/












































































































































































































































































































































































































































Author: Калинкин И.П. Булатов М.И.
Tags: спектральные методы анализа оптические методы анализа химия аналитическая химия
Year: 1986




Text
М. И. БУЛАТОВ, И. П. КАЛИН КИН
ПРАКТИЧЕСКОЕ РУКОВОДСТВО ПО
ФОТОМЕТРИЧЕСКИМ МЕТОДАМ АНАЛИЗА
Издание пятое, переработан ное
Ленинград
«Химия»
Ленинградское отделение 1986
643 Б907
УДК 543.42
Рецензент: Засл. деят. науки и техники РСФСР проф. д-р хим. наук Ю. А. Клячко
~Б И Б Л И О ТЕ К Л
р ЛН’.Ы» у и и в • • а
УДК 543.42
Булатов М. И., Калинкин И. П.
Практическое руководство по фотометрическим методам анализа. — 5-е изд., перераб. — Л.: Химия, 1986. — 432 с.
В пятом издании (4-е изд. вышло в 1976 г.) обновлен и переработан материал, посвященный чувствительности фотометрических определений, аппаратурному оформлению методов и расчетам. Описаны условия фотометрического определения веществ, аппаратура и методы измерения свето-поглощення растворов в видимой и ультрафиолетовой областях спектра. Специальные главы посвящены метрологическим характеристикам и математической обработке экспериментальных данных
Предназначена работникам исследовательских и заводских химических лабораторий Полезна также студентам и преподавателям хнмико-аналитических специальностей вузов
Табл. 36. Ил. 123. Библиогр. список 425 назв.
1804000000-066
050 (01)-86
66-86
Б
© Издательство «Химия», 1976 © Издательство «Химия», 1986, с изменениями
ПРЕДИСЛОВИЕ К ПЯТОМУ ИЗДАНИЮ
Коренное повышение качества продукции является делом первостепенной важности, как отмечено в «Основных направлениях экономического и социального развития СССР на 1986 — 1990 годы и на период до 2000 года». Решению этой проблемы должно способствовать совершенствование методов аналитического контроля. Дальнейшее развитие фотометрических методов анализа (двухволновая спектрофотометрия, экстракционно-фотометрический метод), особенно с применением разнолигандных комплексов, потребовало изложения ряда дополнительных вопросов теории и практики фотометрического анализа и существенного обновления материала книги. Кроме того, за период после выхода четвертого издания (1976 г.) аналитические и метрологические характеристики, используемые в фотометрическом анализе, претерпели определенные терминологические и смысловые изменения, что также потребовало соответствующих уточнений и исправлений.
В пятом издании значительно переработан весь материал книги; в нем отражена литература по наиболее принципиальным вопросам фотометрического анализа до 1985 г. Главы 3—7 практически написаны заново. Главы 1, 4 и 8 дополнены необходимым материалом по применению в фотометрическом анализе разнолигандных комплексов и их исследованию. Рассмотрены новые примеры теоретических обоснований условий фотометрических определений (глава 10). Практические работы вынесены в самостоятельную главу (глава 11).
При переиздании учтены рекомендации ИЮПАК и Научного Совета АН СССР по аналитической химии по вопросам терминологии, определений и обозначений метрологических характеристик фотометрического анализа [1—6], представления результатов фотометрических определений [7—10] и химического анализа [11—14]. Кроме того, по возможности учтены рекомендации ГОСТ 8.04—72 «Показатели точности измерений и формы представления результатов измерений» и ГОСТ 16263—70 «Метрология. Термины и определения».
Исключение в части терминологии сделано в отношении термина «погрешность», вместо которого в ряде случаев по методическим соображениям и в соответствии с терминологией общей теории ошибок использован термин «ошибка».
Во всех случаях, кроме специально оговоренных в тексте, под процентным содержанием компонента в смеси (или в растворе) понимается его массовая доля, выраженная в процентах.
Авторы выражают глубокую благодарность рецензенту книги заслуженному деятелю науки и техники РСФСР, докт. хим. наук проф. Ю. А. Клячко и канд. хим. наук А. Б. Бланку за ценные советы и замечания.
1*
3
ПРИНЯТЫЕ ТЕРМИНЫ И ОБОЗНАЧЕНИЯ
А — оптическая плотность раствора светопоглощающего вещества*, в реакциях — обозначение аниона
а — равновесная активность иона, моль/л
В — атомная масса
С — аналитическая концентрация** вещества, моль/л
[С]—суммарная равновесная концентрация, моль/л
[Л4П+], [7?~], [Л-]—равновесные концентрации стехиометрических 3* форм реагирующих ионов или молекул, моль/л
D — коэффициент распределения вещества (в экстракции)
Е — степень (фактор) извлечения (в экстракции)
f — коэффициент активности
HR, HR'—фотометрический (экстракционный) реагент
1 — ионная сила раствора
№, р° — термодинамические константы равновесий
К, Р — концентрационные константы равновесий К', — условные константы равновесий
Ль Ki,.... Ki, — ступенчатые константы устойчивости
Ка, Къ — константы ионизации
* Под оптической плотностью [1g (Л/7) ] (Absorbance) будем понимать абсорбционную плотность или плотность поглощения, обуслов тенную только поглощением света и не включающую потерю световой энергии вследствие отражения и рассеяния.
** Под аналитической концентрацией понимают общую исходную (или избыточную) концентрацию вещества в растворе [15, 16] без учета долевого распределения его конкретных форм. Во всех случаях, когда молярную концентрацию используют для оценки общего аналитического содержания вещества без указания его конкретной формы, ее отождествляют с формульной [15, с. 58] концентрацией, выраженной числом молей вещества формульного состава в литре раствора [17, с. 34].
3* Под стехиометрическими (координируемыми) формами будем понимать такие формы реагирующих ионов (молекул), которые полностью соответствуют стехиометрическому уравнению или из которых непосредственно состоит рассматриваемое стехиометрическое соединение.
4
/Сн, рн — константы протонирования
KD — константа распределения (в экстракции) К-we — константа экстракции
Kw — ионное произведение воды
I(s — произведение растворимости
L — лиганд, комплексующий реагент
L, Y — маскирующий реагент
М — определяемый ион металла т — определяемый минимум
п — среднее координационное число лигандов (лигандное число, функция образования) рН1/г — значение pH полузыделения (в экстракции)
г — отношение объема органической фазы к водной
S — растворимость осадка, моль/л Sa/b — фактор разделения (в экстракции) 5д/в — фактор обогащения (в экстракции) s — эффективное сечение кюветы, см2 «д — стандартное отклонение оптической плотности
Т — пропускание, % t — температура, °C
V — объем органической фазы
W — объем водной фазы
Zt — заряд иона
а — мольная доля конкретной формы компонента
aq — функция распределения 1/ам — функция закомплексованности
Pnm — константа образования (устойчивости) полиядерного комплекса MTOLn
Pi, р2, ...» Pi, Рй — полные константы устойчивости
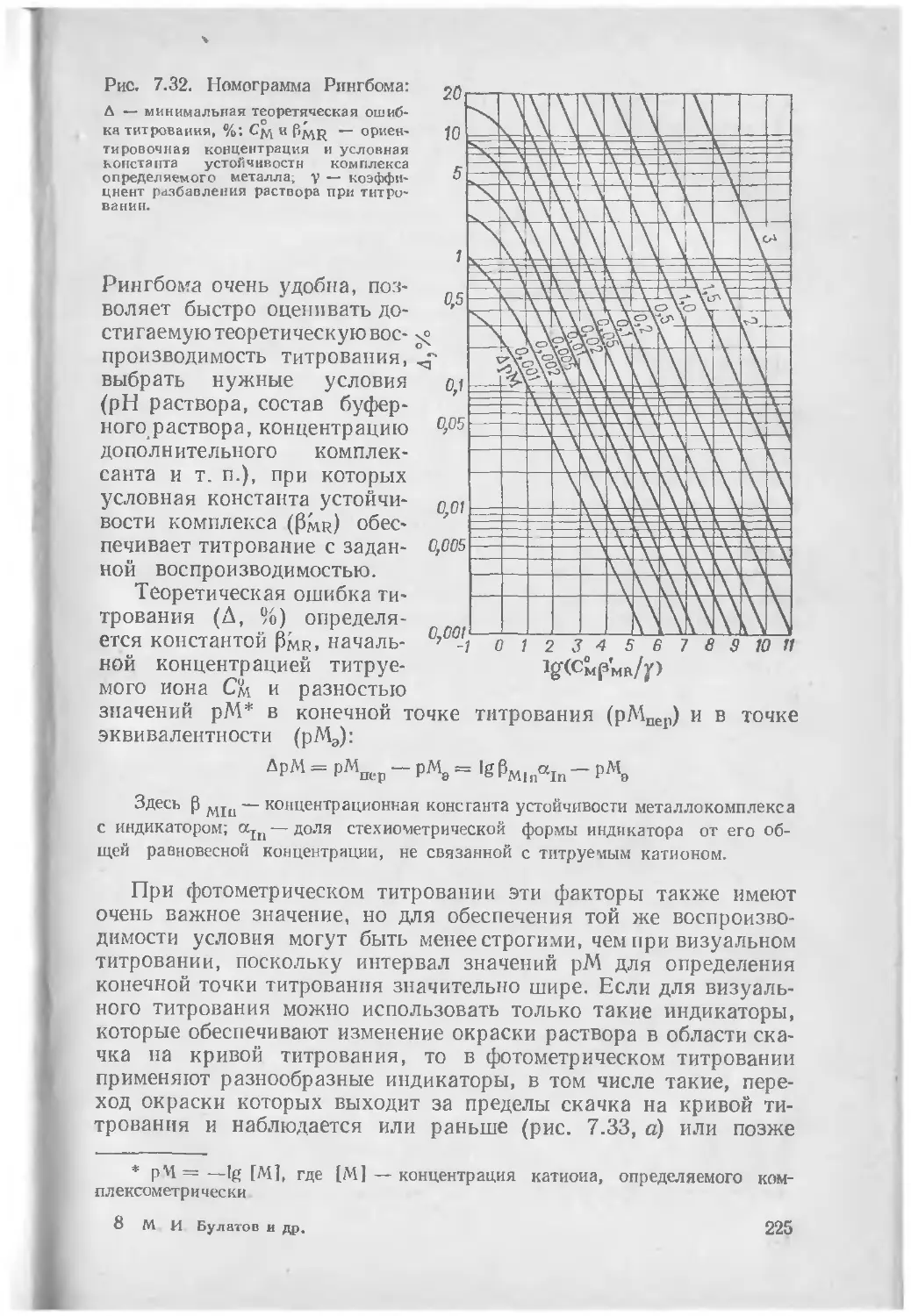
Д — ошибка определения (титрования), % ДрМ, ДрН — разность величин рМ, pH в конце титрования и в точке эквивалентности
S — степень диссоциации комплекса
ez — молярный коэффициент светопоглощения (молярный показатель поглощения), л/(моль-см)
Примечание. Обозначения р, К, С, [с], a, D в тексте снабжены нижними индексами, показывающими вид частиц или равновесий, к которым они относятся (например, См, Cr, Рмкп, Dr и т. д.).
