = — 1g Ка
Наблюдаемые величины рА„ для бензойной и /г-нитробен.зойной кислот [7]
равны 4,20 и 3,42 соответственно.
Вышеприведенное рассмотрение электрических взаимодействий, влияющих
па равновесие, было очень упрощено, и его необходимо расширить. Упроще-
ние состояло в предположении, что поляризация определенных групп может
быть без изменения перенесена из одного соединения в другое; допускалось,
что дополнительная поляризация At по сравнению с А переносится без
изменения из Aj — В в А[ — В', а дополнительная поляризация В' по
сравнению с В переносится без изменения из А — В' в Ai — В', так что
можно было ограничиться рассмотрением только взаимодействия неизменяе-
мых дополнительных поляризаций в А, — В'. В действительности дело
обстоит иначе; как было показано в этой главе в разд. 2,6, поляризации
групп пе являются постоянными и зависят от взаимодействия между ними;
44
Внутримолекулярное и межмолекулярное воанмоиейстиия
связанные с этим изменения, т. е. поляризации взаимодействия, должны
приниматься в расчет. Поляризация группы А изменяется при превраще-
нии А — В в А — В'; взаимодействие, вызывающее это изменение, обуслов-
ливает энергетический и, возможно, энтропийный эффект. Способность
группы Aj к взаимодействию может быть уже иной, и изменение поляриза-
ции этой группы будет различным при превращении At — В в At — В';
поэтому такого рода взаимодействие приведет к иному энергетическому
п. возможно, иному энтропийному эффекту. Представляет интерес различие
между этими энергетическими эффектами и между энтропийными эффектами,
если последние наблюдаются. Это различие показывает, в какой мере поля-
ризация взаимодействия влияет на зависимость положения равновесия
от строения реагирующих молекул. Такое влияние иногда бывает очень
значительным.
Например, если игнорировать поляризацию взаимодействия, можно ожи-
дать, что анисовая кислота ге-СН3ОС6Н4СООН вследствие электроотрица-
тельности кислородного атома метоксильной группы является более сильной
кислотой, чем бензойная, т. е. что первая имеет более высокое значение Кп
и более низкое значение рКа. На самом доле анисовая кислота — более
слабая кислота, чем бензойная, о чем свидетельствуют следующие данные [7]:
С6Н5СООН (рКа = 4,20) и ге-СН3ОС6Н4СООН (РКа = 4.47). Это объяс-
няется поляризацией взаимодействия (а по существу, как мы увидим ниже,
«мезомерным эффектом»), которая наблюдается как в анисовой кислоте, так
и в ее анионе, но сильнее выражена в недиссоциировапной кислоте и, таким
образом, избирательно ее стабилизирует [8].
г. Внутримолекулярное взаимодействие и скорость химических реак-
ций. Кинетика реакций тесно связана с их механизмом: реагенты сталки-
ваются и атомы перегруппировываются, причем старые связи разрываются
и образуются новые: скорость реакции определяется деталями химического
процесса, а не только конечным результатом.
Особого внимания заслуживают термические реакции; процессы перегруп-
пировки атомов в таких реакциях характеризуются двумя особенностями,
настолько общими, что они определяют рассмотрение всех деталей. Первая
особенность заключается в том, что для проведения реакции почти никогда
не требуется энергии, достаточной для того, чтобы разорвать старые связи
полностью перед образованием новых; процессы разрыва и образования
связей протекают синхронно. Это предположение было впервые ясно сфор-
мулировано Льюисом [9] и развито в качестве составной части общей теории
Лондоном [10]. Для многих газовых реакций указанный принцип следует
применять при рассмотрении процесса разрыва и образования ковалентных
связей. Это относится и ко многим реакциям в растворе. Однако при рас-
смотрении большого числа других реакций в растворе из-за наличия сильной
электростатической связи, как, например, при сольватации ионов [11],
требуется более широкая интерпретация этого принципа. Так, например,
разрыв ковалентной связи может происходить синхронно с сольватацией
иона без одновременного образования новой ковалентной связи, и, с другой
стороны, образование ковалентной связи может протекать синхронно с де-
сольватацией иона.
Второй общий принцип, сформулированный Лондоном [10], заключается
в том, что все термические реакции являются «адиабатическими». Это озна-
чает, что в ходе реакции не происходит электронного возбуждения и что
поэтому по мере перегруппировки атомов в соответствии с принципом Фран-
ка — Кондона потенциальная энергия системы изменяется не скачкооб-
разно, а непрерывно.
4-5
Глпва II
Значение этих принципов можно проиллюстрировать на простом примере
реакции в газовой фазе между атомом и двухатомной молекулой
X + Y —Z
X-Y-J-Z
По мере того как X приближается кт — Z, взаимодействие между связы-
вающим электроном X и электронами связи Y — Z приводит к тому, что
расстояние между Y и Z начинает постепенно увеличиваться. Вначале
потенциальная энергия растет, но не в такой мере, как это наблюдалось
бы при увеличении расстояния между Y и Z в отсутствие взаимодействия,
в основном потому, что электрон Y может теперь спариваться двумя спосо-
бами, и этот резонанс уменьшает энергию системы. Аналогичным образом
можно рассматривать и процесс приближения Z к X — Y. Между эти-
ми начальными и конечными состояниями реализуется конфигурация
X - • • Y • • • Z, названная переходным состоянием и характеризующаяся самой
большой энергией по сравнению с любой
промежуточной конфигурацией, через ко-
торые проходит система. Но благодаря
резонансу эта максимальная энергия мень-
ше, чем энергия, необходимая для того,
чтобы вызвать диссоциацию каждой двух-
атомной молекулы *. В переходном состоя-
нии Y частично связан ковалентно как
с X, так и с Z. Если соответствующим
образом выбрать координату, измеряющую
степень перегруппировки атомов, то из-
менение потенциальной энергии на протя-
жении процесса можно представить кривой
общей формы, показанной на рис. 11.
Разность ДА потенциальной энергии между
начальным и переходным состояниями яв-
ляется энергией активации реакции без
учета некоторых поправок. Что касается
поправок, то их необходимо ввести, во-
первых, на разность нулевых колебатель-
Рис. И. Изменение потенциальной
энергии при реакции X +
+ YZ = XY + Z.
Для теплоты реакции ДН и энер-
гии активации ДЕ^ не введены
поправки на нулевую энергию
и тепловую энергию соответ-
ствующих частиц.
ных энергий в начальном и переходном со-
стояниях’и, во-вторых, на разность их тепловых энергий при температуре реак-
ции. Чтобы лучше понять, что подразумевается под термином «координата
реакции», необходимо рассмотреть потенциальную энергию как функцию всех
внутренних координат системы, т. е. (Зге — 6) координат для ге-атомной
системы. Для трехатомной системы имеются три внутренние координаты.
Однако рассмотрение можно упростить, приняв во внимание, что во всех
конфигурациях, приближающихся к конфигурации переходного состояния,
атомы X и Z отталкивают друг друга, так что для переходного состояния
следует учитывать лишь нелинейные конфигурации трех атомов. При этом
ограничении остаются две внутренние координаты, в качестве которых
удобно принять расстояния XY и YZ. В этой системе координат потен-
циальную энергию можно представить в виде поверхности, которая состоит,
как показывает контурная диаграмма на рис. 12, из двух «долин», сходя-
щихся в точке «седловины», соответствующей переходному состоянию.
Координата реакции проходит вдоль дна каждой долины и через седловину,
* Для случая X = Y = Z = Н прирост энергии в переходном состоянии составляет
9 ккал/моль (37,68-103 Дж/моль), тогда как энергия диссоциации равна 103 ккал/моль
(431,24-103 Дж/моль).
46
Внутримолекулярное и межмолекулярное взаимодействия
всегда следуя по паипизшему возможному пути от дна одной долины к дну
другой. Кривая па рис. И является профилем сечения этой поверхности,
взятого вдоль координаты реакции.
Если бы система XYZ представляла собой устойчивую молекулу, то в пере-
ходном состоянии координата реакции соответствовала бы колебательной
координате X — Y — Z. Однако переходному состоянию при изменении
этой особой координаты соответствует энергетический максимум, а не
минимум, как в молекуле. С другой стороны, для ортогональной коорди-
наты X — Y — Z переходное состояние, как и для молекулы, соответствует
энергетическому минимуму. В об-
щем случае реагирующей системы,
включающей п атомов, поверх-
ность потенциальной энергии сле-
дует представлять себе поверхно-
стью в пространстве (Зи — 6) про-
странственных координат и одной
координаты энергии. В такой си-
стеме переходное состояние ста-
бильно в (Зге — 7) колебательных
координатах, по не стабильно по
координате реакции. Для этой по-
следней внутренней степени сво-
боды движение системы можно ско-
рее уподобить поступательному
движению, чем колебательному.
Эти представления лежат в основе
теории переходного состояния [12].
Данная теория исходит из того,
что переходное состояние находит-
Рис. 12. Изменение потенциальной энергии
для линейных конфигураций реаги-
рующей системы X-J-YZ = XY + Z.
Форма энергетической поверхности указа-
на энергетическими контурами: две доли-
ны, соответствующие начальному и конеч-
ному состояниям, сходятся в седловине,
представляющей переходное состояние.
Координата реакции (................) проходит че-
рез энергетический максимум в переход-
ном состоянии. Однако ортогональная ко-
ордината (-------) проходит через мини-
мум энергии в переходном состоянии.
ся в равновесии с начальным со-
стоянием; следовательно, необхо-
димо вычислить относительное ко-
личество систем, находящихся в
переходном состоянии. Это зависит
от энергии и энтропии переходного
состояния по сравнению с энер-
гией и энтропией начального со-
стояния, т. е. от высоты и формы
седловины поверхности потенци-
альной энергии. На низкую седловину подняться легче, чем на высо-
кую той же самой формы. Седловина, которая широко открыта при
подходе не только вдоль координаты реакции, но и вдоль других коор-
динат (допускается, таким образом, большая свобода в расположении ато-
мов), более доступна, чем седловина той же самой высоты, которая сильно
закрыта потенциальными барьерами при подходе вдоль тех же координат,
что сильно ограничивает возможные положения атомов. Скорость реакции
прямо пропорциональна числу систем в переходном состоянии и обратно
пропорциональна среднему времени, которое необходимо для прохождения
через стадию переходного состояния.
Формальное сочетание этих факторов приводит к уравнению для скорости
реакции
Скорость = (JcT/h) e^s h/Re,-mtMrt
47
Глава II
где к — константа скорости в кинетическом уравнении для реакции поряд-
ка а относительно реагента А и Ъ относительно реагента В (суммарный
порядок равен а + Ъ 4- . .
Скорость = к [ А]а [В]ь ...
В первом уравнении к — постоянная Больцмана и h — постоянная Планка;
А//4= (теплота активации) и A.S4- (энтропия активации) представляют
собой разности между соответствующими термодинамическими функциями
переходного и начального состояний. При расчете энтропии переходного
состояния удобно опустить ту ее часть, которая зависит от движения вдоль
координаты реакции. Эту часть, так же как и зависимость скорости от вре-
кТ
мени прохождения через переходное состояние, учитывает множитель —.
Теплота активации приближенно равна энергии активации, причем разница
заключается только в незначительной поправке на кинетическую энергию
и внешнюю работу, если последняя имеется
АЕ А = A7Z* + кТ - pbv*
В элементарной форме, в которой закон действия масс обычно применяется
в кинетике реакций, в приведенном выше уравнении для скорости в квад-
ратных скобках указываются концентрации. Однако в теории, по которой
скорость реакции пропорциональна той доле всех имеющихся систем, кото-
рая находится в переходном состоянии, очевидно, необходимо учитывать
поправку на активности или коэффициенты активностей, если последние
значительно отличаются от единицы. Термодинамическая активность а~--
= c'-rf^ переходного состояния пропорциональна произведению активностей
(«д = сд/д и т. д.) всех реагирующих молекул; следовательно, концентра-
ция систем в переходном состоянии с^=, которой должна быть пропорциональ-
на скорость реакции, равна этому произведению активностей, деленному
па коэффициент активности переходного состояния. Таким образом, ско-
рость реакции выражается уравнением
Скорость = /гсдСв . .. - -А —
где с — концентрации и / — коэффициенты активности, причем /4= — коэф-
фициент активности для переходного состояния. Если реакции протекают
в растворе, то множитель, содержащий коэффициенты активности, должен
учитывать все отклонения от идеального состояния. Это кинетическое урав-
нение было предложено Бренстедом в 1922 г., который, таким образом, уже
тогда предвосхитил некоторые идеи теории переходного состояния [13].
Следует отметить, что ввиду отсутствия достаточных данных об энергети-
ческих поверхностях реакций полное уравнение теории переходного состоя-
ния можно использовать только в редких случаях при строгих вычислениях
скорости реакции. Тем не менее часто можно с пользой применять идеи
этой теории при качественной оценке влияния структуры молекулы или
растворителя на теплоту и энтропию активации и тем самым на скорость
реакции.
Величины, которые обычно вычисляются па основании измерений темпера-
турной зависимости скорости реакции, являются параметрами А и Ед в урав-
нении Аррениуса
к = Ае~Е^пг
48
Внутримолекцлярипе и межлюлекулярное взаимодействия
Сравнение с уравнением теории переходного состояния показывает, что
уравнение Аррениуса является лишь приближенным, поскольку констан-
та А в действительности включает ряд величин, зависящих от температуры.
Однако, как правило, эти величины изменяются с температурой значитель-
но медленнее, чем множитель е-ЕА/нт, так что весьма малые изменения
в величине Ел могут почти полностью компенсировать незначительные
изменения величины А в обычно используемом интервале температур.
По этой причине принято считать аррениусовскую энергию активации Е&
приближением к истинной энергии активации, а логарифм величины А,
фактора частоты по Аррениусу,— приближенной мерой (почти линейной
функцией) энтропии активации; обе эти величины отражают в полуколи-
чественной форме изменения энергии и энтропии активации ряда реакций
или же одной реакции в ряде растворителей.
В связи с такими представлениями об общей природе факторов, определяю-
щих скорость реакции, возникает вопрос о том, как электрические эффекты
могут влиять на энергию и энтропию активации и тем самым на скорость
реакции. Можно допустить, что очень многие реагенты первоначально дей-
ствуют либо на валентные электроны, либо на положительно заряженное
атомное ядро атакуемого атома и что, следовательно, возникновение в пер-
вом случае высокой или во втором — низкой электронной плотности у этого
атома облегчает образование переходного состояния. Другими словами,
в переходном состоянии реакционный центр становится сильно полярным,
что заставляет электроны либо оттягиваться от остальной части молекулы,
либо втягиваться в нее. В любом конкретном случае полярные заместители
должны этому или способствовать, или препятствовать.
Рассмотрим реакцию между А — В и С, в которой С атакует положительный
реакционный центр в В, образуя переходное состояние [А — В ... С]^,
причем электроны смещаются к радикалу А в большей степени, чем в исход-
ной молекуле А — В:
А —Вл-С —> [А—В ... С]' —> А—В' + С'
Наряду с этим рассмотрим аналогичную реакцию, в которой А структурно
изменено до А1; причем At имеет большее сродство к электронам, чем А:
At—В + С —> [Aj—В ••• С]^ —> А1—В' + С'
Тогда на основании аргументов, аналогичных изложенным в разд. 2,в
в связи с влиянием полярных групп на химическое равновесие, можно
утверждать, что энергия активации второй реакции должна быть меньше.
Соответственно скорость второй реакции должна быть больше при часто
выполняющемся условии, согласно которому влиянием структурного изме-
нения па энтропию активации можно пренебречь. Если А структурно изме-
нить и превратить в А2, причем А2 будет иметь меньшее сродство к электро-
нам, чем А. то влияние изменения энергии активации на скорость реакции
окажется обратным, нежели описанное выше. Если реакция принадле-
жит к противоположному полярному типу, т. е. если реагент С атакует
валентные электроны В с образованием переходного состояния, в котором
электроны оттянуты от радикала А более, чем в исходной молекуле, то кине-
тический эффект превращения А в Ai или в А2 будет в каждом случае про-
тивоположен описанному выше.
Пусть, например, А = С6Н5, В = Н, С = NO2, А! = w-NO2C6H4, В' =
= NO2 и С' = Н+. Сравним реакции нитрования бензола и нитробензола
4-0772 49
Глава II
в пара-положение, причем агентом в обоих случаях является ион нитрония
NO2C6H4H + NOJ
C6H5H + NOJ ->
C6I-I5NO2 + H+
2 J
NO2C0H4NO2 + H+
В этих реакциях ион нитрония атакует электроны атома углерода бензола
и образуется переходное состояние (указано в скобках), причем реакцион-
ный центр поляризуется положительно. Присутствие в исходной молекуле
нитрогруппы, обладающей формальным диполем, должно вызывать в пере-
ходном состоянии такое взаимодействие, которое повышает энергию акти-
вации нитрования нитробензола по сравнению с нитрованием незамещенного
бензола. Скорость нитрования нитробензола настолько меньше скорости
нитрования бензола, что обе эти скорости невозможно экспериментально
измерить при одинаковых условиях. На основании известных данных можно
считать, что скорости сравниваемых реакций отличаются по крайней мере
в 10е раз. С теоретической точки зрения можно ожидать, что это различие
почти целиком объясняется разностью в энергиях активации. Поскольку
эти молекулы обладают довольно жесткой структурой, а модификация
строения исходных молекул локализуется на некотором расстоянии от реак-
ционного центра, нельзя ожидать заметных изменений в энтропии актива-
ции. Однако структурное изменение в непосредственной близости к реак-
ционному центру (как при opmo-замещении) или замена растворителя,
в котором протекает реакция, может изменить форму энергетической поверх-
ности в седловине и тогда следует ожидать изменения не только энтропии,
но п энергии активации.
Это предварительное описание электронных кинетических эффектов нуждает-
ся в некотором расширении подобно тому, как первоначальное описание
электронных термодинамических эффектов было расширено в разд. 2.в.
Анализ кинетических факторов до сих пор ограничивался рамками так
называемых эффектов поляризации, вызываемых заместителями, поскольку
не учитывалась дополнительная поляризация, связанная с изменением строе-
ния молекулы при переходе от начального состояния к переходному. В дей-
ствительности в переходном состоянии поляризация, вызываемая атакую-
щими группами, накладывается на поляризацию реакционного центра, ока-
зывая, таким образом, влияние на энергию, а возможно, и на энтропию
активации. Однако это лишь неполное описание электронных кинетических
эффектов, поскольку не учитываются так называемые эффекты поляризуе-
мости. Заряд реакционного центра в переходном состоянии должен дефор-
мировать общую электронную систему, создавая временную поляризацию
взаимодействия. Эта поляризация меняется при любом структурном изме-
нении, влияющем на поляризуемость электронной системы или, иначе
говоря, приводящем к возникновению новой поляризуемости в соответствии
с деформирующим полем. Таким образом, изменение структуры связано
с дополнительной временной поляризацией взаимодействия, не представ-
ленной в начальном состоянии, но имеющейся в переходном состоянии,
когда она должна накладываться на поляризацию, вызываемую реакцион-
ным центром, оказывая, таким образом, влияние на энергию и, возможно,
на энтропию активации данной реакции.
Например, если принять во внимание только эффекты поляризации, можно
было бы ожидать, что вследствие электроотрицательности метоксигруппы
50
Внутримолекулярное и межмолекулярное взаимодействия
анизол должен нитроваться в пара-положение медленнее и с большей энер-
гией активации, чем бензол. В действительности анизол нитруется намного
быстрее, чем бензол, причем различие настолько велико, что эти скорости
невозможно непосредственно сравнивать в идентичных условиях. Соотно-
шение скоростей, по-видимому, превышает 104 [14] *. Такое резкое изме-
нение в противоположную сторону результатов, ожидаемых из рассмотре-
ния поляризации анизола, приписывается эффекту поляризуемости: поло-
жительная полярность, приобретаемая в переходном состоянии, обуслов-
ливает временное оттягивание электронов метоксильной группы к реак-
ционному центру, хотя смещение их в этом направлении находится в про-
тиворечии с обычной полярностью метоксильной группы.
При сравнении этих рассуждений с положениями, сформулированными
в разд. 2,в, видно, что влияние поляризации группы и поляризации взаимо-
действия на химическое равновесие аналогично влиянию поляризации и по-
ляризуемости на скорости реакции. При этом, конечно, имеется некоторое
различие в характере зависимости эффекта от времени. Поляризация группы
и поляризация взаимодействия являются свойствами молекул в нормальных
состояниях; однако поляризационный эффект обусловлен полярностью в нор-
мальном состоянии, а эффект поляризуемости — временное взаимодействие,
не связанное с полярностью в нормальном состоянии. Как показано ниже,
кинетические эффекты поляризуемости имеют большое значение.
д. Внутримолекулярное взаимодействие и стереохимическая форма.
Как уже отмечалось в гл. I, простая связь в соответствии с теорией обла-
дает круговой симметрией относительно оси, проходящей через ядра. Следо-
вательно, вращение вокруг таких простых связей, как СС-связь в этане,
должно быть совершенно свободным, если не считать возможного влияния
внутренних взаимодействий. Отсутствие в таких случаях свободного вра-
щения было показано Кемпом и Питцером [15], а также Кистяковским,
Лечером и Ститтом [16] на основании данных по энтропии и теплоемкости
этана при низкой температуре. При температуре жидкого воздуха вклад
в теплоемкость всех типов колебаний, кроме крутильных, относительно мал
из-за большой величины колебательных квантов. Тот факт, что значение
теплоемкости превышает величину 37?, обусловленную поступательными
и вращательными степенями свободы молекулы, объясняется наличием
крутильных колебаний. Если бы относительное вращение метильной группы
было совершенно свободным, то удельная теплоемкость для этой степени
свободы была бы такая же, как и для вращения (0,5??). Если это вращение
несколько заторможено, то соответствующая теплоемкость должна быть
большей (из-за потенциальной энергии). При сильно заторможенном вра-
щении и соответственно большом значении крутильной частоты удельная
теплоемкость должна быть пренебрежимо малой (из-за большой величины
кванта); в действительности она равна 0,37?. Примерно в то же время такой
же вывод был сделан Говардом [17] на основании анализа вращательных
структур по ИК-спектру этапа. Говард оценил частоту крутильных колеба-
ний, которая оказалась равной приблизительно 230 см-1. Частота, вычис-
ленная из температурных данных, составляет 280 см-1. Анализ сверхтонкой
структуры ИК-спектра, проведенный позже Смитом, дал величину 290 см-1;
и, наконец, прямое наблюдение Вейсом и Лероем [18] очень слабых полос
крутильных колебаний, запрещенных из соображений симметрии, но раз-
решенных кориолисовским взаимодействием, показало, что основная часто-
* При бромировании это отношение, по оценке де ла Мара и Вернона, составляет
1,2-109 [14].
51
Глава II
та равна 289 см-1; она значительно меньше частоты крутильных колебаний
этилена (1027 см-1) [19]. Энергетический барьер, тормозящий внутреннее
вращение в этане, равен 2,93 ккал/моль (12,27 *103 Дж/моль) [и, таким
образом, уровень нулевой энергии на 0,41 ккал/моль (1,72 *103 Дж/моль)
выше минимума потенциальной энергии]. Он весьма велик по сравнению
с долей энергии, приходящейся на одну степень свободы при обычной тем-
пературе [0,6 ккал/моль (2,51 -103 Дж/моль)], поэтому можно не сомневаться,
что почти все молекулы имеют энергетически наиболее благоприятную кон-
формацию. Однако этот барьер слишком мал, если его рассматривать как
энергию активации, что не позволяет надеяться на выделение геометриче-
ских изомеров производных этана, имеющих барьеры аналогичной высоты.
Конформациями называются те формы, которые может принять молекула
при вращении групп вокруг одной или нескольких одинарных (или лучше
говорить «формально» одинарных) связей. Вследствие симметрии молекула
этана должна существовать в одной из двух стабильных конформаций. Пер-
вая конформация, в которой пары метильных атомов водорода при проекции
на поперечную плоскость накладываются друг на друга, называется засло-
ненной конформацией. Вторая стабильная конформация соответствует моде-
ли, в которой в проекции на поперечную плоскость связи одной метильной
группы располагаются симметрично между связями другой метильной
группы. Такая конформация называется заторможенной. В 1949 г. Смит
детально проанализировал ИК-спектр этана и показал, что стабильной кон-
формацией является заторможенная [18].
Немного ранее такое же заключение было сделано при исследовании ста-
бильности циклоалканов. Стабильная заслоненная модель этапа должна
соответствовать такому случаю, когда между СН-связями каждого атома
углерода существует взаимное притяжение. В этом случае циклопентан
был бы более стабилен, чем циклогексан, так как в почти ненапряженной
плоской форме циклопентана все С — С-фрагменты имеют заслоненную
конформацию, а в молекуле циклогексана такая конформация привела
бы к значительному напряжению кольца. С другой стороны, стабильная
заторможенная модель этана подразумевает отталкивание между СН-связя-
ми каждого атома углерода. Такое отталкивание привело бы к меньшей
стабильности циклопентана по сравнению с циклогексаном, поскольку
в молекуле циклопентана все СН-связи одновременно не могут находиться
в заторможенном положении, тогда как в ненапряженной неплоской триго-
нальной форме циклогексана (форме «кресла») все С — С-фрагменты имеют
заторможенную конформацию. Спитцер и Хафман [20] на основании опре-
деления теплот сгорания ряда простых циклоолефинов пришли к заклю-
чению, что циклогексан (единственный циклопарафин, который может иметь
одновременно ненапряженное кольцо и все СН-связи строго в заторможен-
ном положении) — наиболее стабильный углеводород этого ряда.
п в (СН2)П 5 6 7 8
—ДЯ^3°с/«, ккал/моль (103 Дж/моль) 158,7(664,44) 157,4(659,0) 158,3(662,77) 158,8(664,85)
Хассел с помощью рентгеноструктурного анализа, дифракции электронов
и измерения дипольных моментов показал, что простые соединения ряда
циклогексана во всех состояниях, даже в газовой фазе, имеют главным
образом конформацию «кресла». Другая ненапряженная, хотя и менее ста-
бильная конформация циклогексана называется формой «ванны» («лодки»).
В действительности эта форма представляет собой набор одинаковых кон-
формаций, превращающихся друг в друга в результате такого согласован-
ия
Внутримолекулярное и межмолекулярное взаимодействия
ного вращения вокруг связей кольца, при котором каждая из трех пар
наиболее удаленных друг от друга атомов углерода является поочередно
«носом» и «кормой» «лодки» («ванны»), т. е. лежит в плоскости симметрии
молекулы. Каждая из таких конформаций, однако, имеет два С — С-фраг-
мента в заслоненной форме (борта «лодки»), поэтому можно полагать, что
стабильные конформации будут промежуточными между описанными выше
превращающимися друг в друга конформациями. Они не будут иметь пло-
скости симметрии, но будут иметь вращательную ось симметрии второго
порядка, как показано на нижеприведенной схеме:
Конформация «кресла» цик-
логексана (имеет оси сим-
метрии: вращательную треть-
его порядка и перемежаю-
щуюся шестого порядка)
Конформация «ванны» цик-
логексана (метастабильная;
имеет вращательную ось
симметрии второго порядка)
Предполагаемая [23] равно-
весная конформация цикло-
пентана. Отмечены атомы
углерода,лежащиенаО,32 А
(3,2 • 10~2 нм) выше (+) и ни-
же (О) средней плоскости
В циклопентане заслоненная конформация С — С-фрагментов не реали-
зуется, так как кольцо является неплоским, несмотря на то что наименее
напряженная форма должна быть плоской. Этот факт установлен термо-
химическими измерениями [21], а также анализом ИК- и микроволновых
спектров [22] и измерениями электрического двойного лучепреломления [23].
Геометрия стабильной конформации точно не известна, но ее можно изобра-
зить формулой, указанной выше [23].
Для того чтобы определить стабильные конформации, а также высоты энер-
гетических барьеров вращения вокруг простых связей, ряд простых произ-
водных и аналогов этана был исследован термохимическим, спектральным
и другими физическими методами. Если производное этана имеет шесть
заместителей, связанных простыми связями, оно всегда существует пред-
почтительно в заторможенной конформации. Однако если число замести-
телей уменьшить до пяти (один заместитель связан двойной связью), то одна
простая связь заслоняется двойной связью. Эти стабильные конформации
показаны ниже в виде проекций Ньюмена.
Стабильная конформация С — С-фрагмента
с шестью заместителями, связанными про-
стыми связями
Стабильная конформация С — С-фрагмепта
с четырьмя заместителями, связанными
простыми связями, и одним заместителем,
связанным двойной связью
Если по крайней мере один из атомов углерода С — С-фрагмента замещен
таким образом, чтобы сохранить тригональную симметрию, то три барьера,
которые необходимо преодолеть при полном вращении, будут эквивалент-
53
Глава II
ными. В этом случае конформационной изомерии наблюдаться не будет,
и для данного вещества будет существовать только одна характерная высота
барьера вращения. То же самое справедливо для случая, когда один из
атомов углерода связан двойной связью, а также вообще для всякого X — Y-
фрагмента связанных атомов, каждый из которых имеет по октету элект-
ронов, причем X, или Y, или оба эти атома имеют тригональную симметрию.
Только один барьер вращения будет также наблюдаться, если один из ато-
мов связан с двумя другими заместителями. Высоты энергетических барье-
ров вращения для молекул, не имеющих конформационных изомеров, при-
ведены в табл. 4 [24].
Таблица 4
Высоты энергетических барьеров, препятствующих вращению
вокруг простых связей
Связь Высота барьера, ккал/моль (4,187х Х103 Дж/моль) Связь Высота барьера, ккал/моль (4.187Х X1Q3 Дж/моль)
СН3- СН3 2,90 СН3 —SiH3 1,70
ch3-ch2f 3,33 СН3 —SiH2F 1,56
СН3—СН2С1 3,56 СН3 —SiHF2 1,24
СНз—СН2Вг 3,57 СН3—SiF3 1,20
СН3-СН21 3,22 СН3—СеН3 1,20
ch3-ch2cn 3,05 CClg— SiClg 4,3
ch3-chf2 3,18 SiCl3—SiCl3 1,0
CH3-CF3 3,04 СН3 —СН = СН2 1,98
СНз-СС13 2,91 СН3-СН = О 1,16
CF3-CF3 3,02 СН3-CF = O 1,04
CF3 —СС13 6,00 СН3-СС1 = О 1,30
СС13 —СС13 10,8 СНз- C(CN) = O 1,21
ch3-nh2 1,96
СНз-ОН 1,07
CH3-SH 1,27
Следует обратить внимание па то, что высота барьера для С — С-фрагмента
с шестью небольшими по размеру боковыми атомами составляет ~3 ккал/моль
(12,56 -10® Дж/моль) независимо от природы этих атомов. Выше определен-
ной величины суммарного размера заместителей у С — С-связи происходит
увеличение барьера, например в случае CF3CC13 и СС13СС13. Барьер умень-
шается, когда центральный С — С-фрагмент меняется на СХ- или XY-фраг-
мепты с большей длиной связи и с меньшим кручением; кроме того, высота
барьера уменьшается, если в фрагменте СХ число связей X, которые во вре-
мя вращения должны одновременно переходить через заслоненное положе-
ние, уменьшается от трех до двух или до одного. Высота барьера также
уменьшается при наличии двойной связи с углеродом и еще более — с кис-
лородом. Ниже будет показано, что л-электроны карбонильной группы
в основном концентрируются около атома кислорода, и поэтому в приве-
денных примерах не создают особых препятствий вращению вокруг С — С-
связи.
Очевидно, что силы, препятствующие вращению в этане и его производных
с небольшими заместителями, являются силами отталкивания между боко-
54
Внутримолекулярное и межмолекулярное взаимодействия
выми связями С — С-фрагмента; это отталкивание будет максимальным,
когда боковые связи имеют заслоненную конформацию. Отталкивание долж-
но проявляться наиболее сильно между теми парами боковых связей, кото-
рые расположены ближе всего к атомам углерода центрального С — С-фраг-
мента и, таким образом, ближе всего друг к другу. Другими словами, наи-
более сильное отталкивание проявляется между связями, находящимися
в пределах радиуса атома углерода С — С-фрагмеита. Это следует из того
факта, что энергетически барьер вращения почти не зависит от природы
боковых атомов, если эти атомы невелики по размерам. Вероятно, возник-
новение сил отталкивания обусловлено главным образом электронным
обменом, однако определенный вклад могут вносить также квадруполь-
квадрупольное отталкивание и, вероятно, электростатическое взаимодей-
ствие.
Если бы боковые связи, находящиеся внутри радиуса атома углерода С — С-
фрагмента, ничем не отличались бы от одинаково заселенных хр3-орбиталей,
то не существовало бы никакого барьера вращения, так как три такие орби-
тали тетраэдрического атома создали бы электронное распределение, имею-
щее полную круговую симметрию относительно оси четвертой орбитали.
Поэтому следует допустить, что между такими орбиталями и связями, кото-
рые они образуют, существует различие; это различие имеется даже в тех
частях связей, которые находятся в пределах радиуса углерода. Таким
образом, круговая симметрия совокупности трех атомных орбиталей нару-
шается, и совокупность трех связей имеет уже тригональную симметрию,
а распределение электронов похоже на трилистник, лепестки которого,
проходящие через три связи, перпендикулярны линии связи, вокруг кото-
рой происходит вращение. В дополнение к этому необходимо вслед за Полин-
гом предположить, что атомные хр3-орбитали более тесно концентрируются
около их собственных осей симметрии путем включения определенных долей
d- и /-орбиталей. Или, если не хотят использовать описание при помощи
атомных орбиталей при такой высокой степени приближения, так как эти
орбитали в любом случае сильно изменяются при превращении в орбитали
связей, можно просто отметить, что орбитали связей боковых связей под
влиянием ядер образующих их атомов должны становиться более концент-
рированными по сравнению с родительскими атомными хр3-орбиталями угле-
рода. Вследствие этого распределение электронной плотности в сечении,
проходящем через боковые связи, будет давать трилистник, что приведет
к периодическому изменению энергии при вращении вокруг центральной
С — С-связи.
Совершенно очевидно, что, когда одна из боковых связей является двойной,
простая связь будет находиться в заслоненной конформации, так как про-
стая связь в этом случае будет лежать в узловой плоскости л-орбитали
связи, т. е. в плоскости нулевой плотности л-электронов.
Если нп одна из замещенных метильных групп С — С-фрагмента не имеет
тригональной симметрии, возникает конформационная (т. е. вращательная)
изомерия, так как три энергетических минимума не эквивалентны. Изоме-
ры, которым соответствуют разные энергетические минимумы, можно спе-
цифицировать, указав наименьший угол, на который надо совершить пово-
рот по спирали, связывающий две принятые за основу сравнения группы
у разных атомов центральной связи С — С (ось спирали совпадает с осевой
линией связи С — С) [25]. Угол вращения по спирали обозначается зна-
ками плюс или минус или, по более новой классификации, буквами Р (plus)
или М (minus) [26] в зависимости от правого или левого направления витков
спирали. Все изомеры, имеющие спиральные углы, отличные от 0° (0 рад)
55
Глава II
или 180° (3,141 рад), обладают хиральностью * (handedness) и, будучи выде-
ленными в чистом виде, оптической активностью; при этом буквы Р и М
относятся к энантиомерам. Если величины спиральных углов известны
лишь приближенно (а часто, когда и точно известны), то для полуколиче-
ственного описания спиральных углов, отличающихся не более чем на ±30°
(4),523 рад) от идеализированных углов в 0° (0 рад), ±60° (1,047 рад) ±120°
(2,094 рад) и ±180° (3,141 рад), применяют соответственно термины: син-
перипланарный (sp), синклинальный (sc), антиклинальный (ас) и антппери-
планарный (ар) **.
В случае замещенных этанов простейшая система конформационных изоме-
ров возникает, когда каждая из замещенных метильных групп имеет два
одинаковых атома (или две группы) и один — отличающийся от них. Эти
отличающиеся заместители в двух метильных группах принимают за основу
сравнения. В этом случае система изомеров включает один антиперипла-
нарный изомер (ар) и два энантиомерных синклинальных изомера (P-sc
и M-sc). На графике зависимости энергии от торсионного угла (угла кру-
чения) минимум, соответствующий антиперипланарному изомеру, будет
расположен между двумя одинаковыми минимумами, соответствующими
синклинальным энантиомерам; однако антиперипланарный минимум энер-
гии может лежать или ниже или выше синклинальных минимумов.
Существование таких изомеров было доказано [27], в первую очередь, с по-
мощью спектров комбинационного рассеяния и ИК-спектров дихлор- и ди-
бромэтанов. Каждый спектр представлял собой наложение двух спектров,
один из которых постепенно исчезал при низких температурах, что. таким
образом, указывало на наличие равновесия между двумя изомерами различ-
ной относительной стабильности. Низкотемпературные спектры показывают,
что в этих частных случаях более стабильные вращательные изомеры нахо-
дятся в антипорипланарной конформации (центросимметричной). Это заклю-
чение подтверждается измерением дипольных моментов, которые сильно
уменьшаются при понижении температуры. Антиперипланарный изомер
можно выделить в чистом виде при постепенном охлаждении; твердое веще-
ство содержит только этот изомер. Из данных по влиянию температуры
на спектры паров был сделан вывод, что синклинальные изомеры дихлор-
и дибромэтанов соответственно на 1,2 и 1,5 ккал/моль (5,024 ИО3 и 6,280 х
X Ю3 Дж/моль) менее стабильны, чем антиперипланарные изомеры. Высо-
ты энергетических барьеров, разделяющих изомеры, не известны, однако
они должны быть порядка нескольких килокалорий на моль, так как в про-
тивном случае ширина полос в спектре была бы большей.
* Термин «хиральность» (от грет, слова «chier»— рука) означает, что движение по спи-
рали соответствует правилу правой или левой руки.— Прим, перев.
** Приводим значения греческих слов, пз которых образованы термины: peri — вокруг,
syn — рядом, klino — гну, выгибаю.— Прим, перев.
56
Внутримолекулярное и межмолекулярное взаимодействия
Относительная стабильность ряда изомеров приведена в табл. 5 [28]. Она
не всегда соответствует относительной стабильности дигалогенэтанов. И в га-
зовой и в жидкой фазах синклинальные изомеры я-пропилгалогенидов более
Таблица 5
Относительная стабильность вращательных изомеров
Соединение Esc — Еау, ккал/моль (4,187 • Юз Дж/моль) Изомер, со- ставляющий твердое со- стояние
газ жидкость
СН2С1 —СН2С1 ±1,2 ±0,0 ар
СН2Вг —СН2Вг ±1,5 ±0,7 ар
ch3ch2-ch2f ~ ±0,5
СН3СН2-СН2С1 —0,05 ±0,05 ар
СН3СН2—СН2Вг —0,15 ар
СН3СН2-СН21 ар
С1СН2-СН2(ОН) -0,95 -0,95 SC
С1СН2-СНС12 ~ ±0,0 -1,1 SC
СН3СН2-СН2СН3 ±0,8 ар
стабильны, чем антиперипланарные. Изучение микроволновых спектров
газообразных фторидов показывает, что синклинальные изомеры, у которых
угол вращения по спирали у С( — С2-связи равен ±63° (1,099 рад), на
0,47+0,31 ккал/моль (1,968• 103 + 1,298-103 Дж/моль) более стабиль-
ны, чем антипланарные. Антиклинальный барьер между синклинальными
энантиомерами и антиперипланарным изомером равен ~4 ккал/моль
(~16,75-103 Дж/моль), в то время как сами синклинальные энантиомеры
разделены синперипланарным барьером, составляющим ~10 ккал/моль
(~41,87 -103 Дж/моль). Кроме того, вращательные спектры показывают, что
все изомеры имеют барьер, препятствующий вращению метильной группы
вокруг С2 — С3-связи. Для синклинальных изомеров этот барьер равен
2,9 ккал/моль (12,14 -103 Дж/моль), а для антиперипланарного изомера —
2,7 ккал/моль (11,30-103 Дж/моль). Синклинальные изомеры я-пропилхло-
рида в газовой и в жидкой фазах, а также я-пропилбромида в газовой фазе
немного более стабильны, чем антиперипланарные изомеры. Однако твердые
хлорид и бромид являются чистыми антиперипланарными формами. Вероят-
но, это обусловлено тем, что, несмотря на низкую устойчивость в молекуляр-
ном состоянии, эти изомеры имеют более высокую энергию решетки. Синкли-
нальная форма этиленхлоргидрина на 1,0 ккал/моль (4,19-103 Дж/моль)
стабильнее антиперипланарной (в газообразном и жидком состояниях).
В этом случае твердая фаза полностью состоит из синклинального рацемата,
однако не известно, упакованы ли молекулы Р- и 17-энантиомеров вместе
в одинаковые рацемические кристаллы или они раздельно образуют энан-
тиомерные кристаллы, которые могут быть разделены. То же справедливо
и для 1,1,2-трихлорэтана. В табл. 5 знак плюс означает, что антиперипла-
нарный изомер более стабилен, а знак минус — что более стабильны син-
клинальные изомеры.
В пропане и изобутане каждая метильная группа находится в заслоненной
конформации относительно неконцевого атома углерода. Простейшим алка-
ном, обладающим конформационной изомерией, является я-бутан (табл. 5,
последняя строка). В жидком состоянии бутана присутствуют как синкли-
57
Глава II
нальная, так и антиперипланарная формы, причем преобладает последняя.
Твердое состояние состоит из чистого антиперипланарного изомера, угле-
родная цепь которого образует плоский зигзаг, а молекула в целом центро-
симметрична. Все высшие нормальные алканы в твердом состоянии имеют
плоские зигзагообразные конформации, причем соединения с четным числом
атомов углерода являются центросимметричными. В устойчивой конформа-
ции циклогексана все связи кольца являются синклинальными и имеют
попеременно Р- и ^-конфигурации. В стабильной конформации циклоде-
капа [29] две кольцевые связи являются антиперипланарными, а остальные
восемь кольцевых связей образуют совокупность синклинальных связей,
причем количество Р- и 7И-связей одинаково. На формулах, приведенных
ниже, буквы Р и М относятся к конформациям P-sc и M-sc соответственно.
Р М
Совершенно другой тип внутримолекулярных взаимодействий обусловливает
существование вращательной изомерии, например, в случае бутадиена-1,3,
в котором вращение вокруг центральной, формально простой связи затруд-
нено. Это соединение исследовалось спектральным [30] и термохимическим
[31] методами. Данные двух методов совпали, причем последний метод дает
более детальную картину. Изомеры бутадиена образуются не за счет оттал-
кивания между атомами или группами, связанными с двумя внутренними
атомами углерода: такие изомеры были бы неплоскими, энантиомерными
и энергетически эквивалентными друг другу. На самом же деле существуют
два плоских энергетически неэквивалентных изомера, из которых более
стабильным является изомер, имеющий два одинаковых атома в перипла-
нарной конформации. Энергия синперипланарного изомера на 2,3 ккал/моль
(9,63 -103 Дж/моль) выше. Для превращения антиперипланарного изомера
в синперипланарный необходимо преодолеть барьер в 4,9 ккал/моль
(20,52-103 Дж/моль). Известны и другие подобные примеры. С помощью
микроволновых спектров [32] и ультразвукового метода [33] был исследо-
ван акролеин. В случае этого ненасыщенного альдегида и некоторых его
производных более стабильным оказался антиперипланарный изомер; энер-
гия синперипланарного изомера на 2 ккал/моль (8,37 ИО3 Дж/моль) выше,
а величина барьера, препятствующего превращению первого изомера во вто-
рой, составляет пе менее 7 ккал/моль (29,31 -Ю3 Дж/моль). ИК-спектр глиок-
саля [34] показывает, что это соединение существует почти полностью в ап-
типерипланарной форме. Можно полагать, что стабильность синперипла-
нарного изомера более чем на 2 ккал/моль (8,37 -103 Дж/моль) ниже. Высота
барьера неизвестна. Эти и другие данные приведены в табл. 6.
Считается, что, когда формально простая связь находится между двумя
двойными связями, преобладающую роль играет фактор, называемый сопря-
жением. Этот фактор подробнее будет рассмотрен в разд. 3 настоящей главы.
Ненасыщенность двойных связей распространяется на связь, лежащую
между ними, придавая ей некоторый характер двойной связи. При этом
возникает тенденция к плоскому расположению атомов двух двойных связей
и всех атомов, непосредственно связанных с ними, и таким образом возни-
кают препятствия вращению, нарушающему плоскую конфигурацию.
68
Внутримолекулярное и межмолекулярное взаимодействия
Таблица 6
Относительная стабильность и высоты энергетических барьеров,
препятствующих взаимопревращению планарных изомеров
Антиперипланарный Барьер, ккал/моль (4,187х ХЮ3 Дж/моль) Синперипланарный
формула энергия, ккал/моль (4.187Х XI О3 Дж/моль) энергия, ккал/моль (4,187х X103 Дж/МОЛЬ) формула
н сн2
\ //
н н
'с-с/'
н
/
N — О
О
н н
4,9 2,3 ^с-с/ Z X Н2С сн2 н н
7,0 2,1 / \ Н2С о н н
— > 2 / X о о н
11 0,0 \-0 Z \ О н
9 0,5 N —О X \ О н
10 0,5 N -О / \ О сн3
9 0,0 N —О oz \2Н5
Эту концепцию можно расширить. В разд. 3 будет показано, что ненасы-
щенность, которой благодаря наличию неподеленных электронов могут
обладать такие элементы, как кислород или азот, по механизму сопряжения
может взаимодействовать с ненасыщенностью двойной связи через проме-
жуточную формально простую связь. Этот эффект также будет придавать
простой связи некоторый характер двойной связи, включая тенденцию
к плоскому расположению боковых связей. Поэтому возникнет сопротивле-
ние скручиванию плоской конфигурации.
Простейший оксиолефин — виниловый спирт — не существует, поэтому
рассмотрим муравьиную кислоту. По данным ИК-спектроскопии [35], му-
равьиная кислота состоит из двух изомеров, оба из которых имеют плоскую
59
Глава II
конфигурацию. В молекуле более стабильного изомера гидроксильный атом
водорода и карбонильный кислород находятся в синперипланарном поло-
жении друг к другу. Антиперипланарный изомер на 2,2 ккал/моль (9,21 х
X Ю3 Дж/моль) менее стабилен. Барьер между более и менее стабильными
изомерами несколько больше 11 ккал/моль (46,05 •10® Дж/моль). То же
самое наблюдается в случае азотистой кислоты и ее эфиров (табл. 6). Сама
азотистая кислота была исследована микроволновым методом [36], метил-
нитрит — термохимическим методом [37], а метил- и этилнитриты — методом
ЯМР [38]. В этих примерах различие в стабильности изомеров очень
невелико и не всегда изменяется в одном и том же направлении. Однако
высота барьеров вращения составляет примерно 10 ккал/моль (41,87 х
X Ю3 Дж/моль).
Если один из атомов углерода, образующих формально простую связь, имеет
симметрию второго порядка, то конформационная изомерия исчезает, но барь-
ер вращения остается. Симметрию второго порядка имеет фенильная
группа фенола, поскольку ненасыщенность двойных связей бензольного
кольца симметрично распространяется на все кольцо (см. разд. 3). Исследо-
вание микроволновых спектров фенола показало [39], что атом водорода
гидроксильной группы лежит в плоскости фенильного ядра и что барьер
вращения вокруг связи кислород — фенил составляет 3,1 ккал/моль
(12,98 -103 Дж/моль). Нитрогруппа также имеет симметрию второго поряд-
ка, поскольку ненасыщенность двойной связи симметрично распределяется
между двумя NO-связями. Изучение ИК-спектра азотной кислоты показа-
ло [40]. что ее молекулы являются плоскими, а барьер вращения вокруг
связи NO составляет 9 ккал/моль (37,68 -103 Дж/моль). Из микроволновых
спектров метилнитрата следует [41], что эта молекула обладает точно таки-
ми же свойствами, как и азотная кислота (табл. 7).
Таблица 7
Барьер вращения вокруг формально простых связей в плоских молекулах,
не имеющих изомеров
Молекула Барьер, ккал/моль (4,187х XI О3 Дж/моль) 1 Молекула Барьер. ккал/моль (4.187> Х103 Дж/моль)
СН3
XC-NZ
ОХ ^ClL;
СНз СН3
^С —
// \
О сн3
о о
— N '
^'о
7
О Н
О сн3
N —О
12
2,9
Симметрия второго порядка может быть достигнута, если в молекулах,
например анилина или формамида, силы сопряжения будут достаточно
60
Внутримолекулярное и межмолекулярное взаимодействия
сильными, чтобы перекрыть присущую азоту тенденцию образовывать
пирамидальную структуру и таким образом обеспечить совершенно плоскую
конфигурацию молекулы. Мы пе знаем, достаточно ли велики силы сопря-
жения в анилине, однако при изучении микроволновых спектров формамида
[42] было показано, что в этой молекуле сопряжение не в полной мере конт-
ролирует стереохимию молекулы, так как NH-связи выведены из плоскости
NCO примерно на 10° (0,175 рад). Кроме того, спектры ядерного магнитного
резонанса диметилформамида и диметилацетамида показывают [43], что эти
молекулы, не считая атомов водорода метильной группы, плоские и что
имеется энергетический барьер средней величины, препятствующий враще-
нию вокруг связи С — N амидной группы. Числовые данные приведены
в табл. 7.
Каждый атом азота связи N — N в четырехокиси азота обладает симметрией,
и молекула является плоской. Небольшая величина барьера внутреннего
вращения [44] может быть связана с низкой энергией диссоциации связи
N — N, равной 13,7 ккал/моль (57,36-103 Дж/моль).
Над рассмотренными здесь и другими силами, приводящими к возникно-
вению вращательной изомерии плоских молекул, на небольших расстоя-
ниях могут преобладать силы отталкивания, возникающие в результате
электронного обмена. Вследствие этого могут появиться неплоские изомеры.
Вероятно, действие этих сил отчасти проявляется уже в случае простых
производных этана при условии, что суммарный размер заместителей больше
определенного предела, например в CF3CC13 и СС13СС13 (табл. 4). Одна-
ко наиболее яркие примеры можно найти в ряду диарильных соеди-
нений.
В 1922 г. Кристи и Кеннер [45] при проверке оказавшейся несостоятельной
гипотезы о стереохимических формах бифенила расщепили на оптические
антиподы 6,6'-динитродифеиовую кислоту. В 1926 г. правильное объяснение
стереохимии производных бифенила было дано почти одновременно Тэрне-
ром и Лефевром [46], Беллом и Кеньоном [47] и Миллсом [48]; они считали,
что в процессе вращения вокруг связи между обоими ядрами орто-группы
одного кольца настолько близко приближаются друг к другу, что возникает
пространственное отталкивание, которое препятствует сохранению плоских
форм и приводит к такому большому энергетическому барьеру между энан-
тиомерными неплоскими формами, что они не превращаются друг в друга
при обычных условиях *.
Энантиомерные формы 6,6'-динитродпфеновой кислоты
Поворотная изомерия аналогичного типа, обусловленная заторможенным
вращением вокруг простой CN-связи, открыта Миллсом и Эллиотом [50]
* Кун и Альбрехт [49] не обнаружили заметной рацемизации раствора 6,6'-динитро-
дпфеновой кислоты в 2 н. NaOH при нагревании в запаянных трубках в течение несколь-
ких часов при 160° С.
61
Глава 11
на примере М-бензосульфонил-]\-(8-нитро-1-нафтил)глицина
Энантиомерные [формы К-бензосульфонил-]\-(8-нптро-1-пафтил)глицпна
Последующие работы, посвященные этому типу изомерии, состояли глав-
ным образом в детальном изучении влияния различных комбинаций, пре-
пятствующих вращению групп на оптическую стабильность. В ряду бифе-
нила выделены оптические изомеры с тремя, двумя и даже с одним замести-
телем в орто-положении к связи между кольцами, хотя чем меньше замести-
телей имеется в орто-положении, тем большими по размерам они должны
быть *.
Высота барьера заторможенного вращения дается приближенно экспери-
ментальной энергией активации рацемизации, определяемой из температур-
ного коэффициента реакции при помощи уравнения Аррениуса. Уэстхеймер
п другие рассчитали высоты барьеров на основании данных о потенциалах
отталкивания между ограничивающими вращение атомами и группами
и силовых постоянных связей, растяжение и изгиб которых может умень-
шить выигрыш энергии межатомного притяжения в плоской конфигурации.
Результаты расчета были позднее подтверждены другими группами иссле-
дователей. Некоторые из наблюдаемых и вычисленных величин энергии
приведены в табл. 8.
Имеется несколько классов ароматических соединений, в частности нитро-
соединения и амины, относительно которых пришли к выводу, что внутри-
молекулярное взаимодействие, подобное описанному для бутадиена-1,3,
ориентирует атомы заместителя в плоскость бензольного кольца или в близ-
кое к плоскому положение. Доказательством служат данные о том, что
физические и химические свойства, обычно обусловленные нитро- или амино-
заместителем, существенно изменяются при замещении в орто-положении.
Это объясняется отталкиванием того же типа, которое является причиной
изомерии производных бифенила, и препятствует плоскостной или почти
плоскостной ориентации заместителя. Такие эффекты, а также влияние
внутримолекулярного взаимодействия на стереохимическую форму, обна-
руживаемое по измерениям длин связей или валентных углов или при помо-
щи тонких спектроскопических исследований симметрии молекул, будут
рассмотрены ниже (гл. III и IV).
е. Внешняя передача внутримолекулярного взаимодействия. Следует
ожидать, что в общем наблюдаемое взаимодействие между любой группой
в молекуле и другими частями молекулы передается двумя сильно отли-
чающимися способами: через электронную систему самой молекулы или
через пространство вне молекулы. Передача влияния через атомы и связи,
называемая внутренней передачей, в значительной степени и довольно
сложным образом зависит от электронной структуры молекулы. Эта зави-
симость будет рассмотрена в разд. 3. Передача влияния через пространство'
* Единственный известный случай оптической активности, зависящей от одной орто-
группы в бифениле, обнаружен Лессли и Тэрнером [51], которые расщепили З'-бромдпфе-
нил-2-триметиларсониевые соли, но не смогли этого сделать в случае соответствующих.
2-триметиламмониевых солей.
62
Таблица 8
Энергия активации по Аррениусу для рацемизации и вычисленные энергетические
барьеры, ограничивающие вращение вокруг простой связи в производных бифенила
и ариламинов
Соединение Среда Ед (набл.), ккал/моль (4,18 7 X ХЮ3 Дж/моль) Е (вычисл.), ккал/моль(4,18 7 х Х103 Дж/моль) Лите- рату- ра
NH2 СН3 1 X X X X Газ или в (СбНз)2О 45,1 52
СНз NH2 NO, СООН В 2 н. 22,6 53
1 / у X растворе Na2CO3
1 СООН
NO2 СООН В 2 и. 22,6 54
' 1 1 O,N — у X у X „ NO 2 растворе Na2COg
1 СООН
NO, ОСН3 1 С,Н5ОН — 20 55
1 СООН
I I Na-Соль 28,0 29—33 56
в воде
НООС-Х х X Х_СООН
i I
I 1 То же 21,6 21-23 56
НО ОС— /' X У Х>- с ООН
I
Вг
НООС-/ X, У Х_соои 19+0,5 18 57
В г
CH3CONCH3 1 SO.J XX/ 1 1! Na-Соль в воде 22,6 58
1 || X/
CII3
а Большая величина энергии тетраиодкислоты по сравнению с дииодкислотой приписывается
«поддерживающему влиянию» внешнего атома пода в первом соединении па внутренние атомы пода.
63
Глава II
вне .молекулы (внешняя передача') в основном обусловливает взаимодействие
между молекулами, рассмотренное в разд. 1. Пространство в непосредствен-
ной близости от молекулы может быть пустым (в случае газа) или запол-
ненным молекулами растворителя. Полярные или поляризующиеся и таким
образом превращающиеся в полярные молекулы растворителя будут сильно
влиять на силы межмолекулярного взаимодействия, изменяя их так, что
они не будут одинаковыми па передающем и воспринимающем концах транс-
миссионной системы.
Согласно модели межмолекулярного взаимодействия, рассмотренной в разд.
1, необходимо различать три вида сил, ответственных за передачу
влияния от одной части молекулы к другой через внешнюю среду. Во-пер-
вых, существуют электростатические силы дальнего действия, обусловлен-
ные наличием полей и диполей. Эффект, вызываемый этими силами, носит
название эффекта поля. Во-вторых, возможны электрокинетические силы
более близкого действия, которые обусловлены взаимной корреляцией
движения необменивающихся электронных пар. Эти силы являются сла-
быми и проявляются в заметной степени только в редких случаях. Вызывае-
мый ими эффект называется электр акинетическим эффектом. В-третьих,
существует сильное обменное взаимодействие между электронными парами,
действующее на коротких расстояниях. Действие этих сил давно известно
под названием пространственных эффектов или реже под названием первич-
ных пространственных эфнфектов.
На приведенной ниже схеме суммированы внутримолекулярные взаимодей-
ствия, передающиеся через внешнюю среду; влияние этих взаимодействий
па физические свойства и химические реакции будет обсуждаться в после-
дующих главах.
{электростатических сил приводит к эффекту поля
электрокинетических сил приводит к электрокинети-
ческому эффекту
сил обмена приводит к пространственным эффектам
Конечно, внутреннее и внешнее взаимодействия взаимно влияют друг на
друга. Наиболее обстоятельно изучено изменение геометрической формы
молекулы в результате пространственного отталкивания, что в свою очередь
приводит к значительному изменению взаимодействий, передающихся через
внешнюю среду. Мы будем называть это явление вторичным простран-
ственным эффектом.
3. Передача внутримолекулярного взаимодействия
При передаче взаимодействия через электронную систему (и посредством
электронной системы) изменяются положения и движение электронов, нахо-
дящихся между взаимодействующими группами. Таким образом, теория
внутримолекулярных взаимодействий является по cyipecTBj’ теорией элек-
тронных смещений.
а. Механизм электронных смещений и теория внутримолекулярного
взаимодействия. Все теории электронных смещений исходят из электронной
теории атома и валентности, основной вывод которой заключается в том,
что благодаря стабильности определенных электронных групп, особенно
дублета и октета, лимитируются виды электронных смещений, которые
можно было бы считать механизмами миграции внутримолекулярных элек-
трических эффектов. Электронная теория требует, чтобы при смещениях
64
Внутримолекулярное и межмолекулярное взаимодействия
электронов спаренные электроны, октеты или другие стабильные электрон-
ные группировки в атомах по возможности сохранялись.
Установлены два механизма электронных смещений, при которых такие
электронные группировки сохраняются. Первый характеризуется тем, что
все смещаемые электронные дублеты остаются связанными в своих перво-
начальных атомных октетах. Смещения такого рода были предположены
впервые Льюисом [59], который показал, как электрическая асимметрия,
возникающая вследствие неэквивалентного распределения электронов между
разными атомами, т. е. вследствие их электроположительности или электро-
отрицательности, может распространяться вдоль цепи связанных атомов
(одинаковых или неодинаковых) по механизму электростатической индукции.
Это называется индуктивным механизмом электронных смещений и часто
обозначается стрелками, указывающими направление повышения концен-
трации электронов. Таким образом, символ
С1 «— С <— С <— С
показывает, что электроотрицательность хлора и результирующая асим-
метрия связи углерод — хлор заставляет электроны углерод-углеродных
связей концентрироваться в общем направлении к атому хлора. Однако
не следует думать, что электронные смещения, изображенные стрелками,
всегда равны: диполь углерод — хлор наводит меньший диполь в смежной
углерод-углеродной связи, потому что электроны этой связи менее подвиж-
ны; эти диполи наводят еще меньшие диполи в следующей углерод-углерод-
ной связи. Таким образом, влияние будет уменьшаться по мере удаления
от источника возмущения. Каждый атом углерода, теряющий электроны
в большей степени, чем он их приобретает, становится центром маленького
положительного заряда. Это может быть изображено следующим образом:
б- б+ 66+ 666+
С1 <—- С С «—с
где б— и 6+ обозначают малые доли единицы заряда, а сочетание несколь-
ких 6 указывает на последовательное уменьшение заряда.
Льюис рассматривал индуктивный механизм электронных смещений как
постоянное свойство молекул, которое влияет на физические свойства и хи-
мическое равновесие. Так, например, различие в силе хлоруксусной и уксус-
ной кислот он объяснял, исходя из того, что электроны каждой связи в пер-
вой молекуле по сравнению с электронами соответствующей связи второй
молекулы смещены к атому хлора; дойдя до способного к ионизации атома
водорода, этот эффект может обусловить более легкую диссоциацию его
в первой молекуле по сравнению со второй
С1 «- СН2 СО «— О «— И
Н —СН2-СО —О —Н
Если индуктивный механизм электронных смещений используется для опи-
сания нормальных состояний молекул, он называется индуктивным эффек-
том. Его квантовое описание будет дано ниже. Кроме того, будет показано,
что подобный механизм электронных смещений проявляется в процессах
активации, т. е. в образовании переходных состояний при химических
реакциях.
Второй механизм электронных смещений с сохранением электронных дубле-
тов и октетов характеризуется заменой одного дублета другим в том же
атомном октете. Смещения этого типа впервые были рассмотрены Лоури [60],
5-0772
65
Глава II
который показал, как внедрение в октет пеподелеппого дублета соседнего
атома может вызвать удаление другого дублета, который в дальнейшем
может либо стать неподеленным, либо инициировать дальнейший аналогич-
ный обмен в молекуле. Такое смещение путем замены особенно характерно
для ненасыщенных молекул и может быть названо механизмом сопряжения-,
как показано ниже, его можно считать развитием ранних представлений
о сопряжении в ненасыщенных системах. Такие смещения обычно обозна-
чаются изогнутыми стрелками, исходящими от дублетов или из положений,
которые занимают дублеты в соответствии с химической формулой, и направ-
ленными к положениям, к которым происходит смещение. Таким образом,
стрелки в формуле
R2N—С=С—С=О
обозначают смещение дублета, которое может превратить неполярную
структуру в диполярную
R2N-C = C-C= О -» P,2N = C-C = C-0
Сам Лоури не считал, что этот механизм электронных смещений отражает
постоянное состояние молекулы; по мнению Лоури, это разновидность
химического превращения, механизм молекулярной активации, происходя-
щей в процессе химической реакции. Допущение, что механизм сопряжения
может описывать нормальное состояние молекул, сделали Хилда Ипгольд
и автор [61]. Предполагается, что дублеты в такой структуре, которая изоб-
ражена выше, могут быть в одно и то же время частично обобщенными и ча-
стично необобщеппыми или же могут принадлежать одновременно двум
связям. Это позволяет применять теоретический анализ при рассмотрении
физических свойств и химического равновесия. Если механизм сопряжения
применяется для описания электронных смещений в нормальных состоя-
ниях молекул, он называется мезомерным эффектом (или мезомерией).
Квантовое описание будет дано ниже.
Таковы два общих механизма электронных смещений в молекулах с запол-
ненными электронными оболочками. Известны два других механизма, кото-
рые возможны при наличии незаполненных оболочек. Однако эти механиз-
мы имеют ограниченное значение и рассматриваться не будут.
Индуктивный и мезомерный эффекты не зависят от времени, т. е. являются
факторами, определяющими распределение электронов в основном состоя-
нии молекул. Эти эффекты в общем называются поляризационными эффек-
тами (или эффектами поляризации).
Большинство обычно определяемых физических свойств молекул (размеры,
константы упругости, дипольные моменты, по не электронные спектры)
являются свойствами их основных состояний. Поэтому при изучении этих
свойств нужно учитывать только два указанных выше типа внутримолеку-
лярных полярных эффектов, т. е. поляризационные эффекты.
Химические равновесия зависят от разности энергии и энтропии основных
состояний реагентов и продуктов обратимой реакции. Поэтому при изуче-
нии химических равновесий также необходимо учитывать только эффекты
поляризации.
С другой стороны, при изучении скоростей реакций рассматривают разность
энергий начального и переходного состояний. Начальное состояние реак-
ции состоит из основных состояний реагентов, поэтому при его рассмотрении
необходимо учитывать поляризационные эффекты. При описании переход-
ного состояния можно в качестве первого шага поляризационные эффекты
66
Внутримолекулярное и межмолекулярное взаимодействия
в исходных молекулах формально перенести без изменения в переходное
состояние. Однако после этого следует ввести поляризационные взаимодей-
ствия в переходном состоянии, возникающие в процессе активации и обус-
ловленные электрическими свойствами реакционного центра относительно
окружающей поляризуемой структуры. Такая дополнительная поляризация,
которая, как впервые отметили Флоренс Шоу и автор [62], может иметь
большое значение при рассмотрении скоростей реакций, является быстро-
течной и в отличие от поляризации не может быть исследована в основном
состоянии. Ее величина может быть оценена только из электронных условий
реакции и поляризуемости молекулы. По этой: причине два указанных выше
механизма электронных смещений в том случае, когда они действуют во вре-
мя реакции и таким образом зависят от времени, называются эффектами
поляризуемост и.
Эффект поляризуемости, реализуемый ио индуктивному механизму, назы-
вается индуктомерным эффектом. Он был открыт позже |63]. чем не зави-
сящий от времени индуктивный эффект. Эффект поляризуемости, вызывае-
мый механизмом сопряжения, называется электромерным эффектом. Как
уже отмечалось, этот механизм был постулирован Лоури 160J еще до того,
как было высказано предположение о существовании пе зависящего от вре-
мени мезомерного эффекта. Обычно индуктомерпый эффект менее важен,
чем электромерный, поскольку ст-элсктропы. как правило, образуют более
сильные связи и поэтому менее поляризуемы, чем л-электропы, обусловли-
вающие электромерный эффект.
Электрическая поляризуемость молекул, т. е. мобильность их электронов
во внешнем электрическом поле, может быть выяснена при изучении неко-
торых физических свойств (например, диэлектрических и оптических) основ-
ных состояний. Таким образом, поляризуемость (индуктомерпый и электро-
мерный эффекты) в некоторых случаях может быть исследована физически-
ми методами. Поляризуемость зависит от того, насколько легко электроны
из основного состояния могут переходить в разрешенные квантовой химией
возбужденные состояния. Другими словами, поляризуемость определенным
образом связана с возбужденными электронными состояниями, т. е. со спект-
ральными свойствами.
Теперь составим таблицу внутримолекулярных электронных смещений,
в которой отметим два механизма таких смещений и подразделение каждого
из механизмов на зависящую и пе зависящую от времени части (табл. 9).
Отметим также области приложения различных эффектов.
В этой таблице эффекты обозначены стрелками, уже употреблявшимися
выше, и буквами. Буквенные символы в простейших случаях не нужны,
однако они удобны в более или менее сложных случаях. Буквы обозначают
общее направление и механизм электронных сдвигов: знаки плюс или минус
перед буквами соответствуют электроположительному или электроотрица-
тельному сдвигу. Большинство буквенных символов хорошо известно и ши-
роко употребимо, однако буква К, обозначающая механизм сопряжения,
была введена относительно недавно Ола *. Символы Is и I d, относящиеся
к «статическому» и «динамическому» эффектам, были введены Джонсоном
и использованы Ремиком [65].
* См. работу [64]. Старый символ Т (таутомерный) имеет только историческое значение
и. так же как и символ С (сопряжение), очевидно, неудобен для применения в органи-
ческой химии. Ола, написав монографию [64] па немецком языке, употребил начальную
букву (К) немецкого слова Konjugation, которая и используется в данной книге. В англий-
ском языке такая замена символа С па К вполне обычна: в математике, например, для
обозначения константы (constant) наряду с символом С используют символы к пли К.
5* 67
Глава И
Таблица !)
Внутренняя передача электронных взаимодействий.
Разделение эффектов и их приложимость к различным явлениям
Электронный механизм Электронный эффект
поляризация поляризуемость
Индуктивный (—>-) (/) Сопряжение ( ) (К) Индуктивный (Z.) Мезомерный (М) Индуктомерный (1ц) Электромерный (Е)
Область приложения Многие физические свойства Равновесия реакций Скорости реакций Оптические свойства Скорости реакции
б. Доэлектронные представления, развившиеся в теорию внутримоле-
кулярных электронных взаимодействий. Современные теории электронных
смещений в значительной мере основываются на доэлектронных теориях
органической химии. Ввиду огромной роли, которую сыграли эти теории
в истории развития органической химии, им можно посвятить отдельную
книгу. Здесь же мы можем дать лишь очень краткий очерк.
Дуалистическая теория, опубликованная Берцелиусом в 1812 г., никогда
полностью не теряла своего значения в химии, хотя казалось, что это должно
было произойти, поскольку в 1840 г. Дюма и сотрудники показали, что
она не согласуется со многими свойствами органических веществ. Однако
в 1870-х годах дуализм был воскрешен, так как было необходимо объяснить
накопившиеся данные, касающиеся ориентации при ароматическом заме-
щении. Некоторые заместители ориентировали второй заместитель в мета-
положение бензольного кольца, другие же — в орто- и пара-положения.
Кернер [66], Хюбнер [67] и Нельтинг [68] отнесли к летпа-ориентирующим
те группы, которые обычно придавали или увеличивали уже имеющиеся
до их введения кислотные свойства молекул; таким образом, кислотные
свойства, как и во времена Берцелиуса, ассоциировались с представлениями
об отрицательном электричестве, орто- и пара-Ориентирующие заместители
включали такие группы, которые обычно придавали или усиливали основ-
ные свойства; эти свойства Берцелиус связывал с положительным электри-
чеством (гл. VI). В 1890-х годах для того, чтобы схематизировать реакции
металлирования и алкилирования, а также указать направление присоеди-
нения молекул к ненасыщенным кетонам и сложным эфирам, Михаэль [69]
пометил знаками алифатические атомы углерода в этих соединениях (гл. XIII
и XV). Михаэль высказал предположение, что электрополярность пере-
дается по контактному механизму от атома, обладающего этим качеством,
к соседним частям молекулы. Итак, к концу XIX века вновь появившаяся
более «мягкая» форма дуализма была в основном готова для выражения
ее на языке электронной теории валентности. После 1916 г. появились
зачатки теории электронного смещения, осуществляющегося по индуктив-
ному механизму.
68
Внутримолекулярное и межмолекулярное взаимодействия
Знаменитые двенадцать статей Тиле, появившиеся одна за другой в 1898 г.
[70], послужили огромным вкладом в химическую теорию. На многочислен-
ных примерах Тиле показал, что две двойные связи, разделенные простой
связью, ведут себя как единая («сопряженная») система, вступающая в реак-
ции 1,4-присоединения; примером может служить присоединение брома
к бутадиену-1,3 (гл. XIII). Тиле предположил, что ненасыщенность двой-
ных связей распространяется на центральную простую связь; при этом
реакционные центры смещаются к концам «сопряженной системы». Он объяс-
нил пониженную ненасыщенность бензола, представив эту молекулу в виде
сопряженной системы, не имеющей «концов» (гл. IV). Если все это изло-
жить, используя представление, что связь образуется двумя электронами,
то легко видеть, что доэлектронная теория Тиле отражает второй из рас-
смотренных выше механизмов электронных смещений — сопряжение. Более
того, так как Тиле считал перераспределение сродства по связям постоянным
свойством сопряженных молекул, его теория имеет близкое отношение
к основам теории мезомерного эффекта.
В теории Тиле отсутствовало лишь одно важное обобщение: в ней не учиты-
валась ненасыщенность скрытых валентностей, например таких, которыми
обладают, как мы в настоящее время говорим, неподеленные пары электро-
нов или незаполненные валентные оболочки. Теория Тиле не учитывала
их участие в сопряжении. Однако этот недостаток уже отсутствовал в тео-
рии Флюршейма, предложенной в 1902 г. [71] для объяснения влияния
заместителей на ориентацию при ароматическом замещении (гл. VI). Важное
место в своей теории Флюршейм отвел потенциальным валентностям, напри-
мер, трехлигапдного азота, силы связывания которых распространяются
по системе сопряжения. То же самое мог бы сделать Тиле, если бы он опре-
делил скрытые валентности как единицы ненасыщенности, способные сопря-
гаться с другими единицами ненасыщенности, например с двойными свя-
зями. Теория Флюршейма была также теорией перманентного перераспре-
деления сродства, что было установлено при применении ее к проблемам
химического равновесия [72] и зависимости силы кислот и оснований от их
строения (гл. XIV). Указанные работы явились наиболее важными иссле-
дованиями, предшествовавшими теории мезомерного эффекта.
Со временем в эти теории постепенно вошли представления об электрополяр-
ности. В теории Тиле электрополярность не рассматривалась. Флюршейм
уже применял эти представления, объединив представления Михаэля со
своими собственными. Однако Флюршейм не связывал полярность с пере-
распределением валентности с помощью сопряжения. Значительно позже,
в 1924 г., появилась теория Арндта, в которой строение пирона рассматри-
валось как промежуточное между формально диполярной ароматической
и неполярной ненасыщенной структурами. В этой теории, как и в теории
Флюршейма, применялось представление о перераспределении парциаль-
ных валентностей, однако Арндт более определенно связывал изменение
полярности с перераспределением валентностей [73]. Таким образом, теория
Арндта особенно тесно связана с теорией мезомерии, появившейся два года
спустя. Теория Арндта не получила широкого распространения; она не вы-
ходила за рамки ряда пиронов и тиопиронов, которые правильно рассмат-
ривала как ангидроосновапия оксония и сульфония (гл. XI). В 1925 г.
Робинсон распространил эту теорию на другие ароматические оксониевые
и аммониевые ангидрооснования [74]. Он использовал электронное рассмот-
рение, но придерживался таких представлений *, которые в настоящее время
* Предполагалось, что число электронов в ароматических соединениях должно быть
кратным шести, так что нафталин, например, должен был иметь 12 электронов.
69
Глава 11
нельзя считать правильными. Кроме того, это исследование также ограни-
чилось рассмотрением лишь ангидрооснований (гл. XI).
Возвращаясь к началу столетия, следует упомянуть о теории полярной
активации молекул во время реакции Лэпуорта 1751, которая принадлежит
к доэлектроиным разновидностям теории электромерного эффекта. Он пред-
положил, что парциальные валентности возникают в процессе активации,
когда начинается ионизация; отсюда вытекало заключение о полярной при-
роде парциальных валентностей, которые Лэпуорт описывал в духе теории
Тиле. В более поздней работе [76] Лэпуорт принял для временно возникаю-
щих парциальных валентностей модель силовых трубок по Фарадею, однако
в общих чертах теория осталась той же самой. Большая заслуга теории
Лэпуорта заключается в том, что она свела воедино представление поляр-
ности с представлением о валентности. Лэпуорт пришел к неправильном}^
выводу о чередующей полярности не вследствие объединения этих пред-
ставлений, а вследствие неправильной формы объединения. Однако Лэпуорт
уже в 1901 г. понимал, что сущность химических реакций заключается
в электрическом взаимодействии, связанном с избирательной полярностью
(гл. VI, XIII и XV).
Более поздней важной фазой в развитии теории электронных смещений
явилось объединение различных полярных эффектов в единую теоретиче-
скую картину (табл. 9). Первый шаг в этом направлении сделали Лукас
и сотрудники, которые в 1924 г. объединили ранее применявшиеся отдельно
представления об индуктивном и электромерном эффектах [77]. Они пока-
зали, что первый эффект может способствовать и определять направление
второго. Например, активация олефина при реакциях присоединения осу-
ществляется следующим образом:
СНз—СнХсн
Предполагается, что в этом примере метильная группа обусловливает индук-
тивный эффект, который инициирует электромерную активацию двойной
связи, обеспечивающую присоединение протона бромистого водорода к кон-
цевому атому углерода с образованием в качестве продукта реакции изо-
пропилбромида (гл. XIII). Возможность подобных комбинаций индуктивно-
го и электромерного эффектов допускалась в 1926 г. Робинсоном и сотр.
[78], особенно при объяснении ориентации при ароматическом замещении.
В 1926 г. автор и Хилда Ипгольд ввели понятие о мезомерпом эффекте [61],
а в 1927 г. Флоренс Шоу применила представления о совместном действии
индуктивного, мезомерного и электромерного эффектов [62] для объяснения
ориентации и реакционной способности в ароматическом замещении (гл.
VI). Несколько лет спустя [63] было введено понятие об ипдуктомерном
эффекте, построение теории электронных смещений было завершено, и ее
уже можно было применять ко многим явлениям (табл. 9).
В последующих четырех разделах будут обсуждены четыре компонента,
из которых слагается полярное влияние. Однако в этих разделах будут
рассмотрены только общие принципы без примеров, так как последним
посвящена вся оставшаяся часть книги.
в. Индуктивные эффекты связанных атомов и групп. Отталкивание
и притяжение электронов внутри молекул наиболее целесообразно рассмат-
ривать относительно какого-либо стандарта, в качестве которого обычно
выбирают водород. Можно считать, что группа электроположительна, если
опа при прочих равных условиях отталкивает электроны сильнее, чем водо-
70
Внутримолекулярное и межмолекулярное взаимодействия
род. Так, группа X в соединении XCR3 электроположительна, т. е. оттал-
кивает электроны, если электронная плотность в остатке CR3 больше, чем
в соединении HCR3. При помощи физических методов невозможно указать,
где в связях X — С или Н — С кончаются атомы X или II и начинается
атом С, но это не является серьезным затруднением, так как атом-
ные радиусы в простых связях углерода приблизительно постоянны
10,77 А (7,7 -10~2 нм)]. Для сравнительных целей в качестве границы атома
вполне можно пользоваться этой величиной. Точно так же группу Y в YCR3
можно считать электроотрицательной или притягивающей электроны, если
электронная плотность CR3 меньше, чем в Н —CR3. С помощью принятых
обозначений это можно представить следующим образом:
X —> CR3 Н —CR3 Y CR3
+ Z-Эффект Стандарт — Z-Эффект
Неоднократно отмечалось, что поляризация групп не строго постоянна
и пе характеристична; она зависит от поляризации взаимодействия, возни-
кающей под влиянием молекулярного окружения. Проблему классификации
групп по их поляризации удобно представить в такой форме, чтобы иска-
жения были минимальными. Это условие будет соблюдено, если предполо-
жить, что рассматриваемые группы являются заместителями в производ-
ных алканов.
Наибольшее электростатическое различие следует постулировать между
формально заряженными и формально нейтральными группами. Анионные
заместители, например
«— О", s-
по-видимому, как правило, сильнее отталкивают электроны по сравнению
с нейтральной группой. Катионные группы, например
-> NR3, -> SR2
должны, как правило, притягивать электроны сильнее, чем нейтральные
группы. Группы с формально биполярными связями, например
—> no2, —> SO2R —> SOR
+ -
всегда соединены своими катионными центрами с остатком молекулы, кото-
рой они принадлежат, а следовательно, должны сильнее притягивать элект-
роны по сравнению с нейтральными группами. Это, конечно, общие законо-
мерности без учета индивидуальных отклонений, которые могут привести
к частичному несоблюдению указанных правил.
Индивидуальные отклонения, зависящие от типа химического соединения,
отчетливо проявляются при рассмотрении ряда различных групп с оди-
наковыми формальными зарядами. Это можно проиллюстрировать на при-
мере нейтральных групп, хотя аналогичные рассуждения применимы и к ани-
онным, катионным или биполярным группам с одинаковыми формальными
зарядами. В изоэлектронном ряду
—> СН3, —> NH2, —> ОН, —> F
полный ядерный заряд становится все более централизованным, и, таким
образом, как уже отмечалось в гл. I, разд. 2,д, группы притягивают элект-
роны все сильнее. Такую же зависимость можно ожидать в других рядах,
центральные атомы которых принадлежат к другим периодам периодической
системы Менделеева, с галогеном в конце ряда.
71
Глава II
Следует ожидать, что электрическая асимметрия, характерная для связан-
ного атома фтора, уменьшается в случае аналогично связанного хлора
из-за компенсирующей поляризации электронного остова последнего. Ком-
пенсация возрастает с увеличением размера электронного остова в более
тяжелых галогенах; таким образом, следует ожидать, что притяжение
электронов галогенами увеличивается с уменьшением атомного номера,
т. е. возрастает в ряду
—>1, —» Вг, —>С1, —>F
Аналогичная зависимость проявляется в рядах с центральными атомами,
принадлежащими к группам кислорода или азота.
Таким образом, выявляется закономерная связь между индуктивным эффек-
том атома и его положением в периодической системе. Если R — неполярная
или слабо полярная группа (например, водород или алкил), то все такие
группы, как
— NR2, —OR, —SR, —Hal, =NR, =0. =N
должны притягивать электроны сильнее, чем метильная группа. Степень
притяжения возрастает с увеличением номера группы периодической систе-
мы и уменьшается с увеличением номера периода. Вследствие больших
возможностей для электронных смещений, обусловленных дополнительными
электронами кратных связей, притяжение электронов атомом с кратной
связью сильнее, чем соответствующим атомом с простой связью.
Дейтерий несколько более электроположителен по сравнению с легким
водородом. Этот факт был экспериментально обнаружен Халеви в 195G г.
при изучении влияния на силу кислот замещения на дейтерий; он согла-
суется с другими подобными исследованиями (гл. XIV) и наблюдаемыми
дипольными моментами (гл. III). Длина связи С — D немного меньше срод-
ней длины С —• Н-связи *; такое сжатие связи может приводить к отталки-
ванию электронов связи по направлению к электронам оставшейся части
молекулы, создавая, таким образом, эффект электроположительности.
Индуктивный эффект всех алкильных групп равен нулю. Для метильной
группы такой вывод вытекает из симметрии и неполярного характера этапа
и метана, а для других алкильных групп — из последовательности анало-
гичных сравнений. Однако указанный вывод связан с тем обстоятельством,
что в целях стандартизации и сведения к минимуму эффектов поляризации
взаимодействия были выбраны молекулы алканов. По существу алкильные
группы в отличие от уже рассмотренных групп, собственная полярность
которых делает их классификацию качественно нечувствительной к иска-
жениям из-за поляризации взаимодействия, проявляют те полярные эффек-
ты, которые вызываются другими полярными группами, имеющимися в мо-
лекуле; другими словами, индуктивная поляризация алкильных групп
является главным образом поляризацией взаимодействия. В этой связи
важным свойством алкильных групп являтся их большая поляризуемость
по сравнению с водородом; таким образом, группа СН3 становится слабо
электроноотталкивающей при сравнении СН3 -> СООН с Н — GOOH или
* Это является следствием нормальной (кривая Морзе) ангармоничности связей, кото-
рая приводит к тому, что растяжение связи происходит легче, чем сжатие, и, следователь-
но, усредненная по времени длина связи значительно увеличивается, так как увеличивается
амплитуда колебаний. При переходе от С — Н к С — D амплитуда колебаний на нулевом
уровне уменьшается, поскольку увеличивается масса, движущаяся в одном и том же
силовом поле; таким образом, усредненная по времени длина связи уменьшается. Подроб-
нее об этом и о других родственных вопросах см. работу [79].
72
Внутримолекулярное и межмолекулярное взаимодействия
СНз СН2С1 с Н — СН2С1. Если наиболее широко распространенные замес-
тители являются в большинстве случаев электронопритягивающими, то ал-
кильные группы обычно действуют как слабые электроноотталкивающие
группы.
Относительно слабого полярного эффекта метильной и, следовательно, всех
алкильных групп можно сказать, что метильная группа в самих алканах
не является строго нейтральной, а обладает очень слабой электроотрица-
тельностью. Автор пришел к этому выводу несколько лет назад па основа-
нии того факта, что спектры поглощения алканов не являются неизменными
в гомологическом ряду. Из этого следует, что электроны не полностью
локализованы на связях, а могут в некоторой степени перемещаться со связи
на связь, т. е. молекулярные орбитали, построенные с учетом этого мигра-
ционного движения, энергетически не эквивалентны составляющим их орби-
талям связей. Следовательно, при переходе к высшим членам гомологиче-
ского ряда электроны будут концентрироваться на концах алкановой цепи,
так что метильная группа по сравнению с водородо.м слегка оттягивает
электроны от остальной части молекулы алкана. По аналогии со сверх-
сопряжением (разд. З.и) можно полагать, что подвижность электронов
метильной группы обусловлена электронами С — Н-связей. В настоящее
время имеется ряд прямых доказательств полярности С — Н-связей, неко-
торые из них будут обсуждены в гл. III, разд,. 1,г.
Теперь рассмотрим такие ненасыщенные углеводородные радикалы, как
винил, фенил и этинил. Как будет показано ниже, различными эксперимен-
тальными способами установлено, что алкильные группы, связанные с нена-
сыщенными группировками, ведут себя так, как если бы они отталкивали
электроны
сн3-^сп=сн2 сн3—>с0н5 сп3—>с = сн
Другими словами, радикалы винил, фенил и этинил являются электропо-
притягиватощими, если они присутствуют в качестве заместителей в пара-
финах. Винильная и фенильная группы, по-видимому, притягивают эле-
ктроны примерно с одинаковой силой, в то время как этинильная группа
притягивает их значительно сильнее. Более того, этинильная группа опре-
деленно оттягивает электроны от фенильной группы, а возможно, и от ви-
пильной группы
НС = С«—СН = СН2 НС = С«— С6Н5
Объяснение этой зависимости по Уолшу приведено в гл. I, разд. 4,6. Соглас-
но Уолшу, атомы углерода в винильиоп и фенильной группах образуют
простые связи, используя плоские тригональные орбитали (строго или
с известной степенью приближения), в то время как этинильный атом угле-
рода образует простую связь с линейной орбиталью. Степень «-гибридизации
тригональных гибридных орбиталей несколько больше, а линейной орбита-
ли значительно больше, чем для обычной тетраэдрической орбитали углеро-
да; это увеличение «-характера вследствие концентрирования электронов
у ядра атома углерода вызывает повышенную электроотрицательность угле-
родного атома.
Возможно, что указанный эффект увеличивает электроотрицательность дру-
гих групп, содержащих атом углерода с двойной пли тройной связью, напри-
мер, в карбонильной и циангруппах.
Данные, иллюстрирующие эти положения, сведены в табл. 10.
Проводя количественную оценку относительных величин индуктивных эффек-
тов групп, полагают, что движущей силой индуктивного смещения эле-
73
Глава II
Таблица 10
Индуктивные эффекты групп
Отталкивание электронов (Л-У)
-D>-H
— NR> —О
- Se>- - S > —О
- C(CH3)3 > - СН(СН3)2 > - СН2СП3 > — Cl Т з
Притяжение электронов (—Г)
- ОК, 5 > —NR3
— NR3 Д > — PRg > —AsR3^ >— SbR3
-or2> > - sr2 > > —S+cR, > —TeR,
— or2> > — OR
=nr2> >-nr3: >-no2> —NR,
-s'o2r- >-s63- > — SOR > — SR
—sr,^ > —SOR
— >-OR> • — NR, > >[-CR3]
— F^ >-Cl > - — Br > > —I
-OR^ > — S!i > — SeR
— SiR3 > —GeR3
= o; > = nr [; > = CR2]
= o; > —OR
N > = NR > •—nr2
C = CR- >-CR = cr, с >-CR2CR3]
-CH,- > —H
ктропов является электростатическое взаимодействие в более или менее клас-
сическом смысле. Несомненно, однако, что действие электростатических сил
находится в соответствии с квантовомеханическими законами, определяю-
щими структуру основных состояний. Допустим, что мы соединили нейтраль-
ный радикал СН3 с нейтральным атомом F путем чистой коллигации, в ре-
зультате чего получили строго гомеополярную молекулу СН3 — F, и затем
«позволили электронам двигаться». Гомеополярная молекула не соответ-
ствует стационарному состоянию CH3F, поэтому «освобожденные» электроны
будут действительно «двигаться», создавая диполь, до тех пор, пока не будет
достигнуто стационарное состояние. Эта стадия образования нормальной
дипольной молекулы СН3 F является спонтанным процессом и, следо-
вательно, сопровождается уменьшением энергии. Теперь допустим, что
мы соединили СНз с F- посредством чистой координации, которая приводит
к образованию электровалентной ионной пары CH3F“, и затем «позволили
электронам двигаться». Опять-таки электроны будут «двигаться», и мы
получим нормальную полярную молекулу СН3 —F с существенно кова-
лентной связью. При этом энергия будет уменьшаться на большую величину,
чем в первом случае.
Все сказанное можно описать при помощи квантовохимического резонанса.
Это, конечно, слишком упрощено, так как все химические связи, начиная
от молекулярного иона водорода, уже гибридны. Тот факт, что рассматри-
вается резонанс в данной системе, вовсе не обусловлен какими-то естествен-
74
Внутримолекулярное и межмолекулярное взаимодействия
ными особенностями системы. Просто вводится чисто искусственное при-
ближение, которое выбрали для того, чтобы описать характер связи.
Если чисто умозрительно вначале построить гомеополярную молекулу CH3F
или же сначала представить фтористый метил в виде ионной пары, то для
перехода от одной из этих структур к нормальной молекуле необходимо
будет прибегнуть к концепции квантовомехапического резонанса. При
таком приближении к описанию полярной ковалентной связи возникает
энергетический эффект, связанный с резонансом, который был описан Полин-
гом [80] на примере теплот образования двухатомных молекул. Полинг,
например, показал, что теплоты образования всех галогенидов водорода
больше среднего арифметического или, что теоретически более приемлемо,
среднего геометрического между теплотами образования водорода и соответ-
ствующих галогенов.
Для простоты примем, что образование диполя связи во фтористом метиле
является двухэлектронной задачей, т. е. будем пренебрегать влиянием
сдвига электронов на другие электроны. Допустим, что два рассматриваемых
электрона вначале находятся на орбитали связи гомеополярпой структу-
ры А. В этом случае двухэлектронная волновая функция ЧД (квадрат кото-
рой характеризует вероятность распределения электронов в пространстве)
будет нестационарной вследствие существования у фтора орбитали (заня-
той пеподеленпыми электронами в структуре В), на которую «утекают»
электроны связи структуры А, когда образуется ионная структура В
(A) СН3 —F CHJF- (В)
Итак, электроны, первоначально помещенные на орбиталь связи А, с вол-
новой функцией Чга могут спонтанно захватываться атомной орбиталью
фтора, которая занята в В и имеет волновую функцию ЧД; поскольку
такой захват обратим, ни ЧД, ни ЧД не соответствуют стационарным состоя-
ниям. Стационарное состояние должно включать взаимодействие между ЧД
и ЧД и в первом приближении может быть представлено нормализованной
совокупностью ЧД и Чгв, причем основное стационарное состояние имеет
форму аЧД Д /?ЧД, где а — больший из двух положительных коэффициен-
тов смешивания а и Ъ. При переходе от нестационарной неполярной струк-
туры ЧД к стационарном у полярному состоянию аЧД + бЧД будет проис-
ходить уменьшение энергии на величину энергии резонанса, обусловлен-
ного полярностью связи. Это должно происходить вследствие того, что при
смешении волновых функций движение электронов становится более раз-
машистым, и поэтому в соответствии с принципом неопределенности пулевая
энергия электронов электрополярного основного состояния уменьшается.
Таким образом, энергия индуктивного эффекта, как и каждый другой фак-
тор химического взаимодействия,— это энергия резонанса. При объяснении
кислотности хлоруксусиой кислоты, на примере которой Лыоис впервые
показал существование индуктивного эффекта, исходят из одной неполярной
и нескольких полярных структур
С.1-СН2-СО-О-11 С1 С11,-СО-О-Н
ci-cn2 со—о—ti ci—сп2—со о—н
С1-СН2 — СО — о н
Волновая функция нормального индуктивно поляризованного состояния
будет представлять собой комбинацию волновых функций всех этих структур.
75
Глава II
Энергия нормального состояния будет меньше энергии любой из этих струк-
тур, даже неполярной.
г. Индуктомерные эффекты связанных атомов и групп. Эффекты поля-
ризуемости являются результатом действия электрических сил, возникаю-
щих в реакционном центре в переходном состоянии реакции, на поляризуе-
мые части молекулы. Знак поляризуемости зависит от того, сопровождается
ли реакция увеличением или уменьшением электронной плотности в реак-
ционном центре, и всегда будет таким, чтобы облегчить протекание реакции.
Величина эффекта поляризуемости зависит как от степени увеличения или
уменьшения электронной плотности в реакционном центре, так и от поля-
ризуемости системы, которую реакционный центр деформирует.
Под индуктомерной поляризуемостью понимают поляризуемость, которую
проявляют в насыщенных соединениях атомы и группы вдоль линий их свя-
зей. Такая поляризуемость в общем должна зависеть от прочности связы-
вания валентных электронов: чем сильнее они связаны, тем меньше они
поляризуются. Для изоэлектронных атомов с заполненными валентными
оболочками индуктомерпая поляризуемость, таким образом, зависит от
электроотрицательности: чем больше электроотрицательность атома, тем
меньше его поляризуемость. Следовательно, во-первых, надо ожидать, что-
индуктомерная поляризуемость атома резко понижается при положитель-
ном ионном заряде и, конечно, сильно повышается при отрицательном
заряде
— О > —OR > —OR2
Во-вторых, можно ожидать, что индуктомерная поляризуемость изоэлект-
ронных атомов, имеющих одинаковый формальный заряд, понижается при
переходе слева направо от одного элемента к другому в одном и том же-
периоде периодической системы
— CR3> — NR2 > —OR > —F
Из оптических данных известно (гл. Ill), что атомные валентные оболочки,
имеющие более высокое главное квантовое число, легче поляризуются, чем
аналогичные оболочки с более низким квантовым числом; очевидно, в ато-
мах с более высоким атомным номером на поляризуемость значительнее
влияет большее расстояние валентных электронов от ядра, нежели большая
неполнота экранирования внутренними электронами. Так как оптические
данные показывают, что связи более поляризуемы вдоль, чем поперек (гл.
III), то можно ожидать, что для аналогичных валентных оболочек изме-
нения индуктомерной поляризуемости повторяют изменения полной поля-
ризуемости. Таким образом, на основании опытных данных следует ожи-
дать, что индуктомерная поляризуемость понижается с уменьшением атом-
ного номера в одной и той же группе таблицы Менделеева
— I > —Вг > —Cl > —F
Наконец, дополнительная индуктомерная поляризуемость вызывается эле-
ктронами кратных связей, которые обусловливают также электромерную
поляризуемость, возникающую вследствие участия кратных связей в сопря-
жении.
Атом водорода, естественно, менее поляризуем, чем любая нейтральная груп-
па, содержащая водород. Согласно Халеви [79], дейтерий поляризуем
немного меньше, чем протий. Этот вывод следует из данных по оптической
рефракции, а также из теоретических рассмотрений. Связь С — D немного
76
Внутримолекулярное и межмолекулярное взаимодействия
короче С — Н-связи, поэтому ядра располагаются несколько ближе к эле-
ктронам связей [79]. Данные, характеризующие индуктомерную поляризуе-
мость различных групп, приведены в табл. 11.
Таблица 11
(±)-Индуктомерная поляризуемость групп
-О
-CR3
- I
S N
-СВ,
-OR
-nr2
— Вт
= NR
-CHR2
-or2
— OR
-Cl
— NR2
-ch2r
И т. д.
> — F и t. д.
> —F и t. д.
и т. д.
> — CII3 >11
При очень малых электронных смещениях индуктомерная поляризуемость
одинакова в обоих направлениях. Но при больших смещениях эта симмет-
рия, по-видимому, нарушается и электроны легче смещаются в одном направ-
лении, чем в другом. В настоящее время нет надежных данных о том,
в каких случаях эта асимметрия появляется и насколько она становится
важной.
Даже в случае небольших смещений механизм поляризуемости не является
классическим. Действие электрического поля пе заключается в смещении
электронов в направлении, обусловленном его действием. К молекуле при-
менимы квантовые ограничения, и электрическое поле, действующее на основ-
ное состояние, может только изменить положение и направление движения
электронов в этом состоянии, обусловливая определенный вклад в эти пара-
метры (при слабых полях пропорциональный интенсивности поля) соответ-
ствующих параметров одного или нескольких возбужденных состояний,
разрешенных квантовой механикой. Таким образом, поляризуемость основ-
ного состояния зависит от количества и энергии возбужденных состояний,
п особенно — от энергий самых доступных возбужденных состояний. Индук-
томерная поляризуемость особенно сильно проявляется при наличии поляр-
ных возбужденных состояний, аппроксимациями которых в первом прибли-
жении могут считаться приведенные выше полярные структуры фтористого
метила и хлоруксусной кислоты. Чем больше доступность этих полярных
состояний, т. е. чем ниже их энергия, тем в большей степени их орбитали
перекрываются с орбиталями основного состояния, что облегчает захват
электронов, т. е. тем больше индуктомерная поляризуемость основного
состояния.
д. Мезомерные эффекты в ненасыщенных системах. Пусть имеется
воображаемая молекула, содержащая формально неполярную сопряженную
ненасыщенную систему, например
R2N — С = С — С = 0
в которой пока отсутствует мезомерный эффект; будем считать, что распре-
деление электронных пар среди атомов строго соответствует теории валент-
ности. Предположим, что затем будут происходить электронные смещения
до тех пор, пока не образуется реальная молекула, т. е. пока внутренняя
-энергия системы не станет минимальной.
А Р~\ П,
(б+) И2М^С=С^С=О («-)
77
Глава II
Согласно теории мезомерного эффекта, распределение электронных пар
в мезомерной молекуле является промежуточным между распределением,
которое требуется по теории валентности для первоначальной непо-
лярной структуры, и распределением для альтернативной диполярной
структуры
R2N = С—С—С—О
Электронные смещения по механизму сопряжения, которые были допущены
в первоначальной неполярной структуре, должны, следовательно, привести
к образованию зарядов на концах мезомерной системы (как это показ,апо
знаками 6-(- и 6—). Более того, в процессе перехода неполярной структуры
в мсзомериое состояние электроны должны двигаться против электростати-
ческих сил. которые все в большей степени возникают по мере их переме-
щения. Мезомерное состояние будет достигнуто тогда, когда электроны уже
пе смогут перемещаться далее против электростатических сил.
При обсуждении степени мезомерных смещений в различных системах про-
ще всего исходить из рассмотрения величины смещения, определяемой рав-
новесием между неэлектростатическими силами, вызывающими смещение
(природа которых будет рассмотрена ниже), и электростатическими силами,
препятствующими смещению. Постулируем пока существование сил, вызы-
вающих смещение; тщательно подберем системы для сравнения, по воз-
можности строго подобные, и рассмотрим интенсивность, с которой электро-
статическое сопротивление смещению может развиваться в различных
случаях. Позднее будут рассмотрены силы, вызывающие смещение, и их
изменения при переходе от одной системы к другой.
Следует также отметить, что в системах, представленных выше, увеличи-
вается ковалентность одного из концевых атомов, который становится
положительно заряженным, и одновременно уменьшается ковалентность
другого атома, который при этом становится отрицательно заряженным.
Очевидно, при обсуждении электростатического сопротивления мезомер-
ному смещению необходимо рассмотреть факторы, которые придают связан-
ному атому с заполненной валентной оболочкой склонность к увеличению
или уменьшению своей ковалентности.
Увеличение ковалентности зависит от обладания пеподелепными электрон-
ными парами и от их способности взаимодействовать с соседним атомным
ядром. Сродство пеподеленных электронов к ядру другого атома обратно
пропорционально силе их связывания, т. е. электроотрицательности атома,
которому они принадлежат. Соответственно следует ожидать общего отличия
между заряженными и формально нейтральными группами: отрицательно
заряженные группы, как правило, и.меют тенденцию увеличивать свою кова-
лентность, становясь почти нейтральными, в большей степени, чем нейтраль-
ные группы, которые при увеличении своей ковалентности становятся
положительно заряженными; нейтральные группы имеют тенденцию
увеличивать свою ковалентность больше, чем положительно заряженные
группы.
— О > - OR > - 0R2
Следует ожидать, что в случае групп с одинаковым формальным зарядом,
например нейтральных групп, существует связь между тенденцией к уве-
личению ковалентности и положением центрального атома в периодической
системе элементов. Как уже отмечалось в гл. I, разд. 2,д, сродство веподе-
лепных электронов к ядру другого атома уменьшается с увеличением эле-
ктроотрицательности в изоэлектронном ряду. Следовательно, склонность уве-
78
Внутримолекулярное и межмолекулярное взаимодействия
личивать ковалентность должна понижаться слева направо в периоде таб-
лицы Менделеева
— NR, >- OR > — F
При сравнении элементов одной и той же группы периодической системы
необходимо принять во внимание и другой фактор, а именно стереохимиче-
ские последствия различий в главном квантовом числе между валентными
оболочками соответствующих атомов и углерода. Увеличение ковалентно-
сти. обусловленное мезомерным эффектом, заключается в образовании
частичной двойной связи; благодаря принципу перекрывания двойные
связи образуются легче всего, когда оба атома, и в особенности р-орбитали
их валентных оболочек, имеют примерно одинаковые размеры. Поэтому
можно понять, почему галогены, как это установлено па основании данных
о дипольных моментах (гл. XIII) и химическом равновесии (гл. XIII и XIV7),
но своей способности увеличивать ковалентность при мезомерпом эффекте
располагаются в ряд
— Fj>—С1> —Вт >-1
Элементы группы кислорода составляют аналогичный ряд
— OR > — SR > — SeR
Если любая из этих групп (обозначаемых в последующем изложении X)
присоединена к ненасыщенному остатку, например к винилу или фенилу,
механизм сопряжения всегда действует таким образом, что уменьшается
электронная плотность групп X и возрастает электронная плотность нена-
сыщенного остатка. По отношению к этому электронному смещению груп-
па X действует электроположительно. Обозначив результирующий мезо-
мерпый эффект +71/, можем написать
(«-)
(ЖХ^-с=0х«-)
(S-)
‘гМ-Эффект
Если группы, которые проявляют +71/-эффект, в отдельности связаны
с одной и той же ненасыщенной системой, сравнительная интенсивность
мезомерпого эффекта должна соответствовать относительной склонности
заместителей увеличивать свою ковалентность.
Рассматриваемое ниже уменьшение ковалентности сопровождается исчезно-
вением кратных связей. На основании электростатических и стереохимиче-
ских данных, аналогичных тем, которые уже приводились, можно прийти
к выводу, что положительный ионный заряд, высокий помер группы в таб-
лице Менделеева и высокий помер периода являются главными факторами,
усиливающими склонность кратно связанного атома к уменьшению его
ковалентности. Таким образом, можно ожидать неравенств
= NR2>^NR
вследствие различия в зарядах
= О> —NR
пз-.за разницы в номерах групп в периодической системе и
= S> = 0
вследствие принадлежности атомов к разным периодам.
79
Глава II
Если какой-либо из этих заместителей Y находится на конце ненасыщенной
системы, группа Y действует электроотрицательно, оттягивая электроны
пз системы. В этом случае мы будем обозначать результирующий мезомер-
пый эффект — М
(«-)^=С-^С=С(г+)
(«+)
—M-Эффект
Если группы Y порознь связаны с одной и той же ненасыщенной системой,
мезомерные поляризации соответствуют относительной склонности заме-
стителей уменьшать свою ковалентность.
Данные, характеризующие мезомерные эффекты различных групп, приве-
дены в табл. 12.
Таблица 12
Мезомерные эффекты групп
Электронное отталкивание, зависящее от увеличения ковалентности
— О > — OR > — 0R2
- S > —SR > — SR,
- I > — IR
— NR 2 > —OR > — F
— OR > —SR > — SeR
— F > -Cl > —Вг> -1
Электронное притяжение, зависящее от уменьшения ковалентности ( — М)
= NR2 > — NR
-О > =NR > -C(i2
= s > — О
E= N > -. CR
Теперь рассмотрим комбинации -\~M- и —М-заместителей в системах
Г\ Г\ /—v Г\
X-ic2=Y и X—С=С—C==Y
Карбоксильная группа и различные ее производные дают ряд интересных
примеров такого рода простейших систем. Рассмотрим, как электронная
структура этих форм должна изменяться при независимом изменении X
и Y. Оставляя Y неизменным и заменяя X последовательно группами с по-
степенно уменьшающимся -{-^/-эффектом, можно построить ряд, в котором
внутреннее мезомерное смещение электронов уменьшается, например
с<г
О
В начале ряда находится карбоксилат-ион, симметрия которого показывает,
что мезомерное смещение здесь имеет максимальную величину, эквивалент-
Внутримолекулярное и межмолекулярное взаимодействие
пую передаче 0,5 электрона от одного кислородного атома к другому. Для
альдегидной группы, находящейся в конце ряда, внутренняя мезомерная
передача заряда равна пулю. Если какая-нибудь из этих групп связана
своим углеродным атомом с таким ненасыщенным остатком, как винил или
фенил, то вся группа в целом действует электроотрицательно, т. е. оттяги-
вает электроны, потому что группа Y (двоесвязанный атом кислорода)
сопряжена с ненасыщенным остатком, тогда как группа X не сопряжена,
ибо опа отделена от ненасыщенного остатка двумя простыми связями
С-*с=С(Ж
О
(5-)Y
({+)
<»-)Y <’+>
-М.-Эффект CX=Y
Заместитель X, будучи другим источником электронов для двоесвязанного
атома кислорода, должен оказывать влияние на —37-эффект всей группы.
Таким образом, сравнительные —37-эффекты этих групп в целом могут
быть представлены обратным рядом, выражающим относительные мезомер-
н ые смещения внутри групп:
— СНО > - СОС1 > — COOR > — CONR2 > - соо-
— М-Эффекты
Следует отметить, что если эти группы связаны с ненасыщенным остатком
не через атом углерода, а через X, то они проявляют Д- 37-эффект, т. е. дей-
ствуют электроположительно. В данном случае X находится в сопряжении,
a Y не находится в сопряжении с ненасыщенным остатком
Y ) ( x-iCiC(J-) Y )
(8-)\ /(«+) («-)Ч /
С С ({_)
+М~Эффект ХС=У
Роль Y здесь заключается в конкуренции с 37-эффектом X путем созда-
ния другого пути распределения неподеленных электронов X. Таким обра-
зом, ацилирование всегда уменьшает 37-эффект гидроксила и аминогруппы.
Однако порядок изменения интенсивности эффектов обратный указанному
выше, например для случая амидов и сложных эфиров
RCONR —> RCOO —
-ф М-Эффекты
Можно построить ряды другого типа, сохраняя X неизменным и последо-
вательно заменяя Y группами с постепенно уменьшающимися —37-эффек-
тами:
NR
NR2
—С—н
<5 — 0 772
81
Г лава II
В приведенном ряду внутреннее мезомерное смещение уменьшается слева
направо: для первого члена ряда оно максимально и соответствует передаче
0,5 электрона от одного атома азота к другому; для последнего члена оно
равно нулю. Если эти группы связаны своим углеродным атомом с ненасы-
щенным остатком, то по уже указанной причине эти группы проявляют
—^/-эффекты. Интенсивность таких эффектов уменьшается только в зави-
симости от величины —7И-эффекта Y, поскольку X, который обусловливает
конкурирующий эффект, является постоянным во всем ряду
— С( = NR2)NR2 > — CONR2 > — С( = NR)NR2
— M-Эффекты
Если группы связаны с ненасыщенным остатком не через углерод, а через
атом X, которым в этих примерах является атом азота, связанный простой
связью, то по вышеуказанной причине группы в целом будут проявлять
-фТИ-эффект. Эти эффекты уменьшаются вследствие влияния мезомерных
эффектов внутри самих групп, однако такое влияние все более и более умень-
шается по мере изменения Y в этом ряду. Следовательно, интенсивность
-фМ-эффектов групп в целом изменяется в следующем порядке:
RCH2NR - > RC( = NR)NR — > RCONR — > R (= NR2)NR -
-фМ-Эффекты
Мезомерные эффекты простых систем X — С = Y должны обнаруживаться
и в более протяженных системах X — С = С — С = Y. Так, свойства кар-
боксильной группы должны проявляться и в р-кетоенольной группе
Н—б^сХо Н—б^-С=^С^-С=О)
Особенность таких р-кетоенолов, как ацетилацетон (который при обычных
условиях находится в основном в енольной форме), заключается в том,
что они мало похожи на карбоновые кислоты, и, в частности, кислотность
их водных растворов намного меньше кислотности карбоновых кислот.
Однако следует заметить, что образование внутренней водородной связи
(гл. I, разд. 2,е) в р-кетоенолах стереохимически вполне возможно. Уста-
новлено, что такие связи образуются в простейших алифатических р-кето-
енолах, которые слишком летучи по сравнению с обычными гидроксильными
соединениями и даже более летучи, чем изомерные р-дикетоны. Такое обра-
зование водородных связей, несомненно, мешает проявлению нормальных
функций концевых групп мезомерной системы
СН3—С = СН —С —СН3
0-Н..-0 (рА'„~8)
Однако если такой вид водородной связи затруднен или совершенно невоз-
можен, например в результате включения р-кетоенольной системы в кольцо,
как в диметилдигидрорезорцине или в «кристаллическом димере метилкете-
на» (которые также являются моноенолами и имеют формулы, приведенные
ниже), свойства рассматриваемых групп становятся значительно более
похожими на свойства карбоновых кислот. Диметилдигидрорезорцин был
первоначально ошибочно принят за карбоновую кислоту. Его кислотность
82
Внутримолекулярное и межмолекулярное взаимодействия
в воде [81] * немного меньше кислотности уксусной кислоты, в то время как
кислотность димера метилкетена, так же как и кислотность двухосновной
кислоты циклобутендиолдиона (так называемой «квадратной кислоты»,
получаемой, например, при гидролизе димера GF2 = СС12), больше, чем
кислотность уксусной кислоты [82]. Соответствующие численные значения
приведены ниже (для уксусной кислоты рКа = 4,20):
СН3\ ,СН2 —сХ
С .СИ
СНд/ \сн2-с/
\он
(р£о = 5,15)
СН3—CH С —СН3 (р£0 = 2,8)
О=С—С—ОН (р£а1 1)
О=С—С—ОН (рЛ’а2 = 2,2)
ОН
Трополоны — пример еще более протяженных систем карбоксильного типа
X — С = С — С = С — С = С — С = Y; впрочем, это относится только
к форме, в которой возможно существование водородной связи, что ясно
из величины рКа [83]
НС д -н
^сн—сн^ \о/
(Р^а = 6,7)
То, что химическая функция сильно не изменяется при введении группы
— С = С — в сопряженную систему, впервые было установлено Анжели
в 1924 г. [84] и является одним из основных принципов химии.
Системы, подобные Ц-А/-мезомерным системам X — С = Y, но с централь-
ным атомом азота, представлены нитро-, азокси-, нитритной, нитрозамин-
ной, диазоокси-, диазоаминной и некоторыми другими более сложными
группами
В нитрогруппе мезомерные смещения максимальны и соответствуют переда-
че 0,5 электрона от одного атома кислорода к другому.
Карбоксилат-ион, амидиний-ион, нитрогруппа и анионы р-кетоенолов могут
служить примерами систем, которым мезомерия придает симметрию, не отра-
жаемую какой-либо валентной структурой. Такая симметрия иногда выра-
жается при помощи изогнутой связи для пары электронов, которые либо
* Внутренняя водородная связь в диметилдигидрорезорцине может быть в некоторой
степени допущена вследствие гибкости шестичлеиного углеродного кольца.
6* 83
Глава II
частично поделены и частично не поделены, либо поделены одновременно
в двух связях
[О-С—О]-
[R2N-C-NR2]+
О—N—О
[olc-^c-Q)]'
Теория мезомерии в случаях как простых +М-систем типа ХС = С и т. д.
или самокомпепсированпьгх +М-систем типа X — С = Y и т. д., так и ана-
логичных систем, но с центральным атомом азота не исключает тех случаев,
когда атом X связан двойной связью в валентной структуре при условии,
что X имеет неподелепные электроны и может образовывать тройную связь.
Кетены и диазосоедипения могут служить примерами соединений, обладаю-
щих Ч-М-эффектом, в результате которого происходит увеличение кова-
лентности первоначальной двойной связи
/-Д
O=ic--CR2
N±N=CR2
Несомненно, именно поэтому кетены и диазосоединения при реакциях так
легко присоединяют протон к концевому атому углерода. В случае кетена
установлено (гл. III), что мезомерные смещения электронов значительны
даже несмотря на то, что сначала они направлены против поля диполя
в карбонильной группе. Цианаты и азиды служат примерами ±7И-эффекта
в системах с кумулированными двойными связями. Здесь мезомерные
смещения, очевидно, могут происходить в любом направлении, причем
менее значительное смещение указано нижними стрелками в следующих
формулах:
С точки зрения симметрии в двуокиси углерода, питроний-ионе и азид-ионе
оба направления электронных смещений одинаково важны. Таким образом,
мезомерия не может вызвать новый диполь, хотя может создать новый квад-
руполь. Мезомерия приводит к топким различиям в электронном распре-
делении, так как электронные пары концевых атомов становятся менее диф-
ференцированными в отношении обобщения и пеобобщения и менее лока-
лизованными, чем это отображается обычными валентными формулами
Ненасыщенные углеводороды представляют наиболее важный тип уравно-
вешенного сопряжения. Простейшим примером может служить бутадиеп-1,3
СН2=сн^СН==СН
2
Здесь также не может возникнуть диполь, по смещение электронов в двух
направлениях приводит к образованию нового квадруполя и делокализует
$4
Внутримолекулярное и межмолекулярное взаимодействия
электронные пары с нарушением строгой дифференциации в их принадлеж-
ности к индивидуальным связям в валентной формуле. Эти смещения, следо-
вательно, имеют тенденцию выравнять среднюю электронную плотность
связей в углеродной цени, что во многом напоминает описываемое теорией
Тиле выравнивание сродства связей.
Сильные ограничения, накладываемые па мезомерные смещения в нейтраль-
ных сопряженных системах из-за образования концевых зарядов, в особен-
ности на атомах углерода, не относятся к замкнутым сопряженным систе-
мам. В такой системе электронные смещения протекают свободно, посколь-
ку это зависит от электростатических факторов, до тех пор (если позволяет
симметрия), пока не будет достигнуто полное равенство электронных плот-
ностей всех связей. Основным примером может служить бензол, и выска-
занное положение можно рассматривать как электронное истолкование
осцилляционной гипотезы Кекуле и формулы бензола по Тиле, основанной
на гипотезе парциальных валентностей (более детально вопрос рассмотрен
в гл. IV).
Теперь следует рассмотреть природу сил, которые вызывают мезомерное
смещение электронов. При приближенном описании химической связи, как
уже было показано для случая индуктивного эффекта, эти силы связывают-
ся с делокализацией электронов, которая в согласии с принципом неопре-
деленности приводит к уменьшению пулевой энергии электронов (умень-
шение энергии называется энергией резонанса).
В 1927 г. Гейтлер и Лондон создали фундаментальную теорию ковалент-
ности, возникающей благодаря обменному резонансу. В 1929 г. Бартон
и автор [85] высказали предположение, что мезомерные эффекты вызываются
делокализацией электронов, обусловленной существованием альтернативных
валентных структур; таким образом была объяснена стабильность свобод-
ных ароматических радикалов и соответствующих ионов. В 1931—1932 гг.
Хюккель [86] весьма основательно рассмотрел проблему бензола, используя
для этого имевшиеся и развитые им новые кваптовомеханические пред-
ставления. В 1932 г. Полинг [87] рассмотрел ряд типично мезомерных моле-
кул, включая двуокись углерода, бензол, макромолекулу графита, а так-
же нитрат- и карбонат-иопы, объяснив их форму и устойчивость квантово-
механическим резонансом. В 1933 г. автор [88] показал, каким образом
сложившиеся в то время представления могут быть включены в общую
теорию мезомерии. В 1933 г. Полинг и Уэлапд [89] развили работы Хюккеля
о строении бензола, разработав более простой математический метод, кото-
рый они смогли приложить и к другим ароматическим углеводородам, вклю-
чая свободные радикалы. Некоторые вопросы, связанные с дальней-
шей разработкой количественной трактовки таких проблем, рассмотрены
в гл. IV.
Чтобы объяснить эти представления, рассмотрим карбоксилат-ион. Пред-
положим, что все электроны, кроме четырех, локализованы на атомных
орбиталях или орбиталях связей и образуют, таким образом, молекулярный
каркас или остов. Остаются только две пары электронов, возможности
обмена которых между орбиталями необходимо исследовать. Сначала допу-
стим, что одна пара этих электронов принадлежит ^-орбитали атома кисло-
85
Глава II
рода, а другая — л-орбитали двойной связи, как этого требует валентная
структура А
(А) 6 — ОО О = С —О (Б)
Допустив, что эти электроны сохраняют свою принадлежность к указан-
ным орбиталям, можно описать их движение четырехэлектронной волновой
функцией Тд (имея в виду, что 2 характеризует вероятность распреде-
ления этих четырех электронов при всех возможных комбинациях четырех
положений в пространстве). Другая энергетически эквивалентная четырех-
электронная волновая функция Чтб аналогичным образом соответствует
валентной структуре Б; однако, согласно постановке задачи, Ч’д является
запятой, а Тв — незанятой функциями *. Движение электронов, дозволен-
ное Ч'а, разрешает электронам различные перемещения и возможность при-
тяжения к оболочке атома углерода или отталкивания от нее, т. е. эле-
ктроны могут самопроизвольно переходить из занятой функции Уд на перво-
начально пе занятую Тв- Поэтому возникает резонанс: пи первоначально
занятая Т-функция, ни первоначально не занятая функция '1Г пе представ-
ляют стационарного состояния. Эти функции .заменяются двумя новыми
четырехэлектронными волновыми функциями системы, каждая из которых
описывает чередующуюся принадлежность электронов к атомным орбита-
лям или орбиталям связи симметричным образом, что ведет к сочетанию
движения электронов с дополнительным движением, допускаемым резонан-
сом. Имеются две такие четырехэлектронные функции: одна, когда она
занята, представляет состояние с пониженной, а другая — состояние с по-
вышенной энергией. Первое состояние является нормальным мезомерным
состоянием. Понижение энергии по сравнению с энергией валентной струк-
туры называется энергией резонанса или, более специфически, энергией
мезомерии. Состояние с повышенной энергией является возбужденным мезо-
мерным состоянием; оно представляет интерес главным образом в связи
с оптическими свойствами.
Таким образом, в основе мезомерии лежит делокализация электронов,
ограниченная электростатическими силами, приводящая к более стабильной
системе и часто вызывающая появление новых диполей, порой противо-
положных постоянным электростатическим диполям. В таких симметричных
системах, как двуокись углерода и бутадиен-1,3, мезомерия не вызывает
постоянных диполей, но формальная нейтральность концевых атомов пред-
ставляет собой усреднение мгновенных состояний заряда, флуктуации
которых происходят в более широких пределах, чем это было бы без мезо-
мерии. В бензоле электронные плотности шести связей, равные вследствие
мезомерии, также являются усреднением сильных флуктуаций электрон-
ных плотностей (гл. IV).
Например, в карбонат- или питрат-ионах, которые имеют три эквивалент-
ные валентные структуры, вследствие мезомерии возникают одно нормаль-
ное и два возбужденных состояния. Функция Т каждого из этих состояний
симметрична относительно ^-функций трех ковалентных структур, и поэто-
му молекула обладает тригональной симметрией, как это показано ниже
для карбонат-иона при помощи изогнутой связи, причем ионный аряд сим-
* При сравнении этой теории с теорией образования ковалентной связи водорода
(гл. I, разд. 3,г) следует иметь в виду, что одна валентная структура соответствует одному
распределению обоих электронов между двумя атомами водорода (по отнюдь пе у одного).
86
Внутримолекулярное и межмолекулярное взаимодействия
метричио поделен между атомами кислорода:
Ковалентные структуры карбокат-
иона
Мезоллерное
состояние
2-
Если ковалентные структуры мезомерной молекулы не являются энергети-
чески эквивалентными, как в недиссоциированной карбоксильной группе,
в сложноэфирпой или амидной группах, в цианатах и азидах, в винилхло-
риде, анилине или бензальдегиде (это действительно общий случай), то
мезомерия приводит к менее эф-
фективной стабилизации электрон-
ной системы по сравнению с рас-
пределением электронов в наибо-
лее стабильной из ковалентных
структур. Однако, если орбитали
атомов и связей менее стабильных
ковалентных структур доступны
в какой-либо степени для элект-
ООО о=с-о
Карбоксилат-ион
О-7._^О
Карбонат-ион
ронов, находящихся первоначаль-
но на орбиталях наиболее ста-
бильной ковалентной структуры,
мезомерия может привести к нор-
мальному состоянию, которое в
известной мере стабильнее, чем
наиболее стабильная ковалентная
структура. В таком случае энер-
гия мезомерии условно рассчиты-
вается по отношению к наиболее
стабильной из валентных структур.
Двуокись углерода и бутадиен-1,3,
а также нитроамины и эфиры азот-
ной кислоты представляют более
специальные частные примеры
общего случая; в этих соедине-
ниях в сопряжении участвуют
более чем две ковалентные струк-
туры, по лишь некоторые из них
являются эквивалентными. Энер-
гетические отношения между ва-
r2n=c-o
I
r2n-c=o
~т~
Алшд
RN-C=O
= RN=C-0
R. RN=O0
Алкилцианат
О=с>о О-С=6
0=00
Двуокись углерода
R2N=N<q-
R2N-N^O“ R N-N*O_
___ \O ________0
Нитроамин
Рис. 13. Схематические энергетические уров-
ни мезомерпых молекул.
Энергии ковалентных структур (сущест-
вование которых можно представить в
качестве начального приближения) пока-
заны пунктиром, а энергии мезомерных
состояний — сплошной линией. Стрелки
обозначают величины условных энергий
мезомерии.
лентными структурами и мезомер-
ными состояниями для некоторых общих и специальных систем качественно
представлены на рис. 13. Как правило, энергии этих состояний распреде-
лены в интервале, перекрывающем как нижнюю, так и верхнюю границы
интервала энергий отдельных структур. Энергия мезомерии будет тем
больше, чем больше число структур, которые лежат достаточно низко
по сравнению с наиболее стабильной структурой. Можно сделать вывод,
что более протяженная сопряженная система стабилизируется большей
энергией мезомерии, чем аналогичная менее протяженная система. Таким
образом, Р-кетоенолят-ион обладает большей энергией мезомерии, чем кар-
57
Глава II
боксилат-ион, а гексатриеп-1,3,5 — большей, чем бутадиен-1,3
X — С С — С-С >Х — С=-С
C = C-^C-C = Y>C=C-C = Y
X —С = С —С—Y >Х —С -Y
С—-С—С = С—С^О > С = С—С- с
е. Мезомерия и стереохимическая форма. Стереохимические следствия
из теории мезомерии выводятся наиболее просто, иногда даже в сверхупро-
щенной форме, из модели двойной связи Хюккеля — Пенни, так как эту
модель легко распространить на сопряженные системы (гл. I, разд. 4,6).
Сопряжение рассматривается как результат бокового перекрывания атом-
ных ^-орбиталей или л-орбиталей, возникших из р-орбиталей. Следова-
тельно, нормальное мезомерпое состояние будет максимально стабильным
Рис. 14. Иллюстрация стабильности плоского расположения атомов и их о-связей в со-
пряженной системе.
А — бутадиен-1,3; Б—амидная группа. Взаимодействующие атомные р -орбитали пред-
ставлены в виде полярных диаграмм: взаимодействия не независимы, так как принцип
Паули должен соблюдаться для каждого атома. 0-Связи представлены осями симмет-
рии — межъядерпыми линиями. Атом кислорода амидной группы имеет несопряженную
р- (показана) и s-орбитали (или, возможно, sp-гибридную орбиталь) (не изображена); обе
заняты неподеленными электронами.
при максимальном перекрывании, т. е. в том случае, когда сопрягающиеся
р- и л-орбитали имеют общую узловую плоскость или, другими словами,
когда все атомы сопряженной системы и все связанные с ними о-связи лежат
в одной плоскости. Для бутадиена и амидной группы это показано на рис. 14.
Эта простая теория объясняет отсутствие свободного вращения и стабиль-
ность планарной или приблизительно планарной конформации центральной
формально простой связи в ряде сопряженных систем, описанных в разд.
2,д, например бутадиена-1,3, муравьиной кислоты, различных амидов,
азотной кислоты, эфиров азотистой и азотной кислот и фенола. С другой
стороны, влиянию мезомерии, приводящему к плоской структуре, могут
в различной степени препятствовать силы, действующие но связям или
через окружающую среду.
В качестве иллюстрации эффекта этих сил рассмотрим трехлигапдный азот.
В этом примере внутриатомные силы приводят к пирамидальной конфигура-
ции связей. Объяснение этого состоит в том, что несвязывающая орбиталь
приобретает дополнительный s-характер за счет связывающих орбиталей,
в результате чего неподеленные электроны приближаются к ядру; энергия,
выделяющаяся в результате этого процесса, превышает энергию ослабления
трех связей, s-характер которых уменьшается. Когда доля s-характера,
переносимого от связей к неподеленным электронам, очень мала, перенос
88
Внутримолекулярное и межмолекуляриое взаимодействия
происходит в большей степени в соответствии с положением энергетического
равновесия. Таким образом, хотя мезомерия обусловливает плоскую конфи-
гурацию амидной группы, все же следует ожидать, что структура любого
амида немного выведена из плоскости. Как отмечалось в разд. 2,д, молекула
формамида действительно не является совершенно плоской.
Наиболее широко и давно известным примером противодействия несвязы-
вающих сил выплащивающему эффекту мезомерии является существование
энантиомеров в ряду диарильных соединений (разд. 2,д). Мезомерия, затра-
гивающая обе арильные группы, стремится привести их в копланарное
состояние. Однако пространственное отталкивание, обусловленное обменным
взаимодействием между заместителями в арильных группах, выводит ариль-
ные группы из общей плоскости, и образуются настолько стабильные кон-
формации, что оптическая активность существует даже при повышенных
температурах.
Пространственное отталкивание, которое нарушает коплапарность не толь-
ко диарилов, но и замещенных нитробензолов, нитроанилинов и других
сопряженных систем, уменьшает сопряжение функциональных групп с бен-
зольным кольцом, т. е. степень делокализации л-электронной системы. Эти
эффекты, обусловливающие изменение физических свойств, например диполь-
ных моментов, а также химических свойств (реакционной способности,
кислотно-основного равновесия, скоростей ароматического замещения) носит
название «вторичных пространственных эффектов» и будет обсуждаться
в гл. Ill, VI и XIV.
ж. Электромерные эффекты в ненасыщенных системах. Электромерным
эффектом называют электронное смещение в мезомерной системе под дей-
ствием сил, которые возникают в реакционном центре в переходном состоянии.
Электромерный эффект является эффектом поляризуемости, и его направ-
ление всегда такое, которое облегчает реакцию.
Если мезомерная система образуется в результате сопряжения одной -}-М-
группы X или одной —ТИ-группы Y (или +7И-группы) с ненасыщенным
углеводородным остатком, как, например, в X —С = С или С = С — С = Y,
то величина электронного смещения, которое переводит рассматриваемую
структуру в нормальное мезомерное состояние, никогда пе превышает малой
доли (почти всегда меньше 10%) величины, достаточной для полного пре-
4- —— 4- _
вращения в диполярную структуру X = С — С или С — С = С — Y.
Это ясно из данных о дипольных моментах (гл. III). Конечно, предполагает-
ся, что энергия мезомерии сравнительно мала. В таких случаях дополни-
тельные электронные смещения по механизму сопряжения возможны глав-
ным образом в сторону диполярной структуры; поэтому сильные электро-
мерные эффекты являются строго однонаправленными, причем Н-ТИ-группы
обладают сильными -(-^-эффектами в реакциях, в которых реакционный
центр носит электроположительный характер, и слабыми —Е’-эффектами
в реакциях с электроотрицательным реакционным центром. Аналогично
—М-группы проявляют сильные —Е’-эффекты в реакциях, в которых эти
силы действуют в прямом направлении, и только слабые -(-Е’-эффекты там,
где эти силы действуют в обратном направлении.
В этих случаях электромерная поляризуемость, как и мезомерная поляри-
зация, ограничена электростатическими силами. Так, например, -фЕ'-эффект
заместителя уменьшается положительным ионным зарядом и электроотри-
цательностью в случае идентично заряженных атомов одного и того же
периода системы Менделеева. Однако, подобно индуктомерной поляризуе-
мости, он будет увеличиваться с возрастанием номера периода в одной
89
Глава II
и той же группе периодической системы *. —^-Эффект заместителя увели-
чивается положительным ионным зарядом при повышении номера группы
в периоде таблицы Менделеева и при возрастании номера периода. Эти
выводы сведены в последних двух разделах табл. 13.
Таблица 13
Электромерная поляризуемость групп
1. + Е-Эффект —X в винил — X и т. д.
— О >—OR > — OR2 и т. д.
— NR2 > — OR > — F п т. д.
— I >—Br> —Cl> —F п т. д.
2. —Е-Эффект = У в винил — С = Y и т. д.
= NR2> = NR и т. д.
= 0 > = NR> = CR2 и т. д.
= S > = 0 п т. д.
3. ф-Е-Эффект —X в карбонил — X и т. 0.
— OR —О п т. д.
— F >—OR >—NR2 и т. д.
— I > — Вг —С1>—F и т. д.
4. —Е-Эффект — Ye R2N — C = Y или RO — C = Y
= NR> = NR2 n т. д.
= NR > = 0 и т. д.
= S > = О и t. д.
5. ±Е-Эффект.ы C — C в (C = C)n и m. a.
C=C-C=C-C=C>C=C-C=C>C=C и т. д.
CeH5-C = C>C = C и т. д.
Как уже отмечалось, изложенные правила выполняются, когда заместители
связаны простой связью с ненасыщенным углеводородным остатком с обра-
зованием систем, имеющих сравнительно малую энергию мезомерии. К систе-
мам, имеющим большую энергию мезомерии, т. е. к системам, в которых
нормальное мезомерное состояние не является близким к одной из валент-
ных структур, применимы другие правила. Чтобы понять это, необходимо
вновь вспомнить, что поляризуемость — квантованное свойство и что,
поскольку квантовая теория разрешает только некоторые состояния эле-
ктронов, электроны в молекуле будут взаимодействовать с приложенными
электрическими силами не так, как они взаимодействовали бы, если бы
молекула имела структуру с зарядом, подчиняющуюся классическим эле-
ктростатическим и электродинамическим законам. Приложенное электриче-
ское поле деформирует нормальное состояние молекулы путем придачи
недеформированному нормальному состоянию электронов в некоторой сте-
* Причины этого снова квантовомеханические: высшие состояния, от которых зависит
поляризуемость основного состояния и которые приближенно можно представить приве-
денными выше дпполярпыми структурами, более доступны, когда, например, X = I,
чем когда X = F, поскольку в молекулах, как и в атомах, электронные состояния менее
разделены друг от друга по энергии, когда главное квантовое число выше.
90
Внутримолекулярное и меж молекулярное взаимодействия
пени (возрастающей с увеличением силы поля) характера одного или несколь-
ких разрешенных возбужденных состояний. Чем более доступно возбужден-
ное состояние в смысле вероятности перехода, определяемой в соответствии
с принципом перекрывания геометрией орбитали, а также чем ниже энергия
возбужденного состояния, тем больше эффект деформирующего поля или,
F=C-d
Винилгало-
генид
r3=c-c|
I
I
RO-OCJ
V —
Виниловый
Эфир
R2N=C-cj
RZN-C=C|
Т~~
Виниламин
_м
RO=C-C
'^zS’S
т*
___J—
r2n-oo
6-00 0=С-0
Б
Галогенан-
гидрид
карбоно-
вой
Эфир
карбоно-
вой
кислоты
кислоты
Амид
карбоно-
вой
кислоты
Карбоксилат-
анион
с-с=с-с с-с=с-с
с-с-с=с с=с-с-с
В
с-с-ос
ос-с-с
Тс=с-с=с
]5ута()иен-193
Рис. 15. Схема ^энергетических уровней мезомерпых систем, иллюстрирующая главные
факторы, которые обусловливают электромерную поляризуемость основных
состояний.
Замечание. Халеви указал автору, что нельзя ограничиваться учетом поляризуемости
лишь одного верхнего состояния, так как это не справедливо для систем с более высокими
главными квантовыми числами, т. е, с более заполненными энергетическими уровнями.
другими словами, тем больше вклад этого возбужденного состояния в поля-
ризуемость нормального состояния. Поляризуемость определяется суммой
термов, каждый из которых пропорционален вероятности перехода, деленной
на энергию, необходимую для соответствующих возбуждений. Терм, соответ-
ствующий низкой энергии возбуждения, может преобладать. Понятно, что
возбуждения в действительности нет; но именно возможность его осуществ-
ления придает основному состоянию свойство поляризуемости.
Почему же мы тем не менее применяем полуклассическое электростатиче-
ское приближение, например, в предыдущей части разд. 3, ж? Это стано-
вится ясным при рассмотрении рис. 15, А. В приведенных в качестве приме-
37
Глава II
ра винильных производных винил — X заместители X относятся ко второй
строке первой части табл. 13. В этих случаях энергия мезомерии (стрелка,
направленная вниз) значительно меньше, чем качественно оцененная раз-
ность энергий между неполярной и диполярной валентными структурами
(стрелка, направленная вверх). Считается, хотя и несколько умозрительно,
что неполярная валентная структура служит хорошим приближением
к реальному мезомерному основному состоянию, а диполярная валентная
структура — к реальному высокополяриому мезомерному возбужденному
состоянию. Это отражено на приведенных диаграммах. Таким образом,
можно предположить, что реальная энергия возбуждения между мезомер-
ными основным и возбужденным состояниями (направленная вверх сплош-
ная стрелка) будет меняться при переходе от одного случая к другому при-
мерно в той же степени, в которой изменяется фиктивная энергия возбуж-
дения (пунктирная стрелка) между валентными структурами. Если пред-
положить, что геометрия орбиталей и, следовательно, вероятность переходов
не сильно изменяются при переходе от одного случая к другому, то электро-
мерная поляризуемость будет приблизительно соответствовать обратной
величине энергии возбуждения, а в более грубом приближении — обратной
величине фиктивной энергии возбуждения. Вот почему при применении уже
рассмотренных простых электростатических представлений получаются каче-
ственно корректные результаты (например, вывод о том, что +2?-электро-
мерпая поляризуемость групп X в структурах винил — X изменяется в ря-
ду F < RO <С R2N). Остальным данным первой части табл. 13 может быть
дано подобное же квантовохимическое подтверждение.
Однако по мере того, как энергия мезомерии становится сравнимой по ве-
личине или превышающей энергетическую разность между валентными
структурами, электростатическая модель перестает быть справедливой, что
ясно следует из рис. 15,Б. Они относятся к карбоксильным соединениям
карбонил — X, в которых соответствующие пары валентных структур
энергетически менее разделены, чем в только что рассмотренной серин.
Поскольку X изменяется от F к ОСН3 и затем к N(CH3)2, энергетические
уровни сближаются и при X, равном О-, совпадают. Однако энергия мезо-
мерии велика и при указанном изменении X становится еще больше, поэто
му мезомерные состояния будут удаляться друг от друга по энергии, что
и показано на диаграммах. Электромерпая поляризуемость будет примерно
обратно пропорциональна разности энергий мезомерпых форм и, таким
образом, в противоположность серии винил — X -j-E'-электромерпая поля
ризуемость X в серии карбонил — X будет убывать в ряду F > OR > NR2 >
Д> О-. Именно такое квантовомеханическое приближение необходимо
принять, чтобы последовательно уяснить данные, представленные в третьем
разделе табл. 13.
Во втором разделе табл. 13 приведены данные для таких систем, как винил —
— С = Y и арил — С = Y, выведенные выше с помощью полуклассическоп
электростатической теории, которая в данном случае применима вследствие
того, что энергия мезомерии таких систем достаточно мала. Это можно
было бы подтвердить с помощью трех диаграмм, точно таких же, как дна
граммы, приведенные в первом ряду рис. 15, по соответствующих по порядку
слева направо соединениям: С = С — С = NR, С = С — С = О иС = С —
+
— С = NR2. Разность энергий мезомерпых состояний, которая связана
с разностью энергий валентных структур, в этом ряду опять-таки умень-
шается, и поэтому —^-электромерпая поляризуемость этих групп возра-
стает в ряду = NR < = О < NR+ в соответствии с данными второго раз-
дела табл. 13.
92
Внутримолекулярное и меж молекулярное взаимодействия
Однако для обоснования выводов, сделанных в четвертом разделе табл. 13,
необходимо более корректное квантовое приближение, так как энергия
мезомерии представленных здесь производных карбоксильного типа велика.
Можно было бы привести схематические энергетические диаграммы, подоб-
ные приведенным на рис. 15, В и соответствующие слева направо соеди-
нениям R2N — С = NR, R2N — С = О и R2N — С = NR*, т. е. амидину,
амиду и иону амидиния соответственно. Из этих диаграмм можно было
бы видеть, что в этом ряду уровни энергии пар валентных структур сбли-
жаются, однако вследствие роста энергии мезомерии разность энергии
между парами мезомерных состояний будет увеличиваться. Таким образом,
—Е-электромерная поляризуемость групп в структурах карбоксильного
типа R2N — С = Y будет уменьшаться в ряду = NR > = О > = NR+2
в соответствии с данными четвертого раздела табл. 13.
Другие проблемы возникают при рассмотрении сопряженных полиенов
(пятый раздел табл. 13). В этих случаях вследствие сильного взаимодей-
ствия между (часто довольно многочисленными) возбужденными состояния-
ми энергия некоторых образующихся таким образом возбужденных состоя-
ний близка к энергии неполярной максимально двоесвязанной структуры.
Благодаря этому разность энергий мезомерии между этими состояниями
и основным состоянием увеличивается. Возбужденные состояния обладают
меньшей степенью двоесвязности, при этом несвязывающие электроны
сосредоточиваются у концов сопряженной системы; поэтому электромерная
поляризуемость основного состояния зависит от легкости, с которой поля-
ризующее поле может способствовать приобретению основным состоянием
некоторого характера всех этих возбужденных состояний или части из них.
Конечно, более низкое возбужденное состояние более доступно и, следова-
тельно, вносит больший вклад в рассматриваемый процесс. Как показано
на рис. 15, В, удлинение цепи сопряжения увеличивает число низколежа-
щих возбужденных состояний и поэтому должно увеличивать электромер-
ную поляризуемость основного состояния вследствие увеличения числа
и уменьшения энергии тех возбужденных состояний, которые уча-
ствуют в поляризации основного состояния под влиянием деформирую-
щего поля.
Таким образом, электромерная поляризуемость гораздо более важна, чем
индуктомерная. Электромерная поляризуемость затрагивает сопряженные
л-электроны, которые расположены на высших занятых орбиталях молеку-
лы, а первым возбужденным состоянием соответствуют низшие незанятые
орбитали. Поэтому эти электроны высокополяризуемы. Индуктомерная
поляризуемость обусловлена ст-электронами, расположенными на низколе-
жащих орбиталях, и первому возбужденному состоянию соответствуют орби-
тали с более высокой энергией, чем энергия любой упомянутой л-орбитали.
Поэтому ст-электроны гораздо менее поляризуемы.
Часто упускают из вида, что дополнительная поляризация, необходимая
для перехода из начального состояния в переходное, не адекватна поля-
ризуемости, т. е. подвижности электронов, которая обусловливает легкость
или трудность возникновения дополнительной поляризации.
з. Стереохимия электромерной поляризуемости [90]. Пути поляри-
зуемости основных состояний предопределены волновыми функциями воз-
бужденных состояний молекул, поэтому полярные реагенты, вызывающие
электромерный эффект, будут предпочтительно атаковать те положения,
в которых в некотором низколежащем возбужденном состоянии имеется
избыток или недостаток электронов в зависимости от требований реагента.
Как было показано в разд. 3,ж, в сопряженных ненасыщенных системах
93
Глава II
такие положения соответствуют концевым или главным образом концевым,
атомам. В гл. XIII будет показано, что присоединение к сопряженным
ненасыщенным соединениям почти всегда начинается с присоединения по
концевым атомам.
Имеется важное различие между поляризуемостью под действием фотона,
являющейся мерой оптической рефракции, и существенной для химической
реакционной способности поляризуемостью под действием полярного реаген-
та. Вследствие высокой частоты волны фотонов они поляризуют только
электронную систему молекулы субстрата; в то же время медленно движу-
щиеся химические реагенты наряду с электронами поляризуют также внут-
реннюю ядерную систему. Таким образом, электромерная поляризуемость
Рис. 16. Распределение электронов и расположение ядер в основном и первом возбуж-
денном состояниях ацетилена.
Выведено из электронной и атомной поляризуемости в основном состоянии (расстояние
дано в А).
имеет стереохимический аспект. Поскольку основные пути поляризации
основного состояния молекулы субстрата определяются низшими возбуж-
денными состояниями, необходимо изучать стереохимию возбужденных
состояний.
К сожалению, по этому вопросу известно очень мало количественных данных,
т. е. данных, позволяющих по структуре ненасыщенного соединения пред-
сказать структурный тип возбужденного состояния. Одним из известных
случаев является ацетилен [91]. Основное состояние ацетилена линейно,
углерод-углеродная связь осуществляется шестью электронами и имеет
размеры, указанные на рис. 16 для основного состояния. Первое возбужден-
ное состояние, переход в которое из основного состояния разрешен по прави-
лу спинов, напоминает основное состояние этилена с удаленными транс-
водородными атомами, на месте которых находятся несвязывающие орбитали,
содержащие (взаимозаменяемо в соответствии с запретом Паули) приблизи-
тельно три электрона, первоначально принадлежавшие тройной связи. Эта
связь становится беднее электронами, и поэтому удлиняется. Такое распре-
деление электронов и экспериментально определенные расстояния между
ядрами представлены на рис. 16 для возбужденного состояния. На рис. 16
для основного состояния при помощи стрелок показано, каким образом
электронодефицитный реагент R+ при достижении одного из концов ацетиле-
новой связи смещает не только л-электроны и соседний протон, но также
и отдаленный протон, причем последний — в направлении, неожиданном
с точки зрения классического рассмотрения. Таким образом, любая после-
дующая реакция, например с X' при присоединении по тройной связи,
если она происходит вслед за начальной атакой R+, приведет к образованию-
траис-аддукта [90].
94
Внутримолекулярное и меж молекулярное взаимодействия
Первое возбужденное состояние формальдегида, переход в которое из основ-
ного состояния разрешен по правилу спинов, также было исследовано коли-
чественно. Оно является пирамидальным с сильно удлиненной СО-связью,
выведенной примерно па 30° (0,524 рад) из плоскости СН2-группы (рис. 17) *.
В возбужденном состоянии электронная конфигурация примерно наполо-
вину изменена в сторону конфигурации молекулы метанола, у которой
удалены два атома водорода, а орбиталь неподеленных электронов, обра-
зующаяся за счет первоначальной двойной связи, теперь находится у атома
Основное
Рис. 17. Распределение электронов и расположение ядер в основном и первом возбуж-
денном состояниях (верхний уровень инверсии) формальдегида (расстояние дано-
в А)
углерода и занята приблизительно одним электроном **. Эта дополнительная
орбиталь в возбужденном состоянии отмечает направление, по которому
происходит атака реагентов по карбонильным группам в их нормальном
состоянии.
Геометрия первого возбужденного состояния этилена недостаточно ясна.
То же можно сказать и о первом возбужденном состоянии бензола; хотя
обычно предполагается, что это состояние имеет две равновесные конфигу-
рации, в одной из которых кольцо удлинено вдоль линии, проходящей
через ияра-положения, а в другой — в перпендикулярном направлении.
В любом случае происходит концентрация электронов в направлении более
острых углов.
и. Гиперконъюгация |94]. Гиперконъюгация представляет собой прибли-
жение второго порядка. Уже говорилось, что классификация электронов
на сил — это приближение и что ст-электропы обладают в остаточной форме
некоторыми свойствами л-электронов. Эти свойства состоят в способности
к сопряжению (если позволяет симметрия) во втором приближении с л-
электропами или создающими их р-электронами и орбиталями и в третьем
приближении — с другими о-электронами. Взаимодействие второго порядка,
которое будет рассмотрено в данном разделе, называется гиперконъюгацией.
Способность а-электронов к такому взаимодействию зависит от распростра-
ненности о-орбиталей в «боковых» направлениях и, таким образом, от кон-
* См. [92]. Приведенные здесь данные получены Пулем, Паркином и Липдсеем при
точном и детальном анализе поглощения формальдегида в близкой ультрафиолетовой
области.
Подобным образом изогнуто первое синглетное возбужденное состояние тиофосгена
CSCB, в котором связь сера — углерод удлинена на 0,10 А (1 -10~2 им) и отклоняется
от плоскости СС1г на 32° (0,558 рад) [93].
** Такое описание не противоречит спектроскопическим данным, согласно которым
при возбуждении формальдегида несвязывающий электрон кислорода частично перехо-
дит к углероду. Частичное расспаривацие эт-электронов, происходящее при выходе моле-
кулы из плоскости, будет, во-первых, компенсироваться спариванием за счет пеподелен-
ных электронов атома кислорода п, во-вторых, вносить вклад в заселенность повой орби-
тали неподеленных электронов на углероде.
95
Глава II
кретного типа с-связи. Связи с атомами водорода, вероятно, имеют большую
протяженность в боковом направлении, о чем можно судить по тому факту,
что СН-связи почти изотропны, тогда как СС-связи и большинство других
связей, в которых не участвует водород, гораздо более поляризуемы в направ-
лении своей длины, чем в поперечном направлении. Эксперимент показывает,
насколько заметно в каждом конкретном случае гиперконъюгация может
изменить физические и химические свойства молекулы.
Гиперконъюгация а-электронов СН-связей, впервые обнаруженная, лучше
всего подтвержденная и, конечно, наиболее важная форма гиперконъюгации,
была открыта Бейкером и Натаном [95] в 1935 г. Они изучили кинетику
присоединения замещенных бепзилбромидов к пиридину с образованием
бензилпиридинийбромидов. Было показано, что этой реакции способствует
перемещение электронов в зону реакции. Бейкер и Натан установили, что
/г-алкильные заместители, будучи группами, отдающими электроны, уско-
ряют реакцию, но не в последовательности, соответствующей индуктивному
механизму электронных смещений:
(СН3)3С - >?(СП3)2СН- > СН3СН2 - > СН3 -
В действительности наблюдался обратный порядок, и поэтому был сделан
вывод, что в и-алкилбензильных системах существует дополнительный
механизм освобождения электронов алкильными группами, который наиболее
сильно проявляется в метильной группе, но становится меньшим при замене
водородов в метильной группе па углеводородные радикалы. Подобная ано-
малия обнаружена не во всех системах, содержащих алкильные группы.
Бейкер и Натан установили, что аномалия исчезает, когда заместители
не находятся в геара-положении, и связали ее с присутствием смежной
сопряженной системы бензольного ядра. Они предположили, что электронная
пара СН-связей в метильной группе значительно меньше локализована, чем
электроны СС-связей, и способна, как и неподеленные электронные пары,
вступать в сопряжение с ненасыщенной системой.
н^Е^с=сКс=О^
Этот механизм электронных смещений действует в том же направлении,
что и индуктивный эффект, но так как новый механизм зависит от количе-
ства соответствующим образом расположенных СН-связей, его эффективность
уменьшается в ряду
СН3 > СН3СН2 > (СН3)2СН > (СН3)3С
Если эти различия более важны, чем различия в индуктивном эффекте,
то алкильные группы вызовут результирующую отдачу электронов в указан-
ном порядке.
Итак, теория сопряжения, предложенная Тиле в 1898 г., была расширена
дважды: сначала в 1902 г. Флюршеймом, который, основываясь на формаль-
ном сходстве в опытах, включил во взаимодействие с парциальными связями
скрытые валентности, и затем в 1935 г. Бейкером и Натаном, включившими
в нее донорное взаимодействие с валентностями, запятыми атомами водорода.
Последние авторы предложили для электронных смещений механизм гипер-
конъюгации (сверхсопряжения), который для СН-связей является электро-
положительным, т. е. +А".
Бейкер и сотрудники сразу же поставили задачу определить, является ли
гиперконъюгация фактором постоянной поляризации, т. е. гипермезомерным
эффектом (~)-М), от которого зависит положение химических равновесий
96
Внутримолекулярное и межмолекулярное взаимодействия
и физические свойства, или это только фактор поляризуемости, т. е. гипср-
электромерный эффект (+-Е), необходимый для стимулирования фотона
или молекулы реагента и поэтому проявляющийся только в оптических
свойствах и скоростях химических реакций, но не влияющий на другие
физические свойства и химические равновесия. В 1935 г. Бейкер, Натан
и Шопи показали, что гиперконъюгация действительно включает гипермезо-
мерный эффект -\-М, потому что она замедляет те реакции, при которых
электроны должны быть удалены из реакционного центра [96]. Известно,
что такой реакцией является катализируемое основаниями прототропное
взаимопревращение диарилазометипов; показано, что это превращение
сильнее всего замедляется тг-метильном группой и слабее всего трет-
бутилыюй
w-RCeH4Cn=--NCH2C6H5 n-CeH4CH2N = CHCeH5
К аналогичному выводу позднее пришли при изучении равновесия между
замещенными бензальдегидами и их циангидринами
n-RCeIl4CJ10 + HCN »-RCeH4CH(OH)CN
Установлено [97], что большая термодинамическая стабильность альдегида
по сравнению с его циангидрином увеличивается при наличии и-алкильных
групп, причем сильнее всего сказывается влияние метильной группы
и менее всего тцет-бутильной. Этого следует ожидать, если мезомерный
эффект алкильных групп превосходит индуктивный, так как в альдегиде
сопряжение распространяется на карбонильную группу в боковой цепи
и, следовательно, стабилизирует альдегид по сравнению с циангидрином.
Более подробные данные о гиперконъюгации приведены в гл. XIII.
Установлено также существование гиперконъюгациопного электромерпого
уЬ’-эффекта алкильных групп. Такой вывод вытекает из наблюдения [98],
согласно которому при таких реакциях, как, например, гидролиз бенз-
гидрилгалогепидов (который, как известно, зависит от предварительного
расщепления молекулы па ионы), кинетический эффект, обусловленный
гиперконъюгацией n-алкильпых заместителей, сильно увеличивается.
n-RC0H4CH(CeII5)Cl n-RC6H4CH(C6H5) + Cl-
11одобные реакции сильно ускоряются алкильньши заместителями и значи-
тельно больше метилом, чем трет-бутилом. Эти реакции рассматриваются
в гл. VII.
Мы не будем рассказывать историю гиперконъюгации. История этого эффекта
содержит многочисленные регулярные экспериментальные подтверждения
его существования [99], множество попыток физической интерпретации
и различные попытки приближенного квантовомеханического истолкования.
Ссылки па гиперконъюгацию, т. е. на электронодонорный эффект СН-связей,
можно будет найти в гл. VI, VII, IX, XI и XIII. В соответствующих местах
упомянут и убедительный пример донорных свойств NH- и ОН-
связей, аналогичный гиперконъюгации типа -\-К и найденный де ла
Маром [99].
Таким образом, участие электронов СН-связей в гиперконъюгационных
эффектах -J-M и -\-Е, приводящих к освобождению электронов, хорошо
обосновано. Можно предполагать, что аналогично ведут себя электронные
нары ОН- и NH-связей, но здесь доказательство затруднено из-за подвиж-
ности протона и присутствия неподеленных электронов.
Для объяснения некоторых химических равновесий было постулировано
1100] участие электронной пары С —Hal-связей в электроотрицательных
7-0772
97
Глава II
гиперконъюгационных электронных смещениях —К (гипермезомерная поля-
ризация —М)
С=С—С==С—С~На1
Сравнительную термодинамическую стабильность взаимно превращаемых
дигал огеполефинов, которые образуются в результате 1,2- и 1,4-присоедине-
ния галогенов к сопряженным диолефинам, можно объяснить с помощью
простого химического рассуждения. Если в дигалогенолефинах отсутствует
обычное сопряжение, которое может искажать картину, всегда оказывается,
что 1,4-дигалогенид термодинамически более устойчив, чем его 1,2-изомер,
даже когда гиперконъюгация водорода должна обусловливать большую
стабильность 1,2-дигалогенида, например:
СНз-СНВг—СН = СН —СНВг-СНз СН3 — СНВг— СНВг-СН = СН-СП3
Следовательно, необходимо допустить, что в данном случае, по-видимому,
имеет место мезомерия, зависящая от гиперконъюгации с участием галогена,
и что она энергетически более важна, чем гиперконъюгация с участием водо-
рода. Доказательства -цЛ/ и —Л/-гиперконъюгационной мезомерии СН-
и CHal-связей соответственно, основанные на длинах связи и дипольных
моментах, приведены в гл. III.
СПИСОК ЛИТЕРАТУРЫ
1. Eisenschitz R., London F., Z. Physik, 60, 491 (1930); London F., ibid., 63, 245 (1930):
Z. physik. Chem., Bit, 222 (1930).
2. Lennard-J ones J. £., в книге Fowler R. H., «Statistical Mechanics», Cambridge Univ.
Press, 1935, Chap. 10; Fowler R. H., Guggenheim E. A., Statistical Thermodynamics,
Cambridge Univ. Press, 1939, Chap. 7; Buckingham R. 4., Proc. Roy. Soc. (London),
A168, 264 (1938); Corner J., Trans. Faraday Soc., 44, 914 (1948).
3. Debye P., Physik Z., 13, 97 (1912); Polare Molekeln, Hirzel, Leipzig, 1929.
4. Thomson J. J., Phil. Mag., 46, 513 (1923).
5. Ingold С. K., Ann. Repts. on Progress Chem. (Chem. Soc. London), 23, 144 (1926).
6. Lewis G. N., Randall M., Thermodynamics, McGraw-Hill, New York, 1923, Chap. 22.
7. Dippy J. F. J., Chem. Revs., 25, 151 (1939); Watson H. B., Modern Theories of Organic
Chemistry, Clarendon Press, Oxford, 2nd Edn., 1941, Chap. 2.
8. Ingold С. К., J. Chem. Soc., 1933, 1124.
9. Lewis G. AL, Valence and the Structure of Atoms and Molecules, Chemical Catalog Co.,
New York, 1923, p. 113.
10. London F., Z. Elektrochera., 35, 552 (1929).
11. Hughes E. D., Ingold С. К., J. Chem. Soc., 1935, 244.
12. Pelzer II., Wigner E., Z. physik. Chem., B15, 445 (1932); Eyring H., J. Chem. Phys.,
3, 107 (193.5); Evans M. G., Polanyi M., Trans. Faraday Soc., 31, 875 (1935): Wynne-
Jones It'. F. K., Eyring II., J. Chem. Phys., 3, 492 (1935); Glasstone S., Laidler K. J.,
Eyring II., Theory of Rate Processes, McGraw-Hill, New York, 1941.
13. Bronsted J. N., Z. physik. Chem., 102, 169 (1922).
14. de la Mare P. B. D., Vernon C. A., J. Chem. Soc., 1951, 1764.
15. Kemp J. D., Pitzer K. S., J. Chem. Phys., 4, 749 (1936); J. Am. Chem. Soc., 59, 276
(1937).
16. Kistiakowsky G. B., Lacher J. R., Stitt F., J. Chem. Phys., 6, 407 (1938); 7, 289 (1939).
17. Howard J. B., J. Chem. Phys., 5, 451 (1937).
18. Smith L.G.J. Chem. Phys., 17, 139 (1949); Weiss S., Leroi G. E., ibid., 48, 262 (1968).
19. Arnett R. L., Crawford B. L., Jr., J. Chem. Phys., 18, 118 (1950).
20. Spitzer R., Huffman H. M., J. Am. Chem. Soc., 69, 211 (1947).
21. Aston J. G., Schumann S. C., Fink II. L., Doty P. M., J. Am. Chem. Soc., 63, 2029
(1941); 65, 541 (1943); Pitzer К. S., Gwinn W. D., ibid., 63,3313 (1941); Kilpatrick J. E.„
Pitzer K. S., Spitzer R., ibid., 69, 2483 (1947).
98
Внутримолекулярное и меж молекулярное взаимодействия
22. Miller F. A., Inskeep R. G., J. Chem. Phys., 18, 1510 (1950); Rathjens G. IE., Jr.,
J. Chem. Phys., 36, 2401 (1962).
23. LeFevre C. G., LeFevre R. J. W., J. Chem. Soc., 1956, 3549.
24. Millen D. J., в книге de la Mare P.B.D., Klyne W., «Progress in Stereochemistry»,
Butterworths, London, 1962, Vol. 3, p. 138; Marino Y., Hirota E., J. Chem. Phys.,
28, 185 (1958).
25. Klyne W., Prelog V., Experientia, 16, 521 (1960).
26. Chan R. S., Ingold С. K., Prelog V., Angew. Chem., Internat. Edit., 5, 383 (1966).
27. Mizushima S., Structure of Molecules and Internal Rotation, Academic Press, New
York, 1954.
28. Millen D. J., в книге de la Mare P.B.D., Klyne W., «Progress in Stereochemistry»,
Vol. 3, p. 138; Hirota E., J. Chem. Phys., 37, 283 (1962).
29. Dunitz J. D., Prelog V., Angew. Chem., 72, 896 (1960).
30. Bradaco K., Kahavec L., Z. physikal. Chem., B48, 63 (1940); Mulliken R. S., Rev.
Mod. Physics, 14, 265 (1942); Rasmussen R. S., Tunnicliff D. D., Brettain R. R.,
J. Chem. Phys., 11, 432 (1943); Richards С. M., Nielsen J. R., J. Optical Sci. Am.,
40, 438 (1950).
31. Scott R. B., Meyers С. H., Rands R. D., Jr., Brickwedde F. G., Bekkendahl N.,
J. Research Nat. Bur. Standards, 35, 39 (1945); Aston J. G., Szasz G. J., Wooley W. H.,
Brickwedde F. G., J. Chem. Phys., 14, 87 (1946).
32. Wagner R., Fine J., Simmons J. W., Goldstein J. H., J. Chem. Phys., 26, 634 (1957).
33. de Groot M. S., Lamb J., Proc. Roy. Soc., A242, 36 (1957).
34. Cole A. R. H., Thompson H. W., Proc. Roy. Soc., A200, 10 (1949).
35. Miyazawa T., Pitzer K. S., J. Chem. Phys., 30, 1076 (1959).
36. Jones L. II., Badger К. M., Moore G. E., J. Chem. Phys., 19, 599 (1951).
37. Gray P., Pratt M. W., J. Chem. Soc., 1958, 3403.
38. Piette L. II., Anderson W. A., J. Chem. Phys., 30, 899 (1959); Gray P., Reeves L. W.,
ibid., 32, 1878 (1960).
39. Kajima, T., J. Phys. Soc. Japan, 15, 284 (1960).
40. Cohn II., Ingold С. K., Poole H. G., J. Chem. Soc., 1952, 4272.
41. Dixon W. B., Wilson E. B., J. Chem. Phys., 35, 191 (1961).
42. Costain С. C., Dowling J. W., J. Chem. Phys., 32, 158 (1960).
43. Gutowsky H. S., Iloln С. H., J. Chem. Phys., 25, 1228 (1956).
44. Snyder H. G., Hisatsune I. C., J. Mol. Spect., 1, 139 (1957).
45. Christie G. II., Kenner J., J. Chem. Soc., 121, 614 (1922).
46. Turner E. E., LeFevre R. J. W., Chem. & Ind., 45, 831, 883 (1926).
47. Bell F., Kenyon J., Chem. & Ind., 45, 864 (1926).
48. Mills W. II., Chem. & Ind., 45. 884, 905 (1926).
49. Kuhn R., Albrecht O., Ann., 455, 272 (1927).
50. Mills W. II., Elliot К. A. C., J. Chem. Soc., 130, 1291 (1928).
51. Lesslie M. S., Turner E. E., J. Chem. Soc., 1933, 1588.
52. Kistiakowsky G. B., Smith W. R., J. Am. Chem. Soc., 58, 1043 (1936).
53. Kuhn R., Albrecht O., Ann., 455, 272 (1927).
54. Kuhn R., Albrecht O., Ann., 458, 221 (1927).
55. Li С. C., Adams R., J. Am. Chem. Soc., 57, 1565 (1935).
56. Reiger M., Wesiheimer F. II., J. Am. Chem. Soc., 72, 19 (1950); cp. Ling С. С. K.,
Harris M. M., J. Chem. Soc., 1964, 1825.
57. Westheimer F. H., Mayer J. E., J. Chem. Phys., 14, 733 (1946); Wesiheimer F. H.,
ibid., 15, 252 (1947); Harris M. M., Proc. Chem. Soc., 1959, 367; Harris M. M., Mit-
chell R. K., J. Chem. Soc., 1960, 1905.
58. Mills W. II., Kelham R. M., J. Chem. Soc., 1937, 274.
59. Lewis G. N., Valence and the Structure of Atoms and Molecules, p. 139.
60. Lowry T. M., J. Chem. Soc., 123, 822, 1886 (1923); Nature, 114, 376 (1925).
61. Ingold С. K., Ingold E. II., J. Chem. Soc., 1926, 1310.
62. Ingold С. K., Shaw F. R., J. Chem. Soc., 1927, 2918.
63. Ingold С. K., J. Chem. Soc., 1933, 1120.
64. Olah G. A., Einfiihrung in der Theoretische Organische Chemie, Akademie-Verlag,
Berlin, 1960, pp. 180—181.
65. Johnson J. R. в книге Gilman H., «Organic Chemistry», Wiley, New York, 1938, p. 1015;
Remick A. E., Electronic Interpretations of Organic Chemistry, Wiley, New York,
1st Edn., 1943.
66. Koerner W., Gazz. chim. ital., 4, 305, 446 (1874).
67. Iliibner H., Ber., 8, 878 (1875).
68. Noelting E., Ber., 9, 1797 (1876).
69. Michael A., J. prakt. Chem., 46, 204 (1892) и последующие работы; 60, 286 (1899)
и последующие работы.
7* 99
Глава II
70. Thiele J., Ann., 306, 87 (1898).
71. Flurschelm B., J. prakt. Chem., 66, 321 (1902).
72. Flurscheim B., J. Chem. Soo., 95, 722 (1909); 97, 91 (1910); J. Soc. Chem. Ind. (Lon-
don), 44, 246 (1925).
73. Arndt F., Scholz E., Nachtway F., Bor., 57, 1903 (1924).
74. Armit J. W., Robinson R-, J. Chem. Soc., 126, 1604 (1925).
75. Lapworth A., J. Chem. Soc., 73, 445 (1898); 79, 1265 (1901).
76. Lapworth A., Proc. Manchester Lit. Phil. Soc., 64, № 3 (1920); J. Chem. Soc., 121,
416 (1922); Nature, 115, 625 (1925).
77. Lucas H. J., Jameson A. Y., J, Am. Chem. Soc., 46, 2475 (1924); Lucas H. J., Moy-
se H. W., ibid., 47, 1459 (1925); Lucas II. J., Simpson T. P., Carter J. M., ibid., 47,
1462 (1925).
78. Allan J., Oxford A. E., Robinson R., Smith J. С., J. Chem. Soc., 1926, 401.
79. Halevi E., Prog. Phys. Chem., 1, 109 (1963).
80. Pauling L., Nature of the Chemical Bond, 3rd Edn., 1960, Chap. Ill, p. 64.
81. Von Schilling V., Ann. 308, 193 (1900).
82. Woodward R. B., SmallG., J. Am. Chem. Soc.,72, 1297 (1950); MonksG., Hegenberg P.,
Angew. Chem., Internat. Edit., 5, 888 (1966).
83. Doering W. von E., Knox L. H., J. Am. Chem. Soc., 73, 828 (1951).
84. Angeli A., Atti. R. Acad. Lincei, [v], 33, 109 (1924).
85. Burton II., Ingold С. K., Proc. Leeds Phil. Lit. Soc., Sci. Sect. 1, 421 (1929),
86. Hiickel E., Z. Physik, 70, 204 (1931); 72, 310; 76, 628 (1932).
87. Pauling L., Proc. Nat. Acad. Sci. (U.S.), 18, 293 (1932).
88. Ingold С. К., J. Chem. Soc., 1933, 1120.
89. Pauling L., Wheland G. W., J. Chem. Phys., 1, 322 (1933).
90. Ingold С. K., J. Chim. Phys., 53, 473 (1956); Burnell L., Tetrahedron, 20, 2403 (1964);
21, 49 (1965).
91. Ingold С. K., King G. W., Nature, 169, 1101 (1952); J. Chem. Soc., 1953, 2702;
Innes К. К., J. Chem. Phys., 22, 863 (1954).
92. Brand J. C. D., J. Chem. Soc., 1956, 858; Robinson G. N., Digiorgio V. E., Canad.
J. Chem., 36, 31 (1958).
93. Brand J. C. D., Callomon J. H., MouleD. C., Tyrrell J., Goodwin T. H., Trans. Faraday
Soc., 61, 2365 (1965).
94. Baker J. W., Hyperconjugation, Oxford University Press, Oxford, 1962.
95. Baker J. W., Nathan W. S., J. Chem. Soc., 1935, 1844.
96. Baker J. W., Nathan W. S., Shoppee C. W., J. Chem. Soc., 1935, 1847.
97. Baker J. W-, Hemming M. L., J. Chem. Soc., 1942, 191.
98. Hughes E. D., Ingold С. K., Taher N. A., J. Chem. Soc., 1940, 949.
99. de la Mare P. B. D., Tetrahedron, 5, 107 (1959); Berliner E., ibid., p. 202.
100. Lora-Tamayo M., Cons. Sup. Inv. Cien., Inst. Alonso Barba, Madrid, 1948, 3; de la
Mare P. B. D., Hughes E. D., Ingold С. К., J. Chem. Soc., 1948, 17.
ГЛАВА HI
ФИЗИЧЕСКИЕ СВОЙСТВА МОЛЕКУЛ
1. Электрические дипольные моменты......................102
2. Теплоты образования и тепловой эффект реакций .... 115
3. Электрическая поляризуемость ....................... 122
4. Симметрия и размеры молекул .........................138
5. Упругие свойства молекул.............................148
Список литературы ...................................152
Теорию химических связей, которая описана в двух предыдущих главах,
можно проще всего применить при рассмотрении физических свойств молекул
в их основных электронных состояниях. Этому вопросу посвящена настоящая
и некоторые последующие главы. Из таких свойств будут рассмотрены элек-
трическая полярность, электрическая и диамагнитная поляризуемости,
внутренняя энергия, геометрия и упругость молекул. Указанные свойства
не могут быть рассмотрены без обсуждения ароматических свойств, поэтому
начало гл. IV следует рассматривать как продолжение настоящей. В гл. IV
также обсуждаются вопросы напряжения в кольце и другие факторы, влияю-
щие на внутреннюю энергию. Все выводы относятся к основным электрон-
ным состояниям, однако в связи с вопросом об электрической поляризуемости
основных состояний приходится упоминать и возбужденные состояния.
Рассмотрение физических свойств возбужденных состояний и в связи с этим
электронных переходов и явлений цветности — поглощения света, флуорес-
ценции и фосфоресценции — увело бы пас слишком далеко от темы, и поэто-
му в данной книге не приводится.
1. Электрические дипольные моменты
а. Экспериментальные данные. Измерение электрических дипольных
моментов молекул представляет собой один из важнейших методов, позво-
ляющих получать сведения о распределении электронов в молекуле. Принци-
пы измерений дипольных моментов были установлены Дебаем [1]. Предло-
женный им абсолютный метод основан на определении температурного коэф-
фициента диэлектрической проницаемости вещества в газообразном состоя-
нии. Дебай разработал также приближенный метод, основанный на измере-
нии диэлектрической проницаемости при какой-либо температуре в сочетании
с определением показателя преломления. Второй из этих методов часто
применяется не к газообразным веществам, а к разбавленным растворам
в неполярном растворителе, например в бензоле. Однако результаты, полу-
ченные при измерениях в растворах, неизбежно приближенны, потому
что установленная таким образом величина является в лучшем случае сум-
мой момента молекулы с моментом, наведенным ею в окружающем раствори-
теле, а поправку, обусловленную наведенным моментом, которая может
увеличить или уменьшить наблюдаемый момент молекулы, трудно опреде-
лить, и ее обычно не вносят. В 'то же время ввиду экспериментальных затруд-
нений моменты многих соединений в парообразном состоянии не могут быть
измерены или поддаются определению с недостаточной точностью; поэтому
часто приходится довольствоваться приближенными значениями моментов,
полученными при измерениях в растворах.
В ходе дальнейшего изложения для сравнения приводятся оба значения
величин дипольных моментов молекул, а именно величины дипольных момен-
тов веществ в газообразном состоянии, определенные по методу, который
основан на измерении температурного коэффициента (условно обозначены
буквой Г), и величины, выведенные на основании определения коэффициента
преломления в бензольном растворе (обозначены буквой Б). Первый метод
может дать лишь экспериментальные погрешности; во втором главные погреш-
ности обусловлены ошибками самого метода; ошибки, связанные с раствори-
телем, можно до некоторой степени уменьшить, применяя один и тот же
растворитель.
Дипольные моменты могут быть определены еще одним методом, который,
если его можно применить, дает большую точность, а именно по смещению
вращательных линий в микроволновом спектре (эффект Штарка). Главное
ограничение этого метода состоит в необходимости анализа спектра и иденти-
102
Физические свойства молекул
фикации нужных вращательных переходов. Дипольные моменты, полученные
таким образом, относятся к газовой фазе. Дипольные моменты выражаются
в единицах дебаях (Д); 1Д равен 10-18 эл. ст. ед. - см в системе CGS, или
3,3-10-30 Кл-м в системе СИ.
б. Дипольные моменты соединений с диполярными связями. Дипольные
моменты соединений, имеющих диполярные связи, как правило, значительно
больше моментов соединений, не содержащих этих связей. Таким образом,
следует считать, что если имеется диполярная связь, то она главным образом
и определяет величину наблюдаемого момента. Величины дипольных
Таблица 14
Дипольные моменты [Д (3,3-10~30 Кл-м)] молекул
с диполярными связями
Серия 1 Метод
(CH3)3NO (CH3)3NBF3 CH3NBC13 C2H5H2NA1C13
5,02 5,76 6,23 6,94 Б
](С2Н5)2С)ЫЗеС12 (C2H5)2OBF3 (C2H5)2OBC13 (C2H5)2OA1C13
6,71 5,29 5,98 6,68 Б
(СН3)3РВС13 (CeH5)3PBCl3 (C2H5)2SBC13
7,03 7,01 6,00 Б
Серия 2
c2h5n = с CeH5N = C CH3NO2 CeH5NO2
3,54 4,19 Г
3,47 3,53 3,15 4,03 Б
Серия 3 / 4
(СвН5)3РО (CeH5)3PS (СвН5)2Йе (CH3)2S-C x|=/
\Z \
4,28 4,73 4,83 6,2 4 Z Б
+ — +—
(u3o-C4H9)2SO (CeH5)2SO (CH3)2SO2 (CeHs)2SO2
4,44 Г
3,90 4,00 5,14 Б
(CeH5)3AsO (CeH5)2SeO (CeH5)3SbS (n-CH3CeH4)2T eO
5,50 4,44 5,40 3,93 Б
моментов для соединений с диполярными связями приведены в табл. 14, где
эти соединения разделены на три серии *.
Все моменты соединений, относящихся к серии 1, лежат в пределах 5 . . . 7 Д
(16,5-10~30. . . 23,1-10"30 Кл-м.) Рассмотрим сначала дипольный момент
окиси триметиламина, почти полностью обусловленный наличием NO-связи.
Длина NO-связи равна 1,36 А (13,6-10-а нм) [2], и если электрон с зарядом
* Моменты соединений серии 1 и 3 взяты из работы Филлипса, Хантера и Саттона
12], а моменты соединений серии 2 — по сводке Вессона [3].
103
Глава III
—4,80-Ю-10 эл. ст. ед. (15,84-10-20 Кл-м) переместится на это расстояние,
то дипольный момент должен быть равен 6,53 Д (21,55-10~30 Кл-м). Разность
между этой величиной и опытной [5,02 Д (16,57-IO-30 Кл-м)[ вызывается
обратной поляризацией NO-связи, индуцированной ее собственным диполь-
ным моментом. Однако эта разница дает заниженное значение обратной
поляризации, так как незначительные наведенные моменты в метильных
группах также входят в наблюдаемый дипольный момент. Данное явление
является общим для моментов диполярных связей: наблюдаемые моменты
меньше вычисленных для перемещения электрона между неполяризуемыми
атомами. Более высокие значения моментов других соединений серии 1
должны быть приписаны частично моменту, вызванному электроотрицатель-
ным атомом галогена, и частично большей длине связей в соединениях фосфо-
ра, серы и алюминия. Если пренебречь малыми эффектами, то дипольный
момент приведенного в таблице соединения бериллия можно считать вектор-
ной суммой моментов двух диполярных связей, которые, будучи направлены
к вершинам тетраэдра, имели бы величины 5,81 Д (19,17-10~30 Кл-м); эта
величина включает возмущение, вносимое одним атомом хлора и одной
этильной группой.
Моменты соединений серии 2 значительно меньше. При длине тройной связи
в изонитрилах, равной 1,17 А (11,7-10'2 нм), ее вычисленный момент для
полного переноса электрона равен 5,62 Д (18,55-10~30 Кл-м) [4]. Необычно
большое отклонение величин измеренных моментов от рассчитанных может
быть, по-видимому, приписано большой обратной поляризации легко поля-
ризуемой тройной связи в этих соединениях.
Иначе обстоит дело с нитросоединениями. Здесь следовало бы ожидать
незначительного момента переноса электрона вследствие мезомерного рас-
пределения отрицательного заряда между двумя кислородными атомами,
разделенными большим валентным углом; при длине NO-связи [5], равной
1,21 А (12,1 -10'2 нм), и угле ONO, составляющем 127° (2,216 рад), вычислен-
ный момент равен лишь 2,59 Д (8,55-10-30 Кл-м), тогда как наблюдаемые
моменты аномально велики. Это можно объяснить двумя причинами. Первая
и менее важная причина — вероятная низкая поляризуемость нитрогруппы,
обусловленная стабилизирующим эффектом энергии мезомерии, большая
величина которой связана с высокой степенью симметрии группы (гл. II,
разд. 3,ж и разд. 3,в настоящей главы). Вторая причина имеет большее
значение; она заключается в том, что нитрогруппа не только вызывает индук-
тивную поляризацию углеводородного остатка, но и приводит либо к гипер-
конъюгации, либо к дополнительному мезомерному сопряжению, что в свою
очередь ведет к увеличению дипольных моментов (гл. II, разд. 3, д и е):
Естественно, что сопряженная система в нитробензоле вызывает больший
дипольный момент, чем гиперконъюгационная система в нитрометане. Ни
в одной из других рассмотренных молекул не может быть обнаружено такого
эффекта, даже в изонитрилах, вследствие того что углеродный атом уже несет
отрицательный заряд. Действительно, дипольные моменты триметил- и три-
фенилфосфинбортрихлоридов одинаковы. Моменты этил- и фенилизонитрилов
104
Физические свойства молекул
также равны между собой, в то время как моменты нитрометана и нитробен-
зола резко различаются.
Соединения серии 3 отличаются от предыдущих. Во всех уже рассмотренных
соединениях положительный атом диполярной связи не может увеличить
ковалентность из-за отсутствия d-орбиталей в валентной оболочке (азот,
кислород) или же, если такая возможность и имеется (фосфор, сера), отри-
цательный атом (бор) не имеет электронов для образования дополнительной
связи. В веществах, относящихся к серии 3, положительные атомы (фосфор,
мышьяк, сурьма, сера, селен, теллур) имеют незаполненные орбитали,
а отрицательные атомы (кислород, сера, селен, углерод) — неподеленные
электроны; следовательно, такие диполи могут в большей или меньшей
степени нейтрализоваться дополнительным полным или частичным обобще-
нием электронов, осуществляемым за счет d-орбиталей положительных атомов
и неподеленных электронов отрицательных атомов. Такое обобщение приво-
дит к полному или частичному образованию двойной связи, второй компонент
которой (л-связь) возникает за счет р-орбиталей отрицательного атома
и р-орбитали положительного атома. Такая р — d л-связь, естественно,
отличается по своим физическим и химическим характеристикам от р — р л-
связи, обычной двойной связи в органических соединениях. Полинг и Бро-
куэй [6] впервые высказали мысль, что двойные связи с участием d-орбиталей
представлены в кислородсодержащих кислотах тяжелых элементов; под-
тверждение таких взглядов заключалось главным образом в том, что ХО-
связи в этих кислотах значительно короче ожидаемых величин для диполяр-
ных простых связей. Филлипс, Хантер и Саттон [2], основываясь на данных,
характеризующих соединения серии 3, убедительно доказали возможность
частичного образования таких связей.
Вычисленный момент при полном переносе электрона равен 7,44 Д
(24,55-Ю-30 Кл -м) для окисей фосфинов, 6,86 Д (22,64-Ю-30 Кл-м) и 7,90 Д
(26,07-Ю-30 Кл-м) для сульфонов (при допущении двух независимых пере-
мещений электронов вдоль направлений к вершинам тетраэдра). Наблюдае-
мые моменты составляют лишь немногим более половины вычисленных
величин; такая же или большая разница обнаружена и в большинстве других
случаев. Это может быть объяснено частичным образованием р,d-двойных
связей. Диметилсульфонийфлуоренилид представляет «исключение, под-
тверждающее правило». В этом случае атом серы имеет вакантные орбитали,
а атом углерода — неподеленные электроны, так что по строению это соедине-
ние должно быть включено в серию 3, хотя в соответствии с величиной диполь-
ного момента [6,2 Д (20,46-10-30 Кл-м)1 его следовало бы отнести к серии 1.
Как полагают Филлипс, Хантер и Саттон [2], здесь, очевидно, невозможны
смещения электронов углерода к атому серы из-за мезомерного включения
их в ароматическую систему * (гл. IV).
в. Дипольный момент и электроотрицательность. Изменения дипольных
моментов соединений, в которых метильные группы связаны с атомами
различных элементов одного периода, а также с атомами элементов одной
и той же группы периодической системы Менделеева, видны из табл. 15**.
Величины, приведенные в первом ряду, иллюстрируют изменение дипольных
моментов при переходе слева направо в одном периоде таблицы Менделеева.
* Крейг (частное сообщение) считает, что даже в этом соединении образуется заметная
двойная связь с участием d-орбиталей атома серы, ибо в противном случае отрицательный
заряд должен был бы распределиться в результате мезомерного эффекта по ароматическому
ядру и момент был бы значительно больше 6,2 Д (20,46-Ю-30 Кл-м).
** Данные, приведенные в этой и следующих таблицах разд. 1, взяты из сводки Вес-
сона [3].
105
Глава III
Таблица 15
Дипольные моменты [Д(3,3-1О_зо Кл-м)] метилсодержащих
соединений
Серия 1 сн3сн3 ch3nh2 CH3OH CH3F Метод
0,0 1,32 1,69 1,81 Г
1,46 1,66 Б
(CH3)2NH 1,02 (СН3)2О 1,29 Г
1,17 Б
(CH3)3N 0,65 Г
0,86
Серия 2 CH3F CH3C1 СН3Вг СН31
1,81 1,83 1,79 1,64 Г
1,86 1,82 1,48 Б
По мере того как заряд ядра в изоэлектронном ряду — СН3, — NH2, — ОН,
— F возрастает, дипольный момент увеличивается, причем первый интервал
намного превышает остальные. Это, по-видимому, обусловлено нелинейной
конфигурацией молекул метиламина и метилового спирта. Можно ожидать,
что при переходе к следующей группе периодической системы должны равно-
мерно увеличиваться только составляющие моментов, направленные вдоль
связи с заместителем. Среди соединений верхнего ряда табл. 15 только
метиламин и метиловый спирт имеют поперечную составляющую момента,
которая увеличивает наблюдаемый общий момент молекулы.
Весьма маловероятно, чтобы уменьшение моментов при переходе от метил-
амина к триметиламину и от метилового спирта к диметиловому эфиру про-
исходило бы главным образом вследствие изменений углов между связями.
Хотя точные величины этих углов неизвестны, они, по-видимому, отличаются
друг от друга лишь на несколько градусов: в триметиламине и диметиловом
эфире они соответственно равны 108 и 111° (1,885 и 1,937 рад) с точностью
до 3° (0,0523 рад), в аммиаке угол равен 107° (1,867 рад), а в воде — 105°
(1,832 рад). Наиболее вероятной основной причиной изменений моментов
является то, что NH- и ОН-связи имеют больший момент, чем связи NC
и ОС. Если предположительно приписать связям NC и ОС продольные момен-
ты, равные соответственно 0,6 и 1,2 Д (1,98-10~30 и 3,96-10~30 Кл-м) (т. е.
равные одной трети и двум третям момента фтористого метила при игнори-
ровании малых различий в длине NC-, ОС- и FC-связей), а связям NH
и ОН — 1,3 и 1,5 Д (4,29-10-30 и 4,95-10~30 Кл-м) (соответствующие момен-
там аммиака и воды) и если считать все углы в метильных соединениях
тетраэдрическими, то вычисленные моменты окажутся равными: CH3NH21,25;
СН3ОН 1,58; (CH3)2NH 1,05; (СН3)2О 1,38 и (CH3)3N 0,60 Д (4,12-Ю-30,
5,21-10-30, 3,46-10~30, 4,55-10—30 и 1,98НО-30 Кл-м соответственно). (Здесь,
как и всюду в этом разделе, моменты, обусловленные наличием необобщен-
ных электронов, рассматриваются как распределенные по связям.)
Менее правильная закономерность наблюдается для соединений серии 2:
моменты фтористого, хлористого и бромистого метилов примерно одинаковы,
106
Физические свойства молекул
в то время как иодистый метил обладает заметно меньшим моментом. По-види-
мому, такие данные являются результатом одновременного действия двух
противоположных влияний, а именно: индуктивного эффекта, уменьшающе-
гося от фторида к иодиду (гл. II, разд. 3,в), и удлинения связи углерода
с атомами галогенов при переходе от фтора к иоду.
г. Дипольные моменты алканов. В гл. II, разд. 3,в, было указано, что
вследствие некоторой делокализации электронов о-связей метильные группы
в алканах в принципе должны быть электроотрицательными по отношению
к метиленовым и метановым группам.
Дипольные моменты пропана и изобутана были измерены Лайдом [7] микро-
волновым методом и оказались равными 0,083 и 0,132 Д (27,3-10-32
и 43,56-10~3° Кл-м) соответственно с ошибкой измерения около ±0,001 Д
(1-10-33 Кл-м). Нас, конечно, интересует направление дипольного момента,
однако сначала выясним его составляющие. Момент пропана, для которого,
как известно, угол между связями ССС равен 112,4° (1,961 рад), можно разло-
жить на два момента связей СН3 — СН2 по 0,073 Д (24,09-Ю-32 Кл-м).
Три момента таких связей, направленных друг к другу под углом 111,15°
(1,940 рад) (угол ССС в изобутане), дают молекулярный момент 0,133 Д
(43,89-10"32 Кл-м), который хорошо согласуется с наблюдаемой
величиной.
По данным Лайда можно определить и направление этих моментов. Из влия-
ния дейтерирования на константы кислотности следует, что дейтерий более
электроположителен, чем обычный водород. Лайд установил, что дипольный
момент 2-дейтеробутана по величине больше момента обычного изобутана.
Применение правила сложения векторов приводит к выводу, что в изобутане
метильные группы по отношению к метановому атому углерода являются
электроотрицательными, что уже было предположено теоретически.
СНзч
СН3-(-С-Н
сн3^
0,132 Д (43,56.10-за Кл-м)
СН3
СН3-«—С<—D
сн3/
0,141 Д (46,53-10-32 Кл-м)
Это заключение подтверждается последующими микроволновыми измерения-
ми дипольных моментов, выполненными Лаури и Ментером [8]. Во-первых,
они подтвердили электроположительность дейтерия, отметав различие
в моментах недейтерированных и дейтерированных молекул известной
полярности, например фтористого метила:
CH3F CD3F
1,847 Д (6,09.10-3° Кл-м) 1,858 Д (6,13-10-3° Кл-м)
Затем они исследовали пропан и два дейтеропропана и получили следующие
результаты:
СН3|^ СН3. CD3t.
/СНг /CD2 /СН2
CH3Z СН/ CD/
0,085 Д (28,05-10-32 Кл-м) 0,095 Д (31,35-10-32 Кл-м) 0,076 Д (25,08.10~32 Кл-м)
Из этих данных следует, что момент связи СН3 — СН2 равен 0,075 Д
(24,75-Ю-32 Кл-м) и имеет направление, указанное в формулах
107
Глава III
стрелками. Эта величина близка к ранее полученной Лайдом [0,073 Д (24,09 X
X Ю-32 Кл -м)].
Работа Лайда позволила количественно оценить степень электроположитель-
ности дейтерия, которая, будучи выраженной через разность моментов связей
СН и CD, оказалась равной 0,009 Д (29,7-10'33 Кл-м). Из данных работы
Лаури и Ментера такую оценку можно провести двумя способами, если счи-
тать, что угол DCD является тетраэдрическим (что несомненно неточно).
В результате этих оценок получаются значения 0,008 и 0,009 Д (26,4-10-33
и 29,7-10-33 Кл-м).
д. Дипольные моменты гомологов. В табл. 16 приведены дипольные
моменты некоторых замещенных парафинов, образующих гомологические
Таблица 16
Дипольные моменты [Д (3,3-10“30 Кл-м)] в гомологических рядах
некоторых замещенных парафинов
(в каждом случае верхняя цифра относится к газообразному состоянию,
а нижняя — к раствору в бензоле)
Серия 1
СН3С1 C2HSC1 и-С3Н7С1 H-C4H9CI изо-С3Н7С1 т/>ет-С4Н8С1
1,83 1,86 2,01 2,04 1,94 2,04 1,93 2,15 2,04 2,13
СНдВГ C2H5Br и-С3Н7Вг и-С4Н9Вг изо-С3Н7Вг mpem-C4IIgBr
1,79 2,01 2,15 2,15 2,19
1,82 1,88 1,93 1,93 2,04 2,21
СН31 1,64 C2H5I 1,87 и-С3Н71 1,97 H-C4H9I 2,08 изо-С3Н71 трет-С4Н81
1,48 1,78 1,84 С 1,88 ерия 2 1,84 2,13
CH3NO2 C2H5NO2 b-C3H7NO2 h-C4H9NO2 из o-C3H7NO2 mpem-C4H8NO2
3,54 3,15 3,70 3,19 3,72 (3,35) 3,29 3,37 3,71
CHgCN 3,94 c2h5cn 4,03 b-C3H7CN 4,05 h-C4H9CN 4,09 U3O-C3H7CN тр«т-С4НдСК
3,51 3,57 3,57 С 3,57 ерия 3 3,61 3,65
CHgOH C2H5OH и-С3Н7ОН И-С4Н9ОН изо-С3Н7ОН mpem-C4HgOH
1,69 1,69 1,64 1,63 1,68
1,66 1,66 1,71 1,66 1,70 1,66
ch3nh2 1,33 1,46 c2h5nh2 1,38 b-C3H7NH2 С b-C4H9NH2 1,40 ерия 4 u30-C3H7NH2 mpem-C4HgNH2 1,29
CHgCHO 2,69 C2H5CHO 2,73 и-С3Н7СНО 2,72 и-С4Н8СНО uao-C3H7CHO трет-С^НдСНО
2,49 2,54 2,57 2,57 2,56
ряды. Таблица по вертикали делится на три части: левая и средняя части,
взятые вместе, содержат моменты гомологов нормального строения от метила
до нормального бутила, а средняя и правая части совместно показывают
влияние разветвления в радикале (от метила до тпретп-бутила). По горизонта-
108
Физические свойства молекул
ли таблица имеет четыре раздела, в которых приведены данные о дипольных
моментах соединений следующих типов:
Серия 1 — молекулы без поперечного момента с заместителем, не содержа-
щим кратных связей.
Серия 2 — молекулы без поперечного момента с заместителями, содержащи-
ми кратные связи.
Серия 3 — молекулы, обладающие поперечным моментом и имеющие заме-
стители с простыми связями.
Серия 4 — молекулы, обладающие поперечным моментом и имеющие заме-
стители с кратными связями.
Данные серии 1 этой таблицы показывают общее увеличение момента в гомо-
логическом ряду моногалогенопроизводных; в случае гомологов изо-строения
наблюдается более заметное увеличение момента, чем для нормальных
гомологов, что находится в соответствии с представлениями об индуктивном
эффекте (гл. II, разд. 3,а).
Из серии 2 табл. 16 видно, что здесь наблюдается значительно меньшее
увеличение моментов, несмотря на то что можно было бы ожидать вдвое
большего увеличения моментов поскольку величины моментов метильных
производных примерно в 2 раза больше, чем в серии 1. Более того, наблю-
дается лишь некоторое увеличение момента при переходе от метила к этилу
и отсутствие заметного изменения при переходе к mpem-бутилу. Эта анома-
лия может быть объяснена гиперконъюгацией. Нитрометан, как уже было
отмечено, имеет дипольный момент, увеличенный вследствие гиперконъюга-
ции. Это должно быть справедливо также и для ацетонитрила
H^C^-C=N
Влияние гиперконъюгации уменьшается в гомологическом ряду, в то время
как индуктивный эффект возрастает, вследствие чего наблюдается лишь
незначительное изменение результирующего момента.
Следует также отметить, что неожиданное направление моментов связей
элементов IV группы
(6 + )CH3-SiH3(6-) (б + )СН3-GeH3(6 —)
0,735 Д (2,43-Ю-зо Кл-м) 0,642 Д (2,12-10-30 Кл-м)
определенное Лаури и Ментером [8] методом замещения на дейтерий, может
быть обусловлено гиперконъюгацией электронов СН3-группы с й-орбиталями
атомов Si и Ge.
Данные для соединений серии 3 свидетельствуют о наличии компенсирующих
влияний иных типов. Моменты рассматриваемых соединений обусловлены
как продольными, так и поперечными составляющими. Наведенные в угле-
водородном остатке моменты должны усиливать продольные и уменьшать
поперечные составляющие моментов. Таким образом, суммарно индукция
не ведет к значительным изменениям дипольных моментов спиртов, но замет-
но уменьшает моменты и в гомологическом ряду аминов, у которых, как
уже отмечалось, продольная составляющая момента значительно меньше,
чем у спиртов.
В случае соединений, относящихся к серии 4, можно ожидать наличия обоих
видов компенсаций, наблюдаемых отдельно в соединениях, приведенных
в сериях 2 и 3; данные таблицы согласуются с этим предположением.
109
Глава III
е. Дипольные моменты и сопряжение. Еще до получения опытных дан-
ных было предсказано [9], что мезомерные системы могут обладать электри-
ческим моментом, противоположным и иногда даже превосходящим по вели-
чине электростатический момент; в последнем случае общее смещение
электронов может происходить в обратном направлении. Это иллюстрируют
следующие структуры:
(«+)А1К—►NRs(S-) (З-)Аг—CnR2(5+)
Хьендал [10] одним из первых определил моменты, обусловленные наличием
заместителей в бензольном кольце, в случае как моно-, так и полизамещенных
бензолов, используя при этом приближенный принцип векторной аддитив-
ности Томсона (гл. II, разд. 2,6). При этом было установлено, что группа
NH2 в анилине является положительным концом диполя.
Саттон [11] предложил более общий метод исследования обусловленных
мезомерией моментов ароматических соединений. Он показал, что даже в тех
случаях, когда, как это часто бывает, мезомерный эффект не превосходит
индуктивного эффекта, наблюдаются значительные различия в значениях
дипольных моментов двух молекул алкил — Ви арил — R, где группы R
одинаковы. Независимо от направления индивидуальных моментов разность
в моментах, т. е. момент ароматического соединения минус момент алифати-
ческого соединения, всегда соответствует направлению ожидаемого мезомер-
ного эффекта в ароматическом соединении. Для иллюстрации можно при-
писать моментам знак плюс или минус в зависимости от того, обладает ли
заместитель электроположительным или электроотрицательным характером.
Тогда алкилфториды и фторбензо л будут иметь отрицательные моменты,
хотя абсолютное значение момента фторбензола будет меньше; следовательно,
дополнительный момент арилфторида является положительным, что согла-
суется со структурно возможным +1И-эффектом атома фтора (гл. II, разд.
3,д). Нитрилы жирного ряда и бензонитрил также имеют отрицательные
моменты, но абсолютное значение момента бензонитрила больше, а, следова-
тельно, его дополнительный момент отрицателен, и это согласуется с ожи-
даемым —M-эффектом заместителя (гл. II, разд. 3,д).
{-I, + М) Аг-O’ AP^CN (-Д - М)
Другие примеры приведены в табл. 17. Здесь значения дипольных моментов
метильных и треш-бутильных производных сравниваются с аналогичными
значениями для фенильных производных. Чтобы обеспечить возможность
сравнения, были выбраны такие заместители, для которых направления
связей являются осями симметрии; в этом случае мезомерный и индуктивный
моменты лежат вдоль оси симметрии, так что их величины можно складывать
или вычитать. Такое упрощение справедливо для одноатомных и тригональ-
ных заместителей. Это условие выполняется в случае алифатических и аро-
матических нитрогрупп, а также и для ароматической диметиламиногруппы
как стереохимическое следствие мезомерии (гл. II, разд. 3,е). Подобное
упрощение не может быть сделано для алифатических соединений с диметил-
аминогруппой; однако в данном случае наблюдаемые моменты отличаются
от вычисленной автором величины продольного момента [0,6 Д (1,98 X
X 10-30 Кл -м)] настолько мало, что практически не имеет значения, какая из
величин берется для сравнения с резко отличающимся значением момента
ароматической молекулы.
110
Физические свойства молекул
Таблица 17
Дипольные моменты [Д (3,3-10“30 Кл-м)] некоторых алифатических
и ароматических соединений
R Дипольные моменты Метод Разность
СНзН mpem-CjHgR CeHgR С6Н5Н-СН3К CfjHsR—mpem-CiHgR
N(CH3)2 -0,65 4-1,61 г 4-2,26
-0,86 4-1,58 Б 4-2,44
сн3 0,0 (0,0) 4-0,37 Г 4-0,37 4-0,37
(0,0) (0,0) 4-0,34 Б 4-0,34 +0,34
F — 1,81 —1,57 Г 4-0,24
С1 -1,83 —2,13 —1,70 Г 4-0,13 +0,43
-1,86 -1,57 Б 40,29
Вг -1,78 —1,71 Г 4-0,08
—1,82 -2,21 —1,55 Б 4-0,27 +0,66
I -1,48 -2,13 -1,38 Б 4-0,10 +0,75
СС13 -1,57 -2,07 Б -0,50
CN -4,03 —4,39 Г -0,36
-3,51 -3,65 —3,94 Б -0,43 —0,29
no2 -3,54 —3,71 -4,19 Г —0,65 -0,48
-3,15 —4,03 Б —0,88
Для группы N(CH3)2 характерна очень большая разница в моментах *,
т. е. большой +М-мезомерный момент. Для галогенов наблюдаются малые
мезомерные моменты того же знака +М; их величина уменьшается, как это
и должно быть (гл. II, разд. 3,д), от F к I. Группы CN и NO2 характеризуют-
ся мезомерными моментами противоположного знака —М. По предположе-
нию все эти моменты возникают в результате сопряженного смещения эле-
ктронов. Для группы СН3 в разность моментов, по-видимому, входит мезомер-
ный момент гиперконъюгации -\-М, хотя данный случай и нуждается в даль-
нейшем изучении. В то же время для группы СН3 разность моментов в основ-
ном зависит от момента гиперконъюгации —М (гл. II, разд. 3,е)**.
Следует подчеркнуть, что все эти мезомерные моменты малы по сравнению
с моментами [И—26 Д (36,3 -10-30—85,8 -10“30 Кл -м)], которые возникали бы
в результате полного превращения в ионные структуры следующих типов:
(CH3)2N =\ или N = C=^ >+
Это указывает на то, что электростатические силы сравнительно рано пре-
кращают мезомерные смещения электронов (гл. II, разд. 3,Д). Данное
обстоятельство придает электромерному эффекту в этих системах сильную
асимметрию, которая становится почти односторонней в случае больших
деформирующих сил (гл. II, разд. 3,ж) ***.
* При сравнении с стему R =N (СН3)2, NHCH., МН2’флифатическими аминами
Смит [12] заключил, что мезомерные моменты этих групп R в фенил — R составляют
-[-2,06, 4-1,93 и 4-1,67 Д (6,80-10“30, 6,37-10“30 и 5,51-10“30 Кл-м) соответственно.
** Для соединений R=CF3 найдены следующие дипольные моменты (в бензоле): для
трифторметилпиклогексана —2,40 Д (—7,92-10“30 Кл-м), для бензотрифторида
—2,60 Д (—8,58.10“30 Кл-м); разность - 0,20 Д (—0,66-IO-30 Кл-м) [13].
*** Подобные оценки могут быть в некоторых случаях проведены физическими мето-
дами: например, для циркулярно симметричных заместителей, имеющих ядерный квадру-
польиый момент (например, С1),— с помощью констант ядерного квадрупольного расщеп-
111
Глава III
Число сравнимых между собой парафиновых и олефиновых соединений,
имеющих оси симметрии, весьма ограниченно. Характерным примером могут
служить кетен и ацетон, данные для которых приведены в табл. 18. Если
допустить, что, подобно бутадиену-1,3 (гл. II, разд. 2,д), стабильной будет
транс-форма акролеина, то любой возможный мезомерный момент окажется
под весьма малым углом к общему моменту, и можно в первом приближении
применить обычные упрощенные рассуждения для сравнения акролеина
и ацетальдегида. Как показывают данные табл. 18, карбонильный кислород
Таблица 18
Дипольные моменты [Д (3,3-10~30 Кл-м)] некоторых насыщенных
и ненасыщенных соединений, содержащих карбонильную группу
Соединение Дипольный момент Соединение Дипольный момент Метод Разность
СН3 -2,85 -1,45 г +1,40
О_=с/ О = С = СН2
СН3 —2,74 -1,43 Б -1,31
о = сн—сн3 —2,69 О^СН-СН=СН2 -3,04 Г -0,35
—2,49 -2,85 Б -0,38
в кетене производит +Л/-эффект *, а в акролеине оказывает —.[/-эффект,
что соответствует теоретическим представлениям
б^с^сн2 6Хсн-^сн^=снг
ж. Дипольные моменты и вторичный пространственный эффект. Хэмпсон
и сотр. [16] обнаружили явление, весьма хорошо иллюстрирующее стерео-
химическую необходимость копланарности для мезомерных систем (гл. II,
разд. 3,е). Они установили, что если в производном бензола две метильные
группы в орто-положении препятствуют многоатомному +М- или —М-заме-
стителю (например, диметиламино- или нитрогруппе) располагаться в одной
плоскости с бензольным кольцом, то мезомерный момент почти исчезает;
в этом случае величина дипольного момента, обусловленная заместителем,
уменьшается и приближается к ее значению в алифатических соединениях.
В заметной степени это не наблюдается в случае незамещенной аминогруппы,
которая по своим размерам недостаточно велика, чтобы орто-метильные
группы могли воспрепятствовать ее расположению в одной плоскости с ядром
лснпя, получаемых из микроволновых спектров. Так, если бы не было мезомерного смеще-
ния электронов от С1 к NO2 в нитрилхлориде С1 — NO2, распределение плотности элект-
ронов у ядра хлора имело бы циркулярную симметрию относительно оси связи С1 —
— N и ядерный квадруполь, определяемый как ансамбль зарядов + +, налагающийся
на сферический положительный заряд, не имел бы никакой предпочтительной ориентации
относительно линии связи. Однако мезомерное смещение уменьшает электронную плот-
ность около ядра С1 выше и ниже плоскости NOa-группы, и, следовательно, ядра имеют
различную энергию в зависимости от того, положительные или отрицательные заряды
квадруполя будут лежать в этой плоскости. Разность энергий этих состояний, определяе-
мая из расщепления линий в микроволновом спектре, показывает, что от атома хлора
к нитрогруппе переходит 5% одного л-электрона [14].
* Это подтверждено Хакинсом и Лефевром, которые измерили моменты высших кете-
нов и сравнили полученные результаты с данными для кетонов [15].
112
Физические свойства молекул
(по крайней мере приблизительно); это совершенно не наблюдается в случае
одноатомных заместителей, например брома.
Наблюдение Хэмпсона позволило открыть общее явление, заключающееся
в том, что стерическое отталкивание нарушает сопряжение, выводя из плоско-
сти часть сопряженной системы, в результате чего мезомерный и электромер-
ный эффекты резко ослабевают или вовсе исчезают. Такого рода влияние
пространственных факторов на сопряжение, как бы оно ни проявлялось,
можно назвать вторичным пространственным эффектом. Это явление более
подробно рассматривается в настоящей главе, а также в гл. VI и XIV.
Таблица 19
Сравнение дипольных моментов [Д(3,3-1О-Зо Кл-м)] в бензоле
производных мезитилена и дурола с аналогичными производными бензола
Заместители Производные Заместители Производные
мези- тилена дурола бензо- ла мези- тилена дурола бензо- ла
Монозамещенн N(CH3)2 no2 nh2 Br ые про 1,03 3,67 1,40 1,52 изводил. 3,62 1,39 1,55 ie 1,58 4,03 1,54 1,55 п-Дизамещен N(CH3)2 и no2 NH2 и NO2 Вг и NO2 чые пр оизводн 4,11 4,98 2,36 ые 6,87 6,10 2,65
В левой части табл. 19 приводятся некоторые данные Хэмпсона. Для
наглядности проведено сравнение соответствующих значений для производ-
ных мезитилена, дурола и аналогичных производных бензола. Моменты,
возникающие в результате прямого взаимодействия метильных групп с бен-
зольным ядром, можно не учитывать вследствие симметричного расположе-
ния метильных групп.
Если сильные -\-М- и —M-заместители занимают ^-положения в бензольном
кольце, например в тг-нитроанилине, то результирующий дипольный момент
особенно велик не только потому, что каждый из заместителей находится
в сопряжении с бензольным кольцом (что ведет к усилению момента), но
и потому, что оба заместителя через ядро находятся в сопряжении друг
с другом, следствием чего является дополнительное увеличение общего
момента.
В n-нитродиметилдуридине'все эти эффекты в значительной степени подавле-
ны, так как четыре метильные группы препятствуют занятию диметиламино-
и нитрогруппами положений, необходимых для сопряжения. При этом наблю-
дается резкое уменьшение момента по сравнению с моментом п-нитродиметил-
анилина. В тг-нитродуридине два из трех эффектов уничтожаются, в то время
как аминогруппа, имеющая малые размеры, может проявлять свой мезомер-
ный эффект; поэтому здесь обнаруживается меньшее уменьшение момента
по сравнению с моментом тг-нитроанилина. Бром проявляет очень слабую
8-0772
113
Глава III
способность к сопряжению, и, следовательно, при переходе от п-бромнитро-
дурола к n-бромнитробензолу происходит еще меньшее уменьшение величины
момента, подобно тому как это наблюдается в случае соответствующих соеди-
нений, не содержащих брома. Высказанные соображения подтверждают
данными, приведенными в табл. 19.
з. Дипольные моменты ненасыщенных углеводородов. Ниже приведены
дипольные моменты ряда ненасыщенных углеводородов, включая аромати-
ческие углеводороды. В каждом случае указано известное или предполагаемое
направление поляризации.
СН3—>СН2 = СН2 СН3 —»СН = СН — СН = СН2
+ 0,35 Д (1,15.10-30 Кл-м) (газ) +0,68 Д (2,24-1О~зо Кл-м) (газ)
СН3-^СвН5 (СН3)3С —> С6Н5
+0,34 Д (1,12-Ю-зо Кл-м) (в бензоле) +0,56 Д (1,85-10“30 Кл-м) (в бензоле;
СН3—»С = СН
+ 0,75 Д (2,47-10-зо Кл-м) (газ)
НС = С«—С6Н5
— 0,78 Д (—2,57-10-30 Кл-м) (в бензоле)
Направление поляризации соответствует представлениям об ее индуктивной
природе. Поляризация возникает вследствие первоначально неодинаковой
гибридизации атомных орбиталей, из которых образуется связь, обозначен-
ная стрелкой. Согласно этим представлениям, электроны связи между двумя
подобными атомами, т. е. между атомами с одинаковыми атомными номерами,
будут смещены в сторону атома с более высоким s-характером первоначаль-
ной атомной орбитали, т. е. в сторону более ненасыщенного из двух атомов
(гл. I, разд. 4,6). Направление поляризации в молекулах толуола и фенил-
ацетилена было определено экспериментально путем введения в пара-поло-
жение бензольного кольца заместителей с известным направлением поляри-
зации (галогены, нитрогруппа) и измерения моментов замещенных углеводо-
родов. Для расчета использовался приближенный метод сложения век-
торов.
Что касается величин моментов, то больший момент ацетиленовых соедине-
ний не является неожиданным, так как в этом случае асимметрия гибридиза-
ции наибольшая. Труднее объяснить большую величину момента пиперилена
(пентадиена-1,3) по сравнению с пропиленом и больший момент mpem-бутил-
бензола по сравнению с толуолом: в обоих случаях моменты изменяются
в ожидаемом направлении, однако разность моментов слишком велика.
Можно предположить, что в случае алкилзамещенных ненасыщенных угле-
водородов определенный вклад в величину дипольных моментов вносит
гиперконъюгация, эффект которой наибольший для метильных производных.
При таком предположении эффект удлинения ненасыщенной цепи, например
при переходе от пропилена к пиперилену, очевиден: вследствие гиперконъю-
гации происходит смещение электронов от одной связи к другой, и это сме-
щение наибольшее в более длинных системах.
Применяя те же представления о влиянии природы алкильной группы,
которые были использованы для объяснения изменения дипольного момента
при переходе от толуола к mpem-бутилбензолу, необходимо предположить,
что в случае алкилцианидов и нитроалканов более выраженный эффект
гиперконъюгации метила по сравнению с mpem-бутилом приблизительно
уравновешивается более сильным индуктивным эффектом mpem-бутила
(разд. 1,д), тогда как в алкилбензолах такая компенсация не достигается.
114
Физические свойства молекул
Это, возможно, объясняется тем, что в алкилцианидах и нитроалканах
полярность алкильной группы возникает вследствие полярности связанной
с ней группы (гл. II, разд. 3,в), т. е. в результате действия внешнего источ-
ника поляризации, и поэтому в этих соединениях индуктивный и гипер-
конъюгациоиный эффекты в достаточной степени независимы друг от друга,
и в первом приближении их можно считать аддитивными. С другой стороны,
в ал кил замещенных ненасыщенных углеводородах эти эффекты ни в коей
мере не являются независимыми, так как индуктивная поляризация зависит
от различия в гибридизации, которое уменьшается при наличии гиперконъю-
гации. Так, например, в толуоле гиперконъюгация уменьшает индуктивную
компоненту электрического момента, но в то же время вносит и свой вклад,
так что суммарный эффект нельзя предугадать. С этой точки зрения можно
довольно изящно объяснить, почему момент толуола значительно меньше
чисто индуктивного момента mpem-бутилбензола.
2. Теплоты образования и тепловой эффект реакций
а. Величины констант энергии связи. Теплоту образования соединения
из атомов можно вычислить на основании закона Гесса по теплоте сгорания
соединения, теплотам образования продуктов сгорания из элементов и тепло-
там атомизации элементов. Все используемые значения должны относиться
к нормальным состояниям молекул и атомов. В идеальном случае все тепло-
ты должны быть приведены к О К, но данные, необходимые для такого приве-
дения, часто недоступны, и для большинства целей такая поправка несуще-
ственна.
Фаянс [17] первым пытался представить теплоты образования многоатомных
молекул при помощи аддитивных постоянных для каждой связи. Существен-
ным здесь является то, что энергия диссоциации связи в многоатомной моле-
куле включает энергию реорганизации, связанную с изменением внутреннего
строения диссоциированных осколков, т. е. атомов или свободных радикалов.
По этой причине различны, например, теплоты четырех стадий атомизации
метана
СН4—>СН3-|-Н—> СН2-]-2Н—>СН+ЗН—>С4-4Н
Обычно среднюю величину этих теплот, т. е. одну четвертую часть теплоты
атомизации (или теплоты образования из атомов) метана, называют «энергией
связи» СН. Мы будем называть эту величину «константой энергии связи-»,
ибо она не является энергией разрыва отдельной связи, но, подобно кон-
станте молекулярной рефракции, имеет значение средней величины, получен-
ной исходя из принципа аддитивности и для иллюстрации этого принципа.
Подобно другим аддитивным принципам, этот принцип также является неточ-
ным, так как иначе и не может быть (гл. II, разд. 2,6), тем не менее он поле-
зен как отправной пункт для изучения тех отклонений, которые настолько
сильны, что никакое усреднение не может их затмить.
Данный пример показывает наиболее прямой способ получения констант
энергии тех связей многоатомных молекул, для которых энергия простой
связи XY может быть принята равной i/n теплоты образования соедине-
ния XYn. Для соединений углерода этот метод применялся в основном для
связей СН и CHal, содержащих атомы водорода или галогенов.
Константы энергии других простых связей, например CN, СО и CS, вычис-
ляются иным путем. Они выражаются разностью между теплотой образования
молекулы, содержащей одну такую связь, и константами энергии других
связей, определяемых описанным выше способом. Константа простой связи
8* 115
Глава III
CN равна, таким образом, величине остатка, полученного после вычитания
из теплоты образования метиламина констант энергии трех СН-связей и
констант энергии двух NH-связей, вычисленных, как описано выше, из
теплот образования метана и аммиака. Разумеется, эта разность не будет
представлять энергию диссоциации CN-связи в метиламине, но, будучи выве-
дена на основе принципа аддитивности, она удобна для его иллюстрации
и анализа.
Константы энергии всех двойных и тройных связей в многоатомных органи-
ческих молекулах (С = С, С = С, С = О, С = N, С = N и т. д.) также можно
определять, пользуясь этим методом.
Чтобы придать возможно более приемлемую форму принципу приблизитель-
ной аддитивности констант энергий связи, Полинг [18] применил прием,
подобный тому, который используется при обработке данных по рефракции
и другим аддитивным константам. Сначала из рассмотрения исключаются все
молекулы, которые содержат формальные заряды и сопряженные цепи, т. е.
такие молекулы, которые обусловливают наибольшие отклонения от принци-
па аддитивности. Затем на основании экспериментальных данных подсчиты-
вают, пользуясь принципом аддитивности, константы энергии связей в дру-
гих молекулах. Эти молекулы наиболее наглядно иллюстрируют аддитив-
ность констант энергий связи. Для лучшего согласия с опытными данными
по аналогии с классическим аддитивным описанием молекулярной рефракции
для карбонила и циангруппы допускается несколько разных величин, т. е.
молчаливо признается неаддитивность. Некоторые из определенных Полин-
гом значений констант энергии связей (при 25 °C) приведены в табл. 20.
Таблица 20
Константы энергии связей (по данным Полинга)
Связь Е, ккал/моль (4,187X XI03 Дж/моль) Связь Е, ккал/моль (4,187х Х103 Дж/моль) Связь Е, ккал/моль(4,187X Х103 Дж/МОЛЬ)
с—с 83,1 I Простые с Н-Н вязи 104,2 С-Н 98,8
C-N 69,7 | N —N 38,4 N —Н 93,4
С-0 84,0 0 — 0 33,2 О —Н 110,6
C-S 62,0 S-S 50,9 S-H 81,1
С —F 105,4 F —F 36,6 F —Н 134,6
С-С1 78,5 С1-С1 58,0 С1 —Н 103,2
С —Вг 65,9 Вг —Вг 46,1 Вг-Н 87,5
С-1 57,4 I-I 36,1 I-H 71,4
Двойные связи
С = С I 147 || С = О(СН2О) I 164 || C = O(R2CO) | 174
C = N | 147 II C = O(RCHO) | 171 1 C = S | 174
Т ройные связи
С = С I 194 || С = N(HCN) 1 207 |1 С = N(RCN) | 213
116
Физические свойства молекул
В дальнейшем весьма важно иметь представление о степени точности, кото-
рую могут обеспечить эти константы при подсчетах энергий образования
при условии отсутствия больших отклонений. Проверка была сделана Росси-
ни и сотр. [19] путем точных измерений теплот сгорания углеводородов. Эти
данные для парафинов приведены в табл. 21. Они представляют собой тепло-
ты образования (—\Н) соответствующих соединений (в качестве исходных
Таблица 21
Теплоты образования парафинов (по данным Россини)
Парафин -ДИ, ккал/моль (4,187-103 Дж/моль) Разность для СН2
сн4 17,86 2 33
сн3сн3 20,19 4 56
сн3сн2сн3 24,75 4 96
СН3СН2СН2СН3 29,71 5 03
СН3СН2СН2СНСН3 34,74
СН3СН2СН3 24,75 6 60
СН3СНСН3 31,35
сн3
СН3СН2СН2СН3 29,71 6 96
сн3снсн2сн3 36,67
сн3
сн3 31,35
СН3СНСН3 8,06
СН3
СН3ССН3 39,41
сн3
веществ применяют графит и молекулярный водород при температуре 25 °С2
а не атомы углерода и водорода при О К); состояние молекулы не имеет
отношения к постоянству или к отклонениям от постоянства разностей
теплот образования соединений, формулы которых отличаются друг от друга
одинаковым образом. Хотя поправка на температуру уменьшила бы все
разности теплот, эти поправки [0,4 . . . 0,6 ккал/моль (1,675 -103 . . .
2,512-103 Дж/моль)] лишь незначительно превышают (примерно вдвое)
ошибки опыта. Очевидно, что теплота образования не увеличивается на
постоянную величину каждый раз, когда вводится одна СС- и две СН-связи
при увеличении формулы на СН2: увеличение теплоты составляет пример-
но 2(8,37-103), 5(20,93-103), 7(29,31-103) или 8(33,49-103) ккал/моль
(Дж/моль) в зависимости от того, замещает ли новая СН3-группа водород
в СН4, СН3-, СН2- или СН-группах, т. е. постоянство наблюдается лишь
в пределах ±3 ккал/моль (12,56-103 Дж/моль) —величины, которая для
этих соединений является степенью точности применяемого принципа адди-
тивности. Эти данные ничего не говорят о том, зависят ли отклонения глав-
ным образом от свойств замещенной СН-связи или от свойств замещающих
СС-связей. Согласно этим данным, разветвление стабилизирует парафины
(рис. 18). Питцер [20] считает, что этот эффект в основном возникает вслед-
ствие большего электрокинетического притяжения непосредственно не свя-
117
Глава III
занных частей более разветвленного углеводорода. Это может быть важным
фактором. Увеличение стабильности с ростом числа метильных групп можно
объяснить, если считать, что электроны в алканах несколько сдвинуты
О
н-Бутан
-1.64
Изобутан
О
н-Пентан
-1,93
Изопентан
~4,62
Неопентан
О
Бутен-1
-1,77
уис-Бутен-2
-2,72
транс -Бутен-2
-3,58
Изобутилен
Рис. 18. Изменение стабильности в ряду изомер-
ных бутанов, пентанов и бутенов.
Энергии даны в килокалориях на 1 моль.
Нулевые уровни не одинаковы и выбраны
произвольно.
по направлению к метильным
группам (гл. II, разд. 3,в и
разд. 1,г данной главы). Этот
фактор действует в том же
направлении, что и указан-
ный выше, однако трудно оце-
нить, насколько он важен *.
Россини с сотрудниками изу-
чил также олефины; теплоты
гидрирования олефинов точ-
но измерили Кистяковский
и сотр. [21]. Данные Кистя-
ковского вместе с данными
Россини по теплотам образо-
вания парафинов позволили
установить теплоты образо-
вания олефинов. Полученные
значения теплот образования
— ДН приведены в табл. 22.
Они показывают, что влияние
гомологии вдали от двойной
связи такое же, как и у парафинов. Эти данные, кроме того, показывают,
что замещение Н на СН3-группу в ненасыщенных СН2- и СН-группах уве-
личивает теплоту образования на 7 ... 8 ккал/моль (29,31 -103 . . . 33,40 х
X Ю3 Дж/моль), т. е. как раз на столько же, на сколько замещение Н
в СН2- или СН-группах парафинов.
Наглядным примером могут служить бутены (рис. 18). Кроме заметного
различия в стабильности геометрических изомеров бутена-2, наблюдается
общее увеличение стабильности в ряду
С2Н5СН = СН2 СН8СН,= СНСН3 (СН3)2С = СН2
Это можно было бы истолковать как проявление энергии гиперконъюгации
между алкильными группами и двойной связью, но полученные Россини
результаты показывают, что аналогичное увеличение стабильности наблю-
далось и в ряду парафинов
С2Н5—СН —СН2
I I
сн3 сн3
СН3СН снсн8
I I
сн3 сн3
(СН3)2С - сн2
сн8 СН3
Следовательно, энергия гиперконъюгации между алкильной группой и этиле-
новой двойной связью в лучшем случае величина того же порядка, что
и энергия взаимодействия (главным образом электростатического) между
атомами и связями в парафинах, и составляет максимально несколько кило-
калорий на 1 моль.
Более определенные заключения можно сделать на основании теплот обра-
зования ацетиленов; некоторые величины, полученные из теплот гидрирова-
ния [22] в сочетании с теплотами образования парафинов приведены в табл.
23. Разность в теплотах образования между гомологами в данном случае
составляет 9 ... 10 ккал/моль (37,68-103 . . . 41,87-103 Дж/моль), т. е.
* Этот вывод сделан Шаадом из некоторых квантовомеханических расчетов.
118
Физические свойства молекул
Таблица 22
Теплоты образования олефинов (по данным Россини и Кистяковского)
Олефин — ДИ, ккал/моль (4,187 • 103 Дж/моль) Разность для СН2, нкал/моль (4,187-103 Дж/моль)
цис (-) транс цис (-) транс
сн2=сн2 12,56
СН2 = СНСН3 -4,96 7,60
СН2 = С(СН3)2 +3,20 8,16
СН3СН = СН2 СН3СН = СНСН3 + 1,39 -4,96 +2,34 6,35 7,30
СН3СН = С(СН3)2 +9,99 8,60 7,65
сн2=снсн3 -4,96
СН2 = СНСН2СН3 -0,38 4,58
СН2 = СНСН2СН2СН3 +4,64 5,20
СН3СН = СНСН3 СН3СН = СНСН2СН3 СН2 = С(СН3)2 +1,39 +6,40 +3,20 +2,24 +7,35 } 5,01 5,01
СН2СН3 5,22
СН2 = с/ +8,42
сн3
СН2 = СНСН2СН3 -0,38
сн3 6,96
СН2 =снсн/ +6,58
сн3
заметно превышает разности, наблюдаемые в парафиновом и олефиновом
рядах; более того, заместители здесь слишком отдалены друг от друга,
чтобы в сколько-нибудь значительной степени непосредственно взаимодей-
ствовать между собой. Следовательно, эти данные явно указывают на энерге-
тически важное взаимодействие между метильными заместителями и тройной
связью. Однако данные таблицы не определяют ни абсолютного, ни даже
Таблица 23
Теплоты образования ацетиленов (по данным Россини и Кистяковского)
Ацетилен -дн, ккал/моль (4,187* 103 Дж/моль) Разность для СН2, ккал/моль (4,187 • 103 Дж/моль)
нс = сн СН3С = сн СН3С ~ ссн3 -54,87 -44,95 -35,87 9,92 9,08
относительного значения в суммарном эффекте энергии гиперконъюгации,
т. е. делокализации электронов СН-связи метильной группы и энергии
индуктивного электронного смещения (разд. 1, в) в простых СС-связях.
б. Энергия сопряженных ненасыщенных систем. Указанные сомнения
не касаются общего значения термохимических данных для всех видов сопря-
женных ненасыщенных систем. Как показали Полинг и Шерман [23], экзо-
119
Глава III
термичность образования молекул, содержащих такие системы, всегда боль-
ше энергий, вычисленных на основании констант энергии связей, а отклоне-
ния обычно превышают, часто значительно, приближение ±3 ккал/моль
(12,56-103 Дж/моль), в пределах которого константы энергии аддитивны.
Так, экзотермичность образования (—АЯ) карбоновых кислот примерно
на 28 ккал/моль (117,23-103 Дж/моль) больше, чем величина, вычисленная
по принципу аддитивности из констант энергии двойной связи С = О и раз-
личных простых связей в обычной валентной формуле.
Выявляющуюся таким образом дополнительную стабильность нельзя припи-
сать индуктивному эффекту. Константы энергии выведены для всех полярных
связей из теплот образования простых молекул с учетом полярности связей.
Резкое увеличение стабильности систем с сопряженными связями должно
поэтому быть связано с делокализацией электронов в сопряженных системах;
повышение стабильности объясняется энергией мезомерии, обусловленной
сопряжением.
Поскольку формальные заряды вызывают большие искажения, принцип
приблизительной аддитивности может быть применен только к формально
неполярным структурам, которые всегда должны быть стабильнее соответ-
ствующих диполярных структур. Если существует больше одной неполярной
структуры, то более устойчива структура, имеющая большее число двойных
связей. Энергия мезомерии равна разности между истинной теплотой обра-
зования и величиной, вычисленной для наиболее стабильной валентной
структуры. Последняя всегда является обычно принятой структурой, напри-
мер CONH2 для амидной группы и структурой Кекуле для бензола. В табл. 24
Таблица 24
Значения энергии мезомерии, полученные из констант энергий связей
Соединение Энергия мезо- мерии, ккал/моль (4,187 • 103 Дж/моль) Соединение Энергия мезоме- рии, ккал/моль (4,187-103 Дж/моль)
Двуокись углерода 36 Стирол 37 + 5
Сероокись углерода 20 т ранс-Ствлъ§Ы1 2-37 + 7
Сероуглерод И транс, транс-1,4-Дифенил- 2-37+11
Алкилизоцианаты 7 бутадиен-1,3
Карбоновые кислоты 28 Фепилацетилен 37 + 10
Эфиры карбоновых кислот 24 Анилин 37 + 6
Амиды карбоновых кислот 21 Фенол 37+7
Эфиры угольной кислоты 42 Бензальдегид 37 + 4
Мочевина 37 Ацетофенон 37 + 7
Трополон 36 Бензофенон 2-37 + 10
Бензол 37 Бензонитрил 37 + 10
Бифенил 2-37 + 5 Бензойная кислота 37 + 28 + 4
1, 3,5-Трифенилбензол 4-37 + 20
приведены некоторые энергии мезомерии, рассчитанные этим методом. Такая
таблица была дана впервые Полингом и Шерманом [23] и затем пересмотрена
Полингом [18]. В табл. 24 отсутствуют данные для конденсированных и гете-
роциклических систем; они рассматриваются в гл. IV. Однако энергия
резонанса бензола приводится главным образом для того, чтобы по разности
можно было оценить энергетический эффект сопряжения двух фенильных
групп или фенильной группы с боковой цепью +М- и —Л7-типа.
120
Физические свойства молекул
Особенно большими являются величины для двуокиси углерода и ее анало-
гов, для карбоксильной группы во всех ее формах и для ароматического
ядра; все это согласуется с общеизвестными химическими данными, поскольку
свойства двуокиси углерода действительно весьма отличаются от свойств
Таблица 25
Значения энергии мезомерии [ккал/моль (4,187-103 Дж/моль)], полученные
из теплот гидрирования (по данным Кистяковского и сотр.)
Сопряженная система Количе- ство Нг, моли -ДИ Разность Молекула для сравнения Количе- ство Н2, моли -ди
Бутадиен-1,3 2 57,07 3,6 Бутен-1 1 30,34
54,11 4,1 | Бутен-1 1 30,34
Пентадиен-1,3 2 Пентен-2 1 27,95 в
Циклопентадиен 2 50,86 3,0 Циклопентен 1 26,91
Циклогексадп- 2 53,37 3,8 Циклогексен 1 28,59
ен-1,3 Кротоновый аль- 1 25,16 2,9 Бутен-2 1 28,10 г
дегид Этилвиниловый 1 26,74 3,4 Пропилен 1 30,11
эфир 2-Этоксипропен-1 1 25,10 3,3 Изобутилен 1 28,39
2-Метоксибутен-2 1 24,80 2,1 2-Метилбутен-2 1 26,92
Дивиниловый 2 57,24 3,5 Пентадиен-1,4 2 60,79
эфир Винилацетат 1 30,12 0,0 Пропилен 1 30,11
Бензол 3 49,80 36,0 а Циклогексен 1 28,59
Стирол 4 77,50 38,4 6 (Пропилен (Циклогексен 1 1 30,11 28,59
1,2-Дигидронаф- 24,6 4,0 1 Циклогексен 1 28,6
1 3,0 J 1,4-Д игидронаф- 1 27,6
талин талин Д
трапс-Стильбен Д 1 20,6 7,0 тра «с-Бутен-2 1 27,6
uuc-Стильбен Д 1 26,3 2,3 иис-Бутен-2 1 28,6
1,4-Дифенилбу- 2 44,5 10,7» трян.с-Бутен-2 1 27,6
тадиен-1,3 Д> е
а Ср. величину 37 ккал/моль, полученную из констант энергий связей.
6 Ср. величину 42 ккал/моль, полученную из констант энергий связей.
в Величина дана для смеси цис- и транс-изомеров.
г Среднее значение величин для г^ис- и транс-форм. (В опытах с кротоновым альдегидом альде-
гидная группа не гидрировалась.)
Д Измерено при 29 °C в уксусной кислоте и с приближением приведено к газообразному состоя-
нию при 82 °C; к этим же условиям отнесены и другие измерения.
е транс-транс-Изомер.
ж Ср. величину 11 ккал/моль, полученную из констант энергий связей.
дикетона, свойства карбоновых кислот — от свойств кетоспиртов, а свойства
бензола — от свойств триолефинов. Даже значительно меньший энергетиче-
ский эффект сопряжения между кольцом и боковой цепью в анилине и феноле
соответствует вполне заметному влиянию на химические свойства как коль-
ца, так и боковой цепи.
121
Глава III
Другим и, возможно, более точным способом оценки энергий мезомерии
сопряженных систем является измерение не избыточной экзотермичности
их образования, а меньшей экзотермичности деструкции систем. Кистяков-
ский и сотрудники применяли этот метод для оценки сравнительно малых
по величине энергий мезомерии; при этом они использовали гидрирование
как метод разрушения сопряженной системы. Теплота гидрирования системы
сравнивалась с теплотами гидрирования отдельной двойной связи в нена-
сыщенной молекуле, например теплота гидрирования бутадиена-1,3 срав-
нивалась с теплотой гидрирования двух молекул бутена-1, теплота гидриро-
вания бензола — с теплотой гидрирования трех молекул циклогексена,
теплота гидрирования виниловых эфиров — с теплотой гидрирования пропи-
лена и т. д. Данные табл. 25 относятся к газообразному состоянию при 82 °C
[24] без внесения других поправок на разницу в энтальпии; однако эти
поправки невелики.
Значения энергии мезомерии, приведенные в колонке «разность» в табл. 25,
для различных систем приблизительно составляют [ккал/моль(4,187 X
X 103Дж/моль)]
С = С—С= С 3,6 Алкил —О—С=С 2,7
Арил—С = С 3,1 Ацил-О —С = С 0,0
С = С—С = О 2,9
В системе ацил — О — С = С винильная группа уничтожает сопряжение
карбоксила, сама сопрягаясь с атомом кислорода в энергетически эквива-
лентной степени. Для игранс-стильбена наблюдаемая энергия мезомерии
на 0,9 ккал/моль (3,77-103 Дж/моль) больше величины 2-3,1 ккал/моль
(2-12,98-103 Дж/моль), а для 1,4-дифенилбутадиена-1,3— на 0,9 ккал/моль
(3,77-103 Дж/моль) больше, чем 2-3,1 +3,6 ккал/моль (2-12,98-103 +
+ 15,07 -103 Дж/моль). Эту разницу можно считать мерой сквозного сопря-
жения, характеризующей степень, в которой фенильные группы «чувствуют»
друг друга через разделяющую их цепь в этой линейной сопряженной системе.
В меньшей энергии мезомерии щгс-стильбена, несомненно, усматривается
физическое проявление вторичного пространственного эффекта Хэмпсона
(разд. 1, ж). Здесь взаимодействие между фенильными группами выводит
их из плоскости центральной этиленовой связи, нарушая сопряжение
и уменьшая его энергетический эффект на две трети. Эта молекула изоэлек-
тронна и должна быть стереохимически подобна щгс-азобензолу, стереохи-
мия которого точно установлена (см. рис. 23 в гл. III).
3. Электрическая поляризуемость
а. Смысл констант рефракции. Электрическая поляризуемость молеку-
лы, т. е. а, усредненная по сфере, выражается, не считая постоянного мно-
жителя, молекулярной рефракцией
г di
[Я] = — а,
где N — число Авогадро. В свою очередь молекулярная рефракция связана
с показателем преломления света п соотношением Лорентца
«2-1 М
«2 + 2‘ d '
где М — молекулярный вес, ad — плотность при той температуре и давле-
нии, при которых измерен коэффициент лучепреломления. Формула Лорент-
ца предполагает изотропное распределение молекул; она выполняется для
122
Физические свойства молекул
газов, кристаллов кубической симметрии и для тех аморфных жидкостей
и твердых тел, которые если не бесструктурны, то по крайней мере локально
изотропны. Таким образом, эта формула является лишь приближенной
для реальных аморфных жидкостей и твердых тел; однако для очень многих
жидкостей формула служит достаточно хорошим приближением. Поляризуе-
мость зависит от частоты, потому что электроны обладают инерцией, и, сле-
довательно, чтобы получить поляризуемость в статическом поле, следует
перейти к нулевой частоте с помощью дисперсной формулы. Однако для
бесцветных веществ частоты красного и желтого света (например, линии На
или D) достаточно низки, чтобы поправкой можно было пренебречь. Молеку-
лярные рефракции выражаются в кубических сантиметрах и имеют размер-
ность см3/моль.
Предложены две системы аддитивных представлений для молекулярных
рефракций. Обе системы в первую очередь исключают молекулы с большими
и сложными отклонениями от аддитивности (в особенности молекулы с фор-
мальными зарядами и сопряженными системами), а затем анализируют
рефракции других молекул для того, чтобы получить, насколько это возмож-
но, ряд аддитивных констант.
На основании классического метода Ландольта, Брюля, Эйзенлора и других
удалось рассчитать значения атомных констант. В то же время было уста-
новлено, что даже при указанных ограничениях недостаточно приписывать
одно определенное значение константы данному атому, потому что ненасы-
щенность, а также вид связи атома с другим атомом изменяют величину
константы. Влияние, обусловленное этиленовыми и ацетиленовыми связями,
учитывается дополнительными константами (т. е. по существу принимают
два значения поляризуемости для одинаковых электронов), причем влияние
других кратных связей отражается специальными константами для одного
или обоих связанных атомов, а влияние, обусловленное различными типами
атомов, связанных с данным атомом, отражается другими специальными
константами для данного атома (Брюль приводит более 30 таких констант
для азота). Таким образом, простая идея аддитивности оказалась на практике
весьма усложненной. В такой форме эта идея создала хорошую основу для
учета больших отклонений, вызываемых полярностью и сопряжением.
В табл. 26 приведены некоторые значения атомных рефракций по Эйзенлору
[25], а также значения для двойной и тройной углерод-углеродных связей.
Другая система рассуждений, введенная Штейгером и развитая Смитом,
Таблица 26
Константы рефракции (см3/моль) атомов и кратных связей
Атом или связь На D Атом или связь На D
н 1,092 1,110 N (первичные амины) 2,309 2,322
с 2,413 2,418 N (вторичные амины) 2,475 2,499
СС (двойная связь) 1,686 1,733 N (третичные амины) 2,807 2,840
•СС (тройная связь) 2,328 2,398 Fa 1,088 1,090
О (спирты) 1,639 1,643 Cl 5,933 5,967
•О (эфиры) 1,522 1,525 Br 8,803 8,863
О (карбонил) 2,189 2,221 I 13,757 13,900
а Более поздние данные, включенные для сравнения с другими галогенами.
123
Глава III
Фаянсом, Кнорром и Денби, основывается на следующем: так как поляри-
зуемость почти полностью обусловлена валентными оболочками атомов
Таблица 27
Константы рефракции (см3/моль) октетов
для D-линии натрия
Октет Константа рефракции Октет Константа рефракции
С в С(Н4) 6,80 Si в Si(C4) 9,20
С в С(С4) 4,84 Р в Р(Н3) 11,8
N в N(H3) 5,65 Р в Р(С3) 11,04
N в N(H2C) 5,13 S в S(H2) 9,57
N в N(HC2) 4,81 S в S(HC) 9,40
N в N(C3) 4,65 S в S(C2) 9,18
О в О(Н2) 3,76 Cl в С1Н 6,68
О -в О(НС) 3,23 Cl в С1(С) 6,57
О в 0(62) 2,85 С1 в Cl(Si) 7,04
F в F(C) 1,65 Аг 4,20
Ne 1,00
и только в очень малой степени внутренними прочно связанными электрона-
ми, казалось бы более правильным разделить молекулярные рефракции
на составные части, обусловленные
Группы периодической системы
Рис. 19. Константы рефракций октетов
нейтральных атомов, образую-
щих связи с углеродом.
связями и пеподеленными электронными
парами. Такая система, естественно,
более гибка, чем атомная система,
потому что число констант при этом
автоматически возрастает: число типов
простых связей между п типами ато-
мов равно п (п + 1)/2, а каждый тип
двойной и тройной связей выражается
особой константой [26]. Трудность до-
ведения этих рассуждений до логиче-
ского конца заключается в отсутствии
способа определения констант для обоб-
щенных и для неподеленных электронов
данного атома. Поэтому Смит предло-
жил константы рефракции октетов, т. е.
суммарную рефракцию обобщенных и
неподеленных пар октета. В табл. 27
приведены некоторые величины, полу-
ченные Смитом. Денби делит констан-
ту неподеленной пары по связям, вклю-
чая часть этой константы в константу
каждой связи. Любой метод восполне-
ния логического пробела приводит к
системе, не менее удовлетворительной, чем атомная система, и вполне могу-
щей служить базой для рассмотрения влияний полярности и сопряжения.
Неподеленные электроны в кратных связях оказывают весьма значительное
влияние на поляризуемость, и это можно качественно показать несколькими
124
Физические свойства молекул
способами, например сравнением атомных рефракций или рефракций октетов
в таком изоэлектронном ряду, как CR4, NR3, OR2, FR, Ne или SiR4, PR3,
SR2, G1R и Ar (рис. 19). Как правило, константы рефракции уменьшаются
с увеличением электроотрицательности, однако константы атомов группы
азота не обладают этим свойством, а константы атомов группы кислорода
и даже галогенов (исключение представляет фтор) снижаются не настолько,
насколько можно было бы ожидать, принимая во внимание увеличение
заряда ядра. Эти явления, очевидно, обусловлены влиянием неподеленных
электронов.
б. Влияние полярности на рефракцию. Влияние электроотрицательно-
сти на рефракцию наиболее просто иллюстрируется константами рефракций
одноатомных ионов, выведенными Фаянсом и другими [27] на основании
показателей преломления ионных растворов и кристаллов. Данные для ионов
[Таблица 28
{Константы рефракции (см3/моль) одноатомных ионов с заполненным октетом
02- 7,0 F- 2,4 Ne 1,0 Na+ 0,5 Mg2+ 0,3 Sc3+ 0,9
S2- 15 ci- 9,0 Аг 4,2 К+ 2,2 Са2+ 1,3 La3+ 3,3
Se2- 16 Вг- 12,6 Кг 6,4 Rb+ 3,6 Sr2+ 2,2
Те2- 24 I- 19,0 Хе 10,4 Cs+ 6,1 Ва2+ 4,2
с заполненным октетом приведены в табл. 28. Согласно этим данным, значе-
ния рефракции плавно уменьшаются в изоэлектронном ряду по мере того,
как возрастает заряд ядра.
Точно такой же электростатический эффект обнаруживается в некоторой
более усложненной форме при сравнении констант рефракции октетов одного
и того же атома, обладающего различными формальными зарядами (табл. 29).
Таблица 29
Рефракция октета (в см3/моль для D-линии натрия) для октетов
ХНИ с нейтральным и заряженным атомом X
N 0 Cl Br I
X+ X (нейтральный) x- X2- 4,3 5,6 3,1 3,8 5,1 7,0 6,7 9,0 9,1 12,6 13,7 19,0
По мере того как атом становится формально более положительным, т. е.
по мере того как неподеленная электронная пара в его октете постепенно
заменяется обобщенной парой, константа рефракции октета уменьшается.
В диполярных связях влияние формального положительного заряда, как
и следовало ожидать, преобладает, так как он занимает более центральное
положение. Этот эффект может быть выявлен сравнением константы рефрак-
ции группы, содержащей диполярную связь, с константами рефракции
изомерной группы без формальных зарядов; установлено, что первая кон-
станта рефракции меньше. Сравнивая рефракцию целой молекулы или груп-
125
Глава I JI
пы, содержащей диполярную связь, с рефракцией другой молекулы или
группы, в которой диполярная связь разрушена отрывом атома, можно
показать, что, хотя потеря атома и должна бы вызывать уменьшение рефрак-
ции, тем не менее рефракция, как это установлено, не уменьшается, а иногда
может даже возрастать. Примеры обоих способов сравнения [28] приведены
в табл. 30.
Таблица 30
Константы рефракции (см3/моль для линии На)
различных групп с диполярной связью
Группа Соединение [«la Группа Соединение [«А
Сравнение с изомерными группами
no2 Нитросоединения 6,52 no2 Алкилнитриты 7,21
so3 Алкилсульфонаты 9,78 so3 Диалкилсульфиты 11,08
Сравнение с группами, содержащими на один атом кислорода меньше
ClgPO Хлорокись фосфора 25,24 С13Р Треххлористый фосфор 26,31
РО4 Триалкилфосфаты 10,64 РО3 Триалкилфосфиты 11,87
so2 Диалкилсульфоны 8,58 so Диалкилсульфоксиды 8,53
so4 Диалкилсульфаты 11,07 so3 Диалкилсульфиты 11,08
в. Рефракции сопряженных ненасыщенных систем. Такие системы, как
С = С — С = С, С = С — С = 0 и N = С — С = N, имеющие сопряженные
кратные связи, а также системы Аг — С = О и Аг — С = С, в которых арил
находится в сопряжении с кратной связью, характеризуются экзальтацией
рефракции, т. е. обладают большей молекулярной рефракцией, чем вычислен-
ная на основе принципа аддитивности с включением всех поправок на нена-
сыщенности отдельных атомов, составляющих сопряженную систему [25].
Экзальтация рефракции наблюдается очень часто, хотя она и не является
универсальным свойством сопряженной ненасыщенной системы в обобщен-
ном смысле, т. е. мезомерной системы. Это положение можно иллюстрировать
для А 717-системы
X^cZc^-
используя метод, при помощи которого Саттон установил мезомерные
дипольные моменты, а именно метод сравнения ароматических и алифатиче-
ских соединений, имеющих один и тот же единственный заместитель X. Такое
сравнение проводится в табл. 31. Константы рефракции заместителя X
в ароматическом веществе вычислены из молекулярной рефракции по раз-
ности с использованием эмпирической константы для фенильной группы
(молекулярная рефракция бензола минус константа атома водорода). Есте-
ственно, что значения экзальтации (последняя колонка таблицы) относятся
только к сопряжению заместителя X с фенильной группой и не включают
ту часть, которая обусловлена самой фенильной группой.
Для таких систем, в которых мезомерное электронное смещение мало (разд.
1,е), так же как и энергия мезомерии, обусловленная сопряжением
с заместителем (разд. 2,6), неполярная валентная структура является
приближением к нормальному состоянию, в то время как диполярные валент-
126
Физические свойства молекул
Таблица 31
Атомные константы рефракции (см3/моль для На)
+ M-заместителей в ароматических и алифатических соединениях (й = алкил)
Ароматические соединения Алифатические соединения A[RX]
система [Rx] система I«xl
— N c6h5-nr2 4,22 R-NR2 2,81 1,41
— N C6HS-NHR 3,69 R-NHR 2,47 +1,22
— N C6H5-NH2 3,24 r-nh2 2,31 +0,93
-О c6h5-or 2,18 R —OR 1,64 +0,54
-О C6H5 — он 1,76 R —ОН 1,52 +0,24
— S C6H5-SH 8,13 R-SH 7,64 +0,49
—F c6h5-f 0,92 R-F 1,05 -0,13
-С1 C6H5-C1 6,03 R-Cl 5,93 +0,10
— Вг C6H5 —Вг 8,88 R —Br 8,80 -f-0,08
-I С6Н5-1 13,94 R-I 13,76 +0,18
— N — c6h5-nr-c6h5 5,23 R — NR — R 2,81 + 2,42
-О- С6Н5—О —С9Н5 2,84 R —0 —R 1,64 + 1,20
— S — CeH5 —S —С6Н5 9,45 R-S-R 7,92 -rl,53
>- Свн5ч ;р-с6н5 С6Н5' 12,70 1 cc 9,23 +3,47
ные структуры — приближениями к возбужденным мезомерный состояниям
(гл. II, разд. 3,д). Учитывая, что влияние возмущающего электрического
поля должно деформировать нормальное состояние в сторону возбужденного
состояния (с частичным участием Т-функции последнего), следует ожидать
зависимости электромерной поляризуемости таких мезомерных систем от
стабильности дипольных структур (гл. II, разд. 3,ж). При этом, чем ниже
они лежат на энергетической шкале, тем больше будет доля электромерной
поляризуемости системы в общей поляризуемости молекулы, т. е. тем больше
должна быть экзальтация, как это и показано в табл. 31. Стабильность
диполярных структур типа X = С — С будет наивысшей в случае амино-
соединений и самой низкой для галогенопроизводных. Отсюда можно прийти
к выводу о существовании общей тенденции в изменении экзальтаций
в последовательности N > О, S > Hal. Если можно написать множество
эквивалентных диполярных структур, соответствующих сопряжению
в альтернативных направлениях, то электромерная поляризуемость (гл. II,
разд. 3,ж) * таких молекул будет высокой и поэтому следует ожидать, что
экзальтации, обусловленные одинаковым замещающим атомом, возрастают
в такой последовательности: тройная > двойная > простая связи, что частич-
но подтверждается опытными данными.
Малые отрицательные экзальтации, обусловленные атомом фтора в арома-
тическом ядре, могут быть результатом влияния его электроотрицательности.
* Следует помнить, что многочисленность эквивалентных наиболее стабильных
валентных структур может дать обратный результат (гл. II, разд. 3,ж п далее).
127
Глава III
Фтор сильно притягивает электроны (—Z-эффект), и, следовательно, можно
ожидать, что, как и в случае положительного заряда, фтор уменьшает поля-
ризуемость связанной с ним группы. Весьма вероятно, что поляризуемость
легко поляризуемой группы уменьшается резче, чем поляризуемость
менее поляризуемой алкильной группы. Несомненно, что индуктивный
эффект сказывается во всех случаях, приведенных в табл. 31. Он обуслов-
ливает отрицательные экзальтации, величины которых понижаются в ряду
Hal 2> О, S > N; однако лишь в случае фтора эта отрицательная экзаль-
тация, обусловленная индуктивным эффектом, превышает положитель-
ную экзальтацию, вызываемую электромерным эффектом.
Экзальтации, возникающие в сопряженных системах с концевым заместите
лем типа — М, широко изучены в работах Врюля, Ауверса и Эйзенлора [25].
Г*
С=С—C=Y
В табл. 32 сопоставляются ароматические молекулы, имеющие —М-системы,
и алифатические соединения, содержащие такие же заместители, а также
проводится аналогичное сравнение олефинов и парафинов. Как и раньше,
Таблица 32
Атомные константы рефракции (в см3/моль для На)
—M-заместителей в ароматических, олефиновых и параф инов ь:х£Соединепиях(Д- алкил)
Y Ароматическое соединение или олефин Парафин A [Hy]
система [йу] система [Яу]
= 0 С6Н5-СН = О 3,37 R —СН = О 2,19 +1,18
= 0 C6H5-CR = O 3,08 R — CR = O 2,19 +0,89
= 0 C6H5-C(OR) = O 2,90 R — C(OR) = O 2,19 +0,71
= N С6Н5-С= N 3,92 R —C = N 2,98 +0,94
= 0 CHR = CH —СН = О 3,48 R — CH = O 2,19 +1,29
= 0 CHR = CH-CR=O 3,10 R—CR = O 2,19 +0,91
= 0 CHR = CH — C(OR) = O 2,75 R — C(OR) = O 2,19 +0,56
константа фенильной группы установлена эмпирически; это же относится
к константе карбоксильной группы. Сделано это для того, чтобы выделить
эффект рассматриваемой формы сопряжения.
В перечисленных —M-системах, подобно 4-М-системам, мезомерные элек-
тронные смещения и энергии мезомерии малы (разд. 1,е и 2,6), а следова-
тельно, их неполярные валентные структуры должны приблизительно соот-
ветствовать’их нормальным мезомерным состояниям, тогда как диполярные
валентные структуры должны соответствовать возбужденным мезомерным
состояниям (гл. II, разд. 3,ж). Таким образом, электронная поляризуемость
в общем должна зависеть от стабильности диполярных структур типа
X — С = С — Y. Такая последовательность экзальтаций СН = О >> CR =
= О > C(OR) = О, найденная как для ароматических производных, так
и для производных олефинов, по-видимому, правдоподобна. Можно ожидать,
что стабильность диполярных валентных структур уменьшается в том же
порядке, так как для получения таких структур необходимо раз-
рушить цепь гиперконъюгации во втором и цепь сопряжения в третьем
случаях.
128
Физические свойства молекул
В ±Л/-системах, представленных производными карбоновых кислот, экзаль
тацип уменьшаются в ряду
СОС1 > — COOR > — CONR2 > СОО-
причем в двух последних случаях значения экзальтации становятся отрпца
тельными. Это подтверждается данными в табл. 33 для производных карбо-
новых кислот.
Таблица 33
Положительные и отрицательные экзальтации рефракций (в см3/моль для На),
обусловленные карбоксильной группой (R - алкил)
G в G — СХ = О Карбоксильная форма --СХ = О
СОС1 - СООН - СООН CONR-2 а —соо-6
н — 4-0,17 4-0,10 -0,16 -0,7
СН3- 4-0,38 + 0,00 —0,14 —0,36 —0,9
RO- —0,23 —0,39
а Амиды с группами CONHR и CONH2 имеют экзальтации на 0,2—0,5 смз/моль более положи-
тельные, чем величины, приведенные для CONR2. Это, возможно, происходит благодаря таутомерии,
так как формы С(ОН) = NR и С(ОН) = NH должны иметь молекулярную рефракцию примерно на
1,0 смз/моль большую, чем изомерные им формы CONHR и CONH2.
® Эмпирические молекулярные рефракции для анионов рассчитаны с помощью закона смешения
по данным показателей преломления (линия D) водных растворов формиатов и ацетатов щелочных
металлов. Это можно сделать только приблизительно, потому что поправка на влияние ионных заря-
дов на изменение прелом тения окружающей воды несколько неопределенна. Аддитивно рассчитанные
значения рефракции быт получены на основании константы Фаянса для отрицательного атома
кислорода п константы Эйзенлора для карбонильного кислорода и других атомов. Таким образом,
разности, указанные как отрицательные экзатьтации, должны отражать эффект сопряжения карбо-
ксилат-иона. Переход от данных для линии D к данным для линии Н не представляет труда.
Общий результат получается именно такой, какой и следовало ожидать
(гл. II, разд. 3,е и ж). До тех пор пока мезомерные смещения электронов
и энергии мезомерии малы (как это и должно быть в группе — GOC1), наблю-
дается положительная экзальтация, как в случае уже рассмотренных систем.
Но по мере того как мезомерия становится все более выраженной, как
в — COOR, — CONR2 и, наконец, — ООО-, валентные структуры все мень-
ше отражают мезомерное состояние. Поскольку нижнее и верхнее состояния
все больше расходятся под влиянием резонанса, внешнему электрическому
полю все труднее обеспечивать суперпозицию верхнего и нижнего состояний.
Короче говоря, электромерная система становится постепенно все менее
поляризуемой.
В двуокиси углерода подобное влияние сильно выраженной мезомерии позво-
ляет полностью уничтожить положительную экзальтацию, обычную для
сопряженных систем: наблюдаемое значение [_R]a = 6,71, аддитивно вычис-
ленное [1?]а = 6,79; А [7?]а = —0,08 см3/моль.
Брюль, Ауверс и Эйзенлор на многих примерах показали, что сопряжение
между двойными этиленовыми связями, между двойной или тройной связью
и фенильными группами в ненасыщенных углеводородах всегда вызывает
положительные экзальтации рефракций. Наиболее отчетливая особенность
подобного рода экзальтаций заключается в том, что величина ее сильно
возрастает по мере того, как расширяется система сопряжения. Примеры
приведены в табл. 34; как и раньше, для рефракции фенильной группы
принята эмпирическая величина.
9-0772
129
Глава III
Таблица 34
Экзальтации рефракции сопряженных ненасыщенных углеводородов
Углеводород Д [П]а, смЗ/молз
; сн2=с(си3)-с(сн3)=сн2 J-0,72
t СН2=СН-СН=СН-СН=СН2 —2,07
J СвН5-СН = СН2 4-1,20
I С6Н5-СН = СН-СН = СН2 -3,66
Г С6Н5-С = СН -J-1,30
t С6Н5 —С = С —С = С —С6Н5 -L10,7
Для простейшего ненасыщенного углеводорода возможны не менее двух
диполярных структур; даже если их энергии велики, они одинаковы или
очень близки друг к другу, в результате чего энергия одного из возбужден-
ных мезомерных состояний сильно смещается, приближаясь к энергии нор-
мального мезомерного состояния. Чем больше удлиняется сопряженная
система, тем больше таких диполярных структур и тем больше в результате
их наложения смещается низшее возбужденное мезомерное состояние в сторо-
ну нормального состояния (гл. II, разд. 3,ж). Это ведет к повышенной
электромерной поляризуемости, а следовательно, к повышенной экзальтации
общей поляризуемости.
В случае бензола установлена незначительная отрицательная экзальтация:
наблюдаемое значение [7?]а = 25,93, аддитивно вычисленное [Д]а = 26,09,
А [7?]а = —0,16 см3/моль.
Приведенные данные вновь иллюстрируют стабилизирующий эффект мезо-
мерии, который в этом случае сильно снижает энергию нормального состоя-
ния, но не оказывает столь большого влияния на возбужденные состоянии
с большим весом полярных структур [29].
Гетероциклические ароматические молекулы также проявляют отрицатель-
ную экзальтацию, что иллюстрируется следующими данными (см3/моль
для красной На-линии):
Пиридин Пиррол Фуран Тиофен
— 0,97 —0,85 —0,80 —1,34
г. Поляризуемость как функция направления. Поляризуемость являет-
ся симметричным тензором. В любой молекуле, даже в несимметричной,
при классическом рассмотрении существуют три взаимно перпендикулярных
направления, каждое из которых обладает тем свойством, что электрическое
поле по данному направлению может перемещать электроны лишь вдоль
этого направления, но не под углом. Эти направления называются главными
осями поляризуемости, а поляризуемости вдоль этих осей — главными поля-
ризуемостями и обычно обозначаются а, Ъ и с. Величины и направления а,
b и с полностью определяют поляризуемость как функцию направления.
Если молекула имеет поворотную или зеркально-поворотную ось симметрии
или в случае нескольких осей симметрии имеет одну особую ось, то одна из
трех главных осей поляризуемости совпадает с этим направлением. Если
молекула имеет плоскость симметрии или плоскость, связанную с зеркально-
поворотной осью, то две из трех главных осей симметрии лежат в этой плоско-
сти. В случае четырех или более поворотных осей третьего или высшего
порядка, как, например, в таких тетраэдрических молекулах, как СС14,
130
Физические свойства молекул
Р4О10, (CH2)6N4, в таких октаэдрических молекулах, как Fe(CN)<_, или
в таких сферических молекулах, как Hg, Вг-, все три значения главных
поляризуемостей будут равны, т. е. а = Ъ = с. В этом случае их оси могут
быть выбраны произвольно при условии взаимной перпендикулярности.
Если молекула не удовлетворяет этому условию, но имеет одну поворотную
или зеркально-поворотную ось третьего или высшего порядка, как, например,
в СНС13 (поворотная ось третьего порядка), СО2- (третьего порядка), IF5
(четвертого порядка), СН2 = С = СН2 (зеркально-поворотная ось четвертого
порядка), Ni(CN)2- (четвертого порядка), IF7 (пятого порядка), С2Н6 (зер-
кально-поворотная ось шестого порядка), С6Н0 (шестого порядка), С12 (беско-
нечного порядка), то два из трех значений главных поляризуемостей равны
(а Ъ = с); в этом случае ось а совпадает с осью третьего или высшего
порядка, в то время как оси равной поляризуемости Ьис могут быть выбра-
ны произвольно при условии, что все три оси будут взаимно перпендикуляр-
ны. Если структура молекулы не удовлетворяет этим условиям и обладает
еще тремя поворотными осями второго порядка, как в п-С6Н4Х2, то все три
значения главных поляризуемостей будут не равны (а =/= Ъ =£ с), однако их
оси будут совпадать с осями симметрии. Для всех низкосимметричных, в том
числе и для несимметричных молекул, а =/= Ъ =/= с. Однако, как правило,
если есть единственная ось симметрии, то она совпадает с одной из главных
осей поляризуемости, а две другие главные оси поляризуемости лежат
в плоскости симметрии. Это правило полностью определяет направление
осей, если одна из осей второго порядка находится в плоскости симметрии,
как, например, в фенантрене; направление осей поляризуемости определяет-
ся частично, если одна из осей второго порядка перпендикулярна плоскости
симметрии, как, например, в хризене, или если имеется только плоскость
симметрии, как в хинолине, или же только поворотная ось второго порядка,
как, например, в замещенных алленах ХНС = С = СНХ. Наконец, в тех
случаях, когда имеется лишь центр симметрии или налицо полная асиммет-
рия молекулы, главные оси поляризуемости не могут быть ориентированы
относительно структуры молекулы.
Молекула будет обладать неисчезающим дипольным моментом при соблюде-
нии одного из следующих условий: 1) молекула несимметрична, как, напри-
мер, холестерин; 2) имеется одна поворотная ось симметрии, как в 2,2'-
диаминодифениле; 3) имеется одна плоскость симметрии, как в NOC1, НСООН,
С6Н5ОН, кумарине; 4) имеется одна поворотная ось n-го порядка, лежащая
в п плоскостях симметрии, как в Н2О, СН2О, C6H5NO2 (п = 2) или в NH3,
CH3I, хинуклидине (п = 3). Во втором и четвертом случаях направление
дипольных моментов совпадает с осями симметрии, а следовательно, и с одной
из главных осей поляризуемости.
Число независимых функций а, Ь и с, которые должны быть определены
экспериментально для того, чтобы полностью описать поляризуемость как
функцию направления, очевидно, будет равно одной, двум или трем соответ-
ственно в зависимости от того, равны ли все три, только два значения или
значения главных поляризуемостей вообще различны.
Средняя поляризуемость а определяется по уравнению
За = а + b Ц- с
Мерой ее служит молекулярная рефракция.
Величина, называемая анизотропией поляризуемости и обозначаемая у2,
определяется по уравнению
4у2 = (а — Ъ)2 - г (Ь — с)2 + (с — а)2
9* 23J
Глава III
и в равной степени зависит от разностей между а, Ъ и с. Ее можно измерить
методом, сущность которого заключается в следующем.
Обычный свет, перпендикулярно рассеянный в конусе Тиндаля, является
полностью плоскополяризованным, если молекула изотропна (у2 = 0). Это
связано с тем, что вынужденные колебания электронов, которые обусловли-
вают рассеянное излучение, строго перпендикулярны плоскости рассеяния,
т. е. направлению распространения обоих пучков света (рис. 20, Л). Однако
излучение, рассеянное неизотропными молекулами, содержит второй плоско-
поляризованный компонент, т. е. компонент, поляризованный в плоскости
Рис. 20. Перпендикулярное рассеяние природного света.
А — изотропной молекулой; Б — анизотропной молекулой в
произвольной ориентации.
рассеяния: беспорядочность ориентаций неизотропных молекул приводит
к тому, что вынужденные колебания совершаются под различными углами
к падающему переменному электрическому полю. Направления этих колеба-
ний почти всегда не совпадают с направлением какой-либо из главных осей
поляризуемости, и такие колебания имеют компоненту, параллельную
направлению распространения излучения (рис. 20, Б). Следовательно, сте-
пень деполяризации рассеянного света, которую можно измерить, будет
зависеть от анизотропии поляризуемости молекул и может быть исполь-
зована для определения этой величины.
Третье соотношение между а, b и с может быть установлено только в том
случае, если молекула обладает неисчезающее малым дипольным моментом и,
направление которого совпадает с направлением главной оси поляризуемости,
например оси а. Тогда величина % представляет собой избыток поляризуемо-
сти в направлении диполя по сравнению со средним значением поляризуемо-
сти в перпендикулярной плоскости. Она может быть названа полярной анизо-
тропией и иметь положительное или отрицательное значение. Величина
полярной анизотропии определяется по уравнению
2% = 2а — b — с
Ее можно определить измерением эффекта Керра.
Если вещество, обладающее дипольным моментом, поместить в сильное
электростатическое поле, то молекулы ориентируются и поляризуются
проходящим плоскополяризованным лучом по-разному в зависимости от того,
поляризован ли он параллельно или перпендикулярно электрическому
полю (рис. 21). Возникающее таким образом двойное лучепреломление (или
разность показателей преломления) зависит от ц, %, а также от температуры,
плотности и напряженности электрического поля. Таким образом, если изве-
стен дипольный момент, измерение разности показателей преломления, т. е.
U2
Физические свойства молекул
эффекта Керра, может быть использовано для определения полярной анизо-
тропии.
Даже неполярные молекулы, если они анизотропны, могут вызвать в некото-
рой мере двойное лучепреломление в сильном электростатическом поле,
поскольку наведенные диполи будут иметь в молекулах предпочтительные
направления, что и обусловит упорядоченную ориентацию молекул в электри-
ческом поле. Это явление зависит от анизотропии молекул (у2), а также
от температуры, плотности и напряженности электрического поля. Путем
Рпс. 21. Двойное электрическое лучепреломление анизотропной молекулой (ориенти-
рованной строго по направлению электрического поля Е).
Анизотропная молекула более поляризуема в направлении диполя (компонента а), чем
в поперечном направлении (компонента Ь). Выходящий луч а-поляризованного света
вследствие большей поляризации при проходе через среду замедляется (т. е. прелом-
ляется) в большей степени, чем выходящий луч b-поляризованного света.
измерения эффекта Керра величину у2 для неполярных молекул можно опре-
делить более точно, чем путем измерения деполяризации тиндалевского рас-
сеяния. Для молекул с постоянным дипольным моментом влияние дипольных
моментов, наведенных полем, также имеет место, но оно лишь незначительно
изменяет наблюдаемое двойное лучепреломление. Если у2 измерена методом
деполяризации, то при определении % должна быть введена соответствующая
поправка.
д. Направленные поляризуемости связей. Данные о молекулярной
поляризуемости как функции направления установлены главным образом
в результате экспериментальных исследований Стюарта [30] и Лефевров [31].
В табл. 35 приведены некоторые данные для линейных молекул и для моле-
кул, которые можно считать линейными, если не принимать во внимание ато-
мы водорода метильных групп. В табл. 36 содержатся данные для плоских
молекул или молекул, которые можно считать плоскими, если не принимать
во внимание метильные группы [30, 32]. Поляризуемость молекул для удоб-
ства выражена в кубических ангстремах на молекулу.
Приведенные данные показывают, что, как правило, связи сильнее поля-
ризуются в продольном, нежели в поперечном направлении. Почти всегда
линия или плоскость, которая содержит все или большинство связей или
хотя бы большинство поляризуемых связей, имеет наибольшие главные
поляризуемости. Исключение представляет молекула водорода; однако поля-
ризуемость водорода настолько мала, что водородные атомы независимо от
того, лежат ли они на линии или в плоскости других атомов, не изменяют
общего распределения молекулярной поляризуемости.
Как уже отмечалось в разд. 3,а, нельзя дать удовлетворительного анализа
молекулярной поляризуемости с помощью аддитивных констант связи, если
электроны связи и неподеленные электроны находятся в одной и той же
валентной оболочке. Это не относится к СС- и СН-связям; аддитивные кон-
станты [29, 31] для направленных поляризуемостей указанных связей при-
ведены в табл. 37.
133
Глава III
Таблица 35
Направленные поляризуемости [А3/молекула (10-3 нм3/молекула)]
для D-линии в линейных и сравнимых молекулах
Ось а является линией, соединяющей атомы
(исключая атомы водорода метильных групп)
Соединение а Ь — с Соединение а Ъ = с
н2 0,68 0,89 cs2 15,14 5,54
n2 2,43 1,43 HCN 3,92 1,92
О2 2,43 1,19 N,0 5,32 1,83
С12 6,60 3,62 С2Н2 5,12 2,43
НС1 НВг 3,13 4,23 2,39 3,32 c2n2 7,76 3,64
HI 6,58 4,89 CH3CH3 5,50 4,05
со 2,60 1,62 СН3С1 5,42 4,14
со2 4,49 2,14 СН3Вг 6,85 4,90
Таблица 36
Направленные поляризуемости [ А3/молекула (10~3 нм3/молекула)]
для D-линии в плоских и почти плоских молекулах
Ось а — ось диполя. Оси а и Ь лежат в плоскости,
содержащей атомы данной молекулы а (исключая атомы водорода метильных групп)
Молекула а ъ С Молекула а ъ С
H2s 4,01 4,04 3,44 (СН3)2О 4,88 6,30 4,31
С6н6 12,31 12,31 6,35 (СН3)2СО 7,08 7,10 4,82
С6Н5С1 15,93 13,24 7,58 CeHjCHg 15,64 13,66 7,48
c6h5no2 17,76 13,25 7,75 лг-СбН4(СН3)2 16,16 17,83 8,55
c5h5n 10,84 11,88 5,78
а Для бензола локализация в плоскости атомов оси Ь, а не оси с следует из симметрии, при
которой требуется наличие двух равных поляризуемостей в данной плоскости. Для других молекул
это следует из величин поляризуемости в соответствии с приближенным принципом аддитивности.
Хотя большинство связей менее поляризуемы в поперечном направлении,
чем в продольном, GH-связь почти изотропна. Способность СН-связей
к гиперконъюгации с л-электронами можно с уверенностью связать с необыч-
но высокой относительной поляризуемостью в поперечном направлении,
поскольку при сопряжении происходит боковое по отношению к линии связи
перекрывание орбиталей (гл. II, разд. 2,д и 3,и).
Все СС-связи в 3—4 раза более поляризуемы в продольном направлении,
чем в поперечном. Двойные связи поляризуются в двух поперечных направле-
ниях, для двойной СС-связи поляризуемость в этих двух направлениях почти
одинакова.
В нижней части табл. 37 приведены данные для сильно поляризуемых связей
между атомами, один из которых имеет неподеленные электроны; однако
134
Физические свойства молекул
Таблица 37
Продольные и поперечные поляризуемости
связей [А3/молекула (10-3 нм3/молекула)]
для D-линии
Связь Продольная Поперечная
С —н 0,65 0,65
С—С. 0,97 0,26
с = с 2,80 0,77; 0,73
с = с 3,79 1,26
С —F(CII3F) 1,25 0,4
С— С1(СН3С1) 3,2 2,2
С-Вг(СН3Вг) 4,6 3,1
С —1(СН31) 6,8 4,7
С — О (триоксан) 0,89 0,46
О=0[(СН3)2С0]- 2,30 1,40; 0,46
N —H(NII3) 0,50 0,83
N-C1(CH3)3N] 0,57 0,69
приведенные значения вычислены из данных молекул такой высокой сим-
метрии, что не остается сомнений относительно степени, в которой поляри-
зуемость неподеленных электронов включена в поляризуемость, приписан-
ную рассматриваемым связям. Поляризуемость, приписанная связям С —
— Hal, включает поляризуемость всех неподеленных электронов галогенов.
Поляризуемость, приписанная карбонильной двойной связи, включает поля-
ризуемость всех неподеленных электронов атома кислорода. Если считать,
что по аналогии с электронами большинства связей поляризуемость рас-
сматриваемых систем больше по осям орбиталей, чем в поперечном направле-
нии, то можно понять большое различие между поляризуемостью карбониль-
ной двойной связи в двух поперечных направлениях. Поляризуемость
каждой NH- и NC-связи включает одну треть поляризуемости неподеленных
электронов азота. Если эти электроны анизотропны, как уже было предпо-
ложено для неподеленных электронных пар кислорода, то можно объяснить,
почему поляризуемость NH- и NC-связей в поперечном направлении так
велика по сравнению с поляризуемостью этих связей в продольном направ-
лении.
е. Направленная поляризуемость сопряженных систем и систем с гипер-
конъюгацией. Как было показано в гл. II, разд. 3,ж, возбужденные состоя-
ния, определяющие направления электромерной поляризуемости основного
состояния сопряженных систем двойных связей, могут быть в первом прибли-
жении представлены как резонансные гибриды пар полярных структур
с обращенным распределением зарядов; в этом случае заряды располагаются
по концам системы. При таком представлении необходимо, чтобы экзальтация
поляризуемости преобладала в одном направлении, а именно вдоль цепи
сопряжения.
Лефевр и Лефевр исследовали электрическое двойное лучепреломление
и светорассеивающую деполяризацию в сериях сравнимых ароматических
и алифатических соединений, содержащих заместители, которые могут сопря-
135
Глава III
гаться с бензольным кольцом. Так, они сравнили С6Н5Х и СН3Х с одинако-
выми заместителями X типа -{-Е, имеющими неподеленные электроны,
а также С6Н5У и CH3Y с одинаковыми заместителями Y типа —Е, содержа-
щими электроотрицательные ненасыщенные группы, способные к сопряже-
нию. Это позволило им определить зависимость экзальтации поляризуемости
от направления, обусловленной сопряжением заместителей с ароматическим
кольцом. Применялся метод вычитания, аналогичный методу, использован-
ному при составлении табл. 31 для оценки экзальтации сферически усреднен-
ной поляризуемости, определенной из молекулярной рефракции. Эти данные
[33] приведены в табл. 38.
Таблица 38
Направленная поляризуемость [А3 молекула (5О-3 нм3/молекула)] для D-лпнпп
одинаковых заместителей в алифатических и ароматических соединениях
Ось а--ось диполя. Ось Ъ лежит в плоскости кольца ароматического соединения
или в плоскости нптрогруппы нитрометана
•Заместитель а ъ С а ъ С Sa дь Дс
4-Е-Заместители X
X СНзХ С6н5х Экзальтации в ( бХ;)Х
F 1,2 0,4 0,4 0,8 0,8 0,3 —0,4 4 0,4 -0,1
С1 3,2 2,2 2 2 4,3 2,0 1,5 -г-1,1 -0,2 —0,7
Вг 4,6 3,1 3,1 6,3 2,5 2,2 0-1,7 -0,6 —0,9
I 6,8 4,7 4,7 9,2 5,4 3,3 —2.4 — 0,7 — 1,4
— E-Заместители Y
Y СНзУ С6н5х Экзальтация в
СХ 3,5 1,8 1,8 5,7 1,1 1,4 -^2 2 -0,7 -0,4
хо2 3,4 2,8 2,3 5,7 1,5 1,9 -2,3 -1,3 -0,4
Лефевры установили, что в соответствии с ожидаемым экзальтации поляри-
зуемости вдоль оси диполя (Аа), которая во всех случаях является и осью
сопряжения, положительна по величине. Как и ожидалось, фтор в качестве
заместителя дает небольшую отрицательную экзальтацию Аа; однако, как
уже объяснялось (разд. 3,в), влиянием электроотрицательности фтора
на поляризуемость связанных с ним групп, вероятно, пренебречь нельзя.
В гл. II, разд. 3,г, табл. 11, уже отмечалось, что индуктомерная поляри-
зуемость галогенов уменьшается в ряду I > Вт > CI > F. Результаты,
полученные Лефеврами, показывают, что в этом же ряду уменьшается поло-
жительная экзальтация поляризуемости вдоль оси сопряженных систем,
в которые входят галогены. Это объясняется тем, что, как уже говорилось
выше, поляризуемость зависит главным образом от энергии возбуждения
подходящих возбужденных состояний: энергия возбуждения уменьшается,
когда в возбуждении участвуют электроны с более высоким главным кванто-
вым числом (гл. II, разд. 3,е). В молекулах, как и в атомах, энергетические
136
Физические свойства молекул
уровни электронов сближаются при возрастании главного квантового
числа.
Наиболее поразительным результатом, следующим из данных табл. 38,
является то, что положительная экзальтация в продольном направлении Да
сопровождается отрицательными экзальтациями Д& и Дс в поперечных
направлениях. Экзальтация поляризуемости в двух поперечных направле-
ниях подчиняется разным правилам. Рассмотрим сначала поперечную экзаль-
тацию Д& в плоскости кольца.
В основном состоянии бензола ароматическая л-система обладает полной
гексагональной симметрией (гл. IV): электроны могут двигаться с одинаковой
легкостью по периферии кольца во всех направлениях, параллельных плоско-
сти кольца. В основных состояниях рассматриваемых здесь замещенных
бензолов мезомерное смещение электронов мало, и поэтому свойства л-систе-
мы бензольного кольца не будут сильно отличаться от свойств л-спстемы
самого бензола. Однако в полярных возбужденных состояниях, которые
предопределяют пути поляризуемости основного состояния, л-электроны
в значительной степени локализованы на противоположных сторонах кольца.
Это очевидно, так как полярные состояния приближаются по структуре
к диполярной валентной хиноидной форме
Такая неравномерность в распределении кратных связей кольца должна
уменьшать подвижность электронов в поляризующем поле, под действием
которого электроны движутся от одной стороны кольца к другой в направле-
нии Ъ.
Отрицательная экзальтация поляризуемости в направлении, перпендикуляр-
ном плоскости кольца, т. е. в направлении с, невелика, за исключением
случаев, когда в молекуле имеются электроны с главным квантовым числом
больше 2, например в хлор-, бром- и иодбензолах. В этих случаях отрица-
тельная экзальтация возрастает с увеличением главного квантового числа
валентной оболочки галогена, что можно объяснить степенью обобществ-
ления, которое должны претерпевать Зр-, 4р- и 5р-орбитали хлор-, бром-
и иодбензолов при переходе в герл-состояние, сопряженное с 2рл-электронами
бензола. Это обобществление увеличивается в ряду С1 <; Вг <; I, и поскольку
оно осуществляется в направлении с, то и дает отрицательную экзальтацию
в этом направлении, увеличивающуюся в ряду С1 < Вг <; I.
В табл. 39 приведены данные Лефевров для игреиг-бутилхлорида, трет-
бутилбромида и игреиг-бутилиодида. По сравнению с метилгалогенидами для
mpem-бутилгалогенидов наблюдается экзальтация поляризуемости вдоль
оси а, т. е. вдоль линии связи углерод — галоген. В хлориде экзальтация
несколько меньше, но для бромида и иодида — довольно велика. Ось а сов-
падает с направлением гиперконъюгации. Так же как и для галогенбензолов,
экзальтация увеличивается при увеличении атомного веса галогена. Таким
образом, снова необходимо предположить, что полярные возбужденные
состояния для галогенов больших размеров лежат ниже по энергии.
ш
Глава III
Таблица 39
Поляризуемость [А3/молекула (10~3 нм3/молекула)] для D-линии
галогенов в метил- и трет-бутилгалогенидах
в различных направлениях
Ось а —ось диполя. Перпендикулярные к ней оси Ь и с
эквивалентны
X СН3Х mpem-CziHgX Экзальтация в трет- С4Н9Х
а Ъ — с а Ь = с Да ДЬ = Дс
С1 3,2 2,2 3,9 1,6 +0,7 -0,6
Вг 4,6 3,1 6,0 2,6 +1,4 -0,5
I 6,8 4,7 8,8 4,2 +2,0 -0,5
Подобно галогенбензолам, положительная продольная экзальтация сопро-
вождается отрицательной экзальтацией в поперечном направлении. Однако
в алифатических системах отрицательная экзальтация во всех направлениях
невелика. В частности, она не увеличивается при повышении атомного веса
галогена, так как только рпо-э.тектроны галогенов, а не пр„, как в аромати-
ческой системе, участвуют в гиперконъюгационном взаимодействии с метиль-
ной группой.
4. Симметрия и размеры молекул
а. Симметрия сопряженных ненасыщенных молекул. Наиболее важными
методами, позволяющими получать сведения о геометрии молекул, являются
методы, основанные на изучении инфракрасных, микроволновых спектров,
спектров комбинационного рассеяния, а также дифракции электронов
и рентгеновских лучей. Спектроскопия — не только чувствительный метод
изучения симметрии молекулы; она позволяет очень точно установить разме-
ры малых молекул. Использование дифракции дает менее точные величины,
но применимо для значительно большего числа молекул.
Спектроскопические измерения основаны на том, что симметрия молекул
приводит к внутренней компенсации взаимодействий электромагнитного
излучения с различными «частями» симметричной молекулы и, таким образом,
обусловливает нулевой эффект, точно так же как две «половины» молекулы
мезовинной кислоты, взаимно компенсируя друг друга, делают всю молекулу
оптически неактивной.
Молекула, способная колебаться определенным образом, будет поглощать
свет, частота которого равна частоте собственного колебания молекулы.
Поглощение будет происходить в том случае, если колебательное движение
молекулы будет создавать переменный электрический момент (возможно,
наложенный на постоянный дипольный момент, хотя это и не имеет суще-
ственного значения), как, например, при продольных колебаниях полярных
связей. Если имеются две связи, растягивающиеся и сжимающиеся в одной
фазе, если они совершенно идентичны по длине, упругости, массе, полярности
и изменению полярности по длине и если они направлены одна по отношению
к другой точно под углом 180° (3,141 рад), тогда и только тогда будет проис-
ходить полное погашение переменных моментов, а следовательно, не будет
наблюдаться никакого поглощения и полосы будут отсутствовать там, где
они должны были бы появляться. То же явление будет наблюдаться в случае,
138
Физические свойства молекул
если три совершенно идентичные связи направлены одна к другой под углом
120° (2,094 рад).
Первоначально полагали, что карбонат- и нитрат-ионы не обладают плоско-
тригональными конфигурациями, а имеют по одной двойной и по две простые
связи. Позднее в соответствии с теорией мезомерии им была приписана пло-
скотригональная симметрия, причем в каждой связи участвовало 8/3 электро-
на. Шефер [34], применяя описанный метод, установил, что карбонат-
и нитрат-ионы действительно имеют тригональную симметрию. Полученные
из спектра комбинационного рассеяния частоты полносимметричных валент-
ных колебаний («breathing frequency») отсутствуют в инфракрасном спектре
поглощения.
Спектры комбинационного рассеяния этих ионов показывают также, что
рассматриваемые ионы плоские (правда, данный факт менее интересен, так
как он не подвергался сомнению). Взаимодействие между излучением
и молекулой, дающее спектр комбинационного рассеяния, отличается от
взаимодействия, приводящего к поглощению: колебание приводит к комби-
национному рассеянию в том случае, если оно способно создать в молекуле
осциллирующую поляризуемость (конечно, наложенную на постоянную
поляризуемость). Допустим сначала, что карбонат-ион в равновесной кон-
фигурации имеет пирамидальное строение. Тогда величина поляризуемости
будет зависеть от высоты пирамиды, причем постоянное значение поляри-
зуемости будет соответствовать тому моменту, когда высота пирамиды ста-
новится равной нулю, т. е. когда углеродный атом в своем колебании прохо-
дит через плоскость трех атомов кислорода. Следовательно, если атом угле-
рода совершает небольшие колебания относительно равновесного положения
вверх и вниз по отношению к тригональной оси симметрии, то изменения
поляризуемости будут наблюдаться лишь в том случае, если равновесная
конфигурация будет пирамидальной, а не плоской, и линии комбинационно-
го рассеяния появятся только в первом случае. Действительно, частота
внеплоскостного колебания, которая обнаруживается в инфракрасном спект-
ре, отсутствует в спектре комбинационного рассеяния. То же самое справед-
ливо и для нитрат-иона.
Аналогичным образом было показано, что бензол имеет плоскую строго
гексагональную симметрию (гл. IV), а не более низкую симметрию, такую,
как у структуры Кекуле или у не предполагавшейся одно время искаженной
гексагональной структуры. Формы внутренней компенсации, которыми раз-
личаются эти типы симметрии, несколько более сложны, чем для рассмотрен-
ных выше случаев, но общий принцип остается тем же.
б. Размеры несопряженных молекул. Наиболее точные размеры молекул
установлены в результате анализа вращательных частот в спектрах; чаще
всего пользуются инфракрасными и микроволновыми спектрами. Анализ
позволяет непосредственно определить три главных момента инерции (в
линейных молекулах один из них почти равен нулю, а в плоских молекулах
сумма почти равна третьему). В симметричных молекулах два или все три
момента инерции могут быть одинаковы: законы симметрии так же строго
определяют равенство величин моментов и локализацию осей, как и в случае
электрической поляризуемости (разд. 3, г). Исходя из измеренных независи-
мых величин главных моментов инерции, можно рассчитать межатомные
расстояния. Если число независимых несимметрично направленных расстоя-
ний, необходимых для определения геометрии молекулы, превышает число
независимых главных моментов инерции, можно увеличить число измеряе-
мых моментов инерции, повторяя спектроскопические измерения с изотопны-
ми модификациями молекул: изотопный заместитель изменит массу, а сле-
139
Глава III
довательно, и моменты инерции, не меняя при этом геометрии молекулы. Так,
HCN (линейная молекула) имеет два равных конечных главных момента
инерции, на основании которых можно рассчитать только одну величину,
в то время как необходимо определить два независимых межатомных рас-
стояния. Оба эти расстояния можно найти, если исследовать HCN и DCN.
Таким образом, например, было установлено [35], что углы между связями
НХН в молекулах Н2О, H2S и H2Se соответственно равны 105, 92 и 91°
(1,832, 1,605 и 1,588 рад), в NH3, РН3, AsH3, SbH3-107, 94, 92 и 91е (1.867,
1,640, 1,605 и 1,588 рад), а в этилене и формальдегиде — 117 и 116° (2,042
и 2,024 рад) соответственно. Найдено также, что длина связи СН уменьшается
в ряду этан, этилен, ацетилен [длина связи соответственно равна 1,102,
1,085 и 1,059 А (11,02 НО-2, 10,85-Ю-2 и 10,59-10-2 нм)], а длина связи СС
резко уменьшается в этом же ряду [1,543, 1,338 и 1,205 А (15,43-10"2,
13,38 НО-2 и 12,05 НО-2 нм)]. Наиболее важные значения длин и углов связей,
полученные путем спектроскопических измерений, приводятся в табл. 40
[35, 36].
Таблица 40
Некоторые длины п углы связей, измеренные
с помощью спектральных методов
Соединение Длина Хв или XY, А (0,1 нм) Длина ХН, А (0,1 нм) Угол НХН, град (17,45-10-3 рад)
н,о 0,957 105,0
NH3 1,014 106,8
СН4 [37] 1,092 109,5
СН3-СН3 [37] 1,536 1,091 108,0
СН3—ОН а 1,434 1,093 109,5
СН2 = СН2 1,338 1,085 117,4
СН2==О [39] 1,206 1,117 115,8
СН = СН 1,205 1,059
СН = N 1,157 1,063
а По данным Иваша и Деннисона [38], длина связи ОН составляет
0,937 А (9,37 10-2 нм), а угол СОН — 105,9° (1,848 рад). Было найдено,
что ось симметрии СНз-группы проходит между О- и Н-атомами гидрок-
сила на расстоянии 0,079 к (7,9-Ю-з нм) от атома кислорода, т. е
С — О-связь повернута на 3,0° (0,052 рад) относительно тетраэдрического
направления.
Множество других значений длин связи было получено дифракционными
методами, причем величины, полученные при помощи дифракции рентгенов-
ских лучей в твердых телах, имеют точность ±0,02 А (2-10-3 нм) [в лучшем
случае ±0,01 А (1-10-3 нм)], а величины, полученные при помощи дифракции
электронов в газовой фазе, имеют точность от ±0,2 до ±0,04 А (от ±2 -Ю-2
до ±4-10“3 нм); по-видимому, при измерениях в кристаллах наблюдаемые
длины связей несколько искажены в результате действия сил кристалличе-
ской решетки.
Наиболее общий вывод из многочисленных дифракционных измерений
заключается в том, что длина связи, определенная в пределах установленной
ошибки опыта, но иногда и менее точно, служит характеристикой типа
связи (т. е. пары атомов, образующих связь) и кратности связи (т. е. пока-
140
Физические свойства молекул
зывает, является ли связь простой, двойной или тройной) при условии, что
молекулы не содержат сопряженных ненасыщенных или некоторых гипер-
конътогационных систем.
Что касается влияния природы двух связанных атомов на длину связи
данной кратности, было замечено, что при отсутствии сопряженных систем
длина связи АВ часто приближается к среднему арифметическому значению
длин связей АА и ВВ; это означает, что длины связей могут быть представле-
ны как суммы радиусов, которые характеризуют атомы и кратность связей
и в сочетании дают длины соответствующих связей. Наилучшие значения
ковалентных, радиусов связей даны Полингом [18] (стр. 221), некоторые из
них приведены в табл. 41. Следует помнить, что правило аддитивности выпол-
няется лишь приблизительно, однако оно весьма полезно, тем более что адди-
тивность часто бывает довольно точной.
Таблица 41
Ковалентные радиусы атомов [А (0,1 нм)) по Полингу
Связь С N О F
Простая Двойная Тройная 0,772 0,667 0,603 0,70 0,60 0,55 0,66 0.55 0,64
Si p s Cl
Простая Двойная Тройная 1,17 1,10 0.93 1,04 0,94 0,99
Ge As Se Pr
Простая 1,22 1,21 1,17 1,14
Sn Sb Те I
Простая 1,40 1,41 1.37 1,33
Влияние кратности связи на длину связи между двумя одинаковыми атомами
иллюстрируется данными табл. 40 для СС-связей в этане, этилене и ацетиле-
не. Каждому атому, образующему кратную связь, приписываются различ-
ные радиусы в соответствии с кратностью данной связи (табл. 41).
в. Размеры сопряженных молекул. В ряду этан, этилен, ацетилен про-
исходит укорочение СС-связи, степень которого пропорциональна логарифму
кратности связи. Аналогичное укорочение СО-связи происходит при переходе
от метанола к формальдегиду (рис. 22). В данной книге не используется
термин «порядок связи», так как при различных квантовохимических при-
ближениях порядок связи получается разным. Термин «кратность связи»
141
Глава III
имеет отношение лишь к общепринятому выражению «кратная связь» и под
ним понимают половинное количество электронов, образующих связь.
Если вследствие мезомерии молекула становится симметричной, то кратность
связей уже не выражается целым числом. Например, кратность СС-связей
в бензоле и графите составляет 3/2 и 4/3 соответственно. Такие же значения
кратности связей СО приписаны формиат- и карбонат-ионам. При переходе
от этана или метанола к этим соединениям СС- или СО-связи укорачиваются
в большей степени, чем это следует из полулогарифмических линейных
графиков кратность — длина связи. Это объясняется тем, что дополнительные
электроны обеспечивают более
Рис. 22. Зависимость между длиной и крат-
ностью связи.
1 — этан; 2 — метанол; 3 — графит; 4 — бен-
зол; 5 — карбонат-ион; 6 — этилен; 7 —фор-
миат-ион; 8 — формальдегид; 9 — ацетилен.
прочную связь между атомами
не только вследствие увеличе-
ния своего числа, но, кроме
того, и вследствие делокализа-
ции, которая еще более увели-
чивает прочность связи.
Относительного большого уко-
рочения связи можно ожидать
и в том случае, когда дополни-
тельная делокализация элек-
тронов вследствие мезомерии
относительно мала и точно не
может быть определена, так как
она не вносит новых элементов
симметрии. Это обусловливается
двумя одинаковыми, усиливаю-
щими друг друга взаимодей-
ствиями. Когда кратность фор-
мально двойной или формаль-
ной тройной связи вследствие
мезомерии несколько умень-
шается и в то же самое время (также вследствие мезомерии) увеличивается
степень делокализации других электронов, нельзя ожидать значитель-
ного увеличения длины связи, так как обеднение электронами и делокализа-
ция действуют в противоположных направлениях. Если вследствие гипер-
конъюгации уменьшается электронное содержание простой связи, то наиболее
вероятным является существенное увеличение ее длины, поскольку происхо-
дит отклонение простой связи в сторону «несвязывающей»; степень этого
отклонения определить трудно. Некоторые качественные заключения могут
быть выведены из кривых линий на рис. 21. Экспериментальные данные,
относящиеся к СС-связям, приведены в табл. 42 [36]. Если вследствие сопря-
жения на простых СС-связях делокализуются дополнительные электроны,
связь укорачивается на 0,05 . . . 0,16 А (5 -Ю-3 ... 16 -10-3 нм). Сопряжение
обедняет электронами двойные и тройные связи, но увеличивает степень
делокализации электронов этих связей, и, таким щбразом, длина связи
изменяется незначительно, не более чем на 0,005 А (5 -Ю-4 нм).
Было сделано предположение, что наблюдаемое укорочение связей не имеет
никакого^отношения к распределению и делокализации электронов, а цели-
ком обусловлено изменением гибридизации (например, от sp3 к sp2 или sp)
орбиталей углерода, участвующих в образовании связей. Конечно, такое
изменение гибридизации атомных орбиталей происходит, однако если бы
молекулярные явления были связаны исключительно с изменением гибриди-
зации, то в ряду — С — А, = С — A, hsC — А укорочение связей С — А
112
Физические свойства, молекул
Таблица 42
Влияние роста цепи сопряжения и обеднения электронами
на длину [А (0,1 нм)] формально простой, двойной и тройной СС-связеп
Стандартная связь Стандартная связь Стандартная связь
СН3—СН3 1,543 сн2=сн2 1,338 СН = СН 1,205
Усиленная простая связь Усиленная двойная связь Обедненная тройная связь
сн2=сн-сн=сн2 1,483 СН = С— СН = О 1,446 СН2 = СН —С = N 1,426 CH = С —С = N 1,378 сн2=с=с=сп2 1,284 CH=C-CsN 1,205
Обедненная двойная связь
си2=сн- сн = сн2 1,337 СН2 = СН-С = N 1,339
не зависело бы от природы А (при условии, что можно использовать понятие
«ковалентные радиусы»). На самом же деле это не так. Если же пользоваться
понятием ковалентных (связевых) радиусов, то необходимо допустить, что
влияние строения молекулы на длины связей целиком обусловливается чисто
атомными свойствами — гибридизацией орбиталей. Точно известно [36],
что длина СН-связи (тип А — Н) зависит от природы соединения (табл. 43).
Таблица 43
Влияние ненасыщенности углерода на длину
СН-связей [А (0,1 нм)]
Н-СН2-СН3
1,0914
н—сн = сн2
1,085
Н —С6Н5
1,084
__ -______
н—с = сн
1,059
Н-С = N
1,063
--- >
0,007
0,023
0,030
Отсюда при интерпретации с помощью орбиталей следует, что простая связь,
расположенная между двумя двойными, двойной и тройной или между двумя
тройными связями, должна быть укорочена на 0,014, 0,04 и 0,06 А (1,4 -10~3,
4-10~3 и 6-10-3 нм) соответственно. Однако наблюдаемое укорочение связей
(табл. 42) в 3—4 раза больше этих величин и составляет 0,06,'Aj,12 ii 0,17 А
(6 -10~3, 12-10-3 и 17-10"3 нм) соответственно *.
* См. работу [40]. Длину простой СС-связи в газообразном бутадиене измерить труд-
но. Если использовать данные по длинам соответствующих связей в кристаллах, получен-
ные рентгеноструктурным анализом для полностью тронс-сорбамида, метилмуконата
и мукононитрила [44], то получается, что укорочение простой связи составляет 0,09 А
(9«Ю-3 нм), а не 0,06 А (6-Ю-3 нм).
из
Таблица 44
Длина связей в соединениях с сопряженными связями
Соединение
Рассчитано
лз радиусов а5
А (0,1 НМ)
Найдено. Л (0,1 нм)
Мстод^'
Дите-
э 1 гура
Ненасыщенные галогеносоединения.
( СвН5С1 СС1 1,76 1,69+0,02 дэ 43
< о-, м-1 /г-СбН4С12 cci 1,76 (1,69.. .l,71)+0,02 ДЭ 43
1 С6С16 CC1 1,76 1,70+0,02 ДЭ 43
Г СН.,-=СНС1 CCI 1,76 1,69+0,02 до 44
1 1,1- и 1,2-С2И4С12 CCI 1,76 (1,67. . .1,71)^0,02 Д:-» 44
1 С2С14 CCI 1,76 1,73+0,02 ДЭ •44
1 СН = СС1 CCI 1,76 1,68+0,04 ДЭ 45
1 СН = СВг CBr 1,91 1,80+0,03 ДЭ 45
( N - СС1 CCI 1,76 1,629 с 46
1 N .-S СВг CBr 1,91 1,790 с 46
Нитржоединения
CH3NO2 { N — 0 1,36 1,22+0,02 ДЭ 47
N = O 1,15 1,22+0,02 ДЭ 47
C(NO,)4 1 N-0 1,36 1,22+0,02 ДЭ 48
N = O 1,15 1,22+0,02 ДЭ 48
Ка рбонсила m-uou
HCOONa| C-0 1,43 1,27+0,01 Р 49
C = O 1,22 l,27±0,01 р 49
C —0 1.43 1,26+0.02 в 50
KII3CH2COO- | C=O 1,22 1,26+0,02 р 50
Карбонат- и нитрат- ионы
CciCO с-о 1,43 1,31 + 0.01 Р 51
C +) 1,22 1.31+0,01 р 51
N-0 1,36 1,21+0,01 р 51
N = O 1,15 1,21+0,01 р 51
N2O5(NO3-non) N-0 1,36 1,243+0,01 р 52
N = O 1,15 l,243+o,(H р 52
Карбоксамид
CH3CONH, | 1,47 1,38+0,05 р 53
c = o 1,22 1,28+0,05 р 53
Карбамид
CO(NH2)2 { c%° 1,25+0,01 1,38+0,05 р Р 54 54
Кратная связь азотистых соединений,
mpanc-Cell^ = NC6H6 { C-N 1,47 1,41+0,03 Р 55
N — N 1,20 1,23+0,03 р 55
!juc-CeH5N NCbH5 C-N N - N 1,47 1,20 1,46+0,03 1,23+0,03 р Р 56 56
Физические свойства молекул
Продолжение табл. 44
Соединение Связь Рассчитано из радиусов а, А (0,1 мн) Найдено, А (0,1 нм) Метод® Лите- ратура
Двуокись углерода; азид- и нитроний-ионы
о = с = о c = o 1,22 1,632+0,003 c 57
NaN3, KN3, NH4N3 N = N 1,20 (l,15...1,16)+0,02 p 58
no2cio4 N = O 1,15 l,10±0,01 p 52
d Значения радиусов взяты из табл. 43.
0 ДЭ — дифракция электронов, Р— рентгеноструктурный метод, С — спектроскопия.
Данные табл. 44 также иллюстрируют вышеуказанную точку зрения. Про-
стые связи, которые вследствие сопряжения приобретают частичную крат-
ность, всегда значительно укорочены, тогда как двойные связи, которые
вследствие этого процесса частично теряют кратность, или совсем не удли-
няются, или удлиняются на небольшую величину.
Выше уже было дано определение термина «вторичный пространственный
эффект» (гл. II, разд. 2, е) и показано, как он проявляется в величинах
дипольных моментов (разд. 1, ж) (благодаря чему он и был открыт Хэмпсо-
Рпс. 23. Геометрия транс- и г^ис-азобензолов.
тиранс-Соединение плоское, но бензольные
кольца в -tyuc-соединении не лежат в одной
плоскости (подобно трехлопастному пропел-
леру, у которого удалили одну лопасть),
что приводит к увеличению расстояния между
ортио-положениями почти до величины, рав-
ной расстоянию между слоями в графите.
Скручивание молекулы препятствует сопря-
жению между кольцами и атомами азота, и,
таким образом, связи CN не укорочены, как
в тирляс-изомепе.
ном). Кроме того, было отмечено, что этот эффект косвенно проявляется в теп-
лотах реакций (разд. 2, б). Теперь посмотрим, как этот эффект отражается
на длинах связей, взяв в качестве иллюстрации длину связи CN в цис-
та. транс-азобензолах (табл. 44). Теория будет такой же, как и при рассмот-
рении теплот гидрирования цис- и транс-стильбенов (разд. 2, б), транс-
Азобензол — плоская молекула, так как дополнительная делокализация
электронов на формально простых СИ-связях^вследствие мезомерии не встре-
чает стереохимических препятствий. Поэтому CN-связи должны укорачи-
ваться, что и наблюдается в действительности: укорочение составляет около
0,06 А (6 -10-3 нм). Однако цис-азобензол не может быть плоским вследствие
отталкивания opmo-CH-групп бензольных колец. Анализ структуры кристал-
лов показывает, что бензольные кольца выведены из плоскости атомов
С — N — N — С (рис. 23) на угол 56° (0,977 рад), поэтому просвет между
10-0772
145
Глава III
орпго-углеродными атомами составляет 3,3 А (33-10-2 нм), что почти равно
расстоянию между плоскими слоями в графите [3,4 А (34-10-2 нм)]. Такое
расположение колец препятствует мезомерному перераспределению электро-
нов между азогруппой и бензольными кольцами, и в соответствии с этим
Хэмпсон и Робертсон установили, что CN-связи укорачиваются в небольшой
степени.
Вопрос о размерах молекул ароматических соединений будет рассматривать-
ся также в гл. IV.
г. Размеры молекул с гиперконъюгацией. Как показывают данные
табл. 45, простая СС-связь между метильной группой и углеродом, связанным
двойной или тройной связью, несколько короче связи между метильной груп-
пой и алкильным остатком. Этот факт для случая двоесвязанного углерода
был адекватно установлен на примере ацетальдегида (№ 1 в табл. 45)
Таблица 45
Длины простых СС-связей в молекулах с возможной
гиперконъюгацией СН-связи (4ДГ)
Номер соедине- ния Примеры соединений ф Длина, А (0,1 нм) Укорочение, А (0,1 нм) Метод а
Простая связь между метилом и алкилом
| СН3-СН3 1 1,543 | | с
Простая свя зъ между метилом и двойной связью
1 | СНз-СН =0 | 1,501 | 0,042 | с
Простая связь между метилом и тройной связью
2 СН3-С = СН | 1,46+0,02 1,459 0,084 ДЭ с
3 СН3 — C = N | 1,49+0,03 1,458 0,085 ДЭ с
4 СН3-С = С-С = N 1,454 0,089 с
5 сн3 —С = С —сн3 1,47+0,02 ~ 0,07 ДЭ
6 СН3-С = С-С = С-СН3 1,47+0,02 -0,07 ДЭ
а С — спектроскопия [36], ДЭ — дифракция электронов [59].
с помощью микроволновых спектров. В этом случае СС-связь укорочена
на 0,042 А (4,2 -10-3 нм). Полинг, Спрингалл и Палмер [59] с помощью диф-
ракции электронов на примере соединений 2, 3, 5 и 6 (табл. 45) установили,
что аналогичное укорочение простой связи в случае углерода с тройной
связью больше по величине. Данные микроволновых спектров, полученные
Трамбаруло, Горди и др., позволяют проверить точность дифракционных
измерений (№ 2, 3 и 4 в табл. 45). Сравнение значений, полученных для
примеров № 2 и 3, подтверждает тот факт, что точность метода дифракции
электронов соответствует указанной, поэтому доверие к результатам № 5 и 6
возрастет. Точно измеренное укорочение связей составляет 0,084 А
(8,4-IT3 нм) для метилацетилена и 0,089 А (8,9-10-3 нм) для метилцианаце-
тилена.
146
Физические свойства молекул
Выше уже приводились доказательства существования гиперконъюгации
при рассмотрении физических свойств, например дипольных моментов
(разд. 1, б и д) и теплот реакций (разд. 2, а). Вероятно, таким же образом
лучше всего интерпретировать изменение длины связей, наблюдающееся,
к примеру, в случае метилацетилена.
нСсЛс^сн
Дополнительная делокализация электронов на простой СС-связи укорачивает
связь и ответственна, вероятно, более чем за половину наблюдаемого укоро-
чения. Легко понять большее укорочение в случае тройной связи по сравне-
нию с двойной. Нельзя с уверенностью ожидать, что обеднение электронами
СН-связей будет конкурировать с делокализацией связей в боковом направ-
лении и таким образом объяснять наблюдаемые изменения длин связей; ни
в одном случае такого эффекта не было обнаруя^ено.
В связи с этим вновь представляется случай интерпретировать наблюдаемое
укорочение связей изменением гибридизации атомных орбиталей углерода
без учета гиперконъюгации, т. е. без учета перераспределения и делокализа-
ции электронов. Против этого можно возразить так же, как это было сделано
выше, а именно: если справедлива такая точка зрения, то укорочение связи
при переходе от — С — А к = С — А и далее к == С — А не должно зави-
сеть от А. Однако наблюдаемое укорочение более чем в 2 раза превышает
эффект, ожидаемый при таком предположении. Таким образом, необходимо
принять, что наблюдаемое укорочение связей в значительной степени обус-
ловлено гиперконъюгацией, а не гибридизацией атомных орбиталей
углерода *.
В табл. 46 приведены данные Полинга, Горди и Сэйлора [60] по дифракции
Таблица 46
Длина простых связей [А (0,1 нм)] в молекулах с возможной
гпперкоиъюгацией СН- н CHal-групп ( >/ и —
Пример Длина связи СС Укорочение 0 Длина связи Cllal Укорочение °
С1СН2С = сн 1,47+0,02 -0,07 1,82+0,02 ~ 0,06
ВгСНоС = сн 1,47+0,02 - 0,07 1,95+0,02 ~ 0,04
1СН2С - сн 1,47+0,02 ~ 0,07 2,13+0,03 ~ 0,03.
а Данные получены методом дифракции электронов [56].
б Отклонения в длинах связей рассчитаны исходя из сумм ковалентных радиусов
(табл. 41).
электронов на хлористом, бромистом и иодистом пропаргиле. В этих соеди-
нениях простые связи СС имеют одну и ту же длину 1,47 ± 0,02 А
(14,7-10”2± 2-10-3 нм). Таким образом, они значительно укорочены. Учи-
* Такова была точка зрения Крейга в 1951 г., когда еще не были опубликованы
точные данные по длинам связей, полученные с помощью микроволновых спектров.
ю* 747
Глава III
тывая ошибку измерения, можно сказать, что степень укорочения СС-связей
в этих соединениях мало отличается от величины, ожидаемой на основании
того факта, что в молекуле имеются две СН-связи, способные к гиперконъю-
гации (4-М). Поскольку в этих молекулах, помимо 4-М-гиперконъюгации
СН-связей, существует —М-гиперконъюгация CHal-связи, необходимо сде-
лать вывод, что обе эти формы гиперконъюгации вносят вклад в укорочение
СС-связи.
Н2
Наличие —М-гиперконъюгации с участием атомов галогена следует из
наблюдаемого удлинения связей CHal; в двух случаях оно больше экспери-
ментальной ошибки (табл. 46). Вероятно, удлинение связей CHal главным
образом обусловлено обеднением их электронами, так как при гиперконъю-
гации связывающие электроны частично становятся несвязывающими.
5. Упругие свойства молекул
а. Физический смысл констант упругости. Наблюдаемые в спектрах
колебательные частоты можно использовать для оценки «жесткости» молекул.
Основной количественной мерой «жесткости» является константа закона
Гука, или модуль упругости, обычно называемый силовой постоянной. Закон
Гука применим к особым типам малых деформаций.
Для двухатомных молекул физический смысл силовой постоянной ясен:
константа к для малых деформаций в соответствии с законом Гука является
коэффициентом пропорциональности в выражении для потенциальной энер-
гии деформации V как функции изменения (Аж) внутренней координаты х
(в данном случае длины связи):
27 = к (Аж)2
Иными словами, константа к — возвратная сила, отнесенная к единице
изменения длины связи. Она непосредственно определяется частотой коле-
бания v согласно уравнению
4л 2v2 — — = к ( — 4- —)
т \ иц т2 J
где т —«приведенная масса» молекулы, а тг и т2— массы атомов. Если
амплитуда колебания велика, то следует делать поправку на «ангармонич-
ность», т. е. на отклонения от закона Гука; эти отклонения заключаются
в том, что V частично зависит от более высоких степеней Аж.
В общем случае потенциальная энергия и-атомной молекулы является
функцией всех (Зи — 6) внутренних координат *, и, так как по закону Гука
она является квадратичной функцией, потенциальная энергия равна сумме
(Зи — 6) (Зп — 5)/2 членов типа к (Ах)2 или к' (Аж Ау), причем каждый член
будет иметь свою силовую постоянную к. Число колебательных частот
* В линейных молекулах внутренних координат на одну больше.
J48
Физические свойства молекул
(Зга — 6) (даже если они все определены) недостаточно * для того, чтобы
определить все постоянные к. Существует два выхода из этого затруднения.
Наиболее удовлетворительный метод — измерение колебательных частот
нескольких изотопных модификаций молекулы с целью получения по край-
ней мере стольких колебательных частот, сколько существует силовых
постоянных (которые, разумеется, одинаковы для различных изотопных
модификаций). Однако обычно пользуются другим и часто единственным
доступным способом. В уравнении потенциальной энергии пренебрегают
теми членами, которые считаются наименее существенными, оставляя столько
членов, сколько известно частот, а иногда при разумном выборе, обеспечи-
вающем хороший расчет частот, даже меньше.
Если, как это чаще всего делают, выбирают приближенную функцию потен-
циальной энергии, соответствующую так называемой модели силового поля
валентных сил, то остается по одному члену к (Дж)2 на каждую связь (в этом
выражении Дж — изменение длины связи, а к — силовая постоянная данной
связи). В отличие от двухатомных молекул силовые постоянные связей мно-
гоатомных молекул являются весьма приближенными, поэтому не следует
пытаться выражать их с большой точностью. Кроме члена к (Дж)2, они зави-
сят от составляющих потенциальной энергии, которыми мы пренебрегли,
а также и от других членов. Наличие в полной функции таких членов, как
к' (ДхАу), показывает, что энергия деформации не может быть аддитивно
распределена по связям.
В потенциальную функцию модели силового поля валентных сил входит
также и другой ряд членов. Они имеют вид 6 (Д0)2 и относятся к валентным
углам, причем Д0 является деформацией угла, а константа 6 представляет
возвратный момент, отнесенный к единице изменения угла, и называется
константой момента или, более часто, деформационной силовой постоянной.
Если существует сопротивление крутильному колебанию, то добавляется
член у<р2, где ф — угол кручения, у — возвратный момент на радиан угла
кручения, называемый крутильной силовой постоянной. Эти угловые силовые
постоянные еще более приближенные и менее отражают структуру молекулы,
чем силовые постоянные связи.
Метод модели силового поля валентных сил был подробно разработан, что
позволяет применять его для характеристики весьма специфических отноше-
ний; например, практиковалось добавление члена, характеризующего энер-
гию отталкивания близко расположенных, но непосредственно не связанных
атомов, таких, как хлор в четыреххлористом углероде.
б. Силовые постоянные связей и типы связей [35, 61]. Силовые постоян-
ные связей выражаются в динах на сантиметр. Полученные величины лежат
в пределах 104 . . . 106 дин/см (10 . . . 103 Н/м), причем большинство из них
близки к верхнему пределу. Величину, равную 10-5 к, или 105 дин/см
(102 Н/м), принимают за «практическую единицу» силовой постоянной.
Силовая постоянная связи приближенно характеризует связь, т. е. пару
связанных атомов и кратность связи. Она зависит от состояния системы,
от окружающих атомов, особенно от их полярности и ненасыщенности;
следует подчеркнуть также влияние этих факторов на кратность исследуемой
связи.
Зависимость силовых постоянных связей от химической природы атомов
можно иллюстрировать силовыми постоянными ХН-связей в гидридах ХНП.
Как показывают цифры, имеется явная зависимость от положения'атома X
* Число различных силовых постоянных, как и число различных колебательных
частот, уменьшается в симметричных молекулах, однако всегда первое число превышает
второе.
149
Глава III
в таблице Менделеева
' В 3,6* С 5,0 N 6,5 О 7,6 F 9,7
хн < Si 2,7 Р 3,1 S 4,0 Se 3,1 Cl 5,2 Вг 4,1 I 3,1
Закономерность, существующая в ряду галогенов, сохраняется в ряду их
метильных производных
ХС ... F 5,6 С1 3,4 Вг 2,9 I 2,3
Простейшей иллюстрацией влияния полярности ХН-связи на силовую
постоянную связи является сравнение постоянных связи NH в аммиаке
и ионе аммония:
NH ... NH3 6,5 NHJ 5,4
Как показывают величины силовых постоянных метильных производных,
индуцированная полярность, по-видимому, оказывает заметное влияние
на силовые постоянные СН-связей:
[ СН3СН3 НСН3 ICH3 ВгСН3 CICH3 FCH3
Н ( 5,1 5,0 5,0 4,95 4,9 4,7
Приведенные ниже значения с тловых постоянных показывают влияние нена-
сыщенности углеродных атомов или совместное влияние ненасыщенности
и полярности на силовые постоянные связи:
СН
НСН3ь/
5,0
нсн = сн2 нс = сн
5,1
5,9
НС6Н5
5,1
НСН = 0 НС = N
4,4 5,9
Эти данные нелегко истолковать; увеличение силовых постоянных в ряду
метан, этилен, ацетилен может быть обусловлено изменением гибридизации
(гл. I, разд. 4,6). Однако разности между постоянными удивительно неодина-
ковы, хотя в то же время величины для этилена и бензола строго согласуют-
ся. Не ясно, почему силовые постоянные связи в случае этилена и формальде-
гида различны, тогда как соответствующие величины для ацетилена и циани-
стого водорода одинаковы.
Но все это имеет несравненно меньшее значение, чем действие других факто-
ров, а именно кратности связей. Влияние кратности связей на силовые
постоянные можно видеть из следующих данных:
[ Н3С —СН3 Н2С = СН2 нс = сн
СС { j
I 4,5 9,8 15,6
СО>
Н2С = О
12,3
CN-|
НС = N
'18,1
Величины постоянных простой, двойной и тройной связей относятся друг
к другу как 1 : 2,2 : 3,5. Надежные данные для силовых постоянных простых
* Приведена величина для четырех внешних атомов водорода в молекуле В2Н6.
150
Физические свойства молекул
CN- и GO-связей отсутствуют, но если предположить, что их значения лежат
между значениями постоянных для СС- и CF-связей, т. е. приблизительно
равны 5, то примерно такое же соотношение, как и приведенное выше, будет
выражать влияние кратности на силовые постоянные CN- и СО-связей.
в. Силовые постоянные, сопряжение и гиперконъюгация. Явная зави-
симость силовых постоянных от кратности связей позволяет ожидать, что
эти постоянные будут отражать изменения кратности связей в мезомерных
системах и, возможно, увеличение жесткости связи, точно так же как укоро-
чение связи в случаях значительной мезомерной энергии.
Имеющиеся в распоряжении данные подтверждают эти предположения.
Ниже приведен ряд значений силовых постоянных связей СС, расположен-
ных, насколько это оказалось возможным, в последовательности увеличения
кратности связей:
Н3ССН3 СН3С = СН CH3CN NCCN свнв Н2С = СН2
СС 1 Кратность 1 1 + 1 + 1++ 1,5 (м) 2
( Силовая постоянная 4,5 5,1 5,3 6,7 7,6 9,8
Н2С=СН2 н2с=с=сн2 О=С=С=С=О СН [3 — С= СН НС=СН
СС J Кратность 2 2 2 (м) 3- 3
( Силовая постоянная 9,8 9,7 14,9 15,3 15,6
Верхний ряд содержит две молекулы с гиперконъюгацией (1+) и одну,
в которой обычное сопряжение (1 ++) должно увеличивать кратность про-
стой связи. Видно, что при наличии сокращения или гиперконъюгации связи
становятся более жесткими, однако нельзя различить, обусловлено это увели-
чением электронной плотности на связи или изменением гибридизации орби-
талей. Впрочем, это затруднение не влияет на заключения, которые сделаны
для примеров, приведенных ниже. Молекула бензола благодаря мезомерии
(обозначенной символом м) оказывается более жесткой, чем этого можно было
бы ожидать для номинальной кратности 1,5. Нижний ряд содержит несопря-
женный аллен (увеличения жесткости двойной связи нет), сопряженную
недоокись углерода (м) (большое увеличение жесткости при отсутствии увели-
чения кратности GG-связей, так как центральный атом углерода не имеет
соответствующих орбиталей) и метилацетилен, в котором гиперконъюгация
должна уменьшить кратность тройной связи (3—) и несколько снизить
константу силы связи. В этих случаях не может возникнуть никаких вопро-
сов об изменении гибридизации.
Силовые постоянные связей карбонильных соединений, по-видимому, нахо-
дятся в аналогичных отношениях. Можно было бы ожидать, правда, что
постоянная для кетена будет занимать более точно среднее положение между
постоянными для ^'формальдегида и недоокиси углерода:
Н2С= О
( Кратность 2
С О| \
{ Силовая постоянная 12,3
Н2С=С= О
2+ +
12,3
о=с=с=с=о
2-|—р (м)
14,2
о = с.= о
2 (м)
15,5
Большая жесткость связи в двуокиси углерода, по-видимому, обусловливает-
ся очень большой энергией мезомерии данной молекулы (м). Кратность ее
не может увеличиться, так как атом углерода не имеет соответствующих
орбиталей.
151
Глава III
Силовые постоянные циангруппы указывают на интересный факт влияния
гиперконъюгации с метилом и на наличие влияния сопряжения со второй
циангруппой или галогеном. Все эти виды гиперконъюгации (3—)или сопря-
жения (3 — —) снижают кратность формально тройной связи и сопровож-
даются уменьшением силовых постоянных. В любом случае такое уменьше-
ние нельзя объяснить изменением гибридизации без смещения электронной
плотности.
СН = СН H3C-C = N N=C-C=N I-C = N BrC=N C1-C=N
Г Кратность 3 3— 3----3--------3-----3-------
t Силовая постоянная 18,1 17,5 17,5 16,8 16,8 16,7
Отметим, что хотя теория гиперконъюгации первоначально была введена
лишь для объяснения зависимости скоростей реакций и положения равнове-
сий (гл. II, разд. 3,и) от строения молекулы, в настоящее время она помогает
интерпретировать влияние структуры на некоторые физические свойства
(разд. 1,6 и д, 2,а, 4,г и данный раздел). Несмотря на некоторую кри-
тику в адрес этой теории, она приносит большую пользу, чтобы от нее от-
казаться.
В качестве заключительного примера можно привести значения силовых
постоянных связей С — Hal в галогенцианах и сравнить их со значениями
силовых постоянных метилгалогенидов
СНа!
Cl—CN 5,3 |Br-CN 4,211— CN 2,9
С1—СН3 3,4|Вг —СН3 2,9U —СН3 2,3
Из приведенных данных видно, что силовые постоянные для галогенцианов
больше. Частично это может быть обусловлено полярностью. Однако разница
в значениях настолько велика для хлоридов и так резко снижается при
переходе к иодидам, что, по-видимому, главной причиной, обусловливающей
различие, является сопряжение (с увеличением кратности связи) по крайней
мере в случае хлоридов, ибо сопряжение должно оказывать тем большее
влияние, чем меньше размер атома галогена (гл. II, разд. 3,д).
СПИСОК ЛИТЕРАТУРЫ
1. Debye Р., Physik. Z., 13, 97 (1912); Polare Molekeln, Hirzel, Leipzig, 1929.
2. Phillips G. M., Hunter J. S., Sutton L. E., J. Chem. Soc., 1945, 146.
3. Wesson L. G., Tables of Electric Dipole Moments, Technology Press, Cambridge, Mass.,
1948.
4. Brockway L. 0., J. Am. Chem. Soc., 58, 2516 (1936).
5. Brockway L. 0., Beach J. Y., Pauling L., J. Am. Chem. Soc., 57, 2693 (1935); Sto-
sick A. J., ibid., 61, 1127 (1939).
6. Pauling L., Brockway L. O., J. Am. Chem. Soc., 59, 13 (1937).
7. Lide D. R., Mann D. E., J. Chem. Phys., 29, 914 (1959); Lide D. R., ibid., 33, 1514,
1519 (1960).
8. Laurie V. W., Muenter J. S., J. Am. Chem. Soc., 88, 2883 (1966); J. Chem. Phys., 45,
825 (1966).
9. Ingold С. K., Ann. Repts. on Progress Chem. (Chem. Soc. London), 23, 144
(1926).
10. Hojendahl K., Studies of Dipole Moments, Bianco Lunos Bagtrikkeri, Copenhagen,
1928.
11. Sutton L. E., Proc. Roy. Soc. (London), A133, 668 (1931).
12. Smith J. W., J. Chem. Soc., 1961, 81.
13. Roberts J. D., Webb R. L., McElhill E. A., J. Am. Chem. Soc., 72, 408 (1950).
14. Millen D. J., Sinnott К. M., J. Chem. Soc., 1958, 380.
152
Физические свойства молекул
15. HukinsA. A., LeFevre R. J. W., Nature, 164, 1050 (1949); Angyal С. L., Barclay G. A.,
Ilukins A. A., LeFevre R. J. W., J. Chem. Soc., 1951, 2583.
16. Birtles R. H., Hampson G. C., J. Chem. Soc., 1937, 10; Ingham С. E., Hampson G. C.,
ibid., 1939, 981; см. также Few A. V., Smith J. W., ibid., 1949, 2663; Kopod H., Sut-
ton L. E., de Jong W. A., Verkade P. E., Wepster В. M., Rec. trav. chim., 71, 521 (1952);
Kopod H., Sutton L. E., Verkade P. E., Wepster В. M., Rec. trav. chim., 78, 790
(1959).
17. Fajans K., Ber., 53, 643 (1920); 55, 2836 (1922).
18. Pauling L., Nature of the Chemical Bond, Cornell University Press, Ithaca, New York,
3rd Edn., 1960, pp. 73, 188.
19. Rossini F. D., Chem. Revs., 27, 1 (1940).
20. Pitzer K. S., Catalano E., J. Am. Chem. Soc., 78, 4844 (1956); Pitzer K. S., Advances
in Chemical Physics, Interscience Publishers, New York, 1959, Vol. 2, p. 59.
21. Kistiakowsky G. B., Romeyn H., Jr., Ruhoff J. R., Smith H. A., Vaughan W. E., J. Am.
Chem. Soc., 57, 65 (1935); Kistiakowsky G. B., Ruhoff J. R., Smith H. A., Vaughan W. E.,
ibid., p. 876.
22. Conn J. B., Kistiakowsky G. B., Smith E. A., J. Am. Chem. Soc., 61, 1868 (1939).
23. Pauling L., Sherman J., J. Chem. Phys., 1, 606 (1933).
24. Kistiakowsky G. B., Ruhoff J. R., Smith H. A., Vaughan W. E., J. Am. Chem. Soc.,
58, 146 (1936); Dolliver M. A., Gresham T. L., Kistiakowsky G. B., Vaughan W. E.,
ibid., 59, 831 (1937); Dolliver M. A., Gresham T. L., Kistiakowsky G. B., Smith H. A.,
Vaughan W. E., ibid., 60, 440 (1938); Williams R. B., J. Am. Chem. Soc., 64, 1395
(1942).
25. Eisenlohr F., Spektrochemie organischer Verbindungen, Enke, Stuttgart, 1912.
26. Von Steiger A. L., Ber., 54, 1381 (1921); Smyth С. P., Phil. Mag., 50, 361 (1925); Dielec-
tric Constant and Molecular Structure, Chemical Catalog Co., New York, 1931, Chap.
8; Fajans K., Knorr C. A., Ber., 59, 249 (1926); Denbigh K. G., Trans. Faraday Soc.,
36, 936 (1940).
27. Wasastjerna J. A., Z. physik. Chem., 101, 193 (1922); Soc. Sci. Fennica, Commentatio-
nes Phys. Math., 1, 37 (1933); Fajans K., Joos G., Z. Physik, 23, 1 (1924); Born M.,
Heisenberg W., ibid., 23, 388 (1924); Fajans K., Kohner H., Geffcken W., Z. Electrochem.,
34, 1 (1928); Fajans K., ibid., p. 502; Smyth С. P., Dielectric Constant and Molecular
Structure, Chap. 8.
28. Strecker W., Spitaler P., Ber., 59, 1754 (1926).
29. LeFevre R. J. W., J. Proc. Roy. Soc. N.S.W., 95, 1 (1961); LeFevre R. J. W., Orr B. J.,
Ritchie G. L. D-, J. Chem. Soc., B273, 281 (1966).
30. Stuart H. A., Molekulstruktur, Springer, Berlin. 1934.
31. LeFevre R. J. W., in «Advances in Physical Organic Chemistry», ed. V. Gold,
Vol. 3, p. 1.
32. Cm. [30]; Denbigh K. G., Trans. Faraday Soc., 36, 936 (1940).
33. LeFevre C. G., LeFevre R. J. W., J. Chem. Soc., 1945, 1577; LeFevre R. J. W., Rao В. P.,
ibid., 1958, 1465.
34. Schaefer C., Trans. Faraday Soc., 24, 841 (1929); Schaefer C., Bormuth C., Z. physik,
62, 508 (1930).
35. Herzberg G., Infra-red and Raman Spectra, Van Nostrand, New York, 1945,
Chap. 4.
36. Costain С. C., Stoicheff В. P., J. Chem. Phys., 30, 777 (1959).
37. LepardD. W., Sweeney D. M. C., Welsh L. H., J. Chem. Phys., 40, 1567 (1962).
38. Ivash E. V., Dennison D. M., J. Chem. Phys., 21, 1804 (1953).
39. Oka T., J. Phys. Soc. Japan, 15, 2274 (1960).
40. Coulson C. A., Volume Commemoratif Victor Henri, Maison Desoir, Liege, 1948,
p. 15.
41. Filippakis S. E., Leiserowitz L., Schmidt G. M. J., J. Chem. Soc., B290, 297, 305 (1967).
42. Pauling L., Nature of the Chemical Bond, 3rd Edn., p. 221.
43. Brockway L. O., Palmer K. J., J. Am. Chem. Soc., 59, 2181 (1937).
44. Brockway L. O., Beach J. Y., Pauling L., ibid., 57, 2690 (1935).
45. Brockway L. O., Coop I. E., Trans. Faraday Soc., 34, 1429 (1938).
46. Towers С. H., Holden A. N., Merritt F. R., Phys. Rev., 74, 113 (1948).
47. Rogowski F., Ber., 75, 244 (1942).
48. Stosick A. J., J. Am. Chem. Soc., 61, 1127 (1939).
49. Pauling L., Brockway L. O., Proc. Nat. Acad. Sci. (U.S.), 20, 336 (1934).
50. Zachariasen W. H., Phys. Rev., 53, 917 (1938).
51. Elliot N., J. Am. Chem. Soc., 59, 1380 (1937).
52. Grison P. E., Ericks K., de Vries, Acta Cryst., 3, 290 (1950); Truter M. R., Cruick-
shank D. J., Jaffery G. A., ibid., 13, 855 (1960).
53. Senti F., Harker D., J. Am. Chem. Soc., 62, 2008 (1940).
153
Глава III
54. Wykoff R. W. J., Cory R. B., Z. Krist., 89, 462 (1934).
55. de Lange J. J., Robertson J. M., Woodward I., Z. Krist., A171, 398 (1939); Robert-
son J. M., J. Chem. Soc., 1939, 232.
56. Hampson G. C., Robertson J. M., J. Chem. Soc., 1941, 409.
57. Barker E. F., Adel A., Phys. Rev., 44, 185 (1933); Herzberg G., Infra-red and Raman
Spectra, pp. 394, 398.
58. Frevel L. K., 7.. Krist., 94, 197 (1936); J. Am. Chem. Soc., 58, 779 (1936).
59. Pauling L., Springall H. D., Palmer K. J., J. Am. Chem. Soc., 61, 927 (1939).
60. Pauling L., Gordy W., Saylor J. H., J. Am. Chem. Soc., 64, 1733 (1942).
61. Crawford B. L., Jr., Brinkley S. R., J. Chem. Phys., 9, 69 (1941); Noether H. D., ibid.,
10, 664 (1942); Linnett W., ibid., 8, 91 (1940); Trans. Faraday Soc., 37, 469 (1941);
Quart. Rev. Chem. Soc., 1, 13 (1947).
ГЛАВА IV
АРОМАТИЧНОСТЬ И СВЯЗАННЫЕ С НЕЙ ФИЗИЧЕСКИЕ СВОЙСТВА
1. Теория ароматических соединений...........................156
2. Симметрия, размеры и упругие свойства ароматических молекул 178
3. Энергия алифатических и ароматических колец...............183
4. Диамагнитная восприимчивость алифатических и ароматиче-
ских молекул....................................................188
Список литературы ........................................197
1. Теория ароматических соединений
а. Развитие теории строения бензола *. В 1825 г. Фарадей выделил
бензол из коксового газа. В 1834 г. Митчерлих получил бензол перегонкой
бензойной кислоты с известью. Эти исследователи не могли предвидеть, что
полученное ими вещество будет признано родоначальником наиболее много-
численного класса органических соединений. Быстрое развитие химии аро-
матических соединений в течение последующих десятилетий основывалось
на успехе первых попыток создания теории строения бензола.
В начале шестидесятых годов прошлого века органическая химия базирова-
лась на теории типов. Усилиями Франкланда, Вильямсона, Кекуле и других
были установлены правила валентности и структурные формулы, главным
образом в приложении к соединениям алифатического ряда. Однако суще-
ствовала тогда еще не очень большая группа «ароматических» соединений,
которые по составу и поведению отличались от алифатических соединений
и, казалось, представляли собой исключение из правил структуры и валент-
ности. В 1865 г. Кекуле показал, что эту аномалию можно устранить и пове-
дение ароматических соединений можно привести в соответствие с установлен-
ными правилами, если допустить, что все такого рода соединения содержат
общее «ядро», состоящее из шести углеродных атомов [2]. В развитие этой
гипотезы Кекуле предположил, что атомы углерода, образующие бензольное
кольцо, связаны между собой попеременно простыми и двойными связями,
причем каждый атом сохраняет по единице сродства. Он полагал, хотя и не
мог этого строго доказать, что все шесть положений в кольце эквивалентны.
Следующие десятилетия характеризовались интенсивным развитием основ
химии ароматических соединений. В 1874 г. Ладенбург доказал правиль-
ность постулированной Кекуле равноценности каждого из шести положений;
эти представления были уточнены и упрощены Вроблевским в 1878 г.
и Хюбнером в 1879 г. [3].
Работы Вроблевского остаются одним из лучших примеров методов и логики
определения структуры органического соединения. Он приготовил пять
теоретически возможных монобромбензойных кислот, чтобы выяснить разли-
чия между ними. Исходным веществом для синтезов служил тг-толуидин,
метильная группа которого в дальнейшем определяла положение карбоксиль-
ной группы. Метод Вроблевского заключался во введении брома непосред-
ственно или через нитрогруппу, а затем в использовании брома, нитрогруп-
пы (или продукта превращения последней, например аминогруппы) или иода
с целью блокирования одного или нескольких положений; одновременно
в другое место молекулы вводили бром или заместитель, который можно
заместить бромом, после чего все блокирующие группы заменялись на водо-
род. Таким образом было блокировано сначала одно положение, затем первое
и второе, далее первые два и третье и, наконец, первые три и четвертое.
Из пяти конечных продуктов две пары оказались идентичными. Ладенбург
до этого показал, что наличие двух пар эквивалентных положений для вто-
рого заместителя может служить строгим доказательством эквивалентности
всех шести положений для первого заместителя. Так, три оксибензойные
кислоты дают один и тот же фенол при декарбоксилировании и бензойную
кислоту при восстановлении, а фенол можно превратить через бромбензол
в бензойную кислоту. Таким образом было показано, что для первого заме-
стителя четыре положения эквивалентны. Далее было известно, что две
из оксибензойных кислот характеризуются тем, что каждая содержит гидро-
ксил в одном из двух эквивалентных положений. Эквивалентность для вто-
рого заместителя должна сохраниться, когда первый замещается на водород,
* Прекрасный обзор ранних теорий имеется в работе Лахмапа [1].
156
Ароматичность и связанные с ней физические свойства
и, таким образом, второй заместитель становится первым; этот результат
указывает на то, что пятое и шестое положения в бензоле эквивалентны четы-
рем остальным.
Именно в этот период была в принципе решена проблема определения отно-
сительных положений заместителей, хотя вначале и имелись неувязки.
Кекуле предположил, что все соединения, в которых заместители рассматри-
вались как находящиеся в таких же относительных положениях, как в стан-
дартном «орто-соединении», должны быть названы «орто-соединениями»
и т. д. Первоначально эти обозначения применялись без указания на поло-
жения, которым они в действительности соответствовали. Позднее было
найдено, что большинство так называемых орто-соединений содержит заме-
стители в 1,2-положениях, поэтому приставка «орто» была сохранена за 1,2-
производными бензола; аналогично обстояло дело и с другими приставками.
Предложенный принцип классификации отличался простотой. Сначала сле-
довало установить определенные стандарты, что можно было сделать лишь
в ограниченном числе благоприятных случаев. Так, Гребе в 1869 г. устано-
вил формулу нафталина и на этом основании сделал вывод, что фталевая
кислота является 1,2-производным бензола. Отсюда следовало, что такие
дизамещенные бензола, у которых заместители могут быть генетически
связаны с карбоксильными группами фталевой кислоты, являются 1,2-про-
изводными. Хотя этот принцип и прост, он часто оказывался явно неудовлет-
ворительным. Дело в том, что некоторые реакции, которые применялись
для установления генетической связи, особенно введение гидроксила путем
сплавления сульфокислот со щелочами и введение циангруппы действием
цианидов щелочных металлов на галогеннитросоединения, часто сопро-
вождаются перегруппировками (причины которых и в настоящее время не
вполне понятны). Такие осложнения было трудно установить и предотвратить.
В 1874 г. Грисс, Салковский и Кернер [4] независимо друг от друга предло-
жили надежный метод определения ориентации при замещении в бензольном
ядре. Он заключался в сравнении ди- и тризамещенных бензолов, а не только
дизамещенных. Так, например, из шести известных диаминобензойных кислот
при декарбоксилировании две давали один и тот же диаминобензол, три
другие — второй и, наконец, последняя — третий. Исходя из этого первый
должен быть орто-, второй — мета-, а третий — иара-фенилендиамином.
Изучение таких отношений не зависело от перегруппировок. Последующие
работы по определению положения заместителей в бензольном ядре базиро-
вались на этом методе.
Таким образом, в первое десятилетие после появления классической работы
Кекуле были решены проблемы симметрии и ориентации в бензольном ядре.
В этот же период началась разработка более существенной проблемы, кото-
рая на многие годы привлекла к себе внимание химиков. Она заключалась
в установлении характера и распределения тех шести углеродных валентно-
стей в бензоле, которые не требуются для сохранения кольца и удерживания
атомов водорода.
Распределение этих шести валентностей на три двойные связи, которое сделал
Кекуле в 1865 г., было предварительным, так как тогда еще не был решен
вопрос о симметрии бензольного кольца. В 1872 г. Кекуле [5] выдвинул
дополнительную гипотезу, согласно которой происходит осцилляция между
двумя возможными положениями двойных связей:
/Ч /\
I! I-IJ
157
Глава IV
Основанием для такой гипотезы послужило отмеченное уже Ладенбургом
отсутствие изомерии орто-производных бензола, допускаемой первоначаль-
ной формулой бензола по Кекуле. Вскоре стало ясно, что для объяснения
наблюдаемого числа изомеров необходимо учитывать дополнительную сим-
метрию, обусловленную осцилляцией.
Вслед за первоначальной теорией, но еще до ее уточнения были предприняты
две другие попытки объяснения симметрии бензольного кольца в соответствии
с наблюдаемым числом изомерных производных бензола. Так, Ладенбург [6]
в 1869 г. предложил призматическую формулу бензола, которая вполне
соответствовала числу существующих различных производных бензола.
Однако свойства симметрии замещаемых положений таковы, что требуют
следующей корреляции с простой кольцевой формулой (см. нумерацию поло-
жений на приведенной ниже схеме):
орто-Положения (6 пар) находятся на концах диагоналей плоскостей призмы,
лета-положения (2 тройки)—на углах плоскостей треугольников, а пара-
положения (3 пары) — на концах боковых ребер призмы. Таким образом,
в призматической формуле мета- и тгара-положения связаны непосредствен-
но, а орто-положения непосредственно не связаны между собой (в отличие
от простой кольцевой формулы). Это приводило к довольно громоздкой фор-
муле для нафталина.
В 1886 г. Байер начал ряд экспериментальных исследований, чтобы выяснить
распределение валентностей в бензоле. Метод Байера заключался в восста-
новлении производных бензола в производные циклогексадиена, циклогексе-
на и циклогексана, а затем в установлении строения продуктов восстановле-
ния обычными методами химии алифатических соединений [7]. Эта работа
опровергла призматическую формулу бензола. Из орто-, мета- и пара-
производных бензола были получены соответственно 1,2-, 1,3- и 1,4-произ-
водные циклогексана, а из этилового эфира 2,5-диокситерефталевой кислоты,
в котором все три пары иара-положений были фиксированы, получен после
восстановления этиловый эфир сукциноянтарной кислоты, имеющий все
заместители в тех положениях, в которых их следовало ожидать в соответ-
ствии с простой кольцевой формулой.
Другой ранней формулой бензола, находящейся в согласии с симметрией,
была диагональная формула Клауса [8], предложенная в 1867 г.
Первоначально Клаус допускал, что связи между орто- и между пара-
положениями идентичны, так что каждый атом углерода связан с тремя
другими совершенно одинаково. (Затруднение, связанное с необходимостью
привести допущение в согласие с эвклидовой геометрией, не играло суще-
ственной роли, так как в то время формула рассматривалась лишь как символ
химических отношений независимо от истинных геометрических характери-
158
Ароматичность и связанные с ней физические свойства
стик молекулы.) В 1882 г. Клаус предположил, что связи между zzapa-поло-
жениями коренным образом отличаются от всех известных типов связей.
Таким образом, использование для таких связей специального символа яви-
лось первым шагом на пути признания того факта, что существуют трудно-
сти, не разрешимые в рамках установившихся представлений [9].
По существу та же мысль вашла более ясное отражение в центрической
формуле бензола.
Впервые ее предложил Армстронг [10], затем она была принята Байером [И]
в качестве наилучшего символа, соответствующего его взглядам, и, наконец,
признана Клаусом как наиболее подходящее выражение его представлений
о строении бензола.
Первоначально центрическая формула отражала симметрию бензола, но
не давала возможности объяснить другие его свойства. Однако уже в 1891 г.
Бамбергер [12] использовал эту формулу при обсуждении вопроса о стабиль-
ности бензола. Бамбергер пытался установить связь между стабильностью
различных ароматических соединений и наличием центрированной шести-
валентной группировки, которая, как он предполагал, является существен-
ным фактором стабильности. По мнению Бамбергера, пиридин может образо-
вать шестивалентную группировку без использования валентностей атома
азота, участвующих в образовании солей, а пиррол может создать такую
группировку только в случае использования всех валентностей азота; учи-
тывая это, можно понять, почему пиридин имеет свойства третичного амина,
а пиррол практически не обладает основными свойствами.
Такое рассуждение совершенно правильно и соответствует фактическому
положению вещей. В настоящее время, чтобы выразить ту же мысль, следует
заменить термин «валентность» термином «электрон».
Из ранних попыток наиболее удачной была попытка объяснения стабильно-
сти ароматических соединений с удовлетворением условий симметрии, сде-
ланная Тиле [13] в 1898 г. Изучение ненасыщенных соединений привело
Тиле к допущению, что лабильные валентности двойных связей в принципе
могут делиться любым образом и что в большинстве случаев они частично
связаны и частично свободны. Тиле показал, каким образом в цепи чере-
дующихся простых и двойных связей возможность деления связей на части
может привести к их «сопряжению», т. е. к более равномерному распределе-
нию сродства по двойным связям и промежуточным простым связям, вслед-
ствие чего реакция протекает главным образом по концам цепи, где имеется
свободное некомпенсированное сродство. Таким образом было объяснено,
почему происходит присоединение к концам сопряженной ненасыщенной
системы в алифатическом ряду. Далее Тиле подчеркнул, что в кекулевской
структуре бензола сопряжение происходит по замкнутому кольцу; цепь
159
Глава IV
сопряжения не имеет концов, и дополнительная реакционная способность
бензола не может быть велика. Условие симметрии выполнялось при допу-
щении осцилляции по Кекуле и совершенно равномерного распределения
сродства, при котором все связи кольца эквивалентны.
Выдающееся значение этой теории заключалось в том, что в ней рассматри-
валась стабильность ароматических соединений на базе представлений,
сложившихся при изучении других классов соединений.
Структурная теория должна отражать основные общие черты ароматических
систем: их симметрию, стабильность и превращения. Центрическая формула
отражает симметрию, но не объясняет стабильности. Формула Тиле отражает
симметрию бензола и объясняет его устойчивость. В то же время рассмотре-
ние способов разрушения бензоидных систем приводит к признанию форму-
лы Кекуле, конечно, с учетом динамической гипотезы, позволяющей удовлет-
ворить требования симметрии. Превращения, которые вызывают разрушение
ароматической системы без разрыва кольца, протекают с образованием
продуктов присоединения {орто- или геара-хиноидных производных); все
подобные превращения более понятны, если их интерпретировать на основе
формулы Кекуле, а не какой-либо другой. Таким образом, последняя
доэлектронная теория строения бензола явилась продолжением динамической
гипотезы Кекуле. Вскоре после работ Кекуле Дьюар [14] предложил для
бензола «мостиковую» статическую формулу, однако она совершенно не
удовлетворяла требованию симметрии. В 1922 г. автор книги [15] обратил
внимание на эту формулу, рассматривая иара-связь как качественно сравни-
мую с реакционноспособным компонентом двойной связи; автор предложил
объединить дьюаровские формулы с кекулевскими (каждую с двумя или
тремя ориентациями, необходимыми для выполнения условий симметрии)
в более сложную динамическую систему
В этом предложении особое внимание уделялось превращениям ароматиче-
ских систем; главной целью привлечения двух типов структур была одновре-
менная интерпретация орто- и ияра-хиноидных превращений. Однако при
этом не рассматривали проблему стабильности, если не считать идеи, заклю-
чающейся в том, что достаточно быстро протекающее взаимопревращение
форм может обеспечить возможность реакции в любом направлении.
Таким образом, воззрения классической химии привели в тупик: условие
симметрии можно удовлетворить разными способами, но ни один из них
одновременно не мог удовлетворительно объяснить и стабильность, и пре-
вращения ароматических систем. В зависимости от того, какая из этих
характеристик взята в качестве исходной, можно следовать по одному из
двух указанных путей; исторически они привели к двум различным пред-
ставлениям: к теории Тиле и к расширенной динамической гипотезе. Десять
лет спустя после обоснования последней электронная теория химической
связи примирила эти казавшиеся далекими друг от друга представления,
160
Ароматичность и связанные с ней физические свойства
показав, что обе теории могут дополнять одна другую и отражать проявления
одних и тех же внутренних физических факторов.
б. Мезомерия в бензольном ядре. Для сохранения стройности изло-
жения здесь уместно напомнить об открытии электронной теории валент-
ности (1916 г.), о физической природе ковалентной связи (1927 г.), о мезомерии
как общем явлении (1926 г.) и о трактовке мезомерии на основе расширенной
интерпретации ковалентной связи (1929 г.), хотя обо всем этом говорилось
в гл. I и II.
Мезомерию бензола впервые детально и основательно рассмотрел Хюккель
[16] в 1931—1932 гг.; при изучении данной проблемы он применил два при-
ближенных формальных метода. Первый, называемый методом молекулярных
орбиталей, был разработан несколько раньше Леннард-Джонсом и в даль-
нейшем развит Малликеном и другими. В этом методе недооценивалась
ковалентная природа связи в молекулах, и по этой причине метод не очень
хорошо согласовывался с валентными представлениями. Второй метод, пред-
ложенный Хюккелем, известен под названием метода валентных схем. Он
приводил к другой крайности — переоценке ковалентного характера связи,
и именно по этой причине он был весьма удобен для интерпретации валентных
представлений. Технику этого метода упростили Полинг и Уэланд [17],
которые применили его при изучении бензола и других ароматических
углеводородов.
В ходе разработки этой проблемы Хюккель, а также Полинг и Уэланд исхо-
дили из допущения, что для образования молекулярной группы из шести
л-электронов каждый углеродный атом в бензоле предоставляет один атом-
ный р-электрон с осью симметрии, перпендикулярной плоскости молекулы.
Остальные электроны участвуют в образовании простых о-связей, состав-
ляющих правильный шестиугольник из шести СН-центров. Предполагают,
что каждый л-электрон движется в общем потенциальном поле шестиуголь-
ного «каркаса» и в выравненном поле остальных л-электронов. Нетрудно
показать, что плоскость атомных ядер является узловой плоскостью всех
л-электронов, а это ведет к распределению плотности электронов, имеющей
гексагональную симметрию, как показано на рис. 24.
Общей целью этих методов является расчет мезомерной или резонансной
энергии ароматической системы, т. е. оценка дополнительной стабильности,
вызываемой тем, что электроны не локализованы в трех статических двойных
связях, предусмотренных формулой Кекуле. Результат зависит от допуще-
ний, сделанных в отношении дополнительного движения, дозволенного для
этих электронов. В методе молекулярных орбиталей при вычислении орби-
талей, доступных одному из таких электронов при его движении среди ато-
мов в общем молекулярном поле, взаимодействием между л-злектронами
пренебрегают. Лишь затем учитывают принцип Паули, распределяя электро-
ны попарно по наиболее стабильным из рассчитанных орбиталей, однако
взаимодействие л-электронов не принимают во внимание ни на одной стадии
расчета. Может показаться удивительным, что такой расчет дает хороший
результат. Однако вычисленные энергии выражаются через особую единицу
энергии, обозначаемую Р, численное значение которой не рассчитывается.
В принципе Р представляет собой выигрыш энергии в результате того, что
один атомный р-электрон распространяет свое движение до соседней р-орби-
тали; на практике р — произвольная постоянная, при помощи которой
можно избавиться от многих погрешностей метода, подбирая ее таким обра-
зом, чтобы получить наилучшее совпадение с экспериментальными данными.
Вычисленные энергии для бензола, нафталина, антрацена и фенантрена
соответственно равны 2,00р, 3,68р, 5,32р и 5,45р. Эти коэффициенты при-
1 1—0772
161
Глава IV
близительно правильно передают наблюдаемые соотношения. Если 0 считать
равной 20 ккал/моль (82,74-103 Дж/моль), то получается, как показывает
Рис. 24. Распределение заряда л-электронной оболочки в бензоле.
табл. 47, неплохое соответствие термохимических и вычисленных энергий
мезомерии. При таком сопоставлении следует помнить, что так называемые
Таблица 47
Рассчитанные энергии мезомерии [ккал/моль (4187-103 Дж/моль)]
ароматических углеводородов; сравнение с термохимическими данными
Соединение Наблюдаемые Рассчитанные
аддитивная схема гидрирование метод моле- кулярных орбиталей (Р = 20) метод валент- ных схем (а = 39)
Бензол 37 36 40 39
Нафталин 75 74 71
Антрацен 105 106 108
Фенантрен 110 109 110
«наблюдаемые» энергии мезомерии связаны с некоторыми факторами произ-
вольного характера (гл. III, разд. 2,6).
В методе валентных схем принцип Паули и взаимодействие электронов
учитываются с самого начала. Предполагается, что электроны остаются
в виде трех пар со спаренными спинами, никогда не принадлежащих (по
интерпретации Хюккеля или Полинга и Уэланда) менее чем двум атомам.
Они действительно представляют собой связи, характеризующиеся опре-
деленными значениями внутренней энергии и взаимным отталкиванием,
присущим двухэлектронным связям; но они являются подвижными связями,
162
Ароматичность и связанные с ней физические свойства
непрерывно перемещающимися между определенными положениями, на
которые указывалось еще в ранних динамических представлениях о структу-
ре бензола. Для установления пределов подвижности подвижных связей
оказалось целесообразным учитывать не только структуры Кекуле, но
и структуры Дьюара; все остальные структуры исключаются, ибо связи
пересекаться не могут *. Следовательно, волновая функция М нормального
мезомерного состояния определяется соответствующими двумя кекулевскими
и тремя дыоаровскими структурами (К и D), отвечающими требованию
симметрии. Коэффициенты Полинга и Уэланда для этих функций определяют-
ся следующим уравнением: М =0,625 (Ki -\-К2) + 0,271 (Z^ -}-D2 + А).
Эти коэффициенты показывают, что структуры Кекуле имеют большее значе-
ние, чем структуры Дьюара, но все же последними нельзя пренебречь при
характеристике мезомерного состояния. На долю структур Дьюара прихо-
дится около 20% вычисленной энергии мезомерии. Энергия мезомерии
выражается в единицах энергии, обозначаемых а; в принципе а представляет
собой выигрыш энергии в результате обмена двух соседних атомных р-
электронов, как и при образовании л-составляющей статической двойной
связи. На практике а — произвольная постоянная, подбираемая для наилуч-
шего совпадения рассчитанных значений с экспериментальными данными.
Вычисленные энергии для бензола, нафталина, антрацена и фенантрена
соответственно равны 1,11а, 2,04а, 3,09а и 3,15а, причем при расчете послед-
них двух величин допущены более грубые приближения. Приведенные выше
коэффициенты также приблизительно правильно передают истинные соотно-
шения. Из данных табл. 47 видно, что наблюдается неплохое совпадение
рассчитанных значений с экспериментальными данными при а, равном
35 ккал/моль (146,54-Ю3 Дж/моль). В данном случае также создается поло-
жение, при котором не только рассчитанные величины существенно зависят
от эмпирических данных, но и наблюдаемые величины частично зависят
от принимаемой интерпретации.
Метод валентных схем был значительно усовершенствован, однако еще не
удалось избежать применения произвольных постоянных (неэмпирический
расчет а чрезвычайно затруднителен). Главным несоответствием в ранних
расчетах по этому методу было то, что оценка мезомерного состояния дела-
лась с учетом только ковалентных структур, тогда как при рассмотрении
необходимо учитывать и диполярные структуры; как правило, чем более
полно учтены такие структуры, тем лучший результат должен получиться.
Возможны два типа однократно диполярных структур с зарядами на соседних
атомах: А (12 структур) и В (12 структур). Существуют три типа с зарядами
не на соседних атомах: Р (6 структур); Q (24 структуры) и R (6 структур),
а также двукратно и трехкратно диполярные структуры. Ниже эти типы
структур приводятся в порядке уменьшения их значения.
+ + +
Включение таких полярных структур при расчете по методу валентных схем
было начато Скларом [18] и развито Крейгом [19], который решил эту пробле-
* Точнее, в результате пересечения получаются такие схемы взаимного насыщения
спинов, которые уже учитывались в структурах без пересечения.
и*
Глава IV
му, дополнив пять структур типа К и D двадцатью четырьмя структура-
ми А и В.
Волновая функция бензола в нормальном мезомерном состоянии имеет,
по Крейгу, следующий вид:
М = 0,39 (Kt + К2) + 0,17 (Di + D2 + D3) + 0,07 (Л + Л2 + . .. Л12) +
—/—0,03 (BiA-B^A-... Bf%)
Вычисленная энергия оказывается на 17 ккал/моль (71,18-103 Дж/моль)
меньше энергии нормального мезомерного состояния по Полингу и Уэланду.
Это показывает, что расчет, основанный только на неполярных структурах,
связан с довольно значительными погрешностями. Кроме того, становится
ясным, что дальнейшие уточнения изменили бы энергию всего лишь на не-
сколько килокалорий на моль. В настоящее время близкого совпадения
с опытом можно добиться только при использовании экспериментальных
данных.
Метод молекулярных орбиталей развивался путями, представляющими
теоретический интерес, поскольку он дает в принципе возможность неэмпи-
рического расчета энергий л-электронов. В настоящее время точность метода
еще недостаточна для того, чтобы получить количественное совпадение
с экспериментальными данными; однако для бензола этот метод все-таки
дал качественно удовлетворительную картину энергетических уровней
и оказался полезным для характеристики некоторых возбужденных электрон-
ных состояний, обнаруженных спектроскопически. Первые шаги в этом
направлении были сделаны Гопперт-Майером и Скларом [20]. Сначала они
образовали из произведений волновых функций, рассчитанных с помощью
обычного метода молекулярных орбиталей, «антисимметричную» линейную
комбинацию. Это соответствовало ограничению, согласно которому у одного
атома не может быть более двух л-электронов, и тем самым удовлетворяло
запрету Паули для атомов. Затем, распределив электроны попарно по трем
наинизшим орбиталям, они придавали волновой функции в соответствии
с принципом Паули полностью антисимметричный характер и вычисляли
энергию взаимного отталкивания л-электронов, усреднив ее по всем орбита-
лям. Склар и Лиддейн [21] и Парр и Кроуфорд [22] теоретически вычислили
необходимые для расчета энергетические термы. Следующий шаг, сделанный
Крейгом [23], основывался на допущении, что энергия взаимодействия
л-электронов вызывает изменение их движения; отсюда следовало, что было
бы неправильным усреднять энергию по невозмущенным орбиталям. Действи-
тельно, л-электронные пары не локализованы строго в трех антисимметрич-
ных орбиталях и способны двигаться в пределах всех орбиталей, а это при-
водит к изменению общей электронной волновой функции, подобно тому как
это происходит в каждой резонансной системе, до тех пор пока энергия не
станет минимальной. В принципе данный метод, применимый для «конфигу-
рационного взаимодействия» и впервые предложенный для молекулярных
систем Крейгом, передает молекулярные волновые функции не хуже любого
иного метода, исходящего только из р-атомных волновых функций.
Легко понять, почему, несмотря на мезомерию ароматического ядра, валент-
ные формулы, особенно сравнительно устойчивые структуры Кекуле, ока-
зываются полезными для понимания превращений ароматического ядра,
почему, например, имеет смысл вводить изогнутые стрелки в формулы Кеку-
ле, хотя такие формулы сами по себе не могут объяснить устойчивости аро-
матического ядра. Если в химическом превращении, затрагивающем арома-
тическое ядро, или в переходном состоянии такого превращения электрон-
ные пары рассматриваются как преимущественно локализованные, то удобно
164
Ароматичность и связанные с ней физические свойства
при трактовке механизма реакции рассматривать эти электронные пары
так, как будто они только что смещены до локализованных положений.
Такое представление возможно, если расчленить общее превращение на сле-
дующие два накладывающихся друг на друга процесса: 1) возбуждение
нормального мезомерного состояния до валентной структуры и 2) обычная
химическая реакция этой валентной структуры. Составляющие процессы,
как и процессы разрыва и образования связей в бимолекулярных реакциях
замещения в насыщенных соединениях (гл. II, разд. 2,г), следует рассма-
тривать как протекающие одновременно, а не последовательно; при этом
не требуется вся энергия мезомерии для завершения процесса 1 до начала
процесса 2. Однако энергия активации любого процесса, который оконча-
тельно или временно разрушает мезомерную систему, возрастает вследствие
мезомерии, хотя бы и не на полную величину энергии мезомерии. Таким
образом, теория мезомерии удовлетворяет трем главным требованиям пробле-
мы ароматического ядра, а именно объясняет его симметрию, устойчивость
и превращения.
Теория строения бензола, соответствующим образом модифицированная,
служит основой для качественных выводов о строении пиридина. Однако
удовлетворительный количественный расчет энергетических состояний
пиридина в настоящее время невозможен, так как из-за наличия гетероатома
необходимо вычислять новые неизвестные интегралы взаимодействий.
Наблюдения показывают, что введение гетероатома мало изменяет размер
и форму молекулы (разд. 2,6). Другие свойства, особенно энергия мезо-
мерии (разд. 3,6) и экзальтация диамагнитной восприимчивости (разд. 4.д),
которые обусловлены делокализацией л-электронов, также изменяются
незначительно. Однако гетероатом создает дипольный момент [у пиридина
р. = —2,26Д (—7,46-10~30 Кл-м)], отрицательный знак которого означает,
что в соответствии с ожидаемым эффектом электроны смещены по направле-
нию к более электроотрицательному атому азота [24]. Основываясь на про-
веденном в гл. III, разд. 1,в и г анализе дипольных моментов простых
алкиламинов и учитывая влияние удлинения углеродной цепи, можно было
бы ожидать, что дипольный момент пиридина, обусловленный лишь влия-
нием электроотрицательности азота на о-электроны, будет составлять только
около —1,0Д (—3,3-10~30 Кл-м). Необходимо, таким образом, предполо-
жить, что дополнительный момент, равный —1,2Д (—3,96-10“30 Кл-м),
возникает вследствие смещения электронов л-системы; в среднем это смеще-
ние составляет 0,04 А (4-10~3 нм), т. е. в несколько раз превышает среднее
смещение о-электронов. Дипольный момент хинолина равен —2,29Д
(—7,56-10-30 Кл-м), а момент изохинолина составляет —2,73Д
(—9,01 -10“30 Кл-м) [24]. Эти моменты также обусловлены электроотрица-
тельностью атома азота, влияние которой в изохинолине распространяется
на электроны более удаленного кольца.
в. Конденсированные полициклические бензоидные системы. При пере-
ходе от бензола к нафталину л-электронная система делается менее симме-
тричной и проблема становится значительно более сложной. В соответствии
с двумя структурами Кекуле для бензола можно написать подобные три
формулы нафталина: одну с центром симметрии (А) и две без него, отличаю-
щихся друг от друга лишь положением кратных связей и Бг).
/'\/\ /К/\
I II I I HI III I
Ч/\/ Х/Ч/ \z\z
A Bi в2
165
Глава IV
Возможны также 39 неполярных структур с трансаннулярными связями
и значительно большее число полярных структур. В целом система настоль-
ко сложна, что возникают сомнения в полезности ее рассмотрения с помощью
крайне грубых упрощений. Наиболее серьезным упрощением является
исключение из рассмотрения всех «мостиковых» и полярных структур,
несмотря на их многочисленность. Но и при этом остается проблема количе-
ственной оценки вкладов структур А и Б в смешанной волновой функции;
однако после всех допущений легко сделать следующий шаг и принять,
что вклады их одинаковы. При этом можно прийти к интересному заклю-
чению, что кратность 1,2-связей нафталина равна 8/3 и превышает кратность
всех остальных связей (4/3). Отсюда следует, что 1,2-связи нафталина более
похожи на двойные, чем связи в бензоле, а также что в целом 1,2,3,4-система
нафталина в известной мере подобна бутадиену-1,3 и, возможно, еще более
близка 1,4-дифенилбутадиену-1,3.
4/3 5/3
Эти выводы во многом подтверждаются известными фактами. Нафтол-2
галогенируется и сочетается в положение 1, т. е. его фенольная система
более похожа на енольную систему с двойной связью в положении 1,2,
нежели на систему с двойной связью в положении 2,3. Нафталин присоеди-
няет хлор труднее, чем олефины, но значительно легче, чем бензол *. Нафта-
лин восстанавливается натрием в спирте точно так же, как и 1,4-дифенил-
бутадиен-1,3; при этом присоединяются два атома водорода в положения 1,4,
а двойная связь в положении 2,3 не затрагивается. Геометрические доказа-
тельства подобного распределения кратностей связей даны ниже.
Фенантрен представляет собой другой пример, когда такие простые рассуж-
дения приводят к химически правильным результатам. Для фенантрена
возможны пять структур типа кекулевских четырех разных типов, причем
две из них различаются только ориентацией. Если веса всех структур при-
нять равными, то кратность связей окажется распределенной следующим
образом:
9,10-Связь имеет кратность 9/5 и, следовательно, должна быть очень близка
к двойной. Неугловые связи боковых колец, как и в нафталине, имеют
чередующуюся кратность, хотя различия здесь менее резко выражены.
Заключение, сделанное в отношение 9,10-связи, находится в полном согла-
сии с химическим опытом. Фенантрен присоединяет бром в положения 9, 10.
Он легко восстанавливается, давая 9,10-дигидропроизводное и продукты
дальнейшего гидрирования, и легко окисляется в положении 9,10, образуя
фенантренхинон и дифеновую кислоту. Подобное химическое поведение
9,10-связи очень похоже на поведение двойной связи стильбена.
* В более старой литературе сообщалось, что нафталин образует дихлорид, однако
попытки повторить этот синтез к успеху не привели. На самом деле нафталин дает несколь-
ко стереомерных тетрахлоридов (гл. XIII).
166
Ароматичность и связанные с ней физические свойства
В случае антрацена наблюдается противоположная картина. Если вывести
кратности связей для четырех кекулевских структур двух типов, каждый
из которых имеет две ориентации, и допустить равные веса всех структур,
то можно получить следующий результат:
Приведенные данные свидетельствуют о том, что способность к присоеди-
нению в боковых кольцах, в которых кратности связей изменяются более
резко, чем в нафталине, должна полностью перекрыть реакционную способ-
ность центрального кольца, в котором связи СН-групп такие же, как в бен-
золе.
Это не подтверждается опытом. Антрацен восстанавливается амальгамой
натрия с образованием 9,10-дигидроантрацена. Хлор, бром и нитрующие
агенты замещают в антрацене водороды последовательно в положении 9 и 10.
Однако при нитровании в уксусной кислоте можно выделить 9-нитро-
9,10-дигидроанатрил-10-ацетат, который в дальнейшем легко превращается
в 9-нитроантрацен. Антранол-9 и антрон-9 можно разделить, но они тауто-
мерии и легко превращаются один в другой, так же как и 9,10-антрагидро-
хинон и 9-оксиантрон-10.
Таким образом, хотелось бы иметь тест, позволяющий определить, насколь-
ко качественно применимо простое приближение, при котором в одинаковой
степени смешиваются только структуры Кекуле. Однако, проводя этот тест
и заранее зная, что теория несостоятельна, необходимо также знать, насколь-
ко заслуживают доверия опытные данные. Указанное приближение нельзя
применять в качестве основы для теоретического рассмотрения опытных
данных, хотя в некоторых случаях это было сделано. Были предприняты
попытки рассчитать распределение электронов в полициклических арома-
тических системах. Иногда такие расчеты заманчивы, но ни одному из них
нельзя полностью доверять, так как допущения, принятые в приближенных
методах расчета, почти всегда по порядку величины сравнимы с определяе-
мыми величинами.
Можно ожидать, что в общем взаимоотношение между структурами нафта-
лина и хинолина или изохинолина будет таким же, как и между структурами
бензола и пиридина, описанными в конце разд. 1,6.
г. Небензоидные замкнуто-сопряженные системы *. Уже в первых
работах по применению теории молекулярных орбиталей к бензолу Хюк-
кель [16] показал, что при последовательном заполнении молекулярных
л-орбиталей циклической системы вырожденными электронными парами
вначале заполняется одна из орбиталей, а затем орбитали с более высокой
энергией. Другими словами, Хюккель высказал идею о молекулярных
электронных уровнях, аналогичных электронным уровням в атомах. Запол-
ненные оболочки могут содержать два л-электрона (этилен), шесть л-элек-
тронов (бензол) и в общем 4п + 2 электронов, однако четыре (циклобута-
диен), восемь (циклооктатетраен) или в общем 4п л-электронов не образуют
заполненных оболочек. Таким образом, бензольное кольцо, содержащее
шесть л-электронов (ароматический секстет), является представителем замк-
нутой л-электронной системы, стабилизированной за счет делокализации
двойных связей. Идея о стабильности ансамбля из шести связывающих элек-
* Более полно этот вопрос изложен в гл. 1 монографии Крейга [25].
167
Глава IV
тронов (связывающий секстет), отражающая тенденцию системы к построе-
нию шестиэлектронной ячейки, сразу же была принята, так же как ранее
было принято эмпирическое предположение Бамбергера (разд. 1,а), посколь-
ку на ее основе можно было объяснить ароматичность пятичлеиных гетеро-
циклов. Идея Хюккеля очень рациональна.
Правило Хюккеля (4п + 2) применимо только к моноциклическим л-элек-
тронным системам, хотя некоторые авторы применяли его к полицикличе-
ским системам, к которым оно может не иметь никакого отношения. Напри-
мер, это правило не объясняет стабильности аценафтилена, который имеет
12 л-электронов. В ряду моноциклических соединений это правило приме-
нимо вплоть до восьмичленного кольца; учитывая простоту теории, из кото-
рой выведено правило Хюккеля, вероятно, не следует ожидать от него
чего-то большего. Циклобутадиен настолько неустойчив, что не может
храниться. Циклооктатетраен может храниться, но является очень реак-
ционноспособным неплоским полиолефином. Моноциклические сопряженные
соединения от десятичленного и выше недостаточно изучены; вероятно,
правило Хюккеля для таких циклов не является главным фактором, обуслов-
ливающим их стабильность или неустойчивость.
Хюккелевская классификация л-электронных систем основывается на эле-
ментарной теории молекулярных орбиталей, которая не учитывает оттал-
кивания между электронами. Пытаясь учесть этот эффект, Коулсон и Раш-
брук пришли к другой полезной классификации, согласно которой л-элек-
тронные системы делятся на «альтернантные» (сокращенно «альт.»), если
спиновые метки (а и 0) л-электронной системы непрерывно чередуются,
и «неальтернантные» («неа.») в противном случае. Смысл этой классификации
состоит в том, что в неальтернантных углеводородах распределение л-элек-
тронов между л-центрами является нечетным, причем заряд на одном конце
связи увеличивается за счет уменьшения заряда на другом конце. Так,
в азулене л-электроны не только смещены от семичленного цикла к пяти-
членному (если представить, что вначале каждый цикл имел по шесть л-элек-
тронов), обусловливая появление дипольного момента, но также, согласно
расчетам, в каждом кольце они сконцентрированы у чередующихся атомов.
Можно ожидать, что такая концентрация зарядов на некоторых л-центрах
будет мешать делокализации; однако этот эффект невелик. В настоящее
время известно много неальтернантных молекул. Энергия мезомерии этих
молекул понижена не сильно. Альтернантность, вообще говоря, не связана
с ароматичностью.
Примеры такой классификации, а также классификации по другому призна-
ку, описываемой ниже, приведены на схеме на стр. 169.
Расчеты по методу молекулярных орбиталей и по методу валентных схем,
выполненные Хюккелем, в случае бензола приводят к достоверным резуль-
татам, однако в случае 4п-систем с незаполненными оболочками получаются
неверные данные. Так, согласно расчету по методу валентных схем, цикло-
бутадиен и циклооктатетраен должны обладать большой энергией мезоме-
рии. При улучшении метода расчета это несоответствие с опытными данными
должно уменьшаться, что уже было показано для случая бензола (разд. 1.6).
Крейг исследовал факторы, обусловливающие наблюдаемые отклонения,
и показал, что если проводить расчеты по методу молекулярных орбиталей
или валентных схем, не делая тех приближений, которые допускаются
в простейших методах, то энергия мезомерии для циклобутена и цикло-
октатетраена получается очень низкой. Хотя такие расчеты возможны только
в простейших случаях, Крейг нашел более общий критерий, по которому
можно распознать системы с низкой энергией мезомерии (и таким образом
168
Ароматичность и связанные с ней физические свойства
а
Циклобутадиен,
^незаполненная
оболочка, альт.,псев.
Бензол,
6=заполненная
оболочка, альт.,аром.
Циклооктатетраен,
8=незаполненная
оболочка, альт.,псев.
Пентален,
8, неа., псев.
Азулен,
10, неа., аром.
Гептален,
12, неа,, псев.
£
аJl#а
р р р
Бензодипентадиен,
12, неа., псев.
классифицировать л-электронные системы) и который предназначен специ-
ально для определения степени ароматичности. Этот критерий основан
на свойствах симметрии.
Критерий Крейга применим к молекулам, имеющим по крайней мере одну
ось симметрии второго порядка, проходящую не меньше чем через два л-элек-
тронных центра. Это правило можно проследить с помощью любой из приве-
денных выше диаграмм. Вначале пометим спины в молекуле, как описано
выше, с тем лишь дополнительным условием (выполненным на диаграммах),
что каждое неизбежное нарушение альтернантности должно происходить
на простой связи в структуре кекулевского типа. Затем выберем какую-
нибудь ось второго порядка, проходящую через атомы (вертикаль или
горизонталь на диаграммах), и мысленно перевернем молекулу, вращая
ее вокруг этой оси. Теперь подсчитаем количество л-центров, поменяв-
шихся местами друг с другом, и количество таких взаимопревращений,
которые привели к перемене спиновой метки. Если сумма этих двух чисел
нечетная, то молекула классифицируется как «псевдоароматическая» («псев.»).
Если сумма четная, то молекула является «ароматической» («аром.»). Эта
классификация не имеет никакого отношения к числу л-электронов или
к числу колец. Однако все моноциклические системы класса 4п (по Хюккелю)
являются псевдоароматическими.
Суть описанного теста состоит в выяснении вопроса, имеет ли многоэлек-
тронпая волновая функция основного состояния более низкую симметрию,
чем ядерный остов, или нет. Если имеет, то по аналогии с появлением узлов
в орбиталях следует ожидать, что более низкая симметрия будет препят-
ствовать общей делокализации л-электронов и таким образом приведет
к уменьшению энергии мезомерии. Можно придумать большое число молекул
с несимметричными основными состояниями, однако, за исключением цикло-
октатетраена, все они крайне неустойчивы или вообще не получены, несмотря
на ряд попыток их синтезировать. Все такие молекулы не обладают арома-
169
Глава IV
тической стабильностью и могут быть названы удачным термином «псевдо-
ароматические». С помощью простейших методов молекулярных орбиталей
и валентных схем основным состояниям этих молекул могла бы быть приписа-
на различная симметрия, и, таким образом, нельзя было бы ожидать соот-
ветствия рассчитанных энергий. Согласно Крейгу, главная цель состоит
в том, чтобы сделать расчет возможно менее эмпирическим, т. е. менее зави-
сящим от численных данных, полученных опытным путем на других молеку-
лах, которые (при расчете псевдоароматических молекул) всегда отличаются
по симметрии от истинно ароматических молекул.
Тест на симметрию применим только в том случае, когда молекула симме-
трична. Он не может быть применен, например, к фенантрену — типично
ароматическому соединению, к стабильному, не обладающему свойствами
олефина дифенилену или к типичному диолефину, дибензопенталену. Ни одна
из этих молекул не имеет оси симметрии второго порядка, проходящей через
атомы углерода. Последние два соединения изображены ниже *.
Дифенилен
(12, альт.)
Д ибензопентален
{16, неа.)
В этих случаях можно лишь попытаться оценить стабильность более простых
и более симметричных «родственных» соединений. Можно полагать, что
устойчивость двух приведенных выше соединений обусловлена бензольными
кольцами, т. е. что бензольные кольца не сильно сопряжены друг с другом.
В случае дифенилена важным дестабилизирующим фактором является
«напряжение» кольца; более подробно этот фактор будет рассмотрен в разд.
3,а. Напряжение в принципе может существовать во всех кольцах,
включая л-электронные системы. Преобладающим компонентом является
напряжение изгиба связей, которое в кольцевых сопряженных системах
максимально для четырехчленного цикла. Иногда важную роль играет
трансаннулярное несвязывающее взаимодействие между атомами кольца;
это взаимодействие максимально проявляется также в случае четырехчлен-
ного цикла. Третий фактор, имеющий значение только в случае больших
колец,— это трансаннулярное несвязывающее взаимодействие между атома-
ми, связанными с кольцом; этот фактор проявляется во внутрикольцевом
взаимодействии атомов в циклах определенных размеров.
Теперь кратко остановимся на экспериментальных данных для ряда соеди-
нений, теоретически рассмотренных выше.
* Приведенные простые кекулевские структуры, вероятно, наиболее важны, так
как они лучше всего отражают внутренние углы небензоидных колец. (Угол между двумя
простыми связями, не связанными с двойной связью, меньше, чем угол между простой
и двойной связями.) Миллс и Никсон [26] доказали это тем, что инданол и тетралол всту-
пают в реакцию азосочетания по енольной группировке (показано стрелками).
Н0\/ч/СНг\
II I /сн
но / /СН2
у у хсн2
сн2
170
Ароматичность и связанные с ней физические свойства
Циклобутадиен в свободном состоянии получен не был, однако он существует
в виде некоторых производных, а именно в виде комплексов с металлами,
например (G4H4)Fe(GO)3. При попытке выделить углеводород были получены
только продукты полимеризации или присоединения [27]. Эти реакции
полностью стереоспецифичны, откуда следует, что основное состояние цикло-
бутадиена синглетно, т. е. это соединение не является бирадикалом.
Циклооктатетраен впервые был получен Виллыптеттером [28] из псевдо-
пельтьерина (см. ниже) путем удаления азотного мостика с помощью двух-
стадийного исчерпывающего метилирования. Циклооктатетраен — высоко-
реакционноспособный полиолефин, обладающий свойствами, удивлявшими
всех. Поэтому работа Виллыптеттера была повторена и, чему никогда не будет
оправдания, поставлена под сомнение (обзор см. [29]). Впоследствии Реп-
пе [30] получил это соединение полимеризацией ацетилена в присутствии
цианида никеля в тетрагидрофуране; этот метод позволил получать цикло-
октатетраен в больших количествах. Реппе подтвердил данные Виллыптет-
тера; его работы и более поздние работы Коупа и сотр. значительно расши-
рили наши знания о физических свойствах и химических превращениях
циклооктатетраена [31]. И Коуп и другие авторы, чтобы установить истину,
получили это вещество также и методом Виллыптеттера [32].
СН2-СН---СН2
III г А=/
СН2 NCH3 СО \ ~7
I I I \__________________/
сн2-сн—сн2 • -—
Псевдопельтьерин Циклооктатетраен
Циклооктатетраен имеет форму седла, построенного из четырех' плоских
почти ненапряженных этиленовых фрагментов, л-орбитальное перекрывание
между которыми очень слабо выражено [33]. Благодаря такому строению
молекула не напряжена; при переходе молекулы в плоское состояние типа
бензола потеря ненапряженности ничем не компенсируется, так как энергия
мезомерии в несимметричном основном состоянии л-электронной системы
плоской молекулы мала. Как показывает рентгеноструктурное исследова-
ние монокарбоновой кислоты [34], связи GG кольца поочередно имеют длину
1,322 и 1,470 А (13,22-Ю-2 и 14,70-1СН2 нм), ошибка составляет ±0,005 А
(5-10-4 нм). Углы между связями в кольце равны 126,4 ± 0,4° (2,206 ±
± 0,0070 рад). Частота валентных колебаний группы G = G (1639 см~1)
соответствует изолированным, т. е. несопряженным двойным связям [35].
Экзальтация рефракции очень мала [30]. Кроме того, не наблюдается экзаль-
тации диамагнитной восприимчивости [36], наличие которой служит тестом
на делокализацию электронов в цикле (разд. 4,д). Теплоты образования
и реакций соответствуют очень низкой энергии мезомерии [37], даже мень-
шей, чем ожидаемая энергия мезомерии нециклического сопряженного тетра-
ена. Циклооктатетраен представляет собой желтую жидкость, однако наблю-
даемая окраска обусловлена затяжкой колебательного «хвоста» абсорбцион-
ной полосы, максимум которой расположен в ультрафиолетовой области,
и вторжением ее в видимую область спектра.
Циклооктатетраен при нагревании изомеризуется до стирола и вообще при
действии многих реагентов превращается в производные бензола. Например,
при реакции с окисью хрома образуется терефталевая кислота *. Цикло-
* Обзор химических свойств циклооктатетраена см. [38].
171
Глава IV
октатетраен каталитически восстанавливается до циклооктана. Он также
восстанавливается щелочными металлами и присоединяет галогеноводороды
и галогены. Виллыптеттер отметил, что при присоединении образуются
мостики в цикле. Некоторые из этих реакций были тщательно исследованы
Реппе и Коупом (см. [39], а также гл. XI, разд. 3). Явление интрааннуляр-
ной валентной таутомерии рассмотрено в гл. XI, разд. 3,г. Анет с сотр.
и Робертс с сотр. [40] с помощью ядерного магнитного резонанса показали,
что в самом циклооктатетраене и во многих его замещенных, например
во фтор- и дейтерокарбэтоксипроизводных, двойные связи движутся по коль-
цу; поскольку при этом происходит деформация молекулы, то возникает
барьер активации, препятствующий миграции связей.
Энергия активации такого процесса для самого циклооктатетраена состав-
ляет 13,7 ккал/моль (57,36-103 Дж/моль), время жизни наиболее стабильных
конформаций при 0 °C исчисляется сантисекундами. У некоторых замещен-
ных циклооктатетраенов энергетический барьер выше, и время жизни инди-
видуальных конформеров при 0 °C составляет несколько секунд.
Зондхеймер и сотр. [41] методами окислительной циклизации или цикло-
поликонденсации алкен-а,(о-диинов получили ряд больших сопряженных
циклов. Наиболее известны соединения Ci4H14 и С18Н18, названные автора-
ми [14]- и [18]-аннуленами. Эти углеводороды относятся к типу 4и -f- 2.
Спектры ЯМР этих соединений показывают, что они могут принимать пло-
скую конформацию. В случае [14]-аннулена плоская конформация устой-
чива при —60 °C, а в случае [18]-аннулена — при комнатной температуре.
При повышении температуры для [14]-аннулена до комнатной, а для [18]-
аннулена до 60 °C плоские конформации становятся нестабильными. Этот
факт, а также большую устойчивость плоской конформации [18]-аннулена
можно объяснить взаимным отталкиванием внутренних атомов водорода,
которое проявляется сильнее в случае [18]-аннулена. Энергия мезомерии
[18]-аннулена, определенная по теплоте сгорания, составляет 100 ккал/моль
(418,68-103 Дж/моль). [24]-Аннулен относится к типу 4и, но [30]-аннулен —
к типу 4и 2. Оба они неустойчивы. Вероятно, в случае таких неконцен-
трированных л-электронных систем, к которым принадлежат эти макроцик-
лы, правило Хюккеля становится малопригодным.
Пентален и бензодипентадиен до сих пор не получены, несмотря на множе-
ство попыток, однако гептален был синтезирован и представляет собой
окрашенный неустойчивый легко полимеризующийся полиолефин [42].
Азулен хорошо известен как родоначальник большой группы веществ,
широко распространенных в природных эфирных маслах. В твердом виде
азулен синий. Данные рентгеноструктурного анализа показывают, что его
172
Ароматичность и связанные с ней физические свойства
молекулы являются плоскими;
длины связей (А) приведены ниже:
Средняя длина связи, как и в бензоле, составляет 1,40 А (14,0-Ю-2 нм),
но мостиковая связь, которая в обеих кекулевских структурах является
простой, значительно длиннее [1,48 А (14,8-10“2 нм)]. Дипольный момент
азулена равен 1,0 Д (3,3-10-30 Кл-м); данные для 2-хлор-, 2-бром- и 2-циан-
азуленов показывают, что в азулене электроны смещены от семичленного
кольца к пятичленному. Для азулена обнаружена экзальтация диамагнит-
ной восприимчивости почти такая же, как для изомерного ему нафталина
(разд. 4,в) [44]. Это означает, что электроны делокализованы в циклах азулена
и нафталина почти одинаково. Азулен имеет высокую, хотя и не вполне
точно известную, энергию мезомерии. Ее оценивают на 30 ккал/моль
(125,6-103 Дж/моль) меньше, чем энергию мезомерии нафталина. Если при-
нять для нафталина величину Полинга, то энергия мезомерии азулена будет
составлять 45 ккал/моль (188,41-103 Дж/моль) [45]. Однако независимое
определение теплоты гидрирования дает величину 31 ккал/моль (129,79-
-103 Дж/моль) [37]. Вероятно, можно считать, что неисправленная величина
энергии мезомерии азулена составляет примерно половину энергии мезо-
мерии нафталина. Для получения правильного результата эту величину
необходимо уменьшить примерно на 10 ккал/моль (41,87-103 Дж/моль),
чтобы таким образом учесть напряжение в кольце.
Азулен вступает в большинство типичных реакций электрофильного арома-
тического замещения: ацилирование по Фриделю — Крафтсу, нитрование,
сульфирование и азосочетание, однако для этих синтезов необходимо тща-
тельно подбирать условия, так как сильные реагенты могут разрушить
молекулу [46]. Замещение идет преимущественно в положения 1 и 3, т. е., как
и следовало ожидать, в пятичленный цикл, который, как мы знаем, имеет
частичный отрицательный заряд. Это подтверждается расчетом, согласно
которому наибольшей плотностью л-электронов обладает пятичленное кольцо.
Физические свойства аценафтилена еще мало изучены. Известно, что это
очень устойчивый углеводород, образующийся при многих высокотемпера-
турных реакциях, включая пиролитическое дегидрирование дигидропроиз-
водного (с мостиком СН2 — СН2) при 700 °C. Это соединение можно также
окислить до аценафтилена мягкими окислителями. Аценафтилен образуется
при щелочном десульфировании сульфо- и дисульфокислот аценафтена,
при этом происходит необычная реакция отщепления водорода. Мостик
— СН = СН — в аценафтилене очень похож на аналогичный мостик в фенан-
трене, по которому главным образом идут реакции присоединения, например
галогенов; продукты замещения образуются только в результате последую-
щего элиминирования. При действии сильных окислителей образуется нафти-
левая кислота.
Дифенилен впервые был получен Лотропом [47] из 2,2'-дииодбифенила и окиси
меди(1) при 350 °C. Из теплоты сгорания следует, что он обладает значитель-
ной энергией мезомерии — около 22 ккал/моль (92,11-103 Дж/моль) [48].
Если учесть, что энергия напряжения цикла составляет не менее 30 ккал/моль
(125,60-103 Дж/моль), то можно заключить, что энергия собственно делока-
лизации л-электронов составляет более 50 ккал/моль (209,34-103 Дж/моль),
173
Глава IV
т. е. не очень сильно отличается от энергии делокализации в бифениле [около
79 ккал/моль (330,76-103 Дж/моль)]. По данным электронных спектров
в дифенилене имеется довольно сильное взаимодействие между бензольными
кольцами [49].
Относительно химических свойств дифенилена Бейкер [50] пишет: «При рабо-
те с дифениленом постепенно приходишь к выводу, что почти во всех отноше-
ниях он является типичным полиядерным ароматическим соединением».
Почти все известные реакции дифенилена относятся к типу реакций обычного
электрофильного ароматического замещения [51]. Его можно галогениро-
вать, нитровать, динитровать нитрующей смесью, ацетилировать, а с избыт-
ком реагентов — диацетилировать и дисульфировать. Первый заместитель
входит только в положение 2, а второй — в положение 6.
1,2,4,5-Дибензопентален, описанный Бладом и Линстедом [52], представляет
собой вещество совершенно другого типа. Все его известные реакции затра-
гивают центральную бутадиеновую группировку. Эта группировка гидри-
руется по концам амальгамой натрия, полностью гидрируется палладием
на угле и претерпевает полную деструкцию до бензил-2,2'-дикарбоновой
кислоты при озонолизе. Во всех этих реакциях бензольные кольца не затра-
гиваются.
д. Ароматические ядра с нечетным числом членов в цикле. Валент-
ные связи ароматических структур с нечетным числом членов в цикле не могут
быть замкнуто-сопряженными, поэтому тип мезомерии в пятичленных гетеро-
циклах, например в пирроле, качественно отличается от мезомерии в шести-
членных ядрах типа бензола и пиридина. Диполярные структуры, которые
при теоретической трактовке строения бензола должны учитываться только
во втором приближении, являются основными в мезомерии ядер типа пирро-
ла. Главные структуры, необходимые для отображения мезомерии пиррола,
следующие:
Эти структуры допускают полную циркуляцию л-электронов по всему
кольцу, однако одна пара электронов постоянно остается неподеленной.
Таким образом, движение электронов по часовой стрелке смещает неподелен-
ную пару против часовой стрелки, как указано стрелками на второй формуле:
Это означает, что в мезомерной состоянии неподеленные электроны распро-
страняются по всему кольцу, так что атомы углерода имеют частично анион-
ный характер, а атом азота — в значительной степени катионоподобен.
Аналогичное рассуждение применимо и к другим пятичленным ароматиче-
ским ядрам (тиофену, фурану, глиоксалину, оксазолу и т. д.) и к пятичлен-
ным кольцам конденсированных систем (индолу, карбазолу, тионафтену,
пурину и т. д.).
174
Ароматичность и связанные с ней физические свойства
Энергия мезомерии пиррола велика, однако меньше, чем бензола и пиридина.
Такой же вывод следует из сравнения аналогичных полиядерных углеводоро-
дов, например индола с нафталином или хинолином. Аналогичные различия
существуют и для гетероциклов с другим гетероатомом, таких, как фуран
и тиофен (разд. 3,6). Экзальтация диамагнитной восприимчивости для
пиррола не была измерена; однако известно, что для фурана экзальтация
велика, хотя и меньше, чем для бензола и пиридина. В случае тиофена
экзальтация так же велика, как и в случае бензола и пиридина (разд. 4,г).
Пиррол имеет дипольный момент +1,80 Д (5,94-Ю-30 Кл-м), направление
которого противоположно направлению дипольного момента пиридина [53].
Величину момента пиррола можно приближенно проанализировать, как
указано на схеме, приведенной ниже. Анализ показывает, что момент,
обусловленный мезомерным переносом электронов азота в кольцо, способ-
ствующим возникновению отрицательного заряда на атомах углерода и поло-
жительного заряда на азоте (см. приведенные выше полярные структуры),
составляет +2,3 Д (7,59-10-30 Кл-м).
—2,3 1,8
Для пиррола
NH-Связь а Электроотри- цательность б Мезомерный перенос в
1,3 —0,8.2,3 = = -1,8 2,3
а Момент связи, вычисленный из момента аммиака.
б Влияние электроотрицательности вычислено из дипольного момента пиридина .
Множитель 0,8 в грубом приближении отражает различие в числе л- и а-электронов и
разную геометрию пиррола и пиридина.
в Сложение векторов.
Пиридин, хинолин и акридин являются основаниями, сравнимыми по силе
с анилином, однако их пятичленные аналоги — пиррол, индол и карбазол —
практически не обладают основностью. Мезомерия объясняет этот факт
аналогично теории Бамбергера тем, что неподеленные электроны азота
включены в ароматическую систему. Атомы водорода иминогруппы пиррола,
индола и карбазола обладают кислотными свойствами аналогично имидным
водородным атомам сукцинимида и фталимида. Теория мезомерии также
объясняет этот факт, так как, согласно этой теории, атомы азота в указан-
ных соединениях должны иметь положительный заряд.
Атомы углерода пиррольного ядра значительно более реакционноспособны,
чем в пиридине или бензоле. В одном из учебников по органической химии
отмечается (дважды на одной странице): «Следует подчеркнуть большую
реакционную способность метиновых атомов водорода в пирроле. Эти атомы
могут замещаться на другие атомы или группы с легкостью, сравнимой или
даже большей, чем в случае иминного водорода». Именно это и следует
из теории, предполагающей распределение неподеленной пары атома азота
по кольцу. Одним из примеров может служить высокая экзотермичность
реакции пиррола с галогенами. Для проведения этой реакции в препаратив-
ных целях необходимо работать в очень разбавленных растворах, но даже
в этих условиях обычно замещаются четыре или пять атомов водорода. Этим
пиррол сильно отличается от бензола и еще сильнее от пиридина.
175
Глава IV
Как было показано *, 2,5-диметилпиррол и 1,2,5-триметилпиррол при озоно-
лизе дают как глиоксаль, так и метилглиоксаль. Отсюда следует, что ненасы-
щенность углерода пе локализована на 2,3- и 4,5-связях, а распределена
по кольцу согласно формулам, приведенным выше.
Очевидно, что с этим типом мезомерии связана также способность цикло-
пентадиена образовывать стабильные соли с натрием и другими щелочными
металлами (это свойство используется для выделения циклопентадиена
из легкой фракции каменноугольной смолы). Сам углеводород не является
ароматическим — его энергия мезомерии почти такая же, как у бутадиена.
Однако анион обладает ароматичностью вследствие тех же причин, что
и пиррол. Этот анион гораздо более устойчив, чем анионы других угле-
водородов. Как показывают приведенные ниже валентные структуры, заряд
в анионе равномерно распределен между пятью атомами углерода.
Инден и флуорен также образуют соли со щелочными металлами. При
подщелачивании водных растворов солей 9-флуорениламмония или 9-флуоре-
нилсульфония образуется глубоко окрашенное слегка разлагающееся соеди-
нение. Окрашенное вещество, образующееся из солей диметил-9-флуоренил-
сульфония, изучено довольно подробно и представляет собой диметилсуль-
фопий-9-флуоренилид [56].
/Ч /Ч
у\ + «и- у\_ +
CH-S(CH3)2 ---> C-S(CH3)2
А/
Причина образования илида состоит в том, что анионный центр стабилизи-
рован мезомерией циклопентадиенидного типа. Большой дипольный момент
указанного илида (гл. III, разд. I, б) подтверждает вывод о том, что непо-
деленная пара углерода не переходит к атому серы, хотя последний и имеет
незанятые орбитали. Кролльпфайфер и Шнайдер [57] получили соответ-
ствующий пиридиениевый илид, а Виттиг и сотр. [57] — триметиламмоние-
вый, триметилфосфониевый и триметиларсониевый илиды.
(CeH4)2CH-N^ > (С6Н4)2СН - Х(СН3)3
(X = N, Р, As)
У азотсодержащих илидов вопрос об обобщении илидной пары электронов
азотом даже не может быть поставлен.
* См. [54]. В работе [55] показано, что 2,6-диметилпирон при озонолизе дает глиокси-
ловую кислоту и метилглиоксаль, что указывает на правильность предположения Арндта
(гл. II, разд. 3,6) о частичном превращении пирона в пприлиевый ион.
176
Ароматичность и связанные с ней физические свойства
Свободный анион циклопентадиена представляет собой абсолютно правиль-
ный пятиугольник, что было доказано рентгеноструктурным анализом его
железосодержащего производного ферроцена (C5H5)2Fe. Ферроцен, впервые
полученный Кили и Посоном [58], имеет конфигурацию пентагональной
антипризмы с атомом железа в центре симметрии [59, 60].
Существование изомерии у производных ферроцена создает впечатление,
что барьер вращения отдельных колец вокруг оси симметрии пятого порядка
невелик. Ферроцен летуч, устойчив при нагревании, легко окисляется,
по не восстанавливается. Он не склонен к реакциям присоединения, но легко
вступает в реакции электрофильного замещения, например сульфирования
и ацилирования по Фриделю — Крафтсу [61]. В этих реакциях первые два
заместителя входят в разные кольца. Однако при наличии в одном из колец
алкильного заместителя ацильная группа частично входит в это же кольцо
в положения 2 и 3. Отсюда можно сделать вывод, что ферроцен обладает
ароматичностью. Пока не совсем ясно, каким образом анионный заряд колец
переходит к железу, не нарушая делокализации. Очевидно, что этот процесс
происходит с участием Зй-орбиталей атома железа.