5
ВВЕДЕНИЕ
Методы анализа, основанные на поглощении световой энергии атомами и молекулами анализируемых веществ, представляют обширную группу абсорбционных оптических методов, получивших очень широкое распространение как на промышленных предприятиях, так и в научно-исследовательских лабораториях. При поглощении света атомы и молекулы поглощающих веществ переходят в новое, энергетически возбужденное состояние. Избыточная энергия атомов и молекул в одних случаях расходуется на повышение их поступательной, вращательной или колебательной энергии, в других — выделяется в виде вторичного излучения или расходуется на фотохимические реакции. В зависимости от вида поглощающих частиц и способа трансформирования избыточной энергии возбуждения различают:
1. Атомно-абсорбционный анализ, основанный на поглощении световой энергии атомами анализируемых веществ.
2. Молекулярный абсорбционный анализ, т. е. анализ по поглощению света молекулами анализируемого вещества и сложными ионами в ультрафиолетовой, видимой и инфракрасной областях спектра (спектрофотометрия, фотоколориметрия, ИК -спектроскопия).
3. Анализ по поглощению и рассеянию световой энергии взвешенными частицами анализируемого вещества (турбидиметрия, нефелометрия).
4. Люминесцентный (флуорометрический) анализ, основанный на измерении излучения, возникающего в результате выделения избытка энергии возбужденными молекулами анализируемого вещества.
Все эти методы иногда объединяют в одну группу спектрохимических [15] или спектроскопических [18] методов анализа, хотя они и имеют существенные различия. Фотоколориметрия и спектрофотометрия основаны на взаимодействии излучения с однородными системами, и их обычно объединяют в одну группу фотометрических методов анализа*.
* В данном руководстве термин «фотометрический анализ» относится только к фотоколориметрическим и спектрофотометрическим методам анализа.
6
Из методов молекулярного абсорбционного анализа наибольшее распространение получили фотометрические. Турбидиметрические и нефелометрические методы используют гораздо реже, обычно лишь в тех случаях, когда для определяемого вещества не удается подобрать хороших фотометрических реагентов. Флуорометрический (люминесцентный) анализ, обладающий очень высокой чувствительностью [предел обнаружения около 1 х X 10-8%], также имеет ограниченное применение вследствие того, что лишь небольшая часть соединений флуоресцирует с достаточной интенсивностью.
В фотометрических методах используют избирательное поглощение света молекулами анализируемого вещества. В результате поглощения излучения молекула поглощающего вещества переходит из основного состояния с минимальной энергией Et в более высокое энергетическое состояние Е2. Энергия возбуждения распределяется по отдельным энергетическим колебательным уровням молекулы, превращаясь в тепловую. Механизм этого превращения сложен и недостаточно глубоко изучен. Электронные переходы, вызванные поглощением строго определенных квантов световой энергии, характеризуются наличием строго определенных полос поглощения в электронных спектрах поглощающих молекул. Причем поглощение света происходит только в том случае, когда энергия поглощаемого кванта совпадает с разностью энергий ДЕ между квантованными энергетическими уровнями в конечном (Е2) и начальном (Е^ состояниях поглощающей молекулы:
hv — ЕЕ = Еа — Ег
Здесь Л —постоянная Планка (й = 6,625-10-м Дж-с); v — частота* поглощаемого излучения, которая определяется энергией поглощенного кванта и выражается отношением скорости распространения излучения с (скорости световой волны в вакууме с = 3- 1О10 см/с) к длине волны X**: v = с/Х.
Энергия излучения характеризуется электромагнитным спектром (рис. 1), охватывающим область от километровых радиоволн до десятых долей ангстрема у-излучения и космических лучей. Для характеристики участка спектра часто используют также и волновое число v, которое показывает, какое число длин волн приходится на 1 см пути излучения в вакууме, и определяется соотношением: v = 1/Х.
Природа полос поглощения в ультрафиолетовой (10—400 нм) и видимой (400—760 нм) областях спектра одинакова и связана главным образом с числом и расположением электронов в поглощающих молекулах и ионах. Б инфракрасной области (0,8—
* Частота излучения "V измеряется в обратных секундах (с-1), герцах (Гц); 1 Гц = 1 с-1.
** Длина волны X измеряется в ангстремах (1 А = 1-10~8 см), микрометрах или микронах (I мкм = 1 мк = 1-Ю-6 м), нанометрах или миллимикронах (1 нм = 1 ммк = 10 А = 1 • 10~9 м).
7
CO
Типы переходов,
Тип поглощаемого
вызванные поглощением
Г“
Переходы внутренних электронов
I
1019-
излучения
ПЛТШТППТТ I Рентгеновское 1 11 излучение ! Дшшшии
f
Переходы внешних электронов
♦ А
S 10*s'
Ультрафиолетовое и видимое'
11 J JIU J1 *j V J'i1!1' J. I U '
Молекулярное колебание
Инфракрасное
UJIIILIIILIIII
io-2
Молекулярное вращение
10‘1
(ЭПР)
1
Изменение спинового состояния - под действием
магнитного 1' поля
Л
(ЯМР)
-
'//ТУ, в о инов ое '/7/7/
107.-
Радиоизлучение II II (ЯМР)|| II
-102
.10“
Длина волны, см
Увеличенная область ультрафиолетового и видимого излучения
111111111II111
Ультрафиолетовое 1
IIIIII I I ! и I
Фиолетовое | Синее | Зеленое hi । п 111 Желтое IIIIIHII Оранжевое I (Красное I lliiiiiliiil
г-200
-400
-600
L-800
S
s
Рис. I. Электромагнитный спектр излучения
1000 мкм) она в большей степени связана с колебаниями атомов в молекулах поглощающего вещества.
Интенсивность полос поглощения в электронных спектрах поглощающих молекул зависит от химической природы металла-комплексообразователя и координируемых им лигандов и объясняется с различных позиций, соответствующих разным представлениям (теориям) о природе химической связи. Наиболее интенсивные полосы поглощения наблюдаются в тех случаях, когда образование молекулы поглощающего соединения сопровождается переносом заряда, т. е. переносом электрона (или смещением электронной плотности) от иона металла к лиганду или наоборот. В зависимости от используемой аппаратуры в фотометрическом анализе различают спектрофотометрический метод — анализ по поглощению монохроматического* света и фотоколори метрический — анализ по поглощению полихроматического (немонохроматического) света. Оба метода основаны на общем принципе — существовании пропорциональной зависимости между светопогло-щением и концентрацией поглощающего вещества.
Фотометрические методы, в которых измеряют светопоглощение окрашенных растворов, иногда называют «колориметрическими». Однако это название, применяемое к оценке интенсивности окраски, справедливо лишь для немногих визуальных определений по интенсивности окрашенных растворов. Колориметрический анализ используют сравнительно редко, главным образом в не приспособленных для аналитических определений условиях (например, в геологических экспедициях, в полевых работах и т. и.). «Точность» колориметрического анализа невысокая [погрешность определения составляет ±10% (отн.)].
Фотоколориметрические методы, использующие сравнительно несложную аппаратуру, обеспечивают достаточную точность [погрешность определения составляет 1—3% (отн.) и широко применяются для определения концентрации растворов.
В спектрофотометрических методах применяют более сложные приборы — спектрофотометры, позволяющие проводить анализ как окрашенных, так и бесцветных соединений по избирательному поглощению монохроматического света в видимой, ультрафиолетовой и инфракрасной областях спектра. Наиболее совершенные спектрофотометрические методы анализа характеризуются высокой точностью [погрешность определения 1—0,5% (отн.)]. Это, прежде всего, относится к дифференциальной спектрофотометрии и спектрофотометрическому титрованию, применяющимся для определения веществ в широком интервале концентраций, особенно при больших содержаниях. При соответствующих условиях эти методы практически не уступают по точности классическим методам анализа и применяются при аттестации аналитических методик и стандартных образцов.
* Излучение, с котором все волны имеют одинаковую частоту v (или длину волны X), называют монохроматическим.
9
Спектрофотометрические методы по сравнению с фотоколори-метрическими позволяют решать более широкий круг задач:
1. Количественное определение содержания элементов и органических веществ в широком интервале длин волн от 185 до 1100 нм.
2. Количественный анализ многокомпонентных систем (одновременное количественное определение нескольких элементов) .
3. Определение состава светопоглощающих комплексных соединений.
4. Определение констант устойчивости светопоглощающих комплексных соединений и констант диссоциации органических реагентов.
5. Изучение химических равновесий и определение фотометрических характеристик светопоглощающих соединений.
Нижняя граница определяемых содержаний элементов в фотометрических методах, как правило, составляет 10"4—10-6% [18— 22]. Возможности фотометрических методов, их место в ряду других спектроскопических и физических методов анализа и сравнительные количественные и метрологические характеристики рассмотрены в работах [18—21].
Несмотря на интенсивное развитие других аналитических методов, по-прежнему эффективно и широко используют фотометрические методы. Это обусловлено следующими обстоятельствами :
1. Наличием различных фотометрических методик анализа [22—34 ] практически на все элементы периодической системы и многочисленные органические вещества.
2 Возможностью использовать относительно недорогую и общедоступную аппаратуру для проведения фотометрических определений с достаточно высокой точностью.
3. Широким выбором фотометрических методов и методик, позволяющих проводить определение элементов в интервале содержаний от 100 до 10-6%, включая анализ веществ высокой степени чистоты [25, 27—31 ] и микрообъектов (пленок) [23, 26].
Основными направлениями в развитии современных фотометрических методов анализа по-прежнему являются: повышение их чувствительности и селективности, обеспечение высокой воспроизводимости и правильности результатов. Большое значение при этом придается созданию автоматизированных спектрофотометрических комплексов, снабженных микро-ЭВМ, позволяющими экспрессно изучать и анализировать сложные многокомпонентные и дисперсные системы, определять следовые количества элементов, микропримесей и т. д. В последнее время в литературе по фотометрическим методам особое внимание уделяется четырем направлениям [18, 27, 30]:
метрологии фотометрических измерений и определений;
ю
развитию высокочувствительных, селективных и экспрессных экстракционно-фотометрических методов, особенно с применением разнолигандных комплексов;
созданию различных типов спектрофотометров, оснащенных компьютерами и микропроцессорами, позволяющими автоматизировать фотометрические методы и шире внедрять в практику селективные и точные методы двухволновой и производной спектрофотометрии ;
созданию принципиально новых классов приборов — фото-акустических спектрофотометров — и фотоакустической спектрофотометрии, которая сможет снизить предел обнаружения примерно на два порядка.
Высокая специфичность, возможность широкого выбора полос поглощения, сравнительная легкость и высокая точность измерений, достигаем! я современной аппаратурой, обеспечивают фотометрическому анализу широкое применение в настоящее время и благоприятные перспективы на будущее для его использования в различных областях науки, техники и производства.
Часть I
ОБЩИЕ ПОЛОЖЕНИЯ И АППАРАТУРА ФОТОМЕТРИЧЕСКОГО АНАЛИЗА
Г лава 1
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ОКРАШЕННЫХ* СОЕДИНЕНИЙ
В фотометрическом анализе содержание вещества (элемента) определяется по светопоглощению его окрашенного раствора. В зависимости от типа химической реакции, применяемой для образования светопоглощающего соединения определяемого элемента, фотометрические методы подразделяют на прямые и косвенные [31—33, 351. В прямых методах определяемый ион М с помощью реагента R переводят в окрашенное соединение MR, а затем измеряют светопоглощение раствора этого соединения. При косвенных определениях используют вспомогательные окрашенные соединения MiR', которые при взаимодействии с определяемым ионом либо разрушаются сами, либо образуют новые светопоглощающие соединения. Для этих целей применяют следующие приемы:
1. Образование окрашенного соединения по реакции катионного обмена (или в сочетании с ней):
М + MtR' ч=^ MR' +М1 определяемый бесцветный окрашенный
ион комплекс комплекс
Mi + R
MiR окрашенное соединение
2. Разрушение окрашенного соединения по реакции катионного или анионного обмена:
А + MjR' 4=t МХА -р R'
определяемый окрашенное бесцветное
ион соединение соединение
м + MjR' MR' +Mj
определяемый окрашенное бесцветное
ион соединение соединение
* В общем случае под окрашенными соединениями будем понимать соединения, избирательно поглощающие свет не только в видимой, но и в ультрафиолетовой и инфракрасной областях спектра.
12
3. Выделение определяемого иона в виде малорастворимого соединения и последующее определение эквивалентного количества осадителя в виде окрашенного соединения:
А 4-м МА определяемый осадок
нон
М 4- R MR
осадитель окрашенное
соединение
4. Образование окрашенных соединений определяемых ионов (или окрашенных форм реагента) по реакциям окисления — восстановления.
5. Образование окрашенных соединений в результате синтеза или разрушения сложных органических соединений в присутствии (или с участием) определяемых ионов.
6. Проведение каталитической индикаторной реакции (с участием определяемого иона в качестве катализатора) между двумя веществами, одно из которых окрашено или может быть превращено в окрашенное соединение.
Косвенные методы, основанные на разрушении окрашенных соединений, применяют в основном для определения галогенид-и сульфат-ионов и некоторых других анионов [30—33]. Для определения анионов используют также и другие приемы (п. п. 3— 5) [35 J. Каталитические реакции используют в кинетических методах анализа в сочетании с фотометрическими для определения как катионов, так и анионов [36, 37 ]. Основными затруднениями при косвенных определениях являются ограниченная специфичность и различные побочные процессы. Косвенные методы трудоемки, и, как правило, менее точны. Однако в ряде случаев, особенно когда необходимо отделение определяемого иона осаждением, использование косвенных определений вполне оправдывается. Кроме того, при таких косвенных определениях может быть достигнута более высокая чувствительность.
В фотометрическом анализе применяют различные типы окрашенных соединений. Из однороднолигандных комплексов используют преимущественно комплексные и внутрикомплексные (хелатные) соединения ионов металлов с органическими реагентами. Для ряда металлов находят применение ацидокомплексы с неорганическими лигандами (SCN", С1", Вг_, I"), пероксидные комплексы и гетерополисоединения (As, Ge, Mo, Р, Si, V, W). Разнолигандные комплексы могут быть со смешанной координационной сферой и типа ионных ассоциатов; все они содержат катионные или анионные хромофорные реагенты, а иногда и поверхностно-активные вещества (ПАВ). Светопоглощение окрашенных растворов зависит от природы светопоглощающих соединений, условий их образования и состава среды. Ниже рассмо
13
трены основные физико-химические характеристики окрашенных соединений, условия их образования и фотометрического определения.
1.1. ПРОЧНОСТЬ ОКРАШЕННЫХ СОЕДИНЕНИЙ
Количественной характеристикой устойчивости любого комплексного соединения MRn является его термодинамическая константа у с т о й ч и в о с т и * р = aMR(j/aMaR (о — активность иона, указанного индексом), которая зависит от температуры и давления. В аналитической практике для количественной оценки устойчивости соединений часто пользуются также концентрационными, или кажущимися, константами устойчивости Рмкп —
— [MRn ]/[М1 IR ]п (в квадратных скобках указаны равновесные концентрации окрашенного комплекса, определяемого катиона М и реагента R), зависящими не только от температуры, но и от ионной силы раствора.
При благоприятных условиях, когда образующие окрашенное соединение ионы находятся практически полностью в стехиометрической форме и на их взаимодействие не накладываются какие-либо побочные, сопряженные реакции, с помощью термодинамических или концентрационных констант устойчивости можно получи 1ь количественную информацию о равновесных концентрациях реагирующих веществ и степени образования окрашенного соедигения, обосновать выбор того или иного фотометрического реагента и условия его аналитического применения.
Состояние равновесия в растворе разнолигандных комплексов характеризуют с помощью двух констант [381. Одна из них является константой образования (устойчивости) разнолигандного комплекса и характеризует его устойчивость в реакциях сольватированного катиона М с лигандами R и R':
М-J-/R + <R ^=>MR;R- (11)
, = |.MRyR;]/([M] [R]z [R'F) (1.2)
Другая константа, которую называют константой сопропорционирования, характеризует устойчивость разнолигандного комплекса к однороднолигандным (для рассматриваемых лигандов):
Z/mMRm + t/mMR^ <=> MR/R) (m = / + i) (1.3)
kMR/R; = lMRX]/([MW/m [MR;],/m) (i.4)
* Прочность окрашенных соединений часто характеризуют также константами нестойкости, которые являются обратными величинами констант устойчивости (Амяп — l/₽MRn)-
14
Взаимосвязь между константами образования и сопропорционирования выражается уравнением:
PmRjRJ- = О-5)
Здесь и ₽ич;п ~~ константы устойчивости однороднолигандных
комплексов MRm и MR^
Константа сопрэпорционирования одновременно характеризует и меру совместимости разнородных лигандов в координационной сфере комплекса. Если лиганды R и R' несовместимы, tg разнолигандный комплекс менее устойчив, чем однороднолигандные комплексы [38]. Константа сопропорционирования в этом случае меньше единицы, а константа устойчивости разнолигандного комплекса меньше среднего геометрического из констант устойчивости однороднолигандных комплексов. При совместимости лигандов R и R' константа сопропорционирования больше единицы 138—41 ].
Прочность окрашенного соединения и его устойчивость в водных растворах увеличиваются с возрастанием констант устойчивости. Чем выше прочность окрашенного комплекса MR„, тем полнее определяемый ион М связывается с фотометрическим реагентом R в окрашенное соединение, тем выше могут быть точность и чувствительность фотометрического определения, меньше влияние посторонних ионов, присутствующих в растворе. Так, степень связывания Fe (III) в роданидный комплекс заметно уменьшается в присутствии хлорид-ионов, а при наличии фосфорной кислоты происходит полное обесцвечивание раствора роданида железа. Между тем большие количества хлорид-ионов практически не влияют на определение Fe (III) в виде более прочного салицилатного комплекса; в значительно меньшей степени сказывается также присутствие фосфорной кислоты и фосфат-ионов. Поэтому реагенты для фотометрического анализа необходимо выбирать таким образом, чтобы окрашенное соединение определяемого иона было бы достаточно устойчивым и значительно более прочным, чем возможные соединения этого реагента с другими ионами, содержащимися в анализируемом растворе. При этом следует иметь в виду, что для количественной оценки образования окрашенного соединения определяемого иона термодинамические или концентрационные константы в обычном виде можно использовать лишь при отсутствии побочных реакций.
К сожалению, в реальных условиях проведения анализа определяемые ионы и фотометрические реагенты, как правило, участвуют в побочных, сопряженных равновесиях, которые часто оказывают очень сильное влияние на относительную устойчивость окрашенных соединений определяемых ионов. Нередко бывают случаи, когда окрашенное соединение с большой термодинамической константой устойчивости в условиях проведения анализа оказывается относительно малоустойчивым из-за того, что реаги
15
рующие вещества одновременно участвуют в сопряженных реакциях с другими компонентами, содержащимися в растворе. Например, известно, что сульфосалицилатный комплекс Fe (III) (lg Pfessai = 14,6) значительно прочнее роданидного комплекса (lg 0FescN2+= 3,1), однако в кислой среде из-за сильной про-тонизации сульфосалицилат-ионов относительная устойчивость сульфссалицилатного комплекса Fe (III) может оказаться меньше, чем относительная устойчивость роданидного комплекса. Так, если в 1 М растворе НС1 смешать равные объемы 0,02 М раствора FeClj с 0,02 М растворами сульфосалициловой кислоты и роданида аммония, то при этих условиях в растворе NH4SCN появляется интенсивная кроваво-красная окраска, свойственная роданидному комплексу Fe (III), в то время как раствор сульфо-салициловой кислоты остается бесцветным и сульфосалицилатный комплекс Fe (III) практически не образуется.
Поэтому в условиях проведения анализа, когда на основное равновесие накладываются побочные, сопряженные реакции, для количественной оценки относительной устойчивости окрашенных соединений необходимо использовать условные (эффективные) константы устойчивости [42—47 ].
1.1.1. УСЛОВНЫЕ КОНСТАНТЫ УСТОЙЧИВОСТИ
Под условной (эффективной) константой устойчивости понимают такую характеристику, которая является производной концентрационной константы, но в отличие от нее содержит в своем выражении коэффициенты, учитывающие влияние побочных, сопряженных равновесий. Условная константа устойчивости комплекса MR„ выражается соотношением:
Pmr„ = /([См1 [Ск]") (1.6)
Здесь [MRn] —равновесная концентрация исследуемого окрашенного соединения, моль/л; [СдУ1 и [CR ] — суммарные равновесные концентрации всех ионных форм определяемого катиона и реагента, не связанных в окрашенное соединение MRn, моль/л.
В присутствии конкурирующих (маскирующих) комплексан-тов L равновесные концентрации стехиометрических форм катиона М и реагента R, не связанных в комплекс MR,,, составляют лишь долю от их общих равновесных концентраций во всех ионных формах, т. е. [М]/[См] = ам и [R ]/[CR] = aR. .
Суммарная равновесная концентрация всех форм определяемого иона, не связанного в комплекс MRn, будет слагаться из равновесных концентраций всех его незакомплексованных и закомплексованных форм:
[См] = [М] + [MLJ + [MLJ + • + [MLp]... (1.7)
Выражая составляющие равновесия (1.7) через константы устойчивости комплексных ионов ML4 и вычисляя для катиона М
16
Тфункцию закомплексованности Фронеуса [48], получим:
1/«м = [См]/[М] = ([М] + [М] [L] -Ь р£ [М] [L]2 +
р
+ • + Рр [М] [L]P)/[M] = 2 [LF ПРИ 1 = 0 = 1 (п0 условию)
1=0
Здесь pL — полная константа устойчивости комплекса МЦ-; [L]—равновесная концентрация конкурирующего комплексанта, моль/л; р — максимальное координационное число иона М в соединениях ML;.