Циклогепт ат риен (тропилиден) — не очень стабильный полиолефин. Однако
Деринг и Нокс [61] показали, что он присоединяет Вг2 с образованием аддук-
та, от которого отщепляется НВт и образуются ионы брома и циклогептат-
риенилия (тропилия) — исключительно стабильного карбониевого иона *
Ч~Вг;^
—НВг
Ион тропилия имеет полную гептагональную симметрию с делокализован-
ным л-секстетом. Немостиковые эквивалентные валентные структуры приве-
дены ниже.
Большинство карбониевых ионов (гл. VII) мгновенно и необратимо реаги-
руют с водой и любыми другими нейтральными или анионными основаниями.
Однако ион тропилия в воде довольно устойчив и реагирует с ней обратимо,
качественно напоминая катионы псевдооснований (гл. XI). Степень смещения
равновесия гс образованием псевдооснования — циклогептатриенола (тропе-
нола) довольно мала:
С7Н++Н2О С7Н7ОН + Н+
* Название карбониевых ионов и карбанионов образуются путем добавления оконча-
ний «ий» и «ид» к названию радикала:
СН3
метил
СН+
метилий
CHj
метилид
12-0772
177
Глава IV
Даже в разбавленных растворах существует свободный ион тропилия.
В этом ярко проявляется стабилизирующий эффект делокализации л-элек-
тронного секстета.
В настоящее время еще не открыт большой ряд семичленных гетероаромати-
ческих систем типа боратропилена и бериллатропилена — электронодефи-
цитных аналогов пиррола и фурана.
В— Н
Боратропилен
Бериллатропилен
2. Симметрия, размеры и упругие свойства ароматических
молекул
Разделы 2—4 данной главы являются продолжением гл. III, посвященной
физическим свойствам молекул. Ниже будет рассмотрена связь этих свойств
с ароматичностью. Данные по дипольным моментам (гл. III, разд. 1 и заме-
чания в разд. 1, д) и по электрической поляризуемости (гл. III, разд. 3)
дают мало информации об ароматичности. В настоящем разделе будут рас-
смотрены симметрия, размеры и упругие свойства ароматических молекул,
а в последующем — энергия ароматических и алифатических колец. Затем:
будет рассмотрена диамагнитная поляризуемость алифатических и арома-
тических молекул, которая не обсуждалась в гл. III.
а. Симметрия, размеры и упругие свойства молекулы бензола.
Молекула бензола строго плоская и представляет собой правильный шести-
угольник (этого не было бы, если бы строение бензола строго соответствовало
структуре Кекуле). Такой вывод первоначально основывался на результа-
тах, полученных методом дифракции электронов, который не очень точен [62|.
Одно время полагали, что отклонение от симметрии можно обнаружить
спектроскопически, однако в колебательно-вращательном спектре паров [63]
не было обнаружено частот, которые противоречили бы столь строгой сим-
метрии. Бензол имеет 20 фундаментальных частот, однако вследствие симме-
трии молекулы многие из них запрещены, за исключением четырех частот
в ИК-спектре и семи частот в спектре комбинационного рассеяния. Таким
образом, девять частот вообще запрещены. Считалось, что отклонение от стро-
го гексагональной симметрии можно установить по появлению этих запре-
щенных частот. Однако оказалось, что появление этих частот связано с очень
малыми деформациями, обусловленными вандерваальсовыми силами в жидком
веществе [64]. Наблюдаемые в конденсированной фазе частоты, появление-
которых обусловлено межмолекулярными силами, оказались идентичными
частотам, рассчитанным по спектрам асимметрично дейтерированных бен-
золов [63]. В твердом состоянии межмолекулярные силы дают такой же
эффект, правда, спектр имеет более тонкую структуру. Структура молекулы
бензола в кристаллическом состоянии еще очень близка к планарной гекса-
гональной, однако окружающие молекулы в кристалле располагаются
таким образом, что симметрия каждой данной молекулы понижается и они
становятся только центросимметричными. Вследствие этого в ИК-спектре
можно наблюдать шесть из девяти запрещенных частот [65]. Эффективным
способом выявления запрещенных частот является исследование бензола
в растворителях типа сероуглерода, которые оказывают сильное электро-
кинетическое действие [66]. В ИК-спектре можно наблюдать все 20 частот
178
Ароматичность и связанные с ней физические свойства
бензола. Из этих линий 16 полностью исчезают в спектре газообразного
бензола.
Наиболее точные размеры бензола получены из спектров комбинационного
рассеяния [67]. Длины связей GG и GH составляют 1,397 и 1,084 А (13,97 НО-2
и 10,84-10"2 нм) соответственно.
Из колебательных частот при допущении, что энергию деформации можно'
представить шестью константами закона Гука, можно рассчитать упругие
свойства бензола [68], т. е. силовую константу CG-связи (F), силовую кон-
станту СН-связи (/), а также две константы изгибающих моментов в плоско-
сти молекулы, одна из которых (А) относится к деформации угла между
связями кольца, а другая (6) — к плоскостному отклонению СН-связи
от биссектрисы угла в кольце. Можно также определить две константы
моментов вывода из плоскости молекулы Г и у, первая из которых соот-
ветствует скручиванию CG-связи, обусловленному несовпадающими вне-
плоскостными изгибающими колебаниями атомов водорода, связанных
с атомами углерода этой связи, а вторая — изгибающими колебаниями
СН-связи, выводящими ее из плоскости ближайших трех атомов углерода.
Рассчитанные константы моментов приведены ниже:
^ = 7,61 -105 дин/см (7,61 -102 Н/м)
А = 13,7-10"12 дин-см/радиан (13,7-Ю'19 Н-м/рад)
Г = 1,02- 10"12 дин-см/радиан (1,02-10"19 Н-м/рад)
/ = 5,06-105 дин/см (5,06-Ю2 Н/м)
6 = 8,0-10"12 дин-см/радиан (8,0-10-19 Н-м/рад)
у = 2,62-10~12 дин-см/радиан (2,62-10"19 Н-м/рад)
Эти силовые константы и константы моментов в целом довольно хорошо
воспроизводят фундаментальные частоты бензола и его различных дейтеро-
производных, однако для одного типа колебаний получается неверная
частота. Отклонение частот, рассчитанных методом наименьших квадратов,
для бензола составляет 8%, если не учитывать одну частоту, для которой
ошибка равна 40%. Нетрудно рассчитать частоту (992 см"1), которая соот-
ветствует особому виду колебаний и зависит только от одной силовой посто-
янной — константы растяжения СС-связи (Е) в шестиконстантной системе
сил, рассмотренной выше. Величина этой константы, приведенная выше,
была рассчитана из частоты «пульсации» («breathing»), т. е. колебания, при
котором все шесть СС-связей растягиваются одновременно (см. схему).
Если полученную величину применить для расчета частот тригонального
колебания, при котором три CG-связи растягиваются, тогда как три другие
сжимаются, а все углы в кольце сохраняются равными 120° (2,094 рад)
и молекула в целом остается плоской, то рассчитанная частота составляет
1837 см-1. Наблюдаемая частота равна 1310 см"1 [65].
Такое большое несоответствие, очевидно, связано с тем обстоятельством,
что в результате тригональных колебаний ядерный остов изменяется, при-
ближаясь к каноническим структурам — структурам, которые могли бы
12* 17д
Глава IV
существовать, если бы не было делокализации л-электронов. Эта структура
является кекулевской, в ней чередуются длинные и короткие связи, но не
изменяются углы в кольце и не нарушается планарность. Можно предполо-
жить, что в результате изменения ядерного скелета бензола будет происхо-
дить и изменение электронных волновых функций, и таким образом будут
образовываться связи чередующейся кратности, имеющие разные силовые
постоянные. В результате силы упругости будут уменьшаться, что приведет
к снижению частоты. Силы упругости уменьшаются в (1310/1837)2 раз,
т. е. примерно в два раза.
Согласно данным, приведенным в разд. 1,6, силовые константы СС-связей
примерно пропорциональны кратности связей. Если приписать СС-связи
бензола кратность 1,5, то определенная выше силовая константа для бензола
будет укладываться в соответствующий ряд:
Кратность СС-связи 1 1,5 2 3
F, 105 дин/см (102 Н/м) 4,5 7,6 9,8 15,6
Наиболее замечательная особенность констант изгибающих моментов состоит
в низких значениях внеплоскостных констант. Константа момента скручи-
вания в бензоле [1012Г = 1 дин-см/радиан (10-7 Н-м/рад)] равна лишь
одной пятой константы скручивания для этилена (5,3 в тех же единицах) [69],
тогда как внеплоскостная константа изгиба СН-связи бензола [1012у =
= 2,7 дин-см/радиан (2,7-IO-7 Н-м/рад)] составляет только одну треть
соответствующей плоскостной константы бензола. Планарность бензола
подчеркивается так часто, что иногда забывают, насколько легко эта моле-
кула подвергается внеплоскостной деформации. Например, всеобщее удив-
ление вызвал тот факт, что га-диксилилен (формула приведена ниже) оказался
стабильным соединением. Вандерваальсово расстояние между двумя парал-
лельными бензольными кольцами должно быть примерно равным расстоя-
нию между параллельными слоями в графите [3,4 А (34-10-2 нм)], поэтому
для замыкания циклофанового цикла в и-дик-силилене необходима значи-
тельная внеплоскостная деформация бензольных колец. Результаты рентге-
ноструктурного анализа, проведенного Брауном [70], приведены ниже.
Установлено, что бензольные кольца в этой молекуле сближены по сравнению
с графитом на 0,3 А (3-10-2 нм). Однако бензольные кольца не являются
плоскими и образуют два двугранных угла по 10,7° (0,187 рад) (см. схему).
Связи с метиленовыми группами изогнуты в том же направлении еще на угол
13,9° (0,243 рад). Такая деформация кольца точно соответствует деформации,
обусловливающей появление самой низкой из фундаментальных колебатель-
ных частот бензола (400 см-1); классическая амплитуда этого колебания
на нулевом уровне равна одной четверти деформации, обнаруженной в и-ди-
ксилилене.
Н2С сн2
Н2С сн2
180
Таблица 48
Длины углерод-углеродных связей в ароматических углеводородах
Углеводород
Длина СС-связи,А(0,1мм)
Кратность
связи
Бензол
г Нафталин
Антрацен
1,2,5,бДи6енз-
антрацен
Пирен
Перилен
Коронен
Овален
Графит
3/2
181
Глаза IV
б. Размеры ароматических молекул. Робертсон с сотр. [71] недавно
провел серию измерений длин СС-связей в многоядерных ароматических
углеводородах рентгеноскопическим методом с точностью до ±0,02 А
(2-10-3 нм). Для нафталина [72], антрацена [73], коронена [74] и овалена [75]
определены положения всех атомов углерода относительно друг друга.
Для пирена [76], 1,2,5,6-дибензантрацена [77] и перилена [75] это сделано
только для некоторых атомов углерода, а для других атомов длины выведены
на основании предполагаемой молекулярной симметрии. При этом было
установлено, что в любом из этих углеводородов не все связи между углерод-
ными атомами идентичны. Интересное сравнение наблюдаемых отклонений
с вычисленными кратностями связи (без учета межкольцевых связей, которые
рассматриваются в разд. 3,в) дано в табл. 48.
В нафталине, перилене, коронене и овалене наблюдается качественное
соответствие между измеренными длинами связей и кратностью связей,
вычисленной простейшим способом. При этом в нафталине наблюдается
чередование длин внешних связей каждого кольца, а в коронене длина
внутренних связей каждого кольца равна длине СС-связи в графите, в то
время как внешние связи могут быть различной длины в зависимости от типа
связи, но приблизительно равны длине СС-связей в бензоле- В антрацене
такого соответствия не наблюдается, поскольку 2,3-связь короче, чем можно
Таблица 49
Размеры гетероароматических колец
Соединение Длина связей, А (0,1 нм) Гетероугол, град (17,45 • 10-з рад) Литература
Пиридин Пиразин /\ 1,400+0,005 1,390+0,005 \/ 1,340+0,005 N N /4—1,341 116,7 80
| || 1,335 Ч/ -1,341 N 116 82
Пиррол 1! II \// 1,42+0,02 N 1 Н 105+4 79
Фуран 1,440+0,016 || ||— 1,354+0,016 \ Z — 1,371+0,016 О || II 106,1+0,6 81
Тиофен 1,74+0,03 S 91±4 79
182
Ароматичность и связанные с ней физические свойства
было бы ожидать. В 1,2,5,6-дибензантрацене и пирене имеются большие
количественные расхождения, в особенности в случае связи, которая соот-
ветствует 9,10-связи фенантрена; она должна быть почти двойной связью,
однако длина ее практически такая же, как и длина GC-связей в бензоле.
Размеры некоторых гетероциклических ароматических ядер приведены
в табл. 49. Данные этой таблицы понятны, за исключением того, что СС-связи
в пиразине неожиданно оказываются очень короткими. В шестичленных
кольцах длины связей такие же, как в бензоле, если ввести поправку на мень-
ший радиус азота по сравнению с радиусом углерода. В пятичленных циклах
связи, формально простые в неполярных структурах, значительно меньше
суммы ковалентных радиусов, а формально двойные связи — несколько
больше, чем сумма радиусов двоесвязанных атомов. Эти факты объясняются
делокализацией электронов по кольцу и согласуются с данными Вибо
(стр. 176), который показал, что СС-связи в пирроле способны расщепляться
при озонолизе.
Как уже упоминалось в разд. 1, г, длины СС-связей в азулене равны длине
связей в бензоле. Исключение составляет мостиковая связь [1,48 А (14,8 х
хЮ"2нм)1, которая ненамного короче простой связи. Такое распределение
длин связей соответствует тому обстоятельству, что система сопряжения
распространяется по десятичленному циклу, но не по пятичленному или
семичленному циклам азулена; это согласуется также с тем, что азулен аро-
матичен по тесту симметрии Крейга. В циклооктатетраене связи имеют
чередующуюся длину и соответствуют слегка укороченной простой [1,47 А
(14,70-10'2 нм)] и стандартной двойной [1,32 А (13,2-10-2 нм)] связям.
Из этого следует, что степень функционального сопряжения мала, что под-
тверждается неплоским строением этой молекулы и, как будет показано
ниже, низкой энергией мезомерии. По электронной симметрии циклоокта-
тетраен принадлежит к псевдоароматическим молекулам.
3. Энергия алифатических и ароматических колец
а. Пространственная энергия алифатических колец. В гл. II,
разд. 2, д, было показано, что теплоты сгорания циклоалканов в гомологи-
ческом ряду возрастают не аддитивно, как в нормальных алканах с открытой
цепью, если молекулы последних достаточно велики, и эффектом концевых
групп можно пренебречь. Изменение теплот сгорания циклоалканов показы-
вает, что эта энергия включает член, зависящий от размеров цикла. Спитцер
и Хафман впервые показали, что в ряду от циклопентана до циклооктана
минимальную энергию имеет циклогексан. Если принять энергетический
вклад, обусловленный размерами кольца, равным нулю для циклогексана,
то для других колец он будет положительным.
С тех пор появилось много данных по теплотам сгорания циклоалканов.
Они были получены главным образом Купсом, который подтвердил данные
Спитцера и Хафмана. Приняв энергию циклогексана за нулевую точку,
можно вычислить энергию колец Ег для разных циклоалканов СпН2п, от цик-
лопропана до циклогептадекана. Эти данные [ккал/моль (4,187 -103 Дж/моль)]
приведены ниже [83] и графически представлены на рис. 25.
п 3 456789 10
ЕТ 27,9 26,6 6,0 0,0 5,9 9,7 12,1 12,8
п И 12 13 14 15 16 17
FT 12,3 5,1 7,1 3,6 5,0 4,3 4,5
183
Глава IV
Можно указать три причины увеличения энергии образования алифатических
колец, каждая из которых проявляется в циклах различных размеров
(рис. 25). В 1885 г. Байер ввел понятие о напряжении связей, обусловленном
отклонением углов в кольце от тетраэдрического [84]. Кроме двойных и трой-
ных связей, которые Байер рассматривал как циклы, он рассмотрел трех-
и четырехчленные циклы, которые менее стабильны, чем пяти- и шести-
членные циклы. Байер довел свое сравнение только до шестичленных циклов;
однако, когда в 1890 г. Заксе предложил ненапряженные конформации
для циклогексана [85], теория Байера стала в общем неприменимой. Однако
Рис. 25. Энергия циклоалканов и размеры
циклов.
Ошибка для п = 17 составляет
0,5 ккал/моль и уменьшается прибли-
зительно пропорционально п.
для трех-, четырех- и пятичлен-
ных циклов теория Байера приме-
нима, если не обращать внимания
на тот факт, что, согласно накоп-
ленным экспериментальным дан-
ным, четырехчленные циклы не-
намного более стабильны по срав-
нению с трехчленными [86]. Таким
образом, возникла идея, что раз-
личную стабильность циклов нуж-
но объяснить какими-то другими
причинами, неизвестными Байеру.
Байер назвал явление, увеличи-
вающее энергию циклов, терми-
ном «spannung», эквивалентом ко-
торого в английском языке должно
было быть слово «stress» («нажим»,
«напряжение»), но который был
неверно переведен как «strain»
(«натяжение»); последний термин
и стал общепринятым в англий-
ской литературе *. Однако в на-
стоящее время, когда выявлены
другие стереохимические причины
увеличения энергии колец, это
явление принято называть «байе-
ровским напряжением».
Затем был открыт эффект, связанный с различной стабильностью затормо-
женных и заслоненных конформаций связей колец. Как указывалось в гл. II,
разд. 2, д, энергетическое различие между этими конформациями в случае
этана было установлено в 1936 г. Кемпом и Питцером. В 1947 г. Спитцер
и Хафман впервые определили величину этого эффекта для циклоалканов.
Они пришли к выводу, что более стабильной является заторможенная кон-
формация, так как минимальную энергию в гомологическом ряду цикло-
алканов имел циклогексан — единственный гомолог, для которого возмож-
на полностью заторможенная конформация, свободная от байеровского
напряжения. Конформация колец циклопентана и циклогептана должна
быть таковой, чтобы свести к минимуму сумму байеровского напряжения
и энергии незаторможенных конформаций. Последний энергетический член
получил название «питцеровское напряжение». Эта форма напряжения
также является источником нестабильности, с одной стороны, циклобутана
* Аналогичная путаница в словах «stress» и «strain» породила термин «electronic strain»
(что означает «смещение электронного облака») (1926 г.).
184
Ароматичность и связанные с ней физические свойства
и циклопропана, а с другой, циклооктана и других высших гомологов цикло-
гептана, хотя в очень больших циклах этот эффект постепенно затухает.
Еще до открытия «питцеровского напряжения» был обнаружен еще один
эффект, который обусловливает относительную нестабильность 9-, 10- и 11-
членных циклов. Первое указание на него появилось в работе Ружички
1920-х годов по получению макроциклических кетонов путем пиролиза
ториевых или других солей алкан-а, w-дикарбоновых кислот: выход резко
уменьшался почти до нуля при переходе от шестичленных кетонов к 9—11-
членным и затем медленно возрастал при переходе к большим циклам [87].
В 1930-х годах Циглеру удалось значительно повысить выходы макроцик-
лических кетонов путем использования реакции Торпе * в условиях исчер-
пывающего разбавления. Такие условия были выбраны для того, чтобы
подавить конкуренцию межмолекулярной конденсации, которая прояв-
ляется особенно сильно в том случае, когда при циклизации образуются
большие циклы [88]; тем не менее Циглеру не удалось получить высоких
выходов «средних циклов», т. е. циклов с числом атомов от восьми до двенад-
цати.
Проанализировав предложенные пути синтеза и свойства средних циклов,
которые уже в это время были получены с хорошими выходами при помощи
ацилоиновой конденсации **, Прелог в 1950 г. предположил, что средние
циклы неустойчивы вследствие трансаннулярного отталкивания атомов,
связанных с кольцом. Чтобы понизить энергию других видов напряжения,
эти атомы вынуждены при образовании цикла повернуться внутрь кольца.
В малых циклах внутренние атомы взаимно не перекрываются [89]. Затем
этот эффект был подтвержден термохимическими измерениями [83], а суще-
ствование трансаннулярного взаимодействия — «напряжения Прелога» —
нашло отражение в большом числе физических и химических свойств.
Напряжение Прелога химически доказывается трансаннулярными реакция-
ми между атомами или группами, которые не реагировали бы друг с другом,
если бы они находились в разных молекулах и могли бы сближаться
* Реакция Торпе — это катализируемая основаниями циклизация динитрилов в имино-
нитрилы, гидролизующиеся до циклических кетонов:
** При реакции с натрием две сложноэфирные группы разных молекул или одной
и той же молекулы могут конденсироваться друг с другом, образуя ацилоины с открытой
цепью или циклические. Ацилоины с большими циклами легко получаются и без высо-
кого разбавления. Возможный путь реакции представлен ниже:
/СО2С2Н5
(Н2С)„
ХСО2С2Н5
2Na
----> (Н2С)
.СО 2Na
(Н2С)»\1, ---------
/С — ONa
(Н2С)„. ||
'С —ONa
Н2О .СНОН
185
Глава IV
на такое же расстояние, как в цикле. В случае производных циклодекана
известно, что реакции, протекающие через карбониевые ионы (гл. VII и IX),
например нуклеофильное замещение и элиминирование при ацетолизе и-толу-
олсульфонатов (X = О8О2СвН4СНз в формуле, приведенной ниже) в уксус-
ной кислоте и при дезаминировании аминов азотистой кислотой, включают
трансаннулярный гидридный перенос. С помощью изотопной метки было
доказано, что входящий заместитель (Y = ОСОСН3, ОН) или возникающая
двойная связь в значительной степени находятся не в том месте, где нахо-
дилась уходящая группа (X) [90]. Детальный механизм этих процессов
не известен, однако сущность этих реакций выражается следующей схемой:
1 +
/СЩСНХСНгСНг, ,СН2СНСН2СН2 ч
сн2< >сн2 —> сн2< >сн2 —*
YCHaCHsCHsCHs/ \СН2СН,СН2СН2/
5 6
,СН2СН,СН2СН2Ч
-» СН2< )СН2 —»
^СШСНСНгСНг/
,СН2СН2СН2СН2.
сн2< >сн2
xCH2CHYCHoCH./
5
/СН2СН2СН2СН2х
сн2< >сн2
\сн2сн=снсн/
I 5 6
Подобные трансаннулярные реакции, вероятно, характерны для циклов,
расположенных на центральном горбе рис. 25. Что касается производных
циклододекана (начиная с этого цикла напряжение Прелога должно исче-
зать), то для них установлены только «локальные», т. е. абсолютно нормаль-
ные, реакции замещения и элиминирования [91].
Наиболее важное физическое доказательство трансаннулярного взаимодей-
ствия было получено Дунитцем и сотр. [92] при рентгеноструктурном иссле-
довании структуры хлоргидрата циклонониламина, дихлоргидрата транс-
1,6-диаминоциклодекана и циклододекана. В 9- и 10-членных циклах шесть
атомов водорода находятся внутри кольца, и поэтому в 9-членном цикле
в трех парах в положении 1,4 или 1,5, а в 10-членном цикле ов двух парах
в положении 1,5 атомы водорода находятся на расстоянии 1,8 А (18 -10-2 нм)
друг от друга, т. е. па 0,4 А (4-10-2 нм) ближе, чем расстояние, на котором
они «касаются» друг друга [2,2 А (22-10-2 нм)]. Условное изображение 10-
и 12-членного циклов дано ниже:
Теперь очень просто объяснить дилемму начала столетия, почему цикло-
бутан оказывается более неустойчивым, чем можно ожидать исходя из вели-
чины байеровского напряжения. Этот факт установлен с помощью термо-
химических данных, приведенных выше. В циклобутане должно существо-
вать трансаннулярное отталкивание между самими кольцевыми атомами;
это соединение является единственным циклоалканом, в котором это взаимо-
действие выражено сильно. Трансаннулярное перекрывание атомов углерода
186
Ароматичность и связанные с ней физические свойства
по диагонали циклобутана составляет только около 0,2 А (2 -10"2 нм). В цик-
лобутане центры трансаннулярного сжатия двух противоположных пар
атомов углерода находятся в одной точке (или почти в одной точке, если
кольцо неплоское *), благодаря чему общая энергия отталкивания более
чем в два раза выше, чем в случае двух трансаннулярных атомов.
Единственной кратной связью, способной существовать в малых циклах,
является цис-двойная связь; однако средние и большие циклы могут содер-
жать тройные и даже транс-двойные связи. Если средний цикл достаточно
велик, чтобы в нем могла существовать транс-двойная связь, но не достаточно
велик для обеспечения свободного вращения вокруг боковых простых свя-
зей, служащих цапфами, то могут образовываться хиральные энантиомеры,
настолько стабильные, что их можно получать и хранить. Как показал
Коуп [94], такой оптической стабильностью обладает транс-циклооктен;
однако стабильность быстро уменьшается с дальнейшим увеличением разме-
ров цикла: транс-циклононен можно разделить на оптические изомеры толь-
ко при низкой температуре, а при комнатной температуре он рацемизуется.
б. Энергия мезомерии ароматических колец. Основное значение
термохимических измерений на ароматических системах состоит в том, что
с их помощью можно оценить энергии мезомерии этих систем. В гл. II,
разд. 2,6, указывалось, что первоначальная оценка энергии мезомерии
может проводиться по теплотам гидрирования и по теплотам сгорания.
В первом случае проводится сравнение с теплотами гидрирования молекул,
содержащих такое же число несопряженных двойных связей. Некоторые
величины энергий мезомерии, определенных таким путем, приведены
в табл. 50. Числа в графе «разность» отражают энергию мезомерии, неисправ-
ленную на напряжение в циклах; единственной молекулой из приведенных
Таблица 50
Энергии мезомерии, определенные исходя из теплот гидрирования
Аромати- ческое соединение Коли- чество н2, моли —ЬН, ккал/моль (4.187Х ХЮ» Дж/моль) Разность, ккал/моль (4.187Х ХЮ3 Дж/моль) Соединение для сравнения Коли- чество н2, моли —ДН, ккал/моль (4.187Х X10 3 Дж/моль)
Бензол [95] 3 49,8 36,9 Циклопентен 1 26,9
Фуран [96] 2 36,6 17,2 Циклогексен 1 28,9
Азулен [97] 5 99,0 32,5 Циклогептен 1 25,9
в табл. 50, для которой это исправление существенно, является азулен.
Однако и для него поправка не превышает 10 ккал/моль (41,87 -103 Дж/моль).
Второй метод оценки энергии мезомерии состоит в сравнении теплот обра-
зования, выведенных из теплот сгорания с суммой энергий связей. Некото-
рые величины, полученные таким путем Полингом, приведены в табл. 51.
Единственным соединением, для которого требуется введение большой
поправки в полученную вышеуказанным способом энергию мезомерии,
является дифенилен. Необходимая поправка в этом случае составляет около
30 ккал/моль (125,6 -103 Дж/моль).
* Данные по дифракции электронов, спектрам комбинационного рассеяния, ИК-
и ЯМР-спектрам показывают, что цикл в равновесной конфигурации не является плоским,
а образует двугранный угол около 160° (2,792 рад) с барьером инверсии около 1 ккал/моль
(4,187 -103 Дж/моль) [93].
187
Глава IV
Таблица 51
Энергии мезомерии, выведенные из энергий связей
Соединение Энергия мезомерии, ккал/моль (4,187 103 Дж/моль) Соединение Энергия мезомерии, ккал/моль (4,187-103 Дж/моль)
Бензол 37 Пиридин 43
Нафталин 77 Хинолин 69
Антрацен 105 Пиррол 31
Фенантрен 110 Индол 54
Циклоокта- 5 Карбазол 91
тетраен Фуран 23
Азулен 45 Тиофен 31
Дифенилен 22
(см. [98])
Наиболее существенные выводы из приведенных данных состоят в следую-
щем: введение азота в шестичленное ароматическое кольцо вносит лишь
небольшое изменение в энергию мезомерии, энергия мезомерии конденси-
рованных шестичленных циклов постепенно увеличивается с ростом числа
колец (или числа электронов) и энергия мезомерии пятичленных гетеро-
циклов меньше соответствующей энергии шестичленных гетероциклов,
но все же еще достаточно велика. Небензоидные углеводороды, циклоокта-
тетраен, азулен и дифенилен каждый в отдельности обсуждались в разд. 1, г.
4. Диамагнитная восприимчивость алифатических
и ароматических молекул
а. Природа диамагнитной восприимчивости. Атом или молекула
могут обладать постоянным магнитным моментом в двух основных случаях:
во-первых, если имеется один или несколько электронов с неспаренными
спинами; во-вторых, если электроны движутся по своим орбиталям с резуль-
тирующим угловым моментом. Магнитный момент молекулы кислорода
обусловлен только первой причиной (два неспаренных спина). Окись азота
обладает магнитным моментом, возникающим в результате комбинации
обеих причин (один неспаренный спин и один квант орбитального углового
момента *).
Когда магнитное поле воздействует на вещество, молекулы которого обла-
дают постоянным магнитным моментом, молекулярные моменты ориенти-
руются в направлении поля и наблюдается явление парамагнетизма, т. е.
положительная магнитная восприимчивость. Парамагнитная восприимчи-
вость велика по сравнению с восприимчивостью, которая возникает в резуль-
тате рассматриваемых ниже магнитных эффектов.
* Окись азота не обладает магнитным моментом при низкой температуре, потому что
в этих условиях оба эффекта взаимно компенсируются. Влияние температуры сказывается,
потому что в нормальном состоянии окись азота включает два состояния, отличающихся
друг от друга на 0,344 ккал/моль (1,44 -103 Дж/моль). Наличие двух состояний обусловлено
тем, что момент спина может либо совпадать, либо быть противоположным орбитальному
моменту. В последнем случае магнитные моменты компенсируются. Состояние с противо-
положными моментами является более низко лежащим, и при низкой температуре
осуществляется лишь это состояние.
188
Ароматичность и связанные с ней физические свойства
Почти все устойчивые органические молекулы (исключение составляют сво-
бодные радикалы с неспаренными спинами электронов) не имеют постоянного
магнитного момента и поэтому не обнаруживают парамагнетизма. Однако
они обладают остаточным, весьма слабым эффектом противоположного знака;
этот эффект в принципе представлен во всех веществах, хотя он проявляется
только при отсутствии постоянных молекулярных магнитных моментов.
Такое явление называется диамагнетизмом и измеряется отрицательной
магнитной восприимчивостью. Лармор, а затем более детально Ланжевен
показали, что диамагнетизм вызывается тем, что магнитное поле наводит
круговой ток, магнитное поле которого противоположно наводящему полю.
Это происходит даже в изолированном атоме, в котором под действием поля
электроны начинают вращаться в плоскости, перпендикулярной полю.
В большей степени это происходит тогда, когда токи проводимости возникают
в многоатомной системе. Каждый электрон, действующий как весьма малый
круговой ток, дает отдельную составляющую наведенной магнитной поля-
ризации вещества. Легко показать, что такая составляющая пропорциональ-
на величине средней площади орбиты, которую описывает электрон. (Наве-
денное движение электрона естественно накладывается на невозмущенное
движение; однако можно выделить из общего движения часть, обусловлен-
ную магнитным полем, и говорить о ней как о движении по «орбите».)
Измеряемая магнитная восприимчивость выражается восприимчивостью
на 1 см. Если эту величину разделить на плотность и помножить на моле-
кулярный вес, получим восприимчивость на 1 моль — так называемую
молярную восприимчивость %.
Для одноатомных веществ со сферическими атомами теория Лармора —
Ланжевена приводит к формуле
у= у
“ втс2 j/j г
п
где N — число Авогадро, т — масса и е — заряд электрона, с — скорость
света, a rf — среднее значение квадрата радиуса магнитной орбиты i-ro
электрона, причем сумма берется для всех п электронов атома. Следует
привести другую геометрическую модель, которая потребуется в дальней-
шем. Если атомы представлять себе не в виде сфер, а в виде круговых цилинд-
ров или пластинок с тем же радиусом и если пластинки расположены перпен-
дикулярно полю, то средние площади, описываемые круговыми токами,
очевидно, будут больше во столько раз, во сколько объем цилиндра больше
объема вписанной сферы; тогда формула должна иметь вид
л 4 me2 г
п
Магнитная восприимчивость отдельной молекулы является тензорной вели-
чиной. Диамагнитные сферические атомы, тетраэдрическая молекула XY4
и некоторые другие молекулы являются магнитно изотропными: их магнит-
ная восприимчивость в расчете на одну молекулу одинакова во всех направ-
лениях
1 = 7JN
189
Глава IV
Большинство диамагнитных молекул не изотропно: их магнитные воспри-
имчивости измеренные в газах, жидкостях и мелко раздробленных твердых
веществах, являются средними магнитными восприимчивостями и обычным
образом связаны с тремя главными магнитными восприимчивостями х, у, z,
направленными вдоль трех взаимно перпендикулярных главных осей маг-
нитной восприимчивости
3^ = ^ + y + z
Правила симметрии для локализации главных осей точно такие же, как
и в случае электрической поляризуемости (гл. III, разд. 3,г). Для начала
мы ограничимся рассмотрением средних магнитных восприимчивостей.
б. Принцип аддитивности Паскаля; диамагнитные константы.
В насыщенных многоатомных молекулах и в молекулах с изолированными
кратными связями электроны в основном локализованы; магнитное поле
заставляет их циркулировать в основном локально, т. е. в пределах атомов
или связей, которым принадлежат электроны. Если бы они циркулировали
строго внутри невзаимодействующих магнитно изотропных атомов или дру-
гих локализованных частиц, то магнитную восприимчивость можно было бы
точно вычислить сложением величин отдельных частиц. В 1910 г. Паскаль
экспериментально установил, что средние молярные магнитные восприим-
чивости диамагнитных молекул, подобно молекулярным рефракциям, могут
быть выражены посредством аддитивных констант атомов с внесением кон-
ститутивных поправок *.
Ввиду того что молярная магнитная восприимчивость х в диамагнитных
молекулах отрицательна, удобнее пользоваться положительной величиной
— X, которую можно назвать молярной диамагнитной восприимчивостью.
Диамагнитные восприимчивости представляют собой величины порядка
10-6 . . . 10-4 эл.-магн. ед./моль, поэтому проще пользоваться величинами
— 106х, т. е. принимать 10-6 эл.-магн. ед./моль за практическую единицу
диамагнитной восприимчивости. Эти единицы и применяются в дальнейшем.
Атомные константы и константы кратных связей, по Паскалю, приведены
в табл. 52 в несколько измененном виде. Для атомов даны константы, которые
Таблица 52
Диамагнитные константы атомов и кратных связей
Атом - 106% Кратная связь (исключая атомы) — 10вх
н 2,9
с 6,0 (С)-(C) -5,5
N 5,6 (C) = (N) -8,2
О 4,6 (С)-(О) -6,3
S 15,0 (N) = (N) -4,0
F 6,3 (N) = (O) -8,0
С1 17,2
Вт 26,5 (С) = (С) -0,8
I 40,5 (С) (N) -0,8
* См. три наиболее подробные работы Паскаля [99] и его статьи, опубликованные
в разное время в Bull. soc. chim. France, Ann. chim. и Compt. rend., а также гл. 4 моногра-
фии Бхатнагара и Матхура [100].
190
Ароматичность и связанные с ней физические свойства
были или могут быть вычислены из полученных Паскалем эксперименталь-
ных данных для насыщенных алифатических соединений, причем для каж-
дого атома дается только одна константа. Константы кратных связей при-
бавляются к сумме атомных констант, включая константы кратно связанных
атомов *. Для констант приведены значения с точностью до второго знака;
погрешности, обусловленные применением принципа аддитивности, и веро-
ятные ошибки опыта не оправдывают более высокую точность. При наличии
ионных зарядов, формальных диполярных зарядов и других источников
сильной полярности, например при накоплении атомов галогенов или при
наличии некоторых видов сопряжения, особенно сопряжения в ароматиче-
ских системах, наблюдаются отклонения, которые выходят за обычно допу-
стимые пределы, о чем будет речь ниже.
Диамагнитную восприимчивость свободного водородного атома можно точно
вычислить [101]; она равна 2,37. Нельзя ожидать, что для связанного атома
водорода диамагнитная константа будет такой же. В случае констант других
атомов имеется очевидная связь с размерами атома и особенно с номером
периода в таблице Менделеева. В некоторых случаях, как показано ни-
же, константы могут быть связаны с атомным радиусом, т. е. с возможной ве-
личиной магнитных орбит, и притом приближенно в количественной
форме.
Обсуждение констант кратных связей приводится в разд. 4,г.
в. Диамагнитные константы ионов; влияние полярности. Простей-
шей иллюстрацией влияния полярности на диамагнитную восприимчивость
может служить сравнение диамагнитных констант моноатомных, изоэлек-
тронных атомов и ионов. Эти данные приведены в табл. 53 **. Увеличение
* При сравнении приведенных в табл. 52 констант с данными Паскаля необходимо
учитывать также следующее:
Во-первых, Паскаль в качестве стандартного соединения брал воду, причем ошибка
измерений ее магнитной восприимчивости составляла —4%, поэтому во все ранние
экспериментальные данные Паскаля должна быть введена поправка —4%.
Во-вторых, Паскаль вывел большинство атомных констант (кроме констант N, О и F)
из измерений магнитных восприимчивостей свободных элементов. Полученные таким
образом величины для углерода (из тростникового сахара) и для серы хорошо согласуются
с константами, полученными из измерений для органических соединений. Другие вели-
чины, особенно константы С1, Вг и I, плохо согласуются со значениями, которые можно
вывести на основании измерений магнитной восприимчивости алкилгалогенидов. Кон-
станты элементарных галогенов согласуются с величинами, полученными при определении
магнитной восприимчивости арилгалогенидов; поэтому Паскаль рассматривал арилгало-
гениды как магпитнопормальные, а алкилгалогениды — как аномальные, между тем
правильным является обратный подход ввиду сопряжения галогена с ароматическим
ядром и d-орбитального взаимодействия, которое, вероятно, имеет место в молекулах
С12, Вг2 и 12.
В-третьих, Паскаль трактовал ненасыщенность так же, как Брюль, при обработке
данных о молекулярных рефракциях. Паскаль ввел константы для двойных и тройных
углерод-углеродных связей, однако в случае других кратных связей Паскаль приписы-
вал специальные константы одному из атомов, связанных кратной связью (например,
для кислорода в СО). В таблице для каждого атома приведена только одна константа;
при наличии кратной связи к сумме атомных значений должна быть прибавлена дополни-
тельная константа. Далее Паскаль ввел некоторые специальные константы для атомов,
в системах, которые можно рассматривать как мезомерные (02 в карбоксиле, азот, связан-
ный с арилом, и т. д.). По-видимому, предпочтительнее принять для всех атомов и крат-
ных связей значения, характерные для них при отсутствии мезомерии, а затем наблюдае-
мую разность считать обусловленной мезомерией. Особенно это важно для ароматических
систем, для которых Паскаль принимал центрическую формулу, не приписывая, однако,
центрическим валентностям магнитного эффекта.
** Экспериментальные данные для инертных газов собраны Селвудом [102]. Данные
для ионов приведены Ангусом [103].
191
Глава IV
заряда ядра уменьшает размеры электронной оболочки и, следовательно,
площадь магнитных орбит и магнитную восприимчивость.
Таблица 53
Диамагнитные константы (— 106 X) одноатомных ионов и атомов
инертных газов
Не 1,9 Li+ 1,0
F- 9,0 Ne 7,6 Na+ 6,5 Mg2+ 5,0
CI+ 23,5 Аг 19,2 К+ 15,0 Ca2+ 11
Вг- 34,5 Кг 28,5 Rb+ 22,5 Sr2'*' 19
I- 50,5 Хе 43,2 Cs+ 35,0 Ba2* 30
г. Влияние ненасыщенности на диамагнетизм. Как показывают
данные табл. 52, диамагнитные константы любых кратных связей отрица-
тельны: помимо возможного влияния сопряжения, которое еще не было
рассмотрено, ненасыщенность вызывает уменьшение диамагнетизма. Сами
по себе константы характеризуются двумя величинами различного порядка:
константы двойных связей значительны и отрицательны, величина их (без
учета знака) того же порядка, что и атомные константы; константы тройных
связей отрицательны и малы по величине, они приблизительно на порядок
меньше, чем атомные константы.
Эти соотношения можно качественно связать с геометрическими формами
электронных облаков в двойной и тройной связях (см. гл. 1, рис. 8). Размеры
ЛСоболочек атомов слишком малы, чтобы соответствующие магнитные
орбиты обладали достаточной величиной; почти весь диамагнетизм атомов
обусловлен их валентной оболочкой. Можно было бы ожидать, что благодаря
сжатию за счет л-компоненты в двойной связи ^-составляющая должна
иметь несколько меньшее влияние на диамагнетизм, чем в случае обычной
простой связи. Важно, однако, то, что л-составляющая не благоприятствует
образованию магнитных орбит атомного размера, потому что она рассекается
надвое плоскостью нулевой электронной плотности и через эту плоскость
электроны свободно проходить не могут. Таким образом, диамагнитная
восприимчивость резко уменьшается при образовании двойной связи из про-
стой.
В тройной связи сжатие а-орбиталей вдоль межъядерной линии, не-
сомненно, больше, чем в двойной связи. Однако здесь л-оболочка пред-
ставляет собой не плоскость, а линию нулевой электронной плотности, что
является значительно меньшим ограничением. Следовательно, диамагнит-
ная восприимчивость значительно увеличивается при образовании тройной
связи из двойной. Конечно, это лишь приближенная картина; при отсут-
ствии данных о направленных магнитных свойствах несопряженных двойных
и тройных связей попытка количественной трактовки проблемы весьма
затруднительна.
д. Влияние сопряжения на диамагнетизм. Влияние такого рода
удобно описывать посредством выражаемых в —10в% экзальтаций величин,
вычисляемых суммированием констант атомов и кратных связей валентной
структуры, например структуры Кекуле для бензола, структуры с двумя
двойными связями для фурана, R2N — С — О для амидной группы, 0“ —
— N + = О для нитрогруппы и т. д. Вычисленные таким способом экзаль-
тации приведены в левой части табл. 54. Если одна из приведенных систем
включается в более длинную цепь сопряжения или две такие системы нахо-
дятся в сопряжении между собой, то можно суммировать константы атомов
192
Ароматичность и связанные с ней физические свойства
и кратных связей, как и раньше, и добавлять к ним экзальтации, приведен-
ные в левой части табл. 54, не принимая во внимание только такие влияния,
которые могут возникнуть в результате нового или более широкого сопря-
жения. Наблюдаемое повышение величины —10в% по сравнению с величи-
ной, вычисленной, как указано выше, называется дополнительной экзаль-
Таблица 54
Экзальтации диамагнитной восприимчивости для сопряженных систем
Система Экзальтация, —108 Система Дополнительные экзаль- тации, — 10в %
С = С —С=С +0,5 Аг — Аг а +0,5
С == С — с = с 0 Аг —С = С +1
N =С-С = N 0 Аг —С = С +1,5
Бензол +18,0 Аг —NH2
Пиридин +18,5 Ar-NHR ! -1
Нафталин +36 Аг—NR2 )
Антрацен +55 Аг—ОН 1 -4 1
Фенантрен +53,5 Аг-OR J +1
Хризен +67,5 Аг-Cl +2,5
Фуран +14 Аг—Вг +3,5
Тиофен +18 Аг —I +3,5
Циклооктатетраен +2,5 Аг-СНО
Азулен +35,5 Ar—COR
НО-С =0 Ar—CO2R +1,5 6
+5
но-с = о 1 Аг—CONH2 J
1 h2n—с=о +3,5 6
+
-0 —N = O 1 + 2 « Ar—NO2 +0,5 в
а Аг — арильный остаток (фенил или замещенный фенил).
б Величины для амидов частично обусловлены таутомерией.
в Эти величины включают влияние, обусловленное формальными зарядами в NOj-rpynne.
тацией, обусловленной дополнительным сопряжением. Дополнительные
экзальтации, полученные таким способом, приводятся в правой части табл. 54.
Величины экзальтации округлены с точностью до 0,5, так как в большей
точности нет необходимости.
Прежде всего бросается в глаза незначительное влияние сопряжения крат-
ных связей в открытых цепях. Можно сделать вывод, что уменьшение крат-
ности одних и увеличение кратности других связей дает приблизительно
компенсирующиеся эффекты и что под действием магнитного поля электроны
в каждый данный момент времени циркулируют в основном в пределах
одной связи.
Совершенно иная картина характерна для полностью замкнутых сопряжен-
ных систем, например ароматических молекул, в диамагнетизме которых
наблюдаются большие положительные экзальтации. Эти эффекты рассма-
13-0772
193
Глава IV
триваются как результат способности электронов л-оболочки циркулировать
по всей молекуле, вследствие чего орбиты становятся значительно больше
атомных размеров. Паскаль показал, что в случае таких конденсированных
ароматических систем, как нафталин или пирен, наблюдаются исключитель-
но высокие экзальтации. Это можно объяснить тем, что некоторые электроны
в таких системах способны создавать орбиты, охватывающие несколько
колец. Следует отметить, что в случае шестичленных циклических углеводо-
родов экзальтация постепенно увеличивается по мере увеличения числа
колец. В пятичленных ароматических кольцах экзальтация так же или
почти так же велика, как в бензоле, в циклооктатетраене имеет тривиальную
величину, а в азулене — такую же величину, как в изомерном ему нафта-
лине. Эти влияния были подробно изучены с учетом изменения диамагнитной
восприимчивости по направлениям, что описано в разд. 4,е.
Единственными другими значительными экзальтациями, представленными
в левой части табл. 54, являются намного меньшие по величине экзальтации
различных производных карбоксильной группы, а также питрогруппы.
Здесь уместно заметить, что обе атомные р-орбитали и л-орбитали двойной
связи рассекаются пополам узловой плоскостью. Можно предположить,
что перекрывание р- и л-орбиталей в сопряженной системе приводит к неко-
торому расширению магнитных орбит.
Среди величин дополнительных экзальтаций, приведенных в правой части
табл. 54, только одна отрицательна, а все остальные положительны и могут
быть качественно объяснены увеличением магнитных орбит благодаря пере-
крыванию ароматических л-орбиталей с р- или л-орбиталями боковых
цепей. По-видимому, можно допустить, что положительные экзальтации,
вызываемые указанной причиной, имеются и в особом случае производных
анилина, когда наблюдаются отрицательные экзальтации, что объясняется
наложением на положительную экзальтацию большей (по абсолютной
величине) отрицательной экзальтации, обусловленной совсем иными при-
чинами.
Возможной причиной является измененная гибридизация орбитали неподе-
ленных электронов атома азота. Атомные константы азота вычислены на осно-
вании магнитных восприимчивостей алифатических аминов, в которых непо-
деленные электроны находятся на тетраэдрических орбиталях: такие орбитали
весьма неравномерно делятся их конической узловой поверхностью (гл. I,
разд. 4,а, рис. 6); они имеют одну область, в которой могут образоваться
магнитные орбиты приблизительно атомных размеров. Однако в аромати-
ческих аминах происходит изменение гибридизации, которое можно описать
как двухстадийное: сначала тетраэдрическая орбиталь превращается в р-
орбиталь, которая рассекается надвое узловой плоскостью, затем р-орбиталь
вступает в сопряжение с ароматическими л-орбиталями. Первый этап может
вызвать отрицательную экзальтацию, как при образовании двойной связи
из простой (разд. 4,г), второй — положительную; наблюдаемый эффект
является результирующим. Подобные рассуждения можно приложить и к
производным фенола, допустив, что именно количественная разница обу-
словливает наблюдаемые незначительные величины положительной экзаль-
тации. Такого рода рассуждения неприложимы к галогенопроизводным
бензола (а также к производным тиофенола), поскольку пеподеленные элек-
троны галогена (и серы), по-видимому, находятся на почти негибридизован-
ных р-орбиталях. Довольно значительные положительные экзальтации
арилгалогенидов вполне согласуются с приведенными объяснениями. Следу-
ет помнить также, что большие размеры валентных оболочек галогенов,
делают магнитную восприимчивость чувствительной к любому простран-
194
Ароматичность и связанные с ней физические свойства
ственному расширению этих оболочек благодаря сопряжению с ароматиче-
ской системой.
е. Анизотропия диамагнетизма в ароматических системах. Паскаль
принял для диамагнитной константы углерода в насыщенных алифатических
соединениях величину, равную 6,0. Он рассчитал это значение из магнитной
восприимчивости элементарного углерода, полученного из тростникового
сахара. Позднее экспериментальным путем были определены соответствую-
щие величины для углерода в алмазе, которые практически совпадали
с приведенным выше значением. Если к этой величине приложить формулу
Лармора — Ланжевена, считая атом углерода сферическим и имеющим четы-
ре магнитно эффективных электрона, то можно показать, что средний квад-
ратичный радиус магнитных орбит равен 0,73 А. (7,3-Ю-2 нм). Введение
поправок, учитывающих влияние электронов остова, может уменьшить
рассчитанное значение радиуса только до величины 0,72 А (7,2-Ю-2 нм).
Таким образом, расчет дает величину, приближающуюся к ковалентному
радиусу углерода [0,77 А (7,7 -10~2 нм)], а следовательно, напрашивается
вывод о том, что в данном случае магнитные орбиты близки к атомным раз-
мерам.
Графит обладает гораздо большей диамагнитной восприимчивостью, чем
аморфный углерод или алмаз; диамагнитная константа атома углерода,
выведенная из восприимчивости графита, равна 42 единицам. Если вычи-
слить значение радиуса магнитных орбит исходя из этой величины, то радиус
будет в /7 раз больше, чем ранее вычисленный, т. е. намного больше, чем
радиус атома углерода. Это представляет собой частный случай общего
вывода Паскаля об аномально высоком диамагнетизме ароматических
молекул.
Однако качественное истолкование этих влияний, а именно вывод, что
л-электроны не ограничиваются магнитными орбитами в пределах атомов,
но способны описывать значительно большие орбиты вокруг колец, не может
быть облечено в количественную форму без учета направленности молеку-
лярного диамагнетизма. Согласно теории, этот избыточный диамагнетизм
неравномерно распределен по направлениям; он ограничивается только одной
из трех главных диамагнитных восприимчивостей молекулы, которые усред-
няются в диамагнитных измерениях, обсужденных в предыдущих раз-
делах.
Зависимость магнитной восприимчивости молекулы от направления можно
изучать, измеряя восприимчивость монокристаллов при условии, что струк-
тура кристалла известна и является подходящей, т. е. молекулы не ориен-
тированы таким образом, что их индивидуальные анизотропии взаимно
уничтожаются в кристалле в целом. Измерения такого рода были выполнены
Раманом, Кришнаном и Лонсдейлом. Значение этих измерений рассмотрено
Полингом и Лонсдейлом [104].
Исходя из магнитных данных, Лонсдейл рассчитал среднеквадратичные
радиусы магнитных орбит, описываемых каждым из двух главных типов
валентных электронов ряда ароматических молекул. Некоторые данные
Лонсдейла приведены в табл. 55. При этом были сделаны следующие упро-
щающие допущения: 1) магнитным эффектом внутренних электронов можно
пренебречь; 2) о-электроны рассматриваются как магнитно изотропные,
а радиусы магнитных орбит отвечают формуле Лармора — Ланжевена для
случая изотропии; 3) л-электроны характеризуются незначительным маг-
нетизмом в плоскости молекулы, так что их магнитные орбиты, возникаю-
щие только в плоскости молекулы под влиянием полей, нормальных к этой
13* 195
Глава IV
плоскости, должны подсчитываться по формуле Лармора — Ланжевена,
модифицированной для плоских молекул (см. разд. 4,а). Первое допущение
вполне правомерно. Второе и третье, по-видимому, включают взаимно ком-
пенсирующиеся погрешности, поскольку можно ожидать, что о-электроны
СС-связей под сжимающим влиянием оболочки имеют уменьшенную диамаг-
нитную восприимчивость по отношению к полям, ориентированным в пло-
скости молекулы. л-Электроны должны иметь определенную, хотя бы и ма-
лую, восприимчивость по отношению к полям, ориентированным в плоскости
молекулы. Несмотря на принятие ряда допущений, общий результат расче-
тов вполне удовлетворителен.
Таблица 55
Радиусы магнитных орбит электронов ряда ароматических молекул
(по данным Лонсдейла)
с-Каркас
р-Электроны л-Электроны
ЧИСЛО радиус, А (0,1 нм) ЧИСЛО .радиус, А (0,1 нм)
24 -0,71 6 -1,51
46 0,70 12 1,53
68 0,70 18 1,52
90 0,68 24 1,57
38 0,71 10 1,64
52 0,69 14 1,75
66 0,68 18 1,72
58 0,70 16 1,86
оо -0,79 ОО 7,80
Для всех типов ароматических ядер среднее значение радиуса магнитных
орбит о-электронов близко к 0,70 А. (7 -10~2 нм). Эта величина совпадает
со средним ковалентным радиусом атома углерода в ароматических системах.
Для бензола и неконденсированных многоядерных углеводородов (дифени-
ла, ге-дифенилбензола и ди-ге-дифенилила) среднее значение радиуса магнит-
ных орбит л-электронов в плоскости молекулы почти постоянно и равно
196
Ароматичность и связанные с ней физические свойства
1,53 А (15,3-10 2 нм). Это, как и следовало ожидать, несколько больше, чем
радиус бензольного кольца, измеренного для углеродного скелета бензола
[1,40 А. (14,0-Ю'2 нм)]. Очевидно, л-электроны в бензоле циркулируют
полностью вокруг кольца, а л-электроны неконденсированных многоядерных
углеводородов циркулируют главным образом вокруг собственных колец
и не могут в заметной степени под влиянием магнитного поля перемещаться
от одного кольца к другому.
Для конденсированных многоядерных углеводородов (нафталина, антра-
цена, хризена и пирена) значение среднего радиуса магнитных орбит л-элек-
тронов в плоскости молекулы возрастает с увеличением числа колец. Однако
оно возрастает не так быстро, как квадратный корень из числа электронов,
что наблюдалось бы, если бы все л-электроны передвигались вокруг всей
молекулы. Следует сделать вывод, что некоторые л-электроны циркулируют
вокруг собственных колец, в то время как некоторые другие описывают
орбиты, охватывающие несколько колец. Сравнение пирена с хризеном
показывает, что для данного числа колец значение среднего радиуса возра-
стает с увеличением степени конденсации. В более конденсированных систе-
мах большая часть л-электронов перемещается из кольца в кольцо под
влиянием магнитного поля, описывая, таким образом, орбиты, охваты-
вающие несколько колец. Предел достигается, когда конденсированная
ароматическая «сетка» становится неопределенно большой, как в графи-
те, для которого магнитные данные указывают на среднюю площадь маг-
нитных орбит электронов, равную приблизительно 30 бензольным
кольцам.
СПИСОК ЛИТЕРАТУРЫ
1. Lachmann A., The Spirit of Organic Chemistry, Macmillan, New York, 1899,
Chap. 3.
2. Kekule F. A., Bull. soc. chim. (France), 3, 98 (1865); Ann., 137, 169 (1866); Lehrbuch
der organische Chemie, Enke, Erlangen, 2, 493 (1866).
3. Ladenburg A., Ber., 2, 140 (1869); Ann., 172, 344 (1874); Wroblewsky E., Ber., 5, 30
(1872); Ann., 168, 153 (1873); 192, 196 (1878); Hiibner H., Peterman A., ibid., 149,
130 (1869); Hiibner H., ibid., 195, 1 (1879).
4. Griess P., Ber., 7, 1226 (1874); Salkowsky H., Ann., 173, 66 (1874); Koerner W., Gazz.
chim. ital., 4, 305 (1874).
5. Kekule F. A., Ann., 162, 77 (1872).
6. Ladenburg A., Ber., 2, 141, 272 (1869).
7. V. Baeyer A. et al., Ann., 245, 103 (1887); Ber., 19, 1797 (1888); Ann., 251, 257 (1889);
Ber., 23, 1277 (1890); Ann., 256, 1 (1890); Ann., 266, 169 (1892); Ann., 276, 259
(1893).
8. Claus A., Theoretische Betrachtungen und deren Anwendung zur Systematik der orga-
nischen Chemie, Freiburg, 1867, p. 207.
9. Claus A., Ber., 15, 1407 (1882); 20, 1423 (1887).
10. Armstrong H. E., J. Chem. Soc., 51, 254 (1887).
11. v. Baeyer A., Ann., 245, 118 (1887).
12. Bamberger E., Ber., 24, 1758 (1891); 26, 1946 (1893); Ann., 273, 373 (1893).
13. Thiele J., Ann., 306, 125 (1898).
14. Dewar J., Proc. Roy., Soc. (Edinburgh), 1866, 84.
15. Ingold С. K., J. Chem. Soc., 121, 1133 (1922).
16. Hiickel E., Z. Physik, 70, 204 (1931); 72, 310; 76, 626 (1932).
17. Pauling L., Wheland G. W., J. Chem. Phys., 1, 362 (1933); Sherman J., ibid., 2, 488
(1934); Wheland G. W., ibid., 3, 356 (1935).
18. Sklar A. L., J. Chem. Phys., 5, 669 (1937).
19. Craig D. P., Proc. Roy. Soc. (London), A200, 272, 390, 401 (1950).
20. Goeppert-Mayer M., Sklar A. L., J. Chem. Phys., 6, 645 (1938).
21. Sklar A. L., Lyddane R. H., ibid., 7, 374 (1939).
22. Parr R. G., Crawford B. L., Jr., ibid., 16, 1049 (1948).
197
Глава IV
23. Craig D. P., Proc. Roy. Soc. (London), A200, 474 (1950); Parr R. G., Craig D. P.,
Ross I. G., J. Chem. Phys., 18, 1561 (1950).
24. Buckingham A. £>., Chau J. Y. H., Freeman H. C., LeFevre R. J. W., Rao D. A. A. S. N.,
Tardif J., J. Chem. Soc., 1956, 1405.
25. Крейг Д. в книге «Небензоидные ароматические соединения», гл. 1, ИЛ, М.,
1963.
26. Mills W. Н., Nixon I. G., J. Chem. Soc., 1930, 2510.
27. Emerson G. F., Watts L., Petitt 77., J. Am. Chem. Soc., 87, 153 (1965); Emerson G. F.,
Watts L., Petitt R., Fitzpatrick J. D., ibid., 3253, 3254; Watts L., Fitzpatrick J. D.,
Petitt R., ibid., 88, 623 (1966).
28. Willstatter R., Weser E., Ber., 44, 3442 (1911); Willstatter R., Heidelburger M., Ber.,
46, 517 (1913).
29. Baker W., J. Chem. Soc., 1945, 258.
30. Reppe J. W., B.I.O.S. Reports, 137, № 22 (1935).
31. Cope A. C. and coworkers in more than 20 papers in J. Am. Chem. Soc., since
1950.
32. Cope A. C., Overberger C. G., J. Am. Chem. Soc., 70, 1433 (1948).
33. Tanaka I., Shuda S., Bull. Chem. Soc. Japan, 23, 54 (1950); Pearson W. B., Pimen-
tel G. C., Pitzer K. S., J. Am. Chem. Soc., 74, 3437 (1952).
34. Shoemaker D. P., Kindler H., Sly W. G., Srivastava R. C., J. Am. Chem. Soc., 87,
482 (1965).
35. Lippincott E. R., Lord R. C., McDonald R. S., J. Am. Chem. Soc., 73, 3370
(1951).
36. Pink R. C., Ubbelohde A. R., Trans. Faraday Soc., 44, 708 (1948).
37. Turner R. B., Meador W. R., Doering W. von E., Knox L. H., Mayer J. R., Wiley D. W.,
J. Am. Chem. Soc., 79, 4127 (1957) и ссылки па литературу, приведенные в этой
работе.
38. Рафаэль Р. в книге «Небензоидные ароматические соединения», гл. 8, ИЛ, М.,
1963.
39. Reppe J. W., Schlichting О., Klager К., Toepel Т., Ann., 560, 1 (1948).
40. Anet F. A. L., J. Am. Chem. Soc., 84, 671 (1962); Anet F. A. L., Bourn A. S. R.,
Lin Y. S., ibid., 86, 3576 (1964); Gwynn D. E., Whitesides G. M., Roberts J. D., ibid.,
87, 2862 (1965).
41. Sondheimer F., Amiel У., Wolovsky R., J. Am. Chem. Soc., 79, 4247 (1957); Sondhei-
mer F., Wolovsky R., ibid., 81, 1771, 4755 (1959); Sondheimer F., Wolovsky R., Guo-
mi У., ibid., 82, 755 (1960); Sondheimer F., Guomi У., ibid., p. 5765; Sondheimer F.,
Wolovsky R., Amiel У., ibid., 84, 274 (1962); Sondheimer F., Wolovsky R., Aniel J.,
Jackman L. M., Ben-Effraim D. A., Guomi У., Bolhner-By A. A., ibid., p. 4307; Guo-
mi У., Sondheimer F., Proc. Chem. Soc., 1964, 299; Guomi У., Sondheimer F., Male-
ra A., Wolovsky R., ibid., p. 397; Beezer A. E., Mortimer С. T., Springall H. D., Sond-
heimer F., Wolowsky R., J. Chem. Soc., 1965, 216.
42. Dauben H., Bertelli D. J., J. Am. Chem. Soc., 83, 4659 (1961).
43. Robertson J. M., Shearer H. M. M., Sim G. A. Watson D. G., Acta Cryst., 15, 1
(1962).
44. Klemm W., Ber., 90, 1051 (1957).
45. Heilbronner E., Wieland K., Helv. Chim. Acta, 30, 947 (1947).
46. Anderson A. G., Jr., Nelson J. A., J. Am. Chem. Soc., 72, 3824 (1950); Anderson A. G.,
Jr., Nelson J. A., Tazuma J. J., ibid., 75, 4980 (1953).
47. Lolhrop W. C., J. Am. Chem. Soc., 63, 1187 (1941).
48. Cass R. C., Springall H. D., Quincey P. G., J. Chem. Soc., 1955, 1188.
49. Carr E. P., Pickett L. W., Voris D., J. Am. Chem. Soc., 63, 3031 (1941).
50. Бейкер У., Мак-Оми Дж. в книге «Небензоидные ароматические соединения», гл. 2,
ИЛ, М., 1963.
51. Baker W., Boarland М. Р. V., McOmie J. F. W., J. Chem. Soc., 1954, 1476.
52. Blood С. T., Linstead R. P., J. Chem. Soc., 1952, 2663.
53. Kofod H., Sutton L. E., Jackson J., J. Chem. Soc., 1952, 1467.
54. Wibaut J. P., Gulje A. R., Proc. Koninkl. Akad. Nederland. Wetenschap, B54, 330
(1951); Wibaut J. P., ibid., B68, 117 (1965).
55. Wibaut J. P., Herzberg S., Proc. Koninkl. Akad. Nederland. Wetenschap., 56, 333
(1953).
56. Ingold С. K., Jessop J. A., J. Chem. Soc., 1929, 2357; 1930, 713.
57. Krollpfeiffer F., Schneider K., Ann., 530, 34 (1937); Wittig G., Felletschin F., Ann.,
555, 133 (1944); Wittig G., Laib H., Ann., 580, 57 (1953).
58. Kealy T. J., Pauson P. L., Nature, 168, 1039 (1951).
59. Dunitz J. D., Orgel L. E., Nature, 171, 121 (1953).
60. Pauson P. L , Quart. Revs., 9, 391 (1955).
198
Ароматичность и связанные с ней физические свойства
61. Doering W. von Е., Knox L. Н., J. Am. Chem. Soc., 76, 3203 (1954).
62. Wierl R., Ann. Physik, 8, 521 (1931); Jones P. L. F., Trans. Faraday Soc., 31, 1036
(1935); Schomaker V., Pauling L., J. Am. Chem. Soc., 61, 1769 (1939).
63. Angus W. R., Bailey C. R., Hale J. B., Ingold С. K., Leckie A. H., Raisin C. G.,
Thompson J. W., Wilson C. L., J. Chem. Soc., 1936, 912, et seq.; Bailey C. R., Best A. P.,
Carson S. C., Gordon R. R., Hale J. B., Herzfeld N., Holden J. W., Ingold С. K.,
Leckie A. H., Poole H. G., Weldon L. H. P., Wilson C. L., ibid., 1946, 222,
et seq.
64. Angus W. R., Bailey C. R., Hale J. B., Ingold С. K., Leckie A. H., Raisin C. G., Wil-
son C. L., J. Chem. Soc., 1936, 966.
65. Mair R. D., Homig D. F., J. Chem. Phys., 17, 1236 (1949).
66. Thomson J. K., Pullin A. D. E., J. Chem. Soc., 1957, 1658; Ferguson E. E., J. Chem.
Phys., 26, 1265 (1957).
67. Langseth A., Stoicheff В. P., Canadian J. Phys., 34, 350 (1956).
68. Wilson E. B., Physical Rev., 45, 706 (1934); Bell R. P., Trans. Faraday Soc., 41,
293 (1945); Garforth F. M., Ingold С. K., Poole H. G., J. Chem. Soc., 1948,
492, 508.
69. Arnett R. L., Crawford B. L., Jr., J. Chem. Phys., 18, 118 (1950).
70. Brown C. J., J. Chem. Soc., 1953, 3265.
71. Robertson J. M., White J. G., Proc. Roy. Soc. (London), A190, 329 (1947).
72. Abrahams S. C., Robertson J. M., White J. G., Acta Cryst., 2, 233, 238 (1949); Cruik-
shank D. W. J., Sparks R. A., Proc. Roy. Soc., A258, 270 (1960).
73. Mathieson A. M., Robertson J. M., Sinclair V. C., Acta Cryst., 3, 245 (1950); Sin-
clair V. C., Robertson J. M., Mathieson A. M., ibid., p. 251; Cruikshank D. W. J.,
Sparks R. A., Proc. Roy. Soc., A258, 270 (1960).
74. Robertson J. M., White J. G., J. Chem. Soc., 1945, 607.
75. Donaldson D. M., Robertson J. M., White J. G., Proc. Roy. Soc., A220, 157
(1953).
76. Robertson J. M., White J. G., J. Chem. Soc., 1947, 358.
77. Robertson J. M., White J. G., J. Chem. Soc., 1937, 1001.
78. Donaldson D. M., Robertson J. M., White J. G., Proc. Roy. Soc., A220, 311
(1953).
79. Schomaker V., Pauling L., J. Am. Chem. Soc., 61, 1769 (1939).
80. Bak B., Hansen L., Rastrup-Andersen J., J. Chem. Phys., 22, 2013 (1954).
81. Bak B., Hausen L., Rastrup-Andersen J., Disc. Faraday Soc., 19, 30 (1955).
82. Innes К. K., Merritt J. A., Tincher W. C., Tilford S. G., Nature, 187, 500 (1960).
83. Данные для C3H6: Knowlton J. W., Rossini F. D., J. Res. Nat. Bur. Standards, 43,
113 (1949); для C4HS: Kaarsenmaker S., Coops J., Rec. trav. chim., 69, 1364 (1950);
для С5Н10 — CSH16: Spitzer R., Huffman II. M., J. Am. Chem. Soc., 60, 211 (1947);
для C4H8 — CQH1S: Coops J., Kaarsenmaker S., Rec. trav. chim., 71, 261 (1952);
для СщНго — С17Нз4: Coops J., van Kamp H., Lambregts W. A., Visser B. J.,
Dekker H., ibid., 79, 1226 (1960); Крэйг Д. в книге «Небензоидные ароматические
соединения», гл. 2, ИЛ, М., 1963; Sicher J. в книге «Progress in Stereochemistry»,
Editors de la Mare P. B. D. and Klyne W., Butterworths, London, 1962, Vol. 3,
p. 202.
84. v. Baeyer A., Ber., 18, 2277 (1885).
85. Sachse II., Ber., 23, 1362 (1890).
86. Perkin W. H., Simonsen J., J. Chem. Soc., 91, 816 (1907).
87. Ruzicka L., Stoll M., Schinz H., Helv. Chim. Acta, 9, 249 (1926).
88. Ziegler K., Emde II., Ohlinger II., Ann., 504, 94 (1933).
89. Prelog V., J. Chem. Soc., 1950, 420.
90. Prelog V., Urech II. J., Bothner-By A. A., Wiinsch J., Helv. chim. Acta, 38, 1095
(1955); Urech H. J., Prelog V., ibid., 40, 477 (1957).
91. Kiing W., Prelog V., Croat. Chim. Acta, 29, 357 (1957).
92. Bryan R. F., Dunitz J. D., Helv. Chim. Acta, 43, 3 (1960); Huber-Buser E., Dunitz J. D.,
ibid., p. 760; Dunitz J. D., Shearer II. M., ibid., p. 18.
93. Dunitz J. D., Schomaker V., J. Chem. Phys., 20, 1703 (1952); Rathfens G. W., Free-
man N. K., Gwinn W. D., Pitzer K. S., J. Am. Chem. Soc., 75, 5634 (1953); Lam-
bert J. B., Roberts J. D., ibid., 87, 3884 (1965).
94. Cope A. C., Ganellin C. R., Johnson H. W., Jr., J. Am. Chem. Soc., 84, 3191 (1962);
Cope A. C., Ganellin C. R., Johnson H. W., Jr., van Auken T. W., Winkler H. J. S.,
ibid., 85, 3276 (1963); Cope A. C., Banholzer K., Keller H., Pawson B. A.,
Whang J. H., Winkler J. S., ibid., 87, 3644 (1965); Cope A. C., Pawson B. A.,
ibid., p. 3649.
95. Kistiakowsky G. B., Ruhoff J. R., Smith H. A., Vaughan W. E., J. Am. Chem. Soc.,
58, 146 (1936).
199
Глава IV
96. Dolliver М. A., Gresham Т. Е., Kistiakowsky G. В., Smith Н. A., Vaughan W. Е.,
J. Am. Chem. Soc., 60, 440 (1938).
97. Тtimer R. В., Meador W. R., Doering W. von E., Knox L. H., Mayer J. R., Wiley D. W.,
J. Am. Chem. Soc., 79, 4127 (1957).
98. Cass R. C-, Springall H. D., Quincey P. G.. J. Chem. Soc., 1955, 1188.
99. Pascal P., Ann. chim. phys., 19, 5 (1910); 25, 289 (1912); 29, 218 (1913).
100. Bhatnagar S. S., Mathur К. M., Physical Principles and Applications of Magneto-
chemistry, Macmillan, London, 1935, Chap. 4.
101. van Vleck J. H., The Theory of Electric and Magnetic Susceptibilities, Clarendon Press,
Oxford, 1932, p. 206.
102. Seiwood P. W, Magnetochemistry, Interscience, New York, 1943, p. 33.
103. Angus W. R., Ann. Repts. on Progress Chemistry (Chem. Soc. London), 36, 33
(1941).
104. Raman С. V., Krishnan K. S., Proc. Roy. Soc. (London), A113, 511 (1927); Lonsdale K..
Krishnan K. S., Proc. Roy. Soc. (London), A156, 597 (1936); Pauling L., J. Chem
Phys., 4, 673 (1936); Lonsdale K., Proc. Roy. Soc. (London), A159, 149 (1937).
ГЛАВА V
КЛАССИФИКАЦИЯ РЕАГЕНТОВ И РЕАКЦИЙ
1. Классификация реагентов ........................202
2. Классификация реакций ..........................207
Список литературы...............................225
1. Классификация реагентов
Химические реакции являются в сущности электрическими процессами.
В соответствии с этим можно представить себе, что действие реагента на моле-
кулу органического соединения осуществляется благодаря тому, что реагент
имеет некоторое остаточное сродство к атомному ядру или к электронам;
возможно, конечно, и одинаковое сродство к ядру и электронам, т. е. сродство
к атомам. Большинство реагентов, которые обладают четным числом элек-
тронов, принадлежит к одному из первых двух указанных типов; реагенты
с нечетным числом электронов принадлежат к третьему, более обособлен-
ному типу.
а. Обобщенная концепция восстановления и окисления. Удобным
и исторически оправдавшим себя подходом к классификации реагентов
с четным числом электронов, т. е. реагентов, атакующих ядра или электроны,
является последовательное развитие представлений о восстановлении и окис-
лении. Эти взгляды высказывались еще в период доэлектронных теорий
Михаэля и Лэпуорта. После возникновения электронной теории атома
и электронной теории валентности одними из первых, кто применил и раз-
вивал такого рода представления, были Фрай и Штиглиц [1].
Задолго до введения электронной теории валентности термины «восстановле-
ние» и «окисление» перестали ограничиваться реакциями отщепления и при-
соединения кислорода. Водород или металл окисляются при превращении
в катион, соответствующий окислу, т. е. в кислоту или в соль; катион вос-
станавливается, если он превращается в водород или металл. Аналогично
галоген или другой неметалл восстанавливаются, превращаясь в галоген-ион
или в другой анион кислоты или соли; анион окисляется, превращаясь
в галоген или в другой неметалл. Электронная формулировка подобного рода
процессов восстановления — окисления на этой ступени более расширенного
толкования термина показывает, что восстановительный агент отдает элек-
троны окислительному агенту. В некоторых реакциях действительно наблю-
дается перенос электронов, например:
Fe (металл)-)-Cu2+ —> Fe2+-|-Cu (металл)
В случае других восстановительно-окислительных процессов происходит
только частичный перенос электронов; восстанавливающий агент уступает
свои электроны, не теряя связи с ними, в то время как окисляющий агент
получает электроны, не присваивая их целиком; для иллюстрации можно
привести следующий пример:
I- + C1—Cl —> I —С1 + С1-
Эта форма обобщенной восстановительно-окислительной концепции долгое
время была общепринятой. При развитии электронной интерпретации до
уровня широких обобщений было бы нелогично останавливаться на рассмот-
рении только случаев передачи и получения электронов. Так, процесс
нейтрализации
ОН- + Н —ОН2 —> НО —Н + Н2О
в доэлектронный период не мог рассматриваться как восстановление —
окисление. Однако процесс нейтрализации можно описать в рамках перво-
начальной концепции окисления — восстановления с помощью рассуждений,
лишь несколько более сложных, чем те, которые потребовались, например,
в приведенном выше обмене галоген — галогенид-ион. Ион гидроксила
202
Классификация реагентов и реакций
превращается в связанный гидроксильный радикал, который, как и свобод-
ный гидроксильный радикал, может считаться окислительным агентом, хотя
и в ограниченном смысле. Поэтому ион гидроксила подвергается изменению,
близко соответствующему окислению, и ведет себя подобно восстановитель-
ному агенту; следовательно, эти термины могли быть распространены и на
данный случай. Таким же образом сольватированный ион водорода связы-
вается атомом водорода, который, подобно свободному атому водорода,
является признанным восстановительным агентом. Следовательно, можно
считать, что ион водорода способен к восстановлению или что он ведет себя
как окислительный агент. Такую аргументацию можно развить и перенести
на процесс присоединения
F-4-BF3 —> BFj
в котором связь с окислением — восстановлением в первоначальном смысле
выражена несколько более косвенно. При этом следует прежде всего под-
черкнуть, что ион фтора превращается в связанный атом фтора, который,
как и свободный атом фтора, в соответствии с прежними представлениями
по окислительным свойствам можно рассматривать как эквивалент гидро-
ксильного радикала.
При рассмотрении ряда примеров, незначительно отличающихся один
от другого, можно в известной степени расширить пределы основной кон-
цепции восстановления — окисления. Очевидно, нет оснований для установ-
ления четких границ, которые охватывали бы все случаи передачи и полу-
чения электронов. В этом состоит приближение к применяемой в дальнейшем
классификации реагентов по их полярности. Ниже будут показаны два других
подхода, которые ведут в сущности к этой же классификации.
б. Нуклеофильные реагенты. Реагенты, которые участвуют в реак-
циях, отдавая свои электроны или разделяя их с другими атомными ядрами,
называются нуклеофильными реагентами. В табл. 56 приведены некоторые
нуклеофильные реагенты и данные, характеризующие некоторые общие
особенности химии таких реагентов.
Реагенты, приведенные в табл. 56, делятся на две основные группы, причем
в одну входят первые три, а в другую — остальные шесть реагентов. Реаген-
Таблица 56
Некоторые нуклеофильные реагенты
Номер реаген- та Реагент Обозна- чение a Число активных электронов
1 [Fe(CN)e]i- R 1 1
2 Na (мет.) R 1 ! полностью передаются
3 Sn2+ R 2 J
4 so2 R 1 пара X
5 s2- R, В 1, 2, 3, 4 пары I
6 CN- (R),B 1 пара
7 он- В -1 о ттогчгт г частично, но передаются 1, нары 1
8 NHg В 1 пара |
9 [Co(H2O)5(OH)]2+ В 1 пара J
а R — восстановительный агент согласно доэлектронным представлениям (см. текст); В — основа-
ние по определению Бренстеда и Лоури (см. текст).
203
Глава V
ты, указанные под номерами 1—3, при реакциях полностью отдают один
или более электронов; эти реагенты являются типичными восстановитель-
ными агентами в ограниченном смысле этого термина. В выбранных примерах
эти три реагента обладают совершенно различными электрическими заряда-
ми. Приведенные примеры показывают, что нуклеофильный характер опре-
деляется не только зарядом, но зависит в гораздо более сложной степени
от электронного строения реагентов.
Деление веществ по их восстановительной способности в обычном доэлек-
тронном смысле этого термина не совпадает с основным делением таблицы
на две группы. Восстановительными свойствами обладает и часть соединений
второй группы реагентов, которые реагируют только с обобщением одной
или более неподеленных электронных пар. Реагенты № 4 и 5 обладают вос-
становительными свойствами в обычном смысле, № 6 является промежуточ-
ным, а реагенты № 7—9 не обладают восстановительными свойствами.
Вторая группа включает в себя и другую подгруппу, а именно вещества
№ 5—9, к которой относится также и часть восстановительных реагентов.
Одинаковое поведение таких реагентов по отношению к присоединению
протона было отмечено Бренстедом [2] и Лоури [3], которые придали рас-
ширенное толкование терминам «кислота» и «основание» по аналогии с рав-
новесиями типа
S2- + H+ дД SH-
CN- + H+ ДД HCN
ОН- + Н+ Н2О
NH3 + H+ ДД NH+
[Со(Н2О)5(ОН)]2+ + Н+ ДД [Со(Н2О)в]3+.
Определяющим уравнением является следующее:
Основание-)-Н+ Кислота.
Всякое основание и кислота, между которыми существует такое соотноше-
ние, называются «сопряженными». Из оснований, представленных в левой
части приведенных уравнений, только NH3 мог быть в прежнее время причи-
слен к основаниям. Среди кислот, перечисленных в правой части, только
HCN и занимающая промежуточное положение вода могли быть прежде
признаны кислотами. Бренстед и Лоури отметили, однако, что различие
в электрическом заряде не имеет существенного значения по сравнению
со сходством в химическом поведении, положенным в основу их определения,
которое теперь почти повсеместно принято. Основность и кислотность не свя-
заны непосредственно с зарядами. Оба эти свойства зависят гораздо более
сложным образом от электронного строения реагентов.
Таким образом, в подгруппе, в которую входят реагенты № 5—9, представ-
лены типичные основания. Естественно, что все основания нуклеофильны.
Основность, т. е. сродство к водородным ядрам, является особым выраже-
нием нуклеофильного характера, или нуклеофильности, т. е. сродства к атом-
ному ядру вообще. Нуклеофильность в любой форме, в том числе основность,
является ярко выраженным свойством строения, не всегда соответствую-
щим заряду.
в. Электрофильные реагенты. Реагенты, действие которых связано
с приобретением электронов или с частичным приобретением электронной
пары, которая ранее принадлежала полностью другой молекуле, называют
204
Классификация реагентов и реакций
электрофильными реагентами или электрофилами. В табл. 57 перечислены
некоторые электрофильные реагенты.
Таблица 57
Некоторые электрофильные реагенты
Номер реагента Реагент Фрагменты Обозначение а Активные электроны
перенесенный освобожден- ный
1 S2O|- 2S( )?- Ох 2 1
2 [Fe(CN)e]3- Без расщепления Ох I полное приоорете-
3 [Fe(H2O)e]3+ To же Ох J ние
4 О3 О О2 Ох 1 пара А
5 so2- О sor Ох 1 пара
6 Cl2 С1+ С1- Ох 1 пара
С1+ 1 ОН- 1
7 HOCl Н+ 1 ОС1- } Ох, А 1 пара
Г 0 HNO2 1
8 HNOg ОН- У Ох, А 1 пара
1 н+ NO3 J частич-
9 H3O+ Н+, 2Н+ н2о, он- А . п ное при- 1,2 пары обрет£_
10 HC1 Н+ ci- А 1 пара ние
11 HSOj н+ SO2- А 1 пара
12 HgClj Без расщепления 1 пара
13 A1C13 То же 1 пара
14 SnCl4 » 2 пары
15 NO| » » 1 пара
16 Be2+ » 4 пары
17 Co3+ » » 6 пар
а Ох — окислительный агент согласно доэлектронным представлениям; А — кислота по опреде-
лению Бренстеда и Лоури.
В этой таблице следует различать две главные группы: первые три реагента
составляют одну группу и все остальные — другую. Реагенты № 1—3 при
реакциях полностью присваивают электроны молекул, с которыми реаги-
руют. Все они являются типичными окислительными агентами в обычном
смысле. Приведенные примеры, в которых рассматриваются реагенты, харак-
теризующиеся весьма различным состоянием электронного заряда, могут
служить иллюстрацией того положения, что электрофильный характер
не просто соответствует заряду.
Рассматриваемая первая основная группа реагентов содержит две подгруп-
пы. Это деление зависит от того, заполнена ли полностью электронная
оболочка приобретающего электроны реагента или нет. Первый случай
представлен реагентом № 1; приобретение электронов здесь связано с рас-
щеплением реагента. В случае реагентов № 2 и 3 присоединение электронов
возможно без расщепления.
Вторая группа в таблице включает реагенты № 4—17, которые при реакции
приобретают лишь частично некоторые электроны другой молекулы (обоб-
щение неподеленных пар). Окислительные свойства характерны также и для
реагентов этой группы, но не для всей группы полностью. Реагенты № 4—8
являются окислителями в обычном смысле, в то время как реагенты № 9—14,
за исключением немногочисленных особых случаев, не рассматриваются
как окислители.
20,5
Глава V
Вторая группа в свою очередь делится на две подгруппы в зависимости
от того, заполнены или не заполнены электронные оболочки реагентов.
Например, в случае реагентов № 4—11 приобретаемые электроны могут
быть получены только путем расщепления реагента; поскольку реагент
требует лишь частичного присоединения электронов, то при его расщеплении
часть реагента переносится и связывается в продуктах реакции, в то время
как другая часть остается свободной. Переносимая часть может быть любым
молекулярным осколком, способным к включению новой электронной пары
в свою валентную оболочку. Примерами могут служить нейтральный кисло-
родный атом, протон, хлорипий-ион и нитроний-ион. Если переносимая
часть является протоном, то реагент ведет себя как кислота по определению
Бренстеда и Лоури. Таким образом, кислоты принадлежат к числу электро-
филов. Способность к переносу протона, очевидно, представляет собой част-
ный случай способности переноса атомного ядра с меньшим числом электрон-
ных пар, чем оно может иметь в своей валентной оболочке. Поэтому кислот-
ность является особым проявлением электрофильного характера, или
электрофильности, т. е. сродства к внешним электронам вообще. Электро-
фильные реагенты этой подгруппы в зависимости от того, действуют ли они
с переносом такого нейтрального атома, как кислород, или такого положи-
тельного иона, как протон, могут различаться по зарядности. То, что Брен-
стед и Лоури отметили для кислот, справедливо, по-видимому, для более
широкой группы реагентов, для которых химическая активность не выра-
жается простым отношением к заряду.
В последней подгруппе, представленной реагентами № 12—17, дополнитель-
ные электронные пары могут быть включены в электронные оболочки, поэто-
му такие реагенты соединяются с атакуемой молекулой без расщепления.
Нормальное взаимодействие состоит в координации с неподеленными элек-
тронными парами атома или с л-электронными парами ненасыщенной моле-
кулы. При этом, как и в предыдущих случаях, аналогию в поведении этих
реагентов нельзя объяснить только состоянием зарядов.
г. Другие подходы к классификации реагентов. Наряду с пред-
ставлениями Фрая и Штиглица об электронной природе процессов окисле-
ния, которым в дальнейшем изложении отдано предпочтение, были высказа-
ны и подтверждены два других положения, которые ведут или могут при-
вести к тому же конечному результату.
Один из таких подходов выдвинут Лэпуортом [4], который исходил из ион-
ных представлений. Лэпуорт был одним из немногих, кто еще в начале
нынешнего столетия полагал, что многие органические реакции протекают
как ионные. Он уподоблял все реагенты либо анионам, либо катионам.
«Анионоидным» он считал реагент, который ведет себя подобно аниону,
а «катионоидным» — реагент, который ведет себя как катион. Недостаток
этой концепции состоит в излишнем подчеркивании роли зарядов, при кото-
ром не учитываются представления Бренстеда и Лоури о том, что близкая
реакционная способность может наблюдаться и при различии в зарядах.
Например, нельзя сказать с полной уверенностью, что ион перманганата
в качестве окислительного агента ведет себя как катион или что ион двух-
валентного олова при восстановлении ведет себя подобно аниону. Такие
представления об анионоидном или катионоидном характере реагентов
необходимо уточнить, указав, что анионоидным или катионоидным стано-
вится реагент, который ведет себя так, как должен был бы вести себя анион
или катион, если бы заряд играл доминирующую роль в реакционной спо-
собности. В такой трактовке классификация Лэпуорта эквивалентна клас-
сификации, данной в предыдущем разделе: «анионоидный» становится сино-
206
Классификация реагентов и реакций
нимом «нуклеофильного» и «катионоидный» — синонимом «электрофильного»
реагентов.
Другой подход был предложен Льюисом [5], исходившим из классической
теории кислот и оснований; он стремился расширить существовавшие пред-
ставления. Как отмечалось, Бренстед и Лоури отбросили исходное ограни-
чение по отношению к нейтральным веществам, оставив в качестве суще-
ственного ограничения перенос протона. Льюис предложил не ограничивать-
ся реакциями с участием протона и применил название «кислота» к каждому
электрофилу, образующему связь, т. е. ко всякому веществу, которое спо-
собно принимать часть электронов от другой молекулы. Согласно его опре-
делению, «основное вещество предоставляет электронную пару для химиче-
ской связи, а кислотное вещество принимает такую пару». Соответствующие
примеры имеются во вторых главных группах табл. 56 и 57. Льюис касался
главным образом вопроса о способности реагентов к координации; было
естественно сделать следующий шаг с тем, чтобы несколько расширить
понятия «основание» и «кислота» и включить те реагенты, которые полностью
отдают или принимают электроны. Льюис, вероятно, допускал такое рас-
ширенное толкование, которое в явной форме не иллюстрировалось им,
но все же соответствует по крайней мере одному из данных Льюисом альтер-
нативных определений. Примеры, подтверждающие это, можно найти в пер-
вых разделах табл. 56 и 57. В таком более широком смысле термин «основа-
ние» становится синонимом «нуклеофильного» реагента, а термин «кислота» —
синонимом «электрофильного» реагента. Цепь рассуждений Льюиса трудно
критиковать; однако определения «оснований» и «кислот» по Бренстеду
и Лоури весьма полезны и их нельзя не учитывать. В дальнейшем изложении
понятие «оснований» и «кислот» применяется без особых объяснений в смыс-
ле, данном Бренстедом и Лоури, а термины «нуклеофильный» и «электро-
фильный» применяются для описания более широких классов реагентов *.
2. Классификация реакций
а. Гомолитические и гетеролитические реакции. При химических
реакциях образуются и разрушаются связи; наиболее важным видом связи,
которая может образоваться или разрушиться, является ковалентная связь.
Как отмечалось в гл. I, возможны два пути создания или разрушения кова-
лентной связи.
Во-первых, она может образоваться вследствие обобщения электронов,
поставляемых каждым участвующим в процессе реагентом (коллигация).
Эта связь может разрываться при обратном процессе — гомолизе.
Коллигация
А-+В- ( > А: В
Гомолиз
В реакциях этого типа А- и В- являются атомами с нечетным числом элек-
тронов, такими, как Н- или С1-, или свободными радикалами, такими, как
НО- или СН' (где точка означает наличие одного неспаренного электрона).
Известно небольшое число молекул с нечетным числом электронов, стабиль-
ных в обычных условиях, хотя многие из них можно на короткое время
* Можно было бы расширить значение терминов «восстановление» и «окисление»
настолько, чтобы они стали синонимами «нуклеофильный» и «электрофильный» соответ-
ственно. Но это не предлагалось по аналогичной причине, а именно потому, что представ-
ления, которые еще с доэлектронного периода связывались с терминами «восстановление»
и «окисление», придавали этим словам определенный смысл, и поэтому было нецелесо-
образно применять их в более широком смысле.
207
Глава V
получить при термической, фотолитической или радиационной диссоциации
ковалентной связи. Каталитические явления на поверхности также могут
служить причиной такой диссоциации (например, диссоциации водорода
на платине), хотя продукты диссоциации (в данном примере атомы водорода
или ионизированные атомы) остаются связанными с поверхностью. Реакции
с образованием и разрывом ковалентных связей путем коллигации и гомолиза
соответственно называются гомолитическими реакциями. Многие такие
реакции начинаются с одноэлектронного термического, радиационного или
катализируемого активной поверхностью гомолиза. Некоторые реакции,
по-видимому, инициируются взаимным гомолизом двух связей с образова-
нием более прочной связи или связей. Если в начальной стадии процесса
возникают частицы с нечетным числом электронов, то в дальнейшем они
не только соединяются одна с другой, но также атакуют молекулу с четным
числом электронов, вызывая тем самым цепные реакции.
Во-вторых, ковалентная связь может быть образована координацией и раз-
рушена обратным процессом — гетеролизом
Координация
А : +В ~» А : В
Гетеролиз
В процессах этого типа частицы А: и В, как правило, представляют собой
молекулы с четным числом электронов, в большинстве случаев стабильные
при обычных условиях. Реакции, в которых образуются и разрываются
связи вследствие координации и гетеролиза, известны под названием гете-
ролитических реакций. Большинство низкотемпературных реакций, за исклю-
чением тех, которые инициируются окислительно-восстановительным про-
цессом, термической, радиационной или поверхностнокатализируемой дис-
социацией, являются гетеролитическими реакциями. В простейшем случае
образуется только одна связь, т. е. реакция включает одиночный акт коор-
динации или гетеролиза. Но во многих случаях вещества взаимодействуют
с образованием двух или более связей и включают как координацию, так
и гетеролиз. Сейчас, когда прошло столетие после открытия структурной
теории, можно сказать, что в общем органическая химия развивалась на осно-
ве представлений о гетеролитическом разрыве и образовании связей. Тем
не менее в последнее время широкое внимание привлекают и гомолитические
реакции. Поэтому здесь уместно перейти к более детальной классификации
гетеролитических реакций.
б. Нуклеофильные и электрофильные реакции. Различию в функ-
циях и противоположному характеру молекул, обозначенных А: и В в при-
веденном выше уравнении для координации и гетеролиза, очевидно, должно
соответствовать в классификации реагентов с четным числом электронов
деление на нуклеофильный и электрофильный типы, причем А: принадлежит
к первому типу, В — ко второму.
Совершенно ясно, что можно лишь чисто условно считать одно из двух
взаимодействующих веществ «реагентом», а другое —«субстратом», т. е. веще-
ством, на которое воздействует реагент.
Однако во многих реакциях органической химии определенное разграничение
реагирующих веществ является обоснованным. При реакции брома с нафта-
лином бром рассматривается как реагент; если фенилгидразин взаимодей-
ствует с ацетоном, первый обычно считается реагентом. Если нет сомнений
в том, какое вещество является «реагентом», то реакции удобно обозначать
в соответствии с типом, к которому реагент принадлежит. Так как бром
представляет собой электрофил, бромирование нафталина называют злектро-
208
Классификация реагентов и реакций
филъной реакцией, причем нафталин рассматривается как атакуемая молеку-
ла; фенилгидразин является нуклеофилом, и превращение ацетона в его
фенилгидразон классифицируется как нуклеофильная реакция, причем ацетон
рассматривается как атакуемая молекула.
в. Нуклеофильное и электрофильное замещение. Приведенная выше
классификация реагентов особенно легко применима для реакций заме-
щения, так как в этом случае не требуется знания механизма реакции.
Рассмотрим образование метилэтилового эфира при действии алкоголят-
иона на иодистый метил
С2Н5О"+СН31 CH3OC2H5+I- (SN)
Реакция, очевидно, является замещением: иод замещается этоксильной
группой. Ион алкоголята рассматривается как реагент, который в данном
случае является замещающим агентом. Независимо от механизма реакции
замещающий агент предоставляет оба электрона для создания связи, т. е.
ведет себя как нуклеофильный реагент. Поэтому реакцию правильно назвать
реакцией замещения нуклеофильным реагентом или, короче, нуклеофильным
замещением. Часто такое обозначение сокращается введением символа SN.
Это делается с целью создания основы сокращенного обозначения механиз-
ма реакции, который можно записать более подробно, если применить допол-
нительные символы; такая система записи будет применяться в дальнейшем
изложении.
Теперь рассмотрим образование нитробензола при действии нитроний-иона
на бензол
NOJ+CeH5-Н С6Н5-NO24-H+ (Se)
Эта реакция является замещением, а нитроний-ион — замещающим аген-
том. Оставляя в стороне детали механизма реакции, можно сказать, что
новый заместитель — нитрогруппа — связывается в продукте реакции парой
электронов, которые ранее принадлежали атакуемой молекуле бензола;
иначе говоря, замещающий агент является электрофильным. Данную реак-
цию, следовательно, можно классифицировать как электрофильное замеще-
ние и обозначить символом Se-
Посмотрим, насколько широко можно обобщить эти простые соображения.
В приведенном примере нуклеофильного замещения механизм реакции
является одним из простейших механизмов, с помощью которых галоген
в алкилгалогениде можно заместить на группу типа OR. В качестве второго
примера можно рассмотреть превращение хлористого mpem-бутила в водном
растворе в mpem-бутиловый спирт
Н2О трет C4Hg - С1 —> трет-СЩд — ОН Н+ С1~
Механизм этой реакции более сложен, чем в рассмотренном выше случае,
однако для широко обсуждаемой классификации нет необходимости знать
этот механизм. Хотя данная реакция может состоять из многих стадий
и многие молекулы могут быть какими-либо способами попутно вовлечены
в этот процесс, все же остается справедливым, что образование связи с новыми
заместителями происходит за счет электронов, получаемых от замещающего
агента. Такая реакция, следовательно, также относится к реакциям нуклео-
фильного замещения. Даже если ускорить данную реакцию путем добавки
солей серебра или ртути, приведенное выше утверждение сохраняет силу,
поскольку катализирующий катион не является замещающим агентом.
В общем случае реакции замещения галогена в алкилгалогенидах или в доста-
точно реакционноспособных арилгалогенидах на группы ОН, OR, SH, SR, SR*,
14-0772
209
Глава V
SeRj и т. д. при действии воды, спиртов, сернистого водорода, меркаптанов,
диалкилсульфидов, диалкилселенидов и других или же, если возможно,
анионов этих веществ относятся к типу нуклеофильного замещения. Анало-
гично к нуклеофильному замещению относятся реакции, в которых связан-
ный галоген замещается на группу NH2, NHR, NR2, NRJ, PR*, AsRJ и дру-
гие при действии аммиака, аминов, амидов, имидов, фосфинов, арсинов
и т. д. или, если это возможно, анионов этих веществ. Замещение галогена
изотопом того же галогена или другим галогеном при действии галоген-
ионов также представляет собой один из видов нуклеофильного замещения.
Такой же реакцией является замещение галогена на CN, СН(СООС2Н5)2 и пр.,
происходящее под действием соответствующих анионов.
Приведенная выше реакция гидролиза хлористого mpem-бутила в кислой
среде представляет равновесную реакцию. Все сказанное о прямой реакции
без изменения приложимо и к обратной реакции
трет-С^Н.д—ОН-j-Н+ + С1~ —> трет С4Н8 - С1-]-Н2О
Эта реакция также представляет собой нуклеофильное замещение. Взаимо-
действие простых эфиров с алкилгалогенидами, как в анализе по Цейзелю
СН3— OR+H++I- —> СН3 —I-j-HOR
также соответствует нуклеофильному замещению. Вообще те реакции,
в которых гидроксильные группы в спиртах или алкокси- или арилокси-
группы в эфирах замещаются на галоген, являются реакциями нуклео-
фильного замещения.
Реакции, в которых из алкилгалогепидов образуются сульфониевые, селепо-
ниевые, аммониевые и фосфониевые ионы, представляют собой обратимые
процессы. И в этом случае соображения, которые относились к прямым реак-
циям, сохраняют силу и для обратных реакций. Таким образом, замещение
ионной сульфониевой группы на галоген в таких реакциях, как
R—SRJJ-C1' —> R—Cl-|- SR2 (R = алкил)
является нуклеофильным замещением, в котором анион соли представляет
собой замещающий агент, а катион выступает в роли субстрата. Очевидно,
что аналогично протекают процессы замещения ионных ониевых групп
на гидроксил, алкокси-, арилокси- или ацильную группу при разложении
ониевых оснований, алкоголятов, фенолятов или карбоксилатов, например
R — SRJ + OH~ —> R—OH4-SR2 (Н=алкил)
Замещающим агентом не обязательно должен быть противоанион ониевого
катиона; разложить соль, имеющую слабо нуклеофильный анион, можно
добавкой более сильного нуклеофильного, но нейтрального реагента, как
в следующем примере:
R —SRJ + NR3 —> R—NRJJ-SRj (Ц = алкил)
Все подобные реакции являются реакциями нуклеофильного замещения.
Вообще реакции, в которых ионные ониевые группы замещаются галогеном,
OR, NR2, NRJ и т. д., представляют собой процессы нуклеофильного заме-
щения.
Ранее каждая из приведенных выше реакций рассматривалась в соответ-
ствующем разделе курса по органической химии, так как в них участвуют
различные классы веществ. Теперь эти реакции объединены в одном разделе
по признаку общности механизма, что было установлено и стало общепри-
нятым (хотя первоначально общность механизма вызывала сомнение) в 1933—
1935 гг. [6].
210
Классификация реагентов и реакций
Приведенный пример электрофильного замещения, а именно нитрование
бензола нитроний-ионом (в предположении, что последний представлен
в ионизированной соли нитрония), является по механизму одним из простей-
ших; следует заметить, однако, что для применяемой классификации это
обстоятельство не имеет существенного значения. Если нитрующим агентом
служит азотная кислота, то механизм процесса осложняется стадиями,
предшествующими нитрованию. Но в этом случае нитрогруппа связана
в продукте реакции электронной парой, принадлежавшей ранее не азотной
кислоте, а бензолу, т. е. атакуемому веществу, а не замещающему агенту
C6H5-H + HNO3 -» CbH5-NO2 + H2O
Таким образом, нитрование также представляет собой электрофильное
замещение. Вообще нитрование, если оно принадлежит к числу реакций
гетеролитического замещения, как в случае нитрования ароматических
соединений, конечно при обычных условиях, представляет собой электро-
фильное замещение.
С помощью аргументов, абсолютно аналогичных изложенным выше при
рассмотрении нуклеофильного замещения, можно показать, что в ряду
ароматических соединений не только нитрование, но и нитрозирование,
галогенирование, сульфирование, реакция Фриделя — Крафтса (ацилиро-
вание и алкилирование), водородный обмен между ароматическим соеди-
нением и сильной кислотой, азосочетание и меркурирование относятся
к типу электрофильного замещения независимо от того, известен или не изве-
стен их механизм.
Классификация реакций гетеролитического замещения на нуклеофильные
п электрофильные реакции дана в табл. 58. В этой таблице нуклеофильное
замещение иллюстрируется только примерами из химии алифатических
соединений, однако, как будет показано в гл. VI, существует важная группа
реакций нуклеофильного замещения и в ароматическом ряду. Аналогично,
хотя электрофильное замещение представлено примерами из ароматического
ряда, однако электрофильное замещение у насыщенного атома также суще-
ствует и будет рассмотрено в гл. VII.
г. Нуклеофильное и электрофильное присоединение. В отличие
от замещения гетеролитическое присоединение вообще нельзя классифици-
ровать без знания или хотя бы без определенных предположений о механизме
присоединения. Однако при этом не возникает больших трудностей, как
это может показаться на первый взгляд. Во-первых, механизм некоторых
наиболее важных и типичных реакций присоединения установлен специаль-
ными экспериментальными исследованиями; во-вторых, многие другие реак-
ции присоединения настолько аналогичны процессам присоединения, меха-
низм которых доказан, что не остается сомнений относительно их характера;,
в-третьих, из общего химического поведения реагентов можно сделать важ-
ные выводы относительно тех или иных реагентов. В случае новых реакций
присоединения можно подойти к их классификации, применив такого же
рода аргументацию, но в обратном порядке.
Для того чтобы лучше объяснить метод решения вопроса, рассмотрим сле-
дующий пример. Предположим, что адденд А — В присоединяется к акцеп-
тору X : Y с образованием продукта присоединения А — X — Y — В_
Следует выяснить, образуется ли связь А с X за счет электронов А — В
и связь В с Y за счет электронов X — Y или наоборот. Кроме того, необхо-
димо ответить на вопрос, присоединяется ли А — В вследствие своей нуклео-
фильности (т. е. начинается ли реакция взаимодействием электронов А — В
с ядрами X — Y) или же вследствие электрофильности А — В, когда ядро
14*
Глава V
Таблица 58
Некоторые реакции нуклеофильного и электрофильного замещения а
Реагент Субстрат (атакуемая частица) Продукты замещения
Нуклеофильное замещение
Hal- AlkHal' AlkHal
OR-, OHR AlkHal AlkOR
sr2 AlkHal AlkSR+
nhr2 AlkHal AlkNR2
NR3 AlkHal AlkNRj
CN- AlkHal AlkCN
HHal AlkOR AklHal
Hal- AlkSRf, AlkNRJ и т. д. AlkHal
OR-, OHR AlkSRj, AlkNRJ и т. д. AlkOR
NR3 AlkSRJ AlkNRJ
NOHal AlkNH2 AlkHal
NOOR AlkNH2 AlkOR
Электрофильное замещение
NO}, HNO3 и т. д.
На1+, На12 и т. д.
SO3, H2SO4 и т. д.
RCO+, RCOC1 ит. д.
Ar'NJ
NO+, HNO2 и т. д.
Alk+, AlkHal и т. д.
Дейтерокислоты
HgX2, соли
ArH, R2NH, ROH
ArH, R2NH, ROH
ArH, R2NH, ROH
ArH, R2NH, ROH
ArH, R2NH, ROH
ArH, R2NH, ROH
ArH, R2NH, ROH
ArH
ArH
ArNO2, R2NNO2, RONO2
ArHal, R2NHal, ROHal
ArSO3H, R2NSO3H, ROSO3H
ArCOR, R2NCOR, ROCOR
ArN2Ar', R2NN2Ar', RON2Ar'
ArNO, R2NNO, RONO
ArAlk, R2NAlk, ROAlk
ArD
ArHgX
a Aik — почти любая алкильная группа, включая арилалкильную группу; в некоторых немно-
гих случаях может дополнительно означать замещенную (обычно о- или n-нитро) арилалкильную
группу; Аг — почти любая арильная группа; R — водород или алкил, включая арилалкил, а в неко-
торых случаях ацил или арил.
А — В атакуют электроны X — Y, причем остальной процесс и в том и в дру-
гом случае протекает со снижением энергии. Общая химическая классифи-
кация А — В дает предположительный ответ на второй из этих вопросов,
что обычно означает и ответ на первый вопрос. Таким образом, выясняется,
как следует классифицировать процессы присоединения; за этим следуют
поиски аналогии и, возможно, специальные исследования.
В качестве первого примера рассмотрим присоединение аммиака к ацеталь-
дегиду с образованием ацетальдегидаммиака
.ОН
NH3 + CH3CH = O СН3СН< (AdN)
XNH2
Адденд (аммиак) представляет собой нуклеофильный реагент (см. разд. 1,6,
табл. 56). Вероятно, он присоединяется вследствие своей нуклеофильности;
неподеленные электроны азота атакуют ядро атома углерода карбонильной
группы; в дальнейшем реакция завершается благодаря легкости переноса
протона к неподеленному электрону атома кислорода. Таким образом, реак-
212
Классификация реагентов и реакций
цию можно классифицировать как нуклеофильное присоединение. Сделанное
заключение подтверждается аналогией с более подробно изученными приме-
рами. За основу символического обозначения особенностей механизма при-
соединения принимают символ AcIn-
В качестве второго примера рассмотрим присоединение хлористого водорода
к пропилену с образованием изопропилхлорида
НС1+СН3СН = СН2 —> СН3СНС1-СН3 (AdE).
Адденд (хлористый водород) относится к электрофильным реагентам (см. разд.
1,в, табл. 57). Предположим, что он присоединяется вследствие своей
электрофильности, соединяясь своим протоном и л-электронами атома
углерода: при этом реакция заканчивается благодаря легкости присоедине-
ния хлор-иона к другому атому углерода, которому теперь не хватает элек-
трона. Поэтому процесс можно классифицировать как электрофильное
присоединение. Это подтверждается аналогией с более подробно изученными
сходными процессами. Такого рода процессы присоединения обозначают
символом Айк-
В этих рассуждениях подразумевается, что карбонильная группа является
преимущественно электрофильным реагентом, а этиленовая система пред-
ставляет собой нуклеофильный реагент. Следует иметь в виду, что речь идет
о ненасыщенных несопряженных двойных связях. Эти выводы, естественно,
согласуются с общим химическим опытом, свидетельствующим о том, что
карбонильные соединения особенно склонны к взаимодействию со многими
нуклеофильными реагентами (растворенными металлами, растворенными
восстановительными агентами, цианид- и сульфид-ионами, аминами, гидра-
зинами, гидроксиламином), в то время как олефины особенно чувствительны
ко многим электрофильным реагентам (всякого рода окислительным агентам,
галогенам, сильным кислотам и пр.). Эти особенности могут быть связаны
с различиями в строении, состоящими в том, что электроны карбонильной
группы, включая л-электроны, сильно смещены к атому кислорода, вслед-
ствие чего ядра углерода становятся слабо экранированными; с другой
стороны, ядра атомов углерода этиленовой группы хорошо экранируются
их внешним двойным слоем л-электронов.
В то время как восстановление такими растворенными металлами, как
натрий, представляет собой нуклеофильное присоединение и осуществляется
в случае карбонильной и циангрупп, но не идет с олефинами, гидрирование
олефинов элементарным водородом на поверхности таких переходных метал-
лов, как платина, представляет собой гомолитический процесс и, вероятно,
зависит от поверхностноабсорбированных, возможно, ионизированных ато-
мов водорода. Окисление олефинов элементарным кислородом, так назы-
ваемое аутоокисление, подобным же образом является гомолитическим
процессом и зависит от бирадикальной природы молекулярного кислорода,
каждый атом которого обладает неспаренным электроном.
Более детально некоторые специфические случаи рассмотрены ниже, а пока
следует ограничиться табл. 59, в которой приведен перечень наиболее важных
нуклеофильных и электрофильных реакций присоединения. Подобранные
примеры могут служить иллюстрациями, но отнюдь не претендуют на исчер-
пывающую полноту.
Последняя реакция, приведенная в первой части таблицы, нуждается в неко-
торых пояснениях. Эта реакция включает альдольную конденсацию, которая,
как показывают исследования, зависит от склонности к присоединению
одной карбонильной молекулы к аниону другой. Как известно, из продуктов
присоединения в дальнейшем часто выделяется вода с образованием а,0-
213
Глава V
Таблица 59 Некоторые реакции нуклеофильного и электрофильного присоединения а
Адденд Акцептор Продукт присоединения
Реакции нуклеофильного присоединения
Растворенные металлы R2C = O; RC = N r2choh, rch2nh2
Сг2+. Н2О RC = N rch2nh2
Реактивы Гриньяра R2C = O Н2С(ОМеталл)Алкил
он-, Н2О r2c = o R2C(OH)2[2: R2CO]
он-, Н,0 RC = N RCOj
H2S RC = N rcsnh2
NH..X R2C=O R2C(OH)NHX[-+ R2C = NX]
CN-, ROH r2c = o R2C(OH)CN
{CR2Y]-, ROH r2c=o R2C(OH)CR2YH r2c = CR2]
Реакции электрофильного присоединения
MnOj. Н2О R2C — CR2 R2C(OH)CR2(OH)
Н3О<- R2C--CR2 R2CHCR2(OH)
H2SO4 r2c = cr2 R2CHCR2(OSO3H)
Hal2 r2c=cr2 R2CHalCR2Hal
,Ual2. ROI1 r2c=cr2 R2CHalCR2(OH)
HOHal. H3O+ r2c=cr2 R2CHalCR2(OH)
HHal r2c = cr2 R2CHCR2Hal
NOHal r2c = cr2 R2C(NO)CR2Hal
R = Н, алкил или арил; X — R, OR или NR2; Y — COR, CN или NO2*
ненасыщенных карбонильных соединений. Последний пример во второй
части таблицы представляет собой реакцию присоединения по Тильдену.
Образующиеся нитрозосоединения при этом обычно подвергаются таутомер-
ному превращению в оксимы.
Если двойные связи в олефинах и карбонильных соединениях сопряжены
друг с другом, в системе С = С — С = О углерод-углеродная двойная
связь приобретает электрофильные свойства, частично в дополнение и частич-
но вместо ее нормального нуклеофильного характера. Эти факты можно
объяснить заметным смещением всей сопряженной л-оболочки в направле-
нии к кислородному атому карбонильной группы с частичным обнажением
ядер атомов углерода на другом конце системы. Следовательно, ряд реакций
нуклеофильного присоединения к простой карбонильной группе протекает
также и в случае сопряженной системы, концевой углеродный атом которой
действует подобно карбонильному углеродному атому. Так, альдольной
конденсации простых карбонильных соединений соответствует реакция
присоединения к сопряженной системе с концевыми карбонильными группа-
ми (реакция Михаэля).
.ОН
R,C = O-|-H-CR2COR —* r2c<
\cr2cor
(альдольная конденсация)
/CHR—CR=O
R2C = CR — CR = O + H-CR2COR -» R2C<
xCR2COR
(реакция Михаэля)
214
Классификация реагентов и реакций
д. Классификация реакций отщепления. В принципе реакции отщеп-
ления можно классифицировать в таком же плане, но в данном случае это
вряд ли необходимо, потому что все наиболее важные и лучше исследован-
ные реакции соответствуют одному общему типу. Во всех случаях выделяется
НВ, причем Н переходит в виде протона к веществам основного характера,
специально введенным или служащим растворителем, в то время как В осво-
бождается, не образуя в процессе реакции новую ковалентную связь, хотя
она в последующем и может образоваться. Эти же положения могут быть
применимы к большой группе реакций с отщеплением олефинов. Некоторые
примеры таких реакций представлены следующими уравнениями:
С2Н5О- + (СН3)2СНВг —> С2Н5ОН-|-СНзСН = СН2 + Вг-
Н2О + (СН3)3СВг Н3О+ + (СН3)2С = СН2 +Вг-
он-+C2H5N(CH3)3 -> H2O + CH2 = CH2 + (CH3)3N
H2O + (CH3)3CS(CH3)2 -» H3O+ + (CH3)2C = CH2 + (CH3)2S
Как видно из уравнений, единственной новой ковалентной связью, обра-
зующейся при этих процессах между внешней молекулой и фрагментом
выделяющегося вещества, является связь между протоном и нуклеофильной
молекулой, которая присоединяет протон. В соответствии с принятой систе-
мой эти реакции можно классифицировать как нуклеофильные; однако нет
необходимости подчеркивать это в номенклатуре. Поэтому для обозначения
процессов указанных типов можно применять символ Е вместо более точ-
ного En.
В механизмах процессов отщепления с образованием карбонильных веществ
имеются различия, которые, однако, не затрагивают классификацию по поляр-
ности. Речь идет о реакциях, которые, как показано в следующих примерах,
противоположны образованию продуктов присоединения к карбонильным
соединениям, например циангидринов, альдолей или кетолов:
OH- + HOC(CH3)2CN H2O-|-O = C(CH3)2-|-CN-
ОН- + НОС(СН3)2СН2СОСН3 -» Н2О + О = С(СН3)2+[СН2СОСН3]-
На основе наших знаний о механизме соответствующих реакций присоеди-
нения и представлений о том, что одна и та же поверхность реакции и одна
и та же координата реакции применимы к прямой и обратной реакциям
(так называемый принцип микроскопической обратимости) *, можно утвер-
ждать, что в начальной стадии этих процессов отщепления наряду с перено-
сом протона к основанию происходит освобождение анионов. Эти анионы
представляют собой умеренно сильные или очень сильные основания (наибо-
лее слабый из них — цианид-ион — приблизительно сравним с триметил-
амином), и, следовательно, они имеют возможность присоединять протоны.
Вот почему они, возможно за исключением цианид-иона, обычно могут быть
выделены в виде соответствующих кислот. Однако при реакциях элимини-
рования, сопровождающихся образованием карбонильных соединений,
создается только одна ковалентная связь с внешним реагентом, являющимся
нуклеофильным, так же как в реакции отщепления с образованием
олефина.
* Этот принцип в общей форме состоит в следующем: если имеется много путей реак-
ции от данных исходных к данным конечным продуктам, то при равновесии по каждому
из отдельных путей в прямой реакции участвует столько же молекулярных систем, сколько
и в обратной реакции. Для нас важно лишь то, что наиболее легкий путь для прямой
реакции является наиболее легким путем и для обратной реакции.
215
Глава V
Реакция элиминирования, аналогичная реакциям, связанным с образова-
нием карбонильных соединений, происходит при образовании этиленовых
двойных связей, сопряженных с карбонильными группами. Сюда относится
реакция, обратная процессу присоединения по Михаэлю, вполне аналогич-
ная обратной реакции альдольной конденсации. Следующий пример иллю-
стрирует это соотношение:
СООС2Н5 СООС2Н5
I I
С2Н5О- 4- НСН — С(СН3)2СН(СООС2Н5)2 —> С2Н5ОН + СН = СН(СН3)2 + [СН(СООС2Н5)2]-
Все эти реакции являются реакциями нуклеофильного элиминирования.
е. Получение радикалов для гомолитических реакций. Единствен-
ным легко доступным и очень важным свободным радикалом для проведения
гомолитических реакций является молекулярный кислород. Большинство
других радикальных реагентов необходимо получать непосредственно в реак-
ционной смеси, так как время их жизни чрезвычайно мало. Существует
ряд процессов, с помощью которых можно получать свободные радикалы.
Для создания контролируемой концентрации радикалов широко исполь-
зуют одноэлектронные окислительно-восстановительные реакции. Примером
может служить одноэлектронное восстановление перекиси водорода в гидро-
ксильные радикалы, происходящее в смеси перекиси водорода с солью
двухвалентного железа, давно известной под названием реактива Фентона.
Свойства этой смеси были изучены Хабером и Вейссом [7]
е (из Fe2+)-pHO —ОН —► НО~ + НО-
Аналогично соли Fe(II) и Со(П) реагируют с ионами динадсерной кислоты
и органическими гидроперекисями.
Радикалы могут образовываться путем переноса электрона на электроде.
Образующиеся радикалы обычно реагируют, не успев далеко уйти от поверх-
ности электрода в раствор. Классическая реакция Кольбе, состоящая в диме-
ризации алкильных групп путем анодного окисления карбоксилатных ионов,
почти наверняка идет через радикалы.
RCOO" —> е (к аноду)-|-RCOO« —► CO2-|-R —> R2
Для получения многих типов радикалов может быть применен термический
гомолиз ковалентных связей. Температурный режим зависит от прочности
разрывающейся связи. По грубой оценке для разрыва связей с энергией
диссоциации 40 ... 50 ккал/моль (167,47-Ю3 . . . 209,34-103 Дж/моль)
требуется температура 200 . . . 400 °C, например:
Вг2 —► 2Вг-
12 -> 2Ь
НО—NO2 HO-+NO2
СН3—N=N—СН3 CH3.+N2 + CH3.
а для разрыва связей с энергией 30 ... 40 ккал/моль (125,60-103 . . .
. . . 167,47 -103 Дж/моль) необходима температура 50 . . . 200 °C, например:
F2 —> 2F-
(СН3)зСО-ОС(СН3)з 2(СН3)3СО- 2СН3.+2(СН3)2СО
С6Н5СОО —ОСОС6Н5 —> 2СвН5СОО. —> 2СвН5. + 2СО2
(CH3)2C(CN) — N = N — C(CH3)2CN -* 2(CH3)2C(CN)-|-N 2
CH3(CeH5)2C—С(СвНд)2СНз > 2CH3(CeH5)2C •
216
Классификация реагентов и реакций
Энергии диссоциации некоторых связей, полученные при термическом обра-
зовании радикалов, приведены в табл. 60 *.
Таблица 60
Энергии диссоциации [ккал/моль(4,187.103 Дж/моль)] некоторых связей
Связь Энергия Связь Энергия
н — н С-Hal
н—н 104 Н3С —F 108
н —с F3C — F 121
Н —СНз 101 Н3С — Cl 80
н—СН2СН3 96 С13С-С1 68
Н-СН(СНз)2 92 СН2 = СНСН2-С1 60
Н—С(СН3)3 89 Н3С —Вг 67
Н-Свн5 102 С13С —Вг 50'
Н-СН2СеН5 83 С6Н5-Вг 71
н—сно 76 СвН5СН2—Вг 51
Н -N СН2 = СНСН2 —Вг 46
h-nh2 102 н3с—i 54
Н —О свн5-1 61
н-он 117 СвН5СН2-1 39
Н —ООН 90 СН2 = СНСН2-1 36
Н —S N — N
H-SH 90 nh2-nh2 60
Н - Hal N —О
Н —F 135 O2N-OH 40
Н -С1 103 o2n-och3 38
Н—Вт 87 0-0
H—I 71 НО —он 48
с —с НО —ОС(СН3)3 42
Н3С-СН3 83 (СНз)зСО- ОС(СН3)3 37
Н3С-С„Н5 87 СН3СОО —ОСОСНз 30
Н3С-СН2СвН5 63 О — Hal
Н3С — С112СН сн. 61 НО-С1 60
н2с — сн2 ~ 152 НО —Вг 56
С—N S-S
H3C-NO2 57 CH3S-SCH3 69
H3C-N = NCH3 46 Hal - Hal
(CH3)2(CN)C-N=NC(CN)(CH3)2 31 F —F 37
С-0 F-Cl 61
Н3С — он 90 Cl-Cl 58
нсо—он 90 Cl —Br 52
СН3СО — он 90 Cl-1 50
С — S Br —Br 46
СН3 —SH 70 Br-I 42
(CH3)3C-SH 65 I-I 36
* Для двухатомных и некоторых трехатомных молекул приведены точные значения,
а для других молекул — приближенные. Большинство данных заимствовано из работы
[8]. Данные для галогенов см. в работе [9]. Энергии диссоциации СН-связей боковых
цепей толуола и связи С — I иодбензола приведены в статье [10]. Энергии связей С — N
217
Глава V
Каталитическое гидрирование металлами VIII группы, имеющими запол-
ненные tZ-оболочки (никель, палладий, платина), почти наверняка связано
с поверхностным гомолизом молекул водорода. Механизм реакции не ясен:
в реакции, по-видимому, участвуют атомы металла слабой связи металл —
металл, которые образуют две слабые связи металл — водород. При этом
должна разрываться прочная связь в молекуле водорода, поэтому в реакцию
вступают только те соединения, у которых связь, присоединяющая водород,
слаба. Известно, что некоторые квадратные комплексы металлов VIII группы
при комнатной температуре поглощают 1 моль водорода, при этом два атома
водорода занимают свободные положения октаэдра. Возможно, что эти
примеры являются молекулярной моделью того процесса, который проис-
ходит на поверхности металла.
Другим важным методом получения радикалов является иеионизирующий
фотолиз молекул. Фотолизирующий свет должен иметь спектральные харак-
теристики (видимая или близкая ультрафиолетовая область), обусловливаю-
щие диссоциацию молекул в высшем электронном состоянии. Адсорбционные
полосы, связанные с диссоциацией, имеют все четыре галогена, которые
таким образом могут быть атомизированы фотолитически. Происходит воз-
буждение одного из неподеленных (несвязывающих) рл-электронов без
инверсии или с инверсией спина; этот'электрон переходит на разрыхляющую
ро-орбиталь, т. е. на орбиталь такой же симметрии, как высшая орбиталь,
описанная в гл. I, разд. 3,г, для возбужденной молекулы водорода. Эта
орбиталь имеет круговую симметрию относительно связи, но узловую пло-
скость, находящуюся между ядрами. Точно так же, как и в случае водорода,
разрыхляющий эффект электрона, находящегося на разрыхляющей орбитали
возбужденных молекул галогенов, настолько велик, что происходит спонтан-
ная диссоциация этих молекул. При диссоциации возможны синглетные
и триплетные состояния. В случае легких элементов инверсия спина мало-
вероятна (так как в этих молекулах магнитные поля, инвертирующие спин,
слабы), и поэтому триплетные состояния, более устойчивые, чем синглетные,
в соответствии с правилом Гунда (гл. I, разд. 3,6), в случае фтора и хлора
встречаются редко. С другой стороны, при возбуждении высших галогенов —
брома и иода — преобладает инверсия. Однако независимо от того, триплет-
ным или синглетным будет возбужденное состояние, оно все равно диссо-
циирует.
hy
С12 (нормальный) —> С12 (возбужденный, синглет) —> 2С1-
hy
Вг2 (нормальный) —► Вг2 (возбужденный, триплет) —> 2Вг-
Примерами более сложных молекул, способных диссоциировать на радикалы
при фотолизе, могут служить азометан и бромтрихлорметан:
hv
СН3—N=N — СН3 —> 2CH3-+N2
hv
С13С — Вг —> С13С.+Вг.
Единственным способным диссоциировать возбужденным состоянием моле-
кулы водорода является низшее триплетное состояние (гл. I, разд. 3, г).
в азо-бис-(изобутиронитриле) nN — О в азотной кислоте, представляющие собой прибли-
зительные энергии активации гомолиза, определены ван Хуком и Тобольским [11] и Джон-
соном с сотр. [12] соответственно. Величины для связи N — О в метилнитрите определены
в работе [13], а для связи О — О в перекиси водорода и гидроперекиси mpem-бутила
приведены в статье [14]. Все данные для связей С — S и S — S получены Палмером
и Лоссингом [15].
218
Классификация реагентов и реакций
однако это состояние не достигается при поглощении света, так как в таком
небольшом атоме инверсия спина очень затруднена. Триплетное возбужденное
состояние может быть получено с помощью электронного удара. В результате
электронного удара молекула водорода может перейти в ряд возбужденных
синглетных и триплетных состояний. Все возбужденные синглетные состоя-
ния путем излучения превращаются в основное состояние, а все триплетные
состояния с более высокой энергией, чем низшее триплетное состояние,
излучая энергию, переходят в низшее триплетное состояние. Один из элек-
тронов в этом состоянии молекулы находится на разрыхляющей 2ро-орби-
тали, в результате чего молекула диссоциирует (гл. I, разд. 3, г). В общих
чертах по такому механизму происходит образование атомов водорода в водо-
родной разрядной трубке и в водородной дуге Ленгмюра или паяльной
лампе
Н2 (нормальный) ----
Н2 (высшие триплеты)
Н2 (низший триплет) —>
2Н-
При действии ионизирующего излучения, например рентгеновских лучей,
можно получить самые разнообразные радикалы и катионы, даже из простей-
ших алканов. Механизм процесса изучен плохо. Считается, что в первой
стадии происходит разряд электрона, при котором образуется катион с нечет-
ным числом электронов, который распадается на катион и радикал. Распад
может происходить по одному пути, например в случае воды:
Н2О----> Н2О+ —> ОН- + Н+
или по двум путям, как, например, в случае метана:
|->СН3.4-Н+
сн4 —> сщ-------------------------
L>CHJ + H.
Для метана преобладающим является второй путь распада. Высшие алканы
распадаются, кроме того, на различные «осколки», включая катионы с нечет-
ным числом электронов и даже нейтральные молекулы с четным числом элек-
тронов, например олефины.
ж. Гомолитическое замещение. Замещение с участием радикала может
быть двух типов в зависимости от того, является ли субстрат насыщенным или
ненасыщенным соединением. Если субстрат насыщен, то замещение может
начинаться только с отщепления атома: при этом создается радикальный
центр, впоследствии вступающий в коллигацию. Если субстрат ненасыщен,
то радикальный реагент может вскрыть л-электронную систему без разрыва
<т-связей. Если радикальный реагент не может взаимодействовать с л-элек-
тронпой системой (как обычно бывает в случае кислорода), то реакция осу-
ществляется по первому механизму, а именно начинается с разрыва о-связи.
Назовем реакцию, осуществляющуюся по первому механизму, т. е. начинаю-
щуюся с отрыва атома, насыщенным гомолитическим замещением. Чаще
всего отщепляется атом с внешней стороны молекулы, т. е. одновалентный
атом. Обычно первоначально отщепляется атом водорода, однако наблюда-
лось также отщепление галогена. Вопрос о том, сможет или не сможет спе-
циально добавленный «инициирующий радикал» отщепить определенный
атом от субстрата, решается с помощью термодинамических данных; если
термодинамически этот процесс возможен, то затем рассматривают кине-
тические ограничения. С точки зрения термодинамики перенос атома должен
219
Глава V
быть экзотермичным или лишь слабо эндотермичным. Данные для выяснения
этого вопроса могут быть получены из приближенных значений энергий
диссоциации связей, приведенных в табл. 60. Например, можно видеть,
что отщепление атома водорода от метана гидроксильным радикалом или
атомом хлора термодинамически выгодно, а тиольным радикалом или атомом
брома термодинамически невыгодно. Однако, даже если процесс термоди-
намически выгоден, могут возникать кинетические препятствия. Процессы
разрыва и образования связей могут быть энергетически сбалансированы или
даже выгодны, но тем не менее бывает необходима дополнительная энергия
для того, чтобы они начались. Таким образом, экзотермическая реакция
может иметь определенную энергию активации, а эндотермический процесс
может иметь энергию активации, превышающую его эндотермичность.
Если внешний источник производит лишь небольшое количество радикалов,
то для протекания второй стадии замещения необходимо, чтобы радикал,
образующийся при отщеплении атома от субстрата, обладал способностью
отрывать радикал от какой-нибудь молекулы. Возможность или невозмож-
ность этого процесса с точки зрения термодинамики можно проверить с помо-
щью данных табл. 60, как это уже было описано ранее.
Если в результате второй стадии образуется тот же самый радикал, что и при
отрыве атома от субстрата на первой стадии замещения, то первая стадия
может повториться и затем одна стадия будет следовать за другой чередую-
щимся образом. Порождение радикала радикалом приведет к развитию
кинетической цепи, скорость реакции будет велика, а количество субстрата,
превратившегося в продукт реакции, значительно превзойдет количество
инициирующих радикалов. По такому механизму происходит знаменитая
реакция — прототип гомолитического хлорирования — хлора с водородом,
для которой на основе данных Боденштейна и др. Нернст [16] впервые пред-
ложил цепной механизм, который впоследствии был подтвержден всеми
исследователями [17]. Механизм реакции представлен на следующей схеме
(R = Н):
RH + C1- —> R- + HC1 I тт
R.+C12 — RC1 + CI. } ЦепЬ
При R = Н эта цепная реакция термодинамически выгодна, так как энергия
диссоциации НС1 больше, чем энергия диссоциации Н2 и С12. Если R =
= алкил, т. е. в случае хлорирования алканов, цепная реакция также термо-
динамически выгодна, так как не только энергия диссоциации НС1 больше
энергии диссоциации связи R — Н, но также и энергия диссоциации связи
R — С1 больше энергии диссоциации С12. Эти выводы следуют из данных
табл. 60.
Кинетические цепи реакции водорода с хлором, а также реакции хлориро-
вания алканов очень длинны. Длина цепи зависит от частоты процессов
обрыва, т. е. процессов, приводящих к уничтожению радикалов, регенери-
рующих друг друга при развитии цепи. Примером гомолитической реакции
с малой длиной кинетической цепи является нитрование алканов азотной
кислотой в газовой фазе. Укорочение цепи обусловливается конкурирующей
побочной реакцией окисления.
В реакциях аутоокисления происходит замещение молекулой кислорода;
процесс состоит из двух стадий, совершенно аналогичных стадиям только
что рассмотренных реакций, и, кроме того, включает дополнительную ста-
дию, необходимость которой вызвана тем, что молекула кислорода представ-
ляет собой бирадикал (триплетная молекула) и поэтому первичный продукт
замещения сам является радикалом. Как уже было отмечено, по не совсем
220
Классификация реагентов и реакций
понятным причинам молекула кислорода мало склонна реагировать с запол-
ненной л-электронной оболочкой. Характерно, что аутоокисление начи-
нается с отщепления слабо связанных атомов водорода — одного в газовой
фазе или одного либо двух в жидкой фазе. В частности, особенно легко про-
исходит отщепление атома водорода от бензильного и аллильного углерода,
а также от углерода, связанного с кислородом, особенно от СНО-группы
(гл. VII): изопропилбензол, высыхающие масла и альдегиды дают соответ-
ственно радикалы бензильного, аллильного или ацильного типов. Образую-
щийся при этом радикал затем захватывает вторую молекулу кислорода.
На этом заканчивается собственное замещение Н на О2. Однако продуктом
замещения является радикал гидроперекиси, и поэтому он вступает в после-
дующую реакцию, при которой отщепляется атом водорода от молекулы
субстрата и образуются гидроперекись и радикал, идентичный исходному.
Таким образом, возникает цепная реакция
RH + O2 R-+HOO-
2RH-pO2 —2R-+HOOH
R-+02-*R00. "I
ROO-+RH -> ROOH-pR. j ЦепЬ
Часто кинетические цепи при окислении очень длинны. Образующиеся гидро-
перекиси обычно довольно неустойчивы в условиях реакции. Они разла-
гаются гомолитически с образованием инициирующих цепь радикалов
и вследствие этого становятся главным источником радикалов даже при
очень низких степенях окисления. Таким образом, гидроперекиси даже при
очень низких концентрациях будут более эффективными источниками ради-
калов, чем прямая реакция кислорода с субстратом. Вследствие того что
радикалы, образующиеся при разложении продукта цепной реакции, сами
инициируют цепную реакцию, процесс окисления является автокаталитиче-
ским и его скорость увеличивается во времени по мере увеличения степени
окисления. Таким образом аутоокисление представляет собой разветвлен-
ную цепную реакцию, которая, судя по форме кинетической кривой, может
привести к взрыву, если не ограничить скорость поступления реагентов
в зону реакции.
Теперь вернемся к второму типу гомолитического замещения, который
назовем ненасыщенным гомолитическим замещением. В этом случае субстрат
содержит оболочку л-электронов, например двойную или тройную связь
или бензольное кольцо, и атакующий радикал вступает в коллигациюс одним
из электронов л-связи. Образующийся радикал может затем или отщепить
какой-нибудь атом, или захватить второй радикал. В первом случае произой-
дет реакция замещения, а во втором — реакция присоединения.
Экспериментальные данные показывают, что ароматические субстраты могут
вступать как в реакции замещения, так и в реакции присоединения. Вопрос,
в какую именно реакцию они вступят в данных условиях, зависит от того,
к чему более склонен реагент: отщепить какой-либо атом от первоначально
образующегося радикала или присоединить к нему второй радикал. В прин-
ципе в реакцию замещения могут вступать и олефины, однако до настоящего
времени такие реакции не известны: согласно экспериментальным данным,
олефины всегда вступают только в реакции присоединения. В данном разделе
мы рассматриваем замещение и поэтому обратимся к ароматическим суб-
стратам.
Когда ароматические субстраты реагируют с метильными, высшими алкиль-
ными, фенильными или замещенными фенильными радикалами (все эти
221
Глава V
радикалы можно получить количественно, например пиролизом перекисей
или азосоединений), замещение происходит в две стадии, первая из которых
является медленной:
Z\ Z\ZR
I II+R---------* I Ьн
| || Медленно | |
\/ ZZ'
z\zR z\zR
| |\н +R. | || +RH
%/ ZZ
Реакции присоединения рассмотрены в следующем разделе.
Иногда нам будет удобно обозначать гомолитическое замещение символом
SH, после которого можно написать дополнительные символы, указывающие
на механизм.
з. Гомолитическое присоединение. Известны три вида гомолитиче-
ского присоединения, два из которых совершенно различны, а третий
является смесью двух первых. Назовем первый тип присоединения гомолити-
ческим моноприсоединением. Эти реакции происходят, когда из ненасыщенно-
го субстрата после захвата радикала образуется радикал, который вместо
отщепления атома под действием внешнего радикала, как при замещении,
захватывает второй радикал, отщепляющийся от реагента, завершая при-
соединение. Если радикал, оставшийся от реагента после реакции присоеди-
нения, идентичен начальному, то происходит цепная реакция. По цепному
механизму происходит фотохимическая реакция присоединения хлора
к бензолу
Образующееся дихлорпроизводное далее присоединяет хлор по такому же
механизму; в результате образуется гексахлорбензол.
Олефины вступают исключительно в реакцию гомолитического присоеди-
нения, а не замещения. Механизм фотохимического присоединения хлора
к этилену такой же, как и в случае бензола. По такому же механизму проис-
ходит присоединение бромистого водорода в присутствии перекисей, при
действии которых из НВг образуются инициирующие цепь атомы брома:
СН2 = СН2 + Вг- ВгСН2-СН2. 1
ВгСН2 — СН2- Д-НВг —> ВгСН2-СН2Н + Вг- J 4
Радикал ВгСН2 — СН2- должен отрывать от НВг не Вт-, а Н-; это следует
из данных табл. 60.
В рассматриваемых реакциях в качестве аддендов могут выступать орга-
нические молекулы, например бромтрихлорметан (присоединение С13С и Вг)
999,
Классификация реагентов и реакций
и альдегиды (присоединение RCO и Н). Бромтрихлорметан может присоеди-
няться к олефинам аналогично хлору с помощью фотохимического иницииро-
вания или аналогично бромистому водороду с помощью химических ради-
кально-цепных инициаторов, добавленных или образующихся в небольших
количествах. В качестве химических инициаторов могут выступать, напри-
мер, метильные радикалы, продуцируемые любым стандартным путем.
Из данных табл. 60 ясно, что метильный радикал способен отщеплять бром
от бромтрихлорметана и, таким образом, давать инициирующий цепь трихлор-
метильный радикал. Независимо от способа инициирования — фотохими-
ческого или химического — гомолитическое моноприсоединение происходит
по следующему стандартному пути:
СН2 = СН2 + С13С- -> С13ССН2-СН2. •)
сс13сн2-сн2-+с13свг —> с13ссн2—сн2вт+с13с- j епь
В результате инициируемого химически метильными или фенильными ради-
калами присоединения альдегидов к олефинам образуются кетоны.
Второй из трех типов гомолитического присоединения к олефиновым суб-
стратам можно назвать гомолитическим полиприсоединением *. В результате
реакции образуются полимеры **. Гомолитическое полиприсоединение
является одной из форм самоприсоединения. Оно происходит в том случае,
когда в результате химического инициирования первоначально образовав-
шийся радикал вместо того, чтобы отщепить второй радикал от какого-нибудь
соединения, присоединяется к другой молекуле олефинового субстрата.
В результате образуется новый радикал, который обладает теми же свой-
ствами, что и его предшественник, и поэтому присоединяется к другой
молекуле олефина. Таким образом, путем последовательного присоединения
олефиновых молекул могут образовываться длинные структурные цепи,
причем процесс роста цепи обрывается только тогда, когда растущий радикал
реагирует с каким-нибудь другим радикалом. Описанными свойствами обла-
дают многие олефины, особенно с концевыми метиленовыми группами. Ниже
приведена схема полимеризации стирола.
R. Д-СН2=СН —> RCH2—сн-
I I
CeH5 CjHg
НСН2-СН-+СН2 = СН -» rch2—сн-сн2 — сн-
I I II
[С6н5 с6н5 свн5 свн5
RCH2— СН — СН2— СН.-1-СН2 = СН —>
I I I
С6Н5 СвН5 С6Н5
RCH2- СН - СН,— СН - СН2 — СН.
I I I
СвН5 СвН5 СвН5
RCH2- СН — [СН2 - СН]п - CH2CHR'
I I I
свн5 С6Н5 С6Н5
* Этот термин позволяет объединить под термином «полимеризация» как процесс
полипрпсоедпнения, так и процесс поликонденсации, что уже становится общепринятым.
** Собственно говоря, продукты поликонденсации называются «поликонденсатами»,
но их очень часто называют также «полимерами».
223
Глава V
Кинетически эта реакция не является цепной в первоначальном понимании
этого термина. Однако последовательные стадии настолько схожи между
собой, что можно расширить терминологию и назвать эту реакцию цепной;
такой же вывод следует из математического анализа ее кинетики. Конечно,
термин «цепная реакция» не имеет никакого отношения к тому факту, что
в процессе данной реакции происходит рост структурной углеродной цепи.
Третий тип гомолитического присоединения к олефинам мы назовем гомо-
литическим телеприсоединением. Продукт реакции называется теломером».
Гомолитическое телеприсоединение происходит в тех случаях, когда после
инициирования радикальной цепи первоначально образующийся радикал
имеет одинаковую возможность отщепить радикал от источника радикалов,
как при моноприсоединении, или присоединиться к другой молекуле олефи-
нового субстрата, как при полиприсоединении. При таких обстоятельствах
между первой и второй (конечной) стадиями моноприсоединения может
произойти несколько стадий полиприсоединения. Возможен и другой вариант,
когда начинаются цепи полиприсоединения, но они, будучи еще очень корот-
кими, так быстро обрываются, что существенную часть образующегося низ-
комолекулярного полимера (теломера) составляют концевые группы, кото-
рые присоединились бы к молекуле олефина при реакциях моноприсое-
динения.
В качестве примера можно указать на присоединение четыреххлористого
углерода и ацетальдегида к этилену при инициировании фенильными ради-
калами. В результате реакции теломеризации образуются следующие
продукты.
С13С-СН2СН2[СН2СН2]ПСН2СН2С1 1
CH3COCH2CH2[CH2CH2]nCH2CH2H J ’ ’ •
Гомолитическое присоединение можно обозначить символом Айн-
и. Некоторые другие реакции. Единичные процессы гетеролиза и гомо-
лиза имеют довольно простые механизмы, если рассматривать их в рамках
органической химии. Такие процессы позволяют исследовать равновесия,
которые вследствие простоты отношения между начальными и конечными
веществами дают основную информацию об относительной стабильности,
т. е. разности свободных энергий молекул, ионов и радикалов.
Полученные таким путем данные можно использовать при корреляции
структурных эффектов с реакционной способностью в уже изученных реакци-
ях замещения, присоединения и отщепления.
Молекулярные перегруппировки происходят в значительной мере в резуль-
тате процессов замещения, присоединения и отщепления. Ниже будет пока-
зано, что перегруппировки удобно классифицировать в первую очередь
согласно тому, в какой системе они происходят — насыщенной, ненасыщен-
ной или ароматической. Внутри каждого из этих классов перегруппировки
подразделяются на нуклеофильные и электрофильные. Эти термины, как
обычно, отражают природу реагента, обусловливающего реакцию, в которой
происходит перегруппировка, и мы будем их использовать. Однако для
классификации перегруппировок в ненасыщенном ряду мы используем
эквивалентные исторически сложившиеся названия, которые обращают
внимание на природу мигрирующей группы, а не на природу реагента, напри-
мер прототропия и анионотропия.
Некоторые реакции присоединения и отщепления, а также некоторые моле-
кулярные перегруппировки нельзя классифицировать ни как нуклеофильные,
ни как электрофильные, ни как гомолитические не потому, что недостаточно
224
Классификация реагентов и реакций
точно известен их механизм, а по причинам принципиального характера.
Эти реакции происходят без участия внешнего реагента и проходят через
циклическую промежуточную конфигурацию, расположенную по координате
реакции не обязательно в области, соответствующей максимуму или мини-
муму энергии. В этой конфигурации происходит перераспределение связей;
примером может служить схема, приведенная ниже. Известно, что такое
перераспределение связей включает движение электронов по периферии
промежуточной циклической конфигурации; однако не известно, остаются ли
электроны спаренными или во время перегруппировки они разъединяются
и снова спариваются; нельзя также узнать, какой путь они проходят при
движении от мест локализации старых связей к местам локализации новых.
/CH2 HC+ CHR IKY /CH2, +HR
1 + 11 ~ II 1
HC. CHR' HCX ZCHR'
Хн2 чсн/
H2cZ
СН
\cHR
/СН.
|Н2с+ 4CHR
|нА Z.CHR'
ХСН+
I
Н2СЧ /CHR'
^CHZ
Мы рассмотрим только гомогенные реакции в растворе и газовой фазе.
Количественно эти реакции описываются энергией и законами вероятности,
управляющими поведением большой совокупности молекулярных систем.
Мы не будем рассматривать реакции в твердой фазе, на поверхности твердых
веществ или реакции с участием ферментов. В этих реакциях по крайней
мере некоторые из взаимодействующих атомов постоянно находятся в закреп-
ленных положениях, поэтому в них не наблюдается зависимости от произве-
дения вероятности благоприятного расположения и скорости таких атомов
и наиболее важными становятся топохимические факторы. К топохимическим
реакциям относятся многие классы, вернее, несколько больших классов
органических реакций, однако их рассмотрение выходит за пределы данной
книги.
СПИСОК ЛИТЕРАТУРЫ
1. Fry Н. S., The Electronic Conception of Valence and the Constitution of Benzene, Long-
mans, Green, and Co, London and New York, 1921; Stieglitz J., J. Am. Chem. Soc., 44,
1293 (1922).
2. Bronsted J. N., Rec. trav. chim., 42, 718 (1923).
3. Lowry T. M., J. Soc. Chem. Ind. (London), 42, 43 (1923).
4. Lapworth A., Mem. and Proc. Manchester Lit. & Phil. Soc., 69, p. xviii (1925)- Nature
115, 625 (1925).
5. Lewis G. N., Valence and the Structure of Atoms and Molecules, Chemical Catalog Co.,
New York, 1923, p. 141; J. Franklin Inst., 226, 293 (1938).
6. Hughes E. D., Ingold С. K., Patel C. S., J. Chem. Soc., 1933, 526; Hughes E. D.,
Ingold С. K., ibid., 1933, 1571; Gleave J. L., Hughes E. D., Ingold С. K., ibid., 1935’
235; Hughes E. D., Ingold С. K., ibid., 1935, 244.
7. Haber F., Weiss J., Proc. Roy Soc., A147, 332 (1934); cp. Urt N., Chem. Rev. 50 375
(1952).
8. Cottrell T. L., Strengths of Chemical Bonds, Butterworths Scientific Publications
London, 1958, Chap. 9, p. 173.
15-0772
226
Глава V
9. Pauling L., Nature of the Chemical Bond, Cornell University Press, Ithaca, New York,
3rd Edn., 1960, Chap. 3, pp. 79, 83.
10. Szwarc M., Ann. Rev. Phys. Chem., 8, 439 (1957).
11. Van Hook J. P., Tobolsky A. V., J. Am. Chem. Soc., 80, 779 (1958).
12. Johnston H. S., Foering L., Tao Y. S., Moserly G. S., J. Am. Chem. Soc., 73, 2319
(1951).
13. Gray P., Williams A., Chem. Rev., 59, 239 (1959).
14. Benson S. W., J. Chem. Phys., 40, 1007 (1964).
15. Palmer T. F., Lossing F. P., J. Am. Chem. Soc., 84, 4661 (1962).
16. Nernst W., Z. Elektrochem., 24, 335 (1918).
17. Hinshelwood C. N., Kinetics of Chemical Change in Gaseous Systems. Clarendon Press,
Oxford, 1933, p. 1001; Semenoff N. N., Chemical Kinetics and Chain Reactions, Claren-
don Press, Oxford, 1935, p. 89.
ГЛАВА VI
ЗАМЕЩЕНИЕ В АРОМАТИЧЕСКОМ РЯДУ
1. Развитие теории ароматического замещения.................228
2. Ориентация при электрофильном замещении в ароматическом
ряду ...................................................234
3. Реакционная способность в электрофильном ароматическом за-
мещении ......................................................245
4. Соотношения между орто- и иора-изомерами при электрофиль-
ном ароматическом замещении...................................259
5. Механизм нитрования ароматических соединений.............270
6. Механизмы других реакций электрофильного ароматического
замещения ..............................................285
7. Нуклеофильное ароматическое замещение....................318
8. Гомолигическое ароматическое замещение...................333
Список литературы ......................................342
15*
В этой главе мы начнем обсуждение механизмов органических реакций,
широко применяя классификацию, изложенную в гл. V. Сначала будут
рассмотрены реакции замещения, а именно реакции электрофильного заме-
щения в ароматическом ряду, по той причине, что исторически изучение
этих реакций позволило сформулировать электронную теорию взаимодей-
ствий органических соединений; это особенно касается идеи об одновремен-
ном действии эффектов поляризации и поляризуемости, каждый из которых
содержит индуктивную и конъюгативную составляющие (гл. II). При электро-
фильном ароматическом замещении главными факторами являются ориента-
ция и скорость замещения. Изучение этих факторов привело к открытию
ряда закономерностей (гл. II) еще за много лет до того, как в результате
применения кинетических методов стал выясняться действительный механизм
электрофильного замещения
1. Развитие теории ароматического замещения
а. Ранние правила ориентации. Одним из первых результатов введения
в 1874 г. надежных методов определения положения заместителей в бензоль-
ном ядре явилось открытие закономерностей ориентации при замещении.
Как было установлено, имеются два совершенно различных типа ориентаций:
один — в орто- и ияра-положения и другой — в меига-положение. Кроме
того, оказалось, что природа ранее введенного заместителя определяет
положения, в которые вступает новый заместитель. Уже в середине 70-х
годов были сделаны попытки обобщить известные к тому времени факты.
Представление о том, что атомы и группы обладают электрохимическим
характером, вытекало из учения Берцелиуса. Первые три предложенных
правила ориентации при ароматическом замещении — Кернера [1], Хюбне-
ра [2] и Нольтинга [3] — в основном мало отличались одно от другого; все
они исходили из того, что характерные свойства ориентирующих групп
определяются их электрополярной природой. Правило гласило, что отрица-
тельные группы, т. е. те, которые придают или усиливают кислотные свой-
ства, при достаточной электроотрицательности направляют новый замести-
тель в меига-положение, а положительные группы, т. е. те, которые придают
или усиливают основные свойства, а также нейтральные и даже слабо отри-
цательные группы направляют новый заместитель в орто- и пара-положения.
Уже в этот период был отмечен, но отнесен к случайным тот факт, что заме-
стители, которые можно легко ввести вместо водорода с соблюдением опре-
деленной ориентации, сами носят ясно выраженный отрицательный харак-
тер, как, например, галогены, нитро- и сульфогруппы.
Такого рода правила способствовали последующему развитию теории:
они предвещали ту роль, которую сыграет индуктивный эффект в современ-
ной теории ориентации при замещении в ароматическом ядре. К электро-
отрицательным группам относятся такие группы, которые электростатически
притягивают электроны, уменьшая тем самым электронную плотность
в остатке молекулы, и оказывают таким образом влияние, вызываемое теперь
отрицательным индуктивным эффектом (—7). Электроположительные группы
электростатически увеличивают электронную плотность в остатке, т. е.
оказывают положительный индуктивный эффект (+7). Из правил Кернера,
Хюбнера и Нольтинга следует, что группы, которые сильно притягивают
электроны (—7), направляют заместители в мета-положение, в то время
как другие группы направляют заместители в орто- и пара-положения, что
вполне согласуется с опытом.
В последующие десятилетия были выдвинуты другие правила ориентации;
они прямо или косвенно связали ориентацию с состоянием насыщенности
или ненасыщенности имеющегося заместителя. В 1887 г. Армстронг [4]
228
Замещение в ароматическом ряду
отметил, что заместители, соединенные с бензольным кольцом через атом,
содержащий кратную связь, обладают мета-ориентирующим эффектом,
в то время как другие заместители ориентируют замещение в орто- и пара-
положения. Позднее Форлендер [5] особо подчеркивал значение по существу
такого же утверждения. В 1892 г. Крам Браун и Гибсон [6] выдвинули свое
знаменитое правило, состоящее в том, что заместитель X может быть мета-
ориентирующим, если НХ можно непосредственно окислить в НОХ; все
прочие заместители являются орто- и иара-ориентирующими. Несмотря
на то что ни одно из рассматриваемых правил не было точным, последнее
являлось более строгим.
Хотя природа существующей между указанными правилами связи до сих
пор не выяснена, они очень близки между собой; в них предвосхищена роль,
которую играет мезомерный эффект в современной теории ориентации при
ароматическом замещении. Вещество ХН легче окисляется в НОХ, если
последнее характеризуется по сравнению с первым повышенной стабиль-
ностью, например представляет собой весьма стабильную мезомернуювсисте-
му, для которой характерно наличие неподеленных электронов; это условие
выполняется, когда — X представляет собой группу — Y = Z с кратной
связью при условии, что Z характеризуется высоким сродством к электрону.
Н—6^-yXz
Эти группы являются мета-ориентирующими, если способность системы
притягивать электроны ароматического ядра по аналогичному механизму
достаточно велика.
аД-уД
Правила Армстронга и Форлендера указывают на ненасыщенность как на
условие лета-ориентации; само по себе это условие не является достаточ-
ным. В правиле Крама Брауна и Гибсона выдвинуто другое условие, заклю-
чающееся в том, что ненасыщенность сопровождается притяжением электро-
нов; сочетание этих факторов обусловливает мета-ориентацию, а следова-
тельно, это правило отличается большей точностью. Но и это правило все же
не вполне точно, потому что, хотя оба условия вместе достаточны, они не
необходимы; сильное притяжение электронов, даже без ненасыщенности,
вызывает мета-ориентацию.
б. Доэлектронные и переходные теории ориентации. В 1902 г. Флюр-
шейм [7] выдвинул первую серьезную теорию (в отличие от правил) ориента-
ции. Он использовал идею Вернера о том, что химическое сродство, даже
если оставить в стороне ненасыщенные соединения, может частично расхо-
доваться на образование связи, а частично оставаться свободным. Флюр-
шейм допустил, что такой атом, как «двухвалентный» кислород, со свой-
ственной ему способностью увеличивать свою валентность, требует в случае
связи с углеродом ароматического ядра больше сродства от углерода, умень-
шая тем самым сродство углерода к другим атомам и свободное сродство.
Этот атом соответственно требует меньше сродства от следующих атомов;
таким образом, это нарушение распространяется дальше, в результате чего
возникает чередующееся распределение связей и свободного сродства, как
это показано на.приведенной ниже схеме Флюршейма (Л). Такое распределе-
ние приводит к орто- и пара-ориентации при допущении, что замещающий
229
Глава VI
агент притягивается преимущественно к положениям, имеющим наибольшее
свободное сродство. Замещающий атом в своем высшем валентном состоянии,
например «пятивалентный» азот в нитрогруппе, рассматривался как атом,
запас сродства которого почти истощен: он способен дать малое сродство
па связь с атомом углерода ароматического ядра. Все это создает иное рас-
пределение сродства, ведущее к .чета-ориентации, как показано ниже на
схеме Б.
Схема Флюршейма. Связи с высоким и низким сродством обозначены соответственно
полужирными и тонкими линиями; атомы с большим и малым свободным сродством
отмечены длинными и короткими пунктирными линиями. А — орто- и пара-ориентация;
Б — .мета-ориентация.
Атом углерода рассматривался как имеющий постоянную валентность;
следовательно, поведение углеродного атома, непосредственно связанного
с ядром, определяется связанным с ним атомом переменной валентности.
В соответствии с этим бензотрихлорид, бензальдегид и бензонитрил должны
быть преимущественно .мета-ориентирующими (что и наблюдается), а фенил-
нитрометан — преимущественно орто- и пара-ориентирующим (чего не
наблюдается). Углеродный атом, соединенный с ядром и связанный с другим
атомом углерода кратной связью, как, например, в стироле, может как нена-
сыщенный атом требовать от бензольного кольца высокого сродства и таким
образом вызывает орто- и пара-ориентацию (как это и наблюдается вопреки
правилу Армстронга — Форлендера). Если атом углерода связан простыми
связями только с водородом и атомами углерода, как, например, в толуоле
и этилбензоле, то свойства такого атома углерода определяются главным
образом ненасыщенностью самого бензольного кольца; это должно привести
к орто- и пара-ориентации (что соответствует опыту).
Представления Флюршейма сыграли выдающуюся роль в развитии совре-
менной теории.
Во-первых, они конкретизировали общие идеи о роли ненасыщенности и
сопряжения, увидев в способности атома увеличивать свою валентность
(обобщать свои неподеленные электроны, по современной терминологии)
условие, которое может, подобно двойной и тройной связям, нарушать
распределение сродства у соседних атомов.
Во-вторых, они совершенно правильно описали перераспределение сродства,
обусловленное таким видом ненасыщенности, и объяснили орто- и пара-
ориентацию. Эти представления могли быть (и были) непосредственно
переведены на электронные понятия; таким образом, как показано ниже,
представления Флюршейма учитывают роль электромерного эффекта в совре-
менной теории ароматического замещения.
Однако в случае меша-ориентирующих влияний возникают затруднения.
Уже в 1902 г. стало известно одно меша-ориентирующее соединение, а именно
230
Замещение в ароматическом ряду
фенилнитрометан, которое не укладывалось в рамки теории; Флюршейм
дал этому случаю крайне неубедительное объяснение (и, как было позднее
доказано, неверное [8]). После 1920 г. было выявлено много других примеров
такого рода, которые не могли быть удовлетворительно объяснены. Причина
затруднений заключается частично в том, что доэлектронная теория Флюр-
шейма истолковывала электрохимический характер как свойство, обособлен-
ное от распределения валентности, и частично в том, что, согласно этой
теории, основанной на вернеровском представлении о валентности, не при-
знавали большой разницы в подвижности, которая существует между насы-
щенными и ненасыщенными валентностями (т. е. между л- и a-свя-
зями) .
Голлеман предложил важный критерий, позволивший согласовать между
собой все теории ориентации. В своей книге [9], появившейся в 1910 г.,
Голлеман обработал известные факты ориентации, включая собственные
многочисленные количественные наблюдения. Данная им теоретическая
интерпретация основана на предположении, что первым шагом при замеще-
нии является присоединение к кекулевской двойной связи, а также что
ориентирующий эффект заместителя заключается в изменении реакционной
способности двойных связей к присоединению. Однако Голлеман не пытался
дать законченной теории ориентации, учитывающей влияние на реакционную
способность строения заместителей. Важной стороной в представлениях
Голлемана было утверждение о связи между ориентацией и реакционной
способностью ^’ароматического ядра. Голлеман показал, что орто- и пара-
ориентирующие заместители обычно увеличивают скорость дальнейшего
замещения, тогда как .чета-ориентация, несомненно, сопровождается умень-
шением скорости замещения.
Следовательно, ни одну теорию нельзя признать удовлетворительной, если
она рассматривает мета-углеродный атом при мета-ориентации точно
так же, как орто- и napa-углеродные атомы при орто- и napa-замещении.
Теория Флюршейма не удовлетворяет этому требованию, так как она пред-
полагает активацию мета-углеродных атомов при мета-замещении, тогда
как соотношение скоростей реакций показывает, что все атомы ядра дезак-
тивируются.
В течение следующих пятнадцати лет внимание привлек ряд весьма близких
теорий, часто известных в отдельности или собирательно под названием
«теории альтернирующих полярностей» [10]. В первоначальной форме
«теория альтернирующих полярностей» была выдвинута Фраем, который
предположил, что атомы бензола имеют чередующиеся заряды в двух
быстро взаимопревращающихся формах; такие атомы Фрай называл «элек-
тромерами».
Электромеры бензола по Фраю
В большинстве производных бензола преобладает один из электромеров;
при замещении предпочтительно реагирует положительный атом водорода,
и поэтому определенный электромер обусловливает преимущественную ориен-
231
Глава VI
тацию, как это показывает следующая схема:
OR
+
"О О"
Предпочтительный
электромер для орто-
пара- ориентации
Предпочтительный
электромер
для мета-ориентации
Размещение чередующихся зарядов, которому благоприятствует данный
заместитель, устанавливается по правилу Крама Брауна и Гибсона сле-
дующим образом. Окисление обусловливает высвобождение электронов;
следовательно, легкое протекание реакции НХ -> НОХ означает, что X
стремится зарядить связанный с ним атом отрицательно. Например, тот
факт, что реакция окисления HNO2->HNO3 идет легко, свидетельствует
о том, что нитрогруппа отталкивает электроны к соседнему атому, в частно-
сти к соседнему углеродному атому ядра, заряжая его отрицательно, как
показано на приведенной выше схеме. Интересно отметить, что такое на
первый взгляд правдоподобное соображение приводит к неправильному
выводу; это объясняется игнорированием эффекта ненасыщенности и вызы-
ваемой им мезомерии, которые обусловливают легкость окисления азотистой
кислоты в азотную. Однако это неправильное исходное положение в сочета-
нии с также неправильным допущением чередующейся полярности приводит
к правильным выводам об ориентации.
Форлендер и Лэпуорт пришли к аналогичному выводу о предпочтительном
распределении зарядов в бензольном кольце, допустив, что атом в заместите-
ле, обладающий наиболее резко выраженной электрохимической природой,
названный Лэпуортом «ключевым» атомом, определяет порядок чередования
зарядов. Так, например, поскольку атомы кислорода в — OR и — N02
отрицательны, атом азота в NO2 становится положительным; атомы водорода
в СН3 положительны, и поэтому атом углерода становится отрицательным.
Форлендер выдвинул дополнительное предположение о том, что знак ионно-
го заряда на атоме, непосредственно связанном с бензольным кольцом, опре-
деляет полярность. Форлендер внес большой вклад в теорию ориентации,
показав экспериментально [11], что такие катионоидные заместители, как
— N(GH3)3, являются меига-ориентирующими, в то время как было известно,
что анионоидный заместитель — О- ориентирует в орто- и пара-поло-
жения.
Что касается механизма ориентации, то Лэпуорт и Лоури высказали пред-
положение о возникновении зарядов только в процессе реакции. Лоури счи-
тал, что именно углеродные, а не водородные атомы бензольного кольца
приобретают заряды. Между тем Кермак и Робинсон предложили электрон-
ную (но лишенную физических оснований) картину образования зарядов
в результате чередующегося сжатия и расширения октетов.
Эта теория столкнулась со многими трудностями. Форлендер указал на ее
неудовлетворительность, когда Малхэрб [12] установил, что пгрепг-бутилбеп-
зол ориентирует замещение главным образом в пара-положение. Кроме того,
232
Замещение в ароматическом ряду
выяснилось [13], что а-стириловый эфир замещается в орто- и пара-положе-
ния; очевидно, атом кислорода в заместителе — G(OR) = СН2 не является
«ключевым» атомом. Далее было установлено [14], что наиболее ярко выра-
женный по своей электрохимической природе заместитель — F не определяет
ориентации при конкуренции с менее электроотрицательными заместителями
— OR и — NR2 и что — OR так же ведет себя при конкуренции с — NR2.
При этом всегда необходимо прибегать к критерию Голлемана и учитывать,
что теорию нельзя признать правильной, если она связывает мета-ориента-
цию с активацией мета-положений и, следовательно, считает мета-положе-
ния при мета-ориентации измененными точно так же, как и орто- и пара-
положения при орто- и пара-ориентации. По этой причине теория альтерни-
рующих полярностей была заранее обречена на неуспех. Ретроспективно
эту теорию можно рассматривать как выражение глубокого убеждения, что
каким-то образом электрохимические факторы имеют фундаментальное
значение в ориентации и что теория должна отражать это обстоятельство.
в. Возникновение электронной теории ориентации. В 1926 г. трактовка
правил ориентации приняла то направление, которого она придерживается
до сих пор. Этот вопрос рассматривался как составная часть более широкой
проблемы влияния строения на реакционную способность вообще. Вместе
с тем изучение ориентации в ароматическом ряду стало в течение нескольких
лет главным объектом исследования, поскольку позволяло подойти к реше-
нию указанной более широкой проблемы.
К этому времени накопилось много опытных данных, которые давали воз-
можность построить общую теорию влияния строения на реакционную спо-
собность. Льюис установил, что определенный тип электронных смещений
является постоянным свойством молекул, что теперь известно под названием
индуктивного эффекта. Лоури постулировал и обосновал на примерах выдви-
нутый им тип электронных смещений, возникающих в результате активации,
что теперь называют электромерным эффектом (гл. II, разд. 3,а). При рас-
смотрении механизма присоединений к олефинам Лукас объединил индуктив-
ный эффект с электромерным и показал, как первый может усиливать второй
эффект и придавать ему определенное направление (гл. II, разд. 3,6). Все
реагенты классифицировались по признаку их сродства к электронам или
к ядрам; в современной терминологии они разделяются на электрофильные
и нуклеофильные. При этом было отмечено, что реагенты, принимающие
участие в ароматическом замещении, к которым относятся правила ориента-
ции, являются электрофильными (гл. V, разд. 1 и 2,в). Примерно в это же
время Робинсон и автор с сотрудниками начали разрабатывать на основе
такого рода идей теорию ориентации при замещении в ароматическом ядре
[15]. В самом начале было введено представление о постоянных электронных
смещениях по механизму сопряжения — о мезомерном эффекте (гл. II,
разд. 3,а). Несколько позднее в общую теорию было введено представление
об активирующих смещениях по индуктивному механизму — об индуктомер-
ном эффекте (гл. II, разд. 3,а).
Оставалось только проверить и критически оценить выдвинутую теорию.
Это было сделано, как показано ниже, в результате получения данных,
позволяющих проникнуть глубже в механизм ориентации. Во-первых, было
изучено количественно соотношение изомеров, получающихся при замеще-
нии, и таким образом продолжены работы в направлении, впервые исследо-
ванном Голлеманом. Во-вторых, для более глубокого проникновения в суть
проблемы была изучена зависимость между направлением замещения и ско-
ростью реакции, что ведет к выяснению взаимоотношений ориентации и
реакционной способности; на важность этой взаимосвязи указывал еще
233
Глава VI
Голлеман. Развиваемая теория ориентации и указанные две стороны ее весь-
ма тесно связаны между собой, и поэтому для удобства они будут рассматри-
ваться совместно.
2. Ориентация при электрофильном замещении
в ароматическом ряду [16]
а. Ориентирующее влияние полярных заместителей. Форлендер [17]
сделал важное открытие, состоящее в том, что свободный положительный
ионный центр в заместителе, непосредственно связанном с бензольным ядром,
ориентирует преимущественно или исключительно в метла-положение неза-
висимо от того, является ли элемент, несущий заряд, легким или тяжелым,
металлом или неметаллом *. Примерами могут служить нитрование нитрата
фенилтриметиламмония или дифенилиодония, динитратов дифенилсвинца
или трифенилвисмута. Челленджер и Ротштейн [19] показали, что ряд метал-
лов, а именно ртуть, таллий и олово, в виде положительного заряженного
ионного центра, связанного непосредственно с ароматическим кольцом, силь-
но ориентируют замещение в метла-положение. В дальнейшем число таких
заместителей увеличилось в результате включения заместителей, содержа-
щих элементы V и VI групп таблицы Менделеева; к числу изученных катио-
нов, ориентирующих в метла-положение, относятся следующие:
C6H5Hg+ (С6Н5)2Т1+ (C6H5)2Sn2+ (С6Н5)2РЬ2+ (C6H6)sBi2+ (С6Н5)21+
CeH5N(CH3)3 С6Н5Р(СН8)3 C6H5As(CH3)3 C6H5Sb(CH3)3 C6H5S(CH3)2 CeHsSe(CH3)2
Заместители положительного ионного типа принадлежат к сильнейшим
метла-ориентирующим группам. Однако степень метла-ориентации непрерыв-
но понижается с увеличением длины углеродной цепи, расположенной между
положительным зарядом и бензольным ядром. Этот вывод был установлен
при сравнении реакций нитрования фенил-, бензил-, 2-фенилэтил- и 3-фенил-
л-пропилтриметиламмоний-ионов [20]. Выход метла-продуктов нитрования
в процентах указан в табл. 61. Динитропроизводное не образуется, и поэтому
остальное составляют орто- и лара-нитропроизводные. Ниже будет обсужде-
на зависимость между ориентацией и скоростью нитрования, однако уже
сейчас вслед за Риддом и другими авторами [21] можно указать, что ско-
рость мононитрования монотонно повышается с удлинением цепи (табл. 61).
В предпоследнем примере таблицы положительный полюс влияет на бензоль-
ное кольцо лишь очень незначительно, и поэтому метла-продукт образуется
лишь в немного большем количестве, чем при нитровании толуола (3%).
Ряд данных отчетливо показывает, что влияние, обусловливающее чередова-
ние в типе замещения, не передается через боковую цепь. Любая теория
чередования эффекта, .например теория альтернирующих полярностей Флюр-
шейма, исходя из того, что фенилтриметиламмоний-ион является метла-ориен-
тирующим, требует, чтобы бензилтриметиламмоний-ион был бы преимуще-
ственно орто- и лара-ориентирующим. Подобное положение имеет место
и в уже упоминавшемся случае нитробензола и фенилнитрометана, при
объяснении которого Флюршейм испытывал затруднения.
* Элементы второго периода таблицы Менделеева, способные образовывать ониевые
соединения, у которых остаются неподеленные электроны, могут составлять исключение.
В то же время как (СвН5)2С1+ и (С6Н5)21+ дают почти нацело .мета-продукт, (C6HS)3O+,
не имеющий пустых d-орбиталей, способных к — /С-сопряжению, нитруется преимуще-
ственно в пара-положение [18].
234
Замещение в ароматическом ряду
Таблица 61
Влияние удлинения боковой цепи на относительные
скорости мононитрования ароматических аммониевых
ионов и выход .мета-продуктов нитрования
Ароматический аммониевый ион Скорость реакции Выход мета- продукта, %
CBHSN(CH3)3 1 89
CeH6CH2N(CH3)3 3,6-103 85
CeHj(CH2)2N(CH3)3 4,63-10в 19
CeHs(CH2)3N(CH8)3 5,10-107 5
CeH6CH3 4,69-108 3,5
Влияние положения элемента, дающего положительный ионный заряд,
в таблице Менделеева иллюстрируется сравнением фенил- и бензилполи-
метилониевых ионов элементов V и VI групп. Экспериментальные данные
и в этом случае относятся к нитрованию. Относительное содержание мета-
изомеров в продуктах нитрования приведено в табл. 62.
Таблица 62
Количества мета-производных, образующихся при мононитровании
фенил- и бензилониевых ионов
/4pN(CH8)3
\Z
89%
— Р(СН3)3
V
97%
As(CH3)3
96%
Sb(CH3)3
\z
86%
S(CH3)2
\Z
90%
Se(CH8)2
\z
Высокий выход
|A- CH2N(CH8)3
\z
85%
CH2As(CH3)8
\z
7%
CH2S(CH3).
\z
52%
|Pj-CH2Se(CH3)2
\Z
16%
Все эти данные подтверждают, что мета-ориентация является результатом
оттягивания электронов к ориентирующему заместителю и обусловлена
электроотрицательностью, а это соответствует представлениям ученых,
создавших ранние правила ориентации. Электронное смещение вызывается
отрицательным индуктивным эффектом (гл. II, разд. 3,а), воздействие кото-
рого на ароматическое ядро уменьшается вдоль цепи насыщенных углерод-
235
Глава VI
ных атомов, становясь едва заметным через три углеродных атома.
вбб+ 66+ 6+ +
СвН6—> СН2—> СН2—> СН2—> N(CH3)3 (-/-Механизм)
Во всех случаях главной причиной притяжения электронов является катион-
ный заряд на ониевом атоме. При данном заряде существенное значение
имеет также внутренне присущая электроотрицательность элемента. Как
правило, мета-ориентирующая способность увеличивается в изоэлектронном
ряду атомов (гл. II, разд. 3, б) слева направо в таблице Менделеева, т. е.
с увеличением полного заряда ядра
+ + + +
S > Р и Se > As
Кроме того, мета-ориентирующая способность уменьшается по мере увели-
чения атомного веса в данной группе таблицы Менделеева (гл. II, разд. 3,6),
т. е. уменьшается с усилением экранирования ядер
+ + + + +
N>P>As и S>Se
Аномальными свойствами обладают фенилониевые соли. Наиболее ярко это
проявляется в том, что количество мета-нитроизомера при нитровании
фенилтриметиламмония меньше ожидаемого на основании теоретических
соображений. Этого не наблюдалось в ранних работах; однако в 1962 г.
Ридд, Ротштейн и др., используя более точные методы анализа изомеров,
получили данные, приведенные в табл. 62. Наибольший интерес представля-
ют данные по нитрованию иона фенилтриметиламмония.
Необходимо было объяснить две аномалии. Первая касается необычного
положения азота в ряду других элементов V группы, входящих в состав
фенилониевых солей. Ридд объяснил это, предположив, что в молекулах всех
солей, кроме аммонийных, осуществляется особый вид сопряжения (—К-
сопряжение), которое обусловлено наличием пустых d-орбиталей у электро-
отрицательных элементов. л-Электроны ароматического кольца сопрягаются
с пустыми Зй-орбиталями фосфора, 4й-орбиталями мышьяка и Sd-орбиталями
сурьмы; азот не имеет валентных d-орбиталей, поэтому — А-сопряжение
невозможно. Наиболее сильно такое сопряжение проявляется в случае
фосфора, так как его d-орбитали малы и должны меньше деформироваться
для обеспечения эффективного сопряжения, чем большие d-орбитали мышья-
ка и сурьмы. d-Орбитали деформируются (сжимаются) под влиянием поло-
жительного заряда. Тот факт, что сжатие d-орбиталей в потенциальном поле
может в значительной мере облегчить dn — рл-связывание, был впервые
отмечен Крейгом [22]. Подобные эффекты должны проявляться и в фенил-
ониевых ионах элементов VI группы — серы и селена.
Вторая аномалия состоит в том, что различие в мета-ориентирующих свой-
ствах фенил- и бензилтриметиламмониевых ионов очень мало; мы, однако,
отложим обсуждение этого явления до тех пор, пока не выясним общей
зависимости между ориентацией и скоростями замещения.
Сильный орто-пара-ориентирующий эффект свободных отрицательных ион-
ных центров аналогичным образом подтверждается исключительным орто-
пар a-замещением фенолов в щелочной среде, например при азосочетании или
при хлорировании щелочными гипохлоритами. Сравнение опытных данных,
характеризующих ориентирующую способность различных анионных эле-
ментов (О, S, Se и т. д.) или одного и того же элемента в различных положе-
ниях относительно ароматического ядра, пока еще не проводилось.
236
Замещение в ароматическом ряду
Предполагается, что ориентация в фенолят-ионах обусловлена отталкивани-
ем электронов от анионного центра к ароматическому кольцу; это результат
влияния электроположительности. Несомненно, здесь налагается положи-
тельный индуктивный эффект
С8 Н 5 «— О (-|- /-Механизм)
хотя одновременно действует и играет важную роль и + Х-эффект (сопряже-
ние неподеленных пар кислорода с л-электронами кольца). Эти эффекты
сопряжения будут рассмотрены ниже.
б. Ориентирующие влияния диполярных связей. Теория октета при-
писывает нитрогруппе одну диполярную связь (гл. I, разд. 2,г); хотя по
мезомерным представлениям диполь распределен по двум связям (гл. II,
разд. 3,в), в молекуле присутствует только один электрохимический экви-
валент диполярной связи. Физические исследования показывают (гл. III,
разд. 1,6), что, хотя электроны в нитрогруппе сильно смещены к положи-
тельному атому, все же в значительной мере сохранившийся дипольный
характер связи оказывает влияние на остаток молекулы. Кроме того,
в нитрогруппе имеется ненасыщенность, также распределенная между двумя
связями. Хотя, строго говоря, не следует игнорировать влияние ненасыщен-
ности при трактовке явлений ориентации, ориентирующий эффект диполяр-
ных связей настолько велик, что в первом приближении ненасыщенностью
можно пренебречь.
Октетная теория приписывает сульфогруппе две диполярные связи и полное
отсутствие ненасыщенности (гл. I, разд. 2,г). Физические исследования
показывают, что в сульфогруппе электроны сильно смещены к атому серы
(гл. III, разд. 1,6) и что сера в известной мере увеличивает свою ковалент-
ность вследствие частичного образования двойных связей за счет d-орбиталей.
При этом все же в значительной мере сохраняется диполярный характер
связей, что влияет на реакционную способность остальной части молекулы.
В этом случае, также в первом приближении, можно пренебречь ненасыщен-
ностью.
Любая диполярная группа, например NO2 или SO2R, всегда соединена
с соседним остатком положительным концом своей диполярной связи или
связей. Следовательно, можно ожидать, что влияние таких групп качествен-
но подобно влиянию положительного заряда, количественно уменьшенного
вследствие противоположного влияния более отдаленного отрицательного
конца диполя.
Нитрогруппа — одна из сильнейших лге/па-ориептирующих групп, не имею-
щих явного ионного заряда. В табл. 63 приведены данные, показывающие
содержание мета-изомеров в продуктах нитрования нитробензола [23],
фенилнитрометана [24] и 2-фенил-1-нитроэтана [20]. (Данные Бейкера для
фенилнитрометана подтверждают ранние качественные опыты Голлема-
на [25], показавшего, что при нитровании фенилнитрометана преобладает
мета-ориентирующий эффект, так как образуется более 50% мета-изомера.
Учитывая эти данные наряду с другими, Флюршейм в 1902 г. счел необходи-
мым выступить в защиту своей теории; об этом говорилось в гл. V, разд. 1,6.)
Данные таблицы относятся к мононитрованию, а разность до 100% соответ-
ствует выходу орто- и пара-нитропродуктов.
Суммарный эффект обусловлен притяжением ароматических электронов
положительным концом диполярной связи, действующим либо непосредствен-
но, либо через углеродную цепь, как и в упоминавшемся выше случае три-
метиламмоний-ионов. мета-Ориентация нитрогруппы заметно слабее, чем
в случае триметиламмоний-иона с идентичной углеродной цепью, что можно
237
Глава VI
Таблица 63
Количества .мета-производных, образующихся
при мононитровании нитробензола и его гомоло-
гов с нормальными боковыми цепями
Соединение
Выход мета -продукта. %
93
55
13
объяснить влиянием отрицательного кольца диполярной связи, включая
влияние, которое имеет форму электронных смещений при обратной поляри-
зации в самой нитрогруппе. Однако в целом нитрогруппа является электро-
отрицательной, оказывающей во всех случаях сильный —/-эффект. Ненасы-
щенность нитрогруппы, обусловливающая —Х-сопряжение, действует в том
же направлении.
К сильным мета-ориентирующим группам относится и сульфоновая группа:
фенилалкилсульфон [26] нитруется почти исключительно в мета-положе-
ние, бензолсульфокислота [27] в значительно меньшем количестве дает
мета-продукт при нитровании. Указанное различие в степени мета-заме-
щения подтверждено количественным изучением нитрования бензилсульфо-
соединений [28]. Данные приведены в табл. 64.
Таблица 64
Количества мета-производных, образующихся при моно-
нитровании фенил- и бепзилсульфосоединений
Соединение Выход мета- продукта, % Соединение Выход мета- продукта, %
/4/SO2C2H5 II ч| 89 /4/CH2SO2C2JI5 II х| 22
и /X/SO2OCH3 || х| — II 1 \z ^/СНгЗОгОСНз II '1 32
ll 1 \z /4/SO2O- II 1 60 ll 1 \z /4/CH2SO2O- II 1 14
II 1 \z II 1 \Z
238
Замещение в ароматическом ряду
Эти данные можно объяснить, если принять допущение, что сульфокислота
нитруется в виде аниона. В сульфоне и сложном эфире сульфокислоты частич-
ная компенсация .чета-ориентирующего влияния заряженного атома серы
обусловлена двумя формально отрицательно заряженными атомами кислорода
в сульфогруппе; в сульфокислоте благодаря ионной диссоциации возникает
третий такой же атом кислорода, что ведет к изменению ориентирующей
способности группы.
в. Ориентирующее влияние формально нейтральных групп без неподе-
ленных электронов или с незаполненными электронными оболочками. При
обсуждении сильного ориентирующего влияния ионных и диполярных связей
можно в известном приближении пренебрегать ненасыщенностью, которая
имеется в разных случаях либо в виде необобщенной пары электронов, или
вакантных валентных орбиталей, например в диметилсульфониевой группе,
либо в виде двойной связи, например в нитрогруппе. Однако такой упрощен-
ный подход недопустим даже при предварительном рассмотрении более сла-
бых ориентирующих влияний нейтральных групп. Необходимо с самого
начала принять во внимание по крайней мере сопряжение неподеленных
электронных пар или пустых валентных орбиталей с ароматическим ядром
с тем, чтобы позже в качестве приближения заняться подробным рассмотре-
нием сопряжения кратных связей. В соответствии с этим сначала будет рас-
смотрено ориентирующее влияние нейтральных групп, которые не обладают
неподеленными электронами или валентными орбиталями, сопряженными
с ядрами, а затем разобраны другие случаи, обусловленные таким сопря-
жением.
На основании химического опыта и определения дипольных моментов твердо
установлено, что замещение водорода в ароматическом ядре алкильной
группой приводит к слабому смещению электронов от заместителя к ядру;
следовательно, алкильная группа является слабо электроположительной.
Точно так же установлено, что замещение водорода типичными электроотри-
цательными элементами, например галогенами, карбонильной группой,
неионизированными формами карбоксильной группы или циангруппой,
приводит к резкому смещению электронов в сторону заместителя; следова-
тельно, все эти группы являются сильно электроотрицательными. Такое
деление соответствует принятой классификации групп на основании их
ожидаемых индуктивных эффектов (гл. II, разд. 3,в).
«—Aik -Н —»Hal —» COR —» CN
+1 (стандарт) — I
Отсюда следует, что у алкильных групп должен преобладать орто-пара-
ориентирующий эффект, а карбонильная, карбоксильная и циангруппы
должны быть преимущественно мета-ориентирующими, что в действитель-
ности и наблюдается, как это подтверждается данными табл. 65. (Ориенти-
рующее влияние галогенов, непосредственно связанных с ядром и имеющих
неподеленные электронные пары в сопряжении с ароматическим кольцом,
рассматривается ниже. В ходе последующего изложения будет отмечено
также, что у карбонильной, карбоксильной и циангрупп, а также нитро-
группы, непосредственно соединенных с ароматическим ядром, мета-ориен-
тирующее влияние частично обусловлено сопряжением их кратных связей
с ядром.)
Из изложенной выше электрополярной классификации следует, что замеще-
ние водорода в заместителе в любом положении на алкильную группу снижа-
ет л«ета-ориентирующий эффект заместителя; с другой стороны, замещение
водорода галогеном, карбонильной, карбоксильной или циангруппой усили-
239
Глава VI
Таблица 65
Количества мета-производных, образующихся при мононитровании замещенных
в боковой цепи толуолов
/\_ СОО ч- СН8
Ч/
73% [29]
С0° СНг СНз
Ч/
68% [29]
'%- соон
I II
82% [29, 40]
7% [33]
29% [36]
СН2 -> С1
Ч/
14% [33, 34, 37]
55% [37]
84% [36]
3% [30-32] 14% [33, 34]
_СООС2Н5
Z\ _ СН
I II \соосЛ
23% [33, 38]
Z\,_ соон
Ч/
Z\___сн,___н
Ч/
3% [30-32]
СОСНз
\/
68% [39]
CN
Ч/
81% [41]
СН2 -> СООС2Н6
Ч/
13% [37]
CONH2
70% [40]
CH2CN
Ч/
20% [37]
82% [29, 40]
СН(СООС2Н5)2
23% [33, 38]
^,СООС2Н5
44“ С СООС2Н5
I ^соос2н,
~ч/
57 % [33]
Z\_ coci
\о% [40]
СН2СН(СООСН3)2
ЧЧ
8% [42]
вает мета-ориентирующее влияние заместителя. Эти эффекты играют особо
важную роль в тех случаях, когда замещение осуществлено в непосредствен-
ной близости к ароматическому ядру, так как индуктивное влияние сильно
снижается при передаче по цепи. При замещении в одном и том же положе-
нии величина эффекта зависит от степени электроотрицательности замещаю-
щей группы. Все эти соображения подкрепляются данными (относящимися
к нитрованию), приведенными в табл. 65, в которой указано процентное
содержание мета-нитроизомера в мононитропродуктах. Каждый горизон-
тальный ряд в таблице отражает изменение полярности заместителя опре-
деленного типа, за исключением последнего ряда, который относится к двум
независимым парам гомологов.
.240
Замещение в ароматическом ряду
Атом дейтерия, связанный с бензольным кольцом, не оказывает заметного
ориентирующего эффекта [43]. Дейтерий несколько более электроположите-
лен, но несколько менее поляризуем, чем протий; эти два эффекта, вероятно,
компенсируются. Возможно также, что эти эффекты не компенсируются,
но настолько малы, что не оказывают влияния на ориентацию. Триметил-
силильная группа (CH3)3Si оказывает незначительное ориентирующее влия-
ние. При нитровании фенилтриметилсилана образуется нитробензол и смесь
нитротриметилсилилбензолов, которая содержит 40% .мета-изомера (стати-
стическое количество) [44].
г. Ориентирующее влияние формально нейтральных групп, содержа-
щих неподеленные электронные пары. Ненасыщенность оказывает наиболь-
шее влияние в случае формально нейтральных заместителей с пеподеленными
электронами, способными к сопряжению с ароматическим ядром; такие заме-
стители являются орто-геара-ориентирующими независимо от направления
их индуктивного эффекта. Это влияние соответствующим образом располо-
женных неподеленных электронных пар недостаточно для того, чтобы обра-
тить .мета-ориентацию таких заместителей с формальным положительным
зарядом, как, например, сульфоний- и иодоний-ионы. Однако это влияние
превосходит .мета-ориентацию, которую можно ожидать от нейтральных
групп, например от таких электронопритягивающих заместителей, как
галогены и алкоксильные группы. Такой эффект приписывается возникнове-
нию отрицательных зарядов в орто- и геара-положениях вследствие сопря-
женного смещения неподеленных электронных пар заместителя по -\-К-
механизму (гл. II, разд. 3,а).
6+хД^^р S— (+К-Механизм)
Такая трактовка * оставляет открытым вопрос о том, является ли сопряже-
ние, вызывающее указанное воздействие, по существу постоянным свойством
молекулы (-|-М-эффект) или оно представляет собой форму активации (+£'-
эффект). Этот вопрос рассматривается ниже. Однако тот факт, что даже
галогенбензолы оказывают орто- и геара-ориентирующий эффект, несмотря
на то что они, как известно, сильно смещают электроны от кольца к гало-
гену, показывает, что смещения, которые играют такую важную роль
в наблюдаемой ориентации, в действительности являются временным
фактором.
Ориентация данного типа присуща элементам (в нейтральной форме) V,
VI и VII групп таблицы Менделеева. В изоэлектронном ряду сила ориента-
ции изменяется в соответствии с реакционной способностью неподеленных
электронов, а следовательно, уменьшается с увеличением номера группы
(гл. I, разд. 2,д). Такой вывод сделан [45] на основании изучения конкури-
рующего влияния двух различных групп при нитровании (табл. 66).
При сравнении азота и кислорода необходимо иметь в виду, что ацилирова-
ние атома азота, цель которого заключается в предотвращении образования
соли с нитрующей смесью, снижает орто-геара-ориентирующее влияние
ацилированной группы. Это вытекает из многих известных опытных данных.
Так, ацетилгваякол нитруется исключительно в геара-положение по отноше-
* О применении в этом случае формулы Кекуле см. гл. IV, разд. 1,6.
16-0772
241
Глава VI
Таблица 66
Конкурирующая ориентация N, О и F при нитровании дизамещенных бензолов
NHCOCH3 орто и пара по отношению к Г NHCOCH3 76% (выделено)
У-°СН3 1 ОСН3 13% (выделено)
ZS- N(CH3)COCH3 орто и пара по отношению к Г N(CH3)COCH3 69% (выделено)
IN- 0СН3 Т ОСН3 4% (выделено)
/Ч NHCOCH3 орто и пара по отношению к Г NHCOCH3 71% (выделено)
ll\ J- ОСОСНз tОСОСН3 0% (выделено)
,А- ОСНз орто и пара по отношению к f ОСН3 97% (по анализу)
II I— F I F 3% (по анализу)
\/
2%
13% -/\-NHCOCII3
Пример:
74% “\Z“ 0СНз
0%
Цифры указывают количество образующихся
мононитропродуктов (общий выход 89%)
нию к метоксильной группе [46].
ОСН3 |А|- ОСН3
I L ОСОСН3 । L ОСОСН3
o2NZ \/
Далее, в то время как однократного ацилирования азота (например, в ацето-
га-толуидине) недостаточно, чтобы сделать ориентирующую силу ациламино-
группы ниже ориентирующей силы метильной группы, двойное ацилирова-
ние (например, во фталоил-га-толуидине) вызывает необходимое снижение
ориентирующей силы, и нитрование этих соединений протекает в направле-
нии, указанном на следующей схеме [47]:
СН3- \ S-NHCOCH3
Этот эффект вполне понятен с теоретической точки зрения. Ацилирование
атома кислорода или азота приводит к образованию заместителя типа карбо-
ксильной группы X — С = Y, собственная мезомерия которой мешает ориен-
тации, вызываемой сопряжением X с арильной системой (гл. II, разд. 3,д).
Несмотря на помехи, причиняемые ацилированием, атом азота определяет
ориентацию, если он конкурирует с кислородом; точно так же кислород
определяет ориентацию, если он конкурирует с фтором. Таким образом,
242
Замещение в ароматическом ряду
можно прийти к ряду N Z> О > F, как того и следовало ожидать на основа-
нии +М- и -|-Е-эффектов (гл. II, разд. 3,д и е).
Электроны двойной связи в стироле сопряжены с ароматическим ядром
во многом аналогично неподеленным электронам. Возникающая орто-пара-
ориентация не нарушается даже такими концевыми электронопритягивающи-
ми группами, какие имеются в коричной кислоте [48], стирол-со-сульфохлори-
де [48] и ю-нитростироле [49]; даже это последнее соединение дает при нитро-
вании только 2% лгта-нитропроизводного:
CeH5CH = CHCOOH CeH5CH = CHSO2C1 CeH5CH = CHNO2
0% jiema-NO2 <2% .iitw-NO, ~ 2% люта-КО2
Таким образом, здесь положение сходно с положением в ряду галогенбензо-
лов, оршо-иара-ориентация которых не нарушается электроотрицательностью
галогенов. Как и раньше, это происходит потому, что смещения электронов,
приводящие к орто-геара-замещению в производных стирола, должны быть
эффектами поляризуемости, что подтверждается данными, которые рассмотре-
ны ниже.
Более тонко сбалансированные эффекты наблюдаются в соединениях бен-
зильного типа, которые являются преимущественно оршо-пара-ориенти-
рующими, несмотря на наличие сильно электроотрицательных заместителей
в боковой цепи (например, хлористый бензил или бензилцианид, табл. 65)
[50]. Вместо сопряжения в этих системах надо рассматривать гиперконъюга-
цию. Анализ этих эффектов будет дан после рассмотрения скоростей
реакций.
д. Ориентирующее влияние связанных с ядром групп, имеющих вакант-
ные орбитали в валентной оболочке. Гарвей и Норман [51] в результате
исследования фенилборной кислоты значительно расширили соответствую-
щий класс ориентантов, о котором ранее было известно очень мало. Нитрова-
ние фенилборной кислоты смесью азотной и серной кислот дает .и-нптроизо-
мер с выходом 73%. Остаток борной кислоты обладает слабыми электрополо-
жительными свойствами, поэтому очевидно, что причина сильной мета-
ориентации состоит в оттягивании ароматических электронов от кольца
путем сопряжения с вакантной 2р-орбита'лью атома бора *.
^~В (ОН)2 (—К~ Механизм)
Как уже отмечалось в разд. 2,а, сопряжение ароматических л-электронов
с вакантными Зй-орбиталями в ионах фенилтриметилфосфония и фенилтри-
метилсульфония отчасти обусловливает сильную жета-ориентацию, которая
наблюдается в этих ионах. Подобные, но, вероятно, более слабые эффекты
—^-сопряжения могут вносить вклад в жеша-ориентацию аналогичных
катионов высших элементов V и VI групп таблицы Менделеева.
е. Ориентационные эффекты в ионогенных системах. Кажущиеся исклю-
чения из правила, согласно которому нейтральный атом азота превосходит
атом кислорода по своей способности вызывать ортпо-иара-ориентацию, обна-
руживаются при возникновении ионных зарядов на одном из этих атомов
* См. работу [51]. Если не удалять основание, то оно может координироваться с элек-
троноакцепторной орбиталью бора. Авторы показали, что при нитровании в уксусном
ангидриде фенилборпая кислота проявляет преимущественно opmo-rcapa-ориентацию;
они предположили, что в этих условиях реакция частично протекает через аддукт
С6Н5В(ОН)2 - О(СОСН3)2.
16* 243
Глава VI
в условиях замещения. Теоретический порядок, в котором убывает орто-
иа/ш-ориентирующая сила азота и кислорода в нейтральной и заряженной
— +
формах, таков: О > NR2 > OR > NR3. Этот порядок, как будет ясно из
дальнейшего изложения, является также теоретическим порядком скоростей
замещения. Поэтому фенолы проявляют тенденцию к замещению в форме
анионов, а производные анилина — в их нейтральной форме, даже если
более реакционноспособная форма присутствует в незначительном количе-
стве. Так, Н-кислота, как известно, вступает в реакцию азосочетания
в оршо-положение к гидроксилу в щелочной среде и в оршо-положение
к аминогруппе в кислой среде, образуя, таким образом, два различных азо-
красителя. Реакция первого вида подтверждает неравенство О > NR2,
второе направление реакции подтверждает неравенство NR2 > OR:
НО NH2
I I
(Щелочной раствор)-------------- (Кислый раствор)
НО3
Положения присоединяемых заместителей в Н-кислоте
В псевдокислотных системах отрицательный заряд, возникающий в результа-
те отщепления протона, оказывается распределенным, что обусловлено мезо-
мерными свойствами аниона. Если часть анионного заряда достигнет атома,
соединенного с ароматическим кольцом, то в результате этого усиливается
оршо-нара-ориентация. Бейкер [52] показал это на примере нитрования
фенилнитрометана и его производных; при нитровании этих псевдокислот
в виде солей наблюдается резко увеличенный выход оршо-пара-производных.
Очевидно, анион фенилнитрометана ориентируется в оршо-геара-положения;
так, например, если литиевую соль фенилнитрометана добавлять к азотной
кислоте, нитрование аниона протекает достаточно быстро и происходит до
того, как вследствие захвата протона успеет восстановиться преимуществен-
но жеша-ориентирующая, но медленнее замещающаяся псевдоформа ней-
тральной молекулы
С6Н5—CR=N—О (анион псевдокислоты)
В псевдоосновных системах положительный заряд, возникающий в результа-
те захвата протона или потери аниона, становится распределенным в связи
с мезомерными свойствами катиона. Если часть заряда катиона достигает
атома, присоединенного к бензольному кольцу, то в результате этого усили-
вается жеша-ориентация. Это позволило объяснить тот факт, что при нитро-
вании бензальдегида, ацетофенона и бензилиденимина в концентрированной
серной кислоте образуются продукты с очень высоким содержанием мета-
изомеров (около 90%) [53].
С6Н5— CH==NHR
(катион псевдооснования)
Другого рода примеры приведены Лефевром [54], который показал, что
2-фенилбензпириллий- и 2-фенил-1-метилхинолинийнитраты нитруются
244
Замещение в ароматическом ряду
практически исключительно в лета-положение бензольного кольца
3. Реакционная способность в электрофильном
ароматическом замещении [55]
а. Предмет и метод исследования. Реагенты, которые осуществляют
рассматриваемые направленные замещения в ароматических соединениях,
принадлежат к числу электрофильных (гл. V, разд. 2, в). Можно полагать,
что они или непосредственно, или, находясь в том или ином преобразованном
виде, преимущественно атакуют те положения ядра, в которых электроны
являются наиболее доступными.
На основании изучения ориентаций, описанного в разд. 2, могут быть сдела-
ны три общих вывода. Из влияния электрополярности заместителей (-(-/-эф-
фекты) следует, что, во-первых, орто-иара-ориентация связана с передачей
электронов от заместителя к ароматическому кольцу; во-вторых, мета-
ориентация обусловлена удалением электронов от кольца. Из описанного
ориентирующего влияния ненасыщенности (-(-/С-эффект) вытекает третий
вывод: активирующие отрицательные заряды избирательно передаются
в орто- и геара-положения при введении орто-иара-ориентирующих замести-
телей. По-видимому, вполне естественно дополнить эти положения четвертым
выводом: положительные заряды избирательно дезактивируют орто- и пара-
положения, когда электроны удаляются из кольца при введении мета-
ориентирующих заместителей. Это означает, что .мета-ориентированное
замещение, если оно происходит, имеет место не потому, что лета-положе-
ния становятся более реакционноспособными под влиянием ориентирующего
заместителя, а потому, что дезактивируются орто- и wapa-положения. Такой
вывод находится в соответствии с правилом Голлемана о низкой реакционной
способности, характерной для лета-ориентации. Сформулированные три
вывода и одно дополняющее допущение схематически можно представить
следующим образом:
рорто-пара
мета
(Выведено)
г-
орто-пара,
мета
(Допущено)
Еще на сравнительно ранней стадии изучения проблемы стало ясно, что для
углубленного понимания сущности ориентации при замещениях необходимо
более детально рассмотреть вопрос о реакционной способности. Недостаточно
знать соотношения, в которых образуются изомеры; необходимо знать, как
ориентирующий заместитель влияет на те скорости замещения в различные
положения, от которых зависит соотношение количеств образующихся изоме-
ров. Особенно важно знать, в какой степени скорость атаки в каждое поло-
жение бензольного ядра увеличивается или уменьшается в присутствии
ориентирующего заместителя по сравнению с незамещенным бензолом.
245
Глава VI
Например, при нитровании толуола образуются орто-, пара- и жеша-изомеры
в количестве 57, 40 и 3,2% соответственно.
Однако эти результаты сами по себе не могут решить вопроса о том, по какому
из трех возможных механизмов ориентирует метильная группа: 1) посред-
ством активации орто- и тгара-положений и дезактивации жеша-положений
(альтернирование); 2) активацией орто- и гаара-положений при отсутствии
влияния на жета-положения или 3) путем сильной активации орто- и пара-
положений при менее сильной активации жеша-положений или же одним
из двух других возможных путей. Чтобы решить этот вопрос, необходимо
в дополнение к данным об ориентации знать соотношения скоростей нитрова-
ния толуола и бензола.
Это можно определить одним из двух способов. Наиболее очевидным является
кинетический метод’, можно изучить кинетику нитрования бензола и толуола
в отдельности и затем сравнить константы скорости. Однако этот метод
ненадежен, если механизм реакции не вполне ясен: в большинстве замещений
в ароматическом ряду, как и в случае других реакций, исходные реагенты
превращаются в конечные продукты через ряд стадий, из которых по суще-
ству лишь одна, а именно атака некоторых активных положений в арома-
тическом кольце, имеет отношение к реакционной способности последнего.
Так, например, процесс нитрования азотной кислотой состоит из четырех
стадий, из которых только третья имеет отношение к обсуждаемому вопросу.
Следовательно, необходимо быть уверенным, что ни одна из сравниваемых
скоростей реакции не определяется какой-либо из не относящихся к ориента-
ции стадий реакции и что соотношение найденных опытным путем констант
скоростей идентично соотношению скоростей именно тех стадий реакции,
которые представляют интерес при изучении данного вопроса. Кроме того,
необходимо быть уверенным, что отсутствуют активные частицы, которые
могут обусловить протекание множества относящихся к делу стадий. Этим
методом можно пользоваться лишь при соблюдении тщательных мер предосто-
рожности.
Более простым и надежным является некинетический способ, называемый
методом конкурирующих реакций [56]. Он совершенно не зависит от влияния
и даже от неравных степеней влияния на скорость суммарной реакции стадий,
не относящихся к ориентации. В этом методе сравнимые вещества, например
бензол и толуол, в одном и том же гомогенном растворе конкурентно реаги-
руют с общим реагентом, например азотной кислотой, причем количество
этого общего реагента должно быть очень мало (в принципе бесконечно мало).
В дальнейшем, не изучая процесс во времени, по окончании реакции опре-
деляют соотношения продуктов реакции, в данном случае нитробензола
и всех нитротолуолов. При этом можно ввести легко оцениваемую поправку,
обусловленную тем, что практически количество исходного реагента хотя
и мало, но не бесконечно мало, и, следовательно, соотношения конкури-
рующих веществ несколько изменяются ввиду того, что одно реагирует
быстрее другого. Главное допущение при этом методе заключается в том, что
определяющая ориентацию стадия имеет один и тот же порядок реакции
по отношению к обоим сравниваемым веществам. Поскольку вряд ли возмож-
но, чтобы в этих условиях порядок реакции отличен от первого, это пред-
положение представляется довольно оправданным. В этом методе исполь-
зуется также допущение об отсутствии нескольких параллельных, опреде-
ляющих ориентацию стадий, зависящих от наличия различных активных
частиц; в большинстве случаев для нитрования это является вполне оправ-
данным допущением, ибо указанная реакция полностью преобладает благо-
даря легкости образования и высокой электрофильности нитроний-иона.
246
Замещение в ароматическом ряду
Важное условие состоит в том, чтобы добиться полностью гомогенного смеше-
ния реагентов перед реакцией (иногда пытаются использовать этот метод,
не обращая достаточного внимания на это условие).
б. Результаты и их истолкование без учета эффекта сопряжения. Неко-
торые сравнительные скорости нитрования, измеренные методом конкури-
рующих реакций, приведены в табл. 67. В таблице указаны суммарные ско-
Таблица 67
Относительные скорости мононитрования производных бензола а
Соединение Скорость Литера- тура Соединение Скорость Литера- тура
СвН5-Н 1,00 CeH5-CH2CN 0,345 63
С0Н5-СН3 24,5 55, 62,63 CeH5 —CH2NO2 0,122 63
Св1Т5-С(СН3)3 15,5 62, 63 c6h5-ch2so2c2h5 0,24 64
С6Н3—соос,н5 0,0037 58
C6H5-SO2C6H5 0,0035 67 C6H5-CH2N(CH3)3 0,000026 66
С6Н5-К 0,15 59 С6Н5-СН2Р(СНз)з 0,0066 64
СВН3 —С1 0,033 59 +
СвН5-Вг 0,030 59 СвНэ — CH2As(CH3)3 0,0127 64
с6н5-1 0,18 59 ,СвН5-К(СН3)з 1,2-10-в 66
СвН5-СН = СНСО2Н 0,11 60 i С6Н5 - Р(СН3)з 5,8-10-8 66
С6Н5 - no2 6-10-в 65
С6Н5-СН2ОСН3 6,5 63 СвН5-А8(СНз)з 4,6-10-’ 66
С6Н5-СН2СООС2Н5 3,75 61, 63 C(iH5 -Sb(CH:i)3 0,000018 66
СвИ5-СН2С1 0,71 63
Примечание. Данные, сильно отличающиеся от приведенных и очень близкие к единице, были
опубликованы Ола и сотрудниками начиная с 1961 г. Они были получены при нитровании
солями нитрония обычно в тетраметиленсульфоне (сульфолане) в качестве растворителя. Эти
данные не стоит принимать во внимание, так как метод конкурирующих реакций был исполь-
зован без соблюдения элементарных условий метода, заключающихся в том, что скорость
реакции должна быть меньше скорости смешения реагентов. См. также [67]. Вопрос детально
обсуждался Риддом [68]; скоро также выйдет работа Р. J. Christy, J. И. Ridd, N. Sears
рости нитрования во все положения ядра, выраженные в условных величи-
нах, причем скорость нитрования бензола принята за единицу. Условия
нитрования не абсолютно одинаковы во всех случаях, но опыт показывает,
что эти сравнительные скорости малочувствительны к изменению условий
реакции.
Если скорости умножить на величины, характеризующие соотношения
изомеров, то можно прийти к сравнительным величинам для относительных
скоростей атаки в каждое отдельное положение ядра, т. е. в орто-, мета-
ть гаара-положения для каждого соединения, для которого имеются необхо-
димые данные. Удобно пользоваться такой единицей скорости, при которой
суммарная скорость нитрования бензола равна шести (вместо единицы),
а единица соответствует скорости атаки в одно из возможных положений
в бензоле. Тогда величины, относящиеся к отдельному положению в ядре
замещенного бензола, если они больше единицы, обозначают активацию
данного положения заместителем, а если они меньше единицы — дезактива-
цию данного положения. Рассчитанные таким образом значения называются
факторами парциальных скоростей. Данные, полученные в результате такого
сочетания относительных скоростей и соотношений изомеров, приведены
в конце этого и в следующем разделе.
247
Глава VI
Для удобства рассмотрения этих данных заместители разбиты на четыре типа
(табл. 68) в соответствии с полярными влияниями, посредством которых
Таблица 68
Классификация ориентирующих заместителей
т Электронный механизм Примеры Влияние
на ориентацию на реакционную способность
(1)+/ С6н5 R С6Нз — СНз орто-пара Активация
(2) -I ^6^5 -R С6Н5-СО2С2Н5 мета Дезактивация
С6Нз — S(CH3)2 Как и Для (2)
(3)-Л + К С6н5Хц С6н5-С1 орто-пара Дезактивация
С6Н5-ОСН3 Как и для (4)
(4) + Д + К c6h5Zr С6Н5-О- орто-пара Активация
они предположительно оказывают воздействие на ориентацию. Классифика-
ция упрощена, так как два вполне возможных типа -j-I, —К и —I, —К
не приведены; заместители неприведенных классов включены в типы -]-7
и —I соответственно, исходя из того что — /<-эффект играет второстепенную
роль. Это упрощение обосновывается ниже.
В заключение этого раздела рассмотрим интерпретацию ориентирующего
влияния заместителей типа (1) и (2). Сначала выпишем факторы парциальных
скоростей для реакции нитрования толуола и этилбензоата.
СН3
I
42,/\42
2,2^2,2
59
СООС2Н5
I
0,002б/\),0026
O,OO79^Jo,OO79
0,0009
Данные приводятся с точностью только до второго знака, так как парциаль-
ные скорости в некоторой степени зависят от условий реакции, особенно-
от температуры. Приведенные значения получены при обычной температуре.
С повышением температуры содержание изомера с низким выходом, как
правило, увеличивается, а содержание изомера с высоким выходом пони-
жается. Это обусловлено тем, что с увеличением абсолютной температуры
уменьшается различие в энергиях активации, так как аррениусовская экспо-
нента зависит от Еа1Т. Экспериментально это было четко показано лишь
в некоторых случаях, например при нитровании толуола [69], при котором,
содержание лета-изомера повышается от менее 3% при —30 °C до более
4% при 60 °C.
Если обратиться к толуолу как к примеру соединения с заместителем типа (1),
то станет ясно, что вопрос, оставшийся открытым на основании данных
о соотношении изомеров, получил разрешение благодаря данным о скоростях:
метильная группа активирует все положения ядра, причем сильно орто-
и геара-положения и слабее лета-положения. По-видимому, это обычное
действие заместителей типа (1). Объяснение заключается в том, что -|-7-эф-
фект создает отрицательный заряд в «-положении, который далее распро-
248
Замещение в ароматическом ряду
страняется по механизму сопряжения преимущественно в орто- и пара-
положения и в меньшей степени в лета-положения, вследствие чего наблю-
дается орто- и пара-ориентация и общая активация ядра. Ниже приведена
схема поляризации:
Ъ--88’
(54-) (+1~Эффект)
55-
Данные для этилового эфира бензойной кислоты показывают, что влияние
заместителей типа (2) как раз обратное: они дезактивируют все положения,
причем орто- и пара-положения больше, чем мета, результатом чего являет-
ся жига-ориентация, сопровождаемая общей дезактивацией ядра.
а+ <534-
(-1-Эффект)
54- 554-
Можно привести некоторые простейшие выводы, вытекающие из этих пред-
ставлений. Во-первых, главная причина столь частого применения нитрова-
ния для количественного изучения замещения в ароматических соединениях
заключается в том, что нитрогруппа является сильно дезактивирующим
заместителем типа (2), благодаря чему при нитровании особенно легко раз-
граничить стадии последовательного замещения, что необходимо делать
при работах количественного характера, проводимых с целью изучения
соотношения образующихся изомеров. Такое строгое разграничение значи-
тельно труднее провести в других случаях. Во-вторых, в таких гомоцикли-
ческих многоядерных системах, как бифенил, нафталин или антрахинон,
активирующие заместители типа (1) ведут к дальнейшим замещениям в уже
замещенном кольце, в то время как дезактивирующие заместители типа (2)
направляют замещение в другое ароматическое кольцо. Так, 1-метилнафта-
лин нитруется в положение 4, в то время как 1-нитронафталин нитруется
в положения 5 и 8. В-третьих, атом азота в пиридине, находящемся в форме
свободного основания, при галогенировании в определенных условиях
ведет себя как заместитель типа (2). Так, пиридин галогенируется преиму-
щественно в положение 3 (0-положение). В кислой среде (обычной при нитро-
вании или сульфировании) катионный заряд пиридиний-иона очень сильно
дезактивирует все ядро, хотя дезактивация наименее сильно выражена
в положении 3. Действительно, пиридин нитруется и сульфируется очень
трудно и именно в положение 3. В-четвертых, в хинолиний-ионе дезактиви-
рующий эффект катионного заряда направляет замещение в бензольное
кольцо. Так, хинолин нитруется и сульфируется без труда в положения 5
и 8. В солях 2-фенилбензпирилия и 2-фенил-1-метилхинолиния конденсиро-
ванное бензольное кольцо, соседнее с положительным полюсом, дезактиви-
руется сильнее, чем неконденсированное кольцо. Поэтому замещение
происходит в лета-положение последнего, как уже отмечалось в
разд. 2,е.
Типичными дезактивирующими и жтпа-ориентирующими группами типа (2)
являются карбоэтокси- и этилсульфонильная группы; их влияние на ско-
рость нитрования проиллюстрировано в табл. 67. Косвенно было оценено
влияние групп на скорость хлорирования и бромирования молекулярными
249
Глава VI
галогенами в органических растворителях [70]. Группы типа (2) по влиянию
на реакционную способность располагаются в ряд
(Н) > СОС6Н5> СООС2Н5> СООН > CN > NO2
В этом ряду иногда происходит изменение скорости в миллион раз. В табл. 69
приведены логарифмы факторов парциальных скоростей для лета-замеще-
Таблица 69
Логарифмы факторов парциальных скоростей для мета -замещения в ряду
монозамещенных бензолов с ориентирующими заместителями типа (2)
Реагент Ориентирующий заместитель
СОС6Н5 соос2н5 СООН CN NO2
HNO3 в (СН3СО)2О С12 в СНдСООН Вг2 в CH3NO2 —2,64 —2,10 —2,82 -3,11 -6,05 -5,3 — 7,0
ния. Можно сделать предварительный вывод, что скорости реакций увели-
чиваются в последовательности: нитрование, хлорирование (С12), броми-
рование (Вг2).
в. Эффект сопряжения, аномалия Голлемана и различие в эффектах
поляризации и поляризуемости. Особое значение заместителей типа (3), т. е.
заместителей класса —I, -\-К, состоит в том, что вследствие противоположно-
го действия индуктивного эффекта и эффекта сопряжения в ситуации, когда
эти эффекты более или менее сбалансированы, можно определить различие
в механизме их действия.
Различия в механизме действия —I- и /-/Г-эффектов проявляются в так
называемой «аномалии Голлемана». Как указывалось в разд. 1,6, в 1910 г.
Голлеман выдвинул глубоко верный принцип, согласно которому орто-
-иара-ориентация связана с активацией, а лета-ориентация — с дезактива-
цией. Исключение составляли галогены; в 1927 г. было показано [56], что
в галогенбензолах орто-геара-ориентация сочетается с дезактивацией. Затем
было выяснено, что в случае электрофильного замещения такое поведение
свойственно заместителям типа (3). Противоположная аномалия, состоящая
в лета-ориентации в сочетании с активацией, никогда не наблюдалась.
Наблюдаемая аномалия указывает на проявляющееся в определенный момент
времени раздельное влияние поляризации и поляризуемости (гл. II,
разд. 3,а).
Распределение реакционной способности по кольцу, обусловленное замести-
телями типа (3), в том случае, когда индуктивный эффект и эффект сопряже-
ния близки по величине, иллюстрируется факторами парциальных ско-
ростей, приведенными ниже для нитрования хлор- и бромбензолов. Все
положения дезактивированы, и более всего — лета-положение.
С1 Вг
I I
0,029,029 0, ОЗЗ,/\о, 033
0,0009JJo ,0009 0,0011J Jo, 0011
0,137 0,112
Допустим, что заместители типа (3) расположены в ряд, который начинается
+
группой — S(CH3)2 с преобладающим —/-эффектом и очень слабым -\-К-
250
Замещение в ароматическом ряду
эффектом. Затем следуют несколько промежуточных соединений типа галоге-
нов и со-карбоксивинила, в которых важную роль играют оба вида смещений.
Ряд заканчивается заместителем типа — ОСН3, обладающим слабым — /-эф-
фектом и очень сильным -|-А-эффектом. В начале такого ряда проявляется
мета-ориентация, связанная с дезактивацией, а в конце ряда наблюдается
орто-пара-ориентация в сочетании с активацией ядра. В середине ряда
переход от преобладающей мета- к орто-пара-ориентации происходит рань-
ше, чем переход от дезактивации ядра к его активации. Между этими двумя
переходными точками окажется несколько групп, и среди них гало-
гены и со-карбоксивинил, для которых характерно нарушение правила
Голлемана ввиду сочетания орто-па-
ра-ориентации с дезактивацией ядра
(см. табл. 68).
При анализе этих фактов можно раньше
всего прийти к заключению [56], что
+ А-эффект значительно строже огра-
ничивается действием на орто- и па-
ра-положения, чем —/-эффект, и в отли-
чие от последнего мало влияет на ме-
та-положения. Этот вывод можно про-
верить с помощью схематического ана-
лиза, пример которого дан на рис. 26.
Группы типа (3) расположены по гори-
зонтали, образуя описанный выше ряд.
Активация и дезактивация ядра отсчи-
тывается вверх или вниз по логарифми-
ческой шкале от нуля, который пред-
ставляет собой логарифм скорости
замещения в одно из положений бен-
Рис. 26. Схематический анализ совме-
стного действия —/-и+йГ-эф-
фектов на ориентацию и реак-
ционную способность при элек-
трофильном замещении в аро-
матическом ряду.
зола, принятой за единицу.
Сначала рассмотрим, как можно определить влияние только одного —/-эф-
фекта. Как уже отмечалось, этот вид смещений дезактивирует все положе-
ния в ядре, причем орто- и пара-положения в большей степени, чем мета-
положения. Такое влияние должно сильнее сказываться для групп, распо-
ложенных в левой части схемы, чем для групп, находящихся в правой части.
Затем можно построить А, на которой находятся точки, соответствующие
дезактивации орто- или (альтернативно) пара-положений, так как влияние
на них заместителей весьма сходно. Между этой кривой и нулевой осью
лежит значительно менее наклонная кривая Б, соответствующая дезактива-
ции мета-положений. В этом случае должна преобладать мета-ориентация
(потому что мета-кривая Б расположена над орто-пара-кривой А) и, следо-
вательно, дезактивация, поскольку более высокая кривая лежит ниже нуле-
вой оси. Степень снижения кривой Б ниже нулевой оси по направлению
к кривой А служит мерой величины передачи влияния на реакционную
способность от орто-пара-положений к мета-положениям.
Теперь можно рассмотреть, как эти кривые должны смещаться при учете
накладывающегося +А-эффекта. В этом случае, конечно, увеличивается
реакционная способность орто- и пара-положений, что должно сильнее
сказываться для групп правой части диаграммы, чем для групп левой части.
Следовательно, кривая А должна быть повернута вверх от исходной точки
в левой части; получается новая кривая В. мета-Кривая Б должна быть
повернута качественно аналогичным образом. Однако (и в этом заключается
основной пункт рассуждения), чтобы отразить нарушение правила Голлема-
251
Глава VI
на, результирующая лета-кривая Г должна пересечь ортпо-пара-кривую В
ниже нулевой оси. Только таким образом можно получить центральную
область, в которой ориго-пара-кривая лежит выше лепга-кривой, причем обе
кривые лежат ниже нулевой оси, т. е. область ортпо-пара-ориентаций в соче-
тании с общей дезактивацией ядра. На диаграмме указаны две точки, кото-
рые обозначают разделение всего поля на три части. В левой части кривая Г
лежит выше кривой В, а верхняя кривая лежит ниже нулевой оси; здесь
наблюдается лета-ориентация, как всегда, в сочетании с дезактивацией.
Справа кривая В лежит выше кривой Г, а верхняя кривая лежит выше нуле-
вой оси; здесь ортпо-пара-ориентация связана, как обычно, с активацией.
В центре расположена аномальная область.
Из этих данных вытекает, что кривая Б должна быть смещена очень незначи-
тельно, чтобы дать результирующую лета-кривую Г. Один способ обеспече-
ния указанного выше аномального взаимоотношения ориентации с реакцион-
ной способностью заключается в том, что угол между Б и Г должен составлять
меньшую долю от угла между А и В, чем угол наклона Б относительно угла
наклона А. Другими словами, аномалия показывает, что -(-Л’-механизм
действует с заметной специфичностью на орто- и пара-положения и в отличие
от индуктивного механизма —I мало влияет на лета-положения.
Далее следует рассмотреть причину ортпо-пара-селективности между сопря-
женным и индуктивным механизмами. Можно сделать вывод, что это указы-
вает на то, что смещения, возникающие в соответствии с двумя механизмами,
различны и зависят от времени, в то время как индуктивный механизм —I
отражает преимущественно постоянное состояние поляризации молекулы
—Is', +Л’-механизм оказывает главное влияние на временный процесс -(-.Е,
который происходит, когда проявляется роль атакующего агента в поляри-
зуемости системы.
На основании изучения влияния заместителей типа (1) и (2) уже был сделан
вывод, что свободные заряды, которые благодаря +/-эффектам размещены
в орто- и пара-положениях, частично передаются и в лета-положение. Если
+Л’-механизм действительно обусловливает появление свободных зарядов
в ориго-пара-положениях, то эти заряды неизбежно частично передаются
в лета-положения, и в этом случае -J-Л’-механизм обусловливает не
большую ортпо-пара-специфичность, чем —/-механизм. Следует сделать
вывод, что заряды, создаваемые -(-Л’-механизмом, не свободны и по мере созда-
ния локально нейтрализуются. Это равносильно утверждению, что смеще-
ния электронов по +Л’-механизму являются электромерными -|-£-смещения-
ми под влиянием электрофильных реагентов и что они, следовательно, при-
обретают значение только тогда, когда реагент уже находится у орто-
и пара-положений и атакует эти положения.
Таким образом, можно ожидать существенного различия в селективности,
обусловленной факторами поляризации и поляризуемости: последний прояв-
ляется только в том месте, где имеется реагент, и только до тех пор, пока
реагент там присутствует. При электрофильном ароматическом замещении
смещение электронов путем сопряжения с реакционным центром в орто-
или пара-положениях в основном обусловлено эффектом поляризуемости.
Можно, как это иногда делают, рассматривать влияние сопряжения в началь-
ном и переходном состояниях; при этом следует учесть, что легкость перехода
системы из начального в переходное состояние зависит от электромерного
эффекта.
Заместители типа (3) при введении в бензольное кольцо изменяют его реак-
ционную способность в широких пределах. В этом можно убедиться при
сравнении скоростей реакций хлорирования и бромирования (многие из
252
Замещение в ароматическом ряду
которых получены косвенным путем) молекулярными галогенами в уксусной
кислоте и других растворителях. Эти данные приведены в правой части
табл. 67. Вычисленные факторы парциальных скоростей для пара-галогени-
рования охватывают интервал 1019; логарифмы этих величин приведены
в табл. 70 [70]. Сильно активирующие заместители располагаются в ряд:
Таблица 70
Логарифмы факторов парциальных скоростей для тгара-галогенирования
монозамещенных бензолов, содержащих заместители ориентационного типа (3)
Ориентирующий заместитель Хлорирова- ние Бромирова- ние Ориентирующий заместитель Хлорирова- ние Бромирова- ние
N(CH3)2 NHCOCHa N(CH3)COCH3 n = nc6h5 он +6,4 +3,3 +2,3 +7,7 +19,5 +9,1 +6,15 +11,7 ОСН3 ОС6Н5 ОСОС6Н5 F +2,0 +0,8 +9,8 +7,9
N > О > F; активация уменьшается при N- и О-ацилировании (гл. II,
разд. 3,д и е). Замещение атома водорода в ОН- и NH-группах на метильную
группу несколько уменьшает активацию. Робертсон, де ла Мар и Сведлунг
предположили, что этот эффект связан с гиперконъюгационным электро-
мерным эффектом NH- и ОН-связей.
Из данных табл. 67 следует, что скорости нитрования в серии галогенбензо-
лов проходят через минимум: F > С1 « Вг < I. Из этих данных можно
заключить, что +7Оэффекты галогенов изменяются следующим образом:
в случае фтора -[-^-эффектом можно пренебречь, +М-эффект максимален,
поэтому доминирует +М-эффект, который обычно оказывает малое влияние
на скорости. В случае иода максимален +Е-эффект, который поэтому пре-
обладает. Такая же последовательность в расположении галогенов была
найдена Иллюминати [71] при бромировании молекулярным бромом в нитро-
метане. В этой работе круг исследованных заместителей был расширен
включением элементов не только VII, но и VI группы таблицы Менделеева.
В результате было установлено, что изменение скорости в ряду заместителей
VI группы (СН3О > CH3S) аналогично изменению скорости в ряду галогенов
(F > С1), однако эти заместители в отличие от галогенов являются сильно
активирующими. Эти данные получены при изучении бромирования метокси-
и метилтиодурола молекулярным бромом в уксусной кислоте. Ниже приве-
дены скорости бромирования этих соединений, отнесенные к скорости броми-
рования в одно из возможных положений дурола:
ОСН3
СНзх/[х/СН3
III
сн3/4</\сн3
164 000
SCH3
СН3\у1\/,СНз
СН3/^/\сН3
970
Для заместителей типа (4) сравнительная количественная информация
отсутствует, однако качественные данные хорошо согласуются с теоретически
ожидаемыми. Для этих групп существенны оба механизма смещения электро-
253
Глава VI
нов, обозначаемые символами -\-1 и -}-К, эти группы очень сильно электро-
положительны, поэтому нужно ожидать очень высоких скоростей и исключи-
тельно орнго-нара-ориентацию. Скорости электрофильного замещения фено-
лят-иона так велики, что до сих пор не удалось найти подходящего метода
их измерения и сравнения со скоростями реакций других производных бензо-
ла. Ориентация исключительно орто-пара. На следующей схеме изображен
+.Е-эффект, вызываемый электрофильным агентом, атакующим орто- или
пара-положения (реагент обозначен знаком
(+Е~Эффект при атаке орто- и пара-положений)
На основании высказанных соображений можно объяснить упрощение, при-
нятое при обосновании классификации, согласно которой ориентирующие
агенты делятся на четыре типа. Как известно из физических данных (гл. III),
Д-Х-механизм электронных смещений включает частично -ДМ-эффект, кото-
рый представляет собой постоянную поляризацию молекулы. Как только что
было сказано, другая часть — эффект -\-Е, т. е. эффект временной поляри-
зуемости, вызываемый электрофильным реагентом,— имеет особое значение
при замещении в ароматических соединениях. Следовательно, становится
ясным, почему в приведенной классификации ориентирующих групп опущен
—A-механизм. Этот механизм также состоит в принципе из двух частей:
из —М- и —jE-эффектов. Однако электрофильные реагенты не вызывают
—jE-эффекта. Следовательно, остается только —7И-эффект, который для
упрощения считался уже учтенным посредством добавления (или вычитания)
его величины к более значительной постоянной поляризации, обусловленной
-\-1 и —/-эффектами типов (1) и (2). В более детальной классификации оба
эти типа должны быть разбиты на подтипы, как это указано в табл. 71.
Таблица 71
Расширенная классификация ориентирующих заместителей
Тип а Электронный механизм Пример Влияние
на ориента- цию на реакционную способность
(1) + / С„н5 R с„н5«-сн3 орто-пара Активация
(Г)+Д - м СдО-R орто-пара Активация или дез-
или мета активация
(2)-/ c6h5->r С6Н5 -> NH+ мета Дезактивация
(2')-Д --М с6н^со2с2н5 мета То же
а Типы (3) и (4), как и в табл. 6 8.
254
Замещение в ароматическом ряду
Ниже приведены иллюстрации электронных смещений, включающих Ц-7И-
и —М-эффекты:
5- 55-
(г+) г-
' ь-ы-
+М-Эффект
55-f-
5 +
54" 554-
—М-Эффект
Вовсе не важно, что смещения электронов по механизмам сопряжения или
индуктивному, которые при определенном сбалансировании обусловливают
аномалию Голлемана, первоначально возникают у одного и того же атома
(например, у атома галогена). Эффект сопряжения -\-К должен начинаться
у атома, связанного с ароматическим кольцом, однако индуктивный эффект
—I может возникать у более далекого атома и транслироваться к атому,
связанному с кольцом. Именно такое явление наблюдается для боковой цепи
коричной кислоты. Это соединение нитруется почти исключительно в орто-
и иара-положения, но с дезактивацией. Этиленовая двойная связь оказывает
-[-Х-эффект, а электроотрицательность карбоксильной группы сообщает
олефиновому центру —/-характер, достаточный для возникновения аномалии
Голлемана. К этому же типу относятся стирол-ю-сульфокислота и ю-нитро-
стирол. Боковые цепи в этих трех соединениях относятся к заместителям
типа (3); легко показать, что подобное влияние должны оказывать замести-
тели типа (4), обладающие согласованными ф.I- и -[-Х-эффектами, например
ионизированная карбоксильная группа.
г. Гиперконъюгация и аномалия Голлемана. Как показали Ноулс
и Норман, а также Рилей и Ротстейн [50], в бензильных соединениях индук-
тивная передача электроотрицательности может уравновешиваться электро-
положительной гиперконъюгацией СН-групп, и этого достаточно для воз-
никновения аномалии Голлемана другого типа по сравнению с описанной
выше. Заместители типа СН2Х, где X — какие-нибудь группы, расположен-
ные в порядке уменьшения электроотрицательности, образуют ряд, в котором
происходит изменение ориентации и скоростей, например:
c6h5-ch2no2 c6h5-ch2cn С6Н5-СН2С1 С6Н5—СН2ОСН3
л«ета-Ориентация, ориго-пара-Ориептация, орт о-пара-Ориентация,
дезактивация дезактивация активация
+
Этот ряд качественно подобен ряду S(CH3)2, . . . Hal, . . . ОСН3, приве-
денному на рис. 26, однако в первом ряду средние изменения скоростей
и направления ориентации выражены гораздо слабее. В этом ряду для двух
средних членов наблюдается аномалия Голлемана. Факторы парциальных
скоростей приведены ниже (ради экономии места орто- и лета-факторы
приведены только один раз).
ch2no2
I
//>0,082
\ Jo, 200
0,167
ch2cn
I
//>0, 25
\Jo,21
1.15
СН2С1
/\о,72
\Jo,3O
2,24
СН2ОСН3
//,10,0
\/1,3
16,3
Очевидно, что для объяснения закономерностей этой серии можно применить
те же аргументы, что и в предыдущем разделе. Из приведенных данных
255
Глава VI
неизбежно следуют выводы, уже сделанные упомянутыми выше авторами.
Влияние протонов этих —I, ^-^-заместителей типа (3) по механизму гипер-
конъюгации -\-К специфически передается в орто- и пара-положения, тогда
как —Z-индуктивный эффект передается от орто- и пара-положений к мета-
положению. Отсюда следует, что в то время как -/-эффект в основном являет-
ся эффектом поляризации, -(-//-эффект гиперконъюгации — это преимуще-
ственно эффект поляризуемости, действующий тогда и только тогда, когда
электрофильный замещающий агент находится у орто- или пара-положений.
д. Применение понятия об эффекте поля. Эффект поля, вызываемый
электрополярными заместителями, согласуется по знаку с индуктивным
эффектом, однако имеет иное пространственное распределение, уменьшаясь
с увеличением кратчайшего расстояния от источника поля до точки субстра-
та, в которой он проявляется. Следует ожидать, что эффекты поля будут
сильно зависеть от диэлектрических свойств среды, конечно, при условии,
если по крайней мере одна полярная способная переориентироваться молеку-
ла растворителя находится на пути действия поля.
В случае электрофильного ароматического замещения эффект поля трудно
отличить от индуктивного эффекта, потому что, во-первых, знаки обоих
эффектов совпадают и, во-вторых, после того как эти эффекты вызвали сме-
щения электронов в ароматическом кольце, дальнейшее перераспределение
электронной плотности с большей эффективностью происходит по системе
сопряжения самого ароматического кольца. Относительно того, где искать
доказательства существования эффекта поля, в первом издании этой книги
было сказано, что эффект поля «мог бы, если бы он был достаточно велик,
произвести обращение от мета- к пара-ориентации или наоборот, если эта
ориентация ожидается при наличии только индуктивных эффектов». «Одна-
ко,— было сказано затем,— эффект поля не является достаточно сильным».
Тем не менее в недавней работе Ридда и сотр. [72] были приведены доказа-
тельства действия эффекта поля в предсказанном направлении.
Индуктивный эффект и эффект поля в своей простейшей форме проявляются,
когда они обусловлены ионным зарядом. В рассмотренном Риддом случае
распределение заряда по кольцу вследствие действия этих эффектов, обуслов-
ленных положительным зарядом в боковой цепи, представляется следующим.
Индуктивный эффект от а-углеродного атома распространяется почти без
ослабления (на рис. 27 без изменения) в орто- и пара-положения, а затем
из этих положений со значительным ослаблением — в лета-положения.
Эффект поля возникает главным образом на орто-углеродных атомах (влия-
нием поля на другие положения ядра можно пренебречь), от которых он
распространяется без заметного ослабления (на рис. 27 без изменения)
в орто- и пара-положения по отношению к этим атомам (т. е. в а- и лета-
положения к заместителю), а отсюда со значительным ослаблением — в един-
ственное оставшееся положение кольца (пара-положение к заместителю).
Таким образом, индуктивный эффект и эффект поля обусловливают противо-
положное влияние на ориентацию в мета- и пара-положения, но оба дезакти-
вируют орто-положения.
6+ бб +
X---/ >6 +
6 + ^+-66 +
Индуктивный эффект
+ б+ б +
Х-.-<^ ^>66 +
б+ 6 + ~6 +
Эффект поля
Ридд и сотрудники сравнили нитрование фенил- и бензилтриметиламмо-
ниевых ионов нитрующей смесью. В противоположность более ранним рабо-
там, согласно которым катион фенилтриметиламмония образует только
256
Замещение в ароматическом ряду
лета-нитропроизводное, было найдено, что оба катиона дают некоторое
количество пара-изомера, а именно 89 и 85% .мета-нитроизомеров и 11 и 13%
пара-изомеров. Однако, несмотря на почти одинаковое соотношение про-
дуктов, скорости реакции сильно отличаются. В табл. 72 приведены полу-
ченные результаты; в нее включены также данные [72], которые будут обсуж-
даться ниже.
Предполагается, что в ионе фенилтриметиламмония эффект поля хотя
и велик, но все же несколько меньше индуктивного эффекта. Сильное прояв-
ление эффекта поля объясняется затруднением внедрения молекулы способ-
ного к переориентации растворителя в пространстве между аммонийным
Рис. 27. Диаграмма свободных энергий
для нитрования фенил- и бензил-
триметиламмониевых ионов, иллю-
стрирующая, каким образом силь-
ный эффект поля в фенильном
ионе может увеличить дезактива-
цию, одновременно уменьшая вы-
ход лета-продукта до величины,
характерной для менее дезакти-
вированного бензильного иона.
Для простоты небольшие эффекты
поля и гиперконъюгации в бен-
зильном ионе не приведены.
полюсом и орто-положениями, на которых этот эффект проявляется главным
образом, вследствие чего не происходит диэлектрического ослабления интен-
сивности поля. С другой стороны, в ионе бензилтриметиламмония поле,
возникающее на заряженном центре, прежде чем достигнуть орто-положе-
ний, пересекает часть полярной среды и таким образом значительно
ослабляется. В этом случае в первом приближении можно пренебречь этим
относительно слабым эффектом поля, так же как и слабым эффектом гипер-
конъюгации [40]. Учитывая только сильные эффекты, можно начертить
упрощенную схематическую диаграмму свободных энергий (рис. 27), которая
показывает, каким образом сильный эффект поля в фенилтриметиламмониевом
катионе обусловливает такое же соотношение мета- и пара-изомеров, как
при нитровании бензилтриметиламмониевого катиона, несмотря на большую
разницу в дезактивации ядра в этих двух случаях. Этот пример доказывает
существование эффекта поля. При увеличении числа метиленовых групп
между кольцом и ионным центром внутренний индуктивный эффект и куло-
новский эффект поля быстро ослабевают, однако первый — по экспоненте,
т. е. быстрее, в результате чего на кольцо действует в основном лишь слабый
эффект поля.
Те же авторы изучили нитрование ионов анилиния нитрующей смесью (т. е.
протонированных, а не метилированных) ионов фениламмония. Некоторые
данные представлены в табл. 72. Показано, что протонированные катионы
нитруются как таковые, а не в виде сопряженных оснований. Последние
присутствуют в таком незначительном количестве, что даже если замещение
происходит при каждом их столкновении с реагентом, его скорость будет
вносить исчезающе малый вклад в наблюдаемую скорость процесса. В случае
протонированных катионов наблюдается более слабая дезактивация и более
слабое лета-ориентирующее действие. Ридд приписал этот эффект удалению
от катионного центра метильных групп, обладающих низкой диэлектрической
257
17-0772
Глава VI
Таблица 72
Относительные скорости и состав продуктов нитрования фенилониевых
и бензилониевых ионов и фенилнитрометана
Соединение Скорость (CgHg = 1) Количество изомера, %
орто мета пара
CeH5N(CH3)3 1,2-10-s 0,05 89 11
c6h5nh3 9,4-10-7 1,5 62 37
c6h5ch2no2 12,2-10-2 22 55 23
C6H5CH2N(C h3)3 4,3-10-5 2 85 13
С6Н5СН2Р(СН3)з 6,6-10-3 13 19 68
C6H5CH2A+s(CH3)3 12,7-Ю-з 17 7 76
проницаемостью, т. е. увеличению дисперсии эффективного заряда и его
поля в среде с более высокой диэлектрической проницаемостью.
Ридд также сравнил данные по скоростям и ориентации, полученные Рилеем
и Ротстейном для ионов бензилтриметилфосфония и бензилтриметиларсония,
с одной стороны, с данными Ноулса и Нормана для фенилнитрометана —
с другой. Хотя нитросоединение менее дезактивировано, чем любая из этих
двух ониевых солей, при нитровании нитросоединения образуется большее
количество лета-изомера (табл. 72). Ридд в соответствии с общими электро-
статическими принципами объяснил это более быстрым ослаблением с рас-
стоянием эффекта поля, вызванного диполем, по сравнению с эффектом
поля, вызванного монополем (катионом или анионом). Как видно из рис. 27,
при исчезновении или сильном ослаблении эффекта поля должны увеличи-
ваться скорость и выход лета-изомера.
Мы обсудили эффект поля субстрата, который обусловлен заместителями,
расположенными далеко от реакционного центра. Заместители должны
оказывать влияние на атакующий реагент; это влияние можно назвать
эффектом поля реагента. Этот эффект, способствуя притяжению или отталки-
ванию полярного реагента, изменяет его локальную концентрацию вблизи
потенциальных центров реакции и, следовательно, влияет на скорость
реакции у этих центров. Этот тип эффекта поля впервые был описан Бьерру-
мом в связи с изучением кислотно-основного равновесия (гл. XIV).
Эффект поля реагента, вероятно, имеет значение при нитровании, когда
реагентом является не нейтральная частица, а ион нитрония. При рассмотрен-
ном выше нитровании фенил- и бензиламмониевых ионов поле аммониевых
ионов будет отталкивать нитрониевые ионы, и поэтому в этом случае будет
заметно понижаться скорость нитрования в орто-положении и немного
понижаться скорость нитрования в мета- и иара-положения. Кинетические
эффекты в этом случае будут качественно такими же, как и при влиянии
поля субстрата. В настоящее время эти компоненты влияния поля на ско-
рость ароматического замещения разделить нельзя, несмотря на то что
в принципе рассматриваемые два эффекта должны проявляться по-разному.
Однако можно ожидать, что в других системах, в которых не содержится
бензольного кольца, способного путем сопряжения передавать к реакцион-
ному центру любое внешнее влияние, оказываемое на кольцо, эффект поля
реагента окажется более важным, чем эффект поля субстрата. Эти случаи
будут разобраны на конкретных примерах в гл. XIV и XVI.
258
Замещение в ароматическом ряду
4. Соотношения между орто- и пара-изомерами
при электрофильном ароматическом замещении
а. Факторы, влияющие па соотношение между орто- и пара-изомера-
ми. Теория, рассматривающая орто- и пара-ориентацию совместно в проти-
вовес л/епга-ориентации, учитывает лишь влияние общих факторов строения.
При переходе от этого вопроса к вопросу о конкуренции орто- и /гара-заме-
щения, а также об орпго-пара-ориентирующих группах и об орто-пара-
изомерах, образующихся в качестве побочных продуктов при преимуще-
ственной лета-ориентации, было установлено, что здесь следует учитывать
более тонкие влияния. Например, природу замещающей группы нельзя
уже считать сравнительно маловажным фактором.
Общий анализ данных, касающихся конкуренции орто- и пара-замещения,
произведен в 1926 г. автором данной книги, который показал существование
предсказанного Голлеманом [73] пространственного фактора, а также индук-
тивного эффекта и эффекта сопряжения [74]. Эти эффекты проявляются
в следующем.
Первичный пространственный эффект уменьшает реакционную способность
в орто-положении
Реакционная способность
„ „ , у Г — /-Эффект уменьшает 1 .
Индуктивный эффект 1 , !• орто > пара
• [-[-/-Эффект увеличивает]
— Л/-Эффект уменьшает ]
, лп,у [ пара > орто
+ Е-Эффект увеличивает]
В 1928 г. было высказано предположение [75], что индуктивные эффекты
влияют не только сами по себе, но и посредством связанных с ними эффектов
поля. В 1949 г. было сделано дополнительное предположение [76], что эффек-
ты сопряжения возникают вследствие квантовохимического различия в ста-
бильности орто- и пара-хиноидных структур. В дальнейшем мы будем при-
держиваться указанной классификации, анализируя сначала опытные дан-
ные, а затем рассматривая физические механизмы полярных эффектов.
б. Доказательства первичного пространственного эффекта. В отличие
от ориентации, обусловленной полярными факторами, которая почти цели-
ком зависит от природы ориентирующего, а не входящего заместителя,
ориентация, обусловленная пространственными факторами, возникает вслед-
ствие пространственного взаимодействия между ориентирующим заместите-
лем и переносчиком входящей группы, и поэтому пространственный эффект
зависит от эффективного объема каждой из этих групп. Развивая эти взгля-
ды, Голлеман [73] привел ряд данных, которые собраны в табл. 73 и. как
Таблица 73
Влияние входящего заместителя на процентное содержание орто-
п па /и,-изомеров, образующихся из некоторых производных бензола
Входящая группа Замещенные соединения и содержание изомеров (%)
толуол хлорбензол бромбензол фенол
орто пара орто пара орто пара орто пара
С1 39,0 55,0 45,1 52,5 49,8 50,2
no2 56,0 40,9 30,1 69,9 37,6 62,4 40,0 60,0
Вг 39,7 60,3 11,2 87,2 13,1 85,1 9,8 90,2
SO3H 31,9 62,0 0,0 100,0 0,0 100,0
259
Глава VI
видно, согласуются с высказанным предположением о влиянии эффективного
объема входящей группы и заместителя. Как показывают эти данные, неза-
висимо от вида уже присутствующего заместителя относительное содержание
оршо-изомеров уменьшается, а относительное содержание пара-изомеров
возрастает в следующем ряду входящих заместителей: Cl, NO2, Вг, SO3H.
Этот ряд не соответствует ряду полярности заместителей, а определяется
увеличением пространственных затруднений в рассматриваемых реакциях
(что и было предположено Голлеманом).
Более четкое разделение первичного пространственного и полярного эффек-
тов возможно в том случае, когда ориентирующая группа лишь слабо поляр-
на и размеры ее могут быть значительно увеличены без существенного изме-
нения полярности. В этой связи были широко изучены алкилбензолы. Лефевр
особо подчеркнул роль пространственных факторов, препятствующих орто-
замещению и ведущих к высокому выходу пара-изомеров при нитровании
гомологов толуола с разветвленной боковой цепью [77]. Эти данные, впослед-
ствии дополненные, приведены в табл. 74.
Таблица 74
Количества (%) продуктов, образующихся
при мононитрованип моноалкилбензолов
М опоалкил бензол Замещение в положение.
орто мета пара
। .11 1 Щ [78] 57 3,2 40
С6Н6СН2СНз [79, 80] 45 6,5 48
С6Н5С.Н(СН3)2 [80, 81] 30 7,5 62
С6Н5С(СН3)3 [62] 12 8,5 70
Для толуола и игреиг-бутилбензола соотношение мета- и пара-нитропроиз-
водных примерно одинаково. Наибольшие различия наблюдаются в соотно-
шении орто- и мета-, а также орто- и пара-продуктов. Эти данные указы-
вают на то, что различие связано главным образом с ориго-положением.
Простейшее объяснение заключается в том, что полярные влияния двух
алкильных групп мало различаются и что изменение в содержании изомеров
обусловлено преимущественно избирательным подавлением (по-видимому,
пространственного характера) замещения в ориго-положение в случае большей
алкильной группы.
Лефевр аналогичным образом объяснил преимущественное замещение
в ориго-положение к метильной группе в случае n-алкилтолуолов. Такое
замещение обнаружено при нитровании и-этилтолуола [82], при нитровании,
галогенировании и сульфировании п-цимола [77] и при нитровании п-трет-
бутилтолуола [83]
СН3 S— СН2СН3
сн3 -
>- СН(СН3)2
сн3 -
С(СН3)3
До последнего времени не было строгой необходимости в признании такого
толкования, в особенности в отношении n-алкилтолуолов. Не исключалось
и другое объяснение, заключающееся в том, что из всех алкильных групп
метильная группа обладает благодаря гиперконъюгации наиболее сильным
орпго-иара-ориентирующим влиянием. Однако Кон, Хыоз, Джонс и Пилинг
опровергли эту точку зрения, показав, что скорости мононитрования толуола
Замещение в ароматическом ряду
и mpem-бутилбензола относятся как 100 : 64. Если эти величины сопоставить
с относительными количествами изомеров (табл. 74) и со скоростью нитрова-
ния толуола (табл. 73), то можно установить относительные скорости атаки
в каждое положение ядра толуола и mpem-бутилбензола; соответствующие
величины приводятся ниже (скорость замещения в одно положение бензола
принята за единицу):
СП3 С(СН3)3
I I
42г/'х42 5,5^5,5
2,2^2,2 4,0^4,0
59 75
Это показывает, что, несмотря на гиперконъюгацию, mpem-бутильная группа
немного сильнее, чем метильная, активирует все положения ядра, за исклю-
чением орто-положений. С другой стороны, mpem-бутильная группа значи-
тельно сильнее по сравнению с метильной группой подавляет орто-замеще-
ние. Такое избирательное подавление едва ли можно объяснить иначе, как
проявлением пространственного эффекта.
Де ла Мар, Браун и другие авторы [84] изучили состав продуктов и скорости
реакций галогенирования толуола и mpem-бутилбензола двумя хлорирующи-
ми (С1+и С12) и двумя бронирующими (Вг+ и Вг2) агентами. Суть этих работ
состояла в том, чтобы, скомбинировав подход Голлемана (изменение заме-
щающего агента) с подходом Лефевра — Хьюза (изучение ориентации
и скоростей замещения в ряду слабополярных алкилбензолов), отличить
пространственные эффекты от полярных. В отличие от электрофильного
нитрования, при котором почти всегда атакующим агентом является ион
нитрония, электрофильное хлорирование в зависимости от условий может
протекать под действием различных агентов, включая хлориниевый ион
и молекулярный хлор. Подобно этому, электрофильное бромирование может
осуществляться и через броминиевый ион, и через молекулярный бром.
В разд. 6,а мы обсудим, какие частицы, образующиеся из реагента, являются
активными; здесь же примем, что участие указанных двух частиц в процессе
галогенирования установлено экспериментально. Ионы галогениния суще-
ствуют в кислых водных растворах, а галогены в молекулярной форме —
в полярных органических растворителях, из которых наиболее часто употреб-
ляются содержащая немного воды уксусная кислота и нитрометан.
Соотношение изомерных продуктов при галогенировании толуола и трет-
бутилбензола приведено в табл. 75, в которую включены также данные по
нитрованию этих углеводородов. Отметим, что во всех этих реакциях для
Таблица 75
Количества (%) изомеров, образующихся при замещении толуола
и mpem-бутилбензола различными электрофильными агентами
Электрофиль- ный реагент Толуол mpem-Бутилбензол
орто мета па ра орто мета пара
С1+ 75 2,2 23 42
Вг+ 70 2,3 28 38 7 55
С12 60 0,5 39,5 22 2,1 76
NOJ 57 3,2 40 12 8,5 79
Вг2 33 0,2 67 <8 92
261
Глава VI
mpem-бутилбензола выход орто-изомера ниже, а выходы мета- и пара-изоме-
ров выше, чем для толуола. Кроме того, в ряду, соответствующем порядку
расположения реагентов в таблице, выход орто-изомеров уменьшается.
Порядок расположения реагентов в таблице соответствует увеличению их
эффективного размера, но не соответствует увеличению или уменьшению
полярности.
В табл. 76 приведены скорости реакций толуола и mpem-бутилбензола с раз-
личными агентами, отнесенные к скорости замещения в бензольном кольце.
Порядок расположения замещающих агентов здесь несколько изменен,
Таблица 76
Относительные скорости реакций замещения толуола и /пре/п-бутилбензола с раз-
личными электрофильными агентами
(скорость замещения в бензоле принята равной единице для каждого реагента)
Замещающий реагент Толуол третп-Кутил- бензол •Чамещ ающий реагент Толуол трет-Бутил- бензол
С1+ 60 С12 345 110
Вг+ NOt 36 24,5 12 15,5 Вг2 605 125
чтобы показать, что ориентирующие свойства заместителей более способству-
ют замещению нейтральными реагентами, чем катионными. При действии
нейтральных реагентов соотношение изомеров определяется в основном
природой заместителя, т. е. имеет место почти исключительно орто-пара-
ориептация (табл. 75). Можно предположить, что такая ориентация под
действием нейтральных агентов имеет электромерную природу [85]; электро-
мерный эффект сам по себе, как мы увидим в разд. 4,г, должен способство-
вать /гара-замещению за счет opmo-замещения и. таким образом, менять
местами С12 и NO7 (ср. табл. 75 и 76). В самом деле, в случае менее простран-
ственно затрудненного толуола орто- и геара-изомеры образуются в соотно-
шении, близком к 1 : 1.
Объединив данные табл. 75 и 76, можно определить факторы парциальных
скоростей для реакций галогенирования толуола и mpem-бутилбензола, т. е.
Бромирование Вг+
Хлорирование С12
Бромирование Вг2
СН3
I
76|/\,76
2,5^2,5
59
СН3
I
62О,/Ч-,62О
\/5
820
СН3
600./\б00
5,51^5,5
2400
С(СН3)3
14/\14
2,в'. /2,6
39
С(СН3)з
I
72/\?2
7\/7
505
С(СН3)3
<зо,/\<зо
750
262
Замещение в ароматическом ряду
скорости атаки реагента по каждому индивидуальному положению в ядрах
этих молекул, отнесенные к скорости атаки по каждому положению в бензоле.
Выше приведены полные схемы для двух из четырех галогенирующих аген-
тов и неполная схема для третьего из них.
Наиболее примечательным является то, что при переходе от метильной
к mpem-бутильной группе скорость орто-бромирования или хлорирования
при действии Вг+ уменьшается в 5 раз, при действии С12 — в 9 раз, а при
действии Вг2 — более (вероятно, даже гораздо более) чем в 20 раз.
в. Доказательства индуктивного эффекта. Общепризнано существование
полярных эффектов, благодаря которым -(-/-заместители увеличивают реак-
ционную способность орто-положения больше, чем пара-положения, в то
время как —/-заместители уменьшают реакционную способность орто-
положения больше, чем пара-положения. Однако в отношении -(-/-групп
имеются лишь доказательства, основанные на примере толуола. Соотношение
между орто- и пара-замещенными толуола, образующимися при нитровании,
равное 1,5, несомненно, превышает соотношение в случае большинства орто-
пара-ориентирующих веществ; однако это соотношение меньше 2, т. е. соот-
ношения, соответствующего числу возможных положений каждого типа.
Предполагается, что оно было бы больше 2, если бы метильная группа не
экранировала в какой-то степени орто-положения [86]. Четкое доказатель-
ство этой точки зрения было получено де ла Маром при изучении хлорирова-
ния толуола ионом С1+ и его бромирования ионом Вг+ (табл. 75). В случае
таких, как мы полагаем, относительно небольших замещающих агентов
соотношение орто/пара повышается до 3,3 и 2,5 соответственно.
В то же время существенным является подтверждение того, что —/-группы
дезактивируют орто-положения больше, чем пара-положения. В табл. 77
Таблица 77
Количество (%) изомеров, образующихся при мононитровании некоторых аромати-
ческих соединений с электроотрицательными заместителями
C6H5F[87] С6Н5С1[88, 97] С6Н5Вг[89, 97] С6Н51[90, 97]
орто 12 30 37 38
мета 0,9 1,2 1,8
пара 87 69 62 60
C6H5CH2F[91] СвН5СН2С1[92] C6H5CH2CN[93] С6Н5СН2СО2С2Н5[94]
орто 28 32 17 32
мета 18 14 14 10
пара 54 54 69 58
CeH5CH3[95] С6Н5СН2С1[92] С6Н5СНС12[96] СвН5СС13[96]
орто 57 32 23 6,8
мета 3,2 14 34 64
пара 40 54 43 29
для некоторых случаев приведены соотношения£изомеров, образующихся
при нитровании соединений определенного вида. В ряду CeH5F, С8Н5С1,
С6Н5Вг, С8Н51 наблюдается увеличение содержания орто-нитропроизводного.
Эти данные противоположны результатам, которые можно было бы ожидать,
учитывая пространственные затруднения, однако они согласуются с пред-
ставлением об избирательной полярной дезактивации орто-положений гало-
генами, интенсивность которой соответствует их электроотрицательности
(F > С1 > Вг > I), следствием чего является подавление влияния простран-
ственных затруднений. Увеличение содержания орто-продуктов наблюдается
263
Глава VI
также в случае нитрования GeH5GH2F и GeH5GH2Gl, хотя здесь, как этого
и можно было ожидать, отмечается значительно меньшее различие. Эти
данные подтверждают, что наблюдаемые явления связаны именно с —/-эф-
фектом, а не с -(-^-эффектом галогенов. Если обратиться к данным нитрова-
ния других групп производных, например CeH5GH2GN, СвН5СН2СООС2Н5,
с заместителями соответствующей электроотрицательности и размеров, то-
станет ясно, что обсуждаемый эффект не специфичен только для галоген-
производных. Естественно, необходимы подходящие соотношения между
электроотрицательностью и объемом заместителя. Уменьшение содержания
орто-производного в ряду СвН6СН3, СвН5СН2С1, С8Н5СНС12, С8Н5СС13
представляет собой случай отсутствия отмеченного выше соответствия между
электроотрицательностью и размерами заместителя. Эти данные рассматри-
вались как доказательство избирательной ортпо-дезактивации вследствие про-
странственных затруднений, а также электроотрицательности заместителей.
Однако именно потому, что указанные данные допускают возможность обоих
объяснений, они не могут служить доказательством какого-либо из них.
Подобно тому как при преимущественной оршо-пара-ориентации, обусловлен-
ной электроотрицательными заместителями, образуется главным образом
пара-производное, в случае бензотрихлорида, в котором электроотрицатель-
ность боковой цепи достаточно велика для того, чтобы образовалось главным
образом летпа-нитропроизводное, основным побочным продуктом является
пара-нитропроизводное. Этот результат не полностью определяется простран-
ственной формой боковой цепи; так, при нитровании фенил- и бензилтри-
метиламмониевых ионов, а также ионов анилиния и бензиламмония главным
побочным продуктом будет пара-производное (разд. 3, д). По-видимому, это-
является правилом для всех летпа-ориентирующих соединений, которые
образуют заметные количества побочных продуктов. Исключение составляет
одна группа соединений, которую мы сейчас и рассмотрим.
г. Доказательства влияния сопряжения. —Jf-Эффект. Исключениями
являются все случаи, когда летпа-ориентирующие заместители содержат
кратную связь, расположение которой обусловливает возникновение —М-
эффекта, причем заместитель относится к типу —I,— М, т. е. к типу (2')
в расширенной классификации, приведенной в табл. 71. К группам такого
вида относятся — COR, — COOR, — CN, — NO2 и т. д. Как показывают
данные табл. 78, главным побочным продуктом при этом является орто~
производное.
Таблица 78
Количества изомеров, образующихся при мононитровании
некоторых производных бензола с ненасыщенными электро-
отрицательными заместителями
Соединение Количество изомера, %
орто мета пара
С6Н5СНО[98] ~19 72 -9
С6Н5СОСН3[99] -30 68
СвН5СООН[ЮО] 18,5 80,2 1,3
С6Н5СООС2Н5[ЮО] 28,3 68,4 3,3
C6H5CONH2[101] 27 70 ~3
СвН5СОС1[101] 8 90 <2
CeH5CN[102] 15 83 2
CeH5NO2[103] 6,4 93,4 0,3
264
Замещение в ароматическом ряду
Наблюдаемое явление может быть объяснено тремя способами. На основании
данных, приведенных в таблице, автор книги допустил, что причина заклю-
чается в затрудненности пара-замещения (из-за —М-эффекта заместителя)
[104]. Лэпуорт и Робинсон [105] предложили другое объяснение более легкого
opmo-замещения, допустив, что кратная связь в боковой цепи присоединяет
реагент и передает его внутримолекулярно в орто-положение. Однако дан-
ные, по-видимому, свидетельствуют скорее в пользу специфической пара-
дезактивации, а не специфической opmo-активации. Причина заключается
в том, что допустимое увеличение скорости opmo-замещения не может снизить
содержания пара-изомера до наблюдаемой очень малой величины и сохранить
наблюдаемое очень высокое содержание лета-изомера. Простой арифмети-
ческий расчет показывает, что соотношения изомеров, приведенные в табл. 78,
нельзя вывести из соотношения изомеров, обусловленного ориентацией насы-
щенных заместителей с учетом соответствующих парциальных скоростей
opmo-замещения, протекающего по предложенному Лэпуортом и Робинсоном
механизму.
Третье объяснение было дано сравнительно недавно; оно подтверждается
квантовомеханическими расчетами, в общем довольно приближенными.
Согласно этим расчетам, высокое отношение орто/пара должно наблюдаться
в случае двойной связи, сопряженной с кольцом, и это не требует каких-либо
других специальных объяснений. Уменьшение этого соотношения в других
случаях является следствием пространственных препятствий [106]. Де ла Мар
и Ридд отвергли этот аргумент [107], отметив, что соотношения 1/2 (орто)/пара
и 1/2 (мета)/пара параллельно уменьшаются в ряду заместителей, содержа-
щих двойную связь, сопряженную с ядром, и что оба соотношения равны
единице в одной и той же точке зависимости от природы заместителя, если
последние меняются от сопряженных электроотрицательных к сопряженным
электроположительным заместителям (от —М к Данные, полученные
этими авторами, приведены в табл. 79. Трудно предположить, что представ-
Таблица 79
Соотношение изомеров при нитровании производных бензола,
содержащих ориентирующие заместители с двойными связями,
сопряженными с кольцом
Ориентирующий заместитель 1/2 (орто)/пара 1/2 (мета)/пара Ссылка
no2 11 135 Табл. 78
соон 7 31 То же
conh2 4,5 12 » »
СООС2Н5 4,3 10 » »
сно 1,6 4 » »
CH(NO2) = CHC6H4NO2-» 0,32 0,22 [108]
спсп\о2 0,23 0,015 [108]
ленные результаты являются случайным стечением обстоятельств. Причиной,
обусловливающей указанные соотношения, не могут быть пространственные
препятствия, т. е. оба ряда соотношений обусловливаются взаимодействием
ориентирующего заместителя с пара-положением. Из строения этих замести-
телей следует, что рассматриваемое взаимодействие должно представлять
по существу селективный эффект сопряжения.
265
Глава VI
Таким образом, аргументация данного явления за истекшие 30 лет претерпе-
ла полный оборот по кругу, упрочив нашу уверенность в справедливости
первоначального заключения о том, что в противоположность —/-группам
—I, —7И-группы оказывают на соотношение орто- и пара-изомеров именно
полярное влияние, обусловленное —Л/-характером групп и проявляющееся
в селективной пара-дезактивации.
д. Доказательство влияния сопряжения. ф/7-Эффект. Мы частично
коснулись этого влияния в двух последних строках табл. 79. Для проверки
предположения о возможности преобладающего влияния поляризуемости
в форме электромерного -{-/[-эффекта необходимо рассмотреть окси- и амино-
замещенные. Ориентация под влиянием этих заместителей почти полностью
обусловлена чистым эффектом поляризуемости, и поэтому можно ожидать,
что в этом случае особое значение имеет природа замещающего агента.
Факт зависимости ориентации от замещающего агента в случае окси- и амино-
соединений впервые был отмечен Лэпуортом и Робинсоном [105]. Они ссыла-
лись на исследования Гаттермана и Либермана [109], которые обнаружили,
что 1-нафтол-З- и 1-нафтол-5-сульфокислоты, а также 1-нафтиламин-З-
и 1-нафтиламин-5-сульфокислоты сочетаются с отрицательно замещенным
(например, нитрозамещенным) диазоний-ионом в положение 4, а с незамещен-
ным диазоний-ионом главным образом в положение 2:
(NH2)
диазоний-иоп |
он
(NH2)
ОН
| Незамещенный
/\/\Z
к Z I
Отрицательно замещенный диазоний-ион SO3H
Пространственные затруднения не могут служить причиной этого явления.
Лэпуорт и Робинсон отметили, что электрофильная реакционная способность
диазоний-иона увеличивается при введении в него отрицательной группы *;
такое увеличение, например, наблюдается в ряду
C6H5Nt O2NC6H4Nt, (O2N)2C6H3N2
Следовательно, можно прийти к заключению, что переход от орто- к пара-
сочетанию связан с увеличением активности реагента.
Рассмотрим, однако, в качестве примера соединение с противоположной
тенденцией. Фенол нитрозируется главным образом в пара-положение;
орто-изомер образуется в количестве около 8% [110]. Однако при нитрова-
нии фенола в условиях, при которых участие азотистой кислоты в реакции
сведено к минимуму, образуется не менее 40% орто-изомера [111]. Каче-
ственно сходная картина наблюдается и в случае диметиланилина
NO+
и его носители
* Качественно это было известно давно, а недавно подтверждено количественно
(разд. 6,а).
266
Замещение в ароматическом ряду
Этот эффект также не может быть обусловлен и пространственными затрудне-
ниями; что же касается реакционной способности, то наблюдается обратное
явление. Это качественно очевидно, так как многие ароматические соеди-
нения легко нитруются, но не нитрозируются, и нитрующие агенты вообще
более активны, чем нитрозирующие, а следовательно, электрофильная актив-
ность возрастает в ряду N0 + , NO^. Миллен [112] подтвердил этот вывод
спектроскопически, показав, что при наличии эквивалентных концентраций
нитроний- и нитрозоний-катионов в серной кислоте нуклеофильный реа-
гент — вода почти полностью разрушает нитроний-катион, существенно
не изменяя концентрацию нитрозоний-катиона. Вышеизложенные факты
обобщены в следующей схеме:
Реакционная способность уменьшается
Усиление замещения ( (NO2)2c6H3N+>N02C6H4N+>c6h5n+__>
в иаро-положение Реакционная способность увеличивается
NO+<NO+
Ослабление замещения
в ивра-положенпе
В одном отношении реагенты обоих классов, сильнее замещающие в пара-
положения, сходны: —М-мезомерные системы, образованные группами,
вводимыми в ароматическое ядро, в результате рассматриваемых реакций
сильнее взаимодействуют с ф-Я-ориентирующим заместителем, чем более
слабые napa-замещающие реагенты. Для диазогрупп это теоретически под-
тверждается схемой, показывающей, как нитрогруппа увеличивает сопря-
жение в цепи:
Для нитрозо- и нитрогрупп теоретическое обоснование совершенно аналогич-
но приведенному ранее для простых карбонильной и карбоксильной групп
(гл. II, разд. З.д); нитрогруппа (как и карбоксильная) содержит внутреннюю
мезомерную систему, лимитирующую степень, в которой данная группа
в целом может быть в сопряжении с ароматической системой, тогда как для
нитрозогруппы (как и для карбонильной) таких ограничений не существует *.
Таким образом, можно прийти к заключению, что -фЯ-заместители, акти-
вирующие как орто-, так и пара-положения, избирательно активируют
пара-положения, причем тем сильнее, чем эффективнее сопряжение всту-
пающей группы через ароматическое кольцо с ориентирующим заместителем.
е. Механизм индуктивного влияния на соотношение между орто-
и пара-изомерами. Эффект поля. Теперь следует перейти к вопросу о физи-
ческих механизмах, лежащих в основе полярных влияний на соотношение
между орто- и пара-изомерами; мы начнем с щ/-эффектов.
Общепринято, что влияние индуктивных эффектов заместителей на соотно-
шение opmolnapa в действительности имеет составляющую эффекта поля [114],
т. е. прямого действия электростатического поля заместителей (гл. II, разд.
2,а и разд. 1,д настоящей главы).
* Экспериментальное подтверждение различия нитро- и нитрозогрупп дано Лефевром
267
Глава VI
Как было показано в разд. 3,е, и индуктивный эффект ориентирующего
заместителя, обусловливающий реакционную способность различных поло-
жений молекулы субстрата, и связанный с ним эффект поля, который действу-
ет через внешнюю среду, оба имеют электростатическую природу. Поэтому
оба эффекта при переходе от одной группы к другой изменяются по знаку
и по величине в одном и том же направлении и часто рассматриваются вместе
под обобщенным названием «индуктивный эффект». Однако при благоприят-
ных обстоятельствах индуктивный эффект в своем более специфическом
смысле и эффект поля феноменологически различимы вследствие их различно-
го распределения по реакционноспособным положениям. При электрофиль-
ном ароматическом замещении эффект поля наиболее сильно влияет на орто-
положения, слабее на лета-положения и очень слабо на пара-положение.
Влияние поля на соотношение opmolnapa не зависит от того обстоятельства,
что теоретически сам эффект поля состоит из двух компонентов (разд. 3,е),
так как эти два компонента влияют на реакционноспособные положения
качественно одинаковым образом, и поэтому в настоящее время не могут
быть разделены.
Электроположительный эффект поля на орто/иара-соотношение состоит
в том, что ориентирующий эффект заместителей -[-/-типа будет способство-
вать селективной активации орто-положений. Это, как мы видели в разд. 4,в,
можно наблюдать на опыте, несмотря на противодействующее влияние пер-
вичного пространственного эффекта. Электроотрицательный эффект поля
на орто/пара-соотношение состоит в том, что ориентирующие заместите-
ли—/-типа способствуют дезактивации орто-положений. В разд. 4,в было
показано, что последний эффект также можно наблюдать на опыте, даже
в тех случаях, когда в том же направлении действует первичный простран-
ственный эффект (однако для этого необходимо сильно варьировать природу
заместителей) или когда определенный класс ненасыщенных заместителей,
рассмотренный в следующем разделе, селективно дезактивирует пара-поло-
жения.
ж. Механизм влияния мезомерии на соотношение между орто- и пара-
изомерами. Мезомерный эффект представляет собой избирательный внутри-
молекулярный полярный эффект. Мезомерная поляризация —М, преобладая
над связанным с ней эффектом поля, который селективно дезактивирует
орто-положения, избирательно дезактивирует в основном пара-положения
(разд. 2,г), поэтому —71/-эффект вызывает в пара-положении более сильные
положительные заряды, чем в орто-положениях. Другими словами, мезо-
мерный эффект создает мезомерное состояние, содержащее неполярную
структуру а и диполярные структуры б и в, причем структура б входит
с большим весом, чем структура в.
а
Это заключение особенно подчеркивалось де ла Мэром [115]. Допустимое
теоретическое обоснование заключается в том, что резонансная система
а — б, в которой только одна л-связь может избежать делокализации при
переносе заряда, характеризуется большей неопределенностью положения
электрона, чем резонансная система а — в, в которой две л-связи могут
оставаться локализованными в процессе переноса. Следовательно, в соответ-
ствии с принципом неопределенности энергия минимальна при большем весе
структуры б (по сравнению со структурой в) в ме.зомерной системе. (Анало-
268
Замещение в ароматическом ряду
гичным образом можно понять большую стабильность re-бензохинона по
сравнению с о-бензохиноном *.)
Вывод о том, что механизм этого рода вызывает дезактивацию, подтверждает-
ся также опытными данными об ориентации. Например, хорошо известно,
что если оршо-пара-ориентирующий заместитель типа -у-Е и сильный мета-
ориентирующий заместитель типа —I, —М находятся в лшша-положении
один по отношению к другому, то, вопреки пространственным затруднениям,
третий заместитель часто вступает в положение между ними. Так, ж-нитро-
анизол нитруется главным образом в положение 2 и частично в положения 4
и 6 [116], в то время как м-оксибензальдегид хлорируется главным образом
в положение 2 [117], нитруется в положения 2, 4 и 6 [118] и бромируется
в положения 4 и 6, причем главным образом в последнее положение [119].
ОСНд
I
4
Замещения в положение 2 можно было бы ожидать, если бы такие диполяр-
ные структуры, как б' общего типа б, играли особенно важную роль в мезо-
мерном состоянии этих молекул, так как электромерное электронное смеще-
ние, возникающее в метоксильной или гидроксильной группе, должно
было бы проявляться главным образом в положении 2.
(5’)
Тот факт, что хлорирование протекает почти исключительно в положение 2,
а нитрование частично и бромирование почти целиком в положения 4 и 6,
может быть обусловлен влиянием пространственных затруднений. Ряд
Cl < NO2 < Вг представляет собой часть ряда Голлемана 01 < NO2 <
< Вг < SO3H для случаев, когда проявляются пространственные затрудне-
ния в зависимости от природы вступающей группы, причем различное пове-
дение обычно близких друг другу галогенов служит подтверждением пред-
ставлений Голлемана.
з. Механизм электромерного влияния на соотношение между орто- и
/iapa-изомерами. Ясно, что ориентация, вызываемая эффектом поляризуе-
мости-)- Е, может рассматриваться только в связи с реагентом, который сти-
мулирует данный эффект. Как уже подчеркнул Уотерс [120], переходное со-
стояние при замещении и исходное состояние ароматической системы должны
при изучении ориентации рассматриваться раздельно; необходимость соблю-
дения такого строгого подхода возрастает, если ориентирующая группа типа
+2? создает особые различия между переходным и начальным состоя-
ниями.
При соответствующем обращении знаков и с учетом высказанных положений
можно применить для установления влияния электромерного -J-E-эффекта
* Этот вывод следует из различных квантовохпмических расчетов, согласующихся
друг с другом, однако вследствие их приближенного характера они мало что добавляют
к выводам, следующим пз качественного рассмотрения.
269
Глава VI
на соотношение между орто- и геара-изомерами физическую теорию, являю-
щуюся простым расширением теории, изложенной в предшествующем разделе
в связи с мезомерный эффектом —М.
Необходимо объяснить общую тенденцию -^-ориентированных замещений
к образованию преимущественно пара-производных, а также усиление этой
тенденции, если вводимая группа имеет сильный —УИ-характер. По причине,
указанной в предыдущем разделе, -J-E-свойства ориентирующей группы
и —7И-характер вводимой группы могут совместно эффективно вызвать обра-
зование стабильного мезомерного состояния, если эти группы находятся
в «ара-положениях, так что кратчайший путь через сопряженную систему
является максимальным; если даже они находились бы в орто-положениях,
кратчайший путь был бы меньше. Часть подобных различий в стабильности
конечных орто- и «ара-продуктов проявляется как результат различий
в стабильности переходных состояний при орто- и пара-замещениях. Таким
образом, получает объяснение предпочтительная пара-ориентация. Если
введенная группа обладает более сильным —^/-характером, например при
переходе от фенилазо- к п-нитрофенилазо- или от нитро- к нитрозогруппе,
можно ожидать, что все энергетические эффекты, обусловленные сопряже-
нием между ориентирующей и вступающей группами, увеличиваются, вклю-
чая различия в энергиях переходных состояний при орто- и пара-замещении.
Этим объясняется усиленная тенденция более сильных —М-заместителей
к пара-ориентации.
5. Механизм нитрования ароматических соединений
После обсуждения факторов, которые определяют направление электро-
фильного ароматического замещения, остается рассмотреть конкретные при-
меры таких процессов замещения. Каждый конкретный вид замещения пред-
ставляет собой отдельную проблему, хотя следует ожидать значительного
сходства в различных реакциях, оправдывающего их объединение в общий
класс реакций электрофильного замещения. Нитрование сыграло столь
выдающуюся роль в развитии теории ориентации, что вполне естественно
принять этот процесс в качестве типичного примера при изучении механизмов
замещения.
а. Существование нитроний-иона. Несколько раз уже упоминалось
о нитровании через нитроний-ион NO]». Это старая теория, которая была
предложена Ойлером в 1903 г. и неоднократно выдвигалась на основании
косвенных данных [121]; однако до 1946 г. эти представления не были дока-
заны и даже существование нитроний-иона окончательно не было подтвержде-
но [122]. С 1946 г., как показано ниже, установлены многие детали процесса
нитрования.
Существование нитроний-иона доказано четырьмя способами, которые не
зависят от поведения этого иона при нитровании. Образование нитроний-иона
в больших концентрациях в некоторых растворах, особенно в серной кислоте,
доказано следующими методами: 1) криоскопическими измерениями; 2) спек-
троскопическим изучением растворов. В ионных кристаллах нитроний-ион
идентифицирован как катион 3) в результате получения и спектроскопи-
ческого изучения кристаллических солей нитрония и 4) рентгеноскопическим
исследованием солей нитрония. Рассмотрим соответствующие доказатель-
ства:
1. Ганч подробно изучил влияние добавок на температуру замерзания сер-
ной кислоты; одной из добавок, особенно интересовавшей Ганча, была азот-
ная кислота, которая, как он предположил, превращается в протонированные
270
Замещение в ароматическом ряду
формы
HNO3 + H2SO4 HaNOJ + HSOj
HNOs4-2H2SO4 —> H3NO2+-!-2HSO4-
Гапч определил, насколько понижается температура замерзания серной
кислоты при растворении в ней азотной кислоты. При этом были получены
значения, в три раза превышающие вычисленные для идеальных растворов
[123]. На основании полученных результатов Ганч пришел к заключению,
что азотная кислота в избытке серной кислоты существует главным образом
в виде двухвалентного катиона H3NO3+. Такие же криоскопические данные
получены и другими авторами [124]. Вывод Ганча подтвержден результатами
изучения ультрафиолетовых спектров поглощения [125] и электропроводно-
сти [126]. Однако при применении усовершенствованной техники криоско-
пических исследований было найдено, что понижение температуры замерза-
ния серной кислоты в результате добавления азотной кислоты соответствует
не трехкратной, а примерно четырехкратной величине депрессии идеального
раствора [127]; это безошибочно свидетельствует о превращении азотной
кислоты в нитроний-ион по уравнению
HNO3 + 2H2SO4 —» NO+ + H3O+ + 2HSOj-
Иначе четырехкратная депрессия возникнуть не может. Небольшое отклоне-
ние от строго четырехкратной величины объясняется тем, что незначительное
влияние на температуру замерзания растворов имеет межионное притяже-
ние [128], однако вода хотя и является сильным основанием по отношению
к серной кислоте, но все же не бесконечно сильным [129], так что наряду
с образованием ионов, указанных в правой части уравнения
H2O + H2SO4 —> H3O+4-HSOj
происходит их частичная рекомбинация. На основании константы равнове-
сия этой реакции, выведенной независимыми способами, вычислено, что
депрессия, вызываемая азотной кислотой с учетодг поправки на малое коли-
чество вновь ассоциированных ионов воды, становится точно равной четырех-
кратной идеальной депрессии. Следовательно, превращение азотной кислоты
в питроний-ион носит количественный характер. Таким же образом было
найдено [130], что пятиокись, тетраокись и трехокись азота вызывают депрес-
сии, близкие к шестикратной депрессии идеального раствора, и что при
введении аналогичной поправки эти депрессии становятся точно шестикрат-
ными. Данные показывают, что окисли азота количественно превращаются
в нитроний- и нитрозоний-катионы в соответствии с уравнениями:
N2O5 + 3H2SO4 2NOJ + H3O+ + 3HSO4
N2O4 + 3H2SO4 дД NO2+ + NO+ + H3O+ + 3HSOj
N2Os + 3H2SO4 2NO+ + HsO++3HSO4
Первое из приведенных уравнений с учетом реакции азотной кислоты с сер-
ной, по-видимому, характеризует суммарную реакцию, предполагающую
ионную диссоциацию пятиокиси азота
n2o5 ;± noj+no3-
2. Спектроскопическая идентификация нитроний-иона производится по двум
частотам (1400 и 1050 см-1), которые в спектрах комбинационного рассеяния
271
Глава VI
некоторых смесей, содержащих азотную кислоту, появляются с такой интен-
сивностью, ито не могут быть приписаны молекуле азотной кислоты. Медар
[131] первым обнаружил такие частоты в спектре смеси азотной и серной
кислот. Суз и Бринер [132] установили их также в спектрах растворов пяти-
окиси азота в азотной кислоте. Шеден [133] широко изучил вопрос, и боль-
шинством наших знаний об этих частотах мы обязаны ему. Он обнаружил
подобные частоты малой интенсивности в спектре самой азотной кислоты,
тем самым показав, что они принадлежат продуктам взаимодействия молекул
азотной кислоты между собой. Шеден доказал, что источником их являются
продукты дегидратации азотной кислоты и что при действии воды указанные
частоты исчезают вследствие регенерации азотной кислоты. Эти данные сви-
детельствуют о том, что источником указанных частот является пятиокись
азота. Однако Шеден установил, что спектры растворов пятиокиси азота
в апротонных растворителях совершенно иные. Поэтому был сделан вывод,
что эти две частоты обусловлены особыми формами пятиокиси азота. Пред-
полагается, что такими особыми формами могут быть ионизованные формы
[134], причем частота 1400 см-1 принадлежит нитроний-иону, а 1050 см-1 —
питрат-иону или в присутствии серной кислоты — гидросульфат-иону в соот-
ветствии с известными спектрами этих анионов. Дальнейшие спектроскопи-
ческие исследования [135] подтвердили эти предположения. Другие частоты
комбинационного рассеяния нитрат- и гидросульфат-ионов были найдены
в спектрах других растворов. При смешении азотной кислоты с хлорной или
селеновой кислотами появляется интенсивная частота 1400 см-1 без одно-
временного появления частоты 1050 см-1. При этом наблюдались спектры
хлорат- или гидроселенат-ионов; однако они не содержат частот, близких
к частоте 1050 см-1. Наконец, более подробное исследование спектров в самых
различных условиях показало, что источник частоты 1400 см-1 имеет
в спектре комбинационного рассеяния только эту частоту. Поэтому этот
источник может быть двухатомной или линейной трехатомной молекулой
с одинаковыми атомами на концах. Единственной такой стабильной молеку-
лой с еще неизвестным спектром, которая может получиться из элементов
азотной кислоты, является нитроний-иОн. Таким образом, была создана
основа для спектроскопического обнаружения и определения нитроний-иона.
По этому методу получены подтверждения правильности приведенных урав-
нений ионизации азотной кислоты и пятиокиси азота в серной кислоте 1136].
Кроме того, установлено, что в азотной кислоте пятиокись азота является
сильным электролитом, причем единственными частицами в растворе являют-
ся нитроний- и нитрат-ионы [137].
3. Ганч первым показал, что азотная и хлорная кислоты взаи.модействуют
с образованием твердых солеобразных соединений [138], которые, как он
полагал, представляют собой перхлораты протонированных катионов азотной
кислоты (H2NO3)+(CIO4)~ и (H3NO3)2+(CIO4)2. Эти же эксперименты были
повторены с применением усовершенствованной техники. Таким образом
было установлено [139], что эти твердые вещества в действительности пред-
ставляют собой смеси нитроний- и гидроксопийперхлоратов (NO2) +(С1О4)~
и (Н3О)+(СЮ4)_, образовавшихся в результате реакции
НХО3з-2НС1О4 N0J-PHg0+4-2C104
Пятиокись азота и хлорная кислота образуют те же продукты, но в других
соотношениях:
N2O3 жзнсЮд —» 2N0+-!-H30+-p3C107
272
Замещение в ароматическом ряду
Кристаллизацией из нитрометана получена также чистая соль нитрония
(NO2)+(G1O4)“. Получены соли нитрония * (NO2) +(FSO3)~, (NO2)+(HS2O7)~
и (NO2)+(S2O7)a~. Все они имеют ионное строение, причем спектр комбина-
ционного рассеяния кристаллов представляет собой наложение уже изве-
стных спектров этих ионов [141]. Как показали Миллен, Пул и автор этой
книги [142] на основании спек-
тральных данных Шедена и Прадье
[143], кристаллическая пятиокись
азота представляет собой нитро-
нийнитрат (NO2)+(NO3)~. При ра-
створении пятиокиси азота в азот-
ной кислоте не наблюдается изме-
нения ковалентности, но проис-
ходит такой же простой распад
на ионы, как и при растворении
хлористого натрия в воде. Одна-
ко, как и хлористый натрий **,
пятиокись азота испаряется с со-
хранением ковалентной структу-
ры. Другие достаточно устойчи
вые соли нитрония позднее были
получены Шмайссером и Элише-
ром, но особенно следует отме-
тить работу Ола И сотр. [144]. Не- Рис- 28- Схематическое изображение струк-
г „ туры N2O4.
которые из этих солеи в настоящее
время являются продажными ре-
активами, например (NO2)+(BF4)- (NO2)+(PF8)", (NO2)+(AsF8)~h (NO2)+(SbFe)".
4. В двух случаях, а именно для нитронийперхлората [145] и нитронийнитра-
та (пятиокись азота) [146] ионное строение солей нитрония подтверждено
рентгеноструктурным анализом. В обоих случаях подтверждено линейное
строение нитроний-иона. Структура пятиокиси азота по Гризону, Эриксу
и де Фризу [146] воспроизведена на рис. 28.
б. Эффективность нитроний-иона. Для выяснения вопроса, что является
атакующим агентом при нитровании ароматических соединений и может ли
нитроний-ион быть таким агентом, необходимо изучить кинетику нитрования.
В этом отношении о многом говорит порядок реакции.
Мартинсену [147] первому удалось установить порядок реакции. В качестве
растворителя он применял серную кислоту. Он нашел, что нитрование
нитробензола смесью азотной и серной кислот представляет собой реакцию
второго порядка. Этот результат подтвержден последующими исследования-
ми [148]; второй порядок реакции установлен в случае нитропроизводных
бензола, бензойной кислоты, бензолсульфокислоты и некоторых производных
антрахинона.
Скорость в H2SO4 = A2[ArH][HNO3]
С этим результатом можно связать наблюдения [149], относящиеся к другой
сильной кислоте, применяемой в качестве растворителя, а именно к азотной
* Гиллеспи криоскопически и Миллен спектроскопически [140] установили, что
олеум содержит по крайней мере две высшие по сравнению с H2SO4 и H2S2O7 кислоты,
а именно H2S3O)0 и H2S4O13, а также ионы всех этих кислот. Получена соль нитрония
и одной из этих высших кислот (NO2)£ (S3O10)2-.
** Об энергетических соотношениях, в особенности при различной летучести, см.
ранее цитированную работу Миллена [136].
18 — 07 7 2
273
Глава VI
кислоте; нитрование в этом растворителе представляет процесс первого
порядка, что установлено для нитрозамещенных бензолов и антрахинонов
Скорость в HNO3 = Ад[АгН]. (2)
Данное уравнение вытекает из уравнения (1), если азотная кислота постоянно
находится в избытке, т. е. когда она служит растворителем.
Эти результаты показывают, что атакующий реагент либо является азотной
кислотой, либо образуется из нее настолько быстро, что всегда сохраняется
постоянное соотношение. Однако из одних кинетических данных нельзя
установить, велико это соотношение или мало.
Изучение нитрования в органических растворителях, особенно в нитрометане
и уксусной кислоте при постоянном избытке азотной кислоты по сравнению
с ароматическим соединением, позволяет сделать более широкие выводы [150].
В этом случае порядок реакции, установленный методом конкурирующих
реакций (разд. 3,а), зависит от реакционной способности производного
бензола. Для ароматических соединений, более реакционноспособных, чем
бензол, а при некоторых условиях и для самого бензола порядок реакции
нулевой. Это означает, что нитрование протекает с постоянной скоростью
независимо от концентрации ароматического соединения, сразу же прекра-
щаясь при полном израсходовании последнего. Все соединения, которые
подчиняются этому закону: бензол, толуол, этилбензол, n-ксилол, мезитилен
и и-хлоранизол при сравнении показали одинаковую скорость нитро-
вания.
Скорость нитрования достаточно реакционноспособных ароматических
соединений в органическом растворителе = A0(HNO3 в постоянном избытке) ' '
При некоторых условиях в случае бензола и всегда при нитровании фтор-
и иодбензолов, которые на основании определений по методу конкурирую-
щих реакций несколько менее активны, чем бензол (разд. 3,а и табл. 67),
скорость реакции лишь в слабой степени зависит от концентрации, а в слу-
чае хлор- и бромбензолов, которые еще менее реакционноспособны (см.
табл. 67), эта зависимость выражена сильнее. Таким образом, степень зави-
симости увеличивается по мере уменьшения реакционной способности, по
до определенного предела. Предел достигается в случае двух дезактиви-
рующих атомов галогена, например в дихлорбензоле. Этот предел имеет
вполне определенную величину и не превышается, например, в случае три-
хлорбензола. Предел может быть достигнут при наличии одного более сильно
дезактивирующего заместителя, как карбэтоксильная группа в этиловом
эфире бензойной кислоты (см. табл. 67). Эта предельная зависимость скоро-
сти от концентрации является простой пропорциональностью, т. е. протекают
реакции первого порядка. Естественно, константы скорости для тех соеди-
нений, которые подчиняются этому закону, различны, причем они находятся
в соотношениях, которых следовало бы ожидать, исходя из дезактивирующих
влияний заместителей, в соответствии с данными метода конкурирующих
реакций.
Скорость достаточно нереакционноспособных ароматических соединений ,,,
в органических растворителях = Ai[ArH](HNO3 в постоянном избытке) ‘И
Из уравнений (1) — (4) непосредственный ответ на вопрос о механизме реак-
ции дает уравнение (3). Нулевой порядок реакции означает, что медленно
протекающий процесс, скорость которого измеряется, таков, что аромати-
ческое соединение в нем не принимает участия; возникающий при этом реа-
гент связывается по мере образования ароматическим соединением, которое
274
Замещение в ароматическом ряду
при этом подвергается нитрованию. Медленный процесс неспецифичеп для
данного растворителя, потому что химически различные растворители вызы-
вают одинаковые кинетические явления. Следовательно, этот процесс обус-
ловлен азотной кислотой. Это не может быть просто переносом протона,
потому что, как можно полагать, такой процесс в кислородсодержащих
кислотах происходит мгновенно. Рассматриваемый процесс характеризуется
определенной энергией активации, а следовательно, он обусловлен разрывом
связи в азотной кислоте. Далее процесс является гетеро литическим, посколь-
ку создается электрофильный нитрующий агент, т. о. осколок с четным чис-
лом электронов, содержащий группу NO2. с недостатком электронов у атома
азота. Остается единственная возможность — резко выраженный гетеролиз,,
как это показано па схеме, а реагент, который связывается по мере его обра-
зования ароматическим соединением, представляет собой нитроний-ион *.
Н—О
О
So
—> но-Ж ко*
Соотношения, выражаемые уравнениями (1), (2) и (4), согласуются с этим
заключением, хотя сами по себе они не могут привести к нему. В сильно-
кислом растворителе нитроний-ион образуется быстро и составляет постоян-
ную часть вводимой в реакцию азотной кислоты. Из физических данных
известно, что в серной кислоте эта часть близка к единице, в то время как
в азотной кислоте она близка к нулю. Однако эта разница не вытекает из
кинетических данных **. Кинетический порядок реакции зависит только
от скорости превращения [уравнения (1) и (2)].
Однако в органических растворителях нитроний-ион образуется с измеримой
скоростью, т. е. как раз с такой, которую можно измерить при любых реакци-
ях нулевого порядка. В случае нитрования достаточно нереакционноспособ-
ных ароматических соединений можно даже при такой скорости создать
постоянную концентрацию, из которой ароматическое соединение потребляет
необходимое количество нитрующего агента в соответствии со своей кон
центрацией и реакционной способностью [уравнение (4)].
Вода в качестве растворителя влияет в основном так же, как рассмотренные
выше органические растворители. Важными общими свойствами этих раство-
рителей являются следующие: во-первых, они полярны и поэтому способны
сольватировать ионы, а, во-вторых, будучи более слабыми кислотами по
сравнению с азотной кислотой, эти растворители представляют собой среды,
в которых образование нитропий-иона может происходить достаточно мед-
ленно, и скорость этого процесса можно измерить. Чтобы определить, какая
* Перенос протона, который происходит после медленно протекающей стадпп. кине-
тически но играет существенной ролл. Поэтому при гетеролизе одного из двух атомов
кислорода с образованием О2- и HNO|+ мгновенный перенос протона сопровождался бы
регенерацией NO^. Следовательно, остается в силе положение о том, что активных?
реагентом является NOJ.
** Ли п Миллен [151] при научении электропроводности аналитически чистой азот-
ной кислоты показали, что она содержит продукты диссоциации в следующих концентра-
циях:
гмп+т гмп-1 пчш f 0.51 моль/л при -10 °C
[NO+]^[NO3] = [H2O]=--’0 61 моль/л при __20ос
•Это означает, что 4—5% по весу аналитически чистой и безводной азотной кислоты дис-
социирует при этих температурах по уравнению
2HNOS —> NO+ + NO7 + H2O
18* .175
Глава VI
частица в водной среде является нитрующим агентом, необходимо работать
с ароматическими соединениями, которые, помимо растворимости в воде,
обладали бы достаточной реакционной способностью. В этом случае, если
нитрующим агентом служит нитроний-ион, ароматическое соединение будет
захватывать эту частицу быстрее, чем произойдет превращение нитроний-
иона в азотную кислоту под действием избытка воды. Такой довольно тонкий
эксперимент был проведен Бантоном и Халеви [152]. В качестве среды эти
авторы применили смесь около 40 молей азотной кислоты с 60 молями воды,
а в качестве водорастворимых субстратов — аралкансульфонаты натрия,
например мезитилен-1а-сульфонат и изодурол-2а-сульфонат, которые содер-
жали несколько активирующих метильных групп:
СН3 СН3
I__ ___I
/ /— CH2SOg СНз—/ /— CH2SO3
I I
СН3 СНз
Кинетика нитрования этих соединений зависит от концентрации субстрата.
При миллимолярных концентрациях наблюдается первый кинетический поря-
док относительно субстрата, а при децимолярных концентрациях порядок
реакции по субстрату равен нулю.
Скорость при низкой концентрации субстрата /г,
в водной азотной кислоте = Zc1[ArH](HNO3 в постоянном избытке) ' '
Скорость при высокой концентрации субстрата
в водной азотной кислоте = Aq(HNO3 в постоянном избытке) ' '
Эти результаты показывают, что даже в водной среде (в которой стационарная
концентрация нитроний-иона так низка, что этот ион не может быть обна-
ружен спектрально) нитрующим агентом все же является ион нитрония. При
низких концентрациях эти субстраты не могут улавливать все нитроний-
ионы по мере их возникновения [уравнение (5)] и большинство этих ионов
становится «добычей» молекул воды. Однако при высоких концентрациях
[уравнение (6)1 субстрат улавливает почти все ионы нитрония, несмотря
на присутствие воды. В этом случае скорость нитрования соответствует ско-
рости чисто неорганической реакции разрыва связи в азотной кислоте с обра-
зованием нитроний-иона.
В гл. VIII будет показано, что скорость этой реакции нулевого порядка
соответствует скорости обмена 18О между азотной кислотой и водой. Это
служит независимым доказательством того, что в азотной кислоте разрывается
связь N — О и что нитрующий агент возникает именно в процессе этой
реакции.
в. Механизм образования нитроний-иона. При отсутствии прямых дока-
зательств можно предположить, что нитроний-ион образуется из азотной
кислоты в результате одностадийного бимолекулярного гетеролиза.
HNO3-|-HNO3 -> NO+ + NOj + H2O
Переходное состояние такого расщепления можно легко себе предста-
вить. Однако было показано, что на самом деле это расщепление протекает
® две стадии:
Быстро
HNO3+HNO3 Т-------» H2NOj + NOg
Медленно
H2NO+-------> H2O + NO£
276
Замещение в ароматическом ряду
Первая стадия схемы представляет собой особый случай более общего меха-
низма. Роль каждой из двух молекул азотной кислоты различна: первая
поставляет протон для второй, которая служит действительным источником
нитроний-иона. Таким образом, первую молекулу можно заменить молекулой
другой достаточно сильной кислоты, при этом механизм по существу остается
тем же. Вторую стадию можно схематически представить следующим образом:
O2N-OH2 O2N++H2O
Эта схема выражает мономолекулярный гетеролиз, аналогичный гетеролизу
органических соединений, который приводит, как показано в гл. VII, к обра-
зованию карбоний-ионов.
Подлинным доказательством сказанного служат результаты изучения влия-
ния добавок сильных кислот [149], ионизованных нитратов, а также воды
на скорости нитрования при реакциях нулевого и первого порядка в органи-
ческих растворителях. Основные результаты следующие: 1. Очень малые
добавки серной кислоты сильно ускоряют обе реакции, причем скорости
увеличиваются пропорционально концентрации добавляемой кислоты.
2. Очень малые количества нитрат-ионов сильно замедляют обе реакции,
причем обратные величины скоростей в определенном интервале концен-
траций возрастают пропорционально концентрации добавляемого нитрата.
3. В случае нитрования, протекающего по реакции нулевого и первого
порядка, большие изменения скоростей, обусловленные добавкой кислоты
или нитрата, не сопровождаются нарушением порядка' реакции. 4. По срав-
нению с этими эффектами вода незначительно влияет на скорость реакции
нулевого порядка; однако добавление значительных количеств воды
уменьшает скорость реакции и попутно с сильным уменьшением скорости
реакции нулевого порядка переходит в реакцию первого порядка.
Отмеченные в пунктах 1 и 2 результаты могут соответствовать либо одно-
стадийному, либо двухстадийному механизмам образования нитроний-иона.
Эти данные просто показывают, что общий процесс включает стадию обра-
тимой реакции, которая в прямом направлении сопровождается захватом
одного протона с образованием одного нитрат-иона. Для решения!вопроса
о механизме реакции следует обратить особое внимание наважнейшее из четы-
рех положений, отмеченное в пункте 3. Имеет значение то, что нитрат-ионы
сильно замедляют реакцию нулевого порядка, не нарушая ее характера.
Отсюда сразу вытекает вывод, что образование нитроний-иона, являющееся
лимитирующей стадией, в свою очередь происходит в другой*стадии. Стадия
реакции, при которой получается нитрат-ион, осуществляется легко и сильно
обратима. Однако другая стадия реакции, а именно образование нитроний-
иона, необратима. Для нулевого порядка нитрованиягважно, чтобы суще-
ственная часть образовавшегося нитроний-иона не превращалась в исходные-
реагенты, а расходовалась на нитрование ароматического соединения. Следо-
вательно, в этом случае процесс должен состоять из двух стадий, на первой
из которых образуется нитрат-ион, а на второй конечной стадии — ион нит-
рония. Эти условия полностью стехиометрически соответствуют приведенным
уравнениям двухстадийного механизма; остальные показатели вполне отве-
чают этому механизму. Вода не образуется на легко обратимой стадии; следо-
вательно, вода в отличие от нитрат-иона не влияет на скорость реакции.
Однако при добавлении большого количества воды вторая стадия становится
обратимой и нитрование сильно замедляется; процесс превращается, как
это и должно быть, из реакции нулевого порядка в реакцию первого
порядка.
277
Глава VI
Эти наблюдения доказывают, что ион H2NOg существует независимо. Этот
ион, следовательно, является возможным нитрующим агентом, хотя и неэф-
фективным в описанных условиях нитрования. Пока еще не было доказано
присутствия иона H2NOg в каких-либо растворах в высокой концентрации;
не выделены и его соли.
г. Механизм атаки нитроний-ионом. Предложены две теории связыва-
ния интротгий-иона с ароматическим кольцом с отщеплением протона. Одна
: . ;:па заключается в том, что введение нитроний-иона и передача протона
гжепшсму основанию представляют собой тримолекулярпый процесс, проис-
>. >дящ1ш в одну стадию, который можно обозначить SE3. При этом связываю-
щее протон основание можно обозначить А-, учитывая, что в кислой среде
наиболее доступным основанием обычно является анион кислоты.
NOj + ArH + A- ArNO2 + HA (SE3)
Эта в принципе допустимая теория в настоящее время пе пользуется призна-
нием *. Согласно другой принятой теории, допускаются две стадии [154],
а именно медленное связывание нитроний-иона с последующим быстрым
отщеплением протона. Скорость реакции определяется бимолекулярной
стадией, и механизм процесса может быть обозначен SE2.
Медленно / ТТ X
NOJ + ArH-------> Аг+;
zn
Аг ж А--------------> Ar.NO,-и НА j
'•С ' " J
«.•дна из причин, почему тримолекулярпый механизм фактически никогда
не отстаивали, заключается в том, что в соответствии с описанной и гл. VII
хорошо разработанной теорией о влиянии растворителя па скорость про-
цесса SE3, в котором исчезают ионные заряды, он должен замедляться поляр-
ным растворителем, а в то же время известно, что ароматическое нитрование
особенно хорошо протекает в полярных растворителях. Правда, указанный
эффект в значительной мере обусловлен влиянием растворителя на процессы,
которые вызывают образование нитроний-ионов; однако это не может опре-
делять влияния растворителя на общий процесс нитрования, если на послед-
ней или на любой важной предшествующей стадии такое влияние противо-
положно. Более детальный анализ [149], основанный па изучении влияния
растворителя на реакции нулевого и первого порядка, показал, что увеличе-
ние полярности растворителя в действительности сильно ускоряет образова-
ние нитроний-иона и оказывает сравнительно незначительное влияние на ско-
рость атаки ароматической молекулы нитроний-иона. Эти данные находятся
в соответствии с механизмом SE2, при котором на стадии, определяющей
скорость реакции, не образуются и не исчезают ионные заряды; с другой
стороны, на основании указанных данных исключается механизм SE3.
Меландеру [154] принадлежит изящное доказательство того, что с кинетиче-
ской точки зрения перенос протона является несущественной стадией при
атаке нитроний-ионом. Он использовал то обстоятельство, что связи с изотоп-
ными атомами водорода значительно различаются по нулевой энергии,
л то время как только часть этой разницы обнаруживается в переходных
состояниях реакций с разрывом связей, поскольку частично разорванные
связи обладают меньшими колебательными квантами; это ведет к различиям
в энергиях активации и в скоростях. Экспериментальным путем точно уста-
* Имеются указания, что вторичные явления, которые привлекались для поддержки
тримолекулярпого механизма, можно объяснить иначе [153].
278
Замещение в ароматическом ряду
новлепо, что протий и дейтерий замещаются в одинаковых соединениях
в самых разнообразных реакциях с весьма различными скоростями, причем
протий замещается в 3—10 раз быстрее, чем дейтерий. Аналогично и скорости
замещения протия и трития различаются в 5—25 раз. Меландер использовал
тритий. Его метод заключался в нитровании ароматического соединения,
содержащего тритий в одпом из эквивалентных положений, например^бепзола
с одним атомом трития, толуола с одним атомом трития в орто-положении
и т. д. В пределах ошибок опыта Меландер установил отсутствие предпо-
чтительного замещения протия перед тритием при нитровании. Этим было
убедительно доказано, что отщепление протона от молекулы ароматического
соединения не входит в определяющую скорость стадию атаки нитроиий-
иопом. Это было подтверждено в работе Боннера, Бойера и Гвин Уильямса
[1541, которые показали, что скорости нитрования нитрующей смесью нитро-
бензола и иептадейтеронитробензола равны.
Что касается геометрической формы промежуточно образующегося катиона,
то о ней можно лишь делать предположения; по-видимому, наиболее вероятно,
что группы II п NO2 связаны тетраэдрическими связями.
Н
Следовательно, образуется ион циклогексадиенилия, в котором ионный заряд
распределен между орто- и пара-положениями ядра. Другими словами,
ион является мезомерный со следующими главными валентными структурами:
Впервые предположение об образовании такого рода промежуточных соеди-
нений при ароматическом замещении было высказано Пфейфером и Вицин-
гером [155], а теоретически обосновано Уэландом [156]. В некоторых реак-
циях электрофильного ароматического замещения при низких температурах
подобные промежуточные катионы были выделены в виде солей. Так, при
нитровании бензотрифторида фтористым нитрилом в присутствии трехфто-
ристого бора (образуется борфторид нитрония). Ола и Куп [157] получили
указанное ниже промежуточное соединение в виде желтой кристаллической
соли, устойчивой ниже —50 °C, но при повышении температуры разлагаю-
щейся с отщеплением протона и образованием м-нитробензотрифторида.
bf4-
Существовапие подобных промежуточных продуктов присоединения ни в коем
случае нельзя рассматривать как подтверждение механизма Se2, который
характеризуется только тем, что вложено в его символ: при электрофильном
замещении в переходном состоянии образуется частичная ковалентная связь
279
Глава VI
между двумя реагентами. Употребляемый символ SE2 ничего не говорит
ни о числе стадий процесса, ни, если их несколько, о том, какая из них
определяет скорость. Этот символ умышленно не отражает еще и очень мно-
гих других деталей процесса и, подобно другим таким символам, определяет
лишь границы класса [реакций, не касаясь различий между механизмами
внутри данного общего класса.
Подводя итоги, можно сказать, что при нитровании азотной кислотой суще-
ственную роль играют следующие четыре стадии:
HA + HNO3^H2NOJ + A- (1)
H2NOt=₽tNO+4-H2O (2)
NO£-|-ArH=p*ArHNO+ (3)
ArHNO+-f-A-4=t ArNO2 + HA (4)
Кислота НА является наиболее сильной из имеющихся кислот; это азотная
кислота, если нет более сильных кислот. В тех случаях, когда в качестве НА
берется такая кислота, как фторсульфоновая, хлорная, хлорсульфоновая,
серная или селеновая [157], то вода, образующаяся по стадии (2), частично
или полностью ионизуется
на-;-н2о^н3о++а- (5)
Азотная кислота не настолько сильна, чтобы в значительной степени реаги-
ровать с водой как с основанием в условиях нитрования; однако ион H2NO*
является такой кислотой, и равновесие
H2NO+ + H2O = H3O+ + HNO3 (6)
как уже отмечалось, имеет малое, но заметное влияние на кинетику нитро-
вания [149].
д. Переносчики нитроний-иона. Частицы с более сложной структурой,
чем ион NO*, могут атаковать ароматическое ядро и вводить в него нитро-
группу; такие частицы можно назвать переносчиками нитроний-иона. Можно
ожидать, что переносчик NO2X будет тем лучшим нитрующим агентом, чем
сильнее X будет притягивать электроны, и поэтому ряд
NO+ NO2OHJ, NO2ONO2, NO2OCOCeH5, NO2OH
отражает последовательно убывающую способность к нитрованию. Инте-
ресно проследить, как в действительности изменяется нитрующая способ-
ность в этом ряду; трудность заключается в том, чтобы отличить прямое
нитрование посредством NO2X от нитрования через нитроний-ион, образую-
щийся из NO2X.
Как уже говорилось, ацидониевый ион азотной кислоты, вероятно, не уча-
ствует в нитровании в обычных условиях. Его роль состоит только в генери-
ровании ионов нитрония.
Ковалентная пятиокись азота в четыреххлористом углероде, по-видимому,
является нитрующим агентом, хотя при доказательстве этого возникают
затруднения вследствие легкости, с которой эта молекула в присутствии
сольватирующих веществ дает нитроний-ион. Здесь возможна простая реак-
ция второго порядка [158]:
Скорость нитрования N2O5 в CC14 = fc2[N2O5][ArH]
Однако обычно эта реакция перекрывается аутокаталитическим процессом
или даже рядом аутокаталитических процессов более высокого порядка,
зависящих от образования азотной кислоты в процессе нитрования, что-
280
Замещение в ароматическом ряду
соответствует кинетическому уравнению
Скорость нитрования N2O5—HNO3 в СС14 = /с[Х2О5][АгН][НХО3]п
где п обычно равно 3 и, по-видимому, колеблется в пределах от 2 до 4. Реак-
цию второго порядка можно отделить от катализируемых реакций при приме-
нении низких концентраций (по очевидным причинам) и не слишком низких
температур (так как реакции высокого порядка имеют низкие температурные
коэффициенты). В этих условиях происходит прямое нитрование молекуляр-
ной пятиокисью азота. Каталитические процессы могут приобретать исклю-
чительное значение при добавлении азотной кислоты. Предполагается, что
при этом нитрующим агентом является нитроний-ион, который возникает,
если образовавшаяся или добавленная азотная кислота ионизует некоторую
часть пятиокиси азота. Минимальное число молекул азотной кислоты, кото-
рое может эффективно ионизовать одну молекулу пятиокиси азота, равно
двум, т. е. по одной молекуле на «сольватацию» каждого иона. Общее число
молекул, которое необходимо для сольватации пары ионов в азотной кислоте
как растворителе, равно четырем [158]. Таким образом, установленные вели-
чины показателя степени п представляются вполне обоснованными.
По-видимому, пятиокисью азота заканчивается ряд непосредственно нитрую-
щих агентов. Изучение [160] бензоилнитрата, который, как кажется, вполне
аналогичен ацетилнитрату, показало, что он действует не сам по себе, а как
регулятор, поддерживающий низкую концентрацию пятиокиси азота, обра-
зующейся вследствие диспропорционирования, катализируемого кислотами
2NO2OCOC6H5 N2O5+(C6H5CO)2O
В свою очередь нитрование пятиокисью азота, как показано выше, происхо-
дит почти полностью через ионизацию с образованием иона нитрония. С этим
согласуется наблюдение Поля [161], который отметил резкое уменьшение
скорости нитрования азотной кислотой в уксусном ангидриде, т. е. ацетил-
нитратом, при добавлении нитрата натрия.
Отсутствуют сведения, подтверждающие, что молекулярная азотная кислота
способна нитровать соединения в каких бы то ни было условиях опыта.
Можно сделать вывод, что нитрование соединениями NO2X происходит
преимущественно или исключительно через стадию ионизации
NO2X —> NOJ + X-
NOf + АгН —» ArNO2+H+
Протекание реакции через общее промежуточное соединение катионного типа
независимо от природы замещающего агента характерно только для арома-
тического нитрования и ни в коем случае не относится к другим процессам
замещения.
е. Влияние азотистой кислоты на нитрование нитроний-ионом. «Азоти-
стой кислотой» считают такой продукт в нитрующем растворе, который при.
разбавлении водой можно определить в виде азотистой кислоты. Влияние
добавленной или имеющейся в виде примеси азотистой кислоты на скорость
нитрования нитроний-ионом осложняет реакцию; этого влияния можно
легко избежать, применяя среду, свободную от азотистой кислоты. Вот
почему об этом влиянии до сих пор не упоминалось. Однако азотистая кис-
лота все же связана с определенной группой процессов нитрования, под-
лежащих теперь рассмотрению.
Азотистая кислота замедляет нитрование, протекающее через стадию обра-
зования нитроний-иона. До того как этому явлению было дано объяснение,,
чисто эмпирически было обнаружено [150], что замедление нитрования при
281
Глава VI
низких концентрациях азотистой кислоты можно описать включением в кине-
тические уравнения реакций нулевого и первого порядков некоторого общего
множителя.
Скорость = Ло(1 + <z[HNO2]1/2)-i,
Скорость = Л1[АгН](1 + a[HNO2]1/2)-1
При этом азотная кислота должна быть в постоянном избытке (HNO2 — ана-
литически обнаруживаемая HNO2).
Причина этого антикатализа теперь выяснена [149]. Эффект зависит от сле-
дующих свойств неорганических азотистых соединений, которые были уста-
новлены независимо [162] частично на основании спектроскопических данных,
частично измерениями электропроводности и другими измерениями. В избытке
азотной кислоты азотистая кислота существует преимущественно в виде
тетраокиси азота. Частично тетраокись азота гомолитически превращается
в двуокись азота; однако для рассматриваемого вопроса это существенного
значения не имеет. Тетраокись азота также подвергается гетеролизу, т. е.
иопизуется почти полностью в чистой азотной кислоте как растворителе
и является лишь очень слабым электролитом в органических растворителях,
содержащих азотную кислоту; при этом образуются нитрозоний- и нит-
рат-ионы
2NO2 N2O4 7^ NO+ + NOJ
Образовавшиеся нитрат-ионы отщепляют при нитровании протон от H2NO2,
который является предшественником нитроний-иона, как это было описано
в случае добавки ионизованных нитратов; азотистая кислота, таким образом,
замедляет нитрование. Легко убедиться, что этот механизм процесса приводит
к указанным выше уравнениям.
При более высоких концентрациях азотистой кислоты и в присутствии неко-
торого количества воды наступает более резко выраженный антикатализ
азотистой кислотой. Объяснение заключается в том, что при указанных усло-
виях малое, но кинетически эффективное количество азотистой кислоты
находится в виде трехокиси азота, образовавшейся по реакции
2N2O4 + H2O Zl N2O3 + 2HNO3
Трехокись азота также подвергается в слабой степени гомолизу и в значи-
тельной мере гетеролизу с образованием нитрозоний- и нитрит-иопов:
NO4-NO2 N2O3 no+ + no2
Нитрит-ион, как более сильное основание, эффективнее, чем нитрат-ион,
отщепляет протон от H2NO3 и тем самым снижает скорость нитрования. Рас-
четы показывают, что это дополнительное влияние на нитрование может
быть отражено, если добавить некоторую величину к антикаталитическому
множителю с тем, чтобы множитель в скобках в правой части уравнения
принял вид
(l+a[HNO2]1/2 + 6[HNO2]3/2)-i
При соответствующем выборе концентраций можно достигнуть такого поло-
жения, что последний член полинома будет настолько превосходить осталь-
ные, что ими можно пренебречь. Такая упрощенная форма расширенного
кинетического уравнения подтверждена экспериментально.
ж. Нитрование через нитрозирование. Уже давно известно, что суще-
ствуют две группы ароматических соединений, при нитровании которых
282
Замещение в ароматическом ряду
азотистая кислота часто ведет себя как катализатор, в противоположность
тому, что установлено для нитрования ароматических соединений вообще.
В эти две группы входят производные (в том числе продукты алкилирования)
фенола и анилина. Возникают два вопроса: как это происходит и почему
данное явление характерно для производных фенола и анилина?
Пре;кде чем на основании опытных данных ответить на первый из этих
вопросов, следует устранить мешающие осложнения. Во-первых, часть аро-
матических соединений, нитрование которых катализируется азотистой кис-
лотой, в значительной степени окисляются в условиях нитрования. Эти
побочные реакции, которые пока не рассматриваются, приводят к образо-
ванию азотистой кислоты, благодаря чему положительный катализ превра-
щается в аутокатализ. Так ведет себя фенол и в меньшей степени анизол.
Однако при определенной дезактивации ядра не происходит окисления
в результате замещения, но положительный катализ азотистой кислотой
сохраняется. Это наблюдается, например, в случае n-нитрофенола и тг-хлора-
низола.
Изучение кинетики нитрования в уксусной кислоте [163] этих и подобных
соединений азотной кислотой при постоянном ее избытке показало, что сум-
марная реакция, на первый взгляд запутанная, в действительности состоит
из двух конкурирующих реакций, каждая из которых достаточно проста.
Эти реакции при надлежащем выборе условий могут протекать почти в чистой
форме. Одна из них соответствует уже знакомому уравнению
Скорость — Л-о(1 -palHNOa]1/2)-1
Это уравнение характерно для нитрования через нитроний-ион; вследствие
высокой реакционной способности таких ароматических соединений следует
ожидать реакции нулевого порядка. Другая новая реакция имеет следующую
кинетическую характеристику:
Скорость = /c2[ArH][HN02]
(И в этом случае HNO2 — аналитически обнаруживаемая HNO2.) Очевидно,
это именно такая реакция, для которой наблюдается положительный катализ
азотистой кислотой. Каталитический эффект можно свести к минимуму
возможно более полным вытеснением азотистой кислоты и увеличением кон-
центрации азотной кислоты до такой степени, чтобы нитроний-ион мог
легко образоваться. С другой стороны, каталитическое влияние азотистой
кислоты можно сделать доминирующим, если работать с большим количе-
ством азотистой кислоты и снижать концентрацию азотной кислоты.
Считают, что кинетическая форма катализируемого процесса убедительно
подтверждает теорию, которая часто обсуждалась без каких-либо определен-
ных выводов, а именно теорию, согласно которой каталитическое нитрование
состоит в первоначальном нитрозировании с последующим быстрым окисле-
нием нитрозосоединения с регенерацией азотистой кислоты, расходуемой
в первой медленной стадии.
Медленно
ArH + HNO2--------> ArNO + H2O
Быстро
ArNO + HNOa-------> ArNO2+HNO2
Возникает вопрос о механизме нитрозирования и особенно о том, какие
из носителей нитрозоний-иона реагируют в условиях кинетического исследо-
вания. A priori можно признать, что следует рассмотреть такой ряд:
NO+, H2NO+, N2O4i N2O3 и HNOa
283
Глава VI
Хотя в этом ряду свободный нитрозоний-ион наиболее реакционноспособный,
тем не менее и другие реагенты также могут присутствовать и приобретать
преобладающее кинетическое значение.
В разд. 5,е мы отмечали, что в органических растворителях, содержащих
избыток азотной кислоты, аналитически определяемая азотистая кислота
присутствует главным образом в виде N2O4. Этот смешанный ангидрид пере-
ходит в другие частицы, присутствующие в растворе в небольших количе-
ствах, посредством следующих равновесных реакций:
n2o4
N2O4 -р н2о
NO+4-NOj
HNO2 + HNO3
N2O4-f-H2O
2N2O4 + H2O
H2NO+ + NO3
N2O3+2HNO3
Нитрозирующие агенты, очевидно, можно идентифицировать путем изуче-
ния влияния добавок нитрат-ионов и воды на константу скорости реакции
второго порядка к2. Небольшие добавки воды не оказывают влияния,
но относительно малые добавки нитрат-ионов заметно уменьшают константу
скорости. Это означает, что только первое из четырех написанных выше урав-
нений регулирует концентрацию нитрозирующего агента. Математическая
форма зависимости константы скорости от концентрации нитратных ионов
имеет вид
А2 = AJ-J-Zc^NOg]-1
Это выражение показывает, что каталитическая скорость слагается из кон-
станты к'2, которая соответствует атаке молекулами N2O4 на ароматические
соединения, и константы к2, обусловленной атакой ионами NO+, которые,
хотя и присутствуют в значительно меньшей концентрации, гораздо более
реакционноспособны, чем преобладающие в растворе молекулы N2O4 [164].
Остается невыясненным вопрос, почему положительный катализ азотистой
кислотой играет роль лишь при нитровании ароматических соединений,
содержащих такие заместители, как OR и NR2. Можно полагать, что при-
чина заключается не в прямом участии этих заместителей, а в том, что аро-
матические ядра в этих системах чрезвычайно реакционноспособны по отно-
шению к электрофильным реагентам. Поэтому для получения мононитро-,
а не полинитропроизводных, необходимо придерживаться условий, при кото-
рых нитроний-ион образуется с трудом, в результате чего открывается воз-
можность реакции с менее активными (см. разд. 4, д), но и имеющимися
в гораздо большем количестве носителями нитрозоний-иона. Два явления
могут служить дополнительными доводами в пользу этих представлений.
Ro-первых, соединения типа ArOR и ArNR2 могут вести себя по отношению
к нитроний-иону так же, как и другие ароматические соединения, если,
как уже отмечалось, исключается конкуренция с носителями нитрозоний-
иона. Во-вторых, установлено, что ароматические соединения иного типа,
чем ArOR и ArNR2, например ароматические углеводороды, при достаточной
реакционной способности ядра, обусловленной замещением, ведут себя при
нитровании подобно фенолам и аминам. Так, например, при нитровании
мезитилена азотной кислотой в органических растворителях азотистая кис-
лота оказывается сильным положительным катализатором. Кинетические
исследования подтвердили, что при нитровании как и-хлорани.зола и подобных
ему соединений, так и мезитилена протекают два конкурирующих процесса,
284
Замещение в ароматическом ряду
каждый из которых при определенных концентрациях может быть домини-
рующим, и что оба эти процесса подчиняются одинаковым кинетическим
уравнениям *.
6. Механизмы других реакций электрофильного
ароматического замещения
а. Хлорирование, бромирование и иодирование. Различные галогени-
рующие агенты можно рассматривать как «переносчики» галогеноний-иона
На1+; один из наиболее интересных вопросов, касающихся механизма гало-
генирования ароматических соединений, заключается в том, чтобы выяснить,
протекает ли замещение через стадию предварительного образования сво-
бодного галогений-иона или же галоген передается ароматическому ядру
переносчиком, например гипогалогенацидиевым ионом НаЮН*, элементар-
ным галогеном На12, гипогалогенной кислотой НаЮН или даже полигало-
генными анионами типа Hal^.
В работах Нойеса и Штиглица [166] был впервые поставлен вопрос о суще-
ствовании галогений-иона На1+. В поддержку существования иодиний-иона 1 +
выступил Льюис [167], который показал, что жидкий иод обладает значитель-
ной электропроводностью. Одно время полагали [1681, что голубое парамаг-
нитное вещество, образующееся при добавлении окислителей (йодата или
персульфата) к раствору иода в олеуме, представляет собой 1+. Однако Гил-
леспи и Майлн [169] с помощью взаимно подтверждающих физических мето-
дов показали, что в этих условиях образуется ион 1+ с нечетным числом
электронов. Они обнаружили также небольшое количество I+, но не обнару-
жили 1+. Для других галогенов, однако, такого прямого подтверждения
не было получено.
При изучении кинетики электрофильного галогенирования Сопер и Смит
[170] в 1926 г. открыли два переносчика хлора, а именно хлорноватистую
кислоту и молекулярный хлор. Они изучили скорость хлорирования в водной
среде фенола гипохлоритом в определенном интервале рН^и получили сле-
дующее кинетическое уравнение:
Скорость-Л[С1ОН][СвН5О~] (1)
Альтернативное выражение, в которое в качестве реагентов входил бы
CIO- и СвН5ОН, хотя и согласуется с кинетикой, но противоречит электро-
фильной природе замещения и поэтому рассматриваться не будет. Те же
авторы обнаружили, что скорость реакции увеличивается при использовании
хлорноватистой кислоты, которая количественно образует элементарный
хлор. В этом случае реакция описывается уравнением (2):
Скорость == fc[Cl2][C6H5O-] (2)
* Распространенное мнение, что нитрование производных фенила и анилина в ядро
обусловлено прямым взаимодействием между боковыми цепями и нитрующим агентом,
вероятно, связано с часто наблюдаемым отщеплением одной алкильной группы при нитро-
вании эфиров фенола, а также диалкиланилинов. Однако изучение этих реакций привело
к заключению, что они протекают в конкуренции или вслед за нитрованием в ядро, опре-
деляющим скорость процесса. Существуют два механизма отщепления. Один из них —
неокислительный, реализуемый преимущественно в случае эфиров фенола с отщеплением
алкильных групп, главным образом в виде карбониевых ионов R+, так как при примене-
нии в качестве растворителя уксусной кислоты ион карбония связывается в виде эфира
ROCOCH3, другой — окислительный механизм, характерный для диалкиланилинов,
при котором алкильная группа, по-видимому, отщепляется в виде радикал-катиона
(например, CHJ), поскольку он может быть обнаружен в виде альдегида. Детальнее этот
вопрос рассмотрен в работах Бантона, Хьюза и Ингольда [165].
285
Глава VI
Эта работа показывает, что молекулярный хлор — гораздо более эффектив-
ный переносчик хлора при хлорировании, чем молекулярная хлорноватистая
кислота.
В 1950 г. кинетика ароматического хлорирования в водной среде была вновь
исследована де ла Маром, Хьюзом и Верноном [171]. Эти авторы изучили
хлорирование ароматических соединений водной хлорноватистой кислотой,
подкисленной хлорной или серной кислотами ине содержащей хлорид-ионов,
а следовательно, и молекулярного хлора, от которых освобождались добав-
лением ионов серебра. Такая система обладала сильным хлорирующим
действием; Дербишайр и Уотерс [172], а также де ла Мар и сотр. [173] пока-
зали, что в случае не очень реакционноспособных ароматических соединений,
например толуол-со-сульфокислоты, бензола и толуола, кинетика хлориро-
вания в этой системе соответствует следующему уравнению:
Скорость = Л[АгН][СЮН][Н+] (3)
Если бы больше ничего не было известно, то можно было бы предположить,
что хлорирующей частицей в данном случае является СЮН* или ион хло-
риния С1+, однако результаты исследований показали, что приемлема лишь
последняя интерпретация. Де ла Мар, Хьюз и Вернон [171, 173] постепенно
увеличивали реакционную способность ароматического субстрата до тех
пор, пока фактор [АгН] не исчез из выражения для скорости и последнее
превратилось в уравнение
Скорость = &[С1ОН][Н+] (4)
Примерами соединений, скорости хлорирования которых не зависят от их
концентрации, являются анизол, диметиловый эфир гидрохинона и фенол.
В работе [171] было также установлено, что указанные растворы хлорнова-
тистой и хлорной кислот могут быть применены для присоединения элементов
хлорноватистой кислоты к олефинам и что реакционноспособные олефины,
например аллилфторид и аллилэтиловый эфир, вступают в реакции присоеди-
нения со скоростями, не зависящими от концентрации и природы олефинового
субстрата. (Более того, скорость присоединения к олефинам была такая же,
как и скорость замещения в ароматических соединениях.
Кинетические явления, отображенные уравнениями (3) и (4), абсолютно
тождественны явлениям, обнаруженным в случае образования нитроний-иона
и нитрования им. Следующий за предравновесной протонизацией молекулы
хлорноватистой кислоты медленный и поэтому определяющий энергию акти-
вации чисто неорганический процесс разрыва связи в неорганической моле-
куле обусловливает появление хлорирующего агента. Де ла Мар. Хьюз
и Вернон [171] представили этот процесс следующим образом:
СЮН + НзО+ С1ОН2+ + Н2О
Быстро
Ci-Qw------> С1+ + Н2О
Медленно
(5)
Как указал Миллен (частное сообщение), не следует удивляться тому, что
катионы С1+ и СЮЩ могут сосуществовать в водных растворах в качестве
медленно взаимопревращающихся частиц, хотя по строению один из них
является гидратом другого. Свободный ион хлориния по правилу Гунда
(гл. I, разд. 3, б) должен находиться в триплетном состоянии с двумя элект-
ронами на разных орбиталях с параллельными спинами (подобно атому кис-
лорода; табл. 2). Таким образом, он пе может непосредственно координи-
286
Замещение в ароматическом ряду
роваться с молекулой воды. Чтобы такая координация стала возможной,
ион хлориния должен перейти в состояние, более богатое энергией, в котором
все электроны имеют спаренные спины, а одна из Зр-орбиталей не занята.
Энергия возбуждения служит источником энергии активации гидратации,
однако последняя несколько меньше, благодаря действию адиабатического
принципа Лондона (гл. II, разд. 3, г).
Такое объяснение не является общепризнанным, так как существуют рас-
четы [174], предпринятые для того, чтобы показать, что свободная энергия,
необходимая для образования иона хлориния, настолько велика, что этот
катион не может образоваться. История повторяется: именно на основании
очень большой вычисленной энергии, необходимой для образования карбо-
ниевых ионов, концепцию карбопиевых ионов не принимали в 1930-х годах,
так же как не принимали теорию диссоциации Аррениуса в 1890-х годах
и позднее. Вероятно, как и в указанных двух случаях, в конце концов дело
будет улажено учетом энергии сольватации.
Трудно предложить альтернативное правдоподобное объяснение указанных
кинетических закономерностей. Первая попытка в этом направлении [175] —
предположение о медленном переносе протона с образованием в качестве
хлорирующего агента НОС1Н+ — оказалась неудачной, вследствие того что,
во-первых, НОС1Н+ не является хлорирующим агентом и, во-вторых, в этом
случае скорость образования хлорирующего агента замедлялась бы в тяже-
лой воде. Свейн и Кетли [176] показали, что в D2O реакция ускоряется
в 2 раза, что соответствует нормальному ускорению реакции предравновес-
ного переноса протона [уравнение (5)]. Второе предположение [177], заклю-
чающееся в допущении медленного переноса положительного хлора
от CIOHJ к AgCl с образованием в качестве хлорирующего агента иона AgCl*,
также можно отвергнуть [178], поскольку строение хлористого серебра
и в растворе, и в твердом состоянии не соответствует формуле AgCl, содер-
жащей только ионы серебра и хлора, и скорости реакций хлорирования,
имеющих нулевой порядок по субстрату, практически не зависят от присут-
ствия в небольших концентрациях ионов серебра в растворе.
Де ла Мар, Хьюз и Вернон [171, 173] нашли, что в случае наиболее реак-
ционноспособных субстратов концентрация субстрата снова начинает вхо-
дить в выражение для скорости, т. е. кинетика реакции частично или пол-
ностью вновь начинает соответствовать уравнению (3). Авторы объяснили
полученные данные, предположив, что высокоактивные субстраты отнимают
хлор непосредственно от протонированной хлорноватистой кислоты, не дожи-
даясь, пока произойдет медленное образование иона хлориния. Это явление
наблюдалось для ряда простых эфиров фенола; аналогично происходит при-
соединение хлора к некоторым реакционноспособным олефинам, например
к изобутилену.
Существуют еще два способа определения природы переносчика ионного
хлора. Первый связан с тем обстоятельством, что способность сильной кис-
лоты отдавать протон слабому основанию, растворенному в воде, является
функцией кислотности системы. Гаммет назвал эту функцию при низкой
концентрации кислоты h0 = [Н+], однако выше определенной концентрации
кислотность повышается более круто, чем ]Н+]. Таким образом, скорость
реакции, включающей предравновесный захват протона, должна изменяться
симбатно с изменением h0, но не [Н+] (когда эти величины не равны). На
каком бы примере это ни проверялось, всегда оказывалось, что в случае,
когда h0 и [Н+] сильно отличаются, в приведенном выше кинетическом урав-
нении множитель [Н+] нужно заменить на h0 [173, 179]. Таким образом,
подтверждается первая стадия уравнения (5).
287
Глава VI
Другой способ (разд. 2,6 и в) состоит в определении орто/шгра-соотношения
при взаимодействии ароматических углеводородов, в особенности толуола
и mpem-бутилбензола, с агентами различной электрофильности — катионом
хлора (или брома) и молекулярным хлором (или бромом). Наблюдаемые
соотношения нельзя было объяснить, если бы катионный хлорирующий
агент, для которого предполагается формула С1+, на самом деле имел бы
строение НОС1Н+ или AgClJ- Таким образом, подтверждается справедливость
второй стадии уравнения (5).
Итак, при хлорировании в водном растворе возможно образование четырех
электрофильных переносчиков хлора. Их ожидаемая активность уменьшается
в ряду
С1+, C1OHJ, С1-С1, С1-ОН
В неводных средах наиболее активным хлорирующим агентом является
молекулярный хлор, однако и другие переносчики играют важную роль.
Хорошо известен ацетат хлора С1ОСОСН3, образующийся в уксусной кислоте
из окиси хлора или из хлора и ацетата ртути [180].
Френсис [181] установил, что бромная вода бронирует л-нитрофенол
в 1000 раз быстрее, чем эквимолярный раствор бромноватистой кислоты
при pH 3. Френсис ошибочно предположил, что свободный ион Вг+ может
быть эффективным реагентом в бромной воде. Шилов и Каняев [182] сделали
важное открытие, что водная бромноватистая кислота в присутствии сильных
кислот, например хлорной, становится значительно более активным брони-
рующим агентом, чем бромная вода; для бромирования анизол-л1-сульфокис-
лоты подкисленным водным раствором бромноватистой кислоты Шилов
и Каняев вывели уравнение скорости
Скорость = /с|АгН][НОВг][Н+] (6)
Произведение [НОВг][Н+], очевидно, пропорционально концентрации иона
BrOHJ; поскольку активность воды постоянна, это произведение, как ука-
зали авторы, может служить мерой стационарной концентрации броминий-
иона Вг+. Вильсон и Сопер [183] отметили, что при бромировании Вг2 должна
наблюдаться другая зависимость скорости реакции от концентрации бромид-
иона, чем при бромировании через BrOHJ или Вг+. Исследуя эту зависимость
на примере бромирования Л1-нитроанизола бромной водой, они показали,
что активным агентом в бромной воде является Вг2. При бромировании
.м-нитроанизола подкисленным водным раствором бромноватистой кислоты
они подтвердили уравнение Шилова — Каняева; уравнение Шилова —
Каняева подтвердили также Дербишайр и Уотерс [184] при бромировании
бензилсульфокислоты и бензойной кислоты тем же реагентом. Все исследо-
ватели признают, что данное уравнение указывает на бромирование с уча-
стием BrOHj или Вг+, но не позволяет остановиться на одной из этих воз-
можностей. Бромирование ароматических соединений бромом проводилось
в уксусной кислоте как растворителе [185] и в 70%-ном водном растворе
уксусной кислоты [188]. В этих условиях молекулярный бром, несомненно,
один из эффективных носителей брома, однако пока еще не ясно, играет ли
роль вероятный носитель (ацетилгипобромит) в процессах бромирования
и, если играет, то в чем она заключается.
Имеются данные, позволяющие считать, что в исследованных условиях
катионным бромирующим агентом является Вг+, а не ВгОНз, т. е. что пред-
равновесная стадия включает дегидратацию ВгОН2.
BrOH4-H3O+ BrOHt-f-H2O
BrOHf Br+ + H2O
288
Замещение в ароматическом ряду
Энергетический барьер между BrOHJ и Вг+, вероятно, должен быть меньшим,
чем между CIOHJ и С1+, так как энергия перехода триплет — синглет, безус-
ловно, должна быть значительно ниже для 4р-электронов брома, чем для
Зр-электронов хлора.
Одно из доказательств бромирования посредством иона Вг+ можно получить,
исследуя влияние кислотности среды. Снова необходимо учесть то обстоя-
тельство, что скорость реакции, включающей предравновесную стадию пере-
носа протона [уравнение (5) и (7)], в том случае, когда это единственное
предравновесие, влияющее на кинетику данной реакции, должна возрастать
с увеличением кислотной функции Гаммета h0 (при таких концентрациях
кислоты, когда h0 увеличивается быстрее, чем [Н+]). Голд предложил еще
одну меру кислотности — функцию j0. При переходе от низких концентра-
ций кислоты к высоким эта функция вначале изменяется как [Н+], затем
как h0 и, наконец, при еще более высоких концентрациях — даже более
круто, чем /г0; /0 характеризует кислотность среды в том смысле, насколько
она влияет на скорость реакции, связанной как с переносом протона, так
и с дегидратацией, например две стадии уравнения (7). Дербишайр и Уотерс
[184] провели кинетическое исследование бромирования бензойной кислоты
в сильнокислых растворах бромноватистой кислоты и показали, что скорость
линейно зависит от h0. Однако при очень высоких концентрациях кислоты
наблюдалось отклонение от линейной зависимости в сторону j0. Чтобы окон-
чательно доказать существование зависимости скорости от ;0, необходимо
провести измерения при еще большей кислотности среды.
Второй аргумент аналогичен аргументу, примененному для случая хлори-
рования; согласно интерпретации, данной в разд. 2, б и в, соотношение орто-
и тгара-изомеров в случае толуола и шреш-бутилбензола не выдерживалось
бы, если бы бронирующим агентом являлся не Br+, a BrOHJ-
Итак, в водной среде активность бронирующего агента снижается при пере-
ходе от катионной формы брома, которая, вероятнее всего, является ионом
броминия Вг+, к молекулярной форме галогена Вг2. В бромной воде и раст-
ворах брома в неводных растворителях наиболее важным бронирующим
агентом является молекулярный бром.
При иодировании создается до некоторой степени аналогичное положение.
Скорость иодирования фенола в водном растворе при различных значениях
pH изучалась Сопером и Смитом [187] в 1927 г. Они установили, что скорость
иодирования может быть выражена уравнением
Скорость = MIOH]]CeH5OH] (8)
Выдвинутая в то время интерпретация заключалась в том, что молекулярная
иодноватистая кислота атакует молекулу фенола, причем пришлось сделать
неожиданный вывод, что иодноватистая кислота —- более сильный иодирую-
щий агент, чем молекулярный иод. Впоследствии Пэйнтер и Сопер [188]
показали, что приведенное выше уравнение можно изобразить в следующем
виде:
Скорость = Л'[1ОЩ][СвН5О-] (9)
В такой форме уравнение можно истолковать, как указание на то, что ион
IOHJ атакует фенолят-ион. Далее, поскольку активность воды постоянна,
то соотношение иодиний-ион, если он имеется, и lOHJ постоянно, и, следо-
вательно, уравнению можно придать и такой вид:
Скорость = /с''[1+][С6Н5О~] (10)
Данное уравнение позволяет сделать вывод, что иодиний-ион атакует фено-
лят-ион. Однако одна из этих трех возможностей исключается, если учесть
19—0772
289
Глава VI
результаты недавно произведенных Берлинером [189] исследований скорости
иодирования анилина в водной среде. Кинетика этих процессов весьма сход-
на, если принять ион анилиния как аналог молекулы фенола; при отсутствии
других данных результаты кинетических определений можно истолковать
аналогично как следствие взаимодействия любых из трех пар реагентов,
которые обозначены в виде произведений концентраций в следующих экви-
валентных кинетических уравнениях:
Скорость = /c[IOH][CeH5NH3]
Скорость = /c'[IOH+][CeH5NH2]
Скорость /c"[I+][CeH5NH2]
(11)
(12)
(13)
Однако, как известно, иодирование через ион анилиния должно было бы
протекать как леига-ориентирующий процесс, тогда как на самом деле иоди-
рование анилина представляет собой легко протекающий процесс ориго-
тгора-замещения, и, следовательно, допустимы только две последние воз-
можности [уравнения (12) и (13)]. Если для иодирования фенола применимы
уравнения (9) и (10), то в соответствии с теорией скорость реакции фенолят-
иона с иодирующей частицей (1+ или ГОЩ) должна значительно превышать
скорость реакциии этой же частицы с молекулой анилина.
Берлинер изучил также кинетику иодирования анизола и некоторых произ-
водных анилина хлористым иодом в водной хлористоводородной кислоте
[190]. Он вновь сделал вывод, что иодирующей частицей является катионная
форма иода — 1+ или ЮНД
Таким образом, достаточно хорошо установлено, что электрофильным иоди-
рующим агентом служит катионная форма иода. Является ли эта активная
форма ионом иодиния или протонированной иодноватистой кислотой, пока
не установлено, однако по аналогии с бромированием можно предположить,
что в предравновесной стадии образуется свободный ион иодиния 1+, который
и служит наиболее эффективным иодирующим агентом.
Теперь перейдем к механизму взаимодействия галогенирующих агентов
с ароматическими кольцами. Какими доказательствами мы располагаем,
чтобы сделать вывод о том, что атака ионом галогенония напоминает атаку
катионом нитрония (разд. 5, г)? Нужно сказать, что захват иона галогенония
происходит на первой стадии процесса, в результате чего образуется ион
циклогексадиенилия (аддукт Пфейфера — Вицингера), который на второй
стадии теряет протон с гораздо большей скоростью по сравнению со скоростью
его обратного распада на исходные реагенты:
.Hal
НаВ-АгН <> Аг+ф
Медленно Н
.Hal
Аг+Д -----------—> АгНа1-|-11+
'Н Быстро
При этом условии последняя стадия не влияет на скорость галогенирования,
которая, таким образом, определяется или скоростью образования иона
циклогексадиенилия, или скоростью образования иона галогения, когда
этот процесс медленный. Таким образом, если происходит отщепление не
протона, а дейтерона или тритона, это не должно сказываться на скорости
галогенирования. Доказательство быстрого отщепления протона было полу-
чено де ла Мэром, Данном и Харвеем [191] для случая бромирования катион-
ным бромом. Авторы показали, что скорости бромирования бензола и гек-
290
Замещение в ароматическом ряду
(16)
садейтеробензола в кислом растворе бромноватистой кислоты одинаковы, что
согласуется с кинетическим уравнением (6) и схемой (14).
Вероятно, по такому7 же механизму происходит и хлорирование ионом хло-
риния, но иодирование катионом иода может происходить по-другому, так
как ароматическое иодирование в значительной степени обратимо и поэтому
отщепление иона 1+ от катионного аддукта [уравнение (14)] может происхо-
дить быстрее, чем отщепление протона [192].
Ароматическое хлорирование молекулярным хлором в воде [170] и в уксус-
ной кислоте [193] подчиняется кинетическому уравнению (15), приведенному
ниже. Аналогично может протекать бромирование молекулярным бромом
в воде [194] и при достаточном разбавлении (с'Л7/100) — в уксусной кислоте
[195].
Скорость = &[АгН][Х2] (15)
Из этого уравнения следует, что процесс захвата молекулярного галогена
и образование нейтрального циклогексадиенилдигалогенида должен проте-
кать быстро. Дальнейшие стадии, вероятно, описываются схемой (16):
/Х-Х /X
Аг+/ —> Аг+/ -|-Х
ХН Н
,Х
Аг+< —> АгХ + н+
ХН
Как показал Робертсон [196], бромирование бромом в уксусной кислоте при
его высоких концентрациях (до Л//40) является реакцией третьего порядка:
Скорость = &[АгН][Вг2]2 (17)
В случае хлорирования не известно ни одной реакции, подчиняющейся подоб-
ному кинетическому уравнению, поэтому7 можно предположить, что роль
второй молекулы брома состоит не в отщеплении ароматического протона;
участие второй молекулы брома обусловлено сильно выраженной способ-
ностью высших галогенов к самосоединению с расширением валентной элект-
ронной оболочки, особенно при наличии отрицательного заряда, как, напри-
мер, в пергалогенид-ионах. На основании этого Гвин Уильямс предложил
[197] интерпретировать уравнение (17) с помощью следующей схемы:
7Вг—Вг /Вг
Аг+/ +Вг2-----------------> Аг+: +Brj
'Н Медленно '•ц
/Вг
Аг+'------------> ArBr'/'-H*
4II
В еще более концентрированных растворах брома в уксусной кислоте (до
М/5) Робертсон и сотр. [198] наблюдали реакцию четвертого порядка
Скорость = А [АгН][Вг2]3 (19)
Следует отметить, что эффективный порядок по брому повышался при добав-
лении к уксусной кислоте неполярных растворителей, например хлороформа,
и понижался при добавлении воды. Остается неясным, как интерпретировать
уравнение (19): расширением ли полигалогенидной схемы Гвин Уильямса или
предположением об атаке ароматического субстрата первоначально обра-
зующимся димером брома Вг4.
Факторы парциальных скоростей для галогенирования монозамещенных
бензолов приведены в разд. 4, б и 6, к.
(18)
19* 291
Глава VI
Механизм галогенирования фенолов катионными или молекулярными гало-
генами аналогичен механизму реакций, связанных с прототропией (гл. XI)
и в особенности с галогенированием кето-енольных систем (фенол подобен
енолу). Как было показано, галогенирование фенолов в воде протекает через
арилоксидный анион, который быстро образуется, быстро снова протони-
руется и может быстро галогенироваться. Если галогенирование на самом деле
происходит быстро, то самой медленной стадией стадийного процесса гало-
генирования может быть конечная стадия отщепления протона. Это было
доказано Цоллингером и сотр. [199] на примере бромирования G-кислоты
в водной среде. Скорость реакции с подкисленной бромноватистой кислотой
не зависела от pH (что соответствует реакции Вг+ с АгО”) и была равна ско-
рости бромирования элементарным бромом; эта скорость не зависела от кон-
центрации даже при избытке реагента. Очевидно, промежуточный аддукт
образуется быстро и обратимо, как только в растворе появляется бронирую-
щий агент. Этот аддукт был действительно получен в сравнительно высокой
концентрации; время его жизни достаточно велико, что позволило установить
его строение с помощью УФ- и ЯМР-спектров, которое, как и ожидалось,
соответствует броменону:
HO3S
HO3S
G Кислота
он /Q£q- Вг+пл«Вг2
Быстро Быстро
Медленно
(20)
б. Сульфирование. Кинетика ароматического сульфирования была изу-
чена в трех системах, которые будут рассмотрены отдельно. Первая система
представляет собой слегка разбавленную водой серную кислоту. Эта система
впервые изучалась Стаббсом, Вильямсом и Хиншелвудом [200], которые
в качестве растворителя использовали нитробензол, однако полученные
результаты оказались весьма сложными вследствие эффектов обратимости.
Коудрею и Дейвису [201] удалось обойти эти трудности при изучении суль-
фирования re-нитротолуола в серной кислоте (92—99 вес. %) при большом
постоянном ее избытке. Давенпорт и Хыоз (частное сообщение) использовали
нитробензол как растворитель, но проводили сульфирование толуола и о-кси-
лола постоянным избытком различных водных кислот (95—99 вес. %
H2SO4), что исключало обратимость. Результаты последних двух исследова-
телей согласуются между собой и приводят к заключению, что активной
частицей, содержащейся в сульфирующей среде этого типа, является мономер
трехокиси серы или, возможно, его сернокислотный сольват.
Возможными активными частицами, которые уже рассматривались в этой
или в другой связи, являются следующие: 1) ион H3SO+; 2) мономер SO3
или его сольват; 3) ион HSOg, по-видимому, включая его возможные сольваты,
и 4) димер трехокиси серы S2Oe. Известно, что реагенты (1) и (2) присутствуют
в чистой серной кислоте: моляльная концентрация H3SO+ в чистой серной
кислоте, по данным Гиллеспи [202], равна 0,013, а сольвата дисерной кислоты
или мономера трехокиси серы — 0,0083. Реагент (3), как мы увидим, содер-
292
Замещение в ароматическом ряду
жится в олеуме. Реагент (4) встречается в олеуме в виде сольвата трисерной
кислоты.
Все эти возможные реагенты обусловливают замедление сульфирования
при добавлении воды; однако вызываемое при этом замедление должно под-
чиняться различным законам. Находящаяся в серной кислоте вода в очень
большой степени, а в данном случае можно считать, что полностью, иони-
зована в соответствии с уравнением
Н2О + H2SO4 Н3О+ + HSO4
Число ионов воды, которое должно получиться в процессе образования одной
молекулы или иона любого из этих возможных реагентов, как это мож-
но видеть из приведенных ниже уравнений, соответственно равно 1, 2, 3
и 4:
(1) 2H2SO4 = H3SOt + HSO|
(2) 2H2SO4 = SO3 + H3O+-|-HSO4
(3) 3H2SO4 = HSO++H3O+ + 2HSO4-
(4) 4H2SO4 = S2Oe + 2H3O+ + 2HSC>7
Следовательно, скорость атаки ионом H3SO4 обратно пропорциональна пер-
вой степени концентрации воды, SO3 — обратно пропорциональна квадрату,
HSOg — обратно пропорциональна кубу, a S2Oe — обратно пропорциональ-
на четвертой степени концентрации воды. Экспериментальные данные одно-
значны: скорость прямо пропорциональна первой степени концентрации
ароматического соединения и обратно пропорциональна квадрату концен-
трации воды. Как видно из уравнений, влияние добавления ионизующего
гидросульфата можно использовать для выяснения различия между первыми
двумя возможностями и двумя последними; все результаты согласуются с вы-
водом о том, что активной частицей является мономер трехокиси серы. В этих
условиях переходное состояние реакции сульфирования состоит из АгН
и SO3.
Сульфирование в олеуме протекает иначе. Бранд и Хорнинг [203] провели
сульфирование нитробензола, иона фенилтриметиламмония и их н-метиль-
ных и n-галогенпроизводных в олеуме, содержащем 4—10% «свободной»
трехокиси серы. Они обнаружили, что логарифмы констант скоростей этих
реакций первого порядка являются линейной функцией суммы двух лога-
рифмов — логарифма функции кислотности Гамметта h0 (—Но) и логарифма
парциального давления (т. е. термодинамической активности) трехокиси серы;
наклон прямой близок к единице. Было учтено, что среда служит постав-
щиком как трехокиси серы, так и протонов; обе частицы входят в переходное
состояние, которое таким образом имеет состав АгН + SO3 + Н+.
Таким образом осуществляются два механизма сульфирования в зависи-
мости от кислотности среды; при более высокой кислотности в переходное
состояние входит также протон.
(Коудрей и Дейвис)
/\/Н Н-ЗОя+Н* /\/5ОзН _НФ /Ч so н
—- * хн ^=1
' -SO3-H+ +н+ 1
(Бранд и Хорнинг)
293
Глава VI
Сульфирование — обратимая реакция, равновесие которой сдвигается впра-
во при высокой концентрации кислоты; при низкой кислотности происходит
обратный процесс, т. е. десульфирование. Если знать механизм одного направ-
ления обратимого процесса, то из него можно вывести механизм и обратной
реакции, так как для обеих реакций переходное состояние будет одним и тем
же (принцип микроскопической обратимости). Лонг и Поль [204] отметили,
что, приняв механизм Коудрея и Дейвиса, нужно предположить, что
в 92—99%-ной серной кислоте десульфирование происходит без кислотного
катализа. Изучению этого вопроса мешает то обстоятельство, что при таких
концентрациях кислоты равновесие полностью сдвинуто в одну сторону;
однако в более разбавленной серной кислоте, скажем 10—50%-ной, отчетливо
проявляется кислотный катализ этой реакции. Использ>гя данные Крафтса
[205] и свои данные, Голд и Сэтчелл [206], а также Лонг и Поль показали,
что в 15—45%-ной серной кислоте скорость зависит от функции Гамметта /г о,
что указывает на участие протона в предкинетическом равновесии. Голд
и Сэтчелл отметили, что такая зависимость должна наблюдаться в рамках
механизма Коудрея — Дейвиса в том случае, если сульфокислота находится
в растворе в основном в виде сульфонат-иона. При концентрации серной
кислоты выше 50% (lg h0 > 3) сульфокислота переходит из ионной формы
в недиссоциированную, и зависимость от h0 должна исчезать. Используя
данные Пинноу [207] и Лантца [208], Голд и Сэтчелл показали, что при кон-
центрации серной кислоты выше 50 % зависимость скорости десульфирования
от h0 гораздо менее заметна. Впоследствии Фуллер и автор книги установили,
что зависимость скорости десульфирования 2,4-диметоксибензолсульфокисло-
ты в водной хлорной кислоте от h0 при lg h0 = 3 начинает исчезать и почти
полностью исчезает при lg h0 = 4,3 [209]. Таким образом, почти полностью
исчезает кислотный катализ десульфирования.
Об изотопном эффекте водорода при сульфировании в водной серной кислоте
сведений не имеется, однако небольшой эффект наблюдался при сульфирова-
нии в олеуме. При замене водорода в пара-положении на тритий скорость
сульфирования бромбензола олеумом в нитробензоле в качестве растворителя
уменьшается более чем наполовину [210]. Отсюда следует, что энергия пере-
ходного состояния второй стадии механизма Бранда—Хорнинга лишь немно-
го меньше энергии переходного состояния первой стадии. Скорость сульфи-
рования иона n-толилтриметиламмония в растворе олеума, содержащем 7 %
«свободной» трехокиси серы, уменьшается примерно на три четверти перво-
начальной величины, если вместо серной кислоты для приготовления олеума
использовать дидейтеросерную кислоту [211]. Этот факт можно объяснить,
например, тем, что компоненты адденда в механизме Бранда — Хорнинга
первоначально не были соединены друг с другом и собственно перенос протона
к молекуле трехокиси серы осуществляется в процессе присоединения послед-
ней к молекуле субстрата.
Кинетика сульфирования производных бензола трехокисыо серы в нитро-
бензоле как растворителе была изучена Хиншелвудом и сотр. [212]. Наблю-
даемые скорости пропорциональны концентрации ароматического соединения
и квадрату концентрации трехокиси серы. Было сделано заключение, что,
хотя, несомненно, в реакции принимают участие две молекулы трехокиси се-
ры, активной частицей является предварительно образующийся димер S2O0.
Однако, если процесс сульфирования, как и другие процессы ароматического
замещения, имеет две стадии, то соотношение скоростей в апротонном раство-
рителе может быть таким, что общий кинетический эффект взаимодействия
одной мономерной молекулы SO3 в каждой стадии привел бы к наблюдаемому
закону скорости.
294
Замещение в ароматическом ряду
в. Водородный обмен. Качественное изучение введения дейтерия в аро-
матическое ядро с помощью дейтерокислот привело к заключению [213],
что водородный обмен, осуществляемый таким образом, представляет собой
типичное электрофильное замещение. Разумеется, это замещение обратимо
и полностью равновесно, если не считать изотопные различия.
Доказательства таковы. Во-первых, установлено, что дейтерирующие кисло-
ты по силе располагаются в ряд
D2SO4 > D2SeO4 > D3O+ > C6H5OD > D2O
который, очевидно, идентичен ряду кислотности. Как правило, любой член
соответствующего ряда протиевых кислот будет легко передавать протон
сопряженному основанию кислоты, стоящей справа от него, и лишь в малой
степени — сопряженному основанию кислоты, стоящей слева от него.
Во-вторых, ароматические заместители так изменяют реакционную способ-
ность ароматического ядра в отношении таких реагентов, что дают после-
довательность скоростей, идентичную с последовательностью скоростей
хорошо изученных реакций нитрования, хлорирования и азосочетания.
-О> — NR2> — OR> (Н)> — SO3H
Вследствие больших различий не все реагенты можно было использовать
препаративно для дейтерирования производных бензола. Бензолсульфокис-
лота не дейтерировалась серной кислотой в условиях, в которых сам бензол
легко дейтерировался серной и селеновой кислотами. Анизол, анилин и диме-
тиланилин легко дейтерировались водной кислотой, т. е. дейтероксоний-
ионом. Фенол дейтерировался в водном растворе щелочи. Изучение зависи-
мости скорости от pH показало, что фенолят-ион одновременно подвергался
быстрому дейтерированию в результате атаки молекулами фенилдейтерокси-
да и медленному дейтерированию при атаке молекулой дейтероксида, причем
дейтероксид-ионы таким действием не обладали.
В-третьих, было получено подтверждение, что водородный обмен подчи-
няется обычным правилам ориентации при реакциях электрофильного арома-
тического замещения. Оказалось, что фенол, анилин и анизол подвергаются
легкому дейтерированию только в три положения ядра; это число было опре-
делено по суммарному поглощению дейтерия, причем контролировалось
дейтерирование боковой цепи. Бест и Вильсон [214] формально доказали
замещением дейтерия на бром в случае фенола и анилина, что этими тремя
обменивающимися положениями являются орто- и гецра-положения. Даль-
нейшее доказательство [215] было дано для анилина отщеплением амино-
группы после дейтерирования, а затем спектроскопически было показано,
что полученный бензол является чистым 1,3,5-тридейтеробензолом.
В 1950-х годах было выявлено, что интерпретировать механизм переноса
протона от кислоты к ароматическому субстрату не так просто. Дело в том,
что скорости ароматического водородного обмена приблизительно соответ-
ствуют йо-гамметовской мере способности среды к равновесной передаче
протона слабому электронейтральному основанию. Существование такой
корреляции привело к выводу, что перенос протона осуществляется в пред-
равновесной стадии и поэтому является полным; протон утрачивает связь
со своим первоначальным носителем и приобретает связь с реагирующей
системой до того, как последняя достигнет переходного состояния. Пере-
ходное состояние в этом случае должно иметь состав: субстрат плюс протон,
но не должно содержать кислотного остатка, от которого отщепился протон.
Вот почему трудно было представить, что же является медленной стадией
водородного обмена, т. е. когда система проходит через переходное состояние.
295
Глава VI
Невозможно придумать два взаимопревращающихся изомера протонирован-
ного ароматического соединения, медленный процесс внутримолекулярного
переноса протона между которыми определял бы скорость реакции. Эта
теория никогда не считалась пригодной. Выражение для скорости реакции
при предполагаемой выше структуре переходного состояния должно вклю-
чать, кроме концентрации субстрата, только концентрацию ионов водорода
(как и при специфическом катализе ионами водорода) и не должно включать
концентрации недиссоциированной кислоты, которая служит источником
протонов. Однако даже в ранних исследованиях ароматического водород-
ного обмена с кислотами было показано, что не только ион водорода, но
и каждая кислота, присутствующая в смеси кислот, вносит собственный вклад
в скорость реакции (как при общем кислотном катализе). Например, в выра-
жение для скорости обмена в присутствии фенолят-иона важный вклад вносит
член, содержащий концентрацию недиссоциированных молекул фенола,
действующего как кислота. Непосредственное участие недиссоциированной
кислоты в ароматическом водородном обмене было вновь и гораздо более
полно показано в 1959 г. Кресге и Чиангом [216] на примере 1,3,5-тримето-
ксибензола и в 1960 г. Колапиетро и Лонгом [217] на примере азулена. Эти
исследования показали, что в буферных растворах молекулы недиссоциирован-
ной кислоты вносят существенный вклад в скорость обмена. Отсюда следует,
что в переходное состояние обмена входит вся молекула кислоты, от которой
и происходит отщепление протона. Другими словами, это означает, что мед-
ленной стадией является собственно перенос протона, что можно предста-
вить следующей схемой (для введения дейтерия):
„Н
ArH + DX “Д Аг+< +X“T±ArD+HX
XD
Причины первоначальных заблуждений были вскрыты Паннеттом при изу-
чении факторов, контролирующих кислотность. Функция Гаммета /г0, опре-
деляемая измерением равновесного содержания соответствующих протониро-
ванных азотистых оснований, удовлетворительно применима для большинст-
ва азотистых и кислородных оснований, но неприменима к углеродным
основаниям. То, что она оказалась применимой к частичному переносу
протона в переходном состоянии водородного обмена у атома углерода
ароматических углеводородов, следует рассматривать как чистую случай-
ность [218].
Образование солей циклогексадиенилия АгН*Х~ (см. приведенную выше схему)
было доказано измерением электропроводности ароматических углеводоро-
дов в жидком фтористом водороде [219], понижением давления паров трех-
фтористого бора в растворе фтористого водорода, содержащем ароматиче-
ские соединения, и опытами по распределению этих соединений между
«-гептаном и смесью фтористого водорода и трехфтористого бора [220]. Опыты
по экстракции позволили оценить относительную основность ароматических
углеводородов. В табл. 80 приведены данные по относительной электропро-
водности и относительной основности ароматических соединений (без поправ-
ки на различную растворимость неионизованных углеводородов) [221].
Очевидно, что метильные заместители способствуют захвату протона и обра-
зованию соли ArHJBF-, особенно находясь в орто- и пара-положениях,
т. е. метильные группы в лета-положении друг к другу взаимно усиливают
свое влияние. Из данных для гексаметилбензола следует, что протон может
присоединяться не только к углероду, связанному с водородом, но и к угле-
роду, связанному с метильной группой. При переходе от толуола к высшим,
гомологам основность настолько увеличивается, что соли циклогексадие-
296
Замещение в ароматическом ряду
Таблица 80
Относительная электропроводность в HF, относительная основность в HF — BF3
и относительные скорости водородного обмена в CF3COOH для некоторых
ароматических углеводородов
Углеводород Электропровод- ность в HF Основность в HF — BF3 Скорость водо- родного обмена В CF3COOH
Бензол 0,0012
Толуол 0,20
ге-Ксилол 1 1 1
о-Ксилол 1,1 2 1,2
л-Ксилол 26 20 37
Псевдокумол(1,2,4) 63 40 100
Гемимеллитол(1,2,3) 69 40 127
Дурол(1,2,4,5) 140 120 330
Пренитол(1,2,3,4) 400 170
Мезитилен(1,3,5) 13 000 2 800 11 300
Изодурол (1,2,3,5) 16 000 6 500 30 000
Пентаметилбензол 29 000 8 700 32 000
Гексаметилбензол 97 000 89 000
нилия можно выделить при —80 °C в виде желтых или оранжевых твердых
соединений [222]. При более высоких температурах (от —70 до —10 °C)
они превращаются в расплавы, обладающие электропроводностью, и затем
медленно разлагаются. При разложении такой соли, полученной из ме-
зитилена, фтористого дейтерия и трехфтористого бора образуется 60% фтори-
стого водорода и 40% фтористого дейтерия. Такой первичный изотопный
эффект дейтерия свидетельствует о том, что дейтерон связывается с субст-
ратом в степени, сравнимой со степенью связанности собственного протона
ароматического субстрата (см. формулы, приведенные ниже). На примере
системы пентаметилбензол — фтористый водород — трехфтористый бор с по-
мощью ядерного магнитного резонанса было показано наличие метиленовой
группы (см. нижеприведенные формулы) [223].
Клит и Лангсет [224] первые применили для ароматического водородного*
обмена систему Фриделя — Крафтса. Они препаративно продейтерировали
бензол, используя хлористый дейтерий и хлористый алюминий, а при по-
вторной процедуре получили гексадейтеробензол.
В табл. 80 содержатся данные Лауэра и Стедмана [225] по относительным
скоростям дейтерирования ароматических углеводородов в трифторуксусной
кислоте, содержащей окись дейтерия. Примечательно, что скорость дейте-
рирования изменяется в том же порядке, что и равновесная степень прото-
нирования.
297
Глава VI
Степень активации или дезактивации заместителями различных положений
бензольного кольца иллюстрируется факторами парциальных скоростей,
приведенными на следующей схеме:
СН3
I
/''83
\/1,9
83
ОСН3
I
//,23 ООО
Jo,25
55 000
СН3
!
/ХС250
\/4
420
трет-С^Нд
I
//,200
\/7
490
Н па D, води. H2SO4 или води. НС1О4 [226]
D па Н, CF3COOD [227]
трет-С^Нд
I
/J170
. /9
\/
180
С1
i
//,0,035
Jo, 002
0,160
I
I
//.0,043
х Jo, 003
0,112
Н на Т, водн. H2SO4 или водн. СН3СООН— H2SO4 [228]
Влияние заместителей аналогично их влиянию при нитровании и галоге-
нировании (разд. 3,6 и в и 4,6). Пространственные затруднения орто-
обмену, вероятно, незначительны, даже при обмене с трифторуксусной кис-
лотой, когда замещающим агентом может быть молекулярная форма этой
кислоты; в водных кислотах, в которых переносчиком протона является Н3О+,
эти затруднения проявляются в еще меньшей степени. Отметим дезактивацию
ж/м-положепий в анизоле: она согласуется с классификацией метоксила
как —I, Ч-А-заместителя (разд. 3,с и рис. 24 в первом издании этой книги,
которое вышло еще до появления прямых экспериментальных доказательств).
г. Алкилирование. Эти реакции замещения обычно происходят под
действием алкилгалогенидов и катализаторов Фриделя — Крафтса. Послед-
ние представляют собой электронодефицитные галогениды, например хло-
ристый алюминий, а вообще говоря, галогениды бора, алюминия, титана,
железа, цинка, галлия и т. д., которые имеют склонность образовывать ком-
плексные галогениды. Кроме того, алкилирование может быть проведено
действием олефина, катализатора Фриделя — Крафтса и так называемого
сокатализатора, в качестве которого служит протонная кислота; в результате
реакции образуется или галогеноводород, или вода. В принципе в каждой
из этих систем может образовываться ион алкилия
СН3СН2С1+А1С13 —> СН3СН.®4-А1С1г
СН2 = СН24-НС1 + А1С1з CH3CHJ+AICI7
Обычно предполагается, что частицей, атакующей и алкилирующей арома-
тическое кольцо, является ион алкилия — свободный или в виде ионной пары.
Эта идея была высказана в 1927 г. Меервейном [229]. В настоящее время
получено огромное число доказательств в пользу этой идеи, хотя еще не под-
тверждено, может ли образование карбониевого иона быть стадией, контро-
лирующей скорость алкилирования.
Хлористый алюминий нерастворим в малоосновных растворителях — алка-
нах и сероуглероде, слабо растворим в бензоле и низших аренах и хорошо
растворим в основных растворителях — эфире, кетонах, нитрилах * и ни-
тросоединениях. Бромистый алюминий растворим во всех растворителях.
* Автор, по-видимому, имеет в виду л-основпость.— Прим, перев.
298
Замещение в ароматическом ряду
В растворе эти галогениды, вероятно, находятся в одинаковых состояниях:
в алканах и сероуглероде — в виде А12Х6, в низших аренах — в виде окра-
шенных л-комплексов или комплексов с переносом заряда арен • • -A^CIb,
связь в которых подобна связи между бензолом и пикратом, в основных
растворителях — в виде высокополярных координированных аддуктов,
например (С2Н5)2ОА1С13 (гл. III, разд. 1,а). Аналогично ведут себя соот-
ветствующие галогениды галлия. Эти различные формы в растворах галоге-
нидов алюминия и галлия могут рассматриваться как различные источники
образования активных частиц, т. е. мономерных некоординированных А1Х3
и GaX3. При добавлении алкилгалогенидов немедленно устанавливается
равновесие, в рез}тльтате чего образуется диполярный координационный
+ — -1-
аддукт RX — А1Х3 или RX — GaX3. Когда галогенид металла МХ3 перво-
начально слабо закомплексован (М2Хв или арен •••М2Хв), новое равновесие
+ —
.может сдвигаться в сторону нового комплекса RX • • • МХ3; это следует из изме-
рений давления паров RX. Если, однако, первоначальный комплекс прочен,
как в основных растворителях, положение нового равновесия может быть
сдвинуто в сторону старого комплекса. Все эти процессы осуществляются
быстро. Затем происходят дальнейшие изменения — медленно в случае
метил- и первичных алкилгалогенидов, но гораздо быстрее в случае вторич-
ных и третичных алкилгалогенидов. Эти изменения могут быть зарегистри-
рованы с помощью перехода изотопной метки при обмене галогена в смеси
алкилгалогенида и галогенида металла [230]. Может осуществляться обмен
и разных галогенидов, например хлора и брома; при участии галогенидов
алюминия обмен галогена может происходить между двумя алкилгалоге-
нидами с разными алкильными группами и разными атомами галогена.
Наиболее просто эти процессы обмена галогена можно объяснить, предполо-
жив ионизацию координированного аддукта по схеме
— + —
Растворитель с основными свойствами*• • • AlX3-]-RX RX—А1Х3 R+ + A1Xj
Галогениды алюминия и галлия склонны к самоассоциации, подобно трехоки-
си серы и метафосфорной кислоте; и поэтому в некоторых условиях в выше-
приведенное уравнение необходимо добавить еще одну молекулу А1Х3,
например
Арен- • • А12Х6+ RX RX —Al2Xe R-j-A^Xy
Мы уже рассматривали обстоятельства, при которых в первом из приведенных
уравнений равновесие должно быть сдвинуто влево или вправо. О втором
равновесии мало что известно. Можно ожидать сдвига равновесия вправо
при увеличении стабильности карбониевых ионов в ряду метил-катион,
первичный, вторичный, третичный алкил-катионы (см. гл. VII). Оно должно
сдвигаться вправо при переходе к высшим пергалогенполиалюминатам или
полигаллатам. Равновесные смеси обладают малой электропроводностью;
этого и следует ожидать в любом случае, так как в применяемых растворите-
лях ионы существуют почти полностью в виде ионных пар.
При добавлении к этой системе ароматического углеводорода происходит
типичная реакция алкилирования по Фриделю — Крафтсу. Алкилирова-
ние — процесс обратимый. При протекании прямой реакции образуется
галогеноводород, который является агентом дезалкилирования и водородно-
го обмена. Дезалкилирование подобно водородному обмену; как мы уже
отмечали (разд. 6,в), кислотная система Фриделя — Крафтса может при-
299
Глава VI
соединять протон не только к незамещенному, но и алкилзамещенному
ароматическому атому углерода. Такое рассмотрение помогает описать меха-
низм алкилирования. Из принципа микроскопической обратимости следует,
что ион циклогексадиенилия, образование которого в качестве промежуточ-
ного соединения было установлено при изотопном обмене, в частности, для
случая алкилпроизводпых, должно быть общим как для алкилирования,
так и для дезалкилирования
RX — А1Х3
п
R+AlXj
,R ( H+AlXp
т +1 И +А1Х"
I (НХ + А1Х3
Во многих системах Фриделя — Крафтса, например содержащих бромиды
алюминия или галлия, алкилирование ароматических соединений происхо-
дит в гомогенном растворе как в неосновных, так и в основных растворителях.
Однако в системах, содержащих трехфтористый бор и фтористый водород,
а также хлористый алюминий и хлористый водород, если в качестве рас-
творителей использовать углеводороды или другие неосновные соединения,
образуются два жидких слоя. Один из этих слоев является кислотно-солевым
и содержит все взятое количество бора или алюминия, именно в этом слое
происходит реакция. Второй слой — органический, он содержит органиче-
ские реагенты и продукты реакции, если они не слишком сильно основны.
Способность арена переходить в кислотно-солевой слой зависит от основности
арена, а также от температуры, так как при повышении температуры умень-
шается солеподобная сольватация арена, и последний возвращается в угле-
водородный слой (частное сообщение Д. Пирсона).
Возвращаясь к механизму реакции, отметим, что имеется препаративное
доказательство предполагаемого промежуточного образования циклогексадие-
нилия [231]. При алкилировании толуола, л-ксилола и мезитилена фтори-
стым метилом, этилом, изопропилом и щреш-бутилом в присутствии в каче-
стве катализатора трехфтористого бора при низких температурах Ола и Кун
получили ряд тетрафторборатов соответствующих продуктов присоединения,
которые представляют собой твердые вещества желтого, оранжевого или
красного цвета. При температурах между —110 и —50 °C из этих солей обра-
зуются проводящие ток расплавы, которые при дальнейшем повышении
температуры разлагаются с образованием алкильных производных арома-
тических соединений. Деринг и сотрудники показали, что конечным продук-
том полиметилирования бензола хлористым метилом при катализе хлори-
стым алюминием является довольно стабильный тетрахлоралюминат гепта-
метилциклогексадиенилия. В воде, если среда не очень кислая, от катиона
отщепляется протон и образуется метиленгексаметилциклогексадиен. Депро-
тонирование обратимо, а образующийся олефин настолько основен, что его'
можно «экстрагировать» 4 н. водной соляной кислотой, т. е. регенерировать
первоначальный катион.
300
Замещение в ароматическом ряду
Отличительной особенностью алкилирования по Фриделю — Крафтсу являет-
ся то, что эта реакция сопровождается изомеризациями. Изомеризации,
конечно, происходят вследствие обратимости реакции, т. е. вследствие чере-
дующегося дезалкилирования и алкилирования. Такое чередование парал-
лельно приводит к диспропорционированию — передаче алкильных групп
от одной ароматической молекулы к другой: толуол, например, можно пре-
вратить в смесь бензола и ксилолов. Более интересна внутримолекулярная
изомеризация, происходящая часто при низких температурах (частное сооб-
щение Д. Пирсона) и по характеру напоминающая перегруппировку Ваг-
нера — Меервейна; в этих условиях не происходит диспропорционирования.
Эти перегруппировки служат тестом на механизм с участием карбониевых
ионов в качестве промежуточных частиц, так как для их протекания необ-
ходимо, чтобы первоначально образовался карбониевый ион. Они состоят
в смещении водорода или другого заместителя, обычно метильной группы,
•со своими электронами связи от атома углерода, с которым эти группы были
первоначально связаны, к карбониевому центру, при этом место первона-
чальной локализации группы становится новым карбоний-ионным центром
(гл. X). Ксилолы, например, претерпевают внутримолекулярную перегруп-
пировку под действием кислоты, содержащейся в катализаторе и сокатали-
заторе Фриделя — Крафтса. Таким путем можно осуществить изомеризацию
некоторых ксилолов в другие изомеры. Исследование кинетики этой реакции
показало [232], что происходит только 1,2-алкильный сдвиг; это характерно
для перегруппировок Вагнера — Меервейна. Определены четыре константы
скоростей, соответствующие реакциям: орто мета и пара мета, однако
попытка определить еще две константы, соответствующие процессу орто
/ пара привела к выводу, что обе эти константы равны нулю- Карбониевым
моном, в котором по требованию должна происходить перегруппировка,
в данном случае, конечно, является ион циклогексадиенилия, через который
происходит алкилирование по Фриделю — Крафтсу. Ниже приведена схема
лара-леша-изомеризации:
В углеводородных растворителях в присутствии лишь небольших количеств
катализатора Фриделя — Крафтса равновесная смесь ксилола содержит
около 60% л^еша-изомера и по 20% каждого из других изомеров. Однако,
поскольку .и-ксилол гораздо более основен, чем оба других изомера (табл. 80),
равновесие можно «обмануть», используя избыток сильно кислого реагента
Фриделя — Крафтса, который будет селективно связывать наиболее основный
изомер. Таким образом, препаративный выход лг-ксилола можно довести поч-
ти до 100%.
Кинетика алкилирования изучалась Брауном и сотр. [233]. Скорость реак-
ции в случае алкилирования бензола, толуола и .м-ксилола 3,4-дихлорбен-
зилхлоридом и тг-нитробензилхлоридом в нитробензоле в присутствии хло-
ристого алюминия выражалась следующим образом:
Скорость ос [ArH][RCl][AlCl3]
301
Глава VI
Подобным уравнением третьего порядка описываются также реакции бензола
и толуола с бромистым этилом и бромистым алюминием или с метил- и этил-
бромидами и бромистым галлием в 1,2,4-трихлорбензоле в качестве раство-
рителя. Кинетическое уравнение свидетельствует о том, что в этих раствори-
телях в переходное состояние входят оба компонента: RX + МХ3, которые-
в начальном состоянии являлись кинетически независимыми частицами,
вероятно вследствие того, что одна из них образует прочный комплекс с рас-
творителем. Таким образом, приведенное ниже равновесие сдвинуто влево:
— ______________________________________+ —
Основной растворитель4- —МХз-f-RX <— Растворитель-|-RX—МХ3
Однако в углеводородных растворителях кинетическое выражение полу-
чается иным, и смысл его пока не ясен.
То, что алкилирующим агентом в реакции Фриделя — Крафтса служит
одна из форм карбониевого иона (а если это так, то по кинетическим данным
этой формой является ионная пара), доказывается наличием перегруппиров-
ки типа Вагнера — Меервейна в самой вводимой алкильной группе, если,
конечно, ее структура позволяет эту перегруппировку наблюдать. Согласно
теории перегруппировок Вагнера — Меервейна, если алкильная группа изо-
меризуется, то она должна в какой-то момент реакции пройти через стадию
карбониевого иона. Используя этот критерий, можно показать, что карбо-
ниевые ионы образуются из алкилгалогенида и катализатора Фриделя —
Крафтса без участия ароматического субстрата. Отсюда вовсе не следует,
что наблюдаемая перегруппировка Вагнера — Меервейна обязательно будет
происходить до того, как первоначально образующийся карбониевый ион
прореагирует с исходным ароматическим субстратом; будет происходить
изомеризация или нет — это зависит от строения алкильной группы. Так,
н-пропилхлорид при обработке хлористым алюминием дает изопропилхлорид;
при добавлении хлористого дейтерия в продукте реакции дейтерий не обна-
руживается [234]. Таким образом, изомеризация происходит не через элими-
нирование с образованием пропилена, а как перегруппировка Вагнера —
Меервейна, включающая сдвиг водорода из положения 2 в положение 1
и-пропилиевого иона, в результате чего образуется изопропилиевый ион:
СН3СН2СН2С1 CH3CH2CHJ 7» СН3СНСН3 ^7 СН3СНС1СН3
Однако, если алкилировать бензол и-пропилхлоридом и хлористым алюми-
нием, продуктом реакции будет и-пропилбензол. Вероятно, этот первичный
алкил-катион настолько реакционноспособен, что его реакция с бензолом
происходит раньше, чем 2 -> 1-гидридный сдвиг. Если же ослабить связь
атома водорода с углеродом в положении 2, перейти от и-пропил к изобутил-
хлориду, то последнее соединение будет не только перегруппировываться
в mpem-бутилхлорид при обработке хлористым алюминием, но также и при
реакции с бензолом в присутствии А1С13 будет давать трет-бутилбензол.
Аналогично предыдущему случаю, здесь также происходит перегруппировка
Вагнера — Меервейна:
(СН3)2СНСН2С1 (СН3)2СНСН+ 7^ (СН3)2ССН3 (СН3)3СС1
В данном примере гидридный сдвиг происходит значительно быстрее, чем
атака бензольного кольца даже первичным алкил-катионом, так что алкили-
рующим агентом становится более стабильный, хотя и медленнее реагирую-
щий третичный карбониевый ион. Эти примеры иллюстрируют крайние слу-
чаи двух противоположных влияний строения алкильной группы [235] на
время жизни и реакционную способность взаимопревращающихся ионов?
302
Замещение в ароматическом ряду
легкость образования карбониевых ионов увеличивается, но реакционная
способность уменьшается в ряду первичный <; вторичный <; третичный.
Взаимопревращение карбониевых ионов может осуществляться как путем
перехода водорода, так и путем перехода метильной группы; обычно самой
медленной стадией является перемещение водорода или метила от вторичного
атома углерода. Вторичные бутилгалогениды дают смеси втор- и трет-
бутилбензолов, в то время как изобутил- и треиг-бутилгалогениды — только
пгрепг-бутилбензол
,СН3
+ 4" /
СН3СН2СНСН3 —> СН2 —СН
I . „ Хснз
Продукт
+ /сн3
СНз-Сб
хсн3
Агн|
Продукт
Неопентил-, сдагор-изоамил- и третп-амилгалогениды образуют идентичные
смеси emop-изоамил- и пгрепг-амилбензолов
(СН3)3ССН2 (СН3)2ССН2СН2 (СН3)2ССНСН3
|АгН ]АгН
Продукт Продукт
И так далее. По-видимому, вторичные и третичные изомерные карбониевые
ионы находятся в подвижном равновесии друг с другом, и выход продуктов
алкилирования определяется балансом между временем жизни и реакционной
способностью.
Реакционная способность различных положений бензольного кольца при
алкилировании по Фриделю — Крафтсу качественно подобна уже рассмот-
ренной для случая нитрования и галогенирования, за исключением того, что
различие между отдельными положениями при алкилировании проявляется
в меньшей степени. Рассматривая алкилирование и сравнивая его с алкили-
рованием бензола, можно сказать, что толуол слабо активирован, а хлор-
бензол слабо дезактивирован; в обоих субстратах различие между орто-,
мета- и пара-реакционной способностью выражено слабее и больше обра-
зуется леша-изомера, чем при нитровании. Эти данные были получены Брау-
ном и сотр. [236], которые теоретически объясняли их, предположив, что
более активные реагенты менее селективны как по отношению к разным суб-
стратам, так и по отношению к различным положениям одного и того же суб-
страта. Если взять один и тот же субстрат — толуол, то при метилировании,
этилировании и изопропилировании алкилбромидами в присутствии броми-
стого галлия скорость реакции, согласно Аллену и Ятсу [236], увеличивает-
ся в отношении 1 : 14 : 20 000. Вероятно, этот ряд обусловлен или возрас-
танием скорости образования карбониевого иона, или увеличением его равно-
весной концентрации (или двумя факторами вместе), а скорость реакции
более зависит от концентрации карбониевых ионов, чем от скорости его взаи-
модействия с субстратом.
В табл. 81 приведены некоторые данные, полученные Брауном и сотрудни-
ками, по скоростям замещения в производных бензола, отнесенные к бензолу,
а также данные Брауна и Аллена и Ятса по соотношению изомеров, обра-
зующихся при алкилировании производных бензола [236]. Данные Брауна
и сотрудников получены в условиях, указанных в заглавии таблицы, а данные
Аллена и Ятса (цифры, выделенные курсивом) — в различных системах,
изменение которых, однако, мало влияет на соотношение изомеров. Реакции
303
Глава VI
Таблица 81
Относительные скорости и соотношение изомеров при алкилировании
главным образом алкилбромидами и бромистым галлием (Браун, Аллен)
Субстрат Замещение Раствори- тель (25 °C) Относи- тельная скорость (СбНб= 1) Количество изомера, %
орто мета пара
Метилирование 5,7 56, 60 10, 14 34, 26
С6Н5СН3 | Этилирование СбНбСНз 2,5 38, 48 21, 18 41, 34
Изопропилирование 1,8 26, 42 27, 22 47, 36
€6H5F 0,28 43 14 43
СвН5С1 Этилирование С6Н4С12 0,215 42 16 42
С6Н5Вг 0,133 24 22 54
проводили в тщательно контролируемых условиях, чтобы исключить эффекты
изомеризации продуктов замещения.
Ниже приведены факторы парциальных скоростей, полученные Брауном
при этилировании. Данные для толуола качественно нормально согласуются
с влиянием заместителя типа (1) (+/), а для галогенбензолов — с влиянием
заместителей типа (3) (—I, +/Q, которые проявляют аномалию Голлемана.
Однако различие между разными положениями в данном случае гораздо
менее выражено, чем в случае нитрования (разд. 3,6 и в):
СН3
I
6,0
F
I
/\о,36
\/0,12
0,74
С1
I
/Jo,27
^Jo,i6
0,39
Вт
I
/\),ю
\ Jo, 087
0,43
Для ароматического алкилирования можно использовать и другие пути
получения карбониевых ионов. Один из них состоит в дегидратации прото-
нированных спиртов, например бензгидрола
(СвН5)2СНОН + Н+ (СвН6)2СН+ + Н2О
Бетел и Голд [237] показали, что скорость бензгидрилирования мезитилена,
анизола и других ароматических соединений бензгидролом в уксусной кис-
лоте, содержащей серную кислоту, зависит от меры кислотности /о Голда,
характеризующей способность среды поставлять протоны для отщепления
гидроксильного иона от оксисоединения (см. выше). Другой метод, пред-
ложенный Пирсоном и сотр. [238], состоит в дезаминировании первичных
алифатических аминов, например изопиламина, азотистой кислотой
(CH3)2CHNH2 + HNO2+H+ (CH3)2CH+ + N2+2H2O
При изопропилировании с помощью этого метода бензола или алкилбензолов
получаются низкие выходы, так как в применяемой среде карбониевые ионы
претерпевают ряд других превращений. Однако соотношение изомерных
цимолов, образующихся из толуола, почти совпадает с полученным Алленом
и Ятсом в обычных условиях алкилирования.
д. Ацилирование. Ионы ацилия RCO+ гораздо более стабильны, менее
реакционноспособны и поэтому образуются легче, чем ионы алкидия.
В 1937 г. Трефферс и Гаммет [239] при помощи криоскопии показали, что
304
Замещение в ароматическом ряду
мезитиленкарбоновая кислота в растворе серной кислоты количественно
гетеролизована согласно уравнению
С6Н2(СН3)зСООН + 2Н28О4 С6Н2(СН3)зСО+ + Н3О+ + 2Н8О7
Бензойная кислота таким свойством не обладает, она просто протонируется;
то же относится и к уксусной кислоте. Однако Гиллеспи [240] показал, опять
с помощью криоскопии, что бензойный ангидрид и уксусный ангидрид коли-
чественно гетеролизуются в серной кислоте:
CeH6COOCOCeH6 + 2H2SO4 CeH6CO+ + CeH6COOHJ + 2HSOr
По данным Бартона и Прайла [241], растворы этих ангидридов в серной или
хлорной кислоте можно использовать для ароматического ацилирования.
Авторы показали, что раствор, полученный смешением хлористого ацетила
и перхлората серебра в нитрометане, после отфильтровывания хлорида сереб-
ра содержит алкилирующий агент, способный, например, перевести анизол
в п-метоксиацетофенон быстро и с высоким выходом. В этой системе ацили-
рующим агентом может быть только ион ацетилия СН3СО+, конечно коор-
динированный с нитрометаном.
Важную роль в ацилировании по Фриделю — Крафтсу играют соли ацилия
комплексных пергалогенкислот, выделенные в кристаллическом состоянии
и охарактеризованные криоскопически. Сил [242] в 1943 г. получил и оха-
рактеризовал одну из них — борфторид ацетилия CH3CO+BFr. Множество
ацилиевых солей, содержащих анионы BFy, PF", AsF", SbF" и SbCl", получили
Ола и сотр. [242]. Ряд из них авторы использовали для О-, N- и S-ацилирова-
ния; для ароматического ацилирования применялись гексафторантимонат
и гексахлорантимонат ацетилия CH3CO+SbF" и СН3СО+ЗЪС1".
Кинетика ацилирования по Фриделю — Крафтсу была изучена Брауном
и сотр. [243]. Скорость бензоилирования бензола и алкилбензолов бензоил-
хлоридом и хлористым алюминием в избытке хлористого бензоила или в ди-
хлорэтане как растворителе, а также скорость ацетилирования этих угле-
водородов хлористым ацетилом и хлористым алюминием в дихлорэтане опре-
деляется следующим выражением:
Скорость ос [АгН][А1С13]
Во всех случаях хлористый алюминий вначале образует почти стехиометри-
ческий комплекс с ацилхлоридом, который берется в избытке и является
наиболее основным соединением из присутствующих в системе. Вероятно,
образуется ионизующийся аддукт
КС = 6 — А1С13 RCO+ + AICI7
Cl
Кинетические данные показывают, что именно этот аддукт * в диполярной
или ионной форме, а не ацилиевый ион, образующийся при диссоциации,
входит в переходное состояние ароматического ацилирования.
Одной из главных причин, позволяющей считать, что активным агентом
является ионная пара, состоит в том, что, как мы увидим ниже, относительная
реакционная способность различных ароматических соединений или раз-
личных положений ароматического кольца при ацилировании по Фриделю —
Крафтсу в тех системах, которые были изучены кинетически, очень мало
* Результаты рентгеноструктурных исследований показывают, что бензоилхлорид
в твердом состоянии находится в диполярной форме [244].
20-0772
305
Глава VI
отличается от относительной реакционной способности при ацилировании зара-
нее полученными солями ацилия, например гексафторантимонатом в инерт-
ном, но полярном растворителе. Обычно считают, однако, что происходит
образование и перенос ацилиевого иона: сначала этот ион присоединяется
к ароматической молекуле с образованием иона ацилциклогексадиенилия,.
который затем депротонируется:
/Ч_ н /\/G0R COR
RCO++ I + |\н I +н+
До сих пор ни одной хорошо охарактеризованной соли, включающей ион
ацилциклогексадиенилия, не было выделено. Относительная реакционная
способность различных ароматических молекул и распределение реакционной
способности по ароматическому кольцу при ацилировании были изучены
Брауном, а также Ола с сотрудниками. Браун изучал бензоилирование и аце-
тилирование ацилхлоридами и хлористым алюминием в различных раство-
рителях, т. е. использовал реагенты Фриделя — Крафтса [236]. Ола изучал
ацетилирование, используя те же реагенты и, кроме того, заранее получен-
ные соли: гексафторантимонат и гексахлорантимонат ацетилия в нитрометане-
[245]. Полученные двумя методами результаты отличались мало. Некоторые
данные этих авторов, отнесенные к бензолу, приведены в табл. 82. Очень
Таблица 82
Относительные скорости и соотношение изомеров при бензоилировании
бензоилхлоридом и хлористым алюминием и при ацетилировании подобным образом
или гексагалоантимонатами ацетилия (Браун, Ола)
Субстрат Реагент Растворитель (25 °C) Относи- тельная скорость (СбНб —1) Количество изомера, %
орто мета пара
Бензоилирование
с3н5сн3 С3Н6СОС1 110 9,3 1,4 89
С6Н5СН3 С2Н4С12 117
CgHsCziHg-mpem > С6Н6СОС1+А1С13 С2Н4С1.2 76 0,0 5,4 95
c6h5f С2Н4С12 0,25 0,0 0,0 100-
С6Н5С1 С6Н5СОС1 0,0115 0,0 0,0 100
Ацетилирование
свн5сн3 СН3СОС1+А1С13 C2H4C12 128 1,2 1,2 98
свн5сн3 CH3CO+SbF3 CH3NO2 125 1,4 0,9 98
СдЩСНз CH3CO+SbClj ch3no2 121 0,8 0,9 98
CgHgCijHg-mpezn CH3COCI + А1С13 ch3no2 114 0,0 3,8 96
CeHsC4H9-mpem CH3CO+SbFs ch3no2 74 0,0 5,7 94
c6h5f CH3CO+SbF« ch3no2 0,51
С6Н5С1 CH3COCl-j-А1С13 C2H4C12 0,021 0,0 0,5 99
С6Н5С1 CH3CO+SbF7 CH3NO2 0,016
С6Н5С1 CH3CO+SbFe ch3no2 0,02
С6Н5Вг CH3COC1 + A1C13 C2H4C12 0,014 0,0 0,0 100
С6Н6Вг CH3CO+SbF3 CH3NO3 0,01
интересно, что отличие ацилирования от нитрования качественно противопо-
ложно отличию алкилирования от нитрования. Следует сказать, что актива-
306
Замещение в ароматическом ряду
ция и дезактивация более выражена, а соотношение изомеров более нерав-
номерное при ацилировании, чем при нитровании, и еще более, чем при алки-
лировании. Браун связывает эти эффекты с понижением реакционной спо-
собности ионов ацилия по сравнению с ионами нитрония и алкилия.
Такой же вывод можно сделать при рассмотрении факторов парциальных
скоростей. Некоторые примеры, относящиеся к ацетилированию ацетил-
хлоридом и хлористым алюминием в дихлорэтане как растворителе, приве-
дены ниже:
СНз С(СН3)з С1
1 1 1 /\о,О
\/6 \/ 3 1о,оооз
750 660 0,125
Стадией, частично определяющей скорость, может быть и отщепление заме-
щаемого протона. Этот вывод сделан на основании наблюдения [245], что
скорости ацилирования готовыми гексафтор- и гексахлорантимонатами аце-
тилия уменьшаются при замещении ароматического дейтерия. Скорости ацети-
лирования гексадейтеробензола примерно в 2,2 раза меньше скоростей аце-
тилирования обычного бензола. Аналогичное уменьшение скорости наблю-
дается в случае ацилирования толуола и мезитилена. Вероятно, энергетиче-
ские барьеры, разделяющие предполагаемый промежуточный ион ацетил-
циклогексадиенилия от исходных соединений (отщепление ацилия) и конеч-
ных продуктов (отщепление протона) сравнимы по величине.
е. Азосочетание. На основании теории ориентации уже с 1926 г. было
ясно, что активной частицей при азосочетании является электрофильный
агент, который едва ли может быть чем-нибудь иным, кроме диазоний-иона
ArNj. Однако формально подтвердить это не удавалось до 1941 г.
Первыми обширными работами по кинетике азосочетания фенолов были иссле-
дования Конанта и Петерсона [246]. Эти авторы считали скорость пропор-
циональной [ArN2OH][Ar'OH] и рассматривали процесс как бимолекулярную
реакцию между этими молекулами. Однако, так как концентрация воды
постоянна, кинетические законы удовлетворяются и в том случае, если одна
молекула воды удаляется из переходного состояния, причем скорость будет
пропорциональна [ArN+][Ar'O“], что соответствует представлению о бимо-
лекулярной реакции между диазоний- и фенолят-ионами. Уистар и Бартлет
[247] и Хаузер и Бреслоу [248] расширили кинетические исследования, вклю-
чив сочетания с ароматическими аминами. Они показали, что допущение,
согласно которому [ArN+][Ar'O“] является соответствующим произведением
концентраций для сочетания фенолов, a [ArN+][Ar'NR2] — для аминов, лишь
в общих чертах соответствует процессам сочетания.
Большинство диазониевых ионов в кислых и слабощелочных водных рас-
творах термодинамически стабильнее неионизованных диазогидратов.
За пределами этого интервала pH скорость азосочетания увеличивается
с ростом pH, причем так, как это должно было бы наблюдаться, если бы един-
ственным эффектом уменьшенной кислотности или увеличенной основности
была бы замена малореакционноспособной молекулы Аг'ОН на высоко-
реакционноспособный анион Аг'О- (при сочетании с фенолами) или замена
нереакционноспособного катиона Ar'NHR* на реакционноспособную моле-
кулу Ar'NR2 (при сочетании с аминами). Но при высоком значении pH равно-
весие заметно сдвигается влево, уменьшая концентрацию диазоний-
иона и, следовательно, уменьшая ускоряющее влияние основности на азо-
20* 307
Глава VI
сочетание.
Ar'N = NOH Ar'N++OH~
Бензолдиазоний-ион — довольно слабый электрофил. Это доказывается тем,
что он может реагировать с фенолят-ионом или молекулой анилина, но не
с молекулой менее реакционноспособного анизола или еще менее реакционно-
способных соединений бензола. Однако реакционная способность диазоний-
иона увеличивается в результате введения электроотрицательных замести-
телей. Это доказано величинами скоростей, измеренных Конантом и Петер-
соном, приведенных и обработанных Гамметом [249]. Ниже даны сравнитель-
ные скорости сочетания для пара-замещенных бензолдиазоний-ионов с пятью
различными фенолами (отклонения значений для различных фенолов не пре-
вышают 25 %):
— NO2 —SOj -Вг Н --СН3 —ОСН3
1300 13 13 (1) 0,4 0,1
Именно таких соотношений и следовало ожидать, потому что нитрогруппа
должна увеличивать, а метоксигруппа уменьшать положительный заряд,
который несет диазогруппа, как это показано на следующей схеме:
[o2n-^^>^n=n]+
Мейер и сотр. [250] эмпирически установили, что нитрозаместители способ-
ствуют расширению границ применения реакции азосочетания. Бензолди-
азоний-ион легко сочетается с эфирами флороглюцина, но не сочетается с ме-
нее реакционноспособными эфирами фенола. п-Нитробензолдиазоний-ион
сочетается с эфирами резорцина, но все же не сочетается с эфирами фенола.
Однако 2,4-динитробензолдиазоний-ион сочетается с анизолом и фенетолом,
а 2,4,6-тринитробензолдиазоний-ион — с мезитиленом (который сравним
по реакционной способности с анизолом с электроотрицательным заместите-
лем — n-хлоранизолом). Эти результаты * с очевидностью показывают, что
в принципе диазоний-ион является общим реагентом для ароматического
электрофильного замещения.
Еще в прошлом веке было хорошо известно [251], что легкость сочетания диа-
зониевых ионов с М,М-диалкиланилинами уменьшается при введении в по-
следние молекулы opmo-заместителей. Причина этого стала ясна только
после открытия вторичного пространственного эффекта в 1937 г. (гл. III,
разд. 1): отталкивание от opmo-заместителя выводит диалкиламиногруппу
из плоскости кольца и нарушает активирующее сопряжение с кольцом
(разд. 6,и).
Цоллингер [252] показал, что азосочетание происходит по бимолекулярному
механизму SE2 через промежуточный ион циклогексадиенилия
(1) /N2Ar'
Ar'N±4-ArH , Ar+<
/NaAr'
Ar+( В
---> ArN2Ar'4-HB+
(2)
(Se2)
* Приведенные Жданные показывают также, что нитрозаместители могут расширить
азосочетание в такой мере, что оно включит электрофильное замещение олефинов. Таким
образом, хотя бензолдиазоний-ион не может сочетаться с простейшими производными
бутадиена-1,3, n-нитробензолдиазоний-ион удовлетворительно сочетается с 2,3-диметил-
бутадиеном-1,3, но не сочетается с менее реакционноспособными бутадиенами, в то
время как 2,4-динитробензолдиазоний-ион сочетается с 2-метилбутадиеном-1,3 и даже
с самим бутадиеном-1,3. Во всех случаях сочетание идет в положение 1.
308
Замещение в ароматическом ряду
но не по одностадийному тримолекулярному механизму SE3
Ar'NJ4-ArH4-B Ar'N2Ar+HB+ (SE3)
Доказательства основывались на кинетическом эффекте, обусловленном
введением дейтерия вместо протона, а также на том, что в реакции иногда
проявляется общий основной катализ. Обычно при азосочетании не наблю-
дается ни кинетического изотопного эффекта, ни заметного основного ката-
лиза (если субстрат полностью находится в реакционноспособной форме,
например фенол в форме фенолят-иона). Это свидетельствует против меха-
низма SE3, но может быть согласовано с механизмом SE2, если предположить,
что стадия (2) настолько более быстрая, чем стадия (—1), что отщепление
протона не влияет на скорость реакции, которая, таким образом, определя-
ется только стадией присоединения (1).
Однако в ряде случаев, особенно когда по соседству с центром, по которому
происходит сочетание, находится в качестве заместителя сульфонат-ион,
при введении дейтерия скорость уменьшается и параллельно начинает про-
являться основной катализ. Катализ является общим основным, так как
вклад в увеличение скорости вносит каждое основание смеси, например не
только вода, но и ацетат-ионы ацетатного буфера. Эти эффекты означают, что,
по крайней мере частично, определяющей скорость стадией является отщеп-
ление протона. В серии из трех соединений, приведенной ниже, кинетические
изотопные эффекты возрастают, т. е. увеличивается роль стадии отщепления
протона. Данные относятся к сочетанию с диазотированным тг-хлоранилином
в положения, помеченные атомом дейтерия.
о-
I D
mz
\/\/
I
SOJ
Ащ/Ащ 1,04
3,10
6,55
Наблюдаемые эффекты нельзя объяснить участием механизма SE3. Если бы
было так, то связанный с концентрацией основания член в выражении для
каталитической скорости линейно возрастал бы с ростом концентрации осно-
вания, тогда как в действительности он возрастает не линейно, а по закону,
указанному ниже. Альтернативное объяснение состоит в том, что сульфонат-
ион настолько понижает скорость отщепления протона от ионного аддукта,
образующегося при механизме SE2, что она становится сравнимой (или даже
ниже) со скоростью обратной реакции распада аддукта. При этом будет
возрастать изотопный эффект (см. выше) и появляться основной катализ.
Скорость катализируемой реакции в этом случае будет связана с концентра-
цией основания множителем [В]/(с + [В]), где с — константа, равная отно-
шению констант скоростей реакций образования и исчезновения аддукта
(ki/k2). Таким образом, с ростом концентрации основания скорость азосоче-
тания возрастает медленнее, чем в случае линейного закона. Следовательно,
все данные поддерживают механизм SE2.
ж. Меркурирование. Если имеются некоторые сомнения относительно
электрофильной природы этой реакции в неполярных растворителях, то в
полярных растворителях этот процесс определенно относится к электро-
фильному типу. Клапрот и Уэстхеймер [253] показали, что меркурирование
перхлоратом ртути в воде в присутствии хлорной кислоты приводит к соот-
309
Глава VI
ношению изомеров (табл. 83), близкому к соотношениям, получающимся при
других реакциях электрофильного замещения. Свободная кислота препят-
ствует гидролизу, т. е. депротонированию водной координационной ячейки
катиона ртути, и сильно увеличивает скорость замещения. Последний эффект
Таблица 83
Ориентация при меркурировании
Меркурирование Hg(C104)2 Количество изомеров, %
ортпо пара мета
Толуол (в 40%-ной водной НС1О4, 25 °C) 19 74 7
Нитробензол (в 60%-ной водной НС1О4, 23 °C) И 89
не связан с кислотным катализом, так как добавление перхлората натрия
также увеличивает скорость. Перрен и Уэстхеймер [254] показали, что этот
эффект обусловлен активацией иона ртути, происходящей, аналогично акти-
вации ионов водорода, при удалении воды из сольватной оболочки. Термо-
динамическая активность растворителя (воды) уменьшается при добавлении
электролита. Авторы доказали, что при постоянной температуре скорость
меркурирования — функция давления паров воды над раствором, какой бы
электролит ни добавлялся.
В воде меркурирование подчиняется следующему кинетическому закону:
Скорость ос [АгН] [Hg2+]
Перрен и Уэстхеймер установили, что скорость меркурирования гексадей-
теробензола в 4,9—6,7 раза меньше скорости меркурирования бензола.
Механизм реакции не установлен, однако наличие изотопного эффекта пока-
зывает, что если реакция относится к типу SE2 и в ней образуется циклогекса-
диенилиевый аддукт, то определяющей скорость стадией должно быть отщеп-
ление протона от аддукта.
Браун и сотр. [236] определили распределение реакционной способности
по различным положениям кольца в реакции меркурирования замещенных
бензолов ацетатом ртути в уксусной кислоте, определив факторы парциальных
скоростей для ряда замещенных бензолов. Некоторые из полученных резуль-
татов приведены ниже:
СН3
/\б,7
\/’2
23
С(СН3)3
/у о
\/3,4
17
осн3
I
/\186
\/ 1
2300
С1
/\),075
Jo, 060
0,36
Качественно эти результаты согласуются с данными для других реакций
электрофильного замещения, однако различие между реакционной способ-
ностью различных субстратов и различных положений одного и того же суб-
страта в случае меркурирования выражено несколько слабее, чем, например,
в случае нитрования или водородного обмена.
з. Десилилирование, дегермилирование, дестаннирование и деплюм-
бирование. Иборн и сотр. [255] показали, что связанные с ароматическим
кольцом группы XR3, содержащие элементы IV группы (X = Si, Ge, Sn,
Pb; R = алкил), можно заместить на водород, бром, ртуть и т. д., действуя
310
Замещение в ароматическом ряду
соответствующими электрофильными агентами, теми же, какие применяются
для замещения ароматического водорода. Авторы широко исследовали заме-
щение элементов IV группы на водород при действии сильных кислот; резуль-
таты этих исследований позволили провести новые сравнения. Предшествую-
щие исследования электрофильного ароматического замещения позволяют
сравнивать влияние замещающего агента и заместителей в кольце на скорость
замещения одной и той же группы, а именно атома водорода.
Ацидолиз производных элементов IV группы позволяет выяснить, как влияет
природа уходящей группы на скорость реакции с одним и тем же замещающим
агентом и как эта скорость меняется при введении заместителей.
Ацидолиз любого из упомянутых соединений обычно проводят действием
хлорной или серной кислот в водном органическом растворителе. Стехиомет-
рия реакции соответствует уравнению
ArXR3 + H3O+ АгН + XRJ + Н20
с тем лишь дополнением, что отщепившийся остаток XR3 в этих условиях
превращается в гидроокись, алкоголят или соль.
Все факторы — природа замещающего агента, стехиометрия реакции и влия-
ние заместителей на реакционную способность — ясно указывают на то, что
реакция является электрофильным замещением. Детальный механизм точно
не выяснен, однако Иборн и сотр. показали, что для всех четырех элементов
IV группы при переходе от воды к окиси дейтерия скорость реакции умень-
шается в 1,6—3,1 раза. Это означает, что, по крайней мере частично, ско-
рость реакции контролируется скоростью присоединения протона, поэтому
в случае 8Е2-механизма медленной стадией является образование замещенно-
го иона циклогексадиенилия
XR3 Н
Н30++ | XR3 медленно + j\R | +Й3Х+
Такой вывод соответствует данным Уэстхеймера, который показал,
что при ароматическом меркурировании медленной стадией будет от-
щепление протона; в данном случае реакция происходит в обратном
направлении.
Скорость ацидолиза групп R3X зависит от положения X в таблице Менделее-
ва: она возрастает при переходе к высшим периодам. Иборн и Панде уста-
новили (в некоторых случаях косвенно вследствие огромного интервала изме-
нения скоростей) следующий порядок относительной реакционной способ-
ности:
н SiR3 1 GeR3 35 SnR3 350 000 PbR3 200 000 000
1 104 105,5 109,5 1012
Скорости реакции, отнесенные к скорости водородного обмена в бензоле,
приведены в нижней строке схемы. Природа алкильных групп R3 относи-
тельно не существенна: Иборн изучал триметил-, триэтил- и трициклогексил-
производные и показал, что скорость в этом ряду уменьшается, однако все
изменение лежит в пределах одного порядка.
Общее представление об активирующем и дезактивирующем влиянии заме-
стителей в пара-положении ароматического кольца можно получить при
рассмотрении данных табл. 84. Интервал изменения скоростей очень велик;
наибольшие изменения наблюдаются в случае триалкилсилильной группы,
затем при переходе к производным германия, олова и свинца они постепенно
311
Глава VI
Таблица 84
Влияние ««/^-заместителей на относительную скорость
ацидолитического замещения групп XR3 (Иборн)
Реагент
п-Заместитель НС1О4, Н2О, СНзОН, 51 °C HCIO4, Н2О, 25 °C
отщепляющаяся группа
Si(CH3)3 &е(С2Н6)з SnfCgHiih РЬ(С6Нц)з
N(CH3)2 30 000 000 20 000 000 20000
он 10000 2 700
ОСНз 1500 540 63 21
СН3 21 14 6 3,4
mpem-C4Hg 16 И 7
н 1 1 1 1
F 0,75 0,92 0,62
С1 0,13 0,17 0,19 0,32
Вт 0,10 0,17 0,15
I 0,10 0,13 0,14
СООНа 0,0015 0,0052 0,030
N(CH3)a 0,00038 0,0011 0,0064
NOa 0,00012 0,00037
а Для соединений Si и Ge использовали следующие реагенты, H2SO4, Н2О, СНзСООН, 50 °C»
уменьшаются. Однако даже при десилилировании кинетические эффекты мень-
ше, чем при водородном обмене (разд. 6,в). Так, введение га-метоксигруппы
увеличивает скорость ацидолитического дедейтерирования в 55 000 раз,
а скорость десилилирования — только в 1500 раз.
Данные табл. 84, а также упомянутые в разд. 3,в показывают, что группа ОН
имеет более сильный активирующий эффект, чем ОСН3. Это явление очень
распространено в реакциях электрофильного ароматического замещения
и наиболее удачно интерпретируется как эффект гиперконъюгации НО-
связи, который будет далее обсуждаться в разд. 6,к.
Ниже приводятся относительные скорости ацидолитического замещения
групп XR3 в различных положениях бензольного кольца, имеющего в каче-
стве заместителей метильную, метоксильную, фенильную группы и хлор.
Отметим, что в согласии с обобщением Брауна при увеличении легкости
замещения ХН3-группы т. е. при увеличении силы замещающего агента,
активация или дезактивация, вызываемая заместителем, становится менее
выраженной и поэтому различия в реакционной способности сглаживаются.
Во всех реакциях метильная группа проявляет нормальный (+/) эффект
заместителя типа (1); она активирует все положения бензольного кольца,
правда, лета-положения очень слабо. Метоксильная группа относится к заме-
стителям типа (3) (—Z, +-/Г), однако в этом экстремальном случае эффект
сопряжения преобладает, поэтому в рассматриваемых реакциях, так же
как при водородном обмене (разд. 6,в), наблюдается сильная орто- и пара-
активация и слабая лета-дезактивация. Такое влияние следовало бы ожидать,
при условии, если бы сопряжение этой группы проявлялось бы как чистый
312
Замещение в ароматическом ряду
эффект поляризуемости, который влияет исключительно на орто- и пара-
положения (разд. 3,в), не затрагивая лета-положения, на которое, таким
образом, влияет только слабый дезактивирующий индуктивный эффект,
обусловленный электроотрицательностью метоксигруппы. Фенильный заме-
ститель также относится к типу (3), однако для него нет такого резкого раз-
личия между электроотрицательным индуктивным эффектом и электрополо-
жительным эффектом сопряжения; тем не менее электроотрицательность
фенила еще слишком мала (или сопряжение слишком велико), чтобы могла
наблюдаться аномалия Голлемана. Таким образом, в этом случае распре-
деление реакционной способности по кольцу качественно соответствует слу-
чаю метоксигруппы, однако различие между разными положениями выражено
менее резко. Фенильная группа также активирует орто- и пара-положения,
но в противоположность метильной группе дезактивирует лгета-положение.
Хлор относится к заместителям типа (3), проявляющим аномалию Голлемана,
т. е. он дезактивирует все положения бензольного кольца, однако таким
образом, что более реакционноспособными остаются орто- и пара-положения.
Хотя экспериментальных данных недостаточно, не может быть сомнений,
что в рассматриваемых реакциях влияние хлора соответствует этому общему
положению. На схемах, приведенных ниже, над стрелками указаны отщепля-
ющиеся группы, а под стрелками — ацидолизирующий агент: символ а
обозначает систему НС1О4, Н2О, СН3ОН, 51 °C, символ б — НС1О4, Н2О,
С2Н5ОН, 25 °C, и символ в - H2SO4, Н2О, СН3СООН, 50 °C.
Si(CH3)3
сн3
I
/\7,8
\/2,3
21,3
Ge(C2H5)3
а
8п(СбНц)з
б
Si(CH3)3
в
Si(CH3)3
а
ОСНд
/\335
^Jo,23
1510
Ge(C2Hs)3
f---------
а
ОСН.з
I
/Лго?
\Jo,51
540
ОСНд
I
^/*0,85
63
с6н5
/^5,85
^Jo,33
2,83
С1 С1
I I
/\?
Si(CH3)3 Г ве(С2Н5)з Г
\у0,012 * в . Jo,019
0,19 0Д7
СГ С1
I I
8п(СбНц)з Г РЬ(СбН,1)3 Г
< в \Jo,O39 * в \Jo,125
0,19 0,32
и. Нитрозирование. В разд. 5,ж было сказано, что сильно реакционно-
способные ароматические соединения, а именно фенолы, анилины и их О-
и N-алкилпроизводные, нитрозируются азотистой кислотой в избытке' азот-
ной кислоты в орто- и пара-положения, образуя нитрозосоединения, которые
сразу окисляются азотной кислотой до нитросоединений. Было также отме-
чено, что аналитически определяемая азотистая кислота существует в таких
кислотных смесях главным образом в виде азотистого ангидрида и лишь
в незначительной степени в виде иона нитрозония; обе эти частицы являются
переносчиками нитрозогруппы.
Кинетика ароматического нитрозирования в условиях, когда образующееся
нитрозосоединение остается неизменным, была исследована Риддом и Кьюре-
ши [256] на примере диметиланилина. В водных растворах сильной хлорной
313
Глава VI
кислоты кинетическое уравнение в точности соответствует уравнению, описан-
ному более полно в гл. VIII для диазотирования и некоторых других реакций
азотистой кислоты: при низкой кислотности, например при [Н+] =
=0,002моль/л, скорость не зависит от концентрации субстрата и реакция имеет
второй порядок [уравнение (1)1; при более высокой кислотности, например
при [Н+] = 0,075 моль/л, в кинетическое выражение начинает входить кон-
центрация субстрата и реакция имеет общий третий порядок [уравнение (2)]:
Скорость oc[HN02]2 (1)
Скорость cc[C6H5N(CH3)2][HNO2]2 (2)
Интерпретировать результаты можно следующим образом. При низкой кис-
лотности, когда выполняется уравнение (1), которое отражает скорость обра-
зования азотистого ангидрида, скорость общей реакции определяется скоро-
стью этого процесса: в растворе имеется достаточное количество амина в виде
свободного основания, которое захватывает N2O3 с такой скоростью, с какой
она образуется. На схеме, приведенной ниже, реакция (1) является медлен-
ной, а реакция (2) — быстрой, гораздо более быстрой, чем реакция (—1):
(1)
2HNO2 N2O3 + H2O
(-1)
N2O3 + CeH5N(CH3)2 - --> Продукты
При достаточно высокой кислотности среды амина в виде свободного основа-
ния содержится настолько мало, что скорость реакции (2) становится меньше
скорости реакции (—1). В этих условиях устанавливается равновесная кон-
центрация азотистого ангидрида [реакции (1) и (—1)1, который медленно
расходуется на реакцию с ароматическим соединением. Вследствие этого
в уравнении (2) появляется концентрация субстрата, а квадратичный множи-
тель в этом случае соответствует стационарной концентрации азотистого
ангидрида. Следует заметить, что концентрационные факторы в уравнениях
(1) и (2) точно соответствуют тому, что написано: [C6H5N(CH3)2] — это кон-
центрация амина в виде свободного основания, a [HNO2] — концентрация
неионизованной азотистой кислоты, а не ее аналитически определяемое
количество.
В рассматриваемом случае переносчиком нитрозогруппы, конечно, является
азотистый ангидрид, т. е. нитрозилнитрит. Однако если необходимую кис-
лотность создать не с помощью хлорной кислоты, анион которой не может
образовывать ковалентного соединения с нитрозилом, а какой-либо другой
кислоты, анион, которой обладает таким свойством, то переносчиком нитрозо-
группы будут также другие нитрозильные соединения. Так, нитрозирование
катализируется ионами брома, и Ридд и Кьюреши показали, что это обуслов-
лено образованием нитрозилбромида.
HNO2 + H+-|-Br- МОВг-фЩО
NOBr4-C6H5N(CH3)2 —> Продукты
Основным продуктом нитрозирования независимо от переносчика является
n-нитродиметиланилин. Однако реакция сопровождается окислением, осо-
бенно значительным при высоком разбавлении. При низких кислотностях
это не влияет на скорость исчезновения диметиланилина: общая скорость,
т. е. скорость нитрозирования плюс скорость окисления, просто равна ско-
рости образования переносчика нитрозогруппы. Поэтому побочная реакция
314
Замещение в ароматическом ряду
окисления происходит после завершения процесса, определяющего скорость,
а переносчик нитрозогруппы служит и нитрозирующим и окисляющим аген-
том. Предполагается, что первичный продукт, образующийся при реакции
-с переносчиком, может отщеплять или протон с образованием нитрозосоедине-
ния, или молекулу окиси азота с образованием продукта одноэлектронного
окисления — радикала, димеризующегося в тетраметилбензидин (который
был выделен). Все процессы нитрозирования сопровождаются отщеплением
окиси азота и одного электрона, далее процесс повторяется до тех пор, пока
от двух молекул диметиланилина не отщепятся четыре электрона; в качестве
конечного продукта образуется ион тетраметилдибензохиниминия
(CH3)2N-<^__1
+ XNO )
(CH3)2N NO + H+ + X-
Как показал Фридлендер [251], диметиланилины, имеющие орто-заместители,
не нитрозируются. По данным Хиккинботтома [257], не нитрозируется также
Х-метил-Х-треш-бутиланилин. Эти эффекты, проявляющиеся также при
азосочетании (разд. 6,е), как показали Феркаде и Вепстер [258] путем срав-
нения физических и термодинамических свойств этих оснований, обуслов-
лены пространственным взаимодействием между N- и орто-заместителями
(даже между о-Н и N-заместителем, если последний достаточно велик).
Благодаря этому происходит поворот основной группы и нарушается ее сопря-
жение с кольцом, активирующее замещение (вторичный пространственный
эффект).
к. Дальнейшее обсуждение эффектов гиперконъюгации. В данном раз-
деле мы продолжим начатое в разд. 3,в и г обсуждение ориентирующих
эффектов заместителей, способных к электроположительному сопряжению
•с кольцом или обладающих гиперконъюгацией, используя данные, приве-
денные в предыдущих параграфах разд. 6. Мы рассмотрим гиперконъюгацию
связей N — Н, О — НиС — Н. Выше было показано, что при электрофиль-
ном ароматическом замещении не только эффект сопряжения, но и эффект
гиперконъюгации по своей сути являются в основном электромерными эффек-
тами, связанными с поляризуемостью (+-Е').
В 1953 г. Робертсон, де ла Мар и Сведлунг [259] предложили расширенную
концепцию гиперконъюгации, включив в нее, кроме С — Н-связей, также
связи N — Н и О — Н. С тех пор, главным образом, благодаря работам де
ла Мара и Иборна, было получено множество доказательств гиперконъю-
гации этих связей; де ла Мар, кроме того, отверг альтернативные объясне-
ния наблюдаемых эффектов.
В табл. 85 приведены некоторые примеры, иллюстрирующие тот факт, что
-скорости реакций пара-замещения в зависимости от природы заместителя
уменьшаются в рядах: ХНСОСН3 > Х(СН3)СОСН3 и ОН > ОСН3, где
знак неравенства отражает изменение скоростей галогенирования в пределах
315
Глава VI
Таблица 85
Логарифмы факторов парциальных скоростей для пара -замещения
в C6H5R [сравнение NHCOCH3 с N(CH)3COCH3 и ОН с ОСН3]
R Бромирование Вг2 в СНзСООН (табл. 7 0) Хлорирование С12 в СНзСООН (табл. 70) Десилилирова- ние Н3О+, водн. СНзОН (табл. 84) Дегермилирова- ние НзО+, ВОДН. С2Н5ОИ (табл. 84)
NHCOCH3 N(CH3)COCH3 ОН ОСН3 9,1 6,1 11,7 9,8 6,4 3,3 4,0 3,2 3,4 2,7
двух-трех порядков, а кислотного десилилирования и дегермилирования
в пределах одного порядка.
Кинетические данные показывают, что большие скорости замещения соеди-
нений, содержащих группы NHCOCH3 и ОН, не связаны с ионизацией и про-
теканием реакции через анион: отщепление протона не происходит, и он вхо-
дит в переходное состояние замещения.
Трудно предположить, что электроположительная метильная группа, если
она связана с ароматическим атомом углерода, станет электроотрицательной,
если она связана с азотом или кислородом. В тех случаях, когда отсутствует
сопряжение между заместителем и удаленным от него реакционным центром,
метилированный заместитель — более сильный донор электронов, чем неме-
тилированный. Так, л-метоксибензойная кислота — более слабая, чем м-
оксибензойная. Метильная группа в СН3 — N и СН3 — О обладает поло-
жительным индукционным эффектом.
На примере галогенирования де ла Мар [261] показал, что скорости заме-
щения в случае указанных водородсодержащих заместителей нельзя считать
нормальными, а скорости замещения при наличии метилированных заме-
стителей представлять как аномально заниженные (на 2—3 порядка) вслед-
ствие вторичных пространственных эффектов, обусловленных отталкиванием
между N- и О-метильной группой и вызывающих нарушение сопряжения
с бензольным кольцом. Если поменять местами орпго-водородный атом и ме-
тильный заместитель у гетероатома, то по сравнению с неметилированным
соединением скорость уменьшится гораздо менее значительно — только
в 5 раз, что соответствует ожидаемому изменению при введении метильной
группы в кольцо (в мета-псложение к галогену).
Точку зрения де ла Мара разделяют не все исследователи, в основном вслед-
ствие распространненого не совсем обоснованного предрассудка, состоящего
в следующем: обычно думают, что различные пары электронов, вносящие
вклад в общее сопряжение, допустим две неподеленные пары и пара электро-
нов связи ОН фенольного гидроксила, с точки зрения пространственной
направленности не зависят друг от друга. Если электронные пары ведут себя
не как единое целое, а независимо и аддитивно и все независимые компоненты
действуют в одном направлении, то при повороте ОН-группы относительно
бензольного ядра гиперконъюгация О — Н-связи может усиливаться только
при одновременном ослаблении сопряжения неподеленной пары. Однако
теория гиперконъюгации не рассматривает электронные пары как независи-
мые аддитивные составляющие. Не зависящая от нашего выбора природа
вещей такова, что мы не можем различить эффекты поляризуемости поделен-
316
Замещение в ароматическом ряду
ных и неподеленных электронов одного и того же атома (гл. III, разд. 3,а);
поэтому, согласно учению Эйнштейна, мы не имеем права вводить подобное
подразделение в теорию. Если стереохимия атома благоприятствует сопря-
жению, то будет наблюдаться совместное перераспределение всех электронов,
приводящее к переносу заряда.
Указанное выше явление впервые было отмечено де ла Мэром [261], который
также показал, что ОН-группа сильнее активирует ядро, чем OD-rpynna,
при введении которой скорость бромирования уменьшается на величину
7«0H//c0d = 1,85. Это означает, что хотя по кинетическим данным в переход-
ном состоянии ОН-группа остается недиссоциированной, ее внутренняя связь
существенно ослабляется, как будто происходит частичная утрата электронов
связи.
По сравнению с NH- и ОН-связями гиперконъюгация СН-связей выражена
слабее. В случае алкильных групп эффект гиперконъюгации по порядку
величины приблизительно такой же, как индуктивные эффекты, от которых
его можно отличить только по обращенному ряду влияния разветвленности
гомологов на скорость, да и то лишь в том случае, если эффекты гиперконъю-
гации превышают индуктивные эффекты. Разделить эти эффекты трудно
еще и потому, что индуктивный и гиперконъюгационный эффекты не явля-
ются независимыми друг от друга: первый эффект зависит от различия в нена-
сыщенности, т. е. орбитальной гибридизации (гл. II, разд. 3,в) связанных
атомов, но это различие сглаживается эффектом гиперконъюгации, и, таким
образом, эффект гиперконъюгации всегда кажется большим, чем он есть
на самом деле (гл. III, разд. 1,з). При такой ситуации вовсе не удивляет
тот факт, что взамен гиперконъюгации предлагаются альтернативные объяс-
нения. Тщательно рассмотрев эти альтернативы, Берлинер [262], ссылаясь
не только на ароматическое замещение, но также на большое число других
гетеролитических органических реакций, пришел к выводу, что ни одна из
этих альтернатив не может с такой полнотой, как концепция гиперконъюга-
ции, объяснить огромный комплекс накопленных фактов.
Прежде чем приступить к обзору данных по ароматическому замещению,
необходимо остановиться еще на двух характерных особенностях гиперконъю-
гации. Первая состоит в том, что для проявления гиперконъюгации необ-
ходимо, чтобы сопряженная система включала реакционный центр: если
это условие нарушается, то эффект гиперконъюгации должен исчезать.
Вторая особенность состоит в том, что в рассматриваемых реакциях гипер-
конъюгация в основном проявляется как эффект поляризуемости, поэтому
при переходе от одного заместителя к другому величина эффекта гиперконъю-
гации будет изменяться в значительно более широких пределах, чем величина
индуктивного эффекта.
Без особого вреда для принципа мы можем ограничиться рассмотрением
только метильного и mpem-бутильного заместителей. Индуктивный эффект
этих групп, активирующий все положения кольца, уменьшается в ряду трет-
С4Н9 > СН3. Эффект гиперконъюгации, активирующий орто- и пара-поло-
жения, уменьшается в обратном порядке: СН3 > mpem-CJlg; гиперконъю-
гация не должна активировать лета-положение. В табл. 86 собраны данные
по факторам парциальных скоростей для различных реакций замещения
в мета- и пара-положения бензольного ядра, имеющего метильные и трет-
бутильные заместители. Данные для орто-положений не приводятся,
так как влияние пространственных факторов усложнило бы рассмотрение
явления.
Сразу видно, что для всех случаев лета-замещения наблюдается порядок
активации, соответствующий индуктивным эффектам: mpem-C4H9 > СН3.
317
Глава, VI
В случае пара-замещения такого однозначного вывода сделать нельзя, и поэто-
му необходимо ввести какие-то дополнительные факторы, влияющие на
скорость реакции. Для девяти реакций наблюдаемое увеличение скорости
пара-замещения соответствует ряду гиперконъюгации СН3 > mpe/n-C4H9.
Таблица 86
Факторы парциальных скоростей для пара- и мета-замещения
в C6H5R (сравнение СН3 и нгреш-С4Н9)
Номер реак- ции Реакция замещения, реагент пара-Замещение лита-Замещение
R = СН3 R = mpem-CiHa Н = СНз R трст-С^Нд
1 Нитрование, NOJ 59 75 2,2 4,0
2 Бромирование, Вг+ 59 39 2,5 2,6
3 Хлорирование, С12 820 500 5,0 6,9
4 Бромирование, Вга 2420 750 5,5
5 H-Обмен, CF3CO2H 420 490 3,8 7,0
6 H-Обмен, Н3О+ 250 180 5,0 9,3
7 Ацетилирование, СН3СОА1С14 750 660 4,8 13,1
8 Бензоилирование, СеН5СОА1С14 580 430 4,6 12,3
9 Меркурирование, Hg2+ 23,0 17,2 2,2 3,4
10 Десилилирование, Н3О+ 21,3 11,9 2,3 3,0
11 Дегермилирование, Н3О+ 14,0 11,5 2,1 3,3
12 Дестаннирование, Н3О+ 5,6 7,0 1,85
Для трех остальных реакций (№ 1, 5 и 12) общая пара-активация соответ-
ствует индуктивному ряду, однако при переходе от СН3 к mpem-CJlg ско
рость изменяется значительно меньше, чем при л«еига-замещении (№ 1 и 5).
Можно сделать вывод, что даже в тех случаях, когда скорость пара-заме-
щения соответствует индуктивному ряду заместителей, проявляется также
и эффект гиперконъюгации, по величине сравнимый, но однако не превышаю-
щий величину индуктивного эффекта.
7. Нуклеофильное ароматическое замещение*
К числу реакций нуклеофильного ароматического замещения относятся те
процессы замещения, в которых нуклеофильные реагенты, такие, как Вг”,
SR- или NR3, присоединяются к углероду ароматического ядра, а ранее
имевшиеся заместители типа Cl, NO2 или NJ удаляются совместно со связую-
щими электронами.
Даже водород с его связующими электронами (т. е. в виде Н~) может быть
удален, но это сопряжено со значительными трудностями. В бимолекулярном
нуклеофильном замещении электроноакцепторные заместители, особенно
заместители, сопряженные с ароматической системой, такие, как нитриль-
ная, карбонильная, циан- или сульфонильная группа, облегчают атаку
реагента; гетероатомы в положении 2 или 4 в пиридине действуют подобным
же образом. Общая природа нуклеофильного замещения в ароматических
* В данном разделе широко использован материал из статьи Баннета и Цалера [263].
318
Замещение в ароматическом ряду
соединениях становится понятной из следующих примеров:
N02 /N02
N02-<^ X- Вг + СНзО- O2N ОСН3 + Вг-
/ч /ч
I I +NH3->|| |
Уч v~NH2+hg1
N Cl N
\~N£ + I~ I + N2
Нуклеофильное замещение в ароматическом ряду может протекать по раз-
личным механизмам. Наиболее вероятны мономолекулярный и бимолекуляр-
ный механизмы; возможны и другие, менее известные механизмы. Мономоле-
кулярный механизм ограничен замещением таких заместителей, которые
достаточно свободно связаны с ядром и могут подвергаться самопроизволь-
ному гетеролизу в растворе. Бимолекулярный механизм является более
общим, несомненно, потому, что он в меньшей степени зависит от свойств
отщепляемой группы. Известно много случаев замещения в ароматическом
ряду при действии нуклеофильных реагентов, характеризующихся тем,
что вводимые группы не занимают места, ранее занятого отщепившейся
группой; такие замещения, следовательно, связаны с перемещением водорода.
В ходе дальнейшего изложения последовательно рассматриваются все эти
проблемы.
а. Мономолекулярное замещение. Хорошо известным примером нуклео-
фильного замещения в ароматическом ряду, осуществляемого по мономоле-
кулярному механизму SN1, является некаталитическое разложение ионов
диазония в гидроксилсодержащих растворителях с образованием фенолов
или фениловых эфиров, часто сопровождаемое образованием арилгалоге-
нидов или других подобных продуктов замещения, если в растворе имеются
необходимые нуклеофильные анионы:
Медленно д
ArN$-------> Ar+ + N2 1
Быстро
Аг+ + Н2О------> АгОН + Н+
Быстро (SjjlY
Ar+ + ROH------> ArOR + H+
Быстро
Ar+4-Cl- ------> ArCl
Мелвин-Хьюзом и Джонсоном [264], а также Уотерсом [265] высказано
предположение, что эти реакции протекают по механизму S^l- В наиболее
ясной, развернутой форме эту точку зрения аргументировали Баннет и Цалер
[263]. Как было показано [26-4, 266], разложение хлористого бензолдиазония
в воде протекает как реакция первого порядка. Ее скорость не зависит от
природы или концентрации анионов, связанных с катионом диазония, даже
если этот анион в значительной мере переходит в продукт реакции [266, 267].
Скорость образования фенола оказывается почти одинаковой как в D2O,
так и в Н2О [266]. Кинетическое влияние заместителей в ароматическом ядре
[266] является характерным и полностью отличным от любой наблюдаемой
формы бимолекулярного нуклеофильного замещения (разд. 7,6). Этот харак-
терный эффект можно легко понять, если допустить, что реакция протекает
по мономолекулярному механизму.
319
Глава VI
Теоретические предпосылки, касающиеся кинетических эффектов замести-
телей при мономолекулярном нуклеофильном замещении, состоят в следую-
щем. Во-первых, заместители, которые поставляют электроны, должны
увеличивать, а заместители, оттягивающие электроны,— замедлять скорость
гетеролиза, определяющего скорость всего процесса и, следовательно, изме-
ряемую скорость реакции. Эти эффекты, как правило, должны быть более
сильными, когда заместители находятся в орто- или пара-положениях,
и менее сильными, когда они расположены в .мета-положениях. При этом
не учитываются некоторые особые обстоятельства, касающиеся ионов диазо-
ния; указанный вопрос будет рассмотрен ниже. Далее, первичный прост-
ранственный эффект opmo-заместителей должен ускорять гетеролиз и, сле-
довательно, измеряемую скорость реакции. В случае орто-заместителей,
сопряженных с ядром, вторичный пространственный эффект может ослабить
любой полярный эффект, возникающий вследствие сопряжения.
В табл. 87 приведены данные, полученные Кросли, Кинли и Бенбруком
[266]. Рассматривая сначала мета-заместители, можно сделать вывод, что
+7- и Х-группы, включая некоторые группы (С6Н5, OR), которые относятся
Таблица 87
Влияние заместителей на скорость разложения солей
бензолдиазония по реакции первого порядка в воде при 28,8 °C
Заместитель 107fel, С-1
орто мета пара
он 6,8 9100 0,93
ОСНз 3400 0,11
Сен5 1100 1700 37
сн3 3700 3400 91
н 740 740 740
со он 140 410 91
SOg 91 150 42
Cl 0,14 31 1,4
no2 0,37 0,69 3,1
к типу (—1, +К) с сильным +.К-характером, увеличивают скорость реакции,
в то время как —I и — К-группы, включающие группу (С1), принадлежащую
к типу (—I, +£) с сильным —/-характером, затрудняют реакцию. Этого
результата можно было ожидать. Если рассматривать вопрос, действуют ли
те же заместители аналогично, но более сильно в орто- и пара-положениях,
можно установить, что электроноакцепторные орто- и пара-заместители дей-
ствительно в значительной степени замедляют реакцию, а электронодонорные
заместители вместо более сильного ускорения в орто- и пара-положениях
обычно замедляют реакцию тем сильнее, чем больше они предположительно
должны были бы ее ускорять.
Несмотря на то что замедляющее действие орто- и пара-заместителей типа
-\-К не было теоретически предсказано, Хьюз (цитировано по работе [263])
указывал, что такого эффекта следовало ожидать из-за своеобразной комби-
320
Замещение в ароматическом ряду
нации заряда и ненасыщенности группы диазония. орто- и пара-Заместители
типа +К, вызывая +2И-эффект в первоначальном состоянии иона диазония,
будут действовать в направлении достижения нейтральности N2-rpynnbi
в целом, но это будет происходить за счет увеличения ковалентности арил-
азотной связи, которая вследствие этого становится более устойчивой к гете-
ролизу. Например, +2И-эффект пара-метоксигруппы может в некоторой сте-
пени вызвать включение волновой функции структуры
RO N = N
в мезомерную волновую функцию нормального иона диазония. Любая соот-
ветствующая модификация волновой функции переходного состояния гете-
ролиза должна быть меньшей, а следовательно, реакция будет замедляться.
Метильные группы могут действовать качественно подобным же образом
из-за гиперконъюгации с участием водорода. В действительности орто-
метильная и орпго-фенильная группы не замедляют реакции, тогда как те же
группы в пара-положении вызывают замедление реакции, вероятно, из-за
первичного пространственного ускорения, т. е. пространственного выталки-
вания группы N2. В случае орпго-фенильного заместителя вторичный прост-
ранственный эффект, вызывая поворот фенильной группы, будет действовать
в том же направлении.
б. Бимолекулярное замещение. Большинство процессов нуклеофильного
замещения, в том числе отщепление нейтрального заместителя, особенно
галогена из ароматического ядра при не очень высокой температуре, протека-
ет по бимолекулярному механизму SN2. Это подтверждается вторым порядком
реакции, что отмечено во многих исследованиях. В качестве примера можно
привести одно из ранних кинетических исследований Лулофса [268], который
изучал реакцию между 2,4-динитрохлорбензолом и метилатом и этилатом
натрия в метиловом или этиловом спирте. Можно привести также более
позднее исследование Сегдена и Лу [269] по скоростям реакции между 2,4-
динитробромбензолом и бромистым литием (с радиоактивным бромом) в эти-
лендиацетате.
(NO2)2C„H3C1 + C2H5O- -> (NO2)2CeH3OC2H5 + Cl-
(NO2)2C6H3Br + Br > (NO2)2CeH3Br-]-Br
В замещениях этого типа скорость атаки различными реагентами одной
и той же ароматической молекулы соответствует степени их нуклеофильности.
К такому заключению пришли Баннет и Цалер [263] после сопоставления
данных, собранных из многих источников. Дополнительные данные получили
Баннет и Дэвис [270]. Для скорости замещения хлора в 2,4-динитрохлорбен-
золе установлен следующий порядок скорости введения нуклеофильных
групп:
C6H5S- > СН3О~ > СвН5О“ > ОН- > пиперидин > анилин > 1“ > Вг-
Кинетический эффект заместителей в ароматической молекуле при бимоле-
кулярном нуклеофильном замещении был теоретически рассмотрен автором
книги [271], который сопоставил ожидаемые полярные эффекты с наблюдаемы-
ми при бимолекулярном электрофильном замещении в ароматических соеди-
нениях. Чрезвычайное ускорение реакции должны вызывать группы типа
—I, —К в орто- и пара-положениях, в особенности если они несут реальный
или формальный положительный заряд в сочетании с ненасыщенностью,
как в N* и NO2. Более слабые ускоряющие эффекты должны оказывать те же
группы в жеша-положении. В общем более слабые ускоряющие эффекты
21—0772
321
Глава VI
должны вызывать группы типа —I, например NR3, с качественно подобной
же зависимостью от положения заместителя. Слабые ускоряющие эффекты
можно ожидать при .иеига-положении групп типа —I, '\К с достаточно сла-
быми + 7£-свойствами, как в С1. Замедляющие эффекты можно ожидать от
групп типа +/ или Д-Г, +2И, например СН3, или от групп типа —I, + М
со слабыми —/-свойствами, например NR2, в орто- или пара-поло-
жениях.
В этих процессах замещения возможен замедляющий первичный простран-
ственный эффект, так как они являются бимолекулярными; этот эффект
не должен быть очень сильным из-за того, что атака реагента направлена
сбоку к плоскости ароматического ядра. При бимолекулярном нуклеофиль-
ном замещении возможно также проявление вторичного пространственного,
фактора, т. е. нарушение сопряжения.
Большинство этих предположений может быть подтверждено имеющимися
наблюдениями. Выдающееся значение иона диазония как заместителя, уско-
ряющего нуклеофильное замещение галогена, а также нитро- или алкоксиль-
ной группы в орто- или пара-положениях, хорошо известно; Баннет и Цалер
считают этот эффект «досадно большим». При диазотировании ароматических
аминов указанные группы могут замещаться гидроксилом при взаимодействии
с водой или хлором, если при диазотировании употребляется соляная кислота.
Так, из 4,5-динитро-о-анизидина при диазотировании в уксусной кислоте
образуется 1,4-диазоокись [272].
NO2 NO2 /NO2
h2n no2 N2 no2 n2 0-
CH3C) CH3OZ CH3O
Сильное ускорение обсуждаемых процессов замещения под влиянием нитро-
групп, находящихся в орто- и пара-положениях, хорошо известно. 2,4,6-Три-
нитрогалогенбензолы очень легко реагируют [273] с водой, спиртами, аммиа-
ком, с первичными и вторичными аминами, образуя пикриновую кислоту,
ее эфиры или амиды. Вероятно, ни одна из этих реакций не является мопомо-
лекулярной; реакция 2,4,6-тринитрохлорбензола с этиловым спиртом, хотя
и протекает очень быстро, сильно ускоряется при добавлении этилата натрия
(неопубликованные данные Грэхема, Хьюза и Ингольда). 2,4-Динитрогало-
генбензолы взаимодействуют с подобными реагентами медленнее, и реакции
с алкоголят-ионами в спиртовых растворах представляют собой реакции
второго порядка [268, 274]. орто- и иара-Мононитрогалогепбензолы подверга-
ются аналогичной реакции еще более медленно, причем в кинетически изу-
ченных случаях было установлено протекание реакции второго порядка;
л«-нитрогалогенбензолы реагируют еще медленнее [275]. При действии раство-
ра метилата натрия в метиловом спирте на .и-нитрохлорбензол не происходит
заметного отщепления хлора при температуре ниже той, при которой нитро-
группа подвергается восстановлению с образованием л4,л«'-дихлоразоксибен-
зола. При действии этого же реагента на .и-нитрофторбензол наблюдается
нормальное замещение галогена с образованием .и-нитроанизола; реакция
протекает гораздо медленнее, чем в случае изомерных о- и и-нитрофторсое-
динений.
Как было показано еще в 1890 г., группы —GHO, —СОС8Н5, —GOOR, —СОО-,
—CN, —SO2NH2, — SOg в орто- или пара-положениях, хотя и слабо, но так-
же активируют бимолекулярное нуклеофильное замещение галогена в аро-
матическом ядре. Если какая-нибудь из этих групп вводится в орто- или
322
Замещение в ароматическом ряду
иара-положение к галогену в нитрогалогенбензоле, то скорость замещения
галогена при действии аммиака или анилина увеличивается [276]. Количе-
ственные данные для ряда групп в иара-положении, в том числе для некото-
рых сильно активирующих групп, некоторых слабо активирующих и дезак-
тивирующих галогенов и некоторых дезактивирующих алкильных, гидрок-
Таблица 88
Относительные скорости нуклеофильного замещения галогенов
R R-^ Br + + пиперидин в бензоле при 99 °C R-/ S-Cl + NaOCHg в
\o2 СНзОН при 25 °C
^no2 = ЮОО *no2 =1000
no2 1000 1000 170 000
SO3CH3 53 106 18 000
N(CH3)3 33 5 500
CN 31
COCH3 13 12 2100
Cl 0,07 11
H 0,006 1
сильных, алкоксильиых, амино- и диалкаминогрупп, были получены
Баннетом и сотр. [277] (табл. 88), а также Берлинером и Моттеком [2781
(табл. 89).
Таблица 89
Относительные скорости нуклеофильного
замещения брома
R —— Вг в избытке пиперидина нрп25 °C
Xno2
R ftH= 1 R ftH= 1
Br 7,82 трет-СцНс) 0,170
Cl 5,59 сн3 0,146
I 5,42 OCH3 0,0180
COOH 2,52 OC2H5 0,0151
N(CH3)2 0,00121
H 1 OH 0,00059
F 0,260 nh2 0,00013
В этих таблицах группы расположены в соответствии с их кинетическим
+
эффектом. То обстоятельство, что N(GH3)3 стоит ниже NO2, указывает на важ-
ность для реакции —^-эффекта, который не может быть вызван первой груп-
пой. Положение группы SO2CH3 указывает на наличие —^-эффекта, что.
соответствует и многим другим данным, показывающим (гл. III),- что серц
21* ,323
Глава VI
в сульфонильной группе более чем четырехковалентна. Хлор по сравнению
с другими атомами тяжелых галогенов активирует менее значительно, чем
можно было бы ожидать, учитывая его электроотрицательность, что, по-
видимому, обясняется достаточно большим +2И-эффектом хлора. Дезакти-
вирующая роль фтора, несомненно, объясняется сильным -j-M-эффектом.
Порядок, в котором расположены две дезактивирующие алкильные группы,
может быть приписан мезомерной гиперконъюгации с участием метильной
группы. Сильно дезактивирующие группы OR и NR2 являются сильными
Д-М-группами; ОН-группа выпадает из ряда, вероятно, вследствие того,
что в щелочной среде она существует частично в виде О-.
Может быть выявлен также слабый первичный пространственный эффект.
Полуколичественпые данные [279] показывают, что если в и-нитрогалоген-
бензол ввести один и затем второй такой же атом галогена в орто- положение
по отношению к замещаемому атому галогена, то наблюдается следующий
кинетический эффект. Первый атом хлора, брома или иода в орто-положении
ускоряет реакцию, второй атом хлора или брома в орто-положении пе вызы-
вает существенного дальнейшего ускорения; второй же атом иода уменьшает
скорость реакции. Может быть выявлен также вторичный пространственный
эффект. Так, было найдено [280], что при реакции замещенных о-нитро-,
n-нитро- и 2,4-динитрохлорбензолов с метилатом натрия в метиловом спирте
хлор в качестве заместителя нормально ускоряет реакцию, однако ускоряю-
щий эффект выражен слабее, чем следовало бы ожидать для данного поло-
жения хлора относительно замещаемой группы, в том случае, когда хлор
находится в орто-положении к активирующей нитрогруппе. Если при такой
ситуации с другой стороны нитрогруппы также имеется орто-заместитель,
то реакция может даже замедлиться. Было также обнаружено [281],
что алкильный заместитель в .мета-положении к атому хлора в 2,4-ди-
нитрохлорбензоле уменьшает скорость реакции с пиперидином или метилат-
ионом довольно сильно, если алкильная группа находится в положении 5,
и очень сильно, если она находится в положении 3 (т. е. между нитро-
группами); оба эффекта проявляются сильнее при переходе от метила
к mpem-бутилу.
Таблица 90
Относительные скорости нуклеофильного
замещения фтора
X
ХО2—X %—F-J-Y- в метаноле при 0 °C
И
СНз
Вг
он-
сн3о-
ch3s-
1
36
48
1
23
2о8
1
47
298
В табл. 90 приведены данные, которые, согласно справедливому предпо-
ложению Баннета [282], иллюстрируют эффект электрокинетического при-
тяжения: атака более поляризуемым агентом облегчается при введении по
соседству с реакционным центром более поляризуемого заместителя.
324
Замещение в ароматическом ряду
Гегероатом в пиридине оказывает активирующее влияние на нуклеофильное
замещение в положении 2 и 4. Это действие несколько слабее, чем активи-
рующий эффект нитрозаместителя в бензоле при замещении в орто- и пара-
положениях. Манджини и Френгелли [283] показали, что реакция 2-хлор-
5-нитропиридина с некоторыми аминами и алкоголят-ионами протекает
гораздо медленнее, чем соответствующие реакции 2,4-динитрохлорбензола.
Хотя теоретически сравнение этих соединений отнюдь не является простым,
разницу в поведении хлорпитропроизводных ряда бензола и пиридина можно,
по-видимому, объяснить меньшей электроотрицательностью, т. е. меньшим
—^-эффектом пиридиновой системы.
В случае перегруппировки Смайлса [284] в щелочной среде от атома углерода
в ароматическом ядре отделяется электроотрицательный мостик —X —,
который замещается атомом Y — сопряженным основанием некоторого кис-
лотного центра YH, расположенного по другую сторону мостика в исходной
молекуле. Эта перегруппировка может рассматриваться как внутримолеку-
лярное нуклеофильное замещение в ароматических системах
X хн
Rz ^Ar Ос110ванив RZ Ar
^YH
Мостиком — X — может быть — SO2 —, — SO —, — S — или — О —,
а кислотным центром YH может быть группа — ОН, — SH, — NHR,
— GONHR или — SO2NHR. Обычно требуется активация надлежащим обра-
зом расположенной нитро- или другой подобной группой. В качестве типич-
ного примера приведем следующую реакцию [285]:
no2 no2
СН3 SO2 I СН3 SO2H I
Изучение сравнительного влияния различных заместителей в различных
положениях кольца на легкость перегруппировки Смайлса подтверждает
ее принадлежность к реакциям нуклеофильного замещения.
Скорость бимолекулярного нуклеофильного замещения зависит, естественно,
не только от степени нуклеофильности атакующего реагента и от полярного
и пространственного влияния заместителей, но также и от природы отщепляе-
мой группы. Вопрос этот достаточно сложен. Баннет и Цалер па основании
очень тщательного изучения литературы установили указанную ниже после-
довательность легкости отщепления групп:
F > NO2 > Cl, Br, I > N3 > SO3R > f\'R3 > OAr > OAlk > SR > SO2R > NR2
Баннет и Цалер подчеркнули, что предположенный ими порядок является
лишь приблизительным и может в ряде случаев изменяться в зависимости
325
Глава VI
ют природы замещающего агента и заместителей в кольце. Они пришли к пра-
вильному выводу, что стабильность аниона облегчает отщепление, но что
здесь имеют важное значение различные виды сопряжения, в которые заме-
щаемая группа вовлечена до отщепления, а в некоторых случаях и после
отщепления.
По мнению Баннета и Цалера [263], бимолекулярное нуклеофильное заме-
щение происходит через стадии присоединения и отщепления с промежуточ-
ным образованием циклогексадиенилидного аддукта, существование которо-
го во многих случаях было доказано.
(Sn2)
Если замещающим агентом служит нейтральный первичный или вторичный
амин, то предполагаемый аддукт представляет собой диполь, от которого
отщепляется протон или до, или в процессе второй стадии, приведенной выше;
таким образом, в этом случае наблюдается незначительное видоизменение меха-
низма. Промежуточный циклогексадиенилид в любом случае является мезо-
мерным ионом, в котором отрицательный заряд распределен в соответствии
с тремя валентными структурами.
Главный аргумент в пользу этого механизма состоит в том. что выделить
можно лишь тот промежуточный циклогексадиенилид, который наиболее
стабилен (или наименее нестабилен) относительно исходного ароматического
соединения и продукта замещения. Стабилизации благоприятствует, во-
первых, сильное увеличение способности системы удерживать отрицательный
заряд при наличии нитрогруппы, и, во-вторых, такая комбинация X иУ,
когда пи X, ни Y как анионы не обладают достаточной стабильностью, и по-
этому они отщепляются от аддукта в виде анионов недостаточно быстро. Эти
данные показывают, что последовательность присоединение — отщепление
действительно возможна, однако они не доказывают общности указанного
механизма.
Чтобы доказать это, вспомним, что уже в начале столетия Мейзенгеймер
[286] выяснил природу окрашенных аддуктов, образующихся из ароматиче-
ских полинитросоединений и алкоголятов или цианидов металлов, показав,
что как из 2,4,6-тринитроанизола и этилата калия, так и из 2,4,6-тринитро-
фепетола и метилата калия образуются одинаковые продукты; более того,
в обоих случаях полученные аддукты дают при обработке кислотой смесь
трипитроанизола и тринитрофенетола. Соответствующая реакция приво-
дится ниже, причем положение атома калия обозначено произвольно:
СН3О ОС2Н5
O2N I NO2 СН3О ОС2Н5 O2N I no2
02к
326
Замещение в ароматическом ряду
Крэмптон и Голд [287] с помощью спектров протонного магнитного резо-
нанса установили, что циклогексадиенилид-анионы, образующиеся при при-
соединении метилата калия к метилпикрату и 1,3,5-тринитробензолу, имеют
мезомерную структуру
СН3О осн3
NO2\^/NO2
no2
СН3О н
NO2\/\/NO2
no2
В ряду гетероциклических соединений присоединение реагентов Гриньяра
или алкиллития к пиридинам или хинолинам, а также карбаниона металлоор-
ганического соединения к четвертичному ониевому иону, возникшему из аро-
матического основания, ведет к особенно стабильным аддуктам. Это не вызы-
вает удивления, так как обе группы, которые могут выделиться как анионы,
а именно водород и алкил, в этом состоянии обладают очень низкой ста-
бильностью. Их стабильности в качестве анионов резко различаются между
собой; поэтому нуклеофильное замещение в сущности протекает преиму-
щественно в одном направлении, а именно происходит алкилирование с поте-
рей гидрид-иона. Например, н-бутиллитий и пиридин образуют кристалличе-
ский продукт присоединения, из которого при высокой температуре отщеп-
ляется гидрид лития [288]:
Реакция пиридина с амидом натрия при температуре около 200 °C (обычные
условия получения 2-аминопиридина) может протекать по сходному пути,
за исключением того, что равновесие, по-видимому, смещено в сторону исход-
ных веществ, так как промежуточный продукт присоединения легче теряет
ион амида, чем гидрид-ион. Однако такое равновесие будет нарушаться вслед-
ствие переноса катиона между аминопиридином и гидридом натрия. При
этом образуется водород, с выделением которого реакция завершается:
C5H4NNH2 + NaH C5H4NNHNa + H2
Наиболее обычный прием для преодоления трудности выделения гидрид-
иона из промежуточного продукта присоединения при нуклеофильном заме-
щении сводится к добавлению окислительного агента. Так, реакция между
хинолином и амидом натрия не идет сколько-нибудь заметно в жидком аммиа-
ке, но при добавлении нитрата натрия образуются с хорошим выходом 2-
и 4-аминохинолины [289].
327
Глава VI
При реакциях ароматических нитросоединений с такими сильными нуклео-
фильными реагентами, как OR-, NR-или GN-, некоторые нитросоединения
сами действуют как окислительные агенты. Это наблюдается, например, в слу-
чае образования нитрофенола из нитробензола и твердого едкого кали [290],
тг-нитротрифениламина из нитробензола и дифениламида натрия [291] или
З-нитро-2-циананизола из .м-динитробензола и цианистого калия в метиловом
спирте [292]. Расход нитросоедииения по этой причине часто можно устранить
и получить лучший выход, если в реакцию вводить независимый окисли-
тельный агент. Это достигается, например, при превращении спльи-тринитро-
бензола в пикриновую кислоту при действии водного раствора красной кровя-
ной соли [293]. Если ароматическое соединение не может легко действовать
как окислительный агент, то вводить таковой особенно желательно, и это
делается, например, в производстве ализарина нагреванием антрахинон-
2-сульфокислоты с едким кали или натром в присутствии нитрата или хло-
рата [294].
Несмотря на существование промежуточных продуктов присоединения,
к этим реакциям можно применять термин «бимолекулярное нуклеофильное
замещение» или символ 3^2. Этот символ означает лишь то, что происходит
нуклеофильное замещение, переходное состояние которого включает оба реа-
гента, связанные ковалентной связью, однако этот символ ничего не говорит
о числе стадий реакции и о том, какая из них контролирует скорость сум-
марного процесса. Подобно другим случаям, символ SN2 относится к классу
реакций в целом и не отражает частных различий внутри данного класса.
в. Цианирование нитросоединений (реакция Рихтера). Эта реакция отно-
сится к одному из нескольких типов нуклеофильного замещения в аромати-
ческом ряду, общая характеристика которых состоит в том, что нуклеофиль-
ный реагент вступает в молекулу не в то место, которое раньше занимала
отщепляющаяся группа. Очевидно, такое замещение сопровождается пере-
носом водорода из одного положения ароматического ядра в другое.
В гл. IV, разд. 1,а уже отмечалось, что одна из реакций, которая исполь-
зовалась еще в 1870 г. при попытках классифицировать производные бензола
по относительным положениям, в которые вступают заместители, представ-
ляла собой замещение нитрогруппы на циангруппу с последующим ее омыле-
нием в карбоксильную группу. Эта реакция была открыта Рихтером [295],
который осуществлял ее обработкой нитросоединений цианистым калием
в этиловом спирте при температуре 120—270 °C. Сначала предполагалось,
что при реакции имеется соответствие в положениях нитрогруппы и карбок-
328
Замещение в ароматическом ряду
сильной группы, но в 1875 г. Рихтер [296] сделал неожиданное заключение,
что карбоксильная группа оказывается не на месте, ранее занятом нитрогруп-
пой. Наблюдения Рихтера были подтверждены и расширены Голлеманом
[297], а также недавно Баннетом, Кормаком и Мак-Кеем [298].
Общие черты реакции Рихтера, как она была названа Баннетом, можно
видеть па следующих примерах:
этиловом спирте при 150 °C —>
no2
I
j/S /^,-СООН НООС-/^
И | ~pK.CN в этиловом спирте при 150 °C —> | -р I I
\z_ Вг \z Вг \zL Вг
no2
Br
+ KCN
в этиловом спирте при 150 °C —> Кислота не образуется
Подобные результаты были получены с Cl, I и СН3О вместо Вг в приведенных
выше примерах. Ни в одном из этих случаев не замещается атом галогена.
Цианид-ион, по-видимому, имеет только небольшую тенденцию к атаке угле-
рода, несущего атом галогена, хотя он обладает значительным сродством
к углероду, несущему водород в оршо-положении и, возможно, в пара-
положении по отношению к нитрогруппе.
Баннет и Цалер [263] рассмотрели эту реакцию и предложили ее механизм,
первая стадия которого состоит в присоединении цианид-иона в р-положение
двойной связи углерод — углерод, сопряженной с нитрогруппой; в алифа-
тическом ряду аналогично происходит ^-присоединение цианид-иона к аф-
ненасыщенным эфирам. Авторы предполагают, что процесс замещения завер-
шается после захвата протона из спиртовой среды отщеплением азотистой
кислоты и гидролизом циангруппы. Первая стадия этого механизма — при-
соединение цианид-иона — в настоящее время принята всеми, однако меха-
низм стадии отщепления азотсодержащей группы, вероятно, иной.
Розенблюм [299] установил, что азот отщепляется главным образом в виде N2,
а не HNO2 и NH3. Проводя реакцию Рихтера в присутствии меченого аммиака
15NH3, Розенблюм показал, что N2 образуется не из нитрита аммония, так
как выделяющийся азот не содержал изотопа 15N. Самюэль использовал
меченный 18О растворитель, оставив немеченым кислород нитрогруппы, и уста-
новил, что карбоксильная группа образуется пе путем гидролиза, так как
только один из двух атомов кислорода карбоксильной группы был меченым,
т. е. образовался при реакции с растворителем; второй атом кислорода, оче-
видно, ранее входил в состав нитрогруппы. На основании этих фактов можно'
предложить приведенный ниже механизм реакции, идею которого Розенблюм
приписывает Вудварду. Предполагаемый механизм подтверждается данными
Ульмана и Барткуса, показавшими, что синтетический бензпиразолон в усло-
329
Глава VI
виях реакции Рихтера претерпевает аналогичные изменения.
г. Замещение через элиминирование (образование дегидробензола). Заме-
щение галогена аминогруппой при действии амидов металлов на ароматические
галогениды ведет часто, но не всегда, к образованию продуктов перегруп-
пировки. Кажется, что амид-ион может атаковать или углерод ядра, несущий
галоген, или углерод, несущий водород, находящийся в орто-положении
по отношению к галогену. Основное исследование этих процессов было выпол-
нено Кимом и Хейсерманом [300]. Ким установил, что при нагревании п-
дибромбензола с «-толуидином и натронной известью образуется Х,Х'-ди-«-
толил-лг-фенилендиамин. Хейсерман наблюдал, что о-, м- и и-дихлорбензолы
при взаимодействии с дифениламидом калия превращаются в N,N,N',N'-TeTpa-
фенил-л«-фенилендиамин, хотя «ара-дигалогенид образует также некоторое
количество ««pa-диамина. Представление об этой реакции основано главным
образом на систематических исследованиях Гильмана и сотр. [301], которые
изучали взаимодействие амида натрия в жидком аммиаке или диэтиламида
лития в эфире с рядом ароматических моногалогенозамещенных. Общую
схему реакций можно иллюстрировать следующими примерами:
Х = С1, Вг, ОС2Н5; На1 = С1, Вг
Х = С1, CF3; Hal = Cl
330
Замещение в ароматическом ряду
Х = С1, Вг, I, OCHg, SCH3, N(CH3)2, сн3, cf3,
SO2CH3;
Hal = Cl, Вг, I
Hal = F (может давать оба
соединения);
Hal = Cl, Вг, I (дает только
2-замещенное
соединение)
Hal = F, Cl, Вг, I
Механизм этих реакций был выяснен Робертсом и сотр. [302, 304]. Авторы
показали, что реакция протекает через предварительно промотируемое
основаниями элиминирование с образованием в качестве промежуточного
соединения дегидробензола или, как еще называют это соединение, бензина,
имеющего короткое, но вполне определенное время жизни. К дегидробензолу
затем присоединяются элементы амина, анион которого инициирует элими-
нирование, отщепляя протон в орто-положении к атому галогена.
Сначала Робертс показал, что хлор- и иодбензолы, меченные 14С по углероду,
связанному с галогеном, при обработке амидом калия образуют анилин,
в котором метка 14С равномерно распределена (с учетом ошибки эксперимента
и ожидаемого небольшого изотопного эффекта) между углеродом, с которым
связана аминогруппа, и углеродом в орто-положении к этой группе
NH2
Дегидробензол
nh2
Птот механизм подтверждается при изучении влияния орто-дейтерирования.
Эри аминировании о-дейтерированного хлорбензола в указанных условиях
было найдено, что хлорбензол еще до завершения его превращения в анилин
обменивает дейтерий на протий из растворителя. Однако в этих же условиях
оставшийся бромбензол не вступал в реакцию обмена водорода. Таким обра-
зом, для этих двух соединений нужно предположить разные механизмы элими-
нирования. Хлор во время развития анионного центра в орто-положении
может оставаться связанным с бензольным кольцом, так как амид-ион являет-
ся достаточно сильным основанием, чтобы при этих обстоятельствах могло
произойти образование аниона. Таким образом, процесс элиминирования
описывается двухстадийным механизмом, который впоследствии мы отнесем
к типу мономолекулярного элиминирования с образованием основания, сопря-
женного субстрату; этот механизм обозначается символом Е1сВ.
Н
331
Глава VI
С другой стороны, связь углерод — бром при развитии заряда в орто-поло-
жеиии не может сохраниться, и поэтому происходит отщепление аниона
брома, уносящего отрицательный заряд. Этот процесс происходит в одну
стадию и относится к типу бимолекулярного элиминирования, обозначае-
мого символом Е2;
В каждом случае образовавшийся дегидробензол быстро реагирует с эле-
ментами аммиака, вероятно вначале с амид-ионом, а затем с протоном. Опыты
с хлорбензолом показывают, что аминирование включает первоначальное
отщепление орто-протона. В случае бромбепзола отщепление брома проис-
ходит в определяющей скорость стадии, поэтому введение в орто-положение
дейтерия вместо протия замедляет аминирование в = 5,7 раза. Кроме
того, замещение с перегруппировкой и без перегруппировки замедляется
одинаковым образом.
Промежуточное образование дегидробензола подтверждается опытами, в ко-
торых он улавливается добавленными нуклеофилами. Нуклеофилы, сами
по себе не реагирующие с галогенбензолами в жидком аммиаке, замещают
галоген в присутствии небольшого количества амида натрия. Это явление
первоначально наблюдал Бергстром [303], а затем объяснил Робертс
в работе [304].
Так, в присутствии фенолят- или фенилтиолат-ионов в жидком аммиаке
вместо анилина, нормального продукта аминирования, в значительном коли-
честве образуется диэтиловый эфир или дифенилсульфид. Единственным
анионом в этой системе, который может отщеплять ароматический протон,
является амид-ион. Однако при последующем присоединении к ненасыщенно-
му атому углерода, фенолят- и тиофенолят-ионы успешно конкурируют
с амид-ионом, причем наибольшей реакционной способностью обладает тио-
фенолятный ион.
Вероятно, по дегидробензольному механизму частично происходит также
гидролиз арилгалогенидов водными растворами щелочей при высоких тем-
пературах. Так, .м-хлортолуол дает смесь о-, м- и и-крезолов, а и-хлортолуол—
смесь м- и и-крезолов. Однако обычно с механизмом элиминирования
конкурирует несколько механизмов прямого замещения — SN1 и SN2, кото-
рые не приводят к образованию продуктов перегруппировки; в случае про-
стых фенилгалогенидов перегруппировка происходит лишь на 50%. Подан-
ным Робертса [305], хлорбензол, меченный 14С по углероду, связанному с гало-
геном, при гидролизе 4 н. водным раствором едкого натра при 340 °C дает
58% фенола с меткой в замещенном положении и 42% фенола с меткой в орто-
положении к гидроксилу. Из того факта, что при уменьшении основности
среды происходит и уменьшение количества перегруппированного продукта,
следует, что с механизмом элиминирования конкурирует главным образом
механизм SN1.
Дегидробензол был получен также и другими путями, помимо промотируе-
мого основаниями элиминирования, например при пиролизе или фотолизе
о-иодфенилмеркуриодида o-IC6H4HgI [306, 310] или диазотированной антра-
ниловой кислоты, т. е. внутренней соли о-П+СДЮСОд [307, ЗЮ]. При терми-
ческом образовании в присутствии фурана дегидробензол присоединяется
в положения 2,5, а к антрацену — в положения 9, 10; при этом образуются
332
Замещение в ароматическом ряду
приведенные ниже соединения [307, 308]:
1,4-Диокись 1,4-дигидронафталина Триптицен
При получении в бензоле дегидробензол дает три изомерных углеводорода:
аддукт с бензолом — бифенил, 1,4-аддукт и валентный изомер 1,2-аддукта,
приведенный ниже [309]:
Бензо(>ицикло-[2,2,2]-окгпа-
триен
Не выделен
Бензоциклооктате-
траен
При импульсном фотолизе кристаллических о-иодфенилмеркуриодида или
о-диазонийбензоата в вакууме через 10 мкс можно зафиксировать спектр
поглощения дегидробензола, имеющий интенсивную, но без внутренней
структуры полосу в области 230—270 ммк; через 1 мс этот спектр сменяется
спектром дифенилена, этот углеводород накапливается в твердой фазе и может
быть идентифицирован независимым образом [310].
Со спектральными данными согласуются результаты масс-спектров; согласно
последним, в промежуток времени, в котором происходит изменение спектра
поглощения, масса 76 заменяется на массу 152 [311].
Мы не можем с уверенностью утверждать, что неожиданно обнаруженные
перегруппировки, которые (кроме реакции Рихтера) были источником огром-
ных затруднений, возникших перед исследователями 70-х годов прошлого
века при попытке установить строение дизамещенных бензолов путем
генетических корреляций (гл. IV, разд. 1,а),— например, превращение
п-бензолдисульфокислоты и n-бромфенола под действием щелочи в резорцин
или аналогичное превращение м- и о-бромфенолов в смеси пирокатехина
и резорцина — протекают по механизму с образованием дегидробензола.
Однако это вполне возможно, так как во всех случаях реакции проводились
в сильнощелочных условиях.
8. Гомолитическое ароматическое замещение*
Возможность протекания ароматического замещения по третьему типу,
который характеризуется тем, что замещающий агент не является ни элект-
рофилом, ни нуклеофилом, а радикалом, впервые была рассмотрена Хеем
* При написании этой главы были широко использованы обзорная статья Хея [312]
и монография Уильямса [313].
333
Глава VI
[314, 315] в 1934 г. В то время к числу реакций, протекающих по такому меха-
низму, можно было отнести лишь реакцию арилировапия.
а. Арилирование. Хей исследовал синтез диарилов по методу Гомберга,
т. е. термическим разложением водных растворов солей диазония в присут-
ствии нерастворимых в воде производных бензола. По причинам, упомяну-
тым ниже, Хей интерпретировал эти реакции как гомолиз ковалентных псев-
досолей, которые находятся в водной фазе в равновесии с ионизированными
солями и экстрагируются из нее неводной ароматической фазой, где они и раз-
лагаются с образованием арильных радикалов, способных замещать водород
в молекуле ароматического растворителя:
ArN++X" Аг—N = N —X Ar-+N2 + X-
RCeH4Ar и другие продукты (Х = С1, ОСОСН3, ОН пт. д.)
Грив и Хей [314] улучшили этот метод, исключив водную фазу путем исполь-
зования перегруппировки ациларилнитрозаминов в ковалентные диазоаце-
таты (X = ОСОСН3), которую можно проводить непосредственно в бензоль-
ном растворе или растворе ароматического углеводорода. Таким образом,
растворитель арилируется в гомогенной среде
Ar — n —N = O —> Аг —N = N — О
I I
COCHg СОСНз
Даже этот метод приводил к образованию сложных смесей, которые в то время
было трудно проанализировать, поэтому каждый выделял лишь то, что мог.
Однако, несмотря на недостаточно полное знание состава реакционных
смесей, Хей сумел доказать, что в этом виде замещения заместители, отли-
чающиеся по всем полярным характеристикам, например метил, бром и нитро-
ксил, тем не менее ориентируют реагент в одни и те же положения. Кроме
того, и реакционная способность бензольного кольца с указанными заме-
стителями была почти одинаковой. (Такое явление наблюдалось потому,
что медленной стадией реакции является изомеризация; однако общий вывод
Хея, к счастью, оказался верным.) Хей [315] изучил также две другие реак-
ции, в которых образуется фенил, с точки зрения их пригодности для фени-
лирования производных бензола, а именно пиролиз фенилазотрифенилметана
и перекиси бензоила:
RGeHs
СеН5 —N = N —С(СеН5)3 -» Сен5. +N2+ .С(СеН5)з->
—> RCeH4C6H5 и т. д.
RCeHg
(СеН5СОО)2 —> 2СеН5СОО. —> 2С6Н5-Ц-2СО2 -> RCeH4C6H5 и т. д.
В этих реакциях наблюдались примерно такие же закономерности, как
и при арилировании ароматических растворителей ациларилнитрозаминами:
полярное влияние присутствующих заместителей почти не проявлялось.
Такое замещение не может быть ни электрофильным, ни нуклеофильным;
оно является гомолитическим.
Изучен также ряд других реакций фенильного радикала и показано, что
эти процессы арилирования происходят точно таким же образом, как описан-
ные выше. К таким процессам относятся пиролиз фенилдиметилтриазеиа
С6Н5 — N = N — N(CH3)2 [316], фенилиодозодибензоата С8Н51(ОСОС6Н5)2
[317] и тетрабензоата свинца РЬ(ОСОС6Н5)4 [318], а также фотолиз три-
фенилвисмута (C6H5)3Bi [319].
334
Замещение в ароматическом ряду
Однако наиболее важное значение имеют работы, опубликованные начиная
с 1952 г. в основном Хеем и Уильямсом, по количественному изучению ари-
лирования различных монозамещенных бензолов с целью установить соот-
ношение образующихся изомерных моноарилпроизводных. Из этих данных
можно вычислить факторы парциальных скоростей для арилирования разных
положений ароматического кольца, т. е. те факторы, которые отражают акти-
вирующее или дезактивирующее влияние заместителей на разные положения
бензольного кольца. Такие данные характеризуют гомолитическое арома-
тическое замещение; они показывают на его очень сильное отличие от электро-
фильного и нуклеофильного замещения.
Первое, что необходимо при проведении количественных исследований,
это четко установить стехиометрию реакции. В качестве источника радикалов
с большим или меньшим успехом применялось большинство из упомянутых
выше соединений, однако чаще всего использовалось разложение перекиси
бензоила и ацетилфенилнитрозамина. Последующая реакция замещения,
а также другие реакции, приводящие к продуктам с относительно высоким
молекулярным весом, проводились в как можно более разбавленных раство-
рах, чтобы уменьшить их скорость; при этом главная проблема состояла в том,
чтобы избежать инициирования реакций посторонними инициаторами. В этом
смысле удобным реагентом является перекись бензоила, так как при ее разло-
жении образуются только два фенильных радикала. Однако первоначально
возникающие бензоилокси-радикалы, вероятно не способные отщеплять
водород от бензола и его простейших замещенных (гл. V, разд. 2,е, табл. 60),
могут отщеплять водород от нафталина, фенолов и анилинов, а также бен-
зильный, а-алкоксильный, альдегидный и, вероятно, третичный водород
боковых цепей. Тем не менее в случае многих ароматических субстратов
стехиометрия реакции с перекисями ароилов приблизительно соответствует
уравнению
(Аг'СО2)2 + АгН Аг—Аг' + Аг'СООН + СО2
и это означает, что только один арильный радикал из молекулы перекиси
реагирует с субстратом, а отрыв протона происходит при действии арилокси-
радикала. В этих реакциях наряду с Аг — Аг' не образуется Аг2; это означает,
что замещение и отрыв протона арилокси-радикалом не являются независи-
мыми процессами. В противоположность этому в случае нафталина количе-
ство образующейся кислоты сильно превышает требуемое по приведенному
выше уравнению, и, помимо арилнафталина, образуются изомерные динаф-
тилы и нафтилбензоаты. При арилировании толуола количество продукта
замещения в боковую цепь довольно мало, и его легко можно учесть. Однако
при арилировании перекисным методом этилбензола, изопропилбензола и ани-
зола реакции в боковой цепи преобладают. В этих случаях следует перейти
к другим источникам арил-радикалов. Фенилирование нафталина изучалось
при использовании ацетилфенилнитрозамина.
Применение современных методов анализа, основанных на спектроскопии
и хроматографии, подтвердило ранее установленный факт, что при замещении
одним и тем же радикалом, полученным из разных источников при одной
и той же температуре, ориентация одинакова. Так, при фенилировании
нитробензола при 125 °C образуются нитробифенилы в количестве 56%
орто-, 15% мета- и 29% пара-изомеров (±1%) независимо от того, из какого
соединения получены фенильные радикалы: из перекиси бензоила, фенил-
азотрифенилметана или фенилиодозодибензоата.
Для определения относительных скоростей арилирования необходимо исполь-
зовать метод, учитывающий то, что медленной стадией реакции является
335
Глава VI
одна из стадий образования арильного радикала, а не атака последнего на
молекулу ароматического соединения. Поэтому относительные скорости
определялись методом конкурирующих реакций (разд. 3,а): составлялась
смесь субстратов, являющаяся растворителем, в котором происходило обра-
зование радикалов, а затем определялось соотношение продуктов арили-
рования.
Некоторые данные по ориентации и относительным скоростям арилирования,
а также факторы парциальных скоростей приведены в табл. 91.
Таблица 91
Количество изомеров, относительные скорости и факторы парциальных скоростей
для гомолитического ароматического фенилирования
Субстрат Количество изомеров а, 0/ /о Относительная скорость (С6Н6 = 1) Факторы парциальных скоростей д
орто или а мета или р пара или v А* 6 Б в В г орто или СС мета или |3 пара или v
Пиридин 58 28 14 1,04 1,0 1,8 0,87 0,87
C6H5F 54 31 15 1,03 1,7 0,95 0,86
С6Н5С1 50 32 18 1,06 1,4 1,5 1,6 1,0 1,2
С6Н5Вг 50 33 17 1,29 1,4 1,6 1,9 1,3 1,3
С6Нэ1 52 31 17 1,32 1,7 2,0 1,3 1,3
С6Н5СН3 67 19 14 1,23 е 1,9 2,5 0,71 1,0
c6h5cf3 29 41 30 1,0 0,87 1,2 1,8
СеН5С(СН3)з 24 49 27 0,64 0,46 0,94 1,0
CeH5Si(CH3)3 31 45 24 1,1 1,0 1,4 1,5
C6H5SO3CH3 53 33 14 1,5 2,4 1,5 1,3
c6h5cn 60 10 30 3,7 6,5 1,1 6,5
c6h5no2 62 10 28 2,94 3,0 5,5 0,86 4,0
CeH5COOCH3 58 17 25 1,78 3,1 0,93 2,7
CeHsCgHg 49 23 28 2,94 3,0 2,1 1,0 2,5
Нафталин 79 21 9,9 11,7 3,1
а Выходы изомеров взяты из обзорной статьи Хея [512] и монографии Уильямса [313], данные
по бензотрифториду были опубликованы позднее [320]. Во всех случаях фенильные радикалы полу-
чались из перекиси бензоила при 80 °C, за исключением бензонитрила, арилирование которого про-
водилось при 70 °C. Приведены в основном данные Хея, Уильямса и сотр. (1952—1964 гг.); резуль-
таты для пиридина, бром- и иодбензолов и для метилбензолсульфоната получены Дэнли, Греггом
и сотр. (1952 г.), а для фенилтриметилсилана — Рондесведтом и Бланчардом (1956 г.)
6 Данные, приведенные в этой колонке, определены Хеем, Уильямсом и сотр. (1952—196 4 гг.)
и пересмотрены Хеем, Орманом и Уильямсом [321]; включены исправленные данные Хея, Сондерса
и Уильямса для метилбензоата [322]. Фенильные радикалы получались из перекиси бензоила при 80°С
(начальная концентрация 0,01 моль/л).
в Данные, приведенные в этой колонке, получены в 1954 г. Дэнли и Греггом [312, 313]. Ради-
калы получались из перекиси бензоила (начальная концентрация 0,03- .0,06 моль/л при 70 °C).
г Данные, приведенные в этой колонке, за исключением нафталина, получены Хыозгеном
и Граши (1950, 1957 гг.) [312, 313]. Радикалы генерировались из ацетилфенилнитрозамина при 20 °C.
Данные для нафталина получены Дэвисом, Хеем и Уильямсом [323] с тем же источником радикалов
при 25 °C.
Д Для вычисления факторов парциальных скоростей использовались данные по скоростям из ко-
лонки А, а если их не было, то из колонки Б; данные для нафталина взяты из колонки В. Они полу-
чены при температуре 70 80 °C, кроме данных для нафталина, которые измерялись при 25 °C,
поэтому точное сравнение невозможно. Показано [322], что изменение растворителя не влияет ни на
скорости реакций, ни па соотношение изомеров, поэтому от него не зависят и факторы парциальных
скоростей.
е Относительная скорость исправлена с учетом реакции в боковой цепи (13%).
336
Замещение в ароматическом ряду
Эти данные подтверждают ранние представления о том, что выход изомерных
продуктов реакции примерно одинаков и на него не влияют группы, сильно
отличающиеся по полярности. Независимо от природы заместителя скорости
реакции имеют примерно одинаковую величину, т. е. нет ярко выраженной
активации или дезактивации. Неполярный характер замещения подтверж-
дается тем, что бензол и пиридин, которые сильно отличаются по смещению
заряда по СН-связям, ведут себя аналогично. Факторы парциальных скоро-
стей также одинаковы: исключая последние пять субстратов (табл. 91),
все они порядка единицы; при этом .чепга-факторы изменяются в пределах
0,7. . .1,5, пара-факторы — в пределах 0,9.. .1,8 и орто-факторы — в пре-
делах 0,5...2,5. В последних четырех примерах при арилировании произ-
водных бензола лгета-фактор совпадает с предыдущими, однако почти
все пара- и орто-факторы, а также а- и p-факторы для нафталина повы-
шаются.
Общей структурной особенностью орто- и пара- или а- и Р-активированных
соединений является а,Р-ненасыщенность в 2рл-системе, сопряженной с коль-
цом, в которое происходит замещение. (Включение в эту серию бифенила,
вероятно, связано с тем, что эта молекула скручена вдоль связи, соединяющей
бензольные кольца.) Следовательно, можно предположить, что при арили-
ровании стадией, на которую влияют заместители, является присоединение
арильного радикала. Образующийся радикальный аддукт может стабили-
зироваться путем сопряжения с делокализацией радикального центра по трем
положениям бензольного кольца; при отсутствии заместителей с а,р~2рл-
сопряжением делокализация этим и ограничивается, как показано ниже
для фторбензола, и, таким образом, стабильность орто-, мета-и пара-аддук-
тов примерно одинакова. Однако в случае субстратов с упомянутым выше
сопряжением между заместителем и кольцом только в мета-аддукте делока-
лизация ограничена тремя положениями в бензольном кольце, для орто-
и пара-аддуктов бензола или а- и р-аддуктов нафталина возникает дополни-
тельная стабилизация вследствие увеличения степени делокализации за счет
роста цепи сопряжения, включающей уже, кроме бензольного кольца, также
и заместитель. Ниже это иллюстрируется на примере бензонитрила. На при-
веденных ниже схемах предполагаемое положение радикального центра
обозначается точками, указано также и перераспределение связей в 2рл-
системе. Скорости реакций, конечно, связаны со стабильностью переходных
состояний, а не промежуточных продуктов, однако качественно оба эти
фактора при переходе от одного соединения к другому, вероятно, изменяются
симбатно.
Хей и Уильямс [312, 313, 324] рассмотрели влияние заместителей различной
полярности в арильных радикалах на скорости арилирования замещенных
бензолов. Полученные данные показывают, что в этой реакции наблюдается
лишь незначительное влияние указанных факторов на соотношение продуктов
и относительные скорости.
22-0772
337
Глава VI
Уже отмечалось, что в условиях, считающихся удобными для количественно-
го изучения арилирования, отщепление замещающегося водорода не пред-
шествует захвату арильного радикала, оно должно или сопутствовать при-
соединению, или следовать за ним. Из эффектов сопряжения в этой реакции
мы заключили, что отщепление водорода следует за медленной стадией при-
соединения радикала. Эти выводы подтверждаются отсутствием изотопного
эффекта водорода в субстрате: замещение и протия, и трития идет с одина-
ковой скоростью [325]. Отсюда следует, что отщепление водорода осуще-
ствляется не в определяющей скорость стадии, а в конечной быстрой
стадии.
Ароматическое арилирование представляет собой типичный пример ненасы-
щенного гомолитического замещения (по классификации, данной в гл. V,
разд. 2,ж). Если при его описании учитывать, как обычно, молекулярность
определяющей скорость стадии, то следует назвать эту реакцию бимолекуляр-
ным гомолитическим ароматическим замещением и обозначить символом SH2.
(SH2)
Очевидно, механизм Sh2 аналогичен механизмам SE2 и SN2. Мезомерный
промежуточный циклогексадиенил-радикал соответствует мезомерным ионам
циклогексадиенилия и циклогексадиенилида — промежуточным соединениям
при гетеролитических реакциях.
б. Алкилирование. В растворах, в которых образуются алкильные
радикалы, ароматические соединения вступают в реакции, включающие
алкилирование. Для этой цели могут применяться источники алкильных
радикалов, аналогичные источники арильных радикалов: ацетилалкилнитроз-
амины, перекись ацетила, фенилиодозодиацетат, тетраацетат свинца и его
гомологи; радикалы могут образовываться на аноде при электролизе карбо-
новых кислот и при фотолизе алкильных соединений тяжелых металлов.
Для получения некоторых радикалов применяют специальные методы,
например разложение ди-трет-бутилперекиси, в результате которого обра-
зуются метильные радикалы и ацетон.
В большинстве случаев выходы моноалкильных замещенных ароматических
субстратов довольно низки, так как попутно образуется большое число других
продуктов. Если из источника радикалов сначала получаются ацетокси-
радикалы, то они могут входить в субстрат в виде сложноэфирной группы.
Известно, что при действии тетраацетата свинца на нафталин образуются
не метилнафталины, а только нафтилацетаты. Причина гораздо большей
распространенности нежелательных реакций при ароматическом алкилирова-
нии по сравнению с арилированием заключается в том, что при введении
алкильной группы вводится бензильный водород, поэтому образующиеся
алкильные радикалы расходуются на дегидрирование и последующее алки-
лирование боковой цепи.
Некоторые данные относительно соотношения изомеров при монометили-
ровании представлены в табл. 92. В более ранних работах, в которых метиль-
ные радикалы получались пиролизом диацетил- или ди-шреш-бутилперекисей,
338
Замещение в ароматическом ряду
выходы смесей изомеров были малы или вообще не оценивались, поэтому
нельзя быть уверенным в том, что на эти данные не накладывают отпечаток
последующие реакции. В работе Корбета и Уильямса [328] метильные ради-
Таблица 92
Количества изомеров, образующихся при гомолитическом
ароматическом метилировании
Субстрат Количество изомера, % При- меча- ние Субстрат Количество изомера, % При- меча- ние
орто мета пара орто мета пара
CeH6F 57 37 6 а 45 18 37 а
62 28 10 а 57 26 17 в
64 25 11 б Сб^ОСНз 74 15 И б
62 28 10 а CeH5CN 48 9 43 б
СеН5Вг { 67 23 10 б C9H5NO2 66 6 28 5
а См. работу [328]. Источник радикалов CHjHgl + Av при температуре кипения ароматического
субстрата.
6 См. работу [327]. Источник радикалов ди-трет-бутилперекись при 130 .. . 145 °C.
в См. работу [326]. Источник радикалов (СНзСООЦ при 110 °C.
калы получались фотолизом метилмеркуриодида и выход изомерных метил-
замещенных аренов достигал выхода метана, как и требуется по уравнению
СНз-Н-АгН —» СН4 + Аг.
СН3-+Аг- —> СН3 —Аг
поэтому найденное соотношение изомеров должно соответствовать соотноше-
нию, в котором они первоначально образуются.
В реакции с анизолом частично происходит деметилирование боковой цепи.
В реакции с толуолом частично метилируется боковая цепь, причем, согласно
Илиелу и сотр. [326], количество этилбензола равно количеству лг-ксилола.
В общем можно сделать вывод, что наблюдаются такие же закономерности,
как и при фенилировании.
Благодаря работам Шварца и сотр. [329] были получены данные по относи-
тельным скоростям реакций различных ароматических соединений с метиль-
ными радикалами, хотя эти скорости и не связаны с определяемым соотно-
шением продуктов. Метод Шварца состоит в изучении способности каждого
ароматического соединения конкурировать за радикалы, образующиеся при
разложении перекиси ацетила, с одним и тем же стандартным алифатическим
растворителем (изооктаном). В чистом растворителе эти радикалы отрывают
водород, образуя в качестве газообразных продуктов двуокись углерода
и метан (наряду с небольшим количеством этана, образующегося при диме-
ризации метильных радикалов). В присутствии ароматического субстрата
количество метана, выделяющееся вместе с двуокисью углерода, уменьшает-
ся, так как метильные радикалы реагируют с ароматическим соединением.
Последующие реакции сводились к минимуму при использовании разбавлен-
ных растворов. Типичные данные приведены в табл. 93. Главная трудность
их интерпретации состоит в том, что не было проведено анализа даже общего»
22* <339
Глава VI
Таблица 93
Относительные скорости ароматического метилирования
перекисью ацетила в изооктане при 65 или 85 °C (по данным Шварца)
Соединение Относитель- ная скорость Соединение Относитель- ная скорость Соединение Относитель- ная скорость
С6н6 1 С6Н5СН3 1,7 СвН6СООС2Н5 5,2
С6Н5Р 2,2 Свн5осн3 0,65 С6Н5ХО2 10
С6Н5С1 4,2 CtiH5CN 12 С6Н5СОС6Н5 11
СеН5Вг 3,6 СеН5СОСН3 2,4 СеН5С6Н5 5
Нафталин 22 Антрацен Фенантрен 820 27 Тетрацен Хризен 9230 57
Пиридин 3 Хинолин Изохинолин 29 36 Карбазол 420
выхода продуктов (кроме анализа смесей образующихся изомеров), поэтому
ничего нельзя сказать о стехиометрии изученных реакций.
Скорости метилирования производных бензола напоминают относительные
скорости фенилирования, и, возможно, это можно объяснить аналогично
объяснению, данному в разд. 8,а. Специфическая особенность рассматривае-
мых данных состоит в том, что сродство к метильному радикалу резко воз-
растает при переходе к конденсированным, особенно к линейным поликонден-
сированным ароматическим системам. Аналогичные данные были опублико-
ваны Смидом и Шварцем [330] для этилирования, н-пропилирования и изо-
пропилирования. Укажем, что в общем такие же результаты были ранее полу-
чены Коойманом и Фаренхорстом [331] для относительных скоростей реакции
ароматических соединений с трихлорметильным радикалом. Теоретическое
рассмотрение, проведенное Коулсоном [332], показывает, что скорость реак-
ции определяется главным образом той степенью, в которой первоначально
образующийся радикальный аддукт (механизм Sh2, см. разд. 8,а) может
стабилизироваться делокализацией радикального центра по сопряженной
2рл-системе. В настоящее время это, вероятно, наиболее весомое доказа-
тельство 8н2-механизма для алкилирования.
в. Гидроксилирование. При ароматическом гидроксилировании гидрок-
сильные радикалы получают главным образом из реактива Фентона или
путем радиолиза воды (гл. V, разд. 2,е). То, что эти реагенты приводят к обра-
зованию одних и тех же продуктов реакции ароматического гидроксилирова-
ния, впервые было показано Стейном и Вейссом [333]. Далее было показано,
что в реакции гидроксилирования может применяться фотолиз перекиси водо-
рода [334] и фотоиндуцируемый межионный перенос электрона в гидроокиси
железа [335].
Трудностей, возникающих при установлении ясной и простой стехиометрии,
при гидроксилировании в общем больше, чем при арилировании и алкилиро-
вании, так как, помимо множества последующих реакций, гидроксилирова-
нию также предшествует ряд реакций [336]. Из-за этого, как показали Стейн
и Вейсс, прочность связей не может служить критерием преимущественного
направления реакции. Связь С6Н5 — ОН, вероятно, довольно прочна вслед-
ствие сопряжения. Связь Н — ОН очень прочна, примерно на 15 ккал/моль
340
Замещение в ароматическом ряду
(62,8-IO3 Дж/моль) более прочная, чем связь Н —С6Н5 (гл. V, разд. 2,е,
табл. 60). Некоторые из предшествующих реакций приводят к замещению
заместителя на гидроксил, например нитробензол превращается в фенол
и в три нитрофенола, причем нитрогруппа отщепляется в виде азотной кис-
лоты [336]. В результате других предшествующих реакций получаются ди-
арилы — димеры арильных радикалов, образующихся при отщеплении
водорода от субстрата [336]. Это означает, что реакция проходит через стадию
отщепления водорода с образованием арильного радикала, который частично
димеризуется, прежде чем присоединением гидроксильного радикала закон-
чится процесс замещения. Отметим, однако, что реакция гидроксилирования
протекает не только по этому механизму. Указанный механизм, как видно,
отличается от Зн2-механизма арилирования и алкилирования, первой стадией
которых является присоединение, а последующей стадией — отщепление
водорода. При гидроксилировании преобладает отщепление водорода из боко-
вой цепи, что можно объяснить большой теплотой образования воды. Из толуо-
ла образуется не только продукт окисления боковой цепи — бензальдегид,
но также и дибензил [336]. Фенол, по-видимому, вначале теряет водород
ОН-группы, однако затем образующийся радикальный центр перераспреде-
ляется между орто- и пара-положениями, по которым и происходит дальней-
шее гидроксилирование и окисление в хиноидные соединения [337].
Все другие исследованные монозамещенные бензолы дают три оксипроиз-
водных, однако вследствие сложной стереохимии определенное соотношение
изомеров имеет лишь полуколичественное значение. Величины, представ-
ленные в табл. 94, определены с точностью около 5%. Данные Даунеса [338]
Таблица 94
Количества изомеров, образующихся при гомолитическом
ароматическом гидроксилировании
Субстрат Источник •ОН Количество изомера, % Литера- тура
орто или а мета или пара
С6Н5С1 ( Реагент Фентона 45 25 30 ЗЗЗв
[ Н2О, рентгеновские лучи 30 30 40 ЗЗЗв
С6Н5СООН ( Н2О, рентгеновские лучи 50 30 20 338-
<! Н2О2, hv 50 25 25 334
[ Fe3+OH~, hv 40 40 20 335
c6h5no2 Г Реагент Фентона 25 50 25 333а
]. Н2О, рентгеновские лучи 30 30 40 3336
Нафталин Г Реагент Фентона 80 20 334
1_ Н2О2, hv 75 25 334
для бензойной кислоты и облученной воды получены с использованием пре-
цизионных методов.
Полученные данные показывают, что не существует резкого различия в ско-
ростях реакции гидроксилирования ароматических соединений, что согла-
суется с данными табл. 91 и 92. Это подтверждает общепринятую точку зре-
ния, что рассмотренные реакции относятся к гомолитическому замещению.
Что касается деталей процесса, то в настоящее время мы можем сказать
лишь то, что отщепление водорода не является конечной стадией.
341
Глава VI
СПИСОК ЛИТЕРАТУРЫ
1. Koerner W., Gazz. chim. ital., 4, 305, 446 (1874).
2. Hiibner H., Ber., 8, 873 (1875).
3. Noelting E., Ber., 9, 1797 (1876).
4. Armstrong H. E., J. Chem. Soc., 51, 258 (1887).
5. Vorldnder D., Ann., 320, 122 (1902).
6. Crum Brown A., Gibson J., J. Chem. Soc., 61, 367 (1892).
7. Fliirscheim B., J. prakt. Chem., 66, 321 (1902); 71, 497 (1905); Ber., 39, 2015 (1906);
Chem. and Ind., 44, 246 (1925); J. Chem. Soc., 1926, 1562.
8. Baker J. W., Ingold С. K., J. Chem. Soc., 1926, 2462; 1929, 423.
9. Holleman A. F., Die direkte Einfiihrung von Substituenten in den Benzolkern, Veit,
Leipzig, 1910.
10. Fry H. S., J. Am. Chem. Soc., 34, 664 (1912); 36, 248, 262, 1038 (1914); 37, 855 (1915);
The Electronic Conception of Valence and the Constitution of Benzene, Longmans
Green and Co., London and New York, 1921; Vorldnder D., Ber., 52, 263 (1919); 58,
1893 (1925); Lapworth A., Mem. Prac. Manchester Lit. and. Phil. Soc., 64, iii, 2 (1920);
J. Chem. Soc., 121, 416 (1922); Kermack W. O., Robinson R., ibid., p. 427; Lowry T. M.,
ibid., 123, 826 (1923).
11. Vorldnder D., Siebert E., Ber., 52, 282 (1919); Vorldnder D., ibid., 58, 1893
(1925).
12. Malherbe D. F. du T., Ber., 52, 319 (1919).
13. Ingold С. K., Ingold E. H., J. Chem. Soc., 127, 870 (1925).
14. Ingold С. K., Ingold E. H., J. Chem. Soc., 1926, 1310; Holmes E. L., Ingold С. K.,
Ingold E. H., ibid., p. 1328.
15. A lien J., Oxford A. E., Robinson R., Smith J. C., J. Chem. Soc., 1926, 401; Ingold С. K.,
Ingold E. H., ibid., p. 1310; Ing H. R., Robinson R., ibid., p. 1365; Goss F. R.,
Ingold С. K., Wilson I. S., ibid., p. 2440; Ingold С. K., Ann. Bept. on Progress Chem.
(Chem. Soc. London), 23, 129 (1926); Ingold С. K., Shaw F. R., J. Chem. Soc., 1927,
2918.
16. Ingold С. К., Bee. trav. chim., 48, 797 (1929).
17. Vorldnder D., Siebert E., Ber., 52, 283 (1919); Vorldnder D., ibid., 58, 1893
(1925).
18. Несмеянов A. H., Толстая T. П., Исаева Л. С., Гриб А. В., ДАН СССР, 133, 602
(1960).
19. Challenger F., Rothstein E., J. Chem. Soc., 1934, 1258.
20. Goss F. R., Ingold С. K., Wilson I. S., J. Chem. Soc., 1926, 2440; Goss F. R., Han-
hart W., Ingold С. K., ibid., 127, 250; Ingold С. K., Wilson I. S., ibid., p. 810;
Ridd J. H., J. Tennessee Acad. Sci., 40, 92 (1965).
21. Ingold С. K., Shaw F. R., Wilson I. S., J. Chem. Soc., 1928, 2030; Baker J. W., Mof-
fitt W. G., ibid., 1930, 1722; Brickman M., Johnson S., Ridd J. H., Proc. Chem. Soc.,
1962, 228; Riley F. L., Rothstein E., J. Chem. Soc., 1964, 3872; Brickman M.,
Utley J. H. P., Ridd J. H., ibid., 1965, 6851; Gastaninza A., Modro T. A., Ridd J. H.,
Utley J. H. P., ibid., 1968, B, 534.
22. Craig D. P., Bev. Pure Appl. Chem., 4, 4 (1954); Craig D. P., Magnusson E. A.,
J. Chem. Soc., 1956, 4895.
23. Holleman A. F., de Bruyn B. R., Bee. trav. chim., 19, 79 (1900); Baker J. W., J. Chem.
Soc., 1929, 2255; Knowles J. R., Norman R. О. C., ibid., 1961, 2938.
24. Baker J. W-, Wilson I. S., J. Chem. Soc., 1927, 842.
25. Holleman A. F., Bee. trav. chim., 14, 123 (1895).
26. Twist R. F., Smiles S., J. Chem. Soc., 127, 1248 (1925); Riley F. L., Rothstein E.,
J. Chem. Soc., 1964, 3860.
27. Obermiller J., J. prakt. Chem., 69, 70 (1914).
28. Ingold С. K., Ingold E. H., Shaw F. R., J. Chem. Soc., 1927, 813; Riley F. L., Roth-
stein E., J. Chem. Soc., 1964, 3860.
29. Holleman A. F., Bee. trav. chim., 18, 267 (1899).
30. Holleman A. F., Vermeulen J., de Mooy W. J., Bee. trav. chim., 33, 1 (1914).
31. Ingold С. K., Lapworth A., Rothstein E., Ward D., J. Chem. Soc., 1931, 1959.
32. Cohn H., Hughes E. D., Jones M. H., частное сообщение.
33. Fliirscheim В., Holmes E. L., J. Chem. Soc., 1928, 1067.
34. Ingold С. K., Shaw F. R., J. Chem. Soc., 1949, 575.
35. Ingold С. K., Ingold E. H., J. Chem. Soc., 1928, 2249.
36. Baker J. W., Ingold С. K., J. Chem. Soc., 1926, 2462.
37. Knowles J. R., Norman R. О. C., J. Chem. Soc., 1961, 2938.
38. Baker J. W., Ingold С. K., J. Chem. Soc., 1927, 832.
39. Baker J. W., Moffitt W. G., J. Chem. Soc., 1931, 314.
342
Замещение в ароматическом ряду
40. Cooper К. Е., Ingold С. К., J. Chem. Soc., 1927, 836.
41. Baker J. W., Cooper К. E., Ingold С. К., J. Chem. Soc., 1928, 426; Wibaut J. P.,
van Strick R., Rec. trav. chim., 77, 316 (1958); Hammond G. S., Douglas K. J., J. Am.
Chem. Soc., 81, 1184 (1959).
42. Baker J. W., Eccles A., J. Chem. Soc., 1927, 2125.
43. Lauer W. M., Noland W. E., J. Am. Chem. Soc., 75, 3689 (1953); Bonner T. G.,
Bowyer F., Williams Gwyn, J. Chem. Soc., 1953, 2650; de la Mare P. B. D., Dunn T. M.,
Harvey J. T., ibid., 1957, 923.
44. Speier J. L., J. Am. Chem. Soc., 75, 2930 (1953).
45. Ingold С. K., Ingold E. H., J. Chem. Soc., 1926, 1310; Holmes E. L., Ingold С. K.,
ibid., p. 1328.
46. Reverdin F., Crepieux P., Ber., 36, 2257 (1903); Ingold С. K., Ingold E. H., J. Chem.
Soc., 1926, 1310.
47. Brady O. L., Quick W. G. E., Welling W. F., J. Chem. Soc., 127, 2264 (1925).
48. Bardwell F. C., Rohde К., J. Am. Chem. Soc., 70, 1191 (1948).
49. Baker J. W., Wilson I. S., J. Chem. Soc., 1927, 842.
50. Knowles J. R., Norman R. О. С., I. Chem. Soc., 1961, 2938; Riley F. L., Rothstein E.,
J. Chem. Soc., 1964, 3860, 3872.
51. Harvey D. R., Norman R. О. С., J. Chem. Soc., 1962, 3822.
52. Baker J. W., J. Chem. Soc., 1929, 2257.
53. Baker J. W., Ingold С. К., J. Chem. Soc., 1930, 431; Baker J. W., Moffitt W. G., ibid.,
1931, 314.
54. LeFevre R. J. W., J. Chem. Soc., 1929, 2771; LeFevre R. J. W., Mathur F. C., ibid.,
1930, 2236; LeFevre R. J. W., LeFevre C. G., ibid., 1932, 1988.
55. Ingold С. K., Shaw F. R., J. Chem. Soc., 1927, 2918; Ingold С. K., Rec. trav. chim.,
48, 797 (1929); J. Chem. Soc., 1933, 1120.
56. Ingold С. K., Shaw F. R., J. Chem. Soc., 1927, 2918.
57. Ingold С. K., Lapworth A., Rothstein E., Ward D., J. Chem., Soc., 1931, 1959.
58. Ingold С. K., Smith M. S., J. Chem. Soc., 1938, 905.
59. Bird M. L., Ingold С. К., J. Chem. Soc., 1938, 918.
60. Bardwell F. G., Rohde K., J. Am. Chem. Soc., 70, 1191 (1948).
61. Ingold С. K., Shaw F. R., J. Chem. Soc., 1949, 575.
62. Cohn H., Hughes E. D., Jones M. H., Peeling M. G., Nature, 169, 291 (1952).
63. Knowles J. R., Norman R. О. С., J. Chem. Soc., 1961, 2938.
64. Riley F. L., Rothstein E., J. Chem. Soc., 1964, 3860, 3872.
65. Tillett J. G., J. Chem. Soc., 1962, 5142.
66. Ridd J. H., Utley J. H. P., Proc. Chem. Soc., 1964, 24; Ridd J. H., Utley J. H. P.,
Brickman M., J. Chem. Soc., 1965, 6851; частное сообщение д-ра Ридда (Ridd J. H.).
67. Tolgesi W. S., Canad. J. Chem., 43, 343 (1965); Caille S. Y., Corriu J. P., Chem. Comm.,
1967, 1251.
68. Ridd J. H., Studies in Chemical Structure and Mechanism, Methuen, London, 1966,
Chap. 7.
69. Hoheman A. F., van der Arend J. E., Rec. trav. chim., 28, 408 (1909); Hollemann A. F.,
Vermeulen J., de Mooy W. J., ibid., 33, 1 (1914); Jones W. W., Russell M., J. Chem.
Soc., 1947, 921; RobertsR. M., Heilberger P., Watkins J. D., Browder H. P., Kobe К. A.,
J. Am. Chem. Soc., 80, 4285 (1958).
70. Orton K. J. L., Bradfield A. E., J. Chem. Soc., 1927, 896; Bradfield A. E., Brynmor Jones,
Trans. Faraday Soc., 37, 726 (1941); Robertson P. W., de la Mare P. B. D., Swed-
lund В. E., J. Chem. Soc., 1953, 782; Aromatic Substitution, de la Mare P. B. D. and
Ridd J. H., Butterworths Scientific Pubs., London, 1959, pp. 140 and 146; de la
Mare P. B. D., Vernon C. A., J. Chem. Soc., 1943, 246; Stock L. M., Brown H. B.,
J. Am. Chem. Soc., 82, 1942 (1960).
71. Illuminati G., Marino B., J. Am. Chem. Soc., 78, 4975 (1956); Illuminati G., ibid., 80,
4941, 4945 (1958).
72. Knowles J. R., Norman R. О. C., J. Chem. Soc., 1961, 2938; Brickman M., Johnson S.,
Ridd J. H., Proc. Chem. Soc., 1962, 228; Brickman M., Ridd J. H., J. Chem. Soc.,
1965, 6845; Brickman M., Ridd J. H., Ultey J. H. P., ibid., p. 6851; Riley F. L.,
Rothstein E., ibid., 1964, 3860, 3872; Ridd J. H., Tetrahedron, 1964, 43; частное
сообщение д-ра Ридда (Ridd J. H.).
73. Holleman A. F., Chem. Revs., 1, 218 (1925).
74. Ingold С. K., Ann. Repts. on Progress Chem. (Chem. Soc. London), 23, 140
(1926).
75. Ingold С. K., Vass С. C. N., J. Chem. Soc., 1928, 417; Lapworth A., Robinson R., Mem.
Proc. Manchester Lit. & Phil. Soc., 72, 243 (1928).
76. De la Mare P. B. D., J. Chem. Soc., 1949, 2871.
77. LeFevre R. J. W., J. Chem. Soc., 1933, 980; 1934, 1501.
343
Глава VI
78. Holleman A. F., Vermeulen J., de Mooy W. J., Rec. trav. chim., 33, 1 (1914);
Ingold С. K., Lapworth A., Rothstein E., WardD., J. Chem. Soc., 1931, 1959; Cohn II.,
Hughes E. D., Jones M. H., Peeling M. G., Nature, 169, 291 (1952).
79. Cline E. L.,Reid E. E., J. Am. Chem. Soc., 49, 3150 (1927); Brown H. C., Bonner W. H..
ibid., 76, 605 (1954).
80. Vavon G., Collier A., Bull. soc. chim. (France), 41, 357 (1927).
81. Cohn H., Hughes E. D., Jones M. H., Reeling M. G., Nature, 169, 291 (1952).
82. Brady 0. L., Day J. N. E., J. Chem. Soc., 1934, 114.
83. Battegay M., Haeffely P., Bull. soc. chim. (France), 35, 981 (1924).
84. de la Mare P. B. D., Harvey J. T., J. Chem. Soc., 1956, 63; 1957, 131; de la
Mare P. B. D., Harvey J. T., Hassan M., Varma S., ibid., 1958, 2756; Brown II. C.,
Stock L. M., J. Am. Chem. Soc., 79, 1421, 5175 (1957); Aromatic Substitution, by de
la Mare P. B. D., Ridd J. H., Butterworths Scientific Pubs., London, 1959, Chaps. 8—
10; Stock L. M., Himoe H., J. Am. Chem. Soc., 83, 1937 (1961).
85. de la Mare P. B. D., в книге «Progress in Stereochemistry», Editors Klyne W. and
de la Mare P. B. D., Butterworths Scientific Pubs. London, 1958, Vol. 2, p. 72, and
references (since 1949) there cited.
86. de la Mare P. B. D., J. Chem. Soc., 1949, 2871.
87. Holleman A. F., Rec. trav. chim., 24, 140 (1905).
88. Holleman A. F., de Bruyn B. R., Rec. trav. chim., 19, 189 (1900).
89. Holleman A. F., de Bruyn B. R., Rec. trav. chim., 19, 364 (1900).
90. Holleman A. F., Rec. trav. chim., 32, 134 (1913).
91. Ingold С. K., Ingold E. H., J. Chem. Soc., 1928, 2249.
92. Holleman A. F., Vermeulen J., de Mooy W. J., Rec. trav. chim., 33, 1 (1914); Fltir-
scheim B., Holmes E. L., J. Chem. Soc., 1928, 1607; Ingold С. K., Shaw F. R., ibid.,
1949 575.
93. Baker J. W., Cooper К. E., Ingold С. К., J. Chem. Soc., 1928, 426.
94. Fliirscheim B., Holmes E. L., cm. [92]; Baker J. W., Ingold С. К., J. Chem. Soc., 1927,
832.
95. Holleman A. F., Vermeulen J., de Mooy W. J., cm. [92]; Ingold С. K., Lapworth A.,
Rothstein E., WardD., J. Chem. Soc., 1931, 1959; Cohn H., Hughes E. D., Jones M. H.,
частное сообщение.
96. Holleman A. F., Vermeulen J., de Mooy W. J., cm. [92]; Fliirscheim B., Holmes E. L.,
cm. [92].
97. Roberts J. D., Sanford J. K., Sixma F. L. J., Cerfontain H., Zeigt R., J. Am. Chem.
Soc., 76, 4525 (1954).
98. Brady O. L., Harris S., J. Chem. Soc., 123, 484 (1923); Baker J. W., Moffitt W. G.,
ibid., 1931, 314.
99. Camps R., Arch. Pharm., 240, 1 (1901); Baker J. W., Moffitt W. G., cm. [98].
100. Holleman A. F., Rec. trav. chim., 18, 267 (1899).
101. Baker J. W., Cooper К. E., Ingold С. К., J. Chem. Soc., 1927, 836.
102. Baker J. W., Ingold С. K., J. Chem. Soc., 1928, 436; Wibaut J. P., van Strik R., Rec.
trav. chim., 77, 316 (1958).
103. Holleman A. F., de Bruyn B. R., Rec. trav. chim., 19, 79 (1900).
104. Ingold С. K., Ann. Repts. on Progress Chem. (Chem. Soc. London), 23 140
(1926).
105. Lapworth A., Robinson R., Mem. Proc. Manchester Lit. & Phil. Soc., 72, 243
(1928).
106. Roberts J. D., Streitwieser A., J. Am. Chem. Soc., 74, 4723 (1952); Brown R. D., ibid.,
75, 4077 (1953).
107. De la Mare P. B. D., Ridd J. H., Aromatic Substitution, Butterworths Scientific
Pubs., London, 1959, p. 82.
108. Baker J. W., Wilson I. S., J. Chem. Soc., 1927, 842.
109. Gattermann L., Liebermann H., Ann., 393, 198 (1912).
110. Veibel S., Ber., 63, 1577 (1930).
111. Bunton C. A., Hughes E. D., Ingold С. K., Jacobs D. I. H., Jones M. H., Minkoff G. J.,
Reed R. I., J. Chem. Soc., 1950, 2628.
112. Millen D. J., J. Chem. Soc., 1950, 2600.
113. LeFevre R. J. W., J. Chem. Soc., 1931, 810.
114. Ingold С. K., Vass С. C. N., J. Chem. Soc., 1928, 417; Lapworth A., Robinson R.,
Mem. Manchester Lit. & Phil. Soc., 72, 243 (1928).
115. de la Mare P. B. D., J. Chem. Soc., 1949, 2871.
116. Holleman A. F., Rec. trav. chim., 22, 263 (1903).
117. Hodgson H. H., Beard H. G., J. Chem. Soc., 1926, 147.
118. Friedlander P., Schenk O., Ber., 47, 3040 (1914); Hodgson H. H., Beard H. G., см.
[117].
344
Замещение в ароматическом ряду
119. Hodgson Н. Н., Beard Н. G., J. Chem. Soc., 127, 876 (1925); 1926, 147.
120. Waters W. A., J. Chem. Soc., 1948, 727.
121. Euler H., Ann., 330, 280 (1903); Z. angew. Chem., 35, 580 (1922); Walden P., ibid.,
37, 390 (1924); Ri T., Eyring H., J. Chem. Phys., 8, 433 (1940); Price С. C., Chem.
Revs., 29, 51 (1941); Westheimer F. H., Kharasch M. S., J. Am. Chem. Soc., 68,.
1871 (1946); Bennett G. M., Brand J. C. D., Williams G., J. Chem. Soc., 1946
869.
122. Hughes E. D., Ingold С. K., Reed R. I., Nature, 158, 448 (1946); Gillespie R. J., Gra-
ham J., Hughes E. D., Ingold С. K., Peeling E. R. A., ibid., p. 480; Ingold С. K.,
Millen D. J., Poole H. G., ibid., Goddard D. R., Hughes E. D., Ingold С. K., ibid.
For a review see Gillespie R. J., Millen D. J., Quart. Rev. Chem. Soc., 2 277
(1948).
123. Hantzsch A., Z. physik Chem., 65, 41 (1908).
124. de Robles C. R., Moles E., Anales fis. у quim. (Madrid), 32, 474 (1935).
125. Hantzsch A., Rer., 58, 941 (1925).
126. Усанович M., Успехи физической химии, 2, 239 (1935); 3, 703 (1935).
127. Gillespie R. J., Graham J., Hughes E. D., Ingold С. K., Peeling E. R. A., Nature, 158
480 (1946).
128. Gillespie R. J., Hughes E. D., Ingold С. К., J. Chem. Soc., 1950, 2473.
129. Gillespie R. J., J. Chem. Soc., 1950, 2493.
130. Gillespie R. J., Graham I., Hughes E. D., Ingold С. K., Peeling E. R. A., J. Chem.
Soc., 1950, 2504.
131. Medard L., Compt. rend., 199, 1615 (1934).
132. Susz B., Briner E., Helv. Chim. Acta, 18, 378 (1935).
133. Chedin J., Compt. rend., 200, 1397 (1935); 201, 552, 714 (1935); 202, 220, 1067 (1936);
203, 772, 1509 (1936); Ann. chim., 8, 243 (1937).
134. Bennett G. M., Brand J. C. D., Williams G., J. Chem. Soc., 1946, 869.
135. Ingold С. K., Millen D. J., Poole H. G., Nature, 158, 480 (1946); J. Chem. Soc., 1950,
2576.
136. Millen D. J., J. Chem. Soc., 1950, 2600.
137. Ingold С. K., Millen D. J., J. Chem. Soc., 1950, 2612.
138. Hantzsch A., Ber., 58, 958 (1925); Hantzsch A., Berger K., Ber., 61, 1328 (1928).
139. Goddard D. R., Hughes E. D., Ingold С. K., Nature, 158, 480 (1946); J. Chem. Soc.,
1950 2559.
140. Gillespie R. I., J. Chem. Soc., 1950, 2516; Millen D. J., ibid., p. 2589.
141. Millen D. J., Goddard D. R., Hughes E. D., Ingold С. K., Nature, 158, 480 (1946);
Millen D. J., J. Chem. Soc., 1950, 2606.
142. Ingold С. K., Millen D. J., Poole H. G., Nature, 158, 480 (1946).
143. Chedin J., Pradier J. C., Compt. rend., 203, 722 (1936).
144. Schmeisser M., Elischer S., Z. Naturforsch., 7b, 583 (1952); Kuhn S. J., Olah G. A.,
J. Am. Chem. Soc., 83, 4570 (1961).
145. Cox F. G., Jeffery G. A., Truter M. R., Nature, 162, 259 (1948); Truter M. R., Cruiks-
hank D. J., Jeffery G. A., Acta Cryst., 13, 855 (1960).
146. Grison P. E., Ericks K., de Vries J. L., Acta Cryst., 3, 290 (1950).
147. Martinsen H., 7.. physik. Chem., 50, 385 (1904); ibid., 59, 605 (1907).
148. Klemenc A., Scholler K., Z. anorg. u. allgem. Chem., 141, 231 (1924); Lauer K., Oda R.,
J. prakt. Chem., 144, 176 (1936); Ber., 69, 1061 (1936); Oda R., Ueda U., Bull. Inst.
Phys. Chem. Research (Tokyo), 20, 335 (1941); Westheimer F. H., Kharasch M. S.,
J. Am. Chem. Soc., 68, 1871 (1946); Bennett G. M., Brand J. C. D., James D. M.,
Saunders T. J., Williams G., J. Chem. Soc., 1947 , 474.
149. Hughes E. D., Ingold С. K., Reed R. I., J. Chem. Soc., 1950, 2400.
150. Benford G. A., Ingold С. K., J. Chem. Soc., 1938, 929; Hughes E. D., Ingold С. K.,
Reed R. I., Nature, 158, 448 (1946); J. Chem. Soc., 1950, 2400.
151. Lee W. H., Millen D. J., J. Chem. Soc., 1956, 4463.
152. Bunton C. A., Halevi E. A., Llewellyn D. R., J. Chem. Soc., 1952, 4913; Bunton C. A.,
Halevi E. A., ibid., p. 4917; Bunton C. A., Stedman G., ibid., 1958, 2420.
153. Gillespie R. J., Millen D. J., Quart. Rev. Chem. Soc., 2, 277 (1948); Gillespie R. J.t
Hughes E. D., Ingold С. K., Millen D. J., Reed R. I., Nature, 163, 599 (1949); Gil-
lespie R. J., Norton D. G., J. Chem. Soc., 1952, 971.
154. Melander L., Nature, 163, 599 (1949); Acta Chem. Scand., 3, 95 (1949); Arkiv Kemi,
2, 213 (1950); cf. Bonner T. G., Bowyer F., Williams G., J. Chem. Soc., 1953,
2650.
155. Pfeiffer P., Wizinger R., Ann., 461, 132 (1928).
156. Wheland G. W., J. Am. Chem. Soc., 64, 1900 (1942).
157. Olah G. A., Kuhn S. J., J. Am. Chem. Soc., 80, 6541 (1958).
158. Gold V., Hughes E. D., Ingold С. K., Williams G. H., J. Chem. Soc., 1950, 2452.
345
Глава VI
159. Gillespie R. J., Hughes E. D., Ingold С. К., J. Chem. Soc., 1950, 2552.
160. Gold V., Hughes E. D., Ingold С. К., J. Chem. Soc., 1950, 2467.
161. Paul M. A., J. Am. Chem. Soc., 80, 5329 (1958); cf. Ridd J. H., Studies in Chemical
Structure and Mechanism, Methuen, London, 1966, Chap. 7.
162. Goulden J. D. S., Millen D. J., J. Chem. Soc., 1950, 2620.
163. Runton C. A., Hughes E. D., Ingold С. K., JacobsD. I. H., Jones M. H., Minkoff G. J.,
Reed R. I., J. Chem. Soc., 1950, 2628.
164. Rlackall E., Hughes E. D., Ingold С. К., J. Chem. Soc., 1952, 28.
165. Runton C. A., Hughes E. D., Ingold С. K., Jacobs D. I. H., Jones M. H., Min-
koff G. J., Reed R. I., cm. [163]; Glazer J., Hughes E. D., Ingold С. K., James A- T.,
Jones G. T., Roberts E., J. Chem. Soc., 1950, 2657; Hughes E. D., Jones G. T., ibid.,
p. 2678.
166. Noyes W. A., J. Am. Chem. Soc., 23, 460 (1901); Stieglitz J., ibid., p. 797.
167. Lewis G. N., J. Am. Chem. Soc., 38, 762 (1916).
168. Arotsky J., Mishra H. C., Symons A. C. R., J. Chem. Soc., 1961, 15.
169. Gillespie R. J., Milne J. M., Chem. Comm., 1966, 158.
170. Soper F. G., Smith G. F., J. Chem. Soc., 1926, 1582.
171. de la Mare P. R. D., Hughes E. D., Vernon C. A., Research, 3, 192, 242 (1950).
172. Derbyshire D. H., Waters W. A., J. Chem. Soc., 1951, 73.
173. de la Mare P. R. D., Ketley A. D., Vernon С. A., J. Chem. Soc., 1954, 1290; de la
Mare P. R. D., Harvey J. T., Hassan M., Varma S., ibid., 1958, 2756.
174. Rell R. P., Gelles E., J. Chem. Soc., 1951, 2734.
175. Mulliken R. S., quoted and discussed by de la Mare P. B. D. and Ridd J. H., Aromatic
Substitution, Butterworths Scientific Pubs., London, 1959, p. 118.
176. Swain C. G., Ketley A. D., J. Am. Chem. Soc., 77, 3410 (1955).
177. Шилов E. А., Еайнштейн Ф. M., Ясников А. А., Кинетика и катализ, 2, 214 (1961);
Arotsky J., Symons M. C. R., Quart. Rev. Chem. Soc., 16, 194 (1962).
178. Ridd J. H., Ann. Repts. Chem. Soc., 58, 163 (1961).
179. de la Mare P. R. D., Ridd J. H., Aromatic Substitution, Butterworths Scientific Pubs.,
London, 1959, p. 116.
180. de la Mare P. R. D., Ketley A. D., Vernon C. A., Research, 6, 12S (1953); de la
Mare P. R. D., Hilton I. C., Vernon С. A., J. Chem. Soc., 1960, 4039; de la Ma-
re P. R. D., Hilton I. C., Varma S., ibid., p. 4044.
181. Francis A. W., J. Am. Chem. Soc., 47, 2340 (1925).
182. Шилов E. А., Каняев H. П., ДАН СССР, 24, 890 (1939).
183. Wilson W. J., Soper F. G., J. Chem. Soc., 1949, 3376.
184. Derbyshire A. E., Water W. A., Nature, 164, 446 (1949); J. Chem. Soc., 1950, 564,
574.
185. Robertson P. W., de la Mare P. R. D., Johnston W. T. G., J. Chem. Soc., 1943, 276;
de la Mare P. R. D., Robertson P. W-, ibid., 1948, 100.
186. Rradfield A. E., Davies G. I., Long E., J. Chem. Soc., 1949, 1389; Rranch S. J.,
Jones R., ibid., 1954, 2317.
187. Soper F. G., Smith G. F., J. Chem. Soc., 1927, 2757.
188. Painter R. S-, Soper F. G., J. Chem. Soc., 1947, 342.
189. Rerliner G., J. Am. Chem. Soc., 72, 4003 (1950); 73, 4307 (1951).
190. Rerliner E., J. Am. Chem. Soc., 78, 3632 (1956); 80, 856 (1958).
191. de la Mare P. R. D., Dunn T. M., Harvey J. T., J. Chem. Soc., 1957, 923.
192. de la Mare P. R. D., Ridd J. H., Aromatic Substitution, Butterworths Scientific Pubs.,
London, 1959, p. 121.
193. Orton K. J. P., Rradfield A. E., J. Chem. Soc., 129, 966 (1927).
194. Rertoud A., Mosset M., J. Chim. phys., 36, 272 (1934).
195. Walker I. K., Robertson P. W., J. Chem. Soc., 1939, 1515.
196. Robertson P. W., Clare N. T., McNaught K. J., Paul G. W., J. Chem. Soc., 1937, 335;
Robertson P. W., de la Mare P. R. D.t Johnston W. T. G., J. Chem. Soc., 1943,
276.
197. Williams G., Trans. Faraday Soc., 39, 729 (1941).
198. Robertson P. W-, de la Mare P. R. D., Johnston W. T. G., J. Chem. Soc., 1943,
276.
199. Christen M., Zollinger H., Helv. Chim. Acta, 45, 2057, 2066 (1962); Christen M.,
Koch W., Simon W., Zollinger H., ibid., p. 2077.
200. Stubbs F. J., Williams C. D., Hinshelwood C. N., J. Chem. Soc., 1948, 1065.
201. Cowdrey W. A., Davies D. S., J. Chem. Soc., 1949, 1471.
202. Gillespie R. J., J. Chem. Soc., 1950, 2516.
203. Rrand J. C. D., Horning W. C., J. Chem. Soc., 1952, 3622.
204. Long.F. A., Paul M. A., Chem. Rev., 57, 935 (1957).
205. Crafts J. M., Ber., 34, 1350 (1901); Bull. soc. chim. (France), 1, 917 (1907).
346
Замещение в ароматическом ряду
206. Gold V., Satchell D. Р. N., J. Chem. Soc., 1956, 1635.
207. Pinnow J., Z. Elektrochem., 21, 380 (1915); 23, 243 (1917).
208. Lantz R., Bull. soc. chim. (France), 12, 253, 1004 (1945).
209. Fuller M. W., Ingold С. К., неопубликованные данные.
210. Melander L., Acta Chim. Scand., 3, 95 (1949); Arkiv. Kemi, 2, 213 (1950); Berglund-
Larsson U., Melander L., Arkiv. Kemi, 6, 219 (1953).
211. Brand J. C. D., Jarvie J. W. P., Horning W. С., J. Chem. Soc., 1959, 3844.
212. Vicary D. R., Hinshelwood C. N., J. Chem. Soc., 1939, 1372; Wadsworth K. D.,
Hinshelwood C. N., ibid., 1944, 469; Dresel E., Hinshelwood C. N., ibid.,
p. 649.
213. Ingold С. K., Raisin C. G., Wilson C. L., Nature, 134, 734 (1934); J. Chem. Soc.,
1936 915
214. Best A. P., Wilson C. L., J. Chem. Soc., 1938, 28.
215. Bailey C. R., Best A. P., Gordon R. R., Hale J. B., Ingold С. K., Leckie A. H.,
Weldon L. H. P., Wilson C. L., Nature, 139, 880 (1937); Bailey C. R., Hale J. B.,
Herzfeld AL, Ingold С. K., Leckie A. H., Poole H. G., J. Chem. Soc., 1946,
255.
216. Kresge A. J., Chiang Y., J. Am. Chem. Soc., 81, 5509 (1959); 83, 2877 (1961); Proc.
Chem. Soc., 1961, 81.
217. Colapietro J., Long F. A., Chem. and Ind., 1960, 1056.
218. Bunnett J. F., J. Am. Chem. Soc., 83, 4056 (1961).
219. Kilpatrick M., Lubarsky J. E., J. Am. Chem. Soc., 75, 577 (1953).
220. McCaulay D. A., Lien A. P., J. Am. Chem. Soc., 73, 2013 (1951).
221. McCaulay D. A., Lien A. P., Tetrahedron, 5, 186 (1959).
222. Olah G. A., Kuhn S. J., J. Am. Chem. Soc., 80, 6535 (1958); Olah G. A., ParlethA. E.,
Olah J. A., ibid., p. 6540.
223. MacLean C., van der Waals J. H., Mackor E. L., Mod. Phys., 1, 247 (1958).
224. Klit A., Langseth A., Nature, 135, 956 (1935); Z. Physik. Chem. A176, 65
(1936).
225. Lauer W., Stedman G., J. Am. Chem. Soc., 80, 6439 (1958).
226. Gold V., Satchell D. P. N., J. Chem. Soc., 1956, 2743; Satchell D. P. N., ibid., p. 3911.
227. Meakor E. L., Smith P. G., van der Waals J. H., Trans. Faraday Soc., 83, 1309 (1957);
Lauer W., Matson G., Stedman G., J. Am. Chem. Soc., 80, 6433, 6437 (1958); Baker R.,
Earborn C., Taylor R., J. Chem. Soc., 1961, 4927.
228. Eaborn C., Taylor R., J. Chem. Soc., 1961, 247, 2388.
229. Meerwein H., Ann., 455, 327 (1927).
230. Fairbrother F., J. Chem. Soc., 1937, 313; 1941, 293; 1945, 503; Trans. Faraday Soc.,
37, 763 (1941); Siizma F. L. J., Hendricks H., Holzapffel D., Rec. trav. chim., 75, 127
(1956).
231. Olah G., Kuhn S., Olah J., J. Chem. Soc., 1957, 2174; Olah G. A., Kuhn S. J., J. Am.
Chem. Soc., 80, 6541 (1958); Doering W. von E., Saunders M., Boyton H. G., Ear-
hart H. W., Wadley E. F., Edwards W. R., Tetrahedron, 4, 1178 (1958).
232. Allen R. H., Turner A., Yates L. D., J. Am. Chem. Soc., 81, 42 (1959); Allen R. H.,
Yates L. D., ibid., p. 5289; Allen R. H., Yates L. D., Early D. S., ibid., 82, 4853 (1960);
Allen R. H., ibid., p. 4856.
233. Brown H. C., Grayson M., J. Am. Chem. Soc., 75, 6285 (1953); Jungk H., Smoot C. R.,
Brown H. C., ibid., 78, 2185 (1956); Smoot С. M., Brown H. C., ibid., pp. 6245, 6249;
Choi S. U., Brown H. C., ibid., 81, 3315 (1959); 85, 2596 (1963).
234. Топчиев А. В., Крейцелъ Б. А., Андреев Л. Н., ДАН СССР, 92, 781 (1953).
235. Calloway N. О., Chem. Rev., 17, 237 (1935); Gilman Н., Meals R. N., J. Org. Chem.,
8, 126 (1943); cf. also Schmierling L., West J. P., J. Am. Chem. Soc., 76, 1917 (1954).
236. Stock L. M., Browin H. С., в книге «Advances in Physical Organic Chemistry», Editor
Gold V., Academic Press, London and New York, 1963, vol. 1, p. 35; Allen R. H.,
Yates L. D., J. Am. Chem. Soc., 83, 2799 (1961).
237. Bethel R. D., Gold V., Chem. and Ind., 1956, 741; J. Chem. Soc., 1958, 1905; Bet-
hel R. D., Gold V., Riley T., ibid., 1959, 313.
238. Pearson D. E., Breder С. V., Craig J. С., J. Am. Chem. Soc., 86, 5054 (1964).
239. Treffers H. P., Hammett L. P., J. Am. Chem. Soc., 59, 1708 (1937).
240. Gillespie R. J., J. Chem. Soc., 1950, 2997.
241. Burton H., Praill P. F. G., J. Chem. Soc., 1950, 1203, 2034; 1951, 522, 529, 726.
242. Seel F., Z. anorg. Chem., 250, 334 (1943); Olah G. A., Kuhn S. J., Tolgyesi W. S.,
Baker E. B., J. Am. Chem. Soc., 84, 2733 (1962); OlahG. A., Kuhn S. J., Tolgyesi W. S.,
Moffatt M. E., ibid., 85, 1328 (1963).
243. Brown II. C., Jenson F. R., J. Am. Chem. Soc., 80, 2291, 2296 (1958); Brown H. C..
Marino G., ibid., 81, 3308 (1959); Brown H. C., Marino G., Stock L. M., ibid.,
p. 3210.
347
Глава VI
244. Rasmussen S. E., Broch N. S., Chem. Comm., 1965, 289.
245. Olah G. A., Moffatt M. E., Kuhn S. J., Hardie В. A., J. Am. Chem. Soc., 86, 2198
(1964); Olah G. A., Kuhn S. J., Flood S. H., Hardie B. A., ibid., p. 2203.
246. Conant J. B., Peterson W. D., J. Am. Chem. Soc., 52, 1220 (1930).
247. Wistar R., Bartlett P. D., J. Am. Chem. Soc., 63, 413 (1941).
248. Hauser C. R., Breslow D. S., J. Am. Chem. Soc., 63, 418 (1941).
249. Hammett L. P., Physical Organic Chemistry, McGraw-Hill, New York, 1940,
p. 314.
250. Meyer К. H., Leinhardt S., Ann., 398, 66 (1913); Meyer К. H., Irschick A., Schlosser H.,
Ber., 47, 1741 (1914); Meyer К. H., Schaller V., Ber., 52, 1468 (1919); Meyer К. H.,
Tochtermann H., Ber., 54, 2283 (1921).
251. Friedlander P., Monatsh., 19, 627 (1898); cf. van Hoek T. C., Verkade P. E., Wep-
ster В. M., Bee. trav. chim., 77, 559 (1958).
252. Zollinger H., Helv. Chim. Acta, 38, 1597, 1617, 1632 (1955); Stamm O. A., Zollin-
ger H., ibid., 40, 1955 (1957); Hodson H. F., Stamm O. A., Zollinger H., ibid., 41, 1816
(1958); Ernst R.. Stamm O. A., Zollinger H., ibid., p. 2274; Zollinger H., Angew. Chem.,
70, 2021 (1958).
253. Klaproth W. J., Westheimer F. H., J. Am. Chem. Soc., 72, 4461 (1950).
254. Perren C., Westheimer F. H., J. Am. Chem. Soc., 85, 273 (1963).
255. Eaborn С., J. Chem. Soc., 1956, 4856; Deans F. B., Eaborn C., ibid., 1959, 2299;
Deans F. B., Eaborn C., Webster D. E., ibid., p. 3031; Eaborn C., Lasocki Z., Web-
ster D. E., ibid., p. 3034; Eaborn C., Moore R. C., ibid., p. 3640; Eaborn C., Pan-
de К. C., ibid., 1960, 1566; 1961, 297, 3715, 5082; Eaborn C., Waters J. A., ibid.,
p. 542; Bott R. W., Eaborn C., Greaseley P. M., ibid., 1964, 4804; Bott R. W., Eaborn C.,
Walton D. R. M., J. Organmetal. Chem., 2, 154 (1964).
256. Ridd J. H., Qureshi E., J. Chem. Soc., в печати.
257. Hickinbottom W. J., J. Chem. Soc., 1933, 496.
258. Burgers J., Hoefnagel M. A., Verkade P. E., Visser H., Wepster В. M., Rec. trav. chim.,
77, 481 (1958); van Hoek R. C., Verkade P. E., Wepster В. M., ibid., p. 559; Brou-
wers J. A. C. Th., Biflma S. C., Verkade P. E., Wepster В. M., ibid., p. 1080; van
Loon A., Verkade P. E., Wepster В. M., ibid., 79, 977 (1960).
259. Robertson P. W., de la Mare P. B. D., Swedlund В. E., J. Chem. Soc., 1953, 782.
260. Chuchani G., J. Chem. Soc., 1959, 1763; 1960, 325; de la Mare P. B. D., Tetrahedron,
5, 107 (1959).
261. De la Mare P. B. D., el Dusouque О. M. H., Tillett J. G., Zellner M., J. Chem. Soc.,
1964, 5306; de la Mare P. B. D., el Dusouque О. M. H., ibid., 1967B, 251.
262. Berliner E., Tetrahedron, 5, 202 (1959).
263. Bunnett J. F., Zahler R. E., Chem. Revs., 49, 273 (1951).
264. Moelwyn-Hughes E. A., Johnson M., Trans. Faraday Soc., 36, 948 (1940).
265. Waters W. A., J. Chem. Soc., 1942, 266.
266. Crossley M. L., Kienle R. H., Benbrook С. H., J. Am. Chem. Soc., 62, 1400
(1940).
267. Cain J. C., Ber., 38, 2511 (1905); Lewis E. S., Cooper J. E., J. Am. Chem. Soc., 84,
3847 (1962).
268. Lulofs P. K., Rec. trav. chim., 20, 292 (1901).
269. Le Roux J. J., Lu C. S., Sugden S., Thomson R. W. K., J. Chem. Soc., 1945,
586.
270. Bunnett J. F., Davies G. T., J. Am. Chem. Soc., 76, 3011 (1958).
271. Ingold С. K., Rec. trav. chim., 48, 797 (1929).
272. Forest H. S., Walker Л, J. Chem. Soc., 1948, 1939.
273. Pisani F., Compt. rend., 39, 852 (1854); Austin P. T., Ber., 8, 666 (1875); van de
Vliet P. G., Rec. trav. chim., 43, 606 (1924).
274. Holleman A. F., ter Weel J., Rec. trav. chim., 35, 41 (1915).
275. Holleman A. F., Beekman J. Q., Proc. Koninkl. Nederland. Akad. Wetenschap., 6, 327
(1903); Holleman A. F., ibid., 6, 659 (1904); Holleman A. F., Beekman J. W., Rec.
trav. chim., 23, 225 (1904); Holleman A. F., de Mooy W. J., ibid., 35, 5 (1915); Bun-
nett J. F., Levitt A., J. Am. Chem. Soc., 70, 2778 (1948); Bevan C. W. L., Bye G. C.,
J. Chem. Soc., 1954, 3091.
276. Schopff M., Ber., 22, 3281 (1889); 23, 3440 (1890); 24, 3771 (1891); Grohmann A., Ber.,
23, 3445 (1890); 24, 3808 (1891); Fischer P., Ber., 24, 3785 (1891).
277. Bunnett J. F., Levitt A., J. Am. Chem. Soc., 70, 2778 (1948); Bunnett J. F., Moe H.,
Knutson D., ibid., 76, 3936 (1954).
278. Berliner E., Mottek L. С., J. Am. Chem. Soc., 74, 1574 (1952); cf. Heppolette R. L.,
Miller J., ibid., 75, 4205 (1953).
279. Sandin R. B., Liskear M., J. Am. Chem. Soc., 57, 1304 (1935).
280. Крюгер E. А., Беднова M. С., Ж0Х, 3, 67 (1933).
348
Замещение в ароматическом ряду
281. Capon В., Chapman N. В., J. Chem. Soc., 1957, 600.
282. Bunnett J. F., J. Am. Chem. Soc., 79, 5969 (1957); Bunnett J. F., Richardson J. D
ibid., 81, 315 (1959).
283. Mangini A., Frenguelli B., Gazz. chim. ital., 69, 86 (1939).
284. Smiles S. и сотр., более десяти статей в J. Chem. Soc. с 1931 до 1938 г.; первая статья:
J. Chem. Soc., 1931, 914.
285. Levy A. A., Rains Н. С., Smiles S., J. Chem. Soc., 1931, 3264.
286. Meisenheimer J., Ann., 323, 205 (1902).
287. Crampton M. R., Gold V., J. Chem. Soc., 1964, 4293.
288. Ziegler K., Zeiser H., Ber., 63, 1874 (1930).
289. Bergstrom F. W., J. Org. Chem., 2, 411 (1937).
290. Wohl A., Ber., 32, 3486 (1899).
291. Bergstrom F. W., Granera I. M., Erickson V., J. Org. Chem., 7, 98 (1942).
292. Lobry de Bruyn C. A., Rec. trav. chim., 2, 205 (1883).
293. Hepp P., Ann., 215, 344 (1882).
294. Герм. пат. 186526.
295. von Richter V., Ber., 4, 21, 459, 553 (1871); 7, 1145 (1874); 8, 1418 (1875).
296. von Richter V., Ber., 8, 1418 (1875).
297. Holleman M., Rec. trav. chim., 24, 194 (1905).
298. Bunnett J. F., Cormack J. F., McKay F. С., J. Org. Chem., 15, 481 (1950); Bunnett J. F.,
Rauhut M. H., Knutson D., Russell G. E., J. Am. Chem. Soc., 76, 5755 (1954); Bun-
nett J. F., Rauhut M. H., J. Org. Chem., 21, 934, 939, 944 (1956).
299. Rosenblum M., J. Am. Chem. Soc., 82, 3797 (1960); Samuel D., J. Chem. Soc., 1960,
1318; Ullman E. F., Bartkus E. A., Chem. and Ind., 1962, 93.
300. Kym O., J. prakt. Chem., 51, 325 (1895); Haeussermann O., Ber., 33, 939 (1900): 34
38 (1901).
301. Gilman H., Avakian S., J. Am. Chem. Soc., 67, 349 (1945); Gilman H. Grounse N. N.,
Massie S- P-, Benkeser R. A., Spatz S. M., ibid., p. 2106; Gilman H., Kyle R. H.,
Benkeser R. A., ibid., 68, 143 (1946); Gilman H., Kyle R. H., ibid., 70, 3945 (1948);
Benkeser R. A., Severson R. G., ibid., 71, 3838 (1949); Martin G. A., Iowa State Coll
J. Sci., 21, 38 (1946); Chem. Abs., 41, 952 (1947).
302. Roberts J. D., Simmons H. E., Carlsmith L. A., Vaughan C. W., J. Am. Chem. Soc.,
75, 3290 (1953): Roberts J. D., Semenow D. N., Simmons H. E., Carlsmith L. A., ibid.,
78, 601 (1956).
303. Bergstrom F. W., Wright H. E., Chandler C., Gilkey W. H., J. Org. Chem., 1, 170 (1936);
Wright H. E., Bergstrom F. W., ibid., p. 179; Bergstrom F. W., Agostinho R., J. Am.
Chem. Soc., 67, 2152 (1945); Dirstine P. H., Bergstrom F. W., J. Org. Chem., 11, 55
(1946).
304. Jenny E. F., Caserio M. C., Roberts J. D., Experientia, 14, 349 (1958); Roberts J. D.,
Spec. Pubs. Chem. Soc., 12, 115 (1958).
305. Bottini A. T., Roberts J. D., J. Am. Chem. Soc., 79, 1458 (1957).
306. Wittig G., Ebel H. F., Angew. Chem., 72, 554 (1960).
307. Stiles M., Miller R. G., J. Am. Chem. Soc., 82, 3802 (1960).
308. Stiles M., Miller R. G., Burkhardt U., J. Am. Chem. Soc., 85, 1792 (1963).
309. Miller R. G., Stiles M., J. Am. Chem. Soc., 85, 1798 (1963).
.3 10. Berry R. S., Spokes G. N., Stiles M., J. Am. Chem. Soc., 82, 5240 (1960); 84, 3570
(1962).
311. Berry R. S., Clarty J., Schafer M. E., J. Am. Chem. Soc., 86, 2738 (1964).
312. Hey D. H., Vistas in Free Radical Chemistry, Pergamon Press, London, 1959, p. 209.
313. Williams G. H., Homolytic Aromatic Substitution, Pergamon Press, London,
1960.
314. Grieve W. S. M., Hey D. H., J. Chem. Soc., 1934, 1797.
315. Hey D. H., J. Chem. Soc., 1934, 1966.
316. Elks J., Hey D. H., J. Chem. Soc., 1943, 441.
317. Hey D. H., Stirling C. J. M., Williams G. H., J. Chem. Soc., 1956, 1475.
318. Hey D. H., Stirling C. J. M., Williams G. H., J. Chem. Soc., 1954, 2747.
319. Hey D. H., Shingleton D. A., Williams G. H., J. Chem. Soc., 1963, 5612.
320. Hey D. H., Saunders F. G., Williams G. H., J. Chem. Soc., 1961, 554.
321. Hey D. H., Orman S-, Williams G. H., J. Chem. Soc., 1961, 565.
322. Hey D. H., Saunders F. C., Williams G. H., J. Chem. Soc., 1964, 3409.
323. Davies D. I., Hey D. H., Williams G. H., J. Chem. Soc., 1961, 3112.
324. Hey D. H., Moulden H. N., Williams G. H., J. Chem. Soc., 1960, 3769; Hambling J. K.,
Hey D. H., WilliamsG. H., ibid., p. 3782; 1962, 487; Hambling J. K., Hey D. H. Wil-
liams G. H., Orman S., ibid., 1961, 3108; 1965, 101.
325. Chang Shih, Hey D. H., Williams G. H., J. Chem. Soc., 1959, 1871.
326. Eliel E. L., Rabindran K., Wilen S. H., J. Org. Chem., 22, 829 (1957).
349
Глава VI
327. Cowley В. R., Norman R. О. С., Waters W. A., J. Chem. Soc., 1959, 1799.
328. Corbett G. E., Williams G. H., J. Chem. Soc., 1964, 3437.
329. Levy M., Szwarc M., J. Chem. Phys., 22, 1621 (1954); J. Am. Chem. Soc., 77, 1949
(1955); Szwarc M., J. Polymer Sci., 16, 367 (1955); Buckley R. P., Leavitt F., Szwarc M.,
J. Am. Chem. Soc., 78, 5557 (1956); Hellman W. J., Rembaum A., Szwarc M., J. Chem.
Soc., 1957, 1127; Szwarc M., J. Phys. Chem., 61, 40 (1957).
330. Smid J., Szwarc M., J. Am. Chem. Soc., 78, 3322 (1956); 79, 1534 (1957); J. Chem.
Phys., 29, 432 (1958).
331. Kooyman E. C., Farenhorst E., Trans. Faraday Soc., 49, 58 (1953).
332. Coulson C. A., J. Chem. Soc., 1955, 1435; cf. Binks J. A., Gresser J., Szwarc M., ibid.,
1960, 3944.
333. (a) Loebl H., Stein G., Weiss J., J. Chem. Soc., 1949, 2074; (6) 1950, 2704; (в) John-
son G. R. A., Stein G., Weiss J., ibid., 1951, 3275.
334. Boyland E., Sims P., J. Chem. Soc., 1953, 2966.
335. Betts H. G. C., Evans M. G., Uri N., Nature, 166, 869 (1950); Betts H. G. C., Uri N.,
J. Am. Chem. Soc., 75, 2750 (1953).
336. Merz J. H., Waters W. A., J. Chem. Soc., 1949, 2427.
337. Steine G., Weiss J., J. Chem. Soc., 1951, 3264.
338. Downes A. M., Austral. J. Chem., 11, 154 (1958).
ГЛАВА VII
ЗАМЕЩЕНИЕ В АЛИФАТИЧЕСКОМ РЯДУ *
1. Механизм нуклеофильного замещения у насыщенного атома
углерода...................................................352
2. Влияние полярных эффектов на механизм и скорость нуклео-
фильного замещения.........................................357
3. Влияние растворителя..................................379
4. Температурные эффекты при нуклеофильном замещении . . . 390
5. Каталитические эффекты................................394
6. Солевые эффекты.......................................400
7. Пространственная ориентация при нуклеофильном замеще-
нии ........................................................419
8. Пространственные эффекты при нуклеофильном замещении . 447
9. Нуклеофильное замещение у олефинового атома углерода . . 460
Ю. Электрофильное замещение в алифатическом ряду.........463
11. Гомолитическое замещение в алифатическом ряду.........478
Список литературы ......................................497
* В основу этой главы положены трн обзора Хьюза [Hughes Е. D., Trans. Faraday Soc..
34, 185, 202 (1938); 37, 603 (1941)].
1. Механизм нуклеофильного замещения у насыщенного
атома углерода
а. Нуклеофильное замещение в алифатическом ряду. Рассматриваемые
реакции, объединенные общим уравнением
Y-|-Rj — X —> RY-|-X (SN)
характеризуются тем, что новая связь образуется за счет координации,
а старая гетеролитически разрывается по месту, указанному пунктирной
линией. При этом происходит перенос электронов от замещающего агента Y
к центру замещения в алкильном радикале, а затем к отщепляемой группе X;
таким образом, в результате замещения формальный положительный заряд Y
и отрицательный заряд X возрастают на одну единицу. Кроме того, не суще-
ствует никаких ограничений в зарядном типе исходных и конечных продуктов
реакции.
К числу реакций нуклеофильного замещения относятся многие хорошо
известные реакции (табл. 58), например реакция Финкельштейна
I--L-RC1 —» RI4-C1"
реакция Меншуткина
RgN-i RC1 —* RNR3 +Cl-
ri различные типы гофмановского разложения:
OH- + RNRJ —» ROH+R3Y
I--HRNRJ ri + r;n
Другими примерами могут служить реакции кислотного или щелочного гид-
ролиза и алкоголиза алкилгалогенидов, а также гидролиз сложных эфиров —
алкилнитратов, алкилсульфатов и алкилсульфопатов:
H2O + rc1 ROH-pHCl
C2H5O- + RCI —> ROC2H5+C1-
По типу нуклеофильного замещения протекают также процессы превращения
алкилгалогенидов в нитрилы, алкилирование фенолов и тиолов, алкилиро-
вание эфиров малоновой кислоты и ацетоуксусного эфира и т. п. Этот тип
реакций замещения включает почти все процессы алкилирования, в которых
алкилирующими агентами служат алкилгалогениды, алкиловые эфиры кисло-
родных кислот типа алкилсульфатов или алкилсульфонатов, полностью
алкилированные аммониевые или другие ониевые ионы и промежуточно
образующиеся ионы алкилдиазония. Приведенное ниже уравнение является
примером реакции ониевого обмена, распределение зарядов в которой отли-
чается от всех вышерассмотренных реакций:
r;n+rsr2 rnr;+r2s
Дезаминирование при действии водных растворов азотистой кислоты также
можно рассматривать как процесс нуклеофильного замещения:
H,O-|-RN+ —> ROH + N2 + H+
В этих процессах могут участвовать любые замещенные алкильные группы,
а также ненасыщенные группы.
До 1927 г. реакции, примеры которых были приведены выше, не рассматри-
вались с единой точки зрения; считалось, что они принадлежат разным обла-
стям органической химии. Впоследствии, однако, они были объединены в один
352
Замещение в алифатическом ряду
класс реакций, известный под названием «нуклеофильное замещение у насы-
щенного атома углерода». Определенные связи расщепляются по одному
и тому же механизму. Единое толкование этих процессов стало возможным
благодаря изучению их механизма.
б. Развитие теории нуклеофильного замещения. Еще до развития пред-
ставлений об электронном характере ковалентной связи высказывались
предположения о механизме нуклеофильного замещения у насыщенного атома
углерода. Согласно одной точке зрения [1], замещение начинается с обра-
зования продукта присоединения замещающего агента к атакуемому соеди-
нению с последующим отщеплением замещаемой группы от образовавшегося
молекулярного соединения. Иное представление, выдвинутое Ле-Белем [2],
сводится к тому, что введение атакующей и отщепление замещаемой групп
представляют собой взаимозависимые синхронные процессы; позднее эта
теория была возрождена в электронной форме Льюисом [3], а физическая
интерпретация была дана Лондоном [4] (гл. П, разд. 2,г). Наконец, возможен
третий подход, заключающийся в том, что первоначально происходит диссо-
циация замещаемого соединения; такая теория была сформулирована Лоури
[5], который впервые употребил электронные представления и предположил,
что в результате диссоциации может образоваться ион карбония, способный
реагировать впоследствии с атакующим агентом.
В 1927—1930 гг. Ханхарт, Бартон, Ротстейн, Патель и автор книги трак-
товали различные процессы нуклеофильного замещения, придерживаясь тео-
рий синхронного замещения и предварительной диссоциации, иногда отдавая
предпочтение одной из них, иногда допуская, что существенное значение
имеют обе теории. К таким процессам относятся, например, гофмановское
разложение [6], реакции обмена в анионотропных системах [7], замещение
в насыщенных боковых цепях ароматических соединений [8]. Несколько
позднее реакции замещения у насыщенного атома углерода изучали Меер
и Поляньи [9], а также Олсон, Лонг и Фог [10], которые придерживались
представлений о синхронном механизме * [11]. Детальное исследование этих
реакций стало возможным после работ Хьюза и автора [12—14], показавших
в 1933—1935 гг. общность механизма всех реакций нуклеофильного замеще-
ния и установивших зависимость между синхронным механизмом и процессом
предварительной диссоциации.
в. Бимолекулярный и мономолекулярный механизмы. Основное поло-
жение состоит в том, что нуклеофильное замещение (SN) как гетеролитиче-
ская реакция в растворе может протекать по одному из двух механизмов.
Первый из них включает только одну стадию, причем изменение ковалент-
ности связей происходит одновременно в двух реагирующих молекулах;
этот механизм можно назвать бимолекулярным и обозначить SN2. Этот меха-
низм сходен с механизмом гомолитического замещения по Лондону, поскольку
образование и разрыв связей происходят одновременно; кроме того, подобно
гомолитическому замещению, одновременность изменения ковалентности
связей является основным фактором, снижающим энергию активации про-
цесса. Однако в отличие от механизма Лондона, при котором изменения
ковалентности обусловлены образованием ковалетной связи и гомолизом,
при бимолекулярном нуклеофильном механизме связи изменяются вследствие
координации и гетеролиза; при этом происходит перенос электрона от ата-
* К этому времени относится попытка доказать, что по энергетическим соображениям
образование и разрыв ковалентных связей всегда происходят синхронно, откуда следует,
что механизм предварительной диссоциации неприемлем. Однако при этом не учиты-
вали, что энергии сольватации и десольватации ионов в полярных растворителях могут
быть одного порядка с энергиями образования и разрыва ковалентных связей.
23-0772
353
Глава VII
кующего агента к месту замещения, а оттуда к отщепляемой группе. Это
может быть изображено схемой
Y^ Alk-Gc -> Y—Aik + X (Sn2)
в которой не указаны ионные заряды, поскольку последние могут быть рас-
пределены по-разному.
Оба механизма можно описать, пользуясь теорией переходного состояния.
В механизме гомолитического замещения, предложенном Лондоном, в пере-
ходном состоянии имеются две парциальные связи, длина которых больше,
чем длина образующихся ковалентных связей. Переходное состояние при
нуклеофильном замещении, протекающем по бимолекулярному механизму,
характеризуется наличием двух парциальных и частично ионных связей,
которые отличаются от образующихся связей по длине и полярности. В пере-
ходном состоянии «симметричного замещения», например при действии бро-
мид-иона (который можно пометить изотопной меткой) на бромистый алкил,
претерпевающие изменение связи можно представить как наполовину ионные,
наполовину ковалентные; каждый атом-ион брома удерживается связью
с кратностью 0,5 и несет половину величины отрицательного заряда (индук-
ционное влияние не учитывается).
( — г/2е)Вг ... Aik ... Вт ( — 1/2е)
Другое важное отличие лондоновского механизма гомолитического замеще-
ния в газовой фазе от бимолекулярного механизма нуклеофильного замеще-
ния в растворе состоит в том, что в последнем случае значительную роль
играет сольватация. Когда бы ни происходило изменение зарядов — в началь-
ной стадии, в переходном состоянии или в конечной стадии замещения,—
в любом полярном растворителе всегда наблюдается значительное влияние
электростатических сил сольватации. В связи с тем что сольватационные силы,
являющиеся силами электростатическими, не характеризуются направленно-
стью, их не обозначают в формулах, где указываются только связи. Однако
по своей энергии эти силы не менее важны, чем связи, и должны учитываться
как первостепенный фактор, определяющий величину энергии акти-
вации.
Второй возможный механизм нуклеофильного замещения включает две ста-
дии: медленный гетеролиз атакуемого соединения с последующей быстрой
координацией между образующимся карбоний-ионом и замещающим агентом.
Скорость всего процесса определяется первой стадией. Поскольку на этой
стадии только в одной молекуле происходит изменение ковалентности, этот
механизм называют мономолекулярным и обозначают SN1
Aik—X-----> Alk+ + X
Медленно
О Alk+----> Y—Aik
Быстро
(SN1)
И в этом случае знаки зарядов у X и Y опущены, так как заряды могут быть
распределены по-разному.
Следует отличать энергию первоначального гетеролиза от энергии сольвата-
ции ионов (обоих ионов, а не одного, как ошибочно утверждалось). Раз-
ность этих энергий приближается к величине теплоты ионизации в растворе
354
Замещение в алифатическом ряду
(рис. 29). Поэтому механизм SN1 осуществляется практически легко только
в достаточно полярных растворителях (что не всегда в полной мере учиты-
валось).
Предполагается, что в процессе ионизации система проходит через переходное
состояние, отвечающее максимальной энергии и характеризующееся нали-
чием растянутой парциальной связи и частичного перемещения заряда,
что может быть изображено следующим образом:
( + Se)R ... Br(-Se)
Предполагается, что исходная молекула и сольватированные ионы отделены
друг от друга энергетическим барьером такой формы, что существует не
только энергия активации ио-
низации, но и определенная
энергия активации рекомбина-
ции ионов (рис. 29). Сущест-
вование такого барьера под-
тверждается некоторыми экспе-
риментальными данными (разд.
6,г). Теоретическая интерпре-
тация данного явления заклю-
чается в следующем: электрон-
ному переносу должно предше-
ствовать растяжение связей в
молекуле, протекающее без за-
метных изменений электронной
волновой функции. Сильное из-
менение волновой функции про-
исходит позднее при приближе-
Рпс. 29. Энергетическая диаграмма ионизации
ковалентной молекулы в полярном рас-
творителе.
нии к переходному состоянию,
когда происходит перенос электрона, сопровождающийся дальнейшим разде-
лением атомов. Затем в результате переноса в общий энергетический баланс
реакции в быстро возрастающей степени начинает входить энергия сольва-
тации образующихся ионных зарядов, благодаря чему наступает момент,
когда дальнейшее увеличение расстояния между ионами сопровождается
понижением суммарной энергии системы.
г. Кинетические критерии механизма. Замещению, происходящему по
бимолекулярному механизму, отвечает кинетика реакции второго порядка
в том случае, если оба реагирующих компонента находятся в малых и кон-
тролируемых концентрациях.
Скорость = k2 [Y] [RX] (SN2, типичный случай)
Это уравнение является типичным для выражения кинетики реакций, проте-
кающих в соответствии с S ^-механизмом.
Однако подобное уравнение нельзя рассматривать как универсальное для
выражения S ^-механизма: при определенных условиях реакции, проте-
кающие по этому механизму, могут отвечать кинетике реакций первого поряд-
ка. Это наблюдается, например, в том случае, если один из реагентов нахо-
дится постоянно в большом избытке, когда замещающий агент является
одновременно одним из основных ингредиентов растворителя- Такое же
положение может создаться, если один из компонентов служит буфером при
равновесной реакции или же, наконец, если оба компонента не являются
кинетически независимыми, как это имеет место при образовании ионных
23* 355
Глава, VII
пар или других электростатически связанных групп в неполярных раство-
рителях. Конечно, возможно и промежуточное состояние, когда порядок
реакции нс является целочисленным и постоянным. Таким образом, кинетика
первого порядка — это предельный случай.
Скорость = fcj [RX] (Sn2, предельный псевдомономолекулярный случай)
Реакция замещения, протекающая по мономолекулярному механизму, также
может проходить по закону реакций первого порядка при общей скорости
процесса, равной скорости гетеролиза. Однако это также можно рассматри-
вать как предельный случай, приближенно осуществляющийся, если ско-
рость процесса, обратного гетеролизу, значительно меньше скорости коорди-
нации карбоний-иона с атакующим агентом. Такое приближение часто, хотя
и не всегда, достаточно верно, если замещающий агент присутствует в зна-
чительном избытке, например если он представляет собой один из основных
ингредиентов растворителя.
Скорость = Ад [RX] (SN1, предельный случай)
Более общим случаем является такой, при котором скорость процесса,
обратного гетеролизу, сравнима со скоростью взаимодействия между кар-
боний-ионом и замещающим агентом. Отношение соответствующих констант
скорости можно выразить некоторой константой а в кинетическом уравнении
вида
Скорость — к [RX] {1 -j-a[X]/[Y]}-1 (SN1, типичный случай)
Это уравнение выражает типичную кинетику реакции, протекающей но
механизму SN1; оно весьма характерно для данного механизма реакции,
если его можно достаточно четко отличить от более простых предельных
форм.
Приведенные выше уравнения показывают, что не всегда имеется стандарт-
ное однозначное соответствие между механизмом реакции и ее порядком.
При выборе механизма, кроме порядка реакции, следует принимать во вни-
мание все другие имеющиеся данные.
Для установления механизма реакции применяются различные критерии.
Каждый из них имеет определенные границы применения и свои слабые
стороны. Кинетический анализ является наиболее широко применимым кри-
терием. Соответствующие данные, правда, не позволяют прийти к определен-
ному заключению, если замещающий агент одновременно играет роль рас-
творителя и наблюдаемая кинетическая картина не отклоняется значительно
от реакции первого порядка. В этом случае на основании только кинетиче-
ских данных трудно судить о механизме реакции и решить вопрос, отвечает
ли кинетическая картина предельному случаю механизма SN2 благодаря
большому избытку атакующего агента или имеет место предельный случай
механизма SN1 вследствие недостаточной обратимости гетеролиза. На прак-
тике обычно применяются все возможные критерии, причем лишь в немногих
случаях не удается сделать определенных выводов.
д. Обозначения механизмов реакций. В гл. VI, разд. 5,в и г, а также
в настоящей главе, разд. 1,в и г, уже упоминались названия и символы ряда
механизмов реакций в соответствии с обычно применяемой в настоящее время
системой, согласно которой механизм бимолекулярного электрофильного
замещения обозначается символом Se2, механизм мономолекулярного нуклео-
фильного замещения — символом SN1 и т. п. Этот общий способ обозначения
механизма реакций был предложен в 1928 г. [15]; заслуживает упоминания
то, что, несмотря на повторное разъяснение вопроса [16], не всегда учиты-
356
Замещение в алифатическом ряду
валось [17], что цифры в символическом обозначении и в не меньшей степени
названия относятся к молекулярности реакции, а не к кинетическому порядку
реакции. Поскольку обозначения предназначены для отличия механизмов
реакции, молекулярность реакции играет первостепенную роль, в то время
как кинетический порядок реакции представляет собой только одну из изме-
римых количественных ее характеристик и зависит частично от механизма
реакции, а частично от факторов, не связанных с механизмом.
При применении понятия молекулярности для номенклатуры и обозначения
механизма реакции следует иметь в виду следующие обстоятельства. Сложную
реакцию можно условно характеризовать молекулярностью стадии, опре-
деляющей скорость реакции. Молекулярность той или иной стадии реакции
представляет собой число молекул, в которых обязательно происходят изме-
нения ковалентности связей. Этому определению, используемому с 1933 г.
[12] и прочно вошедшему в обиход, надо отдать предпочтение перед опреде-
лением, основанным на числе всех молекул, «принимающих участие» в реак-
ции или «вступающих» в реакцию. Несмотря на то что необходимость в такой
трактовке в свое время специально подчеркивалась [18], в некоторых работах
[19] она не была учтена; поэтому хотелось бы еще раз отметить роль молеку-
лярности для того, чтобы данное нами определение было достаточно общим
и пригодным для реакций в растворе. Обычно в подобных реакциях, в большей
или меньшей степени, участвует неизвестное число молекул растворителя,
особенно молекул полярного растворителя, а иногда и полярные молекулы
растворенного вещества. Эти молекулы оказывают электростатическое влия-
ние на взаимодействующие ионы и полярные молекулы, на процессы гетеро-
лиза и координации. Если в понятие молекулярности включить подобные
электростатически участвующие молекулы, то в этом случае почти все реакции
в растворах должны были бы описываться как «полимолекулярные»; подоб-
ная попытка была бы неудачной и привела бы к значительным затруднениям
в выработке классификации. Поэтому при определении молекулярности учи-
тываются только те молекулы, в которых происходит изменение ковалентности
связей, а электростатические процессы с участием других молекул часто
объединяются понятием сольватация. Изменение связей должно рассматри-
ваться как важнейшая характеристика механизма реакции; поэтому моле-
кулярность в приведенном выше смысле представляет собой наиболее прием-
лемую основу для первоначальной классификации механизма реакций.
Однако изменения, связанные с сольватацией, зависят от большого числа
еще мало изученных факторов; поэтому лучше считать, что такие изменения
обусловливают отклонения от механизма реакций, фундаментально характе-
ризуемых изменениями ковалентности связей, т. е. молекулярностью.
2. Влияние полярных эффектов на механизм и скорость
нуклеофильного замещения
а. Влияние строения органического радикала: алкильные радикалы.
Способность алкильных групп отдавать электроны зависит от места в гомо-
логическом ряду и от разветвленности алкильных радикалов. Во многих
случаях и индуктивный эффект, и гиперконъюгация возрастают при переходе
к высшим гомологам, при этом легкость отдачи электронов в рядах простей-
ших нормальных и разветвленных гомологов увеличивается в следующем
порядке:
СН3 < С2Н5 ~ Н-С3Н7 и высшие нормальные радикалы
СН3 <С2Н6 < ИЗ0-С3Н7 ^трет-С^Ну
357
Глава VII
Рис. 30. Изменение механизма, по-
рядка и скорости реакции
нуклеофильного замещения
RX + У = RY + X в за-
висимости от природы ал-
кильных групп (располо-
жены в порядке возраста-
ния электроположительно-
сти).
Бимолекулярное нуклеофильное замещение (SN2) включает одновременное
перемещение электрона от замещающего агента к алкильной группе и от
последней к отщепляемой группе. Как правило, эти перемещения электронов
не полностью сбалансированы в переходном состоянии, в связи с чем можно
ожидать влияния эффекта на скорость реакции. Роль этого эффекта невелика,
так как он зависит только от неполного сбалансирования перемещения элект-
ронов; для определения направленности этого эффекта необходим детальный
анализ. Как будет показано в разд. 8, группы атомов алкильного радикала,
которые находятся достаточно близко к центру замещения, также обуслов-
ливают стерический эффект. Однако, за исключением некоторых особых
структур, рассматриваемый эффект также
невелик по сравнению с описываемым ниже.
В стадии, определяющей скорость мономо-
лекулярного нуклеофильного замещения
(SN1), происходит перенос электрона от
алкильной группы к отщепляемой группе
без компенсирующего перехода электрона
к алкильному радикалу. В этом случае
следует ожидать значительного влияния
полярности на кинетику реакции. Направ-
ление этого эффекта не вызывает сом-
нения: электронодонорные заместители
должны увеличивать скорость замещения.
Предположим, что исследуется влияние
различных алкильных групп на скорость
и механизм нуклеофильного замещения,
например щелочного гидролиза алкилбро-
мидов, причем опыты проводятся в одном
и том же растворителе при одинаковой
температуре. Предположим также, что
изучаются поочередно алкильные группы,
расположенные в ряд в порядке возраста-
ния их способности к отдаче электрона.
Обычно в случае метильной группы, нахо-
дящейся в начале ряда, наблюдается
бимолекулярный механизм замещения (SN2). Но у последующих членов
ряда повышение легкости отдачи электронов влияет на скорость реакции
по мономолекулярному механизму (SN1) настолько сильно, что возмож-
ность таких реакций все увеличивается, пока, наконец, начиная с некото-
рого ряда, мономолекулярный механизм не станет преобладающим.
На рис. 30 показано, что с изменением молекулярного механизма наблюда-
ются, во-первых, изменения в абсолютной скорости реакций, во-вторых,
меняется порядок реакции. Вплоть до того места, где в ряду алкильных групп
происходит изменение механизма реакции, наблюдается медленное изменение
(чаще всего уменьшение) скорости реакции. В дальнейшем происходит значи-
тельное увеличение скорости. С другой стороны, при удачно выбранной кон-
центрации до «критической» точки протекают реакции второго порядка,
но начиная с момента изменения механизма реакции характер реакции
изменяется и в зависимости от атакующего агента наблюдается типичная
кинетическая картина, характерная для мономолекулярного механизма или
одной из предельных форм, например реакции первого порядка (разд. 1, г).
Такое влияние строения было предсказано еще тогда, когда по этому вопросу
имелись лишь отдельные отрывочные сведения [19]. В последующие годы
358
Замещение в алифатическом ряду
это влияние твердо установлено в первую очередь для важного ряда
R = CH3, С2Н5, U30-C3H7, /npem-C^Hg
для двух реакций замещения: гидролиза алкилбромидов разбавленной
щелочью в водном спирте [22] и гофмановского обменного разложения раз-
бавленных водных растворов гидроокисей сульфония [21]
ОН- + А1кВг —» AlkOH-f-Br-
OH" + AlkSR; —* AlkOH-hR'S
Аналогичная картина установлена и в случае других алкильных радикалов.
Абсолютная скорость двух указанных выше реакций при постоянной кон-
центрации реагентов в одном и том же растворителе и постоянной темпера-
туре при переходе от метильной к этильной группе немного понижается.
Далее, при переходе от этильной к изопропильной группе в зависимости от
концентрации щелочи скорость реакции незначительно повышается или
понижается, а при переходе от изопропильной к mpem-бутил ьной быстро
повышается. В случае метильной и этильной групп и сравнительно разбав-
ленных растворов обе реакции отвечают реакциям второго порядка; в слу-
чае изопропильной и mpem-бутильной групп при тех же условиях протекает
реакция первого порядка. Эти изменения иллюстрируются данными [21],
приведенными в табл. 95 для гидролиза алкилбромидов в 80%-ном водном
Таблица 95
Скорости замещения алкилбромидов
Среда: 80% С2Н3ОН [-20% Н2О (по объему); температура 55 °C
Алкилбромид Константа скорости реакции второго порядка, л-моль-i-с-1 Константа скорости реакции первого порядка, с-1 Удельная скорость а в 0,01 н. NaOH
СНдВг 2140 21,4
С2Н5ВГ 170 1,7
изо-С3Н7Вг 4,7 0,24 0,29
mpe/e-C^HgBr 1010-10-5 1010-10~5
а Удельная скорость определяется тем количеством остающегося алкилбромида, которое под-
вергается превращению в течение 1 с.
растворе этилового спирта, а также данными по гидролизу сложных алкило-
вых эфиров, например алкилнитратов [22].
Четыре группы — метильная, этильная, изопропильная и mpem-бутильная —
могут служить примерами для иллюстрации наиболее характерных явлений,
происходящих при реакциях нуклеофильного замещения в насыщенных
соединениях жирного ряда, за исключением некоторых обсуждаемых ниже
особых эффектов, которые наблюдаются для более разветвленных групп,
например неопентильной, три-шреш-бутилкарбинильной и некоторых алици-
клических групп. Пока можно ограничиться указанием на то, что высшие
нормальные группы ведут себя в основном аналогично этильной, незначи-
тельно отличаясь лишь по скорости реакций (высшие гомологи реагируют
несколько медленнее, чем этильные соединения). Порядок реакции при ана-
359
Глава VII
логичных условиях остается неизменным. Так, реакция замещения первичных
бромистых алкилов с алкоксильными ионами в безводном этиловом спирте
всегда является реакцией второго порядка; скорости этой реакции [23]
при 55 °C для разных RBr, отнесенные к скорости реакции метилбромида,
соответственно равны:
СН3 17,6; С2Н5 1,00; н-С3Н7 0,31; н-С4Н9 0,23; н-С5Н14 0,21
Аналогичные реакции алкилиодидов с фенолят-ионами также относятся
к реакциям второго порядка. Сегаллер [24] получил количественные данные
для относительных скоростей реакции при 42,5 °C и установил, что они
мало зависят от температуры
СН3 4,84; С2Н5 1,00; н-С3Н7 0,40; н-С4Н9 0,39;
н-С3Н13 0,36; н-С7Н15 0,35; н-С8Н17 0,34; н-С1вН33 0,33
Высшие вторичные алкильные группы — 2-«-бутильная, 2-«-амильная,
3-«-амильная и 2-«-октильная — ведут себя в основном подобно изопропиль-
ной группе. По механизму реакций эти группы являются пограничными:
наблюдаемые замещения протекают как реакции либо первого, либо второго
порядка. Опытные данные [25] показывают, что при этом скорость замещения
при реакциях как первого, так и второго порядка остается примерно одина-
ковой (колебания не более чем в два раза). Например, относительные скоро-
сти реакций второго порядка между бромистыми алкилами и этилат-ионом
в безводном спирте при 25 °C соответственно равны:
изо-С3Н7 1,00; 2-н-С4Н9 1,29; 2-н-С5Ни 1,16; 3-и-С5Ни 0,93
Ниже приведены значения относительных скоростей реакции первого порядка
между бромистыми алкилами и 60%-ным водным раствором этилового спирта
при 80 °C:
изо-С3Н7 1,00; 2-и-С4Н9 1,04; 2-м-С5Н14 0,79; 3-и-С5Нц 0,85; 2-н-С8Н17 0,99
Высшие третичные алкильные группы ведут себя аналогично третичной
бутильной группе, проявляя, как и последняя, сильную тенденцию к реак-
циям мономолекулярного замещения [26, 27]. Большинство измеренных
скоростей отвечает реакциям первого порядка. При переходе от одной тре-
тичной группы к другой эти скорости изменяются, но, за исключением очень
сильно разветвленных или полициклических групп, которые пока не обсуж-
даются, они различаются между собой незначительно. Эти положения могут
быть иллюстрированы данными Шортера и Хиншелвуда [27] по скорости
сольволиза (реакции первого порядка) многих третичных хлористых и йоди-
стых алкилов в 80 %-ном водном растворе этилового спирта. Ниже приведены
относительные скорости реакций при 35 °C для ряда третичных галоген-
производных с двумя метильными группами и одной более сложной:
СН3С(СН3)2 С2Н6С(СН3)2 н-С3Н7С(СН3)2 и-С4Н9С(СН3)2 н-С5НпС(СН3)2
Хлориды! 1,00 1,61 1,41 1,33 1,21
Йодиды: 1,00 1,85 1,77 1,75 1,53
Следующие данные по относительным скоростям реакций при 35 °C пока-
зывают влияние последовательной замены в третичной алкильной группе
метильных радикалов на этильные:
С(СН3)3
Хлориды: 1,00
Йодиды: 1,00
С2Н6С(СН3)2
1,61
1,85
(С2Н6)2ССН3
2,33
3,14
С(С2Н6)3
2,60
4,39
360
Замещение в алифатическом ряду
Карбониевый ион, образующийся во время первоначального акта гетеролиза,
может, далее, либо присоединять анион, либо терять протон. В результате
каждой из этих реакций может образоваться несколько продуктов: соответ-
ствующие спирт, эфир, а также один или несколько олефинов. Однако скоро-
сти всех этих одновременных реакций зависят от общей для них медленной
стадии ионизации, поэтому приведенные данные, основанные на скорости
выделения галогенид-иона, отражают скорость ионизации.
Таким образом, очевидно, что в ряду метил, любой первичный, любой вто-
ричный и любой третичный алкилы механизм нуклеофильного замещения
может измениться от бимолекулярного к мономолекулярному; это изменение
в рассмотренных реакциях обычно имеет место между первичной и вторичной
алкильными группами, а для некоторых реакций, особенно таких, как реак-
ции алкилгалогенидов в растворителях, содержащих воду,— ближе к вто-
ричным группам. Таким образом, эту точку, первоначально для простоты
названную критической «точкой» изменения механизма реакции, лучше было
бы назвать «областью». Переход от одного механизма к другому нерезок;
в достаточно полном ряду должны содержаться группы радикалов, которые
реагируют в легко осуществимых условиях по обоим механизмам.
Это можно обосновать с точки зрения простых макрохимических, а вероятно,
и молекулярных представлений. Во-первых, скорость бимолекулярного заме-
щения пропорциональна концентрации атакующего агента, в то время как
скорость мономолекулярного замещения обычно не пропорциональна этой
концентрации. Поэтому изменение концентрации замещающего агента может
изменить относительную скорость реакции по обоим механизмам. Далее,
скорость мономолекулярного замещения часто гораздо сильнее зависит
от ионизующей способности растворителя, чем скорость бимолекулярного
замещения. Это особенно справедливо для реакций алкилгалогенидов:
например, муравьиная кислота, будучи сильно ионизующим растворителем,
способствует реакциям мономолекулярного замещения даже в случае пер-
вичных алкилгалогенидов; водный этиловый спирт и водный ацетон обладают
меньшей ионизующей способностью, а безводные спирт и ацетон — еще
менее ионизующие растворители. Иная картина, которая будет рассмотрена
в разд. 3, а, наблюдается при разложении ониевых солей, когда оба реагента
находятся в ионной форме: скорость реакции бимолекулярного замещения
в отличие от мономолекулярного весьма чувствительна к изменениям среды.
Однако и для этих реакций, как и для реакций алкилгалогенидов, справедли-
во, что оба механизма значительно отличаются друг от друга по их зависи-
мости от растворителя, природа которого может менять относительное зна-
чение обоих механизмов. Температура оказывает менее сильное влияние
на механизм реакции. При определенной реакции в случае алкильной груп-
пы, расположенной близко к критической «точке», в которой изменяется
механизм реакции (т. е. в случае группы, тенденция которой к реакциям
по одному из механизмов не проявляется достаточно четко), путем изменения
условий реакций, в особенности концентрации реагирующих веществ и раст-
ворителя, можно добиться того, что преобладающим будет один из механизмов.
Такие изменения легко осуществляются для реакций вторичных алкилга-
логенидов. В слабоионизующих растворителях, например в спиртах, и при
высокой концентрации активных нуклеофильных реагентов, например алко-
голят-, азид- или тиолат-ионов, происходит бимолекулярное замещение;
однако в сильноионизующих растворителях, например в муравьиной кис-
лоте, и при относительно небольшой концентрации активных анионов реак-
ция протекает по мономолекулярному механизму, на что указывают кине-
тические и другие характеристики процесса. Короче говоря, вторичные алкил-
361
Глава VII
галогениды находятся в пограничной «области» изменения механизма реакции.
Из этих соображений вытекает возможность осуществления реакций, соот-
ветствующих пунктирной части кривой SN2 на рис. 30. Например, в реак-
циях алкилгалогенидов можно применять настолько слабоионизующие раст-
ворители, что вторичные и, возможно, даже третичные алкилгалогениды
не будут ионизованы, и потому если и смогут реагировать, то только по SN2-
механизму. Аналогичные рассуждения применимы и в отношении кривой SN1.
Естественно, изменению механизма реакции будет содействовать использова-
ние значительных концентраций сильного нуклеофильного реагента. Ниже
приведены три примера в качестве иллюстрации рассмотренного эффекта.
Для алкилгалогенидов слабоионизующим растворителем является безводный
спирт. Вислиценус [28] использовал его для изучения взаимодействия алкил-
галогенидов с натрацетоуксусным эфиром
СОСН3 СОСН3
КаСц/ +RX —> RCH^ +NaX
\юос2н5 \юос2н5
Вислиценус установил, что скорости реакции уменьшаются в следующем
порядке:
СН3> С2Н5> изо-С3Н7> трет-С^Нд
причем в случае mpem-C4H9 скорость реакции настолько мала, что не под-
дается измерению. Постепенное уменьшение скоростей реакции в приведен-
ном ряду соответствует спадающему участку кривой S ^-механизма (рис. 30).
Меншуткин [29] исследовал реакции второго порядка между триэтиламином
и иодистыми алкилами в ацетоне
(C2H5)3N + RI -> RN(C2H5)3 + I-
Отпосительные скорости реакции при 100 °C, по данным Меншуткина, сле-
дующие:
СН3 100; С2Н5 88; н-С3Н7 1,7; и-С4Н9 1,2; н-С7Н15 0,9;
(н-С3Н17 0,9; изо-С3Н7 0,18
Исследование реакций mpem-бутилиодида оказалось невозможным из-за
выделения олефина; по-видимому, и при реакции с изопропилиодидом также
частично образуется олефин. Но, во всяком случае, полученные Меншутки-
ным результаты соответствуют следующему ряду уменьшения скоростей
для 8м2-механизма: СН3 > С2Н5 > высшие нормальные радикалы > изо-
С3Н7.
Реакции второго порядка между ионами иода и хлористыми алкилами в аце-
тоне (реакции Финкельштейна) были впервые кинетически исследованы
Конантом и Кирнером [30]
I-+RC1 —> RI-|- Cl-
Впоследствии де ла Мар, Фауден, Хьюз и др. [31] исследовали шесть из девя-
ти возможных реакций обмена
Y--J-RX —» RY + X-
где X и Y = С1, Вг, I. Если X = Y, то применялся метод меченых атомов.
Изменения скоростей в зависимости от строения алкильной группы для
всех этих реакций были качественно одинаковыми; количественные резуль-
таты этих исследований будут подробно рассмотрены ниже. Пока же сле-
362
Замещение в алифатическом ряду
дует отметить, что относительные скорости реакций простейших алкилбро-
мидов с радиоактивным бромом при 25 °C имеют следующие значения:
СНд 100; С2Н9 1,31; «-С3Н7 0,81; ггзо-СдНу 0,015; mpem-C4H9 0,004
На основании этих данных для реакций Финкельштейна имеет место после-
довательность скоростей, характерная для S ^-механизма: СН3 > С2Н5 >
> высшие нормальные радикалы > изо-С3П7 > mpem-C4H9.
Картина, обратная описанной в этих примерах, наблюдается при приме-
нении настолько хорошо ионизующих растворителей и таких слабых нук-
леофильных реагентов, что даже первичные алкилгалогениды реагируют
по ионизационному механизму SN1. Можно представить себе, что кривая
SN1 на рис. 30 при этом расположена настолько высоко, что всю ее можно
рассматривать независимо от кривой SN2.
Такого рода эффект рассмотрен в работе Бейтмана и Хьюза. Для изучения
реакций алкилгалогенидов указанные авторы использовали в качестве
растворителя муравьиную кислоту, показав на нескольких примерах, что
для этих соединений она является лучшим ионизующим растворителем,
чем вода [32]. Эти исследователи изучили гидролиз бромистых алкилов
водной муравьиной кислотой
H2O + RBr ROH + H+4-Br-
Даже в случае первичных алкилгалогенидов процесс является в основном
реакцией первого порядка, скорость которой лишь слабо зависит от нез-
начительной концентрации воды. Относительные скорости реакции [33]
при 100 °C равны
СН3Вг 1,00; С2Н5Вг 1,71; изо-С3Н7Вг 4,47; mpem-C4H9Br ~ 108
и группы можно расположить в ряд
СН3 < С2Н5 < U30-C3H7 m/?em-C4H9
В этих случаях наблюдается значительное и непрерывное увеличение ско-
рости реакции в соответствии с кривой SN1 на рис. 30.
В особых случаях механизм SnI может наблюдаться даже у первичных
алкилгалогенидов в водно-спиртовых растворах; это происходит тогда,
когда структура алкильной группы такова, что механизм SN2 стерически
невозможен. Простейшим известным примером может служить реакция
неопентилбромида (разд. 9).
С точки зрения представлений о молекулярности также нельзя ожидать
резкого перехода от бимолекулярного механизма к мономолекулярному;
другими словами, нельзя классифицировать каждый молекулярный акт
как чисто бимолекулярный или строго мономолекулярный, не учитывая
возможности промежуточного состояния. Это непосредственно следует
из рассмотрения элементарной картины обоих процессов. В одном из них
отщепляемая группа освобождается в результате действия атакующего
агента, в другом отщепление этой группы происходит без его участия. В то же
время следует допускать и возможность промежуточного состояния с уча-
стием атакующего агента и гетеролизующей системы [34]. Этот случай будет
рассмотрен в следующем разделе.
б. Влияние строения органического радикала. Алкильные радикалы,
содержащие заместители. Подобно тому как алкильные радикалы как про-
изводные метила получаются путем введения сх-алкильных заместителей,
аралкильные радикалы образуются в результате введения сх-арильных заме-
стителей. В алкильных соединениях отделение группы с ее обобщенными
электронами облегчается индуктивным эффектом: образующийся карбо-
363
Глава VII
ниевый ион стабилизируется в лучшем случае лишь вследствие гиперконъю-
гации. В аралкильных соединениях, содержащих сх-фенильные заместители,
отделение замещаемой группы облегчено более мощным механизмом элек-
тронного сопряжения. Образующийся карбониевый ион стабилизируется
вследствие сопряжения. Поэтому, как и следовало полагать, установлено,
что сх-фенильные заместители в аралкильных радикалах оказывают значи-
тельный полярный эффект как на би-, так и на мономолекулярные механизмы
нуклеофильного замещения. Особенно сильно это сказывается в случае
мономолекулярного механизма. Весьма полезно приближенное правило,
согласно которому по влиянию на механизм реакции один сх-фенильный
радикал соответствует примерно двум алкильным заместителям. Так, в слу-
чае реакций аралкилгалогенидов бензильная группа в бензилгалогенидах
является первичной, но по механизму реакции эти соединения принадлежат,
как и вторичные алкилгалогениды, к пограничной области; сх-фенилэтильный
радикал, хотя и представляет собой вторичный радикал, все же, как и тре-
тичные радикалы, в значительной степени способствует мономолекулярным
реакциям.
С целью определения механизма нуклеофильного замещения достаточно
подробно исследованы два ряда аралкильных радикалов
R = CH3, СН2С6Н5, СН(СвН5)2, С(С6Н5)3
R = CH2CgH5, СН(СН3)С6Н6, С(СН3)2СвН5
При этом первый ряд изучен в отношении обеих указанных ниже реакций
в водных растворах, а второй — только в отношении замещения галогена
OH" + RHal ROH + Hal"
OH-4-RNR3 —» ROH4-RJN
В каждом из рядов изменение механизма происходит у бензильного ради-
кала или по соседству с ним; члены ряда, расположенные справа, подвергают-
ся мономолекулярным реакциям.
Оливье [35] показал, что гидролиз хлористого бензила 50%-ным водным
раствором ацетона в присутствии щелочи можно приближенно рассматри-
вать как сумму двух реакций, из которых одна не зависит от концентрации
иона гидроксила, а вторая пропорциональна его концентрации. Он также
показал, что при использовании в качестве растворителя воды реакция
становится в еще большей степени независимой от концентрации гидроксиль-
ного иона. Подробно изучен [36] гидролиз бензгидрилхлорида. Процесс
гидролиза протекает в водном спирте, содержащем ион гидроксила, незави-
симо от концентрации последнего. Более того, детальное изучение реакции
в водном спирте и водном ацетоне показало, что кинетически этот процесс
только приблизительно соответствует реакции первого порядка, в действи-
тельности кинетика процесса соответствует общему случаю мономолеку-
лярного сольволиза. Гидролиз сх-фенилэтилхлорида в 80%-ном водном рас-
творе ацетона, содержащем ионы гидроксила, также протекает независимо
от концентрации последних [37]. Трифенилметилхлорид гидролизуется в вод-
ных растворителях настолько быстро, что реакцию нельзя точно изучить.
Большая скорость этой реакции может служить доказательством того, что
процесс отвечает мономолекулярному механизму.
В реакциях гидроокисей алкилзамещенного аммония изменение механизма
происходит в области между бензильной и бепзгидрильной группами [38].
Метильная и бензильная группы в ионах аммония отщепляются в виде соот-
ветствующих спиртов со скоростью, приблизительно пропорциональной
364
Замещение в алифатическом ряду
концентрации гидроксильных ионов. В то же время бензгидрильный радикал
отщепляется в виде бензгидрола со скоростью, не зависящей от концентрации
гидроксильных ионов. Следует полагать, что в случае первых двух радикалов
реакция идет по бимолекулярному, а в случае бензгидрильного радикала —
по мономолекулярному механизму; имеются косвенные доказательства
того, что в случае трифенилметильной группы реакция протекает по моно-
молекулярному механизму.
Гидролиз и алкоголиз аллилгалогенидов похожи на соответствующие реак-
ции бензилгалогенидов и представляют собой другой пример подобных реак-
ций алкилгалогенидов; в муравьиной кислоте гидролиз мономолекулярен [39].
Влияние и-алкильных заместителей в бензильном соединении на скорость
различных бимолекулярных и мономолекулярных процессов замещения
показано в исследовании Бивана, Хьюза и автора книги [40]. Помимо нук-
леофильного замещения в алифатических соединениях, в этих работах изуча-
лись некоторые аналогичные реакции нуклеофильного замещения по кар-
боксильной группе и в ароматическом кольце. Относительные скорости этих
реакций приведены в табл. 96. Установлено, что в тех случаях, когда
Таблица 96
Относительная скорость некоторых процессов нуклеофильного замещения
арилалкильных и арильных соединений и их н-метил- и п-трет-бутильных
производных (по данным Бивана и Хьюза)
Темпе- рату- ра, °C Моле- куляр- ность Относительная скорость
Реагенты Растворитель п-Н п-СНз п-трет- с4н9
СН2Вг + С6Н5№ (СН3)2СО 20 2 1 1,66 1,35
СН2Вг + 1- (СНз)2СО 0 2 1 1,45 1,35
-/ CH2Br-UC2H5O- С,Н5ОН 25 2 1 1,48 1,36
СН2Вг-Ь (СН3)3СО- (СНз)зСОН 60 2 1 1,32 1,18
—/ у— СН2С1 + С,Н5О- с,н5он 25 2 1 1,58 1,47
СН2Вг+Н2О нсоон 25 1 1 57,9 28,0
—/ CH2NC5H5 + C2H3O- С,Н5ОН 20 2 1 0,507 0,629
—/ СООС2Н5 у но- X Z 85%-ный водный С2Н5ОН | 25 2 1 0,436 0,616
-/ СОС1 + С2Н3ОН NO, С2Н5ОН 0 2 1 0,531 0,667
^-С1ГС,Н5О- с2н6он 50 2 1 0,173 0,310
\о2
а Данные о реакции бромистого бензила с пиридином получены
Бейкером и Натаном [41].
365
Глава VII
и-метильный заместитель в бензильной группе увеличивает скорость реакции
(во всех реакциях, сопровождающихся удалением электронов от реакцион-
ного центра), и-щрепг-бутильный радикал также увеличивает скорость
реакции, но в меньшей степени; если же и-метильный заместитель снижает
скорость реакции (в реакциях, идущих с притяжением электронов к реак-
ционному центру), и-щрет-бутильный радикал также снижает скорость
реакции, но не столь значительно. Относительная величина влияния этих
двух алкильных радикалов находится в соответствии с теорией, согласно
которой гиперконъюгационное взаимодействие с бензольным кольцом уве-
личивается или уменьшается в зависимости от изменения полярности реак-
ционного центра молекулы в переходном состоянии. Резкое различие в кине-
тических эффектах заместителей при переходе от бимолекулярного заме-
щения к мономолекулярному, протекающему с удалением электронов,
иллюстрирует не только различия в электронных характеристиках обоих
механизмов, но также и значение электромерного Н-Е-полярного эффекта
алкильных групп.
Кинетические эффекты гиперконъюгации в случае мономолекулярной реак-
ции при несколько более разнообразных алкильных радикалах можно
проиллюстрировать па примере [42] гидролиза и-алкилбензгидрилхлоридов
в 80%-ном водном растворе ацетона. Как показано в табл. 97, алкильные
Таблица 97
Скорости и аррениусовские энергии активации гидролиза
n-алкилбензгидрилхлоридов в 80 %-ном водном растворе ацетона
п ара-3 аместител ь 106 ki при 0 °C (fei, с-1) Е, ккал/моль (4,1 87-103 Дж/моль)
н 2,82 21,0
сн3 83,5 18,9
С2н5 62,6 19,4
изо-СдНу 47,0 19,8
35,9 20,1
группы по величине скоростей реакции соответствующих соединений обра-
зуют следующий ряд:
Н < {СН3 > С2Н5 > U30-C3H7 > mpem-CaHg},
причем аррениусовские энергии активации изменяются в обратном порядке.
Точно такая же последовательность изменения скоростей реакций найдена
[43] для мономолекулярного сольволиза 1,1-диметилпропаргилхлорида и его
3-алкилзамещенных RG = СС(СН3)2С1 в водном этаноле, где R = Н, СН3,
С2Н5, изо-С3Н7, wipe//z-C4II9.
Как уже отмечалось (гл. II, разд. 3,в и г), по индуктивному эффекту дейте-
рий более электроположителен, чем легкий водород, и поэтому менее элек-
троположителен по электромерному эффекту гиперконъюгации. Можно
ожидать, что гиперконъюгация будет способствовать ионизации трет-
бутилгалогенидов и других подобных соединений
н/с^с-^с’
Наблюдаемое влияние замещения водорода на дейтерий в трепг-алкилгало-
генидах на скорость мономолекулярных реакций согласуется с указанными
представлениями.
366
Замещение в алифатическом ряду
Как показал Шайнер [44], скорости мономолекулярного сольволиза таких
галогенидов при введении дейтерия в положение, в котором может прояв-
ляться гиперконъюгация с ионизующимся центром, уменьшаются обычно
на несколько процентов. Относительные скорости сольволиза шреш-амилхло-
рида в 80%-ном водном растворе этанола при О °C составляют:
С1
I
СН3СН2С(СН3)2
100
С1
I
CH3CH2C(CD3)2
56
Cl
CH3CD2(!(CH3)2
71
Cl
СН3СО2(1(СО3)2
41
Таким образом, каждый атом дейтерия в метиленовой группе уменьшает
скорость сольволиза на 16%, а каждый атом дейтерия в сх-метильных груп-
пах — на 9 %. Влияние гиперконъюгации с реакционным центром было
проиллюстрировано в работе Льюиса [45], который показал, что замена СН3
на GDз в и-СН3СвН4СН(СН3)С1 уменьшает скорость сольволиза этого хлори-
да в уксусной кислоте на 10%, тогда как соответствующее изотопное замеще-
ние в л-СН3СвН4СН(СН3)С1 не влияет на скорость. Стереохимическое усло-
вие, которое, как предполагали, необходимо для передачи эффекта гипер-
конъюгации от единственного атома водорода и состоит в том, что связь,
образуемая этим атомом, не должна лежать в плоскости образующегося
карбониевого иона *, было элегантно подтверждено Шайнером [46] на при-
мере дибензобициклооктилхлорида, формула которого приведена ниже.
Плоскость возникающего 2-карбониевого иона является плоскостью мостика,
отмеченного жирными линиями; 1- и 4-водородные атомы у головы моста
лежат в этой плоскости. Шайнер обнаружил, что, в то время как введение
двух атомов дейтерия в 3-СВ2-группы уменьшает скорость сольволиза
в 60 %-ном водном растворе этанола на 12%, введение атомов дейтерия
в положения 1 и 4, т. е. 1-GD- и 4-GD-rpynn, на скорость не влияет.
В качестве одного из примеров изменения механизма при введении неугле-
водородного заместителя в алкильный радикал можно привести соединения,
содержащие индуктивно электроноотталкивающий карбоксилат-ион СО2.
* Если СН-связь лежит в этой плоскости, то орбиталь этой связи будет ортогональной
к «пустой» орбитали карбониевого иона, т. е. интеграл перекрывания будет равен нулю.
Строго говоря, это должно относиться к переходному состоянию образования карбо-
ниевого иона, а не к самому иону. Однако, как мы увидим позднее, в переходном состоя-
нии подобного типа происходит почти полный перенос электрона, поэтому переходное
состояние можно аппроксимировать самим карбониевым ионом.
367
Глава VII
Рассмотрение этого примера важно в связи с пространственными отноше-
ниями при замещении, которые будут рассмотрены ниже. Эксперименталь-
ный материал по этому вопросу относится к сольволизу сх-галогенацилат-
ионов в воде и в водном спирте. Сольволиз бромацетат-иона изучен Даусоном
с сотр. [47], сх-бромпропионат-иона— Сентером и Вудом [48], броммалонат-
иона — Мадсеном [49], сольволиз этого же иона и сх-бромметилмалонат-иона—
Хьюзом и Тагером [50]. Последние установили, что происходит изменение
механизма в случае двух указанных ниже рядов. Основным доказательством
служит то, что гидролиз бромсодержащих радикалов, расположенных слева
от разделительных линий, ускоряется щелочами, в то время как соответ-
ствующие реакции соединений, содержащих радикалы, расположенные
справа от этой линии, не ускоряются
(S 2) J СНз СН2СОО“ I сн(соо-)2 -)
N J [СН2СН3|СН3СН(СОО-) CH3C(COO-)2J
Наиболее раннее определение области изменения механизма в случае вто-
рого ряда находится в соответствии с известным положительным влиянием
метильных заместителей на мономолекулярный механизм; сх-метильный заме-
ститель и сх-карбоксилат-ион, по-видимому, также способствуют мономоле-
кулярному замещению. Грипенберг, Хьюз и автор книги [51] впоследствии
изучили два следующих ряда карбоксилированных бромидов, для которых
преобладает мономолекулярный механизм:
(СН3)3С (СН3)2С(СОО~) СН3С(СОО-)2 т .
J. (Sn 1 во всех случаях)
(СН3)3ССН2 (СН3)3ССН(СОО-) (СН3)3СС(СОО-)2 J
Указанные авторы пришли к выводу, что каждая введенная метильная груп-
па увеличивает скорость мономолекулярного процесса примерно в 104, а каж-
дый введенный карбоксилат-ион — примерно в 103 раз. Однако рассмотрение
данных по скоростям в соответствии с уравнением Аррениуса указывает
на значительные различия в роли энергии и энтропии активации в этих двух
отношениях скоростей — различия, которые могут быть сопоставлены
с изменениями энергии и энтропии сольватации заместителей, содержащих
заряженные карбоксилат-ионы.
Можно ожидать, что мономолекулярный механизм должен быть резче выра-
жен в реакциях тех галогенидов, где заместитель, содержащий карбокси-
лат-ион, входит в аралкильную группу, например в радикал СН(С6Н5)(СОО~).
Действительно, Сентер и Таккер [52] установили, что гидролиз сх-хлор-
и сх-бромфенилацетат-ионов в воде протекает исключительно по мономолеку-
лярпому механизму.
Переходя от заместителей, содержащих карбоксилат-ионы, к галогензаме-
щенным, для которых характерен конъюгационный сдвиг электронов к кар-
бониевому центру, можно сначала рассмотреть проведенное Оливье и Вебе-
ром [53] сравнительное исследование гидролиза бензилхлорида, бензальхло-
рида и бензотрихлорида в 50%-ном водном растворе, содержащем щелочь.
Естественно, в двух последних случаях происходит последовательное заме-
щение атомов хлора; однако результаты показывают, что отщепление второго
и третьего атомов хлора происходит тотчас же после отщепления первого,
к которому поэтому в каждом случае и относится измерение скорости. Гид-
ролиз крайнего члена рассматриваемого ряда — бензилхлорида — в приме-
ненном растворителе в основном является бимолекулярным процессом,
в значительной степени зависящим от концентрации ионов гидроксила. Бенз-
альхлорид гидролизуется быстро со скоростью, не зависящей от концен-
трации гидроксильных ионов. Бензотрихлорид гидролизуется еще быстрее;
368
Замещение в алифатическом ряду
в этом случае скорость гидролиза также не зависит от концентрации ионов
гидроксила. Гидролиз бензгидрилхлорида в общем происходит так же;
это же относится и к бензгидрилиденхлориду, который, однако, реагирует
быстрее. Как показали Бенсли и Констэм [53], во всех случаях введение
хлора уменьшает аррениусовскую энергию активации гидролиза. Эти резуль-
таты позволяют предположить, что присутствующие, помимо фенильного
радикала, второй и третий атомы хлора поставляют электроны в реакцион-
ный центр, тем самым способствуя ионизации первого атома хлора и моно-
молекулярному характеру гидролиза. Каждый атом хлора будет действовать
в этом направлении; однако, как только в результате гидролиза первый
атом хлора заместится атомом кислорода, кислород также будет поставлять
электроны по такому же электромерному механизму, но значительно сильнее
ОС* р
С1—С—Cl RO—С—С1
Это приводит к тому, что вторая и третья стадии гидролиза наступают тотчас
вслед за первой.
В отсутствие фенильной группы максимально возможное накопление а-ато-
мов хлора, как, например, в СС14, все же недостаточно для осуществления
ионизации одного из них в такой степени, чтобы сделать этот процесс в водных
или спиртовых растворах преобладающим механизмом щелочного гидролиза
или алкоголиза. Для полигалогенпроизводных метана характерен особый
механизм, который будет рассмотрен в гл. IX, разд. 7.
Эффект, который не могут вызвать несколько атомов галогенов в метил-
галогенидах, осуществляется в присутствии одного метоксильного замести-
теля; метоксиметилхлорид в водных и спиртовых растворах подвергается
очень быстрому гидролизу и алкоголизу с образованием формальдегида или
формаля. На основании того, что подобного рода процессы протекают с боль-
шой скоростью, еще в 1935 г. пришли к выводу, что рассматриваемые реак-
ции являются мономолекулярными; впоследствии этот вывод был подтвер-
жден и кинетически [54]. Стадией, определяющей скорость реакции, которая
по оценкам происходит по крайней мере в 1014 раз быстрее, чем соответствую-
щая реакция хлористого метила, является ионизация атома хлора с обра-
зованием мезомерного катиона, который является частично ионом оксония,
а частично ионом карбония и быстро присоединяет соответственно гидрокси-
или алкокси-анион из молекулы растворителя
СН,6^-СН2-С1------>СН3б=СН2 + С1
Медленно
с,н5он +сн3б=сн2------> СН3ОСН2ОС2Н5+Н
Быстро
Если в качестве растворителя используется вода, то при аналогичном моно-
молекулярном замещении образуется полуацеталь формальдегида
ch361ch2-Qji —---->СН3б=СН2 + ci
медленно л
Н2О +СН3б=СН2 QCH3O сн2он + н
24-0772
369
Глава VII
После переноса протона происходит еще более быстрая вторая стадия гидро-
лиза с образованием формальдегида. Процесс в основном проходит по подоб-
ному же механизму:
-O^CHs-^ОНСНз-» о=сн2 + СН3ОН
ге-Метоксигруппа в бензил- или бензгидрилгалогенидах или аренсульфона-
тах также способствует мономолекулярному сольволизу в таких раствори-
телях, как низшие спирты или водный ацетон; скорость возрастает примерно
в 104 раз. Подобным, хотя и более слабым эффектом обладает и-фенокси-
группа: скорость возрастает в несколько сотен раз. л-Метоксигруппа не ока-
зывает такого влияния даже в малой степени. Все это [55] согласуется
с предложением о первостепенной важности электромерного эффекта в про-
цессе активации ионизации связи СХ, который стабилизует потенциальный
карбониевый ион. Более слабая по сравнению с и-метоксигруппой актива-
ция n-феноксигруппой объясняется конкурирующим сопряжением незаме-
щенной фенильной группы с неподеленной парой кислорода. Отсутствие
эффекта у л-метоксигруппы объясняется тем, что в этом случае электромер-
ный эффект не может передаваться к реакционному центру.
n-Метокси- и n-феноксибензильные системы были использованы Констэмом
[56] для установления сущности механизма реакций, протекающих в погра-
ничной области S N1 — 8 n2. Его метод, один из лучших кинетических мето-
дов, позволяющих отличить сольволитические реакции типа SN1 от реакций
типа Sn2, будет рассмотрен в разд. 4. Согласно этому и другим тестам,
гидролиз n-метокси- и n-феноксибензилхлоридов в 70 %-ном водном растворе
диоксана является реакцией SN1. В этих реакциях замещение происходит
под действием различных анионов, включая очень слабо нуклеофильные
анионы. Все анионы, более нуклеофильные, чем тетрафторборат и перхло-
рат, вступают в реакцию и дают вклад в общую скорость в виде дополни-
тельных членов второго порядка, по величине превышающие нормальные
солевые эффекты, которые могут быть учтены. Основной вопрос состоит в том,
как учесть степень участия электронов этих анионов в переходном состоянии,
в котором происходит смещение электронов от атома углерода к отщепляю-
щемуся атому хлора.
При мономолекулярном сольволизе возникающий положительный заряд
стабилизуется исключительно внутримолекулярно с помощью поляризуемой
n-оксибензильной системы. Если изменить эту систему, перейдя, например,
от и-метоксибензильной к менее электронодонорной и-феноксибензильной
системе, свободная энергия активации ионизации связи углерод — хлор
возрастет, что и отражается на соотношении скоростей: /с1(ОСН3)//с1(ОС6Н5) =
= 135 при 20 °C. Если атакующий анион, принимающий участие в бимоле-
кулярной реакции, лишь в незначительной степени снабжает электронами
центр гетеролиза и эту задачу, как и выше, выполняет внутренняя электро-
мерная система, изменение энергии активации должно быть почти таким же,
как и в указанном выше случае, и для этой бимолекулярной реакции отно-
шение /с2(ОСН3)//с2(ОСвН5) будет близко к 135 при 20 °C. Если рассмотреть
другой крайний случай, когда в переходном состоянии почти вся электрон-
ная плотность поставляется к центру гетеролиза от атакующего аниона,
а не за счет внутримолекулярного смещения, то в этом случае должно
наблюдаться лишь небольшое различие между n-метокси- и и-фенокси-
замещенными, т. е. соотношение скоростей бимолекулярных реакций
Й2(ОСН3)//с2(ОСвН5) должно быть близким к единице. Таким образом, полу-
370
Замещение в алифатическом ряду
чается шкала соотношений скоростей, ограниченная двумя указанными
случаями; подобная шкала не может быть составлена, если принимать
во внимание сами константы скоростей, а не их соотношение. С помощью
этой шкалы, переходя от слабо нуклеофильных анионов к сильно нуклео-
фильным, можно выяснить, в какой степени эти нуклеофилы поставляют
свои электроны при образовании переходного состояния: если переноса
электрона не происходит, то указанное соотношение равно 135, а если
наблюдается полный перенос, то это соотношение равно единице.
Именно таким образом Констэм интерпретировал данные табл. 98. Несмотря
на то что бензолсульфонат- и нитрат-ионы очень реакционноспособны
Таблица 98
Константы скоростей (лмоль-1-с~1) реакций замещения
n-метокси- и n-феноксибензилхлоридов различными анионами
в 70 %-ном водном растворе ацетона при 20 °C
(по данным Констэма)
Анион 104h2 (ОСНз) 10% (ОСвН5) (ОСНз)/й2 (ОСвН5)
CeH5SO3- 2,0 1,6 125
NOj 3,2 3,2 139
F" 4,2 4,9 86
Радиоактивный Cl~ 6,6 7,6 87
Вг- 7,9 50,9 15,5
Ns 34,5 710,3 4,9
в бимолекулярных реакциях, при наличии внутренней электромерной систе-
мы они вносят минимальный вклад в обеспечение электронами реакционного
центра. Фторид- и хлорид-ионы в несколько большей степени поставляют
электроны реакционному центру. Сильно нуклеофильные бромид- и азид-
ионы почти полностью отдают свои электроны при образовании переходного
состояния. Все эти реакции бимолекулярны. Однако, очевидно, они обра-
зуют целый спектр механизмов, один край которого лежит в области, погра-
ничной с мономолекулярный механизмом.
Важную группу реакций нуклеофильного замещения составляют реакции
с предварительной протонизацией субстрата; они, однако, в принципе
не отличаются от уже рассмотренных реакций нейтральных молекул и ионов.
Простые эфиры получаются из спиртов под действием сильных кислот,
а гидролизуются в водных растворах кислот: очевидно, в этих случаях
замещение происходит в ионных сопряженных кислотах спиртов и эфиров
AlkOH* и AlkOHR+; оксониевая группа аналогично SR+ и NRJ легко заме-
щается под действием нуклеофилов. В реакциях замещения субстратов
в присутствии достаточного количества другого нуклеофила, например
иодид-иона, входящая группа не обязательно должна ранее принадлежать
молекуле спирта или воды. В качестве примера можно привести реакцию
Цейзеля для определения ароматически связанной метоксигруппы, когда
под действием водной иодистоводородной кислоты происходит образование
йодистого метила. В случае алифатических спиртов эти реакции происходят
медленно, иногда лишь при высоких температурах. Как отмечалось в разд. 2,г,
эта реакция в зависимости от строения реагирующих компонентов и от усло-
вий может происходить по разным механизмам, причем не всегда ясно —
по моно- или бимолекулярному. В случае пгрепг-бутилового спирта в качестве
24* у?!
Глава VII
субстрата и серной кислоты как растворителя механизм является мономоле-
кулярным, так как при отсутствии замещающего агента наблюдается ультра-
фиолетовый спектр тпретп-бутил-катиона (очень напоминающий спектр изо-
электронного триметилбора) и, таким образом, можно следить за скоростью
образования этого карбониевого иона [54]. Вероятно, тпретп-бутилбисульфат
в серной кислоте в значительной степени ионизован.
Ацетали, устойчивые в щелочных растворах, легко подвергаются гидролизу
в водных растворах кислот. Эти реакции простейших ацеталей протекают
намного быстрее, чем аналогичные реакции простейших простых эфиров:
по данным Скрабала [58], этоксильная группа в диэтилацетале СН3СН(ОС2Н5)2
гидролизуется примерно в 1011 раз быстрее, чем этоксигруппа в диэтиловом
эфире СН3СН2ОС2Н5. Поэтому с известной степенью достоверности можно
предположить, что реакции ацеталей в конечном счете являются реакциями
мономолекулярного замещения соответствующих сопряженных кислот
(Sn1cA)
СН36^СН2-О)НСН3------>сн3о=сн2 + СН3ОН
Медленно
н2о +сн3б=сн2------СН3ОС1Т2ОН + Н+
Бысгпро
Процесс аналогичен описанному ранее гидролизу метоксиметилхлорида;
как и в последнем случае, образовавшийся полуацеталь, вероятно, еще
быстрее во второй стадии гидролиза превращается в альдегид.
Как впервые было предположено Бренстедом и Винни-Джонсом [59], при
гидролизе ацеталей проявляется специфический катализ водородными иона-
ми, т. е. скорость гидролиза зависит от предравновесия захвата протона.
Дальнейшие наблюдения подтвердили это предположение, так как при доста-
точно высокой кислотности скорость зависит от функции кислотности Гам-
мета h0, а не от стехиометрической кислотности [60], и применение в качестве
растворителя вместо воды D2O приводит к увеличению скорости в три раза
[61]. О’Горман и Лукас обнаружили, что формали оптически активных вто-
ричных спиртов гидролизуются без рацемизации [62], и поэтому пришли
к выводу, что при гидролизе происходит разрыв связи кислорода с централь-
ным углеродным атомом, а не с алкильной группой. Это подтверждается тем,
что 18О, содержащийся в водной среде, не обнаруживается в спиртах, обра-
зующихся при гидролизе ацеталей [63].
Влияние алкильных заместителей на скорость гидролиза ацеталей подтверж-
дает предположение о мономолекулярном механизме. Для ацеталей пента-
эритрита и формальдегида, ацетальдегида или ацетона установлено следую-
щее соотношение скоростей гидролиза [63] в соляной кислоте при 25 °C:
Н2 1; НСН3 6000; Н3ССН3 10 000 000
Каждая метильная группа увеличивает скорость примерно в 103-5 раза.
Этот значительный кинетический эффект можно объяснить тем, что метиль-
ный заместитель присоединен непосредственно к атому углерода, который
в мономолекулярном процессе становится катионом. Он аналогичен другим
известным эффектам а-метильных заместителей на скорость подобных про-
цессов, например на скорость замещения а-карбоксилатоалкилгалогенидов
(см. ниже).
в. Влияние строения входящей группы. Другой подход, который можно
использовать для того, чтобы различить бимолекулярный и мономолекуляр-
ный механизм замещения, основан на том, что скорость бимолекулярного
372
Замещение в алифатическом ряду
процесса зависит от нуклеофильной силы замещающего агента, а скорость
мономолекулярной реакции от этого фактора может не зависеть. В послед-
нем случае можно быть уверенным (не учитывая специфических обстоятельств,
описанных в разд. 6,з), что замещающий агент не входит в переходное состоя-
ние и поэтому в переходном состоянии не происходит завязывания ковалент-
ной связи.
На многих примерах было показано, что при мономолекулярной замеще-
нии различные замещающие агенты реагируют с одинаковой скоростью.
Если щретп-бутилхлорид гидролизовать в водном растворе муравьиной кис-
лоты, то скорость гидролиза не увеличивается в присутствии формиата и хлор-
ацетата кальция, несмотря на то, что в продуктах реакции в значительных
количествах содержатся щретп-бутилформиат и щретп-бутилхлорацетат, кото-
рые образуются не путем этерификации первоначально образующегося
пгретп-бутилового спирта [65]. Скорость гидролиза п,п'-диметилбензгидрил-
хлорида в водном растворе ацетона также не зависит от добавок азида натрия
(если учесть солевой эффект, разд. 6), хотя получается значительное количе-
ство п,п'-диметилбензгидрилазида, который не может образоваться из спир-
та [65]. Это находится в резком противоречии с реакцией азида натрия
с n-метокси- и n-феноксибензилхлоридами, для которых реакция со слабыми
нуклеофилами идет по механизму SN1, ас азид-ионом — по механизму SN2
(табл. 98). Установлено, что в двуокиси серы в качестве растворителя бенз-
гидрилхлорид с одинаковой скоростью реагирует с триэтиламином, пириди-
ном и фторид-ионом; то же самое можно сказать относительно реакции
л-хлорбензгидрилхлорида с пиридином, иодидом и фторидом. В последнем
случае наблюдалось специфическое отклонение от кинетического уравнения
первого порядка [67]; это уравнение в разд. 1 ,г было названо «SN1, типичный
случай», и, как было указано, если оно четко применимо, это служит дока-
зательством мономолекулярной реакции.
На этих примерах можно проиллюстрировать второй критерий для выбора
механизма. Если различные замещающие агенты приводят к образованию
разных продуктов замещения с неодинаковыми скоростями, мы обычно
делаем вывод (как было показано в предыдущем разделе, не всегда справед-
ливый), что рассматриваемая реакция происходит по бимолекулярному меха-
низму. Однако если различные замещающие агенты приводят к образованию
разных продуктов замещения (или даже одинаковых продуктов) с одной
и той же скоростью, то считается вероятным, что реакция происходит по моно-
молекулярному механизму.
На ранних стадиях развития теории нуклеофильного алифатического замеще-
ния этот критерий широко применялся как доказательство мономолекуляр-
ного механизма тех гидролитических реакций галогенидов, кинетика гидро-
лиза которых даже в щелочном растворе соответствовала первому порядку
в пределах точности измерений, доступной в то время. Например, было
показано, что тпрепг-бутилхлорид и бензгидрилхлорид в водном ацетоне
гидролизуются по реакции примерно первого общего порядка и скорость
гидролиза почти не изменяется независимо от того, в кислой, нейтральной
или щелочной среде проводится реакция: в частности, реакция не ускоряется
гидроксильными ионами.
Отсюда был сделан вывод, что реакции являются мономолекулярными и их
скорость зависит от скорости ионизации галогенидов. То, что наблюдаемые
скорости не связаны с атакой молекул воды на галогениды, следует из дан-
ных по гидролизу в присутствии гидроксильных ионов. Если в растворе
содержится достаточное количество гидроксильных ионов — гораздо более
сильных нуклеофилов, чем вода, то ускорения реакции все равно не наблю-
373
Глава VII
дается в противоположность фактам, известным для других случаев, напри-
мер для метил- или этилгалогенидов.
Теперь рассмотрим очень запутанный вопрос: что же определяет нуклео-
фильную силу реагента? Трудность, возникающая при попытке дать точное
определение силы нуклеофила, состоит в том, что рассматриваемое свойство
определяется не только нуклеофильностью, но и другими причинами, отчасти
рассмотренными выше. Порядок нуклеофильности может изменяться в зави-
симости от выбранной реакционной системы, например при переходе от одно-
го субстрата к другому или от одного растворителя к другому при одинако-
вом субстрате. Если субстрат является катионом, то его реакции с анионами
будут идти легче, чем реакции с нейтральными нуклеофилами (хотя этот
эффект относительно не велик [68, 72]). Важнейшее свойство растворителя —
его способность сольватировать катионы и анионы [69]. Различие возникает
также вследствие того, оценивается нуклеофильная сила по положению рав-
новесия или по скорости реакции: очевидно, в необходимых случаях нужно
учитывать метод оценки нуклеофильности (термодинамическая и кинетиче-
ская нуклеофильность).
Когда нуклеофил отщепляет протон, то говорят, что он ведет себя как осно-
вание; в таком случае сила нуклеофила соответствует его силе как основа-
ния, т. е. основности. Кинетическая и термодинамическая основности изме-
няются примерно параллельно друг другу; об этом свидетельствует широ-
кая применимость каталитического закона Бренстеда. Это соотношение
может не наблюдаться, если нуклеофил не отщепляет протона, а атакует
насыщенный или ненасыщенный атом углерода или другого элемента.
Например, термодинамическая основность в гидроксилсодержащих рас-
творителях уменьшается в рядах F_ > С1~ и т. д. и RO_>RS_ и т. д.
(R = Н, алкил, арил), однако в этих же растворителях скорость нуклео-
фильного замещения у насыщенного атома углерода в случае нейтральных
субстратов изменяется в обратном порядке. С другой стороны, известно, что
скорости замещения у четырехлигандного бора, фосфора и серы соответ-
ствуют основности нуклеофилов [70, 73].
В^общем сила нуклеофила зависит от сочетания некоторых его свойств;
в реакциях с одними субстратами эти свойства сочетаются лучше, а в реак-
циях с другими субстратами — хуже. Главные свойства нуклеофила — это,
конечно, его поляризация и поляризуемость. Важными факторами являются
также сольватация и часто, хотя и не во всех случаях, пространственное
взаимодействие с субстратом.
Что касается поляризации, то, согласно представлениям Бренстеда, общий
электрический заряд у реакционного центра во время тесного сближения
реагентов не играет такой существенной роли, как высокоинтенсивное
локальное поле орбиталей, обусловливающих нуклеофильные свойства
частицы. Такое представление автоматически учитывается в работах Эдвард-
са и др. [71, 73], согласно которым сила нуклеофила во всех реакциях опреде-
ляется прежде всего его термодинамической основностью. Предполагается,
что вследствие небольших тепловых эффектов кислотно-основных равнове-
сий или вследствие относительно малого перекрывания между орбиталями
водорода и нуклеофила поляризуемость вносит относительно небольшой
вклад в термодинамическую основность. Кроме того, нужно учесть допол-
нительную поляризуемость, которая играет важную роль в про-
цессах активации, т. е. при различных других необратимых реакциях
нуклеофилов.
Эдвардс [71] предложил простой способ учета поляризуемости, введя в урав-
нение свободной энергии активации член, пропорциональный логарифму
374
Замещение в алифатическом ряду
молекулярной рефракции нуклеофила. Это уравнение имеет вид
lg (knlkaq) = р (1,74 + рКа) + а 1g (7?п/7?а9),
где kjkaq — соотношение констант скоростей реакции субстрата с нуклео-
филом и водой, взятой в качестве стандартного нуклеофила, рКа — сила
кислоты, сопряженной нуклеофилу, RnIRaq — отношение молекулярных
рефракций нуклеофила и воды, 0 и a — константы, характеризующие отно-
сительную важность факторов поляризации и поляризуемости соответствен-
но в условиях реакции с данным субстратом.
Это уравнение достаточно хорошо согласуется с результатами эксперимента.
Если использовать значения рефракций ионов в разных растворителях,
можно в определенной степени проследить влияние сольватации анионов
на их поляризуемость [69]. Полного количественного согласия ожидать
нельзя, так как учитываемая таким образом поляризуемость представляет
собой среднюю изотропную величину, в то время как для реакции более
важной является анизотропная направленная поляризуемость орбитали
нуклеофила в направлении реакционного центра. Если данные по такой
субмолекулярной поляризуемости отсутствуют, единственное, что можно сде-
лать, это исследовать зависимость между структурой нуклеофилов и сте-
пенью отклонения от уравнения Эдвардса.
Важным этапом в исследовании этого эффекта послужило открытие Дженк-
сом и Карриуоло [72] группы нуклеофилов, включающей ОС1~, ООН",
ООСНз, N5, SO*3-, NH2OH, (CH3)2NOH, N-оксипиперидин, N-оксифталимид,
ацетоксим, салицилальдоксим, ацетилгидроксамовую кислоту, изонитро-
зоацетон, изонитрозоацетилацетон и 1-окись 4-аминопиридина, обладающих
аномально высокой нуклеофильной реакционной способностью. Как отме-
тили затем Эдвардс и Пирсон [73], аномальная реакционная способность
этих нуклеофилов проявляется во всех изученных реакциях. Они назвали
это явление «a-эффектом», принимая во внимание наличие неподеленных
электронов у атома, названного «a-атомом» и соседнего с нуклеофильным
центром. Аналогичным эффектом обладают электроны кислорода в метокси-
метилхлориде, которые ускоряют ионизацию хлорида *.
Отнесение наблюдаемого эффекта на счет неподеленных a-электронов не сов-
сем корректно. Это следует из ряда наблюдений, например для азид-иона
и 1-окиси 4-аминопиридина.
Аналогия с метоксиметилхлоридом недостаточно точна, поскольку в отличие
от электронов связи углерод — хлор, которые уходят от атома углерода,
освобождая орбиталь для электронов кислородного атома, электроны нук-
леофила никогда не покидают своей орбитали, и поэтому а-электроны
не могут ее занять. Этот случай представляет собой еще один пример того
общего положения (один пример уже рассматривался в связи с гиперконъю-
гацией ОН-группы; гл. VI, разд. 6,к), когда описание с помощью метода
валентных схем даже с учетом мезомерии не может быть достаточно обосно-
ванным теоретически даже для качественного описания. Причина состоит
в том, что движение электронов между соприкасающимися, но не сопряжен-
ными орбиталями атомов и связей сравнительно более важно, чем движение
внутри орбителей: в этом случае нет иного пути, как рассматривать молекулу
с позиций теории молекулярных орбиталей.
a-Эффект будет проявляться в том случае, когда высшая занятая орбиталь,
расположенная главным образом у нуклеофильного атома, является раз-
* В одной из лекций автор книги использовал термин «антисопряжение», чтобы указать
на повышение энергии при взаимодействии с разрыхляющими электронами и на пони-
жение энергии при взаимодействии со связывающими электронами, т. е. при сопряжении.
375
Глава VII
рыхляющей с узловой плоскостью, перпендикулярной связи между этим
атомом и а-атомом. Именно электроны обусловливают неоднородную поля-
ризуемость при взаимодействии с любым субстратом, способным использо-
вать поляризуемость нуклеофила. Так, гипохлорит-ион имеет 14-электрон-
ную валентную ячейку, состоящую из двух о-связанных (обусловливающих
связь О — С1), четырех о-несвязывающих (две свободные пары на продол-
жении линии связи), четырех л-связывающих электронов и обладающих
наивысшей энергией четырех л*-разрыхляющих электронов. Энергия пони-
жается, когда эти л*-электроны «снимаются» с разрыхляющих орбиталей;
для двух из них это возможно при перекрывании со становящейся пустой
орбиталью электрофильного субстрата. Перекисные анионы имеют два
электрона на разрыхляющей орбитали, а азид-ион — четыре электрона,
расположенных по два на каждом из двух концевых атомов. Большинство
других нуклеофилов, приведенных выше, имеют по два разрыхляющих
электрона, а если больше, то только два из них участвуют в рассматривае-
мых процессах, например два электрона, расположенные главным образом
у кислорода 1-окиси 4-аминопиридина, или два электрона сульфит-иона,
заполняющие одновременно разрыхляющие орбитали всех трех связей.
(В качестве легкого и полезного упражнения можно рассмотреть вопрос
о том, какие стационарные электронные волны «уходят» в потенциальную
яму, создаваемую симметрией сульфит-иона, и сколько из них с наимень-
шим количеством узлов, особенно с наименьшим количеством узлов между
соседними атомами, будут заняты имеющимися электронами.)
г. Влияние строения уходящей группы. Легкость отщепления галоге-
нов от алкилгалогенидов как в SN2-, так и в 8к1-реакциях возрастает в ряду
F<Cl<Br<I
В этом же ряду уменьшается различие в реакционной способности галоге-
нидов. Так, в гидроксилсодержащих растворителях хлориды более реак-
ционноспособны, чем фториды, в несколько тысяч раз, бромиды реакционно-
способнее хлоридов в несколько десятков раз, а иодиды более реакционно-
способны, чем бромиды, в несколько раз. При переходе от механизма SN2
к механизму SN1, т. е. при соответствующем изменении алкильной группы
или замещающего агента, указанные различия проявляются в более высо-
кой степени. В случае реакций SN2 приведенные соотношения скоростей
обычно возрастают при переходе от протонных к апротонным растворителям;
предполагается [74], что это обусловлено сольватацией галогенов протон-
ными растворителями, приводящей к более легкому отщеплению в виде анио-
на галогена с более низким атомным весом, для которого зарождающаяся
в переходном состоянии водородная связь выражена более заметно. Про-
странственные влияния, по-видимому, оказывают очень малое влияние
на зависимость скорости реакции от природы уходящей группы.
Для сравнения первоначально нейтральных отщепляющихся групп с перво-
начально катионными аммониевыми и сульфониевыми группами очень труд-
но выбрать подходящий растворитель, так как условия сольватации и ее
изменение при переходе от начального через переходное к конечному состоя-
нию сильно отличаются для нейтральных и заряженных отщепляющих
групп. Известно, что в гидроксилсодержащих растворителях легкость отщеп-
ления алкилированных аммониевых и сульфониевых групп возрастает
в последовательности
R3N R2S
Сложноэфирные группы составляют класс первоначально нейтральных
отщепляющих групп, способных замещаться нуклеофильными агентами.
376
Замещение в алифатическом ряду
Широко исследованы сульфонилоксигруппы эфиров арен- и алкансульфокис-
лот. Они замещаются подобно галогену в алкилгалогенидах, например:
Ar'S_-|-ROSO2Ar —> RSAr'+ ArSO2O“
С2Н5ОН +ROSO2Ar ROC2H5 + ArSO2OH
и по аналогии с алкилгалогенидами при замещении может происходить кон-
курирующая реакция отщепления олефинов. Механизм реакций замещения
в случае алкилсульфонатов, как и в случае алкилгалогенидов, может быть
бимолекулярным или мономолекулярный; изменение механизма в зависимо-
сти от строения алкила, замещающего агента и растворителя также совер-
шенно аналогично в этих двух случаях. Однако сульфонилоксигруппа
не может быть поставлена в один ряд с галогенами. Гофман [74] показал,
что скорость отщепления сульфонилоксигруппы может быть меньше скорости
отщепления брома или гораздо больше скорости отщепления иода — это зави-
сит от природы алкила, замещающего агента и растворителя. В табл. 99
Таблица 99
Соотношение &отз//сВг (или ^OBs^Br) констант скоростей бимолекулярного замещения
и мономолекулярного замещения и элиминирования п-алкилтолуол-
или n-алкилбензолсульфонатов (ROTs или ROBs) и алкилбромидов с различными
нуклеофилами в разных растворителях (по данным Гофмана)
Алкил Кине- тиче- ский поря- док Предпола- гаемый меха- низм Нуклеофильный анион Растворитель Тем- пера- тура, °C ftOTs/ftBr
СНз 2 Sn2 Cl- (СН3)2СО 25 0,42
СНз 2 Sn2 п-СНзСвН48- С2Н5ОН 0 0,36
с2н5 2 Sn2 п-СН3С6Н48- 1 С2Н5ОН 50% Н2О-С2Н5ОН 0 0 0,40 0,39
етор-СЩд 2 Sn2 п-СН3С6Н48- 92%С2Н5ОН—Н2О 0 2,3
СНз 2 Sn2 с2н5о- С2Н5ОН 0 5,4
СНз 2 Sn2 он- Н2о 0 6,3
СНз 1 Sn2 С2Н5ОН 50 16
с2н5 1 Sn2 С2Н5ОН 50 15
U30-C3II7 1 Смешанный С2Н5ОН 50 73
тргт-СШэ - 1 Sn1 + E1 С2н5он 50 > 3000
С6Н5(СН3)СН 1 SN1 С2н5он 50 845
СНз 1 Sn2 н20 50 18а
C2Hs 1 S;y2 н2о 50 17а
И-С3Н7 1 Sn2 Н2о 50 17а
U30-C4H9 1 Sn2 Н2о 50 12а
нео-С5Нц 1 Sn1 + E16 Н2о 50 5,6а
U30-C3H7 Смешанный н20 50 105а
СНз 1 Sn2 нсоон 95 24
с2н5 1 Смешанный нсоон 95 41
H-C3H7 1 Sn2 нсоон 95 47
ИЗО-С4Н9 1 Sn2 нсоон 95 58
иео-СзНц 1 sni + ei нсоон 95 90
ИЗО-С3Н7 1 Sn2 нсоон 95 360
трет-С^Н.^ 1 El CH3CN 0 5130
С6Н5(СН3)СН El CH3CN 0 470
d Отношения ^OBs^Br определены Робертсоном (ссылки см. в работе Гофмана [74]).
6 При сольволизе в воде происходит перегруппировка Вагнера-Меервейна (р-ю<-метильный перенос).
377
Глава VII
приведены отношения констант скоростей для реакций соответствующих
л-толуолсульфонатов (и бензолсульфонатов) и алкилбромидов. Эти кон-
станты соответствуют сольволитическим реакциям первого порядка, проте-
кающим по механизму SN2, или сольволитическим реакция первого порядка
или приблизительно первого порядка, которые могут происходить как по
механизму SN2, так и по механизму SN1. В последнем случае замещение
и элиминирование олефинов по механизму Е1 кинетически не различимы,
так как измеряется скорость ионизации — стадии, общей для обоих процес-
сов. В некоторых случаях в результате реакций SN1 и Е1 могут образовы-
ваться продукты перегруппировки Вагнера — Меервейна.
В табл. 99 приведены лишь некоторые из многочисленных данных, собран-
ных Гофманом. Как видно, соотношение скоростей сульфонат/бромид может
изменяться от значений, меньших 1, до значений, больших 1000; в изменении
этого соотношения можно проследить определенную закономерность. При
бимолекулярном замещении анионами с высокой нуклеофильностью к угле-
роду (например, RS-) отношение Aots/^вг составляет примерно 0,5, анионами
с более низкой нуклеофильностью (например, ОН-) — примерно 5, а при
замещении нейтральными молекулами гидроксилсодержащих раствори-
телей (С2Н5ОН, Н2О, НСООН) это отношение равно примерно 20. При моно-
молекулярном сольволизе в тех же растворителях и в ацетонитриле, в кото-
ром происходит только элиминирование, отношение констант скоростей
изменяется в широком диапазоне, начиная от указанных значений до зна-
чений, равных нескольким тысячам, при этом скорость мономолекулярных
реакций изменяется от низких до высоких значений. В случае мономолеку-
лярных реакций самое низкое соотношение Аотв/^вг наблюдается для пер-
вичных алкильных групп. В случае неопентильной группы, когда медленная
мономолекулярная реакция идет только потому, что бимолекулярная реак-
ция еще более замедлена из-за пространственных затруднений (разд. 8),
указанное соотношение равно 6 для реакции в воде, сопровождающейся
перегруппировкой Вагнера — Меервейна, и 90 в более ионизирующем рас-
творителе — муравьиной кислоте. Соединения с вторичными алкильными
группами, например с изопропильной, сольволизуются в муравьиной кис-
лоте значительно быстрее, а отношение к0^/кЪт равно 360. Еще быстрее
реагируют соединения, содержащие вторичную 1-фенилэтильную группу,
для которых указанное соотношение составляет 500...800. Наконец, еще
быстрее реагируют третичные соединения, представленные в таблице
mpem-бутилпроизводными; в этом случае соотношение Aots/^вг превыша-
ет 3000.
Гофман интерпретировал эти данные следующим образом. Если субстрат
и условия реакции таковы, что для отщепления уходящей группы необхо-
димо, чтобы она приняла значительную долю заряда электрона, т. е. если
в переходном состоянии происходит значительный перенос заряда, то суль-
фонилоксигруппа, в которой этот заряд может распределяться по трем ато-
мам кислорода и бензольному кольцу вследствие мезомерии, будет отщеп-
ляться с меньшей энергией активации, т. е. быстрее, чем моноатомные груп-
пы, включая большие атомы галогенов. Таким образом, согласно этой теории,
величина соотношения скоростей сульфонат/бромид указывает на степень
переноса электрона от углерода, связанного с отщепляющейся группой,
к этой группе при образовании переходного состояния.
На основании этого можно понять и объяснить получающиеся результаты.
При бимолекулярных реакциях «положение невозможного возврата» элек-
трона, переносимого от атома углерода к отщепляющейся группе, будет
достигнуто раньше в том случае, если «возврату» электрона будет препят-
378
Замещение в алифатическом ряду
ствовать необратимый переход электронов к этому атому углерода от ата-
кующего сильного нуклеофила. Положение невозможного возврата будет
достигаться все более и более позднее *, если постепенно уменьшать силу
нуклеофильного агента. С другой стороны, мономолекулярные реакции
активируются главным образом по механизму поляризуемости, т. е. путем
делокализации электронов внутри субстрата; чем лучше происходит такая
делокализация (т. е. перенос электронов и их возврат), тем быстрее будет идти
реакция и тем большей степени разделения зарядов будет соответствовать
положение, в котором невозможен возврат электронов. Таким образом,
зависимость скорости бимолекулярных реакций от степени переноса заряда
будет противоположна аналогичной зависимости для мономолекулярных
реакций. Некоторые перегруппировки типа Вагнера — Меервейна могут
происходить раньше, чем достигнуто положение невозможного возврата;
необходимым условием для этого является синхронность смещения алкиль-
ной группы (которая переходит со своими связывающими электронами,
т. е. как нуклеофил; гл. X) к атому углерода, участвующему в переносе
электрона.
В разд. 6 будет описан метод, позволяющий в подходящих случаях количе-
ственно оценить степень переноса электрона при образовании переходного
состояния. При сольволизе тпретп-бутилхлорида в воде степень переноса
заряда составляет около 0,6 электрона. Если иметь еще несколько таких
точек, то вполне возможно, что удастся построить калибровочную шкалу
для соотношения Гофмана.
3 Влияние растворителя
а. Влияние растворителя на скорость реакции. В предыдущих разделах
было установлено, что многие общие реакции, например гидролиз алкилга-
логенидов и распад четвертичных аммониевых солей, до определенного вре-
мени не считавшиеся однотипными, в действительности оказались настолько
сходными, что вполне подтвердили нашу классификацию, согласно которой
они относятся к группе реакций нуклеофильного замещения. Тем не менее
более детальная классификация этих реакций в зависимости от типа заряда
(75] позволяет выявить некоторые их особенности на фоне общего сходства.
Эти отличия проявляются особенно отчетливо при рассмотрении роли раство-
рителей.
При нуклеофильном замещении электрон перемещается от замещающего
агента к зоне замещения, а оттуда к отщепляемой группе. Замещающий
агент до реакции может быть заряжен отрицательно или быть нейтральным,
становясь после реакции соответственно формально нейтральным или поло-
жительно заряженным. Отщепляющаяся группа может быть до реакции
формально нейтральной или заряженной положительно, становясь впослед-
ствии отрицательно заряженной или нейтральной. Эти возможные случаи
взаимно не связаны, и поэтому реакции нуклеофильного замещения по типу
зарядов можно подразделить на четыре типа, указанных в табл. 100.
Сольватация представляет собой в основном электростатическое явление.
Когда ион или полярная молекула попадает в полярный растворитель, они
ориентируют и притягивают молекулы растворителя, тем самым производя
электростатическую работу. В результате энергия системы понижается
и система становится более стабильной. Энергия сольватации иона в поляр-
ном растворителе может быть очень большой; часто она того же порядка,
* По координате реакции.— Прим, перев.
379
Глава VII
Таблица 100
Классификация реакций нуклеофильного замещения
по зарядному признаку
Тип
Тип
1. Первоначально реагент отрицателен, группа нейтральна
„ fHO’ + RCl —> ROH4-C1-
Примеры: | j-^-Rd _> Ri + ci-
Первоиачально реагент нейтрален, группа нейтральна
2.
Тип
3.
R3N4-RCI—» rnr;+ci-
NH3+RCI -» RNH34-C1_(—>- RNH2)
lH2O + RCl —> ROH2 + Cl-(—> ROH)
Первоначально реагент отрицателен, группа положительна
Примеры: <
Тип
4.
„ HO- + RNR3 ROH + R3N
Примеры: J +
I I-4-RNR3 RI4-R3N
Первоначально реагент нейтрален, группа положительна
Примеры: <
R3N+RSR2 RNR^ + SR"
NH3+RSR" rnh3+sr;(-> RNH2)
vh2o+rsr; roh2+sr~(^ roh)
что и прочность ковалентной связи *. Следовательно, переход от менее
полярного к более полярному растворителю связан с увеличением или
уменьшением экзотермичности реакции в зависимости от того, будут ли
продукты реакции более или менее полярны, чем исходные вещества. Воз-
можно и противоположное изменение энтропии: сольватация, хотя и умень-
шает энергию, может увеличить упорядочение молекул растворителя, тем
самым уменьшая априорную вероятность сольватированного состояния.
Однако предположение о том, что изменение энергии преобладает над изме-
нением свободной энергии, хорошо обосновано; в соответствии с этим равно-
весие должно сдвигаться в сторону более полярных продуктов в более поляр-
ных растворителях.
Соответствующая приведенным представлениям теория [75], трактующая
влияние растворителя на кинетику реакции, состоит в том, что переход
к более полярному растворителю способствует уменьшению или увеличе-
нию теплоты активации (по отношению к которой аррениусовская энергия
активации является обычным приближением) в зависимости от того, являет-
ся ли переходное состояние более или менее полярным, чем исходное. Воз-
можны противоположные изменения энтропии активации, приближенно
соответствующей аррениусовскому предэкспоненциальному множителю,
но качественная теория предполагает, что изменения скорости должны
в основном определяться изменениями энергии, и поэтому более полярный
растворитель будет ускорять или замедлять реакции в зависимости от того,
являются ли реагенты в переходном состоянии более или менее полярными,
чем в первоначальном состоянии. Конечно, даже для качественных целей
* Полуколичественный расчет, см. [76], особенно гл. 1.
380
Замещение в алифатическом ряду
необходимо определить главные факторы, объединенные термином «поляр-
ность». Что касается степени сольватации, ожидаемой в случае наличия
электрически заряженных частиц, то в этом отношении приняты три сле-
дующих положения, касающихся исходных состояний:
1. Сольватация увеличивается с увеличением заряда.
2. Сольватация уменьшается с увеличением рассредоточения данного
заряда.
3. Уменьшение сольватации благодаря рассредоточению заряда меньше,
чем в случае исчезновения заряда.
Что касается растворителей, то в этом отношении следует принять, что
полярность, т. е. способность к сольватации ионов растворенных веществ,
во-первых, увеличивается с увеличением дипольного момента растворителя
и, во-вторых, уменьшается с увеличением толщины экранирующего слоя
дипольных зарядов. Таким образом, «протонные» растворители, в которых
протон гидроксильной или амидной групп слабо экранирован, например сер-
ная кислота, вода, метанол и формамид, составляют класс в общем наиболее
-сильно сольватирующих растворителей. Особенно сильно они сольвати-
руют анионы, причем небольшие анионы сильнее, чем большие. Полярные
апротонные растворители — двуокись серы, диметилсульфоксид, тетраме-
тиленсульфон (сульфолан), диметилформамид, нитрометан и ацетонитрил —
относятся к умеренно и менее специфически сольватирующим растворителям;
они склонны сольватировать катионы. В ряду ацетон, уксусная кислота
(которая в основном димеризована), бензол и гептан постепенно уменьшаются
полярность и сольватирующая способность.
Эти аргументы и допущения могут быть использованы для качественного
предсказания влияния полярности растворителя на скорость всех гетероли-
•тических реакций, механизм которых известен. Для нуклеофильного заме-
щения главными факторами являются тип заряда и механизм. Прежде всего
рассмотрим бимолекулярный механизм. В случае мономолекулярного меха-
низма непосредственное значение имеют исходное и переходное состояния
в стадии, определяющей скорость реакции. Это иллюстрируется данными,
приведенными в табл. 101. В трех средних колонках для каждого типа заряда
Таблица 101
Предсказанное влияние растворителей на скорость нуклеофильного замещения
Расположение заряда Влияние увеличения поляр- ности растворителя на скорость
Тип заряда первона- чальное состояние переходное состояние Влияние активации на заряды
1 2 3 4 Л 1 и 2 3 и 4 Y- + RX Y+RX Y + RX Y + RX гономолекуля RX RX Бимоле в- в- Y...R...X в+ в- Y. ..R...X б- в+ Y...R...X б+ 6+ Y. . ,R.. .X рный механизм 6+ й- R...X б+ 6+ R ...X кулярный механизм Рассредоточение Увеличение Уменьшение Рас средоточе ние стадия, определяющая с Увеличение Рассредоточение Небольшое уменьшение Большое увеличение Большое уменьшение Небольшое уменьшение корость процесса) Большое увеличение Небольшое уменьшение
381
Глава VII
и для каждого механизма показано, что происходит с зарядами при переходи
от исходного к переходному состоянию. В последней колонке приведен пред-
сказанный кинетический эффект. Термины «большой» и «небольшой» имеют
только относительное значение; они даны на основании теории, согласно
которой влияние рассредоточения заряда значительно меньше, чем эффект
его возникновения или исчезновения.
Имеется много доказательств правильности этих предположений. Де Брайн,
и Штегер [77] показали, что скорость щелочного гидролиза метил- и этилио-
дидов в водном этиловом спирте уменьшается с увеличением содержания
воды. Хьюз [78] установил, что скорость гидролиза шреш-бутилхлорида
(не зависящая от прибавления щелочи) с увеличением содержания воды
в спирте увеличивается. Ле Ру и Сегден [79] показали, что скорость обмена
брома между бромид-ионом (из бромистого лития) и н-бутилбромидом в аце-
тоновом растворе уменьшается при добавлении к раствору 10% воды. Дейвис
и Льюис [80] установили, что скорости присоединения диэтиланилина и неко-
торых арилдиэтилфосфинов к иодистому этилу в ацетоновом растворе увели-
чиваются при добавлении 10% воды. Меншуткин [29] показал, что присоеди-
нение триэтиламина к иодистому этилу протекает быстрее в спиртах, чем
в углеводородах. Для этой реакции Меншуткин установил следующую-
последовательность растворителей в порядке уменьшения скорости реакции:
СНдОН > С2Н5ОН > (СН3)2СО > С6Н6 > С6Н14
Аналогичная общая закономерность для реакций метилгалогенидов или
первичных алкилгалогенидов с другими аминами или сульфидами была
подтверждена рядом последующих исследователей [81, 82].
Гальбан [83] установил, что распад триэтилсульфонийбромида в спиртах
протекает медленнее, чем в ацетоне. Глив, Хьюз и автор книги [84] показали,,
что скорость щелочного гидролиза триметилсульфоний-катиона в водно-
спиртовом растворе уменьшается с увеличением концентрации воды. Хьюз
и автор книги [85] нашли, что скорость гидролиза щре/п-бутилдиметилсуль-
фоний-иона (не зависящая от щелочности) при соответствующем изменении
растворителя также уменьшается.
Все это было известно в 1935 г., когда была опубликована теория влияния
растворителей. Однако в то время не было известно ни одной реакции с типом
заряда 4, существование которых и вид кинетического эффекта растворителя
оставались не подтвержденными вплоть до 1960 г., когда Хьюз и Уайттинг-
хэм описали некоторые примеры таких реакций [86]. Эти авторы показали,
Таблица 102
Относительные скорости и аррениусовские параметры для реакции
+ +
(CH3)3N + CH3S(CH3)2 —» (СН3)3Г<СНз-(-8(СНз)2 в четырех растворителях
(по данным Хьюза и Уайттингхэма)
Растворитель Относительная скорость (45 °C) 1g А Еа, ккал/моль (4,187-103 Дж/моль)
Н2о 1 4,90 23,1
СН3ОН 6 3,05 21,6
С2Н5ОН 10 0,94 20,6
ch3no2 119 0,19 18,0
382
Таблица ЮЗ
Влияние растворителей на скорость нуклеофильного замещения
Тип Пример Константа3 скорости Содержание воды в водном С2Н5ОН, % (по объему) Предсказан- ное влияние
0 10 20 30 40 50 60 100
Бимолекулярный механизм б
1 изо-С3Н7Вг+ОН- 105*2(55 °C) 6,0 4,9 3,0 Небольшое
уменьшение
2 изо-СзЩВг-]- Н2О 10**1(55 °C) 1,73 23,6 66,7 Большое
увеличение
3 (CH3)3S + OH- 10**2(100°С) 7240 178 15,1 0,37 Большое
уменьшение
4 (CH3)3S4- (CH3)3N 105*2(45 °C) 6,67 0,05 Небольшое
уменьшение
Мономолекулярный механизм в
1 и 2 mpem-CiHgCl 106*i(25 °C) 103*i(55 °C) 106*i(25 °C) 104*i(25 °C) 0,20 0,11 1,71 9,14 13,2 14,5 5,8 40,3 126 148 367 12941 Большое увеличение
mpem-CiHgBr трет-С5НцС1 трет-С6НцВг ч
3 и 4 ' mpem-C4HgS(CH3)2 . mpem-C5H1)S(CH3)2 105*i(50 °C) 103*i(65 °C) 1,90 1,24 0,62 0,45 0,60 Небольшое уменьшение
а Константа скорости второго порядка *3 выражается в л-моль-1-с-1, константа скорости первого порядка hi— в с-1, температура указана
в скобках.
б Константы скорости, приведенные для реакций с бимолекулярным механизмом, относятся только к 8^2-замещению, сопровождающему процесс
элиминирования олефина (Е2). Причина заключается в том, что бимолекулярное элиминирование протекает независимо от бимолекулярного замещения;
эти реакции не представлены в каком-либо общем процессе. Предполагается, что сольволиз изопропилбромида в нейтральной или кислой среде является
в основном бимолекулярной реакцией.
в Константы скорости для реакций с мономолекулярным механизмом являются в целом константами реакции первого порядка и включают как
замещение, так и элиминирование (Sj^l+El) потому, что первая стадия мономолекулярного элиминирования идентична первой стадии мономолекулярного
замещения. Именно к этой стадии относятся измерения и теоретические предсказания.
Глава VII
например, что скорости реакций второго порядка между триметиламином
и триметилсульфониевым ионом в четырех растворителях возрастают в ряду
Н2О < СН3ОН < G2H5OH < CH3NO2- Таким образом, при уменьшении
полярности растворителя скорость увеличивается. Очевидно, что начальное
состояние более сольватировано, чем переходное. В табл. 102 приведены
относительные скорости и аррениусовские параметры активации для этой
реакции в четырех растворителях. Данные показывают, что в указанном
ряду растворителей уменьшение энергии сольватации перекрывает противо-
действующий эффект энтропии сольватации.
Аналогичный вывод можно сделать на основании данных [86, 87] табл. 103,
в которой приведена зависимость скоростей замещения от состава водно-
спиртовой смеси, взятой в качестве растворителя. Скорости (четвертая
колонка) как бимолекулярных, так и мономолекулярных реакций различных
типов зарядов изменяются в предсказанном направлении (пятая колонка).
Более того, имеется заметное различие в величинах наблюдаемых эффектов
в соответствии с предсказанными ранее «большими» и «небольшими» эффек-
тами. В одних случаях при переходе от чистой воды к чистому этанолу
скорости отличаются в несколько раз, а в других случаях в 104 раза.
Различие в сольватации ионов протонными и апротонными растворителями
было рассмотрено Паркером [69]. В табл. 104 приведены два примера, иллю-
Таблица 104
Относительные скорости реакций
(A) CI-4-CH3I CH3CI+I-
(Б) Nj + n-NO2C6H4F -> n-NO2CeH4N3 + F~
в пяти растворителях при 25 °C (по данным Паркера)
Растворитель Реакция А Реакция Б
СН3ОН 1 1
hconh2 12,5 5,6
HCONHCH3 45,3 15,7
HCON(CH3)2 1 200000 24 000
СН3СОМ(СН3)2 7400 000 84 000
стрирующие этот эффект. Эти примеры относятся к S N 2-реакциям в алифати-
ческом и ароматическом рядах. (Теория кинетических эффектов растворите-
лей хотя и была разработана для замещения в алифатическом ряду, тем
не менее применима ко всем другим гетеролитическим реакциям в растворе.)
Исходное состояние обеих реакций более сольватировано, чем переходное;
кроме того, анион, вступающий в реакцию, более сольватирован, чем обра-
зующийся анион, особенно в протонных растворителях. При переходе от про-
тонных к апротонным растворителям (табл. 104) происходит постепенное
уменьшение полярности, что должно приводить к увеличению скорости
реакций. Это и наблюдается в действительности. Три растворителя — сред-
ние члены ряда — являются N-гомологами, поэтому на первый взгляд
можно было ожидать лишь небольшого изменения скорости при переходе
от одного из этих растворителей к другому. В действительности же при
переходе от метилформамида — потенциального донора водородных связей—
к диметилформамиду наблюдается резкий скачок скорости. По-видимому,
384
Замещение в алифатическом ряду
главным фактором, определяющим скорость этих реакций, является сольва-
тация анионов, особенно сильно проявляющаяся в протонных растворителях.
б. Влияние растворителя на соотношение продуктов реакции. Если два
замещающих агента Yt и Y2 в одном растворе конкурируют между собой
при взаимодействии с одним и тем же субстратом RX, образуя продукты
RYt и RY2, то имеются две независимые кинетические величины, которые
определенным образом зависят от среды. В качестве этих величин возьмем
суммарную скорость распада RX и отношение количеств образующихся про-
дуктов RY1/RY2. Этот выбор удобен для демонстрации различий между
бимолекулярным и мономолекулярный механизмами замещения. При бимо-
лекулярном механизме имеется лишь одна стадия, в связи с чем как суммар-
ная скорость, так и соотношение продуктов реакции определяется этой ста-
дией. При мономолекулярном механизме скорость реакции определяется
медленно протекающей стадией гетеролиза RX. Однако в этом случае соотно-
шение продуктов реакции определяется совершенно независимым фактором,
а именно конкуренцией последующих быстрых стадий. На этом отличии
может базироваться критерий механизма, в котором используются кинетиче-
ские эффекты растворителя. При этом следует установить, связаны ли между
собой влияния среды на суммарную скорость реакции и на соотношение про-
дуктов реакции в указанной системе или эти влияния независимы.
Этот критерий механизма наиболее ценен при изучении таких реакций пер-
вого порядка, в которых замещающий агент, являющийся растворителем,
сохраняет постоянную активную массу. До сих пор этот критерий приме-
нялся главным образом к реакциям такого типа, как гидролиз и алко-
голиз алкилгалогенидов в первоначально нейтральных водно-спиртовых
растворах.
Простым примером может служить сольволиз бензгидрилхлорида в перво-
начально нейтральном водном растворе этилового спирта с образованием
этилового эфира бензгидрола и самого бензгидрола [88]. Если в сухой этило-
вый спирт, взятый в качестве растворителя, добавлять воду, то суммарная
скорость реакции увеличивается. Однако этот результат нельзя объяснять
большей скоростью образования бензгидрола, так как он образуется лишь
в незначительном количестве; в основном влияние добавки воды приводит
к увеличению скорости образования бензгидрилэтилового эфира. Очевидно,
в данном случае скорость и соотношение продуктов реакции определяются
независимо друг от друга. Подобное положение совместимо с мономолекуляр-
ным, но не бимолекулярным механизмом [89].
Количественно вопрос можно решить по этому методу с помощью аддитивной
схемы. Если известна скорость реакции при двух крайних соотношениях
в составе бинарного растворителя, то путем интерполяции можно подсчитать
скорость для промежуточных соотношений. В одной из таких формул в каче-
стве параметров состава использованы парциальные давления паров [90].
Скорость = (kaPa-vkwpw) рпх (1)
где ка и кw — константы скоростей, ра, pw и pRX — давления паров соответ-
ственно спирта, воды и алкилгалогенида. Предложена также и модифициро-
ванная формула *. От любого такого двухчленного выражения, основанного
на принципе аддитивности, требуется лишь, чтобы на основании двух кон-
стант скоростей ка и kw, измеренных при двух соотношениях компонентов
растворителя, можно было вывести значения скоростей при других соотно-
* Вместо давления паров Бартлет [91] предложил использовать мольные доли раство-
римых компонентов.
25 — 0772
385
Глава VII
шениях этих компонентов. Если предположить, что это так, т. е. что сумма
двух членов именно таким образом описывает суммарную скорость процесса,
то определение механизма реакции состоит в выяснении, описывает ли соот-
ношение двух множителей соотношение продуктов реакции, другими сло-
вами, можно ли соотношение продуктов реакции определить при помощи
следующей формулы, основанной на кинетически измеренных константах
ка и kw-.
ROAlk/(ROH + ROAlk) = kapj (kapa + kwpw) (2>
Это возможно, если реакция протекает по бимолекулярному механизму.
Если же реакция протекает по мономолекулярному механизму, то константы
скоростей ка и kw нельзя использовать для установления количественного
соотношения продуктов реакции.
Данный способ использован в случае сольволиза типичных первичного и тре-
тичного алкилгалогенидов, а именно н-бутилбромида и шреш-бутилхлорида
в первоначально нейтральных растворах воды, метилового или этилового
спиртов [92]. Найдено, что во всех случаях константы скорости удовлетво-
ряют уравнению (1); однако уравнение (2), как это следует из данных
табл. 105, правильно выражает состав продуктов реакции н-бутилбромида,
Таблица 105
Тест для выяснения зависимости механизма сольволиза
от природы растворителя
Количество эфира 100-ROAlk/(ROAlk+ROH), образующегося при сольволизе алкилгалогенидов
в водном метиловом и этиловом спирте, вычислено с учетом изменения общей скорости сольволиза
при изменении состава растворителя
Растворитель Температура, °C Количество Н2О, мол. % Количество ROAlk, %
найдено вычислено
н-Бутилб ромид
Смесь СН3ОН и Н2О 59,4 14,1 88 86
26,3 74 75
30,8 72 72
48,3 61 61
60,1 51 53
Смесь С2Н5ОН и Н2О 75,1 25,8 63 63
56,2 50 45
73,7 35 39
трет-Бутилбромид
Смесь СН3ОН и Н2О 25,0 16,4 83 43
28,4 68 29
40,0 49 17
Смесь С2Н5ОН и Н2О 25,0 26,4 53 18
44,7 33 И
68,3 18 8
но не может быть использовано для определения состава продуктов реакции
тнретн-бутилхлорида. Этот вывод согласуется с тем, что гидролиз (или соль-
волиз) первичных алкилгалогенидов в водно-спиртовой среде протекает
по бимолекулярному, а третичных алкилгалогенидов — по мономолекуляр-
ному механизму.
386
Замещение в алифатическом ряду
Подобный метод [93] применялся в случаях, когда в состав растворителя
входит только один из конструирующих агентов. Метод можно использовать
также и в том случае, когда реагенты не являются составной частью раство-
рителя; в этом случае механизм обычно определяется проще. Как уже указы-
валось, при добавлении азида натрия к водному ацетону, в котором происхо-
дит гидролиз п,п'-диметилбензгидрилхлорида, скорость этой реакции,
не считая солевого эффекта, не изменяется (разд. 6), хотя значительное
количество исходного вещества, которое могло превратиться в п,п'-диметил-
бензгидрол, присутствует в продуктах реакции в виде п,п'-диметилбензгид-
рилазида. Если использовать водный ацетон постоянной концентрации
и увеличивать концентрацию растворенного азид-иона, то увеличивается
относительное количество алкилазида. Это совпадает с предположением,
что скорость образования органических азидов пропорциональна концен-
трации азид-иона, между тем как скорость конкурирующей реакции гидроли-
за остается постоянной. Этот вывод иллюстрируется данными, помещенными
в левой части табл. 106. При определенной концентрации азид-иона (допу-
стим, 0,05 М) с увеличением содержания воды в водном ацетоне от 10 до 50%
Таблица 106
Тест для выяснения зависимости механизма сольволиза от природы
растворителя и растворенного замещающего агента
Скорости и соотношения продуктов гидролиза п,и'-диметилбензгидрилхлорида
в водном ацетоне, содержащем азид натрия, при 0 °C. Содержание воды
и концентрация азид-иона варьировались независимо друг от друга
Количество Н^О, об.% Концентра- ция NaNs Количество образо- вавшегося RNaa> % Количе- ство НаО, об.% Концен- трация NaN3 Общая скорость б Количество образовавше- гося RN3, %
найдено вычислено
50 0,0512 60,2 60,3 10 0,051 1,59 60,3
50 0,1001 75,8 76,5 15 0,051 7,05 59,6
50 0,2002 85,8 87,7 20 0,052 22,4 61,2
50 0,4867 95,8 94,8 50 0,052 Очень быстро 60,0
а Определено как 100 RNa/IRNs+ROH), где П=(п-СНзСвН4)2СН.
б Измерено в присутствии соли: 1 Ottj, где kj выражено в с-1.
скорость суммарной реакции увеличивается значительно, в то время как
скорость образования диметилбензгидрилазида остается постоянной (это
иллюстрируется данными, приведенными в правой части табл. 106).
Поскольку при постоянной скорости реакции могут образоваться различ-
ные продукты и, наоборот, при переменной скорости может образоваться
одно соединение, нет необходимости в особых расчетах для того, чтобы
видеть, что, как и при мономолекулярном механизме, скорость реакции
и образующиеся продукты определяются независимо
Медленно
RC1 -----------> R+
---> ROH
Быстро
Стадия, определяющая
скорость реакции
Стадии, определяющие
выход продуктов реакции
25* 387
Глава VII
Можно прийти к следующему заключению: в присутствии достаточного
количества воды первоначально образующийся карбониевый ион полностью
ею сольватируется и совершенно не сольватируется ацетоном. Таким обра-
зом, скорость, с которой карбониевый ион ковалентно связывается с одной
из сольватирующих молекул воды, не зависит от состава преобладающей
массы среды *. В то же время скорость взаимодействия карбониевого иона
с азид-ионом зависит от скорости, с которой они проникают через сольват-
ную оболочку воды, и, следовательно, обычным образом зависит от концен-
трации азида во внешней среде.
Этот метод установления механизма реакции может быть дополнен другим.
При мономолекулярном механизме состав продуктов реакции определяется
конкурентным взаимодействием нуклеофилов с карбониевым ионом. При
бимолекулярном же механизме состав продуктов реакции определяется кон-
курентным взаимодействием с исходной молекулой алкилгалогенида. Поэто-
му если раздельно ввести два алкилгалогенида с одинаковыми радикалами,
но различными атомами галогенов (например, алкилхлорид и соответствую-
щий алкилбромид) в одинаковый по составу раствор, например в смесь
ацетона с водой, имеющую определенный состав и содержащую растворен-
ный азид-ион также в определенной концентрации, то в случае мономоле-
кулярного механизма состав продуктов реакции будет одинаковым даже
в том случае, если скорости реакций весьма различны:
RC1----- h^._^Roh
RBr ----- ——------------------------> RNg
Стадия, определяющая Стадия, определяющая
скорость реакции выход продуктов реакции
Этот метод был проверен на примере гидролиза бензгидрилхлорида и бенз-
гидрилбромида в водном ацетоне в присутствии растворенного азида натрия
[94]. Соответствующие данные приведены в табл. 107. Хотя скорости реак-
ции значительно отличаются между собой, соотношение продуктов реакций
Таблица 107
Тест для выяснения зависимости механизма сольволиза
от природы замещаемого соединения
Скорости и соотношения продуктов гидролиза
бензгидрилхлорида и бензгидрилбромида в 90%-ном водном
ацетоне, содержащем 0,101 М азида натрия при 50 °C
RX Й1, С-1 кон, % RN3, %
(СвН5)2СНС1 12,4-10-5 66,0 34,0
(СвН5)2СНВг 416-10-5 66,5 33,5
одинаково, а это в сочетании с другими данными служит подтверждением
того, что реакция замещения подобных галогенидов в растворителях, содер-
жащих воду, протекает по мономолекулярному механизму.
* Обычное заключение, что сольватационная ячейка состоит лишь из молекул воды,
не обязательно справедливо.
388
Замещение в алифатическом ряду
в. Механизм в полярных апротонных растворителях. После того как
механизм нуклеофильного замещения в несольволитических и сольволитиче-
ских условиях в гидроксилсодержащих растворителях был подразделен
на два предельных случая, были проведены исследования, в которых труд-
ности, возникающие вследствие сольволиза при изучении мономолекуляр-
ного замещения, были устранены благодаря применению апротонных рас-
творителей. Эти растворители сами не могут вступать в реакцию, однако
полярность их достаточно велика и способствует ионизации субстрата,
от которой зависит скорость мономолекулярного замещения и элимини-
рования.
Первым растворителем, использованным для этой цели, была двуокись серы,
диэлектрическая проницаемость которой равна примерно 15 (зависит от тем-
пературы). В этом растворителе происходит ионизация трет-бутил- и бенз-
гидрилгалогенидов с образованием карбониевых ионов в низкой концен-
трации, однако в этом случае проявляются солевой эффект и эффект поляр-
ности растворенных соединений, которые будут описаны ниже. Это, однако,
не влияет на общий вывод о том, что в жидком SO2 замещение происходит
по механизмам SN2 и SN1, причем последний не сопровождается сольволизом.
Реакция между иодистым метилом и пиридином по своей кинетике относит-
ся к SN2-THny. Реакции mpem-бутилбромида и .м-хлорбензгидрилхлорида
с ионами фтора фтористого тетраэтиламмония имеют кинетику SNl-THna
с большим солевым эффектом, характерным для такого механизма (разд. 6, е).
Те же субстраты, судя по кинетике, реагируют с пиридином и триэтиламином
в основном по SNl-THny, однако в этом случае наблюдаются некоторые откло-
нения от чистого S ^-процесса, вероятно, вследствие действия дальних
электростатических сил в этой среде [95].
При исследовании реакций в нитрометане—растворителе с более высокой
диэлектрической проницаемостью (около 40), благодаря чему устраняются
возмущения, связанные с силами дальнего действия, в 1954 г. были сфор-
мулированы два новых принципа. В этой среде /прет-бутилбромид вступает
в реакции замещения с ионами радиоактивного брома, хлора и нитрит-ионом
(все из тетраэтил аммониевых солей), которые могут сопровождаться, а могут
и не сопровождаться элиминированием. Реакции имеют первый кинетиче-
ский порядок по субстрату и нулевой по замещающим агентам. Наблюда-
лись кинетические солевые эффекты, соответствующие мономолекулярный
реакциям, причем все три реакции имели одинаковую скорость при низких
концентрациях солей. Тот же субстрат вступает в реакции замещения
первого порядка с водой, этанолом и фенолом; эти реакции имеют одинако-
вую скорость при низких концентрациях реагентов, близкую к скорости
замещения указанными выше анионами. Очевидно, что скорость всех шести
реакций определяется общей стадией ионизации. Однако при повышении
концентрации гидроксилсодержащих реагентов скорость реакции возрастает
по линейному закону, т. е. в этом случае в выражении для скорости имеется
член второго порядка. Это не означает, что при высокой концентрации
реагентов реакция частично идет по механизму SN2, так как наклон кривой
скорость — концентрация реагентов, т. е. константа скорости реакции
второго порядка, не увеличивается при повышении силы гидроксилсодер-
жащего нуклеофила. Этот наклон зависит от кислотности нуклеофила. Соот-
ношение скоростей в ряду С8Н5ОН > Н2О > С2Н5ОН составляет 5,5 : 2 : 1.
Это является следствием общего кислотного катализа при S ^-замещении
в алкилбромидах. Переходное состояние этой реакции имеет вид:
б+ д—
mpem-CiHg •. • Вг • • • Н •. • OR
389
Глава VII
В гидроксилсодержащих растворителях наблюдается кислотный катализ
только при замещении фтора (разд. 5,6), но не других галогенов. Вероятно,
кислотный катализ нуклеофильного замещения в принципе возможен для
всех алкилгалогенидов; это зависит от основности растворителя и его способ-
ности конкурировать с алкилгалогенидом в реакции с донором прото-
нов [96].
Второй новый принцип, сформулированный в 1954 г., касается нового меха-
низма нуклеофильного замещения — нового в том смысле, что хотя он
состоит из тех же двух химических стадий, что и механизм SN1, но опреде-
ляющей скорость является вторая стадия. Поэтому в опытах, поставленных
согласно данным выше правилам для определения механизма (разд. 1,д),
этот механизм должен проявляться особым образом. Такой механизм вклю-
чает медленную бимолекулярную реакцию замещающего агента с карбоние-
вым ионом, который быстро образуется из субстрата в предравновесной
стадии. Этот механизм можно обозначить символом SN2(C+). Указанный меха-
низм возможен, конечно, лишь в том случае, когда карбониевый ион очень
стабилен, легко образуется и медленно реагирует с нуклеофилом. Приме-
ром может служить реакция трифенилметилхлорида в нитрометане с гидро-
ксилсодержащими замещающими агентами:
(С6Н5)8С —С1 ----* (С6Н5)3С+ + С1-
Выстро SN2(C+)
(с(,щ)!,с++вон;----* (ceH5)8coR+ н+
Медленно '
При растворении трифенилметилхлорида в нитрометане, судя по спектру,
немедленно образуются ионы трифенилкарбония, а в присутствии радио-
активных хлорид-ионов мгновенно устанавливается равновесие обмена.
Были изучены реакции с водой, этанолом и фенолом [96]. Эти реакции обра-
тимы и в типичных условиях дают равновесную смесь, соответствующую
10...20% превращения. Их можно провести на 100%, если добавлять пири-
дин для связывания выделяющегося хлористого водорода. При зтом про-
исходит изменение механизма; скорость не зависит от избытка пиридина.
Спектральные данные показывают, что первоначально происходит образова-
ние комплекса трифенилкарбониевого иона с пиридином, вероятно, в виде
иона пиридиния, в котором заряд рассредоточен между атомом азота и арал-
кильной группой. Затем происходит реакция второго порядка, скорость кото-
рой возрастает в соответствии не с кислотностью, а с нуклеофильностью
замещающих агентов. Отношение скоростей в ряду С2Н5ОН > Н2О >
> СвН5ОН равно 20 : 5 : 1. Очевидно, что реагенты выступают в роли нук-
леофилов, откуда следует вывод, что они реагируют именно с комплексным
карбониевым ионом.
Рассмотрение механизма в малополярных или неполярных растворителях
мы пока отложим до разд. 6,ж и з, так как этот вопрос во многом связан
с солевыми эффектами, которые будут рассмотрены в разд. 6.
4. Температурные эффекты при нуклеофильном замещении
а. Энтропия активации. Измеряя скорости реакции в каком-то интер-
вале температур, при помощи уравнения для скорости реакций теории
абсолютных скоростей (гл. II, разд. 2,г) можно определить теплоту акти-
вации АЯ" и энтропию активации А5*. Сами эти величины также зависят
390
Замещение в алифатическом ряду
от температуры, поэтому полученные значения применимы только к дан-
ному интервалу температур.
Энтропия активации зависит от многих факторов. Когда при бимолекуляр-
ной реакции две начальные частицы соединяются вместе, образуя переходное
состояние, трансляционная и вращательная энтропия двух частиц умень-
шается до значений, соответствующих единой частице; незначительное возра-
стание колебательной энтропии недостаточно для компенсации этого эффек-
та. Эти факторы дают отрицательный вклад в энтропию активации. Если
в процессе образования переходного состояния создаются (или исчезают)
заряды, сольватирующие молекулы полярных растворителей становятся
более компактно (или свободно) связанными, и поэтому энтропия этих соль-
ватирующих молекул уменьшается (или увеличивается). Это дает отрица-
тельный (или положительный) вклад в энтропию активации. Если некото-
рые связи в реагентах при образовании переходного состояния частично
ослабевают, колебания, обусловленные силами возврата, дают выигрыш
в энтропии и, таким образом, положительный вклад в энтропию активации.
Рассмотрев первый из перечисленных факторов, Лонг [97] предположил,
что в общем 8к2-реакции должны обладать менее положительной или более
отрицательной энтропией активации по сравнению с аналогичными SN1-
реакциями, и это различие должно служить критерием механизма, особенно
необходимым в случае трудно интерпретируемых сольволитических реак-
ций. Лонг проиллюстрировал это различие в области кислотно-катализи-
руемого гидролиза, например ацеталей и эфиров карбоновых кислот, сопря-
женные кислоты которых являются субстратами нуклеофильного замещения.
Он подчеркнул, что предложенный критерий механизма должен использо-
ваться очень осторожно, так как даже при одном и том же механизме значе-
ния энтропии активации могут меняться в широких пределах. В табл. 108
Таблица 108
Энтропия активации (кал• моль-1 • °C-1) сольволиза алкилгалогенидов
и алкилсульфонатов при 50 °C (по данным Констэма, Робертсона)а
Замещаемая группа Растворитель Предполагаемый S^l Предполагаемый Sj^-2
примеры алкилов интервал примеры алкилов интервал Д8^
CI или Вг н2о трет-С4Н9, Н^О-CgH и От +3 до +9 СНд, С2Н5, U30-CgH7 От —9 до —5
50% Н2О — 50% С2Н5ОН mpem-C^Hg, (С6Н5)2СН
50 — 80%-ный водный ацетон п-СН3ОС7Нв, СвН5СНС1, С6Н5СС12 От —14 до —9 С2н5, изо-С3Н7 От —23 до —19
CHgSOg ИЛИ n-CHgCgHsSOg н20 USO-CgHy, нео-СбНц От -5 до —3 От —14 до —12
В таблице приведены интервалы изменения энтропии, полученные на основании многих точных
данных Робертсона для воды и Констэма для водно-органических растворителей; цитируется по обзору
Констэма [98] и последующим публикациям [99, 100].
391
Глава VII
приведены данные, иллюстрирующие как общий принцип, так и интервал
изменения величин энтропии активации при некатализируемом сольволизе-
алкилгалогенидов и алкилсульфонатов. Данные были получены Констэмом,
Робертсоном и сотрудниками. Несмотря на значительный разброс данных,
можно видеть, что при бимолекулярном сольволизе энтропия активации
на 9... 14 кал/°С (37,68...57,62 Дж/°С) более отрицательна, чем энтропия
активации мономолекулярного сольволиза почти таких же по структуре
соединений, что соответствует общему принципу Лонга.
Разброс значений энтропии может быть обусловлен многими причинами,
но важной является упомянутый выше второй фактор, а именно сольватация.
Согласно данным таблицы, и при мономолекулярном и при бимолекулярном
механизме энтропия активации выше (т. е. более положительна или менее
отрицательна) в воде по сравнению с водно-органическими смешанными
растворителями. Энтропия воды (сольватирующей полярное переходное
состояние) несомненно будет ниже в чистой воде, чем в смешанном раство-
рителе, где к ней прибавляется энтропия смешения. Однако детальная кар-
тина сольватационной ячейки начального и переходного состояний для
каждого механизма и каждого индивидуального субстрата в данном раство-
рителе будет различной, что и обусловливает изменения энтропии активации
при переходе от одного случая к другому.
б. Теплоемкость активации. Если очень точно измерять скорость реак-
ции в данном интервале температур, можно определить температурный гра-
диент теплоты активации dAH^tdT, известный под названием теплоемкости
активации АС*. В принципе и эта величина является функцией температуры;
однако, поскольку она уже получена дифференцированием по температуре,
ее собственную зависимость от температуры обнаружить трудно.
Констэм [101] предположил, что для данного механизма и данного раство-
рителя безразмерное отношение должно быть менее чувствитель-
ным к изменению субстрата, чем сами величины AS* и АС*. Поэтому для
данного растворителя указанное соотношение и служит лучшим тестом
на механизм, чем величины AS* и АС*. Констэм подтвердил это предположе-
ние данными по сольволизу алкилгалогенидов и алкилсульфонатов в 50 %-ном
водном растворе этанола и 50-, 70-, 80- и 85%-ных водных растворах ацето-
на [101]. Некоторые данные для 50%-ного водного ацетона приведены
в табл. 109. Метод Констэма не так хорошо применим к чистой воде, в кото-
рой величины AS* мономолекулярного замещения часто близки к нулю,
и поэтому отношение &C+I&S* становится очень большим и очень чувстви-
тельным к изменению субстрата.
По величине отношения субстраты, приведенные в табл. 109, мож-
но разделить на две группы, точно так же как их можно разделить по меха-
низму сольволиза, предполагаемому по другим соображениям: мы полагаем,
что первые четыре субстрата (табл. 109) реагируют по бимолекулярному,
а остальные пять — по мономолекулярному механизмам. Аналогичная двой-
ственность механизма сольволиза была установлена и для других водно-
органических растворителей. Величины как для бимолекулярных,
так и для мономолекулярных реакций зависят от природы растворителя,
однако при мономолекулярном механизме это отношение всегда значительно1
больше, чем при бимолекулярном.
Существование различия в величинах отношений характеризую-
щее бимолекулярные и мономолекулярные реакции, вероятно, означает,
что хотя обе отрицательные величины АС* и AS* отражают ограничение,
накладываемое в переходном состоянии на положение и движение входя-
щих в него молекул воды, они отражают это ограничение по-разному, причем
392
Замещение в алифатическом ряду
Таблица 109
Теплоемкость и энтропия активации для сольволиза
алкилгалогенидов в 50%-ном водном растворе ацетона
при 50 °C (по данным Констэма)а
Алкилгалогенид -де*’ кал/°С -дс^> кал/°С ДС^ де* Среднее
н-С3Н7Вг 26,8 23,4 1,13 1
н-С4Н9Вг СвН5СН2С1 20,8 22,4 27,6 21,6 1,32 0,95 > 1,13
С6Н5СН2Вг 20,8 23,6 1,13
m/^m-C^TTgCl 11,3 29,7 2,63 Л
СвН5СНС12 11,3 29,4 2,62
п-СН3С6Н4СНС12 12,0 39,3 3,27 2,89
п-КО2С6Н4СН(С6Н5)С1 10,8 33,5 3,10
С6Н5СС13 16,2 46,1 2,83 J
а Данные из работ, цитированных в примечании к табл. 108.
результаты были получены для алкил-п-толуолсульфонатов [102].
Согласующиеся
это различие зависит от структуры переходного состояния. При образова-
нии переходного состояния бимолекулярной реакции потеря определенных
классических степеней свободы одной молекулы воды, входящей в переход-
ное состояние, будет уменьшать как энтропию, так и теплоемкость. Допол-
нительное уменьшение этих величин обусловливается электрострикцией
сольватной воды; однако вследствие того, что бимолекулярное переходное
состояние малополярно, этот вклад будет не так велик, как в случае, обсуж-
даемом ниже. В переходное состояние мономолекулярной реакции не входит
ковалентно связанная молекула воды. Однако энтропия и теплоемкость
уменьшаются вследствие сильной электрострикции молекул воды сольвата-
ционной ячейки высокополярного переходного состояния, причем, как будет
объяснено ниже, теплоемкость уменьшается сильнее. Причина состоит в том,
что энтропия — это усредненная по температуре теплоемкость, определяе-
мая для низких температур *, и что сольватация путем образования водо-
родных связей в переходном состоянии мономолекулярной реакции должна
в сильной степени затруднять колебания водорода, которые вследствие своей
относительно высокой частоты не являются возбужденными при низких
температурах ни в исходном, ни в переходном состояниях, а обладают лишь
немного повышенной энергией. Поэтому энтропийный эффект, связанный
с изменением сольватации, должен особенно влиять на теплоемкость акти-
вации. Это приводит к высоким значениям отношения в том слу-
чае, когда упорядоченность переходного состояния обусловлена главным
образом электростатической сольватацией водой (8к1-механизм) и к низким
значениям когда упорядоченность обусловлена главным образом
ковалентным связыванием воды (Эк2-механизм). Такое обобщение, конечно,
является эмпирическим, однако оно показывает, каким образом по величине-
• Точнее
т
S = j
о
С
-jC dT.
393
Глава VII
отношения можно судить о механизме, не рассматривая строение
субстрата. Указанный подход имеет большее значение для мономолеку-
лярного, а не для бимолекулярного механизма.
5. Каталитические эффекты
а. Растворители в роли катализаторов. Под термином «катализ» обычно
понимают ускорение реакции при добавлении веществ, которые сами не испы-
тывают химических изменений независимо от того, включает ли механизм
катализа ковалентное или электровалентное участие катализатора. Так,
катализом можно считать явление ускорения реакции (протекающей в дан-
ном растворителе медленно) путем добавления незначительных количеств
другого растворителя, в котором реакция протекает быстро. Однако меха-
низм в этом случае носит электростатический характер и сводится к дости-
жению лучшей сольватации полярного переходного состояния реакции.
Из-за ошибочного взгляда, что порядок реакции является синонимом моле-
кулярности, нигде не пришлось столкнуться с таким большим количеством
трудностей, как при рассмотрении вопроса о влиянии растворителя при
нуклеофильном замещении. Впервые этот вопрос был поставлен в 1937 г.
[103]. Бейтман и Хьюз [104] показали, что скорость гидролиза шреш-бутил-
хлорида в mpem-бутиловый спирт водой в муравьиной кислоте не зависит
от концентрации (незначительной) воды. Согласно взглядам указанных
исследователей, вода, хотя и является замещающим агентом, ковалентно
не участвует в стадии, определяющей скорость этой реакции с предполагае-
мым мономолекулярным механизмом. Не вступает в реакцию вода и электро-
статически, поскольку муравьиная кислота — лучший сольватирующий
агент. С другой стороны, была высказана точка зрения [103], что, поскольку
гидролиз шреш-бутилбромида и бензгидрилхлорида во влажном ацетоне
ускоряется при добавлении воды по (как это было описано) асимптотически
линейному закону (хотя линейность и не была доказана), замещение должно
быть бимолекулярным (по утверждению авторов, как и любые другие про-
цессы замещения всех алкилгалогенидов). Следует указать, что независимо от
того, являются ли реакции во влажном ацетоне действительно бимолекуляр-
ными (что возможно), указанное выше предположение не вытекает из опыт-
ных данных [105]. Как показано в табл. 101, SN2- и S ^-механизмы замещения
зарядного типа 2 сильно ускоряются полярными растворителями; скорость
реакции увеличивается при переходе от ацетона к воде примерно в 104...
10е раз. Поэтому кривая зависимости скорости от состава бинарного раство-
рителя для любой, даже не гидролитической, реакции типа заряда 2, вероят-
но, начиная с чистого ацетона, поднимается так круто, что порядок реакции
относительно воды становится равным по крайней мере единице. Это может
происходить просто потому, что вода — настолько более благоприятный
растворитель, чем ацетон, что при избытке последнего ведет себя как ката-
лизатор. Иными словами, ацетон не способен препятствовать воде участво-
вать в электростатической сольватации полярного переходного состояния
при Sn2- и S ^-механизмах замещения.
После 1948 г. этот вопрос был поставлен снова, но уже в несколько ином
плане [106]. Было предположено, что «бимолекулярное» и «мономолекуляр-
ное» замещения имеют одинаковый механизм, а именно все процессы замеще-
ния в алкилгалогенидах в действительности тримолекулярны. Например,
было показано, что скорость реакции бромистого метила с пиридином, при-
водящая к образованию N-метилпиридинийбромида, для которой ранее
предполагался бимолекулярный механизм, в бензоле, содержащем метанол,
394
Замещение в алифатическом ряду
пропорциональна произведению [С5Н5К][СН3Вг][СН3ОН]. Конечно, подоб-
ная кинетическая картина могла бы наблюдаться и в случае 8к2-механизма
в определенном интервале концентраций потому, что, во-первых, эта реак-
ция с типом заряда 2 сильно ускоряется полярными растворителями,
л во-вторых, метанол гораздо более полярен, чем бензол. Таким образом,
множитель [СН3ОН] может отражать электростатический катализ раство-
рителем (cosolvent). Другим примером может служить реакция между
метиловым спиртом и трифенилметилхлоридом в бензоле, содержащем пири-
диновое основание, для которой ранее предполагался мономолекулярный
механизм. Вначале было установлено, что скорость метанолиза пропорцио-
нальна выражению [СН3ОН]а[(С8Н5)3СС1], причем пиридиновое основание
не оказывало никакого влияния, кроме того, что оно связывало хлористый
водород. Впоследствии пришли к выводу [106, 107], что скорость реакции
как в присутствии, так и в отсутствие пиридина не имеет постоянной квадра-
тичной зависимости от концентрации метанола. Кажущийся порядок по мета-
нолу при повышении его концентрации постепенно растет от единицы или
менее единицы до трех и более, проходя через значение, равное двум, без
какой-либо видимой задержки. Это позволяет отвергнуть концепцию уни-
версальной тримолекулярности. Рассматриваемая реакция, вероятно,
является одним из вариантов S гД-механизма. Однако ионы, образующиеся
при первоначальной ионизации, остаются рядом друг с другом и вследствие
большого радиуса действия электростатических сил в бензоле притягивают
к себе в разном количестве полярные молекулы, присутствующие в растворе;
таким образом образуются мультиполярные агрегаты, внутри которых
заканчивается нуклеофильное замещение [108]. Доказательства, что такая
физическая агрегация в растворителях с низкой диэлектрической прони-
цаемостью может происходить быстрее, чем изменения ковалентности внут-
ри агрегатов, будут даны в разд. 6. Б присутствии пиридинового основания
замещающий агент, вероятно, взаимодействует с трифенилметилпиридинием,
а не с карбониевым ионом, поскольку последний, судя по спектральным дан-
ным, полученным в других растворителях, тотчас же реагирует с пиридино-
выми основаниями (разд. 3,а).
б. Кислотный катализ. При сольволизе первичных, вторичных и тре-
тичных алкилхлоридов, алкилбромидов и алкилиодидов в первоначально
нейтральной или кислой среде в гидроксилсодержащих растворителях
не наблюдалось никаких признаков катализа добавленной или образую-
щейся в ходе реакции кислотой. Иная картина наблюдается в случае алкил-
фторидов. В общем случае сольволиз этих соединений в указанных выше
условиях катализируется кислотами. Кислотный катализ при сольволизе
алкилфторидов был открыт Миллером и Бернстейном на примере сольволиза
различных замещенных фтористых бензилов в 70%-ном водном растворе
этанола [109]. Чапман и Леви [НО] обнаружили это явление при сольволизе
алифатических первичных, вторичных и третичных алкилфторидов в водном
этаноле; они также указали на некоторые кинетические особенности ката-
лиза.
В случае первичных и вторичных фтористых алкилов, например н-амил-,
2-метил-н-бутил- и циклогексилфторидов, реакция катализируется не только
первоначально добавленным хлористым водородом, но также аутокатали-
зируется образующимся фтористым водородом. В случае третичных алкил-
фторидов, например mpem-бутил- и шреш-амилфторидов, аутокатализ не про-
является, и кинетика этого относительно быстрого каталитического соль-
волиза соответствует реакции первого порядка, величина константы скоро-
сти которой пропорциональна концентрации добавленной сильной кислоты.
395
Глава VII
Все исследователи отмечают, что катализ обусловлен высокой основностью'
фторид-иона, легко образующего водородные связи и исключительно сильно'
сольватируемого протонными растворителями. Эти свойства в несколько,
ослабленной форме будут проявляться и в случае частичного образования
иона фторида в переходном состоянии сольволиза фтористых алкилов. Кине-
тические различия в поведении, с одной стороны, первичных и вторичных,
а с другой стороны, третичных алкилфторидов, вероятно, можно объяснить,
если предположить, что первые реагируют по SN2-, а последние—по S^-меха-
низму. Согласно принципу Гофмана, при гетеролизе связи углерод — гало-
ген по механизму SN2 в переходном состоянии происходит перенос электро-
нов в меньшей степени, чем при механизме SN1. В переходном состоянии
типа Sn2 потенциальный фторид-ион может быть достаточно отрицательно,
заряженным и поэтому достаточно основным, чтобы образовывать водород-
ную связь с кислотой, однако он не может оторвать протон от кислоты.
Поэтому переходные состояния можно представить следующим образом:
Н2О ... R ... F ••• Н ... ОН+
Н2О ... R ... F ••• Н ... F
Этот случай соответствует общему кислотному катализу (включая ауто-
катализ). С другой стороны, в переходном состоянии типа SN1 более отрица-
тельно заряженный и поэтому более основный атом фтора (потенциальный
фторид-ион) может полностью отщепить протон. Поэтому можно предполо-
жить, что переходное состояние имеет вид
б+ б-
R • • F•• • Н*
В данном случае проявляется специфический катализ водородными ионами,
исчезает аутокатализ и можно ожидать, что удельная скорость этой реак-
ции первого порядка при низкой кислотности будет пропорциональна кон-
центрации водородных ионов, а при высокой кислотности — функции h0
Гаммета; последний случай до сих пор не был подтвержден эксперимен-
тально.
Такое же различие в реакциях между фторидами и другими галогенидами
наблюдается в случае ацилгалогенидов [111]. Сольволиз ацилфторидов в гидро-
ксилсодержащих растворителях катализируется кислотами, а ацилхлоридов
не катализируется кислотами.
Кислотный катализ при гидролизе ацеталей обсуждался в разд. 2,6. Кислот-
ный катализ при сольволизе сложных эфиров в гидроксилсодержащих
растворителях будет рассмотрен в гл. XV.
Как отмечалось в разд. 3, в, в апротонных растворителях с настолько низкой
основностью, что они не способны конкурировать как основания с алкилга-
логенидами, может наблюдаться кислотный катализ не только в реакциях
алкилфторидов, но и в реакциях других галогенидов. Покер и сотр. [112]
исследовали катализ хлористым водородом изотопного обмена хлора между
1-фенилэтилхлоридом и радиоактивным 35С1 в нитрометане. Авторы пока-
зали, что выражение для скорости реакции замещения состоит из двух сла-
гаемых
V = JtjtRCl] + jfc2[RCl] [ НС1]
(SN1) (SN1-HC1)
Первое слагаемое представляет собой скорость некатализируемой иониза-
ции 1-фенилэтилхлорида; эта скорость была измерена в других реакциях
и были получены сходящиеся результаты. Например, ее можно измерить
как скорость мономолекулярного элиминирования с образованием стирола;
396
Замещение в алифатическом ряду
это превращение 1-фенилэтилхлорид претерпевает в нитрометане при добав-
лении пиридина для подавления обратной реакции присоединения. Таким
образом, первое слагаемое соответствует чистому механизму SN1. Дальней-
шее исследование с применением стереохимического метода, детальное
объяснение которого будет дано в разд. 7, показало, что второй член в выра-
жении для скорости не относится к механизму SN2, а выражает скорость
отщепления хлорид-иона от 1-фенилэтилхлорида под действием молекулы
хлористого водорода; другими словами, этот член отражает общий кислот-
ный катализ, определяющий скорость стадии ионизации SNl-npopecca, кото-
рой соответствует следующее переходное состояние:
в+ 6-
R ... С1 • • • Н • • • С1
Этот каталитический мономолекулярный механизм можно обозначить сим-
волом SN1-HC1.
в. Катализ солями металлов. Общим методом превращения алкилгало-
генидов AlkHal в спирты AlkOH является кипячение первых с водной суспен-
зией окиси серебра. Если в качестве растворителя использовать спирт ROH,
а не воду, то вместо спирта AlkOH образуется простой эфир AlkOR. Те же
продукты гораздо быстрее получаются, если вместо окиси серебра взять
растворимую соль серебра, например нитрат или ацетат; в этом случае
наряду со спиртом или эфиром получаются соответствующие сложные эфиры
AlkNO3 или А1кОСОСН3.
Кинетические исследования [ИЗ] показали, что во всех подобных реакциях
в определяющей скорость стадии участвуют алкилгалогенид и ион серебра.
Стадия реакции, определяющая ее скорость, протекает в основном на поверх-
ности любой присутствующей нерастворимой серебряной соли, в том числе
и на образующемся и осаждающемся галогениде серебра, и на первоначаль-
но внесенном нерастворимом продукте, например на окиси серебра.
Эти реакции, описываемые как S ^-подобное замещение, инициируются
ионизацией галогенсодержащего соединения на поверхности серебряной
соли при помощи адсорбированных ионов серебра. Завершающий акт реак-
ции состоит в захвате аниона
ионом (R = алкил)
RHaI+Ag+
из раствора образовавшимся карбониевым
AgX
поверхность
R+4-AgHaI
R+ + H2O
R+ + R'OH
R+4-Y"
ROH + H+
ROR' + H+
RY
Позднее такое объяснение процесса было подтверждено следующими тремя
наблюдениями. Во-первых, было показано, что влияние изменения в строе-
нии на скорость реакции с качественной стороны одинаково как для замеще-
ния, катализируемого ионом серебра, так и для мономолекулярного замеще-
ния; таким образом, в обоих случаях по скорости реакций алкилгалогениды
располагаются в следующий ряд: третичные >> вторичные > первичные.
Во-вторых, пространственное течение реакции замещения, катализируемой
ионом серебра, следует, как было показано [114], правилам, применимым
к S^-замещению, а не отличным от них правилам Эк2-замещения (разд. 7, д).
Наконец, в-третьих, S^-замещение должно приводить (через изомеризацию
R+) к образованию продуктов с перегруппировкой атомов, a S^-замеще-
ние — к продуктам нормального строения. В результате реакций, катали-
зируемых ионом серебра [115], образуются продукты с перегруппировкой
атомов (гл. X).
397
Глава VII
Катализ при замещении алкилгалогенидов солями ртути [116] не сопровож-
дается выпадением осадка, а следовательно, не наблюдается поверхност-
ного катализа. Однако в другом отношении этот процесс является более
сложным, поскольку возможен катализ частицами Hga+, HgX+ и HgX2;
было показано, что в разных случаях наиболее эффективен катализ первой
и последней частицами, между которыми несомненно должна существовать
конкуренция [116—118]. Однако нет сомнения в том, что и в этих случаях
наиболее вероятен механизм типа SN1. Во-первых, было показано, что вслед-
ствие обратимости определяющей скорость начальной стадии наблюдается
отклонение от кинетики первого порядка, обусловленное законом действия
масс (разд. 6, а) [117]. Во-вторых, было установлено, что пространственное
течение процессов замещения следует правилам 8к1-реакций [117, 118].
В-третьих, было замечено, что если при 8к1-реакциях образуются продук-
ты с перегруппировкой атомов, то они образуются и при реакциях, ката-
лизируемых азотнокислой ртутью(П) [115].
Многие соли серебра растворимы в ацетонитриле. Покер и Кевилл [119]
исследовали электропроводность нитратов, нитритов и перхлоратов тетра-
этиламмония и серебра в ацетонитриле, а также изучили кинетику, состав
и стереохимию продуктов реакций замещения этими солями и элиминиро-
вания олефина для случая бромистого и хлористого 2-октила. Установлено,
что все соли представляют собой слабые электролиты с константами диссо-
циации порядка 10а моль/л. Однако нитрит серебра обладает особыми свой-
ствами, образуя при ионизации стабильный анионный комплекс Ag(NO2)2
и лишь небольшое количество нитрит-ионов NO2.
Реакция нитрата тетраэтиламмония с 2-октилбромидом — обратимый про-
цесс второго порядка, протекающий по механизму SN2 с обращением конфи-
гурации (разд. 7,г).
NOJ-J-RBr RNO34-Br (SN2)-
В противоположность рассматриваемой ниже реакции в данном случае-
не происходит конкурирующего образования октенов. Реакция нитрата
серебра с 2-октилбромидом протекает гораздо быстрее и имеет кинетиче-
ский порядок, равный 2,5 (1 по субстрату и 1,5 по соли). При добавлении нит-
рата тетраэтиламмония порядок по соли изменяется и становится равным
1 по нитрату серебра и 0,5 по общему количеству нитрата. Реакция завер-
шается выпадением в осадок бромида серебра, однако в присутствии раство-
ренной соли серебра нельзя обнаружить гетерогенного катализа выпавшим
осадком. То же самое относится и к реакции нитрата серебра с 2-октилхло-
ридом. В обеих реакциях образуются октены, однако их количество различ-
но: 7,9% при 45 °C в случае бромида и 3,3% при 45 °C для хлорида. Обра-
зующийся 2-октилнитрат имеет обращенную конфигурацию с частичной
рацемизацией; последнее свойственно реакциям типа SN1 (разд. 7,д).
2-Октилбромид при 100 °C дает 16% октенов и 84 % инвертированного 2-октил-
нитрата, потерявшего 87% оптической чистоты. Эти показатели не зависят
от концентрации нитрата серебра в присутствии или в отсутствие нитрата
тетраэтиламмония.
Кинетические данные были интерпретированы следующим образом. Поло-
винный порядок указывает, что в переходное состояние входит нитрат-ион,
образующийся из солей, которые главным образом находятся в виде ионных
пар; общий состав переходного состояния можно выразить как NO3 +
+ RHal + AgNO3. Состав продуктов реакции указывает на промежуточное
образование карбониевого иона R+, который реагирует, еще находясь под
398
Замещение в алифатическом ряду
влиянием галогенид-иона. Общий вывод состоит в том, что RHal ионизи-
руется только в момент атаки по концам связи электрофилом AgNO3 и нук-
леофилом NO3. Этот механизм можно обозначить символом NOJ-Sn1-
AgNO3. Этот механизм применим и в случае других серебряных солей,
поэтому ниже мы приводим его в общем виде. Например (при X = NO3):
RHal+Ag+X- \ RHal ••• Ag+X"
Медленно
Х“ + RHal • • • Ag+X"--->
X-R+Hal-Ag+--------►
X-R+Hal-Ag+ + X-
V (X--SNl-AgX)
RX+AgHal
Олефин+ HX-|-AgHal -
В этой схеме карбониевый ион входит в состав ионного квадруплета; с таким
предположением мы еще встретимся в разд. 6,ж и з.
Реакции 2-октилбромида с нитритами тетраэтиламмония и серебра во всех
отношениях аналогичны реакциям с соответствующими нитратами, если
учесть специфический способ ионизации нитрита серебра. 2-Октилбромид
реагирует с нитритом серебра не быстрее, а значительно медленнее, чем
с нитритом тетраэтиламмония, что, очевидно, обусловлено очень низкой
концентрацией свободного нитрит-иона, образующегося из нитрита серебра.
Скорость реакции с нитритом серебра при добавлении эквивалентного коли-
чества нитрита тетраэтиламмония мало увеличивается вследствие того,
что добавленные нитрит-ионы образуют комплексные анионы Ag(NO2)2-
Однако эти данные не мешают сделать вывода, что реакция с солями серебра
и при X = NO2 относится к типу X“-SNl-AgX.
Перхлорат тетраэтиламмония, однако, почти не действует на 2-октилбромид
при тех температурах, когда происходит реакция с перхлоратом серебра,
в которой первоначально образуются 2-октилперхлорат и октены. Реакция
с серебряной солью имеет простую кинетику только при очень низкой кон-
центрации соли, при этом порядок как по субстрату, так и по соли равен 1,0
(а не 1,5). Добавление перхлората тетраэтиламмония лишь немного увели-
чивает скорость. Очевидно, что не входящие в ионные пары перхлорат-ионы
обладают недостаточной нуклеофильностью и не могут содействовать иони-
зации субстрата. Было предположено, что эту функцию выполняют моле-
кулы ацетонитрила, поэтому механизм реакции можно обозначить симво-
лом CH3CN-SNl-AgC104.
Перхлорат серебра растворим в бензоле, и в нем он быстро реагирует с алкил-
галогенидами. Покер и Кевилл [120] изучили кинетику и состав продуктов
этой реакции для случая 2-октилбромида. Эта реакция имеет первый порядок
по субстрату и полуторный по соли при ее низкой концентрации. Добавление
перхлората тетра-и-бутиламмония заметно увеличивает скорость. Перво-
начально образуются 2-октилперхлорат и октены, выход которых при 25 °C
был равен 61 и 38% соответственно; это соотношение не зависело от концен-
трации солей. Как было показано с помощью обратной реакции с ионами
брома в условиях механизма SN2 (разд. 7,г), образующийся 2-октилперхло-
рат имел преимущественно обращенную конфигурацию, однако частично
он был рацемизован, что соответствует S ^1-механизму (разд. 7,д). Все это
согласуется с сформулированным выше общим механизмом, который в дан-
ном случае можно обозначить символом G104-SNl-AgC104. Остается неясным,
как связать высокую скорость с очень низкой равновесной концентрацией
перхлорат-ионов, не входящих в ионные пары, поэтому действительный
механизм может быть отличным от указанного общего типа.
399-
Глава VII
6. Солевые эффекты
а. Эффекты, обусловленные законом действия масс и ионной силой
11211- В разд. 1 отмечалось, что в условиях, при которых бимолекулярный
механизм отвечает реакции первого порядка, мономолекулярный механизм
имеет обычно в известной мере сложную кинетическую форму, которая
не обязательно сводится к реакции первого порядка. Это происходит, напри-
мер, в том случае, когда замещающий агент играет роль растворителя.
Причиной этого служит обратимость первоначального гетеролитического
процесса при мономолекулярных реакциях, что можно проиллюстрировать
на примере гидролиза’хлористого алкила в растворителе, содержащем воду:
Н2О
RCI ~R++C1----------> ROH + H+ + C1-
(2) (3)
Чем больше становится концентрация хлорид-иона за счет реакции (3),
тем большее значение приобретает реакция (2). Таким образом, стадия
ионизации, определяющая скорость реакции, значительно замедляется
за счет обратимости процесса. При этом не обязательно, чтобы вся реакция
в целом была обратима, напротив, реакция может представлять собой
совершенно необратимый гидролиз; скорость реакции все больше и больше
замедляется по сравнению со скоростью реакции первого порядка. Это мож-
но выразить, видоизменив уравнение реакции S f^l-типа при условии постоян-
ной активной массы замещающего агента. В этом случае уравнение имеет
следующий вид:
dx/dt = ki (а — х) (1 -j-ccx)-1,
где а — исходная концентрация алкилгалогенида, х — концентрация хло-
рид-иона, образующегося при гидролизе, множитель (1 + сся:)-1 описывает
возрастающее уменьшение «удельной скорости» (dx/dt)/(а — х)', константа а
выражает способность хлорид-иона к конкуренции с водой в реакции с кар-
бониевым ионом.
Аналогичного эффекта, естественно, следует ожидать в том случае, если
вместо определения образовавшейся соляной кислоты заранее ввести в реак-
цию не которое количество НС1, хлористого натрия или другого способного
к ионизации хлорида. Различие состоит в том, что в данном случае скорость
реакции с самого начала уменьшается согласно уравнению
dx/dt = к1 (а — х) {1 -f-cc (с-рх)}'1
где с — концентрация заранее внесенного хлорид-иона.
Константы а в принципе меняются при изменении условий реакции, в кото-
рой происходит конкуренция между анионами и растворителем за карбоние-
вый ион. Они резко меняются при изменении субстрата, возрастая при уве-
личении относительной стабильности карбониевого иона. От природы рас-
творителя они зависят в меньшей степени.
Такое влияние солей на мономолекулярные реакции проявляется в случае
«солей с общими ионами», т. е. солей, анионы которых идентичны анионам,
образующимся при гидролизе. «Соли без общих ионов», т. е. соли с каким-
либо другим анионом, например азид натрия в случае гидролиза алкилхлори-
да, не способны к подобному влиянию, поскольку они вступают п реакции
400
Замещение в алифатическом ряду
только после завершения стадии ионизации, определяющей скорость реакции.
(1)
RC1 R+ + C1-
(2)
(3)
КОН + Н*+С1-
(4)
----> RN3 + C1-.
N3
Таким образом, согласно закону действия масс, подобные соли совершенно
не влияют на первоначальную скорость расщепления алкилгалогенида, хотя
благодаря конкурирующей реакции с карбониевым ионом они могут ослабить
тормозящее действие образующегося хлорид-иона на гидролиз исходного
алкилгалогенида.
Бимолекулярное замещение приводит к совершенно иным результатам.
В случае необратимого гидролиза хлорида, протекающего по схеме
H2O4-RC1 —> ROH 4-Н+ + Cl-
закон действия масс не может оказать никакого кинетического влияния
ни на образование хлорид-иона, ни на образование любой добавляемой
соли «с общим ионом». С другой стороны, добавляемая соль «без общего иона»,
такая, как NaN3, способна с самого начала, если она вообще вступает в реак-
цию, ускорять расщепление молекулы алкилгалогенида вследствие осуще-
ствления реакции по схеме
N3+RC1 -> RN3 + C1-
б. Предсказываемое влияние ионной силы [121]. Все указанные выше
выводы являются следствием закона действия масс. Кроме того, всегда сле-
дует считаться и с электростатическими эффектами, обусловленными ионной
силой растворов. Теория этих явлений качественно аналогична теории влия-
ния растворителя. Как при мономолекулярной ионизации нейтральной
молекулы, так и при бимолекулярной реакции между нейтральными моле-
кулами растворитель повышенной полярности ускоряет реакцию- Это про-
исходит вследствие того, что полярное переходное состояние стабилизуется
за счет притяжения полярных молекул растворителя. Увеличение скорости
этих реакций может происходить и за счет инертных солей, поскольку заря-
ды в переходном состоянии могут лучше стабилизоваться вследствие при-
тяжения окружающих ионов. Подобно тому как влияние растворителя при
мономолекулярном механизме больше, чем при бимолекулярном, из-за
большего в последнем случае рассредоточения заряда в переходном состоя-
нии, ускоряющее влияние ионной силы более заметно в случае мономолеку-
лярного механизма. Такие электростатические эффекты солей, естественно,
зависят от диэлектрической проницаемости растворителя: чем меньше диэлек-
трическая проницаемость, тем сильнее влияют электростатические силы
на ионы и через ионы и тем сильнее их кинетическое влияние.
Взяв в качестве примера вновь мономолекулярный гидролиз алкилхлорида,
можно, во-первых, отметить возрастание в процессе реакции ионной силы.
Следовательно, электростатическое влияние ионной силы должно привести
к прогрессивному увеличению константы скорости реакции. Если сначала
внести в реакционную смесь любую соль с общим ионом или без него,
то с самого начала благодаря электростатическому эффекту должна уве-
личиться скорость реакции.
В случае бимолекулярного механизма гидролиза электростатические эффек-
ты качественно аналогичны описанным, но значительно слабее.
26-0772
401
Глава VII
Рассмотренные положения объединены в табл. 110. Эффекты ионной силы
мы будем рассматривать в основном тогда, когда они действуют в одном
направлении с эффектом действия масс.
Таблица 110
Предсказанные влияния закона действия масс и ионной силы
на мономолекулярный и бимолекулярный сольволиз алкилгалогенидов
Закон действия масс + Ионная сила
Образующиеся ионы Прогрессирующее уменьшение удель- ной скорости + Прогрессирующее увели- чение удельной ско- рости
SN1 Соль с общим ионом Замедление + Ускорение
Соль без общего иона Отсутствие первона- чального эффекта •ф- Ускорение
Образующиеся ионы Не влияют + Небольшое прог рссирую- щее увеличение удель- ной скорости
Sn2 Соль с общим ионом Не влияет + Небольшое ускорение
Соль без общего иона Ускорение Небольшое ускорение
Наиболее интересными из приведенных в табл. НО положений являются те,,
которые (выделены курсивом) основаны на влиянии закона действия масс
на мономолекулярный механизм. Однако на этот эффект накладывается
существенный по величине и противоположный по направлению эффект
ионной силы. Поэтому мы должны уметь аналитически разделять эффекты,
связанные с законом действия масс и с изменением ионной силы. В разд. 6,а
была дана приближенная количественная теория эффекта закона действия
масс. Для целей анализа мы должны уделить внимание и приближенной тео-
рии эффекта ионной силы.
Рассмотрим теорию [121], основанную на предположении, которое будет-
критически проверено ниже, что растворы достаточно разбавлены для того,
чтобы можно было учитывать лишь электростатические силы дальнего дей-
ствия. Эти силы заставляют окружающие ионы образовать вокруг централь-
ного диполя, который, допустим, представляет собой переходное состояние
ионизации при мономолекулярном механизме, слабую «ионную атмосферу».
Если эта слабая ионная атмосфера образуется вокруг центрального одноза-
рядного иона, то, согласно теории Дебая, можно определить коэффициент
активности с помощью следующего уравнения:
”ln = /W ^1/2eV1/2 (DkT)-3/2
где N — число Авогадро, е — заряд электрона, к — постоянная Больцмана,
р, — ионная сила раствора, D — диэлектрическая проницаемость раствори-
теля. Аналогично можно вычислить (если подобрать правильное решение
уравнения Пуассона; с этой трудностью сталкивается теория Дебая) коэф-
фициент активности центрального диполя, состоящего из двух точечных
зарядов ±ze, разделенных расстоянием d; этот коэффициент выражается
402
Замещение в алифатическом ряду
следующей формулой:
- In fd = AW ф (DkT)~2
Таким образом, чтобы рассчитать кинетический эффект ионной силы, един-
ственное, что необходимо сделать — это ввести указанные коэффициенты
в бренстедовское уравнение для скорости (гл. II, разд. 2, г). В частности,
в случае обратимой ионизации
fei
RX R++X-
Й2
в соответствующие выражения необходимо ввести и
Скорость диссоциации = fc1[RX]/(j
/?
Скорость рекомбинации ионов = Л-2[Н+][Х—1 -Д
ld
В эти уравнения в неявном виде входит один параметр, называемый «кон-
стантой ионной силы» п, которая определяется следующим образом:
n = z2d
В наших расчетах по влиянию закона действия масс на скорость мономоле-
кулярной реакции мы использовали «константу закона действия масс» а,
определенную как отношение k2/k3. Таким образом, наши расчеты эффектов
ионной силы включают такую же, но никак не большую степень произвола
и могут быть применены для анализа налагающихся эффектов.
Константа ионной силы п имеет размерность длины. Если предположить,
что z — доля электрона, переходящая от углерода к галогену при активации
с образованием переходного состояния ионизации,— не слишком мала,
то величина п должна быть близка к длине связи, что и соответствует дей-
ствительности, так как п выражается в ангстремах. Ее величина не меняется
при переходе от 70- к 90%-ному водному раствору ацетона, хотя и может
меняться при более значительном изменении растворителя. Величина о
зависит от строения субстрата и заметно возрастает, когда вследствие сопря-
жения или гиперконъюгации центры возникающих зарядов диполя рас-
полагаются дальше друг от друга, или, другими словами, когда возрастает d.
Описанные выше представления о влиянии ионной силы, т. е. о влиянии
дальних электростатических сил окружающих ионов, справедливы только
при условии, если можно пренебречь локальными эффектами поля заряда
иона, которые называются «специфическими» солевыми эффектами и харак-
теризуют природу каждого данного иона. С точки зрения термодинамики
общий солевой эффект (кулоновский плюс специфический) состоит в том,
что окружающие ионы путем увеличения когезии увеличивают «внутреннее
давление» в системе, что приводит (если отсутствуют уравновешивающие
или преобладающие специфические силы притяжения между окружающими
ионами и растворенным веществом) к «выдавливанию» органического рас-
творенного вещества, например к его выходу из сольватационной ячейки
или вообще к образованию отдельной органической фазы; в общем это озна-
чает увеличение коэффициента активности органического вещества. Мы учи-
тывали разницу во влиянии на начальное и переходное состояния лишь той
части общего солевого эффекта, которая зависит от дальнодействующего
кулоновского поля зарядов окружающих ионов. Остается учесть специфи-
26* 403
Глава VII
ческую часть эффекта, т. е. ту часть, которая зависит от локальных взаимо-
действий и обусловлена иными свойствами ионов, чем просто их заряд;
поэтому эти свойства изменяются при переходе от одного иона к другому,
имеющему тот же заряд. Характеризует ли этот специфический эффект толь-
ко начальное состояние или только переходное состояние либо, как есть
в действительности, он характеризует различие между этими двумя состоя-
ниями — вот те вопросы, которые нужно выяснить.
Эти вопросы изучали Кларке и Тафт [122] на примере гидролиза тпретп-бутил-
хлорида в воде — реакции, для которой влияние закона действия масс
на кинетику пренебрежимо мало. Метод состоял в сравнении влияния добав-
ленных солей на начальное давление паров растворенного /гареиг-бутилхло-
рида и на скорость его гидролиза; данные, полученные таким образом,
позволяли оценить отдельно влияние каждой соли на исходное и переходное
состояния. Результаты показали, что общий солевой эффект как на исход-
ное, так и на переходное состояние в значительной степени обусловливался
специфическим взаимодействием, которое изменяется при переходе от одной
соли к другой, однако в случае простых неорганических солей их специфи-
ческое влияние на оба состояния было одинаково. Это должно означать,
что в данном случае специфические солевые эффекты в действительности
обусловлены локальным взаимодействием ион — растворитель, и поскольку
можно полагать, что объемы исходного и переходного состояний малы, внут-
реннее давление, обусловленное этими эффектами, в обоих случаях пример-
но одинаково. Таким образом, различное влияние солей на исходное и пере-
ходное состояния в основном является следствием кулоновских сил, которые
можно вычислить согласно уравнениям, приведенным выше. Кларке и Тафт
подтвердили неспецифическую природу этого эффекта и зависимость его
только от макроскопической ионной силы на гораздо большем ряде солей,
чем это было сделано ранее, включив в этот ряд 2 — 1- и 1 — 1-электролиты.
Ниже мы будем ссылаться на сделанный ими вывод о том, что при мономоле-
кулярном гидролизе тпретп-бутилхлорида в переходном состоянии происходит
увеличение дипольного характера этой молекулы.
Предположения, выделенные курсивом в табл. 110, следуют из закона дей-
ствия масс, который влияет на мономолекулярный механизм таким образом,
что удельная скорость процесса по ходу реакции в отсутствие добавленных
солей уменьшается, а соли с общим ионом (но не другие соли!) замедляют
реакцию с самого ее начала. В последующих разделах эти эффекты будут
проиллюстрированы на экспериментальных примерах.
в. Влияние образующихся ионов на кинетику сольволиза. Сначала
рассмотрим пример мономолекулярного сольволиза, а именно гидролиз
нгретп-бутилбромида в водном ацетоне [123], когда эффект закона действия
масс настолько мал, что можно в отдельности рассмотреть эффект ионной
силы. При этом в ходе реакции константа скорости увеличивается. Это можно
видеть на рис. 31, хотя на нем представлена кривая для интегральной кон-
станты скорости первого порядка i-1 In [al{а — ж)], наклон которой из-за
усредняющего влияния интегрирования в два раза меньше наклона кривой
константы скорости (dx/dt'j/ia — ж). На графике отображены результаты
двух экспериментов, проведенных с растворами, содержащими различное
количество воды, а следовательно, обладающими заметно отличными диэлек-
трическими проницаемостями. Полученные кривые находятся в соответствии
с теорией разбавленных растворов. Константу ионной силы о для трет-
бутилбромида можно вычислить либо на основании данных, полученных при
проведении подобных экспериментов в разных условиях, либо на основании
наблюдаемого кинетического эффекта при добавлении солей без общих ионов
404
Замещение в алифатическом ряду
(разд. 6, г). Таким образом, теоретические кривые построены без учета
опытных данных, с которыми они сравниваются на рис. 31. Наблюдаемое
соответствие подтверждает применимость теории влияния ионной силы.
В частности, подтверждается, что свойством растворителя, влияющим
на кинетический солевой эффект, является только его диэлектрическая
Рис. 31. Кинетика гидролиза
mpem-бутил бромид а в
водном растворе ацетона.
Среднее значение константы
скорости первого порядка
(константа скорости в нача-
ле реакции принята за еди-
ницу) в зависимости от хода
реакции (в процентах). Кри-
вая 1 и точки ф относятся к
реакциям в 90%-ном ацето-
не, а кривая 2 и точки
О — к реакциям в 70 %-ном
ацетоне, в обоих случаях
при 25 °C. Точки соответст-
вуют экспериментальным
данным, кривые вычислены
на основании электростати-
ческой теории влияния ион-
ной силы с помощью неза-
висимо рассчитанного зна-
чения константы ионной
силы о.
100х/а
проницаемость. Это означает, что константа ионной силы о в приведенном
примере не зависит от природы растворителя.
В качестве дополнительного примера рассмотрим кинетику гидролиза
гещ'-диметилбензгидрилхлорида в водном растворе ацетона. В этой реакции
проявляется значительный эффект ионной силы, но в еще большей степени
Рис. 32. Кинетика гидролиза пщ'-диме-
тилбензгидрилхлорида в вод-
ном растворе ацетона.
Среднее значение константы скоро-
сти первого порядка (константа
скорости в начале реакции принята
за единицу) в зависимости от хода
реакции (в процентах). Кривая 1 и
точки ф относятся к 90%'Ному, а
кривая 2 и точки О — к 80 %-ному
ацетону, в обоих случаях при 0 °C.
Точки соответствуют эксперимен-
тальным данным, кривые построе-
ны по расчетным данным.
эффект закона действия масс. В результате этого по мере протекания реак-
ции удельная скорость сильно уменьшается. На рис. 32 показано уменьше-
ние интегральной константы скорости первого порядка и тем самым про-
иллюстрировано то, что было предсказано для мономолекулярного меха-
низма в табл. 110. Существование эффекта ионной силы, хотя он и превос-
ходит эффект закона действия масс, проявляется в различии скоростей
уменьшения константы для растворов ацетона с различным содержанием
воды. Эффект ионной силы зависит от диэлектрической проницаемости,
а следовательно, от содержания воды в реакционной среде; в то же время
405
Глава VII
эффект закона действия масс не зависит от содержания воды в ацетоне,
поскольку было показано, что влияние азида также не зависит от содержа-
ния воды (разд. 3,6). Весьма вероятное предположение о том, что влияние
хлорид-иона также не зависит от содержания воды, согласуется с количест-
венными расчетами кинетики гидролиза. Кривые, приведенные на рис. 32.
вычислены теоретически при помощи констант ионной силы (ст) и закона
действия масс (из значений а) для диметилбензгидрилхлорида. Константа а
оценена по кинетическому эффекту добавленных солей без общего иона
(разд. 6,г). Константу а можно определить либо по данным эксперимента
указанного типа, либо по кинетическому эффекту при добавлении солей
с общим ионом (разд. 6,г). Следует отметить, что соответствие между теоре-
тическими кривыми и экспериментальными данными сильно зависит от пред-
положенной молекулярности быстрой реакции между карбониевым ионом
и водой. Этот вопрос подробнее обсуждается далее в разд. 6,д.
г. Влияние добавляемых солей на начальную скорость сольволиза.
В табл. 111 приведены некоторые данные о влиянии добавляемых солей
Таблица III
Влияние добавляемых солей на первоначальную скорость
гидролиза алкилгалогенидов в водном растворе ацетона
Водный раствор ацетона Температура, °C Соль Концент- рация соли, моль/л Изменение скорости, %
трет-Бутилбромид [123] 90%-ный 50 NaN3 0,10 +40
90%-ный 50 LiCl 0,10 +39
90%-ный 50 *LiBr 0,10 +42
Бензгидрилхлорид [124] 80%-ный 25 ♦LiCl 0,10 -13
80%-ный 25 LiBr 0,10 + 17
Бензгидрилбромид [124] 80%-ный 25 LiCl 0,10 +27
80%-ный 25 *LiBr 0,10 -13
п^п'-Диметилбензгидрилхлорид [125] 90%-ный 0 NaN3 0,05 +69
85%-ный 0 NaN3 0,05 +48
85%-ный 0 ♦LiCl 0,05 -46
85%-ный 0 LiBr 0,05 +46
85%-ный 0 (CH3)4NNO3 0,05 +52
80%-ный 0 NaN3 0,05 +36
на начальную скорость мономолекулярного гидролиза четырех алкилгалоге-
нидов в водном растворе ацетона. Во всех случаях измерялась скорость
образования иона галогена из алкилгалогенида; это позволило определить
скорость ионизации. Если анион добавляемой соли может вступать в реак-
цию с образованием устойчивого продукта, то скорость образования кислоты
не является мерой скорости ионизации. Применялись соли в одной из двух
стандартных концентраций. Необходимо также указать, что абсолютно все
соли без общего иона (не отмеченные в таблице звездочками) увеличивают
скорость, и это соответствует теоретическим предсказаниям, приведенным
406
Замещение в алифатическом ряду
в табл. НО. Соли же с общим ионом (отмеченные звездочкой), за исключе-
нием тре/га-бутилбромида, на гидролиз которого, как известно, мало влияет
закон действия масс, уменьшают первоначальную скорость гидролиза;
это подтверждается торможением за счет закона действия масс, предсказан-
ным для мономолекулярного механизма (табл. 110).
Из данных типа приведенных на рис. 31 и 32 и в табл. 111 были вычислены
[121] константы ионной силы и закона действия масс (п и ос) для сольволиза
ряда mpezzz-бутил-, бензгидрил- и алкилбензгидрилгалогенидов в водных
органических растворителях, главным образом в водном растворе ацетона.
Полученные результаты приведены в табл. 112. В случае треттг-бутилброми-
да а ~ 1 л/моль, т. е. слишком мала для того, чтобы влиять на кинетику
Таблица 112
Константы ионной силы [<г, А (0,1 мм)] и закона действия масс
(а, л/моль) для сольволиза алкилгалогенидов в водном растворе ацетона
Алкилгалогенид Раство- ритель - водный ацетон, % (ПО объему) Темпе- ратура, °C а а
из ки- нети- ческих данных из влия- ния солей без обще- го иона из кине- тических данных ИЗ влия- ния солей с общим ионом
m/>em-C4HgCl 80 а 35 0,25
трет-СдНдВг 70 ... 90 25 ... 50 0,75 0,8
(СеН5)2СНС1 70 ... 90 25 ... 50 1,6 10 13
(С6Н5)2СНВг 80 ... 90 25 1,75 50 70
4-тр em-Ci Н 9Св Н 4
и СН(СеН5)С1 80 ... 90 25 ... 50 2,2 20
4-СН3С6Н4СН(СеН5)С1 80 25 35 6 316
(4-СН3СеН4)2СНС1 80 ... 90 0 2,75 70 90
а В водном растворе метанола [126]. Последующие данные для бензгидрилхлорида в 70%-ном
водном ацетоне при 0 “С вновь подтвердили, что электроположительные заместители увели-
чивают а [127].
6 Рассчитано для а=2,2 А (22-10-2 нм).
сольволиза или обусловливать различие между кинетическими эффектами
солей с общим и без общего иона. Поэтому из данных по кинетике сольво-
лиза и кинетическим эффектам солей можно получить величины о. Однако,
изучая скорость появления радиоактивности в трет-бутил хлориде при его
реакции с радиоактивным хлорид-ионом, Бантон и Нейак [126] получили
для трет-С4Н9С1 величину ос = 0,2...0,3 л/моль. В случае бензгидрильной
и алкилбензгидрильной систем имеют влияние как константа о, так и а,
поэтому вначале из эффекта солей с общим ионом определялась ц, а затем
с использованием полученных значений ц из данных по кинетике сольволиза
и влиянию солей с общим ионом вычислялась константа а.
Используя двухточечнозарядную модель молекулы, можно вычислить,
в какой степени должен осуществляться перенос заряда во время активации
на расстояние, равное длине связи СВг, в переходном состоянии при соль-
волизе mpem-бутилбромида в водном растворе ацетона, чтобы это соответ-
•ствовало величине ц = 0,8 А. (8 ИО-2 нм). Рассматривая кривую Морзе,
характеризующую растяжение связи при активации, примем, что длина
связи СВг в точке, соответствующей энергии активации, равна
407
Глава VII
2,4 А (24-10“2 нм). Отсюда получаем 0,6 электрона. Кларке и Тафт [122],
используя ту же модель и исходя из данных по влиянию солей отдельно
на исходное и переходное состояния гидролиза щрепг-бутилхлорида в воде,
Получили следующую картину: в исходном состоянии от С к С1 перенесено
0,2 электрона, а в переходном состоянии — 0,8 электрона. Они отметили,
что такой прием оценки степени переноса заряда несколько искусствен.
Если провести подобные вычисления степени переноса заряда, сопровож-
дающего образование переходного состояния сольволиза 4,4'-диметилбенз-
гидрилхлорида в водном растворе ацетона, то, используя наблюдаемое зна-
чение о = 2,75 А (27,5-Ю-2 нм), получим 1,1 электрона, т. е. более чем
нужно для полной ионизации галогена. Конечно, дело в том, что центроид
положительного заряда не расположен на а-углеродном атоме, а сдвинут
внутрь алкильной группы. Кроме того, кривая Морзе дает лишь низший
предел длины связи в переходном состоянии (рис. 29). Помимо этого, двух-
точечнозарядная модель теоретически применима лишь к очень разбавлен-
ным растворам и не очень хороша в случае растворов умеренной концентра-
ции, в которых проводились измерения. Тем не менее данные табл. 112 пока-
зывают, что величина о тем больше, чем на большее расстояние происходит
перенос электрона, сопровождающий активацию (об этом можно качественно
судить, сравнивая молекулы с разной структурой).
Константы закона действия масс а отражают реакционную способность пол-
ностью образовавшегося карбониевого иона; они указывают, какова ско-
рость реакции карбониевого иона с галогенид-ионом, отнесенная к скорости
реакции с водой. Из данных табл. 112 следует, что отношение этих скоростей
сильно возрастает, когда строение субстрата изменяется в сторону стабили-
зации карбониевого иона путем делокализации заряда. Причина этого,
вероятно, в том, что, несмотря на уменьшение общей реакционной способ-
ности вследствие делокализации, делокализованный карбониевый ион относи-
тельно быстрее реагирует с анионом, так как последний лучше, чем нейтраль-
ная молекула, притягивает делокализованный заряд, т. е. в случае аниона
в большей степени проявляется фактор поляризуемости карбониевого иона.
д. Молекулярность быстрых стадий мономолекулярного замещения.
Рассмотрим мономолекулярное сольволитическое замещение, состоящее
из процессов (1) —(3), причем процесс (1) протекает медленно, а процессы
(2) и (3) — быстро
О)
(1)
RC1 R+4-C1- —
(2)
-----г- 11113-f-v.x
NJ
Можно допустить, что в присутствии анионного реагента, участвующего
в быстром процесс (4), одновременно идет и несольволитическое замещение,
включающее процессы (1), (2) и (4). Преимущество такого предположения
заключается в том, что тем самым выделяется процесс (4) того же общего
типа, что и процесс (2), конкуренцию которого с другими быстрыми процес-
сами можно изучить.
В предыдущих разделах было приведено много доказательств в пользу
того, что процесс (1) является мономолекулярным в соответствии с приведен-
ным общим определением молекулярности (разд. 1 ,д).
Теперь можно попытаться доказать, что процесс (2), а также процесс (4)
бимолекулярны. Как описано в разд. 3,6, на основании соотношения обра-
зующегося алкилазида при изменении концентрации азид-иона процесс (4|
н2о
(4)
408
Замещение в алифатическом ряду
относительно концентрации азид-иона мономолекулярен и имеет такую же
молекулярность (предположительно первого порядка), как и все конкури-
рующие быстрые процессы с участием карбониевых ионов. Следовательно,
процесс (4), а по аналогии и процесс (2) являются бимолекулярными. Что
касается процесса (2), то такой вывод подтвержден, как показано ниже,
количественными данными по кинетике чисто сольволитического заме-
щения.
Кроме того, можно показать, как это и следует из теории ионизации (рис. 29),
что процессы рекомбинации ионов (2) и (4) обладают конечной энергией
активации, хотя в настоящее время и нельзя более строго количественно
установить низшие пределы действительных величин энергий активации.
Необходимо измерить температурный коэффициент влияния необщего иона
или температурный коэффициент уменьшения скорости реакции, вызывае-
мого добавлением общего иона. В качестве примера можно привести иссле-
дованный Хаудоном, Хьюзом и автором [128] гидролиз тг,п'-диметилбензгид-
рилхлорида при различных температурах в водном растворе ацетона в при-
сутствии азида натрия. При этом отдельно измеряли скорости образования
спирта и алкилазида. На основании результатов опытов можно, применяя
уравнение Аррениуса, подсчитать энергию активации процесса (1), а также
разность энергий активации процессов (4) и (3) Е% — E'f. Эта разность
имеет положительное значение и равна 4,0 и 3,9 ккал/моль (16,75-103
и 16,33-103 Дж/моль) соответственно для 90- и 85%-ных водных растворов
ацетона. Таким образом, хотя пока не известно, обладает ли процесс (3)
конечной энергией активации, все же можно утверждать, что энергия акти-
вации процесса (4) составляет не менее 4 ккал/моль (16,75 -103 Дж/моль). Ана-
логичное заключение справедливо и для процесса (2).
Наконец рассмотрим [121] молекулярность процесса (3). Обсуждение осно-
вывается на том, что любая количественная трактовка влияния как ионной
силы, так и закона действия масс на кинетику мономолекулярного сольво-
лиза (см., например, кривые на рис. 32) зависит от молекулярности всех
процессов. Это происходит потому, что ионная сила изменяет суммарную
скорость реакции не только путем изменения скорости медленного процес-
са (1). Ионная сила действует и на быстро протекающие процессы (2) и (3),
влияя на распределение образующихся ионов карбония между реакциями (2)
и (3); эффект ионной силы в любом процессе зависит от строения переходного
состояния, а следовательно, влияет на молекулярность. На основании ска-
занного можно принять, что в отношении карбониевого иона процессы (1)
и (2) являются соответственно моно- и бимолекулярными, а процесс (3) —
мономолекулярный. Остается определить молекулярность процесса (3)
в отношении воды; для определения этой недостающей характеристики
мономолекулярного замещения можно использовать количественную форму
кинетики сольволиза.
Наиболее вероятными, но в то же время требующими проверки являются
следующие два предположения: первое, что процесс (3) по отношению к воде
мономолекулярен, и второе, что процесс (3) по отношению к воде полимоле-
кулярен. Первая гипотеза предполагает простое переходное состояние
(R- • -ОН2)+, коэффициент активности которого, как и для R+, определяется
законом Дебая (разд. 6,6). Она приводит к совершенно неправильной кине-
тике, которая не отвечает опытным данным, подобным тем, которые приве-
дены на рис. 32. Согласно второй гипотезе, переходное состояние можно
представить в виде {R • •-(ОН2)П}+, где значение п достаточно велико; наи-
более важная кинетическая особенность переходного состояния состоит
в значительном пространственном (в трех измерениях) распределении поло-
409
Глава VII
жительного заряда. Это означает, что мы не можем вычислить коэффициент
активности по закону Дебая. Из-за большой диффузности заряда в этом
случае применимо более общее уравнение Дебая — Хюккеля, в которое
входит дополнительный множитель, повышающий коэффициент активности
до значения, близкого к единице. Если придерживаться этой гипотезы,
то кинетические кривые имеют правильную форму; кривые, вычисленные
на основании этой гипотезы (значение п предполагается бесконечным), сов-
падают с найденными точками на рис. 32.
Элементарная интуиция подсказывает, что процесс (3) полимолекулярен
по отношению к воде. Кажется естественным, что карбониевый ион, образую-
щийся внутри водной сольватационной ячейки, должен реагировать с водой
с распадом ячейки; энергия этого процесса не велика, но вследствие высокой
молекулярности энтропия очень отрицательна. Трудно предположить, что
с ионом карбония реагирует молекула воды с высокой энергией, которая
проходит внутрь сольватационной ячейки без нарушения последней (что
справедливо в случае атаки анионом), поскольку для карбониевого иона
более доступны молекулы воды внутренней сольватационной сферы.
Полимолекулярность процесса (3) по отношению к воде своеобразно подтвер-
ждается изучением состава продуктов реакции, в которой происходит конку-
ренция между каким-либо анионом и водой. В разд. 3,6 упоминалось, что
скорость превращения щп'-диметилбензгидрильного катиона в спирт
в водно-ацетоновых растворах с различным содержанием воды и одинаковым
содержанием азид-ионов не зависит от концентрации воды. Это указывает
на то, что в реакции участвует карбониевый ион и его сольватная оболочка,
а не карбониевый ион и кинетически независимая молекула воды.
е. Влияние солей на мономолекулярное замещение в сернистом ангидриде
как растворителе. Как было показано в разд. 2,в, бензгидрилхлорид в этом
растворителе подвергается замещению с одинаковой скоростью при действии
триэтиламина, пиридина и фторид-иона; л-хлорбензгидрилхлорид также с оди-
наковой скоростью реагирует с пиридином, иодид- и фторид-ионами. Можно
предположить, что все эти процессы являются мономолекулярными. Детально
была изучена кинетика одного из этих процессов, а именно взаимодействия
хлорбензгидрилхлорида с фторид-ионом, вводимым в реакцию в виде тетра-
метиламмонийфторида [129]. До определенного этапа этот процесс проте-
кает в прямом направлении, не нарушаемом обратным процессом. Для него
характерен значительный ускоряющий эффект ионной силы и весьма боль-
шой замедляющий эффект закона действия масс. Эту реакцию можно изо-
бразить следующим образом:
(1) F-
RC1 ~---* R+4-C1- —-> RF + C1-
(2) (3)
Ускоряющий эффект соли без общего иона можно проиллюстрировать тем,
что при 0,05 М концентрации тетраэтиламмонийфторида скорость реак-
ции в три раза выше, чем предельная скорость при низких концентрациях
соли. Соль с общим ионом оказывает сильный замедляющий эффект. Если
реакция проводится в присутствии специально добавленного тетраэтилам-
монийхлорида (0,047 7И), то начальная скорость замещения фторид-ионом
уменьшается в 150 раз. В отсутствие добавок хлорида реакция сильно замед-
ляется образующимися в процессе реакции хлорид-ионами: вначале она
идет быстро, а затем медленно. Такие очень сильные эффекты, обусловлен-
ные законом действия масс, являются мерой конкуренции замещающего
агента с общим ионом, которая меньше конкуренции замещающего агента
410
Замещение в алифатическом ряду
с гидроксилсодержащим растворителем, с которым общий ион успешно
конкурирует в реакциях сольволиза. Результаты исследований в жидком
SO2 показывают, что доказательство мономолекулярного механизма соль-
волиза можно получить не только при исследовании самого сольволиза.
ж. Солевые эффекты при мономолекулярном сольволизе в уксусной кислоте.
Ключевой момент в понимании солевых эффектов в уксусной кислоте состоит
в том, что в этом растворителе электростатические силы действуют на боль-
ших расстояниях. Диэлектрическая проницаемость уксусной кислоты состав-
ляет около 6. В этом растворителе два одновалентных противоположно
заряженных иона притягиваются друг к другу с энергией, равной средней
кинетической энергии каждого из них на расстоянии 150 А. (15 нм). При
обычно используемых концентрациях солей эти ионы даже при случайном
распределении будут находиться друг от друга на расстояниях, меньших
150 А. (15 нм), и поэтому они должны главным образом соединяться в пары
или другие агрегаты с более коротким радиусом действия сил. Еще в 1930-х го-
дах было известно [130], что в уксусной кислоте соли существуют главным
образом в виде ионных пар, находящихся в равновесии с небольшим коли-
чеством свободных ионов, ионными тройниками и, возможно, с квадру-
полями.
Определение ионной пары — это в значительной степени семантическая
проблема, во многом аналогичная, например, тому, в каком положении
открытая дверь уже не может считаться «полуоткрытой». В качестве услов-
ной границы между ионными парами и диссоциированными ионами приняли
такую степень разделения (с кулоновской энергией 2кТ), при которой коли-
чество ионных пар противоположно заряженных ионов меньше, чем при
любой другой степени разделения [131]. Для 1-1-электролитов в уксусной
кислоте это расстояние равно 40 А (4 нм). При уменьшении расстояния
между ионами количество ионных пар увеличивается: кривая этой зависи-
мости имеет довольно крутой подъем. Таким образом, «дверь диссоциации»
в уксусной кислоте может быть не только «закрытой» или «открытой»,
но в значительном интервале степеней разделения может быть и «полуоткры-
той»; количество ионных пар с разной степенью разделения определяется
кривой распределения Больцмана, и каждая из ионных пар имеет собствен-
ную, ей присущую химическую и электрохимическую активность, поэтому
все измерения определенным образом усредняются.
Уинстейн и сотр. [132] изучили солевые эффекты при сольволизе в уксусной
кислоте таких алкилгалогенидов и аренеульфонатов, для которых при изу-
чении стереохимии (разд. 7) был доказан мономолекулярный механизм,
т. е. механизм, включающий предварительную стадию ионизации (но не дис-
социации на ионы) субстрата. Они описали два кинетических солевых эффек-
та. Для всех субстратов был обнаружен нормальный солевой эффект, когда
скорость линейно возрастала с увеличением концентрации соли часто вплоть
до довольно высоких значений, например до 0,1 М. Во многих случаях,
например для неофил- и пинаколилгалогенидов или аренсульфонатов, дру-
гих эффектов не наблюдалось. В некоторых случаях, например при сольво-
лизе 1-и-анизил-2-пропил-и-толуолсульфоната, проявлялся дополнительный
«специальный» солевой эффект, состоящий в резком ускорении реакции
при первоначальном внесении в систему 10-3 М соли; при достижении кон-
центрации соли ЗЛО-3 М наблюдаемая крутая кривая зависимости кон-
станты скорости от концентрации соли плавно переходила в нормальную
пологую почти линейную зависимость. На рис. 33—35 приведены такие кри-
вые с отнесением разных участков к разным эффектам для случая холестерил-
477
Глава VII
и-толуолсульфоната, изученного Топсомом [133], который вслед за Уинстей-
ном детально исследовал специальный солевой эффект. Уинстейн обнаружил,
Рис. 33. Скорости сольволиза холестерил-м-толуолсульфоната при 50,2 °C в уксусной
кислоте, содержащей соли.
1 — нормальный эффект; г — специальный эффект.
что специальный эффект перхлората
фоната подавлялся дополнительным
арилсульфоната лития и что эта соль
может уменьшить скорость реакции
лития при сольволизе алкиларенсуль-
введением соли с общим ионом —
в отсутствие каких-либо других солей
до величины, меньшей, чем скорость
Рис. 34. Левая часть кривой, приведенной
на рис. 33 для LiC104, в сильно
увеличенном масштабе, а именно:
ордината X 2 и абсцисса X 250.
Рис. 35. Скорости сольволиза и обмена
аниона для холестерпл-н-то-
луолсульфоната при 50,2 °C в
уксусной кислоте, содержащей
радиоактивную литиевую соль
п-толуолсульфокислоты.
в отсутствие любых солей. Он предположил, что субстрат диссоциирует
на ионы по стадиям, образуя вначале «внутреннюю ионную пару», обозна-
чаемую, R+X_, затем «разделенную растворителем» ионную пару, обозна-
чаемую R + ||X~, в которой между ионами имеется мономолекулярный слой
растворителя, и, наконец, диссоциированные ионы. Любая из этих дискрет-
ных частиц может реагировать с растворителем.
(1) (2) (3)
RX ; * R+X- R+ЦХ- < * R+ + X-
(-1) (-2) (-3)
I I I
Продукты Продукты Продукты
412
Замещение в алифатическом ряду
Нормальный солевой эффект обусловлен влиянием соли на стадию иониза-
ции (1). Специальный солевой эффект был описан как «очищающий» эффект,
удаляющий анион из разделенной растворителем ионной пары и, таким
образом, подавляющий стадию (—2). Соль с общим ионом не способна выз-
вать такого эффекта. Однако соль с общим ионом может промотировать ста-
дию (—3) и, таким образом, ингибировать сольволиз, проходящий через
стадию диссоциации на ионы.
В работе Топсома [133] дано альтернативное объяснение. Автор исследовал
сольволиз холестерилхлорида и холестерил-и-толуолсульфоната, для кото-
рых наблюдался специальный солевой эффект, а также сольволиз бензгид-
рилгалогенидов и некоторых производных, в случае которых этот эффект
не был обнаружен. Изучались соли четырех типов: соли без общего иона,
которые на дают стабильных продуктов, отличных от продуктов сольволи-
за, соли без общего иона, которые образуют такие продукты, соли с общим
ионом и соли с лиат-ионом.
Форма специального эффекта солей, которые, как, например, перхлорат
лития, не дают продуктов, отличающихся от продуктов сольволиза, для
случая холестерил-и-толуолсульфоната приведена на рис. 34. В узком
интервале концентраций, где проявляется специальный солевой эффект,
полученные данные описываются уравнением
1 . Ь
кв— ко а' [Соль]
где ks и к0 — константы скорости первого порядка для сольволиза в присут-
ствии и в отсутствие соли соответственно. Это уравнение выражает конкурен-
цию между солью и растворителем в реакции с неустойчивым промежуточ-
ным соединением. Если принять, что это промежуточное состояние представ-
ляет собой ионную пару в рамках определения Бьеррума, т. е. без разгра-
ничений на подчастицы, и если обозначить ионную пару холестерил-и-толуол-
сульфоната R+OTs~, то, учитывая форму специального солевого эффекта
можно написать следующую схему реакции: ’
ROTs -» R+OTs-
СНзСООН
--------> КОСОСНз
| СНз ООН
UX * Q
где Q — еще одно неустойчивое промежуточное соединение, природу кото-
рого предстоит рассмотреть.
Соли других классов в общем ведут себя аналогичным образом. Соли без
общего иона, например хлористый литий, которые дают устойчивый про-
дукт реакции, влияют на сольволиз холестерил-п-толуолсульфоната анало-
гично перхлорату лития, если при расчете степени превращения субстрата
учесть, что он превращается не только в ацетат, но также и в хлорид. Соли
с общим ионом, например и-толуолсульфонат лития, проявляют такой же
эффект, если ввести изотопную метку в и-толуолсульфонатный ион соли
и суммировать скорость изотопного обмена со скоростью сольволиза
(рис. 35). Такая же форма специального солевого эффекта была получена
при использовании соли с лиат-ионом — ацетата лития.
Изучая специальные солевые эффекты солей, добавленных попарно, Топсом
смог определить относительные скорости их реакций с неустойчивым про-
413
Глава VII
межуточным соединением. Например, при сольволизе холестерил-га-толуол-
сульфоната был получен следующий ряд:
LiOCOCH3 70, LiBr 20, LiCl 10. LiC104 4. LiOTs 1
При сольволизе холестерилхлорида не только значения относительных
скоростей были другими, но даже и сама последовательность солей была
иной. Это показывает, что неустойчивым промежуточным соединением
является ионная пара, включающая карбониевый ион, а не свободный ион
холестериния. Эти данные в противоположность обсуждаемым ниже под-
тверждают образование (но не участие в последующих реакциях) промежуточ-
ного соединения, обозначенного на приведенной выше схеме конкурирую-
щих реакций символом Q.
Путем экстраполяции прямой, соответствующей нормальному солевому
эффекту (рис. 33), Топсом смог определить значения скоростей, которые
наблюдались бы при максимальном проявлении специального солевого
эффекта при отсутствии нормального солевого эффекта. В случае холестерил-
«-толуолсульфоната, для которого скорость сольволиза в отсутствие солей
составляет 13,2-10-5 с-1 при 50,2° С, полученные путем экстраполяции зна-
чения скоростей сольволиза в присутствии солей были значительно выше,
а именно:
LiC104 33,3, ЫОСОСНз 31,0, LiBr 28,5, LiCl 27,5, LiOTs 25,0
Ясно, что при такой экстраполяции определялась скорость суммарного
расхода субстрата, который превращался в результате реакции в несколько
конечных продуктов.
Тот факт, что для каждой соли полученные значения не совпадают, пока-
зывает, что максимальный специальный солевой эффект не может быть
объяснен подавлением обратной стадии, предполагаемой в схеме Уинстейна.
Кроме того, величина максимального ускорения не коррелируется со спо-
собностью солей конкурировать с растворителем в реакции сольволиза.
Это означает, что ускорение, по крайней мере частично, зависит от скорости
стадии, иной, чем стадия образования промежуточного соединения Q. Оче-
видно, эта стадия следует за стадией образования Q, т. е. соответствует пре-
вращению Q в конечный продукт. Таким образом, скорость превращения
субстрата в продукт сольволиза может контролироваться не только ско-
ростью его первоначальной ионизации.
Топсом обнаружил, что небольшие добавки нитрометана к уксусной кислоте
(растворитель) селективно подавляют специальные солевые эффекты, и,
таким образом, подтвердил, что последние возникают вследствие низкой
диэлектрической проницаемости уксусной кислоты.
Идея о том, что в растворителях, достаточно проницаемых для электроста-
тических сил, может осуществляться такой тип мономолекулярного
механизма, в котором общую скорость определяет не только стадия
ионизации, но и последующая стадия, была выдвинута несколько ранее
на совершенно иных основаниях для объяснения реакций замещения
в еще менее полярном растворителе — бензоле [134]. Топсом использовал
аналогичную схему для замещения в растворах уксусной кислоты, пред-
положив, что промежуточное соединение Q представляет собой ионный агре-
гат, названный квадруплетом, который претерпевает медленную геометри-
ческую реорганизацию, в результате чего образуются несколько конечных
продуктов. В следующей схеме:
ш
Замещение в алифатическом ряду
Рис. 36. Зависимость свободной энер-
гии от координаты реакции
согласно теории специаль-
ного солевого эффекта в ре-
акциях 8л1-сольволиза в
уксусной кислоте; случай
соли с общим ионом.
все стадии могут влиять на скорость, за исключением стадий (2) и (2')г
которые обычно контролируются диффузией и практически протекают мгно-
венно, так как в указанных растворителях электростатические силы дейст-
вуют на дальние расстояния. В тех случаях, когда наблюдается специальный
солевой эффект, на схеме потенциальных энергий для случая общего иона
(рис. 36) пики А, В и В' довольно близки по высоте, поэтому, даже если
они не равны, препятствия, создаваемые
ими на пути реакции, сравнимы по вели-
чине. Если пик В и поэтому В' много мень-
ше Л, то будет иметь место механизм SN1
без заметного специального солевого эф-
фекта. Если пик В' и поэтому В значи-
тельно больше А, то будет наблюдаться ме-
ханизм Sn2 (С+), аналогичный описанному
в разд. 3,в для случая растворителя нит-
рометана. Проявление специального со-
левого эффекта, таким образом, указы-
вает на механизм, промежуточный между
SN1 и Sn2(C+).
Вероятно, энергия активации реорганиза-
ции (3) первоначально возникшего квад-
руплета в конфигурацию, обеспечиваю-
щую образование второго продукта, обу-
словлена электростатической работой по-
тери контакта между противоположно
заряженными ионами, которая достаточно
велика, и поэтому обеспечивает существо
з. Солевые эффекты при мономолекулярном замещении в бензоле. В этом
растворителе сольволиз не может происходить, а осуществляется замеще-
ние растворенными нуклеофилами. В этом случае снова ключевым моментом
является большая проницаемость бензола (диэлектрическая проницаемость
2,25) действию электростатических сил. Влияние закона действия масс
в химической кинетике определяется статистикой независимых друг от друга
двух видов квадруполей.
противоположно заряженных частиц, т. е. частиц, которые практически
все время находятся вне досягаемости действия сил каждой другой части-
цы. В случае высоко полярных частиц, растворенных в бензоле, это условие
выполняется редко. Обычно взаимодействующие частицы не являются неза-
115
Глава VII
ВИСИЛ1ЫМИ, и это необходимо учитывать при изучении кинетики реакций
в бензоле.
Два одновалентных противоположно заряженных иона притягиваются друг
к другу в бензоле с энергией, равной средней кинетической энергии каждого
на расстоянии 500 А (50 нм). На языке теории столкновений их диаметры
столкновения будут поэтому иметь порядок 500 А (50 нм). При наиболее
разбавленных из обычно употребляемых концентраций растворов наиболь-
шее разделение будет составлять лишь 50 А (5 нм). Ясно, что при этих раз-
бавлениях система ионов будет иметь уже «столкнутую» (collided) струк-
туру, так] как любое разделение ионов приведет к повышению энергии.
БолыпинствоТионов должно соединяться в пары или другие агрегаты с более
Рис. 37. Зависимость электропроводности растворов солей в бензоле от концентрации
соли и теоретические кривые для простых ионов и ионных тройников в равно-
весии с большим избытком ионных пар.
коротким радиусом действия силового поля. Однако даже ионные пары име-
ют, если по аналогии так можно назвать, «диаметр столкновения», сравни-
мый с расстоянием между соседними частицами, поэтому даже ионные пары
могут переходить в «столкнутое» состояние.
Природа частиц, присутствующих в растворах солей в бензоле, может быть
установлена измерением электроповодности растворов [136]. Логарифми-
ческий график зависимости электропроводности от концентрации тетра-
н-бутиламмониевых солей приведен на рис. 37. Характерна форма кривых.
При концентрациях от меньших чем 10~5 М до 10~2 М соли находятся глав-
ным образом в виде ионных пар. При концентрации 10~5 М несколько мил-
лионных долей соли диссоциированы на простые ионы, которые переносят
большую часть тока: если бы весь ток был обусловлен движением ионов,
наклон графика был бы равен —1/2. При концентрации 10~4 М ионы при-
соединяются к парам, образуя ионные тройники, опять-таки в количестве
нескольких миллионных долей от общей концентрации соли; в этом случае
большая часть тока переносится ионными тройниками: если бы весь ток
был обусловлен движением ионных тройников, наклон графика был бы
равен д/2- (Прямые с наклоном +V2 приведены на графике.) Однако,
прежде чем это происходит, ионные тройники сами превращаются в неза-
ряженные ионные квадруполи, поэтому при концентрации около 10 ~3 М
наклон графика начинает уменьшаться. Наконец, при концентрациях около
416
Замещение в алифатическом ряду
10-2 М во все большем количестве образуются заряженные агрегаты более
высшего порядка, чем квадруполи. Небольшие количества простых ионов
и ионных тройников обусловливают ряд заметных кинетических эффектов.
Однако выше концентрации 10 ~2 М, когда преобладают высшие агрегаты,
трудно ожидать, что реакция с солями будет подчиняться простым кинети-
ческим закономерностям-
В указанных условиях могут проявиться два кинетических эффекта [134].
Наиболее интересный состоит в следующем. Когда субстрат RX ионизиро-
ван до ионной пары R+X~ в бензоле, содержащем реагирующую соль M+Y~
в умеренной концентрации, карбониевый ион ионной пары сразу же стал-
кивается с реагентом, так как солеобразный реагент в момент зарождения
карбониевого иона уже будет находиться в поле действия его сил в реак-
ционноспособной форме. Вероятность столкновения при этом равна едини-
це, и увеличение концентрации реагента не может ее повысить. Но если,
как в случае специального солевого эффекта в растворителе — уксусной
кислоте (разд. 6,ж), ионы, составляющие реагенты, после первоначального
контакта успевают передислоцироваться до того, как произошло образова-
ние ковалентной связи, то скорость реакции, будучи, как мы видели, неза-
висимой от концентрации реагентов, может зависеть от того, одинаковы
или нет анионы, входящие в состав обоих реагентов, поскольку для кине-
тики может иметь значение вторая стадия активации. В дополнение к это-
му, если имеется смесь реагирующих солей M+Y~ и M+Z_, скорость реак-
ции с каждой из них будет уменьшаться с уменьшением их мольной доли,
а общая скорость будет связана с изменением состава смеси растворенных
солей от чистого M+Y~ к чистому M+Z~ зависимостью, близкой к линейной
(однако скорость не будет зависеть от общей концентрации солей). Такое
сочетание независимости от концентрации реагента с зависимостью от его
состава никогда не наблюдалось в случае реакций в высоко полярных раство-
рителях. Это явление может наблюдаться только в «столкнутом» состоянии
и, вероятно, только тогда, когда реагенты имеют подходящие полярность
и пространственное строение.
Второе следствие, вытекающее из наличия в бензольных растворах солей
сил большого радиуса действия, состоит в широком проявлении в этой
среде электростатического катализа, не имеющего определенного кинети-
ческого порядка. Окружающие полярное переходное состояние полярные
частицы будут оказывать кинетический эффект, зависящий от их числа
и пространственного расположения, причем эффект проявляют и частицы,
относительно далеко расположенные от места локализации переходного
состояния, так как этот эффект не является специфическим свойством близ-
лежащих частиц. Можно подсчитать, что такой катализ окружающими
ионными парами будет проявляться уже при концентрации 10~2 М и возрас-
тать при увеличении концентрации реагентов. Однако количественной
теории катализа окружающими ионными парами в настоящее время не
существует.
Оба указанных положения можно проиллюстрировать на примере реакции
нуклеофильного замещения хлора в трифенилметилхлориде в бензоле под
действием меченного по хлору хлорида тетра-и-бутиламмония или азида
тетра-м-бутиламмония [136]. При концентрации соли ниже 10-8 М реакция
имеет первый порядок по субстрату и нулевой — по соли. Азид тетра-и-бутил-
аммония растворим примерно до концентрации 10~2 М; при приближении
к этой концентрации наблюдается электростатический катализ, и наблюдае-
мый кинетический порядок постепенно возрастает (рис. 38). Поразительно,
что скорости реакции при нулевом порядке по соли различны для двух исследо-
27-0772
417
Глава VII
ванных солей', эти данные для 30 °C приведены ниже:
(C6H5)3CC1 + (C4H9)4N+C1- (C6H5)3CC1 + (C4H9)4N+C1- (*! =0,50-10-5 С"1)
(C6H5)3CC1+(C4H9)4N+N3 (C6H5)3CN7 + (C4H9)4N+C1- (/с1 = 2,50-10-5 С"1)
Скорость реакции со смесями обеих реагирующих солей при нулевом порядке
по их суммарной концентрации изменяется при изменении состава смесей.
Так, для смеси состава 50 : 50 в указанных условиях константа скорости
обмена хлора равна 0,25-10“5 с-1, т. е. составляет половину от указанной
выше для чистого хлорида, а константа скорости реакции с азидом равна
1,25-IO-5 с-1, т. е. опять-таки составляет половину от приведенной выше;
Рис. 38. Зависимость скорости реакции
трифенилметилхлорида с ази-
дом тетра-и.-бутиламмония от
концентрации соли в бензоле
при 30 °C (J. Chem. Soc., 1957,
1232).
Короткие прямые линии — теорети-
ческие кривые для реакций, кине-
тика которых отвечает нулевому,
первому и второму порядку по соли.
Рис. 39. Схематическое изображение-
зависимости свободной энер-
гии от координаты реакции
согласно теории 8^1-реакций
замещения солями в растворе-
бензола.
при этом общая константа скорости превращения субстрата равна 1,50 х
X IO-5 с-г Увеличение концентрации обеих солей вдвое не изменяет ни ско-
рости реакции, ни состава реакционной смеси. Добавление других солей,
например перхлората тетра-н-бутиламмония, который не образует устойчи-
вого продукта реакции, не имеет никакого кинетического эффекта.
Предложенная интерпретация этих эффектов [134] в общем аналогична
интерпретации специальных солевых эффектов в уксусной кислоте (разд. 6.ж).
В этих реакциях типа SN1 определяющая скорость ионизация RX при-
водит к ионной паре R+X“, которая мгновенно связывается с ближайшей
ионной парой соли, образуя квадруполь R+X~M+Y~. Образование квадру-
поля не требует энергии активации, для разных 1-1-зарядпых солей оно
примерно одинаково экзотермично. Однако первоначально образовавшийся
квадруполь должен претерпевать реорганизацию в R+Y~M+X~, имеющую
энергию активации. Реорганизация необходима для того, чтобы располо-
жение ионов в квадруполе стало удобным для образования ковалентной свя-
зи, т. е. продукта реакции
F.X R+X- R+X-M+Y-
RY дД R+Y- R+Y-M+X-
418
Замещение в алифатическом ряду
Второй барьер активации, обусловленный реорганизацией квадруполя,
по энергии может лежать немного выше или немного ниже барьера иониза-
ции, поэтому разные соли при нулевом кинетическом порядке реагируют
с разной скоростью (рис. 39) *. Если энергия реорганизации очень велика,
то будет наблюдаться реакция второго порядка, происходящая по механизму
SN2(C+). Если энергия реорганизации значительно ниже, то будет наблю-
даться S гД-реакция с нулевым кинетическим порядком, как в полярных
растворителях. Таким образом, концепция второго барьера активации пред-
полагает механизм, промежуточный между SnI и Sn2(C+). Как и ранее,
предполагается, что энергия активации реорганизации квадруполя обуслов-
лена электростатической работой, необходимой для разъединения противо-
положно заряженных ионов. Грубый расчет по закону Кулона для случая
обмена хлора дает разность в величинах энергий активации двух стадий,
равную 4 ккал/моль (16,75-Ю3 Дж/моль). Можно думать, что эта величина
завышена.
Несколько иные закономерности наблюдаются в случае полярных, но не ион-
ных замещающих агентов, например в случае спиртов. Для них была полу-
чена плавная логарифмическая зависимость скорости от концентрации,
подобная приведенной на рис. 38, однако имеются некоторые сомнения
насчет предельных наклонов кривой по ее краям**. Эти реакции ускоряются
солью с необщим ионом, не приводящей к образованию устойчивого про-
дукта,— перхлоратом тетра-н-бутиламмония. Соль с общим ионом — хлорид
тетра-«-бутиламмония — подавляет этот эффект, а в отсутствие перхлората
уменьшает скорость алкоголиза до величины, меньшей, чем соответствую-
щая скорость в отсутствие солей. Эти явления качественно напоминают
специальный солевой эффект при сольволизе в уксусной кислоте, хотя
и несколько отличаются по форме проявления. Тем не менее возможно, что
они должны рассматриваться аналогичным образом.
7. Пространственная ориентация при нуклеофильном
замещении
а. Проблема вальденовского обращения [140].{В 1895 г. Вальден [141]
закончил изучение ряда приведенных ниже превращений. Исходное опти-
чески активное вещество при этих процессах превращается тем или иным
путем в один из двух оптических энантиомеров.
(— [-Аспарагиновая кислота
_______________________________I______________________________
HNO2 |_________________________NOBr
( —[-Яблочная кислота (— [-Бромянтарная'кислота
* Свейн [137] не смог описать эти данные, применяя теорию стационарного состояния.
То же возражение выдвинул и Уипстейн [138]. Однако с самого начала пояснялось (1957 г.),
что предполагается «столкнутое» состояние, в котором системы не являются независи-
мыми, и поэтому нельзя ожидать применимости теории стационарного состояния.
** См. [139]. Авторы последней из приведенных в ссылке [139] работы показали, что
в присутствии пиридина и других третичных аминов при реакции с метанолом кинети-
ческий порядок по метаполу при его низких концентрациях равен единице. Учитывая,
что по спектральным наблюдениям карбониевый ион исчезает при добавлении пиридина
(разд. 3,в), эти авторы предположили, что метанол реагирует с четвертичной аммониевой
солью. С другой стороны, нет четких доказательств, что в отсутствие третичных аминов
порядок по метанолу меньше единицы, поскольку степень разбавления растворов была
очень близка к предельно возможной для проведения измерений.
27* 419
Глава VII
СНзОН |
(-J-Метиловый эфир
яблочной кислоты
РВг5 |
(+ )-Метиловый эфир
бромянтарной кислоты
| сн3он
(— )-Метиловый эфир
бромянтарной кислоты
Затем на приведенном ниже примере с использованием различных простых
процессов замещения Вальден получил аналогичные результаты. Он дока-
зал возможность (см. схему) превращения оптически активного вещества
в его энантиомер в результате двух процессов замещения при асимметри-
ческом атоме углерода.
(— )-Яблочная кислота
| hno2
(— )-Аспарагиновая
кислота
|noci
Ag2O
Н2О
РС16
^кон
(+ )-Хлорянтарная кислота
Ag2O, нао
кон
(— )-Хлорянтарная кислота <
РС15
(—)-Яблочная кислота
По крайней мере в одном пункте в первой из этих схем и в двух — во второй
во время замещения происходит превращение асимметрического атома вместе
с тремя связанными с ним группами в его энантиомерную конфигурацию.
Подобное превращение называется валъденовским, или оптическим, обраще-
нием. Не менее чем в двух пунктах второй из приведенных схем при асим-
метрическом центре может происходить замещение, во время которого
сохраняется исходная конфигурация. Таким образом, замещение не обяза-
тельно сопровождается инверсией. В действительности имеется два типа
замещения: один, при котором происходит обращение конфигурации, и вто-
рой, протекающий без обращения конфигурации. Цикл превращений, вклю-
чающий энантиомеры, называются валъденовским циклом.
Приведенные данные не позволяют установить, какая из реакций замеще-
ния приводит к инверсии. Общеизвестно, что одинаковый знак вращения
для различных соединений недостаточен для утверждения о сходстве конфи-
гурации. Даже оптически активные кислоты и их анионы, а также амины
и их катионы могут иногда обладать противоположным вращением. Еще
меньше дают эти результаты для понимания условий, приводящих к инвер-
сии,'причин инверсии и того, как она протекает.
С самого начала проблема казалась весьма загадочной. В 1907 г. Фишер
считал открытие Вальдена наиболее изумительным исследованием в области
оптической активности со времени фундаментальных работ Пастера. Новые
примеры, полученные в последующие десятилетия, и в особенности в первом
десятилетии нашего века, в работах самого Фишера, а также Мак-Кензи,
не сняли покрывала загадочности с этих процессов. Схемы обращения,
подобные предложенной Вальденом для веществ группы яблочной кислоты,
были разработаны и для соединений группы молочной кислоты, группы
миндальной кислоты и для ряда других групп. В своей книге «Оптическое
обращение», опубликованной в 1919 г., Вальден привел более 20 подобных
схем, большинство которых могут служить доказательством существова-
ния обращения Вальдена.
420
Замещение в алифатическом ряду
Эта проблема (выяснение вопроса о том, в каких случаях, как и почему про-
исходит вальденовское обращение) оставалась невыясненной в течение 40 лет.
Теперь легко видеть, что эту проблему нельзя было разрешить до открытия
в 1933—1935 гг. двух мехнизмов нуклеофильного замещения в алифати-
ческом ряду.
В идеальном случае общую проблему можно разбить на два требующих после-
довательного разрешения вопроса. Первый заключается в строгом сопостав-
лении конфигурации исходных и конечных продуктов замещения при асим-
метрическом центре. Затем, выявив случаи вальденовского обращения,
необходимо установить условия, которые вызывают обращение, а следова-
тельно, выяснить механизм процесса; эти вопросы соответственно рассматри-
ваются раздельно в дальнейшем изложении. Однако необходимо отметить,
что оба эти вопроса неизбежно в известной мере переплетаются, ибо точ-
ность методов определения конфигурации продуктов реакции связана с уста-
новлением механизма реакции. Поэтому сначала можно ограничиться при-
менением надежного, но не вполне строгого метода изучения первого вопро-
са, с тем чтобы впоследствии путем уточнений сделать этот метод и достаточно
строгим.
б. Соотношение конфигураций. Первые попытки определения конфигура-
ций исходного и конечного продуктов замещения основывались на теориях
механизма замещения. Вальден считал замену галогена в галогенсодержа-
щем соединении оксигруппой при действии щелочи более простой реакцией,
чем гидроксилирование при помощи влажной окиси серебра, которая, как
он предполагал, образует молекулярное соединение с органическим веще-
ством. В соответствии с этим Вальден полагал, что замещение щелочами
протекает с сохранением конфигурации, в то время как замещение с помощью
окиси серебра сопровождается инверсией [142]. Фишер [143] заметил, что
некоторые галогенсодержащие кислоты дают при щелочном гидролизе один
и тот же изомер, если исходить как из самой галогенокислоты, так и из ее
эфира или амида, в то время как при омылении окисью серебра образуется
один энантиомер в том случае, когда карбоксильная группа свободна, и дру-
гой — когда она защищена. На основании этого Фишер пришел к выводу,
что при щелочном гидролизе конфигурация сохраняется, а при действии окиси
серебра на свободную галогенокислоту происходит инверсия. Сейчас извест-
но, что все эти заключения ошибочны.
Для отнесения многих относительных конфигураций Франкланд [144] осно-
вывался на наблюдаемых закономерностях во влиянии специфических реа-
гентов на изменение или сохранение знака вращения при переходе от исход-
ного к конечному продукту реакции. Он предполагал, что если при приме-
нении данного реагента почти всегда образуется определенный продукт
с сохранением или изменением знака вращения, то это характерно для дан-
ного реагента. В настоящее время установлено, что большинство предполо-
жений Франкланда является правильным.
Основная идея Клоу [145] заключалась в том, что с изменением физических
условий (среды, концентрации, температуры и длины волны света) соединения
одинаковой конфигурации претерпевают одни и те же изменения в отно-
шении величины вращения. Этот метод, применяемый без достаточной осмо-
трительности, приводил к многим ошибочным заключениям. Однако Фрейден-
берг [146], проведший достаточно обширное исследование границ приложи-
мости этого метода, использовал его со значительным успехом. Предполо-
жения Фрейденберга, которые впоследствии подвергались проверке, почти
все оказались правильными.
421
Глава VII
Насколько известно в настоящее время, все эти методы (или по крайней мере
преобладающее большинство их) основаны на принципах, надежность кото-
рых не может быть доказана. Следовательно, ни один из обобщающих выво-
дов не является в достаточной степени достоверным. По-иному обстоит
дело с методом Кеньона и Филлипса [147], который может быть пояснен
на примере 2-н-октильных соединений. Из четырех приведенных ниже реак-
ций одна (если не три) должна сопровождаться инверсией, что следует
из общего результата. Единственной реакцией, которая может протекать
таким образом, является реакция вытеснения сульфонатной группы ацетат-
ионом, протекающая в этиловом спирте (реакция 1), поскольку именно она —
единственная реакция из четырех приведенных, которая вызывает изме-
нение связей асимметрического атома углерода
RSOC1
( + )-С8Н17ОН------> ( + )-C8H17OSOR (—)-С8Н17ОН
Окисление (СНзССДаО
СНзСОО"
(реакция 1) ( + )-C8H17OSO2R ---> ( — )-С8Н17ОСОСИ3
В настоящее время предполагается, что атака сульфонатной группы хлорид-
ионом в этиловом спирте (реакция 2) должна протекать по аналогичному
механизму и поэтому, подобно атаке ацетат-ионом, должна вызывать обра-
щение конфигурации. Установлено, что алкилхлорид, полученный действием
хлорид-иона на алкилсульфонат, обладает обратным знаком вращения по сра-
внению с исходным спиртом, из которого был получен сульфонат
С1-
(реакция 2) ( + )-C8Hi7OSO2R -> ( —)-С8Н17С1
Следовательно, если 2-н-октиловый спирт и 2-н-октилхлорид обладают оди-
наковым знаком вращения, то они имеют одну и ту же конфигурацию.
Наиболее слабое место подобного довода в действительности не так уж сла-
бо: недоказанным является лишь предположение о том, что поведение ацетат-
ионов аналогично поведению хлорид-ионов. Если сделать дальнейший шаг
и предположить, что существует аналогия в поведении октилсульфоната
и октилхлорида, то можно прийти к правильному выводу (разд. 7,в) о вза-
имодействии радиоактивного иодид-иона и 2-и-октилиодида (реакция 3).
Действительно, путем сравнения скорости приобретения радиоактивности
алкилгалогенидом со скоростью уменьшения его оптической активности можно
показать (разд. 7,г), что в этом примере каждый молекулярный акт заме-
щения приводит к обращению конфигурации
(реакция 3) ( + )-С8Н171 —> ( — )-С8Н171
Как будет показано в следующих разделах, этот метод может считаться
вполне строгим, если привлечь легко доступные кинетические данные, дока-
зывающие, что все рассмотренные аналогичные реакции представляют собой
бимолекулярные процессы.
До того как стали известны абсолютные конфигурации, пространственное
строение энантиомеров устанавливалось отнесением к произвольно выбран-
ному Фишером глицериновому альдегиду. Энантиомерам глицерина, имею-
щим (+)- и (—)-вращение, приписывались конфигурации D (правая) и ь (ле-
вая) соответственно. Эти конфигурации приведены ниже в виде фишеровских
422
Замещение в алифатическом ряду
проекций на плоскость (группы, расположенные слева и справа, находятся
перед, а группы, изображенные сверху и снизу,— за плоскостью проекции)
ОНО ОНО
Н-------ОН
СН2ОН
( + ) “D - (Я)
НО------н
СН2ОН
(-)-l-GS)
В общем случае необходимо привести в строгое соответствие расположение
четырех групп у какого-нибудь асимметрического центра и расположение
групп у асимметрического центра глицеринового альдегида; тогда этому
асимметрическому центру можно приписать, безотносительно к знаку
•оптического вращения вещества, конфигурацию D или ь в зависимости
от того, какой конфигурации глицеринового альдегида соответствует
этот центр.
В настоящее время конфигурации большинства хорошо известных органи-
ческих соединений соотнесены с конфигурацией глицеринового альдегида
и, следовательно, друг с другом. Это было сделано путем установления гене-
тической связи между рядом последовательных процессов, в которых про-
исходило изменение групп или без нарушения связей у асимметрического
атома, или путем замены этих связей на другие при помощи реакций с извест-
ным механизмом и известной стереохимией (разд. 7,г) *.
Для того чтобы знать пространственную направленность реакций, необхо-
димо установить лишь относительную конфигурацию исходных и конечных
веществ. Впоследствии мы часто будем употреблять символы d и l, обозна-
чающие относительную конфигурацию, для описания пространственного
течения реакций замещения.
В действительности мы обычно знаем и абсолютные конфигурации реагентов
и продуктов. Предположение Фишера о конфигурации глицеринового альде-
гида было произвольным и имело равные шансы быть верным или неверным.
Однако оно оказалось правильным. Это было установлено Бийво [1491 при
помощи рентгеноструктурного анализа в 1951 г. Поэтому, зная цепь конфи-
гурационных корреляций, восходящую к глицериновому альдегиду, мы,
таким образом, знаем и абсолютные конфигурации.
Часто более удобно установить абсолютные, а не относительные конфигура-
ции. Трудность в обобщении относительных конфигураций состоит в том,
чтобы правильно отразить соответствие между четырьмя группами, связан-
ными с каким-либо асимметрическим центром, и четырьмя группами глице-
ринового альдегида. Абсолютные конфигурации устанавливаются с помощью
правила последовательности [150]. В случае асимметрического центра Cabcd
группы abed располагаются в последовательности, соответствующей умень-
шению атомного номера первого атома группы или, если они одинаковы,
в соответствии с уменьшением атомного номера второго атома группы и т. д.;
* Первым важным шагом, сделанным в 1950 г., явилось соотнесение конфигураций гли-
церинового альдегида, который до этого уже успешно использовался в качестве вещества
сравнения для установления конфигурации углеводородов и их производных с конфи-
гурацией серина, использовавшегося как независимое вещество сравнения для отнесения
конфигураций аминокислот и их производных. Такая связь между двумя стандартами
дозволила скоррелировать конфигурации больших групп соединений друг с другом
[148]. С тех пор в единую корреляционную систему были сведены большинство терпенов,
стероиды, некоторые алкалоиды и множество других соединений.
423
Глава VII
далее нужно смотреть на тетраэдр со стороны, противоположной группе dT
и если при этом последовательность а —> Ь -> с соответствует движению
по часовой стрелке, конфигурация описывается как R (rectus, правая), а если
против часовой, то как 5 (sinister, левая). В глицериновом альдегиде после-
довательность групп такова: ОН, СНО, СН2ОН, Н, причем атом О группы
СНО считается за два, так как он связан двойной связью. Обозначение абсо-
лютных конфигураций будет зависеть от последовательности этих групп,
рассмотренной по правилам, данным выше. Такая система применима ко всем
органическим молекулам, обладающим хиральностью (сходство с правой
или левой рукой) и, следовательно, потенциальной оптической активностью
независимо от того, имеют они асимметрический атом или молекула асим-
метрична в целом. Хиральность соответствует малой симметрии, а именно
наличию вращательных осей симметрии без ограничения их мультиплет-
ности или числа. Структуры, обладающие или не обладающие хиральностью,
называются хиралъными или ахиральными соответственно.
в. Механизм замещения и пространственная ориентация. В большин-
стве ранних теорий пространственного замещения, особенно в теории Фишера
[151], Вернера [152] и Пфейфера [153], было постулировано предварительное
образование продукта присоединения, в котором входящая группа присоеди-
няется к асимметрическому атому либо со стороны замещаемой группы,
либо с противоположной стороны. Теории Кадамера [154] и Мейзенгеймера
[155] развивают представления этого же типа.
Представление о том, что присоединение входящей и отщепление уходящей
группы может протекать синхронно, было впервые выражено Ле-Белем
[15б]; Льюис [157] первый объяснил, каким образом вальденовское обраще-
ние может быть осуществлено. Эта концепция была развита Олсоном и Лон-
гом [158], которые пришли к выводу, что каждое замещение с изменением
только одной связи сопровождается инверсией, а также Меером и Поляньи
[159], которые усматривали различия в замещении при действии анионов
и катионов, причем допускали, что атака первых сопровождается обраще-
нием, в то время как при атаке катионов конфигурация сохраняется.
Другое возможное объяснение механизма вальденовского обращения, заклю-
чающееся в предварительной диссоциации, было предложено Лоури [160],
который предполагал, что замещение, протекающее с промежуточным обра-
зованием карбониевого иона, может происходить с преобладающим сохране-
нием конфигурации. С другой стороны, Кеньон и Филлипс [161] предпола-
гали, что такое замещение должно приводить к обращению конфигурации.
Обе интерпретации основывались на предположении, что замещение закан-
чивается прежде, чем карбониевый ион успевает принять плоскую конфи-
гурацию.
Когда стало ясно, что двойственность механизма является общей для нуклео-
фильного замещения в алифатическом ряду, Хьюз и автор книги [162] начали
развивать теорию синхронного присоединения и диссоциации и теорию пред-
варительной диссоциации, относя первую к бимолекулярной, а вторую —
к мономолекулярной формам замещения. На основании физических данных
указанные авторы предположили, что бимолекулярное замещение всегда
сопровождается обращением, в то время как моиомолекулярное замещение
может в зависимости от обстоятельств, которые следует рассмотреть деталь-
но, включать инверсию, рацемизацию или сохранение конфигурации.
г. Пространственная ориентация при S^-замещении. Предполагалось,
что для бимолекулярного механизма замещения (Sn2) обращение конфигура-
ции является правилом, поскольку переходное состояние I, приводящее
к инверсии, обладает гораздо меньшей внутренней энергией, чем состояние!!,
424
Замещение в алифатическом ряду
соответствующее сохранению пространственной конфигурации. Это обуслов-
лено тем, что в переходном состоянии I связь X- • • -Y, удерживающая
входящую и уходящую группы, должна характеризоваться почти плоской
поверхностью нулевой электронной плотности (плоской, если Х= Y). В этой
плоскости могут лежать три CR-связи; такое строение уменьшает положи-
тельную обменную энергию между изменяющимися и сохраняющимися
связями * и между электронными парами, становящимися связывающей
и несвязывающей. Состояние II не допускает такого устойчивого строения
(D
(II) >
Выявить переходное состояние I можно проще всего с помощью метода мече-
ных атомов, предложенного Хьюзом с сотрудниками для замещения гало-
гена в реакции между галогенид-ионами и молекулами алкилгалогенидов.
Ранее было известно, что оптически активный алкилгалогенид, в котором
единственным центром асимметрии является атом углерода, связанный с гало-
геном, может обычно подвергаться рацемизации при обработке в растворе
галогенидом щелочного металла, содержащим тот же галоген, что и в алкил-
галогениде. На основании этого факта можно сделать следующий вывод:
если рацемизация происходит в результате замещения галогена в алкилгало-
гениде ионом галогенида, то по крайней мере некоторые из индивидуальных
молекулярных актов замещения включают обращение. Однако вывод можно
распространить гораздо дальше, если использовать алкилгалогенид с радио-
активным атомом галогена; в этом случае можно не только устано-
вить факт замещения, но и исследовать кинетику процесса, а также сравнить
скорости замещения со скоростью уменьшения оптической активности.
Эти положения были проверены [163] на трех примерах: в случае рацемиза-
ции 2-н-октилиодида иодистым натрием, 1-фенилэтилбромида бромистым
литием, а также а-бромпропионовой кислоты бромистым литием. В каждом
случае в качестве растворителя использовали сухой ацетон (звездочками отме-
чены радиоактивные атомы).
Г + С6Н13СН1СН3 —> С6Н13СН1СН3 + 1-
Вг- + С6Н5СНВгСН3 —> С6Н5СНВгСН3 + Вг-
Вг"+НО2ССНВгСН3 —> НО2ССнЙгСН3-|-Вг-
В каждом примере было показано, что, во-первых, кинетически замещение
соответствует реакции второго порядка и поэтому проходит по бимолеку-
лярному механизму; во-вторых, скорость уменьшения оптической активности
в два раза превышает скорость изотопного обмена, т. е. каждый индивиду-
альный акт бимолекулярного замещения изменял конфигурацию, проходя,
таким образом, через переходное состояние I.
Все эти примеры относятся к реакциям с типом заряда 1 (отрицательно заря-
женный реагент, нейтральный субстрат). Это обстоятельство позволяет вы-
двинуть альтернативное предположение, а именно что полученный результат
зависит не от обменных, а от электростатических сил, поскольку диполь
* Предполагается, что входящие электроны не могут занять высшие орбитали углерода
до ухода уходящих электронов. Возможно, что при нуклеофильном замещении у более
тяжелых элементов дело обстоит по-иному.
425
Глава VII
углерод — галоген в алкилгалогениде может направить замещающий агент
анионного типа к положительному концу диполя, т. е. к стороне углерод-
ного атома, более удаленной от атома галогена. Однако можно по-
казать, что это объяснение не правильно; так как при переходе к реак-
циям с типом заряда 3 (отрицательно заряженный реагент, положи-
тельно заряженный субстрат) направление диполя связи между
углеродом и уходящей группой может измениться, но замещение
все же происходит с обращением конфигурации. Когда формулировался
основной принцип обращения конфигурации при бимолекулярном замеще-
нии, в литературе имелось указание против электростатического объясне-
ния. Оно относилось к стереохимии замещения в ряду ментана и было рас-
смотрено в первом издании этой книги (стр. 304). Однако имеющиеся данные
не позволяли сделать окончательного вывода вследствие наличия в моле-
куле несколько асимметрических центров и в еще большей степени из-за
того, что не было кинетических доказательств механизма реакции. С тех
пор Хьюзом и сотрудниками были получены более надежные данные.
Один из примеров, подтверждающих общность пространственной направлен-
ности 5к2-реакции с типом заряда 3 и 1, приведен ниже [164]:
( + )-С6Н5СН(СН3)С1
Sjj2 (тип 1)
sh-
Ng
Sn2 (тип 1)
( —)-CeH5CH(CH3)SH
-)-CeH5CH(CH3)S(CH3)2
H2/Pt
CeH5CH(CH3)N3--------> (- )-CeH5CH(CH3)NH2
Ng H2/Pt
4 9-,;—47» CeH5CH(CH3)N3 ----------> ( + )-C6H6CH(CH3)NH2
DjqZ (ТИП 0)
1-Фенилэтилхлорид с помощью 5к2-реакций с типом заряда 1 был превра-
щен в тиол, который был прометилирован без затрагивания асимметрического
центра до сульфониевого иона, и в азид, который был восстановлен опять-
таки без затрагивания асимметрического центра до амина. С помощью изо-
топного метода было установлено, что 8к2-реакции с типом заряда 1 про-
исходят с обращением конфигурации; таким образом, указанные выше суль-
фониевый ион и амии имеют сходную конфигурацию. Затем сульфониевый
ион был превращен в азид по механизму Sn2 (тип заряда 3), азид при восста-
новлении дал другой энантиомерный амин. Это показывает, что замещение
с типом заряда 3 происходит с обращением конфигурации, несмотря на проти-
водействующие электростатические силы.
В другом примере коррелируется пространственная направленность SN2-
замещения с типом заряда 2 (нейтральный реагент, нейтральный субстрат)
и 8к2-замещения с типом заряда 1. Нейтральным реагентом в работе Гоф-
мана и Хьюза [165] была тиомочевина. Корреляционная схема имеет следую-
щий вид:
( + )-С6Н5СН(СН3)Вг —
Sn2 (тип 1)
---—-----> (-)-CeH5CH(CH3)SH
SC(NH2)2 +
4-77—47* (-)-c6h5ch(Ch3)sC(NH2)2
1-Фепилэтилбромид был превращен с помощью Б^-процесса с типом заряда
1 в тиол инвертированной конфигурации и с помощью 8к2-реакциис типом
426
Замещение в алифатическом ряду
заряда 2 — в тиурониевый ион, который при гидролизе без затрагивания
асимметрического центра дает оптически идентичный тиол. Поэтому заме-
щение с типом заряда 2 должно происходить с обращением конфигу-
рации.
Последний из примеров Гофмана и Хьюза [166] касается реакций с типом
заряда 4 (нейтральный реагент, положительно заряженный субстрат).
Схема превращений приведена ниже:
+ SN 2 (тип 4) +
(—)-C6H5CH(CH3)S(CH3)2 ( + )-C6H5CH(CH3)SC(NH2)2
&C(iNtl2)2
|н2О
+ CH3OTs
( + )-C6H5CH(CH3)S(CH3)2 <------( + )-C6H5CH(CH3)SH
1-Фенилэтилдиметилсульфониевый ион был превращен с помощью реакции
5к2-замещения с типом заряда 4 в тиурониевый ион, который при гидролизе
до тиола и последующем метилировании, проходящих без затрагивания
реакционного центра, дает снова 1-фенилэтилдиметилсульфониевый ион,
имеющий угол вращения такой же по величине, но противоположный по зна-
ку. Следовательно, реакция замещения с типом заряда 4 происходит с обра-
щением конфигурации.
Хотя было проведено множество исследований по стереохимии З^-реакций,
примеры, приведенные в этом разделе, служат основой для формулирования
Зк2-правила. Это правило было опубликовано в 1937 г. и базировалось
на принципе Паули. Оно явилось первым практически ценным правилом,
которое связало пространственную направленность химических реакций
с их механизмом [162]. Любопытно, что указанное правило было принято
практически безоговорочно и широко и корректно применялось для кор-
реляции конфигураций задолго до того, как в 1964 г. оно было окончательно
подтверждено.
Sn2-Правило. Замещение по механизму S N2 происходит с обращением конфи-
гурации независимо от каких-либо деталей строения.
Это правило является настолько простым и определенным и в такой мере
лишено ограничений, что представляет собой наиболее общий и надежный
метод определения конфигурации при замещении у отдельного асимметри-
ческого центра одной группы другой. При использовании этого правила основ-
ная экспериментальная задача всегда состоит в том, чтобы доказать, что
нуклеофильное замещение в действительности протекает по бимолекуляр-
ному механизму. Многие конфигурационные отношения были установлены
по этому методу [167].
Подтверждением Зк2-правила служат наблюдения Бартлета, что замещение
галогена у мостикового углерода не может протекать по механизму SN2.
Очевидно, причина данного явления заключается в том, что мостиковая
структура препятствует образованию переходного состояния I. В качестве
примеров приведем 1-хлорапокамфан и 1-бромтриптицен [167, 168]
СН2-СС1—СН2
С(СН3)2
СН2-СН — СН2
д. Типы пространственной ориентации при SnI-замещении. Простран-
ственная направленность мономолекулярного нуклеофильного замещения
менее однозначна. При Зк2-замещении, как уже было показано, всегда наблю-
дается инверсия конфигурации. При S ^-замещении результаты можно
427
Глава VII
подразделить на две группы. В случае так называемого «нормального» заме-
щения, которое осуществляется при отсутствии в молекуле некоторых групп
(они будут рассмотрены ниже), получаются смеси инвертированного и раце-
мизованного продуктов в любых отношениях, вплоть до предельных. Эта
отношение зависит от природы субстрата и среды, особенно от стабильности
карбониевого иона, обусловленной строением субстрата, и от способности
растворителя к захвату иона карбония. При «аномальном» замещении, осу-
ществляющемся при наличии в молекуле некоторых определенных групп,
получаются смеси продуктов с сохраненной конфигурацией и рацемизован-
ных продуктов в любых отношениях, вплоть до предельных. Это отношение
опять-таки зависит от строения субстрата и природы растворителя.
Итак, пространственная направленность реакций нуклеофильного замеще-
ния может быть трех типов: один из них соответствует механизму SN2, а два
других —механизму SN1, причем применимость каждого из двух послед-
них зависит от особенностей строения субстрата. Такое отношение между
стереохимией и механизмом было выведено в 1937 г. на основе эксперимен-
тальных данных, собранных в табл. 113. Это было ответом на стоявший
около 40 лет вопрос о причинах вальденовского обращения.
Первые два случая, приведенные в табл. ИЗ, относятся к вторичным алкил-
галогенидам [169]. Они показывают, что при 5к2-механизме наблюдается
практически полная инверсия; когда механизм точно переходит или когда
можно полагать, что переходит к нормальному механизму SnI или к меха-
низму SN1 с катализом серебром, наблюдается небольшая рацемизация.
Следующие два случая относятся к вторичным аралкилгалогенидам, в кото-
рых образующийся карбониевый ион может сопрягаться с арильной груп-
пой [170]. В этих случаях в 5к2-реакциях наблюдается полная инверсия,
а в реакциях, для которых предполагается SNl-механизм или каталити-
ческий механизм SN1, происходит инверсия со значительной рацемизацией.
По определению механизма SN1 в переходном состоянии ионизации суб-
страта не происходит образования ковалентной связи с замещающим аген-
том. вВ этом состоянии уходящий анион отходит лишь на приблизительно
0,5 А (5-Ю-2 нм). Если бы замещающий агент образовывал ковалентную
связь, когда анион удалился на расстояние, немного большее, чем указан-
ное, например на 1,0 А (0,1 нм), или на расстояние, необходимое для образо-
вания тесной ионной пары, замещение оставалось бы мономолекулярный,
но происходило бы с инверсией конфигурации, так как одна сторона карбо-
ниевого иона была бы экранирована уходящим анионом. Однако должны
существовать ионы с большими степенями разделения, когда противоион
все еще будет экранировать одну из сторон карбониевого иона в заметно
большей степени; при этом хотя и не исключается замещение с сохранением
конфигурации, все же оно будет менее вероятным, чем замещение с инверси-
ей. В этом случае будет наблюдаться инверсия с рацемизацией. Только в
том случае, когда карбониевый ион настолько стабилен, а реакционная
способность среды настолько низка, что уходящий анион имеет время
продиффундировать от карбониевого иона до атаки замещающего
агента, можно ожидать полной рацемизации *. Ясно, что степень
* Стереоспецифичность, обусловленную экранированием 8^1-замещения, время от вре-
мени объясняют как замещение в ионных парах, включающих ион карбонпя. На самом
деле она относится и ко многим другим случаям. Однако, учитывая любое точное опре-
деление ионной пары, было бы слишком большим ограничением объяснить условия полной
пли частичной стереоселективности через экранирование. Например, можно ожидать,
что в воде существенное экранирование будет распространяться далеко за пределы рас-
стояния Бьеррума.
428
Таблица ll'i
Отношение эффекта вращения к механизм}' замещения
Галогенозамеще1гное Вступаю- щая группа Замещающий агент Среда Механизм Конфигурация Оптиче- ски я чистота, %
С6н13 )СНС1 сн3/ ос2н5 С2Н5О- C2H5OH Sn2 Инверсия -100
СвН 1з )СНВг сн/ он он ос2н5 ос2н5 ОС2Н5 он ОС2Н5 он ос2н5 он- н2о С2Н5О- С2Н5ОН С2Н5О- Ag1, AgBr Ag+, AgBr Ag+, AgBr, Ag2O Ag+, AgBr, Ag2O 60%-ный води. C2H3OH 60%-ный водн. С2Н5ОН 60 %-ный води. С2Н5ОН 60%-пый водн. С2Н5ОП С2Н5ОН н2о С2Н6ОН Н2О С2Н5ОН Sn2 SN1 Sn2 SN1 SN2 Ag+-SN1 Ag+-SN1 Ag+-SN1 Ag+-SN1 Инверсия Рацемизация, инверсия Инверсия Рацемизация, инверсия Инверсия Рацемизация, инверсия То же » » » » -100 66 -100 74 -100 72 94 74 94
С6Н5 >СНС1 сн/ он он он ОСНз осн3 ос2н5 ос2н5 он H2O H2O H2O CH8O- CH3OH C2H5O- C2H6OH Ag+, AgCl, Ag2O Н2О 60 %-ный водн. (СН3)2СО 80%-ный води. (СН3)2СО СН3ОН СНдОН С2Н5ОН С2Н5ОН н2о SN1 SN1 SN1 Sn2 SN1 SN2 SN1 Ag+-SN1 » » » » » » Инверсия Рацемизация, инверсия Инверсия Рацемизация, инверсия То же 17 5 2 Высокая Низкая Высокая Низкая 3 ... 29
СвН5. >СНВг сн/ он H2O н2о SN1 Рацемизация, инверсия Низкая
П родолжение табл. 113
Га л о гено замещенное Вступаю- щая группа Замещающий агент Среда Механизм Конфигурация Оптиче- ская чистота, %
ноос.
)СНВг он Н2О, H2SO4 H2O Sn2 Инверсия -100
сн3/
CH3OOC. ОСНз CH3O- CH3OH Sn2 Инверсия -100
>СНВг ОСНз CH3OH CH3OH Sn2 » -100
СН3Х ОСН3 Ag\ AgBr CH3OH Ag+-SN1 Рацемизация, инверсия 89
hoocch2nhco4
)СНВг ОН Ag+, AgBr, Ag2CO3 H2O Ag+-SN1 Рацемизация, инверсия 73
СНз'
он OH- I/O Sn2 Инверсия -100
он H2O, OH- H2O sni Сохранение -100
-оос. ОСНз CH3O- CH3OH Sn2 Инверсия -100
\CHBr ОСНз CH3OH, CH3O- CH3OH sni Сохранение -100
сн/ ОН Ag+, AgBr H2O Ag+-SN1 Рацемизация, сохранение 87
ОН Ag+, AgBr, Ag2O H2O Ag+-SN1 То же 28 ... 58
он AgF, AgBr, Ag2CO3 I/O Ag+-SN1 » » 36
Замещение в алифатическом ряду
рацемизации будет возрастать с увеличением стабильности карбониевого
иона и с уменьшением склонности среды к захвату этого иона, т. е. при уве-
личении времени жизни карбониевого иона в данной среде.
Если, как, например, при переходе от вторичных алкилгалогенидов к вто-
ричным аралкилгалогенидам, время жизни карбониевого иона увеличивается
вследствие мезомерии, количество рацемизованного продукта будет воз-
растать (табл. ИЗ). Такое же явление наблюдалось [171] при переходе
от чисто алифатических вторичных алкилгалогенидов к алифатическим
третичным алкилгалогенидам, например к метилэтилизогексилкарбинил-
хлориду. Как и следовало ожидать, в сходных условиях продукт
из третичного соединения рацемизован в гораздо большей степени.