Если в растворе содержится одновременно N конкурирующих комплексантов L, то при вычислении функции закомплексованности необходимо учитывать равновесия со всеми N комплексан-тами:
1/«м = 1+U £₽hL']‘ о-8)
I 11 1
Суммарная равновесная концентрация [CR ] фотометрического реагента, не связанного в комплекс MRn, будет слагаться из равновесных концентраций его стехиометрической формы R и различных протонированных форм:
[Cr] = [R] + [HR] -р .. • + [HqR] (1.9)
Здесь q — максимальное координационное число, проявляемое реагентом R при координации с ионами водорода (включая и протонированные формы кислоты).
Функцию закомплексованности для фотометрического реагента вычисляют аналогично:
<7
l/aR = 5? Р/1 [Н+]‘ при i = 0 fig = 1 (по условию) (1.10) (=0
Здесь [Н+] — равновесная концентрация иоиов водорода в растворе, моль/л; — полная константа устойчивости * протонного комплекса фотометрического реагента RHJ--9.
Если стехиометрической формой реагента является его частично протонированная форма RH« то при заданном значении pH раствора вычисляют мольную долю а этой формы:
|/9 \
«RHOT=₽"[H+]'n/ 2₽«[Н+Г (1.11)
I \ 1=0 /
Заменяя в выражении условной константы устойчивости суммарные равновесные концентрации [См ] и [CR ] равными им значениями концентраций стехиометрических форм этих компонен-
* Фотометрические реагенты обычно характеризуют ступенчатыми коц-стантами кислотной ионизации Ка1, Ка2.. Кад< при этом следует иметь в виду,
что рн = pH = 1/(^Ка v_1); рн = 1/Ка1Ка2... Кач.
£ ШОТ р -ш ОТ—
м/э ^/7
14 р
тов, получим формулу для вычисления условной константы устойчивости окрашенного комплекса MRn при любых заданных условиях:
",г)
Аналогичные выражения имеют условная константа устойчивости разнолигандного комплекса MR;RJ
6 - = Р аа'а1, (1-13)
'mR;-R£ HMR;Rj М R R ' '
и условная константа сопропорционирования:
MRjRj PMR;Ri/ V MRm* MR(J ' '
1.1.2. ОСНОВНЫЕ КОЛИЧЕСТВЕННЫЕ ЗАВИСИМОСТИ
С помощью условной константы устойчивости можно найти количественную зависимость между концентрацией определяемого иона М и реагента R, когда определяемый ион практически полностью связывается в фотометрируемый комплекс MRn.
Если образуется комплекс состава MRn, то практически полное связывание определяемого иона в окрашенное соединение будет происходить в тех случаях, когда равновесная концентрация комплекса составит не менее 99% от общей концентрации определяемого иона М ([MRn] 0,99 С^. В условиях равновесия при стехиометрическом соотношении компонентов М и R концентрация реагента, не связанного в комплекс, будет в п раз больше концентрации иона М (CR = пСм).
Вводя эти условия в выражение условной константы устойчивости, получим
= риц = = °-99См
MRn 0,01CM(n-0,01CM)'‘ ""(О-О’СмГ1
откуда:
₽мепСм>Ю2(п+,)/лп (1.15)
Следовательно, если неравенство (1.15) соблюдается, то фото-метрируемое соединение настолько устойчиво в водном растворе, что даже в отсутствие избытка реагента определяемый ион практически полностью переходит в окрашенный комплекс.
Когда реакция комплексообразования протекает в присутствии р-кратного избытка реагента (CR = рпСм), то для тех же 18
условий, обеспечивающих образование комплекса не менее, чем на 99%, получим
|MRn] 0’99См
R" LCM1 ICr]" 0,01Cm (PnCM - o,99ncMf
°'99CM
0.01CM(nCM)'‘(p-0,99)"
откуда
₽MRnCM > °’99‘ Ю2/(лр — 0,99/1)"
ИЛИ
РмвпС^>107п"(р-1)" (1.16)
Соблюдение неравенства (1.16) свидетельствует о практически полном переходе определяемого иона М в окрашенное соединение, который достигается при р-кратном избытке реагента.
При невысокой относительной устойчивости комплекса, когда неравенства (1.15) и (1,16) не соблюдаются, необходимая степень связанности определяемого иона в окрашенное соединение обеспечивается избытком фотометрического реагента. Полагая, как и ранее, [MRn 1 0,99См, из выражения условной константы
устойчивости найдем, что:
(МО1См1 =0,99См/(0,01См)«1-102
Следовательно,
₽MRn[CR]">l-103 (1-17)
При этих условиях определяемый ион М практически полностью будет связан в окрашенный комплекс MRn. Из неравенства (1.17) легко вычислить равновесную концентрацию фотометрического реагента, необходимую для практически полного перехода определяемого иона в окрашенное соединение:
[CR] > (ЮО/р^)1'" (1.18)
Общая концентрация реагента будет складываться из равновесной и координационно связанной концентраций: CR = [CR1 + + /гСм (где п — среднее координационное число)
На рис. 1.1 представлена диаграмма зависимости равновесной концентрации фотометрического реагента, необходимой для перевода определяемого иона М в окрашенное соединение, от условной константы устойчивости комплекса MRn. Из диаграммы видно, что наименьшее значение условной константы устойчивости комплекса, при котором практически все количество определяемого иона можно перевести в окрашенное соединение, должно составлять не менее 1- 102 (Рмип 1- Ю2).
19
Рис. 1.1. Диаграмма зависимости равновесной концентрации фотометрического реагента от значения условной константы устойчивости комплекса.
В некоторых случаях, когда при фотометрическом определении не добиваются полного перехода определяемого иона в окрашенное соединение, минимальное значение условной константы устойчивости может быть и меньше, чем 1. 102. Но при этом следует иметь в виду, что фо-тометрирование всех растворов необходимо проводить при строго по-
стоянном избытке реагента и pH раствора. Погрешности фото-
метрического определения в условиях неполного связывания
определяемого иона в окрашенное соединение, как правило, значительно возрастают.
Для разнолигандного комплекса MR/RJ полнота связывания катиона М в комплекс определяется соотношением
[ЛШЛ]/[см] = PMR/R; [cRf [CR'J‘ > ь1°2
(1.19)
которое можно использовать для вычисления концентраций лигандов R и R', необходимых для полного перевода катиона М в разнолигандный комплекс Если принять, что минимальный избыток лигандов R и R' должен превышать концентрацию определяемого металла в k раз, то минимальное значение условной константы устойчивости разнолигандного комплекса, при котором все количество определяемого металла будет закомплексовано, составит:
Р«К/К;>107(‘см)м
(1.20)
Здесь См — ориентировочная концентрация определяемого металла, моль/л.
Для комплекса MRR' состава 1 i 1 i 1 при k — 10 и См = = Ь 10~4 моль/л минимальное значение Pmrr* должно составлять 1.108.
Если смешанный комплекс содержит в своем составе Н+-или ОН’-ионы, то условие его количественного выхода будет выражаться соблюдением неравенств *
Pmhr 1Д 1^ 1-Ю2
Pmohr 1 1 • 102
(1.21)
(1.22)
Pmhr — Pmhr^hrSAmr» Pmohr — Pmohr/^mr-
20
В тех случаях, когда для прямого фотометрического определения не удается подобрать хорошего фотометрического реагента, применяют косвенные методы, основанные на различии относительной устойчивости комплексов [35, 43, 49].
Определение катионов по реакции замещения. К определяемому иону М добавляют избыток комплекса вспомогательного металла М' с маскирующим реагентом (M'Y), и стехиометрическое количество замещенного вспомогательного иона определяют по фотометрической реакции с соответствующим реагентом R. При этом необходимо обеспечить количественное протекание как реакции замещения, так и реакции образования фотометрируемого соединения. Количественное протекание реакции замещения (^99%)
M + M'Y MY+M' (1.23)
возможно при значении условной константы равновесия (1.23) при См'у ~ 2См не менее чем 1.102, а в общем случае, когда
K>v/l»M-y> 0*24)
При вычислении условных констант устойчивости реакцию между ионами М, М' и реагентом R следует рассматривать как побочную, поэтому неравенство (1.24) можно переписать в таком виде:
Pmy/Pm'y ‘ 10*яМ'/ям I* ’2^
Чем выше закомплексованность вспомогательного иона М' реагентом R, тем меньше необходима разница в константах устойчивости Pmy и Pm'y для количественного протекания реакции замещения.
Второе необходимое условие состоит в обеспечении строгого соответствия концентраций светопоглощающего комплекса M'R и определяемого иона М. Для этого фотометрический реагент R не должен вытеснять ион М' из его соединения с маскирующим реагентом M'Y более чем на 1%.
_ Если косвенное определение проводят при См'?1См = а и CR/CM = b, то условная константа равновесия
M'Y-pR^±M'R + Y (1.26)
при а = b = 2 не должна превышать 1.10~2, а в общем случае
Pm'r/Pm'Y = Рм'Как7(Рм'тат) 1 -10~®/[(« — 1) (Ь — 1)] (1-27)
Количественный же переход замещенного иона М' в светопоглощающий комплекс M'R обеспечивается соблюдением неравенства (1.17). Все эти условия выполняются, например, при косвенном определении Fe (III) с использованием комплекса Си (II) с этилендиаминтетраацетатом натрия и пиридилазонафтолом в качестве фотометрического реагента [43].
21
Определение анионов по реакции замещения. К определяемому иону А добавляют избыток вспомогательного светопоглощающего комплекса M'R, и по уменьшению его светопоглощения определяют неизвестное содержание иона А. Здесь так же, как и в предшествующем случае, желательно обеспечить количественное замещение иона R на А:
M'R + A М'А + R (1.28)
При 100%-ном избытке комплекса M'R условная константа равновесия (1.28) должна удовлетворять неравенству!
(1-29)
1.2. ПОСТОЯНСТВО СОСТАВА ОКРАШЕННЫХ СОЕДИНЕНИЙ
Окрашенное соединение можно считать удобным для применения в фотометрии, если оно имеет постоянный состав, отвечающий определенной химической формуле. Постоянный состав окрашенного соединения обусловливает постоянство интенсивности окраски раствора и является одним из основных факторов, влияющих на точность фотометрического определения. Однако в практике нередко наблюдается непостоянство состава. Главные причины этого рассмотрены ниже.
Изменение состава окрашенного комплекса в связи со ступенчатым характером его образования и диссоциации. Определяемый ион М, образуя окрашенное соединение MRn, может быть связан с различным числом ионов (или молекул) реагента R; поэтому в растворе часто находятся в равновесии различные по составу комплексные ионы (MR, MR2, MR3, ••)> имеющие, как правило, одинаковую окраску разной интенсивности. Например, Fe3+ образует с SCN- ряд комплексных ионов кроваво-красного цвета различной интенсивности в зависимости от избыточной концентрации роданид-иона — С, моль/л:
С = 5-10-' Fe3+ 4- SCN- [FpSCN]2+
С = 1,2 IO-2 Fe3+ 4- 2SCN- [Fe (SCN)2]+
С = 4 IO"2 Fe3+ 4- 3SCN- «=> [Fe (SCN)3]
C = 1,6 10 1 Fe5+ 4- 4SCN- «=> [Fe (SCNJJ-
C = 7- IO"1 Fe + 4- 5SCN- ;«=> [Fe (SCN), ]«-
В условиях избытка реагента концентрации равновесных форм анализируемого иона определяются избыточной концентрацией реагента.
Чтобы избежать больших ошибок из-за непостоянства интенсивности окраски анализируемых растворов, необходимо вы
22
бирать такие реагенты, с которыми определяемый ион давал бы прочное комплексное соединение, состоящее лишь из какого-то одного комплексного иона. Если такой реагент выбрать невозможно, то определение следует проводить при одинаковых избыточных концентрациях реагента в стандартном и исследуемом растворах. Несоблюдение этого условия приводит к получению окрашенных растворов различной интенсивности и, следовательно, к ошибкам.
Разложение окрашенного соединения во времени. Многие соединения изменяют интенсивность своей окраски во времени. В некоторых случаях из-за малой скорости реакции образования окрашенных соединений интенсивность окраски развивается постепенно и лишь по истечении определенного времени (10— 20 мин) достигает своего максимального и постоянного значения. В других случаях, наоборот, интенсивность окраски развивается очень быстро, но ее постоянное значение сохраняется сравнительно недолго. Спустя некоторое время интенсивность окраски начинает уменьшаться либо потому, что происходит окислительно-восстановительное взаимодействие между реагирующими ионами, либо в результате постепенного разрушения окрашенного соединения под влиянием присутствующих в растворе посторонних веществ, изменения pH среды, явлений протолиза, ассоциации и т. п.
В фотометрическом анализе можно использовать только такие окрашенные соединения, которые сохраняют устойчивую окраску не менее 10—15 мин. Если же устойчивых окрашенных соединений получить не удается, то используют стойкие имитирующие растворы, одинаковые с ними по окраске, либо к исследуемому окрашенному раствору добавляют специальные стабилизирующие вещества: желатин, крахмал, гуммиарабик, некоторые органические растворители.
Для большинства окрашенных соединений в литературе описано изменение интенсивности окраски во времени, однако, если приходится иметь дело с неизученным соединением, то перед фотометрированием необходимо убедиться в возможности получения стойкой окраски исследуемого раствора. Для этого приготавливают 2—3 пробы окрашенного раствора и следят за изменением интенсивности его окраски во времени, сравнивая со свежеприготовленными окрашенными растворами той же концентрации или периодически измеряя оптическую плотность. Постоянство значений оптической плотности окрашенного раствора в течение определенного времени свидетельствует о постоянстве интенсивности его окраски.
Присутствие посторонних веществ, взаимодействующих с определяемым ионам М или с выбранным для фотометрирования реагентом R. Посторонние ионы, присутствующие в анализируемом растворе одновременно с определяемым ионом, часто оказывают значительное влияние на результаты фотометрического
23
анализа. Особенно необходимо учитывать комплексообразующую способность посторонних ионов.
Если определяемый ион М образует с реагентом R относительно менее прочное соединение, чем с посторонним ионом, то определение иона М с помощью этого реагента становится практически невозможным. В этом случае необходимо либо удалить посторонний ион, либо подобрать другой реагент, либо изменить условия проведения анализа таким образом, чтобы относительная устойчивость фотометрнруемого соединения была бы значительно выше, чем относительная устойчивость бесцветного комплекса определяемого иона с посторонним конкурирующим комплексан-том (лигандом). Например, при определении Ёе (III) при pH л 3 присутствие небольших количеств фторид-ионов вызывает заметное обесцвечивание раствора роданида железа (Pfescn2+ = 5,2 X X 102), так как ионы Fe (III) связываются в более прочный фторидный комплекс (|3peF2+ = 1,6- 105) и ни при каких значениях pH раствора влияние фторид-ионов устранить не удается.
В присутствии фторид-ионов Fe (III) можно определять с помощью другого реагента, например салициловой кислоты, с которой ионы Fe (III) образуют более прочный салицилатный комплекс. Правда, при pH = 3,0 относительная устойчивость моно-салицилатного комплекса Fe (III) сравнительно невелика (PpeSai+ = = 1,1. 105) и мешающее влияние фторид-ионов можно устранить только в том случае, если общая концентрация салициловой кислоты будет превышать концентрацию фторид-ионов не менее чем в 1,5. 104 раз. Если же фотометрическое определение Fe (III) проводить в виде трисалицилатного комплекса при pH = 10,0 (PpesaiT ~ 2. 1012), то мешающее влияние фторид-ионов удается пол<юстью устранить. Минимальную избыточную концентрацию, необходимую для полного связывания Fe (III) в салицилатный комплекс в присутствии фторид-ионов, можно вычислить по формуле (1.18).
Даже в тех случаях, когда окрашенное соединение определяемого иона М с реагентом R прочнее, чем бесцветное соединение с посторонним ионом, мешающее влияние последнего все же может сказываться и уменьшать выход окрашенного соединения. Мешающее влияние в этом случае может быть сведено к минимуму или полностью устранено введением в раствор соответствующего избытка фотометрического реагента. Избыточная концентрация реагента легко может быть найдена по уравнению (1.18) или по диаграмме (рис. 1.1). При этом, вычисляя условную константу устойчивости светопоглощающего комплекса MRn> необходимо учитывать не только сопряженное комплексообразование катиона, но и участие реагента в сопряженном комплексообразовании с посторонним катионом М':
1 q п'
= -Т5Т- = S₽" 1Н+Ё + £ lCM'l «М- IR]'-1 (1 -30)
1 * О 1
й
V
Здесь 1/ам, = У Pj" [L]‘; [L] —равновесная концентрация дополнитель-о
ного комплексанта, маскирующего посторонний ион М'; [См,] — максимально возможная концентрация постороннего иона М' в анализируемом растворе.
В простейшем случае, когда комплексант L отсутствует и комплекс M'R,r имеет состав 1:1, уравнение (1.30) упрощается-
1/«R = У РР 1н+Г' + Р-.1-.ч [См,] (1.31)
о
При более сложном составе комплекса M'R„- (п > 1) следует использовать общее уравнение (1.30), которое удобнее решать методом последовательных приближений (методом итерации).
Взаимодействие посторонних веществ с определенным ионом М или реагентом R приводит иногда к образованию новых окрашенных соединений, которые значительно затрудняют, а иногда и полностью исключают возможность фотометрического определения иона М.
1.3. ВЛИЯНИЕ pH РАСТВОРА
НА ОБРАЗОВАНИЕ ОКРАШЕННЫХ СОЕДИНЕНИЙ
Влияние pH раствора на окрашенные комплексы выражается в различных формах, однако в большинстве случаев оно сводится к разрушению или изменению состава окрашенного соединения. Иногда оно способствует образованию окрашенных комплексов с посторонними ионами, присутствующими в растворе, обусловливает изменение растворимости окрашенных соединений, влияет на состояние окислительно-восстановительного взаимодействия.
Ниже рассмотрены основные случаи влияния кислотности среды на воспроизводимость и правильность фотометрических определений.
Окрашенные комплексы с анионами сильных кислот. Анионы сильных кислот (Cl-, I-, SCN- и др.) даже при очень высокой концентрации Н+ в растворе не связываются ими в молекулы кислоты, поэтому концентрация анионов в растворе изменяется незначительно. При повышении кислотности раствора протонизация реагента протекает в незначительной степени и условная константа устойчивости комплекса MRn практически остается без изменений. Следовательно, повышение кислотности раствора не приводит к разрушению окрашенного соединения, образованного анионом сильной кислоты.
При уменьшении кислотности среды, т. е. при повышении pH раствора, катионы металла, как правило, взаимодействуют с ОН-ионами, образуя в конечном счете малорастворимые гидроксиды или основные соли. Окрашенное соединение при этом разрушается. Малорастворимое соединение может и не образоваться, тем не менее участие определяемых катионов в сопряженном комплексо
25
образовании с ОН-ионами значительно уменьшает условную константу устойчивости окрашенного комплекса и, следовательно, приводит к уменьшению степени связанности определяемого иона в окрашенное соединение. Особенно сильное влияние наблюдается для малопрочных комплексов, которые при увеличении pH раствора могут быть разрушены полностью.
Поэтому реакции образования окрашенных, соединений ионов металлов с анионами сильных кислот целесообразно проводить в достаточно кислых средах, где условная константа устойчивости окрашенного комплекса сохраняет свое наибольшее значение.
Окрашенные комплексы с анионами слабых кислот. Когда в качестве реагентов используют слабые органические кислоты HR (салициловая кислота, ализарин, диметилглиоксим и др.), изменение pH раствора оказывает очень сильное, хотя внешне и не всегда заметное, влияние. Полнота связывания иона М в окрашенное соединение MRn зависит от концентрации в растворе анионов реагента R-, которая в свою очередь зависит от концентрации Н+ в растворе. В кислых растворах концентрация R-бывает невелика, так как равновесие ионизации слабой кислоты HR сильно смещено в сторону недиссоциированной (кислотной) формы реагента. Увеличить концентрацию R- путем повышения общей концентрации реагента не всегда удается, поскольку слабые органические кислоты часто имеют ограниченную растворимость. В этом случае концентрацию увеличивают повышением pH раствора, которое смещает равновесие ионизации кислоты в сторону его солевой формы R“.
Таким образом, реакции образования окрашенных соединений ионов металлов с анионами слабых кислот следует проводить по возможности в менее кислых средах. Однако уменьшение концентрации Н+ необходимо осуществлять очень осторожно, так как при повышении pH раствора может происходить образование основных солей или гидроксидов определяемых металлов; может изменяться состав окрашенного соединения вследствие ступенчатости комплексообразования. В некоторых случаях, когда влияние конкурирующего комплексообразования ОН-ионов преобладает над влиянием депротонирования реагента, повышение pH раствора может привести к противоположным результатам, т. е. к уменьшению степени связанности иона М в окрашенное соединение. Поэтому максимальный выход светопоглощающего комплекса будет наблюдаться только в определенном интервале значений pH раствора.
Оптимальную кислотность раствора, при которой необходимо прово_.ить анализ, можно определить по максимальному значению условной константы устойчивости окрашенного комплекса (рис. 1.2). При уменьшении кислотности среды вследствие депротонирования реагента условная константа устойчивости комплекса MR„ увеличивается, достигая максимального значения в тот момент, когда влияние депротонирования реагента уравно-
26
Рис. 1.2. Зависимость условной константы устойчивости комплексона-тов различных металлов от pH раствора.
вешивается влиянием сопряженного комплексообразования катисна ОН-ионами. При дальнейшем повышении pH раствора вследствие преобладания комплексообразования катиона металла ОН-ионами условная константа устойчивости, а следовательно, и выход окрашенного комплекса будут уменьшаться. Наиболее благоприятной кислотностью среды при проведении фотометрической реакции с бесцвет-
ным реагентом можно считать тот интервал pH раствора, в котором условная константа устойчивости окрашенного комплекса
достигает своего максимального значения.