Аномальное положение возникает в том случае, когда имеется заместитель,
который спустя некоторое время после того, как отделяющаяся группа уже
выйдет из промежуточного состояния гетеролиза, но прежде, чем эта группа
отойдет на достаточно большое расстояние, образует слабую связь (обычно
преимущественно электростатического характера) с карбониевым центром
со стороны, отдаленной от удаляющейся группы. В дальнейшем эта сторона
будет защищена от атаки замещающего агента до тех пор, пока отщепляю-
щаяся группа не удалится настолько, что будет возможна атака с ее собствен-
ной стороны; эта атака освобождает защищающую группу и) способствует
замещению с сохранением конфигурации. Если защищающая группа экра-
нирует слабо, то преобладающее сохранение конфигурации может сопро-
вождаться в большей или менявшей степени рацемизацией. При этом необ-
ходимо (хотя в гл. X эти ограничения будут сняты), чтобы защищающие
группы обладали ненасыщенностью, т. е. имели бы неподеленные электроны
или кратные связи (р или л), которые могли бы легко взаимодействовать
с карбониевым ионным центром. Необходимо также, чтобы защищающая
группа не была настолько нуклеофильна и была настолько благоприятно
расположена, что ее участие в процессе могло бы происходить до того, как
наступит переходное состояние гетеролиза. Таким образом, замещение
с сохранением конфигурации зависит от ряда условий, в том числе от поляр-
ности, а также от стереохимической вероятности реакции.
С исторической и других точек зрения наиболее важной группой, способ-
ствующей сохранению конфигурации, является а-карбоксилат-ион а-СО”,
например а-бромпропионат-ион ~ООССН(СН3)Вг. В данном случае сохра-
нение конфигурации при замещении частично можно объяснить электроноот-
талкивающими свойствами этой группы; подобно а-алкильному заместите-
лю, а-карбоксилат-ион способствует 8к1-замещению (разд. 2,6), тем самым
обеспечивая одно из условий, необходимых для сохранения конфигурации.
Далее, группа СОО~ является нуклеофильной и имеет необходимые неподе-
ленные электроны. Однако расположение а-карбоксилат-иона позволяет
ему образовывать лишь слабую связь: заместитель с возникающим карбоние-
вым ионом образует довольно длинную, но слабую, по существу электроста-
тическую, связь, тем самым сохраняя конфигурацию до тех пор, пока заме-
щающий агент не займет положения, которое занимала отделяющаяся груп-
па. Это проиллюстрировано в последних четырех примерах (особенно в пос-
леднем) в табл. ИЗ. а-Бромпропионовая кислота, ее эфир и амид качественно
ведут себя подобно другим вторичным алкилгалогенидам, однако а-бром-
пропионат-ион — совсем по-ионому [172]. В щелочном растворе а-бромпро-
пионат-ион может гидролизоваться в соответствии с механизмом, который
по кинетическим данным является бимолекулярным SN2; образующаяся
молочная кислота, как и следовало ожидать, имеет обращенную конфигура-
цию. Однако а-бромпропионат-ион может гидролизоваться даже в разбавлен-
431
Глава VII
ном щелочном растворе по механизму, который, по кинетическим данным,
является мономолекулярный SN1; в этом случае образование молочной
кислоты происходит с преобладающим сохранением конфигурации. Схема-
тично этот процесс можно представить следующим образом:
О- _ о-
I + н2о I
О = ССН(СН3)Вг I ;-СНСНз + Вг- -> О = ССН(СН3)ОН
о=с/
В случае фенилбромацетат-иона ~ООССН(С6Н5)Вг образующийся карбоние-
вый ион дополнительно стабилизуется за счет мезомерии; поэтому даже
в сильнощелочном растворе преобладает мономолекулярный механизм.
Однако преобладающее сохранение конфигурации, обусловленное данным
механизмом, сопровождается значительной рацемизацией [173].
Важное значение слабой связи с участием карбоксилатного иона можно
наблюдать, поместив эту группу в £5-, у- и б-положения по отношению к отще-
пляющему галогену. При этом обычно происходит внутримолекулярное
8к2-замещение. Реакция протекает быстрее, чем следовало бы ожидать для
SN 1-замещения галогена. В некоторых случаях реакция протекает даже
быстрее, чем катализируемое ионами серебра 8м1-замещение, и поэтому
рассматриваемая реакция не чувствительна к ионам серебра. В результате
реакции обычно образуется |3-, у- или б-лактон с обращенной конфигура-
цией. Подобные лактоны могут быть гидролизованы различными путями,
но особенно легко в щелочном растворе. При этом происходит разрыв связи
СО — О не в первоначальном месте замещения, где по необходимости конфи-
гурация лактона сохраняется. Таким образом, и исходные галогенкислоты
превращаются в щелочном растворе в оксикислоты с полным обращением
конфигурации, что не случилось бы при мономолекулярном замещении,
при котором после гетеролиза конфигурация сохраняется.
Легко понять, почему удалось обнаружить вальденовское обращение
и почему почти все ранние работы по этому вопросу были выполнены в области
реакций а-замещенных карбоновых кислот. Трудность обнаружения валь-
деновского обращения была связана с его широкой распространенностью *.
Прежде чем «открыть» вальденовское обращение, необходимо было устано-
вить гораздо менее общие явления замещения, протекающие с сохранением
конфигурации, и тем самым доказать существование стереохимически проти-
воположных процессов. Ионизованная карбоксильная группа в «-положе-
нии не только способствует мономолекулярному замещению, но также
обладает рядом свойств, позволяющим ей образовать эффективную, но доста-
точно слабую связь. Поэтому реакции а-галогенкислот дали широкие воз-
можности для наблюдения вальденовского обращения.
Рассмотренные принципы можно суммировать в виде второго правила про-
странственной ориентации при нуклеофильном замещении. Это правило
менее однозначно, чем 8к2-правило, и в приведенной ниже форме применимо
только к замещению у ациклического атома углерода.
3 ^-Правило. Замещение по механизму SN1 происходит с обращением конфи-
гурации, сопровождающейся рацемизацией в любых соотношениях вплоть
до крайних случаев, если не существует слабого внутримолекулярного взаимо-
действия карбониевого центра с нуклеофильным заместителем-, в последнем
случае наблюдается сохранение конфигурации с рацемизацией, соотношение
которых может быть любым, включая оба крайних случая.
* Если бы весь мир был голубым, мы бы не знали, что такое голубизна.
432
Замещение в алифатическом ряду
Как будет показано, вывод о механизме реакции, основанный лишь на стерео-
химических данных без достаточных кинетических доказательств (например,
строгие кинетические доказательства часто трудно получить для реакций
сольволиза), не является достаточным. Если даже мы и можем принять
на веру, что а) происходит нуклеофильное замещение и б) его механизм соот-
ветствует одному из уже встречавшихся ранее (хотя имеются и другие меха-
низмы), мы все же должны остерегаться делать окончательные выводы.
Наблюдаемое полное обращение конфигурации само по себе не дает воз-
можности сделать выбора между механизмами SN1 и SN2. Наблюдаемая
полная или частичная рацемизация не позволяет сделать выбора в пользу
Sк1-механизма до тех пор, пока не будет показано, что указанная стерео-
химия обусловлена единственной необратимой стадией замещения и отсут-
ствуют конкурирующие, предшествующие или последующие процессы.
Наблюдаемое полное или частичное сохранение конфигурации не позволяет
остановиться на 8к1-механизме по тем же самым причинам. В общем стерео-
химия без поддержки кинетическими данными мало что дает для выяснения
механизма: начинать с нее — это значит начинать не с того конца.
е. Обращение конфигурации с рацемизацией: дальнейшие детали.
В этом разделе мы рассмотрим два крайних случая. Сначала обсудим реакции
сольволиза типа SN1, в которых образующийся карбониевый ион настолько
стабилен, а растворитель настолько плохо с ним реагирует, что если замеще-
ние происходит только у асимметрического центра, продукт сольволиза
будет полностью рацемическим. В некоторых таких случаях, хотя и не во
всех, скорость потери оптической активности при сольволитическом заме-
щении превышает скорость появления рацемического продукта сольволиза.
Это, конечно, означает, что субстрат, выделенный до завершения сольво-
лиза, будет частично рацемизован.
Увеличение скорости рацемизации может быть объяснено любым из несколь-
ких способов, большинство которых мы сейчас рассмотрим. Например,
образующиеся при сольволизе анионы могут вступать в бимолекулярную
реакцию с еще не сольволизованным субстратом, в результате чего обра-
зуется некоторое количество инвертированных молекул субстрата, которому
соответствует в два раза большее количество рацемата. Этот случай можно
распознать кинетически: если сольволиз — реакция первого порядка, раце-
мизация будет не первого порядка вследствие постепенного накопления
рацемизующих анионов. Диссоциированные ионы, образовавшиеся на пер-
вой стадии мономолекулярного замещения, могут также рекомбинироваться
с карбониевыми ионами, образуя таким образом рацемический субстрат.
Этот случай также можно распознать кинетически: если рацемизация имеет
первый порядок, то сольволиз не будет первого порядка, так как образова-
ние анионов постепенно будет увеличивать скорость рекомбинации ионов
и, следовательно, скорость сольволиза будет со временем все более умень-
шаться ниже значений, соответствующих первому порядку (разд. 6,а).
Если образуется рацемический продукт, сольволиз имеет первый порядок и ра-
цемизация происходит по первому порядку; большая скорость рацемизации
может быть, кроме того, обусловлена перегруппировкой, сопровождающей
сольволиз, при которой промежуточный карбониевый ион проходит через
симметричную конфигурацию. Этот случай можно обнаружить при анализе
состава продуктов реакции. Мы рассмотрим его в последних главах книги.
Увеличение скорости рацемизации первого порядка, сопровождающей
сольволиз первого порядка, в результате которого образуется рацемичес-
кий, но не перегруппированный продукт, наблюдалось Уинстейном и сотр.
[174] в 1960 г. Они установили, например, что п-хлорбензгидрилхлорид
28-0772
433
Глава VII
в уксусной кислоте, содержащей ацетат лития, при 25 °C рацемизует-
ся в 30...70 раз быстрее, чем превращается в п-хлорбензгидрилацетат.
Тот же субстрат в 80%-ном водном растворе ацетона рацемизуется в 5 раз
быстрее, чем превращается в бензгидрол.
Более высокая по сравнению со скоростью сольволиза скорость рацемиза-
ции, естественно, обусловлена обратимостью контролирующей скорость
стадии ионизации, которая, как показывает кинетический первый порядок,
успевает пройти несколько раз в прямом и обратном направлении, прежде
чем противоположно заряженные ионы станут кинетически независимыми.
. Н2О
RR'CHCl RR'CH+C1- RR'CH+ + C1" ---------> RR'CHOH + HCI
Не предполагая равновесия между небольшим количеством дискретных
подчастиц, объединяемых названием «ионные пары», можно сказать в общем
виде, что после прохождения барьера активации ионизации вследствие
диффузии противоположно заряженных ионов друг от друга образование
каждой подчастицы будет связано с определенной степенью вероятности,
которую можно измерить, лишь интегрируя по всем степеням разделения.
Существует определенная асимптотически увеличивающаяся с увеличе-
нием степени разделения вероятность того, что с карбониевым ионом будет
взаимодействовать растворитель. Существует также определенная вероят-
ность, что ионы будут рекомбинировать; эта вероятность уменьшается
до низкого значения при увеличении степени разделения. Кроме того, после
достижения какой-то небольшой критической степени разделения появится
определенная растущая при увеличении разделения вероятность, что карбо-
ниевый ион перед рекомбинацией с анионом сделает примерно пол-оборота
вокруг оси, перпендикулярной линии, соединяющей ионы. Избыточная
скорость рацемизации (первого порядка) по сравнению с скоростью сольво-
лиза (первого порядка), ведущего к образованию рацемата, обусловлена
произведением последних двух вероятностей, определенных интегрирова-
нием по всем степеням разделения.
Гёринг [175] предложил метод, позволяющий изучить поведение противоио-
нов при степенях разделения, меньших, чем степени разделения, дающие
стереохимически ощутимые результаты, обусловленные вращением карбо-
ниевых ионов. В этом методе используются эфиры карбоновых кислот, один
из атомов кислорода которых имеет изотопную метку. При некотором раз-
делении противоположно заряженных ионов заряд аниона распределяется
между двумя атомами кислорода и после рекомбинации ионной пары обра-
зуется эфир, в котором изотопная метка одинаковым образом распределена
между обоими атомами кислорода. Так, при гидролизе бензгидрил-и-нитро-
бензоата, содержащего 18О в карбонильной группе, в 90%-ном водном
растворе ацетона при 118,6 °C скорость усреднения изотопной метки в три
раза превышает скорость гидролиза; это означает, что из общего числа
молекул, ионизующихся до относительно малой степени разделения,
25% гидролизуются, а 75% возвращаются в исходное соединение. При
гидролизе в той же среде при 99,6 °C оптически активного и-хлорбензгид-
рил-и-нитробензоата, меченного 18О по карбонильной группе, из числа моле-
кул, ионизовавшихся до расстояния, на котором обнаруживается усредне-
ние изотопной метки по атомам кислорода, 28% гидролизовались,
а из остальных, вернувшихся в исходное состояние, 56% сохранили перво-
начальную конфигурацию и 16% после пол-оборота карбониевого иона
дали исходное соединение с обращенной конфигурацией.
434
Замещение в алифатическом ряду
Фава и сотр. [176] изучили некоторые реакции различных замещенных
бензгидрилтиоцианатов в ацетонитриле, показав, что каждая проходит
через стадию ионизации. Были изучены обмен с меченым изоцианатным
ионом 38SCN~, проходящий через ионную диссоциацию, изомеризация
в изотиоцианаты, для которой требуется только небольшое разделение
ионных пар, и рацемизация, сопровождающая все эти процессы. Для
изомеризации необходимо, чтобы анион повернулся так, чтобы один его
конец занял положение другого, а для рацемизации — чтобы перед реком-
бинацией катион повернулся на пол-оборота. В изученных примерах пары
ионов, достаточно разделенные, чтобы рекомбинировать без вращения или
с вращением аниона, или без вращения или с вращением карбониевого иона,
имеют больше шансов рекомбинировать любым из указанных путей, чем
полностью диссоциировать и, таким образом, вступить в обмен. В случае
оптически активного n-хлорбензгидрилтиоцианата в ацетонитриле при 70 °C
ионная пара реагирует в нескольких направлениях, приблизительное коли-
чественное соотношение между которыми приведено ниже:
46
(-)-RSCN <-----; 64
( + )-RSCN
( — )-RSCN
R+(SCN)-
(-)-RNCS
( + )-RNCS
(±)-R++ SCN"
Установлено, что тиоцианат и изотиоцианат с одинаковыми знаками враще-
ния имеют подобные конфигурации; таким образом, мы видим, что перевора-
чивание аниона и вращение катиона может происходить на одной и той же
стадии разделения противоионов.
Теперь рассмотрим другой крайний случай 8м1-замещения, протекающего
с частичным обращением и рацемизацией, когда карбониевый ион является
настолько короткоживущим, что трудно определить, образуется ли он
вообще, т. е. трудно отличить механизм SN1 с участием карбониевого иона,
почти все время экранированного противоионом, от механизма SN2. Грюн-
вальд, Хеллер и Клейн [177] нашли, что 1-фенилэтиловый спирт в 18О-воде,
содержащей 0,017 н. хлорной кислоты, при 30 °C претерпевает рацемизацию,
скорость которой в 1,22 раза больше скорости обмена кислорода с раствори-
телем
Ha18O-f-RR'CH —OHJ —* RR'CH^i8OH2+4-H2O
Этот случай не является предельным из возможных. Из отношения ско-
ростей следует, что 61 % актов замещения приводит к обращению конфигу-
рации, а 39% — к сохранению, или, иначе говоря, 22% актов замещения
приводят к инверсии конфигурации, а 78% — к рацемизации. Не прихо-
дится сомневаться, что в этом примере основным механизмом замещения
является SN1, причем субстрат берется в виде сопряженной кислоты спирта.
Рассмотрим теперь результаты тех же опытов с чисто алифатическим вторич-
ным спиртом. Бантон, Конашевич и Ллевиллин [178] обнаружили, что втор-
бутиловый спирт в 18О-воде, содержащей различные малые концентрации
хлорной кислоты, при 100,8 °C рацемизуется в два раза быстрее, чем происхо-
дит кислородный обмен с растворителем. Здесь каждый акт замещения
приводит к обращению конфигурации, и нельзя сказать, можно ли вообще
исключить S N1-механизм, объяснив это отсутствием фенильной группы,
стабилизирующей карбониевый ион, или результаты можно объяснить
в рамках 8м1-механизма с учетом уменьшения времени жизни карбониевого
28* 435
Глава VII
иона, в течение которого он почти все время остается экранированным. Эту
проблему можно решить, используя конкурирующие реакции.
Рассмотрим влияние изменения среды. Вайнер и Снин [179] * отметили, что
при гидролизе 2-октилметансульфоната при 65 °C в 50 %-ном водном растворе
диоксана 94% образующегося октанола-2 имеют обращенную конфигура-
цию, а 6% сохраняют конфигурацию. Авторы также наблюдали, что при
гидролизе этого эфира в чистой воде образуется полностью инвертирован-
ный октанол~2. Механизм гидролиза в чистой воде логически вывести нельзя,
однако можно представить такой случай, когда и в 50 %-ном водном растворе
диоксана и в чистой воде осуществляется механизм SN1, а различие в стерео-
химии обусловлено превосходящим сродством чистой воды к карбониевому
иону, которая реагирует с ним еще до того, как противоион отойдет на доста-
точное расстояние.
Эта интерпретация может объяснить другое резкое стереохимическое разли-
чие, обнаруженное в указанной работе, а именно то, что при гидролизе
2-октил-и-бромбензолсульфоната при 65 °C в 75%-ном водном диоксане
88% образующегося октанола-2 имели обращенную конфигурацию
и 12 % октанола-2 — сохраненную конфигурацию. В присутствии же доста-
точного количества азида натрия, когда образуется 30% октанола-2 и 70%
2-октилазида, оба эти соединения были полностью инвертированными.
Можно опять-таки предположить, что увеличение реакционной способности
среды по отношению к карбониевому иону, достигаемое в данном случае
добавлением азид-иона, не дает аниону времени отойти от карбониевого
иона на расстояние, достаточное для того, чтобы последний по-настоящему
дезэкранировался **.
До сих пор в этом разделе мы рассматривали в основном сольволиз. Некото-
рые из рассмотренных явлений наблюдались и в несольволитических реак-
циях замещения, для которых в общем легче провести кинетические иссле-
дования, т. е. легче установить механизм. Покер и сотр. [112] исследовали
некоторые реакции замещения 1-фенилэтилхлорида в нитрометане и, исполь-
зуя кинетические и стереохимические данные, установили, что можно раз-
личить три механизма, а именно мономолекулярный, бимолекулярный
и кислотно-катализируемый мономолекулярный.
При 99,8 °C константа скорости первого порядка для рацемизации 1-фенил-
этилхлорида в нитрометане (1,2 ПО-5 с-1) была в 4,2 раза больше скорости
обмена хлорид-иона (SN1), а в отсутствие источника хлорид-ионов кон-
станта скорости элиминирования с образованием стирола (первого поряд-
ка, Е1) составляла 0,29 •10-бс-1. Избыточная скорость рацемизации, как
было указано ранее, обусловлена рекомбинацией той части ионных пар,
* Объяснение авторов принять нельзя. Оно состоит в том, что сохранение конфигурации
обусловлено двумя последовательными стадиями типа Sjsj2, каждая из которых приводит
к обращению конфигурации, причем первоначально в роли замещающего агента высту-
пает молекула диоксана, которая затем вновь отщепляется. Азид-ион вступает в реакцию
на второй стадии, и, таким образом, двухстадийный процесс образования октанола-?
не сопровождается сохранением конфигурации.
.__„ н2о
/О ________ _____________> R — ОН
R —X ---=-----> о/ ^О+ —R —
X----/ NJ
----> R —N3
Если бы было так, то 2-октилазид, образующийся вместо 12% октанола-2, имеющего
сохраненную конфигурацию, имел бы также сохраненную конфигурацию. В действитель-
ности же образуется полностью инвертированный 2-октилазид.
** См. сноску на стр. 428.
436
Замещение в алифатическом ряду
в которых успевает произойти вращение карбониевого иона с образованием
инвертированного продукта.
При использовании в качестве источника замещающих хлорид-ионов
(C2H5)4N38C1 замещение частично происходило по реакции второго порядка,
скорость которой равна 6,1 -IO-3 [RC1] [звС1_] моль-л-1-с-1. Одновременно
с этой реакцией происходила рацемизация (второго порядка), скорость кото-
рой была равна 12,4-IO-3 [RC1] [зеС1_] моль-л-1-с-1. Соотношение этих
скоростей, равное 1,97, а также кинетика второго порядка позволяют пред-
положить, что в этом случае реакция происходит по механизму SN2. Н38С1
выступает, с одной стороны, в качестве источника замещающих хлорид-
ионов, а с другой стороны, как было указано выше, в качестве общекислот-
ного катализатора ионизации субстрата; обмен хлора имеет второй порядок,
и скорость его равна 1,52-10~3 [RC1] [НзвС1] моль-л-1-с-1. Параллельно
с этим обменом происходит рацемизация (второго порядка), скорость кото-
рой составляет 1,50 «Ю-3 [RC1] [Н38С1]. Отношение этих скоростей, равное
0,99, а также наблюдаемая кинетика второго порядка показывают, что
в этом случае реакция происходит по мономолекулярному механизму,
катализируемому молекулой кислоты (HC1-SN1); кроме того, молекула
НС1 удаляет ионизованный С1_ (связывая его в виде очень стабильного
комплексного аниона С1НС1-), в результате чего карбониевый ион пол-
ностью освобождается от экранирования противоионами и поэтому обра-
зуется полностью рацемизованный продукт замещения.
ж. Сохранение конфигурации и рацемизация; дальнейшие детали.
В разд. 7,в указывалось, что открытие вальденовского обращения было
обусловлено наблюдением над некоторыми относительно редко встречаю-
щимися процессами замещения, которые, как было показано позже, проис-
ходят с преимущественным сохранением конфигурации. Указанное стерео-
химическое свойство было обусловлено двумя причинами: во-первых, соеди-
нения, которые обладали этими свойствами, реагировали по механизму SN1,
и, во-вторых, они содержали в подходящем положении заместитель, обла-
дающий свойством закреплять конфигурацию промежуточного карбоние-
вого иона. Закрепление конфигурации асимметрического центра приводит
к сохранению конфигурации при замещении. Сохранение может быть
не полным, если строение карбониевого иона делает возможным сопряже-
ние, которое стабилизирует карбониевый ион и переводит его в состояние,
близкое к плоскому.
Результаты всех работ по вальденовскому обращению в первые 42 года
после его открытия (с 1895 г. и до его объяснения в 1937 г.) зависели от нали-
чия или отсутствия лишь одного заместителя, способного образовать связь
с карбониевым ионом, обладающую свойством в процессе 8к1-замещения
сохранять энантиомерную конфигурацию при переходе от субстрата к про-
дукту реакции. Таким заместителем был а-карбоксилат-ион. В более широком
смысле этот заместитель содержал в у-положении атом с неподеленными
электронами. С 1942 г. Уинстейн провел большую работу по обобщению
влияния p-заместителей (у-атомов) на сохранение конфигурации при заме-
щении типа SN1. В настоящее время к таким заместителям относятся сле-
дующие: а-СО~, р-С1, p-Br, р-ОСОСН3, р-ОСН3, p-NHCOC6H5. Другие авто-
ры [180] показали, что к этому же классу принадлежат p-SR-группы.
Другие у-атомы с неподеленными электронами, например карбонильный
кислород при 8к1-замещении в а-галогенкарбонильных соединениях,
вероятно, влияют так же; однако в этих случаях отсутствуют исчерпываю-
щие экспериментальные доказательства. Можно обсудить несколько приме-
ров из работ Уинстейна.
437
Глава VII
Некоторые данные были получены при сольволизе органических галогени-
дов в уксусной кислоте при катализе солями серебра. Замещение, вероятно,
происходит по каталитическому механизму Ag+-SN1. Так, из эритро- и трео-
форм 2-бром-З-метоксибутана образуются соответственно эритро- и трео-
формы 2-ацетокси-З-метоксибутана [181]. Кроме того, хотя это и не было
установлено для указанного случая, на основании аналогичных исследова-
ний, которые будут приведены в следующем разделе, несомненно, что если
исходный /прео-изомер был оптически активным, скажем D-трео, то продукт
реакции представляет собой рацемат п.ь-щрео.
Эти данные показывают, что реакция должна проходить через ахиральную
конфигурацию, поэтому был сделан вывод, что для обеспечения симметрии
системы связи метоксильного атома кислорода с [5- и а-углеродными атома-
ми должны проходить через состояние эквивалентности, становясь затем
снова неэквивалентными при разрыве с любой стороны.
СН3 СН3
ОСН3
с'-----с
(D или L)
н н
(D,L)
СН3 СН,
ОСН3
с'
С
сн3сосг
н
Н
К подобным же результатам приводит исследование замещения CH3C6H4SO2O
на СН3СОО при сольволизе алкиловых эфиров и-толуолсульфокислоты
в уксусной кислоте без добавления других реагентов. Для этой реакции
Уинстейн, Мак-Касланд, Лукас и другие [182] установили, что в случае
группы р-СН3СОО или p-C6H6CONH происходит замещение a-CH3C6H4SO2O
на ОСОСНз с сохранением конфигурации. Так, щракс-1-ацетокси-2-и-толуол-
сульфонилоксициклогексан превращается в /пранс-1,2-диацетоксициклогек-
сан. Предполагается, что реакция протекает через стадию мезомерного ахи-
рального пятичленного гетероциклического катиона с атомом кислорода
в цикле, содержащего пары эквивалентных частично электростатических
связей, что представлено одной асимметричной ковалентной структурой:
сн 2-сн2-сн-сн2
СНзСОО
н
(D или L)
438
Замещение в алифатическом ряду
сн2-сн2-сн2-сн2
I
ОС(СН3) j -о
с-----------с
0^..(СН3)СО
н н
D,L
До 1946 г. все известные группы, удерживающие конфигурацию, содержали
неподеленные электроны, которые, как можно было думать, взаимодей-
ствуют с карбониевым ионом с частичным переносом заряда; затем, однако,
Шоппи [183] показал, что в подобной ситуации аналогичным образом
проявляют себя электроны двойной связи. Он установил, что холестерил-
хлорид и холестерин подвергаются многим реакциям замещения, например
ацетолизу хлорида с образованием холестерилацетата, с сохранением кон-
фигурации, в то время как их дигидропроизводные, холестанилхлорид
и холестанолпроизводные, обычно подвергаются замещению с обращением
конфигурации в месте замещения. Было сделано заключение, что л-электро-
ны 5,6-двойной связи способствуют сохранению конфигурации; при замеще-
нии в положении 3 промежуточный карбониевый ион, по-видимому, обла-
дает положительным зарядом, распределенным между положениями 3 и 6.
Уинстейн и Адамс [184] показали, что ацетолиз холестерил-п-толуолсуль-
фоната, приводящий к образованию холестерилацетата, также протекает
с сохранением конфигурации, причем скорость замещения в уксусной кислоте
не зависит от концентрации ацетат-ионов (независимо от солевых эффектов).
Достаточно высокая скорость реакции позволила предположить, что участие
двойной связи проявляется еще до того, как образовалось переходное состоя-
ние ионизации, стабильность которого поэтому будет выше вследствие
перераспределения заряда иона [184]. Далее Уинстейн и сотр. [185] пока-
зали, что л-электронная система фенильной группы также может способ-
ствовать сохранению конфигурации, например при сольволизе 1-фенил-
2-пропил-п-толуолсульфоната СвН5СН2СН(СН3)ОТз, который в муравьиной
кислоте происходит с преобладающим сохранением конфигураций.
з. Замещение ОН на Hal. Все реакции, механизм которых кинетически
можно было установить уже в 1937 г. и поэтому с помощью 8к2-правила
поляриметрически определить относительную конфигурацию исходных
439
Глава VII
и конечных веществ, относились к замещению галогена или оксиэфирных
групп на различные галогены, кислород-, серу- и азотсодержащие группы,
включая ОН и NH2. Найденные конфигурационные корреляции сразу
позволили выяснить пространственное направление некоторых обратных
процессов замещения, для которых трудно непосредственным кинетическим
исследованием выяснить механизм; в этих процессах происходило замещение
группы ОН или NH2 в субстрате. Так, некоторые работы 1937 г. [162] ставили
целью выяснить, насколько хорошо выведенные пространственные факторы
соответствуют SN2- и 8к1-правилам, чтобы таким образом с помощью стерео-
химических данных делать заключение о механизме реакций. Один из наи-
более четких результатов, полученных при изучении реакций замены ОН
на Hal, состоит в открытии того факта, что стереохимия некоторых из этих
реакций не согласуется ни с одним из этих двух правил, т. е. необходимо
допустить какой-то новый механизм. Было предположено, что эти процессы
осуществляются внутримолекулярным переходом через циклическое пере-
ходное состояние в комплексе, образованном субстратом и реагентом. Такой
механизм был назван внутримолекулярным нуклеофильным замещением
и обозначен символом SNi.
Примеры пространственной направленности реакций заменены ОН на Hal
приведены в табл. 114; они относятся к замещению спиртовой группы
на галоген при действии хлорангидридов или хлорокисей кислот фосфора
и серы или хлористого водорода. После того как исследование этого типа
реакций в основном было закончено, подобные результаты были получены
и для бромидов в аналогичных реакциях. В приведенной таблице указаны
только преобладающие стереохимические результаты; в большинстве слу-
чаев при этом происходит частичная рацемизация, но трудно определить,
Таблица 114
Пространственное течение процесса замещения ОН на С1
(Ру—пиридин; I—инверсия; R—сохранение)
Оксипроизводное РС13 РС13+Ру РОС1з РОС1з+Ру РС15 РС15-|-Ру SOCI2 SOCIa+Py HC1
2-н-Октиловый спирт I I I I I I I
1-Фенилэтиловый I I I I I I R I I
эфир Молочная кислота I I I I
и ее эфир Яблочная кислота I I
и ее эфир Миндальная кислота I I R I
и ее эфир а-Окси-а-фенилпро- I R
пионовая кислота
и ее эфир а-Окси-Р-фенилпро- I I
пионовая кислота
и ее эфир р-Окси-р-фепилпро- I I I R I I
пионовая кислота
и ее эфир
440
Замещение в алифатическом ряду
насколько она присуща самому процессу замещения, так как получающийся
алкилгалогенид всегда подвергается рацемизации под влиянием либо одно-
временно образующихся ионов галогена, либо образующегося или исполь-
зуемого галогеноводорода.
Эти результаты можно обобщить следующим образом: галогеноводород
и галогенпроизводные фосфора во всех случаях вызывают преобладающее
обращение конфигурации; тионилхлорид иногда, но не всегда, приводит
к таким же результатам. В этом случае сохранению конфигурации способ-
ствует наличие электронодонорных заместителей у центра замещения (ранее
сделанное утверждение, что для этого необходим а-фенильный заместитель,
относится к частному случаю *), а реагенты, подобные третичным ос-
нованиям, продуцируют хлорид-ионы и уменьшают степень сохранения
конфигурации **.
Предложенные механизмы [162], отражающие общую ситуацию, состоят
в следующем.
Было предположено, что первая ступень всегда состоит в образовании ком-
плекса сложного эфира с галогенидом, если можно включить в это понятие
и комплексы с галогеноводородом, содержащие водородную связь: ROPCl*,
ROPOC12, ROPCI2, ROSOC1, R0---HC1h т. д. Далее предполагалось, что
комплекс может либо ионизоваться с образованием галогенид-иона, либо
претерпевать внутримолекулярную перегруппировку (SNi). За ионизацией
галогена может следовать либо S ^-замещение ионом галогенида с обраще-
нием конфигурации, либо S ^-замещение обычно с преобладанием инверсии,
но с сохранением конфигурации в присутствии заместителей, способствую-
щих сохранению конфигурации. Перегруппировка может включать полное
расщепление на ионы С0-связи комплекса без разделения образующихся
ионов, которые могут взаимодействовать с переносом галогенид-иона
и сохранять при этом исходную конфигурацию. Все это учитывается при
рассмотрении реакции с тионилхлоридом (см. ниже).
Было предположено, что комплексы с галогеноводородом и галогенидами
фосфора обладают большей тенденцией к выделению галогенид-иона и мень-
шей тенденцией к гетеролизу СО-связи и перегруппировке, чем эфиры хлор-
сернистой кислоты. Пиридин и другие третичные амины могут способство-
вать отделению иона галогенида вследствие образования комплексного
пиридиниевого или какого-либо другого аммониевого галогенида; наличие
сильно электронодонорного заместителя, например фенила, у места замеще-
ния может способствовать СО-гетеролизу
и поэтому облегчать перегруппировку.
Упомянутые выше механизмы введения галогенида являются, очевидно,
более гипотетическими и менее определенными, чем хорошо установленные
механизмы SN2- и S ^-замещения галогена. Модифицированный механизм,
учитывающий дополнительные данные, был предложен Льюисом и Бузером
[187], однако некоторые данные ему противоречат. Поэтому в настоящее
время предлагаемый механизм рассматриваемой реакции лучше считать
грубым приближением к истине. Указанные авторы выделили вторичные
алкилхлорсульфиты в оптически активном состоянии и показали, что пере-
* Впервые на это указал Франкланд [144].
** Влияние пиридина и других третичных аминов впервые наблюдали Кеньон и Фил-
липс ([186] и несколько последующих статей).
441
Глава VII
ходное состояние процесса их разложения настолько полярно, насколько
это возможно, что, однако, не следует из общих представлений о механизме
реакции. Пространственная направленность реакции разложения сложным
образом зависела от природы растворителя.
\ А х
-)€/ XSO —--------------> \c-Cl+SO2 (SNi)
/ pi/ Сохранение /
конфигурации
-7COSO+4-CI----------> ci-cA +so2
/ Инверсия х.
I -> Cl-C^ +SO2
—C+-LSO,+ С1~ Обычная инверсия.
(Sn2)
(SN1)
~С —Cl-}-SO2
Сохранение конфигурации
в отдельных случаях
Механизм типа SNi для этих процессов замещения доказан экспериментами
Лукаса и Уинстейна, которые на отдельных примерах показали, что при
наличии ^-расположенного атома галогена замещение гидроксила галогеном
протекает с сохранением конфигурации [188]. Так, эритро- и щрео-формы
2-бромбутанола-З при обработке бромистым водородом или трехбромистым
фосфором образуют соответственно мезо- и рацемическую формы 2,3-дибром-
бутана. Кроме того, на примере реакции бромзамещенного спирта с бро-
мистым водородом было установлено, что оптически активный щрео-изомер
спирта дает полностью рацемизованный щрео-дибромид, тем самым подтвер-
ждая механизм, схематически изображенный ниже. Этот механизм идентичен
механизму мономолекулярного замещения галогена, протекающего в при-
сутствии p-групп с неподеленными электронами.
Шоппи [183] показал, что холестерин при действии пятихлористого фосфора
и тионилхлорида (с добавкой пиридина или без него) превращается в холе-
стерилхлорид с сохранением конфигурации в месте замещения. Механизм
этих реакций, несомненно, близок к механизму гидролиза хлорида, который,
как уже отмечалось, протекает с сохранением конфигурации. Это служит
дополнительным доказательством того, что двойная связь в холестерине
способствует сохранению конфигурации.
4:42
Замещение в алифатическом ряду
Механизм SNi характерен для других реакций. В качестве примера приведем
реакцию октанола-2 с фосгеном, взятую из работы Кеньона и Филлипса 1147].
Вторая стадия этой реакции протекает с сохранением конфигурации, оче-
видно, вследствие внутримолекулярного процесса
С8Н13-СН — СН3 —» СвН13—СН —СН3 —> СвН13—СН-СН3+СО2
I \ 1
ОН С1 О С1
и. Замещение NH2 на OR или Hal. Эти процессы дезаминирования,
которые проводят действием азотистой кислоты или других нитрозильных
соединений на первичные амины, составляют вторую группу реакций,
к которым с целью предсказания пространственной направленности обрат-
ных процессов замещения были применены сведения об относительных
конфигурациях, полученные при применении 8м2-правила. Учитывая про-
странственную направленность реакций дезаминирования и SN2- и S ^-пра-
вила, оказалось возможным сделать вывод о механизме этих реакций [189].
Главный вывод состоял в том, что все процессы дезаминирования предста-
вляют собой реакции 8к1-замещения в первоначально образующихся диазо-
ниевых ионах. Кинетика процесса замещения не поддается изучению, так
как по сравнению с образованием диазониевого иона эта стадия является
быстрой. Кинетика образования ионов диазония была выяснена в 1950 г.
методом конкурирующих реакций: как мы увидим в гл. VIII, разд. 1, в,
скорость образования нитрозамина, диазотирования и дезаминирования
определяется контролирующей скорость стадией N-нитрозирования.
Вернемся к стереохимическим данным. Некоторые поляриметрические дан-
ные о реакциях дезаминирования были известны уже из ранних работ Фишера
и Мак-Кензи. В дальнейшем подобные исследования были проведены для
разных групп В на примерах следующих реакций:
RNH24-HNO24-кислота 4-растворитель Н2О —> ROH
RNH24~HNO24-кислота-|-растворитель С2Н5ОН —> ROC2H5
НКН24-КОС14-НС14-растворитель Н2О —> RC1
RNH24~N2O3 или NOC14-НВг4-растворитель Н2О —> RBr
Основные выводы, которые были получены путем уже известных конфигу-
рационных корреляций для исходных и конечных веществ и которые дают
представление о пространственной направленности реакций, приведены
в табл. 115.
Данные, приведенные в таблице, со всей очевидностью показывают, что
дезаминирование во всех его формах подчиняется S гД-правилу простран-
ственной ориентации при замещении. Кроме того, можно сделать вывод, что
последние стадии дезаминирования носят характер SNl-npopecca, включаю-
щего промежуточное образование карбониевого иона. По-видимому, процесс
начинается с N-нитрозирования, за которым следует перегруппировка
и ионизация нитрозамина, протекающая в свою очередь с образованием
сначала иона диазония, а затем карбониевого иона, и заканчивается соеди-
нением карбониевого иона с находящимся в растворе анионом (например,
С1~, Вг- или ОН-, С2Н6О_), которые могут образоваться из молекул раство-
рителя
RNH2 —» RNHNO -* RNJ —> R+-^RX
Тип Sfjl
443
Глава VII
Таблица 115
Пространственное течение процесса дезаминирования
(R—сохранение, Rac—рацемизация, I—инверсия)
Исходные вещества RNHa Продукты реакции
ROH ROC2H5 RC1 RBr
C2H6CH(NH2)CH3 Rac + 1 Rac -1 Rac-}- I
h-C6Hi3CH(NH2)CH3 Rac-1 Рас1 Rac - I Rac-}- I
C6H5CH(NH2)CH3 Raca +1 Pac-|-1 Rac;il-1 Raca-j-1
CH3CH(NH2)COOH R R R
CH3CH(NH2)COOC2H5 Rac-|- I
C6H5CH(NH2)COOH Raca-|-1 Raca 1 Paca+ I
а В этих реакциях наблюдается очень сильная рацемизация.
к. Ориентация замещения в алициклических системах. В свободной
от напряжений заторможенной конфигурации циклогексана (форма кресла),
являющейся стабильной конформацией, шесть внекольцевых конформа-
ционно эквивалентных связей называются аксиальными (а, параллельны оси
третьего порядка), а шесть других, также эквивалентных связей, назы-
ваются экваториальными (е, направлены почти радиально наружу)
Наиболее стабильными конформациями замещенных циклогексанов будут те,
в которых заместитель или самый больший заместитель, если их несколько,
занимает экваториальное положение, так как при этом отсутствует про-
странственное 1,3-отталкивание между двумя аксиальными заместителями
(аа или а'а') или между одним заместителем и водородом. Когда замещаемая
группа, например галоген или этерифицированная оксигруппа, в основной
конформации занимает аксиальное положение, в этой конформации она
может замещаться по механизму SN2 с инверсией конфигурации. Однако,
когда замещаемая группа занимает экваториальное положение, в этой
конформации она труднее замещается по механизму SN2 вследствие затруд-
нения инвертирующей конфигурацию атаки нуклеофила из-за экранирую-
щего влияния кольца. Конформация субстрата должна сначала превра-
титься в менее устойчивую. Пространственная ориентация замещения
не будет меняться, так как 8к2-правило совершенно однозначно, но скорость
замещения уменьшится.
Это положение было проиллюстрировано Илиелом и Ро [190] на примере
реакции замещения второго порядка между фенилтиолат-ионом C6H5S“
и цис- и транс-изомерами 4-третн-бутилциклогексил-и-толуолсульфоната.
/СН2СН2. СН2СН2.
mpem-C4H9CH )CHOTs mpem-C4H9CH< )CHSCeH5
\сн2сн/ хсн2сн/
444
Замещение в алифатическом ряду
Эта реакция происходит с обращением конфигурации: транс-эфир дает
цис-сульфид и, наоборот, цис-эфир приводит к транс-сульфиду. В 95%-ном
водном растворе этанола при 25 °C транс-эфир реагирует в 19 раз быстрее,
чем цис-эфир. Илиел и Ро объяснили это, резонно предположив, что
mpem-бутильная группа в стабильной конформации всегда занимает
экваториальное положение. Поэтому в транс-эфире тозилатная группа будет
аксиальной, т. е. более склонной к S ^-замещению. Однако в цис-эфире
тозилатная группа в основной конформации будет экваториальной, и для
осуществления S ^-замещения потребуется некоторая энергия искажения
конформации.
Замещение в производных циклогексана по механизму SN1 имеет иные
закономерности ввиду того, что в этом случае пространственная направлен-
ность определяется конкуренцией экранирования с двух сторон карбоние-
вого иона; в этом случае экранирование, обусловленное строением кольца,
конкурирует с обычным экранированием уходящей группой. Кроме того,
трудно предположить, что чисто алифатический вторичный карбониевый
ион, образующийся в гидроксилсодержащей среде, будет иметь время жизни,
достаточное для перехода в равновесную конфигурацию. Следовательно,
диастереомерные субстраты, которые могли бы дать один и тот же равновес-
ный карбониевый ион, если бы последний был достаточно стабилен и достигал
равновесной конфигурации, будут в действительности давать различные
продукты S ^-замещения или смесь продуктов.
Особенно ясно эти закономерности были проиллюстрированы на примере
дезаминирования; как мы уже видели, эта реакция протекает по механизму
Эм1-замещения. Ее специфическая особенность состоит в малой степени
экранирования отщепляющейся молекулы азота и в том, что последняя рано
освобождается от своей гидроксильной сольватной ячейки. Поэтому чисто
алифатический карбониевый ион, образующийся таким путем, имеет очень
малое время жизни и реагирует практически по мере своего образования;
пространственная направленность реакции в основном определяется экрани-
рованием карбониевого центра углеродным скелетом молекулы.
Эти выводы впервые были сделаны Миллзом [191] при анализе наблюдений
В. Хюккеля [192] и Корнуберта [193] по дезаминированию в водных раство-
рах аминопроизводных алкилированных циклогексанов, а также цис-
та транс-декалинов. Миллз отметил, что в любой паре эпимерных амино-
ментанов одно соединение дает почти исключительно спирт с сохранением
конфигурации, а другое — ментен и спирты с преобладающим обращением
конфигурации. При сравнении конформаций оказывается, что первое харак-
терно для экваториальной аминогруппы, а последнее — для аксиальной.
Так, ментиламин и неоментиламин были выбраны потому, что они имеют
основные конформации (приведенные ниже), в которых изопропильная
группа занимает экваториальное положение. Поэтому аминогруппа в мен-
тиламине экваториальна; этот эпимер дает ментол сходной конфигурации.
В неоментиламине аминогруппа занимает аксиальное положение, и поэтому
из этого соединения образуется ментен и ментол в несколько большем коли-
честве, чем неоментол. Такое же различие было найдено для серии декали-
нов, однако в этом случае конформационные отношения более запутанны.
Если замещаемую группу перевести от вторичного кольцевого атома угле-
рода к углероду у головы моста, влияние структуры кольца на замещение
этой группы будет более ярко выраженным. Как уже говорилось (разд. 7, в),
в 1939 г. Бартлет показал, что в этом случае 8к2-механизм полностью исклю-
чается. Он продемонстрировал это на примерах 1-хлорапокамфана и 1-бром-
триптицена (формулы которых приведены на стр. 427). С тех пор его откры-
445
Глава VII
тие было подтверждено на ряде других систем, и поэтому нет сомнения,
что этот принцип является общим.
х~положение ЫН2 в ментиламине и ОН в ментоле
у-положение NH2 в неоментиламине и ОН в неоментоле
Замещение по механизму SN1 при атоме углерода у головы моста должно
происходить с полным сохранением конфигурации вследствие полного
экранирования углеродным скелетом. Что касается скоростей замещения,
то Бартлет установил, что даже S ^-замещение происходит с трудом. Он
сделал вывод, что вследствие невозможности образования плоского карбо-
ниевого иона у головы моста гетеролиз связи сильно затрудняется. В даль-
нейшем это также было подтверждено; этот вопрос был исследован Шлейе-
ром [194], который обнаружил ряд интересных примеров. В 80 %-ном этаноле
при 25 °C скорости сольволиза бромпроизводных с бромом у головы моста:
бицикло-[2,2,1]-гептана (норборнана), бицикло-[2,2,2]-октана и адамантана,
отнесенные к треш-бутил бромиду, составляют следующий ряд:
Эти отношения скоростей, вероятно, не зависят от природы уходящей группы:
точные величины отношения (mpem-бутил)/(адамантил) для галогенидов
в указанных условиях составляют: для хлоридов 1210, для бромидов 817,
для иодидов 1080. Они, по-видимому, не зависят и от сольволизующей
среды: соотношение (адамантил)/(норборнил) составляет 1010 для бромидов
в водном растворе этанола и 1011 для n-толуолсульфонатов в уксус-
ной кислоте.
Шлейер [194] отметил, что эти данные можно интерпретировать с помощью
концепции Бартлета, согласно которой напряжение, возникающее в кольцах,
препятствует образованию плоского карбониевого иона. В норборнане
сопротивление переходу атома углерода у головы моста в плоское состояние
больше, чем в бицикло-[2,2,2]-октане, а в последнем — больше, чем в ада-
мантане. Однако ион адамантилия существует и может быть изучен в растворе
SbF5 — SO2, где противоионом является гексафторантимонат-ион. Шлейер,
Ола и сотр. [195] изучили протонный магнитный резонанс этой системы.
Степень дезэкранирования (лишения электронов) протонов не умень-
шается, как обычно, с увеличением расстояния от положительного заряда:
третичные протоны в положениях 3, 5 и 7 дезэкранированы несколько больше,
чем ближние протоны в положениях 2, 8 и 9 (найденные химические сдвиги
для 2-Н, 3-Н и 4-Н составляли 4,50, 5,42 и 2,67 м. д. соответственно). В связи
446
Замещение в алифатическом ряду
с этим было предположено, что катионный центр в положении 1 заметно
стабилизуется за счет поляризации электронов третичных СН-связей в поло-
жениях 3, 5 и 7, которые притягиваются к этому центру внутри адаманта-
нового скелета. В соответствии с этим было найдено, что скорости сольво-
лиза 1-адамантилбромида уменьшаются при введении заместителей в поло-
жение 3, заметно уменьшаются электроотрицательными заместителями
и довольно слабо — метильными группами (табл. 116). Хотя последний
эффект не достаточно велик для того, чтобы точно раскрыть его причину,
он, как отметили Форт и Шлейер [194], согласуется со сделанным на других
примерах выводом (гл. II, разд. 3,в, и гл. III, разд. 1,г) о том, что метильная
группа в алканах обладает слабо выраженными электроотрицательными
свойствами.
Таблица. 116
Относительные скорости сольволиза замещенных
1-адамантилбромидов (по данным Форта и Шлейера)
Заместитель Водный раствор диоксана Температура, °C Относительная скорость
н 3-СООН 2-Вг 70%-пый 100 1 0,016 2-0,0015
Н 3-СНз 3,5-(СН3)2 3,5,7-(СН3)3 ч 80%-ный У 70 1 0,69 0,45 0,31а
а Величина, полученная Гробом и цитированная Фортом и Шпейе-
ром [194].
8. Пространственные эффекты при нуклеофильном замещении
а. Концепция пространственных затруднений. Хотя общая теория
реакционной способности органических соединений была дана в современном
виде только в 1930 г., задолго до этого существовали некоторые более огра-
ниченные теории, выдвинутые для объяснения аномальных или специальных
эффектов. К таким теориям следует отнести теорию пространственных
затруднений, предложенную в прошлом столетии для того, чтобы объяснить,
почему при определенных структурных особенностях некоторые общие
реакции не протекают с обычной легкостью. Выдвинутое объяснение было
очень простым: для осуществления реакции в сложной молекуле необходимо
доставить реагент к предназначенному месту; это может быть затруднено
в том случае, когда такое место хорошо экранировано благодаря разветвлен-
ности структуры.
Эта мысль возникла еще у Гофмана [196], который в 1870 г. показал, что
такие полиметилированные анилины, как, например, диметилмезидин,
не способны реагировать с иодистым метилом. После некоторых специальных
исследований, проведенных в девяностых годах прошлого столетия, эта
идея стала руководящей. Так, Клаус [197] установил, что замещенные
в орто-положении арилцианиды и бензамиды исключительно стойки по отно-
447
Глава VII
шению к гидролизу обычными реагентами. Керман [198] показал, что реак-
ции каждой из карбонильных групп в бензохиноне с гидроксиламином
протекают в ограниченной мере при наличии одного и особенно двух орто-
заместителей; эффект, очевидно, в меньшей степени зависит от химической
природы заместителей, чем от их близкого расположения к карбонильной
группе. Мейер [199] с особой тщательностью подтвердил это, показав, что
ppmo-дизамещенные бензойные кислоты труднее этерифицируются и что их
эфиры труднее гидролизуются, чем при отсутствии оршо-заместителей,
в то время как opmo-дизамещенные фенилуксусные кислоты, в которых
место реакции смещено на один атом дальше от места разветвления струк-
туры, легко этерифицируются, а их эфиры легко гидролизуются. И в этом
случае химическая природа заместителей имеет меньшее значение, чем их
расположение вблизи места реакции.
Классическая теория пространственных затруднений проста, и основная
идея ее правильна. Однако в течение 50 лет эта идея, несмотря на многие
дополнительные данные, не развивалась далее, что объясняется глубокими
причинами. Приблизительно с 1890 по 1940 г. пространственные затрудне-
ния рассматривались только с геометрической точки зрения; при этом непра-
вильно ограничивались нормальными молекулами и в основном тетраэдри-
ческой моделью атома углерода. Пространственные затруднения как кинети-
ческое явление связаны, с одной стороны, с начальным состоянием исходных
реагентов, а с другой — с переходным состоянием реакции; именно послед-
нее играет решающую роль в тех случаях, когда в этой стадии необходимо
геометрически сблизить между собой большее число атомов, чем в любой
обычной молекуле в исходном состоянии. Пространственное строение пере-
ходного состояния другое, чем у обычных молекул; как отмечалось, углерод-
ный атом в зоне реакции не обязательно соответствует тетраэдрической
модели. Только с того времени, как подробное изучение вальденовского
обращения раскрыло конфигурации переходного состояния при различных
механизмах реакции (разд. 7), появилась возможность рассматривать про-
странственные затруднения с геометрически правильной точки зрения.
Пространственные затруднения зависят от пространственной ориентации
реакции, последняя же зависит от механизма реакции. Легко понять, почему
изучение пространственных затруднений, как и пространственной ориента-
ции, не могло быть успешным вплоть до 1935 г., когда впервые было полу-
чено достаточное количество данных о механизме реакций и в связи с этим
появилась возможность рассматривать эти явления.
В 1937 г. проблема пространственной ориентации была рассмотрена так,
как это указано выше. В 1941 г. Хьюз [200] расширил рамки дискуссии о про-
странственных затруднениях, развил высказанные выше положения; он
показал, что эта теория пространственных затруднений в правильной гео-
метрической интерпретации может проложить путь к количественной дина-
мической трактовке проблемы. Успех в этой последней области лимити-
руется в основном нашими пока еще скудными знаниями (гл. II, разд. 1,в)
о силах отталкивания между непосредственно не связанными атомами.
Известно, что между парой сближающихся не связанных между собой атомов
действуют сначала слабые силы притяжения, а затем слабые силы отталки-
вания до тех пор, пока расстояние между ними не достигнет определенной
небольшой величины, когда обменные силы отталкивания быстро возра-
стают. Поэтому кажется, что представления о пространственных затрудне-
ниях приложимы только к особым случаям. Принципиально энергия оттал-
кивания несвязанных атомов присуща каждой многоатомной молекуле
или переходному состоянию, но она очень часто остается настолько малой,
448
Замещение в алифатическом ряду
что не оказывает качественно заметного влияния на скорость реакции.
Если, однако, благодаря усложнению (возможно, весьма малому) строения
реагента в переходном состоянии некоторые не связанные между собой
атомы принуждены находиться на небольшом расстоянии друг от друга,
энергия отталкивания может внезапно стать соизмеримой с энергией изме-
нения связей. Качественно при этом наблюдается появление кинетических
эффектов, которые описываются как пространственные затруднения.
Пространственные эффекты, которые непосредственно зависят от различий
в энергиях отталкивания несвязанных атомов, можно назвать, когда их
необходимо выделить, первичными пространственными эффектами. В данном
разделе затрагиваются только первичные пространственные эффекты. Иногда
возникает необходимость учитывать другую группу пространственных
эффектов, о которых уже говорилось в гл. III и IV. Эти эффекты прояв-
ляются косвенно в тех реакциях, в которых вследствие отталкивания
несвязанных атомов нарушается внутримолекулярная передача полярных
эффектов сопряжения. Эффекты такого рода можно назвать вторичными
пространственными эффектами. Они будут рассмотрены в последующих
разделах.
Пока обсуждаются только первичные пространственные эффекты, которые
непосредственно зависят от определенных различий в энергиях отталкива-
ния. Если существует различие между первоначальным и переходным
состояниями реакции, то наблюдаются кинетические пространственные эффек-
ты. Наиболее важной группой подобных эффектов является та, в которой
энергия отталкивания в переходном состоянии больше, чем в исходном.
Сюда относятся кинетические эффекты, объединяемые термином «простран-
ственные затруднения», который естественно сохранен по соображениям
историческим, хотя было бы удобнее заменить его более точным термином
«пространственное замедление», чтобы, таким образом, было ясно, что
речь идет о «затруднении» кинетическом, а не термодинамическом. Следует
полагать, что в некоторых реакциях, особенно в тех, которые зависят от ско-
рости гетеролиза, определяющего скорость реакции, энергия отталкивания
в переходном состоянии меньше, чем в исходном. В этом случае возможен
второй тип кинетического пространственного эффекта, а именно простран-
ственное ускорение.
б. Пространственное замедление и замедление вследствие эффекта утя-
желения при бимолекулярном замещении. Бимолекулярное нуклеофильное
замещение
Y-4-CabcX -> YCabc+X-
характеризуется переходным состоянием, которое, как правило, по своему
виду сходно с показанным на рис. 40. Если симметрия такова, как это пока-
зано, то переходное состояние должно быть именно таким, как на приведен-
ной схеме *. Следует выяснить, до какой степени энергия возрастает вслед-
ствие отталкивания пар атомов, непосредственно не связанных между собой,
сначала в исходной молекуле СаЬсХ, а затем, что особенно важно, в пере-
ходном состоянии {Y • •-СаЬс-•’X}-, поскольку любое повышение энергии
отталкивания в переходном состоянии увеличивает энергию активации
реакции и соответственно уменьшает ее скорость.
В качестве простого примера рассмотрим симметричное замещение, напри-
мер реакцию обмена брома, которую можно изучить только с применением
* Предложены, однако, без всяких оснований, также две другие структуры переход-
ного состояния с плоскостью симметрии.
29-0772
449
Глава VII
меченых атомов:
Вг“ + СаЬсВг —> ВгСаЬс 4~ Вг-
В бимолекулярном переходном состоянии^ {Вг • •-СаЬс • •-Вг}- центральный
атом углерода имеет точную конфигурацию тригональной бипирамиды,
а анионный заряд равномерно распределен между атомами брома. Те связи,
которые в процессе реакции не образуются и не разрушаются, могут рас-
сматриваться до получения более точных сведений как связи, имеющие
нормальную длину. Наполовину образовавшиеся и наполовину расщепив-
шиеся связи с атомами брома имеют длины, приблизительно на 0,35 А
Рис. 40. Модель переходного состояния
в реакциях бимолекулярного
нуклеофильного замещения.
(3,5-Ю-2 мм) превышающие длину
полностью образовавшейся связи
С—Вг *.
Имеющиеся эмпирические сведения о
равновесных расстояниях между не
связанными между собой атомами осно-
ваны главным образом на рентгено-
структурных измерениях расстояний
между молекулами в кристаллах. По-
скольку эти расстояния относятся к
твердому телу, в рассматриваемом слу-
чае они не могут считаться точными;
все же эти сведения позволяют сделать
определенные полуколичественные зак-
лючения. Так, можно прийти к выводу,
что в наиболее простой из рассматри-
ваемых обменных реакций, т. е. в ре-
акции метилбромида, как в нор-
мальной молекуле СН3Вг, так и в
переходном состоянии {Вг • • -СН3 • • -Вг}- энергия отталкивания незна-
чительна, расстояния между парами непосредственно не связанных между
собой атомов превышают расстояния, при которых отталкивание начинает
становиться большим. Поэтому в данном случае пространственные затрудне-
ния не оказывают влияния на энергию активации. Таким образом, реакцию,
в которой отсутствует пространственное торможение, можно использовать
в качестве стандартной для сопоставления и оценки пространственного тор-
можения в реакциях гомологического ряда алкилбромидов.
Главные особенности влияния структуры алкильного радикала на про-
странственное торможение можно сформулировать на основании сравнения
гомологов двух типов, а именно а- и ^-метилированных рядов.
Метил, этил, изопропил, трет-бутил (а-ряд)
Этил, и-пропил, изобутил, неопентил (0-ряд)
При рассмотрении экспериментальных данных следует помнить, что в реак-
циях радикалов а-ряда можно предполагать значительный полярный эффект,
в то время как в реакциях радикалов p-ряда полярный эффект варьируемой
части радикала лишь в незначительной степени будет достигать реакцион-
ного центра.
* То есть району энергии активации на кривой Морзе соответствует существенное растя-
жение связей. Приведенная величина, вероятно, занижена, ио нужно учесть, что ее
легко вычислить и что она позволяет одним и тем же способом проводить сравнение раз-
ных реакций замещения.
450
Замещение в алифатическом ряду
Характерные формы переходного состояния при реакциях этихг радикалов
показаны на рис. 41. В метильной группе а, & и с — атомы водорода, которые
в a-рядах постепенно замещаются метильными группами. В этильной группе
d, е и / — атомы водорода, которые в 0-рядах замещаются метильными груп-
пами. Предполагается, что та из указанных форм, которая имеет наименьшую
энергию отталкивания, является эффективной; если поместить атомы водо-
рода в точках^ и е (когда это возможно), то форма, изображенная слева,
Рис. 41. Конфигурации алкильных групп в переходных состояниях реакций бимолеку-
лярного замещения.
должна быть эффективной для всех указанных групп, за исключением изобу-
тильной группы, которая должна использовать другую форму с метиль-
ными группами при d и е.
Оценив вначале, исходя из геометрии молекул и переходных состояний,
расстояния между несвязанными отталкивающимися атомами, затем по фор-
муле, приведенной в гл. II, разд. 1, в, соответствующие энергии отталкивания
и, наконец, уменьшение энергии отталкивания при таком расположении
входящего и уходящего галогенов, когда общая (связывающая + несвя-
зывающая) энергия минимальна, можно вычислить вклад пространственных
факторов в общую энергию активации замещения. Эти данные для реакций
обмена брома приведены в табл. 117 в строке, обозначенной АЯ-‘ (про-
странств.) (рассч.).
Ввиду принятых допущений эти величины имеют лишь полуколичественный
характер; поэтому более целесообразно сначала рассмотреть качественную
сторону этих величин. Во-первых, в а-рядах этильная группа обладает
незначительной пространственной энергией активации, которая в изопро-
пильной и mpem-бутильной группах становится соответственно удвоенной
и утроенной. Это объясняется в основном тем, что отталкивание между
каждым а-метильным заместителем и каждым атомом брома в переходном
состоянии реакции взаимно независимо. Во-вторых, в 0-рядах простран-
ственная энергия чрезвычайно быстро возрастает до значительной величины
в случае неопентильной группы, которая, таким образом, проявляет
свойства, характерные для явления пространственных затруднений. Объяс-
нение заключается в том, что, в то время как единственный 0-метильный
заместитель в н-пропильной группе в переходном состоянии может повер-
нуться так, что будет удален от атомов брома, в случае изобутила две метиль-
ные группы могут сделать это только частично, а в случае неопентильного
радикала две из трех метильных групп обязательно расположены к атомам
брома ближе предела, ниже которого энергия отталкивания становится
29* 451
Глава VII
Таблица 117
Рассчитанные и наблюдаемые влияния структуры алкильной группы
на энергии активации [ккал/моль (4,187-103 Дж/моль)]
обмена бромид-иона в бромистых алкилах
.Алкильная группа &НТ= (пространств.) (рассч.) ДН^ (полярн.) (предп.) Энергия активации
общая (теор.) наблюдаемая (в ацетоне)
а-Ряд
СН3 0,0 (0,0) 0,0 (0,0)
С2Н5 0,8 1,о 1,8 1,7
«ао-СзНу 1,6 2,0 3,6 3,9
трет-СдНд 2,5 3,0 5,5
fl-Ряд
с2нд 0,8 1,0 1,8 1,7
H-C3H7 0,8 1,0 1,8 1,7
2,3 1,0 3,3 3,1
7,3 1,0 8,3 6,2
значительной. Как правило, отталкиванием в исходном состоянии, по абсо-
лютной величине довольно значительным, можно пренебречь по сравнению
с отталкиванием в переходном состоянии.
По аналогии с открытыми В. Майером пространственными затруднениями
в реакциях карбоксильной группы в opmo-дизамещенных бензойных кисло-
тах (в отличие от гомологов фенилуксусной кислоты) можно было бы пред-
полагать более значительные пространственные затруднения в бимолеку-
лярных реакциях шреш-бутилгалогенидов, содержащих галоген ближе
к месту разветвления, чем в соответствующих неопентилгалогенидах, где
галоген находится на один атом дальше от места разветвления. Однако такая
аргументация ошибочна. Сделанный выше противоположный вывод основан
на том, что в первую очередь геометрия переходного, а не нормального
состояния молекулы важна для объяснения пространственного замедления.
Подобные расчеты были произведены и для других пар входящих и уходя-
щих галогенов. На первый взгляд может показаться странным, что рассчи-
танные величины, характеризующие пространственное отталкивание,
не сильно зависят от размеров входящих и отщепляющихся групп, которые
могут быть и не идентичными, хотя расчеты гораздо легче сделать в случае
одинаковых групп. Причина этого заключается в компенсирующих факто-
рах. Так, если в реакции обмена брома бромид-ион заменить на иодид-ион,
то больший галоген в переходном состоянии будет находиться дальше
от центрального атома углерода, и поэтому все расстояния между ато-
мом иода и несвязанными с ним атомами будут увеличиваться. Однако
из-за больших размеров иодид-иона допустимые пределы расстояний, ниже
которых отталкивание становится значительным, также увеличиваются.
В результате этого отталкивание остается примерно одинаковым и энергии
отталкивания имеют одинаковый порядок величины. Так, величина А/7*
(пространств.) (рассч.) для обмена Вг~ Д- RBr равна 7,3 ккал/моль
(30,56-103 Дж/моль), а для обмена RBr + I- эта величина составляет
7,8 ккал/моль (32,66-103 Дж/моль).
В общем случае для бимолекулярного замещения величина пространствен-
ного отталкивания в переходном состоянии, обусловленная объемом входя-
452
Замещение в алифатическом ряду
щей и отщепляющейся групп, зависит от геометрии переходного состояния.
Как упоминалось, для нуклеофильного замещения алифатических соедине-
ний степень отталкивания гораздо больше зависит от размера постоянно
присутствующих групп, участвующих в отталкивании, чем от размеров
замещаемой и атакующих групп. С другой стороны, при электрофильном
замещении ароматических соединений (гл. VI), как это легко можно вычис-
лить, при допущении приблизительно тетраэдрической связи в точке заме-
щения в переходном состоянии (гл. VI, разд. 5,г), величина отталкивания
лишь немного больше зависит от размеров оршо-группы, чем от размеров
вступающей группы; важность обоих этих факторов в ароматическом замеще-
нии, по-видимому, подтверждается приведенными в гл. VI, разд. 4,6,
результатами наблюдений. Однако влияние opmo-заместителей при бимоле-
кулярном нуклеофильном замещении в бензилгалогенидах описывается
другими соотношениями. Как было показано в детальных исследова-
ниях Чарлтона и Хьюза [201], эти эффекты могут быть скоррелированы
со специфическими особенностями геометрии начального и переходного
состояний.
Отмеченное выше теоретическое положение, касающееся бимолекулярного
замещения для нуклеофильного замещения алифатических соединений, было
впервые выдвинуто в 1941 г. Хьюзом [200], который получил и экспери-
ментальные кинетические данные по этому вопросу. Впоследствии Достров-
ский и Хьюз [202] распространили исследование на большинство упомяну-
тых выше групп в различных процессах замещения. Тем самым были каче-
ственно объяснены ранее известные факты из области химии неопентилгало-
генидов, которые вплоть до этого времени считались аномальными.
Уитмор [203] обратил внимание на заметную пассивность неопентилгалоге-
нидов. За исключением реакции с Na и Mg, неопентилхлорид не подвер-
гается другим общим реакциям алкилгалогенидов. Даже неопентилиодид
обнаруживает незначительную тенденцию реагировать или вовсе не реаги-
рует с этилат-, окси-или цианид-ионами. Если прибегать только к аналогии
с классическими примерами пространственных затруднений и не учитывать
механизма процесса, то трудно сделать вывод, что причина этого явления
заключается в пространственных затруднениях.
Достровский и Хьюз [202] установили, что различие в реакционной способ-
ности неопентилгалогенидов и обычных алкилгалогенидов носит скорее
количественный, чем качественный характер; так, неопентилбромид реаги-
рует с этилат-ионами в этиловом спирте и с ионами иода в ацетоне в 104—
105 раз медленнее, чем простейшие первичные алкилбромиды. Достровский
и Хьюз показали также, что эти заметные отличия в реакционной способ-
ности свойственны бимолекулярному механизму и что в различных реак-
циях мономолекулярного замещения неопентилбромид по реакционной
способности вполне сходен с другими первичными алкилбромидами. Более
детально этот вопрос будет рассмотрен в следующем разделе. Наблюдаемые
величины скоростей реакций второго порядка между метилбромидами и пер-
вичными алкилбромидами с этилат-ионами в этиловом спирте при 55 °C
могут служить иллюстрацией особого положения неопентилбромида при
бимолекулярном замещении. В большинстве случаев, хотя и не всегда,
различие в скорости реакции находит отражение в аррениусовских энергиях
активации, как это показано в табл. 118.
Можно сравнить эти данные только с рассчитанными пространственными
инкрементами энергии активации ДЯ* (пространств.) (рассч.), приведен-
ными в табл. 117. К ним нужно прибавить полярные инкременты, т. е. инкре-
менты, связанные с изменением индуктивных эффектов при замене метильной
453
Глава VII
Таблица 118
Реакция второго порядка метилбромида и других первичных
алкилбромидов с этилатом натрия в этиловом спирте
(fc2 и Л в л-моль-1-с-1)
Алкильная группа 103й2 при 5 5 °C Е, ккал/моль (4,187-103Дж/моль) 10-пд
сн3 34,4 20,0 7,2
С2Н5 1,95 21,0 2,1
и-С3Н7 0,547
иао-С4Н9 0,058 22,8 0,95
Heo-CgH)! 8,26-IO'6 26,2 0,0023
группы в гомологическом ряду на высшие алкильные группы. Все, что мы
можем сказать о полярных эффектах, опираясь на существующую теорию,
сводится к тому, что вклады а-метильных заместителей в первом приближе-
нии должны быть равными, а вклады 0-метильных заместителей — пренебре-
жимо малыми. Взяв значение 1 ккал/моль (4,187 -103 Дж/моль) для одной
а-метильной группы и нулевое значение для 0-метильной группы, можно
получить величины А// (полярн.) (предпол.) (табл. 117). Согласно данным
таблицы, сумма указанных теоретически рассчитанных пространственных
и полярных энергий, т. е. инкременты влияния строения на энергию акти-
вации, очень близки к наблюдаемым величинам. Наибольшее отличие между
этими данными наблюдается в случае неопентильной группы, имеющей
определенную гибкость, не учтенную в расчетах. Игнорирование возможной
деформации приводит к тому, что пространственное отталкивание кажется
более существенным, чем оно есть на самом деле. Подобные расчеты и их
сравнение с экспериментальными данными были проведены и для других
входящих и уходящих галогенов. В общем они подобны данным, получен-
ным для обмена бромида.
Приведенные в табл. 117 теоретические и экспериментальные данные взяты
из последней работы Хьюза и сотр. [204], посвященной реакции Финкель-
штейна. Они рассчитали также энтропии активации (см. ниже) и сравнили
их с опытными данными.
Как уже отмечалось, при расчете пространственных энергий активации
требуется минимизировать энергию связывающих и несвязывающих взаимо-
действий при изменении положения галогена в переходном состоянии,
т. е. на языке геометрии необходимо рассчитать какую-то энергетическую
поверхность. Эта поверхность имеет форму бассейна. Высота, на которой
находится дно бассейна, представляет собой энергию переходного состояния,
а крутизна его стенок определяет энтропию этого состояния. Исходя
из формы бассейна, по стандартной формуле можно рассчитать вклад про-
странственных факторов в энтропию активации.
В этих расчетах возникает непредвиденный эффект. Энтропия энергетиче-
ского бассейна зависит не только от формы бассейна, но также и от масс
и 'моментов инерции находящихся в нем кинетических систем: чем больше
эти внутримолекулярные факторы, тем ближе друг к другу располагаются
соответствующие энергетические уровни в бассейне и тем больше будет его
«вместительность», логарифм которой соответствует его энтропии. При
таком простом рассмотрении требуется ввести понятие об еще одном классе
структурных эффектов, который ранее подробно не рассматривался. Этот
454
Замещение в алифатическом ряду
эффект не является ни стерическим, ни полярным, т. е. он не зависит от рас-
пределения объемной массы или заряда. Он носит название «эффекта утяжеле-
ния» (ponderal effect), так как зависит от распределения «удельной массы»
(mass), но не объемной массы (bulk) или заряда. Нейтроны имеют удельную
массу, но не имеют объемной массы и заряда, поэтому если в систему доба-
вить нейтрон, т. е. произвести изотопное замещение, то должен наблюдаться
кинетический эффект утяжеления. Нет сомнения, что эффект утяжеления
часто остается незамеченным и примешивается к пространственным и поляр-
ным эффектам.
Таблица 119
Рассчитанные и наблюдаемые эффекты строения алкильной группы
на энтропию активации [кал-моль-1-°C-1 (4,187 Дж.моль_1.°С-1)]
обмена бромид-иона с алкилбромидами
Алкильная группа AS^ (масс.) (рассч.) AS^ (пространств.) (рассч.) Энтропия активации
общая (теор.) наблюдаемая (в ацетоне)
а-Ряд СНз (0, 0) (0, 0) (0, 0) (0,0)
С2н5 -1,45 -0,66 -2,11 —2,7
Jiao-CgHy -2,30 -1,52 -3,82 -4,7
трет-СЩд -1,95 +0,83 -1,12
$-Ряд с2н5 -1,45 -0,66 -2,11 -2,7
н-С3Н7 -2,64 -0,67 -3,31 -4,1
ИЗО-СдНд -3,66 -1,06 -4,72 -5,0
нео-С5Нц -4,39 -1,86 -6,25 -9,6
Указанный инкремент энтропии активации был рассчитан как влияние
на энтропию активации такого изменения в строении алкила, в результате
которого не происходит изменения энергетической поверхности. Общий
энтропийный эффект обусловлен изменением и массы, и энергетической
поверхности. Таким образом, с учетом энтропии изменения массы был
вычислен пространственный инкремент энтропии активации. Эти данные
приведены в табл. 119. Они показывают, что все эффекты, связанные с изме-
нением массы, отрицательны (уменьшают скорость) и что их величина воз-
растает с увеличением массы алкила и с ростом расстояния добавляемой
в переходном состоянии массы от места расположения массы галогена.
Энтропийные пространственные эффекты также отрицательны для всех
алкильных групп, кроме щретп-бутильной группы.
В табл. 119 проведено сравнение рассчитанных данных с наблюдаемым
влиянием алкильной гомологии на энтропию активации. Наблюдаемые
значения представляют собой просто разность значений логарифмов предэк-
споненциальных факторов Аррениуса, причем эта разность умножена
на 4,575 для перевода в температурные единицы. Рассчитанные энтропийные
эффекты изменяются параллельно наблюдаемым, однако первые всегда
меньше по абсолютной величине. Наиболее вероятным источником ошибки
является пренебрежение эффектом растворителя. Можно было полагать,
что, поскольку каждое значение получено путем двух процессов вычита-
ния — параметров начального состояния из параметров переходного
455
Глава VII
состояния и параметров метильной группы из параметров высших алкиль-
ных групп, энергии и энтропии сольватации в разности разностей сокра-
тятся; однако, возможно, это не так. Трудно предположить, что ошибка
связана с пренебрежением при расчетах вкладом энергии активации утяже-
ления масс; она должна быть небольшой вследствие большой массы кинети-
ческих частиц. Мало вероятно, чтобы заметная ошибка была обусловлена
и неучетом полярных энтропий: они должны быть очень небольшими вслед-
ствие слабой полярности алкильных групп. Необходимо отметить, что энер-
гетические поверхности, а отсюда и пространственные энергии, простран-
ственная энтропия и энтропия изменения массы — все эти величины вычис-
лены без использования каких-либо имеющихся в распоряжении констант
скоростей. Все необходимые константы были получены в результате измере-
ний совсем других параметров: длин связей, прочностей связей, колебатель-
ных частот, сжимаемости и т. д.
Комбинация рассчитанных энергетических и энтропийных факторов позво-
ляет получить относительные скорости реакций, которые можно непосред-
ственно сравнить с наблюдаемыми значениями. Такое сравнение для обмена
бромид-иона в ацетоне при 25 °C проведено в табл. 120. Можно видеть,
что рассчитанные и наблюдаемые константы в интервале изменения, равном
5 000 000, отличаются не более чем в 5 раз.
Таблица 120
Наблюдаемые и рассчитанные структурные эффекты
на скорости реакций Br_-|-RBr —> RBr4-Br~ в ацетоне
при 25 °C
Относительные скорости
R
рассчитанные
наблюдаемые
а-Ряд
СНз
с2н5
иао-С3Н7
тргт-С4Н9
$-Ряд
С2Н5
H-C3H7
Ц30-С4Нд
иео-С5Нц
1
0,017
0,00035
0,000053
0,017
0,0090
0,00035
0,000000037
1
0,013
0,00014
0,000039
0,013
0,0085
0,00044
0,00000020
а Кук и Паркер [205] в статье, которая вышла слишком поздно
и поэтому здесь не обсуждается, показали, что кажущиеся скорости
реакций Финкельштейна для mpem-бутилгалогенидов (но не для других
алкилгалогенидов) слишком высоки вследствие конкурирующего элимини-
рования с последующим присоединением. Поэтому соответствие между
вычисленными и наблюдаемыми скоростями в этом случае хуже, чем сле-
дует из приведенных значений.
в. Кинетические пространственные эффекты при мономолекулярном
замещении. Пространственное замедление может проявляться при бимоле-
кулярном замещении потому, что атомы более сближены в переходном
состоянии, чем в исходном; в некоторых структурах такое сближение может
обусловливать повышение энергетического барьера. Однако при мономоле-
456
Замещение в алифатическом ряду
кулярном замещении пространственное торможение играет незначительную
роль, поскольку в этом случае переходное состояние образуется с участием
электростатических сил сольватации более дальнего действия, в результате
чего атомы, наиболее близко расположенные к зоне реакции, оказываются
сближенными не более, чем раньше [206].
Хьюз и Достровский [207] использовали эти положения при изучении заме-
щений в алкилгалогенидах. Несмотря на то что Уитмор даже в жестких
условиях не добился большого успеха при попытке гидролизовать неопен-
тилгалогениды действием таких реагентов, как концентрированная спирто-
вая щелочь, Хьюз и Достровский показали, что гидролиз можно осущест-
вить простым добавлением воды к спирту. Причиной является то, что алкил-
галогениды в принципе могут гидролизоваться по механизму как SN2,
так и SN1; в случае большинства первичных алкилгалогенидов легче осуще-
ствляется SN2-MexaHH3M; однако в особом случае, когда этот механизм
пространственно значительно затруднен, алкилгалогениды могут легче
гидролизоваться в условиях, способствующих ионизации по SгД-механизму,
так как на этот механизм не влияют пространственные затруднения.
Таблица 121
Относительные скорости бимолекулярных или мономолекулярных реакций
замещения первичных и третичных алкилбромидов
Алкилбромид RBr+ С2Н5О- в С2Н5ОН при 25 °C НВг-|-50%-ный водный С2Н5ОН при 95 °C RBr+НгО в НСООН при 95 °C
СН3СН2Вг (1) (1) 1
СН3СН2СН2Вг (0,28) (0,58) 0,69
(СН3)2СНСН2Вг (0,030) (0,080)
(СНз)3ССН2Вг (0,0000042) (0,0064) 0,57
RC18 0%-ный водный %RBr --80-ный водный RI4-8 0%-ный водный
C2H5OH при 25 °C С2Н5ОН при 25 °C С2Н5ОН при 25 °C
СН3С(СНз)2Вг 1 1 1
СН3СН2С(СН3)2Вг 1,68 1,78 2,00
(СН3)2СНС(СН3)2Вг 0,87 1,22 1,62
(СНз)3СС(СНз)2Вг 1,16 1,68 2,84
Это положение можно подтвердить данными, приведенными в двух верхних
рядах табл. 121, характеризующих относительные скорости гидролиза или
алкоголиза четырех первичных алкилбромидов 0-ряда. В этой таблице
в скобках приведены значения скорости, о которых предположительно или
точно известно, что они относятся к бимолекулярным реакциям; остальные
величины соответствуют скоростям мономолекулярного замещения. Верх-
ний ряд, приведенный для сравнения, охватывает данные уже обсужденной
реакции второго порядка с этилат-ионами в сухом спирте; эти реакции,
несомненно, бимолекулярны, и вдоль ряда отмечается сначала постепенное,
а затем резкое увеличение пространственных затруднений. Второй ряд
содержит данные о сольволизе в 50 %-ном водном растворе этилового спирта.
Обращает на себя внимание тот факт, что три первые величины изменяются
457
Глава VII
в пределах того же порядка, что и в первом ряду, а четвертая уменьшается
по сравнению с третьей до порядка 101 вместо 104, как это наблюдается
в первом ряду. Объяснение состоит в том, что первые три величины пред-
ставляют собой скорости бимолекулярных реакций с молекулами раствори-
теля, а четвертая (примерно в тысячу раз превышающая ожидаемую вели-
чину) — скорость мономолекулярной реакции первичного алкилгалогенида.
Высказанное предположение подтверждается тем фактом, что, в то время
как реакции этил-, к-пропил- и изобутилбромидов в 50%-ном водном спирте
значительно ускоряются при добавлении щелочи, реакция неопентилбро-
мида не чувствительна к щелочам. Кроме того, замечено, что в то время как
реакция второго порядка между неопентилбромидом и этилат-ионами ведет
к нормальному продукту — неопентилэтиловому эфиру, реакция неопен-
тилбромида в 50%-ном водном растворе спирта приводит к образованию
mpem-амилового спирта и его этилового эфира, а также амиленов. Наблю-
даемая перегруппировка, как отмечено в гл. X, может служить кри-
терием мономолекулярного механизма с участием этой особой алкильной
группы.
Наиболее известным растворителем для мономолекулярного замещения
в алкилгалогенидах является муравьиная кислота, которая достаточно
легко ионизует даже первичные алкилбромиды. Достровский и Хьюз
использовали ее в качестве растворителя для изучения реакций гидролиза;
найденные относительные скорости приведены в третьем ряду табл. 121.
Все приведенные величины — одного порядка, как и следовало ожидать,
в том случае, если они соответствуют скоростям мономолекулярной реакции,
когда пространственными затруднениями и полярным эффектом р-замещения
можно пренебречь.
В нижней части табл. 121 представлены реакции трех рядов третичных
алкилгалогенидов (с таким же увеличением цепи вдоль ряда, как и в случае
первичных бромистых алкилов) в водном растворе спирта [208]. Имеются
веские доказательства того, что все эти реакции мономолекулярны. Харак-
терно, что все скорости в каждом ряду имеют величину одного порядка,
а это находится в соответствии с гипотезой, согласно которой мономолеку-
лярные реакции не чувствительны к пространственным затруднениям
и полярный эффект ^-алкилирования незначителен. На основании сравнения
данных любых строк с верхней строкой табл. 121 можно сделать убедитель-
ный вывод, что бимолекулярные и мономолекулярные процессы замещения
характеризуются различными состояниями, т. е. различия в механизмах
весьма существенны.
Первичные, вторичные и третичные алкилгалогениды типа (СН3)3ССН2Х,
(СН3)3ССН(СН3)Х и (СН3)3СС(СН3)2Х, для которых вследствие блокировки
затруднены S ^-реакции, реагируют предпочтительно или исключительно
по S ^1-механизму; при этом вдоль указанного гомологического ряда ско-
рость заметно возрастает, а энергия активации уменьшается. Найдено [209],
что при сольволизе соответствующих бромидов в 80%-ном водном растворе
этанола энергии активации составляют 30,0, 26,7 и 23,3 ккал/моль
(125,6-103, 111,79-103 и 97,55-103 Дж/моль) соответственно. Уменьшение
энергии на 3...4 ккал/моль (12,56-103...16,75-103 Дж/моль) при введении
каждой следующей а-метильной группы наблюдается для хлоридов, а также
в других органических растворителях.
Хотя пространственное замедление не играет никакой роли при мономоле-
кулярном нуклеофильном замещении простых алкилгалогенидов, оно в неко-
торой степени проявляется в реакциях вторичных аралкилгалогенидов,
имеющих два opmo-заместителя. Это было установлено Чарлтоном и Хьюзом
458
Замещение в алифатическом ряду
1210]. Объяснение этих авторов иллюстрируется приведенными формулами
ди-орто-замещенных а-арилэтилхлорида и образующегося из него карбо-
ниевого иона.
В исходном состоянии реакционноспособная боковая цепь имеет тетраэдри-
ческую конфигурацию и все ее заместители находятся вне плоскости бен-
зольного кольца, тем самым избегается значительное взаимодействие
с opmo-метильными группами. В карбониевом ионе силы связывания, если
это возможно, пытаются перевести связи, показанные на схеме, в плоскость
кольца; при этом создается максимальное взаимодействие между метильной
группой боковой цепи и одной из opmo-метильных групп. Конфигурация
переходного состояния при мономолекулярной механизме, приводящем
к образованию карбониевого иона, близка к конфигурации карбониевого
иона, поэтому в переходном состоянии будет частично проявляться про-
странственное отталкивание, свойственное структуре иона. Это простран-
ственное отталкивание обусловливает пространственное замедление моно-
молекулярной реакции.
Нуклеофильное замещение в ди-орто-замещенных а-арилэтилгалогенидах
в полярных растворителях, по полярности не ниже, по крайней мере,
ацетона, происходит преимущественно мономолекулярно. Мономолеку-
лярный механизм сольволиза был доказан для а-мезитилэтилхлорида
2,4,6-(СН3)3С6Н2СН(СН3)С1 в водном и безводном этаноле. Безусловно, аро-
матические метильные группы оказывают полярное влияние на эти реакции.
Это влияние Чарлтон и Хьюз оценили путем сравнения скоростей этих реак-
ций со скоростями реакций изомерных низших гомологов. Пространственный
эффект не наблюдался до тех пор, пока в молекулу не вводилась какая-либо
третья из трех метильных групп, указанных на приведенных выше форму-
лах. В этих случаях скорости внезапно становились меньше, чем можно
было предполагать, исходя лишь из полярных эффектов; степень уменьшения
скоростей соответствовала увеличению энергии переходного состояния
ионизации за счет несвязывающих взаимодействий приблизительно
на 1...1,5 ккал/моль (4,19-103...6,28-Ю3 Дж/моль).
Возможность пространственного ускорения при мономолекулярной нуклео-
фильном замещении, т. е. возможность ситуации, когда внутримолекуляр-
ное отталкивание несвязанных групп в основном состоянии исчезает благо-
даря растягиванию связи в переходном состоянии гетеролиза, обсуждалась
Брауном и Хьюзом и их сотрудниками [211]. Это предположение очень
правоподобно и, вероятно, его можно доказать и для замещения, как это
уже доказано для реакций элиминирования (гл. IX); однако доказательства,
полученные до настоящего времени, не достаточно удовлетворительны.
Трудность состоит не в том, чтобы найти неожиданное увеличение скорости
мономолекулярного замещения, а в том, чтобы найти процесс, в котором
ускоряющее действие оказывало бы только взаимодействие между несвя-
занными атомами, а не взаимодействие связанных атомов того типа, кото-
рый классифицируется как циклосинартетический (разд. 7, е) или синар-
тетический (гл. X) эффект.
459
Глава VII
9. Нуклеофильное замещение у олефинового атома углерода
а. Кинетический характер. Нуклеофильное замещение у олефинового
атома углерода
Y- + RR'CH = CR" —X —> RR'CH = CR"—Y-р Х~
подобно нуклеофильному ароматическому замещению при отсутствии элек-
троноакцепторных заместителей, таких, как нитрогруппа, сульфонильная,
карбонильная или карбоксильная группа, происходит очень медленно.
Так, реакция Финкельштейна между иодид-ионом и бромистым винилом
или со-бромстиролом в спиртовых растворителях лишь при температуре
около 200 °C достигает скорости, удобной для изменения, тогда как соот-
ветствующие реакции иодид-иона с цис- и /прайс-со-бром-н-нитростиролами
идут относительно легко [212].
Все детальные исследования кинетики нуклеофильного олефинового замеще-
ния проводились с субстратами, содержащими активирующие электроотри-
цательные группы. Модена и сотр. [213] изучили следующие процессы
замещения:
Y- + C6H5SO2CH = CH — Cl —> C6H5SO2CH = CH—Y + C1-
Y- + C6H5SOCH = CH-C1 C6H5SOCH=CH- Y +Cl-
где Y- = CH3O~, C2H5O“, C6H5O~, CH3S“, N". Кинетика реакций всегда
соответствовала общему второму порядку, первому по каждому из реагентов.
То же самое наблюдали Вернон и сотр. [214] для следующих процессов:
Y- + С2Н5О2СС = С(СН3)~ С1->С2Н5ОаСНС = С(СН3) — Y + Cl-
где Y- = С2Н5О~, С6Н5О , C2H5S , CgH5S .
Такая кинетика соответствует механизму типа SN2, вероятно, аналогичному
общему механизму нуклеофильного ароматического замещения в присут-
ствии активирующих электроотрицательных заместителей (гл. VI, разд. 8).
Можно также предположить механизм отщепления — присоединения, когда
реакция проходит через промежуточное образование ацетилена по аналогии
с процессами нуклеофильного ароматического замещения, происходящими
через промежуточное образование дегидробензола. Однако при таком меха-
низме гщс-транс-изомерные субстраты должны давать один и тот же продукт
или одинаковую смесь продуктов, чего, как мы увидим, не наблюдается.
Кроме того, Вернон провел реакцию этилтиолат-иона с этиловыми эфирами
цис- и шраис-р-хлоркротоновых кислот в дейтероэтаноле и не обнаружил
дейтерия в продуктах замещения. Отсюда следует, что реакция происходит
по S ^-механизму.
Однако Модена и сотр. [215] показали, что в случае сильноосновных нуклео-
филов, например СН3О_ в метаноле, реакция цмс-р-аренсульфонилвинил-
галогенидов частично осуществляется по механизму отщепления — при-
соединения, конкурирующего с прямым нуклеофильным замещением. В слу-
чае же транс-изомер а осуществляется исключительно механизм прямого
замещения.
ArSO2. .Cl
)С = С<
СНзО“ '
СНзОИ
ArSO2 — С= С-Н
СН3О-
СНзОН
ArSO2. .ОСН3
> /С = С<
Hz ХН
Образование ацетиленового промежуточного соединения из гщс-изомера
было обнаружено спектрофотометрически. Этот механизм становится пре-
460
Замещение в алифатическом ряду
обладающим при повышенных температурах. Так, в вышеуказанной реак-
ции при Аг=ге-СвН4СН3 по механизму отщепления — присоединения обра-
зуется половина продукта замещения при О °C и три четверти при 25 °C.
Если реакция проводится с менее сильными нуклеофилами, например пер-
вичными и вторичными аминами, то даже цмс-изомер реагирует только
по механизму прямого нуклеофильного замещения типа SN2.
Другие доказательства механизма прямого замещения следуют из стерео-
химических данных.
б. Пространственная направленность. При решении вопроса о про-
странственной направленности рассматриваемых процессов необходимо
избегать ошибок, связанных с возможностью предшествующей или после-
дующей изомеризации. Несмотря на некоторые сомнения относительно
возможности предшествующей изомеризации, Миллер и Ионан [212] все же
сделали вывод, что реакция цис- и шранс-со-бром-п-нитростирола с ио дид-
ионами происходит почти с полным сохранением геометрической конфигу-
рации. Модена [213] установил, что замещение в сульфонах и сульфоксидах
(формулы приведены выше) всегда происходит с общим сохранением гео-
метрической конфигурации. Вернон не сделал вывода о пространственной
направленности изученных им реакций этилат-иона с этиловыми эфирами
цис- и шрпкс-р-хлоркротоновых кислот (формулы приведены выше), так как
оба изомера дают один и тот же этоксипродукт и, кроме того, изомер этого
продукта вообще неизвестен, что делает невозможным контроль за после-
дующей изомеризацией первоначально образующегося изомера. Однако
реакция замещения тех же эфиров с тиолат-ионами происходит так, что,
хотя в основном продукты реакции в каждом случае имеют ту же геометри-
ческую конфигурацию, что и исходные соединения, некоторая часть про-
дуктов реакции имеет обращенную конфигурацию [214]. Эти данные приве-
дены в табл. 122. Таким образом, в рассматриваемых реакциях в некоторой
степени происходит геометрическая инверсия, причем изомерные субстраты
дают смесь продуктов неодинакового состава. Доказано, что предшествую-
щая и последующая изомеризации в этих случаях отсутствуют.
Таблица 122
Продукты реакции этилтиолат- и фенилтиолат-ионов
с этиловыми эфирами цис- и шрамс-р-хлоркротоновых кислот
в этаноле при О °C (по данным Вернона)
Э тил-р-хлоркротонат Замещающий агент Продукты замещения, %
транс цис
цис C2H5S- 85 15
транс C2H5S- 9 91
цис c6H5s- 88 12
транс C6H6S- 36 64
«С помощью этих данных можно попытаться вслед за Верноном выяснить
последовательность внедрения и отщепления групп и, таким образом, попы-
таться представить себе строение промежуточных частиц в 8м2-реакциях
этого типа. Среди всех возможных ситуаций выделяются две главные. Во-пер-
вых, возможен случай, когда присоединение и отщепление перекрываются
во времени, т. е. либо нацело, либо частично происходят синхронно. В этом
случае первоначально двойная связь всегда остается по кратности выше
461
Глава VII
простой и дискретного промежуточного соединения не образуется. Во-вто-
рых, возможен случай, когда присоединение и отщепление разделены во вре-
мени, и в промежутке между этими стадиями существует промежуточный
продукт присоединения с чисто простой связью, обладающий или коротким,
или неопределенно долгим временем жизни. Естественно, что эти два меха-
низма могут перекрываться друг с другом, так как всегда существует некото-
рый статистический разброс в поведении каждой индивидуальной моле-
кулы, тем не менее стереохимия, типичная для каждого случая, будет раз-
личной. Это различие следует из схемы, приведенной на рис. 42. Если при-
соединение и отщепление во времени перекрываются, как на схеме слева,
'С1
Переходное Промежуточное
состояние соединение
Конечный продукт
(с сохраненной кон-
фигурацией)
Конечный продукт
(с обращенной кон-
фигурацией)
Рис. 42. Стереохимия бимолекуляр-
ного нуклеофильного заме-
щения у олефинового атома
углерода в случае механиз-
мов с присоединением и с
отсутствием промежуточно-
го продукта присоединения.
В примерах, описанных в тек-
сте, Х=нуклеофильный анион I-,
RO-, RS- или N3, R = Н или
СН3; в ^ио-изомерах х =
n-NO2CeH4, CeH6SO2, CeH5SO
или СООС2Н8, а у—Н; в транс-
изомере х=Н, а у =n-NOoCeH4,
C6H5S02,CeH6SO,COOC2H5B слу-
чае групп n-N02CeH4, CetI5SO2H
CeH6SO замещение протекает с
сохранением геометрической
конфигурации, в то время как в
случае СООС2Н6 как активиру-
ющей группы образуется также
небольшое количество продукта
с обращенной конфигурацией;
цис- и транс-субстраты дают
различные смеси продуктов.
то должен образовываться продукт с полным сохранением геометрической
конфигурации. Вероятно, такой механизм наблюдали Миллер и Ионан при
замещении в ге-нитростирилбромиде и Модена при замещении в непредель-
ных сульфонах и сульфокислотах. Если присоединение и отщепление во вре-
мени разделены (схема справа на рис. 42), то промежуточный карбанион
с простыми связями, который при своем образовании первоначально должен
иметь конформацию, соответствующую цис- или транс-конфигурации суб-
страта, будет переходить в свою наиболее стабильную конформацию; поэтому
один или оба изомерных субстрата в результате замещения будут давать
определенное количество геометрически инвертированного продукта,
т. е. состав продуктов реакции будет в большей или меньшей степени прибли-
жаться к среднему. По-видимому, подобный случай наблюдал Вернон при
изучении реакции замещения между тиолат-ионом и этил-р-хлоркротона-
тами. Если промежуточный карбанион с простыми связями будет жить
достаточно долгое время и примет равновесную конформацию, то цис-
и транс-изомеры будут давать одну и ту же смесь продуктов замещения.
В указанных экспериментах этого не наблюдалось. Можно ожидать, что
положение каждого индивидуального механизма в ряду механизмов будет
зависеть от электроотрицательности активирующего заместителя, т . е. заме-
стителя с выраженными электроноакцепторными свойствами, которые облег-
462
Замещение в алифатическом ряду
чают образование промежуточного карбаниона и способствуют реакции
замещения.
Ясно, что в общем существует аналогия между указанными реакциями
и нуклеофильным ароматическим замещением, хотя сама по себе аналогия,
конечно, не может служить основой для формулирования механизма.
10. Электрофильное замещение в алифатическом ряду [216]
а. Область применения и наблюдаемые механизмы. Электроны насы-
щенного углерода по сравнению с ароматическим атомом углерода менее
доступны, поскольку они должны оттягиваться с локализованной о-связи.
В связи с этим не удивительно, что реагенты, обычно используемые при
ароматическом электрофильном замещении, не действуют подобным образом
на производные алканов.
Более позитивные предсказания можно сделать при сравнении реакций
нуклеофильного и электрофильного замещения в алифатическом ряду.
В одном процессе при уходе отщепляющейся группы освобождается место
для электронной пары реагента, а в другом — электронная пара при уходе
отщепляющейся группы остается. В реакциях первого типа входящая и ухо-
дящая группы субстрата перемещаются со своими электронами связи и обыч-
но являются анионами, такими, как окси-анионы или галогенид-ионы;
в реакциях второго типа эти группы входят и уходят без своих электронов
связи и поэтому обычно являются катионами, такими, как ионы металлов
или водорода. Вследствие того что множество простейших катионов пред-
ставляет собой катионы металлов, можно ожидать, что электрофильное
замещение в алифатическом ряду распространено в химии металлооргани-
ческих соединений, точно так же как нуклеофильное замещение распро-
странено в химии соединений, которые по контрасту мы могли бы назвать
неметаллоорганическими. Все металлы, связанные подходящим образом
с другими группами, являются потенциальными участниками таких реак-
ций, поэтому можно ожидать, что к электрофильному замещению в алифа-
тическом ряду будут относиться главным образом реакции замещения
металла на металл в алкилах металлов, а также представляющие особые
случаи реакции замещения водорода на водород.
Изучение механизма электрофильного замещения в алифатическом ряду
началось интенсивно проводиться только в конце 1950-х годов. Основой
для выяснения закономерностей электрофильного замещения в алифатиче-
ском ряду явилась (и почти не утратила этого значения в настоящее время)
реакция замещения ртути на ртуть; подобное же значение имеет изучение
нитрования для электрофильного ароматического замещения, проводив-
шееся в 1920—1940-х годах. В 1958 г. было найдено, что алкильные соеди-
нения ртути могут быть разделены на оптические антиподы, что обеспечи-
вало возможность исследовать стереохимию реакций замещения ртути.
Первым примером стабильной оптически активной молекулы, содержащей
один асимметрический атом углерода, у которого одним из четырех замести-
телей — металл, был втор-бутилмеркурбромид emop-C4H9HgBr; он был раз-
делен на оптические изомеры через манделат, затем подобным же образом
были разделены другие ртутьорганические соединения [217]. Это стерео-
химическое открытие привело к тому, что замещение в ртутьалкилах стало
исходным пунктом исследования закономерностей электрофильного замеще-
ния. Наибольший интерес представляет применение этих соединений при
изучении реакций замещения ртути на ртуть с применением меченых соеди-
463
Глава VII
нений, поскольку, когда входящий и уходящий атомы представляют собой
одни и те же элементы, отпадает необходимость дополнительного исследова-
ния с целью корреляции знаков вращения с относительной конфигурацией,
чего нельзя избежать при изучении замещения одного элемента на другой.
С помощью подобных исследований Хьюз и сотр. на 20 лет ранее установили
стереохимическое S ^-правило (разд. 7, г).
В это же время [218] было предсказано, что должны существовать три,
и только три, стехиометрически различные электрофильные реакции обмена
ртути, а именно: одно-, двух- и трехалкильные замещения, представленные
ниже. Двухалкильная реакция в то время была известна; вскоре были
открыты и две другие.
XjHg ^R^-HgX XHgR + HgX2 {одноалкильная)
XjHg ^SX-HgR XHgR + XHgR (двухалкильная)
XRHg^R)—HgR RHgR + HgRX (трехалкильная)
В это время были предсказаны три основных механизма электрофильного
алифатического замещения по аналогии с известными механизмами нуклео-
фильного замещения [218], а именно бимолекулярный (SE2), мономолеку-
лярный (SE1) и внутримолекулярный (SEi), приведенные ниже для стехио-
метрического случая одноалкильного обмена. Один из них — бимолеку-
лярный механизм — был открыт и полностью доказан почти сразу же.
Другие с некоторыми предсказанными видоизменениями были выявлены
в течение последующих нескольких лет.
X'Hg HgX"------> X'Hg—R + HgX" (Se2)
R—HgX" —----> R + HgX'
Медленно
X'Hg + R ----> X'HgR
Быстро
HgX"
*9 CY
X'Hf
R HgX"
I + I
X'Hg ¥
(SE1)
(SEi)
Эти механизмы выявлены кинетически. ЭЕ2-Механизм обусловливает второй
кинетический порядок. В этом случае переходное состояние является «откры-
тым». 8Е1-Механизм также обусловливает второй кинетический порядок, однако
в этом случае переходное состояние является «закрытым». Наиболее очевидное
кинетическое различие между этими механизмами состоит в том, что если в за-
мещающем агенте X'HgY'первым ионизуется Y', то по всем причинам ско-
рость ЭЕ2-реакции в ряду ртутных солей с уменьшающейся силой коорди-
нации, например HgBr2, Hg(OCOCH3)2, Hg(NO3)2, Hg(C104)2, должна сильно
возрастать, тогда как скорость ЭЕ1-реакции будет равна нулю, если Y' пол-
ностью ионизован. Поскольку координационная сила Y' способствует
оттягиванию отщепляющегося атома ртути, в том же самом ряду скорость
реакции должна сильно уменьшаться. Механизм SB1 можно сразу опреде-
лить по кинетической форме: первый порядок по субстрату и нулевой
по замещающему агенту.
464
Замещение в алифатическом ряду
Что касается предсказания стереохимии [218], то 3Е2-'правило не может
быть основано на принципе Паули, поскольку при бимолекулярном электро-
фильном замещении на изменяющейся атомной орбитали имеется лишь два
электрона, т. е. нет избыточных электронов, способных с ними конкуриро-
вать, в отличие от нуклеофильного замещения, где наличие этого фактора
заставляет располагаться обменивающиеся группы порознь друг от друга,
что приводит к бимолекулярному замещению с инверсией. Каков бы ни был
действительный путь бимолекулярного замещения, он должен соответ-
ствовать балансу сил, которые a priori невозможно оценивать простым
и достаточно определенным способом. Что касается пространственной напра-
вленности реакций мономолекулярного электрофильного замещения,
то в этом случае можно ожидать рацемизации, хотя в действительности
точная геометрия карбаниона неизвестна, а если она пирамидальна, то неиз-
вестна частота инверсии пирамиды. Внутримолекулярное электрофильное
замещение должно происходить с полным сохранением конфигурации,
поскольку переходное состояние циклическое, а цикл слишком мал, чтобы
включать углы, обеспечивающие инверсию.
б. 8Е2-Механизм обмена ртути. В 1952—1957 гг., т. е. до того, как
был в основных чертах выяснен механизм обмена ртути, появились сообще-
ния Райта, Несмеянова, Уинстейна, Реутова и др. [219], посвященные стерео-
химии этих реакций. Выводы были сделаны, однако они не были хорошо
обоснованы, во-первых, потому, что в качестве примеров были взяты ртуть-
замещенные терпены и другие соединения, обладающие несколькими асим-
метрическими центрами в оптически активной форме, так что каждая реак-
ция с участием атома ртути происходила при постоянном влиянии асим-
метрических центров. Вторая причина, не позволяющая удовлетвориться
сделанными выводами, была фундаментальной и состояла в том, что про-
странственная направленность любой реакции является свойством меха-
низма, а не стехиометрии, и поэтому доказать стереохимию без доказатель-
ства механизма — это все равно, что найти утерянную шляпу и не найти
ее владельца. Вначале должна быть выяснена кинетика кинетически простой
реакции и лишь затем будет польза от стереохимических исследований,
которые необходимо скоррелировать с кинетикой. С 1958 г. стереохимия
обмена ртути была изучена на примере соединений с одним асимметрическим
центром, и данные были сопоставлены с более ранними кинетическими иссле-
дованиями Реутова и сотрудников и Хьюза с сотрудниками.
Первая такая работа была посвящена замещению двухалкильного стехио-
метрического типа [220], например реакции солей ртути с ди-втор-бутил-
ртутыо в ацетоне или этаноле
emop-(C4H9)2Hg4-HgX2 —> 2emop-C4H9HgX (SE2)
В ацетоне или этаноле при X = Br, ОСОСН3, NO3 реакция имела первый
порядок по каждому из реагентов, т. е. общий второй порядок. Скорость
замещения в указанном ряду анионов X увеличивалась так быстро, что для
установления кинетической зависимости при переходе к каждому следую-
щему аниону нужно было понижать температуру. В этаноле скорости имели
следующие значения: для HgBr2 при 25 °C 0,39, для Hg(OCOCH3)2 при
0 °C 5,3 и для Hg(NO3)2 при —47 °C 7,6 л-моль"1-с-1. В случае Hg(C104)2
при —50 °C скорость была настолько велика, что кинетику нельзя было
исследовать. Эти данные соответствуют механизму SE2.
Затем было установлено, что при таком механизме конфигурация сохра-
няется. Оптически активный втор-бутилмеркурбромид с помощью втор-
30-0772
465
Глава VII
бутильного соединения Гриньяра (рацемического, поскольку соединения
Гриньяра оптически нестабильны) был превращен в ди-втор-бутил ртуть,
которая поэтому имела оптически активную метку лишь в одной из двух
в прочих отношениях эквивалентных алкильных группах. В приведенных
ниже уравнениях знак градуса обозначает оптическую метку
emop-CaHgHgBr + emop-CiiHgMgCl —> emop-C^H-gHgC^g-emop 4-MgClBr
Это соединение затем было использовано в качестве субстрата для исследо-
вания обмена ртути с солями ртути в определенных кинетических условиях.
В случае бромида ртути(П) обмен должен приводить, как видно из уравнения
о о
emop-C4H9HgC4Hg-emop4-HgBr2 —> emop-C4H9HgBr4-emop-C4H9HgBr
к втор-бутилмеркурбромиду, имеющему удельную активность, равную
половине активности вещества, введенного в реакцию Гриньяра. Точнее
говоря, удельная активность должна составлять половину активности
исходного ewzop-бутилмеркурбромида при условии, если оптическая метка
на алкильной группе сохраняется во время замещения атома ртути, свя-
занного с этой группой, на другой атом, т. е., другими словами, если при
замещении конфигурация количественно сохраняется. Если замещение
должно приводить к рацемизации, то индивидуальные молекулярные акты
замещения будут давать каждый энантиомерный продукт с равной вероят-
ностью и конечная удельная активность будет составлять одну четверть
от первоначальной. Если замещение происходит с инверсией (как бимолеку-
лярное нуклеофильное замещение), то конечная активность должна быть
равной нулю. Как показывают данные табл. 123, наблюдаемая оптическая
Таблица 123
Стереохимия 8Е2-реакций двухалкильного обмена ртути
между солями ртути и ди-вшор-бутилртутью
Исходный emop-C4H9HgBr [а$ -15,2°
Конечный emop-C4HgHgBr
при сохранении конфигурации [а]Ь° — 7,6°
при рацемизации [«]Ь° — 3,8°
при инверсии [а]Ь° ± 0,0°
Замещающий агент Растворитель [а]о конечного emop-CiHeHgBr, град.
HgBr2 C2H5OH — 7,6
HgBr24-3LiBr C2H6OH — 7,8
Hg(OCOCH3)2 C2H6OH -7,5
Hg(NO3)2 C2H5OH — 7,8
Hg(NO3)2+ HNO3 50%H2O- 50%C2H5OH -7,2
активность составляет половину от исходной для реакций с HgBr2,
Hg(OCOCH3)2 и Hg(NO3)2, причем в продуктах последних двух реакций
для поляриметрических измерений была проведена замена аниона на бромид-
ион. Аналогичные результаты были получены для 5-метил-2-гексильного
соединения, в котором алкильная группа имела асимметрию у атома угле-
рода, связанного со ртутью.
466
Замещение в алифатическом ряду
Если оптически активный втор-бутилмеркурбромид смешать в этаноле
с оптически неактивной ди-втор-бути л ртутью, последняя приобретает актив-
ность. Если втор-бутилмеркурбромид, содержащий 203Hg, смешать в этаноле
с обычной ди-втор-бутилртутью, то последняя становится радиоактивной.
Такие переходы меток, приведенные на следующих схемах (звездочка обо-
значает радиоактивную метку), свидетельствуют в пользу трехалкильного
обмена ртути
RHgR-f-RHgBr —> RHgR + RHgBr
RHgR + RHg*Br RHg*R + RHgBr (Se2)
Эти уравнения показывают, что переходы обеих меток должны происходить
с одинаковой скоростью, что и наблюдается в действительности [221].
В принципе переходы меток можно было бы представить и как результат
двух последовательных стадий двухалкильного обмена, однако в этом случае
оптическая метка переходила бы в два раза быстрее, чем радиоактивная
метка, согласно схеме:
2RHgBr
R9Hg 4~HgBr2
Медленно о о
-------> RHgR + HgBr,
Быстро
-------> 2RHgBr
Медленно
2RHg*Br ------------> RHg*R + Hg*Bra
R2Hg + Hg*Br2 RHg*Br + RHgBr
(не обнаружено)
Одинаковые скорости распределения обеих меток свидетельствуют в пользу
простого трехалкильного обмена. С помощью радиометрических измерений
в этаноле был установлен общий второй порядок, первый по каждому
из реагентов. При переходе от emop-C4H9HgBr к emop-C4H9HgOCOCH3
и затем к emop-C4H9HgNO3 скорость реакции, как и в предыдущем случае,
резко возрастала. Очевидно, реакция происходит по механизму SE2, как
указано выше. Итак, сравнение радиометрического и поляриметрического
измерений кинетики показывает, что трехалкильное замещение также
характеризуется полным сохранением конфигурации.
Одноалкильный обмен ртути, например в этаноле
RHgBr + Hg*Br2 —> RHg*Br + HgBra (SB2)
был обнаружен при использовании солей радиоактивной ртути еще до того,
как была понята его природа: первоначально его рассматривали как резуль-
тат двух стадий двухалкильного замещения [222]:
Медленно
2RHgBr -------> R2Hg4-HgBr2 "J
Быстро ? (не Реализуется)
R2Hg-[-Hg*Br2 ---—RHg*Br-f-RHgBr J
Однако кинетические исследования показывают [223], что реакция пред-
ставляет собой одноалкильную стехиометрическую форму обмена ртути,
проходя через стадию, которая в этаноле имеет первый порядок по каждому
из реагентов, т. е. общий второй порядок. Это было установлено для алкил-
ртутных солей с такими алкильными группами, как метильная и втор-
бутильная (а впоследствии и с другими группами), и с такими анионами,
как I", Вг-, СН3СОО“, NO"; эти же анионы входили в состав солей ртути.
В указанном ряду анионов скорость реакции опять-таки сильно возрастала.
Наконец, с помощью оптически активных солей emop-бутилртути было
зо* 467
Глава VII
показано, что обмен, исследованный радиометрически, далеко проходит за
полупериод без потери оптической активности. Это показывает, что меха-
низм реакции снова относится к типу Se2 и что этот механизм и в случае
одноалкильной стехиометрической формы обмена приводит к полному сохра-
нению конфигурации.
Влияние размера и строения алкильной группы на бимолекулярный электро-
фильный обмен ртути между алкильными соединениями ртути и ионами
ртути можно понять на основе первичного пространственного эффекта.
Однако этот эффект проявляется совсем иным образом, чем при нуклео-
фильном замещении в алифатическом ряду (разд. 8). Причина состоит в том,
что геометрическая форма переходного состояния, приводящая к сохране-
нию конфигурации при бимолекулярном электрофильном замещении, сильно
отличается от соответствующей формы переходного состояния бимолекуляр-
ного нуклеофильного замещения, при котором происходит обращение кон-
фигурации. Пространственное замедление при бимолекулярном электро-
фильном замещении по сравнению с нуклеофильным замещением довольно
мало, что и следовало ожидать, учитывая меньшую концентрацию электро-
нов у места замещения при электрофильном замещении. Хьюз и Фольгер
[224] установили, что в реакциях алкилмеркурбромидов с бромидом радио-
активной ртути(П) а-метилирование алкильной группы приводит к умень-
шению скорости в 3...7 раз на каждой ступени при переходе от метильной
к первичной алкильной и далее к вторичной алкильной группе; однако
влияние |3-метилирования на скорость выявить трудно. В частности, неопен-
тильная группа не занимает особого положения в ряду первичных алкиль-
ных групп: скорость обмена ртути в неопентилмеркурбромиде составляет
около 3/4 скорости обмена в этилмеркурбромиде. Чего-то в этом роде и следо-
вало ожидать, поскольку в переходном состоянии бимолекулярного электро-
фильного замещения, приводящего к сохранению конфигурации, обе обме-
нивающиеся группы расположены по одну сторону области замещения,
а другая сторона, где располагается алкильная группа, остается полностью
свободной.
Реутов и сотр. [225] изучили полярные влияния заместителей в аралкил-
ртутных солях на скорость их обмена с солями ртути, имеющего второй
порядок. Эти эффекты также оказались очень небольшими; они меняли свое
направление в зависимости от природы взятого соединения. При обмене
n-замещенных бензилмеркурбромидов с бромидом ртути(П) в хинолине
электроположительные заместители слегка ускоряли, а электроотрицатель-
ные — слегка замедляли реакцию. Полярные влияния на скорость имею-
щего второй порядок обмена н-замещенных а-карбэтоксибензилмеркурбро-
мидов с бромидом ртути(П) в 80%-ном водном растворе этанола также были
невелики, но имели противоположное направление. Очевидно, здесь имеет
место тонкий баланс между импортом и экспортом электронов у области
замещения в переходном состоянии бимолекулярного электрофильного
замещения.
в. Sgi-механизм при обмене ртути. При изучении кинетики обмена ртути
между алкилртутными солями и солями ртути в этаноле и ацетоне наблю-
дался катализ анионами.[Наличие катализа свидетельствует о каком-то новом
механизме. Каталитическая эффективность уменьшалась в ряду 1~ > Вг- >
> Cl~^> СН3СОО-> NO3 [226]. Эта последовательность соответствует меха-
низму типа Sgi (разд. 10,а). Кинетическая форма катализа для обмена между
метилмеркурбромидом и бромидом ртути(П) при катализе бромистым литием
в ацетоне приведена на рис. 43. Скорость линейно возрастала с ростом кон-
центрации бромистого лития, затем кривая резко меняла направление
468
Замещение в алифатическом ряду
и далее линейность сохранялась во всем исследованном интервале кон-
центраций. Наклон прямой после изменения ее направления не во всех
случаях был больше первоначального. В случае неопентильной группы после
излома наклон уменьшался (рис. 44). В случае emop-бутильной группы
наклон после излома был меньше при катализе бромидом и гораздо меньше
при катализе ацетатом [226]. Однако излом на прямой всегда имел место
при соотношении концентраций реагентов и аниона, равном 1:1; второго
излома не наблюдалось.
Указанные кинетические характеристики соответствуют механизму, прояв-
ляющемуся в каждом случае. Имеются два каталитических процесса.
Рис. 43. Влияние концентрации
LiBr на скорость реак-
ции замещения второго
порядка CH3HgBr +
Hg*Br2 (каждого по
0,095) в этиловом спирте
при 0 °C (J. Chem. Soc.,
1961, 1144).
Рис. 44. Влияние концентрации LiBr
на скорость реакции замеще-
ния второго порядка
(CH3)3CCH2HgBr+HgBr2 (ка-
ждого по 0,092) в этиловом
спирте при 100 °C (J. Chem.
Soc., 1961, 2361).
При низких концентрациях катализатора в предравновесной стадии к реа-
генту присоединяется один анион, который затем участвует в образовании
переходного состояния ртутного обмена. При более высоких концентра-
циях катализатора один из реагентов в предравновесной стадии захваты-
вает второй анион, который затем также входит в переходное состояние.
Эти два каталитических процесса, в переходное состояние одного из кото-
рых входит один, а другого — два дополнительных аниона, носят на-
звания одноанионного и двуханионного катализа одноалкильного обмена
(рис. 43 и 44).
При одноанионном катализе бромид-ионом предравновесие имеет следующий
вид:
HgBr2+Br~ HgBrj
Равновесие, очевидно, сдвинуто вправо, так как, если оно сдвинуто влево,
от него не было бы никакой пользы, а на кривой в точке 1,1-эквивалентности
не наблюдалось бы перегиба. Если бы равновесие находилось где-то посере-
дине, то не было бы линейности. Вторым предравновесием при двуханионном
катализе могла бы быть реакция
RHgBr4-Br- Т» RHgBrj
469
Глава VII
Также очевидно, что это равновесие сдвинуто влево, так как, если бы оно
было сдвинуто вправо, на прямой должен был бы получиться второй пере-
гиб в точке 2,1-эквивалентности, а этого не наблюдается; если бы положение
равновесия находилось посередине, не наблюдалось бы линейности.
Способы, которыми дополнительные анионы могли бы быть включены в пере-
Переходное состояние
для SEi-механизма
(одноанионный катализ)
Переходное состояние
для SEi-механизма
(двуханионный катализ)
Циклическая природа этих переходных состояний также следует из кине-
тических данных. Если рассмотреть случай X = Y = Вг, то на схеме пере-
ходного состояния для одноанионного катализа Y должен, по уже рас-
смотренным причинам, переходить от нижнего из двух атомов ртути,
т. е. от входящего атома. Достаточно только написать химическое уравнение
обмена, чтобы увидеть, что Y должен переходить к верхнему из двух атомов
ртути, т. е. к уходящему атому. Поэтому структура переходного состояния
должна отображать переход аниона Y от одного атома ртути к другому,
т. е. Y должен быть мостиком между двумя атомами ртути. Таким образом,
имеем циклическое переходное состояние и механизм одного из общих пред-
сказанных типов, который после его открытия был назван SEi. При двух-
анионном катализе дополнительный анион должен переходить к тому
из атомов ртути, который координационно менее насыщен. Этот механизм
представляет собой другой частный случай общего механизма SEi.
Очевидно, что эти переходные состояния должны приводить к сохранению
стереохимической конфигурации при замещении. Обе формы катализа
можно с некоторой вероятностью отличить друг от друга подбором соот-
ветствующих условий; в случае в/пор-б утильной группы было показано,
что при обоих механизмах конфигурация количественно сохраняется.
Эти механизмы изучались также с точки зрения кинетических эффектов
гомологии алкильных групп [224]. В случае простых алкильных групп,
вплоть до неопентильной, эти эффекты качественно сходны с эффектами,
обнаруженными в Эк2-обменах. Однако для высших гомологов уменьшение
скорости в общем более значительно при одноанионных ЭЕ1-обменах и менее
значительно при двуханионных 3Е1-обменах. Этот факт можно объяснить
отталкиванием анионов, не лежащих в плоскости переходного состояния.
Грубые расчеты пространственного отталкивания, основанные на предпола-
гаемых структурах переходного состояния, дали результаты, согласую-
щиеся с опытными данными.
г. 8Е1-Механизм обмена ртути. Кинетически установленный мономоле-
кулярный обмен ртути наблюдался почти одновременно Реутовым и сотр.,
а также Хьюзом, Робертсом и автором на одном и том же примере замещения
ртути в а-карбэтоксибензилмеркурбромиде при действии бромида радио-
активной ртути(П) в диметилсульфоксиде [227]. Работа Хьюза и сотр. [228]
была опубликована позже, однако она включала разделение субстрата
на оптические изомеры, стереохимию замещения и изучение катализа.
470
Замещение в алифатическом ряду
В основу обоих исследований легла одна и та же мысль, а именно: реакция
по механизму SE1 будет облегчаться при увеличении легкости образования
карбаниона, а этого можно добиться, используя вместо простых алкильных
групп группы с электроотрицательными заместителями и применяя раство-
ритель, способный сольватировать карбанион, не разрушая его. (Природа
растворителя очень важна для механизма: та же реакция происходит
по механизму SE2 в водном растворе ацетона [228], в водном растворе этанола
[229] и пиридине [230].) Реутов использовал тот же принцип и в случае
другого субстрата — в реакции ге-нитробензилмеркурбромида с бромидом
радиоактивной ртути(П) в диметилсульфоксиде [231]. Замещение также
происходило по механизму SE1, однако в этом случае нельзя было легко
осуществить стереохимические исследования.
Ключевым моментом является первый порядок замещения по субстрату
и нулевой по замещающему агенту. Это и все косвенные доказательства
позволяют сделать вывод, что реакции этих субстратов в диметилсульфоксиде
происходят по механизму SE1, например:
c6H5s Медленно CgH54 ч
;ch- HgBr - > )CH~+HgBr+
C2H3OOC' C2H5OOC'
CeH5x * CeHjx *
>CH- + HgBr2 -> )CH-HgBr + Br-
СгЩООС/ CaHjOOCZ
(SE1)
После разделения субстрата на оптические антиподы было найдено, что
указанный процесс замещения происходит с полной рацемизацией. Поляри-
метрическая и радиометрическая скорости обмена были равны; это позво-
лило сделать вывод, что каждый молекулярный акт замещения приводит
с равной вероятностью к любой энантиомерной форме продукта замещения.
д. 8Е1-2Х"-Механизм обмена ртути. Замещение а-карбэтоксибензил-
меркурбромида бромидом радиоактивной ртути(П) в диметилсульфоксиде
сильно катализируется бромид-ионом. Этот катализ проявляется на контро-
лирующей скорость стадии образования промежуточного карбаниона,
и поэтому этот случай является одной из форм мономолекулярного меха-
низма. Однако катализ означает проявление нового механизма [228], и этот
механизм, четвертый из известных для обмена ртути, не был четко пред-
сказан в 1958 г. Очень интересно, что скорость этого обмена пропорцио-
нальна квадрату концентрации бромид-иона и что, хотя в принципе должен
быть член, линейно связанный с концентрацией бромид-ионов, этот член
был слишком мал и не был обнаружен. Таким образом, каталитический
механизм, обозначаемый SEl-2Br- или, в более общем виде, SE1-2X-, вклю-
чает два бромид-иона, связанные с субстратом. Очевидно, что аддукт с одним
анионом недостаточно нестабилен, чтобы быть хорошим промежуточным
соединением каталитической реакции, однако аддукт с двумя ионами доста-
точно нестабилен, вероятно, вследствие того, что двойной отрицательный
заряд может при последующем гетеролизе расщепляться, давая два взаимно
отталкивающихся аниона, один из которых является карбанионным проме-
жуточным соединением. Этот механизм приводит к полной рацемизации.
Быстро Быстро
RHgBr ......—» RHgBr2 .......RHgBr|”
Медленно
RHgBr?,"-------> R- + HgBrj
* Быстро *
R~ + HgBr2-------> RHgBr+Br~
(Se1-2X-)
471
Глава VII
Этот каталитический механизм также, вероятно, впервые был обнаружен
Реутовым и сотр. [232] на примере мономолекулярного обмена между п-нит-
робензилмеркурбромидом и бромидом радиоактивной ртути(П) в диметил-
сульфоксиде. Имеется лишь одно кинетическое отличие от предыдущего
случая, которое можно интерпретировать, предположив, что в отличие от
предыдущих случаев, в которых положение предравновесия было сдвинуто
влево, в данном случае первое предравновесие находится ближе к середине,
тогда как второе — еще сдвинуто влево. Наблюдения показывают, что ско-
рость реакции первого порядка при увеличении концентрации бромид-ионов
постепенно возрастает в соответствии с плавной кривой, однако эта кривая
сразу же становится гораздо более крутой, когда концентрация бромид-иона
становится близкой к концентрации бромида ртути(П).
е. Другие реакции замещения металла на металл. Мы только слегка
коснемся этой большой темы, приведя несколько примеров. К одной катего-
рии можно отнести изучение реакций соединений других металлов группы Б
с соединениями ртути или с другими реагентами. К другой категории отно-
сятся реакции соединений переходных металлов четвертого периода.
Замещение золота(1) на ртуть(П) было изучено на примере комплексных
соединений золота с фосфинами с использованием солей ртути или алкил-
ртути в качестве замещающих агентов. В случае соединений алкилзолота(1)
с простыми алкильными группами в таких растворителях, как диоксан,
водный диоксан и диметилсульфоксид, наблюдалась реакция с кинетикой
второго порядка, первого по каждому из реагентов [233]. Путем измене-
ния аниона в замещающем агенте в сторону менее ковалентно связанных,
т. е. более ионизованных анионов, и добавления общих анионов удалось
выяснить, что в этих реакциях замещения реализуется механизм SE2, на-
пример:
Диоксан _
(CeH5)3P-AuCH3+HgBr2 (CeH5)3p.AuBr+CH3HgBr (SE2)
дмсо /г,
(С6Н5)3Р. AuC2H5+CH3HgBr -----> (C6H5)3P.AuBr+C2H5HgCH3 (SE2)
Замещение золота(Ш) на ртуть(П) в случае фосфиновых комплексов золото-
органических соединений с простыми алкильными группами, вероятно,
происходит аналогично замещению одновалентного золота [233]. Механизм
реакций также относится к типу SE2, например:
(CeH5)3P.Au(CH3)34-HgBr2 ---> (C6H5)3P-Au(CH3)2Br + CH3HgBr (SE2)
Мономолекулярный механизм SE1 наблюдается при наличии в золотооргани-
ческом соединении электроотрицательных групп [233]. Реакция (С6Н5)3Р-
•АиС(СК)(СООС2Н5)(СН2)зСНз с CH3HgCl в диметилсульфоксиде при О °C
шла мгновенно. Хотя этот факт сам по себе ни о чем не говорит, можно
быть вполне уверенным, что в этом случае реакция происходит по меха-
низму SE1. При использовании смешанного растворителя, содержащего
только 10% диметил сульфоксида и 90% диоксана, скорость реакции в доста-
точной степени замедляется, и поэтому можно измерить кинетику реакции.
Кинетические исследования показывают, что в этом растворителе реакция
происходит по механизму SE1. Реакция имеет первый порядок по 1-циан-
1-карбэтокси-н-амилзолоту и нулевой — по метилмеркурхлориду. При
замене метилмеркурхлорида на этилмеркурхлорид или метилмеркурацетат
472
Замещение в алифатическом ряду
абсолютная скорость не меняется:
CN
| Медленно
(СвН5)3Р. Аи(СН2)3СН3 ------>
СООС2Н5
r-CN -л-
| Быстро
С(СН2)3СН3 +CH3HgCl------------>
-СООС2Н5 -
Быстро
[(СвН5)3РАи]+ + С1----->
[(С6Н5)3РАи]+ +
r-CN -л
I
С(СН2)зСН3
-dooc2H5 -
CN
CH3HgC(CH2)3CH3+Cl-
СООС2Н5
(CeH5)3PAuCl
(SE1)
Реакция замещения ртути(П) на олово(1У) в случае субстрата диметил-
ртути и замещающего агента триметилстаннилбромида имеет второй кинети-
ческий порядок в ацетонитриле, диметилформамиде и диметилсульфок-
сиде [234];
CH3HgCH3+(CH3)3SnBr -> CH3HgBr+(CH3)3SnCH3 (S£2)
Аналогичную кинетику имеет реакция обмена олова(1У) на олово(1У) между
тетраметилоловом и ионизированной солью трифторацетатом триметил-
олова, изученная радиометрически в ацетонитриле [234]:
(CH3)3SnCH3+(CH3)JSn+CF3COO- —> (CH3)£SnCH3 + (CH3)3Sn+CF3COO- (SE2)
Солевые эффекты свидетельствуют в пользу 3Е2-механизма.
Замещение таллия(Ш) на ртуть(П) в случае триалкилталлиевых соединений
и солей алкилртути в диметилформамиде происходит слишком быстро, так
что кинетику изучить нельзя. Хотя механизм этих реакций не установлен,
вероятно, эти процессы относятся к типу SE1 [235]. Самая медленная стадия
процесса — отщепление карбаниона R~ от триалкилталлия R3T1 — с прак-
тической точки зрения слишком быстра, но тем не менее, вероятно, именно
она и является стадией, определяющей скорость
(С2Н5)3Т1
e7nop-C4H9HgNO3
С2Н? + emo p-C4H9Hg+
Быстро, но определяет
скорость
ZZTt С2Н3 + (С2Н5)2Т1+
Очень быстро
--------------------> eznop-C4HgHg+ + NO3
Очень быстро
--------------------> eTnop-C4H9HgC2H5
(SE1)
Замещение таллия(Ш) на таллий(Ш), изученное радиометрически, в случае
реакции триалкилталлия с ионом диалкилталлия * в диметилформамиде
также происходит очень быстро. R этом случае механизм реакции также
не доказан, однако вполне вероятно, что он относится к типу SE1.
Быстро, но определяет |
скорость
(С2Н5)3Т1 <............ > С2Н3 (С2Н5)2Т1+ (SE1)
* Очень быстро [
С2Ну + (С2Н5)2Т1+ > (С2Н5)3Т1* J
Несмотря на путаные ранние данные, касающиеся молекулярного веса
реагентов Гриньяра, в настоящее время удалось выяснить, что эти соеди-
* Ион диалкилталлия R2T1+ (который изоэлектронен диалкилртути) очень стабилен.
Диалкилталлиевые соли сильных кислот представляют собой сильные электролиты
в воде, если они в ней растворимы, и довольно сильные электролиты в диметилформ-
амиде.
473
Глава VII
нения, особенно бромиды, в сильнокоординирующихся эфирных раствори-
телях, вероятно, мономерны, однако в менее координирующихся раствори-
телях они могут димеризоваться (особенно хлориды) путем образования
галогенных мостиков [236]. В кристаллическом виде реактивы Гриньяра
содержат две молекулы эфира на один атом магния, причем оказалось, что
в тех соединениях, которые были исследованы рентгеноструктурным методом,
а именно в CeH5MgBr-[О(С2Н5)2]2 и C6H5MgBr>[О(С2Н5)2]2, атом магния
является тетраэдрическим [237]. Робертс и сотр. [238] показали, что в эфире
реактивы Гриньяра находятся в виде стабильных компонентов равновесия
Шленка (равновесие двухалкильного обмена магния, гл. XIII, разд. 2,в)
и что это равновесие в отличие от аналогичного равновесия обмена цинка
устанавливается быстро.
R2Mg + MgX2 2RMgX
Радиоактивный магний, введенный в такую систему, статистически распре-
деляется быстрее, чем можно провести разделение компонентов [238].
Хотя механизм этой обратимой реакции точно не выяснен, согласно стерео-
химическим данным Робертса и сотр. [238], полученным при помощи метода
протонного магнитного резонанса, можно предполагать, что реализуется
механизм SE1, т. е. определяющей скорость, хотя и быстрой, стадией
является образование карбаниона. Оказалось, что в растворе неопентиль-
ного соединения Гриньяра (CH3)3CCH2MgCl в эфире метиленовые атомы
водорода инвертируются; этот процесс привел бы к рацемизации, если бы
а-углеродный атом имел асимметрию и был бы центром оптической актив-
ности. Время инверсии было порядка миллисекунд при комнатной темпера-
туре. При добавлении к эфирному раствору диоксана осаждался хлористый
магний, и поэтому приведенное выше равновесие сдвигается влево и,
таким образом, в растворе накапливается почти чистый диалкилмагний
[(CH3)3CCH2]2Mg. В этом соединении метиленовые атомы водорода также
претерпевают стереохимическую инверсию, время которой в этом случае
составляет порядка секунд при комнатной температуре. С этими данными
согласуется следующий механизм установления равновесия двухалкиль-
ного обмена магния:
RMgCl R--|-MgCl+
RMgCl RMg+ + Cl-
Cl- + MgCl+ MgCl2
R_-|-RMg+ RzMg
К второй группе реакций электрофильного замещения металла на металл
относятся реакции октаэдрических комплексов переходных металлов, напри-
мер хрома(Ш) и кобальта(Ш). Эти реакции изучал Джонсон, который
использовал водорастворимые алкильные группы, имеющие заместители
с катионным зарядом, что позволяло в качестве растворителя применять
воду. Алкильная группа была связана с ионом переходного металла, кото-
рый был акватирован и имел октаэдрическую конфигурацию. Алкильные
группы принадлежали к пиридиометильному типу, например 4-пиколиниевые
группы
HN^ СН2— и CH3N^__________________СН2 —
и их 2- и 3-изомеры.
474
Замещение в алифатическом ряду
Таким образом Кумбс и Джонсон [239] изучили, например, ряд реакций
замещения хрома(Ш) на ртуть(П) в воде. Кинетически были исследованы
следующие реакции (Рут+ = 4-пиридиометил, Х_ = С1~, Вг- или NO”):
Pym+-Crnl(H2O)i+ + HgX2 -> Pym+-HgX + Cr(H2O)3+ + X-'
Pym+-Crni(H2O)i+ + HgX? -> Pym+-HgX+Cr(H2O)|+H-2X- (SE2)
Pym+ — Crln(H2O)i+ + HgXr -> Pym+-HgX+Cr(H2O)’++3X- J
Согласно кинетическим данным, все реакции имеют механизм SE2 даже
тогда, когда замещающим агентом являются комплексные анионы ртути(П).
Ни одна из реакций не относилась к типу SEi, при котором один из анионов,
отщепляющихся от ртути, должен координироваться с атомом хрома. Кроме
кинетических данных, это следовало из того факта, что неорганическим
продуктом реакции всегда являлся Cr(H2O)f+ даже в тех условиях, когда
образовавшиеся Сг(Н2О)5С12+ и Сг(Н2О)5Вг2+ могли бы быть достаточно
устойчивыми. Другой факт, характеризующий эти реакции, состоит в том,
что при X” = С1” скорости всех трех реакций равны в пределах порядка,
а при X” = Вг- в пределах двух порядков. Если бы замещаемая группа
была нейтральной из-за электрохимических причин, можно было бы ожи-
дать, что скорость электрофильного замещения резко уменьшалась бы при
увеличении отрицательного заряда замещающего агента. Однако, очевидно,
что, когда замещаемая группа имеет большой положительный заряд, влия-
ние кулоновского притяжения противоположно заряженных субстрата
и замещающего агента на кинетику реакции не проявляется.
ж. Замещение металла на водород. Для этих реакций характерны
механизмы SE2, SE1 и SE1-2X", а также другие механизмы, не обнаружен-
ные в реакциях замещения металла на металл.
Сначала приведем несколько примеров кислотного протолиза соединений
металлов побочных групп. Описан ряд реакций кислотного протолиза солей
алкилртути [240], диалкилртутных соединений [241] и тетраалкилсвинца
[242], т. е. замещения типа Н(1) — Hg(II) и Н(1) — Pb(IV). Реакции имеют
второй кинетический порядок и почти наверняка протекают по механизму
Se2, например:
Водный диоксан
CH3HgI + HC104 --------------> CH4+HgI+
Вода
(CH2 = CHCH2)2Hg+HC104 ------> CH2 = CHCH3 + CH2 = CHCH2Hg+
Уксусная
(C2H5)4Pb + нсю4 ——> С2Н6 + (С2Н5)3РЬ+
KMLJ1O1 d
(Se2)
Учитывая данные Робертса [238], согласно которым реактивы Гриньяра
и диалкилмагниевые соединения, вероятно, обменивают магний в эфире
или диоксане по механизму SE1, можно предполагать, что эти соединения
протолитически разлагаются также по этому механизму.
Механизм SE1-2X_ был найден Кодом и Джонсоном [243] при протолизе
4-пиридиометилмеркурхлорида в присутствии хлорид-ионов в воде. Был
обнаружен конкурирующий механизм SE1-X”, при котором в реакции уча-
ствует только один хлорид-ион и который не реализуется в реакциях обмена
металла на металл. В предравновесной стадии происходит захват анионов,
а затем от образующихся комплексных ионов спонтанно, но медленно отще-
пляется хлормеркургруппа и образуется карбанионный центр на углероде,
475
Глава VII
который быстро протонируется. Можно записать следующие два механизма:
Pym+HgCl ;•» Pym+HgClj 7^ Pym+HgCl|-
Медленно , Л
Pym+HgClj-----------> Pym± + HgCl2 > (SE1-X“)
Pym±_|-H+-----------> HPym+ (4-пиколиний) J
Медленно >
Pym+HgCl’^----------> Pym±4-HgClj I (Se1-2X“)
Pym±4-H+ -----------> HPym+ (4-пиколиний)J
Скорость реакции связана с концентрацией хлорид-ионов линейно или
квадратично в зависимости от механизма, но в каждом случае не зависит
от кислотности, т. е. пиколиновый остаток остается в катионной форме.
Особый механизм, включающий прототропное изменение, применим к про-
толизу а-карбэтоксибензилмеркурхлорида (или бромида) в водных органи-
ческих растворителях [244]. Чистый субстрат не изменяется при кипячении
в разбавленной хлорной кислоте, однако небольшие концентрации галоге-
нид-ионов приводят к быстрому протолизу даже при О °C. Скорость реак-
ции зависит от квадрата концентрации галогенид-ионов и квадрата кон-
центрации ионов водорода; она также зависит от обратной величины
концентрации неорганической ртути(П). В приводимой интерпретации пред-
полагается, что за предравновесным захватом двух хлорид-ионов следует
предравновесие протолиза ртутного остатка. Это обеспечивает второй поря-
док по хлорид-ионам, первый по ионам водорода и обратный по ртути(П).
Органический продукт автоматически перегруппировывается, в резуль-
тате чего образуется енольная форма карбэтоксигруппы. Возврат в обыч-
ную форму, вероятно, происходит не быстро (гл. XI) и катализируется
ионами водорода. Это объясняет, почему в выражение для скорости
входит концентрация ионов водорода во второй степени. Превращение
енола в обычную форму карбоксила рассматривается как контролирующая
скорость стадия в цепи последовательных реакций, которая до образования
енольного соединения, т. е. до стадии ацидолиза ртути, представляет собой
катализуемый двумя анионами бимолекулярный протолиз с перегруппи-
ровкой SE2'-2X~; этот механизм не известен в случае замещения металла
на металл.
с2н5оосх с2н5оосх с2н5оосх
^CH-HgCl-^± CH-HgCl,'^ THHgClr
свн5/ с6н/ свн5/
С2Н5ООСХ С2Н5О(ОН)СХ
Н++ ;cH-Hgcii- 7--------------* Чн + HgClj
СвНд' С6Н5/
(Se2'-2X-)
н+
С2Н5О(ОН)С = СНСвН5—---------> С2Н5ООССН2СвН5
Медленно
В качестве примера ацидолиза соединения переходного металла можно
отметить протолиз соединений кобальта(Ш) по механизму SE1, изученный
Джонсоном, Тобом и Уонгом [245]. 4-Пиридиометильная группа снова
обозначена символом Руш+. Подбором кислотности можно установить
равновесие между пентациан- и тетрацианаквопиридиометильными ком-
плексами кобальта в любом положении. Истинным субстратом является
тетрацианаквокомплекс. Этот комплекс спонтанно, хотя и медленно, теряет
карбанион, в действительности представляющий собой бетаин, поскольку
476
Замещение в алифатическом ряду
он содержит катионный заместитель; затем бетаин быстро протонируется.
Pym+Conl(CN)r Pym+Conl(CN)4(H2O)2-
ттт Медленно ттт
Pym+Conl(CN)4(H2O)2-----------> Pym± + CoIII(CN)4(H2O)- *
Pym±-[-H+ ----------> HPym+ (4-пиколиний)
(SeI)
Скорость реакции пропорциональна равновесной концентрации тетрациан-
аквопиридиометильного комплекса, но не зависит от кислотности, если
только пиколиновый остаток остается в катионной форме.
з. Замещение, связанное с наличием ароматического кольца. Иллюми-
нати и сотр. [246] отметили, что при действии С12 или Вг2 в уксусной кислоте
в темноте без катализаторов на полизамещенные бензолы, содержащие
алкильные заместители, а также полярные группы, галогенирование в ядро
сопровождается в небольшой степени а-галогенированием алкильных групп.
Галогенирование в боковые цепи происходит всегда независимо от реак-
ционной способности кольца, которую можно изменять введением полярных
заместителей; при этом оказалось, что обе реакции в одинаковой степени
чувствительны к влиянию полярных факторов. Это было бы невозможным,
если бы реакция в ядро была электрофильной, а в боковую цепь — гомоли-
тической. В случае гексаметилбензола, когда реакция по кольцу исключена,
абсолютная скорость галогенирования гексаметилбензола в боковую цепь
была больше, чем скорость замещения в ядро пентаметилбензола или любого
низшего алкилбензола, и значительно больше скорости, которую можно
было предположить для гомолитического замещения в этих условиях. Кине-
тически хлорирование представляет собой реакцию второго порядка, первого
по субстрату и первого по молекулярному хлору. Константы скоростей,
приведенные в табл. 124, иллюстрируют чувствительность замещения
в боковую цепь к полярным влияниям.
При замещении не происходит ни присоединения галогена, ни отщепления
какой-либо боковой алкильной группы. При хлорировании (но не при броми-
ровании) в образующемся аралкилгалогениде содержится небольшое коли-
чество соответствующего ацетата, который образуется параллельно с гало-
генидом, а не путем последующей реакции.
Таблица 124
Константы скоростей второго порядка монохлорирования
в боковую цепь гексазамещенных бензолов хлором
в уксусной кислоте (по данным Иллюминати)
X в С6(СНз)5Х fe2
при 18 °C при 3 0 °C
сн3 3,90-Юв
С1 1,27-105
Вг 7,90-104
CN 1,06-102 2,64-102
Первая стадия предполагаемого механизма [246, 247] состоит в образовании
иона циклогексадиенилия: эта стадия является общей как для замещения
в ядро, так и для замещения в боковую цепь, если происходят обе эти реак-
ции. Замещение в боковую цепь заканчивается перегруппировкой, что
477
Глава VII
иллюстрируется ниже на примере хлорирования гексаметилбензола
Сб(СН3)(;
СН3 -|А= СНз
СН3 JH-СНз
I
сн3
СН3
I
СНз-ZS- СН2С1
СНз-СНз
I
СНз
СНзСОО-
СН3
СНз^/^/СЩОСОСНз
Детали перегруппировки не выяснены, однако вполне вероятно, что за отще-
плением протона (аналогично иону гептаметилциклогексадиенилия Дёринга;
гл. VI, разд. 6,г) происходит анионотропная перегруппировка (гл. XI).
При таком механизме возможно участие лиатного иона, как показано выше
для ацетата.
11. Гомолитическое замещение в алифатическом ряду
а. Неорганические прототипные реакции. Как отмечалось в гл. V,
разд. 2,ж, историческим прототипом многих реакций гомолитического
замещения в насыщенных органических соединениях является реакция
в газовой фазе между хлором и водородом. Именно для этой реакции
на основании экспериментальных данных Боденштейна и других Нернст
в 1918 г. предложил первый специальный цепной механизм, который под-
твердился во всех последующих работах.
Инициирование цепи происходит через атомы хлора, которые первоначально
образуются в малых концентрациях из молекулярного хлора термически,
фотохимически или путем химической реакции с некоторыми предвари-
тельно образованными радикалами
Cl —Cl —> 2С1-
(1)
Две стадии, которые путем чередующегося повторения регенерации атомов
водорода и хлора обусловливают развитие цепи, и представляют собой
собственно гомолитическое замещение. Первая из них (2) — замещение
атома водорода в молекуле Н2 на атом хлора
С1- + Н-Н -> С1-Н + Н. (2)
Как видно из величин энергий связей (табл. 60), эта реакция почти термо-
нейтральна. Была измерена общая скорость реакции между хлором и водо-
родом и кинетика проанализирована так детально, что в настоящее время
известна скорость этой индивидуальной стадии. Выражение для константы
скорости второго порядка в интервале температур от 0 до 770 °C имеет
вид [248]
к2 (С1 • + Н2) = 7,9 +101» ехр (—5500/277’) (л. моль-1 . с"1)
Вторая стадия (3), которая повторяется по мере развития цепи, состоит
в реакции атома водорода с молекулой хлора:
H.-I-C1-C1 -> H-Cl-pCl. (3)
478
Замещение в алифатическом ряду
Как следует из данных табл. 60, этот процесс экзотермичен [45 ккал/моль
(188,41-103 Дж/моль)]. Это очень быстрая реакция—настолько быстрая,
что обратимость термонейтральной стадии не удается обнаружить.
Много лет назад Лондон, Поляньи и Эйринг теоретически доказали,
что переходное состояние гомолитических процессов замещения того
класса реакций, к которым принадлежат реакции (2) и (3), должно быть
линейным.
Обрыв цепи обусловлен любым процессом, способным вызвать исчезновение
переносящих цепь радикалов, например коллигацией (4), реакцией радика-
лов с посторонним веществом или захватом радикалов поверхностью
2С1- —> Cl2 2Н-—>Н2 Н-+С1- -> НС1 (4)
Общая реакция сильно ингибируется кислородом, вероятно, вследствие
того, что последний обрывает цепь, препятствуя атомам водорода участво-
вать в стадиях развития цепи. Кислород соединяется с атомами Н-, образуя
радикалы НО2-, а окончательно —перекись водорода.
Общая скорость реакции гомолитического хлорирования молекулярного
водорода определяется совместно частотой зарождения цепей, скоростью
определяющей скорость стадии одного звена цепи (2) и числом звеньев,
содержащихся в цепи, которое зависит от скорости совокупности процессов
обрыва цепи. Цепи неингибированных реакций длинны и обычно содержат
несколько тысяч звеньев. Кинетика суммарного процесса поэтому зависит
не только от кинетики определяющей скорость стадии цепной реакции,
но также и от кинетики стадий инициирования и обрыва цепи, несмотря
на то, что стехиометрически в этих процессах часто участвует лишь неболь-
шая доля превращаемого материала.
б. Галогенирование молекулярными галогенами. Хлорирование алканов
и замещенных алканов молекулярным хлором в газовой фазе кинетически
настолько сходно с газофазным хлорированием водорода, что не остается
сомнений в общности их механизма [249]. Кинетические цепи такие же длин-
ные, они часто содержат 104 и более звеньев. На первой стадии образуются
атомы хлора. Было изучено хлорирование, инициируемое термически и фото-
химически [250].
Первая стадия переноса цепи, которая, по крайней мере в случае простых
алканов, контролирует скорость развития цепи, состоит в атаке атома хлора
на молекулу алкана, отрыве атома водорода и образовании алкильного
радикала. Это иллюстрируется уравнением (5) для случая метана
С1- + Н—СН3 —> С1— Н4-СН3. (5)
Этот тип так называемого «отрыва водорода» («hydrogen abstraction») очень
распространен в качестве переносящей цепь стадии гомолитического заме-
щения в насыщенных соединениях. Собственно говоря, происходит замеще-
ние у атома водорода, которое несомненно протекает через линейное пере-
ходное состояние. При хлорировании метана эта стадия реакции, как следует
из данных табл. 60, термонейтральна; для гомологичных алканов она слабо
экзотермична. Скорость стадии взаимодействия атома хлора с метаном
по порядку величины такая же, как скорость его взаимодействия с водоро-
дом. Это позволило Тротман-Диккенсону и его сотрудникам, проведшим
конкурентное хлорирование водорода и метана, путем сопоставления скоро-
стей с известной абсолютной скоростью хлорирования водорода получить
абсолютные значения скоростей взаимодействия атома хлора с метаном.
Затем с помощью конкурентного хлорирования метана и его производных
479
Глава VII
были получены абсолютные скорости реакции атома хлора с производными
метана [251]. Некоторые из этих данных приведены в табл. 125.
Таблица 125
Аррениусовские параметры и скорости
взаимодействия атома хлора с алканами
Алкан В, Л МОЛЬ-1-с-1 ЕА, ккал/моль (4,187•103 Дж/моль) /12 при 100 °C, Л • МОЛЬ-1•С-1
сн4 2,6-ЮЮ 3,85 1,8-10»
С2н6 12,0-lQio 1,00 310-10»
(СН3),СН2 17,6-ЮЮ 0,67 710-10»
(СН3)3СН 19,6-lOW 0,86 613-10»
Вторая стадия единичного звена цепи состоит в атаке алкильного радикала
па молекулу хлора, как показано в уравнении (6) для случая метильного
радикала. В результате образуется хлорсодержащий продукт (хлористый
метил) и атом хлора, который вновь вступает в реакцию (5)
СН3-+С1-С1 СН3 —C1-J-C1- (6)
По данным табл. 60, это экзотермическая реакция [22 ккал/моль
(92,11 -103 Дж/моль)]. Она очень быстрая — настолько быстрая, что нельзя
обнаружить никаких признаков обратимости почти термонейтральной
реакции (5).
Обрыв цепи, как и в предыдущем случае, происходит в результате коллига-
ции радикалов или любым другим путем, при котором удаляются перенося-
щие цепь радикалы. Хлорирование алканов ингибируется кислородом,
который, вероятно, захватывает алкильные радикалы, образуя радика-
лы ВО2-. При увеличении парциального давления кислорода можно перейти
от ингибируемого кислородом хлорирования к катализируемому хлором
окислению алкана, а при промежуточных парциальных давлениях можно
получить одновременно хлорированные и окисленные продукты, например
фосген из метана.
Доказательства, касающиеся механизма гомолитического хлорирования
молекулярным хлором в растворе, менее четки, однако, насколько можно
судить, этот механизм в основном такой же, как в газовой фазе, хотя кинети-
ческие цепи часто могут быть более короткими.
Первую обширную сводку продуктов гомолитического хлорирования угле-
водородов в газовой и жидкой фазах дали Хасс и сотр. [252]. Они отметили
отсутствие перегруппировок углеродного скелета, однако ниже будет ука-
зано одно исключение из этого правила. Все атомы водорода алкана под-
вержены конкурентному замещению, скорость которого обычно возрастает
в ряду первичный < вторичный < третичный; при повышении температуры
это различие уменьшается. Указанные авторы ввели очень полезную кон-
цепцию «селективности», которая определяется относительными значениями
отношений скоростей атаки на разные типы атомов водорода в молекуле
к числу атомов водорода каждого типа. Так, если пропан подвергся па 60%
а-мопозамещению и на 40% р-монозамещению, соотношение селективностей
вторичный/первичный будет равно 2.
Поскольку реакция начинается с отрыва водорода, относительные скорости
хлорирования в разные положения алкана будут контролироваться разли-
480
Замещение в алифатическом ряду
чием в скоростях отрыва разных атомов водорода атомом хлора в процессе
типа (5) (см. выше). Температурный эффект, наблюдавшийся Хассом, дол-
жен означать, что скорости отрыва водорода частично контролируются энер-
гией активации; наблюдавшийся порядок изменения скоростей (первичный <
вторичный < третичный) должен поэтому означать, что в этом ряду умень-
шаются соответствующие энергии активации, т. е. реакция становится более
экзотермической, что и можно было ожидать на основании сравнения энергий
С — Н-связей. Тем не менее данные табл. 125 показывают, что, кроме метана,
для которого энергия активации равна 4 ккал/моль (16,75-IO3 Дж/моль),
энергии активации отрыва водорода атомом хлора во всех гомологах
алканов, составляют около 1 ккал/моль (4,187 -103 Дж/моль). Вследствие
этого не наблюдается большого различия в скоростях. Относительные ско-
рости отрыва атома водорода от групп СН3, СН2 и СН при 100 °C составляют
примерно 1 : 4,3 : 7,0 в газовой фазе и 1 : 2,0 : 3,0 в жидкой фазе [253, 254].
Относительная скорость отрыва водорода от СН4 равна примерно 0,01.
Бромирование насыщенного углеродного атома очень сходно с более полно
исследованным хлорированием. Обычно бромирование в газовой фазе или
в растворе — цепная реакция, однако с гораздо более короткими цепями,
часть которых может включать лишь одно звено, реакция может уже не быть
цепным процессом. Причина этого в основном энергетическая: короткие
цепи характерны для замещения сильно связанного водорода. При низких
температурах реагентам трудно приобрести энергию, необходимую для акти-
вации процесса переноса цепи, с которым конкурирует безактивационный
обрыв цепи путем коллигации.
Как показывают данные табл. 60, контролирующая скорость стадия (7)
при бромировании метана атомами брома является эндотермической
[14 ккал/моль (58,62-103 Дж/моль)]; таким образом, энергия активации дол-
жна быть больше этой величины. И в самом деле, энергия активации, опре-
деленная Кистяковским и сотр. [255], оказалась равной 17,8 ккал/моль
(74,52-103 Дж/моль)
Вг- + Н — СН3 -> ВгН+СН3. (7)
Исходя из этого значения энергии активации, можно было ожидать, как
и оказалось на самом деле, что бромирование метана при температуре около
180 °C в основном нецепной процесс.
По данным табл. 60, последующая стадия (8) является экзотермической
[19 ккал/моль (79,55-103 Дж/моль)], поэтому энергия активации этой стадии
мала.
СН3-+Вг — Вг —> СН8Вг-]-Вг. (8)
Вследствие этого не наблюдается обратимости стадии (7), и общий процесс
бромирования необратим. Это также подтверждается экспериментально.
В случае гомологов метана и циклоалканов первоначально рвущиеся СН-
связи более слабы, поэтому появляются кинетические цепи. Джост [256]
обнаружил, что фотохимическое бромирование циклогексана представляет
собой цепную реакцию с длиной цепи около 2 при комнатной температуре
и 12-37 при 100 °C.
В случае разрыва еще менее прочных СН-связей, например в хлороформе,
реакция становится обратимой. Салливан и Дэвидсон [257] установили, что
при бромировании хлороформа в газовой фазе при 150—180 °C константа
равновесия
К = [ ВгСС13][ НВг ]/[ НСС13][Вг2]
31-0772
48]
Глава VIJ
равна приблизительно 2. Наблюдаемая кинетика удовлетворяет следующей
схеме:
Вг2 т--> 2Вг-
9
Вг-+Н—CC13^Z± Вг-Н {-С13С. (9)
9
10
С13С- +Вг-Вг ;---> ВгСС13 + Вг. (10)
-10
Прямая реакция (10) лишь в 25 раз быстрее, чем обратная реакция (—9).
Гомолитическое бромирование алканов по сравнению с хлорированием более
селективно: вторичные положения более реакционноспособны, а третичные —
еще более реакционноспособны, чем первичные [258]. Так, при газофазном
бромировании изобутана образуется только третичный бромид, а изопентан
дает третичный бромид с выходом, в 20 раз большим по сравнению с выходом
вторичного бромида.
Несмотря на легкость термического образования атомов иода, иодирование
алканов иодом не идет. Сказываются неблагоприятные термодинамические
факторы: иодистые алкилы легко восстанавливаются иодистым водородом.
Кинетические факторы также неблагоприятны, так как стадия взаимодей-
ствия атома иода с алканом слишком эндотермична.
Фторирование алканов и их производных фтором идет так быстро и настолько
экзотермично, что трудно детально исследовать механизм этих реакций.
Состав продуктов фторирования был изучен в основном Бигелоу [259] и Мил-
лером [260], которые работали или с газами, сильно разбавленными азотом,
или с растворами в перфторированных растворителях. В случае метана обра-
зуются все возможные фторированные метаны, а также C2F6 и C3F8. Этап
дает моно-, ди-, три-, пента- и гексафторэтаны, а также CF4. Из хлорофор-
ма образуются фторхлороформ и С2С16. Таким образом, помимо обычных про-
дуктов замещения, получаются и продукты димеризации, и продукты
распада.
Фтор может также легко термически распадаться па атомы, как и иод; тепло-
ты атомизации фтора и иода составляют 37 и 36 ккал/моль (154,91 -10s
и 150,72-Ю3 Дж/моль) соответственно. Цепная реакция, развивающаяся
путем взаимодействия атома фтора с алканом с образованием алкильного
радикала, который затем атакует молекулу фтора, энергетически очень выгод-
на; экзотермичность первой контролирующей скорость стадии составляет
34 ккал/моль (142,35-103 Дж/моль), но даже при этом важное значение имеет
коллигация радикалов, обрывающая цепь, поскольку всегда можно выде-
лить в заметных количествах продукты димеризации. Таким образом, можно
прийти к выводу, что кинетические цепи должны быть короткими, тем пе
менее фторирование происходит спонтанно и быстро при —80 °C в темноте
в отсутствие химических инициаторов цепной реакции. Это заставляет усом-
ниться в том, что инициирование цепи связано с термическим образованием
атомов фтора, и склониться к принятию предположения Миллера, согласно
которому инициирование цепи происходит непосредственно при действии
молекул фтора, присутствующих в смеси в больших количествах. В этой
реакции могут образовываться как атом фтора, так и алкильный радикал
путем взаимодействия
RH + F2 —> R.+HF-FF.
которое почти термонейтрально: ДЯ для метана равна 3 ккал/моль
(12,56-Ю3 Дж/моль), а для этана составляет—2 ккал/моль
482
Замещение в алифатическом ряду
(—8,37 -103 Дж/моль). Механизм образования продуктов расщепления,
например CF4, не выяснен; возможно, что происходит бимолекулярное
гомолитическое замещение 8н2-типа *
F-+CF3CF3 —> FCF3+ -CF3 (SH2)
(гл. V, разд. 2,ж).
Утверждение Хасса о том, что хлорирование не сопровождается скелетными
перегруппировками, имеет одно известное исключение, настолько резонное,
что оно «подтверждает правило». Необходимым условием для перегруппиров-
ки в реакции насыщенной молекулы является образование промежуточного
соединения с незаполненной электронной ячейкой, т. е. карбониевого иона
или нейтрального углеводородного радикала. Эти механические условия
выполняются в принятом механизме хлорирования, который в самом деле
включает углеводородный радикал. Однако, хотя это условие необходимо,
оно не является достаточным. Необходимо, чтобы также выполнялись термо-
динамические условия, т. е. изменение свободной энергии должно происхо-
дить в направлении, благоприятствующем перегруппировке, и кинетические
условия, т. е. время жизни радикалов должно быть достаточно большим,
чтобы могла произойти миграция. В настоящее время перегруппировкам
через карбониевые ионы посвящено большое число работ; очевидно, пере-
группировки через карбониевые ионы происходят легче, чем перегруппиров-
ки через углеводородные радикалы. Вероятно, кинетическое условие, соглас-
но которому время жизни промежуточного соединения должно превышать
время, необходимое для перегруппировки, гораздо легче осуществляется
в случае карбониевых ионов, чем в случае радикалов; невыполнимость этого
условия может служить причиной того, почему Хасс никогда не наблюдал
перегруппировки. Однако, по крайней мере в некоторых реакциях, могут
образовываться радикалы, которые обладают необходимой стабильностью,
и поэтому может произойти перегруппировка путем внутримолекулярной
миграции достаточно подвижной группы. Простейшим достоверно установ-
ленным радикалом этого типа является «неофильный» (2-фенилизобутиль-
ный) радикал С6Н6(СН3)2ССН2-, который был получен Урри и Харашом [262]
при восстановлении неофилхлорида реактивом Гриньяра и хлоридом кобаль-
та(П). Образование радикала было доказано выделением димера; Урри
и Хараш показали, что некоторое количество этих радикалов в применен-
ных условиях путем миграции фенильной группы перегруппировывается
в вероятно более стабильный2-бензилизопропильный радикал С6Н5СНС(СН3)2,
также выделенный в виде димера. Недимеризованными продуктами реакции
являются соединения, образующиеся путем окислительно-восстановительного
диспропорционирования перегруппировавшегося радикала, а именно со, со-
* Существовавшее ранее предубеждение против бимолекулярного гомолитического
замещения, за исключением замещения у концевого и одновалентного атома, например
водорода, было в значительной мере ослаблено открытием ряда реакций радикального
замещения у внутренних и поэтому многовалентных атомов. Так, связи S — S в дисуль-
фидах [которые довольно прочны — 69 ккал/моль (288,89-108 Дж/моль) в диметилди-
сульфиде] и в тиосульфонатах разрываются при действии алкильных или арильных
радикалов, которые соединяются с каждым фрагментом субстрата, где они и могут быть
затем обнаружены наряду с димеризованными радикалами [261], например:
ArS—SO2Ar CgHgCHg*
ArS — СН2С6Н5 + . SO2Ar
ArS-+C6H5CH2-SO2Ar
—> Димер
Димер
Димер
31* 483
Глава VII
диметилстирол и 2-бензилпропан
С.Н5С(СН3)2СН2С1, )
г и М Р1 г гч С6Н5С(СНз)2СН2 -> (СН3)2ССН2С6Н5 -> (СН3)2С = СНС6Н5
C6H5MgCl, CoCI2 J
Димер Димер (СН3)2СНСН2С6Н5
Неофильный радикал — это радикал, который, согласно радикально-цепно-
му механизму хлорирования, вероятно, образуется в качестве основного про-
межуточного радикала при гомолитическом хлорировании тдет-бутилбен-
зола. Подтверждением этого служит то, что при газофазном хлорировании
этого углеводорода хлором при 190...245 °C на свету [263] продукты реакции
содержат не только неперегруппированный 2-фенилизобутилхлорид (нео-
филхлорид) и продукты его дальнейшего хлорирования, но и продукты
перегруппировки, например 2-бензилизопропилхлорид и родственные ему
перегруппировавшиеся олефины.
С6Н5С(СН3)3,
Cl2, hv
С6Н5С(СН3)СН2 -» (СН3)2ССН2С6Н5
| (СН3)2С=СНСвН5
С6Н5С(СН3)2СН2С1 (СН3)2С(С1)СН2С6Н5
Многие радикальные реагенты проявляют в некоторой степени полярные
свойства, которые особенно заметны у атомов галогенов. Из разности в ста-
бильностях С1_ и С1+ можно заключить, что реагент СЬ должен обладать
заметной электрофильностью: этот реагент отщепляет нейтральный атом
водорода тем лучше, чем большая электронная плотность на этом атоме
водорода. В противоположность этому алкильные и арильные радикалы
(например, СН3-,С6Н5-) не обладают в заметной степени ни электрофильно-
стью, ни нуклеофильностью. Очень часто заместители влияют на отрыв водо-
рода радикалами с высоким и низким сродством к электрону по-разному. Это
можно проиллюстрировать данными Теддера [254] по отрыву водорода от про-
пионовой кислоты радикалами CI- и СН3- (табл. 126). Так, хлор преиму-
Таблица 126
Селективность отрыва водорода
от пропионовой кислоты
Радикал СНз СН2 — соон
СН3 1 7,8
С1 1 0,03
щественно атакует положение, удаленное от карбоксильной группы, в то
время как метил предпочитает атаковать соседнее «-положение. Подобно
этому другие электроотрицательные заместители, такие, как галогены, кар-
бонильная и циангруппы, также затрудняют хлорирование соседних атомов
углерода, но облегчают атаку метильными радикалами.
Такое влияние этих заместителей на скорость взаимодействия с электро-
фильными радикалами обусловлено индуктивными эффектами.
Подобные явления наблюдались также при отрыве водорода от боковой цепи
толуола. Некоторые типичные данные по относительным скоростям отрыва
атома водорода от замещенных в кольце толуолов приведены в табл. 127.
484
Замещение в алифатическом ряду
Таблица 121
Относительная реакционная способность замещенных толуолов
по отношению к некоторым радикалам
4-Заместитель Cl «при 40 °C [264] Вг- при 80 °C [265] С6Н5- при 60 °C [267]
СН3О 11,7
С6Н5О 2,50 0,97
СНз 1,57 2,56 1,47
н 1,00 1,00 1,00
С1 0,79 0,80 1,07
CN 0,11 [266J
no2 0,32 0,11 0,81
На основании этих данных электронодонорные заместители увеличивают,
а электроноакцепторные — уменьшают относительную скорость галогениро-
вания, однако эти заместители сравнительно мало влияют на отрыв водорода
фенильным радикалом.
Если заместитель находится у того же атома улерода, от которого отщепля-
ется атом водорода, то индуктивный эффект не всегда оказывается преобла-
дающим. Например, хлористый метил хлорируется быстрее, чем метан.
В качестве еще одного примера можно привести данные Теддера [254] по гало-
генированию 1-фторбутана (табл. 128). Бромирование (наиболее селективный
Таблица 128
Селективность при галогенировании] 1-фторбутана]
(по данным Теддера)
Галогенирование ch2f сн2 — сн2 — СНз
Фторирование при 20 °C 0,3 0,8 1,0 1
Хлорирование при 78 °C 0,9 1,7 3,7 1
Бромирование при 146 °C 10 9 88 1
из всех процессов галогенирования) в положение 2 сильно затруднено по
сравнению с бромированием в положение 3 вследствие индуктивного эффекта
атома фтора. Однако в положение 1 бром вступает быстрее, чем в положение^.
Это обусловлено тем, что относительно слабый отщепляющий агент наилуч-
шим образом осуществляет свое действие в переходном состоянии, в котором
СН-связь значительно растянута и ослаблена. Такое ослабление связи
в положении 1 обусловливается способностью атома фтора сопрягаться
с атомом водорода. Это облегчает отрыв водорода, несмотря на противо-
положно действующий индуктивный эффект атома фтора
F-tC-ЧГ 'В г
Стрелки с одним зубцом означают одноэлектронный перенос. Атом кисло-
рода простых эфиров особенно склонен к такому виду сопряжения. Перво-
начально в значительной мере преобладает атака атома хлора по а-углерод-
485
Глава VII
ному атому эфира [268], хотя образующиеся при этом продукты обычно
неустойчивы.
Замедляющий эффект групп COF и COG1 при хлорировании метиленовых
групп алкильной цепи уменьшается вдоль цепи в соответствии с ослаблением
индуктивного эффекта групп, хотя не легко понять, почему это ослабление
с увеличением расстояния от полярной группы очень сходно в полярных
растворителях, неполярных растворителях и в газовой фазе. Эти данные,
полученные Сингхом и Теддером [269], приведены в табл. 129.
Таблица 129
Селективность хлорирования н-гексаноилфторида в газовой фазе
и н-гексаноилхлорида в растворе при 50... 52 °C (по данным Сингха и Теддера)
Условия FCO сн2 — сн2 -СН2 СН2 — СНз
В газовой фазе 0,16 1,5 4,0 4,4 1
С1СО сн2 — сн2 - СН2 сн2 — СНз
В СС14 0,17 0,58 1,6 2,0 1
BCH3CN 0.16 0,51 1,6 2,2 1
Следует также отметить роль растворителя в радикальных реакциях: глу-
бокие эффекты наблюдались при хлорировании молекулярным хлором.
Одно из правил Хасса состоит в том, что относительные скорости хлорирова-
ния в жидкой фазе сравнимы с относительными скоростями, полученными
при гораздо более высоких температурах в газовой фазе. Отсюда следует,
что при одинаковой температуре селективность реакции в жидкой фазе мень-
ше, чем в газовой. Это можно объяснить, предположив, что атом хлора, столк-
нувшийся с любой частью молекулы углеводорода, удерживается раствори-
телем в контакте с этой частью молекулы достаточно долго для того, чтобы
могла произойти реакция, даже если в другом месте молекулы имеется более
реакционноспособный центр [253] *.
В 1957 г. Рассел [2721 сообщил о более значительном эффекте растворителя;
он наблюдал, что на селективность жидкофазного хлорирования может
сильно влиять природа растворителя. Например, при фотохимической
реакции с чистым 2,3-диметилбутапом при 25 °C третичный атом водорода
отщепляется в 4,2 раза быстрее, чем первичный. Это соотношение селективно-
стей третичный/первичный возрастает до 20 в случае 4 М раствора диметил-
бутана в бензоле, до 35 в 4 М растворе в mpe/n-бутилбензоле и до 225 в 12 М
растворе в сероуглероде. Изменение селективности было приписано комплек-
сообразованию атома хлора с растворителем. К растворителям, образующим
комплексы, относятся ряд ароматических соединений, сероуглерод и N,N-
диметилформамид. Указаний на образование комплексов атома хлора с али-
фатическими углеводородами, нитробензолом, олефинами и сложными эфи-
* См. также [270]. Возможно, что это объяснение недостаточно, однако можно указать
на небольшое различие в параметрах уравнения Аррениуса для газо- и жидкофазного
хлорирования, наблюдавшееся Галибой, Теддером и Уолтоном [271]. Уравнение Арре-
ниуса — довольно грубый инструмент для анализа влияния строения на кинетику,
и, с точки зрения автора, такие небольшие различия объяснить не просто.
486
Замещение в алифатическом ряду
рами обнаружить не удалось. Отщепление водорода от алканов сильно за-
комплексованным атомом хлора является не экзотермическим, а эндотерми-
ческим процессом, поэтому селективность этих реакций приближается
к селективности бромирования в газовой фазе. Увеличение селективности
хлорирования в ароматических растворителях можно связать с основностью
этих растворителей, мерой которой служит константа равновесия взаимо-
действия ароматических соединений с хлористым водородом при —78 °C
[272]. Рассел предположил, что основным фактором, обусловливающим вли-
яние растворителя, является образование комплекса с переносом заряда
между электроотрицательным атомом хлора и растворителем. Согласно Уол-
лингу [273], при жидкофазном хлорировании могут также иметь значение
такие факторы, как полярность и поляризуемость растворителя.
Рассел и сотр. [274] опубликовали ряд важных данных по фотохлорированию
более основных ароматических углеводородов. Эти углеводороды выступа-
ют как специфические комплексообразующие агенты для атома хлора.
Высокая селективность, наблюдавшаяся в отсутствие растворителя, умень-
шается при разбавлении инертным некомплексующим растворителем. Напри-
мер, при 40 °C отношение селективностей третичный/первичный для кумола
без растворителя равно 42; оно уменьшается до 10 для 3 М раствора кумола
в нитробензоле, до 5 для 1,5 М раствора кумола в нитробензоле и, по оцен-
кам, должно быть равно 3,5 при исчерпывающем разбавлении этим раство-
рителем.
Менее выраженные эффекты растворителя наблюдались при отрыве атома
водорода радикалами, отличными от атома хлора, например алкокси- [273 —
275] и перокси-радикалами [276, 277]. В этих случаях несомненно более
важными свойствами являются полярность и поляризуемость растворителя,
а не его основность.
в. Галогенирование другими реагентами. Препаративная ценность ради-
кально-цепного галогенирования, описанного в предыдущем разделе, весьма
ограниченна. Исключая незначительные вариации, вызванные изменением
температуры или растворителя, можно сказать, что строение продукта
реакции предопределено природой используемого галогена. Это ограничение
во многом устраняется при использовании ряда специфических переносчи-
ков галогена (S — X), которые дают продукты, отличающиеся от образую-
щихся при молекулярном галогенировании, так как атом водорода отщепля-
ется другим радикалом (S-, а не Х-). Несколько наиболее важных перенос-
чиков галогена приведены в табл. 130. Развитие цепи можно представить
схемой:
S —X + R. —> R —X + S-
S.+RH SH+R.
Таблица 130
Галогенирование специфическими реагентами
Переносчик галогена Отщепляющийся радикал Продукт
Сульфурилхлорид [253, 278] SO2C1- и С]. Хлорид
трет-Бутилгипохлорит [279] (СН3)зСО. »
Четыреххлористый углерод [253] СС13- »
Смесь С12 —Вг2 [253] сь Бромид
Трихлорбромметан [280] СС13. »
N-Бромсукцинимид (см. текст) Вг- »
487
Глава VII
Поскольку строение продуктов определяется только природой отщепляю-
щегося радикала, для разных галогенов, связанных с одним и тем же пере-
носчиком цепи, должно наблюдаться одно и то же распределение изомеров.
Уоллинг и Падва [281] подтвердили это на примере галогенирования ряда
углеводородов трет-бутил гипохлоритом и терет-бутилгипобромитом. Иден-
тичность состава продуктов была также использована для доказательства
того, что при бромировании N-бромсукцинимидом реакционноспособным про-
межуточным соединением является атом брома, а не сукцинимидильный
радикал (табл. 130).
Рассел и Ито [278] исследовали продукты, образующиеся при фотохлориро-
вании хлорциклопентана рядом хлорирующих агентов. Полученные ими
результаты приведены в табл. 131. Относительная реакционная способность
Таблица 131
Относительные скорости хлорирования
хлорциклопентана в a-, 0- и у-положения
(по данным Рассела и Ито)
Переносчик галогена
С12 в СС14 0,50 0,29
С12 в С6н6 0,43 0,36
SO2CI2 0,54 0,42 б
С12 в CS2 0,48 0,61
CC18SO2C1 0,58 1,1 б
(СН3)зСОС1 0,81 2,4
а На один атом водорода при 4 0 °C.
6 При 80 °C.
Р- и у-положений (т. е. к^/к^) служит мерой чувствительности атакующего
радикала к индуктивному эффекту атома хлора в a-положении. Поскольку это
отношение реакционных способностей более или менее одинаково для всех
радикалов, резонно предположить, что мерой чувствительности атакующего
радикала к прочности разрывающейся связи углерод — водород является
относительная реакционная способность «-положения (т. е. fca/fc?). Наименее
прочная углерод-водородная связь в рассматриваемой молекуле находится
в «-положении, поэтому доля реакции в это положение возрастает при
уменьшении реакционной способности атакующей частицы. Отсюда становит-
ся понятным, почему чувствительность реакции к энергии диссоциации связи
наименьшая для «свободного» атома хлора и наибольшая для mpem-бутокси-
радикала, в то время как атомы хлора в комплексе с растворителем занимают
промежуточное положение.
Необходимо специально упомянуть о N-бромсукцинимиде, так как этот
широко используемый бронирующий агент функционирует совершенно иначе,
чем галогенирующие агенты, приведенные в табл. 130. Реакция N-бромсук-
цинимида с олефинами в кипящем четыреххлористом углероде дает главным
образом аллилбромиды, а не продукт присоединения — дибромид, который
всегда образуется при использовании в качестве галогенирующего агента
самого брома. Первоначально был предложен свободнорадикальный цепной
механизм, включающий в качестве переносчика цепи сукцинимидильный
488
Замещение в алифатическом ряду
радикал [253, 282]
S —Вг —> S. + Br.
S>-|—СН2СН=СН--> SH-J- — СН — СН = СН —
SBr+-CHCH=CH-----> S-4— СНВгСН=СН-
СН2-СО
I
сн2—ссг
Радикально-цепной характер этой реакции был хорошо доказан в последую-
щих исследованиях [283]. Однако в работе Голдфингера [284] по аллильному
хлорированию N-хлорсукцинимидом сделан вывод, что переносчиками цепи
служат атомы галогена, а не сукцинимидильные радикалы. С помощью кине-
тического анализа он показал, что при низких концентрациях галогена про-
текает предпочтительно замещение, а не присоединение. Функция N-бром-
сукцинимида состоит в том, чтобы поддерживать низкую постоянную кон-
центрацию молекулярного брома, образующегося при быстрой гетеролити-
ческой реакции с НВг
S —Br+НВг —> SH + Br2
Затем происходит цепная реакция
Вг. + —СН2—СН = СН---> НВг+—СНСН = СН —
Вг2+-СНСН = СН----> Вг.-]-— СНВгСН = СН —
наряду с побочным процессом гомолитического присоединения брома по двой-
ной связи. Этот процесс не является главным вследствие обратимости первой
стадии присоединения
Вг.-|-—СН2СН = СН— — СН2СН —СНВг —
Вг2- + -СН2СН-СНВг Вг«+ -СН2-СНВг-СНВг-
Таким образом, при низкой концентрации брома, устанавливающейся в при-
сутствии N-бромсукцинимида, отрыв водорода более выгоден, чем присоеди-
нение. Механизм Голдфингера в настоящее время подтвержден огромным
количеством фактов, полученных при изучении как аллильного [285, 286],
так и бензильного [265, 266, 286—288] замещения. Не может быть ни малей-
шего сомнения в том, что бромирование бромсукцинимидом не протекает
путем образования сукцинимидильных радикалов в заметной степени.
г. Аутоокисление. Это название носит процесс окисления молекулярным
кислородом, в частности, содержащимся в воздухе. В 1832 г. Либих и Вёлер
[289] открыли аутоокисление бензальдегида. В 1897 г. Ёриссен [290] отме-
тил, что эта реакция обусловлена образованием в смеси окисляющего агента,
который является преходящим, и что в присутствии избытка уксусного ангид-
рида поглощается двойное по сравнению с обычным количество кислорода.
Объяснение, данное Байером и Виллигером [291] в 1900 г., состояло в том,
что в реакции вначале образуется в то время неизвестное вещество — над-
бензойная кислота, которая далее восстанавливается другой молекулой бенз-
альдегида до бензойной кислоты. Они предположили, что в присутствии из-
бытка уксусного ангидрида надбензойная кислота превращается в стабильное
соединение — смешанный ангидрид надбензойной и уксусной кислот. В от-
сутствие подобных агентов надбензойная кислота является первым продуктом
окисления бензальдегида, который можно выделить, что и было сделано
в 1926 г. Ёриссеном и Ван дер Бееком [292]. Затем те же авторы, а также Бэк-
стрем заметили, что истинный короткоживущий окисляющий агент — более
сильный, чем надбензойная кислота, которая из него образуется. Этот агент
может окислять антрацен и даже четыреххлористый углерод. В настоящее
489
Глава VII
время имеется ряд доказательств того, что эта реакция происходит по ради-
кально-цепному механизму: аутоокисление может промотироваться светом
и химическими инициаторами, образующими радикалы, например надбензой-
ной кислотой, и может ингибироваться такими «антиоксидантами», как
гидрохинон и другие легко окисляющиеся фенолы и ароматические амины.
Из квантового выхода, полученного при фотоинициировании, Бэкстрем
[293] определил, что кинетические цепи могут включать до 10 000 ...
15 000 звеньев. В 1934 г. Бэкстрем [294] кинетически установил, что разви-
тие цепи состоит из следующих стадий (R = СвН6):
RCO.-|-O2 = RCOO — ОО.
RCO —OO-+RCHO = RCOOOH + RCO.
Вторая стадия контролирует скорость. Аутоокисление ацетальдегида (R =
= СП3), открытое в 1835 г. Либихом и Вёлером [295], получило аналогичное
объяснение в 1930 г. благодаря кинетическим исследованиям Бовепа и Титца
[296]. Абсолютные скорости стадий развития цепи и процессов обрыва цепи
для окисления бензальдегида и декапаля были определены в 1952 г. Мелвил-
лем [297] при использовании метода прерывистого фотоипициирования, кото-
рый Мелвилл предложил одновременно с Бартлетом за несколько лет до
этого. Обе стадии развития цепи во всех случаях являются экзотермически-
ми; однако вторая из приведенных выше стадий имеет заметную энергию
активации [5...8 ккал/моль (20,93-Ю3 ... 33,49-103 Дж/моль)].
Изучение окисления ненасыщенных и ацилированных углеводородов нена-
сыщенных сложных эфиров и алкиловых и аралкиловых простых эфиров
проводилось в общем по такой же методике. Первичными выделенными про-
дуктами окисления в большинстве случаев были так называемые «мольокси-
ды», которые затем были идентифицированы как гидроперекиси. Некоторые
из них можно было выделить с хорошим выходом, другие только с низким
выходом, преимущественно при малых степенях превращения, так как сами
гидроперекиси разлагаются. В 1928 г. была выделена гидроперекись цикло-
гексена [298], а в 1932 г.— гидроперекись тетрамина [299]; в последующие
10 лет Криги, Хок и Фармер [298] доказали, что эти гидроперекиси содержат
функциональную группу в аллильном или бензильном положениях
ООН ООН
/\ /\/\
I !1 И I
\/ ч/\/
В течение 1940-х и 1950-х годов были выделены многие другие гидроперекиси.
Легко образуется гидроперекись из кумола; она так гладко разлагается на
фенол и ацетон при действии кислот, что реакция получила промышленное
развитие [300].
(Н+)
С6Н5СН(СН3)2 -+ С6Н5С(СН3)2-ООН--> С6Н5ОН + (СН3)2С = О
Подробно исследованы перекиси этиллинолеата [301]. Водород наиболее лег-
ко отщепляется от метиленовой группы, расположенной между двумя двой-
ными связями, однако выделенная смесь гидроперекисей содержала главным
образом сопряженные изомеры, приведенные ниже, очевидно, образовавшие-
ся путем перегруппировки. Строение этих соединений было доказано вос-
490
Замещение в алифатическом ряду
становлением до этиловых эфиров 9- и 13-оксистеариновых кислот
СН3(СИ2)2СН=СН —СН2 —СН = СН(СН2)7СООС2Н5
ООН
СН3(СН2)2(!н —СН = СН —СЯ = СН(СН2)7СООС2Н5
, ООН
1 I
СН3(СН2)2СН = СН — СП - СП — СН(СН2)7СООС2Н5
В общем насыщенные соединения аутоокисляются с большим трудом, чем
ненасыщенные; они атакуются преимущественно по третичным положениям.
Так, перекиси
ООН
ООН ООН
/\ /\ сн3х | | сн3
и ^ССН2СН2С/
\/\/ СН3/ \сн3
были синтезированы из декалина [302] и диизобутила [303] соответственно.
Как пример аутоокисления вторичного водорода была получена гидроперекись
из циклогексана [304], н-декан образует смесь вторичных гидроперекисей
[305]. Простые эфиры, содержащие «-водород, аутоокисляются очень легко
по «-положению. Дигидроперекись
ООН ООН
(СН3)2С —О —С(СН3)2
была получена из диизопропилового эфира [306].
Соединения с олефиновыми двойными связями не обязательно в качестве
единственного или даже основного первичного продукта должны давать
гидроперекись: вместо такого замещения может происходить присоединение
[307]. Майо и сотр. [308] показали, что олефины, способные полимеризовать-
ся или сополимеризоваться, легко дают с почти количественным выходом
1 : 1-сополимер мономерного олефина с кислородом, т. е. полиперекись
R'OO. + RCH^CH2 RCH — СИ, OOR'
RCH —СН2 —OOR' + O2 RCH — СН, ОО1Г
О-О.
RCH —CH2-OOR' + RCH=CH2 R —СН —СН2—OOR'
О-О- 00 —СН2 —CHR и т. д.
Полиперекиси состава приблизительно 1 : 1 были получены из ряда олефи-
нов, например стирола, метилметакрилата и винилацетата. Олефины могут
также реагировать с перекисными радикалами с образованием эпокисей
[307-309]
О
— Cz=C—+ROO. — С—^С— +RO-
Эта реакция была обнаружена при окислении многих простых олефинов,
например пропилена, бутена-2, 2,4,4-триметилпентена-1, циклогексена
и циклооктена.
491
Глава VII
Кинетика аутоокисления, например циклогексена [310], тетралина [311],
кумола [312], этиллинолеата [313] и дибензилового эфира [314, 315], исследо-
валась в тех же аспектах, что и кинетика аутоокисления альдегидов. Перво-
начально было установлено, что независимо от способа инициирования —
термического, светового или химического под действием таких инициаторов,
как перекись бензоила или а,а'-азо-бнс-изобутиронитрил,— каждое звено
цепи содержит две стадии развития цепи, обозначенные буквами р и q, из
которых определяющей скорость является вторая стадия q
(р) r. + o2-»roo.
(q) ROO. + RH ROOH + R.
Принцип, лежащий в основе всех кинетических опытов, состоит в следующем.
Общее выражение для скорости таких цепных реакций несколько усложнено
вследствие того, что необходимо учитывать три реакции обрыва цепи, обозна-
ченные ниже буквами г, s и t
(г) 2R.
(s) ROO«4-R« —> > Неактивные продукты
(i) 2ROO. —» J
Скорости этих процессов обрыва зависят соответственно от нулевой, первой
и второй степеней давления кислорода: поэтому при увеличении давления
кислорода выше некоторой величины, которая зависит от конкретного слу-
чая, но обычно не выше 100 мм, можно сделать так, что преобладающим будет
процесс £, а другими процессами обрыва (г и s) можно пренебречь. В этом слу-
чае общая скорость цепной реакции становится не зависящей от давления
кислорода, а пропорциональной только первой степени концентрации суб-
страта. Эта скорость связана с корнем квадратным из скорости инициирова-
ния Rt, которую можно измерить независимым образом, например для
инициирования перекисью бензоила, по количеству выделяющейся двуокиси
углерода. Таким образом, выражение для скорости в случае «высокого»
давления кислорода настолько просто и настолько характеристично, что его
применимость является тестом на указанный механизм. Обозначив симво-
лами кд и kt константы скоростей второго порядка кинетически важных ста-
дий q и t соответственно, получим выражение для скорости
Скорэсть = Л„[НН] Т/
При низких давлениях кислорода скорости процессов обрыва г и s могут
стать сравнимыми со скоростью процесса t. Для тех нескольких случаев,
которые были изучены, константы скоростей этих трех процессов были вели-
чинами одного порядка.
Описанные до этого параграфа методы позволяют определять лишь относи-
тельные скорости стадий, т. е. можно получить только отношения констант
скоростей. Абсолютные константы скоростей могут быть определены методом
импульсного фотоинициирования Бартлета и Мелвилла, что было сделано-
для ряда уже упомянутых примеров [295, 311—313]. Константа скорости
первой стадии развития цепи р, к примеру, может быть почти в миллион раз
больше, чем константа скорости контролирующей скорость стадии q. Таким
образом, она может на один-два порядка быть выше скорости, контролируе-
мой диффузией. Точно так же константы скоростей трех стадий обрыва цепи
могут быть примерно в миллион раз больше константы скорости стадии q.
Например, для окисления этиллинолеата при 25 СС константы скорости вто-
492
Замещение в алифатическом ряду
рого порядка имеют следующие значения: kp = 1 -IO7, kq — 5, кТ = 2-107,.
ks = 5-107 и kt = 2-107 л-моль-1-с*1.
Систематическое изучение влияния строения на абсолютные константы
скоростей отрыва водорода от углеводородов перекисными радикалами (kq)
и на константы скоростей саморазложения перекисных радикалов (kt) было
проведено Говардом и Кейтом Ингольдом [316]. Скорости отрыва атома
водорода, как правило, возрастали в ряду первичный, вторичный, третичный
атом водорода. Скорости также возрастали при увеличении числа л-электрон-
ных систем (этиленовых или ароматических), связанных с атомом углерода,
имеющим водород, который, таким образом, становился бензильным или
аллильным. Кроме того, выяснилось, что л-электронные системы делают
атом водорода более подвижным, когда вся аллильная или бензильная систе-
ма является частью кольца, а не открытой цепью. Константы скорости
саморазложения двух перекисных радикалов в общем увеличивались в ряду:
третичные перекисные радикалы < вторичные аллильные с открытой цепью <
< циклические вторичные С вторичные бензильные с открытой цепью <
< первичные перекисные радикалы < гидроперекисные радикалы.
До сих пор рассматривалась только первая фаза аутоокисления, в результа-
те которой обычно образуются гидроперекиси. Вторая общая фаза включает
разложение этих соединений. Обычно при аутоокислении наблюдается ауто-
катализ вследствие того, что первоначально образующиеся гидроперекиси
разлагаются, давая радикалы, которые начинают новую цепь окисления.
Таким образом, аутоокисление — это разветвленная цепная реакция. Раз-
ложение гидроперекиси на радикалы может происходить как мономолеку-
лярно, так и бимолекулярно [317]
ROOH -» RO. + -ОН
ROOH + HOOR —> RO2-+H2O + RO.
Разложение на радикалы ускоряется ионами металлов переменной валентно-
сти, которые могут вступать в реакции одноэлектронного переноса [253, 317].
Ионы металлов могут действовать или как восстанавливающие агенты (двух-
валентное железо), или как окисляющие агенты (четырехвалентный церий)
Fe2+ + ROOH Fe3+ + RO. 4-ОН-
Ce4+-|-ROOH Ce3+ + ROO. + H+
Ионы кобальта и марганца действуют по обоим механизмам, т. е. ионы низ-
шей валентности восстанавливают гидроперекись, а ионы высшей валентно-
сти — окисляют ее. Поэтому ионы таких металлов являются настоящими
катализаторами разложения гидроперекисей. Радикалы, образующиеся при
таком разложении, способствуют окислению, образуя дополнительное коли-
чество гидроперекиси, которая в свою очередь разлагается ионами металлов.
Таким путем ионы некоторых переходных металлов функционируют как
катализаторы аутоокисления.
Аутоокисление органического соединения часто можно сильно замедлить
добавками небольших количеств веществ, известных под названием анти-
оксиданты. Антиоксиданты делятся на две основные категории: (1) первичные
антиоксиданты, которые разрушают переносящие цепь радикалы и поэтому
часто называются ингибиторами радикалов, и (2) вторичные антиоксиданты,
которые разрушают разветвляющие цепь гидроперекиси и поэтому могут быть
названы разрушителями перекисей.
Ингибиторами радикалов обычно являются фенолы или вторичные аромати-
ческие амины, содержащие легко отщепляющийся атом водорода, после
потери которого они образуют стабилизованный мезомерным распределени-
493
Глава VII
ем неспаренного электрона относительно мало реакционноспособный радикал.
Таким образом, если АН — ингибитор, то первую стадию ингибирования
фенолами [318] и аминами [319] можно представить схемой
RO2. + AH -» ROOH + A.
Если эта реакция успешно конкурирует с нормальной реакцией перекисных
радикалов (развитие цепи), то она будет уменьшать концентрацию радикалов.
Общая скорость окисления будет уменьшать концентрацию радикалов.
Общая скорость окисления будет поэтому уменьшаться при условии, если
радикал А- менее способен продолжать цепь, чем замененный им перекис-
ный радикал. Скорость этой первой стадии ингибирования увеличивается
при введении электронодонорных заместителей в ароматическое кольцо
ингибитора [318, 319]. В случае сильных ингибиторов радикал А- будет
разрушаться, взаимодействуя с другим радикалом, например, путем колли-
гации с самим собой или со вторым перекисным радикалом.
А • А • А2
ROO-+A. —> ROOA
Продукты этих реакций были идентифицированы в некоторых реакциях типа
фенол — перекисный радикал [320, 321]. В случае более слабых ингибито-
ров радикал А- может продолжить цепь окисления, отрывая атом водорода от
субстрата [322] или его гидроперекиси [323].
A-+RH AH+R.
A-+ROOH —» AH + ROO-
Такие слабые ингибиторы, участвующие в реакциях переноса цепи этого
типа, иногда называются замедлителями аутоокисления. Их участие в ре-
акциях приводит к гораздо более сложной кинетической форме, чем наблю-
даемая в случае сильных ингибиторов [322—324].
Разрушителями перекисей обычно служат соединения, содержащие серу,
селен или фосфор в форме, которую можно превратить в сульфоксид, селено-
ксид или окись фосфина. Превращение гидроперекиси в нерадикальный про-
дукт можно представить следующим общим уравнением (De ==s decomposer):
ROOH+De —» ROH + DeO
Например, реакция между насыщенными сульфидами и некоторыми гидро-
перекисями приводит к сульфоксиду
ROOH + R'R"S -» ROH + R'R"SO
Простейший из предложенных механизмов основан на том факте, что реакция
имеет второй порядок по гидроперекиси в углеводородных растворителях, но
первый порядок по гидроперекиси в спиртовых растворителях [325]. В свя-
зи с этим предполагается, что в реакции участвует гидроперекись, связанная
водородными связями:
R __ о - О + SR'R" R — О
] В углеводородах |
н н ----------------:------> и
К z (второй порядок
по гидроперекиси)
R -О — ОД-SR'R'' R — О
; I В AlkOH I
Н II -----:------> н
\ z (первый порядок
Ajk п0 гидроперекиси)
О<—SR'R"
Н
O^-OR
О<—SR'R"
Н
dC-Alk
494
Замещение в алифатическом ряду
Эта идея дает очень правдоподобное объяснение, как может произойти отрыв
кислорода из середины формально линейной молекулы гидроперекиси.
Устойчивость органических соединений к окислению часто может быть улуч-
шена в большей степени, чем это соответствует простой аддитивности, путем
добавления совместно ингибитора радикалов и разрушителя перекисей.
Каждый антиоксидант в этом случае, как говорят, является синергистом по
отношению к другому. Ингибитор радикалов препятствует возникновению
длинных реакционных цепей, однако он не может полностью подавить обра-
зование гидроперекисей. Разрушение этих гидроперекисей в реакции с раз-
рушителем перекисей препятствует инициированию новых реакционных
цепей путем образования радикалов из гидроперекисей. Таким образом, оба
антиоксиданта дополняют и усиливают друг друга.
д. Нитрование. Нитрование алканов и циклоалканов разбавленной
азотной кислотой при температуре ее кипения или при более высоких тем-
пературах (концентрированная азотая кислота в этих условиях действует
только как окислитель) известно с 1880 г. [326]. Одно время думали или по
крайней мере надеялись, что нитрование алканов и их производных разбав-
ленной азотной кислотой при повышенных температурах можно развить
в такой же важный метод получения исходных веществ для многих синтезов,
каким является нитрование ароматических соединений при низких темпера-
турах концентрированной азотной кислотой или нитрующей смесью. В дей-
ствительности же нитрование алканов почти потеряло значение, хотя
и используется еще в отдельных случаях.
Первое систематическое изучение продуктов нитрования алканов было начато
в 1936 г. Хассом [327], который использовал газовый поток при температуре
иногда значительно выше 150 °C. Хасс сделал вывод, что реакция происходит
в газовой фазе, поскольку силикагель не оказывал на нее влияния. Про-
дукты реакции содержали смесь всех мононитросоединений, которые могли
получиться при введении нитрогруппы вместо любого атома водорода или
любого низшего алкильного радикала в исходный алкан; кроме того, полу-
чались все альдегиды и кетоны, которые могли образоваться при захвате
кислорода с отщеплением водорода или низшей алкильной группы, а также
продукты окисления этих альдегидов и кетонов. По легкости отщепления
различные атомы водорода располагались в ряд третичный > вторичный >
> первичный, причем по мере повышения температуры различия несколько
уменьшались. Перегруппировки углеродного скелета не наблюдались. Это
произвело такое сильное впечатление на Хасса, что он сделал это первым из
13 «правил», выведенных им из своих исследований.
Специальное изучение продуктов было проведено в 1959 г. Хьюзом и сотр.
[328] с целью проверки возможности включения алкильных радикалов в ме-
ханизм реакции. Показано, что правило Хасса, запрещающее перегруппиров-
ки, нарушается при нитровании в боковую цепь mpem-бутилбензола, кото-
рый дает заметное количество продукта перегруппировки — неофильный
радикал С6Н5С(СН3)2СН2 • (разд. 11,6). Водная азотная кислота (4—7%)
нитрирует mpem-бутилбензол в газовой фазе при 325...530°C только в боко-
вую цепь, которая расщепляется, образуя не только неперегруппированные
продукты, такие, как ацетофенон, но также и продукты перегруппировки —
бензальдегид и а>,а>-димети л стирол
СеН5С(СН3)з > СвН5С(СН3)2СН2. > (СН3)2ССН2СвН5
1 i i I
CeH5C(CH3)2CH2NO2 CeH5COCH3 C6H5CHO (СН3)2С = СНС6Н5
4.95
Глава VII
Для осуществления перегруппировки необходимо использовать разбавлен-
ную кислоту, поэтому был сделан вывод, что пары воды действуют главным
образом как инертный разбавитель, способствующий увеличению времени
жизни радикала и, таким образом, перегруппировке.
В том же 1959 г. Шей и Гибер [329] опубликовали результаты количественно-
го исследования образования нитрометана из метана и азотной кислоты при
280...490 °C в избытке азота, служившего разбавителем. Они обнаружили,
что отношение вкладов нитрования к окислению равно 35 : 65 и это отноше-
ние сохраняется постоянным в ходе реакции. При этих температурах нитро-
метан все-таки разлагается, хотя и относительно медленно, однако Шей
и Гибер смогли измерить последующие реакции нитрометена и принять их
в расчет. При окислении образуются формальдегид, муравьиная кислота
и двуокись углерода.
Первый теоретический механизм, поддержанный Хассом [330], состоял
в том, что радикалы, получающиеся термически из азотной кислоты или из
нестабильных продуктов! окисления, отрывают водород от алкана, образуя
алкильные радикалы, которые начинают цепную реакцию
R-+HNO3 —> rno2 + -oh
R-H+.OH -» H2O + R
Установлено, что известные радикальные инициаторы — кислород и хлор —
способствуют нитрованию, а известный ингибитор радикалов — окись азо-
та — замедляет его.
Позднее Хасс и Бахман [331] предложили нецепной радикальный механизм
HNO3 -» ho. + no2
RH4-.OH —> R-+H2O
R-+NO2 —> RNO2
указав, однако, что возможны дополнительные стадии. Было предположено,
что некоторая часть двуокиси азота, соединяясь с радикалами, образует
алкилнитрат, который, как известно, при разложении дает алкильный ради-
кал и карбонильное соединение, образование которого обусловливает наблю-
даемый выход продукта окислительной деструкции
R3C —ONO —» R'--j-RjCO-f-NO
Кинетика газофазного нитрования метана, разбавленного азотом до общего
давления 1 атм (9,8 ИО4 Па) при 350 °C, была изучена Хьюзом и сотр. [332].
Данные можно суммировать в виде следующей схемы:
ХЕ, ккал/моль (4,187-103 Дж/моль)
(г) HNO3 -» HO-+NO2 +40
(р) НО.+СН4 -» Н2О + СН3- 1 -16
• цепь
(?) CH3. + HNO3 CH3NO2 + .OH J —17
(t) CH3- + HNO3CH3OH + NO2 —50
Вторая стадия звена цепи (q) определяет скорость развития цепи. Выражение
для скорости цепной реакции первого порядка имеет вид
- ^™°3l=-~^- + MHNO3] = fc; {2 + ~р} [HNO3]
Cl I QI J
496
Замещение в алифатическом ряду
где kt — константа скорости первого порядка, a kq и kt — константы скоро-
сти второго порядка стадий, приведенных выше в химических уравнениях.
Величина kt определялась в отдельных опытах, в которых азотная кислота
подвергалась пиролизу в отсутствие метана. Величина kq/kt оказалась рав-
ной 0,8, откуда следует что -у , или 44%, прореагировавшего метана превра-
щается в нитрометан, а 56% — в продукты окисления. Шей и Гиберт привели
значения 35 и 65 % соответственно, однако то, что по кинетическим данным
мы называем «нитрометаном», на самом деле включает параллельно образую-
щийся метилнитрит, который, будучи нестабильным при температурах экспе-
римента. будет давать дополнительное количество продуктов окисления.
Таким образом, рассчитанные 44% так называемого нитрометана на самом деле
могут содержать 35 % действительно нитрометана и 9 % метилнитрита в со-
ответствии с наблюдаемым составом продуктов реакции.
Алкильные радикалы атакуют молекулу азотной кислоты по внутренней
связи, в близкой к равновероятной степени, соединяясь как с нитроксиль-
ным, так и с гидроксильным остатками. Этот вывод согласуется с данными
Дегани и Тунда [261], которые показали, что алкильные и арильные радикалы
атакуют связи S — Sb дисульфидах и тиосульфонатах, в каждом случае
соединяясь с любым остатком этих субстратов. Это означает, что такой
механизм взаимодействия алкильного радикала с молекулой азотной кислоты
и является главной причиной, объясняющей, почему гомолитическое нитро-
вание алканов всегда сопровождается их окислением. Это предположение
объясняет случайное открытие Хассом небольших количеств спиртов в про-
дуктах газофазного нитрования алканов. Кинетически цепная реакция,
инициируемая стадией диссоциации (г), обрывается на стадии окисления (Z).
Значительная степень окисления обусловливает довольно малую длину
цепей. В приведенном случаэ средняя длина цепи составляет 1 -j- kq/kt =
= 1,8 звеньев.
СПИСОК ЛИТЕРАТУРЫ
1. Fischer Е., Ann., 381, 123 (1911); Werner А., Вег., 44, 873 (1911); Ann., 386, 1 (1912);
Pfeiffer Р., ibid., 383, 92 (1911); Gadamer J., J. prakt. Chem., 87, 312 (1913); Meisen-
heimer J., Ann., 456, 126 (1912).
2. Le Bel J.-A., J. chim. phys., 9, 323 (1911).
3. Lewis G. N., Valence and the Structure of Atoms and Molecules, Chemical Catalog Co.,
New York, 1923, p. 113.
4. London F., Z. Elektrochem., 35, 552 (1929).
5. Lowry T. M., Inst, inter, chim. Solvay, Conseil chim. (Brussels), 1925, 130.
6. Hanhart W., Ingold С. К., J. Chem. Soc., 1927, 997.
7. Burton H., Ingold С. K., J. Chem. Soc., 1928, 904.
8. Ingold С. K„ Rothstein E., J. Chem. Soc., 1928, 1217; Ingold С. K., Patel C. S.,
J. Indian Chem. Soc., 7, 95 (1930).
9. Meer N., Polanyi M., Z. physik. Chem., B19, 164 (1932).
10. Olson A. R., J. Chem. Phys., 1, 418 (1933); Olson A. R., Long F. A., J. Am. Chem.
Soc., 56, 1294 (1934); Olson A. R., Voge H. H., J. Am. Chem. Soc.,
56, 1690 (1934).
11. Hughes E. D., Ingold С. K., J. Chem. Soc., 1935, 244.
12. Hughes E. D., Ingold С. K., Patel C. S., J. Chem. Soc., 1933, 526.
13. Hughes E. D., Ingold С. K., J. Chem. Soc., 1933, 1571; Gleave J. L., Hughes E. D..
Ingold С. K., ibid., 1935, 235.
14. Hughes E. D., Ingold С. К., J. Chem. Soc., 1935, 244; Hughes E. D., ibid., p. 255.
15. Ingold С. K., Rothstein E., J. Chem. Soc., 1928, 1217.
16. Bateman L. C., Cooper К. E., Ingold С. К., J. Chem. Soc., 1940, 925.
17. Alexander E. R., Principles of Ionic Organic Beactions, Wiley and Sons New York,
1950, pp. 81, 84.
32—0772
497
Глава VII
18. Bateman L. C., Church M. G., Hughes E. D., Ingold С. K., Taher N. A., J. Chem. Soc.,
1940, 1008.
19. Swain C. G., J. Am. Chem. Soc., 70, 119 (1948) and later.
20. Hughes E. D., Ingold С. K., J. Chem. Soc., 1935, 244; Hughes E. D., ibid., p. 255;
Hughes E. D., Ingold С. K., Shapiro U. G., ibid., 1936, 255; Bateman L. C-, Cooper K. A.
Hughes E. D., Ingold С. K., ibid., 1940, 925.
21. Hughes E. D., Ingold С. K., J. Chem. Soc., 1933, 1571; Gleave J. L., Hughes E. D.,
Ingold С. K., ibid., 1935, 236.
22. Baker J. W., Easty D. M., J. Chem. Soc., 1952, 1192, 1208.
23. Dostrovsky I., Hughes E. D., J. Chem. Soc., 1946, 157, et seq.; Dhar M. L., Hughes E. D.,
Ingold С. K., Masterman S., ibid., 1948, 2055.
24. Segaller D., J. Chem. Soc., 103, 1154 (1913); 105, 106 (1914).
25. Hughes E. D., Ingold С. K., Shapiro U., J. Chem. Soc., 1936, 225; Hughes E. D., Sha-
piro U. G., ibid., 1937, 1177, 1192; Dhar M. L., Hughes E. D., Ingold С. K., ibid.,
1948, 2058.
26. Cooper K. A., Hughes E. D., J. Chem. Soc., 1937, 1183; Cooper K. A., Hughes E. D.,
Ingold С. K., ibid., p. 1280; Hughes E. D., MacNulty B. J., ibid., p. 1283; Dhar M. L.,
Hughes E. D., Ingold С. K., ibid., 1948, 2065; Hughes E. D., Ingold С. K., Woolf L. I.,
ibid., p. 2084; Brown II. C., Fletcher R. S., J. Am. Chem. Soc., 71, 1845 (1949);
Brown H. C., Stern A., ibid., 72, 5068 (1950).
27. Shorter J., Hinshelwood C. N., J. Chem. Soc., 1949, 2412.
28. Wisliscenus J-, Ann., 212, 232 (1882).
29. Menschutkin N., Z. physik. Chem., 5, 589 (1890).
30. Conant J. B., Kirner W. R., J. Am. Chem. Soc., 46, 235 (1924).
31. de la Mare P. B. D., J. Chem. Soc., 1955, 3169; Hughes E. D., Ingold С. K., Mac-
kie J. D. H., ibid., pp. 3173, 3177; de la MareP. B. D., ibid., p. 3180; Fowden L. H.,
Hughes E. D., Ingold С. K., ibid., pp. 3187, 3193; de la Mare P. B. D., ibid., p. 3196;
Hughes E. D., Ingold С. K., Parker A. J., ibid., 1960, 4400.
32. Bateman L. C., Hughes E. D., J. Chem. Soc., 1937, 1187; 1940, 935, 940, 945; Doslrov-
sky I., Hughes E. D., ibid., 1946, 171.
33. Bateman L. C., Hughes E. D., J. Chem. Soc., 1940, 945.
34. Gleave J. L., Hughes E. D., Ingold С. К., J. Chem. Soc., 1935, 236.
35. Olivier S. C. J-, Weber A. P., Rec. trav. chim., 53, 869 (1934); Olivier S. C. J., ibid.,
p. 891.
36. Ward A. M., J. Chem. Soc., 1927, 2285; Farinacci N. T., Hammett L. P., J. Am. Chem.
Soc., 59, 2542 (1937); Bateman L. C., Hughes E. D., Ingold С. K., ibid., 60, 380 (1938);
Hughes E. D., Ingold С. K., Taher N. A., J. Chem. Soc., 1940, 949; Church M. G.,
Hughes E. D., Ingold С. K., ibid., p. 966.
37. Ward A. M., J. Chem. Soc., 1927, 445; Hughes E. D., Ingold С. K., Scott A. D., ibid.,
1937, 1201.
38. Hughes E. D., Ingold С. K., J. Chem. Soc., 1933, 69; Hughes E. D., J. Chem. Soc.
1933 75.
39. Vernon C. A., J- Chem. Soc., 1954, 423.
40. Bevan C. W. L., Hughes E. D., Ingold С. K., Nature, 171, 301 (1953).
41. Baker J. W., Nathan W. S., J. Chem. Soc., 1935, 1844.
42. Hughes E. D., Ingold С. K., Taher N. A., J. Chem. Soc., 1940, 949.
43. Burowoy A., Spicer E., J. Chem. Soc., 1954, 3654.
44. Shiner V. J., J. Am. Chem. Soc., 75, 2925 (1953).
45 Lewis E. S., Johnson R. R., Coppinger G. M., J. Am. Chem. Soc., 81, 3140
(1959).
46. Shiner V. J., J. Am. Chem. Soc., 82, 2655 (1960).
47 Dawson H. M., Dyson N. В., J. Chem. Soc., 49, 1133 (1933); Brooke H., Dawson H. M.,
ibid., 1936, 497.
48. Senter G., J. Chem. Soc., 95, 1827 (1909); Senter G., Wood H., ibid., 107, 1070 (1915);
109, 681 (1916); Cowdrey W. A., Hughes E. D., Ingold С. K., J. Chem. Soc., 1937,
1208.
49. Madsen E. H., Z. physik. Chem., 86, 538 (1914).
50. Hughes E. D., Taher N. A., J. Chem. Soc., 1940, 956.
51. Gripenberg J., Hughes E. D., Ingold С. K., Nature, 164, 480 (1948).
52. Senter G., J. Chem. Soc., 107, 908 (1915); Senter G., Tucker S. H., J. Chem. Soc., 109,
’ 690 (1916).
53. Olivier S. C. J-, Weber A. P., Rec. trav. chim., 53, 869 (1931); Hine J. S., Lee D. E.,
J. Am. Chem. Soc., 73, 22 (1951); Bensley B., Kohnstam G., J. Chem. Soc., 1956,
287.
54. Ballinger P., de la Mare P. B.D., Kohnstam G., Prestt В. M., J. Chem. Soc., 1955,
3641.
498
Замещение в алифатическом, ряду
55. A Itscher S., Baltzly R., Blenheim S. W., J. Am. Chem. Soc., 74, 3649 (1952); Kochi J. K.,
Hammond G. S., ibid., 75, 3445 (1953); Simonetta M., Farini G. J., J. Chem. Soc.,
1954, 1840; Cowie G. R., Fox J. R., Pitcher M. J. H., Hooton K. A., Hunt D. M., Kohn-
stam G., Shillaker B., Proc. Chem. Soc., 1961, 222; Fox J. R., Kohnstam G., ibid., 1964,
115.
56. Kohnstam G., Queen A., Riber T., Chem. and Ind., 1962, 1287.
57. Rosenbaum J., Symons M. C. R., Proc. Chem. Soc., 1959, 92.
58. Skrabal A., Skrabal R., Z. physik. Chem., 181, 449 (1938).
59. Bronsted J. N., Wynne-Jones W. F. K., Trans. Faraday Soc., 25, 39 (1929).
60. McIntyre D., Long F. A., J. Am. Chem. Soc., 76, 3240 (1954).
61. Orr W. J. C., Butler J. A. V., J. Chem. Soc., 1937, 330.
62. O’Gorman J. M., Lucas H. J., J. Am. Chem. Soc., 72, 5489 (1950).
63. Stasink F., Sheppard W. A., Bourns A. N., Canad. J. Chem., 34, 123 (1956).
64. Skrabal A., Zlatewa M., Z. physik. Chem., 122, 349 (1926).
65. Bateman L. C., Hughes E. D., J. Chem. Soc., 1940, 935.
66. Bateman L. C., Hughes E. D., Ingold С. K., J. Chem. Soc., 1940, 974; Bateman L. C.,
Church M. G., Hughes E. D., Ingold С. K., Taker N. A., J. Chem. Soc., 1940, 995,
998.
67. Bateman L. C., Hughes E. D., Ingold С. K., J. Chem. Soc., 1940, 1011, 1017.
68. Swain C. G., Scott С. B., Lohmann К. H., J. Am. Chem. Soc., 75, 136 (1953).
69. Miller J., Parker A. J., J. Am. Chem. Soc., 83, 117 (1961); Parker A. J., J. Chem. Soc.,
1961, 1328, 4398; Quart. Rev. Chem. Soc., 16, 163 (1962).
70. Bunnett J. F., Ann. Rev. Phys. Chem., 14, 271 (1963).
71. Edwards J. O., J. Am. Chem. Soc., 76, 1540 (1954); 78, 1819 (1956).
72. Jencks W. P., Carriuolo J., J. Am. Chem. Soc., 82, 1778 (1960).
73. Edwards J. O., Pearson R. G., J. Am. Chem. Soc., 84, 16 (1962).
74. Hoffmann M. H. R., J. Chem. Soc., 1965, 6253, 6762.
75. Hughes E. D., Ingold С. K., J. Chem. Soc., 1935, 244.
76. Gurney R. W., Ions in Solution, Cambridge Univ. Press, 1936, особенно гл. 1.
77. de Bruyn C. A. L., Steger A., Rec. trav. chim., 18, 41, 311 (1899).
78. Hughes E. D., J. Chem. Soc., 1935, 255.
79. LeRoux L. J., Sugden S., J. Chem. Soc., 1939, 1279; LeRoux L. J., Sugden S., Thomp-
son R. H. K., ibid., 1945, 586.
80. Davies W. C., Lewis W. P. G., J. Chem. Soc., 1934, 1599.
81. Carrara G., Gazz. chim. ital., 24, 180 (1894); Hemptinne A., Bekaert A., Z. physik.
Chem., 28, 225 (1899); von Halban II., 2. phyzik. Chem., 84, 128 (1913); Сох II. E.,
J. Chem. Soc., 119, 142 (1921); Hawkins J. A., J. Chem. Soc., 121, 1170
(1922).
82. Мухин Г. E., Гинсберг P., Моисеева К., Укр. хим. журнал, 2, 136 (1926); Essex Н.,
Gelormini О., J. Am. Chem. Soc., 48, 882 (1926); McCombie II., Scarborough H. A.,
Smith F. F. P., J. Chem. Soc., 1927, 802.
83. von Halban II., Z. physik. Chem., 67, 129 (1909).
84. Gleave J. L., Hughes E. D., Ingold С. K., J. Chem. Soc., 1935, 236; о механизме см.
Pocker Y., Parker A. J., J. Org. Chem., 31, 1526 (1966).
85. Hughes E. D., Ingold С. К., J. Chem. Soc., 1933, 1571.
86. Hughes E. D., Whittingham D. J., J. Chem. Soc., 1960, 806.
87. Cooper R. A., Dhar M. L., Hughes E. D., Ingold С. K., MacNulty B. J., Woolf L. D.,
J. Chem. Soc., 1948, 2043.
88. Farinacci N. T., Hammett L. P., J. Am. Chem. Soc., 59, 2544 (1937).
89. Bateman L. C., Hughes E. D., Ingold С. K., J. Am. Chem. Soc., 60, 3080 (1938).
90. Olson A. R., Halford R. S., J. Am. Chem. Soc., 59, 2644 (1937).
91. Bartlett P. D., J. Am. Chem. Soc., 61, 1630 (1939).
92. Bateman L. C., Hughes E. D., Ingold С. K., J. Chem. Soc., 1938, 881; Bird M. L.,
Hughes E. D., Ingold С. K., ibid., 1943, 255.
93. Bateman L. C., Hughes E. D., Ingold С. K., J. Chem. Soc., 1940, 974.
94. Church M. G., Hughes E. D., Ingold С. К., J. Chem. Soc., 1940, 966.
95. Bateman L. C., Hughes E. D., Ingold С. K., J. Chem. Soc., 1940, 1011, 1017;
Bird M. L., Hughes E. D., Ingold С. K., ibid., 1954, 634; Bunton C. A., Greenstreet С. H.,
Hughes E. D., Ingold С. K., ibid., pp. 642, 647.
96. Gelles E., Hughes E. D., Ingold С. K., J. Chem. Soc., 1954, 2918; de la Mare P. B. D.,
Hughes E. D., Ingold С. K., Pocker Y., ibid., p. 2930.
97- Long F., Pritchard J. G., Stafford F. E., J. Am. Chem. Soc., 79, 2362 (1957); Lond F. A.,
Schaleger L. L., Advances in Physical Organic Chemistry, Editor Gold V., Academic
Press, London and New York, 1963, Vol. 1, p. 1.
98. Kohnstam G., Spec. Pubs. Chem. Soc., 16, 179 (1962).
99- Cowie G. R., Fitches H. J. M., Kohnstam G., J. Chem. Soc., 1963, 1585.
32* 499
Глава VII
100. Moelwyn-Hughes Е. A., Robertson R. Е., Sugamori S., J. Chem. Soc., 1965,
1965.
101. Bensley B., Kohnstam G., J. Chem. Soc., 1957, 4747; Kohnstam G., ibid., 1960, 2066;
Konhstam G., Chem. Soc. Special Pubs., 16, 179 (1962); Cowie G. R., Fitches H. J. M.,
Kohnstam G., J. Chem. Soc., 1963, 1585.
102. Kohnstam G., Tidy D., Chem. and Ind., 1962, 1193.
103. Taylor W., несколько статей; ссылки и обсуждение см. J. Chem. Soc., 1940, 899,
et seq.
104. Bateman L. C., Hughes E. D., J. Chem. Soc., 1937, 1187.
105. Bateman L. C., Cooper K. A., Hughes E. D., J. Chem. Soc., 1940, 913; Church M. G.,
Hughes E. D., ibid., p. 920.
106. Swain C. G., J. Chem. Soc., 1957, 1206, et seq.
107. Swain C. G., Pegues E. E., J. Am. Chem. Soc., 80. 812 (1958).
108. Hughes E. D., Ingold С. K., Mok S. F., Patai S., Packer У., J. Chem. Soc., 1957, 1265.
109. Miller W. T., Bernstein J., J. Am. Chem. Soc.. 70, 3600 (1948).
110. Chapman N. B., Levy J. L., J. Chem. Soc., 1952, 1677.
111. Bevan C. W. L., Hudson R. F., J. Chem. Soc., 1953, 2187.
112. Pocker Y., Mueller W. A., Naso F., Tocki G., J. Am. Chem. Soc., 86, 5011, 5012
(1964).
113. Senter G., J. Chem. Soc., 97. 346 (1910); 99. 95 (1911): Baker J. W., ibid., 1934, 987;
Kappanna A. N., Proc. Indian Acad. Sci., 2, 512 (1935); Hughes E. D., Ingold С. K.,
Masterman S., I. Chem. Soc., 1937, 1236; Cowdrey W. A., Hughes E. D., Ingold С. K.,
ibid., 1937, 1243.
114. Hughes E. D., Ingold С. K., Masterman S., cm. [113]: Cowdrey W. A., Hughes E. D.,
Ingold С. K., cm. [113].
115. Whitmore F. C-, Whittle E. L., Popkin A. H., J. Am. Chem. Soc., 61, 1586 (1939);
Dostrovsky I., Hughes E. D., J. Chem. Soc., 1946, 166, 169.
116. Nicholet В. H., Stevens D. R.. J. Am. Chem. Soc., 50, 135, 212 (1928); Roberts I., Ham-
mett L. P., ibid., 59, 1063 (1937); Benfey О. T., ibid., 70, 2165 (1948).
117. Saramma K., Ananta Raman R. I., Proc. Indian Acad. Sci., 49, 111 (1959); J. Am.
Chem. Soc., 82, 1574 (1960); Saramma K., Thesis, Kerala, 1965.
118. Bodendorf K., Bohtne H., Ann., 516, 1 (1935); Ananta Raman R. I., Bunton C. A.,
Hughes E. D., частное сообщение.
119. Pocker У., Kevill D. N., J. Am. Chem. Soc., 87, 4760, 4771, 4778 (1965).
120. Pocker У., Kevill D. N., J. Am. Chem. Soc., 87, 5060 (1965).
121. Bateman L. C., Church M. G., Hughes E. D., Ingold С. K., Taher N. A., J. Chem. Soc.,
1940, 979.
122. Clarke G. A., Williams T. R., Taft R. W., J. Am. Chem. Soc., 84, 2292 (1962); Clar-
ke G. A., Taft R. W., ibid., p. 2295.
123. Bateman L. C., Hughes E. D., Ingold С. К., J. Chem. Soc., 1940. 960.
124. Benfey О. T., Hughes E. D., Ingold С. К., J. Chem. Soc., 1952, 2488.
125. Batejnan L. C., Hughes E. D., Ingold С. К., J. Chem. Soc., 1940, 974.
126. Bunton C. A., Nayak B., J. Chem. Soc., 1959, 3854.
127. Bailey J. H., Fox J. R-, Jackson E., Kohnstam G., Queen A., Chem. Comm., 1966,
122.
128. Hawdon A. R., Hughes E. D., Ingold С. K., J. Chem. Soc., 1952, 2499.
129. Bateman L. C., Hughes E. D., Ingold С. К., J. Chem. Soc., 1940, 1017.
130. Kolthoff I. M., Willman A., J. Am. Chem. Soc., 56, 1007 (1934); Jones M. M., Gris-
wold E., ibid., 76, 3247 (1954).
131. Bferrum N., Kgl. danske Vidensk. Selskab., Mat.-fys. Medd., 7, № 9 (1926).
132. Winstein S. et al., J. Am. Chem. Soc., 76, 2597 (1954).
133. Topsom R. D., Thesis, London, 1959; Topsom A. D., Ingold С. K., Pocker У., неопуб-
ликованная статья.
134. Hughes E. D., Ingold С. K.. Mok S. F., Patai S., Pocker У., J. Chem. Soc., 1957, 1265;
Ingold С. K., Proc. Chem. Soc., 1957, 279.
135. Fuoss R. M., Krauss C. A., J. Am. Chem. Soc., 55, 2387, 3614 (1933); J. Am. Chem,
Soc., 57, 1 (1935); Hughes E. D., Ingold С. K., Patai S., Pocker У., J. Chem.
Soc., 1957, 1206.
136. Swain C. G., Kreevoy M. M., J. Am. Chem. Soc.. 77, 1122 (1955); Hughes E. D.,
Ingold С. K.. Mok S. F., Patai S., Pocker У., J. Chem. Soc., 1957, 1220; Hughes E. D.,
Ingold С. K., Patai S., Pocker У., ibid., p. 1230.
137. Swain C. G., J. Am. Chem. Soc., 80, 816 (1958).
138. Winstein S., J. Am. Chem. Soc., 83, 893 (1961).
139. Hughes E. D., Ingold С. K., Mok S. F.. Pocker У., J. Chem. Soc., 1957, 1238; Hu-
ghes E. D., Ingold С. K., Patai S., Pocker У., ibid., p. 1256; Swain C. G., Pegues E. E.,
J. Am. Chem. Soc., 80, 812 (1958).
500
Замещение в алифатическом ряду
140. Chattaway F. D., Smiles S., Ann. Repts on Progress Chem. (Chem. Soc. London), 8,
60 (1911); Frankland P., J. Chem. Soc., 103, 717 (1913); Walden P., Optische Umkehrer-
scheinungen, Vieweg, Braunschweig, 1919.
141. Walden P., Ber., 28, 1287, 2766 (1895).
142. Walden P., Ber., 32, 1848 (1899).
143. Fischer E., Ber., 40, 489 (1907).
144. Frankland P., J. Chem. Soc., 103, 717 (1913).
145. Clough G. W., J. Chem. Soc., 105, 49 (1914); 107, 96, 1059 (1915); 113, 526 (1918);
1926, 1674.
146. Freudenberg K., Brauns F., Siegel H., Ber., 56, 193 (1923); Freudenberg K., Markert L.,
Ber., 60,2447 (1927); Freudenberg K., Lux A., Ber., 61, 1083 (1928); Freudenberg K.r
Kuhn W., Ber., 64, 703 (1931).
147. Phillips H., J. Chem. Soc., 123, 44 (1923); Kenyon J., Phillips H., Turley H. G., ibid.,
127, 399 (1925); Houssa A. J. H., Kenyon J., Phillips H., ibid., 1929, 1700; Kenyon J.,
Phillips H., Trans. Faraday Soc., 26, 451 (1930); Kenyon J., Phillips H., Taylor F. M. H.
J. Chem. Soc., 1933, 173; Kenyon J., Phillips H., Pittman V. P., ibid., 1935, 1072;
Kenyon J., Phillips H., Shutt G. R., ibid., p. 1663; Bean С. M., Kenyon J., Phil-
lips H., ibid., 1936, 303; Hughes E. D., Ingold С. K., Masterman S., ibid., 1937, 1196;
Hughes E. D., Ingold С. K., Scott A. D., ibid., p. 1201; Houssa A. H. J., Phillips H.,
J. Chem. Soc., 1932, 108; Hayford M. B., Kenyon J., Phillips H., J. Chem. Soc.,
1933 179.
148. Brewster P., Hughes E. D., Ingold С. K., Rao P. A. D., Nature, 166, 178 (1950).
149. Bifvoet B. J., Peerdeman A. F., van Bommel A. J., Nature, 168, 271 (1951).
150. Cahn R. S., Ingold С. K., J. Chem. Soc., 1951, 612; Cahn R. S., Ingold С. K.,
Prelog V., Experientia, 12, 81 (1956); Angew. Chem. Internal. Edit., 4, 385
(1966).
151. Fischer E., Ann., 381, 126 (1911).
152. Werner A., Ber., 44, 873 (1911); Ann., 386, 70 (1912).
153. Pfeiffer P-, Ann., 383, 123 (1911).
154. Gadamer J., J. prakt. Chem., 87, 372 (1913).
155. Meisenheimer J., Ann., 456, 126 (1927).
156. Le Bel J.-A., J. chim. phys., 9, 323 (1911).
157. Lewis G. N., Valence and the Structure of Atoms and Molecules, p. 113.
158. Olson A. R., J. Chem. Phys., 1, 418 (1933); Olson A. R., Long F. A., J. Am. Chem.
Soc., 56, 1294 (1934); 58, 393 (1936).
159. Meer N., Polanyi M., Z. physik. Chem., B19, 164 (1932).
160. Lowry T. M., Inst, intern, chim. Solvay, conseil chim. (Brussels), 1925, 130.
161. Kenyon J., Phillips H., Trans. Faraday Soc., 26, 451 (1930); Kenyon J., Lipscomb A. G.,
Phillips H., J. Chem. Soc., 1930, 415.
162. Hughes E. D., Ingold С. К., J. Chem. Soc., 1935, 254; Cowdrey W. A., Hughes E. D.t
Ingold С. K., Masterman S., Scott A. D., ibid., 1937, 1252.
163. Hughes E. D., Juliusberger F., Masterman S., Topley B., Weiss J., J. Chem. Soc., 1935,
1525; Hughes E. D., Juliusberger FScott A. D., Topley B., Weiss J., ibid., 1936, 1173;
Cowdrey W. A., Hughes E. D., Nevell T. P., Wilson C. L., J. Chem. Soc., 1938,
209.
164. Brewster P., Hiron F., Hughes E. D., Ingold С. K., Rao P. A. D., Nature, 166, 178
(1950); Hiron F., Hughes E. D., J. Chem. Soc., I960, 795; Harvey S. H., Hoye P. A. T.r
Hughes E. D., Ingold С. K., ibid., p. 800; Hoffmann H. M. R., Hughes E. D., ibid.,
1964, 1244; Hoffmann H. M. R., ibid., p. 1249.
165. Hoffmann H. M. R., Hughes E. D., J. Chem. Soc., 1964, 1252.
166. Hoffmann H. M. R., Hughes E. D., J. Chem. Soc., 1964, 1259.
167. Bartlett P. B., Knox L. H., J. Am. Chem. Soc., 61, 3184 (1939).
168. Bartlett P. D., Lewis E. S., J. Am. Chem. Soc., 72, 1005 (1950).
169. Hughes E. D., Ingold С. K., Masterman S., J. Chem. Soc., 1937, 1196, 1236.
170. Hughes E. D., Ingold С. K., Scott A. D., J. Chem. Soc., 1937, 1201; Hughes E. D.,
Ingold С. K., Masterman S., ibid., p. 1236.
171. Hughes E. D., Ingold С. K., Martin R. J. L., Meigh D. F., Nature, 166, 679 (1950);
Doering W. von E., Zeiss H. H., J. Am. Chem. Soc., 75, 4733 (1953).
172. Cowdrey W. A., Hughes E. D., Ingold С. K., J. Chem. Soc., 1937, 1208.
173. Church M. G., Hughes E. D., частное сообщение.
174. Winstein S., Gall J. S., Hofo M., Smith S., J. Am. Chem. Soc., 82, 1010 (1960).
175. Goering II. L., Levy J. F., J. Am. Chem. Soc., 84, 3853 (1962); Goering H. L., Levy J. F.,
Brody R. G., ibid., 85, 3059 (1964).
176. Iliceto A., Fava A., Mazzucato U., Rossetto О., J. Am. Chem. Soc., 83, 2729 (1961);
Fava A., Iliceto A., Ceccon A., Koch P., ibid., 87, 1015 (1965); Ceccon A., Papa I.,
Fava A., ibid., 88, 4643 (1966); Fava A., Iliceto A., Ceccon A., Tetrahedron Letters, 11,
501
Глава VII
685 (1963); Fava A., Tonnelato U., Congiu L., Tetrahedron Letters, 22, 1657
(1965).
177. Grunwald E., Heller H., Klein F. S., J. Chem. Soc., 1957, 2604.
178. Bunton C. A., Konasiewicz A., Llewellyn D. R., J. Chem. Soc., 1955, 604.
179. Weiner H., Sheen R. A., J. Am. Chem. Soc., 87, 287, 292 (1965); Sheen R. A., Lar-
sen J. W., ibid., 88, 2513 (1966).
180. Peters A. R., Walker E., Biochem. J., 17, 260 (1923); Fuson R. C., Price С. C., Bur-
ness D. M., J. Org. Chem., 11, 477 (1946); Bartlett P. D., Swain C. G., J. Am. Chem.
Soc., 71, 1406 (1949).
181. Winstein S., Buckles R. E., J. Am. Chem. Soc., 64, 2780, 2787 (1942); Winstein S.,
Henderson R. R., ibid., 65, 2096 (1943); Winstein S., Seymour D., J. Am. Chem. Soc.,
68, 118 (1946).
182. Winstein S., Hess H. V., Buckles R. E., J. Am. Chem. Soc., 64, 2796 (1942); Win-
stein S., Buckles R. E., ibid., 65, 613, (1943); Winstein S., Hanson C., Grunwald E.,
ibid., 70, 812 (1948); Winstein S., Grunwald E., Buckles R. E., Hanson C., ibid.,
p. 816: Winstein S., Grunwald E., Ingraham L. L., ibid., p. 821; Winstein S., Grun-
wald E., ibid., p. 828; McCasland G. E., Clark R. K., Jr., Carter H. E., ibid., 71, 638
(1949); Lucas H. J., Mitchell F. W., Jr., Garner H. K., ibid., 72, 2138 (1950).
183. Shoppee C. W., J. Chem. Soc., 1946, 1138 1147.
184. Winstein S., Adams R., J. Am. Chem. Soc., 70, 838 (1948).
185. Winstein S., Brown M., Schreider К. C., Schlesinger A. H., J. Am. Chem. Soc., 74,
1140 (1952).
186. Kenyon J., Phillips H., J. Chem. Soc., 1930, 415.
187. Lewis E. S., Boozer С. E., J. Am. Chem. Soc., 74, 308 (1952); 75, 3182 (1953); 76, 794
(1954).
188. Winstein S., Lucas H. J., J. Am. Chem. Soc., 61, 1576, 2845 (1939); Lucas H. J.,
Gould C. W., Jr., ibid., 63, 2541 (1941); Winstein S., J. Am. Chem. Soc., 64,
2791 (1942).
189. Brewster P., Hiron F., Hughes E. D., Ingold С. K., Rao P. A. D., Nature, 166, 178
(1950).
190. Eliel E. E., Ro R. S., J. Am. Chem. Soc., 79, 5995 (1957).
191. Mills A. J., J. Chem. Soc., 1953, 280.
192. Hiickel W., Ann., 533, 1 (1938).
193. Cornubert R., Bull. Soc. chim. (France), 1951, C23.
194. Schleyer P. von R., Nicholas R. D., J. Am. Chem. Soc., 83, 2700 (1961); Fort R. C.,
Jr., Schleyer P. von R., Chem. Rev., 64, 277 (1964).
195. R. Shleyer P. von R., Fort R. C., Jr., Watts W. E., Comisarow M. P., Olah G. A., J. Am.
Chem. Soc., 86, 4195 (1964).
196. Hofmann A. W. Ber., 5, 704 (1872); 8, 61 (1875).
197. Claus A., Herbaborg J., Ann., 265, 364 (1891); Claus A., Siebert R., ibid., p. 378;
Claus A., Baysen C., ibid., 266, 223 (1891); Claus A., Beck L., ibid., 269, 207 (1892);
Claus A., Weil A., ibid., p. 216.
198. Kehrmann F., Ber., 21, 3315 (1883); Kehrmann F., Messinger J., Ber., 23, 3557 (1890);
Kehrmann F., ,T. prakt. Chem., 40, 257 (1889); J. prakt. Chem. Soc., 42, 134
(1890).
199. Meyer V., Ber., 37, 510 (1894); Meyer V., Sudborough J. J., ibid., pp. 1580, 3146;
Meyer V., Ber., 28, 182, 1251, 2773, 3197 (1895); Kellas A. M., Z. physik. Chem., 24,
221 (1897).
200. Hughes E. D., Trans. Faraday Soc., 37, 603 (1941); Evans A. G., Polanyi M., Nature,
149, 608, 653 (1942).
201. Crossley M. L., Kienle R. H., Benbrook С. H., J. Am. Chem. Soc., 62, 1400 (1940);
Cohn H., Hughes E. D., Jones M. H., Peeling M. G., Nature, 169, 291 (1952); Charl-
ton J. C., Hughes E. D., J. Chem. Soc., 1956, 855.
202. Dostrovsky I., Hughes E. D., J. Chem. Soc., 1946, 157, 161, 164, 166, 169, 171; Dostrov-
sky I., Hughes E. D., Ingold С. K., ibid., p. 173.
203. Whitmore F. C., Fleming G. H., J. Am. Chem. Soc., 55, 4161 (1933); Whitmore F. C.,
Whittle E. L., Popkin A. H., ibid., 61, 1586 (1939); Whitmore F. C., Popkin A. H.,
Bernstein H. I., Wilkins J. P., ibid., 63, 124 (1941).
204. de la Mare P. B. D., England B. D., Fowden L., Hughes E. D., Ingold С. К., J. Chim.
Phys., 45, 236 (1948); de la Mare P. B. D., Fowden L., Hughes E. D., Ingold С. K.,
Mackie J. D. H., J. Chem. Soc., 1955, 3169 et seq. (8 papers); Ingold С. K., Quart. Rev.
Chem. Soc., 11, 1 (1957).
205. Cook D., Parker A. J., J. Chem. Soc., 1968B, 142.
206. Hughes E. D., Trans. Faraday Soc., 37, 620 (1941); Day J. N. E., Ingold С. K., ibid.,
p. 699.
207. Hughes E. D., Trans. Faraday Soc., 37, 620 (1941); Dostrovsky I., Hughes E. D., J. Chem.
502
Замещение в алифатическом ряду
Soc., 1946, 166, 169, 171; Dostrovsky I., Hughes E. D., Ingold С. K., ibid.,
p. 190.
208. Hughes E. D., J. Chem. Soc., 1935, 255; Cooper K. A., Hughes E. D., Ingold С. K.,
ibid., 1937, 1280; Hughes E. D., MacNulty B. J., ibid., p. 1283; Shorter J., Hinshel-
wood С. H., ibid., 1949, 2412; Brown H. C., Fletcher R. S., J. Am. Chem. Soc., 71,
1845 (1949); Brown H. C., Stern A., ibid., 72, 5068 (1950); Hughes E. D., Ingold С. K.,
Martin R. J. L., Meigh D. F., Nature, 166, 679 (1950).
209. Hughes E. D. et al., Nature, 166, 679 (1950).
210. Charlton J. C., Hughes E. D., J. Chem. Soc., 1954, 2939; 1956, 850.
211. Brown H. C., Fletcher R. S., J. Am. Chem. Soc., 71, 1845 (1949); Brown H. C., Stern A.,
J. Am. Chem. Soc., 72, 5068 (1950); Brown F., Davies T. D., Dostrovsky I., Evans O. J.,
Hughes E. D., Nature, 167, 987 (1951); Hughes E. D., Quart. Rev. Chem. Soc., 5,
245 (1951).
212. Miller S. I., Yonan P. K., J. Am. Chem. Soc., 79, 5031 (1957).
213. Montenari F., Bull. Soc. Fac. Chim. ind. (Bologna), 31, 16 (1958); Modena G., Ricerca
sci., 28, 341 (1958); Maioli L., Modena G., Gazz. Chim. Ital., 89, 854 (1959); Mode-
na G., Todesco P. E., ibid., p. 866; Modena G., Todesco P. E., Tamti S., ibid., p. 878;
Ghersetti S., Lugli G., Melloni G., Modena G., Todesco P. E., Vivarelli P., J. Chem. Soc.,
1965 2227.
214. Jones D. E., Vernon C. A., Nature, 176, 791 (1955); Jones D. E., Morris D. O., Ver-
non C. A., White R. F. M., J. Chem. Soc., 1960, 2349.
215. Di Nunno L., Modena G., Scorrazo G., J. Chem. Soc., 1966, 1186.
216. ReutovO. A., Record Chem. Progress, 22, 1 (1961); Ingold С. K., ibid., 25, 145 (1964);
Helv. Chim. Acta, 47, 1191 (1964).
217. Charman H. B., Hughes E. D., Ingold С. K., Chem. and Ind., 1958, 1517; J. Chem. Soc.,
1959, 2523; Реутов О. А., Углова E. В., Изв. АН СССР, отд. хим. наук, 1959, 757;
Jensen F. R., Whipple L. D., Keddergaertner D. K., Landgrebe J. A., J. Am. Chem.
Soc., 82, 2466 (1960); Hughes E. D., Ingold С. K., Roberts R. M. G., J. Chem. Soc.,
1964, 3900.
218. Charman H. B., Ingold С. K., J. Chem. Soc., 1959, 2523.
219. Wright G. F., Canad. J. Chem., 30, 268 (1952); cp. Brook A. G., Wright G. F., Acta
Cryst., 4, 50 (1951); Wright G. F., Ann. New York Acad. Sci., 65, 436 (1957); Несмея-
нов A. H., Реутов О. А., Поддубная С. С., Изв. АН СССР, отд. хим. наук, 1953, 649;
Winstein S., Traylor T. G., Garner C. S., J. Am. Chem. Soc., 77, 3711 (1955); Winstein S.,
Traylor T. G., ibid., 78, 2597 (1956); Реутов О. А., Белецкая И. П., Менделишви-
ли Р. Е., ДАН СССР, 116, 617 (1957).
220. Charman Н. С., Hughes Е. D., Ingold С. К., J. Chem. Soc., 1959, 2530; Реутов О. А.,
Углова Е. В., Изв. АН СССР, отд. хим. наук, 1959, 1691.
221. Charman Н. В., Hughes Е. D., Ingold С. К., Thorpe F. G., J. Chem. Soc., 1961,
1121.
222. Неведов В. Д., Синотова Е. Н., ЖФХ, 30, 2356 (1956).
223. Hughes Е. D., Ingold С. К., Thorpe F. G., Volger Н. С., J. Chem. Soc., 1961, 1133;
ср. Reutov О. A., Angew. Chem., 72, 198 (1960).
224. Hughes Е. D., Volger H. С., J. Chem. Soc., 1961, 2389.
225. Реутов О. А., Смолина Д. А., Калявин В. А., ДАН СССР, 139, 389 (1961); Реу-
тов О. А., Соколов В. И., Белецкая И. П., Рябокобылко Ю. С., Изв. АН СССР, сер.
хим., 1963, 965.
226. Charman Н. В., Hughes Е. D., Ingold С. К., Volger Н. С., J. Chem. Soc., 1961, 1142.
227. Реутов О. А., Прайснар Б., Белецкая И. П., Соколов В. И., Изв. АН СССР, сер.
хим., 1963, 970.
228. Hughes Е. D., Ingold С. К., Roberts R. М. G., J. Chem. Soc., 1964, 3900.
229. Реутов О. А., Соколов В. И., Белецкая И. П., Рябокобылко Ю. С., Изв. АН СССР,
сер. хим., 1963, 965.
230. Реутов О. А., Соколов В. И., Белецкая И. П., Изв. АН СССР, отд. хим. наук, 1961,
1213.
231. Калявин В. А., Смолина Т. А., Реутов О. А., ДАН СССР, 156, 95 (1964).
232. Калявин В. А., Смолина Т. А., Реутов О. А., ДАН СССР, 157, 919 (1964).
233. Gregory В. J., Ingold С. К., J. Chem. Soc., в печати.
234. Ingold С. К., Keiner J. A., J. Chem. Soc., в печати.
235. Hart С. R., Ingold С. К., J. Chem. Soc., 1964, 4372; Jensen F. R., Heyman D., J. Am.
Chem. Soc., 88, 3438 (1966).
236. Vreugdenhil A. D., Blomberg C., Rec. trav. chim., 82, 453 (1963); Ashby E. C., Bec-
ker W. S., J. Am. Chem. Soc., 85, 118 (1963); Ashby E. C., Smith M. B., ibid., 86,
4363 (1964).
237. Stucky G. D., Rundle R. E., J. Am. Chem. Soc., 85, 1002 (1963); Geggenberger L. J.,
Rundle R. E., ibid., 86, 5344 (1964); 90, 5375 (1968).
503
Глава VII
238. Cowan D. О., Hsu J., Roberts J. D., J. Org. Chem., 29, 3688 (1964); Whitesides G. M.,
Witanowsky M., Roberts J. D., J. Am. Chem. Soc., 87, 2854 (1965).
239. Coombes R. G., Johnson M. D., J. Chem. Soc., 1966A, 1805.
240. Kreevoy M. M., J. Am. Chem. Soc., 79, 5927 (1959).
241. Winstein S., Traylor T. G., J. Am. Chem. Soc., 77, 3747 (1955); Kreevoy M. M., Stein-
wand P. J., Kayser W. W-, ibid., 86, 5013 (1964); 88, 124 (1966).
242. Robinson G. C., J. Org. Chem., 38, 843 (1963).
243. Coad J. R., Johnson M. D., J. Chem. Soc., 1967B, 635.
244. Coad J. R., Ingold С. K., J. Chem. Soc., 1968B, 1455.
245. Johnson M. D., Tobe M. L., Wong L. У., J. Chem. Soc., 1967A, 491.
246. Illuminati G., Siegel F., Ric. Sci. Rend., 7, 458 (1964); Baciocchi E., Illuminati G.,
ibid., p. 462; Baciocchi E., Illuminati G., Siegel F., Spec. Pubs. Chem. Soc., 19, 158
(1965); Baciocchi E., Ciana A., Illuminati G., Pasini C., J. Am. Chem. Soc., 87 3953
(1965).
247. Baciocchi E., Illuminati G., Tetrahedron Letters, 1962, 6371.
248. Ashmore P. G., Chanmugam J., Trans. Faraday Soc., 49, 254 (1953).
249. Semenov N. N., Chemical Kinetics and Chain Reactions, Clarendon Press, Oxford,
1935, p. 122; Steacie E. W. R., Atom and Free Radical Reactions, Reinhold Publishing
Corp., New York, 1934, p. 667; cm. [253].
250. Pease R. H., Walz G. F., J. Am. Chem. Soc., 53, 3728 (1931); Coehn A., Cordes H.,
Z. phys. Chem., B9, 1 (1930); Jones J. R., Bates J. R., J. Am. Chem. Soc.. 56, 2282
(1934); Tamura M., Rev. Phys. Chem. Japan, 15, 86 (1941).
251. Pritchard H. O., Pyke J. B., Trotman-Dickenson A. F., J. Am. Chem. Soc.. 77, 2629
(1955).
252. Hass H. B., McBee E. J., Weber P., Ind. Eng. Chem., 27, 1190 (1935); 28, 333 (1936).
253. Уоллинг Ч., Свободные радикалы в растворе, ИЛ, И., 1960.
254. Tedder J. М., Quart. Revs. Chem. Soc., 14, 336 (1960).
255. Anderson H. C., Kistiakowsky G. B., Van Artsdalen E. R., J. Chem. Phys., 10, 305
(1942); 11, 6 (1943); 12, 469 (1944).
256. Jost W., Z. phys. Chem. Bodenstein Festband, 1931, 291.
257. Sullivan J. H., Davidson N., J. Chem. Phys., 19, 143 (1951).
258. Kharasch M. S., Hered W., Mayo F. R., J. Org. Chem., 6, 818 (1941); Eckstein В. H.,
Sheraga H. A., Van Artsdalen E. R., J. Chem. Phys., 22, 28 (1954); Russell G. A.,
Brown H. C., J. Am. Chem. Soc., 77, 4025 (1955).
259. Bigelow L. A., Chem. Revs., 40, 51 (1947).
260. Miller W. T., Jr., J. Am. Chem. Soc., 62, 341 (1940); Hadly E. T., Miller W. T.. Jr.,
ibid., p. 3362; Miller W. T., Jr., Dittman A. L., ibid., 78, 2793 (1956); Miller W. T.,
Jr., Koch S. D., McLafferty F. W., Jr., ibid., p. 4492.
261. Degani J., Tundo A., Annali Chim., 51, 543 (1961); Degani J., Tundo A., Tiecco M.,
ibid., p. 550; Degani J., Tundo A., Tiecco M., Gazz. Chim. ital., 92, 1204, 1213
(1962).
262. Urry И7. H., Kharasch M. S., J. Am. Chem. Soc., 66, 1438 (1944).
263. Backhurst J. D., Hughes E. D., Ingold С. К., J. Chem. Soc., 1959, 2742.
264. Russell G. A., Williamson R. C., Jr., J. Am. Chem. Soc., 86, 2357 (1964).
265. Walling C., Rieger A. L., Tanner D. D., J. Am. Chem. Soc., 85, 3129 (1963).
266. Pearson R. E., Martin J. C., J. Am. Chem. Soc., 85, 354, 3142 (1963).
267. Bridger R. F., Russell G. A., J. Am. Chem. Soc., 85, 3754 (1963).
268. Singh H., Tedder J. M., J. Chem. Soc., 1966B, 612.
269. Singh H., Tedder J. M., J. Chem. Soc., 1966B, 605.
270. Walling C., Mayahi M. F., J. Am. Chem. Soc., 81, 1485 (1959).
271. Galiba I., Tedder J. M., Walton J. C., J. Chem. Soc., 1966B, 604.
272. Russell G. A., J. Am. Chem. Soc., 79, 2977 (1957); 80, 4987, 4997, 5002 (1958); Tetra-
hedron, 8, 101 (1960).
273. Walling C., Wagner P. J., J. Am. Chem. Soc., 86, 3368 (1964).
274. Russell G. A., Ito A., Hendry D. G., J. Am. Chem. Soc., 85, 2976 (1963).
275. Russell G. A., J. Org. Chem., 24, 300 (1959).
276. Howard J. A., Ingold K. U., Can. J. Chem., 42, 1250 (1964).
277. Hendry D. G., Russell G. A., J. Am. Chem. Soc., 86, 2368 (1964).
278. Russell G. A., Ito A., J. Am. Chem. Soc., 85, 2983 (1963).
279. Walling C., Jacknow В. B., J. Am. Chem. Soc., 82, 6108, 6113 (1960).
280. Huyser E. S., J. Am. Chem.' Soc., 82, 391, 394 (1960); J. Org. Chem., 26,3261
(1961).
281. Walling C., Padwa A., J. Org. Chem., 27, 2976 (1962).
282. Bloomfield G. F., J. Chem. Soc., 1944, 114.
283. Dauben J., Jr., McCoy L. L., J. Am. Chem. Soc., 81, 4863 5404 (1959); J. Org. Chem.
24, 1577 (1959).
504
Замещение в алифатическом ряду
284. Adam J., Gosselain Р. A., Goldfinger Р., Nature, 171, 704 (1953); Bull. Soc. chim.
Beiges, 65, 533 (1956).
285. Sixma F. L. J., Riem. R. H., Koninkl. Ned. Akad. Wetenschap. Proc., 61B, 183
(1958).
286. McGrath В. P., Tedder J. M., Proc. Chem. Soc., 1961, 80.
287. Russell G. A., DeBoer C., Desmond К. M., J. Am. Chem. Soc., 85, 365 (1963).
288. Russell G. A., Desmond К. M., J. Am. Chem. Soc., 85, 3139 (1963).
289. Wohler F., Liebig J., Ann., 3, 253 (1832).
290. Jorissen W. R., Z. physik. Chem., 22, 34 (1897).
291. v. Baeyer A., Villiger V., Ber., 33, 1575 (1900).
292. Jorissen W. R., Van der Beek P. A. A., Rec. trav. chim., 45, 245 (1926); 46, 43 (1927);
49, 139 (1930).
293. Backstrom H. L. J., J. Am. Chem. Soc., 49, 1460 (1927); Backstrom H. L. J., Beat-
ty H. A., J. Phys. Chem., 35, 2530 (1931).
294. Backstrom H. L. J., Z. physik. Chem., B25, 99 (1934).
295. Liebig J., Wohler F., Ann., 14, 139 (1835).
296. Bowen E. J., Tietz E. L., J. Chem. Soc., 1930, 234.
297. Cooper H. R., Melville H. W., J. Chem. Soc., 1951, 1994; Ingles T. A., Melville H. W.,
Proc. Roy. Soc. (London), A218, 175 (1952).
298. Stephens H. N., J. Am. Chem. Soc., 50, 558 (1928); Criegge R., Ann., 522, 75 (1935);
Criegee R., PilzH., Flygare H., Ber., 72, 1799 (1939); Hock H., Schrader O., Naturwiss.,
24, 159 (1936); Angew. Chem., 39, 565 (1936); Hock H., Ganieke K., Ber., 71, 1430
(1938); 72, 2516 (1939); Farmer E. H., Sundralingham A., J. Chem. Soc., 1942
121.
299. Hartmann M., Seiberth M., Helv. Chim. Acta, 15, 1390 (1932); Hock H., Susemihl W-,
Ber., 66, 61 (1933).
300. Hock H., Lang S., Ber., 77, 257 (1944).
301. Bolland J. L., Koch H. P., J. Chem. Soc., 1945, 445; Khan N. A., J. Chem. Phys.,
21, 952 (1953); Khan N. A., Lundberg W. O., Holman R. T., J. Am. Chem. Soc., 76,
1779 (1954).
302. Criegee R., Bor., 77, 22 (1944); Иванов К. И., Савинова В. К., ДАН СССР, 48, 31
(1945).
303. Wibaut J. Р., Strang A., Konink Ned. Akad. Wetens. Proc., B5, 102 (1951).
304. Farkas A., Passaglia E., J. Am. Chem. Soc., 72, 3333 (1950).
305. Иванов К. И., Савинова В. К., ДАН СССР, 59, 493 (1948).
306. Rieche A., Koch К., Вег.. 75, 1016 (1942); Иванов К. И., Савинова В. К., Михайло-
ва Е. Г., ЖОХ, 16 , 65, 1003 (1946).
307. Twigg G. Н., Spec. Suppl. Chem. Eng. Science, 3, 5 (1954).
308. Mayo F. R., J. Am. Chem. Soc., 80, 2465, 2497 (1958); Mayo F. R., Miller A. A.,
ibid.,78, 1017, 1023 (1956); 80, 2480,2493 (1958); Mayo F. R., Miller A. A., Russell G. A.,
ibid., p. 2560; van Sickle D. E., Mayo F. R., Arluck R. M., ibid., 87, 4824, 4832
(1965).
309. Brill W. F., J. Am. Chem. Soc., 85, 141 (1963); Brill W. F., Indicator N., J. Org. Chem.,
29, 710 (1964).
310. Bateman L., Gee G., Proc. Roy. Soc. (London), A195, 376 (1948).
311. Bamford С. H., Dewar M. J. S., Proc. Roy. Soc. (London), A198, 252 (1949); Wood-
ward A. E., Mesrobian R. B., J. Am. Chem. Soc., 75, 6189 (1953).
312. Melville H. W-, Richards S., J. Chem. Soc., 1954, 944.
313. Bateman L., Quart. Revs. Chem. Soc., 8, 147 (1954).
314. Debiais L., Niclause M., Letort M., Compt. rend., 239, 539 (1954).
315. Debiais L., Niclause M., Letort M., Horstmann P., Compt. rend., 239, 587
(1954).
316. Howard J. A., Ingold K. U., Can. J. Chem., 45, 703 (1967).
317. Ingold K. U., Chem. 'Revs., 61, 563 (1961).
318. Howard J. A., Ingold K. U., Can. J. Chem., 40, 1851 (1962); 41, 1744, 2800 (1963);
42, 1044 (1964).
319. Brownlie I. T., Ingold K. U., Can. J. Chem., 44, 861 (1966).
320. Bickel A. F., Kooyman E. C., J. Chem. Soc., 1953, 3211.
321. Horswill E. C., Ingold K. U., Can. J. Chem., 44, 263, 269 (1966).
322. Mahoney L. R., Ferris F. C., J. Am. Chem. Soc., 85, 2345 (1963).
323. Thomas J. R., J. Am. Chem. Soc., 85, 2166 (1963); 86, 4807 (1964).
324. Howard J. A., Ingold K. U., Can. J. Chem., 42, 2324 (1964); 43, 2724 (1965).
325. Bateman L., Hargrave K., Proc. Roy. Soc. (London), A224, 389, 399 (1954).
326. Beilstein F., Kurbatov A., Ber., 13, 1818, 2028 (1880); cp. Ellis C., The Chemistry of
Petroleum Hydrocarbons, Chemical Catalog Co., New York, 1937, Vol. 2,
p. 1087.
505
Глава VII
327. HassH. В., Hodge Е. В., Vanderbilt В. М., Ind. Eng. Chem., 28, 339 (1936); Hass H. В.,
Paterson J. A., ibid., 30, 67 (1938); Seigle L. W., Hass H. B., ibid., 31, 648 (1939);
Hibsham H. H., Pierson E. H., Hass H. B., ibid., 32, 427 (1940); Hass H. B., Dorsky J.,
Hodge E. B., ibid., 33, 1138 (1941); Hass H. B., Riley E. F., Chem. Revs., 32, 376
(1943); Hass II. B., Ind. Eng. Chem., 35, 1146 (1943); Hass H. B., Howe A. P., ibid.,
38, 251 (1946); HassH. B., Shechter H., ibid., 39, 817 (1947); Hass H. B., Alexander L. G.,
ibid., 41, 2266 (1949).
328. Duffin H. C., Hughes E. D., Ingold С. K., J. Chem. Soc., 1959, 2734.
329. Schay G., Giber J., Tamas J., Soos D., Magyar Kem. Folyoirat, 65, 311 (1959);
Giber J., Meisel T., Acta Chim. Akad. Sci. Hungary, 22, 455 (1960).
330. McCleary R. F., Dagering E. F., Ind. Eng. Chem., 30, 64 (1938); Hass H. B., Pater-
son J. A., ibid., p. 67.
331. Bachman G. B., Hass H. B., Addison L. M., Hewett J. V., Kohn L., Millikan A., J. Org.
Chem., 17, 906 (1952) et seq. (5 papers); Bachman G. B., Atwood N. T., Pollack M.,
ibid., 19, 312 (1954); Bachman G. B., Standish N. W., J. Org. Chem., 26, 570
(1961).
332. Godfrey T. S., Hughes E. D., Ingold С. K., J. Chem. Soc., 1965, 1063.
ГЛАВА VIII
ЗАМЕЩЕНИЕ У ГЕТЕРОАТОМОВ
1. Электрофильное замещение у атомов азота и кислорода .... 508
2. Нуклеофильное замещение у атомов элементов третьего
периода.......................................... 527
Список литературы ............................... 535
1. Электрофильное замещение у атомов азота и кислорода
а. N-Нитрование. Электрофильное замещение было открыто и перво-
начально исследовано на примере ароматического замещения, при котором
замещающий агент атакует ненасыщенную электронную оболочку субстрата,
т. е. сопряженные 2р-электроны углерода. Поскольку в таком сопряжении
активное участие могут принимать 2р-электроны атомов азота и кислорода
ароматических аминов и фенолов, можно ожидать, что 2р-электроны трех-
лигандного азота и двухлигандного кислорода будут достаточно походить на
2р-электроны углерода в смысле уязвимости к атаке электрофильными аген-
тами. При этом, конечно, следует учитывать возможные кинетические и термо-
динамические ограничения. Ожидаемый механизм таких процессов заме-
щения будет, по-видимому, в общих чертах одинаковым для С-. N- и
О-замещения.
Нитрование представляет собой одну из простейших форм электрофильного
ароматического замещения, поскольку оно происходит преимущественно по
одному и тому же главному механизму с участием иона нитрония. Этот
четырехстадийный механизм (гл. VI, разд. 5,г) приведен ниже, где аромати-
ческий субстрат обозначен RH, а не АгН, для того чтобы можно было распро-
странить его, что и будет сделано, на другие классы субстратов
+1
HNO3 + HNO3 ----->
-1
H2NO3 4-NO3
(быстро)
+2
H2NOJ —
-2
NO+ + RH —
4
NO2RH++NC>3----->
NO+4-H2O
no2rh+
NO2R + HNO3
(медленно)
(медленно)
Стандартный
механизм
(быстро)
Механизм доказан кинетически. Первая стадия, прямое направление +1
которой соответствует захвату протона, и стадия 4, на которой происходит
компенсирующая отдача протона, обычно являются быстрыми. Положение
предравновесия (±1) может быть изменено, поскольку одна из двух молекул
азотной кислоты выполняет только кислотную функцию; эту функцию может
выполнять специально добавленная более сильная кислота, например серная,
которая, как и следовало ожидать, ускоряет нитрование. Аналогично не-
большие добавки нитрат-ионов, как и предполагали, замедляют нитрование.
Стадии ±2 и 3 в принципе протекают медленно, и вопрос о том, какая из них
определяет скорость общего процесса, будет зависеть от соотношения скоро-
стей реакций иона нитрония, образовавшегося на стадии + 2 с двумя ну-
клеофилами, способными его присоединять, а именно с водой на стадии —2
и с субстратом на стадии 3. Меняя либо концентрацию воды, либо концентра-
цию или собственную реакционную способность субстрата, можно сделать
стадию 3 или настолько быстрой по сравнению со стадией —2, что скорость
нитрования будет контролироваться скоростью образования иона нитрония
(нулевой порядок по субстрату), или настолько медленной по сравнению со
стадией —2, что ион нитрония будет образовываться в равновесной концен-
трации (равновесие ±2), так что скорость нитрования будет контролироваться
стадией 3 (первый порядок по субстрату). При соответствующем подборе
ароматических субстратов механизм реакции может изменяться между дву-
мя указанными возможностями в зависимости от природы растворителя,
который изменялся от полярных растворителей, таких, как нитрометан,
содержащих очень небольшое количество воды, до нитрующих смесей с высо-
ким содержанием воды (гл. VI, разд. 5).
508
Замещение у гетероатомов
Все эти кинетические тесты могут быть применены и основные результаты
воспроизведены при N-нитровании аминов до нитроаминов, если устранить
усложняющие факторы, связанные с основностью и другими факторами,
влияющими на реакционную способность аминов. Впервые это было установ-
лено Блэколлом и Хьюзом [1] в 1952 г. Чтобы упростить корреляцию с аро-
матическим нитрованием, необходимо избежать протонизации неподеленных
электронов атома азота, которые необходимы для захвата иона нитрония.
Нитрование метилпикрамида (2,4,6- тринитро-Х-метиланилина) до тетрила
(Х,2,4,6-тетранитро-Х-метиланилина)
представляет собой N-нитрование, в условиях которого не могут происходить
никакие последующие реакции, а первоначально имеющиеся нитрогруппы
настолько уменьшают основность, что протонизация в заметной степени не
происходит ни в нитрующей смеси, ни в более кислой среде, чем 3 М раствор
азотной кислоты в нитрометане. В этих условиях можно воспроизводить
многие кинетические явления, наблюдавшиеся при ароматическом нитро-
вании, что подтверждает общность механизмов обоих процессов [1]. При
нулевом порядке по субстрату абсолютные скорости для этил-, и-пропил-
и и-бутилпикрамидов были одинаковыми и равны скоростям нитрования
бензола, толуола, этилбензола и т. д. в тех же условиях. Таким образом,
скорости всех этих реакций равны скорости образования иона нитрония
на стадии 4-2 приведенного выше стандартного механизма. Константа скоро-
сти первого порядка по субстрату зависит от скорости стадии 3 и поэтому
для каждого субстрата различна; например, для метилпикрамида константа
скорости N-нитрования первого порядка по субстрату в 1,4 раза больше, чем
константа скорости С-нитрования бензола.
В тех же условиях, а также в более кислых средах нитрование более основ-
ных аминов сопровождается следующими двумя явлениями. Первое выводит-
ся автоматически: в этом случае в заметной степени происходит предвари-
тельная протонизация амина, и поэтому в растворе содержится лишь неболь-
шое количество амина в форме свободного основания, способной реагировать
с ионом нитрония. Второе явление, которое обсуждали Халеви, Рон и Спен-
сер [2], указывает на необходимость расширения стандартного механизма
нитрования ионом нитрония при его применении к N-нитрованию. Необходи-
мое расширение стандартного механизма состоит в следующем.
В противоположность ароматическим нитросоединениям нитроамины спо-
собны к ацидолизу при действии сильных кислот; другими словами, в то
время как ароматическое С-нитрование необратимо, N-нитрование аминов
обратимо в заметной степени. Чтобы включить N-нитрование в общий меха-
низм. необходимо предусмотреть обратимость стадии 3, т. е. нельзя по ана-
логии с ароматическим нитрованием считать, что обратная стадия —3 всег-
да будет медленной по сравнению с конечной стадией 4 отрыва протона. Это
необходимое обобщение можно записать следующим образом:
4-1
HA + HNO3 7 * H2NO+ + A-
-1
д-2
h2noj ;—» no+ + h2o
-2
(быстро)
(медленно) I
)
Механизм
Халеви
509
Глава VIII
+3
NO+ + RH —z!: NO2RH<-
-з
NO2RH+ + A- ——» NO2R+HA
1
(медленно)
(медленно)
Механизм Халеви
Халеви и сотрудники экспериментально исследовали нитрование метилпикр-
амида [RH = 2,4,6-(NO2)3CeH2NHCH3] азотной кислотой в водной серной
кислоте; концентрация последней была более 50%. Оба реагента брали
в низких концентрациях, однако азотная кислота присутствовала в постоян-
ном избытке. На этом примере было показано, что при увеличении кислотно-
сти среды, т. е. при уменьшении способности среды принимать протон от
нитро-аммониевого аддукта [NO2RH+ = 2,4,6-(NO2)3C6H2N(CH3)(NO2)H+],
кинетический контроль реакции переходит от стадии 3 (когда стадия —3
медленнее стадии 4) к стадии 4 (когда стадия 4 медленнее стадии —3). Отсю-
да следует вывод, что в применении ко всем классам субстратов любая стадия
механизма, включающего ион нитрония (2, 3 или 4), может стать определяю-
щей скорость.
Халеви, Рон и Спейсер сделали этот вывод на основании наблюдения первич-
ного изотопного эффекта, т. е. с использованием метода, с помощью которого
Меландер доказал, что при ароматическом С-нитровании стадия 4 является
быстрой, а определяющая скорость стадия осуществляется не позднее ста-
дии 3 (гл. VI, разд. 5,г). Метод состоит в наблюдении кинетического эффек-
та, вызываемого замещением на дейтерий того атома водорода, который
участвует в стадии 4. Халеви и сотрудники сравнили скорости нитрования
2.4,6-(NO2)3CeH2NHCH3 и 2,4,6-(NO2)3C6H2NDCH3 в водном растворе сер-
ной кислоты (реакция первого порядка). Авторы столкнулись с трудностью,
которой не было в первоначальной работе Меландера по ароматическому
С-нитрованию, состоящей в том, что в противоположность С-водороду N-
водород быстро обменивается с протонами среды. Поэтому было необходимо
проводить измерения в системах ArNHCH3 — H2SO4/H2O и ArNDCH3 —
—D2SO4/D2O, а затем при сравнении скоростей исключить из наблюдаемого
общего эффекта определенный таким образом кинетический изотопный эффект
среды, чтобы учесть только изменение скорости стадии 4. Поэтому авторы
сравнивали скорости в растворах H2SO4/H2O и D2SO4/D2O не точно экви-
валентного состава, но равной нитрующей силы, которая была откалиброва-
на по скорости С-нитрования стандартного ароматического соединения, кото-
рым по ряду причин практического характера была со-толуолсульфокислота.
Были получены следующие результаты. Б водной серной кислоте, концен-
трация которой была не выше 50 %\ изотопного эффекта водорода не наблю-
далось, т. е. отношение констант скоростей первого порядка было
равно единице. Такой же результат получил Меландер (он использовал три-
тий) для ароматического С-нитрования (гл. VI, разд. 5,г). При повышении
концентрации кислоты до 56 % указанное отношение скоростей, т. е. изотоп-
ный эффект, постепенно возрастало от 1,0 до 4,8. При еще более высокой
концентрации кислоты это отношение оставалось равным 4,8. Повышение
значения от 1,0 до 4,8 связано с различием в нулевой энергии между началь-
ным и переходным состояниями переноса водорода на стадии 4, что указывает
на ослабление связи субстрата с этим атомом водорода в переходном состоя-
нии. Это отношение становится максимальным, когда кислотность среды
достигает такого значения, что скорость уже полностью контролируется
стадией 4. Во всей серии измерений, таким образом, происходит постепенное
510
Замещение у гетероатомов
смещение кинетического контроля от стадии 3 (когда стадия 4 намного быст-
рее стадии —3) к стадии 4 (когда увеличение кислотности уменьшает ско-
рость стадии 4 и последняя становится медленнее стадии 3).
б. О-Нитрование. Первое доказательство того, что этерификация спир-
та азотной кислотой в действительности представляет собой О-нитрование,
было получено Клейном и Ментсером [3] в 1951 г. на примере превращения
целлюлозы в нитрат целлюлозы (нитроцеллюлозу). С помощью кислорода-18
они показали, что в каждой группе СН — ОН происходит замещение Н на
NO2, а не ОН на NO3, т. е. в продукте реакции содержатся два, а не три ато-
ма кислорода азотной кислоты. Доказательство механизма, позволившее
установить, что в реакциях моно- и полиатомных спиртов, включая также
другой промышленно важный пример — превращение глицерина в глицерин-
тринитрат (нитроглицерин), участвуют ионы нитрония, впервые было полу-
чено Хьюзом и Блэколлом [4] в 1952 г.
Метод был аналогичен примененному для изучения N-нитрования. например,
метилпикрамида, т. е. на О-нитровании воспроизводились кинетические зако-
номерности, ранее установленные для ароматического С-нитрования. Для
достаточно реакционноспособных спиртов было показано изменение кине-
тического порядка от нулевого до первого. Это означает, что кинетический
контроль переходит от стадии 2 к стадии 3 стандартного механизма нитро-
вания ионом нитрония. Эти данные были получены для нитрования 2...4 М
азотной кислотой в нитрометане метилового спирта, п-нитробензилового
спирта, этиленгликоля, триметиленгликоля и глицерина, причем в послед-
нем случае только для а- и сх'-гидроксильных групп. Для всех этих спиртов
константы скорости нулевого порядка, рассчитанные на одну гидроксильную
группу (например, считая 1 моль этиленгликоля за 2 моля участвующих
в реакции гидроксильных групп), были одинаковы и равны константе скоро-
сти реакции нулевого порядка нитрования алкилпикрамидов, бензола или
любого их гомолога в тех же условиях. Константы скоростей для реакции
первого порядка, конечно, отличались для каждого спирта; так, метило-
вый спирт реагировал в 30 раз быстрее бензола (в 1,25 раза быстрее
толуола).
Некоторые спирты, подобно некоторым ароматическим субстратам, доста-
точно реакционноспособны, чтобы захватывать все образующиеся ионы
нитрония, и поэтому кинетика реакции имеет точно нулевой порядок по
субстрату в нитрометановом растворе азотной кислоты (см. выше). Напри-
мер, неопентиловый спирт нитруется медленнее, чем любой из до сих пор
упомянутых спиртов, поэтому эта реакция имеет кинетический порядок,
промежуточный между нулевым и первым по субстрату. Согласно косвенной
оценке, константа скорости первого порядка для реакции этого спирта
в нитрометановом растворе азотной кислоты, содержащем некоторое коли-
чество воды, составляет примерно 10-1 от соответствующей величины для
бензола. Поведение неопентилового спирта, таким образом, аналогично пове-
дению хлорбензола. В этих же условиях глицерин-а,а'-динитрат нитруется
еще медленнее, кинетика соответствует первому порядку по субстрату. Можно
оценить, что константа скорости этой реакции составляет примерно 10 2
от соответствующей величины для бензола. Такое поведение сравнимо с пове-
дением и-дихлорбензола или этилбензоата. Если в качестве субстрата для
нитрования взять глицерин, то вслед за относительно быстрым нитрованием
по а- и сх'-гидроксильным группам (нулевой порядок) происходит медленное
нитрование p-гидроксильной группы (первый порядок).
Одним из основных примеров нитрования является нитрование воды, кото-
рое можно выявить изотопным обменом кислорода (меченый кислород в при-
511
Глава VIII
веденной ниже схеме отмечен звездочкой) между азотной кислотой и водой
НО —NO2-j-H —ОН —» HO-H + NO2-OH
Кислородный обмен в водной азотной кислоте происходит со скоростью,
которая сильно возрастает с ростом концентрации кислоты и имеет удобные
для измерения значения при концентрациях азотной кислоты около 35...
40 мол. %. Бантон, Халеви и Ллевиллин [5] показали это, применив 18О;
они также измерили скорости обмена при других составах водной азотной
кислоты.
Если применить для нитрования воды механизм с участием ионов нитрония,
то стадии 3 и 4 при RH = Н2О будут идентичны стадиям —2 и —1 соот-
ветственно
NO2-OH-lhNO3-----> NO2-OHJ + NOj
1
NO2 — OHJ---> NOJ + OH2 (медленно)
2
NO.J + OH, — у» NO2- -OJ1+
NO2OHJ4-NOj—N02-0H + HN03
В этом случае определяющей скорость стадией может быть только стадия 2,
поскольку пи один из присутствующих в смеси реагентов не конкурирует
с водой за образующийся ион нитрония. Поэтому если наблюдаемый кислород-
ный обмен в самом деле означает нитрование воды ионом нитрония, то ско-
рость обмена должна быть равной скорости образования ионов нитрония на
стадии 2, а эта скорость может быть независимо определена как скорость
нитрования ароматических соединений, достаточно реакционноспособных
для того, чтобы нитрование в водной кислоте, для которой измерялась ско-
рость кислородного обмена, имело нулевой кинетический порядок.
Главным образом с целью провести такое сравнение Бантон, Халеви и Стед-
ман [6] определили скорости нитрования нулевого порядка некоторых суль-
фированных в боковую цепь полиалкилбензолов, которые растворимы в воде
и достаточно реакционноспособны, чтобы их нитрование в подходящем интер-
вале концентраций азотной кислоты имело нулевой кинетический порядок.
В качестве ароматических субстратов брались анионы мезитилен-сх-сульфо-
кислоты, изодурол-2сх-сульфокислоты и 2-мезитилэтансульфокислоты. В пре-
делах погрешности опытов скорости нитрования нулевого порядка этих
трех субстратов были одинаковыми и равными скорости изотопного обмена
кислорода в этой среде. Состав среды (36,4...39,4 мол. % HNO3) соответство-
вал трехкратному интервалу измерения скоростей. Эти опыты не оставляют
сомнений, что кислородный обмен между азотной кислотой и водой в водной
азотной кислоте представляет собой О-нитрование воды нитрониевым ионом.
в. N-Нитрозирование, диазотирование и дезаминирование. В данном
разделе будут даны краткое последовательное описание этих реакций и два
общих вывода, которые следуют из изучения их кинетики.
Первый вывод указывает на причину, по которой мы будем совместно рас-
сматривать три упомянутые реакции. Она состоит в том, что все эти реакции
содержат контролирующую скорость стадию N-нитрозирования, которая
может также быть лишь первой стадией процесса. В случае вторичных аминов
реакция на зтом заканчивается. В случае первичных ароматических аминов
последующая быстрая ионизация приводит к иону диазония, который доста-
точно стабилен и может рассматриваться как конечный продукт. В случае
первичных алифатических аминов этот продукт неустойчив, так как быстрое
512
Замещение у гетероатомов
отщепление азота приводит к образованию карбониевого иона и, следова-
тельно, всех продуктов его реакций с присутствующими в смеси подходящи-
ми реагентами. Это было постепенно выяснено при изучении кинетики всех
трех реакций. Однако исследования, позволившие раскрыть наиболее важ-
ные закономерности, раньше всего были проведены на примере реакции
диазотирования. Поэтому в обзоре кинетических данных будет рассмотрена
реакция диазотирования, чтобы проследить общую нить доказательств, а дру-
гие реакции будут приведены главным образом для того, чтобы отметить
определенные сходства или отличия.
Второй общий вывод, к которому приходят на основании экспериментальных
данных, состоит в том, что теоретически нитрозирование сильно отличается
от нитрования. Нитрование может осуществляться любым теоретически
возможным переносчиком нитрония NO2X, где X-— основание любой
основности от бесконечно малой (когда оно отсутствует) до верхнего возмож-
ного предела, например:
NO+, NO2OH£, NO2Bt, NO2NO3, NO2NO2, NO2OCOCH3, N020H
В действительности нитрование осуществляется почти исключительно ионом
NO2, а главная функция любого другого нитрильного соединения состоит
в генерировании NO*. Подобно этому, нитрозирование теоретически может
осуществляться любым переносчиком нитрозония NOX, например:
NO+, NO —OHJ, NOBr, NONO3 [7], NONO2, NO —ОСОСН3, NO-ОН
Различие состоит в том, что нитрозирование почти всегда осуществляется
именно этими переносчиками, которые во многих случаях взаимно превра-
щаются друг в друга.
Максимальное упрощение эксперимента достигается исключением возможно
большего числа анионов, которые образуют ковалентные нитрозильные
соединения, например при работе с разбавленным водным раствором нитри-
та, слегка подкисленным хлорной кислотой. В этих условиях число возмож-
ных переносчиков нитрозония уменьшается до четырех: NO+, NO— ОЩ,
NONO2, NO — ОН. Можно написать кинетические выражения для нитро-
зирования каждым из этих переносчиков [8]. В общем случае необходимо
принять во внимание следующие возможности: во-первых, рассматриваемый
переносчик всегда имеется в распоряжении, и определяющей скорость
стадией является его взаимодействие с амином, и, во-вторых, переносчик
образуется из исходных соединений медленно, и его образование определяет
скорость общей реакции, при этом реакция с амином происходит быстро.
Однако для некоторых переносчиков вопрос об их медленном образовании
даже не может возникнуть. Два таких переносчика, существующие в упомя-
нутых выше упрощенных условиях, — это молекулярная азотистая кислота,
которую вводят в реакцию в нужном количестве, и ее ацидиевый ион, кото-
рый, как полагают, образуется из нее в быстрой предравновесной стадии.
Для этих переносчиков ожидаемые кинетические выражения даны уравне-
ниями (1) и (2), приведенными ниже:
HNO2, медленная атака:
Скорость ос [Amhh][HNO2] (1)
H2NOJ, медленная атака:
Скорость ос [Amhh][HNO2][H+] (2)
Ясно, что в этих и других формулах берется истинная, а не стехиометрическая
концентрация. Так, «амин» означает амин в виде свободного основания.
33 — 0772
513
Глава VIII
a <<HNO2»— недиссоциированную молекулу кислоты. Единственной особен-
ностью строения, которая никак не отражена в формулах, является сольва-
тация; поэтому ион водорода обозначен Н+.
Если переносчиком служит азотистый ангидрид, то необходимо учесть воз-
можность как его медленного взаимодействия с амином, так и медленного его
образования. В первом случае нужно предположить существование пред-
равновесия
2HNO2 N2O3+H2O
Вследствие этого необходимо принять, что скорость контролируется прямой
реакцией, и предположить, что все атомы, участвующие в равновесии, входят
в переходное состояние. Тогда выражения для скорости будут следующие:
N2O3, медленная атака:
Скорость ос [Amhh][HNO2]2 (3)
N2Oa, медленное образование:
Скорость ос [HNO2]2 (4)
Наконец, когда нитрозирующим агентом служит NO+, скорость его медлен-
ного взаимодействия с амином будет зависеть от следующего предравновесия:
hno2+h+ no++h2o
в то время как скорость его медленного образования будет зависеть или от
медленного отщепления воды от образовавшегося в предравновесии иона
ацидия, или от медленного отщепления нитритного иона от образовавшегося
в предравновесии азотистого ангидрида
I I
NO|----ОН2+ NOI---NO2
I I
Соответствующие кинетические уравнения (5)—(7) приведены ниже:
NO+, медленная атака:
Скорость ос [Amhh][HNO2][H+] (5)
NO+. медленное образование из H2NOJ:
Скорость ос [HNO2][H+] (6)
NO+, медленное образование из N2O3:
Скорость ос [HNO2]2 (7)
Следует отметить, что некоторые уравнения повторяются: уравнение (2)
неотличимо от уравнения (5), а уравнение (4) — от уравнения (7).
Кинетика диазотирования впервые была описана Ганчем и Шуманом [9]
в 1899 г. Работа проводилась в 0,002 н. НС1. Авторы получили кинетические
кривые второго порядка и без дальнейшей проверки предположили, что этот
порядок соответствует первому порядку по каждому из реагентов, что было
действительно справедливо для любой реакции второго порядка, исследован-
ной до того времени. Авторы также полагали, что амин реагирует в форме
соли, но это не отражается на кинетике при постоянной кислотности. Таким
образом, этой интерпретации соответствует уравнение (1). Полученные дан-
ные привели авторов к выводу, за который позднее их критиковали, что раз-
личные ароматические амины реагируют с одинаковой скоростью. В 1913—
1920 гг. Тассили [10] и в 1920 г. Бёзекен и сотр. [11] вновь исследовали эту
514
Замещение у гетероатомов
реакцию и снова пришли к выводу, что кинетика лучше соответствует вто-
рому порядку, первому по каждому из реагентов [уравнение (1)]. Однако
Бёзекен, применяя несколько более сильную кислоту (около 0,01 н.), чем
Ганч, наблюдал различие в скоростях диазотирования разных аминов, что
дало ему право критиковать сделанный ранее Ганчем противоположный
вывод. Тем не менее в 1935 г. Рейли и Драмм [12] снова исследовали скорости
диазотирования в очень разбавленной кислоте (около 0,002 н.) и показали,
что константы скоростей второго порядка для трех аминов, специально подоб-
ранных таким образом, что для них можно было ожидать различной реак-
ционной способности аминогруппы, одинаковы. Эти данные заставили Бёзе-
кена [13] вновь выразить свое сомнение по этому поводу.
Однако к этому времени были получены принципиально новые результаты.
В 1928 г. Тейлор [14] наблюдал третий кинетический порядок [уравнение (3)]
при дезаминировании первичных алифатических аминов, а в 1929 г. Тейлор
и Прайс [15] получили тот же результат для N-нитрозирования вторичных
алифатических аминов. В 1931 г. Абель [16] установил, что реакция самого
аммиака с азотистой кислотой также имеет третий кинетический порядок.
Наконец, в 1936 г. Шмид [17] обнаружил третий кинетический порядок при
диазотировании анилина, однако в более кислой среде, чем до него исполь-
зовалась, а именно в 0,2 н. кислоте. Шмид писал так, будто ему ничего не
было известно о втором кинетическом порядке диазотирования, о котором
все время сообщалось с 1899 по 1935 г.; вслед за ним другие авторы до 1950 г.
поддерживали этот «заговор молчания»: даже Гаммет [18], который первый
в 1940 г. понял, что уравнение (3) может означать нитрозирование азотистым
ангидридом, не упомянул, что указанное расхождение требует объяснения.
Некоторые авторы вплоть до 1951 г. утверждали, что уравнение (3) экспери-
ментально ошибочно, так как оно включает «не нужную» молекулу азотистой
кислоты [19]; однако на самом деле это уравнение не является ошибочным [20].
Противоречие было разрешено в 1950 г., когда Хьюз, Ридд и автор книги
[21] показали, что, во-первых, кинетический порядок диазотирования дей-
ствительно уменьшается от третьего до второго при уменьшении кислотности
от децинормальных до миллинормальных концентраций, и, во-вторых, в от-
личие от реакции третьего порядка, которая правильно отражалась уравне-
нием (3), реакция второго порядка на самом деле имела не первые порядки
по каждому из реагентов, а нулевой порядок по амину и второй по азотистой
кислоте [уравнение (4)] [21]. Наблюдавшийся переход от нулевого к первому
порядку по субстрату при постоянном втором порядке по азотистой кислоте
полностью доказывает, что переносчиком нитрозония служит N2O3. Эти
аргументы соответствуют аргументам, впервые примененным для доказатель-
ства того, что при ароматическом нитровании «переносчиком» нитрониевого
иона является сам этот катион: изменение кинетической формы обусловлива-
лось изменением судьбы субстрата, который конкурировал с водой в реак-
ции с переносчиком. В настоящем случае при миллинормальной кислотности
в растворе так много амина в форме свободного основания, что амин полно-
стью подавляет конкуренцию воды и поэтому скорость диазотирования пред-
ставляет собой скорость чисто неорганического процесса [уравнение (4)],
приводящего к азотистому ангидриду. Отсюда ясно, почему Ганч и другие
нашли, что все амины диазотируются с одной и той же скоростью. Отсюда
также ясно, почему Бёзекен на наблюдал этого: в его случае не было чистого
второго порядка, так как сантинормальная кислотность должна соответ-
ствовать переходной кинетической области. Однако при децинормальной
кислотности амина в форме свободного основания так мало, что вода пред-
ставляет собой гораздо более сильный конкурент за переносчика нитрозония;
33* 515
Глава VIII
это приводит к тому, что благодаря предравновесию в растворе появляется
постоянный «фонд» азотистого ангидрида, который медленно расходуется на
реакцию с субстратом со скоростью, определяющейся уравнением (3). Этот
«фонд» часто можно обнаружить по бледно-зеленому окрашиванию (азотистый
ангидрид имеет глубокую синюю окраску, а нитрит-ион — слегка желтова-
тую) при диазотировании в этих условиях. При такой кинетической форме
разные анилины не будут диазотироваться с одинаковой скоростью, что
ясно из данных табл. 132. Таким образом, участие N2O3 в качестве эффек-
Таблица 132
Константы скоростей третьего порядка [уравнение (3)] (л2-моль-2.с-1)
для дезаминирования, нитрозированпя и диазотирования через предравновесное
образование азотистого ангидрида в разбавленной водной азотистой кислоте
Амин Темпера- тура, °C Ю-5й3 Амин Темпера- тура, °C 10"3А3
NH3 ch3nh2 k-C3H7NH2 N-7 (CH3)2NH ’заминирован 25 25 25 I итрозироваъ 25 ие 0,04 4,8 2,8 те 4,0 Диазот C0H5NH2 n-CH3OCeH4NH2 ;i-C1C6H4NH2 7Z-(CH3)JNC6H4NH2 грование 25 0 0 0 и 27 3,11 3,56 0,92 0,14
тивного переносчика нитрозония в этих условиях можно считать дока-
занным [22].
Такое же изменение кинетики наблюдали Калацис и Ридд [23] при N-нитро-
зировании N-метиланилина. Вновь при повышении кислотности до 0,1 н.
наблюдался полный переход от кинетического уравнения (4) к уравнению (3).
Дальнейшее исследование в разбавленной водной хлорной кислоте позволило
открыть другой переносчик нитрозония [24]. Эффект состоял в увеличении
скорости, которое не могло быть объяснено уравнениями (3) и (4), так как,
согласно этим уравнениям, уменьшение концентрации азотистой кислоты
должно резко уменьшать значения слагаемых скорости, зависящих от
[HNO2]2- Роль этого нового процесса в сравнении с квадратичными членами
также возрастала с увеличением концентрации кислоты (которая все время
оставалась разбавленной) и при использовании в качеств субстратов несколь-
ко менее основных аминов, таких, как о-хлор- и и-нитроанилины. Эта реак-
ция катализируется ионами водорода, а ее кинетика удовлетворяет уравне-
нию третьего порядка, которое дважды встречалось выше [уравнения (2)
и (5)]. Поэтому реакция должна соответствовать нитрозированию или ациди-
евым ионом азотистой кислоты H2NO2, или нитрозониевым ионом NO+.
образующимся в предравновесной стадии. Выбор между этими альтернатива-
ми был сделан при изучении конкуренции между кислотами в таком простом
каталитическом процессе и путем сравнения катализа кислотой и галогенид-
ионом, который будет рассмотрен ниже.
Уже давно известно, что хлорид-ион представляет собой мягкий катализатор
диазотирования, бромид-ион — более сильный катализатор, а иодид-ион —
еще более сильный. Шмид [25] показал (для хлорида и бромида), что в при-
сутствии галогенид-ионов выражение для скорости содержит член типа
уравнения (8), приведенного ниже (X- = С1“, Вг-, I-). Гаммет [18] пред-
576
Замещение у гетероатомов
положил, что это уравнение могло бы означать участие нитрозилгалогенида
в качестве переносчика галогена. Хьюз и Ридд [26], начав с диазотирования,
протекающего согласно этому уравнению, смогли (для бромида и иодида)
путем применения несколько более слабых основаншг в очень большой кон-
центрации настолько увеличить реакционную способность субстрата по срав-
нению с водой, что переносчик нитрозония полностью захватывался субстра-
том. В этом случае скорость перестала зависеть от концентрации субстрата
и контролировалась только теми неорганическими процессами, в которых
образуется переносчик; при этом наблюдался переход от уравнения четвер-
того порядка (8) к уравнению третьего порядка (9). Такое изменение кинети-
ческой формы доказывает, что переносчиком действительно служит нитро-
зилгалогенид NOX.
NOX, медленная атака:
Скорость ос [Амин] [HNO2][H+][X~] (8)
NOX, медленное образование:
Скорость ос [HNO2][H+][X~] (9)
Аргументация совпадала с таковой для идентификации в качестве переносчи-
ка азотистого ангидрида, т. е. нитрозилнитрита. Уравнения (3) и (4) можно
перевести в форму, соответствующую уравнениям (8) и (9) соответственно:
тогда X- в последних уравнениях будет соответствовать NO2.
Точная интерпретация неоднозначного уравнения (2)/(5) в настоящее время
не может быть осуществлена [27]. Поскольку первоначально образующийся
ион NO + и координирующийся ион, скажем Вг-, могут моментально дать
NOBr, предельная скорость диазотирования по уравнению (9), которое отра-
жает скорость образования NOBr, вероятно, будет равна скорости образова-
ния NO+. Если же диазотирование по уравнению (2)/(5) зависит от пред-
равновесной стадии образования NO+, то скорость этой реакции не могла
бы превысить скорость реакции (9). Однако было найдено, что уравнение
(2)/(5) и уравнение (9) вносят совершенно не зависимый друг от друга вклад
в общую скорость и, кроме того, каждый член в выражении для скорости
путем соответствующего изменения концентрации можно сделать превышаю-
щим другой. Это показывает, что ион нитрозония не входит ни в уравнение
(2)/(5), ни в уравнение (9). Поэтому первое уравнение соответствует уравне-
нию (2), т. е. нитрозированию через ацидиевый ион азотистой кис-
лоты. Последнее уравнение соответствует образованию нитрозилгалогенида
через H2NO2+.
Имеется и другой путь, с помощью которого анионы, отличные от нитрит-
иона, или, как их можно называть, «чужие» анионы, могут участвовать
в нитрозировании: они могут катализировать образование азотистого ангид-
рида, который является истинным нитрозирующим агентом. Наиболее легко
наблюдать такое действие в случае анионов слабых кислот — буферных анио-
нов типа ацетата или фталата. Эти анионы в контролирующей скорость ста-
дии могут образовывать нитрозильные соединения, например нитрозилаце-
тат, а затем либо прямо нитрозировать амин, либо взаимодействовать с нит-
рит-ионом с образованием азотистого ангидрида, который далее нитрозирует
амин; при условии, что все эти реакции замещения быстрые, получим кинети-
ку, согласующуюся с уравнением (9). Однако если первоначально образовав-
шееся нитрозильное соединение атакует нитрит-ион и если также при умень-
шении концентрации нитрита концентрация нитрозильного соединения дости-
гает равновесной, так что контролирующей скорость становится его атака
на нитрит-ион, а последующее нитрозирование азотистым ангидридом все
517
Глава VIII
еще остается быстрым, то будет наблюдаться переход к кинетической форме,
выражаемой уравнением (10)
NZO3, медленное образование из NOX:
Скорость ос [HNO2][H+][X-][NO2] I
Скорость ос [HNO2]2[X-] |
Переход от уравнения (9) к уравнению (10) наблюдается, когда X- = ацетат
или фталат [28]. Скорость, выражаемая уравнением (10), соответствует ско-
рости реакции нитрозилацетата или нитрозилфталата с нитрит-ионом с обра-
зованием N2O3.
Если в качестве предшествующего равновесия рассматривать перенос про-
тона между кислотами в воде, то уравнение (9) может означать только бимо-
лекулярную атаку нуклеофила Х“ на первоначально образующийся ион
ацндия, а нитрозирование по уравнению (9) соответствует следующему неор-
ганическому Sjy2-npopeccy
X- + NO-Q)H2+ —-> X—NO + ОН2 (9а)
Sn2
Уравнение (4) представляет собой особый случай — самодегидратацию (4а)
азотистой кислоты путем бимолекулярного нуклеофильного замещения сопря-
женным основанием кислоты в сопряженной кислоте кислоты
NOr + NO—X-----> NO2—NO + X-
Sn2
Другой специальный случай, изученный Стедманом [29], состоит в атаке
азид-ионами, которая приводит к нитрозилазиду, известному при низкой
температуре, но выделяющему азот и закись азота мгновенно при комнатной
температуре
N3~ + NO-Q)H2+ > N3—NO + OH2 -> N2 + N2O + H2O
Sn2
Еще~0Д1гим] специальным случаем является нитрозирование ионом ацидия
азотистой кислоты, протекающее по уравнению (2). В этом случае роль нукле-
офила выполняет амин
RNH2 + NO-Q)H2+ > RNH2NO+ + ОН2
Sn2 [
RNHNO +H+
(2a)
Другие специальные случаи, когда нуклеофилом служит оксисоединение,
претерпевающее О-нитрозирование, будут рассмотрены в следующем
разделе.
Уравнение (9) выражает скорость превращения первоначально образующе-
гося нитрозирующего агента H2NOj во второй нитрозирующий агент, т. е.
нитрозилгалогенид или нитрозилкарбоксилат. Уравнение (10) выражает
скорость превращения этого второго нитрозирующего агента в третий
NOr + NO-Q)H2+-----> NO2-NO + ОН3 (Юа)
Sn2
Нитрозирование всеми этими косвенно образующимися нитрозирующими аген-
тами [уравнения (8) и (3)], подобное неорганическим SN2-npon;eccaM, представ-
518
Замещение у гетероатомов
лено здесь в форме (8а)
RNH2 + NCM-k-->RNH2NO + X-
Sn2 j
RNHNO +H+
(8a)
Такие превращения путем бимолекулярного нуклеофильного замещения,
вероятно, являются общим свойством нитрозильных соединений. Все кине-
тические данные, относящиеся к диазотированию или другим процессам
N-нитрозирования в разбавленных водных растворах кислот, можно сум-
мировать в виде схемы (рис. 45) взаимного превращения по механизму Sn2
переносчиков нитрозония, любой из которых может нитрозировать субстрат
18, 21]. Каждая реакция на этой схеме, включая нитрозирование субстрата,
Общий
кислотный катализ
NOX
Отсутствие катализа
Рис. 45. Нитрозирование как неорганическая реакция SN2-Tnna.
может быть сделана контролирующей скорость и изучена кинетически; циф-
ры у стрелок соответствуют номерам уравнений, приведенных выше, которые
выражают кинетику реакций контролируемых стадий, к которой относится
данная стрелка. Первой стадией, конечно, является мгновенный захват
протона азотистой кислотой, в результате чего в предравновесной стадии
образуется ион ацидия. В этом ионе молекула воды занимает место, которое
при нитрозировании должно быть занятым молекулой амина. Замещение
воды амином может произойти непосредственно. Однако быстрее происходит
замещение воды нитрит-ионом и далее замещение нитритного иона амином.
Аналогично, если имеются некоторые «чужие» анионы X", они могут заме-
щать воду быстрее, чем нитрит-ионы, а затем они сами будут замещаться
амином.
Ридд [22] из опытных данных сделал вывод, что, хотя механизм нитрозирова-
ния с участием H2NO2 наиболее прост, он очень легко сменяется механиз-
мом с участием N2O3 в отсутствие «чужих» ионов или механизмом с общим
кислотным катализом в присутствии таких ионов. Он также объяснил дру-
гой экспериментальный факт, состоящий в том, что механизм с участием
H2NOj легче всего наблюдать для производных анилина типа о-хлор- или
и-нитроанилина, которые обладают ослабленными как основными, так,
вероятно, и нуклеофильными свойствами вследствие наличия электроотри-
цательных заместителей. В табл. 133 приведены константы скоростей третьего
порядка для реакций, скорость которых контролируется N- или О-нитро-
зированием и другими формами нитрозирования ряда нейтральных и анион-
ных нуклеофилов (Nuc) в соответствии с уравнением
CKopoCTb = /ca[Nuc][HNO2][H+] (2)
519
Глава VIII
Таблица. 133
Константы скоростей третьего порядка [уравнение (2)]
(л2-моль~2-с-1) для реакций, скорость которых контролируется
нитрозированием ионом H2NOf в разбавленной водной кислоте
при О °C [22]
Соединение ,!з Соединение «з
N-Нитрозирование Нитрозирование анионов
о-Хлоранилип 175 Нитрит-ион 1893
о-Н итроанилин 145 Тиоцианат-ион 1440
п-Нитроапилип 161 Азид-ион 2340
2,4-Динитроанилин 3,7 Хлорид-ион 975
Бромид-ион 1170
О-Н ит розировани е Иодид-ион 1370
Аскорбиновая кислота 63 Ацет ат-ион 2200
Вода ~4а Аскорбат-ион ~ 2000
а Величина для воды, которая являлась также
для [Н2О1=5 5 М и поэтому, конечно, условна.
растворителем,
рассчитывалась
В настоящее время основность HNO2 неизвестна, и поэтому зти константы
третьего порядка нельзя превратить в константы второго порядка ([уравне-
ние (2')]:
CKopoCTb=fc2[Nuc][H2NOJ] (2Z)
которые соответствуют контролирующему скорость процессу типа SN2.
Однако известно, что умножение просто на константу равновесия может
настолько изменить всю серию констант, что и серия констант второго поряд-
ка, и серия констант третьего порядка, приведенные в табл. 133, будут под-
разделяться на две группы в соответствии с зарядом нуклеофильного суб-
страта; напомним, что в ряде случаев заряд оказывает лишь небольшое влия-
ние. Следовательно, можно предположить, что скорости бимолекулярных
реакций могут достигать скоростей реакций, происходящих при любом столк-
новении реагентов, энергия активации которых не выше 4...5 ккал/моль
(16,75-103 ... 20, 93-103 Дж/моль), т. е. величины, необходимой для удаления
растворителя перед образованием частиц реагента. Главным эффектом стро-
ения в этом случае было бы влияние анионного заряда на увеличение сечения
столкновения с катионным реагентом, т. е. увеличилась бы скорость столкно-
вения. Поэтому на вышеприведенной схеме абсолютные скорости реакций,
обозначенных цифрами 4 и 9, в общем будут велики по сравнению со скоро-
стью реакции 2.
Константы скоростей третьего порядка, определяемые уравнением
CKopocTb = Aa[Nuc][HNO2]2 (3)
можно, как это сделано в табл. 132, приближенно выразить через константы
скорости второго порядка контролирующего скорость S ^-замещения
[уравнение (3')]:
Скорость = A2[Nuc][N2Oa] (3Z)
Константа равновесия [N2O3]/[HNO2]2 известна приблизительно. Для нитро-
зирования анилина азо тистым ангидридом в воде при 20 °C к2 имеет величину
0,9-Ю7 л-моль"1 -с'1. Это на несколько порядков меньше константы скоро-
520
Замещение у гетероатомов
сти реакции, идущей при каждом столкновении. Поэтому нитрозирование
N2O3 должно быть реакцией, имеющей среднюю энергию активации, которая
может изменяться при изменении нуклеофильной силы субстрата при вве-
дении полярных заместителей. Поэтому несмотря на то, что примеры, при-
веденные в табл. 132 и 133, мало перекрываются, для того чтобы непосред-
ственно проверить теорию, Ридд сделал вывод, что реакция, описываемая
уравнением (3'), будет более чувствительной к полярным влияниям, чем
реакция, подчиняющаяся уравнению (2'). Таким образом, можно понять,
почему последняя реакция становится относительно более важной, в то вре-
мя как первая замедляется электроотрицательными заместителями типа
нитрогруппы.
Катализируемое кислотами зависящее от положения предравновесия обра-
зование нитрозилгалогенидов должно быть очень сходным с реакциями,
проходящими при каждом столкновении. Шмид [30] перевел константы чет-
вертого порядка катализируемого хлористым водородом диазотирования,
которому соответствует уравнение (8)
CKopocTb = fc4[Nuc][HN02][H+][Cl-] (8)
в константы второго порядка для контролирующей скорость 8к2-стадии,
которая соответствует уравнению типа (8'),
CKopoCTb = fe2[Nuc][NOCl] (8')
используя известную константу равновесия [NOG1]/[HNO2][H+][G1-]. Кон-
станты, приведенные в табл. 134, на один или два порядка меньше констант-
скоростей столкновения двух молекул.
Таблица 134
Константы скоростей второго порядка [уравнение (8а)] (л-моль-1 -с-1)
диазотирования; скорость контролируется нитрозированием
нитрозилхлоридом в разбавленной водной кислоте при 25 °C [30J
Амин Й2 Амин Й2
Анилин о-Толупдин .и-Толуидпн п-Толуидин 2,60-100 2,44-109 2,70-109 3,0-109 о-Хлоранилин ж-Хлоранилин л-Хлоранилин 1,16-109 1,63-109 1,89-109
Попутно отметим контраст между нитрозированием, которое происходит
через множество переносчиков, взаимно превращающихся друг в друга
в результате неорганических реакций, подобных S ^-замещению (рис. 45),
и нитрованием, схематически представленным на рис. 46, при котором каж-
дый из возможных переносчиков вначале претерпевает гетеролиз типа S^l
с образованием одного и того же эффективного переносчика — иона нитро-
ния. Очевидно, что класс нитрозильных соединений труднее гетеролизуется
до иона нитрозония, чем класс нитрильных соединений до иона нитрония.
Лидстоун [31] отметил, что подобное различие существует в легкости дегидра-
тации муравьиной и угольной кислот, которым изоэлектронны ионы H2NO2
и H2NO|, если считать, что термодинамически стабильный изомер первого
иона протонирован по азоту, вследствие чего необходим предварительный
+ 4*
эндотермический сдвиг протона О = NH(OH) О = N — ОН2, теплота
которого входит в энергию активации дегидратации.
Все рассмотренные выше механизмы диазотирования или нитрозирования
применимы к разбавленным водным растворам кислот. При расширении
521
Глава VIII
этих исследований на область более сильных кислотных сред Ридд и сотр. [32]
открыли еще два механизма N-нитрозирования, в обоих из которых субстра-
том являлся не амин, а ион аммония.
Вышеописанный механизм диазотирования можно применять для любого
производного анилина при концентрации кислоты ниже 0,5 н. Однако для
слабо нуклеофильных анилинов, таких, как нитроанилины, механизм с уча-
стием иона H2NOj [уравнение (2)] применим для концентрации кислоты до
NO2X
н+
HNO3s=i H2NO3+s=i:NO2+ >RHNO2+ »RNO2
Быстро H2O^- RH Быстро
n2cv
Рис. 46. Нитрование через первоначальный 8ц2-гетеролиз потенциальных переносчиков
иона нитрония.
3 н. [32]. При диазотировании же более сильно нуклеофильных анилинов —
самого анилина и толуидинов и при N-нитрозировании N-метиланилина при
концентрации кислоты между 0,5 и 3 н. осуществляется новый механизм.
В этом интервале кислотности в случае анилинов промежуточной нуклео-
фильности типа хлоранилинов новый механизм прекрывается с механизмом,
которому соответствует уравнение (2). Если принять в расчет солевые эффек-
ты, кинетическое уравнение для этого нового механизма будет следую-
щим [32]:
Скорость a [ArNHJ][HNO2]/i0 (И)
Уравнение имеет такой вид вследствие того, что оно применимо к области
кислотности, когда существуют различия между [Н+], Ао и /0. Произведение
[HNO2]/»q пропорционально [H2NO2] и означает участие этого иона, обра-
зующегося в предравновесной стадии. (Если бы нитрозирующим аген-
том был NO+, то соответствующее произведение имело бы вид [HNO2]j0 [33].)
В рассматриваемой области кислотности в случае аминов, к которым приме-
нимо уравнение (11), абсолютные скорости слишком велики — гораздо выше
средней скорости столкновений, и это не позволяет считать, что нитрозиро-
вание происходит через свободный амин. Таким образом, уравнение (11)
можно заменить уравнением (И'):
Скорость ос [ArNHJ][H2NOJ] (И')
Первое, что можно предположить, — это то, что при таком механизме ско-
рость контролируется одностадийным ЗвЗ-нитрозированием ионом H2NO2
с выбросом протона от анилинового атома азота; однако изучение влияния
заместителей в кольце анилина [34] свидетельствует в пользу значительно
большей вероятности атаки реагента на смежную л-систему. Замещение
заканчивается перегруппировкой, т. е. общий механизм аналогичен механиз-
му, предложенному Иллюминати (гл. VII, разд. 10,ж) для хлорирования
и бромирования гексаметилбензола и других полиметилбензолов в боковую
цепь бромом в уксусной кислоте. В пользу этого свидетельствует также ряд
относительных скоростей диазотирования замещенных анилинов в 3 н. хлор-
ной кислоте
Н п-СН3 .м-СП3 и-С1 .и-С1 п-ОСН3 .и-ОСП3
1 7,4 6,8 0,20 0,41 9,5 18,6
622
Замещение у гетероатомов
Наиболее сильную активацию оказывает ж-метоксигруппа, и в общем мета-
заместители имеют кинетический эффект, сравнимый с эффектом пара-заме-
стителей. Было предположено, что ион нитрозония, передаваемый ионом
H2NO2, присоединяется в а- или орто-положение, образуя нитрозоаммоний-
циклогексадиенильный ион, который путем перегруппировки, детали кото-
рой в настоящее время не выяснены, превращается в соединение с нитрозо-
группой у аммонийного атома с последующим отщеплением протона
+ + Медленно + /NO +
ArNH3-4-NO —ОН2---------* Аг/ + —» ArNH2NO + H+
XNK3 I (11а)
ArN+ ArNHNO + H+
Второй механизм с участием иона анилиния в качестве субстрата, также
открытый Чаллисом и Риддом [35], наблюдался в приблизительно 60%-ном
водном растворе хлорной кислоты и в 70% ной водной серной кислоте. Его
специфическая особенность состоит в определяющем скорость переносе про-
тона от питрозированного субстрата к среде, протоноакцепторные свойства
которой понижены; этот механизм аналогичен механизму N-нитрования при
высокой кислотности, предложенному Халеви (разд. 1,а).
Данные по нитрозированию при этих кислотностях состоят в следующем.
При любой одной и той же кислотности скорости диазотирования анилина,
n-толуидина и п-нитроанилина пропорциональны произведению [стехио-
метрический амин] [стехиометрическая азотистая кислота]. При указанной
кислотности первый множитель соответствует полному образованию ArNH*
который и должен служить субстратом, поскольку абсолютные скорости
слишком велики, чтобы рассматривать в качестве субстрата свободное осно-
вание. При этой кислотности стехиометрическая азотная кислота в основном
находится в виде иона нитрозония [36], а поскольку этот ион не только при-
сутствует в преобладающем количестве, но и должен быть наиболее актив-
ным из специфических нитрозирующих агентов, практически точно можно
сказать, что второй множитель эквивалентен [NO+], т. е. происходит взаимо-
действие NO+ с ионом анилиния. При увеличении кислотности скорость
диазотирования уменьшается примерно в соответствии с квадратом обратной
величины /г0, т. е. уравнение для скорости имеет следующий вид:
Скорость a [ArNHJ][NO+]/iQ2 (12)
Это означает, что два протона, соответствующие протонам, отщепляющимся
согласно механизму (11а), или оба отщепляются в предравновесной стадии
обратимого процесса, или первый протон отщепляется в этой стадии, а вто-
рой — в определяющей скорость стадии реакции и медленнее при более
высокой кислотности, когда способность среды принимать протон умень-
шается. Чаллис и Ридд отдали предпочтение второй альтернативе, поскольку
скорость реакции в D2SO4/D2O была в 10 раз меньше, чем в H2SO4/H2O.
Такой большой изотопный эффект ясно указывает на медленный перенос
протона, поэтому можно предложить следующий механизм:
ArNH3 + NO+ Т--» ArNH2NO++H+
Быстро I
Медленно (12а)
ArNJ <— ArNHNO + H+
Первую стадию уравнения (12а) можно описать более детально, хотя доказа-
тельства механизма еще не опубликованы; т. е., пользуясь механизмом (11а),
523
Глава VIII
можно включить начальную стадию образования иона нитрозоциклогекса-
диенилия и последующую стадию миграции нитрозогруппы в боковую цепь.
Возможно, что стадия присоединения, идентичная первой стадии ароматиче-
ского С-нитрозирования, могла бы быть легко обратимой [37].
Что касается быстрых реакций, следующих за образованием нитрозопроиз-
водных первичных аминов, то единственным свидетельством об их механизме
служит строение, включая стереохимию, выделяемых продуктов. В аромати-
ческом ряду ион диазония является достаточно стабильным, чтобы его можно
было рассматривать как конечный продукт реакции. Как будет отмечено
в гл. XI, Ганч выделил ароматические первичные питрозамипы, а также диа-
зогидраты. Основываясь главным образом на результатах этой работы, обыч-
но считают, что в водных кислотных растворах быстрый процесс превращения
нитрозаминов проходит через стадию диазогидратов до ионов диазония
и что этот процесс осуществляется путем прототропной изомеризации через
ионы с последующей псевдоосновной ионизацией
_ н+ Н+
ArNHNO <----r-t ArI^-N=Q)- ?=±
Н+ -Н+
П с- Н+ +
z=zzz±ArN=VN-On----» ArN=N + Н2О
В алифатическом ряду ионы диазония нестабильны. В апротонных раствори-
телях они могут и не образовываться, и конечным продуктом может быть
ангидрооснование псевдооснования (гл. XI), а именно диазоалкан. Показано,
что метилнитрозамин, полученный в эфире при низких температурах, пре-
вращается в диазометан [38]
CH3NHNO -» CHSN = N —ОН -> CH8=N = N + H2O
Однако конечные продукты дезаминирования первичных алифатических
аминов в гидроксилсодержащих растворителях не могут образовываться
через стадию диазоалкана, поскольку Стрейтвизер и Шеффер [39] показали,
что указанная реакция протекает без изотопного обмена между а-водородом
и водородом растворителя. Бейкер, Купер и автор [40] в 1928 г. и Уитмор [41]
в 1932 г. предположили, что дезаминирование в обычных протонных средах
происходит через ион диазония и карбониевый ион
RNH —NO —> RN=NOH —* RNJ —> R+ —> Продукты
Эта идея первоначально не была принята, однако в 1950 г. [42] она была под-
тверждена стереохимическими данными, уже обсуждавшимися в гл. VII.
разд. 7, и, и поэтому в настоящее время не вызывает серьезных возражений,
хотя было много дискуссий о степени сольватации и времени жизни иона
диазония и карбониевого иона.
Продукты простейшей реакции дезаминирования — взаимодействия азоти-
стой кислоты с метиламином в разбавленной водной кислоте — были описаны
Остином [43] в 1960 г. В одной из серий экспериментов в растворе 0,5 моля
хлоргидрата метиламина и 1,5 моля нитрита натрия в 650 мл воды при мед-
ленном добавлении 0,4...0,6 моля 0,3 н. серной кислоты образуются следую-
щие продукты: метиловый спирт 6...25%, метилнитрит 45...35%, нитроме-
тан 6%, метилнитроновая кислота 10...12%, хлористый метил 13%; количе-
ство выделившегося азота на 3% превышало теоретическое, но не за счет
небольших примесей двуокиси углерода, а также цианистого водорода
и аммиака. Эта работа останется в истории органической химии по той
причине, что во всех более ранних описаниях реакции ничего не говорилось
524
Замещение у гетероатомов
о составе продуктов и изобретательные авторы учебников заполняли этот
пробел догадками, как часто бывает, не точными, но кажущимися вполне
правоподобными, чтобы скрыть существующий пробел. Никто до Остина не
обнаружил и даже не догадался, что главным продуктом является метил-
иитрит, который, будучи газом, выделяется вместе с хлористым метилом
и азотом, несмотря на попытки сконденсировать его. Метиловый спирт,
который, судя по учебникам, должен быть главным продуктом, обнаружен
лишь в небольших количествах: он образовывался бы в больших количествах,
если бы путем О-нитрозирования не превращался в метилнитрит. Дополни-
тельное количество метилнитрита и немного нитрометана образуются при
прямом захвате нитрит-иона ионом карбония. Нитрометан образовывался
бы в большем количестве, если бы он далее не превращался путем С-нитро-
зирования в метилнитроловую кислоту. Определенная часть этого вещества
разлагается до ожидаемых продуктов гидролиза его продукта дегидратации
NOS — CN (это соединение не известно), т. е. до HCN и продуктов, произо-
шедших из HCNO, а именно СО2 и NH3. Схема Остина приведена ниже:
Н2О С1-
СН3ОН <----(CHJ)-----> СН3С1
hno2 | NO£
CH3ONO CH3N02 hon=ch-no2—» hcn,co2,nh3
При дезаминировании высших первичных алифатических аминов карбоние-
вый ион имеет большие возможности: он может не только вступать во вторую
стадию замещения (разными способами), но также терять протон (реакция
элиминирования) либо перегруппировываться с миграцией водорода, или
метила и после этого присоединять нуклеофил или отщеплять протон. Все
это действительно происходит. Элиминирование будет рассмотрено в гл. IX.
а перегруппировки — в гл. X. Что касается непосредственно замещения
аминогруппы, то важным результатом является происходящая при этом
рацемизация, конкурирующая с инверсией (эти вопросы уже рассматрива-
лись в гл. VII, разд. 7,и). Например, н-бутиламин [39, 44] дает н-бутильные
соединения, н-бутены, emop-бутильные соединения (миграция водорода),
а при наличии а-дейтерия, создающего асимметрический а-углеродный атом,
образующиеся н-бутильные соединения рацемизуются и инвертируются
в сравнимой степени.
г. 0-Нитрозирование и связанное с ним окисление. Кинетика нитрозиро-
вания воды была исследована Бантоном, Ллевиллином, Стедмэном и другими
авторами [45] с применением изотопной метки 18О (на схеме отмечен звез-
дочкой)
HN02+H0H NO-6h + H2O
При таком обмене кислорода проявляются различные кинетические формы,
описанные выше для диазотирования, нитрозирования и дезаминирования
аминов в разбавленных водных растворах кислот; некоторые модификации
обусловлены изменением строения субстрата. Так, те кинетические факторы
N-нитрозирования, в которых не участвует амин, проявляются без измене-
ния, а те факторы, в которые входит концентрация амина, проявляются без
нее, так как новый субстрат — вода, будучи растворителем, имеет постоян-
ную концентрацию.
При низкой кислотности и высоких концентрациях (0,5...2,5 М) азотистой
кислоты скорость обмена выражается кинетическим уравнением (4)
Скорость ос [HNO2]2 (4)
525
Глава VIII
Скорость обмена равна скорости диазотирования при тех же условиях [45].
Это объясняется тем, что нитрозирование растворителя — воды (с отмечен-
ным звездочкой атомом кислорода на приведенной ниже схеме) происходит
через азотистый ангидрид и его скорость определяется скоростью образова-
ния N2O3
NO-OH4-H+ у -» NO-OH+
Медленно
NOi + NO-OH+-----------> NO2-NO-}-OH2 ,
* *
NO2-NO + OH2 --------> NO2+NO-OHJ
NO-OHJ----------> NO---- OH + H+
При низкой кислотности и при низких концентрациях азотистой кислоты
(около 10-3 М) кинетическое уравнение имеет иную форму [46]:
Скорость ос [HNO2][H+] (2")
Это уравнение соответствует вырожденному уравнению (2), когда субстратом
является растворитель, как при обмене кислорода. Уравнение соответствует
нитрозированию воды ионом H2NO2:
Н2О + NO-Q)H2+ Н2О—NO+ + ОН2
НО—NO + Н+ . (2а").
При введении азид-ионов, конкурирующих с водой, выделяющаяся закись
азота не содержала изотопа кислорода-18, т. е. азид-ион нитрозировался
частицей, не обменявшей свой кислород с кислородом воды; это доказывает,
что нитрозирование не включает предравновесного образования иона нитро-
зония [уравнение (5)], так как в этом случае образовавшийся ион нитрозония
должен был содержать 18О.
Сравнительно активные «чужие» анионы в достаточной концентрации могут
служить субстратами нитрозирования вместо нитрит-иона или воды. В этом
случае кислородный обмен происходит в соответствии с уравнением (9)
Скорость ос [HNO2][H+][X~] (9)
Это было показано для «чужого» ацетатного иона [47]. В таком случае ско-
рость контролируется стадией (9а) (при X- = (Л1...СОО’), причем обмен
протекает через нитрозилацетат (ацетилнитрит)
X- + NO-Q>H2+ -► X—NO + ОН2 (9а)
Последующие стадии аналогичны указанным выше для кислородного обмена
через азотистый ангидрид.
0-Нитрозирование насыщенных спиртов, вероятно, происходит просто, так
как эфиры азотистой кислоты стабильны. Однако кинетика их образования
пока не изучена.
Продукты О-нитрозирования фенолов и енолов неустойчивы, и при их обра-
зовании выделяется окись азота. В общем этот процесс соответствует одно-
электронному окислению. Первоначальным продуктом окисления моноатом-
ных фенолов или енолов является окси-радикал ВО-, который инициирует
цепные реакции, в результате чего могут образовываться полимерные про-
526
Замещение у гетероатомов
дукты. Однако двухатомные фенолы, такие, как гидрохинон или сопряжен-
ные ендиолы («редуктоны»), например аскорбиновая кислота, очень быстро
вступают во вторую стадию одноэлектронного окисления, давая дикетон
(хинон или дегидроаскорбиновую кислоту), представляющий собой продукт
двухэлектронного окисления. Стехиометрия таких процессов окисле-
ния представлена ниже на примере аскорбиновой кислоты, где R =
= СН2(ОН)СН(ОН)
ОН ОН 0 0
I 1 н II
Ц.-.Т-- с 4-2HN02 —» С----С + 2NО + 2Щ0
II II
RCH — О —СО RCH —О—СО
Бантон, Дан, Лове и их сотр. [48] тщательно изучили кинетику таких процес-
сов окисления, главным образом окисления аскорбиновой кислоты азотистой
кислотой в слабокислых водных или водно-диоксановых растворах без доба-
вок и с добавками «чужих» анионов. Они обнаружили следующие явления.
Два субстрата, присутствующие в сравнимых количествах, например молекула
аскорбиновой кислоты и аскорбат-ион, из которых последний специфически
более склонен к окислению, окисляются независимо, конкурируя между
собой. Окисление каждого из этих субстратов может происходить через
любой переносчик из одной и той же серии, а именно через ион H2NO2, N2O3
и в присутствии галогенид-ионов через нитрозилхлорид и нитрозилбромид,
которые известны в качестве агентов N-нитрозирования, диазотирования,
дезаминирования и кислородного обмена с водой. Из опытов, в которых опре-
деляющим скорость окисления любого субстрата было образование N2O3,
а не его реакция с субстратом, были получены скорости образования N2O3,
близкие к величинам, полученным из данных для различных реакций N-
и О-нитрозирования, приведенным выше. Большинство данных рис. 45
подтверждалось дважды — по одному разу для каждого субстрата. Един-
ственной стадией, не подтвержденной на этих примерах, была стадия 10,
а для аскорбат-иона — и стадия 2 (рис. 45). Бантон, Дан и Лове иллюстри-
ровали это семейство окислительных реакций следующей схемой:
НО ОН ON —О ОН
I I NOX | |
с. с----------> с==с
। । Медленно ] ।
RCH-O-CO RCH-O-CO
О- ONO
-NO I I
<----- c— —c
Быстро । 1
RCH-0 —CO
О- он
I I
RCH-O-CO
NOX Быстро
2. Нуклеофильное замещение у атомов элементов третьего
периода
а. Замещение у атома кремния. Нуклеофильное замещение для триалкил-
или триарилсилильных производных исследовалось кинетически, стерео-
химически или обоими методами одновременно.
527
Глава VIII
В 1957 г. Аллен и Модена [49] показали, что обмен хлора между трифенил-
силилхлоридом и радиоактивным хлоридом тетраэтиламмония в диоксане
имеет второй порядок, который они объяснили механизмом SN2
Cl- + (C6H5)3SiCl -> ClSi(C6H5)3-r ci- (Sn2)
Гидролиз силилхлорида в водном растворе диоксана имел первый порядок
и был чувствителен к анионному катализу; однако кислород в образовав-
шемся силилгидроксиде не содержал изотопной метки из растворителя —
воды. Очевидно, в этом случае не происходит быстрой, обратимой реакции
между силильным производным и водой.
С 1959 г. Соммер и Файр [50] исследовали стереохимию продуктов различных
реакций замещения у атома кремния в метилфенил-а-пафтилсилильных соеди-
нениях CH3(CeH5)(C10H7)SiX. Они описали «вальденовский цикл»
С12 в ССЦ LiAlHi в эфире
' (— )-СН3(С6Н5)(С10Н7)81С1
( + )-CH3(CeH5)(C10H7)SiH
(-)-CH3(CeH5)(C10H7)Sill
L1AIH4 в эфире СЦ в CCl j
( + )-CH3(CeH5)(Ci0H7)SiCl <
который интерпретировали как чередование электрофильного хлорирования
с сохранением конфигурации и нуклеофильного замещения хлора гидрид-
ным ионом с обращением конфигурации. Более интересно отсутствие валь-
деповского цикла при комбинации упомянутого выше нуклеофильного заме-
щения с процессом также нуклеофильного замещения, сопровождающимся
тем же результатом.
ЫА1Н4
(-)-CH3(CeH5)(C10H7)SiCl ——-> (-)-CH3(C6H5)(C10H7)SiH
В ЛрИри
-ОСН3 1АА1Н4
в СНзОН в эфире
----------> (_)-CH3(CeH5)(C10H7)SiOCH3--------
Если верна интерпретация «вальденовского цикла», то эти последние реак-
ции должны приводить к сохранению конфигурации. Соммер и Файр, прове-
дя соотнесения ряда относительных конфигураций, главным образом путем
рассмотрения изоморфизма, пришли к выводу, что вторая из последователь-
ных стадий удовлетворяет этому условию. Это означает, что ион тетрагидро-
алюмината может действовать двумя путями: в случае галогенов, как в цикле
Вальдена, замещение более легко осуществляется через атаку по одному
центру с инверсией конфигурации (S N2), однако в случае кислородсодержа-
щих групп, например метилатной, такая реакция идет труднее, чем двух-
центровое взаимодействие с субстратом, когда замещение происходит через
циклическое переходное состояние с сохранением конфигурации (SNi)
H3AIH + CH3(C6H5)(Cl0H7)SiCl^HSi(CloH7)(CsH5)CH3 + Н3А1С1- (SN2)
н-
CH3(C6H5)(C1OH7)S1 А1Н:
CH3(C6H5)(C10H7)SiH + Н3А1ОСН3
(SN1)
ОСН3
Замещение у гетероатомов
Важно отметить, что ацилоксильные соединения кремния, например
CH3(C6H5)(C10H7)SiOCOCH3, ведут себя подобно галогенным соединениям.
Они легко восстанавливаются ионом тетрагидроалюмината с обращением
конфигурации, а не с трудом, как алкоксипроизводные, приводящие к про-
дукту с сохранением конфигурации.
Показано, что механизм 8^2 является общим для нуклеофильного замещения
в силилхлориде и силилбромиде CH3(CeH5)(C10H7)SiCl и CH3(C6H5)(C10H7)SiBr.
Не только AIH4 и СН3О_, но и нуклеофилы mpem-C4H9O_, циклогексиламин,
Н2О, СН3ОН и ряд других замещают галогены с инверсией конфи-
гурации.
Преобладание стереохимической инверсии и поэтому, как можно предполо-
жить, S ^-механизма при замещении галогена в силилбромиде и силилхлори-
де, не является правилом для фторида. Некоторые реагенты замещают фтор
с сохранением конфигурации, т. е. двухцентровой атакой БтЛ-типа. Тенден-
ция к такому типу замещения больше, когда уходящей группой является
метилатная, и еще больше, когда уходящей группой является гидрид. Сом-
мер и Корте считают, что для данного замещающего агента отщепляющиеся
группы X образуют следующий ряд по уменьшающейся способности к отще-
плению: Cl > F > ОСН3 > Н (в соответствии с уменьшением кислотности
НХ), и поэтому необходимость двухцентровой атаки типа S^i, приводящей
к сохранению конфигурации, увеличивается в ряду Cl < F < ОСН3 < Н.
Для данной уходящей группы механизм будет зависеть от реагента, напри-
мер от того, насколько он сильный нуклеофил, а если это ионизующаяся
молекула, то насколько легко образуется анионная часть. Например, ион-
ность углеводородных соединений лития, вероятно, увеличивается в ряду
простой алкил < аллил или бензил < бензгидрил. Простые алкиллитиевые
соединения имеют наибольшую тенденцию к замещению с сохранением кон-
фигурации, поэтому можно заключить, что в этом случае реакция происходит
по согласованному механизму SNi. Бензгидриллитий имеет наибольшую тен-
денцию к замещению с инверсией и поэтому реагирует, вероятно, по SN2-
механизму. Изменение стереохимии реакции в указанных двух сериях пока-
зано ниже:
Х=С1 F ОСН3 н
Алкиллитий инв. сохр. сохр. сохр.
Аллпллитий инв. инв. инв. сохр.
Бензиллитий инв. инв. инв. сохр.
Бензгидриллитий инв. инв. инв. инв.
Стереохимия реакции, конечно, является свойством ее механизма, поэтому
желательно коррелировать стереохимические данные с независимыми выво-
дами о механизме. Эти выводы содержатся в кинетическом исследовании Ибор-
на и сотр. [51], в котором они шли путем, параллельным более раннему
исследованию Хьюза по изотопному обмену галогенид-иона у атома углеро-
да. Последнее позволило впервые количественно доказать обращение конфи-
гурации при 8х2-замещении у атома углерода (гл. VII, разд. 6,г). Иборн
изучал обмен меченой метоксигруппы в оптически активном метилфенил-а-
нафтилсилилметилате на метоксигруппу в метанольном растворе. Реакция
катализировалась кислотами и основаниями и очень сильно — метилатом
натрия. Метилатная группа субстрата была мечена радиоактивным тритием;
сравнивались скорости детритирования и рацемизации. Во всех условиях
скорость рацемизации была точно в два раза выше скорости детритирования,
т. е. метилатного обмена. Таким образом, не только замещение первоначально
34-0772
529
Глава VIII
образующимся метилат-ионом, но и сольволиз и кислотный сольволиз про-
исходят с количественным обращением конфигурации, например:
сн3о--;-(- )-cH3(c6H6)(C10H7)SiOCH3
—> (-Г )-CH3OSi(CI0H7)(CeII5) СН3 - р -осн3 (Sn2)
Учитывая неудачную попытку Аллена и Модены обнаружить какой-либо
диссоциативный или аддитивный механизм при нуклеофильном замещении
в силилгалогенидах, вероятно, можно сделать вывод, что не только анион-
ный обмен второго порядка, но и сольволиз относятся к S ^-замещению.
б. Замещение у атома фосфора. Фосфор был первым элементом третьего
периода, для которого было исследовано нуклеофильное замещение.
В 1953 г. и позже Достровский и сотр. [52] изучили кинетику сольволиза
в воде, водном и безводном этаноле, кинетику анионного замещения под
действием галогенид-, этилат-и фенолят-ионов, а также кинетику замещения
под действием первичных аминов от метиламина до /пренг-бутиламина в тех
же растворителях на примере ряда галогенсодержащих эфиров фосфора.
Субстратами были фосфинилхлориды и фосфинилфториды HalPOR3, фос-
фонилфториды FPO(R)(OR) и хлор- и фторангидриды эфиров фосфорной
кислоты HalPO(OR)2, содержащие R = метил, этил, изопропил, бензил
и фенил. Типы замещения иллюстрированы ниже, при этом в качестве продук-
тов образовывались фосфинаты, фосфонаты и фосфаты:
Н2О + С1Р0(СН3)2 НОРО(СН3)2 + НС1
OH- + FPO(C2H5)(OC21I5) —> HOPO(C2H5)(OC2H5) + F-
F_-J-C1PO(OC3II7-ujo)2 —> FPO(OC3H7-«3o)2 4- Cl-
Сольволиз имел первый порядок, а замещение под действием растворенных
нуклеофилов — второй, первый по каждому из реагентов. Влияние раствори-
теля и солей во всех случаях не согласовывалось с ионизационным механиз-
мом (SN1), а, как и следовало ожидать, соответствовало одностадийному би-
молекулярному замещению (SN2). Замещение нуклеофилами, растворенными
в Н218О, происходило без обмена кислорода с растворителем. В целом все
данные указывали на 8к2-механизм и свидетельствовали против процесса
диссоциации или образования промежуточного продукта присоединения за
счет расширения валентной оболочки.
С 1959 г. для кислородных эфиров фосфора было найдено несколько «ва.тьде-
новских схем». Согласно Грину и Хадсону [53], от 3-фенантренилметилфос-
финилхлорида можно перейти к двум энантиомерным фосфинатам в одну или
в две стадии замещения:
(4-)-Phen(CH3)POCl ( — )-Plien(CH3)POOCH3
(_)-Phen(CH3)POF (4-)-Phen(CH3)POOCH3
Обращение конфигурации происходит или на каждой стадии, или только на
одной. Вследствие формального сходства всех этих процессов более вероятно,
что обращение происходит на всех стадиях. Подобная схема для фосфонатной
серии была найдена Аароном и сотр. [54]. К этому случаю применимы те же
аргументы
( —)-СН3(ОС3Н7-изо) РОС1 —2S7 (4-)-СН3(ОС3Н7-изо)РООС2Н5
|ch3s-
(4-)-CH3(OC3H7-uao)POSCH3 ill'll. ( — )-СН3(ОС3Н7-изо)РООС2Н5 [55]
530
Замещение у гетероатомон
Менее очевидный пример был найден Грином и Хадсоном на примере омыле-
ния алкилтиофосфонатов с разрывом связи кислород — фосфор; при этом
наблюдалось количественное обращение конфигурации у атома фосфора.
Получен пиротиофосфонат с оптически активным центром только у одного
из двух (в других отношениях эквивалентных) атомов фосфора. Вследствие
симметрии молекулы точно половина всех гидролитических атак, приводя-
щих к продукту реакции — неполному эфиру, происходит у оптически актив-
ных атомов фосфора. Однако этого оказалось достаточно, чтобы произошла
полная рацемизация продукта; это доказывает, что рацемизация происходит
в два раза быстрее замещения, которое поэтому происходит с количествен-
ным обращением конфигурации. В приведенной ниже схеме оптическая метка
обозначена знаком градуса:
(C2H5O)(CH3)PSONa-]-C 1PS;CH3)(OC2H3) —>-
(C2H3O)(CH3)PS-O-PS(CH3)(OC2H3) 2^
—> (HO)(CH3)PS—О —PS(C1I3)(OC2H3)
Уже в 1962 г. Грин и Хадсон [56] применили для замещения у фосфора метод
Хьюза, состоящий в сравнении скоростей изотопного замещения у асиммет-
рического центра, кинетика которого известна, со скоростью, сопутствующей
рацемизации. Они показали, что при замещении метилатной группы, мечен-
ной 14С, в оптически активном метилфосфинате на немеченный метилат-ион
скорость рацемизации в два раза превышала скорость S ^2-замещения и по-
этому при каждом молекулярном акте замещения происходило обращение
конфигурации. Это было первым твердым доказательством стереохимического
8х2-правила при замещении у атома элемента, отличного от углерода. В при-
веденном ниже уравнении радиоактивная метка обозначена звездочкой:
* СНчОН *
СП3О-4-С6Н5(С2Н5)РООСН3 —3_> СН3ОРО(С2Н5)С6Н5 + СН3О-
Причиной отсутствия промежуточного продукта присоединения при замеще-
нии кислородных эфиров фосфора может быть то, что на сопряжение с 277-
электронами атомов кислорода уходит значительная часть связывающей силы
ЗсГорбиталей фосфора. Конечно, ситуация должна измениться в случае реак-
ций тетраалкилфосфониевых ионов. В 1930-х годах Фентон, Хей и автор кни-
ги [57] изучили разложение гидроокисей тетраалкилфосфония до окисей
триалкилфосфинов и алканов. Изучая влияние строения на соотношение
продуктов, они пришли к выводу, что образуется пятилигандный тетраал-
килоксифосфан, который отщепляет одну алкильную группу в виде карбани-
она. В 1954 г. Мак Ивен, Ван дер Верф и сотр. [59] выяснили механизм этой
реакции и более твердо подтвердили указанную концепцию, показав, что
реакция имеет третий кинетический порядок, первый по фосфониевому иону
и второй по гидроксильному иону. Поэтому превращение гидроокиси тетра-
бензилфосфония в окись трибензилфосфина и толуол можно представить или
трехстадийным механизмом
(С6Н5СН2)4Р+ + ОИ- (С3Н3СН2)4РОН
(СКП5СН2)4РОН + ОН- Медл!]1Х С3Н5СН2+(С((Н5СН2)3РО + Н2О
С6Н5СН?+Н2О С8Н5СН3 + ОН-
или четырехстадийным механизмом
(С;!Н5СН^4Р+4-ОН- ; (С3Н5СН2)4РОН
34* 531
Глава VIII
(CeH5CH2)4POH + ОН- , , » (С6Н5СН2)4РО- + Н2О
Медленно
(С6Н5СН2)4РО- ------> СвН5СН2+(С„Н5СН2)3РО
С6Н5СН2+Н2О --------> С6Н5СН34-ОН-
Оба механизма объясняют тот факт, что скорость увеличивалась, когда отще-
пляемая группа содержала электроноакцепторные мета- или тгара-заместите-
ли, которые могли бы ускорять отщепление карбаниона.
В стереохимической работе Мак Ивена и Ван дер Верфа был открыт и другой
механизм этой реакции. Авторы сделали вывод об обращении конфигурации
при образовании окиси фосфина и алкана из гидроокиси фосфония (два меха-
низма, приведенные выше) на основании того, что образующийся продукт
был энантиомером продукта, полученного методом, развитым ими для про-
ведения указанного превращения через циклическое переходное состояние,
когда конфигурация почти наверняка сохраняется. Дело в том, что гидро-
окись (-(-)-метилэтилфенилбензилфосфония дает толуол и окись (—)-метил-
этилфенилфосфина, тогда как тот же (-|-)-фосфониевый ион при отщеплении
протона н-бутиллитием дает фосфонийбензилид, присоединяющий бензальде-
гид с образованием фосфониевого бетаина, который разлагается на стильбен
и окись (+)-фосфина
/СН3
(-|-)-СвН5СН2 — Р С2Н5
ХС6Н5
C4H9Li
сн3 С2Н5 сн3
он- \ / \
---> С6Н5СН2-Р-ОН С6Н5СН3 + (-)-С2Н5-РО
СвН5 С6Н5
/СН3
С6Н5СН = Р С2Н5
\с6н5
С6Н5СНО + / сн3
------> С6Н5-СН-Р(-С2Н5
I \с6н5
С6Н5 —СН —О~
/СН.,
С6Н5СН + ( + )-ОР^-С2Н5
II \с6н5
С0Н5СН
Асимметричные третичные фосфины дают оптически активные стабильные
изомеры. Следует ожидать, что электрофильные аденды, имеющие секстет
электронов, например атом кислорода, атом серы или карбониевый ион,
будут взаимодействовать с валентными неподеленными электронами фосфора
и поэтому присоединяться с сохранением конфигурации. Именно это пред-
положение позволяет интерпретировать данные Хорнера [59], изложенные
ниже, вместе с доказательством инверсии, данным Мак Ивеном и Ван дер
Верфом для разложения гидроокисей фосфония. Хорнер получил оптически
активный метил-н-пропилфенилфосфин электролитическим восстановлением
оптически активной соли метил-н-пропилфенилбензилфосфония. Он пред-
положил, что эта реакция происходит с преимущественным сохранением
конфигурации. Затем Хорнер превратил фосфин в окись фосфина прямым
окислением и в его энантиомер двумя разными путями, включающими кватер-
низацию и последующее разложение образующихся гидроокисей фосфония
СН3
(—)-С3Н7 —Р —СН2С6Н5
С^н5
он- _ + / СН3
----> (_)_0 —рАСзН7
\ С„н5
СвНбСНаВг 2е
ш
Замещение у гетероатомов
СНз СН3
\ s \+
( + )-С3Н7 — Р —>( + )-С3Н7 — Р — S
С6Н5 С6Н5
СНз1
t
СНз
(4- )-С3Н7 — Р — SCH3
С6н5
Н2О2
СН
( + )-С3Н7-Р-О-
С6Н5
в. Замещение у атома серы. Автору не известно ни одного кинетического
исследования механизма нуклеофильного замещения у атома серы, однако
с 1925 г. стало ясно, что эти процессы обычно происходят с инверсией конфи-
гурации. В 1925 г., ко всеобщему удивлению, Филлипс [60] получил н-алкил-
сульфинаты (например, этил-тг-толуолсульфинат) в оптически активной фор-
ме. В ходе работы он показал, что одностадийная переэтерификация этого
эфира и нормального первичного спирта (например, бутилового)
ArSO — OR-LR'OH —> ArSO — OR'+ ROH
всегда сопровождается изменением знака вращения. Филлипс сделал вывод,
что обращение конфигурации происходит у атома серы. Несомненно, ориги-
нальный вывод Филлипса основывался на экспериментальных данных по
влиянию алкильной гомологии на оптическое вращение; однако этот факт
был объяснен, когда постепенно раскрытие физического механизма оптиче-
ской активности [61] привело к выводу, что гомологический структурный
инкремент, собственное оптическое поглощение которого обладает высокой
энергией, соответствующей далекой ультрафиолетовой области, может не
иметь значительного влияния на свойства спирального перехода в суль-
финильной группе в близкой ультрафиолетовой области, от которых зависит
оптическая активность сульфоновых эфиров в видимом свете. Следуя Филлип-
су, можно представить, что переэтерификация происходит по механизму SN2
Аг<"
R'OH + J^S+— OR—>R'O—+ ROH
Очевидно, можно составить вальденовский цикл, включающий подобное пре-
вращение R'-эфира в R''-эфир и вслед за этим в энантиомерный R-эфир, а по-
том повторить эти три превращения в обратном порядке.
Джонсон и сотр. [62] получили вальденовский цикл, основанный на инвер-
сии, которая сопровождает основной гидролиз алкоксисульфониевых ионов
в сульфоксиды. Первый такой пример относится к ахиральным соединениям,
имеющим плоскость симметрии; при этом происходила инверсия геометри-
ческой формы в циклической системе. Таким путем цнс-4-хлорфенилтиан-
окись этилировалась тетрафторборатом триэтилоксония в растворе в хлори-
стом метилене и образующийся этоксисульфониевый ион затем гидролизовал-
ся водным раствором едкого натра до транс-тианокиси, которая аналогичным
533
Глава VIII
этилированием и последующим гидролизом превращалась в гр/с-окись
В качестве хирального соединения [63] был выбран оптически активный бен-
зил-тг-толилсульфоксид, который в четыре стадии с помощью чередующихся
этилирования и основного гидролиза превращался в энантиомер, а затем —
в исходный изомер с 95 %-ной оптической чистотой (приводятся значе-
ния [cc]f>5)
0“ + ОС2Н5
! (C2H5)3OBFJ |
CH3CeH1^S+-CH2CeH5 -----------> CH3CGH4 — S+-CH2C6H5
Исходная-)-94,6е; конечная-(-89,9° Д-2О30
t он" 1 О|Н
1 (C2H5)3O+BFJ *
CH3C0H4-S+-CH2CeH5------------> CH3CcH4-S+-CH2CeH5
I I
OC2H5 0-
— 208° —92,4°
Эфиры сульфиновой кислоты при действии реактивов Гриньяра превращают-
ся в сульфоксиды. Независимо от того, реагируют ли реактивы Гриньяра
как таковые или, что кажется более вероятным, через первоначальную иони-
зацию (гл. VII, разд. 10,д), такие сильные нуклеофилы почти наверняка
будут взаимодействовать с атомом серы по S N 2-подобному механизму с обра-
щением конфигурации, если только на этот случай можно распространить
данные Филлипса и Джонсона
C2H5MgBr«z>CzH5"+ MgBr+
л-СН3С6Н4^^ /^С.Н4СН3-л
C2Hs+ ^S+—OCioH19->CzHrS^^ + ОСюНД
Мислоу и сотр. [64] показали, что это так. В ряде случаев, включающем
и приведенную выше реакцию между этилмагнийбромидом и ментил-и-
толуолсульфинатом, авторы провели корреляцию конфигураций хиральных
исходных соединений и продуктов реакций путем анализа их дисперсии опти-
ческого вращения в ультрафиолетовой области, таким образом доказав
инверсию.
Следующие проблемы, которые необходимо решить в этой области, очевидны.
Было бы очень желательно подтвердить предполагаемый механизм кинети-
чески и путем введения изотопной метки провести тест на наличие обратимо
образующегося промежуточного продукта присоединения.
534
Замещение у гетероатомов
СПИСОК ЛИТЕРАТУРЫ
1. Blackall Е. L., Hughes Е. D., Nature. 170, 972 (1952); Hughes Е. D., Ingold С. К.,
Pearson В. В., J. Chem. Soc., 1958, 4357.
2. Halevi Е. A., Ron Л., Speiser S., J. Chem. Soc., 1965, 2560.
3. Klein R., Mentser M., J. Am. Chem. Soc., 71, 5888 (1951).
4. Blackall E. L., Hughes E. D., Nature. 170, 972 (1952); Blackall E. L., Hughes E. D.,
Ingold С. K., Pearson R. B., J. Chem. Soc., 1958, 4366.
5. Bunton C. A., Halevi E. A., Llewellyn D. R., J. Chem. Soc., 1952, 4913.
6. Bunton C. A., Halevi E. A., J. Chem. Soc., 1952, 4917; Bunton C. A., Stedman G.,
J. Chem. Soc., 1958, 4240.
7. Fateley W. G., Bent H. A., Crawford В., J. Chem. Phys., 31, 204 (1959); Parts L., Mil-
ler J. T., Jr., ibid., 43, 136 (1965).
8. Ingold С. K., Substitution at Elements other than Carbon, Weizmann Science Press,
Jerusalem, 1959, Chap. 2.
9. Hantzsch A., Schumann M., Ber., 32, 1691 (1899).
10. Tassilly E., Compt. rend., 157, 1148 (1913); 158, 335, 489 (1914); Bull. soc. chim. (Fran-
ce), 27, 19 (1920).
11. Boeseken J., Brandsma N. F., Schoutissen II. A. J., Proc. Acad. Sci. Amsterdam, 23,
249 (1920).
12. Reilly J., Drumm P. J., J. Chem. Soc., 1935, 871.
13. Boeseken J., Schoutissen H. A. J., Rec. trav. chim., 54, 956 (1935).
14. Taylor T. W. J., J. Chem. Soc., 1928, 1099, 1897.
15. Taylor T. W. J., Price L. S., J. Chem. Soc., 1929, 2052.
16. Abel E., Schmid H., Schraftrank J., Zeit. phys. Chem. Bodenstein Festschrift, 1931, 510.
17. Schmid II., Z. Eloklrochem., 42, 579 (1936); Schmid H., Muhr G., Ber., 70, 421 (1937).
18. Hammett L. P., Physical Organic Chemistry, McGraw-Hill, New York and London,
1940, p. 294.
19. Earl J. C., Hills II. G., J. Chem. Soc., 1939, 1089; Dusenbury J. H., Powell R. E., J. Am.
Chem. Soc., 73, 3266, 3269 (1951).
20. Austin A. T., Hughes E. D., Ingold С. K., Ridd J. H., J. Am. Chem. Soc., 74, 555
(1952); Ewing G. J., Bauer N., J. Phys. Chem., 62, 1449 (1958); Larkworthy L. F., J.
Chem. Soc., 1959, 3116.
21. Hughes E. D., Ingold С. K., Ridd J. II., Nature, 166, 642 (1950); J. Chem. Soc., 1958,
58, 65, 77, 88; Hughes E. D., Ridd J. H., ibid., pp. 70, 82; Larkworthy L. F., ibid.,
1959 3116.
22. Ridd J. H., Quart. Rev. Chem. Soc., 15, 418 (1961).
23. Kalatzis E., Ridd J. H., J. Chem. Soc., 1966B, 529.
24. Hughes E. D., Ingold С. K., Ridd J. H., J. Chem. Soc., 1958, 77; Larkowrthy L. F.,
ibid., 1959, 3304.
25. Schmid H., Z. Elektrochem., 43, 626 (1937); Schmid H., Muhr G., Ber., 70, 421 (1937).
26. Hughes E. D., Ridd J. II., J. Chem. Soc., 1958, 82.
27. Hughes E. D., Ingold С. K., Ridd J. H., J. Chem. Soc., 1958, 88; cp. Lucien II. W.,
J. Am. Chem. Soc., 80, 4458 (1958).
28. Hughes E. D., Ridd J. II., J. Chem. Soc., 1958, 70; Edwards J. O., Abbott J. R., Elli-
son II. R., Nyberg J., J. Phys. Chem., 63, 359 (1959); Stedman G., J. Chem. Soc., I960,
1702: Ridd J. H., Quart. Rev., 15, 425 (1961).
29. Stedman G., J. Chem. Soc., 1959, 2943.
30. Schmid H., Hallaba E., Monatsh., 87, 56 (1956); Schmid H., Fouad M. G., ibid., 88, 631
(1957); Schmid H., Essler C., ibid., p. 1110.
31. Lidstone A. G., Chem. and Ind.. 1959, 1316.
32. Challis В. C., Ridd J. H., Proc. Chem. Soc., 1961, 173; J. Chem. Soc., 1962, 5197;
Challis В. C., Ridd J. H., Larkworlhy L. F., ibid., p. 5203.
33. Dena N. C., Berkheimer II. E., Evans W. L., Peterson J. H., J. Am. Chem. Soc., 81, 2344
(1959).
34. de Fabrizio E. C. R., Kalatzis E., Ridd J. II., J. Chem. Soc., 1966B, 533.
35. Challis В. C., Ridd J. II., Proc. Chem. Soc., 1960, 245.
36. Bayliss N. S., Watts D. W., Austral. J . Chem., 9, 319 (1956): Singer K., Vamplew P. A .,
J. Chem. Soc.. 1956, 3871; Miescher B., Canad. J. Res., 33, 355 (1955).
37. Ridd J. II., Qureshi E. A., J. Chem. Soc., в печати.
38. Miiller E., Haiss II., Rundel W., Chem. Ber., 93, 1541 (1960).
39. Streitwieser A., Schaeffer W. D., J. Am. Chem. Soc., 79, 2888 (1957).
40. Baker J. W., Cooper К. E., Ingold С. K., J. Chem. Soc., 1928, 426.
41. Whitmore F. C., J. Am. Chem. Soc., 54, 3274 (1932).
42. Brewster P., Hiron F., Hughes E. D., Ingold С. К., Bao P. A. D., Nature, 166, 178
(1950).
535
Глава VIII
43. Austin А. Т., Nature, 188, 1086 (1960).
44. Whitmore F. C., Langlois D. P., J. Am. Chem. Soc., 54, 3441 (1932).
45. Bunton C. A., Llewellyn D. R., Stedman G., Nature, 175, 83 (1955); Spec. Pubs. Chem.
Soc., 10, 113 (1959); J. Chem. Soc., 1959, 568; Bunton C. A., Burch J. E., Challis В. C.,
Ridd J. H., в статье Ridd J. H., Quart. Rev., Chem. Soc., 15, 424 (1960).
46. Bunton C. A., Stedman G., J. Chem. Soc., 1959, 3466.
47. Bunton C. A., Masui M., J. Chem. Soc., 1960, 304.
48. Bunton C. A., Dahn H., Loewe L., Nature, 183, 163 (1959); Helv. Chim. Acta, 43, 320
(1960); Dahn H., Loewe L., Liischer E., Menasse R., ibid., p. 287 et seq. (5 papers).
49. Allen A. D., Modena G., J. Chem. Soc., 1957, 3671.
50. Sommer L. H., Fyre C. L., J. Am. Chem. Soc., 81, 1013 (1959); 82, 3796 (1960); 83, 220
(1961); Sommer L. H., Angew. Chem. Internet. Edit., 1, 143 (1962); Sommer L. H.,
Fyre C. L., Parker G. A., Michael K. W., J. Am. Chem. Soc., 86, 3271, 3276 (1964);
Sommer L. H., Parker G. A., Fyre C. L., ibid., p. 3280; Sommer L. H., Parker G. A.,
Lloyd N. C., ibid., 89, 857 (1967); Sommer L. H., Korte W. D., ibid., p. 5802; Som-
mer L. H., Stereochemistry, Mechanism and Silicon, McGraw-Hill. New York, 1965.
51. Baker R., Bott R. W., Eaborn C., Jones P. W., J. Organometal. Chem., 1, 37 (1963).
52. Dostrovsky I., Halman M., J. Chem. Soc., 1953, 502, 508, 511, 516; 1956, 1004; Hal-
man M., ibid., 1959, 305; cp. Drago R. A., Mode V. A., Kay J. G., Lydy D. L.. J. Am.
Chem. Soc., 87, 5010 (1965).
53. Green M., Hudson R. F., Proc. Chem. Soc., 1959, 227; J. Chem. Soc., 1963, 540; Angew.
Chem. Internet. Edit., 2, 11 (1963).
54. Aaron H. A., Oyeda R. T., Frack H. F., Miller J. H., J. Am. Chem. Soc., 64, 617 (1962).
55. Green M., Hudson R. F., J. Chem. Soc., 1963, 3883.
56. Green M., Hudson R. F., Proc. Chem. Soc., 1962, 307; J. Chem. Soc., 1963. 540.
57. Fenton G. W., Ingold С. К., J. Chem. Soc., 1929, 2342; Hey L., Ingold С. K., ibid.,
1933, 531.
58. Kumli K. F., McEwen W. E., VanderWerf С. A., J. Am. Chem. Soc., 79, 3805 (1957):
Blade-Font A., VanderWerf C. A., McEwen W. E., ibid., 80, 2396. 2646 (1960); Pari-
sek С. B., McEwen W. E., VanderWerf C. A., ibid., p. 5503; McEwen W. E., Bladё-
Font A., VanderWerf C. A., ibid., 84, 677 (1962); McEwen W. E., Blade-Font A., Van-
derWerf C. A., Kumli K. F., Zanger A., ibid., 86, 2378 (1964); McEwen W. E., Axel-
rad G., Zanger M., VanderWerf C. A., ibid., 87, 2948 (1965).
59. Horner L., Pure and Applied Chemistry, 9, 225 (1964).
60. Phillips H-, J. Chem. Soc., 1925, 2552.
61. Mason S. F., Quart. Rev. Chem. Soc., 17, 20 (1963); Proc. Roy. Soc. (London), A297,
62. Johnson C. R., J. Am. Chem. Soc., 85, 1020 (1963).
63. Johnson C. R., Sapp J. B., Am. Chem. Soc., Meeting, New York, Sept. 1963, abstracts
p. 230, and personal communication from Dr. Carl R. Johnson.
64. Mislow K., Green M. M., Laur P., Alelillo J. T., Simmons T., Ternay A. L., Jr., J. Am.
Chem. Soc., 87, 1958 (1965).
ГЛАВА IX
ОТЩЕПЛЕНИЕ С ОБРАЗОВАНИЕМ ОЛЕФИНОВ, АЦЕТИЛЕНОВ
И КАРБЕНОВ
1. Гетеролитические механизмы 1,2-отщепления................. 538
2. Влияние строения на ориентацию и скорость 1,2-элиминирова-
ния............................................................. 542
3. Влияние окружающей среды на скорость и соотношение про-
дуктов 1,2-элиминирования ...................................... 561
4. Пространственная направленность 1,2-отщепления............ 567
5. Мономолекулярное 1,2-отщепление в газовой фазе............ 578
6. Фрагментации Гроба ....................................... 587
7. 1,1-Отщепление с образованием карбенов.................... 592
Список литературы....................................... 595
1. Гетеролитические механизмы 1,2-отщепления [1]
Нуклеофильное замещение и отщепление (элиминирование) олефина так
часто наблюдаются одновременно, что возникает мысль о том, что двойствен-
ность механизма, являющаяся ключом к трактовке реакций замещения,
может быть в известной мере характерной также и для случаев реакций отще-
пления. Такое предположение подтвердилось.
а. Бимолекулярный механизм. В 1927 г. Хенхерт и автор данной книги
[2] признали бимолекулярный механизм общим механизмом отщепления
олефинов. Бимолекулярный механизм наиболее распространен в реакциях
отщепления и может быть представлен следующим образом. Реагент Y. обла-
дающий нуклеофильными, в частности основными, свойствами, захватывает
атом водорода в виде протона. Одновременно с этим электронооттягивающая
группа X отделяется вместе с теми электронами, которыми она ранее облада-
ла в виде поделенных электронов. Так как в первоначальной молекуле водо-
родный атом и группа, притягивающая электроны, связаны с соседними
углеродными атомами, то электронные оболочки атома могут оставаться
заполненными в процессе химической реакции; таким образом, возникает
содействующий реакции кооперативный эффект: рвутся две связи, но каждое
расщепление способствует другому и оба вместе представляют единый син-
хронный акт. Механизм можно схематически изобразить следующим обра-
зом:
Y 4- Н ДжХсК2Дк —»YH + CR2=CR2 + X (Е2)
Стрелки указывают, как происходит обмен зарядов между Y и X, хотя сами
заряды могут иметь различные комбинации знаков. Первоначально Y может
быть либо заряженным отрицательно, либо нейтральным, а X может быть
формально нейтральным или положительным. Для осуществления реакции
необходимо только, чтобы после перемещения формальный заряд Y стал бы
на единицу более положительным, а заряд X — на единицу более отрицатель-
ным, чем до реакции.
До настоящего времени бимолекулярный механизм был доказан на примерах
образования олефинов из тетраалкиламмониевых солей [2], некоторых солей
тетраалкилфосфония [3]. солей триалкилсульфония [4], сульфонов [5], нитро-
алканов [6], галогенидов [7], включая фториды [8], сульфонатов [9] и карбок-
силатов [10]. Применялись нейтральные или отрицательно заряженные нукле-
офильные реагенты, основность которых колебалась в пределах от воды до
алкоголят- или амид-ионов. Ниже перечислены способные отщепляться
группы:
X —NR3, PR3, SR2, 6н2, SO2R, NO2, Hal, OSO,R, OCOR
Y- JI2(), (CH3)3N, OH-, RO", CH3COO-, ArO-, NH2, СО.|-
B качестве типичных можно привести следующие изученные реакции:
(CII3)3X4 ArCH2CH3N(CII3)3 (СH3)3NH + АгСН СН2 + (CH3)3N
O1I- + CH3CII2S(CH3)2 H2O + CH2-CH2 + (C1I3)2S
OH-H-CH3CH2SO2C2Hs Н,О + СН2 CH2-]-C2H5SO2
С2Н3О- + АгСН2СН2Вг С2Н5ОН +АгСН = СН2 +Bl-
Доказательства такого механизма состоят: 1) в необходимости участия силь-
ного основания; 2) в демонстрации с помощью меченых атомов протекания
реакции в одну стадию; 3) во влиянии структурных факторов; 4) во влиянии
538
Отщепление с образованием олефинов, ацетиленов и карбеиО:
среды на легкость протекания и направление реакции. Доказательства пер-
вых двух типов обсуждены в этом разделе, а третьего и четвертого — в по-
следующих разделах.
Необходимость в сильном основании при указанных выше реакциях отщеп-
ления качественно ясна. Количественно это выражается кинетическим урав-
нением второго порядка
Скорость-=&2[Y][HCR2CR2X]
Второй порядок был формально установлен практически во всех приведен-
ных выше случаях. Типичные реакции, подчиняющиеся уравнению второго
порядка, приведены ниже (из них первая протекает в воде, а вторая — в эти-
ловом спирте)
OH- + CeH5CH2CH2N(CH3)3 H2O-f-CelI5CH = СН2-(-(CH3)3N
С2115О- + СН3СН(СН3)Вг —> С2Н5ОН + СН2-СНСН3+В1-
Необходимость в сильном основном реагенте, о которой свидетельствует
кинетическая картина реакции второго порядка, согласуется не только
с бимолекулярным отщеплением Е2, но и с мопомолекулярным отщеплением
от сопряженного основания субстрата (Е1сВ) (цифра 1 в этом символе отно-
сится к молекулярности элиминирования, а не образования сопряженного
основания). Этот механизм преобладает при отщеплении с образованием
карбонильной группы или олефиновой двойной связи, сопряженной с кар-
бонильной группой или другой электроотрицательной ненасыщенной группой
(гл. XIII), однако он встречается чрезвычайно редко при элиминировании
с образованием простых олефинов.
При помощи изотопного метода Скелл и Хаузер [И] показали, как можно
различить бимолекулярное и мономолекулярное отщепление от сопряжен-
ного основания. Согласно данным этих исследователей, вполне типичное
элиминирование олефина, а именно образование стирола из 2-фенилэтил-
бромида и этилат-иона, которое, как показано, подчиняется кинетике второго
порядка, действительно протекает по бимолекулярному механизму
С2Н6О- + С3Н5СП2СИ2Вг —> C2H5OH+C6H5CH--CH2 + Br- (Е2)
Можно было бы предположить другую возможную интерпретацию реакции,
а именно предварительное образование сопряженного основания — карба-
ниона и последующее мономолекулярное отщепление иона брома
С2Н5О- + СвН5СН2СН2Вг С2Н5ОН + С6Н5СНСН2Вг ]
С6Н5СНСН2Вг —» С6Н5СН = СН2 + В1- J (ElcB)
Однако при этом механизме первая стадии должна быть обратимой, и поэтому
если реакцию проводить в дейтерированном спирте C2H5OD, то бромид,
оставшийся после того, как часть его превратится в стирол, должен был бы
содержать дейтерий. В действительности Скелл и Хаузер установили, что
бромид, выделенный после такого частичного превращения, не содержит
дейтерия — результат, который не соответствует предположению о том,
что реакция проходит через стадию образования сопряженного основания.
Мы говорим «пе соответствует», так как, конечно, эти отрицательные данные,
строго говоря, не являются отрицательным доказательством; однако, чтобы
доказать такой механизм, желательно было бы получить ясные положитель-
ные данные. Был сделан ряд попыток «открыть» механизм ElcB при образо-
539
Глава IX
вании простых олефинов. Этот механизм, вероятно, не имеет общего значе-
ния, хотя возможно, что в ограниченных случаях он применим *.
б. Мономолекулярный механизм. В 1935 г. этот механизм впервые был
предложен Хьюзом в качестве общего механизма выделения олефинов [15].
Главная его особенность состоит в образовании карбониевого иона, происхо-
дящем в результате отрыва электрофильной группы X вместе с парой ранее
поделенных электронов под влиянием сил сольватации, но без взаимодей-
ствия X с реагентом Y, акцептором протонов; карбониевый ион затем отдает
протон растворителю или другому акцептору протонов. Таким образом,
реакция протекает в две стадии, из которых первая — гетеролитическое
отщепление X — определяет скорость реакции. Поэтому скорость всей
реакции не зависит от добавления основания. Механизм можно представить
следующим образом:
О
И—CR2-CR2—X----------> Н—CR2-CR2 + X
Медленно
+ + (Ь1)
Н—CR2—CR2------->Н + CR2=CR2
Быстро
где группа X до реакции может быть либо формально нейтральной, либо
положительно заряженной и становится в результате реакции соответственно
либо отрицательно заряженной, либо нейтральной.
К настоящему времени этот механизм установлен или является вероятным
для многих случаев элиминирования олефина из солей сульфония [16],
галогенидов [17] и эфиров сульфокислот [181, т. е. для следующего ряда
электрофильных групп:
X = SR2, Cl, Вг, I, OSO2R
Ниже приведены реакции отщепления, для которых мономолекулярный
механизм является преобладающим:
СНзС(С11з)28(СНз)2 Н~-]-СН2 = С(СН3)2-|-(CH3)2S
СН3С(СН3)2Вг —> H+-i-CH2 = C(CH3)2 + Br-
Доказательства этого механизма основываются на следующем: 1) процесс
не зависит от присутствия основания, 2) аналогия с конкурирующим моно-
молекулярным замещением, 3) влияние структурных факторов и 4) влияние
среды на скорость и направление реакции. В этом разделе рассмотрены
два первых пункта, а пункты 3) и 4) рассматриваются в последующих
разделах.
Кинетическая картина реакции отражает независимость мономолекуляр-
ного отщепления от наличия оснований. В первом приближении эти про-
* Указания на этот механизм содержатся в работе [12], однако кинетика изученных
реакций не была простой и строение продуктов не было установлено. Возможно, что
этот механизм наблюдался в работе [13], однако для объяснения кинетики необходимо
было знать некоторые дополнительные константы. Механизм Е1сВ, вероятно, был реали-
зован Бордуэллом и сотр. [14], которые наблюдали легкое ({uc-элиминирование уксусной
кислоты от 1-нитро-2-ацетокси-2-фенилциклогексана при действии пиперидина в смеси
хлороформ — этиловый спирт. Однако они получили не простой олефин, а ненасыщен-
ное нитросоедпнение. Механизм Е1сВ является стандартным при образовании а, Р-нена-
сыщепных карбонильных соединений (гл. XIII), поэтому не может быть сомнений, что
он имеет место и в случае а, р-пенасыщенных нитрилов и питросоединений.
540
Отщепление с образованием олефинов, ацетиленов и карбенов
цессы характеризуются уравнением реакции первого порядка
Скорость = /^[НСЙгСйгХ]
что было доказано для следующей реакции, скорость которой в этиловом
спирте почти не изменяется при прибавлении этилата натрия
СН3С(СН3)2Вг —> Н+СН2 = С(СН3)2 + Вг-
Во втором приближении возможны отклонения от кинетического уравнения
первого порядка, которые объясняются солевыми эффектами (об этом гово-
рилось в гл. VII, разд.6) и которые весьма характерны для мономолекуляр-
ных процессов. Как было установлено, такие отклонения встречаются
в процессах мономолекулярного замещения и элиминирования.
Мономолекулярное замещение (SN1) и мономолекулярное отщепление (Е1)
имеют общую медленную стадию; стереохимия этих процессов определяется
альтернативными последующими быстрыми стадиями:
HCR2CR2—Y (SN1)
у_^
HCR2CR2-^-X----► HCR2CR2+ Быстро
Медленно \
Y~\
CR2=CR2 + HY (El)
Константа скорости суммарной мономолекулярной реакции
kl = -|- I'(E 1)
соответствует скорости гетеролиза субстрата, однако отношение /c(El)//q,
равное доле образующегося олефина, определяется долей участия карбоние-
вого иона в каждой из этих реакций. Если бы можно было рассматривать
анион вне радиуса действия экранирующих сил, то в серии субстратов с одной
и той же алкильной группой, но различными уходящими группами X сум-
марная скорость могла бы сильно меняться, однако относительное количе-
ство образующегося олефина должно было остаться постоянным. Это послу-
жило одним из аргументов при доказательстве El-механизма отщепления
от вторичных и третичных алкилгалогенидов и сульфониевых ионов в водном
растворе этилового спирта, который был сформулирован в конце 1930-х годов.
Однако впоследствии, поскольку стереохимические исследования показали,
что с экранированием необходимо считаться, было указано, что постоянство
относительного выхода олефина является лишь приблизительным. Данные
[19], приведенные в табл. 135, показывают, что при фиксированной алкиль-
ной группе изменение уходящей группы может изменить скорость в 100 раз
или более, причем изменение относительного выхода олефина происходило
не более чем в 1,3 раза.
При переходе к менее ионизующим растворителям, например к безводному
этанолу, или к менее ионизующим и менее основным растворителям типа
безводной уксусной кислоты упомянутое выше правило, имеющее ограничен-
ное применение, нарушается. Ионные пары более стабильны, чем диссоции-
рованные ионы, поэтому важное значение приобретает экранирование про-
тивоионом, т. е. разные противоионы будут участвовать в стадии, заканчи-
вающей замещение, в различной степени. Кроме того, противоион может
лучше отщеплять протон, чем растворитель, и, таким образом, разные
противоионы будут облегчать отщепление по сравнению с присоединением
в разной степени. Этот второй эффект также имеет стереохимическое след-
ствие, которое будет рассмотрено в разд. 4,6. Здесь же оно будет проиллю-
541
Глава IX
Таблица 135
Влияние X в мопомолекулярных реакциях AlkX па общую скорость (к, с-1)
и на выход олефинов [A- (El)/А|]
Р а с т в о р и т р л ь — в о д - ный этиловый спирт Темпера- тура, °C Алкил X 1 05k,
60 %-ны ii 100,0 2-Октпл С1 В г 0,805 26. 8 0,13 0,14
80 % -iibiii 65,3 ш/ют-Бутил S(CTI3)2 Cl 11,8 89,7 0,357 0,363
60%-пый 25,0 шрелг-Бутп.т Cl В г I 0,854 37,2 90,1 0,168 0,126 0,129
8О°о-нып 50,0 трет-Амил S(CH3)2 Cl 6.66 28,5 0.478 0,403
8о "о -11 ы ii 25,2 трет-Амил Cl Вг I 1,50 58,3 174 0,333 0,262 0,260
стрировано данными Уипстейпа и Койсевера [20], обнаруживших, что
ш/^етл-бутилхлорид, шретп-бутилбромид и ион диметилсульфония в безводном
этиловом спирте при 75 °C дают 44. 36 и 18% олефина соответственно, а в без-
водной уксусной кислоте — 73, 69 и 12% олефина соответственно. Хлорид-
и бромид-иопы, конечно, гораздо бэлее осповпы, чем диметилсульфид, отще-
пляющийся от сульфониевого иона.
Известен и другой механизм элиминирования олефинов через карбониевые
ионы. Он обозначается символом Е2(С+) и, вероятно, также родствен
.механизму Е1, как механизм Е1сВ механизму Е2. Он характеризуется тем,
что в этом случае вместо медленного образования карбопиевого иона и быстро-
го отщепления протона карбониевый ион образуется в быстрой предравно-
весной стадии, и поэтому скорость реакции контролирует стадия атаки про-
тона основанием
И-СИ2 — СВ,-X ----------> Н — CR2 — CR+4-X--)
Быстро I Е2(С+)’
Медленно I
х + н-CR2 — CR+ -------> YH-!-CR2=--CR2 ’
В гл. VI1, разд. 3, в был приведен карбокатионный механизм нуклеофильного
замещения, обозначенный символом SN2(C+). Этот механизм включал пред-
варительное образование иона карбония и последующую медленную реком-
бинацию его с основанием, т. е. этот механизм является двойником рас-
сматриваемого механизма элиминирования. Хотя можно сказать, что послед-
ний механизм существует, точные доказательства его здесь не приводятся.
Он упомянут для того, чтобы показать главные механизмы 1,2-элиминирова-
ния, однако более удобно обсудить его в гл. XIII, разд. 1,а и 2,а.
2. Влияние строения на ориентацию и скорость 1,2 элими-
нирования
а. Правила ориентации и их смысл. Анализ влияния строения
па направление и скорость реакций отщепления начали проводить в прош-
лом столетии, когда были сделаны наблюдения, позволившие установить
542
Отщепление с образованием олефинов, ацетиленов и карбенов
правила отщепления от различных классов субстратов, а именно от тетраал-
киламмониевых солей и алкилгалогенидов. Это было сделано задолго до того,
как стало ясным, что эти процессы элиминирования принадлежат к одному
семейству реакций.
В 1851 г. Гофман [21] отметил, что гидроокиси четвертичных аммонийных
соединений, содержащие разные алкильные группы, разлагаются с образо-
ванием главным образом этилена, если в них содержится этильная группа.
Гораздо позднее, в 1927 г., Ханхарт и автор [23] показали, что это правило
можно обобщить па четвертичные гидроокиси аммония, содержащие лишь
первичные алкильные группы (что очень важно); в этом случае образуется
в основном этилен, имеющий наименьшее число алкильных заместителей.
Это правило можно назвать «общим правилом Гофмана».
В 1875 г. Зайцев [22] показал, что при элиминировании олефина из вторич-
ных и третичных алкилгалогенидов в наибольших количествах образуется
тот олефин, для образования которого требуется отщепление водородного
атома от наименее гидрогенизированного атома углерода. Это правило можно
сформулировать иначе: из вторичного или третичного алкилгалогенпда
получается олефин с наибольшим количеством алкильных заместителей.
До тех пор пока правила Гофмана и Зайцева рассматривались просто как
обобщение эмпирических наблюдений в различных областях органической
химии, никто и не думал их сравнивать. Но после 1927 г., когда была дана
трактовка распада четвертичных аммониевых оснований, объединившая
этот процесс в один класс с реакциями алкилгалогенидов, возникла необ-
ходимость выяснить, почему различные видоизменения одной и той же
реакции подчиняются двум противоположным правилам, совместно обоб-
щающим все данные об ориентации при элиминировании. Предполагалось
(и предполагается), что соответствующие влияния должны быть одинако-
выми, т. е. должны проявляться во всех органических реакциях, а не только
в узкой области простых алкильных субстратов. Оба типа влияния строения
должны иметь место; они должны быть независимыми, т. е. действовать или
в одном, или в разных направлениях. Когда их влияние противоположно,
получаются противоположные правила в зависимости от того, какой эффект
преобладает. Важная проблема состояла в идентификации структурных
эффектов, так как от способа их комбинации зависит относительная важ-
ность того или иного эффекта.
Этот вопрос был решен наполовину в 1927 г., когда было предположено, что
справедливость правила Гофмана для алкильных групп в ониевых соедине-
ниях связана главным образом с влиянием полярности заместителей на кис-
лотность Р-протона. Однако па другую часть вопроса ответа нельзя было
дать до 1935 г., когда была открыта гиперконъюгация.
б. Бимолекулярное отщепление (особенно от алкилониевых ионов);
эффект полярности, преобладание индуктивного эффекта. Роль индуктивной
поляризации в создании ситуации, отражающейся в общем правиле Гоф-
мана, в 1927 г. была описана следующим образом [23]. Субстрат должен
содержать сильную электроноакцепторную группу, которая затем отще-
пляется. Эта группа, индуцируя положительные заряды на всех окружаю-
щих атомах, ослабляет связи протонов, причем достаточное ослабление
связи Р-протона обусловливает элиминирование по механизму Е2. Любая
группа, которая путем отдачи электронов имеет тенденцию нейтрализовать
индуцированный положительный заряд на Р-углеродном атоме и тем самым
усилить его связь с Р-протоном, должна тормозить реакцию. Такое влияние
оказывает концевая метильная группа //-пропильной группы, что в соот-
ветствии с правилом Гофмана объясняет относительно слабую способность
543
Глава IX
«-пропильной группы расщепляться с выделением олефина в присутствии
этильной группы, склонной к элиминированию этилена.
Н (_____Н____
СНз -»СН—СН2—1О-1 СНз-^СН-Н
pi q ^Cil Отщепляется
Аналогично изобутильная группа почти не будет участвовать в элиминиро-
вании, если присутствует «-пропильная и тем более этильная группа. Есте-
ственно, картина не меняется, если концевые метильные радикалы «-про-
пильной и изобутильной групп заменены на этильный или высший радикал
гомологического ряда. Такой вывод соответствует упоминавшемуся расши-
ренному правилу Гофмана о преимущественном образовании наименее
алкилированного олефина.
Дальнейшее обобщение вытекает из представлений о том, что защитное
смещение электронов может передаваться p-углеродному атому от несосед-
них атомов, хотя и со значительно меньшей интенсивностью. Так, защитный
эффект, наблюдаемый в случае «-пропильной группы, увеличивается для
«-бутильной группы, но в меньшей степени, чем для изобутильной. Как
правило, в гомологическом ряду защитный эффект увеличивается до опреде-
ленного предела, но при условии, что радикал с разветвленной цепью всегда
оказывает большее влияние, чем изомерный радикал с нормальной цепью.
Можно ожидать, что электроноакцепторные заместители, если не принимать
во внимание размеры углеводородных радикалов, способствуют элиминиро-
ванию и влияние их особенно сильно, если они непосредственно связаны
-с p-углеродным атомом.
____Н_____ И
"J гу+ I
С1 «- СН^СНз 1-^—N—СНз—СН—н
Отщепляется ^СН
Эти теоретические выводы были проверены на реакциях алкоголятов с аммо-
ниевыми и сульфониевыми основаниями, содержащими первичные алкильные
или замещенные алкильные группы, а также на примерах разложения анало-
гично построенных сульфонов. Позднее такие наблюдения распространили
на вторичные и третичные алкильные группы.
В течение некоторого времени после 1927 г. исследовалась только ориентация
первичных алкильных групп при реакциях отщепления. Сущность одного
из вариантов заключалась в получении ониевых ионов или сульфонов с раз-
личными алкильными группами в одной и той же молекуле (как показано
в предыдущей формуле) и в определении соотношения различных олефинов
в продуктах реакции после ее завершения. Таким образом, было показано
[24], что при разложении сульфониевых оснований первые четыре первичные
алкильные группы по легкости отщепления олефинов располагаются в сле-
дующем порядке:
этил > и-пропил > «-бутил > изобутил
Другим более ранним методом изучались ониевые ионы, содержащие метиль-
ные группы и лишь одну алкильную группу, способную давать олефин; при
этом предполагалось, что изменения в группе, образующей олефин, не влияют
заметно на тенденцию метильной группы к отщеплению в процессе замещения.
Таким образом, легкость элиминирования в случае различных групп, спо-
544
Отщепление с образованием олефинов, ацетиленов и карбенов
собных отщепиться в виде олефина, по сравнению с этим почти стандарт-
ным процессом замещения была принята за меру способности этих групп
давать олефин. Этим методом изучались [23, 25] продукты разложения
аммониевых оснований. При этом первичные алкильные группы по легкости
образования олефина располагаются в следующем порядке:
этил и-пропил и-бутил и-амил ~ «-октил > изоамил изобутил
После того как были даны указанные объяснения и примеры, появился ряд
работ, посвященных составу продуктов элиминирования ониевых соедине-
ний. Одной из наиболее значительных является опубликованная в 1952 г.
работа Смита и Франка [26], в которой смесь олефинов, образующаяся при
разложении четвертичных аммониевых гидроокисей с различными алкиль-
ными группами, анализировалась масс-спектрометрически. Некоторые
Таблица 136
Выход (%) олефинов из гидроокисей тетраалкиламмония
(по данным Смита и Франка)
Алкильные группы в R4N+ Алкильная цепь после Количество олефина
найдено в пересчете на одинаковое количество групп
6? бу НИЗ- ШИЙ ВЫС- ШИЙ низ- ший ВЫС- ШИЙ
(С2Н5)2, (м-С3Н7)2 с- 96 4 96 4
(н-С3Н7)2, (н-С4Нд)2 С- С-С- 62 38 62 38
Н-С3Н7, (н-С4Н9)3 С- С-С- 83 17 61 39
(н-СзН7)3, И-С4Н9 с- С-С- C. 36 64 62 38
(н-СдНд^, (иЗО-С5Нц)2 с-с- >с- Сх с. 67 33 67 33
И-С4Н9, uao-CjHn, (СН3)2 с-с- С\ >С- cz С\ 66 34 66 34
иао-С5Нц, нео-С6Н13, (СН3)2 >С- cz С-С- с 91 9 91 9
результаты представлены в табл. 136. Данные, приведенные в двух послед-
них колонках, учитывают статистическую поправку на разное число одина-
ковых групп в исходной молекуле. Они показывают, что различные алкиль-
ные группы в ионах аммония ведут себя независимо друг от друга. Кроме
того, данные показывают, что ориентирующий отщепление эффект затухает
вдоль цепи аналогично индуктивному эффекту. у-Углеродный атом, т. е. бли-
жайший атом к кислому [1-протону, является, согласно правилу Гофмана,
сильным ориентантом, тогда как первый и второй 6-углеродные атомы оказы-
вают одинаково слабый эффект. В противоположность индуктивному эффекту
первичный пространственный эффект не ослабляется. Вследствие соответ-
ствующей формы кривой зависимости энергии взаимодействия несвязанных
групп от расстояния этот эффект быстро возрастает при увеличении плот-
ности атомов вокруг реакционного центра. Введение третьего 6-углеродного
атома (неогексильная группа) оказывает гораздо более сильный эффект, чем
два 6-угле родных атома изоамильной группы. Смит и Франк [26] отметили,
545
Глава IX
что, вероятно, первичный пространственный эффект оказывает наиболее
сильное влияние при увеличении разветвления в этом месте углеродной цепи.
После 1935 г., когда было открыто мономолекулярное элиминирование,
появилась необходимость подтвердить кинетическими данными впослед-
ствии оказавшиеся правильными представления о том, что уже изученные
реакции с участием первичных алкильных групп являются реакциями бимо-
лекулярными. Кроме того, используя кинетический метод, можно было бы
попытаться, в частности, установить, согласуются ли индивидуальные
скорости таких бимолекулярных процессов также хорошо с требованиями
теории структурных влияний, как согласуются их ориентационные соотно-
шения с правилом Гофмана. Теория предсказывает, что увеличение числа
алкильных групп после p-углеродного атома должно уменьшать скорость
Таблица 137
Константы скорости (А’2, л-моль-1-с-1) бимолекулярного отщепления
от этилатов алкилониевых соединений в этиловом спирте
A: RN(CH3)3OC2H5 —> Олефин -|- С2Н5ОН -|- (CH3)3N
Б: RS(CH3)2OC2Ht Олефин4-С2Н5ОН + (CH3)2S
1 05^2
R
реакция А при 104 °C реакция Б при 64 СС
Этил 71,3 79
и-Проппл 5,16 29
и-Бутпл 2,82 21
Изобутил 1.68 10
R Реакция Б, 10.>k2
более длинная цепь более короткая цепь общая
Изопропил а 520 520 1040
eniop-Бутил а 185 510 695
/гарет-Бутил 6 27 27 80
mpem-Лмил 6 8 24 57
а При 64 °C. 6 При 24 “С.
реакции. Данные, представленные в верхнем ряду табл. 137, подтверждают
эту теорию [27].
Новые подтверждения были получены при изучении вторичных и третичных
алкилониевых ионов. Ранее было известно, что правило Гофмана приложимо
к первичным алкильным группам в ониевых ионах, а правило Зайцева —
к вторичным и третичным алкильным группам в алкилгалогенидах. Однако
не было известно, объясняется ли отличие этих правил различием в типе
алкильных групп, в типе электроноакцепторных групп или в механизме
элиминирования. В кинетически контролируемых условиях бимолекуляр-
546
Отщепление с образованием олефинов, ацетиленов и карбенов
ного отщепления, как оказалось, реакции вторичных и третичных алкиль-
ных групп в сульфониевых ионах аналогично реакции первичных алкиль-
ных групп подчиняются правилу Гофмана. Этот факт в случае вторичных
и третичных групп был установлен на основании того, что двойная связь
образуется преимущественно в более короткой из неодинаково развет-
вленных групп и что различие в скоростях, определяющее ориентацион-
ный эффект, обусловлено не увеличением скорости образования двойной
связи в менее разветвленной цепи, а уменьшением скорости этой реакции
в более сильно разветвленной части алкильной группы. Это проиллюстри-
ровано в последних двух разделах табл. 137. Так, этилат втор-бутилдиметил-
сульфония и в указанных условиях дает 26% бутена-2 и 74% бутена-1,,
и, как показывают данные таблицы, это является следствием уменьшения
скорости образования двойной связи в более длинной ветви етор-бутиль-
ной группы, которая становится меньше скорости образования двойной
связи в одной из ветвей симметричной гомологичной изопропильной груп-
пы [28]. Аналогично этилат mpem-амилдиметилсульфония дает 14% 2-метил-
бутена-2 и 86% 2-метилбутена-1, и основная причина состоит в уменьшении
скорости реакции в длинной ветви по сравнению со скоростью реакций
в одной ветви низшего гомолога — треттг-бутильной группы [29].
Были изучены также эффекты гомологии нормальных и разветвленных
первичных алкильных групп на скорости разложения ионов алкилдиметил-
сульфония и алкилтриметиламмония под действием этилат-ионов в этиловом
спирте и игрет-бутилат-ионов в mpem-бутиловом спирте [27]. Цель исследо-
вания состояла в том. чтобы более точно определить, отразится ли на ско-
ростях ожидаемое затухание индуктивного эффекта с ростом цепи, связан-
ной с Р-протоном, а если отразится, то будет ли наблюдаться соответствие
с полярными эффектами в других реакциях и будут ли эти эффекты согласо-
вываться с ожидаемыми на основании известной электроотрицательности
полярных концевых групп. Гомологические серии подбирались таким обра-
зом, чтобы показать, на какой стадии разветвления появляются простран-
ственные затруднения.
Таблица 138
Константы скорости второго порядка (fc2, л-моль-1-с-1)
реакции отщепления олефинов от этилатов алкилдиметилсульфония
в этиловом спирте при 64 °C и различие в свободной энергии активации
R в Н8(СН3ДсС2Н3 10о/?2 ДЛЯ Е2 AG^, ккал/моль (4,187- 103 Дж/моль) AG^ для последней введенной СНу-группы, ккал/моль (4,187 -10» Дж/моль) AG^ (рассч.), ккал/моль (4,187 -1СН Дж/моль)
СТТ3СН2- 7!) 0 0
СН3СН2СН2- 29 0,67 0,67 0,70
(СН3)2СНСН2- 10 1,38 0,71 1,60
СН3СН2СН2СП2- 21 0,88 0,21 0,80
(СН3)2СНСН,СН2- 16 1,06 0,18 0,83
(СН3)3ССН2СП2- 0,43 3,49 2,43 0,87
Все изученные серии свидетельствуют об общих закономерностях. Данные
для одной из них приведены в табл. 138. Уменьшение скорости при удлине-
нии цепи удобно выразить через инкременты свободной энергии актива-
ции AGZ. Введение одного атома углерода в у-положение увеличивает сво-
бодную энергию на 0,7 ккал/моль (2,93 -103 Дж/моль), введение второго у-угле-
родного атома также увеличивает AG* на 0,7 ккал/моль (2,93-103 Дж/моль).
35* 517
Глава IX
Введение первого и второго атома углерода в 6-положение приводит к уве-
личению свободной энергии еще на 0,2 ккал/моль (0,84-103 Дж/моль) на
каждый атом углерода. Однако введение третьего 6-углеродного атома,
как в неогексильной группе, увеличивает AG=^ не на 0,2 ккал/моль
(0,84-10s Дж/моль), а на 2,4 ккал/моль (10,05-103 Дж/моль). Очевидно,
на этой стадии разветвление приводит к появлению пространственных пре-
пятствий, которые ранее не проявлялись. Это точно соответствует выводу
Смита и Франка, сделанному при изучении состава смеси олефинов, обра-
зующихся из ионов аммония со смешанными алкильными группами.
Поскольку электростатическая энергия пропорциональна заряду, можно
сказать, что фактор ослабления поляризации при передаче через атом угле-
рода равен 3,5. Вследствие эффектов поля (гл. VI, разд. 3,д и 4,е) точное
значение этого фактора зависит inter alia от среды, в качестве которой в этих
работах использовался этиловый спирт. Расчеты, основанные на поляризуе-
мости атомов и связей, дают величину, приблизительно равную 5, однако
они относятся к ионам в газовой фазе. Много лет назад Бренч и Калвин [30],
исходя из влияния гомологии на силу карбоновых кислот в воде, вывели
величину 2,8.
Рассчитав по данным поляризуемости величины заряда, создаваемого поляр-
ной концевой группой на всех атомах алкильной группы, и приняв, что
энергия индуцирования заряда на p-водородном атоме в поле этилат-иона
в переходном состоянии соответствует электростатической части свободной
энергии активации, можно получить значения, приведенные в последнем
столбце табл. 138. За исключением неогексильной группы, эти значения
по порядку величины соответствуют наблюдаемым, и, таким образом, из этих
грубых расчетов следует, что на основании индуктивных эффектов конце-
вого ониевого иона на алкильную цепь можно правильно предсказать изме-
нение скорости реакции и, следовательно, наблюдаемый состав продуктов
реакции.
В случае неогексильной группы необходимо предположить, что энергия
пространственного отталкивания в переходном состоянии равна 2,2 ккал/моль
(9,21 -103 Дж/моль). Не вызывает сомнения, что это отталкивание осуще-
ствляется между Р-пгретп-бутильным остатком и а-сульфониевой группой.
Этот эффект нельзя рассчитать методом, указанным в гл. VII, разд. 8, б,
поскольку геометрия переходного состояния достаточно точно не известна.
Однако этим методом можно рассчитать, что энергия пространственного
отталкивания не должна превышать 6,5 ккал/моль (27,21 -103 Дж/моль).
а если принять, что в переходном состоянии эти группы находятся на пол-
пути между начальным и конечным положением, где они располагаются
в плоскости двойной связи, то рассчитанная энергия отталкивания будет
равна около 2 ккал/моль (8,37 -103 Дж/моль) *.
Необходимо сделать предостережение, что теория правила Гофмана не пол-
ностью отражает закономерности ориентации двойной связи и изменения
скоростей бимолекулярного отщепления, даже в случае ониевых ионов.
Так, серия этил, изопропил, пгреиг-бутил характеризуется постепенным
увеличением числа а-метильных заместителей, что должно оказывать слабый
* В 1950—1956 гг. в литературе появилось множество предположений (см. [27]) о том,
что правило Гофмана и скорость Е2-реакций в ониевых ионах обусловлены исключи-
тельно пространственными препятствиями, однако эта идея Брауна па самом деле не отра-
жает известных опытных данных. Через десять лет эта идея появилась вновь без упоми-
нания противоречащих ей примеров (см. [27]). При этом для объяснения противополож-
ного влияния размеров галогенов был введен новый неопределенный параметр, назван-
ный «пространственными требованиями» [31].
548
Отщепление с образованием олефинов, ацетиленов и карбенов
защитный эффект на p-протоны, и если это так, то скорость реакции в этом
ряду должна немного уменьшаться. В действительности же, как показы-
вают данные табл. 139, скорость значительно возрастает, что не объясняется
Таблица 139
Скорости и выходы олефинов при бимолекулярных реакциях
алкилсульфониевых ионов
Реакция с С2Н5О- в С2Н5ОН при 45 °C;
скорости fc(Sjsi2) и /ДЕ 2) в л-моль-1-с-1
R в HS(CH3)2 105h(SN2) 105h(E2) Выход олефина, %
Этил 37 5,0 12
Изопропил 73 114 61
mpem-Бутил 90 2930 97 ... 100
даже учетом различного числа p-протонов. Сопутствующее S ^-замещение
не характеризуется соответствующим возрастанием скорости, и поэтому
выход олефина в этом ряду возрастает.
При удлинении алкильных цепей в p-положении ониевых солей полярность,
приводящая к гофмановскому типу влияния структуры на ориентацию
и скорость, имеет большее значение, чем насыщенность, которая, как будет
показано, является в целом преобладающим фактором при отщеплении
от алкилгалогенидов и приводит к правилу Зайцева. Однако эта дихотомия *
между ониевыми ионами и галогенидами не абсолютна. Как показали Сон-
дерс и сотр. [8], один из галогенов, а именно фтор, обладает преимуществен-
ным гофмановским эффектом. Так, 2-галогенпентаны СН3СНХСН2СН2СН3
при обработке этилатом калия в кипящем этиловом спирте дают следующие
соотношения изомерных пентенов:
F С1 Вг I
Пентен-1 82
Пептен-2 18
36 25 20
64 75 80
Качественно такая же дихотомия и количественно такая же градация наблю-
дались для 2-галоген-2-метилпентанов (СНз^СХСНгСНгСНд.
в. Бимолекулярное отщепление в алкилгалогенидах; влияние ненасы-
щенности, преобладание электромерного эффекта. Полярный механизм,
лежащий в основе правила Зайцева, был в 1941 г. интерпретирован [32J
как смещение электронов при сопряжении или, более точно, как гипер-
конъюгационное смещение, главным образом действующее во время реакции
как электромерный эффект. Для простых алкильных групп механизм может
быть представлен следующим образом:
Изменяемые связи
в переходном состоянии
Типерконъюгация
* Дихотомия (греч.) — в данном случае деление понятий на две части.— Прим, перев.
549
Г лава IX
В переходном состоянии электроны соответствующим образом расположен-
ной СН-связи, например у-СН-связи, находятся в гиперконъюгации с элек-
тронами частично образовавшейся аф-двойной связи. Это уменьшает энер-
гию переходного состояния, что облегчает реакцию.
Подобно тому как правило Гофмана является частным следствием индук-
тивного эффекта (что было показано в предыдущем разделе), одним из след-
ствий электромерного эффекта является правило Зайцева. При этом, как
и в первом случае, теоретические выводы оказываются гораздо шире перво-
начального эмпирического правила. Ожидаемые последствия, к которым
приводит электромерный эффект, можно подразделить на следующие четыре
типа.
Во-первых, вследствие электромерного эффекта алкильный радикал, связан-
ный в P-положении со всей алкильной группой, стабилизует переходное
состояние и, таким образом, облегчает элиминирование этой группы в виде
олефина в противоположность расширенному правилу Гофмана. Это теоре-
тически предсказанное влияние может проявиться в случае реакции как
различных алкильных групп, так и неодинаково разветвленных цепей
одной и той же группы. Например, н-пропильная группа должна отщеплять
пропилен легче, чем этильная — этилен, а симметричная 3-н-амильная
группа должна давать пентен-2 легче, чем изопропильная — пропилен.
Применение этого положения к разветвленным вторичным и третичным
группам и есть правило Зайцева, согласно которому двойная связь пред-
почтительно образуется между более алкилированными углеродными атома-
ми. Так, в случае 2-н-бутильной группы должен отщепляться главным
образом бутен-2, а третичная амильная группа отщепляет в основном три-
метилэтилен.
Во-вторых, поскольку оба конца образующейся двойной связи одинаково
доступны для гиперконъюгации, то радикал, расположенный в а-положе-
нии, в общем должен оказывать такой же электромерный эффект, как
и радикал, находящийся в p-положении. Ясно, что (в отличие от соответ-
ствующего индуктивного влияния а-радикалов на p-протоны) это влияние
имеет преобладающее значение. На самом деле во всех случаях электро-
мерный эффект tz-радикалов оказывает преобладающее влияние. Можно
сделать вывод, что в третичных алкильных группах должно легче происхо-
дить бимолекулярное отщепление, чем во вторичных, а в последних — легче,
чем в первичных. Это должно быть справедливым для ряда ттгреттг-бутил >
> изопропил > этил, а также для ряда трет-амил > отгор-бутил >
> и-пропил.
Третье теоретическое положение является расширением первых двух, так
как оно определяет относительное влияние различных р- или а-алкильных
радикалов. В любом случае наибольший эффект вызывается метильной
группой, имеющей три атома водорода, способных к гиперконъюгации.
В гомологическом ряду углеводородных радикалов этот эффект умень-
шается при переходе от первичных к вторичным и к третичным алкильным
радикалам.
Поскольку приложение теории не ограничено алкильными группами, можно
сделать четвертый вывод. Ненасыщенность влияющей группы и вытекаю-
щая отсюда способность к сопряжению является одним из важных свойств.
Если возможна гиперконъюгация алкильного заместителя в а- или Р-положе-
нии с возникающей двойной связью, что способствует ее образованию,
то расположенные таким же образом ненасыщенные заместители, например
винил или фенил, также способные к сопряжению, будут еще сильнее спо-
собствовать образованию двойной связи.
650
Отщепление с образованием олефинов, ацетиленов и карбенов
В данном разделе приведены кинетические данные о скорости и ориентации
при бимолекулярном отщеплении на примере алкилгалогенидов, подтвер-
ждающие приведенные выше положения. Влияние радикалов в а- и (3-поло-
жениях на скорость элиминирования олефина в различных рядах первич-
ных, вторичных и третичных алкилбромидов приведено в табл. 140 и 141 [33].
Таблица 140
Бимолекулярное образование олефина из алкилбромидов
с различными радикалами в (З-положении
Реакции проводили в С2Н5ОН;
константы скорости /с(Е2) в л-моль-1-с-1
Группа
R или П-2
ККЩЕ2) (25 °C)
Серия 1. RCH2CH2Br + C2H5O- С2Н5ОН + RCH = СН2 +Вг~
R2CHCH2Br + C2H5O- -ч> C2H5OH-LR2C = CH2 + Br-
Этил Н 1.2 а
и-Пропил СН3 5,3 а
н-Бутил с2н5 4,3 а
и-Амил С3Н7 3,5 а
Изобутил (СН3)2 8,6 а
2-Фенилэтил С6н5 561 а
Серия 2. RCH,CH(CH3)Br+C2H5O- -ч>
C2H5OH+RCH- CHCH34-Bi-
Изопропил Н 0,118
втор-Бутил СН3 0,282
2-н-Ампл С2Н5 0,196
Серия 3. RCH2CH(C2H5)Br + C2H5O- —»
—> С2Н5ОН + RCH = СНС2Н5 + Вг-
emop-Бутил н 0,065
2-н-Амил СН3 0,200
Серия 4. RCH2C(CH3)2Br + C2H5O- -ч>
C2H6OH + RCH = C(CH3)2 + B1-
/герет-Бутил Н 1,00
трет- Амил СН3 4,20
а При 55 °C.
Приведенные данные относятся к скоростям образования двойной связи
в определенном положении. Так, в случае образования пропилена из изопро-
пилбромида 105 к = 2-0,118, а в случае образования бутиленов из вторич-
ного бутилбромида 105А: = 0,282 -р 0,065 (л-моль-1-с-1).
Данные табл. 140 показывают, что один |3-метильный радикал увеличивает
скорость бимолекулярного отщепления, два |3-метильных радикала увеличи-
вают скорость сильнее, а один |3-фенильный заместитель еще сильнее.
Из результатов этой таблицы также следует, что высшие алкильные (3-заме-
стители увеличивают скорость бимолекулярного отщепления, но не так
значительно, как (3-метильный радикал. Согласно данным табл. 141, одна
а-метильная группа повышает скорость бимолекулярной реакции, а два
551
Глава IX
Таблица 141
Бимолекулярное образование олефина из алкилбромидов
с различными радикалами в а-положении
Реакции проводили в С2Н5ОН при 25 °C;
константы скорости А(Е2) в л-моль-1-с-1
Группа R или Н2 105fe(E2)
Серия 1. CH3CHRBr+C2H6O- С2Н5ОН-|-СН2 = CHR + Вг~
CH3CR2Br + C2H5O- C2H5OH+CH2 = CR2+Br-
Этил Н 0,025
Изопропил СН3 0,118
в/пор-Бутил С2н5 0,065
2-н-Амил W-C3H7 0,080
mpem-Бутил (СН3)2 1,00
т рет-Амил СН3С2Н5 0,85
1-Фенилэтил Свн5 0,79
Серия 2. CH3CH2CHRBr+C2H5O--» С2Н5ОН+СН3СН=СНВ-гВг-
CH3CH2CR2Br+C2H5O- -> с2н5он+сн3сн-
= СК2+Вг~
к-Пропил Н 0,083
втор-Бутил сн3 0,282
З-н-Амил С2н5 0,200
mpem-Амил (СН3)2 4,20
а-метильных радикала действуют сильнее, так же как и а-фенильный ради-
кал. В то же время высшие а-алкильные заместители увеличивают скорость
в меньшей степени, чем а-метильный радикал. Все это находится в соот-
ветствии с ранее высказанными теоретическими положениями.
Тем не менее некоторые величины в таблицах показывают, что объяснение
влияния заместителей на скорость бимолекулярного отщепления только
на основании электромерного эффекта не является достаточно строгим
и полным. Так, метильный радикал оказывает большее влияние в а-положе-
нии, в то время как фенильный радикал — в ^-положении. Это можно
объяснить одновременным влиянием индуктивного эффекта, значительного
только в случае ^-заместителей. Для отталкивающей электроны |3-метильной
группы индуктивный эффект действует в направлении, противоположном
электромерному эффекту, а для притягивающей электроны ^-фенильной
группы — в том же направлении.
В табл. 142 показано, каким образом в реакциях бимолекулярного элими-
нирования у вторичных и третичных алкилгалогенидов сочетаются эффекты,
влияющие на скорость реакции и ориентацию. В данном случае ранее при-
веденные константы скоростей второго порядка, представляющие собой
скорости элиминирования олефина из различных ветвей алкильных групп,
противопоставлены другим группам, находящимся в разных концах моле-
кулы. Приведенные же величины относятся к радикалам в а- и ^-положениях
образующейся двойной связи.
Сравнение примеров 1 и 2 показывает (левая колонка), что дополнительная
Р-метильная группа в случае 2 ускоряет в согласии с теорией образование
бутена-2 (по сравнению с его низшим гомологом), но замена а-метила а-этилом
552
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблица 142
Некоторые значения скоростей, иллюстрирующие ориентацию
при бимолекулярном образовании олефина из алкилбромидов
Реакции с C2H5ONa в С2113ОН при 25°C; константы скорости k (Е2) в л-моль^-с-1
с=с-сн <
р а
а Р а Р
СН-СВг-СН ---------------СН-С=С
Р а
№ Заместители Скорость для каж- дой цепи 105k (Е2) Скорость для каж- дой цепи 105k (Е2) Заместители
₽ а СС р
1 СН3 0,118 СН3СНВгСН3 0,118 СНз
2 СН3 СН3 0,282 СН3СН2СНВгСН3 0,065 С2Н5
3 с2н5 СНз 0,196 СН3СН2СН2СНВгСН3 0,080 И-С3Н7
4 сн3 с2н5 0,200 СН3СН2СНВгСН2СН3 0,200 С2Н5 СПз
5 (СН3)2 1,00 /СНз СН3СВг< хсн3 1,00 1,00 (СН3)2 (СН3)2
6 сн3 (СН3)2 4,20 СН3 СН3СН2СВг< хсн3 0,85 0,85 сн3с2н5 сн3с2н5
(правая колонка), как и следует ожидать, замедляет образование бутена-1.
И то и другое изменение скорости обусловлено ориентацией в соответствии
с правилом Зайцева
Е2 Е2
(81%) СН3СН = СНСН3 СН3СН2СНВгСН3 —> СН3СН2СН = СН2 (19%)
Аналогичное рассмотрение примеров 2 и 3 позволяет понять, почему в третьем
примере изменения скорости отщепления в двух направлениях приводят
в соответствии с теорией к меньшей степени ориентации по Зайцеву, несмотря
на увеличение асимметрии исходного бромида
Е2 Е2
(71%) СН3СН2СН = СНСН3 <— СН3СН2СН2СНВгСН3 СН3СН2СН2СН = СН2 (29%)
Примеры 5 и 6 сравнивались так же, как и примеры 1 и 2, за исключением
того, что в определение ориентационных соотношений входит удвоенный
статистический вес одного из олефинов
Е2 Е2
(71%) СН3СН = С(СН3)2 <— СН3СН2СВг(СН3)2 -* СН3СН2С(СН3) = СН2 (29%)
Указанный анализ данных по скоростям и составу продуктов Е2-реакций
(например, в табл. 142) был в дальнейшем применен к гомологическим рядам
вторичных алкилбромидов Шинером и сотр. [33]; при этом оказалось, что
электромерный эффект остается преобладающим.
Кстати, здесь же уместно отметить (поскольку это потребуется ниже) раз-
личное влияние структуры алкила на бимолекулярные реакции замещения
и отщепления алкилгалогенидов и на относительное количество образую-
щегося олефина. Как упоминалось в гл. VI, разд. 6,а, в ряду алкилгалоге-
нидов с увеличением числа сс-метильных групп скорость бимолекулярных
реакций замещения уменьшается, а скорость элиминирования возрастает
(см. соответствующие данные в этом разделе); следовательно, должно увели-
читься относительное количество образующегося олефина, что и подтвер-
ждается данными, приведенными в табл. 143.
553
Глава IX
Наконец, следует еще раз указать, что по данным Сондерса [8], алкилфто-
риды не похожи на другие галогениды. Здесь сильна полярность концевой
группы,* а а-ненасыщенность, развивающаяся в переходном состоянии,
Таблица 143
Скорости реакций и выходы олефинов при
бимолекулярных реакциях алкилбромидов
Реакция с C2H5ONa в С2НзОН при 55 °C;
константы скорости к (Sjsj2) и к (Е2) в л-моль-1-с-1
а-Ряд 105Ц (Sn2) 105Й. (Е2) Выход олефина, %
Этил 118,2 1,2 1,0
Изопропил 2,1 7,6 79
mpem-Бутил Малая 50 ~ 100
может быть слабой. Полученные данные показывают, что при расщеплении
алкилфторидов преобладает расщепление по Гофману.
г. Мономолекулярное отщепление от ониевых ионов и галогенидов;
электромерный эффект. Мономолекулярное отщепление наблюдалось в слу-
чае вторичных и третичных ониевых ионов и вторичных и третичных алкил-
галогенидов. Найдено, что в этих случаях применимо правило Зайцева.
Можно сделать вывод, что в этих реакциях электромерный эффект является
определяющим.
Не вызывает сомнения тот факт, что в мономолекулярных реакциях элими-
нирования ониевые ионы и галогениды в противоположность реакциям
бимолекулярного элиминирования ведут себя одинаковым образом. При
бимолекулярных реакциях конкурирующие формы атаки на ониевые ионы
и на галогениды приводят к различным по существу продуктам, а при моно-
молекулярных реакциях как из ониевого иона, так и из галогенида перво-
начально образуется карбониевый ион, который подвергается конкури-
рующим между собой атакам. Однако количественно ориентация по Зай-
цеву более резко выражена при мономолекулярных реакциях элиминирова-
ния от ониевых ионов и галогенидов, чем при бимолекулярных реакциях
отщепления из галогенидов.
Вследствие двухстадийной природы мономолекулярного механизма кон-
станта скорости элиминирования к (Е1) является составной: она представ-
ляет собой произведение скорости первичного гетеролиза к{ на ту часть
от общего количества образовавшихся карбониевых ионов, которая вступает
в реакцию отщепления. В ряде случаев эта часть, т. е. процентное содержа-
ние олефина, образующегося при мономолекулярных реакциях, находится
в более простой зависимости от структуры, чем формальная константа ско-
рости отдельного процесса отщепления.
На примере вторичных алкилбромидов (табл. 144) показано влияние струк-
туры на скорость и относительное количество олефина, образующегося при
мономолекулярной реакции. Из приведенных значений [34] следует, что
как р-метильный, так и р-этильный радикалы способствуют более полному
протеканию элиминирования, причем влияние первого значительно больше.
Эта обычное различие в гиперконъюгации алкильных радикалов находится
в соответствии с теорией реакций, определяемых электромерный эффектом.
Анализ данных по мономолекулярному элиминированию из третичных бутил-
55^
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблииа 144
Мономолекулярная реакция образования олефинов
из вторичных алкилбромидов
Реакции проводили в 60%-ном водном растворе
этилового спирта при 80 °C; константы скорости ki и k (Е1), с-1
Алкильная группа 105/ц 105ft (El) Выход олефина, %
снзх )СН — сн/ СН3СН2, >сн- сн/ сн3сн2. )СН — СН3СН2 сн3сн2сн2Х >СН- сн/ 7,06 7,41 5,97 5,61 0,32 0,63 0,90 0,38 4,6 8,5 15,1 6,8
бромидов и третичных амилбромидов (табл. 145) показывает, каким образом
скорость и степень мономолекулярного элиминирования у различных цепей
Таблица 145
Некоторые значения скоростей, иллюстрирующие суммарный результат
прн мономолекулярной образовании олефинов из алкилбромидов а
Реакции проводили в этиловом спирте при 25 °C; константы скорости /с, и к (Е1) в с-1
а (3 а Р С=С-СН <— СН СВг-СИ —> СН-С=С Ра Ра
Заместители Для каждой цепи Для каждой цепи Заместители
е а выход олефина, % 105fe (Е1) 105ft (El) ВЫХОД олефина, % СС р
сн3 (СН3)2 (СНз)2 6,3 29,6 0,029 0,323 /СН3 СН3-СВг< \сн3 /СН3 СН3СН2СВ/ \сн3 0,029 0,029 0,036 0,036 6,3 6,3 3,3 3,3 (СН3)2 (СН3)2 сн:>с2н5 СН3С2Н5
Суммарные
скорости и количество отщепившегося олефина
1 О'’/i-l
1 05ft (El)
0,086
0,396
тпрстп-Бутил.............................................. 0,45
трегп-Ампл. .............................................. 1,0 9
Суммарная ориентация при отщеплении олефина
СНзСН=С(СНз)2, %
тпретп-Амил........................................... 82
Выход
олефина»
%
19,0
36,3
СНзСН2С(СНз)=СН2, %
18
алкильных групп совместно оказывают влияние на общую скорость отщеп-
ления, на количество образующегося олефина и на ориентацию при элими-
нировании. Эти величины [35] сравнимы с величинами для бимолекулярных
555
Глава IX
реакций соответствующих галогенидов, приведенных в примерах 5 и 6
табл. 142. Аналогичные данные в левой стороне каждой таблицы указывают
на качественное сходство влияния структуры: дополнительный р-метиль-
ный радикал в амильной группе облегчает образование двойной связи в этой
группе. Однако в случае молекулярного механизма такое влияние струк-
туры оказывается сильнее. Это верно не только для скорости, с которой
образуется двойная связь, но и для количеств, в которых она образуется
в определенной части молекулы. Однако величины, приведенные в правой
стороне этих таблиц, сильно отличаются друг от друга. Так, при бимоле-
кулярной реакции в соответствии с требованиями теории электромерного
эффекта замена а-(СН3)2 на а-СН3С2Н5 уменьшает скорость образования
соединения с двойной связью; при мономолекулярной реакции скорость
образования двойной связи в соответствующей алкильной цепи возрастает.
Если принять во внимание различия в механизме реакции, то эти данные
легко объяснить. Так, скорость элиминирования А(Е1) содержит в качестве
множителя — скорость гетеролиза, которая больше для третичного
амилбромида, чем для третичного бутилбромида; на этой стадии молекулы
насыщены и преобладает индуктивный эффект. Однако можно обойтись без
учета этого, не относящегося к делу обстоятельства, если сосредоточить
внимание не на скорости образования олефина, а на его выходах,
т. е. на заключительной стадии мономолекулярного процесса. В завер-
шающих стадиях мономолекулярного отщепления, в согласии с теорией
электромерного эффекта, замена а-(СН3)2 на а-СН3С2Н5 уменьшает степень
образования двойной связи в соответствующей алкильной цепи. В сноске
под табл. 145 показано, каким образом сочетание отдельных скоростей ведет
к суммарной скорости, наблюдаемому выходу олефина, а также к ориента-
ции по Зайцеву.
Указанный анализ скоростей и выходов продуктов в значительной мере был
применен впоследствии к серии третичных алкилхлоридов, т. е. от трет-
бутильной до высших mpem-алкильных групп. Результаты свидетельствуют
Таблица 146
Выходы олефинов, образующихся при мономолекулярных реакциях
вторичных и третичных алкильных соединений в водном растворе
этилового спирта
Алкильные соединения Растворитель - ВОДНЫЙ С2Н5ОН Темпепатура, '°C Выход олефина, %
Вторичные
Пропилбромид 80%-ный 80 4,6
Бутилбромид 60 % -нып 80 8,5
Т ретичные
Бутилхлорид 80%-ный 65 36,3
Бутилдиметил сульфопий 80%-ный 65 35,7
Амилхлорид 80%-ный 60 40,3
Амилдиметилсульфоний 60%-ный 65 39,8
80 % -ный 65 49,4
80%-пып 83 53,4
о том, что преобладающим продолжает оставаться электромерный эффект [36].
В разд. 2,6 отмечалось, что пространственные препятствия играют лишь
556
Отщепление с образованием олефинов, ацетиленов и карбенов
небольшую роль в Е2-отщеплении. Показано [36], что пространственные
препятствия при реакциях Е1 также мало влияют на выход олефинов. Несу-
щественная роль пространственных препятствий при элиминировании
по сравнению с замещением несомненно обусловлена легкой доступностью
протона, отщепляющегося от системы при элиминировании.
Сравнение количеств, в которых образуются олефины при мономолекуляр-
ных реакциях вторичных и третичных алкилгалогенидов, показывает
(табл. 146), что дополнительный а-метильный заместитель сильно способ-
ствует сдвигу мономолекулярного процесса в сторону элиминирова-
ния [37].
д. Дихотомия в реакциях отщепления; сочетание индуктивного и элек-
тромерного эффектов. Первоначально, безотносительно к механизму реак-
ции, правило Гофмана относилось к элиминированию в первичных алкиль-
ных группах в ионах аммония, а правило Зайцева — к элиминированию
во вторичных и третичных группах в алкилгалогенидах. Сначала не было
ясно, что определяет границы применимости этих двух правил — структура
алкила, замещаемая группа или механизм реакции. Как показано в предыду-
щих разделах, определяющим является не строение алкильного радикала,
а природа замещаемой группы и механизм реакции: при бимолекулярном
отщеплении ониевые ионы следуют правилу Гофмана, а алкилгалогениды,
кроме фторидов,— правилу Зайцева, в то время как при мономолекулярном
элиминировании и те и другие следуют правилу Зайцева.
Таблица 147
Некоторые значения скоростей, иллюстрирующие суммарный результат
при бимолекулярном образовании олефинов из алкилсульфониевых ионова
Реакции этилатов сульфония в 97%-ном С2Н5ОН при 24 °C;
константы скоростей к2 и к (Е2) в л-моль-1.с_1
С ='С - сн <— Й а Заместители Для каждой цепи а ₽ СН - С - СН 3 а| +1 S(CH3)2 Для каждой цепи а (3 > СН — С — С Заместители
а выход оле- фина , % 10% (Е2) 10% (Е2) ВЫХОД оле- фина, о/ /о а 13
СН3 (СН3)2 (СН3)2 (33) (14) 27 8 сн3 CH3C[S(CH3)2]/ СН3 СНз CH3CH2C[S(CHs)2]^ сн3 27 27 24 24 (33) (33) (41) (41) (СН3)2 (СН3)2 (СН3)С2Н5 (СН3)С2П5
а Суммарные скорости и выходы олефинов (бимолекулярная реакция)
105йз 10% (Е2) Выход
олефина,
%
mpem-Бутил ..........80 80 100
тргт-Амил............. 58 56 96
Суммарная ориентация (бимолекулярная реакция)
СН3СН = С(СН3)2, % СН3СН2С(СН3) = СН2, %
шреш-Амил.............14 86
J-57
Глава IX
В пограничной области применимости обоих правил зависящие от структуры
отклонения в скоростях реакций, в выходах олефина и в направлении элими-
нирования иллюстрируются данными табл. 147 и 148. Эти данные относятся
Таблица 148
Некоторые значения скоростей, иллюстрирующие суммарный результат
при мономолекулярном образовании олефина из алкилсульфониевых ионов а
Реакции сульфонийиодидов в 97%-пом этиловом спирте при 50 °C;
константы скорости к\ и к (Е1) в с-1
С = С - СН <— 3 а Заместители Для каждой цепи а [3 СН - С - СН ₽ J 3(СНз)2 Для каждой цепи > сн - Заместители а Р с — С
3 а выход оле- фина, 0/ /0 105/; (Е1) 105h (El) выход оле- фина, % а 6
(СН3)2 17 (0,30) СН;; (0,30) 17 (СН3)2
CH3C[S(CH3)2]/
СНз (0,30) 17 (СН3)2
сн3 (0,63) 4 (СН3)С2Н5
сн3 (СН3)2 56 (8,41) CH3CH2C[S(CH3)2]/
сн3 (0,63) 4 (СН3)С2Н6
а Суммарные скорости и выходы олефина
105ki
тирет-Бутич ......... 1,8
mpem-Амил........... 15,0
Суммарная, ориентация (мономолску.
СН3СН
mpeni-Амил...........
(мономолекулярная реакция)
105fe (Е1) Выход олефина, %
0,9 51
9,7 64
пиан реакция)
С(СН3)2, % СН3СП2С(СН3) - сн2, %
7 13
к уже упомянутым двум соединениям: ионам трет-бутил и трет-амил ди-
метилсульфония. Все изменения при переходе от одной таблицы к другой
обусловлены механизмом реакции [37, 38]. Данные табл. 147 относятся
к бимолекулярному механизму; хотя в таблице приведены как скорости
реакции, так и выходы олефинов для случаев элиминирования в различных
ветвях алкильных цепей, все же при этом механизме наиболее простая зави-
симость от структуры существует у скоростей реакций, поэтому выходы
олефинов приведены в скобках. При введении дополнительной метильной
группы в высший гомолог наблюдается уменьшение скорости элиминиро-
вания в длинной цепи mpem-амильной группы при незначительном измене-
нии скорости элиминирования в короткой цепи, как и должно быть при
преобладающем индуктивном эффекте. Общие результаты этих индиви-
дуальных изменений приведены в сноске под табл. 147. В табл. 148 приведены
данные тех же реакций, но для мономолекулярного механизма. И здесь
указаны как скорости реакции, так и выходы олефинов, но так как для этих
реакций наиболее непосредственно от структуры зависит количество обра-
зующегося олефина, то скорости приведены в скобках. Как и следовало
ожидать, поскольку электромерный эффект играет решающую роль, допол-
нительный метильный радикал увеличивает степень элиминирования
558
Отщепление с образованием олефинов, ацетиленов и карбенов
в длинной цепи трет-а&шльной группы и уменьшает скорость в короткой.
Данные, приведенные в сноске под табл. 148, иллюстрируют общий вывод,
заключающийся в качественной противоположности проявления всех струк-
турных влияний в случае мономолекулярного и бимолекулярного меха-
низмов.
Элиминирование в алкильных соединениях другого типа, чем ионы сульфо-
нил и алкилгалогениды, классифицируется (хотя и на менее надежной
экспериментальной основе) таким же образом по механизму реакции и ориен-
тации. На этом основании было сделано заключение, что в общей зависи-
мости от механизма элиминирования и от структуры замещаемых групп
превалирующее влияние на скорость и ориентацию может оказывать либо
индуктивный, либо электромерный эффект. Дихотомия может быть выра-
жена следующим образом [32, 37]:
+ + + 4-_ +__
Индуктивный контроль: механизм Е2; X NR;j, SR,, ОН2, S()2R, NO,, F
Электромерный контроль: механизм Е2; X Cl, Br, I, OSO2B, OCOR
механизм El; X — SR2, ОН2, Cl, Br, 1, OSO2R
Строение алкильного радикала непосредственно не играет роли, но оказы-
вает косвенное влияние, так как от строения зависит скорость при любом
механизме и, следовательно, относительное значение того или иного меха-
низма.
При раздельном рассмотрении областей приложимости индуктивного и элек-
тромерного эффектов следует иметь в виду то обстоятельство, что влияние
оказывают оба эффекта, но один из них в зависимости от рассмотренных
ниже условий преобладает. В разд. 2,6 и в, приведены доказательства неко-
торого изменения суммарного результата, определяемого превалирующим
эффектом, вследствие действия второстепенного эффекта. Для получения
более убедительных доказательств следует оставить в стороне алкильные
группы и исследовать, с одной стороны, более полярные, а с другой. —
более ненасыщенные группы. Можно полагать, что в первом случае решаю-
щую роль должен играть индуктивный, а во втором — электромерный
эффект, замаскированный в случае соответствующих алкильных радика-
лов.
Хорошим примером влияния групп различной полярности на скорости
бимолекулярного отщепления Е2 являются данные Сондерса и Уильямса [39]
по реакции пара-замещенных 2-фенилэтилсульфониевых ионов с этилат-
ионами в этиловом спирте [39]. Некоторые константы скоростей приведены
в табл. 149. При сравнении скоростей в ряду, начинающемся с метокси-
и кончающемся нитрогруппой, качественно видно, что полярные эффекты
действительно оказывают влияние. Из приведенного ряда данных для каждой
серии субстратов следует, что реакция сульфониевых ионов более чувстви-
тельна к полярным эффектам, чем реакция бромидов. Сондерс и Уильямс
предложили количественный тест на полярную природу кинетических
эффектов, состоящий в сравнении этих эффектов с термодинамическими
эффектами тех же пара-заместителей на константы кислотности фенолов.
И при индуктивной и при гиперконъюгационной передаче полярных влияний
кинетическая кислотность p-протонов 2-арилэтилсульфониевьтх ионов
и 2-арилэтилбромидов почти также зависит от природы пара-заместителей,
как термодинамическая кислотность фенолов. Этот метод сравнения поляр-
ных эффектов в разных реакциях будет рассмотрен в общем виде в гл. XVI,
однако здесь достаточно сказать, что идентичность полярных влияний счи-
5-59
Глава IX
Таблица 149
Константы скоростей (fc2, л-моль-1-с-1)
бимолекулярного отщепления
»-RCeH4CII2CH2X re-RCeH4CH —СН2, где X = S(CH3)2
и Вг, в этиловом спирте при 30 °C
(подданным Сондерса и Уильямса)
R 101 ft2 [X = S(CH3)2] 105 k2 (X = Br)
СН3О 11 16
сн3 23 23
и 50 42
Ci 244 191
СН3СО 1720
no2 75200
тается хорошо доказанной, если логарифмы констант скоростей или равно-
весия для одной реакции линейно связаны с логарифмами констант скоростей
или равновесия для другой реакции. С помощью этого теста можно заключить,
что влияние ?гар«-заместителей на скорости бимолекулярного отщепления
определенно осуществляется по такому же механизму, как и их влияние
на равновесную кислотность фенолов.
В качестве дополнительного примера рассмотрим бимолекулярное отщепле-
ние в случае алкилониевых ионов; эти процессы подчиняются правилу
Гофмана в случае простых алкильных групп. Если, однако, ввести в а- или
р-положение группу, подобную фенильной, с малой полярностью и высокой
степенью ненасыщенности, то правило Гофмана оказывается недействи-
тельным, поскольку предпочтительно образуется стирол, а не этилен [23].
Это в основном не является следствием индуктивного эффекта, обусловлен-
ного полярностью боковой цепи, так как качественно одинаковые резуль-
таты наблюдаются при введении фенила как в а-, так и в р-положения.
Скорости отщепления по реакции второго порядка [28, 40] для этилатов
этил-, 1-фенилэтил- и 2-фенилэтилдиметилсульфония в этиловом спирте
при 64 °C относятся как 1 : 100 : 430. Высокие скорости для обоих фенили-
рованных производных обусловлены тем, что сильно ненасыщенные фениль-
ные группы сделали определяющим электромерный эффект, что, естественно,
не произошло бы при введении алкильного радикала. То же самое наблю-
дается при бимолекулярном отщеплении от алкилбромидов, хотя более
очевидным было бы ожидать преобладающего влияния ненасыщенности.
В случае этил-, 1-фенилэтил- и 2-фенилэтилбромидов скорости бимолекуляр-
ного отщепления этилат-ионами в этиловом спирте при 55 °C приблизительно
относятся [36] как 1 : 35 : 350.
Таким образом, при отщеплении олефинов, так же как и при любой другой
гетеролитической реакции, имеют значение и полярность и ненасыщенность,
приводящие к смещению электронов по индуктивному механизму или
по механизму сопряжения, если позволяет структура молекулы. Кроме того,
совместное действие оказывают поляризация и поляризуемость, но, посколь-
ку в процессе реакции создается ненасыщенность, образованию переходного
состояния чаще способствует сопряжение в форме эффекта поляризуемости,
т. о. электромерный эффект. Ясно, что, когда влияющие группы обладают
неодинаковыми полярностью и ненасыщенностью, они будут проявлять
560
Отщепление с образованием олефинов, ацетиленов и карбенов
индуктивный или электромерный эффект в зависимости от своей природы,
и в этих обстоятельствах дихотомия Гофмана — Зайцева уже не имеет
значения.
Правила Гофмана и Зайцева, даже в общей форме, применимы только
к простым незамещенным насыщенным алкильным группам, составляющим
очень небольшую часть групп, влияние которых при элиминировании нас
интересует. Они применимы только к этим случаям потому, что алкильные
группы занимают уникальное положение среди органических групп, так
как сами по себе они лишены тех общих свойств полярности и ненасыщен-
ности, от которых зависят ориентация и скорость отщепления. Эти группы
практически неполярны, введение же некоторых полярных групп приводит
только к потенциальной полярности. Они не обладают врожденной ненасы-
щенностью, а имеют только скрытую ненасыщенность, связанную с гипер-
конъюгацией с ненасыщенными группами. Противоположные правила при-
менимы к алкильным группам потому, что эти группы не имеют свойств,
которые бы влияли на систему элиминирования, кроме тех свойств, которые
индуцируются в них самой системой. Поэтому их поведение определяется
самой системой. Полярность и правило Гофмана будут преобладать, когда
концевой группой является ионный центр или очень электроотрицательная
группа и когда в переходном состоянии Е2-реакции степень развивающейся
ненасыщенности не слишком велика. Гиперконъюгация и правило Зайцева
будут преобладать, когда концевая группа менее полярна и когда в переход-
ном состоянии Е2-реакции к отщепляющейся группе переходит значительная
доля электрона. Гиперконъюгация и правило Зайцева будут еще больше
преобладать, когда перед началом образования продукта реакции к отщеп-
ляющейся группе перейдет целый электрон, т. е. при Е1-механизме.
3. Влияние окружающей среды на скорость и соотношение
продуктов 1,2-элиминирования
а. Влияние среды на механизм. Ранние литературные данные об отщепле-
нии олефинов очень противоречивы, что было обусловлено главным образом
непониманием двойственности механизма. Один из вопросов, к которому
было привлечено большое внимание, заключался в выяснении количеств
образующихся олефинов при действии основных реагентов на различные
алкилгалогениды. Неф [41] в одних и тех же условиях выделил больше оле-
фина из третичных алкилгалогенидов, чем из вторичных, и из последних
больше, чем из первичных (первичные < вторичные < третичные). В про-
водившемся почти одновременно исследовании Брюсова [42] были получены
противоположные результаты: олефина образовывалось больше из вторич-
ных, чем из третичных галогенидов (вторичные > третичные). Впоследствии
Сегаллер [43] подтвердил последовательность вторичные < третичные. Значи-
тельно позже один исследователь, необоснованно предположив, что оба
вывода одновременно не могут быть правильными, решил опытным путем
сделать между ними выбор. Еще позднее [32, 44] кажущееся противоречие
было разрешено следующим образом.
Уже отмечалось, что при любом механизме элиминирования, Е2 или Е1,
количество олефина, образующегося из простых алкильных соединений,
возрастает в ряду первичный < вторичный < третичный (вследствие уско-
ряющего эффекта, оказываемого а-алкильной ветвью на образование
а,Р-двойной связи). Данные также показывают, что для любого типа алкиль-
ных групп выход олефина возрастает при переходе от Е1- к Е2-механизму.
36 — 0772
561
Глава IX
Еще одной истиной является тот факт, что, как и следовало ожидать, при
механизме Е2 выход олефина возрастает при увеличении основности реа-
гента, который отщепляет [3-протон. Если при увеличении концентрации
реагента или его основности или при уменьшении ионизующей силы раство-
рителя по отношению к алкилгалогениду происходит изменение механизма
от Е1 до Е2, то для первичных, вторичных и третичных алкилгалогенидов
это изменение будет происходить на разных стадиях изменений указанных
условий. В случае первичных галогенидов механизм Е2 может реализоваться
уже до начала изменения условий, в случае вторичных галогенидов измене-
ние механизма до Е2 будет происходить на относительно ранних стадиях,
а в случае третичных галогенидов — на относительно поздних стадиях изме-
нения условий. Поэтому в определенном интервале изменения любого из этих
свойств окружающей среды вторичные галогениды могут реагировать
по механизму Е2, а третичные — по механизму Е1. Если реагентом служит
сильное основание, например гидроксил- или этилат-ионы, то из вторичных
галогенидов образуется больше олефина, чем из третичных. В этих условиях
работал Брюсов в водном растворе этилового спирта. Неф использовал этилат-
ионы в сухом этиловом спирте; в этом случае все субстраты реагировали
по механизму Е2. Сегаллер, а также Мережковский [45] применяли относи-
тельно слабоосновные реагенты — фенолят- и ацетат-ионы.
б. Влияние растворителя на скорость реакции и на выходы олефинов при
неизменном механизме реакции [32, 46J. В общих чертах теоретическая
обработка данных о влиянии растворителя на кинетику нуклеофильного
замещения дана в гл. VII, разд. 3,а, где проведено сравнение опытных дан-
ных с результатами, получающимися при применении этой теории к моно-
и бимолекулярному нуклеофильному замещению. В настоящем разделе
рассмотрено приложение указанной теории к бимолекулярному и мономоле-
кулярному отщеплению олефинов; полученные выводы сопоставлены с экспе-
риментальными данными.
Как следует из разд. 1, механизмы нуклеофильного замещения и элиминиро-
вания чрезвычайно близки между собой. В обоих случаях при бимолекуляр-
ных реакциях перемещение электронов от реагента к отщепляемой группе
одинаково, но при элиминировании оно протекает через более длинную цепь
атомов, чем при замещении. В обоих случаях мономолекулярные реакции
имеют общую стадию, определяющую скорость всей реакции. Следующие
быстрые стадии тоже одинаковы, за исключением того, что при элиминиро-
вании перемещение электронов происходит через большее число атомов, чем
при замещении.
Рассматриваемые реакции разделим, как и раньше, на четыре типа, отли-
чающиеся по типу заряда (табл. 150). Как при реакциях отщепления, так
и при замещении остается верным, что сходство этих типов значительно
важнее, чем их различия; однако зависимость кинетики этих реакций от при-
роды растворителей показывает, что в обоих случаях четыре типа реакций
отличаются между собой.
Применяемая теория исходит из того, что кинетическое влияние раствори-
теля является главным образом результатом различий энергий сольватации
начального и переходного состояний и что такие различия влияют на энер-
гию активации. Эта же теория предполагает, что сольватация в любом состоя-
нии. зависящая от электрических зарядов, увеличивается с увеличением
заряда и уменьшается при его рассредоточении, причем величина заряда
играет более важную роль, чем его распределение. Наконец, теория допус-
кает, что сольватирующая способность растворителя возрастает с увеличе-
нием его дипольного момента и уменьшается с увеличением экранирования
562
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблица 150
Типы зарядов при отщеплении с образованием олефина
Тип 1. Первоначально акцептор протона отрицательный, замещаемая группа ней-
тральная.
Пример-. ОН + НСН2СН(СН3)Вг -+ Н2О 4- СН2 = СНСНз4-Вг-
Т п п 2. Первоначально акцептор протона нейтральный, замещаемая группа ней-
тральная.
Пример-. С2Н5ОП + НСН2С(СН3)2С1 С2И5ОН2 + СН2 = С(СН3)2 + Cl-
Тип 3. Первоначально акцептор протона отрицательный, замещаемая группа поло-
жительная.
Пример: С2Н5О- + HCH(CH3)C(CH3)2S(CH3)2С2Н5ОН 4-СН3СНС(СН3)2+ (CH3)2S
Т п п 4. Первоначально акцептор протона нейтральный, замещаемая группа поло-
жительная.
Пример: Н2О -L HCH(Ar)CH2N(CH3)3Н3О 4-АгСН = СН24- (CH3)3N
дипольных зарядов. Так, растворители Н2О, С2Н5ОН, (СН3)2СО, С6Н6 распо-
ложены в порядке уменьшения сольватирующей силы.
Применение высказанных положений к бимолекулярным реакциям показано
в верхней части табл. 151. Приведено также распределение зарядов при акти-
вации и кинетические выводы. Для предсказания влияния растворителя
на скорость бимолекулярного элиминирования необходимы только данные,
относящиеся к реакциям Е2. Для более точных выводов о влиянии природы
растворителя на выход олефина, образующегося при суммарной бимолеку-
лярной реакции, требуется раздельное рассмотрение кинетических данных
реакций SN2 и Е2; поэтому в таблицу7 включены данные для реакций SN2.
Теоретически различие состоит в том, что в переходном состоянии при отще-
плении заряд рассредоточен в большей степени, чем в промежуточном состоя-
нии при замещении. Из этого следует (если можно показать, что величина
зарядов в переходных состояниях в обоих случаях одинакова), что в реак-
циях типа 1 и 4 различия в распределении заряда служат причиной пред-
сказанного кинетического эффекта, который количественно больше в случае
отщепления, а не замещения. Наконец, это положение приводит к выводу
о влиянии растворителя на выход образующегося олефина. Слова «большое»
и «небольшое» в двух последних столбцах табл. 151 означают, что влияние,
обусловленное величиной заряда, важнее влияния, обусловленного рас-
пределением заряда.
В табл. 151 приведена также характеристика мономолекулярных реакций.
Рассматривается влияние растворителя на скорость суммарной мономоле-
кулярной реакции SN1 + Е1, т. е. на скорость первичного гетеролиза.
Таким образом, необходимо учитывать переходное состояние этого про-
цесса. Отмечено влияние растворителя на выход образующегося олефина,
причем отдельно рассматривается влияние растворителя на скорости неизме-
римо более быстрых процессов, завершающих реакции SN1 и Е1. Таким
образом, необходимо учитывать переходные состояния этих быстрых про-
цессов. Характерной их чертой является большее рассредоточение заряда
в переходном состоянии быстрой стадии отщепления. Если величины зарядов
в этих обоих переходных состояниях одинаковы (как в реакциях типов 2 и 4),
36* 563
Глава IX
Таблица 151
Предсказанное влияние растворителя на скорости и выход олефина,
образующегося при бимолекулярном и мономолекулярном отщеплении
Тип заря- да Размещение зарядов Влияние активации на заряды Влияние разбавления
Реак- ция начальное состояние переходное состояние на скорость на выход олефина
Бимолекулярный механизм', скорости и выход олефина
1 { Sn2 Е2 Y--4-RX Y- + RX 6- Y- б- Y---H- б+ Y- б+ Y- • -Н- 6~ Y- 6- Y--H- б+ Y- 6+ R. • • Стт б- • X б- ТС---Х Рассеива- ние Небольшое уменьшение Небольшое уменьшение
2 { 3 { 4 | SN2 Е2 Sn2 Е2 SN2 Y-|-RX Y-I-RX Y--LRX+ Y- + RX+ Y4-RX+ R. • СТ" R. • • Стт • R. • • X 6- ТС...Х 6+ •X б+ ТС---Х •вХ+ 6+ 1 J Увеличение Уменьше- ние Рассеива- Большое увеличение Большое уменьшение Небольшое 0 ? Небольшое
E2 ¥ П.\ ' Y--H. • СТТ ТС- • -X ние уменьшение уменьшение
Мономолекулярный механизм', скорости
SN1
+Е1
SN1
-f-El
И
Л
Увеличе-
ние
Уменьше-
ние
Большое
увеличение
Небольшое
уменьшение
Мономолекулярный механизм; выход олефина
1 f 2 1 SN1 El Y~i.IT Y~ + R+ 6- Y- 6- Y-- •H- 6+ 6+ • R Cttty1’ > Уменыпе- J шение
2 Г 4 1 SN1 El Y-LR + Y-pR+ <>+ Y- Y-- •H- •R ^+ CT7TC > Рассеива- J ние
Небольшое
уменьшение
?
то это различие в распределении зарядов позволяет сделать вывод о влиянии
растворителя на количество образующегося олефина.
В табл. 152 приведены данные, показывающие влияние природы раствори-
теля на скорости бимолекулярного отщепления Е2. В качестве растворите-
лей использованы смеси этилового или н-пропилового спирта с водой, в кото-
рых вода, конечно, является наиболее полярным компонентом. То, что
реакции типов 1 и 3 бимолекулярпы, доказывает их кинетический порядок,
а величины, приведенные в таблице, представляют собой константы скоростей
реакций второго порядка. Реакции типа 4 — сольволитические, и поэтому
приведенные в таблице величины являются константами скоростей реакций
первого порядка. Однако вследствие большой чувствительности скоростей
к добавлению оснований (даже слабых) можно полагать, что реакция
в основном бимолекулярна. Этот вывод подтверждается сильным уменьше-
нием скорости при замене отщепляющегося водорода на тритий [47].
56‘4
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблица 152
Экснериментальные данные, характеризующие влияние природы
растворителя на скорость бимолекулярного отщепления (Е2)
Тип Пример а Тем- пера- тура, °C Константа скорости 6 при содержа- нии Н2О в C2H5OH, % Предсказанное влияние
0 10 20 30 40 100
1 ОН + изо-С3Н7Вг в 55 1,46 0,71 0,47 Небольшое
3 ОН + (C2Hs)3S г 100 2050 210 2,4 уменьшение Значительное
4 H2O + Ar(CH2)2N(CH3)3 Д 100 2,10 1,40 0,39 уменьшение Небольшое
уменьшение
а Реакции типов 1 и 3 проводили в водном этиловом спирте, а типа 4, где Ar = 71-NO2C6H4,—
в водном н-пропиловом спирте.
6 Константы скорости второго порядка k% выражены в л-моль-1 • с-1, а константы скорости
первого порядка Л, — в с-1. Численные значения взяты из работы [46].
Б 104 ft2.
г 105 fc2,
Д 104 hj.
В последнем столбце таблицы для сравнения с опытными данными повторно
приводятся качественные предсказания, сделанные на основании теории.
Результаты эксперимента отвечают ожидаемым различиям между «значитель-
ным» и «небольшим» эффектами. Во всем интервале состава растворителей
величины изменяются либо в тысячи и больше, либо только в несколько раз.
В табл. 103 (гл. VII, разд. 3) были приведены результаты подобной же теоре-
тической трактовки влияния природы растворителя на суммарную скорость
мономолекулярной реакции, т. е. на суммарную скорость процесса SN1 + El.
Таблица 153
Экспериментальные данные, характеризующие влияние природы растворителя
на выход олефина при бимолекулярном и мономолекулярной отщеплении
Тип Пример а Выход олефина, % при содержании воды в водном С2Н5ОН, % 0 | 20 | 40 | 100 Предсказанный эффект
Бимолекулярный механизм
1 ОН + мо-С3Н7Вг (55° С) 71 59 54 Небольшое уменьшение
3 OH + (C2Hs)3S+ (100° С) 100 100 86 ?
Мономолекулярный механизм
2 mpem-C4HgBr (25° С) 19,0 12,6
т/>ет-С5НцС1 (25° С) 33,0 25,7 Небольшое уменьшение
mpem-CjHiiBr (25° С) 36,3 26,2
4 трет-С5НцЗ(СН3)2 (50° С) 64,4 47,8
7npem-C5H11S(CH3)2 (65° С) — 49,4 39,8 Небольшое уменьшение
а Числовые значения взяты из работы [46].
565
Глава IX
Во всех случаях эффект совпадает с ожидаемым. И опять предсказанное
«большое» увеличение скорости при переходе от спирта к воде проявляется
в увеличении в тысячи раз, в то время как при предсказанном «небольшом»
уменьшении скорости наблюдается уменьшение в несколько раз.
В табл. 153 сведены данные о влиянии состава водно-спиртовой смеси на коли-
чество олефина, образующегося при бимолекулярных и мономолекулярных
реакциях алкилгалогенидов и алкилониевых ионов. С увеличением содер-
жания воды в растворителе, как правило, наблюдается умеренное понижение
количества образующегося олефина. Во всех проверенных случаях теорети-
ческие предсказания согласуются с опытом. В последнем столбце табл. 153
для сравнения с опытными данными приведены теоретические предсказания.
в. Влияние температуры на скорость отщепления и выход олефина [48].
В большинстве известных случаев энергия активации бимолекулярного
отщепления олефинов от алкилхлоридов, алкилбромидов и алкилиодидов
при действии гидроксил- или этилат-ионов составляет 20...24 ккал/моль
(83,74-103... 100,48-103 Дж/моль), тогда как для алкилдиметилсульфоние-
вых ионов соответствующие величины несколько выше и обычно равны
24...26 ккал/моль (100,48-103... 108,86-103 Дж/моль). С заметным постоян-
ством энергия активации Е2-элиминирования для любого субстрата в любом
исследованном растворителе на 2 ± 1 ккал/моль (8,37 -103 ± 4,187 х
X Ю3 Дж/моль) больше, чем энергия активации сопровождающего элимини-
рование 8х2-замещения. Это означает, что при увеличении температуры
выход олефинов возрастает. Так, из изопропилбромида при бимолекулярной
реакции с лиат-ионами бОо-б-ного водного этилового спирта при 45 °C полу-
чается 53% пропилена, а при 100 °C — 64 % пропилена; в этом проявляется
типичное влияние температуры.
Одна из интерпретаций, аналогичная интерпретации влияния растворителя
на выход олефинов, состоит в том, что в переходном состоянии Е2-элиминиро-
вания единичный заряд распределяется среди большего числа атомов, чем
в переходном состоянии S^-замещения, поэтому переходное состояние
элиминирования будет менее сильно сольватировано и для достижения пере-
ходного состояния будет требоваться большая энергия активации, чем при
замещении. Соответствующие аргументы нельзя применить к алкилсульфо-
ниевым ионам, поскольку в этом случае заряд переходного состояния при
элиминировании и замещении может быть не одинаковым. Однако из опыт-
ных данных известно, что влияние растворителя на выход олефина для алкил-
галогенидов и сульфониевых ионов качественно подобно, и поэтому если
влияние температуры на относительный выход продуктов действительно
контролируется сольватацией, то логично было бы ожидать качественно
одинакового влияния в алкилгалогенидах и сульфониевых ионах, что
и было обнаружено.
При мономолекулярном отщеплении и замещении измеряемая энергия
активации представляет собой энергию активации начальной медленной
стадии, общей для обеих реакций. Для большинства шреш-алкилхлоридов
и шретп-алкилбромидов в сухом или водном этиловом спирте известные энер-
гии активации мономолекулярных реакций составляют 22...24 ккал/моль
(92,11 -IO3... 100,48-103 Дж/моль). Для ионов шретп-алкилсульфония эти
величины гораздо выше — 31...33 ккал/моль (129,79-103...138,16 X
' 103 Дж/моль). Это почти наверняка обусловлено сильной сольватацией
ионного центра в исходном состоянии сульфониевого иона. Причина того,
что такое большое различие в энергиях активации не наблюдалошэ при
бимолекулярном элиминировании или бимолекулярном замещении в алкил-
галогенидах и сульфониевых ионах, может состоять в том, что в бимолеку-
566
Отщепление с образованием олефинов, ацетиленов и карбенов
лярных реакциях дополнительная энергия, необходимая на десольватацию,
в значительной мере компенсируется кулоновской энергией притяжения
анионного реагента. Подтверждением объяснения, основанного на сольва-
тации, является то обстоятельство, что при мономолекулярной замещении
в алкилбромидах, содержащих ионизованный а-карбоксилатный замести-
тель в воде или водном этиловом спирте (гл. VII, разд. 2,6), энергия
активации, которая составляла бы 22...24 ккал/моль (92,11 -103...
...100,48-103 Дж/моль), если бы а-карбоксилатный заместитель был замещен
на а-метильный заместитель, повышается до 29...33 ккал/моль (121,42 х
XЮ3...138,16-103 Дж/моль) [49].
В мономолекулярных реакциях выход олефина определяется относитель-
ными скоростями альтернативных быстрых стадий разложения карбониевого
иона. Наблюдения показывают, что для любого субстрата, который вступает
в конкурирующие реакции мономолекулярного отщепления и замещения,
при повышении температуры увеличивается выход олефина. Так,
нгрет-бутилбромид при сольволизе в сухом этиловом спирте дает 19% изо-
бутилена при 28 °C, но 28% при 55 °C, а ион тпретп-амилдиметилсульфония
при сольволизе в 80 %-ном водном этиловом спирте дает 48% амиленов при
50 °C и 54% при 83 °C. Хотя нельзя измерить энергии активации быстрых
процессов, определяющих указанное соотношение продуктов, рассмотрение
соотношений продуктов реакций и их зависимости от температуры показы-
вает, что энергия активации процесса, которым завершается отщепление,
должна быть всегда на 2 ± 1 ккал/моль (8,37 -IO3 ± 4,187-103 Дж/моль)
выше, чем энергия активации стадии, завершающей замещение.
Указанные процессы являются быстрыми реакциями первоначально образо-
вавшегося карбониевого иона, и одно из объяснений различного темпера-
турного эффекта состоит в том, что в переходном состоянии реакции, завер-
шающей отщепление, единичный положительный заряд распределяется между
большим числом атомов (что приводит к меньшему уменьшению энергии
сольватации), чем в переходном состоянии реакции, завершающей конку-
рирующее замещение.
4. Пространственная направленность 1,2-отщепления
а. Пространственная направленность отщепления с образованием
ацетиленов. В старой литературе содержится ряд наблюдений, указы-
вающих на существование в алициклических соединениях и в олефинах
таких положений, которые стереохимически благоприятны и неблагоприятны
для отщепления. Михаэль [50] в своих исследованиях показал, что хлор-
фумаровая кислота при действии щелочей превращается в ацетилендикарбо-
новую кислоту в 50 раз быстрее, чем хлормалеиновая. Шаванн [51] наблюдал,
что ywc-дихлорэтилен под влиянием щелочей превращается в хлорацетилен
в 20 раз быстрее, чем ттг/шяс-изомер.
ООС — С — С1 он- он- С1—С—соо-
|| _ ----> -оосс = ССОО- <-||
Н-С-СОО 50 1 Н — с— соо-
JI — С — CI он- он- С1 — С—Н
II НС СС1 - II
H-C-CI 20 1 Н-С-С1
Позднее Кристол и сотр. [52] изучили другие примеры. Так, цис-п-нитро-
стирилбромид в присутствии этанольного раствора этилата натрия претер-
певает элиминирование в 2300 раз быстрее, чем происходит альтернативная
567
Глава IX
реакция присоединения к транс-изомеру
n-NO2C6H4—С—Н с2н5о-
| ----> ra-NO2C6H4C = С — Н
Вг—С —Н
n-NO2C6H4-С-Н С2н5о-
П -------> zi-NO2C6H4CH2CH(OC2H5)2
Н-С-Вг
Очевидно, транс-элиминирование, т. е. элиминирование водорода и гало-
гена, расположенных в транс-положении, происходит легче, чем цас-элими-
нирование. Иногда различие в скоростях мало, но иногда транс-элиминиро-
вание является практически единственным процессом. Можно вновь указать,
что по данным Модены (гл. VII, разд. 9,а) замещение галогена на метокси-
группу в цнс-р-арилсульфонилвинилхлоридах происходит через транс-эли-
минирование ацетиленового производного по механизму Е2 с последующим
транс-присоединением
ArSO2 — С —Н
11
CI-C-H
ArSO2 — С = С —Н —>
ArSO2 — С— Н
II
сн3о—с—н
тогда как соответствующая замена С1 на СН3О в транс-|3-аренсульфонил-
винилхлоридах происходит только как прямое Зк2-замещение.
Все эти реакции показывают, что стереохимически предпочтительным
является транс-элиминирование, хотя может происходить и цнс-элиминиро-
вание. В 1948 г. было отмечено, что гетеролитическое элиминирование вклю-
чает [36] внутримолекулярное замещение типа Sn2 у а-углеродного атома,
для которого справедлив принцип, состоящий в том, что новая пара электро-
нов входит со стороны, удаленной от места отщепления старой электронной
пары (гл. VII, разд. 7). Согласованно с этим процессом происходит замеще-
ние типа Se2 у Р-углеродного атома. Стереохимия такого замещения, вероят-
но, не ограничена каким-либо твердым принципом. Однако при таком замеще-
нии обычно сохраняется конфигурация (гл. VII, разд. 10), поэтому согласо-
ванная реакция происходит с транс-элиминированием, как представлено
ниже. Если, однако, в переходном состоянии участвующие в реакции элек-
троны достаточно освобождены вследствие большой степени отщепления
протона и смещены за атом углерода, то может произойти цнс-элиминиро-
вание [53]
/3
С1
транс-Элиминирование
цис-Элиминирование
б. Пространственная направленность отщепления олефинов. Стерео-
химия отщепления олефинов от алкильных производных будет зависеть
от конформации. В этом случае может происходить или антиперипланар-
ное (ар) элиминирование, если при Е2-механизме изменения связей так
синхронизированы, что в переходном состоянии перенос протона не чрез-
мерно выражен, или синперипланарное (sp) элиминирование, когда в пере-
568
Отщепление с образованием олефинов, ацетиленов и карбенов
ходном состоянии Е2-процесса перенос протона значителен [53]
ар-Элиминирование
sp-Элиминирование
В нормальной заторможенной конформации атомов у С — С-связи анти-
перипланарная (ар) конформация отщепляющихся атомов позволяет осуще-
ствляться антиперипланарному элиминированию. Можно ожидать, что
оно будет предпочтительным, потому что в таком случае будет осуществляться
сочетание обычной стереохимии Зв2-замещения с необходимой стереохимией
8х2-замсщсния [36, 53]. Другая возможная конформация отщепляющихся
групп является синклинальной (sc). Эта конформация в меньшей степени
способствует отщеплению, чем даже синперипланарная (sp), для которой
обычно возможно синперипланарное элиминирование, хотя и более затруд-
ненное по сравнению с антиперипланарный элиминированием. Когда общая
конформация является заслоненной (в ряде специальных случаев), отще-
пляющиеся атомы могут быть или антиклинальными (ас), или синперипла-
нарпыми (sp). В первом случае элиминирование сильно затруднено по срав-
нению с ар-формой, а во втором может происходить довольно легко, по лег-
кости находясь на втором месте после ар-элиминирования. sp- и ар-Формы
элиминирования, таким образом, осуществляются легче всего, причем
ар-элиминирование происходит с большей легкостью.
CI
Н(ар)
С1
Начальная заторможенная
конформация
Начальная заслоненная
конформация
В циклоалканах конформационные и геометрические факторы связаны друг
с другом. В простейших циклогексанах, для которых наиболее стабильной
является конформация кресла, все связи кольца имеют заторможенные кон-
формации, поэтому экзоциклические связи делятся на две группы, а именно:
на аксиальные (а) и экваториальные (е) связи. Две 1,2-транс-связи могут
быть или антиперипланарными (ар), когда обе связи аксиальны (а), или
синклинальными (sc), когда обе связи экваториальны (е), как показано ниже.
1,2-^мс-Связи должны содержать одну аксиальную и одну экваториальную
связи, т. е. неизбежно быть синклинальными (sc). С другой стороны, в про-
стом циклопентановом кольце связи кольца в той степени, в какой это кольцо
можно рассматривать как плоское, будут иметь приблизительно заслоненные
конформации, и поэтому 1,2-транс-связи будут антиклинальными (ас),
а 1,2-^нс-связи — приблизительно синперипланарными (sp). Таким образом,
в то время как простейшие производные циклогексана должны вступать
преимущественно в реакции транс-элиминирования (при условии, что
отщепляющиеся атомы находятся или могут легко переходить в аксиальное
положение), простейшие производные циклопентана должны вступать как
569
Глава IX
в транс-, так и в цис-элиминирование с небольшим преобладанием первого
процесса.
Лее транс-конформации циклогексана цис-Конформация циклогексана
Одним из простейших примеров большого различия в легкости Е2-элими-
нирования цис- и /«ракс-а томов в производных циклогексана является случай
гексахлорциклогексанов. Эти соединения известны в нескольких стерео-
химических формах, каждая из которых при действии щелочи претерпевает
трехстадийное дегидрохлорирование до соответствующего трихлорбензола
[54]. Для некоторых изомеров медленной является лишь первая стадия
дегидрохлорирования, тогда как для других медленными будут две первые
стадии. Все изученные реакции отщепления имеют кинетический второй
порядок и энергию активации в диапазоне 19. ..21 ккал/моль (79,55 ЛО3...
Таблица 154
Аррениусовские энергии активации [ккал/моль (4,187-IO3 Дж/моль)]
щелочного дегидрохлорирования гексахлорциклогексанов в водном растворе
этилового спирта по реакции второго порядка
Изомер гексахлор- циклогексана Структура: цис-С! Стадия реакции Водный С2Н5ОН
90%-ный (Пастернак) 80%-ный (Кристол)
сс 1:2:4 1 19,0 18,5
3 1:3:5 1 32,3 31,0
Л’ 1 : 4 1 20,4 } (20,6)
2 20,1
6 1 : 3 1 21,6
2 21,1
е 1:2:3 1 21,4
87,92-103 Дж/моль) (табл. 154). Исключение составляет лишь одна реак-
ция, а именно первая стадия реакции ^-изомера, имеющая энергию актива-
ции 32 ккал/моль (133,98 ИО3 Дж/моль). Это впервые было обнаружено
в 1947 г. и первоначально считалось, что в этом случае реализуется другой
механизм, однако никакого изменения механизма не было доказано; следо-
вательно, явление имеет стереохимическое объяснение. Конфигурации
различных стереоизомеров гексахлорциклогексана (обозначены греческими
буквами в табл. 154) характеризуются числом цис-атомов хлора, располо-
женных по одну сторону циклогексанового кольца. Очевидно, что триго-
нально-симметричный p-изомер имеет только цис-1,2-пары атомов Н и С1.
Все атомы хлора экваториальны, а все атомы водорода аксиальны. Таким
образом, должно происходить цис-элиминирование из sc-положений, кото-
рое сильно затруднено даже по сравнению с sp-элиминированием.
570
Отщепление с образованием олефинов, ацетиленов и карбенов
Де Пуи и сотр. [55] показали, что стереохимическое различие в Е2-элими-
нировании производных циклопентана гораздо менее заметно, чем у произ-
водных циклогексана. Так, для цис- и тра«с-1-фенилциклогексил-2-га-толуол-
сульфонатов, которые реагируют с mpem-бутилат-ионом в mpem-бутиловом
спирте
/J^C6H5
п \hJ-OTs
тра/ю-элиминирование от цис-изомера происходит лишь в 14 раз быстрее
цис-элиминирования от mpauc-изомера. Отношение скоростей для реакций
соответствующих циклогексановых субстратов равно 104. Такое различие
в стереоспецифичности Е2-процессов можно ожидать на основании конфор-
мационного анализа, данного выше. Де Пуи и сотр. выяснили, что как
цис-, так и транс-элиминирование имеют механизм Е2.
Зихср и сотр. [56] описали примеры из ряда циклодекана, в которых одно-
временно или в сравнимой степени происходит анти-элиминирование,
т. е. элиминирование, близкое к теоретически предпочтительному ар-про-
цессу, и снн-элиминирование, близкое к sp-элиминированию. Иногда син-
элиминирование происходило быстрее анта-элиминирования в несколько
(до десяти) раз. В качестве примеров были взяты реакция 1,1,4,4-тетраметил-
циклодекан-7-га-толуолсульфоната с znpem-бутилатом калия в диметилформ-
амиде, для которой обе формы элиминирования наблюдались в сравнимых
количествах, и реакция соответствующего 7-триметиламмониевого иона
с метилат-ионами в метиловом спирте, для которой преобладало син-элими-
пирование. В результате каждой из этих реакций образовывались цис-
пли транс-6,7-олефины и цис- или транс-1.8-олефины.
цис или транс цис или транс
Путем введения дейтериевой метки в 6-положение цис- и транс-изомеров
было доказано, что атом водорода, отщепляющийся при образовании
7,8-ена, всегда находился в транс-положении к отщепляющейся группе X
(этот атом водорода указан на схеме). Отсюда следует, что цис-олефин обра-
зуется при анти-элиминировании, а транс-олефин — при син-элиминиро-
вании (после скручивания конформации). Скорость син-элиминирования
от аммониевого иона, в результате которого образуется транс-1,8-олефин,
примерно в 10 раз больше скорости анти-элиминирования с образованием
цис-7,8-олефина.
Зихер и сотр. [56] распространили это исследование на ациклический ряд,
что имеет довольно важное значение, поскольку все уже обсуждавшиеся дан-
ные по пространственной направленности элиминирования олефинов были
получены в ациклическом ряду. Вопрос состоял в том, в каком положении —
син или анти — должны находиться атомы (Н) и (X), чтобы они могли
отщепляться в процессе бимолекулярного элиминирования от RHC(X) —
— C(H)HR', и какой олефин образуется в каждом случае — цис или транс!
В изученном примере X = Х(СН3)з, R = неопентил и R' = и-бутил; при
этом любой из атомов водорода группы — C(H)HR' стереоспецифически
571
Глава IX
замещался на дейтерий, и таким образом были получены трео- и эритро-
монодейтеропроизводные, являвшиеся субстратами при проведении отще-
пления. Из формул, приведенных ниже, можно видеть, что если в самом деле
тракс-олефин образуется при син-элиминировании, а цис-олефин — при
aumu-элиминировании, то при образовании как цис-, так и транс-олефина
трео-изомер должен терять дейтерий, а эритро-изомер не должен его терять.
Это в общем соответствует экспериментальным данным.
транс цис
транс цис
Когда в качестве основания, необходимого для осуществления этих бимоле-
кулярных реакций отщепления, применялся метилат-ион в метиловом спирте,
количество транс-олефина, образовавшегося при син-элиминировании,
и количество цис-олефина, образовавшегося при анти-элиминировании,
были сравнимыми, а их отношение было равно 5 : 4 в случае трео-аммоние-
вого иона и 3 : 4 в случае эритро-иона. Однако при использовании трет-
бутилат-иона в mpem-бутиловом спирте или диметилформамиде реакция
более чем на 90% имела характер сни-элиминирования, приводящего к обра-
зованию транс-олефииа. Отсюда был сделан вывод, что общим стереохими-
ческим правилом расщепления по Гофману является син-элиминирование.
Кроме того, было отмечено, что син-элиминирование особенно преобладает
при действии более сильных оснований; это согласуется с идеей о том. что
син-элиминирование ускоряется при увеличении степени переноса протона
в переходном состоянии бимолекулярного механизма [27, 53].
В дальнейшем этот вывод был подтвержден. Бантроп, Хыоз и автор [27]
пришли к выводу, что при Е2-элиминировании в одинаковых условиях сте-
пень переноса протона в переходном состоянии в случае аммониевых ионов
больше, чем в случае ионов сульфония, и что эта степень для любого иона
возрастает при переходе от этилат-иона в этиловом спирте к mpem-бутилат-
иону в mpem-бутиловом спирте. Завада и Зихер [56] установили, что те же
правила контролируют образование цис- вместо транс-нонена-4
из и-СзЩСНгСНХС^д-н. В этиловом или метиловом спирте авторы полу-
чили главным образом цис-олефин, причем из 4-нонилтриметиламмония цис-
олефина получалось больше, чем из иона 4-нонилдиметилсульфония. Однако
оба соединения в mpem-бутиловом спирте дают в основном тракс-олефин.
Ниже приведены данные по образованию цис-олефина (%) из 4-нонилтри-
метиламмония и 4-нонилдиметилсульфония.
mpem-C4H9O-/mpem-C4H9OH С2Н5О-/С2Н5ОН СН3О-/СН3ОН
X = N(CH3)3 26 74 81
X = S(CH3)3
9
64
572
Отщепление с образованием олефинов, ацетиленов и карбенов
Таким образом, многие данные подтверждают точку зрения о том, что сын-
элиминирование происходит легче, во-первых, при усилении степени переноса
[5-протона по сравнению со степенью переноса ос-электрона (при Е2-меха-
низме) и, во-вторых, при увеличении степени приближения конформации
алкильной группы к той конформации, которая соответствовала бы точному
sp-элиминированию. Такое влияние этих факторов ожидается также и из тео-
ретических предпосылок [53].
В неопубликованной статье Зихер и Завада отмечают, что наличие корреля-
ции сын-элиминирования с т^ннс-олефиновым продуктом и ннты-элими-
пирования с цис-продуктом означает, что в обоих формах отщепления преиму-
щественно отщепляется один и тот же индивидуальный атом водорода
Р-СН2-группы. Эти два атома водорода первоначально неэквивалентны
вследствие влияния асимметрического и-углеродного атома. Почему эта
неэквивалентность коррелирует процесс с продуктом именно указанным,
а не другим возможным образом — остается не ясным.
Влияние дополнительных ненасыщенных или полярных заместителей на
стереохимию было исследовано в ряду циклогексана. Бордуэлл, Кристол
и др. [57] показали, что при катализируемом основаниями элиминировании
ненасыщенность [5-фенильного заместителя в циклогексановом субстрате,
действуя совместно с полярным эффектом ос-аммонийного полюса, может
вызвать uwc-элиминирование, которое в этом ряду по необходимости должно
быть синклинальным. Подобный эффект может вызвать также сильно аци-
дифицирующая сульфоновая группа в [5-положении.
Н^С6Н5 он~
(CH3)3N+J- Н
SO2Ar
Одно время считалось, что в таких примерах может происходить изменение
механизма от согласованного Е2 до постадийного Е1сВ, однако Шайнер [58]
показал, что это не так. С другой стороны, из этих данных довольно четко
следует, что достаточная ацидификация [5-протона приводит к увеличению
степени спи-элиминирования. И действительно, так и должно быть, если
учесть, что в рамках Е2-механизма увеличение кислотности [5-протона приво-
дит к тому, что компонента электрофильного замещения Е2-процесса приво-
дит к обращению конфигурации.
В 1940 г., ранее чем была предложена изложенная выше теория и получены
данные по стереоспецифичности Е2-реакций, Хюккель [9] впервые четко
высказал предположение, что стереохимия отщепления зависит от механизма
реакции. Изучая соотношение изомеров в продуктах элиминирования от
стереоизомерных алициклических субстратов (здесь впервые были примене-
ны сильпоосновные и почти нейтральные условия), Хюккель сделал заклю-
чение, что, во-первых, элиминирование по механизму Е2 стереоспецифично
в том смысле, что в ряду производных циклогексана предпочтительно про-
исходит тп/ншс-элиминировапие, и, во-вторых, элиминирование по механизму
Е1 не является стереоспецифическим. Хюккель не применял кинетический
контроль для подтверждения предполагаемых механизмов, однако он принял,
что отщепление в сильноосновных условиях, вероятно, происходит по бимо-
лекулярному механизму, а отщепление в почти нейтральных условиях
в сильноионизующих растворителях — по мономолекулярному механизму.
Наиболее обширные и информативные исследования Хюккель выполнил на
ментил- и неоментилхлоридах и триметиламмониевых ионах. Эта работа была
573
Глава IX
проверена и завершена Хьюзом и сотр. [59, 60], которые применили кинети-
ческий контроль и иногда меняли условия, чтобы разделить два механизма,
если они реализовывались одновременно. Полученные ими результаты при-
ведены в табл. 155.
Таблица 155
Пространственная и полярная ориентация элиминирования
в ментилхлоридах и триметиламмонисвых ионах
СН2-СН2-СНСН3 СН2-СН2-СНСН3 СН2-СН2-СНСН3
I I -> I 1+1 I
пзо-С3Н7С(И) — СН(Х)СН2 изо-С3Н7С=СН —СН2 изо-С3Н7СН — СН = СН
(4) (3) (2) (4) (3) (3) (2)
Бимолекулярное элиминирование', X —С1 (растворитель С2Н5ОН)
. Г Неоментихлорид + СзНзО" —> Ментен-3+ Ментеп-2
1 t транс-(Н) (X) Е2 ~ 78 % ~ 22 %
Ориентация по правилу Зайцева
9 Г Ментилхлорид + СаНзО-Мептен-Зф-Ментен-2
“ I дш--(Н)(Х) Е2 0% 100%
Ориентация против правила Зайцева
Бимолекулярное элиминирование', X = N(CH3)3+ (растворитель Н2О)
,, ( Неоментил-Х(СН3)£ф-ОН"Мептен-Зф-Мептен-2
' { транс-(Н) (X) Е2 ~ 88 % ~ 12 %
Ориентация по правилу Зайцева (аномально)
, Г Ментил-К(СН3)3 ф- ОН- Мептен-3+Мептеп-2
4 I гщс-(Н)(Х) Е2 0% 100%
Ориентация строго по правилу Гофмана
Бимолекулярное элиминирование; X = N(CH3)J (растворитель С2Н5ОН)
о, f HeoMCHTji.4-N(CH3)J4-C2H5O_ Ментен-3 ф-Ментеп-2
° { траис-(Н) (X) Е2 65 % 35 %
Ориентация по правилу Зайцева (аномально)
f Ментпл-Х(СН3)3 + С2Н5О_ —> Мептен-Зф- Мептеп-2
4 Кс-(Н)(Х) Е2 0% 100%
Ориентация строго по правилу Гофмана
Мономолекулярное элиминирование; Х = С1 (растворитель 80%-ньш водный CoHgOH)
г f Неоментилхлорид Ментен-Зф-Мептен-2
° I транс-(Н) (X) Е1 98,8 % 1,2 %
Г Ментилхлорпд->-Ментен-Зф-Ментеп-2
° [. цис-(Н) (X) Е1 68 % 32 %
Ориентация по правилу Зайцева
Мономолекулярное элиминирование; X=-N(CH3)J (растворитель Н.,О)
г. ( Неоментил-Х(СН3)£ —>- Ментеп-Зф-Ментен-2
‘ { транс-(Н) (X) Е1 98,1% 1,9%
Ориентация по правилу Зайцева
о ( Ментил-Х(СН3)£ —> Ментен-Зф-Мептен-2
I 1щс-(Н)(Х) Е1 68% 32%
Ориентация по правилу Зайцева
Во всех случаях происходило отщепление группы Х[С1 или N(CH3)g] из
положения 3 вместе с протоном от 2-углеродного атома, с которым связано
два протона — один в цис-, а другой в щранс-положении к X, или с единствен-
ным протоном в положении 4. В табл. 155 цис- или транс-положение этого про-
574
Отщепление с образованием олефинов, ацетиленов и карбенов
тона по отношению к X указано сразу же под наименованием субстрата.
Реакция 1 — это Е2-элиминирование от неоментилхлорида, для которого
возможно стереоспецифическое транс-элиминирование в любом альтернатив-
ном направлении. Поэтому наблюдаемое направление реакции контролирует-
ся теми факторами, которые способствуют протеканию Е2-процесса по пра-
вилу Зайцева. И действительно, ориентация в этом случае соответствует
правилу Зайцева. В реакции 2 субстратом служит ментилхлорид, содержащий
единственный протон в грс-положении. Поэтому вследствие стереоспецифич-
ности Е2-механизма элиминирование вынуждено идти по направлению,
противоположному направлению, ожидаемому на основании правила Зай-
цева [59].
Реакция 3 соответствует Е2-отщеплению от неоментилтриметиламмониевого
иона, который имеет транс-протон с каждой стороны аммониевой группы,
и поэтому направление элиминирования не имеет стереохимических ограни-
чений. Бимолекулярное отщепление в случае ионов аммония обычно подчи-
няется общему правилу Гофмана, т. е. преимущественно образуется наименее
алкилированный этилен, однако в данном случае реакция подчиняется пра-
вилу Зайцева, т. е. приводит к образованию наиболее алкилированного
этилена. Этот случай действительно является аномальным и не объясняется
теорией, которая разъяснялась выше; этот вопрос еще будет обсуждаться
в разд. 4,в. Сейчас же возвратимся к таблице. Реакция 4 соответствует
Е2-отщеплению от иона ментилтриметил аммония, при котором стереохими-
ческие ограничения приводят к реализации правила Гофмана. Однако в этом
случае правило Гофмана выполняется очень строго, так как образуется
исключительно ожидаемый по правилу Гофмана олефин. Реакции 3' и 4'
подобны реакциям 3 и 4 соответственно и отличаются лишь тем, что вместо
воды в качестве растворителя используется этиловый спирт. Наибольший
интерес представляет то, что аномальность реакции 3 в реакции 3' уменьшает-
ся, однако полностью не исчезает [60].
Остальная часть таблицы относится к реакциям тех же субстратов, происхо-
дящим по механизму Е1. Первоначальная концепция Хюккеля состояла
в том, что при таком механизме вследствие промежуточного образования
карбониевого иона стереоспецифичность должна быть небольшой. Данные,
которые имелись в распоряжении Хюккеля, были недостаточно полными,
однако, если бы Хюккель имел результаты, соответствующие реакциям 5—8,
он несомненно объяснил бы их тем, что соответствующие хлориды и ионы три-
метиламмония дают один и тот же ион карбония, который, вероятно, являет-
ся одним и тем же в случае и ментил- и неоментилпроизводных, и поэтому
строение олефина, образующегося из любого субстрата, должно находиться
в соответствии с правилом Зайцева. И действительно, все эти реакции под-
чиняются правилу Зайцева [59, 60]. Из неоментилхлорида и неоментилтри-
метиламмония образуются смеси олефинов практически одинакового состава
с большим преобладанием того олефина, образование которого соответствует
правилу Зайцева (реакции 5 и 7). Также идентичны и смеси олефинов, обра-
зующиеся из ментилхлорида и иона ментилтриметиламмоиия. однако в этом
случае состав смесей более сбалансирован (реакции 6 и 8). Создается впечат-
ление, будто бы карбониевые ионы в момент потери протона «не помнят»,
каким путем они образовались, но «помнят», какой протон должен отщепить-
ся. Другими словами, хотя экранирующим действием уходящей группы
можно пренебречь, карбониевые ионы не достигают полного конформацион-
ного равновесия.
Если при мономолекулярной отщеплении Е1 уходящая группа отщепляется
в виде отрицательно заряженного иона, а растворитель обладает очень низ-
575
Глава IX
кой основностью, то этот противоион может быть наилучшим акцептором
протона от иона карбония; а если при этом растворитель имеет также и низ-
кую диэлектрическую проницаемость, что обусловливает некоторую ста-
бильность ионных пар, то мономолекулярный механизм может приобрести
стереоспецифичность, так как протон будет иметь тенденцию находиться по-
соседству с образовавшимся, но еще не отошедшим на большое расстояние
противоионом. Таким образом, синперипланарное или синклинальное эли-
минирование (или, как еще можно сказать, син-элиминирование) будет пре-
обладать над аити-перипланарным или антиклинальным, т. е. над анти-
элиминированием. Это было показано Скеллом и сотр. [61] при измерении
содержания дейтерия в цис- и транс-бутенах-2, образующихся из дритро-
и трео-З-дейтеробутил-2-га-толуолсульфонатов. Как следует из приведенных
ниже формул, сан-элиминирование от эрнтро-субстрата приводит к дейтери-
рованному транс-бутену или (после конформационного вращения) к недей-
тированному цис-бутену, тогда как при снн-элиминировании от трео-субстра-
та дейтерий должен содержаться в цис-бутене. При анти-элиминировании,
естественно, должен получиться противоположный результат.
Некоторые экспериментальные данные этих авторов приведены в табл. 156.
Для сравнения в таблицу включены данные по Е2-элиминированию. «Соль-
волитическое» элиминирование обозначено символом Е1. Как видно, относи-
Таблица 156
Относительная важность син-элиминирования, при котором
происходит образование цис- и транс-бутенов-2 из стереоизомерных
З-дентеробутил-2-п-толуолсульфонатов. Оценено по содержанию
дейтерия в олефинах (по данным Скелла)
Механизм и условия Субстрат син-Элиминирование (%) при образовании
транс- бутена-2 иис-бутснэ-2
Е2, С2Н5О- в С2Н5ОН эритпро 0 0
80%-пый води. С2Н5ОН эритпро 34 16
I Уксусная кислота эритпро 82 65
Нитробензол эритпро 98 95
v Нитробензол тпрео 89 99
тельная стереохимическая неспецифичность этих реакций исчезает при
использовании в качестве растворителя нитробензола. Имеется косвенное
576
Отщепление с образованием олефинов, ацетиленов и карбенов
указание, что в сухих растворителях эти «сольволитические» реакции отно-
сятся к типу Е1, однако, кроме того, это следует из самих приведенных
данных, особенно если сравнить их с El-реакциями в газовой фазе (разд. 5),
в которых «растворитель», т. е. пустое пространство, имеет предельно низкие
основность и диэлектрическую проницаемость, и поэтому ионные пары очень
стабильны и никакая частица, кроме противоиона, не может отщеплять
протона.
в. Стереохимия и стадийность изменения связей при Е2-элиминировании.
Вернемся к реакциям элиминирования в диссоциирующих растворителях,
например в растворителях, содержащих воду, и в частности к бимолекуляр-
ному элиминированию.
Стадийность изменения связей при Е2-элиминировании частично рассматри-
валась в предыдущем разделе, поскольку без этого невозможно обсуждать
стереохимию Е2-процессов. Поэтому в этом разделе будут сделаны лишь
некоторые дополнительные замечания.
Хотя отщепление (5-протона и перенос а-электрона при Е2-элиминировании
и происходят согласованно, эти стадии не точно синхронны: их несинхрон-
ность определяется зарядом и степенью ненасыщенности переходного состоя-
ния. Эта мысль была высказана Ханхартом и автором [2] в 1927 г., однако
свое развитие она получила лишь недавно и наиболее четко — в обзоре Бан-
нета [62], опубликованном в 1962 г. Она имеет стереохимическую подоплеку,
поскольку стереоспецифичность Е2-реакций зависит от степени «сцепленно-
сти» изменений связей: при наибольшей степени «сцепленности» этих стадий
обе связи рвутся приблизительно синхронно, а при ослаблении степени
«сцепленности» разрыв одной связи сильно опережает разрыв другой. Эти два
крайних случая асинхронности, т. е. «несцепленности», реализуются при
механизме ElcB, при котором перенос протона происходит на отдельной
предварительной стадии, и при механизме Е1, при котором аналогичным
образом происходит полный электронный перенос. Можно представить, что
между этими предельными случаями находится область различных механиз-
мов Е2, в которой изменение связей происходит хотя и согласованно, но
может иметь место «отклонение» от точной синхронизации в любом направ-
лении и поэтому изменяться влияние полярных факторов на скорость.
В предыдущем разделе были приведены данные, показывающие, что если
изменение строения достаточно ацидифицирует (5-протон, то в реакциях цик-
логексана и других соединений может происходить цис-элиминирование,
хотя эти реакции и сохраняют Е2-характер. В этих случаях можно уверенно
считать, что Е2-механизм «отклоняется» в сторону механизма ElcB с пре-
обладанием отщепления протона в степени, которая позволяет происходить
цис-элиминированию, если ацидифицированный протон находится в цис-
положении.
Отклонение в другую сторону, вероятно, было найдено Хьюзом и Уилби [60]
(реакции 3 и 3' в табл. 155) при Е2-элиминировании от неоментилтриметил-
аммониевого иона. Трудность состоит в том, что эти реакции в чистой форме
не наблюдаются даже в очень сильных водных растворах щелочей, так как на
этот процесс накладывается исключительно легко протекающее Е1-отщепле-
ние. Эта El-реакция, подобно всем другим таким реакциям (табл. 155),
следует правилу Зайцева. Однако сопутствующая реакция типа Е2, которая,
будучи реакцией аммониевого иона, должна была бы следовать правилу
Гофмана, также подчиняется правилу Зайцева, хотя и менее строго. Хьюз
и Уилби приняли, что легкость El-реакции обусловлена легкостью отщепле-
ния группы Х(СН3)з (вероятно, вследствие того, что она аксиальна, т. е.
испытывает конформационное отталкивание) и что это свойство передается
577
37-0772
Глава IX
Е2-реакции, т. е. отщепление триметиламмониевой группы может опережать
перенос протона. Это приводит к тому, что переходное состояние в определен-
ной степени начинает иметь характер иона карбония, и если этот фактор пре-
обладает, то ориентация соответствует правилу Зайцева.
5. Мономолекулярное 1,2-отщепление в газовой фазе [63]
а. Влияние строения на кинетику. Очень интересно было бы изучить
элиминирование в газовой фазе, т. е. в отсутствие растворителя. Необходимо
также совсем исключить катализ поверхностью, однако обычно это можно
сделать, лишь нанося на внутреннюю поверхность сосуда подходящие покры-
тия. Наблюдающаяся при этом гомогенная реакция может включать ради-
кальные процессы, которые, если они не играют большой роли, могут быть
подавлены добавками ингибиторов, служащих ловушкой для радикалов.
Остающаяся реакция в этом случае является реакцией молекул в газовой
фазе.
Молекулярное элиминирование в газовой фазе, главным образом элиминиро-
вание от алкилхлоридов, было изучено Бартоном и сотр. [64] в 1949 г.,
а реакции алкилбромидов и других алкилгалогенидов, алкилацетатов и дру-
гих сложных эфиров — Макколлом и сотр. во многих работах [65] начиная
с 1953 г. Эти реакции протекают с удобными для измерения скоростями при
различных температурах, в основном в интервале между 250 и 500 °C. Все эти
процессы мономолекулярны, их кинетика соответствует первому порядку
и во всех случаях, где это специально проверялось, наблюдалось линдема-
новское уменьшение скорости при низких давлениях *. Аррениусовский
частотный фактор во всех реакциях близок к теоретическому (для мономоле-
кулярных реакций 1013 с-1). Таким образом, различие в скоростях между
двумя реакциями почти полностью соответствует различию в энергиях акти-
вации.
Первые данные Макколла и сотр. [65] по мономолекулярному дегидрогало-
генированию алкилбромидов показали, что энергия этих реакций почти
целиком зависит от энергии разрыва связи СВг и мало зависит от энергии
разрыва связи СН. Если в молекулу этилбромида последовательно вводить
метильные группы, так, чтобы СВг- и СН-центры становились независимо
первичными, вторичными и третичными, то относительные скорости будут
изменяться, как показано в верхней части табл. 157. Очевидно, скорость
определяется главным образом заместителями у связи СВг. Эффект замеще-
ния у СН-центра настолько велик, что трудно определить, действительно ли
он обусловлен влиянием на связь СН или передается через цепь на связь СВг.
Это же явление иллюстрируется значениями энергий активации, приведен-
ными в нижней части табл. 157.
Наблюдения Макколла и Томаса [65] показывают, что эти и другие данные по
энергиям активации газофазного мономолекулярного галогенирования не
соответствуют энергиям гомолитической диссоциации связей углерод — гало-
ген, но соответствуют энергиям гетеролитической диссоциации этих связей.
Это иллюстрируется данными табл. 158, причем энергии активации дегидро-
бромирования взяты из табл. 157, а соответствующие данные для дегидро-
хлорирования — из более ранней работы Бартона [64].
* Это явление хорошо известно и состоит в резком уменьшении скорости, сопровож-
дающемся изменением кинетического порядка до второго, которое происходит при умень-
шении давления до той величины, когда частота столкновений становится ниже предела,
необходимого для поддержания распределения Максвелла, и часть молекул субстрата
с высокой энергией не участвует в реакции.
578
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблица 157
Мономолекулярное дегидробромированпе в газовой фазе алкилбромидов
R R
ХС— С , где R = I-I или СН3
/ ! 1\
R Н Br R
СН СВг
первичный вторичный третичный
Относительные скорости при 380 СС
Первичный 1 170 32 000
Вторичный 3,5 380 46 000
Третичный 6,3 130 000
Аррениусовская энергия активации, ккал/моль (4,187-Ю-3 Дж/моль)
Первичный 53,9 47,8 42,2
Вторичный 50,7 46,5 40,5
Третичный 50,4 39,0
Таблица 158
Сравнение энергий активации газофазного мономолекулярного
дегидрогалогенирования с энергиями гомолитической и гетеро литической
диссоциаций связей углерод —галоген [ккал/моль (4,187-103 Дж/моль)]
Субстрат Аррениусовская Еа D (R-+Br ) о (R+ + Вг-)
С2Н5Вг 53,9 67,2 183,7
uao-CgHyBr 47,8 67,6 153,6
mpem-CjHgBr 42,2 63,8 140,3
С2Н5С1 60,2 80,9 192,8
U30-C3H7C1 50,5 82,2 166,3
пгрет-СдНдС! 41,5 78,3 150,2
Макколл и Томас затем показали наличие близкой аналогии во влиянии
строения алкилгалогенида на скорости газофазного мономолекулярного отще-
пления и на скорости гетеролитических мономолекулярных реакций заме-
щения и элиминирования SN1 и El в водных и других сильнополярных
растворителях. Последующее расширение этих исследований, проведенное
указанными авторами независимо друг от друга, показало, что подобная кор-
реляция всегда выдерживается. Можно привести два примера, основанных
на данных Томаса.
Один из возможных путей для выяснения степени разрыва СН-связи в пере-
ходном состоянии газофазного мономолекулярного элиминирования состоит
в изучении реакций субстратов, в которых эта связь сильно ослаблена, напри-
мер когда она является аллильной или бензильной. Это соответствует введе-
нию в молекулу алкилгалогенида |3-винильной или |3-фенильной группы.
Томас [66] ввел в молекулу алкилбромида |3-винильный заместитель. Эффект
ненасыщенности винильной группы был выявлен путем сравнения с соот-
ветствующим ^-метильным соединением. Как можно видеть из данных
37* 579
Глава IX
табл. 159, никакого эффекта не наблюдалось. Ясно, что в переходном состоя-
нии этой мономолекулярной реакции СН-связь не участвует. Очевидно также.
Таблица 159
Влияние р-винильной группы на скорость газофазного мономолекулярного
дегидробромирования
Субстрат Аррениусов- ская А, с~1 Аррениусов- ская ккал/моль (4,187х Х103 Дж/моль) Относитель- ная скорость при 380 °C
р СП3 — СН а -СН -СНз Ю13.53 46,5 1,00
1 н сн2 = сн 1 Вг — СН -СН-СНз 1012.М 44,7 1,05
1 н 1 Вг
что в переходном состоянии не происходит частичного образования двойной
связи между атомами углерода, т. е. переходное состояние «не знает», что оно
находится па пути превращения в сопряженный диен. Подобные же резуль-
таты были получены Макколлом и Стивенсоном [67] с [5-фенильным замести-
телем.
Одним из наиболее сильных эффектов, ускоряющим SN1- или El-реакции
алкилгалогенидов в водных или других ионизующих растворителях, обла-
дает ос-метоксигруппа. Увеличение скорости так велико, что его можно толь-
ко косвенно оценить. Принятое объяснение состоит в том, что и-метоксигруп-
па хорошо приспособлена к сопряжению с карбониевым ионом, образующим-
ся на первой стадии гетеролиза.
СНз—б^СН2-^С1----->СН3—6=-СН2---->
Медленно
(SnD
Н2()
---->СН3—О—СН2ОН
Быстро )
Второй пример Томаса позволяет установить, действительно ли газофазное
мономолекулярное элиминирование является гетеролитической реакцией
типа Е1, т. е. действительно ли первоначально образуется ион карбопия,
так как в этом случае должно проявляться аналогичное резкое увеличение
скорости.
СНз-0-CH-Cl-------> сн3-6=сн —>
сн2—н снДн
(Е1)
---->СНз-О-СН
Быстро ||
сн2
Томас [66] измерил скорость газофазной реакции а-метоксиэтилхлорида
и установил, что она имеет измеримую величину при температурах на 200 °C
,580
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблица 160
Влияние а-метоксигруппы на скорость газофазного мономолекулярного
дегидрохлорирования; сравнение с сольволизом
Субстрат А, с-1 ккал/моль (4.187Х ХЮЗ Дж/моль) Относительная скорость
при 330 °C при 0 °C сольволиза типа Sjsjl при 0 °C (рассч.)
сн2-сн-н 10Ы,в 60,8 1 — 1 1
1 ! Н С1
СП2-СН-СН3 Ю13,40 50,5 -- 1 — 1 - 1
1 i Н С1
сн2--сн—осн3 1 Н С1 Ю11»46 33,3 106,8 104,3 1018,9 1011,9 - 1017 1011. . .1012
ниже, чем температуры, которые необходимо применять в случае хлористых
алкилов, содержащих вместо СН3О группы Н или СН3, т. е. этил- и изопро-
пилхлоридов. Сравнение проводилось с последним соединением, поскольку
метильная группа может участвовать в гиперконъюгации с обедненным элек-
тронами а-углеродным атомом. Сравнение скоростей и параметров Аррени-
уса для этих хлоридов дано в табл. 160. Относительные скорости вначале
рассчитывались для 330 °C — температуры, лежащей между интервалами
температур, в которых проводилось изучение наименее и наиболее реакцион-
носпособных хлоридов. Эта температура была выбрана по той причине, что-
бы минимизировать дальность экстраполяции. Затем были рассчитаны отно-
сительные скорости при 0 °C, чтобы можно было сравнить их с относитель-
ными скоростями сольволиза при 0 °C, приведенными в нижней части
таблицы. Эти расчеты менее надежны, поскольку экстраполяция проводилась
в большом интервале температур. Значения скоростей сольволиза получены
косвенным путем и поэтому довольно неточны. Тем не менее, несмотря на
неточность данных при 0 °C, их сравнение не оставляет сомнения в том, что
эти процессы, идущие в таких разных условиях, по существу одинаковы, так
как они резко ускоряются при наличии метоксигруппы.
Далее другие примеры не будут так детально рассматриваться, однако следует
отметить, что Макколл и сотрудники провели тесты такого рода почти для
всех простых заместителей в алкилгалогенидах, для которых был известен
эффект на скорости SN1- и El-реакций. Например, хотя, как было показано,
Р-винильная или р-фенильная группа не оказывает заметного влияния на
скорость газофазной реакции, «-фенильная группа и при мономолекуляр-
ном элиминировании в газовой фазе, и при сольволитических SN1- или El-
реакциях оказывает сильное ускоряющее действие — более сильное, чем одна
метильная группа, и, вероятно, такое же сильное, как две а-метильные груп-
пы. Скорости реакций и в газовой фазе, и в растворителе возрастают в сравни-
мой степени, если учесть различие в температурах, при которых проводились
эти две реакции. Кроме того, в газофазной реакции атом хлора в р-положении
оказывает слабый замедляющий эффект, а в «-положении — слабый ускоря-
ющий эффект, гораздо более слабый, чем эффект «-метильной группы; все
эти эффекты были сравнимы по величине с эффектами, хорошо известными
в мономолекулярных сольволитических реакциях [68].
Глава IX
Газофазное дегидрогалогенирование и другие подобные реакции рассматри-
вались также с точки зрения предполагаемого «четырехцентрового переход-
ного состояния»
RzC.—_СЙ2
Й-—
Подразумевалось, что все четыре связи изменяются одновременно и что
нигде не возникает сильной локальной полярности. Открытие Макколлом
и Томасом того факта, что газофазные реакции дегидрогалогенирования —
двух стадийные процессы типа Е1, при которых первой и определяющей ско-
рость стадией является ионизация,
r2c-cr2 —> (r2c—cr/i —-> r2c=cr2 + нх
I К Медленно) | + Быстро
Н
Н
(Е1)
было неожиданным и поставило под сомнение традиционную точку зрения,
согласно которой все газофазные процессы — всегда гомолитические, энер-
гия гетеролиза в газовой фазе не может быть обеспечена и ионы в газовой
фазе не могут появляться ниже температуры пламени. Единственно, о чем
забывали при традиционном подходе,— это то. что, хотя энергия гетеролиза,
равная 140...190 ккал/моль (586,15-103...795,49-103 Дж/моль) (табл. 158).
действительно недостижима при температурах реакций, большая часть этой
энергии необходима для диссоциации образовавшихся ионов, а для Е1-элими-
нированпя диссоциации вовсе пе требуется [65].
Необходимо рассмотреть вопрос о том, является ли анион в только что обра-
зовавшейся ионной паре связанным электростатическими силами с ионным
карбониевым центром и атомом водорода, в конце концов отщепляющимся
в виде протона, или этот анион притягивается также и всеми другими атома-
ми водорода, которые приобретают положительные заряды, отдавая элек-
троны путем гиперконъюгации с карбониевым ионным центром. Макколл
придерживается последней точки зрения и считает, что такова принципиаль-
ная основа этих реакций, однако нужно согласиться, что вследствие различия
в пространственном расположении разные атомы водорода будут обладать
различным действием. Макколл совместно с Хардингом и Россом [69] пока-
зали. что при гомогенном газофазном дегидрохлорировании оптически актив-
ного 2-октилхлорида при 327...358 °C скорость потери оптической активности,
которая имеет первый порядок, равна, но не больше, скорости дегидрохлориро-
вания. также имеющего первый порядок. Это означает, что хлорид-ион
в ионной паре не может мигрировать на другую сторону карбониевого иона,
а это в определенной мере согласуется с предположением, что хлорид-ион
удерживается протонным полем атомов водорода соседних групп. Эта точка
зрения находится в соответствии также с некоторыми данными, приведен-
ными в разд. 5,в.
Помимо дегидрогалогенирования, составляющего наиболее полно исследо-
ванный класс газофазных реакций мономолекулярного элиминирования,
были изучены также и некоторые другие реакции подобного типа, в частно-
сти дегидрокарбоксилирование ацетатов и других сложных эфиров. Из
опубликованных данных можно составить таблицу (табл. 161) по тому же
принципу, что и табл. 157 для бромидов. Из данных табл. 161 следует тот же
вывод: скорость определяется почти полностью ближайшим окружением
атома углерода, связанного с ацетатной группой, а не окружением углерода,
582
Отщепление с образованием олефинов, ацетиленов и карбенов
Таблица 161
Мономолекулярное газофазное дегидрокарбоксилирование алкилацетатов
R R
С — С , где R = H или СП3. Приведены относительные скорости
R I I
и ососщ
при — 400 °C [70] а
С-ОСОСНз
СН
первичный
вторичный
третичный
Первичный
Вторичный
Третичный
1
0,91
0,46
24 (25)
38 (24)
2000 (1660)
5000 (2490)
6800
Значения, выделенные курсивом, взяты из работы [71].
связанного с отщепляющимся водородом. Это согласуется с двухстадийной
реакцией, первая стадия которой, определяющая энергию активации, соот-
ветствует отщеплению ацетата. Для обоснования предположения о гетероли-
тической диссоциации и для того, чтобы удостовериться, участвует ли второй
атом кислорода сложноэфирпой группы в процессе образования переходного
состояния, конечно, необходимы другие исследования. Небольшое различие
в числовых данных, приведенных в табл. 161 и 157, указывает, что переходное
состояние при элиминировании ацетата менее полярно, чем при элиминиро-
вании бромид-иона. Однако независимо от того, одинаков ли механизм в этих
двух случаях или пет, такое различие может быть вызвано меньшей электро-
отрицательностью ацетатной группы по сравнению с атомом брома.
б. Ориентация и стереохимия. Склонность газофазных реакций моно-
молекулярного отщепления к цис-элиминированию впервые была отмечена
Бартоном [72] при изучении производных циклогексана. Он показал, что от
ментилхлорида атом хлора отщепляется на 73% вместе с 4-водородным ато-
мом. который находится в цис-положении, т. е. является синклинальным по
отношению к атому хлора. Макколл и Бэмкоул [73] подтвердили эти данные
и, кроме того, установили, что от неоментилхлорида, в котором 4-водородный
атом находится в mpauc-положении, т. е. конформационно антиперипланарен
по отношению к атому хлора, на 85% отщепляется один из вторичных атомов
водорода в положении 2. Ниже приведены стабильные конформации ментил-
и неоментилхлоридов, в которых все боковые группы или, если это невоз-
можно, наибольшие из них являются экваториальными.
Ментилхлорид
, Неоментил хлорид
(27% ментена-2 и 73% ментена-3) (85%ментена-2 и 15% ментена-3)
583
Глава IX
Можно думать, что такие реакции гомогенного дегидрохлорирования,будучи
процессами типа Е1, протекают согласно правилу Зайцева, если вследствие
стереохимических причин не начинает преобладать синклинальное элимини-
рование. Стереохимическую предпочтительность такого элиминирования
можно рассматривать как результат применимости к реакциям в газовой
фазе принципа Скелла (предыдущий раздел), согласно которому при El-
элиминировании в растворе в случае основных и способствующих диссоци-
ации на ионы растворителей стереоспецифичность обычно мала, по при пере-
ходе к менее основным и менее диссоциирующим растворителям стереоспеци-
фичность возрастает таким образом, что более предпочтительным становится
синклинальное элиминирование. В неосновных и недиссоциирующих раство-
рителях, как и в газовой фазе, образующийся недиссоциирующий анион под
действием сил притяжения остается у наиболее доступного протона, которым
в циклогексановых системах всегда является синклинальный протон *.
в. Отщепление с перегруппировкой. Одно из наиболее четких и быстрых
доказательств промежуточного образования карбониевого иона при моно-
молекулярном процессе состоит в обнаружении сопутствующей этому про-
цессу перегруппировки Вагнера. Перегруппировка Вагнера связана с перво-
начальным образованием электронодефицитного центра, который, за исклю-
чением очень небольшого числа случаев, когда этот центр может иметь ради-
кальный характер, является ионным карбониевым центром. Перегруппиров-
ка состоит в смещении группы (обычно атома водорода или алкила) вместе
с ее электронами связи к первоначальному карбониевому ионному центру,
при этом положительный заряд переходит в более стабильное положение.
Подобное смещение происходит до того, как разрушается карбониевый
ион, т. е. до того, как заканчивается процесс замещения или отщепле-
ния.
В 1961 г. Макколл и Свинборн [74] впервые обнаружили перегруппировку
Вагнера в газовой фазе, которая происходила при дегидрохлорировании нео-
пентилхлорида. Этот хлорид не имеет ни одного p-водородного атома, однако
скорость его разложения при 444 °C составляет около одной четверти скоро-
сти разложения хлористого этила. На 25% разложение происходит путем
фрагментации, в результате которой происходит отщепление хлористого
метила и метана с образованием изобутена и изомерных хлоризобутенов.
Однако на 75% разложение происходит путем дегидрохлорирования с ваг-
неровским смещением метильной группы, в результате которого образуется
более стабильный третичный ион карбонил, который теряет один из своих
протонов и превращается в смесь изоамиленов. По приведенному ниже соста-
ву смеси изоамиленов ничего нельзя сказать об относительных скоростях
отщепления разных протонов, так как состав смеси является равновесным;
хотя образующийся наряду с изоамиленами хлористый водород при темпе-
ратурах реакции не присоединяется к амиленам в заметной степени, при
этих температурах он, однако, катализирует их быстрое взаимопревраще-
ние.
Тем не менее можно сказать, что относительная стабильность образующихся
олефинов такова, что ее можно связать с эффектом гиперконъюгации, поэто-
му можно предположить, что стабильность переходных состояний образова-
ния любых из них из одного и того же карбониевого иона под действием одно-
го и того же акцептора протона будет соответствовать относительной стабиль-
* Бартон, Хед и Уильямс [72] отметили, что направление элиминирования может изме-
ниться под влиянием катализа поверхностью. Поэтому нет необходимости обсуждать
ориентацию и стереохимию отщепления, не зная, является ли оно гомогенным.
584
Отщепление с образованием олефинов, ацетиленов и карбенов
пости этих олефинов
снзх Медленно СН3
СН3/С —СН2-С1--------> сн3
СН3/ сн3
/с-сн2 СНз\с-сн2.-сн3
/ СНз' I
Быстро I
СНЗХ сн3х сн3.
>с-сн,-сн3 >с=сн-сн, >сн-сп-сн2
сн/ СП/ СН3/
30% 65% 5%
Хорошо известная в растворах реакция отщепления НС1 от борнил- и изо-
борнилхлоридов, протекающая без перегруппировки или с перегруппиров-
кой, была проведена Макколлом и Бикнеллом [74] в газовой фазе и оказа-
лась мономолекулярной. Авторы предложили схему, в которой состав про-
дуктов реакции контролируется стереохимическими факторами. В частности,
образовавшийся, но не диссоциированный хлорид-ион занимает положение
между соседним с ним атомом водорода и атомами водорода, следующими за
соседними и расположенными по ту же сторону кольца, что и хлорид-ион.
Эта кольцевая система имеет три стороны, однако будут рассмотрены только
две из них, называемые эндо и экзо. э«Эо-2-Хлорид-ион, образовавшийся из
борнилхлорида, имеет три возможности для взаимодействия: с эндо-3-водо-
родом, с энЭо-6-водородом и с водородом метильной группы в голове моста,
имеющим э«Эо-конформацию. В последнем случае присходит вагнеровское
1,2-смещение 6-метиленовой группы (которое превращает вторичный 2-кар-
бониевый ион в более устойчивый третичный 1-карбониевый ион), эндо-
Хлорид-ион реагирует по всем трем этим возможным направлениям, образуя
борнилен, трициклен и камфен в соотношениях, указанных ниже, экзо-2-
Хлорид-ион, образовавшийся из изоборнилхлорида, может реагировать толь-
ко по двум направлениям, а именно: атаковать или экзо-3-водород, или
водородный атом метильной группы в голове моста, находящийся в экзо-
конформации. В последнем случае происходит та же самая перегруппиров-
ка Вагнера. экзо-Хлорид-ион реагирует по обоим из этих возможных путей,
образуя борнилен и камфен в соотношении, указанном на приведенной ниже
схеме:
Камфен
585
Глава IX
Перегруппировка Вагнера в карбониевом поне (С1 отщепляется только с атомами II,
расположенными по ту же сторону молекулы, имеющей три стороны)
г. Гомогенный кислотный катализ. В случае газофазного мономолеку-
лярного элиминирования от алкилгалогенидов или сложных эфиров не
наблюдается никакого катализа. Однако в случае спиртов и простых эфиров
сильными катализаторами являются галогеноводороды. Первое сообщение
обэтом сделали Макколл и Стимсон в 1960 г., затем Стимсон изучил это явле-
ние более подробно [75].
Исследованные реакции происходили при разных температурах в интервале
от 240 до 520 °C. Они имели кинетический второй порядок (скорость ос [ROH
или ROMe] [НХ]); однако галогеноводород в реакции не расходовался, хотя
его концентрация и входила в кинетическое уравнение. По своей ката-
литической силе галогеноводороды располагались в ряд HI > НВт > НС1,
причем обычно соотношение скоростей в этом ряду, скажем, при 380 °C было
равно 150 : 25 : 1. Отсюда следует, что каталитическое поведение связано
с кислотностью катализатора. Кинетический эффект структурных изменений
в алкильной группе при катализируемых реакциях был качественно сходен
с эффектом, наблюдавшимся при дегидрогалогенировании алкилгалогенидов,
и почти одинаков с эффектом, наблюдавшимся при дегидрокарбоксилирова-
нии сложных алкиловых эфиров. Последовательное введение метильных
групп в положение, с которым связана отщепляющаяся гидроксильная
группа, приводило к существенному возрастанию скорости, например,
в соотношении 1 : 25 : 1600 в серии этиловый, изопропиловый и трет-бути-
ловый спирты при 440 °C. Однако последовательное введение метильных
групп в положение, с которым связан отщепляющийся атом водорода, изме-
няло скорость не более че.м в 2 раза. Естественно думать, что активация свя-
зана с разрывом связи углерод — кислород и, кроме того, что при катализе
газообразными кислотами связь углерод — кислород разрывается аналогич-
но связям С — Hal пли С — ОСОСП3, т. е. гетеролитически. Механизм
реакции .можно изобразить следующей схемой:
СНз СНз
СНз-С^Ън а СНз-С+
у . Медленно; /
СН,' : СН/ —
[ I
Br—Н Н Вг-
(СН3)2С=СН2
+
НВг
Общая концепция газофазных кислотно-катализируемых реакций отщепле-
ния была подтверждена Кроссом и Стимсоном на следующем примере. Изве-
стно, что изобутилен в присутствии кислот типа серной или борфтористо-
водородной — достаточно сильных, чтобы превратить его в ион mpem-бути-
лия, способен захватывать окись углерода с образованием иона пивалилия, из
которого можно препаративно получить пивалиновую кислоту путем после-
довательных обратимых реакций
н+ со н2о
(сн3)2с-сн2 —z± (сн3)3с+ ~> (сп3)3ссо+ ,±zzr.
(CH3)3C —СО —OI1 + Т» (СН3)3ССОО11ЖЦ+
586
Отщепление с образованием олефинов, ацетиленов и карбенов
В гомогенных газофазных условиях происходит обратная реакция. Авторы
показали, что в присутствии бромистого водорода при 340...460 °C пивалино-
вая кислота распадается на изобутилен, окись углерода и воду. В этих усло-
виях происходит гомогенная газофазная реакция, подчиняющаяся следую-
щему кинетическому уравнению:
Скорость оС[(СН3)3ССООН][НВг]
однако бромистый водород при этом не расходуется. Ниже приведен механизм
реакции, который соответствует механизму образования олефинов из спиртов:
СН3 \ Медленно СН3 \ СН3\
СН3С —СО —ОН------------> СН3-^С —СО+ -> сн3-фс+
СИ;/ : СН3/ сн2/
: Вг- I Вг-
Вг-Н H
-> (СН3)2С = СН2
НВг
В приведенной интерпретации элиминирования в условиях газофазного
катализа медленную стадию можно разделить на две, если рассматривать
в качестве второго промежуточного продукта оксониевую ионную пару.
Однако такое детализирование, вероятно, не так важно для формулирова-
ния принципиальных сторон механизма рассмотренных здесь реакций.
6. Фрагментации Гроба [76]
а. Типы фрагментаций. Эти реакции в основном представляют собой
процессы отщепления, в которых роль p-водородного атома, участвующего
в обычных реакциях элиминирования, играет группа атомов, потенциально
стабильная в виде катиона и в таком виде способная отщепляться от осталь-
ной части молекулы. В 1955 г. Гроб впервые отметил, что реакции этого типа
широко распространены в органической химии. Ниже приведены некоторые
примеры.
Указанная аналогия особенно заметна в случае алкилгалогенидов, содержа-
щих р-трет-бутильную группу. В этом случае вместо (или параллельно)
обычного элиминирования галогеноводорода наблюдается отщепление трет-
бутилгалогенида [77]
(сн3)3с-4ш(сн3р- с(сн3)2 Cci CH3CH=C(CHS)2 + (СН3)3 С-С1
В данном случае можно считать, что развитию катионного характера потен-
циально катионной p-группы способствует гиперконъюгация и поэтому эта
группа отщепляется. Однако более существенное содействие может оказы-
вать чистое сопряжение, например, в у-оксиалкилгалогенидах. В приводи-
мом примере происходит отщепление остатка гликолевой кислоты, связан-
ного с циклогексановым кольцом, вместе с галогеном из другой боковой
цепи [78]
СО,Н СО2Н СНСО2Н
+ О=СНСО2Н + НВг
587
Глава IX
Если расщепляющаяся связь углерод — углерод является частью кольца, то
углеродные атомы, конечно, не будут отщепляться от молекулы; это можно
проиллюстрировать следующим примером, взятым из химии стероидов [79]:
С1бНзо
н—О
mpem-C4HgO
> С16НЗО
3-0ксикопростанил -Зп-толуол-
сульфонат
Аналогично этому не будет происходить и отщепления потенциального
аниона, если последний образует двойную связь, например карбонильную
группу. По такому пути происходят многие процессы декарбоксилирования,
например декарбоксилирование малоновой кислоты [80]
О
ОН
н^о^со-снг-с'
сн2=с
+ о=со
он
он
СНз—СОгН
Таким же образом идут и другие типы реакций, включая обратный распад
альдоля, например изобутиральдоля до изомасляного альдегида *
Н—ЦЩ-СН [СН (СН3)2] —AJ (СН3)Р-СН=й ->
-> (СН3)2С =СН—ОН + О=СНСН (СН3)2
I
(СН3)2СН — сн=о
и реакция, обратная реакции Михаэля, например распад этилового эфира
|3-фенил-а-карбоксиглутаровой кислоты до коричной и малоновой кислот **
ОН
сн=с
+ СН=СНС6Н5
H-'CH-CHCsHs) —f СН-С
СО2С2Н5 ОС2Н5 СО2С2Н5 ’ ОС2Н5 СО2С2Н5
СН2(СО2С2Н5)2
СО2С2Н5
у-Аминоалкилгалогениды в рассматриваемом смысле подобны у-оксигало-
генидам. Множество примеров решающего влияния аминогруппы на рас-
щепление связей углерод — углерод можно найти в химии алкалоидов.
В приведенном ниже примере субстратом является аддукт бромистого водо-
рода с хинидином, который при обработке водным нитратом серебра дает
нихидин, т. е. происходит раскрытие кольца и (что не было замечено внача-
* См. работу [81]. Эта реакция была первым примером; затем было показано, что она
имеет общее значение.
** См. работу [82]. Здесь приводится одна из двух реакций, на примере которых была
установлена общность этой схемы для многих реакций, однако в гораздо более ранней
литературе [83] можно найти еще одну реакцию, которая уже забыта.
588
Отщепление с образованием олефинов, ацетиленов и карбенов
ле) отщепление одного атома углерода в виде формальдегида [84]
СН—Вг
/О1Г
СН
СЛ,0 СНз
+ СН=СН
Htf /ОН
4—СН
^-6-Метокси-4-ха.нолил} (Гтотетическии)
Нихидин
Итак, можно сгруппировать в одно семейство множество реакций, о родстве
которых раньше и не думали; Гроб предложил их называть фрагментациями.
Гроб предположил, что все эти реакции можно связать вместе не только по
структурной форме, но также и по общности механизма. Он попытался выяс-
нить этот механизм на примере ряда реакций у-аминогалогенидов и у-амино-
сульфонатов.
б. Механизм фрагментации [76]. Гроб рассматривал два механизма.
Первый аналогичен Е2-элиминированию с тем лишь исключением, что в этих
фрагментациях реагент с основными свойствами «встроен» в молекулу. Процесс
является мономолекулярным, но идет в одну стадию аналогично Е2-реак-
циям, при этом все связи изменяются согласованно. Этот процесс обозначается
символом F1(E2). Можно полагать, что он должен иметь следующие отли-
чительные признаки:
1. Будет образовываться лишь один продукт, так как отсутствует стадия
образования промежуточного продукта, на которой могло бы произойти
разветвление пути реакции.
2. Процесс будет стереохимически более выгодным, когда связь с отщепляю-
щейся группой, фрагментирующая связь, а также «ось» (отмечена пунктиром)
основных электронов будут параллельны друг другу (жирные линии)
(SK2) (Sn2)
В=су + СУ=С„ -I- X
LF1(E2)]
(Se2)
Дело в том, что составляющие нуклеофильного замещения уС?иСк должны
приводить к обращению конфигурации, а составляющая электрофильного
замещения у Ср будет преимущественно сохранять конфигурацию, и поэтому
предпочтительные конформации у связей BCV и СрСа должны быть антипери-
планарными {ар). Конформация у связи СрСт не важна.
3. На скорость реакции будут оказывать влияние структурные изменения
основной группы В и ближайшее окружение Си.
4. При таком механизме не может происходить перегруппировка Вагнера.
Второй механизм Гроба аналогичен Е1-элиминированию и, подобно этому
механизму, состоит из двух стадий: за медленным образованием Си-карбопне-
вого иона следует быстрая фрагментация последнего. Этот механизм можно
обозначить символом F1(E1).
В-С7-Сд-Сл£х—>в-с -с,-с. + X-
Медленно
в-с7-^с?-са-----► вст +
Быстро
[F1(E1)]
C(S—С,
589
Глава IX
Он должен иметь следующие особенности:
1. Могут получаться различные продукты, поскольку промежуточно образует-
ся карбониевый ион. В общем одновременно могут образовываться про-
дукты нуклеофильного замещения, элиминирования и фрагментации.
2. Конформации групп у связей ВС7 и СрСи будут иметь гораздо меньшее
значение.
3. Структурные изменения в основной группе В будут иметь небольшое вли-
яние на скорость суммарной реакции, которая существенно будет зависеть
от ближайшего окружения Си.
4. Может происходить перегруппировка Вагнера.
На разных примерах Гроб наблюдал все наиболее значительные из пере-
численных особенностей этих двух реакций, что позволило ему сделать
отнесение механизмов, обсуждаемое ниже.
в. Механизм, родственный Е2-элиминированию. К этому типу можно
отнести приведенные ниже реакции (1) — (3), поскольку эти фрагментации
идут нацело, стереоспецифичпо и их скорость зависит от основности у-группы.
Зв - Тропанилхлорид
н
3]3-Нортропанилхлорид
Отн. скорость
53 000*
•4-Хлорхинуклидин
а Скорости отнесены к хлорциклогексану в 80 %-пом водном растворе С2Н5ОН при
82 СС. реакция которого приводит только к замещению и элиминированию.
6 Скорость отнесена к 1-хлорбицикло-[2,2,2]-октану в 80%-ном водном растворе
CoHjOH при 40 =С, который вступает только в реакцию замещения.
Реакции Зр-тропанил- и Зр-нортропанилпроизводных стереоселективны.
Геометрический изомер субстрата (1), т. е. За-тропанилхлорид. в котором С1
находится в г;г/с-положении по отношению к СН2СН2-мостику. а СрСа-связь
в цепи С,,СрСаХ конформационно синклинальна, вступает только в реакции
нуклеофильного замещения и элиминирования, которые не сопровождаются
фрагментацией.
г. Механизм, родственный El-отщеплению. При гидролизе водным рас-
твором едкого натра субстраты (4)—(7), приведенные ниже, образуют про-
дукты замещения, элиминирования и фрагментации. Очевидно, что состав
смеси продуктов реакции зависит от основности у-заместителя. Влияние
оказывают также заместители, связанные с Са; в этих системах вклад реакции
590
Отщепление с образованием олефинов, ацетиленов и карбенов
фрагментации снижается до нуля при переходе от вторичного Са к первичному
Н,
(4) J>N —СН2— СН2— С(СН3)2— С1—> 20% фрагментации
СН3\
(5) >N-CH2CH2C(CH3)2 —С1 —> 45% фрагментации
сн3/
/С(СН3)2 —Вг
(6) СНз — N / \ > 60% фрагментации
(7)
—»82% фрагментации
Однако основность у-заместителя и даже замена его на не обладающий основ-
ностью изоэлектронный углеводородный остаток почти не оказывает влияния
на скорость реакции. Это иллюстрируется примерами (8) и (9):
Отн. скорость
(8)H2N — СН2СН2С(СН3)2 — С1—> 20% фрагментации 0,99а
(9)(CH3)2NCH2CH2C(CH3)2— Cl —> 50% фрагментации 0,75а
а Относительно (СН3)2СНСН2С(СН3)2С1, скорость реакции которого в 80 %-ном водном
С2Н5ОН при 56 °C принята за 1,00. В случае этого соединения происходит только
замещение и отщепление.
При сольволизе хинуклидин-4-карбинил-п-толуолсульфоната [пример (10)}
в 80 %-ном водном растворе этилового спирта происходит перегруппировка
Вагнера с расширением одного кольца и фрагментацией другого
(Скорость= 0,22)
В тех же условиях соответствующий гомоциклический субстрат дает только
продукт замещения с расширением цикла
(Скорость^, 00)
Если принять скорость этой реакции за единицу, то скорость фрагментации
хинуклидина составит лишь 0,22 (в 80%-ном водном этиловом спирте при
116 °C) и поэтому, несмотря на высокий выход продукта фрагментации, состав
смеси продуктов реакции не может определяться на контролирующей ско-
рость стадии. Таким образом, реакция хинуклидина имеет механизм F1(E1),
что согласуется с наблюдаемой в этом случае перегруппировкой Вагнера.
591
Глава IX
7. 1,1-Отщепление с образованием карбенов
Идея о том, что щелочной гидролиз хлороформа, ведущий к образованию
окиси углерода и формиат-иона, может протекать через «двуххлористый
углерод», впервые была высказана Гейтхером [85] в 1862 г. В 1897 г. она
была поддержана Нефом [86], распространившим ее на множество подобных
реакций. Однако эта схема была доказана только в 1950 г. работами Хай-
на [87], который также установил детальный механизм таких реакций. Сде-
лав ряд обобщений, Хайн и сотрудники показали, что 1,1-элиминирование
может протекать по двум механизмам.
а. Мономолекулярное элиминирование в сопряженном основании. Изве-
стно, что атом хлора в «-положении всегда увеличивает скорость SN1-
замещения, но обычно уменьшает скорость 8к2-замещения. Так, щелочной
гидролиз хлористого метилена, имеющий второй кинетический порядок,
идет значительно медленнее, чем гидролиз хлористого метила. Однако
скорость гидролиза хлороформа в 103 раз выше, чем скорость гидролиза хло-
ристого метилена, и, кроме того, значительно выше скорости гидролиза
четыреххлористого углерода. Максимальная скорость гидролиза галофор-
мов — общее явление для всех рядов галогенометанов. Например, скорость
реакции всех галоформов, содержащих бром, примерно в 103 раз выше, чем
скорость реакции хлорбромметана или бромистого метилена.
Это явление не наблюдается при замещении второго порядка такими реаген-
тами, как фенилтиолат-ион C6H5S_, которые обладают высокой нуклеофиль-
ностью при замещении у атома углерода, но невысокой основностью, т. е.
низкой нуклеофильностью по отношению к водороду. В этих реакциях заме-
щения галоформы реагируют медленнее дигалометанов. Однако реакции
галоформов сильно катализируются гидроксильными ионами.
Непосредственное доказательство того, что относительно быстрые реакции
галоформов в присутствии сильных оснований обусловлены высокой кислот-
ностью атомов водорода в галоформах, было получено при изучении катали-
зируемого основаниями обмена водорода на дейтерий в водных растворите-
лях. Для хлороформа и вообще для всех галоформов, не содержащих фтора,
скорость водородного обмена была в 10—1000 раз выше скорости гидролиза.
Очевидно, что первой стадией гидролиза является стадия водородного обмена,
т. е. стадия предравновесного образования сопряженного основания гало-
форма (в случае хлороформа — трихлорметилид-иона СС1“).
Судьбу этого карбаниона можно проследить при изучении солевых эффектов
при щелочном гидролизе хлороформа. Добавки хлористого натрия умень-
шали скорость, и контрольные опыты с другими солями, например с пер-
хлоратом патрия. показали, что наблюдаемое уменьшение скорости в присут-
ствии хлорид-ионов является типичным проявлением закона действия масс
при мономолекулярном замещении (гл. VII, разд.6). Аналогичные солевые
эффекты наблюдались и в случае других галоформов.
Все эти факты, установленные Хайном и сотр. [87], согласуются с объясне-
нием, данным Хайном в 1950 г., а именно что в случае хлороформа и других
галоформов, содержащих не более одного атома фтора, в предравновесной
стадии происходит образование сопряженного основания — тригалометилид-
ного иона, затем вступающего в мономолекулярную реакцию, первой и наи-
более медленной стадией которой является отщепление галогенид-иона с обра-
зованием дигалогенметилена, т. е. соединения двухвалентного углерода,
относящегося к большому классу соединений, известных под названием кар-
бенов. Затем карбен быстро захватывается каким-нибудь реагентом: в вод-
ной среде — водой с образованием окиси углерода, а в присутствии гидро-
592
Отщепление с образованием олефинов, ацетиленов и карбенов
ксильных ионов — с образованием формиата. Так происходит 1,1-отщепле-
ние путем мономолекулярной реакции сопряженного субстрату основания;
этот механизм удобно обозначить символом 1,1-Е1сВ.
(О
СНХз + ОН- ~ ---------* CXj+H2O
(-1)
(2) Медленно
СХ3 СХ2+Х- [ (1, 1-ElcB)
(-2)
СХ2-ЦН2О -----------> СО, НСО2 и X-
он- J
Такой механизм 1,1-отщепления применим не только к реакциям галофор-
мов. В следующем примере вместо одного из атомов галогена имеется суль-
фоновая группа, которая отщепляется от первоначально образующегося
сопряженного основания с образованием карбена, а затем продуктов его
реакции с растворителем [88]
C6H5SO2CHF2 + CH3O- C6H5SO2CFj + CM3OH
CeH5SO2CFy * CF2 + C6H5SO2
CF2 + CH3OH -> CH3OCHF2
В примере, приведенном ниже, ион сульфония, не содержащий галогена,
быстро и обратимо превращается при действии водного раствора щелочи
в сопряженное основание, которое медленно отщепляет диметилсульфид,
превращаясь в ароматический карбен, обнаруженный в конце реакции в виде
стильбенового димера [89]
В 1954 г. Миллер указал автору, что имеется ряд реакций, относительно
которых утверждается, что они протекают через образование карбенов
38-0772
593
Глава IX
путем 1,1-отщепления, но для которых не удалось получить ясных доказа-
тельств отсутствия бимолекулярного нуклеофильного замещения аниона
(X-) в первоначально образовавшемся карбанионе (CX YZ-), т. е. не удалось
доказать, что процесс протекает по мономолекулярному механизму с отщеп-
лением этого аниона и образованием карбена. В своих ранних работах Хайн
очень осторожно высказывался по этому поводу (см. выше). Однако такая
осторожность не всеми соблюдается, и поэтому возможно, что роль карбено-
вых реакций слишком переоценивается и недооцениваются бимолекулярные
процессы, которые имеют место на самом деле.
Дёринг и Гофман [90] обнаружили, что карбены очень легко присоединяются
к олефинам с образованием производных циклопропана. Эта реакция позво-
ляет строго установить факт образования карбенов при любом процессе.
Если карбены образуются путем 1,1-отщепления, то таким путем можно дока-
зать механизм 1,1-Е1сВ. Естественно, что лучше всего этот метод срабатывает
в том случае, когда отсутствуют другие соединения, служащие «ловушкой»
для карбенов, т. е. в инертном растворителе, содержащем олефин, или в рас-
творе самого олефина. В качестве иллюстрации можно привести следующую
реакцию, осуществленную в растворе тетраметилэтилена [91]:
CIIBr(CN)2 + (C2H5)3N 7Г CBr(CN)7 + (C2H5)3NH
CBr(CN)2 -> C(CN)2-|-Br-
(СН3)2С^
C(CN)2+(CH3)2C = C(CH3)2 (CH3)2C-C(CN)2
б. Бимолекулярное отщепление. Хайн обнаружил, что введение одного
атома фтора в галоформ (CHFC12, CHFCIBr или CHFBr2) оказывает заметный
кинетический эффект. Щелочной гидролиз галоформов, не содержащих фто-
ра, имеет второй кинетический порядок; константа скорости реакции равна
константе скорости второго порядка кг стадии (1) механизма 1,1-Е1сВ, умно-
женной на kzKk-i + &2) ~ kzlk_i, последняя небольшая величина отражает
степень перехода образовавшегося сопряженного основания в карбен вместо
возврата в исходный галоформ. Введение одного атома фтора уменьшает
константу ki и настолько увеличивает константу к2, что последняя становится
сравнимой с величиной к^, а в некоторых случаях — даже больше ее.
В результате величина к21(к^ &2) так сильно увеличивается, что. несмотря
на уменьшение кь общая скорость возрастает. Другими словами, начинается
смещение контроля общей скорости от стадии (2) к стадии (1); поэтому можно
ожидать, что при введении второго атома фтора это смещение завершится
и общая скорость будет целиком определяться скоростью стадии (1), т. е.
уменьшаться с уменьшением константы к{.
Однако опытные данные показали [92]. что дифторсоединения CHF2C1,
CHF2Br и CHF2I подвергаются щелочному гидролизу с гораздо более высо-
кой скоростью, чем можно ожидать на основании указанной выше причины.
Реакции протекали слишком быстро для того, чтобы их можно было отнести
к бимолекулярному нуклеофильному замещению SN2 и их скорости зависели
от основности реагента, а не от его нуклеофильности по отношению к углеро-
ду. Кроме того, в отличие от щелочного гидролиза других галоформов в дан-
ном случае водородный обмен между субстратом и средой имел пренебрежи-
мо малую скорость. Хайн и сотрудники объяснили эти данные, предположив,
что в данном случае образование сопряженного основания не происходит,
594
Отщепление с образованием олефинов, ацетиленов и карбенов
а реакция идет по одностадийному механизму бимолекулярного 1,1-отщепле-
ния, который можно обозначить символом 1,1-Е2
НО-4 Н — С1'2--Х---->- Н2О + СР2л-Х- )
он- } (1, 1-Е2)
CF2+H2O------> СО, НСО2 и F- J
Теоретически можно предсказать и возможность отщепления карбенов по
механизму однако этот механизм пока не был реализован.
СПИСОК ЛИТЕРАТУРЫ
1. Banthorpe D. V., Reaction Mechanisms in Organic Chemistry, Editor Hughes E. D.,
Vol. 2, «Elimination Reactions», Elsevier Publishing Co., London, 1962; Studies in
Chemical Structure and Reactivity, Editor Ridd J. H., Methuen, London, 1966,
Chap. 3.
2. Hanhart W., Ingold С. K., J. Chem. Soc., 1927, 997; Ingold С. K., Vass С. C. N., J.
Chem. Soc., 1928, 3125; v. Braun J., Teuffert W., Weissbach K., Ann., 472, 121 (1929);
v. Braun J., Buchmann E. R., Ber., 64, 2610 (1931); v. Braun J., Hamann K., Ber.,
65, 1580 (1932); Ingold С. K., Patel C. S., J. Chem. Soc., 1933, 68; Hughes E. D.,
Ingold С. K., J. Chem. Soc., 1933, 523; Hughes E. D., Ingold С. K., Patel C. S.,
J. Chem. Soc., 1933, 526; Hodnett E. M., Flynn J. J., Jr., J. Am. Chem. Soc., 79
2300 (1957).
3. Fenton G. W., Ingold С. K., J. Chem. Soc., 1929, 2342; Hey L., Ingold С. K., ibid.,
1933 531
4. Green L., Sutherland B., J. Chem. Soc., 99, 1174 (1911); Ingold С. K., Jessop J. A.,
Kuriyan К. I., Mandour A. M. M., ibid., 1933, 533; Ingold С. K., Kuriyan К. I., ibid.,
p. 991; Hughes E. D., Ingold С. K., ibid., p. 1571; Gleave J. L., Hughes E. D.,
Ingold С. K., ibid., 1935, 236; Cooper K. A., Dhar M. L., Hughes E. D., Ingold С. K.,
MacNulty B. J., Woolf L. I., ibid., 1948, 2043; Cooper K. A., Hughes E. D., IngoldC. K.,
Maw G. A., MacNulty B. J., J. Chem. Soc., 1948, 2049; Hughes E. D., Ingold С. K.,
Maw G. A., J. Chem. Soc., 1948, 2072; Hughes E. D., Ingold С. K., Woolf L. I.,
J. Chem. Soc., 1948, 2077; Hughes E. D., IngoldC. K., Woolf L. I., J. Chem. Soc.,
1948 , 2084; Hughes E. D., IngoldC. K., Mandour A. M. M., J. Chem. Soc., 1948,
2090.
5. Fenton G. W-, Ingold С. K., J. Chem. Soc., 1928, 3127; Fenton G. W., Ingold С. K.,
J. Chem. Soc., 1929, 2338; Fenton G. W., Ingold С. K., J. Chem. Soc., 1930,
705.
6. Jones W. H., Science, 166, 387 (1953).
7. Olivier S. C. J., Weber A. P., Rec. trav. chim., 53, 1087 (1934); Olivier S. C. J., ibid.,
p. 1093; Hughes E. D., J. Am. Chem. Soc., 57, 708 (1935); Hughes E. D., Ingold С. K..
J. Chem. Soc., 1935, 244; Hughes E. D., Ingold С. K., Shapiro V. G., ibid., 1936, 225;
Hughes E. D., Shapiro U. G., ibid., 1937, 1177, 1192; Hughes E. D., Ingold С. K., Master-
man S., MacNulty B. J., ibid., 1940, 899; Cooper K. A., Dhar M. L., Hughes E. D.,
Ingold С. K., MacNulty B. J., Woolf L. I., ibid., 1948, 2043; Cooper K. A., Hughes E. D.,
Ingold С. K., Maw G. A., MacNulty B. J., J. Chem. Soc., 1948, 2049; Dhar M. L..
Hughes E. D., Ingold С. K., Masterman S., J. Chem. Soc., 1948, 2055; Dhar M. L.,
Hughes E. D., Ingold С. K., J. Chem. Soc., 1948, 2058, 2065.
8. Saunders W. H., Jr., Fahrenholz S. R., Caress E. A., Lowe J. P., Schreiber M., J. Am.
Chem. Soc., 87, 3401 (1965); Saunders W. H., Jr., Schreiber M. R., Chem. Comm., 1966,
145.
9. Htickel W., Тарре W., Legutke G., Ann., 543, 191 (1940).
10. Hauser C. R., Shivers J. C., Shell P. S., J. Am. Chem. Soc., 67, 409 (1945).
11. Shell P. S., Hauser C. R., J. Am. Chem. Soc., 67, 1661 (1945); Hill D. G., Stewart B..
Kantor S. WT., Judge W. A., Hauser C. R., J. Am. Chem. Soc., 76, 5129
(1954).
12. Hine J., Miesbach R., Ramsay О. B., J. Am. Chem. Soc., 83, 1222 (1961).
13. Crowell T. I., Hill A. T., Kemp A. T., Lutz R. E., J. Am. Chem. Soc., 85, 2521 (1963).
14. Bardwell F. G., Arnold R. L., Banowski J. B., J. Org. Chem., 28, 2496 (1963).
15. Hughes E. D., J. Am. Chem. Soc., 57, 708 (1935).
16. Cooper K. A., Hughes E. D., Ingold С. K., MacNulty B. J., J. Chem. Soc., 1940, 2038:
Cooper K. A., Dhar M. L., Hughes E. D., Ingold С. K., MacNulty B. J., Woolf L. I.,
J. Chem. Soc., 1940 , 2043; Cooper K. A., Hughes E. D., Ingold С. K., Maw G. A..
MacNulty B. J., J. Chem. Soc., 1940, 2049; Hughes E. D., Ingold С. K., Woolf L. I.,
38* 595
Глава IX
J. Chem. Soc., 1940, 2084; Hughes E. D., Ingold С. K., Mandour A. M. M., J. Chem.
Soc., 1940, 2090.
17. Hughes E. D., J. Am. Chem. Soc., 57, 708 (1935); Hughes E. D., Ingold С. K., Scott A. D.,
Nature. 138, 120 (1936); J. Chem. Soc., 1937, 1271; Hughes E. D., Ingold С. K., Sha-
piro U. G., ibid., 1937, 1277; Cooper K. A., Hughes E. D., Ingold С. K., ibid., p. 1280;
Hughes E. D.. MacNulty B. J., ibid., p. 1283; Cooper K. A., Hughes E. D., Ingold С. K.,
Maw G. A., MacNulty B. J., ibid., 1940, 2049; Dhar M. L., Hughes E. D., Ingold С. K.,
J. Chem. Soc., 1940, 2058, 2065; Hughes E. D., Ingold С. K., Mandour A. M. M.,
J. Chem. Soc., 1940, 2090.
18. Htickel W., Тарре W., Ann., 537, 113 (1939); Hiickel W., Тарре W., Legutke G., ibid.,
543, 191 (1940).
19. Hughes E. D., Ingold С. K., Shapiro U. G., J. Chem. Soc., 1937, 1277; Cooper K. A.,
Hughes E. D., Ingold С. K., ibid., p. 1280; Hughes E. D., MacNulty В. J., ibid., p. 1283;
Cooper K. A., Hughes E. D., Ingold С. K., MacNulty B. J., J. Chem. Soc., 1940,
2038.
20. Winstein S., Coicevera M., J. Am. Chem. Soc., 85, 1702 (1963).
21. Hofmann A. W., Ann., 78, 253 (1851); 79, 11 (1852).
22. Saytzeff A., Ann., 179, 296 (1875).
23. Hanhart W., Ingold С. K., J. Chem. Soc., 1927, 997.
24. Ingold С. K., Jessop J. A., Kuriyan К. I., Mandour A. M. M., J. Chem. Soc., 1933,
533.
25. Ingold С. K., Vass С. C. N., J. Chem. Soc., 1928, 3125.
26. Smith P. A. S., Frank S., J. Am. Chem. Soc., 74, 509 (1952).
27. Banthorpe D. V., Hughes E. D., Ingold С. К., J. Chem. Soc., 1960, 4050.
28. Hughes E. D., Ingold С. K., Maw G. A., Woolf L. I., J. Chem. Soc., 1948, 2077.
29. Hughes E. D., Ingold С. K., Woolf L. I., J. Chem. Soc., 1948, 2084.
30. Branch G. E. K.. Calvin M., The Theory of Organic Chemistry, Prentice-Hall, Englewood
Cliffs, N.J., 1941, Chap. 6.
31. Brown H. C., Klimisch R. L., J. Am. Chem. Soc., 88, 1425 (1966).
32. Hughes E. D., Ingold С. K., Trans. Faraday Soc., 37, 657 (1941).
33. Hughes E. D., Ingold С. K., MacNulty B. J., J. Chem. Soc., 1940, 899; Dhar M. L.,
Hughes E. D., Ingold С. K., Masterman S., ibid., 1948, 2055; Dhar M. L., Hughes E. D.,
Ingold С. K., ibid., pp. 2058, 2065; Shiner V. J., Jr., Boskin M. J., J. Am. Chem. Soc.,
77, 5515 (1955).
34. Dhar M. L., Hughes E. D., Ingold С. K., J. Chem. Soc., 1948, 2058.
35. Dhar M. L., Hughes E. D., Ingold С. K., J. Chem. Soc., 1948, 2065.
36. Hughes E. D., Ingold С. K., Shiner V. J., Jr., J. Chem. Soc., 1953, 3827.
37. Dhar M. L., Hughes E. D., Ingold С. K., Mandour A. M. M., Maw G. A., Woolf L. I.,
J. Chem. Soc., 1948, 2109.
38. Hughes E. D., Ingold С. K., Woolf I,. I., J. Chem. Soc., 1948, 2084.
39. DePuy С. II., Froemsdorf D. H., J. Am. Chem. Soc., 79, 3710 (1957); Saunders W. H.,
Jr., Williams R. A., ibid., p. 3712.
40. Hughes E. D., Ingold С. K., Maw G. A., J. Chem. Soc., 1948, 2072.
41. Nef J. U., Ann., 309, 126 (1899); 318, 1 (1901).
42. Brussoff S., Z. physik. Chem., 34, 129 (1900).
43. Segaller D., J. Chem. Soc., 103, 1421 (1913).
44. Hughes E. D., Ingold С. K., Masterman S., MacNulty B. J., J. Chem. Soc., 1940,
899.
45. Mereshkowsky В. K., Ann., 431, 231 (1923).
46. Cooper K. A., Dhar M. L., Hughes E. D., Ingold С. K., MacNulty B. J., Woolf L. I.,
J. Chem. Soc., 1948, 2043.
47. Hodnett E. M., Flynn J. J. Jr., J. Am. Chem. Soc., 79, 2300 (1957).
48. Cooper K. A., Hughes E. D., Ingold С. K., Maw G. A., MacNulty B. J., J. Chem. Soc.,
1948, 2049.
49. Hughes E. D., Taher N. A., J. Chem. Soc., 1940, 956; Gripenberg J., Hughes E. D.,
Ingold С. K., Nature, 161, 480 (1948).
50. Michael A., J. prakt. Chem., 52, 308 (1895).
51. Chavanne G., Bull, soc., chim. (Beiges), 26, 287 (1912).
52. Cristol S. J., Begoon A., Norris W. P-, Ranly P. S., J. Am. Chem. Soc., 76, 4558
(1954).
53 .Ingold С. K., Proc. Chem. Soc., 1962, 265; cp. Charman H. B., Hughes E. D., Ingold С. K.,
J. Chem. Soc., 1959, 1523, 1530.
54. Cristol S. J., J. Am. Chem. Soc., 69, 338 (1947); Hughes E. D., Ingold С. K., Paster-
nak R., J. Chem. Soc., 1953, 3832.
55. DePuy С. H., Thum R. D.. Morris G. F., J. Am. Chem. Soc., 84, 1314 (1962):
596
Отщепление с образованием олефинов, ацетиленов и карбенов
DePuy С. Н., Morris G. F., Smith J. S., Stuart R. J., J. Am. Chem. Soc., 87, 242
(1965).
56. Zavada J., Sicker J., Proc. Chem. Soc., 1963, 96; Zavada J., Svoboda M., Sicker J.,
Tetrahedron Letters, 1966, 1627; Zavada J., Krupicka J., Sicher J., Chem. Comm.,
1967, 66; Pankova M., Sicher J., Zavada J., ibid., p. 394.
57. Arnold R. T., Richardson P. N., J. Am. Chem. Soc.. 76, 3649 (1954); Weinstock J.,
Pearson R. G., Bardwell F. G., J. Am. Chem. Soc., 76. 4748 (1954); 77, 6706 (1955);
78, 3468, 3473 (1956); Bordwell F. G., Kern R. J., J. Am. Chem. Soc., 77, 1141
(1955).
58. Shiner V. J., Jr., Smith M. L., J. Am. Chem. Soc., 80, 4093 (1958); Weinstock J., Ber-
nardi J. L., Pearson R. G., ibid., p. 4961.
59. Hughes E. D., Ingold С. K., Rose J. B., J. Chem. Soc., 1953, 3839.
60. Hughes E. D., Wilby J., J. Chem. Soc., 1960, 4094.
61. Shell P. S., Hall W. L., J. Am. Chem. Soc., 65, 2851 (1963).
62. Bunnett J. F., Angew. Chem. internet. Edit., 1, 225 (1962); Ingold С. K., Proc. Chem.
Soc., 1962, 265.
63. Maccoll А., в книге «The Chemistry of Alkenes», Editor S. Patai, Interscience Publishers,
London, 1964, Chap. 3; «Gas-phase Heterolysis» in «Advances in Organic Chemistry»,
Editor V. Gold, Academic Press, London, Vol. 3, 1965, p. 91; Studies in Chemical
Structure and Reactivity, Editor J. IL, Ridd, Methuen, London, 1966,
Chap. 4.
64. Barton D. H. R., Howlett К. H., J. Chem. Soc., 1949, 155, 165; Barton D. II. R.,
Onyon P. F., Trans. Faraday Soc., 45, 725 (1949); Barton D.H.R., Head A. T.,
J. Chem. Soc., 1950, 46, 114; Williams R. J. W., J. Chem. Soc., 1952, 453; 1953,
1715.
65. Green J. II. S., Maccoll A., Thomas P. J., J. Chem. Phys., 21, 178 (1953); Maccoll A.,
Thomas P. J., Nature, 176, 392 (1955); Маккол А., в книге «Теоретическая органиче-
ская химия», ИЛ, М., 1963, стр. 289. Spec. Pubs. Chem. Soc., 16,158 (1962); Ingold С. K.,
Proc. Chem., Soc., 1957, 297.
66. Thomas P. J., J. Chem. Soc.. 1959, 1192; 1961, 136.
67. Stephenson B., Thesis, University of London, 1959; Maccoll А., см. в ссылке [65].
68. Hojjmann II. M. R., Maccoll A., J. Am. Chem. Soc., 87, 3774 (1965).
69. Harding C. J., Maccoll A., Ross R. A., Chem. Comm., 1967, 289.
70. Scheer E. С., Kooyman E. C., Sixma F. L. J., Rec. trav. chim., 82, 1123 (1963).
71. Maccoll A., J. Chem. Soc., 1958, 3398; Emovon E. U., Maccoll A., ibid., 1962,
335
72. Barton D. II. R.. J. Chem. Soc., 1949, 2197; Barton D. II. R., Head A. J., Williams R. J.,
ibid., 1952, 453; 1953, J715.
73. Bamkole T., Thesis, University of London, 1964; Maccoll А., см. в* ссылке [65],
p. 111.
74. Maccoll A., Swinbourne E. S., Proc. Chem. Soc., 1960, 409; J. Chem. Soc., 1964, 149:
Bicknell R. C., Thesis, University of London, 1962; Maccoll А., см. в ссылке [65],
p. 110.
75. Maccoll A., Stimson V. R., J. Chem. Soc., 1960, 2386; Lewis К. G.. Stimson V. R., ibid.,
3087; Ross R. A., Stimson V. R., ibid., p. 3090; Failes R. E., Stimson V. R., J. Chem.
Soc., 1962, 653; Stimson V.R., Watson E.J., J. Chem. Soc., 1960, 3920; 1961, 1392;
1963, 1524; Cross J. T. D., Stimson V. R., Chem. Comm., 1966, 360; Maccoll A.,
см. в ссылке [65].
76. Grob C. A., Baumann W.. Helv. Chim. Acta, 38, 594 (1955); Grob C. A., Experion-
tia, 13, 126 (1957); «Теоретическая органическая химия», ИЛ, М., 1963, стр.
146.
77. Brown F., Davies Т. D., Dostrovsky I., Evans О. J., Hughes E. D., Nature, 167, 897
(1951); Whitmore F. C., Stahly E. E., J. Am. Chem. Soc., 55, 4153 (1933); 67, 2158
(1945).
78. Beesley R. M., Ingold С. K., Thorpe J. F., J. Chem. Soc., 107, 1080 (1915); Searles S.,
Gostatowsky M. J., J. Ain. Chem. Soc., 75, 3030 (1953); Lukes R., Plesek J., Chem.
Listy, 45, 1826 (1955).
79. Clayton R. B., Henbest II. B., Smith M., J. Chem. Soc., 1957, 1982.
80. Hentzel C., Ann., 139, 132 (1866).
81. Usherwood E. II., J. Chem. Soc., 123, 1717 (1923).
82. Ingold С. K., Powell W. J., J. Chem. Soc., 119, 1976 (1921).
83. Vorlander D., Ber., 33, 3185 (1900).
84. Gibbs E. M., Henry T. A., J. Chem. Soc., 1939, 240.
85. Geuther A., Ann., 122, 121 (1862).
86. Nef J. U., Ann.. 298, 202 (1897).
87. Hine J., J. Am. Chem. Soc., 72, 2438 (1950); Hine J., Peeks C. R., Oakes B. !)., ibid.,
597
Глава IX
76, 827 (1954); Hine J., Dowell A. M., ibid., p. 2688; Hine J., Thomas С. H., Emer-
son S. J., ibid., 77, 3886 (1955); Hine J., Dowell A. M., Singley J. E., ibid., 78, 479
(1956); Hine J., Emerson S. J., Brader W. H., ibid., p. 2282; Hine J., Burske N. W..
ibid., p. 3337; Hine J., Burske N. И7., Hine M., Langford P. B., J. Am. Chem. Soc.,
79, 1406 (1957); Hine J., Butterworth R., Langford P. B., J. Am. Chem. Soc., 80,
819 (1958).
88. Hine J., Porter J. J., J. Am. Chem. Soc., 82, 6178 (1960).
89. Swain C. G., Thornton E. R., J. Am. Chem. Soc., 83, 4033 (1961).
90. Doering W. v. E., Hoffmann A. K., J. Am. Chem. Soc., 76, 6162 (1954).
91. Swenson J. S., Rapoport R., J. Am. Chem. Soc., 87, 1397 (1965).
92. Hine J., Langford P. B., J. Am. Chem. Soc., 79, 5497 (1957); Hine J., Porter J. D.,
ibid., p. 5493; Hine J., Ketley A. D., J.Oi’g. Chem., 25, 606.
ГЛАВА X
ПЕРЕГРУППИРОВКИ В НАСЫЩЕННЫХ СИСТЕМАХ
1 Нуклеофильные перегруппировки в насыщенных системах 600
2 . Электрофильные перегруппировки в насыщенных системах . . 648
Список литературы........................................ 652
1. Нуклеофильные перегруппировки в насыщенных системах
В настоящей главе рассматриваются перегруппировки при изомеризации,
замещении или отщеплении в насыщенных углеродных, углерод-азотных
и углерод-кислородных системах. Термин «насыщенная система» не означает,
что молекула, претерпевающая перегруппировку, всегда полностью насы-
щена. Скорее это означает, что при рассматриваемых перегруппировках,
если и имеется кратная связь или же ароматические циклы, то они оказывают
лишь незначительное влияние. Для краткости такие процессы можно назвать
насыщенными перегруппировками.
Перегруппировки, при которых происходит перемещение группы, несущей
избыток электронов, к какому-либо центру с недостатком электронов, назы-
ваются нуклеофильными перегруппировками. В этом разделе рассматривают-
ся нуклеофильные перегруппировки в насыщенных системах. Из многих
таких перегруппировок сначала в общем виде рассматриваются лишь два типа
(разд. 1,а и б), затем трактуются происходящие при этом процессы (разд. 1,в).
рассматриваются некоторые другие аналогичные перегруппировки (разд. 1.г)
и, наконец, детально обсуждается механизм процесса (разд. 1,д — з).
а. Перегруппировки, сопровождающиеся образованием карбонильной
группы; пинаколиновая и родственные перегруппировки. В 1859 г. Фиттиг [1]
получил пинакон, а в 1860 г. он же отметил превращение пинакона под
действием серной кислоты в кетон пинаколин
(СН3)2С(ОН)С(СН3)2ОН СНяСОС(СН3)з
Отличительная черта этого процесса — перемещение метильной группы от
одного гидроксилсодержащего углерода к другому. С 1860 г. было исследо-
вано много подобных превращений, и стало ясно, что реакция Фиттига
является типичным примером довольно общего процесса, характерного для
замещенных 1,2-гликолей. При действии сильных кислот, например серной,
такие гликоли превращаются в карбонильные соединения с перемещением
заместителя от атома углерода, который при этом усиливает степень своего
связывания с кислородом, образуя карбонильную группу, к углеродному
атому, теряющему кислород. Такие превращения носят названия пинаколи-
новых перегруппировок.
При этом не обязательно, чтобы мигрировали именно метильная или другая
алкильная группа; мигрировать может также фенильный или какой-либо
другой арильный радикал. Более того, не обязательно, чтобы мигрирующий
радикал принадлежал двутретичному гликолю: аналогичный процесс проте-
кает и в случае третично-вторичных или двувторичных гликолей, что видно
из следующих примеров:’
(СвН5)2С(ОН)С(С6Н5)2ОН -> С6Н5СОС(СвН5)8 [2]
(СН3)2С(ОН)С(С6Н5)2ОН -> СН3СОС(СвН5)2СН3 [3]
С6Н5СН(ОН)С(СНз)(С6Н5)(ОН)й-> ОНСС(С6Н6)2СН3 14}
С6Н5СН(ОН)СН(СвН5)ОН -* ОНССН(СвН5)з 15}
Другие превращения некоторых третично-вторичных и двувторичных глико-
лей и большинства третично-первичных и вторично-первичных гликолей
относятся к этомуже типу при условии, что перемещается атом водорода, как
в приведенных ниже примерах (Ап = n-анизил; То1 = п-толил):
СН3СН(ОН)С(С6Н5)2ОН —> СН3СОСН(СвН5)2 [6}
С6Н5СН(ОН)С(С6Н5)2ОН -> СвН5СОСН(С3Н5), [7]
600
Перегруппировки в насыщенных системах
С6Н5СН(ОН)СНАпОН - -» С6Н5СОСН2Ап [8]
СН3СН(ОН)СН(С6Н5)ОН - * СН3СОСН2СвН5 [9]
СН2(ОН)С(С6Н5)2ОН - » ОНССН(С6Н5)2 [Ю]
СН2(ОН)СН(СН3)ОН - ОНССН2СН3 [11]
СН2(ОН)С(СвН5)ТоЮН ОНССН(СвН6)То1 [12]
Мислоу и Зигель [12] показали, что в последнем случае оптически активное
исходное соединение дает оптически активный продукт, т. е. переход водоро-
да, связанного с углеродом первичной спиртовой группы, к атому углерода,
от которого отщепляется гидроксил, происходит стереоспецифично; таким
образом, не происходит отщепления первичного водорода и третичного гид-
роксила в виде воды и не образуется винилового спирта, который мог бы
перегруппировываться.
Пинаколиновую перегруппировку подробно изучали главным образом Дани-
лов и Венус-Данилова, Тиффно, Орехов, Леви, а также Бахман с сотруд-
никами. Основная цель исследований заключалась в выяснении, какая из
групп мигрирует или мигрирует преимущественно, если структура допу-
скает несколько возможностей. Названные авторы стремились выяснить,
насколько прочна связь различных групп, путем определения их «сравни-
тельной способности к миграции». Однако результаты этих эксперименталь-
ных работ надо рассматривать в тесной связи с механизмом перегруппировки.
В общем случае направление перегруппировки гликоля
RR'C(OH)C(OH)R"R"'
зависит от двух чрезвычайно существенных обстоятельств, которые и должны
быть последовательно рассмотрены. Во-первых, следует выяснить, какая из
гидроксильных групп отщепляется, так как этим определяется, какая из
двух пар радикалов дает радикал для замещения гидроксильной группы.
Закономерность отщепления гироксильной группы [13] обоснована и хорошо
подтверждается экспериментом: уход гидроксила из RR'C(OH) облегчается
индуктивным эффектом и высвобождением электронов по механизму сопря-
жения (+/ и Ч-К) из групп RR', точно так же как и при обычном мономоле-
кулярном замещении или отщеплении (SN1 и El). Ясно, что следует обсудить
вопрос о кислотном катализе при отщеплении гидроксила в результате гете-
ролиза с образованием карбониевого иона, подобно тому, как это происходит
в сопряженных кислотах спиртов при мономолекулярных реакциях
I А I
RR'C—ОН2+—> RR'C+ + Н2О
При этом легкость отщепления гидроксильной группы в зависимости от RR'
соответствует ряду
RR' = п-анизил > фенил > алкил > Н
Эта последовательность относится к приведенным выше примерам пинаколи-
новой перегруппировки, и ее можно легко понять.
После того как установлено, какая из гидроксильных групп отщепляется,
остается решить вопрос, какая из двух имеющихся групп займет место ушед-
шего гидроксила. При этом вопрос о миграции водорода можно не рассматри-
вать ввиду уже отмеченной неопределенности происходящего процесса.
Если говорить о других группах, то основной принцип, который должен при-
меняться, заключается в том, что наиболее подвижной следует считать груп-
601
Глава X
пу, обладающую большей способностью отдавать электроны (+/ и +К).
Так, если в гликоле имеется и арильная и алкильная группы, то мигрирует
арильная группа [14], например:
СН3(СвН5)С(ОН)С(ОН)(СН3)СвН5 -> СН3СОС(СН3)(С6Н5)2
Если в соединении имеются различные арильные группы, как в симметрич-
ном пинаконе Бахмана
АгАг'С(ОН)С(ОН)АгАг'
то мигрирует группа, более активная по отношению к электрофильным аген-
там, что иллюстрируется следующим рядом сравнительной подвижности [15]:
п-анизил > п-толил > л-толил > л-апизил > фенил >
> п-хлорфенил > о-анизил > л-хлорфепил
Аномальное положение занимает только о-анизильная группа, что можно
связать с пространственными факторами.
Смысл указанного правила подвижности с точки зрения механизма реакции
состоит в том, что мигрирующая группа использует свои электроны для
первоначального связывания с соседним электронодефицитным центром,
и этот процесс происходит до того, как эти электроны становятся полностью
высвобожденными в результате полного разрыва связи.
Показано, что скорость превращения пинакона в пинаколин зависит от
кислотной функции Гаммета h0 и характер этой зависимости соответствует
специфическому катализу водородными ионами, но не общему кислотно-
му катализу, т. е. первой стадией процесса является предравновесное обра-
зование сопряженной кислоты гликоля [16, 17]. При сравнении скорости
превращения в Н38О со скоростью внедрения изотопа 18О было также пока-
зано, что дегидратация сопряженной кислоты обратима и скорость обратной
реакции, состоящей в присоединении воды к карбониевому иону, сравнима со
скоростью миграции метильной группы в карбониевом ионе [17].
Существует ряд перегруппировок, вполне аналогичных пинаколиновой.
Одна из них, а именно перегруппировка замещенной окиси этилена в карбо-
нильные соединения, происходит под влиянием кислот. Правила о месте раз-
рыва связи атома кислорода с одним из атомов углерода и о перемещающей-
ся группе, по-видимому, те же, что и в случае пинаколиновой перегруппи-
ровки. По данным одного исследования [18], окиси
О
(СвН5)гС-СНЙ, где R = CH3, С2Н5, н-С3Н7, изо-С3Н7, и-С4Н9, emop-C4H9, СвН5СН2, СвН5
превращаются в бензгидрилкетоны, причем атом кислорода уходит от
(С6Н5)2С-группы. Если же R = n-анизил, то расщепляется другая кисло-
родная связь и образуется фенилкетон
О
(СвН5)9С'^-'ЬнИ (CeH5)2CHCOR (R=anKM или фенил)
О
(CeH5)2CZ——^CHR —► CeH5COCH(CeH5)R (R = я-анизил)
Иная перегруппировка обсуждаемого типа, часто называемая ацилоиновой,
состоит в превращении а-оксикетонов и а-оксиальдегидов под влиянием
602
Перегруппировки в насыщенных системах
спиртового раствора серной кислоты в изомерные а-оксикетоны. При этом
кислородсодержащие функциональные группы меняются местами и происхо-
дит перемещение одного радикала, например:
f*O <ОН
изо-С3Н7- С—С-СН3-
СН3
<0 ГОН
Ч ч
н—с—с-с6н5
с6н5
он о
I II
изо-С3Н7—С—С-СН3
СНз
он о
I II
—♦ Н-С—C-C6HS
с6н5
[19]
[20]
Бензиловую перегруппировку впервые наблюдал Либих. Она обычно происходит
под влиянием водно-спиртовых растворов щелочей [21]. Впоследствии был
выделен продукт присоединения С6Н5СОСОС6Н5,КОН и показано, что он
претерпевает перегруппировку в калиевую соль бензиловой кислоты
(СвН5)2С(ОН)СООК [22]. Кроме того, выделено соединение С6Н5СОСОС6Н5,
C2H5ONa, которое претерпевает перегруппировку только после добавления
воды, в результате чего образуется натриевая соль бензиловой кислоты [23].
В водно-щелочных растворах скорость превращения бензила в бензиловую
кислоту пропорциональна произведению [бензилПОН-]. Следовательно,
в данном примере имеет место специфичный катализ гидроксильным ионом,
а не частный случай общего основного катализа [24]. Все эти наблюдения
согласуются с теорией [25], согласно которой перегруппировка идет через
промежуточное образование комплекса с гидроксильным ионом и с электрон-
ными смещениями, аналогичными сформулированным выше для ацилоино-
вой перегруппировки
О
II
О=С—С-С6н5------->
I (ОН-)
с6н5
С6Н5
О 0-
- II I
О-С-С-С6Н5-----►
Известны и такие перегруппировки, при которых возникновение карбониль-
ной группы определяется наличием электроположительного атома углерода,
образующегося при отщеплении не кислорода, а, например, галогенов или
азота.
Примером подобного превращения служит найденная Тиффно иодгид-
ринная перегруппировка при реакции третичных p-иодированных спиртов
с серебряными или ртутными солями, которые, как известно, катализируют
различного рода реакции отщепления галогена, носящие характер моно-
молекулярного гетеролиза (гл. VII, разд. 5,в). Тиффно показал, что надле-
жащим образом замещенные иодгидрины можно превратить в карбонильные
603
Глава X
соединения с перемещением заместителя, как, например
с6н5 С6Н5
н-^-с(сн3)-сн2Л: -> о=с(сн3)сн2 126]
сн3
о=с—сн
сн2 \зн2
I I
сн2—сн2
[27]
В случае аналогичных соединений, где вместо иода стоит гидроксил, а реа-
гентом служит водная серная кислота, направление перегруппировки не
соответствует ожидаемому: в данном случае происходит отщепление третич-
ной, а не вторичной или первичной гидроксильной группы. В других отно-
шениях реакция характеризуется обычным порядком подвижности групп::
С6Н5 > алкил и высший алкил > СН3.
Аналогичная перегруппировка, названная пинаколиновым дезаминировани-
ем, происходит, как показал Мак-Кензи, при дезаминировании третичных
Р-аминоспиртов под действием азотистой кислоты. Процесс, по-видимому,
заключается в отщеплении азота от первоначально образующегося диазоние-
вого иона и соответствует мономолекулярной реакции (гл. VII, разд. 6,и).
Несколько примеров подобного рода приведено ниже (Ап = «-анизил):
НОС(СН3)2СН(С6Н5)МН2 -» СН3СОСН(СН3)С6Н5 [28]:
НОС(СН3)(С6Н5)СН(СвН5)КН2 -» СН3СОСН(С6Н5)2 [29]'
НОС(СвН5)2СН(СвН5)КН2 -> СвН5СОСН(С6Н5)2 [30]
HOC(CeH5)(An)CH2NH2 -> С6Н5СОСН2Ап [31]
Опять-таки, по крайней мере в трех последних примерах, при замене амино-
группы гидроксильной и при применении сильной кислоты направление про-
цесса было бы иным — молекула теряла бы третичную гидроксильную груп-
пу. Во всех других отношениях реакция характеризуется обычным порядком
подвижности: Ап > С8Н5 > СН3.
б. Перегруппировки в реакциях отщепления олефинов, нуклеофильно-
го замещения и присоединения олефинов; реакции Вагнера — Меервейна
и родственные процессы. Перегруппировки рассматриваемого типа были
впервые описаны в 1899 г. Вагнером, который считал их важными для пони-
мания отношений между различными членами ряда бициклических терпе-
нов; Вагнер наблюдал такие перегруппировки и при превращении пинена
в борнильные соединения. Предлагая общепринятую в настоящее время
формулу камфена, Вагнер допускал такие перегруппировки при образова-
нии камфена из соединений борнила и при обратном превращении камфена
в борнильные соединения. Меервейну принадлежит дальнейшее развитие
представлений Вагнера; Меервейн расширил область изучения и применения
этой перегруппировки настолько, что она теперь признана одним из общих
явлений органической химии и характерна не только для химии терпенов.
Более того, как показано ниже, Меервейн сделал большой, единственный
в своем роде вклад в разъяснение механизма перегруппировки.
Область бициклических терпенов богата примерами перегруппировок Ваг-
нера — Меервейна, и некоторые из них приведены ниже. Многие перегруп-
пировки включают изменение структуры кольца вследствие перемещения
одной из связей углерода, образующего центральный мостик, в соседнее
604
Перегруппировки в насыщенных системах
положение, где первоначально имелся электроотрицательный заместитель
или же двойная связь, при присоединении к которой кислоты такой замести-
тель мог бы образоваться. Однако некоторые другие перегруппировки вклю-
чают только перемещение метильной группы в соседнее положение, что удо-
влетворяет одному из сформулированных условий. В любом случае характер
перегруппировки остается одним и тем же.
Перегруппировки обсуждаемого типа можно также разбить на группы в зави-
симости от суммарной стехиометрии реакции. Некоторые из них являются
реакциями отщепления олефина, другие — нуклеофильным замещением,
в частном случае с изомеризацией ив еще более особом случае с рацемизацией;
имеются перегруппировки, включающие присоединение к олефину. Если,
Камфен [32,33]
Изоборнилхлорид
Сантен [34]
Камфенилилхлорид
Изоборнил хлорид
[33,35]
[36,37]
З’ац 'м изация камфенгидрохлорида (а следовательно, камфена и изоборнилхлорпда)
605
Глава X
следуя указанию Меервейна, представить все эти реакции как протекающие
через карбониевый ион, образующийся в результате потери аниона или же
захвата протона, то можно показать, что все они обладают общими чертами,
несмотря на разнообразие в перегруппировках.
Фенхиловый спирт
[32,33,38]
В случае изоборнилхлорида элиминирование с перегруппировкой с образова-
нием камфена было изучено в гомогенной газовой фазе; об этом уже упомина-
лось в гл. IX, разд. 5,в.
Меервейн расширил представления о таких перегруппировках за рамки’ряда
бициклических терпенов [39]. Так, например, он показал, что 2,2-диметил-
циклогексанол при кислотной дегидратации превращается в смесь изопропили-
денциклопентана и 1,2-диметилциклогексена. Эти реакции являются моно-
циклическими моделями — первая для превращения изоборнеола в камфен,
а вторая для превращения камфенилола в сантен.
Моноциклической моделью перегруппировки, которая инициируется йоорди-
нацией протона с двойной связью, а не отщеплением аниона от насыщенной
молекулы (или аналогичным гетеролизом), может служить давно известное
превращение а-камфолитовой кислоты в изолауронолевую кислоту при дей-
ствии разбавленной серной кислоты [40]
СН3
Нзолауронолеваа кислота
Простой моделью в ациклическом ряду служит давно известное превращение
пинаколинового спирта при кислотной дегидратации в тетраметилэтилен [41].
606
Перегруппировки в насыщенных системах
Последнее соединение является основным продуктом реакции, хотя более
поздняя работа [42] показала, что образуется также 1-метил-1-изопропил-
этилен
/>ОН2+
СНЗХ *4 СН3.+
>С—СН-СН3-> >С—СН-СН3->
СН/ н сн/ |
СН3 СН3
СН3
'>С=С-СН3
СНз ^нз
сн2.
>с-сн-сн3
сн/ I
сн3
Существует еще более простая группа примеров, изучение которой Уитмо-
ром с сотрудниками, а также Достровским и Хьюзом приобрело важное
значение для понимания механизма обсуждаемой перегруппировки. Сюда
относятся превращение неопентилового спирта при действии бромистого
водорода в трет-амилбромид [43] и неопентилиодида в mpem-амиловый спирт
при действии серебряных или ртутных солей [44], а также превращение нео-
пентилбромида в водном этиловом спирте в смесь трети-амилового спирта,
mpem-амилэтилового эфира и триметилэтилена [45]
(СН3)3С-СН2-0)Н2
Х^СНз +
^С-СН2
* СНз СН3
О .
(СН3)3С—СН3—Hal
Вг~
-----> (СН3)2СВгСН2СН3
Н2О
-----> (СН3)2С(ОН)СН2СН3
с2н5он ,
-------> (СН3)2С(ОС2Н5)СН2СН,
О£«£8ДД^ (,СН3)2С=СНСН3
Из перегруппировок, подобных перегруппировке Вагнера — Меервейна, но
обусловленных карбониевым ионом, образующимся при отщеплении иных,
чем галогены или кислород, групп, наиболее важными являются перегруп-
пировки, связанные с дезаминированием первичных аминов при действии
азотистой кислоты. Они обычно называются перегруппировками Демьянова.
Заслуга Демьянова заключается в том, что он показал возможность исполь-
зования таких перегруппировок для расширения и, по крайней мере в од-
ном случае, для сужения алициклических колец.
Давно было известно, что при обработке н-пропиламина азотистой кислотой
образуется немного w-пропилового спирта наряду с большим количеством
изопропилового [46]. Остюда следует, что большая часть карбониевых ионов,
образующихся при отщеплении азота от первоначального диазониевого иона
(гл. VIII, разд. 1,в), претерпевает 2-> 1-смещение водорода, прежде чем
присоединить гидроксил от растворителя.
Н Н
hno2 I + +1
ch3ch2ch2nh2-------> сн3—сн—сн2 —* сн3—сн—сн2
СН3СН2СН2ОН СН3СН(ОН)СН3
Используя 14С, Робертс [47] показал, что при дезаминировании этиламина
2-> 1-смещение водорода происходит в малой степени: только 1,5% образу-
607
Глава X
ющегося этилового спирта содержали гидроксил у атома углерода, с которым
первоначально была связана аминогруппа [47].
Робертс также установил, что «-пропиловый спирт, образующийся в качестве
одного из продуктов при дезаминировании н-пропиламина, в небольшой
степени перегруппирован. На это указывал тот факт, что образующийся спирт
частично терял метку 14С, которая первоначально находилась в положении
1 амина. Реутов [48] обнаружил некоторую потерю радиоактивной метки из
положения 3 спирта, и, таким образом, открыл 3 —> 1-сдвиг водорода, о кото-
ром речь еще будет идти ниже.
Н Н
I + + I
сн2-сн2—сн2 —> СН2-СН2-СН2
Однако в последующих работах Карабацоса и Орцеха, а также Ли, Крагера
и Уонга было показано, что при дезаминировании с образованием «-пропило-
вого спирта перегруппировка проходит на 3—4% [49]. Метки 14С или тритие-
вая во время дезаминирования частично теряются из положения 1 и в образу-
ющемся w-пропиловом спирте могут быть обнаружены в положении и 2 и 3.
Это могло бы означать, что происходит 3 —> 1-сдвиг водорода, сопровождаю-
щийся 2 -> 1-сдвигом метила; с последней перегруппировкой мы еще встре-
тимся *.
СН3 СН3 _ СЩ
СН2-^СН2 СН2-СН2 СН2-СН3
(На этих схемах идентичность атомов подчеркнута их одинаковым расположе-
нием, хотя номера атомов по химической номенклатуре при изомеризации
изменяются.)
Проведя тщательный анализ продуктов дезаминирования аминов с нормаль-
ной цепью, Витинг и сотр. [50] установили факт смещения водорода вдоль
длинных цепей атомов углерода. Например, при дезаминировании 1-октил-
амина образуются следующие изомерные октилацетаты:
1- (4,6%), 2- (18,5%), 3- (2,8%), 4- (0,19%)
и следующие октены:
. ._ _0/. (цис-2- (2,1%), цис-3- (0,52%), цис-i- (не образуется)
’ ° ’ (транс-2- (7,7%), транс-3- (0,95%), транс-i- (0,08%)
Пока не известно, происходят ли дальние сдвиги непосредственно или они
являются следствием нескольких последовательных более коротких сдвигов.
Однако 1,5-сдвиг, наблюдавшийся Прелогом при дезаминировании цикло-
дециламина, показывает, что одностадийный дальний сдвиг может иметь
место, если расстояние между участвующими атомами углерода достаточно
мало (гл. IV, разд. 3,а).
* Авторы считают, что промежуточно образуется «протонированное циклопропановое
кольцо» (см. стр. 643), т. е. три таких кольца, взаимопревращающихся друг в друга
путем сдвига дополнительного протона от одной связи к другой вокруг кольца. Мы думаем,
что имеющиеся данные не могут достаточно прочно обосновать ни такое предположение,
ни предположение, указанное в тексте. Например, с любой точки зрения трудно объяс-
нить, почему, согласно опубликованным данным, атомы D в CD2-rpynne остаются вместе
у одного атома углерода при перегруппировке, сопровождающей дезаминирование
CH3CD2CD2NH2, но в значительной мере разъединяются при перегруппировке, сопро-
вождающей дезаминирование CH3CH2CD2NH2 и CH3CD2CH2NH2. Следует напомнить,
что вследствие небольшого процента перегруппировки и незначительного количества
«-пропилового спирта, образующегося при дезаминировании, экспериментальная работа
очень трудна.
ш
Перегруппировки в насыщенных системах
Витипг рассмотрел вопрос о том, имеет ли первоначально образовавшийся ион
диазония время жизни, достаточное для того, чтобы противоион успел про-
диффундировать в раствор прежде, чем произойдет отщепление азота и обра-
зование карбониевого иона. Оказалось, что это зависит от стабильности кар-
бониевого иона, нуклеофильности аниона и ионизующей силы растворителя.
При дезаминировании насыщенных первичных алкиламинов, например
1-октиламина, в растворителях, полярность которых изменялась от воды до
уксусной кислоты, диазониевый ион безусловно живет достаточно долго,
и достигается равновесие диссоциации. Однако, если образующийся карбони-
евый ион является вторичным, то, судя по составу продуктов реакции,
исходный диазониевый ион переходит в ион карбонил на той стадии, когда
противоион находится очень близко; при этом сам ион диазония вряд ли может
существовать в свободном виде.
Неопентиламин при обработке азотистой кислотой превращается в трет-
амиловый спирт [51]. В данном случае при дезаминировании наблюдается
перемещение метильной группы
СН3 СН3 НО СН3
I HNO2 k + ||
(Ch3)2c-ch2nh2-------> (сн3)/-сн2 (сн3)2с-сн2
Карабацос, Орцех и Мегерсон [52] с помощью изотопных меток показали,
что в этом случае происходит просто смещение метильной группы, так как
три атома метиленовой группы неопентиламина входят в состав метиленовой
группы образующегося mpem-амилового спирта.
Ниже приведены примеры изменения кольца при дезаминировании цикличе-
ских систем: из циклобутиламина образуются циклобутанол и циклопропил-
карбинол [53]; циклопропилметиламин подобным же образом дает циклопро-
пилкарбинол и циклобутанол [54], а циклобутилметиламин — циклобутил-
карбинол и метиленциклобутан, циклопентанол п циклопентен [55]
fCH2\ Л | )СНСН2ОН
СН2Х I )chch,nh2 —» сн/ 1 ^н/ 1 /СН2Х 1 СН / 2\снон СНа V сн2 J
/СН2. сн/ >chch2nh2 - сн/ ( /СН2. /СН2. сн/ >СНСН2ОН и сн/ >С = СН2 1 \сн/ хсн/ * ) /СН2СНОН ХСН2СН сн/ ] и сн/ II 1 \сн2сн2 ХСН2СН
Аналогично циклопентилметиламин [56], циклогексилметиламин [57], цикло-
гептилметиламин [56] и циклооктилметиламин [58] соответственно дают цикло-
гексанол, циклогептанол, циклооктанол и циклононанол, причем всегда
наряду с другими продуктами, которые в последнем случае содержат цикло-
октилкарбинол и 1-метилциклооктен.
В качестве примера реакции, обратной расширению цикла по Демьянову,
можно привести превращение циклопропиламина в аллиловый спирт. В про-
цессе реакции а-дейтериевый атом в амине остается в том же положении, где
он был, а именно в ^-положении аллильного остатка
СН2 СН2
\ HNO2 X
CDNH2 ---->• CD+
СН^ СН2
HOCH,/
39-0772
609
Глава X
Вновь было отметено, что карбониевый ион, образующийся из иона диазония,
более склонен к перегруппировке, чем ион, образующийся из алкилсульфона-
та или другого эфира или галогенида путем отщепления аниона. Так, при
сольволизе я-бутил-п-бромбензолсульфоната в уксусной кислоте из бутило-
вых эфиров образуется только w-бутилацетат, тогда как при дезаминировании
w-бутиламина в том же растворителе — 70% м-бутилацетата и 30% втор-
бутилацетата [59]. Наиболее подходящее объяснение, которое, как будет
показано ниже, объясняет и стереохимические данные, состоит в том, что
карбониевый ион, образующийся при отщеплении аниона, в большей степе-
ни успевает достигнуть той степени стабильности, которая обусловлена
равновесием сольватации, по сравнению с карбониевым ионом, получающим-
ся при отщеплении нейтральной молекулы азота. В карбониевом ионе, обра-
зующемся последним путем, перегруппировка может конкурировать с закры-
тием сольватационной ячейки и с достижением конформационного равно-
весия внутри нее.
в. Теория пинаколиновой перегруппировки и перегруппировки Вагне-
ра — Меервейна. Наиболее раннее объяснение механизма пинаколиновой
и родственных ей перегруппировок сводилось к предположению об образова-
нии в ходе реакций промежуточного трехчленного кольца. Примерно в 1880 г.
Броер и Цинке [60] высказали мысль, что при пинаколиновой перегруппиров-
ке образуются в качестве промежуточных продуктов циклические окиси;
Эрленмейер [61J предположил, что в данном случае получаются циклопропа-
ны, например:
(СН3)2С — С(СН3)2 и СН3С(ОН) —С(СН3)2
0 cnf
Для критической оценки этих предположений в то время имелось слишком
мало сведений о свойствах такого рода веществ.
В 1907—1924 гг. Тиффно и Мак-Кензи с сотрудниками опровергли окисную
гипотезу главным образом на основании двух положений.
Во-первых, в перегруппировках, сопровождающихся либо дегидратацией
гликолей, либо дегидрогалогенированием [3-иодспиртов, либо дезаминирова-
нием (3-аминоспиртов, из продуктов не доведенной до конца реакции не уда-
ется выделить циклических окисей. Кроме того, оказалось, что приготов-
ленные по независимым методикам такие окиси настолько устойчивы, что их,
несомненно, можно было бы выделить, если бы они действительно образова-
лись в процессе реакции.
Во-вторых, несостоятельность окисной теории доказывается тем, что при
дегидрогалогенировании [3-иодспиртов и дезаминировании (3-аминоспиртов
направление перемещения группы часто отлично от ожидаемого переме-
щения группы в случае промежуточного образования окиси (настоящая
глава, разд. 1,а).
Циклопропановая гипотеза была опровергнута сначала Монтанем и Акри
(1905 г.) на некоторых частных примерах и затем в более общем виде Меер-
вейном и ван Эмстером (1920 г.). Согласно гипотезе Эрленмейера, предпола-
галось, что происходит отщепление водорода от мигрирующей группы перед
образованием новой связи.
Монтань [62] и Акри [63] показали, что, по крайней мере в случае перемеще-
ния арильных групп, неверно предположение, что водород уходит из орто-по-
ложения, куда становится перемещающаяся группа. Если бы это было так, то
в результате перегруппировки первоначальный пара-заместитель превратился
610
Перегруппировки в насыщенных системах
бы в лета-заместитель
(п-С1СвН4)С(ОН)С(ОН)(С6Н4С1-п)2
С1
(п-С1СвН4)С0-С(С6Н4С1-п)2
I
\ 4
I
С1
в то время как в действительности всегда сохраняется пара-заместитель.
Циклопропановая гипотеза первоначально привлекла к себе внимание благо-
даря тому, что она могла быть применена как к пинаколиновой, так и к пере-
группировке Вагнера — Меервейна. В последнем случае применение этой
гипотезы было несколько проще. Типичный пример применения ее к пере-
группировке Вагнера — Меервейна состоит в когда-то широко принятом
предположении, что известный углеводород трициклен является промежу-
точным при взаимном превращении изоборниловых эфиров в камфеновые
эфиры или камфен. Однако Меервейн и ван Эмстер [64] показали, что это
предположение неверно по двум причинам. Одна из них заключается в том,
что хотя трициклен можно превратить в камфен или его производные, однако
этого не происходит при обработке 33%-ной серной кислотой, несмотря на
то, что в этих условиях изоборнеол гладко превращается в камфен.
Изоборниловые
эфиры
НХ
Трициклен
Другая причина состоит в том, что хотя взаимное превращение изоборнило-
вых эфиров и камфена, катализируемое кислотами, в условиях, обычно
вызывающих эту реакцию, и сопровождается рацемизацией, последняя
в действительности является результатом независимого процесса (перемеще-
ния метильного радикала, как показано в разд. 1,6); этот процесс можно
осуществить в результате применения кислотных катализаторов средней
силы, как, например, муравьиной кислоты. При этом оптически активный
камфен превращается в активные изоборниловые эфиры, что не могло бы
произойти, если бы промежуточным продуктом реакции был ахиральный
трициклен.
Другая, также отвергнутая гипотеза, выдвинутая Тиффно [65] в 1907 г.
и широко применявшаяся Мак-Кензи в 20-х годах текущего столетия, пред-
полагала образование свободных радикалов в качестве промежуточных про-
дуктов, например в случае пинаколиновой перегруппировки — бирадика-
ла типа
R2C(O)-CR2
Эта теория появилась в то время, когда о свойствах свободных радикалов
было известно слишком мало, чтобы можно было сразу же критически оценить
ее. Но несмотря на то, что специально ничего не было предпринято для
опровержения этой теории, она постепенно вышла из употребления. Это
отчасти произошло потому, что в 1922 г. Меервейн и ван Эмстер предложили
гораздо лучшую теорию, а частично потому, что растущие знания о свойствах
свободных радикалов привели к выводу, что если бы теория Тиффно была
39* 611
Глава X
верной, то, вопреки экспериментальным фактам, перегруппировка сопрово-
ждалась бы цепными реакциями, полимеризацией, гомолизом растворителей
и другими характерными для свободнорадикальных процессов явлениями.
В 1922 г. вторая работа Меервейна и ван Эмстера [66] положила начало
современной теории обсуждаемых перегруппировок. Меервейн и ван Эмстер
изучили кинетику некоторых перегруппировок, а именно перегруппировку
камфенгидрохлорида в изоборнилхлорид, и привели убедительные факты,
показывающие, что эта перегруппировка обусловлена ионизацией, причем
изменения в углеродном скелете происходят в первоначально образующемся
карбониевом ионе.
Камфенгид-
рохлорид\
Камфен
Потеря С1 г д30^0рНил.
Захват СГ \ хлорид
Для^подтверждения этого взгляда были приведены два основных довода.
Первый состоит в том, что скорость перегруппировки по реакции первого
порядка зависит от природы растворителя, а именно от его ионизирующей
способности. Скорости реакций для ряда растворителей уменьшаются в сле-
дующем порядке:
SO2 > CH3NO2 > CH3CN > CeH5NO2 > CeH5CN >
> CeH5OCH3> CeH5Br> C2H5Br> C,H5C1
> CeHe > петролейный эфир > эфир
Этот порядок в основном соответствует уменьшению ионизующей способности
растворителей по отношению к трифенилметилхлориду. Другой довод состоит
в том, что рассматриваемая перегруппировка катализируется так называе-
мыми электрофильными катализаторами, например галогеноводородами
в неосновных растворителях (гл. VII, разд. 5,6). Мощными катализаторами
перегруппировки являются способные к координации хлориды металлов
HgCl2, FeCl3, SbCl3, SnCl4, SbCl5, каждый из которых образует продукт при-
соединения с трифенилметилхлоридом; в то же время такие хлориды, как
РС13 и SiCl4, не дающие продуктов присоединения с трифенилметилхлоридом,
не обладают и каталитической активностью.
Несколько позже Меервейн с сотр. [67] привел третий довод в защиту своей
теории; он показал, что ряд эфиров камфена претерпевает превращение в изо-
борниловые эфиры со скоростью, которая возрастает с увеличением стабиль-
ности аниона эфирного радикала и отвечает силе соответствующих кислот.
Ряд производных 2-хлорцимола-5
сульфонат > бромид > хлорид > трихлорацетат > лгнитробензоат
характеризует собой сравнительную тенденцию различных эфирных групп
к переходу в форму аниона. Несколько позже автор книги добавил четвер-
тый аргумент [68], а именно: структурные изменения, при которых увеличи-
вается электронная плотность в положении, из которого должен уйти анион,
действительно ведут к ускорению перегруппировки. Данные, подтверждаю-
щие изложенное, взятые главным образом из литературы по перегруппиров-
кам типа пинаколиновой, изложены в разд. 1,а. Ниже приведены также
и другие аргументы, выдвинутые позднее.
612
Перегруппировки в насыщенных системах
Теория Меервейна, достаточно хорошо обоснованная в 1928 г., точно указы-
вает на обязательную первичную ступень в обсуждаемых перегруппировках.
К тому же времени автором книги и Шоппи [69] были ясно сформулированы
общие положения об электронных изменениях при перегруппировках. Таким
образом, были сопоставлены различные типы перегруппировок, приведенные
в основном в двух предыдущих разделах, обсуждено направление перегруп-
пировок и проблема миграционной подвижности в указанных ниже направ-
лениях:
R
~О^С-С=0)
R
Н р
Н—С—С—С—х
R
н-^-с—c-Gc
R
В течение 30-х годов текущего столетия Уитмор и сотр. [70] значительно
расширили представления о перегруппировке Вагнера — Меервейна в аци-
клическом ряду. Они изучали главным образом простые превращения неопен-
тильных соединений в трет-амильные. Уитмор с сотрудниками нашел, что
неопентиловый спирт и неопентилгалогениды лишь с большим трудом под-
вергаются типичным формам нуклеофильного замещения. Все же такое
замещение наряду с элиминированием легко происходит при применении
некоторых особых методов, а именно при обработке галогенидов водными
растворами серебряных или ртутных солей; в этих условиях в результате
перегруппировки образуются соединения с mpem-амильной группой, или
изоамилены. В 1947 г. Достровский и Хьюз [71] сделали определенные выводы
из этих фактов. На основании кинетических данных они показали, что
трудно протекающее замещение представляет собой S к2-замещение и что при
этом не происходит перегруппировки, как, например:
С2Н5О” в С2Н5ОН
(СН3)3ССН2Вг------—------> (СН3)3ССН2ОС2Н5
Они показали также, что легкое нуклеофильное замещение или элиминиро-
вание представляет собой мономолекулярный или катализируемый моно-
молекулярный процесс и что во всех случаях, когда установлен этот тип
процессов, наблюдается перегруппировка
5 0%-ный водный С2Н5ОН ( (СН3)2С(ОН)СН2СН3
(СН3)3ССН2Вг -----—— ---------> (СН3)2С(ОС2Н5)СН2СН3
sn1+e1 I (СН3)2С=СНСН3
Эта работа в области кинетики привела к установлению связи между изуче-
нием перегруппировок и общим исследованием мономолекулярных реакций
замещения и отщепления, что позволило при разработке проблемы перегруп-
пировок использовать всю совокупность относящихся к механизму моно-
молекулярных реакций данных, приведенных в гл. VII и IX.
При протолизе неопентильных соединений Гриньяра выделен только неопен-
тан и не образуется изопентана. Этот и другие аналогичные процессы почти
наверняка протекают через карбанионы (гл. VI, разд. 8, е). Уитмор [72]
исследовал эти и другие реакции металлических производных неопентана,
которые могут протекать или на самом деле протекают через образование
карбанионов, и ни в одном случае не получил доказательств перегруп-
пировки.
613
Глава X
Хараш, подробно изучивший химию свободных радикалов, широко исполь-
зовал реактивы Гриньяра в сочетании с хлоридом кобальта(И) для восста-
новления алкилгалогенидов в свободные алкильные радикалы и совместно
с Ури [73] установил, что при такой обработке неофилхлорида образуются
измененные в результате перегруппировки углеводороды наряду с некоторы-
ми их димерами; образование последних служит одним из доказательств
того, что реакции протекают при участии свободных радикалов:
Димер Димер
t . Л
(СН3)2С(СеН5)СН2С1 -> (СН3)2С(С6Н5)СН2 -> (СН3)2ССН2С6Н5
(СН3)2СНСН2С6Н5 + (СН3)2С = СНС6Н5
Уитмор [74] первый пришел к выводу, что перегруппировки некоторых азот-
содержащих систем, а именно перегруппировки Гофмана, Лоссеня и Курциу-
са, о которых будет сказано ниже, принадлежат к тому же классу процессов,
что пинаколиновая перегруппировка и перегруппировка Вагнера — Меер-
вейна. Эти перегруппировки, подобно реакции Вагнера — Меервейна, про-
текают с предварительным образованием в результате гетеролиза обедненного
электронами центра с секстетом электронов. Уитмор избегал называть обед-
ненный электронами углерод карбониевым ионом. Он решительно возражал
[75] против теории Меервейна и ионного механизма перегруппировки. Но
все же у Уитмора [76] можно найти описание неопентильных перегруппиро-
вок как процессов, протекающих через «неопентильную систему с 30 элек-
тронами». Следовательно, концепция Уитмора, если подсчитать валентные
электроны, по-видимому, очень близка к «карбониевому иону» Меервейна *.
В 1946 г. Достровский, Хьюз и автор книги [77] положили в основу своих
представлений о механизме перегруппировок следующие два постулата:
1) для осуществления перегруппировки необходимо, чтобы при перво-
начальном разрыве связи образовался атом с незаполненным октетом (кине-
тическое условие);
2) когда таким образом создаются условия для осуществления перегруппи-
ровки по определенному механизму, перегруппировка может происходить
только при соответствующем изменении свободной энергии системы (термо-
динамическое условие).
Кинетическое условие необходимо, но недостаточно. Дополнительная необ-
ходимость в выполнении термодинамического условия иллюстрируется сопо-
ставлением следующих трех систем:
(СН3)3ССН2Х -> (СН3)2С(Х')СН2СН3
(СН3)3ССН(СвН5)Х -> (СН3)2С(Х')СН(С6Н5)СН3
(СвН5)3ССН(СвН5)Х -> (С. Н5)2С(Х')СН(СвН5)2
При удовлетворении в каждом случае условий для мономолекулярного гете-
ролиза X первая и третья перегруппировки происходят, а вторая нет [78].
Эта работа завершает определенную ступень в развитии теории перегруп-
пировок насыщенных соединений. Она служит подтверждением и обобщением
механизма Меервейна. Остается решение в деталях различных геометриче-
ских и динамических вопросов, что сделано в разд. 1, д — з.
г. Перегруппировки Вольфа, Бекмана, Гофмана, Лоссеня, Курциуса
и Шмидта. Из общего положения, заключающегося в том, что при перво-
* Впоследствии в письме к Хьюзу Уитмор объяснил, что к такой двусмысленности его
привело сильное давление со стороны существовавшей в то время оппозиции концепции
карбониевых ионов как промежуточных частиц в органической химии.
6U
Перегруппировки в насыщенных системах
начальном разрыве связи образуется атом с незаполненным октетом, выте-
кает возможность промежуточной стадии, помимо образования карбоние-
вого иона. Электронный секстет в оболочке с главным квантовым числом 2
имеется в трехвалентном положительном атоме углерода, двухковалентном
нейтральном атоме углерода, двухковалентном положительном атоме азота
и одноковалентном нейтральном атоме азота; септетом электронов обладают
трехковалентный нейтральный атом углерода и двухковалентный нейтраль-
ный атом азота. Известны перегруппировки, которые можно объяснить почти
всеми этими промежуточными состояниями, однако доказательства правиль-
ности такого объяснения основаны в ряде случаев на формальной аналогии
с более подробно изученными перегруппировками. В табл. 162 суммируется
Таблица 162
Типы перегруппировок в насыщенных системах
Предполагаемые промежуточные
соединения
Перегруппировки
Описание
-Ас+ (секстет)
С (секстет)
N+ (секстет)
— N (секстет)
(септет)
Пинаколиновая и другие
Вагнера —Меервейна и другие
Вольфа
Бекмана, Шмидта
Гофмана, Лоссеня, Курциуса
Ури —Хараша
Разд. 1, а
Разд. 1, б
> Разд. 1, г
Разд. 1, в
все вышесказанное. Следует учесть, что не всегда перегруппировка насыщен-
ных соединений упоминается под своим названием. Так, перегруппировка
Демьянова включена под рубрикой реакции Вагнера — Меервейна. Рас-
смотрим теперь отдельные наиболее интересные случаи.
Перегруппировка Вольфа состоит в превращении а-диазокетона при нагрева-
нии в растворителе в присутствии поверхностноактивных катализаторов или
без них в кетен и азот или в продукты взаимодействия кетена с окси- или
аминосоединениями, присутствующими в растворе:
O=C-CHN2 —>
О = С = СН<
R <
R
4-Н2О —> HOOCCH2R
+ R'OH R'OOCCH2R
4-NHg —> NH2COCH2R
+ R'NH2 -> NHR'COCH2R
Так как а-диазокетоны обычно готовятся из хлорангидридов кислот и диазо-
метана, то перегруппировка Вольфа [79] является одной из стадий реакции
Арндта — Эйстерта [80], заключающейся в превращении кислот в их сле-
дующий гомолог:
RCOOH -> RCOC1 -> RCOCHN2 -> HOOCCH2R
Предполагается следующий механизм процесса:
R R R Н2О НО R
I + _ н | —>11
О=с—CH=N=N О=СТСН О=С=СН О=С—СН2
615
Глава X
Доказательством правильности этого механизма служит то, что диазосоеди-
нение несомненно теряет азот и поэтому возможно образование карбена.
Затем происходит перемещение к атому углероду рядом стоящей группы, что
подтверждается как строением образующихся продуктов, так и при помощи
изотопного замещения атомов углерода, между которыми идет перемещение
групп [81]. Кроме того, при наличии апротонных условий могут быть выде-
лены кетены, которые представляют собой первые стабильные продукты
перегруппировки [82]. В следующем разделе рассмотрены некоторые стерео-
химические доказательства приведенной выше схемы.
Ясное представление о механизме бекмановской перегруппировки кетокси-
мов [83] при каталитическом действии сильных кислот, галогеноводородов
или пятихлористого фосфора можно вывести при изучении перегруппировок,
происходящих не в самих оксимах, а в их ацильных производных, которые
уже обладают необходимой способностью к ионизации.
Оксим —>
R ОАцил
R'_ C = N
R R НО R
!->+—>- + I —> 1 I —> Амид
R'C = N R'C = N R'C = N
Как установил Кухара, реакция идет через ацильное производное. Работа
Кухара затем была расширена Чапманом [84]. В этих исследованиях приве-
дены доказательства того, что ионизация ацильного производного определяет
скорость перегруппировки. Кухара с сотрудниками показал, что сульфониль-
ные производные оксимов после тщательной очистки претерпевают в ней-
тральном растворителе без добавки кислотных катализаторов быструю пере-
группировку в О-сульфонильные производные амидов. Так, эфир бензосуль-
фокислоты и бензофеноноксима переходит в изомерное соединение, которое
легко гидролизуется в бензанилид и бензолсульфокислоту:
СвН5 СвН5 С6Н5
I I I
C6H5C = N С6Н5 —C = N C6H5CONH + HOSO2C6H5
I I
OSO2CeH5 OSO2C6H5
Кухара и Чапман изучали скорость бекмановской перегруппировки. Куха-
ра показал, что с увеличением силы кислоты скорость изомеризации выделен-
ных эфиров оксимов возрастает. Так, установлен следующий ряд относитель-
ных скоростей перегруппировки в зависимости от природы кислоты:
C6H5SO3H > СН2С1СООН > С8Н5СООН > СН3СООН
Изучая изомеризацию пикриловых эфиров оксимов бензофенона и ацетофено-
на в N-пикрилпроизводные бензанилида и ацетанилида, Чапман нашел, что
скорость процесса сильно зависит от полярности растворителя. Он устано-
вил следующую сравнительную скорость перегруппировки при добавлении
небольших количеств полярных веществ в неполярную среду, например
в четыреххлористый углерод:
CH3CN > CH3NO2 > (СН3)2СО > С6Н5С1 > неполярные растворители
Все сказанное характерно для процессов, скорости которых определяются
ионизацией.
Хьюзген и сотр. [85] показали, что введение электронодонорных заместите-
лей в мигрирующую фенильную группу бензолсульфоната оксима ацетофе-
нона ускоряет перегруппировку, а введение электроноакцепторных замести-
телей замедляет ее. Этот эффект характерен для миграции арила к электро-
нодефицитным центрам в насыщенных нуклеофильных перегруппировках.
616
Перегруппировки в насыщенных системах
Изучая каталитическое действие НС1, Чапман установил, что N-хлоримины
не претерпевают перегруппировки, но что хлористый водород в отличие от
большинства сильных кислот не является простым катализатором: для
осуществления перегруппировки требуется одновременное присутствие ами-
да. В отсутствие первоначально добавленного амида катализ хлористым
водородом носит аутокаталитический характер. Индукционный период,
вероятно, представляет собой время, которое требуется для образования
небольших количеств амида, получающихся в результате перегруппировки,
происходящей под влиянием случайных катализаторов. Чапман высказал
правдоподобное предположение, что при катализе хлористым водородом
претерпевающий перегруппировку эфир оксима превращается в сопряжен-
ную кислоту R2C = NOCR = NHR+, возникающую из имидольного произ-
водного оксима, образующегося при взаимодействии оксима с имидохлоридом,
который в свою очередь раньше получается из хлористого водорода и под-
вергающегося перегруппировке амида:
HC1 Оксим тт+
RCO — NHR-----> C1CR = NR----> R2C = NOCR = NR Катион
Подобным образом можно объяснить каталитическое действие пятихлористого
фосфора; этот эффект можно также объяснить промежуточным образованием
хлорфосфата оксима. Бродский и Миклухин [86] показали, что при после-
довательной обработке бензофеноноксима пятихлористым фосфором и водой
с меченым изотопом кислорода в образующемся бензанилиде, который сам по
себе в условиях опыта не обменивается кислородом с водой, содержится
меченый кислород. Это находится в согласии с механизмом, предположен-
ным Кухара и Чапманом, согласно которому требуется полное отщепление
кислорода из молекулы первоначально взятого оксима.
Уитмор [74] впервые отметил, что к гофмановской перегруппировке амидов
[87] (при действии галогенов и щелочей) в изоцианаты или в продукты их
взаимодействия с компонентами системы, например в амины в присутствии
воды, можно применить общую для всех обсуждаемых перегруппировок схе-
му. Процесс можно изобразить следующим образом:
R R Н R
I -> I / - I-
О=С—NH2 О=С—N O=C-r-N
\ и
Hal
RNH2
O=C=N + СО2
R
Однако детали отщепления галогеноводорода, в результате которого сначала
из-за потери протона, а потом иона галогена образуется промежуточный
одновалентный азот, все еще до некоторой степени гипотетичны.
Главным доказательством приведенного механизма является возможность
осуществления перегруппировки путем обработки основанием первоначально
полученного N-галогенамина и выделение изоцианатов — первых стабиль-
ных продуктов в апротонных условиях. Отсюда следует, что должно про-
изойти отщепление галогеноводорода, которое влечет за собой образование
соединения с одновалентным азотом; при этом, несомненно, происходит
миграция группы. Проссер и Илиел [88] показали, что при проведении пере-
группировки Гофмана в смеси двух амидов, один из которых содержал дей-
терий в остатке R, а другой — 15N, не происходит перекрестного перераспре-
деления меток, т. е. миграция группы происходит внутримолекулярно.
Найдено [89], что реакция ускоряется введением электроноотталкивающих
617
Глава X
заместителей в мета- и^ «ара-положения мигрирующей фенильной груп-
пы (R) и замедляется введением в эти же положения электронопритягиваю-
щих групп; это показывает, что миграционная подвижность подчиняется
правилам, которые применимы к пинаколиновой и другим родственным ей
перегруппировкам, для которых они проверялись. Как показано ниже, име-
ются определенные доказательства внутримолекулярного перемещения групп.
Перегруппировка Лоссеня весьма близка перегруппировке Гофмана: при
разложении ацильных производных, например бензоильных производных
гидроксамовых кислот, предпочтительно с применением оснований в качест-
ве катализаторов, образуются изоцианаты или соответственно продукты
взаимодействия изоцианатов с компонентами системы, например амины
в присутствии воды.
R Н R R RNH2
I / Н I н2о
О=С—N — О=С—-N — O=C=N ----->
\ Ъ
ОАцил СО2
Доказательства механизма по стадиям точно соответствуют тем, которые при-
менимы к перегруппировке Гофмана, но в определенном отношении несколько
расширены [90]. Установлено, что реакция Лоссеня, подобно перегруппиров-
ке Гофмана, ускоряется введением электроноотталкивающих и замедляется
введением электронопритягивающих заместителей в мета- и пара-положения
мигрирующей фенильной группы (R). Кроме того, дополнительно найдено,
что перегруппировка Лоссеня замедляется при наличии электроноотталки-
вающих и ускоряется при наличии электронопритягивающих заместителей
в мета- и пара-положениях отщепляющейся бензоилоксигруппы (ОСОСеН5).
Эти данные служат дальнейшим подтверждением теории, согласно которой
процесс, определяющий скорость, включает гетеролитическое отщепление
OCOR в виде ОСОБ-.
Перегруппировка Курциуса представляет собой несколько более отдаленный
вариант перегруппировок Бекмана и Лоссеня: азиды кислот при нагревании
в растворителях теряют азот (как ацилдиазометаны при перегруппировке
Вольфа), а в результате реакции образуется изоцианат (вместо кетена, полу-
чающегося при перегруппировке Вольфа) или продукты его превращений.
R R R Н2О (RNH2
I + - - Н - I ----------------->
O=C-N=N=N O=C-r-N O=C=N + СО2
Рассмотрим также реакцию Шмидта, являющуюся модификацией разбирае-
мой перегруппировки и заключающуюся в том, что в присутствии серной
кислоты из карбоновой и азотистоводородной кислот образуется ацилазид,
который без выделения подвергают разложению и перегруппировке. После
разбавления водой обычно получают амин, образующийся при последователь-
ной обработке изоцианата серной кислотой и водой. При этом в дополнение
к приведенной выше схеме [91] следует указать на то, что серная кислота
значительно ускоряет разложение ацилазидов. Можно предположить, что
отщепление азота происходит легче в сопряженной кислоте азида, чем в са-
мом азиде. По аналогии с перегруппировкой Бекмана и в этом случае должен
образоваться положительно заряженный атом азота, имеющий во втором
618
Перегруппировки в насыщенных системах
квантовом уровне секстет электронов:
R Н+ R R
I + - -------* I + ---► I-*+ —►
О=С—N=N=N О=С—NH—N=N O=C-t-NH
V
R Н2О (RNH2
' о=с=i * i+ coj
Доказательства механизма перегруппировки Курциуса, включая и модифи-
кацию Шмидта, в основном те же, что и для перегруппировок Гофмана
и Лоссеня. Ботнер-Бай и Фридман [92], используя 15N, показали, что азот,
связанный с углеродом в RCON = N = N, становится связанным с R в про-
дукте перегруппировки. Это приводит к выводу, что миграция R происходит
внутримолекулярно. Кинетическое влияние заместителей в реакции Шмидта
было изучено только для .чета-заместителей в мигрирующей фенильной
группе [931; однако ряд относительных скоростей перегруппировки (СН3 >
>Н > ОН > Cl > NO2) ясно показывает влияние полярности на подвиж-
ность мигрирующей группы, аналогичное найденному для других перегруп-
пировок. Кроме того, как показано ниже, реакция и в стереохимическом
отношении подобна перегруппировкам Гофмана и Лоссеня.
д. Стереохимия нуклеофильных перегруппировок в насыщенных систе-
мах. При рассмотрении общей схемы электронных смещений во всех разби-
R
раемых перегруппировках возникают три следующих вопроса, относящихся
к стереохимии всего процесса: 1) каково влияние на конфигурацию R замены
его связи с А связью с В; 2) каково влияние на конфигурацию В замены его
связи с X связью с R; 3) каково влияние на А замены его связи с R связью
с Y? Такова суть проблемы, хотя в действительности не все эти вопросы
в различных перегруппировках играют решающую роль.
Указанные вопросы будут рассмотрены поочередно. Для удобства будем
называть R — мигрирующей группой, В — конечным местом миграции или
местом диссоциации и А — началом миграции или местом ассоциации.
Стереохимический эффект перегруппировки, поскольку он влияет на кон-
фигурацию мигрирующей группы R, представляет собой вопрос, относящий-
ся ко всем перегруппировкам, в которых R — алкильная или замещенная
алкильная группа, но, конечно, не относится к таким перегруппировкам,
когда R — арильная группа. Ни пинаколиновая, ни перегруппировка Ваг-
нера — Меервейна, ни родственные им реакции не позволяют дать прямого
ответа на этот вопрос. Однако эту проблему можно решить косвенно, исполь-
зуя данные по синартезису (см. следующий раздел), т. е. данные по образо-
ванию общей связи А и В с электронами связи R во время миграции R, кото-
рая не могла бы образоваться, если бы не сохранялась конфигурация R. Кро-
ме того, известны многие перегруппировки бициклических и полицикличе-
ских соединений, которые были бы невозможны, если бы конфигурация
мигрирующего остатка не сохранялась. На поставленный вопрос можно отве-
тить на основании данных по другим перегруппировкам, а именно перегруп-
пировке Вольфа, сопровождающейся перемещением групп от углерода
к углероду, и перегруппировкам Бекмана, Гофмана, Лоссеня, Курциуса
619
Глава X
и Шмидта, при которых происходит перемещение группы от углерода
к азоту. Все перегруппировки к азоту тесно связаны между собой: подтвер-
ждения этому будут приведены в первую очередь.
В 1926—1933 гг. Уоллис с сотрудниками наблюдал сохранение оптической
активности при перегруппировке некоторых производных а-бензилпропионо-
вой кислоты в а-бензилэтиламин по Курциусу [94], Гофману [95] и Лоссе-
ню [96]. Браун и Фримельт [97] отметили, что это положение справедливо
и для реакции Шмидта. Оптическое вращение указывает на то, что стерео-
химический эффект четырех процессов одинаков. В 1935 г., когда Кеньон
и Филлипс создали метод определения относительных конфигураций по зна-
ку вращения (гл. VII, разд. 7, б), а Питман [98] удачно применил этот метод,
стало совершенно ясно, что общий стереохимический результат, наблюдав-
шийся Уоллисом и Брауном, свидетельствует о сохранении конфигу-
рации.
свн5сн2 лтт ч свн5сн2
। fПерегруппировки ; ।
СН3 — С — СО —> < Гофмана, Лоссеня, > —> СН3 — С — NH2 + CO2
I I (Курциуса, Шмидта; I
В 1940 г. это заключение получило дальнейшую поддержку в обзорной работе
Арчера [99], относящейся, в частности, к перегруппировке Гофмана. Моно-
амид 0-камфорной кислоты должен быть г{мс-соединением вследствие его
связи с камфорной кислотой и, следовательно, с камфорой. 1-Аминодигидро-
а-камфолитовая кислота, получаемая из моноамида по Гофману, также дол-
жна быть zfwc-соединением вследствие ее легкого превращения в лактам.
Следовательно, при реакции Гофмана конфигурация сохраняется.
В 1941 г. Кеньон и Янг [100] включили перегруппировку Бекмана в упомяну-
тое выше обобщение. Они превратили оптически активную 3-н-гептанкарбоно-
вую кислоту в 3-н-гептилметилкетон; затем по методу Гофмана перегруппи-
ровали амид этой кислоты, а по методу Бекмана — оксим кетона. Оба про-
цесса ведут к одному и тому же оптическому изомеру 3-н-гептиламина.
Поскольку перегруппировка Гофмана не сопровождается изменением конфи-
гурации, то, следовательно, это положение относится и к перегруппировке
Бекмана.
Амид, по
( + )-к-С4Н9(С2Н5)СНСООН Гофма^> ( + )-«-C4H9(C2H5)CHNH2
Ацилбромид,
реакция с
Cd(CH3)2
Ацетилирование
Гидролиз
Оксим, по
( + )-и-С4Н9(С2Н5)СНСОСН3 —--—> (—)-m-C4H9(C2H5)CHNHCOCH3
Бекману
В 1946 г. Кеньон с сотр. [100, 101] на примере а-фенилэтильных соединений
изучил пять родственных перегруппировок и на основании данных о степени
620
Перегруппировки в насыщенных системах
оптической чистоты (см. величины ниже) показал, насколько полно в каждом
случае сохраняется асимметрия * [R = С6Н5(СН3)СН]
( RC( = NOH)CH3 (Бекман) — > ( —)-RNHCOCH3 (99,6%)
I RC0NH2 (Гофман) — » ( —)-RNH2 (95,8%)
(-H-RCOOH _ » J RCONHOCOC6H5 (Лоссень) — > (-)-RNH2 (99,2%)
RCON3 (Курциус) — » (-)-RNH2 (99,3%)
[ RCON3 (Шмидт) — » (—)-RNH2 (99,6%)
Перегруппировка Вольфа — единственная из обсуждаемых перегруппировок,
в которой происходит перемещение к углероду. Лейн и Уоллис [102] показа-
ли, что она также протекает с сохранением конфигурации. Они превратили
а-к-бутилгидротроповую кислоту в следующий высший гомолог методом
Арндта — Эйстерта, который, как было показано выше, включает перегруп-
пировку Вольфа. Затем Лейн и Уоллис превратили полученную кислоту
в исходный низший гомолог по методу Барбье — Виланда, заключающемуся
в действии на эфиры кислот реактива Гриньяра и окислении образующегося
при этом третичного спирта. Полученная таким образом кислота оказалась
идентичной первоначально взятой (хотя карбоксильная группа имеет другое
происхождение); сохранение оптической чистоты по завершении цикла ока-
залось равным 99,5%.
С6Н5 СеН5 С6Н5 СООН
\ гппп Хлорангидрид + \ Перегруппировка \
н-СдНп — С — СООН-------------> н-СдНо—С — СО --------------> н-СлНч —С —сн2
/ +CH2N2 / | Вольфа /
СН3 СН3 CHN2 СН3
СгОз
CgHg С(С6Н5)2ОН
\ I
Н-С^Нд—С — СН2
СНз
Метиловый эфир
+C6H5MgBr
В работах Кеньона и Уоллиса была достигнута высокая степень сохранения
асимметрии, и это приводит к строгому выводу о том, что мигрирующая
группа не покидает системы. Такое положение было подтверждено создани-
ем условий, при которых миграция групп из системы при условии, что она
происходила на атомных расстояниях за время, равное продолжительности
столкновения, снимала бы заторможенность вращения, и оптическая а тив-
ность, обусловленная отсутствием свободного вращения, должна была бы
быть потеряна. Уоллис и Мойер [103] показали, что при гофмановской пере-
группировке замещенного о-а-нафтилбензамида этого не происходит; Белл
[104] установил, что это явление не наблюдается при перегруппировках заме-
щенного о-фенилбензамида и соответствующего бензоилазида по Гофману
и Курциусу. Далее Лейн и Уоллис [105], используя соответствующий заме-
щенный диазоацетофенон, доказали, что оптическая активность не исчезает
и при перегруппировке Вольфа. Исходные вещества имели следующее
* Наблюдаемую незначительную рацемизацию, сопровождающую гофмановскую пере-
группировку гидротропамида, можно объяснить применением в этом случае водной
щелочи — мощного рацемизующего агента для прототропных соединений с подвижным
водородом в центре асимметрии, какими являются во всех перегруппировках исходные
вещества, так же как и промежуточно образующийся изоцианат. Другие реакции не свя-
заны с применением сильнощелочных агентов.
621
Глава X
строение:
Г офман
---•---------------ч Курциус Вольф
NO2 (Ьелл)
(Лейн и Уоллис)
(Уоллис и Мойер)
Второй из поставленных вопросов относится к конфигурационному эффекту
при перемещении группы к конечному месту миграции. Химически это важ-
но только в том случае, если в соответствии с принятыми обозначениями
ковалентность (лигандность) В не может стать меньше трех для углерода
и меньше двух для азота. Сюда относятся три важных случая: пинаколиновая
перегруппировка и близкие к ней процессы; реакция Вагнера — Меервейна
и другие близкие реакции; перегруппировка Бекмана (табл. 162).
Что касается пинаколиновой и родственных ей перегруппировок, то эта
проблема решалась двумя методами. Бартлет и сотр. [105] применили цикли-
ческие стереоизомерные соединения с ограниченной возможностью переме-
щения и отщепления группы лишь в определенные характерные положения;
в этих условиях сравнивалась легкость реакции. Они установили, что цис-
1,2-диметилциклогександиол-1,2 при обработке 20 %-ной серной кислотой
чрезвычайно гладко переходит в 2,2-диметилциклогексанон-1, а цпс-1,2-
диметилциклопентандиол-1,2 таким же образом превращается в 2,2-диметил-
циклопентанон-1. В то же время транс-1,2-диметилциклогександиол-1,2
в тех же условиях дает 1-ацетил-1-метилциклопентан, а транс-1,2-диметил-
циклопентандиол-1,2 превращается в смолу. Как цис-, так и транс-1,3-
дифенилаценафтендиол-7,8 при обработке водным раствором серной кислоты
дает 8,8-дифенилаценафтенон-7; однако реакция идет лучше в случае цис-
изомера. В условиях, в которых транс-диол претерпевает перегруппировку,
происходит предварительная изомеризация в цис-диол
Следовательно, пинаколиновая перегруппировка протекает более гладко
в том случае, когда мигрирующая группа R подходит к конечному месту
миграции В со стороны, противоположной той, из которой уходит группа X,
чем когда она подходит с той же стороны. Это возможно только при реакци-
ях с инверсией в конечном месте миграции.
Обсуждаемый вопрос можно решить и другим методом: атакуя В из любого
положения при асимметрическом В и сопоставляя его конфигурацию до
622
Перегруппировки в насыщенных системах
и после перегруппировки. Бернстейн и Уитмор на основании данных Мак-
Кензи и сотрудников применили этот метод к дезаминированию. Исходя из
оптически активного аланина, Мак-Кензи, Роджер и Уиле [107] приготовили
при помощи синтеза Гриньяра приведенный ниже оптически активный амино-
спирт и затем азотистой кислотой в уксуснокислом растворе дезаминировали
его в оптически активный кетон. Этот же кетон в оптически активной форме
впоследствии был синтезирован Конантом и Карлсоном [108] методом Гринь-
яра из хлорангидрида гидротроповой кислоты. Исходный продукт Мак-
Кензи, Роджера и Уилса — аланин — был синтезирован Лейсом [109]
путем окисления фенильной группы в оптически активном а-фенилэтиламине
при защищенной аминогруппе. Оставалось завершить генетический цикл,
связав гидротроповую кислоту с а-фенилэтиламином; это сделали Бернстейн
и Уитмор [110], превратив по реакции Курциуса кислоту в амин. Как пока-
зано на схеме, этот генетический цикл превращений оптически активных
веществ гладко замыкается (в скобках даны начальные буквы фамилий ис-
следователей):
HNOa в холодной
(~НСвН5)2С - СНСНз СНзСООН (М р7> ( + )-СвН5СОСН(СН3)С,Н5
он nh2
Действие реактива '' Действие реактива ''
Гриньяра на слож- Гриньяра на хлор-
ный офир (М. Р. У) ангидрид (К. К.)
(+)-НООС-СН(СПз)С6Н5
Курциус
(Б. У.)
Окисление бензоль-
( + )-H2NCH(CH3)COOH <----------------— (-)-H2NCH(CH3)CeH5
' 1 ' ' ного производного (Л.) ' V а/ о о
Важность этого цикла превращений легко видеть, так как только в двух
случаях, когда меняется связь в асимметрическом центре (а именно при
дезаминировании и реакции Курциуса), новая группа занимает положение,
которое занимала старая. Так, исходя из аланина (1) через стадии дезамини-
рования (2), (3) и реакции Курциуса (4), (5) снова приходят к аланину (6)
(1)
(2)
(3)
(4)
(5)
(6)
nh2
CH3CHZ
\соон ) ,
? Гриньяр
/NH2 J *
СН3С1Г
XC(C6H5)2OH 1
? Дезаминирование I
/С6Н5 ) *
CH3CHZ
\сос6н5 ) „ А
” ° I Гриньяр
/С6Н5 / 1
CH3CHZ
\соон ) .
NH2 J Курциус 1
CH3CHZ
\с6Н5 ) „ I
У Окисление
ЛН2 >
CHgCff
\соон
623
Глава X
При этом две группы меняются местами, что должно привести к инверсии;
следовательно, генетический цикл не должен замкнуться. Тот факт, что на
самом деле генетический цикл замыкается, показывает, что дезаминирование
или реакция Курциуса также сопровождаются инверсией. Но так как опре-
деленные доводы (см. выше) свидетельствуют, что реакция Курциуса не
вызывает инверсии, то следует сделать вывод, что последняя происходит при
дезаминировании. Кроме того, из наблюдений Мак-Кензи, Роджера и Уилса
можно сделать вывод, что инверсия протекает не количественно. Эти иссле-
дователи выделили перегруппированный кетон почти с количественным выхо-
дом, но он сохранил оптическую чистоту лишь на 77 % или, другими словами,
произошла инверсия на 88%. Ни исходный, ни конечный продукты не могут
претерпевать рацемизации при действии реагента, и, следовательно, рацеми-
зация наступает в процессе превращения. Общий вывод: пинаколиновое
дезаминирование вызывает в конечном месте миграции главным образом
инверсию наряду с некоторой рацемизацией.
Причина частичной рацемизации, т. е. некоторый вклад миграции с сохране-
нием конфигурации у конечного места миграции, была выяснена в результате
ряда последующих исследований. Во-первых, на многих примерах было уста-
новлено, что в структурах, содержащих менее подвижную группу в стерео-
химически более благоприятных условиях по отношению к конечному месту
миграции, пространственная специфичность замещения в этом месте может
привести к кажущемуся исключению из правила миграционной подвижно-
сти. Этим, несомненно, объясняются результаты изученного Мак-Кензи
и Милсом [111] случая дезаминирования, приведенного ниже и рассматри-
вавшегося как исключение из правила полярности. Так, Поллак и Кертин
[112] недавно подтвердили, что фенильный радикал почти всегда мигрирует
предпочтительно перед га-анизильным
С6Н5 С6Н5 С6Н5
\ / hno2 /
С — С ---------> п-СН3ОС6Н4СОСН
п-СН3ОС6щ| ''свЩ
он nh2
Следует отметить, что поскольку имеются два асимметрических центра, то
существуют два рацемических диастереомерных аминоспирта. Один из них
был синтезирован из дезиламина и анизилмагнийбромида. Впоследствии Мак-
Кензи и Вуд [ИЗ] показали, что оба диастереомерных аминоспирта при деза-
минировании могут перегруппировываться различным образом
С6Н5 С6Н5
С — С—н
с10н71 I
он nh2
hno2
0-Форма----->
hno2
а-Форма----->
сен5
с6н5
С6Н5СОСН
С10Н7
С1ОН7 —
нафтил
Такие пары диастереомеров получаются путем двухстадийного гриньяров-
ского синтеза аминоспиртов из эфиров аминокислот через аминокетоны.
Общий синтез протекал стереоспецифично на 98—99%. Миллс и Поллак
и Кертин получили таким образом множество диастереомерных пар амино-
спиртов и показали, что в любом случае предпочтительно мигрирует та
арильная группа, которая была введена при действии первого из двух
624
Перегруппировки в насыщенных системах
арильных реагентов Гриньяра *. Используя это принцип, Бенджамин, Шеф-
фер и Коллинз [114] с помощью двухстадийного гриньяровского синтеза
с фенилмагнийбромидом получили из каждого энантиомера аланина дифенил-
карбиноламин Мак-Кензи, Роджера и Уилса (стр. 623). При этом сначала
первая, а затем вторая фенильная группы были помечены изотопом 14С.
В обоих случаях подтвердились данные Мак-Кензи, Роджера и Уилса,
согласно которым при дезаминировании миграция фенильной группы про-
исходит на 88% с обращением и на 12% с сохранением конфигурации у конеч-
ного места миграции. Затем инвертированный продукт был отделен от раце-
мического продукта и путем расщепления было установлено положение
радиоактивной метки. В результате этого удалось выяснить, что продукт,
образование которого происходит с инверсией конфигурации (выход 88%),
образуется только путем миграции фенильного остатка, который был введен
в молекулу первым (при синтезе Гриньяра), а остальные 12% продукта,
образование которого происходит с сохранением конфигурации, образуются
только путем миграции второй фенильной группы [114].
Эти результаты можно объяснить на основе ряда принципов, уже выведен-
ных на независимых основаниях. Во-первых, согласно общей электронной
схеме, развитой и описанной в разд. 1,в и вновь процитированной в начале
этого раздела, процесс отщепления X и присоединения R к конечному месту
миграции В является нуклеофильным замещением, поскольку и входящая
и уходящая группы перемещаются вместе со своими электронами связи. Если
изменение связей происходит достаточно синхронно и поэтому локальный
процесс носит характер S х2-замещения (хотя общий процесс замещения
относится к SN1-Tnny), то стереохимическим результатом реакции должна
быть инверсия конфигурации у конечного места миграции. Однако, если
вначале отщепляется X, т. е. локальный процесс подобен 8х1-замещению,
то будет происходить рацемизация, степень которой будет зависеть от того,
насколько далеко успеет продиффундировать X- перед началом стадии ми-
грации (полная рацемизация произойдет, когда X- успеет удалиться на рас-
стояние, превышающее радиус действия экранирующих сил). Во-вторых,
в гл. VII, разд. 7,и, и в гл. VIII, разд. 1,в указывалось, что стереохимия
реакций дезаминирования соответствует общему 8х1-механизму, т. е. отщеп-
ление X (в данном случае NJ) происходит без участия постороннего нуклео-
фила. Кроме того, необходимо вспомнить тот вывод, который был сделан
в разд. 1,6; образующийся при отщеплении карбониевый ион вначале не
полностью сольватирован, т. е. находится в высокореакционноспособной
форме, в которой может происходить миграция группы до момента достиже-
ния конформационного равновесия.
Данные Бенджамина, Шеффера и Коллинза суммированы на приведенной
ниже схеме. Слева приведена проекция Ньюмена для диазониевого иона,
образующегося из одного из диастереомерных меченых аминоспиртов. Кри-
вые стрелки в этой формуле показывают, что, если миграция меченной 14С
фенильной группы и отщепление NJ мало разделены во времени, будет обра-
зовываться инвертированный продукт и что этот инвертированный продукт
образуется за счет миграции только той фенильной группы, которая была
меченой. Если в другом крайнем случае образующийся карбониевый ион
имеет очень долгое время свободного существования — в частности, доста-
точно долгое по сравнению со временем, требующимся для конформацион-
ного вращения, то будет образовываться рацемический продукт и, более
* В первом издании этой книги были приведены известные примеры таких превраще-
ний; все эти реакции очень сходны с реакциями, приведенными выше.
40-0772
625
Глава X
того, карбониевые ионы, образовавшиеся из каждого из энантиомеров, будут
представлять собой равновесную смесь двух изомеров положения, в одном из
которых будет происходить миграция обычного, а в другом — меченого
фенильного остатка. Как отметили Бенджамин, Шеффер и Коллинз, в их опы-
тах такая ситуация не возникала: напротив, нужно предположить, что время
жизни образующегося карбониевого иона мало по сравнению со временем
конформационного вращения. Поэтому карбониевый ион реагирует в той
конформации, которая близка к конформации иона, только что образовав-
шегося из иона диазония (см. схему). Этот карбониевый ион нарисован
дважды, чтобы можно было (с помощью кривых стрелок) указать два пути его
распада. Главный продукт реакции будет иметь инвертированную конфигу-
рацию и будет содержать только тот изомер, который соответствует миграции
меченой фенильной группы. Второй продукт будет иметь сохраненную кон-
фигурацию и будет содержать только другой изомер положения, соответ-
ствующий миграции немеченой фенильной группы.
Мигрирует меченый СвН5,
конфигурация инверти-
руется (88%)
с6н5
Мигрирует немеченый С6Н5,
конфигурация сохраняется
(12 %)
Другой случай, который следует обсудить,—перегруппировка Вагнера —
Меервейна. Примером может служить превращение изоборнилхлорида
в камфен. Как ясно из формул, приведенных на стр. 605, углеродный атом,
теряющий хлор, образует новую связь со стороны, противоположной той,
которую раньше занимал хлор. Действительно, не только изоборнилхлорид,
но и борнилхлорид в ионизующих растворителях и в присутствии оснований,
сила которых больше, чем у воды, превращается в камфен. Но превращение
изоборнилхлорида протекает значительно легче, чем превращение борнил-
хлорида, где новая связь должна образоваться со стороны углеродного атома,
от которого отщепляется хлор. Локальный процесс у конечного места мигра-
ции может иметь характер S х2-замещения только в реакции изоборнилхлори-
да, поэтому только в этом случае происходит обращение конфигурации.
Показано, что при дезаминировании оптически активного сс-дейтеронеопентил-
амина в результате происходящей одновременно миграции метильной группы
наблюдается обращение конфигурации при конечном месте миграции [115].
Из сказанного следует, что при перегруппировке Вагнера — Меервейна
инверсия в конечном месте миграции является преобладающей формой
реакции.
Последний из рассматриваемых примеров — бекмановская перегруппировка
оксимов. Эта перегруппировка представляет собой яркий пример ошибочно-
626
Перегруппировки в насыщенных системах
сти структурных выводов, сделанных без учета механизма реакции. Ганч
и Вернер [116], предложившие в 1890 г. геометрическую интерпретацию
изомерии оксимов, пользовались для определения конфигурации изомеров
двумя методами. В случае альдоксимов была использована легкость превра-
щения ацильных производных только одного из изомеров в соответствующий
нитрил при действии оснований. В случае кетоксимов они основывались на
различии в продуктах бекмановской перегруппировки. Во всех случаях
основное допущение заключается в том, что отщепившиеся или обменявшиеся
местами группы расположены в цис-положении. (СН3СО)2О К2СОз R —С —Н > R —С —H > RCN II II ит-д' N-OH N — ОСОСН3 РС15 R — C-R' > R-C-OH Tt R —С = О II ит-д' II 1 N —ОН N— R' NH— R'
Схемы Ганча и Вернера для установления конфигураций
На основании изложенного принципа в течение 30 лет относили конфигура-
ции большого числа оксимов. Однако Мейзенгеймер [117] опроверг это для
кетоксимов, получив при расщеплении изоксазола «неправильный» стерео-
изомер; озонирование трифенилизоксазола приводит к бензоилбензил-р-моно-
оксиму, а р-монооксим является тем изомером, который претерпевает пере-
группировку Бекмана, давая фенилглиоксанилид
С6Н5-С-С-С6Н5 С6Н5СО —С —с6н5
II II Оз II
CeH5-C N -----------> С6Н5СОО —N
''о7'
C6H5COCI
<-------
КОН
С6Н5СО —С —С6Н5
и
НО —N
РС15
---->
С6Н5СО-С—ОН С6Н5СО-С=О
II I
С6Н5—N C6H5-NH
Несколько позднее Бреди и Бишоп [118] подобным же образом опровергли
конфигурацию, принятую для альдоксимов, показав, что из двух изомеров
«неправильный» легче превращается в изоксазол. Так, например, та форма
2-хлор-5-нитробензальдоксима, ацильное производное которого дает нитрил,
легко претерпевает превращение при действии щелочей, вероятно, через
нитробензизоксазол, который в данных условиях переходит в нитросали-
цилонитрил
C6H3(NO2)-C-H
I II
Cl НО —N
Параллельным изучением циклизации Мейзенгеймер [117] подтвердил непра-
вильность ранее принятых конфигураций кетоксимов. Форма 2-хлор-5-
нитробензофеноноксима, которая при бекмановской перегруппировке дает
хлорнитробензанилид (с перемещением С6Н5), является той формой, которая
при действии щелочи превращается в нитрозамещенный фенилбензизоксазол
CeH3(NO2)C- свн5
С1 Н0-Г4
40* 627
Глава X
Из этих наблюдений можно сделать два вывода. Первый состоит в том, что
предпочтительной формой отщепления в случае образования нитрила из
оксима, как и вещества с тройной связью из олефинов (гл. IX, разд. 4,а),
является транс-элиминирование. Второй вывод, ближе относящийся к обсу-
ждаемому вопросу, заключается в том, что при бекмановской перегруппиров-
ке мигрирующая группа расположена по отношению к атому азота со сто-
роны, противоположной той, от которой уходит кислородсодержащая
группа.
Третий стереохимический вопрос относится к влиянию перегруппировки на
конфигурацию начального места миграции А. Этот вопрос важен только
тогда, когда А четырехковалентен как до, так и после перегруппировки.
Единственным важным случаем такого рода является перегруппировка Ваг-
нера — Меервейна при замещении, включающая особый вид замещения
отщепленной группой, т. е. изомеризацию.
Ответ на этот вопрос вытекает из формул, уже приведенных для некоторых
из этих превращений (стр. 605, 606). Метиловый эфир камфена и камфенгид-
рохлорид непосредственно превращаются в изоборнилхлорид. В этом экзо-
изомере атом хлора по отношению к циклу находится со стороны, противо-
положной той, где ранее находилась переместившаяся связь. Пиненгидро-
хлорид непосредственно превращается в борнилхлорид; при этом образуется
эндо-изомер, в котором атом галогена также находится со стороны кольца,
противоположной той, где ранее находилась переместившаяся связь.
Эти примеры показывают, что для перегруппировки Вагнера — Меервейна.
сопровождающейся замещением и изомеризацией, как правило, характерна
инверсия в начальном месте миграции. Другим подтверждением может слу-
жить заметная обратимость превращения камфенгидрохлорида в изоборнил-
хлорид [66]. Отсюда вытекает предположение, что перегруппировки Вагне-
ра— Меервейна, как правило, обратимы. Так как при обратной реакции в пер-
вом процессе конечное место миграции становится начальным, то на основа-
нии принципа микроскопической обратимости можно ожидать, что как
к конечному, так и к начальному месту миграции применимы одинаковые
стереохимические правила. Действительно, детальное исследование под-
тверждает, что у обоих центров инверсия — предпочтительная форма реак-
ции. Результаты всех рассмотренных стереохимических исследований можно
суммировать следующей общей формулой:
R
у А преобладает инверсия,
А—В у R — сохранение конфигурации,
гг К у В преобладает инверсия
Ly х*
е. Ионные промежуточные соединения. Ненасыщенные мигрирующие
группы. Циклизация. Природа промежуточных частиц в нуклеофильных
алифатических перегруппировках — карбониевых ионов — имеет множест-
во различных аспектов *: эти частицы будут рассмотрены в этом и последую-
щих двух разделах. Рассмотрение удобно начать с работ, посвященных
вальденовскому обращению (гл. VII, разд. 7, особенно разд. 7,ж и з). В этих
* Эта область была предметом длительной полемики. Вместо того чтобы прослеживать
запутанную дискуссию, мы просто ее опустим и представим общую картину, которую
автор считает верной. Автору очень помогла книга Бартлета «Неклассические ионы»
<W. A. Benjamin, Inc., New York, 1965), которая содержит перепечатку 75 оригиналь-
ных статей и дополнительные комментарии и является ценным руководством в этой
области.
ш
Перегруппировки в насыщенных системах
разделах указывалось, что 8х1-замещение протекает с сохранением конфи-
гурации, когда p-заместитель, т. е. у-атом, обладает р- или л-электронами,
с помощью которых он может координироваться с образовавшимся или обра-
зующимся карбониевым ионом, в результате чего возникает трехчленный цикл,
в котором новая связь часто довольно слаба. Если на конечной стадии заме-
щения происходит раскрытие этого цикла с разрывом новой связи, то наблю-
дается замещение без перегруппировки, но с сохранением конфигурации.
Однако некоторые циклические катионы могут переходить в такое состояние
(не обязательно наиболее стабильное), в котором старая и новая связи у-
атома симметричны; в этом случае цикл может раскрыться с любой стороны.
При этом наблюдается как замещение без перегруппировки с преобладаю-
щим сохранением конфигурации, так и замещение с перегруппировкой
и значительной инверсией конфигурации у конечного места миграции.
Рассмотренное ранее (гл. VII, разд. 7,з) превращение оптически активного
трео-2-бромбутанола-З под действием бромистого водорода в рацемический
2,3-дибромбутан проходит через промежуточное состояние, в котором атом
брома, первоначально связанный с положением 2, находится в плоскости
симметрии, и вследствие этого происходит его миграция в степени, близкой
к 50%. Вначале удобнее рассмотреть аналогичный пример, изученный Кра-
мом. При сольволизе оптически активной эритро-формы З-фенилбутил-2-
и-толуолсульфоната в уксусной кислоте образуется активный эритро-З-
фенилбутил-2-ацетат подобной конфигурации, а при сольволизе оптически
активной трео-формы толуолсульфоната образуется рацемическая трео-
форма ацетата. Это можно объяснить тем, что в случае эритро-изомера в кати-
онном промежуточном соединении фенильная группа располагается на вра-
щательной оси симметрии второго порядка, а в случае трео-изомера — в пло-
скости симметрии. В каждом случае степень миграции фенильной группы
составляет 50% [119].
СН3
I .-С6н5
с——
I TsO’
н
(,2S)-3pumpo
ГСН3 Н 1+
I ,'СвН<1
с------с
СН3 |_н
сн.
СН3 н
UCeH5 I
С-- *с
I АсО
Н
сн3
.с—
АсО*<
сн3 н
(2S)-3/mmpo
Н
1,-СвН5
с
I
сн3
сн3 сн3
c"CeHs
LH
(,2R)-mpeo
СН3 СН, СН, СН.
I^CSH5 | |
с------>с + с-
I Acer I Ас О<|
Н Н Н
(2R)-mpeo
(2S)-mpea
Н
Возникает вопрос, что же представляют собой ионные частицы с трехчленным
циклом? Являются ли эти частицы переходными состояниями обратимых
взаимопревращений двух изомерных карбониевых ионов (как указано слева
на приведенной ниже схеме), в одном из которых заряд локализован на
начальном, а в другом — на конечном месте миграции? Или, может быть,
такие частицы представляют собой истинные промежуточные соединения,
в которых имеется значительный перенос заряда и его мезомерная дело-
кализация? На приведенной ниже схеме справа представлена классическая
формула такого промежуточного соединения, обоснованная тем, что миграция
фенильной группы представляет собой внутримолекулярное электрофильное
ароматическое замещение, в процессе которого образуется карбониевы й
629
Глава X
ион. Первой стадией любой такой реакции является присоединение с образо-
ванием иона циклогексадиенилия, который во многих случаях может быть
выделен в виде солей; поэтому можно ожидать, что ион циклопропанспи-
роциклогексадиенилия будет достаточно стабильным (гл. VI, разд. 5,в,
6,в и г).
На эти вопросы нельзя ответить в общем виде, так как ответ зависит от каж-
дого конкретного примера, в частности, от степени распределения ионного
заряда на начальном и конечном месте миграции, с одной стороны, и на
мигрирующей группе — с другой.
Боннер и Коллинз [120] изучили катализируемую кислотами обратимую
изомеризацию 1.2,2-трифенилэтилацетата в уксусной кислоте с тройной
изотопной меткой
(С6Н5)2СН-СНС6Н5 С6Н5СН-СН(СвН5)2
I 1
ОСОСНз ОСОСНз
Они установили, что метка, первоначально находившаяся в одной фенильной
группе, равномерно распределяется по всем трем группам со скоростью,
равной скорости распределения метки на одном из этановых атомов углерода
между обоими атомами этой группы, а также равной скорости обмена мече-
ной ацетатной группы с ацетатными группами среды. Такая относительно
высокая скорость перераспределения метки между фенильными группами
не может быть объяснена с помощью предположения о промежуточном
образовании циклогексадиенилия, который образуется при каждом переносе
фенильной группы. Наблюдаемое соотношение между скоростями перехода
трех меток можно объяснить, только предположив, что после ухода ацетат-
ной группы, т. е. в карбониевом ионе, происходит множество переходов
фенильной группы в прямом и обратном направлениях. Таким образом, при
такой перегруппировке образуются два промежуточных иона карбония,
в одном из которых заряд находится на конечном, а в другом — на началь-
ном месте миграции; равновесие между этими ионами устанавливается быст-
рее, чем происходит их исчезновение путем захвата ацетатного иона. В этом
случае предпочтение следует отдать структурам, в которых заряд сохраняет-
ся или в начальном или в конечном месте миграции; оба эти центра принад-
лежат к бензильному типу, и поэтому заряд на каждом из них в действитель-
ности рассредоточен по соседним фенильным группам, что стабилизирует
катион (мезомерный эффект).
В работе Касерио, Левина и Робертса [121] предпочтение было отдано про-
тивоположной точке зрения. Указанные авторы работали с системой, не
содержавшей других ненасыщенных групп, кроме единственной мигрирующей
фенильной группы. Кроме того, для дополнительной локализации положи-
тельного заряда (с образованием оксониевой структуры) в фенильное кольцо
был введен кислородсодержащий заместитель. С помощью специального дат-
чика авторы показали, что в этой системе происходит образование промежу-
точного циклопропанспироциклогексадиенилия с относительно большим
временем жизни. В качестве субстрата использовался 2-и-(со-оксифенэтил)этил-
амин, который дезаминировался азотистой кислотой. В положение 1
630
Перегруппировки в насыщенных системах
этиламиногруппы вводилось два атома дейтерия, переход которых в положе-
ние 2 служил тестом на перегруппировку Вагнера — Меервейна. В ©-поло-
жение оксиэтильной группы также были введены два атома дейтерия, рас-
пределение которых между двумя атомами углерода этой группы служило
тестом на перегруппировку этой группы, происходящую через обратимое
образование циклического ацеталя в карбониевом ионе, претерпевающем
перегруппировку Вагнера — Меервейна. Перегруппировка оксиэтильной
группы может происходить только в том случае, если карбониевый ион
существует достаточно долгое время, в течение которого может образоваться
циклический ацеталь. Оказалось, что обе пары атомов дейтерия перераспре-
деляются между двумя соседними атомами углерода. Это показывает, что
циклопропанспиро-ион существует достаточно долгое время, в течение
которого завершается другая реакция, а именно образование и разрыв
этиленацеталя. Таким образом, спиро-ион в этом случае является истинным
промежуточным соединением, а не симметричным переходным состоянием
реакции изомеризации карбониевых ионов.
I
он
Логичным завершением этой идеи является применение заместителя О~,
при наличии которого обычный промежуточный катион превращается в ней-
тральный циклогексадиенон. Бэрд и Уинстейн [122] при превращении 2-п-
оксифенилэтилбромида в метиловый эфир под действием раствора метилата
натрия в метиловом спирте смогли спектрофотометрически следить за воз-
растанием и уменьшением концентрации промежуточно образующегося
циклопропанспироциклогексадиенона; кроме того, его удалось выделить.
Максимальной стабильностью это соединение обладало при pH 12, однако
при pH выше 14 происходило быстрое присоединение метилат-иона
о-
СН2СН2Вг
ж. Ионные промежуточные соединения. Насыщенные мигрирующие
группы. Синартезис. Перегруппировки Вагнера — Меервейна и пинаколи-
631
Глава X
новая большей частью связаны с миграцией групп, не имеющих р- или л-
электронов, т. е. с миграцией насыщенных групп типа метильной или выс-
ших алкильных, а также атома водорода, имеющих лишь о-электроны.
В конце 1930-х годов было замечено, что, несмотря на мономолекулярный
характер общей реакции, скорость которой контролируется стадией иониза-
ции, при таких перегруппировках, так же как и при перегруппировках
с миграцией ненасыщенных групп, локальный процесс у конечного места
миграции согласуется с одним из двух типов реакций. Он может соответ-
ствовать типу SN1, когда ионизация происходит перед образованием пере-
ходного состояния, через которое осуществляется миграция группы
R R R
I -» I + -» + I
Л —В—X А—В А—В
или типу Sn2, когда миграция и ионизация происходят совместно
R R
I —> + I
А —В —X А-В
Эти процессы имеют два наблюдаемых отличия. Первое отличие является
стереохимическим: при локальном процессе типа SN1 в большей или меньшей
степени должна наблюдаться рацемизация, а при локальном процессе типа
Sn2 —стереоспецифическая инверсия у конечного места миграции. Второе
отличие является кинетическим: при 8к1-реакции контролирующий ско-
рость гетеролиз, не ускоряясь внутримолекулярным взаимодействием, должен
иметь такую же скорость, как и гетеролиз той же связи с тем же локальным
окружением в более простых молекулах, в которых не происходит перегруп-
пировки. При реакции же типа SN2 гетеролиз будет ускоряться благодаря
участию в процессе мигрирующей группы. Такие аномально быстрые пере-
группировки уже давно известны в ряду терпенов. Возникает вопрос, что
является, как часто говорят, «движущей силой» миграции насыщенных
групп, например метильной или высших алкильных? Считается, что это
обусловлено образованием иона, более стабильного, чем каждый из сле-
дующих:
R R
+ I I +
А—В или А — В
Стабилизация обусловлена резонансом между этими двумя каноническими
структурами, т. е. делокализацией мигрирующих сг-электронов между двумя
атомами, между которыми распределяется и положительный заряд.
R
А---В
Впервые эта идея появилась в 1940 г. [123]. Она была эффективно исполь-
зована в 50-х годах, особенно в начале важных исследований Уинстейна
и Робертса [124], к которым мы еще вернемся. В этот период это предположе-
ние обсуждалось Хьюзом [125], Уинстейном [125], а также Хьюзом, Брау-
ном, Смитом и автором [126], особенно в связи с кинетическими эффектами
миграции, связанной с ионизацией. Последние авторы указали на аналогию
между делокализацией п-связей в карбониевых ионах и в соединениях бора
и предложили называть такую делокализацию п-связей «синартезисом»
632
Перегруппировки в насыщенных системах
(совместным ускорением) в противоположность делокализации л-связей,
называемой «мезомерией» *.
Для иллюстрации характера кинетики перегруппировок, включающих заме-
щение типа SN1 у места диссоциации, вначале рассмотрим сольволиз неопен-
тилбромида, в результате которого образуются mpem-амиловый спирт и дру-
гие продукты, содержащие mpem-амильную группу:
(СН3)3ССН2Вт _> (CH3)2C(OR)CH2CH3 и др.
Скорость отщепления иона галогена от этого первичного бромида по реакции
первого порядка примерно равна скоростям протекающих в таких же усло-
виях мономолекулярных реакций наиболее простых первичных бромидов,
например этилбромида **:
Гидролиз при 95 °C во влажной ( Этилбромид, Ац = 2,70-10~’с-1
НСООН t Неопентилбромид, Ад = 1,53-10”’с”1
Очевидно, мигрирующая метильная группа не способствует отщеплению
галогена несмотря на то, что миграция должна происходить вскоре после
отщепления галогена и прежде чем с карбониевым ионом (даже первичным)
успеет прореагировать растворитель. Этот вывод основан на том, что ни
в одном случае не было обнаружено большого количества неперегруппирован-
ного продукта, однако этот вывод был сделан еще до разработки хроматогра-
фического метода [122]. Сольволиз пинаколилхлорида также ведет исключи-
тельно к продуктам перегруппировки
(СН3)3ССНС1СН3 _» (CH3)2C(OR)CH(CH3)2 и др.
Этим иллюстрируются те же самые отношения и в случае вторичного галоге-
нида. Скорости гетеролиза при этом того же самого порядка, как и при
мономолекулярной сольволизе таких простых вторичных хлоридов, как
изопропилхлорид [130].
Сольволиз при 80 °C в 80%-ном ( Изопропилхлорид, Ад ча 5-10”’с”1 '
водном С2Н5ОН X Пинаколилхлорид, Ад = 1,94-10”’ с-1
Миграция и в этом случае не ускоряет гетеролиза, несмотря на то что она
происходит вскоре после завершения гетеролиза.
Давно известным примером, показывающим резкое различие в стереохимии
и кинетике между перегруппировками, в которых локальные процессы
у места диссоциации имеют механизм SN1 или S N2, является сольволиз бор-
нильных или азоборнильных галогенидов или эфиров. Продуктами перегруп-
пировки борнилхлорида при сольволизе по реакции первого порядка явля-
ются соединения ряда камфена: камфенгидрат, эфиры камфена и сам кам-
фен [126]. Скорости в этом случае того же порядка, что и при сольволизе
* Одновременно Робертс и Ли [127] ввели термин «неклассический», который с тех пор
применяется для выражения любой делокализации заряда в карбониевом ионе (мезо-
мерной и синартетической). Этот термин можно использовать на коротком промежутке
времени, поскольку он не является позитивным, однако если принять его и использовать
длительное время, то оп становится «классическим», т. е. уже неверным. «Мезомерия» —
старый общепринятый термин. Таким же может стать и термин «синартезис».
** См. работу [128]. Недавно был проведен хроматографический анализ продуктов соль-
волиза различных неопентильных систем. Гофман и Фразер [129] провели детальный
анализ продуктов сольволиза га-толуолсульфоната в различных по составу смесях эти-
лового спирта с водой. Они показали, что, кроме преобладающих mpem-амильных и ами-
леновых продуктов, образуются и неопентильные производные в количестве 3—10%,
а также побочный продукт — 1,1-диметилциклопропан в количестве 0,0—0,3%.
633
Глава X
пинаколилхлорида, а следовательно, того же самого порядка, как и ско-
рость мономолекулярного сольволиза простых вторичных хлоридов, таких,
как изопропилхлорид. Однако сольволиз изоборнилхлорида, ведущий к тем
же продуктам перегруппировки, протекает со скоростями, в среднем
в 30 000—100 000 раз большими, чем упоминавшиеся ранее.
Сказанное подтверждается следующими данными:
( Пинаколилхлорид, ki = 1,94 • 1О'7 с~ 1
Сольволиз при 80 °C в 80%-ном I БорннлхлориД) « 1,5.10-7 с-1
водном С2Н5ОН [ Изоборнилхлорид, 1,4-10-2 с-1
Большая скорость сольволиза изоборнилхлорида, безусловно, должна быть
приписана синартетическому, а не пространственному ускорению. Для
изоборнилхлорида, который имеет экзо-структуру, перемещение связи может
начаться раньше ионизации атома хлора, находящегося в переходном состо-
янии. Следовательно, перемещение связи может иметь ускоряющее влияние
на скорость реакции. В то же время в борнилхлориде (эиЗо-хлориде) пере-
мещение связи не может начаться, пока атом хлора не начнет перемещаться,
т. е. происходит немного позднее образования переходного состояния. Таким
образом, перемещение связи не может влиять на скорость реакции. Короче
говоря, эти хлориды иллюстрируют перегруппировки, включающие неуско-
ренное мономолекулярное и ускоренное бимолекулярное «замещение» в месте
диссоциации.
Борнилхлорид Изоборнилхлорид
Концепция синартезиса приводит к выводу, что реакции замещения или
элиминирования, которые не сопровождаются перегруппировками, также
могут ускоряться, в общем таким же самым образом, как реакции с перегруп-
пировками. Следовательно, если скорость замещения или отщепления кон-
тролируется ионизацией и если образующийся карбониевый ион может
в значительной степени стабилизоваться синартезисом, некоторая доля этой
дополнительной стабильности может передаваться переходному состоянию
этих реакций, т. е. этот ион будет образовываться быстрее. Однако будет ли
этот ион, синартетическое строение которого потенциально способствует
перегруппировке, перегруппировываться в действительности — это зави-
сит от термодинамических факторов. Если продукт перегруппировки будет
менее стабильным, чем неперегруппировавшийся изомер, то будет образо-
вываться последний и скорость его образования будет повышенной, т. е.
именно образование синартетического иона, а не другая стадия будет опре-
делять скорость общей реакции.
Ускорение реакции без перегруппировки можно проследить на следующем
примере. Скорость гидролиза камфенгидрохлорида в камфенгидрат и кам-
фен в растворителях, содержащих воду, настолько велика, что измерить ее
нелегко; она явно больше соответствующей скорости гидролиза простых
третичных алкилхлоридов. Скорость алкоголиза камфенгидрохлорида была
измерена [126] и оказалась почти в 6000 раз больше скорости алкоголиза
634
Перегруппировки в насыщенных системах
трет-бутилхлорида.
Сольволиз при О °C ( mpem-Бутилхлорид, /ц » 7-10-9 с-1
в сухом С2Н5ОН X Камфенгидрохлорид, Ад =4,0-10~6с-1
Общие схемы, объясняющие ускорение реакций мономолекулярного заме-
щения и отщепления (как с перегруппировкой, так и без нее), обусловлен-
ное синартезисом в карбониевом ионе, который образуется на стадии,
Рис. 47. Схематическое изображение мономолекуляр-
пых реакций, не сопровождающихся пере-
группировкой.
------реакция, проходящая через левый барьер до
обычного иона, а затем также через левый барьер
до конечного продукта, соответствует мономолеку-
лярному замещению без перегруппировки, идущему
с нормальной скоростью, например сольволиз трет-
амилхлорида; ---- реакция, проходящая через
левый барьер до синартетического иона, а затем
также через левый барьер до конечного продукта,
соответствует мономолекулярному замещению без
перегруппировки, например сольволиз камфенгидро-
хлорида. Реакция, проходящая слева направо, опи-
сывает ускоренную перегруппировку, например кам-
фенгидрохлорида в изоборнилхлорид.
определяющей скорость, приведены на рис. 47 и 48, взятых из работы [126].
Вначале выясним, что означают пунктирные линии. На схемах, как обычно,
приведена зависимость энергии от координаты реакции. Начальные и конеч-
ные состояния не представлены, так как по энергии они могут располагаться
в любой последовательности, что не влияет на механизм, но, однако, опре-
деляет, будет ли этот механизм иметь преимущество перед каким-либо иным
механизмом или нет. Предполагается, что изменение координаты реакции
ион
Рис. 48. Схематическое изображение мономолекулярных
реакций, сопровождающихся перегруппировкой.
----------------реакция проходящая через левый барьер и
заканчивающаяся также с левой стороны этого барьера,
описывает мономолекулярное замещение, идущее с нор-
мальной скоростью, например сольволиз неопентилбро-
мида;----------- реакция, полностью проходящая слева
направо, описывает ускоренную перегруппировку,
например гидролиз изоборнилхлорида до камфенгид-
рата и камфена. Если реакция проходит через левый
барьер, а затем также через левый барьер до конеч-
ного продукта, то она соответствует ускоренной побоч-
ной реакции без перегруппировки.
на каждой диаграмме начинается слева, и реакция заканчивается, перейдя
через максимум, соответствующий более низко лежащему переходному
состоянию; другими словами, по крайней мере в принципе, реакция имеет
мало шансов закончиться на той стороне барьера, который соответствует
меньшей энергии активации перехода через этот барьер. Поэтому реакция,
приведенная на рис. 47, останавливается на первом минимуме. Это означает,
что образуется первый карбониевый ион, который затем рекомбинируется
с некоторым анионом без перегруппировки, как, например, в случае соль-
волиза mpem-амилбромида. Реакция, приведенная на рис. 48, заканчивается
на правом минимуме, т. е. первоначально образующийся карбониевый ион
переходит во второй карбониевый ион, т. е. в продукт перегруппировки,
как, например, при мономолекулярном сольволизе неопентилбромида. Цен-
тральное переходное состояние, обозначенное на рис. 47 и 48 пунктирной
линией, соответствует ситуации, выполняющейся в приведенных примерах
635
Глава X
В обоих случаях, как при упомянутом замещении без перегруппировки, так
и при перегруппировке, ускорения реакции не наблюдается, т. е. ни потен-
циально возможное (в первом примере), ни осуществляющееся в действитель-
ности (во втором примере) перемещение группы не влияет на скорость.
Предполагается, что энергетический барьер в центре достаточно высок,
чтобы устранить энергетически сколько-нибудь существенный резонанс
между первым и вторым ионами. Отсюда следует, что ни потенциальная воз-
можность образования, ни действительное образование второго иона не
влияет на энергию первого иона и, следовательно, на энергию образования
промежуточного состояния первого иона, т. е. на то, от чего зависит
скорость.
Теоретически наиболее простым объяснением ускоренного замещения,
происходящего как с перегруппировкой, так и без нее, может служить путь,
согласно которому необходимо абстрагироваться от всех побочных процессов
и предположить, что препятствующий резонансу барьер между первым
и вторым ионами полностью устраняется. Тогда ионные структуры будут
взаимодействовать, образуя нормальный ион, более стабильный, чем любая
из этих структур, так называемый синартетический ион. Следовательно,
имеется не только стабилизованный карбониевый ион, но также стабилизо-
ванное переходное состояние ионизации, и поэтому происходят или ускорен-
ные мономолекулярные реакции без перегруппировки, или же ускоренные
перегруппировки. (Сказанное объясняется тем, что промежуточные состоя-
ния, электронные волновые функции которых могут быть приблизительно
определены как среднее значение электронных волновых функций исходных
и конечных продуктов, отражают устойчивые изменения, происходящие
в исходных и конечных продуктах.)
Сплошные линии на рис. 47 и 48 графически отражают это положение.
Реакция, проходящая через левый барьер до синартетического иона, а затем,
также через левый барьер, до конечного продукта, соответствует ускоренной
мономолекулярной реакции без перегруппировки, например сольволизу
камфенгидрохлорида. Видно, что не только в реакциях с перегруппировкой,
но и в мономолекулярных реакциях без смещения группы может возникать
синартетическое ускорение. Реакция, полностью проходящая в направлении
слева направо на обеих диаграммах, описывает ускоренную перегруппиров-
ку. При энергетическом профиле, приведенном на рис. 47, реакция идет
слева направо лишь в небольшой степени, однако если общая реакция все
время, хотя в малой степени, идет по этому пути, то при благоприятном
соотношении между термодинамическими параметрами исходного и конеч-
ного вещества может наблюдаться перегруппировка. Реакция, приведенная
на рис. 48, в основном проходит слева направо и соответствует ускорен-
ной перегруппировке типа гидролиза изоборнилхлорида до камфенгидрата.
В данном разделе будут рассмотрены внутренние структурные превращения
синартетического карбониевого иона класса норборнильных соединений,
поскольку именно на них были получены данные, наиболее близкие к указан-
ной выше точке зрения. Превращение экзо-2-норборнилхлорида в его энан-
тиомер соответствует превращению изоборнилхлорида в камфенгидрохлорид
(в последнем случае молекулы содержат на три метильные группы больше),
поэтому перегруппировку изомерных порборнилхлоридов можно изучать
поляриметрически. В этой системе отсутствуют метильные группы, которые
являются меткой у соответствующих атомов углерода, но зато, как будет
показано ниже, можно ввести гораздо более информативные изотопные метки.
Важная особенность этой системы состоит в том, что при образовании синар-
тетического иона последний будет иметь плоскость симметрии (симметрия Cs).
636
Перегруппировки в насыщенных системах
что, безусловно, отразится на стереохимии. Для того чтобы легче было это
уяснить, структура экзо-2-норборнилгалогенида или соответствующего эфи-
ра приведена ниже сначала в обычной проекции, а затем в проекции, полу-
ченной после вращения предыдущей модели до тех пор, пока положение 1 не
окажется впереди, и последующего переворачивания молекулы экзо-стороной
книзу. Другие формулы отображают превращение Вагнера — Меервейна
в энантиомер и строение возможно образующегося промежуточного карбоние-
вого иона симметрии Cs
7 ЭКЗО
5-^4
Перегруппи-
ровка
Вагнера-
Меервейна
экзо-2-Норборниловый эфир
или галогенид
Cs~ Симметрия синар-
тетического иона
Уипстейн’и Трайфан [131] показали, что экзо-2-норборнильные эфиры претер-
певают сольволитическое замещение гораздо быстрее, чем их эиЗо-изомеры,
хотя различие в скоростях в этом случае не так велико, как в борнильной
серии. Так, экзо-2-ге-бромбензолсульфонат в уксусной кислоте превращается
в экзо-ацетат в ~350 раз быстрее, чем в эиЗо-сульфонат, при этом скорость
последней реакции примерно равна скорости превращения циклогексил-2-ге-
бромбензолсульфоната в циклогексилацетат. Уинстейн и Трайфан предполо-
жили, что реакция экзо-сульфоната «чувствует движущую силу», т. е. согла-
сованно с ионизацией начинает происходить смещение группы до заверше-
ния образования переходного состояния. И из экзо- и из эиЗо-сульфонатов
образовывался чистый экзо-ацетат. Этот продукт, образовавшийся из экзо-
сульфоната, был рацемизован по крайней мере па 99,95%. Продукт, образо-
вавшийся из экЭо-сульфопата. был рацемизован на 92%, а остальные 8%
соответствовали инверсии конфигурации. При сольволизе оптически актив-
ного бромбепзолсульфоната скорость его рацемизации значительно превыша-
ла скорость сольволиза (например, в два раза). Это показывает, что в обра-
тимой стадии происходит образование оптически неактивного промежуточ-
ного соединения, которое может превращаться вновь в исходное соединение
или в конечный продукт с рацемизацией.
Робертс, Ли и Сондерс [132] синтезировали норборнеол (присоединением по
Дильсу — Альдеру винилацетата к циклопентадиену и т. д.), меченный 14С
в положениях 2 и 3, как показано на приведенных выше формулах. После
ацетолиза экзо-2-п.-бромбензолсульфоната авторы провели ступенчатую
деградацию образовавшегося экзо-ацетата путем чередующихся окисления
и перегруппировки Курциуса с целью обнаружения распределения метки по
углеродным атомам.
I ОСОСНз
КСО2Н _ 2СО, +
СО2Н
2,3
CL NHl
1,7,4,5,6
637
Глава X
/СО2Н /^NH2
->С02+ ^,С02Н _>2СОг + ^Snh2
7 1,4,5,6 1,4 5,6
Результаты приведены ниже, причем значения в скобках были вычислены на
основании определенных предположений.
Перегруппировка Вагнера — Меервейна, проходящая через промежуточ-
ный, имеющий плоскость симметрии синартетический ион (см. выше), должна
вызывать равномерное распределение меток, первоначально находившихся
в положениях 2 и 3, между этими положениями и положениями 1 и 7. Если
можно предположить, что метка, обнаруженная (после деградации) в поло-
жениях 1,4, полностью находится в положении 1, то ввиду совпадения рас-
считанных и опытных данных можно считать, что за время жизни карбониево-
го иона распределение метки достигает равновесного значения. Однако
наблюдается также дальнейшее перераспределение метки в положения 5
и 6, причем перераспределение не достигает равновесия за время жизни
карбониевого иона. Следовательно, нужно предположить, что происходит
а,у-сдвиг водорода, т. е. 6 ->• 2-сдвиг, в общем виде установленный Реуто-
вым при дезаминировании w-пропиламина (разд. 1,6). В данном случае пере-
ход метки из положений 2, 3, 1 и 7 в положения 5 и 6 обусловлен переходом
водорода из положения 6 в положение 2 или 1, который сопутствует уже рас-
смотренному переходу углерода в положении 6 из положения 1 в положение 2
и обратно.
Существуют две точки зрения на промежуточные частицы, образующиеся
в этих реакциях. Согласно первой, карбониевый ион симметрии Cs, уже при-
водившийся выше и вновь представленный ниже в виде формулы I, путем
водородных сдвигов переходит в два других эквивалентных иона II и III,
также с симметрией Cs. Скорость цикла взаимопревращений I II III,
как предполагается, сравнима по величине, но меньше скорости взаимодей-
ствия I, а на самом деле — любого из этих эквивалентных ионов карбония
с нуклеофильными замещающими агентами.
I II III IV
638
Перегруппировки в насыщенных системах
В равновесии циклически взаимопревращающиеся системы I, II и III дол-
жны быть функционально тригональными, и, следовательно, можно пред-
положить существование единого иона симметрии СЗР. Этот ион представлен
формулой IV, и его можно рассматривать как нортрициклен с дополни-
тельным протоном на оси С3. При такой картине должны быть усредненные
связи между тремя атомами углерода и связанным с ними протоном.
Положительный заряд равномерно распределяется между тремя атомами
углерода и в какой-то степени на протоне. Чтобы объяснить наблюдаемое
перераспределение метки, нужно предположить, что вначале образуются Cs-
ионы I, которые частично реагируют с растворителем или другим нуклеофи-
лом, а частично переходят в С3!,-ионы IV, далее вступающие в реакцию
с нуклеофилом.
Выбор между этими двумя гипотезами, которые одно время приводились
вместе, вероятно, можно сделать, исходя из работ Шлейера, Ола и сотр.
[133] по спектрам протонного магнитного резонанса норборнилгалогенидов,
растворенных в смеси пятифтористой сурьмы с двуокисью серы. При поме-
щении в эту смесь экзо-2-иорборнилфторида он полностью превращался
в гексафтораптимонат норборнилия; карбониевый ион имел сравнительно
долгое время жизни и был достаточно стабилен, чтобы можно было изучать
его спектрально вплоть до 40 °C. Ниже —60 °C спектр ПМР имел три острых
пика с относительной интенсивностью 6, 1 и 4; этот спектр более тонко не
разрешался до —120 °C. Так и должно было быть по теории в случае трех
эквивалентных синартетических ионов симметрии Cs, взаимопревращающих-
ся друг в друга путем а, у-водородных сдвигов, которые могут происходить
быстрее, чем спиновые переходы даже при —120 °C. В случае С3ю-модели
карбониевого иона IV должны были бы наблюдаться четыре пика с относи-
тельными площадями 6, 1, 3 и 1; таким образом, спектры ПМР не свидетель-
ствуют в пользу существования иона IV.
Выше —60 °C ион норборнилия медленно изменяется, что приводит к упро-
щению спектра протонного магнитного резонанса. С ростом температуры три
пика уширяются и сливаются, а затем объединенный пик утоньчается,
и при —5 °C спектр содержит один резкий пик. Теперь все атомы водорода
эквивалентны, и это при учете уже рассмотренных а,р-углеродных и а,у-
водородных миграций может только означать медленную перегруппировку,
в процессе которой каждый атом успевает побывать в голове моста. Вероятнее
всего, происходит а,р~водородный сдвиг, аналогичный сдвигу метильной
группы в перегруппировке Наметкина в химии терпенов, т. е. 3,2-, 5,6-
или 7,1-переходы водорода в ионах I, II и III.
Из данных по влиянию температуры на форму спектральных линий Сондерс,
Шлейер и Ола определили скорость 3,2-перехода и других подобных процес-
сов а,р-миграции водорода. Энергия активации составляет 10,8 ккал/моль
(45,22-103 Дж/моль), что по уравнению Аррениуса, kt = 1012-3
exp (—10 800/RT) с-1, приводит к значению константы скорости 2,5 НО4 с-1
при 25 °C.
Основываясь на том факте, что даже более медленная из двух быстрых пере-
группировок — а,у-водородный сдвиг, судя по спектрам, не может быть
остановлена даже при —120 °C, Бартлет * рассчитал минимальные скорости
* См. работу [134]. Идея о синартетическом ускорении была подвергнута жестокой кри-
тике Брауном [135], с которым можно согласиться в том, что в ряде случаев наблюдаемое
ускорение было отнесено за счет синартезиса без достаточных оснований. Однако Бартлет
пояснил, что это не дискридитирует верных кинетических, стереохимических, спектроско-
пических и изотопных доказательств в пользу синартезиса.
639
Глава X
обеих перегруппировок. Константа скорости при этой температуре должна
составлять по крайней мере 3-10s с-1, и, предполагая, что предэкспонен-
циальный фактор равен 1012>3 с-1, можно вычислить, что энергия активации
не должна превышать 4,8 ккал/моль (20,10-Ю3 Дж/моль); это приводит
к значению скорости 6-108 с”1 при 25 °C.
Далее Бартлет напомнил, что скорость реакции иона норбориилия с уксус-
ной кислотой в работе Робертса, Ли и Сондерса была в 3,3 раза выше скоро-
сти 6,2- и других а,у-водородных сдвигов; кроме того, более низкий предел
степени рацемизации при ацетолизе (Уинстейн, Трайфан) экзо-норборниль-
ного эфира показывает, что скорость смещения углерод-углеродной связи
(если есть такая отдельная стадия) по крайней мере в 2000 раз больше ско-
рости реакции этого иона с растворителем. Перемножив эти факторы, два
из которых соответствуют нижним пределам, Бартлет вычислил минималь-
ную скорость перемещения углерод-углеродной связи (предполагая, что
Синартетический
ион
Рис. 49. Схематическое изображение
карбониевого иона с двумя
симметрично расположен-
ными зарядами и происхо-
дящего отсюда синартети-
ческого иона, имеющего пло-
скость симметрии, напри-
мер иона норбориилия.
существует такая отдельная стадия), которая оказалась равной 4-1012 с-1
при 25 °C. Такая величина скорости, очевидно, означает отсутствие энер-
гии активации и соответствует не чему иному, как частоте колебаний свя-
зей.
Следовательно, в период между имеющими энергию активации переходами
водорода ион норбориилия не может быть изображен как находящаяся
в равновесии система первый ион — второй ион, в которых заряды лока-
лизованы по-разному и которые разделены между собой энергетическим
барьером. Поэтому лучше рассматривать этот ион как один из эквива-
лентных синартетических ионов симметрии Cs, как представлено на
рис. 49.
з. Ионные промежуточные соединения. Смешанная ол-делокализация.
Обобщение синартезиса. Первый шаг к расширенной теории был сделан
в 1955 г. Уинстейном, Шатавским, Нортоном и Вудвардом [136]. Основанием
послужил тот хорошо известный факт, что 7-норборнилгалогениды и 7-нор-
борниловые эфиры очень медленно сольволизуются. Этот эффект все припи-
сывали трудности возникновения карбониевого иона вследствие жестко
закрепленного угла в кольце в положении 7, так как известно, что легкость
образования карбониевого иона в голове моста чрезвычайно чувствительна
к геометрии и жесткости молекулы (гл. VII, разд. 7,ж). Новые данные состо-
яли в том, что awmu-7-норборненильные эфиры, содержащие двойную связь
между положениями 2 и 3, сольволизовались очень легко. Впоследствии
было найдено, что еще более легко сольволизуются 7-норборнадиенильные
эфиры, в которых имеется еще одна двойная связь в положении 5, 6 [137].
Соотношение скоростей при ацетолизе ге-толуолсульфонатов приблизительно
640
Перегруппировки в насыщенных системах
следующее:
7-Норборпил 10-7
jHdo-5-Норборнепил 10-1
Циклогексил 1
зкзо-5-Норборненил 103
анты-7-Норбориенил 104
7-Норбориадиенил 10"
Эти данные привели к мысли, что боковые л-электроны могут двигаться за
пределы связи, таким образом ускоряя образование карбоний-ионного
центра в положении 7.
На предположение о том, что интегральное изменение ковалентности пере-
мещает заряд в положение 3 особенно стабильного «классического» иона
с 1,2,7-циклопропановым кольцом, всегда существует несколько возраже-
ний; простейшее па сегодняшний день, вероятно, состоит в том, что, как
показали Стари и Сондерс [138], в спектре протонного магнитного резонанса
тетрафторбората 7-норборнадиепила в двуокиси серы или в нитрометане не
обнаруживается 7 неэквивалентных протонов. Кроме того, не обнаруживает-
ся и трех серий сигналов от протонов в положениях 1, 2 и 4, которые наблю-
дались бы, если бы обе двойные связи изменялись одинаково с образованием
иона, имеющего две плоскости симметрии (С2„). В действительности спектр
содержит четыре сигнала от протонов в положениях 1, 2, 2 и 2, что указывает
на преобладающее участие одной двойной связи, в результате чего образует-
ся ион с одной плоскостью симметрии (Cs); эта симметрия сохраняется доль-
ше, чем по крайней мере время, необходимое для изменения спина протонов.
Приведенные выше диаграммы с кривыми стрелками объясняют большое
ускорение реакций и согласуются с указанной симметрией.
Как уже было показано, синартезис может проявиться в кинетике и стерео-
химии реакций, происходящих без перегруппировки или с перегруппиров-
кой, при этом тип конечного продукта зависит от термодинамических факто-
1 ров. Данные Уинстейна — Вудварда иллюстрируют синартезис в общей фор-
ме (которая будет дана ниже), но без перегруппировки. Обобщенная форма
синартезиса с перегруппировкой была впервые показана в работе Ле Най
[139] в 1960 г. Она обнаружила, что наличие двойной связи в А4-циклогеп-
тенилметил-га-бромбензолсульфонате увеличивает скорость его ацетолиза
в 30 раз по сравнению со скоростью ацетолиза насыщенного аналога и что
продуктом ацетолиза этого ненасыщенного моноциклического эфира являет-
ся насыщенный бициклический эфир 2-экзо-бицикло-[3.2,1]-октилацетат
(OBs — га-бромбензолсульфонат)
OBs
Дгт
ососн3
Аналогичная циклизация, приводящая к порборнильным соединениям, была
описана Лаутоном и изучена Бартлетом и сотр. [140]. Исходным соединением
служил р-(А3-циклопептенил)этиларенсульфонат. Его сольволиз в уксусной
4 1-0772
641
Глава X
кислоте, содержащей ацетат натрия, имел первый порядок и скорость его
в 75—95 раз превышала скорость сольволиза соответствующего насыщенного
эфира; ненасыщенный эфир сольволизовался с циклизацией, давая экзо-2-
норборнилацетат
АгЗОгО
ОСОСН,
Ниже приведены некоторые относительные скорости реакций, изученных
Бартлетом, и указан класс образующихся продуктов (ONbs — и-нитро-
бензолсульфонат). Эти данные ясно показывают, что критическими фактора-
ми в обусловленном синартезисом ускорении циклизации являются наличие
двойной связи, а также размер цикла и длина боковой цепи. Они также
указывают, что ускоряющий эффект двойной связи кумулятивно и аддитивно
усиливается при введении концевых метильных групп, как и можно было бы
ожидать, предполагая нуклеофильное участие л-электропов двойной связи
СН2 — СН2 — ONbs
CH2CH2ONbs
1,5
CH2CH2ONbs
~ 1
СН3
CH2CH2CH2ONbs | CH2CH2CH2ONbs
сн?
~ 1 ~ 1
( —> экзо-норборнил)
СНз
? ?— CH2CH2ONbs
600
(—> СН3-экао-норборпил)
сн3
|| '?-- CH2CH2ONbs
сн?
300
(—> (СНз)2-эк,эо-порборнпл)
С Нз
|f4'-CH2CII2CH2ONbs ,| CH2CH2CH2ONbs
СН?
~ 1 4
(без циклизации) (немного бициклического олефина)
В 1959 г., когда уже было известно ускорение боковыми л-электропами,
открытое Уинстейном и Вудвардом, но когда еще не появилось сообщения
о превращении таких л-электронов путем перегруппировки в о-электроны,
Робертс и сотр. [141], побуждаемые экспериментальными результатами,
которые будут рассмотрены, пришли к выводу о необходимости расширения
642
Перегруппировки в насыщенных системах
картины синартезиса. Первоначально синартетическую связь разделяли на
две парциальные связи, направленные к атомам, которые мигрирующий атом
связывает мостиком и которые поэтому делят между собой карбониевый
положительный заряд. Часто это является хорошим первым приближением,
однако в общем, и это принципиально, необходимо допустить частичное
смещение синартетической пары электронов от мостикового атома, которое
усиливает связь между двумя другими атомами, и таким образом некоторая
часть заряда па этих атомах переходит к мостиковому атому. В результате
добавляется третья каноническая структура, соответствующая третьей исход-
ной структуре (использованной впоследствии Ле Най, Лаутоном и Бартле-
том), ведущей к образованию синартетического иона, а именно структура,
в которой сбоку от карбоний-ионного центра находится л-связь. Поэтому,
следуя представлениям Робертса 1959 г. о «неклассических ионах», можно
представить, что трехцентровое связывание синартетической парой электро-
нов на самом деле более симметрично, поскольку эта электронная пара рас-
пределяется не по двум, а по трем парциальным связям. Такую трехцентро-
вую двухэлектронную связь удобно изображать точечным треугольником:
А---В А----------В А=В
Канонические структуры синартетического иона
R
А^—В
Синартетический ион
Простейшим синартетическим ионом, уже давно известным [142], конечно,
является ион Н3 Дж. Дж. Томсона; вероятно, подобное строение имеют
и другие «неклассические» ионы, обнаруживаемые масс-спектрометрически,
например СН5
СН,
А Л
Если синартетический ион возникает при 1,2- или 1,3-гидридном сдвиге (что
нельзя, считать само собой разумеющимся и очень трудно доказать), то его
можно представить следующими структурами:
. Н
Такие умозрительные структуры иногда называют «протонированной двой-
ной связью» или «протонированным циклопропановым кольцом». Из масс-
41* в4з
Глава X
спектрометрии хорошо известны ионы винилия («протонированная тройная
связь»), аллилия и циклопропенилия, два последних немного известны и из
обычных химических данных. Строение этих ионов, вероятно, следующее:
Н
нс===сн
Экспериментальные исследования [141], в которых впервые были развиты
эти идеи, касались скелетных изомеризаций, сопровождавших нуклеофильное
замещение в циклопропилметильных, циклобутильных и гомоаллильных
соединениях. Робертс и Мазур показали, что в большинстве случаев гидро-
лиз и ацетолиз соответствующих хлоридов, превращение спиртов в галоге-
ниды при помощи галогенидов фосфора и галогеноводородов, а также деза-
минирование соответствующих аминов азотистой кислотой в определенной
мере сопровождается скелетной изомеризацией. Бергстром и Зигель [143]
показали, что этаполи.з циклопропилметилбензолсульфоиата даже в при-
сутствии этилат-ионов является реакцией первого кинетического порядка
и происходит быстро — приблизительно в 10 раз быстрее соответствующей
реакции аллилбензолсульфоната. Касерио, Грэем и Робертс [144] далее
установили, что при этаполизе циклопропилметилхлорида и циклобутилхло-
рида в водном спирте с почти одинаковой скоростью образуется одна и та же
смесь продуктов, состоящая из шести соединений, а именно циклопропил-
метилового, циклобутилового и гомоаллилового спиртов и трех соответ-
ствующих этиловых эфиров. Вероятно, существует общая тер,модинамиче-
ская тенденция к переходу от циклопропилметильных к гомоаллильным
изомерам
X
хотя соответствующие константы равновесия не очень велики, например 36
для циклопропилметил- и циклобутилхлоридов при 25 °C (катализ хлори-
стым цинком). В общем равновесие устанавливается быстрее слева направо,
чем справа налево; так, циклопропилметапол при действии разбавленной
соляной кислоты количественно превращается в циклобутаиол, а при дей-
ствии концентированной соляной кислоты и хлористого цинка (реактив Лу-
каса) — в гомоаллилхлорид. Естественно, что образование многих продук-
тов не определяется термодинамикой, поскольку нестабильные промежуточ-
ные частицы могут реагировать с растворителем быстрее, чем изомеризо-
ваться в более стабильные частицы.
Вследствие сильного изгиба связей в циклопропановом кольце эти связи
в значительной степени имеют л-характер, даже связи циклобутанового
кольца будут обладать некоторым л-характером. Таким образом, паше обыч-
ное подразделение связей на а и я в этих системах несколько условно. Цик-
лопропильная группа обладает несколько более сильным ускоряющим эф-
фектом в 8к1-реакциях по сравнению с винильной группой; это показывает,
что способность к сопряжению не целиком обусловлена л-компонентой свя-
зи. Харт и сотрудники показали, что такой ускоряющий эффект циклопро-
644
Перегруппировки в насыщенных системах
пильных групп обладает кумулятивностью *; при последовательной замене
изопропильных остатков в т/>мс-(изопропил)карбинил-и-питробензолсуль-
фонате па циклопропильные остатки относительные скорости сольволиза
в 80%-ном водном диоксапе увеличиваются следующим образом:
В описанной выше работе 1959 г. Робертса и сотрудников циклопропил-
метильная группа, содержащая 14С в боковой цепи, путем нуклеофильных
реакций замещения превращалась в продукты, при расщеплении которых
определялось распределение метки 14С [142]. Были исследованы два возмож-
ных случая. Первый иллюстрируется превращением циклопропилметанола
под действием реактива Лукаса в равновесную смесь продуктов, преобладаю-
щим компонентом которой является гомоаллилхлорид. Распределение метки
в этом соединении было следующим:
СН2
\ * НС1
CII-C1L-OH ------> С1
/ 1 ZnCI2
СН2 4
СН2—сн2 —сн = сн2
100%
I
60,5% 0,1О/о 33,4%
Равномерное распределение метки между тремя метиленовыми группами,
которое можно предположить, исходя из этих данных, указывает на то, что
или образуется только одно промежуточное соединение, молекула которого
имеет ось симметрии третьего порядка (С3) и, вероятно, также еще три пло-
скости симметрии (С3г,), или образуются три эквивалентных взаимопревра-
щающихся друг в друга промежуточных соединения, каждое из которых име-
ет более низкую симметрию или, может быть, Cs).
Далее необходимо исследовать неравновесные продукты, состав которых
определяется реакциями первоначально образовавшегося промежуточного
соединения, которые происходят настолько быстро, что это промежуточное
соединение пе успевает достигнуть положения равновесия с другими про-
межуточными соединениями, если возможно их образование. Имеется много
указаний на то, что карбониевый ион, образующийся при отщеплении азо-
та от диазониевого иона при дезаминировании в водной среде, очень быстро
реагирует с водой (разд. 1,6). Поэтому было продолжено изучение дезамини-
рования циклопропилметиламииа в воде. При этом получались три спирта
в соответствии, приведенном ниже, которое, очевидно, далеко от равно-
весного. Распределение метки, первоначально находившейся в боковой цепи,
* См. работу [145]. Степень изгиба связей, которая обусловливает сопряжение цикло-
пропанового кольца (рх-К), выявляется из карты распределения электронной плотности,
полученной при рентгепоструктурном исследовании цис-1,2,3-трицианциклопропапа:
отчетливый максимум проявляется па расстоянии 0,32 А (3,2 -Ю'-2 нм) от середины сто-
роны треугольника, образованного межъядерными линиями кольца [146].
645
Глава X
в молекулах двух главных
продуктов дезаминирования приведено ниже:
i«C
Метка
СН2 |
yCH-CH,-NH2
CHi
Очевидно, что наивысшая концентрация метки находится в том месте, где
она была первоначально, т. е. в боковой цепи. Что касается других положе-
ний, то содержание метки выше равновесной величины обнаруживается
только в положениях 2 и 4 циклобутанола. В кольцевых метиленовых груп-
пах циклопропилметанола и в 3-метиленовой группе циклобутанола содер-
жание метки ниже равновесных значений. Очевидно, здесь наблюдаются
последовательные процессы изомеризации карбониевого иона, скорости
которых сравнимы со скоростью реакции этого иона с водой, являющейся
растворителем, и со скоростью последовательных процессов, через которые
протекает реакция. В момент гибели карбониевого иона содержание меченого
углерода в боковой цепи циклопропильного соединения снижается от 100%
до величины, лежащей между 100 и 33% (равновесное значение), что обуслов-
лено прежде всего переходом метки к метиленовым группам циклобутанола
в положениях 2 и 4, в которых содержание меченого углерода повышается
от 0% до величины, превышающей равновесную. Это означает, что последо-
вательные переходы метки происходят со скоростями, сравнимыми, по не
сильно превышающими скорость указанных первоначальных переходов
метки, и поэтому образуется ряд соединений, расположение метки в которых
соответствует направлению первичных переходов. В продуктах, образовав-
шихся в результате последовательных переходов, в момент гибели иона содер-
жание меченого углерода лежит между 0% и равновесными значениями.
Очевидно, что это соответствует серии промежуточных карбониевых ионов,
а не единственному универсальному иону.
В первоначально образовавшемся ионе циклопропилметилия * могли бы
протекать три последовательные перегруппировки Вагнера — Меервейна.
Представленные ниже проекции получаются, если смотреть на третичный
атом углерода сзади из точки, лежащей на продолжении линии СН-связи.
Атомам углерода удобно дать буквенные обозначения, как показано на пер-
вой формуле. Естественно, что все взаимопревращения обратимы, однако
* Предполагается, что этот ион имеет симметрию С3 , что в случае стабильного третичного
гомолога — иона цпклопропилдиметилкарбипилия (СН2)2СНС(СНз)^ — доказано спект-
ром протонного магнитного резонанса в растворителе Ола (FSO3H/SbF5/SO2) [147]. В этом
ионе карбониевый центр, конечно, стабилен в третичном положении. Плотность сим-
метрии пересекает кольцо, и в ней лежит атом углерода боковой цепи. В этой плоскости
лежат даже метильные атомы углерода, поскольку в такой конформации достигается
максимальное сопряжение с изогнутыми связями кольца.
646
Перегруппировки в насыщенных системах
для простоты на диаграмме искривленные стрелки, показывающие направле-
ние изменения связей, а также стрелки, показывающие направление реак-
ции, направлены только слева направо, т. е. соответствуют реакциям, про-
исходящим до установления равновесия («первая волна последовательных
реакций»). Видно, что радиоактивная метка вначале переходит одновременно
двумя параллельными реакциями в положении 2 и 4 иона циклобутилия,
затем двумя параллельными путями в кольцевые метиленовые группы иона
циклопропилметилия, и, наконец, оба параллельных процесса сходятся
и метка переходит в положение 3 иона циклобутилия. Таким путем можно
хорошо объяснить наблюдающееся распределение метки *
Каждая стадия изомеризации происходит через соответствующий синарте-
тический ион. Эти ионы приведены ниже. Каждый ион в отдельности асим-
метричен (симметрия Ct), однако они образуют три пары зеркально противо-
положных ионов; эти пары совмещаются друг с другом путем поворота на
угол 2л/3. Таким образом, если бы эти ионы всегда находились в равновесии,
их химическое поведение было бы неотличимым от поведения одного иона
с осью симметрии третьего порядка и тремя плоскостями симметрии (C3V) **.
* Тот факт, что в момент гибели иона содержание меченого углерода в метиленовой
группе в положении 3 циклобутилиевого иона достигает близкого к равновесному зна-
чения, можно объяснить на основании того, что па «первой волве последовательных
реакций» ион циклобутилия, приведенный справа, образуется очень поздно. Наблю-
даемое содержание метки можно рассчитать, исходя из уравнений реакций; при этом
получаются значения, близкие к экспериментальным.
** Робертс и сотр. обсудили возможную роль иона С3в и пришли к выводу, что оп может
возникать (way point) на пути превращения Cj-ионов, но сам по себе не может являться
647
Глава X
В двух из этих синартетических ионах связь, промежуточная между простой
и двойной, находится в положении ad, в двух других — в положении ас
и в двух оставшихся —в положении ab. Если бы в каждом случае делокализо-
ванные пары электронов локализовались бы с образованием двойной связи,
то получались бы гомоаллильные ионы с различной перестановкой атомов
углерода. Учитывая это, Робертс предсказал тип превращения, позднее осу-
ществленный в обратимых условиях Ле Най, Лаутоном и Бартлетом. Имен-
но необходимость такого предположения послужила причиной, которая при-
вела Робертса к пересмотру концепции синартетических карбониевых ионов.
Гомоаллильные ионы образуются различными путями в точках, отмеченных
черточками на стрелках, характеризующих протекание циклической изо-
меризации (см. схему, приведенную выше). Соответствующими цифрами
обозначены синартетические ионы, которые при этом могли бы возникать.
Можно видеть, что гомоаллильные ионы, первоначально образующиеся из
синартетических ионов 1 и 2, будут иметь радиоактивную метку в положе-
нии 4; гомоаллильные ионы, образующиеся из синартетических ионов 3 и 4,
будут иметь метку в положении 2 и, наконец, гомоаллильные ионы, обра-
зующиеся из синартетических ионов 5 и 6, будут иметь метку в положении 1.
Метки в положении 3 ожидать не следует. Итак, при протекании «волны
обратимых реакций» слева направо радиоактивная метка должна появляться
в гомоаллильпых конечных продуктах в последовательности 4, 2, 1. К сожа-
лению, распределение метки в гомоаллиловом спирте, который образуется
с выходом 5%, до сих пор не определено.
2. Электрофильные перегруппировки в насыщенных системах
Очевидно, можно ожидать существования группы гетеролитических пере-
группировок, которые во всех отношениях диаметрально противоположны
перегруппировкам, описанным в разд. 1. При этом отщепление аниона или
какой-либо другой процесс, ведущий к образованию карбониевого иона или
другого обедненного электронами центра, должно быть в обсуждаемых пере-
группировках заменено отщеплением протона или другого катиона. Такая
реакция поведет к образованию карбаниона или другого центра, обладающего
активной неподеленной парой электронов. Мигрирующая группа при этом
перемещается не в виде нуклеофильного остатка с полным октетом, а в виде
электрофильного остатка с секстетом электронов, оставляя электроны на
месте старой связи и образуя путем координации новую связь с активной
неподеленной парой электронов. В то же время оставшиеся электроны могут
присоединять протон. Если этот тип перегруппировки, подобно другим, не
зависит от наличия кратных связей, то его можно назвать электрофильной
перегруппировкой в насыщенных системах. Электронные смещения, сопровож-
дающие эту перегруппировку, могут быть представлены следующим образом:
R
n/a—В^-М
истинным промежуточным соединением. Кроме того, они ввели в боковую цепь цикло-
пропилметиламина метильную группу, получили хиральный центр в оптически активной
форме и показали, что при образовании гомолога циклопропилкарбинола происходит
почти полная рацемизация, подтвердив, таким образом, что промежуточно образуется
не только один неизомеризованный (7,-пон [148].
648
/7ерегруппировки в насыщенных системах
Обычно М и N — протоны, хотя ими могут быть любые катионы. Перегруп-
пировки такого рода известны, но до сих пор они изучены менее подробно,
чем нуклеофильные перегруппировки в насыщенных системах.
а. Перегруппировка Стивенса. В этой перегруппировке А — ониевый
атом, В — углерод. В настоящем разделе прводится несколько примеров
этой перегруппировки. Первый пример [149] типичен и для большинства
других реакций, изученных впоследствии Стивенсом с сотрудниками при
исследовании обсуждаемой перегруппировки. Гидроокись фепацилбензил-
диметиламмония в водном растворе гладко переходит в со-диметиламипо-ю-
бепзилацетофепоп с перемещением бензильной группы от аммониевого азота
к фенацильному углероду. Предвосхищая механизм реакции, ее можно изоб-
разить в предположении, что промежуточно образующийся карбанион пред-
ставляет собой мезомерный апиоп кето-еполыюй системы (гл. XI)
СН2С6Н5 _ СН2С6Н5 СН2С6Н5
/ г~\ он г/ - .. \
(CH3)2N-СН—Н —>(CH3)2N-----CH->(CH3)2N—сн
СОС6Н5 COCGH5 ioc6H5
В случае карбониламмониевых соединений реакция имеет общий характер.
Например, группа, к которой происходит миграция (а именно фенацильная
группа в приведенном выше примере), была заменена ацетонильной группой.
Мигрирующая группа (бензил в приведенном выше примере) может быть
заменена па аллил, 1-фенэтил, бензгидрил, 9-флуоренил, 3-фенилпропаргил
и фенацил. Группа, от которой идет перемещение (диметиламмоний в приве-
денном примере), может быть заменена на пиперидиний. Более того, известны
аналогичные реакции и в случае сульфониевых соединений [150]
СН2С6Н5 СН2С6Н5 СН2С6Н5
/ он- г/ - .. \
CH3S--СН—Н --->- CH3S--CH->CH3S—сн
+ I + I I
сос6н5 сос6н5 сос6н5
Установлено, что хотя наличие кетонной группировки во всех вышеприведен-
ных примерах, несомненно, ускоряет реакцию, тем не менее это условие не
является необходимым. Перемещение группы может происходить и в простом
углеводородном остатке при условии, что он достаточно устойчив в виде
аниона. Так, при обработке бромистого бензгидрилтриметиламмопия фенил-
литием происходит перемещение метильной группы к бензгидрильной груп-
пе [151].
СН3
(cn3)2N^c’(c^Vn
+ I
С'бПг
C6H5Li,
CHj
(CH3)2N—CCGH5
I
C6H5
В перегруппировке такого типа, проводимой в апротонных растворителях,
промежуточное дипольное состояние, вероятно, не образуется. Стереохими-
ческие данные показывают, что происходит синхронное отщепление протона
и переход алкила от азота к углероду (см. ниже).
Во всех легко идущих реакциях мигрирующая группа перемещается от
ониевого атома к кето-еполыюму углеродному атому, т. е. от центра, пред-
расположенного к потере катиона, к углеродному центру, способному сохра-
Глава X
нять отрицательный заряд и реагировать в форме аниона (гл. XI). Этот факт
может служить убедительным доказательством приведенного механизма.
Во всех случаях наблюдалась зависимость скорости процесса от присутствия
гидроксильных ионов в водных растворах или алкокси-ионов в спиртовых
растворах. На примере гидроокиси фенацилбензилдиметиламмония Томпсон
и Стивенс [152] нашли, что скорость реакции увеличивается с увеличением
концентрации щелочи, стремясь к пределу при ее избытке. Следовательно,
при наличии достаточного избытка щелочи полностью образуется анионный
центр, что по крайней мере для веществ этого класса подтверждает двух-
стадийный характер реакции.
Внутримолекулярный характер перегруппировки был установлен Стивен-
сом [153], который показал, что при совместном проведении двух перегруп-
пировок
(CH3)2NG1CH2COG2 -> (CH3)2NCHG1COG2
(CH3)2NG3CH2COG4 --> (CH3)2NCHG3COG4
скорости которых при раздельном проведении близки (растворитель во всех
случаях один и тот же), образуются только указанные выше продукты без
примеси продуктов перекрестных реакций. В этих реакциях приняты следую-
щие условные обозначения: G1 — ж-бромбензил, G2 — фенил, G3 — бензил,
G4 — и-бромфенил. Гораздо позднее Джонсон и Стивенс [154] повторили этот
опыт, используя более чувствительный метод радиоактивных индикаторов.
В одном из исходных соединений бензильная группа содержала в боковой
цепи 14С. В исходные соединения входили следующие группы: G1 = 14С-беи-
зил, G2 = га-бромфенил, G3 = бензил, G4 = фенил. Скорости перегруп-
пировки этих соединений, взятых в индивидуальном состоянии, составляли
G1G2 : G3G4 = 0,8 : 1,0. При совместной перегруппировке G3G4-npoflyKT был
нерадиоактивен.
На скорость перегруппировки совершенно определенное влияние оказывает
введение заместителей в мигрирующую бензильную группу фенацилбензил-
диметиламмоний-иона; электроноакцепторные заместители ускоряют пере-
группировку [152, 153]. Как длязгетиа-, так и для иа/эа-заместителей сохра-
няется следующая последовательность скоростей:
СН3О<Н, СН3<С1, Вг, I<NO2
На основании этого Стивенс предположил, что бензильная группа мигрирует
со своим полным октетом и это компенсируется переходом пары электронов
от отрицательного атома углерода к азоту. Однако Беннетт и Чапман [155]
отметили, что наблюдаемые полярные эффекты согласуются также с теорией,
по которой группа мигрирует только с секстетом электронов, если допустить,
что мигрирующая группа начинает образовывать новую связь до освобожде-
ния от электронов старой. В нуклеофильных перегруппировках в насыщен-
ных системах зависимость от полярности, как и должно быть, обратная.
Имеется много доказательств, что при нуклеофильных перегруппировках
в насыщенных системах группа мигрирует с полным октетом и что ее пере'
мещение облегчается электронодонорными заместителями.
Изучено также влияние заместителей в фенацильной группе на скорость
перегруппировки [156]. Этот эффект слабее, но и здесь электроноакцепторные
заместители затрудняют миграцию. Для иа/эа-заместителей в фенацильном
кольце сохраняется следующая последовательность скоростей’
СН3>Н>СН3О, С1, Вт, I>NO2
650
Перегруппировки в насыщенных системах
Здесь влияние этих заместителей на образование анионного центра и на его
реакционную способность должно быть качественно противоположно и долж-
но складываться в результате действия двух эффектов. На основании нали-
чия предела на кривой зависимости ускорения реакции от концентрации
щелочей можно предположить, что во всех фепациальпых производных ани-
онный центр образуется в достаточно полной мере; этим объясняется приве-
денный выше ряд влияния полярности заместителей.
Что касается стереохимии перегруппировки Стивенса в случае карбонилсодер-
жащих соединений, то хорошо известно, что мигрирующая группа переме-
щается с сохранением конфигурации [157]. Кемпбелл, Хаустон и Кеньон
установили, что в случае оптически активной гидроокиси фенацил-(1-фенил-
этил)диметиламмопия 1-фенилэтильная группа переходит без рацемизации.
Впоследствии Брюстер и Клайн расщепили продукт перегруппировки до
оптически активной у-фенилмасляной кислоты, конфигурация которой была
известна, тем самым показав, что в перегруппировке Стивенса конфигурация
1-фенилэтильной группы сохраняется.
Для случая более трудно идущей перегруппировки соединений, в которых
отсутствует карбонильная группа, Хилл и Чен [158] смогли установить, что
при перегруппировке Стивенса оптическая активность, центром которой
первоначально являлся аммонийный азот в ионе аллилбензилметилфенил-
аммония, сохраняется и в продуктах реакции, в которых этот центр должен
располагаться на атоме углерода. Это ясно показывает, что процесс идет
в одну стадию:
с6н5 СН2С6Н5
N СН—Н
с6н5 сн2с6н
mpem-C4HqOK \ \
------ 9 » N------СН
[ 6 (CH3)2SO
сн=сн2
сн=сн2
5
б. Перегруппировка Виттига [159]. В этом процессе атом А на общей
схеме — нейтральный атом кислорода, В — атом углерода. Простые эфиры
при металлировании или другом аналогичном воздействии изомеризуются
в спирты с перемещением одной алкильной группы в сх-положение другой.
Только такие группы, как бензил или аллил, образующие достаточно устой-
чивые анионы, т. е. способные легко подвергаться металлированию, могут
в применявшихся до сих пор условиях вызывать миграцию. Ниже приведено
несколько примеров:
ОН
[160]
С6Н5СИ2ОСН3 —» С6Н5СН
Чсн3
сн2=сн-сн2 сн2=сн—сн-он
'"'О -»
СН2 = СН-СН2 сн2 = сн-сн2
[161]
[162]
651
Глава X
По методу Виттига такая перегруппировка происходит в результате обработки
эфиров фениллитием. Первая стадия процесса состоит в замещении а-вод о род-
ного атома бензильной или другой реакционноспособной алкильной группы
на литий. В этих металлических производных и происходит перегруппи-
ровка.
Виттиг предположил, что в металлическом производном либо уже имеется,
либо легко образуется карбанион, к отрицательному центру которого пере-
мещается в виде электрофильного остатка другая алкильная группа, име-
ющая только шесть электронов в оболочке с главным квантовым числом 2.
СН, СНз СНз СНз СНз
О—СНз^Чо—CH^-Li -4-СН — О—СН НО—СН
I I III
С6Н5 с6н5 с6н5 с6н5 С6Н5
Хаузер и Кантор [162] осуществили эту же реакцию путем обработки эфиров
амидом калия в жидком аммиаке с последующим добавлением этилового эфи-
ра. Условия этой реакции (среда и структурные особенности исходных эфи-
ров) соответствуют предложенному Виттигом механизму реакции.
Имеются некоторые качественные данные по относительным скоростям мигра-
ции различных алкильных групп. В случае 9-флуорениловых эфиров Вит-
тиг и сотрудники наблюдали следующую последовательность скоростей:
аллил, бензил > метил, этил > фенил [163]. Из исследования Хаузера
и Кантора [1621 следует, что emop-бутил > неопентил. Виттиг и Шлор [164[
установили следующий порядок: метил > mpem-бутил. Тот факт, что в ряду
простых насыщенных алкильных групп неопентильная и mpem-бутильная
группы обладают относительно низкими скоростями миграции, указывает
па согласованное изменение связей с мигрирующей группой, точно такое же,
как и при перегруппировке Стивенса.
СПИСОК ЛИТЕРАТУРЫ
1. Fittig R.. Ann., 110, 17 (1859): 114, 54 (1860).
2. Thorner W., Zincke T., Ber.. 10, 1473 (1877).
3. Parry W., .1. Chem. Soc., 107, 108 (1915).
4. Tiffeneau M.. Dorlencourt, Ann. chim. phys., 16, 237 (19U9).
5. Brenner A., Zincke T., Ann., 198, 141 (1879).
6. Stoermer R., Ber., 39, 2288 (1906).
7. Tiffeneau M., Compt. rend., 148, 29 (1908).
8. Orekhor A., Tiffeneau M., Bull soc. chim. (France), 37, 1410 (1925).
9. Tiffeneau M., Ann. chim. phys., 10. 328 (1907).
10. Stoermer R., cm. [6|: Tiffeneau M.. Ann. chim. phys.. 10, 328 (1907).
11. Kbtz A., Richter K.. J. prakt. Chem., Ill, 373 (1925).
12. Mislow K., Siegel M., J. Am. Chem. Soc., 74, 1060 (1952).
13. Ingold С. K., Ann. Repts. on Progress Chem. (Chem. Soc. London), 25, 133
(1928).
14. Thorner W.. Zincke T.. Ber., 13, 641 (1880).
15. Bachmann IV. E., Moser Е.П., J. Am. Chem. Soc., 54, 1124 (1932); Bachmann W. E..
Stemberger II. R., J. Am. Chem. Soc., 55, 3821 (1933); 56, 170 (1934); Bach-
mann W. E., Perguson J. W., J. Am. Chem. Soc., 56, 2081 (1934).
16. Duncan. J. F., Lynn K. R., J. Chem. Soc., 1956, 3512, 3519, 3674.
17. Bunton C. A.. Iladwick T., Llewellyn D. R., Packer Y.. J. Chem. Soc., 1958. 403; Bun-
ton C. A., Carr M. I)., ibid., 1963. 5854.
652
Перегруппировки в насыщенных системах
18. Lagrave R., Ann. chim., 8, 363 (1927).
19. Oumnow A. I., Bull. soc. chim. (France). 43, 568 (1928).
20. Danilov S., Ber., 60, 2390 (1927).
21. Liebig J., Ann., 25, 27 (1838); Zenin N., ibid., 31, 329 (1839); Jena A., ibid., 155. 77
(1870).
22. Scheuling G., Ber., 56, 252 (1923).
23. Lachmann A., J. Am. Chem. Soc., 45, 1509 (1923).
24. Westheimer F. II., J. Am. Chem. Soc., 58, 2209 (1936).
25. Ingold С. K., Shoppee C. W., J. Chem. Soc., 1928, 371.
26. Tiffeneau M., Compt. rend., 145, 593 (1907).
27. Le Brazidec M., Compt. rend., 159, 774 (1914).
28. McKenzie A., Lesslie M. S., Ber., 62, 288 (1929).
29. McKenzie A., Roger R., J. Chem. Soc., 125, 844 (1924).
30. McKenzie A., Richardson A. C., J. Chem. Soc., 123, 79 (1923).
31. Orekhov A., Roger R., Compt. rend., 180, 70 (1925).
32. Вагнер Г., ЖРФХО, 31. 680 (1899).
33. Meerwein H., van Emster К., Ber., 53, 1815 (1920); 55, 2500 (1922).
34. Meerwein II., Ann., 405, 129 (1914); Komppa G., Hintikka S. K., Bull. soc. chim.
(France), 21, 13 (1917).
35. Meerwein II., Gerard L., Ann., 435, 174 (1923): Meerwein II., Hammel O., Serini A.,
Vaster J., ibid., 453. 16 (1927).
36. Simonsen J. L., Owen L. N., The Terpenes, 2nd Edn., Cambridge Univ. Press, 2, 290
(1949).
37. Roberts J. D., Vining J. A., J. Am. Chem. Soc., 75, 3165 (1953); Roberts J. D., Per-
ry R., Jr., ibid., p. 3168.
38. Wagner G., Bricknar W., Ber., 32, 2302 (1899).
39. Meerwein II.. Unkel W., Ann., 376, 152 (1910); Meerwein H., ibid., 396, 200 (1913);
405, 129 (1914): 417, 255 (1918).
40. Shoppee C. W., Proc. Leeds Phil. Soc., Sci. Sect., 1, 301 (1928).
41. Zelinsky N.. Zelikov J., Ber., 34, 3249 (1901).
42. Whitmore F. C., Rothrock II. S., J. Am. Chem. Soc., 55, 1100 (1933).
43. Whitmore F. C., Rothrock H. S., J. Am. Chem. Soc., 54, 3431 (1932).
44. Whitmore F. C.. Wittie E. L., Popkin A. II., J. Am. Chem. Soc., 61, 1586 (1939).
45. Dostrovsky I., Hughes E. D., J. Chem. Soc., 1946, 166.
46. Linnemann E., Siersch A., Ann., 144, 129 (1867); Linnemann E., ibid., 161, 15
(1872).
47. Roberts J. D., Vancey J. A., J. Am. Chem. Soc., 74, 5943 (1952); Roberts J. D.,
Regan С. M., ibid., 75, 2069 (1953): Roberts J. D., Ilalmann M., J. Am. Chem. Soc.,
75. 5759 (1953).
48. Реутов О. А., Шашкина T. H., ДАН СССР, 133, 606 (1960); Tetrahedron, 18, 237
(1962).
49. Кarabatsos G. J., Orzech С. Е., Jr.. J. Am. Chem. Soc., 84, 2839 (1962); ibid., 87, 4394
(1965); Lee С. C... Kruger J. E., Wong E. W. C., J. Am. Chem. Soc., 87, 3986,
3987 (1965).
50. Maskill II., Southam R. M., Whiting M. C., Chem. Comm., 1965, 496.
51. Freund M.. Lenze F.. Ber., 24, 2150 (1891).
52. Karabatsos G. B., Orzech С. E., Megerson F. S., J. Am. Chem. Soc., 86, 1994 (1964).
53. Zelinsky A'., GuttJ.. Ber., 40, 4744 (1907): Demjanov N., ibid., p. 4961: Demjanov A.,
Dojarenko M., Ber., 41, 43 (1908).
54. Демьянов II., ЖРФХО. 35, 375 (1903); ibid., 39, 1077 (1907): Ber., 40, 4393
(1907).
55. Демьянов II., Лучников M., ЖРФХО, 35, 26 (1903).
56. Wallach О.. Ann., 353, 318 (1907).
57. Демьянов II., ЖРФХО, 36, 166 (1904).
58. Ruzicka L.. Brugger W., Helv. Chim. Acta, 9, 318 (1926).
59. Streitwieser A., Jr.. Schaeffer W. D., J. Am. Chem. Soc., 79, 2888 (1957).
60. Breuer A., Zincke T., Ann., 198, 141 (1879).
61. Erlenmeyer E., Ber., 14, 322 (1881).
(52. Montagne P. S., Rec. trav. chim., 24, 105 (1905); Rec. trav. chim., 25, 411
(1906).
13. Acree S. F., Am. Chem. J., 33, 180 (1905).
44. Meerwein II., van Emster K., Ber., 53, 1815 (1920).
65. Tiffeneau M., Bull. soc. chim. (France), 1, 1221 (1907).
66. Meerwein II., van Emster K., Ber., 55, 2500 (1922).
67. Meerwein IT.. Hammel O., Serini A., Vorster J., Ann., 453, 16 (1927).
68. Ingold С. K.. Ann. Repts. on Progress Chem. (Chem. Soc. London), 25, 133 (1928).
653
Глава X
69. Ingold С. К., Shoppee С. W., J. Chem. Soc., 1928, 365; Shoppee C. W., Proc. Leeds
Phil. Lit. Soc., Sci. Sect., 1, 301 (1928); Ingold С. K., Ann. Rept. on Progress Chem.
(Chem. Soc. London), 25, 124, 133 (1928).
70. Whitmore F. C., Rothrock H. S., J. Am. Chem. Soc., 54, 3431 (1932); Whitmore F. C.,
Fleming G. H., ibid., 55, 4161 (1933); Whitmore F. C., Wittie E. L., Popkin A. II.,
ibid., 61, 1586 (1939).
71. Dostrovsky I., Hughes E. D., J. Chem. Soc., 1946 157, 161, 166, 169, 171.
72. Whitmore F. C., Wittie E. L., Harrian B. R., J. Am. Chem. Soc., 61, 1535 (1939);
Whitmore F. C., Popkin A. H., Bernstein H. I., Wilkins J. P., J. Am. Chem. Soc.,.
63, 124 (1941); Whitmore F. C., Zook II. D., J. Am. Chem. Soc., 64, 1783 (1942);
Whitmore F. C.. Weisgerber C. A., Shabica A. C., J. Am. Chem. Soc., 65, 1469
(1943).
73. Urry W. H., Kharasch M. S., J. Am. Chem. Soc., 66, 1438 (1944).
74. Whitmore F. C., J. Am. Chem. Soc., 54, 3274 (1932).
75. Whitmore F. C., Fleming G. H., J. Chem. Soc., 1934, 1269.
76. Wilmore F. C.. Whittle E. L., Popkin A. II., см. [70].
77. Dostrovsky I., Hughes E. D., Ingold С. К., J. Chem. Soc., 1946, 192.
78. Skell P., Hauser C. R., J. Am. Chem. Soc., 64, 2633 (1942).
79. Wolff L., Ann., 394, 23 (1912).
80. Arndt F., Eistert B., Ber., 68, 200 (1935).
81. Huggett C., Arnold R. T.. Taylor T. I.. J. Am. Chem. Soc., 64, 3043 (1942).
82. Schroeter G., Ber., 42, 2326 (1909); 49, 2704 (1916); Staudinger II., Hirzel H., ibid.,
p. 2522.
83. Beckmann E., Ber., 19, 988 (1886).
84. Kuhara M., Matsumiya K., Matsumami N., Met. Coll. Sci. Kyoto Imp. Univ., 1, 105
(1914); Kuhara M., Watanabe H., ibid., 1, № 9, p. 349 (1916). Chapman A. W.,
Howis С. C., J. Chem. Soc., 1933, 806; Chapman A. W., ibid., 1934, 1550; 1935, 1223;
Chapman A. W., Fidler F. A., ibid., 1936, 448.
85. Huisgen R., Witte J., Wilz H., Jira W., Ann., 609, 191 (1957).
86. Бродский А. И., Миклухин Г. П,, Изв. АН СССР, 32, 558 (1941); Миклухин Г. П.т
Бродский А. И., XcTt. физической химии, 16, 63 (1942).
87. Hofmann A. W., Вег., 15, 762 (1882).
88. Prosser Т. J., Eliel Е. L., J. Am. Chem. Soc., 79, 2544 (1957).
89. Hauser С. R., Renfrow W. В., Jr., J. Am. Chem. Soc., 59, 121 (1937).
90. Renfrow W. B., Jr., Hauser C. R., J. Am. Chem. Soc., 59, 2308 (1937); Bright R. D.„
Hauser C. R., ibid., 61, 618 (1939).
91. Newman M. S., Gildenhorn H. L.. J. Am. Chem. Soc., 70, 317 (1948).
92. Bothner-By A. A., Friedman L., J. Am. Chem. Soc., 73, 5391 (1951).
93. Briggs L. H., Lyttleton J. W., J. Chem. Soc., 1943, 421.
94. Jones L. W., Wallis E. S., J. Am. Chem. Soc,, 48, 169 (1926).
95. Wallis E. S., Nagel S. C., J. Am. Chem. Soc., 53, 2787 (1931).
96. Wallis E. S., Dripps R. D., J. Am. Chem. Soc., 55, 1701 (1933).
97. v. Braun J., Friehmelt E., Ber., 66, 684 (1933).
98. Kenyon J., Phillips H., Pittman V. P., J. Chem. Soc., 1935, 1072.
99. Archer S., J. Am. Chem. Soc., 62, 1872 (1940).
100. Kenyon J., Young D. P., J. Chem. Soc., 1941, 263.
101. Arcus C. L., Kenyon J., J. Chem. Soc., 1939, 916; Campbell A., Kenyon J., ibid., 1946-
25.
102. Lane J. F., Wallis E. S., J. Am. Chem. Soc., 63, 1674 (1941).
103. Wallis E. S., Moyer W. W., J. Am. Chem. Soc., 55, 2598 (1933).
104. Bell F., J. Chem. Soc., 1934, 835.
105. Lane J, F., Wallis E. S., J. Org. Chem., 6, 443 (1941).
106. Bartlett P. D., Pockel I., J. Am. Chem. Soc., 59, 820 (1937); Bartlett P. D., Bavley A.r
J. Am. Chem. Soc., 60, 2416 (1938); Bartlett P. D., Brown R. F., J. Am. Chem.
Soc., 62, 2927 (1940).
107. McKenzie A., Roger R., Wills G. О., J. Chem. Soc., 1926, 779.
108. Conant J. B., Carlson G. II., J. Am. Chem. Soc., 54, 4048 (1932).
109. Leithe W., Ber., 64, 2827 (1931).
110. Bernstein H. I., Whitmore F. C., J. Am. Chem. Soc., 61, 1324 (1939).
111. McKenzie A., Mills A. K., Ber., 62, 1784 (1929).
112. Pollak P. I., Curtin D. Y., J. Am. Chem. Soc.. 72, 961 (1950).
113. McKenzie A., Wood A. D.. Ber., 71, 358 (1938).
114. Benjamin В. M., Schaeffer II. J., Collins C. J., J. Am. Chem. Soc., 79, 6160
(1957).
115. Guthrie R. D.. J. Am. Chem. Soc., 89, 6718 (1967).
116. Hantzsch A., Werner A., Ber., 23, 11 (1890); Hantzsch A., Ber., 24, 13 (1891).
654
Перегруппировки в насыщенных системах
117. Meisenheimer JВег., 54, 3206 (1921); Meisenheimer J., Zimmerman Р., v. Kummer М.,
Ann., 446, 205 (1926).
118. Brady О. L., Bishop G., J. Chem. Soc., 127, 1357 (1925).
119. Cram D. J., J. Am. Chem. Soc., 74, 2129 (1952).
120. Bonner W. A., Collins C. J., J. Am. Chem. Soc., 77, 99 (1955); Bonner W. A., Col-
lins C. J., Lester С. T., ibid., 81, 466 (1959).
121. Caserio M. C., Levin R. D., Roberts J. D., J. Am. Chem. Soc., 87, 5651 (1965).
122. Baird R., Winstein S., J. Am. Chem. Soc., 85, 567 (1963).
123. Nevell T. P., de Salas E., Wilson C. L., J. Chem. Soc., 1939, 1188; частное сообщение
Ингольда, приведенное в работе Watson Н. В., Ann. Reports on Progress Chem. (Chem.
Soc. London), 36, 197 (1939), и в книге Watson H. В., Modern Theories of Organic
Chemistry, Oxford Univ. Press, 2nd, Edn., 1941, p. 208.
124. Winstein S., Trifan D. S., J. Am. Chem. Soc., 71, 2953 (1949); Roberts J. D., Ben-
nett W., Armstrong R., ibid., 72, 3329 (1950).
125. Hughes E. D., Bull. soc. chim. (France), 18, p. C41 (1951); Winstein S., ibid.,
p. C55.
126. Brown P., Hughes E. D., Ingold С. K., Smith J. F., Nature, 168, 65 (1951).
127. Roberts J. D., Lee С. С., J. Am. Chem. Soc., 73, 5009 (1951).
128. Hughes E. D., J. Chem. Soc., 1946, 164, 166, 171.
129. Hoffmann H. M. R., Fraser G. M., Chem. Comm., 1967, 561.
130. Martin R. J. L., Thesis, London, 1949; Hughes E. D., Ingold С. K., Martin R. J. L.,
Meigh D. F., Nature, 166, 679 (1950).
131. Winstein S., Trifan D., J. Am. Chem. Soc., 71, 2953 (1949); 74, 1147, 1152
(1952).
132. Roberts J. DLee С. C.. J. Am. Chem. Soc., 73, 5009 (1951); Roberts J. D., Lee С. C.,
Saunders W. H., Jr., ibid., 76, 4501 (1954).
133. Schleyer P. von R., Watts W. E., Fort R. C., Jr., Comiserow M. B., Olah G. A., J. Am.
Chem. Soc., 86, 5679 (1964); Saunders M., Schleyer P. von R. Olah G. Л., ibid.,
p. 5680.
134. Bartlett P. D., Non-classical Ions, Benjamin W. A., Inc., New York, 1965,
p. 525.
135. Brown II. C., Special Pubs. Chem. Soc., 16, 140 (1962).
136. Winstein S., Shatavsky M., Norton C., Woodward R. D., J. Am. Chem. Soc., 77, 4183
(1955).
137. Winstein S., Ordronneau C., J. Am. Chem. Soc., 82, 2084 (1960).
138. Story P. R., Saunders M., J. Am. Chem. Soc., 84, 4876 (1962).
139. Le Ny G., Compt. rend., 251, 1526 (1960).
140. Lawton R. G., J. Am. Chem. Soc., 83, 2399 (1961); Bartlett P. D., Bank S., ibid.,
p. 2591; Bartlett P. D., Bank S., Crawford R. J., Schmid G. H., ibid., 87, 1288 (1965);
Bartlett P. D., Sargent G. D., ibid., p. 1277; Bartlett P. D., Clessen W. D., Cogdell T. J.,
ibid., p. 1308; Bartlett P. D., Trahanovsky W. S., Boldon D. A., Schmid G. H., ibid.,
p. 1314.
141. Roberts J. D., Mazur W. H., J. Am. Chem. Soc., 73, 2509 (1951); Mazur W. H., Whi-
te W. N., Semenov D. A., Lee С. C., Silver M. S., Roberts J. D., J. Am. Chem. Soc.,
81, 4390 (1959).
142. Thomson J. J., Phil. Mag., 21, 225 (1911); 24, 209 (1912).
143. Bergstrom C. G., Siegel S., J. Am. Chem. Soc., 74, 145 (1952).
144. Caserio M. C., Graham W. H., Roberts J. D., Tetrahedron, 11, 171 (1960).
145. Hart H., Sandri J. M., J. Am. Chem. Soc., 81, 320 (1959); Hart H., Law P. A., ibid.,
84, 2462 (1962).
146. Hartman A., Hirshfeld F. L., Acta Cryst., 20, 80 (1966).
147. Pitman C. U., Jr., Olah G. A., J. Am. Chem. Soc., 87, 5123 (1965).
148. Vogel M., Roberts J. D., J. Am. Chem. Soc., 88, 2262 (1966).
149. Stevens T. S., Creighton E. MGordon A. B., MacNicol M., J. Chem. Soc., 1928, 3193;
Stevens T. S., ibid., 1930, 2107; Stevens T. S., Sneddon W. W., Steller E. T., Thomp-
son T., ibid., p. 2119; Thompson T., Stevens T. S., ibid., 1932, 55, 1932; Dunn J. L.,
Stevens T. S., ibid., p. 1926; ibid., 1934, 279.
150. Thompson T., Stevens T. S., J. Chem. Soc., 1932, 69.
151. Thompson T., Stevens T. S., J. Chem. Soc., 1932, 1932; Hughes E. D., Ingold С. K.,
J. Chem. Soc., 1933, 71; Wittig G., Mangold R., Felletshin G., Ann., 580, 116
(1948).
152. Thompson T., Stevens T. S., J. Chem. Soc., 1932, 55.
153. Stevens T. S., J. Chem. Soc., 1930, 2107.
154. Johnson R. A. W., Stevens T. S., J. Chem. Soc., 1955, 4887.
155. Bennett G. M., Chapman A. W.. Ann. Repts. on Progress Chem. (Chem. Soc. London)
27, 123 (1930); cp. [162].
655
Глава X
156. Dunn J. L., Stevens T. S., J. Chem. Soc., 1932, 1926.
157. Campbell A., Houston A. H. L., Kenyon J., J. Chem. Soc., 1947, 93: Brewster J. II.,
Kline M. W., J. Am. Chem. Soc., 75, 5179 (1953).
158. Hill R. K., Chan T.-K., J. Am. Chem. Soc., 88, 866 (1966).
159. Wittig G., Experientia, 14, 389 (1958).
160. Wittig G., Lohman L., Ann., 550, 260 (1942).
161. Wittig G., Happe W., Ann., 557, 205 (1947).
162. Hauser C. R., Kantor S. W., J. Am. Chem. Soc., 73, 1437 (1951).
163. Wittig G., Clausnizer R., Ann., 588, 145 (1954); Wittig G., Stahnecker E., ibid., 605,
69 (1957).
164. Wittig G., Schlor II., Suomen Kemistilehti, 31, 2 (1958).
ГЛАВА XI
ПЕРЕГРУППИРОВКИ НЕНАСЫЩЕННЫХ СОЕДИНЕНИЙ
1. Катионотропия ....................................... 658
2. Псевдоосновность и анионотропия...................... 691
3. Валентная таутомерия................................. 710
Список литературы .................................. 731
42-0772
1. Катионотропия
а. Развитие концепции таутомерии. После того как Гейтер [1] в 1863 г.
получил ацетоуксусный эфир, развернулась дискуссия о строении этого-
вещества [2], в которой участвовали, с одной стороны, Гейтер, а с другой —
Франкланд и Дюппа. Этот вопрос не был разрешен и в 1877 г., когда Вис-
лиценус [3] изучил отдельные стадии присоединения металлов к ацетоуксус-
ному эфиру и его алкилирования. В поисках аналогичных явлений Конрад
и Бишоф [4] обнаружили в 1880 г. наличие таких же свойств у малонового
эфира; позже подобные свойства были установлены и у многих других кето-
нов и эфиров не вполне выясненного строения, обладающих способностью
алкилироваться. Вопрос о структуре подобных соединений стал рассматри-
ваться с иных позиций, после того как Михаэль [5] в 1887 г. показал, что
как О-, так и С-производные ацетоуксусного эфира можно получить из его
соответствующих металлических производных. Поэтому с учетом многих
аналогий был сделан вывод, что на основании этих реакций нельзя точно
установить положение атома металла. Даже если удавалось надежно уста-
новить структуру исходного вещества, строение его металлических производ-
ных оставалось неясным.
В то же время при исследовании не только кетонов и сложных эфиров, но
и других классов органических соединений также были обнаружены случаи
аналогичного характера. В 1877 г. Бутлеров [6], изучая изомеризацию
диизобутиленов, объяснил перемещение двойной связи присоединением
и отщеплением воды. Он считал, что вследствие легкости такого обратимого
изомерного превращения вещество может вступать в реакцию то в виде одной,
то в виде другой структуры в зависимости от природы реагента и условий
реакции
СН3
(СН3)3С—СН = С//
СНз
он
| СНз
(СН3)зС-СН2-С\
CH3J
сп2
(СНз)дС-СН2— С
СН3
В 1882 г. Байер и Экономидес [7] установили, что обычный изатин дает как
О-, так и N-производные. Байер предложил объяснить это явление при
помощи представлений о псевдомерии. В соответствии со взглядами Байера
вещество может образовываться в процессе реакций в виде нескольких струк-
тур, которые легко взаимопревращаются. Причина того, что может быть
выделена только одна форма вещества, заключается в большой устойчивости
этой формы по сравнению с другими формами. Изатину Байер несколько
произвольно приписал имидольную структуру.
В 1874 г. Грисс [8] синтезировал «-бромдиазоаминобензол двумя способа-
ми, которые, казалось, должны были привести к образованию изомеров.
В 1884 г. Гольдшмидт [9] установил, что «-нитрозофенол, ранее полученный
Байером и Каро действием азотистой кислоты на фенол, также можно полу-
чить действием гидроксиламина на «-бензохинон.
В 1885—1886 гг. Лаар [10] опубликовал свои интересные взгляды по вопросу
строения подобных соединений. Он заметил, что неясность в строении веществ
обсуждаемого типа можно объяснить наличием нескольких формальных
структур, которым присуща неопределенность положения одного атома
водорода и по крайней мере одной двойной связи, как в случае структур
HXY = Z и X = YZH; Лаар назвал подобную систему триадной. Он счи-
тал, что такие две структуры нельзя полностью отделить одну от другой;
они представляют собой лишь крайние положения внутримолекулярного
колебательного состояния в одном веществе. Такое явление он назвал
658
Перегруппировки ненасыщенных соединений
таутомерией', этот термин широко применяется до настоящего времени для
описания подобных явлений, однако при этом представления Лаара не счи-
таются абсолютно верными.
Большинству современников Лаара было ясно, что он не опроверг теорию
Бутлерова и Байера, заключающуюся в том, что каждое вещество представ-
лено одной структурой, а легкость изомерного превращения в любом направ-
лении либо позволяет образоваться любому из изомеров из другого, либо
допускает реакцию, которой должна предшествовать изомеризация с образо-
ванием другого изомера. Впоследствии, примерно через десять лет, когда
были выделены в чистом виде легко переходящие друг в друга изомерные
формы многих таких веществ, как р-дикетоны, 0-кетоэфиры, 0-альдоэфиры
и жирные нитросоединения, эта теория получила весьма убедительное под-
тверждение.
В 1896 г. Кляйзен [11] установил, что ацетилдибензоилметан и трибензоил-
метан можно выделить в двух кристаллических формах. Одна из этих форм
представляет собой кислоту умеренной силы, которая легко образует окра-
шенную комплексную соль железа, соль меди и растворимую в воде натрие-
вую соль. Вторая форма представляет вещество, которое Ганч отнес к классу
псевдокислот: оно непосредственно не образует солей, но по истечении неко-
торого времени, которое можно принять за время, необходимое для превра-
щения псевдокислоты в изомерную истинную кислоту, появляется способ-
ность к солеобразованию. Такое же различие во времени наблюдается и при
обратных процессах, а именно: из солей можно быстро получить исходную
кислоту, превращение которой в псевдокислоту протекает во времени. Такие
превращения вещества в расплавленном или растворенном состоянии ука-
зывают на возможность обратимости подобных процессов, что приводит
к равновесию, если кристаллизация не ведет к выделению одной из форм.
Равновесие между формами в растворах зависит от характера растворителя:
в общем в более полярных растворителях оно сдвинуто в сторону образования
псевдокислот. Кляйзен правильно считал, что псевдокислота имеет кетон-
ную, а изомерная кислота — енольную структуру:
С6Н5СОСН(СОС6Н5)2 С6Н5С(ОН) = С(СОС6Н5)2
Псевдокислота Кислота
Таким образом, рассматриваемый случай является примером кето-енолъной
таутомерии. Компоненты этой системы взаимопревращаемы, но могут быть
выделены в виде веществ определенной структуры: кетона и енола.
В те же годы Вислиценус начал исследование изомерии метилового и этило-
вого эфиров формилфенилуксусной кислоты, каждый из которых был получен
в жидкой и кристаллической формах [12]. Обе формы представляли собой
кислоты, причем кристаллическая форма оказалась более кислой; окрашен-
ный комплекс с железом(Ш) образует только менее кислая жидкая форма.
Вначале Вислиценус предполагал, что жидкая форма имеет альдегидную
структуру, а кристаллическая — оксиметнленовую, хотя, исходя из такого
предположения, трудно было понять образование комплекса с хлоридом
железа(Ш) только из жидкой формы. Но дальнейшие исследования, прове-
денные Вислиценусом, Мейером и Дикманом [13] в 1912—1920 гг., показали,
что жидкая форма в действительности является смесью альдегидной формы
и одного из стереоизомеров оксиметиленовой структуры. Несомненно,
последний представляет собой г^нс-изомер с внутримолекулярной водородной
связью, обладающий меньшей кислотностью, но способный образовывать
внутримолекулярное комплексное соединение (гл. П, разд. 3,д). Более кис-
лая кристаллическая форма, которая образует соль железа, а не его коорди-
42* 659
Глава XI
нацпопное
мерой
соединение, является, несомненно,
/пракс-оксиметиленовым
изо-
сно
Cell JjHCOOR
неон
С0Н5ССООП
поен
С6Н5ССООВ
Первоиачально этим фактам не придавали должного значения, однако Вис-
лицепусу уже в 1896 г. была ясна общая склонность таких изомеров к обра-
тимым взаимопревращениям в растворах.
В том же году Кнорр [14] опубликовал результаты исследования в области
изомерии этиловых эфиров дибензоил- и диацетилянтарных кислот; каждый
из них был получен в нескольких формах, причем последний — пе менее
чем в пяти. Две из таких форм были идентифицированы как возможные
стереоизомерные дикетоны, две — как стереоизомерные моноенолы и послед-
няя — как один из трех возможных стереоизомеров диенолов; значительно
позже такой вывод подтвердили Кнорр и Кауфман [15].
СН3СОСНСООС2Н5
С.Н3СОСНСООС2Н5
мезо п рацемический
СН3С(ОН)^ССООС2П5
I
СН3СОСИСООС,Н5
цис и транс
СН3С(ОН) = ССООС2П5
I
СН3С(ОН) = ССООС2Н5
(известна одна форма
из трех возможных)
В своих ранних работах Кнорр установил, что если растворить любую из
этих форм, то происходит взаимопревращение изомеров и образуется их
равновесная смесь. Впоследствии это было также подтверждено Кауфма-
ном [16], которому удалось аналитическими методами проследить за пре-
вращениями и определить количественный состав равновесной смеси изо-
меров.
Как Вислиценус, так и Кнорр еще в первоначальной стадии исследования
высказали интересные предположения о механизме изомеризации, полагая,
что подвижный атом водорода перемещается в виде иона. Этот вопрос более
подробно будет рассмотрен ниже.
Еще в 1896 г. Ганч и Шульц [17] выделили вторую форму фенилпитрометана.
Вместо хорошо известного желтого масла, слаборастворимого в водном рас-
творе щелочи, они при осторожном подкислении щелочного раствора выдели-
ли бесцветное, кристаллическое, более легко растворимое в воде кислое
вещество, которое со временем, а иногда и быстро полностью превращалось
в желтое масло. Маслянистое вещество представляло собой псевдокислоту,
а кристаллический изомер, который Ганч назвал azfu-формой, является кис-
лотой в обычном смысле:
c6h5ch2no2 c6h5ch=nooh
Псевдокислота Кислота
Ганч [18] показал, что по уменьшению электропроводности водных растворов
можно проследить за самопроизвольным переходом аг/п-формы в псевдо-
кислоту.
После этих важных экспериментальных доказательств казалось странным
придерживаться гипотезы внутримолекулярных колебаний Лаара; тем не
менее она не только сохранилась, но даже разрабатывалась в течение десяти
лет. Взгляды Лаара были признаны бесплодными и отвергнуты еще до 1911 г.
когда Кнорру, Роту и Авербеку [19] удалось разделить давно известное тауто-
мерное вещество — ацетоуксусный эфир на его кетонную и енольную формы.
Согласно теории Лаара, выделение изомеров в чистом виде в случае такого
Перегруппировки ненасыщенных соединений
типичного таутомерного вещества было невозможно. Свойства выделенных
изомеров были полностью аналогичны свойствам изомеров, полученных ранее
в случае других кето-енольных систем. Однако вследствие того, что в данном
случае взаимное превращение протекает значительно быстрее, выделение
отдельных изомеров оказалось необходимым производить при более низких
температурах, чем примененные, например, Кляйзеном при выделении изо-
меров ацетилдибензоилметана. Кето-форма ацетоуксусного эфира кристал-
лизуется из петролейного эфира при —80 °C. Енольная форма была выделена
при обработке хлористым водородом избытка суспензии натриевого произ-
водного в петролейном эфире также при —80 °C и с последующим фильтро-
ванием и отгонкой растворителя. При этом было показано, что между указан-
ными формами в жидком ацетоуксусном эфире и его растворах устанавли-
вается равновесие, очень легко сдвигающееся при действии кислот и осно-
ваний; это обратимое взаимопревращение является реакцией первого порядка.
Впоследствии Мейер и сотр. [20] приготовили чистые кетоэфир и енол более
удобным способом — перегонкой ацетоуксусного эфира под вакуумом в от-
сутствие катализаторов. Во избежание щелочной реакции стекла они поль-
зовались кварцевой аппаратурой. Более летучим изомером оказался енол.
С 1911 г. благодаря тому, что Лаар объединил между собой множество'
несвязанных наблюдений над этим явлением, стало ясно, что концепция тауто-
мерии должна рассматриваться с позиций Бутлерова — Байера, т. е. как
обратимое превращение изомеров. Проблема выделения и доказательства
идентичности таутомеров стала делом экспериментальной техники и приме-
няемой температуры.
Благодаря разработанному Мейером [21] в 1911 г. простому объемному
методу определения содержания енола в кето-енольной смеси удалось более
точно выяснить картину равновесия между молекулами резко различной
структуры. Этот метод основан на том, что енолы, но не кето-формы быстро
реагируют с бромом, образуя бромкетоны. Избыток брома связывают нафто-
лом-2, бромкетон восстанавливают иодистым водородом, а выделяющийся
иод оттитровывают тиосульфатом.
-СОН -С=О —С = О
II 7,7^ I эд? I + Н
В табл. 163 приведены значения равновесного содержания енола (в процен-
тах), определенные Мейером в жидких кето-енолах при обычной температу-
ре. Другие подобные данные приведены в разд. 1,г.
Мейер изучал также содержание енола в растворах кето-енолов в равно-
весном состоянии. Теоретически более полярный растворитель должен
благоприятствовать образованию более полярного таутомера, так как при
этом может возрастать энергия сольватации. Мейер установил, что более
полярные растворители благоприятствуют образованию кето-форм 0-дикето-
на и p-кетоэфиров; некоторые данные Мейера, касающиеся растворов ацето-
уксусного эфира, приведены в табл. 164. На основании этих данных можно
сделать вывод, что, согласно Мейеру, енолы менее полярны, чем кето-формы.
Как известно, енолы более летучи, чем кето-формы. Оба указанных факта
подтверждают предположение о большей полярности кето-форм, обычно
объясняемое (гл. I, разд. 2,е) значительным уменьшением полярности енолов
благодаря наличию внутримолекулярной водородной связи *. Однако, как
* Мейер установил, что для растворов многих кето-енолов в ряде растворителей равновес-
ное соотношение кетон/енол определяется двумя факторами, причем один зависит от при-
роды кето-енола, а другой — от природы растворителя. В соответствии с этим свободная
энергия изомеризации любого кето-енола в любом растворителе приблизительно равна
661
Глава XI
Таблица 163
Содержание енола в жидких кето-енолах (по данным Мейера)
Кето-енол Содержание енола, % Кето-енол Содержание енола, %
Метиловый эфир ацетоуксус- ной кислоты 4,8 Этиловый эфир ацетондикар- боновой кислоты 16,8
Ацетоуксусный эфир 7,5 Ацетилацетон 80,4
Метиловый эфир бензоилук- сусной кислоты -Этиловый эфир бензоилуксус- ной кислоты 16,7 29,2 Бензоилацетон 98,0
Таблица 164
Содержание енола в ацетоуксусном эфире в различных растворителях при 18° С
(по данным Мейера)
Растворитель Содержание енола, % Растворитель Содержание енола, %
Вода 0,40 Ацетон 7,3
25%-ный метиловый спирт 0,83 Хлороформ 8,2
50%-ный метиловый спирт 1,52 Нитробензол 10,1
50%-ный этиловый спирт 2,18 Этилацетат 12,9
Метиловый спирт 6,87 Бензол 16,2
Этиловый спирт 10,52 Диэтиловый эфир 27,1
Пропиловый спирт 12,45 Сероуглерод 32,4
Амиловый спирт 15,33 Гексан 46,4
показал Вассерман [22], в случае кето-енолов, в енольных формах которых
не легко возникают внутримолекулярные водородные связи, растворитель
оказывает противоположное влияние. В качестве примера Вассерман при-
водил дикетен («димерный кетен»), который в растворителях, не содержащих
гидроксила, проявляет себя как кетон, а в водной среде ведет себя как енол:
СН2 = С-СН2 СН2 = С —СН
II I II
о-с=о о-с—он
сумме свободной энергии изомеризации кето-енола в стандартном растворителе и разности
свободных энергий сольватации кетона и енола по сравнению со стандартным кето-енолом
в этом растворителе. Эти выводы явились одним из первых примеров применения прин-
ципа аддитивности свободной энергии, имеющего в настоящее время большое значение.
В данном случае этот принцип следует учитывать, поскольку разности энергий сольва-
тации должны быть почти независимыми от тех составных частей молекул, которые не пре-
терпевают изменений в процессе реакции, а сольватационные силы в высокой степени
локализуются на растворенных молекулах. Экспериментальные данные согласуются
с законом Вант-Гоффа — Димрота; этот закон можно выразить следующим образом:
^раствор = ХгазгГенри>
где К — константы равновесия для процесса изомеризации в растворителе и в газооб-
разном состоянии; г — отношение растворимостей (т. е. константа Генри) газообразных
изомер ов в растворителе.
662
Перегруппировки ненасыщенных соединений
За несколько лет до того, как Мейер разработал способ определения равно-
весных концентраций енолов, Лэпуорт показал, что бромирование позво-
ляет определить скорость образования енола из карбонильного соединения,
находящегося в равновесии с очень малым количеством енола. В 1904 г.
Лэпуорт в основной работе установил, что скорость бромирования ацетона
в кислых водных растворах пропорциональна концентрации ацетона и концен-
трации водородных ионов и не зависит от концентрации брома [23].
Он объяснил это енолизацией кетона, катализируемой кислотами:
Н* Вгг
iCH3COCH3 -----------> СН3С(ОН) = СН2 —-------> СН3СОСН2Вг
“° Медленно ' ' Быстро
В 1912 г. Мейер [24] показал, что скорость катализируемого кислотами бро-
мирования малоновой кислоты в водной среде также не зависит от концен-
трации галогена. В 1926 г. Даусон изучил иодирование ацетона в водной
среде, катализируемое различными кислотами и основаниями, и показал,
что при любом катализаторе скорость реакции не зависит от концентрации
галогена [25]. Белл и Лонге-Хиггинс установили аналогичные факты при
хлорировании и бромировании ацетона, т. е. при процессах, катализируе-
мых гидроксильными ионами [25]. Эти исследования подтвердили правиль-
ность объяснения, данного Лэпуортом. Единственное изменение, которое
следовало сделать, связано с тем, что при катализе основаниями сама ено-
лизация представляет собой сложный процесс, скорость которого опреде-
ляется ионизацией, а ионизация равным образом определяет скорость гало-
генирования.
СН3СОСН3 ---------> СН3С(О) = СН2 —---> СНзСОСНоВг
Медленно Быстро
Однако при кислотном катализе выше определенной кислотности среды
быстрой реакцией может служить галогенирование молекулярного енола.
Такой вывод сделан в недавней работе Белла и сотр. [26].
На примерах главным образом хлорирования или бромирования этилмало-
ната и ацетона авторы показали, что при уменьшении концентрации гало-
гена до 10 ~4 /I/ и ниже скорость его реакции с анионом или енолом может
быть меньше скорости образования аниона или енола, т. е. определяющей
скорость становится атака галогеном, и поэтому общая скорость становится
пропорциональной концентрации галогена. На примере галогенирования
этилмалоната, изучая зависимость скорости реакции при таких концентра-
циях от pH, авторы затем показали, что при условиях, близких к ней-
тральным, и при разбавлении кислоты до pH 3 галоген взаимодействует
с анионом, но при более высокой кислотности галогенирование протекает
главным образом через молекулярный енол.
б. Аналогия между таутомерией и реакцией присоединения. Коль-
чато-цепная таутомерия. Вывод о том, что каждый таутомер существует
самостоятельно и что таутомерия является одним из типов химических
реакций, привел со временем к представлениям о наличии аналогии между
таутомерными превращениями и другими химическими реакциями. В част-
ности, Хилда Ингольд [27] высказала предположение, согласно которому
таутомерные превращения можно сравнить с обратимыми реакциями при-
соединения. Такой подход оказался полезным направлением в работах
начала 20-х годов нашего столетия, посвященных так называемой кольчато-
цепной таутомерии.
В качестве примера можно привести альдольную конденсацию: адденд
Н—С, с которым должна быть связана «активирующая» группа X (напри-
663
Глава XI
мер, карбонильная группа), присоединяется к ненасыщенному акцептору
С = О следующим образом: Н к О и С к С, т. е.
II , I I
Н — СХ + С = О ХС — С-0- н
II II
Уравнение дано в обратимой форме, так как Хилда Ингольд доказала общую
обратимость альдольных конденсаций. При этом было отмечено, что наибо-
лее ярко выраженной внутримолекулярной формой этой реакции является
кето-енольное превращение; группа НС кето-структуры «активизируется»,
становясь аддендом под влиянием соседней группы С = О, действующей
также как акцептор
I I , I I
н-с-с = о т» с=с—о-н
На основании интерполяции был сделан вывод, что могут встречаться
и менее ярко выраженные формы этого основного процесса; к их числу
относится обратимое взаимное превращение изомеров с открытой цепью
и циклического соединения, показанное на примере кето-циклолъной тауто-
мерии
I । , I I
Н —СХ С = 0 7^СХ — с —о —н
(CRaJra (CRa)»
Торп и сотр. [28] изучили ряд систем такого типа. В качестве кетонов при-
менялись сс-кетоглутаровые кислоты с различными P-заместителями, а цикли-
ческими изомерами служили циклопропаноловые кислоты
СОСООН
R2C
\н2соон
С(ОН)СООН
r2c
снсоон
Такие системы были не очень подвижными, и для ускорения взаимного
превращения добавлялась щелочь: таким образом устанавливалось равно-
весие между анионами кислот в водном растворе.
По Михаэлю, присоединение является исходным пунктом для рассмотре-
ния другой группы аналогичных процессов: адденд Н — С, несущий акти-
вирующую группу X, присоединяется к акцептору С = С, несущему акти-
вирующую группу Y подобного же типа, причем Н присоединяется к угле-
роду. связанному с Y, а С — к другому концу этиленовой связи
II I I I
H-CX + C = CY CX-C-CY-H
II II
Форлендер [29] доказал обратимый характер этой реакции. Для наиболее
ярко выраженной внутримолекулярной формы этой реакции характерно
перемещение водородного атома между крайними атомами пропиленовой
системы, связанными с активирующими группами.
I I I
Н —СХ —C=CY
I I I
СХ = С—CY — Н
В этом случае имеется так называемая «трехуглеродная» триадная система,
широко изученная Торпом на примере глутаконовых кислот и их эфиров,
664
Перегруппировки ненасыщенных соединений
в которых активирующими группами X и Y являются карбоксильные груп-
пы. Он показал, что в асимметрично замещенных глутаконовых кислотах
и эфирах атом водорода очень легко перемещается с одного конца пропиле-
новой системы на другой. Соответствующую трехуглеродную колъчато-цеп-
ную систему можно изобразить в общем виде следующей схемой:
1 । । , I II
Н-СХ C = CY СХ——С —CY—Н
(CR2)n (CR2)n
Торп и сотр. [30] при изучении стадий процесса димеризации встретились
с примером таутомерии такого типа, которую обнаружили Гутцейт [31]
в карбоксиглутаконовых эфирах и Феркаде [32] в цианглутаконовых эфи-
рах; этот процесс в значительной степени катализировался основаниями.
Установлено, что при действии оснований на этиловый эфир а, а'-дикарбо-
ксиглутаконовой кислоты образуются два близких по структуре димера
(стереоизомерные различия не учитываются). Первоначально образуется
вещество, жидкое при комнатной температуре, которое представляет собой
ненасыщенный эфир с открытой цепью, имеющий строение нормального
продукта присоединения по Михаэлю а-СН-группы одной молекулы глу-
таконового эфира к двойной связи Ср = Cv другой молекулы.
(С2Н6ООС)2СНСН = С(СООС2Н6)2 (С2Н5ООС)2СНСНСН(СООС2Н5)2
(С2Н5ООС)2С = СНСН(СООС2Н5)2 (С2Н5ООС)2С = СНС(СООС2Н5)2
Такое строение этого соединения было доказано гидролизом, термическим
декарбоксилированием и последующим окислением до метантриуксусной
и щавелевой кислот. Жидкий димер очень быстро изомеризуется в твердый
насыщенный димер. Это превращение катализируется основаниями. Строе-
ние димера как производного циклобутана было установлено декарбокси-
лированием продуктов гидролиза с образованием насыщенной тетракарбо-
новой кислоты, полученной в пяти возможных стереоизомерных формах,
характеризующихся, как и их различные ангидриды, генетическими связя-
ми, обусловленными указанной структурой. Если в процессе изомеризации
жидкого димерного эфира заставить кристаллизоваться твердый изомер, то
превращение протекает полностью. Однако без дополнительного вмеша-
тельства в жидкой фазе устанавливается определенное равновесие, при кото-
ром ненасыщенный и насыщенный изомеры содержатся в следующем соот-
ношении:
(С2Н5ООС)2СН СН = С(СООС2Н5)2 (С2Н5ООС)2С-СНСН(СООС2Н5)2
II I \
(С2Н5ООС)2СНСН —С(СООС2Н5)2 (С2Н6ООС)2СНСН-С(СООС2Н5)2
20% 80%
Особый интерес представляет случай равновесного присоединения воды или
спиртов к карбонильным соединениям с образованием полуацеталей
। , I
Н —OR + C = O RO —С —О-Н
I 1
Аналогичная триадная система предполагается в таутомерном превраще-
нии карбоксильной группы
। , I
Н —О — С=О О = С—ОН
665
Глава XI
Кольчато-цепным аналогом таких таутомерных превращений является так-
же кето-лакгполъная система, которую можно представить следующим
образом:
I ' I
Н-0 С=О — с— о-н
(СВг)п (СВг)п
На основании приведенных данных Якобсон и Штельцнер [33] предсказали
таутомерию этого типа и отметили ее значение для химии сахаров. Сахара
с восстановительными свойствами обладают типичными свойствами альде-
гидов и кетонов, в то время как их изомерия и отношение к гликозидам
свидетельствует о том, что они являются лактолами. Эти факты легко объяс-
нить возможностью обратимых превращений описанного типа. Две стерео-
изомерные лактольные формы сахара не должны непосредственно превра-
щаться друг в друга, но могут достигать равновесного состояния с участием
их изомеров, имеющих открытую цепь. Этот факт позволяет объяснить
явление мутаротации, которое наблюдается в случае свежеприготовленных
растворов практически всех сахаров с восстановительными свойствами.
Эти идеи были развиты в 1913 г., но не привлекали внимания до 1924 г.
В этот период обсуждались два предположения, сделанные еще в 1903 г.,
и было показано, что они не согласуются с опытными данными. С другой
стороны, общий обзор [34] соединений класса сахаров, как обладающих,
так и не обладающих свойством мутаротации, показывает, что их строение
соответствует требованиям теории Якобсона и Штельцнера, а именно системе
R
О-----С—X —н
Х(СН2)/
-с любым электроотрицательным X, причем в качестве R может быть любой
радикал.
в. Развитие концепции прототропии. Представление о подвижном
атоме водорода, перемещающемся в виде протона, возникло еще в 1896 г.
К этому времени начали накапливаться факты, указывающие на существо-
вание определенной связи между скоростью достижения таутомерного рав-
новесия и влиянием среды на степень диссоциации. Кляйзен [35] установил,
что лабильность известных форм ацетилдибензоилметана — наибольшая
в водном спирте, меньше в сухом спирте или ацетоне и чрезвычайно мала
в бензоле. Аналогичные наблюдения Кнорра [36] легли в основу его теоре-
тической концепции, которую Вислиценус [37] в обзоре о таутомерии, опубли-
кованном в 1898 г. принял и сформулировал в следующем виде:
«Это явление невозможно рассматривать, не став на ту точку зрения, что
взаимопревращение форм связано с диссоциацией. Можно считать, что эти
вещества всегда до некоторой степени диссоциированы, причем освобожде-
ние подвижного водорода в виде катиона происходит даже при незначи-
тельной степени диссоциации. В анионе изменения связей будут происходить
значительно легче, так как они в этом случае не связаны с перемещением
атома водорода».
Вислиценус считал, что медленное протекание таутомерных превращений
объясняется одновременной диссоциацией лишь небольшого количества
молекул.
Этой теории придерживались только исследователи, выдвинувшие ее. При-
знание и развитие ее шло медленно; в течение четверти века она не подви-
666
Перегруппировки ненасыщенных соединений
нулась вперед. Причина этого очевидна: если атом водорода диссоциирует
в виде положительного иона, который может занимать различные положе-
ния, то отрицательный заряд, оставшийся в старом положении, должен
тоже перемещаться в новое положение. Как происходит это перемещение,
нельзя было точно представить без электронной теории валентности, в соот-
ветствии с которой перемещение связи является смещением электрического
заряда.
В 1922 г. Пиго и автор настоящей книги [38] предприняли исследование
с целью установления связи между подвижностью формально одинаковых
триадных систем и их структурой, а следовательно, и их способностью алки-
лироваться, присоединять ионы металлов и проявлять другие химические
свойства, зависящие от способности к ионизации. Авторы исследовали «сим-
метричные» триадные системы типа RXHY = XR', на большую подвиж-
ность которых указывает как идентичность синтезированных описанным
способом веществ с взаимозамещенными R и R', так и два ряда реакций
отдельных таутомеров, полученных любым путем. Первоначально сравни-
вались четыре формальные системы, которые можно построить при замене
X или Y на N или С, a R и R' — на арильные группы
ArNHN = NAr' ArCH2N = CHAr'
ArNHCR = NAr' ArCH2CR = CHAr'
Две из этих систем были изучены значительно раньше.
Грисс [39] первый открыл явление таутомерии в диазоаминосоединениях
ArNHN=NAr' ArN = NNHAr'
R 1874 г. он синтезировал «-бромдиазоаминобензол сначала из иона бензол-
диазония и «-броманилина, а затем из иона «-бромбензолдиазония и анили-
на, причем в обоих случаях были получены продукты, не отличающиеся
по своим свойствам.
CeHsNJ+BrCeHaNJ^ —> C6H5N = NNHC6H4Br по свойствам
BrCeH4NJ4-C6H6NH2 —> BrC6H4N = NNHCeH5 j не отличаются
Кроме того, Грисс изучил вещества, содержащие метильную группу вместо
брома в качестве заместителя в «ара-положении. Впоследствии Нельтинг
и Биндер [40] расщепили метильное производное, выделенное в единствен-
ной форме обычным восстановлением его до амина и гидразина и получили
смесь двух аминов и двух гидразинов:
CeH6NH2, CH3C6H4NH2, c6h5nhnh2, ch3c6h4nhnh2
Наконец, Мелдола и Стритфилд [41] изучили этилирование ряда диазоами-
носоединений этилатом натрия и иодистым этилом, в результате которого
образуются нетаутомерные конечные продукты. Из каждого несимметрично
замещенного диазоаминобензола ArNHN = NA/, выделенного в одной-
единственной форме, они получили смесь двух этильных производных:
ArN(C2H5)N = NAr' и ArN = NN(C2H6)Ar'
Эти производные были выделены из смеси и идентифицированы синтезами
из соответствующих ионов диазония и N-этилариламинов.
Подобным образом в 1895—1899 гг. Пехман [42], Марквальд [43], а также
Уилер и Джонсон [44] доказали наличие таутомерии в амидиновой системе
ArNHCR = NAr' ArN = CRNHAr'
Пехман установил идентичность Х-фенил-Х'-«-толилбензамидина, синтези-
рованного из N-фенилбензимидохлорида и «-толуидина и из N-и-толилбен-
667
Глава XI
зимидохлорида и анилина.
CeH5N = C(C6H5)Cl + CH3C6H4NH2 —» CeH5N = C(C6H5)NHC6H4CH3 | по свойствам
CH3C6H4N = C(C6H5)Cl + CeH5NH2 —> CH3C6H4N = C(C6H5)NHC6H5 J не отличаются
Марквальд осуществил гидролитическое расщепление этого вещества и полу-
чил два амина и два амида
c9h5nh2, ch3c6h4nh2, c6h5nhcoc6h5, ch3c6h4nhcoc6h5
Пехман блокировал таутомерную систему этилированием и выделил два
различных этильных производных:
CeH5N = C(C6H5)N(C2H5)C6H4CH3 и CH3CeH4N = C(CeH5)N(C2H5)CeH5
строение которых было подтверждено синтезом из соответствующих имидо-
хлоридов и N-этиларил аминов. Уилер и Джонсон установили по существу
то же самое в других случаях, особенно на примере формамидинов, которые
они синтезировали, исходя из имидоэфиров и аминов с соответствующими
фенильными и и-толильными радикалами:
CeH5N = CHOC2H5 + CH3CeH4NH2 —> C6H6N = CHNHC6H4CH3 1 по свойствам
CH3CeH4N = CHOC2H5 + C6H5NH2 —» CH3C6H4N = CHNHC6H5 J не отличаются
В отличие от подвижных диазоаминовой и амидиновой систем диарилиро-
ванная у различных атомов углерода метиленазометиновая система чрез-
вычайно малоподвижна [38]. Изомерные формы
ArCH2N = CHAr' и Ar'CH2N = CHAr
могут быть получены в отдельности конденсацией бензиламинов и бензаль-
дегидов при соответствующих заместителях. Как было показано впослед-
ствии [45], эти изомеры могут взаимно превращаться друг в друга лишь в при-
сутствии такого сильноосновного катализатора, как этилат-ион, но при этом
реакция протекает медленно. Такую же систему представляет собой арили-
рованная у концевых атомов углерода трехуглеродная система, изомерные
формы которой
АгСН2СН = СНАг' и Аг'СН2СН = СНАг
можно получить конденсацией 0-арилпропионатов и ароматических альде-
гидов [38]. Эти изомеры проявляют слабую склонность к взаимопревраще-
ниям [46], однако, как будет показано ниже, при действии этилата натрия
они могут медленно превращаться друг в друга.
В то время как диазоаминосоединения и амидины легко алкилируются,
замещенные метиленазометиновые и пропиленовые соединения не алкили-
руются. Подвижность систем, очевидно, находится в связи с их способно-
стью алкилироваться и зависит от электроотрицательности атома, от кото-
рого в каждой реакции отщепляется атом водорода. Отщепление протекает
легко, если этим атомом является азот, и трудно в случае углерода; природа
центрального атома триадной системы оказывает незначительное влияние.
Эти выводы остаются справедливыми при переходе от трехуглеродной систе-
мы, на концах которой находятся две арильные группы, к трехуглеродной
системе, па конце которой находится одна оршо-арилеповая группа, как
в случае индена [47]. Метоксииндены синтезированы обычными методами
через метокси-1-гидринданамины, представленные ниже в виде двух форм,
668
Перегруппировки ненасыщенных соединений
которые после их выделения оказались идентичными:
СН2
При окислении этого вещества, существующего лишь в одной форме, была
получена смесь двух метоксигомофталевых кислот
сн3о -/Y- СН2СООН jAj- СН2СООН
СООН СН3О —СООН
В результате блокирования таутомерной системы конденсацией с пиперо-
налем была получена смесь производных пиперонилидена, которую можно
разделить
С = СНС6Н3(ООСН2)
сн
С=СНС6Н3(ООСН2)
Инден благодаря своим особенностям, описанным в гл. IV, разд. 1,д, отно-
сится к одному из редких типов углеводородов, легко образующих металли-
ческие производные. Таким образом, высокая подвижность трехуглеродной
системы индена становится важным подтверждением ионизационной теории
таутомерии. Очевидно, таутомерной подвижностью обусловлена способность
алкилироваться и легкость образования металлических производных неза-
висимо от других общих химических свойств, например от способности
вещества реагировать либо как кислота, подобно большинству кето-енолов,
либо как основание, подобно диазоаминобензолу, либо обладать свойствами
углеводорода, как инден.
Вскоре после того, как были сделаны эти наблюдения, в ионную теорию
таутомерии на основе подвижного атома водорода или прототропии (как
назвал ее Лоури, убежденный в том, что она связана с переносом протона),
являющуюся по существу частным случаем катионотропии, было включено
предположение о смещении электронов по механизму сопряжения, сопро-
вождающем перенос протона. Развитие этой теории шло в двух направле-
ниях. Согласно первому, разрабатываемому Шоппи, Торпе и автором этой
книги [48], предполагалось наличие двухступенчатого механизма, пред-
ставленного ниже:
* ifi-Y-Ql- в xWi
Этот механизм обозначается символом В-SeI', что означает катализи-
руемое основанием мономолекулярное электрофильное замещение, сопро-
вождающееся перегруппировкой. В дальнейшем этот механизм для крат-
кости будет часто называться мономолекулярным механизмом прототроп-
ного превращения, несмотря на то что в соответствии с принятым нами общим
правилом определения молекулярности (гл. VII, разд. 1, д) приведенный выше
669
Глава XI
механизм является бимолекулярным. Такого рода неточность в обозначении
допускалась и раньше, когда некоторые реакции замещения алкилгало-
генидов, катализируемые ионом серебра, были обозначены символом Ag+-
SnI, в котором особо выделена роль катализатора и игнорируется числен-
ное выражение молекулярности для того, чтобы сохранить важную анало-
гию с обозначением некатализируемых реакций SnI. Причины, по которым
пришлось это сделать, частично вызываются обычно удобным, хотя и неточ-
ным, представлением о том, что ион водорода в отличие от других ионов
никогда не образуется в процессе реакции и не разрушается, а всегда толь-
ко переносится. Это означает, что он никогда не находится в свободном
состоянии, а всегда ковалентно связан с какими-либо атомами. Несмотря
на это все же можно явно обнаружить аналогию между высвобождением иона
водорода из перегруппировывающейся системы, как в случае, приведенном
выше, и высвобождением иона металла или иного катиона, которые могут
существовать, и даже длительное время, в свободном состоянии. Особое
свойство водорода заключается в его способности образовывать водородную
связь, что часто вызывает неопределенность числа молекул, входящих
в состав основания, катализирующего реакцию. Таким образом, приведен-
ное выше уравнение может представлять собой обычную ионизацию карбо-
новой кислоты в водной среде. От одного атома кислорода этой кислоты
отщепляется протон с образованием при этом мезомерного карбоксилат-иона,
высвободившийся протон может вновь связаться с любым атомом кислорода.
В этом случае катализирующим основанием может быть молекула воды
или чаще полимерная группа с неопределенным числом молекул, связан-
ных водородной связью. Для того чтобы избежать трудностей в этом вопро-
се, мы предпочли при определении молекулярности исключить катализи-
рующее основание. Однако главная причина этого будет выяснена позже:
дело в том, что определение прототропии сохраняет аналогию с анионотро-
пией (являющейся формой нуклеофильного замещения, протекающего с пере-
группировкой). Таким образом, возможно параллельное рассмотрение про-
тотропии и анионотропии как случаев обычных электрофильного и нуклео-
фильного замещений, протекающих без перегруппировок; при этом сохра-
няется удобное соответствие механизмов рассматриваемых реакцией.
В приведенном выше мономолекулярном механизме реакции таутомерной
системе придана триадная форма, однако предлагаемый механизм охваты-
вает как пентадные и вообще многоатомные сопряженные системы, так
и кольчато-цепные. Рассматривался также вопрос о том, почему исходное
вещество может при некоторых условиях предварительно превращаться
в измененную форму, например в сопряженную кислоту, которая
затем претерпевает таутомерные превращения: последовательные депрото-
низацию и соответствующую перегруппировку, согласно приведенному
механизму.
В дополнение к таким уже отмеченным влияниям на подвижность систем,
которые согласуются с механизмом B-SeI', было установлено, что можно
ожидать и других эффектов, также отвечающих опыту. Один из таких эффек-
тов заключается в катализе процесса прототропии. В соответствии с при-
веденным механизмом необходимо наличие основного катализатора. Опыт
показывает, что в тех случаях, когда подвижность системы достаточно мала
и позволяет уловить влияние катализатора, основание представляет собой
универсальный катализатор, а основность характеризует силу катализа-
тора. Более того, если подвижность системы очень мала, как в случае диари-
лированных метиленазометинов и пропиленов, а также некоторых кольчато-
цепных систем, описанных в предыдущем разделе, то основания являются
670
Перегруппировки ненасыщенных соединений
единственными эффективными катализаторами. Если подвижность системы
столь высока, что не удается проследить за скоростью превращения, то
можно считать, что растворитель или само вещество обладает достаточной
основностью для связывания перемещающегося протона. Если кислота
оказывает катализирующее действие (когда система содержит необобщенные
электроны), то можно предположить, что исходная система, например кето-
енол или мутаротирующий сахар, начинает превращаться (с предваритель-
ным присоединением протона) в сопряженную кислоту. Эта кислота представ-
ляет собой по существу новую прототропную систему, подвижность которой
настолько больше исходной, что растворитель или некоторая часть непрото-
низированного исходного вещества может действовать как основание в соот-
ветствии с приведенным выше механизмом. При более слабой степени дис-
социации исходная система может за счет водородных связей образовывать
с ковалентной кислотой продукт присоединения, обладающий повышенной
подвижностью.
Такой взгляд на характер действия кислотного катализатора представляет
собой частный вывод, вытекающий из общей ионной теории прототропии;
он состоит в том, что подвижность системы должна увеличиваться за счет
наведенной электроотрицательности, т. е. под влиянием индуктивного эффек-
та —/-заместителей; притягивание электронов из зоны диссоциации должно
ускорять изомерные превращения, а отталкивание их к центру диссоциации
тормозит эти превращения. В 1926 г., когда был предложен этот механизм,
такой вывод соответствовал многочисленным фактам; впоследствии Шонии
[49] при изучении влияния заместителей в мета- и иара-положении на под-
вижность диарилированных метиленазометинов и заместителей в пара-
положении на подвижность аналогичных трехуглеродных систем более систе-
матически подтвердил этот вывод. В табл. 165 приведены найденные им
Таблица 165
Влияние заместителей на скорость взаимопревращения арилированных триадных
прототропных систем (по данным Шоппи). Приведены значения 105 (/с4-/с_4),
причем fcj и выражены в с-1
Система I -( rc6h4ch2n = снс6н5 rc6h4ch = nch2c6h5 fe_l в 0,145 н. растворе C2H5ONa в С2Н5ОН при 82° С ( СООС2Н5 и СООС2Н5 1 kl 1
Система II < RC6H4CH2C = СНС6Н5 rc6h4ch = ССН2С6Н5 fe-1 . в 1,45 н. растворе C2H5ONa в С2Н5ОН при 85° С
Система К
N(CH3)2 СНз ОСНз I Вг С1 NO-?
I, R в жета-положении I, R в пара-положении II, R в пара-положении 16,8 1,47 Мало 30,6 8,95 69,6 15,2 1,51 204 189 10,8 263 197 17,7 296 217 28,3 4080
значения скоростей реакций первого порядка при состоянии, близком к рав-
новесному. Шоппи установил, что если для каждой системы расположить
эти скорости соответственно дипольным моментам заместителей в аромати-
ческом кольце, то образуется монотонный ряд.
671
Глава XI
Другим примером влияния наведенной электроотрицательности на подвиж-
ность трехуглеродной системы может служить случай, когда на концах
такой системы находятся группы ионов аммония. Хотя соли катионов
(CH3)3NCH2CH = CHN(CH3)2C2H5 и (CH3)3NCH = CHCH2N(CH3)2C2H5
можно выделить как индивидуальные соединения, между ними в разбав-
ленных водных растворах щелочей или в изопропиловом спирте при ком-
натной температуре легко устанавливается равновесие [50].
Выводы, которые были сделаны в 1930 г. из положений ионной теории тауто-
мерии на основе концепций подвижного атома водорода и в особенности
из положений теории мономолекулярного механизма, сводятся к тому, что
соответствующие теоретические положения согласуются с многочисленными
фактами, относящимися к прототропной подвижности систем. Эти факты
можно расположить в хронологическом порядке следующим образом:
1. Подвижность водорода увеличивается с ростом ионизирующей способно-
сти растворителя (1896 г.).
2. Подвижность водорода связана со способностью алкилироваться и легко-
стью присоединения металла (1922 г.).
3. Подвижность сильно зависит от электроотрицательности атома, от кото-
рого происходит отщепление водорода (1922 г.).
4. Подвижность во всех случаях зависит от катализа основаниями, но неко-
торые системы подвергаются и катализу кислотами (1926 г.).
5. Подвижность водорода увеличивается при наличии электроноакцептор-
ных заместителей (1929 г.).
В дополнение к этим фактам в обзоре [51], опубликованном примерно в то же
время, подчеркиваются еще два обстоятельства. Первое состоит в следую-
щем: аналогия с реакциями присоединения, предложенная Хилдой Ингольд,
позволяет предположить, что и обсуждаемые реакции протекают согласно
тем же механизмам. Имелось также независимое доказательство связи рас-
сматриваемых реакций присоединения с ионизацией атома водорода. Второе
обстоятельство состояло в некотором соответствии механизма рассматривае-
мых реакций анионотропии, которая, как было недавно установлено, являет-
ся общим процессом и, по данным, которые уже начали накапливаться в то
время, протекает по ионному механизму.
Как уже было сказано, теория прототропии развивалась по двум направ-
лениям, причем второе направление было развито Лоури [52] в 1927 г.
В соответствии с механизмом Лоури реакция протекает в одну стадию и вы-
свобождение протона на одном конце системы сопряжено с одновременным
присоединением ее к другому концу
В? И Jl G'? н^в2
Согласно приведенному объяснению, этот механизм обозначается символом
B-Se2', что означает бимолекулярное электрофильное замещение, катали-
зируемое основанием и протекающее с перегруппировкой. Для краткости
будем называть его бимолекулярным механизмом. Для прямой реакции
основание В4 обозначено в символе как В, а участие кислоты НВ+ в этом
символе выражено числом, указывающим молекулярность.
Когда впервые были предложены мономолекулярный и бимолекулярный
механизмы прототропии, считалось, что они друг друга взаимно исключают.
.672
Перегруппировки ненасыщенных соединений
В некоторых случаях предпочтение отдавалось мономолекулярному меха-
низму. Бейкер [53] выступал против бимолекулярного механизма, основы-
ваясь на том, что полярные заместители не в такой степени влияют на мута-
ротацию некоторых производных сахаров, как можно было бы ожидать,
если бы процесс протекал в соответствии с этим механизмом. Но бимолеку-
лярный механизм довольно сложен, и на основании этого механизма нельзя
дать однозначного ответа на вопрос о влиянии заместителей. Педерсен [54]
возражал против бимолекулярного механизма, исходя из анализа скоростей
превращения некоторых прототропных систем в водной среде, а именно мута-
ротации глюкозы и енолизации ацетона, скорость которой определялась
по скорости галогенирования. Оказалось, что в выражение для скорости
прототропного превращения системы S в присутствии кислоты НВ и осно-
вания В- входят такие члены, как [S], [S][HB] и [S][B“], каждый из которых
может представлять либо мономолекулярную, либо бимолекулярную про-
тотропную изомеризацию, поскольку для реакций, протекающих в водной
среде, в выражение для скорости реакции всегда можно включить допол-
нительные множители [Н2О]. Вместе с тем в уравнении для скорости реак-
ций нет ожидаемого множителя [S][HB][B_], как можно было ожидать *
в случае бимолекулярного механизма. Однако, как указал Свейн [55], это
возражение недостаточно обосновано. Дело в том, что кинетические неопре-
деленности, например то обстоятельство, что нельзя точно различить **
члены [S][HB][H2O] и [S][H3O+][B~], не дают возможности сказать, являет-
ся ли член [S][HB][B_] кинетически важным. Таким образом, доводы Педер-
сена не исключают возможности бимолекулярного механизма прототропии.
Доводы Свейна, естественно, также не исключают мономолекулярного меха-
низма. Белл и Клани [56] подчеркнули это обстоятельство, применив аргу-
ментацию Свейна к случаю гидратации ацетальдегида — реакции обрати-
мого протолитического присоединения, аналогичного кольчато-цепной
системе мутаротирующей глюкозы (см. разд. 1,6). Они исследовали значи-
мость членов, содержащихся в уравнении скорости реакции [57], и, сделав
допущения, связанные с кинетическими неопределенностями, показали, что
в соответствии с бимолекулярным механизмом обязательно следует ожидать
значительно большего вклада члена [S][HB][B_], чем наблюдалось; до сих
пор роль этого члена еще не установлена. По-видимому, по крайней мере
в этом случае, бимолекулярный механизм непригоден, и поэтому нельзя
вообще исключить мономолекулярный механизм для протолитических реак-
ций, протекающих в водной среде.
Из приведенного краткого исторического обзора можно сделать вывод, что
все попытки исключить мономолекулярный или бимолекулярный механиз-
мы прототропии не имели успеха. В разд. 1,е мы увидим, что недавно был
вновь поднят вопрос о невозможности бимолекулярного механизма Лоури
даже в приложении к некоторым специальным системам. Однако оконча-
тельно этот вопрос еще не решен, так как доказать всеобщую непригодность
всегда очень трудно.
Катализируемую кислотами прототропию удобно рассматривать как пред-
варительное превращение исходного вещества под действием кислотного
катализатора в более подвижный таутомер, в частности в ионную сопряжен-
ную кислоту, которая обладает значительно более сильным кислым харак-
тером и поэтому легче депротонируется в нужном положении, чем исходное
* На основе эмпирических правил о приблизительной аддитивности свободной энергии
в связи с кинетическими эффектами различных катализаторов.
** По закону действия масс в случае переноса протона НВ -f- Н2О = В~ -f- Н30+ про-
изведение [НВ] [Н20] пропорционально [В~][Н3О+].
43-0772
673
Глава XI
вещество. По-видимому, большинство ионных сопряженных кислот обла-
дает достаточной кислотностью, чтобы способствовать мономолекулярному
механизму перегруппировки. Таким образом, суммарный процесс можно
описать как мономолекулярное электрофильное замещение, катализируемое
основанием и сопровождающееся перегруппировкой в сопряженную кислоту
исходного таутомерного вещества B-SeI'cA.
г. Аналогия с реакциями отщепления; положение равновесия и скорость
реакций. В 1941 г. Хьюз предложил иную схему для сопоставления прото-
тропных превращений с другими хорошо известными реакциями. В первона-
чальной схеме, предложенной Хилдой Ингольд, прототропные превращения
рассматривались как внутримолекулярные реакции присоединения; Хьюз
предложил; рассматривать их как внутримолекулярные реакции отщепления.
Приведем наглядное сравнение первой схемы, представляющей реакцию
енолизации, катализируемую основанием, и второй, представляющей отщеп-
ление олефина по механизму Е2.
Основание.. .Н Основание... Н
К Р К ГУ + +
R—С—С==О и R—С^С—X (X = N, S,Hal)
II II
Формальное различие этих двух схем состоит лишь в том, что группа СО
не расщепляется, а группа С — Х0 расщепляется. Как было показано
в гл. IX, заместители оказывают влияние на реакцию отщепления вслед-
ствие как индуктивного эффекта, так и эффекта сопряжения. Любой из этих
эффектов может стать доминирующим и привести к ориентации типа Гофмана
или Зайцева; тип ориентации определяется полярностью и ненасыщенностью
заместителей, а в случае алкильных заместителей — полярностью группы X.
Согласно схеме Хьюза, можно ожидать именно таких двух типов структур-
ных влияний на прототропные превращения. На примере структурных влия-
ний как на равновесие, так и на скорость прототропных превращений он
показал, что такая же двойственная электронная концепция хорошо опи-
сывает влияние факторов строения на прототропные превращения [58]. Рас-
смотрим с этой точки зрения некоторые термодинамические и кинетические
данные.
Имеющиеся сведения о равновесии кето-енолъных систем основываются
главным образом на применении уже упомянутого метода определения содер-
жания енола бромированием по Мейеру. Этот метод широко использовался
как самим Мейером, так и Дикманом, Ауэрсом, Конантом, Шварценбахом
и другими. Впоследствии метод был значительно усовершенствован Шварцен-
бахом и сотр. [59], которые, применив метод потока и электрометрические
измерения, разработали способ, позволяющий определять значительно
меньшие концентрации енола, чем это можно было сделать обычным объем-
ным методом. В табл. 166 приведены полученные указанными авторами зна-
чения равновесных концентраций енола при 25 °C в газовой фазе, в жидком
кето-еноле, в разбавленных растворах кето-енола в гексане и в воде. Для
удобства таблица разбита на разделы, обозначенные буквами.
Ацетон и другие простые кетоны находятся в равновесии преимущественно
в кето-форме. Как видно из данных, приведенных в разделах А и Б табл. 166,
введение карбэтоксильного заместителя увеличивает равновесное содержа-
ние енола, а ацетильная группа еще в большей степени смещает равновесие
в сторону енола. Такое влияние заместителей на равновесные концентрации
енола можно объяснить двумя причинами: во-первых, тем, что индуктивный
эффект (—I) электроотрицательного карбэтоксильного или ацетильного
674
Таблица 166
Равновесное содержание енола (%) в кето-енольных системах
Система Состояние Растворитель Литература
газ ЖИДКОСТЬ гексан вода
А СН3СОСН3 СН3СОСН2СООС2Н5 С2Н5ООССН2СОСН2СООС2Н5 49 0,00025 7,5 17 49 59 21, 62, 63 21
Б с2н5ооссн2соос2н5 СН3СОСН2СОСН3 92 0,0 80 92 15 21 21, 61-63
В СН3СОСН(СН3)СООС2Н5 СН3СОСН(С2Н5)СООС2Н5 СН3СОСН(С3Н7-изо)СООС2Н5 СН3СОСН(С6Н5)СООС2Н5 С6Н5СОСН2СООС2Н5 С6Н5СОСН(С2Н5)СООС2Н5 14 10 6,1 80 4 3 5 30 21 4 12 14 6 67 62 62 62 62 60, 61 61
Г НСОСН(СООС2Н5)2 СН3СОСН(СООС2Н5)2 С2Н5СОСН(СООС2Н5)2 94 69 44 61 61 61
д С2Н5СОСН2СОСН3 СН3СОСН(СН3)СОСН3 СН3СОСН(С2Н5)СОСН3 44 35 72 33 28 59 26 2,8 61 59, 61, 62 61, 62
Е СН3СОСН2СОС6Н5 С2Н5СОСН2СОС3Н5 99 94 21, 61 61
Ж Цпклопентанон Циклопеитанон-2-СООС2Н5 Циклопентанон-2-С ОСН3 Циклопентанон-2-СН О 27 0,0048 4,6 15 41 59 60, 63 59 59
3 Циклогексанон Циклогексанон-2-СО ОС2Н5 Циклогексанон-2-СОСНз Циклогексанон-2-СНО Диметилдигидрорезорцин 91 0,020 74 100 29 48 95 59 60, 63 59 59, 61 59
43*
Глава XI
заместителя ослабляет связь С — протон и тем самым уменьшает термоди-
намическую устойчивость кето-формы; во-вторых, тем, что для проявления
эффекта сопряжения (—М) ненасыщенного карбэтоксильного или ацетиль-
ного заместителя требуется должным образом расположенная двойная
связь, что увеличивает относительную термодинамическую устойчивость
енола. Можно предположить, что различие между влиянием карбэтоксиль-
ной или ацетильной групп обусловлено в основном второй причиной,
так как при этом указанные группы сильно отличаются, а по их —M-эффек-
там ацетильная группа влияет сильнее, чем карбэтоксильная (гл. II,
разд. 3,д).
Последующие разделы таблицы (В, Г, Д и Е) иллюстрируют различное влия-
ние простых алкильных групп, уменьшающих равновесное содержание енола
в p-кетоэфирах и р-дикетонах. Следует согласиться с Хьюзом, что для про-
стых алкильных групп в таких кето-енольных системах превалирует индук-
тивный эффект (+/), защищающий С-протон и тем самым увеличивающий
термодинамическую устойчивость кетона. По аналогии с реакциями отщеп-
ления карбонильную группу по ее способности выдвигать на передний план
индуктивный эффект алкильных групп в р-кето-енольных системах можно
сравнивать скорее с ониевыми группами, чем с атомами галогенов как заме-
стителями.
Как следует из сравнения данных для ацетона, циклопентанона и циклогек-
санона, алкильные группы увеличивают равновесное содержание енольной
формы в простых кетонах. Это указывает на превалирующее влияние гипер-
конъюгационного эффекта (+М). Можно предположить, что двойная связь
в еноле более склонна к гиперконъюгации с алкильным остатком при отсут-
ствии второй карбонильной группы, с которой возможно сопряжение, как
в случае |3-кето-енолов.
Как свидетельствуют величины, приведенные в разделах В и Е табл. 166,
электрополярный эффект подавляется эффектом ненасыщенности фениль-
ной группы, способность которой участвовать в самой длинной из возмож-
ных цепей сопряжения позволяет ей увеличивать относительную термодина-
мическую устойчивость енольных изомеров Р-дикетонов и [3-кето-
эфиров.
Данные, приведенные в разд. Ж и 3 той же таблицы для производных цикло-
пентана и циклогексана, показывают, что большие размеры кольца способ-
ствуют образованию енольных форм. Причина этого явления не установ-
лена; однако, вероятно, крайний атом водорода почти ненапряженного зигза-
гообразного остатка —(СН2)4— в циклогексене лучше подчиняется стерео-
химическим требованиям, необходимым для гиперконъюгации с группой
С = С в цикле, чем крайний атом водорода почти ненапряженного и пло-
ского остатка —(СН2)3— в циклопентене.
Что касается кинетики процесса енолизации, то, как установлено методом
бромирования Лэпуорта, алкильные группы уменьшают скорость енолиза-
ции, катализируемой основаниями. Это видно из данных, полученных Эван-
сом и Гордоном [64] для бромирования фенилалкилкетонов CeH5COCHRR'
в 75%-ной водной уксусной кислоте, содержащей в качестве катализатора
ацетат-ионы. Относительные скорости этого процесса при 45 °C приводятся
ниже:
RR' = H,H СН3,Н С2Н5,Н изо-С3Н7,Н СН3,СН3
100 15,4 12,0 5,9 3,0
На эти данные ссылался Хьюз [58] для подтверждения своего тезиса о том,
что индуктивный эффект является главной частью общего кинетического
676
Перегруппировки ненасыщенных соединений
полярного эффекта алкильных групп в простых кето-енольных системах,
так же как в реакциях бимолекулярного отщепления от ионов аммония
и сульфония.
Кардуэлл [65] подчеркнул, что иодирование первичных алкилметилкетонов
RCH2COCH3, катализируемое основаниями, приводит препаративно через
RCH2COCI3 к образованию RCH2COOC2H5, что указывает на наличие гоф-
мановской ориентации при галогенировании и, следовательно, при еноли-
зации, обусловливающей скорость процесса в соответствии с приведенными
кинетическими представлениями.
Кардуэлл и Килнер [66] отметили, что при катализируемом кислотами бро-
мировании кетонов возникают совершенно иные кинетические условия,
заключающиеся в том, что во влиянии алкильных групп преобладает эффект
гиперконъюгации; поэтому в данном случае наблюдается ориентация по Зай-
цеву и соответствующие изменения скоростей реакции. Из препаративных
данных известно, что кетоны, формулы которых приводятся ниже, в основ-
ном подвергаются бромированию, катализируемому кислотами, в месте,
отмеченном звездочками, что указывает на размещение енольной двойной
связи соответственно обобщенному правилу Зайцева.
СН3СН2СОСН3 СН3СН2СН2СОСН3 (сн3)2снсосн3
сн3 СН3 СОСН3 СОСН(СН3)2 1 1* 1
|//=0 0=о 0 0 *
Кардуэлл и Килнер использовали данные, приведенные в работе Кетча
и других [67], и количественно проанализировали соотношения а- и а'-моно-
бромирования несимметричных кетонов при кислотном катализе; они сопо-
ставили наблюдаемые ориентационные соотношения с данными Доусона
[68] по суммарным скоростям катализируемого кислотами иодирования кето-
нов, для того чтобы рассчитать (а это возможно, так как скорости иодиро-
вания и бромирования в равной степени определяются енолизацией) относи-
тельные парциальные скорости образования енольных двойных связей
в алкильных группах различных кетонов. Результаты их вычислений при-
ведены в табл. 167, где показано, как алкильные заместители влияют на поло-
жение двойной связи С = С енола. Из рассмотрения первых двух строк
Таблица 167
Относительные парциальные скорости катализируемой кислотами
енолизации кетонов, измеренные по скорости галогенирования
(по данным Кардуэлла и Килнера)
/& = С —Сн/ ^СН — С — сн/ -> /сн — с = с/
11 он 11 н l н н он н
Заместители Скорость Кетон Скорость Заместители
а а
сн3 50 сн3сосн3 50 СН3
сн3 сн3 76 С2Н5СОСН3 28 С2Н5
с2н5 сн3 59 н-С3Н7СОСН3 35 н-С3Н7
сн3 С2н5 41 С2Н5СОС2Н5 41 С2н5 СН3
677
Глава XI
этой таблицы видно, что дополнительный [3-метильный заместитель в метил-
этилкетоне ускоряет енолизацию в этильной ветви, а при замене а-метиль-
ного на а-этильный заместитель скорость енолизации в метильной ветви
метилэтилкетона заметно снижается. Подобные соотношения можно видеть
и при рассмотрении остальных строк таблицы; выявленные соотношения
указывают на определенные гиперконъюгационные влияния алкильных
заместителей на скорость енолизации. Действительно, эти соотношения
очень хорошо согласуются с данными, приведенными в табл. 142 (гл. IX)
для реакции бимолекулярного отщепления олефинов от алкилбромидов,
скорость которой определяется гиперконъюгацией в соответствии с обобщен-
ным правилом Зайцева (см. стр. 553).
Объясняя, почему гиперконъюгация играет решающую роль во влиянии
алкильных групп на скорость енолизации, катализируемой кислотами,
хотя в случае енолизации, катализируемой основаниями, такую же роль
играет индуктивный эффект, Кардуэлл и Килнер с полным основанием
высказывают предположение, что при частичном образовании иона карбо-
ния более сильная ненасыщенность наступает в промежуточном состоянии
енолизации, катализируемой кислотами, а не в реакции, катализируемой
основаниями. В особом случае катализа водородным ионом эффективным
исходным таутомером будет не сам кетон, как при катализе основанием,
а сопряженная кислота кетона — мезомерный ион, соответствующий валент-
ным структурам
:сн-с=он и сн — с—он
Как и в случае мономолекулярного отщепления олефина, которое протекает
по^обобщенному правилу Зайцева, образование иона карбония должно (как
указано в^гл. IX, разд. 2,д) вызвать сильную гиперконъюгацию алкильных
групп в переходном состоянии образования углерод-углеродной двойной
связи.
Рассмотрим теперь вопрос о равновесиях в первичных и вторичных нитропара-
финах. Эти вещества существуют обычно в виде нормальных нитросоедине-
ний в равновесии с очень малыми количествами агрг-изомеров. Тернбул
и Мэрон [69] определили содержание этих изомеров в водных растворах про-
стейших представителей указанного ряда. Они определяли кондуктометри-
ческим методом сначала константу кислотности йщн-соединения, что воз-
можно благодаря медленному превращению его в нитроформу, а затем кон-
станту кислотности нитроизомера, что возможно, учитывая почти полное его
образование. Поскольку ионы этих двух форм являются общими, то из отно-
шения констант кислотности можно установить равновесное содержание
изомеров в смеси.
Из данных, приведенных в табл. 168, видно, что метильные заместители
во всех случаях увеличивают термодинамическую устойчивость йщн-изомера,
а также, хотя и в меньшей степени, устойчивость аци-антна по сравнению
с нормальным нитроизомером и его анионом. Это, по-видимому, происходит
за счет гиперконъюгационного +7И-эффекта метильного радикала, дейст-
вующего на полностью образованную CN-двойную связь между атомами С
и N в ацн-структуре (формула приведена ниже) и на ту частичную двойную
связь, которая расположена между атомами С и N в мезомерном анионе.
Такие эффекты отличаются от эффектов, относящихся к [3-кетоэфирам и [3-
дикетонам; в этом случае для проблемы равновесия важно, что имеется пол-
ностью образованная двойная углерод-углеродная связь, способная к гипер-
конъюгации с алкильной группой, но все же, несмотря на это, превалирует
678
Перегруппировки ненасыщенных соединений
Таблица 168
Константы кислотности и равновесное содержание нормальных
и «дн-форм алифатических нитросоединений в воде при 25 °C
(по данным Тернбула и Мэрона)
Нормальная форма Содержание а-ци-формы, %
а^и-формы нормальной формы
CH3NO2 3,25 10,21 0,000011
ch3ch2no2 4,41 8,46 0,0089
(CH3)2CHNO2 5,11 7,68 0,275
индуктивный эффект алкильных групп. При равновесии в системе нитро —
аци-нитросоединение, по-видимому, большее значение эффекта гиперконъю-
гации, чем индуктивного эффекта алкильных групп (несмотря на иное соот-
ношение для кето-енолов), можно отнести за счет сочетания положитель-
ного заряда с ненасыщенностью системы нитро — ацм-нитро, а также за счет
уже обсуждавшегося конкурирующего сопряжения в кето-енолах
о-
НзС—CH=N+
ОН
Сильная гиперконъюга-
ция
RC=O
H^-cXcR'
\)Н
Более слабая гипер-
конъюгация
Изучая кинетику изомеризации нитропарафинов, Мэрон и Ла Мер [70]
определили с помощью кондуктометрического метода скорость нейтрализа-
ции некоторых простых алифатических нитросоединений в водных растворах.
Оказалось, что скорость превращения нитросоединения в его ацм-изомер
определяется скоростью нейтрализации. Таким образом, скорость нейтрали-
зации имеет то же значение для нитро — ацн-нитро-систем, какое для кето-
енолов имеет скорость бромирования кетонов при катализе основанием.
Относительные скорости превращения нитро-формы в ацм-форму при О °C
для некоторых простых нитросоединений RR'GHNO2 приведены ниже:
RR' = H,H СН3,Н С2Н5,Н СН3,СН3
100 16,4 12,3 0,87
Из рассмотрения этих данных видно, что алкильные заместители замедляют
отщепление протона от нитросоединений, так же как зто происходит в слу-
чае кетонов. Можно сделать вывод, что это является следствием индуктив-
ного эффекта алкильных групп, который определяет скорость отщепления
протона от нитросоединений, как и от кетонов. Тот факт, что индуктивный
эффект главным образом влияет на скорость отщепления протона, а гипер-
конъюгационный эффект алкильных групп — на равновесие в процессе
отщепления протона от нитросоединений, можно объяснить тем, что в пере-
ходном состоянии процесса переноса протона двойная связь в CN выражена
недостаточно для того, чтобы преобладал эффект гиперконъюгации в алкиль-
ных заместителях.
Альдегидные свойства альдолактольных систем пиранозных сахаров, обла-
дающих восстановительными свойствами, связаны с высокими скоростями
679
Глава XI
установления равновесий, включающих очень небольшое равновесное содер-
жание альдегидной формы.
О---СН—ОН НО СН = О
---
(СН2)П (СН2)П
Диастереомерные а- и fi-формы Альдо- или 7-форма
В равновесном растворе глюкозы при pH 6,9 содержание глюкозы (альдо-
формы) оценивается в 0,0026% [71].
Кон, Линстед и сотрудники * подробно изучили равновесия в а,0- и (3,у-
ненасыщенных карбоксилат-ионах, эфирах карбоновых кислот, кетонах
и нитрилах. В этом случае типичными являются следующие системы:
( сн3сн2сн2сн = снсоо- СН3СН2СН = СНСН2СОСГ
I СН3СН2СН2СН=СНСООС2Н5 СН3СН2СН = СНСН2СООС2Н5
(а, ₽) „ (В, V)
СН3СН2С(С2Н5) = СНСОСН3 7^ СН3СН = С(С2Н5)СН2СОСН8
(СН3)2СНСН = CHCN 7^ (CH3)2C = CHCH2CN j
Действие основных катализаторов приводит к равновесной смеси изомеров,
представленных в этих системах; гидроксильные ионы в воде образуют обыч-
ную равновесную систему для карбоксилат-ионов, а этилат-ионы в этиловом
спирте эффективны для эфиров карбоновых кислот, метилкетонов и нитри-
лов. Особенно подвижно равновесие для кетонов, для которых часто доста-
точно наличия следов основания, чтобы вызвать их изомеризацию. Подвиж-
ность указанных систем, несомненно, зависит решающим образом от нена-
сыщенности атомов кислорода и азота концевых групп. Таким образом, эти
системы можно рассматривать скорее как пентадные кето-енолъные или
пентадные циан-иминные системы, чем как трехуглеродные системы. Одна-
ко енолы и имины находятся в равновесной смеси в чрезвычайно малых коли-
чествах, которые еще не определены. Поэтому можно рассматривать лишь
равновесное содержание карбоксильных, карбонильных или циан-форм,
которое значительно отличается от равновесного содержания в пропилено-
вой системе. По этой причине эти системы часто называют «трехуглерод-
ными», а концевую группу, содержащую ненасыщенный электроотрица-
тельный атом, в отдельности обозначают как «активирующую группу».
Из наблюдений Кона и Линстеда можно сделать следующий вывод. Равно-
весие между а,Р~ и р,у-ненасыщенными формами любой из этих трехугле-
родных систем мало зависит от природы активирующей группы, но очень
сильно зависит от положения заместителей в пропиленовой системе. Если
у-положение прототропной системы или каждое у-положение (если их боль-
ше одного) остается незамещенным, как в случае анионов, зфиров или нитри-
лов кротоновой, р,р-диметилакриловой и [3-метилкоричной кислот, то при
условиях равновесия в системе присутствует почти исключительно «^-нена-
сыщенное соединение.
ch3cr=chcoo; CH2=CRCH2COO
I(R = H. СН3, С6Н5)
Как показывает опыт, замена [3-водорода на р-метильную или [3-фенильную
группу не вносит изменений в это правило. Однако введение одной или двух
* Работы этих исследователей рассмотрены в обзоре автора настоящей книги [72]; более
широко вопрос рассмотрел Бейкер [73]. Линстед опубликовал свои данные в табличной
форме в книге Гилмана [74]. В этих работах содержатся полные ссылки на многочислен-
ные оригинальные исследования.
680
Перегруппировки ненасыщенных соединений
у-метильных или других у-алкильных групп смещает реакцию в сторону
образования р,у-ненасыщенного изомера, который в этом случае становится
важной и часто преобладающей составляющей равновесной смеси
сн3сн2ск=снсоо- ch3ch=crch2coo-
В этом же смысле у-фенильная группа оказывает значительно большее влия-
ние, и в равновесии находится исключительно Р,у-изомер
СвН5СН2СК = СНСОО_ c6h5ch = crch2coo-
Высшие у-алкильные группы оказывают меньшее влияние, чем р-метильные,
как это можно видеть на примере следующей системы:
RCH2C(CH2R) = CHCOO- 7^ RCH = C(CH2R)CH2COO-
Для этой системы имеются такие данные:
у-Заместитель (R) Н СН3 С2Н5 изо-С3Н7
Содержание £,у-формы в равновесии, % 0 79 67 51
Введение метильной группы в a-положение должно способствовать образо-
ванию а,р-ненасыщеиных форм, однако таких примеров встречается мало.
Влияние такого замещения невелико и, по-видимому, неоднозначно; влия-
ние даже р-фенильной группы также мало и неоднозначно. Высшие р-алкиль-
ные заместители обусловливают у-замещение системы. Введение метила в со-
положение способствует образованию а,0-ненасыщенных форм. Для системы.
CH3CHRCH = CR'COO- 7» CH3CR' = CHCHR'COO-
это подтверждается следующими данными:
Ион пентеновой у-СН3 Незамещенный а-СН3
кислоты (пенте-
ноат-ион)
Содержание Р,у- 78 32 19
формы в равно-
весии, %
Этим фактам была дана следующая интерпретация [75]. Незамещенные в у-
положении соединения существуют в равновесной смеси практически только
в их а,р-ненасыщенных формах, так как сопряжение между пропиленовой
двойной связью этой формы и ненасыщенной активирующей группой увели-
чивает относительную термодинамическую устойчивость а,0-ненасыщенных
изомеров. Для p-заместителей характерно сопряжение или гиперконъюга-
ция с пропиленовой двойной связью в любом положении, и поэтому они
по-разному влияют на относительную устойчивость изомеров. В у-метиль-
ных соединениях энергия сопряжения между пропиленовой связью и акти-
вирующей группой а,р-ненасыщенной формы в значительной степени компен-
сируется энергией гиперконъюгации между пропиленовой связью и у-ме-
тильной группой р,у-ненасыщенной формы. В у-фенильных соединениях
налицо сверхкомпенсация в том же направлении из-за большей энергии
мезомерии (сопряжения между пропиленовой двойной связью и фенильной
группой) р,у-ненасыщенного изомера. Более слабое влияние у-алкильных
групп в заместителях, больших, чем метильная группа, в соответствии с ря-
дом Н < {СН3 > С2Н5> пзо-С3Н7} является нормальным следствием гипер-
конъюгации (гл. II, разд. 3,и). Наконец, подобно тому как для у-метильной
681
Глава XI
группы характерна гиперконъюгация с р,у-двойной связью, способствую-
щая образованию р,у-ненасыщенных соединений, для а-метильной группы
характерна гиперконъюгация с аф-двойной связью, способствующая обра-
зованию а,р-ненасыщенных изомеров.