Оптимальное значение pH раствора при проведении фотометрического определения можно найти и аналитическим путем. Для этого необходимо условную константу устойчивости фотометри-руемого комплекса представить как функцию концентрации Н-ионов (функция должна проходить через максимум) и первую производную этой функции приравнять к нулю:
₽MRn = ₽mr aM«R = Pmr /[f [Н+Г') (£p«[H+]'
" n I L \ о / \ о
Здесь — полная константа устойчивости гидроксокомплекса М (ОН);« /Си, — ионное произведение воды.
Следовательно, оптимальное значение концентрации Н-ионов можно найти решением уравнения:
[Н+Г‘] ( £р?[НЧ‘'
о / \ о
= 0
(1.32)
Взяв производную и раскрыв знаки сумм, получим уравнение в общем виде:
(oOHiz 9(-»ОН rz2 \
+ + • • J (’ + IH+1 + P2H tH+l2 +-)-
(ftOHr оОНдг2 \
,+-WL + + ” J № + 2₽”tH+1 + < fH+l2 +•) = 0
(1-33)
27
Для простейшего случая, когда светопоглощающее соединение имеет состав 1 > 1 и сопряженные реакции протонизации фотометрического реагента и гидроксокомплексообразования катиона протекают только по первой ступени, т. е. когда п — q = j = 1, уравнение (1.33) упрощается-
оОН /z /
^^-(1+йГ [НП)-(м
oOH с-Pi pFT
₽»=0
Отсюда оптимальное значение [Н+}:
(1-34)
Если стехиометрической формой реагента HqR является его частично протонированная форма то переменная соста-
вляющая условной константы устойчивости, зависящая от концентрации Н-ионов, будет определяться произведением:
£ Р?н<,[Н+Г
О
Q
£ ₽« [н+]'7(₽« [Н+Г)
(1.35)
Взяв первую производную этого выражения по концентрации Н-ионов и приравняв ее к нулю, после преобразования получим: (Ррх. 1Н+Г2 + 2Р?Н№Ш [Н+]-3 +...)(+
pH [H+j 1-т pH \
+-----Zh----+----------+•••!-
Pm Pm '
(- n (i + ррх [н+]-1 + р°х [н+]'2 +...)
т [H+]-(m+I) оН Pm
+
(1 — т.) Р“ [Н+)"п (2 — т) р2н [Н+]‘-т
6** + Р“
Нт Нт
(1.36)
Решение уравнения (1.36) позволяет найти оптимальное значение pH раствора, при котором обеспечивается максимальный выход комплекса с протонированной формой HmRm~'7 реагента.
Однако на практике чаще используют более широкий интервал pH раствора, где обеспечивается практически полное связывание определяемого иона М в светопоглощающее соединение MRn, т. е. где |3мип [CR ]" 1. 102 (рис. 1.3).
Более сложно решается вопрос о выборе значения pH раствора для проведения фотометрического определения в тех случаях, когда при оптимальном значении pH значительная часть фотометрического реагента находится в такой форме, спектр поглощения которой очень близок к спектру анализируемого соединения.
28
Рис. 1.3. Графическое определение оптимального интервала pH в фотометрическом анализе (комплексы MR2 или MRR', [CR] = = [CR,]= 1-10~3 моль/л).
— значение pH максимального выхода r M8KG светопоглощающего соединения; рНг — рН2 оптимальный интервал pH ..при фотометрическом определении.
В этих случаях проведение анализа при pH раствора, обеспечивающем максимальный выход светопоглощающего соединения, из-за большого поглощения самого реагента сопряжено со значительными погрешностями, особенно при определении малых количеств анализируемого элемента. В этих условиях фотометрирование растворов целесообразнее проводить при меньших значениях pH раствора, при которых хотя и не достигается максимальный выход комплекса, но содержание светопоглощающей формы реагента также не превышает 10—15% от светопоглощения анализируемого комплекса. Избыточная концентрация реагента при этом должна быть по возможности небольшой, но обеспечивающей практически полный перевод определяемого элемента в светопоглощающее соединение (₽мкп [CR 1" 1.102).
Окрашенные комплексы с реагентами, проявляющими индикаторные свойства. Многие окрашенные реагенты, являющиеся слабыми органическими кислотами, проявляют индикаторные свойства (ализарин, дитизон, арсеназо, торон и другие). Такие реагенты способны изменять свою окраску с изменением концентрации Н+. Это связано с состоянием равновесия, в котопом кислотная (молекулярная) форма реагента-индикатора HR отличается по структуре и окраске от солевой формы R" этого же реагента. При повышении pH раствора происходит более полное превращение кислотной формы реагента в солевую. Понижение pH раствора приводит к увеличению концентрации слабой кислоты.
Таким образом, несоблюдение постоянства pH среды вызовет изменение не только интенсивности окраски раствора, но даже и самой окраски. Например, алюминий в слабокислой среде при pH 4,5 образует с ализарином комплексное соединение красного цвета; сам ализарин при этом значении pH имеет желтый цвет. Однако при pH > 5,5, вследствие преобладания солевой формы ализарина, раствор приобретает красно-фиолетовый цвет, который легко можно принять за окраску комплекса алюминия с ализарином. Поэтому определение алюминия с ализарином рекомендуется проводить в интервале pH = 4,54-5,0. Следова-
29
тельно, при использовании реагентов, проявляющих индикаторные свойства, реакции образования окрашенных соединений следует проводить при строго определенной кислотности раствора.
Нужный интервал pH среды обеспечивается, как правило, применением буферных растворов.
Разнолигандные комплексы. Для разнолигандных комплексов состава MR/RJ оптимальный интервал pH раствора будет определяться максимальным значением его условной константы устойчивости, а точнее, максимальнььм значением произведения а-кээффи-циентов: амоДак.. На рис. 1.3 показан допустимый интервал pH при фотометрическом определении катионаМ в виде разнолнганд-ного комплекса MRR'. Если катион металла образует смешанный протон- или гидроксидсодержащий комплекс MHR или MOHR и константы устойчивости этих комплексов известны, то можно легко определить интервал pH доминирования смешанных комплексов. Так, количественный выход протонированного комплекса MHR будет наблюдаться, когда
[MHR]/[MR] = ₽«HR [Н+] > 1 • 102 (1.37)
т. е. при pH < 1g рДня — 2, а при pH 1g Pmhr + 2 его концентрация становится несоизмеримо малой по сравнению с концентрацией комплекса MR.
Совершенно аналогично количественный выход смешанного комплекса MOHR наблюдается при условии, когда
[MOHR]/[MR] = Pmohr [ОН’] >1-102 (1.38)
или при pH 16 — lg Pmohr - при pH < 12 — 1g Pmohr содержание смешанного комплекса MOHR несоизмеримо мало и его образованием можно пренебречь.
1.4. КОЛИЧЕСТВО
ФОТОМЕТРИЧЕСКОГО РЕАГЕНТА
Реакция образования окрашенного соединения MR„ требует некоторого избытка фотометрического реагента. В одних случаях для полного связывания иона в окрашенное соединение достаточно прибавить небольшой избыток (20—30%) фотометрического реагента по сравнению со стехиометрически рассчитанным количеством. Это имеет место, когда окрашенное соединение характеризуется высокой прочностью (Pmr]„См > 102("+1)/п") И в растворе отсутствуют компоненты, реагирующие с определяемым ионом или реагентом. В других случаях, когда прочность окрашенного соединения невелика (PmrhCm < 10?("+п/л'г) и вследствие его значительной диссоциации или конкурирующего комплексообразования некоторое количество определяемого иона остается не связанным в окрашенный комплекс, применяют боль-30
(1.39)
м"
шой избыток реагента, превышающий стехиометрическое количество в 10 раз и более. Кратность избытка реагента, необходимого для полного связывания определяемого иона в окрашенное соединение, можно вычислить по формуле (1.39), зная состав и условную константу устойчивости окрашенного соединения:
p>i + po7₽MRJ‘^
В ряде случаев [50], если комплексообразование протекает ступенчато и в условиях избытка реагента образуются окрашенные комплексы различного состава, фотометрирование можно проводить при некотором недостатке реагента (CR/CM т 0,84-0,9), когда образуется только простейший комплекс состава 1:1. Для обеспечения постоянного соотношения концентраций определяемого металла и реагента в стандартных и анализируемых растворах необходимый объем раствора реагента при определении неизвестного количества металла устанавливают предварительно по кривой насыщения или кривой фотометрического титрования (см. оазд. 8.1.3 и 8.1.4).
Глава 2
ОПТИЧЕСКИЕ СВОЙСТВА
ОКРАШЕННЫХ СОЕДИНЕНИЙ В РАСТВОРАХ
2.1. ПОГЛОЩЕНИЕ СВЕТА РАСТВОРАМИ ОКРАШЕННЫХ СОЕДИНЕНИЙ
При прохождении света через слой окрашенного вещества часть его отражается, часть поглощается и часть света проходит через слой вещества (рис. 2.1).
Интенсивность падающего светового потока при прохождении через поглощающий раствор разлагается на составляющие:
1« — I + /ц + /от
Рис. 2.1. Прохождение светового потока через окрашенный раствор.
31
женной части светового потока за счет рассеяния при работе
Рис. 2.2. Графическое выражение закона Бугера—Ламберта.
Здесь I — интенсивность светового потока, прошедшего через слой вещества; /п — интенсивность светового потока, поглощенного окрашенным веществом, /от — интенсивность отраженного светового потока.
При сравнительных измерениях поглощения света различными растворами пользуются одинаковыми кюветами, для которых интенсивность отра-постоянна и мала; потеря света с истинными растворами стано
вится также ничтожно малой. Ослабление света происходит глав-
ным образом за счет поглощения (абсорбции) световой энергии /п окрашенным раствором. Интенсивности падающего светового потока /0 и прошедшего через раствор / могут быть
определены экспериментально.
Связь между интенсивностями падающего светового потока /0 и светового потока, прошедшего через слой окрашенного вещества /, устанавливается законом Бугера — Ламберта, согласно которому однородные слои одного и того же вещества одинаковой толщины поглощают одну и ту же долю падающей на них световой энергии (при постоянной концентрации растворенного вещества). Математически этот закон выражается уравнением экспоненциальной зависимости:
I = /ое-а/
Здесь е — основание натуральных логарифмов; а — коэффициент поглощения; / — толщина поглощающего слоя.
На рис. 2.2 показана экспоненциальная кривая, выражающая графическую зависимость между интенсивностью светового потока, прошедшего через слой поглощающего вещества, и толщиной слоя.
Связь между концентрацией поглощающего раствора и его оптической плотностью 1g (IJI) выражается законом Бера, согласно которому оптическая плотность раствора прямо пропорциональна концентрации растворенного вещества при постоянной толщине слоя:
lg(Z0'7) = *10
Здесь kr — коэффициент пропорциональности, С — концентрация растворенного вещества.
82
2.1.1. ОСНОВНОЙ ЗАКОН СВЕТОПОГ ЛОЩЕНИЯ*
(ОБЪЕДИНЕННЫЙ ЗАКОН БУГЕРА-ЛАМБЕРТА-БЕРА)
Зависимость интенсивности монохроматического светового потока, прошедшего через слой окрашенного раствора, от интенсивности падающего потока света, концентрации окрашенного вещества и толщины слоя раствора определяется следующим уравнением:
1 = l„-lO-kCl (2.1)
Здесь k — коэффициент светопоглощения, зависящий от природы растворенною вещества, температуры, растворителя н длины волны света.