Из приведенного выше объяснения вытекает, что для равновесий в рас-
сматриваемых системах гиперконъюгация алкильных заместителей имеет
большее значение, чем индуктивный эффект; в этом случае имеется большое
сходство с равновесиями в нитро — a^u-нитро-системах, чем с равновесия-
ми в р-дикетонах. Вероятной причиной является то, что если в этих пентад-
ных кето-енольных системах действительно содержится енольная форма, то
можно приближенно считать, что индуктивные эффекты сказываются равно-
мерно в пропиленовой части системы. Оказываемое группой R влияние
(-[-/-эффект) удерживает протон как в R — СН — С = С, так и в R - С =
С — СН *, однако если R — ненасыщенная группа, то только вторая систе-
ма испытывает стабилизирующее влияние сопряжения, а если R — алкиль-
ная группа, то только вторая система участвует в гиперконъюгации.
Кон и Линстед приводят данные, характеризующие влияние заместителей
в пропиленовой системе на скорость превращения этих систем; это влияние
весьма сложно, и, пожалуй, более или менее точно можно сказать лишь то,
что введение алкила в любое положение замедляет превращение. Теоретиче-
ски следовало ожидать, что этот вопрос окажется сложным, так как подвиж-
ность системы зависит в основном от природы активирующих групп; поэтому
заместители в пропиленовой системе влияют на ее подвижность не прямо,
а вследствие эффектов, передаваемых от активирующих групп.
R отношении скорости образования таутомеров из общего аниона сущест-
вует аналогия с простыми псевдокислотами. Так, равновесие между этило-
выми эфирами циклопентилиденмалоновой кислоты и циклопентенилмало-
яовой кислоты полностью сдвинуто в сторону первого эфира
СН2---С = ССООС2Н5 СН = С----------СНСООС2Н5
I 11 I II
СН2СН2СН2 СООС2Н5 СН2СН2СН2 СООС2Н5
Однако Хью и Кон [77] установили, что если натриевые производные эфи-
ров осторожно подкислить (применяли бензойную кислоту в смеси этилового
и петролейпого эфиров), то можно выделить неустойчивый циклопентенило-
вый эфир или по крайней мере смесь, богатую этим изомером, который
постепенно переходит в устойчивый циклопентилиденовый эфир.
Таким образом, любую из рассмотренных в этом разделе систем можно
использовать для иллюстрации общего правила, сформулированного Кэтч-
полом, Хьюзом и автором [78] для реакций, идущих через промежуточное
неустойчивое соединение, т. е. для мономолекулярного механизма прото-
тропии. Это правило состоит в следующем: если высвобождаемый кислотами
протон переходит от кислот к мезомерному аниону слабо ионизированных
таутомеров с заметно отличающейся устойчивостью, то наиболее быстро
образующийся таутомер обладает наименьшей термодинамической устой-
чивостью", он является также и тем таутомером, который наиболее легко
* То же наблюдали Оссорио и сотр. [76] в случае метиленазометинов. При последова-
тельном переходе от R = СН3 к R = С2Н6, иао-СзН7, трет-С4Н9 в системе
Н Н
। Ч ।
(I) R - С(С6Н5) - N = СНС6Н5 R-C(C6H5) = N - СНС6Н5 (II)
fe-i
В этанольном растворе этилата скорости и постепенно в сравнимой степени умень-
шаются в 40 раз, в то время как константа равновесия изменяется лишь в интервале
от 1,22 до 1,47.
•682
Перегруппировки ненасыщенных соединений
отдает протон основаниям. Обычно это положение иллюстрируется свой-
ствами псевдокислот. Если подкислить соль фенилнитрометана, то сначала
образуется термодинамически неустойчивый изомер, представляющий собой
/щп-нитросоединение, которое может быть выделено при быстрой обработке
в отсутствие сильных катализаторов. В конечном счете, естественно, обра-
зуется термодинамический более устойчивый изомер, представляющий собой
нитросоединение; ощп-нитросоединение образует с основаниями соли зна-
чительно быстрее, чем нитросоединение.
Диаграмма энергий, приведенная на рис. 50, поясняет изложенное выше.
Кинетика процесса образования таутомеров в определенных соотношениях
при переходе протона к мезомерному аниону определяется двумя верхними
Переходное
состояние
Переходное
состояние
Ионы (один
.мезомерный)
Неустойчивый
таутомер
Стабильный
таутомер
Рис. 50. Энергетическая диаграмма,
иллюстрирующая различие
между кинетическим и тер-
модинамическим контролем
за количеством образую-
щихся туатомеров при ассо-
циации ионов, а также раз-
личия между скоростями и
степенью ионной диссоциа-
ции таутомеров.
уровнями, термодинамический фактор их окончательных соотношений регу-
лируется двумя нижними уровнями. Таким образом, из ионов сначала обра-
зуется менее устойчивый таутомер, который в дальнейшем снова становится
ионизованным и переходит в более устойчивый таутомер. Следовательно,
когда в результате нескольких последовательных реакций путем ассоциации
ионов образуются таутомеры, соотношения таутомеров будут в основном
зависеть от того, что определяет течение реакции — кинетические или тер-
модинамические факторы, т. е. позволяют ли условия установиться равно-
весию между образовавшимися таутомерами. Как показано ниже, это поло-
жение имеет значение для реакций присоединения. Что касается ионной
диссоциации таутомеров, то из схемы видно, что менее устойчивый таутомер
будет ионизоваться не только в большей степени, но и быстрее, чем более
устойчивый таутомер.
Эти соотношения понятны, если учесть, что наименее устойчивой формой
молекулы является ее ионизованная форма, для превращения которой
в энергетически мало отличающийся неустойчивый таутомер требуется менее
значительное перераспределение электронов с меньшей энергией активации,
чем в устойчивый таутомер, сильно отличающийся как энергетически, так
и по распределению электронов. Такое объяснение применимо к любой
реакции, идущей через промежуточное состояние с высокой энергией. Это
состояние быстрее будет переходить в конечное состояние, более богатое
энергией, так как для этого требуется меньшая электронная реорганизация.
Этот вывод строго применим только к электронной энергии, однако большая
часть энергии молекул в любом состоянии представляет собой электронную
энергию, так что изменение электронной энергии почти точно соответствует
изменению общей энергии вдоль координаты реакции *.
* Хэммонд [79] использует этот принцип в более широком смысле, а именно: богатое
энергией промежуточное состояние «по структуре» ближе к более богатому энергией
конечному продукту. Если, как делают многие, под «структурой» понимать определен-
ную конфигурацию и конформацию, то этот принцип становится неверным, поскольку
683
Глава XT
д. Мономолекулярный механизм прототропии. Вернемся к вопро-
су о механизме прототропного превращения, о котором шла речь в конце
разд. 1,а. Примерно к 1930 г. концепция прототропии находилась на сле-
дующей стадии развития: во-первых, были развиты представления о моно-
молекулярном механизме, которые были подтверждены тщательным сравне-
нием требований этого механизма с наблюдаемым влиянием среды и строе-
ния на подвижность систем; во-вторых, был предложен бимолекулярный
механизм, против которого были выдвинуты некоторые возражения. В тече-
ние 30-х годов нашего столетия в этом вопросе произошли изменения в двух
направлениях, которые будут описаны в этом и в следующих разделах:
во-первых, доказательства, подтверждающие мономолекулярный механизм,
становились все более четкими, и, во-вторых, были обнаружены определен-
ные границы приложимости бимолекулярного механизма. Это произошло
после введения двух новых методов, позволяющих распознавать механизм
прототропии и основанных на изучении изменения оптической активности,
сопровождающей прототропное превращение, и на изучении водородного
обмена, которое стало возможным благодаря применению только что начав-
шегося использоваться в то время изотопа — дейтерия.
В данном разделе рассмотрены те работы 30-х годов, которые были направ-
лены на подтверждение двухстадийного мономолекулярного механизма про-
тотропии B-Se1'. Следует отметить, что в этих работах рассматривались
малоподвижные прототропные системы, главным образом триадные кето-
енольные системы или пентадные циан-иминные системы. Кроме того, были
изучены еще два дополнительных случая. В одном случае исследуемым изо-
мером был термодинамический устойчивый член таутомерной системы, напри-
мер кето-форма кето-енольной системы простого кетона. Во втором случае
исследовался термодинамически неустойчивый таутомер, например р,у-нена-
сыщенный нитрил, который мог практически полностью превращаться в его-
а,0-ненасыщепный изомер.
Для объяснения принципов, применяемых при изучении термодинамически
устойчивых таутомеров, рассмотрим поведение псевдокислотного кетона,
вытекающее из мономолекулярного механизма. Особо следует учитывать
катализ прототропии основаниями и кислотами. Сначала рассмотрим более
простой случай катализа основаниями, который можно представить следую-
щим выражением:
в +
’^-с-^Ьу
нс—с=о^вн +
в 4- С=С—ОН
Для данного случая относительная термодинамическая устойчивость кетона,
ионов и енола представлена соответственно в правой части, центре и левой
части диаграммы, приведенной на рис. 50.
Лэпуорт показал, что, несмотря на термодинамическую устойчивость псевдо-
кислотного кетона, можно измерить скорость его енолизации косвенным-
путем по скорости бромирования. При этом скорость бромирования зависит
от природы катализатора, а не от галогена. Двухстадийный мономолеку-
лярный механизм описывает бромирование и енолизацию как процессы,
скорость которых в равной степени определяется одним и тем же начальным
соответствующие энергии составляют лишь небольшую часть общей энергии, и поэтому
нельзя гарантировать, что вдоль координаты реакции между изменениями конфигурации
и конформации и общей энергией будет наблюдаться соответствие (ср. 181]). Этот
принцип, согласно Хайну, можно рассматривать как частный случай «принципа наи-
меньшего действия» Райса и Теллера [82].
684
Перегруппировки ненасыщенных соединений
высвобождением протона. В случае катализа основанием в соответствии
с этим механизмом анион, а не енол служит реакционноспособной частицей
по отношению к электрофильным реагентам независимо от того, являются ли
они галогенами или кислотами, поставляющими протон. Предполагается,
что в случае бромирования анион связывается галогеном, а поскольку анион
бромируется быстро и необратимо, то скорость бромирования равна скорости
образования аниона. Следует принять во внимание, что при енолизации, как
это видно из энергетической диаграммы (рис. 50), присоединение протона
происходит значительно быстрее к кислороду аниона, чем к углероду.
Поэтому скорость образования енола также равна скорости образования
аниона и может измеряться по скорости бромирования.
Эту интерпретацию можно проверить, так как существуют два других мето-
да измерения скорости образования аниона. Один из них основан на следую-
щем: если углеродный атом, несущий подвижный атом водорода, является
асимметрическим центром оптической активности и присутствует в молекуле
в единственном числе, то в одной из валентных структур, в которой этот
атом углерода связан двойной связью, должна наблюдаться потеря оптиче-
ской активности мезомерного аниона. Поэтому, предполагая, что условия
асимметрии выполнены, скорость образования аниона должна измеряться
скоростью рацемизации, которая в свою очередь равна измеряемой скорости
связывания брома при прочих равных условиях: одинаковой температуре,
одном и том же растворителе и катализаторе (основание). Следует отметить,
что указанная эквивалентность измеряемых скоростей сама по себе еще
не подтверждает механизма реакции, так как образуемый любым способом
енол лишен оптической активности и может сразу бромироваться: так, если,
например, он образуется за счет внутримолекулярного переноса атома водо-
рода, то скорость рацемизации может все же оказаться равной скорости свя-
зывания добавляемого брома. Однако, как показано ниже, такое равенство
скоростей составляет необходимое звено в цепи доказательств.
Второй метод измерения скорости образования аниона не требует столь про-
странных пояснений. Он состоит в следующем. Кетон вместе с основанием,
служащим катализатором, растворяется в растворителе, содержащем дей-
тероксигруппы, так что любые анионы, образующиеся вследствие отщепления
протона, могут присоединить только дейтроны. Из рассмотрения относи-
тельной термодинамической устойчивости кетонов, ионов и енола, указанных
на энергетической диаграмме (рис. 50), следует, что в стационарном состоя-
нии лишь очень небольшая часть общего количества кето-енола существует
в виде аниона или енола. Поэтому в любое время после короткого начального
периода, необходимого для установления стационарного состояния, ско-
рость образования аниона равна скорости превращения его в главный про-
дукт, а именно в дейтерокетон; иными словами, скорость притока дейтронов
в любой период времени равна скорости высвобождения протонов. Таким
образом, если приведенное объяснение правильно, то измеряемая скорость
дейтерирования должна быть равна измеряемым скоростям бромирования
и рацемизации. Конечно, при сравнении требуется соблюдение одинаковых
условий (температура, растворитель и катализатор). Следует отметить, что
природа растворителя оказывает значительное влияние на реакцию * при
измерении скорости дейтерирования.
В случае кислотного катализа теория более сложна, но выводы из обеих
теорий аналогичны. В случае общего кислотного катализа первая стадия
* Как правило, скорость реакции в гидроксильных и дейтероксильных растворителях
не одинакова, так как энергия частично зависит от нулевой энергии водорода.
685
Глава XI
процесса заключается в превращении первоначальной прототропной системы
в солеобразный комплекс, который затем подвергается депротонизации; ско-
рость последней^ реакции определяет скорость всего процесса
НС—С=О г НВ НС—с=о,нв
В' + НС—С=О,НВ «=♦ В'Н+ + г с—с—оу,НВ
В'Н+ + С=С—ОН + в-
Именно депротонированный комплекс может мгновенно реагировать с гало-
генами и с донорами протонов или дейтронов; он также является первым про-
межуточным соединением, в котором может происходить потеря оптической
активности. Скорость образования апротонного комплекса из кетона пред-
ставляет собой составную величину; но так как кетон — термодинамически
устойчивый таутомер, то именно эта скорость определяет измеряемые ско-
рости бромирования, рацемизации и присоединения дейтерия. В частном
случае катализа водородным ионом сначала протекает превращение в сопря-
женную кислоту которая в соответствии с мономолекулярным механизмом
подвергается депротонизации с непосредственным образованием енола
Н —С-С = О + HB,yt H-C-C = OH + Bk
В' + Н —С-С=ОН В'Ы + С = С—ОН
JZ \ I __ Z \ Г
При этом специфическом типе катализа енол представляет собой наиболее
активную депротонизированную форму, скорость образования которой соот-
ветствует скорости бромирования, рацемизации и дейтерообмена.
Экспериментально найденные скорости можно сравнивать двумя способами.
Во-первых, сравнивают скорость галогенирования термодинамически устой-
чивого кетона или другого подобного таутомера и скорость его рацемизации
(при этом должны быть соблюдены необходимые условия асимметрии) при
одинаковых условиях — температуре, растворителе и катализаторе. Снача-
ла были проведены такие сравнения для процессов кислотного катализа.
Рамберг и Мелландер [83] сравнили скорости бромирования и рацемизации
сульфонокислоты (I) в воде, содержащей бромистоводородную кислоту
в качестве катализатора. Рамберг и Хедлунд [84] изучали бромирование
сульфонокислоты (II) в подобных же условиях. Вильсон и автор этой книги
[85] исследовали бромирование кетона III, сравнивая скорости этого про-
цесса и рацемизации в водной уксусной кислоте, содержащей бромистый
водород. Бартлет и Стауфер [86] изучали поведение кетона (IV), сравнивая
скорости его иодирования и рацемизации в уксусной кислоте, содержащей
азотную кислоту. Во всех случаях скорость галогенирования равнялась
скорости рацемизации. Сюй и Вильсон [87] рассмотрели более простой в тео-
ретическом отношении случай катализа основанием, сравнивая скорости
бромирования и рацемизации кетона IV в водном растворе уксусной кислоты,
причем в качестве катализатора служили ацетат-ионы. И в этом случае
686
Перегруппировки ненасыщенных соединений
оказалось, что скорости рассматриваемых двух процессов были равны,
c2H5so2
/СП-СО2Н
СН3
С2н5
СОС6Н5
СНз
IV
СП —СО2П
С6н5
/СН —СО —сн3
МентилО2С
VI
СНз
/СО
СНз
V
Как уже указывалось выше, такое сравнение скоростей само по себе не позво-
ляет выявить стадию, определяющую скорость всего процесса. Дело в том,
что любой способ удаления подвижного атома водорода от асимметрического
центра, будь то отщепление его от молекулы или внутримолекулярное пере-
мещение, может вызвать рацемизацию. Однако результаты второго метода
сравнения скоростей, а именно скорости присоединения дейтерия из раство-
рителя, содержащего дейтероксигруппу, и скорости бромирования или раце-
мизации в том же растворителе и в идентичных условиях свидетельствуют
о переходе атома водорода к растворителю. Рейтц [88] показал, что для про-
цессов кислотного катализа начальная скорость бромирования «легкого»
ацетона (СН3)2СО в тяжелой воде (D2O) при катализаторе D3O+ равна
начальной скорости присоединения дейтерия к ацетону в отсутствие брома.
Для процессов, катализируемых основаниями, Сюй, Вильсон и автор этой
книги [89] сравнивали скорости рацемизации и дейтерирования кетона (IV)
в смеси D2O —диоксан, где в качестве катализатора служил OD~-hoh,
и установили, что скорости этих процессов равны.
Из такого сравнения скоростей можно сделать следующие выводы. Для псев-
докислотных кетонов при катализе основаниями:
Скорости
галогенирования
рацемизации
водородного обмена
в
в
в
с
«легком» растворителе )
I равны (IV)
«легком» растворителе J
«тяжелом» растворителе ) ,тхтч
I равны (IV)
«тяжелым» растворителем J
Для псевдокислотных кетонов при кислотном катализе:
рацемизации в «легком» растворителе } .
I (в «легком» растворителе • Равны
Скорости { галогенирования I
] I в «тяжелом» растворителе
' водородного обмена в «тяжелом» растворителе
} равны (V)
Таким образом, скорости галогенирования, рацемизации и водородного
обмена термодинамически устойчивых псевдокислот с гидроксилсодержащи-
ми растворителями при кислотном и щелочном катализе равны. Скорости
этих процессов определяются скоростью одной и той же стадии, а именно
стадии депротонизации псевдокислоты.
Для рассмотрения вопроса о скоростях изомеризации нужно перейти к тер-
модинамически уравновешенным или неустойчивым таутомерам. Сравнение
скоростей рацемизации и изомеризации системы, для которой можно изме-
рить положение равновесия, было проведено Кимбаллом [90] на примере
687
Глава XI
соединения VI; при перекристаллизации (—)-ментил-(±)-а-фенилацетоаце-
тата из метанола выделяется один из диастереомеров, который в бензоле,
содержащем следы пиперидина, претерпевает мутаротацию, т. е. рацемиза-
цию фенилацетоацетатного остатка, со скоростью, примерно в два раза пре-
вышающей скорость енолизации указанного кетоэфира. При енолизации,
согласно методу Мейера, образуется равновесная смесь, содержащая 71%
енола. Очевидно, оптическая активность теряется на стадии аниона, обра-
зующегося при депротонировании пиперидином. Этот анион вновь протони-
руется до кетона и енола, скорости образования которых сравнимы по вели-
чине, и поэтому скорость изменения оптической активности превышает ско-
рость енолизации.
Сравнение скоростей водородного обмена и изомеризации было проведено
для случая системы со смещенным равновесием на примере ненасыщенных
нитрилов — циклогексенил- и циклогексенилиденацетоиитрилов, формулы
которых даны ниже. Такое же сравнение скоростей было проведено для водо-
родного обмена и изомеризации циклопентенил- и циклопентилиденмалоно-
вых эфиров Хыо и Кона, упомянутых на стр. 682; р,у-ненасыщенный
нитрил может быть выделен, но он термодинамически абсолютно неустойчив
и при действии щелочного катализатора почти полностью переходит в а,р-
ненасыщенный таутомер
CH2CH=C-CHaCN СН2СН2 —C = CHCN
(М I I I I (a,p)
CH2CH2 —CH2 CH2CH2—CH2
Кроме того, проведено экспериментальное сравнение [91] скорости изомери-
зации, катализируемой основанием, и скорости водородного обмена любого
изомера со средой. Катализаторами процесса изомеризации в дейтероксилсо-
держащем растворителе C2H5OD служили этилат-ионы, а скорость изомери-
зации сравнивалась со скоростью присоединения дейтерия к любому из тауто-
меров. В результате было установлено, что скорость дейтерирования 0,у-не-
насыщенного нитрила несравненно выше скорости его изомеризации
и что в свою очередь эта скорость несравненно выше скорости дейтерирова-
ния а,р-ненасыщенного нитрила: водородный обмен (Р,у) превращение
(Р,у —>-а,р) водородный обмен (а,р).
Далее, если не происходит образования промежуточного продукта между
§,у- и а,р-ненасыщенными нитрилами, т. е. если у-водородный атом может
быть введен только во время удаления а-водородного атома, то должно быть
соответствие между скоростью водородного обмена и скоростью изомериза-
ции. Найденные сильно различающиеся соотношения скоростей указывают
на то, что образование промежуточного продукта в состоянии равновесия
вызывает водородный обмен со средой: последний может происходить и без
полного превращения одного таутомера в другой. Это согласуется с гипо-
тезой промежуточного образования аниона, причем наблюдаемые соотноше-
ния скоростей можно понять из рассмотрения диаграммы энергии, приведен-
ной на рис. 50. Как легко видеть, только небольшая часть анионов, образо-
вавшихся из термодинамически неустойчивого р,у-соединения, преодолевает
высокий энергетический барьер для превращения в термодинамически устой-
чивый а,р-продукт; большая же часть анионов превращается в исходный
Р,у-изомер, преодолевая лишь низкий барьер. Превращение анионов не влия-
ет на скорость изомеризации; однако каждый образовавшийся анион при-
водит к водородному обмену. Таким образом, водородный обмен протекает
быстрее, чем изомерное превращение. Более того, изомерное превращение
протекает быстрее, чем водородный обмен продукта превращения. Как хоро-
688
Перегруппировки ненасыщенных соединений
шо видно из диаграммы энергии (рис. 50), для полного перехода р,у-нена-
сыщенного исходного соединения в сх,р-продукт, т. е. для процесса изомери-
зации, необходимо преодолеть меньший энергетический барьер, чем при
частично обратимом превращении а,р-ненасыщенного продукта в ионы —
процессе, необходимом для водородного обмена.
е. Бимолекулярный механизм прототропии. Наличие бимолекулярного
механизма B-SE2' было установлено подобными же методами на примере
метиленазометиновой системы, содержащей арильные группы в крайних
положениях. Такого рода соединения относятся к одной из самых малопод-
вижных триадных систем, но могут быть приведены в состояние равновесия
при действии таких сильных оснований, как этилат-ионы в этиловом спирте.
Применялись два метода. Один из них состоит в сравнении скорости изомери-
зации, определяемой химическим путем, со скоростью рацемизации оптиче-
ски активного таутомера. Второй основан на сравнении любой из этих ско-
ростей со скоростью водородного обмена между таутомерами и средой.
В табл. 169 приведены изученные системы. Все они термодинамически более
или менее сбалансированы и образуют доступные для измерения равновес-
ные количества обоих таутомеров.
Таблица 169
Равновесие метиленазометинов в этиловом спирте, для которых сравниваются
скорости водородного обмена, рацемизации и изомеризации,
катализируемых основанием
(I)
R R"
^CHN = C/
R' R'"
OC2H5 R R"
-—> 2c=n-ch<^
R' R'"
(П)
При- мер R R' R" R'" Равновесие I : II
1 Свн5 сн3 С6н5 п-С1С6Н4 50 : 50 при 85 °C
2 С0Н5 сн3 С6н5 с6н5 68 :32 при 85 °C
3 м-С6Н5С6Н4 С6Н5 С6Н5 н 56 : 44 при 25 °C
4 п-СН3ОС6Н4 н С6Н5 н 24 : 76 при 74 °C
(1) КО(СН2)гОН в HO(CH2)2OH; (2) mpem-C4HgOK в mpem-СдНдОН
5 | С6Н5 | СН3 | я-С1СвН4 | м-С1С0Н4 150: 50 при 75 °C (2)
Соотношение между скоростью рацемизации и скоростью изомеризации,
определяемой химическим путем, изучалось [92] на примерах 1—3. Во всех
случаях при изомеризации в этиловом спирте, катализируемой этилат-иона-
ми, оптически активный изомер I дает неактивный изомер II; это верно
даже для случая 2, когда соединение II имеет центр асимметрии. Следова-
тельно, можно было определить три скорости, а именно: скорость реак-
ции I II, устанавливаемую химическими методами, скорость обратной
реакции II -> I и скорость процесса потери оптической активности тауто-
мером I. Необходимо было решить, происходит ли потеря оптической актив-
ности таутомера I со скоростью, соответствующей превращению в неактив-
ную форму II и регенерации из формы II, или же скорость потери активности
превышает скорость указанных двух последовательных процессов. Согласно
бимолекулярному механизму B-SE2', скорость рацемизации не может быть
столь большой; скорость потери оптической активности должна быть равна
вычисленной на основании скоростей изомеризации в обоих направлениях,
44-0772
689
Глава XI
определяемых химическими методами. Мономолекулярному механизму
B-Se1z соответствует промежуточный анион, потеря оптической активности
которого происходит так, что полный цикл превращения в форму II и обрат-
но может быть сокращен обратимым превращением промежуточных ионов,
что приводит к более быстрой рацемизации. Опыт показывает, что в каждом
случае при катализе этилат-ионами в спирте скорость рацемизации, сопро-
вождающей изомеризацию, является такой, какой она была бы в том слу-
чае, если бы не происходило сокращения цикла. Это свидетельствует о том,
что в процессе реакции не происходит обратимого образования промежуточ-
ного продукта, теряющего оптическую активность, и поэтому или не обра-
зуется промежуточного аниона, или его образование в заметной степени,
необратимо, т. е., образуясь из соединения I, этот анион при протонизации
во всех случаях дает только соединение II, что в случаях 1 и 2 соответствует
протонизации бензгидрильного, а в случае 3 — бензильного положения.
Авторы считают вторую возможность маловероятной и останавливаются
на одностадийной изомеризации по механизму Лоури B-Se2'.
На примерах 3—5 табл. 169 были изучены соотношения между скоростями
изомеризации в обоих направлениях или соответствующей скоростью раце-
мизации и скоростью водородного обмена между таутомерной системой и сре-
дой. В случае мономолекулярного механизма образуется промежуточный
анион, и поэтому скорость водородного обмена должна быть больше скорости
любого непосредственно измеряемого процесса изомеризации. При бимоле-
кулярном механизме должны быть равны начальные скорости изомеризации
одного таутомера и присоединения водорода к изомеризующейся системе,
в частности дейтерия из дейтероксилсодержащего растворителя. Очевид-
но, что если растворитель может поставлять только дейтроны, а не про-
тоны, то при превращении каждой исходной молекулы по бимолекулярному
механизму к системе присоединяется один дейтрон *.
С2Н56^Н D—OC2HS С2Н5О-Н D С2Н5О’
RR'C—N=^CR"R"' RR'C=N-CR"R'"
Первая попытка подтвердить на этом принципе бимолекулярный механизм
была сделана де Саласом и Вильсоном [93] применительно к системе 4
(табл. 169). Скорости изомеризации, определяемые химическими методами,
сравнивались со скоростью присоединения дейтерия всей системой из не пол-
ностью дейтерированного гидроксилсодержащего растворителя. Результаты
исследований показали лишь, что начальные скорости изомеризации и водо-
родного обмена — величины одного и того же порядка, относительные же
скорости переноса протонов и дейтронов из растворителя к растворенному
веществу не были установлены **. Однако недавно Оссорио и Хьюз [94] при-
* После начального равенства скоростей в процессе реакции скорость дейтерирования
должна превышать скорость изомеризации. Дело в том, что в то время как в начале изо-
меризации соединения I практически весь образовавшийся продукт II возникает в ре-
зультате простого процесса I -+ II, который позволяет только одному атому дейтерия
перейти в молекулу II, в последующее время будет присутствовать некоторое количество
соединения II, прошедшее более сложный путь I —II —I —II, что ведет к переходу
более чем одного атома дейтерия в окончательно образовавшуюся молекулу II.
** Протоны и дейтроны переносятся с различными скоростями, отношение которых прин-
ципиально различно для каждого процесса переноса, поскольку он зависит от нулевой
энергии водорода в начальном и переходном состояниях. Не зная этого отношения,
нельзя определить скорость водородного обмена с помощью только изотопа обмени-
вающегося водорода.
690
Перегруппировки ненасыщенных соединений
менили исчерпывающее дейтерирование к системе 3 (табл. 169). Они сравни-
ли скорости изомеризации со скоростью рацемизации. Оссорио и Хьюз сопо-
ставляли скорость потери оптической активности со скоростью присоедине-
ния дейтерия в процессе изомеризации соединения I, катализируемой эти-
лат-ионами в чистом дейтероксилсодержащем растворителе C2H5OD. Они
установили, что начальные скорости потери оптической активности и при-
соединения дейтерия точно равны между собой *. Из этого следует, что
начальные скорости изомеризации и присоединения дейтерия равны и что,
следовательно, в простом процессе I -> II присоединяется точно один атом
дейтерия, как того требует одностадийный бимолекулярный механизм
B-Se2'.
Крам и Гатри, изучая систему 5, сделали противоположные заключения.
Как указано в табл. 169, они применяли другие основание и растворитель,
однако в этом случае прототропная система очень напоминала систему 1.
В работе Крама и Гатри не приводится кинетических данных**, однако
авторы доказали следующие соотношения скоростей: Н-обмен в II пре-
вращение II -> I Н-обмен в I. Отсюда следует, что в этих условиях обмен
соединения II не связан с его превращением в I. По мнению авторов, обмен
в II происходит через карбанион, через этот же карбанион, но по мономоле-
кулярному механизму изомеризации B-Se1' соединение II превращается
в I, а тот факт, что эта изомеризация происходит гораздо медленнее, чем
обмен, объясняется почти полной протонизацией карбаниона по бензгидриль-
ному положению с возвратом к соединению II, а не с превращением
в I.
Необходимо указать на различие между этими данными и данными Оссорио
и Хыоза по системе 3, в случае которой карбанион должен был бы протони-
роваться только по бензильному положению. Следует надеяться, что это
кажущееся противоречие будет разрешено последующими кинетическими
исследованиями.
2. Псевдоосновность и анионотропия
а. Псевдоосновность. Ганч первый обнаружил особую форму таутомер-
ного превращения в случае обратимой реакции между ионизированными чет-
вертичными аммониевыми основаниями, содержащими азот с двойной свя-
зью, и изомерными неионными карбинолами. Последние он назвал псевдо-
основаниями.
Одним из первых примеров псевдооснований могут служить гидроокись и соли
5-фенил-10-метилакридиния [96]. Его соли с такими сильными кислотами,
как соляная, являются типичными четвертичными аммониевыми солями.
Если водный раствор такой соли, например хлорида, подщелочить окисью
серебра, то полученный раствор сначала обладает свойствами раствора чет-
вертичного аммониевого основания; он отличается высокой электропровод-
ностью и сильнощелочной реакцией. Но со временем электропроводность
уменьшается и раствор становится почти нейтральным: образующийся кова-
* После того как изомеризуется около 10% вещества, скорость дейтерирования значи-
тельно превышает скорость рацемизации, а это указывает на то, что несколько атомов
дейтерия присоединяется к молекуле в процессе изомеризации.
** См. работу [95]. Аргументы авторов основывались на «константах скоростей, опреде-
ленных по одной точке», однако одной точки недостаточно для доказательства, что опре-
деленная таким образом величина является константой.
44* 691
Глава XI
лентный карбинол может выкристаллизоваться.
Такое медленное образование нейтрального карбинола из катиона и иона
гидроксила напоминает медленное образование нормального фенилнитро-
метана из его аниона и иона водорода. Псевдоосновный карбинол соответ-
ствует псевдокислотному фенилнитрометану, а ионы, образующие спирт,
соответствуют ионам нитросоединения. В этой аналогии отсутствует вторая
ковалентная, очень легко ионизирующаяся форма основания, соответствую-
щая ковалентной ацм-форме нитросоединения. В псевдоосновности «изомер-
ное превращение» происходит только между электровалентной ионной парой
и ковалентным карбинолом.
При обработке такого карбинола сильными кислотами акридиний-ион реге-
нерируется с отщеплением иона гидроксила. Таким образом, можно пред-
положить, что указанное превращение обратимо. Устойчивые анионы, напри-
мер перхлораты или хлориды, благоприятствуют образованию ионных пар.
В присутствии такого сильного нуклеофильного аниона, как гидроксил,
образуется преимущественно ковалентная молекула. К ее образованию при-
водит не только ион гидроксила; циан-ион, например, обладает достаточной
нуклеофильностью для образования ковалентного цианида. Ганч назвал
такие соединения псевдосолями.
Псевдооснование
или псевдосоль
Х~ = С1~, С1О4_ и т.д. + }—
, ~ -+С.Н—ИУ >
X- = ОН-, CN- и mA >=<
5 7
С6Н5+ X
Аммониевое основание
или соль
Дальнейшие признаки псевдоосновных систем были выявлены Ганчем
на других примерах, а именно на производных N-алкилхинолиния. Соли
этого катиона при подщелачивании дают щелочные растворы, которые с тече-
нием времени теряют щелочную реакцию и образуют псевдооснования.
Последние могут быть окислены феррицианидом в а-хинолоны [97] и поэтому
рассматриваются как 2-карбинолы
692
Перегруппировки ненасыщенных соединений
Большую термодинамическую устойчивость 2-карбинолов по сравнению
с 4-карбинолами можно объяснить тем, что только в первых сохраняется
сопряжение фенильной и винильной групп. Такой же тип структуры сохра-
няется в случае эфиров (например, приведенных ниже), которые легко обра-
зуются из карбинола, вероятно, путем обмена анионов в реакции со спиртом-
растворителем или самопроизвольной реакции с другой молекулой карби-
нола в отсутствие другого спирта
Образование псевдосоли можно обнаружить при взаимодействии солей
метилхинолиния с различными источниками карбанионов, например с реак-
тивами Гриньяра [98], с псевдокислотными карбонильными или с нитросоеди-
нениями [99]
СИзМц!
,/\/ч
I I! СНСНз
СН3
CH3NO2
СН3
Реакция псевдосолей с цианид-ионом приводит исключительно к образова-
нию 4-цианида, а не 2-цианида, что можно доказать последовательным
окислением, деметилированием и гидролизом до цинхониновой кислоты
[100]. Можно допустить, что выделенный цианид действительно является
прототропным таутомером 4-псевдосоли; в литературе не встречается дока-
зательств обратного. Этот таутомер, обладая большим сопряжением, термо-
динамически более устойчив, чем сама 4-псевдосоль или чем 3-псевдосоль
или ее любой прототропный таутомер:
CN
I
/\/\
L 'I /щ
'/''г/
I
СН3
Свободные циклические псевдооснования, образованные путем координации
гидроксила с положением 2, содержат кольчато-цепную прототропную систе-
му; циклические 2-карбинолы таутомерны по отношению к дециклизованным
693
Глава XI
аминокарбонильным соединениям
Действительно, соли метилхинолиния реагируют с гидроксиламином
и с фенилгидразином с образованием соответственно оксима и фенилгидра-
зона, которые сами являются членами кольчато-цепных прототропных си-
стем, и, по-видимому, более устойчивы в их циклической гидроксиламино-
и фенилгидразино-формах точно так же, как соответствующие производные
способных к мутаротации сахаров (разд. 1,6) [101].
Н^^СНз
До сих пор еще не установлено, образуются ли эти соединения путем обмена
анионов в циклическом псевдоосновании или через карбонильные реакции
аминоальдегидов с открытой цепью.
Как указали Ганч и Кальб [102], ионы алкилпиридиния имеют значительно
меньшую способность переходить в ковалентные псевдоформы, чем ионы
алкилхинолиния. При подщелачивании раствора метилпиридиниевой соли
щелочная реакция и электропроводность раствора сохраняются, а на суще-
ствование некоторого количества псевдооснования в равновесии с ионной
парой указывает образование а-пиридона при окислении феррицианидом
Это отличие от хинолиновых аналогов, возможно, означает, что большая
потеря ароматической устойчивости обусловлена скорее частичным насы-
щением простого пиридинового кольца, чем насыщением пиридиновой части
хинолиновой системы, в которой устойчивость бензольного кольца, вероят-
но, увеличивается за счет разрушения ароматического характера гетеро-
циклического кольца.
Некоторые псевдоосновные системы обладают другим свойством, которое
можно проиллюстрировать на примере а- и у-пиридонов. Существо вопроса
состоит в том, что если в четвертичном ионе аммония существует кислотный
центр, например фенольная или иная кислотная группа, то гидроокись
аммония подвергается дегидратации типа внутримолекулярной нейтрализа-
ции кислоты основанием; при этом кислотный центр депротонизуется и обра-
зуются так называемые ангидрооснования. Например, если 2-оксипиридин,
или а-пиридон, как его обычно называют по его прототропному таутомеру,
694
Перегруппировки ненасыщенных соединений
алкилировать обычным способом как пиридин, то образующийся ион четвер-
тичного аммония переходит в его ангидрооснование — N-алкил-а-пиридон [103]
(fj-OH+OH-
+ N
I
R
Этому основанию можно приписать, как показано, бетаиноподобную или
карбонильную структуру, как это предполагалось ранее, но подлинная
структура его, конечно, мезомерна между этими структурами.
Ганч и Кальб [102] рассматривали котарнин как пример псевдоосновности
в гидроароматической аммониевой системе. Соли котарнина с сильными кис-
лотами определенно являются ионными, но гидроокись (собственно котар-
нин) представляет собой псевдооснование, тогда как его цианид — ковалент-
ную псевдосоль. Катион реагирует с любым источником реакционноспособ-
ного аниона, например со спиртами, меркаптанами [104], с псевдокислотными
карбонильными [105] и нитросоединениями, с инденом [106] и с реактивами
Гриньяра [107] с образованием ковалентных продуктов обычного типа:
Котарнин как циклический псевдоосновный 2-карбинол является членом
кольчато-цепной прототропной системы, а его таутомером будет аминоаль-
дегид [108]
Действительно, котарнин реагирует с гидроксиламином [109] и анилином
[110] с образованием соответственно оксима и анила, которые сами представ-
ляют собой кольчато-цепные таутомеры и, возможно, более устойчивы в их
циклических формах, как наблюдается в других подобных случаях. Вопрос
о том, образуются ли они из карбинола котарнина путем обмена аниона или
через альдегидную форму котарнина, обсуждался, но до сих пор не сделано
никаких определенных выводов.
695
Глава XI
Псевдоосновный 2-карбинол может и не быть циклическим, но в случае
циклических соединений имеется кольчато-цепная прототропная система,
а в случае их ациклических соединений — протолитическая система. Кар-
бинол, несомненно, является неустойчивым аддуктом в обратимой реакции
присоединения, так же как в случае присоединения аминов к альдегидам
или кетонам
>n=c + он <=* \Qc^qh \nh + С=О
I / о /
Высказанные соображения обосновывают реакцию Манниха, в которой
аммиак, первичный или вторичный амин и формальдегид реагируют с анио-
ном, который достаточно доступен и нуклеофилен для того, чтобы вытеснить
ион гидроксила из псевдооснования. Обычными источниками анионов слу-
жат псевдокислотные карбонильные соединения, но применялись и псевдо-
кислотные нитросоединения, а также спирты и с полученными таким обра-
зом псевдоалкоголятами или эфирами реактивы Гриньяра. Некоторые при-
меры этих реакций приведены ниже:
C6HbCOCH3 + CH2O4-(CH3)2NH —> CeH5COCH2CHN(CH3)2
NO2CH2CH3 + CII2O-HCH2)5NH NO2CH(CH3)CH2N(CH2)5
h-C4H9OH + CH2O + (C2H5)2NH —> «-C4H9OCH2N(C2H5)2
CH2=CHCH2MgCl-h»-C4H9OCH2N(C2H5)2 —> CH2 = CHCH2CH2N(C2H5)2
Такие трифенилметановые красители, как малахитовый зеленый и параро-
занилин, считались Ганчем и Освальдом [111] псевдоосновными системами.
Эти системы формально несколько отличаются от ранее рассмотренных.
Центр координации расположен дальше от атома азота; в соответствии
с принятой нумерацией (N = 1) эти карбинолы представляют собой 6-кар-
бинолы. В данном случае не ионы аммония, а ковалентные псевдоформы
носят вполне ароматический характер. Устойчивость и окраска катионов
обусловлена их мезомерной формой, вследствие которой эти катионы в зна-
чительной степени хиноидны. Однако в большинстве реакций эти системы
ведут себя подобно ранее рассмотренным системам. Сами по себе красители
являются электровалентными аммониевыми солями таких сильных кислот,
как, например, соляная кислота. При прибавлении щелочи к раствору любо-
го из них, например кристаллического фиолетового, окраска постепенно
бледнеет; цвет, электропроводность и щелочная реакция исчезают одновремен-
но, а образующийся бесцветный ковалентный карбинол, являющийся псевдо-
основанием, может быть осажден. При добавлении к красителю цианид-иона
образуется бесцветный’ковалентный цианид, т. е. псевдосоль. Другие источ-
ники реакционноспособных анионов, например реактивы Гриньяра [112],
взаимодействуют с катионом красителя с образованием ковалентных про-
дуктов аналогичного строения.
696
Перегруппировки ненасыщенных соединений
(CH3)2N ___\
(CH3)2N -<f
(CH3)2N
Кристаллический фиолетовый и его псевдоформы (приведены только валентные структуры).
В мезомерном состоянии катиона заряд поделен между тремя атомами азота,
а также в некоторой степени между тремя а-, шестью мета- и одним
а>-углеродным атомами
Терджон и Ла Мер [ИЗ] показали, что реакция катиона кристаллического’
фиолетового и гидроксильного иона имеет кинетический второй порядок
и отрицательный солевой эффект, свойственный реакции однозарядных про-
тивоионов, приводящей к образованию нейтральных продуктов.
В химии ароматических диазосоединений Ганч [114] обнаружил случай
псевдоосновности, сопровождающейся координацией не с атомом углерода,
а со вторым атомом азота. Диазониевые соли сильных кислот представляют
собой аммониевые ионные формы этой системы. Диазогидроокиси, которые
должны быть ковалентными вследствие образования их из ионов диазония
и гидроксила, являются псевдооснованиями этой системы, даже если впо-
следствии они ионизированы как кислоты в щелочной среде и образуют
диазотаты. Ковалентные диазоцианиды и диазосульфонаты представляют
собой примеры псевдосолей.
+ Х-=С1~ и т. д.
Ar—N = N < ' ' . Ar—N = N —X
х~=он_, с:ги т. д.
Наличие тройной связи в ионной форме и вследствие этого двойной связи
в псевдоформах приводит, как показал Ганч, к определенным следствиям.
Один из выводов состоит в том, что псевдоосновная диазогидроокись являет-
ся членом прототропной системы, а не протолитической системы — аддукта
обратимого присоединения. Имея в виду умеренную кислотность диазогидро-
окисей, т. е. легкость образования диазотат-ионов, можно с уверенностью
сказать, что прототропные взаимопревращения протекают по ионному меха-
низму B-SE1'.
Аг -N=N-OH^ [Ar-N—N- Of + Ar -NH-N=O
Тот факт, что протоны легко отщепляются от атомов азота и кислорода,
а также наличие аналогии между таутомерией диазоаминосоединений, ами-
дов п амидинов, свидетельствуют о том, что диазогидроокись — нитрозами-
новая система, подобно другим упомянутым прототропным системам, очень
подвижна, и поэтому чрезвычайно трудно, а в ряде случаев и невозможно
выделить и сохранить отдельные изомеры.
Вторым важным следствием ненасыщенности псевдоформ является их склон-
ность в геометрической изомерии, подобной изомерии оксимов и азобензола.
Тезис, впервые выдвинутый Ганчем, который он отстаивал до конца своей
жизни, состоял в том, что существование двух рядов диазотатов, диазоциани-
дов и диазосульфонатов можно объяснить этой способностью к геометриче-
ской изомерии. Несмотря на многочисленные возражения, выдвигавшиеся
против этого тезиса, в настоящее время он общепризнан [115].
Хорошо известны не только аммониевые, по также и оксониевые псевдоос-
697
Глава XI
новные системы. Ксантгидрол образует ионный трехбромистый ксантилий
и комплекс с FeCl3 [116]. Однако на меньшую относительную устойчивость
ксантилий-иона по сравнению с N-алкилакридиний-ионом указывает тот
факт, что хлористый и бромистый ксантгидрилы выделены в виде ковалент-
ных псевдосолей [117]. Сам карбинол представляет собой полностью кова-
лентное псевдооснование. Однако он претерпевает большинство обычных
реакций обмена анионов, характерных для псевдооснований, например
с такими кето-енолами, как ацетилацетон, и с псевдокислотами различного
типа [118]
Н
С
/Ч/ Х-=Вг£, FcClJ
I II I Нх~ -------------------------------->
Х/\ Х"=СГ, ОН-, СН(СОСНг)2 и т. д.
о
+
В качестве простейших ароматических ионов оксония,
центр и поэтому образующих ангидрооснования, можно привести а- и у-
пироны. Они образуют с кислотами соли, катионами которых являются 2-
или 4-оксипирилий-ионы [119]. Они легко депротонируются слабыми осно-
ваниями, например водой, образуя ангидрооснования, т. е. собственно
пироны, которым, подобно другим ангидрооснованиям, может быть при-
писана бетаиноподобная или карбонильная валентная структура, а в дей-
ствительности они мезомерны между этими структурами. Соотношение этих
структур в общем виде может быть выражено следующим образом:
+ 4-
О 0 0
CHg—I'f —СН3 Основание СНз——СН3 СН3——СН3
имеющих кислотный
Оксипирилий-ион Валентные структуры пирона
Строение антоцианинов подобно строению многоядерных ионов оксипирилия,
в частности ионов оксифлавилия и соответствующих ангидрооснований. Кис-
лотные центры в катионах расположены большей частью в кольцах, не содер-
жащих оксониевых центров, так что ангидрооснования имеют межъядерный
хиноидный характер, как это показано ниже для дракорходина:
Валентные структуры
ангидроосновапия дракорходина
698
Перегруппировки ненасыщенных соединений
Здесь приведен очень простой пример: обычные антоцианины, производные
агликонов пеларгонидина, цианидина и дельфинидина, имеют несколько
кислотных центров; отсюда следует, что ангидрооснования не только мезо-
мерны, по и прототропны. Например, ангидрооснование каллистефина, т. е.
пеларгонидин-3-глюкозид, может существовать в трех изомерных формах,
отличающихся распределением фенольных протонов, причем каждый тауто-
мер является мезомерным между бетаиноподобной и хиноидной валентными
структурами, как это показано ниже. Подвижность этой прототропной систе-
мы, по-видимому, чрезвычайно велика, и поэтому нельзя выделить и сохра-
нить отдельные таутомеры.
Q о и ______
YY но-(у ||-<=>-°и
>/^/\0С6Н1105 Y^^OCeHnOs
ОН ' о
+
о
но -/V S-(r
I II I
y\^'\oceH11o5
OH
0
ho-^/ °
Y^^OCeHnOs
I OH
Валентные структуры трех прототропных форм ангидроосновапия каллистефина
Известны также сульфониевые псевдоосновные системы. Тиоксантгидрол
образует сульфониевые соли, теряя при этом гидроксильную группу, точно
так же, как ксантгидрол образует соли оксония [120]. Соли 2-фенилбензди-
тиилия при подщелачивании дают ковалентный карбинол [121]
S S
/\/ \ X-=NO3, C10Z и т. д. Z\/ \
I С-Свн5+Х- . ________________;сх-с6н5
кА/ х-он кА/
б. Анионотропия. Развитие концепции. Мономолекулярный меха-
низм *. В 1922 г. Жилле [123] обратил внимание на формальное сходство
некоторых перегруппировок, в которых происходит миграция электроотрица-
* Подробный обзор этого аспекта анионотропии дан де ла Маром в книге [122].
699
Глава XI
тельных групп. Он назвал их «отрицательными миграциями», хотя и не вкла-
дывал в это понятие представления о том, что в этом процессе участвуют
отрицательные ионы. Жилле применил термин «отрицательный» в том же
смысле, как его применяли в 70-х годах прошлого века: атом галогена или
карбонильная группа были «отрицательными» и оба были склонны к мигра-
ции. Однако Жилле приводил данные о многих перегруппировках, которые
теперь рассматриваются как анионотропные, включая давно известные
взаимные превращения гераниола и линалоола или их эфиров, происходящие
с перемещением гидроксила или ацилокси-радикала:
(СН3)2С = СНСН2СН2С(СН3) = СНСН2ОН (СН3)2С = СНСН2СН2С(СН3)(ОН)СН = СН2
Гераниол Линалоол
В 1927 г. Прево [124] рассматривал такие перегруппировки, исходя из пред-
ставлений теории, названной им синионизмом. Согласно этой теории, допу-
скалась ионизация перегруппировывающейся молекулы, причем важно то,
что одна из частиц должна была быть триполярным ионом. Последнее поло-
жение теории ограничивало возможность ее применения для объяснения
химических процессов, происходящих в перегруппировывающихся систе-
мах, т. е. влияния структурных особенностей и условий среды на их под-
вижность. В следующем году Бартон и автор этой книги [125] теоретически
охарактеризовали анионотропию как явление, дополняющее прототропию;
они предложили для анионотропии ионный механизм, аналогичный ион-
ному механизму прототропии; факторы, влияющие на анионотропную под-
вижность, они объяснили исходя из этого механизма.
В самой простой форме для подробно изученной трехуглеродной системы
этот механизм можно представить в следующем виде:
[С-С-С]+
С-<?=С С=О-С
х) Сх
(М')
[X]-
Главная черта этого механизма заключается в допущении промежуточного
образования мезомерного катиона. Механизму придан символ SN1' частично
для того, чтобы подчеркнуть аналогию с механизмом прототропии B-Se1z,
согласно которому промежуточно образуется мезомерный анион. Однако
для введения символа SnI' имеется и другая причина. Очевидно, механизм
процесса не должен существенно измениться, если после отделения одного
аниона к системе присоединяется другой, даже и отличный анион. Если
этот новый анион присоединяется по месту отделения первого аниона, то
весь процесс можно назвать мономолекулярным нуклеофильным замеще-
нием и обозначить символом SnI. Если же новый анион присоединяется
к другому концу мезомерного катиона, то естественно рассматривать такое
замещение, сопровождающееся подобной перегруппировкой, как разно-
видность замещения без перегруппировки и обозначать символом SnI'-
Во всяком случае нелогично исключить из данного определения особый слу-
чай, в котором присоединяющийся к системе анион идентичен отщепившему-
ся аниону. Иными словами, анион X - в приведенной схеме должен рассмат-
риваться как анион, который на различных стадиях процесса может быть
идентичным или не идентичным отщепляющемуся аниону. Имея в виду эти
соображения, ионный механизм анионотропной перегруппировки обозначи-
ли символом SnI'.
700
Перегруппировки ненасыщенных соединений
Вышеприведенный механизм был предложен на основании четырех аргумен-
тов [126], к которым позже были присоединены и другие. Во-первых, для
определенной трехуглеродной системы с различными мигрирующими груп-
пами подвижность заметно возрастает при увеличении устойчивости этих
групп в виде аниона. Как было известно, гераниол и линалоол взаимно пре-
вращаются друг в друга в воде только при температурах, близких к 200 °C,
в то время как их ацетаты взаимно превращаются друг в друга в уксусном
ангидриде при более низких температурах. Установлено, что практически
необратимые превращения производных 1-фенилаллила в циннамиловые
(3-фенилаллиловые) соединения
С6Н5СН(Х)СН = СН2 —> СвН5СН = СНСН2(Х)
протекают с большим трудом со спиртами (в отличие от их сопряженных
кислот), легче с ацетатами и настолько легко с бромидами, что в последнем
случае невозможно выделить менее устойчивый изомер. По подвижности эти
системы можно расположить в ряд X = Вг > ОСОСН3 > ОН.
Во-вторых, для различных трехуглеродных систем с одной и той же мигри-
рующей группой, а именно ацетоксильной, подвижность заметно увеличи-
вается в случае структурных изменений, которые могут увеличить приток
электронов к центру, от которого должна отщепиться мигрирующая группа.
Так, в случае 1-фенилаллиловой системы, приведенной выше (X = ОСОСН)3,
подвижность возрастает при введении в пара-положение метильной группы
и в еще большей степени хлора. При замене фенильной группы метильной
подвижность системы очень сильно уменьшается. Сравнительная подвиж-
ность при концевых группах, присоединенных к трехуглеродной системе,
выражается рядом *
п-С1С0Н4 » п-СН3СсН4 > С8Н5 > СН3 > Н
В-третьих, исходя из ионного механизма легко понять влияние растворите-
лей и катализаторов. В отсутствие значительного катализа растворенным
веществом приведенное выше превращение 1-фенилаллилацетата (X =
= ОСОСН3) в циннамилацетат протекает в ряде растворителей со скоростями,
порядок которых согласуется с диэлектрической проницаемостью растворите-
лей: бензонитрил > уксусный ангидрид >> хлорбензол > и-ксилол. Типич-
ным примером катализа анионотропного превращения растворенным вещест-
вом может служить обычный катализ такими сильными кислотами, как H2SO4
или НВг, или сольватированным ионом водорода. Катализирующее действие
их тем сильнее, чем большей основностью обладает подвижная группа; оно
велико, например, в случае спиртов и мало в случае эфиров. Кук [127]
приводит следующий пример, иллюстрирующий противоположное отношение
прототропии и анионотропии к каталитическим эффектам. Приведенная
ниже в центре структура способна к изомеризации в двух направлениях:
одно из них связано с анионотропным превращением, другое — с прототроп-
ным. Для спирта этого ряда, его метилового эфира и ацетата (X = ОН, ОСН3,
ОСОСН3) осуществлены анионотропные превращения, катализируемые силь-
ными кислотами. В случае спирта (X = ОН) протекает также прототропное
* Для катализируемой кислотами изомеризации замещенных аллиловых спиртов был
получен иной порядок, но, конечно, разные спирты образуют различные сопряженные
кислоты в различных равновесных соотношениях. Поэтому в данном случае нельзя непо-
средственно сравнивать подвижности таких систем.
701
Глава XI
превращение, катализируемое сильными щелочами:
С6Н5
С1 СН2
I I
Z\Z\Z4
с6н5
I
С1 сн
! II
Z\Z\/\
\z\z\z
I I
х С1
н3о+
С6н5
I
С1 снх
I I
\Z\Z\Z
I I
Н С1
н ХС1
В-четвертых, Бартон показал, что изомеризация 1-фенилаллилового эфира
и-нитробензойной кислоты в ее циннамиловый эфир в таких растворителях,
как бензонитрил или уксусный ангидрид, при прибавлении тетраметилам-
монийацетата может быть в значительной степени сведена к замещению, про-
текающему с перегруппировкой. Предложенная схема реакции приводится
ниже:
С6Н5СН(Х)СН=СН2 -> [С6Н5СН—СН—СН2]+ + Г
С6Н5СН=СНСН2 X
Y- L С6Н5СН=СНСН2(¥)
X=OCOC,H<NOrn; Y = OCOCH3
Впоследствии Бартон [128], сравнивая скорости реакций, подтвердил, что
обратная реакция не является ступенчатой перегруппировкой и замещением
и должна протекать одновременно с прямой изомеризацией.
Увеличение электронной плотности по механизму сопряжения, при помощи
которого галогены в орто- или пара-положениях в арилкарбинильном соеди-
нении обусловливают подвижность некоторых приведенных выше систем,
свидетельствует о том, что псевдоосновность и анионотропия суть перекры-
вающиеся явления. Два анионотропных карбинола, активированных таким
образом галогеном, метоксильной или некоторыми другими группами (X),
обладающими требуемыми пеподеленными электронами, могут рассматривать-
ся как псевдооснования одного и того же ониевого иона
Другие доказательства в пользу SN1'-механизма были получены после 1928 г.
при изучении кинетики реакций в сочетании с установлением состава продук-
тов реакции, включая и изучение кинетики реакций при помощи меченых
атомов, а также при исследовании изменения оптической активности, сопро-
вождающем перегруппировку.
Ряд кинетических измерений непосредственно подтверждает механизм. Де
ла Мар и Вернон [129] показали, что данные Брауде, Джонса и Штерна [130]
по относительным скоростям перегруппировки
RC6H4CH(OH)CH = CHCH3 —> RC6H4CH = CHCH(CH3)OH
702
Перегруппировки ненасыщенных соединений
в подкисленном водном растворе этилового спирта, а именно
R = n-OCH3 п-СН3 .и-СП3 Н n-F n-Cl .w-Cl
105 9 1,2 1 0,8 0,22 0,17
очень хорошо коррелируются со скоростями нуклеофильного замещения
этих же соединений в боковую цепь, связанную с кольцом. Де ла Мар и Вер-
нон [129] отметили, что данные Вернона [131] по влиянию заместителей на
скорости сольволиза аллилхлоридов в водном этиловом спирте или в мура-
вьиной кислоте ясно указывают на необходимость смещения электронов
к а- или у-углеродному атому (табл. 170).
Таблица 170
Относительные скорости сольволиза аллилхлоридов
(по данным де ла Мара и Вернона)
Заместитель Положение заместителя
а 0 тран c-v
Н 1 1 1
С1 3,1
трет-С^Нд 2300
сн3 5900 0,5 3600
свн5 - 5-105
(СН3)2 -8-107 - 1,5-10’
В кинетическом исследовании другого рода было установлено, что катали-
зируемая кислотой перегруппировка 1-фенилаллилового спирта до циннами-
лового (3-фенилаллилового) спирта происходит по механизму SnI'. Эта пере-
группировка была проведена в воде, меченной кислородом-18, Бантоном,
Даном и Поккером [132], а также Герингом и Дилгреном [133]. Основной
результат состоял в том, что кислород в образовавшемся циннамиловом спир-
те полностью или почти полностью поступал из воды 18О, а неперегруппиро-
вавшийся фенил аллиловый спирт частично содержал 18О. Судя по относи-
тельным скоростям обмена кислорода и перегруппировки, 60% образовав-
шихся ионов карбония дают исходный спирт.
СН2 = СН-СН(С6Н5)-ОН+ [СН2-СН-СН(СЬН5)]+ -> Н2О-СН2-СН = СНС6На
Такой механизм был в общих чертах доказан Кеньоном и сотрудниками,
которые изучили изменение оптической активности при анионотропной пере-
группировке. Таким путем авторы доказали общность механизма SnI'-
В большом числе случаев наблюдалась рацемизация, которую и следовало
ожидать, учитывая, что в процессе реакции образуется трехуглеродная асим-
метрическая система. Данные по перегруппировкам без изомеризации и с изо-
меризацией удобно рассмотреть по очереди.
В случае перегруппировок без изомеризации основным фактом является то,
что оптическая активность, зависящая от асимметрии молекулы и исчезаю-
щая при ее ионизации, теряется в процессе перегруппировки. В случае
симметричного соединения, например при со льволитическом замещении
1,3-диметилаллилового моноэфира фталевой кислоты в муравьиной или уксус-
ной кислотах или в метиловом спирте, невозможно доказать химическими
703
Глава XI
методами, что произошла перегруппировка.
СН3СНСН = СНСН3 СН3СНСН = СНСН3 СН3СН = СНСНСН3
I I = I
ОСОС6Н4СООН OR OR
Однако Кеньон с сотр. [134] показал не только то, что продукт сольволиза
полностью или почти полностью рацемизирован, но также что исходный слож-
ный эфир, выделенный при не дошедшей до конца реакции, в значительной
степени рацемизирован, как если бы большая часть его была ионизована.
Важным фактом является то, что в случае таких наиболее ионизующих рас-
творителей, как муравьиная кислота, продукт реакции лишен оптической
активности, а в случае таких слабо ионизующих растворителей, как «-бути-
ловый спирт, рацемизация протекает в незначительной степени (наибольшая
наблюдаемая активность составляла всего 9%). Такая небольшая остаточная
активность, несомненно, объясняется тем, что главная реакция SN1' сопро-
вождается побочной реакцией SN2, т. е. бимолекулярным замещением без
перегруппировки. Знак остаточной активности всегда указывает на обраще-
ние конфигурации в исходном центре асимметрии. Кроме того, доказано [135],
что при сольволизе хлористого 1-п-пропил-З-метилаллила и соответствующе-
го моноэфира фталевой кислоты в одинаковых растворителях остаточная
активность в некоторых случаях (максимум 2%), несомненно, обусловлена
замещением без перегруппировки, поскольку оптическая активность исчеза-
ет при гидрировании, нарушающем асимметрию в перегруппировавшейся
молекуле (из-за образования второй пропильной группы), а не в перегруп-
пировавшейся молекуле.
В качестве примеров перегруппировок с изомеризацией [136] можно приве-
сти превращение четырех 1-фенил-З-метилаллиловых соединений, а именно
спирта в кислых растворах (в предположении, что активным началом являет-
ся сопряженная кислота), эфиров уксусной кислоты, эфира «-нитробензой-
ной кислоты и моноэфира фталевой кислоты в их 1-метил-З-фенил-изомеры.
При проведении этих реакций в разбавленных растворах (водная кислота
для спирта, уксусная кислота для эфира уксусной кислоты, хлороформ для
эфира «-нитробензойной кислоты и сероуглерод для эфира фталевой кисло-
ты; испытаны и другие растворители) было установлено, что продукты
реакции не обладают или почти не обладают оптической активностью, за
исключением моноэфира фталевой кислоты, который сохраняет активность
вплоть до 58%.
Последний результат может быть обусловлен проявлением другого механиз-
ма, который будет рассмотрен в разд. 2,г.
в. Бимолекулярный механизм анионотропии. Хьюз [138] впервые пред-
ложил для анионотропных превращений бимолекулярный механизм Sn2',
который, с одной стороны, аналогичен бимолекулярному механизму прото-
тропии B-Se2', а, с другой стороны, является формой бимолекулярного
нуклеофильного замещения SN2. Как и раньше, X" представляет анион
в общем виде, так что в данном уравнении оба X могут и не быть химически
идентичными.
сД^=С С=^-С (Sn2')
х? <х- X?
По поводу этого механизма можно высказать некоторые соображения на
основании изучения кинетики реакции и состава продуктов реакции. Ранние
попытки обнаружить таким путем бимолекулярный механизм перегруппиров-
704
Перегруппировки ненасыщенных соединений
ки привели к получению новых доказательств мономолекулярного механизма
и к выводу, что бимолекулярная перегруппировка не играет существенной
роли в изученных системах. Достаточно будет привести следующие примеры.
Одной из наиболее тщательно изученных групп реакций [138] является пре-
вращение без изомеризации хлористого 1-метилаллила и хлористого кротила
(3-метилаллила) при взаимодействии с этиловым спиртом, содержащим
этилат натрия, в 1-метилаллил- и кротилэтиловые эфиры; исходные вещества,
несмотря на способность к взаимопревращениям, достаточно устойчивы
и могут быть изучены раздельно. Этилат натрия вводился в реакцию либо
в малых концентрациях, что приводило к реакции первого порядка, либо
в больших концентрациях, что вело к реакции второго порядка. Установле-
но, что при замещении по реакции первого порядка оба хлорида дают такую
смесь эфиров, которую можно было ожидать, если бы в соответствии с моно-
молекулярными механизмами SnI и Sn1z образовался бы с перегруппиров-
кой или без нее общий промежуточный ион. При замещении по реакции вто-
рого порядка каждый хлорид давал только неперегруппировавшийся эфир.
Короче говоря, в этом случае наблюдались механизмы SN1, SN1' и S N2,
но не механизм SN2'.
Второго
порядка
-------> СНзСН(ОС2Н5)СН — СН2 (исключительно)
СН3СН(С1)СН = СН2
СН3СН = СНСН2(С1)
Первого
порядка
------> С СН3СН(ОС2Н5)СН = СН2 (-13%)
Первого г
порядка |
------> I СН3СН = СНСН2(ОС2Н5) (-87%)
Второго
порядка
-------> СН3СН = СНСН2(ОС2Н5) (исключительно)
На основании этого был сделан вывод, что замещение, при котором входящий
заместитель является производным псевдокислоты, происходит по механиз-
му Sn2'. При обработке ацетоуксусным эфиром и основными катализаторами
циннамиловый спирт (через сложные эфиры) превращается в 1-фенил-
аллилацетон, а 1-фенилаллиловый спирт аналогичным образом — в цин-
намилацетон [139]. При обработке малоновым эфиром и этилатом натрия
1-метилаллилхлорид превращается частично в продукт нормального замеще-
ния и частично в перегруппированный продукт — этиловый эфир кротил-
малоновой кислоты [140]. Показано, что 1-этилаллилхлорид претерпевает
такие же превращения. Однако, как указал Дьюар [141] (его аргументация
оспаривалась, но не была опровергнута), эти данные не убедительны, так
как псевдокислота имеет два реакционных центра и поэтому продукт, обра-
зовавшийся при О-аллилировании псевдокислоты по механизму S N2 и после-
дующей перегруппировке Кляйзена (разд. 3), будет идентичен продукту
С-аллидирования, происходящему по механизму SN2'. Делались также
и другие подобные нереальные предположения.
Причиной тех трудностей, с которыми пришлось столкнуться при установ-
лении механизма Sn2\ является не то, что данный механизм не существует,
а то, что при условиях, необходимых для бимолекулярного замещения, этот
механизм всегда оказывается замаскированным из-за более быстрой реак-
ции Sn2. Этого можно избежать при применении следующего метода [142].
Аллильное соединение, например бромистый 1-метилаллил, вводится в реак-
цию при условиях, приводящих только к бимолекулярному замещению.
45-077?
705
Глава XI
с анионом, например с ионом меченого брома, отличающимся от аниона,
образуемого аллильным соединением, только по своей радиоактивности.
При этом создаются условия, при которых в каждом элементарном молеку-
лярном акте замещения по механизму S N2 исходная молекула превращается
в идентичную молекулу; таким образом, исходное вещество не расходуется
по механизму SN2 и при требуемой температуре сохраняется возможность
неограниченное время следить за изомеризацией по механизму SN2'. Изучать
кинетику последней реакции и определять ее скорость можно обычными хими-
ческими методами. Скорость реакции SN2' можно сравнить со скоростью
реакции обмена SN2 при помощи меченых атомов. Ингленд и Хьюз исследо-
вали взаимодействие бромистого 1-метилаллила и бромистого кротила с бро-
мистым литием в ацетоне. Полученные ими данные о кинетике этих процессов
приведены в табл. 171.
Таблица 111
Константы скорости и аррениусовские параметры для бимолекулярной
реакции обмена брома (S^2) и для бимолекулярной изомеризации (Sk?')
бромистых аллилов при действии бромистого лития в ацетоне
[по данным Ингленда и Хьюза; fc2 и Л2, л-моль-1-с-1;
JEA, ккал/моль (4,187-103 Дж/моль)]
Бромид Реакция 106Й2 (25 °C) lg л2 ЕЛ
. . ( Sn2 879 9,06 16,5
1-Метилаллил Sn2' 14,9 9,40 19,4
Кротил { Sn2 Sn2' 141 000 5 9,93 ~ 9 14,7 ~ 19
Из данных табл. 171 очевидно, что оба изомерных галогенида претерпевают
обе бимолекулярные реакции. В случае бромистого 1-метилаллила SN2-
реакция протекает в 60 раз быстрее, чем процесс SN2', а для бромистого кро-
тила реакция обмена по механизму SN2 протекает в 28 000 раз быстрее, чем
изомеризация по механизму SN2'. Легко понять, почему так трудно обнару-
живается механизм SN2', если сопутствующее ему замещение SN2 может его
маскировать. Ясно также, что различие между двумя отношениями скоростей
Sn2/S n2' в основном объясняется разностью в скоростях реакций SN2.
Таким образом, тот факт, что в случае 1-метилаллила процесс SN2' наблю-
дать труднее, чем в случае бромистого кротила, объясняется главным обра-
зом локальным эффектом метильного заместителя в положении 1 на локаль-
ную реакцию SN2. Если в качестве заместителя в положение 1 вводить боль-
ший радикал, то, вероятно, этот эффект будет сильнее и при достаточных
размерах радикала S м2-реакция может вовсе не идти. Влияние мог бы оказы-
вать локальный полярный эффект, однако такие эффекты в S ^-реакциях
довольно слабы. Эта точка зрения была достаточно хорошо подтверждена
в работах де ла Мара, Вернона и их сотрудников. При обработке аллил-
иденхлорида (1-хлораллилхлорида) этилат-ионами в этиловом спирте про-
исходит реакция второго порядка, приводящая к образованию равных коли-
честв этоксильных соединений — продуктов SN2- и S ^'-реакций; первый
продукт, как и следовало ожидать, далее превращается в ацеталь [143]
С1 СН2 = СН-СН(ОС2Н5)С1 СН2 = СНСН(ОС2Н8)2 (Sn2)
СН2 = СН —СН-С1<ОС2Н8
*С2Н8ОСН2СН = СНС1
(Sn2')
706
Перегруппировки ненасыщенных соединений
В этих же условиях 1-тпреиг-бутилаллилхлорид дает только этокси-изомер,
образовавшийся по реакции SN2' [144]
С2Н5О-
тргтС4Н9 -------> C2H5OCH2CH = CHC4H9-mpem (SN2')
СН2 = СН-(^Н-С1
Де ла Мар и Вернон привели еще несколько примеров реакций, частично или
полностью протекающих по механизму SN2'; при этом соединения выбира-
лись так, чтобы уменьшить роль механизма SN2 [145].
г. Внутримолекулярный механизм анионотропии. Одно из ранних пред-
положений, касающихся такого механизма, основывалось на данных Кеньона
и сотр. [136] по изменению оптической активности, сопровождавшему анионо-
тропную перегруппировку. Как отмечалось в разд. 2, авторы указали на
одно исключение из правила общей рацемизации, которое наблюдалось
в случае перегруппировок 1-фенил-З-метилаллиловых эфиров. Аномально
вел себя моноэфир фталевой кислоты, и поэтому было высказано предположе-
ние *, что в процессе изомеризации участвует свободная карбоксильная
группа, которая действует как внутримолекулярный кислотный катализа-
тор, способствующий расщеплению старой СО-связи, а также как нуклео-
фильный реагент при образовании новой связи СО
R—СН-^СН^=СН—R' R—СН=СН—СН—R'
К Г ।
О ’ Н-0 о—н о
II II
СО—С6Щ—СО CO-CJK—со
Этот тип внутримолекулярного механизма обозначается символом SNi',
поскольку он родствен с механизмом замещения SNi, т. е. с внутримолеку-
лярной перегруппировкой, ведущей к замещению группы у единичного
атома углерода (гл. VII, разд. 7,з), точно так же как механизм SN2' родствен
с механизмом SN2 или механизм SN1' с SN1.
Другие данные, свидетельствующие в пользу механизма SNi', были получены
при изучении процессов превращения замещенного аллилового спирта при
действии галогеноводородов, галогенидов фосфора или тионилхлорида
в один или оба анионотропных галогенида. Такие реакции 1- и 3-этилаллило-
вого спирта изучались Мейзенгеймером и Линком [148], а реакции 1- или 3-
метилаллилового спирта — Янгом и Лейном [149]. Как установили Янг
и Лейн, реакции с галогеноводородами и с галогенидами фосфора отвечают
механизмам SN1 и SN1', которые протекают с образованием общего иона
карбония, и одновременно механизму SN2, включающему атаку ионом гало-
генида сложного эфира или комплекса, образованного с участием водородной
связи (гл. VII, разд. 7,к). Как и в случае насыщенных спиртов, для этих
аллиловых спиртов в реакции с галогенидами фосфора присутствие пиридина
способствует протеканию бимолекулярных процессов; таким образом, для
механизма SN2 создаются благоприятные условия и перегруппировки прояв-
ляются в меньшей степени, т. е. механизм SN2' играет лишь незначительную
роль. Мейзенгеймер и Линк, используя тионилхлорид, способствующий
осуществлению механизма SNi в реакциях насыщенных спиртов (гл. VII,
* См. работу [146]. Это предположение относится в основном к перегруппировкам в рас-
творах. Кеньон и сотрудники нашли несколько примеров сохранения оптической актив-
ности в случае верастворенных веществ в процессе анионотропных перегруппировок,
протекающих иногда при действии случайных катализаторов (см., например, работу
[147]). Однако механизм этого процесса в таких условиях совершенно неясен.
45* 707
Глава XI
разд. 7,з), получили, однако, необычный результат: первичный спирт давал
преимущественно вторичный хлорид, а вторичный спирт — главным образом
первичный хлорид. В эфирном растворе, согласно последующим данным Янга
и сотр. [150], перекрестные превращения происходят практически нацело.
Очевидно, перегруппировка является внутренним свойством этого механиз-
ма, независимо от относительной устойчивости продуктов реакции. Как
отмечают Робертс, Уинстейн и Янг [151], это ясно указывает на перегруп-
пировку образующегося вначале хлорсульфинового эфира в соответствии
с механизмом SNi'
R-CH^CH^=CH-R' R—СН=СН—СН—R' (SNi')
lx x*- —> SO, + I
0-*—SO—Cl Cl
Геринг и сотр. [152] установили, что при превращении цис- и щра«с-5-метил-
циклогексен-2-олов-1 в хлориды под действием хлористого тионила в эфире
сохраняется оптическая активность, а также геометрическая конфигурация
производных циклогексана
Этого и следует ожидать при механизме SNi', при котором конфигурация
исходных соединений сохраняется
Другой тип реакций с механизмом SNi' наблюдали Янг, Уэбб и Геринг [153].
При обработке 1-метилаллилхлорида диэтиламином в бензольном растворе
образуется кротилдиэтиламин. Первоначально эта реакция рассматривалась
как пример механизма SN2', однако оказалось, что в ней существенную роль
играет аминный атом водорода и поэтому лучше всего рассматривать эту
реакцию как протекающую через циклическое переходное состояние, т. е. по
механизму типа SNi':
сн,сн-^сн=сн2
X* -------------->
С1---Н—N(C2H5)2
СН3СН=СН—СНг
НС1 + I
N(C2H5)2
Сторк и Уайт [154], используя в качестве замещающего агента пиперидин,
а в качестве анионотропной системы сложный циклогексениловый эфир
708
Перегруппировки ненасыщенных соединений
аллильного типа, показали, что эта реакция происходит с сохранением кон-
фигурации. Чтобы замедлить реакцию S N2, в молекулу вводилась метильная
или высшая алкильная группа (R). Эти группы служили также стереохими-
ческой меткой, по отношению к которой можно было определить, находятся ли
отщепляемая (X = 2,6-дихлорбензоат) и входящая пиперидиновая группы
в zfizc-положении или они занимают транс-положение. Реакция проводи-
лась при 130 °C без растворителя или в ксилольном растворе; в ксилоле
реакция имела второй кинетический порядок. Хотя входящая и уходящая
группы имели разное пространственное расположение по отношению друг
к другу, они всегда располагались в одинаковом геометрическом положении
(транс) по отношению к группе, служившей стереохимической меткой. Сторк
и Уайт предположили, что осуществляется механизм SN2', однако, исходя
из того, что по данным Янга, Узбба и Геринга в таких реакциях важную
роль играет наличие в молекуле амина атома водорода [153] (см. выше),
можно сделать вывод, что механизм реакции Сторка и Уайта также относит-
ся к типу SNi'.
Принадлежность реакции к типу SN2' обычно устанавливается очень легко,
однако различить механизмы S Nl' и SNi' иногда очень трудно. Предельная
ситуация возникает, когда реакция, в полярных растворителях несомненно
имеющая механизм SN1', проводится в растворителях с такой низкой ди-
электрической проницаемостью, что ионы, если они образуются, в заметной
степени рекомбинируются или даже вообще не происходит диссоциации. В та-
ком случае эти ионы, связанные друг с другом флуктуирующими, не имею-
щими определенного направления, но достаточно сильными электростатиче-
скими силами, могут рекомбинироваться с перегруппировкой. Продукт пере-
группировки альтернативно может также образовываться по механизму SNi'
с циклическим упорядоченным переходным состоянием, в котором связи име-
ют в определенной степени ковалентный характер, и перемещающаяся группа
образует мостик между двумя положениями, в конце концов заменяющими
друг друга. Чтобы различить эти два случая, применяется тест на потерю
оптической активности (который часто трудно применить); большая степень
потери оптической активности, а также положительный результат теста на
полярность переходного состояния, которая определяется по кинетическому
солевому эффекту и эффекту растворителя, указывают, что более вероятным
будет механизм SNl', а не SNi'.
Случай такого неясного механизма был описан Янгом, Уинстейном и Герин-
гом [155] в 1951 г. Сольволиз 1,1-диметилаллилхлорида в уксусной кислоте
(диэлектрическая проницаемость около 6) в значительной степени сопровож-
дается изомеризацией с образованием 3,3-диметилаллилхлорида; присут-
ствующие в растворе галогенид-ионы не участвуют в этой изомеризации.
Показано, что это явление связано с применением в качестве растворителя
уксусной кислоты и при переходе к этиловому спирту уже не наблюдается.
Геринг и сотр. [156] исследовали это явление на примере сольволиза цис-
и шронс-5-метилциклогексен-2-ил-1-хлоридов (формулы соответствующих
спиртов приведены на стр. 708) в уксусной кислоте и этиловом спирте.
709
Глава XI
В результате реакции образуются смеси цис- и транс-продуктов одинакового
состава, однако потеря оптической активности происходит быстрее, чем поте-
ря геометрической чистоты. Изучение изомеризации цис- и транс-5-метил-
циклогексен-2-иловых-1 моноэфиров фталевой кислоты в ацетонитриле [157]
также показывает, что оптическая активность исчезает быстрее (в этом случае
гораздо быстрее), чем цис-транс-специфичность молекул. Добавление 3-
нитрофталатных ионов приводит к нитрофталатным эфирам, т. е. эти ионы
участвуют в реакции. Все это означает, что реакция происходит по механиз-
му SN1' и что в рамках этого механизма для того, чтобы циклогексеновое
кольцо было способно повернуться на 180° (3,14 рад) относительно аниона,
необходима большая степень разделения противоионов, чем для того, чтобы
анион смог достигнуть другого конца трехуглеродной системы с той же сторо-
ны циклогексенового кольца (что ведет к рацемизации). Но, конечно, оста-
ется открытым вопрос о том, образуются ли ионы в действительности или
перед их образованием возникает мостиковая ковалентная трехуглеродная
система, обусловливающая наблюдаемые результаты. Может даже быть, что
ответы на этот вопрос будут различными для хлорида и монофталатов, по-
скольку в последнем случае подвижная группа более приспособлена к обра-
зованию мостика типа предложенного Хьюзом (стр. 707).
д. Анионотропные равновесия. Такие равновесия могут, конечно, рас-
сматриваться совершенно независимо от механизма перегруппировки. В рав-
новесии между изомерными коричными и 1-фенилаллиловыми соединениями
последние находятся всегда в очень малых количествах:
(100%) CeH5CH = CHCH2X С8Н5СНХСН = СН2 (0%)
Этого можно ожидать, если учесть дополнительную стабильность производ-
ных коричной кислоты благодаря наличию сопряжения бензольного ядра
с винильной группой. [Приняв разность свободных энергий равной
6 ккал/моль (25,12-Ю3 Дж/моль), получим для отношения 1-фенилаллил/цин-
намил при обычной температуре величину е-вооо/воо _. e-io = Ю-4’3 =
= 1/20 000.] Равновесие между хлористым кротилом и хлористым 1-метил-
аллилом, а также между аналогичными бромидами сдвинуто в том же напра-
влении, но не столь резко, и поэтому соотношение изомеров можно легко
определить. Точное соотношение зависит от природы галогенида, растворите-
ля и температуры, но лежит в указанных ниже пределах:
(75-85%) СН3СН = СНСН2Х СН3СНХСН = СН2 (15-25%)
В этом случае главное энергетическое различие между рассматриваемыми
изомерами обусловлено гиперконъюгацией с метильной группой; во всяком
случае, можно сказать, что имеющиеся скудные сведения о таких энергети-
ческих факторах не противоречат данным о равновесии [если допустить для
разности свободных энергий значение 1 ккал/моль (4,187-103 Дж/моль), то
равновесное отношение окажется равным 1:5].
3. Валентная таутомерия
а. Перегруппировка Кляйзена. Эта реакция состоит в перегруппировке
О-аллиловых эфиров в opmo-аллильные производные фенолов и О-аллиловых
эфиров в С-аллильные производные кето-енолов [158], включающие пере-
710
Перегруппировки ненасыщенных соединений
группировки винилаллиловых эфиров в аллилацетальдегиды [159]:
СН2
\н
I II
С сн2
/ ч
СН3 СН
С2Н5ООС
сн2
ч
сн
сн2
но \н
I I
с сн2
/ ч /
СНз с
I
СгЩООС
сн2 сн2 сн2
/ ч ч ч
о сн о сн но сн2
I II II I I I
нс сн2 НС сн2 НС сн2
^СНа \н2 ^СН
Приведенные выше перегруппировки происходят без катализатора при на-
гревании О-аллильных соединений до температуры около 200 °C либо в виде
чистых жидкостей, либо в таких инертных растворителях, как дифениловый
эфир или диэтиланилин. В процессе перегруппировки эфиров фенолов аллиль-
ная группа направляется в свободное орто-положение; ни в одном из изу-
ченных простых примеров не было обнаружено даже частичного перемещения
аллильной группы в пара-положение [160].
Исходя из соединений с замещенной аллильной группой, было показано, что
миграция аллильных групп от атома кислорода к орто-атому углерода в фе-
ноле и от кислорода к углероду в кето-еноле всегда сопровождается переме-
щением двойной связи в аллильной группе из положения 1 в положение 3.
Так, О-коричный и О-кротиловый эфиры фенола дают о-1-фенилаллилфенол
и о-1-метилаллилфенол [161]. О-Циннамильное производное ацетоуксусного
эфира при перегруппировке превращается в G-1-фенилаллильное производ-
ное этого эфира [162].
В отношении этих перегруппировок было установлено следующее. Во-пер-
вых, превращение аллил-п-крезилового эфира в о-аллил-п-метилфенол
в дифениловом эфире [163] является реакцией первого порядка. Кроме того,
превращение аллилвинилового эфира в газовой фазе в аллилацетальдегид
представляет собой гомогенную газовую реакцию первого порядка [164].
Это указывает на то, что в каждом индивидуальном молекулярном акте
перегруппировки участвует только одна молекула. Во-вторых, если проис-
ходят совместно две перегруппировки, они протекают независимо друг от
друга и при этом не получается перекрестных продуктов. Так, из смеси
2-нафтилаллилового и фенилкоричного эфиров при нагревании в отсутствие
растворителя образуется только 1-аллилнафтол-2 и о-1-фенилаллилфенол
[165]. Поэтому ясно, что исходные эфиры не расщепляются на две части.
В-третьих, перемещение двойной связи в аллильной группе из положения 1
в положение 3 может происходить не только в направлении образования
более устойчивого продукта, но и другим путем; так, из фенил-3-этилаллило-
711
Глава XI
вого эфира образуется о-1-этилаллилфенол [166], фенил-1-этилаллиловый
эфир превращается в о-3-этилаллилфенол [167]. Это указывает на то, что
в механизм перегруппировки обязательно включается перемещение двойной
связи в аллильной группе. В-четвертых, оптически активный 1-3-диметил-
аллилфениловый эфир превращается в активный о-1,3-диметилаллилфенол
[168]. Этот факт свидетельствует о том, что образование связи в орто-поло-
жении начинается до того, как происходит полный разрыв эфирной связи.
Из этих данных ясно, что скелетная перегруппировка, за которой иногда
может следовать прототропное превращение, представляет собой одностадий-
ную мономолекулярную реакцию, протекающую через циклическое шести-
атомное переходное состояние, соответствующее согласованной миграции трех
(расположенных через одну связь) связей в оставшиеся три положения
(формулы на стр. 711).
Далее можно задать вопрос: насколько полярно это переходное состояние?
Вероятно, оно не очень полярно. Об этом свидетельствует множество данных,
показывающих, что введение сильно полярных заместителей в ароматическое
кольцо мало влияет на скорость перегруппировки Кляйзена. Так, согласно
Герингу и Якобсону [169], скорости перегруппировки в дифениловом эфире
при 185 °C в большом ряду монозамещенных фенилаллиловых эфиров, содер-
жащих ускоряющие реакцию n-диметиламино-, п-амино- и и-метоксигруппы
и замедляющие реакцию n-циан- и .и-нитрогруппы, изменяются лишь в
16 раз.
Были сделаны попытки выяснить стереохимию перегруппировки Кляйзена
и, в частности, ответить на вопрос, какая из конформаций шестичленного
кольца (при отсутствии специфических структурных особенностей) в пере-
ходном состоянии более стабильна — кресло или ванна.
Схематическое изображение кресловидной (слева) и подобной ванне (справа) конформаций,
которые могут возникать в процессе перегруппировки Кляйзена.
Существующие недостаточно полные доказательства свидетельствуют в поль-
зу кресловидной формы [170]. Этот вывод косвенно подтверждается аналогией
с перегруппировкой Коупа (разд. 3,6), стереохимию которой определить
проще.
Кляйзен и Эйслеб [158] показали, что, если аллилариловый эфир, имеющий
заместители в обоих орто-положениях, нагревать как таковой или в раство-
рителях при температуре порядка 200 °C, то происходит миграция аллиль-
ной группы в пара-положение. При использовании замещенных аллильных
групп было показано, что в противоположность орто-миграции при пара-
миграции 1,3-смещения связей не происходит [171]. пара-Миграция пред-
ставляет собой реакцию первого порядка [172]. Оптически активный 1,3-
диметилаллил-2,6-диметилфениловый эфир дает оптически активный про-
дукт пара-перегруппировки [168].
Хард и Поллак [166] первыми высказали правильную мысль о механизме
таких пара-миграций. По их мнению, первая стадия заключается в орто-
миграции с образованием циклогексадиена, в котором вследствие того, что
прототропное превращение в фенол исключено из-за отсутствия орто-водо-
рода, происходит миграция аллильной группы через новое циклическое пере-
ходное состояние в пара-положение. При этом вначале также образуется
712
Перегруппировки, ненасыщенных соединений
дйеной, который в конце концов путем прототропного превращения перехо-
дит в фенол.
О ОН
Таким образом, наблюдаемое отсутствие 1,3-изомеризации аллильной груп-
пы в пара-положении обусловлено такой изомеризацией на каждой из двух
стадий процесса.
Этот механизм был основательно изучен несколькими способами. Шмид,
Хегель и Шмид [173] показали, что в продукте перегруппировки аллил-
2,6-диаллилфенилового эфира — 2,4,6-триаллилфеноле — метка 14С, перво-
начально содержавшаяся в a-положении только одной орто-группы, распре-
деляется между ориго-а- и пара-ю-положениями. Это доказывает, что реакция
идет через триаллилциклогексадиенон, указанный в скобках:
ОСН2СН = СН2
1
СН2= СНСН2 —СН2СН = сн2
\/
сн2=снсн
II *
/\^СН2СН — СН2
II рсн2сн = сн2
\/
он
СН2 = СНСН2 ~|'|/''гр СН2СН = сн2
\z
I *
СН2СН = СН2
Аналогичные данные были получены при применении в качестве метки
в аллильных группах обычных заместителей [174]. При пара-перегруппиров-
ке аллил-2,6-диметилфенилового эфира Конрой и Файрстоун [175] смогли
обнаружить промежуточный циклогексадиенон в виде аддукта Дильса —
Альдера с малеиновым ангидридом; строение выделенного аддукта было
доказано синтезом. Вряд ли существуют более веские доказательства.
Общая обратимость этих циклических реакций, проявляющаяся по крайней
мере на одной или на обеих стадиях перегруппировок Кляйзена, если она не
нарушается прототропными превращениями, была изящно доказана Кал-
берером, Шмидом и Шмидом [176]. Они показали, что аллил-2,6-диметил-4-
аллилфениловый эфир, содержащий метку 14С в положении 4-аллильной
группы, при нагревании до 168 °C дает химически тот же самый эфир, в кото-
ром, однако, метка 14С распределена между исходным положением и со-
713
Глава XI
положением О-аллильной группы
ОСН2СН = СН2 ОСН2СН = СН2
I I
СНз -|А- СНз ? СН3-|Р|-СН3
I * I
СН2СН = СН2 СН2СН = СН2
Можно считать, что существенной и определяющей характеристикой пере-
группировки Кляйзена является протекание процесса через циклическое
переходное состояние, содержащее атом кислорода и пять атомов углерода.
Очевидно, что примеры таких реакций должны быть как в алифатическом,
так и в ароматическом ряду. Однако при пара-перегруппировке Кляйзена,
которая наблюдается только в ряду ароматических соединений, за собственно
перегруппировкой Кляйзена следует подобная же, но уже другая перегруп-
пировка, протекающая через циклическое переходное состояние с шестью
атомами углерода. Эта перегруппировка имеет самостоятельное значение
и ее не надо рассматривать как связанную с собственно перегруппировкой
Кляйзена и только в ароматических системах. Такая перегруппировка будет
рассмотрена в следующем разделе.
б. Перегруппировка Коупа. Перегруппировка, протекающая через цик-
лическое переходное состояние с шестью атомами углерода, была открыта
Коупом и Харди в 1940 г. Основные закономерности этой перегруппи-
ровки были установлены Коупом и сотр. [177] в течение нескольких следую-
щих лет.
Вначале были изучены перегруппировки винилаллилзамещенных малоновых
эфиров, циануксусных эфиров и малононитрилов. Винильные и аллильные
заместители часто содержали алкильные группы. При нагревании этих
эфиров и нитрилов без растворителя или с растворителем при температурах
между 135 и 200 °C происходит перегруппировка с переходом аллильной
группы от малонового атома углерода к ^-углероду винильного заместителя
и образуется алкенилиденмалоновый эфир или нитрил. Если, как часто было,
аллильный заместитель содержал алкильные группы, то можно было уста-
новить, что этот заместитель в процессе перегруппировки претерпевает
1,3-изомеризацию
СН2 = С(СН3) — С(СО2С2Н5)2 СН2 — С(СН3) = С(СО2С2Н5)2
СН3СН = СН-(^Н2 сн3^н-сн = сн2
Кинетически перегруппировки имели первый порядок. Если в одном и том
же растворе проводить перегруппировку двух соединений, которые перегруп-
пировываются со сравнимыми скоростями, то можно показать, что обе пере-
группировки протекают независимо и каждая приводит к собственному про-
дукту реакции без каких-либо признаков образования перекрестных про-
дуктов.
Вначале сложилось впечатление, что перегруппировка облегчается электро-
отрицательными («активирующими») группами GN и СО2С2Н5, но затем
выяснилось, что активирующее действие этих групп обусловлено не столько
их электроотрицательностью, сколько их ненасыщенностью и способностью
сопрягаться с этиленовыми двойными связями в конечном продукте. Если
эти сильно полярные группы заменить на фенильную группу, то перегруп-
пировка проходит почти так же легко при температурах ниже 200 °C.
714
Перегруппировки ненасыщенных соединений
сн2 = СН — снсбн5 СН2—сн = снсбн5
I I
СН2 = СН —СН2 сн2—сн = сн2
СН2 = СН —СНС6Н5 сн2—СН=СНС6Н5
СН3СН = СН — сн2 СН3СН — сн = сн2
Таким образом, ясно, что переходные состояния при перегруппировках
Коупа и Кляйзена сходны между собой и не очень полярны. Нет сомнения,
что способная к сопряжению фенильная группа в этих и других подобных
случаях уменьшает энергию продукта относительно энергии исходных соеди-
нений, тем самым создавая теормодинамические предпосылки для протека-
ния реакции слева направо. При качественном сравнении скоростей стано-
вится очевидным также, что фенильная группа уменьшает энергию пере-
ходного состояния, отнесенную к исходному состоянию, таким образом уско-
ряя реакцию, идущую слева направо.
При замене фенильной группы на метильную происходят ожидаемые измене-
ния. Реакция еще идет, но медленнее, и уже требуется температура 300 °C.
В этом случае реакция до конца не доходит, и, вероятно, можно показать,
что в системе имеет место равновесие
СН2 = СН-СНСН3 СН2-СН = СНСН3
СН2=СН-СН2 СН2-СН = СН2
Если исключить из этого примера метильный заместитель, то получится
«чистая» перегруппировка Коупа, которая является симметричной и, веро-
ятно, должна идти еще медленнее, давая равновесную смесь состава 1 : 1
химически идентичных соединений
СН2 = СН-СН2 ? СН2-СН = СН2
СН2 = СН—СН2 СН2-СН = СН2
Равновесие могло бы быть показано с помощью изотопных меток, а возможно
и по спектрам ядерного магнитного резонанса, снятым при достаточно высо-
кой температуре (принцип описан в разд. 3,г). Насколько известно автору,
такие исследования в настоящее время не проведены.
Стереохимия перегруппировки Коупа и, в частности, вопрос о конформации
циклического переходного состояния, были изучены Дерингом и Ротом [178].
Был исследован 2,3-диметилгексадиен-1,5, который, обладая двумя экви-
валентными асимметрическими атомами углерода, существует в рацемиче-
ской и мезо-формах. Эти формы претерпевают перегруппировку Коупа при
180 и 225 °C соответственно, давая равновесную смесь, в которой преобладают
продукты перегруппировки, т. е. изомерные октадиены-2,6
СН2 = СН —СНСНз ? сн2 — сн = снсн3
СН2 = СН-^НСН3 *" сн2-сн = снсн3
Рацемическая и мезо цис-цис, транс-транс
и цис-транс
Тест на конформацию переходного состояния заключается в следующем:
если это кресловидная конформация, то рацемический реагент должен да-
вать цис-цис и трпис-шраис-продукты, а мезо-реагент — только цис-транс-
продукт (см. диаграммы). Если же конформация переходного состояния
подобна ванне, то рацемический реагент будет давать только цис-транс-
продукт, а мезо-реагент — цис-цис- и шраис-шрянс-продукты. В действитель-
на
Глава XI
ности рацемический реагент давал 10% цис-цис-продукта и 90% транс-
mpuuc-продукта, а мезо-реагент только цис-пгранс-продукт. Это четко сви-
детельствует в пользу кресловидного переходного состояния *.
Дей конформера одного энан- цис-цис- транс-транс-
тиомера рацемического ре- Продукт Продукт
агента (другой энантиомер дает
такие же неактивные продукты)
Два конформера мезореагента Два конформера цис транс-продукта
в. Кольчато-цепная валентная таутомерия. В двух предыдущих разде-
лах рассматривалась валентная таутомерия в системах с открытой цепью,
которая оставалась открытой после валентных изменений. В этом разделе
будет рассмотрена валентная изомеризация, в результате которой происходит
раскрытие цикла или его образование. Для начала рассмотрим два случая
настолько экстремальных, что их можно было бы обсуждать как в предыду-
щем, так и в данном разделах. Оба примера найдены Фогелем и относятся
к случаю перегруппировки Коупа, в которой боковые цепи небольших али-
фатических циклов (последние немного схожи с двойными связями) включа-
ются в образующиеся циклы большего размера. цис-Форма 1,2-дивинил-
циклобутана при 100 °C быстро превращается в цис-циклооктадиен-1,5.
В этих условиях mpuuc-дивинильный изомер в реакцию не вступает, а при
240 °C расщепляется на две молекулы бутадиена-1,3 [180]. Еще более
поразительно, что цис-форма 1,2-дивинилциклопропана настолько легко
переходит в цис-циклогептадиен-1,4, что первое соединение невозможно
выделить. транс-Изомер этого дивинильного соединения можно выделить,
он переходит в тот же самый циклогептадиен только при нагревании
до 200 °C [181].
* Теоретическое рассмотрение причин большей стабильности именно этой конформации
переходного состояния приведено в работе [179].
716
Перегруппировки, ненасыщенных соединений
В этих реакциях циклизации боковых цепей в ^wc-положении обычно более
стабильная кресловидная форма переходного состояния реализовываться не
может. Реакции должны происходить через переходное состояние, имеющее
конформацию типа ванны; однако они идут довольно легко, и их скорость
возрастает при переходе от циклобутанового к циклопропановому исходному
соединению.
Эти наблюдения можно объяснить тем, что кольцевые связи циклобутанового
кольца изогнуты и поэтому в некоторой степени имеют л-характер. Кольцевые
связи циклопропанового кольца изогнуты гораздо сильнее и имеют хорошо
выраженный л-характер. Известно, что циклопропановые связи могут сопря-
гаться аналогично двойным связям с другими двойными связями, если по-
следние расположены относительно циклопропанового кольца надлежащим
образом. Поэтому лишь чисто формальной экстраполяцией является переход
к системе, в которой вместо циклобутанового и циклопропанового колец,
как в предыдущих примерах, содержатся двойные связи, или даже переход
к системе, в которой вовсе нет колец, а винильные двойные связи сопрягают-
ся непосредственно друг с другом. Таким образом, переходим к процессам
циклизации, названным Вудвардом и Гофманом электроциклическими реак-
циями [182], которые включают реакции образования простой связи между
концевыми атомами линейной сопряженной системы и обратные реакции рас-
щепления циклов. Эти реакции рассматриваются здесь как кольчато-цепные
процессы вследствие того, что они в принципе всегда обратимы, поэтому вопрос
о том, как удобнее их рассматривать — как образование или как расщепление
цикла, связан с термодинамическими факторами, не зависящими ни от раз-
личий в механизме, ни от любых факторов, связанных с механизмом, например
от стереохимических характеристик процесса.
Вудвард и Гофман [182] объяснили стереохимию этих реакций, предположив,
что для их осуществления необходимо, чтобы участвующие молекулярные
орбитали подходили друг другу с точки зрения симметрии и, что наиболее
важно, чтобы новая о-связь, возникающая при циклизации, образовывалась
из высшей занятой сопряженной л-орбитали *. Объясняя эту теорию, рас-
смотрим вначале только термические изомеризации, и поэтому, согласно
принципу Лондона, адиабатические взаимопревращения основных электрон-
ных состояний.
Рассмотрим превращение бутадиена-1,3 в циклобутен (на самом деле эта
реакция практически нацело идет в противоположном направлении). Четыре
л-электрона бутадиена занимают попарно две наиболее низкие молекуляр-
ные л-орбитали. Наиболее низко лежащая из всех орбиталей имеет един-
ственную узловую плоскость, совпадающую с плоскостью сопряжения. Ее
симметрия совпадает с симметрией л-орбитали двойной связи в начальный
момент образования циклобутена, и эта орбиталь должна рассматриваться
как прародительница орбитали двойной сязи в продукте циклизации. Выс-
шая из двух занятых л-орбиталей бутадиена имеет такую же узловую
плоскость, а также вторую узловую плоскость, которая лежит в плоскости
* Авторы указали, что, согласно Хавинге и Шлатману [183], предположение о том, что
па стереохимию кольчато-цепной валентной таутомерии может оказывать решающее
^влияние симметрия орбиталей, было высказано Остергоффом.
717
Глава XI
симметрии молекулы под прямым углом к плоскости сопряжения. Положе-
ние обеих узловых плоскостей, обусловливающих существование двух осей
симметрии второго порядка, а также соответствующие знаки волновой функ-
ции у атомов углерода приведены на диаграмме, изображенной ниже. Эта
орбиталь должна быть прародительницей п-связи циклобутена. Очевидно,
что эта орбиталь должна концентрироваться по концам системы и менять фазо-
вый знак путем скручивания таким образом, чтобы увеличить степень пере-
крывания между концевыми атомами бутадиена. Необходимое для изменения
фазового знака вращение орбиталей (в данном случае в одну и ту же сторону
у каждого концевого атома) описывается Вудвардом и Гофманом как «кон-
ротация»
Стереохимическое следствие конротации состоит в том, что две пары групп
А,В и C,D, связанные с первым и четвертым атомами углерода бутадиена,
в циклобутене будут иметь такое расположение, которое обусловлено
конротацией при замыкании цикла. Если происходит раскрытие циклобуте-
нового цикла, то вследствие конротации естественно должен образовываться
бутадиен с соответствующим расположением этих групп.
Известно, что такого рода превращения имеют высокую стереоспецифичность.
Во всех примерах, изученных до настоящего времени, стереохимия соответ-
ствует описанной теории. Так, при нагревании до 120 °C метиловый эфир
?/ис-циклобутен-3,4-дикарбоновой кислоты (А == С = СО2СН3; В = D = Н)
из трех хорошо известных возможных метилмуконатов (цис-цис, транс-транс
и цис-транс} дает только г/ыс-транс-муконат [184]. В соответствии с этим
цис- и шранс-1,2,3,4-тетраметилциклобутены при нагревании до 200 °C
дают соответственно цис-транс- и г/ыс-г/ыс-3,4-диметилгексадиены-2,4 [185].
Все эти процессы являются конротационными.
Теперь рассмотрим превращение г/пс-гексатриена-1,3,5 в циклогексадиен-1,3
(обычно это направление реакции термодинамически более выгодно). Заме-
тим, что шесть л-электронов гексатриена будут занимать три низшие моле-
кулярные л-орбитали. Самая низшая из них имеет единственную узловую
плоскость, совпадающую с плоскостью сопряжения, вторая из низших
орбиталей имеет такую же узловую плоскость, а также другую узловую плос-
кость, расположенную в плоскости симметрии молекулы, перпендикулярной
к плоскости сопряжения. Эти две орбитали по всем элементам симметрии
соответствуют двум орбиталям, составляющим л-электронную систему бута-
диенового фрагмента, содержащегося в молекуле образующегося циклогек-
садиена (выше это пояснялось на примере самого бутадиена), и поэтому эти
две орбитали гексатриена могут рассматриваться как прародительницы
л-системы бутадиенового фрагмента. Таким образом, высшая занятая орби-
таль гексатриена, третья по энергии из трех л-орбиталей, должна рассмат-
118
Перегруппировки ненасыщенных соединений
риваться как прародительница о-связи, замыкающей циклогексадиеновое
кольцо. Эта орбиталь гексатриена имеет одну узловую плоскость, совпадаю-
щую с плоскостью сопряжения, и две другие узловые поверхности, которые
перпендикулярны плоскости сопряжения и проходят через линии связей,
как показано на схеме
Таким образом, орбиталь, замыкающая цикл, имеет плоскость симметрии,
а не ось симметрии второго порядка, как соответствующая орбиталь бутади-
ена, и поэтому фазовые отношения между разными частями орбитали будут
иметь чередующиеся знаки.
Чтобы образовалась замыкающая цикл о-связь, эта л-орбиталь гексатриена
должна концентрироваться по концам системы и вращаться таким образом,
чтобы фазовое перекрывание между концевыми атомами было наилучшим.
Вудвард и Гофман назвали необходимый для этого способ вращения «дисро-
таторным» (в противоположных направлениях у каждого концевого атома).
Отсюда следует, что две пары групп А,В и G,D, связанные с первым и шестым
атомами углерода гексатриена, при замыкании соответствующего цикло-
гексадиенового цикла будут двигаться дисротационно; очевидно, что обрат-
ная реакция также будет дисротаторной.
Циклизации такого рода высокостереоспецифичны; стереоспецифичность
в известных простых случаях соответствует предсказанной. Так, транс-цис~
пгр«нс-1,6-диметилгексатриен-1,3,5 (А = D = СН3, В = С = Н) дает цис-
1,2-диметилциклогексадиен-3,5, тогда как из цпс-цпс-пгранс-Цб-диметил-
гексатриена-1,3,5 (В = D = СН3, А = С = Н) образуется пгранс-диметил-
циклогексадиен [186]. Очевидно, эти реакции происходят дисротационно.
Другие примеры относятся к химии витамина D. При нагревании до 150—
200 °C прекальциферол (получаемый фотохимически из эргостерина) пре-
вращается в смесь двух изомеров эргостерина, в одном из которых — пиро-
кальцифероле — группы 10-СН3 и 9-Н находятся в цпс-а-положении («за»
плоскостью бумаги на формуле, приведенной ниже), а в другом — изопиро-
кальцифероле — те же группы находятся в цис-р-положении («перед» пло-
скостью листа бумаги) [187]. Вначале эти группы находились в плос-
кости сопряжения прекальциферола, и поэтому должна происходить
дисротация. у——
К 3 \Ъ I_____I
j
Прекальциферол
719
Глава XI
Прекальциферол
Еще один аргумент в пользу рассматриваемой теории, приведенный в пер-
вой статье Вудварда и Гофмана, состоит в том, что стереохимические правила
для кольчато-цепной валентной таутомерии основных состояний л-электрон-
ных систем будут обращаться при соответствующих фотохимических пре-
вращениях. Первая сильная (т. е. разрешенная различием в симметрии заря-
дов) полоса поглощения в ультрафиолетовом спектре сопряженной л-элек-
тронной системы соответствует переходу одного электрона с высшей занятой
на низшую незанятую л-орбиталь. Эта новая орбиталь этого электрона будет
иметь на одну нодальную поверхность, пересекающую линии связей, боль-
ше, чем старая орбиталь. В таком возбужденном электронном состоянии
соотношение симметрии по концам л-системы для высшей занятой орбитали
будет противоположным симметрии высшей занятой орбитали основного
электронного состояния; и поэтому в том случае, если стереохимия неадиаба-
тической обратимой циклизации, проходящей через возбужденное состояние,
контролируется орбитальной симметрией (заранее это предвидеть нельзя),
стереохимические правила должны быть противоположны правилам, при-
менимым к соответствующим реакциям, идущим через основное состояние.
Другими словами, при фотохимической реакции бутадиен-1,3 должен пере-
ходить в циклобутен дисротаторно, а гексатриен-1,3,5 должен переходить
в циклогексадиен-1,3 конротаторно.
Некоторые примеры для первого типа соединений будут приведены в следу-
ющем разделе. Вудвард и Гофман [182] приводят ряд примеров реакций вто-
рого типа. Например, транс-^ис-пгранс-1,6-диметилгексатриен-1,3,5 при
облучении дает транс-1,2-диметилциклогексадиен-3,5 [183, 188]. Известен
ряд примеров из области полиненасыщенных стероидов; например, как эрго-
стерин, в котором группы 10-СН3 и 9-Н находятся в транс-положении друг
к другу (Р-СН3 и а-Н; СН3 «перед» и Н «за» плоскостью бумаги), так и его
стереоизомер люмистерин, в котором эти группы также находятся в транс-
положении, но конфигурация соответствует а-СН3 и |3-Н («за и «перед»
плоскостью бумаги соответственно), при облучении [например, ртутной лам-
пой с длиной волны 2537 А (253,7 нм)] превращаются в прекальциферол,
720
Перегруппировки ненасыщенных соединений
причем в случае эргостерина реакция обратима [189]. Очевидно, все эти сте-
реохимические процессы происходят коиротаторно.
Вудвард и Гофман отметили, что описываемый контроль орбитальной сим-
метрии над стереохимией может не осуществляться при наличии других
факторов. Так, хотя, как было показано, цикл г^ис-1,2,3,4-тетраметилцикло-
бутена при 200 °C гладко раскрывается путем конротации, родственное соеди-
нение диметилбициклогептен, формула которого приведена ниже, не способ-
ное к такой реакции по стереохимическим причинам, претерпевает дисрота-
торное раскрытие цикла, однако только при 400 °C и довольно медлен-
но [190].
Конротаторно при 200°С
до цис-транс-3,4-диметил-
гексадиена-24
Дисротаторно при 400°С
до 1,2-диметилциклогепта-
диена-2,7
Можно предвидеть, что в специфических л-электронных системах эти про-
стые стереохимические правила могут не соблюдаться, например в некото-
рых системах с перекрестным сопряжением, в которых внеплоскостные узлы
пересекаются внутри молекулы, и таким образом может получиться «непра-
вильное» число узлов между концами бутадиеновой системы. Всегда может
существовать одна из общих причин несоблюдения этих правил, состоящая
во влиянии колебаний ядер на характеристики различных электронных сос-
тояний, поэтому, чем выше температура, тем менее строгими становятся сте-
реохимические правила.
г. Интрааннулярная валентная таутомерия. Эта проблема имеет более
чем столетнюю историю и восходит еще к Кекуле, охватывая целый период
предквантовых попыток понять структуру бензола (гл. IV, разд. 1). В’связи
с этими исследованиями возник глубокий интерес к проблеме взаимопревра-
щений кекулевских структур бензола, и вследствие этого интерес, с одной
стороны, к циклобутадиену, циклооктатетраену и высшим аннуленам,
а, с другой стороны, к гипотетической бициклогексадиеновой структуре
бензола (Дьюар) и ее низшему мостиковому ненасыщенному аналогу бици-
клопентену. Исследования в этой области велись еще до того, как было
выявлено различие между мезомерией, т. е. валентными изменениями, про-
исходящими со скоростью перемещения электронов и поэтому по существу
отражающими движение электронов, и валентной таутомерией, т. е. пере-
распределением валентности, происходящим с гораздо более низкими скоро-
стями, сравнимыми со скоростями движения ядер (что приводит к сущест-
вованию изомеров). Это одна из причин (другая, конечно, состоит в низком
уровне экспериментальной техники, существовавшей в то время), объясняю-
щая, почему ранние исследования, несмотря на их многочисленность, яви-
лись лишь этапом на пути более позднего развития и довольно в малой сте-
пени использовались позднее.
46-0772
721
Глава XI
Одно из ранних представлений, получивших дальнейшее развитие, состояло,
в следующем. Фармер и автор настоящей книги [191] в 1920 г. установили,
что кето-енольная дикарбоновая кислота, которая была синтезирована
Перкином и Торпом [192] при одновременном замыкании трехчленного,
и четырехчленного колец с целью получения производных бициклопентена
и которая при декарбоксилировании давала кетомонокарбоновую кислоту,
впоследствии синтезированную Тойвоненом [193] замыканием пятичленного,
цикла, при окислении в зависимости от окисляющего агента превращалась,
или в транс-кароновую кислоту, или в сх,сх-диметилаконитовую кислоту
СО2С2Н5
•С-СН(СО2С2Н5)2
СН—- со2с2н5
СО2Н
I
/СО сн3
(СН3)2С< I
хсн2-со
Конденсация
Дикмана и гидролиз
Альдольная
конденсация
и отщепление Н2О
СО2Н
СО2Н
/С---С-СО2Н
(СН3)2С< II
(СН3)2с/
С = С-СО2Н
I
сн = с—он
СО2Н
I
/С—сн
-> (СН3)2С< I
\сн2— со
K3Fe(CN)e | KMnO4
/СН —СО2Н
<1
ХСН — СО2Н
/С(СО2Н) = СН
(СН3)2С< I
хсо2н СО2Н
В этой и других подобных системах бициклический и моноциклический изо-
меры не были разделены, поэтому возможность двух путей синтеза кислоты
и двух путей ее расщепления была рассмотрена как указание на существо-
вание интрааннулярной валентной таутомерии (название предложено.
Торпом), достаточно мобильной и поэтому препятствующей разделению
постулированных валентных изомеров.
Аналогичный пример валентной таутомерии в системе циклопентадиен —
бициклопентен был описан недавно ван Тамеленом и сотр. [194]. При облу-
чении в этиловом спирте циклопентадиен частично превращается в бицикло-
пентен, который выделялся разгонкой, а затем был очищен газовой хромато-
графией. Спектр ядерного магнитного резонанса показывает, что образую-
щееся соединение содержит четыре сорта атомов водорода, а не три, как
циклопентадиен. При восстановлении диимином (получающимся из азоди-
карбоновой кислоты) образуется бициклопентан. В темноте бициклопентен
вновь переходит в циклопентадиен (с примесью полимера); при комнатной
температуре время жизни бициклопентена составляет около 2 ч
Замедленность реакции в темноте, не согласующаяся с термодинамически-
ми факторами, которые способствуют такому превращению, объясняется
необходимостью дисротаторного изменения связей, которое не согласуется
722
Перегруппировки ненасыщенных соединений
с орбитальной симметрией основного состояния
Следующий важный случай относится к взаимопревращению бензола
и бициклогексадиена, имеющего структуру дьюаровского бензола, но гео-
метрию, сильно отличающуюся от геометрии бензола. Автор книги [195J
пытался получить производное бициклогексадиена в 1922 г. замыканием
четырехчленного цикла с другим четырехчленным циклом по методу Дикма-
на, но получил только производное бензола (орцинол)
СН3
CH2-(L СН2- СО2С2Н5
I I
СО — сн2
сн2—С — сн2
I I I
Leo— СН-СО
Недавно Куксон и сотр. [196] при добавлении метилового эфира ацетилен-
дикарбоновой кислоты к тетраметилциклобутадиену также получили только
бензоидный продукт (метиловый эфир тетраметилфталевой кислоты)
СН3С = ССН3
I I
СН3С = ССН3
ССО2СН3
+ Hi
ССО2СН3
СН3С —С-ССО2СН3
II I II
СН3С-С — ССО2СН3
I
СНз
сн3
I
СНз-/^.- СО2СН3
CH3-IN- СО2СН3
I
сн3
Бициклобутадиеновые соединения были успешно выделены в 60-х годах.
В 1963 г. ван Тамелен и Паппас [197] опубликовали первую работу, в которой
эта система хотя не была получена, но являлась промежуточным соединени-
ем в стадийном синтезе, осуществленном исходя из моноалициклического
соединения. На первой стадии синтеза при облучении г^с-1,2-дигидрофтале-
вого ангидрида образовывался бициклогексеновый ангидрид, содержавший
одну двойную связь и дававший спектр ЯМР, по которому можно было иден-
тифицировать мостиковую структуру
Это интрааннулярное валентное превращение по типу гомологично фото-
превращению циклопентадиена в бициклопентен. В этом процессе вместо
трехчленного образуется четырехчленный цикл. Как и в более простых систе-
мах, превращение разрешено при определенной симметрии первой возбуж-
денной л-орбитали.
Превращение мостикового ангидрида в бициклогексадиен было осуществлено
при помощи окислительного удаления ангидридной группы тетраацетатом
свинца в пиридине. При перегонке бициклогексадиен отгоняется с первой
фракцией и дает спектр ядерного магнитного резонанса, соответствующий
его структуре. Диимин восстанавливает его в известный бициклогексан.
Бициклогексадиен спонтанно превращается в бензол при комнатной темпера-
46* 723
Глава XI
туре с полупериодом около двух дней, а при 90 °C превращение заканчивает-
ся за полчаса. Следовательно, такой процесс имеет высокую энергию акти-
вации, несмотря на ожидаемую большую экзотермичность
Петтит и сотр. [198] показали, что реакции циклобутадиена, в свободном
состоянии быстро димеризующегося, могут быть исследованы, если его
применять в виде стабильного железотрикарбонильного комплекса, из кото-
рого он выделяется ионами церия. Они также установили, что этот угле-
водород может захватываться производными ацетилена, включая фенил-
ацетилен, метилпропиолат и метиловый эфир ацетилендикарбоновой кисло-
ты, с образованием производного бициклогексадиена. Несмотря на то что
эти соединения можно выделить, при нагревании они более или менее легко
превращаются в производные бензола
Се4+
(CO)3Fe(C4H4) -►
RC=CR'
Производные второго валентного изомера бензола были получены Фихе
и сотр. [199] в ходе работы по исследованию спонтанной полимеризации трет-
бутилфторацетилена. Продукт на две трети состоит из тримера и на одну
треть из тетрамера. Примерно половину тримерной фракции составляет
производное бициклогексадиена. Его строение, представленное ниже, следу-
ет из спектра ядерного магнитного резонанса, а также из того факта, что при
100 °C он быстро и нацело переходит в 1,2,3-три-игретп-бутилтрифторбензол.
Другая половина тримерной фракции содержит производное трициклогек-
сена, которое Фихе назвал «бензваленом». Предложенная структура этого
тримера хорошо объясняет его спектральное и химическое поведение. Этот
тример стабильнее другого, но при 220 °C быстро ароматизуется, превраща-
ясь в 1,2,4-три-игретп-бутилтрифторбензол
тдет-С4Н9
mpem-C4H9
mpem-CJtig^ СдНд-трет
F
3 nf
С
। '^лтрет-С4Нд
F
Т'' 'wC4H9-mpem
F
100°С
С4Н9-трет
220°С
трет-СдНд
F
F
F
Первый бициклогексадиен был получен при фотоизомеризации производного
бензола. В 1962 г. Паппас и ван Тамелен [200] облучили светом с длиной
волны 2537 А (253,7 нм) 1,2,4-три-игретп-бутилбензол и получили изомер бици-
клогексадиена, который превращался в исходное производное бензола в тем-
ноте при 200 °C. Эта работа была расширена Вильцбахом и Капланом [201],
которые показали, что при облучении [2537 А (253,7 нм)] 1,2,4- или 1,3,5-
три-игреиг-бутилбензола образуется фотостационарная смесь трех валентных
724
Перегруппировки ненасыщенных соединений
изомеров. Один из них, бициклогексадиен, как указано выше, при нагрева-
нии превращается в 1,2,4-три-тпреиг-бутилбензол. Второй изомер, трицикло-
гексен, или бензвален, при нагревании также дает 1,2,4-три-игреиг-бутилбен-
зол. Третьим изомером является тетрациклогексан, или призман, имеющий
призматическую структуру бензола Ладенбурга. Этот изомер при нагрева-
нии при 115 °C в течение часа через промежуточно образующиеся изомеры
бициклогексадиена, которые можно выделить, превращается в смесь 1,2,4-
и 1,3,5-три-игреиг-бутилбензолов. Ниже приведены формулы этих валентных
изомеров.
ТПрвТП~ С4Нд
трет~ C4Hg
C4H9-mpem
mpezn-C4Hg
mpem-CJlg C4H9-mpem
zn/je/rz-C4H9
tp
I C4H9-mpem
mpem-C^Hg
Три-трет-бутил-
бициклогексадиен
Три-трет-бутил~ Три-трет-бутил—
бензвален призман
При облучении ди- и триметилбензолов ультрафиолетовым светом метильные
группы меняют свое расположение относительно друг друга. Из о-ксилола
образуется смесь м- и n-ксилолов, а мезитилен дает псевдокумол [202].
В последнем случае с помощью изотопной метки было доказано, что перегруп-
пировывается не одна из метильных групп, а один из кольцевых атомов
углерода вместе со своей метильной группой (звездочками указано положе-
ние изотопной метки 14С)
СН3 СН3
Фотопревращение 1,3,5-три-игреиг-бутилбензола в мостиковые валентные
изомеры и термическое превращение этих изомеров в 1,2,4-три-щретп-бутил-
бензол моделируют эти процессы изомеризации. Эти процессы могут проте-
кать или через бензвален, или через призман, образующийся из одного изо-
мера бициклогексадиена и превращающийся в производное бензола через
другой изомер.
Циклопропановое кольцо обладает заметной тенденцией к сопряжению
с двойной связью. Поэтому можно ожидать, что двойная связь и циклопро-
пановый цикл в шестичленном кольце а-туйена будут способными к сопря-
жению в кольце. Это сопряжение можно рассматривать аналогично цикли-
ческому сопряжению в системе циклопентадиен—бициклопентен с тем лишь
отличием, что в данном случае место двойной связи циклопентадиена зани-
мает циклопропановый цикл
з
СН
3Н7-изо
?±СН
дНу-КЗО
з
Деринг и Ламберг [203] показали, что именно по такому механизму про-
исходит рацемизация а-туйена при 250 °C. Они ввели дейтерий в положение 3
и после проведения рацемизации обнаружили, что он распределен между
положениями 1 и 3. Рацемизация происходит потому, что двугранный угол
725
Глава XI
между кольцами во время перегруппировки становится равным 180 °C.
Перейдем к семичленным циклам. Входящее в такой цикл циклопропановое
кольцо может сопрягаться с двумя двойными связями. Этот цикл можно
рассматривать как подобный кекулевской структуре бензола, за исключе-
нием того, что место одной двойной связи бензола в данном случае занимает
циклопропановое кольцо. Такого рода таутомерами являются норкарадиен
и тропилиден. Положение равновесия в этом частном случае полностью сдви-
нуто в сторону тропилидена, так как при синтезе из бензола или циклогепта-
диена получается продукт, спектр ядерного магнитного резонанса которого
соответствует только структуре тропилидена [204]
Норкарадиен
Тропилиден
Противоположный крайний случай соответствует 7,7-дицианоноркарадиену,
который также является единственным изомером в своей системе, но уже
имеет мостиковую, а не тропилиденовую структуру [205]. Можно думать, что
в этом случае обращение знака разности свободных энергий обусловлено
квазисопряжением циклопропанового цикла с циангруппами.
Первым примером системы, находящейся в истинном равновесии, были 7-
циан-7-трифторметильные производные, полученные Цыганском [206]. В этой
системе взаимопревращение происходит настолько быстро, что спектр про-
тонного магнитного резонанса имеет диффузную структуру при комнатной
температуре и недостаточно разрешен даже при —80 °C. Равновесие по-настоя-
щему «замораживается» при —112 °C, при этом смесь таутомеров имеет
состав, указанный ниже:
60% 40%
Мостиковая структура подтверждается наличием в продуктах каталитиче-
ского гидрирования наряду с производным циклогептана и циклогексилмета-
на также и производного норкарана.
Другая равновесная система окись бензола — оксепин была открыта Фоге-
лем и сотр. [207]. Кинетику этого превращения изучил Гюнтер [208] методом
ядерного магнитного резонанса. При 0 °C положение равновесия соответст-
вует следующему составу смеси:
Константы скоростей имели следующие значения: kr= 14,1-Ю6 с-1 и =
= 2,29-108 с-1. Таким образом, время жизни изомеров составляет доли
ми к р осе к у нд ы.
126
Перегруппировки ненасыщенных соединений
В восьмичленном цикле ненасыщенность может распределяться по-разному.
Рассмотрим вначале систему, гомологичную превращению норкарадиена
в тропилиден, в которой вместо циклопропанового кольца содержится цикло-
бутановый цикл. Следует ожидать, что высшая гомологичная интрааннуляр-
ная таутомерная система будет отличаться от низшей двумя аспектами:
во-первых, термодинамическое различие между изомерами будет менее рез-
ким и, во-вторых, подвижность системы будет гораздо меньшей. Циклобута-
новый цикл обладает меньшей ненасыщенностью по сравнению с циклопро-
пановым. Описываемый случай исследовали Коуп и сотр. [209], которые
установили, что первый продукт гидрирования циклооктатетраена — цик-
лооктатриен — при кратковременном нагревании до 80—100 °C частично
превращается в мостиковый изомер
При комнатной температуре таутомеры можно разделить. (Триен образует
комплекс с нитратом серебра.) Строение мостикового таутомера доказано его
восстановлением в бициклооктан и окислением в цнс-циклобутан-1,2-дикарбо-
новую кислоту.
Фогель, а также Хыозген и их сотрудники показали, что сам циклооктатет-
раен находится в интрааннулярной валентной таутомерии с менее стабиль-
ным бициклооктатриеном. Фогель и сотр. получили бициклооктатриен дебро-
мированием соответствующего синтетического дибромида при низкой тем-
пературе [210]. Строение продукта было установлено восстановлением в уже
известный бициклооктан, а также с помощью спектров ядерного магнитного
резонанса. При температурах около —20 °C бициклооктатриен спонтанно
и нацело превращается в циклооктатетраен
При 0 °C константа скорости kl этого термодинамически выгодного процесса
составляет 7,5-10-4 с-1; эта величина соответствует полупериоду, равному
15 мин. Энергия активации этой реакции составляет 18,7 ккал/моль
(78,29-10® Дж/моль). Константа равновесия [циклооктатетраен]/[бициклоок-
татриен] известна не точно, но, вероятно, равна ~103>3 при 0 °C и ~104
при 100 °C. Однако Хьюзгену и Митцшу [211] удалось измерить константу
изомеризации циклооктатетраена k-t в термодинамически не благоприятном
направлении при 100 °C и более высоких температурах. Измерение прово-
дилось методом быстрого перевода продукта изомеризации в нереакционно-
способную форму. Авторы показали, что диенофилы могут присоединяться
к бициклооктатриену, образующемуся из циклооктатетраена. В частности,
аддукт с малеиновым ангидридом имеет следующее строение:
Малеиновый ангидрид недостаточно реакционноспособен, чтобы служить
ловушкой для образующегося бициклооктатриена, но при переходе к таким
727
Глава XI
диенофилам, как тетрацианэтилен и дицианмалеимид, которые, как известно,
реагируют с простыми диенами в несколько тысяч раз быстрее, чем малеи-
новый ангидрид, указанную изомеризацию можно изучать количественно.
Измеряемые скорости присоединения становятся не зависящими от кон-
центрации и природы диенофила и равны скорости изомеризации, величина
которой определяется константой к^. При 100 °C в диоксане константа k_t
имеет величину 4,4-Ю-1 с-1. Энергия активации аналогичной реакции фенил-
циклооктатетраена составляет 27,2 ккал/моль (113,88-10® Дж/моль). Видно,
что в обоих направлениях реакция имеет довольно высокую энергию акти-
вации; это находится в соответствии с фактом значительного изменения гео-
метрии при таутомерном превращении.
В восьмичленном цикле возможна валентная таутомерия с участием цикло-
пропанового кольца; в результате такого превращения число двойных связей
не уменьшается. Такая система является винилогом сх-туйена; отличие состо-
ит в том, что в цикле содержатся вместо одной две двойные связи. Простей-
шим примером может служить гомотропилиден, полученный Дёрингом
и Ротом в 1963 г. действием диазометана и хлорида меди(1) на тропилиден
[212]. Спектр протонного магнитного резонанса свидетельствует о быстром
таутомерном переходе между эквивалентными таутомерами
Основы спектроскопического определения наличия быстрого взаимопрев-
ращения эквивалентных таутомеров состоят в следующем. В любом спектре
(например, инфракрасном), если вследствие принципа неопределенности
Д7? -Az ~ h время наблюдения становится гораздо более коротким, чем
время жизни таутомера, будут наблюдаться спектральные частоты одного
таутомера и вид спектра не будет зависеть от температуры. Однако в случае
протонного магнитного резонанса энергии настолько малы, что время наблю-
дения становится относительно большим — от миллисекунд до секунд. Если
при некоторой температуре время жизни таутомера сравнимо со временем
наблюдения сигнала, то ниже этой температуры будет наблюдаться дискрет-
ный спектр, не меняющийся при изменении температуры. В спектре будут
наблюдаться сигналы протонов, входящих в состав индивидуальных таутоме-
ров. Выше определенной температуры будет наблюдаться другой спектр,
также дискретный и не зависящий от температуры, однако сигналы протонов
будут такими, которые наблюдались бы, если бы имелась дополнительная
симметрия (плоскость симметрии), обусловленная усредненной структурой,
т. е. «размазыванием» подвижных связей. В интервале между этими поро-
говыми температурами будет наблюдаться диффузный спектр, меняющийся
с температурой. В описываемом случае разные дискретные спектры, соот-
ветствующие теории, были получены при —50 °C и при 180 °C, а в интервале
между этими температурами спектр был диффузным и зависел от температуры.
Хотя эти спектры количественно не анализировались, качественно из них
следует, что время жизни таутомера при 0 °C должно быть порядка санти-
секунды.
Дёринг и Рот предположили, что гомотропилиден должен существовать
в двух конформациях, одна из которых напоминает кресло и является более
стабильной, а другая напоминает ванну и менее стабильна, однако в ней
валентные изменения осуществляются легче. Поэтому концентрация наибо-
725
Перегруппировки ненасыщенных соединений
лее устойчивой конформации индивидуального таутомера будет довольно
низкой. В соответствии с этим можно предсказать, что введение 1,5-мости-
ка, стабилизующего ванноподобную конформацию гомотропилиденового
остатка, приведет к более подвижной таутомерной системе. Этот вывод был
подтвержден Шредером [213] и другими на ряде примеров, в частности на при-
мере представленного ниже дигидробульвалена, имеющего мостик СН2СН2:
Дигидробульвален
Спектр протонного магнитного резонанса этого соединения анализировался
количественно, и была определена константа скорости взаимопревращения
таутомеров. При О °C константа первого порядка была равна 330 000 с-1,
что соответствует времени жизни таутомера порядка 3 мкс. С другой стороны,
валентные изменения в этом случае зависят от движения ядер, которому
оказывает сопротивление жесткая структура, поэтому энергия активации
равна 12,6 ккал/моль (52,75-103 Дж/моль).
Дёринг и Рот сделали и другое предположение. Оно состояло в том, что если
в качестве 1,5-мостика использовать группу — СН = СН —, то образуется
представленная ниже тригональная симметричная структура, еще до своего
открытия названная «бульваленом», число эквивалентных таутомеров которой
равно 1 209 600, так как все десять атомов углерода путем последовательного
изменения связей гомотропилиденового типа могут в разные периоды времени
занимать разные положения.
Это обусловлено следующим. При единичном изменении голова моста 1
может переходить в положения 4, 5 или 6, при последующем изменении —
в положения 2, 8 или 9 и, наконец, далее — в положения 3, 7 или 10. Таким
образом, любой из десяти атомов углерода может находиться в голове моста.
Может также происходить последовательное взаимопревращение двух ато-
мов углерода, связанных между собой через третий атом. Так, посредством
двух гомотропилиденовых валентных изменений может происходить смеще-
ние связей атомов 2 и 4 от атомов 1 и 5, 6 соответственно к атомам 1, 9 и 6,
а затем к атомам 9 и 6, 7 соответственно без каких-либо других изменений
в типе связей. Другая пара таких валентных изменений приводит к смещению
связей атомов 2 и 4 посредством другого ротационного процесса к атомам 10
и 7,8; еще одна пара валетных изменений приводит к смещению этих связей
к атомам 5 и 8,1 соответственно. Еще одно валентное изменение, седьмое по
счету, приведет к образованию связей с атомами 5, 6 и 1 соответственно без
729
Глава XI
изменения других связей. При этом атомы 2 и 4 поменяются местами. Вслед-
ствие симметрии подобное взаимопревращение может происходить между
атомами 9 и 5 и атомами 8 и 6; при этом атом 1 остается в голове моста, а
поскольку каждый атом углерода может занять положение в голове моста,
все пары сх,у-атомов могут взаимно меняться местами.
Далее, путем последовательных сх,у-обменов может происходить взаимо-
превращение двух соседних атомов. Так, путем описанного а,у-обмена можно
сделать так, что атом 2 перейдет в положение 4, а затем, соответствующим
образом изменив положение головы моста, перевести его в положение 10
и далее аналогично в положение 1; последний процесс является также первой
из трех стадий, необходимых для аналогичного перехода атома 1 в положение
10, затем в положение 4 и затем в положение 2. Сдвиг положения головы
моста, происходящий во время пятикратного изменения семи связей, при
котором каждый раз происходит сх,у-обмен (2-4, 4-10, 10-1, 4-10, 2-4), соот-
ветствует схеме 1 —>- 9 -> 2 -> 3 -> 1 или 1 -> 9 -> 5 -> 9 -> 1; число гомо-
тропилиденовых превращений, необходимых для таких смещений положения
у головы моста, равно или двум, четырем, двум и четырем, или двум, четырем,
четырем и двум соответственно. Общее число последовательных гомотропи-
лиденовых превращений поэтому равно 47; в результате все атомы, кроме
меняющихся атомов 1 и 2, остаются там, где они находились ранее. Оче-
видно, то же самое можно проделать с атомами 1 и 8 и с атомами 1 и 9; при
этом атом 9 остается в голове моста, а поскольку в голове моста могут нахо-
диться все атомы углерода, все сх,р-пары атомов могут взаимно меняться
местами.
Кроме того, может происходить перемена мест атомов, находящихся друг
от друга через два других атома углерода. Так, при соответствующем сме-
щении головы моста в интервале между стадиями а,у-обмена можно пере-
местить атом из положения 5 в положение 9, а затем в положение 2, при
этом атом 2 переходит в положение 9, а затем в положение 5. В общем в ре-
зультате таких последовательных перегруппировок (29 индивидуальных
гомотропилиденовых перегруппировок) все атомы во всех положениях,
за исключением положений 2 и 5, взаимно меняются местами. Как и в пре-
дыдущем случае, отсюда следует, что все а,6-пары атомов могут меняться
местами. В бульваленовой системе не существует атомов, отделенных друг
от друга более чем двумя атомами углерода.
Таким образом, в конце концов (находясь в цейтноте по шахматной терми-
нологии) мы приходим к выводу, что в любой паре атомов углерода, выбранной
из всех десяти атомов углерода бульвалена, может происходить взаимная
перемена мест атомов, т. е. все десять атомов могут свободно меняться местами.
Поэтому число эквивалентных изомеров равно факториалу 10, деленному на
число симметрии 3, т. е. 1,2 миллиона. Отсюда следует, что выше темпера-
туры, при которой время жизни таутомера становится много короче времени
наблюдения сигнала, в спектре протонного магнитного резонанса все десять
атомов водорода будут эквивалентными и спектр будет состоять только из
одной линии. В низкотемпературном спектре должны проявляться четыре
сорта протонов, но наиболее сильно будут отличаться друг от друга шесть
олефиновых и четыре циклоалкановых атома водорода.
Через несколько месяцев после опубликования этих теоретических рассмот-
рений Шредер [214], применив неожиданный и высокопрактичный метод,
получил бульвален, установил его строение и подтвердил предсказание
Дёринга и Рота. Он установил, что легкодоступный димер циклооктатетраена,
образующийся в качестве главного продукта при термической реакции цик-
лооктатетраена, является 1,2—1,5-аддуктом и, таким образом, как показано
730
Перегруппировки ненасыщенных соединений
ниже, 1,5-мостиковым гомотропилиденом. Спектры протонного магнитного
резонанса показывают, что это соединение обладает соответствующими тау-
томерными свойствами. При фотолизе оно дает с высоким выходом бульвален
и бензол.
Строение бульвалена было доказано химически, во-первых, восстановлением
двойных связей с помощью диимида (образующегося при катализируемом
ионами меди аутоокислении гидразина) последовательно до дигидро-, тет-
рагидро- и гексагидропроизводных и, во-вторых, озонолизом двойных связей
с последующим восстановлением до г/нс-1,2,3-три(оксиметил)циклопропана.
При 120 °C спектр протонного магнитного резонанса состоял из одной линии.
При —85 °C спектр содержал две пары частично перекрывающихся линий,
при этом соотношение интенсивностей этих пар было равно 4 : 6. Менее
интенсивная пара линий состояла из неравных компонентов, а более интен-
сивная — из почти равных компонентов. Естественно, что первый из этих
дублетов относится к циклоалкановым протонам и соотношение интенсивно-
стей его компонентов должно быть равным 1:3. Второй дублет обусловлен
олефиновыми протонами, и его компоненты должны быть одинаковыми по
интенсивности.
Подвижность таутомерной системы бульвалена занимает промежуточное
положение между подвижностью систем гомотропилидена и 1,5-мостикового
тропилидена. Константа скорости таутомерного превращения в бульвалене
при 0 °C составляет 790 с-1 [215], что соответствует времени жизни таутомеров
около миллисекунды. Таким образом, скорость таутомерного превращения
в бульвалене в 400 раз меньше, чем в дигидробульвалене. Энергия активации
валентного изменения имеет величину 11,7 ккал/моль (48,99-10 Дж/моль),
т. е. на 0,9 ккал/моль (3,77 ПО3 Дж/моль) меньше, чем для дигидробульвалена.
Таким образом, основная причина более низкой скорости внутримолекуляр-
ного превращения в бульвалене, по-видимому, обусловлена энтропийным
фактором. Это может быть следствием того, что спектрометр ядерного маг-
нитного резонанса измеряет скорость достижения эффективной эквивалент-
ности между всеми десятью атомами водорода, а эквивалентность может
быть достигнута в результате лишь некоторых валентных изменений, т. е.
лишь небольшой части из всех возможных превращений.
СПИСОК ЛИТЕРАТУРЫ
1. Geuther A., Gottinger Anzeigen, 1863, 281; Arch. Pharm., 106, 97 (1863); Jahresber.,
1863 323
2. Frankland E., DuppaB. F., Ann., 135, 217 (1865); 138, 204, 328 (1866); Geuther A.,
Z. Chem., 1868, 58.
3. Wislicenus J., Ann., 186, 161 (1877); 190, 287.
4. Conrad M., Bischoff C. A., Ann., 204, 121 (1880).
5. Michael A., J. prakt. Chem., 37, 473 (1887).
6. Butlerow A., Ann., 189, 44 (1877).
7. v. Baeyer AOekonomides S., Ber., 15, 2093 (1882); v. Baeyer A., Ber., 16, 2188 (1883).
8. Griess P., Ber., 7, 1618 (1874).
9. Goldschmidt H., Ber., 17, 213 (1884).
10. Laar C., Ber., 18, 648 (1885); 19, 730 (1886).
11. Claisen L., Ann., 291, 25 (1896).
12. Wisliscenus W., Ann., 291, 147 (1896); 312, 34 (1900); Ber., 32, 2837 (1899).
731
Глава XI
13. Wisliscenus W., Ann., 389, 265 (1912); 413, 206 (1916); 421, 119 (1920); Meyer К. H..
Ber., 45, 2843 (1912); Dieckmann W., Ber., 49, 2213 (1916); 50, 1375 (1917).
14. Knorr L., Ann., 293, 70 (1896); 306, 322 (1899).
15. Knorr L., Kaufmann H. P., Ber., 55, 232 (1922).
16. Kaufmann H. P., Ber., 55, 2255 (1922); Ann., 429, 247 (1922).
17. Hantzsch 4., Schultze 0. W., Ber., 29, 699, 2251 (1896).
18. Hantzsch A., Ber., 32, 575, 3066 (1899); Hantzsch A., Volt A., ibid., p. 607.
19. Knorr L., Rothe 0., Averbeck H., Ber., 44, 1138 (1911).
20. Meyer К. H., Schoeller V., Ber., 53, 1410 (1920); Meyer К. H., Hopff H., Ber., 54, 579
(1921).
21. Meyer К. HAnn., 380, 212 (1911); Meyer К. HKoppelmeier PBer., 44, 2718 (1911);
Meyer К. H., Ber., 45, 2843 (1912); 47, 826 (1914).
22. Wassermann A., J. Chem. Soc., 1948, 1323.
23. Lapworth A., J. Chem. Soc., 85, 30 (1904).
24. Meyer К. H., Ber., 45, 2864 (1912).
25. Dawson H. M., Carter J. S., J. Chem. Soc., 1926, 2282; Dawson H. M., Dean N. C..
ibid., p. 2872; Dawson H. M., Hoskins C. R., ibid., p. 3166; Bell R. P., Longuet-Hig-
gins H. C., ibid., 1946, 636; Bell R. P., Jones P., ibid., 1953, 88.
26. Bell R. P., Spiro M., J. Chem. Soc., 1953, 429; Bell R. P., Yates K., ibid., 1962, 1927;
2285; Bell R. P., Davis G. G., ibid., 1964, 902.
27. Ingold E. H., Chem. & Ind., 1923, 1246; J. Chem. Soc., 123, 1717 (1923).
28. Deshapande S. S., Thorpe J. F., J. Chem. Soc., 121, 1430 (1922); Bains L., Thorpe J. F.,
ibid., 123, 1206 (1923); Baker J. W., 127, 1678 (1925).
29. Vorldnder D., Ber., 33, 3185 (1900); Ingold С. K., Powell W. J., J. Chem. Soc., 119,
1976 (1921); Ingold С. K., Perren E. A., ibid., 121, 1414 (1922).
30. Ingold С. K., Perren E. A., Thorpe J. F., J. Chem. Soc., 121, 1765 (1922).
31. Guthzeit M., Ber., 31, 2753 (1898); Ber., 34, 675 (1901); Guthzeit M., Weiss A., Schaf-
fer W., J. prakt. Chem., 80, 393 (1909).
32. Verkade P. E., Proc. Koninkl. Nederland. Akad. Wetenschap., 27, 1133 (1919).
33. Jacobson P., Stelzner R. в книге Meyer V., Jacobson P. «Lehrbuch der Organischen
Chemie», Veit, Leipzig, 2nd Edn. 1913, I, 2, 886, 910, 915, 927.
34. Baker J. W., Ingold С. K., Thorpe J. F., J. Chem. Soc., 125, 268 (1924); Gilmour R.,
ibid., p. 705.
35. Claisen L., Ann., 291, 80, 86 (1896).
36. Knorr L., Ann., 293, 38 (1896).
37. Wisliscenus W., Uber Tautomerie, Ahrens Sammlung, Stuttgart, 2, 187 (1898).
38. Ingold С. K., Piggott H. A., J. Chem. Soc., 121, 2381 (1922).
39. Griess P., Ber., 7, 1618 (1874); Ann., 137, 60 (1886).
40. Noelting E., Binder F., Ber., 20, 3004 (1887).
41. Meldola R., Streatfield F. W., J. Chem. Soc., 51, 102, 434 (1887); 53, 664 (1888); 55,
4122 (1889); 57, 785 (1890); Smith C., Watts С. H., ibid., 97, 562 (1910).
42. v. Pechmann H., Ber., 28, 869, 2362 (1895); 30, 1779 (1897).
43. Marckwald W., Ann., 286, 343 (1895).
44. Wheeler L. H., Johnson T. B., Ber., 32, 35 (1899); Roberts R. M., J. Am. Chem. Soc.,
72, 3603 (1950).
45. Ingold С. K., Shoppee C. W., J. Chem. Soc., 1929, 1199.
46. Ingold С. K., Shoppee C. W., J. Chem. Soc., 1929, 447.
47. Ingold С. K., Piggott H. A., J. Chem. Soc., 123, 1469 (1923).
48. Ingold С. K., Shoppee C. W., Thorpe J. F., J. Chem. Soc., 1926, 1477.
49. Shoppee C. W., J. Chem. Soc., 1930, 968; 1931, 1225; 1932, 696.
50. Ingold С. K., Rothstein E., J. Chem. Soc., 1931, 1666.
51. Ingold С. K., Shoppee C. W., J. Chem. Soc., 1929, 1199.
52. Lowry T. M., J. Chem. Soc., 1927, 2554.
53. Baker J. W., J. Chem. Soc., 1928, 1583, 1979; 1929, 1205.
54. Pedersen K. J., J. Phys. Chem., 38 , 581 (1934).
55. Swain C. G., J. Am. Chem. Soc., 72, 4578 (1950).
56. Bell R. P., Clunie J. C., Nature, 167, 363 (1951).
57. Bell R. P., Darwent B. de B., Trans. Faraday Soc., 46, 34 (1950).
58. Hughes E. D., Nature, 147, 812 (1941).
59. Schwarzenbach G., Felder E., Helv. Chim. Acta, 27, 1044 (1944); Schwarzenbach G.,
Witwer C., ibid., 30, 656 (1947).
60. Dieckmann W., Ber., 55, 2470 (1922).
61. v. Auwers K., Jacobson H., Ann., 426, 161 (1922).
62. Conant J. B., Thompson A. F., J. Am. Chem. Soc., 54, 4039 (1932).
63. Schreck R., J. Am. Chem. Soc., 71, 1881 (1949).
64. Evans D. P.,’Gordon J. J., J. Chem. Soc., 1938, 1434.
732
Перегруппировки ненасыщенных соединений
65. Cardwell Н. М. Е., J. Chem. Soc., 1951, 2442.
66. Cardwell Н. М. Е., Kilner А. Е. Н., J. Chem. Soc., 1951, 2430.
67. Catch J. R., Elliot D. F., Hey D. H., Jones E. R. H., J. Chem. Soc., 1948, 272;
Catch J. R., Hey D. H., Jones E. R. H., Wilson W., J. Chem. Soc., 1948,
276.
68. Dawson H. M., Wheatley R., J. Chem. Soc., 97, 2048 (1910); Dawson H. M., Ark H.,
ibid., 99, 1740 (1911).
69. Turnbull D., Maron S. H., J. Am. Chem. Soc., 65, 212 (1943).
70. Maron S. H., La Мег V. K., J. Am. Chem. Soc., 60, 2588 (1938).
71. Los J. M., Simpson L. B., Weissner K., J. Am. Chem. Soc., 78, 1564 (1956).
72. Ingold С. K., Ann. Repts. on Progress Chem. (Chem. Soc. London), 24, 109 (1927);
ibid., 25, 119 (1928).
73. Baker J. W. в книге «Tautomerism», Routledge, London, 1934, Chap. 9.
74. Linstead R. P. в книге Gilman H. «Organic Chemistry», Wiley, New York, 1938,
p. 819.
75. De la Mare P. B. D., Hughes E. D., Ingold С. K., J. Chem. Soc., 1948, 22.
76. Perez Ossorio R., Herrera F. G., Hidalgo A., Ann. Real. Soc. Esp. Fis. Quim., 52, 123
(1956); 54, 471 (1958).
77. Hugh W. E., Kon G. A. R., J. Chem. Soc., 1930, 775.
78. Catchpole A. G., Hughes E. D., Ingold С. K., J. Chem. Soc., 1948, 11.
79. Hammond G. S., J. Am. Chem. Soc., 77, 334 (1955).
80. Ingold С. K., The Transition State, Spec. Publ. Chem. Soc., 16, 119 (1962).
81. Hine, J. Org. Chem., 31, 1236 (1966).
82. Rice F. O., Teller E., J. Chem. Phys., 6, 489 (1938); 7, 199 (1939).
83. Ramberg L., Mellander A., Arkiv Kemi Mineral. Geol., Bll, № 31 (1934).
84. Ramberg L., Hedlund I., Arkiv Kemi Mineral. Geol., Bll, № 41 (1934).
85. Ingold С. K., Wilson C. L., J. Chem. Soc., 1934, 773.
86. Bartlett P. D., Stauffer С. H., J. Am. Chem. Soc., 37, 2580 (1935).
87. Hsu S. K., Wilson C. L., J. Chem. Soc., 1936, 623.
88. Reitz 0., Z. physik. Chem., 179, 119 (1937).
89. Hsu S. K., Ingold С. K., Wilson C. L., J. Chem. Soc., 1938, 78.
90. Kimball R. H., J. Am. Chem. Soc., 58, 1963 (1936).
91. Ingold С. K., de Salas E., Wilson C. L., J. Chem. Soc., 1936, 1328.
92. Ingold С. K., Wilson C. L., J. Chem. Soc., 1933, 1493; Ingold С. K., Wilson C. L.,
J. Chem. Soc., 1934, 93; Hsu. S. K., Ingold С. K., Wilson C. L., J. Chem Soc.,
1935, 1774.
93. De Salas E., Wilson C. L., J. Chem. Soc., 1938, 319.
94. Ossorio R. P., Hughes E. D., J. Chem. Soc., 1952, 426.
95. Cram D. J., Guthrie R. D., J. Am. Chem. Soc., 87, 397 (1965); J. Am. Chem. Soc.,
88, 5760 (1966).
96. Hantzsch A., Ber., 32, 575 (1899); Hantzsch A., Kalb M., ibid., p. 3109.
97. Decker H., Ber., 25, 443 (1892); J. prakt. Chem., 47 , 28 (1893).
98. Freund M., Ber., 37, 4666 (1904); Freund M., Richard L., Ber., 42, 1101 (1909).
99. Kaufmann 4., герм. пат. 250154 (1912).
100. Kaufmann A., Albertini A., Ber., 42, 3776 (1909); Kaufmann A., Widmer R., Ber.,
44, 2058 (1911).
101. Kaufmann A., Struber P., Ber., 44, 680 (1911); Decker H., Kaufmann A., 3. prakt.
Chem., 84, 219 (1911).
102. Hantzsch A., Kalb M., Ber., 32, 3109 (1899).
103. v. Pechmann H., Baltzner 0., Ber., 24, 3144 (1891).
104. Freund M., Bamberg P., Ber., 35, 1739 (1902).
105. Liebermann C., Glawe A., Ber., 37, 2738 (1904); Kropf F., ibid., p. 2744.
106. Hope E., Robinson R., J. Chem. Soc., 99, 2119 (1911); 103, 361 (1913).
107. Freund M., Ber., 36, 4257 (1903).
108. Decker H., Ber., 33, 2273 (1900).
109. Baser W., Ann., 254, 334 (1889).
110. Freund M., Becker F., Ber., 36, 1522 (1903).
111. Hantzcsh A., Osswald G., Ber., 33, 278 (1900).
112. Freund M., Beck H., Ber., 37, 4679 (1904).
113. Turgeon J. C., La Мег V. K., J. Am. Chem. Soc., 74, 5988 (1952).
114. Hantzsch A., Ber., 32, 3132 (1899).
115. LeFevre R. J. W., Roper R., Reece I. H., J. Chem. Soc., 1959, 4104.
116. Werner A., Ber., 34, 3301 (1901); Hewitt J. T., ibid., p. 3820.
117. Gomberg M., Cone L. H., Ann., 376, 188 (1910).
118. Fosse R., Bull. soc. chim. (France), 35, 1005 (1906).
119. Collie J. N., Tickle T., J. Chem. Soc., 75, 710 (1899); Werner A., Ber., 34, 3300 (1901);
733
Глава XI
Hantzsch А., Вег., 52, 1535 (1919); Arndt F., Scholtz E., Nachtwey P., Ber., 57, 1903
(1924).
120. Werner A., Ber., 34, 3301 (1901); Hilditch T. P., Smiles S., I. Chem. Soc., 99, 160
(1911).
121. Hartley W. R. H., Smiles S., J. Chem. Soc., 1926, 1821.
122. De la Mare P. B. D., в книге «Molecular Rearrangements», Editor P. de Mayo, Inter-
science, New York, 1963, Chap. 2.
123. Gillet A., Bull. soc. chim. (Beiges), 31, 366 (1922).
124. Prevost C., Compt. rend., 185, 132 (1927).
125. Burton H., Ingold С. K., J. Chem. Soc., 1928, 904.
126. Cm. [125]; Burton H., J. Chem. Soc., 1928, 1650; Ingold С. K., Ann. Repts. on Progress
Chem. (Chem. Soc. London), 25, 127 (1928).
127. Cook J. W., J. Chem. Soc., 1928, 2798.
128. Burton H., J. Chem. Soc., 1934, 1268.
129. De la Mare P. B. D., Vernon C. A., Studies in Chemical Structure and Mechanism,
Editor J. H. Ridd, Methuen, London, 1966, Chap. 2.
130. Braude E. A., Jones E. R. H., Stern E. S., J. Chem. Soc., 1946, 396.
131. Vernon C. A., J. Chem. Soc., 1954, 423.
132. Bunton C. A., Dahn H., Pocker Y., Chem. and Ind., 1958, 1516.
133. Goering H. L., Dilgren R. E., J. Am. Chem. Soc., 82, 5744 (1960).
134. Balfe M. P., Hills H. W., Kenyon J., Phillips H., Platt В. C., J. Chem. Soc., 1942,
556.
135. Arcus C. L., Kenyon J., J. Chem. Soc., 1938, 1912.
136. Kenyon J., Partridge S- M., Phillips H., J. Chem. Soc., 1937, 207.
137. Hughes E. D., Trans. Faraday Soc., 34, 194 (1938).
138. Catchpole A. G., Hughes E. D., Trans. Faraday Soc., 37, 629 (1941); J. Chem. Soc.,
1948, 4.
139. Carroll F. M., J. Chem. Soc., 1940, 266; Wilson C. L., Trans. Faraday Soc., 37, 631
(1941).
140. Kepner R. E., Winstein S., Young W. G., J. Am. Chem. Soc., 71, 115 (1949).
141. Dewar M. J., Bull. soc. chim. (France), 18, C43 (1951).
142. De la Mare P. B. D., England B. D., Fowden L., Hughes E. D., Ingold С. K., J. chim.
phys., 45, 236 (1948); England B. D., Hughes E. D., Nature, 168, 1002 (1957); Eng-
land B. D., J. Chem. Soc., 1955, 1615.
143. De la Mare P. B. D., Hughes E. D., Vernon C. A., Nature, 169, 672 (1952); De la
Mare P. B. D., Vernon C. A., J. Chem. Soc., 1952, 3325.
144. De la Mare P. B. D., Hughes E. D., Merriman P. C., Pichat L., Vernon C. A., J. Chem.
Soc., 1958, 2513.
145. De la Mare P. B. D., Vernon C. A., J. Chem. Soc., 1952, 3331, 3628; J. Chem. Soc..
1953 3555.
146. Hughes E. D., Trans. Faraday Soc., 37, 725 (1941); Catchpole A. G., Hughes E. D.,
Ingold С. K., J. Chem. Soc., 1948, 13.
147. Airs R. S., Balfe M. P., Kenyon J., J. Chem. Soc., 1942, 18.
148. Meisenheimer J. M., Link J., Ann., 479, 211 (1930).
149. Young W. G., Lane J. F., J. Am. Chem. Soc., 59, 2051 (1937); J. Am. Chem. Soc.,
60, 847 (1938).
150. Caserio F. F., Dennis С. E., de Wolfe R. H., Young W. G., J. Am. Chem. Soc., 77,
4182 (1955).
151. Roberts J. D., Young W- G., Winstein S., J. Am. Chem. Soc., 64 , 2157 (1942).
152. Goering H. L., Newitt T. D., Silversmith E. F., J. Am. Chem. Soc., 77, 4042
(1955).
153. Young W. G., Webb I. D., Goering H. L., J. Am. Chem. Soc., 73, 1076 (1951).
154. Stork G., White W. N., J. Am. Chem. Soc., 75, 4119 (1953); J. Am. Chem. Soc.,
78, 4609 (1956).
155. Young W. G., Winstein S., Goering H. L., J. Am. Chem. Soc., 73, 1958 (1951).
156. Goering H. L., Newitt T. D., Silversmith E. F., J. Am. Chem. Soc., 77, 5026
(1955).
157. Goering H. L., Blanchard J. P., Silversmith E. F., J. Am. Chem. Soc., 76, 5409
(1954).
158. Claisen L., Ber., 45, 3157 (1912); Ann., 418, 69 (1919); Claisen L., Eisleb 0., 401, 21
(1913); Claisen L., Tietze E., Ber., 58, 275 (1925).
159. Hurd C. D., Pollack M. A., J. Am. Chem. Soc., 60. 1905 (1938); J. Org. Chem., 3, 550
(1938).
160. Lauer W. M., Leekly R. M., J. Am. Chem. Soc., 61, 3042 (1939).
161. Claisen L., Tietze E., Ber., 58, 275 (1925); 59, 2344 (1926); Hurd C. D., Cohen F. L.,
J. Am. Chem. Soc., 53, 1917 (1931).
734
Перегруппировки ненасыщенных соединений
162. Lauer W. М., Kilburn Е., J. Am. Chem. Soc., 59, 2580 (1937).
163. Kincaid J. F., Tarbell D. S-, J. Am. Chem. Soc., 61, 3085 (1939).
164. Schuler F. W., Murphy G. W., J. Am. Chem. Soc., 72, 3155 (1950); Stein L., Mur-
phy G. W., ibid., 74, 1041 (1952); Frey H. M., Pope В. M., J. Chem. Soc., 1966B,
209.
165. Hurd C. D., Schmerling L., J. Am. Chem. Soc., 59, 107 (1937).
166. Hurd C. D., Pollak M. A., J. Org. Chem., 3, 550 (1937).
167. Lauer W. M., Filbert W. F., J. Am. Chem. Soc., 58, 1388 (1936).
168. Alexander E. R., Kluiber R. W., J. Am. Chem. Soc., 73, 4304 (1951).
169. Goering H. L-, Jacobson R. R., J. Am. Chem. Soc., 80, 3277 (1958).
170. Marwell E. N., Stephenson J. L., J. Org. Chem., 25, 676 (1960); Marwell E. N.,.
Stephenson J. L., Ong J., J. Am. Chem. Soc., 87, 1267 (1965).
171. Masson 0., Hornhardt H., Diederichsen J., Ber., 72, 100 (1939); Masson 0., Diederich-
sen J., ibid., 1523; Ryan J. P., O’Connor P. R., J. Am. Chem. Soc., 74, 5866 (1952);
Conroy H., Firestone R. A., ibid., 75, 2530 (1953); Rhoads S. J., Raulins R.,
Reynolds R. D., ibid., 2531.
172. Tarbell D. S., Kincaid J. F., J. Am. Chem. Soc., 62, 728 (1940).
173. Schmid K., Haegele W., Schmid H., Experientia, 9 , 414 (1953); Helv. Chim. Actar
37, 1080 (1954).
174. Marvell E. N., Teranishi R., J. Am. Chem. Soc., 76, 6165 (1954); Curton D. У., John-
son H. W., Jr., ibid., 78, 2611 (1956).
175. Conroy H., Firestone R. A., J. Am. Chem. Soc., 75, 2550 (1953); J. Am. Chem. Soc.,
78, 2290 (1956).
176. Von Kalberer F., Schmid K., Schmid H., Helv. Chim. Acta, 39, 555 (1956).
177. Cope A. C., Hardy E. M., J. Am. Chem. Soc., 62, 441 (1940); Cope A. C., Hoyle К. E.,
Heyl D., ibid., 63, 1843 (1941); Cope A. C., Hardy E. M., Horstmann С. M., ibid.,
p. 1852; Whyte D. E., Cope A- C., ibid., 65, 1999 (1943); Levy H., Cope A. C., ibid.,
66, 1684 (1944); Forster E. G., Cope A. C., Daniels F., J. Am. Chem. Soc.,
69, 1893 (1947).
178. Doering W7. von E., Roth W. R., Tetrahedron, 18, 67 (1962).
179. Hoffmann R., Woodward R. B., J. Am. Chem. Soc., 87, 4389 (1965).
180. Vogel E., Ann., 615, 1 (1958).
181. Vogel E., Angew. Chem., 72, 21 (1960); Vogel E., Ott К. H., Ann., 644, 172
(1961).
182. Woodward R. B., Hoffmann R., J. Am. Chem. Soc., 87, 395 (1965).
183. Oosterhoff L. J., Havinga E., Schlatmann J. L. M. A., Tetrahedron, 16, 151
(1961).
184. Vogel E., Ann., 615, 14 (1958).
185. Criegee R., Noll K., Ann., 627, 1 (1959).
186. Vogel E., Marvell E., частное сообщение; см. [182].
187. Havinga E., Schlatmann L. L. M. A., Tetrahedron, 16, 146 (1961).
188. Fonken G. J., Tetrahedron, 1962, 549.
189. Havinga E., de Kock R. J., Rappolatt M. P., Tetrahedron, 11, 276 (1960).
190. Criegee R., Furrer H., Chem. Ber., 97, 2949 (1964).
191. Farmer E. H., Ingold С. K., J. Chem. Soc., 117, 1302 (1920); Farmer E. H., Ingold С. K.,
Thorpe J. F., ibid., 121, 128 (1922).
192. Perkin W. H., Jr., Thorpe J. F., J. Chem. Soc., 79, 729 (1901).
193. Toivonen N. J., Ann., 419, 176 (1919).
194. Braumann J., Ellis L. E., van Tamelin E. E., J. Am. Chem. Soc., 88, 846 (1966).
195. Ingold С. K., J. Chem. Soc., 121, 1143 (1922).
196. Berkhoff С. E., Cookson R. C., Hudee J., Williams R. 0., Proc. Chem. Soc., 1961.
312.
197. Von Tamelen E. E., Pappas S. P., J. Am. Chem. Soc., 85, 3297 (1963).
198. Watts L., Fitzpatrick J. D., Pettit R., J. Am. Chem. Soc., 87, 3253 (1965); Burt G. D.,
Pettit R., Chem. Comm., 1965, 517.
199. Viehe H. G., Merenyi R., Oth J. F. M., Senders J. R., Valange P., Angew. Chem. inter-
net. Edit., 3, 755 (1964); Viehe H. G., Angew. Chem. internet. Edit.. 4,
746 (1965).
200. Pappas S. P., van Tamelen E. E., J. Am. Chem. Soc., 84, 3789 (1962).
201. Wilzbach К. E., Kaplan L., J. Am. Chem. Soc., 87, 4004 (1965).
202. Wilzbach К. E., Kaplan L., J. Am. Chem. Soc., 86, 2307 (1964); Wilzbach К. E.,
Kaplan L., Brown W. G., Yan S. S., ibid., 87, 675 (1965).
203. Doering W. von E., Lambert J. B., Tetrahedron, 19, 1989 (1963).
204. Anet F. 4. L., J. Am. Chem. Soc., 86, 438 (1964); Jenson F. R., Smith L. A., ibid.,,
p. 956.
205. Ciganek E., J. Am. Chem. Soc., 87, 652 (1965).
735
Глава XI
206. Ciganek Е., J. Am. Chem. Soc., 87, 1149 (1965).
207. Vogel E., Boll W. A., Gunther H., Tetrahedron Letters, 1965, 609.
208. Giinther II., частное сообщение Шредеру [213].
209. Cope A. C., Haven A. C., Ramp F. L., Trumbull E. R., J. Am. Chem. Soc., 74, 4867
(1952).
210. Vogel E., Kiefer H., Roth W. R., Angew. Chem. internat Edit., 3, 442 (1964).
211. Huisgen R., Mietzsch F., Angew. Chem. internat. Edit., 3, 83 (1964).
212. Doering W. von E., Roth W., Tetrahedron, 19, 715 (1963).
213. Schroder G., Angew. Chem. internat. Edit., 4, 752 (1965).
214. Schroder G., Angew. Chem. internat. Edit., 2, 481 (1963); Chem. Ber., 97, 3130, 3140
(1964); Merenyi R., Oth J. F. M., Schroder G., ibid., p. 3150.
215. Gilles J. M., Oth J. F. M., неопубликованная работа, цитируется в работе Шреде-
ра [213].
ГЛАВА XII
ПЕРЕГРУППИРОВКИ В АРОМАТИЧЕСКОМ РЯДУ
1. Электрофильные перегруппировки ароматических соединений . . 738
2. Нуклеофильные перегруппировки ароматических соединений . . 748
3. Внутримолекулярные перегруппировки ароматических соедине-
ний ......................................................751
Список литературы...................................779
47-0772
1. Электрофильные перегруппировки ароматических
соединений
В настоящей главе рассматриваются главным
следующих типов:
Кислота
>—NRX -------->
и
где Х = С1, Вг, I, N2Ar, NO, алкил, ОН, NO2i
образом перегруппировки
NRAr.
Vox —™ S-ОН и
— ОН
где Х = алкил.
Очевидно, существует большое число таких перегруппировок, и приведенный
выше перечень реакций можно расширить. Несомненно также, что все эти
реакции формально очень сходны между собой: заместитель X из боковой
цепи замещает водород ароматического кольца, а водород из кольца переходит
в боковую цепь; в исходной молекуле между мигрирующей группой и кольцом
находится лишь один атом, обладающий неподеленными электронами; миг
рирующая группа переходит только в орто- или иара-положение, и все эти
реакции катализируются кислотами.
Анализируя механизм этих реакций, можно заметить тем не менее, что они
не во всем столь схожи между собой, как это кажется на первый взгляд.
Первое их различие состоит в типе полярности. Совершенно ясно, что во
многих перегруппировках такого типа мигрирующая группа уходит из боко-
вой цепи в виде электрофильной частицы без электронов, которыми она была
связана в боковой цепи, и связывается с ароматическим атомом углерода
при помощи электронов, которые предоставляются этим атомом углерода.
Мы будем называть такие реакции электрофильными перегруппировками
ароматических соединений, а указанное свойство таких реакций рассматри-
вать как их главный классификационный признак. Эти реакции обсуждаются
в настоящем разделе.
Среди перечисленных выше перегруппировок имеется другой класс: мигри-
рующие группы переходят в виде нуклеофильных частиц, уносящих с собой
электроны, которыми они были связаны в боковой цепи, и используют эти
электроны для образования связи с атомом углерода ароматического кольца.
Такие реакции мы будем называть нуклеофильными перегруппировками аро-
матических соединений', они будут рассмотрены в разд. 2.
Названные выше перегруппировки часто, но не вполне логично относят
к «межмолекулярным» перегруппировкам. Это делают для того, чтобы отли-
чить их от внутримолекулярных перегруппировок ароматичееких соединений,
которые будут рассмотрены в разд. 3. Последние протекают через цикличе-
ское переходное состояние, в котором невозможно определить, каким образом
перемещаются электроны: гетеролитически парами или гомолитически с нару-
шением пар и созданием новых. В самом деле, согласно принципу неопре-
деленности, невозможность познания пути перемещения электрона является
одним из законов природы, вследствие которого циклическое переходное
состояние настолько устойчиво, что возможна внутримолекулярная пере-
группировка. Таким образом, классификация внутримолекулярных пере-
группировок на основе признаков исключительной электрофильности и нук-
леофильности или даже гетеролитичности и гомолитичности не имеет физи-
ческого смысла: типично внутримолекулярная реакция будет обладать всеми
738
Перегруппировки в ароматическом ряду
этими признаками, хотя могут быть случаи, когда один из них оказывается
преобладающим.
Сделав эти предварительные замечания, можно перейти к основному пред-
мету обсуждения — электрофильным перегруппировкам ароматических сое-
динений.
а. Перегруппировки галогенаминов по Ортону. Типичным примером
такой перегруппировки может служить превращение N-хлорацетанилида
в смесь о- и и-хлорацетанилидов в присутствии соляной кислоты и, как пра-
вило, в таких гидроксилсодержащих растворителях, как уксусная кислота,
вода или водная уксусная кислота [1].
/С1 нс1 /Н
CeH5N< ----------> о- и n-ClCeH4N<
хсосн3 \сосн3
До 1909 г. перемещение галогена из боковой цепи в ядро, примером которого
служит приведенная реакция, относили к чисто внутримолекулярным пере-
группировкам. Но в 1909 г. Ортон и Джонс [2] развили иной взгляд на эту
реакцию, согласно которому сначала происходит обратимый ацидолиз
N-хлорсоединения с образованием ацетанилида и элементарного хлора, а затем
злементарный хлор атакует ацетанилид по обычной схеме С-хлорирования
ароматических соединений:
/С1 (1) /Н (з)
CeHsN< + НС1 CeHsN< + С12
хсосн3 7^7 \сосн3
/И
—» о- и n-ClCeH4N< + НС1
ХСОСН3
Такой взгляд на механизм реакции сохранил свое значение и до сегодняшнего
дня, хотя часто подвергался критике. Вместе с тем он нуждается в некоторых
дополнениях.
В ранних работах Ортона и Джонса было показано, что наблюдаемые изо-
мерные превращения специфически катализируются соляной кислотой,
а общий кислотный катализ в этих условиях почти не проявляется. Из рас-
твора, в котором проводили перегруппировку N-хлорацетанилида, были выде-
лены ацетанилид и элементарный хлор. При переходе от N-хлорацетанилида
к его производным с дезактивированным кольцом (например, к N-хлор-
2,4-дихлорацетанилиду) было найдено, что выделяющийся хлор можно
использовать для хлорирования некоторых более реакционноспособных
ароматических соединений, например ацетанилида или анизола. В дальней-
шем было показано [3], что в одном и том же растворителе и при одной и той
же температуре соотношение орто- и пара-изомеров хлорацетанилида в про-
дуктах реакции получается одинаковым независимо от тоге, исходят ли из
N-хлорацетанилида и соляной кислоты или из ацетанилида и хлора.
Здесь появляется одна тонкость. Механизм реакции Ортона можно разделить
на отдельные стадии (1), (2), (3) и добавить к ним гипотетическую внутримо-
лекулярную перегруппировку (4).
CeHsNClCOCH3
(1)
CtH5NHCOCH3
+ НСК_ (4)
(2) (о-, n-)ClC.H4NHCOCH3 + НС1
+ Ch (3)
Если бы равновесие (1) — (2) всегда устанавливалось значительно быстрее,
чем происходила изомеризация, то невозможно было бы определить, по како-
47* 739
Глава XII
му из двух путей — (3) или (4) — образуется конечный продукт изомериза-
ции. Реакции (4) соответствуют такие же исходные соединения, продукты
и кинетические закономерности, как и последовательности реакций (1) + (3),
реакции (3) соответствуют те же реагенты, продукты и кинетические зако-
номерности, что и последовательности реакций (2) (4). Однако реакции (1)
и (2), как правило, протекают не очень быстро по сравнению с реакцией (3).
Положение равновесия (1)—(2) сильно зависит от природы растворителя:
вводе стабильной системой является C6H5NC1COCH3 -j- НС1, что безусловно
объясняется сильной ионной сольватацией НС1; в уксусной кислоте ста-
бильной системой является CGH5NHCOCH3 -|- С12. Скорость реакций (3)
можно изменять в широких пределах путем введения заместителей в арома-
тическое кольцо.
В своей первой работе Ортон и Джонс показали, что в водном растворе
уксусной кислоты при концентрации последней менее 65% скорость изоме-
ризации N-хлорацетанилида меньше скорости С-хлорирования ацетанилида
хлором. Это свидетельствует о том, что общая скорость процесса, по крайней
мере частично, определяется скоростью реакции (1). Участие внутримоле-
кулярной реакции (4) в общем процессе привело бы к тому, что общая скорость
изомеризации стала бы больше скорости хлорирования. Впоследствии Сопер
[4] показал, что в воде в качестве растворителя реакция (1) становится пол-
ностью лимитирующей скорость, причем скорость изомеризации N-хлор-
ацетанилида в присутствии соляной кислоты точно равна скорости образо-
вания хлора. Ортон, Сопер и Уильямс [5] установили, что для некоторых
замещенных в кольце ацетанилидов, а именно о-, м- и п-хлорацетанилидов,
и-бромацетанилида и ацето-о- и ацето-тг-толуидинов, можно измерить скоро-
сти реакций (2) и (3) в данном растворителе; скорость реакции (1) в 40%-ной
водной уксусной кислоте по сравнению со скоростями реакций (2) и (3)
пренебрежимо мала. Далее, было показано, что если исходными веществами
служат ацетанилид и хлор, то в любой момент времени соотношение между
выходами N- и С-хлорированных продуктов постоянно. Это удовлетворяет
критерию Вегшейдера для двух одновременно протекающих реакций одина-
кового порядка. Одновременно протекают реакции (2) и (3), однако реак-
ция (2) отличается тем, что за ней следует реакция (4). Таким образом, в слу-
чае реакции Ортона перегруппировка хлорамина в гидроксилсодержащем
растворителе осуществляется по межмолекулярному (1) — (2) —(3), а не внут-
римолекулярному (4) механизму.
Эти представления были подтверждены Олсоном и сотр. [6], которые изу-
чали изомеризацию N-хлорацетанилида в присутствии соляной кислоты
с радиоактивным изотопом хлора и установили, что общая радиоактивность
хлора, связанного с ароматическим кольцом, примерно соответствует радио-
активности неорганического хлора реакционной среды.
Как уже отмечалось, соляная кислота является специфическим катализа-
тором изомерного превращения N-хлорацетанилида. Однако аналогичные
превращения вызывают и другие галогеноводородные кислоты: бромистово-
дородная кислота дает о- и и-бромацетанилиды, а иодистоводородная кисло-
та — о- и тг-иодацетанилиды [7]. Эти превращения можно объяснить на осно-
ве механизма Ортона, согласно которому промежуточно должны получаться
хлористый бром и хлористый иод. Эти соединения участвуют затем в реакции
в качестве бромирующего и иодирующего агентов соответственно.
Из реакций (1), (2) и (3), входящих в механизм Ортона, реакция (3) вряд
ли нуждается в комментариях. Реакция С-хлорирования ароматических сое-
динений была рассмотрена в гл. VI, разд. 6, а. Известно, что в молекулярном
хлоре сочетается термодинамическая стабильность с электрофильной реак-
740
Перегруппировки в ароматическом ряду
ционной способностью, что делает его весьма эффективным хлорирующим
агентом. Поэтому, если хотят изучить хлорирование такими реагентами,
как С1+ или СЮЩ, специфически более реакционноспособными, чем моле-
кулярный хлор, хотя и менее устойчивыми, необходимо принять все меры
к тому, чтобы из реакционной среды были удалены даже следы молекулярного
хлора. Эффективность хлора как хлорирующего агента является, по нашему
мнению, одним из доводов в пользу приведенного выше механизма перегруп-
пировки хлораминов по Ортону.
Однако реакция (1) нуждается в дополнительных комментариях. Из нося-
щего качественный характер факта специфического действия соляной кислоты
как катализатора следует, что соляная кислота вступает в реакцию в виде
ионов. Этот вывод кажется правдоподобным, так как большинство реакций
изучалось в сильно разбавленных водных растворах. Такой вывод подтверж-
дается кинетическими данными: реакция имеет третий порядок [8], т. е.
первый по хлорамину и второй по соляной кислоте или, если отдать пред-
почтение ионной интерпретации, первый по хлорамину, первый по иону во-
дорода и первый по иону хлора:
'Скорость ос р<лорамин][НС1]2 ос [Хлорамин][Н+][С1~]
Ричардсон и Сопер [7] привели следующее подтверждение ионной интерпре-
тации механизма Ортона. Они исследовали кинетику превращения N-хлор-
ацетанилида в о- и n-бромацетанилиды под действием бромистого водорода
в сильно разбавленных водных растворах и при условиях, в которых опреде-
ляющей ск орость оказывается реакция (1). Реакция (1) в данном случае
ведет к о бразованию хлористого брома, а реакция (3) бромирования арома-
тического кольца с выделением соляной кислоты протекает мгновенно.
Медленно 'Бистро
iC6H4NCICOCH3+ НВг ------> СвЩКНСОСНз+ВгСР-------—>
BrC6H4NHCOCH3+HCl
Скорость этой реакции записывается в виде выражения
Скорость <х [Хлорамин][Н+][Вг~],
которое исключает любую альтернативную молекулярную форму, так как
протоны поставляют обе кислоты, в то время как чрезвычайно реакционно-
способный анион, а именно анион брома, дает только одна кислота.
Исходя из вышесказанного можно рассматривать (частное сообщение Хьюза)
реакцию (1) как бимолекулярное нуклеофильное замещение у атома хлора
в ионе хлораммония галогенид-ионов, т. е. замещение 8х2-типа в ониевой
соли, но у хлора, а не у углерода.
Hal- + Cl-^-NH(CHaCO)Ar —> Hal—Cl + NH(CHsCO)Ar
Если принять эти представления, то, согласно принципу микроскопической
обратимости, реакцию (2) можно рассматривать как бимолекулярное нуклео-
фильное замещение одного атома галогена в молекуле галогена молекулой
ацетанилида.
Реакцию (1), следовательно, нельзя считать, как это иногда делают, гидро-
лизом. Однако Сопер и Прайд [8] показали, что в водных растворах в незна-
чительной степени протекает побочная реакция гидролитического характера;
при этом образуется хлорноватистая кислота, которая реагирует с присут-
ствующей в растворе соляной кислотой с образованием хлора. Доказатель-
ством этого Сопер и Прайд считают тот факт, что скорость ацидолитического
741
Глава XII
замещения галогена в N-хлорацетанилиде в водных кислотах, помимо соля-
ной, возрастает с увеличением силы кислоты и одинакова в случае таких
сильных кислот, как азотная, серная и хлорная. Ясно, что эта реакция про-
текает с участием иона водорода, в то время как анионы сильных кислот уча-
стия в ней не принимают. Однако, если рассматривать реакцию в присутствии
соляной кислоты, то следует принять во внимание образование хлора, хотя
его количество не превышает нескольких процентов. Механизм этого срав-
нительно медленного гидролитического процесса не известен. Как и в пре-
дыдущем случае, он, по-видимому, представляет собой замещение типа Sn2
в хлораммониевом ионе, причем замещающим агентом является вода:
Н2О + С1-^Н(СН3СС^Аг-> Н2б—Cl + NH<PH3CO)Ar
Возможно также и замещение типа SN1
Cl-GlH(CH3C(Mr -»Cl + NH(CH3CO)Ar
H2O + Cl — H26—Cl
Все же следует считать, что соляная кислота является специфическим ката-
лизатором перегруппировки не только потому, что ацидолиз в присутствии
хлорид-иона протекает значительно быстрее, чем гидролиз в присутствии
воды, но и потому, что при полном отсутствии ионов хлора образуется хлор-
новатистая кислота. Последняя представляет собой слабый хлорирующий
агент. Равновесие ее превращения в активную катионную форму с образова-
нием хлорирующего агента, сравнимого по эффективности с хлором, полу-
ченным из хлорид-ионов, сдвинуто влево.
В 1912 г. Ортон [2] писал о перегруппировке хлораминов: «Несмотря на то,
что возможность внутримолекулярного превращения при определенных
условиях еще не доказана, не следует полагать, что такая возможность исклю-
чена». Ортон был одним из первых исследователей механизма реакции; он
отвергал представления, разделяемые некоторыми современными авторами,
о том, что нет реакций, протекающих более чем по одному механизму.
Взгляды на механизм перегруппировки хлораминов остаются до сегод-
няшнего дня почти такими же, как они были сформулированы в свое время
Ортоном.
Белл [9] на примерах N-хлорацетанилида, N-бромацетанилида, N-бромбен-
занилида и N-иодформанилида привел доказательства того, что перегруппи-
ровки галогенаминов в таких апротонных растворителях, как хлорбензол,
представляют собой одностадийные внутримолекулярные процессы, проте-
кающие в условиях общего кислотного катализа, причем наблюдаемую ско-
рость реакции нельзя связать с аналитической концентрацией галогена
в системе. Сопер с сотр. [10] дал другое объяснение, предположив, что реакция
идет через ацилгипогалогенит НаЮСОСН3, который несомненно образуется
в этих условиях и является хорошим галогенирующим агентом. Несколько
раз высказывался по поводу этой проблемы Дьюар, который сначала разде-
лял точку зрения Белла [И], а позднее — Сопера [12]. Но до сих пор вопрос
находится в том состоянии, в каком его оставил Ортон: внутримолекулярная
перегруппировка является возможной.
Не исключена также возможность существования других механизмов реак-
ции, отличных от предложенного Ортоном, даже если точно не известно,
каковы они. Например, можно допустить существование некоторого гомо-
742
Перегруппировки в ароматическом ряду
литического механизма, так как известно, что под действием света происходит •
ускорение перегруппировки N-хлорацетанилида [13].
б. Перегруппировки диазоаминосоединений. Типичной иллюстрацией
такой перегруппировки может служить превращение диазоаминобензола
в п-аминоазобензол
HG1 или
C6H6N =N -NHC6H5 ----- „ n-NH2CeH4N=NC6H5
JtICl-J-CeHsIN rig
Реакцию можно провести, например, под действием спиртового раствора
соляной кислоты или лучше анилина вместе с солянокислым анилином
или другими солями анилина [14]. В большинстве примеров азогруппа мигри-
рует в пара-положение. Миграция в ортпо-положение происходит труднее,
но она протекает, если пара-положение занято.
Несмотря на то что эта реакция исследована менее подробно, чем перегруп-
пировка хлораминов, противоречий в ее истории гораздо меньше, и начиная
с 1885 г. никто серьезно не сомневался в том, что диазоамино — аминоазо-
превращение представляет собой межмолекулярную реакцию. В то время
Фрисуэлл и Грин [14] предположили, что катализируемая кислотами реакция
проходит ступенчато: за ацидолизом — реакцией, обратной образованию
диазоаминосоединения, следует обычный процесс азосочетания. Механизм
зтой реакции, который будет рассмотрен ниже, оставляет открытым воп-
рос — участвует ли в ацидолизе, как и в механизме Ортона, анион катализи-
рующей кислоты. Другими словами, не известно, что является первичным
продуктом ацидолиза — ковалентная диазопсевдосоль или ионизированная
соль диазония!
(1)
CeH5N = N — NHCeH5 + HX ДД
(2)
• C6H6N = NX
It
.С6н5щ+х-
> -f- c6h5nh2
CeH6N+ + c6h5nh2 nh2c6h4n = nc6h5
Первые наблюдения, имеющие большое значение для толкования механизма
реакции, были сделаны Ницким [15], показавшим, что диазогруппа от пере-
группировывающегося диазоаминосоединения может переходить к молекуле
постороннего ароматического амина; так, обработка п-диазоаминотолуола
солянокислым анилином или о-толуидином ведет к образованию следующих
продуктов:
/СН3
СН3N = N —/ У- NН2 и СН3-^ У~ NH2
Позднее было показано [16], что посторонняя молекула, связывающая пере-
ходящую азогруппу, не обязательно должна быть ароматическим амином,
а может быть и фенолом. Из диазоаминобензола и фенола были получены
4-бензолазофенол и анилин
C8H5N=N-NHC6H5+<^ он c„h5n = n
-/ S- OH + C6H5NH2
Гольдшмидт [17] предложил модификацию механизма Фрисуэлла — Грина,
объясняющую, в частности, катализирующее влияние анилина и других
оснований в сочетании с кислотами. Влияние анилина и других оснований
отмечают все исследователи, и в соответствии с механизмом Гольдшмидта
можно предположить, что ароматические основания действуют точно так
же, как и анионы катализирующей кислоты в механизме Фрисуэлла —
743
Глава XII
Грина. Однако если анилин выполняет эту функцию, то не остается неясно-
стей в вопросе, образуется ковалетное или ионное азосоединение: оно являет-
ся ковалентным так же, как и конечный продукт. Поэтому вторая стадия
общего механизма Фрисуэлла — Грина отпадает.
NH2-^ \ +
N — NH2C6H5
II
n-c6h5
-* NH2-Z ^-N +C6H5NH24-H+
II
n-c6h5
Итак, механизм Гольдшмидта можно рассматривать как частный случай
механизма Фрисуэлла — Грина, в котором вместо галогеноводородных кис-
+
лот НХ катализатором служит ион анилиния CeH5NH3. Именно такой по су-
ществу была окончательная точка зрения Гольдшмидта. Теоретически можно
допустить, что механизм диазоаминовой перегруппировки по Фрисуэллу —
Грину совершенно аналогичен механизму перегруппировки хлораминов
по Ортону. Подобно тому как в механизме Ортона допускается, что ацидолиз
хлора включает на стадии (1) S ^-замещение хлор-ионом атома хлора в хлор-
аммониевом ионе, так и в перегруппировке диазоаминов на стадии (1) аци-
долиз азогруппы представляет собой S ^-замещение азогруппы в азоаммо-
ниевом ионе нуклеофильным сопряженным основанием.
Это положение подтверждено убедительными кинетическими исследованиями
Гольдшмидта с сотр. [17]. В качестве растворителя они использовали главным
образом анилин и некоторые другие аналогичные основания. При этом было
установлено, что наблюдается общий кислотный катализ и реакция имеет
первый порядок по диазоаминосоединению. При использовании в качестве
катализаторов таких сильных кислот, как соляная, бромистоводородная
и азотная, порядок реакции по кислоте равен примерно единице. Эти три кис-
лоты проявляют не совсем одинаковый кинетический эффект, и более тща-
тельное исследование показывает, что реакция частично имеет второй поря-
док по этим кислотам. Далее, при замене сильных кислот на слабые кислоты,
сила которых уменьшается в следующем ряду: 3,5-динитро-, о-нитро-, м-
нитро- и о-бромбензойные, порядок реакции в отношении катализирующей
кислоты возрастает (при не слишком низкой ее концентрации) и по мере
уменьшения силы кислоты приближается к двум. Все это соответствует
положению, согласно которому для образования переходного состояния
ацидолиза требуется диазоаминосоединение, протон и нуклеофильный реа-
гент. Если в качестве катализатора используют сильные кислоты со слабо
нуклеофильными анионами, то нуклеофильные функции выполняет главным
образом анилин, который служит и растворителем, а ионы галогена выполня-
ют эту функцию лишь частично. Если же катализатором служат слабые кис-
лоты с сильно нуклеофильными анионами, то нуклеофильные функции вы-
полняют в большей степени эти анионы, а не анилин. Такая интерпретация
механизма и кинетики данной реакции позволяет проводить широкие ана-
логии с хлораминовой перегруппировкой.
в. Перегруппировка нитрозаминов по Фишеру — Хеппу. Фишер и Хепп
[18] обнаружили, что некоторые ароматические нитрозамины при их обра-
ботке кислотами, в особенности соляной и бромистоводородной, перегруппи-
ровываются с образованием изомеров и переходом нитрозогруппы в арома-
тическое кольцо
нш
C6H5N(CH3)NO----> n-NOC6H4NHCH3
В простейших случаях бензольного ряда в основном образуются п-нитрозо-
соединения, в то время как Х-нитрозо-Х-алкил-2-нафтиламины перегруппи-
744
Перегруппировки в ароматическом ряду
ровываются в 1-нитрозо-Х-алкил-2-нафтиламины [19]. Реакция обычно про-
водится в спиртовом растворе в присутствии катализатора — соляной или
бромистоводородной кислот. В качестве растворителя вместо спирта можно
использовать диэтиловый эфир, уксусную кислоту и воду.
Фишер и Хепп сначала предположили, что эта изомеризация представляет
собой внутримолекулярную перегруппировку. Но в 1912 г. Фишер [20]
показал, что образование побочных галогенированных продуктов, которое
обычно наблюдается, можно объяснить действием свободного галогена,
выделяющегося при окислении катализирующей галогеноводородной кисло-
ты азотистой кислотой. Последняя также образуется в процессе реакции.
В 1913 г. Губен [21] показал, что в некоторых случаях, когда выход продукта
перегруппировки невелик, его можно значительно повысить путем добавления
в реакционную систему нитрита натрия. Значение этих наблюдений для выяс-
нения механизма перегруппировки, конечно, не велико, однако на этом
примере Губен развил теорию о том, что реакция является межмолекулярной
и протекает следующим образом: вначале происходит ацидолитическое депи-
трозирование — процесс, обратный обычному образованию нитрозамина,
а затем идет прямое нитрозирование вторичного амина в ядро одновременно
образующимся нитрозилгалогенидом или, возможно, продуктом его пре-
вращения, например азотистой кислотой:
(1) х (3)
CeH5NRNO + HX > C6H5NHR + NOX -------> NOC6H4NHR + HX
(2)
Впоследствии были получены дополнительные данные, подтверждающие эту
теорию. Небер и Раушер [22] установили, что хлористый и бромистый водо-
роды — более эффективные катализаторы, чем другие сильные кислоты.
Это подтверждает ту точку зрения, что нитрозилхлорид и нитрозилбромид,
обладая большой стабильностью в сочетании с высокой реакционной способ-
ностью, являются агентами, особенно пригодными для нитрозирования.
Было найдено также [23], что при ацидолизе ароматических нитрозаминов
в присутствии мочевины образуется не С-нитрозоизомер, а только вторичный
амин. Было опубликовано несколько сообщений о переходе нитрозогруппы
к посторонней ароматической молекуле. Так, при обработке Х-нитрозо-Х-
метиланилина хлористым водородом в растворе этилового спирта в присут-
ствии диметиланилина образуются метиланилин и п-нитрозодиметиланилин
[22]. Аналогичные продукты образуются при обработке Х-нитрозо-Х-метил-
2,4-динитроанилина хлористым водородом в эфирном растворе в присутствии
диметиланилина [24]. Для того чтобы надежно обосновать механизм реакции,
необходимо провести кинетическое исследование перегруппировки в целом
и по возможности отдельных ее стадий (1) — (3) так же, как это сделано
на примере хлораминовой перегруппировки.
г. Перегруппировка алкиланилинов по Гофману — Марциусу. Гофман
и Марциус открыли перегруппировку, состоящую в том, что бромгидраты
или хлоргидраты Х,Х-диалкил- и N-алкиланилинов при термическом раз-
ложении дают соли алкилированных в ядро вторичных или первичных произ-
водных анилина [25].
нх
C6H5NR2 ----> о- или n-RC6H4NHR
нх
C6H5NHR-----> о- или n-RCeH4NH2
Дальнейшее исследование этой перегруппировки проводил в основном Хик-
кинботтом.
745
Глава XII
Для протекания перегруппировки обычно требуется высокая температура:
около 250—300 °C, если алкильная группа является первичной, и ниже
200 °C, если алкильная группа является вторичной или третичной. Алкильные
группы мигрируют главным образом в пара-положение, а если оно занято,
то в ориго-положение; пара-миграция в незначительной степени сопровож-
дается миграцией в ориго-положение. Так, бромгидрат N-метиланилина при
нагревании перегруппировывается в смесь, содержащую соли п-толуидина
и в небольшом количестве о-толуидина. Может протекать также полиалкили-
рование, и не только путем последовательных превращений третичных аминов
через вторичные в первичные, ио и путем перегруппировки вторичных аминов.
В последнем случае должен происходить перенос алкильных групп: если
до реакции в каждой молекуле основания было по одной алкильной группе,
то после реакции некоторые молекулы не содержат ни одной, а некоторые
содержат две или более алкильных групп. Например, N-w-бутиланилин пере-
группировывается через хлоргидрат преимущественно в n-w-бутиланилин,
наряду с которым в небольших количествах образуются анилин и N-n-ди-н-
бутиланилин и в еще меньших количествах более высоко бутилированные
анилины [26]. Хиккинботтом [27] покаеал, что в процессе перегруппировки
образуются алкилгалогениды, а в случае этил- и высших алкиламинов также
олефины, которые можно выделить, естественно с низким выходом, и иденти-
фицировать. Он установил также, что в процессе перегруппировки алкильные
группы изомеризуются таким образом, как это можно было бы предположить
при промежуточном образовании карбониевых ионов. Так, если бромгидрат
N-изобутиланилина перегруппировывается в конечный продукт — п-трет-
бутиланилин, то при этом можно выделить изобутилбромид и изобутилен
[28]. При аналогичной перегруппировке N-изоамиланилина образуются изо-
амилбромид, триметилэтилен и п-тпретп-амиланилин [29]. Следует отметить,
что алкильные группы в алкилбромидах не перегруппировываются в отличие
от олефинов и алкильных групп в С-алкиланилинах, которые подвергаются
перегруппировке. Хиккинботтом [29] показал, что такой легко ионизующийся
алкилгалогенид, как трифенилметилхлорид, можно использовать для вве-
дения трифенилметильной группы в пара-положение молекулы диметилани-
лина. И наконец, Хиккинботтом установил, что различные олефины, обра-
зующиеся в процессе перегруппировки, могут при этом конденсироваться
с анилином в присутствии его бромгидрата. Например, из промежуточного
изобутилена образуется п-игреиг-бутиланилин [29].
Перегруппировка Гофмана — Марциуса долгое время считалась внутримо-
лекулярной, и эта точка зрения имеет своих сторонников до настоящего вре-
мени. Разделяя ее, Дьюар [11] считает, что эта реакция подтверждает его
теорию «л-комплекса» *. Но аргументы Дьюара нельзя считать убедитель-
ными.
Противоположную точку зрения, согласно которой реакция является меж-
молекулярной, впервые высказал Михаэль [30]. Он считал, что алкилгалоге-
ниды являются активными промежуточными продуктами, и эта идея полу-
чила поддержку [31]. Хиккинботтом предположил, что алкильная группа
отщепляется от анилиний-иона в виде иона карбония, который может всту-
пать затем в различные независимые реакции: соединяться с галогенид-
ионом, образуя алкилгалогенид, терять протон, превращаясь в олефин, или
атаковать бензольное кольцо, давая продукты перегруппировки. Карбоние-
вый ион может реагировать в перегруппированной форме, если он способен
перегруппировываться. Эта теория объясняет все факты, кроме одного, а имен-
* В последних работах эта точка зрения защищается не так безоговорочно.
746
Перегруппировки в ароматическом ряду
но: если алкильная группа перегруппировывается во время миграции, то она
могла бы остаться в неперегруппированной форме и в выделяемом алкил-
галогениде; однако и в олефине и в ге-алкиланилине она присутствует исклю-
чительно в перегрупированной форме. Хьюз (частное сообщение) предложил
механизм, который объединяет механизмы Михаэля и Хиккинботтома и соот-
ветствует общим представлениям о нуклеофильном замещении и отщеплении.
Если вспомнить, что галогенид-ион сильно нуклеофилен по отношению
к углероду, но не по отношению к водороду, то можно допустить, что раз-
ложение соли анилиния при высокой ее концентрации протекает в соответ-
ствии с механизмом SN2
C6H5NH2R + Hal —> C6H5NH2 + RHal
Это делает первую стадию перегруппировки алкиланилинов аналогичной
первой стадии перегруппировки по Ортону, а, возможно, также перегруп-
пировки диазоаминов и перегруппировки Фишера — Хеппа. Следует отме-
тить, что если R представляет собой алкильную группу, претерпевающую
внутримолекулярную перегруппировку (например, изобутил в тпретп-бутил),
то такая перегруппировка все же происходит не в молекуле алкилгалогенида.
Далее, поскольку соль анилиния при высокой температуре должна рассмат-
риваться как сильнополярная среда, предполагается, что алкилгалогенид
взаимодействует с анилином по S ^-механизму и что при этом промежуточ-
но образуется карбоний-ион, который также служит источником олефина,
образующегося, согласно этой теории, при обратимой побочной реакции
RHal R+ + Hal-
R+ + C6H5NH2 Олефин+ C6H5NHs
r++c6h5nh2 rc6h4nh2 + h+
h+4-c6h5nh2 c6h5nh3
д. Перегруппировки алкилариловых эфиров. При обработке алкилари-
ловых эфиров либо такими сильными кислотами, как серная, либо катали-
заторами реакции Фриделя — Крафтса, например хлористым алюминием,
или под действием сильных электрофильных реагентов, как правило, про-
исходит дезалкилирование эфиров, но в ряде случаев дезалкилирование
частично или полностью заменяется перегруппировкой. При этом алкильная
группа входит в ароматическое кольцо в орто- или пара-положение
Сильная
CfiH5OR--------> о- или n-RC6H4OH
6 ° кислота ° *
В указанных условиях перегруппировка эфиров с третичной алкильной
группой в качестве заместителя протекает очень легко, вторичные замести-
тели мигрируют труднее, а простейшие первичные алкильные группы пере-
ходят с трудом или совсем не переходят даже в присутствии наиболее сильных
катализаторов. Хартман и Гаттерман [32] установили, что в присутствии
хлористого алюминия анизол и фенетол дезалкилируются нацело, а изобутил-
фениловый эфир — частично, образуя ароматические продукты, содержащие,
помимо фенола, бутилфенол. Последний был идентифицирован позднее как
п-тпретп-бутилфенол [33].
Ряд обстоятельств указывает на то, что каталитическая перегруппировка
фениловых эфиров по своему механизму более или менее близка к реакции
Гофмана — Марциуса, в особенности потому, что включает образование
747
Глава XII
алкилкарбониевого иона. Во-первых, имеются опытные данные, указываю-
щие, что легче мигрируют те алкильные группы, которые образуют более
устойчивые ионы карбонил. Во-вторых, при использовании в качестве ката-
лизатора хлористого алюминия наряду с продуктами перегруппировки побоч-
но образуются только алкилхлориды; при перегруппировке 2-бутилфени-
лового эфира в растворе уксусной кислоты в присутствии серной кислоты
как катализатора побочно образуются олефины [34]. В-третьих, наблюдались
реакции переноса алкильных групп. Перегруппировка изопропилфенилового
эфира в присутствии хлористого алюминия сопровождается образованием
не только изопропилфенола, но и других фенольных продуктов — фенола
и диизопропилфенола [33, 35]. Если ту же перегруппировку проводить в при-
сутствии дифенилового эфира, образуется некоторое количество и-изопропил-
фенилфенилового эфира, а если ее проводить в избытке бензола, получается
некоторое количество изопропилбензола [33]. В-четвертых, те алкильные
группы, которые обычно легко перегруппировываются в виде карбоний-иона,
содержатся в продуктах реакции в перегруппированной форме. Так. если
2-бутилфениловый эфир при обработке хлористым алюминием дает п-2-
бутилфенол, то изобутилфениловый и шретп-бутилфениловый эфиры образуют
обычный продукт перегруппировки — и-тпретп-бутилфенол [33]. На осно-
вании вышесказанного совершенно очевидно, что в процессе катализируемой
кислотами перегруппировки О-алкильная группа уходит от атома кислорода
в виде карбониевого иона или превращается в карбониевый ион на какой-то
стадии миграции.
Алкильные группы третичных алкилфениловых эфиров переходят в кольцо
при нагревании обычно до 200—250 °C. Так, тпретп-бутилфениловый эфир
может быть термически перегруппирован в га-тпретп-бутилфенол [33], а нео-
пентилдиметилкарбинилфениловый эфир — в п-неопентилдиметилкарбипил-
фенол (СНз)зССН2С(СНз)2С6Н4ОН. В последнем случае было показано [36].
что в присутствии дифенилового эфира не происходит перехода алкильной
группы к молекуле дифенилового эфира, т. е. идет процесс, обратный тому,
который должен был бы происходить при каталитической перегруппировке:
по-видимому, пиролитическая перегруппировка протекает по иному меха-
низму. Было найдено [37], что при нагревании до 200 °C оптически активного
1-фенилэтилфенилового и 2,6-ксилилового эфиров среди прочих продуктов
образуются не полностью рацемизованные о-1-фенилэтилфенолы и и-1-фенил-
этил-2,6-ксилолы соответственно. Отсюда можно сделать вывод, что пиро-
литическая перегруппировка отчасти является внутримолекулярным процес-
сом. Однако детали этого процесса не выяснены.
2. Нуклеофильные перегруппировки ароматических
соединений
а. Перегруппировка гидроксиламинов. Бамбергер [38] впервые наблю-
дал, что при обработке фенилгидроксиламина разбавленным водным раство-
ром серной кислоты в качестве основного продукта образуется п-аминофенол.
Кислота
C6H5NHOH ------> n-H0C6H4NH2
Дальнейшее исследование этой и ряда аналогичных реакций выполнено
главным образом Бамбергером [39].
Вопрос о механизме этих процессов вызвал противоречивые толкования.
Бамбергер [40] рассматривал их как «межмолекулярные» процессы с про-
межуточным образованием соединения одновалентного азота ArN. Доводы
748
Перегруппировки в ароматическом ряду
Бамбергера будут приведены ниже, и, как мы увидим, его выводы очень
близки к современным представлениям. Когда в 1921 г. Бамбергер предложил
свою теорию, вопрос о классификации перегруппировки на основе полярности
еще не возникал. Классифицировал эту перегруппировку намного позднее,
в 1949 г., Дьюар [11], во-первых, как внутримолекулярную и, во-вторых,
как электрофильную, полагая, что она иллюстрирует его теорию «л-ком-
плекса». Доводы Дьюара состояли в том, что перенести гидроксил от арил-
гидроксиламина к иному амину или фенолу не удается и что перегруппировка
в водном растворе не сопровождается образованием перекиси водорода. Однако
доводы Дьюара нельзя считать убедительными, и они вступают в противоречие
с существующими в настоящее время представлениями *.
Современная точка зрения сформулирована Юкавой [41] в 1950 г. и состоит
в том, что перегруппировка гидроксиламинов является межмолекулярной
нуклеофильной реакцией и не имеет ничего общего с теорией «л-комплекса».
Представляя, как принято, мезомерную молекулу в виде одной валентной
структуры, эту точку зрения для случая пара-перегруппировки можно
изобразить схематически следующим образом:
х—v + + /=\ У:
___J>-NHOH2 Н— /_____2=nh
S=NH Y-Z S-NH.
Н/\=/ \=/
В самом изомерном превращении нуклеофильный реагент Y: представляет
собой молекулу воды, но, принимая во внимание связь между процессами
замещения без изомеризации и с перегруппировкой, можно полагать, что
в роли Y: может выступать любая сравнительно легко доступная и реакцион-
носпособная нуклеофильная молекула или анион. В соответствии с наблю-
даемым кислотным катализом реакции можно утверждать, что она начинает-
ся с образования ионной сопряженной кислоты арилгидроксиламина, кото-
рая изображена в форме, подвергшейся указанному гетеролизу, а не в воз-
можно более стабильной форме с дополнительным протоном у азота. Продукт
гетеролиза, представленный второй формулой, имеет заряд карбониевого
иона, который может находиться не только в пара-положении, как показано
на схеме, но и в ориго-положении, так что, используя для карбониевого иона
различные валентные структуры, можно объяснить в общем случае образо-
вание и орто-, и пара-продуктов. Переход от третьей формулы к четвертой
представляет собой обычное прототропное изменение, изображенное здесь
без расшифровки механизма. Если Y: содержит гидроксильную группу,
протон, конечно, может отщепляться от продукта, указанного на схеме
последним.
Первые две стадии приведенной выше схемы отображают гетеролиз с обра-
зованием мезомерного карбониевого иона, который затем присоединяет
нуклеофильный реагент в положение, отличное от положения первоначаль-
ного гетеролиза. Это обычная форма мономолекулярного нуклеофильного
замещения с перегруппировкой SN1'. Вполне допустимо, что при некоторых
обстоятельствах обе стадии сливаются в одну, что соответствует бимолеку-
лярному нуклеофильному замещению с перегруппировкой SN2'. Механизм
реакции в этом случае можно выразить так:
Н S- NHOH2 i Y'4></ \= NH Y NH2
\=Z
* В последних работах эти доводы опровергнуты.
749
Глава XII
Оба механизма хорошо известны как анионотропные процессы (гл. IX,
разд. 3,6 и в) и относятся к широко распространенному классу органиче-
ских реакций; перегруппировка алкилгидроксиламинов может служить
примером таких реакций в ароматическом ряду. В настоящее время нельзя
сделать выбор между двумя механизмами этих нуклеофильных реакций,
поскольку методы, позволяющие различить эти два механизма, не были
применены. Можно лишь предположить, что наиболее вероятен меха-
низм SN1'.
Имеющиеся данные в пользу этого механизма основаны на изучении про-
дуктов и кинетики реакции. Значительная часть работы по исследованию
состава продуктов реакции была проделана много лет назад главным обра-
зом Бамбергером [39]. Эти исследования были дополнены и развиты сравни-
тельно недавно Юкавой [41]. Перегруппировка фенилгидроксиламина под
действием разбавленной водной серной кислоты дает в основном ге-амино-
фенол. Однако, если для разбавления кислоты использовать этиловый спирт,
образуются о- и га-фенетидины, а при проведении реакции в растворе мети-
лового спирта — анизидины; при замене серной кислоты на соляную полу-
чаются о- и re-хлор анилины. При добавлении в реакционную среду фенола
в продуктах реакции содержатся n-HOCeH4CeH4NH2-re (и некоторое коли-
чество C6H5NHCeH4OH-n), а при добавлении анилина — небольшое количе-
ство CeH5NHCeH4NH2-n. Иногда образование n-аминофенола сопровождает-
ся также образованием эфира n-NH2CeH4OCeH4NH2-re. Тот факт, что при
перегруппировке алкилгидроксиламина в аминофенол место гидроксильной
группы могут занимать многие фрагменты, убедительно свидетельствует
о внутримолекулярном механизме реакции. Кроме того, поскольку все
фрагменты являются нуклеофильными
Y; = Н2О, С2Н5ОН, СН3ОН, С1“, С6Н5ОН, CeH5NH2, n-NH2C6H4OH
можно допустить образование активных электрофильных промежуточных
соединений.
Бамбергер провел в одинаковых условиях широкое сравнительное иссле-
дование продуктов, получающихся из арилгидроксиламинов и соответствую-
щих арилазидов при отщеплении последними азота. Он обнаружил, что
состав продуктов обеих реакций в общем (что поразительно) один и тот же, что
заставило его предположить существование одинакового промежуточного сое-
динения, а именно соединения одновалентного азота ArN. Если предполо-
жить, что ArN присоединяет дополнительный протон, о чем свидетельствуют
+
кинетические данные, то образуется ArNH, представляющий собой одну
из валентных структур мезомерного карбониевого иона, существование кото-
рого как активного электрофильного промежуточного продукта было допу-
щено ранее.
Как показал Бамбергер, перегруппировка я-толилгидроксиламина в водном
растворе кислоты при низкой температуре приводит к образованию метил-
имииохинола и метилхинола
СНзч/=х
но/\=/
= NH
и продуктов их дальнейшего превращения. В этом случае дальнейшие реак-
ции не происходят, так как конечное прототропное превращение из-за нали-
чия метильной группы невозможно.
Следует упомянуть, что среди продуктов, получаемых из фенилгидроксил-
амина, Бамбергер обычно находил анилин и азоксибензол. Однако эти веще-
750
Перегруппировки в ароматическом ряду
ства образуются, по-видимому, в результате побочного окислительно-вос-
становительного превращения фенилгидроксиламина.
На основе результатов кинетического исследования реакции установлено
[42], что окислительно-восстановительное превращение представляет собой
цепную реакцию, легко начинающуюся при кратковременном соприкос-
новении с атмосферным кислородом и легко приостанавливающуюся, если
такое соприкосновение с кислородом отсутствует. Катализируемая кислотой
перегруппировка фенилгидроксиламина представляет собой совершенно иную
реакцию, не зависящую от присутствия кислорода или других окислителей.
Кинетическое исследование показало также, что скорость перегруппировки:
зависит от концентрации сопряженной кислоты фенилгидроксиламина.
При низкой кислотности скорость реакции пропорциональна кислотности,
но эта пропорциональность исчезает, как только количество кислоты оказы-
вается достаточным для почти полной ионизации основания.
По-видимому, эта реакция нуждается в дальнейшем исследовании, хотя
вполне вероятно, что общий характер перегруппировки арилгидроксиламинов.
изложен правильно.
3. Внутримолекулярные перегруппировки ароматических
соединений
а. Перегруппировка нитроаминов. Бамбергер [43] установил, что фенил-
нитроамин, фенилметилнитроамин и подобные арилнитроамины при обработке
водными растворами сильных кислот или хлористым водородом в среде орга-
нического растворителя перегруппировываются с образованием главным
образом о-нитроанилина или его производных с небольшой примесью п-
нитроанилина и его производных
NHNO2 nh2
Z\ /\__JJ 0
| || Кисвюта । || u2
Учитывая результаты параллельных серий исследований, показывающие,
что обработка первичных и вторичных аминов нейтральными или не сильно
кислыми нитрующими агентами, такими, как пятиокись азота, часто приводит
к N-нитрованию, Бамбергер [44] предположил, что С-нитрование этих аминов
сильно кислыми нитрующими агентами состоит в N-нитровании с последую-
щей катализируемой кислотами внутримолекулярной перегруппировкой
N-нитросоединения в С-нитросоединение.
В настоящее время, очевидно, нет необходимости в такой гипотезе так назы-
ваемого «непрямого нитрования». На основании тех же фактов можно было
бы в равной мере выдвинуть другие гипотезы, аналогичные предложенным
для хлораминов или диазоамино — аминоазо-перегруппировки. Например,
нитроаминовую перегруппировку можно рассматривать не как внутримо-
лекулярную, а как одну из так называемых «межмолекулярных перегруппи-
ровок», представляющую собой сложный процесс, включающий ацидолиз
N-нитрогруппы с образованием нитрующего агента, за которым следует обыч-
ный процесс нитрования ароматического соединения образовавшимся нит-
рующим агентом. В рамках приводимой ниже схемы вместо предположения
о «непрямом нитровании», т. е. вместо того, чтобы считать, что реакция (3)
в действительности представляет собой последовательность реакций (2) + (4),
751
Глава XII
Бамбергер мог бы предположить «межмолекулярную перегруппировку»,
т. е. считать, что реакция (4) является результатом двух реакций (1) + (3).
C6H6NHNO2
(1) (2) +
C6H6NH2 + NO2X-'"'"’(3)
Можно с одинаковой вероятностью допустить третью альтернативную гипо-
тезу, считая, что первые две неверны и что реакции (3) и (4) являются неза-
висимыми, а реакции (1) и (2) вообще не участвуют в этом процессе.
Ортон пытался проверить вторую гипотезу, согласно которой изомеризация
нитроамина может считаться «межмолекулярной перегруппировкой», т. е.
гипотезу о том, что реакция (4) представляет собой последовательность реак-
ций (1) + (3). При этом Ортон руководствовался результатами собственного
исследования хлораминовой перегруппировки (разд. 1, а). Однако Ортон
и сотр. [45] очень быстро нашли, что нитроаминовая перегруппировка явля-
ется совершенно иным процессом. Они отметили, что изомеризация нитро-
аминов катализируется любой сильной кислотой, но главный их вывод состо-
ял в том, что хотя нитроамины в особых случаях в кислой среде и могут нит-
ровать другие ароматические соединения, действуя как нитрующие агенты,
в данных условиях «в системе обычно не образуются нитрующие агенты, в то
время как нитроамины претерпевают изомеризацию».
Впоследствии Хьюз и Джонс [46] исследовали этот вопрос на двух примерах
и получили дополнительные данные, которые дают материал для дальнейшей
проверки предположений Ортона. Первым примером служила перегруппи-
ровка п-нитрофенил-Х-метилнитроамина в 2,4-динитро-Х-метиланилин
4-NO2CeH4N(CH3)NO2 —> 2,4-(NO2)2CeH3NHCH3
в присутствии различных по силе кислот от муравьиной до серной и в раз-
личных растворителях, включая воду, этиловый спирт, уксусную кислоту
и этиловый эфир. Однако авторы установили, что денитрования нитроамина
ни в каких условиях обнаружить нельзя, так как не образуется и-нитро-
Х-метиланилин в присутствии легко нитрующего постороннего соединения
и само это соединение не нитруется. Вторым примером служила перегруппи-
ровка 2,4-динитрофенил-Х-метиланилина
2,4-(NO2)2CeH3N(CH3)NO2 2,4,6-(NO2)3C6H2NHCH3
в 80- или 100 %-ной серной кислоте. В этих условиях денитрование нитроамина
идет легко, о чем свидетельствуют два факта. Во-первых, при добавлении
легко нитрующихся веществ, таких, как и-ксилол, фенол или диметиланилин,
можно выделить продукт денитрования — 2,4-динитро-Х-метиланилин. Во-
вторых, из продуктов реакции можно выделить продукты нитрования этих
добавленных веществ. Вместе с тем Хьюз и Джонс показали, что в условиях,
при которых перегруппировка легко протекает, ни образующиеся нитрующие
агенты, ни азотная кислота, добавленная в эквивалентном количестве, не
способны осуществить нитрование продуктов денитрования (2,4-тринитро-
Х-метиланилина). Таким образом, Хьюз и Джонс установили, что в рамках
приведенной выше схемы для первого случая реакцию (4) нельзя заменить
реакциями (1) + (3), так как слишком медленной является реакция (1),
в то время как для второго случая этого нельзя сделать потому, что если
реакция (1) идет достаточно быстро, то слишком медленной будет реакция (3).
752
Перегруппировки в ароматическом, ряду
Таблица 171
Соотношение изомеров в продуктах нитрования анилина
и перегруппировки фенилнитроамина
Количество изомера, %
13еакция
Условия
орто мета пара
Голлеман, Хартогс и Линден (1911 г.)
Нитрование CeH5NH3NO3 95%-ная водн. H2SO4, —20 °C 4 39 56
Нитрование C6H6NH3NO3, 85%-ная водн. HNO3 5 32 62
Нитрование C6H5NH3NO3, (СН3СО)2О 82 3 15
Перегруппировка CeH6NHNO2, 74%-ная водн. H2SO4, —20°C 93 1,5 3,5
Хьюз и Джонс (1950 г.)
Нитрование C6H5NH3NO3, 85%-ная водн. H2SO4, 10 °C 6 34 59
П ерегруппировка C6H5NHNO2, 85%ная води. H2SO4, 10 °C 93 0 7
Голлеман, Хартогс и ван дер Линден [47] пытались проверить гипотезу
о «непрямом» нитровании путем измерения соотношения орто-, мета- и пара-
нитропродуктов, получаемых, с одной стороны, при нитровании анилина,
а с другой стороны, при действии кислот на фенилнитроамин. Они получили
результаты, приведенные в табл. 171, и пришли к выводу, что С-нитрование
анилина не всегда протекает через предшествующее N-нитрование. Осторож-
ность такого вывода объясняется тем, что условия нитрования и перегруппи-
ровки не были одинаковыми, а соотношение изомеров, образующихся при
нитровании, зависит от условий проведения реакции. Хьюз и Джонс [46]
провели подобные исследования, в которых условия нитрования и перегруп-
пировки были одинаковыми. Их результаты, приведенные в табл. 171, не
оставляют никаких сомнений в том, что реакции (4) и (3) являются независи-
мыми.
Отсюда следует, что из трех приведенных выше гипотез следует выбрать
третью: реакции (3) и (4) являются независимыми.
На этой стадии исследований основным был вопрос, почему нитроамины всту-
пают в реакцию внутримолекулярной перегруппировки с легкостью, не при-
сущей ни одному из соединений, рассмотренных в предыдущих двух разделах
главы. Предполагали, что объяснить это можно тем же, чем и чрезвычайно
высокий выход оршо-изомеров при перегруппировке нитроаминов (точно
так же, как при перегруппировке Кляйзена): мигрирующие группы содержат
ненасыщенную трехатомную систему * *, которая может образовывать мостик
с оршо-положением. Небольшое количество пара-изомеров может получаться
в результате межмолекулярного процесса нитрования или в результате
повторного образования внутримолекулярной мостиковой связи. Браун-
штейн, Бантон и Хьюз [48] исследовали перегруппировку N-нитроанилина
в водной серной кислоте, содержащей 15Х-азотную кислоту. Азот-15 ни
в орто-, ни в пара-продуктах перегруппировки обнаружен не был, и это
позволило авторам сделать вывод, что оба изомера образуются в результате
внутримолекулярного процесса. Этот вывод подтверждает высказанное не-
сколько ранее Хьюзом мнение ** об образовании мостиковой связи нитрито-
* Мигрирующая группа в диазоаминовой перегруппировке содержит такую же систему,
но ее третий атом является а-бензоидным и, вероятно, нереакционноспособным.
* * См. пе рвое издание этой книги, стр. 505 русского перевода.
48-0772
753
Глава XII
группами [48].
Первая стадия этой схемы описывает реакцию, специфически катализируе-
мую ионом водорода. Открытым остается вопрос о времени жизни промежу-
точного Х-нитритосоединения. На стадии превращения С-нитрито- в С-
нитросоединение, сопровождающейся ароматизирующим депротонированием,
наряду с другими остается нерешенным вопрос о последовательности стадий.
Банторп, Хьюз и их сотрудники [49] установили некоторые дополнительные
закономерности в перегруппировках N-питроанилина, Х-нитро-1-нафтила-
мина и Х-нитро-Х-метил-1-нафтиламина [49]. Во всех этих случаях в основном
образуются орто- или 2-нитропродукты перегруппировки наряду с неболь-
шими количествами пара- или 4-нитропродуктов. Они доказали, что во всех
случаях специфическим катализатором перегруппировки служит ион водо-
рода, а не общая кислота. В качестве доказательства были применены оба
известных стандартных теста. Во-первых, было найдено, что скорость реак-
ции зависит от функций Гаммета /г0, а не от [Н+]. Наклон прямой зависимости
log к от —Но примерно равен 1,3; однако величина наклона, большая еди-
ницы, может быть вызвана солевым эффектом, так как в аналогичном случае
катализируемой бензидиновой перегруппировки (разд. 3, б) наклон ста-
новится нормальным при определении функции Гаммета в присутствии солей,
т. е. в условиях проведения перегруппировки. Во-вторых, было найдено,
что скорость реакции в среде дейтерированной кислоты выше. Это указывает
на то, что в предравновесии происходит захват протона, а в этой реакции
дейтрон более активен, так как D3O+ в D2O является более сильной
кислотой, чем Н3О + в Н2О. Такие же точно результаты были получены
Уайтом и сотр. [50] при исследовании перегруппировки Х-нитро-Х-метила-
нилина.
Второе наблюдение Банторпа и Хьюза, хотя и совпадает с первоначальными
данными Ортона [45] и Хьюза и Джонса [46], противоречит общим представ-
лениям, основанным, вероятно, на поведении Х-нитропроизводных поли-
нитроанилинов, таких, как пикрамид, согласно которым кислотный гете-
ролиз Х-нитрогрупп Х-нитроанилина или его простейших производных дол-
жен приводить к образованию азотной кислоты. В противоположность этому
оказалось, что образуется азотистая кислота, а также продукты взаимодей-
ствия с ней, например ионы диазония, наряду с фенолами, окрашенными про-
дуктами окисления и смолами, которые получаются в присутствии азотистой
кислоты из о- или и-аминофенола. Перегруппировка Х-нитроанилина с высо-
ким выходом происходит при довольно высокой кислотности среды (напри-
мер, в 75%-ной водной серной кислоте). При более низкой кислотности пере-
группировка идет с меньшим выходом, уменьшающимся до 40% за счет
образования продуктов отщепления боковых цепей. Реакции отщепления
боковых цепей становятся доминирующими также и при очень высокой кис-
754
Перегруппировки в ароматическом ряду
лотности среды; они сопровождаются бурным выделением окислов азота.
Согласно механизму Хьюза, нитрит-ионы могут легко отщепляться на раз-
личных промежуточных стадиях перегруппировки N-нитроанилина *.
Третий раздел работы посвящен выяснению внутримолекулярной природы
этих перегруппировок с помощью изотопа 15N в широком диапазоне изме-
нения кислотности среды, включая такие ее значения, при которых парал-
лельно протекающее отщепление боковых цепей становится уже значитель-
ным. Изотоп 15N вводили в реакционную среду в виде нитритов и нитратов,
но ни в том, ни в другом случаях он не был обнаружен в продуктах пере-
группировки.
Четвертая группа результатов, полученных этими исследователями, связана
с дейтерированием ароматического субстрата в те положения, в которые
мигрирует нитрогруппа. Дейтерирование N-нитроанилина в 2,4,6-положения
и Н-нитро-1-нафтиламина в 2,4-положения не влияло на скорость перегруп-
пировки. Отсюда следует, что конечная стадия отщепления протона и конку-
рирующее с ней орпго-пара-превращение нитритосоединения являются быст-
рыми стадиями, следующими за лимитирующей скорость стадией в механизме
Хьюза. В реакции Х-нитро-1-нафтиламина 2,4-дейтерирование сильно уско-
ряет процесс отщепления боковых цепей. Это позволяет считать, что отщепле-
ние нитрит-иона происходит в механизме Хьюза главным образом от С-
нитритов; поэтому такое элиминирование конкурирует с реакцией потери
протона, приводящей к ароматизации, и становится существенным, когда
отщепление протона затрудняется при замене его дейтроном. В реакции
N-нитроанилина орто- или пара-дейтерирование направляет перегруппи-
ровку в другое положение, причем влияние пара-дейтерирования оказы-
вается большим. Это можно понять, если учесть, что в бензольном ряду пара-
хиноидная структура более устойчива, чем opnw-хиноидная (гл. VI, разд.
4, г), и для ароматизации требуется большая энергия активации, что при-
водит к большей потере нулевой энергии и большему первичному изотоп-
ному эффекту.
Все это свидетельствует о том, что лимитирующая стадия в механизме Хьюза
находится где-то между стадией образования первой сопряженной кислоты
нитроамина и первого С-нитрита.
Известен также ряд других исследований, из которых следует, что отщепле-
ние боковых цепей, т. е. отщепление нитрогрупп от ароматического кольца,
при оптимальной кислотности среды происходит с минимальной скоростью.
Исходя из этого Банторп, Хьюз и Уильямс предположили, что введенный
в систему протон должен удерживаться в ней так долго, чтобы замедлить
отщепление нитрит-иона от С-нитритов, однако слишком высокая кислот-
ность не должна препятствовать ароматизирующему отщеплению протона из
орто- и пара-положений»
* Дьюар [И] высказался против этого механизма, жалуясь на то, что в этом механизме
предполагается очень легкое отщепление нитрита. Возможно, он не знал о доказатель-
ствах в пользу того, что лабильная нитрогруппа отщепляется в виде нитрит-иона и что
при этом образуются смолы и другие продукты, которые Дьюар предсказал на основе
механизма Хьюза; нитрогруппа отщепляется не в виде нитрониевого иона, способного
перейти в нитрат-ион (за исключением полинитросоединений). Дьюар также возражал
против постулированной нитрит — нитро-изомеризации, основываясь на том, что такая
изомеризация не обнаружена в ряду алкилнитритов. Однако вопреки этому такие превра-
щения должны осуществляться, поскольку точно известно, что нитрат-ион может соеди-
няться с карбониевым ионом, образуя или нитросоединения, или нитрит, и поэтому
обратный процесс, при котором происходит достаточная поляризация нитритной группы
в направлении образования нитрит-иона, мог бы привести к термодинамически более
стабильной нитроформе.
48* 755'
Глава XII
Были выдвинуты также другие теории перегруппировки нитроаминов.
Уайт с сотр. [50] предположил, что происходит реакция гомолиза сопряжен-
ной кислоты нитроамина в ароматический катион-радикал кислоты и моле-
кулу двуокиси азота, которые до своей рекомбинации удерживаются раство-
рителем друг около друга. Они также установили, что отщепление боковых
цепей ускоряется добавкой веществ, способных разрушать радикалы, на-
пример иода. Нитриты могут быть чувствительными к таким добавкам,
а нитраты, которые должны получаться в эквивалентном количестве из дву-
окиси азота, должны сохраняться. Однако ускорение иодом не было обна-
ружено.
Банторп с сотрудниками па разных примерах пытался обнаружить сиг-
налы электронного парамагнитного резонанса промежуточных радикалов
или установить влияние предполагаемых радикалов на кинетику радикаль-
ной цепной полимеризации. Эти попытки окончились неудачей.
Дьюар [11] применил свою теорию л-связи для объяснения этой, как и мно-
гих других, внутримолекулярной перегруппировки. В данном конкретном
случае он считал, что нитрогруппа мигрирует в виде нитроний-иона в л-
комплексе. Уход этого иона из системы должен поэтому приводить к обра-
зованию нитрата, но не нитрита, а это противоречит наблюдениям. Неясно
также, почему л-комплекс нитрониевого иона с молекулой анилина может
вести себя так различно (см. табл. 171) в зависимости от того, образуется ли
этот л-комплекс непосредственно из этих частиц или из протонированного
N-нитроанилина в одной и той же среде.
Перегруппировка нитроамипов в нейтральной среде может протекать иначе,
чем в условиях кислотного катализа. Банторп и Томас [51] получили неко-
торые сведения относительно перегруппировки Н-нитро-Н-метил-1-нафтил-
амина в нейтральной среде при температуре 100 °C и при фотохимическом
промотировании при температуре 20 °C. Механизм реакции в этих условиях
изучен пока еще недостаточно.
б. Бензидиновая перегруппировка. Гидразобензол был открыт в 1863 г.
Гофманом [52], который тогда же впервые наблюдал его превращение в бен-
зидин под действием кислот. Бензидин был уже известен Фиттигу [53], иден-
тифицировавшему его как диаминодифенил. Положение аминогрупп было
позднее установлено Шульцем [54]. Шмидт и Шульц [55] отметили, что наря-
ду с бензидином образуется в небольшом количестве второй диаминодифенил,
так называемый дифенилин, и вместе со Штрассером [56] определили поло-
жение в нем аминогрупп. Дальнейшие исследования этой и близких к пей
перегруппировок были выполнены главным образом Якобсоном [57], который
установил образование двух других продуктов изомеризации не только из
гидразобензола, но и из многих других бензоидных гидразосоединений.
Этими продуктами были два аминодифениламина, обычно называемые о-
и «-семидинами.
Под термином «бензидиновая перегруппировка» обычно объединяют ряд
перегруппировок, в которых ароматическое гидразосоедипение под действием
кислот образует либо диаминодифенилы, либо аминодифениламины в резуль-
тате орто- или «ара-сочетания двух ариламинных остатков, которые можно
рассматривать как составные части гидразосоединения. Продуктами пере-
группировки могут быть 2,2'-, 2,4'- и 4,4'-диаминодифенилы, известные
соответственно как о-бензидины, дифенилины и бензидины, а также 2- и 4-
аминодифениламины, известные как о- и «-семидины соответственно. Не все
эти продукты образуются в ходе каждой реакции, но все они были найдены
в различных известных реакциях, и, следовательно, их соотношение можно
представить в виде общей схемы, представленной ниже, в которой А и В явля-
756
Перегруппировки в ароматическом, ряду
ются заместителями:
о-Семидин
NH-C В >NH
п-Семидин
NH2 NHi
о-Бензидин
Бензидин
Наряду с приведенными процессами изомеризации может протекать диспро-
порционирование: некоторая часть гидразосоединения восстанавлива-
ется в анилиновое основание («расщепленные амины»), а некоторая часть
окисляется в азосоединение
^A^NH-NH^B "S
^A.^NH2 + NH2^ В^
\A/N=N^B/
Поскольку для протекания перегруппировки необходима кислота, реакцию
часто проводят в разбавленных водных или водно-спиртовых растворах соля-
ной или серной кислот. Можно использовать также хлористый водород
в среде органического растворителя. С такими сильными кислотами пере-
группировка обычно протекает быстро. В некоторых случаях реакция про-
ходит со слабыми кислотами, такими, как уксусная. Метод, широко приме-
няемый ввиду его удобства для анализа продуктов перегруппировки гидра-
зосоединений, состоит в восстановлении соответствующих, обычно легко
доступных азосоединений кислыми восстанавливающими агентами. При этом
получается гидразосоединение, которое тотчас же перегруппировывается,
хотя некоторое количество восстанавливаемого вещества расщепляется с об-
разованием первичных аминов в результате конкурирующей с перегруппи-
ровкой реакции.
При перегруппировке самого гидразобензола получается смесь изомеров,
состоящая на 70% из бензидина и на 30% из дифенилина. Другие изомеры
образуются в количествах, которые можно обнаружить лишь хроматогра-
фически. Среди продуктов перегруппировки соединений бензольного ряда
получается значительное количество некоторых о-бензидинов, хотя также
нередко образуются и о-семидины, дифенилины и и-семидины. В противо-
положность этому в нафталиновом ряду выделены продукты типа о-бензи-
динов, но изомеры дифенилина как основные продукты не образуются.
Значительное влияние на направление перегруппировки гидразобензолов
оказывают заместители. Они играли бы еще большую роль, если бы эти
перегруппировки не были очень «жесткими»; в них часто происходит отщеп-
ление заместителя, препятствующего перегруппировке. Группы SO3H и СО2Н
отщепляются легче, чем большинство других групп, С1 и СН3СОО — труднее,
a OR — еще труднее; группы NRCOCH3, NR2 и алкильные не отщепляются
совсем. Например, гидразобензол-4-карбоновая кислота перегруппировы-
вается с высоким выходом в бензидин с отщеплением карбоксильной группы,
в то время как 4-ацетоксигидра.зобензол превращается в бензидин только
Глава XII
с низким выходом, при этом перегруппировка направляется заместителем
главным образом в сторону образования дифенилина, в котором заместитель
сохраняется. Имеются данные, что в бензольных кольцах при перегруппи-
ровке реализуются оба вида полярности. Для групп SO3H и СО2Н, отщеп-
ляющихся в виде H2SO4 и Н2СО3, характерна потеря связевых электронов
при отщеплении. С другой стороны, группам С1 и СН3СОО, отщепляющимся
в виде НС1 и СН3СООН, свойственно сохранять свои связевые электроны
при отщеплении.
Наличие двух трудно отщепляемых заместителей в пара-положениях гид-
разобензола исключает вообще любой вид известных перегруппировок, кроме
тех, которые ведут к образованию о-семидина. Один трудно отщепляемый
заместитель в пара-положении исключает образование бензидина; в этом
случае главным продуктом перегруппировки будет дифенилин или о-семидин
в зависимости от природы пара-заместителя. и-Семидины обычно образу-
ются в небольших количествах. Наличие заместителей в гидразобензоле
в орто- или .чета-положении значительно влияет на направление перегруп-
пировки. Большой объем качественных и полуколичественных данных по
составу продуктов перегруппировки гидразобензолов был получен Якобсоном
[57]. Несмотря на необходимость осторожного подхода при интерпретации
экспериментальных данных, еще до того, как была понята сложность меха-
низма бензидиновой перегруппировки, из этих данных был сделан вывод,
что наиболее важным ориентирующим свойством заместителей является их
полярность, как и в большинстве других гетеролитических реакций арома-
тических соединений.
Вопросом о механизме бензидиновой перегруппировки стали интересоваться
еще в начале этого столетия. Труднее всего было объяснить, каким образом
молекула гидразобензола может вывернуться наизнанку, не распадаясь при
этом на части. Самые ранние теории (1903 г.) предполагали, что распад на части
фактически происходит, но вслед за этим происходит воссоединение этих
частей.
Тихвинский [58] предположил, что сначала протекает гомолиз связи N — N
гидразина. Он записывал механизм перегруппировки следующим образом:
C6H5NH CeH4NH2-HCl
| +2НС1 2( —C6H4NH2-HC1) —> |
C6H5NH CeH4NH2-HCl
Необходимость участия кислоты и ее точная роль являются в этой теории
второстепенными факторами, а наиболее существенно предположение о дис-
социации на радикалы, в которых затем радикальные центры переходят
в орто- или пара-положения. Против этой теории Якобсон выдвинул два
серьезных аргумента [57]. Во-первых, он привел работу Виланда [59] по тетра-
арилгидразинам, которые в растворе легко диссоциируют на свободные
радикалы, находящиеся в равновесии с исходным веществом, но не претер-
певают бензидиновой перегруппировки в этих условиях
(C6H5)2NN(CeH5)2 2(C6H5)2N.
Во-вторых, Якобсон указал на то, что при исследовании продуктов пере-
группировки многих несимметрично замещенных гидразосоединений АВ им
были выделены только несимметричные бензидины АВ и не было получено
симметричных бензидинов АА или ВВ. Этот аргумент впоследствии был
подтвержден на примере систем, в которых легко можно было бы обнаружить
небольшие количества симметричных бензидинов, если бы таковые полу-
чались [60, 61].
758
Перегруппировки в ароматическом ряду
Вторая теория предполагает такое расщепление NN-связи, которое в совре-
менных терминах можно интерпретировать как гетеролиз. Впервые эти
идеи были развиты Штиглицем [62] в 1903 г. в следующей форме:
C6H5NHNHC6H54-HX CeH5N <^4-XH3NCeH5
CeH5NH2+\ceH4 = NH —> H2NCeH4CeH4NH2
Смысл этой схемы становится понятным с точки зрения электронной теории
валентности, если представить себе, что сопряженные кислоты из промежу-
точно образующихся ионов одновалентного азота и двухвалентного углерода
являются лишь различными валентными структурами продукта гетеролиза
(С6Н5 = NH)+, который может, по-видимому, соединяться с другим продук-
том гетеролиза C6H5NH2
н+
CeH5NHNHCeH5 —> (CeH5 = NH)+4-CeH5N2 —* Бензидин и г. д.
Аргументы Якобсона против теории гомолитической диссоциации, даже
второй аргумент, практически не могут служить доказательством против
теории Штиглица, так как при гетеролизе несимметричного гидразобензола
АВ в наиболее благоприятном направлении образуются функционально
дополняющие друг друга фрагменты, скажем А+ и В”, которые должны сое-
диняться в несимметричный бензидин АВ. Однако в 1933 г. Кидд и автор
книги [63] исследовали перегруппировку двух очень сходных симметричных
гидразобензолов АА и ВВ в одном растворе. Продукты реакции представляли
собой смесь симметричных бензидинов АА и ВВ и не содержали несиммет-
ричного бензидина АВ, который мог бы образоваться, если бы гидразосоеди-
нения претерпевали любой вид диссоциации — гомолитический или гетеро-
литический. Таким образом, после 1933 г. стало общепризнанным, что
в процессе бензидиновой перегруппировки молекула гидразобензола не распа-
дается на фрагменты. В течение первых 30 лет после опубликования ранних
теорий перегруппировки просто не обращали внимание на трудность в пони-
мании стереохимии этой перегруппировки, которая вытекала из сущест-
вовавших теорий.
В 1933 г. на примере простой реакции было показано, что 2,2'-диметокси-
и 2,2'-диэтоксигидразобензолы
NH— NH NH —NH
II II
CH3O-/S /S-ОСНз С2Н5О-/\ /\-ОС2Н5
и и и и
в присутствии кислоты перегруппировываются в одном и том же растворе
с образованием смеси 3,3'-диметокси- и 3,3'-диэтоксибензидинов
Отсутствие в продуктах реакции несимметричного продукта перекрестного
взаимодействия 3-меток си-3'-этоксибензидина было доказано исследова-
нием диаграммы плавкости [63]. Несколько позднее Уэланд и сотр. [64]
разработали в принципе аналогичный, но более точный метод доказательства
759
Глава XII
отсутствия в продуктах перегруппировки даже следов продукта перекрест-
ного взаимодействия. Гидразобензол АА (2,2'-диметилгидразобензол) был
перегруппирован вместе с гидразобензолом А*В (2-14С-метилгидразобензол),
в котором фрагмент А*, отмеченный звездочкой, является радиоактивным.
Полученный бензидин АА (3,3'-диметилбензидин) оказался нерадиоактив-
ным. Еще позднее, после того как было установлено разнообразие механизмов
бензидиновой перегруппировки и была показана важность исследования
перегруппировки гидразонафталинов для выяснения механизма бензидино-
вой перегруппировки, Банторп [65] привел дополнительные доказательства.
Он осуществил перегруппировки парных смесей трех изомерных гидразонаф-
талинов, а именно 1,1'-, 1,2'- и 2,2'-изомеров, в одном растворе и снова не
получил продуктов перекрестного взаимодействия. Для обнаружения малых
концентраций продуктов он использовал метод бумажной хроматографии,
который позволяет обнаружить десятые доли процента вещества. После
большого числа убедительных доказательств было бы напрасным тратить
усилия на поиски очень малых следов продуктов, так как невозможно дока-
зать, что таковые образуются в результате основной реакции, а не в резуль-
тате независимой второстепенной.
В течение нескольких лет после 1933 г. проблема бензидиновой перегруппи-
ровки обсуждалась главным образом с теоретической точки зрения. Необ-
ходимо было понять, каким образом проявляется во внутримолекулярной
перегруппировке стереохимический фактор. Здесь заслуживают внимания
три теории.
Первая — теория полярного переходного состояния, предложенная в 1941 г.
Хьюзом и автором этой книги [66] и дополненная в 1946 г. с учетом поло-
жений, развитых Хаммиком и Месоном [67]. Согласно этой теории, образу-
ется единственное промежуточное соединение, имеющее одинарную бензо-
идную или хиноидную связь. Вместе с тем теория предполагает, что переход-
ное состояние имеет определенные связи сильно полярного характера. Эта
сильная электростатичность будет сравнительно сильно увеличивать длину
связей и очень сильно (по сравнению с обычной связью) уменьшать изги-
бающие силовые постоянные. Можно показать, что такие свойства этих
связей способствуют энергетически легкому резкому изменению формы моле-
кулы, достаточному для того, чтобы создать стереохимически необходимые
условия для протекания реакции. Промежуточно возникающие полярные
связи напоминают связи, промежуточно образующиеся при мономолекуляр-
ном сольволизе (гл. VII, разд. 6, г).
Другая теория, предложенная в 1946 г. Дьюаром,— это теория л-комплекса
[11]. Она была модифицирована в некоторых деталях, но основное ее поло-
жение состоит в том, что перегруппировка осуществляется через протони-
рованный л-комплекс. Этот комплекс представляет собой промежуточное
соединение и не является переходным состоянием. В комплексе перво-
начальная связь N — N заменяется делокализованной ковалентной, так
называемой л-связью с ароматическим кольцом. Эта л-связь удерживает аро-
матические кольца в параллельных плоскостях и не препятствует их относи-
тельному вращению. Продукты получаются после того, как л-связь заме-
няется локализованной межатомной связью.
Еще одна теория «клеточной диссоциации», которая неоднократно обсужда-
лась после 1950 г., впервые серьезно была обоснована лишь в 1960 г. Вечера
и сотр. [68]. Эта теория предполагает гомолитическое расщепление протони-
рованной гидразо-молекулы на протонированные радикалы, которые ока-
зываются структурно независимыми, но сохраняются в кинетически зави-
симом состоянии в «клетке» растворителя. Большая часть возражений против
760
Перегруппировки в ароматическом ряду
этой теории сводится к тому, что трудно понять отсутствие взаимодействия
со стенками клетки, точнее, множества разных клеток. Для большого числа
бензидиновых перегруппировок, исследованных во многих растворителях,
в продуктах перегруппировки не было найдено ни одного характеристичного
фрагмента растворителя. Другим возражением служит то, что чувствитель-
ные методы обнаружения промежуточных радикалов не позволили ничего
обнаружить [69]. Критика этой теории Дьюаром [11] была основана на ее
несоответствии составу продуктов перегруппировки. Теория сталкивается
и с другими трудностями, связанными, как будет показано ниже, с кинетикой
перегруппировки [70]. В настоящее время эта теория больше не поддержи-
вается Вечера [68].
Кинетика кислотной перегруппировки гидразобензола была впервые иссле-
дована ван Лооном [71] в 1904 г., который установил, что реакция имеет
второй порядок относительно кислоты:
^[Гидразобензол]/^; , , ГГ1,
---[Гидразобензол] = kl = 1
Это наблюдение долгое время оставалось забытым, пока в 1950 г. вновь не-
было сделано Хэммондом и Шайном [72]. Многие исследователи тотчас при-
нялись обобщать этот результат, и за несколько лет он был подтвержден
на ряде замещенных гидразобензолов в большом числе растворителей. В ходе-
этих исследований Карлин и Одиозо [73] обнаружили аномальную перегруп-
пировку 2,2'-диметилгидразобензола (о-гидразотолуола) в 95%-ном водном
этиловом спирте, имевшую порядок по кислоте, равный не 2, а 1,6. Тогда
начались поиски пределов применимости кинетического уравнения Лоона —
Хэммонда — Шайна. Но эта проблема решается не так легко: в самом деле,
чтобы решить ее, необходимо руководствоваться определенной теорией.
В течение почти десятилетия результат Карлина и Одиозо, согласно которому
порядок реакции по кислоте меньше двух, оставался единственным в своем
роде.
Эти данные широко обсуждались. Блекэддер и Хиншельвуд [74] увидели
в них указание на то, что бензидиновая перегруппировка в принципе может
иметь второй механизм, который кинетически соответствует первому порядку
по кислоте. Этот механизм редко наблюдался при кинетических исследова-
ниях, но в примере Карлина и Одиозо по непонятным причинам внес суще-
ственный вклад в скорость суммарной реакции
^[Гидразобензол]/^;
[ Гидразобензол]
==&!== Л2[Н+] + Л3[Н+]а
Дьюар [11] обсудил этот случай с позиций теории л-комплекса; он считал,
что реакция протекает не по двум, а по одному механизму, а именно по меха-
низму с участием л-комплекса с двумя потенциально лимитирующими ско-
рость реакции стадиями. Для образования л-комплекса требуется один протон,
а другой протон необходим для его разложения в продукты. Поэтому в зави-
симости от того, какая стадия — первая или вторая — определяет скорость
реакции, суммарный порядок по кислоте будет равен единице или двум.
Если же лимитирующими’ скорость реакции оказываются обе стадии, то кине-
тический порядок будет иметь промежуточное значение; при этом общее
уравнение для скорости реакции будет иметь вид
[Гидразобензол] _ 1 _ 1 1
^[Гидразобензол]/^; ~ /ц ~/са[Н+] ' МНГ]2
Это уравнение, суммирующее обратные скорости, имеет следствия, отлич-
ные от следствий, которые вытекают из уравнения Блекэддера — Хиншельву-
761
Глава XII
да, суммирующего просто скорости. Теория Дьюара не объясняет, почему
скорость реакции должна контролироваться в сравнимой степени нескольки-
ми стадиями и почему это проявляется в примере Карлина и Одиозо.
Банторп, Хьюз и автор книги приняли приближение Блекэддера — Хин-
шельвуда, но задались вопросом, почему пример Карлина — Одиозо остается
единственным. Более чем восьмилетние настойчивые попытки найти второй
такой же случай, когда порядок реакции по кислоте был бы меньше двух,
оказались безрезультатными. Вероятно, это можно объяснить тем, что при
попытках найти второй такой пример не пользовались теорией полярного
переходного состояния, хотя эта теория указывает, в каком случае следует
искать низкий порядок по кислоте, а именно в реакциях гидразобензолов,
имеющих заместители, которые, во-первых, смещают электроны к фенила-
миновым кольцам этих соединений и, во-вторых, ослабляют силу анилинового
основания в результате нарушения сольватации заряда * (гл. XIV, разд. 1, д).
Каждый из этих факторов мог бы способствовать поляризации протонирован-
ной связи N — N, а эта поляризация в процессе перегруппировки может
передаваться на другие связи в соответствии с теорией полярного переход-
ного состояния. Любой из этих факторов, если он достаточно значим, может
оказывать влияние без помощи другого. Однако в случае орто-метильного
заместителя оба фактора действуют совместно, и, следовательно, любой
другой заместитель, в котором оба эффекта проявятся сильнее, может
уменьшать кинетический порядок более эффективно. Таким образом, можно
написать следующий ряд убывающей эффективности арильных групп в гид-
разосоединениях:
а-нафтил > (З-пафтил > о-толил > фенил
о-анизил > о-толил > фенил
Первые члены ряда были исследованы в 1962 г., и на этих примерах был
полностью выявлен однопротонный механизм бензидиновой перегруппировки
[75]. Было показано, что перегруппировка 1,1'-гидразонафталина представ-
ляет собой реакцию строго первого порядка по кислоте при изменении кон-
центрации ионов водорода в 104 раз. Для 1,2'-гидразонафталина и 2,2'-
гидразонафталина закономерность во многом такая же, за исключением того,
что для второго соединения порядок реакции по кислоте увеличился до 1,2
в области наибольшей кислотности. Однако для Х-1-нафтил-Х'-фенилгид-
разипа и Х-2-нафтил-Х'-фенилгидразина кинетическая картина является
характерно переходной: порядок реакции по кислоте увеличивается примерно
с единицы в области низкой кислотности до почти 2 в области высокой кис-
лотности. Идентичная картина была экспериментально получена и для
примера Карлина и Одиозо с 2,2'-диметилгидразобензолом: увеличение
порядка реакции примерно с 1,3 до 2,0 с повышением кислотности. Карлин
и Одиозо получили для порядка реакции по кислоте величину 1,6, так как
работали при среднем значении кислотности среды.
Эти результаты приведены в табл. 172, в которую вошли данные Бантона
[76], Карлина [73, 77] и Шайна [78] и их сотрудников, а также большое
число данных, полученных Банторпом и сотр. [79] после 1962 г.
Последние данные подтверждают вторую из предсказанных последователь-
ностей, т. е. о-анизильная группа оказывает большее влияние в промоти-
ровании однопротонного механизма, чем о-толильная. В кинетическом смыс-
* Причина последнего эффекта (гл. XIV) состоит в том, что opmo-заместитель уменьшает
степень нейтрализации катионного заряда молекулами растворителя (вследствие элек-
трострикции диполярного растворителя), увеличивая, таким образом, сродство заряда
к внутренним электронам молекулы.
762
Перегруппировки в ароматическом ряду
Таблица 172
Кинетический порядок реакции превращения гидразобензолов по иону водорода
RNHNHR' Водный органический растворитель, % Область [11+] Порядок по Н+ Лите- рату- ра
R R'
60 диоксан ( 0,05-1,0 75
С6н5 Свн5 | 95 этиловый < 0,05-0,10 77
спирт 1
1-с10н7 1-С10Н7 60 диоксан 0,000003—0,03 1 75
1-С10Н7 2-Сц)Н7 60 диоксан 0,003—0,04 1 75
2-CjoH7 2-СюН7 60, 70 диоксап 0,001-0,2 1,0-1,2 75
1-с10н7 Сон5 60 диоксан 0,001—0,3 1,0—1,9 75
2-С10Н7 СвН5 60, 70 диоксап 0,01-0,3 1,2—1,9 75
60 диоксан ( 0,02—0,5 1,4-2,0 75
2-СН3СбН4 2-СН3С6Н4 95 этиловый < 0,03-0,1 1,6 73
спирт 1
3-СН3СвН4 з-сн3с6н4 95 этиловый 0,05—0,1 2 77
спирт
4-СН3СвН4 4-СН3С6Н4 Г 60 диоксан ( 0,005-0,07 76
< 95 этиловый s 0,007-0,03 77
спирт 1
4-mpfm-C4HgC(;H4 4-трет-С4НдСвН3 95 этиловый 0,01-0,05 2 78
спирт
2-СН3ОС6Н4 2-СН3ОСвН4 60 диоксан 0,0001—0,05 1 79
2-СН3ОС6Н4 СвН5 60 диоксан 0,002-0,3 1,1-2,0 79
4-СН3ОСвН4 СвН5 60 диоксан 0,000007—0,005 1 79
4-CH3CONHC6H4 С6н5 60 диоксан 0,007-0,1 1 79
2-FCgH4 2-FC6H4 60 диоксан 0,1—0,8 2 79
2-С1С6Н4 2-С1С6Н4 60 диоксан 0,8-2,8 2 79
4-С1С6Н4 4-С1С6Н4 60 диоксан 0,1-1,0 2 79
4-С1С6Н4 свн5 60 диоксан 0,07-1,0 2 79
2-ВгС6Н4 2-ВгС6Н4 60 диоксан 0,2-2,0 1,2—1,9 79
4-ВгС8Н4 4-ВгС6Н4 60 диоксан 0,1-0,5 2 79
2-1С6Н4 2-1С6Н4 60 диоксан 0,7—1,6 1 79
4-1С6Н4 4-1С6Н4 60 диоксан 0,05-0,5 2 79
2-С6Н5СвН4 2-С6Н5С6Н4 60 диоксап 0,9-1,6 2 79
4-С6Н5С6Н4 4-С6Н3С3Н4 95 этиловый 0,01-0,5 2 78
спирт
4-С3Н5С3Н4 С6Н5 60 диоксан 0,004-0,6 2 79
4-NO2C6H4 С6Н5 60 диоксан 2,0-4,0 2 79
ле одна о-анизильная группа действует так же, как и две о-толильные. Две о-
анизильные группы полностью обеспечивают однопротонный механизм.
Из этой же серии данных следует, что два поляризационных фактора дей-
ствуют независимо друг от друга. Единственная n-анизильная группа и един-
ственная n-ацетаминофенильная группа дают чистый однопротонный кине-
тический результат. Это объясняется, очевидно, тем, что заместители в этих
арильных группах довольно сильно отталкивают электроны от пара-поло-
жения. Это же вытекает и из теории, согласно которой, как будет показано,
пара-положение является наиболее эффективным для высвобождения элек-
763
Глава XII
тронов. Две о-бромфенильные группы дают промежуточную кинетическую'
картину, а две о-иодфенильные — чистую однопротонную картину. Это мож-
но объяснить, очевидно, тем, что при таком изменении размеров заместите-
лей происходит нарушение сольватации заряда (уменьшение силы основания)1
в том положении, которое, как мы увидим ниже, является наименее склонным
удерживать второй присоединенный протон в переходном состоянии. Во
всех случаях разного типа, в которых наибольшую ответственность за умень-
шение кинетического порядка реакций несет лишь один фактор, действие
другого фактора либо вызывает сомнение, либо слабо и противоположно
по направлению.
Рассмотрим теперь некоторые данные, которые по тем или иным причинам
не включены в табл. 172. Имеются данные о переходной кинетической картине
при перегруппировке N-метилгидразобензола в 25 %-ном водном растворе
метилового спирта в области кислотности от 0,0003 до 0,06 н. [80]. Описана
перегруппировка 4,4'-дивинилгидразобензола, катализируемая соляной кис-
лотой в 95%-ном водном этиловом спирте, которая имеет первый порядок
по кислоте [81]. Продукт этой реакции, правда, представляет собой полимер.
Кинетически обратному первому порядку по кислоте подчиняется пере-
группировка 3,3'-диаминогидразобензола в 95 %-ном водном этиловом спирте
[82]. Если допустить, что амин находится в дважды протонированной форме,
то можно считать, что перегруппировка протекает по однопротонному меха-
низму.
Кинетические исследования имеют важное значение для выяснения меха-
низма реакции [70]. Достоверно установленный факт, что в переходной обла-
сти кинетический порядок реакции по кислоте возрастает с увеличением
кислотности, со всей очевидностью вытекает из уравнения Блекэддера —
Хиншельвуда, на основе которого фактически и проводилась количественная
обработка всех кинетических результатов, полученных для переходной обла-
сти. Эти результаты не согласуются с уравнением Дьюара, выведенным из его
теории л-комплекса, согласно которому порядок реакции по кислоте должен
уменьшаться с увеличением кислотности. Более того, л-комплекс с нафта-
линовым остатком должен быть более устойчивым, чем с бензольным остатком,
и при действии протона должен превращаться в конечные продукты труднее.
Кинетический порядок по кислоте при переходе от гидразобензола к гидразо-
нафталину не должен уменьшаться, а скорее должен был бы возрасти. Теория
клеточной диссоциации также не может объяснить эти данные, поскольку
при таком механизме необходимо участие второго протона для создания
электрической симметрии и ослабления связи N — N, что требуется для
осуществления гомолиза. Объяснить с помощью этой теории легкую конку-
ренцию одно- и двухпротонного механизмов перегруппировки было бы не-
разрешимой задачей. В то же время теория полярного переходного состояния
затруднений не встречает; реакция начинается с гетеролитического ослаб-
ления N — N-связи. Этот механизм предусматривает к тому же необходи-
мость присоединения одного протона — наиболее эффективного активатора
перегруппировки. Присоединение второго протона имеет менее существен-
ное значение, так как его действие проявляется только вследствие различия
в его влиянии (в противоположных направлениях) на нарушение асимметрии
и ослабление связи. Конкуренция одно- и двухпротонного механизмов, таким
образом, легко укладывается в рамки этой теории.
Все катализирующие протоны, в том числе и второй, если реакция идет с уча-
стием двух протонов, присоединяются во время предравновесной стадии.
Это означает, что кислотный катализ является специфическим катализом,
вызываемым ионами водорода. Присоединения протона в переходном состоя-
764
Перегруппировки в ароматическом ряду
нии перегруппировки, иными словами общего кислотного катализа пере-
группировки, не происходит. Это справедливо даже в отношении присоеди-
нения второго протона для всех условий, в которых была исследована кине-
тика перегруппировки. Первое доказательство этого было дано в 1957 г.
Бантоном, Мала и автором книги [76]. Несколько позднее такие же дока-
зательства, но в значительно большем числе привел Банторп в рабо-
те [75].
Разработано два независимых метода доказательств, результаты которых
взаимно дополняют друг друга. Во-первых, показано, что скорость бензи-
диновой перегруппировки совпадает с термодинамической мерой кислотности
по Гаммету. Так, при двухпротонном катализе скорость реакции соответст-
вует квадрату функции равновесной кислотности Ло, что характерно для
равновесного переноса протона, а не квадрату концентрации [Н+], что было
бы более справедливо для чисто кинетической формы присоединения протона.
Установлено, что здесь совершенно не подходит и смешанная функция Л0[Н +].
Второе наблюдение состоит в том, что при замене катализа протоном Н +
в растворе Н2О на катализ дейтроном D + в растворе D2O реакция идет быст-
рее. Если бы присоединение протона происходило в переходном состоянии
лимитирующей стадии, то при замене протона на дейтрон проявлялся бы
первичный изотопный эффект и реакция протекала бы медленнее. Однако
изотопный эффект проявляется в предравновесной стадии, где он имеет обрат-
ную величину, поскольку дейтрон переносится от D3O+ в D2O легче, чем
протон от Н3О+ в Н2О. Наблюдаемое ускорение бензидиновой перегруппи-
ровки при переходе от протонного растворителя к дейтронному равно при-
мерно двум па каждый катализирующий протон. Обычно фактор ускорения
для однопротонной перегруппировки 1,1'-гидразонафталина равен 2,3, а для
двухпротонной перегруппировки гидразобензола составляет 4,8 = (2,2)2.
На основании вышесказанного можно сделать краткое заключение, которое
будет сводиться к тому, что теперь мы знаем, когда происходит столкновение
катализирующего протона с молекулой, вызывающее перегруппировку.
Это происходит в самом начале *. Во всех исследованных случаях реакция
начинается с присоединения первого протона, за которым часто следует
присоединение второго протона с образованием в предравновесном состоянии
первой или второй сопряженной кислоты гидразосоединения.
Вернемся теперь к отрыву протонов от перегруппировывающейся системы,
который происходит, как было установлено, на последней стадии процесса.
Для образования любой одной диарильной связи необходима потеря двух
ароматических протонов. Можно исследовать этот процесс при помощи дей-
терирования ароматического кольца в положение, по которому предполо-
жительно произойдет образование диарильной связи. Проиллюстрируем это
результатами двух исследований.
Перегруппировка гидразобензола в водно-органическом растворе с квад-
ратичной зависимостью от концентрации ионов водорода дает 73—76% бен-
зидина с присоединением в положение 4,4' и 27—24% дифенилина с присоеди-
нением в положение 2,4'. Точная картина зависит от природы растворителя,
но не зависит от кислотности среды. Установлено [84], что, когда все четыре
орто-положения гидразобензола заняты атомами дейтерия, скорость реакции
и соотношение продуктов остаются такими же. Точно так же не изменяются
* Тем не менее Дьюар [11] поддерживал идею общего кислотного катализа на стадии
присоединения второго протона вплоть до 1963 г., по скорее на основе теоретического
толкования механизма, чем на основе эксперимента. После этого теория л-комплекса
была пересмотрена и стала настолько гибкой, что, опираясь на нее, нельзя делать ника-
ких определенных прогнозов [83].
765
Глава XII
ни скорость реакции, ни соотношение продуктов при введении атомов дейте-
рия в оба пара-положения.
Вторая серия результатов относится к 1,1'-гидразонафталину и лишь отчасти
имеет отношение к предыдущей. Это соединение перегруппировывается
в водно-органическом растворе со скоростью, линейно зависящей от кон-
центрации ионов водорода, и образует три продукта, соотношение между
которыми незначительно зависит от природы растворителя и не зависит
от кислотности среды. В 60%-ном водном растворе диоксана получается
смесь продуктов, состоящая на 64% из нафтидина, связанного в положении
4,4', на 18% — из связанного в положении 2,2' диарилдиамина, называе-
мого динафтилином, и на 18% из связанного в положении 2,2' диарилимина,
которым оказывается, конечно, дибензкарбазол *.
Установлено [85], что введение атомов дейтерия в 4- и 4'-положения молекулы
исходного соединения не изменяет ни суммарной скорости перегруппировки,
ни соотношения продуктов. Однако при введении атомов дейтерия в 2- и 2'-
положения исходной молекулы суммарная скорость перегруппировки оста-
ется неизменной, а соотношение продуктов изменяется следующим образом:
количество 4,4'-присоединенного нафтидина и сумма 2,2'-присоединенных
продуктов остаются неизменными, а соотношение двух 2,2'-присоединенных
продуктов между собой, т. е. диамина и имина, изменяется в 5 раз (если
быть точным, то несколько более 5) в пользу имина. Совершенно ясно, что
в этом проявляется первичный изотопный эффект, препятствующий потере
ароматического атома дейтерия при конкуренции с реакцией отщепления
аммиака.
Нафтидин Динафтилин ДибензокарбаЗОЛ
Нафтидин, % Динафтилин, % Дибензокарбазол, %.
Без D2 64 18 18
4,4'-D2 63 18 19
2,2'-D2 64 6 30
Эти результаты иллюстрируют последовательность событий при бензидино-
вых перегруппировках. Оба ароматических протона, о которых шла речь,
отщепляются после того, как будет преодолен главный энергетический барьер,
и после того, как путь реакции разветвится по тем направлениям, которые
в конце концов приведут к образованию диарильных связей в разных поло-
жениях. Потеря обоих протонов должна происходить не только па конечной
стадии, но также и со значительно меньшей энергией активации, чем все
предшествующие стадии. Мы принимаем, что отщепление аммиака пред-
ставляет собой внутримолекулярное нуклеофильное замещение, а это озна-
чает, что один атом азота (тот, который отщепляется) протонирован, в то вре-
мя как другой (замещающий нуклеофил) не протонирован. Это означает, что
отщепление двух ароматических протонов не может происходить одновре-
менно, а должно осуществляться последовательно, а именно при отщеплении
второго протона проявляется изотопный эффект в конкуренции с отщепле-
* Показано, что имин является независимым продуктом перегруппировки и не полу-
чается из диамина; диамин может быть дезаминирован, но не в условиях этой перегруп-
пировки [75].
766
П ерегруппировки в ароматическом ряду
нием аммиака. Отщепление этого протона должно протекать с энергией акти-
вации, чтобы можно было наблюдать изотопный эффект, т. е. отщепление
этого протона происходит от истинного промежуточного соединения с энер-
гетической ямой, достаточно глубокой, чтобы содержать несколько энерге-
тических уровней.
Эти выводы относительно начальных и конечных стадий перегруппировки
можно выразить через формулы предполагаемых хиноидных промежуточных
соединений. Покажем это для однопротонной перегруппировки, приводящей
к образованию 2,2'-связанных диарилов — диамина и имина, и для двух-
протонной перегруппировки, в результате которой образуется 4,4'-связанный
диамин. Переходные состояния образуются на стадиях, отмеченных словом
«медленно», но пока не расшифровываются, так как их конфигурация будет
обсуждаться ниже.
Аг NHNH Ai-
Теперь следует выяснить, какие процессы происходят в центральной области
координаты реакции, в особенности в той ее области, где происходит обра-
зование переходного состояния. В этом определенную помощь оказы-
вает исследование кинетики и еще большую — исследование продуктов
реакции.
Наиболее важные результаты, полученные при исследовании кинетики, свя-
заны с солевыми эффектами [70]. Как отмечают почти все исследователи
кинетики бензидиновой перегруппировки, в этих реакциях проявляются
очень большие положительные солевые эффекты. Сначала они были уста-
767
Глава XII
новлены для двухпротонного механизма, но, как это стало ясно затем, они так-
же свойственны и однопротонному механизму. Солевые эффекты означают, что
переходное состояние более полярно, чем исходное. Такие большие эф-
фекты наблюдались в мономолекулярных реакциях нейтральных соединений,
которые распадаются на ионы (гл. VII, разд. 6). Но в однопротонной бензи-
диновой перегруппировке исходное состояние уже содержит ионный заряд,
а в двухпротонном механизме исходное состояние содержит два ионных заря-
да. Таким образом, соответствующие переходные состояния должны
быть намного более полярными, чем даже такие заряженные исходные
состояния.
Как раз это и предсказывает теория полярного переходного состояния. Те-
перь рассмотрим не промежуточные соединения, которые, согласно приведен-
ной выше теории, имеют тривиальную структуру с обычными бензоидной
или хиноидной связями, а переходное состояние, которое, согласно теории,
должно иметь электростатическую природу. Место локализации диполяр-
ных связей будет смещаться при переходе системы через переходное состоя-
ние, по солевые эффекты связаны именно со структурой переходного состояния.
Большая величина солевого эффекта является одним из наиболее убедитель-
ных аргументов против теории клеточной диссоциации. В соответствии с этой
теорией разделенные ионные заряды исходного состояния заменяются в пере-
ходном состоянии прилегающими друг к другу радикал-ионами, на которых
располагаются заряды. При этом должен проявляться небольшой солевой
эффект. Даже знак солевого эффекта вызывает сомнения в справедливости
клеточного механизма.
Вторым ключом к разгадке механизма, предоставляемым кинетическими ис-
следованиями, является факт влияния структуры на скорость. Некоторые
значения констант скоростей перегруппировки приведены в табл. 173. Из
значений констант скоростей к3 двухпротонных реакций видно, что п-метиль-
ные группы в гидразобензоле обладают намного большим ускоряющим
эффектом, чем это можно было бы ожидать от такого среднего по силе поляр-
ности заместителя, и намного большим, чем это наблюдается тогда, когда
метильный заместитель занимает либо мета- либо орто-положение. Это
означает, что в переходном состоянии двухпротонного механизма катионный
заряд концентрируется на пара-углеродном атоме, который, подобно карбо-
ниевому иону, более стабилен, будучи третичным, чем вторичным. Такого же
рода различия в скоростях реакций наблюдаются для соединений, имеющих
галогензаместители в пара- или орто-положениях, которые, несмотря на свою
индуктивную электроотрицательность, вызывают электроположительный
эффект сопряжения.
Когда однопротонный механизм становится преобладающим или доминиру-
ющим вследствие сильного отталкивания электронов заместителями, изме-
нение кинетической формы связано с повышением абсолютной скорости, как
это наблюдается при наличии бензо-, n-ацетоамино- и о- или п-метоксизаме-
стителей. Если доминирующим фактором, вызывающим изменение кинетиче-
ской формы, является уменьшение основности, то это изменение бывает
связано с понижением абсолютной скорости, как это происходит в присут-
ствии о-бром- или о-иодзаместителей. о-Метильный заместитель занимает
промежуточное положение, связывая оба эти класса.
При рассмотрении данных табл. 173 следует иметь в виду кинетический эф-
фект, обусловленный ионной силой, а также тот факт, что некоторые кон-
станты скоростей определены с использованием значений [Н+], а другие
константы скоростей — с использованием h3. Это различие в таблице огово-
рено специально.
768
Таблица 173
Константы скорости двухпротонного (fc3) и однопротонного (fc2)
механизмов перегруппировки в ряду гидразосоединений (ц —формальная ионная сила)
HNHNHH' В 103fe3, д2. МОЛЬ-2 -С-1 Лите- ратура
йз = Й1/[Н+]2 ЙЗ = Й1/Й2
к 1 В' 0 °C 0 °C 25 °C
В 6 0%-ном водном растворе диоксана, содержащем НС1О4
Свн5 с6н5 0,1 1,6 1 75,
Сон5 С6Н5 1,0 30 а 1 580 30 000 б J 76
1-С10Н7 С6н5 0,1 130 75
2-cloH7 С6н5 0,1 5,0 75
2-СН3СвН4 2-СН3С6Н4 0,1 8,5 75
2-СН3С6Н4 2-СН3С6Н4 0,5 53 а 8 400 75
4-СН3СвН4 4-СН3С6Н4 0,05 1400 76
2-СН3ОС6Н4 С6н5 0,4 1350 79
2-FC6H4 2-FC6H4 1,0 47 79
2-С1С6Н4 2-С1СвН4 1,0 0,002а 0,034 79
4-С1С6Н4 С6Н5 0,1 0,066 79
4-С1С6Н4 С6н5 1,0 2,9 а 150 79
4-С1С6114 4-С1СвН4 1,0 0,24 а 12,8 79
2-ВгС6Н4 2-ВгСвН4 1,0 0,60 79
4-ВгС6Н4 4-ВгС6Н4 1,0 102 79
2-1С6Н4 2-1С6Н4 2,0 > 0,01 в 79
4-1С6Н4 4-1С6Н4 0,5 230 79
2-С6Н5С6Н4 2-С6Н5С6Н4 >1 4,8 79
4-СвН5С6Н4 С6н5 1,0 140 а 12 000 79
4-NO2c6H4 С6н5 > 2 0,0041 79
В !.'5% -ном водном растворе этилового спирта, содержащем НС1
Свн5 В3Нв 0,1 2,4 77
2-СН3С6Н5 2-СН3С6Н4 0,05 7,5 73
3-СН3С6Н4 3-СНдС6Н4 0,05 15 77
4-СН3С6Н4 4-СНдС6Н4 0,05 1240 77
4-т pern -С4НдС6Н4 4-mpem-C4HgC6H4 0,05 1200 78
4-С6Н5С6Н4 4-С6Н5С6Н4 0,05 380 78
RNHNHR' 102^2, Л-МОЛЬ-1-С-1 Лите- ра ура
Й2=Й1/[НЧ | k2=ki/h0
R | R' 0 °C | 0 °C | 25 °C
В 60%-ном водном растворе диоксана, содержащем НС1О4
С6н5 С6н5 0,05 <0,002 76
1-С10Н7 1-С10Н7 0,05 180 г 75
1-С10Н7 2-Сц)Н7 0,05 100 75
2-С10Н7 2-СюН7 0,06 46 75
49-0772
Глава XII
Л родолжение табл. 173
RNHNHR' ц 103&2, л-МОЛЬ-1-С-1 Лите- ратура
A2 = /!1/[H+] й2 = Л|,'7ц
R 1 н' 0 °C 0 сс 25 °C
1-с10н7 С6н5 0,1 2,0 75
2-С10Н7 С6Н5 0,1 0,050 75
2-СН3С6Н4 2-СН3С6Н4 0,1 0,021 75
2-СН3С6Н4 2-СН3С6Н4 0,5 0,14 а 75
4-CH3CONHC6H4 с6н5 0,1 46 78
2-СН3ОС6Н4 с6н5 0,4 1,0а 78
2-СН3ОС6Н4 2-СН3ОС6Н4 0,1 170 г 78
4-СН3ОС6Н4 С6Н5 0,1 480 г 78
2-ВгС6Н4 2-ВгС6Н4 1,0 0,008 78
2-1С6Н4 2-1С6Н4 2,0 0,00015 1! 0,0054/ 78
В 95%-ном водном растворе этилового спирта, содержащем НС1
2-СН3СвН4 ! 2-СН3С6Н4 I 0,05 I I 0,025 I I 73
а При низкой кислотности.
б Экстраполировано к данной температуре.
в НС1О4 заменена на НС1.
г При некоторых значениях кислотности использован буфер.
Состав продуктов бензидиновых перегруппировок был предметом несколько
курьезных и вызвавших споры дискуссий. Наиболее известной является за-
гадка Якобсона, т. е. противоположная ориентация при образовании диари-
лов из гидразобензолов и гидразонафталинов. И в тех и в других оба остатка
имеют свободные положения 2 и 4. В этих двух сериях образуются различные
смеси изомеров диарилдиаминов. В бензольной серии 4,4'-связанный диарил
(бензидин) является основным продуктом, а 2,4'-связанный диарил (дифени-
лин) — главным побочным продуктом. Образование этих продуктов обычно
не сопровождается образованием 2,2'-связанного о-бензидина или каких-либо
других продуктов в количествах, превышающих уровень, необходимый для их
экспериментального обнаружения. В нафталиновых сериях образуются три
сравнимых по выходу продукта: 4,4'-связанный диарил — нафтидин, 2,2'-
связанпый диамин, называемый динафтилином, и соответствующий 2,2'-
связанпому диарилу имин, т. е. карбазол (формула на стр. 766). Эти продук-
ты не сопровождаются 2,2'-связанным диарилдиамином или какими-либо
другими соединениями, образующимися в количествах, превышающих уро-
вень их обнаружения. Сказанное выше типично для гидразобензола и 1,1'-
гидразонафталина. Смешанный нафтилфенилгидразин реагирует так, как
если бы он принадлежал к нафталиновой, а не к бензольной серии. Так,
например, из РМ-нафтил-Н'-фенилгидразина образуются точно такие же
продукты, как из 1,1'-гидразонафталина. 2,2'-Гидразонафталин образует
только 1,1'-связанный диарилдиамин и его имин; Н-2-нафтил-Н'-фенилгидра-
зин ведет себя совершенно так же.
Ни один из этих результатов, даже относящийся к гидразобензолу, нельзя
объяснить относительной реакционной способностью 2- и 4-положений
индивидуальных колец. Эти результаты показывают, что ориентация
должна зависеть от внутреннего взаиморасположения колец, т. е. от всей
молекулярной системы в целом.
770
Перегруппировки в ароматическом ряду
Единственными известными случаями в бензольной серии, когда наблюдается
образование о-бензидина в количестве до нескольких процентов, являются
перегруппировки 3,5,3',5'-тетрагалоген- и тетраалкилгидразобензолов [86].
Однако причина такого их поведения остается неясной.
В тех немногих случаях, для которых можно было исследовать кинетику
в переходной области, состав продуктов зависел от механизма. Так, в случае
нафтилгидразинов, когда кинетический порядок реакции по кислоте с увели-
чением кислотности возрастает от 1 до 2, доля 4,4'-связанного диарила
постепенно увеличивалась, а суммарная доля opmo-связанных продуктов,
обнаруживаемых в виде имина, резко снижалась до нуля. Последнее понять
легко. Как уже отмечалось, отщепление аммиака в процессе перегруппи-
ровки требует, чтобы один атом азота (нуклеофильный) находился в форме
свободного основания, а другой (тот, который отщепляется) —в аммониевой
ионной форме.
Поэтому истинное число присоединившихся протонов, достаточное для обра-
зования имина, равно одному: двух протонов для этого слишком
много.
Перейдем теперь к примерам из бензольной серии, в которой одно 4-положе-
ние занято заместителем. Следует прежде всего отметить, что заместители
СООН и SO3H могут отщепляться от 4-положения подобно атомам водорода
и поэтому основным продуктом является 4,4'-связанный бензидин. Замести-
тели отщепляются без своих связевых электронов. Можно предположить, что
эти электроны необходимы для того, чтобы хиноидное промежуточное соеди-
нение могло вновь вернуться в бензоидное соединение.
Наличие одного прочно связанного заместителя в положении 4 исключает
возможность образования бензидина. 2,4'-Связанный дифенилин может тогда
оказаться основным продуктом перегруппировки, если вместо него частично
или полностью будет образовываться 2,К'-связанный о-семидин. Замена дифе-
нилина на семидин будет частичной в том случае, если заместитель в поло-
жении 4 будет электроноакцепторным (например, галогены), но будет пол-
ной, если этот заместитель обладает сильными электронодонорными свой-
ствами (например, OR). В последнем случае основным продуктом перегруп-
пировки становится о-семидин. В обоих случаях, однако, в продуктах пере-
группировки присутствует другой изомер — К,4'-связаштый n-семидин, в не-
больших, но все же заметных количествах *. И наконец, в продуктах пере-
группировки обоих типов обычно встречаются в существенных количествах
продукты диспропорционирования, т. е. расщепленные амины и азосоеди-
нения.
Наличие заместителя в положении 4, очевидно, обусловливает образование
дифенилина, связанного в положениях 2,4', и и-семидина, связанного в поло-
жениях N,4'. о-Семидин, который может иметь связь либо 2,N', либо N,2',
во всех известных случаях оказывается 2,№-изомером. В известных случаях
полярность заместителя R, в особенности если он обладает свойством усили-
вать или ослаблять основность, не влияет на конечный результат. В приве-
денных ниже формулах нумерация такая же, как и в исходных гидразобен-
золах.
* Предположение, выдвинутое Хаммиком и Мунро [67], состоит в том, что п-семидины
не образуются в результате бензидиновой перегруппировки, если отсутствуют ионы
тяжелых металлов. Это предположение было впоследствии поставлено под сомнение
Вечера с сотр. [87] и Шайном и Стенли [78]. Банторп и Купер [79] подтвердили, что п-
семидин является обычным, хотя и второстепенным продуктом одно- и двухпротонной
перегруппировок таких гидразобензолов, которые имеют единственный прочно связан-
ный заместитель.
49* 771
Глава XII
Дифенилин (2,4')
о-Семидин (2,14')
п-Семидин (N,-?')
Два прочно связанных заместителя, занимающих положение 4 и 4' в гидразо-
бензоле, исключают образование всех продуктов, кроме о-семидина. В этом
случае в сравнимой с перегруппировкой или преобладающей над ней степени
происходит диспропорционирование.
При наличии одного или двух заместителей в 2- или 2'-положениях в продук-
тах перегруппировки содержится почти исключительно 4,4'-связанный бен-
зидин. Диспропорционирование при этом происходит лишь в небольшой сте-
пени или не идет вообще.
Тот факт, что ускоряющие реакцию заместители обладают сильным положи-
тельным электромерным эффектом, приводит к селективной электромерной
л»ета-активации; это явление противоречит общей теории ароматического
электрофильного замещения (гл. VI, разд. 3,в). Примером может служить
метоксильный заместитель. Если он занимает положение 4, то будет ускорять
перегруппировку с образованием продукта, замещенного в 2-положение
своего собственного кольца, и, наоборот, если метоксильный заместитель
занимает положение 2, то он будет ускорять перегруппировку с образовани-
ем 4-замещенного продукта.
Однако такая трактовка отмеченного выше факта не верна, поскольку в ней
две ароматические частицы считаются независимыми реагентами, тогда как
в действительности дело обстоит не так и ориентация касается всей моле-
кулы в целом. В частности, электронодонорный заместитель способствует
смещению электронов в другое ароматическое кольцо, а не в то, в котором
он находится.
Если перегруппировка протекает по какому-либо одному кинетическому
механизму, то состав продуктов реакции не будет зависеть от кислотности
среды. Это впервые было показано Карлином и сотр. [77] на примере обра-
зования бензидина и дифенилина из гидразобензола и на примере образования
о-семидина и продуктов диспропорционирования из 4,4'-диметилгидразо-
бензола. Впоследствии это было подтверждено Банторпом и Купером [791
для всех типов продуктов, образующихся из замещенных гидразобен-
золов.
Большинство качественных данных о составе продуктов перегруппировки
было получено в начале столетия Якобсоном [57], который исследовал около
восьмидесяти перегруппировок. О двойственности механизма реакции в то
время ничего не было известно, так же как и о необходимости обращать вни-
мание на условия реакции. Способы разделения продуктов были несовершен-
ны, поэтому Якобсон установил только различие между образованием про-
дуктов в больших, малых и следовых количествах, обозначив это символами
« + + +», «++» и «+» соответственно.
В настоящее время мы понимаем необходимость учитывать условия прове-
дения реакции. С помощью спектроскопии в сочетании с хроматографией
можно проанализировать продукты намного точнее, чем это мог сделать
Якобсон. Некоторые из примеров Якобсона были исследованы вновь уже
772
Таблица 174
Выходы (%) продуктов перегруппировки в ряду гидразосоединений
(в тех случаях, когда не было уверенности в данных,
использованы условные обозначения Якобсона)
А. Гидразобензолы
Заместители в фенильных группах Среда а Поря- док по Н+ 4,4'- Соеди- нение 2,4'-Со- единение 2, N'-Co- единение N, 4'- Соеди- нение Дис- про- пор- циони- рова- ние Лите- ратура
д 2 76 24
э 2 73 27 84
2-СНз, 2-СНз д 1,2 100 75
э 1,2 100 73
3-СН3, S-СНз э 2 100 77
4-СН3, 4-СНз э 2 40 60 77
k-mpem-Cifi^, i-mpem-C^H-Q э 2 49 50 78
2-СН3О, 2-СН3О д 1 95 5 78
2-СН3О д 1,2 - 100 + 78
4-СН3О д 1 55 24 20 78
4-CH3CONH д 1 б, г +++б ++г 70 78
2-F, 2-F д 2 86 14 79
2-С1, 2-С1 д 2 94 6 79
4-С1, 4-С1 д 2 22 75 79
4-С1 д 2 -19 30 20 31 79
2-Вг, 2-Вг д 1,2 95 5 79
4-Вг, 4-Вг д 2 - 30 -70 79
2-1, 2-1 д 1 100 79
4-1, 4-1 д 2 100 79
2-СвН5, 2-СвН5 д 2 90 10 79
4-СвН5, 4-СвН5 э 2 25 75 78
4-СвН5 д 2 +++ + + +в 38 79
4-NO4 д 2 +++ + + +в — 20 -40 79
Б. Гидразон афтали вы и ? i-нафт вл-N '-фен илгидрази ны
Арильные группы Среда Поря- док по Н+ 4,4'-Со- единение 2,4'-Со- единение ортпо-Соедине- ние Диспро- порциони- рование Лите- ратура
диамин ИМИН
1-Нафтил, 1-нафтил д 1 64 18 18 75
1-Нафтил, 2-пафтил д 1 1(?) 55 44 75
2-Нафтил, 2-нафтил д 1 94 6 75
1-Нафтил, фенил д 1 45 43 И 75
2-Нафтил, фенил д 1 99 0,5 75
а Д — 60%-ный водный раствор диоксана с НСЮ4; Э—95%-ный водный раствор этилового
спирта с НС1.
б Не подтверждено сравнением с синтетическим изомером.
в Не установлено, образуется ли 2,N'- или М,2'-соединение.
г Только при значении кислотности, значительно превышающей кинетическую область.
Глава XII
с применением этих современных методов анализа. Полученные результаты
приведены в табл. 174, кроме того, в этой таблице даны некоторые при-
меры, не исследованные ранее Якобсоном.
Теперь рассмотрим, каким образом наиболее правильная теория бензидино-
вой перегруппировки, которой мы располагаем, а именно теория полярного
переходного состояния, решает вопросы ориентации [66, 70]. Начнем с голо-
воломки, заданной Якобсоном.
Согласно двухпротонному механизму перегруппировки гидразобензола,
после протонирования ковалентная связь N — N по мере приближения
к переходному состоянию гетеролитически превращается в электростати-
ческую и, следовательно, становится более длинной и гибкой. Одна из двух
слабо связанных половин ароматической системы при этом приобретает
почти две единицы положительного заряда, а другая половина остается
с почти нулевым суммарным зарядом. Важно отметить, что два заряда в ква-
зикатионной части молекулы в основном локализованы: один из них являет-
ся зарядом иона аммония и это электростатически усиливает квантовое
направление другого заряда *, который находится на электронодефицитном
атоме углерода в пора-положении. Отсюда видно, насколько важна сольва-
тация аммониевого ионного заряда для стабилизации дважды протонирован-
ного переходного состояния: протон, который создает заряд на атоме азота
в квазикатионе, может отщепиться, если в этом месте сильно нарушится
сольватация. Квазимолекулярная часть молекулы в переходном состоянии
будет обладать сильно рассредоточенными частичными диполярными заря-
дами, как в молекуле анилина. Из приведенного ниже распределения зарядов
при переходе системы через переходное состояние следует, что единственны-
ми положениями, в которых сильно электростатическая связь может стать
ковалентной по мере укорачивания, являются 4,4'- и 2,4'-положения; 2,2'-
связывание может происходить лишь в небольшой степени. В переходном
состоянии должно происходить некоторое продольное перемещение сходя-
щихся друг с другом колец, насколько это позволяет способность их связи
к изгибу, поскольку локализованный электронодефицитный центр будет
перемещаться по направлению к центру отрицательных зарядов другого
кольца
Противоположная ситуация возникает в однопротонном механизме перегруп-
пировки 1,1'-гидразонафталина. Квазикатионная часть этой системы при
достижении переходного состояния будет содержать только электроно-
дефицитный заряд, который будет рассредоточен между 2- и 4-положе-
ниями **.
* Именно по квантовым причинам в неконденсированных бензольных кольцах пара-
хиноидная структура стабильнее орто-хиноидной (гл. VI, разд. 4,ж).
** Это также объясняется квантовой причиной: в бензольном кольце, которое содер-
жится внутри нафталиновой молекулы, а- и Р-хиноидные структуры одинаково устой-
чивы.
774
Перегруппировки в ароматическом ряду
Отрицательный заряд дополняющей квазимолекулярной части переходного
комплекса будет распределен аналогично предыдущему случаю. Кроме того,
кольца будут сходиться друг с другом без продольного перемещения, и такая
согласованность будет способствовать образованию связей только в положе-
ниях 4,4' и 2,2' и в очень незначительной степени в положениях 2,4'.
Точно такие же рассуждения можно привести для перегруппировки N-1-
нафтил-К'-фенилгидразина.
Вернемся теперь к бензольной серии для того, чтобы более детально обсу-
дить влияние заместителей. В реакции любого несимметрично замещенного
гидразобензола существует проблема направления N — N-поляризации.
Предположим, что электроны N — N-связи сместились от заместителя, кото-
рый может поставлять электроны (-j-/?-эффект), в направлении электроно-
дефицитного центра. При таком допущении и наличии лишь одного пара-
заместителя кольцо, в котором он находится, становится квазикатионной
частью переходного комплекса. Это будет верно, если переходное состояние
образуется либо по двухпротонному, либо по однопротонному механизму.
И в этом, и в другом переходном состоянии электронодефицитный заряд на
заместителе будет в значительной мере превращаться в положительный заряд
заполненной оболочки ониевого типа. Таким образом, этот заряд будет
выходить за пределы кольца, а подобное смещение заряда будет увеличивать
продольное перемещение связанных колец. Это может привести к получению
(даже преимущественному) о-семидина 2,И'-типа, так как связь будет обра-
зовываться не с ониевым центром, а с электронодефицитным центром, способ-
ным к образованию ковалентной связи
n-Семидины не являются главными продуктами перегруппировки как при
однопротонном, так и при двухпротонном катализе [79] (табл. 174), но могут
частично образовываться из 4-монозвмещенных гидразобензолов. Среди про-
дуктов гомогенной кислотно-катализируемой перегруппировки гидразона-
фталинов они также не встречаются. Это означает, что фактором, обус-
ловливающим их появление, может быть сравнительно высокая стабиль-
ность пара-хиноидной структуры в неконденсированном бензольном
кольце *.
* См. сноску на стр. 774.
775
Глава XII
По этой причине следует отметить, что любая существующая электрононена-
сыщенность и связанный с нею положительный заряд будут концентрировать-
ся в положении 4. Это в свою очередь означает, что n-семидин может образо-
ваться, когда N — N-поляризация смещает связывающие электроны к 4-за-
мещенному кольцу. При наличии 4-нитрозаместителя и двухпротонном
катализе такое направление поляризации является предпочтительным;
при наличии 4-хлорзаместителя оно обусловлено индуктивным эффек-
том.
При наличии 4-метоксизаместителя такое направление поляризации будет ма-
ловероятным, если не считаться с тем фактом, что единственный эффективно
катализирующий протон должен вначале присоединяться к замещенному,
т. е. более основному анилиновому остатку, что, вероятно, в небольшой степе-
ни и происходит в действительности. Необходимо также серьезно рассмо-
треть возможность неблагоприятной поляризации, так как всегда имеется
противоположное влияние на стабильность и реакционную способность:
более затрудненная поляризация будет (в относительном смысле) в боль-
шей степени активировать перегруппировку. Поэтому приближенно можно
изобразить переходное состояние следующим образом:
На этой перспективной диаграмме обозначенная прерывистой линией связь
N—пара-положение может, например, иметь длину около 3 А (0,3 нм). Изоли-
рованная пара единичных зарядов на таком расстоянии друг от друга имеет
энергию 111 ккал/моль (464,73-103 Дж/моль), так что полярная связь, сос-
тавляющая, скажем, одну четвертую часть или меньше от максимума элек-
тровалентной силы, может оказаться вполне достаточной, чтобы совершенно
приостановить диссоциацию в процессе перегруппировки.
Итак, для различных продуктов перегруппировки можно написать различные
схемы переходных состояний, некоторые из них были приведены выше. Сле-
довательно, то, что мы часто коротко называем «переходным» состоянием пере-
группировки, при отнесении к одному из продуктов может быть представлено
единственным значением энергии, однако если рассматривать вместе не-
сколько продуктов, то лучше представить себе, что переходное состояние
характеризуется набором значений энергии, отличающихся друг от друга на
величину, кратную кТ. Зная влияние температуры на соотношение изомеров
в продуктах перегруппировки, можно точнее определить эту картину (или
установить ограничение).
Диспропорционирование гидразобензолов с образованием азосоединений
и расщепленных аминов протекает по механизмам основного и кислотного
катализа, а также по некаталитическому механизму. О некаталитическом
и основном механизмах катализа известно очень мало. Благодаря последнему
многие перегруппировки, катализируемые кислотами, сопровождаются дис-
пропорционированием, что видно из данных табл. 174. Механизмы кислот-
ной перегруппировки и диспропорционирования тесно связаны друг с дру-
гом.
776
Перегруппировки в ароматическом, ряду
Реакции диспропорционирования, так же как и сами перегруппировки, име-
ют первый кинетический порядок по гидразосоединению. Это необходимо
отметить, так как стехиометрически перегруппировка включает только одну
молекулу гидразосоединения, а диспропорционирование —две молекулы.
Кинетика этого процесса показывает, что в лимитирующей стадии диспро-
порционирования участвует только одна молекула из двух. Вторая молекула
должна включаться в последующую стадию, т. е. должно существовать про-
межуточное соединение, характерное для реакций диспропорционирова-
ния.
Аналогично перегруппировкам, реакции диспропорционирования могут
иметь либо первый, либо второй порядок по ионам водорода. Фактически
кинетическая дихотомия перегруппировки применима также к реакциям
диспропорционирования, причем кинетика последних целиком совпадает
с кинетической формой перегруппировок, которые они сопровождают [79].
Кинетические данные показывают, что кислотный механизм диспропорциони-
рования может быть связан с механизмом перегруппировки в той точке коор-
динаты реакции, где уже произошло присоединение катализирующего про-
тона. Переходное состояние должно затем переходить в промежуточное
соединение, которое включает вторую молекулу гидразосоединения, присое-
динившуюся в быстрой стадии. Эта стадия включает окислительно-вос-
становительный процесс, характерный для реакций диспропорционирова-
ния.
Восстанавливающим агентом окислительно-восстановительной системы долж-
на быть целая молекула гидразоарена, так как она превращается в соответ-
ствующий азоарен без перехода арильных остатков от одной частицы к дру-
гой [79]. Окисляющим агентом в этой системе должно быть производное дру-
гой молекулы гидразоарена, а если используется смесь гидразоаренов, то не
обязательно (или не исключительно) молекула того соединения, которое слу-
жит источником восстанавливающего агента (и становится таким образом
азосоединением) [78, 79]. Точная идентификация окисляющего агента в оки-
слительно-восстановительной системе окончательно не произведена, но.
согласно современным исследованиям Банторпа и Винтер, им определенно не
является ни одно из промежуточных хиноидных соединений, постулируемых
в перегруппировках, а он может появляться в результате N — N-расщепле-
ния без параллельного замыкания хиноидной связи. Одним из таких продук-
тов расщепления будет ароматический амин, а другим — одно- или двух-
зарядный катион в зависимости от механизма реакции
или
В окислительно-восстановительном процессе катион будет окислительным
агентом и, приняв два электрона от восстанавливающей молекулы гидразо-
арена, будет образовывать вторую из двух молекул ароматического
амина.
Существует беспротонный механизм бензидиновых перегруппировок. В этом
случае перегруппировки протекают без кислоты в протонных или апротон-
ных растворителях, таких, как этиловый спирт, ацетон и бензол, при темпе-
ратуре 80—130 °C. Они были открыты Кроликом и Лукашевичем [88] и впер-
вые количественно исследованы Шайном [89]. В 1956 г. Шайн обнаружил, что
Глава XII
реакция имеет первый порядок по гидразосоединению и протекает с большей
скоростью в спирте, чем в апротонных растворителях. Эти исследования
были развиты Лукашевичем и Кроликом [90], а также Банторпом и Хью-
зом [91]. Последние авторы исследовали перегруппировки смесей, которые
должны были бы давать продукты перекрестного взаимодействия по механиз-
му диссоциации. Однако таких продуктов не было получено, что позволило
сделать заключение о внутримолекулярном характере механизма некатали-
тической перегруппировки, как и в обоих катализируемых кислотами реак-
циях.
В кинетическом плане здесь установлены обычные закономерности: порядок
реакции по гидразосоединению равен единице как в спиртовых растворах, так
и в полярных апротонных растворителях, таких, как ацетонитрил или ацетон.
Более сложная и пока не объясненная кинетическая картина наблюдается
в растворе бензола. Электронодонорные заместители повышают скорость
перегруппировки. Увеличение полярности растворителя увеличивает ско-
рость перегруппировки.
Все исследователи отмечают тождественность продуктов некаталитической
перегруппировки и перегруппировки, катализируемой кислотой, особенно
протекающей по однопротонному механизму. Так, гидразобензол дает 4,4'-
и 2,4'-(но не 2,2')диамины диарильного ряда, в то время как 1-нафтилгидра-
зины образуют 4,4'- и 2,2'-связанные диамины и 2,2'-связанные имины и не
образуют 2,4'-связанных диаминов. Главные и второстепенные продукты из
пяти гидразинов нафталинового рода, перечисленные в нижней части
табл. 174, качественно идентичны продуктам однопротонной реакции: смесь
продуктов этих реакций не содержит ни одного лишнего и имеет все необхо-
димые вещества. Вообще говоря, наблюдаемое небольшое количественное
расхождение не имеет существенного значения, так как в обоих механизмах
соотношение между образующимися продуктами зависит от растворителя.
Некоторые же важные особенности этих реакций будут рассмотрены
ниже.
Это соответствие в составе продуктов различных реакций является главным
доводом в пользу предположения, что механизм некаталитической перегруп-
пировки относится к тому же классу гетеролитических реакций, в который
входят и катализируемые кислотами перегруппировки, представленные дву-
мя примерами [70]. От двух- и однопротонного механизмов можно прийти
к беспротонному механизму, для которого также применима теория поляр-
ного переходного состояния. Соотношение между тремя предполагаемыми
механизмами можно выразить, указав приблизительное распределение заря-
да между двумя частями ароматического гидразосоединения, которые входят
во все три переходных состояния:
(2+)(±) (4-)(±) ( + )( — )
Двухпротонный Однопротонный Боспротонный
механизм механизм механизм
Можно ожидать, что беспротонный механизм будет отличаться от однопротон-
ного больше, чем однопротонный от двухпротонного, поскольку, как мы уже
видели, присоединение первого протона в большей степени активирует пере-
группировку, чем присоединение второго. Это вытекает из эксперименталь-
ных данных. В отличие от двух- и однопротонного механизмов, которые, как
это можно наблюдать, конкурируют друг с другом, реакции, идущие по
беспротонному и однопротонному механизмам, имеют настолько различные
775
Перегруппировки в ароматическом ряду
скорости и, следовательно, наблюдаются при столь различных температурах,
что наблюдать их одновременно, т. е. изучить кинетику в переходной обла-
сти, совершенно невозможно.
Можно привести еще несколько наблюдений по некаталитическим перегруп-
пировкам. Одно из них состоит в том, что скорость перегруппировки умень-
шается в 6 раз при введении дейтерия в те положения, из которых он должен
быть отщеплен при образовании диарильной связи [91]. Как мы уже отмеча-
ли, такое уменьшение скорости реакции не может происходить при любом
каталитическом механизме перегруппировки. Такой кинетический изотопный
эффект указывает на то, что в некаталитической реакции отщепление первого
протона приводит к смещению системы по координате реакции к переходно-
му состоянию: перенос протона связан с образованием диарильной
связи.
Причина этого очевидна. Часть ароматической молекулы, которая в переход-
ном состоянии поставляет электрон, необходимый для образования диариль-
ной связи, будет наиболее отрицательной. Только при беспротонном меха-
низме эта часть молекулы является квазианионной. Электроны, которые она
поставляет, будут компенсироваться электронами связи С — Н, что может
быть реализовано за счет согласованного слабого смещения протона по на-
правлению к главному анионному центру, расположенному рядом с атомом
азота. В других механизмах такая возможность отсутствует.
Среди перегруппировок гидразосоединений нафталинового ряда основное
различие в соотношении продуктов, как было отмечено [70], обусловлено
однопротонным и беспротонным механизмами реакции. Беспротонные реак-
ции дают низкий выход 4,4'-связанных продуктов и высокий выход 2,2'-
связанных. Это можно приписать большей жесткости связывания в квази-
противоионном переходном состоянии беспротонного механизма.
Можно упомянуть и другое наблюдение, а именно: образование иминов
(т. е. карбазолов) подавляется при добавлении в систему сильных основа-
ний [90]. Если перегруппировку проводить в растворе этилового спирта
и в присутствии этилат-ионов, то продукт, который в отсутствие добавлен-
ного основания можно идентифицировать в виде имина, превращается в до-
полнительное количество о/?то-диамина. Причин для этого более чем доста-
точно.
Как уже отмечалось, образование иминов зависит от оптимальной сте-
пени протонирования азота: двух дополнительных протонов слишком много,
а ни одного протона — слишком мало. При некаталитическом механизме
реакция начинается без протонизации азота. Но немедленно после прохож-
дения через переходное состояние протонизация становится возможной,
поскольку происходит отщепление первого ароматического протона. Обычно
протонизация способствует завершению образования имина с отщеплением
второго ароматического протона. Это происходит, если среда не способству-
ет немедленному депротонированию системы. Если среда достаточно основна,
то депротонизация будет происходить очень быстро.
СПИСОК ЛИТЕРАТУРЫ
1. Bender G., Вег., 19, 2272 (1886); Chattaway F. D., Orton К. J. Р., J. Chem. Soc., 75,
1046 (1899); Armstrong Н. Е., ibid., 77, 1047 (1900).
2. Orton К. J. Р., Jones W. J., Proc. Chem. Soc., 25, 196, 233, 305 (1909); J. Chem. Soc.,
95, 1456 (1909); Brit. Assoc. Advancement Sci., Rept., 1910, 85; Orton K. J. P., ibid.,
779
Глава XII
1911, 94; 1912, 116; 1913, 136; 1914, 105; 1915, 82; Orton К. J. Р., King Н., J. Chem.
Soc., 99, 1185 (1911).
3. Orton K. J. P., Bradfield A. E., J. Chem. Soc., 1927, 986.
4. Soper F. G., J. Phys. Chem., 31, 1192 (1927).
5. Orton K. J. P., Soper F. G., Gwyn Williams, J. Chem. Soc., 1928, 998.
6. Olson A. R., Porter C. W., Long F. A., Halford R. S., J. Am. Chem. Soc., 58, 2467
(1936); Olson A. R., Halford R. S., Hornel J. C., ibid., 59, 1613 (1937); Olson A. R.,
Hornel J. C., J. Org. Chem., 3, 76 (1938).
7. Bradfield A. E., Orton K. J. P., Roberts I. C., J. Chem. Soc., 1928, 782; Richardson M.,
Soper F. G., ibid., 1929, 1873.
8. Blanksma J. J., Rec. trav. chim., 22, 290 (1903); Rivett A. C. D., Z. physik. Chem.,
82, 201 (1913); Hamed H. S., Seitz H.,1. Am. Chem. Soc., 44, 1475 (1922); Soper F. G..
Pryde D. R., J. Chem. Soc., 1927, 2761; Dawson H. M., Millet H., ibid., 1932,
1920.
9. Bell R. P., Proc. Roy. Soc. (London), A143, 377 (1934); Bell R. P., Levinge R. V. H.,
A151, 211 (1935); Bell R. P., J. Chem. Soc., 1936, 1154; Bell R. P., Brown J. F.,
J. Chem. Soc., p. 1520; Bell R. P., Danckwerts P. V., J. Chem. Soc., 1939,
1774.
10. Israel G. C., Tuck A. W. N., Soper F. G., J. Chem. Soc., 1945, 547.
11. Dewar M. J. S., Nature, 156, 784 (1946); J. Chem. Soc., 1946, 406; Disc. Faraday Soc.,
2, 50 (1947); Electronic Theory of Organic Chemistry, Clarendon Press, Oxford. 1949,
p. 233; Bull. soc. chim. (France), 1951, p. 71C.
12. Dewar M. J. S., Scott J. M. W., J. Chem. Soc., 1957, 2676; Теоретическая органиче-
ская химия, ИЛ, М., 1963; Molecular Rearrangements Р. de Mayo, ed., Interscience,
New York, 1963, p. 295.
13. Blanksma J. J., Rec. trav. chim., 21, 366 (1902); Mathews J. M., Williams R. V., J. Am.
Chem. Soc., 45, 2574 (1923).
14. Griess P., Martius C. A., Zeit. Chem., 2, 132 (1866); Kekule R. J., ibid., p. 688;
Witt O. N., Thomas E. G. B., J. Chem. Soc., 43, 112 (1883); Friswell R. J., Green A. G.,
ibid., 47, 917 (1885).
15. Nietzski R., Ber., 10, 662 (1877).
16. Hewmann K., Oeconomides A., Ber., 20,372 (1887); Fischer B., Wimmer H., ibid., p. 1579;
Kidd H. V., J. Org. Chem., 2, 198 (1937).
17. Goldschmidt H., Ber., 24, 2317 (1891); Goldschmidt H., Bardack B., Ber., 25, 1347 (1892);
Goldschmidt HReinders R. U., ibid., 29,1369, 1899 (1896); Goldschmidt H., SalcherR. M.,
Z. physik. Chem., 29, 89 (1899); Goldschmidt H., Johnsen S., Overwien E., ibid., 110,
251 (1924).
18. Fischer O., Hepp E., Ber., 19, 2291 (1886).
19. Fischer O., Hepp E., Ber., 20, 1247, 2471 (1887); Morgan G. T., Evens F. D., J. Chem.
Soc., 115, 1142 (1919).
20. Fischer O., Ber., 45, 1098 (1912).
21. Houben J., Ber., 46, 3984 (1913).
22. Neber P. W., Rauscher H., Ann., 550, 182 (1942).
23. Macmillen W., Reade T. H., J. Chem. Soc., 1929, 585.
24. Glazer J., Hughes E. D., Ingold С. K., James A. T., Jones G. T., Roberts E., J. Chem.
Soc., 1950, 2657.
25. Hofmann A. W., Martius C. A., Ber., 4, 742 (1871); Hofmann A. W., Ber., 5, 704, 720
(1872); 7, 526 (1874).
26. Reilly J., Hickinbottom W. J., J. Chem. Soc., 117, 103 (1920).
27. Hickinbottom W. J., Ryder S. E., A., J. Chem. Soc., 1931, 1281.
28. Hickinbottom W. J., Preston G. H., J. Chem. Soc., 1930, 1566.
29. Hickinbottom W. J., J. Chem. Soc., 1932, 2396; 1934, 1700; 1935, 1279; J. Chem. Soc.,
1937, 404.
30. Michael A., Ber., 14, 2105 (1881); J. Am. Chem. Soc., 42, 787 (1920).
31. Beckmann E., Correns E., Ber., 55, 852 (1922).
32. Hartmann C., Gattermann L., Ber., 25, 253 (1892); Beilstein, Handbuch der Organischen
Chemie, 4th Edn., 6, 143.
33. Smith R. A., J. Am. Chem. Soc., 55, 3718 (1933); J. Am. Chem. Soc., 56, 717
(1934).
34. Sprung M. M., Wallis E. S., J. Am. Chem. Soc., 56, 1715 (1934).
35. Sowa F. J., Hinton H. D., Nieuwland J. A., J. Am. Chem. Soc., 55. 3402 (1933);
Smith R. A., ibid., 56, 717 (1934).
36. Natelson S., J. Am. Chem. Soc., 56, 1583 (1934).
37. Hart H., Eleuterio H. S., J. Am. Chem. Soc., 76, 519 (1954).
38. Bamberger E., Ber., 27, 1347, 1548 (1894).
39. Cm. [38]; Bamberger E., Ann., 424, 233, 297 (1921); 441, 207 (1925).
780
Перегруппировки в ароматическом ряду
40. Bamberger Е., Ann., 424, 233 (1921).
41. Yukawa У., J. Chem. Soc. (Japan), 71, 603 (1950).
42. Heller H. E., Hughes E. D., Ingold С. K., Nature, 168, 909 (1951).
43. Bamberger E., Landsteiner K., Ber., 26, 485 (1893); Bamberger E., Ber., 27, 359 (1894)-
30, 1248 (1897).
44. Bamberger E., Ber., 27, 584 (1894); 28, 399 (1895).
45. Orton K. J. P., Smith A. E., I. Chem. Soc., 87, 389 (1905); Orton K. J. P., Pearson C.,
ibid., 93, 725 (1908); Orton K. J. P., Brit. Assoc. Advancement Sci., Rept., 1912, 116;
Chem. News, 106, 236 (1912); Bradfield A. E., Orton K. J. P., J. Chem. Soc. 1929,
915.
46. Hughes E. D., Jones G. T., J. Chem. Soc., 1950, 2678.
47. Holleman A. F., Hartogs J. C., van der Linden T., Ber., 44. 704 (1911).
48. Brownstein S., Bunton C. A., Hughes E. D., Chem. and Ind., 1956, 981; J. Chem. Soc.
1958, 4354.
49. Banthorpe D. V., Hughes E. D., Williams D. L. H., J. Chem. Soc., 1964, 5349; Bant-
horpe D. V., Thomas J. A., Williams D. L. H., ibid., 1965, 6135; Banthorpe D. V.,
Thomas J. A., ibid., p. 7149.
50. White W. N., Klink J. K., LazdinsD., Hathaway C., Golden J. T., White H. S., J. Am.
Chem. Soc., 83, 2024 (1961).
51. Banthorpe D. F., Thomas J. A., J. Chem. Soc., 1965, 7158.
52. Hofmann A. W., Proc. Roy. Soc. (London), 12, 576 (1863).
53. Fittig R., Ann., 124, 282 (1862).
54. Schultz G., Ann., 174, 227 (1874).
55. Schmidt H., Schultz G., Ber., 11, 1754 (1878).
56. Schmidt H., Schultz G., Ann., 207, 320 (1881); Schmidt H., Schultz G., Strasser H., ibid.,
p. 348.
57. Jacobson P., many papers from 1892 to 1922, including the summarising paper Ann.
428, 76 (1922).
58. Тихвинский M., ЖРФХ0, 35, 667 (1903).
59. Wieland H., Ann., 392, 127 (1912); Ber., 48, 1095 (1915).
60. Wheland G. W., Schwartz T. R., J. Chem. Phys., 17, 425 (1949).
61. Bloink G. J., Pausacker К. H., J. Chem. Soc., 1950, 950.
62. Stieglitz J., Am. Chem. J., 29, 62—63 (1903).
63. Ingold С. K., Kidd H. V., J. Chem. Soc., 1933, 984.
64. Smith D. H., Schwartz J. R., Wheland G. W., J. Am. Chem. Soc., 74 2282
(1952).
65. Banthorpe D. F., J. Chem. Soc., 1962, 2413.
66. Hughes E. D., Ingold С. K., J. Chem. Soc., 1941, 608; 1950, 1638; Ingold С. K., Chem.
Soc. Special Pubs., 16, 118 (1962).
67. Hammick D. LI., Mason S. F., J. Chem. Soc., 1946, 638; Hammick D. LI., Munro D. C.
ibid., 1950, 2049.
68. Vecera M., Synek L., Sterba J., Coll. Czech. Chem. Comm., 25, 1992 (1960); ibid 31
3486 (1966).
69. Banthorpe D. V., Bramley R., Thomas J. A., J. Chem. Soc., 1964, 2900.
70. Banthorpe D. F., Hughes E. D., Ingold С. K., J. Chem. Soc., 1964, 2864.
71. Van Loon J. P., Rec. trav. chim., 23, 62 (1904).
72. Hammond G. S., Shine H. J., J. Am. Chem. Soc., 72, 220 (1950).
73. Carlin R. B., Odioso R. C., J. Am. Chem. Soc., 76, 100 (1954).
74. Blackadder D. A., Hinshelwood C., J. Chem. Soc., 1957, 2898.
75. Banthorpe D. V., Hughes E. D., Ingold С. K., J. Chem. Soc., 1962, 2386, 2418; Bant-
horpe D. V., Hughes E. D., J. Chem. Soc., 1962, 2402; Banthorpe D. F., J. Chem. Soc.,
1962, 2407, 2429; Banthorpe D. V., Ingold С. K., Roy J., Somerville S. M., J. Chem.
Soc. 1962, 2436; Banthorpe D. V., Hughes E. D., Ingold С. K., Roy J. J Chem
Soc., 1962, 3294.
76. Bunton C. A., Ingold С. K., Mhala M. M., J. Chem. Soc., 1959, 1906.
77. Carlin R. B., Nelb R. G., Odioso R. C., J. Am. Chem. Soc., 73, 1002 (1951); Carlin R. B.,
Odioso R. C., ibid., 76, 2345 (1954); Carlin R. B., Wich G. S., ibid. 80 4023
(1958).
78. Shine H. J., Chamness J. T., J. Org. Chem., 52, 901 (1967); Shine H. J., Stanley J. P.
ibid., p. 905.
79. Banthorpe D. V., Cooper A., Ingold С. K., Nature, 216, 232 (1967).
80. White W. N., Preisman R., Chem. and Ind., 1961, 1752.
81. Shine H. J., Chamness J. T., J. Org. Chem., 28, 1222 (1963).
82. Hammond G. S., Clovis J. S., Tetrahedron Letters, 1962, 945.
83. Dewar M. J. S., Marchand A. P., Ann. Rev. Phys. Chem., 16, 338 (1965).
84. Banthorpe D. V., Hughes E. D., J. Chem. Soc., 1962, 3308.
781
Глава XII
85. Banthorpe D. V., Hughes E. D., Ingold С. K., Humberlin R., J. Chem. Soc., 1962,
3299.
86. Carlin R. B., Forshay W. 0., Jr., J. Am. Chem. Soc., 72, 793 (1950); Carlin R. B., Hes-
singer S. A., ibid., 77, 2272 (1955).
87. Vecera M., Gasparic J., Petranek J., Coll. Czech. Chem. Comm., 22, 1603 (1957).
88. Кролик Л. Г., Лукашевич В. О., ДАН СССР, 65, 37 (1949).
89. Shine Н. J., J. Am. Chem. Soc., 78, 4807 (1956); Shine Н. J., Trisler J. C., J. Am.
Chem. Soc., 82, 4054 (1860); Shine H. J., Huang F.-T., Snell R. L., J. Org. Chem.,
26, 380 (1961).
90. Лукашевич В. О., Кролик Л. Г., ДАН СССР. 147, 1090 (1962).
91. Banthorpe D. V., Hughes Е. D., J. Chem. Soc., 1964, 2849, 2860; Banthorpe D. V., ibid.,
p. 2854.
ГЛАВА XIII
ПРИСОЕДИНЕНИЕ И ОБРАТНЫЕ РЕАКЦИИ
1. Электрофильное присоединение к олефиновым соединениям 784
2. Нуклеофильное присоединение к карбонильным и олефиновым
соединениям............................................... 820
3. Гомолитическое присоединение к олефиновым соединениям . . 858
4. Циклоприсоединение к олефинам........................... 874
Список литературы . ................................... 900
1. Электрофильное присоединение к олефиновым
соединениям [1]
В данном разделе рассмотрены реакции присоединения электрофильных
агентов к простым и сопряженным ненасыщенным системам, а именно к оле-
финам и полиолефинам. Для краткости мы будем называть такие реакции
электрофильным присоединением и обозначать символом Ads- Электрофиль-
ные адденды удобно разделить на два класса: кислые электрофильные аген-
ты, представляющие собой сильные кислоты и присоединяющиеся в виде
Н — X, и некислые электрофильные агенты, такие, как галогены, присоеди-
няющиеся в виде X — Y. Кроме того, присоединение к простым и сопряжен-
ным ненасыщенным системам удобно рассматривать отдельно.
а. Присоединение сильных кислот, главным образом алкилгалогенидов
и иона гидроксония, к моноолефинам. Ориентация в реакциях присоединения
к олефинам начала привлекать внимание раньше, чем ориентация в реакциях
замещения в ароматическом ряду. К 1870 г. было установлено несколько
фактов: было известно, например, что пропилен и иодистый водород образу-
ют изопропилиодид [2]. В 1870 г. Марковников предложил правило ориента-
ции при присоединении галогеноводородов к олефинам, согласно которому
присоединение происходит таким образом, что галоген присоединяется
к менее гидрогенизированному атому углерода [3]. Следует отметить, что
Марковников сформулировал свое правило только для реакций присоедине-
ния к углеводородам. Присоединение хлористого и бромистого водородов
к винилбромиду с образованием этилиденгалогенидов было открыто пример-
но в то же время [4], однако Марковников классифицировал эти случаи как
исключение из правила.
При дальнейшем исследовании реакций присоединения галогеноводородов
к олефиновым соединениям встретилось затруднение, вызванное тем обстоя-
тельством, что бромистый водород мог присоединяться по двойной связи не
только «нормальным» образом, т. е. согласно правилу Марковникова, так,
как показано на примере присоединения хлористого и йодистого водородов,
но также и «аномальным» образом, в результате которого происходит обрат-
ная ориентация бромистого водорода. Долгое время условия, вызывающие
такую аномальную ориентацию, оставались неясными; они были окончатель-
но выяснены Харашем и Майо в 1933 г. Эти авторы показали, что аномальное
присоединение бромистого водорода обусловлено наличием перекисей, почти
всегда присутствующих в олефиновых соединениях, если последние в тече-
ние какого-то времени находились на воздухе. При этом происходит следую-
щее [5]: перекись водорода окисляет бромистый водород с выделением атома
брома, который начинает гомолитическую цепную реакцию присоединения
бромистого водорода (разд. 3,а). Хлористый и иодистый водороды обычно не
принимают участия в подобной гомолитической цепной реакции, даже если
перекиси специально введены в систему *. Это обусловлено тем, что атомы
хлора образуются с большим трудом, а легко образующиеся атомы иода
недостаточно реакционноспособны. Установив причину аномального при-
соединения бромистого водорода, Хараш и сотрудники нашли способ исклю-
чить эту реакцию: процесс следует проводить в темноте в отсутствие воздуха
и перекисей; на практике же обычно работают в присутствии антиоксидан-
тов. Благодаря этому впервые стало возможным проведение точных исследо-
ваний ориентации в условиях нормального присоединения бромистого
водорода.
* Все же имеются сведения о влиянии перекисей на присоединение хлористого водо-
рода к некоторым олефинам [6].
784
Присоединение и обратные реакции
Пропилен и иодистый водород образуют исключительно изопропилиодид [7].
Присоединение йодистого водорода к бутену-1 и неопентилэтилену протекает
аналогично [7]. Если подавить реакцию аномального присоединения, то
пропилен и бромистый водород образуют изопропилбромид [8]. При этих
же условиях аналогично протекает присоединение бромистого водорода
к бутену-1 и изобутилену [9]. Эти результаты получаются как в присутствии
растворителя, так и без него. Правило Марковникова соблюдается во всех
случаях, и если олефин имеет концевую двойную связь, то галоген присое-
диняется к неконцевому атому углерода
нх
RCH = CH2 -> RCHX —СН3
Подобная ориентация наблюдается и в случае аллилбромида; 1,2-дигалогенид
образуется при присоединении йодистого водорода [7], а также при при-
соединении бромистого водорода, если подавлена реакция аномального
образования 1,3-дигалогенида [10]. В этом случае ориентация также не
зависит от того, проводят ли реакцию без растворителя или в среде апротон-
ного или гидроксилсодержащего растворителя
нх
ВгСН2—СН = СН2----> ВгСН2-СНХ —СН3
Присоединение элементов бромистого водорода к винилбромиду приводит
к образованию этилиденбромида при нормальном течении реакции [8];
в присутствии же промотирующей перекиси образуется дибромэтан. Добав-
ление йодистого водорода к винилхлориду и при нормальных условиях бро-
мистого водорода к винилхлориду ведет к образованию этилиденгалогени-
да [11]
нх
Hal—СН = СН2----► Hal —СНХ —СН3
В 1870 г. Линнеман и отчасти Вислиценус [12] показали, что добавление
хлористого, бромистого и йодистого водородов к акриловой кислоте приво-
дит во всех случаях к образованию ю-галогенпропионовых кислот
нх
НО2С —СН = СН2---> НО2С-СН2—СН2Х
Этот тип ориентации, как подчеркнул Тиле [13], настолько интересен, что
заслуживает особого внимания. В своей первой посвященной сопряжению
статье, опубликованной в 1898 г., Тиле высказал предположение, что перво-
начально НХ присоединяется к сопряженной системе О = С — С = С
в положение 1,4, причем галоген присоединяется к углеродному атому, а во-
дород — к кислородному вследствие большего сродства к нему. Затем атом
водорода переходит в положение, обеспечивающее воссоздание карбо-
ксильной группы.
Такое объяснение, конечно, нельзя распространить на случай присоединения
йодистого водорода к нейрину, приводящего, как установил Шмидт [14],
к образованию ю-иодозамещенного аммониевого иона
+ HI +
(CH3)3N-CH = CH2---->- (CH3)3N-CH2-CH2I
Хенне и Кей [15] показали, что трифторметилэтилен присоединяет хлори-
стый и бромистый водороды так же, как и нейрин, но не как пропилен
нх
CF3 — СН = СН2 -► CF3 —СН2—СН2Х
Электронная интерпретация правила Марковникова впервые была дана
в приемлемой форме в 1924 г. Лукасом и Джеймсоном [16]. Они предполо-
50-0772 785
Глава XIII
жили, что электроположительная алкильная группа способна так поляризо-
вать соседнюю двойную связь, что атом углерода, наиболее удаленный от
алкильной группы, может очень легко отдавать свои л-электроны для свя-
зывания с протонной частью электрофильного адденда. В дальнейшем эта
теория получила широкое распространение и смогла объяснить другие ори-
ентационные эффекты.
X—н
сн3->снЛ5н2
X—н
л
сн=сн2
Н—X
НО2С <- сн=4=сн2
Я—X
(ch3)3n «- снХсн2
F3C <- СН=СН2
Н—X
Принимая во внимание трудности, связанные с детальным определением
направления перемещения кето-енолыюго протона, можно считать, что меж-
ду этим объяснением и взглядами Тиле на механизм присоединения к аф-
ненасьпценным кислотам нет больших различий. В то же время трактовку
присоединения к галогенэтиленам по Лукасу и Джеймсону можно рассмат-
ривать как расширение основной идеи Тиле, состоящей в том, что сопряжение
может определить направление присоединения к олефиновой связи в сопря-
женной системе. Это аналогично тому, что происходит при электрофильном
замещении в ароматическом ряду, когда электроотрицательные (—I) гало-
генные заместители уменьшают скорость замещения, но не препятствуют
предпочтительной ортпо-иара-ориентации, соответствующей их электрополо-
жительному поляризующему эффекту (~\-Е), ответственному за сопряжение
с ароматической системой. Точно так же как и-нитростирол представляет
собой случай —I, -\-Е и поэтому сходен с бромбензолом в реакции аромати-
ческого замещения, так и n-нитростирол сходен с винилбромидом (— I, -{-Е)
в реакции присоединения по двойной связи. Илиел и сотр. [17] показали, что
введение n-нитрогруппы в стирол сильно уменьшает скорость присоединения
бромистого водорода, но не изменяет ориентации, которая остается такой же,
как в стироле.
НВг
n-NO2CeH4—СН = СН2----> NO2CeH4—СНВг—СН8
Кроме галогеноводородных, другие сильные кислоты также присоединяются
по двойной связи олефина. Наиболее важными примерами могут служить
случаи присоединения иона гидроксония и молекулы серной кислоты с образо-
ванием соответственно ионной сопряженной кислоты спирта и кислого эфи-
ра серной кислоты; в присутствии воды во всех случаях образуется
спирт. Порядок присоединения при этом остается таким же, как и в случае
галогеноводородных кислот. Таким образом, правило Марковникова соблю-
дается и здесь, причем гидроксильная группа спирта присоединяется к ме-
нее гидрированному атому углерода исходного олефина. Кислотная гидра-
тация олефинов до спиртов часто оказывается существенно обратимой.
Таким образом, наблюдается связь между этими реакциями присое-
динения и рассмотренными в гл. IX реакциями отщепления, приводящими
к олефинам.
Примеры, приведенные в предыдущем разделе, были известны еще Бутлеро-
ву [18], который в 1877 г. показал, что «диизобутилен» (1,1-диметил-2-треш-
бутилэтилен) в присутствии разбавленной серной кислоты находится в равно-
весии с так называемым «диизобутолом» (диметилнеопентилкарбинолом)
786
Присоединение и обратные реакции
или с кислым сульфатом последнего
Разб. H2SO4
(СН3)2С=СН-С(СН3)3 -------* (СН3)2С(ОН)СН2С(СН3)3
Значительно позже Эберц и Лукас [19] количественно исследовали равно-
весие между изобутиленом и тпретп-бутиловым спиртом в присутствии избыт-
ка 0,2 н. водной азотной кислоты. При обычной температуре это равновесие
сдвинуто в сторону спирта
Разб. HNO3
(СН3)2С=СН2 \ - -*• (СНз)2С(ОН)СН3
Значительные усилия были затрачены на исследование кинетики присоеди-
нения галогеноводородов и иона гидроксония к олефинам. Кинетика присое-
динения хлористого водорода к изобутилену [20] и бромистого водорода
к пропилену [21] в растворе гептана была исследована Майо с сотрудни-
ками. Они установили, что во всех случаях протекает реакция первого
порядка по олефину и неопределенного порядка, в среднем примерно
третьего, по галогеноводороду. Такого результата можно было ожидать
по следующей причине (гл. VII, разд. 5,а). В любой гетеролитической реак-
ции переходное состояние всегда является высокополярным. В случае
неполярного растворителя молекулы наиболее полярного растворенного
вещества будут группироваться вокруг молекул, находящихся в переходном
состоянии. Если более полярным веществом оказывается сам реагент, порядок
реакции будет высокий и, возможно, неопределенный, как было сказано
выше. Если более полярным веществом окажется продукт реакции, то реак-
ция будет аутокаталитической и, вероятно, с высоким или неопределенным
порядком по катализатору (об этом говорилось в гл. VI. разд. 5,д).
Менее сложная кинетическая картина наблюдается при использовании
полярного растворителя, как в исследованиях Лукаса и сотрудников при
определении скорости катализируемой кислотами гидратации изобутилена
[22] и триметилэтилена [23] в водном растворе. Они установили, что реакция
имеет первый порядок как по кислоте, так и по олефину. Вполне вероятно,
что главной, если не единственной, эффективной кислотой в водном растворе
является ион гидроксония
Скорость ос [Олефин][Н3О+]
Эти кинетические исследования не позволяют ответить на вопрос, как про-
текает реакция: присоединяется ли протон сильной кислоты, т. е. иона
гидроксония, раньше присоединения сопряженного основания, т. е. моле-
кулы воды, и значит весь процесс является двухстадийным, или же протон
и сопряженное основание присоединяются одновременно к двойной олефино-
вой связи. Однако, поскольку присоединение — реакция обратимая, его
нельзя рассматривать в отрыве от отщепления: согласно принципу микро-
скопической обратимости, наиболее легкий путь через потенциальный барьер
от исходного вещества к продукту является в то же время наиболее легким
путем в обратном направлении — от продуктов к исходным веществам. В на-
стоящее время можно считать доказанным *, что замещение и отщепление,
протекающие через ионы сопряженных кислот вторичных и третичных спир-
тов, представляют собой двухступенчатые мономолекулярные процессы типа
SN1 и Е1. Это означает, что присоединение, т. е. реакция, обратная реакции
* См., в частности, гл. VII, разд. 2, г и 3, д.
5о* 787
Глава XIII
отщепления, также является двухстадийным процессом
RChXch2 + НОН2+ -» R-СН—СНз + ОН2
RCH—СН3 + ОН2 —> RCH(OH2)CH3
Уитмор и Джонстон [24] убедительно показали, что подобное заключение
применимо и к реакциям присоединения галогеноводородов. Они установили,
что присоединение хлористого водорода к изопропилэтилену в отсутствие
растворителя ведет к образованию изоамилхлорида и значительного количе-
ства тпреш-амилхлорида, причем последний образуется не из первого,
поскольку изоамилхлорид в условиях реакции стабилен. Следовательно, обра-
зование шретп-амилхлорида может происходить только в результате перегруп-
пировки какого-либо промежуточного продукта присоединения, которым
едва ли может быть что-нибудь, кроме иона карбония. Реакция протекает по
ступенчатому механизму:
(сн3)2снснХсн2 + на -*
-> (СН3)2СНСНСНз 4- CI-—> (СН3)2СНСНС1СНз
+ !
(СН3)зССН2СНз +С1--* (СНз)2 СС1СН2СНз
В результате присоединения йодистого водорода к третп-бутилэтилену также
образуются два иодида: неперегруппированный (СН3)3ССН1СН3 и перегруп-
пированный (СН3)2С1СН(СН3)2 [25]. Согласно исследованиям Хьюза и Пилин-
га (частное сообщение), при проведении реакции при температуре —80 °C
и без растворителя получается по 50% каждого изомера.
Особенность механизма, которую всегда пытаются выяснить для любой реак-
ции, связанной с присоединением протона, состоит в том, заканчивается ли
передача протона в предравновесной стадии, т. е. до того, как система перей-
дет в переходное состояние лимитирующей стадии (специфический катализ
ионом водорода), или перенос протона является медленным процессом, иду-
щим в переходном состоянии (общий кислотный катализ). После почти десяти-
летних исследований эта проблема была решена, когда в 1952 г. Тафт [26]
установил, что скорости кислотной гидратации олефинов зависят от функции
Гаммета Ао более точно, чем от концентрации [Н+]. Шкала h0 Гаммета построе-
на как мера кислотности для равновесного переноса протона к азотистым
основаниям. Вывод, который следовало бы сделать из наблюдений Тафта
(практически автоматически), состоит в том, что скорости гидратации олефи-
нов зависят от равновесного переноса протона (специфический катализ ионом
водорода). Первая трудность, которая затем возникла, состояла в необходи-
мости сделать приемлемое допущение относительно того, что же является
медленной стадией процесса, приводящей к образованию переходного состоя-
ния. Предположения, которые были сделаны (вероятно, единственно возмож-
ные), оказались все же неприемлемыми. Строго говоря, все попытки прове-
рить вывод о существовании предшествующего равновесия с помощью других
методов не смогли подтвердить его. Тафт, как и другие авторы, использовал
несколько независимых подходов.
Исследован кинетический эффект дейтерированного растворителя. Скорость
реакции зависит от предравновесного присоединения протона, которое в дей-
терированной воде происходит в 2—3 раза быстрее, чем в обычной воде, глав-
ным образом потому, что D3O+ — более сильная кислота в D2O, чем Н3О +
в Н2О. Контролирующая скорость стадия переноса протона идет мед-
755
Присоединение и обратные реакции
леннее, когда она соответствует переносу дейтрона в тяжелой воде. Установ-
лено, что кислотная гидратация олефинов в тяжелой воде замедляется
[27, 29, 30].
Тафт рассмотрел все возможные взаимопревращения перед гидратацией двух
олефинов, которые должны давать один и тот же карбониевый ион и один
и тот же спирт
СН3ч (1) CIU (—1) СНз
/=сн-сн, ~сн2—сн3 хс—сн2-сн3
СН3/ (-1) СН3/ (1) сн/
(2)
он
СНзч I
/-СН2—СН3
сн/
Если перенос протона происходит в предравновесной стадии, то переходное
состояние реакции должно соответствовать стадии, отмеченной цифрой (2),
и почти все образовавшиеся карбониевые ионы будут обратно превращаться
в олефины, и лишь небольшая их доля, преодолев энергетический барьер на
пути к переходному состоянию, перейдет в спирт. Поэтому олефины будут
претерпевать взаимопревращение гораздо быстрее, чем гидратацию; однако
опыты показывают, что при гидратации олефинов на глубину 50% их взаимо-
превращения не происходит [28]. Эквивалентным тестом является проведение
реакций гидратации в дейтерированной воде. Если протонирование —
предравновесный процесс, то внедрение дейтерия в исходные олефины должно
идти намного быстрее, чем гидратация. Экспериментально установлено, что
внедрения дейтерия в олефины не происходит [27]. Из этого можно сделать
только один вывод, что реакция (—1) не протекает; таким образом, система
проходит через переходное состояние в то время, когда образуется карбоние-
вый ион, т. е. лимитирующей стадией является перенос протона.
Это означает наличие общего кислотного катализа. Если в состав переход-
ного комплекса входит, помимо олефина, кислота Н3О+, которая служит
переносчиком протона к олефину, то следует допустить возможность суще-
ствования других переходных комплексов, содержащих какие-нибудь другие
кислоты НА. При специфическом катализе ионом водорода переходный ком-
плекс содержит субстрат и протон кислоты, так что любая кислота ведет
к образованию одного и того же переходного комплекса. Обычной трудно-
стью, препятствующей обнаружению мультиплетностипереходных комплексов,
является то, что зависимость скорости от природы кислоты часто бывает
слишком малой, чтобы ее можно было заметить. Хотя это было установлено
уже после того, как первоначально выяснилось, что скорость гидратации
зависит от кислотной функции Гаммета, следует заметить, что Шуберту
с сотр. [30] при использовании очень реакционноспособного олефина, и-ме-
токси-а-метилстирола, и формиатного буфера удалось установить, что гидра-
тация может катализироваться молекулами муравьиной кислоты. Отсюда
становится очевидным, что гидратация олефинов является в принципе реак-
цией общего кислотного катализа.
Наряду с этой проблемой, связанной со способом и временем переноса прото-
на при катализуемой кислотами гидратации олефинов, существует подобная
же проблема, связанная с кислотным катализом при водородном обмене
в ароматических соединениях, уже описанная в гл. VI, разд. 6,в. Оказалось,
что скорость зависит от h0, а не от [Н+], хотя с момента открытия этой реак-
789
Глава XIII
ции и было известно (на что многие не обращали внимания), что она представ-
ляет собой реакцию общего кислотного катализа. В 1961 г. Баннет [31]
решил обе эти проблемы. Он показал, что шкалы кислотности, применяемые
к равновесию в водных растворах кислот, зависят в большой мере от изме-
нения активности растворителя — воды и, следовательно, от количества
связанной воды, включая сольватационную, которое высвобождается или
связывается с растворенным веществом в результате переноса протона.
Высвобождение связанной воды более благоприятствует течению реакций
при высокой кислотности, когда раствор менее богат свободными молекулами
растворителя — воды. Шкала h0 была построена для выражения равновесия
переноса протона от сильно гидрофильных молекул воды к умеренно гидро-
фильным органическим азотистым основаниям. В этих процессах некоторое
количество связанной воды высвобождается, и это служит главной причиной
того, почему шкала h0 с ростом кислотности растет более круто, чем шкала
[Н+]. Шкала h0 прекрасно отражает равновесный перенос протона к умерен-
но гидрофильным органическим азотистым и органическим кислородным осно-
ваниям. Однако, когда высвобождается слишком большое количество свя-
занной воды, что бывает при переносе протона от воды к гидрофобным угле-
родным основаниям, тогда функция h0 становится неприменимой. В этом
случае для того, чтобы описать равновесие переноса протона к углероду,
нужна функция, более круто растущая, чем функция h0. Баннет пришел
к заключению, с которым следует согласиться, что функция Гаммета h0
иногда все же дает приближенное описание псевдоравновесного частичного
переноса протона и связанного с этим переносом частичного высвобождения
воды, протекающих при образовании переходного состояния переноса про-
тона к углероду.
Если записать механизм реакции по Баннету
R2C = CR2 + H3O+ 7 R2CH-CRJ + H2O
Медленно
R2CH-CRJ+H2O 7"..R2CH-CR2-OH£
R2CH-CR2OH£+H2O -----» R2CH-CR2OH + H3O+
и читать его с конца, то получим механизм реакции отщепления олефина от
сопряженной спирту кислоты. Несмотря на то что эта реакция идет через
промежуточное образование карбониевого иона, ее механизм не будет меха-
низмом типа Е1, так как в состав переходного комплекса наряду с карбоние-
вым ионом входит также и молекула воды. Здесь имеет место бимолекуляр-
ный механизм, в основе которого лежит предравновесное образование карбо-
ниевого иона; такой механизм обозначается символом Е2(С+). Он является
копией хорошо известного механизма отщепления заместителя SN2(C+), рас-
смотренного в гл. VII, разд. 3,в. Его можно записать в следующем виде:
R2CHCR2OH + H3O+
r2chcr2oh + h3o+
r2chcr2oh+h3o+
; ~» R2CHCR2OH£
....r2chcr£+н2о
; > R2C = CR2
Медленно
> Е2(С+)
Характерная черта этого механизма состоит в том, что лимитирующей стадией
здесь является последняя стадия, обозначенная словом «медленно».
Возвращаясь к стереохимии реакций присоединения галогеноводородов
и иона гидроксония к циклоолефиновым соединениям, прежде всего следует
обратить внимание на такой факт [32]: циклические а,р-ненасыщенные
790
Присоединение и обратные реакции
кислоты, такие, как циклогексен-1-карбоновая кислота, присоединяют хло-
ристый водород в траис-положение
Трудность интерпретации такого примера состоит в том, что неизвестно, име-
ют ли дело с реакцией, локализованной при олефиновой связи, или, согласно
представлениям Тиле (см. следующий раздел), первоначально водород
присоединяется к карбоксильному атому кислорода с последующей прото-
тропной миграцией к а-углеродному атому, у которого его в конце концов
и находят.
Такая трудность в реакциях присоединения к олефиновым углеводородам не
встречается. Здесь были получены результаты двух типов. Первый можно
иллюстрировать примером Коллинса и Хэммонда по присоединению воды
к 1,2-диметилциклогексену в водном растворе азотной кислоты с образова-
нием почти равных количеств стереоизомерных спиртов в результате цис-
и транс-присоединения [33]. В этом случае не может возникнуть сомнений
в том, что изомеризация происходит уже после образования аддукта.
К этой реакции можно с уверенностью применить уже разобранную кинети-
ческую схему гидратации олефинов, которая показывает, что лимитирующей
стадией является стадия переноса протона, а в качестве промежуточного про-
дукта образуется карбониевый ион. Такая стереохимия, кроме того, согла-
суется с ожидаемой для механизма обратного отщепления Е1, в котором про-
межуточно образующийся карбониевый ион располагает достаточным вре-
менем для того, чтобы принять равновесную конфигурацию, так что асиммет-
рическое экранирование здесь не важно.
Второй тип стереохимии можно показать на примере изученной Коллинсом
и Хэммондом реакции присоединения хлористого водорода к 1,2-диметил-
циклопентену в растворе пентана [34]. В результате этой реакции образуется
транс-i,2-гидрохлорид, который при соответствующей температуре изоме-
ризуется в равновесную смесь цис- и транс-1,2-гидрохлоридов. Однако пер-
воначально получается только транс-изомер. В настоящее время кинетика
этого присоединения неизвестна, и любые предположения, которые мы пыта-
лись бы высказать, делались бы на весьма ненадежной основе. Исследования
Майо по кинетике присоединения бромистого водорода в примерно таких же
условиях дают основания полагать, что порядок реакции по хлористому
водороду должен быть равен примерно двум. Если это подтвердится, то тогда
можно будет предположить, что процесс является согласованным и его
стереохимия соответствует стереохимии отщепления типа Е2. Когда хлор
791
Глава XIII
присоединяется к молекуле, происходит локальное S ^-замещение с инвер-
сией, а когда присоединяется водород, происходит локальное ЗиЗ-замещепие
с сохранением конфигурации. В результате наблюдается тра«с-присоединение
О подобном транс-присоединении сообщалось в работе [35], в которой описа-
на реакция бромистого водорода с 1,2-диметилциклогексеном и другими
циклогексенами в среде пентана или уксусной кислоты. Хотя стереохимия
реакций является свойством их механизма, а не стехиометрии, все же авторы
не провели никаких кинетических исследований, чтобы связать их с этими
стереохимическими данными.
б. Присоединение сильных кислот, главным образом галогеноводород-
ных, к сопряженным полиолефиновым соединениям. В этом и последующих
разделах будут рассмотрены различные аспекты реакций присоединения
к сопряженным ненасыщенным системам. Поэтому целесообразно сделать
вступление ко всему кругу рассматриваемых вопросов и начать с описания
развития исследований в этой области.
Наиболее важным событием в исследовании реакций присоединения к сопря-
женным ненасыщенным системам явилась серия статей Тиле [36], опублико-
ванная в 1898 г. В первой статье он предложил свою теорию остаточного
сродства, возникающего у атома углерода, связанного двойной связью, вслед-
ствие того, что цепь сопряженных двойных связей оказывается насыщенной,
за исключением концевых атомов, остающихся ненасыщенными. Первоначаль-
но Тиле развивал свою теорию не только для того, чтобы объяснить, но и для
того, чтобы показать, каким образом происходит присоединение к концевым
двойным связям при наличии сопряженных двойных связей. Он пытался
объяснить исключения из этого правила, т. е. случаи 1,2-присоединения при
наличии структурной возможности 1,4-присоединения, предположив, что
1,4-присоединение может сопровождаться последующим таутомерным пре-
вращением. В качестве примера он приводил реакцию восстановления бен-
зила в бензоин
С6Н5СОСОС6Н5 С6Н5С(ОН) = С(ОН)С,Н5 —> С,Н5СОСН(ОН)С6Н5
В течение следующего десятилетия теория Тиле подверглась сильной крити-
ке, особенно со стороны Михаэля [37] и Гинриксена [38]. Эта критика своди-
лась прежде всего к теоретическому замечанию о том, что теория Тиле слиш-
ком мало внимания придает особому сродству между атомами присоединя
ющего агента и ненасыщенной молекулой, которое может быть представлено
как проявление электрохимических факторов. G практической стороны эта
критика сводилась к тому, что исключения из правила присоединения по
концам цепи накапливались гораздо быстрее, чем для этого можно было бы
найти объяснения. Например, коричный альдегид образует обычный
циангидрин с цианистым водородом, а не продукт 1,4-присоединения; этило-
вый зфир циннамилиденуксусной кислоты присоединяет малоновый эфир
792
Присоединение и обратные реакции
не по концам сопряженной цепи, а по двойной а,|3-связи
ОН
С6Н5СН = СНСНО —> с6н5сн=снсн;
\CN
С6Н5СН = СНСН = СНСООС2Н5 —> С6Н5СН = СНСНСН2СООС2Н5
(1н(СООС2Н5)2
Очевидно, необходимо было допустить, что если присоединяющийся агент
или ненасыщенная система являются несимметричными и высокополярными
или оба эти вещества обладают этими свойствами, то присоединение может
протекать несимметричным образом в силу особого, вероятно, электрохими-
ческого сродства между реагентами, что приводит к продукту присоедине-
ния, отличному от ожидаемого по теории Тиле. Но в 1909 г. стало ясно, что
даже эта уступка не спасает теорию Тиле: Штраус [39] показал, что симмет-
ричная молекула брома присоединяется к симметричному диолефину 1,4-
дифенилбутадиену несимметричным образом и получается исключительно
1,2-дибромид
СвН5СН = СНСН-СНС6Н5 -» С6Н5СНВгСНВгСН = СНС6Н5
Таким образом, следует признать, что, хотя теория Тиле и может объяснить
наблюдаемое присоединение по концам сопряженной системы, она не может
ни предсказать случаи, когда это правило не соблюдается, ни объяснить их.
Короче говоря, представления Тиле нельзя было считать ни теорией ориен-
тации, ни даже правилом ориентации для реакций присоединения к сопря-
женным ненасыщенным системам, которое по своему значению и определен-
ности можно было бы сравнить с правилами замещения в ароматическом
ряду, установленными к тому времени. Такое положение сохранялось до
1928 г., когда Бартон и автор этой книги [40] опубликовали первую статью
об анионотропных превращениях. В этой работе было предположено, что
присоединение электрофильного агента к сопряженному полиену может
приводить к образованию анионотропных таутомеров, и если присоединение
протекает по наиболее простому двухстадийному механизму, то вторая ста-
дия присоединения — соединение двух ионов, идентична второй стадии обыч-
ного SnI'-механизма анионотропной изомеризации. Это можно показать на
следующем примере:
СН2-=СН-СН=СН2 + НВг -> [СН3-СН-^СН^-СН2]+ + Вг-
СНгСНВг-СН=СН2 и СНгСН=СН-СН2Вг
С этой точки зрения авторы статьи и рассмотрели проблему ориентации в ре-
акциях присоединения к сопряженным системам. Позднее была предложена
более совершенная трактовка этого вопроса [41], которая будет рассмотрена
ниже.
В настоящем разделе будут рассмотрены только реакции присоединения типа
Айи кислых электрофильных реагентов к сопряженным полиолефинам
и теория ориентации для таких реакций. Затем будут обсуждены экспери-
ментальные данные по реакциям присоединения галогеноводородов.
Теория реакций присоединения должна рассматриваться с двух сторон в со-
ответствии с двумя стадиями процесса. Первая заключается в установлении
положения, в которое присоединяется протон в сопряженной системе. Мож-
но допустить, что место присоединения протона, как и в случае его присое-
динения к простым моноолефинам, рассмотренного в предыдущем разделе,
793
Глава XIII
непосредственно определяется электрохимическими принципами ориентации
в реакциях присоединения к ненасыщенным системам. Адденд, будучи элек-
трофильным, атакует в первую очередь атомы, богатые электронами. Следо-
вательно, в случае, например, бутадиена каждая из винильных групп явля-
ется электроположительной по отношению к другой и обладает 4-^-эффек-
том, поэтому электроны концевых атомов становятся наиболее доступными
для присоединяющегося протона. Если концы такой системы неэквивалентны
из-за наличия алкильных или арильных заместителей, то присоединение про-
исходит к тому концу молекулы, в котором сам заместитель может создавать
гиперконъюгационный или конъюгационный Ч-Е-эффект. Так, в случае
2-метилбутадиена-1,3 следует ожидать электрофильного присоединения про-
тона в положение 1, а в случае 1-фенилбутадиена-1,3 —в положение 4 и т. д
(1) СНз (4)
снДс—СН=СН2 С6Н?^-СН=^СН^-СН=^СН2
Экспериментальным доказательством правильности приведенных рассужде-
ний служит то, что не было выделено никаких аддуктов, образование кото-
рых опровергло бы сказанное. Так, в случае бутадиена возможно образование
трех моногидрохлоридов: 2-гидро-2-хлорида, 1-гидро-4-хлорида и 2-гидро-1-
хлорида. Из них только первые два действительно образуются. Для 2-метил-
бутадиена (изопрена) возможны шесть гидрохлоридов, но из них в результате
присоединения протона в положение 1 образуются только два, которые в дей-
ствительности и обнаружены. Аналогично 1-фенилбутадиен может образо-
вать шесть гидрохлоридов, но получаются только два из них в результате
первоначального присоединения в положение 4. Единственный аддукт, выде-
ленный в опытах, является одним из этих двух гидрохлоридов. Несколько
позднее мы снова вернемся к рассмотрению этих примеров.
Другая сторона проблемы ориентации является продолжением первой
и касается положения, в которое происходит присоединение сопряженного
основания присоединяющейся кислоты. Например, в случае присоединения
галогеноводородов эта часть проблемы связана с вопросом: какова судьба
предположительно образующихся ионов, а именно анионотропных галогенидов?
Прежде всего образование этих ионов определяется кинетическими факторами:
изомер, образующийся с большей скоростью, будет присутствовать в боль-
шем количестве. Но если галогениды могут легко переионизоваться, то их
соотношение будет определяться термодинамическими факторами: более
устойчивый изомер будет получаться в большем количестве.
Обычно в простых случаях можно предсказать, какой из двух анионотроп-
ных изомеров более устойчив (на основании сравнения структур с учетом
таких факторов устойчивости, как сопряжение и гиперконьюгация). Так, из
двух гидрогалогенидов бутадиена, которые могут образоваться путем при-
соединения протона к концевым атомам углерода, 1-гидро-4-галогенид
СН3СН = СНСН2Х несколько более устойчив, чем 1-гидро-2-галогенид
CHgGHXCH = СН2, вследствие возможного участия метильной группы пер-
вого из аддуктов в гиперконъюгации. Из двух гидрогалогенидов изопрена,
которые могут образоваться при присоединении протона в положение 1,
1-гидро-4-галогенид (СН3)2С = СНСН2Х значительно более устойчив, чем
1-гидро-2-галогенид (СН3)2СХСН = СН2, вследствие гиперконъюгации с уча-
стием каждой из двух метильных групп в первом аддукте. Из двух гидрогало-
генидов 1-фенилбутадиена, которые могут образоваться при присоединении
протона в положение 4, 4-гидро-З-галогенид С6Н5СН = СНСНХСН3 гораздо
794
Присоединение и обратные реакции
более устойчив, чем 4-гидро-1-галогенид С6Н5СНХСН = СНСН3, так как
только в первом случае возможно сопряжение фенильной и винильной групп.
Приведенные заключения затрагивают лишь качественную сторону ориента-
ционной проблемы. Они справедливы, если анионотропная система в усло-
виях реакции достигает равновесного состояния.
Однако предсказания относительно структуры основного продукта реак-
ции могут оказаться менее точными, если анионотропная система не стре-
мится к равновесному состоянию. В этом случае ориентация во второй ста-
дии присоединения будет определяться кинетическими факторами. Вполне
вероятно, что для анионотропных систем будет справедливо правило, при-
веденное в гл. XI, разд. 1,г, для прототропных систем. Согласно этому пра-
вилу, неустойчивая ионная форма таутомерной системы переходит в менее
устойчивую форму быстрее, чем в более устойчивую ковалентную форму
Таким образом, преобладающим продуктом реакции окажется менее устой-
чивый таутомер, что иллюстрировалось в предыдущем разделе.
Какой из двух факторов — кинетический или термодинамический — опре-
деляет последнюю стадию ориентации, будет зависеть от способности анионо-
тропных продуктов к ионизации и условий реакции. Факторами, определяю-
щими термодинамический контроль реакции, являются: наличие +£'-заме-
стителей (фенил > метил), которые способствуют отщеплению анионов (в ча-
стности, бромид > хлорид), и увеличение полярности растворителя.
Учитывая вышесказанное, рассмотрим теперь некоторые эксперименталь-
ные данные. Хараш и сотр. [42] исследовали присоединение хлористого водо-
рода к бутадиену-1,3 в отсутствие растворителя и в уксусной кислоте как
растворителе. Как уже упоминалось, при этом выделены 1-гидро-4-хлорид
и 1-гидро-2-хлорид и не обнаружен 2-гидро-1-хлорид. Содержание 1-гидро-
4-хлорида составляло 20—25% и не зависело от температуры в интервале
от —80 до 25 °C. В условиях реакции оба хлорида не претерпевают заметных
взаимопревращений. Однако при длительной обработке хлористым водо-
родом при 25 °C наблюдается изомерное превращение, в результате которого
образуется равновесная смесь обоих аддуктов, содержащая 75% 1-гидро-
4-хлорида. Таким образом, первоначально сказывалось влияние кинетиче-
ских факторов, приводящее к образованию в основном менее устойчивого
1-гидро-2-хлорида. При достаточной продолжительности реакции и срав-
нительно высокой температуре преобладают термодинамические факторы,
влияние которых приводит к образованию большего количества более устой-
чивого 1-гидро-4-хлорида.
В случае присоединения бромистого водорода к бутадиену-1,3 образуется
более подвижная анионотропная система. Хараш с сотрудниками получил
смесь продуктов, состав которой определялся кинетическими факторами.
Реакцию проводили только при низкой температуре (—80 °C) и во избежание
аномального присоединения бромистого водорода при тщательном удалении
катализирующих окислителей. Полученная смесь содержала 20% 1-гидро-
4-бромида. При более высокой температуре это соединение получается с боль-
шим выходом, а выделенные изомеры могут претерпевать взаимные пре-
вращения, что ведет к образованию равновесной смеси, которая, как было
показано [43], содержит около 80% 1-гидро-4-бромида.
Ультре [44] доказал, что в реакции присоединения хлористого водорода
к 2-метилбутадиену (изопрену) также существует кинетическая стадия.
Более ранние сообщения об этой реакции содержат противоречивые сведе-
ния. Некоторые авторы утверждают, что образуется только 1-гидро-4-хлорид,
а один автор нашел только 1-гидро-2-хлорид, в то время как другая группа
исследователей отмечает образование обоих веществ. Ультре показал, что
795
Глава XIII
в случае реакции при низкой температуре и при недостатке хлористого водо-
рода образуется преимущественно 1-гидро-2-хлорид. Однако этот аддукт
легко изомеризуется под каталитическим влиянием хлористого водорода
с образованием 1-гидро-4-хлорида. Этим объясняется выделение 1-гидро-4-
хлорида при избытке хлористого водорода. Таким образом, первоначальное
влияние кинетических факторов приводит к образованию менее устойчивого,
а последующее термодинамическое влияние — более устойчивого из двух
аддуктов, возможных при присоединении протона в положение 1 под ориен-
тирующим влиянием 2-метильного заместителя. Как и предполагалось, исхо-
дя из теоретических предпосылок, термодинамическая устойчивость 1,4-
аддукта относительно больше в случае изопрена, чем бутадиена. Результатом
этого является неизмеримо малое содержание 1,2-аддукта в равновесной
смеси в случае изопрена.
Изучено также присоединение бромистого водорода к изопрену, 2,3-диме-
тилбутадиену-1,3 и 1,4-диметилбутадиену-1,3 [45]. Ни в одном случае не
проявляется влияние кинетических факторов. В качестве основного про-
дукта всегда образуется, как и следовало ожидать, более устойчивый изомер
из числа тех, которые получаются согласно правилу присоединения протона.
Так, из изопрена образуется только 1-гидро-4-бромид, из 2,3-диметилбута-
диена-1,3 — только 1-гидро-4-бромид; в то же время в случае 1,4-диметил-
бутадиена получается главным образом 1-гидро-2-бромид СН3СН2СНВгСН =
~ СНСН3 и незначительное количество 1-гидро-4-бромида СН3СН2СН =
= СНСНВгСН3. В последнем случае можно было ожидать образования
1-гидро-2-бромида несколько более устойчивого, чем другой изомер, так
как эффективность гиперконъюгации с участием метильной группы больше,
чем с этильной группой.
Присоединение хлористого водорода к 1-метилбутадиену (пиперилену) ведет
к образованию только 4-гидро-З-хлорида или 4-гидро-1-хлорида [46]; оба
эти изомера имеют одну и ту же формулу СН3СНС1СН = СНСН3. Отсюда
следует совершенно ясный вывод, что присоединение протона идет, как
и следовало ожидать, в положение 4.
В случае 1-фенилбутадиена исследовалось присоединение как хлористого
[47], так и бромистого водорода [48]. И здесь также не удалось обнаружить
стадию, свидетельствующую о кинетическом влиянии. В каждом случае
единственным продуктом реакции оказывается 4-гидро-З-галогенид
С6Н5СН = СНСНХСН3. Он является более устойчивым из двух галоге-
нидов, которые могут образоваться при присоединении протона в по-
ложение 4.
Введение к концевому атому бутадиена электроноакцепторного заместителя,
такого, как карбоксил в сорбиновой кислоте, лишает образующиеся гидро-
хлориды свойства анионотропности [49]. Поэтому структура этих продуктов
определяется механизмом реакции, а не термодинамикой. Присоединение
протона происходит, согласно теории Тиле, к электроотрицательному заме-
стителю, например к кислороду карбоксильной группы, но впоследствии
происходит прототропная миграция в a-положение, где его в конце концов
и обнаруживают. Хлорид-ион присоединяется по месту максимальной эле-
ктронной недостачи, а именно к другому концу сопряженной системы в д-
положение. В результате из сорбиновой кислоты и аналогичных систем полу-
чается а-гидро-6-хлорид [49]
СН3СН = СНСН = СНСООН -» СН3СНС1СН = СНСН2СООН
Кинетика катализируемой кислотами 1,4-гидратации была подробно иссле-
дована на примере превращения фурана в сукциндиальдегид, вероятно, в ре-
796
Присоединение и обратные реакции
зультате кольчато-цепной и кето-енольной прототропии через 1,4-диоксибу-
тадиен
СН-СН НСН=СН НСН=СН
н/ СН _?л-‘™ нс^ Хсй НС7 ^снон -»
\0^
НСН = СН СН-СН сн2-сн2
I/ \ / ч / Ч
-» носн сно -» носн снон —> сно сно
Стемхаус, Дрент и Ван дер Берг [50] показали, что скорость этой реакции
скорее зависит от /г0, чем от [Н+], и убывает в дейтерированной воде
(/cH//cD = 1,7) так же, как и при кислотной гидратации простых олефинов
{гл. XII, разд. 3, а). Из этого следует такой же вывод: скорость реакции
зависит от лимитирующей стадии присоединения протона к ненасыщенному
атому углерода. Кинетических исследований по 1,4-присоединению галоге-
новодородов пока не опубликовано.
Сшереохшпия кислотной 1,4-гидратации также еще не исследована, но данные
по 1,4-присоединению галогеноводорода имеются. Стереохимия должна,
конечно, зависеть от механизма и особенно от того, является ли реакция ста-
дийной или нет. Согласно уже приведенным рассуждениям, согласованное
тримолекулярное трансЛ,2-присоединение хлористого водорода можно рас-
сматривать как согласованное тримолекулярное цис-1,4-присоединение
Первичными продуктами присоединения бромистого дейтерия к циклогекса-
диену-1,4 в растворе пентана являются г{ггс-1,4-гидробромид (80%) и транс-
1,2-гидробромид (20%) [51]. Кинетика и механизм этого превращения неиз-
вестны.
в. Присоединение электрофильных реагентов некислого характера,
главным образом галогенов, к моноолефиновым соединениям. Важнейшую
группу некислых электрофильных аддендов составляют галогены — хлор,
бром и иод, но не фтор. К ним относится также ряд других соединений, таких,
как хлористый нитрозили ацетилнитрат. (Реакции с фтором являются исклю-
чительно гомолитическими.) Присоединение хлора и брома к олефиновым
соединениям можно промотировать фотохимически или с помощью радикалов,
полученных в результате химической реакции. Тогда присоединение ока-
жется гомолитической цепной реакцией. Однако в данном разделе мы рас-
смотрим реакции, идущие в темноте и в отсутствие инициирующих радикалов.
Необходимость полярного растворителя для протекания таких реакций ясно
свидетельствует об их гетеролитическом характере. Известно, что так назы-
ваемое газофазное присоединение хлора и брома к этилену в действительно-
сти протекает на поверхности, которая к тому же должна быть полярной [52].
Установлено также, что присоединение брома и иода к олефинам в среде непо-
лярного растворителя весьма чувствительно к наличию полярной поверх-
ности [53] и таких полярных растворимых добавок, как галогеноводороды
[54] и вода [55]. Значительно проще обстоит дело в случае гетеролитического
процесса в таких обычных полярных растворителях, как вода, метиловый
спирт или уксусная кислота. В этих условиях исследовать механизм гетеро-
литического процесса легче всего.
797
Глава XIII
Действуя в качестве гетеролитических аддендов, галогены ведут себя как
электрофильные реагенты по отношению к ненасыщенным системам, за
исключением совершенно особых случаев, которые рассмотрены ниже. Такое
заключение вытекает из общей классификации, согласно которой галогены
представляют собой электрофильные реагенты (гл. V). Этот вывод особенно
подкрепляется данными, полученными при сравнении реакций присоедине-
ния галогенов к производным этилена [56]. Так, скорость реакций увеличи-
вается при введении в олефин электронодонорных заместителей типа фенила
СеН5 и метила СН3. В то же время электропоакцепторные заместители типа Вг,
+
СООН и N(CH3)3 затрудняют присоединение. Это же подтверждают данные
Робертсона и сотрудников * по исследованию кинетики в сопоставимых
условиях для большого числа реакций присоединения галогенов к олефинам.
Мы снова вернемся к этим работам несколько позднее.
Присоединение галогенов к олефинам в полярных растворителях можно
представить двухступенчатым механизмом с промежуточным образованием
карбониевого иона; этот процесс вполне аналогичен реакциям присоедине-
ния сильных кислот к олефинам, уже рассмотренным ранее
СН2 = СН2 + Вг2 СН2СН2Вг + Вг
СН2СН2Вг+Вг ВгСН2СН2Вт
Хотя первая и лимитирующая стадия этого механизма в некоторых случаях
нуждается в тщательном уточнении, все же такой порядок присоединения
доказывается тем, что вторую стадию реакции можно исключить при введении
достаточно большого количества каких-либо активных анионов. Такие анионы
можно вводить в виде соли, что доказано экспериментами Френсиса [57].
Так, например, реакции между бромом и этиленом в водных растворах
хлорида, иодида или нитрата натрия протекают с образованием следующих
продуктов:
Г NaCI-Н2О
•--------> СН2С1СН2Вг
Вг2 + СН2— СН2 <
NaI-НгО
NaNO3-Н2О
СН21СН2Вг
CH2(NO3)CH2Br
Кроме того, в реакции могут принимать участие анионы гидроксилсодер-
жащего растворителя: гидроксильные ионы воды, метилат-ионы метилового
спирта, ацетат-ионы уксусной кислоты. Так, для присоединения к олефино-
вой двойной связи элементов гипогалогенитной кислоты используется хлор-
ная или бромная вода [58], для присоединения элементов метил- или ацетил-
гипогалогенитов — растворы галогенов в метиловом спирте [59] или в уксус-
ной кислоте [60]
н2о
Вг2 + СН2 = СН2-----> Н+ + СН2(ОН)СН2Вг
СНзОН
Вг2 + С6Н5СН = СНС6Н5------> Н+ + С6Н5СН(ОСНз)СНВгСвН5
СНзСООН
С12+СН2 = СН2-------> Н+ + СН2(ОСОСН3)СН2С1
Следует показать, что эти реакции протекают согласно приведенным урав-
нениям, а не являются реакциями присоединения видоизмененного адденда,
* Робертсон и сотрудники опубликовали более двадцати статей главным образом в J. Chem.
Soc. Полученные ими данные по константам скоростей приведены в монографии де ла
Мара и Болтона [1].
798
Присоединение и обратные реакции
т. е. молекулярного гипогалогенита, образовавшегося при сольволизе гало-
гена
Hal2 + ROH ROHal 4-Н+ +Hal-
Необходимо учитывать, что образование оксигалогенидов во всех подобных
процессах сольволитического галогенирования олефинов сопровождается
образованием дигалогенидов. Подробное изучение этого вопроса возможно
путем исследования соотношения выделенных продуктов и скоростей их
образования. Найдено, что в случаях галогенирования в водной среде коли-
чество галогенгидрина в продуктах реакции не зависит от кислотности.
Очевидно, что влияние кислотности сказывалось бы, если образование гало-
генгидрина было бы связано с присоединением молекулярной кислоты типа
НОС1, получающейся согласно написанной выше обратимой реакции [57,
58]. Вместе с тем количество галогенгидрина уменьшается (как и следует
ожидать во всех случаях) при введении анионов галогена [58]. На примере
бромирования стильбена в метиловом спирте Бартлет и Тарбел [59] показали,
что скорость образования метоксибромида не зависит от кислотности. Более
того, фактически получаемый выход метоксибромида также не зависит от кис-
лотности, но уменьшается с добавлением бром-аниона, что с количественной
стороны может быть объяснено тем, что бромид-ион конкурирует с метиловым
спиртом в реакции с промежуточно образующимся карбониевым ионом.
Реальность существования таких промежуточных продуктов была доказана
Тарбелом и Бартлетом [59] путем получения р-лактонов при реакциях нат-
риевых солей диметилмалеиновой и диметилфумаровой кислот с хлорной
и бромной водой. Получение этих лактонов из образовавшихся галогенгид-
ринов невозможно, так как общеизвестно, что р-лактоны невозможно полу-
чить дегидратацией р-оксикислот или каким-либо другим вариантом метода
дегидратации
СНз СОО- СНз О —СО
-ООСС(СН3) = С(СН3)СОО- Н-а-12» ХС-С-Hal—> ^C-C-Hal
-ООС СНз -оос7 ^СНз
Эти полученные ранее выводы были подтверждены всеми последующими рабо-
тами. В качестве примера можно привести работу Белла и Принга [61] по
исследованию кинетики и продуктов реакции бромной воды с различными
олефиновыми соединениями с добавлением и без добавки нуклеофильных
агентов — анионов хлора и брома. Результаты работы ясно свидетельствуют
о том, что, вообще говоря, связи между кинетикой и продуктами реакции
не существует. Скорость реакции определяется строением субстрата, как
и следует ожидать для реакций, скорость которых контролируется атакой
электрофильного агента. Состав же продуктов зависит от конкуренции между
нуклеофильными агентами в быстро протекающей последующей реакции, что
не может влиять на скорость. Из этого правила найдено лишь одно исключе-
ние — этилфумарат, который, с одной стороны, взаимодействует с молекулой
брома, а с другой стороны, реагирует с водой и бромид-ионом, причем атака
двух последних реагентов происходит одновременно со скоростью, опреде-
ляющей скорость всего процесса. Этот исключительный пример не должен
казаться невероятным: сродство фумаратной структуры к нуклеофилам
вполне достаточно для того, чтобы вступить в реакцию с нуклеофильным
агентом без промежуточного образования карбониевого иона.
799
Глава XIII
Изучение кинетики присоединения галогенов указывает на то, что наиболее
простым является случай присоединения хлора к олефинам в растворителях,
полярность которых не ниже полярности уксусной кислоты. Как было уста-
новлено Робертсоном с сотрудниками, эти реакции для широкого ряда оле-
финовых соединений, главным образом в уксусной кислоте, протекают как
реакции второго порядка
Скорость ос [Олефин][На12]
Кинетические данные указывают, что агентом, атакующим олефин, явля
ется молекула галогена. Прибавление воды и ионизованных солей приводит
к увеличению скорости реакции, что находится в соответствии с картиной
тетеролитического процесса с полярным переходным состоянием. Влияние
изменений в структуре олефина на скорость присоединения (табл. 175)
указывает на то, что галоген действует на олефин как электрофильный реа-
гент.
Таблица 115
Сравнительные скорости присоединения хлора к олефинам типа
RC6H4CH = CHR' в уксусной кислоте
Заместитель Заместитель R'
R СОС6Н5 СООН сно CN no2 SO2ci
л-СН3 800 103
И 61 4,9 1,8 0,022 0,020 0,001
л-С1 23 2,5
n-NO2 0,001
Присоединение брома к олефинам в воде, метиловом спирте, муравьиной кис-
лоте и водных растворах низших спиртов и кислот [62, 63] также подчиня-
ется второму порядку при низкой (порядка 0,001 моль/л) концентрации бро-
ма. Как будет показано ниже, при других условиях кинетика бромирования
имеет более сложный вид. Робертсон и другие объясняют это, исходя из пред-
положения, что олефин и бром образуют предравновесный молекулярный
комплекс, который разрушается различными катализаторами с разной скоро-
стью. При отсутствии таких катализаторов этот предравновесный процесс
должен сохраняться, но в этом случае вряд ли лимитирующей стадией может
быть что-нибудь иное, чем ионизация комплекса. Согласно ряду исследова-
ний, присоединение аниона никак не отражается на скорости реакции при-
соединения
А-фВг2 А-Вг2 А-Вг2 —> АВг+ +Bi-
Этот взгляд был поддержан Дубойсом [63], который привел спектроскопиче-
ские доказательства существования предравновесного состояния, а также
доказательства, основанные на значительном влиянии растворителей на
скорость реакций, контролируемых ионизацией. Таким образом, первая
стадия первоначального двухстадийного механизма присоединения брома
в среде высокополярных растворителей распадается на две стадии, приведен-
800
Присоединение и обратные реакции
ные ниже:
R2C=CR2 + Br2«=±:R2C—;CR2
_B’r—Вг
R2C—cr2 MedJieHHO R2C—cr2
-к-вг —' k + Br'
Точная структура предравновесного комплекса остается пока неясной.
При тех же условиях, в которых происходит присоединение хлора и брома,
присоединение иода, как правило, заметно обратимо. Однако установлено,
что в воде прямая реакция имеет тот же второй порядок [62].
Присоединение брома в уксусной кислоте, но не в более полярном растворителе
в определенном интервале концентраций (около 0,025 моль/л) протекает как
реакция третьего порядка
Скорость сс [Олефин][На12]2
При разбавлении, повышении температуры или прибавлении воды к раствору
проявляется тенденция к снижению порядка реакции от третьего ко второму.
Влияние изменений в строении олефина на скорость присоединения брома,
происходящего по закону третьего порядка, поразительно сходно с аналогич-
ным влиянием на скорость присоединения хлора в уксусной кислоте, проис-
ходящего по закону второго порядка; действительно, соотношение скоростей
этих двух процессов представляет собой почти постоянную величину. Поэто-
му повышенный на единицу порядок реакции с бромом не означает, что про-
исходит существенное изменение механизма присоединения, хотя несомненно,
что другая молекула галогена участвует в переходном состоянии стадии,
определяющей скорость реакции.
Присоединение иода в уксусной кислоте также отвечает реакции третьего
порядка [1]. Аналогично присоединяются хлористый бром, хлористый иод
и бромистый иод. Сравнительные скорости присоединения различных гало-
генов по указанному кинетическому закону выражаются следующими вели-
чинами [1]:
I2 IBr Br2 IC1 ВгС1
Скорость 1 3000 10 000 100 000 4000 000
Последовательность IC1 > IBr > I, соответствует иодирующей способности,
а ряд ВгС1 > Вг2 — бронирующей. Это находится в соответствии с прин-
ципом, что X — Y — тем лучший электрофильный «Х-ирующий» агент, чем
сильнее Y притягивает электроны. Приведенное выше соотношение скоро-
стей заставляет предположить, что присоединение начинается с электро-
фильного введения наиболее электроположительного атома галогена.
Насколько это удалось установить, при присоединении высших галогенов
(брома, иода) независимо от того, имеет реакция второй или третий порядок,
структурные эффекты проявляются аналогично тому, как в реакции при-
соединения хлора: электронодонорные заместители ускоряют, а электроно-
акцепторные заместители замедляют реакцию. Сходство состоит также в на-
блюдаемом положительном солевом эффекте; единственное отличие состоит
в том, что ионы галогена могут усиливать специфический каталитический
эффект, о чем говорилось выше.
То обстоятельство, что реакция третьего порядка не характерна для хлора
и что для нее необходимо наличие в молекуле галогена хотя бы одного атома
51-0772
801
Глава XIII
брома или иода, приводит к предположению, что при реакциях третьего по-
рядка используется более высокая способность к координации, характерная
для более тяжелых галогенов. Обычно считают [64], что молекула галогена
обратимо присоединяется за счет расширения октета одного из атомов гало-
гена с образованием мало устойчивого продукта присоединения в незна-
чительной стационарной концентрации. Этот аддукт сам не способен к отщеп-
лению второго атома галогена в виде аниона, что возможно только в том
случае, если другая молекула галогена участвует в завершении процесса
присоединения (А = олефин)
А+Х2 А-Х2 А"Х2-|-Х2 —> Продукты реакции
Эту формальную схему можно проиллюстрировать следующим примером:
R2C=CR2 + Br2^=^R2C—CR2
“Вг—Вг
R2C—CR2 Медленно —CR2
|/ + Вгг. Медле.я^ I I +Вг2
~Вг—Вг Вг Вг
Вторая стадия этой реакции в действительности может состоять из двух ста-
дий.
Присоединение брома в уксусной кислоте к некоторым олефиновым соедине-
ниям, а именно винил- и аллилгалогенидам, катализируется бромид- и хло-
рид-ионами. Этот каталитический процесс был открыт Нозаки и Оггом [65]
и в дальнейшем исследован Сведлундом и Робертсоном. Кинетика его описы-
вается уравнением
Скорость ос [Олефин][Вг2][На1~]
В случае ряда соединений, для которых проявляется отмеченное каталити-
ческое влияние, скорость присоединения точно так же зависит от структуры,
как и для некаталитического процесса третьего порядка, что иллюстрируется
приводимыми ниже данными по сравнительным скоростям присоединения
брома в уксусной кислоте при 25 °C. При этом олефин и бром применялись
в концентрации 0,0125 моль/л, а бромид и хлорид лития — в концентрации
0,05 моль/л.
СН2 = СНСН2С1 ch2=chch2cn CH2 = CHBr
Вг2 1,6 0,23 0,001
Br2 + LiBr 4 1,2 0,005
Br2 + LiCl 10 2,4 0,007
Из приведенных данных видно, что каждая реакция характеризуется резким
уменьшением скорости (примерно в 1000 раз) при переходе от аллил- к винил-
галогенидам. Это указывает на то, что обсуждаемые каталитические реакции
остаются электрофильными процессами и поэтому не зависят от наличия
предварительно образовавшихся пергалогенид-ионов CIBrg или Вгд, кото-
рые должны обладать нуклеофильным характером. Следовательно, можно
предполагать, что рассматриваемые каталитические реакции являются только
вариантами некаталитических процессов третьего порядка; они протекают,
если ион галогена значительно быстрее, чем молекула галогена, способен
завершить вторую стадию присоединения
А + Вг2 А-Вг2 А-Вг2|-На1“ —> Продукты реакции
802
Присоединение и обратные реакции
Эту формальную схему можно проиллюстрировать следующим примером:
R2C— CRa "Ь Вга <—— RaC CRa
-Вг—Вг
R2C—.CR2 + Медленно ^Ra
"Вг—Br Вг X
Вторая стадия этой реакции также может быть двухстадийной.
Галогены являются типичными электрофильными реагентами. Однако можно
предвидеть, что если олефиновая связь в достаточной степени поляризована
электроноакцепторной группой, то можно осуществить нуклеофильное при-
соединение галогена AdN. Убедительным доказательством возможности этого
служат примеры присоединения хлора и брома к а,^-ненасыщенным альдеги-
дам и кетонам в уксусной кислоте в присутствии сильных кислот [66]. В отсут-
ствие сильных кислот присоединение протекает медленно, но оно энергично
катализируется сильными кислотами; водой катализ подавляется. Зависи-
мость скорости каталитического присоединения от структуры совершенно
иная, чем в случае обычного электрофильного присоединения, например
присоединения хлора по реакции второго порядка. Это иллюстрируется сле-
дующим рядом сравнительных скоростей катализируемого серной кислотой
присоединения брома и некаталитического присоединения хлора; при этом
все реагенты, включая серную кислоту, применялись в концентрациях
0,0125 моль/л в уксусной кислоте:
С6Н5СН
II
нссосбн5
Br2+H2SO4 32
С12 61
С6н5сн
I
НССООС2Н5
<0,02
10
С6н5сн
II
нссно
27
1,8
СН3СН
II
нссно
> 1000
0,41
с6н5сн
II
HCNOj
0,005
0,020
Приведенные данные указывают па то, что как поляризация двойной связи,
так и основность исходной ненасыщенной молекулы являются необходимыми
условиями для высокой чувствительности к катализу. Предполагается,
что лимитирующей стадией процесса будет нуклеофильная атака молекулой
галогена карбониевого иона, образовавшегося в результате присоединения
протона к основной группе исходной молекулы по следующему уравнению:
. + ‘ Вг2
RCH = СН-CR' = О + Н+ 7^ RCH-CH=CR'-OH —
RCH-CH = CR'-ОН
| Продукты реакции
+Вг—Вг
В качестве кислотных катализаторов для подобных случаев присоединения
галогенов бромистый и хлористый водороды более эффективны, чем другие
кислоты, чего нельзя ожидать, исходя из известной сравнительной силы
кислот в уксусной кислоте
НС1О4> НВг’> H2SOd'> HC1>1HNO3
Так, относительные скорости присоединения брома к коричному альдегиду
в уксусной кислоте при концентрации реагентов 0,0125 моль/л и концентра-
51* 803
Глава XIII
ции катализирующей кислоты 0,03 моль/л выражаются следующими вели-
чинами:
НСЮ4 НВг H2SO4 НС1 HNO3
7,3 300 3,4 24 0,8
Предполагается, что каталитическое действие галогеноводородных кислот
проявляется не только вследствие уже описанного присоединения протона
к коричному альдегиду, но также и в результате образования сложных анио-
нов Вг3, С1ВГ2, Cl2Br~, C1J. Эти анионы являются нуклеофильными реаген-
тами, и по своему действию при присоединении галогена к иону карбония
они более эффективны, чем нейтральные молекулы галогенов.
Рассмотрев некоторые вопросы кинетики и катализа, вернемся к продуктам
некаталитического присоединения галогенов и прежде всего к проблеме ориен-
тации, не затрагивая, однако, возможной стереохимической специфичности
реакций.
Ориентация несимметричных галогенов протекает так, что более электропо-
ложительный атом, например атом иода в монохлористом иоде, направляется
преимущественно туда, куда присоединился бы атом водорода при нормальном
присоединении галогеноводорода [67]. Это иллюстрируется примерами
СН3СН = СН2 —* СН2СНС1СН21 CH2 = CHSO3H —» CH2C1CHISO3H
С6Н5СН = СН2 -> С6Н5СНС1СН21 СН3СН = СНСООН -» СН3СНС1СШСООН
Такая ориентация не является количественно однозначной, но проявляется
достаточно ясно для того, чтобы принять механизм, согласно которому иод
участвует в реакции как электрофильная частица 1+, а хлор — как нуклео-
фильная частица С1~.
В соответствии с двухстадийным механизмом образования галогенгидринов
при действии на олефины водных растворов галогенов можно предположить,
что атом галогена и гидроксильная группа присоединяются соответственно
таким же образом, как и атомы водорода и галогена при электрофильном
присоединении галогеноводорода. В некоторых случаях это так и происходит.
Так, изобутилен, который ничего, кроме mpem-бутилхлорида, не образует
при присоединении хлористого водорода, в результате взаимодействия с хло-
ром в воде и присоединения элементов хлорноватистой кислоты дает только
хлор-щрет-бутиловый спирт. Но с уменьшением электроположительности
групп на одном конце этиленовой связи ориентация в этих двух примерах
электрофильного присоединения становится разной, а на определенной ста-
дии — качественно противоположной. Это иллюстрируют данные, взятые из
разных источников де ла Мэром и Болтоном [1] * и суммированные в табл. 176.
Во всех приведенных в таблице примерах присоединение галогеноводорода
происходит таким образом, что водород присоединяется к концевому атому
олефина, а для хлорноватистой кислоты происходит изменение ориентации
присоединения от полного присоединения хлора к концевому атому до почти
полного его присоединения к неконцевому атому углерода.
Очевидно, это нельзя объяснить исходя из предположения, что различие меж-
ду предпочтительной и непредпочтительной поляризацией олефинов обуслов-
лено различием в силе электрофильных агентов. Необходимо исходить из не-
* Следует иметь в виду, что некоторые из старых данных могут быть неточными; это
особенно относится к присоединению НХ.
804
Присоединение и обратные реакции
которого фундаментального различия между присоединяющимися водородом
и хлором; это различие состоит, по-видимому, в том, что только хлор обладает
неподеленными электронами. В результате присоединения галогена обра-
зуется Р-галогенкарбониевый ион, который должен иметь конфигурационные
Таблица 176
Присоединение (%) водорода к концевому атому в реакциях
электрофильного присоединения галогеноводородов к олефинам
и присоединение (%) хлора из хлорноватистой кислоты к концевым
атомам олефина в реакциях олефинов с хлорной водой
Присоединяю- щийся атом сн3. ;с=сн2 CH3Z СНз. ;с=сн2 н7 носн2. >с=сн2 н7 С1СН2. Ч.С=СН2 Н7 С12СН >с=сн2 н7
Концевой Н из НХ 100 100 100 100 100
Концевой С1 из НОС1 100 91 73 30 2
свойства, описанные в гл. VII, разд. 7,ж. Согласно представленной в этом
разделе картине, образующийся Р-хлоркарбониевый ион должен иметь в сво-
ей структуре длинную ослабленную сильно электростатическую связь.
Благодаря этому атом хлора становится легко подвижным, что позволяет
осуществиться Р — a-переходу и приводит к обратной ориентации. В подоб-
ной ситуации водород не обладает подвижностью. Проиллюстрируем это
на примере; сначала, согласно де ла Мару и Притчарду [68], представим
первоначально образующийся карбониевый ион формулой (Л) и будем считать,
что хотя в пределах своего времени жизни этот ион и может превращаться
в изомер Б, последний термодинамически менее устойчив
R-QH—CHj-^R—СН—РН2
Х''С1 if' Б
Малая доля изомера Б в равновесной смеси (если предположить, что рав-
новесие достижимо) не препятствует непропорционально большой доле при-
соединения по пути Б. Изомер Б, как первичный карбониевый ион, будет
атаковываться растворителем или любым анионом с большей скоростью, чем
вторичный карбониевый изомер А.
Такое описание хорошо подтверждается критическим исследованием де ла
Мара, Нейлора и Ллин Уильямса [69], посвященным реакциям присоедине-
ния хлорноватистой кислоты к аллилгалогенидам, включая такой важный
случай, как присоединение к аллилхлориду, содержащему изотопную метку
36С1. Во всех случаях эти исследователи получили три продукта: 1,3-дигало-
генпропанол-2, не содержащий атома галогена у среднего атома углерода,
что соответствует иону карбопия А; 1,2-дигалогенпропанол-З, содержащий
атом галогена, присоединившийся к среднему атому углерода от адденда,
что соответствует карбониевому иону Б; 1,2-дигалогенпропанол-З, получаю-
щийся вследствие миграции концевого атома галогена молекулы олефина
в центральное положение. Это подтверждает реальность процесса миграции
805
Глава XII/
галогена. Кроме того, соотношение продуктов показывает, что промежуточ-
ное образование симметричного соединения не является преобладающим на-
правлением реакции. В примере с хлористым аллилом, меченным 36С1, и ад-
дендом, содержащим немеченый С1, можно написать различные проме-
жуточные структуры, в которых 36С1 и С1 совершенно эквивалентны (не считая
изотопной метки) и в которых один атом хлора находится на одном конце
молекулы, а второй — на другом ее конце. Однако состав продуктов реакции
показывает, что вновь присоединившийся атом хлора более склонен к пере-
мещению к среднему атому углерода, чем уже имевшийся атом 36С1. Таким
образом, нельзя предполагать, что первичным продуктом присоединения
хлора будет открытый карбониевый ион, который должен быть симметрич-
ным; если бы в симметричном ионе образовывалась сильно электростатиче-
ская связь, то происходила бы изомеризация в двух равноценных направле-
ниях и получалось бы 50% карбониевого иона (4) и 50% его изотопной моди-
фикации (Л')
СН2—СН—СН2 СН2—СН—СН2 СН2—,СН—СН2
I I I \ I + I ” I
S’CI Cl 36С1 Cl 36С1 Cl
А’
При этом доли миграции С1 и 36С1 к среднему атому углерода были бы равны,
чего на самом деле не наблюдается. Далее, нельзя принять, что первоначаль-
но образующийся карбониевый ион А обладает эффективной симметрией
в результате равновесного быстрого перемещения связей
А А'
Нельзя также предполагать, что А переходит в симметричное электростати-
чески связанное промежуточное соединение
СН2—СН—СН2
I, X |
36ci ci
которое могло бы в этом случае быть в равновесии с А и А'. Состав продуктов
показывает, что симметрии, к которой ведут все изложенные выше идеи, на
самом деле нет. Все это согласуется с первоначальной картиной де ла Мара
и Притчарда: первоначально образовавшийся карбониевый ион является
несимметричным (Л) и имеет несимметричную электростатическую связь,
которая сохраняется в нем в том же виде, в каком она существовала в момент
ионизации.
Состав продуктов присоединения хлорноватистой кислоты к аллилхлориду-
36С1 в водном растворе, найденный де ла Мэром, Нейлором и Ллин Уильям-
сом, приводится ниже: 70% составляет продукт, в котором атом хлора мигри-
ровал от концевого атома углерода к среднему. За короткое время жизни
первоначально образующийся карбониевый ион Л успевает изомеризоваться
в А' всего лишь на 6%, а не на 50%, как было бы при достижении равнове-
сия. За это время 94% иона Л либо захватываются растворителем, либо изо-
меризуются в Б, который еще быстрее захватывается растворителем, вероятно,
806
Присоединение и обратные реакции
настолько быстро, что не успевает снова превратиться в А.
СН2—СН—СН2
I I /' Н2О
I I/' ---> 1-36С1-2-С1-3-ОН
36С1 С1
(66%)
сн2—сн—сн2
\ I I Н2о
I --------------> 1-С1-2-36С1-3-ОН
36С1 С1
В случае брома и иода следует ожидать увеличения способности к образова-
нию связи с катионным центром иона и, следовательно, большей доли мигра-
ции галогена и большего перехода такого взаимодействия от галогена субст-
рата к галогену присоединяемой галогеноватистой кислоты. Кларк и Ллин
Уильямс [69] исследовали присоединение бромноватистой кислоты к броми-
стому аллилу в водной среде при действии водного брома, подкисленного
хлорной кислотой. В качестве изотопной метки они использовали 82Вг,
вводя его или в аллибромид, или в бром; результат был один и тот же. Они
установили, что миграция брома проходит на 70%, а изомеризация А ->
-+А', которая меняет функцию двух атомов галогена,— на 30%. Схемати-
чески это можно выразить следующим образом:
В реагенте
\СН2—СН—СН2 СН2—СН—СН2
| \ | 55°Z? | | / Нг0
82Вг Вг 82Вг Вг'
1-82Вг-2-Вг-3-ОН
(55%)
СН2—СН—СН2
\ I | Н2О
82Вг Вг
1-Вг-2-82Вг-3-ОН
(24%)
807
Глава XIII
При присоединении хлорноватистой кислоты к аллилбромиду или аллилиоди-
ду следует ожидать еще большего увеличения тенденции к переходу электро-
статической связи в направлении А ->-А'. Эти реакции были исследованы
де ла Маром и Ллин Уильямсом [69]. В настоящее время мы не располагаем
сравнительно простым принципом установления химической идентичности
изотопов, который позволял бы рассчитать полную схему превращения, одна-
ко можно определить верхний и нижний пределы. Степень смещения электро-
статической связи А ->А’, происходящего до того, как все карбониевые ионы
будут захвачены растворителем, при переходе от хлора к брому изменяется
от 28 до 60% и при переходе от хлора к иоду — от 48 до 79%. Очевидно,
скорость связывания растворителем слишком велика, чтобы могло уста-
новиться равновесие между различными карбониевыми ионами.
Де ла Мар и Галандауер 170] исследовали состав продуктов реакций при-
соединения бромноватистой кислоты и хлористого брома к пропилену. Эти
реакции протекают совместно при обработке углеводорода водным раствором
брома в соляной кислоте. С увеличением концентрации соляной кислоты
от 1 до 3 н. концентрация образующегося бромхлорпропана возрастает с 30
до 59%. Это означает, что отношение суммарного присоединения ВгС1 к сум-
марному присоединению ВгОН увеличивается от 0,43 до 1,44. Именно такого
увеличения (в 3,35 раза) можно ожидать. Вместе с тем доля образующегося
бромхлорпропана, в котором атом брома занимает концевое положение, оста-
ется постоянной и равной 54% во всей области изменения концентрации хло-
рида; постоянным, но несколько более высоким (79%) остается и содержание
бромгидрина с концевым атомом брома. Таким образом, для вторичного кар-
бониевого иона типа А (с бромом в качестве галогена) соотношение между
продуктами присоединения хлорид- и гидроксил-ионов из молекул воды
растворителя хотя и меняется с изменением концентрации хлорида, все же
постоянно в 3,1 ± 0,1 раза меньше, чем аналогичное соотношение для пер-
вичного карбониевого иона типа Б. Таким образом, сравнивая каждый кар-
бониевый ион с другим, можно сказать, что вторичный ион типа А предпоч-
тительно присоединяет гидроксил-ионы, а первичный ион типа Б — хлорид-
ионы. Это может означать только то, что система вторичного алкила ближе,
чем система первичного алкила, к предельному случаю псевдооснования,
когда галогены являются ионными соединениями, а оксисоединения —
неионными.
До 1954 г. широко было распространено мнение, основанное на предполо-
жении Робертса и Кимбалла [74], которое будет цитироваться ниже. Они счи-
тали, что для объяснения присоединения галогенов достаточно предположить,
что существует лишь одно промежуточное соединение — катион
С1СН2 — СН2 илп]| СН3СН-СН2
''СГ ХВг+
где галоген находится в ониевой форме, образуя мостик между атомами,
к которым происходит присоединение. Развивая свою теорию двух карбо-
ниевых ионов (Л и Б), де ла Мар и Притчард отметили, что свойства, которые
можно приписать иону Робертса — Кимбалла, аналогичны свойствам эпок-
сидов *, таких, как
С1СИ2СН--С112 или СН3СН-СН2
'б7
* В настоящем издании они не рассматриваются в целях сокращения объема книги;
см. первое издание, стр. 275 и сл.
808
Присоединение и обратные реакции
Раскрытие кольца эпихлоргидрина и окиси пропилена при действии нуклео-
фильных агентов идет преимущественно или исключительно в направлении,
которого следует ожидать в случае замещения типа SN2.
Мы можем рассмотреть модифицированные ионы Робертса — Кимбалла,
допустив, что они являются синартетическими, т. е. допустив наличие неко-
торого заряда на атомах углерода
С1СН2СН—сн2
или
С1
СНзСН—сн2
Ъг
Тогда можно говорить, что большая часть заряда атомов углерода сосредото-
чена на вторичном атоме углерода, хотя такой заряд на первичном атоме
углерода оказывает непропорционально большое влияние на результирую-
щую реакционную способность. Таким образом, можно сделать вполне обос-
нованные предположения, которые воспроизводят все основные выводы
де ла Мара и Притчарда. Единственным недостатком этой теории является
сам принцип, который особенно критиковал Браун *; дело в том, что не
стоит рассматривать ион как синартетический, если в этом нет крайней необ-
ходимости. Точка зрения Брауна является обоснованной: рисковать не стоит,
поскольку переоценка роли синартетических ионов стала обычным явлением,
что вызывает множество дискуссий.
Со стереохимической точки зрения электрофильное присоединение галогенов
или реакции присоединения, инициированные галогенами, как правило
(но не всегда), протекают с образованием mpawc-продуктов. Присоединение
хлора и брома в гидроксилсодержащих растворителях к малеиновой кислоте
дает боЛее 80% рацемической дигалогенянтарной кислоты, а к фумаровой
кислоте — более 80% мезоизомера [72]. В результате присоединения хлора
в отсутствие растворителя к цис- и тра«с-бутену-2 образуется почти исклю-
чительно рацемический и мезо-2,3-дихлорбутаны соответственно [73]. Все эти
примеры иллюстрируют т/щнс-присоединение. Согласно Робертсу и Кимбал-
лу [74], конфигурация продуктов определяется тем, что первый присоеди-
нившийся атом галогена удерживает конфигурацию промежуточного катиона,
который, как уже упоминалось, они рассматривали как ион галогенония:
* Гл. X, разд. 1,ж, сноска на стр. 639. Наблюдение единственного сигнала в спектре
ЯМР раствора тетраметилэтилена и хлора в смеси пятифтористой сурьмы и двуокиси
серы при температуре—60 °C [71] не исключает возможности внутримолекулярного прев-
ращения асимметричных ионов со скоростью, меньшей, чем скорость их захвата молеку-
лами водных растворителей.
809
Глава XIII
Как малеат-ион, так и фумарат-ион, присоединяя хлор и бром в воде, дают
мезодигалогенсукцинат-ион [72]. Первая реакция служит примером цис-
присоединения. Робертс и Кимбалл [74] высказали предположение, что
в этом случае происходит разрыв связи, удерживающей конфигурацию моле-
кулы, так что структура может испытывать внутреннее вращение под влия-
нием сил, действующих между ионными цис-карбоксилатными группами.
Такое объяснение, по-видимому, все же нельзя считать достаточным, так как
Белл и Принг наблюдали цис-присоединение брома в воде к этилмалеату [61].
Конкуренция между первоначально присоединившимся галогеном и карбок-
силат-ионом или карбонильной группой в несении ответственности за кон-
фигурацию молекулы может отчасти играть роль. Такие связи, сохраняю-
щие конфигурацию молекулы, не всегда будут прочными, так как в против-
ном случае присоединение было бы более стереоспецифичным, чем оно есть
в действительности.
Преобладающее цис-присоединение молекулярных галогенов, или по край-
ней мере хлора, встречается редко, и в тех случаях, когда карбониевый
ионный центр, который может образовываться в результате начальной атаки
галогеном, оказывается сопряженным с ненасыщенной цепью полисопряжен-
ной системы. Возможны и другие причины, например кинетического харак-
тера или определяемые влиянием растворителя на диссоциацию ионов, но
говорить об этом сейчас с достоверностью нельзя. Другое затруднение состоит
в том, что сама молекула олефина может претерпевать в процессе присоеди-
нения изомерное превращение в результате, например, обратимости первой
стадии присоединения. Так, присоединение хлора к цис-стильбену в раст-
воре дихлорэтана не является стереоспецифичным и дает в большом количе-
стве продукт цис-присоединения — мезодихлордибензил [75]. Вместе с тем
известно, что в реакции присоединения брома, которая также не является
стереоспецифичной, образуется некоторое количество транс-стильбена [76].