Это соотношение, известное как закон Бугера — Ламберта — Бера, является основным законом светопоглощения и лежит в основе большинства фотометрических методов анализа.
Если концентрация С выражена в молях на литр, а I — в сантиметрах, то k становится молярным коэффициентом светопоглощения и обозначается е7_. Уравнение (2.1) в этом случае будет иметь вид:
/ = /о.1О-Е^с/ (2.2)
После преобразований и логарифмирования уравнений (2.1) и (2.2) получим выражения основных фотометрических величин (табл. 2.1).
При соблюдении основного закона светопоглощения оптическая плотность раствора прямо пропорциональна молярному коэффициенту светопоглощения, концентрации поглощающего вещества и толщине слоя раствора:
А = екС1 (2.3)
При графическом изображении зависимости оптической плотности от концентрации (при постоянном значении /) получается прямая линия. Эта прямая проходит через начало координат при отсутствии поглощения света растворителем и систематических погрешностей (рис. 2.3).
Уравнения (2.2) и (2.3) выведены для монохроматического света, т. е. света определенной длины волны, который может быть
2 М И. Булатов и др.
33
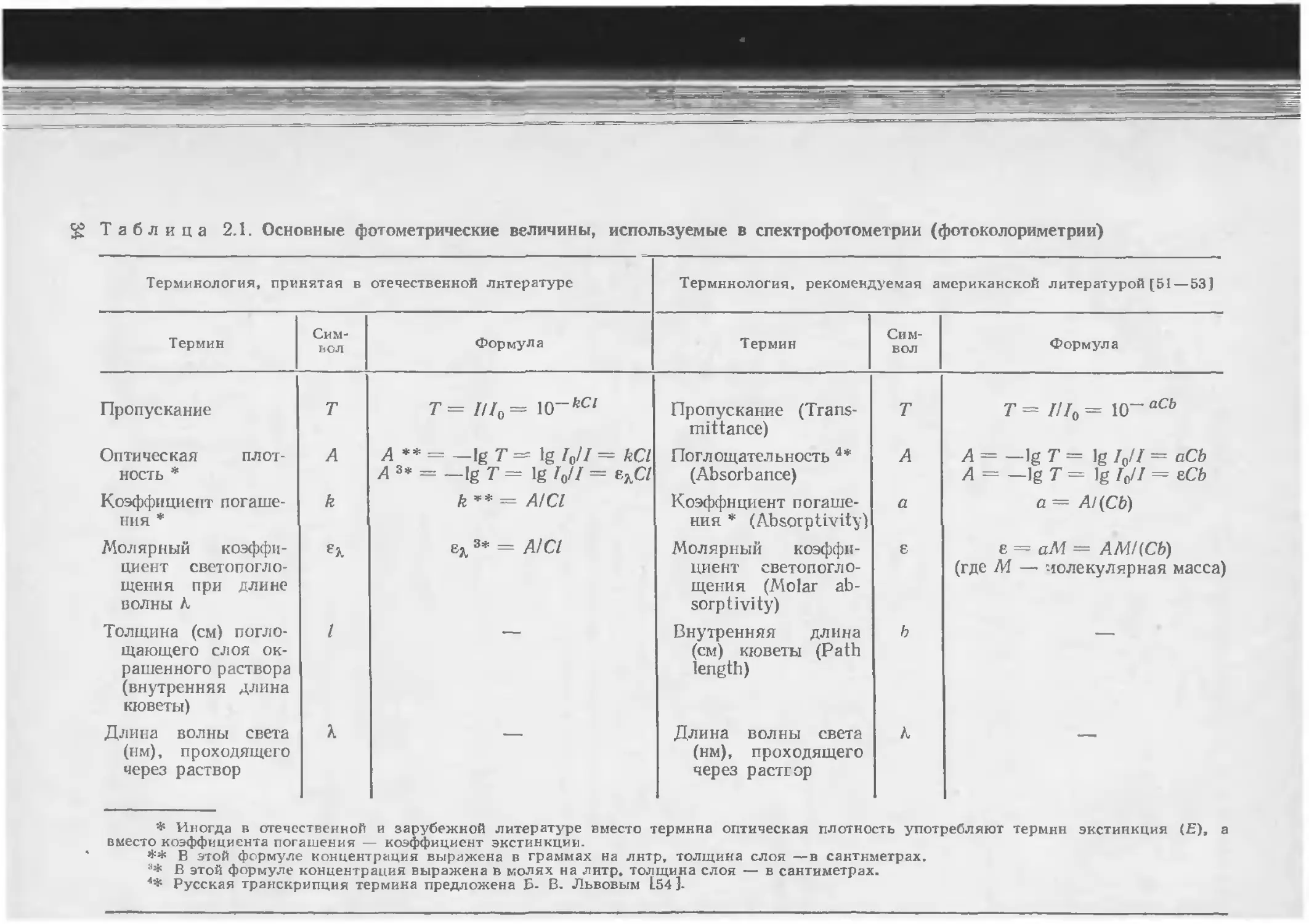
Таблица 2.1. Основные фотометрические величины, используемые в спектрофотометрии (фотоколориметрии)
Терминология, принятая в отечественной литературе Терминология, рекомендуемая американской литературой [51—53]
Термин Символ Формула Термин Символ Формула
Пропускание т т= ///f, = w~kCl Пропускание (Transmittance) т Т = Z/Zo = 10~ аСЬ
Оптическая плот- ность * А А ** = —lgT== lg/0// = kCl А з* = _lg т = 1g I0U = ezC7 Поглощательность 4* (Absorbance) А А = —1g Т = 1g 10Н = аСЬ А = —1g Т = 1g Zo/Z = tCb
Коэффициент погашения * k k ’* = AICl Коэффициент погашения * (Absorptivity) а а = Al(Cb)
Молярный коэффициент светопогло-щения при длине волны Л е?. ex 3* = A1CI Молярный коэффициент светопогло-щения (Molar absorptivity) Е е = аМ — АМКСЬ) (где М. — молекулярная масса)
Толщина (см) поглощающего слоя окрашенного раствора (внутренняя длина кюветы) / Внутренняя длина (см) кюветы (Path length) ь
Длина волны света (нм), проходящего через раствор X — Длина волны света (нм), проходящего через растюр к —
* Иногда в отечественной и зарубежной литературе вместо термина оптическая плотность употребляют термин экстинкция (Е), а вместо коэффициента погашения — коэффициент экстинкции.
** В этой формуле концентрация выражена в граммах на лнтр, толщина слоя —в сантиметрах.
3* В этой формуле концентрация выражена в молях на лнтр, толщина слоя — в сантиметрах.
Русская транскрипция термина предложена Б- В. Львовым 154 ]-
хроматическом, а в полихроматическом свете, т. е. на довольно широком участке спектра — в интервале длин волн 20—100 нм. В этом случае в уравнение (2.3) вместо молярного коэффициента светопоглощения следует подставлять значения среднего молярного коэффициента светопоглощения (ё), зависящие от характеристики светофильтра (ё < еА).
2.1.2. МОЛЯРНЫЙ КОЭФФИЦИЕНТ СВЕТОПОГЛОЩЕНИЯ
Из уравнения (2.3) следует, что А = ех при С = 1 моль/л и I = = 1 см, т. е. молярный коэффициент светопоглощения представляет собой оптическую плотность 12VT раствора, помещенного в кювету с толщиной слоя 1 см. Молярный коэффициент светопоглощения е? зависит от длины волны проходящего света, температуры раствора и природы растворенного вещества и не зависит от толщины поглощающего слоя и концентрации растворенного вещества. Молярный коэффициент светопоглощения отражает индивидуальные свойства окрашенных соединений и является их определяющей характеристикой. Для разных веществ он имеет различное значение. Так, для слабоокрашенных веществ (таких, как хромат калия) ех = 400-4-500, для сильноокрашенных, например соединений кадмия или цинка с дитизоном, ех достигает значений 85 000 и 94 000 соответственно. Значения е?, наиболее интенсивно окрашенных соединений, как правило, не превышает 100 000—120 000. Брауде [55] на основе квантово-химических расчетов показал, что значения е выше 100 000 являются маловероятными *.
Значения ек определяют различными спектрофотометрическими методами по экспериментальным данным (см. гл. 8). Иногда находят из экспериментальных данных по уравнению (2.3). Однако при этом следует иметь в виду, что объективные значения е?_ можно получить лишь при фотометрировании достаточно прочных комплексов в условиях, обеспечивающих количественный переход определяемого иона в окрашенное соединение постоянного состава. Если эти условия не соблюдаются, то таким способом можно найти лишь условный молярный коэффициент светопоглощения е', значение которого будет зависеть от условий фотометрировапия (pH раствора, концентрации конкурирующих комплексующих реагентов и т. п.).
2.1.3. ОТКЛОНЕНИЯ ОТ ОСНОВНОГО ЗАКОНА СВЕТОПОГЛОЩЕНИЯ
Объединенный закон Бугера — Ламберта — Бера многократно проверялся на опытах, и его можно считать строго установленным. Однако на практике могут наблюдаться отклонения, которые
* При образовании сложных разнолигандных ассоциатов молярный коэффициент светопоглощения может н превышать указанные значения [38].
2* 35
Рнс. 2.4. Влияние степени монохроматичности поглощаемого света на соблюдение основного закона свето погл ощени я:
1 — монохроматическое излучение; 2 — полихроматическое излучение.
происходят за счет несоблюдения закона Бера. Закон Бера справедлив для весьма разбавленных растворов, и поэтому область его применения ограничена, которой начинается заметное отклонение
Концентрация, при
от основного закона светопоглощения, зависит от природы поглощающего вещества, степени монохроматичности поглощаемого света (рис. 2.4), точности измерения и присутствия посторонних веществ. Присутствие посторонних электролитов вызывает деформацию молекул или комплексных окрашенных соединений, вследствие чего изменяется интенсивность окраски и светопогло-
щение.
При разбавлении концентрированных окрашенных растворов электролитов изменяется степень диссоциации их на ионы, что также вызывает отклонения от закона Бера. Кроме диссоциации и ассоциации молекул на светопоглощение раствора оказывают влияние явления протолиза, комплексообразования, образования промежуточных продуктов и коллоидов, таутомерные превращения. Сольватация (гидратация) также сказывается на светопог-лощении растворов, так как с изменением концентрации процесс сольватации протекает неодинаково. Эти процессы часто связаны с концентрацией Н+ в растворе, температурой, действием солнечного света и т. п. Изменение концентрации Н+ в растворе приводит к различной степени связанности определяемого иона в окрашенное соединение, к изменению состава последнего или даже к его разрушению (см. разд 1.5).
Особенно большие отклонения от закона Бера могут наблюдаться при разбавлении растворов малоустойчивых окрашенных соединений. Диссоциация светопоглощающего соединения и влияние сопряженных реакций, протекающих в водном растворе, являются одной из основных «химических» причин нарушения закона Бера.