С другой стороны, присоединение хлора к транс-стильбену в растворе ди-
хлорэтана не стереоспецифично, но дает в большом количестве рацемический
дихлордибензил — продукт цис-присоединения [75]. Маловероятно, что это
можно объяснить превращением транс-стильбена в цис-стильбен в процессе
реакции, так как это противоречило бы термодинамике процесса.
Это затруднение можно обойти путем включения двойной связи, по которой
происходит присоединение, в цикл, достаточно малый, чтобы эта связь могла
находиться только в цис-конфигурации. Известно, что аценафтилен при-
соединяет хлор в растворе бензола с образованием цис-1,8-дихлорида [77],
а фенантрен присоединяет хлор в растворе уксусной кислоты с образова-
нием цмс-9,10-дихлорида [78]
В обоих этих случаях, так же как и в примере транс-стильбена (который
имеет плоскую конфигурацию, а его цис-изомер — не плоскую [75]; гл. III,
разд. 2, б и 4, в), любой карбониевый ионный заряд, образующийся на пер-
вой стадии присоединения хлора, будет рассредоточен по всей ароматической
системе. Поэтому следует ожидать ослабления, а возможно, и разрыва любой
810
Присоединение и обратные реакции
связи, ответственной за сохранение конфигурации, которая могла бы уста-
новиться между присоединившимся хлором и ближайшим карбониевым ион-
ным центром.
Присоединение хлора к фенантрену в растворе уксусной кислоты было иссле-
довано де ла Маром с сотр. [78] достаточно подробно, что позволило высказать
некоторые соображения относительно его механизма. Кинетически реакция
имеет второй порядок: первый относительно фенантрена и первый относи-
тельно хлора. Применение радикальных ингибиторов не обнаружило гомо-
литического характера процесса. В продуктах реакции содержится 34%
основного продукта замещения 9-хлорфенантрена, 42% основного продукта
присоединения 9,10-дихлорида, который полностью или почти полностью
состоит из цис-изомера, и 14% 9-хлор-10-ацетоксифенантрена — продукта
присоединения, который преимущественно является шраис-аддуктом с не-
большой примесью цис-изомера. При осуществлении этой реакции в при-
сутствии хлористого лития соотношение между продуктами присоединения
и замещения не увеличивается.
Предположим, что сопряжение с ароматической системой незначительно
влияет на конфигурацию первоначально образующегося монохлор-катиона.
Такой катион должен либо присоединить анион с образованием продукта
присоединения, либо потерять протон с образованием продукта замещения.
Поскольку количество продуктов присоединения не увеличилось за счет
уменьшения выхода продуктов замещения при введении в систему дополни-
тельных хлорид-ионов хлористого лития, можно сделать вывод, что свобод-
ные хлорид-ионы не могут входить в молекулу дихлорида и что второй атом
хлора в молекуле дихлорида появляется из присоединяющейся молекулы
хлора. Этот атом хлора принимает электрон у другого атома хлора, но после
этого не отдаляется от первоначально присоединившегося атома хлора,
а образует противоион ионной пары. Известно, что ионные пары в уксусной
кислоте стремятся уплотниться, так как она обладает низкой диэлектриче-
ской проницаемостью (около 6; гл. VII, разд. 6,ж). Можно понять поэтому,
почему образующийся дихлорид является цис-аддуктом. Иначе образуется
хлорацетокси-аддукт, поскольку в этом случае две части адденда первона-
чально входили в разные молекулы. В рассматриваемом случае хлорид-ион,
оставаясь некоторое время неприсоединенным, экранирует карбониевый ион
с одной стороны, препятствуя его реакции с растворителем — уксусной кис-
лотой. Однако с другой стороны растворитель может свободно реагировать
с карбониевым ионом. Таким образом, происходит конкуренция между
взаимодействием карбониевого иона с хлорид-ионом ионной пары и взаимо-
действием карбониевого иона с растворителем — уксусной кислотой. В ре-
зультате такой конкуренции одновременно образуются цис-дихлорид и хлор-
ацетат, который преимущественно имеет шраис-конфигурацию *.
г. Присоединение электрофильных реагентов некислого характера, глав-
ным образом галогенов, к сопряженным полиолефиновым соединениям.
Вопросы, связанные с присоединением галогенов к сопряженным полиенам,
в частности порядок присоединения и связь его с равновесием и подвижностью
анионотропных систем, рассмотрены в уже упомянутых статьях Бартона,
де ла Мара, Хьюза и автора книги.
Теория ориентации при присоединении типа AdE к сопряженным системам
исходит из тех же общих принципов, что и теория ориентации при присоеди-
нении к полиенам галогеноводородов (гл. XII, разд. 3,6). Считается, что
* Эта теория принадлежит авторам исследования, за исключением несущественной
детали: они считают межгалогенную связь в промежуточном соединении не ионной,
а ковалентной, что требует расширения валентной октетной оболочки.
811
Глава XIII
первый атом галогена, присоединяющийся к углероду, ведет себя как электро-
фильная частица и, следовательно, занимает положение, где находился бы
протон при присоединении галогеноводорода, т. е. всегда в конце сопря-
женной системы; если концы системы отличаются между собой, то атом гало-
гена присоединяется к концевому атому углерода, который характеризуется
большим -{-^'-эффектом. Как и в предыдущих случаях, это правило подтверж-
дается тем, что в продуктах реакции присоединения отсутствуют соединения,
образование которых противоречило бы этому правилу.
Второй атом галогена может присоединиться к сопряженному диену в двух
местах, в случае триена — в трех местах и т. д. Образующиеся при этом
продукты представляют собой анионотропные изомеры. Если в условиях
присоединения анионотропная система малоподвижна, то соотношение изо-
мерных аддуктов будет определяться кинетическими факторами: основным
продуктом реакции будет тот, который быстрее образуется в последней стадии
присоединения. Если же в условиях реакции система подвижна, то соот-
ношение изомеров будет зависеть от термодинамических факторов: преобла-
дающим продуктом реакции будет более устойчивый изомер.
Решение вопроса об относительной устойчивости возможных изомеров дости-
гается обычно путем сравнения их структур с учетом основных общепри-
знанных факторов устойчивости, особенно сопряжения и гиперконъюгации.
Учитывать следует гиперконъюгацию с участием как алкильных групп,,
так и галогена
Н^-сХс и C^Q-C-Qr
В рассмотренных в гл. XII, разд. 3, б, случаях присоединения галогеново-
дородов можно было пренебрегать гиперконъюгацией с участием галогена,
так как в сравнивавшихся системах она всегда уравновешивалась. Однако-
в продуктах присоединения галогенов такого уравновешивания не наблю-
дается. Так, следует ожидать, что из двух дибромидов бутадиена
ВгСН2СН = СНСН2Вг п ВгСН2СНВгСН = СН2
1,4-дибромид более устойчив, так как он содержит две гиперконъюгационные-
системы с участием галогенов, тогда как в 1,2-изомере имеется только одна
такая система. Для 2-метилбутадиена (изопрена) возможно образование двух
дибромидов
СН3 СНз
ВгСН2С = СНСН2Вг и ВгСН2СВгСН = СН2
если первый атом брома присоединится в положение 1. Из этих аддуктов
1,4-дибромид также более устойчив, однако причины этого более глубоки,
чем в предыдущем случае: 1,4-изомер содержит три гиперконъюгационные
системы (две с участием галогенов и одну с участием метильной группы),
тогда как 1,2-дибромид содержит только одну гиперконъюгационную систе-
му (с участием галогена). Большое значение обычного сопряжения можно-
видеть на примере гексатриена-1,3,5. При обычной реакции с первоначальным
присоединением к концевому атому углерода возможно образование трех
изомеров: 1,6-, 1,4- и 1,2-дибромидов
Вг Вг Вг Вг
I III
СН2СН = снсн = снсн2 СН2СН = снснсн = сн2
Вг Вг
сн2 - снсн снсн = сн2
812
Присоединение и обратные реакции
Однако из них только в 1,6- и 1,2-изомерах сохраняется бутадиеновое сопря-
жение. Следовательно, они более устойчивы, чем 1,4-дибромид. Аналогично
наиболее устойчивым из дибромидов 1-фенилбутадиена, которые образуются
при первоначальной атаке в положение 4, является 3,4-дибромид — единст-
венный из возможных изомеров, который сохраняет сопряжение стирольного
типа. Наконец, из двух возможных дибромидов 1,4-дифенилбутадиена более
устойчивым по этой же причине будет 1,2-дибромид, а не 1,4-изомер.
Однако следует ожидать, что возможны такие реакции присоединения, при
которых на определенной стадии ориентация зависит от кинетических фак-
торов; это происходит до того, как достигается равновесие между образовав-
шимися продуктами. В соответствии с общей закономерностью в этом случае
предпочтительно будет образовываться термодинамически менее устойчивый
изомер.
Влияние кинетических факторов на ориентацию наиболее легко проявляется
в реакциях присоединения к бутадиену и его 1- или 1,4-алкильным произ-
водным с образованием аддуктов — первичных или вторичных алкилга-
логенидов. Значительно меньше это влияние проявляется для 2- и 2,3-алкил-
производных бутадиена; менее устойчивыми аддуктами являются легко
ионизующиеся и потому легко изомеризующиеся третичные галогениды.
Едва ли можно наблюдать влияние кинетических факторов при присоедине-
нии к 1- и 1,4-фенилбутадиенам, в которых фенильный заместитель сильно
облегчает ионизацию галогена. В случаях присоединения хлора влияние
кинетических факторов выражено сильнее, чем для брома. Это влияние долж-
но быть более заметно в слабо ионизующих растворителях по сравнению с хо-
рошо ионизующими.
Рассмотрим теперь экспериментальные данные. Присоединение хлора к бута-
диену-1,3 исследовалось Маскатом и Нортропом [79], данные которых были
подтверждены и дополнены Пудовиком [80]. Смесь продуктов присоединения
содержит около 40% 1,4-дихлорида и 60% 1,2-изомера. В условиях образо-
вания оба дихлорида не претерпевают никаких взаимных превращений.
Однако при нагревании до 200 °C или при обработке хлористым цинком при
более низких температурах образуется равновесная смесь, содержащая около
70% 1,4-дихлорида и 30% 1,2-изомера. Таким образом, соотношение обра-
зующихся изомерных аддуктов, несомненно, определяется кинетическими
факторами. Как и ожидалось, основным продуктом в равновесной смеси
является 1,4-дихлорид.
Присоединение брома к бутадиену было впервые изучено Гринером [81],
который выделил жидкий 1,2-дибромид и наблюдал его дальнейшее превра-
щение в кристаллический 1,4-дибромид. Позднее Тиле [36] установил, что
в подобных условиях непосредственно образуется 1,4-дибромид. Впоследствии,
как сообщается в статье Штрауса [82], Тиле признал, что 1,2-дибромид также
образуется непосредственно при реакции. Фармер, Лоуренс и Торп [83],
а вслед за ними и Хатч, Гарднер и Джильберт [84] предприняли тщательное
исследование этой реакции. Они установили, что в условиях, исключающих
изомеризацию, дибромид образуется в количестве, полностью определяемом
природой растворителя. Если реакция сама достигает равновесного состояния
или доводится до равновесия намеренно, в конечной смеси содержится до 80%
1,4-изомера. Приведенные данные указывают на значительное влияние рас-
творителя на определяемое кинетикой соотношение продуктов реакции, на
большую подвижность бромидов по сравнению с хлоридами, на первона-
чальное образование большого количества термодинамически менее устой-
чивого продукта и на образование в конечном итоге того изомера, который
предположительно является более устойчивым.
813
Глава XIII
Присоединение иода к бутадиену дает 1,4-изомер [85]. В этом случае анионо-
тропная система должна была быть очень лабильной, что, вероятно, должно
приводить к образованию равновесной смеси продуктов. Присоедине-
ние хлористого иода к бутадиену в воде дает смесь, состоящую из 80%
С1СН2СН = СНСН21 и 20% СН2 == СНСНС1СН21 [86]. Эта смесь, вероятно,
также является равновесной.
Исследовалось присоединение брома к алкилированным по концам цепи
бутадиенам: 1,4-диметилбутадиену-1,3 [87], циклопентадиену [36, 88] и цик-
логексадиену-1,3 [36, 88]. В последних двух случаях образуются смеси 1,2-
и 1,4-дибромидов без каких-либо признаков равновесия между ними. Все рав-
новесные смеси содержат преимущественно 1,4-изомеры.
Исследовалось также присоединение хлора [89] и брома [90] к изопрену,
а также брома к 2,3-диметилбутадиену [87, 91]. Во всех случаях были выде-
лены равновесные смеси, содержащие преимущественно 1,4-дигалогениды.
В случае присоединения хлора к 2-метилбутадиену (изопрену), кроме 1,4-
дихлорида, образуется небольшое количество 1,2-дихлорида и еще меньшее
количество 3,4-дихлорида.
Если бы это были бромиды, то почти наверняка можно было бы считать, что
они образуются как продукты равновесия. Возможно, что в случае хлоридов
эти продукты образовываться не будут.
Присоединение брома к гексатриену-1,3,5 приводит к образованию только
1,2- и 1,6-дибромидов [92]. Как и следовало ожидать, эти продукты являются
наиболее устойчивыми из возможных дибромидов.
Установлено, что присоединение хлора [93] и брома [82] к 1-фенилбутадиену
приводит к образованию исключительно 3,4-дигалогенидов. При присоедине-
нии брома к 1,4-дифенилбутадиену образуется только 1,2-дибромид [82].
Все эти дигалогениды, несомненно, находятся в состоянии равновесия.
Хлорирование бутадиена в метиловом спирте приводит к образованию смеси,
содержащей 70% 1-хлор-2-метоксибутена-3 и 30% 1-хлор-4-метоксибутена-2.
Аналогично при хлорировании в этиловом спирте или при бромировании
и иодировании в этиловом или и-бутиловом спирте образуется в преобладаю-
щем количестве 1,2-изомер [94]. Вследствие малой подвижности алкоксильных
групп соотношение продуктов этих реакций, по-видимому, определяется
кинетическими факторами. Арбузов и Зороастрова [95] установили, что
основным продуктом хлорирования бутадиена в уксусной кислоте, содер-
жащей ацетат натрия, является 1-хлор-2-ацетоксибутен-3, а побочным про-
дуктом — 1-хлор-4-ацетоксибутен-2. Кадеш [96] показал, что при присоеди-
нении хлорной кислоты к бутадиену в воде образуется в основном 1-хлор-2-
оксибутен-3. Образование этих продуктов, очевидно, также определяется
кинетическими закономерностями.
Маскет и Гримсбай [97] показали, что в качестве основного продукта при
присоединении хлорной и бромной кислот к 1-фенилбутадиену в воде обра-
зуется фенил-4-галоген-3-оксибутен-1. Среди возможных продуктов этой
реакции он является термодинамически наиболее устойчивым, хотя его обра-
зование (вероятно, в меньшем количестве, чем обусловлено термодинамикой)
несомненно подчиняется кинетическим закономерностям *.
В области стереохимии реакций 1,4-присоединения галогенов следует рас-
смотреть два вопроса. Первый относится к углеродному скелету и связан
* Граммит и Вене [98] подтвердили данные Маскета и Гримсбая по присоединению
хлорной кислоты и добавили к этому, что в реакции образуется также небольшое количество
другого изомера — 1-фенил-1-окси-2-хлорбутена-3. Однако этот вывод основап не на пря-
мых определениях, а на результате последующей реакции со щелочью, которая может
протекать сложнее, чем предполагается.
814
Присоединение и обратные реакции
только с присоединением к открытым цепям или диенам с большим кольцом.
Вопрос состоит в том, имеют ли образующиеся моноолефины цис- или транс-
строение. Как показали Мислоу и Хеллман [99], присоединение хлора
к бутадиену в среде четыреххлористого углерода дает 1,4-дихлорид, пред-
ставляющий собой дихлорпроизводное тпраис-бутена-2. Атомы хлора в этой
структуре находятся настолько далеко друг от друга, что они должны были
присоединиться от разных молекул хлора. Это свидетельствует о том, что
реакция либо должна быть тримолекулярной, либо она протекает в две
стадии. Кинетических данных, которые внесли бы ясность в эти стереохими-
ческие наблюдения, пока нет.
Второй вопрос состоит в том, подходят ли два присоединяющихся атома
хлора к плоскости сопряжения с одной стороны (цис-присоединение) или
с противоположных сторон (шраис-присоединение). Янг и сотр. [100] тща-
тельно исследовали реакции присоединения хлора к циклопентадиену в лег-
ком петролейном эфире и хлороформе. Основными продуктами реакции были
продукты 1,4-присоединения и среди них главным был цис-i,4-изомер, хотя
он сопровождался транс-i,4-изомером. В результате реакции образуется
также некоторое количество 1,2-дихлорида, а именно транс-i, 2-изомер.
Основные стереоизомерные продукты 1,4- и 1,2-присоединения являются
цис-i,i- и транс-i,2-изомерами и могут образоваться только в результате
тримолекулярной или двухстадийной реакции. Тот факт, что 1,4-присоедине-
ние не является стереоспецифическим, указывает на двухстадийность про-
цесса, однако кинетические доказательства отсутствуют.
Хлорирование нафталина, так же как и фенантрена (разд. 1, в), включает
конкурирующие реакции присоединения и замещения. Основным продуктом
замещения является 1-хлорнафталин. Что касается продуктов присоедине-
ния, то считается, что они состоят из дихлоридов. Однако последние еще
плохо идентифицированы. Хорошо охарактеризованы лишь 1,2,3,4-тетра-
хлориды. Из шести возможных стереоизомеров описано пять, а для четырех
определена конфигурация [101, 102]. Строение образующихся стереоизоме-
ров зависит от условий проведения реакции и, в частности, от условий, опре-
деляющих гомолитичность или гетеролитичность присоединения. Гетероли-
тическая реакция в уксусной кислоте и темноте была исследована де ла Ма-
ром и Джонсоном с сотр. [102]. Две трети субстрата превращается в продукт
замещения, одна треть — в продукт присоединения; из суммы продуктов
присоединения две трети составляют тетрахлориды, а одну треть — ацетокси-
полихлориды, главным образом ацетокситрихлориды. Тетрахлориды на две
трети состоят из а-изомеров, т. е. (1,2/3,4)- или цис-шраис-цис-тетрахлоридов,
и на одну треть из 6-изомеров — (1,2,3/4)- или цис-т/шис-тпраис-тетрахлори-
дов. Основным ацетокситрихлоридом является 4-ацетокси-(1,2,4/3)-1,2,3-
трихлорид. Их строение будет показано ниже. Если хлорирование проводить
в присутствии хлористого лития, доля а-тетрахлорида снижается, а доля
6-изомера увеличивается: можно даже сделать так, что последний изомер
станет преобладающим.
815
Глава XIII
Объяснение может состоять в том, что а-тетрахлорид образуется по реакции,
аналогичной процессу присоединения хлора к фенантрену. Первая молекула
хлора начинает присоединяться в положение 1 (конец бутадиеновой системы);
так как сопряжение отодвигает образовавшийся заряд карбониевого иона
далеко от положения 2, то первый присоединившийся атом хлора не образует
электростатической связи с этим положением, а второй атом хлора не диссо-
циирует, а присоединяется в ^wc-положение. Вторая молекула -хлора начина-
ет присоединяться в положение 3 (конец стирольной системы), и присоедине-
ние заканчивается таким же образом в положение 4. Поскольку заряд перво-
начально образовавшегося карбониевого иона оттягивается от положения 4
менее сильно, происходит частичная диссоциация хлорид-иона; поэтому про-
исходит образование б-тетрахлорида путем шраис-присоединения хлорида
в количестве, которое может увеличиваться при добавлении хлорид-ионов;
ацетокситрихлорид образуется в результате аналогичного шряис-присоеди-
нения ацетат-иона из растворителя — уксусной кислоты.
д. Инициируемая кислотами полимеризация олефиновых соединений
путем самоприсоединения. Еще в прошлом столетии было известно *, что
некоторые олефиновые соединения могут превращаться в высокомолекуляр-
ные вещества под действием весьма малых количеств сильных протонных
кислот или катализаторов Фриделя — Крафтса. Все эти катализаторы,
согласно представлениям Льюиса, являются кислотами, т. е. сильно электро-
фильными агентами. К олефиновым соединениям, которые могут полимери-
зоваться наиболее легко, относятся виниловые эфиры, изобутилен, стирол,
а-метилстирол, бутадиен и вообще такие алкены, в которых заместители
создают избыток электронов на другом конце двойной связи. Очевидно,
механизм этих процессов невозможно было понять до 1920 г., когда Штаудин-
гер [104] открыл цепной характер полимеризации. Специфическая природа
этих особых форм полимеризации не могла быть понята в течение некоторого
времени после этого, а именно до тех пор, пока общая теория гетеро лити-
ческих реакций в органической химии не достигла некоторого прогресса.
Начало ее быстрого развития относится к 1926 г.
Примерно в 1940 г. стало ясно, что если бы богатый электронами атом угле-
рода действительно соединялся с электрофильным катализатором, то атом
углерода, стоящий на замещенном конце двойной связи, превратился бы
в карбониевый ион. Поэтому можно сказать, что он сам превратился бы
в сильно электрофильную частицу, способную, вероятно, соединяться с бога-
тым электронами атомом углерода второй олефиновой молекулы и вызвать
таким образом первую стадию роста, которая могла бы стать непрерывной
полимеризацией. Эти процессы, катализируемые протонными кислотами,
можно иллюстрировать следующим образом:
H2SO4+CH2=C(CH3)2 —> CH3C(CH3)£}HSOj —> СН3С(СН3)2СН2С(СН3)2 }HSOj —>
CH3C(CH3)2CH2C(CH3)2CH2C(CH3)J}HSOj .
Для полимеризации, катализируемой реактивами Фриделя—Крафтса, можно
написать аналогичную схему, которая, как это будет показано ниже, допу-
скает различные модификации
BF3 + CH2 = C(CH3)2 BF3CH2C(CH3)2 —> BF3CH2C(CH3)2CH2C(CH3)2
-> BF3CH2C(CH3)2CH2C(CH3)CH2C(CH3)2 —> ...
* В 1839 г. Девилль сообщал о превращении «терпентинного спирта» в канифоль при
действии серной кислоты [103].
816
Присоединение и обратные реакции
К протонным кислотным катализаторам относятся как наиболее сильные
неорганические кислоты, такие, как НС1О4 и H2SO4, так и менее сильные,
например Н3РО4, а также наиболее сильные органические кислоты вплоть
до по крайней мере дихлоруксусной кислоты. При работе со слабыми кисло-
тами требуется большее их количество, и они будут эффективными только
при полимеризации наиболее легко полимеризующихся олефинов. Все силь-
ные катализаторы Фриделя — Крафтса оказываются эффективными: BF3,
А1С13, А1Вг3, TiCl4, TiBr4, SbCl5, FeCl3, SnCl4 и т. д.
Полимеризация обычно проходит быстро. Например, изобутилен в присут-
ствии BF3 или А1С13 при температуре —100 °C полимеризуется мгновенно,
т. е. сразу же после смешивания реагентов; степень полимеризации достигает
порядка миллиона (среднее число единиц мономера в полимере). В 1946 —
1947 гг. Эванс и Поляни [1051 открыли, что катализаторы полимеризации
Фриделя — Крафтса не действуют совсем, если в системе не присутствует
сокатализатор, которым часто служит протонная кислота или в общем слу-
чае вода. Так, при смешивании изобутилена и трехфтористого бора никакой
реакции не происходит, если сами реагенты и реакционные сосуды совершен-
но сухие. Это открытие оспаривалось, но, несомненно, оно правильно. Эванс
и Поляни приписали катализирующее действие предельно сильным протон-
ным кислотам, которые образуются из катализатора и сокатализатора,
и предложили следующую схему полимеризации по Фриделю —
Крафтсу:
H2O+BF3 + CH2 = C(CH3)2 CH3C(CH3)t}BF3OH-
-> CH3C(CH3)2CH2C(CH3)2+}BF3OH-
CH3C(CH3)2CH2C(CH3)2CH2C(CH3)t}BF3OH- -^ ...
Из приведенного механизма вытекает возможность существования катализа
третьего типа. На каждой стадии развития цепи происходит реакция с уча-
стием электрофильного карбониевого иона, который поэтому должен быть
способным инициировать аналогичную стадию инициирования цепной реак-
ции; следовательно, заранее полученные карбониевые ионы можно исполь-
зовать в качестве катализатора. Это было впервые осуществлено Илеем
и Ричардсом [106]; они использовали трифенилметилхлорид в растворе
.м-крезола, в котором он образует ион трифенилметилия, для полимеризации
винил-2-этилгексилового эфира.
С тех пор реакция была проведена с разными карбониевыми ионами, включая
mpem-бутилий-, ацетилий- и бензоилий-ионы, вводившиеся в виде перхлора-
тов [107], и ацетилий-ион, вводившийся в виде тетрафторбората [108]. Типич-
ное уравнение реакции записывается так:
(CeH5)3C + CH2 = CHOR —> (CeH5)3CCH2CHOR+ —> (C6H5)3CCH2CH(OR)CH2CHOR+ —>
—> (C6H5)3CCH2CH(OR)CH2CH(OR)CH2CHOR+ —> ...
Любой алкилхлорид будет действовать как сокатализатор реакции Фриде-
ля — Крафтса, вероятно, вследствие того, что первоначально хлорид пре-
вращается в карбониевый ион
RC1 + A1C13 R+}A1C1j
Колклоу и Дейнтон [109] показали это на примере полимеризации стирола
в присутствии хлорида олова(1У), а в качестве сокатализатора — различных
тщательно высушенных алкилхлоридов. Применение в качестве сокатализа-
тора шцеш-бутилхлорида наиболее эффективно, изопропилхлорид сравни-
тельно менее эффективен, а эффективность этилхлорида самая низкая. Кроме
того, Кеннеди и Томас [110] показали, что изобутилен полимеризуется при
52-0772
817
Глава XIII
низкой температуре в присутствии хлористого алюминия, растворенного
в этилхлориде и даже в метилхлориде, при абсолютном отсутствии воды и что
использование 14С-метилхлорида в качестве растворителя и сокатализатора
приводит к образованию полимера, содержащего 14С.
С помощью ЯМР-спектроскопии Кеннеди и Томас [111] исследовали полиме-
ризацию изопропилэтилена и подтвердили, что развитие цепи происходит
за счет промежуточно образующихся карбониевых ионов. Они обнаружили,
что повторяющаяся структурная единица полимера не содержит одного
вторичного и двух третичных атомов углерода, т. е. образуется не за счет
1,2-присоединения
СН2=СНСН(СН3), —> СН2СНСН(СН3)2 —> СН2СНСН(СН3)2
I I I
СН2СНСН(СН3)2
а за счет 1,3-присоединения с образованием двух вторичных и одного тре-
тичного атомов углерода. Это означает, что в каждой новой группе вторично-
го карбониевого иона, образующейся, как было показано выше, путем при-
соединения новой молекулы мономера, до того как продолжится рост молеку-
лы полимера, происходит смещение гидрид-иона, что свойственно таким
карбониевым ионам, с образованием третичного иона. Дальнейшее такое же
двухстадийное присоединение мономера и смещение гидрида приводит к росту
цепи
I
СН2СН2С(СН3)2
СН2СНСН(СН3)2 СН2СН2С(СН3)2 СН2СН2С(СН3)2
I I I
Рост полимера заканчивается потерей протона и образованием двойной свя-
зи в том месте, которое было растущим концом полимерной цепи. Это было
установлено Дейнтоном и Сазерлендом [112] для низкомолекулярных поли-
меров изобутилена с помощью исследования их инфракрасных спектров. Эти
исследователи установили, что концевыми группами являются не только
(СН3)3С-группы, но и группы — СН2С(СН3) = СН2.
Если рассматривать полимеризацию, инициируемую кислотным катализа-
тором или комплексом катализатор — сокатализатор в отсутствие основного
растворителя, то наиболее подходящим основанием для присоединения отще-
пившегося протона будет противоанион, который получается из катализи-
рующей кислоты в процессе роста полимера и образует ионную пару с катион-
ным центром в точке роста. Для того чтобы отличить этот процесс от других,
его называют «обрывом цепи». Другие возможные процессы состоят в том,
что протон захватывается мономерной молекулой и таким образом начинается
рост новой полимерной молекулы. Этот процесс называют «переносом цепи».
Он характеризуется тем, что оба вида процессов — роста и обрыва цепи,
которые катализируются кислотой, не зависят от ее протона, не входящего
в полимерную молекулу, и, кроме того, тем, что одна молекула катализатора
может произвести много молекул полимера. Это наблюдалось эксперимен-
тально [112, ИЗ]. Характерно также то, что кислотный протон захватывает-
ся полимером. Показано, что низкомолекулярные полимеры изобутилена,
полученные при добавлении комплекса BF3 — D2O, содержат дейте-
рий [112].
При инициируемой кислотами полимеризации образуются высокомолекуляр-
ные полимеры, так как случаи обрыва цепей встречаются за время роста
цепи полимера редко. Обрыв цепей происходит сравнительно редко, но не
818
Присоединение и обратные реакции
по той причине, как при радикальной полимеризации, при которой обра-
зующиеся в низкой концентрации два промежуточных радикала соединяются
друг с другом. Обрыв цепей представляет собой внутримолекулярный процесс
по отношению к растущим ионным парам и является медленным, так как
его энергия активации выше, чем энергия активации процесса роста цепи.
Отношение скоростей процессов роста и обрыва цепи, которое представляет
собой степень полимеризации, будет, следовательно, уменьшаться с ростом
температуры. Это одна из характерных черт инициируемой кислотами поли-
меризации. Так, например, степень полимеризации изобутилена в присут-
ствии катализатора трехфтористого бора будет уменьшаться с 5 миллионов
до 50 тысяч при увеличении температуры от —100 до 0 °C [114]. Если нанести
на график зависимость логарифма степени полимеризации от обратной
абсолютной температуры, то можно определить для приведенного примера,
что энергия активации обрыва цепи превышает энергию активации роста
цепи на 4,6 ккал/моль (19,26-Ю3 Дж/моль) [115]. Несомненно, что такой
расчет подразумевает протекание процесса обрыва цепи по одному механиз-
му: если обрыв цепи путем ее переноса, продолжающий процесс полимериза-
ции, по скорости оказывается сравнимым со скоростью «настоящего» обрыва
цепи, то ожидать хороших результатов из графической зависимости Аррениу-
са для степени полимеризации нельзя. Более детальные кинетические иссле-
дования разных реакций показали, что энергия активации развития цепи
в общем случае имеет низкое значение [116].
Если обозначить символом В; скорость инициирования полимеризации моно-
мера М в присутствии кислотного катализатора НА и если Bt определяется
выражением
Bi = ki [НА][М]
скорость роста цепи Вр в ионной паре НМ*}А" — уравнением
ЯР = ЛР[НМ*}А-] [М]
скорость обрыва цепи Bf — выражением
т?, = мнм;} АД
а скорость переноса цепи BtT — уравнением
Btr = ktT [НМГ+) АД [М]
то, предполагая, что kt кр, несложные преобразования приведенных выше
уравнений дают для скорости полимеризации В выражение
7? = ±^[НА][М]2
и для степени полимеризации DP
kt + kfr [М]
Отсюда следует, что главным процессом прекращения роста цепи оказывается
обрыв или перенос, а степень полимеризации пропорциональна концентрации
мономера или не зависит от нее.
Одно из первых подтверждений справедливости выведенных уравнений было
дано в 1949 г. Пеппером [117] на примере полимеризации стирола в стацио-
нарных условиях в присутствии катализатора хлорида олова(1У). В этом
случае перенос цепи как способ прекращения ее роста имел большее значение.
На разных примерах Пеппер показал, что с увеличением диэлектрической
52* 819
Глава XIII
проницаемости растворителя растут скорость и степень полимеризации. Этого
следовало ожидать, так как инициирование включает разделение зарядов,
а обрыв цепи включает исчезновение зарядов. К тому же времени относятся
исследования Идея и Ричардса [117] по полимеризации винил-2-этилгексило-
вого эфира в присутствии катализатора хлорида олова(1У), подтверждающее
выведенные выше кинетические закономерности.
Как установил в 1954 г. Пеппер [118], в отдельных случаях могут встретиться
самые различные отклонения от общих и довольно простых кинетических
закономерностей, выведенных для случая кислотной полимеризации. Одну
такую группу отклонений выявили Хейес и Пеппер [118]. Она относится
к полимеризации стирола, катализируемой сильной, но устойчивой молеку-
лярной кислотой, такой, как, например, серная или хлорная. Инициируемая
серной кислотой полимеризация проходит вначале очень быстро, но и быстро
затухает еще до того, как израсходуется весь мономер. «Выход» полимера при
этом хорошо определяется начальной концентрацией кислоты. Если эту
полимеризацию инициировать хлорной кислотой, то уменьшение выхода
не наблюдается; взамен этого скорость расходования мономера подчиняется
простому закону первого порядка на протяжении всей реакции. Оба эти
результата можно объяснить, исходя из одного и того же предположения,
а именно что стационарные условия в этом случае будут обратными, т. е.
удельная скорость развития цепи вместо того, чтобы намного превышать
скорость инициирования, на самом деле намного меньше ее: кг кр. При
таких условиях немедленно устанавливается постоянная концентрация
растущих карбониевых ионных центров, равная начальной концентрации
катализирующей кислоты [НА]0, а скорость образования полимера Y и сте-
пень полимеризации DP будут определяться выражениями
R = кр [НА]0 [М] exp (— ktt)
, 1 кР
1-Y МНА]0
1 ktr , kt In [М]о-In [М]
DP кр^ кр [М]о — [М]
Эти две кислоты будут отличаться по их кислотности или, другими словами,
по основности их анионов. При инициировании серной кислотой kt имеет
большую величину, и, следовательно, фактор затухания ехр (—ktt) в урав-
нении скорости будет оказывать большое обратное влияние, и выход Y ста-
новится намного ниже единицы. В случае инициирования хлорной кислотой
к> имеет малую величину, вероятно, потому, что перхлорат-ион обладает
малым сродством к протону, поэтому и фактор затухания равен единице,
а уравнение скорости имеет первый порядок, в то время как выход равен
единице, а степень полимеризации лимитируется только процессом переноса
цепи.
2. Нуклеофильное присоединение к карбонильным
и олефиновым соединениям
Обычно для нуклеофильных аддендов типично присоединение к карбониль-
ным группам и к олефиновым связям, поляризованным за счет сопряжения
с карбонильной группой или другими группами — М-типа, например циан-
или нитрогруппами, которые облегчают доступ к углеродному атому с крат-
520
Присоединение и обратные реакции
НОЙ связью
Х’С=О
С=^Э-С=£3
Такие процессы присоединения для краткости называют нуклеофильным
присоединением и обозначаются символом Ad к- В частности, к нуклеофиль-
ному присоединению относятся реакции Гриньяра, где X — карбанион R"
из реактива Гриньяра RMgHal.
I I н2о |
R--pC = O —> R-C-O- -----> R — с-ОН
Наиболее важным классом нуклеофильных аддендов являются некоторые
слабые кислоты, подобные синильной кислоте или аммонийному иону, в том
числе псевдокислоты типа ацетона или малонового эфира. Они присоеди-
няются как НХ либо по карбонильной группе с образованием СХОН, либо
по двойной связи, сопряженной с карбонилом, образуя СХСНС = О. Однако
эти реакции начинаются с атаки основанием, сопряженным кислоте НХ,
а не с атаки протоном; это и придает адденду нуклеофильный характер.
Такие реакции присоединения к обычной карбонильной группе или к сопря-
женной системе, включающей карбонил, обычно легко обратимы. Поэтому
присоединение нельзя изучать в отрыве от тех реакций расщепления, которые
приводят к образованию карбонильных или сопряженных олефин-карбо-
нильных систем; оба вопроса должны рассматриваться совместно. Наиболее
важным нуклеофильным аддендом некислого характера является водород,
который выделяется при растворении металлов в гидроксилсодержащих
растворителях, при восстановлении катионов металлов в подобных средах,
из сложных анионов, способных выделять гидрид-ион и т. д., но не образуется
из молекулярного водорода в присутствии гетерогенных катализаторов.
В обычных условиях присоединение нуклеофильного водорода не является
заметно обратимым. Очень сильные основания, такие, как амид-ионы или
алкилид-ионы, будут присоединяться не только к олефиновым связям,
сопряженным с такими сильно электроотрицательными ненасыщенными
группами, как карбонильная, циан- или нитрогруппы, но также, правда
менее легко, к олефиновым несопряженным связям. Обычно в результате
таких реакций происходит полимеризация.
а. Обратимое присоединение оснований или слабых кислот к обычным
карбонильным соединениям. В 1903 г. Лэпуорт [119] отметил, что на обра-
зование циангидринов из карбонильных соединений и синильной кислоты
оказывают большое влияние катализаторы основного характера. В частности,
он исследовал реакции 1,2-дикето камфана (камфорхинона), воспользовавшись
для этой цели обесцвечиванием при превращении этого дикетона в его циан-
гидрин. Лэпуорт предположил, что стадией, определяющей скорость обра-
зования циангидрина, является присоединение цианид-иона к карбонильной
группе с образованием аниона циангидрина. Лэпуорт учитывал, что образо-
вание циангидрина представляет собой обратимый процесс, и предложил
механизм реакции, являющейся нуклеофильным присоединением Ad N
Медленно /CN
cn-+R2co т —» r2cz
Медленно \q_
/CN Быстро ,CN
r2cz + н+ 7 —» r2cz
XO~ Быстро \qH
821
Глава XIII
Однако предположение, что скорость реакции выражается уравнением
Скорость ОС [Карбонил][С??~]
не нашло количественного подтверждения вследствие большого влияния
незначительных, точно не установленных концентраций цианид-ионов на
скорость реакции. Позднее все же удалось установить механизм процесса
присоединения синильной кислоты к сопряженным ненасыщенным кетонам
[120], о чем будет сказано ниже. До этой работы было проведено важное
исследование кинетики бензоиновой реакции, которая, по сути дела, вклю-
чает и образование циангидрина, и альдольную конденсацию.
Бензоиновая реакция заключается в соединении двух молекул бензальдегида
с образованием бензоина при специфическом каталитическом влиянии цианид-
иона. Лэпуорт изучал эту реакцию в 1903 г. и предположил, что первой
стадией процесса является образование циангидрина бензальдегида, который
затем присоединяется по типу альдольной конденсации ко второй молекуле
бензальдегида; при этом образуется неустойчивый циангидрин бензоина,
немедленно распадающийся на бензоин и синильную кислоту. В 1904 г.
Бредиг и Штерн [121] исследовали кинетику этого процесса и установили, что
его скорость подчиняется уравнению
Скорость ОС [Бензальдегид]2[С№]
Они показали, что на скорость реакции не оказывают влияния неионизован-
ные цианиды типа цианида ртути, а также ионы гидроксила. Механизм
реакции, предложенный Лэпуортом [122] и соответствующий приведенному
кинетическому уравнению, может быть выражен следующей схемой:
CN
cn-+c6h6cho C8HSCH^
\о-
CN _/CN
с8н6сн( C6H6C^
ХО~ ОН
/CN /0-
C6HSC; + C6HSCHO ^2 CSHSCH^ ZCN
XOH XC(C6H5/
N)H
0-
C6H5CH/ zCN /ОН
\C(Csh/ C8H5CH( +CN-
XOH O(C6H5) = 0
Первое из приведенных уравнений соответствует медленной стадии обра-
зования циангидрина. Продукт реакции после прототропного превращения
имеет такое строение, что может принять участие в медленной стадии
процесса альдольной конденсации, представленной третьим уравнением.
Кинетика требует, чтобы либо третья, либо четвертая стадия была наиболее
медленной из всех. Однако, учитывая, что циангидринное равновесие быстро
устанавливается в присутствии таких больших количеств цианид-ионов,
которые обычно применяются в подобных случаях, можно не сомневаться
в том, что самой медленной стадией будет третья.
Несмотря на то что изучение скорости образования обычных циангидринов
затруднительно, при количественном измерении циангидринного равновесия
были получены важные результаты. Лэпуорт и Манске [123] изучили сле-
822
Присоединение и обратные реакции
дующее равновесие:
.ОН
RC6H4CHO + HCN rc6h4ch'
'CN
и определили константы равновесия; на основании этих данных они вычисли-
ли изменения свободных энергий при различных орто-, мета- и «ада-заме-
стителях R при 20 °C в 96%-ном этиловом спирте. Влияние o/wo-заместите-
лей осложнено специфическим внутримолекулярным взаимодействием. В то
же время влияние мета- и «ара-заместителей может быть выражено совершен-
но просто. Влияние «ара-заместителей в общем несколько больше и особенно
значительно в случае ненасыщенных заместителей. Если пренебречь неболь-
шими побочными эффектами, то эти величины свободной энергии соответ-
ствуют дополнительной стабилизации альдегидов, но не их циангидринов,
вследствие возможности сопряжения ненасыщенного заместителя В и нена-
сыщенной группы СНО. Меньшее значение имеет полярность; электроно-
акцепторные группы R склонны уменьшать устойчивость альдегида по срав-
нению с циангидрином, так как сама группа СНО, по-видимому, является
более сильной электроноакцепторной группой, чем CH(OH)CN. Наблю-
даемое влияние л«е«га-заместителей обычно менее значительно, так как в этом
случае отсутствует сопряжение и, следовательно, можно выявить эффект
полярности в чистом виде. Высказанные положения иллюстрируются при-
веденными ниже изменениями свободных энергий при образовании циан-
гидринов из «ара-замещенных альдегидов:
ляря-Заместитель N(CH3)2 ОН ОСН3 СН3 Cl Н
— AG, ккал/моль (4,187 X ’0,5Ь 1,5 2,0 2,7 3,1 3,1
X103 Дж/моль) '
Меньшая величина показывает, что альдегид становится сравнительно устой-
чивым. Можно видеть, что все три группы (кроме хлора), которые способны
к сопряжению (или гиперконъюгации) с альдегидной группой через бензоль-
ное кольцо, способствуют увеличению устойчивости альдегида. В случае
хлора можно предположить, что слабый эффект сопряжения совершенно
исчезает вследствие электроотрицательности хлора. Ниже приведены данные
с влиянии полярности л«е«га-заместителей на изменение свободных энергий:
лстл-Заместитель СН3
— AG, ккал/моль (4,187 X 3,0
Х103 Дж/моль)
Н ОН ОСН3 NO2 Cl
3,1 3,1 3,2 3,4 3,5
Как уже упоминалось в гл. II, разд. 3,и, Бейкер и Хемминг [124] исследова-
ли бензоиновую реакцию, чтобы показать, что гиперконъюгация является
постояннодействующей причиной электронных смещений. Они установили
следующие изменения свободной энергии при различных «а/ш-алкильных
заместителях в 96 %-ном этиловом спирте:
паря-Заместитель Н СНз с2н5 wao-CgHy трет-С^Яд
— AG(35°C), ккал/моль 2,90 2,39 2,48 2,51 2,57
(4,187-103 Дж/моль)
— AG (20 °C), ккал/моль 3,15 2,75 2,80 2,81 2,85
(4,187-103 Дж/моль)
Из этих данных видно, что алкильные группы стабилизуют альдегид по срав-
нению с его циангидрином в следующем порядке: СН3 > С2Н5 > азо-С3Н7 >
2> трет-С^Нд.
823
Глава XIII
Бейкер и Хопкинс изучали циангидринное равновесие с целью сравнения
+М-эффекта галогенов, связанных с бензольным ядром. Такое сопряжение
должно способствовать увеличению стабильности альдегида по сравнению
с циангидрином. Авторы определили изменения свободной энергии при
образовании циангидринов из п- и .w-галогенбензальдегидов в этиловом спир-
те при 20 °C [125]:
Заместитель
— АС, ккал/моль
(4,187-103 Дж/моль)
( пара
I мета
Н F
3,16 2,95
3,16 3,59
Cl Вг I
3,24 3,34 3,36
3,63 3,66 3,62
Легко видеть, что галогены в atenia-положении, обладая индуктивным —I-
эффектом, снижают стабильность альдегида по сравнению с циангидрином.
Также видно, что галогены в «ара-положении в меньшей степени, чем мета-
заместители, относительно дестабилизуют альдегид (один из галогенов,
а именно фтор, в «ара-положении немного стабилизует альдегид). Очевидно,
что мезомерное +7И-влияние галогена, как и следует ожидать, направлено
в сторону повышения относительной устойчивости альдегида. В наибольшей
степени этот эффект проявляется у фтора, в наименьшей степени — у иода.
Короче говоря, проявление мезомерного эффекта, как было указано в гл. II,
разд. 3,д, соответствует ряду F > С1 > Вт > I. В гл. XIV показано, что
такой порядок подтверждается величинами силы соответствующих кислот.
Бейкер, Баррет и Твид на примере бензоиновой реакции показали, что
сравнительное проявление +7И-эффекта нейтральных групп XR (где X —
один из элементов VI группы периодической системы) отвечает ряду OR >
> SR > SeR, указанному в гл. II, разд. 3,д. Они измерили изменения сво-
бодной энергии при образовании циангидринов «ара-замещенных бензаль-
дегидов в этиловом спирте; относительная стабильность альдегидов выра-
жается следующими величинами [126]:
Заместитель н осн3 SCH3 SeCH3
— AG (20 °C), ккал/моль j f пара 3,16 0,81 0,90 1,94
(4,187-103 Дж/моль) 1 L мета 3,16 3,18 3,20 3,22
Такой порядок относительного влияния заместителей также подтверждается
значениями силы кислот, что показано в следующей главе.
Согласно принципу микроскопической обратимости, реакция расщепления
циангидринов с выделением исходного карбонильного соединения протекает
через сопряженное основание циангидрина путем мономолекулярного отщеп-
ления цианид-иона, определяющего скорость всего процесса. Это соответству-
ет механизму отщепления карбонильного соединения, обозначенному симво-
лом ElcB. О приложении этого механизма к реакциям отщепления олефинов
известно мало, хотя он является обычным для реакций отщепления карбо-
нильных соединений.
Известно, что альдольная конденсация аналогична образованию циангидринов
и часто протекает по общему механизму присоединения Лэпуорта. Давно
хорошо известно, что для альдольной конденсации характерна обратимость,
изученная на примере превращения ацетона в диацетоновый спирт [127}
и на ряде других равновесных реакций, включающих простые альдоли
типа изобутиральдоля [128]
(СН3)2С = О+СН3СОСНз (СН3)2С(ОН)СН2СОСН3
(СН3)2СНСН = О + (СН3)2СНСНО (СН3)2СНСН(ОН)С(СН3)2СНО
824
Присоединение и обратные реакции
Эти реакции присоединения экзотермичны, и поэтому термодинамически
предпочтительнее применение низких температур. Во всех случаях бывает
трудно установить существование равновесия, так как многие альдоли
и кетолы легко дегидратируются в а,р-ненасыщенные карбонильные соеди-
нения. Например, ацетальдоль превращается в кротоновый альдегид, диацето-
новый спирт — в окись мезитила. Кроме того, часто многие альдоли и кето-
лы реагируют с карбонильными соединениями с образованием высших аль-
долей и кетолов или же продуктов их дальнейшей дегидратации, например
ацетальдоль димеризуется в «октальдоль», а диацетоновый спирт превращает-
ся в форон.
Альдольная реакция катализируется различными реагентами, но чаще всего
основаниями. Не касаясь вторичных и побочных процессов, присоединение
ацетальдегида к формальдегиду при катализе основаниями может быть
описано следующей схемой:
СН2О + СН3СНО —> СН2(ОН)СН2СНО
Кинетика реакции, как показал Белл [129], выражается уравнением
Скорость ОС [Ацетальдегид][ОН“]
Концентрация формальдегида в этом уравнении отсутствует. Это означает,
что на медленной стадии расходуется только ацетальдегид, а формальдегид
расходуется на следующей быстрой стадии. Это совпадает с общим механиз-
мом Лэпуорта: вторая прямая стадия присоединения оказывается быстрее
по сравнению с обратной первоначальной стадией ионизации
сн3сно + он- сн2сно + н2о
/°"
СН2О+СН2СНО СН2
\сн2сно
0-
СН2 +Н2о СН2(ОН)СН2СНО + ОН
\сн2сно
Иные соотношения между скоростями отдельных стадий наблюдаются в слу-
чае кетолизации ацетона в водно-щелочной среде; можно предположить
следующий механизм реакции:
сн3сосн3+он- СН2СОСН2 +Н2О
,°-
(СН3)2С = О+СН2СОСН3 (СН3)2С/
\СН2СОСН3
/0-
(СН3)2С/ +Н2о (СН3)2С(ОН)СН2СОСН3+ОН-
\сн2сосн3
Исследовалась кинетика реакции, обратной указанной, так как в этом случае
равновесие достигается значительно быстрее [127, 130]. Скорость подчиняется
уравнению
Скорость ОС [Кетол][ОН~]
Скорость декетолизации диацетонового спирта не зависит от концентрации
ацетона. Это означает, что анион СНдСОСНа реагирует с молекулой воды
быстрее, чем с ацетоном; в противном случае последний процесс замедлял бы
декетолизацию. Следовательно, скорость декетолизации определяется либо
825
Глава XIII
первой стадией (последнее из написанных выше уравнений), либо второй.
Однако первая стадия, состоящая в перемещении протона между атомами
кислорода, не может определить скорость всей реакции. Из скорости декето-
лизации и константы равновесия можно вычислить скорость кетолизации
ацетона. Она составляет около 0,001 скорости иодирования или равной ей
скорости дейтерирования ацетона (гл. XI, разд. 1,д), т. е. реакций, скорости
которых определяются скоростью первой стадии кетолизации [131]. Таким
образом, вторая стадия кетолизации протекает медленнее, чем первая или
противоположная ей. Ясно, что вторая стадия определяет как скорость
кетолизации, так и скорость декетолизации. и если бы кинетика кетолизации
была измерена непосредственно, то она выражалась бы уравнением третьего
порядка:
Скорость ОС [Ацетон]2[ОН~]
Альдолизация ацетальдегида в водном растворе щелочи является промежу-
точным звеном между этими двумя кинетическими случаями. Впервые она
была исследована Беллом [132], который установил, что кинетика реакции
описывается уравнением второго порядка. Бенхоффер и Вальтере [131]
показали, что если реакцию проводить в дейтерированной воде, то дейтерий
не захватывается субстратом. Оба эти результата можно понять, если допу-
стить, что вторая стадия прямой реакции проходит достаточно быстро, чтобы
предупредить обратное течение первой стадии (на которой мог присоеди-
ниться дейтерий), и, таким образом, скорость процесса контролируется
первой стадией прямой реакции. Вместе с тем Броше и Джильберт [133]
показали, что этот случай, когда порядок реакции является промежуточным
между вторым и третьим, не носит общего характера; «порядок по ацеталь-
дегиду» в данном случае является приближенным и имеет тенденцию умень-
шаться с увеличением концентрации ацетальдегида. Кроме того, хотя дейте-
рий от тяжелой воды при очень высоких концентрациях ацетальдегида не
присоединяется, при понижении концентрации ацетальдегида присоедине-
ние дейтерия происходит с большей скоростью. Снижание концентрации
ацетальдегида будет замедлять скорость второй прямой стадии по сравнению
со скоростью первой прямой или первой обратной стадий, и в результате
функция лимитирующей стадии будет все больше переходить от первой ко
второй стадии прямой реакции. Беллу и Мактигью [129] при использовании
буферов удалось полностью показать плавный переход от одной к другой
контролирующей скорости стадии, а также наблюдать реакцию, довольно
хорошо описываемую уравнением третьего порядка:
Скорость ОС [Ацетальдегид]2[ОН~]
Найдено, что кинетика альдольного присоединения малонового эфира
к формальдегиду в водном растворе
СН2О + СН2(СООС2Н5)2 —> СН2(ОН)СН(СООС2Н5)2
аналогична кинетике кетолизации ацетона [134]. Реакция, по-видимому,
не останавливается на первой стадии, тем не менее при проведении ее в фос-
фатных буферных растворах была установлена следующая закономерность:
Скорость ОС [Альдегид][Эфир][ОН_]
Это означает, что образование аниона малонового эфира протекает быстро,
а присоединение — медленно.
Изучена также кинетика конденсации бензальдегида с ацетофеноном в эти-
ловом спирте, катализируемой этилат-ионами [135]. При этом был выделен
826
Присоединение и обратные реакции
бензилиденацетофенон; предполагается, что промежуточно образуется кетол
С6Н5СНО + СН3СОС6Н5 —> С6Н5СН(ОН)СН2СОС6Н5 —> С6Н5СН = СНСОС6Н5
Скорость реакции выражается следующим уравнением:
Скорость ОС [Альдегид][Кетон][С2Н5О_]
Это означает, что образование аниона ацетофенона протекает быстро, а его
присоединение или же (что маловероятно) конечная дегидратация — мед-
ленно.
Эти исследования были значительно расширены Патаи и другими. Патаи,
Израэли и Забицки [136, 137] показали, что если кислотные функции слабо-
кислого аддукта выражены в достаточной степени, то в гидроксилсодержащем
растворителе, например в воде или этиловом спирте, он будет реагировать
и без прибавления основного катализатора. В этом случае будет достаточно
основности растворителя. Так, нитрил малоновой кислоты будет присоеди-
няться к бензальдегиду или его замещенным без добавления основного ката-
лизатора. Реакция, конечно, обратима: наличие катализатора никак не
связано с термодинамикой процесса, и образующийся альдоль будет взаимо-
действовать с растворителем и в отсутствие катализатора. Порядок прямой
реакции равен двум
Скорость ОС [Нитрил малоновой кислоты] [Альдегид]
Это уравнение соответствует реакции третьего порядка, протекающей
в присутствии основного катализатора, однако в нем отсутствует концентра-
ция катализатора. Это означает, что ионизация нитрила малоновой кислоты
протекает быстро, а атака альдегида анионом — медленно. Кислотные
свойства этилового эфира циануксусной кислоты выражены слабее, чем
у нитрила малоновой кислоты, но они также проявляются в отсутствие ката-
лизатора, и получаемая кинетическая картина реакции относительно неиз-
менна. Однако в этом случае появляются признаки переходной кинетической
картины; кажущийся порядок реакции для этилового эфира циануксусной
кислоты проявляет тенденцию к снижению с увеличением концентрации
эфира. С увеличением его концентрации скорость стадии присоединения
начинает приближаться к скорости предварительной ионизации, и, таким
образом, функция контроля скорости реакции начинает переходить от второй
стадии механизма к первой. Амид циануксусной кислоты обладает еще более
слабыми кислотными свойствами, и его присоединение протекает в области
переходного кинетического режима. Этиловый эфир малоновой кислоты
проявляет очень слабокислотные свойства и практически реагирует только
в присутствии основного катализатора.
Патаи и сотрудники показали также, что дегидратация альдоля, сопро-
вождающая реакции присоединения, также обратима. Дегидратация в общем
случае катализируется как основаниями, так и кислотами, но, кроме того,
в случае рассмотренных выше реакций дегидратация катализируется раство-
рителем, обладающим либо кислотными, либо основными свойствами.
В соответствии с принципом микроскопической обратимости катализируемая
основаниями реакция, обратная альдольной конденсации, сопровождающая-
ся выделением карбонильного соединения, заключается в мономолекулярном
отщеплении аниона от предварительно образовавшегося сопряженного осно-
вания альдоля. В гл. IX такой механизм отщепления обозначен символом
Е1сВ.
Альдольная конденсация катализируется кислотами. Было выяснено, что
в этом случае применим механизм Лэпуорта, видоизмененный с учетом кислот-
827
Глава XIII
ного катализа, т. е. в первой стадии реакции карбонильное соединение
превращается в соответствующую сопряженную кислоту, являющуюся
частично оксониевым и частично карбониевым ионом. В последней форме
сопряженная кислота — настолько сильный электрофильный реагент, что
способна вызвать замещение в енольной форме карбонильного соединения
(СН3)2 СО + Н+ <=> (СН3)2 С(ОН)
(СН3)2С(ОН) + (£1с(СН3)^0)Н^(СН3)2С(ОН)-СНгС(СН3)=О + Н+
Эти катализируемые кислотами реакции часто протекают дальше: альдоль
или кетол претерпевают дегидратацию. Например, из ацетона образуется
окись мезитила или даже — в результате дальнейшей кетолизации и де-
гидратации — форон.
Нойс и сотр. [138] исследовали кинетику катализируемой кислотами реакции
между ацетоном или метилэтилкетоном с бензальдегидом и «-метокси-
и «-нитробензальдегидом. Полученные ими результаты находятся в соответ-
ствии с общепринятым взглядом на механизм этих реакций: лимитирующей
оказывается стадия присоединения. В этих примерах присоединение альдолей
сопровождается дегидратацией кетолов в бензилиденкетоны, и вся последо-
вательность реакций оказывается обратимой.
Исследовано влияние заместителей на кинетику и термодинамику некоторых
стадий реакции. Принимая во внимание обратимость реакций между бен-
зальдегидом и кетоном, с одной стороны, и кетольным аддуктом — с другой,
было показано, что термодинамическое влияние заместителей аналогично
тому, какое они оказывают на равновесие между бензальдегидами и их циан-
гидринами. В обоих случаях «-метоксизаместители препятствуют присоеди-
нению, что можно понять, так как метоксигруппа сопрягается через бензоль-
ное кольцо с альдегидной группой, в результате чего альдегидная группа
стабилизуется в отличие от циангидрина или кетола, которые стабилизовать-
ся не могут, так как в них ненасыщенность первоначальной альдегидной
функции нарушена в результате присоединения.
Когда влияние заместителей на равновесие реакции было разделено на влия-
ние на скорости противоположных реакций, то оказалось, что «ара-метокси-
заместитель немного ускоряет реакцию присоединения, а обратную реакцию
ускоряет очень сильно. Это свидетельствует о том, что «ара-метоксильная
группа сопряжена с частичным карбониевым центром переходного комплекса
на стадии присоединения в несколько раз сильнее, чем с альдегидной труп-
ной в исходном состоянии. Другими словами, заместитель стабилизует
исходное состояние, но еще сильнее — переходное и не будет стабилизовать
конечное состояние катализируемого кислотами процесса присоединения
альдолей.
Кинетика обратимой дегидратации кетолов в бензилиденкетоны была иссле-
дована только для прямой реакции — дегидратации. И здесь проявился
эффект ускорения реакции при наличии метоксизаместителя. Как будет
показано ниже, этого и следовало ожидать, даже несмотря на то, что мы
не знаем механизма катализируемой кислотами дегидратации.
В этом случае возможны два механизма. Первый состоит в следующем: как
отмечалось в разд. 1,а, кислотная гидратация простых олефиновых соеди-
нений представляет собой пример общего кислотного катализа, и в соответ-
ствии с принципом микроскопической обратимости можно заключить, что
кислотная дегидратация соответствующих простых спиртов проходит через
ион карбония, это соответствует не хорошо изученному механизму Е1,
828
Присоединение и обратные реакции
а менее известному механизму Е2(С+). Если это справедливо в отношении
дегидратации кетола, то можно написать:
+н+ +
С8Н5СН(ОН)СН2СОСН3 -----» С8Н6СН(ОН2)СН2СОСН3
+ — н+
С6Н5СНСН2СОСН3 --------» С6Н5СН = СНСОСНз
Медленно
► Е2(С+)
Самой медленной в этом случае является третья стадия, переходный ком-
плекс которой должен соответствовать частичному образованию карбониевого
иона, так чтобы «-метоксильная группа могла сопрягаться с электрононена-
сыщенным центром. Поскольку такое сопряжение невозможно в исходном
состоянии дегидратации, метоксизаместитель должен оказывать ускоряющее
влияние. Величина этого эффекта должна зависеть от той неопределенной
степени, в которой происходит развитие положительного заряда в переход-
ном комплексе на бензильном углеродном атоме.
Однако нельзя ожидать, что все закономерности, свойственные кислотной
дегидратации простых спиртов, можно полностью перенести на кетолы.
Механизм Е2(С+) обусловлен трудностью отрыва протона от парафинового
углерода, что делает стадию переноса протона самой медленной в этом про-
цессе. При кислотной дегидратации кетолов соответствующей этому процессу
стадией является отрыв протона от прототропного (кето-енольного) углерода.
Это должно протекать намного легче и может происходить достаточно быстро,
так что лимитирующей окажется более ранняя стадия механизма. Тогда
механизм может стать типа Е1, в котором присутствует то же самое проме-
жуточное соединение, но лимитирующей оказывается вторая стадия
С8Н5СН(ОН)СН2СОСН3 —С8Н5СН(ОН2)СН2СОСН3 с ”
+ -н+
С6Н5СНСН2СОСН3 --» С6Н5СН = СНСОСНз
Переходный комплекс в этом случае будет иным, но частично сохранит
характер карбониевого иона, и поэтому «-метоксизаместитель должен уско-
рять дегидратацию, хотя, с другой стороны, величина такого ускорения зави-
сит от той неизвестной доли, в которой катионный заряд принят бензильным
углеродным атомом.
Механизм Е1 соответствует специфическому катализу ионами водорода
гидратации а,Р-ненасыщенных кетонов. Механизм Е2(С+) соответствует
общему кислотному катализу гидратации. Следовательно, определение типа
кислотного катализа в этих реакциях будет решающим фактором при выборе
между двумя механизмами элиминирования.
По-видимому, существует соответствие между ориентацией альдольного
присоединения к асимметричным кетонам и ориентацией при галогенирова-
нии кетонов. На это указывает то, что продукты альдольной реакции зависят
скорее от кинетических, чем от термодинамических факторов; обе реакции
представляют собой электрофильные процессы и протекают при основном
катализе через анион, а при кислотном катализе через енол. В гл. XI,
разд. 1,г, было показано, что метилэтилкетои галогенерируется быстрее
в метильную группу в щелочной среде (контроль ионизации ориентацией
типа Гофмана) и в метиленовую группу в кислой среде (контроль ориентаци-
ей типа Зайцева). Было известно уже в 1902 г., что метилэтилкетои конден-
сируется с бензальдегидом с образованием С6Н6СН = СНСОСН2СН3 в щелоч-
ной среде и образованием СН3СОС( = СНСвН5)СН3 в кислой среде [139].
829
Глава XIII
Третьим типом катализаторов альдольной и подобных ей конденсаций яв-
ляются амины (кроме третичных). Действие этих катализаторов количествен-
но изучалось на примере декетолизации диацетонового спирта [140]. Третич-
ные амины в чистом виде не катализируют эту реакцию; их кажущееся ката-
литическое действие обусловлено наличием в растворах третичных аминов
ионов гидроксила. В то же время аммиак, первичные и вторичные амины
катализируют реакцию по другому механизму, влияя на скорость процесса
следующим образом:
Скорость ос [Кетон][Амин]
Предполагается, что при конденсации аминов с карбонильной группой кетола
образуется иммоний-ион, в котором стадия, определяющая скорость реакции,
происходит быстрее, чем в карбонильном соединении
(СН3)2С< + т=1 (СНз)2С=О + CH2C(=NR2)CH3
(j,:h2C(=nr2)Ch3
По существу, альдольная конденсация состоит в присоединении кето-енола
или другой псевдокислоты HCXYZ к группе С = О альдегида или кетона
с одновременно протекающим процессом дегидратации и часто наблюдаемыми
последующими реакциями. Альдольная конденсация является основой очень
важной группы синтетических методов. Впервые альдольная конденсация
была открыта Вюрцем [141] в 1872 г. Применение щелочи, способствующей
более легкому протеканию альдольной конденсации и основанных на ней
других превращений, было предложено Шмидтом [142] и тщательно исследо-
вано Кляйзеном [143]. Поэтому рассматриваемая реакция, хотя и не совсем
правильно, иногда называется реакцией Кляйзена. Кляйзен также предло-
жил применять кислотные катализаторы для альдольной конденсации
и последующих процессов [144]. Использование аммиака предложено Джап-
пом и Стритфилдом [145], но гораздо детальнее вопрос применения аммиака,
первичных и вторичных аминов был исследован Кнёвенагелем [146] *, после
работ которого реакция, протекающая в этих условиях, стала называться
реакцией Кнёвенагеля.
Еще одной формой альдольной конденсации, которую удобно выделить
отчасти по историческим причинам, отчасти из-за совершенно иных экспери-
ментальных условий, является реакция Перкина. Она была предложена
в 1875 г. Перкином старшим [151] для типичного случая — получения корич-
ной кислоты нагреванием бензальдегида с уксусным ангидридом и ацетатом
натрия при 180 °C. Сначала Перкин [147], а потом и Фиттиг [148] неоднократ-
но пытались выяснить механизм реакции и решить вопрос, участвует ли
в конденсации ангидрид или натриевая соль. Перкин полагал, что решающую
роль играет ангидрид, Фиттиг придерживался иного мнения. Оба исследова-
теля проводили конденсацию, употребляя натриевую соль одной кислоты
в сочетании с ангидридом другой и изучали строение образующегося аналога
коричной кислоты. Оказалось, что результаты зависят от температуры про-
цесса. При этом было сделано открытие [147, 149], сравнительно недавно
подтвержденное [150], заключающееся в легком взаимном превращении
ангидридов и солей
Ангидрид А -|- Соль Б Ангидрид Б -|- Соль А
* См. также ряд статей, на которые ссылается Кнёвенагель во второй из приведенных
в ссылке [146] работе.
830
Присоединение и обратные реакции
Таким образом, стало ясно, что поставленная задача не может быть легко
разрешена указанным путем. Вместе с тем выяснилось, что конденсация
бензальдегида с пропионовым ангидридом и пропионатом натрия или с масля-
ным ангидридом и бутиратом натрия во всех случаях протекает с участием
а-углеродного атома ангидрида или соли кислоты.
Фиттигу удалось получить строгое доказательство того, что первой стадией
реакции является присоединение типа альдольной конденсации. Из бензаль-
дегида, янтарного ангидрида и сукцината натрия при 100 °C был получен
продукт присоединения — у-лактон оксикислоты [151]. Из бензальдегида,
изомасляного ангидрида и изобутирата натрия также при 100 °C Фиттиг
получил в качестве аддукта оксикислоту [152, 153]
С6Н5СНО + СН2(СООН)СН2 —> C6H5CHCH(COOH'iCH2
I I I
СООН о----------со
СвН5СНО + (СН3)2СНСООН СвН5СН(ОН)С(СН3)2СООН
Роль ангидрида и соли оставалась невыясненной до 1928 г. В этом году
Калнин [154] показал, что, в то время как ангидрид действительно участвует
в реакции, натриевая соль карбоновой кислоты может быть заменена многи-
ми другими основаниями, например карбонатом, фосфатом или сульфитом
натрия, триэтиламином, пиридином или хинолином. Таким образом, как
и следовало ожидать на основании общих представлений, ангидрид в этой
реакции является псевдокислотным аддендом, а соль — катализатором основ-
ного характера. По-видимому, протон из ангидрида переходит к аниону
соли, а анион ангидрида присоединяется к карбонильной группе альдегида.
Вернемся теперь к присоединению Н — N к связи С = О и рассмотрим обра-
тимое образование азометинов, оксимов, арилгидразонов и семикарбазонов.
Все эти реакции представляют собой примеры альдольной конденсации:
в результате присоединения образуются карбиноламины или их N-окси-
или N-аминопроизводные; реакции сопровождаются дегидратацией простых
или замещенных карбиноламинов с образованием дважды конденсированных
продуктов. Последовательность реакций, так же как и в альдольной конден-
сации, полностью обратима.
Образование азометинов протекает несколько отлично от других реакций.
С простейшими алифатическими альдегидами или кетонами и первичными
аминами конденсация часто может протекать без участия первоначально
образовавшегося азометина, поскольку способность к присоединению у групп
> С = NR и > С = О одинакова. Вообще говоря, простейшие вещества
могут дать сравнительно сложные продукты, как это прекрасно иллюстрирует
пример реакции между формальдегидом и аммиаком.
Поскольку образование азометина не является одностадийной конденсаций,
равновесие в водном растворе сильно сдвинуто в сторону исходных веществ;
однако в отсутствие воды равновесие сдвигается в сторону образования
продуктов. Полного образования карбиноламинов прежде, чем они начнут
дегидратироваться или переходить вновь в исходные соединения, не проис-
ходит; исключение составляют те случаи, когда в условиях реакции карби-
ноламины выпадают в осадок, например в случае ацетальдегида и аммиака.
В противоположность альдольной конденсации для азометиновой конденса-
ции и обратного ей процесса гидролиза не свойствен основной катализ.
Поскольку амины сами по себе основны, для их присоединения к карбониль-
ной группе депротонирования не требуется. Это не означает, что конденсация
н гидролиз азометинов не могут катализироваться основаниями: катализ
может иметь место при очень высоких значениях pH, как, например, в случае
831
Глава ХШ
образования бензилиденанилина [155]. Хотя амины представляют собой
основания, амид-ионы, образующиеся из них путем депротонирования,
являются еще более сильными основаниями. Однако в интервале pH от
умеренно высоких значений до нейтрального реакции присоединения
и, конечно, вся последовательность, ведущая к образованию азометинов,
и обратные им реакции являются некаталитическими.
По другую сторону от нейтральной точки (в кислой среде) последователь-
ность реакций оказывается катализируемой кислотами; но, как показал
Дженкс [156], кислотный катализ проявляется на стадии дегидратации. Это
вполне естественно, так как карбиноламин представляет собой псевдооснова-
ние и, следовательно, весьма чувствителен к кислотам (гл. XI, разд. 2, а).
С увеличением концентрации кислоты скорость конденсации может пройти
через максимум в некоторой точке, которая соответствует основности при-
соединившегося амина. При высоких значениях кислотности кислота может
так ускорить дегидратацию, что лимитирующей вновь станет стадия присоеди-
нения и это приведет к замедлению процесса, так как избыток кислоты выве-
дет первичный амин из реакции, сведя к минимуму его нуклеофильные свой-
ства.
Если оставить в стороне количественные различия, то эти рассуждения будут
справедливы не только для обратимого образования азометинов, но также
и оксимов, семикарбазонов и, вероятно, арилгидразонов; на основании этого
можно сформулировать общий для всех этих реакций механизм, пользуясь
простым набором формул, в котором нуклеофил обозначим NH2X, где X =
= Н, алкил, арил, ОН, NHAr или NHCONH2-
Катализируемое щелочами присоединение:
nh2x+oh- дД nhx- + h2o
2 /0-
RR'CO + NHX- RR'CZ
\nhx
/О- з /ОН
RR'CZ + Н20 RR'CZ 4-ОН-
\nhx \nhx
Некатализируемое присоединение:
4 ,0~ 5 /ОН
RR'CO+ NH2X RR'CZ RR'CZ
\\H2X+ ^NHX
Бекатализируемая дегидратация:
/ОН в _ 7
RR'COZ RR'C=NHX + OH ^Д RR'C = NX4-H2O
\nhx
Катализируемая кислотами дегидратация:
/ОН 8 /0Н£
RR'CZ 4-Н30+ ~Д RR'CZ +Н20
Nxhx \nhx
/ОНJ 9 + ю
RR'CZ ДД RR'C=NHX+H2O дД RR'C = NX + H3O+
\nhx
832
Присоединение и обратные реакции
Для катализируемого основаниями присоединения скорость конденсации
выражается уравнением
Скорость ос [Карбонильное соединение][Амин][ОН~]
Для некатализируемых реакций присоединения и дегидратации
Скорость ос [Карбонильное соединение][Амин]
При этом не важно, какая стадия — присоединение (стадия 4) или дегидра-
тация (стадия 6) — является лимитирующей. В случае катализируемой
кислотой дегидратации уравнения для скорости могут иметь следующий вид:
Скорость ос [Карбонильное соединение][Амин][НА] или
ДСкорость ос [Карбонильное соединение][Амин][НА]/[А~)
в зависимости от того, является ли кислотный катализ общим (стадия 8
лимитирующая) или специфическим (стадия 9 лимитирующая).
Оба вида кислотного катализа были исследованы на примере гидролиза
разных азометинов в различных растворителях при неодинаковой степени
кислотности, что достигалось с помощью использования буферных растворов
[157, 158]. Вероятно, в соответствующих реакциях азометиновой конденсации
проявляются оба типа катализа, хотя прямых и формальных доказательств
этого не было получено. Если в реакциях конденсации при определенном
значении кислотности лимитирующей стадией оказывается катализируемая
кислотой дегидратация, то при увеличении кислотности эта стадия будет
ускоряться и может стать более быстрой по сравнению со стадией присоеди-
нения и тогда лимитирующей скорость вновь станет стадия присоединения.
Скорость реакции тогда снова перестанет возрастать с увеличением кислот-
ности; если же ввести избыток кислоты, то присоединившийся амин выйдет
из реакции, в которой он реагирует в форме свободного основания, и ско-
рость реакции начнет уменьшаться при дальнейшем увеличении кислотности.
Эти явления хорошо известны [156].
Лимитирующая скорость реакции стадия при образовании или гидролизе
азометина часто может быть выявлена по влиянию на скорость реакции
заместителей. Так, например, при образовании азометина из производных
бензальдегида и-метоксизаместитель в бензальдегиде будет уменьшать ско-
рость присоединения амина по альдегидной группе (реакция 4), но увели-
чивать скорость дегидратации карбиноламина (реакция 6 или реакции 7 и 8).
Такое же противоположное влияние на скорости двух главных стадий общего
процесса будут оказывать и многие другие заместители. Если предположить,
что контролирующей скорость реакции стадией является обратимое при-
соединение (стадия А)
Альдегид + Амин Карбиноламин Азометин
а в
тогда, если рассматривать реакцию, идущую слева направо, т. е. образова-
ние азометина, единственным влиянием заместителя, которое необходимо
учитывать, будет его влияние на стадию А; поэтому влияние заместителя
должно быть достаточно большим в соответствии с природой стадии А. Если,
однако, рассмотреть реакцию, протекающую справа налево, т. е. реакцию
гидролиза азометина, то влияние заместителя на стадию Б, которая в этом
случае является предварительной, может увеличить стационарную кон-
центрацию карбиноламина, когда скорость его распада на стадии А под
влиянием заместителя должна была уменьшиться, или уменьшить стационар-
ную концентрацию, когда скорость распада карбиноламина должна была
53-0772
833
Глава XIII
увеличиваться. Суммарный эффект заместителей на скорость гидролиза,
таким образом, определяется разностью противоположных влияний и, по
всей вероятности, должен быть небольшим и нерегулярным. Вернемся к нача-
лу наших рассуждений и предположим, что лимитирующая стадия соответ-
ствует стадии обратимой дегидратации карбиноламина (стадия Б). Тогда
с помощью тех же аргументов можно показать, что влияние заместителей
на образование азометина должно быть малым и сложным, а на гидролиз
азометина — большим и четко связанным с природой заместителя, как это
предсказывалось для идущей справа налево стадии Б.
Некоторые из этих эффектов можно проиллюстрировать примерами. Пека-
тализируемая конденсация бензальдегида с анилином в растворе бензола
подчиняется закону второго порядка, а влияние заместителе]! на константы
скорости конденсации замещенных бензальдегидов соответствует ожидаемо-
му для того случая образования азометина, когда лимитирующей стадией
является присоединение (стадия 4) [159]. Влияние заместителей на скорость
гидролиза бензилиденапилина и его производных в почти нейтральной среде
или в слабокислом буферном растворе оказывается малым и сложным по
характеру [158]. В обоих примерах лимитирующей скорость является ста-
дия 4. При изучении конденсации замещенных бензальдегидов с 2-бутилами-
ном в метиловом спирте и ацетатном буферном растворе было обнаружено
малое и беспорядочное влияние заместителей, что характерно для случая
лимитирующей стадии 6 [160]. Для гидролиза бензилиденапилина разбавлен-
ной кислотой в водном метиловом спирте без использования буфера были
найдены условия, когда кинетический контроль за ходом реакции осуще-
ствлялся одновременно стадиями 4 и 6, ас увеличением кислотности лими-
тирующая функция переходит от стадии 6 к стадии 4 [161].
Реакции образования оксимов, арилгидразонов и семикарбазонов имеют два
основных отличия от реакции образования азометинов. Одно из них состоит
в том, что в реакциях образования оксимов и гидразонов, включая семикар-
базоны, почти не проявляется тенденции первоначально образовавшихся
продуктов конденсации вступать в реакции последующего присоединения.
Обычно последующие реакции другие. Так, например, альдоксим может
дегидратироваться в нитрил, а арилгидразон — таутомеризоваться в азо-
соединение. Второе общее отличие состоит в том, что даже в среде с высоким
содержанием воды равновесие реакций карбонила с гидроксиламином
и гидразинами, включая семикарбазид, гораздо сильнее смещено в сторону
образования оксима или гидразона или семикарбазона по сравнению с равно-
весием образования азометина. (Поскольку равновесие сдвинуто в одну
сторону, наиболее простым способом выявить обратимость образования окси-
ма или гидразона является связывание гидроксиламина или гидразина,
образовавшихся из их производных в результате гидролиза, избытком друго-
го карбонильного соединения.) Оба эти отличия от реакции образования
азометина можно объяснить с помощью того обстоятельства, что когда осу-
ществляют конденсацию гидроксиламипа или гидразина С карбонильным
соединением, то вводят группу с новой внутренней сопряженной системой
6-^-N==C или bP-N=^C
благодаря чему приобретается термодинамическая стабильность про-
дукта. Этого не происходит в случае конденсации амина с образованием
азометина.
Характерное отличие реакции образования оксимов от реакции образования
гидразонов заключается в ее ярко выраженном основном катализе. Это было
834
Присоединение и обратные реакции
установлено уже на первом примере при исследовании механизма группы
аналогичных реакций С — N-конденсации, проведенном Барретом и Лэпуор-
том [162] в 1908 г. на примере образования оксимов. Они обнаружили суще-
ствование как основного, так и кислотного катализа. Для первого было
высказано предположение, что он протекает по механизму, описанному на
стр. 832. Важность основного катализа для образования оксима можно свя-
зать с тем обстоятельством, что анион гидроксиламина оказывается «анти-
сопряженным» (см. ссылку на стр. 375) и два из его пеподеленных электронов
находятся на разрыхляющей молекулярной орбитали, благодаря чему
анион приобретает очень сильные нуклеофильные свойства (гл. VII, разд.
2,в). Анион гидроксиламина не может выйти из этой ситуации при
помощи скручивания, как это несомненно происходит с анионом гидразина
(гл. VII. разд. 2,в). Малая равновесная концентрация аниона гидроксил-
амина будет, таким образом, оказывать непропорционально большое
влияние на скорость взаимодействия с высокополярным электрофильным
центром.
Кислотный катализ при образовании оксимов [162, 165], арилгидразонов [163]
и семикарбазонов [164—166] следует общим закономерностям. При низкой
кислотности, когда зависимость скорости от концентрации кислотного ката-
лизатора является линейной, скорость расходования карбонильного соеди-
нения соответствует низкой скорости образования оксима или гидразосоеди-
нения. Это означает, что N-замещенный карбиноламин образуется быстро,
а дегидратация продукта его присоединения идет медленно, что служит
доказательством кислотного катализа. С увеличением кислотности линей-
ность исчезает, и скорость реакции проходит через максимум и затем умень-
шается. С ускорением дегидратации лимитирующей вновь будет стадия
присоединения, и по мере дальнейшего повышения кислотности скорость
реакции будет уменьшаться в результате протонирования нуклеофильного
адденда. Для случая образования семикарбазона из замещенных бензальде-
гидов Андерсон и Дженкс [166] показали, что кинетическое влияние замести-
телей при изменении кислотности по обе стороны от максимума скорости
будет различным: со стороны большей кислотности это влияние вызвано
лимитирующим характером стадии присоединения, а со стороны меньшей
кислотности — лимитирующим характером стадии дегидратации псевдоосно-
вания.
Возвращаясь к присоединению Н — Ок карбонильной группе, следует
остановиться на образовании гидратов и алкоголятов карбонильных соеди-
нений. Наиболее изученным примером подобных реакций присоединения
является исследованная Беллом с сотр. [167] гидратация ацетальдегида
СН3СНО + Н2О СН3СН(ОН)2
Они изучили кинетику реакции в обоих направлениях, воспользовавшись
теми фактами, что равновесие значительно сдвинуто в сторону гидрата в вод-
ном растворе и в сторону альдегида в 92%-ном водном растворе ацетона.
В обоих направлениях реакция подвержена как катализу основаниями,
так и общему кислотному катализу. Таким образом, следует различать
механизмы двух типов. Основной катализ характеризуется обратимостью
присоединения гидроксильных ионов к нейтральной молекуле альдегида
/°"
СН3СНО + ОН- СНзСНГ
^он
53* 835
Глава XIII
Эта схема может быть расширена при допущении общего основного катализа,
который может осуществляться, как обычно, несколькими путями. Так
как гидроксильные ионы образуются из растворителя, то наиболее правдо-
подобно предположить, что общий основной катализатор В~ взаимодействует
с водой в качестве растворителя с одновременным образованием гидроксиль-
ных ионов и присоединением последних в тримолекулярной реакции
I Г4 I Л
-В * Н-Ч)1Г С=О В—Н + НО—С—О"
I I
Аналогично кислотный катализ характеризует обратимость присоединения
сопряженной кислоты альдегида к нейтральной молекуле воды
* _ /°Н
сн3снон+н2о ^сн3он;
Х)щ-
Эта схема также должна быть расширена, для того чтобы она могла объяс-
нить общий кислотный катализ. И в этом случае имеется несколько возмож-
ностей. По аналогии с механизмом общего основного катализа можно пред-
полагать, что кислота НА генерирует катион альдегида, который связывает
молекулу растворителя (воды) в синхронной тримолекулярной реакции
A-Lh ^=С <ОН2 —> А + Н-О-С-ОН2
I
Тримолекулярные реакции, в которых принимает участие одна молекула
растворителя, вполне возможны. Если мы выделим из этого механизма
бимолекулярную лимитирующую стадию переноса протона, происходящую
при быстром захвате воды, то получим обратное доказательство справедли-
вости общего механизма катализируемой кислотами дегидратации (стр. 832),
включая общий кислотный катализ.
б. Обратимое присоединение оснований или слабых кислот к сопря-
женным олефиновым карбонильным соединениям. Типичным примером AdN-
присоединения нуклеофильных кислых аддендов может служить присоеди-
нение синильной кислоты к а,^-ненасыщенным кетонам, эфирам или нитри-
лам. Начиная с 1903 г. Лэпуорт [168] исследовал ряд подобных реакций,
например присоединение синильной кислоты к окиси мезитила, форону,
1-цианстильбену и карвону:
(СН3)2С = СНСОСНг —» (CH3)2C(CN)CH2COCH3
[(СН3)2С = СН]2СО -» l(CH3)2C(CN)CH2]2CO
CeH5CH=C(CeH5)CN —> CeH5CH(CN)CH(CeH5)CN
Порядок присоединения во всех случаях один и тот же — циангруппа на-
правляется в p-положение. В большинстве случаев реакция в основном
доходит до конца; ни в одной работе не изучено равновесие при этих реак-
циях. При исследовании скорости каждого из изученных примеров Лэпуорт
показал, что в то время как при смешивании синильной кислоты и ненасы-
836
Присоединение и обратные реакции
щенного вещества присоединение едва ли происходит, оно протекает очень
легко при добавлении цанистого калия. Позднее Джонс [169] изучил кинети-
ку присоединения синильной кислоты к натриевой соли а-цианкоричной
кислоты. Ему удалось установить, что скорость реакции пропорциональна
концентрации свободного цианид-иона
Скорость ос [C6H5CH = C(CN)COO-][CN]
Тем самым Джонс подтвердил вывод Лэпуорта, что стадией, определяющей
скорость процесса, является присоединение цианид-иона в р-положение
а,р-двойной связи
ZCN ,CN
CN4-CeH5CH=C/ —» CeH5CH-CZ
\coo- | ^coo-
CN
Реакция Михаэля, заключающаяся в катализируемом основаниями присоеди-
нении кетонов, эфиров, нитрилов или нитросоединений, обладающих псевдо-
кислотными свойствами, к а, p-двойной связи сопряженного ненасыщенного
кетона, эфира или нитрила сравнима с рассмотренным выше присоединением
синильной кислоты к а,р-двойной связи в такой же степени, как альдольная
конденсация сравнима с образованием обычных циангидринов. Первым
примером реакции Михаэля, открытым в 1887 г. [170], было присоединение
этилмалонового эфира к этиловому эфиру коричной кислоты в присутствии
раствора этилата натрия в этиловом спирте с образованием продукта при-
соединения, из которого после гидролиза и декарбоксилирования была
получена p-фенилглутаровая кислота
zCH2COOC2H5
СвН5СН = СНСООС2Н5 + СН2(СООС2Н5)2 —» CeHjCir
\сн(соос2н5)2
Другим примером, широко использованным Перкином младшим в процессе
синтеза продуктов распада камфоры, служит присоединение этилмалонового
эфира или этилового эфира циануксусной кислоты к этиловому эфиру р,р-
диметилакриловой кислоты с образованием эфиров, которые в результате
гидролиза и декарбоксилирования могут быть переведены в р,р-диметилглу-
таровую кислоту
,СН2СООС2Н5
(CH3)2C = CHCOOC2H5-|-CH2(CN)COOC2H5 —> (СН3)2СХ
X'CH(CN)COOC2H5
Порядок таких реакций присоединения таков, что анион псевдокислотного
адденда всегда направляется в p-положение а,р-двойной связи.
Реакция Михаэля обычно осуществляется по трем общим вариантам. Перво-
начальный способ Михаэля состоял в применении эквимолярного количества
этилата натрия. В другом, так называемом «каталитическом» методе исполь-
зуются лишь очень незначительные количества этилата натрия. Наконец,
в методе Кнёвенагеля [171] в качестве катализатора применяется не этилат
натрия, а вторичный амин типа пиперидина. Реакции в условиях второго
и третьего методов протекают медленнее, чем при применении первого
способа.
Обратимость реакции Михаэля впервые была обнаружена Форлендером [172],
а позднее она изучалась Пауэллом, Перреном и автором книги [173]. Боль-
шинство реакций присоединения Михаэля представляют собой экзотермичные
процессы, и поэтому высокий выход продуктов присоединения наблюдается
837
Глава XIII
только при низких температурах при условии значительного увеличения
длительности реакции. В первых опытах Михаэля с этиловым эфиром корич-
ной кислоты и малоновым эфиром высокий выход продукта присоединения
наблюдался для реакции при комнатной температуре и низкий выход —
при температуре кипения спиртного раствора. Равновесие зависит от способа
проведения реакции, что можно понять, если сделать вполне вероятное пред-
положение, что присоединяющаяся псевдокислота — более сильная кислота,
чем образующийся продукт присоединения, или, другими словами, что анион
псевдокислоты более устойчив. Так, в присутствии 1 экв этилата натрия
возникает равновесное состояние (от которого зависит строение преобла-
дающих продуктов реакции) не между тремя указанными выше эфирами,
а между ненасыщенным эфиром и анионами двух других эфиров. Различие
в термодинамической устойчивости упомянутых анионов препятствует при-
соединению. Предположим далее, что меняются условия реакции и приме-
няются только каталитические количества этилата натрия или же исполь-
зуется метод Кнёвенагеля. Тогда равновесие, определяющее состав конечных
продуктов, установится между тремя эфирами и указанный термодинами-
ческий фактор, препятствующий реакции, не будет играть роли. При этом
по достижении равновесия продукт присоединения получается с лучшим
выходом.
Влияние строения на равновесное состояние изучено сравнительно хорошо.
Алкильные и в еще большей степени арильные группы, находящиеся в а-
или p-положениях в ненасыщенном эфире, термодинамически препятствуют
присоединению. По-видимому, это связано с гиперконъюгацией или сопря-
жением с двойной связью, что способствует ее сохранению- К этому же резуль-
тату может привести и первичный пространственный эффект [173]. Препара-
тивные выходы продуктов присоединения по Михаэлю могут быть достигнуты
при замещении водородов ненасыщенного эфира на одну сопряженную
фенильную или на две гиперконъюгированные метильные группы, например
при реакциях эфиров коричной и р,0-диметилакриловой кислот. При наличии
двух фенильных или трех метильных групп удовлетворительные выходы
не достигаются. Обычно продукты присоединения эфиров циануксусной
кислоты образуются с более высокими выходами, чем из малоновых эфиров,
однако причина этого явления точно не известна.
Кинетика реакции Михаэля изучена на нескольких примерах присоединения
к акрилонитрилу. В этих реакциях присоединение в значительной мере
определяется термодинамическим фактором. Присоединение псевдокислоты
к акрилонитрилу часто называют «цианэтилированием» псевдокислоты.
В качестве примера можно привести присоединение ацетилацетона к акри-
лонитрилу в воде, содержащей едкое кали [174]. Эта реакция подчиняется
написанному ниже уравнению, которое аналогично уравнению Джонса,
найденному им для присоединения цианистого водорода к иону а-цианко-
ричной кислоты
Скорость ОС [CH2=CHCN][(CH3CO)2CH-]
Второй член произведения представляет собой концентрацию ацетилацето-
нат-иона, которая определялась исходя из основности раствора и известных
.значений констант кислотности ацетилацетона. В таком виде кинетическое
уравнение подтверждает теоретический механизм реакции присоединения
Михаэля.
Аммиак, первичные и вторичные амины могут присоединяться к этиленовой
двойной связи а.р-ненасыщенных кетонов и эфиров. Атом азота амина на-
правляется всегда в p-положение. Так, аммиак, метиламин и диметиламин
838
Присоединение и обратные реакции
присоединяются к окиси мезитила с образованием диацетонамина и его
метильных производных [175], а анилин и фенилгидразин присоединяются
к метиловому эфиру бензилиденмалоновой кислоты с образованием аддуктов
аналогичной структуры [176]
(СН3)2ССН2СОСН3 С6Н5СНСН(СООСН3)2
I !
NRR' NHC6H5
Огата и сотр. [174] установили, что присоединение этаноламина к акрило-
нитрилу (цианэтилирование этаноламина) в воде при pH 7,5—9,4 протекает
через присоединение атома азота к p-положению акрилонитрила с образова-
нием в конечном счете HOCH2CH2NHCH2CH2CN.
Найдено, что эта реакция подчиняется уравнению
Скорость ОС [CH2=CH2CN][NH2CH2CH2OH]
Из этих результатов ясно, что присоединение аминов к а,р-ненасыщенным
кетонам, эфирам или нитрилам в умеренно основных растворах проходит
скорее через свободное основание, а не через его анион как нуклеофильную
частицу.
Ненасыщенные кетоны, эфиры и нитрилы также присоединяют спирты в при-
сутствии их алкоголят-ионов. Алкоксигруппа входит в p-положение. Так,
например, бензилиденмалоновый эфир присоединяет метиловый спирт в мета-
нольном растворе метилата натрия [177]. Продукт присоединения имеет
формулу СвН5СН(ОСНз)СН(СООСНз)2.
Показано [168], что кинетика реакций присоединения к акрилонитрилу ряда
простейших алифатических кислот (цианэтилирование спиртов), протекаю-
щая в спиртовых растворах и в присутствии алкоголятов щелочных металлов
как катализаторов, подчиняется уравнению
Скорость a [CH2 = CH2CN][OR-]
Во всех этих случаях, очевидно, протекает нуклеофильное присоединение
алкоголят-иона, а не молекулы спирта.
Основываясь на принципе микроскопической обратимости в применении
к реакции Михаэля, обратимость которой формально доказана, можно сделать
вывод, что процесс отщепления должен начинаться с первоначального отщеп-
ления протона от расщепляющейся молекулы с образованием анионоидного
сопряженного основания, вслед за чем происходит мономолекулярное выде-
ление аниона из сопряженного основания. Таким образом, в этом случае
реализуется механизм отщепления, который в гл. IX был обозначен симво-
лом Е1сВ и который в упомянутой главе не был проиллюстрирован хороши-
ми примерами. Однако в рассматриваемых случаях этот механизм является
общеприменимым и именно по такому механизму образуются двойные связи,
которые, конечно, в данном случае обладают особыми свойствами вследствие
их сопряжения с карбонильной, циан- или нитрогруппой (гл. IX, разд. 1,а,
сноска на стр. 540).
Известно, что присоединение спиртов и воды к а,р-двойной связи, сопря-
женной с карбонилом или карбоксилом, может катализироваться кислотами.
Спирты с хорошими выходами присоединяются к окиси мезитила с образова-
нием эфиров диацетонового спирта при применении в качестве катализатора
незначительного количества серной кислоты [179]. Лукас и сотр. [180] иссле-
довали присоединение воды к а,|3-двойной связи акролеина, акриловой
кислоты и ее гомологов, протекающее при катализе кислотами с образованием
гидроакролеина, гидроакриловой кислоты или ее гомологов. Эти реакции
839
Глава XIII
в значительной степени обратимы
н+
СН2 = СНСНО + Н2О ---» СН2(ОН)СН2СНО
н+
СН2 = СНСООН + Н2О 7--> СН2(ОН)СН2СООН
В водном растворе скорость реакции в обоих направлениях пропорциональна
концентрации сильной кислоты; обычно в качестве кислоты применялись
хлорная или азотная кислота в концентрациях порядка 0,1—1 моль/л.
Скорость прямой реакции'"»: [а, р-Непасыщспное соединение][Н+]
Скорость обратной]реакции ;ос [Р-Оксисоединение][Н+]
Эти результаты находятся в соответствии с предположением, что роль кисло т-
ного катализатора состоит в превращении ненасыщенного карбонильного
соединения в его сопряженную кислоту, которая частично находится в виде
карбониевого иона. Вполне вероятно, что молекула воды непосредственно
атакует карбониевый ион
СН2 = СНСНО + Н+ СН2СН = СНОН
СН2СН=СНОН СН2СН2СНО+Н+
i I
+он2 он
При рассмотрении катализируемых основаниями реакций сопряженных
кетонов, эфиров или нитрилов, например сорбиновой кислоты, циннамили-
денуксусного или циннамилиденмалонового эфиров, возникает проблема
ориентации. В перечисленных сопряженных соединениях как р-, так 6-поло-
жения в принципе поляризованы положительно. Следовательно, они могут
быть местами присоединения нуклеофильного адденда или анионной части
слабокислого или псевдокислого адденда
X- 13-Атака
R—С=С^-С==СЛ-С^
X-' 6-Атака
Рассматриваемые реакции в значительной степени обратимы. Поэтому возни-
кает вопрос, какое положение будет подвергаться атаке и будет связывать
анион адденда.
Если присоединение аниона идет в p-положение, то протон адденда обяза-
тельно присоединяется в a-положение. Енольная форма аддукта предполо-
жительно термодинамически неустойчива
X Н
I I I I I .
—с=с—С—с-с=о
Однако при связывании аниона 6-углеродным атомом протон может направ-
ляться как в а-, так и в у-положения, если предположить, что енол неустой-
чив
X Н X Н
I I I I I I I I I I
—С-С=С-С—С=О и —С—С—с=с—с=о
I I
Вопрос о том, какой иэ этих изомеров будет фактически образовываться,
зависит от кинетических и термодинамических факторов, влияющих на обра-
840
Присоединение и обратные реакции
зование прототропных таутомеров пентадной кето-енольной системы. Этот
вопрос был тщательно изучен Коном и Линстедом. Принципы подхода к ре-
шению этой проблемы рассматривались в гл. XI, разд. 1,г, где было пока-
зано, что в соответствии с требованиями кинетики протон направляется
в «-положение, в то время как термодинамически наиболее устойчивое
положение протона определяется природой заместителей. Поэтому сначала
рассмотрим главным образом вопрос о месте, куда первоначально присоеди-
няется анион, и о месте, где он окончательно остается [181].
Если ориентация присоединения аниона или другой нуклеофильной частицы
к сопряженной полиеновой цепи, содержащей карбонильную или похожую
концевую группу, определяется кинетикой реакции, то нуклеофильная части-
ца будет присоединяться исключительно к другому концу сопряженной
системы. Причина этого аналогична указанной в разд. 1,6 и состоит в том,
что полярность в линейных полиеновых системах всегда имеет наибольшее
значение на концах цепи. Для того чтобы проверить это предположение,
необходимо было бы провести реакцию присоединения с такой степенью
превращения, которая была бы мала по сравнению с равновесной. Но,
насколько известно автору этой книги, исследования продуктов, образую-
щихся в этих условиях, пока не было проведено.
Однако в обычных синтезах реакция Михаэля почти всегда достигает равно-
весия. Поэтому, пытаясь интерпретировать имеющиеся наблюдения, необхо-
димо учитывать порядок присоединения, определяемого термодинамически-
ми факторами, т. е. зависящего от соотношения свободных энергий конечных
продуктов. В некоторых случаях, например при присоединении малонового
эфира к p-винилакриловому эфиру СН2 = СНСН = CHCOOR, необходима
считаться с относительной устойчивостью структур следующих типов:
[fi-Аддукт]- 7» Исходные [б-Аддукт]-
продукты
_!н+
ch2=chchch2coor 'ch2ch=chch2coor сн2сн2сн=chcoor'
CH(C00R)2 (^H(COOR)2 CH(COOR)2
Известны некоторые приближенные количественные данные о соотношениях
Р" и 6-аддуктов, присутствующих в равновесной смеси продуктов присоеди-
нения малонового и циануксусного эфиров к p-винилакриловому эфиру и его
различным метилированным гомологам, а именно к 6-метильному, т. е.
к сорбиновому эфиру, и к а-, Р~ и у-метилсорбиновым эфирам [182]. Эти дан-
ные приведены в табл. 177.
Исключительное 6-присоединение к исходному винилакриловому эфиру
можно объяснить тем, что один из прототропных таутомеров 6-аддукта ста-
билизуется благодаря сопряжению между олефиновой двойной связью
и концевой карбоксильной группой, в то время как в случае р-аддукта такая
стабилизация невозможна. Однако при введении 6-метильного заместителя,
как в сорбиновых эфирах, возникающая гиперконъюгация метильной группы
с 6,у-олефиновой двойной связью р-аддукта придает последнему некоторую
стабильность, вследствие чего в равновесном продукте находится заметное
количество р-аддукта. Этого не наблюдается при введении, помимо 6-, также
а- или Р-метильных заместителей, которые еще более стабилизуют устойчи-
вую форму 6-аддукта за счет гиперконъюгации а- или р-метильных групп
с «,р-олефиновой связью. С другой стороны, при наличии одновременно
841
Глава XIII
Таблица 177
Влияние метильных заместителей в эфирах р-винилакриловой кислоты
на выход (%) р-аддукта при присоединении по Михаэлю
/ б у Р а \
ВАЗ—винилакриловый эфир —С=С —С=С—COOR
\ 1111/
Исходные продукты Винилакри- ловый эфир б-Метил- замещенный а, З-Диметил- замещенный Р, б-Диметил- замещенный V, б-Дим?тил- замещенный
ВАЗ — малонат а 0 9
ВАЗ -J- цпанацетат 6 10 0 0 72
а Использовался метиловый эфир,
б Использовался этиловый эфир.
6- и у-метильных групп р-аддукт приобретает дополнительную устойчивость
благодаря гиперконъюгации, причем этот продукт становится устойчивее
любой формы б-аддукта.
Хорошо изучено влияние б-фенильного заместителя в винилакриловом эфире
на реакцию Михаэля и близкие процессы присоединения.
б-Фенильная группа обусловливает образование в равновесной смеси исклю-
чительно р~аддукта. Так, присоединение синильной кислоты к этиловому эфиру
циннамилиденмалоновой кислоты в присутствии цианистого калия [183],
малонового эфира к этиловому эфиру циннамилиденуксусной кислоты в при-
сутствии этилата натрия [184] и этилового спирта к метиловому эфиру цин-
намилиденмалоновой кислоты в присутствии этилат-ионов в спиртовом
растворе [185] протекает с образованием следующих продуктов присоеди-
нения:
СеН5СН =. СНСНСН(СООС2Н5)2 С6Н5СН = СНСНСН,СООС2Н5
I I
CN СН(СООС2Н5)2
С6Н5СН- СНСНСН(СООСНз)2
ос2щ
Эти результаты, без сомнения, обусловлены превалирующим энергетическим
эффектом сопряжения фенил — винил, которое приводит к образованию
только р-аддуктов.
По данным Фармера и Мартина [186], присоединение метилмалоната к гекса-
триеновому эфиру — метиловому эфиру сорбилиденуксусной кислоты
СН3СН = CHGH = СНСН = СНСООСН3 протекает с образованием р-
и ^-аддуктов, причем первый образуется преимущественно, а никаких следов
б-аддукта не обнаруживается. Отсутствие последнего можно объяснить тем,
что при его образовании нарушалось бы бутадиеноподобное сопряжение,
которое сохраняется в каждом из двух других аддуктов.
в. Присоединение металлалкилов к карбонильным и сопряженным оле-
финовым карбонильным соединениям. Эти реакции приводят к 1,2-присоеди-
нению к карбонильным группам и к 1,2- и 1,4-присоединениям к сопря-
женным олефиновым карбонильным системам
R М
С-0
М2
Присоединение и обратные реакции
R М R М
\ \ I I I х I ! I I
)С = С—C = O + RM -+ >С = С—С —О и )С-С=С-0
/ I I / /
Здесь нет никаких сомнений в том, что такие реакции являются нуклеофиль-
ным присоединением: металлоорганическое соединение отдает свою алкиль-
ную группу в виде карбаниона (предварительно образовавшегося или нет —
это уже другой вопрос) электрофильному центру ненасыщенного соединения,
т. е. либо карбонильному атому углерода, либо p-углеродному атому сопря-
женной олефин-карбонильной группы.
В углеводородных и эфирных растворах, в которых обычно проводят подоб-
ные реакции, некоторая часть карбанионов предварительно уже образуется
в виде ионной пары, находящейся в равновесии с исходным металлооргани-
ческим соединением
RM R-}M+
Ионные пары — очень активные специфичные частицы в отношении нуклео-
фильного присоединения. Но в случае если равновесие сильно сдвинуто
влево, различие концентраций может перевесить специфическую реакцион-
ную способность ионной пары и нуклеофильное присоединение будет проте-
кать за счет ковалентных молекул.
Наиболее широко применяемым алкилметаллическим реагентом для карбо-
нильных соединений является реактив Гриньяра. Важное значение имеют
также литийалкилы. Алкилы металлов I и II групп, более тяжелых, чем
литий и магний, имеют практическое значение для специальных целей.
Сделаем одно замечание общего характера и проведем сравнение между
алкилами щелочных металлов и реактивами Гриньяра. Для данной алкильной
группы ряд активности будет выглядеть следующим образом:
M=MgX<Li<Na<K
Этот ряд основан не на систематических измерениях при одинаковых усло-
виях, а составлен на основе не связанных между собой качественных срав-
нений. Например, на основании того факта, что шреш-бутиллитий не при-
соединяется к гексаметилацетону, в то время как шреш-бутилнатрий образует
(после протолиза аддукта) три-шреш-бутилкарбинол, приходят к заключе-
нию, что Li < Na. Приведенный ряд соответствует теории, поскольку он
отражает ту степень, в которой при данной алкильной группе в данном
растворителе положение равновесия молекула ч=ь ионная пара будет сдвинуто
в сторону очень реакционноспособной ионной пары. Вероятно, в этом же ряду
возрастает и легкость разделения противоионов ионной пары, если для
такого разделения будут созданы подходящие условия.
Очевидно, для данного металла и данного растворителя положение равнове-
сия молекула ионная пара будет зависеть от природы углеводородной
группы R в RM. В качестве примера рассмотрим частный случай одного
металла и одного растворителя: если R = фенил или небольшая алкильная
группа, то равновесие будет сильно сдвинуто в сторону молекулы, а если
в качестве R взять 9-флуоренил или трифенилметил, анионы которых гораздо
более устойчивы, чем анионы незамещенных арил- или алкилгрупп, то равно-
весие должно сдвинуться в сторону образования ионной пары. Большие раз-
меры и делокализация заряда способствуют увеличению устойчивости аниона,
что также ослабляют связь в ионной паре, поэтому в этих случаях даже мож-
но считать, что в присоединении участвуют разделенные ионы.
Физические исследования равновесия молекула ионная пара металлалки-
лов еще не достигли большого прогресса, и поэтому все утверждения отно-
843
Глава XIII
сительно общих закономерностей процесса нуждаются в специальных под-
тверждениях. Однако можно попытаться объяснить главные различия, на
которые указывают многие исследователи, между алкилами щелочных
металлов и реактивами Гриньяра. Имеются литературные данные о выходе
продуктов (табл. 178) 1,2- и 1,4-присоединения для каждого вида реагентов,
если оба типа присоединения возможны при взаимодействии с сопряженными
олефиновыми соединениями. Такое сравнение проведено для обоих видов
реагентов в табл. 178.
Таблица 178
1,2- и 1,4-Присоединение реактивов Гриньяра и органических соединений
Li и Na к а, р-ненасыщенным кетонам
Адденд а, ^-Ненасыщенный кетон Раствори- тель Тип при- соеди- нения Вы- ход а, % Лите- рату- ра Вид реагирую- щего металлоор- ганического соединения
C6H5MgBr Халкон в (С2н5)2о 1,4 94 188 Молекула
u3O-C3H7MgBr Циклогек- сен-2-он-1 (С2н5)2о 1,4 65 189 190 } ’
Пентен-З-он-2 (С2Н5)2О 1,4 66 190 »
Гексен-З-он-2 (C2Hs)2O 1,4 57 190 »
9-Флуоренилмагний- Окись мезитила Ксилол 1,2 30 191 Ионная пара
бромид Трифенилметилмаг- Коричный аль- Бензол 1,2 ? 192 То же
нийхлорид дегид
C6HsLi Халкон (С2Н5)2О (С2Н5)2О 1,2 1,4 69 13 J 188 В основном ионная пара
CeH5Na Халкон (C2H5)2O 1,2 1,4 60 14 } 188 То же
9-Флуорениллитий Окись мезитила Лигроин 1,4 32 191 Отдельные
ионы
Бензальацетон » 1,4 17 191 То же
Халкон » 1,4 ? 191 » »
9-Флуоренилнатрий Халкон С2н5он 1,4 27 192 » »
Трифенилметилнат- Коричный аль- ? 1,4 ? 187 » »
рий дегид
а Препаративный выход продукта, т. е. нижний предел выхода. Если второй продукт не упоми-
нается, его присутствие не было установлено.
б Предположительно (см. текст).
в Халкон = у-бензилиденацетофенон (старое короткое название настолько удобно, что вряд ли
стоит употреблять название по принятой номенклатуре).
Реактивы Гриньяра с незамещенными фенильными или алкильными группа-
ми дают при взаимодействии с аф-ненасыщенными кетонами главным обра-
зом 1,4-аддукты. Одно время считалось, что это можно объяснить тем,
что реактив Гриньяра соединяет атомы 1 и 4 сопряженной системы в пере-
ходном состоянии с помощью шестичленного цикла. Для этого сопряженная
двойная связь исходного кетона должна иметь в переходном состоянии
^ис-конфигурацию, что должно исключать 1,4-присоединение к циклогексен-
544
Присоединение и обратные реакции
2-ону-1, в котором двойная связь имеет шранс-конфигурацию. На самом
же деле 1,4-присоединение в этом случае происходит так же легко, как и к от-
крытым цепям пентен-З-она-2 и гексен-З-она-2, в которых двойная связь
может принять ^нс-конфигурацию, если это потребуется. Все это заставляет
предположить (табл. 178), что реактивы Гриньяра реагируют в виде молекул,
которые развивают большой положительный заряд в 0-положении кетона,
затем отщепляют свой карбанион, тем самым освобождая MgBr+-KaTHOH,
который координируется с кислородным атомом карбонильной группы.
Реактивы Гриньяра, содержащие флуоренильную и трифенилметильную груп-
пы, могут также присоединяться в виде ионной пары через промежуточное
образование катиона только к карбонильному атому кислорода, если силы,
действующие в ионной паре, будут препятствовать присоединению карбанио-
на к карбонильному атому углерода.
Вернемся к ряду алкилов щелочных металлов. В табл. 178 предполагается,
что фениллитий и фенилнатрий реагируют в виде ионной пары и таким образом
их катионы координируются с атомом кислорода карбонильного соединения.
Мы должны предположить, что любой частичный положительный заряд
в молекуле субстрата может лишь с трудом разделять ионы ионной пары
и поэтому нуклеофильная атака вынуждена осуществляться по месту рас-
положения карбонильного углеродного атома. Для флуоренильного и три-
фенилметильного аддендов лития и натрия можно допустить, что они атакуют
субстрат либо в виде ионной пары, либо в виде диссоциированных ионов
(в зависимости от использованного растворителя), но в углеводородном рас-
творителе ионы ионной пары будут разделяться на зарядах субстрата, т. е.
во всех случаях будет происходить нуклеофильное присоединение отдельных
анионов; в случае большого положительного заряда на субстрате присоеди-
нение пойдет в положение 4. Развивать такие рассуждения на основе пред-
положений, конечно, трудно, пока нет определенных электрохимических
данных по этому вопросу.
Относительно присоединения реактивов Гриньяра к карбонильным соедине-
ниям можно сказать больше. Как обсуждалось в гл. VI, разд. 8,д, не вы-
зывает сомнения, что простые алифатические и ароматические соединения
Гриньяра имеют тетраэдрическую мономерную координационную структуру
типа [MgnRX[O(C2H5)2]2]0, прочно координированную с эфирным растворите-
лем. Хлориды проявляют заметную тенденцию к димеризации, может быть
за счет установления галогенной мостиковой связи, но в бромидах и иодидах
эта тенденция сравнительно слаба. Все гриньяровские галогениды участвуют
в подвижном равновесии (называемом равновесием Шленка) [194], которое
записывается в следующем виде:
2RMgX R2Mg+MgX8
Отсюда возникает проблема: реакция Гриньяра может идти либо через RMgX,
либо через R2Mg, даже если равновесное содержание последнего очень мало.
Для диалкильных соединений любых металлов II группы отщепление кар-
баниона протекает намного легче, чем для моноалкилгалогенметаллов (гл. VI,
разд. 8).
Имеется и другая проблема, вероятно, кажущаяся более сложной, чем есть
на самом деле. С помощью спектроскопии было показано [195], что замещен-
ные бензофеноны и метилмагнийбромид в этиловом эфире частично ассоции-
рованы в комплекс и что при обычном течении процесса одновременно рас-
ходуются незакомплексованный и закомплексованный кетоны; при этом
скорость реакции подчиняется первому порядку, когда реактив Гриньяра
845
Глава XIII
имеется в большом избытке *. Очевидно, кетон частично замещает одну моле-
кулу эфира во временно установившемся координационном предравновесии.
Это не должно вносить никаких существенных изменений в присоединение
реактива Гриньяра. Наблюдаемая константа скорости будет средней между
константами скоростей реакций присоединения координированного с эфиром
и координированного с эфиром и кетоном реагента, но скорости обеих этих
реакций по своей величине очень близки друг к другу и, следовательно,
к средней скорости. Установлено, что константа скорости первого порядка
увеличивается с ростом концентрации реактива Гриньяра, присутствующего
в избытке, линейно — до концентрации 0,1 моль/л, а затем менее круто. Это
свидетельствует о том, что в разбавленном растворе кинетика суммарного
процесса подчиняется второму порядку. Тенденция кинетического порядка
реакции уменьшаться в более концентрированных растворах отражает,
по-видимому, тенденцию бромидов Гриньяра димеризоваться.
Кинетические исследования присоединения реактивов Гриньяра проводи-
лись разными исследователями. Некоторые из этих данных противоречат
ДРУГ другу, но эти противоречия здесь рассматриваться не будут. Антеунис
[197] установил, что взаимодействие метилмагнийбромида и метилмагний-
иодида с бензофеноном в смеси эфир — бензол и с пинаколином в эфирном
растворе имеет третий порядок
Скорость ос [Кетон][Стехиом. RMgX]2
Если же реакцию метилмагнийбромида с пинаколином в растворе эфира
проводить с самого начала в избытке бромида магния, то порядок реакции
меняется па второй
Скорость ОС [Кетон][Стехиом. RMgX]
Бикалс и Беккер исследовали присоединение метилмагнийбромида к бензо-
фенону в растворе тетрагидрофурана [198] и получили результаты, подчи-
няющиеся уравнению второго порядка. Правда, этот результат был получен
только для малых глубин реакции, а на больших ее глубинах скорость резко
падала. В приведенных выше уравнениях член [Стехиом. RMgX] означает
стехиометрическую концентрацию реактива Гриньяра, которая вычислена
без учета равновесия Шленка.
Равновесие Шленка охватывает три кинетических случая, между которыми
возможны следующие переходные ситуации: (а) реактив Гриньяра сохра-
няется в основном в виде алкилмагнийгалогенида, но реагирует в минималь-
ной равновесной концентрации диалкилмагний; (б) реактив Гриньяра нахо-
дится почти полностью в виде алкилмагнийгалогенида и в этой же форме
вступает в реакцию; (в) реактив Гриньяра находится в основном в виде диал-
килмагния и в этой же форме вступает в реакцию. Очевидно, не может быть
случая, когда реактив Гриньяра находится в виде диалкилмагния, а реагиру-
ет в виде алкилмагнийгалогенида, так как диалкилмагний обладает наи-
большей реакционной способностью. В случае (а) и (б) реакция записывается
следующим образом:
R;C--O + R2Mg RjCR —OMgR
а в случае (в)
RjC —ОД-RMgX —> R2CR — OMgX
* Кинетическая характеристика присоединения избытка метилмагнийбромида к аце-
тону в этиловом эфире, идущего через стадию предравновесного образования комплекса
реактива Гриньяра и ацетона, была получена Эшби, Дюком и Нейманом [196].
846
Присоединение и обратные реакции
Если имеет место случай (а) и если концентрация галогенида магния в рав-
новесии Шленка по каким-либо причинам поддерживается постоянной, на-
пример за счет насыщения раствора этой солью, как в примере Антеуниса,
когда эфирно-бензольный раствор можно было насытить бромидом магния
или за счет того, что после начального периода концентрация бромистого
магния настолько возрастает, что ее относительное изменение становится
небольшим, то [R2Mg] a [RMgXP № [Стехиом. RMgX] и должна наблюдать-
ся реакция третьего порядка, что фактически и было в опытах Антеуниса.
Допустим, однако, что в раствор эфира, в котором бромистый магний легко
растворим, его добавлено столько, что образование диметилмагния подавлено
и мы имеем случай (б). Тогда форма, в которой реактив Гриньяра сохраняет-
ся в растворе, и форма, в которой он реагирует, будут идентичными и реак-
ция будет протекать по второму порядку, который Антеунис и наблюдал
в таких условиях.
Предположим теперь, что реакцию проводят в тетрагидрофуране, в котором
бромистый магний растворяется настолько мало, что равновесие Шленка
смещается в сторону диалкилмагния. Тогда будет иметь место случай (в),
в котором основная и реагирующая формы реактива Гриньяра будут иден-
тичными, т. е. реакция будет иметь второй порядок, что фактически и наблю-
далось в работе Бикалса и Беккера. Но здесь появляется различие, состоя-
щее в том, что одна из двух алкильных групп будет содержаться в остатке —
OMgR аддукта до тех пор, пока не присоединится другая молекула кетона
и не отщепит ее. Бикалс и Беккер установили, что подчинение реакции закону
второго порядка прекращается, как только в реакцию вступит около 40%
кетона, а затем реакция продолжается с гораздо меньшей скоростью. К мо-
менту вступления в реакцию примерно 60% кетона скорость реакции будет
составлять всего 1% от ее первоначальной величины. Такое низкое значение
остаточной скорости объясняется присоединением RMgOGCRR^ как реактива
Гриньяра к RJGO [187].
г. Гетеролитическое присоединение водорода к карбонильным и сопря-
женным олефиновым карбонильным соединениям. Любое соединение, способ-
ное быть переносчиком гидрид-иона Н~ к положительно поляризованному
углероду, каким является углерод карбонильной группы, будет выступать
как нуклеофильный восстанавливающий агент. Сюда можно сразу при-
числить гидриды металлов, такие, как алюмогидрид лития, и металлалкилы,
содержащие p-водород, например соответствующие реактивы Гриньяра, при-
менение которых привело к тому, что ряд исследователей напрасно тратили
труднодоступные кетоны, восстанавливая их этими реактивами
М-Ф с=о->м+ + нс—о-
М—CE2-CR2—И С=О М+ + CRirCRi + НС—О-
Щелочные растворы альдегидов представляют собой обширный класс вос-
станавливающих агентов. Они будут восстанавливать карбонильные группы
и, в частности, сам альдегид
ОН R ОН R
I I 1 I
RCHO + OH-?±R—С-ЧГ С=О—>R—С +НС-0~
Г1 I II I
^о- н он
847'
Глава XIII
Следующая реакция носит имя Канниццаро:
он-
2RCHO-----> RCH20H + RC00-
Онабыла открыта в 1853 г. Канниццаро [199] на примере бензальдегида.
Реакция протекает в случае альдегидов, не вступающих в альдольную кон-
денсацию и не подвергающихся под влиянием щелочи иным превращениям.
Типичными примерами таких альдегидов могут служить формальдегид,
триметилацетальдегид, бензальдегид и его замещенные, фурфурол.
Механизм реакции в наиболее простой форме может быть рассмотрен при
осуществлении реакции в гомогенной среде в гидроксилсодержащих раство-
рителях в отсутствие поверхностных катализаторов. Растворителем служит
главным образом вода (если альдегид растворим в ней), метиловый спирт
или водный диоксан. В подобных условиях и исследовалась кинетика реакции
Канниццаро на примерах бензальдегида и некоторых его замещенных, фурфу-
рола и формальдегида. Оказалось, что в зависимости от концентрации щелочи
скорость реакции подчиняется двум различным кинетическим законам.
Реакция может протекать по закону третьего порядка
Скорость ос [Альдегид]2[ОН~]
или иметь четвертый порядок
Скорость ОС [Альдегид]2[ОН-]2
В случае бензальдегида в метиловом спирте, в водном метиловом спирте или
диоксане при обычных концентрациях щелочи реакция протекает согласно
уравнению третьего порядка [200]. Реакция фурфурола в воде или в водном
метиловом спирте протекает в соответствии с уравнением четвертого порядка
[201], но в водном диоксане кинетика реакции характеризуется смешанным
порядком, причем при определенных условиях можно приблизиться к пре-
дельной форме реакции третьего порядка [202]. В случае формальдегида
реакция может протекать согласно предельному закону четвертого порядка
[203]; возможно снижение порядка реакции вплоть до третьего [204]. Ско-
рость внутримолекулярной реакции Канниццаро, в которой фенилглиоксаль
под влиянием раствора щелочи в водном метиловом спирте превращается в мин-
дальную кислоту, пропорциональна произведению [Фенилглиоксаль] [ОН-],
что соответствует реакции третьего порядка для обычной межмолекулярной
реакции Канниццаро [205]. При изучении кинетики некоторых реакций
было показано, что перекиси и антиоксиданты не оказывают влияния на ско-
рость.
Влияние заместителей в бензальдегиде на скорость реакции Канниццаро
качественно сказывается следующим образом: электроположительные груп-
пы +7- или 4-М-типа замедляют реакцию, в то время как электроотрица-
тельные группы —I- или —М-типа ускоряют ее, что видно из ряда [200,
206]
n-(CH3)2N С п-СН3О < п-алкил < Н < п-С1 < л«-С1 < jt-NO2
Фреденхаген и Бонхоффер [207] показали, что при реакции Канниццаро
в случае формальдегида и бензальдегида в тяжелой воде продукты реакции
не содержат связи углерод — дейтерий. Это справедливо и для внутримоле-
кулярной реакции Канниццаро — при превращении глиоксаля в водном
щелочном растворе в гликолевую кислоту. Проведенные опыты показы-
вают, что переход водорода из одной молекулы в другую или из одной альде-
гидной группы в другую осуществляется непосредственно, т. е. без обмена со
средой.
848
Присоединение и обратные реакции
На основании этих данных Гаммет [208] в 1940 г. предложил механизм, кото-
рый в настоящее время является общепризнанным для некаталитической
гомогенной реакции в водных растворах. Предполагается, что под влиянием
гидроксильных ионов альдегид превращается в два аниона, обладающих
восстановительной способностью, причем первый из них образуется легче
второго. Первый анион, образующийся в результате простого присоедине-
ния гидроксильного иона к альдегиду, обладает умеренной склонностью
к отдаче гидридных ионов. Второй — с более сильной восстановительной
способностью образуется из первого аниона путем перехода протона к како-
му-либо другому гидроксильному иону. Восстановительная способность
этих гипотетических промежуточных продуктов может быть выражена сле-
дующим образом:
R-CU-Й R-сХЙ
I /Н
он Со-
Предполагается, что эти ионы образуются при быстро устанавливающемся
равновесии между молекулой альдегида и гидроксильными ионами; поэтому
концентрации одно- и двухзаряженных ионов пропорциональны соответ-
ственно [Альдегид][ОН~] и [Альдегид][ОН~]2. Считается также, что отщепле-
ние гидрид-иона от каждого из этих анионов не может происходить путем
мономолекулярной реакции; перенос гидрид-ионов возможен лишь бимоле-
кулярным путем к соответствующему акцептору, особенно к карбонильному
углеродному атому другой молекулы альдегида. Наблюдаемым скоростям
реакции соответствуют следующие уравнения:
/О" Реакция 3 порядка у.0
RCpiI + RCHO-------------> RQf +RCH20-
\он Х)Н
/О- Реакция 4 порядка
RC^H + RCHO--------------> RCf +RCH2O-
\о- \о-
Внутримолекулярная реакция Канниццаро, например превращение фенил-
глиоксаля в присутствии щелочи в миндальную кислоту, протекает как
нуклеофильная перегруппировка насыщенных соединений того же типа,
что и обсуждаемые в гл. X, в частности перегруппировка бензила в бензи-
ловую кислоту, отличающаяся от рассматриваемой здесь лишь наличием
фенила вместо водорода в качестве перемещающейся группы.
Н Н
п <1 л - I
О=С—СА-О- -> О-С-С=О
II II
н5с6 он н5с6 он
На первый взгляд восстановление металлами, растворяющимися в протонной
среде, кажется процессом, совершенно отличным от уже рассмотренных
реакций переноса гидрид-иона, однако это различие лишь кажущееся.
Сразу же приходит мысль, что восстановление растворяющимися металлами
аналогично реакциям гидридного переноса является нуклеофильным при-
соединением; на это указывает тип субстратов, восстанавливающихся рас-
творяющимися металлами — карбонильные и сопряженные олефиновые кар-
бонильные соединения и аналогичные циан- и нитросоединения.
54-0772
849
Глава XIII
Мысль о том, что водород, образующийся на поверхности растворяющихся
металлов, является особой химически активной формой водорода, была
высказана весьма давно. В первые три десятилетия текущего столетия были
широко распространены представления о том, что «выделяющийся» таким
образом водород представляет собой водород in statu nascendi. В то время
не были известны ни способы проверки гомолитических реакций, ни способы
проверки и классификации гетеролитических процессов в зависимости от
природы субстрата, с которым реагент мог бы легко взаимодействовать
(гл. V). В 1929 г. Бартон и автор книги [209] высказали предположение, что
водород, выделяющийся при растворении металлов, состоит не из предвари-
тельно образовавшихся атомов водорода, а из электронов и протонов, источ-
ники которых различны.
Металл рассматривался как модель электронного газа, ограниченного потен-
циальными стенками. Предполагается, что электроны могут проникнуть через
достаточные бреши в стенках, создающиеся вследствие адсорбции электро-
фильного катиона или молекулы. Электроны могут поэтому направляться
к адсорбированному иону водорода, из которого при этом образуется атомар-
ный и затем молекулярный водород. С другой стороны, электроны могут
направляться к иону водорода через адсорбированную ненасыщенную и пото-
му обладающую электропроводностью органическую молекулу, которая
при этом восстанавливается. Эта молекула должна быть электрофильной;
типичной является необходимость в такой —/Г-группе, как карбонил, карбок-
сил, циан-, нитрозо- или нитрогруппа, которая в состоянии создать на
металле требуемый для адсорбции положительно поляризованный электро-
фильный центр, обеспечивающий возможность утечки электронов проводи-
мости.
Необходимая степень электрофильной реакционной способности в боль-
шой мере зависит от природы металла. Так, для металлов типа натрия
при их растворении в спирте даже арильная группа, находящаяся на конце
ненасыщенной системы, например в стироле или в стильбене, достаточно
поляризована и способствует эффективной адсорбции олефиновой молекулы.
Известно, что эти и подобные им ненасыщенные ароматические углеводороды
могут восстанавливаться растворяющимся натрием [210]. Для металлов
типа магния арильная группа уже недостаточно электрофильна, но карбо-
нильная группа достаточно электрофильна, на что указывает восстановление
ацетона магнием до магниевой соли пинакона [211]. В случае металлов типа
амальгамированного цинка или олова необходимым электронным сродством
не обладает даже карбонильная группа, если процесс не проводится в при-
сутствии сильных кислот. Вполне вероятно, что сопряженная кислота кар-
бонильного соединения является сильной электропоакцепторной группой,
как в примере восстановления альдегида или кетона по методу Клемменсена
[212].
Независимо от того, когда первый протон вступает в восстанавливаемую
систему — до, одновременно или после перехода электронов из металлов,
наличие протона обычно необходимо для удержания переместившихся элек-
тронов в процессе десорбции. Это может произойти после захвата только
одного электрона, например при образовании пинакона. Чаще всего это
происходит, когда присоединяются два электрона, заполняются электронные
октеты и органическая система приобретает отрицательный заряд. В подобной
реакции, протекающей на поверхности, происходит присоединение компо-
нентов гидрид-иона Н“, а именно Н+ +2е; реакция может рассматриваться как
гетеролитическая или, более точно, как нуклеофильное присоединение AdN.
После десорбции восстановление завершается гомогенным присоединением
850
Присоединение и обратные реакции
второго протона Н+ из среды
СЖ2=СГ Н+
CHR2-OH
На основании рассмотренной картины восстановительного процесса были
сделаны выводы о причинах ориентации при восстановлении сопряженных
ненасыщенных систем. Восстановление растворяющимися металлами рас-
сматривается как нуклеофильное присоединение. Первоначальное присоеди-
нение Н_ соответствует присоединению CN- при образовании циангидрина
и присоединению СН(СООС2Н5)2 в реакции Михаэля. Присоединение Н +
в конце процесса носит тот же характер, что и заключительная стадия при
других нуклеофильных присоединениях. Однако между восстановлением
и другими процессами присоединения имеется существенное различие: после
того как гидрид-ион присоединяется к восстанавливаемой системе, он остает-
ся в ней. Более того, ни гидрид-ион, ни присоединяющийся в конце про-
цесса протон обычно не могут вновь ионизоваться или переместиться в усло-
виях восстановления. Например, при восстановлении амальгамой натрия
муконовой кислоты в воде или 1,4-дифенилбутадиена-1,3 в спиртовой среде
в процессе реакции не создается основной среды или достаточно высокой
температуры, что могло бы привести к прототропному равновесию продуктов
восстановления. Следовательно, ориентация при восстановлении сопряжен-
ных систем растворяющимися металлами обычно зависит от кинетических,
а не от термодинамических факторов. В этом состоит отличие восстановле-
ния от образования циангидринов и от реакции Михаэля [213].
Значение этого вывода становится особенно ясным, если вспомнить историю
восстановления ненасыщенных сопряженных систем. Впервые вопрос был
поставлен Байером в связи с исследованием строения бензольного ядра
(гл. IV, разд. 1,а). При изучении восстановления терефталевой кислоты
амальгамой натрия Байер [214] показал, что первоначально образуется 2,5-
диеновая кислота с присоединением двух атомов водорода к двум углеродным
атомам, связанным с карбоксильными группами. Однако это дигидросоедине-
ние не является наиболее термодинамически устойчивым, так как при обра-
ботке щелочью оно претерпевает изомеризацию, по-видимому, протекающую
в две стадии:
СООН СООН СООН СООН
I I ! I
СООН СООН СООН СООН
Позднее Байер и Рупе [215] исследовали муконовую кислоту — простейшее
производное бутадиена с расположенными по концам цепи двумя карбок-
сильными группами. При восстановлении амальгамой натрия образуется
Р,у-ненасыщенное дигидросоединение, в котором водородные атомы также
присоединены к атомам углерода, связанным с карбоксильными группами.
Это дигидросоединение также неустойчиво, поскольку при действии щелочи
оно превращается в а,|3-ненасыщенный изомер
СН = СНСН=СН Na—Hg СН2СН = СНСН2 он- СН = СНСН2СН2
I I----------> I I -------> I I
СООН СООН СООН СООН СООН СООН
54* 851
Глава XIII
Немного позднее было показано, что цинпамилиденуксусная кислота [216],
а также циннамилиденмалоновая кислота [217] при восстановлении амаль-
гамой натрия в почти нейтральном растворе превращаются в |3,у-ненасыщен-
пые дигидрокислоты, причем атомы водорода присоединяются к углеродным
атомам, связанным с карбоксильной и фенильной группами. Эти продукты
термодинамически неустойчивы и при действии щелочи превращаются в cc,fJ-
ненасыщенные изомеры
Na—Hg ОН-
С6Н5СН = СНСН = СНСООН -----> СвН5СН2СН=СНСН2СООН------->
—> С6Н5СН2СН2СН = СНСООН
Na—Hg ОН-
С6Н5СН = СНСН = С(СООН)2 ---> С6Н5СН2СН= СНСН(СООН)2 ----->
—> С6Н5СН2СН2СН = С(СООН)2
Известно [218], что нафталин восстанавливается натрием в кипящем этиловом
спирте с образованием 1,4-дигидропроизводного. Штраус [219] показал,
что 1,4-дифенилбутадиен при восстановлении амальгамой натрия в спирте
превращается в 2,3-ненасыщенное дигидросоединение, в котором водородные
атомы присоединены к атомам углерода, связанным с фенильными группами
С6Н5СН = СНСН = СНСвН5 СвН5СН2СН = СНСН2СвН5
Несмотря на то что этот продукт реакции не изомеризуется, можно не сом-
неваться в том, что он термодинамически неустойчив, так как может быть
полностью превращен действием сильных оснований при высокой темпера-
туре в его 1,2-ненасыщенный изомер. Наконец, Кун и Винтерштейн [220]
показали, что 1,6-дифенилгексатриен, 1,8-дифенилоктатетраен и 1,10-дифенил-
пентадекаен при восстановлении амальгамой натрия в спирте превращаются
соответственно в их 1,6-, 1,8- и 1,10-дигидропроизводные; в каждом случае
атомы водорода присоединяются к атомам углерода, связанным с фенильными
группами.
Все эти примеры показывают, что ориентация восстановления определяется
кинетикой процесса. Полиеновая цепь по причинам, уже рассмотренным
в случае других процессов восстановления, положительно поляризована
по одному из ее концов и поэтому способна адсорбироваться растворяющимся
металлом одним или другим концом. В случае соединения типа циннамили-
денуксусной кислоты адсорбция осуществляется у концевого 6-углерода.
Первый протон, захватывающийся полиеновой цепью, будет при этом при-
соединяться к другому (неадсорбированному) концу; второй протон при-
соединится к первому концу после освобождения этого конца в результате
десорбции. Такое связывание протона за счет координации может происходить
значительно быстрее, чем в каких-либо иных условиях. Это объясняется
тем, что, как в уже рассмотренных случаях прототропии и некоторых других
процессах сопряженного присоединения, переходные состояния при коор-
динации (и гетеролизе) у атома углерода, связанного с сильно электроноак-
цепторной группой, стабилизуются за счет электронного резонанса с этой
группой. Этим обусловлено более быстрое протекание реакции в данном
положении по сравнению с другими.
Влияние метильных групп на направленность процесса восстановления
при помощи амальгам даже качественно не похоже на влияние фенильных
групп. Это может быть проиллюстрировано некоторыми данными [209], при-
веденными в табл. 179. Они показывают относительные количества а,|3-
и а,6-дигидропроизводных, образующихся при восстановлении р-винил-
акриловой кислоты СН2 = СНСН = СНСООН и некоторых ее метилирован-
ных гомологов амальгамой натрия в водном растворе углекислого натрия
852
Присоединение и обратные реакции
Таблица 179
Влияние метильных заместителей в 0-винилакриловой кислоте
или ее анионе на выходы (%) а, дигидропроизводного,
образующегося при восстановлении амальгамой натрия в водной среде
/ 6 у 0 а \
ВАК — винилакриловая кислота — С = С — С — С - СООН
\ 1111/
Среда ВАК б-Метил-ВАК а, б-Диметил-ВАК р, 6, б-Триметил-ВАК
Щелочная Кислая 0 18 40 55 28 38
или в водной уксусной кислоте. Первый атом водорода во всех случаях при-
соединяется в a-положение, а второй — в 0- или 6-положение. Таким образом,
можно видеть, что при переходе от винилакриловой к сорбиновой кислоте,
в которой имеется 6-метильная группа, происходит далеко не преимуществен-
ное а,6-присоединение, наблюдаемое при наличии 6-фенильной группы,
на самом деле второй атом водорода присоединяется не только в 6-, но частич-
но и в 0-положение. При введении дополнительного 0-метильного замести-
теля место присоединения второго атома водорода частично переходит опять
к 6-положению, в то время как введение еще одной 6-метильной группы
опять вызывает присоединение в 0-положение.
Необходимо теоретически рассмотреть с точки зрения кинетики реакционную
способность десорбированного аниона. Он может реагировать либо в форме,
в которой его заряд сосредоточен в 6-положении, а двойная связь находится
в 0,у-положении, либо в другой возможной форме с иным распределением
заряда и двойной связи
г- 6-
сДс\-О>-сн2-х
Следует ожидать качественного различия между влиянием фенильной и ме-
тильной групп, расположенных, например, в 6-положении. Так, гиперконъю-
гация метильной группы является однонаправленной; эта группа не может
за счет гиперконъюгации стабилизовать отрицательный заряд на соседнем
атоме, как это осуществляется благодаря сопряжению фенильной группы,
которая, следовательно, может стабилизовать переходные состояния реакций,
в которых создается или исчезает такой заряд. Единственное, в чем может
проявиться влияние 6-метильной группы благодаря гиперконъюгации ее
с двойной связью, заключается в стремлении к локализации двойной связи
в у,6-положении, а отрицательного заряда — в 0-положении. Этим объяс-
няется, почему в случае 6-метильной группы в отличие от 6-фенильной преоб-
ладает присоединение второго атома водорода в 0-положение. Подобные
рассуждения применимы и в отношении 0-метильной группы. Восстановление
в щелочной среде приводит в большей степени к «^-присоединению, чем
в кислой среде. Это различие объясняется тем, что электростатическое влия-
ние карбоксилат-иопа, сказывающееся в щелочной среде более сильно, чем
в кислой, проявляется в стремлении удалить отрицательный заряд на угле-
родном атоме в десорбированном мезомерном карбанионе в более отдаленное
из двух возможных положений.
8-53
Глава XIII
д. Катализируемая щелочами полимеризация олефинов путем самопри-
соединения [221]. Полимеризация олефинов под действием сильных щелочей
была установлена в ряде разрозненных работ, появившихся в начале этого
столетия [222]. Так, например, Пехман полимеризовал этиловый эфир кро-
тоновой кислоты действием этилата натрия, Брайлентс полимеризовал
аллилцианид действием этилата натрия или реактива Гриньяра, Шмидт и Рейтц
полимеризовали нитроолефины действием водного раствора едкого натра
[223]. Причины протекания этих реакций оставались неизвестными до тех
пор, пока в 1920 г. Штаудингер [104] не объяснил цепной характер полиме-
ризации.
Химическую природу инициирования и развития цепных реакций невозмож-
но было оценить до тех пор, пока общая теория гетеролитических реакций
в органической химии, начавшая быстро развиваться в 1926 г., не достигла
определенного прогресса. Фактически только после 1940 г. стало понятным,
что роль сильного основания состоит в нуклеофильном присоединении к моле-
куле мономера, в результате чего образуется карбанион; мономер благодаря
этому становится нуклеофилом и присоединяется ко второй молекуле моно-
мера и т. д.
Полимеризация олефинов под действием щелочных металлов стала известна
в 1910 г. [224]. Важное исследование Гарриса по полимеризации бутадиена
и изопрена в присутствии натрия [225] стало в свое время основой крупно-
тоннажного производства синтетического каучука в Германии. Примерно
в то же время Шленк описал полимеризацию стирола и 1-фенилбутадиена
в присутствии тонкосуспендированного натрия в эфире [226].
Природа реакций полимеризации, инициируемых щелочными металлами,
и аналогичных им по механизму развития цепи реакций полимеризации
при инициировании основаниями и, в частности, металлоорганическими
соединениями была обстоятельно выяснена Циглером [227] в 1930-х годах.
В то время существовало предвзятое мнение, которое разделял, кстати,
и Шленк, что полимеризация, инициированная щелочными металлами, которые
имеют нечетное число электронов, является радикальным процессом раз-
вития цепи. Циглер привел доказательства, что исходная молекула, из кото-
рой начинается рост полимера, содержит два атома щелочного металла.
Такая молекула, следовательно, содержит четное число электронов. Рас-
суждения Циглера можно проиллюстрировать следующей схемой:
2Na + СН2 = СНСН = СН2'-> NaCH2CH = CHCH2Na
2Na+CH2=CHCeH5 NaCH2-CH(C6H5)Na
Молекулы с двумя атомами натрия имеют две точки роста, и развитие цепи
происходит путем присоединения новых молекул мономера к обеим точкам.
При этом предполагалось, что металл-углеродная связь рвется, как только
присоединится новая молекула мономера, и на конце выросшей цепи обра-
зуется новая связь металла с углеродным атомом. Отличие современного
взгляда на механизм такой полимеризации от только что изложенного состоит
в следующем: во-первых, предполагается, что динатриевое соединение обра-
зуется путем димеризации первоначально образовавшихся радикал-анионов,
содержащих, кроме того, в два раза больше углеродных и водородных атомов,
чем в приведенной выше схеме, т. е.
CH2 = CHCH = CH2-L-Na СН2СН = СНСН2 }Na+
2CH2CH = CHCH2}Na+ —> Na+{-CH2CH = CH(CH2)2CH = CHCH2}Na+
854
Присоединение и обратные реакции
Во-вторых, в настоящее время считают, что натриевое соединение присоеди-
няется, подобно любому алкилметаллу, в виде карбаниона, причем проти-
воион натрия движется вместе с отодвигающимся от начала цепи по мере
развития полимеризации отрицательным центром, например:
СН2=СНСвН5 С1[2-СПС,;И5
R-}Na+----------> RCH2CHCGHr}Na+ ---------->
сн2=снс6н5
—> RCH2CH(C6H3)CH2CHCeHj}Na+ --------—>
C1T2-CI[C6II5
—> RCII2CH(CeH5)CH2CH(CeH5)CII2CHCeHr}Na+ -------> и т. д.
Циглер считал, что инициирование металлом является просто удвоенной
формой инициирования металлалкилом.
То, что инициируемая щелочными металлами полимеризация олефинов пред-
ставляет собой частный случай инициирования основаниями, в особенности
такими сильнонуклеофильными анионами, как углерод-, азот- или кислород-
апиопы, впервые стало ясно в 1948 г., когда Бимен [228] исследовал полиме-
ризацию метакрилонитрила при инициировании реактивом Гриньяра, три-
фенилметилнатрием в эфире и натрием в жидком аммиаке. Сандерсон и Хаузер
[229] дополнили этот ряд исследованием полимеризации стирола в присутст-
вии амида натрия в жидком аммиаке. В последнем случае было дано такое
же объяснение: инициирование реакции происходит за счет присоединения
сильнощелочного амид-иона. Показано также, что реакция с амидом натрия
в значительно более слабых по своей ионизующей способности растворителях,
чем жидкий аммиак, не идет.
В 1950 г. сразу в двух лабораториях были проведены исследования по сопо-
лимеризации, в результате которых было очень просто показано различие
между инициируемыми кислотами, щелочами и радикалами реакциями
присоединения — полимеризации олефиновых соединений. Уоллинг, Майо
и сотр. [230] и Лендлер [231] осуществили полимеризацию смеси стирола
н метилметакрилата (в молярном отношении 1 : 1). Выбор именно этих
мономеров объясняется тем, что один из них представляет собой олефиновый
углеводород, весьма чувствительный к электрофильным реагентам, в то
время как другой является аф-ненасыщенным эфиром, чувствительным
к нуклеофильным реагентам. При обработке смеси мономеров трехфтористым
бором, хлоридом или бромидом оловаЦУ) (о сокатализаторе пе упоминалось,
по и ничего пе было сделано для того, чтобы исключить его) продуктом реак-
ции был почти чистый полистирол. Очевидно, реакционноспособный конец
растущей молекулы полимера (вероятно, карбониевый ион) был электрофилен,
он «выбирал» молекулы стирола и «пренебрегал» почти полностью молеку-
лами метилметакрилата. При обработке смеси мономеров натрием или калием
получался совершенно чистый полиметилметакрилат. Растущий конец поли-
мерной цепи в этом случае был нуклеофильным, вероятно, карбанионом
с большим сродством к p-углеродному атому метилметакрилата и сравнитель-
но низким сродством к стиролу. При обработке смеси мономеров перекисью
бензоила получался примерно эквимолярный сополимер. В этом случае
растущие концы полимера должны содержать радикал, легко присоединяю-
щий молекулу мономера любого типа. Результаты этих исследований приве-
дены в табл. 180.
Кинетику инициируемой основаниями полимеризации впервые изучали Абкин
и Медведев [232] в 1936—1940 гг. на примере полимеризации бутадиена в газо-
вой фазе или углеводородных растворителях при инициировании металличе-
ским натрием. Реакция оказалась гетерогенной, по крайней мере на стадии
855
Глава XIII
Таблица 180
Полимеризация эквимолярной смеси стирола и метилметакрилата а
Катализатор Растворитель Темпера- тура, °C Содержание стирола в сополимере [130]а, %
bf3-c2h5oh 30 > 99
Sncn 30 >99
SnBr4 Нитробензол 25 99
К ( Суспендированный 30 <1
Na ( в бензоле металл 30 <1
Na Жидкий аммиак -50 2
(СеН3СО)2О2 60 51
(^6^5^O)2^2 54
аЦифры, набранные курсивом, взяты из работы [131].
инициирования, но из ее качественных кинетических особенностей были
получены некоторые доказательства в пользу механизма Циглера.
В 1952 г. Хиггинсон и Вудинг [233] опубликовали результаты своего кине-
тического исследования, дополненные наблюдениями других авторов,
позволившие получить полную картину механизма промотируемой основа-
ниями полимеризации. В качестве объекта исследования была выбрана поли-
меризация стирола в присутствии амида калия в жидком аммиаке (метил-
метакрилат не был взят потому, что его полимеризация протекает слишком
быстро, чтобы можно было легко провести кинетическое исследование).
Скорость реакции выражается уравнением
Скорость ос [ККН2]1'/з[Стирол]2
а степень полимеризации уравнением
DP ос [Стирол]
Установлено также, что в каждой молекуле полимера имеется аминогруппа
и он не содержит амида калия. Измерение электропроводности амида калия
показало, что в жидком аммиаке он является слабым электролитом. Полу-
чены также значения свободной энергии и приближенные значения теплоты
его ионной диссоциации, а также температурные коэффициенты скоростей
полимеризации.
На основе этих данных можно представить механизм реакции. Он состоит
из стадий инициирования, развития и обрыва цепи. Половинный показатель
степени в уравнении скорости реакции показывает, что инициирование реак-
ции осуществляется диссоциированным ионом амида, а не молекулой амида
калия или его ионной парой. Второй сомножитель в уравнении скорости
указывает, что стадия развития цепи является бимолекулярной. Первая
степень в уравнении для степени полимеризации (DP = kplkt) свидетельст-
вует о том, что обрыв цепи является реакцией первого порядка по полимеру
и что в реакции обрыва цепи в качестве другого реагента может участвовать
только растворитель. То, что в процессе обрыва цепи участвует растворитель,
видно из стехиометрии реакции: обрыв происходит при переносе протона
856
Присоединение и обратные реакции
от молекулы растворителя к растущему аниону полимера
ki
NH2 + CH2 = CHCeH5 —> NH2CH2CHC6Hr
КН2СН2[СН2СНСвН5],.СНСвН5+СН2 = СНСвН5 -4
—> NH2CH2[CH2CHCeH5]r+1CHC6Hr
NH2CH2[CH2CHCeH5]nCHCeH-7 + NH3 —>
—> NH2CH2[CH2CHCeH5]nCHC6H5 + NHa
Для энергии активации инициирования получено значение Et = 13 + 4
ккал/моль (54,43-103 ± 16,75 -103 Дж/моль), для разности между энергиями
активации обрыва и развития цепи Et — Ер = 4 ± 1 ккал/моль (16,75 -103 ±
± 4,187 -103 Дж/моль). Отсюда видно, что в инициируемой основаниями
полимеризации, точно так же как и в полимеризации, инициируемой кислота-
ми, обрыв цепей происходит настолько медленно, что успевает образоваться
полимер с высоким молекулярным весом. Это объясняется тем, что энергия
активации стадии обрыва цепи имеет высокое значение в отличие от процесса
радикальной полимеризации, в котором обрыв цепей осуществляется за счет
рекомбинации радикалов, а скорость реакции, характеризуясь низкой энер-
гией активации, все же низка, так как низка концентрация радикалов.
Аналогичное кинетическое исследование [233] полимеризации стирола при
инициировании процесса металлическим калием в жидком аммиаке привело
к выводу, что механизм реакции соответствует описанному выше, который
является более современной трактовкой механизма Циглера.
Натрийнафталин представляет собой соединение с нечетным числом элект-
ронов и широко применяется вместо натрия или калия в последних работах
по полимеризации олефинов под действием основных инициаторов. Это веще-
ство содержит радикал-анион С10Щ •, в котором лишний электрон находится
на разрыхляющей орбитали. Два таких аниона инициируют полимеризацию
точно так же, как два атома натрия инициируют образование дикарбаниона
из двух молекул олефина. Дикарбанион затем полимеризуется с обоих кон-
цов. Это было доказано Шварцем и сотр. [234] на примере полимеризации а-
метилстирола под действием натрийнафталина в тетрагидрофуране. Они
показали, что степень полимеризации равна 2[мономер]/[инициатор], где
коэффициент 2 представляет собой число точек роста в единичной растущей
молекуле полимера.
Кинетика процесса полимеризации в отсутствие стадии как обрыва, так
и переноса цепи обсуждалась в 1943 г. Флори [235] и позднее Бауэром и Ма-
гатом [236] на примере инициируемой кислотами полимеризации. Но наиболее
ярким примером полимеризации, идущей без обрыва цепей, является поли-
меризация, инициируемая основаниями. В этом случае стадия переноса
цепи часто оказывается невозможной, чего легко добиться, исключив части-
цы, способные путем переноса протона вызвать обрыв цепи.
Такой случай был найден Циглером и Бером [237] в 1928 г. Они установили,
что полимеризация бутадиена, инициируемая натрием, протекает до тех
пор, пока не израсходуется весь мономер; если после этого в систему доба-
вить свежего мономера, полимеризация будет продолжаться с образованием
полимера большего молекулярного веса. Примерно через 30 лет это явление
было вновь открыто Шварцем и сотр. [238], которые использовали его для
получения блок-полимеров и полимеров с заданными концевыми группами,
а также для проведения термодинамических и кинетических исследований,
что будет показано ниже. Они назвали полимеры, имеющие карбанионные
857
Глава XIII
концы, способные к продолжению полимеризации в присутствии мономера,
«живущими» полимерами, а их реакционные концы —«живущими» концами.
Далее они показали, что красный цвет инициируемого основаниями полимер-
стпрола является цветом живущих концов, т. е. бензилид-ионов в ионных
парах, таких, как — CHCeHr}Na+. Под действием оснований стирол может
полимеризоваться до тех пор, пока не вступят в реакцию все его молекулы,
но по мере добавления новых его количеств молекулярный вес полимера будет
расти или, с другой стороны, добавление изопрена приведет к образованию
стирол-изопренового блок-полимера.
Если стадия развития цепи при инициируемой основаниями полимеризации,
которая в принципе является обратимой, обратима практически, то сум-
марный эффект такой обратимости может ограничить размеры, до которых
может расти полимер, а также степень законченности, с которой мономер
может быть исчерпан при полимеризации в отсутствие обрыва или переноса
цепи. Определение равновесия между мономером и полимером с определенной
степенью полимеризации позволяет вывести константу равновесия стадии
развития цепи, а из температурной зависимости этой константы определить
энтропию и энтальпию стадии развития цепи [239]. Для высоких степеней
полимеризации, например в случае стирола, бутадиена или изопрена, эти
величины не зависят от молекулярного веса, но для первых нескольких
стадий образования низкомолекулярного полимера, как, например, в случае
сс-метилстирола [240], они различны.
Необходимо напомнить, что в работе Хиггинсона и Вудинга исследование
кинетики инициируемой амид-ионом полимеризации стирола в жидком аммиа-
ке позволило установить различие между энергиями активации стадии обрыва
и развития цепи. При отсутствии обрыва кинетическое исследование позво-
ляет определить скорость и активационные параметры стадии развития цепи.
Можно выбрать такие начальные условия, при которых число живущих
концов в предварительно образовавшемся полимере будет определяться
количеством введенного инициатора и может быть измерено аналитически
(например, спектрофотометрически). Концентрация живущих концов будет
тогда оставаться постоянной, если полимеризацию осуществлять при добавле-
нии нового количества мономера. Для стирола или а-метилстирола в диокса-
не или тетрагидрофуране такая полимеризация протекает по первому поряд-
ку относительно мономера, а константы скорости первого порядка почти
пропорциональны концентрации живущих концов. Пропорциональная зави-
симость означает, что развитие цепи осуществляется за счет живущих кон-
цов в виде ионных пар — СНС6НДХа+. Однако при полимеризации стирола
в растворе тетрагидрофурана константа скорости второго порядка
Скорость ос [Живущие концы]}Мономер]
заметно возрастает с разбавлением, и это означает, что даже в эфирных
растворителях реакция частично протекает через присутствующие в незна-
чительной концентрации высокоактивные диссоциированные карбанионы.
3. Гомолитическое присоединение к олефиновым
соединениям [2411
В гл. V, разд. 2,з, отмечалось,Дто гомолитическое присоединение можно раз-
делить на два класса. К первому классу относятся реакции моноприсоедине-
ния, в которых аддент XY присоединяется по двойной углерод-углеродной
связи, причем X присоединяется в виде предварительно образовавшегося
радикала с образованием аддукта-радикала ХС — С-, который затем отры-
858
Присоединение и обратные реакции
вает Y от молекулы XY с образованием продукта ХС — CY. Ко второму
классу относятся реакции полиприсоединения, в которых тот же самый перво-
начально образовавшийся аддукт-радикал вместо разложения молекулы XY
присоединяет другую молекулу олефина с образованием высшего аддукт-
радикала ХС — С — С — С-, а из последнего путем дальнейшего такого же
присоединения образуется полимер-радикал. Этот рост продолжается до тех
пор, пока к концу полимер-радикала не присоединится Y с образованием
конечного полимера. Мы рассмотрим эти два типа присоединения отдельно,
но оба они смыкаются с реакциями телоприсоединения — реакциями, полу-
чившими такое название потому, что первоначально образовавшийся аддукт-
радикал ХС — С • присоединяет только несколько молекул олефина до того,
как присоединит концевую Y-группу с образованием молекулы теломера
с низким молекулярным весом ХС(С — C)nY, где п — мало.
а. Реакции моноприсоединения бромистого водорода и сульфидов, гало-
генов, полигалогеналканов и альдегидов к олефиновым соединениям. В нача-
ле 30-х годов считали, что присоединение галогеноводородов к олефинам
подчинаяется правилу Марковникова (разд. 1,а). Вместе с тем имелись
сообщения о реакции присоединения бромистого водорода, которая не следо-
вала этому правилу и в некоторых случаях не подчинялась никаким другим.
В 1933 г. Хараш и Майо [242] тщательно исследовали реакции присоединения
бромистого водорода к аллилбромиду. Они обнаружили, что эта реакция
может идти в разных направлениях в зависимости от условий. В темноте
и в отсутствие кислорода или перекисей или других веществ, неизвестных
в то время, которые мы теперь относим к классу радикальных инициаторов,
присоединение протекает в течение нескольких дней при комнатной темпе-
ратуре по правилу Марковникова с образованием пропилендибромида
А1сдлвнно
СН2 = СНСН2Вг+ НВг ------------> СН3СНВгСН2Вг
в темноте, z
без инициаторов
На свету или в присутствии небольших количеств кислорода или перекисей
присоединение завершается в течение нескольких минут, но с образованием
триметилендибромида
Быстро
СН2=СНСН2Вг + НВг-----------------> СНоВгСН2СН2Вг
свет или инициатор
Спустя четыре года после этого открытия Хей и Уотерс в обзоре, в котором
была действительно внесена ясность в этот новый случай гомолитических
реакций [243], и одновременно Хараш, Энгельман и Майо в работе, разви-
вающей их собственные исследования [244], объяснили эту реакцию. В тем-
ноте и в отсутствие инициаторов эта реакция представляет собой обычную
реакцию электрофильного присоединения, которую начинает протон моле-
кулы бромистого водорода. На свету и в присутствии инициаторов реакция
начинается атомами брома и протекает с большей скоростью благодаря тому,
что в ее механизм входит следующая кинетическая цепочка:
HBr-p/rv —> H’-f-Br" I „
НВг-|-О2—> НО2-+Вг-} Инициирование
Вг* + СН2 = СНСН2Вг —> BrCH2CHCH2Br 1
. } Развитие цепи
ВгСН2СНСН2Вг + НВг ВгСН2СН2СН2Вг + Вг J
С появлением других примеров стало ясно, что атомы брома, если это воз-
можно, всегда присоединяются по концам углеродной цепи. Причина состоит
в том, что образовавшийся электрононенасыщенный радикальный центр имеет
859
Глава XIII
тенденцию располагаться там, где он может лучше всего стабилизоваться за
счет сопряжения или гиперконъюгации, т. е. скорее у вторичного, чем у пер-
вичного атома углерода. Причина, по которой гомолитическое присоединение
бромистого водорода идет вопреки правилу Марковникова, аналогична при-
чине, по которой электрофильное присоединение подчиняется этому правилу,
и состоит в следующем: электрононенасыщенный карбониевый центр, от ко-
торого зависит механизм реакции, оказывается более стабильным при вторич-
ном, а не при первичном атоме углерода (Вг-, так же как и Н+, является
электрофильным агентом).
Если проанализировать написанные выше стадии развития цепи, взяв зна-
чения энергий связи из табл. 60 (гл. V, разд. 2, ж), то можно увидеть, что обе
стадии являются экзотермическими. Стадия развития цепи, как и ожидалось,
оказалась быстрой, что ведет к образованию длинных кинетических цепей.
Если теперь таким же образом проанализировать энергетику этих реакций,
но с другими галогеноводородами, то сразу становится очевидным, что реак-
ция с фтористым водородом будет невозможна, так как связь во фтористом
водороде слишком сильная. Гомолитическое присоединение фтористого водо-
рода не наблюдается.
По таким же причинам гомолитическое присоединение хлористого водорода
должно протекать с трудом. Первая стадия цепного механизма будет сильно
экзотермичной, а вторая — эндотермичной [около 10 ккал/моль (41,87 х
X Ю3 Дж/моль)]. Таким образом будет образовываться радикал-аддукт, одна-
ко он с трудом разлагает хлористый водород; гораздо легче идет присоедине-
ние к нему. Поэтому этот радикал может присоединить новую молекулу оле-
фина — первая из исследованных стадий, ведущих к образованию полимера.
Если на какой-нибудь стадии роста цепи полимер-радикал вместо того, чтобы
присоединить еще одну молекулу олефина, все же разложит молекулу хло-
ристого водорода, то рост этой молекулы полимера прекратится.
C1' + CH2 = CHR —> C1CH2CHR
C1CH2CHR + CH2 = CHR —> C1CH2CHRCH2CHR
ClCH2[CHRCH2]nCHR + HC1 —> ClCH2[CHRCH2]nCH2R +Cl-
Темы настоящего раздела и разд. 3, б частично перекрываются. Однако здесь
следует заметить, что степень полимеризации будет определяться соотно-
шением скоростей, с которыми радикал реагирует с олефиновым мономером
и хлористым водородом; отношение абсолютных скоростей этих реакций
может меняться с изменением относительных концентраций конкурирующих
реагентов. При взаимодействии хлористого водорода с этиленом при высоком
давлении этилена [245] образуется некоторое количество низкомолекуляр-
ного полимера (теломера). Этот факт свидетельствует о не слишком большом
различии удельных скоростей (константы скорости) конкурирующих реаген-
тов.
Реакция гомолитического присоединения йодистого водорода встречает за-
труднения по другой причине. Атомарный иод получается легко, но его при-
соединение к олефину на первой стадии цепного механизма протекает эндо-
термично. Следовательно, открывается более легкий путь реагирования
радикал-аддукта; по мере образования он скорее снова отщепит атом иода,
чем разложит молекулу йодистого водорода или присоединится к другой
олефиновой молекуле. И действительно, гомолитическое присоединение йоди-
стого водорода не наблюдается.
Сероводород и тиолы могут вступать пе только в реакции присоединения,
подобно воде и спиртам, в качестве электрофильных агентов в кислых средах
860
Присоединение и обратные реакции
или в качестве нуклеофильных агентов к сопряженным олефиновым карбо-
нильным соединениям в щелочных средах, но и в такие радикальные реакции
присоединения к большинству типов олефиновых соединений, которые не
требуют присутствия сильной кислоты или сильного основания. Однако,
как установили в 1928 г. Эшворт и Баркхердт [246], скорость этих реакций
зависит от освещения или присутствия воздуха или от обоих факторов сра-
зу. В 1934 г. Баркхердт [247] определил эти реакции как радикальные.
В 1938 г. Хараш, Рид и Майо [248] применили к этой реакции схему цепного
механизма, которая была установлена годом раньше для присоединения
бромистого водорода.
rs-+ch2=cxy —> RSCH2CXY 1
> Цепь
RSCH2CXY + HSR —> rsch2chxy + rs. J
G этого времени стали использовать инициирование реакций с помощью
органических перекисей, впервые примененное Джонсом и Ридом [249]
в 1938 г.
Реакции сероводорода похожи на реакции тиолов, за исключением того,
что первые реакции могут идти как двойное присоединение, например серо-
водород и винилхлорид образуют «горчичный газ» C1CH2CH2SCH2CH2C1.
Как можно видеть из данных табл. 60 (гл. V, разд. 2, е), энергии связей
Н — SH и Н — Вг очень близки. Можно принять, что это, вообще говоря,
верно для связей Н — S в тиолах и Н — Вг в бромистом водороде. Точно
так же очень близки между собой энергии связей С — SH и С — Вг. Таким
образом, обе стадии развития цепи в радикально-цепном присоединении
сероводорода и тиолов к олефиновым соединениям будут экзотермическими,
т. е. будет выполняться основное условие для быстрого развития цепи.
Поэтому можно ожидать, что сероводород и тиолы, так же как и бромистый
водород, будут энергично вступать в реакции гомолитического присое-
динения.
С помощью тех же аргументов, которыми мы пользовались при рассмотрении
гомолитического присоединения галогеноводородов, можно показать, почему
не реализуется гомолитическое присоединение путем разрыва О — Н-связи
в воде и спиртах и почему до сих пор не описаны аналогичные реакции при-
соединения селенида водорода и селенолов, а также теллурида водорода
и теллуролов.
Хотя гомолитическое присоединение тиолов во многом похоже на присоеди-
нение бромистого водорода, наблюдается также отдаленное сходство в гомо-
литическом поведении тиолов и хлористого и йодистого водородов. Такое
сходство с хлористым водородом проявляется в том, что хотя образование
теломера не является нормальным побочным процессом реакции гомоли-
тического присоединения тиола к простым олефинам, оно реализуется
в реакции присоединения к этилену под высоким давлением [250]. Отдаленное
сходство с иодистым водородом, вероятно, проявляется в том, что обратимость
стадии радикального цепного присоединения, которая достаточно велика
для того, чтобы предотвратить гомолитическое присоединение йодистого
водорода, может в реакции присоединения тиола оказаться достаточной
для проявления широко известного эффекта сероводорода в промотировании
грус-тряис-изомеризации олефиновых соединений, медленно вступающих
в реакцию присоединения, например малеиновой кислоты [251].
Качественные наблюдения ясно показывают, что в роли гомолитических
аддендов и бромистый водород, и сероводород, так же как и тиолы, преиму-
щественно являются электрофилами. Можно ожидать, что радикалы, кото-
861
Глава XIII
рым соответствуют стабильные анионы, но нестабильные электронодефицитные-
катионы, являются, по существу, электрофилами. Это можно проиллюстри-
ровать на примере гомолитического присоединения тиогликолевой кислоты
к простым и замещенным стиролам [252],
rc6h4ch=ch2+hsch2co2h —> RC6H4CH2CH2SCH2CO2H
Измерения относительных скоростей расходования стиролов, конкури-
рующих попарно в реакции с тиолом, дают следующие относительные
скорости присоединения тиенильного радикала к замещенным стиролам
при 60 °C:
Заместитель ге-СН3О и-СН3 Н n-Вг n-F
Относительная скорость 100 2,3 1 0,90 0,50
Легкость отрыва атома водорода от тиола аддукт-радикалом может быть,
определена, если каким-либо образом оценить скорость отрыва водорода —
реакции, которая завершает присоединение, при наличии конкуренции
с присоединением к другой молекуле олефина — реакции, которая пред-
ставляет собой одну из стадий образования теломера. В реакции присоеди-
нения тиолов к стиролу и к бутадиену можно оценить степень образования
теломера и на этой основе рассчитать их константы переноса. Эти константы
представляют собой отношение скорости завершения присоединения к ско-
рости продолжения образования теломера. При использовании одного и
и того же олефина, но различных тиолов константы переноса будут соответ-
ствовать относительным скоростям отрыва атома водорода от тиолов. Ряд
таких констант переноса был измерен [253] и систематизирован Уоллингом
[254]. Для незамещенных арил- и алкилтиолов значения констант можно
пязделить на три группы, как показано в табл. 181. Эти данные позволяют
написать ряд относительных скоростей отщепления Н от RSH:
R = арил > первичный алкил > третичный алкил
Таблица 181
Константы переноса для теломеризацип при присоединении тиолов
к стиролу и бутадиену а
R в RSH Темпера- тура, °C Константа R в RSH Темпера- тура, °C Константа
Фенил 60 Очень большая и-Додецил 60 20
н-Бутил 60 22 а-Нафтилметил 50 18
н-Амил 60 18 mpem-Бутил 60 3,6
н-Октил 50 19; 16 mpem-Октил 6 50 4,5; 3,7
а Цифры, выделенные курсивом, относятся к бутадиену.
® 1,1,3,3-Тетраметилбутил.
Арильный радикал стабилизует радикал RS • за счет сопряжения, первичный
алкил — за счет гиперконъюгации, а в случае третичного алкила любая из
этих форм стабилизации отсутствует и, возможно, проявляется стерический
эффект.
Для того чтобы вызвать гомолитическое присоединение галогенов к олефи-
нам, обычно применяют фотохимическое инициирование. Лучше всего эти,
862
Присоединение и обратные реакции
реакции изучены в газовой фазе или в среде неполярных растворителей,
так как в среде полярных растворителей очень легко происходит электро-
фильное присоединение. Эти реакции моноприсоединения не сопровождаются
образованием теломеров.
Шумахер и другие авторы [255, 257] исследовали кинетику фотоинициируе-
мого присоединения хлора и брома к этилену, а также к моно- и полихлор-
этиленам главным образом в газовой фазе и отчасти в растворах. Для реакции
присоединения хлора к этилену уравнение скорости имеет вид
Скорость ос /1/з[С12][С2Н4]1/2
где I — интенсивность поглощаемого света. Это^означает, чтоДтервая стадия
развития цепи
С1-+С2Н4 дД С1СН2СН2-
С1СН2СН2+С12 С1СН2СН2С1 +Cl-
Цепь
является предравновесной, причем обратная реакция’первой стадии проте-
кает быстрее, чем вторая, даже несмотря на то, что обе они экзотермичны.
Обрыв цепи осуществляется за счет взаимодействия атома хлора с хлорал-
кильным радикалом. Длина кинетической цепи достигает нескольких мил-
лионов циклов. В соответствующих реакциях присоединения к винилхлори-
ду, а также к цис- и транс-изомерам ди-, три- и тетрахлорэтиленов концентра-
ция олефина из уравнения скорости исчезает
Скорость ос Д'ДсЦ
Это свидетельствует, между прочим, о том, что обратная реакция первой
стадии цепи в этом случае идет медленнее, чем вторая стадия, т. е.
С?+СН2 = СНС1 —> С1СН2СНС1
С1СН2СНС1 + С12 С1СН2СНС12 +Cl-
Цепь
Приемлемое объяснение состоит в том, что эффект сопряжения (+Л7) перво-
начального заместителя хлора стабилизует электронодефицитиый центр
радикала в радикал-аддукте в большей степени, чем исходный олефин.
В результате прямая реакция первой стадии оказывается не только более
экзотермичной, чем в реакции этилена, но также обусловливает отрицатель-
ный инкремент в ее свободной энергии, несмотря на потерю трансляционной
энтропии. Таким образом, вместо самопроизвольного распада радикал-
аддукт должен разрушать молекулу хлора. Кинетическое уравнение также
показывает, что обрыв цепи проходит с участием двух хлоралкильных ради-
калов, которые, вероятно, либо коллигируются, либо диспропорционируют.
Кинетические цепи имеют достаточно большую длину.
Соответствующие газофазные реакции брома с этиленом и моно- и полихлор-
этиленами включают цепные стадии, которые обе являются еще экзотерми-
ческими, но в гораздо меньшей степени. То, что первая стадия цепного меха-
низма является обратимой, теперь очевидно. В реакции между бромом
и тетрахлорэтиленом суммарное присоединение, идущее в одну сторону
в жидкой фазе при комнатной температуре, оказывается сильно обратимым
в паровой фазе при температуре порядка 100 °C. Тем не менее присоединение
в обоих случаях идет в прямом направлении с образованием довольно плин-
ных кинетических цепей, порядка десятков тысяч циклов. Добавка кислоро-
да уменьшает длину цепей [257]. Очевидно, молекулы кислорода конкури-
руют с атомами брома, усиливая роль процесса обрыва цепей за счет взаимо-
действия с радикал-аддуктом.
863
Глава XIII
Гомолитическое присоединение иода к простым олефинам на первой стадии
цепного механизма должно быть эндотермическим; и в действительности,
суммарный процесс присоединения является термодинамически невыгодным
при комнатной температуре. Однако фотоинициируемое присоединение иода
к этилену, пропилену и четырем изомерным бутиленам реализовано при
температуре —55 °C [256].
Роль обратимости реакции при образовании радикал-аддукта из атомов бро-
ма и олефинов с целью выяснения механизма г^пс-ягранс-изомеризации олефи-
нов была исследована Кетелааром и сотр. [257]. Совместными кинетическими
исследованиями и исследованиями состава продуктов взаимодействия брома
с tywc-дихлорэтиленом в жидкой фазе им удалось показать, что присоедине-
ние брома к олефину и гщс-щодис-изомеризация последнего протекают через
одни н те же промежуточные соединения. Дальнейшая проверка была сделана
Стейнметцем и Нойесом [258], которые использовали радиоактивный бром
для инициирования ^«с-тп/?анс-изомеризации tyiic-дибромэтилена в среде
четыреххлористого углерода при температуре 40—60 °C и смогли показать
с помощью кинетических исследований, что обмен брома, который сопро-
вождает изомеризацию олефина, идет через те же самые промежуточные
соединения, что и изомеризация. Шомакер, Диккенсон и Нойес [259] при-
менили такой же метод для изучения ^ис-транс-изомеризации дихлор-,
дибром- и дииоддекалинов в растворе декалина, инициируемой атомами
иода. Было показано, что эта изомеризация также протекает путем обрати-
мого образования радикал-аддукта из атомов иода и олефина.
Поскольку связь фтор — водород очень прочна, то в реакции любого орга-
нического соединения, содержащего водород, с элементарным фтором про-
исходит более или менее неупорядоченный отрыв водорода. Таким образом,
олефины образуют простые продукты присоединения фтора только в том
случае, когда олефин является пергалогензамещенным. И далее, как пока-
зали Миллер и Кох [260], в этом случае не происходит образования атомов
галогена на первой стадии в отличие от других реакций гомолитического
присоединения галогенов. Связь атомов в молекуле фтора настолько слаба,
а фтор-углеродная связь настолько сильна, что молекулы фтора могут при-
соединяться как таковые с образованием фторированного аддукт-радикала
и атомарного фтора. Атом фтора будет затем присоединяться к соседней
молекуле олефина с образованием другого фторированного аддукт-радикала.
Таким образом, аддукт-радикалы стремятся образовываться локальными
парами, которые наряду с участием в обычном процессе развития цепи всту-
пают во взаимодействие друг с другом с образованием димеров
CX2=CX2-[-F2 •—> FCX2CX2 + F* Инициирование
CX2 = CX2+F. —> FCX2CX£
FCX2CXJ+F2 —> FCX2CX2F4-F*
Развитие цепи
FCX2CX£+F- —> FCX2CX2F
Нормальный обрыв
2FCX2CXJ —> FCX2CX2CX2CX2F Обрыв в результате димеризации
Образование димеров — наиболее характерная особенность реакций при-
соединения фтора. Миллер и Кох продолжили исследования, чтобы доказать
свою теорию образования локальных пар аддукт-радикалов. Они установили,
например, что при обработке фтором смеси двух олефинов с сильно разли-
чающейся реакционной способностью (например, более реакционноспособно-
го CFC1 = CFC1 и менее реакционноспособного CF3CC1 = CC1CF3) более
реакционноспособный олефин монополизирует стадию инициирования
и потому наряду с нормальным продуктом присоединения образуется боль-
864
Присоединение и обратные реакции
шое количество димера, а менее реакционноспособный олефин, который,
вероятно, реагирует только на стадии развития цепи, дает лишь нормальный
продукт присоединения. Образования других побочных продуктов, когда
олефин, например CFC1 = CFC1, содержит хлор, понять легко. Любая реакция,
обратная образованию аддукт-радикала, будет включать потерю высшего
галогена, т. е. в данном примере атома хлора
F* + CFC1 = CFC1 —> CF2C1CFC1 CF2 = CFC1-J-Cl-
Таким образом, происходит замена хлора на фтор. Освобождающийся атом
хлора будет соединяться с аддукт-радикалом
C1- + CF2C1CFC1 —> CF2C1CFC12
Результатом этой реакции будет кажущееся присоединение хлористого фтора.
Эти реакции представляют собой другие побочные процессы.
Полигалогеналканы представляют собой важную группу веществ, способных
к гомолитическому присоединению к олефинам. Как уже отмечалось, гало-
гензаместитель стабилизует соседний углеродный радикал. Радикал, обра-
зующийся в результате отщепления атома от молекулы полигалогеналкана,
должен быть достаточно стабильным, чтобы он мог легко образоваться,
и все же достаточно реакционноспособным, чтобы разорвать л-компоненту
олефиновой двойной связи. Если с одним и тем же атомом углерода связаны
два или три атома галогена, то образование углеродного радикала произойдет
при введении в реакционную систему обычных радикальных инициаторов,
если отщепляемым атомом галогена будет бром или иод. Но если отщепляе-
мым от углерода атомом является хлор или водород, то число атомов галоге-
на должно быть равно трем. Радикал, содержащий два или три атома галоге-
на, связанных с одним и тем же углеродным атомом, обычно будет раскры-
вать олефиновую двойную связь с образованием аддукт-радикала.
Возможность реализации гомолитического присоединения четыреххлористо-
го углерода вытекала из исследований по полимеризации стирола, молеку-
лярный вес которого в присутствии четыреххлористого углерода снижался.
Впервые это показали в 1945 г. Хараш, Йенсен и Урри [261], установив, что
четыреххлористый углерод, а также хлороформ в присутствии радикальных
инициаторов присоединялись к октену-1 с образованием следующих про-
дуктов:
СС14 + СН2=СНСвН13 —> С13ССН2СНС1СвН13
НСС13-}-СН2^СНСвН13 —> С13ССН2СН2С8Н13
В обоих случаях присоединяющийся первым радикал С13С> занимает конеч-
ное положение и образует, таким образом, вторичный алкил-радикал,
присоединение к которому завершается во второй стадии цепного
механизма.
Впоследствии аналогичные реакции присоединения четыреххлористого угле-
рода были осуществлены со многими ациклическими, циклическими, поли-
циклическими и гетероциклическими моно- или полиолефиновыми соедине-
ниями. В качестве инициаторов обычно применяли перекиси ацетила или
бензоила при температуре 60—100 °C. В некоторых случаях обычное при-
соединение сопровождалось образованием теломера.
Другим подробно исследованным тетрагалогенметаном был трихлорбромме-
тан. Он присоединяется в виде фрагментов СС13 и Вг. Этот тетрагалогенид
значительно более реакционноспособен, чем четыреххлористый углерод,
и для осуществления его реакции обычно требуется очень небольшое количе-
ство инициатора. Энергия отрыва атома брома низка, так что для ипицииро-
55-0772
865
Глава XIII
вания реакции достаточно фотооблучения светом с такой длиной волны,
чтобы он мог проходить через стекло пирекс. Присоединение четырехбро-
мистого углерода протекает точно так же. Исследован также трифториодме-
тан. Последовательность реакций в этом случае аналогична последователь-
ности для четыреххлористого углерода
ВгСС1з + СН2=СНСвН13 —> С13ССН2СНВгСвН13
ВгСВг3Ч-СН2=СНС6Н13 —> Вг3ССН2СНВгС6Н13
ICF3 + CH2 = CHCH3 f3cch2chich3
Галоформы, содержащие бром или иод, ведут себя иначе, чем хлороформ,
отщепляя преимущественно атом брома или иода, а не атом водорода.
И в этих реакиях присоединения обычно используют инициирование пере-
кисями.
ВгСНС12 + СН2=СНСН3 —> С12СНСН2СНВгСН3
BrCHBr2+CH2=CHCeH13 Вг2СНСН2СНВгСвН13
1СШ2+СН2=СНСН2ОСОСвН5 12СНСН2СН1СН2ОСОС6Н5
Пергалогенпроизводные некоторых высших парафиновых углеводородов
присоединяются к олефинам обычно в присутствии перекисных инициаторов,
например:
BrCFClCF2Br + CH2 = СНСН3 —> BrF2CCFClCH2CHBrCH3
Общий механизм всех этих реакций присоединения можно проиллюстриро-
вать на примере присоединения трихлорбромметана
С13СВг —> С13С-4-Вг’ Инициирование
C13C. + CH2 = CHR —> С13ССН2СНВ
С13ССН2СНИ + С13СВг —> С13ССН2СНИВг + С13С.
Развитие цепи
С13С- + Вг- —> С13СВг ]
. > Обрыв цепи
Cl3CCH2CHR-f-Br- —» Cl3CCH2CHRBr J
Возможны также и другие процессы обрыва цепи [262].
Присоединение радикалов CXj и CHXj в концевое положение уже свидетель-
ствует о том, что они обладают преимущественно электрофильными свой-
ствами, т. е. что они присоединяются к образовавшемуся углеродному ради-
кальному центру при вторичном атоме углерода, где электронная недоста-
точность может стабилизоваться за счет сопряжения или гиперконъюгации
сильнее, чем это возможно при первичном атоме углерода. Эти соображения
относительно ориентации присоединения подтверждаются величинами отно-
сительных скоростей присоединения, найденными Харашем и сотр. [263, 264]
с помощью метода конкурирующих реакций. Они взяли смесь двух олефинов
СН2 = CRR', конкурирующих в реакции присоединения к радикалу С13С-
C13C.+CH2=CRR' —> C13CCH2CRR'
Полученные результаты в некоторой степени зависят от условий проведения
реакции, но дают следующий порядок изменения скоростей (приведены
R, R'):
СвН5, СН3 > СвН5, ГП> СвН13, Н > СвН5СН2, Н > СН2С1, Н > CH2CN, Н
Здесь проявляется благоприятное влияние электроположительного сопря-
жения (-RM) и неблагоприятное влияние индуктивной электроотрицатель-
ности (—/) на скорость присоединения. Пирсон и Шварц [265] исследовали
подобное присоединение радикала F3C-, который генерировался фотолизом
866
Присоединение и обратные реакции
гексафторазометана
F3C-+CH2=CRR' —> F3CCH2CRR'
Реакции проводили в газовой фазе при температуре от 65 до 180 °C. Для
приведенного выше ряда RR' были получены следующие относительные
скорости и энергии активации:
CeHj, СН3 С6Н5, Н (СН3)2 СНз, Н н2 С1, н F, Н
Относительная q9 скорость (65 °C) 58 39 13 И 5 1
— ДЕ, ккал/моль 9 s (4,187-Ю3 Дж/моль) 2,4 2,5 1,7 1,3 0,8 0
В результате исследований Хуанга [265] по ориентации в реакциях присоеди-
Ф
нения радикала С13С- к таким олефинам, как СН3СН = СНСООС2Н5
Ф
и С6Н5СН = СНСООС2Н5, стало ясно, что стабилизующее влияние замести-
телей на соседние радикальные центры изменяется в ряду
С6Н5> СООС2Н5> СН3
Отсюда вытекает, что даже электроотрицательное сопряжение (—М) стаби-
лизует углеродные радикалы. По-видимому, группа СООС2Н5 сопрягается
с необобщенным электроном радикала. Таким образом, эти пергалогенме-
тильные радикалы, хотя и являются преимущественно электрофильными,
обладают также некоторым нуклеофильным характером.
В заключение этого раздела рассмотрим некоторые кислородные соединения,
в частности альдегиды, как гомолитические адденды. В смеси простого альде-
гида с простым олефином альдегидная связь С — Н обычно является наибо-
лее слабой в системе. Благодаря этому инициирование перекисями будет
давать ацил-радикал RCO •, который будет присоединяться к олефину.
Альдегидная связь С — Н будет, как правило, достаточно слабой, так что
образовавшийся аддукт-радикал может отрывать от альдегида этот атом
водорода. Реакцию моноприсоединения альдегида можно представить сле-
дующей схемой:
R'CO-+CH2=CHR —> R'COCH2CHR-ф
R'COCH2CHR-+R'CHO R'COCH2CH2R+R'CO. J Развитие Чепи
Важным конкурирующим процессом часто является образование теломера.
Уменьшить роль этого процесса можно за счет создания компенсирующей
разности концентраций путем постепенного добавления олефина к избытку
альдегида при энергичном перемешивании раствора, содержащего достаточ-
ное количество инициатора, чтобы обеспечить быстрое протекание реакции.
Моноприсоединение альдегидов к простым олефинам было описано в 1949 г.
Харашем, Урри и Кудерном, а к <х,р-ненасыщенным эфирам и кетонам —
в 1952 г. Патриком [267, 268]. Ниже приведены типичные примеры:
и-С4Н9СНО + СН2 = СНС6Н13 С4Н9СОСН2СН2С6Н13
СН3СОЧ
СНзСНО + ^ис-С2Н5О2ССН = СНСО2С2Н5 —> >СНСН2СО2С2Н5
С2Н5О2С
и-С4Н9СН О + (СН3)2С = СНСОСНз —
» СОС4Н9
(СН3)2ССН2СОСН3
сос4нэ
I
> (СН3)2СНСНСОСН3
(90%)
(10%)
55* 867
Глава XIII
Обычно повышенные выходы продуктов моноприсоединения, получающиеся
в реакциях с а,р-ненасыщенными эфирами и кетонами, такими, как этиловый
эфир малеиновой кислоты и окись мезитила, но не при взаимодействии
с простыми олефинами типа октена-1, можно объяснить тем, что ацильный
радикал ведет себя как очень сильная нуклеофильная частица. Такое объяс-
нение возможно и верно, однако никакие прямые сравнения поведения
ацильных радикалов с другими радикалами автору не известны. Правда,
третий из приведенных выше примеров указывает на то, что ацетильный
заместитель в некоторых случаях служит лучшим стабилизатором соседнего
радикального центра в аддукт-радикале, чем два метильных заместителя.
Этот аргумент использовал Хуанг [266], который в результате исследований
ориентации присоединения н-бутальдегида к различным ненасыщенным
кетонам, кислотам и эфирам вывел следующий ряд эффективности стабилиза-
ции соседнего углеродного радикала:
СН3СО > СООС2Н5 > СООН > СН3
Это еще раз свидетельствует о том, что электроотрицательное сопряжение
(—М) эффективно стабилизует углеродный радикал.
Хорошо известная аутоокисляемость кислородных соединений позволяет
сделать вывод, что в таких соединениях связь <х-СН слаба. Показано, что
способность альдегидов к гомолитическому присоединению с разрывом
а-СН-связи должна быть такой же, как и у спиртов [269]. Так, например,
этиловый спирт и этилен при инициировании перекисями образуют бутанол-2.
И в этом случае наиболее важным конкурирующим процессом будет обра-
зование теломера: в приведенном примере это гексанол-2, октанол-2 и дека-
нол-2. Аналогично ведут себя высшие первичные спирты и вторичные спирты.
а-Ненасыщенные олефины с концевой двойной связью были исследованы
вплоть до додецена-1.
б. Полиприсоединение: инициируемая радикалами полимеризация оле-
финовых соединений путем самоприсоединения. Реакции полимеризации
ненасыщенных соединений в условиях, когда они должны инициироваться
и развиваться при участии радикалов, наблюдались и даже были отнесены
к типу реакции полимеризации еще в прошлом столетии [270] *. Но химиче-
ская природа процесса была вскрыта Штаудингером [104] в 1920 г. Он пока-
зал, что олефиновые молекулы соединяются друг с другом в линейную систе-
му, представляющую собой по сути насыщенную длинную цепную полимер-
ную молекулу
nCH2 = CHR —> (—CH2CHR—)п
В 1935 г. на примере полимеризации стирола Штаудингер и Штейнхофер [272]
показали, что все звенья мономера соединяются по принципу «голова к хво-
сту», поскольку при термическом разложении полистирола при температуре
300 °C получаются, кроме стирола, следующие продукты:
свн5
I
С6Н5 С0Н5 С„Н5 СвН5 С6Н5 |А
СН2СН2СН2 СН2СН2СНСН2СН2 и С„Н5 -1^с„Н5
и не образуется никаких продуктов, имеющих фенильные группы у соседних
углеродных атомов.
* Первое описание таких реакций как реакций полимеризации принадлежит, по-ви-
дпмому, Бертло [271].
868
Присоединение и обратные реакции
Наиболее легко при радикальном инициировании полимеризуются олефины
типа СН2 = CHR или СН2 = CRR', где R и R' способны сопрягаться
(электроположительно или электроотрицательно или обоими этими способа-
ми) с радикальным центром, образующимся у углеродного центра, связанно-
го с радикалами R или R'. Примерами полимеризующихся мономеров с заме-
стителями +7И-типа могут служить винилхлорид, винилацетат, винилиден-
фторид и винилиденхлорид. В качестве мономеров с заместителями —М-типа
используются акрилонитрил, «-метакрилонитрил, метиловые эфиры акрило-
вой и «-метакриловой кислот. Стирол, бутадиен, изопрен и неопрен составля-
ют группу мономеров, при полимеризации которых заместители проявляют
±7И-эффект.
Полимеризация иногда может быть инициирована случайными радикалами
или радикалами, возникающими под действием кислорода или света. Ради-
калы, образующиеся любым из описанных в гл. V, разд. 2, е, способов,
могут инициировать полимеризацию. Сюда относятся, например, такие
окислительно-восстановительные системы, как реактив Фентона, термический
гомолиз перекисей, фотолиз галогенов или алифатических азосоединений
или облучение практически любых органических соединений частицами
с высокой энергией.
Присоединение инициирующего радикала X- к олефиновой молекуле (часто
называемой поэтому мономером) превращает ее в мономерный углеродный
радикал ХС — С-, чем и завершается стадия инициирования полимеризации.
Этот углеродный радикал далее присоединяется ко второй молекуле мономе-
ра, что представляет собой уже первую стадию кинетического развития цепи
и первую стадию структурного роста цепи, в результате которой образуется
димерный углеродный радикал ХС — С—С — С-. После п таких успешных
присоединений растущего аддукт-радикала к новой мономерной молекуле обра-
зуется аддукт-радикал со степенью полимеризации п + 1. Позможная встре-
ча двух таких радикалов со степенью полимеризации т + 1 и п + 1 может
привести к их взаимной аннигиляции. Это может произойти либо путем
присоединения радикальных центров друг к другу (конечным продуктом
будет полимер со степенью полимеризации т + п + 2), либо в результате
переноса атома водорода от одного радикала к другому (в этом случае конеч-
ными продуктами будут одна ненасыщенная и одна насыщенная молекулы
полимеров со степенями полимеризации т + 1 и п + 1). Таким образом,
происходит обрыв цепей за счет коллигации и диспропорционирования
соответственно
вг-
X.4-CH2=CHR-------> XCH2CHR Инициирование
XCH2CHRJ-CH2 = CHR —> X(CH2CHR)CH2CHR I
kP l Развитие
• • •--> ••• ( цепи
X(CH2CHR)n_1CH2CHR+ CH2 = CHR -> X(CH2CHR)nCH2CHR '
X(CH2CHR)mCH2CHR | Обрыв цепи в резупьтате
X(CH2CHR)mCH2CHR X(CH2CHR)nCH2CHR J присоединения радикалов
+ . X(CH2CHR)mCH = CHR 4
X(CH2CHR)nCH2CHR _j_ l Обрыв цепи в результате
----" X(CH2CHRbCH2CH2R f Ди^порциопирования
Обычно весьма трудно определить, каким образом произошел обрыв цепей —
вследствие коллигации или диспропорционирования, даже несмотря на то,
что в результате соединения молекулярный вес полимера удваивается. Кине-
869
Глава XIJI
тически здесь нет различия, и скорость суммы двух процессов можно пред-
ставить в виде простого уравнения второго порядка для констант скоростей
обрыва цепи:
kt = ktc + ktd
Существует еще один способ приостановки структурного роста цепи без
остановки кинетической цепи. Полимерный аддукт-радикал может отдать
свой атом водорода мономерной молекуле, превратив ее в радикал, который
сможет начать рост новой молекулы полимера
X(CH2CHR)nCH2CHR-|-CH2 = CHR —> X(CH2CHR)nCH = CHR + CH3CHR
Кинетически развитие начальной цепи идет при этом совершенно так же,
как и шло бы без передачи атома водорода, но структурный рост цепи оста-
навливается и начинается сначала. Этот процесс называют «переносом цепи».
В том случае когда степень полимеризации нормально связана с кинетикой,
то, как мы увидим ниже, для того, чтобы изменить степень полимеризации,
необходимо изменить скорость реакции, однако наличие процессов переноса
цепи приводит к уменьшению степени полимеризации без изменения ско-
рости.
Этим пользуются на практике для регулирования молекулярного веса
полимера без изменения скорости полимеризации: добавляют в полимери-
зующуюся систему так называемые «агенты, переносящие цепь». В качестве
таких агентов обычно применяют простые соединения YZ, как, например,
четыреххлористый углерод, от которых полимеризующийся аддукт-радикал
может оторвать атом Y так, чтобы образовался новый инициирующий реак-
цию радикал Z-
X(CH2CHR)nCH2CHR+YZ —> X(CH2CHR)nCH2CHYR + Z.
Z- + CH2 = CHR —> ZCH2CHR и t. д.
Кинетическая цепь, первоначально инициированная радикалом Х-, будет
продолжаться дальше, но химическая последовательность цепи приоста-
новится вследствие вмешательства молекулы YZ, которая останавливает
рост старой цепи и начинает рост новой цепи, что при определенных обстоя-
тельствах приведет к уменьшению молекулярного веса полимера.
При расчетах скорости реакции по уравнению, выведенному выше для меха-
низма радикальной полимеризации, можно сделать предположение, что
константы скорости всех стадий развития цепи будут одинаковыми. Это,
очевидно, будет справедливым для всех стадий, за исключением первых трех
или четырех. С учетом такого допущения уравнение скорости принимает
стандартную форму для цепной реакции, приведенную в гл. VII, разд. 11,г,
7?Р = ЫМ] (^)1/2
где Rp — скорость полимеризации, — скорость инициирования, кр —
константа скорости второго порядка стадии развития цепи и kt — суммарная
константа скорости второго порядка обрыва цепи. Уравнение скорости поли-
меризации не отражает скорости переноса цепи.
Кинетическая длина цепи определяется выражением
Rp Rp к*р [МР кр [М]
V- Rt ~ Rt - 2ktRp - -\/2ktR^
870
Присоединение и обратные реакции
При данной концентрации мономера длина кинетической цепи обратно
пропорциональна скорости полимеризации или корню квадратному из ско-
рости инициирования. При данной скорости инициирования она пропор-
циональна концентрации мономера. При отсутствии переноса цепи средняя
степень полимеризации
DP = 2v или v
определяется обрывом цепи в результате коллигации или диспропорциониро-
вания. При наличии переноса цепи степень полимеризации будет меньше
на величину, зависящую от константы скорости процесса переноса цепи
и концентрации введенного в систему специального переносчика цепи, если
он имеется.
Измерения скорости полимеризации при известных скоростях инициирова-
ния дают kp/kt. Индивидуальные константы скоростей кр и kt были определе-
ны с помощью «секторного метода» прекращения фотоинициирования, пред-
ложенного Бартлетом и Мелвиллем, и с помощью других таких же методов,
основанных на релаксации стационарного состояния при внезапном возобнов-
лении или прекращении процесса инициирования (гл. VII, разд. 11,г).
В качестве примера таких определений можно привести данные Кварта,
Бродбента и Бартлета [273], полученные при полимеризации винилового
эфира уксусной кислоты при 25 °C, фотоинициированной в присутствии
ди-щреш-бутилперекиси. В этих условиях кр = 1,0-103 л-моль-1 •с~1 и kt =
= 3,0-107 л-моль-1-с-1. Концентрация полимеризующихся радикалов была
равна примерно 0,5-10-8 моль/л. Полупериод стадии полимеризации был
порядка 10~4 с, средняя степень полимеризации составляла порядка десяти
тысяч, а среднее время роста полимера было равно всего нескольким секун-
дам. Несколько примеров констант скорости развития и обрыва цепи, взятых
из обзора Уоллинга [274], приведено в табл. 182.
Таблица 182
Константы скорости второго порядка
(л-моль-1-с-1) стадий развития
и обрыва цепи при 30° С
Мономер 10-7 kf
Винилхлорид 6800 1200
Винилацетат 990 2,0
Метилакрилат 720 0,22
Метил-а-метакрилат 350 1,5
Стирол 49 0,24
Бутадиен 25
Влияние заместителей в полимеризующемся олефине на скорость его поли-
меризации является очень сложным вопросом, так как здесь превалируют
компенсирующие эффекты. Майо и Уоллинг [275] приложили много усилий
для того, чтобы выяснить этот вопрос. Они установили, что заместитель,
стабилизующий двойную связь олефинового мономера, будет в значительно
большей степени стабилизовать и аддукт-радикал, который образуется из
молекулы олефина в результате присоединения к ней радикала. Кроме того,
он будет ускорять превращение олефина в аддукт-радикал. Вместе с тем
871
Глава XIII
такое сопряжение с радикальным центром в аддукт-радикале будет сильно
замедлять его присоединение к другой молекуле этого же или любого другого
олефина. Тот же самый заместитель влияет, кроме того, на реакционную
способность обоих участников стадии развития цепи гомополимеризации
противоположными способами, но влияние в ненасыщенном радикале будет
больше, чем влияние в ненасыщенном олефине. Таким образом, влияние
заместителя на скорость гомополимеризации должно быть меньше, чем его
влияние на скорость реакции радикала со стандартным олефином, но прибли-
жаться к нему, а не к влиянию заместителя на скорость взаимодействия
олефина со стандартным радикалом. Влияние заместителей можно проанали-
зировать на примере процесса сополимеризации. В этом процессе каждый
аддукт-радикал располагает выбором мономеров, к которым он может при-
соединиться, и каждый мономер располагает выбором радикалов, которые
он может присоединить. Именно поэтому вследствие описанных компенси-
рующих эффектов так легко идет сополимеризация между мономерами,
сильно отличающимися по своей реакционной способности.
На основе исследования состава сополимеров Майо и Уоллинг [275] вывели
ряд эффективности групп R по их влиянию на скорость реакции олефинов
СН2 = GHR с общим присоединяющимся радикалом
R = ОСН3 < ОСОСНз < СНз < Cl < СООСН3 < CN < СОСН3 < СН2-СН < С6Н5
Относительные скорости, которые можно было бы выбрать для иллюстрации
этого ряда, зависят от природы общего присоединяющегося радикала, но
некоторые ряды величин можно привести, пользуясь данными, полученными
Уоллингом [241] для 20 радикалов и более 30 олефинов
ОСН3 ОСОСН3, СН3 Cl СО2СН3 CN, СОСНд СН = СН2, С6Н5
1 1,5-5 3-20 20—60 30-60 50 — 100
Намного труднее получить данные о влиянии заместителей на скорости
присоединения различных аддукт-радикалов к общему олефину, так как
состав продуктов и скорости развития полимеризации должны рассматри-
ваться одновременно. Имеющиеся в литературе сведения позволяют считать,
что для написанного выше ряда величин эти влияния будут изменяться
в обратном порядке:
ОСОСНз, С1 СО2СН3 С6Н5 СН2=СН
100—2000 30—200 1 0,5 —0,7
Во втором случае диапазон изменения величин больше, чем в первом, как
и следовало ожидать, исходя из сказанного в предыдущем параграфе. Сравне-
ние этих двух рядов указывает на большую степень компенсации при гомо-
полимеризации. Это можно показать на примере сравнения полимеризации
винилацетата и стирола. Как олефин, винилацетат реагирует с радикалом
в 50 раз медленнее, чем стирол, но аддукт-радикал винилацетата реагирует
с олефином примерно в 1000 раз быстрее, чем аддукт-радикал стирола. Сум-
марный эффект будет таков: скорость развития полимеризации винилацетата
примерно в 20 раз больше скорости полимеризации стирола.
Полимеризация олефинов может быть ингибирована добавкой различных
веществ. Предполагается, что общий механизм ингибирования включает
рекомбинацию полимеризующегося аддукт-радикала с ингибитором, веду-
щую к образованию либо нерадикального соединения, либо нереакциошю-
способного радикала, которые не могут вести дальше цепь полимеризации
так же легко, как это делает начальный аддукт-радикал. Ингибиторами слу-
жат триарил- или дифенилпикрилгидразил-радикалы. Рекомбинация с ними
872
Присоединение и обратные реакции
аддукт-радикала будет давать нерадикальное соединение. Значительными
ингибирующими свойствами обладает молекулярный кислород. При этом
должен получаться перокси-радикал, который хотя и присоединяется
к некоторым олефинам, например к стиролу, но не может присоединяться
к другим олефиновым соединениям, с которыми приходится встречаться
на практике.
В качестве ингибиторов можно применять, и действительно применяют, много,
веществ нерадикального характера и не находящихся в триплетном состоя-
нии. Все они являются сильно сопряженными, легко восстанавливающимися
электрофильными соединениями ароматического ряда. К ним относятся
в первую очередь хиноны и нитросоединения. Существует два пути, по кото-
рым эти соединения могут выводить полимеризующиеся аддукт-радикалы
из реакции переноса кинетической цепи. Один из них — диспропорциониро-
вание: ингибитор может отбирать у молекулы аддукт-радикала атом водо-
рода. Хиноны в результате такой реакции превращаются в хингидроны
и, возможно, в некоторых случаях в гидрохиноны, а отдавший атом водорода
полимер будет содержать концевую ненасыщенную группу. Другим путем
является простое присоединение всего радикала с образованием такого про-
дукта, в котором радикальный центр будет настолько делокализован, что
он не будет в состоянии присоединять новую молекулу олефина, хотя
и сохранит способность соединяться с другим радикалом.
Общее положение, вытекающее из экспериментальных данных, состоит
в том, что из процессов гомополимеризации наиболее легко будут ингиби-
роваться те, в которых аддукт-радикал особенно реакционноспособен,
а мономер мало реакционноспособен. Примером может служить полимери-
зация винилацетата.
Возможные объяснения ингибирования реакций полимеризации по меха-
низму диспропорционирования или коллигации не исключают друг друга,
но более вероятным представляется обрыв цепей по механизму коллигации.
Среди бензохинонов хлоранил является довольно слабым ингибитором, но.
со стиролом он полимеризуется и дает полимер [276]
С1 С1
! I
- ОСН2СН(С6Н5)О о -
I I
С1 С1
Бензохинон может обрывать растущую полимерную цепь в результате
аналогичного О-алкилирования. Хлоранил не вступает в реакцию С-алки-
лирования, а бензохинон вступает, что доказывается составом полимеров:
при ингибировании полимеризации аллилового эфира уксусной кислоты
бензохиноном происходит как О-, так и С-алкилирование ингибитора [277].
При ингибировании нитробензолом можно ожидать присоединения углерод-
ного радикала в орто- и иара-положения относительно нитрогруппы, но.
можно предположить, что присоединение может идти также и к нитрогруп-
пе [277, 278].
Исследование стехиометрии ингибирования показывает, что в ингибиро-
ванном полимере в зависимости от условий реакции и случайных факторов
на одну молекулу полимера может приходиться либо менее одной, либо
более одной молекулы ингибитора. Конечно, интерпретировать эти резуль-
таты можно по-разному. Здесь следует иметь в виду и мультиплетную атаку,
и диспропорционирование, и сополимеризацию.
873
Глава XIII
4. Циклоприсоединение к олефинам [279]
а. Присоединение карбенов с образованием трехчленных циклов [280].
В гл. IX, разд. 7, говорилось об одном из двух основных методов получения
карбенов — 1,1-отщеплении от замещенных парафинов. Другим важным
методом является пиролиз или фотолиз диазоалканов или кетенов
rr'C = N = N —> RR'C + N2
rr'C = C = O —> RR'C + CO
История циклоприсоединения карбенов возвращает нас к исследованиям
Бюхнера и Курциуса по присоединению диазоалканов к ароматическим
соединениям и олефинам. Из олефинов образуются пиразолины, которые
при пиролизе теряют азот и превращаются в циклопропаны. Эти исследова-
ния были начаты в 1885 г. с бензола и подобных ему соединений. При этом
промежуточно получались не пиразолины [281], а норкарадиены и их более
устойчивая валентно-таутомерная форма — тропилидены. Бюхнер и Хеди-
гер [282] пришли к заключению, что потеря азота происходит не после, а до
присоединения и что эффективным аддендом для таких молекул, как бензол
и нафталин, является карбен.
Простейший карбен — метилен был получен Герцбергом и Шусмитом [283]
из диазометана с помощью метода импульсного фотолиза. Им удалось заре-
гистрировать спектр электронного поглощения, который состоит из двух
отчетливых спектров, т. е. два различных долгоживущих состояния имеют
свои собственные спектры поглощения. Один из этих спектров находится
в вакуумной ультрафиолетовой области [около 1415 А (141,5 нм)]; анализ
его полос показал, что он относится к линейному триплетному состоянию.
Другой спектр находитсяов далекой инфракрасной области [полосы с длинами
волн 6531, 7315 и 8190 А (653,1, 731,5 и 819,0 нм)] и принадлежит напря-
женному синглетному состоянию. При увеличении давления инертного газа
интенсивность системы полос в далекой ультрафиолетовой области увели-
чивается за счет уменьшения интенсивности системы полос в далекой инфра-
красной области. Очевидно, столкновения газовых молекул преодолевают
консервацию спинов, и происходит превращение менее стабильного синглет-
ного состояния в более устойчивое триплетное состояние. Синглетное состоя-
ние имеет неподеленную электронную пару и незанятую р-орбиталь, в то
время как триплетное состояние имеет две однократно занятые р-орбитали
С
ц/ Н-С —Н
Синглет Триплет
В синглете длина СН-связи составляет 1,11 А (11,1 -102 нм) и угол между
связями 102,4° (1,787 рад).
Хотя триплетное состояние более устойчиво, сначала образуется синглетное
состояние из синглетного предшественника, которым является основное
состояние диазометана или кетена или возбужденное состояние, в которое
переходит система из переходного состояния. Более того, синглетное состоя-
ние, если оно образовалось, часто может реагировать как таковое, так как
превращение синглета в триплет является медленным процессом и тре-
бует для своего осуществления большого числа молекулярных столк-
новений.
Известны также более ранние, но менее существенные исследования обра-
зования метилена. В 1938 г. Пирсон, Парселл и Сейх осуществили фотолиз
874
Присоединение и обратные реакции
диазометана в газообразное вещество, которое удаляло с поверхности слой
селенового и теллурового зеркала [284].
Двумя основными реакциями превращения метилена является так назы-
ваемая реакция «включения», состоящая в превращении С — Н в С — СН3,
и реакция циклоприсоединения к олефиновой двойной связи с образованием
циклопропанового кольца. Меервейн с сотр. [285], исследуя в 1942 г. реакцию
включения, отметили, что при фотолизе диазометана в диэтиловом эфире
некоторое количество последнего превращается в этил-н-пропиловый эфир-
и этилизопропиловый эфир. Атака первичных, вторичных и третичных пара-
финовых С — Н-связей, так же как и олефиновых С — Н-связей, происхо-
дит сравнительно легко. При взаимодействии с олефиновым соединением
наиболее важной реакцией может оказаться присоединение к двойной связи.
Так, например, циклогексен, присоединяя метилен, образовавшийся при
•фотолизе диазометана, дает все возможные метилциклогексены и норка-
ран [286]
Некоторые сведения о механизме таких реакций присоединения можно
получить в результате исследования их стереохимии. Если создать такие
условия, в которых первоначально образующийся метилен мог бы вступать
в реакцию присоединения тотчас после своего образования, то реакция
была бы стереоспецифичной. Фрей [287] показал, что при фотолизе диазоме-
тана в растворе транс-бутена-2 при температуре —78 °C получающиеся
продукты имеют исключительно транс-конфигурацию, а при фотолизе
в растворе г^нс-бутена-2 — исключительно ^нс-конфигурацию
СН3 Н
9% 40%
СН3 сн3 сн3 СНз С2Н5 СНз
/ сн2 * \=cz с=с'
н СНз н н н
51%
СН3 СН3
Соотношение между продуктами превращения транс-бутена-2 было измерено
и позволяет сделать заключение, что аддендом служит синглетный метилен,
который вступает в реакции присоединения или включения в одну стадию
без образования какого бы то ни было промежуточного короткоживущего
комплекса, который мог бы изменить конфигурацию
875
Глава XIII
Противоположные результаты получаются при пиролизе диазометана в газо-
вой фазе в присутствии цис-бутена-2, сильно разбавленного аргоном [288].
Первоначально образующийся метилен будет испытывать большое число
столкновений с аргоном до того, как он прореагирует с бутиленом. В ходе
таких предварительных столкновений образовавшийся синглетный метилен
будет переходить в триплетное состояние. Установлено, что при 1000-кратном
разбавлении аргоном теряется вся стереоспецифичность реакций присоедине-
ния и включения. Кроме того, в этих условиях реакция замедляется кисло-
родом, как это часто бывает с радикальными реакциями. В результате реак-
ции образуются следующие продукты:
СН3 СН3
\=cZ -
сн2
сн3 сн3 С2Н5 сн3
\=сх \=с7
сн3 п7
4% 16%
сн3 с2н5 н
сн2=снс—сн3 X'q=c7
-Q7 ^СНз
10% 16%
сн3 сн3
27%
сн3 н
27%
Нестереоспецифическое присоединение по двойной связи можно интерпрети-
ровать исходя из того, что триплетный метилен присоединяется в две стадии
через промежуточный бирадикал, в котором происходят конформационные
изменения
СН2 + С = С —> СН2 —С —С —> Кольцо
Положение дел в реакции включения менее понятно. Существует мнение,
что триплетный метилен как таковой не участвует в реакции включения.
На самом деле он может участвовать в ней так же, как и синглетный метилен,
но менее легко. Если предположить, что триплет включается путем отщепле-
ния водорода (аналогично реакции Н+СН2) с конкурентной коллигацией
с образовавшимся таким образом мезомерный бирадикалом, то в процессе
включения должно изменяться положение и геометрия двойной связи
|сн2—С=С
CH2-f-Н—СН2—С=С —> СН3-• • < . —> Продукты
1СН2=С-С
Алкил- и диалкилкарбены самопроизвольно перегруппировываются, что
эквивалентно реакции внутримолекулярного включения. Включение карбена
в Р-СН-связь ведет к образованию олефина, а в у-СН-связь — к образованию
производных циклопропана [289], как, например:
СН2
СН3 СН3 /\
>СНСН —> >С = СН2 и СН3—СН —СН2
сн/ СН/
Дифенилкарбен, легко образующийся при пиролизе или фотолизе дифенпл-
диазометана, присоединяется к цис- и щраис-бутенам-2 нестереоспецифически
[290]. Смеси, получающиеся из этих изомеров, не одинаковы по своему соста-
876
Присоединение и обратные реакции
ву и должны образовываться по механизму, допускающему некоторые кон-
формационные изменения. Скелл и сотрудники предположили, что легкость
образования триплетного состояния дифенилкарбена такова, что обычно
происходящие реакции являются реакциями именно такого типа. Наблю-
даемые реакции присоединения замедляются кислородом, что вполне согла-
суется с таким предположением.
Карбалкоксикарбены, которые можно получить из алкилдиазоацетатов путем
их термического или фотолитического превращения, вступают в реакцию
циклоприсоединения к олефиновой двойной связи точно так же, как и сам
метилен. Так как в этом случае имеются заместители, в результате реакции
может получаться большее число стереоизомеров, чем из метилена. Так,
например, присоединение этилдиазоацетата к стиролу дает этил-^нс- и этил-
транс-1-фенилциклопропан-2-карбоксилат [291]. Однако если метилдиазо-
ацетат подвергнуть фотолизу в растворе цис- или транс-бутена-2, то полу-
чаются только изомеры циклопропана; в первом случае два изомера, во
втором — один изомер, которые сохраняют цис- или транс-расположение
метильных групп исходного олефинового соединения [292]. Присоединение
оказывается достаточно стереоспецифичным, и это позволяет сделать вывод,
что образующийся карбен реагирует в синглетном состоянии.
Дихлор- и дибромкарбены (СС12 и СВг2) обычно получают по методу Хайна
из галоформов путем 1,1-отщепления (гл. IX, разд. 7). Дёринг и Гофман [293]
показали, что эти карбены могут легко вступать в реакции циклоприсоеди-
нения к олефинам, если исключить конкурирующую реакцию их сольволиза
в формиаты, проводя реакцию в апротонных условиях. Так, например, при
добавлении хлороформа или бромоформа к суспензии mpem-бутилата калия
в растворе циклогексена получаются 7,7-дихлор- или 7,7-дибромноркараны
с хорошим выходом
Эти реакции были распространены на широкий круг олефинов. Реакции
включения не протекают, если в присоединяющем соединении имеется очень
слабая GH-связь, как в кумоле (бензильная СН-группа) [294], аллиловом
[295] или тиоаллиловом [296] эфире (а-СН2-группа). Установлено, что дибром-
карбен, полученный в растворе цис- или транс-бутена-2, присоединяет эти
олефины совершенно стереоспецифическим способом, характерным для при-
соединения метилена в подобных условиях [297]. Очевидно, дибромкарбен
образуется и реагирует в синглетной форме. Весткотт и Скелл [298] получали
дихлоркарбен путем пиролиза четыреххлористого углерода и хлороформа
при температуре 1500 °C и собирали продукты реакции на охлаждаемой жид-
ким азотом поверхности. Они установили присутствие дихлоркарбенов и при
добавлении этилена получили 1,1-дихлорциклопропен. Авторы этой работы
показали, что дихлоркарбен находится в синглетной форме, проведя
реакцию присоединения к цис-и транс-бутену-2. Оказалось, что стереоспеци-
фические характеристики реакции полностью отвечают синглетному состоянию
карбена.
Относительные скорости присоединения дибромкарбена к различным олефи-
нам были определены в сравнительных опытах. Получены следующие резуль-
877
Глава XIII
тэты [297, 299]:
(СН3)2С=С(СН3)2
3,5
Стирол
0,4
(СН3)2С=СНСН3 (СН3)2С = ССН2 Бутадиен
3,2 1 0,5
Циклогексен
Гексен-1
0,4 0,07
Из этих результатов можно сделать вывод, что дибромкарбен обладает сред-
ней электрофильностью, т. е. что его взаимодействие облегчается электро-
положительными заместителями в субстрате. Однако низкая реакционная
способность бутадиена и стирола в приведенном ряду является необычайной.
Хьюзген, Грейши и Зауер [279] рассмотрели это обстоятельство и установили,
что дибромкарбен не имеет общего заряда и может не иметь поля в направле-
нии олефинового субстрата, благодаря чему он не может возбудить электро-
мерный эффект (+7?) в винильном и фенильном заместителях бутадиена
и стирола, хотя и может вызывать электроположительный индуктивный
эффект (+7).
Реакция, Реймера — Тимана [300], в результате которой фенолы превра-
щаются под действием хлороформа и щелочи в орто- и иара-оксибензальде-
гиды, протекает, по-видимому, совершенно так же по механизму образования
на первой стадии дихлоркарбепа. Для случая салицилового альдегида реак-
ция записывается следующим образом:
о- О о- ОН
Последняя из написанных стадий представляет собой суммарное выражение-
гидролиза хлористого бензилидена (гл. VII, разд. 2). Хайн и ван дер Been
[301] показали, что фенолят-ион взаимодействует с хлороформом очень
медленно и что реакция Реймера — Тимана требует дополнительно присут-
ствия ионов гидроксила. Кинетика этого процесса определяется отмеченной
выше медленной стадией. В условиях проведения реакции соотношение
орто- и пара-изомеров равнялось 1,9, но было установлено, что оно зависит
от степени образования ионных пар.
Превращение первичного амина под действием хлороформа и щелочи в изо-
нитрил по Гофману [302] также почти определенно протекает через предва-
рительное присоединение дихлоркарбепа
СС12 + - i---> RN = C
RNH2-----> RNH2.CC12 —> RNHCHCla —
I___» RNHCHO
Это предположение было сделано Нефом [303]. Пока мы не располагаем кине-
тическим подтверждением, но было показано [304], что побочный продукт —
формамид образуется одновременно с изонитрилом, а не после образования
последнего.
Как отмечалось в гл. X, разд. 1,г, важнейшая роль ацилкарбенов состоит
в том, что они являются электронодефицитными промежуточными соедине-
ниями при перегруппировке Вольфа. Они могут получаться в результате
пиролиза или фотолиза а-диазокетона. При их перегруппировке образуется
кетен
О = С — CHN2 —» О = С —СН —> О = С = СН
I ! I
R R R
878
Присоединение и обратные реакции
Особый тип карбенов дает систему спектральных линий («группа 4050»),
которая впервые была обнаружена в спектрах комет. Позднее ее получили
в лабораторных условиях в разряде и пламени. Эти спектры были иденти-
фицированы Дугласом и другими как спектры линейной молекулы С3, т. е.
дикарбена углерода С — С = С [305]. Скелл и сотр. [306] показали, что пары
углерода, полученные в угольной дуге, состоят примерно на 60%
из С3. Другой важной составляющей является С2 — источник голубой
«полосы Свэна», которую может каждый увидеть в портативный спектро-
скоп. Присутствуют также атомы углерода и некоторое количество молекул
тяжелее С3.
Дикарбен углерода С3 можно собрать и хранить в течение нескольких дней
в неопентановых матрицах при температуре —196 °C. Если повысить темпе-
ратуру и добавить олефин, то произойдет циклическое присоединение по
одному или обоим концам молекулы дикарбена [306]. При этом может полу-
читься несколько продуктов, но при взаимодействии с изобутиленом реакция
идет особенно легко и дает только один продукт, идентифицированный как
бис-циклопропилиден
С(СН3)2 (СН3)2С. /С(СН3)2
С = С = С + 2|| -» | >С = С=С< |
СН2 н2с/ ХСН2
б. Присоединение олефинов с образованием четырехчленных циклов.
Штаудингер [307], открывший кетены, много раз наблюдал их способность
к цикло димеризации. Вообще говоря, олефиновая двойная связь одной моле-
кулы кетена может присоединиться либо к карбонильной, либо к олефиновой
двойной связи другой молекулы карбена. Если молекула самого кетена
димеризуется по первому способу, образуется Р-метилен~Р~пропиолактон.
Диалкилкетены димеризуются по второму способу с образованием цикло-
бутандиона-1,3. Моноалкилкетены обычно димеризуются обоими способами,
но образовавшийся дикетодимер почти полностью моноенолизуется с обра-
зованием енола средней кислотной силы, как уже говорилось в гл. II,
разд. 3,д. Примеры этих реакций приведены ниже:
Н2С=С=О
+
н2с=с=о
(СН3)2С = С = О
+
О = С=С(СН3)2
СН3НС —со
I I
СН3НС = С—о
Н2С—со
сн2=с-о
(СН3)2С-СО
I I
ОС-С(СН3)2
СН3НС—СО СН3С = С—оп
II т?- II
ОС-СНСН3 ОС—СНСН,
Вторая группа соединений получила большую известность благодаря своей
склонности к цикло димеризации. К ней относятся перфторолефины и сме-
шанные перфторхлоролефины. Родоначальник этого ряда тетрафторэтилен
легко димеризуется при температуре 200 °C в октафторциклобутан [308].
Хлортрифторэтилен димеризуется «голова к голове», но получающийся
1,2-дихлоргексафторциклобутан представляет собой смесь цис- и транс-
изомеров [309]. 1,1-Дихлор-2,2-дифторэтилен образует только 1,1,2,2-тетра-
хлортетрафторциклобутан, соединенный по принципу «голова к голове»
[310]. Гексафторпропилен димеризуется обоими способами:
F2C — cf2 С12С —cf2
2F2C = CF2 —* | | 2C12C = CF2—> | |
f2c—cf2 C12C—cf2
879
Глава XIII
C1FC — CF2
2C1FC = CF2 —> | | (цис и транс)
C1FC -CF2
f3chc-cf2
2FsCCF = CF2 —» | ] и
F3CHC-CF2
f3chc-cf2
I I
f2c—chcf3
Тетрафторэтилен присоединяется к пропилену [312]. Он также присоеди-
няется при более низкой температуре к бутадиену, но только путем 1,2-
циклоприсоединения с образованием производного циклобутана [312]
F2C = CF2 225 °C F2C-CF2 F2C = CF2 125 “С
+ > I I + *
Н2С=СНСН3 Н2С — снсн3 Н2С = СНСН = СН2
F2C — CF,
-> I г
F2C-CHCH = CH2
Однако циклопентадиен присоединяет тетрафторэтилен главным образом
тем же 1,2-способом и частично по Дильсу — Альдеру путем 1,4-циклопри-
соединепия [312]. Гексафторбутадиен димеризуется в две стадии, как пока-
зано ниже [313]:
160 °C F2C-CFCFCF2 200 “С f2c-cf-cf-cf2
2F2C = CFCF = CF2--> I -----> I I I I
f2c—cfcfcf2 f2c-cf- cf ~cf2
Неисследованным остается вопрос, почему кетены и в особенности полифтор-
этилены должны легко вступать в реакции 1,2-циклоприсоединения, приве-
денные выше. Можно задать вопрос, что общего в особенностях строения
объединяет эти два класса соединений? Ответ на этот вопрос можно дать,
рассмотрев такие случаи присоединения, в которых не участвуют указанные
соединения. Рассмотрим следующие примеры [314—316]:
8СН3
2СН2=СГ
\jN
C2H5OCH = CH2
(NC)2C=C(CN)2
№P6W-C4H9SCH = CH2
F3C CF3
\=C^
NC^ \?N
Комнатная
температура
---------->.
m/>em-C4H9S
CN
В этих примерах очень легко протекающих реакций циклоприсоединения
имеется два рода заместителей. Во-первых, сильные электроноакцепторные
заместители, которые могут или не могут сопрягаться с двойной связью
880
Присоединение и обратные реакции
(CN или CF3). Во-вторых, сильно электронодонорные заместители, которые
определенно сопряжены с олефиновой двойной связью (СН3О и CHgS).
Оба сорта заместителей могут присутствовать в каждой присоединяющейся
молекуле или в одной молекуле могут быть заместители одного рода, а в дру-
гой молекуле — другого. Суммарный эффект можно обозначить уже приня-
тыми символами — I, -\-К. Таким образом, карбонильный кислород кетена
является —I, + /^-заместителем: он индуктивно притягивает электроны
и отталкивает их по механизму сопряжения (гл. II, разд. 3,д, и гл. III,
разд. 1,е). Фтор в молекуле фторолефинов также является —I, Д-Х-замести-
телем: он не только наиболее сильный из всех галогенов индуктивный электро-
ноакцепторный заместитель, но и наиболее сильный из всех галогенов,
отталкивающий электроны по механизму сопряжения (гл. II, разд. 3,в и д).
Неожиданным кажется то, что полярные эффекты двух видов кооперируют
и ускоряют перенос электронов между атомами каждой пары атомов,
которые образуют связь, когда две двойные связи вступают в циклопри-
соединение.
Строение двух стереоизомеров, входящих в состав продуктов третьей из
приведенных выше трех реакций, показывает, что каждая из участвующих
в реакции двойных связей соединяется с другой путем ^пс-присоединения.
В том случае, когда вместо г^пс-дицианолефина в этой реакции используется
транс-изомер, получается исключительно вторая пара циклобутановых
изомеров, для чего снова должно произойти г^пс-^ис-присоединение [316].
Это указывает на то, что 1,2—1,2-циклоприсоединение может протекать
в одну стадию, и действительно было показано, что эти реакции не идут через
образование какого-либо промежуточного соединения с открытой цепью
и достаточным временем жизни для того, чтобы успели произойти конформа-
ционные изменения *.
Необходимо подчеркнуть, что влияние подходящих заместителей на ход
циклоприсоединения состоит лишь в ускоряющем эффекте, но наличие таких
заместителей не является абсолютно необходимым условием для протекания
реакции. Ранее мы уже отмечали некоторые случаи самопроизвольной диме-
ризации и другие быстропротекающие реакции циклоприсоединения высоко-
напряженных полиненасыщенных циклических углеводородов. Так, напри-
мер, циклобутадиен очень быстро димеризуется в трициклооктадиен и при-
соединяет производные ацетилена с образованием бициклогексадиенов
(гл. XI, разд. 3,г).
20 -> ЕШ П + III - ГЛ
Общая трудность классификации таких реакций с участием циклобутадиена
состоит в том, что мы не знаем, каким образом идет циклоприсоединение:
то ли по типу 1,2—1,2, то ли по типу реакции Дильса — Альдера (1,2—1,4).
Однако это не относится к случаю очень быстрой димеризации дегидробензо-
ла в дифенилен (гл. VII, разд. 2,г):
Многие олефиновые соединения, которые не могут димеризоваться под
влиянием температуры, вступают в реакцию циклодимеризации под влияни-
* Монтгомери с сотр. [317] высказал иную точку зрения, основываясь на стереохими-
ческих изменениях, сопровождающих 1,2-циклоприсоединение 1,1-дифтор-2,2-дихлор-
этилена к диенам.
56 — 0772
881
Глава XIII
ем фотохимического воздействия. В качестве примера углеводородов, реаги-
рующих под влиянием облучения в бензольном растворе, приведем аценафти-
лен [318]. Он образует два возможных стереоизомера:
Однако большая часть известных примеров относится к циклоприсоединению
сопряженных ненасыщенных карбонильных, карбоксильных и цианистых
соединений, а также хинонов. Так, например, при облучении халкона
в растворе хлороформа происходит его димеризация по так называемому
принципу «голова к голове»
Аналогичным образом ведет себя дибензилиденацетон С6Н5СН = СНСОСН =
= СНС6Н5 [319]. Большинство реакций фотодимеризации протекает по
типу «голова к голове», но все же некоторая часть идет по типу «голова
к хвосту». Простым сопряженным кетоном, самопроизвольно фотодимери-
зующимся по обоим направлениям, является циклопентенон [320]. В каждом
случае при димеризации образуется только один тпрянс-стереоизомер
Хотя мы и не располагаем определенными сведениями о механизме этих
реакций, можно быть уверенными в том, что геометрическое расположение
молекул реагентов относительно друг друга в тот момент, когда^происходит
передача электронов, является высоко упорядоченным. Об этом свидетель-
ствует тот факт, что фиксированное расположение молекул в кристаллическом
состоянии может либо уничтожать, либо создавать способность к фотодиме-
ризации, и в последнем случае можно ожидать образования соответствующих
продуктов реакции. Шмидт и сотр. [321] провели обширное исследование
этого явления. Для иллюстрации основного положения следует прежде
всего отметить, что если дибензилиденацетон легко фотодимеризуется
в растворе, то в твердом состоянии он устойчив к действию света. Можно
предположить, что реакция в твердом веществе не идет потому, что кристал-
лическая решетка сохраняет двойные связи в определенном положении
и не позволяет им прийти в нужное тесное расположение, необходимое для
фотореакции. Противоположную ситуацию можно проиллюстрировать на
примере тпрянс-коричной кислоты, которая, как известно, не фотодимери-
зуется в растворе, а в твердом состоянии легко образует оба димера — трук-
882
Присоединение и обратные реакции
силловую и изотруксилловую кислоты. Образование этих кислот зависит
от типа облучаемого кристалла: шранс-коричная кислота является диморф-
ной, и одна форма кристалла дает один димер, а другая форма — другой
димер. Можно допустить, что молекулы коричной кислоты в растворе не
образуют пар, геометрическая конфигурация которых могла бы обеспечить
достаточную скорость димеризации, тогда как в решетке кристалла все
молекулы в течение всего времени находятся в виде пар, геометрическая
конфигурация которых удовлетворяет требованиям димеризации.
Шмидт с сотр. [322] исследовали кристаллическую структуру и продукты
фотодимеризации не только щранс-коричной кислоты, но также большого
числа замещенных щранс-коричпых кислот. Большинство из них оказались
полиморфными. Все они имеют какую-нибудь одну или все три типа кристал-
лической упаковки, обозначаемые а, 0 и у. а-Упаковку имеет более устой-
чивая кристаллическая модификация незамещенной тпря/ic-коричной кисло-
ты. В ее кристаллах ближайшие двойные связи находятся на расстоянии
3,56 А (35,6-10-2 нм) от центра, а молекулярные пары расположены сим-
метрично относительно центра инверсии. В результате фотодимеризации
образуется а-труксилловая кислота
НС—СО2Н
Z
С6Н5-СН
НС—С6Н,
Z
но2с—сн
«.-Упаковка (схематически) а-Труксилловая кислота
0-Упаковку имеет менее стабильная из двух кристаллических форм транс-
коричной кислоты, и она характерна для более стабильной, или единствен-
ной, кристаллической формы некоторых замещенных кислот. Расстояние
между двойными связями здесь почти такое же, как и в а-упаковке, но моле-
кулярные пары оказываются связанными. Продуктом фото димеризации
в этом случае является изотруксилловая кислота
НС—СО2Н
С6Н5—ctf
НС—СО2Н
Z
с6н5—сн
р-Упаковка (схемати-
чески)
р-Труксилловая
кислота
Во всех случаях а- и 0-упаковки игранс-коричных кислот кратчайшее рас-
стояние между двойными связями лежит в пределах 3,6 . . . 4,1 А (3,6 • 10-1.. .
4,1 ПО-1 нм); все эти кристаллы дают фотодимеры, в которых имеется
либо центр симметрии, либо плоскость симметрии в соответствии с симметри-
ей а- или 0-упаковки. В третьем виде упаковки — у-упаковке, для которой
найдено всего несколько примеров, кратчайшее расстояние между двойными
связями несколько больше, чем в двух других видах упаковки, и составляет
4,7 . . . 5,1 А (4,7 ПО-1 ... 5,1 -10-1 нм). Эти кристаллы не образуют фото-
димеров.
56* 883
Глава XIII
Аналогичная, правда менее полная, картина была установлена [323] для
некоторых замещенных г/ас-коричных кислот и хинонов. Полученные данные
подтверждают то, что для осуществления циклической фотодимеризации двой-
ные связи должны находиться па расстоянии в пределах 4,0 ± 0,4 А
(0,4 ± 0,04 нм) друг от друга, и, если это условие выполняется, будут обра-
зовываться димеры с сохранением общей симметрии пар соединяющихся
мономеров.
в. Присоединение 1,3-диполей с образованием пятичленных колец.
Некоторые из этих реакций присоединения к олефинам имеют свою историю,
хотя ранее они считались свойственными лишь ограниченному кругу веществ.
Число таких реакций было значительно расширено лишь совсем недавно,
благодаря исследованиям Хьюзгена и сотр. [324]. Адденд для 1,3-цикло-
присоединения должен иметь формальный заряд и, кроме того, может иметь
(и обычно имеет) ненасыщенную группу. Только с гетероатомами азота
и кислорода существует несколько десятков 1,3-аддендов. Некоторые из этих
соединений можно легко выделить, а другие малоустойчивы. Однако соеди-
нения этих типов часто можно получить в присутствии олефиновых соедине-
ний, которые реагируют с ними, не допуская их полимеризации или раз-
ложения.
Давно известными аддендами такого рода являются диазоалканы. Централь-
ный атом их трехатомной ненасыщенной системы обладает псевдооснов-
ностыо и, следовательно, будет делить свой положительный заряд с одним
из двух концевых атомов, в то время как на другом концевом атоме будет
сосредотачиваться такой же по величине отрицательный заряд. Таким обра-
зом, концевые атомы системы будут иметь противоположные по знаку заряды,
причем заряд может распределяться одним из двух возможных способов:
+ +
R.C-fe=N
Эта особенность присуща всем ненасыщенным 1,3-аддендам.
В результате присоединения диазоалканов к ненасыщенным сложным эфи-
рам получаются производные пиразолина, которые легко пиролизуются
с потерей азота и образованием производных циклопропана. Это было впер-
вые показано Бюхнером [325]. Присоединение метилдиазоацетата к метило-
вым эфирам малеиновой и фумаровой кислот дает один и тот же продукт —
А2-пиразолин. Первоначально должны получаться А1-пиразолипы, однако
последующее прототропное превращение приводит к тому, что все стерео-
специфические признаки, отличающие продукты присоединения к эфиру
малеиновой кислоты от продуктов присоединения к эфиру фумаровой кис-
лоты, теряются.
Н
N N
CH3OOCCH = N = N СЩООССН \ СН3ООССН \
+ * I I * I II
СН3ООССН = СНСООСН3 СН3ООССН — СНСООСНз СН3ООССН —ССООСНд
Стереохимия этих реакций была исследована Ауверсом [326] на примере
присоединения диазометана к метиловым эфирам диметилмалеиновой и диме-
тилфумаровой кислот. Автор получил изомерные А^пиразолины, в кото-
рых сохранилось первоначальное расположение цис- и транс-конфигураций
карбоксильных групп. Это свидетельствует о присоединении диполя по типу
884
Присоединение и обратные реакции
г^нс-присоединения, что согласуется с идеей об одностадийное™ процесса
присоединения.
Против альтернативного двухстадийного механизма веские аргументы
выдвинули Хыозген, Грейши и Сойер [279]. Аналогией для такого механиз-
ма могла бы быть гидролитическая и другие реакции диазоалканов, которые,
как полагается, инициируются путем присоединения протона к углеродному
атому. Если бы диазоалканы реагировали подобным образом, т. е. действо-
вали бы как нуклеофильные агенты и присоединялись бы к одному из угле-
родных атомов олефина на первой стадии двухстадийного процесса цикло-
присоединения, то в качестве промежуточного продукта получался бы али-
фатический ион диазония, который, конечно, никогда не смог бы удержать
азот, необходимый для образования пиразолина. Известно, что алифатиче-
ские ионы диазония настолько неустойчивы, что, например, при образо-
вании из первичных аминов и азотистой кислоты они совсем не имеют «сво-
бодного времени жизни», а теряют азот и превращаются в ионы карбония
(гл. X, разд. 1,6).
Хотя реакции присоединения почти наверняка идут в одну стадию, скоро-
сти их таковы, что можно предположить, что диазоалкановые адденды дей-
Таблица 183
Константы скорости реакций присоединения дифенилдиазометана к олефинам
в диметилформамиде при 40 °C по данным Хьюзгена (к2, л-моль-1-с-1)
Олефин 105 fe2 транс-Олефин 1 0^ /<2 цис-Олефины 105 k2
(С6Н5)2С = СН2 0,27 Этиловый эфир ко- ричной кислоты 1,25 Малеиновый 'ангид- рид 5830
С6Н5СН = СН2 1,40 Этиловый эфир кро- тоновой кислоты 2,46 Этиловый эфир кро- 1 тоновой кислоты 0,95
N = ССН = СН2 484 Диметиловый эфир мезаконовой кис- лоты 1,35 Диметиловый эфир цитраконовой кислоты 1,65
СвН5ООССН = СН2 707 Диметиловый эфир фумаровой кисло- ты 2450 Диметиловый эфир малеиновой кис- лоты 68,5
885
Глава XIII
ствуют преимущественно как нуклеофильные агенты. Данные Хыозгена
и сотр. [327], приведенные в табл. 183, показывают, что скорость реакции
присоединения возрастает при введении в молекулу олефина электроноакцеп-
торпого заместителя. Однако наблюдаются и отклонения от общего правила:
не совсем обычным является соотношение между скоростями реакции этил-
акрилата и акрилонитрила; казалось бы совершенно очевидным, что при
переходе эфирных групп из транс- в ^пс-положение скорости реакций долж-
ны заметно уменьшаться. Полностью объяснить эти отклонения из общего
правила не удается.
Одпако преимущественно нуклеофильные свойства диазоалканов в реакциях
присоединения к олефинам подтверждаются ориентацией при присоедине-
нии к а,р-ненасыщенным сложным эфирам, нитрилам и нитросоединениям.
Ауверс и Унгермах [328] установили, что этиловый эфир диазоуксусной
кислоты и этиловый эфир тиглиновой кислоты присоединяются с образова-
нием Д1-пиразолина; диазометан и нитрил коричной кислоты образуют
А2-пиразолин. при этом первоначально образовавшийся аддукт испытывает
прототропное превращение [328].
С2Н5О2ССН = N = N
+
СН3СН = С(СН3)СО2С2Н5
N
н\ / X
С N
С2Н5О2с/ \ /
СН3 —С —С —СО2С2Н5
Н СНз
NH
ch2=n = n
c6h5ch = chcn
\
С6Н5-С-С
~Н CN
Пархам и Хасек [329] установили, что диазометан и 1-нитропропилен дают
Д1-аддукт, который можно выделить и который легко теряет азотистую
кислоту и превращается в 4-метилпиразол
N NH
CH2 = N = N / / '\
+ Н2С N НС N
CH3CH = CHNO2 \ / \ z
с—с с—сн
/I |\ /
СНз Н Н NO2 СНз
Положительно поляризованный p-углеродный атом сопряженного эфира
нитрила или нитросоединения всегда присоединяет углеродный атом диазо-
алкановой системы, как будто последний несет преимущественно отрица-
тельный заряд. Поэтому в качестве эффективной формулы диазоалкана
следует принять правую из двух формул, приведенных на стр. 884.
Нуклеофильные свойства диазоалканов еще раз подтверждаются кинетиче-
скими эффектами, возникающими при введении в молекулу электроноакцеп-
торных групп. Эти группы должны уменьшать реакционную способность
электронов диазоалканов и уменьшать скорость их присоединения. Это
подтверждается данными Хыозгена и Юнга по значениям констант скоростей
реакций различных диазоалканов с одним и тем же гетероциклическим алке-
886
Присоединение и обратные реакции
Таблица 184
Константы скоростей реакций присоединения диазоалканов к этиловому эфиру
5,6-диазонорборнен-5,6-дикарбоновой кислоты в диметилформамиде
при 60 °C по данным Хыозгена и Юнга (fc2, л-моль-1-с-1)
Диазоалкан 105fe2 Диазоалкан 1 05fe2
h2cn2 9700 C6H5COCHNH2 1,8
(CeH5)2CN2 102 n-NO2C6H4COCHN2 0,85
c2h5oocchn2 9,3 CeH5COCN2COCH3 <0,1
ном (неопубликованные экспериментальные данные Хыозгена и Юнга
цитируются по статье [279], стр. 833; табл. 184).
Присоединение азидов к олефиновым соединениям следует общим закономерно-
стям реакций присоединения диазопарафинов. Можно предположить, что ази-
ды вступают в реакцию в одной из следующих крайних структур:
RN===N=N
RN—N=N
В результате присоединения образуются триазолины, которые легко пиро-
лизуются с потерей азота. При пиролизе получаются азиридины и азометины,
что можно показать на примере реакции присоединения фенилазида [330—332]:
0 н хг—хг—хг
100ос C6H5N CjHjN
—> / \ и II
С6Н5Н----Н2 С6Н5С-СНз
c6h5n=n=n n
C6H5CH=CH2 \в___4/
с6н5н н2
c6h5n=n=n
СН2=СНСО2СНз
^НСО2СН3
887
Глава XIII
Скорость отщепления азота от продуктов присоединения ацилазидов. таких,
как бензазид, сравнима со скоростью их образования. В результате присоеди-
нения бензазида к норборнену получаются бензоилазиридин и оксазолин
[332 ]
Фенилазид присоединяется к норборнену с экзо-стороны. Аналогично идет
присоединение к фенхену, но не к апоборнену [330, 333]
Из норборнена Из фенхена.
Апоборнен
Двойная связь норборнена в этом случае чрезвычайно реакционноспособна,
так же как и в большинстве других реакций присоединения. Обычно это
объясняют наличием в циклической структуре байеровского напряжения
(короткий мостик сжимает боковые углы кольца). Непонятно, почему не про-
исходит энЭо-присоединения даже несмотря на то, что в результате экзо-
присоединения метильная группа фенхена может переместиться в эндо-
положение, и несмотря на стерические затруднения (которые можно пред-
положить) экзо-присоединению к апоборнену. Безусловно, в этих примерах
необходимым является щгс-присоединение. В настоящее время не известно,
могут ли происходить конформационные изменения в промежуточном соеди-
нении двухстадийного процесса присоединения, если оно протекает как
цнс-вз аимо действие.
Направление присоединения азида к несимметричным замещенным олефи-
нам было проиллюстрировано выше на примере присоединения фенилазида
к стиролу и этиловому эфиру акриловой кислоты. Эти направления противо-
положны друг другу: фенильный заместитель стирола оказывается в поло-
жении 5, а эфирная группа этилакрилата — в положении 4. Эти результаты
показывают, что из двух мезомерных формул азида, приведенных выше,
большее значение в реакциях циклоприсоединения имеет первая, в которой
отрицательный заряд находится у а-азотного атома. ; Здесь наблюдается
аналогия с диазоалканами, которые присоединяются к сопряженным нена-
сыщенным карбонильным соединениям таким образом, как если бы они
имели отрицательный заряд на атоме углерода.
Хьюзген и сотр. [332] измерили скорости присоединения фенилазида к неко-
торым непредельным соединениям. Полученные ими данные приведены
в табл. 185. Здесь снова проявляется заметное влияние пространственного
888
Присоединение и обратные реакции
строения ненасыщенного субстрата, а также полярной активации электро-
отрицательными заместителями, которая, правда, более слабая, чем в реак-
циях присоединения диазоалканов.
Рассмотрим в качестве третьего примера полярного 1,3-присоединения при-
соединение окисей нитрилов. Общая ненасыщенность этих соединений
Таблица 185
Константы скорости (fc2, л моль-1-с-1) присоединения фенилазида
к непредельным соединениям в четыреххлористом углероде
при 25 °C (по данным Хьюзгена)
Непредельное соединение 107Й.2 Непредельное соединение 10
Гептеи-1 0,24 Диэтилфумарат 8,36
Стирол 0,40 Циклопентен 1,86
Этилакрилат 9,85 Норборнен 188
Этилкротонат 0,27 Диметил малеат 0,34
такая же, как и в первых двух примерах, по ее распределение, как это видно»
из приводимых ниже мезомерных формул, другое:
+
RC==N—О
RC=N
=0
Окись бензонитрила была впервые выделена Виландом [334], который полу-
чил ее методом, ставшим теперь общепринятым. Метод состоит в обработке
соответствующего хлорангидрида гидроксамовой кислоты третичным ами-
ном, который отщепляет элементы соляной кислоты. Выделение окиси нит-
рила представляет собой довольно трудную задачу, так как она очень легко
димеризуется в окись 4,5-дифенилфуразана
z.NOH r3n
с6н5с< -—
С6Н5С = N —О
N
Z \
С6Н5С о
I I
CeH5C=N-O-
Исследование реакции присоединения окисей нитрилов к непредельным
соединениям было бы сильно затруднено из-за упомянутой выше тенденции
окисей нитрилов к димеризации, если бы не возможность их проведения
in situ путем медленного прибавления третичного амина к хлорангидриду
гидроксамовой кислоты. При этом окись нитрила образуется в малой кон-
центрации и тут же связывается непредельным соединением, присутствую-
щим в высокой концентрации. Исчерпывающее объяснение реакций цикло-
присоединения окисей нитрилов дано главным образом Килико и сотр. [335].
Они использовали как выделенные окиси нитрилов, так и в значительно’
большей степени полученные in situ. Ниже приведено несколько примеров
889
Глава XIII
получения изоксазолинов:
Показано [336], что присоединение окиси бензонитрила к метиловым эфирам
малеиновой и фумаровой кислот протекает строго стереоспецифично. Это
служит наиболее простым и сильным аргументом в пользу теории односта-
дийного присоединения. Найдено [337], что окись ацетонитрила присоеди-
няется к этиленам, имеющим только один заместитель, таким образом,
что этот заместитель занимает положение 5 в изоксазоло
СНг=СНХ
CHsCsN-()
С6Н5
X = CN
OC4U9-H
Имеются причины, основанные на аналогии (см. ниже), для того, чтобы
считать, что такая независимость ориентации от полярности не будет сохра-
няться в случае этиленов, замещенных на обоих концах этиленовой цепи.
Однако отметим, что опыты, подтверждающие приведенные данные, до сих
пор не описаны в литературе.
Хыозген и сотр. [338] применили метод конкурирующих реакций для изме-
рения относительных скоростей присоединения окиси бензонитрила к ряду
непредельных соединений. Некоторые результаты их исследования при-
ведены в табл. 186. Они в общем сходны с результатами, полученными для
Таблица 186
Относительные скорости присоединения окиси бензонитрила
к непредельным соединениям в эфире при 25 °C (по данным Хыозгена)
Непредельное соединение Относитель- ная скорость Непредельное соединение Относитель- ная скорость
Гексен-1 Стирол Этилакрилат Винил-н-бутиловый эфир т рянс-Стильбен Этилкротонат 2,6 9,3 66 15 0,24 1,00 Диметилфумарат Циклогексен Циклопентен Норборнол Диметилмалеат 94 0,055 1,04 97 1,61
890
Присоединение и обратные реакции
реакции фенилазида и приведенными в табл. 185: наблюдается сильная зави-
симость скорости реакций от пространственного строения непредельного
субстрата и слабое ускоряющее влияние сопряженных сложноэфирных
групп, а также в данном случае и сопряженных простых эфирных групп.
В качестве представителя класса 1,3-диполярных аддуктов, имеющих более
низкую общую ненасыщенность, чем рассмотренный только что класс соеди-
нений, можно рассмотреть циклоприсоединение нитронов (окисей азометана,
называемых также N-эфирами оксимов). Система связей в них похожа
на систему связей в окисях нитрилов, за исключением того, что ненасыщен-
ность системы снижается при наличии дополнительных заместителей. Для
этих соединений можно написать следующие мезомерные формулы:
RR'C
=NR"—О
Такие вещества присоединяются к непредельным соединениям с образова-
нием изоксазолидинов, как, например [339]:
с6н5
C6H5CH=N+—о-
90°С
СН2=СНСвН5
С6Н5
I
ХК
С6Н5Н6 10 (цис и транс)
V___J
Н2 НС6Н5
Необходимые для реакции нитроны обычно получают из альдегидов и N-алкил-
или N-арилгидроксиламинов; иногда более удобно не выделять их из реак-
ционной смеси, а проводить реакцию в присутствии избытка непредельного
соединения, к которому будет присоединяться нитрон. Так, например,
w-бутиральдегид и N-фенилгидроксиламин, конденсируясь в присутствии
стирола, образуют 2,5-дифенил-З-н-пропилизоксазолиден [337].
Циклические нитроны — весьма удобные реагенты [340], так как в целом
они более реакционноспособны, чем нитроны с открытой цепью. Это объяс-
няется, по-видимому, тем, что циклические нитроны не могут иметь иной
конфигурации, кроме цис, тогда как нитроны с открытой цепью обычно
имеют транс-строение. Грейши показал, что окись 3,4-дигидроизохинолина
присоединяется к метилмалеату и к метилфумарату совершенно стереоспе-
цифично, и это послужило веским аргументом в пользу одностадийного меха-
низма присоединения*:
СНСО2СН3
II
СНСО2СН3
/О —СНСО2СН3
— I
\---СНСО2СН3
Исследовано влияние заместителей в ненасыщенном соединении на направ-
ление реакции присоединения [339]. Если двойная связь ненасыщенного
соединения находится в концевом положении, как, например, в СН2 =
= СНСвН6, СН2 = СНСООС2Н5 и СН2 = С(СН3)СООСН3, то заместитель
оказывается в положении 5 изоксазолидина. Если же оба конца этиленовой
структуры имеют заместители, как, например, в СН3СН = СНСООС2Н5
* Неопубликованные данные, см. работу [279], стр. 863.
891
Глава XIII
и (СН3)2С = СНСООС2Н5, то эфирная группа оказывается в положении 4.
Именно в этом положении и должна находиться сложноэфирная группа,
если учитывать только полярные влияния и если считать, что, подобно ранее
рассмотренным аддендам, нитрон ведет себя как нуклеофильный реагент.
Таблица 187
Константы скорости (к2, л-моль-1-с-1) присоединения С-фенил-Х-метилпитропа
к непредельным соединениям в толуоле [341]
Непредельное соединение 10ё/<2 (120 °C) Непредельное соединение 105h2
85 °C 120 °C
Гептеп-1 2,(54 Метилакрилат 44,0
Циклопентен 0,82 Метилкротонат 3,96 36,4
Норборнен 4,00 Диметилфумарат 72,5
«-Метоксистирол 8,7 Диметилмалеат 24,7
Стирол 11,7 Диметилмезаконат 25,8
«-Нитростирол 51 Диметилцитраконат 3,17 13,4
Большое количество данных по константам скорости присоединения С-фенвл-
N-метилнитрона к различным олефиновым соединениям получено Сейдлом
[341]. Некоторые из них приведены в табл. 187. Эти данные свидетельствуют
о намного более слабой зависимости скоростей реакций от стереохимического
строения присоединяемого непредельного соединения, чем это наблюдается
при присоединении окисей нитрилов. Возможно, менее ненасыщенная систе-
ма оказывается более гибкой. Может быть, главным образом поэтому кине-
тические полярные эффекты проявляются более четко при присоединении
нитронов, чем в случае присоединения окисей нитрилов. Полярные эффекты,
очевидно, свидетельствуют о том, что нитроны проявляют свойства нуклео-
фильных агентов.
Помимо рассмотренных выше четырех типов 1,3-диполей, Хьюзгеп и сотр.
[279] исследовали реакции циклоприсоединения для ряда аналогичных
систем: нитрилиминов, азометиниминов и нитрилилидов.
г. 1,4-Присоединение диенов с образованием шестичленных циклов
[342]. Эта реакция называется реакцией Дильса — Альдера и стала широко
известной благодаря большому значению для синтетической химии. Под
диеном в этом случае подразумевается сопряженный диен, способный при-
соединяться своими 1- и 4-положениями к молекуле другого этиленового
соединения, которое называют диенофилом. В молекуле диенофила вместо
этиленовой группы аналогичную роль может играть ацетиленовая группа.
В случае этиленового диенофила образуется циклогексен, в случае ацетиле-
нового диенофила — циклогексадиен-1,4. При наличии в диенофиле или
диене или как в том, так и в другом дополнительных активных ненасыщенных
групп может произойти дальнейшая циклизация и аддукт окажется поли-
циклическим. Это возможно также в случае некоторых ароматических
систем, содержащих 1,3-бутадиеновые связи большой степени ненасыщен-
ности, как, например, в фурановом кольце и центральном кольце антрацена
в показанных ниже примерах. Ряд ароматических систем может предостав-
лять одну из двойных связей 1,3-бутадиеновой системы; такие примеры рас-
смотрены ниже. Некоторые ароматические системы могут предоставлять,
даже диенофильную двойную связь, как в показанном ниже примере 0-наф-
892
Присоединение и обратные реакции
тола. Приведенные ниже схемы иллюстрируют типичные случаи диенового
синтеза:
Отдельные примеры присоединения диенов были известны и описаны в лите-
ратуре еще в прошлом веке. Так, Цинке [343] открыл димеризацию тетрахлор-
циклопентадиенона и позднее правильно объяснил ее характер. В текущем
столетии Эйлер и Джозефсон [344] определили природу продуктов присоеди-
нения изопрена к и-бензохинону. Однако между этими наблюдениями не уста-
навливалась какая-либо связь, пока в 1928 г. и последующих годах Дильс
и Альдер [345] не показали разнообразные возможности этой реакции.
Присоединение диенов во многих отношениях стереохимически специфично
[346]. Во-первых, по отношению к диену протекает всегда только цис-при-
соединение, на что указывает циклическая структура продуктов реакции,
г. е. диен должен реагировать в синперипланарной конформации централь-
893
Глава XIII
ной формально простой связи. Далее, по отношению к диенофилу также
протекает грс-присоединение. Опытным путем показано, что при двух заме-
стителях в диенофиле в положениях цис или транс по отношению к этилено-
вой связи эти заместители после присоединения сохраняют такое же цис-
или /прайс-расположение по отношению к образовавшемуся алицикличе-
скому кольцу. Так, аддуктами малеинового ангидрида являются ангидриды
циклических цис-1,2-дикарбоновых кислот, а аддуктами фумарилхлорида —
хлорангидриды транс-1,2-дикарбоновых кислот. Далее, конфигурация заме-
стителей диена сохраняется такой же и в аддукте [347]. Так, например,
присоединение малеинового ангидрида к транс-транс-1,4-дифенилбутадие-
пу дает производное циклогексена, в котором фенильные группы занимают
г/нс-положение относительно друг друга
Н н
Согласно этим правилам, носящим имена Альдера и Штайна, происходит
^пс-присоединение реагентов, которые в этом случае сохранят свою
собственную стереохимическую структуру и в аддукте. Исключений из этих
правил не найдено. Следует отметить также, что группы, находившиеся
ранее в диенофиле, в аддукте могут быть направлены либо в сторону двойной
связи, образовавшейся первоначально в диеновой части аддукта, либо
в противоположную сторону от двойной связи. Первый случай обычно
называют эндо-положением, второй — экзо-положением. Наблюдается зако-
номерность, заключающаяся в том, что карбонильная, карбоксильная и дру-
гие ненасыщенные группы, находящиеся в диенофиле, ориентированы обыч-
но исключительно в сторону эндо-аддукта. Ниже приводится схема при-
соединения малеинового ангидрида к циклопентадиену; процесс протекает
с образованием исключительно эндо-аддукта.
эндо
894
Присоединение и обратные реакции
То же правило эндо-присоединения справедливо и для диенов с открытой
цепью, примером чего служит приведенная выше реакция присоединения
малеинового ангидрида к транс-транс-i ,4-дифенилбутадиену. Фенильные
группы занимают не только цпс-положение относительно друг друга, но
и цпс-положение относительно ангидридной группы, которая поэтому должна
ориентироваться в сторону образующейся в процессе присоединения этиле-
новой связи.
Правило эн<Эо-присоединения, однако, имеет ограничения. Первое ограниче-
ние состоит в том, что это правило относится к продуктам, образование
которых определяется кинетическими факторами, т. е. к продуктам, обра-
зующимся по механизму одностадийного необратимого присоединения.
Однако реакция Дильса — Альдера является обратимой и равновесие,
к которому она будет стремиться и которое устанавливается достаточно долго
и при довольно высокой температуре, будет определяться уже термодинами-
ческими факторами, так что продукты равновесной реакции могут и не быть
идентичными первоначально образовавшимся.
Однако со всеми поправками правило эшЭо-присоединения при образовании
первоначального аддукта имеет такие серьезные исключения, что правилом
его до сих пор называют только по историческим причинам [3481- В 1948 г.
Вудвард и Беер установили, что фуран с малеиновым ангидридом в эфиро
образуют экэо-аддукт. Исследование этой быстро протекающей и легко-
обратимой реакции было многократно повторено с целью идентификации
продуктов присоединения, образующихся первоначально, т. е. до начала
протекания обратной реакции. Анет установил, что эти первоначальные
продукты состоят на 33% из эндо- и на 67% из экзо-изомера. При проведении
реакции присоединения фурана к малеиновой кислоте в воде Вудвард и Беер
получили противоположные результаты: главным продуктом оказался
эшЭо-аддукт. Позднее Анет установил, что продукты, образующиеся при
кинетическом контроле этой реакции, содержат 75% эшЭо-аддукта и 25%
экзо-изомера. Если выделение продуктов реакции проводить медленно,
например в течение недели, при комнатной температуре, то можно получить
почти эквивалентную смесь обоих изомеров. Вероятно, доля экзо-изомера
при равновесии существенно превышает 25%.
Альдер и сотр. [349] нашли другие исключения из эшЭо-правила при присоеди-
нении а,|3-ненасыщенных мононитрилов и монокарбоксиэфиров с открытой
цепью к циклопентадиену. Так, например, присоединение к этому диену
акрилонитрила при температуре 100 °C дает, по-видимому, в качестве пер-
вичного продукта почти одинаковые количества эндо- и экзо-аддуктов. Бер-
сон с сотр. [349] количественно исследовали большое число таких реакций
присоединения. Некоторые результаты их исследования приведены
в табл. 188. Показано, что соотношение изомерных продуктов реакции,
образование которых зависит от кинетических факторов, определяется
в первую очередь природой диенофила и в незначительной степени природой
растворителя, а также температурой. Правило экЗо-присоединения в этом
случае играет незначительную роль. Присоединение метилакрилата подчи-
няется эшЭо-правилу во всех растворителях, метил-а-метакрилат в любых
растворителях не подчиняется ему, а метил-кгранс-кротонат занимает про-
межуточное положение. Во всех случаях переход к более полярному раство-
рителю увеличивает долю эшЭо-изомеров в продуктах, образование которых
определяется кинетическими факторами. Это свидетельствует о том, что
переходное состояние при образовании эшЗо-аддукта более полярно, чем
переходное состояние, приводящее к образованию экзо-изомера. Темпера-
тура обычно влияет так, как если бы энтальпийный вклад в изменение сво-
895
Глава XIII
Таблица 188
Содержание .эндо- и экзо-изомеров в продуктах присоединения
диенофилов к циклопентадиепу при кинетическом контроле (по данным Берсона с сотр.)
Раствори- тель сн2 = CHCOOCH3 СН2 С(СНз)СООСНз транс-СНзСН = СНСООСНз
°C эндо, % ЭКЗО, % °C эндо, % ЭКЗО, % °C эндо, % ЭКЗО, %
Декалин -35 80,3 19,7 56 33,0 77,0 3 52,6 47,4
170 74,5 25,5 139 38,2 61,8 66 51,3 48,7
CH3NO2 3 84,6 15,4 3 30,5 69,5 3 62,0 38,0
66 81,4 18,6 66 34,4 65,5 66 57,2 42,8
СН3ОН 0 89,8 18,2 0 41,5 58,5 3 66,4 33,6
56 84,9 15,5 56 40,7 59,3 66 65,0 35,0
бодной энергии активации образования изомеров был больше вклада энтро-
пийного, т. е. увеличение температуры обычно увеличивает долю того изо-
мера, который образуется в меньшем количестве. Различия в значениях
ZAS* и АЯ+ образования двух изомеров были определены из температур-
ных коэффициентов и оказались во всех случаях очень небольшими,
менее 1 ккал/моль (4,187• 103 Дж/моль).
эн<Эо-Правило было первоначально установлено для диенофилов, имеющих
ненасыщенные заместители, которые входили затем и в состав аддукта. Оно
было сформулировано как правило «максимального накопления ненасы-
щенности» в аддукте. Различные объяснения этого правила, которые были
предложены, исходили из предположения о наличии несвязывающего или
вторично связывающего взаимодействия между ненасыщенными замести-
телями (такими, как ангидридная группа малеинового ангидрида), также
входящими в аддукт, и двойной связью диенового остатка, возникающей
при образовании аддукта. Было предложено объяснение, состоящее в посту
лировании несвязывающих взаимодействий, которые включают электро-
кинетическое притяжение между простой и двойной связями в переходном
состоянии образования энЗо-аддукта. Для того чтобы обеспечить преимуще-
ственную энЗо-ориентацию в таком процессе, вовсе не обязательно, чтобы
ненасыщенность при образовании аддукта возникала в обоих партнерах
по присоединению. Известно, например [350], что циклопентен, являющийся
диенофилом, в котором не остается ненасыщенности при образовании аддук-
та, при 200 °C реагирует с циклопентадиеном почти исключительно путем
энЗо-присоединения. Этот процесс наверняка контролируется кинетикой,
так как 200 °C — это самая низкая температура, при которой присоединение
идет со скоростью, достаточно высокой, чтобы ее можно было измерить.
Нашими знаниями кинетики диенового синтеза и обратного ему расщепле-
ния мы обязаны главным образом Вассерману [351]. Полученные им резуль-
таты использованы для составления табл. 189; во-первых, термодинамически
диеновый синтез обратим и экзотермичен, и, следовательно, увеличение
температуры смещает равновесие в сторону диена и диенофила. На примере
трех реакций показано, что — А// примерно равна 17—18 ккал/моль.
Кинетически диеновый синтез является почти уникальной реакцией по лег-
кости, с которой может быть изучена скорость ее протекания в обоих на-
правлениях в гомогенных условиях — как в газовой фазе, так и в растворах
896
Присоединение и обратные реакции
Таблица 189
Параметры Аррениуса для присоединения диенов и обратной реакции расщепления
(по данным Вассермана) аддуктов диенового синтеза
Реагенты Растворитель Присоединение Расщепление
Ig л2 Е2 интервал, °C Ig Е1 интервал, °C
Циклопентадиен-)- +бензохинон С2Н5ОН СНзСООН 7,0 7,5 12,7 11,0 2-30 18-30
Циклопентадиен-]- + циклопента- диен С6н6 cs2 С0н6 cs2 6,5 4,0 6,1 5,7 11,6 8,5 16,4 17,7 8-50 3-39 12-35 0—22 12,6 29 55-79
Парафиновый углево- дород 7,1 17,4 0-17 13,0 34,2 135-175
ЦиклопентадиенЦ- + акролеин В газовой фазе OeHg 6,1 6,1 16,7 13,7 80-150 6—77 13,1 35 27-111
Изопрен-{-акро- леин Бутадиен -{-акро- леин Бутадиен + крото- новый альдегид В газовой фазе То же » » » » 6,2 6,0 6,2 6,0 15,2 18,7 19,7 22,0 108—120 218—333 155—333 242—299 12,3 33,6 102—242
неполярных или полярных растворителей. Как показано в табл. 189, диеновый
синтез в гомогенной газовой фазе протекает как реакция второго порядка:
Скорость присоединения = fc2 [Диен] [Диенофил]
Это же уравнение характеризует скорость реакции в растворе. Расщепление
аддукта в гомогенной газовой фазе протекает как реакция первогощорядка:
Скорость расщепления = к± [Аддукт]
Это же уравнение описывает реакцию в растворе. Зависимость констант
скоростей реакций к2 и Е от температуры Т можно представить в следующем
виде:
к. = А2е-Е^т и к{ = Aie-E^ET
где А2 — в л-моль-1 -с-1, А< — в с-1, Е2 и Е^ — в ккал/моль.
Как видно из данных табл. 189, значения аррениусовских энергий актива-
ции большинства реакций присоединений диенов, включая реакции в газо-
вой фазе и в растворах, лежат в пределах 10...20 ккал/моль (41,87-103 . . .
83,74-103 Дж/моль), а соответствующие предэкспоненциальные множи-
тели примерно в 105 раз меньше, чем в случае обычной скорости соударений
при газовых реакциях. Низкие значения этих величин показывают, что
для образования переходного состояния необходима чрезвычайно специфи-
ческая взаимная ориентация диенов и диенофилов. Значения аррениусов-
ских энергий активации при расщеплении аддуктов находятся в пределах
30...35 ккал/моль (125,60-Ю3...146,54-Ю3 Дж/моль). Предэкспоненциаль-
57-0772
897
Глава XIIT
ные множители в случае реакций либо в газовой фазе, либо в растворах
характеризуются такими же величинами, как и при обычных реакциях
первого порядка в газовой фазе. Ясно, что молекула аддукта не должна.
принять необычную конфигурацию перед возбуждением ее для образования
переходного состояния. Такое сопоставление указывает на то, что переходное
состояние по своей геометрической форме в общем напоминает конфигура-
цию аддукта.
Значения скоростей и параметров Аррениуса для присоединения диена
и расщепления аддуктов диенового синтеза почти одинаковы при взаимодей-
ствии как в газовой фазе, так и в неполярных растворителях. Это показывает,
что степень полярности переходного состояния не очень отличается от поляр-
ности исходных реагентов или от полярности продукта реакции; это пока-
зывает также, что влияние наведенных диполей может вызывать значитель-
ное различие в сольватации. Однако при взаимодействии в неполярных,
и полярных растворителях наблюдается некоторое различие в скоростях
присоединения и заметное различие в величинах параметров Аррениуса.
Таким образом, при превращении исходных реагентов в переходное состоя-
ние наблюдается некоторое изменение полярности, хотя и меньшее, чем в слу-
чае чисто гетеролитических процессов.
Важнейшей проблемой механизма реакции является вопрос о геометриче-
ской конфигурации переходного состояния. Были выдвинуты различные
предположения, но лишь одно из них было достаточно развито. Одна из пред-
ложенных ранее идей исходила из предположения, что реакция протекает
в две стадии через промежуточный бирадикал с открытой цепью. Однако
стереохимическая прочность z^mc-связи между аддендами, каждый из кото-
рых вносит свою собственную стереохимическую конфигурацию в аддукт,
а также отсутствие какого бы то ни было влияния радикалов на реакцию
противоречит идее о двухстадийности общего механизма. Приемлемой оказа-
лась идея Вассермана, развитая им на основе кинетических данных исходя
из предположения об одностадийности реакции присоединения, переходной
состояние которой сходно по геометрической конфигурации с конфигура-
цией аддукта [352]. Это означает, что вновь образующиеся связи между дие-
ном и диенофилом начинают образовываться за счет перекрывания молеку-
лярных л-орбиталей скорее путем концевого, а не бокового перекрывания
соответствующих атомных р-орбиталей. Это положение находится в со-
гласии с диаграммой, приведенной на стр. 894. Из этого механизма следуют
однозначные стереохимические правила. Были сделаны попытки вывести
из этого механизма более сомнительное правило эиЗо-присоединения,
однако нужно признать, что взаимодействие факторов, контролирующих
эндо- или экзо-присоединение, до конца еще не исследовано.
Картина качественно прояснилась в начале 1940-х годов, когда было показа-
но, что электроноакцепторные заместители диенофила и электронодонорные
заместители диена, как правило, ускоряют реакцию диенового синтеза.
Об этом говорилось в первом издании настоящей книги, а в 1955 г. было
сформулировано в виде правила Альдером [353]. Это правило, естественно,
было основано на имевшихся в то время данных. Использованные Альдером
значения констант скорости можно было проверить, что и было впоследст-
вии сделано Зауэром с сотр. [354]. Некоторые из этих значений приведены
в табл. 190.
Данные по присоединению цианзамещенных этиленов к циклопентадиену
и малеинового ангидрида к замещенным бутадиенам, приведенные в верхней
части табл. 190, хорошо подтверждают правило Альдера: с увеличением чис-
ла электроноакцепторных циангрупп в диенофиле скорость реакции быстро
898
Присоединение и обратные реакции
Таблица 190
Константы скорости (к2, л-моль-1-с-1) диенового синтеза
в диоксане (по данным Зауэра)
Этилен Циклопентадиен, 105 fe2 (20 °C) Бутадиен Малеиновый ангидрид 105 k2 (30 °C)
Циан 1,04 2-Хлор 0,69
1,2-Дициан (транс) 81 Незамещенный 6,83
1,2-Дициан(дис) 91 2-Метил 15,4
1,1-Дициан 45 000 1-Метил 22,7
Трициан 483 000 2,3-Диметил 33,6
Тетрациан ~ 43 000 000 1-Метокси 84,1
Присоединение к гексахлорциклопентадиену при 130 °C
Стирол 105 k2 Циклоен 105 к2
п-Метокси 158 Циклопентен 5,9
Незамещенный п-Нитро 79,3 53,8 Малеиновый ангидрид 2,9
растет. При выводе хлорзаместителя из диена или при введении электроно-
донорного метильного или метоксизаместителя в молекулу диена скорость
реакции также значительно возрастает. Однако в нижней части таблицы,
где приведены значения скоростей присоединения некоторых стиролов
и двух пятичленных циклоолефинов к гексахлорциклопентадиену, правило
не соблюдается: при отсутствии электронодонорных метоксильных групп
в диенофиле или введении в его молекулу электроноакцепторных нитро-
или ангидридных групп скорость присоединения диенофила к диену умень-
шается.
Это можно объяснить, по-видимому, следующим образом. Если мы рассмот-
рим, во-первых, присоединение этилена к бутадиену, то следует ожидать,
что при движении системы к переходному состоянию электроны, связываю-
щие адденды в процессе такой активации, будут поставляться главным
образом более нуклеофильным из них, т. е. молекулой бутадиена. В этом
случае мы, конечно, не ставим вопроса, на который для этой реакции и невоз-
можно дать ответа: является ли изменение связи гомолитическим или гете-
ролитическим. Нельзя также определить, точно ли два электрона вносит
каждый реагент для образования двух связей, посредством которых реаген-
ты соединяются друг с другом после перехода через переходное состояние,
когда полностью заканчивается образование продукта. Однако если пред-
почтительное направление смещения электронной плотности при образо-
вании мостика во время процесса активации соответствует предполагаемому,
а именно смещению от диена к диенофилу, то реакция должна облегчаться
при введении электроноакцепторных заместителей в диенофил и электроно-
донорных заместителей в диен. Теперь допустим, что бутадиен уже на-
столько замещен электроотрицательными группами, что он уже не является
более нуклеофильным, чем диенофил. В этом случае остается другая
57* 899
Глава XIII
возможность, а именно мостиковая электронная плотность при активации,
т. е. при переходе от исходного в переходное состояние, будет образо-
вываться главным образом за счет диенофила. Если так, то реакция должна
будет ускоряться электронодонорными заместителями в диенофиле и замед-
ляться при введении в этот реагент электроноакцепторных заместителей.
Вассерман и сотр. [355] подробно изучили роль катализа при диеновом син-
тезе. Оказалось, что реакция мало зависит от катализаторов. Процесс в неко-
торой степени чувствителен к общему кислотному катализу, который даже
применяется при димеризации циклопентадиена, т. е. в случае, когда оба
реагента являются углеводородами. Влияние катализатора обычно прояв-
ляется в том, что реакция протекает по уравнению третьего порядка:
Скорость ОС [Кислота] [Диен] [Диенофил],
Однако в случае димеризации циклопентадиена наблюдается реакция
не только этого, но и на единицу более низкого порядка:
Скорость ос [Кислота] [Циклопентадиен]2,
Скорост3 а [Кислота] [Циклопентадиен]
Это показывает, что катализатор атакует лишь одну из двух соединяющихся
молекул, так как только подобный процесс может определять скорость
реакции. При установлении характера самого воздействия необходимо
учитывать следующие факты. Во-первых, каталитическая активность воз-
растает с увеличением силы кислот в соответствии, например, со следующим
рядом:
СНзСООН <С1СН2СООН <С12СНСООН <СС13СООН <НС1
Сравнительно хорошими катализаторами являются также и фенолы, несмот-
ря на их значительно более низкую кислотность. Во-вторых, катализ наблю-
дается и в случае реакций в газовой фазе. В-третьих, нельзя использовать
растворители, являющиеся по отношению к катализатору основаниями;
например, можно применять четыреххлористый углерод и нельзя применять
диоксан. В-четвертых, слабоосновные нейтральные растворенные вещества
и соли катализирующей кислоты подавляют катализ. Отсюда можно прийти
к выводу, что катализ состоит в присоединении протона кислоты к одному
из реагентов с образованием ионной пары при катализе карбоновыми кисло-
тами и соляной кислотой или комплекса с водородной связью при катализе
фенолом. Затем такой протонированный реагент присоединяет второй реа-
гент, после чего катализирующая кислота снова отщепляется. Имеется ряд
доказательств, что атака протонированным реагентом второго реагента
и является той отличительной чертой катализируемой реакции, благодаря
которой в дальнейшем возникает различие между механизмами катализи-
руемого и некатализируемого присоединений
СПИСОК ЛИТЕРАТУРЫ
1. де ла Мар П., Болтон Р., Реакции электрофильного присоединения к ненасыщен-
ным соединениям, изд-во «Мир», М., 1968.
2. Erlenmeyer Е., Ann., 139, 228 (1866); Butlerow A., ibid., 145, 271 (1868).
3. Markownikoff W., Ann., 153, 256 (1870).
4. Reboul Ann., 155, 29 (1870).
5. Kharasch M. S., Mayo F. R., J. Am. Chem. Soc., 55, 2468 (1933); Mayo F. R., Wal-
ling C., Chem. Revs., 27, 351 (1940); Уоллинг К., Свободные радикалы в растворе,
ИЛ, М., 1960.
6. Raley J. И., Rust F. F., Vaughan W. Е., J. Am. Chem. Soc., 70, 2767 (1948); Ecke G. G.,
Cook N. C., Whitmore F. C., ibid., 72, 1511 (1950).
900
Присоединение и обратные реакции
7. Kharasch М. S., Hannum С., J. Am. Chem. Soc., 56, 1782 (1934).
8. Kharasch М. S., McNab M. C., Mayo F. Я., J. Am. Chem. Soc., 55, 2531 (1933).
9. Kharasch M. S., Hinckley J. A., J. Am. Chem. Soc., 56, 1212 (1934).
10. Kharasch M. S., Mayo Ё. R., J. Am. Chem. Soc., 55, 2461 (1933).
11. Kharasch M. S., Hannum С., J. Am. Chem. Soc., 56, 712 (1934).
12. Linnemann E., Ann., 163, 96 (1872); Wisliscenus J., ibid., 166, 1 (18731.
13. Thiele J., Ann., 306, 87 (1898).
14. Schmidt E., Ann., 267, 300 (1892).
15. Henne A. L., Kaye S., J. Am. Chem. Soc., 72, 3369 (1950).
16. Lucas H. J., Jameson A. Y., J. Am. Chem. Soc., 46, 2475 (1924).
17. Eliel E. L., Goldkamp A. H., Carosins L. E., Eberhardt M., J. Org. Chem., 26, 5188
(1961).
18. Butlerow A., Ann., 189, 76 (1877).
19. Eberz W. F., Lucas H. J., J. Am. Chem. Soc., 56, 1230 (1934).
20. Mayo F. R., Katz J. J., J. Am. Chem. Soc., 69, 1339 (1947).
21. Mayo F. R., Savoy M. G., J. Am. Chem. Soc., 69, 1348 (1947).
22. Lucas H. J., Eberz W. F., J. Am. Chem. Soc., 56, 460 (1934).
23. Lucas H. J., Yun-Ри Liu, J. Am. Chem. Soc., 56, 2138 (1934).
24. Whitmore F. C., Johnston F., J. Am. Chem. Soc., 55, 5020 (1933).
25. Ecke G. G., Cook N. C., Whitmore F. C., J. Am. Chem. Soc., 72, 1511 (1950).
26. Taft R. W., J. Am. Chem. Soc., 74, 5372 (1952); Taft R. W., Purlee E. L., Riesz P.,
de Fazio C. A., ibid., 77, 1584 (1955); Boyd R. H., Taft R. W., Wolf A. P., Christ-
mann D. R., ibid., 82, 4729 (1960).
27. Coe J. S., Gold V., J. Chem. Soc., 1960, 4571; Gold V., Kessick M. A., Proc. Chem.
Soc 1964 295.
28. Levy J. B.' Taft R. W., Hammett L. P., J. Am. Chem. Soc., 75, 1253 (1953).
29. Purlee E. L., Taft R. W., J. Am. Chem. Soc., 78, 5807 (1956).
30. Schubert W. M., Bo Lamm, Keefe J. R., J. Am. Chem. Soc., 86, 4727 (1964).
31. Bunnett J. F., J. Am. Chem. Soc., 83, 4056 (4 papers) (1961).
32. Vaughan W. R. Cravan R. L., Little R. Q., Schoenthaler A. C., ,T. Am. Chem. Soc.,
77. 1594 (1955).
33. Collins С. H., Hammond G. S., J. Org. Chem., 25, 911 (1960).
34. Hammond G. S., Collins С. H., J. Am. Chem. Soc., 82, 5323 (1960).
35. Hammond G. S., Nevitt T. D., J. Am. Chem. Soc., 76, 4121 (1954); Faheg R. C.,
Smith R. A., ibid., 86, 5035 (1964).
36. Thile J., Ann., 306, 87 (1898) et seq. (12 papers); 308, 333 (1899); 314, 298 (1900); 319,
129 (1901).
37. Michael A., J. prakt. Chem., 60, 467 (1899).
38. Hinrichsen F. W., Ann., 336, 168 (1904).
39. Straus F., Ber., 42, 2866 (1909).
40. Burton H., Ingold С. K., J. Chem. Soc., 1928, 910.
41. de la Mare P. B. D., Hughes E. D., Ingold С. K., J. Chem. Soc., 1948, 17.
42. Kharasch M. S., Kritchevsky J., Mayo F. R., J. Org. Chem., 2, 489 (1938).
43. Winstein S., Young W. G., J. Am. Chem. Soc., 58, 104 (1936); England B. D., Hug-
hes E. D., частное сообщение.
44. Ultree A. J., J. Chem. Soc., 1948, 530.
45. Claisen L., J. prakt. Chem., 105, 65 (1922); Farmer E. H., Marshall F. С. В., J. Chem.
Soc., 1931, 129.
46. Henne A. L., Chanan H., Turk A., J. Am. Chem. Soc., 63, 3474 (1941); Пудовик A. H.,
Шавитова H. Б., Ж0Х, 25, 589 (1955).
47. Muskat I. E., Huggins K. A., J. Am. Chem. Soc., 56, 1239 (1934).
48. Riiber C. N., Ber., 44, 2974 (1911).
49. Ingold С. K., Pritchard G. J., Smith H. G., J. Chem. Soc., 1934, 79.
50. Stamhaus E. J.. Drenth W., Van der Berg H., Rec. trav. chim., 83, 107 (1964).
51. Hammond G. S., Warkentin J., J. Am. Chem. Soc., 83, 2554 (1961).
52. Stewart T. D., Edlung K. R., J. Am. Chem. Soc., 45, 1014 (1923); Stewart T. D.,
Fowler R. D., ibid., 48, 1187 (1926); Norrish R. G. W., J. Chem. Soc., 123, 3006 (1923);
Norrish R. G. W., Jones G. G., ibid., 1926, 55.
53. Robertson P. W., Clare N. T., McNaught K. J., Paul G. W., J. Chem. Soc., 1937, 335.
54. Sudborough J. J., Thomas J.. J. Chem. Soc., 87, 715, 2450 (1910); Williams M. D.,
James T. C., ibid., 1928, 343; Hanson N. W., James T. C., ibid., p. 1955; Hanson N. W.
Williams D. M., ibid., 1930, 1059; Williams D. M., ibid., 1932, 2911; Anantakrish-
nan S. V., Venkataraman R., ibid., 1939, 224.
55. Davis H. S., J. Am. Chem. Soc., 50, 2769 (1928).
56. Ingold С. K., Ingold E. H., J. Chem. Soc., 1931, 2354; A natakrishnan S. V., In-
gold С. K., ibid., 1935, 984, 1396.
901
Глава XIII
57. Francis A. W., J. Am. Chem. Soc., 47, 2340 (1925).
58. Biilmann E., Rec. trav. chim., 36, 313 (1917); Gomberg M., J. Am. Chem. Soc., 41,
1414 (1919); Read J., Williams M. M., J. Chem. Soc., 117, 359 (1920); Read J.,
Hook R. G., ibid., p. 1214; Read J., Andrews A. С. P., ibid., 1921, 1774; Read J.,
Hurst E., ibid., 1922, 989, 2550; Read J., Reid W. G., ibid., 1928, 745; Terry E. M.,
Eichelberger L., J. Am. Chem. Soc., 47, 1067 (1925).
59. Conant J. B., Jackson E. L., J. Am. Chem. Soc., 46, 1727 (1924); Jackson E. L., ibid.,
48, 3166 (1926); Meinel K., Ann., 510, 129 (1934); Bartlett P. D., Tarbell D. S., J. Am.
Chem. Soc., 58. 466 (1936); 59, 407 (1937).
60. Backer H- J., Strating J., Rec. trav. chim., 53, 525 (1934); Bockemtiller W., Hoff-
man F. W., Ann., 519, 165 (1935).
61. Bell R. P., Pring M., J. Chem. Soc., 1966B, 1119.
62. Berthoud A.. Mosset M., J. chim. phys., 33, 272 (1936); Bartlett P. D., Tarbell D. S.,
J. Am. Chem. Soc., 58, 466 (1936); Atkinson J. R., Bell R. P., J. Chem. Soc., 1963,
3260.
63. Dubois J. E., Garnier F., Spectrochim. Acta, 23A, 2279 (1967); Chem. Comm., 1968,
241; Boyd R. H., Taft R. W., Wolf A. P., Christman D. R., J. Am. Chem. Soc., 82,
4729 (1960).
64. White E. P., Robertson P. W., J. Chem. Soc., 1939, 1509; Gwyn Williams, Trans. Fara-
day Soc., 37, 749 (1941).
65. N ozaki K., Ogg R.A., Jr., J. Am. Chem. Soc., 64, 637, 704, 709 (1942); Swedlund R. W.,
Robertson P. W., J. Chem. Soc., 1945, 131; 1947 , 630.
66. de la Mare P. B. D., Robertson P. W., J. Chem. Soc., 1945, 888; Rothbaum H. P.,
Ting I., Robertson P. W., ibid., 1948, 980.
67. Ingold С. K., Smith H. G., J. Chem. Soc., 1931, 2742.
68. de la Mare P. B. D., Pritchard J. G., J. Chem. Soc., 1954, 3910, 3990.
69. de la Mare P. B. D., Naylor P. G., Williams D. L. H., J. Chem. Soc., 1962, 443; 1963,
3429; Clarke C. A., Williams D. L. H., ibid., 1966B, 1126.
70. de la Mare P. B. D., Galandauer S., J. Chem. Soc., 1958, 36.
71. Olah G. A., Bollinger J. M., J. Am. Chem. Soc., 89, 4744 (1967).
72. Goldschmidt S., в книге Freudenberg К., «Stereochemie, Deuticke», Vienna, 1933,
p. 520.
73. Lucas H. J., Gould C. W., J. Am. Chem. Soc., 63, 2641 (1941).
74. Roberts I., Kimball G. E., J. Am. Chem. Soc., 59, 947 (1937).
75. Buckles R. E., Knaak D. F., J. Org. Chem., 25, 20 (1960).
76. Buckles R. E., Badar J. M., Thurmeier R. J., J. Org. Chem., 27, 4523 (1962).
77. Cristol S. J., Stermitz F. R., Ramey P. S., J. Am. Chem. Soc., 78, 4939 (1956).
78. de la Mare P. B. D., Klassen N. V., Koenigsberger R., J. Chem. Soc., 1961, 5285; de la
Mare P. B. D., Koenigsberger R., ibid., 1964, 5327.
79. Muskat I. E., Northrop H. E., J. Am. Chem. Soc., 52, 4043 (1930).
80. Пудовик A. H., Ж0Х, 19, 1179 (1949).
81. Griner G., Compt. rend., 116, 723 (1893); 117, 553.
82. Strauss F., Ber., 42, 2872, 2866 (1909).
83. Farmer E. H., Lawrence C. D., Thorpe J. F., J. Chem. Soc., 1928, 729.
84. Hatch L. F., Gardner P. D., Gilbert R. E., J. Am. Chem. Soc., 81, 5943 (1959).
85. Петров А. А., ДАН СССР, 72, 515 (1950).
86. Ingold С. K., Smith H. G., J. Chem. Soc., 1931, 2752.
87. Duden P., Lemme R., Ber., 35, 1335 (1902); Farmer E. H., Lawrence C. D., Scott W. D.,
J. Chem. Soc., 1930, 510.
88. Thiele J., Ann., 314, 296 (1900); Farmer E. H., Scott W. D., J. Chem. Soc., 1929, 172.
89. Jones W. J., Williams H. G., J. Chem. Soc., 1934, 829; Ultree A. J., Rec. trav. chim.,
68, 125 (1949); Hawkins G. E., Philpot M. D., J. Chem. Soc., 1962, 3204.
90. Staudinger H., Muntwyler 0., Kupfer 0., Helv. Chim. Acta, 5, 756 (1922).
91. Kondakow I., J. prakt. Chem., 62, 166 (1900); Macallum A. D., Whitby G. S., Trans.
Roy. Soc. Can., 22, 33 (1928).
92. Farmer E. H., Laroia B. D., Switz T. M., Thorpe J. F., J. Chem. Soc., 1927, 2937.
93. Muskat I. E., Huggins K. A., J. Am. Chem. Soc., 51, 2496 (1929).
94. Петров А. А., Ж0Х, 19, 1046 (1949).
95. Арбузов Б. А., Зороастрова В. M., ДАН СССР, 53, 41 (1946).
96. Kadesch К. G., J. Am. Chem. Soc., 68, 44 (1946).
97. Muskat I. E., Grimsby L. B., J. Am. Chem. Soc., 52, 1574 (1930).
98. Grummitt 0., Vance R. V-, J. Am. Chem. Soc., 72, 2669 (1950).
99. Mislow K., Hellman H. M., J. Am. Chem. Soc., 73, 244 (1951).
100. Young W. G., Hall H. K., Winstein S., J. Am. Chem. Soc., 84, 109 (1956).
101. Lasheen M. A., Acta Cryst., 5, 593 (1952).
102. de la Mare P. B. D., Koenigsberger R., Lomas J. S., Sanchez del Olmo V., Sexton A.
902
Присоединение и обратные реакции
Rec. trav. chim., 84, 109 (1965); de la Mare P. B. D., Johnson M. D., Lomas J. S.,
Sanchez del Olmo V., Chem. Comm., 1965, 483; J. Chem. Soc., 1966, 827.
103. Deville, Ann. chim., 75, 37 (1839).
104. Staudinger H., Ber., 53, 1073 (1920).
105. Evans A. G., Holden D., Plesch P., Polanyi M., Skinner H. A., Weinberger M. A.,
Nature, 157, 102 (1946); Evans A. G., Meadows G. W., Polanyi M., ibid., 158, 94 (1946);
Evans A. G., Polanyi M., J. Chem. Soc., 1947, 252; Evans A. G., Meadows G. W., Trans.
Faraday Soc., 46, 327 (1950).
106. Eley D. D., Richards A. W., Trans. Faraday Soc., 45, 425 (1949).
107. Longworth W. R., Plesch P. H., Proc. Chem. Soc., 1958, 117.
108. Olah G. A., Quinn H. W., Kuhn S. J., J. Am. Chem. Soc., 82, 426 (1960).
109. Colclough R. O., Dainton F. S., Trans. Faraday Soc., 54, 886 (1958).
110. Kennedy J. P., Thomas R. M., J. Polymer Sci., 45, 227, 229 (1960); 46, 481 (1961).
111. Kennedy J. P., Thomas R. M., Makromol. Chem., 53, 28 (1962).
112. Dainton F. S., Sutherland G. В. В. M., J. Polymer Sci., 4, 37 (1949).
113. Colclough R. O., J. Polymer Sci., 8, 467 (1952).
114. Thomas R. M., Sparks W. J., Frolich P. K., Otto M., Mueller-Conradi M., J. Am.
Chem. Soc., 62, 276 (1940).
115. Flory P. J., Principles of Polymer Chemistry, Cornell Univ. Press, Ithaca, New York,
1953, p. 218.
116. Plesch P. H., J. Chem. Soc., 1950, 543.
117. Pepper D. C., Trans. Faraday Soc., 45, 397, 404 (1949); Eley D. D., Richards A. W.,
Trans. Faraday Soc., 45, 436 (1949).
418. Pepper D. C., Quart. Revs. Chem. Soc., 8, 88 (1954); Hayes M. J., Pepper D. C., Proc.
Chem. Soc., 1958, 228; Proc. Roy. Soc. (London), A.263, 63 (1961); Pepper D. C., Frie-
del-Crafts and Related Reactions, Editor G. A. Olah, Interscience, New York, 1964,
Vol. 2, Chap. 30.
119. Lapworth A., J. Chem. Soc., 83, 995 (1903); 85, 1206 (1904).
120. Jones W- J., J. Chem. Soc., 105, 1547, 1560 (1914).
421. Bredig G., Stern E., Z. Elektrochem., 10, 582 (1904); Stern E., Z. physik. Chem., 50,
513 (1905).
122. Clarke R. W. L., Lapworth A., J. Chem. Soc., 91, 694 (1907).
123. Lapworth A., Manske R. H. F., J. Chem. Soc., 1928, 2533.
124. Baker J. W., Hemming M. L., J. Chem. Soc., 1942, 191.
425. Baker J. W., Hopkins H. B., J. Chem. Soc., 1949, 1089.
126. Baker J. W., Barrett S. F. C., Tweed W- T., J. Chem. Soc., 1954, 3831.
127. Koelichen K., Z. physik. Chem., 33, 129 (1900).
428. Ingold E. H., J. Chem. Soc., 123, 1717 (1923); 125, 435 (1924).
129. Bell R. P., McTigue P. T., J. Chem. Soc., 1960, 2983.
130. LaMer V. K., Miller M. L., J. Am. Chem. Soc., 57, 2674 (1935).
431. Bonhoejfer K. F., Walters W. D., Z. physik. Chem., A181, 141 (1938); A182, 265.
132. Bell R. P., J. Chem. Soc., 1937, 1637; Trans. Faraday Soc., 37, 716 (1941).
133. Broche A., Gilbert R., Bull. soc. chim. (France), 1955, 131.
134. Welch K. N., J. Chem. Soc., 1931, 653.
135. Coombs E., Evans D. P., J. Chem. Soc., 1944, 1295.
136. Patai S., Israeli Y., Zabicky J., Chem. and Ind., 1957, 1671.
437. Patai S., Israeli Y., J. Chem. Soc., 1960, 2020, 2025; Patai S., Zabicky J., ibid., p. 2030;
Patai S., Zabicky J., Israeli T., ibid., p. 2038.
138. Noyce D. S., Snyder L. R., J. Am. Chem. Soc., 80, 4033, 4324 (1958); 81, 620 (1959);
JoyceD. C., Reed W. I., ibid., 80, 5539 (1958); 81, 624 (1959); NoyceD. C., Proyor W. A.,
ibid., p. 618.
139. Harries C., Muller G. H., Ber., 35, 966 (1902).
140. Miller J. G., Kilpatrick M., J. Am. Chem. Soc., 53, 3217 (1931); Wesiheimer F. H.,
Cohn H., ibid., 60, 90 (1938).
141. Wurtz A., Jahresber., 1872, 449.
142. Schmidt J. G., Ber., 13, 2342 (1880).
143. Claisen L., Ber., 14, 2468 (1881).
144. Claisen L., Ann., 218, 172 (1883).
145. Japp F. R., Streatfield F. W., J. Chem. Soc., 43. 27 (1883).
146. Knoevenagel E., Ann., 281, 25 (1894); Ber., 37, 4461 (1904).
447. Perkin W. H., Sr., Chem. News, 32, 258 (1875); J. Chem. Soc., 31, 388 (1877); 47, 317
(1886).
448. Fittig R., Ber., 14, 1824 (1881); Ann., 227, 48 (1885).
149. Fittig R., Slocum F. L., Ann., 227, 53 (1885).
150. Hauser C. R., Breslow D. S., J. Am. Chem. Soc., 61, 786 (1939).
451. Fittig R., Jayne H. W., Ann., 216, 97 (1882).
903
Глава XIII
152. Fittig R., Jayne H. W., Ann., 216, 115 (1882); Fittig R., Ott P., Ann., 227, 61 (1885).
153. Breslow D. S., Hauser C. R., J. Am. Chem. Soc., 61, 793 (1939).
154. Kalnin P., Helv. Chim. Acta, 11, 977 (1928).
155. Kesterning B., Halleck L., Melkonian G. A., Z. Elektrochem., 60, 130 (1956).
156. Cordes E. H., Jencks W. P., J. Am. Chem. Soc., 84, 832 (1962); 85, 2843 (1963).
157. Reeves R. L., J. Am. Chem. Soc., 84, 3332 (1962).
158. Willis A. V., Robertson R. E., Can. J. Chem., 31, 361 (1953); Bloch-Chaude 0., Compt.
rend., 239, 804 (1954).
159. Pratt E. F., Kamlet M. J., J. Org. Chem., 26, 4029 (1961).
160. Santerre G. M., Hamstote C. J., Crowell T. L., J. Am. Chem. Soc., 80, 1254 (1958).
161. Willi A. V., Helv. Chim. Acta, 39, 1193 (1956).
162. Barrett E., Lapworth A., J. Chem. Soc., 93, 85 (1908); Olander A., T, phvsik. Chem.,
129, 1 (1927).
163. Bodforss S., Z. physik. Chem., 109, 223 (1924).
164. Conant J. B., Bartlett P. D., J. Am. Chem. Soc., 54, 2881 (1932); Westheimer F. H.,
ibid., 56, 1962 (1934); Price F. P., Jr., Hammett L. P., ibid., 63, 2387 (1941).
165. Jencks W. P., J. Am. Chem. Soc., 81, 475 (1959).
166. Anderson F. M., Jencks W. P., J. Am. Chem. Soc., 82, 1773 (1960).
167. Bell R. P., Higginson W. С. E., Proc. Roy. Soc. (London), A197, 141 (1949); Bell R. P.,
Darwent B. de B., Trans. Faraday Soc., 46, 34 (1950).
168. Lapworth A., J. Chem. Soc., 83, 995 (1903); 85, 1214 (1904); 85, 945 (1906).
169. Jones W. J., J. Chem. Soc., 105, 1547 (1914).
170. Michael A., J. prakt. Chem., 35, 251 (1887).
171. Knoevenagel E., Mottek S., Ber., 37, 4460 (1904).
172. Vorlander D., Ber., 33, 3185 (1900).
173. Ingold С. K., Powell W. J., J. Chem. Soc., 119, 1976 (1921); Ingold С. K., Perren E. A..
ibid., 121, 1414 (1922).
174. Ogata У., Окапи M., Furuya Y., Tabushi L., J. Am. Chem. Soc., 78, 5426 (1956).
175. Hochstetter A., Cohn M., Monatsch., 24, 773 (1903).
176. Blank R., Ber., 28, 145 (1885).
177. Liebermann C., Ber., 26, 1876 (1893).
178. Fett B. A., Zilkha A., J. Org. Chem., 28, 406 (1963).
179. Hoffman A., J. Am. Chem. Soc., 49, 552 (1927).
180. Winstein S., Lucas H. J., J. Am. Chem. Soc., 59, 1461 (1937); Pressman DLucas H. J.,
ibid., 61, 2271 (1939); 62, 2069 (1940); Pressman D-, Brewer L., Lucas H. J., ibid., 64,
1117, 1122 (1942); Pressman D., Lucas H. J., ibid., p. 1953; Lucas H. J., Stewart W. T.,
Pressman D., ibid., 66, 1818 (1944).
181. Bloom J., Ingold С. К., J. Chem. Soc., 1931, 2765; de la Mare P. B. D., Hughes E. D.,
Ingold С. K., ibid., 1948, 17.
182. Farmer E. H., Mehta T. N., J. Chem. Soc., 1931, 1904; Bloom J., Ingold С. K., ibid.,
p. 2765.
183. Thiele J., Meisenheimer J., Ann., 306, 247 (1899).
184. Vorlander D., Groebel P., Ann., 345, 206 (1906).
185. Hinrichsen F. W., Trepel W., Ann., 336, 196 (1904).
186. Farmer E. H., Martin S. R. W., J. Chem. Soc., 1933, 960.
187. Eicher T., Chemistry of the Carbonyl Group, Editor S. Patai, Interscience Publishers,
Chap. 13, p. 621.
188. Kohler E. P., Am. Chem. J., 38, 511 (1907); Gilman H., Kirby R. H., J. Am. Chem.
Soc., 63, 2046 (1941); Sullivan W. I., Swanner F. W., Humphlett W. J., Hauser C. R.,
J. Org. Chem., 26, 2306 (1961).
189. Whitmore F. C., Pedlar G. W., J. Am. Chem. Soc., 63, 758 (1941).
190. Alexander E. R., Coroar G. R., J. Am. Chem. Soc., 73, 2721 (1951).
191. Tucker H. S., Whalley M., J. Chem. Soc., 1949, 50.
192. Taylor R. S., Connor R., J. Org. Chem., 6, 696 (1941).
193. Schmidlin J., Hodgson H. H., Ber., 41, 430 (1908).
194. Schlenk W., Schlenk W., Jr., Ber., 62, 920 (1929).
195. Smith S. G., Tetrahedron Letters, 1963, 409; Smith S. G., Su G., J. Am. Chem. Soc.,
86, 2750 (1964).
196. Ashby E. C., Duke R. B., Neumann H. M., J. Am. Chem. Soc., 89, 1904 (1967).
197. Anteunis M., J. Org. Chem., 26, 4214 (1961); 27, 596 (1962).
198. Bikales N. M., Becker E. I., Can. J. Chem., 41, 1329 (1963).
199. Cannizzaro S., Ann., 88, 129 (1853).
200. Molt E. L., Rec. trav. chim., 56, 233 (1937); Eitel A., Lock G., Monatsh., 72, 392 (1939);
Tommilia E., Ann. Acad. Sci. Fennicae, A59, № 8 (1942).
201. Geib К. H., Z. physik. Chem., A169, 41 (1934).
202. Eitel A., Monatsh., 74, 136 (1942).
904
П рисоединение и обратные реакции
203. v. Euler Н., Lovgren Т., Z. anorg. Chem., 127, 123 (1925).
204. Поль И. И., ЖОХ, 11, 1121 (1941); Pafunen V., Ann. Acad. Sci. Fennicae, A2 № 87
(1950); Martin R. J. L., Austral. J. Chem., 7, 335 (1954).
205. Alexander E. R., J. Am. Chem. Soc., 69, 289 (1947).
206. Weissberger A., Hasse R., J. Chem. Soc., 1934, 535.
207. Freienhagen II., Bonhoeffer K. F., Z. physik. Chem., A181, 379 (1938).
208. Hammett L. P., Physical Organic Chemistry, McGraw-Hill, New York, 1940, p. 350.
209. Rurton H., Ingold С. K., J. Chem. Soc., 1929, 2022.
210. Klages A., Ber., 35, 2642 (1902); Klages A., Keil R., Ber., 36, 1632 (1903).
211. Couturier F., Meunier L., Compt. rend., 140, 721 (1905).
212. Clemmensen E., Ber., 46, 1837 (1913); 47, 51, 681 (1914).
213. de la Mare P. B. D., Hughes E. D., Ingold С. К., J. Chem. Soc., 1948, 17.
214. v. Baeyer A., Ann., 251, 257 (1889).
215. p. Raeyer A., Rupe H., Ann., 256, 1 (1890).
216. Fittig R., Hoffmann T., Ann., 283, 308 (1894).
217. Thiele J., Meisenheimer J., Ann., 306, 225 (1899); Riiber C. N., Ber., 37, 3120 (1904).
218. Ramberger E., Lodter W., Ann., 288, 74 (1895).
219. Strauss F., Ann., 342, 190 (1905).
220. Kuhn R., Winterstein A., Helv. Chim. Acta, 11, 123 (1928).
221. Szwarc M., Smid J., Progress in Beaction Kinetics, Vol. 2, Pergamon Press, London,
1964, p. 219.
222. Pepper D. C., Quart. Bevs. Chem. Soc., 8, 114 (1954).
223. v. Pechmann H., Ber., 33, 3329 (1900); Bruylants P., Bull. Soc. chim. Beiges, 32, 317
(1923); Schmidt E., Rutz G., Ber., 61, 2142 (1928).
224. Matthews F. E., Strange E. H., англ. пат. 24790 (1910).
225. Harries C., Ann., 383, 213 (1911).
226. Schlenk W., Appenrodt J., Michael A., Thai A., Ber., 47, 473 (1914).
227. Ziegler W., many papers since 1928, summarised in Chem. Zeitg., 62, 125 (1938).
228. Beamam R. G., J. Am. Chem. Soc., 70, 3115 (1948).
229. Sanderson J. J., Hauser C. R., J. Am. Chem. Soc., 71, 1595 (1949).
230. Walling C., Briggs E. R., Cummings W., Mayo F. R., J. Am. Chem. Soc., 72, 48 (1950).
231. Landler Y., Compt. rend., 280, 539 (1950).
232. Abkin A., Medvedev S-, Trans. Faraday Soc., 21, 286 (1936); ЖФХ, 13, 705 (1939);
Абкин А., Медведев С., Манонтова О., Усп. хим. физики, 12, 269 (1940).
233. Higginson W. С. Е., Wooding N. S., J. Chem. Soc., 1952, 760, 1178.
234. Waack R., Rembaum A., Coombes J. D., Szwarc M., J. Am. Chem. Soc., 79, 2026
(1957); Brody H., Ladacki M., Milkovich R., Szwarc M., J. Polymer Sci., 25, 221 (1957).
235. Flory P. J., J. Am. Chem. Soc., 65, 372 (1943).
236. Bauer E., Magat M., J. chim. phys., 47, 841 (1950).
237. Ziegler K., Bahr K., Ber., 61, 253 (1928).
238. Szwarc M., Nature, 178, 1168 (1956); Szwarc M., Levy M., Milkovich R., J. Am. Chem.
Soc., 78, 2656 (1956).
239. Brody H., Ladack M., Milkovich R., Szwarc M., J. Polymer Sci., 25, 221 (1957); McCor-
mick H. W., ibid., 448; Worsfold D. J., Bywater S., ibid., 26, 299; Worsfold D. J.,
Bywater S., ibid., 58, 571 (1962); Waack R., Rembaum A., Coombes J. D-, Szwarc M.,
J. Am. Chem. Soc., 79, 2026 (1957); Vraneken A., Smid J., Szwarc M., ibid., 83, 2772
(1961); Trans. Faraday Soc., 58, 2036 (1962).
240. Worsfold D. J., Bywater S., Can. J. Chem., 36, 1141 (1958); Allen G., Gee G., Stretch C.,
J. Polymer Sci., 48, 189 (1960); Geacinov C., Smid J., Szwarc M., J. Am. Chem. Soc.,
84, 2508 (1962).
241. Уоллинг К., Свободные радикалы в растворе, ИЛ, М., 1960; Cadogan J. I. G., Per-
kins М. J., в книге «Chemistry of Alkenes», Editor S. Patai, Interscience Publishers,
London, 1964, Chap. 9.
242. Kharasch M. S., Mayo F. R., J. Am. Chem. Soc., 55, 2468 (1933).
243. Hey D. H., Waters W. A., Chem. Revs., 21, 169 (1937).
244. Kharasch M. S., Engelmann H., Mayo F. R., J. Org. Chem., 2, 288 (1937).
245. Ford T. A., Hanford W- E., Harmon J., Lipscomb R. D., J. Am. Chem. Soc., 74/4323
(1952).
246. Ashworth F., Burkhardt G. N., J. Chem. Soc., 1928, 1791.
247. Burkhardt G. N., Trans. Faraday Soc., 30, 18 (1934).
248. Kharasch M. S., Read A. T., Mayo F. R., Chem. and Ind., 57, 752 (1938).
249. Jones S. O., Reid E. E., J. Am. Chem. Soc., 60, 2452 (1938).
250. Harmon J., пат. США 2390099 (1945).
251. Skraup Z. H., Wien Akad. Sitzungber., 100, 124 (1891).
252. Walling C., Seymour D., Wolfstirn К. B., J. Am. Chem. Soc., 70, 2559 (1948).
253. Smith W. V., J. Am. Chem. Soc., 68, 2059, 2064 (1946); Walling C., ibid., 70, 2561
905
Глава XIII
(1948); Gregg R. A., Aiderman D. M., Mayo F. R., ibid., p. 3740; Meehan E. J., Kohl-
toff I. M., Sinha P. R., J. Polymer Sci., 16, 471 (1955); Pierson R. M., Costanza A. J.,
Weinstein A. H., ibid., 17, 221.
254. Уоллинг К., Свободные радикалы в растворе, ИЛ, М., 1960.
255. Leernakers J. A., Dickenson R. G., J. Am. Chem. Soc., 54, 4648 (1932); Dickenson R. G.,
Carrico J. L., ibid., 56, 1473 (1934); 57, 1343 (1935); Willard J., Daniels F., ibid.,
p. 2240; Muller K. L., Schumacher H. J., Z. Elektrochew., 43, 807 (1937); Z. physik.
Chem., B35, 285, 455 (1937); B42, 327 (1939); Schott C., Schumacher J. H., ibid., B49,
107 (1949); Schmitz H., Schumacher H. J., Jager A., ibid., B51, 281 (1942); Schmitz H.,
• Schumacher H. J., ibid., B52, 72 (1942).
256. Forbes G. S., Nelson A. F., J. Am. Chem. Soc., 59, 693 (1937).
257. Ketelaar J. A. A., Van Verden P. F., Rroers G. H. J., Gersmann H. R., J. Phys. Colloid
Chem., 55, 987 (1951).
258. Steinmetz H., Noyes R. M., J. Am. Chem. Soc., 74, 4141 (1952).
259. Wood R. E., Dickenson R. G., J. Am. Chem. Soc., 61, 3259 (1939); Neyes R. M., Dicken-
son R. G., ibid., 65, 1427 (1943); Neyes R. M., Dickenson R. G., Schomaker V., ibid.,
67, 1319 (1945); Dickenson R. G., Wallis R. F., Wood R. E., ibid., 71, 1238 (1949).
260. Miller W. T., Jr., Koch S. D., Jr., J. Am. Chem. Soc., 79, 3084 (1957).
261. Kharasch M. S., Jensen E. V., Urry W. H., Science, 102, 128 (1945).
262. Tedder J. M., Walton J. W., Trans. Faraday Soc., 60, 1769 (1964).
263. Kharasch M. S., Sage M., J. Org. Chem., 14, 537 (1949).
264. Kharasch M. S., Simon E., Nudenberg W., J. Org. Chem., 18, 328 (1953).
265. Pearson J. M., Szwarc M., Trans. Faraday Soc., 60, 553 (1964).
266. Huang R. L., J. Chem. Soc., 1956, 1749.
267. Kharasch M. S., Urry W. H., Kuderna B. M., J. Org. Chem., 14, 248 (1949).
268. Patrick T. M., Jr., ibid., 17, 1009, 1269 (1952).
269. Urry W. H., Stacey F. W., Huyser E. S., Joveland O. O., J. Am. Chem. Soc., 76, 450
(1954).
270. Simon E., Ann., 31, 265 (1839).
271. Berthelot M., Bull. soc. chim. (France), 6, [ii] 294 (1866).
272. Staudinger H., Steinhofer A., Ann., 517, 35 (1935).
273. Kwart II., Rroadbent H. S., Rartlett P. D., J. Am. Chem. Soc., 72, 1060 (1950).
274. Уоллинг К., Свободные радикалы в растворе, ИЛ, М., 1960.
275. Mayo F. R., Walling С., Chem. Revs., 46, 191 (1950).
276. Rreitenbach J. W., Renner A. J., Can. J. Research, 28B, 509 (1950).
277. Rartlett P. D., Hammond G. S., Kwart H., Disc. Faraday Soc., 2, 342 (1947).
278. Rartlett P. D., Kwart H., J. Am. Chem. Soc., 72, 1051 (1950).
279. Huisgen R., Grashey R., Sauer J., в книге «The Chemistry of Alkanes», Editor S. Patai,
Interscience Publishers, London, 1964, p. 739.
280. Кирмсе В., Химия карбенов, изд. «Мир», М., 1966.
281. Buchner Е., Curtius Т., Вег., 18, 2377 (1885); Buchner Е., Вег., 30, 362 (1897); Dar-
ren W., Buchner Е., Вег., 33, 684 (1900); 34, 982 (1901).
282. Buchner Е., Hediger S., Вег., 36, 3502 (1903).
283. Herzberg G., Shoosmith J., Nature, 183, 1801 (1959); Herzberg G., Proc. Roy. Soc.,
A262, 291 (1961); Herzberg G., Jones J. W. C., ibid., A295, 107 (1966).
284. Pearson T. G., Purcell R. H., Saigh G. S., J. Chem. Soc., 1938, 409.
285. Meerwein H., Rathfen H., Werner H., Ber., 75, 1610 (1942).
286. Doering W. von E., Buttery R. G., Laughlin R. H., Chaudhuri N., J. Am. Chem. Soc.,
78, 3224 (1956).
287. Skell P. S., Woodworth R. C., J. Am. Chem. Soc., 78, 4496 (1956); Doering W. von E.,
La Flamme P., ibid., p. 5447; Frey H. M., ibid., 80, 5005 (1958).
288. Frey H. M., J. Am. Chem. Soc., 82, 5947 (1960).
289. Friedman L., Schechter H., J. Am. Chem. Soc., 81, 5512 (1959).
290. Etter R. M., Skovronek H. S., Skell P. S., J. Am. Chem. Soc., 81, 1008 (1959);
Gloss G. L., Class L. L., Angew. Chem., 74, 431 (1962).
291. Burger A., Yost W. L., J. Am. Chem. Soc., 70, 2198 (1948); Markees D. G., ibid.,
p. 3329; de Waal H. L., Perold G. W., Chem. Ber., 85, 574 (1952).
292. Doering W. von E., Mole T., Tetrahedron, 10, 65 (1960).
293. Doering W. von E., Hoffmann A. K., J. Am. Chem. Soc., 76, 6162 (1954).
294. Fields E. K„ J. Am. Chem. Soc., 84, 1744 (1962).
295. Anderson J. C., Reese С. B., Chem. and Ind., 1963, 575.
296. Pearson W. E., Konces E., J. Am. Chem. Soc., 83, 4034 (1961).
297. Skell P. S., Garner A. Y., J. Am. Chem. Soc., 78, 3049, 5430 (1956).
298. Westcott L. D., Skell P. S., J. Am. Chem. Soc., 87, 1721 (1965).
299. Doering W. von E., Henderson W. A., Jr., J. Am. Chem. Soc., 80, 5274 (1958).
300. Reimer K., Ber., 9, 423 (1876); Reimer K., Tiemann F., ibid., 9, 824, 1285 (1876).
906
Присоединение и обратные реакции
301. Hine J., van der Veen J. M., J. Am. Chem. Soc., 81, 6447 (1959).
302. Hofmann A. W., Ann., 144, 114 (1887); 146, 107 (1888).
303. Hef J. U., Ann., 298, 202 (1897).
304. Smith P. A. S., Kalenda N. W., J. Org. Chem., 23, 1599 (1958).
305. Monriis A., Rosen B., Nature. 164, 724 (1949); Douglas A. E., Astrophys. J., 114, 406
(1951); Clusius K., Douglas A. E., Can. J. Phys., 32, 319 (1954); Ramsey D. A., Ann.
New York, Acad. Sci., 67, 485 (1957).
306. Shell P. S., Westcott L. D., Jr., Goldstein J. P., Engel R. R., J. Am. Chem. Soc., 87,
2829 (1965).
307. Staudinger H., в книге «Die Ketene», F. Enke, Stuttgart, 1912; см. также Enk E.,
Spes H., Angev. Chem., 73, 334 (1961).
308. Atkinson B., Trenwith A. B., J. Chem. Soc., 1953, 2082.
309. Lacher J. R., Thompkin G. W., Park J. D., J. Am. Chem. Soc., 74, 1693 (1952).
310. Henne A. L., Ruh R. P., J. Am. Chem. Soc., 69, 279 (1947).
311. Hauptschein M., Fainberg A. H., Brand M., J. Am. Chem. Soc., 80, 842 (1958).
312. Coffman D. D., Barrick P. L., Cramer R. D., Raasch M. S., J. Am. Chem. Soc., 71,
490 (1949).
313. Prober M., Miller W. T., J. Am. Chem. Soc., 71, 598 (1949).
314. Grundermann K. D., Thomas R., Chem. Ber., 89, 1263 (1956); Grundermann K. D.t
Huchting R., ibid., 92, 415 (1959).
315. Williams S. K., Wiley D. W., McKusick В. C., J. Am. Chem. Soc., 84, 2210, 2216
(1962).
316. Proskow S., Simmons H. E., Cavins T. L., J. Am. Chem. Soc., 85, 2341 (1963).
317. M ontgomery L. K., Schueller K., Bartlett P. D., J. Am. Chem. Soc., 86, 622, 628 (1964).
318. Bowen E. J., Marsh J. D. F., J. Chem. Soc., 1947, 109.
319. Stobbe H., Bremer K., J. prakt. Chem., [2], 123, 1 (1929); Reektenwald G. W.,
Pitts J. N., Letsinger R. L., J. Am. Chem. Soc., 75, 3028 (1953).
320. Eaton P. E., J. Am. Chem. Soc., 84, 2344, 2454 (1962).
321. Cohen M. D., Schmidt G. M. J., J. Chem. Soc., 1964, 1996.
322. Cohen M. D., Schmidt G. M. J., Sonntag F. I., J. Chem. Soc., 1964 (2000);
Schmidt G. M. J., ibid., p. 2014.
323. Bregman J., Osaki K., Schmidt G. M. J., Sonntag F. I., J. Chem. Soc., 1964, 3021;
Rabinovich D., Schmidt G. M. J., ibid., p. 2030.
324. Huisgen R., Proc. Chem. Soc., 1961, 357.
325. Buchner E., Ber., 21, 2637 (1888); Buchner E., Fritsch M., Papendieck A., Witter H.,
273, 214 (1893).
326. von Auwers К., Cauer E., Ann., 470, 284 (1929); von Auwers K., Konig F., Ann., 496,
27, 252 (1932).
327. Huisgen R., Stangl H., Sturm H. J., Wagenhofer H., Angew. Chem., 73, 170 (1961);
Huisgen R., Sturm H. J., Wagenhofer H., Z. Naturforsch., 17b, 202 (1962).
328. von Auwers K., Ungermach 0., Ber., 66, 1198 (1933).
329. Parham W. R., Hasek W. E., J. Am. Chem. Soc., 76, 799 (1954).
330. Alder K., Stein G., Ann., 501, 1 (1933); 515, 165, 185 (1935); Alder K., Rieger H. K.,
Weiss H., Chem. Ber., 88, 144 (1955).
331. Wolff L., Grau G. K., Ann., 394, 68 (1911).
332. Huisgen R., Szeimies G., Mtiller G., Mobius L., неопубликованные данные; см. рабо-
ту [279], стр. 838-843.
333. Alder К., Stein G., Ann., 485, 211, 223 (1931); Diels 0., Konig H., Ber., 71, 1179 (1938).
334. Wieland H., Ber., 40, 1667 (1907).
335. Quilico A., Fusco R., Gazz. chim. ital., 67, 589 (1937); Quilico A., Speroni G., ibid.,
76, 148 (1946); Stagno d’Alcontres G., Grunanger P., ibid., 80, 741, 831 (1950); Stagno
d’Alcontres G., Fenech G., ibid., 82, 175 (1952); Quilico A., Grunanger P., Mezzini R.,
ibid., p. 349; Stagno d’Alcontres G., ibid., p. 627; Grunanger P., ibid., 84, 359 (1954);
Grunanger P., Langella M. R., ibid., 91, 1112 (1961); Grunanger P., Langella M. R.,
Barbulescu N., Quilico A., Tetrahedron Letters, 1961, 89.
336. Quilico A., Stagno d'Alcontres G., Grunanger P., Gazz. chim. ital., 80, 479 (1950);
Quilico A., Grunanger P., ibid., 82, 140 (1952); 85, 1449 (1955).
337. Mukaiyama T., Hoshino T., J. Am. Chem. Soc., 82, 5339 (1960); Bachman G. B.,
Strom L. E., J. Org. Chem., 28, 1150 (1953).
338. Huisgen R., Mack W., Bast K., Kudera E., неопубликованные данные; см. работу
[279], стр. 826.
339. Grashey R., Huisgen R., Leitermann H., Tetrahedron Letters, № 12, 9 (1960); Hauck H.,
Thesis, Munich, 1963.
340. Brown C. W., Marsden K., Rogers M. A. T., Tyler С. M. B., Wright R., Proc. Chem.
Soc., 1962, 254.
341. Seidl R., Thesis, Munich, 1963; см. работу [279], стр. 865.
907
Глава XIII
342. Wassermann A., «Diels — Alder Reaction», Elsever Publishing Co., Amsterdam, 1965.
343. Zincke T., Gunther H., Ann., 272, 243 (1892); Zincke T., Bergmann F., Francke B.,
Prenntzell W., ibid., 296, 135 (1897); Zincke T., Meyer К. H., Ann., 367, 1 (1909);
Zincke T., P t ait endowf W., ibid., 394, 3 (1912).
344. v. Euler H., Josephson K. 0., Ber., 53, 822 (1920).
345. Diels O., Alder K., Ann. 460, 98 (1928).
346. Alder K., Stein G., Angew. Chem., 47, 837 (1934); 50, 510 (1937).
347. Alder K., Schumacher M., Ann., 571, 87 (1951).
348. Woodward В. B., Baer M., J. Am. Chem. Soc., 70, 1161 (1948); Kwart H., Burchuk I.,
ibid., 74, 3094 (1952); Berson J. A., Swidler B., ibid., 75, 1721 (1953); Anet P. A. L.,
Tetrahedron Letters, 1962, 1219.
349. Alder K., Heinbach К., Beube B., Chem. Ber., 91, 1516 (1958); Berson J. A., Hamlet Z.,
Mueller W. A., J. Am. Chem. Soc., 84, 297 (1962).
350. Cristol S. J., Seifert W. K., Solonay S. В., J. Am. Chem. Soc., 82, 2351 (1960).
351. Wassermann A., Trans. Faraday Soc., 34, 128 (1938).
352. Wassermann A., J. Chem. Soc., 1942, 612.
353. Alder K., Experientia, 1955, Suppl. II, 86.
354. Sauer J., Wiest H., Mielert A., Z. Naturforsch., 17b, 203 (1962); Sauer J., Wiest H.,
Mielert A., Lang D., Angew. Chem., 74, 352, 353 (1962); cm. [279].
355. Wassermann A., J. Chem. Soc., 1942, 618; Steiner H., Wassermann A., ibid., 1949, 3046.
ГЛАВА XIV
КИСЛОТЫ И ОСНОВАНИЯ
1. Кислотно-основное равновесие.......................910
Список литературы................ . ............934
Перенос протона в растворе между такими электроотрицательными ато-
мами, как азот, кислород, сера и галогены, происходит настолько быстро,
что кинетику этого переноса можно исследовать только в особых случаях
и с помощью специальных методов. Однако можно определить, и действи-
тельно такие определения проводятся, положение равновесия при переносе
протона. Кислотно-основной перенос протона очень широко распространен
в органической химии, и здесь проведены достаточно точные определения
констант равновесия, что позволяет иметь исчерпывающие сведения о влия-
нии структуры на равновесие. Эти вопросы и станут предметом обсуждения
в данной главе. В отличие от других разделов книги, где больше внимания
уделяется кинетике реакций, в настоящей главе основное внимание будет
уделено термодинамике.
1. Кислотно-основное равновесие
а. Измерение кислотно-основного равновесия. В соответствии с пред
ложением Бренстеда в уравнении
НХ Н++Х-
кислотой считают вещество НХ, а основанием — X-; кислота и основание'
взаимосвязаны, или, как говорят, сопряжены друг с другом независимо
от абсолютных зарядов обеих частиц; знак минус в приведенном случае
указывает лишь на отношение зарядов между кислотой и основанием.
Наиболее часто употребляемым растворителем при изучении кислотно-
основного равновесия является вода. Вода рассматривается как основание
в соответствии с уравнением
Н3О+ Н++Н2О
Путем сочетания двух приведенных выше уравнений можно получить урав-
нение, к которому удобнее применить закон действия масс:
НХ + Н2О н3о+ + х-
Кислота НХ переносит свой протон к основанию Н2О, образуя при этом
кислоту Н3О+ и основание X-. В принципе процесс обратим, и концентра-
цию всех участвующих частиц можно определить. Константа кислотности
кислоты НХ в воде, иногда называемая также константой гидролиза катио-
на НХ (в тех случаях, когда НХ является катионом), представляет собой
константу равновесия реакции, которая в случае воды как растворителя
равна
{Н3О+} {Х~} ~ [Н3О+] [Х~]
а {НХ} ~ [НХ]
Фигурные скобки в этом уравнении указывают на то, что приведенные-
величины являются активностями; их значения легко можно получить, если
концентрации (величины, приводимые в квадратных скобках) настолько,
малы, что коэффициенты активности зависят только от заряда; они равны
единице в случае нейтральных частиц и имеют стандартные значения Дебая —
Мильнера для ионов. Обычная размерность Ка — моль/л. Значения Ка
часто приводятся в виде отрицательных логарифмов рАД:
PXa=-lgXa
Воду можно считать кислотой в соответствии с уравнением
Н2О Н++ОН-
910
Кислоты и основания
В результате комбинации этого уравнения с общим уравнением Бренстеда
можно получить иное выражение равновесия, для которого применим закон
действия масс
Н2О + Х- дД нх + он-
В данном случае кислота Н2О передает свой протон основанию Х-Лобра-
зуя кислоту НХ и основание ОН-. Реакция обратима, и концентрации
частиц могут быть определены. Это уравнение является полезным для сужде-
ний об основании X-, которое, как указывалось выше, может иметь любой
заряд. Константа основности основания X- в воде, иногда называемая
также константой гидролиза аниона X- (когда X- — анион), представляет
собой константу равновесия этой реакции, как это имеет место в том случае,
когда растворителем служит вода
{НХНОН-} [НХПОН-]
ь {X-} ~ [Х-]
Обычная размерность Кь — моль/л.
Сочетание двух приведенных частных уравнений дает уравнение, в котором
вода выступает как в роли кислоты, так и в роли основания
н2о+н2о н3о++он-
К этому уравнению также приложим закон действия масс. Константа
автопротолиза воды представляет собой константу равновесия этой реакции
в случае применения воды в качестве растворителя
*авто={Н3О+} {ОН-} « [Н3О+] [ОН-]
Обычная размерность Хавт0 — (моль/л)2.
Рассмотренные три константы равновесия связаны друг с другом следующим
соотношением:
КаКь = Хавт0
Если величина Хавто известна с достаточной степенью точности, то на прак-
тике обычно константы основности оснований выражают через константы
кислотности их сопряженных кислот, т. е. выражают значения Кь через
величины Ка данной кислотно-основной системы.
Константа кислотности кислоты служит мерой силы кислоты. Кислоту назы-
вают сильной или слабой в зависимости от того, насколько велико или мало
значение константы диссоциации. Термин «сила» по отношению к данной
кислоте часто представляет собой качественный синоним ее константы
диссоциации. Константа диссоциации основания служит мерой силы осно-
вания. Основание называют сильным или слабым в зависимости от величины
константы основности. Термин «сила» в отношении основания часто является
количественным синонимом константы основности основания. Однако из-за
взаимосвязи между Ка, Къ и Хавто деление кислот и оснований на сильные
и слабые не является абсолютным; сильным и слабым кислотам соответ-
ствуют слабые и сильные сопряженные основания. Основания часто клас-
сифицируются в соответствии с силой их сопряженных кислот.
Для определения величины Ка кислоты необходимо измерить концентрацию
или активность по крайней мере одной из трех растворенных частиц, вхо-
дящих в выражение для Ка; концентрации других растворенных частиц
могут быть в этом случае найдены исходя из стехиометрии массы, а также
заряда. Оствальд использовал для этой цели метод измерения электропро-
водности, предложенный Кольраушем для исследования растворов электро-
911
Глава XIV
литов; впоследствии после ряда усовершенствований этот метод был широко
использован для точных определений Ка. Кроме того, активность Н3О+
может быть непосредственно определена путем измерения электродвижущей
силы элемента, содержащего электролит и водородный электрод. Концен-
трация любых неодинаково окрашенных частиц может быть также опре-
делена фотометрически. Имеются также и другие методы, большинство
которых является более косвенным, чем упомянутые.
Если кислота НХ представляет собой сильную кислоту и если ее концентра-
ция определяется только исходя из стехиометрии, как это имеет место в слу-
чае почти всех электронейтральных кислот, то небольшие различия в кон-
центрации кислоты уловить фактически не представляется возможным,
и, следовательно, невозможно найти значение Ка для такой кислоты. Эта
трудность возрастает в случае электронейтральных кислот, имеющих зна-
чения Ка порядка 100 моль/л или более. Так, в случае двух кислот, значи-
тельно отличающихся по силе, например со значениями Ка порядка 100
и 10 000 моль/л, измерения свидетельствовали бы лишь о том, что обе эти
кислоты сильные, но различие в силе между ними неуловимо.
Сравнение сильных электронейтральных кислот, неразличимых в воде,
можно проводить в растворителе, настолько менее основном, чем вода, что
перенос протона от кислоты к растворителю будет в значительной степени
неполным; при этом достигается равновесие, положение которого можно
определить. Хлорная кислота, неизмеримо сильная в воде, является слабой
кислотой на основании определения константы диссоциации в гораздо менее
основном растворителе — в серной кислоте [1]. В свою очередь нейтральные
основания, которые в воде настолько слабы, что их сопряженные кислоты
являются неизмеримо более сильными, другими словами, основания, кото-
рые едва способны захватывать протон от воды, могут присоединять протон
в измеримой равновесной степени от более кислого растворителя. Так,
нитробензол, обладая ничтожной основностью в воде, будет умеренно слабым
основанием с измеримой константой основности в серной кислоте как раство-
рителе [2].
Аналогично в случае равновесия переноса протона от слишком слабых
кислот или к слишком сильным основаниям константы диссоциации кислот
или оснований в воде определить нельзя; однако зти константы можно изме-
рить в более основном или менее; кислом растворителе, например в жидком
аммиаке.
Растворитель, который, подобно воде, может либо терять, либо приобретать
протон, называется амфипротонным растворителем. Если обозначить такой
растворитель LH, то уравнения, определяющие величины Ка, Ке и Хавт0,
остаются такими же, только вместо частных обозначений Н3О+, Н2О
и ОН- соответственно вводятся более общие символы HLH+, LH и L“.
Константы автопротолиза ряда амфипротонных растворителей приведены
в табл. 191-
В печати обсуждался вопрос, всегда ли кислоты и основания располагаются
одинаково в соответствии с их силой в различных растворителях. Очевидно,
на этот вопрос следует ответить отрицательно. Другими словами, если вместо
кислоты или основания рассматривать другую кислоту или другое основа-
ние, образующиеся в результате изменения структуры вблизи от места,
в котором осуществляется отщепление или присоединение протона, то такое
изменение может оказать специфическое влияние (в отношении взаимодей-
ствия с молекулами растворителя). Сказанное относится, например, к вве-
дению орттго-заместителей в бензойную кислоту. С другой стороны, если
изменение структуры происходит достаточно далеко от места, в котором
.912
Кислоты и основания
происходит отщепление или присоединение протона, то можно ожидать,
что в случае кислот и оснований, резко различных по силе, при изменении
растворителя сохраняется порядок их силы. Насколько известно, это спра-
ведливо [5].
Таблица 191
Константы автопротолиза растворителей а
Растворитель Температура, °C Р^авто
Аммиак -33 22
Этиловый спирт 25 19,1
Метиловый спирт 25 16,7
Вода 25 14,0
Вода 100 12,3
Муравьиная кислота 25 6,2
Серная кислота 10 3,24
а Все значения, кроме приведенных в последней строке, взяты
у Гаммета [3]. Значение для серной кислоты определены Гиллеспи [4].
Такие константы равновесия, как константы кислотности, зависят от тем-
пературы; исходя из соответствующих температурных коэффициентов,
можно определить два термодинамических фактора, определяющих равно-
весие,— изменение энтальпии и энтропии. В большинстве известные кон-
станты диссоциации кислот определены с применением воды в качестве
растворителя и при температуре 25 °C. Таким образом, за исключением
немногих случаев, в настоящее время термодинамический анализ провести
нельзя, поскольку имеется слишком мало данных для общего сравнения
отдельных структурных эффектов, от которых зависит константа равнове-
сия. Поэтому в дальнейшем будут сравниваться только константы диссо-
циации кислот, т. е. различия в величинах свободной энергии. В принципе
следует ожидать, что структурные изменения вблизи от места переноса
протона должны оказывать влияние на изменение как энтальпии, так и энтро-
пии, тогда как структурные изменения вдали от места переноса протона
должны заметно влиять только на изменение энтальпии. Это связано с тем,
что изменение энтропии зависит главным образом от изменения конфигу-
рации в результате реакции за счет сольватной оболочки, а также с тем,
что сольватация носит локальный характер и зависит от сил ближнего
действия.
Можно пойти и дальше в оправдании скептического отношения к возмож-
ностям применения термодинамического анализа даже в той ограниченной
степени, в которой он мог бы быть использован. Очевидно, нет никаких
сомнений в том, что изменение знтальпии при температуре опыта не имеет
столь большого значения при рассмотрении структурных влияний, как
изменение свободной энергии или ее эквивалента — константы равновесия.
На основании статистических термодинамических данных Эванс и Полянки
[6] пришли к выводу, согласно которому изменение свободной энергии,
сопровождающее структурные превращения при заданной температуре
в большей степени соответствует изменению энтальпии при абсолютном
нуле, чем изменение энтальпии при той же заданной температуре. Этот
вывод убедительно подтверждается результатами наиболее тщательных
58-0772
913
Глава XIV
опытов, в частности данными, полученными Эверетом и Вайн-Джонсом [7],
предпринявшими попытку использовать изменение энтальпии при темпе-
ратурах опыта. Попытки вычисления изменений энтальпии при нулевой
температуре исходя из наблюдаемых на опыте изменений энтальпии при уме-
ренных температурах связаны со значительными трудностями, поскольку
эти изменения, т. е. изменения теплоемкости, велики (это объясняется глав-
ным образом тем, что вода в ионных сольватах, так же как и лед, имеет
гораздо более низкую теплоемкость, чем жидкая вода). Кроме того, оче-
видно, что изменение теплоемкости нельзя считать постоянным при экстра-
полировании от сравнительно высокой к нулевой температуре. Таким обра-
зом, практически наиболее удобным методом при структурных сопоставле-
ниях является метод, основанный на использовании констант равновесия.
Однако следует иметь в виду, что даже небольшие отклонения могут иметь
существенное значение.
б. Полярные эффекты дальнего действия: эффект поля. Наличие влияния
поля полярной группы на кислотно-основной центр, т. е. электростатиче-
ского влияния группы, воздействующей на данный центр, не связанного
с индуктивным эффектом или с эффектов! сопряжения, передающимися
по соответствующим связям, предвидел Бьеррум. Такой эффект Бьеррум [8]
связал с влиянием поля. Аргументация Бьеррума состояла в следующем.
Сопоставление двух стадий ионизации двухосновной кислоты, например
адипиновой, можно провести, исходя из того, что кислотный компонент
кислотно-основной системы во второй стадии остается тем же, что и в пер-
вой, за исключением того, что кислотный компонент второй стадии имеет
отрицательный заряд, вследствие чего можно не учитывать заряд отщеп-
ляющегося протона.
НО2С(СН2)4СО2Н НО2С(СН2)4СО2+Н+
НО2С(СН2)4СО2 ~О2С(СН2)4СО2 + ВН-
ЕСЛИ этот дополнительный отрицательный заряд не оказывает влияния
на кислотно-основное равновесие, то отношение констант кислотности двух
стадий диссоциации KailKa2 должно быть равно 4, поскольку первая кис-
лота имеет два положения, от которых может отщепиться протон, а ее сопря-
женное основание — одно положение, по которому может присоединиться
протон; в то же время вторая кислота имеет одно положение, от которого
возможно отщепление протона, а ее сопряженное основание — два места
для присоединения протона. Бьеррум предполагал, что электростатическое
влияние дополнительного заряда прибавляется к работе удаления отщеп-
ляющегося протона, способствуя тем самым относительному уменьшению К„2
и, следовательно, увеличению отношения KaJKa2 до значения больше 4.
Все подобные соотношения в случае симметричных дикарбоновых кислот
действительно превышают 4. На основании простого подсчета величины
электростатической работы, которая затем рассматривалась в качестве соот-
ветствующего инкремента в выражении для свободной энергии, Бьеррум
вывел формулу
In (^ai/4/<a2) = Ne^RTDr
где D — эффективная диэлектрическая проницаемость, аг — расстояние
между кислотными атомами водорода. При измерении константы диссо-
циации кислоты в воде при 25 °C Бьеррум принимал величину D равной
диэлектрической проницаемости растворителя, т. е. 80. С помощью этой
формулы Бьеррум подсчитал значения г для многих симметричных двух-
914
Кислоты и основания
основных кислот и получил удовлетворительные результаты. Для адипино-
вой кислоты расчет по Бьерруму приводит к вполне вероятному значе-
нию г = 8 А (0,8 нм).
Гейн и автор книги [9] предприняли количественную проверку теории Бьер-
рума для ряда симметричных двухосновных кислот, включая все гомологи-
ческие кислоты нормального строения вплоть до азелаиновой кислоты,
а также многие ^-алкилированные глутаровые кислоты и алкилированные
малоновые кислоты. Для кислот, у которых длина нормальной цепи больше,
чем в глутаровой кислоте, были получены правдоподобные значения г;
для кислот же с более короткими цепями и всех алкилированных кислот
полученные значения заметно занижены. Такой результат наводит на мысль
о том, что для этих групп кислот принятое значение D слишком велико.
Наблюдаемый результат частично можно объяснить одновременным дей-
ствием индуктивного эффекта; несомненно, этот эффект оказывает влияние-
в случае малоновой и, вероятно, янтарной кислот, однако трудно допустить,
что индуктивный эффект может вызвать значительное различие в свойствах
кислот, содержащих большее число простых связей между карбоксильными
группами, чем в глутаровых кислотах. Значительное, но все же недостаточ-
ное уточнение полученных значений было достигнуто путем учета эффекта
электрического насыщения. Этот эффект состоит в том, что при столь значи-
тельных силах, которые действуют вблизи ионного центра, молекулярный
механизм диэлектрической поляризации начинает играть менее существен-
ную роль, вследствие чего среда физически не способна повышать индукцию
пропорционально полю и величина отношения, выражающего диэлектри-
ческую проницаемость, становится меньше [10]. Однако, как указано ниже,
имеется другая важная причина количественных несоответствий, наблю-
дающихся при использовании уравнения Бьеррума, которая была выяснена
в ряде исследований. В основном она связана с тем, что пространство, кото-
рое в расчете Бьеррума считается занятым молекулами растворителя, в дей-
ствительности частично занято молекулами, эффективная диэлектрическая
проницаемость которых, особенно если они не могут свободно вращаться
по отношению к полю, должна быть ближе к 2, чем к 80.
Эйкен [И] провел расчет, сходный с расчетом Бьеррума, с целью опреде-
ления непосредственного влияния диполя. Принцип расчета может быть
проиллюстрирован на примере влияния поля атома хлора в хлоруксусной
кислоте, увеличивающего константу диссоциации этой кислоты по сравне-
нию с уксусной. В этом случае возникает дополнительный член в выра-
жении для работы, обусловленный диполем связи углерод — хлор, харак-
теризуемым моментом ц и отклонением 0 в направлении диссоциирующего
протона. При учете только этого эффекта отношение констант диссоциации
может быть выражено уравнением
1п(ла /Ка ) = Ne\icosQlRTDr1.
Если в этом уравнении принять величину D равной 80, то для величины г
получается абсурдно малое значение 0,6 A (6-10-2 нм). Однако Смолвуд [12]
считает, что значение D в этих случаях лишь немногим превышает единицу,
поскольку пространство, имеющее наиболее существенное значение для элек-
тростатического расчета, занято в основном частью молекулы кислоты.
Конечно, значительное различие будет наблюдаться в зависимости от того,
принято ли значение D равным 1, 2 или 4; принимая значение D достаточно
низким, можно формально выяснить, возможен ли индуктивный эффект
Льюиса.
58* 915
Глава XIV
Дихотомия значений диэлектрической проницаемости хорошо иллюстри-
руется некоторыми данными Уэланда [13], приведенными в табл. 192. В слу-
чае длинной и тонкой молекулы большая часть пространства, для которого
проводится электростатический расчет, в действительности заполнена раство-
рителем; в этом случае принимается значение D, близкое к верхнему преде-
лу 80. В случае же коротких или толстых молекул пространство, к которому
относится расчет, заполнено главным образом самой молекулой, в связи
с чем более предпочтительным является значение D, близкое к его нижнему
пределу, т. е. к 1.
Таблица 192
Длины молекул, вычисленные при допущении полной зависимости силы кислот
от эффекта поля полярного заместителяа
Кислота Длина рассчитанная Длина модели
D = 80 (по Бьерруму) 0=1 (по Смолвуду) D = 2 и 80 (по Кирквуду и Узст- хеймеру) максималь- ная 6 средняя в
Адипиновая 8,1 635 7,8 9,0 5,6
Хлоруксусная 0,6 5,1 3,0 3,4 3,0
а Уэланд, частное сообщение.
6 Максимально ненапряженные связи.
в Средняя величина с учетом вращения вокруг ненапряженных связей, оцененная для электро-
статического взаимодействия концевых групп.
Кирквуду и Уэстхеймеру [14] принадлежит наиболее удачная попытка
в разработке такой модели, в которой некоторая часть пространства была
бы занята молекулой, а некоторая его часть — растворителем; такая модель
позволяет отдельно вычислить член, отвечающий электростатической части
работы. Кирквуд и Уэстхеймер считали, что полярная группа и центр дис-
социации жестко закреплены в эллипсоидальной молекуле с одинаковой
диэлектрической проницаемостью, равной двум, причем эта молекула окру-
жена средой с диэлектрической проницаемостью, равной 80. Конечно,
допущение, согласно которому молекула имеет точную эллипсоидальную
форму (причем происходит локализация центров полярности и диссоциации
внутри эллипсоида), а также допущение, что диэлектрическая проницае-
мость равна 2, являются произвольными в этой теории; однако такие допу-
щения дают возможность одинаково подходить к рассмотрению различных
по длине и толщине молекул. Результаты некоторых успешных расчетов,
относящихся к рассмотренным выше примерам, приведены в табл. 192.
Все попытки расчета влияния поля в количественном отношении вызывают
сомнение, однако имеется путь, который позволяет качественно судить
о существовании эффекта поля. Необходимо выбрать такую систему, чтобы
эффект поля конкурировал с внутренней передачей полярного влияния по
механизму сопряжения и чтобы было известно, что эффект поля имеет про-
тивоположное направление; тогда при достаточно большой величине эффекта
поля последний может перекрыть эффект сопряжения. Можно привести два
примера, в которых внутренний полярный эффект имеет одно направление,
а эффект поля — противоположное. Такие опыты были проведены Роберт-
сом и сотрудниками. Известно, что передаваемый внутримолекулярно поляр-
ный эффект сопряжения в бензольном кольце проявляется сильнее в более
удаленном мра-положении, чем в более близком мета-положении. Для этих
916
Кислоты и основания
двух положений величина эффекта поля должна изменяться в обратном поряд-
ке. Внутримолекулярно передаваемый эффект и, в частности, различие в его
силе для пара- и .мета-положений могут оказывать очень большое влияние на
сами атомы углерода ароматического ядра. Однако такие эффекты (а также
и различие между ними) могут быть ослаблены введением одинаковых заме-
стителей в оба положения, которое сопровождается удлинением пути внутри-
молекулярной передачи эффекта, что в свою очередь может быть использовано
для изучения полярных эффектов на эти заместители. Это облегчает проявле-
ние эффекта поля (при условии, если он остается достаточно сильным на рас-
стояниях рассматриваемого порядка) и позволяет проиллюстрировать его
отличительные признаки, например величину влияния из мета- и пара-
положений. Наилучшим примером эффекта поля с этой точки зрения являет-
ся влияние свободного заряда, вызывающего сильный эффект дальнего
действия.
В исследованных примерах [15] группа —N+(CH3)3 представляла собой
во всех случаях активный полюс, в то время как заместителями в мета-
ns. пара-положениях были NH, в одном случае и СООН — в другом; полярные
эффекты на эти заместители определялись путем измерения констант
диссоциации.
м- или CHS)3NC6H4NH3 (CH3)sNCeH4NH2 + Н+
м- или n-(CHs)3NC6H4CO2H "7^ [(CHs)sNC6H4COO--|-H+
Результаты исследований приведены в табл. 193. В каждом случае триметил-
аммониевый полюс увеличивает кислотность, однако если он находится
в .мета-положении, то такое увеличение оказывается более сильным, чем
когда Н(СН3)з находится в пара-положении, что и можно было ожидать,
исходя из строения рассматриваемой системы.
Таблица 193
Преобладание влияния положительного полюса на заместители
в мета- и яара-положениях для случая кислотно-основного [равновесия
Кислота
незамещенное вещество •m-(CH3)3N+ n-(CHg)3N +
Ионы анилиния в воде при 25 °C 4,57 2,26 2,51
Бензойные кислоты в 50%-ном спирте при 25 °C 5,71 4,22 4,42
Первое количественное исследование чистого эффекта поля было проведено
Битлстоуном и Ирвином [16]. Авторы определили спектрофотометрически
равновесие ионизации протонированной воды, являвшейся лигандом окта-
эдрического комплекса трехвалентного железа гемов метагемоглобина крови
и дали теоретическое объяснение процессу, исходя из диэлектрической тео-
рии Кирквуда и Уэстхеймера. Известны гемоглобины, отличающиеся только
одним пептидным звеном в определенном положении определенной поли-
пептидной цепи. Так, три гемоглобина крови человека, обозначаемые бук-
вами А (нормальный гемоглобин), S (аномальный гемоглобин, встречающийся
при серповидноклеточной анемии) и С (другая разновидность аномального
гемоглобина), отличаются друг от друга только шестым пептидным звеном,
917
Глава XIV
считая от карбоксильного конца р-полипептидной цепи. В гемоглобине А
этим звеном является глутаминовая кислота
HOOCCH(NH2)CH2CH2COOH
в гемоглобине S — валин (а-аминоизовалериановая кислота)
(CH3)2CHCH(NH2)COOH
в гемоглобине С — лизин (а,е-диаминокапроновая кислота)
HOOCCH(NH2)(CH2)sNH2 1
При значениях pH око ло 8, наиболее подходящих для измерения кислотно-
сти метагемоглобина, свободные кислотные и основные центры пептидных
звеньев глутаминовой кислоты и лизина будут ионизованы и ионные
заряды этих звеньев будут равны —1, 0 и -|-1 соответственно. Ионные заряды
находятся на расстоянии 28,5А (2,85 нм) от атома железа структуры гема;
при замене гемоглобина S на А и С на S вводятся две дополнительные единицы
положительного заряда на расстоянии 30 А (3 им) от ионизирующегося ОН-
центра. На таких расстояниях можно полностью пренебречь межмолекуляр-
ными силами любого типа, которые с расстоянием ослабевают быстрее, чем
кулоновские силы, и таким образом остается чистый эффект поля. Это при-
мер одного из редких случаев в физической органической химии, когда боль-
шая величина молекулярного веса способствует количественному исследова-
нию. Только в случае большой молекулы с жесткой структурой возможны
такие большие внутримолекулярные расстояния, которые можно измерить
с большой точностью. Другим упрощающим обстоятельством является то,
что форма глобулы протеина хорошо аппроксимируется эллипсоидом с осями,
равными 64, 55 и 50 А (6,4, 5,5 и 5,0 нм). Следовательно, диэлектрическую
проницаемость, от которой зависит взаимодействием между переменным
зарядом с известным положением и местом кислотно-основной диссоциации,
можно точно вычислить.
Битлстоун и Ирвин вывели формулу для расчета влияния кулоновского поля
на изменение свободной энергии, энтропии и энтальпии при ионизации мета-
гемоглобина. Кроме того, опи дали теоретическое объяснение случаю, когда
влияние структурного изменения на кислотно-основное равновесие является
полностью кулоновским. Они исходили из того, что потенциал изменяется
обратно пропорционально первой степени расстояния, и любые взаимодей-
ствия с быстро затухающим силовым полем не вносят существенного вклада.
Если имеется структурное изменение такого типа, то изменения в величинах
AG, АН и T\S должны быть пропорциональны друг другу или, другими
словами, зависимость между любыми двумя величинами из AG, \Н и TAS
(например, АН от PAS) должна быть линейной. Это справедливо в случае
гемоглобинов, для которых наблюдается достаточно чистый эффект поля
(гл. XVI).
Для трех гемоглобинов крови человека A, S и С, структурные различия меж-
ду которыми локализуются на расстоянии 30 А (3 нм) от центра ионизации,
такая линейная зависимость действительно существует. При исследовании
поведения некоторых других аномальных гемоглобинов крови человека
с известной структурой, одни из которых, подобно S и Р, имеют специфиче-
ские различия в месте, удаленном от гема, а другие — в месте поблизости
от гема, был сделан вывод, что тесту на чистый эффект поля удовлетворяют
те системы, в которых изменение заряда происходит на расстоянии более
10 А (1 нм) от атома железа. В том случае, когда изменение заряда происхо-
918
Кислоты и основания
дит на меньшем расстоянии от атома железа, чистого эффекта поля не на-
блюдается.
Исследован ряд гемоглобинов крови животных, строение которых детально
не известно. Все они имеют различные термодинамические параметры иони-
зации. Однако, если построить для них график зависимости T&S от кН, то
все точки расположатся на одной прямой, на которой также будут лежать
Рис. 51. Зависимость 7'AS от ДЯ для
ионизации метагемоглобинов,
гемоглобинов животных и че-
ловека в воде при 20 °C и ион-
ной силе 0,05 моль/л [17].
1 — мышь; 2 — крыса; з — обезь-
яна бабуин; 4 — обезьяна Patas;
5 — мартышка липа; 6 — свинья;
7 — собака; 8 — гиена; 9 — обезь-
яна Tantalus; 10 — кошка; 11 —
голубь; 12 — лошадь; 13, 14 и 15—
гемоглобины A, S и С крови че-
ловека; 1в — морская свинка; 17—
ящерица; 18 — летучая мышь;
19 — землеройка; 20 — утка; 21 —
индюк; 22 — курица; 23 — цесар-
ка; 24 — норова; 25 — обезьяна
Mangabey; 2G — кролик.
точки, соответствующие гемоглобинам крови человека A, S и С. Это показано
на рис. 51. Из этого факта был сделан вывод о том, что на расстоянии в пре-
делах 10 А (1 нм) от каждого атома железа находится характеристическая
структура любого нормально функционирующего гемоглобина и структур-
ные изменения на этих расстояниях приводят к серьезным изменениям свойств
и, вероятно, такого жизненно важного свойства, как связывание
кислорода.
В. Полярные влияния, не зависящие от сопряжения: индуктивный
эффект. Многочисленные измерения констант диссоциации карбоновых
кислот, проведенные Оствальдом, показали, что сила кислот в значительной
степени зависит от природы заместителей и связана, очевидно, как с их элек-
трополярностью, так и со степенью их ненасыщенности, а также со степенью
удаленности заместителя от карбоксильной группы. В 1899 г. Генрих [18]
отметил влияние ненасыщенности на силу кислот. В 1902 г. Форлендер [19]
обратил особое внимание на связь между силой карбоновых кислот и элек-
трополярным характером заместителей; в дальнейшем эти данные были сопо-
ставлены с влиянием заместителей на многие химические реакции. Прибли-
зительно к этому же времени относятся попытки Вегшейдера [20] количествен-
но выразить влияние заместителей на силу кислоты. Он вычислил множите-
ли, позволяющие количественно выразить влияние различных заместителей
в различных положениях насыщенной цепи на константы диссоциации кар-
боновых кислот. В 1909 г. Флюршейм [21] изучал силу ароматических кислот
и оснований, устанавливая связь полярных влияний с другими свойствами.
В начальный период развития электронной теории в 1923 г. Льюис [22] пола-
гал, что полярные влияния передаются посредством индукции. В 1933 г.
автор книги [23] связал полярные влияния с мезомерией. В конце тридцатых
и в начале сороковых годов вопрос о полярных влияниях был рассмотрен
более обстоятельно Уотерсом [24], Уотсоном [25], Диппи [26], а также Брен-
919
Глава XIV
чем и Кальвином [27] с учетом различных сторон проблемы. Указанные
исследования составляют и в настоящее время основу изучения внутримолеку-
лярных полярных влияний на силу кислот.
В табл. 194 приведены константы диссоциации жирных кислот. Основную
закономерность можно выразить рядом кислотности
муравьиная > уксусная > высшие кислоты
или, другими словами, замещение атомов водорода в муравьиной или уксус-
ной кислоте метильными группами приводит к ослаблению кислот.
Таблица 194
Термодинамически исправленные константы диссоциации
жирных кислот в воде при 25 °C
Кислота Ю5Ка Кислота Ю5ка
Муравьиная 17,72 Энантовая 1,28
Уксусная 1,75 Каприловая 1,27
Пропионовая 1,33 Пеларгоновая 1,11
и-Масляная 1,50 Изомасляная 1,38
и-Валериановая 1,44 Пивалиновая (триметил- 0,93
Капроновая 1,39 уксусная)
Эти данные согласуются с известной электроположительностью метильной
и других алкильных групп (+7-эффект) в карбоновых кислотах; иными сло-
вами, алкильные группы намного легче, чем водород, отдают свои электро-
ны электроноакцепторному центру молекулы (гл. II, разд. 3,в). Характер
изменения констант диссоциации первых трех гомологов находится в соот-
ветствии с рассмотренным ранее ослаблением индуктивного эффекта через
простую связь. В то же время следует иметь в виду, что гиперконъюгация
должна способствовать значительному увеличению различия между первы-
ми двумя кислотами и незначительному уменьшению различия между
следующими кислотами. Влияние поля едва ли может иметь существенное
значение в таких молекулах, в которых, помимо самого центра диссоциации,
нет полярных заместителей.
Факт незначительных отличий между константами диссоциации высших
гомологов не получил до сих пор удовлетворительного объяснения. Для
высших кислот соответственно наблюдаются незначительные различия в тер-
модинамических или кинетических константах при переходе от одного гомо-
лога к другому. Причина незначительности таких различий является, несом-
ненно, сложной, и этот вопрос не может быть разрешен при современном
уровне знаний.
Введение двойной связи в карбоновые кислоты приводит к заметному повы-
шению их констант диссоциации. Этот эффект проявляется в меньшей сте-
пени при увеличении числа простых связей между ненасыщенной и карбо-
ксильной группами. Сказанное можно иллюстрировать данными, приведен-
ными в верхнем разделе табл. 195. Введение ненасыщенных заместителей
бензоидного типа также приводит к увеличению силы кислоты, причем это
увеличение по порядку величины не отличается от увеличения, наблюдаемого
при введении двойной связи. Увеличение числа простых связей между
карбоксильной и фенильной группами также ослабляет влияние аромати-
920
Кислоты и основания
ческой группировки. Соответствующие примеры приведены во втором
разделе табл. 195. Введение ацетиленовой тройной связи вызывает очень
резкое увеличение силы кислоты, что иллюстрируется последним разделом
таблицы.
Таблица 195
Термодинамически исправленные константы диссоциации
ненасыщенных кислот в воде при 25 °C
Кислота Формула 105Ка
Акриловая сн2=снсоон 5,56
Пропионовая СН3СН2СООН 1,33
В инилуксусная СН2 = СНСН2СООН 4,48
/t-Масляная СН3СН2СН2СООН 1,50
Аллилуксусная СН2 = СНСН2СН2СООН 2,11
«-Валериановая СН3СН2СН2СН2СООН 1,44
Бензойная С6Н5СООН 6,27
Циклогексанкарбоновая СвНиСООН 1,26
Фенилуксусная С6Н5СН2СООН 4,88
Циклогексилуксусная СеНцСНгСООН 1,58
Р-Фенилпропионовая С6Н5СН2СН2СООН 2,19
/t-Масляная СН3СН2СН2СООН 1,50
тракс-Кротоновая СН3СН=СНСООН 2,03
Тетроловая СН3С = ссоон 222,8
Р-Фенилпропионовая С6Н5СН2СН2СООН 2,19
т ракс-Коричная С6Н5СН = СНСООН 3,65
^ис-Коричная С6Н5СН = СНСООН 13,2
Фенилпропиоловая С6Н5С = ссоон 590
Как было указано в гл. II, разд. 3,в, насыщенный углеродный атом образует
о-связи за счет гибридных орбиталей, s-характер которых равен 1/4, тогда
как этиленовый или бензоидный атом углерода образует простые связи за
счет орбиталей, s-характер которых равен 1/3, а ацетиленовый атом углеро-
да — за счет орбиталей с s-характером, равным 1/2. Следовательно, по срав-
нению с насыщенным углеродным атомом этиленовые и бензоидные атомы
углерода являются несколько более электроотрицательными, тогда как
ацетиленовый атом углерода значительно более электроотрицателен. Эти
ненасыщенные атомы вызывают индуктивные эффекты (—Z), которые должны
ослабляться вдоль цепи насыщенных углеродных связей. Качественно эти
положения отражены в табл. 195. Однако имеются некоторые детали, тре-
бующие пояснений.
При сопоставлении констант диссоциации таких кислот, как акриловая,
винилуксусная и аллилуксусная или бензойная, фенилуксусная и Р-фенил-
пропионовая, видно, что различия в первой группе кислот не столь велики по
сравнению с различиями во второй, что соответствует обычному уменьшению
полярных эффектов вдоль цепи простых связей. Это, очевидно, объясняется
следующим. Мезомерный эффект влияет на кислотную силу акриловой и бен-
921
Глава XIV
войной кислот; если бы он был единственным эффектом, то должно было
наблюдаться понижение их кислотной силы по сравнению с соответствую-
щими насыщенными кислотами (разд. 1,д). Наблюдаемое усиление кислот
объясняется тем, что индуктивный эффект преобладает над более слабым
и противоположно направленным мезомерным эффектом.
Сравнение заметно отличающихся констант диссоциации цис- и транс-
коричных кислот подтверждает такую точку зрения. Рассмотренные поло-
жения четко применимы к щракс-коричной кислоте. Однако в ^ис-коричной
кислоте фенильная и карбоксильная группы должны быть повернуты отно-
сительно плоскости связей этиленовой группы так же, как в цис-азобензоле
две фенильные группы выведены из плоскости связей азогруппы (гл. III,
разд. 4,в, рис. 23). Следовательно, в tywc-коричной кислоте отсутствует
сопряжение между функциональными группами, а также мезомерный или
какой-нибудь другой эффект. На примере этой кислоты лучше, чем на дру-
гих подобных примерах, можно показать, как влияет индуктивный эффект
этиленового центра в а,Р-ненасыщенных кислотах.
В любой серии кислот при переходе от насыщенной к а,|3-олефиновой и а,Р-
ацетиленовой кислотам сила кислоты возрастает, причем различие в силе
между второй и третьей кислотами такого ряда значительно выше различия
между силами первой и второй кислот. Это находится в соответствии с выво-
дом, согласно которому изменение содержания s — p-составляющих орби-
талей атомов углерода, участвующих в образовании связи, при переходе от
второй к третьей кислоте указанного ряда вдвое больше, чем при переходе
от первой ко второй. Если не рассматривать tywc-коричную и другие непланар-
ные г{ис-кислоты, то наблюдаемое большое различие в константах диссоциа-
ции может вызываться также другим фактором. Ввиду того что сопряжение
ацетиленовой связи с карбоксильной группой стереохимически возможно
только за счет одной связывающей орбитали л-оболочки, можно предполо-
жить, что мезомерное влияние, действующее в противоположном направле-
нии, чем индуктивный эффект, в случае тройной связи не окажется боль-
шим, чем для двойной. Таким образом, численные данные свидетельствуют
о том, что если бы не было мезомерного эффекта, то сила кислот в ряду жир-
ная, а,р-олефиновая и а,Р-ацетиленовая относилась бы одна к другой как
1 : 10 : 1000 и разность логарифмов констант диссоциации второй и третьей
кислот этого ряда была бы вдвое больше, чем соответствующая разность для
первой и второй кислот, отражая этим удвоение изменений орбитальных
составляющих.
При замещении атома водорода в жирной кислоте таким электроотрицатель-
ным атомом, как хлор, константа кислотности возрастает. С увеличением
расстояния между заместителем и карбоксильной группой такое возрастание
силы кислот становится значительно менее заметным, что иллюстрируется
данными, приведенными в табл. 196 и 197. Приведенные значения констант
диссоциации являются, вероятно, результатом совместного действия индук-
тивного (—7) эффекта и эффекта поля. Для раздельного учета этих двух
влияний необходимы более совершенные, чем имеются в настоящее время,
методы расчета.
Влияние формально нейтральных полярных групп на изменение кислотно-
сти насыщенных кислот зависит от положения замещающего атома в таблице
Менделеева, что видно из табл. 197. Приведенные порядки величин соответ-
ствуют порядкам величины электроотрицательности и уже рассмотренным
рядам (гл. II, разд. 3,в) для индуктивного эффекта (—7)
CR3 < NR2 < OR < F и I < Br < Cl <JF
922
Кислоты и основания
Таблица 196
Константы диссоциации хлорзамещенных жирных кислот
в воде при 25 °C
Кислота 1°»Ка
незаме- щенная а-С1 Р-С1 V-C1 6-С1
Уксусная Пропионовая «-Масляная «-Валериановая 1,75 1,33 1,50 1,44 139 132 106 10,1 8,9 3,0 1,9
незаме- щенная моио- хлор- заме- щештая дихлор- заме- щенная трихлор- замещенная
Уксусная 1,75 139 5500 222 000
Электроположительность дейтерия по сравнению с обычным водородом была
обнаружена при исследовании влияния дейтериевого заместителя на силу
кислот [28]. Это влияние оказалось малым, но постоянным по своему харак-
теру: сила кислот всегда уменьшалась. Некоторые примеры [29, 30] приведе-
ны в табл. 198. Влияние дейтерия в муравьиной, уксусной и пивалиновой
кислотах должно быть преимущественно индуктивным. В бензойной кислоте
и феноле индуктивное влияние будет частично усиливаться сопряжением.
г. Полярные эффекты, включающие сопряжение: мезомерный эффект.
При рассмотрении влияния заместителей у ненасыщенного углеродного ато-
ма в олефиновых или ароматических кислотах можно видеть, что, кроме
индуктивного эффекта и эффекта поля, появляются и накладывающиеся на
них мезомерные эффекты. На это указывают значения констант кислотности *
нескольких групп ароматических кислот, приведенные в табл. 199. В этой
Таблица 197
К онстанты диссоциации замещенных уксусных кислот в воде при 25 °C
X в сщхсоон X в сщхсоон Ю5ка X в СН2ХСООН 105Ка
С6н5сн2 2,2 СН3СН2 1,5 F 260
c6h5nh 3,9 CH3NH — С1 139
С6Н5О 68 СН3О 33 Вг 128
F 260 F 260 I 67
* Данные взяты из статьи Диппи [26]. Измерения констант кислотности для соеди-
нений серий А — Г (табл. 199) проведены Диппи и сотрудниками, а для соединений серии
Д — Бренчем и сотрудниками.
923
Глава XIV
таблице приведены также значения констант кислотности орпго-замещенных
кислот, но обсуждаться они будут ниже. Значения констант диссоциации
мета- и пара-замещенных кислот указывают прежде всего на уже рассмот-
ренные выше общие эффекты полярности: введение электроположительных
алкильных групп всегда приводит к ослаблению кислоты, введение же элек-
троотрицательных заместителей (гидроксильной и алкоксильных групп,
атомов галогенов и нитрогрупп, кроме отдельных особых случаев) — к уси-
лению кислоты, причем влияние галогенов выражено значительно сильнее,
чем влияние гидроксила или алкоксила, а влияние нитрогруппы, имеющей
Таблица 198
Влияние дейтерия в качестве заместителя на силу кислот
в воде при 25° С
Дейтерированные кислоты kh/kd pkd-pkh ApJf на 1 атом дейтерия
DCOOH 1,085 0,035 0,035
CD3COOH 1,033 0,014 0,005
(CD3)3CCOOH 1,042 0,018 0,002
C6D5COOH 1,024 0,010 —
c6d5oh 1,12 0,05
две полярные связи, оказывается наибольшим. Среди леига-замещенных
соединений нет качественных исключений из этого общего правила; однако
они встречаются среди пара-замещенных кислот в случае заместителей,
которые имеют неподеленные электроны, способные к сопряжению с арома-
тической системой. Некоторые из этих пара-заместителей, в частности окси-
и алкоксигруппы, существенно ослабляют кислоты. В тех случаях, когда
введение пара-заместителей, например галогенов, не приводит к ослаблению
кислоты, наблюдаемое увеличение степени диссоциации кислоты является
значительно меньшим, чем при введении тех же заместителей в .меига-псло-
жение; в то же время полярные влияния других нейтральных групп незави-
симо от знака обычно сильнее проявляются в пара-, чем в лгеига-поло-
жении.
Известны также случаи уменьшения кислотности при введении пара-замести-
телей, содержащих неподеленные электроны рядом с бензольным ядром. Это,
несомненно, связано с проявлением мезомерного эффекта -\-М [20], более
сильного в случае кислородных заместителей по сравнению с галогенами,
в соответствии с известной закономерностью OR > Hal для — ЛГ-эффекта
(гл. II, разд. 3,д).
Из данных табл. 199 видно, что для пара-замещенных кислот наиболее зна-
чительное изменение кислотности наблюдается в случае ряда фенилборных
кислот; оно постепенно уменьшается от одного ряда к другому в следующем
порядке:
фенилборная > бензойная > коричная > фенилуксусная/> |3-феиилпропионовая
Такая последовательность находится в строгом соответствии с мезомерный
эффектом. В первых трех случаях происходит сопряжение между влияющей
группой, бензольным кольцом и центром кислотности, причем в первом слу-
924
Таблица 199
Термодинамически исправленные константы диссоциации ароматических кислот
А. Замещенные бензойные кислоты (105ЛГо в воде при 25 °C)
Положение заместителя СНз н ОН ОСНз F NO2
орто 12,3 6,27 105 8,06 54,1 671
мета 5,35 6,27 8,3 8,17 13,6 32,1
пара 4,24 6,27 2,62 3,38 7,22 37,0
Б. Замещенные фенилуксусные кислоты (1057fa в воде при 25° С)
Положение заместителя СНз Н ОСНз С1 NO2
орто 4,88 8,60 9,90
мета 4,88 7,24 10,8
пара 4,27 4,88 4,36 6,45 14,1
В. Замещенные р-фенилпропионовые кислоты (1087fa в воде при 25° С)
Положение заместителя СНз Н ОСНз С1 NO2
орто 2,17 2,19 1,57 2,65 3,13
мо 2,19 2,22 2,60
пара 2,07 2,19 2,04 2,47 3,36
Г. Замещенные транс-коричные кислоты (105А'а в воде при 25 °C)
Положение заместителя СНз Н ОН ОСНз CI NO2
орто 3,16 3,65 2,44 3,45 5,83 7,07
мета 3,61 3,65 4,00 4,21 5,08 7,58
пара 2,73 3,65 — 2,89 3,86 8,99
Д. Замещенные фенилборные кислоты (i№Ka в 25%-ном водном С2Н5ОН при 25 °C)
Положение заместителя СНз н ос2н5 С1 no2
орто 0,261 1,97 0,91 14,0 5,6
мета 1,4 1,97 3,05 13,5 69
пара 1,0 1,97 0,608 6,30 98
Глава XIV
чае система является наиболее, а в третьем — наименее компактной
Х^^Х1в(С)Н)г
— ОН
ch^qj.jY с^
V ОН
В четвертом и пятом случаях сопряжение может быть только между замести-
телем и бензольным кольцом. Следовательно, поляризующий эффект пара-
заместителя ослабляется при наличии между бензольным кольцом и груп-
пой СООН простой связи; еще резче ослабление наблюдается в случае двух
простых связей, как это имеет место в пятом случае.
Данные по константам кислотности позволяют определять характер измене-
ния +М-эффекта в зависимости от периода таблицы Менделеева, к которому
принадлежит заместитель, например в случае группы галогенов. В табл. 200
приведены константы кислотности* моногалогенпроизводных бензойных
Таблица 200
Термодинамически исправленные константы диссоциации галогензамещенных
ароматических кислот и оснований
Положение заместителя н F С1 Вт I
А . Замещенные бензойные кислоты (105Аа в воде при 25 °C)
орто 6,27 54,1 114 140 137
мета 6,27 13,6 14,8 15,4 14,1
пара 6,27 7,22 10,5 10,7
Б. Замещенные бензойные кислоты (1057Сав 50%-ном водном СН3ОН при 18 °C)
орто 0,513 6,61 7,08 7,08 6,6
мета 0,513 1,41 1,45 1,35 1,41
пара 0,513 0,832 1,00 0,933 1,00
В. Замещенные фенилборные кислоты (1010Аа в 25%-ном водном C2HjOH при 25 °C)
орто 1,97 14,0
мета 1,97 11,0 13,5 14,6
пара 1,97 3,66 6,30 7,26
Г. Замещенные фенолы (1010Аа в 30%- пом водном С2Н5ОН при 25 °C)
орпго 0,32 4,27 10,2 9,78 9,12
мета 0,32 1,51 4,90 4,37 3,89
пара 0,32 0,26 1,32 1,55 2,19
Д. Замещенные анилины (1О12.Йф в 30%-ном водном С2Н5ОН при 25 °C)
орто 126 2,95 1,35 1,00 0,36
мета 126 10,5 8,51 7,94 7,59
пара 126 120 28,8 21,9 15,1
* Эти данные тоже взяты из статьи Диппи [26]. Измерение силы кислот проведены:
для серии А Диппи и сотрудниками, для серии Б Купом и Вассерманом, для серии В Брен-
чем и сотрудниками и для серий Г и Д Беннетом, Бруксом и Глесстоном.
926
Кислоты и основания
кислот, фенилборных кислот и фенолов, а также константы основности
моногалогензамещенных анилинов. В таблицу включены также данные для
оршо-галогензамещенных, однако пока эти данные обсуждаться не будут.
Можно заметить, что введение галогена в мета- и иара-положения почти
всегда приводит к усилению кислотности и ослаблению основности, однако
соответствующие эффекты проявляются всегда сильнее в случае мета-
замещенных, чем в случае ипра-замещенных. Атом фтора, введенный в пара-
положение, немного даже уменьшает кислотность фенола, лишь незначи-
тельно усиливает кислотность бензойной кислоты и почти не уменьшает
основности анилина. Эти наблюдения представляют собой частную иллю-
страцию общей закономерности, согласно которой ослабление влияния гало-
генов в геара-положении по сравнению с лгеша-положением должно быть
наибольшим в случае фтора и быстро уменьшаться в ряду галогенов при
переходе от фтора к иоду. Иными словами, уменьшение влияния на кислот-
ность, зависящее от мезомерного эффекта, происходит в последовательно-
сти, уже приведенной выше для -фУИ-эффекта (гл. 1, разд. 3,д):
? > Cl > Br > I
Константы диссоциации галогензамещенных бензойной и фенилборной кис-
лот позволяют сделать вывод о влиянии фтора по сравнению с другими гало-
генами на интенсивность мезомерного эффекта; данные для фенолов и анили-
нов характеризуют влияние всех четырех галогенов. Исторически дело
обстояло таким образом, что указанные соотношения теоретически не были
предсказаны, а были обнаружены при изучении кислотности Бренчем с сотр.
[31] (на примере галогенфенилборных кислот), Диппи, Уотсоном и Уильям-
сом [32] (на примере галогензамещенных ароматических кислот), а также
Беннетом и Глесстоном с сотр. [33] (на примере галогенфенолов и галоген-
анилинов). Позднее Ремик [34] предложил электронную интерпретацию
этого явления (гл. II, разд. 3,д).
Напомним, что эта интерпретация состоит в том, что различие в размерах
атома углерода и замещающего атома ограничивает возможность перекрыва-
ния атомных р-орбиталей, необходимого для увеличения кратности связи
и вызывающего Ц-М-эффект. Из этого следует, что ф-М-эффект будет умень-
шаться с увеличением атомного номера замещающего атома не только среди
галогенов, но и среди элементов других групп системы Менделеева. Это было
подтверждено Бейером и Барретом [35] для ряда кислород, сера, селен при
изучении констант диссоциации производных бензойной кислоты, замещен-
ной в мета- и гаара-положениях группами —ОСН3, —SCH3 и —SeCH3.
Таблица 201
Константы диссоциации (неисправленные) замещенных бензойных кислот
(105Ка в 30%-ном водном С3Н5ОН при 20 °C)
Положение заместител я н ОСНз SCH3 SeCH3
мета 1,57 1,93 1,82 1,84
пара 1,57 0,78 0,97 1,01
В табл. 201 приведены константы диссоциации этих кислот. В то время как
введение этих заместителей в жеша-положение приводит к усилению кисло-
ты, введение их в геара-положепие вызывает ослабление кислотности, при-
927
Глава XIV
чем ослабляющий эффект уменьшается с увеличением атомных номеров. Как
уже указывалось в гл. XIII, разд. 2,а, подобный же вывод был сделан этими
исследователями при изучении равновесия между бензальдегидом и его
циангидрином. Таким образом, как и указывалось в гл. II, разд. 3,д, измене-
ние -J-M-эффекта элементов группы кислорода выражается рядом
ORJ >SI|> SeR.
д. Пространственный и другие эффекты ближнего действия. На степень
диссоциации кислоты, несомненно, оказывают влияние также силы ближнего
действия. В некотором отношении это влияние оказывается сложным; слож-
ность наблюдаемой картины частично обусловлена не столько рассматривае-
мыми эффектами, сколько их сочетанием с эффектом поля, индуктивным
и мезомерным эффектами; эти эффекты, хотя и не являются специфическими
эффектами ближнего действия, однако на малых расстояниях оказывают
особо сильное влияние. Из специфических эффектов ближнего действия,
обусловленных заместителями, можно отметить следующий: 1) первичный
пространственный эффект, т. е. пространственное отталкивание, проявляю-
щееся в различной степени в кислоте и сопряженном ей основании; 2) вто-
ричный пространственный эффект, т. е. скручивание кислотной или основной
группы, сопровождаемое нарушением сопряжения и, следовательно, изме-
нением мезомерного влияния на кислотность; 3) чисто химическое ковалент-
ное взаимодействие заместителя с кислотным или основным центром
или с их сольватной оболочкой, как это наблюдается в случаях водородной
или координационной связей. Этими эффектами не исчерпываются все воз-
можные локальные эффекты; все они объясняются наличием обменных сил,
если не учитывать электрокинетические силы.
Следует ожидать, что все локальные эффекты, вызываемые как силами не
обязательно локального характера, так и силами преимущественно локаль-
ного характера, должны оказывать влияние не только на теплоту реакции,
но также и на изменения энтропии. Однако имеющиеся наблюдения еще
недостаточны для раздельной оценки этих термодинамических факторов.
Флюршейм [36] первый отметил, что все o/mo-заместители, даже электрополо-
жительная метильная группа, увеличивают силу бензойной кислоты. Кон-
станты кислотности, приведенные в табл. 199 и 200, хорошо иллюстрируют
это положение * (особый случай салициловой кислоты рассматривается
ниже). Явление, замеченное Флюршеймом, аналогично тому, которое в на-
стоящее время называется первичным пространственным эффектом. Согласно
наблюдениям Флюршейма, происходит отталкивание иона водорода от боль-
шой по объему группы. Эта идея о возрастании кислотности по такого рода
механизму оказалась, как показано ниже, плодотворной; однако сомнитель-
но, могут ли замещенные бензойные кислоты служить убедительными при-
* Электроположительный характер тр em-бутильной группы был установлен Шосмитом
и Макки [37]. Они приводят следующие (по-видимому, термодинамически не исправлен-
ные) данные для 106 Ка в воде при 25 °C:
С6Н5СООН 6,5
о-(СН3)3СС6Н4СООН 3,5
л4-(СН3)3ССвН4СООН 5,2
га-(СН3)3ССвН4СООН 4,2
При сравнении этих данных с данными табл. 199 видно, что в мета- и пара-положениях
mpem-бутильная группа оказывает такое же влияние, как метильная группа, тогда как
в орто-положении трет-бутильная группа (в соответствии с предполагаемым влиянием
пространственных факторов) значительно в большей степени, чем метильная группа,
влияет на кислотность.
928
Кислоты и основания
мерами. В самом деле, казалось вероятным, что различного рода простран-
ственное отталкивание может действовать в противоположном направлении.
Тот факт, что карбоксилат-ион обладает большим эффективным объемом, чем
карбоксильная группа, отчасти обусловлен увеличением размеров атома при
приобретении отрицательного заряда, а в основном связан с тем, что ионная
группа сольватируется в большей степени, чем нейтральная. С другой сто-
роны, результаты опытов, на которые обратил внимание Флюршейм, можно
объяснить в настоящее время вторичным пространственным эффектом.
Карбоксильная группа, связанная с бензольным кольцом, является группой
—М-типа, и соответствующие ее свойства выражены более резко, чем в слу-
чае карбоксилат-иона (гл. II, разд. 3,д). В результате сопряжения между аро-
матическим кольцом и карбоксильной группой электроны смещены в сторону
карбоксильной группы, причем в недиссоциированной группе это смещение
больше, чем в ее анионной модификации. Следовательно, сопряжение с аро-
матическим ядром ослабляет бензойную кислоту. Поэтому, если простран-
ственное отталкивание выводит карбоксильную группу из плоскости ядра,
то нарушается сопряжение и сила кислоты увеличивается.
Таблица 202
Константы диссоциации некоторых стереоизомерных а, р-олефиновых
кислот (105йГо в воде)
Температура, °C * Кислота Кислота 105Ка
18 СН3СН II нссоон 1,95 НССН3 II НССООН 3,9
18 СНзСН II СН3ССООН 1,1 НССНз II СНзССООН 5,1
25 СН3СН II С1ССООН 72 НССНз II С1ССООН 158
25 С6Н5СН 1! нссоон 3,65 нсс6н5 II НССООН 13,2
18 C1CII II нссоон 22,2 НСС1 II нссоон 47,7
Подобное объяснение можно использовать при рассмотрении различий
в кислотной силе стереоизомерных олефиновых кислот. Некоторые относя-
щиеся сюда данные приведены в табл. 202. Они свидетельствуют о том, что
при изменении пространственного расположения, в результате которого
больший из двух p-заместителей (электроположительная метильная группа
или электроотрицательные фенил либо хлор) переходит из транс- в цис-
положение по отношению к карбоксильной группе, сила кислоты увеличи-
вается. При учете только размеров заместителя следует ожидать, что метиль-
ная или фенильная группа или же хлор в г/мс-положении относитель-
но карбоксильной группы выводят карбоксильную группу из плоскости
этиленовой связи, и поэтому кислота становится более сильной.
59-0772
929
Глава XIV
Доказательством того, что влияние любых орпго-заместителей, даже электро-
положительных, приводящее к повышению кислотности бензойной кислоты,
проявляется лишь на коротких расстояниях, служит отсутствие подобного
эффекта у оршо-метильных групп в рядах 0-фенилпропионовой, а также
коричной кислот, как показано в табл. 199.
В подтверждение своей пространственной теории Флюршейм приводит вто-
рую группу примеров, а именно первичные ароматические амины. Он указы-
вает на то, что все оршо-заместители, даже электроположительные метильные
группы, уменьшают основность анилина. Флюршейм рассматривает орто-
заместитель как группу, отталкивающую ионы водорода и тем самым ослаб-
ляющую основание. Такое объяснение также аналогично первичному
пространственному эффекту. Можно ожидать, что этот эффект действительно
имеет место в аминах ряда анилина. Так как сопряженные кислоты этих
оснований находятся в ионной модификации, то центр кислотности, являясь
сильно сольватированным, всегда имеет больший эффективный объем, чем
аминогруппа в нейтральном основании. Следовательно, различное простран-
ственное отталкивание увеличивает относительную термодинамичскую устой-
чивость основания, т. е. приводит к ослаблению основания или, иными сло-
вами, к усилению его сопряженной кислоты.
Эти выводы противоположны тем, которые могут быть сделаны на основании
вторичного пространственного эффекта. Во-первых, можно показать, что
если бы проявлялся вторичный пространственный эффект, то он действовал
бы в противоположную сторону. В этом случае не кислотный, а основной
центр сопряжен с ароматическим кольцом; так как такое сопряжение при-
надлежит к Ц-УИ-типу, то электроны оттянуты от группы основного характе-
ра. Следовательно, любое нарушение сопряжения за счет деформации моле-
кулы приводит к обратному сдвигу электронов в сторону группы основного
характера, тем самым усиливая основание или, иными словами, ослабляя его
сопряженную кислоту. Во-вторых, работа Хэмпсона по дипольным моментам
(гл. III, разд. 1,е) содержит доказательство того, что вследствие малого раз-
мера первичной аминогруппы вывод заместителя из плоскости кольца, обу-
словливающий вторичный пространственный эффект, либо вовсе отсутствует,
либо реализуется в чрезвычайно слабой степени.
Таким образом, взгляды Флюршейма на силу оснований вряд ли могут быть
заменены другими; в современной трактовке они должны быть сформулиро-
ваны как проявление первичного пространственного эффекта. В табл. 200
приведены некоторые примеры влияния галогенов в ориго-положении на
силу анилиновых оснований. Такие заместители являются сильно электро-
отрицательными, и, следовательно, характер их влияния, изменяясь коли-
чественно, качественно остается неизменным. Однако два примера, приведен-
ные в верхней части табл. 203, ясно указывают на качественно отличные
влияния орпго-заместителей *. При введении метильной группы -(-/-типа
в лгепга-положение кислотность анилиниевого иона слегка уменьшается,
при введении в пара-положение — уменьшается в несколько большей сте-
пени, а при введении в ориго-положение — увеличивается. Метоксильная
группа является группой —/, Ц-М-типа, причем ее +7И-характер может
сильно проявляться лишь в орто- и пара-положениях; введение группы
в лгеига-положение всегда приводит к увеличению кислотности, а введение
в пара-положение — к ее уменьшению; в то же время ориго-замещение при-
* Данные табл. 203 приводятся по статье 'Диппи [26]. Для анилиновых оснований
они установлены Холлом и Спринклом [38], для диметиланилинов — Девисом и Адди-
сом [39].
930
Кислоты и основания
водит к увеличению силы кислоты. Очевидно, opmo-заместитель независимо
от его полярности усиливает кислотность ионов анилиния.
Таблица 203
Константы диссоциации сопряженных кислот некоторых
монозамещенных в ядро анилинов и диметиланилинов
Положение заместителя X СНз н ОСНз
XCeH4NH3 (105Ха в воде при 25 ° С)
орто 4,1 2,4 3,2
мета 2,05 2,4 6,3
пара 0,74 2,4 0,51
XCeH4NH(CH3)2 (106Ха в 50%-ном водном спирте при 20 °C)
орто 0,85 6,2 0,32
пара 1,7 6,2 0,69
Эти представления были проверены на примерах N-замещеппых аромати-
ческих аминов. В противоположность предыдущему метильная и метоксиль-
ная группы при введении их в пара- или орто-положения заметно уменьша-
ют кислотность Х,Х-диметиланилиниевого иона. Это иллюстрируется данны-
ми, приведенными в нижней части табл. 203. Используя данные по дипольным
моментам (гл. III, разд. 1,е), Хэмпсон показал, что диметиламиногруппа
достаточно велика для того, чтобы при введении opmo-метильных заместите-
лей быть выведенной из плоскости ядра, тогда как первичная аминогруппа
слишком мала для подобной деформации. Имеются также и химические дока-
зательства в пользу существования этого эффекта. Много лет назад было
найдено [40], что opmo-заместители, в том числе метильная и этоксильная
группы, сильно уменьшают реакционную способность пара-положения
в Х,Х-диалкиланилинах по отношению к таким электрофильным реагентам,
как ионы диазония, азотистая кислота и формальдегид. Следует отметить,
что эти же заместители (метильная и этоксильная группы) в отсутствие
N-алкильпых групп не оказывают такого влияния. Веркаде, Вепстер
и сотр. [41] расширили эти наблюдения на большое число других реакций
и получили количественные закономерности. Исследуя электронные спектры
аминов, они нашли, что сопряжение связи Х,Х-диметиланилина ослабевает
и исчезает совсем с увеличением размеров или числа алкильных заместите-
лей в орто-положении [41]. Две метильные группы в орто-положениях или
одна трет-бутильная группа в орто-положении практически совершенно
полностью подавляют сопряжение между атомом азота и ароматическим
кольцом. Если эти заместители введены в анилин, то сопряжение не
подавляется. В N-метил- и N-этиланилинах наблюдается промежуточная
картина.
Эти же исследователи измерили константы основности большого числа ани-
линовых оснований. В табл. 204 приведены некоторые из полученных ими
результатов. Измеряя спектральную интенсивность сопряженных связей,
они смогли разработать полуколичественный метод оценки факторов, опре-
деляющих силу оснований. При измерении силы оснований пара- и мета-
алкилпроизводных анилина, N-алкиланилинов и Х,Х-диалкиланилинов
авторы цитируемой работы пришли к выводу, что любая простая алкильная
59* 931
Глава XIV
Таблица 204
Копстанты кислотности сопряженных кислот некоторых алкилированных в кольцо
анилинов и \,\-диметилаиилииов
Заместители рКа в 5 0 %-ном водном этиловом спирте Изменение рК„ за счет
индуктивного эффекта вторичного пространст- венного эффекта первичного пространст- венного эффекта
В анилине
Незамещенный 4,26
2-СН3 4,09 +0,4 — 0.6
3,38 +0,4 —1.3
2,6-(СН3)2 3,49 + 0,8 — 1,5
2,4,6-(СН3)3 4,00 + 1,2 — 1,5
2,6-(СНз)г-4-тр?т-С4Н3 3,88 +1,2 -1,6
2,4-(СН3)2-6-т рет-СдНд 3.40 +1,2 -2,1
2-СН3-4,6-(т ретп-С4Н9)2 3,35 + 1,2 -2,1
2,4,6-(отргт-С4Нд)3 2,20 + 1,2 -3,3
В N, IS -диметиланилине
Незамещенный 4,39
2-СН3 5,15 +0,4 + 1,8 -1,4
2-т рет-СдНд 4,28 +0,4 +2,9 -3,4
2,6-(СН3)2 4,81 +0,8 +2,6 —3,0
2,4,6-(СН3)3 5,19 + 1,2 +2.8 -3,2
2,4-(CH3)2-6-mpem-C4H9 2,93 + 1,2 +2,8 — 5,5
2-СН3-4,6-(/п рет-С411д)2 2,77 +1,2 +2,9 —5,7
группа, если она находится в /гара-положении, увеличивает основность, уве-
личивая рКа сопряженной анилину кислоты па 0,4 единицы; если же алкиль-
ная группа находится в лшта-положелии, то увеличение рКа составляет
0.15 единицы. Эти эффекты следует рассматривать как полярные, а для про-
стых алкильных групп как индуктивные. Можно предположить, исходя из
данных третьего столбца табл. 204, что если алкильные заместители .занимают
орто-положения, то будет проявляться полярный эффект и при отсутствии
специфических локальных изменений прирост величины рКа составит 0,4 еди-
ницы. Можно теперь принять во внимание вторичный пространственный
эффект opmo-заместителей. как это видно из четвертой колонки таблицы,
исходя из предположения, что инкремент пропорционален силе сопряженной
группы. Инкремент, соответствующий ее полному отрыву, равен 3 единицам
рКа. Первичные пространственные влияния opmo-заместителей как в отсут-
ствие N-заместителей, так и при их наличии можно затем вычислить по раз-
ности. Как показано в последней колонке таблицы, все эти инкременты рК0
отрицательны, как и следовало ожидать. По абсолютной величине они боль-
ше для диметиланилинов, чем для анилина, и в обеих сериях возрастают
с увеличением числа и размера opmo-алкильных групп, что и следовало
ожидать. В качестве примера можно сравнить диметиланилин с его 2-метил-
4,6-ди-трет-бутилпроизводпым. Алкильные заместители уменьшают кон-
станту основности триалкилпроизводного анилина в 40 раз. Эта величина
складывается из увеличения в 16 раз за счет индуктивного эффекта, умень-
232
Кислоты и основания
шения в 500 000 раз за счет первичного пространственного эффекта и увели-
чения в 800 раз за счет вторичного пространственного эффекта.
Как было сказано в начале этого раздела, следует ожидать, что из различных
эффектов ближнего действия на силу кислоты оказывают влияние не только
уже рассмотренные первичный и вторичный пространственные эффекты, но
в некоторых особых случаях также и другие эффекты, такие, как водородная
связь и координация. Приведенные выше данные включают некоторые
примеры такого рода взаимодействия.
Из данных табл. 199 видно, что введение в бензойную кислоту метоксильной
группы в орто-положение приводит к увеличению константы кислотности
только в 1,3 раза, тогда как введение гидроксильной группы в орто-поло-
жение увеличивает кислотность в 17 раз. Тот факт, что меньшая из двух
указанных близких по полярности групп вызывает гораздо большее увели-
чение кислотности, не может быть объяснен пространственным эффектом.
Однако это явление можно объяснить наличием внутримолекулярной водо-
родной связи, которая частично по электростатическим причинам, а частично
вследствие ковалентного изменения вызывает новый положительный заряд
в карбоксильной группе и поэтому должна увеличивать кислотность.
ОН
I
С
/ V
U\
о
он
I
с
/\/ Ч)+
о
Большее сомнение вызывает другой из возможных примеров из области арил-
борных кислот, данные для которых приведены в табл. 199. Большую часть
данных в разделе Д этой таблицы можно понять в качественном смысле так
же, как и данные раздела А для бензойной кислоты. Исключение составляет,
как упоминалось, салициловая кислота. Другим важным исключением
является о-нитрофенилборная кислота.
В ряду бензойных кислот более сильно замещенные электроотрицательными
заместителями являются более сильными кислотами, если заместители
занимают орто-положение, а не какое-либо другое. Та же картина
наблюдается и для хлорзамещенных арилборных кислот. Однако для
нитрозамещенных кислот дело обстоит иначе: изменение кислотности в ряду
орто-нитрозамещенных кислот значительно слабее. Правдоподобное объяс-
нение этому уже было дано: как установили Бренч и сотр. [31], орто-нит-
рогруппа обладает такой электронной структурой и таким стереохимическим
расположением, что это позволяет ей координироваться с атомом бора,
в результате чего кислотность ослабляется.
933
Глава XIV
СПИСОК ЛИТЕРАТУРЫ
1. Gillespie R. J., J. Chem. Soc., 1950, 2537.
2. Hantzsch A., Z. physik. Chem., 61, 257 (1907); 62, 626; 65, 41 (1908); Treffers H. P.,
Hammett L. P., J. Am., Chem. Soc., 59, 1708 (1937); Gillespie R. J., J. Chem. Soc.,
1950, 2542.
3. Hammett L. P., Physical Organic Chemistry, McGraw-Hill, New York, 1940, p. 250.
4. Gillespie R. J., J. Chem. Soc., 1950, 2516.
5. Burkhardt G. N., Chem. & Ind., 52, 330 (1933); Dippy J. F. J., .1 Cliem. Soc., 1941, 550.
6. Evans Al. G., Polanyi M., Trans. Faraday Soc., 32, 1333 (1936).
7. Everett D. H., Wynne-Jones W. F. K., Trans. Faraday Soc., 35, 1380 (1939).
8. Bierrum N., Z. physik. Chem., 106, 219 (1923).
9. Gane R., Ingold С. K., J. Chem. Soc., 1928, 1594, 2267; 1929, 1691.
10. Gane R., Ingold С. K., J. Chem. Soc., 1931, 2153; Ingold С. K., ibid., p. 2170;
Ingold С. K. Mohrhenn H. G. G., ibid., 1935, 1482.
11. Eucken A., Angew. Chem., 45, 203 (1932).
12. Smallmood H. M., J. Am. Chem. Soc., 54. 3048 (1932).
13. Wheland G. W-, Advanced Organic Chemistry, Wiley, New York, 1949, Chap. 11.
14. Kirkwood J. G., J. Chem. Phys., 2, 351 (1934); Kirkwood J. G., Westheimer F. H., ibid.,
6, 506, 513 (1938); Westheimer F. II., Shookhoff M. W., J. Am. Chem. Soc., 61, 555
(1939): Tanjord C., Kirkwood J. G., ibid., 79, 5333 (1957).
15. Roberts J. D., Clement R. A., Drysdale J. J., J. Am. Chem. Soc., 73. 2181 (1951).
16. Beetlestone J. G.. Irvine D. II., Proc. Roy. Soc., А2П, 401, 414 (1964); J. Chem. Soc.,
1964, 5086, 5090; 1965, 3271.
17. Beetleston J. G., Irvine D. H., J. Chem. Soc., 1965, 3273.
18. Henrich F., Ber., 32, 668 (1899).
19. Vorldnder D., Ann., 320, 99 (1902).
20. Wegscheider R., Monatsh., 23, 287 (1902).
21. Fliirscheim B., J. Chem. Soc., 95, 722 (1909); 97, 91 (1910).
22. Lewis G. N., Valence and the Structure of Atoms and Molecules, Chemical Catalog Co.,
New York, 1923, Chap. 12.
23. Ingold С. K., J. Chem. Soc., 1933, 1120.
24. Waters W. A., Physical Aspects of Organic Chemistry, Routledge, London, 1st Edn.,
1935, Chap. 9.
25. Watson H. B., Modern Theories of Organic Chemistry, Oxford Univ. Press, 1st Edn.,
1937, Chap. 2.
26. Dippy J. F. J., Chem. Revs., 25, 151 (1939).
27. Branch G. E. K., Calvin M., The Theory of Organic Chemistry, Prentice-Hall, New
York, 1941, Chap. 6.
28. Halevi E. A., Nussim M., Bull. Res. Council, Israel, A5, 263 (1956); ibid., A6, 167
(1957); Halevi E. A., Nussim M., Ron A., J. Chem. Soc., 1963, 966; Halevi E. A., Tetra-
hedron, 1, 174 (1957); Halevi E. A., Prog. Phys. Org. Chem., 1, 109 (1963).
29. Bell R. P., Miller W. T. B., Trans. Faraday Soc., 59, 1147 (1963).
30. Streitwieser A., Jr., Klein H. S., J. Am. Chem. Soc., 85, 2759 (1963).
31. Bettman B., Branch G. E. K., Yabroff D. L., J. Am. Chem. Soc., 56, 1865 (1934).
32. Dippy J. F. J., Watson H. B., Williams F. R., J. Chem. Soc., 1935, 346; Dippy J. F. J.,
Lewis R. H., ibid., 1936, 644.
33. Baddeley G., Bennett G. Al., Glasstone S., Brynmor Jones, J. Chem. Soc., 1935, 1827.
34. Remick A. E., Electronic Interpretations of Organic Chemistry, Wiley, New York,
1st Edn., 1943, p. 66.
35. Baker J. W., Barrett G. F. C.. Tweed W. T., J. Chem. Soc., 1952, 2831.
36. Fliirscheim B., J. Chem. Soc., 95, 718 (1909).
37. Shoesmith J. B., Mackie A., j. Chem. Soc., 1936, 300.
38. Hall F. N., Sprinkle M. R., .1 Am. Chem. Soc., 54, 3469 (1932).
39. Davies W. C., Addis H. W.. 3. Chem. Soc., 1937, 1622.
40. FischerO., Ber., 13, 807 (1880); Weinberg A., Ber., 25, 1610 (1892); Bamberger E., Meim-
berg F., Ber., 28, 1887 (1895); Friedlander P., Monatsh., 19, 627 (1898).
41. Burgers J., Hoefnagel M. A., Verkade P. E., Visier H., Wepster В. M., Rec. trav. chim.,
77, 491 (1958).
ГЛАВА XV
РЕАКЦИИ КАРБОКСИЛЬНОЙ И ФОСФАТНОЙ ГРУПП
1. Гидролиз сложных эфиров и этерификация карбоновых кислот 936
2. Образование и гидролиз амидов карбоновых кислот...... 963
3. Реакции сложных эфиров с карбанионами................ 968
4. Гидролиз эфиров фосфорной кислоты ................... 974
Список литературы . . ............................. 984
Карбоксильная группа характеризуется способностью как к обратимым
реакциям нуклеофильного присоединения, так и к реакциям нуклеофильного
замещения. В этой главе будут поочередно рассмотрены реакции карбоксиль-
ной группы, в которой атакующими нуклеофильными атомами являются
кислород, азот и углерод.
1. Гидролиз сложных эфиров и этерификация карбоновых
кислот [1]
а. Шесть механизмов гидролиза и три механизма этерификации [2].
Этерификация карбоновых кислот и гидролиз сложных эфиров могут про-
текать в соответствии с несколькими механизмами. Их можно классифици-
ровать, исходя из трех особенностей этих реакций.
Во-первых, классификацию можно провести на основании природы реагента.
Известно, что сложные эфиры могут гидролизоваться либо щелочами, либо
кислотами. Многочисленные факты показывают, что к каждому из этих
случаев применимы различные механизмы или даже группы механизмов.
В нейтральных растворах протекает значительно менее известная форма
гидролиза, которая не является просто промежуточной формой между вза-
имодействием в щелочной и кислой средах. Таким образом, может показать-
ся, что существуют три основные группы механизмов; однако мы будем рас-
сматривать только две из этих групп, более близкие между собой и менее
тесно связанные с третьей. Дело в том, что как при омылении щелочами,
так и при гидролизе в нейтральной среде карбоксильная группа реагирует
в виде нейтральной молекулы сложного эфира В'СООВ, а при гидролизе
в кислой среде реагирует ионная сопряженная кислота В'СООНВ+. Прин-
ципиально такая же градация применима и к реакциям этерификации. Устой-
чивость карбоксилат-иона препятствует этерификации в щелочной среде.
Можно предполагать, что этерификация протекает и в нейтральной среде, но
это не установлено; если бы удалось ее осуществить, то карбоксильная груп-
па реагировала бы в форме В'СООН. Катализируемая кислотами этери-
фикация хорошо известна; реагирует в данном случае R'COOHJ. Гидролиз
и этерификацию следует рассматривать совместно, поскольку они являются
процессами, обратными один другому. Реакции кислородного обмена между
кислотами и водой можно считать особыми случаями (группа R заменена на Н)
гидролиза или этерификации. Переэтерификацию эфиров спиртами можно
рассматривать как общий случай (Н заменен на R) гидролиза или этерифи-
кации. Механизмы всех этих реакций можно подразделить, основываясь
на том, происходит или не происходит присоединение протона к карбоксиль-
ному соединению.
Позможно также классифицировать эти реакции на основании другого
признака, а именно места расщепления карбоксильного соединения. Сущест-
вует несколько способов расщепления. Один из них является общим, осталь-
ные же применимы только в определенных случаях, но все они легко осущест-
вимы и дают хорошие результаты. Для гидролиза возможны следующие спо-
собы расщепления:
R'CO;
OR-J-H ; ОЩ
R'COO IR-J-HO | Н
Расщепление связи]'ацил— кислород
Расщепление связи алкил — кислород
При этерификации соответственно возможны следующие способы расщеп ле-
ния:
r'co|oh+h;or
I I
Расщепление связи ацил — кислород
R'COO | Н + НО
।
!R
Расщепление связи алкил — кислород
936
Реакции карбоксильной и фосфатной групп
В настоящее время установлено, что в случае омыления щелочами обычно
происходит расщепление связи ацил — кислород, но возможны и отклонения
от этого правила; в случае гидролиза в нейтральной среде нередко происходит
расщепление связи между алкилом и кислородом. Таким образом, для глав-
ной группы механизмов, в которых атаке подвергается нейтральная молеку-
ла R'COOR, существуют две подгруппы механизмов, отличающихся местом
расщепления сложноэфирной группы. Кроме того, найдено, что при катали-
зируемых кислотами гидролизе и этерификации, когда атакуется ион сопря-
женной кислоты R'COOHR+ или R'COOH* (в зависимости от условий
и структуры соединения), может происходить расщепление связи между
кислородом и ацильной или алкильной группой. Таким образом, перво-
начальная двухступенчатая классификация превращается в четырехступен-
чатую.
После отнесения механизма к определенному классу, основному или кислот-
ному, с расщеплением связи ацил — кислород или алкил — кислород
последний этап классификации состоит в определении механизма при помощи
изучения кинетики реакций, а также продуктов реакций, что позволяет
установить стереохимические или другие особенности изучаемого процесса.
В результате исследований было убедительно показано, что по крайней мере
среди определенных типов соединений существуют два механизма, которые
относятся один к другому как бимолекулярный и мономолекулярный меха-
низмы нуклеофильного замещения или отщепления. По аналогии такие меха-
низмы можно назвать «бимолекулярным» и «мономолекулярный».
Таблица 205
Механизмы гидролиза сложных эфиров н этерификации кислот
Тип механизма Атакуемая форма Наблюдаемые реакции Расщепление связи
ацил — кисло- род алкил—кисло- род
Основной R'COOR Гидролиз Вас2 ВаьД Bal2
Кислотный R'COOHR+или R'COOHf Гидролиз и этерификация Аде! Аде2 AalI
Описываемая классификация приведена в табл. 205. Для удобства «основные»
механизмы, включая механизм реакций в щелочной и нейтральной среде,
обозначаются символом В, а «кислотные»— символом А; расщепление свя-
зей ацил — кислород и алкил — кислород соответственно обозначается АС
и AL *, а молекулярность обозначается, как обычно, цифрами 2 или 1. Указан-
ные три критерия приводят к возможности существования восьми механиз-
мов, из которых шесть, как видно из приведенной таблицы, можно наблюдать
экспериментально.
В разное время делались предположения о существовании гораздо большего
числа механизмов гидролиза и этерификации, но некоторые из этих механиз-
мов вытекают из уже принятых. Введением понятия о дополнительном быст-
ром передвижении протона и другими путями можно прийти к различным
гипотетическим механизмам. Однако нет оснований для обсуждения таких
* В статье [1] использованы менее наглядные символы.
937
Глава XV
о
2 Ц 2
НО ------С--------OR —------->
| Быстро
R'
механизмов, пока не установлено, что они отражают химизм реакции. Поэто-
му целесообразно рассматривать каждый механизм в его простейшей форме.
б. Бимолекулярный основной гидролиз с расщеплением связи ацил —
кислород (механизм ВАС2). Хорошо известно, что при гидролизе сложных
эфиров гидроксил-ионами в водных растворах расщепляется связь ацил —
кислород и протекает процесс, кинетика которого подчиняется закономерно-
стям реакции второго порядка. Теоретически только один механизм согла-
суется с этими наблюдениями, а именно бимолекулярный механизм БАС2.
Этот механизм можно представить как присоединение к карбонильной груп-
пе и отщепление от нее по схеме
О О- О
|| 1 Медленно | Быстро ||
но~+с —OR ------- —> НО — С—OR HO-C + "OR (Вас2)
I Быстро | Медленно I
R' R' R'
(R'COOH + “OR Быетр-^ R'COO-+ROH)
Этот процесс в принципе обратим, но практически вследствие переноса про-
тона от образующейся карбоновой кислоты к щелочи, присутствущей в рас-
творе, равновесие полностью смещено вправо. Этот же механизм можно пред-
ставить и иначе, если использовать модель нуклеофильного замещения
О
|| Медленно
НО-+ С-OR < >
R'
О
—» НО-С+-OR (Вас2)
R'
(R'COOH + “OR ~-blCT-^-» R'COO’+ ROH)
Промежуточный комплекс здесь взят в квадратные скобки и помещен между
стрелками, указывающими направления реакции. Это обусловлено тем, что
если комплекс действительно обладает указанным распределением электро-
нов и не может стать более устойчивым, то он, по-видимому, представляет
собой переходное состояние с максимальной энергией (для перемещения
вдоль координаты реакции), а не молекулу, обладающую минимумом энер-
гии относительно всех нормальных координат. Действительно маловероятно,
что ни один из отрицательных зарядов комплексного иона не находится на
исходном карбонильном атоме кислорода; более вероятно, что на этом атоме
будет сосредоточен весь заряд комплекса, как на схеме, основанной на при-
соединении. Этой схеме соответствует более устойчивый комплекс, который
может быть и истинной молекулой. Реальный комплекс должен быть еще
более устойчивым, так как он мезомерен между комплексами, соответствую-
щими чистому присоединению и чистому замещению. Ниже будет показано,
что существование этого комплекса в виде устойчивой молекулы доказано
экспериментально. Однако мы считаем, что оба представления следует
сохранить как предельные случаи, поскольку каждое из них связано с важ-
ными аналогиями.
Природа щелочного гидролиза сложных эфиров впервые была изучена Гольм-
бергом в 1912 г. Он исследовал сложные эфиры R'COOR, где R — замещен-
938
Реакции карбоксильной и фосфатной групп
ная алкильная группа с асимметрическим атомом углерода, непосредственно
-связанным с карбоксильной группой. Гольмберг полагал, что если в процес-
се реакции группа R отщепляется, то она не сохраняет своей стереохими-
ческой конфигурации. На примере О-ацетиляблочной кислоты CH3COOR,
где R= СН(СООН)СН2СООН, Гольмберг [3] показал, что асимметричная
группа полностью сохраняет конфигурацию в процессе гидролиза щелочами.
На этом основании он сделал вывод, что при омылении сложных эфиров
группа R не отщепляется. При этом была допущена неточность, которую
тогда нельзя было предусмотреть. Дело в том, что если группа R отщепляет-
ся в виде R+, то она может сохранить свою конфигурацию благодаря влиянию
одного из карбоксилат-ионов (гл. VII, разд. 7,д и з).
Впоследствии применялись и другие методы изучения щелочного гидролиза
сложных эфиров. Хилда Ингольд и автор этой книги [4] считали, что если
группа R отщепляется в виде R+, который может быть мезомерным, то долж-
ны образоваться изомерные спирты. Кротил и 1-метилаллил —СН2СН =
= СНСН3 и СН2 = СНСН(СН3) — являются теми R, которые удовлетворя-
ют этому условию. Прево [5] показал, что ацетаты, содержащие такие ради-
калы, гидролизуются щелочами без изомеризации.
Нортон и Куэйл [6] использовали этот же принцип, применив в качестве R
неопентильный радикал —СН2С(СН3)3; если он отщепляется в виде катиона,
то должен реагировать в перегруппированном виде СН2(СН3)С(СН3)2 —
с образованием третичного амилового спирта. Как было установлено, такие
неопентиловые эфиры, как ацетат, подвергаются омылению щелочами без
перегруппировки с образованием неопентилового спирта.
Эти методы исследования ограничены необходимостью выбора особо построен-
ного радикала R. Полянки и Сабо [7] впервые предложили другой метод
вполне общего характера. Они применили в качестве растворителя воду, содер-
жащую изотоп кислорода 18О, и показали на примере гидролиза н-амилацетата
в щелочной среде, что кислород среды не переходит в образующийся спирт
и поэтому должен переходить в кислоту. Эти данные свидетельствуют в поль-
зу расщепления связи ацил — кислород
R'CO;OR + HiOH-----> R'COOH + HOR
I I
Лонг и Фридман [8] получили такие же результаты в случае щелочного омы-
ления у-бутиролактона.
В 1881 г. Вардер [9] на примере гидролиза этилацетата водой показал, что
омыление сложных эфиров в щелочной среде — реакция второго порядка.
Недавно Бендеру [10] удалось решить вопрос, соответствует ли промежуточ-
ное состояние молекуле с продолжительностью жизни, большей периода
соударений, или же только переходному состоянию, возникающему при
таком соударении, которое ведет к реакции.
Бендер исследовал этиловый, изопропиловый и /пре/п-бутиловый эфиры бен-
зойной кислоты, а в качестве растворителей применял либо воду, либо вод-
ный диоксан, причем в обоих случаях вода содержала изотоп кислорода 18О.
Бендер сопоставлял скорость гидролиза и скорость обмена кислорода между
средой и сложным эфиром, определив последнюю на основании анализа
сложного эфира, оставшегося на различных глубинах омыления. При этом
им было найдено, что скорость обмена в зависимости от природы сложного
эфира и среды составляет от 10 до 40% скорости омыления. Это указывает на
то, что продолжительность жизни промежуточного комплекса достаточна,
чтобы произошло необходимое смещение протона, требуемое для того, чтобы
два неалкилированьых кислородных атома комплекса стали эквивалентными
939
Глава X V
и чтобы каждый из них мог отщепиться с дальнейшей регенерацией сложного
эфира. При отщеплении атомов кислорода, связанных с алкильными ради-
калами, образуются продукты гидролиза. Если исходить из модели реакции
присоединения, то вся перестройка о — л-электронного облака происходит
в процессе образования и разрушения комплекса, а не в процессе перемеще-
ния протона; однако, как уже указывалось, последний случай следует рас-
сматривать как предельный.
R'CO2H + OR
Рассмотрим теперь влияние структурных факторов на скорость гидролиза
по механизму ВАс2- Поскольку промежуточный комплекс заряжен отрица-
тельно, следует ожидать, что электроположительные заместители будут
тормозить, а электроотрицательные заместители — ускорять гидролиз слож-
ных эфиров. Кроме того, поскольку лимитирующей стадией является бимо-
лекулярная реакция, следует ожидать пространственных затруднений,
обусловленных заместителями, расположенными вблизи реакционного-
центра.
Таблица 206.
Сравнительные скорости щелочного гидролиза эфиров алифатических кислот
(реакция второго порядка)
А. В воде при 25 °C
CH3COOR J R CH3 с2н5 изо-С3Н7 m/iem-CRL,
( Скорость 1 0,601 0,146 0,0084
R'COOCH3 JR' H СН3 СН2С1 СНС1,
J Скорость 223 1 761 16 000
R'COOCH3 J R' COOCH3 соо- СН2СООСН3 сн2соо-
t Скорость 170 000 8,4 13,7 0,19
R'COOC2H5 j R' CH3 СН2СН3 СОСН3 сн2сосн3
J Скорость 0,601 0,553 10 000 2,66
R'COOC2H5 J R' CHg СН2ОСН3 СН2ОС2Нэ сн2он
t Скорость 0,601 Б. В 88%-ном водном 11,9 6,03 этиловом спирте при 30 °C 6,08
R'COOC2H5 JR' СНз СН2СН3 СН(СН3)2 С(СН3)з
[ Скорость 1 0,470 0,100 0,0105
R'COOC2H5 J R' (СН2)2СН3 (СН2)3СНз СН2СвН5 свн5
"[ Скорость 0,274 0,262 1,322 0,103
Впервые количественные исследования влияния структурных факторов на
скорость омыления сложных эфиров в щелочной среде (реакция второго по-
рядка) были проведены Райхером [И], который, начиная с 1885 г., изучал
сложные жирные эфиры R'COOR с неразветвленными и разветвленными
алкильными группами R и R'. Эти исследования были расширены; результа-
ты изучения жирных эфиров Олсоном [12], Шкрабалом [13] и Киндлером [14]
940
Реакции карбоксильной и фосфатной групп
подтвердили тормозящее действие алкильных заместителей и ускоряющее
действие групп G1, СООСН3, СОСН3, СН2СОСН3 и СН2ОСН3, а также тормо-
зящее влияние отрицательных ионных зарядов *. Соответствующие данные
приведены в табл. 206. Очевидно, влияние полярных эффектов проявляется
независимо от наличия пространственных эффектов.
Таблица 207
Сравнительные скорости щелочного гидролиза эфиров ароматических кислот
(реакция второго порядка)
А. ХС^ЩСООСгНз в 85%-ном водном этиловом спирте при 25 °C
Положение заместителя NHz ОСНз СНз н F С1 Вг I NOz
пара 0,023 0,209 0,456 1 2,03 4,31 5,25 5,05 110
мета 0,697 1 7,52 69,0
орто 0,125 1 3,74 2,24 8,71
Б. ХСДЦСООСцНг, в 60%о-ном водном ацетоне при 25 °C
Положение заместителя NHz СНз н NO3
пара 0,0293 0,403 1 85,1
мета 0,574 0,596 1 47,7
В. ХС6Н4СООС Ц3 в 60%-ном водн 7Л1 ацетоне при 0 ° С
Положение заместителя NHz СНз II NOz
пара 0,510 0,602 1 18,8
мета 0,699 1 13,0
Киндлер [16] впервые подробно изучил влияние заместителей на скорость
реакции щелочного гидролиза второго порядка сложных эфиров R'COOR,
в которых R' или R представляют собой фенильные или замещенные фениль-
ные группы. Исследования Киндлера были продолжены Натаном и автором
этой книги [17], Эвансом, Гордоном и Уотсоном [18], а также Томмила и Хин-
шелвудом [19] главным образом с целью изучения температурного коэффици-
ента реакции и констант Аррениуса. При этом было найдено, что такие
заместители, как СН3, ОСН3 в пара-положении и NH2, замедляют абсолют-
ную скорость реакции, а галогены и группа NO2 ускоряют ее. Эти данные
приведены в табл. 207. Следует отметить, что группа NH2, если она находится
в пара-положении в R', замедляет реакцию сильнее, чем если она находится
в жепга-положении в R' или в пара-положении в R. Сильное влияние этой
группы, находящейся в пара-положении в R', несомненно должно быть
приписано сопряжению между амино- и карбоксильной группами через аро-
матическое кольцо. Такие влияния можно попять, если учитывать, что
* Эти результаты были обобщены и проанализированы Гамметом [15].
941
Глава XV
—I- или —ЛТ-эффекты ускоряют, а -\-1- и +Л/-эффекты замедляют реакцию.
Следует также учитывать, что заместители в орто-положении (за исключени-
ем фтора) в большей степени замедляют или ускоряют реакцию, чем соответ-
ствующие заместители в пара-положении. По-видимому, пространственные
затруднения накладываются на
Рис. 52. Зависимость аррениусовских
параметров ЕА и А от при-
роды заместителей при щелоч-
ном гидролизе эфиров
ХС6Н4СО2С2Н5 в 85%-ном вод-
ном растворе этилового спир-
та [18].
По оси абсцисс — значения 10sfea
(25 °C). Точки, расположенные на
линии, представляют собой ско-
рость, изменяющуюся только под
влиянием Еа; А при этом остается
постоянным. Точки, расположенные
слева от линии, соответствуют
уменьшению значения А.
точек влево от прямой наблюдается
фтора) и, вероятно, означает, что в
измениться под действием не тольк(
ствие передачи полярных эффектов заместителя, энтропия активации изме-
няется только в том случае, если происходит локальное взаимодействие
с реагентом или его составной частью, а также с компонентами сольватной
ячейки переходного состояния (гл. II, разд. 2,г).
Эванс, Гордон и Уотсон [20] применили такой же подход и к случаю сложных
эфиров кислот жирного ряда. Так, они определили температурный коэф-
фициент реакции щелочного гидролиза нормальных и разветвленных насы-
щенных сложных эфиров жирного ряда R'COOC2H5 в 85%-ном этиловом
спирте. Хотя различие в скоростях реакции было в этом случае меньше, чем
в ряду сложных эфиров кислот ароматического ряда, общий результат ока-
зался весьма сходным. Так, оказалось, что разветвление цепи в R' (не при
полярные эффекты всех заместителей
и в меньшей степени заместителей в ор-
то-положении.
При изучении температурных коэффи-
циентов этих реакций было установле-
но, что кинетический эффект мета- и
пара-заместителей проявляется только-
в аррениусовской энергии активации
Еа, а величина предэкспоненциального
множителя А не изменяется под влия-
нием заместителей. Установлено также,
что кинетический эффект заместителей
в орто-положении сказывается не-
только на энергии активации, но и
на величине предэкспоненциального
множителя, который заметно умень-
шается под влиянием всех заместите-
лей в орто-положении, за исключе-
нием фтора. Для гидролиза эфиров
ХСдЩСООСгНд в 85%-ном водном эти-
ловом спирте эта закономерность иллю-
стрируется рис. 52, на котором в коор-
динатах Еа и log 1О5*2 (25 °C) нанесены
соответствующие точки; положение то-
чек относительно прямой, проведен-
ной через точку X = Н, имеющей нак-
лон, равный —2,303 RT (Т = 298 К),
подтверждает сказанное. Эта прямая
соответствует постоянному предэкспо-
ненциальному множителю; величина
сдвига точек влево от этой линии явля-
ется мерой уменьшения предэкспонен-
циального множителя. Такой сдвиг
лишь в случае opmo-заместителей (кроме
то время как энергия активации может
> локальных эффектов, но также и вслед-
tx-углеродном атоме) влияет лишь на аррениусовскую энергию активации,
а предэкспопенциальный множитель при этом остается почти неизменен-
942
Реакции карбоксильной и фосфатной групп
ным. Если же цепь разветвляется при tx-углеродном атоме, то предэкспонен-
циальный множитель уменьшается.
в. Мономолекулярный основной гидролиз с расщеплением связи алкил—
кислород (механизм BAL 1). Молекула сложного эфира R'COOR содержит два
углеродных атома, которые в соответствии с представлениями о нуклеофиль-
ном замещении и присоединении в принципе могут подвергаться атаке
нуклеофильными реагентами. Речь идет об атоме углерода карбоксильной
группы и об а-углеродном атоме алкильной группы. Поскольку первый из
них является ненасыщенным, следует ожидать, что он более реакционно-
способен по отношению к нуклеофильному реагенту. В соответствии с этим
расщепление связи ацил — кислород является общим правилом при основ-
ном гидролизе сложных эфиров. Несмотря на это, можно допустить, что,
когда реагентом является ион гидроксила, параллельно протекают две реак-
ции, которые можно охарактеризовать как атаку ацильной или алкильной
группы, хотя первая реакция протекает быстрее и потому только она прак-
тически наблюдается. Предположим, что ион гидроксила заменяется более
слабыми нуклеофильными реагентами. Тогда скорость обеих реакций умень-
шается, но атака ацильной группы идет быстрее. При определенной структуре
R и соответствующем растворителе устанавливается некоторая скорость
диссоциации эфира R'COOR на R'CO0~ и R+; поскольку нуклеофильный
реагент ослаблен, то скорости атаки алкильной группы, а затем и ацильной
группы становятся ниже скорости ионизации. При этом мы переходим от бимо-
лекулярного расщепления связи ацил — кислород к мономолекулярному
расщеплению связи алкил — кислород, т. е. от механизма Вдс2 к механиз-
му ВдгД- Мы рассматриваем здесь процесс гидролиза, и потому наибольшее
значение имеют ион гидроксила и молекула воды как реагенты. Можно
сделать важный вывод: если описанное изменение механизма происходит до
того, как нуклеофильный реагент настолько ослабляется, что функция нук-
леофильного реагента переходит к молекуле воды, то, несмотря на то что
гидролиз в щелочной среде протекает в соответствии с механизмом ВАС2,
гидролиз в нейтральном растворе будет протекать согласно механизму BAL1.
Механизм BAL1 можно изобразить следующим образом:
Медленно -ч
ROCOR' R+ + -OCOR' ]
Быстр0 I m п
Быстро ( П->АЬГ/
R+ + H2o ~--’ > R — ОН+ I
Медленно *
(ROH2 + "OCOR‘-^^> ROH4-HOCOR')
Если не рассматривать конечный перенос протона, этот механизм соответ-
ствует типичному мономолекулярному нуклеофильному замещению. Реак-
ция в принципе обратима, но практически перенос протона от иона алкил-
оксония к карбоксилат-иону смещает равновесие полностью в сторону гидро-
лиза.
Характерные особенности, позволяющие распознать этот механизм, заклю-
чаются в следующем. Во-первых, этот механизм реализуется в основной сре-
де; наличия кислоты не требуется, процесс протекает в нейтральном или
слабощелочном растворе. Во-вторых, расщепление связи алкил — кислород
может быть установлено обычными методами. Если tx-углеродный атом в ради-
кале R асимметричен, то из оптически активного сложного эфира образуется
943-
Глава XV
рацемический спирт. Если радикал R представляет собой замещенную ал-
лильную группу, то в процессе гидролиза он может изомеризоваться. Если
группа R имеет строение, при котором возможна перегруппировка Вагнера,
как в сложных эфирах камфенового ряда, то такая группа в процессе гидро-
лиза может подвергнуться перегруппировке. Кроме того, при всех условиях
может быть использован метод, основанный на применении изотопа кислорода
18О. В-третьих, кинетика гидролиза должна соответствовать одной из форм,
характерных для мономолекулярного замещения. В-четвертых, электрополо-
жительные группы, особенно 4 TJ-группы, например метоксильная группа,
при надлежащем расположении их в радикале R должны сильно увеличивать
скорость реакции в результате полярного эффекта. Такое же влияние дол-
жны оказывать электроотрицательные группы, например нитрогруппы, при
надлежащем расположении их в радикале R'. В-пятых, эта реакция, как
принадлежащая к мономолекулярным процессам, не должна быть чувстви-
тельной к пространственным влияниям. И наконец, эта реакция должна
сильно ускоряться с увеличением полярности растворителя.
В качестве первого доказательства этого механизма был использован крите-
рий оптической активности. Кеньон и сотр. [21] изучали эту реакцию в свя-
зи с получением оптически активных спиртов гидролизом оптически актив-
ных кислых эфиров фталевой кислоты. В качестве исходного радикала R
они первоначально применили 1,3-дизамещенные аллильные группы, кото-
рые, как известно, склонны к переходу в катионные формы R+. Кеньон
с сотрудниками установил, что при омылении оптически активного 1,3-
диметилаллилового кислого эфира фталевой кислоты и 1-метил-З-фенилал-
лилового кислого эфира фталевой кислоты в слабощелочных водных раство-
рах образуются рацемические спирты, при омылении же концентрированным
спиртовым раствором щелочи образуются оптически активные спирты. На
основании полученных данных были сделаны следующие выводы: во-первых,
при реакции с концентрированной спиртовой щелочью происходит бимо-
лекулярная атака ацильной группы Вдс2, вследствие чего алкильная группа
не может рацемизоваться; во-вторых, скорость этой реакции второго порядка
в разбавленных водных растворах щелочей настолько уменьшается, а ско-
рость мономолекулярного отщепления алкильной группы BAL1 настолько
увеличивается, что наблюдается лишь последняя реакция, которая вслед-
ствие образования катионной формы алкильной группы R+ приводит к раце-
мизации. Впоследствии были поставлены опыты не только с замещенными
аллиловыми эфирами, но главным образом с эфирами бензгидрил- и 1-фенил-
этиловых производных и с некоторыми их аналогами в ряду нафталина.
При этом было установлено, что различные алкильные радикалы значительно
отличаются по способности соответствующих эфиров гидролизоваться по
механизму BAL1, сопровождающемуся рацемизацией, и что это различие
соответствует ожидаемой стабильности соответствующих ионов карбония R+.
Так, оптически активный ге-метоксибензгидриловый кислый эфир фталевой
кислоты образует рацемический спирт даже при обработке 10 н. водным
раствором едкого натра [22], в то время как оптически активные ге-фенокси-
бензгидриловый и 1-тг-анизилэтиловый кислые эфиры фталевой кислоты
образуют такие спирты при применении очень разбавленных щелочей [23],
а незамещенный 1-фенилэтиловый эфир дает рацемический спирт лишь
в почти нейтральной среде [24]. Эти данные находятся в соответствии с ожида-
емой сравнительной способностью заместителей при карбинольном углерод-
ном атоме стабилизовать соответствующий карбониевый ион
СН3О > С6Н5О —/____/__________S- > сн3—
944
Реакции карбоксильной и фосфатной групп
Первое из этих неравенств является следствием конкуренции двух фениль-
ных групп в сопряжении с кислородным атомом феноксигруппы.
Кеньон и его сотрудники подтвердили расщепление связи алкил — кислород
в аллиловых сложных эфирах при гидролизе этих соединений разбавленными
водными щелочами. Наблюдая при этом случаи анионотропных превраще-
ний, они нашли [25], что из 1-фенил-З-метилаллилового кислого эфира фта-
левой кислоты при гидролизе концентрированным раствором щелочи в мети-
ловом спирте образуется лишь исходный спирт, но если применять среду
с большим содержанием воды и особенно более разбавленную щелочь, то
образуется главным образом изомерный 1-метил-З-фенилаллиловый спирт.
На этом основании был сделан вывод, что изменение условий реакции вызва-
ло изменение ее механизма. При наличии концентрированной щелочи и слабо
ионизующего растворителя преобладает механизм Вдс 2, а при слабой щелоч-
ности и хорошо ионизующем растворителе проявляется механизм BAL1.
Те же исследователи на основании выяснения природы продуктов реакции
установили, что у большинства сложных эфиров в обычных условиях их
превращения в простые и сложные эфиры путем сольволиза в спиртах или
в карбоновых кислотах как растворителях расщепляется связь алкил —
кислород. Так, 1,3-диметилаллиловый кислый эфир фталевой кислоты при
обработке метиловым спиртом образует рацемический 1,3-диметилаллил-
метиловый эфир; это результат расщепления связи алкил — кислород
сложного эфира. Тот же сложный эфир при обработке уксусной кислотой
образует рацемический 1,3-диметилаллиловый эфир уксусной кислоты [26]
CH3OH + ROCOR' —> CH3OR + HOCOR'
CH3COOH + ROCOR' —> CH3COOR+ HOCOR'
Аналогично из тг-метоксибензгидрилового кислого эфира фталевой кислоты
и метилового спирта получен рацемический и-метоксибензгидрилметиловый
эфир [27]; 1,1-нафтилэтиловый кислый эфир фталевой кислоты и муравьи-
ная кислота дают рацемический 1,1-нафтилэтиловый эфир муравьиной кис-
лоты [28]; многие сложные эфиры (содержащие 1-фенилэтил- и аналогичные
радикалы), включая эфиры муравьиной, уксусной, бензойной кислоты и кис-
лый эфир фталевой кислоты, дают рацемические простые или сложные эфиры
при сольволизе метиловым или этиловым спиртом, а также муравьиной или
уксусной кислотой [29].
Ни в одном из этих случаев гидролиза по механизму ВАь1 не было подтвер-
ждено, что кинетически они действительно представляют собой мономолеку-
лярные реакции; например, не было показано, что скорости гидролиза или
образования простых эфиров нечувствительны к щелочам. Однако это наблю-
далось в некоторых других случаях. Хэммонд и Радесилл изучили превраще-
ние трифенилметилового эфира бензойной кислоты в смеси этилового спирта
и метилэтилкетона в трифенилметилэтиловый эфир и в бензойную кислоту,
которое должно включать расщепление связи алкил — кислород
С2Н5ОН + (С6Н5)3СОСОСвН5 -» С2Н5ОС(С6Н5)з+СвН5СООН
Хэммонд и Радесилл [30] подтвердили мономолекулярный характер этой
реакции, показав, что ее скорость строго соответствует закономерностям
реакций первого порядка и при введении этилата натрия не увеличивается
(при этом проявляется лишь солевой эффект). Вторым вполне аналогичным
примером может служить превращение трифенилметилацетата в метиловом
спирте в трифенилметилметиловый эфир и уксусную кислоту
СН3ОН + (С6Н5)3СОСОСНз -> СН3ОС(СвН5)з + СНзСООН
60—0772
945
Глава XV
Гомберг и Дэвис [31] установили природу продуктов этой реакции, а Бантон
и Конашевич [32] изучили ее кинетику и показали, что в данном случае про-
текает реакция первого порядка, которая не ускоряется при добавлении
ионов метилата.
Бантон с сотр. [33] изучил гидролиз в щелочной и нейтральной средах ряда
эфиров 2,4,6-трифенилбензойной кислоты, в которых бимолекулярная атака
ацильной группы в значительной степени подавлена из-за пространственных
затруднений. Несмотря на это метиловый, этиловый, н-пропиловый, изобути-
ловый и изопропиловый эфиры гидролизуются щелочами в водном 95 %-ном
метиловом спирте по механизму Baq2: скорости реакции второго порядка,
которые в целом примерно в тысячу раз меньше, чем в случае соответствую-
щих эфиров бензойной кислоты, незначительно уменьшаются в гомологиче-
ском ряду, что характерно для этого механизма (табл. 206). Однако трет-
бутиловый эфир гидролизуется быстрее, чем любой низший эфир, для реак-
ции не требуется щелочи и она имеет первый порядок, который не увеличи-
вается при добавлении щелочи. Отсюда следует, что в этом случае гидролиз
протекает как мономолекулярная реакция. С помощью изотопа кислорода
18О было показано, что при гидролизе расщепляется связь алкил — кислород
* *
Н2О mpem-CaHgOCOCel^CjHj^ —> mpem-C^HgOH (CeHs^CjHgCOOH
Кроме того, поскольку скорость этой реакции должна быть значительно
больше экспериментально не изученной скорости мономолекулярных реак-
ций гидролиза соответствующего метилового, этилового и изопропиловых
эфиров, то этот пример может служить подтверждением ожидаемого ускоря-
ющего действия электроположительного алкильного заместителя и ожидае-
мого отсутствия пространственных затруднений. Как было показано, ско-
рость этой реакции значительно возрастает с увеличением относительного,
количества воды в среде. Таким образом, этот пример со всей полнотой под-
тверждает наличие механизма BAL1.
г. Бимолекулярный основной гидролиз с расщеплением связи алкил —
кислород (механизм ВА12). Если учесть приведенные в начале этого раздела
соображения о соотношениях между механизмами ВАС2 и BAL1, то возмож-
ный в основной среде механизм ВАц2 обычно не поддается обнаружению.
Действительно, этот механизм удалось наблюдать сначала в особой системе,
а затем и в случае обычной системы, но с принятием специальных мер для.
предотвращения маскирующего влияния механизма ВАс2.
Механизм гидролиза BAL2 можно сформулировать следующим образом:
Медленно + _
H2O + ROCOR' с ------> ROH24-OCOR' (Bal2>
Медленно
(ROH2 + OCOR'--------> ROH + HOCOR')
4 г Быстро
Принципиально этот процесс обратим, но перенос протона от иона алкил-
оксония к карбоксилат-иощ/ практически полностью сдвигает реакцию слева
направо.
Экспериментально можно проследить следующие особенности этого механизма:
во-первых, для его осуществления не требуется наличия кислоты; во-вторых,
он сопровождается расщеплением связи алкил — кислород и, в-третьих, он
должен быть бимолекулярным. Две последние характеристики специально-
устанавливать не нужно, если конфигурация асимметричной группы В
в процессе гидролиза полностью обращается. Можно заранее сказать, что на
скорость реакции должны влиять полярные и пространственные эффекты,,
946
Реакции карбоксильной и фосфатной групп
эффект растворителя и солевой эффект. К этому следует добавить, что при-
знак полного обращения, вообще говоря, является ненадежным, так как
этот стереохимический результат может получиться и при мономолекулярной
ионизации в случае очень сильного экранирования (гл. VII, разд. 7,е).
Однако имеющиеся многочисленные экспериментальные данные свидетель-
ствуют о том, что механизм ВдС1 в сходных растворителях сопровождается
рацемизацией. Кроме того, как будет видно ниже, подтверждающим опытом
является анализ продуктов реакции.
Наличие этого механизма было установлено в случае ^-лактоновой системы
на примере лактона яблочной кислоты [34]. В щелочных или в сравнительно
концентрированных кислых растворах этот внутренний сложный эфир гидро-
лизуется по основному и кислотному механизму с расщеплением связи
ацил — кислород и сохранением конфигурации. Однако при некотором про-
межуточном значении pH среды этот сложный эфир медленно гидролизуется
с обращением конфигурации, и, следовательно, реакция идет в соответ-
ствии с бимолекулярным механизмом Вдь2 с расщеплением связи алкил —
кислород. На приведенной ниже схеме (R = СООН) показаны разные места
расщепления; реакция в кислой среде будет рассмотрена позднее.
сн2
Водный гидролиз
Вль2
RHC\. ,\ю
Щелочный гидролиз
вас2
Кислотный гидролиз
Аас1
происходит расщепление связи алкил —
Рис. 53.
Скорости гидролиза двух 0-лактонов
[39].
Но — функция кислотности Гаммета, иден-
тичная pH при не очень высокой кислотно-
сти. Константа скорости h выражена в мин-1
и относится к реакциям в воде при 25 !°С.
1 — р-пропиолактон; 2 — р-бутиролактон.
То же явление наблюдали Олсон и Миллер [35] на примере ^-бутиролактона
(R = СН3). Олсон и Хайд [36] доказали с помощью меченого 18О, что при
гидролизе в нейтральной среде
кислород. Лонг и Перчейз [37]
на примере простейшего члена
этого ряда — р-пропиолактона
(R = Н) — продемонстировали,
что стереохимический критерий
не играет никакой роли, но, как
можно видеть на рис. 53, кине-
тика процесса столь сходна с
кинетикой гидролиза гомологов
лактонов, что и в этом случае
следует допустить такой же ме-
ханизм. В соответствии с таким
выводом Бартлет и Райлендер
[38] показали, что, хотя при
взаимодействии р-пропиолакто-
на с метилат-ионами образуется
оксиэфир НОСН2СН2СООСН3,
лактон реагирует с первона-
чально нейтральным метиловым
спиртом с образованием меток-
сикислоты СН3ОСН2СН2СООН.
Баннету и сотр. [40] в принципе ___
низма Bal2 несмотря на то, что обычно при основном гиролизе этот механизм
маскируется более быстрым механизмом ВАс2. Они изучали реакцию мети-
лата натрия с метиловым эфиром бензойной кислоты в растворе метилового
спирта. Для этой системы реакция Вас 2 является реакцией переэтерификации,
установить общий характер меха-
удалось
60* 947
Глава X V
в процессе которой каждая исходная молекула замещается идентичной. Как
бы быстро ни протекал такой процесс, он не может маскировать реакцию по
механизму BAl2, если она происходит. Это было доказано выделением из
продуктов реакции диметилового эфира с хорошим выходом
СН3О + СН3ОСОСвН5 —> сн3оснз + 6сос6н5
Качественно было показано, что скорость этой реакции зависит от наличия
метилат-иона и поэтому реакция должна протекать в соответствии с механиз-
мом, который представляет собой лишь особый случай S ^-замещения. С по-
мощью этого метода было показано, что метанолиз 2,4,6-трифенилбензоата
также идет по механизму BAL2 [33].
д. Мономолекулярный гидролиз и этерификация в кислой среде с рас-
щеплением связи ацил — кислород (механизм Аас1). Гидролиз сложных
эфиров и этерификация в кислой среде представляют собой взаимно обратные
процессы. При определенных условиях такие процессы в соответствующих
направлениях протекают по идентичным механизмам; при этом соблюдается
принцип микроскопической обратимости. Реакции гидролиза и этерификации
в случае катализа кислотами могут быть отнесены, в частности, к типу про-
мотируемых кислотами реакций кислородного обмена в кислотах и в общем
случае к катализируемой кислотами переэтерификации.
Механизмы, указанные в заглавии этого и следующего параграфов, охваты-
вают обычные случаи гидролиза и этерификации при катализе кислотами.
Для того чтобы установить наличие этих механизмов, необходимо показать,
что реакции сопровождаются расщеплением связи ацил — кислород, хотя
в особом случае кислородного обмена в кислотах это само собой разумеется.
Гольмберг [3] первый установил характер гидролиза в кислой среде. Для этого
он пользовался методом, поволяющим установить наличие изменения конфи-
гурации группы R, асимметричной в месте связи в сложном эфире R'COOR.
Гольмберг показал, что при кислотном гидролизе О-ацетиляблочной кислоты
CH3COOR, где R = СН(СООН)СН2СООН, асимметричная группа сохраняет
свою конфигурацию.
Хилда Ингольд и автор книги [41] применили метод, заключающийся в выбо-
ре такого радикала R, который, отщепившись в виде R+, должен быть мезо-
мерным, что должно приводить при кислотном гидролизе и этерификации
к образованию мезомерпых анионотропных продуктов. Примерами служили
1-метилаллил- и 3-метилаллилацетаты, причем изомеризация не наблю-
далась.
Для обнаружения расщепления связи ацил — кислород при кислотном
гидролизе кислого метилового эфира янтарной кислоты [42], бензгидриль-
ного эфира муравьиной кислоты [43] и -^бутиролактона [44] был использо-
ван метод, основанный на применении изотопа кислорода 180. Доказано, что
этот же тип расщепления наблюдается и в случае этерификации бензойной
кислоты метиловым спиртом [45].
Теоретические соображения и аналогии с другими общими реакциями позво-
ляют предположить наличие двух механизмов гидролиза и этерификации,
катализируемых кислотами и сопровождаемых расщеплением связи ацил —
кислород. Для обоих механизмов характерно наличие предварительно уста-
навливающегося равновесия с присоединением протона, но они отличаются
друг от друга поведением образующегося иона оксония. В случае мономо-
лекулярного механизма AAq1 ион оксония сначала претерпевает гетеролити-
ческое расщепление, определяющее скорость всего процесса, так же как ион
сульфония в случае мономолекулярного нуклеофильного замещения [41].
948
Реакции карбоксильной и фосфатной групп
Таким образом, образуется ион карбония, точнее ацилий-ион R'CO+, кото-
рый затем быстро атакуется гидроксилсодержащей молекулой, как это про-
исходит и в конечной стадии мономолекулярного сольволитического замеще-
ния. Наконец, происходит отщепление протона, эквивалентного протону,
захваченному первоначально. Все процессы переноса протона рассматрива-
ются как мгновенные. Этот механизм, как и в предшествующих случаях,
считается обратимым и пригодным как для гидролиза, так и для этерифика-
ции. Подлинный характер этого механизма (если так можно сказать) опреде-
ляется двумя средними стадиями, и, принимая во внимание соответствие их
двум стадиям мономолекулярного замещения, рассматриваемый механизм
называют мономолекулярным и обозначают символом Аас1.
Быстро +
R'COOR + H+ , -----» R'COOHR
Быстро
+ Медленно + >.
R'COOHR ----------->R'CO-I-ROH
Быстро (Адс1)
+ Быстро +
R'CO-|-H2O .........> R'COOH2
Медленно '
+ Быстро
R'COOH2 Т— —> R'COOH4-H+
Быстро
Бимолекулярный механизм [42, 46] Аас2 выводится из механизма Аас1
точно так же, как механизм бимолекулярного нуклеофильного замещения
выводится из механизма мономолекулярного замещения. Время жизни иона
ацилия уменьшается до продолжительности периода столкновения, так что
те две стадии, которые ранее представляли образование и разрушение этого
иона, в этом случае сливаются в единый бимолекулярный процесс. Началь-
ный захват протона и конечная потеря протона представляют собой; как
и ранее, быстрые обратимые процессы. Для этого процесса характерна сред-
няя стадия, и по аналогии с бимолекулярным замещением этот механизм
описывается как бимолекулярный и обозначается символом Аас2
Быстро +
R'COOR-|-H+ '» R'COOHR
Быстро
+ Медленно +
R'COOHR + H2O ; —» R'COOH2 + ROH (Аас2)
Медленно
+ Быстро
R'COOH2 с —> R'COOH + H+
Быстро
Как и в случае механизма реакции в щелочной среде (ВАС2), стадию, опре-
деляющую скорость рассматриваемого кислотного механизма, можно пред-
ставить гипотетически либо моделью обратимого присоединения к карбо-
нильной группе и распада
О 0"
|| + Медленно + | + Быстро
h2o+c-ohr , > h2o-c-ohr — --»
I Быстро | Медленно
R' R'
О
H20-C + R0H (Aac2)
I
R'
949
Глава XV
либо моделью нуклеофильного замещения
О
II +
Н2О + С—OHR
R'
Медленно
----------->
<----------
Медленно
. + II
---> Н2О —C+HOR
I
R'
(Адс2)
Предполагается, что эта картина соответствует крайним случаям и что
в действительности в промежуточном комплексе имеется некоторое промежу-
точное распределение заряда по сравнению с приведенным выше; этот ком-
плекс обладает большей устойчивостью, чем любая из приведенных струк-
тур, одна из которых соответствует молекуле, а вторая — переходному ком-
плексу. Ниже показано, что промежуточный комплекс действительно являет-
ся молекулой.
Оба механизма, и Аас1, и Аас2, кинетически имеют первый порядок по
эфиру: в обоих случаях константа скорости к\ служит хорошим параметром
реакции при любых значениях кислотности. В разбавленных растворах
кислот оба механизма имеют первый порядок по ионам водорода, но в кон-
центрированных растворах сильных кислот проявляется значительное разли-
чие в зависимости от кислотности. В качестве первого приближения рассмот-
рим прежние представления о различии между этими двумя механизмами.
Механизм AAq1 предполагает предравновесное образование сопряженной
эфиру кислоты, которая затем претерпевает медленный мономолекулярный
гетеролиз. Единственным важным фактором, определящим кинетику реак-
ции, является стационарная концентрация сопряженной кислоты, которая
контролируется термодинамическими факторами. Кроме того, с увеличени-
ем концентрации сильной кислоты в воде функция кислотности Гаммета hG
начинает не совпадать с [Н+], поэтому можно ожидать, что скорость реакции
более точно будет зависеть от величины 7г0. Механизм Аас2 также предпола-
гает наличие предравновесного присоединения протона, но это требует при-
соединения молекулы воды на лимитирующей стадии. С кинетической точки
зрения это эквивалентно присоединению Н30+ в процессе, который лимити-
руется бимолекулярной стадией. Поэтому, как только значения [Н30+] и /г0
начинают заметно расходиться, можно ожидать более точного выражения
скорости реакции с помощью соответствующих значений [Н30+].
Существует довольно хорошая аппроксимация для средних значений кон-
центрации кислоты, но для того чтобы проследить различие между этими
двумя механизмами вплоть до очень высоких концентраций кислоты,
необходима более приближенная теория для выражения параметров кислот-
ности. Путь к созданию такой теории был намечен Баннетом [47] (гл. XII,
разд. 3,а). Функция Гаммета hQ или более удобная ее логарифмическая фор-
ма —Но = lg/i0 введена для описания равновесия переноса протона от водно-
кислотной среды к индикаторным основаниям, которыми служили азотистые
основания. Но в принципе основание любого типа (строго говоря, каждое
основание) нужно описывать своей собственной кислотной функцией. При-
чина состоит в том, что когда мы увеличиваем концентрацию сильной кисло-
ты в воде, мы увеличиваем не только активность ионов водорода, но также,
начиная с некоторого момента, сильно уменьшаем активность воды. Процесс
образования всех сопряженных кислот идет с присоединением протона, как
950
Реакции карбоксильной и фосфатной групп
того требует их строение, но при этом различные сопряженные кислоты зна-
чительно отличаются по своей склонности к гидратации. Так, при образова-
нии относительно гидрофобной сопряженной кислоты и по мере расходова-
ния гидратированных ионов водорода будет освобождаться связанная вода,
и это будет способствовать образованию такой кислоты в сильно концентриро-
ванной кислой среде, где активность воды снижается. Согласно Баннету,
величина, на которую —Но превышает логарифм концентрации ионов водоро-
да, соответствует количеству связанной воды, освобождаемой индикаторной
кислотой, которая применяется для определения функции Но. Если сопря-
женная кислота основного субстрата гидратирована меньше его самого, то
общая кислотная функция —Hs будет расти более круто, чем —Но; если
сопряженная кислота гидратирована больше, чем индикаторная, то —Hs
будет расти менее круто, чем — Но.
Количественная сторона этой теории в форме, удобной для исследования
механизма гидролиза сложных эфиров, была разработана и доказана экспе-
риментально Ятсом и Макклелландом [48] *. Нас интересует скорее сам
метод, чем его результаты, хотя и было бы удобно иллюстрировать его неко-
торыми количественными примерами, полученными авторами метода. Но мы
отложим такую иллюстрацию до того момента, когда перейдем к общему
описанию результатов. Экспериментальная область, выбранная авторами
метода, включает в себя гидролиз различных алкил- и арилацетатов в водном
растворе серной кислоты.
Оба механизма, Аас1 и Аас 2, начинаются с предравновесного образования
сопряженной эфиру кислоты. Поэтому вначале нужно определить кислотную
функцию, которая выражает степень этого термодинамически контролируе-
мого переноса протона. Для всех эфиров, для которых положение предравно-
весия можно было определить с помощью спектрометрии, Ятс и Макклелланд
нашли, что степень переноса протона можно выразить практически одной
и той же функцией —Hs, которая имеет линейную зависимость от—Но,
с наклоном 0,62. Такой наклон, по величине меньший единицы, означает
только, что ониевые ионы эфира гидратированы меньше, чем индикаторные
ионы Гаммета.
Располагая такими функциями кислотности, с помощью которых можно
количественно изучить общую предравновесную стадию обоих механизмов,
Ятс и Макклелланд смогли «выявить» окончательные особенности лимитиру-
ющих скорость вторых стадий этих реакций. Эти вторые стадии отличаются
по числу участвующих в них молекул воды, и поэтому необходимо опреде-
лить, как изменяются скорости этих индивидуальных стадий с изменением
активности воды при увеличении концентрации кислоты. Для этого нужно
построить зависимость log kt от —Hs или, что то же самое, суммы log kt + тН0
(где ти = 0,62) от log аН20. При условии, что равновесное содержание сопря-
женной эфиру кислоты мало, сумма log kt -\-тН0 пропорциональна суммар-
ной свободной энергии активации всего процесса гидролиза и меньше сво-
бодной энергии предравновесной стадии.
В механизме АА(Д во второй стадии эфирный ион, представляющий собой
чистый оксониевый ион, заменяется ацилий-ионом, который частично являет-
ся оксониевым ионом и частично карбониевым ионом. По сути дела, это
катион псевдооснования. Этот катион будет менее гидратирован, чем чистый
оксониевый ион, и уменьшение степени гидратации будет зависеть от того,
насколько его структура будет близка к карбониевому иону. В действитель-
* Автор весьма признателен доктору Ятсу за возможность познакомиться с его статьей
в рукописи до ее опубликования.
951
Глава XV
ности на скорость оказывает влияние не строение самого иона ацилия.
а строение переходного состояния реакции превращения оксониевого иона
эфира в ион ацилия. Однако можно ожидать, что даже переходное состояние
Концентрация H2SO4, %
Рис. 54. Зависимость скорости реакции от
кислотности среды для реакций гид-
ролиза алкилацетатов в водном ра-
створе серной кислоты при 25 °C
(fcj — константа скорости первого
порядка в мин-1; т = 0,62) [48].
1 — метилацетат; 2 — этилацетат; з —
н-пропилацетат.
будет несколько менее гидратиро-
вано, чем исходный оксониевый
ион эфира. Поскольку аН2о с уве-
личением концентрации кислоты
уменьшается, следует ожидать,
что сумма log/Ci -|~тпЯ0 будет
немного возрастать, т. е. график
зависимости logаН2о от log ki + тН0
будет иметь небольшой отрица-
тельный наклон. Как можно ви-
деть из рис. 54, для трех простых
алифатических эфиров уксусной
кислоты в серной кислоте, имею-
щей концентрацию от 85 до 100%
по весу, где преобладает механизм
типа Адс1, приведенная сумма
действительно возрастает при
уменьшении log «h2o и наклон
прямой составляет —0,2. Это оз-
начает, что при превращении ок-
сониевого иона эфира в переход-
ный комплекс, ведущий к обра-
зованию иона ацилия, в раствор
переходит в среднем 0,2 молекулы
первоначально связанной воды.
В концентрированном растворе
серной кислоты нельзя изучить поведение фенилацетата, поскольку проис-
ходит его сульфирование, но поведение п-нитрофенилацетата изучить мож-
но. Процесс протекает при концентрации серной кислоты более 70% по ве-
су по механизму Адс1, и снова на графике зависимости суммы log ki -\-тН0
от log ян2о получается прямая с наклоном —0,2.
В механизме Аас 2 на второй стадии расходуется молекула воды, которая,
очевидно, входит в состав бимолекулярного переходного состояния. Если это
так, то можно ожидать, что, поскольку log ан2о уменьшается с увеличением
концентрации кислоты, сумма log кг -\-тН0 должна испытывать соответ-
ствующее уменьшение с наклоном, равным единице, который можно интер-
претировать как «кинетический порядок» по воде. На самом же деле при этом
механизме зависимость log ан2о от log имеет наклон больший, чем
можно было бы ожидать. Это объясняется тем, что приведенный выше
(стр. 949) механизм реакции слишком упрощен в том смысле, что на схеме не
было указано, что молекула воды, согласно любому механизму кислотного
гидролиза, вначале должна служить переносчиком протона, а затем —
основанием, отщепляющим протон. Вполне возможно, что молекула воды,
которая отщепляет протон от субстрата на третьей стадии механизма Аас 2,
в конце второй стадии будет связана водородной связью с этим протоном;
возможно, что она входит в состав переходного состояния второй стадии,
сопутствуя сосредоточению положительного заряда. Если это так, то наклон
прямой на графике зависимости log ki-^-mHg от log аН20, т. е. «кинетический
порядок» реакции по воде, должен равняться двум. Как видно из рис. 54
для случая тех же трех эфиров в области концентраций серной кислоты от О
952
Реакции карбоксильной и фосфатной групп
до 80%, когда преобладает механизм Аде 2, наклон этой прямой равен 2,0 ±
±0,1. Данные для этилацетата были получены Лейном [49]. Ятс и Макклел-
ланд определили такую же величину наклона для вторичных алкилацетатов
в области концентрации серной кислоты от 0 до 70%, для бензилацетата
в области от 0 до 60% и для фенил-, п-хлорфенил- и п-нитрофенилацетатов
в области от 0 до 50%. Таким образом, механизм Аде 2 преобладает в основ-
ном в среде более разбавленных растворов серной кислоты.
Большое качественное различие между механизмами АдС1 и Адс2 с точки
зрения зависимости константы скорости ki от концентрации сильной кислоты
сводится к следующему: при механизме АдС1 константа скорости с увеличе-
нием концентрации кислоты всегда растет несколько быстрее, чем функция
кислотности, а при механизме АдС2 скорость растет менее круто, чем функция
кислотности; при этом наклон прямой зависимости скорости от концентра-
ции кислоты постепенно уменьшается, скорость гидролиза переходит через
максимум, а затем снижается все быстрее и быстрее, достигая нулевой ско-
рости при активности воды, равной нулю, или какого-то низкого значения
скорости, если возможна перемена механизма гидролиза. Эта часть
кривой зависимости скорости от концентрации кислоты будет приведена
позднее.
Можно предвидеть и другие возможные различия между механизмами Аас1
и Аас2. Скорости гидролиза и этерификации по механизму Аас1 должны
быстро возрастать при введении в ацильную группу электроположительных
заместителей, которые не будут сильно препятствовать присоединению про-
тона к спиртовому атому кислорода, но будут сильно способствовать течению
лимитирующей стадии гетеролиза при ацильном атоме углерода. Этот эффект
должен проявляться значительно сильнее, чем любые кинетические эффекты
таких же заместителей при механизме Аас2. Скорость реакции при механиз-
ме Аас1 не должна быть чувствительной к пространственным затруднениям—
в этом состоит другое его серьезное отличие от механизма Адс2. Можно
наметить и некоторые другие различия. Если сложный эфир гидролизуется по
механизму Аас1 в водной кислоте, содержащей 18О, то этот изотоп не может
перейти в молекулу эфира до того, пока не произойдет гидролиз; в случае же
механизма ААс,2 изотоп 18О может быть обнаружен в эфире. Наблюдения
Лонга (гл. VII, разд. 4,а) показывают, что при мономолекулярном механиз-
ме обычно наблюдается более высокая (более положительная или менее отри-
цательная) энтропия активации, чем при бимолекулярном. В заключение
можно добавить, что скорости гидролиза по механизму AAq1 должны быть
более чувствительными к влиянию растворителя и этот механизм должен
скорее проявляться в таких растворителях, как серная кислота, в которых,
как известно, ионы ацилия устойчивы (гл. VI, разд. 6, д).
Трефферс и Гаммет [50] получили первое прямое доказательство возможно-
сти реакции карбоксильных групп по механизму Аас1; они установили, что
2,4,6-триметилбензойная кислота вызывает четырехкратную депрессию
температуры замерзания растворителя — серной кислоты, что указывает на
превращение этой кислоты в ион мезитоилия
(CH3)3CeH2COOH±H2SO4 -» (CH3)3CeH2COOHJ + HSO7
(CH3)3ceH2cooH+±H2so4 -> (Ch3)3c3h2co++h3o+±hso;
Бензойная кислота вызывает удвоенную депрессию, что указывает на пол-
ноту начального переноса протона; обнаружить дальнейшее превращение
в ион бензоилия криоскопическим методом не удается. Аналогичное поведе-
ние эфиров этих кислот и значение такого рода данных для реакций гидроли-
за было установлено при выливании в воду свежеприготовленного раствора
953
Глава X V
метилового эфира 2,4,6-триметилбензойной кислоты в серной кислоте. При
этом количественно выпадал осадок 2,4,6-триметилбензойной кислоты. При
такой же обработке метилового эфира бензойной кислоты эфир почти полно-
стью остается негидролизованным. Эти результаты находятся в резком
противоречии с известными фактами тормозящего влияния орто-заместите-
лей, в том числе opmo-метильных заместителей, на процесс кислотного гидро-
лиза эфиров бензойной кислоты в растворителях, содержащих воду, и на
этерификацию бензойной кислоты в спиртовой среде. Такое противоречие
ясно указывает на двойственность механизма реакции и находится в соот-
ветствии с представлением о том, что в серной кислоте реакция протекает
по мономолекулярному механизму AAq1, а в водных или спиртовых раство-
рителях — по бимолекулярному механизму Аас2. Серная кислота являет-
ся одним из лучших растворителей, приводящих к образованию из кислот
дегидратированных ониевых ионов. Указанные противоречия свидетель-
ствуют как о сильном полярном влиянии электроположительных метильных
заместителей, так и об отсутствии пространственных затруднений, создавае-
мых заместителями в орто-положении в реакциях, протекающих в серной
кислоте.
Реакции 2,4,6-триметилбензойной кислоты и ее эфиров в серной кислоте
характеризуются быстрым и полным образованием ацилий-иона. Однако
образование такого иона не должно быть ни полным, ни быстрым для того,
чтобы он представлял собой промежуточное соединение при механизме Аас1.
Действительно, в тех случаях, когда удается исследовать кинетику реакций,
оказывается, что ацилий-ион должен образовываться медленно, поскольку он
определяет скорость всего процесса, а стационарная концентрация его может
быть очень мала. Грэхем и Хьюз (частное сообщение) установили это при
изучении кинетики гидролиза метилового эфира бензойной кислоты водой
в серной кислоте. Эта реакция протекает медленно (полупериод 7,7 ч при
20 °C) и является реакцией первого порядка по эфиру и реакцией нулевого
порядка по воде при ее концентрациях до 1 М. Кинетика гидролиза метило-
вого эфира n-толуиловой кислоты весьма сходна с приведенной выше, но
скорость реакции в этом случае в 4 раза больше. Три примера Ятса и Мак-
клелланда по гидролизу алифатических эфиров уксусной кислоты в водном
растворе серной кислоты, идущего по механизму Аас1 при содержании воды
до 15%, приведены на рис. 54. Подобная картина, как упоминалось выше,
имеет место для тг-нитрофенилацетата в среде, содержащей до 30% воды.
Если постепенно разбавлять сильно концентрированный раствор гидролизую-
щей сильной кислоты, например серной, то эфиры 2,4,6-триметилбензойной
кислоты гидролизуются с сохранением механизма AAq1 до более высокой
степени разбавления, чем это происходит в случае эфиров бензойных кислот,
не имеющих opmo-заместителей, или в случае эфиров простейших алифати-
ческих кислот, например ацетатов. Бендер [51] установил, что метиловый
эфир 2,4,6-триметилбензойной кислоты гидролизуется в 3 Л/ водном раство-
ре серной кислоты по механизму AAq1, о чем свидетельствуют два факта:
во-первых, скорость реакции лучше соответствует h0, чем [Н+], и, во-вторых,
в негидролизованный эфир не происходит внедрения изотопа 18О из реакцион-
ной среды. Лонг [52] нашел, что метиловый и а-глицериловый эфиры 2,4,6-
триметилбензойной кислоты претерпевают гидролиз в 4 АГ водном растворе
серной кислоты и 6 АГ водном растворе хлорной кислоты по механизму ААс1,
в то время как в таких же условиях метиловый или а-глицериловый эфиры
бензойной, n-анисовой или 3,4,5-триметоксибензойной кислот гидролизуют-
ся по механизму Аас2. Лонг пришел к такому выводу на основании того, что
в первом случае скорость реакции лучше соответствовала функции h0, а во
954
Реакции карбоксильной и фосфатной групп
втором — [Н+]. Бантон и сотр. [53] исследовали 18О-обмен между водным
раствором сильной гидролизующей кислоты и 2,4,6-триметилбензойной ибен-
зойной кислотами. 2,4,6-Триметилбензойная кислота в смеси диоксан —
вода (в объемном отношении 60 : 40), содержащей 0,4—3,0 М хлорной кисло-
ты, обменивала изотоп 18О со скоростью, соответствующей величине функции
Ао, и имела энтропию активации 4-9 кал/(моль •°C) [37,68 Дж/(моль •°C)];
это означает, что обмен протекает по механизму ААс1. В отличие от этого
бензойная кислота в воде, содержащей 0,4—3,0 М серной кислоты, обмени-
вает изотоп 18О со скоростью, соответствующей величине [Н+], и имеет энт-
ропию активации —30 кал/(моль-°C) [—125,60 Дж/(моль-°C)]; это свидетель-
ствует о протекании реакции обмена по механизму ААс2.
е. Бимолекулярный кислотный гидролиз и этерификация с расщепле-
нием связи ацил — кислород (механизм Аас2). Теория, кинетика и другие
особенности механизма ААс2, позволяющие установить его отличия от меха-
низма AAq1, были разобраны в предыдущем разделе. Это было сделано для
того, чтобы ярче показать многие их отличительные особенности. Из этих
двух механизмов механизм ААс2 более общий, если кислотный катализ осу-
ществляется в разбавленном или умеренно концентрированных растворах
кислоты. Большинство сложных эфиров (за исключением некоторых особых
классов, например сложных эфиров третичных спиртов и ароматических
эфиров с карбоксильной группой, сильно экранированной со стороны
ацильной цепи) в разбавленных или умеренно концентированных водных
растворах сильных кислот, например хлорной или серной, гидролизуются
по бимолекулярному механизму Aaq2. Точно так же кислоты, соответствую-
щие этим эфирам, в разбавленных спиртовых растворах сильных кислот
этерифицируются по механизму AAq2, например по методу Фишера — Спай-
ера при нагревании кислоты в разбавленном спиртовом растворе хлористого
водорода.
Этот механизм привлекал внимание во всех ранних работах, посвященных
исследованию скорости кислотного гидролиза и этерификации,— работах,
которые стали основополагающими в области изучения кинетики вообще
и помогли установить связь кинетики с химической термодинамикой.
В 1862 г. Бертло и Пеан де Сан-Жилль [54] показали, как меняется положе-
ние равновесия при обратимом образовании этилацетата с изменением соот-
ношений реагирующих веществ. Они установили, что скорости обратных
реакций пропорциональны произведению концентраций реагирующих ве-
ществ. Гульдберг и Вааге [55] широко использовали эту работу при обос-
новании закона действия масс в применении к кинетике реакций. Оствальд
[56] установил порядок реакции гидролиза сложных эфиров водными кисло-
тами, показав, что скорость реакции пропорциональна произведению
[Н+] [R'COOR]; отсюда вытекала ясная зависимость скорости реакции от
концентрации сложного эфира и не совсем ясная зависимость скорости реак-
ции от концентрации водородного иона. Что касается последнего, то в 1884 г.
было показано, что скорость гидролиза метилацетата в разбавленных водных
растворах ряда кислот, от сильных до слабых, при одной и той же их кон-
центрации почти пропорциональна (в интервале соотношения скоростей
200 : 1) электропроводности растворов этих кислот. Через три года, после
появления теории электролитической диссоциации, смысл этого наблюдения
стал ясным: скорость реакции пропорциональна концентрации водородных
ионов [Н+], так как электрический ток почти полностью переносится ионами
Н+. Порядок реакций этерификации карбоновых кислот при катализе силь-
ными кислотами в спиртах как растворителях был совершенно четко опреде-
лен Гольдшмидтом [57]. Он установил, что скорость реакции пропорциональ-
955
Глава X V
на произведению концентраций [Н+] [R'COOR]. Поскольку при этерифика-
ции и при кислотном гидролизе сложных эфиров переходное состояние долж-
но быть одним и тем же, приведенный выше результат подтверждает ранее
установленный кинетический закон, по которому происходит гидролиз
в кислой среде; в состав переходного состояния входят либо Н+ + R'COOH-L-
4- ROH, либо Н+ + R'COOR + Н2О. Непосредственное доказательство
кинетического закона, по которому происходит гидролиз сложных эфиров
в кислой среде, было приведено в ряде более поздних работ, в том числе
в исследованиях Друшеля [58] и Паломаа [59].
Кинетические характеристики механизма Аас2 были рассмотрены в предыду-
щем разделе для всей области концентраций, в которой этот механизм реали-
зуется. Теперь мы остановимся на некоторых других характеристиках этого
механизма.
Бендер [60] показал, что в процессе гидролиза по механизму ААс2 комплекс,
образованный сложным эфиром, водой и водородным ионом, представляет
собой устойчивую молекулу, а не просто переходное состояние реакции. Для
этого Бендер воспользовался водой, обогащенной изотопом кислорода 18О,
так же как он это сделал при установлении механизма ВАС2 в щелочной среде.
Бендер исследовал гидролиз этилового эфира бензойной кислоты в воде,
добавляя в качестве катализатора хлорную кислоту. При этом он установил,
что скорость обмена кислорода между сложным эфиром и водой, определенная
по содержанию изотопа 18О в непрореагировавшем сложном эфире на различ-
ных глубинах гидролиза, составляет значительную долю (около 20%) ско-
рости гидролиза. Это означает, что продолжительность жизни промежуточ-
ного комплекса достаточно велика, благодаря чему возможно перемещение
протонов, необходимое для того, чтобы сделать вполне эквивалентными не-
алкилированные атомы кислорода. Это положение можно проиллюстрировать,
как и раньше, на модели реакции, включающей присоединение, хотя ясно,
что эта модель дает лишь ограниченное представление о происходящих изме-
нениях в распределении электронов.
о-
Н2О + О=С—OHRr±
R'
О
* II +
Н2О + С—OHR^
I
R'
R'CO2H2+ + H0R
Биктор Мейер, Садборо и Келасс в работе, проведенной еще в прошлом
столетии, установили сильное влияние пространственных факторов на этери-
фикацию, катализируемую кислотами. В гл. VII, разд. 8, была приведена
ссылка на эту работу, в частности на показанное Мейером замедление про-
цесса этерификации двумя заместителями в орто-положении в бензойной
кислоте, независимо от характера их полярности; в случае же фенилуксус-
ной кислоты эти заместители не оказывают замедляющего действия. Почти
в то же время в начале этого столетия Гольдшмидт [61], Кейлан [62] и Садбо-
ро [63], развивая эту работу экспериментально, определили константы ско-
рости этерификации замещенных бензойных кислот; при этом были обнару-
жены небольшие полярные эффекты мета- и пара-заместителей, а также
956
Реакции карбоксильной и фосфатной групп
Таблица 208
Сравнительные скорости катализируемых кислотами процессов гидролиза
эфиров алифатических кислот и этерификации алифатических кислот
А. Гидролиз под действием НС1 в воде при 25 °C
CH3COOR Г R X Скорость СНз 1 С2Н5 0,97 U30-C3H7 0,53 mpem-C4H9 1,15а
CH3COOR J R сн2с6н5 СН2СН2ОН СН = СН2 свн5
[ Скорость 0,96 0,69 1,20 0,69
R'COOCHg И' н СН3 СООСН3 СН2СН2СООСН3
I Скорость 21,3 1 1,71 0,35
R'COOCH3 f R' сн3 СОСН3 СН2СОСН3 СН2СН2СОСН3
X Скорость 1 1,20 0,16 0,23
Б . Гидролиз под действием CgHgSOg этилового спирта Н в 60%-ном при 60 °C водном растворе
R'COOC2H5 f R' СНз СН2С1 СНС12 СС13
X Скорость 1 0,91 0,40 0,11
В. Этерификация под действием НС1 в спирте при 14,5 °C
R'COOH ГИ' СН3 С2н5 изо-С3Н7 трет-СцН.^
[ Скорость 1 0,83 0,27 0,025
R'COOH И' СН3 СН2С1 СНС12 СС13
1 Скорость 1 0,83 0,017 0,010
R'COOH Г R' СНз СвН5СН2 (С6Н5)2СН (СвН5)3С
X Скорость 1 0,56 0,015 0,000
а Эта величина, вероятно, завышена за счет параллельной реакции гидролиза по механизму
Адь1 <см- РазД- С ЖЬ
заметное тормозящее действие opmo-заместителей, которое, по-видимому,
зависит больше от размеров заместителя, чем от его полярности. Садборо
и сотр. [63], развивая ранние полуколичественные работы Меншуткина, прове-
ли систематическое изучение влияния строения на кинетику этерификации
алифатических кислот; при этом было отмечено тормозящее влияние развет-
вления цепи близ реакционного центра. Скрабал с сотр. [64] в 20-х годах
исследовали кинетику гидролиза эфиров жирных кислот в кислой среде,
причем многие из этих эфиров содержали различные функциональные группы.
Результаты проведенных ими исследований снова показали незначительную
роль полярных эффектов и сильное влияние пространственных затруднений
при наличии заместителей близ карбоксильной группы. В 30-х годах был
опубликован ряд работ, посвященных этим вопросам. Паломаа и сотр. [65]
изучали кинетику гидролиза сложных эфиров жирного ряда с углеводород-
ными и неуглеводородными заместителями, а также кинетику этерификации
кислот такого же строения. Хиншелвуд с сотр. [66] исследовал кинетику
гидролиза и этерификации в алифатическом и ароматическом рядах с целью
выявления влияния заместителей на константы скорости реакции и константы
Аррениуса. Эта работа была расширена в 40-х годах Хартманом и сотр. [67],
которые изучили скорости реакций и параметры Аррениуса на примере этери-
фикации многих замещенных бензойных кислот. Работы в этих направлениях
продолжаются и в настоящее время. При составлении табл. 208 и 209 была
использована лишь небольшая часть данных, относящихся к рассматривае-
957
Глава X V
Таблица. 209
Сравнительные скорости катализируемых кислотами процессов
гидролиза эфиров ароматических кислот и этерификации
ароматических кислот
А. Гидролиз ХС^ЩСООСгНд действием CgHgSOsH в 60%-ном
водном растворе этилового спирта при 100 °C
Положение заместителя ОСНз СНз н Вг no2
пара 0,92 0,97 1 0,98 1,03
мета 1 0,99
орто 1 0,36
Б. Гидролиз СН3СООСвН4Х девствием СвНд8О3Н в 60%-ном
водном растворе этилового спирта при 25 °C
Положение заместителя СНз н СООН NO2
пара 1,06 1 0,89 —
мета 0,97 1 0,86 0,70
В. Этерификация ХС6Щ СООН действием HCI в метиловом спирте при 25 °C
Положение СНз no2
заместителя н Вг
пара 1,03 1 0,64 0,45
мета 1,13 1 0,65 0,38
орто 0,33 1 0,29 0,028
мому вопросу. Однако и приведенных данных достаточно для того, чтобы
показать незначительное влияние полярных эффектов и существенное влия-
ние пространственных факторов на процессы гидролиза и этерификации.
Из приведенных данных видно, насколько мало влияет на скорость реак-
ции в алифатическом ряду замещение электроположительной группы СН3
сильно электроотрицательной группой СОСН3 или СООСНЭ; можно также
заметить, что в ароматическом ряду при введении нитрогруппы в пара-
положение кинетический эффект остается небольшим и в этом проявляется
резкое отличие от влияния таких же заместителей на гидролиз в щелочной
среде (табл. 206 и 207). С другой стороны, в алифатической серии наблюдается
заметное тормозящее действие любого набора из трех заместителей, будь
то (СН3)3, С13 или (СвН5)3, находящихся по соседству с карбоксильным угле-
родным атомом; можно отметить также значительное тормозящее действие
opmo-заместителей в ароматическом ряду. Для развития органической
химии весьма важное значение имел тот факт, что пространственные затруд-
нения были открыты Виктором Мейером именно на примере реакций этери-
фикации. Если бы он выбрал для этой цели гидролиз эфиров карбоновых
кислот в щелочной среде, то при этом также можно было бы выявить роль
пространственных затруднений, но результаты наблюдений было бы труднее
объяснить одной решающей причиной.
958
Реакции карбоксильной и фосфатной групп
Причина, по которой при бимолекулярном кислотном гидролизе и этерифи-
кации полярные эффекты проявляются очень слабо, с качественной стороны
очевидна: любой полярный эффект влияет на обе стадии этого механизма
в почти взаимно компенсирующих противоположных направлениях. Этот
эффект или ускоряет предравновесное протонирование, но задерживает лими-
тирующую стадию атаки сопряженной кислоты водой или спиртом, или по-
давляет протонирование, но ускоряет нуклеофильную атаку. При бимоле-
кулярном щелочном гидролизе эти влияния не компенсированы: единствен-
ным процессом, имеющим кинетическое значение, является нуклеофильная
атака.
ж. Мономолекулярный кислотный гидролиз и этерификация с расщепле-
нием связи алкил—кислород (механизм Aal1). В разд. 1, в было указано,
что при соответствующем выборе радикала R можно наблюдать гидролиз
эфира R'COOR по механизму ВагД, причем стадией, определяющей скорость
реакции, является гетеролиз нейтральной молекулы сложного эфира с обра-
зованием иона карбония
R'COOR —» R'COO~4-R+
Это означает, что при соответствующем выборе радикала R можно осущест-
вить гидролиз и по механизму Aal1, для которого характерен аналогичный,
но несомненно более легко протекающий гетеролиз ионной кислоты, сопря-
женной сложному эфиру. Поскольку ионные кислоты, сопряженные неко-
торым спиртам, способны к гетеролизу до иона карбония, то ясно, что реак-
ции этерификации могут протекать по механизму АдгД- Ниже приводятся
стадии этого^механизма:
Быстро +
R'COOR++ ; > R'COOHR
Быстро
+ Медленно
R'COOHR С ' > R'COOH + R+
Быстро
Быстро +
R+4-H2O — ROH2
Медленно ✓
Быстро
ион2 > roh + h+
Быстро
(Aal1>
Мономолекулярный гетеролиз иона оксония является стадией, определяющей
скорость процесса в обоих направлениях.
Рассмотрим ожидаемые характеристики этого механизма. Катализаторами
реакции должны служить кислоты. Следует ожидать расщепления связи
алкил—кислород, что можно подтвердить применением изотопа кислорода 18О.
В соответствующих случаях должны проявляться признаки образования ио-
на R+. Так, если радикал R асимметричен в месте связи, то он должен под-
вергаться рацемизации, возможно с некоторым обращением конфигурации;
если ион Й+ может распадаться с образованием олефина или претерпевать
вагнеровскую или иную перегруппировку, то гидролиз или этерификация
будут сопровождаться этими процессами. На скорость этой реакции должны
сильно влиять полярные эффекты; в особенности сильно реакция может уско-
ряться под влиянием электронодонорных групп в R. На скорость реакции не
должны влиять пространственные факторы. Энтропийный эффект должен
соответствовать мономолекулярности реакции. Изотоп 18О не должен пере-
ходить в молекулу эфира до ее гидролиза.
959
Глава XV
К числу групп R, способствующих реализации этого механизма, относятся
третичные алкильные группы. По приведенным в табл. 208 данным Скрабала
(первый ряд таблицы) можно сделать предварительный вывод, что этот меха-
низм иногда проявляется и при кислотном гидролизе. Скорость гидролиза
77?/9т-бутилацетата в кислой среде значительно больше, чем можно было
ожидать на основании скоростей гидролиза метил-, этил- и изопропилацета-
тов. Действительно, скорости реакции гидролиза этого ряда проходят через
минимум, сильно напоминающий минимум на кривой скоростей гидролиза
соответствующих алкилгалогенидов; последнее послужило одним из доказа-
тельств двойственности механизма алифатического замещения (гл. VII,
разд. 2, а). Данные Скрабала о скоростях реакции становятся понятными при
допущении, что гидролиз метил-, этил- и изопропилацетатов протекает в соот-
ветствии с механизмом Aaq2, причем влияние пространственных затруднений
в гомологическом ряду усиливается, однако в случае тнрет-бутилацетата
скорость реакции определяется скорее механизмом Aal1, что обусловлено
сравнительно большой устойчивостью трет-бутильной группы в виде иона
карбония.
Этот минимум на кривой зависимости скорости от природы R в развет-
вляющемся гомологическом ряду СН3 > С2Н5 > изо-С3Н7 | < mpem-CJIg
для гидролиза алкилацетатов в разбавленных водных растворах соляной
кислоты отделяет механизм Аас2, реализующийся в левой части приведен-
ного ряда, от механизма АА(Д, реализующегося в правой его части. Другой
минимум, проявляющийся на один член ряда раньше, чем в предыдущем слу-
чае, получен Лейстеном [68] для гидролиза алкилбензоатов в концентриро-
ванной серной кислоте: СН3> С2Н5 | < нзо-С3Н7 < mpem-C4H9. Этот
минимум определяет переход от одного мономолекулярного механизма к дру-
гому: согласно Лейстену, от механизма Аас1 к механизму Aal1 (в указан-
ных выше условиях). Лейстен показал, что кинетический эффект, вызванный
полярными заместителями, такими, как н-нитрогруппа в бензольном кольце,
имеет противоположное направление по разные стороны минимума в най-
денном ряду, точно так же как эффект метильного заместителя в алкильной
части молекулы.
Хьюз, Мастерман и автор этой книги [69] получили некоторые подтвержде-
ния механизма Aal1 в случае реакций этерификации вторичных спиртов.
Они установили, что при этерификации оптически активного 2-и-октилового
спирта в избытке уксусной кислоты и в присутствии серной кислоты как
катализатора образуется сильно рацемизированный сложный эфир.
С помощью изотопа 18О Бантон и Конашевич [32] установили, что трифенил-
метиловый эфир бензойной кислоты гидролизуется в среде водного диоксана
под действием кислотного катализатора с расщеплением связи алкил —
кислород. Они установили также, что эта реакция идет по аналогии с ката-
лизируемыми кислотами реакциями метанолиза, в которых одним из продук-
тов является трифенилметилметиловый эфир:
СН3СООС(С6Н5)3 + Н2О* --> СН3СООН+НОС(СвН5)з
Н"1"
СН3ОСОС(С6Н5)3 + СН3ОН ---> СНзСООН+СН3ОС(С6Н5)3
Н"1"
Несмотря на неполноту кинетических данных для этих реакций, не может
быть каких-либо серьезных сомнений в том, что они проходят по механиз-
му Aal1.
Бантон и сотр. [70] на примере оптически активного третичного алкилацета-
та применили стереохимический критерий для выяснения того, является ли
960
Реакции карбоксильной и фосфатной групп
кислотный гидролиз с расщеплением связи алкил — кислород мономолеку-
лярной или бимолекулярной реакцией. Оптически активный метилэтилизо-
гексилкарбинилацетат подвергался гидролизу в 70%-ном водном растворе
циоксана (в качестве катализатора служила соляная кислота). Образовав-
щийся метилэтилизогексиловый спирт был преимущественно рацемическим
и содержал до 10% оптически инвертированного спирта
(активный) СН3СООС(СНз)(С2Н5)(С6Н1з-изо) ->
н+
CH3COOH + HOC(CH3)(C2H5)(C6H13-uao) (D, L)
Показано, что, хотя сам спирт подвергается рацемизации в водном диоксане
в присутствии кислотного катализатора, эта реакция идет настолько медленно,
что может явиться причиной образования лишь незначительной части раце-
мизированного спирта, получающегося в процессе гидролиза сложного эфира.
На этом основании был сделан вывод, что гидролиз третичных алкилацетатов
водными кислотами протекает по механизму Aal1-
Несколько примеров этого третьего кислотного механизма Aal1 встречается
в исследованиях кинетики кислотного гидролиза алкил- и арилацетатов
в среде водной серной кислоты, проведенных Ятсом и Макклелландом [48].
Они интерпретируют этот механизм следующим образом.
Механизм Aal1, как и другие кислотные механизмы, начинается с равновес-
ного присоединения протона. Эту предварительную стадию можно описать
количественно с помощью кислотной функции — Н$ или ее приближения
—тН0, где т = 0,62. Вторую стадию, являющуюся лимитирующей скорость,
можно «отделить» с помощью функций log/cj—(—Hg) или log kt тН0,
которые определяют свободную энергию активации. Эта стадия является моно-
молекулярным гетеролизом, и ее скорость уменьшается с увеличением
водности среды, так как эта стадия проходит с высвобождением сольватной
воды, и, следовательно, будет легче идти в среде с низкой активностью воды.
На этой стадии сложноэфирный ион оксония превращается в чистый ион
карбония, который гидратируется намного меньше. Состоянием, непосред-
ственно ответственным за скорость реакции, является, конечно, не сам карбо-
ниевый ион, а переходное состояние, через которое идет образование иона
карбония. Но даже этот переходный комплекс должен гидратироваться
существенно меньше, чем исходный сложноэфирный оксониевый ион. Отсю-
да следует, что, поскольку с увеличением концентрации кислоты активность
воды уменьшается, величина log + + тН0 должна сильно возрастать. Таким
образом, природа переходного комплекса, ведущего к образованию алкилий-
иона при механизме Aal1, обусловливает довольно сильно выраженную
отрицательную зависимость log kx + тН0 от log ан2о! по-видимому, угловой
коэффициент этой зависимости больше, чем градиент, равный —0,2, характе-
ризующий переходное состояние, ведущее к образованию ацилий-иона при
механизме Аас1.
Если построить график зависимости log kt от log аН2о для вторичных
алифатических сложных эфиров, например изопропилового и втор-бутило-
вого эфиров уксусной кислоты, то получатся линейные кривые с изломом
такой же формы, какие были получены для метилового и первичных алифати-
ческих сложных эфиров (рис. 54). Первичный арилалкиловый эфир — бен-
зилацетат — дает другую такую же кривую, однако исследование самой
реакции при высокой кислотности затруднено вследствие протекания других
реакций. В левой части таких линейных зависимостей с изломом для всех
соединений наблюдается одинаковый наклон +2,0 ± 0,1, характерный для
механизма ААц2. Такой наклон сохраняется до значения концентрации Сер-
61-0772
961
Глава X V
ной кислоты, равной 70 вес. % в случае вторичных алкилацетатов и 60 вес. %
для бензилацетата. В правой части указанных зависимостей наблюдается
отрицательный наклон кривых, равный для вторичных ацетатов —0,6.
Кривую для бензилацетата можно достаточно далеко продолжить в область
высокой концентрации серной кислоты, этим можно только показать, что
она действительно имеет существенный отрицательный наклон, но не изме-
рить его количественно. Сравнительно большие отрицательные наклоны
кривых, о которых идет речь, характеризуют механизм Aal1. Получить
удовлетворительные результаты для mpem-бутилацетата в данной среде
Рис. 55. Зависимость скорости кислотного гидролиза первичных, вторичных и третичных
алкил- (1—3), бензил- (4) и фенилацетатов (5) от кислотности среды (водная
серная кислота) при различных механизмах [48] (Ац в мин-1 при 25 °C).
невозможно, но данные, полученные по его гидролизу в водной соляной
кислоте, указывают на возможность сильно выраженной отрицательной
зависимости скорости от активности воды. Такая зависимость наблюдается
даже в очень сильно разбавленных растворах кислоты, хотя в этом случае
экспериментальные измерения затруднены в связи с очень слабым изменени-
ем активности воды при изменении кислотности разбавленных растворов.
Механизм ААс2, вероятно, почти не встречается при гидролизе третичных
алкиловых эфиров, в случае которых уже в области малых концентраций
кислоты гидролиз происходит по механизму AAl1.
Из приведенных данных следует качественная характеристика механизма
Aal1: с увеличением концентрации кислоты скорость реакции увеличивает-
ся с постепенно возрастающим градиентом. В случае mpem-бутилацетата это
происходит начиная с неопределенно низких концентраций кислоты. При
кислотном гидролизе вторичных алкилацетатов и бензилацетатов механизм
АдгД будет преобладать над механизмом AAl2 начиная с некоторого зна-
чения концентрации кислоты. До того как произойдет смена механизмов,
реакция по механизму Aal2 достигнет максимума скорости; затем, до того
как скорость снизится до нуля, реакция начнет протекать по механизму AAl1,
и скорость реакции снова начнет очень быстро увеличиваться.
962
Реакции карбоксильной и фосфатной групп
На рис. 55 приведена упрощенная картина зависимости скорости кислотного
гидролиза различных сложных эфиров от кислотности среды при различных
механизмах реакции (по данным Ятса и Макклелланда). Почти совпадающие
для двух вторичных ацетатов кривые заменены одной. То же сделано для
двух алифатических первичных сложных эфиров. Кривая для метилацетата
на рисунке не приведена вовсе: она должна находиться рядом с кривой
для алифатических первичных эфиров, но несколько выше (как это можно
представить себе из рис. 54). Одна кривая приведена также для фенилацета-
тов и замещенных фенилацетатов
Минимум на кривой скорости реакции, о котором уже говорилось выше, для
случая разветвляющихся гомологических эфиров уксусной кислоты разде-
ляет приведенную на рис. 55 диаграмму или, точнее, более полную диаграм-
му, на основе которой построена данная упрощенная диаграмма. Если про-
вести вертикальную прямую в любом месте области концентрации серной
кислоты от 0 до 70%, то получим ряд Скрабала
(Аас2)
СН3 > С2Н5 > ИЗо-С3Н7
mpem-C4Hg
(Аас1)
Если такую линию провести в области концентрации кислоты от 90 до 100%,
то получим минимум Лейстена
(Адс1)
СН3>С2Н5
< U30-C3H7 < трет-С4Н9
(Aal1)
2. Образование и гидролиз амидов карбоновых кислот
а. Обратимое образование амидов из сложных эфиров. В результате
реакций эфиров с аммиаком образуются амиды
R'COOR-|-NH3 R'CONH2 + ROH
Эта реакция широко применяется для получения амидов, однако следует
учитывать, что она, как установил Лотар Мейер [71], обратима. При полу-
чении амидов этим способом следует поддерживать низкую температуру
для того, чтобы амид мог выкристаллизоваться, что, естественно, затрудняет
реакцию в обратном направлении. Кроме того, поскольку образование ами-
дов и эфиров протекает с выделением тепла, высокая температура может
способствовать алкоголизу амида.
Беттс и Гаммет [72] изучали механизм этой реакции на примере взаимодей-
ствия метилового эфира фенилуксусной кислоты с аммиаком в метиловом
спирте; они изучили и два других случая. При этом было показано, что эту
реакцию лишь грубо приближенно можно считать реакцией второго порядка;
в частности, по аммиаку порядок реакции лежит между 1 и 1,5. Они интерпре-
тировали это следующим образом: реакция присоединения аммиака к эфиру
имеет порядок, равный 1, а получаемое сверх первого порядка превышение
вызвано влиянием конкурирущего присоединения амид-иона, образующегося
на предравновесной стадии в результате реакции двух молекул аммиака
2NH3 NHJ + NH^
в количестве, пропорциональном корню квадратному из концентрации аммиа-
ка. Эта идея специфического основного катализа, предполагающая, что
вторая молекула аммиака не входит в состав переходного состояния, вызва-
ла сомнение. Баннет и Дэвис [73] показали, что взаимодействие этилформи-
ата с н-бутиламином с образованием н-бутилформамида в растворе этилового
спирта является реакцией с общим основным катализом, поэтому любое
61* 963
Глава, X V
катализирущее основание должно принимать участие в образовании пере-
ходного состояния; таким образом, основание может катализировать и свою
собственную реакцию. При образовании указанного амида выражение для
скорости не содержало члена, соответствовавшего некаталитической реак-
ции и имевшего порядок по н-бутиламину, равный единице. Выражение для
скорости содержало член с порядком реакции по н-бутиламину, равным 1,5.
Баннет и Дэвис объяснили это каталитическим действием этилат-ионов, обра-
зующихся на предшествующей равновесной стадии
h-C4H9NH2 + C2H5OH h-C4H9NH+ + C2H5O-
в концентрации, пропорциональной корню квадратному из концентрации
амина. Другой член соответствовал порядку реакции по амину, равному 2,0,
что означает, что реакция катализируется самой молекулой амина.
Беттс и Гаммет предположили, что некаталитическая реакция протекает
по механизму Вдс2: начальное бимолекулярное взаимодействие между ами-
ном и сложным эфиром является медленной стадией. Реакцию можно пред-
ставить либо как обратимое присоединение по карбонильной группе, либо
как нуклеофильное замещение в алифатическом ряду, как показано ниже
для реакции с молекулой аммиака
О о-
|| Медленно + | Быстро
NH3 + C—OR ,--------> H3N —С — OR ,
I Быстро ] Медленно
R' R'
О
+ II —
H3N — C + OR
I
R'
(Вас2)
О
11 Медленно
NHa-f-C — OR ; •— ’>
R'
Смысл такого двойственного
H3N
-I О
6- + II _
OR f----------» H3N —C + OR
Медленно |
J R'
(Bac2)
рассмотрения состоит в том, чтобы показать,
что распределение электронов в промежуточном реакционном комплексе,
будь то молекула или переходное состояние, является промежуточным между
двумя приведенными выше. По механизму общего основного катализа Бан-
кета и Дэвиса последняя стадия — отщепление молекулы спирта от ком-
плекса присоединения — рассматривается как медленная стадия, которой
сопутствует основной катализ.
О ОН
|| Быстро |
R"NH2+C — OR > R"NH — С — OR
I I
R' R'
OH ... В
I
R"NH —C —OR
I
R'
-0 H —B+
Быстро | |
> R"NH — C — OR
I
R'
0
Медленно 11
< » R"NH — C
R'
H
I
OR + B
Влияние полярных заместителей на скорость образования амида качественно
соответствует ожидаемому для некаталитической реакции: как известно,
электроноакцепторные заместители ускоряют, а электронодонорные замести-
тели тормозят реакцию. Это будет показано ниже. Полярные эффекты в ре-
акциях общего основного катализа качественно носят двойственный характер
и поэтому непосредственно не согласуются с известными эффектами замести-
телей. Как установил Ганс Мейер [74], сложные эфиры таких сильных кис-
лот, как трихлоруксусная, образуют амиды быстро, в то время как сложные
964
Реакции карбоксильной и фосфатной групп
эфиры таких слабых кислот, как триметилуксусная, — медленно. Гор-
вин [75] полуколичественно показал, что влияние заместителей на скорость
реакций этиловых эфиров бензойной кислоты соответствует следующему
ряду:
n-NO2 > 3t-N02 > Н > п-СН3 > п-СН3О > n-NH2 > л-О"
Гордон, Миллер и Дей [76] изучили скорость реакции различных сложных
эфиров с аммиаком. Они нашли определенную последовательность в измене-
нии величин скоростей и установили некоторые ряды; так, например, ско-
рость гидролиза алкилацетатов CH3COOR уменьшается в ряду
R = С3Н5 > СН2 = СН > СН3 > С2Н5 > U30-C3H7 > znpem-CaHg,
а метиловых эфиров различных кислот R'COOCH3 — в следующей последо-
вательности:
R' = Н > СН3 > С2Н5 > изо-СзН.7 > трепг-С^Нд
Эти результаты отображают, вероятно, совместное влияние полярного и про-
странственного эффектов. Пространственные эффекты, конечно, должны про-
являться при механизме Вдс.2. К сожалению, не известно, в какой степени
реакции, для которых изучался эффект заместителей, будут некаталитически-
ми или каталитическими. Единственным известным случаем влияния заме-
стителей, которое определенно связано с кинетической формой, является
влияние пара-заместителей в реакциях Беттса и Гаммета, т. е. при взаимо-
действии метилфенилацетата и его замещенных в кольце производных с амми-
аком в растворе метилового спирта. В ряду
n-NO2 > n-Cl > Н
соотношение скоростей составляет 3,2 : 1,8 : 1 для некаталитической реак-
ции с первым порядком по аммиаку и 5,9 : 2,1 : 1 для каталитической реак-
ции с порядком 1,5 по аммиаку. Такое сходство во влиянии заместителей
легче понять, если принять механизм, подобный механизму Беттса и Гам-
мета, в котором лимитирующей является первая стадия каталитической или
некаталитической реакции, а не механизм, в котором лимитирующей будет
последняя стадия с основным катализом. Эти вопросы требуют дальнейшего
выяснения.
б. Основной и кислотный гидролиз амидов. Райд [77] изучал гидролиз
амидов водными растворами щелочей и водными растворами сильных кислот.
Он показал, что гидролиз в щелочной среде протекает как реакция второго
порядка, скорость которой пропорциональна произведению [Амид][ОН_],
что ясно указывает на механизм типа ВАС2. Способы изображения реакции
и причины двойственного ее изображения остаются такими же, как и в дру-
гих случаях механизма ВАС2.
О
|| Медленно
ОН +С —NH2 ; >
I Быстро
R'
о- О
| Быстро |[ _
но—С—nh2 ; > НО-C+NH2
| Медленно |
R' R'
О
|| Медленно
но-+с-nh2 ; >
R'
О
6- II 6-
но с nh2
I
R'
О
-------> II _
«------- НО —C4-NH2
Медленно ।
R'
(Вдс2)
(Вас2)
(R'COOH + NH2 —-------> R'COO- + NH3)
Быстро
965
Глава X F
Бендер [10] установил, что промежуточное состояние в реакции основного
гидролиза амида соответствует молекуле, а не переходному состоянию.
Доказательством служит то, что амиды при щелочном гидролизе в воде,
содержащей 18О, связывают некоторую часть этого изотопа еще до заверше-
ния гидролиза.
И в данном случае можно ожидать, что электроноакцепторные группы долж-
ны ускорять, а электронодонорные — тормозить реакцию, которая, кроме
того, должна быть чувствительной к пространственным затруднениям.
Райд исследовал влияние ароматических заместителей на скорость гидро-
лиза замещенных бензамидов в водном растворе гидроокиси бария. Некото-
рые данные по константам скоростей реакций приведены в табл. 210.
Из этих данных видно, что мета- и пара-электроноакцепторные заместители
действительно ускоряют, а электронодонорные заместители тормозят реак-
цию. Кроме того, заместители в орто-положении независимо от их полярно-
сти тормозят реакцию, что согласуется с предположением об их сильном
пространственном влиянии.
Таблица 210
Сравнительные скорости гидролиза замещенных бензамидов щелочами
в воде при 100 °C по реакции второго порядка (по данным Райда)
Положение заместителя 0“ nh2 ОСНз СН3 н I Вг no2
пара 0,20 0,49 0,65 1 1,69 1,91
мета 0,19 0,93 0,83 1 2,60 2,97 5,60
орто 0,064 0,054 1 0,138 0,254
При кислотном гидролизе амидов в не очень концентрированных водных рас-
творах сильных кислот скорость гидролиза, как показал Райд, пропорцио-
нальна произведению [Амид] [Н+]. Поскольку в этих условиях нельзя ожи-
дать, что сопряженная кислота амида будет подвергаться гидролизу до
иона ацилия, то предполагается, что реакция происходит по механизму типа
Адс2. Этот механизм может быть представлен либо моделью обратимой реак-
ции присоединения к карбонильной группе, либо моделью нуклеофильного
замещения в том же смысле, что и в приведенных ранее примерах
О
11 + Медленно
H2O + C-NH3 , »
| Быстро
R'
О
11 + Медленно 6+
Н 2С — N Hg . I Н2О
1 R'
(R'COOH+-lNH3
о-
| + Быстро
Н2О+ —С —NH3 ~
| Медленно
R'
6 +
NH3
------->
Быстро
Медленно
О
II
h2o+-c+nh3
I
R'
О
+ II
H2O-C + NH3
I
R'
R'COOR + NHJ)
(Аас2)
(Аас2)
Как указывалось в разд. 1,е, при механизме Адс2 проявляется слабое поляр-
ное влияние заместителей, но при соответствующем их расположении они
должны оказывать сильное пространственное влияние. Райд изучал влияние
ароматических заместителей на скорость гидролиза замещенных бензамидов
в 0,5 н. растворе соляной кислоты. В табл. 211 приведены полученные им
966
Реакции карбоксильной и фосфатной групп
Таблица 211
Относительные скорости гидролиза замещенных бензамидов 0,5 и. соляной кислотой
в воде при 100 °C (по данным Райда)
Положение заместителя nh2 он СНз н I Вг С1 no2
пара 0,85 0,84 1 0,86 0,91 1,13
мета 0,81 0,87 1 0,94 0,94
орто 0,085 0,208 0,105 1 0,051 0,153 0,026
сравнительные данные. Из этих данных видно, что даже сильно полярные
заместители в мета- и пара-положении оказывают незначительное влияние
на скорость реакции, а заместители в орто-положении независимо от их
полярности заметно тормозят реакцию.
Кинетика катализируемого кислотами гидролиза амидов в воде изучалась
в другой связи. Поскольку скорость кислотного гидролиза амидов невелика,
ее удобно изучать, применяя концентрированные кислоты. Найдено [78], что
по мере увеличения концентрации сильной кислоты скорость реакции про-
ходит через максимум. В случае гидролиза формамида водным раствором
соляной кислоты максимум скорости обнаруживается в 6 н. кислоте. Однако
это значение концентрации несколько отличается для водного раствора сер-
ной кислоты и не остается одним и тем же в случае различных амидов.
Такая картина является обычной для механизма Аас2 в концентрированных
водных растворах кислот. Об этом уже говорилось при описании кислотного
гидролиза сложных эфиров. Картина может стать более сложной в случае
гидролиза амидов, которые склонны к протонированию; таким образом,
протонирование может достигнуть насыщения к тому времени, как актив-
ность начнет уменьшаться при увеличении концентрации сильной кислоты.
Этот вопрос был исследован с количественной стороны Ятсом и сотр. [79].
Сначала определялась кислотная функция На для амидов для того, чтобы
определить степень их предравновесного протонирования. Это позволяло
исследовать зависимость второй определяющей скорость стадии от концен-
трации воды. Положительный угловой коэффициент зависимости logfcj-[-.Яд
от log ад2о в этом случае больше, чем при механизме гидролиза сложных
эфиров Аас2, и соответствует «порядку» реакции по воде, равному скорее 3,
чем 2. По-видимому, переходное состояние содержит не только молекулу
воды, которая является нуклеофильным замещающим агентом у атома угле-
рода, и молекулу воды, которая позднее отдает свой протон, но также и дру-
гую молекулу воды, которая, возможно, способствует отщеплению амино-
группы в виде иона аммония в кислотном растворе.
Имеется доказательство, что к амидам применим и другой из трех известных
кислотных механизмов гидролиза производных карбоновых кислот. Лей-
си [80] показал, что N-mpem-алкилзамещенные амиды в кипящей 30%-ной
или холодной 98%-ной серной кислоте гидролизуются с образованием неза-
мещенных амидов и затем кислот с отщеплением mpem-алкильной группы
в виде третичного спирта или соответствующего ему олефина. Алкильными
группами в этом примере служили mpem-бутильная и «mpem-октильная»
(1,1,3,3-тетраметилбутильная). Были исследованы амиды муравьиной, уксус-
ной и хлоруксусной кислот, а также амиды бензойной кислоты и ее замещен-
ных, содержащих метокси-, хлор- и нитрогруппы в кольце. В ряду замещенных
967
Глава X V
бензамидов установлена следующая последовательность скоростей реакции:
n-CH3O> Н> n-Cl> n-NO2.
Продукты, полученные Лейси, образуются вследствие расщепления связи
алкил — азот, и даже несмотря на отсутствие кинетических исследований,
можно со всей определенностью считать, что гидролиз амидов протекает по
механизму Aal1. Кинетический эффект заместителей в кольце бензамида со-
гласуется с такой интерпретацией. В работе Райда (см. табл. 211) было пока-
зано, что полярные эффекты заместителей в кольце сказываются на гидролизе
амидов с расщеплением связи ацил — азот очень слабо. Это означает, что
влияние полярности на предравновесное протонирование почти сбаланси-
ровано его влиянием на нуклеофильную атаку ацильной группы. Если
место нуклеофильной атаки отодвигается по направлению к алкильной груп-
пе, как того требует механизм Aal1, то следует ожидать, что полярное влия-
ние на скорость гидролиза будет определяться влиянием на предшествую-
щую равновесную стадию протонизации. В результате мы получим такой
полярный эффект, какой наблюдал Лейси.
3. Реакции сложных эфиров с карбанионами
В принципе имеются два важных аспекта этого вопроса. Они связаны с при-
соединением карбанионов к карбонильным соединениям: карбанион может
быть получен либо в результате отрыва протона от псевдокислоты, либо
с помощью металлалкила, например реактива Гриньяра. Присоединение
псевдокислот через их карбанионы к сложным эфирам широко исследова-
лось в течение большого периода времени, хотя и не с точки зрения кинетики
реакций. Эта реакция носит название конденсации Кляйзена. Она имеет
заслуженную историю, которая будет изложена в настоящем разделе. При-
соединение металлалкилов к сложным эфирам широко применяется в синте-
тической химии, но и здесь совершенно упущены вопросы механизма; поэто-
му детально этот механизм рассматривать бесполезно.
а. Конденсация Кляйзена. Эти реакции представляют собой конденса-
цию эфиров карбоновых кислот любого типа с эфирами, кетонами или нитри-
лами, содержащими водород в «-положении. В результате реакции, сопро-
вождающейся отщеплением молекулы спирта, образуются p-кетоэфир или
в случае эфира муравьиной кислоты — p-альдоэфир, кетон или нитрил.
Течению этих реакций способствуют металлический натрий, метилат пли
этилат натрия в твердом виде или в растворах в соответствующем спирте.
Применяются и такие более мощные конденсирующие агенты, как амид нат-
рия, мезитилмагнийбромид, трифенилметилнатрий.
Превращение этилацетата в ацетоуксусный эфир при помощи натрия, откры-
тое Гейтером в 1863 г., представляет собой первый пример кляйзеновской
конденсации. В результате начатого в 1887 г. Кляйзеном изучения химии
и механизма этого процесса были сделаны обобщающие выводы. Начиная
с 1894 г. Дикман изучал применимость этих реакций для замыкания циклов.
Такого рода конденсации известны как реакции Дикмана.
Ниже приведены некоторые примеры конденсации Кляйзена, включающие
и реакцию Дикмана. Как видно, все приведенные исходные соединения содер-
жат достаточное количество водородных атомов в «-положении и поэтому
становится возможным образование кето-енольных таутомеров. Правда,
это не является обязательным условием реакции, но способствует более лег-
кому осуществлению обсуждаемых процессов. Причины этого будут указаны
ниже. В приведенных реакциях кето-енольные соединения представлены для
единообразия в их кето-форме независимо от положения равновесия в про-
968
Реакции карбоксильной и фосфатной групп
тотропных системах этих соединений
СН3СООС2Н5 + СН3СООС2Н5 -» СН3СОСН2СООС2Н5 + С2Н5ОН [81]
С2Н5ООССООС2Н5 + СН3СООС2Н5 -» С2Н5ООССОСН2СООС2Н5+С2Н5ОН [82]
НСООС2Н5 + СН2СООС2Н5 —> НСОСНСООС2Н5 + С2Н5ОН [83]
С6Н5 С6Нд
СН3СН2СООС2Н5+СН2СООС2Н5 —э > СН3СН2СОСНСООС2Н5 + С2Н5ОН [84]
СН3 1 сн3
С6Н5СООС2Н5-|-СН3СОСНз —> - С6Н5СОСН2СОСН3 + С2Н5ОН [85]
СН2СО-СН2 НСООС2Н5+ 1 | -э СН2СН2СН2 НСОСНСО-СН2 | 1 +С3Н5ОН СН2СН2СН2 [86]
СН2СН2СООС2Н5 СН2СН2СО 1 -» 1 1 +с2н5он СН2СН2СООС2Н5 СН2—СНСООС2Н5 [87]
СН2СООСС2Н5 СН2СООС2Н5
1 + 1 ~*
С2Н5ООССН2 С2Н5ОСОСН2
СН2СОСНСООС2Н5
I I +2С2Н5ОН [88]
С2Н5ООССН-СОСН2
СООС2Н5 СН2СООС2Н5 СОСНСООС2Н5
| + ^СН2 -> | \сн2 +2С2Н5ОН] [89]
СООС2Н5 СН2СООС2Н5 СОСНСООС2Н5
Еще в 1901 г. Лэпуорт [90] установил, что свойствами водорода в a-положе-
нии, «активированного» смежной карбоксильной или другой группой подоб-
ного типа, должен обладать также водород в у-положении, если он связан
с такой группой а, р-этиленовой связью. Для иллюстрации этого явления
Лэпуорт показал, что этиловый эфир щавелевой кислоты в присутствии эти-
лата натрия конденсируется по реакции Кляйзена с этиловым эфиром крото-
новой кислоты в у-положение
С2Н5ООССООС2Н5+СН3СН=СНСООС2Н5 —> С2Н5ООССОСН2СН=СНСООС2Н5+С2Н5ОН
Первая серьезная попытка обосновать механизм кляйзеновской конденсации
была сделана Кляйзеном и Лоуменом [91] в 1887—1888 гг. Они предполагали,
что происходит присоединение алкоголятов натрия RON а к эфирам R'COOR,
так как выделили продукты присоединения типа R'C(OR)2ONa и считали,
что эти соединения образуются в качестве промежуточных при конденсации,
конечной стадией которой является отщепление двух молекул спирта
в результате взаимодействия продукта присоединения с молекулой, содержа-
щей два водородных атома в a-положении. Согласно такого рода представле-
ниям, образование ацетоуксусного эфира протекает следующим образом:
CH3COOC2H5 + C2HsONa -> CH3C(ONa)(OC2H5)2
CH3C(ONa)(OC2H5)2 + CH3COOC2H5 CH3C(ONa) = CHCOOC2H5-[-2C2H5OH
В соответствии с этой теорией активным конденсирующим агентом служит
не металлический натрий, а этилат натрия, который образуется сначала из
металлического натрия и следов спирта, а затем в возрастающих количест-
вах из натрия и спирта, возникающего при реакции. Как было установлено,
реакция вначале идет медленно и тем медленнее, чем меньше спирта содержит-
969
Глава XV
ся в исходном этилацетате; по мере протекания реакции скорость ее возра-
стает. Несомненно [92], что Кляйзен и Лоумен правильно установили при-
роду конденсирующего агента. Однако они, по-видимому, ошибочно счита-
ли, что им удалось выделить продукты присоединения, которые они считали
промежуточными соединениями, образующимися при конденсации [93].
Длительное время казалось несомненным, что Кляйзен и Лоумен были не-
правы, допуская возможность существования такого промежуточного продук-
та присоединения, поскольку он не может способствовать введению остатка
молекулы уксусноэтилового эфира в карбоксильную группу, так как в нем
нарушена ненасыщенность последней.
Для подтверждения предложенного механизма реакции Кляйзен приводил
тот факт, что в отличие от этиловых эфиров уксусной, пропионовой, к-мас-
ляной кислот этиловый эфир изомасляной кислоты не способен к аутокон-
денсации при действии натрия или этилата натрия. Даже этиловый эфир
щавелевой кислоты, который особенно легко участвует в конденсации Кляй-
зена, не реагирует с этиловым эфиром изомасляной кислоты. Кляйзен объяс-
нил это тем, что в случае этилового эфира изомасляной кислоты, содержащего
только один атом водорода в «-положении, не может произойти отщепления
двух молекул спирта в соответствии со второй стадией предложенной схемы
реакции. Более удовлетворительное объяснение механизма кляйзеновской
конденсации дал Дикман [94]. В 1900 г. он обнаружил обратимый характер
этой реакции и смещение равновесия алкильными заместителями по сосед-
ству с реакционным центром в сторону исходных веществ. Сам ацетоуксус
ный эфир в малой степени подвержен алкоголизу до этилацетата этилатом
натрия в спиртовой среде; в тех же условиях а-алкилпроизводные ацетоук-
сусного эфира настолько легко подвергаются алкоголизу, что резко снижа-
ется выход такого вещества, как этиловый эфир а-пропиопропионовой кисло-
ты, который может образоваться в результате конденсации, а, а-Диалкил-
производные ацетоуксусного эфира в тех же условиях практически полностью
подвергаются алкоголизу. Так, Дикман установил, что в условиях опыта
невозможность образования этилового эфира а-изобутироизомасляной кис-
лоты объясняется структурными термодинамическими эффектами, а не вы-
звана невозможностью осуществления какой-то стадии, входящей в истинный
механизм конденсации; в этом случае реакция легко протекает не в прямом,
а в обратном направлении. Такие представления о равновесии и о влиянии
строения на равновесные процессы хорошо согласуются с соответствующими
данными, относящимися к альдольной конденсации и реакции Михаэля,
полученными уже к тому времени и частично несколько позже (гл. XIII,
разд. 2,а и б). Однако изменения схемы Кляйзена, введенные Дикманом,
сводились лишь к тому, что этот процесс следует считать обратимым, состоя-
щим из большего числа стадий. Такой подход давал возможность не делать
предположения, что обязательным условием возникновения углерод-угле-
родной связи должно быть отщепление двух молекул спирта. Механизм Кляй-
зена, видоизмененный Дикманом, можно представить следующим образом:
CH3COOC2H5 + C2H5ONa у*~ CH3C(ONa)(OC2H5)2
CH3C(ONa)(OC2H5)2 + СН3СООС2Н5 у* CH3C(ONa)(OC2H5)CH2COOC2H5 + С2Н5ОН
CH3C(ONa)(OC2H5)CH2COOC2H5 у^ CH3COCH2COOC2H5 + C2H5ONa
Неф [95] предложил совершенно иную теорию этого процесса, которая,
подобна теории Кляйзена — Дикмана, также в определенном смысле пра-
вильна. Неф приписывал роль промежуточного продукта металлическому
970
Реакции карбоксильной и фосфатной групп
производному сложного эфира, а не продукту присоединения сложного
эфира к алкоголяту. В своих дальнейших построениях Неф был неправ,
поскольку он считал, что металлическое производное является ненасыщенной
молекулой, присоединяющей другой реагент. Неф * предложил следующую
схему образования ацетоуксусного эфира:
CH2 = C(ONa)(OC2H5) + HCH2COOC2H5 —> HCH2C(ONa)(OC2H5)CH2COOC2H5
CH3C(ONa)(OC2H5)CH2COOC2H5 -> CH3COCH2COOC2H5+C2H5ONa
Слабость этой теории состоит в том, что она неприменима для таких случаев,
как образование этилового эфира бензоилуксусной кислоты.
Взгляды Михаэля [96] были значительно более правильными. Он, подобно
Нефу, считал, что промежуточным продуктом реакции является металличе-
ское производное. Но Михаэль приписал ему правильную функцию, а имен-
но функцию присоединяющегося реагента, а присоединяющейся ненасыщен-
ной системой считал карбоксильную группу другой молекулы.
О Na ONa
II I I
CH3—С + СН2 = С—О —> СН3ССН2-С=О —> CH3COCH2COOC2H5 + C2H5ONa
Ас2н5 ос2н5 ос2н5 ic2H5
Эту схему можно применять во всех случаях конденсации Кляйзена. Однако
теория Михаэля была неполной, чем можно объяснить некоторые его мелкие
ошибки. Так, например, Михаэль возражал против утверждения Кляйзена,
что этилат натрия при образовании ацетоуксусного эфира является лучшим
конденсирующим агентом, чем металлический натрий. В связи с этим Миха-
эль неправильно утверждал, что с металлическим натрием получаются луч-
шие выходы, чем с этилатом натрия. Михаэль, как впоследствии и другие
исследователи, недооценил тот факт, что выходы сами по себе не служат
доказательством механизма, если не доказано, что они обусловлены скоро-
стью основной реакции, а не скоростью других реакций или термодинами-
ческими факторами. Вероятно, в действительности Михаэль отдавал пред-
почтение металлическому натрию, потому что понимал, что натрий лучше
вводить в этилацетат в виде свободного элемента, замещая водород на атом
натрия. Он не имел представления об обратимом отщеплении водородного иона
от этилацетата. Хотя Михаэль раньше других понял важность электрохими-
ческого подхода к органическим реакциям, все же он не пошел настолько дале-
ко, чтобы признать, что специфическая реакционная способность натриевого
соединения зависит от ионов, которые могут из него образоваться-
Недостаток теории Михаэля восполнила теория Лэпуорта [97], которая соот-
ветствует современным представлениям, хотя, конечно, и не была выражена
в понятиях электронной теории валентности. По теории Лэпуорта основание,
например алкоголят-ион, обратимо вытесняет протон из присоединяющегося
сложного эфира, кетона или нитрила; образующийся анион обратимо при-
соединяется к карбоксильному атому углерода, подобно тому как цианид-
ион присоединяется к карбонильному углероду при образовании циангидри-
на, а образующийся таким образом сложный анион обратимо отщепляет
алкоголят-ион. При образовании ацетоуксусного эфира устанавливаются
следующие равновесия:
С2Н5О-+СН3СООС2Н5 С2Н5ОН + СН2СООС2Н5 (1)
* Здесь, как часто и в других работах, Неф описывает активную ненасыщенную связь
как простую связь между атомами, каждый из которых имеет свободную валентность.
971
Глава X V
(Вдс2)
о о-
II , I
С2Н5ООССН24-С-ОС2Н5 С2Н5ООССН2—С-ОС2Н5
I I
СНд СНз
о-
С2Н5ООССН2С-ОС2Н5 С2Н5ООССН2СОСН3 + ОС2Н5
I
СНз
(2)
(3)
С2Н5О- + СН3СОСН2СООС2Н5 СН3СОСНСО2С2Н5 + С2Н5ОН (4)
Современная теория этого процесса отличается от приведенной только тем,
что она оперирует понятиями электронной теории валентности и признает
мезомерный характер участвующих в реакии карбанионов [98].
Приведенная выше равновесная реакция (4) не является постоянной частью
механизма конденсации. Однако эта реакция происходит в том случае (как
в приведенном примере), когда продукт конденсации содержит водород,
способный к ионизации: тогда реакция (4) входит в общее термодинамическое
равновесие, определяющее степень конденсации. Так, ацетоуксусный эфир,
образующийся в результате описанных последовательных процессов, в зна-
чительной степени превращается в анион по уравнению (4), что способ-
ствует смещению вправо равновесных реакций (1) — (3). Равновесная реак-
ция типа (4) не способствует протеканию прямой реакции в случае конденса-
ции этилового эфира изомасляной кислоты с другой молекулой того же эфи-
ра с образованием этилового эфира изобутироизомасляной кислоты.
Образование молекулы спирта в правой части уравнения реакции (1) замед-
ляет протекание равновесных реакций (1) — (3) в прямом направлении. Это,
безусловно, является одной из причин лучших выходов ацетоуксусного
эфира при применении металлического натрия, а не этилата натрия, или,
иными словами, в присутствии небольших количеств этилового спирта.
Существуют некоторые независимые доказательства реальности равновесной
реакции (1). Кеньон и Янг [99] показали, что оптически активные сложные
эфиры типа R'R"CHCOOC2H5 рацемизируются в присутствии этилата натрия.
Браун и Эберли [100] установили, что сложные эфиры, содержащие водород
в a-положении, претерпевают водородный обмен с «тяжелым спиртом»
C2H5OD, содержащим этилат натрия, и что скорость такого обмена харак-
теризует реакционную способность а-водородных атомов этих эфиров в кон-
денсациях Кляйзена.
Уравнения (2) и (3), как показано на модели присоединения к карбонильной
группе, справедливы в случае механизма Вас2- Этот же механизм можно
представить моделью нуклеофильного замещения. Тогда равновесия (2)
и (3) можно заменить одним, проходящим через переходное состояние,
идентичное описанному ранее анионному комплексу присоединения, за
исключением лишь распределения заряда.
° г ° -|
II . в- II в- ,
С2Н5ООССН2+С—ОС2Н5 С2Н5ООССН2 ... С ... ОС2Н5
J:h3 l сн3
о
С2Н5ООССН2-С + С2Н5О-
I
СНз
(Вдс2)
972
Реакции карбоксильной и фосфатной групп
Теоретически можно предположить, что наиболее уплотненный анионный
комплекс действительно обладает промежуточным распределением заряда.
Еще не установлено, обладает ли этот реакционный комплекс достаточной
продолжительностью жизни, чтобы считать его устойчивой молекулой, а не
переходным состоянием, хотя по аналогии можно предположить, что он,
вероятно, является истинной молекулой.
Тот факт, что более сильные, чем этилат-ионы, основания способствуют
конденсации Кляйзена, сначала был установлен эмпирически. Сам Кляйзен
[101] применял амид натрия и показал, что во многих случаях это соединение
является лучшим реагентом, чем натрий или этилат натрия.
Шпильман и Шмидт [102] впервые применили в качестве конденсирующего
агента мезитилмагнийбромид (2,4,6-триметилфенилмагнийбромид) и уста-
новили, что, применяя это соединение, можно провести ряд кляйзеновских
конденсаций, которые ранее не удалось осуществить, включая и самоконден-
сацию этилового эфира изомасляной кислоты. Можно было бы ожидать,
что такой реагент сам атакует карбоксильный атом углерода и это приведет
к реакции Гриньяра. Однако следует полагать, что пространственное влия-
ние метильных заместителей в орто-положениях препятствует этой реакции,
но не мешает взаимодействию мезитил-иона с пространственно более доступ-
ным псевдокислотным протоном.
Исходя из теоретических предпосылок, касающихся приведенных выше
равновесных реакций, Хаузер и Ренфроу [103] предложили использовать
в качестве конденсирующего агента трифенилметилнатрий. Хорошая кон-
денсирующая способность этого соединения обусловлена тем, что вместо
этилат-иона в равновесных реакциях типа (1) участвует трифенилметид-ион,
полностью сдвигающий равновесие вправо, а это ведет к тому, что после-
дующие реакции доходят до полного равновесия. Этот реагент обладает
еще одной особенностью: если продукт конденсации не содержит водород-
ного атома в a-положении, участвующего в равновесной реакции типа (4),
а содержит водородный атом в у-положении, то трифенилметид-ион легко
отщепляет у-протон (алкокси-ион может это сделать лишь в незначительной
степени), что приводит к установлению равновесия, являющегося равнове-
сием типа (4) и отличающегося от последнего лишь местом локализации
переноса протона; благодаря этому происходит сдвиг предшествующих
равновесных реакций вправо. Вероятно, оба эти эффекта и обеспечивают
образование этилового эфира а-изобутироизомасляной кислоты по методу
Хаузера и Ренфроу с лучшими выходами, чем по способу Шпильмана
(СбН5)зС~
(CH3)2CHCOOC2H5 + (CH3)2CHCOOC2H5^ZZZZ^ (СН3)2СНСОС(СН3)2СООС2Н5 + С2Н5ОН
и Шмидта. Конденсация этилового эфира бензойной кислоты с этиловым
эфиром изомасляной кислоты, приводящая к образованию этилового эфира
а-бензоилизомасляной кислоты, в которой проявляется лишь первый фактор
и которую ранее не удалось осуществить, была проведена Хаузером и Рен-
фроу с помощью предложенного ими метода.
(СбНб)зС-
С6Н5СООС2Н5 + (СН3)2СНСООС2Н5 7-- -»
С6Н5СОС(СН3)2СООС2Н5 + С2Н5ОН
Если быстро обработать смесь этилового эфира бензойной кислоты и этило-
вого эфира изомасляной кислоты трифенилметилнатрием, то основным про-
дуктом конденсации будет этиловый эфир а-бензоилизомасляной кислоты;
973
Глава X V
при продолжительной обработке последний продукт исчезает, этиловый эфир
бензойной кислоты регенерируется и основным продуктом конденсации
становится этиловый эфир а-изобутироизомасляной кислоты. Этим можно
объяснить вторую из указанных особенностей трифенилметид-иона как
конденсирующего агента: при длительном воздействии, вероятно, успевает
пройти медленный процесс переноса у-протона от образующегося этилового
эфира а-изобутироизомасляной кислоты.
4. Гидролиз эфиров фосфорной кислоты
Вероятно, покажется довольно нелогичным резкий переход от рассмотрения
механизмов реакций эфиров моноосновных карбоновых кислот к эфирам
трехосновной фосфорной кислоты; однако вследствие биологической важно-
сти органических фосфатов изучение эфиров фосфорной кислоты стало про-
водиться, минуя ряд промежуточных, логически необходимых стадий иссле-
дования менее сложных эфиров. Можно ожидать, что впоследствии будут
подробно исследованы все механизмы реакций эфиров фосфорной кислоты,
то же, что известно в настоящее время, далеко не полно описывает эти реакции.
Фосфорная кислота может образовывать моно-, ди- и триалкиловые эфиры,
из которых два первых могут существовать в виде сопряженных оснований
и все три могут образовывать сопряженные кислоты. Таким образом, эфиры
фосфорной кислоты дают восемь субстратов — и это, если не считать сопря-
женные основания (или кислоты), образующиеся при отщеплении (или при-
соединении) второго протона. Для идентификации механизма, как минимум,
необходимо выяснить три вопроса: 1) что является собственно субстратом
и атакующим нуклеофилом, 2) какая связь разрывается в субстрате
и 3) какова молекулярность процесса? После этого требуется выяснить дру-
гие детали процесса. В данном случае два типа разрыва связи, а именно
алкил — кислород и ацил — кислород, соответствуют разрыву связей угле-
род — кислород и фосфор — кислород.
а. Механизм гидролиза моноэфиров фосфорной кислоты. Общий вид
зависимости скорости гидролиза простых моноалкиловых эфиров фосфорной
кислоты от pH известен уже давно [104]. Она резко отличается от соответ-
ствующей зависимости для эфиров карбоновых кислот. В щелочном растворе
вместо того, чтобы быть быстрым, гидролиз алкилфосфатов происходит очень
медленно; скорость повышается при уменьшении pH в сторону нейтральной
точки и далее примерно до pH 4; около этого значения скорость проходит
через максимум. При дальнейшем повышении кислотности скорость умень-
шается до минимального значения при pH около 0,5, а затем в сильнокислых
растворах вновь возрастает. Бейли и Дезджоберт утверждали, что относи-
тельно высокая скорость в области pH 4 обусловлена реакцией моноаниона
с водой, и сейчас, когда имеются обширные кинетические данные по этой
и другим родственным реакциям, это предположение кажется более прием-
лемым для объяснения кинетической картины, чем кинетически эквивалент-
ное альтернативное предположение, когда реакция происходит между недис-
социированной молекулой и гидроксильными ионами.
Батчер и Уэстхеймер показали, что при гидролизе оптически активного
моно-(1-метокси-2-пропил)фосфата при pH 4 образуется 1-метоксипропа-
нол-2 с полностью сохраненной конфигурацией [105]. Эти данные не имеют
того значения, которое впоследствии им приписали другие авторы, а именно
они не доказывают, что происходит разрыв связи фосфор — кислород,
поскольку 1-метокси-2-пропильная группа содержит метоксизаместитель,
способствующий сохранению конфигурации (гл. VII, разд. 7,д). Однако
974
Реакции карбоксильной и фосфатной групп
то, что в этом случае происходит именно разрыв связи фосфор — кислород,
указанные авторы показали с помощью 18О.
Зависимость скорости от pH была определена Бантоном, Верноном и сотр.
[106] на примере водного гидролиза монометилфосфата (рис. 56). Из формы
этой зависимости следует, что гидролиз осуществляется тремя кинетически
различными путями, для каждого из которых
уравнение, определили константу скорости
и положение или положения разрывающих-
ся связей. Показано, что три кинетически
различимых механизма соответствуют по
крайней мере четырем индивидуальным ре-
акциям. В некоторых случаях оценивалась
молекулярность по воде, служившей так-
же растворителем.
Реакция, которая в интервале pH от 2 до
7 является практически единственной, со-
ответствует какому-то превращению моно-
аниона в воде или его взаимодействию с
водой. Пунктирной линией на рис. 56 обозна-
чен чистый вклад этой реакции в наблюдае-
мую зависимость скорости от pH, который
был рассчитан исходя из концентрации мо-
ноаниона, определенной из известной вели-
чины первой константы диссоциации мо-
ноэфира. С помощью изотопа 18О было по-
казано, что для этой реакции характерно
исключительно расщепление связи фосфор—
авторы вывели кинетическое
Рис. 56. Зависимость скорости
гидролиза монометилфо-
сфата в воде при 100,1 °C
от pH [107].
1 — экспериментальные дан-
ные; 2 —данные, рассчитан-
ные для реакции, протека-
ющей через моноанион.
кислород.
Молекулярность этой реакции по воде не известна. Однако из опытных
данных (разд. 4,6 и г) был сделан вывод, что относительно высокая скорость
гидролиза моноаниона монометилфосфата соответствует скорости кислород-
ного обмена между водой и моноанионом ортофосфорной кислоты, но не
соответствует скорости гидролиза аниона диметилфосфата, т. е. для реакции
необходимо, чтобы фосфорильный атом фосфора был связан как с группой
— ОН, так и с группой — О-. Именно такая структура необходима для
мономолекулярного отщепления аниона с образованием метилового спирта
и мономерного метафосфатного иона, который может сразу же гидратиро-
ваться и образовывать ортофосфат и параллельно полимеризоваться в ста-
бильные три- или другие полиметафосфаты. Триметафосфорная кислота
О О
СН3—Оу-Р-^--> СЙ3ОН + Р=О
H-J-0 0-
(В-АС1)
была обнаружена в реакционной смеси. Отсюда следует, что молекулярность
по воде равна нулю. Если предположить, что молекулярность реакции по
воде равна единице, то возможна другая схема. Уэстхеймер [105] предложил
в циклическое переходное состояние включить молекулу воды, тем самым
заменив четырехзвенное циклическое переходное состояние на шестизвен-
ное. Вернон [106] считает, что атака молекулы воды по атому фосфора могла
бы дополнительно способствовать отщеплению метоксигруппы. Вероятно,
вода может одновременно действовать в обоих этих направлениях. Все эти
975
Глава XV
варианты следует считать открытыми для дискуссии до тех пор, пока не
будет определено число молекул воды, участвующих в процессе.
При более кислых pH преобладающей формой гидролиза являются реакции
ионных сопряженных эфиру кислот. Сопряженная кислота образуется на
предравновесной стадии, об этом свидетельствует небольшая величина
положительного кинетического эффекта, вызываемого заменой воды в раство-
рителе на D2O. Следовательно, определяющей скорость стадией является
замещение в сопряженной кислоте. Общая скорость гидролиза вплоть до
8 н. концентрации хлорной кислоты лучше соответствует [Н+], чем 7i0. Это
показывает, что переходное состояние определяющей скорость стадии
настолько же сильно гидратировано, как и исходное состояние, содержащее
гидратированный ион молекулы моноэфира. Результаты гидролиза водой,
обогащенной изотопом 18О, показывают, что кислотный гидролиз в этой
области pH включает две конкурирующие реакции, имеющие сходную
кинетику: суммарная реакция на 65% происходит с разрывом связи угле-
род — кислород и на 35% — с разрывом связи фосфор — кислород. В срав-
нимой степени кислородный обмен происходит также и в самом моноэфире
до его гидролиза; он происходит по связи фосфор — кислород с отщеплением
неэтерифицированной карбоксильной группы [106].
Эти реакции вполне правдоподобно были представлены как бимолекулярное
нуклеофильное замещение молекулами воды у атома углерода, а не у четырех-
лигандпого фосфора, происходящее в одной или другой ионной форме кисло-
ты, сопряженной монометиловому эфиру. Гидролиз, ведущий к расщеплению
связи углерод — кислород, описывается схемой
Н2О + СН3ОНРО(ОН)2 —> Н2ОСНз+НОРО(ОН)2 (Aal2)
а гидролиз с расщеплением связи фосфор — кислород схемой
Н2О + РО(ОН)2ОНСНз Н2ОРО(ОН)2 + СН3ОН (Аде2)
Аналогично обмен кислорода, предшествующий гидролизу, был представлен
как бимолекулярное нуклеофильное замещение у фосфора в изомерной
ионной сопряженной кислоте
Н2б + РО(ОН)(ОН2)(ОСН3) -» Н2О*РО(ОН)(ОСН3) + Н2О
Все эти процессы бимолекулярного замещения водой соответствуют по край-
ней мере первому порядку по воде.
Следует ожидать, что бимолекулярное нуклеофильное замещение у атома
углерода в описанной только что форме, т. е. кислотный гидролиз по меха-
низму Aal2, будет проявляться в случае алкилфосфатов, содержащих группы
типа метильной или первичных алкильных. Для вторичных и особенно
третичных алкильных групп и даже для первичных алкильных групп,
содержащих электронодонорные a-заместители, гидролиз должен осуще-
ствляться по механизму Aal1. Этот ожидаемый механизм будет проиллю-
стрирован ниже.
При анализе зависимости скорости от pH был найден «нейтральный» меха-
низм, т. е. реакция гидролиза неионизованной молекулы моноэфира моле-
кулой воды. Этот механизм реализуется в области pH, промежуточной
между областью, соответствующей реакции аниона, и областью, соответ-
ствующей реакции двух сопряженных эфиру кислот. При таком механизме
наблюдается сильный положительный солевой эффект, как и следует ожидать
в случае реакции между двумя нейтральными частицами, которая проходит
976
Реакции карбоксильной и фосфатной групп
через полярное переходное состояние и в результате дает пару ионов. Иссле-
дование переноса 18О показывает, что в этой реакции в основном происходит
расщепление связи углерод — кислород. Нельзя исключить и возможность
протекания кинетически аналогичной реакции с расщеплением связи фос-
фор — кислород, однако идентификация такого механизма затруднена вслед-
ствие наложения реакций с участием аниона и сопряженных кислот.
Молекулярность по воде для «нейтральной» реакции до сих пор не определе-
на. Однако вполне резонно было предположено, что гидролиз с расщепле-
нием связи углерод — кислород является бимолекулярным нуклеофильным
замещением у атома углерода под действием молекулы воды
Н2О + СН3ОРО(ОН)2 —> Н2ОСН3+ОРО(ОН)2 (BAL2)
Такой механизм был выведен на основании сравнения скорости этой реакции
со скоростью «нейтрального» гидролиза по механизму BAL1, который будет
рассмотрен ниже.
Опять-таки следует ожидать, что механизм BAL2 не будет реализовываться
в случае «нейтрального» гидролиза моноалкилфосфатов, содержащих алкиль-
ные группы, отличные от метильной или первичных алкильных. Вероятно,
в случае вторичных и особенно третичных алкильных групп гидролиз моно-
фосфатов будет осуществляться по «нейтральному» механизму BAL1, который
ниже будет рассмотрен на примере замещенного первичного моноалкилфос-
фата, алкильная группа которого содержит сильно электронодонорный
а-заместитель.
Бантон и Вернон [108] изучили влияние электронодонорной алкильной груп-
пы на примере одного из имеющих важное биологическое значение фосфа-
тов сахаров. На кривой скорость — pH для гидролиза a-D-глюкопиранозил-
фосфата в воде наблюдается подъем от очень низких скоростей в щелочном
растворе до плато при pH около 5, при котором концентрация моноаниона
достигает максимального значения; однако затем вместо нового спада,
который должен был бы наблюдаться, если бы вследствие увеличения кислот-
ности среды функция гидролизующейся частицы перешла бы к неионизо-
ванной молекуле моноэфира, вновь наблюдается повышение скорости,
которое в области pH от 4 до 0,5, когда происходит постепенное увеличение
доли реакции с участием неионизованной молекулы, осуществляется по
линейному закону. Поэтому в данном случае реакция неионизованной моле-
кулы с водой (или в воде) вместо того, чтобы быть более медленной, чем
реакция аниона (что наблюдается, например, при гидролизе монометил-
фосфата), на самом деле происходит с гораздо большей скоростью. Наряду
с изменением природы субстрата от моноаниона до неионизованной молекулы
происходит и изменение типа расщепляемой связи. Согласно данным изотоп-
ного метода с использованием 18О, в моноанионе разрывается связь фосфор —
кислород, а в неионизованной молекуле — связь углерод — кислород.
Несмотря на то что молекулярность этих реакций по воде не установлена,
очевидно, можно полагать, что гидролиз и моноаниона a-D-глюкопиранозил-
фосфата, и моноаниона метилфосфата имеет одинаковый механизм ВАС 1.
Одинаково также и положение расщепляющейся связи (в обоих случаях
фосфор — кислород), а скорости имеют один и тот же порядок величины.
При гидролизе неионизованных пиранозильного и метилового эфиров также
расщепляется одна и та же связь (в обоих случаях углерод — кислород);
однако большое различие в скоростях очень трудно объяснить влиянием
строения при бимолекулярном механизме BAl2. Скорость гидролиза неиони-
зованного пиранозильного эфира в 105 раз больше скорости гидролиза
62-0772
977
Глава XV
неионизованного метилового эфира. Легко можно прийти к выводу, что-
здесь происходит изменение механизма от BAL2 к BAL1, что согласуется
с известным фактом, что гидролиз пиранозилгалогенидов происходит по-
механизму SN1 [108]. Естественно предположить, что влияние строения,
приводящее к увеличению скорости, связано с наличием эфирной группы,
которая сопрягается с разрывающейся связью точно так же, как в случае
гидролиза хлорметиловых эфиров, и образующийся карбониевый ион частич-
но является оксониевым ионом, т. е. мезомерным катионом псевдооснования,
что значительно увеличивает его стабильность. Таким образом, определяю-
щая скорость стадия гидролиза мономолекулярна, т. е. реализуется меха-
низм BAL1.
В сильнокислом растворе, например в 0,5—3,4 н. хлорной кислоте, начинает
проявляться механизм с участием сопряженной кислоты, константа скорости
этой реакции еще меньше. На основании приведенных выше рассуждений
о мономолекулярной природе реакции неионизованного пиранозильного
эфира можно заключить, что реакция сопряженной этому эфиру кислоты
должна быть мономолекулярной. Дополнительный протон должен сильно*
увеличивать скорость реакции, осуществляющейся по такому механизму,
так как образуется более легко отщепляемая группа. Были найдены спе-
циальные кинетические доказательства этого вывода. С увеличением кислот-
ности среды скорость возрастала приблизительно в соответствии с величиной
функции Гаммета h0, тогда как при гидролизе сопряженной кислоты моно-
метилфосфата скорость возрастает более в соответствии с величиной [Н+].
Следовательно, при гидролизе сопряженной кислоты пиранозильного эфира-
в переходное состояние входит меньшее число молекул воды, чем в переход-
ное состояние гидролиза сопряженной кислоты метилового эфира: при пере-
ходе от сопряженной кислоты метилового эфира к сопряженной кислоте-
пиранозильного эфира происходит изменение механизма от бимолекулярного-
к мономолекулярному [108]. Отсюда следует, что, в то время как сопряженная
кислота метильного соединения претерпевает в одном случае расщепление-
по связи углерод — кислород, а в другом — по связи фосфор — кислород,
в реакции сопряженной кислоты пиранозильного соединения происходит
только расщепление связи углерод — кислород [108]. Таким образом, все
данные свидетельствуют о том, что сопряженная кислота пиранозильного*
соединения должна реагировать по мономолекулярному механизму гидролиза,
типа Aal1.
б. Механизм гидролиза диэфиров фосфорной кислоты. Гидролиз дибен-
зилового эфира фосфорной кислоты изучался Уэстхеймером и сотр. [109]-
978
Реакции карбоксильной и фосфатной групп
Авторы не наблюдали реакции между анионом и водой, но обнаружили
медленное взаимодействие аниона с гидроксильным ионом. Легко протекала
реакция неионизованного кислого эфира с водой, которая преимущественно
проходила путем расщепления связи углерод — кислород. Кроме того,
происходила очень быстрая реакция, катализируемая кислотой. В этой
реакции, вероятно, участвуют сопряженная кислота и вода и происходит
расщепление связи углерод — кислород.
Бантон, Вернон и сотр. [110] нашли аналогичные реакции при изучении
кинетики и продуктов гидролиза диметилфоСфата. Авторы смогли достичь
верхнего предела скорости неизмеримо медленной реакции аниона с водой.
Они установили, что медленная реакция второго порядка между анионом
и гидроксильными ионами происходит с преимущественным расщеплением
связи углерод — кислород. Эта реакция, вероятно, соответствует бимолеку-
лярному нуклеофильному замещению у атома углерода
НО- + СН3ОРО(ОСН3)О--> СН3ОН + ~ОРО(ОСН3)О- (ВХц2)
Согласно данным Уэстхеймера и сотр. [109], эта реакция второго порядка
на 10% происходит путем расщепления связи фосфор — кислород, что
соответствует бимолекулярному нуклеофильному замещению у атома
фосфора
НО- + РО(ОСН3)2О- —> НОРО(ОСН3)О- + СН3О- (ВдС2)
Бантон и Вернон обнаружили, что гидролиз неионизованной молекулы эфира
происходит также по двум параллельным путям: суммарный процесс на 78%
идет с расщеплением связи углерод — кислород и на '22% с расщеплением
связи фосфор—кислород. Авторы рассматривали оба механизма как бимо-
лекулярное замещение, поскольку они получили кинетические доказатель-
ства, что даже в случае протонированного субстрата гидролиз является
преимущественно бимолекулярным.
Н2О + СН3ОРО(ОСН3)(ОН) —> Н2ОСН3 + ОРО(ОСН3)(ОН) (BAL2)
Н2О + РО(ОСН3)2(ОН) -> Н2ОРО(ОСН3)(ОН) + СН3О- (ВАс2)
Гидролиз ионной сопряженной субстрату кислоты также включает две
конкурирующие реакции: общий процесс на 89% происходит путем расщепле-
ния связи углерод — кислород и на И % путем расщепления связи фосфор —
кислород. В районе концентраций хлорной кислоты между 1 и 3 н. суммар-
ная скорость реакции повышается пропорционально [Н+], а не функции
Гаммета h0. Отсюда следует, что в переходное состояние входят все моле-
кулы воды, связанные с начальным состоянием, т. е. вода является реаген-
том, участвующим в медленной стадии процесса. По этой причине этим
реакциям приписывается бимолекулярный механизм.
Н2О + СН3ОНРО(ОСН3)(ОН) -» Н2ОСН3 + НОРО(ОСН3)(ОН) (Aal2)
Н2О + РО(ОН)(ОСН3)(ОНСН3) —> Н2ОРО(ОН)(ОСН3) + СН3ОН (Аас2)
В пределах порядка скорости этих реакций были такими же, как скорости
соответствующих реакций кислотного гидролиза монометилфосфата (ср.
табл. 212).
При гидролизе циклических фосфатов 1,2-гликолей как в кислой, так
и в щелочной средах наблюдался заметный кинетический эффект, вероятно,
обусловленный пространственным строением этих диэфиров фосфорной
62* 979
Глава X V
кислоты. Этот эффект впервые наблюдали Браун и Тодд [111] при изучении
деполимеризации рибонуклеиновой кислоты при ее гидролизе до нуклеоти-
дов. В немного упрощенном виде эту реакцию можно рассматривать как
переэтерификацию, при которой этерифицируется 2-оксигруппа остатка
рибозы и происходит разрыв внутримономерной сложноэфирной связи,
так что образуется мономерный диэфир, в дальнейшем гидролизующийся
до моноэфиров
Чтобы принять эту теорию, необходимо предположить, что циклический
диэфир гидролизуется гораздо быстрее, чем обычные ациклические эфиры.
Тодд и сотр. [112] смогли осуществить синтез таких циклических эфиров
и показали, что эти эфиры на самом деле способны быстро гидролизоваться.
Уэстхеймер и сотр. [109, ИЗ] затратили много усилий, чтобы определить
особенности структур, в которых проявляются эти кинетические эффекты.
Они изучили ряд простых модельных соединений, таких, как этиленфосфат,
и установили, что циклические фосфаты и фосфонаты, содержащие диэфир-
ную группировку — О — РО — О — в качестве части пятичленного цикла,
как в кислой, так и в щелочной среде гидролизуются в 105...108 раз быстрее,
чем аналогичные соединения с открытой цепью. Высокая реакционная спо-
собность, обусловленная циклическим строением эфиров, не ограничивается
лишь гидролитическим раскрытием цикла. В циклическом диэфире типа
этиленфосфата как раскрытие цикла, так и конкурирующий обмен кислорода
между неэтерифицированной гидроксильной группой и растворителем, при
котором, естественно, не происходит раскрытия цикла, идут с высокими
скоростями. Такое совпадение кинетических эффектов кольца в двух конку-
рирующих реакциях, одна из которых происходит с раскрытием этого коль-
ца, а другая — с его сохранением, проявляется как при щелочном, так и при
кислотном гидролизе циклических диэфиров, т. е. в реакции второго порядка
аниона диэфира с гидроксильным ионом и в реакции гидролиза сопряженной
980
Реакции карбоксильной и фосфатной групп
кислоты кислого эфира. Эти быстрые реакции происходят исключительно
путем разрыва связи фосфор — кислород. По сравнению с этими быстрыми
реакциями ни одна из реакций с участием неионизованной формы диэфира,
т. е. ни щелочная, ни кислотная, ни «нейтральная» реакция расщепления
связи углерод — кислород, не имеет скорости достаточно высокой, чтобы
ее можно было обнаружить.
Хааке и Уэстхеймер предложили следующее объяснение наблюдаемых
явлений. Угол связи у атома фосфора в сложноэфирной группе пятичленного
цикла приближается к 90° (1,571 рад) [другие углы в цикле в среднем равны
по 112,5° (1,963 рад)]. В такой структуре в начальном состоянии фосфата
с тетраэдрически гибридизованным атомом фосфора напряжение составляет
лишь несколько килокалорий на моль. С другой стороны, в тригонально-
бипирамидальной конфигурации, предполагаемой для переходного состоя-
ния SN2-Tnna с пятилигандным фосфором, угол связи у атома фосфора,
включающий вновь образующуюся связь, при отсутствии напряжения должен
быть смежным с прямым углом описанной выше модели и, конечно, не
с углами, равными 120° (2,094 рад). Возможность ненапряженной структуры
переходного состояния приводит к существенному уменьшению энергии
активации нуклеофильного замещения. Именно вследствие такого строения
переходного состояния угол между линиями подхода и ухода обмениваю-
щихся групп, обычно равный 180° (3,141 рад), уменьшается приблизительно
до 120° (2,094 рад), и именно поэтому линия приближения входящего лиганда
образует примерно равные углы с линиями удаления двух альтернативно
отщепляющихся лигандов. Отщепление одного из них соответствует рас-
крытию фосфат-эфирного цикла, а отщепление другого, т. е. неэтерифици-
рованной гидроксильной группы, соответствует обмену кислорода. Поэтому
в обоих случаях наблюдается один и тот же кинетический эффект, обуслов-
ленный отсутствием напряжения активации. Для кислотного гидролиза эта
модель приведена ниже:
О \ у \ Альтернативно
\/ V отщепляющиеся
Клиганды
Входящий лиганд \
О
в. Механизм гидролиза триэфиров фосфорной кислоты. Как впервые
показали Блюменталь и Герберт [114], гидролиз триметилфосфата в присут-
ствии щелочи в воде, обогащенной 18О, происходит с расщеплением связи
фосфор — кислород. Бантон, Вернон и сотр. [115] показали, что этот путь
является единственным при гидролизе триметилфосфата и трифенилфосфата
в щелочном растворе. В случае трифенилфосфата тщательное изучение
появления 18О в молекуле эфира до гидролиза (что указывало бы на предва-
рительную обратимую стадию присоединения) показало, что этот изотоп
в молекуле не появляется. Поэтому эти реакции рассматриваются как бимо-
лекулярное нуклеофильное замещение у атома фосфора
HO~ + PO(OR)3 —> HOPO(OR)2+"OR (Вас2)
Бантон и Вернон [115] расширили эти исследования, включив в них гидролиз
в нейтральном и кислом растворах. Триметилфосфат претерпевает гидролиз
первого порядка по воде. Опыты с водой, обогащенной 18О, показали, что
981
Глава X V
эта реакция характеризуется расщеплением связи углерод — кислород.
Для реакции был предложен бимолекулярный механизм
Н2О + СН3ОРО(ОСН3)2Н2ОСН3+ОРО(ОСН3)2 (Bal2)
Скорости гидролиза в ряду моно-, ди-, триметилфосфаты по механизму BAL2
постепенно возрастают. При 100 °C приблизительное соотношение скоростей
составляет 1 : 7 : 70.
Скорость гидролиза триметилфосфата настолько велика, что катализируе-
мую кислотами реакцию трудно изучать вплоть до 3 н. концентрации хлорной
кислоты в воде *. С другой стороны, как показал Тайн [116], кислотный
катализ можно наблюдать в водном диоксане, в котором скорость «нейтраль-
ной» реакции сильно понижена **.
Открытое Уэстхеймером стереохимическое явление проявляется также
в ряду триэфиров [113]. Эфиры типа этиленметилфосфата, содержащие
двухвалентную эфирную группировку — О — РО — О — в пятичленном
цикле, аномально быстро гидролизуются как при катализе гидроксильными
ионами (ВАС2), так и при катализе кислотами, который, вероятно, соответ-
ствует механизму Аас2. Оба типа реакций идут исключительно путем рас-
щепления связи фосфор — кислород. Каждая реакция состоит из двух конку-
рирующих реакций, одна из которых соответствует расщеплению пяти-
членного цикла, а другая — отщеплению ациклической алкоксильной груп-
пы с сохранением цикла. Вторая из этих реакций аналогична реакции обмена
кислорода в циклических диэфирах фосфорной кислоты. Таким образом,
при щелочном и кислотном гидролизе триэфиров ускоряющее действие,
обусловленное наличием цикла, сказывается и на реакции раскрытия цикла,
и на реакции гидролиза ациклической оксигруппы. Все эти результаты
соответствуют теории Уэстхеймера, которая уже излагалась на примере
диэфиров фосфорной кислоты. Так же как и в случае циклических диэфиров,
при гидролизе циклических триэфиров не наблюдалось «нейтральной»
реакции.
В случае гидролиза трифенилфосфата в водно-диоксановых растворах кислот-
ный катализ проявляется. Опыты с водой, меченной 18О, показали, что эта
реакция происходит в значительной степени путем разрыва связи фосфор —
кислород. Зависимость скорости от кислотности среды проходит через макси-
мум в умеренно концентрированном растворе кислоты, например в 1,7 н.
хлорной кислоте в 60%-ном диоксане; точное положение максимума зависит
от соотношения диоксан : вода и добавок солей. Отсюда следует, что для
протекания реакции определенно необходима вода, и скорость гидролиза
уменьшается до нуля, если термодинамическая активность растворителя —
воды будет равна нулю. Это означает, что в данном случае реализуется
механизм ААс2 со следующей контролирующей скорость стадией:
Н2О + РО(ОС6Н5)2(ОНС6Н5) -> Н2ОРО(ОС6Н5)2 + С6Н5ОН (Аас2)
г. Вывод о механизмах гидролиза и обмена кислорода. В табл. 212,
составленной Бантоном, приведены константы скоростей и механизмы гидро-
* Возможно, кроме того, что триэфиры труднее протонируются, чем кислые эфиры,
в которых внутримолекулярная водородная связь облегчает захват протона
Н+ + СГ 'OR НО OHR
^I’dfoR) \^(OR)
** Эти данные были получены для трпэтплфосфата.
982
Реакции карбоксильной и фосфатной групп
Таблица 212
Константы скоростей (100 °C) и механизмы гидролиза эфиров фосфорной кислоты
с указанием расщепляющейся связи (по данным Бантона и Вернона)
Эфир и реагент Моноанион а Нейтральная молекула а Сопряженная кислота а
с - о О- Р С - О о - р С - О О - Р
В~2 AL в1< BAL2 ВАС2 aal2 аас2
<НО)зРО+ Н2О (СН3О)(НО)2РО + Н2О (СН3О)2(НО)РО + Н2О (СН3О)3РО + Н2О (G1u)(HO)2PO в4-Н2О (СН3О)2(НО)РО+ОН- <СН3О)3РО + ОН- 4,03 8,23 <0,001 13,0 0,05 3,3 36,5 1,3 0,9 2,00 0,91 0,55 1,08 0,11
BALJ aal1
30 300 - 40 000 Д 32 (25 °C г)
ВАС2
Медлен- но Очень медленно
а 106 ft!, с-1, при 100 °C и ц = 0.
6 106 й2, л-моль-1-c-l, при 100 “Си ц = 0.
в Glu = a-D-глюкопиранозил.
г Константа скорости при 25 °C и [НСЮ4] = 0,5 моль/л. При более высоких концентрациях
кислоты скорость лучше зависит от ho, чем от [Н+].
Д Это значение соответствует 106 ft2 (л-моль-1-с-1), экстраполированному к 100 °C на основе
известных скоростей при низких температурах.
лиза моно-, ди- и триметилфосфатов и моно-а-глюкопиранозилфосфата в воде
при различных pH среды.
Обмен кислорода между фосфатной группой и растворителем — водой —
является специальным случаем гидролиза, и мы уже не раз отмечали, что
гидролиз кислого эфира сопровождается обменом кислорода неэтерифициро-
ванной гидроксильной группы фосфатного остатка. Для кислородного
обмена фосфатной группы необходимо, чтобы происходило расщепление
связи фосфор — кислород. Наиболее хорошо изученным примером такого
обмена служит обмен в самой фосфорной кислоте; в табл. 212 приведены
константы скоростей обмена, который может происходить по трем раз-
личным механизмам.
Бантон, Ллевиллин, Вернон и Уэлч 1117] измерили скорость обмена кислорода
ортофосфорной кислоты с водой при кислотности среды от pH 8 до 8 н. по
хлорной кислоте. Зависимость скорости от pH в районе pH от 8 до О
настолько похожа на эту зависимость для случая гидролиза монометилфос-
фата, что, если бы на осях координат не было цифровых обозначений, было бы
трудно сказать, к какой из этих двух реакций относится график. На соответ-
ствующей кривой для обмена, абсолютные скорости которого наполовину
меньше, чем скорости гидролиза, максимум скорости приходится на pH 5
вместо 4, а минимум на pH 1 вместо 0,5. Анализ показывает, что в интервале
pH между 3 и 8 в реакцию вступает также моноанион. Как видно из данных
табл. 212, скорость обмена имеет тот же порядок величины, что и скорость
гидролиза этих кислых эфиров, которые в этой области pH имеют одну
983
Глава XV
ионизованную и одну неионизованную гидроксильную группу. Аналогично
связаны между собой и скорости обмена в сопряженной кислоте со скоро-
стями гидролиза сопряженных кислот эфиров, которые гидролизуются путем
расщепления связи фосфор — кислород. Скорость обмена в неионизованпой
молекуле также может быть сравнима со скоростями неионного ацильного
гидролиза, измерению которого, хотя и незначительно, мешает парал-
лельно идущий алкильный гидролиз.
СПИСОК ЛИТЕРАТУРЫ
1. Day J. X. Е., Ingold С. К., Trans. Faraday Soc., 37, 686 (1941).
2. Datta S. C., Day J. N. E., Ingold С. К., J. Chem. Soc., 1939, 838; Hughes E. D.,
Ingold С. K., Masterman S., ibid.,'p. 840.
3. Holmberg B., Ber., 45, 2997 (1912).
4. Ingold С. K., Ingold E. H., J. Chem. Soc., 1932, 758.
5. Prevost C., Ann. chim., 10, 147 (1928).
6. Norton H. M., Quayle O. R., J. Am. Chem. Soc., 62, 1170 (1940).
7. Polanyi M., Szavo A. L., Trans. Faraday Soc., 30, 508 (1934).
8. Long F. A., Friedman L., J. Am. Chem. Soc., 72, 3692 (1950).
9. Warder R. B., Ber., 14, 1361 (1881).
10. Bender M. L., J. Am. Chem. Soc., 73, 1626 (1951); Bender M. L., Ginger R. D.,
Unik J. P., ibid., 80, 1044 (1958).
11. Reicher L. T., Ann., 228, 257 (1885); 232, 103 (1886); 238, 276 (1887).
12. Smith L., Olsson H., Z. physik. Chem., 102, 26 (1922); 118, 99 (1925); Olsson II., ibid.,
p. 107; ibid., 125, 243 (1927); 133, 233 (1928).
13. Skrabal A., Hiigetz A. II., Monatsh., 47, 17 (1926); Skrabal A., Ruckert M. ibid.,
50, 369 (1928).
14. Kindler K., Ann., 452, 90 (1927); Ber., 69, 2792 (1936).
15. Hammett L. P., Physical Organic Chemistry, McGraw-Hill, 1940, Chap. 7.
16. Kindler K., Ann., 450, 1 (1926); 452, 90 (1927); 464, 278 (1928); Ber., 69, 2792 (1936).
17. Ingold С. K., Nathan W. S., J. Chem. Soc., 1936, 222.
18. Evans D. P., Gordon J. J., Watson II. В., J. Chem. Soc., 1937, 1430.
19. Tommila E., Hinshelwood C. N., J. Chem. Soc., 1938, 1801; Tommila E., Ann. Acad.
Sci. Fennicae, A57, № 3 (1941); A59, № 3, 4, 5, 9, 10.
20. Evans D. P., Gordon J. J., Watson H. B., J. Chem. Soc., 1938, 1439.
21. Kenyon J., Partridge S. M., Phillips H., J. Chem. Soc., 1936, 85; Hills W. J., Kenyon J.,
Phillips H., ibid. p. 576.
22. Balfe M. P., Doughty M. A., Kenyon J., Poplett R., J. Chem. Soc., 1942, 605.
23. Balje M. P., Evans A. A., Kenyon J., Nandi K. N., J. Chem. Soc., 1946, 803; Bal-
fe M. P., Kenyon J., Wicks R., ibid., p. 807.
24. Balfe M. P., Bevan G. H., Kenyon J., J. Chem. Soc., 1951, 376.
25. Kenyon J., Partridge S. M., Phillips H., J. Chem. Soc., 1937, 207.
26. Balje M. P., Hills W. J., Kenyon J., Phillips H., Platt В. C., J. Chem. Soc., 1942,
556.
27. Balfe M. P., Doughty M. A., Kenyon J., Poplett R., J. Chem. Soc., 1942, 605; Bun-
ton C. A., Hadwick T., ibid., 1957, 3043.
28. Balje M. P., Downer E. A. W., Evans A. A., Kenyon J., Poplett R., Searle С. E., Tar-
nowey A. L., J. Chem. Soc., 1946, 797.
29. Balje M. P., Bevan G. H., Kenyon J., J. Chem. Soc., 1951, 376.
30. Hammond G. S., Rudesill J. T., J. Am. Chem. Soc., 72, 2769 (1950).
31. Gomberg M., Davies G. T., Ber., 36, 3926 (1903).
32. Bunton C. A., Konasiewicz A., J. Chem. Soc., 1955, 1354.
33. Bunton C. A., Cornyns A. E., Graham J., Quayle J. R., J. Chem. Soc., 1955, 3817.
34. Cowdrey W. A., Hughes E. D., Ingold С. K., Masterman S., Scott A. D., J. Chem.
Soc., 1937, 1264.
35. Olson A- R., Miller R. J., J. Am. Chem. Soc., 60, 2690 (1938).
36. Olson A. R., Hyde J. L., J. Am. Chem. Soc., 63, 2459 (1941).
37. Long F. A., Purchase M., J. Am. Chem. Soc., 73, 3267 (1950).
38. Bartlett P. D., Rylander P. N., J. Am. Chem. Soc., 73, 4273 (1951).
39. Long F. A., Purchase M., J. Am. Chem. Soc., 73, 3271 (1950).
40. Bunnett J. F., Robinson M. M., Pennington F. C., J. Am. Chem. Soc., 72, 2328 (1950).
41. Ingold С. K., Ingold E. H., J. Chem. Soc., 1932, 758.
42. Datta S. C., Day J. N. E., Ingold С. К., J. Chem. Soc., 1939, 838.
984
Реакции карбоксильной и фосфатной групп
43. Bunton С. A., Day J. N. Е., Flowers В. Н., Shell В., Wood J. L., J. Chem. Soc., 1957,
963.
44. Long F. A., Friedman L., J. Am. Chem. Soc., 72, 3692 (1950).
45. Boberts I., Urey H. C., J. Am. Chem. Soc., 60, 2391 (1938).
46. Watson H. B., Modern Theories of Organic Chemistry, Oxford Univ. Press, 1937, p. 130.
47. Bunnett J. F., J. Am. Chem. Soc., 83, 4056 (1961) et seq. (4 papers).
48. Yates K., McClelland В. A., J. Am. Chem. Soc., 89, 2686 (1967).
49. Lane C. A., J. Am. Chem. Soc., 86, 2521 (1964).
50. Treiers H. P., Hammett L. P., J. Am. Chem. Soc., 59, 1708 (1937).
51. Bender M., Ladenheim H., Chen M. C., J. Am. Chem. Soc., 83, 123 (1961).
52. Chmiel С. T., Long F. A., J. Am. Chem. Soc., 78, 3326 (1956).
53. Bunton C. A., James D. H., Senior J. B., J. Chem. Soc., 1960, 3364.
54. Berthelot P. E. M., Pean de Saint Gilles L., Ann. Chim., 65, 385 (1862); 68, 225 (1863).
55. Guldberg С. M., Waage P., Etudes sur les affinites chimiques, Brogger and Cristie,
Christiania, 1867; J. prakt. Chem., 19, 69 (1879).
56. Ostwald W., J. prakt. Chem., 30, 93 (1884).
57. Goldschmidt H., Ber., 28, 3218 (1895).
58. Drushel W. A., Am. J. Sci., 34, 69 (1912); 37, 514 (1914).
59. Polomaa M. H., Ann. Acad. Sci. Fennicae, A4, № 2 (1913); A10, № 16 (1917).
60. Bender M. L., J. Am. Chem. Soc., 73, 1626 (1951).
61. Goldschmidt H., Ber., 28, 3210 (1895); Goldschmidt H., Udby O.,Z. physik. Chem., 60,
728 (1907).
62. Kailan A., Ann., 351, 186 (1907).
63. Sudborough J. J., Lloyd L. L., J. Chem. Soc., 75, 467 (1899); Sudborough J. J., Git-
tinsJ. M., ibid., 93, 210 (1908); Sudborough J. J., Turner M. K., ibid., 101, 237 (1912).
64. Skrabal A., Pjaff E., Airoldi H., Monatsh., 45, 141 (1924); Skrabal A., Hiigelz A. M.,
ibid., 47, 17 (1926); Skrabal A., Zahorka A., ibid., 48, 459 (1927); Skrabal A., Ruc-
kert M., ibid., 50, 369 (1928).
65. Palomaa M. H., Salmi E. J., Jansson J. I., Salo T., Ber., 68, 303 (1935); Palomaa M. H.
Terkimadi K. R., ibid., p. 887; Palomaa M. H., Siitonen T. A., Ber., 69, 1338 (1936).
66. Hinschelwood C. N., Legard A. R., J. Chem. Soc., 1935, 1588; Turner E. W. T., Hin-
schelwood C. N., ibid., 1938, 862; Tommila E., Hinschelwood C. N., ibid., p. 1001.
67. Hartman R. J., Borders A. M., J. Am. Chem. Soc., 59, 2107 (1937); Hartman R. J.,
Storms L. B., Gassmann A. G., ibid., 61, 2167 (1939); Hartman R. J., Gassmann A. G.,
ibid., 62, 1559 (1940); 63, 2393 (1941); Hartman R. J., Hoogsteen II. M., Moede J. A.,
ibid., 66, 1714 (1944).
68. Leisten J. L., J. Chem. Soc., 1956, 1572.
69. Hughes E. D., Ingold С. K., Masterman S., J. Chem. Soc., 1939, 840.
70. Bunton C. A., Hughes E. D., Ingold С. K., Meigh D. F., Nature, 166, 679 (1950).
71. Meyer L., Ber., 22, 24 (1889).
72. Betts R. L., Hammett L. P., J. Am. Chem. Soc., 59, 1568 (1937).
73. Bunnett J. F., Davies G. T., J. Am. Chem. Soc., 82, 665 (1960).
74. Meyer H., Monatsh., 27, 31 (1906).
75. Gorvin J. H., J. Chem. Soc., 1945, 732.
76. Gordon M., Miller J. G., Day A. R., J. Am. Chem. Soc., 70, 1946 (1948); 71, 1245 (1949);
Arnett E. M., Miller J. G., Day A. R., ibid., 72, 5635 (1950).
77. Reid E. E., Am. Chem. J., 21, 284 (1899); 24, 397 (1900); Meloche I., Laidler K. J.,
J. Am. Chem. Soc., 73, 1712 (1951).
78. Beurath A., Z. anorg. Chem. 151, 53 (1926); v. Euler H., Blander A., Z. physik. Chem.,
131, 107 (1928); Taylor T. W. J., J. Chem. Soc., 1930, 2741; Krieble V. K., Holst R. A.,
J. Am. Chem. Soc., 60, 2976 (1938); Edwards J. T., Meacock S. C. R., J. Chem. Soc.,
1957, 2000.
79. Yates K., Stevens J. B., Katritzku A. B., Can. J. Chem., 42, 1957 (1964); Yates K.,
Stevens J. B., ibid., 43, 529 (1965); Yates K., Riordan J. C., ibid., p. 2328.
80. Lacey R. J. Chem. Soc., 1960, 1633.
81. Geuther A., Arch. Pharm., 106, 97 (1863); Jahresber., 1863, 323.
82. Wisliscenus W., Ann., 246, 306 (1888).
83. Wisliscenus W., Ber., 20, 2930 (1887); 291, 147 (1896).
84. Helion R., Oppenheim A., Ber., 10, 699 (1877); Israel A., Ann., 231, 187 (1885).
85. Claisen L., Ber., 20, 655 (1887).
86. Wallach O., Steindorff A., Ann., 329, 109 (1903).
87. Dieckmann W., Ber., 27, 102 (1894).
88. v. Fehling H., Ann., 49, 154 (1844); Duisberg C., Ber., 16, 133 (1883).
89. Dieckmann W., Ber., 27, 965 (1894).
90. Lapworth A., J. Chem. Soc., 79, 1265 (1901).
91. Claisen L., Lowman O., Ber., 20, 651 (1887); 21, 1147 (1888).
985
Глава XV
92. Snell J. M., McElvain В. M., J. Am. Chem. Soc., 53, 2310 (1931).
93. Bunnett J. F.. Robinson M. M., Pennington F. C., J. Am. Chem. Soc., 72, 2328 (1950).
94. Dieckmann W., Ber., 33, 2670 (1900).
95. Nef J. U., Ann., 298, 202 (1897).
96. Michael A., Ber., 33, 3731 (1900); 38, 1922 (1905).
97. Lapworth A., J. Chem. Soc., 79, 1265 (1901); Hahn A. С. O., Lapworth A., Proc.
Chem. Soc., 19, 189 (1903).
98. Arndt F., Eistert B., Ber., 69, 2381 (1936).
99. Kenyon J., Young D. P., J. Chem. Soc., 1940, 216.
100. Brown W. G., Eberly K., J. Am. Chem. Soc., 62, 113 (1940).
101. Claisen L., Ber., 38, 693 (1905).
102. Spielman M. A., Schmidt M. T., J. Am. Chem. Soc., 59, 2009 (1937).
103. Hauser С. R., Benfrow W. B., Jr., J. Am. Chem. Soc., 59, 1823 (1937); Renfrow W. B.,
Jr., Hauser С. R., ibid., 60, 463 (1938).
104. Chevalier J., Compt. rend., 127, 60 (1898); Plummer R. H. A., Scott F. H., J. Chem.
Soc., 93, 1699 (1908); Bailly M. C., Bull. soc. chim. (France), 9, 340, 405 (1942); Fleu-
ry P., Compt. rend., 221, 416 (1945); Desjobert A., ibid., 224, 575 (1947); Bull. soc.
chim. (France), 14, 809 (1947); Bull. soc. chim. biol., 36, 475 (1954).
105. Butcher W. W., Westheimer F. H., J. Am. Chem. Soc., 77, 2420 (1955); Kumamoto J.,
Westheimer F. H., ibid., p. 2515.
106. Vernon C. A., Spec. Publ. Chem. Soc., 8, 17 (1957); Bunton C. A., Llewellyn D. R.,
Oldham K. G., Vernon C. A., J. Chem. Soc., 1958, 3574; cp. Bunton C. A., Keller-
mann D., Oldham K. G., Vernon C. A., ibid., 1966B, 292.
107. Bunton C. A., Llewellyn D. R., Oldham K. G., Vernon C. A., J. Chem. Soc., 1958, 3579.
108. Bunton C. A., Llewellyn D. R., Oldham K. G., Vernon C. A., J. Chem. Soc., 1958, 3588.
109. Kumamoto K., Cox J. R., Jr., Westheimer F. H., J. Am. Chem. Soc., 78, 4858 (1956).
110. Bunton C. A., Mhala M. M., Oldham K. G., Vernon C. A., J. Chem. Soc., 1960, 3233.
111. Brown D. M., Todd A. R., J. Chem. Soc., 1952, 52.
112. Brown D. M., Magrath D. I., Todd A. R., J. Chem. Soc., 1952, 2708.
113. Haake P. C., Westheimer F. H., J. Am. Chem. Soc., 83, 1102 (1961); Kaiser E. T.,
Pamar M., Westheimer F. H., ibid., 85, 602 (1963); Covitz F., Westheimer F. H., ibid.,
p. 1773; Eberhard A., Westheimer F. H., ibid., 87, 253 (1965); Dennis E. A., Westhei-
mer F. H., ibid., 88, 3431, 3432 (1966).
114. Blumenthal E., Herbert J. В. M., Trans. Faraday Soc., 41, 611 (1945).
115. Barnard P. W. C., Bunton C. A., Llewellyn D. R., Vernon C. A., Welch V. A., J. Chem.
Soc., 1961, 2670; Barnard P. W. C., Bunton C. A., Kellermann D., Mhala M. M.,
Silver B., Vernon C. A., Welch V. A., ibid., 1966, 227.
116. Thain E. M., J. Chem. Soc., 1957, 4694.
117. Bunton C. A., Llewellyn D. R., Vernon C. A., Welch V. A., J. Chem. Soc., 1961, 1636.
ГЛАВА XVI
ПОЛЯРНЫЕ ЭФФЕКТЫ
1. Аддитивность и пропорциональность полярных эффектов . . 988
Список литературы ............................. 1015
1. Аддитивность и пропорциональность полярных эффектов
а. Условия, при которых возможны аддитивность и пропорциональ-
ность. В настоящее время еще не создана какая-либо общая и тонная теория
полярных влияний на равновесие или скорости реакций. Однако в предыду-
щих главах уже приводились некоторые полуколичественные данные, кото-
рые показывают, что полярные влияния некоторых видов заместителей
на свободную энергию реакции или на свободную энергию активации неко-
торых реакций могут быть с некоторым приближением аддитивными или
пропорциональными. Рассмотрим два примера.
Во-первых, обратимся к теории Бьеррума о влиянии поля ионного заме-
стителя на равновесие ионизации (гл. XIV, разд. 1,6). Эта теория заклю-
чается в том, что электростатический потенциал поля заместителя в точке,
до которой должен доходить противоионный реагент с зарядом q^, на конеч-
ной стадии реакции будет иметь величину У1, которую можно вычислить.
Инкремент наращивания энергии поля равен Viqi, и Бьеррум рассматривает
его (после статической поправки, входящей в энтропийный член) как добавку
к изменению свободной энергии реакции. Это означает, что если бы ионные
заместители были различными в том смысле, что имели бы разный заряд
(или эффективный заряд) или были бы по-разному расположены относитель-
но друг друга, так чтобы в этой же указанной выше точке возникал потен-
циал У2, то дополнительный вклад в полярную энергию был бы равен V2qi-
Далее, если бы в молекуле присутствовали одновременно первый и второй
указанные выше заместители и были бы расположены на достаточном рас-
стоянии друг от друга, чтобы действовать независимо, то дополнительный
вклад в свободную энергию был бы равен (]Д V2) qi- Таков принцип
аддитивности в простейшей форме. Это означает также, что если вместо
захвата протона, рассмотренного Бьеррумом, рассмотреть захват какого-
либо другого иона, например иона с зарядом q2, то вклады в величину сво-
бодной энергии, обусловленные первым заместителем, вторым заместителем
и обоими заместителями вместе, составляли бы У1д2, V2q2 и (У] Ц- У2) q2-
соответственно. Таков простейший принцип пропорциональности.
Аддитивность и пропорциональность зависят в основном от составляющих
полярного эффекта. Каждая составляющая является произведением двух
факторов, один из которых соответствует какой-либо одной физической
величине одного реагирующего вещества, а другой — одной простой физиче-
ской величине другого реагента. Наиболее существенным является то, что
каждый фактор определяет лишь одну электростатическую величину. Для
двух реагирующих веществ, рассматриваемых вместе, должны быть две
величины, но не обязательно на описанной здесь основе «один к одному».
Если учитывать сравнимые по величине энергии, получаемые при умножении
заряда на потенциал и (точечного) дипольного момента на градиент
потенциала, и, таким образом, вычислять величину двучленного выражения,
например qV р grad У, то пропорциональность может не получаться.
Условия проявления аддитивности и пропорциональности полярных эффек-
тов, влияющих на равновесные реакции, полностью похожи на условия,,
необходимые для проявления этих же свойств в необратимых реакциях.
Согласно теории переходного состояния, конечное состояние, в котором
достигается взаимодействие заряда и потенциала, заменяется соответствую-
щим переходным состоянием. Свойства аддитивности и пропорциональности
в этом случае приписываются свободной энергии активации.
Другим примером, который можно здесь привести, служит теория индук-
ционных эффектов ионных заместителей, скажем, влияние ионного заряда
аммония на скорость отщепления с образованием олефинов по бимолекуляр-
ному механизму Е2 (гл. IX, разд. 2,6). В этом случае вычисляют заряд
988
Полярные эффекты
передаваемый по индуктивному механизму от заместителя к 0-атому водо-
рода. Затем вычисляют потенциал Vj в точке, занимаемой этим атомом
в потенциальном поле основного реагента в переходном состоянии. Резуль-
тирующим энергетическим членом, как и прежде, является qiVi, однако
входящие в него сомножители соответствуют влиянию разных источников
поля. Очевидно, можно рассмотреть изменения в строении полярного заме-
стителя, приводящие к изменениям q, а также изменения природы основного
реагента, приводящие к изменению V, и, таким образом, выявить, как
и ранее, аддитивность и пропорциональность полярных энергий. Условия
для аддитивности и пропорциональности будут такими же, как и рассмотрен-
ные раньше. Этот пример относится к случаю влияния на скорости реакций,
однако можно привести примеры, которые могли бы иллюстрировать ана-
логичный подход и к равновесиям.
Существует возможность гораздо более широкого применения принципа
аддитивности и пропорциональности термодинамических или кинетических
энергетических параметров реакций, чем это показано на двух приведенных
примерах. Отношение между любой парой реагентов является взаимно
обратным — каждое из реагирующих веществ может рассматриваться как
субстрат, а другое — как реагент. Замещение может быть последовательным
и кумулятивным, что приведет в общем к серьезным изменениям химической
структуры. Следовательно, вполне возможно, что некоторые совершенно
различные реакции могли бы коррелироваться указанным выше способом.
При рассмотрении термодинамических свойств реакций в данном контексте
мы будем считать реакции разными, если они имеют различную стехио-
метрию или если физические свойства сред, в которых они проводятся,
различны, особенно при применении неодинаковых растворителей. При рас-
смотрении кинетических свойств реакции считаются различными, если
исходные или переходные состояния неодинаковы, т. е. если имеется раз-
личие в их стехиометрии или механизме или в стадии, лимитирующей ско-
рость; кроме того, реакции считаются различными, если физические свойства
сред, в которых они проводятся, различны.
В соответствии с этими принципами поле действия для перекрестных срав-
нений экспериментальных данных друг с другом получается весьма широ-
ким, но условия, в которых должны наблюдаться аддитивность и пропорцио-
нальность полярных эффектов, строго ограничены. Необходимо исключить
пространственные эффекты и эффекты утяжеления (эффекты утяжеления
всегда являются небольшими). Полярные эффекты одновременно действую-
щих заместителей должны быть независимыми друг от друга. Помимо этого,
каждый полярный эффект должен обладать свойством быть выраженным
в виде простого произведения одних и тех же двух физических величин
на протяжении всей сравниваемой серии реакций. Как будет показано ниже,
этот диапазон применения указанных принципов может быть расширен за
счет использования дополнительных параметров реакции, необходимость
учета которых следует из теории органической химии (гл. II, разд. 3).
Если бы обратимую химическую реакцию, на которую воздействует поляр-
ный эффект заместителя, можно было провести адиабатически, то полярный
эффект, рассматриваемый как электростатическая работа, вошел бы в термо-
динамическую величину изменения внутренней энергии ДП. Однако если
реакция проводится обычным образом в термостате при постоянном объеме,
то происходит эквивалентное изменение теплоты ДЯ. В соответствии с этим
происходит изменение энтропии AS. Изменение свободной энергии AG
состоит из изменения энтальпии ЛЯ и изменения энтропии AS. Подобная
теория может применяться и в том случае, если рассматривается влияние
989
Глава XVI
полярных факторов на скорость реакции. Если предположить, что реакция
проводится изотермически и при постоянном объеме, то полярный эффект
изменит оба параметра активации АЯ* и AG*, а тем самым и свободную
энергию активации AG*.
В своем исчерпывающем обсуждении соотношений между изменением сво-
бодной энергии реакции или энергии активации различных реакций Леффлер
и Грюнвальд[1] рассматривали эффекты, возникающие вследствие зависимо-
сти свободной энергии от температуры, которая выражается уравнением AG =
= АЯ — TAS. Сначала они рассмотрели такой интервал температур, в кото-
ром АЯ можно считать постоянной, при этом АСР равна нулю. Авторы
рассмотрели также более вероятные условия, когда АСР постоянна во всем
диапазоне температур и не обязательно равна нулю. В обоих случаях было
показано, что если вклады (SAG) ряда заместителей в изменение свободной
энергии AG каких-либо двух реакций точно пропорциональны между собой
и если такая пропорциональность выдерживается во всем температурном
диапазоне, то вклады (SAG) заместителей в изменение энтальпии АЯ каждой
реакции будут пропорциональны их вкладам (SAG) в изменение энтропии А5
этой же реакции. Следовательно, любой из этих вкладов (6АЯ или 6AS)
будет пропорционален вкладам (SAG) в изменение свободной энергии AG
этой реакции. Все это будет справедливо для каждой реакции при любой
температуре в данном интервале температур. Таким образом, пометив эти
две реакции цифрами 1 и 2, можно записать пропорциональность между
шестью величинами:
SAG! ос 6АЯ! ос SAGj ос 6AG2 ос 6АЯ2 ос 6AS2.
Первоначально наблюдавшаяся пропорциональность между SAGj и 6AG2
является частью этой общей пропорциональной зависимости. Следовательно,
наблюдаемая пропорциональность между вкладами SAG, вносимыми заме-
стителями в изменение свободной энергии AG двух реакций, обусловливает
такую же пропорциональность между их вкладами (SAG) в энтальпию АЯТ
а также пропорциональность между их вкладами (SAG) в изменение энтро-
пии АА реакций.
Необходимо понять, что из самого факта справедливости этой термодинами-
ческой теоремы вовсе не следует, что в действительности будет наблюдаться
пропорциональная зависимость между любыми из этих величин. Из нее
следует лишь то, что если существует пропорциональность между любыми
двумя из этих величин, то будет наблюдаться пропорциональность и между
другими параметрами. Как уже было сказано, пропорциональность энерге-
тических параметров будет ожидаться только в том случае, если полярные
эффекты могут быть выражены в виде отдельных произведений двух факторов
и если отсутствуют пространственные эффекты и эффекты утяжеления. Экспе-
риментально обнаруженная пропорциональность между 6АЯ и SAG пред-
ставлена на рис. 51. В этом случае выполняются все указанные условия.
Реакция представляет собой равновесия ионизации метагемоглобинов, на
которые влияют ионные заместители, находящиеся на больших расстояниях,
например на расстоянии 30 А (3 нм), от реакционного центра в жесткой
структуре молекулы. На таких расстояниях не могут проявляться никакие
эффекты, кроме кулоновских сил; локальное окружение реакционного цен-
тра является постоянным, и даже масса молекул практически постоянна.
Леффлер и Грюнвальд распространили свои взгляды и на кинетические
параметры. Если оказывается, что вклады SAG* заместителей в кинетиче-
ский параметр AG* для двух реакций точно пропорциональны друг другу
в данном интервале температур, то будет наблюдаться аналогичная пропор-
990
Полярные эффекты
циональность между шестью указанными параметрами, в частности будут
пропорциональными вклады и Условия для обеспечения про-
порциональности этих величин аналогичны указанным выше: слагаемые
полярных эффектов должны представлять собой простые произведения двух
величин и заместители не должны проявлять ни пространственных эффектов,
ни эффектов утяжеления.
б. Пропорциональность полярных эффектов при кислотно-основном
катализе. Впервые пропорциональность структурных вкладов в изменение
свободной энергии реакций была открыта Бренстедом и Педерсеном в 1924 г.
В этом же году они [2] открыли общий основной катализ на примере реакции
разложения нитрамида. В ходе этой работы авторы обнаружили почти всегда
соблюдающуюся характерную зависимость между величинами каталитиче-
ских констант скоростей для различных оснований и силой этих оснований.
При переходе от одного основания к другому, более сильному, прирост лога-
рифма константы скорости каталитической реакции является постоянной
величиной и составляет постоянную долю наращивания логарифма кон-
станты основности оснований [2]. В последующие годы были исследованы
другие подобные реакции, например мутаротация глюкозы и иодирование
ацетона. В ходе этой работы был открыт общий кислотный катализ и анало-
гичная линейная зависимость между логарифмами констант каталитической
скорости и логарифмами констант кислотности катализирующих кислот [3].
Константы основности оснований, разумеется, могут быть выражены как
константы кислотности сопряженных им кислот, и поэтому указанные две
линейные зависимости могут быть представлены в общем виде, который
известен под названием соотношения Бренстеда. Оно может быть .записано
следующим образом:
6 log k = B§ log Ка
где к — каталитическая константа скорости, Ка — константа кислотности
кислотного катализатора или сопряженной кислоты основного катализа-
тора; зйачок 6 обозначает разницу, обусловленную изменением катализа-
тора *. Так как обычно (гл. II, разд. 2,в и г)
, „ AG , , AG'" . , кТ
1OSK 2,303RT И °gt'“ 2,303B7-+1Og Л
зависимость Бренстеда может быть записана как пропорциональность между
структурными вкладами в свободную энергию активации катализируемой
реакции и в свободную энергию кислотно-основного равновесия катализа-
торов
6AG* = BbAGa
В то время, когда Бренстед открыл это соотношение, еще не существовало
теории, объясняющей это явление. Действительно, в 20-е годы до появления
теории переходного состояния для всех было удивительно, что может суще-
ствовать простая зависимость между термодинамикой одной реакции и кине-
тикой другой.
Одним из наиболее глубоких исследований общего кислотного катализа
является работа Белла и Хиггинсона 14] по дегидратации гидрата ацеталь-
дегида в 92%-ном водном растворе ацетона
НА
СН3СН(ОН)2 ---> СН,СНО-4-Н2О
* В константы должны быть внесены статистические поправки на количество эквива-
лентных протонов или мест для протонов в реагирующих веществах.
991
Глава XVI
Зависимость логарифмов скоростей реакции, катализируемой в этом раство-
рителе при 25 °C рядом алифатических и ароматических карбоновых кислот
и фенолов, от констант кислотности катализаторов в воде при 25 °C приве-
дена па рис. 57. Приведенная зависимость охватывает изменение скоростей
в пределах более пяти порядков и изменение силы кислот в пределах десяти
порядков.
Однако эта корреляция недостаточно точна. Если учесть, что логарифмы
представляют собой функции, довольно нечувствительные к изменению
аргумента, то видно, что некоторые из отклонений от прямолинейной зави-
симости для данных незаряженных катализаторов (карбоновых кислот
Рис. 57. Данные Белла и Хиггинсона [4] для общего кислотного катализа дегидратации
гидрата ацетальдегида незаряженными карбоновыми кислотами и фенолами
в 92 %-ном водном растворе ацетона при 25 °C (в том случае, когда реагирующие
вещества содержат эквивалентные протоны или места присоединения протонов,
в К и к введены статистические поправки).
и фенолов) довольно велики. Значительно больших отклонений следует
ожидать, если в этот ряд включить кислоты других типов, например оксимы,
енолы р-дикетонов, аци-формы нитроалканов или заряженные кислоты.
Это может означать либо то, что начинают играть роль структурные эффекты,
отличные от полярных эффектов, либо то, что не соблюдаются условия про-
порциональности полярных эффектов.
При детальном анализе данных можно прийти к выводу, что важной причи-
ной наблюдаемых отклонений являются пространственные эффекты. Про-
странственные эффекты ослабляют силу незаряженных кислот в основном
за счет уменьшения сольватации их анионов; они уменьшают скорость реак-
ции ввиду того, что при образовании переходного состояния бимолекуляр-
ной реакции происходит уплотнение по сравнению с исходным состоянием.
Но эти два эффекта, выраженные как разность энергий, не должны быть
пропорциональны друг другу и естественно не имеют коэффициента пропор-
циональности В, равного наклону линии на рис. 57. В муравьиной, уксусной
и пивалиновой кислотах пространственные эффекты, обусловленные после-
довательным введением метильных групп, оказывают более сильное влияние
на силу кислот, чем на скорость реакций, в результате чего точка для муравьи-
992
Полярные эффекты
ной кислоты находится справа, а точка для пивалиновой кислоты — слева
от линии, проходящей через точку для уксусной кислоты и имеющей наклон,
приведенный на графике. В замещенных бензойных кислотах пространствен-
ные эффекты opmo-заместителей влияют относительно более сильно на ско-
рость реакции, в результате чего точки, соответствующие орто-замещенным
бензойным кислотам, оказываются справа от линии, проведенной так, чтобы
на ней наилучшим образом лежали точки, соответствующие мета- и пара-
замещенным бензойным кислотам, и имеющей наклон, совпадающий с при-
веденным на рис. 57.
Другая общая причина отклонений, как уже отмечалось выше, состоит
в том, что представление электрической энергии полярного эффекта в виде
произведений двух величин, которое приводит к пропорциональным соот-
ношениям как раз потому, что оно является таковым, сильно идеализировано.
В принципе полярная энергия должна выражаться суммой произведений,
так как необходимо принимать во внимание, что свойства поляризации
и поляризуемости не зависят друг от друга. Из данных, полученных для этой
и других реакций, катализируемых кислотами или основаниями, следует,
что значительные отклонения наблюдаются в том случае, когда между
местом локализации структурных изменений и реакционным центром имеется
система сопряженных связей. Именно при таких обстоятельствах можно
ожидать значительных эффектов поляризуемости, которые приводят к появ-
лению новых дополнительных энергетических членов.
в. Аддитивность полярных эффектов при электрофильном замещении
в ароматическом ряду. Аддитивность полярных эффектов, наблюдающаяся
для определенных структурных серий в определенных реакциях, была
открыта также в 20-х годах. Впервые это открытие было сделано Брэдфилдом
и Бринмором Джонсом [5] в 1928 г. Авторы изучали реакцию хлорирования
арилалкиловых эфиров хлором в 99%-ной уксусной кислоте при 20 °C.
Эфиры имели следующие формулы:
Х\
ДО—______/~Х .R°-\ / RO—/________/~Х
где В и X были переменными, так что в молекуле имелись три положения,
в которых одновременно могли происходить структурные изменения. В каче-
стве алкильной группы можно было вводить бензильную группу, которая
сама могла содержать заместители в ароматическом кольце. Установлено,
что, если заместители в ароматическом ядре X сохранять одинаковыми
и изменять лишь природу алкильной группы, соотношения скоростей не
зависят от природы и положения заместителей X (табл. 213). Если группа R
ле меняется, а изменяется структура заместителя X или двух заместителей,
то соотношения скоростей не зависят от природы В (табл. 214).
Существование такой обратно пропорциональной зависимости между ско-
ростями при изменении одновременно присутствующих заместителей озна-
чает, что вклады, вносимые несколькими заместителями в свободную энергию
активации, аддитивны. Пометив заместители значком г, можно записать
следующее выражение:
6AG" = SSW
i
Это уравнение, называемое зависимостью Брэдфилда — Джонса, как изве-
стно, находит широкое применение при электрофильном замещении в арома-
тическом ряду; оно часто используется для приближенного вычисления
63-0772
993
Глава XVI
Таблица 213
Относительные скорости хлорирования арилалкиловых эфиров
ROCeH4X и ROCeH3X2 : влияние изменения R (по данным Брэдфилда и Джонса)
X или Хг R в КОСвЩХ или КОСбНзХг Бензил и замещенный бензил
СНз с2н5 H-C3H7 U30-G3H7 и-С4Нэ II п-С! 34-NO2 71-NO2
п-СООН 100 198 215 444 221 70
n-NO2 100 200 221
п-С1 100 199 225 439 222 67 394 16,0 14,2
о-С1 100 199 14,4
n-Br 100 200 227 438 68 16.0 14,1
o-Br 100 13,8
2,4-Cl2 199 68
2,4-Br2 100 200 64
факторов парциальных скоростей для замещения в ядре при наличии несколь-
ких заместителей; для этих расчетов необходимо знать факторы парциальных
скоростей для замещения в молекуле, имеющей только один заместитель.
Однако соотношение аддитивности не часто выдерживается с такой точно-
стью, какая наблюдалась в первой работе Брэдфилда и Бринмора Джонса;
в этой работе обсуждались относительные скорости, но, если перейти к лога-
рифмам скоростей, различие между данными, относящимися к одному заме-
стителю, еще более сгладятся.
Причину такого точного соответствия данной серии реакций принципу
аддитивности можно легко понять. Группы X обладают отрицательным
индуктивнным эффектом и расположены в лета-положении относительно
центра или центров электрофильного замещения. Их влияние будет обу-
словлено главным образом индуктивной поляризацией —I и связанным
с ней эффектом поля. Группы RO, напротив, обладают положительным
эффектом сопряжения и расположены в орто- и пара-положениях относи-
тельно центра или центров электрофильного замещения. Эти группы уча-
ствуют в активации почти полностью вследствие эффекта сопряжения -\-К,
и, следовательно, в основном проявляется эффект поляризуемости -\-Е
(гл. VI, разд. 3,в). Такое резкое различие в характере смещения электронов,
при помощи которого эти два вида групп вносят свой вклад в общую актива-
цию, способствует тому, что их вклады будут независимыми, а в терминах
полярных эффектов — аддитивными.
Таблица 214
Относительные скорости хлорирования арилалкиловых эфиров
ROCeH4X : влияние изменения X (по данным Брэдфилда и Джонса)
R Заместитель X
и-со2н 71-NO2 п-С1 о-С1 п-Вг
СН3 100 0,674 276 916 281
с2н5 100 0,682 278 925 284
м-СзН? 100 0,691 288 294
изо-С3Н7 100 272 279
и-С4П9 100 283
с6н5сн2 100 269 899 273
994
Полярные эффекты
г. Пропорциональность полярных эффектов ароматических мета-
и пара-заместителей. В 1935 г. Гаммет [6] и Буркхардт Г7] независимо друг
от друга открыли, что различия, вносимые мета- или иора-заместителями
в логарифмы констант равновесия или констант скоростей реакций в боковой
цепи ароматических веществ, пропорциональны для любых двух таких
реакций. Оба исследователя признали, что этой зависимости удовлетворяют
лишь мета- и /гара-заместители в бензольном
кольце, и указали, что opmo-заместители не
подчиняются общей закономерности. Оба ав-
тора подчеркивали эмпирический характер за-
висимости. Так случилось, что в обеих работах
в качестве стандартной реакции, для которой
можно было показать в пределах данной за-
висимости пропорциональность вкладов заме-
стителей в свободную энергию стандартной и
других реакций и поэтому пропорциональность
инкрементов заместителей между собой, выб-
рали равновесие ионизации бензойных кис-
лот в воде.
Рис. 58. Зависимость разности логарифмов констант
скоростей следующих реакций (ордината) от
разности констант кислотности бензойных ки-
слот в воде при 25 °C (абсцисса), обусловлен-
ной наличием мета- и пор «-заместителей [7].
1 — гидролиз в водной кислоте (49 сС) замещенных
фенилбисульфатов (Буркхардт); 2 — щелочной гид-
ролиз в воде (100 °C) амещенных бензамидов
(Райд); 3 — сольволиз в водном этиловом спирте
замещенных бензилхлоридов (Оливье); 4 — щелочной
гидролиз в водном этиловом спирте замещенных
этилбензоатов (Киндлер). Ординаты для каждой
реакции содержат произвольную константу.
Если символом 6О обозначить разницу, вносимую заместителем, и если кр —
константа скорости или константа равновесия какой-то реакции, а К —
константа равновесия ионизации бензойной кислоты, то описанная зависи-
мость может быть представлена следующим уравнением:
бо log Ap = p6CTIog^,
где р — константа, зависящая только от условий реакции.
На рис. 58 представлен один из графиков Буркхардта [7], на котором пока-
зано, что влияние мета- и пара-заместителей на логарифмы констант скоро-
стей ряда реакций в боковой цепи (его собственные или взятые им из лите-
ратуры) линейно коррелируется с логарифмами констант кислотности соот-
ветственно замещенных бензойвых кислот. На представленном графике
наклоны прямых линий соответствуют значениям р.
Не ясно, понимал ли в то время Буркхардт (хотя именно тогда была
опубликована работа, в которой предполагается причина этого [8]), почему
одна из полученных им точек, соответствующая сольволизу бензилхлоридов,
сильно выпадает из корреляции. Сольволиз самого бензилхлорида в водно-
органических растворителях с небольшим содержанием воды протекает по
механизму SN2, но всегда в пограничной области между механизмами SN2
63* 995
Глава XVI
и SN1 (гл. VII, разд. 2,6). Выпадение точки показывает, что при наличии
иара-метильного заместителя, являющегося донором электронов, происхо-
дит настолько сильное смещение механизма от SN2 в направлении SN1,
что «плохая» точка может быть первой точкой на новой, гораздо более крутой
линии, которую можно провести, прдолжив график влево *.
Во время этих исследований еще не было создано теории уравнения Гамме-
та — Буркхардта **. Гаммет постоянно подчеркивал (а в действительности
превозносил) эмпиризм этого уравнения [10]. Однако, как уже было показано
в разд. 1,а, его соотношение имеет теоретическое истолкование. В известном
смысле это соотношение всегда имело приведенный выше физический смысл,
поскольку, как уже объяснялось, истолкование подобного соотношения для
равновесия появилось в теории органической химии с середины 20-х годов,
а для скоростей реакции — с момента появления теории переходного состоя-
ния в 1932—1935 гг. Суть состоит в том, что если всеми эффектами, влияю-
щими на равновесие и скорость реакции, за исключением полярных эффектов,
можно пренебречь (что невозможно при наличии орто-заместителей) и если
каждый полярный эффект может быть представлен как простое произведение
одних и тех же двух электрических величин (что может не соблюдаться из-за
сильной поляризуемости), то должна наблюдаться пропорциональность между
такими произведениями и одним из входящих в них сомножителей, если вто-
рой сомножитель постоянен.
В 1940 г. Гаммет [5] представил зависимость, установленную им и Буркхар-
дтом, в таком виде, в каком она обычно с тех пор и используется. Разность 6О
соответствует различию между данным заместителем и водородом, взятым
в качестве стандарта (последний обозначается нулевым индексом).
1 og к — log к0 = р (log К — log Ко).
Множитель log (К/Ко) зависит только от природы заместителя и обозначает-
ся символом о, а р определяется только типом реакции
Iog I7 = p(J-
Кроме реакций в боковой цепи ароматических соединений, имеется другой
тип реакций, для которых наблюдается приблизительная пропорциональ-
ность между различиями в свободной энергии активации, вызванными введе-
нием мета- и пара-заместителей. К этому типу относятся реакции электро-
фильного замещения в ароматических соединениях. Для этих реакций про-
порциональность свободных энергий была впервые открыта Г. Брауном
и сотр. [11]. И снова opmo-заместители выпадают из корреляции. Если допу-
стить такую пропорциональность различий свободных энергий, например при
нитровании, и рассматривать скорости нитрования С8НЬБ в те положения, по
отношению к которым заместитель R является пара-заместителем, то можно
* Изменение крутизны прямой линии может бить не очень резким, поскольку следует
ожидать, что механизм не будет изменяться очень резко (гл. VII, разд. 2,а). Графики
с плавными кривыми, включающие несколько заместителей, обычно наблюдаются для
реакции SN2, протекающих около границы SN2 — SN1. Хорошим примером таких реак-
ций может служить реакция обмена замещенных бензгидрилтиоцианатов с тиоцианат-
ионом, которая в полярных органических растворителях протекает по конкурентным
Sn2- и Sjq-1-механизмам (оба около границы), причем первый дает исключительно график
с плавными кривыми, а второй — график с крутыми кривыми [9].
** Это соотношение (уравнение) почти всегда связывают лишь с именем Гаммета (не самим
Гамметом, который, как никто другой, имел на это больше всего прав), однако оно было
открыто двумя исследователями, хотя обозначение пр было предложено (позднее) Гам-
метом. (Конечно, каждое «уравнение» можно выразить как словами, так и символами !
996
Полярные эффекты
написать следующее уравнение:
ьнитр
IogT^r = log/“T₽ = ^PHnTp
%-Н
в котором используются факторы парциальных скоростей / (гл. VI, разд. 3,6).
Подобное же уравнение можно написать для скорости нитрования CeH5R
в те положения, по отношению к которым заместитель R является мета-
заместителем. Затем, разделив по частям одно уравнение на другое, можно
получить уравнение, приведенное ниже, в правую часть которого уже не
входит параметр, свидетельствующий о том, что речь идет именно о нитро-
вании, так как рнитР сокращается. Это так называемое «уравнение вкладов»
log fn _
log fM нм
связано с природой заместителя, но не с типом реакции. Для данного замести-
теля левая часть уравнения будет, следовательно, одинаковой для нитрования,
галогенирования, меркурирования, алкилирования по Фриделю — Крафтсу
и других реакций. Это уравнение можно записать в другом виде. Посредством
вычитания обратной величины каждой части уравнения из единицы и замены
функции пп/(оп — сти), которая находится из констант ионизации бензойной
кислоты, одной константой 6r, можно получить следующее уравнение:
log- fn = bR log
J Ml
Это уравнение известно как соотношение селективности Брауна. Индекс R
у константы пропорциональности Ъ напоминает о том, что она зависит от
химического характера (а не положения) заместителя. Однако при одном
и том же заместителе уравнение в целом не связано с типом реакции.
Качественно это свидетельствует о том, что чем сильнее заместитель изменяет
скорость реакции, тем сильнее он будет изменять соотношение скоростей
мета- и пара-замещения или, другими словами, соотношение продуктов
реакции.
Что касается количественной ценности этого соотношения, то оно соответ-
ствует данным по кинетике реакций толуола (R = СН3) и примерно 50 его
замещенных (одинаковые по стехиометрии реакции замещения, проводимые
в различных растворителях, рассматриваются как разные процессы замеще-
ния). Константа селективности &сн3 имеет среднюю величину 1,31 ± 0,10.
Стандартное отклонение (±8%) превышает экспериментальные погрешности.
Если вычислить Ъ исходя из соотношения Ъ = пп/(стп — ол(), то константа
селективности составит 1,68. Эта величина на 27% больше требуемой для
наилучшего совпадения с экспериментальными данными. Преобразование
«уравнения вкладов» в уравнение Брауна уменьшает погрешность. При не-
посредственном использовании «уравнения вкладов» можно найти, что отно-
шение log/n/log/^, которое должно быть постоянным, имеет среднюю величи-
ну 4,0 + 0,5. Стандартное отклонение составляет в этом случае 12,5%. Если
вычислить эту константу из соотношения ап1бм, то получится величина 2,6,
плохо совпадающая с опытной величиной, равной 4,0.
Легко догадаться, почему данные по влиянию мета- и пара-заместителей на
скорость замещения в боковой цепи ароматических соединений и на скорость
электрофильного замещения в ароматическом ядре так сильно не совпадают
при сопоставлении. Обычно и в принципе вклад полярных эффектов замести-
теля в свободную энергию реакции или активации определяется не простым
произведением двух факторов, а суммой нескольких произведений, отличаю-
997
Глава XVI
мета-Положение
о I I 0,111 0,341 0,37 1—0,071—0,101 0,431 0,37 I 0,38 I 0,56 I 0,52 I 0,71 11,76
о* I I 0,05| 0,33| 0,40 I—0,07|—0,0б| О,52|о,37| ]0,5б| |о,67|
пара-Положение
(5 -0,83 -0,27 0,06 0,23 —0,17 -0,20 0,54 0,45 0,50 0,66 0,49 0,78 1,91
а4' —1,7 -0,78 -0,07 0,11 -0,31 -0,26 0,61 0,48 0,65 0,79
О~ 0,67 0,87 1,00 1,05 1,27 3,2
Разность для мря-положений
+ о V о о 0,9 0,51 0,13 0,12 0,14 0,06 -0,07 0,22 0,37 0,34 0,56 0,49 1,3
щихся тем, что величины множителей каждого произведения зависят различ-
ным образом от природы заместителя и от типа реакции. Наиболее простая
ситуация возникает тогда, когда, например, индуктивный эффект и связан-
ный с ним эффект поля настолько преобладают, что другими полярными
эффектами можно пренебречь или, по крайней мере, исключить их посред-
ством соответствующей «обработки» констант, которые должны описывать
лишь влияние самого большого члена суммы произведений. Очень многие
реакции в боковой цепи ароматических соединений приблизительно соот-
ветствуют такой ситуации, хотя во многих случаях наблюдается несовпаде-
ние с ней. Однако при электрофильном замещении в ароматическом кольце,
особенно при наличии пара-заместителей, очень часто наблюдаются значи-
тельные отклонения. Причиной такого различия является то, что при заме-
щении в ядре сопряжение между заместителем и реакционным центром может
быть сильнее выражено, чем в реакциях с участием боковой цепи. При электро-
фильном ароматическом замещении влияние сопряжения проявляется
в основном в виде эффекта поляризуемости (+2?) (гл. VI, разд. 3,в). При
тщательном анализе этого вопроса можно прийти к выводу, что в простейшей
форме эффект поляризуемости вызовет необходимость учета второго энерге-
тического члена, который будет отличаться от первого, и, следовательно, его
зависимость от природы заместителя и типа реакции будет иной. Сумма двух
отличающихся друг от друга энергетических членов, если они сравнимы по
величине, не может быть представлена одним энергетическим членом, в кото-
рый входят две константы.
Исторически сложилось так, что эти и другие подобные затруднения пыта-
лись обойти созданием множества альтернативных рядов констант а (не все
эти константы будут обсуждаться в настоящей главе),.каждый из которых
должен использоваться в какой-либо особой ситуации, а также новых кон-
стант р, соответствующих каждому набору о. Этот метод можно критиковать,
поскольку слишком много усилий было потрачено на сведение опытных дан-
ных воедино, хотя, согласно общепринятому взгляду, сама теория является
слишком приближенной. Несмотря на это, в первом приближении описан-
998
Полярные эффекты
ный выше метод вполне применим, и эти наборы констант способствуют
получению информации для более строгой формулировки.
д. Отклонения полярных эффектов от пропорциональности в случае
ароматических мета- и пара-заместителей. В табл. 215 представлены
примеры констант заместителей для случая мета- и пара-ароматических
заместителей *. Сначала рассмотрим лишь немодифицированные о-константы,
приведенные в верхних строках первого и второго разделов таблицы. Следует
начать с оценки физического смысла этих констант. Набор таких констант,
будучи умноженным на константу р для какой-то данной реакции, описывает
(только для этой реакции) пределы изменения под влиянием заместителей
констант равновесия или скоростей в логарифмической шкале. Константа р
является мерой широты изменения скорости этой реакции относительно
широты изменения констант ионизации бензойной кислоты под влиянием
тех же заместителей (стандартная реакция, для которой р берется равной
единице). Электроотрицательные заместители увеличивают кислотность бен-
зойных кислот, и, следовательно, алгебраически положительные п-константы
выражают электроотрицательность, а алгебраически отрицательные ст-кон-
станты — электроположительность заместителей, за исключением заместите-
лей класса —I, -\-К (и +/, —К', примеры не известны), которые имеют
двоякую полярность. Следует отметить, что метокси-заместитель, относящий-
ся к типу —I, +К, имеет положительную величину о (т. е. является электро-
отрицательным) в .мета-положении, в котором не может происходить сопря-
жения с реакционным центром, и отрицательную величину о (т. е. является
электроположительным), находясь в пара-положении, так как в последнем
случае влияние сопряжения может распространяться достаточно близко
к реакционному центру (если не непосредственно до него). Очевидно, сопря-
жение играет незначительную роль в первом случае и большую роль — во
втором. Положение галогенных заместителей, относящихся к типу —I, +К,
•оказывает менее значительное влияние. В этом случае положительный знак ст
в обоих положениях связан с индуктивной электроотрицательностью, но
увеличение влияния сопряжения в пара-положении уменьшает положитель-
ную величину а. Однако не следует рассматривать разность пара- и мета-
констант как меру сопряжения, так как из разных положений по-разному
будут передаваться индуктивный эффект и эффект поля, что в любом случае
вызовет различие в величинах о для этих положений.
Для того чтобы закончить рассмотрение области применения уравнения
Гаммета — Буркхардта, следует рассмотреть данные, представленные в верх-
ней части табл. 216. В этой таблице приведены некоторые константы реакций
для соединений с мета- и пара-заместителями. Число приведенных констант
невелико, так как многие константы реакции уже приводились. Для одной
и той же реакции, но в другом растворителе, требуется иная константа.
В верхней части таблицы представлены значения р, первоначально получен-
ные при использовании немодифицированных констант заместителей о.
Первой приводится взятая за основу при сравнении реакция — равновесие
ионизации бензойных кислот. Так как для этой реакции величина р принята
равной единице, другие реакции, которые промотируются электроотрицатель-
ными заместителями, будут иметь положительные значения р, а те реакции,
которые ускоряются электроположительными заместителями, будут иметь
отрицательные значения р. Отсюда легко понять, почему наблюдаются
характерные отличия между приведенными значениями, например, почему
* Константы, представленные в табл. 215, и связанные с ними константы реакции, при-
веденные в табл. 216, почти все взяты из работ, указанных в ссылке [12].
999
Глава XVI
Таблица 216
Некоторые константы реакции, полученные при использовании констант
заместителей мета- и пара-замещенных ароматических соединений
(Е — равновесие, R — скорость)
Ароматическое соединение E или R Реакция Константа реакции
АгСОН E Величины p при использовании о Депротонизация, вода, 25 °C (1,00)
АгО- R Реакция с C2H5I(SN2), этиловый спирт, 42 °C -0,99
АгСН2С1 R Реакция с I~(SN2), ацетон, 20 °C 0,78
AtNH2 R Реакция с СвН3СОС1(Зх2), бензол, 25 °C —2,78
ArCOCl R Реакция с C6H5NH2(Sn2), бензол, 25 °C 1,22
ArCOOC2Hs R Щелочной гидролиз (ВАг,2), 85%-ный этиловый 2,46
ArCOOH R спирт, 35 °C Этерификация (AAq2), этиловый спирт, 25 °C -0,47
АгОСОСНз R Кислотный гидролиз (AAq2), 60%-ный ацетон, 25 °C -0,20
ArC0NH2 R Щелочной гидролиз (BAq2), 60%-ный этиловый 1,36
ArCONH2 R спирт, 53 °C Кислотный гидролиз (AAq2), 60?4-ный этиловый -0,48-
ArC(CH3)2Cl R спирт, 52 °C Величины р при использовании <т+ Сольволиз (SN1), 90%-ный ацетон, 25 °C -4,52
Ar3COH E Ионизация сопряженной кислоты, вода, 25 °C -3,64
Ar(CeH5)2CCl R Сольволиз (SN1), 40% С2Н5ОН + 60% (С2Н5)2О, 0 °C —2,34
ArCH = CHCOOH R Присоединение С12, уксусная кислота, 24 °C -4,01
ArH R Нитрование, HNО3, CH3NО2 или (СН3СО)20,0 или 25 °C —6,22
ArH R Бромирование, HOBr-j-H-1-, 50%-ный диоксан, 25 °C —5,78
ArH R Хлорирование, С12, уксусная кислота, 25 °C -8,06
ArH R Бромирование, Вг2, уксусная кислота, 25 °C -12,14
ArOH E Величины р при использовании <Г“ Депротонизация, вода, 25 °C 2,01
ArNHf E Депротонизация, вода, 25 °C 2,77
ArCH2CH2Br R Реакция с С2Н5О~, этиловый спирт, 30 °C 2,15
o-CeH4NO2CI R Реакция с СН3О~, метиловый спирт, 25 °C 3,90
знак р изменяется при изменении заместителей в арильной группе при заме-
щении типа SN2 и почему положительные значения, характерные для ВАс2-
механизма гидролиза сложных эфиров и амидов, уступают место отрицатель-
ным значениям (в общем случае или менее положительным, или отрицатель-
ным) при переходе к соответствующему механизму ААс2.
Окамото и Браун [12] определили два больших класса реакций, на равно-
весие и скорость которых мета- и пара-заместители в ароматическом кольце
влияют в такой степени, что обнаруживаются большие и систематические
отклонения от значений констант скоростей, вычисленных как произведе-
ния рст. Один класс реакций включает реакции в боковой цепи ароматиче-
ских соединений, которые в состоянии равновесия приводят к образованию
а-карбониевого ионного центра или скорость которых контролируется обра-
зованием а-карбониевого ионного центра. В качестве примеров можно при-
1000
Полярные эффекты
вести равновесие образования триарилметильных катионов, скорость соль-
волиза по S j^l-механизму бензильных, бензгидрильных и триарилметильных
соединений, а также присоединение в полярных растворителях молекуляр-
ного хлора к коричной кислоте и стирилкетонам. Другой класс реакций
включает электрофильное замещение в ароматическом ряду. На стадии,
определяющей скорость этих реакций, замещающий реагент занимает такое
положение, что само кольцо принимает ионный карбониевый заряд. Харак-
тер отклонений одинаков для обеих групп примеров, мета- и гаара-Замести-
тели, не способные к сопряжению, не дают значительных отклонений; то же
самое можно сказать об электроотрицательных иора-заместителях, способ-
ных к сопряжению (например, NO2)- Однако в случае электроположительно
сопряженных иара-заместителей (например, СН3О) наблюдаются отклонения,
которые, как показывают факты, увеличиваются в зависимости от силы,
с которой заместитель способен к сопряжению. Отклонения всегда наблю-
даются в одном и том же направлении, а именно: реакция протекает в более
полной степени или быстрее, чем это предсказывает зависимость Гаммета —
Буркхардта.
Окамото и Браун выяснили, что отклонения возникают при наличии сопря-
жения между гаара-з вместите лями и образовавшимся или образующимся
карбониевым ионом: при наличии сопряжения к свободной энергии реакции
или к свободной энергии активации добавляется отрицательный член. Ока-
мото и Браун решили скомпенсировать эти отклонения, предложив новое
уравнение, сформулированное на основе ро-зависимости, но применимое
специально для реакций ароматических а-карбониевых ионов и электро-
фильного замещения в ароматическом ряду. Это уравнение не обязательно
должно быть соотнесено с какой-то реакцией за пределами сферы его при-
менимости, и, следовательно, в качестве стандартной реакции, определяю-
щей константы заместителей, можно выбрать любую реакцию внутри данного
класса, а не обязательно ионизацию бензойных кислот. Следует напомнить,
что константы селективности Брауна плохо коррелируются со значениями о,
выведенными на основе ионизации бензойных кислот. Корреляция экспери-
ментальных данных с новыми константами заместителей, основывающимися
на внутреннем стандарте, как это можно видеть, действительно гораздо лучше.
В качестве внутренней стандартной реакции была выбрана первая из реак-
ций, представленных в средней части табл. 216, а именно гидролиз кумил-
хлорида (а,а-диметилбензилхлорида) в указанных в таблице условиях.
Однако в качестве константы реакции в этом случае была выбрана величина
не +1, а —4,52, так как при таком выборе константы заместителей, обозна-
ченные символом о+ и выведенные из влияния заместителей на скорость
указанной реакции, минимально отличаются от констант о для лета-заме-
стителей, как это показано в верхней части табл. 215. Для гаара-заместите-
лей, как показано в средней части таблицы, также имеется значительное
совпадение между о+ и о, за исключением электроположительно сопряжен-
ных (+Х) заместителей. В случае гаара-заместителей различие между сг+
и п велико для амино- и оксигрупп, мало для галогенов и метильной группы
(а также для первичных и вторичных алкильных групп) и еще меньше (вплоть
до ничтожно малой величины) для других групп. Разность между значе-
ниями п+ и о приводится в нижней части таблицы. Эта разность может рас-
сматриваться как мера силы сопряжения заместителей.
С помощью вычисленных таким образом значений сг+ были рассчитаны вели-
чины р для других реакций в боковой цепи ароматических соединений, иду-
щих через а-карбониевые ионы, а также для реакций электрофильного заме-
щения в ароматическом ряду (средняя часть табл. 216). Все эти реакции вме-
1001
Глава XVI
сте с принятой за основу реакцией гидролиза кумилхлорида облегчаются при
подаче электронов к реакционному центру, т. е. электроположительными
заместителями, и, следовательно, для всех этих реакций значения р являются
отрицательными. Особенно велики по абсолютной величине значения р для
реакций электрофильного ароматического замещения. Для этих реакций
можно наблюдать осуществление принципа де ла Мара (гл. VI, разд. 4,в),
согласно которому электромерные эффекты (+Е) заместителей в пара-
положении оказываются более сильными при электрофильном замещении
о
Рис. 59. Зависимость log (к^^/к-ц) от а
и а+ для пара-заместителей.
Приведены скорости этанолиза
п-монозамещенных трифенил-
метилхлоридов в смеси 40%
этилового спирта и 60% ди-
этилового эфира при 0 °C [13].
1 — сн,о; г — сн3; з — Н; 4 —
F; s — ci; в — no3.
незаряженными переносчиками элект-
рофила по сравнению с катионными
переносчиками того же входящего за-
местителя.
Из верхней диаграммы на рис. 59 сле-
дует, что, для того чтобы на графиках
Гаммета — Буркхардта не было от-
клонений от линейности, связанных
с электроположительно сопряженны-
ми «ара-заместителями, необходимо
несколько изменить некоторые парамет-
ры. На нижней диаграмме показано,
насколько хорошо замена и значениями
сг+ может устранить эти отклонения.
Однако сразу же заметим, что этот при-
мер был выбран только для иллюстра-
ции: константы сг+ не всегда так хоро-
шо исправляют дело, как это можно
видеть.
Имеется другой, менее тщательно иссле-
дованный, но такой же большой класс
реакций, который характеризуется си-
стематическими отклонениями от рп-ли-
нейности. В этом случае отклонения
наблюдаются на другом конце гра-
фика рсг—на том конце, который соот-
ветствует электроноакцепторным (—I,
—К) заместителям. Отклонения опять-
таки существенны только для пара-
заместителей, и снова эти отклонения проявляются в том, что некоторые
заместители действуют более сильно, чем это допускает произведение рсг.
Гаммет впервые отметил такой характер отклонений для случая нитрогуппы.
Затем в 1940 г. он предположил, что эти отклонения могут быть обнаружены
на более широком ряде заместителей, что и было впоследствии найдено
Робертсом, Бордуэллом и др. [14]. Вопрос был решен созданием еще одного
ряда констант, применяющихся в тех случаях, когда наблюдаются отклоне-
ния и когда они бывают существенными. Некоторые из этих констант, соб-
ранные Джаффе [12] и обозначенные значком сг“, представлены во второй
части табл. 215. Эти величины применимы для «ара-заместителей: если откло-
нений не наблюдается, то о" могут рассматриваться как идентичные констан-
там а, а для всех лсеша-заместителей о- могут считаться равными п.
Все факторы, способствующие отклонению этой группы реакций от простой
зависимости Гаммета — Буркхардта, свидетельствуют о том, что отклоне-
ния также возникают лишь при наличии сопряжения между заместителем
и реакционным центром. Это было признано всеми более поздними исследова-
1002
Полярные эффекты
телями. Факты состоят в следующем. Отклонения наблюдаются для замести-
телей в иара-положении. Все они относятся к классу —I, —К, т. е. как
вследствие эффекта сопряжения, так и вследствие индуктивного эффекта
являются электроотрицательными. Во всех случаях, в которых наблюдают-
ся отклонения, реакции промотирутся смещением электронов от реакцион-
ного центра, т. е. значения р всегда являются положительными. Кроме того,
все значения о~ положительны и будут больше соответствующих положи-
тельных значений констант о (если они отличаются от последних). Важнее
всего то, что эти реакции относятся к двум
большим классам, которые, очевидно, яв-
ляются противоположной копией тех двух
классов реакций, для которых были вве-
дены константы сг+. Один из классов ре-
акций, к которым применимы константы
о~, включает реакции в боковой цепи
ароматических соединений, в которых
-обычно (но не обязательно) путем отщепле-
ния протона а-атом боковой цепи приоб-
ретает отрицательный заряд и пару элект-
ронов, которая может сопрягаться через
кольцо с электроноакцепторным пара-
.заместителем. К этой категории реакций
относится депротонирование фенолов и
ионов анилиния. Прототропная а-депро-
тонизация, например, фенилнитрометана
должна также относиться к этой кате-
гории реакций, хотя (насколько это из-
вестно автору) эта область применения
констант о- еще не была проиллюстриро-
вана. Бимолекулярное отщепление, вклю-
чающее а-депротонизацию и приводящее
к образованию стиролов, как показали
•Сондерс и Уильямс, относится к этой же
категории реакций в боковой цепи [15].
Рис. 60. Зависимость log &n_R от о
и о- для пара-заместите-
лей. Константы скорости
^п-R бимолекулярного от-
щепления олефинов от па-
ра-замещенных 2-фенил-
этилбромидов при реакции
с этилат-ионами в этиловом
спирте при 30 °C [15].
1 — СН3О; 2 — СН3; 3 — Н;
4 — С1; 5 — СН3СО; в — NO3.
О зависимость от ст; ф зави-
симость от ст-.
Другой большой класс реакций включает бимолекулярные реакции нуклео-
фильного замещения в ароматическом ряду. Принадлежность этих реакций
к тому же самому классу по типу отклонений от ри-зависимости была
показана Баннетом и его коллегами [16]. Примеры реакций, относящихся
к указанным двум классам, приведены в третьем разделе табл. 216.
Обычно считают, что эти отклонения от линейности ро-зависимости связаны
с эффектами электроотрицательного сопряжения иара-заместителей. Раз-
ность ц” — о, представленная в последней строке табл. 215, может соот-
ветственно рассматриваться как мера силы сопряжения заместителей —ТГ-ти-
па. На рис. 60 приведен пример, когда плохая ро-корреляция в значитель-
ной степени улучшается использованием вместо и констант о~.
е. Энергия полярных эффектов ароматических мета- и пара-заме-
стителей как сумма произведений. Рассмотрим теперь более детально две
главные группы реакций, в которых наблюдаются отклонения от пропорцио-
нальности свободных энергий, и прежде всего те, в которых наблюдается от-
клонение точек, соответствующих иара-заместителям, обладающим электро-
положительным эффектом сопряжения. Эти отклонения в очень заметной
степени проявляются в том районе ро-кривых, которые соответствуют электро-
положительным заместителям (верхняя диаграмма на рис. 59). В этом случае
1003
Глава XVI
применяются константы Окамото и Брауна. Мы используем все время кон-
станты сг+ Окамото и Брауна, так как они применяются значительно чаще,
чем какие-либо другие константы п+. Однако другими исследователями пред-
ложен ряд альтернативных наборов значений и+, каждый из которых имеет
свой собственный ряд значений р. Тот простой факт, что имеется несколько
альтернатив, показывает, что константы Окамото и Брауна в общем случае
не должны рассматриваться как постоянные числовые величины.
Такое отсутствие общей универсальности шкалы п было отмечено в 1959 г.
ван Беккумом, Феркаде и Вепстером [17], а также Юкава и Цуно [18]. Обе
группы исследователей показали, что если в том районе ро-кривой, который
соответствует электроположительным сопрягающимся иара-заместителям,
обнаруживаются положительные отклонения, как показано на верхней
диаграмме на рис. 59, то замена константы п на константу сг+ Окамото и Бра-
уна может привести как к тому, что точки аберрации лягут на линию, как
это показано на этом рисунке, так и к тому (и это чаще всего бывает), что
эти точки не лягут на нее. В том случае, когда отклонения больше приведен-
ных на рисунке, поправка на п+ приведет к размещению точек ближе к ли-
нии, а если отклонения меньше, чем показано на рисунке, то эта поправка
приведет к перемещению точек по другую сторону линии. Для разных реак-
ций степень отклонения от ро-корреляции различна, т. е. наблюдается спектр
отклонений. Ван Беккум, Феркаде и Вепстер построили график, в общем
эквивалентный графику, который можно получить наложением (при этом
надо изменить масштаб по осям) друг на друга прямолинейных участков-
нескольких частично линейных рсг-кривых, подобных кривой, представлен-
ной в верхней части рис. 59. Полученная таким образом диаграмма наложе-
ния кривых имеет вид веера, ручка которого является продолжением одной
из спиц. Влияние сг+-поправки заключается в том, чтобы наклонить опахало
веера таким образом, чтобы ручка была на одной прямой со спицей, находя-
щейся в середине веера.
Юкава и Цуно пошли дальше. Как отмечал профессор Вепстер в своем пись-
ме, присланном им в то время автору, они калибровали створ спиц веера по-
средством поперечной шкалы, при помощи которой численно определяется
положение спиц веера. Это равносильно введению новой константы реакции,
обозначенной символом г, которая характеризует боковое отклонение, т. е.
отклонение от линейности, которое характерно для данной реакции. Вели-
чина г принимается за единицу, когда замена константы о на константу сг+
Окамото и Брауна точно корректирует отклонения. Следовательно, величина
г равна единице для стандартной реакции Окамото и Брауна — гидролиза
кумилхлорида в указанных выше условиях. Отсюда следует, что для этой
реакции поправка в величину о составляет о+ — о. В общем случае поправка,
которую необходимо ввести, равна г(а+ — а). Константа г может быть мень-
ше или больше единицы. Величины г, найденные из опыта, колеблются от
0,2 до более чем 2. При использовании таких символов уравнение Юкава —
Цуно имеет следующий вид:
logT7 = p{a + r^o+~a)}-
Некоторые константы реакции г представлены в табл. 217. Естественно,
параметр г имеет большую величину только для ияра-заместителей, так как
для .мета-заместителей о+ равны о, и поэтому в уравнение г не входит.
Это уравнение хорошо описывает кинетические и термодинамические эффек-
ты мета- и иара-заместителей, но не opmo-заместителей. Юкава и Цуно
в статьях, посвященных этому уравнению, исследовали 35 реакций. Для
1004
Полярные эффекты
Таблица 217
Некоторые константы реакции уравнения Юкава — Цуно
(Е — равновесие, R — скорость)
Ароматическое соединение Е или R Реакция р Г
АгС(СН3)2С1 и Гидролиз, 90%-ный водный ацетон, 25 °C —4,52 (1,00)
АгВ(ОН)2 R Бромдеборирование, 20%-ная уксусная кислота, 25 °C -3,84 2,29
Аг2СНС1 И Метанолиз, метиловый спирт, 25 °C —4,02 1,23
АгН R Бромирование, НОВг+Н+, 50%-ный диок- сан, 25 °C -5,28 1,14
АгН R Нитрование, HNO3, CH3NO2 или СН3СООН, 25 °C -6,38 0,90
АгС(С6Н5)2С1 R Этанолиз, 40% С2Н5ОН + 60% (С2Н5)2О, 0°С -2,52 0,88
ArN=NCeH5 Е Основность, 20%-ный этиловый спирт, 25 °C —2,29 0,85
ArCOCHN2 R Кислотное разложение, уксусная кислота, 40 °C -0,82 0,56
AtC(CH3) = NOH ОН СН3 R Перегруппировка Бекмана, 94%-ная H2SO4, 51 °C -1,98 0,43
АгСНСН = СН R Анионотропия, 60%-ный диоксан, 30 °C —4,06 0,40
Ar2CN2 R Разложение С6Н5СООН, толуол, 25 °C -1,57 0,19
всех этих реакций были получены хорошие линейные графики зависимости
logfc или logl£ от яф г(ц+ — а), настолько же линейные, насколько линеен
нижний график, представленный па рис. 59, который линеен потому, что для
этой реакции величина г случайно оказалась близкой к единице. Однако,
судя по данным табл. 217, величина г может сильно изменяться. Уравнение
Юкава — Цуно с большим успехом использовали другие исследователи,
особенно Иборн [19]. Можно сказать, что уравнение Юкава — Цуно успеш-
но применимо потому, что в него входят четыре константы. Однако они
не обязательно должны определяться все одновременно; сначала, как обычно
и делается, могут быть найдены аир совместно, затем о+ и, наконец, г по-
рознь из неперекрывающихся данных, с помощью которых можно опреде-
лить индивидуальные константы.
В табл. 217 также показано, что Окамото и Браун слишком узко определили
границы той группы реакций, для которой в качестве первого шага необхо-
димо использовать константы о+. Вовсе не обязательно, чтобы конечное
состояние в случае равновесия или переходное состояние в случае необра-
тимой реакции имело карбониевый ионный центр в бензольном кольце или
на атоме, непосредственно связанном с кольцом. Например, отщепление
азота от ароматических производных диазометана скорее связано с образо-
ванием карбеноидного, а не карбониевого ионного интермедиата. Реальной
необходимостью является лишь то, что конечное или переходное состояние
должно содержать электронодефицитный центр любого типа, который дол-
жен быть достаточно сильным для того, чтобы эффективно возбуждать актив-
ное сопряжение геара-заместителя, способного к сопряжению с электрополо-
жительным центром.
1005
Глава XVI
Однако особый интерес уравнение Юкава — Цуно вызывает тем, что оно
является первым уравнением, предложенным для учета влияния полярных
эффектов заместителей на реакционную способность, которое соответствует
положениям теории органической химии, разработанным и появившимся
в середине 20-х годов.
Все исследователи, столкнувшиеся с отклонениями от простых зависимостей
типа произведений per, признают, что эти отклонения связаны главным обра-
зом с сопряжением. Что касается того, каким образом действует сопряжение,
то можно вспомнить два теоретических соображения, выдвинутых в 1927 г.
Первое заключается в том, что полярный эффект в общем включает как
поляризацию, так и поляризуемость. При поляризации заместитель создает
потенциальное поле, под влиянием которого реагент и, следовательно, реаги-
рующая система претерпевают энергетическое изменение. Это изменение энер-
гии может быть в наипростейшем приближении представлено произведением
константы, характеризующей потенциал поля в окончательном или критиче-
ском положении реагента, в соответствии с тем, рассматриваются ли равно-
весные или необратимые реакции, и константы, характеризущей эффектив-
ный заряд, т. е. пространственное распределение зарядов, переносимых
реагентом (в таких интимных ситуациях концепция точечных мультиполей
является несостоятельной). Поляризуемость обусловливает появление дру-
гого энергетического члена совершенно иной структуры. Множитель, учи-
тывающий влияние заместителя, будет совершенно другим, поскольку он
теперь связан с поляризуемостью. Множитель, учитывающий тип реакции,
также будет другим, так как эффективный заряд реагента будет влиять на
поляризуемость, создавая дополнительный потенциал; при этом в реагенте-
и, следовательно, в реагирующей системе будет происходить изменение энер-
гии под влиянием созданного таким образом дополнительного потенциала.
В этом приближении, считая, что поляризуемость является симметричным
тензором, не зависящим от поля и выражающим гармоническое смещение-
электронов, можно написать необходимую зависимость в виде двучлена
с четырьмя константами:
Л к
log или log -j- = Poffo + Р+а+-
Л-о «о
Это уравнение представляет собой уравнение Юкава — Цуно, выраженное-
при помощи иных символов.
Другим давно известным представлением, о котором следует вспомнить,
является представление о связи между принципом полярной двойственности
и сопряжением. Почему сопряжение не может «смешиваться» с индукцией?
Почему при наличии сопряжения принцип полярной двойственности прояв-
ляется особенно ясно? Причина заключается просто в том, что сопряжение,
обусловливая подвижность наиболее слабо связанных электронов в молекуле
и, следовательно, в реагирующей системе молекул, сильно увеличивает ее
поляризуемость. В количественном смысле сопряжение уменьшает энергию
возбуждения. Так как поляризуемость представляет собой сумму членов,
каждый из которых обратно пропорционален энергии возбуждения, то даже
небольшая энергия возбуждения дает большой и часто даже очень большой
вклад в поляризуемость (гл. II, разд. 3,ж). Следовательно, как было показа-
но экспериментально в 1927 г. для полярных эффектов при электрофильном
замещении в ароматическом ряду, заместитель, который обеспечивает увели-
чение степени сопряжения в ходе реакции, сильно увеличивает вклад поляри-
зуемости. Отсюда следует, что маловероятно, чтобы второй член уравнения
Юкава и Цуно, даже если при отсутствии заместителя, обладающего свой-
1006
Полярные эффекты
ством способствовать сопряжению в ходе реакции, он ничтожно мал, был
ничтожно малым при наличии такого заместителя (ср. гл. II, разд. 3,а
и гл. VI, разд. 3,в).
Было бы неправильным считать, что в той группе реакций, в которой наблю-
даются отклонения для электроотрицательных заместителей и поэтому при-
меняются или могут применяться поправки типа о-, в каждом случае
в конечном или переходном состоянии возникают карбанионные или какие-
либо другие полностью сформированные основные центры в бензольном
кольце или на атоме, непосредственно связанном с ним. Например, образо-
вание стиролов, показанное в последнем разделе табл. 216, представляет
собой реакцию Е2, а не реакцию ElcB, т. е. в этой реакции не происходит
промежуточного образования карбанионов. Реальным условием для про-
явления наблюдаемых отклонений является то, что конечное или переходное
состояние должно содержать электронный центр любого типа, достаточно
активный, чтобы эффективно возбуждать электроотрицательное сопряжение
ипрп-заместителя.
Наконец, в основном в целях предсказания вновь рассмотрим отклонения
от линейности pa-зависимости, которые наблюдаются в большой группе
реакций при наличии электроотрицательных иара-заместителей, способных
к сопряжению, и которые особенно велики в случае заместителей (например,
NO2), расположенных у конца рсг-кривых, соответствующего электроотрица-
тельным заместителям. Джаффе [12] предложил для устранения этих откло-
нений применять другие константы заместителей, которые здесь называются
константами и-. Можно считать исторически совершенно случайным тот
факт, что ранее всего были открыты константы сг+, с помощью которых уда-
лось устранить отклонения, наблюдавшиеся для электроположительных
групп, и что именно для этого случая достаточно тщательно исследовалась
дисперсия этих отклонений, которые вначале считались довольно постоян-
ными, чтобы их можно было скомпенсировать однозначными константами сг+.
В результате открытия дисперсии отклонений появилось уравнение Юка-
ва — Цуно. Поэтому следует ожидать, что подобное тщательное исследование
электроотрицательных отклонений для таких реакций, к которым примени-
мы константы о-, приведет к обнаружению аналогичной дисперсии *. Такие
наблюдения могли бы дать эмпирическое обоснование расширенному уравне-
нию Юкава — Цуно
К к
log-=- ИЛИ log -Г- = р {о + г+ (о+ — о) 4- г~ (и- — о)} =
А о #0
= р {о + г (сг+ + ст — 2сг)}
Вторая, более простая форма этой зависимости соответствует случаю, ког-
да г+ и г~ равны между собой, так что в уравнение входит лишь одна кон-
станта г.
Справедливость расширенного уравнения такого типа предсказывается
теоретически. Поскольку поляризуемость, проявляющаяся во время реак-
ции и действующая на коротких расстояниях, вовсе не является не завися-
щим от поля тензором однородной поляризуемости, электронные смещения
могут быть в значительной степени ангармоническими. С 1927 г. известно,
что эффекты поляризуемости имеют двойственную природу (гл. II, разд. 3
* Такое исследование уже начато: скорости основного гидролиза фениловых эфиров,
имеющих пара-заместители типа —К, коррелируются с помощью коэффициентов о,
имеющих вид о + г~ (ц- — о), где г~ = 0,2 — 0,3 [20]. В этом случае роль сопряжения
такова же, как и роль сопряжения, связанная с кислотностью фенолов.
1007
Глава XVI
и гл. VI, разд. 3,в). Следовательно, нельзя думать, чтобы можно было в еди-
ном уравнении представить влияние поляризуемости заместителя менее чем
двумя константами, соответствующими двум видам электрополярности.
Поэтому, развивая предыдущий аргумент, можно получить следующее
уравнение с тремя слагаемыми:
log^ или 1о^’^’ = РоОго + р+т+ + р_о” = Ро(зго + р± КтИ
в которое входит лишь пять констант, если р+ и р~ нет необходимости диф-
ференцировать *. Это уравнение аналогично уравнению Юкава — Цуно,
приведенному выше. Вероятно, уравнения такого типа будут применяться
в будущем.
ж. Пропорциональность полярных эффектов пространственно удален-
ных алифатических заместителей. Три предшествующих раздела ограничи-
вались рассмотрением влияния мета- и норн-заместителей на реакционную
способность ароматических соединений — заместителей, достаточно удален-
ных от реакционного центра, чтобы не рассматривать первичные простран-
ственные эффекты. Вследствие конформационной гибкости алифатических
соединений для них гораздо труднее получить в подобной же степени фикси-
рованную отдаленность заместителя от реакционного центра. Однако это
было осуществлено Робертсом и Морлэндом [21] при использовании соедине-
ний, в которых заместитель и группа, по которой проходила реакция, нахо-
дились у концов мостиковой системы. Авторы изучили реакции 4-замещен-
ных бицикло-[2,2,2]-октан-1-карбоновых кислот и других производных 1-кар-
боновых кислот 4-RG(GH2CH2)3CGO2H и т. д.
При изучении влияния заместителей на константы кислотности этих кислот
в 50 %-ном водном этиловом спирте при 25 °C были определены константы
заместителей, обозначенные и'. Константа реакции р' для этой реакции была
принята равной 1,464, т. е. величине р, найденной для ионизации бензойных
кислот в этом растворителе (р при ионизации в воде равна единице). Фор-
мальное определение а', следовательно, таково:
1,464а'= log (А).
Однако, конечно, сравнение а' с константами о ароматических соединений
будет иметь смысл лишь в том случае, если влияние растворителя, т. е.
влияние замены воды на 50%-ный водный раствор этилового спирта при
25 °C на ионизацию как бицикло-[2,2,2]-октан-1-карбоновых кислот, так
и бензойных кислот будет одинаковым. Это влияние может и не быть точно
одинаковым.
Четыре константы о', определенные Робертсом и Морлэндом, приведены
в верхней части табл. 218. Они сравниваются в таблице с константами амета
и впара ароматических соединений.
В ароматических системах передаче полярных влияний способствует прово-
димость бензольного кольца. Полярный эффект, достигающий бензольного
кольца даже в леша-noложении относительно реакционного центра, частично
передается с помощью сопряжения в положения кольца, ближе расположен-
ные к реакционному центру. Если полярный эффект достигает кольца в пара-
положении, таким же образом он будет передаваться по механизму сопря-
* Автор включил это «теоретическое» уравнение (в других обозначениях) в рукопись
первого издания этой книги, но затем изъял раздел, содержащий это уравнение (раздел,
касающийся полярных эффектов), решив в то время, что лучше подождать эмпириче-
ского развития этого вопроса, которое бы дало свой собственный способ обоснования
уравнения, что впоследствии и было сделано.
1008
Полярные эффекты
жения к тому положению в кольце, которое или является реакционным цент-
ром, или связано с боковой цепью, в которой находится реакционный центр.
В системе Робертса и Морлэнда все такие эффекты ароматического сопряже-
ния исключены, что было сделано специально. При условии что эффектами
утяжеления можно пренебречь (как это обычно и бывает), заместители в поло-
жении 4 этой системы могут влиять на реакцию лишь посредством индук-
тивного эффекта и эффекта поля, причем последний, вероятно, имеет наи-
большее значение.
Таблица 218
Константы заместителей о' и константы реакции р' для реакций 4-замещенных
бицикло-[2,2,2]-октан-1-карбоновых кислот и их производных (Е — равновесие,
R — скорость). Сравнение констант <т' с константами омета и О пара
ароматических соединений (по данным Робертса и Морлэнда)
R СТ' ° мета
ОН 0,28 0,12 -0,27
Вг 0,43 0,39 0,23
СО2С2П5 0,30 0,37 0,45
CN 0,58 0,56 0,66
Ароматическое соединение Е или R Реакция р'
ВС(СН2СН2)3ССО2Н Е Ионизация, 50%-ный этиловый спирт, 25 °C (1,464)
R Реакция с (C6H5)2CN2, С2Н5ОН, 30 °C 0,70
ВС(СН2СН2)3ССО2С2Н5 R Щелочной гидролиз, 87,8%-ный этиловый спирт, 30 °C 2,24
Можно указать на некоторые ожидаемые следствия. Заместители ОН и Вг,
которые при замещении в ароматическом ядре относятся к типу —I, +К,
в системе бициклооктана будут проявлять лишь —/-свойства. Поэтому им
будут соответствовать положительные константы ст', величины которых
будут возрастать в индуктивном порядке ОН < Вг. Если исключается
эффект, который в ароматическом ряду проявляется в виде электроположи-
тельного сопряжения, то соответствующие п' константы алгебраически будут
более положительными, чем даже ароматические константы я мета и гораз-
до более положительными, чем значения впара. Все эти ожидаемые изменения
констант представлены в табл. 218. Заместители СО2С2Н5 и CN, которые при
замещении в ароматическом ядре будут относиться к типу —I, — /^замести-
телей, в мостиковой алифатической системе будут также проявлять лишь
—/-свойства, и, таким образом, им будут соответствовать положительные
константы п'. Однако в этом случае исключается электроотрицательное
сопряжение с ароматическим ядром, и, следовательно, значения о' должны
быть несколько менее положительными, чем Ямета-константы, и значительно
менее положительными, чем Ппара-константы. Как видно из данных табл. 218,
и знаки констант ст', и их разность (в трех случаях из четырех), и большая
величина разности между о' и ъпара, чем между о' и ^мета, соответствуют
ожидаемым. Разность амета—о' для CN имеет «неправильный» знак; однако
64-0772
1009
Глава XVI
она настолько мала, что изменение о' на 5% могло бы переменить знак.
Экспериментальные измерения, вероятно, должны иметь меньшую погреш-
ность, чем это расхождение, однако возникают сомнения, действительно
ли можно гарантировать, что влияние растворителя на ионизацию бицикло-
Рис. 61. Зависимость логарифмов констант
скоростей второго порядка омыле-
ния этиловых эфиров бициклооктан-
карбоновых кислот от логарифмов
констант кислотности соответствую-
щих кислот в условиях, приведен-
ных для этих реакций в табл. 218
[21]. Значения log Л'а, нанесенные
на абсциссе, эквивалентны значениям
о', если не учитывать нулевого по-
ложения и масштаба шкалы.
октанкарбоновых и бензойных кислот, которое не учитывается при выводе
коэффициента 1,464, но с помощью которого определяются п', не отличается
далее в таких небольших пределах.
Если отложить на графике значения log/c для двух реакций против найден-
ных констант заместителей п', то в соответствии с уравнением
ъ
log -f- = ff p
KQ
получаются хорошие прямые линии (рис. 61). Наклоны таких линий дают
значения р', приведенные в нижней части табл. 218.
з. Учет некоторых пространственных эффектов: аддитивность и про-
порциональность остаточных полярных эффектов. Заместители в реакциях
алифатических соединений с открытой цепью и opmo-заместители в реакциях
ароматических соединений обычно обладают, наряду с полярным, заметным
пространственным эффектом. Общего метода анализа результирующих тер-
модинамических или кинетических данных, позволяющего отдельно выде-
лить вклады полярных и пространственных эффектов в общий структурный
эффект, не существует. Тем не менее следует отметить плодотворные иссле-
дования Тафта [23], основанные на ранее предложенном грубом методе [22],
примененном только к гидролизу эфиров карбоновых кислот, а в принципе
применимом к реакциям, идущим по двум механизмам, которые отличаются
лишь количеством протонов в переходном состоянии.
Предположение заключалось в том, что в соотношении скоростей бимолеку-
лярного кислотного и основного гидролиза сложного эфира пространствен-
ные эффекты сокращаются и что, следовательно, изменение этого соотноше-
ния при переходе от одного сложного эфира к другому будет определяться
только полярными эффектами. Это допущение не может быть, конечно, совер-
шенно точным. Справедливо, что два переходных состояния при гидролизе
сложных эфиров являются иэоэлектронными и отличаются лишь двумя
протонами. Однако следует ожидать, что дополнительный протон будет
«сжимать» электронную систему и если «сжатие» будет зависеть от природы
заместителя, т. е. будет проявляться влияние размера заместителя на раз-
ность логарифмов соотношений скоростей, то пространственные эффекты
1010
Полярные эффекты
нельзя полностью исключить *. Однако пространственные эффекты все же
устраняются в достаточно полной степени, и поэтому этот метод можно
попытаться использовать, особенно при отсутствии более обещающей аль-
тернативы.
Тафт выбрал константу аддитивности и константу пропорциональности,
необходимые для связи разности логарифмов соотношений скоростей с кон-
стантой заместителя; последняя была обозначена символом п *. Константа
аддитивности зависит от выбора стандартного эфира. В качестве одной длин-
ной серии эфиров Тафт выбрал ацетаты, в этом случае в качестве замести-
телей рассматриваются группы, замещающие водород ацетильного остатка.
Константа пропорциональности соответствовала константе реакции этого
процесса и была выбрана как средняя величина (2,48) разности между зна-
чениями р для основного и кислотного гидролиза эфиров бензойной кислоты.
Таким образом было получено уравнение Тафта
2,48п* = log
/ kB/kA \
где индексы В и А относятся к основному и кислотному гидролизу, а нуле-
вой индекс соответствует реакции стандартного эфира, например ацетата.
Таблица 219
Константы о * заместителей по Тафту, полученные из скоростей
гидролиза ацетатов, содержащих один заместитель в ацетильной группе
Заместитель в СОСНз a * относи- тельно ацетата 2,2a' Заместитель в COCH3 а * относи- тельно ацетата 2,2а'
N(CH3)J 1,90 CN 1,30 1,27
SO2CH3 1,52 СОСН3 0,60
F 1,10 CH2NO2 0,50
J CI 1,05 СН = СНСН3 0,13
1 CH2CI 0,385 f с„н5 0,215
Br 1,00 1,00 < СН2С6Н5 0,08
I 0,85 1 СН2СН2С6Н5 0,02
oceH5 0,85 СН3 —0,10
OH 0,56 0,62 С2Н5 -0,115
OCH3 0,52 IZ30-C3H7 -0,125
f CF3 0,72 трет-С4Нд -0,165
<! ch2cf3 0,32 СН(СН2)5 -0,06
I ch2ch2cf3 0,12 Si(CH3)3 —0,26
В табл. 219 приведены некоторые константы о *, полученные Тафтом из
скоростей гидролиза сложных эфиров. Эта неполная таблица ограничена
замещенными ацетатами, имеющими лишь один заместитель, замещающий
атом водорода в ацетильной группе. Каждое значение п* относится к одному
и тому же спиртовому радикалу и одинаковым физическим условиям.
С первого взгляда очевидно, что значения п* правильно изменяются в соот-
ветствии с полярностью, однако несколько пунктов заслуживают более
* Предполагалось, что этой процедуре мешает влияние энергии сольватации на разность
логарифмов скоростей, однако, рассматривая разность логарифмов соотношения скоро-
стей, этим фактором можно пренебречь.
64* 1011
Глава XVI
детального рассмотрения. Во-первых, эти значения, которые по самому спо-
собу их вычисления должны представлять почти чистые полярные эффекты,
в тех случаях, когда можно провести сравнение, оказываются пропорцио-
нальными значениям Робертса и Морлэнда, которые по другим причинам
также определяются почти чистыми полярными эффектами. Константа про-
порциональности составляет ц*/а' = 2,2.
Затем, как можно видеть из любого ряда гомологических групп, отмеченных
в табл. 219 фигурными скобками, коэффициент ослабления передачи поляр-
ных эффектов через одну связь G — G, измеряемый соответствующей вели-
чиной а*, равен приблизительно 2,7—2,8. Это совпадает со значением 2,8,
полученным Бранчем и Кальвином при рассмотрении силы алифатических
кислот [24]. Сам факт существования такого коэффициента, даже если он
является лишь приближенным, означает, что происхдит передача влияния со
связи на связь, т. е. что полярный эффект, от которого зависит величина а*,
представляет собой индукционный эффект. Если бы это было справедливо
для бициклической системы Робертса и Морлэнда, то соотношение сг*/а'
было бы равным приблизительно 7 (допуская, что передача влияния проис-
ходит тремя путями через три связи). Тот факт, что указанное соотношение
равно лишь 2,2, совпадает с интуитивным ожиданием того, что основной
полярный эффект, мерой которого служит а', представляет собой эффект
поля.
Наиболее интересной особенностью констант а* для отдельных заместите-
лей является их аддитивность. Это снова показывает, что из этих констант
полностью исключены пространственные эффекты, поскольку последние
ввиду самого характера закона несвязывающего взаимодействия (гл. II,
разд. 1,в) далеко не аддитивны. Мы уже отмечали это в применении к метиль-
ным заместителям неопентильной и неогексильной групп (гл. VII, разд. 8,6
и гл. IX, разд. 2,6): введение третьего метильного заместителя в этих груп-
пах гораздо сильнее замедляет скорость бимолекулярных реакций (SN 2 и Е2
соответственно), чем введение первых двух. Тафт определил значение ц*для
сложных эфиров, имеющих два и три заместителя в ацетильной группе.
Полученные им значения сравниваются в табл. 220 с данными, полученными
исходя из свойств аддитивности отдельных констант заместителей (табл. 219).
Таблица 220
Константы Тафта для ацетатов, содержащих два и три заместителя
в ацетильной группе
Заместители в СОСНз а * (эксп.) а * (аддит.) Заместители в СОСНз а * (эксп.) ст * (аддит.)
F, F 2,05 2,20 СН3 дгрет-С4Н9 —0,28 —0,265
С1, С1 1,94 2,10 СН3, СвН5 0,11 0,115
СН3, СН3 -0,19 -0,20 с2н5, С6Н5 0,04 0,10
с6н5, свн5 0,405 0,43 С1, С1, С1 2,65 3,15
свн5, ОН 0,765 0,775 сн3, сн3, сн3 —0,30 -0,30
Единственным большим расхождением являются данные, полученные для
трихлорацетатов. Это может объясняться, как считает Тафт, эксперимен-
тальной погрешностью. Однако это может быть следствием также и действи-
тельного отсутствия аддитивности. Такое отсутствие аддитивности наблю-
дается в тех случаях, когда накопление полярных заместителей приводит
1012
Полярные эффекты
Таблица 221
Константы о * Тафта для заместителей в ацильной
группе формиатов и акрилатов
Заместитель в СОН о * относи- тельно формиатов Заместитель в СОСН = СН2 а * относи- тельно акрилатов
СН3 -0,49 no2 1,05
сн = сн2 0,16 Cl 0,25
С6н5 0,11 СС13 0,54
СН = СНС6Н5 -0,08 СНС12 0^23
с = СС6Н5 0,86 С6Н5 -0,24
СОСН3 1,16 СНз -0,29
СООСНз 1,51
к значительному приближению к электрическому насыщению индукции.
В табл. 221 представлены другие сложные эфиры, которые исследовал Тафт.
Слева приведены некоторые значения о*, отнесенные к формиатам. В этой
таблице значения а* отнесены к известным или возможным стандартным
соединениям. Так, первое значение отнесено к стандартной серии ацетатов,
влияние заместителей в которых только что обсуждалось. Оно показывает,
что ацетаты можно связать с формиатами, если учесть, что разность соответ-
ствующих значений о* (ацетаты минус формиаты) составляет —0,49. Вторая
строка первой колонки соответствует отнесению к акрилатам; в этом случае
разность о* (акрилаты минус формиаты) составляет 0,16. В правой части
таблицы приведены константы ст* для некоторых mpanc-р-монозамещенных
акрилатов, отнесенные к акрилатам. В обеих сериях значения ст* изменяют-
ся в соответствии с полярными свойствами заместителей. Может показаться
удивительным, что стирильному и фенильному радикалам соответствуют
алгебраически отрицательные значения, которые характерны для электро-
положительных заместителей. Однако следует напомнить, что «испытываемым
объектом» во всех этих молекулах является карбоксильная группа, которая
сама по себе является электроотрицательной. Вследствие ее поляризующего
действия все группы, особенно группы, сопряженные с нею, будут иметь
менее положительные значения ст* (но не обязательно отрицательные, как
в указанных двух примерах), чем в том случае, когда заместители связаны
с неполярной группой.
Другим важным рядом сложных эфиров, исследованным Тафтом, является
серия эфиров бензойной кислоты, содержащих один орто-заместитель.
Связь между полярными эффектами в родоначальной серии бензоатов и в дру-
гих эфирах представлена в третьей строке слева в табл. 221: разность бензоат
минус формиат составляет 0,11. Значения ст* для opmo-замещенных эфиров
бензойной кислоты представлены в табл. 222. Можно полагать, что эти зна-
чения отражают чисто полярные эффекты и, следовательно, их можно срав-
нить с ст-константами мета- и пара-заместителей, которые по другим сообра-
жениям также соответствуют чисто полярным эффектам. В бензольном кольце
имеется внутренняя сопряженная система, и поэтому Тафт проводит сравне-
ние в основном с константами ст для пара-положений, полагая, что вклад
сопряжения в передачу полярного влияния заместителя учитывается в боль-
шей степени при сравнении орто- и пара-заместителей, чем при сравнении
орто- и .мета-заместителей. В табл. 222 сравниваются ст*-константы орто-
и пара-заместителей.
1013
Глава XVI
Таблица 222
Константы о * Тафта для оршо-заместителей в бензоатах
Заместитель X в СОСвН6 а * для орто-Х относительно бензоатов О для пара-Х
ОСН3 -0,39 -0,27
СН3 -0,17 -0,17
F 0,24 0,06
CI 0,20 0,23
Вг 0,21 0,23
I 0,21 0,18
no2 0,78 0,78
Даже если предположить, что пространственные эффекты полностью учте-
ны, все же нельзя ожидать, что две серии констант заместителей будут иден-
тичными, так как, во-первых, из этих двух положений эффект поля будет
влиять различным образом и, во-вторых, способ передачи полярных эффек-
тов, хотя и является аналогичным и в обоих случаях включает сопряжение
и индукцию, никоим образом не является одним и тем же. Однако следует
ожидать, что если эти константы заместителей относятся к чисто полярным
эффектам, то они будут изменяться в одном и том же направлении для обоих
положений. Именно такой качественный вывод и следует из данных табл. 222.
Тафт предположил, что изменения, вызываемые заместителями в log/c или
logA в реакциях, отличных от реакций гидролиза сложных эфиров (для
которых константы заместителей <т* могут быть определены), пропорцио-
нальны значениям а*, определенным из скоростей гидролиза сложных эфи-
ров. Это предположение может быть записано следующим образом:
log(A/A0) или log(k/k0) = p*a*,
где р* — эмпирически определяемая константа пропорциональности для
данной реакции. В графическом выражении это означает, что зависимости
logfc или log# от п* должны давать прямые линии с наклоном р*. Эта зави-
симость не так точна, как уравнение Робертса — Морлэнда, применимое
к узкому кругу реакций, или уравнение Юкава — Цуно, которое имеет
большую или, по крайней мере, такую же точность. Уравнение Тафта не
позволяет заменить известные стандарты на более точные. Некоторые реак-
ции хорошо соответствуют этому уравнению, а некоторые — плохо. Это
уравнение в общем случае не может быть настолько точным, чтобы распро-
страняться на реакции, в которых важную роль играют пространственные
эффекты, поскольку оно связывает logA или logic, которые в принципе зави-
сят от полярных и пространственных эффектов, с константами а*, завися-
щими только от полярных эффектов. Нельзя получить прямую линию или
какую-либо другую линию, строя график зависимости функции двух незави-
симых переменных, имеющих равнозначную важность, от функции лишь
одной из этих переменных.
Это заставляет нас вернуться к историческим истокам линейной зависимо-
сти свободных энергий. Теоретическое обоснование этой зависимости очень
похоже на обоснование зависимости Бренстеда. В последнем случае срав-
ниваются каталитические скорости logA: с кислотностью log А'; эти две вели-
1014
Полярные эффекты
чины в принципе зависят как от полярных, так и от пространственных эф-
фектов, однако различным образом. Тот факт, что, исключая наиболее
плохие корреляции, в общем можно наблюдать удовлетворительную линей-
ную зависимость (рис. 57), означает лишь то, что для этих реакций полярные
эффекты являются гораздо более важными, чем пространственные эффекты,
так что изменение относительных вкладов этих эффектов приводит лишь
к небольшим изменениям абсолютного эффекта. График, подобный приведен-
ному выше, представляет собой прямую линию, которая отражает доминиру-
ющий полярный эффект, на который накладываются пространственные эффек-
ты, приводящие к небольшим или средним по величине отклонениям. Гра-
фики Бренстеда относятся к протолитическим процессам, и в этих процессах
пространственные эффекты в общем имеют довольно небольшое значение, так
же как и при отщеплении олефинов (гл. IX, разд. 2). Причина одна и та же,
а именно: в обоих типах реакций протоны переносятся из относительно про-
странственно неэкранированных положений.
Кроме реакций общекислотного катализа, полярные эффекты преобладают
над пространственными во многих других классах реакций. Можно ожидать,
что до тех пор, пока вклад пространственных эффектов остается небольшим,
некоторая экспериментально измеренная величина, зависящая в основном от
полярных и пространственных эффектов, но практически лишь от полярных
эффектов, будет коррелироваться с величиной, являющейся мерой лишь
полярных эффектов. Многие из реакций, к которым более или менее хорошо
применима зависимость Бренстеда, входят также и в перечень реакций,
предложенных Тафтом в поддержку своего соотношения пропорционально-
сти. То же самое можно сказать и относительно других реакций, в которых
пространственные эффекты играют небольшую роль. Однако, несмотря на
ограниченность применения и низкую точность, зависимость р*о* Тафта
расширяет диапазон реакций, для которых можно количественно показать,
что полярные эффекты являются преобладающими в органической химии
и что небольшие пространственные эффекты также широко распространены.
Однако для проявления сильных пространственных эффектов требуются
особые структурные условия.
СПИСОК ЛИТЕРАТУРЫ
1. Leffler J. Е., Grunwald Е., Rates and Equilibria of Organic Reactions, Wiley, New
York and London, 1963, Chaps. 6 and 9.
2. Bronsted J. N., Pedersen K., Z. physik. Chem., 108, 185 (1924).
3. Bronsted J. N., Duus H. C., Z. physik. Chem., 117, 299 (1925); Dawson H. M., Car-
ter J. S., J. Chem. Soc., 1926, 2282; Dawson H. M., Dean N. C., ibid., p. 2872; Daw-
son H. M., Hoskins С. M., ibid., p. 3166; Bronsted J. N., Guggenheim E. A., J. Am.
Chem. Soc., 49, 2554 (1927); Bronsted J. N., Chem. Revs., 5, 322 (1928); Bell R. P.t
Acid-base Catalysis, Clarendon Press, Oxford, 1941.
4. Bell R. P., Higginson W. С. E., Proc. Roy. Soc., A147, 141 (1949).
5. Bradfield A. E., Jones В., J. Chem. Soc., 1928, 1006, 3073; 1931, 2903.
6. Hammett L. P., Chem. Revs., 17, 225 (1935); Physical Organic Chemistry, McGraw-
Hill Co., New York and London, 1940, Chap. 7.
7. Burkhardt G. N., Nature, 136, 684 (1935); Burkhardt G. N., Ford W. G. K., Singleton E.,
J. Chem. Soc., 1936, 17.
8. Hughes E. D., Ingold С. K., J. Chem. Soc., 1935, 244.
9. Ceccon A., Papa I., Fava A., J. Am. Chem. Soc., 88, 4643 (1966).
10. Hammett L. P., J. Chem. Ed., 43, 464 (1966).
11. Brown H. C., Nelson K. L., J. Am. Chem. Soc., 75, 6292 (1953); Brown H. C., McGa-
ry C. W., ibid., 77, 2300 (1955); Stock L. M., Brown H. C., Advances in Physical Orga-
nic Chemistry, Editor V. Gold, Academic Press, London and New York, Vol. 1, 1963,
p. 35.
1015
Глава XVI
12. Jaffe Н. Н., Chem. Revs., 53, 191 (1953); Okamoto Y., Brown H. C., J. Org. Chem.,
22, 485 (1957); Okamoto Y., Brown H. С., J. Am. Chem. Soc., 80, 4979 (1958); McDa-
niels D. H., Brown Н. С., J. Org. Chem., 23, 420 (1958).
13. Nixon A. C., Branch G. E. K., J. Am. Chem. Soc., 58, 492 (1936).
14. Boberts J. D., McElhill E. Л., J. Am. Chem. Soc., 72, 628 (1950); Bardwell F. G., Coo-
per G. D., ibid., 74, 1058 (1952).
15. Saunders W. H., Jr., Williams В. Л., J. Am. Chem. Soc., 79, 3712 (1957).
16. Bunnett J. F., Draper F., Jr., Bauson P. B., Noble P., Jr., Topkyn B. G., Zahler В. E..
J. Am. Chem. Soc., 75, 642 (1953).
17. Van Bekkum H., Verkade P. E., Wepster В. M., Rec. trav. chim., 78, 85 (1959).
18. Tsumo Y., Ibata T., Yukawa Y., Bull. Chem. Soc., Japan, 32, 960 (1959); Yukawa Y.,
Tsumo Y., ibid., pp. 965, 971.
19. Bott R. W., Eaborn C., J. Chem. Soc., 1963, 2139: Baker B., Bott R. W., Eaborn C ,
ibid., 1964, 627; Bott R. W., Eaborn C., Walton D. R. M., ibid., 1965, 384.
20. Ryan J. J., Humffray A. A., J. Chem. Soc., 1966B, 842; 1967B, 468.
21. Roberts J. D., Moreland W. T., Jr., J. Am. Chem. Soc., 75, 2167 (1953).
22. Ingold С. K., J. Chem. Soc., 1930, 1032.
23. Taft R. W., Jr., J. Am. Chem. Soc., 74, 2729, 3120 (1952); 75, 4231 (1953); Пространст-
венные эффекты в органический химии, ИЛ, М., 1960, гл. 13.
24. Branch G. Е. К., Calvin М., The Theory of Organic Chemistry, Prentice-Hall, Engle-
wood Cliffs, N.J., 1941, Chap. 6.
ГЛАВА XVII
СТАБИЛЬНЫЕ РАДИКАЛЫ
Арилметильные, ариламинильные, ароксильные и другие подобные
радикалы.......................................1081
Список литературы .............................1038
Необходимо объяснить, что означает слово «стабильный», приведенное
в заголовке этой главы. В гл. V были рассмотрены общие методы получения
радикалов. В гл. VI, VII и VIII были обсуждены основные классы реакций,
в которых радикалы реагируют с ароматическими, алифатическими и олефино-
выми соединениями. Исследованные реакции рассматривались с кинети-
ческой точки зрения. Вопрос о термодинамической стабильности радикалов
остался, таким образом, незатронутым. В большинстве своем кинетически
важные радикалы являются совершенно неустойчивыми по сравнению с ис-
ходными и конечными продуктами. Термодинамическая устойчивость ради-
калов может быть количественно оценена в результате определения количест-
ва радикалов, появившихся в процессе равновесия гомолиз — коллигация.
Такой метод, следовательно, применим лишь к радикалам, которые появля-
ются в измеримых равновесных количествах. Применение метода ограничено
теми случаями, когда радикалы являются гораздо более устойчивыми, чем
большинство кинетически важных радикалов; такие радикалы, конечно,
наиболее устойчивы среди известных органических радикалов.
1. Арилметильные, ариламинильные, ароксильные и другие
подобные радикалы
а. Трифенилметильный радикал. В 1900 г. Гомберг [1] при попытке
синтезировать гексафенилэтан из хлористого трифенилметила при реакции
с серебром получил бесцветное твердое вещество, желтый раствор которого
в бензоле обладал высокой реакционной способностью. Раствор сразу же
поглощал кислород, давая перекись (С6Н5)3СООС(С6Н5)3. Точно так же по-
глощалась окись азота, давая нитрозосоединение (CeH5)3CNO, а при погло-
щении иода получался трифенилиодметан (С6Н5)3С1. По этой и другим ана-
логичным причинам Гомберг рассматривал полученное им соединение как
радикал трифенилметил (СеН5)3С-. Молекулярный вес этого радикала, най-
денный по понижению температуры замерзания бензола, оказался, однако,
намного ближе к молекулярному весу гексафенилэтила (СеН5)6С2, чем к мо-
лекулярному весу трифенилметила (СвН5)3С-.
На Гомберга более сильное впечатление произвели цвет и реакционная спо-
собность нового соединения, чем его молекулярный вес. Однако Хайт-
шель [2] и Якобсон [3] сделали попытку согласовать цвет и химические свой-
ства с молекулярным весом, предложив для вещества Гомберга хиноидную
структуру с молекулярным весом гексафенилэтана, но такого необычного
типа, что в то время можно было предполагать, что оно может иметь необхо-
димый цвет и реакционную способность. В этот период оценить справедливость
таких доказательств с помощью других наблюдений, к которым мы вернемся
ниже, было весьма затруднительно, но все же несколько ученых, например
Байер [4], Флюршхейм [5] и Вернер [6], несмотря на все эти сложности,
смогли предугадать диссоциативное равновесие с участием сильно окрашен-
ного очень реакционноспособного радикала, доля которого в равновесии
оказывалась слишком небольшой, чтобы ее можно было измерить криоско-
пическим методом. Затем в 1908 г. наличие равновесия диссоциации было
полностью доказано Шмидлином [7], который обнаружил, что скорость дис-
социации, ведущей к образованию трифенилметила, и скорость реакций это-
го вещества с теми реагентами, которые использовал Гомберг, отличаются
между собой. Шмидлин использовал это различие для иллюстрации того,что
эти процессы являются последовательными
Бензол Оз
(С6Н5)3С-С(С6Н5)з —> (С6Н5)3С. рт- > (СвН5)3СООС(СвН5)3
OblLiPO
Медленно
Бесцветный Желтый Выпадает в осадок
1018
Стабильные радикалы
При встряхивании желтого бензольного раствора на воздухе окраска сразу
же исчезала и перекись выпадала в осадок. Количество осадка было очень
небольшим и составляло всего несколько процентов от количества исходного
гексаарилэтана. Однако при удалении кислорода раствор в течение несколь-
ких минут восстанавливал свою окраску, а затем при новом контакте с кис-
лородом вновь мгновенно обесцвечивался с выпадением в осадок точно такого
же небольшого количества перекиси. Этот цикл можно было неоднократно
повторять.
Одно затруднение, осложнившее первоначальные предположения, возникало
как следствие наблюдений, проведенных Вальденом [8] в 1903 г., который
установил, что гексафенилэтан дает ярко-желтые растворы в двуокиси серы,
хорошо проводящие электрический ток. В этом случае происходит другой
процесс диссоциации:
so2
(С6Н5)3С—С(СЬН5)3 —(С6Н5)3С++(С6Н5)3С-
Катион представлял собой хорошо известный Вальдену катион, возникаю-
щий в растворе трифенилметилхлорида в двуокиси серы. Анион же соответ-
ствовал аниону, возникающему из трифенилметилнатрия в таких основных
растворителях, как, например, аммиак.
Таким образом, необходимо было различать три «трифенилметила»: катион
(С6Н5)3С+, радикал (С6Н5)3С • и анион (СеН5)3С_; сначала не всегда понимали,
что они являются очень разными частицами и что наблюдение и выводы отно-
сительно любой из них не могут быть автоматически перенесены на другие.
Другая большая трудность заключалась в мезомерии всех «триарилметилов»,
особенно радикала и катиона. В 1907 г. Гомберг [9] убеждал себя, что ради-
кал имеет хиноидное строение (что отчасти так и есть). Его доказательством
было то, что атом брома в n-бромфенилдифенилметиле можно отщепить от
молекулы с помощью металлического серебра, как будто атом углерода,
с которым связан бром, не обладает бензоидным характером (отчасти это так
и есть). Подвижность нара-галогенных атомов в соединениях такого типа
была показана на других примерах, что послужило новым подтверждением
хиноидной структуры. Однако в действительности эти примеры описывали
скорее свойства катиона, чем радикала [10]. Доказательством служило то, что
три-(н-бромфенил)метилхлорид, хотя и не претерпевает никаких изменений
в бензольном растворе, находится в равновесии с п-хлорфенилди-(ге-бром-
фенил)метилбромидом в растворе двуокиси серы. Это означает, что перво-
начально диссоциировавший ион хлорида может подойти к nopa-у г л вредно-
му атому, чему способствует мезомерная структура карбониевого иона с об-
разованием молекулы хиноидного строения
В1\ ,--X
£,р/\ С(СвН4Вг-га)2
от которой может отщепиться любой хлорид- или бромид-ион, и эти ионы
в конце концов могут присоединиться к центральному атому углерода, обра-
зуя найденные изомеры.
б. Скорость и степень образования трифенилметила. Существует три
метода определения состава равновесной смеси при равновесной диссоциа-
ции, ведущей к более или менее полному образованию радикалов. Наиболее
распространенным методом исследования соединений триарилметильного
ряда является криоскопическое определение среднего молекулярного веса.
Этот метод, однако, непригоден для определения степени диссоциации гекса-
1019
Глава XVII
фенилэтана, так как погрешность измерения составляет несколько процен-
тов и имеет тот же порядок, что и степень диссоциации при использовании
обычных криоскопических растворителей, например бензола, и при концен-
трациях, удобных для криоскопических измерений.
Вторым методом является колориметрическая оценка концентрации радика-
ла. Этот метод удобен для гексафенилэтана, так как позволяет работать при
гораздо более сильном разбавлении и, следовательно, увеличивать степень
диссоциации. Этот метод естественно вызывает необходимость перехода
к системе с высокой степенью диссоциации, так как в целях калибровки
необходимо знать коэффициент экстинкции радикала. Другими словами,
необходимо бывает связать отклонения от закона Бера с законом разбавления
Оствальда.
Третий метод основан на измерении магнитных свойств неспаренных элек-
тронов в радикале. Более старое приближение к этому методу, например
определение парамагнитной восприимчивости радикала в целом, является
неудовлетворительным, так как в настоящее время невозможно с достаточ-
ной надежностью вычислить диамагнетизм магнитных орбит радикала, на
который накладывается парамагнетизм неспаренного электрона. Однако
измерения методом спектроскопии электронного парамагнитного резонанса
являются надежными, так как они позволяют непосредственно определять
спиновые переходы неспаренных спинов в магнитном поле, накладываемом
извне, и, следовательно, не зависят от магнитных свойств орбитального
движения электронов в целом. Положение линии в спектре ЭПР дает раз-
ность энергии, обусловленную спиновой инверсией в магнитном поле, а соот-
ветствующая калибровочная кривая интенсивности позволяет определить
плотность неспаренных спинов и, следовательно, концентрацию радикалов.
Этим методом можно измерить как очень низкие концентрации радикалов
порядка 10 ~7 М или менее, так и более высокие концентрации. При этом
часто можно получить дополнительную информацию. Магнитное взаимодей-
ствие между неспаренным электроном и не слишком отдаленным ядром, осо-
бенно протонами связанных атомов водорода, проявляется в виде сверхтонко-
го расщепления линии в спектре ЭПР. Оно помогает определить местонахож-
дение неспаренного электрона в радикале. Распределение неспаренного элек-
трона за счет мезомерии по нескольким атомам может привести к появлению
нескольких линий электронного парамагнитного резонанса, каждая из
которых имеет свое характерное сверхтонкое расщепление. Из относитель-
ных интенсивностей можно количественно определить распределение неспа-
ренного электрона среди возможных его положений.
В соответствии с принципом неопределенности короткоживущие радикалы
имеют диффузные уровни энергии и, следовательно, дают широкие линии
в спектре ЭПР. Радикалы с периодом полураспада в пределах 10-10—10~6с
можно определить по ширине линии в спектре ЭПР.
В 1911 г. Пиккард [И] показал, что окраска растворов гексафенилэтана
связана, по-видимому, с его диссоциацией, что вытекает из отклонений от
закона Бера (при разбавлении интенсивность общей окраски возрастала).
На основании этого Циглер и Эвалд [12] в 1929 г. разработали метод калиб-
ровки, применяющийся для определения равновесного состава и, следова-
тельно, для определения констант равновесия. Так, для растворов гекса-
фенилэтана в бензоле при 20 °C зависимость между степенью диссоциации
в процентах (100 а) и разбавлением v = [(CeH5)6C2]_1 можно представить
следующим образом:
v, л/моль 12,5 98 885 25 700 76 000
100а 3,6 9,6 25,8 77,5 90
1020
Стабильные радикалы
Из этих данных можно вычислить константу равновесия:
К = [(СвН3)3С]2/[(СвН5)еС3] =4,1-10-4 Моль/л.
Температурный коэффициент константы равновесия позволяет вычислить
изменение энтальпии, равное 11,3 ккал/моль (47,31 -103 Дж/моль) и изменение
энтропии [23,1 кал/моль -°C (96,72 Дж/моль • °C)]. Увеличение свободной
энергии составляет лишь 4,54 ккал/моль (19,01 -103 Дж/моль). Таким обра-
зом, энтропия является, как всегда, важным фактором, способствующим дис-
социации. В разд. 1 ,г будут рассмотрены работы Циглера и Эвалда по влия-
нию растворителей на эти величины.
Циглер, Эвальд и Орт [13] показали, что реакция гексафенилэтана с иодом
является реакцией первого порядка, скорость которой не зависит от концен-
трации иода. Очевидно, иодирование представляет собой реакцию SrI, ско-
рость которой контролируется медленным предварительным гомолизом
Медленно
(С6Н5)3С — С(СвН5)3 ч(СвН5)3С-+ (СвН5)3С-
SH1
(СвН5)3С- +12
Быстро
> (СвН5)зС1-|-1-
Хотя предварительный гомолиз в значительной степени обратим при отсут-
ствии веществ, захватывающих образующиеся радикалы, в присутствии такого
эффективного акцептора радикалов, каким является иод, процесс идет до
конца. Таким образом, исследуя кинетику реакции радикала с иодом, можно
определить скорость диссоциации гексафенилэтана.
Циглер, Орт и Вебер [14] разработали аналогичный кинетический метод
с использованием окиси азота. При условии что окись азота находится под
давлением, выше некоторого минимального, она может довольно быстро при-
соединять радикалы трифенилметила: происходит реакция первого порядка,
протекающая со скоростью, не зависящей от давления окиси азота. В этом
случае осуществляется другой процесс типа
Медленно
(С6Н5)3С — С(С6Н5)з <.—2Z/
(С8Н5)3С. +(СвН5)3С.
Быстро
(C6H5)3C-+NO
(CeH5)3CNO
SH1
И здесь измеряемая скорость представляла собой скорость предварительного
гомолиза. Она оказалась почти равна скорости реакции радикала с иодом
в том же растворителе и при той же температуре.
Циглер, Эвалд и Лютрингхаус [15] показали, что присоединение кислорода
гексафенилэтаном происходит быстрее, чем присоединение окиси азота. Они
объяснили разницу в скоростях цепной радикальной реакцией автоокисле-
ния, инициируемой образующимися перекисными трифенилметил-радикала-
ми, которые энергично взаимодействуют с недиссоциированными молекулами
гексафенилэтана. Авторы подавили этот процесс путем добавления вещест-
ва, захватывающего перокси-радикалы, например пирогаллола, который
гидрирует их с образованием в качестве конечного продукта гидроперекиси
трифенилметила
Медленно
(СвН5)3С — С(СзН5)3 ____ А. (СвН5)3С- + (С6Н5)3С-
Быстро
(СвНз)зС-+ О2 * (СвНй)зС — О — О-
(С6Н5)3С — О — О-+Н > (С6Н5)3С—о — он >
1021
Глава XVII
Реакция присоединения кислорода в этом случае имеет первый порядок
и не зависит от давления кислорода. Контролируемая той же стадией пред-
варительного гомолиза скорость этой реакции соответствует скорости при-
соединения окиси азота в аналогичных условиях.
На основании полученных таким образом взаимно согласующихся данных
Циглер пришел к выводу, что измерение скорости ингибируемого автоокис-
ления и скорости присоединения окиси азота является наиболее точным ме-
тодом определения скорости диссоциации. Скорость, определяемая с помо-
щью иода, является довольно невысокой, вероятно, ввиду какой-нибудь
неустраненной побочной реакции. Для скорости диссоциации гексафенил-
этана в толуоле при О °C Циглер и его коллеги установили, что = 0,0025 с-1.
Из температурного коэффициента скорости они получили значение фактора
частоты Аррениуса, которое составляет 0,5-10~13 с-1, т. е. обычную величину
для мономолекулярных реакций. Аррениусовская энергия активации равна
19,0 ± 1,0 ккал/моль (79,55-103 + 4,187-103 Дж/моль). Авторы также
исследовали влияние растворителей на кинетические параметры. Эти резуль-
таты будут приведены в разд. 1,г [14—16].
Если принять среднюю величину эндотермичности диссоциации гексафенил-
этана в обычных растворителях равной 11,5 ккал/моль (48,15-103 Дж/моль)
и величину энергии активации гомолиза равной 19 ккал/моль (79,55 х
X Ю3 Дж/моль), то из этого следует, что энергия активации коллигации
радикалов составляет 7,5 ккал/моль (31,4-103 Дж/моль). Следовательно,
перед коллигацией радикалы не находятся близко друг к другу, а приближа-
ются друг к другу во время реакции. Это можно объяснить двумя причинами.
Одной из них является то, что радикальный центр за счет мезомерии дело-
кализован по трем фенильным группам и, чтобы произошел захват радикала,
он должен сконцентрироваться на экзоциклическом атоме углерода. Вторая
причина состоит в том, что центральные связи свободного радикала копла-
нарны и должны изогнуться так, чтобы образовалась пирамидальная конфи-
гурация для обеспечения коллигации. Для осуществления этих изменений
должна затрачиваться энергия, большая часть которой впоследствии ком-
пенсируется благодаря процессу коллигации.
в. Влияние структуры на устойчивость триарилметильных радикалов.
В первом десятилетии этого столетия так и осталась неразрешенной задача
определения степени диссоциации гексафенилэтана или какого-либо про-
стого его замещенного методом криоскопического определения молекуляр-
ного веса. Поэтому важным событием явилось сообщение в 1910 г. Шленка
и его коллег о триарилметилах, равновесная степень образования которых
при диссоциации гексаарилэтанов была большой и в одном случае пол-
ной.
Исследовались системы, полученные при замещении в одной, в двух
и затем во всех трех фенильных группах трифенилметила н-бифенилильными
группами н-СвНьС6Н4. В результате этих исследований было установлено, что
в бензоле при температуре 5 °C и при концентрациях 2.. .3%, наиболее под-
ходящих для криоскопических исследований, степень диссоциации на радика-
лы увеличивалась от величины, меньшей предела чувствительности данного
метода для трифенилметила, до 15, 80 и 100% соответственно при последо-
вательном замещении в трех фенильных группах. Все радикалы, конечно,
имели глубокую окраску. При выпаривании растворов бензола радикал
с одной бифенильной группой снова коллигировался, образуя бесцветный
гексаарилэтан. То же самое происходило с радикалом, имеющим две бифе-
нильные группы, несмотря на высокую степень его образования в растворе.
Однако радикал с тремя бифенильными группами не мог вновь коллигиро-
1022
Стабильные радикалы
ваться и при выпаривании извлекался в виде черного твердого вещества —
первого кристаллического триарилметильного радикала.
Это пионерское исследование ясно показало, что можно увеличить степень
равновесной диссоциации гексаарилэтана, увеличив число арильных групп
и перейдя к полиядерным соединениям. Позднее было показано, что этого
эффекта можно также добиться путем введения нафтильной и фенилзамещен-
ной винильной и других полиядерных групп. Такие примеры приведены
в верхней части табл. 223.
Таблица 223
Примерная степень гомолитической диссоциации гексаарилэтанов в бензоле
при 5 °C и концентрациях 2—3 вес. %
Гексаарилэтан а Степень диссоци- ации, % Лите- ратура Гексаарилэтан а Степень диссоци- ации, % Лите- ратура
{(С6Н5)3С— }2 ~ 5 б {₽-С1оН7(С6Н5)2С-}2 33 18
{п-В iph(C6 Н 5) 2С — }2 15 17 {а-С10Н7(С6Н5)2С — }2 60 18
{n-Biph2(C6H5)C }2 80 17 {(С6Н5)2С==СН — С(С6Н5)2—}2 (С6Н5С = С(С6Н5)Х ) 80 19
{7i-Biph3C—}2 ~ 100 17 1 >С(С6Н5) [с6н5с=С(С6Н5/ }2 100 20
{о-СН3ОСвН4С(С6Н5)2 —}2 26 21 {(jt-CHgOCgH^gC }2 13 22
{(о-СНзОСцН4)2С(С6Н5) — }2 40 22 {л-СН3ОСвН4С(СвН5)>- }2 23 24
{(о-СП;,ОС6Н4),!С-}2 80...100 23 {(n-NO2CeH4)3C-}2 100 25
а n-Biph = бифенилил.
б Вычислено по энтальпии и энтропии (гл. XVI, разд. 1, 6).
Простые алкильные и галогенные замещенные гексафенилэтаны имеют не-
большую степень диссоциации, сравнимую со степенью диссоциации самого
гексафенилэтана. Однако эти замещенные исследованы менее точно. Большая
часть приведенных данных является недостаточно надежной для того, чтобы
их можно было комментировать; различие между любыми приведенными
величинами слишком мало для определения влияния заместителей на поло-
жение равновесия процесса гомолиза. Сильное влияние полярных замести-
телей в гексафенилэтане на положения равновесия, как известно, оказы-
вают орто- и пара-метоксильные и иара-нитрозаместители. Данные для
этих групп представлены в нижней части табл. 223. Метоксигруппа обладает
сильным электроположительным, а нитрогруппа — сильным электроотрица-
тельным эффектами сопряжения. Оба эти эффекта вызывают значительное
увеличение степени диссоциации на радикалы.
Данные, представленные в табл. 223, обсуждались Бартоном и автором этой
книги [26] в 1929 г. с позиций, которые в основном не изменились до настоя-
щего времени. В указанной работе, основные выводы которой частично при-
менимы ко всей теории органической химии, впервые было указано на связь
мезомерии с принципом неопределенности и на тот факт, что движущей
силой распределения электронов по цепи сопряжения является сила кван-
тового происхождения. В этой работе обсуждалось влияние сопряжения
полярных групп на устойчивость карбониевых ионов и карбанионов. Заме-
ститель -j-M-типа путем сопряжения будет стабилизовать карбониевый ион
1023
Глава XVII
благодаря рассредоточению дефицита электронов по молекуле так, что ни
одна из валентных орбиталей не будет полностью незанятой, например
сн3сР- с*
Сопрягающийся заместитель —74'-типа будет стабилизовать карбанион
благодаря делокализации его неподеленных электронов, придавая тем самым
всем сопряженным электронам некоторый характер поделепности, например в
огп-Ог
Основным пунктом наиболее специальной части указанной работы было то,
что увеличение стабильности углеродного радикала, обладающего одно-
электронной недостаточностью и одним неподеленным электроном, обуслов-
ливается обоими этими процессами перераспределения электронной плот-
ности. Особое свойство арильных и сопряженных арилвинильных заместите-
лей состоит в том, что они являются амбиполярными, т. е. заместителями
+ 7И-типа. Этим объясняется их исключительная эффективность как замести-
телей, стабилизующих радикалы
Чем сильнее сопряжение, т. е. чем больше степень делокализации электро-
нов обоих описанных типов, тем выше устойчивость радикалов, как это
наблюдается в случае многоядерных арильных групп (данные, приведенные
в верхней части табл. 223). В настоящее время авторы придерживаются та-
кой же точки зрения, хотя и добавляют, что определенную роль играют про-
странственные эффекты. Наибольшее влияние, однако, должна оказывать
полярность. Пространственные эффекты, например, не могут вызвать боль-
шого увеличения степени диссоциации, которое наблюдается, когда фениль-
ные заместители заменяются на п-бифенильные.
Углерод является элементом, катион и анион которого (когда заряды локали-
зованы) слишком неустойчивы по сравнению с продуктами их превращений,
чтобы обеспечить постоянное наличие этих ионов в растворе. Даже в случае
трифенилзамещенных ионов (С8Н5)3С+ и (СвН5)3С_, которые благодаря рас-
средоточению зарядов могут существовать в растворе, имеются широкие воз-
можности для дальнейшей стабилизации соответствующими полярными заме-
стителями в фенильных группах. Д-М-Заместитель, например орто- или
пара-метоксигруппа, будет, таким образом, стабилизовать анион, тогда как
—М-заместитель, например орто- или пара-нитрогруппа, будет стабилизо-
вать катион. Можно сделать вывод [26], что такое сильное увеличение стабиль-
ности ионов отразится и па влиянии полярных заместителей на устойчивость
радикалов. Для орто- и napa-метокси- и пара-нитрогрупп в нижней части
табл. 223 приведены данные, подтверждающие эту точку зрения.
Если одну фенильную группу в каждой триаде гексафенилэтана заместить на
алкильную (но не на более объемистую, чем фенильная группа), то какого-
либо поддающегося измерению или видимого количества дифенилалкильных
радикалов в состоянии равновесия при комнатной или близкой к ней темпера-
туре не образуется, хотя эти радикалы возникают в качестве кинетических
промежуточных частиц со скоростью, которая может быть определена мето-
дами Циглера. Однако если группа, замещающая фенил, не является алкиль-
1024
Стабильные радикалы
ной, а представляет собой группу —М-типа, например ацильную или циан-
группу, то, как показали Левенхейн и сотр. [27], происходит равновесный
гомолиз, поддающийся количественному измерению. Хотя эти—ЛГ-группы
при наличии двух фенильных групп оказывают определенное промотирующее
влияние на гомолитическую диссоциацию, они не так хороши для этой цели,
как третья ±Л7-фенильная группа. Так, в состоянии равновесия количество
дифенилбензоилметила, образовавшегося из соответствующего димера при
кипячении последнего в толуоле, почти равно количеству трифенилметила,
образовавшегося из соответствующего димера при температуре замерзания
бензола. Ниже приведены примеры констант равновесия гомолитического
образования диарилацилметилов в толуоле при НО °C:
CeHg — С — С6Н5 СвН5 — С — CgHg
сн3 сосвн5
0,0 4,3
6,6 88
Влияние циангрупп на диссоциацию, хотя и проявляется в виде обратимого
появления и исчезновения окраски при нагревании и охлаждении раствора,
все же меньше влияния ацильных групп.
Если фенильную группу в каждом трифенилметильном остатке гексафенил-
этана заместить объемистым алифатическим заместителем, то степень диссо-
циации на радикалы может увеличиться. Это было четко показано Шленком
и Марком [28], которые в качестве заместителя вводили трифенилметильную
группу. Эта группа по типу относится к числу алифатических (аралкильных)
групп и, конечно, непосредственно не сопрягается с центром диссоциации.
Ее влияние заключается, вероятно, в несвязывающем взаимодействии, так
как модели показывают, что в отличие от незамещенного гексафенилэтана
эта группа вызывает пространственное отталкивание во всех конформациях.
Две фенильные группы оказывают более сильное стабилизующее влияние на
радикальный центр на углероде, чем третья фенильная группа. В бензоле при
температуре замерзания декафенил-н-бутан полностью диссоциирует до
пентафенилэтила. Конант и сотр. [29] исследовали влияние mpem-бутила как
замещающей группы. Эта группа способствует стабилизации радикального
центра на углероде, связанном с двумя фенильными группами, но в меньшей
степени, чем фенильная группа. Образование дифенилнеопентильного ради-
кала соответствующего этанового димера происходит в едва заметных коли-
чествах. на что указывает слабое окрашивание раствора при температурах
до 50 °C. (При более высокой температуре вещество разлагается с потерей
окраски.) Если фенильную группу в фенилди-п-бифенилилэтиле Шленка
заместить на треш-бутильную, то степень диссоциации с образованием ради-
кала ди-и-бифенилилнеопентила лишь слегка уменьшается. Ниже примеры
приведены в форме, удобной для сравнения с данными табл. 223:
СвЩ — С С6Н5 СдЩ — С — C6Hs re-CgH5CeH4 — С —
С(СвН5)а С(СН3)3 С(СН3)3
Степень диссоциации 100 0 60 (0,06.1/, С6Н8, 5°С)
в холодных раствори-
телях, %
65-0772
1025
Глава XVII
г. Влияние растворителя на скорость и степень образования трифенил-
метила. В табл. 224 приведены некоторые константы скоростей и равновесий,
определенные Циглером и сотрудниками для гомолиза гексафенилэтанов
в ряде растворителей. Значения константы скоростей [14—16] первого поряд-
ка получены при О °C. Константы равновесия [12] определялись при темпе-
ратуре 20 °C.
Таблица 224
Константы скоростей и равновесий для гомолиза гексафенилэтанов
в различных растворителях (по данным Циглера)
Растворитель 103fei, с-1 (при 0 °C) Растворитель 101К, моль/л (при 20 °C)
Ацетонитрил 1,1 Пропионитрил 1,2
Этилцианоацетат 1,8 Этилбензоат 1,7
Окись мезитила 2,1 Ацетофенон 1,7
Пиридин 2,2 Диоксан 2,5
Толуол 2,5 Бромбензол 3,7
Этиловый спирт 2,8
Бромистый этилен 3,3 Бромистый этилен 3,9
Бензол 4,1
Четыреххлористый угле- 3,9 Хлороформ 6,9
род
Сероуглерод 4,2 Сероуглерод 19,2
Как правило, при измерении констант скоростей и констант диссоциаций
использовались неодинаковые растворители, тем не менее таблица составле-
на так, чтобы в одной строке в обеих колонках таблицы были по возможности
одинаковые растворители и чтобы в каждой колонке соблюдались одинако-
вые последовательности изменения скоростей.
Последовательность изменения скоростей, как следует из данных таблицы,
сильно отличается от всех известных растворителей для гетеро литических
реакций. Растворитель с высокой диэлектрической проницаемостью, напри-
мер ацетонитрил, в котором все гетеролитические реакции с незаряженными
реагирующими веществами проходят с относительно высокими скоростями,
из всех приведенных растворителей наиболее сильно замедляет гомолиз.
Наиболее «быстрыми» растворителями являются неполярный четыреххлори-
стый углерод и сероуглерод. Гидроксилсодержащий растворитель этило-
вый спирт особого положения не занимает. Во всей серии растворителей
скорость реакций изменяется всего в 4 раза. Изменение константы равно-
весия по величине примерно такое же. Равновесная степень образования
радикала является наименьшей в алифатическом нитриле и самой большой
в сероуглероде. В этом случае в общем растворители оказывают более силь-
ное влияние, чем в других, в частности сероуглерод влияет более заметно,
чем в других случаях, в результате чего константы равновесия изменяются
в 16 раз.
Можно видеть, что при переходе от переходного состояния к конечному
состоянию гомолиза различие в сольватации этих соединений и исходного
состояния возрастает и что, следовательно, основным фактором, определяю-
щим влияние растворителя, является сольватация образующегося или в еще
большей степени уже полностью образовавшегося радикала. Приведенные
данные свидетельствуют о связи эффекта растворителя с поляризуемостью
1026
Стабильные радикалы
и показывают, что сероуглерод обладает рядом совершенно особых свойств,
например, как уже отмечалось, он способствует нарушению спектроскопи-
ческой симметрии растворенных в нем молекул бензола (гл. IV, разд. 2.а),
имеет относительно высокое сродство к атомам хлора (гл. VII, разд. 10,в)
и в этом случае исключительно хорошо сольватирует углеродный радикаль-
ный центр. Это можно объяснить тем, что сера представляет собой ненасы-
щенный элемент третьего периода, в результате чего молекула сероуглерода
имеет как низкий потенциал ионизации, так и небольшое сродство к электро-
ну; последнее обусловлено наличием лишь низколежащих пустых орбита-
лей. Учитывая двойственную природу внутренних электронных смещений,
стабилизующих радикальный центр на углероде (разд. 1,в), можно сказать,
что указанная особенность сероуглерода, обусловливающая легкое взаимо-
действие как с его электронами, так и с пустыми орбиталями, могла бы объяс-
нить очевидное сродство сероуглерода к радикалам трифенилметила.
д. Диариламинилы [30]. Тетрафенилгидразин впервые был получен
Виландом в 1911 г. путем окисления дифениламина перманганатом калия
в ацетоне. Оно представляет собой бесцветное вещество, дающее бесцветные
растворы при комнатной температуре, но обратимо окрашивающиеся в зеле-
ный цвет при температуре около 100 “С. Окрашенные растворы не подчиня-
лись закону Бера: изменение интенсивности окраски в зависимости от раз-
бавления показало, что окрашенное вещество получается в результате дис-
социации. Тетрафенилгидразин взаимодействует с окисью азота, давая ди-
фенилнитрозамин (C8H5)2NNO, и с трифенилметилом, давая пентафенилметил-
амин (CeH5)2NC(C6H5)3. Виланд приписывал цвет раствора и реакции ради-
калу дифениламину, образовавшемуся в результате обратимого процесса
(C6H6)2N-N(CeH5)2 ДД 2(CeH5)2N.
В этом случае степень диссоциации была, очевидно, небольшой и количест-
венно не исследовалась. Однако Кейн и Вайзлогль [31] впоследствии для
определения скорости диссоциации тетрафенилгидразина использовали реак-
цию с окисью азота. Эта реакция представляет собой реакцию первого поряд-
ка, и ее скорость выше определенного предела не зависит от давления окиси
азота. Этот процесс представляет собой еще один пример реакции 8н1, и. сле-
довательно, она может применяться для определения скорости диссоциации:
(CeH5)2N — N(C8H5)2 _2Z/ (CeHs)2N-+(CeH5)2N •
Быстро
(C6H5)2N.+NO > (C6H5)2NNO
Исследования проводились с применением в качестве растворителя о-дихлор-
бензола при температуре 75—100 °C. При температуре 100 °C константа
скорости равна 0,0037 с-1. Из зависимости скорости реакции от температу-
ры была определена величина аррениусовского фактора частоты, которая
оказалась равной примерно 101Б с-1, и энергия активации [около 30 ккал/моль
(125,60 -10s Дж/моль)]. Несмотря на невысокую точность определения этих
параметров, все же можно сказать, что такая энергия активации должна
значительно превышать ту величину, при которой реакция будет эндотер-
мичной.
При отсутствии какого-либо реагента система тетрафенилгидразин —
дифениламинил, как показал Виланд [30], вступает в реакцию диспропорцио-
65* 1027
Глава XVII
нирования
СвН5
N
/\/ \/ч
(C6H5)2N-N(C6H5)2 (C6H5)2NH +| || || |
\/\ /\х
N
свн5
Виланд исследовал влияние полярных заместителей в фенильных группах
тетрафенилгидразина на степень его гомолитической диссоциации. В случае
самого тетрафенилгидразина обратимая окраска возникает при кипячении
в толуоле (110 °C), но не при кипячении в бензоле (80 °C). В случае тетра-п-
толилгидразина окраска возникает при кипячении в бензоле. Следовательно,
иара-метильная группа, по-видимому, лишь в малой степени способствует
диссоциации. Но сильные эффекты наблюдаются в случае -|-М-заместителей,
электроположительно сопряженных с центром диссоциации. Тетра-п-анизил-
гидразин образует темно-зеленые растворы при комнатной и более низкой
температурах. Степень диссоциации этого вещества составляет, вероятно,
несколько процентов при криоскопических концентрациях, т. е. недоста-
точно велика, чтобы ее можно было заметить при температуре замерзания
бензола или нитробензола (5—6 °C). Однако тетра-и-диметиламинофепил-
гидразин дает темно-желтые растворы в бензоле и нитробензоле, в которых он
диссоциирует соответственно на 10% при температурах замерзания этих раст-
ворителей. Установлен следующий порядок влияния заместителей на сте-
пень диссоциации тетрафенилгидразинов: h-(CH3)2N> и-СН3О> н-СН3.
Другим крайним случаем является заместитель —M-типа, например группа
h-NO2, ингибирующая диссоциацию. Виланд отметил те случаи, в которых
эти эффекты соответствуют влиянию заместителей при электрофильном
замещении в ароматическом ряду: когда наблюдается несоответствие между
влиянием заместителей на ароматическое электрофильное замещение и на
диссоциацию, приводящую к образованию триарилметилов.
Бартон и автор этой книги [26] рассмотрели эти явления. Они обратили вни-
мание на полярную асимметрию аминильного радикала, указав на сходство
с соответствующими катионом и анионом. В отличие от трехлигандных угле-
родных катиона и аниона, которые при отсутствии делокализации заряда
в растворе не существуют в постоянной концентрации, двухлигандный азот-
ный катион неустойчив в такой же степени, в какой анион относительно
устойчив. Основная причина состоит, конечно, в том, что заряд ядра азота
больше. Следовательно, основным условием структурной стабилизации двух-
лигандного азотного радикала, а также и двухлигандного катиона является
частичное перемещение (и, следовательно, частичное спаривание электронов
электронодефицитиого радикального центра к атому азота). Частичное пере-
мещение неподеленных электронов имеет гораздо меньшее значение. В этом
случае аргументация та же, что и аргументация, с помощью которой при
сравнении родственных катионов и анионов мы пришли к выводу, что, на-
пример, атом брома должен быть и, как известно, это действительно так,
электрофильным радикалом (гл. XIII. разд. 3,а). Двухлигандный азотный
радикал точно так же должен быть электрофильным радикалом. С помощью
таких доводов можно понять, почему пара-заместители по своей способности
обеспечивать термодинамическую стабильность радикалов располагаются
1028
Стабильные радикалы
в ряд, найденный Виландом:
(CH3)2N > СН3О > СН3 > Н > NO2.
е. Ароксилы. Радикалы, которые будут рассмотрены в этом разделе,,
имеют отношение к двум родственным отраслям химии. Во-первых, они при-
надлежат к общему классу кислородных радикалов ВО •, образующихся при
гомолизе перекисей, которые хорошо известны как инициаторы цепных про-
цессов. Их кинетическое значение в этом смысле вытекает из того факта,что
они неустойчивы в тех условиях, в которых они образуются. Во-вторых,
каждый химик знаком с группой относительно устойчивых ароксильных
радикалов, а именно с хингидронами. Не вызывает больших сомнений, что
относительная устойчивость этих радикалов обеспечивается заместителями
-М-типа, а именно орто- или пара-гидроксильными группами. Однако
химия этих радикалов осложнена их ионизацией: они легко образуют реак-
ционноспособные анион-радикалы.
В 1922 г. Гольдшмидт [32] обнаружил, что гваякол (о-метоксифенол) при
окислении перекисью свинца дает зеленый раствор, мгновенно обесцвечи-
вающийся гидрохиноном или трифенилметилом. Гольдшмидт предположил,
что при этом образуется ароксильный радикал и что этот радикал лишь час-
тично ассоциирован с образованием димера; последнее соединение автор не-
правильно принял за перекись диарила. Ни радикал, ни его димер не были
выделены. Однако при работе с дибензогваяколом (9-метокси-10-фенантролом)
и при использовании феррицианидов щелочных металлов в качестве окисляю-
щих агентов Гольдшмидту удалось получить бесцветное твердое вещество
и показать, что оно является димером соответствующего радикала. Растворы
этого вещества были первоначально бесцветными, но через несколько часов
при комнатной температуре они становились окрашенными в глубокий жел-
то-зеленый цвет. Криоскопическое исследование молекулярного веса показа-
ло, что в этих условиях происходит диссоциация примерно на 37%. В любом
случае окраска сразу не исчезала при действии восстановителя, например
гидрохинона или трифенилметила. В результате восстановления был полу-
чен соответствующий гваякол, а продукт реакции с трифенилметилом при
гидролизе давал трифенилкарбинол.
Гольдшмидт считал, что в равновесии гомолиза участвуют ароксильные ради-
калы и перекись диарила 2АгО • =₽*= АтО — ОАг. Однако, как в общем и все
другие предполагаемые перекиси диарилов, эта перекись как таковая не
существует по крайней мере при обычной температуре. Очевидно, что в про-
тивоположность перекисям диалкила и диацила центральная связь перекисей
диарила в основном за счет сопряжения арильного остатка с кислородом
ослаблена так сильно, что происходит ее разрыв. Выделенные же димеры
радикалов ароксила получаются за счет орто- или пара- О — С или С — С
спаривания радикалов, а не за счет образования связи О — О. Если поло-
жение, в котором образуется связь при переходе от радикалов к димеру,
занято заместителем, то происходит в основном образование связи О — С,
что приводит к арилоксициклогексадиенону (представляющему собой сопря-
женный винилог сложного эфира). Конечно, когда таким заместителем явля-
ется атом водорода, он мигрирует, давая фенольный таутомер, и фактором,
определяющим относительную устойчивость, становится ароматизация.
В примере Гольдшмидта димеризация обязательно должна была происходить
по орто-положению и включать образование связи О — С. Следовательно,
правильная схема этого равновесного процесса может быть представлена
1029
Глава XVII
следующим образом [23]:
В термодинамическом смысле ароксильные радикалы, которые получаются
в растворе или в твердом состоянии, должны быть значительно более устойчи-
выми, чем их перекисные димеры, поскольку их устойчивость сравнима или
больше устойчивости наиболее стабильных из изомеров, образующихся при
димеризации с образованием связей с кольцом.
По причинам, объяснявшимся в разд. 1 ,в, следует ожидать, что ароксильные
радикалы, подобно диариламинильным радикалам, должны быть электро-
отрицательными, и следовательно, должны стабилизоваться орто- или пара-
заместителями Ц-М-типа, например метоксигруппой, или -|~7И-заместителя-
ми, например фенильной или бензогруппой, которые могут проявлять
4-Л/-эффект. Оба типа стабилизующих групп представлены в примерах
Гольдшмидта. В общем можно сказать, что ароксилы, тризамещенные в поло-
жения 2, 4 и 6 фенильными, алкоксильными и mpewi-бутильными группами,
наиболее устойчивы из известных радикалов. Все они получены методами
Гольдшмидта, т. е. окислением соответствующих фенолов окислами металлов
или феррицианидом. Стабилизующее влияние бутильной группы с этой точки
зрения является неожиданным, но поскольку стабилизующее действие эти
группы оказывают, занимая орто-положение, очевидно, что очень важным
фактором являются пространственные затруднения димеризации.
2,4, 6-Трифенилфеноксил, исследованный Димротом и сотр. [34], был
получен в твердом состоянии лишь в виде димера, с которым, однако, в раст-
воре при температуре, близкой к комнатной, радикал находится в равновесии.
Например, в сероуглероде при 20 °C константа равновесия, определенная
колориметрическим методом, составляет 2,4-10-6 моль/л. Димер представля-
ет собой продукт гсара-сочетания эфира и кетона
уС6Н5 /СвЩ /С6Н5
2С6Н5-/ У-о. С6н5-/_____------о--------7\ZZ/=0
С 4—( С6Н5/ ч
хс6н5 хс6н5 хс6н5
4-Фенил-2,6-ди-трет-бутилфеноксил, исследованный Мюллером и сотр. [36].
вероятно, в наибольшей степени диссоциирован на свободные радикалы
в растворах при комнатной температуре. Как установлено методом ЭПР,
при температуре 20 °C в 10 %-ном растворе бензола присутствует около 88%
радикалов. При более сильном разбавлении и при применении других мето-
дов определения концентрации радикалов димер, по-видимому, практически
полностью диссоциирован. Строение димера неизвестно; вероятно, он пред-
ставляет собой продукт гсара-сочетания эфира и кетона.
2-Метокси-4,6-ди-трет-бутилфеноксил, исследованный Петранеком и его
коллегами, а также Хаугиллом и Мидлтоном [37], представляет собой ради-
кал голубого цвета, который в растворе находится в равновесии со своим
димером. Как показывает спектр ядерного магнитного резонанса, димер
1030
Стабильные радикалы
почти наверняка представляет собой продукт соединения эфира с кетоном со
связью в положении 4.
2 mpem-C^Hg —<
/ОСН3
О-
'ЧчС4Нд-трет
т рет-СцНд —
О
s= О
СНзО —;
С4118-/п/>ет mpem-C4H8 C4H9-mpezn
Два из наиболее известных ароксильных радикалов, которые абсолютно
устойчивы в твердом состоянии, были открыты одновременно Куком, Мюл-
лером и их сотрудниками. Это 4-метокси-2,6-ди-тпретп-бутилфеноксил [38]
и 2,4,6-три-тпретп-бутилфеноксил [39]. Оба зти радикала представляют собой
черные твердые вещества; первый из них образует красный, а второй —
голубой растворы. Образование димеров не было обнаружено.
/C4Hg-mpem /C4Hg-mpem
/>—О- трет-Сч11д——О-
^ДНд- трет ^С^Нд-трет
Устойчивые ароксильные радикалы
ж. Триарил- и диарилацилгидразилы [40]. В 1920 г. Гольдшмидт обна-
ружил, что гексаарилтетразины более сильно диссоциируют до триарил-
гидразилов, чем тетраарилгидразины Виланда до диариламинилов. При
встряхивании трифенилгидразина с перекисью свинца в диэтиловом эфире
при комнатной температуре получается темно-голубой раствор, который
содержит большое количество радикалов трифенилгидразила. Если опыт
повторить при температуре —60 °C в диметиловом эфире, то в виде светло-
зеленого твердого вещества выделяется гексафенилтетразон. Голубые раст-
воры этого вещества проявляли отклонения от закона Бера, что указывало
на частичную диссоциацию.
(CeH5)2N-N(C6H5)-N(C6H5)-N(C6H5)2 2(CeH5)2N-NC6H5
Голубой раствор немедленно реагировал с гидрохиноном, образуя трифенил-
гидразин, а также с окисью азота, образуя N-нитрозотрифенилгидразин,
более медленно он реагировал с трифенилметилом. При стоянии раствора
вещество разлагалось до дифениламина и бпс-дифенилгидразона хинона
(C6H5)2N - N = С6Н4 = N — N(C6H5)2.
Следует особенно остановиться на еще одном триарилгидразиле ввиду его
особенно высокой устойчивости и ввиду того, что он служит связующим
звеном с классом диарилацилгидразилов, которые будут рассматриваться
ниже. Это дифенилпикрилгидразил, который Гольдшмидт и Ренн [40]
получили путем конденсации несимметричного дифенилгидразина с пикрил-
хлоридом и окисления образовавшегося дифенилпикрилгидразина перекисью
свинца в эфире. Они получили радикал в виде фиолетово-черного твердого
вещества, похожего на перманганат калия. Далее было показано [41], что
радикал обладает парамагнитными свойствами, а его магнитная восприим-
чивость соответствовала магнитной восприимчивости одного неспаренного
спина на молекулу. Детально исследован спектр электронного парамагнит-
ного резонанса этого радикала [42]. Поглощение света фиолетовым раствором
1031
Глава XVII
подчиняется закону Бера, и при охлаждении до —80 °C интенсивность окрас-
ки не уменьшается. Молекулярный вес вещества в растворе соответствует
полностью диссоциированному гидразилу. Вещество не поглощает окись
азота, но мгновенно восстанавливается гидрохиноном до дифенилпикрил-
гидразина. Радикал удобно титровать гидрохиноном, поскольку индикатор
не требуется и по исчезновению окраски раствора судят о конце титрования.
Соответствующий тетразиновый димер не был получен.
При использовании ацетильной, бензоильной и замещенных бензоильных
групп в качестве ацильных заместителей Гольдшмидт и сотр. [40] получили
различные диарилацилгидразины типа Ar2N — NAcH и ArAcN — NArH.
При окислении эти гидразины превращались в тетраарилдиацилтетразины
и (или) продукты их гомолиза — диарилацилгидразилы типа А и Б, пред-
ставленные ниже, которые соответственно называются а,а-диарил-0-ацил-
Р-гидразилами и а,р-диарил-а-ацил-0-гидразилами:
Ar2N—N Ас—N Ас—N Аг2 2Ar2N — N Ac (A)
ArAcN —N Ar—N Ar—N Ar Ac 2ArAcN—NAr (Б)
Почти все эти радикалы устойчивы в том смысле, что они не подвержены
саморазложению, однако они обратимо коллигируются, давая тетразаны.
Поэтому процесс можно детально изучить. Все тетразаны, по крайней мере
частично, диссоциированы в растворе на гидразилы, а некоторые из них дис-
социированы полностью. После растворения тетразанов медленно устанав-
ливалось равновесие (часто в течение нескольких минут) при комнатной
температуре, и после того, как равновесие установилось, его можно «замо-
розить» путем охлаждения до низкой температуры. Что касается реакции, то
большинство радикалов более или менее медленно взаимодействует с окисью
азота и трифенилметилом; но все они мгновенно восстанавливаются даже при
низкой температуре гидрохиноном или гидразобензолом, образуя диарил-
ацилгидразины. Основываясь на таком различии в скоростях (медленное
образование и быстрое восстановление радикалов), Гольдшмидт разработал
титрометрический метод определения степени равновесной диссоциации.
Раствору позволяли достичь состояния равновесия при необходимой темпе-
ратуре. Затем его быстро охлаждали до низкой температуры и титровали
при этой температуре до исчезновения окраски при помощи восстановителя.
Гольдшмидт и сотрудники определили большое число констант равновесия
диссоциации полученных ими тетразанов в различных растворителях при
различных температурах. Примеры полученных ими констант, представлен-
ные в табл. 225, относятся к образованию а,а-диарил-р-ацил-0-гидразилов;
данные свидетельствуют о противоположном влиянии полярных структур-
ных изменений в «ацильных» (включен также и пикрил) и арильных группах.
Из данных табл. 225 совершенно ясно следует, что чем сильнее ацильная
группа притягивает электроны и чем сильнее арильная группа отдает элек-
троны, тем более устойчивы соответствующие радикалы. Эти наблюдения
были обобщены в 1929 г. на этой основе и связаны с вопросами стабильности
других типов радикалов [26], как описано в разд. 1,в—е. Для наших целей
удобно сравнить гидразилы с амилами. Двухлигандные аминильные радика-
лы являются электроотрицательными, как это было установлено, исходя из
гораздо более высокой устойчивости двухлигандного азотного аниона по
сравнению с двухлигандным азотным катионом. Гидразильные радикалы
гораздо менее электроотрицательны, поскольку, в то время как соответ-
ствующий анион будет обладать теми же свойствами, что и предыдущий, соот-
1032
Стабильные радикалы
ветствующий катион, который в этом случае является частично четырех-
лигандным, станет более устойчивым.
N==N-
-N —
.Двухлигандная
структура
Четырехлигандная
структура
Мезомерноь
состояние
Учитывая электроотрицательность аминильных радикалов, можно сказать,
что они будут стабилизоваться электроположительными заместителями, по-
скольку гораздо более важным является рассредоточение электронодефи-
цитного центра (как в соответствующем катионе), чем рассредоточение избы-
точной плотности неподеленных электронов. Используя аналогичные аргу-
Таблица 225
Константы равновесия (К, моль/л) для реакций
Ar2N — NAc — NAc—NAr2 J Ar2N—NAc (по данным Гольдшмидта)
Ацил в радикалах (C6H5)2N - NAc Растворитель Темпера- тура, °C Найденная 104 к 104 к, найденная или вычисленная для толуольного раствора при - 18 °C
Ацил Толуол -18 Небольшая Небольшая
Бензоил » -18 1,14 1,14
п-Нитробензоил » -18 5,3 5,3
Пикрил » -18 100%-ная дис- 100%-ная диссо-
социация циация
R, R в радикалах (n-RCeH4)2N-N(COC6H5)
no2, no2 Хлороформ 0 0,006 - 0,00014
no2, h » -18 0,83 — 0,04
Br, Br Толуол -18 0,145 0,145
Br, H » -18 0,33 0,33
H, H » -18 1,14 1,14
CH3, H » -18 4,3 4,3
CH3, CH3 » -18 17 17
CH3O, H Ацетон -18 35 - 25
CH3O, CH3O » —50 100%-ная дис- 100%-ная диссо-
социация циация
менты, можно сказать, что гидразильные радикалы будут стабилизоваться
как электроположительными, так и электроотрицательными заместителями,
так как в этом случае почти одинаковое значение имеют и рассредоточение
электронодефицитного центра, и рассредоточение неподеленной пары элек-
тронов
1033
Глава XVII
Основными каноническими структурами являются неполярная и диполярная
структуры
N— и \я+ —N —
Вторая из них — диполярная структура — в основном и объясняет стабили-
зующее влияние полярных заместителей. Гольдшмидт установил, что введе-
ние полярных заместителей обоих типов повышает устойчивость гидразила
и что эффективными могут быть оба вида стабилизации одновременно. Пока-
зано [42], что в молекуле дифенилпикрилгидразила 62% спиновой плотности
находится на атоме азота, связанном с пикрильной группой (неполярная
структура), а остальная часть — на другом атоме азота (как в диполярной
структуре) с небольшой дисперсией по фенильному кольцу.
Таблица 226
Энергия [ккал/моль (4,187-103 Дж/моль)] и энтропия [кал/моль.°С (4,187 Дж/моль-°C)]
реакции и активации для реакций
(«-RCeH4)2N-N(COCeH5)N(COCeH5)N(CeH4R-»)2 (2h-RC6H4)2N - NCOC6H5
в ацетоне (по данным Уилмарта и Шварца)
R, R Равновесие Гомолиз Коллигация
дн AS дн* Д8* дн+ Д8*
Вг, В г 11,2 25,1 17,2 --2,7 6,0 -27,8
Вг, Н 10,9 23,2 16,6 -6,6 5,7 -29,8
Н, Н 9,2 23,9 16,6 -7,6 7,4 -31,3
сн3, н 7,9 19,6 15,8 -8,0 8,0 -27,6
СН3,СН3 6,7 18,0 15,0 -10,3 8,3 —28,3
Уилмарт и Шварц [43] определили ряды изменения констант равновесия
и констант скоростей образования иара-замещенныха,а-дифенил-р-бензоил-
0-гидразилов из тетразанов в растворе ацетона при различных температурах,
что позволило вычислить энергии и энтропии реакции и активации. Неко-
торые из полученных данных представлены в табл. 226. Эндотермичность
процессов гомолиза составляет 7 ... И ккал/моль (29,31 -103 ... 46,05 X
X 103 Дж/моль), а энтропия, как и следует ожидать для реакций дис-
социации, сильно возрастает. Энергии активации гомолиза выше примерно на
15...17 ккал/моль (62,80-103...71,18-103 Дж/моль), а энтропия активации
гомолиза имеет небольшую отрицательную величину, поскольку обычно энт-
ропия активации не учитывает изменения энтропии по координате реакции
(гл. II, разд. 2,г). По разности соответствующих величин можно определить,
что энергия активации для коллигации составляет 6...8 ккал/моль (25,12 х
X 1O3...33,49-103 Дж/моль) и что энтропия активации для этого процесса
является сильно отрицательной, что обычно и наблюдается для реакций
ассоциации ввиду значительных потерь в поступательной и вращательной
энтропии. Основной причиной, обусловливающей сам факт наличия энергии
активации процесса коллигации, является необходимость создания некоторой
предварительной концентрации спиновой плотности на коллигирующихся
атомах азота; на этот процесс оказывают влияние соседний атом азота и за-
местители. Влияние растворителя [40] на эти равновесные процессы будет
значительно более сильным, чем влияние растворителя на равновесие гомо-
лиза гексафенилэтана до трифенилметила (разд. 1,г). В случае четырех раст-
1034
1ые радикалы
Таблица 227
Влияние растворителей на константу равновесия и на теплоту и энтропию
диссоциации до а,а-дифенил-р-бензоилгидразила (по данным Гольдшмидта)
Растворитель 104К (при -18 °C), моль/л ДН, ккал/моль (4,187-103 Дж/моль) Д5, ккал/моль (4,187-103 Дж/моль)
Эфир 0,27 9,6 16,9
Толуол 1,14 10,3 22,4
Ацетон 1,48 8,0 13,7
Хлороформ 23,6 5,5 5,7
ворителей, представленных в табл. 227, константы равновесия гомолиза
с образованием а,а-дифенил-р-бензоил-р-гидразила при —18 °C изменяются
в 80 раз. В то время как теплота диссоциации с образованием трифенилметила
относительно нечувствительна к изменению растворителя, теплота диссоциа-
ции с образованием а,а-дифенил-Р-бензоил-0-гидразила изменяется в значи-
тельной степени и всегда в этом же направлении изменяется энтропия.
В этом случае создается сложная ситуация, которая не является неожидан-
ной. При диссоциации углеводородов сольватация будет в общем небольшой
и связана в основном с одним фактором — ненасыщенностью радикала,
однако при диссоциации связей в N — N-цепи все атомы в исходном соедине-
нии и в продукте реакции будут сильно сольватироваться и поэтому термо-
динамические параметры будут отражать лишь небольшую разницу между
большими величинами.
з. Диарил- и диалкиламиноксилы. Большая часть этих радикалов устой-
чива в твердом состоянии и полностью мономерна в растворе даже при низких
температурах. Некоторые из этих радикалов известны; они устойчивы в виде
чистой жидкости и газообразном состоянии. Такие радикалы можно назвать
аминоксилами, как указывалось выше, или окисями аминилов; исследование
спектров их электронного парамагнитного резонанса показывает, что в этих
радикалах спиновая плотность практически поровну распределена между
атомами кислорода и азота. Основные канонические структуры совершенно
аналогичны таким же структурам гидразила. Их можно назвать неполярной
и диполярной структурами.
/N — О и \у+ — б
В мезомерном состоянии концентрация электронов на кислороде несколько
выше, чем на дифенилпикрилгидразиле, связанном с пикрильной группой,
и поэтому спиновая плотность на этом азоте в аминоксилах несколько боль-
ше, чем на дизамещенном атоме азота гидразила. Ни один из возможных
димеров аминоксилов
R2N —О—О —NR2, R2N—О —NR2—б или б — T$R2—NR2— б
в растворе не был обнаружен.
Дифениламиноксил (C6H5)2NO был синтезирован Виландом и Оффенбэхером
[44] в 1914 г. при окислении дифенилгидроксиламина окисью серебра в эфи-
ре. Был получен красный раствор, из которого радикал выкристаллизовы-
вался в виде темно-красных игл с т. пл. 62 °C. Второй метод его получения
по Томасу [45] заключается в окислении дифениламина: радикалы перекиси
1035
Глава XVII
трет-бутила, образующиеся из трет-бутил гидроперекиси и цериевых ионов,
окисляют дифениламин в процессе цепной реакции следующим образом:
mpem-C4H9O2H-|-Ce4+ —> mpem-C4H9O£ +Н+4-Се3+
(C6H5)2NH —mpem-C4H9O —> (C6H5)2N • -|-mpem-C4H9OH 1
(C6H5)2N. +mpem-C4H9O2• (C6H5)2NO.+mpem-C4H9O. J Чепь
Можно вырастить большие кристаллы этого радикала, изоморфные бензо-
фенону, с которыми он образует смешанные кристаллы. При попытке хра-
нить радикалы при комнатной температуре они претерпевают глубокое разло-
жение, однако их можно длительное время хранить при температуре —"15 °C.
Все методы определения молекулярного веса показывают, что вещество моно-
мерно даже при низких температурах. Этот радикал мгновенно восстанавли-
вается рядом восстановителей. Он быстро соединяется с окисью азота и с три-
фенилметилом, хотя продукты, первоначально образующиеся после добавле-
ния этих радикалов, претерпевают сложное разложение.
Большинство из известных замещенных дифениламиноксилов обычно напо-
минают дифениламиноксил: они не очень устойчивые вещества. Однако неко-
торые радикалы этого ряда представляют исключение и гораздо более устой-
чивы. Наиболее простые из них — ди-и-анизиламиноксил, открытый в
в 1919 г. Куртом Мейером и Бильротом [46], и ди-и-нитрофениламиноксил,
полученный в 1920 г. Виландом и Ротом [47]. Первый кристаллизуется в виде
медно-красных пластин и плавится с разложением при 120—150 °C, а вто-
рой — в виде темно-красных кристаллов, т. пл. 109 °C. Эти радикалы, а так-
же их замещенные могут храниться неопределенно долго при обычной тем-
пературе. Во всех исследованных условиях они мономерны, имеют один
неспаренный электрон и ожидаемый спектр ЭПР [48, 49]. Если на основании
этих качественных выводов принять, что сопряжение с метокси- и нитро-
группами оказывает стабилизующее действие, и не обращать внимания на
отсутствие данных по константам равновесия, то очевидна аналогия с гидра-
зидами, поскольку гидразилы, как мы видели, стабилизуются как -|-7И-, так
и —.^/-заместителями. Аналогия не является полной, так как число возмож-
ных положений, в которых могут находиться заместители в аминоксилах,
меньше, чем в гидразидах. Сопряжение с -|-Л/-группой, например с метокси-
группой, и сопряжение с —М-группой, например с нитрогруппой, может
привести к заполнению октета и (или) увеличить связывание, как показано
ниже для случая диполярной структуры радикала:
(СН5б^-СенД-)2ЪГ—о (O2N^H4J^N^4)~
Наиболее интересной особенностью аминоксильных радикалов является то,
что можно получить устойчивые алифатические аминоксиды, в которых фор-
мально отсутствует сопряжение. Известно очень мало таких примеров;
в этих случаях стабильность радикалов увеличивается при введении 4-7
и —/-заместителей, которые достаточно эффективны и обеспечивают сущест-
вование очень устойчивых радикалов [50].
Первым описанным таким радикалом был ди-щреш-бутиламиноксил (трет-
C4H9)2NO. Он был получен в 1961 г. Гофманом и Гендерсоном [51] при дей-
ствии натрия на 2-нитроизобутан. Вместо натрия можно использовать другие
очень сильные основания, например металлалкилы и даже реактивы Гринья-
ра [52]. При нагревании этот радикал не изменяется вплоть до температуры
120 °C. Он устойчив на воздухе и в воде, устойчив к действию разбавленных
водных растворов щелочей. Спектр ЭПР этого радикала показывает, что
1036
Стабильные радикалы
единственный неспаренный электрон, как и в случае ароматических амино-
ксидов, в равной степени распределен между атомами азота и кислорода
[42, 51].
Качественно такую устойчивость этого радикала можно объяснить тем, что
электроположительный эффект (4-7) двух третичных алкильных групп уве-
личивает «заселенность» электронами незаполненной валентной оболочки
и увеличивает его внутреннее связывание. Неполярные и диполярные струк-
туры радикала представлены ниже:
отрет-С4Н8
трет- С4Н9
ЕзС
N—О
ЕзС
трет-С4Н9
^N—О
отрет-С4Н9
Две другие приведенные выше формулы иллюстрируют еще один случай ста-
билизации радикалов электроотрицательным индукционным эффектом (—7)
трифторметильной группы в бпс-трифторметиламиноксиле. Этот радикал был
получен одновременно Блэкли [53] и Макаровым [54] и их сотрудниками
в 1965 г. Он образуется при действии окисляющих агентов (KMnO4, Ag2O,
F2) на бпс-трифторометилгидроксиламин (GF3)2N—ОН, который в свою оче-
редь получается из трифторнитрозометана CF3NO при действии аммиака или
при гидролизе его фотодимера. Радикал (CF3)2NO — газообразное вещество,
окрашенное в пурпурный цвет; он устойчив на воздухе и в воде, а также
в водных щелочах, реагирует с окисью азота, образуя (CF3)2NONO, который
фактически представляет собой фотодимер трифторнитрозометана. При более
низких температурах газообразный радикал конденсируется в темно-пур-
пурную жидкость с т. кип. —25 °C. При еще более низких температурах
жидкость приобретает коричневую окраску и, затвердевая, превращается
в желтое твердое вещество с т. пл. —70 °C. Исследование спектра электрон-
ного парамагнитного резонанса при различных температурах показывает,
что газ при 25 °C состоит целиком из радикалов с одним неспаренным спином
на одну молекулу, но в жидкости при низкой температуре они частично
ассоциированы, и что в твердом состоянии радикалы полностью ассоцииро-
ваны и образуют диамагнитное соединение, которое, вероятно, представляет
собой димер. Теплота диссоциации димера до радикалов оценивается
в 2,5 ккал/моль (10,47 -103 Дж/моль).
Опять необходимо предположить, что полярный эффект (в этом случае
электроотрицательный индуктивный эффект —7) увеличивает связывание,
как показано на приведенной выше схеме, хотя этот эффект должен умень-
шать общее содержание электронов в группе NO. Влияние полярных замес-
тителей на связывание в ди-щреш-бутиламиноксиле и бпс-трифторометил-
аминоксиле может быть выражено в общем виде при помощи теории молеку-
лярных орбиталей. В каждом радикале один электрон группы NO должен
находиться на разрыхляющей орбитали. Если полярный эффект любого
направления приводит к концентрации этого электрона на одном или дру-
гом атоме, то его разрыхляющее влияние, зависящее от степени его при-
надлежности к обоим атомам, будет уменьшаться, что приведет к увеличе-
нию общего связывания.
1037
Глава XVII
СПИСОК ЛИТЕРАТУРЫ
1. Gomberg М., J. Am. Chem. Soc., 22, 757 (1900); Вег., 33, 3150 (1900).
2. Heintschel К., Вег., 36, 320, 579 (1903).
3. Jacobson Р., Вег., 38, 196 (1905).
4. V. Baeyer A., Villiger V., Вег., 35, 1189 (1902).
5. Flurscheim В., J. prakt. Chem., 71, 505 (1905).
6. Werner А., Вег., 39, 1278 (1906).
7. Schmidlin J., Ber., 41, 2471 (1908).
8. Walden P., Z. physik. Chem., 43, 386 (1903).
9. Gomberg M., Ber., 40, 1851 (1907).
10. Gomberg M., Cone H., Ann., 376, 183 (1910); Gomberg M., van Slyke D. D., J. Am.
Chem. Soc., 33, 531 (1911).
11. Piccard J., Ann., 381, 347 (1911).
12. Ziegler K., Ewald L., Ann., 473, 163 (1929).
13. Ziegler K., Ewald L., Orth P., Ann., 479, 277 (1930).
14. Ziegler K., Orth P., Weber K., Ann., 504, 131 (1933).
15. Ziegler K., Ewald L., Seib A., Ann., 504, 162, 182 (1933); Ziegler K., Luttringhaus A.,
ibid., p. 189.
16. Ziegler K., Seib A., Knoevenagel K., Herte P., Andreas F., Ann., 551, 150 (1942).
17. Schlenk W., Weinkal T., Herzenstein A., Ann., 372, 1 (1910); Schlenk W., Herzenstein A.,
Ber., 43, 1753 (1910).
18. Gomberg M., Schoepfle C. S., J. Am. Chem. Soc., 39, 1652 (1917); Gomberg M., Sulli-
van F. W., ibid., 44, 1810 (1922).
19. Ziegler K., Ann., 434, 34 (1923).
20. Ziegler K., Schnell B., Ann., 445, 266 (1925).
21. Gomberg M., Nishida D., J. Am. Chem. Soc., 45, 190 (1923).
22. Bowden S. T., J. Chem. Soc., 1957, 4235.
23. Lund H., J. Am. Chem. Soc., 49, 1346 (1927).
24. Gomberg M., Buckler О. C., J. Am. Chem. Soc., 45, 207 (1923).
25. Ziegler К., Boye E., Ann., 458, 248 (1927).
26. Ingold С. K., Ann. Reports of Progress Chem. (Chem. Soc. London), 25, 164 (1928);
Burton H., Ingold С. K., Proc. Leeds Phil. Soc., Sci. Sect., 1, 421 (1929).
27. Lowenbeim A., Ber., 50, 601 (1925); Lowenbeim A., Gargarin B. F., ibid., p. 2643; Lowen-
beim A., Schmidt H., Ber., 60, 1851 (1927); Lowenbeim A., Schuster L., Ann., 481, 106
(1930).
28. Schlenk W., Mark E., Ber., 55, 2285, 2299 (1922).
29. Conant J. B., Bigelow N. M., J. Am. Chem. Soc., 50, 2041 (1928); Conant J. B.f
Schultz B. F., ibid., 55, 2098 (1933).
30. Wieland H., Ann., 381, 200 (1911); Wieland H., Lecher H., Ber., 45, 2600 (1912); Wie-
land H., Ber., 48, 1078 (1915).
31. Cain С. K., Wiselogle F. Y., J. Am. Chem. Soc., 62, 1163 (1940).
32. Goldschmidt S., Ber., 55, 3194 (1922); Goldschmidt S., Schmidt W., ibid., p. 3197; Gold-
schmidt S., Steigerwald C., Ann., 438, 202 (1924); Goldschmidt S., Vogt A., Bredig M. A.,
Ann., 445, 123 (1925).
33. Muller E., Schurr K., Scheffler K., Ann., 627, 132 (1959).
34. Dimroth K., Kalk F., Neubauer G., Chem. Ber., 90, 2098 (1957); Dimroth K., Neu-
bauer G., Angew. Chem., 69, 95 (1957).
35. Dimroth K., Kalk F., Sell B., Schlomer K., Ann., 624, 51 (1959); Dimroth K., Berudt A.,
Angew. Chem. internet. Edit., 3, 385 (1964).
36. Muller E., Schick A., Scheffler K., Chem. Ber., 92, 474 (1959).
37. Petranek J., Piter J., Doskocilova D., Tetrahedron Letters, 1967, 1979; Hewgill F. B.,
Middleton B. S., J. Chem. Soc., 1967C, 2316.
38. Muller E., Ley K., Chem. Ber., 88, 601 (1955); Cook C. D.,J. Am. Chem. Soc., 78, 2002
(1956).
39. Cook C. D., J. Org. Chem., 18, 291 (1953); Cook C. D., Woodworth В. С., J. Am. Chem.
Soc., 75, 6242 (1953); Miiller E., Ley K., Chem. Ber., 87, 922 (1954); Muller E., Ley K.,
Kiedaisch W., ibid., p. 1605.
40. Goldschmidt S., Ber., 53, 44 (1920); Goldschmidt S., Euler K., Ber., 55, 616 (1922); Gold-
schmidt S., Benn K., ibid., p. 628; Goldschmidt S., Ann., 437, 194 (1924); Goldschmidt S.,
Bader J., Ann., 473, 137 (1929).
41. Muller E., Muller-Bodloff L., Bunge N., Ann., 520, 235 (1935).
42. Buchachenko A. L., Stable Radicals, trans. C.N. Turnon and T. I. Turnon, Consultants
Bureau, New York, 1965, Chap. 4.
43. Wilmarth W. K., Schwartz M., J. Am. Chem. Soc., 77, 4543 (1955).
44. Wieland H., Offenbacher M., Ber., 47, 2111 (1914).
1038
Стабильные радикалы
45. Thomas J. R., J. Am. Chem. Soc., 82, 5955 (1960).
46. Meyer К. H., Billroth H. G., Ber., 52, 1476 (1919).
47. Wieland H., Roth K., Ber., 53, 210 (1920).
48. Cambio L., Gazz. chim. ital., 63, 579 (1933).
49. Бучаченко А. А., Оптика и спектроскопия, 13, 795 (1962).
50. Розанцев Е. Г., Усп. химии, 35, 658 (1966).
51. Hoffmann А. К., Henderson А. Т., J. Am. Chem. Soc., 83, 4671 (1961); Hoffmann A. K.t
Fieldman A. M., Gelblum E., Hodgson W. G., ibid., 86, 639 (1964).
52. Briere R., Rassat A., Bull. soc. chim. (France), 1965, 378.
53. Blackley W. D., Reinhard R. R., J. Am. Chem. Soc., 87, 802 (1965).
54. Макаров С. П., Якубович А. Ю., Дубов С. С., Медведев A. H,, ДАН СССР, 160, 2130
(1965); Мирошниченко И. В., Левин Г. М., Макаров С. П., Видейко А. Ф., Ж. стпукт.
химии, 6, 776 (1965).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адамантан и производные, сольволиз 446,
447
Азиды, реакции присоединения 887—890
Азометины
гидролиз 832, 833
образование 831—834
Азотистая кислота
влияние на нитрование нитроний-
ионом 281—285
и ее эфиры, вращательные изомеры
59, 60
как нитрозирующий агент 313 и сл.
Азотистый ангидрид 313, 314
Азотная кислота
ацидониевый ион 280, 517, 518, 520
и сл.
и ее эфиры, конформации 6
как источник нитроний-иона 271, 272—
281
Азулен 169, 172 и сл.
водородный обмен 296
диамагнитная восприимчивость 193,
194
размеры 183
реакции 173
энергия мезомерии 187, 188
Акролеин
вращательные изомеры 58
дипольный момент 112
Активация
молекулярная теория Лэпуорта 70
энергия 46 и сл.
Алифатические соединения
дипольные моменты 108, 111
направленная поляризуемость замести-
телей 136, 137 и сл.
Алканы
бромирование 481 и сл., 485
галогенирование молекулярными гало-
генами 479—487
дипольные моменты 107 и сл.
конформации 57, 58
нитрование 495 и сл.
теплоты образования 117 и сл.
фторирование 482 и сл., 485
хлорирование 479—481, 484, 485, 487
Алкиланилины, перегруппировка 745—
747
Алкилариловые эфиры, перегруппировка
747 и сл.
Алкилацетаты, дегидрокарбоксилирование
582 и сл.
Алкилбензолы
галогенирование 261, 262
мононитрование 260, 261
Алкилбромиды
дегидробромирование 578, 579
реакции обмена с Вг- 450—456
— отщепления с образованием олефи-
нов 551—557
реакционная способность 452, 453 и сл.
скорости реакций замещения 358, 359
Алкилгалогениды
дегидрогалогенирование 578—587
перегруппировки 633—640
реакции с аминами 362
— с галогенид-ионами 362
— замещения, солевой эффект 400—
408
— мономолекулярного замещения
457—459
— — отщепления 543, 549—557
— с натрацетоуксусным эфиром 362
сольволиз 359—363
— энтропия активации 391, 393
теплоемкость активации 393
Алкилий-ион 298
Алкилониевые
ионы, отщепление с образованием оле-
финов 554, 560 и сл.
соединения, реакции отщепления с об-
разованием олефинов 546 и сл.
Алкилсульфонаты, сольволиз, энтропия
активации 391
Алкилсульфониевые
ионы, отщепление с образованием оле-
финов 558 и сл.
соединения, реакции отщепления с об-
разованием олефинов 547—549
Алкилпиридиния ионы как псевдооснова-
ния 694 и сл.
Алкилы щелочных металлов, сравнение
с реактивами Гриньяра 843 и сл.
Аллилбромиды
изомеризация 706
обмен брома 706
Аллилхлориды, сольволиз 703 и сл.
Альдегиды
восстановление 848 и сл.
присоединение галогенов 803
Альдольная конденсация 214, 824—830
Амиды карбоновых кислот
гидролиз 965—-968
образование из эфиров 963—965
Амфипротонные растворители 912
Ангидрооснования 694 и сл.
Анизол
дейтерирование 295
нитрование 51
Анизотропия
диамагнитизма в ароматических систе-
мах 195 и сл.
поляризуемости 131
полярная 132, 133
Анилин
дейтерирование 295
иодирование 290
нитрование 751—753
— через нитрозирование 282 и сл.
производные как основания 931, 932
1040
Предметный указатель
Апионоидные реагенты 206
Анионотропия 224, 699—740
история 699—710
механизм бимолекулярный 704—707
— внутримолекулярный 707—710
— мономолекулярный 700—704
и псевдоосновность 702
равновесие 710
Аннулены 172
Антиоксиданты 490, 493
синеристы 495
Аптисопряжение 375, 835
Антоцианины
как ангидрооснования 698
как псевдооснования 699
Антрахинон, нитрование 273, 274
Антрацен
диамагнитная восприимчивость 193
размеры 181, 182
реакции 167
энергия мезомерии 162, 167, 188
Аренсульфонаты, реакции нуклеофильного
замещения 377—379
Ариламины
энергетические барьеры вращения 62,63
энергии активации 62, 63
Арилгалогенамины, перегруппировка 739—
743
Арилгидразоны, образование 834, 835
Арндта — Эйстерта реакция 615
Ароксилытые радикалы 1029—1031
Ароматические
аммониевые ионы, нитрование 235
и сл., 256—258
гетероциклические соединения, ре-
фракция 130
молекулы, диамагнитная восприимчи-
вость 193
— радиусы магнитных орбит электро-
нов 195, 196
— размеры 182 и сл.
— теплоты образования и гидрирова-
ния 187, 188
— энергия мезомерии 187 и сл.
соединения, азосочетание 307—309
— алкилирование 298—304
— — гомолитическое 338—340
— арилирование 334—338
— ацетилирование 305
— ацилирование 304—307
— бензоилирование 305, 306
— бромирование 288 и сл., 291
— водородный обмен 295—298
— галогенирование 285—292
— гидроксилирование 340 и сл.
— дейтерирование 296, 297 и сл.
— диазотирование 322
— иодирование 289 и сл.
— меркурирование 309—310
— нитрование 236—239, 263 и сл.,
270—285
— нитрозирование 282—285
— перегруппировки внутримолеку-
лярные 751—779
— поляризуемость заместителей, на-
правленная 135—138
Ароматические
соединения, скорости относительного
замещения галогенов 322—324
— сульфирование 292—294
— теории строения 156—178
— хлорирования 285—288, 291
углеводороды, альтернантные 168
— неальтернантные 168
— энергия мезомерии 162
ядра, гетероциклические, мезомерия
174 и сл.
— — размеры 182
— конденсированные, диамагнетизм
195
— — мезомерия 162, 165 и сл.
---размеры 181, 182 и сл.
— теории строения 156—178
Ароматический характер 156—178
Аррениуса
уравнение 48 и сл.
фактор частоты 49
энергия активации 49
Атомные состояния 20 и сл.
Аутоокисление 213, 489—495
аутокатализ 493
замедлители 494
ингибиторы радикалов 493 и сл.
катализ ионами металлов 493
кинетика 492
разрушители перекисей 493
Ахиральность 424
Аценафтилен 169, 173
циклодимеризация 882
Ацетали, сольволиз 372
Ацетальдегид
присоединение аммиака 212
Ацетилацетоп 82
Ацетилены, теплоты образования 118, 119
Ацетилий-ион 305
Ацетилнитрат, роль в нитровании 281
Ацетон, дипольный момент 112
Ацетонитрил 109
Ацетоуксусный эфир 658, 662
Ацилий-ионы 304 и сл.
Ацилия соли 305
Ацилоиновая
конденсация 185
перегруппировка 602 и сл.
Ацилоины 185
Бекмана перегруппировка 616 и сл., 626—
628
Бензвален и производные 724, 725
Бензидин 756
Бензидиновая перегруппировка 756—779
механизм 758—779
Бензиловая перегруппировка 603
Бензодипентадиен 169
Бензоилий-ион 305
Бензоиновая реакция 822
Бензойная кислота
ионизация 43 и сл.
рАа 45
Бензол
дейтерирование 297
66—0772
1041
Предметный указатель
Бензол
диамагнитная восприимчивость 193,
194
— поляризуемость 195
мезомерия, теория 161 —165
— энергия 120, 162, 187, 188
нитрование 49 и сл.
ориентация в бензольном кольце 156
и сл.
производные, дипольные моменты ИЗ,
114
размеры 178, 180, 181, 182
распределение заряда л-электронной
оболочки 161, 162
— зарядов в ядре 231 и сл.
рефракция 130
силовые постоянные 150, 151, 179
симметрия 139, 156, 178
теории строения 156—160
теория Тиле 159 и сл.
упругие свойства 151, 179 и сл.
формулы 156—160
Бензолдиазоний-иоп 308
разложение 319 и сл.
М-Бензосульфонил-К-(8-нитро-1-нафтил)-
глицин, энантиомеры 62
Бериллатропилен 178
Бифенила производные
заторможенное вращение 61
энергии активации 63
энергетические барьеры вращения 63
Бициклобутановые соединения 723
Бициклогексадиен и производные 723 и сл.,
724, 725
Бицикло-[2,2,1]-гептан (норборнан), соль-
волиз 446
Бицикло-[2,2,2]-октан, сольволиз 446
Бициклооктатриен 727
Бициклопентен и производные 721, 722,
723
Боратропилен 178
Брауна соотношение селективности 997
Бренстеда уравнение для кислотно-основ-
ного катализа 991—993
Броминия-ион 288, 289
Бромониевый ион 288
N-Бромсукцинимид как бронирующий
агент 488 и сл.
Брэдфилда — Джонса соотношение 993
и сл.
Бульвален 729—731
Бутадиен-1,3 84
вращательная изомерия 58
«-Бутан, вращательная изомерия 57 и сл.
Бъеррума теория силы двухосновных кис-
лот 914 и сл., 988
Вагнера — Меервейна перегруппировка 1
301, 302, 604—607, 637, 638 j
карбониевые промежуточные соедине-
ния 630 и сл.
— — — синартетические 732—740
принцип микроскопической обрати-
мости 628
пространственное протекание 626, 628
теория механизма 610—614
1042
Вагнера перегруппировка 584—586
Валентность 12 и сл.
Вальденовский цикл в реакциях замеще-
ния 420
у атома кремния 528 и сл.
— серы 533 и сл.
— фосфора 530 и сл.
Вальденовское обращение 419—447, 628
и сл.
Ван-дер-Ваальса силы 39
Вернер, теория образования связей 15, 16
Виттига перегруппировка 651 и сл.
Включения реакции 875 и сл.
Внутримолекулярные взаимодействия
передача 62, 64—68
и равновесия химических реакций 42—
45
и скорость химических реакции 45—51
и стереохимическая форма 51—62
Водородный обмен
ароматических соединений с кислота-
ми 295—298
прототропных соединений с гидрок-
сильными растворителями 684—691
Вольфа перегруппировка 615 и сл., 621,
878
Восстановление 202 и сл., 847 и сл.
растворяющимися металлами 850—853
— — полиенов, ориентация 851—853
Восстанавливающие агенты 847
Вращение заторможенное
ариламинов 62
бифенилов 61
бутадиена 58
вокруг простой связи 51—62
метильных соединений 54
в этане и производных 51 и сл.
Галогений-ионы 285
Гаммета — Буркхардта уравнение 995—
999
Гексаметилбензол, галогенирование 477
Гексафенилэтан 1018, 1020, 1021
Гексахлорциклогексаны, дегидрохлориро-
вание 570
Гептален 169, 172
Гераниол и его эфиры, анионотропия 700
Гетеролиз 15, 208
Гибридизация 30—32, 33, 34
и полярность 35 и сл.
Гидразобензол, перегруппировка 756, 757—
768
Гидразобензолы, диспропорционирование
776 и сл.
Гидразосоединения ароматические, пере-
группировка 756—779
Гидроксиламипы ароматические, перегруп-
пировка 748—751
Гидроперекиси 490 и сл.
Гиперконъюгацпя 95—98
и направленная поляризуемость 138
в нуклеофильном алифатическом заме-
щении 357, 364, 366
и размеры молекул 146—148
и силовые постоянные связей 151 и сл.
Предметный указатель
Гиперконъюгация
и электрические дипольные моменты
104, 109, 111
и электрофильное ароматическое заме-
щение 255 и сл., 315—318
Гипермезомерный эффект 96
Гиперэлектромерный эффект 97
Гипобромоний-ион 288 и сл.
Гипогалогенацидиевые ионы 285
Гипоиодоний-ион 289 и сл.
Гипохлороний-ион 285—288
Глиоксаль, вращательные изомеры 58
Голлеман
аномалия в электрофильном аромати-
ческом замещении 250—255
правило зависимости ориентации в аро-
матическом соединении и реакцион-
ной способности 231
Гомоаллильные ионы 647, 648
Гомолиз 15, 207
поверхностный молекул водорода 218
термический 216
энергия активации 1022
Гомолитические реакции 207, 208
Г омо литическо е
алифатическое замещение 478—497
— — галогенирование молекулярны-
ми галогенами 479—487
— — — различными агентами 487—
489
— — нитрование углеводородов 495—
497
ароматическое замещение 333—341
— — алкилирование 338—340
— — арилирование 334—338
— — гидроксилирование 340 и сл.
замещение 219—222
присоединение 222—223
Гомотропилиден 728 и сл.
Гофман
перегруппировка 617 и сл.
— стерическое протекание 620—622
правило отщепления олефинов 543—
549
реакция аминов 352, 878
Гофмана — Марциуса перегруппировка
745-747
Графит 181
Гриньяра
реактивы, присоединение к карбониль-
ным соединениям 843—847
— сравнение с металлалкилами щелоч-
ных металлов 843 и сл.
— структура 474, 845
реакция, кинетика 845 и сл.
Гроба фрагментация 587—591
Двуокись серы как растворитель 389
Двуокись углерода, рефракция 129
Дегидратация 829 и сл.
Дегидробензол 331, 332, 333
Дезалкилирование 299
Дезаминирование 443 и сл.
гидридный сдвиг 607 и сл.
диазосоединений 352
Дезаминирование
изменение размеров кольца 609 и сл.
пинаколиновое 604
Демьянова перегруппировка 607—610
Дейтерирование
ароматических соединений кислотами
295
прототропных соединений с дейтерок-
сильными растворителями 685—691
Диазоалканы, реакции присоединения
884—887
Диазоаминобензолы
перегруппировка 743
прототропия 667
Диазоаминосоединения ароматические, пе-
регруппировки 743 и сл.
Диазоний-ион 307
Диазосоединения как псевдооснования 697
Диазотирование у атома азота 512—525
Диалкиламинокспльные радикалы 1036
и сл.
Диамагнетизм 189
Диамагнитная
восприимчивость 188—197
— анизотропия 194—196
— влияние ненасыщенности 192
— — полярности 191
— — сопряжения 192 и сл.
— молярная 190
— направленная 194 и сл.
поляризуемость 188—197
— направленная 195
экзальтация 192, 193
Диамагнитные константы 190
атомов инертных газов 192
ионов 191, 192
Диариламинильные радикалы 1027 и сл.
стабильность в зависимости от струк-
туры 1027, 1028
Диариламиноксильные радикалы 1035—
1037
Диарилацилгидразилы 1031—1035
стабильность 1032—1034
Диарилгидразины, перегруппировки 756—
779
Диарилы 334 и сл., 340
1,2,5,6-Дибензантрацен, размеры 181, 182,
183
Дибензилиденацетон, фотодимеризация 882
Дибензокарбазол 766
1,2,4,5- Дибензопентален 170, 174
Дибромэтан, вращательные изомеры 56
Дигалогенкарбены 877 и сл.
Дигидробульвален 729
Диены, присоединение 892—900
катализ 899 и сл.
конфигурация переходного состояния
898
стереохимия 893—896
термодинамика и кинетика 896—898
Диизобутилены, взаимопревращения 658
Дикарбен углерода 879
Дикмана реакции 968—974
?<-Диксилилеп, размеры 180
Дильса — Альдера реакции 892—900
катализ 899 и сл.
66* 1043
Предметный указатель
Дильса—Альдера реакция
стереохимия 893—896
термодинамика и кинетика 896—898
Диметплацетамид, конформация 61
Диметп.тдпгидрорезорцин 82 и сл.
2,5-Диметилпиррол 176
Диметилсульфоний-9-флуоренилид 176
Диметплформамид, конформация 61
Динафтилин 766
6,6'-Динитродифеновая кислота, оптиче-
ские антиподы 61
Дипольный момент, пеисчезающий, усло-
вия существования 131
Дисперсионные силы 39
Дисротация 719
Дифенплен 170, 173 и сл.
реакции 174
энергия мезомерии 187, 188
Дифенплин 771
Дифенилкарбен 876, 877
Дихлорэтан, вращательные изомеры 56
Длины связей 106
в ароматических молекулах 181
и кратность 142
в молекулах с гиперконъюгацией 146—
148
в молекуле водорода 27
в несопряженных молекулах 140
и пространственный эффект 145
в сопряженных соединениях 142—146
спектрально определенные 139, 140
Дуалистическая теория Берцелиуса 12, 68
Дублетное состояние 24
Дублеты электронов 13
Дурол, производные, дипольные моменты
113, 114
Енолизация кетонов 674—678
равновесие с енолами 674 и сл.
кинетика 676
(Зайцева правило отщепления с образова-
нием олефинов 543, 549—557
Закон действия масс, влияние на нуклео-
фильное алифатическое замещение 400
Замещение
гомолитическое 219—222
— бимолекулярное 338
— мономолекулярное 1021
— насыщенное 219
— ненасыщенное 221
нуклеофильное 209—211, 212
— бимолекулярное 353 и сл.
— мономолекулярное 354 и сл.
— — молекулярность быстрых стадий
408—410
электрофильное 209—211, 212
Замыкание и раскрытие цикла, конрота-
торное и дисротаторное 717—721
Золотоорганические соединения 472 и сл.
Изатин, прототропия 658
Изобутан
вращательные изомеры 57
Изобутан
дипольный момент 107
— — дейтерированного 107
Изобутилен, полимеризация 817, 818, 819
Изонитрилы, дипольные моменты 103, 104
Изомерия вращательная (конформацион-
ная) 55 и сл.
Изомеры вращательные плоские 58—61
высоты энергетических барьеров 59
стабильность 58, 59
Изоэлектронпые ряды 18
Илиды 176
Инден 176
прототропия 669
Индол 175
энергия мезомерии 188
Индуктивный эффект 65, 70—76
квантовомеханический резонанс 74
п сл.
и нуклеофильное алифатическое заме-
щение 357—372
образование циангидринов 823
и отрицательная экзальтация рефрак-
ции 127, 128
п отщепление с образованием олефи-
нов 543—549, 557—561
и скорость бимолекулярного щелоч-
ного гидролиза эфиров карбоновых
кислот 941, 942
и электрофильное ароматическое заме-
щение 234—245, 247—255
— — — соотношение орто/пара 259,
263 и сл., 267 и сл.
Индуктомерный эффект 67, 76—77
Иодиний-ион 289 и сл.
Иодгидринная перегруппировка 603 и сл.
Ионизация ковалентной молекулы в поляр-
ном растворителе, энергетическая диа-
грамма 353
Канниццаро реакция 848 и сл.
Карбазол 175
энергия мезомерии 188
Карбалкоксикарбены 877
Карбен, образование в результате элими-
нирования 592—594
Карбены
получение 874
циклоприсоединение 874—879
— стереохимия 875 и сл.
Карбоксилат-ионы ненасыщенные, прото-
тропия 680
Карбоксильные группы
реакции 936—974
рефракция, поляризуемость 128, 129
Карбониевые ионы как промежуточные
частицы 298, 301 и сл., 304, 361 и сл.,
585, 586, 628—648
Карбониевый ион сипартетический 640—
648
Карбонильные соединения
алкоголоты 835 и сл.
гидраты 835 и сл.
ненасыщенные, прототропия 680 и сл.
присоединение гетеролитическое водо-
рода 847—858
10 и
Предметный указатель
Карбонильные соединения
присоединение кислот и оснований
824—842
— металлалкилов 842—847
Карбоновые кислоты
ароматические, галогензамещенные
926, 927
— дейтерирование 924
— константы кислотности 924, 925
и сл.
насыщенные 919, 920
— хлорзамещенные 922, 923
ненасыщенные, константы диссоциа-
ции 920, 921, 929
этерификация бимолекулярная 955—
959
— механизмы 936—938
— мономолекулярная 948—955, 959—
963
Катионоидные реагенты 206
Катионотропия 658—691
Квантовая механика 20
Квантовые числа 21, 23
Квартетное состояние 24
Кетены
дипольные моменты 112
присоединение к олефинам и карбо-
нильным соединениям 879
Кето-енольпая таутомерия 659
Кето-енольные системы
выделение таутомеров 659
пентадные 680 и сл.
равновесие 661 и сл.
скорости галогенирования, водород-
ного обмена, изомеризации и раце-
мизации 684—691
Кислота, сила 44
Кислотно-основное равновесие 910—933
влияние ненасыщенности 920 и сл.
измерение 910—914
индуктивный эффект 919—923
мезомерный эффект 923—928
пространственные эффекты 928—933
эффекты ближнего действия 933
— поля 914—919
Кислоты сопряженные 204, 910
Льюиса 207
сила 911
Кляйзена
конденсация 968—974
перегруппировка 710—714
реакция 830
Кнёвенагеля реакция 830
Ковалентное связывание 13
Ковалентные радиусы связей 141
Коллигация 14, 207
энергия активации 1022
Кольчато-цепная таутомерия 663—666
кето-лактолов 666
кето-циклолов 664
трехуглеродных систем 664 и сл.
л-Комплекса теория Дьюара 746, 749, 760
и сл.
Конротацпя 718
Константа
кислотности 44, 910
Константа
основности 911
равновесия 42
скорости 48
Константы
автопротолиза растворителей 911, 913
гидролиза анионов 911
— катионов 910
энергии связей 115—119
Конфигурация
стереохимическая, абсолютная, опре-
деление 422, 423 и сл.
— соотношение 421—424
электронная 24, 30 и сл.
Конформация 52 и сл.
ванны 52, 53
заслоненная 52
заторможенная 52
кресла 52, 53
энергетический барьер вращения 53,
54
Координата реакции 46 и сл.
Координация 14, 15 и сл., 208
Коричные кислоты и их производные,
фотодимеризация 883 и сл.
Коронен, размеры 181, 182
Котарнин как псевдооснование 695 и сл.
Коупа перегруппировка 714—716
Кратность связи 140 и сл.
и длина 141 и сл.
силовые постоянные 150—152
Кремнийорганические соединения 527—530
Ксантгидрол как псевдооснованпе 698
Курциуса перегруппировка 618 и сл.
стереохимическое протекание 620—622
Лиганд 15
Линалоол и его эфиры, анионотропия
700
Литийорганические соединения 843, 844
Лондона (электрокинетические) силы 39
Лоссеня перегруппировка 618
стерическое протекание^, 620
Льюис, теория
атомов 13
образования связей 14 и сл.
Магнийорганические соединения см. Гринь-
яра реактивы
Манниха реакция 696
Марковникова правило 784
Мезитилен, производные, дипольные момен-
ты ИЗ
Мезитоплий-иоп 305
Мезомерия
бензола 161—165
и стереохимическая форма 88 и сл.
теория 175, 176
Мезомерные
системы, планарность 88
— симметрия 84, 138
— электрическая поляризуемость
126-130
состояния, связь с валентными струк-
турами 86
1045
Предметный указатель
Мезомерный эффект 66, 70, 77—88
нуклеофильное ароматическое замеще-
ние 321—324
образование циангидринов 824
и равновесие кислота — основание
923—928
резонанс 85—88
силовые постоянные связей 150—152
п скорость бимолекулярного основ-
ного гидролиза эфиров карбоновых
кислот 941, 942
теплота гидрирования 187
— образования 119, 188
и электрический дипольный момент
110—112
электрофильное ароматическое замеще-
ние 250—255
— — — соотношение орто!пара 264
и сл., 268 и сл.
Меншуткина реакция 352, 362
Метан
нитрование газофазное 496 и сл.
фотолиз 219
Метилацетплен, длина связей 146, 147
Метилппкрампд. нитрование 510
Метилен 874 и сл.
Метпленазометины, прототропия 668, 689—
691
Метод
валентных схем для бензола 161, 162
и сл.
— — для цикл обута диена 168
— — для циклооктатетраена 168
конкурирующих реакций 246
молекулярных орбиталей 33, 161, 164
Михаэля реакция 214, 837 и сл.
Молекулярная активация, теория Лэпуорта
70
Молекулярность реакции 357
Мультнплетность 24
правило Гунда 23
Муравьиная кислота, вращательные изо-
меры 59 и сл.
Мутаротация сахаров 666
Напряжение в циклах 183—187
Банера 184
Питцера 185
Прелога 185
Натрппорганпческие соединения 843, 844
Нафталин 173
арилпрование 335
диамагнитная восприимчивость 193
кратность связи 166
размеры 181, 182
реакции 166
энергия мезомерии 162, 166, 188
Нафтидпп 766
Небензоидпые замкнуто-сопряженные
системы 167 —174
Ненасыщенность 58, 59
Неопентплпроизводпые, перегруппировки
607, 613
Нитрилы
ненасыщенные, прототропия 680
Нитрилы
окиси, реакции циклоприсоединения
889 и сл.
Нитроамины ароматические, перегруппи-
ровка 751—756
n-Нитробензойная кислота, ионизация 43
и сл.
Нитробензол
дипольный момент 104
нитрование 49 и сл.
фенилирование 335
Нитрование 49 и сл.
по азоту 508—511
алканов и циклоалканов 495 п сл.
— — газофазное 496 и сл.
ароматических соединений нптронип-
иопом 270—285
— — переносчики нитроний-иона
280-282
— — через нитрозирование 282—
285
по кислороду 511 и сл.
непрямое, теория 751 — 753
Нитрозамины ароматические, перегруппи-
ровка 744 и сл.
Нитрозирование
у атома азота 513—525
— кислорода 525—527
— — воды 526
Нитрозония-иоп 271, 313, 513—525
переносчики 513, 516, 517 и сл.
Нитрозофенолы, прототропия 658
Нитрометан
дипольный момент 104
как растворитель 389, 390
Нитроний-ион 50, 270—285, 508, 509, 511
и сл.
доказательства нитрования нитронип-
ионом 273—276
идентификация 270—273
механизм нитрования 278—280
— образования 276—278
переносчики 280—282, 513
Нитрония соли 272, 273
Нитроны, реакции циклоприсоединения
891 и сл.
Нитропарафины, прототропия
кинетика изомеризации 679
равновесие 678
скорость 678 и сл.
о-Нитрофенилбороновая кислота, сила кис-
лоты 933
Норкарадиен и производные 726
Нуклеофильное алифатическое замещение
352—459
алкиларепсульфонатов 377—379
влияние ионной силы 401—404
— растворителя 379—390
внутримолекулярное 440
гиперконъюгации эффект 366
закон действия масс, кинетические эф-
фекты 379—385
индуктивный эффект 357 — 372
катализ 394—399
кинетический эффект растворителя
379—385
1046
Предметный указатель
Нуклеофильное алифатическое замещение
механизм бимолекулярный 353 и сл.,
357-379
— мономолекулярный 354 и сл.
— обозначения 353, 354, 356 и сл.
несольволитическое в SO2 410 и сл.
переходные состояния 449, 450 и сл.
в полярных апротонных растворителях
384-390
пространственное протекание 419—447
— — алициклические системы 444—
447
пространственные эффекты 444—459
реагенты, кинетические эффекты 372—
376
солевые эффекты 400—419
сольволиз, кинетические эффекты 404—
410
теории 353
тип заряда 380
при углероде в голове моста 445, 446
электромерный эффект 364—372
энтропия активации 390—392
эффект утяжеления 455 и сл.
Нуклеофильное ароматическое замещение
318-333
бимолекулярное 321—328
водорода 327 и сл.
дегидробензольный механизм 330—333
мезомерный эффект в диазосоедине-
ниях 321—324
мономолекулярное 319—321
нитро- или циапгруппы 328—330
промежуточные продукты, природа 326
Нуклеофильное
замещение 209—211, 212
— у атома кремния 527—530
— — серы 533 и сл.
— — фосфора 530—533
— у олефинового атома углерода 460—
463
— пространственное замедление бимо-
лекулярных реакций 449—456
— — — мономолекулярных реакций
458 и сл.
присоединение 211—214, 820—858
Нуклеофильность 204, 374
Нуклеофильные
перегруппировки ароматических соеди-
нений 748—751
— насыщенных соединений 600—648
— ненасыщенных соединений 691—699
реагенты 203 и сл.
реакции 208—216
Обменное взаимодействие 40
Обрыв цепи 818
Овален, размеры 181, 182
Окисление 202 и сл.
фенолов и енолов 526 и сл.
Окислы азота как нитрующие агенты 271
и сл., 282
Окислительно-восстановительные реакции
216
Оксепин, изомеризация 726
Оксимы, образование 834 и сл.
Октеты электронов 13
Олефины
гидратация 789
образование в результате 1,2-отщепле-
ния 538—590
полимеризация, инициируемая кис-
лотами 816—820
— — основаниями 854
— — радикалами 868—873
присоединение азидов 887—890
— альдегидов 867 и сл.
— галогенов 797—811, 862—865
— галогеноводородов 784—792, 859
и сл.
— иона гидроксония 786—792
— карбенов 875—879
— полигалогеналканов 865—867
— тиолов и сероводорода 860 и сл.
теплоты образования 118, 119
цикло димеризация 879—884
Оловоорганические соединения 473
Орбитали 22 и сл.
атомные 22 и сл., 28 и сл.
— гибридные 31
— линейные 31
— перекрывание 29 и сл.
— плоские тригональные 31
— полярные графики 29 и сл.
— тетраэдрические 31, 32
— угловые величины 29—32
молекулярные 25
с вязи л-типа 33
— o'-типа 25, 26
связывающие 25, 27
разрыхляющие 25, 28
эквивалентные 23, 25
Ортона перегруппировка 739—743
Основания
Льюиса 207
сила 911
сопряженные 204, 910
1,1-Отщепление с образованием карбенов
592—594
бимолекулярное 594
мономолекулярное 592—594
1,2-Отщепление
классификация 215
мономолекулярное в газовой фазе
578-587
— — — катализ кислотами 586 и сл.
— — — кинетические эффекты 578—
583
— — — с перегруппировкой Вагнера
584—586
— — — пространственное протекание
583 и сл.
с образованием ацетиленов 567 и сл.
— карбонильных соединений 215
— олефинов 538—590
— — бимолекулярное 538—540, 543—
— — влияние температуры 567 и
сл.
— — индуктивный эффект 543—549,
557—561
1047
Предметный указатель
1,2-Отщепление
с образованием олефинов мономолеку-
лярное 540—542, 554—557
— — правила ориентации 542—543
— — правило Гофмана 543—549
— — — Зайцева 543, 549—557
— — пространственное протекание
568-577
— — тип заряда 562—566
— — электромерный эффект 549—561
— — эффект растворителя 562—566
фрагментация Гроба 587—591
Парамагнетизм 188
Пентален 169
Перегруппировки
в ароматических системах 738—779
— — внутримолекулярные 751—779
— — нуклеофильные 748—751
— — электрофильные 738—748
валентные 710—731
классификация 224
в насыщенных системах, нуклеофиль-
ные 600—648
— — — стерическое протекание
619-628
— — — теория механизма 610—614
— — с нормальной скоростью 632—
640
— — с ускорением 632—640
— — электрофильные 648—652
в ненасыщенных системах 658—731
— — валентные 710—731
— — катионотропные 658—691
— — прототропные 658—691
— — псевдоосновные 691—699
Передача внутримолекулярного взаимо-
действия
внешняя 62, 64
внутренняя 62
Перекиси 489—495
Перекись бензоила, пиролиз 334
Перенос цепи 818
Переходное состояние
реакции 46
термодинамическая активность 48
Перилен, размеры 181, 182
Перкина реакция 830 и сл.
Пинаколиновая перегруппировка 600—602
стерическое протекание 622—626
теория механизма 610—614
Пинаколиновое дезаминирование 604
Пиразин, размеры 182
Пирен, размеры 181, 182
Пиридин
диамагнитная восприимчивость 193
размеры 182
реакции с металлоорганическими со-
единениями 327
энергия мезомерии 188
Пиридоны как ангидроосиования 694 и сл.
Пироны как ангидрооснования 698
Пиррол
дипольный момент 175
диполярные структуры 174
1048
Пиррол
мезомерия 175
основность 175
размеры 182
реакционная способность с галогена-
ми 175
— — при озонолизе 176
Полимеризация олефинов 223
под действием кислот, оснований и ра-
дикалов 854 и сл.
ингибиторы 872 и сл.
инициируемая кислотами 816—820
— основаниями 854—858
— радикалами 868—873
Полимеры 223
живущие 858
Полиолефины, присоединение
галогенов 811—816
галогеноводородов 792—797
Полиперекиси 491
Полифторолефины, присоединение к оле-
финам 879 и сл.
Полиядерпые ароматические молекулы
диамагнетизм 195
мезомерия 162, 165 и сл.
размеры 181, 182
Поляризация 38, 41, 44 и сл., 50, 51
взаимодействия групп 44 и сл., 50, 51
и сила нуклеофила 374 и сл.
и поляризуемость в реакциях элек-
трофильного ароматического заме-
щения 250—255
эффекты 66
Поляризуемость 38 и сл., 42, 67, 89—95,
122—138, 374, 375
анизотропия 131
индуктомерная 76, 77
направленная 130—138
— связей 133 и сл.
— сопряженных систем 135—138
и полярная энергия 1006
распределение групп 42
и сила нуклеофила 374 и сл.
стереохимия 93—95
усредненная по сфере 122—130
эффекты 50, 67
Полярные эффекты 988—1015
аддитивность при электрофильном аро-
матическом замещении 993 и сл.
— и пропорциональность 988—991
Брауна соотношение селективности 997
Бренстеда уравнение 991—993
Брэдфилда — Джонса уравнение 993
Гаммета — Бернхардта уравнение
995—999
остаточные 1010—1015
пропорциональность, идеальная энтро-
пия и энтальпия 989 и сл.
• — для ароматических м- и «-замести-
телей 995—1003
— выражаемая альтернативными па-
раметрами 998—1003
— в кислотно-основном катализе 991 —
993
— пространственно удаленных алифа-
тических заместителей 1008—1010
Предметный у казатель
Полярные эффекты
роль поляризуемости 1006
— сопряжения 1006 и сл.
Тафта уравнение для гидролиза эфи-
ров 1010—1015
Юкаво — Цуно уравнение 1004—
1008
энергия ароматических м- и «-заме-
стителей 1003—1008
Порядок реакции 48
Правила последовательности 423 и сл.
Правило
Гофмана 543—549
Гупда максимальной мультиплетности
23
Зайцева 543, 549—557
Марковнпкова, электронная интерпре-
тация 785 и сл.
SN1 432
Sn2 427
Хюккеля 168
Призман 725
Принцип
аддитивности Паскаля для диамагне-
тизма 190 и сл.
запрета Паули 23, 28
микроскопической обратимости 215,
294, 628, 790, 827, 839
неопределенности 20
Франка — Кондона 45
Присоединение
азидов к олефинам 887—889
аминов к ненасыщенным карбониль-
ным соединениям 838 и сл.
аналогия с таутомерией 663—666
воды к моноолефинам 788 и сл.
— и спиртов к олефинам 839 и сл.
галогенов к моноолефинам 797— 811
— к полиолефинам 811—816
— — стереохимия 814 и сл.
галогеноводородов к моноолефинам
784—792, 859—860
— к сопряженным полиолефинам 792
—797
гетеролитическое водорода к карбони-
лу 847-853
гомолитическое 222—224, 858—873
— альдегидов к олефинам 867 и сл.
— бромистого водорода к олефинам
859 и сл.
— галогенов к олефинам 862—865
— моноприсоединение 859—873
— полигалогеналканов к олефинам
865—867
— сероводорода к олефинам 860 и сл.
— тиолов к олефинам 860 и сл.
диазоалканов к олефинам 884—887
карбенов, реакция внедрения 875 и сл.
металлалкилов к карбонильным соеди-
нениям 842—847
нитронов к олефинам 891 и сл.
нуклеофильное к карбонильным соеди-
нениям 820—853
окисей нитрилов к олефинам 889—891
оснований к карбонильным соедине-
ниям 826—828
Присоединение
оснований к сопряженным полиеновым
карбонильным соединениям 840—
842
реактивов Гриньяра к карбонильным
соединениям 845—847
цианистого водорода к карбонильным
соединениям 821—824
— — к олефиновым карбонильным
соединениям 836—842
циклическое к олефинам 874—900
— — с образованием 3-членных цик-
лов 874—879
— — 4-членных циклов 879—884
— — 5-членпых циклов 884—892
— — 6-членных циклов 892—900
электрофильное к моноолефинам
784-792, 797-911
— — стереохимия 790—792
— и нуклеофильное 211—214
— к полиолефинам 792—797, 811—816
Промежуточные стадии, теория Арндта 69
Пропан
вращательные изомеры 57
дипольный момент 107
н-Пропилгалогениды, вращательные изо-
меры 57
Пропилен, присоединение НС1 213
Пространственная ориентация
алкоголиза и гидролиза галогенидов
424-439
дезаминирования 443 и сл.
при нуклеофильном алифатическом за-
мещении 419—447
превращения аминов в спирты, эфиры
или галогениды 443 и сл.
— спиртов в алкилгалогениды 439—
443
Пространственная энергия в алифатических
кольцах 183—187
Пространственное
замедление 449—456
ускорение 449
Пространственные
затруднения 447—449
эффекты кинетические 449
Пространственный эффект 64, 447—459
вторичный 64, 449, 928
— размеры молекул 145
— и теплота гидрирования 122
— и электрический дипольный момент
112 и сл.
первичный 64, 449, 928
— замедление гидролиза амидов 966
и сл.
— — нуклеофильного алифатического
замещения 449—456
— — образования эфира и гидролиза
956—959
— — — электрофильного ароматиче-
ского замещения 259—263
— ускорение мономолекулярного аро-
матического замещения 321
— в электрофильном ароматическом,
замещении 259—262
— энтропия активации 454, 455
1049
Предметный указатель
Протолиз соединений металлов 475 и сл.
Протонная подвижность 19
Прототропия
амидинов 224, 666—667
аналогия с алкилированием 667—670
— с отщеплением 674—683
— с присоединением 663—666
влияние растворителя на подвижность
666—672
— — на равновесие 661, 674
гиперконъюгационные эффекты и под-
вижность 676
диазоампносистем 667
инденов 669
история 658—663
катализ 670 и сл., 684—686
кето-енольная 659
кольчато-цепная 659, 663—666
— кето-лактолов 666
— кето-циклолов 664
— трехуглеродных систем 661 и сл.
механизм бимолекулярный 670, 672,
689—691
— мономолекулярный 669, 672, 684—
689
питро-аг{и-нитросистем 660
— равновесие 678 и сл.
полярные эффекты на подвижность 671
— — на равновесие 674 и сл.
правило предварительного образова-
ния менее стабильного изомера 682
равновесие в кето-енольных системах
674—676
Псевдоароматическпе молекулы 169, 170
Псевдокпслоты 659
Псевдомерия 658
Псевдооснования 691—699
Псевдоосновность 691—699
Псевдосоли 692 и сл.
Пятиокись азота как нитрующий агент для
ароматических соединений 271, 272, 280
и сл.
структура 273
Равновесие
анионотропное 710
кислотно-основное см. Кислотно-основ-
ное равновесие
Радикалы
в алифатическом замещении 220
в ароматическом замещении 222, 333—
341
образование 216—219
при присоединении к олефинам 222
и сл.
реакции 218—224
стабильность 1018—1037
Размеры молекул 139—148
ароматических 178 и сл., 182 и сл.
с гиперконъюгацией 146—148
несопряженных 139—141
определение спектроскопическое 138,
139
пространственный эффект, влияние 145
сопряженных 141 —146
Рассеяние света 132, 135
Расщепление связи в реакциях карбоксиль-
ных соединений
алкил — кислород 936 и сл.
ацил — кислород 936 и сл.
Рацемизация 433 и сл.
Реагенты
анионоидные 206
катионоидные 206
классификация 202—207
Реакции
адиабатические 45
гетеролитические 208—216
гомолитические 207, 208
классификация 207—225
координата 46 и сл.
нуклеофильные 208—216
термические 45
электрофильные 207—216
Резонанс
квантовый 25 и сл., 85
в мезомерных системах 85—88
при образовании ковалентной связи
25 и сл.
в переходных состояниях 46
Резонансное взаимодействие электронов 27
Реймера — Тимана реакция 878
Рефракция 122—130
карбоксильных производных 129
константы атомов 123
— — с неподеленными электронами
125
— групп с диполярными связями 125,
126
— ионов 125
— кратных связей 123
— октетов 124, 125
— полярности 125
— сопряженных ненасыщенных си-
стем 126—130
молекулярная 42
экзальтация 126—130
Рибонуклеиновая кислота, деполимериза-
ция 980
Рихтера реакция 328—330
Ртутьорганические соединения 463—472,
' 475.
Связь
аксиальная 444
водородная 19 и сл.
гомеополярная 13
двойная 17, 32 п сл.
— модели 32 и сл.
— протонированная 643
— стереохимическая форма 33
диполярная 16, 17
длина в ароматических соединениях
181, 182
— и кратность 140, 141
ковалентная 13, 14 и сл., 20—28
координационная 17
полярность 17 п сл., 35, 69
простая направленная 28—32
л- 33
1050
Предметный указатель
Связь
разрыв гетеролитический 16
— гомолитический 17
тройная 35
— протонированная 644
экваториальная 444
электронная интерпретация 13, 14 и сл.
электростатическая 14
энергия диссоциации 216—217
Семидиновая перегруппировка 771, 774
Семидины 771
Семикарбазоны, образование 834, 835
Серусодержащие соединения 533 и сл.
Силовая постоянная 148 и сл.
в зависимости от структуры 149—152
деформационная 149
крутильная 149
молекулы бензола 179
связи 179 и сл.
— кратность 151
— при сопряжении или гиперконъю-
гации 151—152
Симметрия
бензола 178 и сл.
мезомерных молекул 84
сопряженных ненасыщенных молекул
138 и сл.
Синартезпс 619, 632—648
Синартетическая связь 643
Синартетические ионы 636 и сл., 643, 809
Синартетпческое ускорение 632—640
Синглетное состояние 24, 218
Синергисты 495
Скорость реакции 45 и сл.
константа 48
уравнение 48
эффекты внутримолекулярные 45 и сл.
— поляризации 49
— поляризуемости 50
Смайлса перегруппировка 325
Солевые эффекты
в нуклеофильном алифатическом заме-
щении 400—419
в реакциях бензидиновой перегруппи-
ровки 767 и сл.
солей без общего иона 400, 401
— с общим ионом 400, 401
специфические 403
Сольватация
ионов 45
степень 381
Сольволиз галогенидов 385—419
в апротонных растворителях 389—390
в бензоле 415—419
молекулярность 408 и сл.
в SO2 410 и сл.
в уксусной кислоте 411—415
Сопряжение 58
и полярная энергия 1006
теория Тиле 69, 159 и сл., 791
и экзальтация рефракции 127—130
и электрические дипольные моменты
104, 110—112
Сопряженные
кислоты 204
основания 204
Сопряженные
системы, плоские 88
— размеры 142, 143
— поляризуемость 126—130
— — направленная 135—138
— — электромерная 127—130
— рефракция 126—130
— симметрия 84, 138
— энергия 120
углеводороды, поляризуемость эле-
ктромерная 130
— рефракция 130
Спины электронов 23
Спирты
О-нитрование 511 и сл.
0-нитрозирование 526 и сл.
Стивенса перегруппировка 649—651
Стильбен, присоединение хлора 810
Стирол, полимеризация 223, 819 и сл.,
856 и сл., 868
Субстрат 208
Таллийорганическпе соединения 473
Таутомерия
аналогия с алкилированием 667—670
— с отщеплением 674—683
— с присоединением 663—666
валентная 710—731
история 658 и сл.
кольчато-цепная 663—666
теория Лаара 658 и сл.
Тафта уравнение для гидролиза эфиров
1010—1015
Телеприсоединение 224
Теломеры 224
Теории химического строения 12 и сл.
атомистическая 12
дуалистическая 12
структурная 12
электролитической диссоциации 12
Теория
альтернирующих полярностей 231
атома 13
электронного смещения 68
Тепловой эффект реакций 115—122
Теплота
активации 48
гидрирования 188
— и вторичный пространственный эф-
фект 122
— энергия мезомерии из 121
образования 115—122
— ароматических соединений 187,
188
— ацетиленов 118, 119
— олефинов 118, 119
— парафинов 117
Терпены бициклические, перегруппировка
605 и сл.
Тетраалкиламмонпя гидроокиси, отщепле-
ние с образованием олефинов 538 и сл.,
543—545
Тетрафенилгидразин 1027 и сл.
влияние полярных заместителей в фе-
нильных группах 1028
1051
Предметный указатель
Тиле теория сопряжения 69, 159 и сл.,
792 и сл.
Тпоксантгидрол как псевдооснование 699
Тиофен
диамагнитная восприимчивость 193
размеры 182
экзальтация диамагнитной восприим-
чивости 175
энергия мезомерии 188
Типы зарядов
при нуклеофильном замещении 379—
380
при отщеплении с образованием оле-
фина 562—566
Торпа реакция 185
Трансаннулярное взаимодействие в циклах
186
Триадные прототропные системы 658
взаимопревращения 671
Триарилгидразильные радикалы 1031 и сл.
Триарилметильные радикалы 1018—1027
стабильность, влияния структуры
1022—1025
Триметиламин, окись 17
дипольный момент 103 и сл.
Триметиламмониевые ионы, реакции от-
щепления 574 и сл.
1,2,5- Триметилпиррол 176
Триплетное состояние 24, 218
Трифепилметановые соединения как псев-
доосновапие 696 и сл.
Трифенилметил-анион 1019
Трифенилметил-катион 1019
Трифенилметильный радикал 1018 и сл.
скорость и степень образования 1019—
1022
— — — влияние растворителей
1026—1027
энергия активации образования 1022
1,1,2-Трихлорэтан, вращательные изомеры
57
Тропенол (циклогептатриенол) 177
Тропилиден (циклогептатриен) 177
производные 726
Тропилий-ион (циклогептатриенилий-ион)
Трополоны 83
а-Туйен, изомеризация 725
Угловые величины 29
Углы между связями 29, 30, 34, 35, 106,
140, 182
Узловая поверхность 21
Упругие свойства молекул 148—152
бензола 179
Факторы парциальных скоростей для реак-
ций ароматических соединений 247, 250,
318
алкилирование 304
ацилирование 307
галогенирование 253, 316, 318
меркурирование 310
фенилирование 336, 337
Фенантрен 166, 170
диамагнитная восприимчивость 193
присоединение хлора 810 и сл.
энергия мезомерии 188
Фепилазид, присоединение к непредельным
соединениям 887—889
Фенилазотрифенилметан, пиролиз 334
Фенилдиметилтриазен, пиролиз 334
Фенилиодозодибензоат, пиролиз 334
Фенилнитрометан
нитрование 258
а^и-форма 660
Фенол
галогенирование 292
дейтерирование 295
иодирование 289 и сл.
конформация 60
нитрование 282 и сл.
Ферроцен 177
реакции 177
Филъкинштейна реакция 352, 362, 460
Фиттига реакция 600
Фишера — Хеппа перегруппировка 744
и сл.
Флуорен 176
Флюршейма теория ориентации в аромати-
ческих соединениях 69, 229 и сл.
Формамид, конформации 61
Фосфорная кислота, эфиры, гидролиз 974—
984
диэфиров 978—981
моноэфиров 974—978
суммарный механизм 982—984
триэфиров 981—982
фосфатов сахаров 977—978
циклических эфиров 980 и сл.
Фосфорорганические соединения 530—533
Фотолиз 218
Фриделя — Крафтса
алкилирование 298—304
— с изомеризацией 301
ацилирование 304—307
Фуран
диамагнитная восприимчивость 193
— — экзальтация 175
размеры 182
энергия мезомерии 175, 187, 188
Халкон, циклодимеризация 882
Хинолиния ионы как псевдоосновапия
692 и сл.
Хиральность 56, 424
а-Хлорацетанилиды ароматических соеди-
нений, перегруппировка 739—743
Хлориний-ион 285, 286—288
Хлорциклопентан, хлорирование 488
Хроморганические соединения 474, 475
Циангидрины, образование 821—824
Циан-импнные пентадные системы 680 и сл.
Циклоалканы
бромирование 481
конформации 184 и сл.
нитрование 495
размеры 184
1052
Предметный указатель
Циклоалканы
синтез 185
стабильность 52
теплоты сгорания 183
энергии образования 183, 184
Циклобутадиен 169, 171
Циклобутан
устойчивость 186 и сл.
Циклобутилий-ион 647
Циклобутильные соединения, превращения
644—648
Циклогексадиенилий-ион 300, 308
Циклогексан
конфигурации 444
конформации 52, 53, 570
производные, элиминирование 573
стабильность 52
Циклогексен, энергия мезомерии 187
Циклогептен, энергия мезомерии 187
Циклодекан
конформации 58
производные, реакции элиминирова-
ния 571
Циклодимеризация 875—884
тра«с-Циклононен 187
Циклооктатетраен 169, 171, 727
диамагнитная восприимчивость 193,
194
производные 172
размеры 183
энергия мезомерии 188
Циклооктатриен 727
траис-Циклооктен 187
Циклопентадиен 176
анион 176, 177
Циклопентаи
конформация 53
производные, реакции элиминирова-
ния 571
стабильность 52
Циклопентен, энергия мезомерии 187
Циклоприсоединение 874—900
Цпклопропановое кольцо, протонирован-
ное 643
Циклопропилметилий-ион 646 и сл.
Цпклопропилметильные соединения, пре-
вращения 644—648
Четырехокись азота, стереохимия 61
Шмидта перегруппировка 618 и сл.
пространственное протекание 620
Электрический дипольный момент 102—115
алканов 107 и сл.
ароматических соединений 110—112
и вторичный пространственный эффект
112—114
гиперконъюгировапных соединений
104, 109, 111
гомологов 108 и сл.
групп, распределение 41
— с элементами IV группы 109
Электрический дипольный момент
изотопные эффекты дейтерия 107 и сл.
и индуктивный эффект 109
и мезомерный эффект 110—112
методы измерения 102 и сл.
ненасыщенных углеводородов 114 и сл.
соединений с диполярными связямп
103—105
сопряженных соединений 104, 110—112
электроотрицательности эффект 105—
Электрическое двойное лучепреломление
132, 133, 135
Электровалентность 13
Электрокинетический эффект 64
Электрокинетпческое взаимодействие 39, 64
Электромерный эффект 67, 70, 89—93
в ненасыщенных соединениях 89 и сл.
отщепление с образованием олефинов
549—561
и положительная экзальтация рефрак-
ции 127, 128
сольволиз ацеталей 372
— галогенидов 364—372
— — влияние дейтерия 367
стереохимия 93—95
электрофильное ароматическое заме-
щение 250—255
— — — соотношение орто/пара 266
и сл., 269 и сл.
— присоединение 786
Электромеры 231 и сл.
Электронная
конфигурация 24
плотность, распределение 21 и сл.
— — у атома водорода 21, 22
— — в бензоле 161—165
— — в двойной и тройной связях 33
и сл., 35
Электронное взаимодействие, внутримоле-
кулярное 41 и сл.
л-Электроииые системы, классификация
168 и сл.
Электронные
смещения 65—68
— индуктивный механизм 65
— механизм сопряжения 65, 65
— символы 65 и сл.
— теории 64 и сл.
состояния возбужденные, стереохимия
93—95
уровни молекул 167
Электронный
обмен 26, 28
удар 219
Электроны
л 33
о 33
спарепные, спины 23
эквивалентные 23
Электроотрицательность 18
и электрические дипольные моменты
105—107
Электроотрицательные атомы или группы
Электроположительность 18
1053
Предметный указатель
Электроположительные атомы или группы
19
Электростатическое взаимодействие 38 и сл.
Электрофильное ароматическое замещение
228—318
азосочетание 307—309
азулена 173
алкилирование 298—304
ацилирование 304—307
водородный обмен 295—298
галогенирование 285—392
Голлемана аномалия 250—255
десилидирование, дегерманилирование
дестаннилирование, деплюмбирова-
ние 310—313
дифепилена 173
замещение в гетероциклических систе-
мах 249
— в многоядерных системах 249
меркурирование 309—310
метод конкурирующих реакций 246
нитрование 270—285
нитрозирование 313—315
ориентация, влияние полярных заме-
стителей 234—237
— диполярных связей 237—239
— в ионогенных системах 243—245
— монополями 233—237
— нейтральными группами с незапол-
ненными электронными оболочками
239
— — — с неподеленными электрон-
ными парами 241—243
— — — без неподеленных электрон-
ных пар 239—241
— правила 228 и сл.
— теории 229—233
— электронная теория 233—245
ориентирующие заместители, класси-
фикации 248 и сл., 254
реакционная способность ядра 245—
258
скорость замещения 246, 247
соотношение орто!пара 259—270
— — индуктивный эффект 259, 263,
267 и сл.
— — мезомерный эффект 264—266,
268 и сл.
— — полярные эффекты 259, 263 —
— — пространственный эффект 259—
263
— — электромерный эффект 266—267,
269 и сл.
— — эффект сопряжения 264—269
сульфирование 292—294
факторы парциальных скоростей 247
эффект гиперконъюгацпи 255, 315—
318
— индуктивный 234—245, 248—255
— мезомерный 250—255
— поля 256—258
— сопряжения 236, 250—255
— электромерный 250—255
эффекты поляризации 250—255
— поляризуемости 250—255
Электрофильное алифатическое замещение
463—478
обмен металл — металл 472—475
— ртути, бимолекулярный 465—468
— — внутримолекулярный 468—470
— — — катализ 468 и сл.
— — мономолекулярный 470 и сл.
протолиз солей металлов 475 и сл.
механизмы 464 и сл.
Электрофильное замещение 208—211 см.
также Электрофильное алифатическое
и ароматическое замещение
у атома азота 508—511, 512—525
— — дезаминирование 512—525
— — диазотирование 512—525
— — изотопный эффект 510
— — нитрозирование 512—525
— — нитрование 508—511
— — обобщенный механизм с ионом
нитрония 509 и сл.
— кислорода 511 и сл.
— — нитрозирование 525—527
— — нитрование 511 и сл.
различия между поляризацией и поля-
ризуемостью 250—255
в боковой цепи по соседству с арома-
тическим кольцом 477 и сл.
Электрофильное присоединение 211 — 214,
784-820
Электрофильные ароматические перегруп-
пировки 738—748
алкиланилинов 745—747
алкилариловых эфиров 747 и сл.
галогенаминов 739—743
— ароматических 739—743
диазоаминосоединений 743 и сл.
нитрозаминов 744 и сл.
Электрофильные перегруппировки
насыщенных соединений 648—652
ненасыщенных соединений 658—731
реагенты 204—206
реакции 208—216
Электрофильность 206
Электроциклические реакции 717—721
Энергетические барьеры вращения вокруг
простой связи 53 и сл.
Энергия активации 46 и сл., 451, 452
Аррениуса 49, 63
Энергия мезомерии 85—88, 163
ароматических молекул 162, 173, 187,
188
определение из констант энергий связи
120
— из теплот гидрирования 121, 122
Энергия связи в молекуле водорода 27
Энтальпия 43
Энтропия
активации 48, 455
для равновесия 43
Эпокиси, перегруппировка 602
Этап
вращение вокруг простой связи 51
и сл., 54
замещенные, конформационные изо-
меры 56
конформации 52, 53
1054
Предметный указатель
Этиленхлоргидрин, вращательные изомеры
57
Эфиры карбоновых кислот
бимолекулярный кислотный гидролиз
с расщеплением связи ацил — кис-
лород 955
— основной гидролиз с расщеплением
связи алкил — кислород 946—948
— — — — — ацил — кислород 938—
943
мономолекулярный кислотный гидро-
лиз с расщеплением связи алкил —
кислород 959—963
ацил — кислород 948—
955
— основной гидролиз с расщеплением
связи алкил — кислород 943—946
образование п гидролиз, механизмы
936—938
Тафта уравнение для бимолекулярного
гидролиза 1010—1015
функции кислотности 1148—1151, 1161
х-Эффект 375 и сл.
Эффект поля 64
и кислотность метагемоглобипов 917
и сл., 919
Эффект поля
количественное исследование 917 и сл.
и сила кислот 915—919
в электрофильном ароматическом за-
мещении 256—258
Эффект растворителя
на продукты при нуклеофильном али-
фатическом замещении 385—388
на равновесие при прототропии 661
на скорость при нуклеофильном эро-
тическом замещении 379—390
— при 1.2-отщеплении с образованием
олефинов 562—566
— и степень образования трифенил-
метила 1026 и сл.
Эффект сопряжения
образование циангидрина 823
электрофильное ароматическое заме-
щение 236, 250—255
— — — соотношение орто/пара 264—•
269
Эффекты
поляризации 66
поляризуемости 67
Юкава—Цуно уравнение 1004—1008
уважаемый читатель! Ваши замечания о содержании книги, ее оформлении
и качестве перевода просим присылать по адресу: 129820, Москва, И-110, ГСП,
1-п Рижский пер., д. 2, издательство «Мир».
к. ин гольд
ТЕОРЕТИЧЕСКИЕ основы органической химии
Редактор Г. Б. Шкля“ва. Художник Г. В. Чучелов. Художественный редактор Н. Г. Блинов.
Технический редактор 3. И. Резник. Сдано в набор 17/XI 1972 г. Подписано к печати 29/Ш 1973 г.
Бумага № 1 70х 108/и-33,08 буч. л. Печ. уел. 92,58 в т/ч 1 вкл. Уч.-изд. л. 88,54.
Изд. № 3/6550. Цена 8 р. 73 к. Зак. 0772
ИЗДАТЕЛЬСТВО «МИР» Москва, 1-й Рижский пер., 2
Ордена Трудового Красного знамени Московская типография № 7 «Искра революции» Союзполиграф-
прома при Государственном комитет^ Совета Министров СССР по делам издательств, полиграфии
и книжной торговли. Москва, К-1, Трехпрудный пер., 9