Отклонения от закона Бера в связи с диссоциацией комплекса рассмотрены в работах А. К. Бабко и А. Т. Пилипенко [35, 56]. Согласно этим работам, отклонения от закона Бера (А, %) при разбавлении раствора простейшего монокомплекса определяются уравнением (2 4) в отсутствие избытка реагента и уравнением (2.5) при р-кратном избытке реагента:
Д = Г(К/См)(Кч —1)100
(2.4)
Д= (Л/рСм) (?—!)• ЮО
(2.5)
36
Здесь К — константа нестойкости светопоглощающего комплекса; См —• общая концентрация определяемого иона, моль/л; q — кратность разбавления раствора.
Однако уравнения (2.4) и (2.5) не учитывают влияния условий проведения анализа и поэтому справедливы только в тех случаях, когда ни определяемый ион, ни фотометрический реагент не участвуют в сопряженных равновесиях, что в аналитической практике встречается очень редко, особенно когда реагент является анионом слабой кислоты. В большинстве случаев на равновесие образования светопоглощающего соединения при низких значениях pH накладываются сопряженные равновесия протонизации реагента, а при высоких значениях pH — сопряженные равновесия образования гидроксокомплексов катиона. Если учесть, что в реакциях конкурирующего комплексообразования нередко участвуют компоненты используемого буфера или присутствующие в растворе посторонние комплексанты, то становится очевидным, что количественное отклонение от закона Бера, обусловленное разбавлением светопоглощающего соединения, нельзя рассматривать изолированно от сопряженных равновесий, протекающих в водном растворе.
При наличии сопряженных равновесий степень связанности определяемого иона в светопоглощающее соединение MRn и отклонение от закона Бера при разбавлении количественно можно оценить с помощью условной константы устойчивости [42—471 светопоглощающего комплекса, которая учитывает изменение условий при разбавлении растворов.
Отклонение от закона Бера оценивается относительным изменением оптической плотности окрашенного раствора при его разбавлении, получаемым при неизменном значении произведения концентрации светопоглощающего соединения на толщину слоя (Cl = const).
Пусть имеется окрашенный раствор с концентрацией См и оптической плотностью Ао. После разбавления его в q раз концентрация и оптическая плотность будут равны соответственно C^Jq и Если исходный и разбавленный в q раз окрашенные растворы фотометрировать при одинаковых условиях (/ = const), то отклонение от закона Бера будет определяться соотношением:
Д - inn А0 — дА,{ (1 — бв) — У (См ,')/(!— 6?) _ - 60
Ао елСм/(«-б0) “ 1-6„
(2.6)
Здесь 60 — степень диссоциации комплекса в исходном растворе (мольная Доля в исходном растворе всех ионных форм определяемого иона, не связанных, В светопоглощающий комплекс); — степень диссоциации комплекса в разбавленном растворе.
Выражение для условной константы устойчивости коплекса в отсутствие избытка реагента в тех же обозначениях запи
37
шется следующим образом:
= (1 — 60) СМ = 1-6„
PMRn 6ОСМ (ябоСм)'1 б',+’ (нСм)"
(2.7)
Если при заданных условиях светопоглощающий комплекс характеризуется высокой относительной устойчивостью, т. е. если
Рмп С” > 10О("+1,№ (2.8)
П М
то 60 не превысит 0,01 и, следовательно, с ошибкой не более 1% (отн.) уравнения (2.6) и (2.7) примут более простые выражения:
Л = 100 (6g —60) (2.9)
Рмкп = !/[6S+1 [«См)”] (2-10>
Выразив 60 и 6, в уравнении (2.9) через условную константу устойчивости (2.10) светопоглощающего комплекса в исходном и разбавленном растворах, получим:
ki 1 1
?7₽^м«") "+1 - (1/₽осм«") П+1 ] <2-Н)
При этом следует иметь в виду, что уравнения (2.9)—(2.11) будут справедливы только при соблюдении неравенства (2.8). В противном случае при заданных условиях, зная величины См и ₽MRn, необходимо вычислить для каждого раствора значения 60 и 69 и по уравнению (2.6) без упрощающих допущений найти отклонение от закона Бера. В частном случае, для простейшего комплекса состава 1:1, когда условные константы устойчивости ро и pg одинаковы и равны концентрационной константе устойчивости комплекса Рмпп, уравнение (2.11) становится тождественным уравнению (2.4).
Из рассмотрения уравнения (2.11) видно, что при рд/ро <« (р разбавление раствора приводит к увеличению диссоциации комплекса и Д имеет положительное значение. При pg/Рэ = qr' отклонение от закона Бера, обусловленное разбавлением, равно нулю, так как увеличение диссоциации комплекса при разбавлении уравновешивается его относительным упрочнением вследствие снижения концентрации конкурирующих комплексантов. При Pg/P6 > tf1 разбавление раствора приводит к относительному упрочнению комплекса, отклонение от закона Бера уменьшается и Д имеет отрицательное значение.
При р-кратном избытке реагента условную константу устойчивости светопоглощающего комплекса MRn можно записать в такой форме:
______(1 6р) См____________1 6р______
П 6дСМ (рП + Лбд)™ Сд! 60 (р + бр)” (пСм)”
38
Если степень связанности определяемого иона в комплекс не менее 99%, что бывает при соблюдении неравенства
Pmr^m Ю~1\пп (р—1)п] (2.13)
то 60 не превысит 0,01 и, поскольку 60 1, а р 60) уравне-
ние (2.12) упрощается:
₽MRn = !/L6o (Р«См)"] (2-14)
Выражая значения 60 и 6П в уравнении (2.9) через условную константу устойчивости, описываемую уравнением (2.14), получим расчетное уравнение для вычисления отклонения от закона Бера при ^-кратном разбавлении раствора в условиях р-кратного избытка реагента:
д = юо - ₽;)/[ рор; (PncMf] (2.is)
В тех случаях, когда не соблюдается неравенство (2.13), при известных значениях р, См и величины 60 и 6д следует рассчитывать отдельно для каждого раствора и для вычисления Д использовать общее уравнение (2.6). При рассмотрении уравнения (2.13) видно, что, как и в отсутствие избытка реагента, при
< О'1, fVPo > и fVPo = 0п отклонение от закона Бера при разбавлении получается соответственно положительным, отрицательным и равным нулю. Для простейшего комплекса состава 1 : 1 в отсутствие сопряженных равновесий в водном растворе, когда р, = ро = pMR, уравнение (2.15) превращается в уравнение (2.5).
Минимальное отклонение от закона Бера при разбавлении растворов малопрочных комплексов получается в том случае, когда окрашенный раствор разбавляют не чистым растворителем, а раствором реагента, имеющим концентрацию, равную избыточной концентрации реагента в исходном окрашенном растворе. Тогда разбавление окрашенного раствора происходит при постоянной концентрации реагента. Поэтому степень диссоциации окрашенного соединения остается неизменной и закон Бера практически не нарушается. Недостаточная монохроматичность поглощаемого светового потока обычно вызывает отрицательные отклонения от основного закона светопоглощения. При этом, чем шире интервал длин волн поглощаемого света, тем уже область определяемых концентраций, где соблюдается основной закон светопоглощения (см. рис. 2.4).
2.1.4. ПРАВИЛО АДДИТИВНОСТИ
Если в растворе содержится п светопоглощающих компонентов, которые не вступают друг с другом в химическое взаимодействие, то при условии соблюдения основного закона светопоглощения оптическая плотность такого раствора будет равна сумме пар-
39
циальных оптических плотностей всех содержащихся в растворе светопоглощающих компонентов. В этом проявляется принцип аддитивности (правило аддитивности) оптических плотностей:
Д = EiCjZ -р е2С21 -(-•••+ enCnl = I tfC, (2 16)
1=1
Здесь Bj, es, , еп — молярные коэффициенты светопоглощения компонентов смеси; Ci, Cs, , Cn — молярные концентрации компонентов смеси; I — толщина поглощающего слоя, см.
Принцип аддитивности давно известен и подтвержден экспериментально. На использовании принципа аддитивности основаны все количественные методы спектрофотометрического анализа многокомпонентных систем при одновременном определении содержащихся в них компонентов.
2.2. ЭЛЕКТРОННЫЕ СПЕКТРЫ ПОГЛОЩЕНИЯ
В фотометрическом анализе используют поглощение света молекулами комплексных (координационных) соединений, сольватов, а в ряде случаев и более сложных соединений (ассоциатов, аддуктов и т. п.). Взаимодействие светового излучения с такими сложными многоэлектронными системами описывают с помощью молекулярных спектров поглощения [35, 38, 49], вид которых определяется в основном состоянием электронов внешних орбиталей, участвующих в образовании химической связи.
При взаимодействии молеку лы светопоглощающего вещества со световым излучением энергетические состояния электронов в молекуле изменяются. Молекула поглощает часть падающего излучения, и поглощенная энергия расходуется для перехода электронов из одного из основных состояний в состояние с более высокой энергией. Так как различные электроны в молекуле комплексного соединения обладают неодинаковыми энергиями в основном состоянии, то они возбуждаются излучением разных длин волн, поэтому все светопоглощающие соединения характеризуются избирательным поглощением света. Зависимость интенсивности поглощаемого света от длины волны и характеризует электронный спектр поглощения рассматриваемой молекулы.
В отличие от атомных спектров, представляющих собой отдельные линии, спектр поглощения молекулы состоит из ряда полос поглощения, каждая из которых напоминает гауссову кривую и отличается от других полос интенсивностью и длиной волны максимума поглощения. Полосчатый характер молекулярных спектров поглощения объясняется тем, что вследствие поглощения света наряду с изменением электронных состояний молекул происходит непрерывное перемещение атомных ядер, которое приводит к изменению колебательных состояний молекул. Поэтому каждому электронному переходу фактически соответ-40
ствует целый ряд колебательно-электронных переходов, которые и обусловливают полосчатый характер молекулярных спектров поглощения.
В соответствии с длиной волны поглощаемого света во взаимодействие включаются разные состояния молекулы поглощающего соединения, которые характеризуются различными спектрами поглощения:
Область спектра поглощаемого излучения
Вакуумная ультрафиолетовая
Ультрафиолетовая
Видимая
Ближняя инфракрасная
Дальняя инфракрасная
Микроволновая
Длина волны
10—200 нм
200—400 нм 400—750 нм 0,7—20 мкм
20—1000 мкм 1—1000 мм
Тип спектра поглощения
Электронный » »
Колебател ьный Вращательный
Спектр ЯМР или ЭПР
Электронные спектры обычно выражают зависимостью молярного коэффициента светопоглощения е от длины волны поглощаемого света (рис. 2.5). Длина волны, при которой наблюдается максимальное поглощение света, обозначается через Хмакс, молярный коэффициент светопоглощения — емакс- Область максимального поглощения лучей характеризуется также размытостью максимума поглощения (см. рис. 2.5) — интервалом длин волн (Xi/S макс—X!/, макс), отвечающим половинным значениям максимального молярного коэффициента светопоглощения или максимальной оптической плотности раствора. Положение максимума поглощения света в определенной спектральной области является важной оптической характеристикой вещества, а характер и вид спектра поглощения характеризует его качественную индивидуальность. Спектры поглощения веществ обычно снимают с помощью регистрирующих спектрофотометров (с автоматической записью спектра поглощения), измеряющих оптическую плотность или пропускание растворов в видимой, ультрафиолетовой и инфракрасной областях спектра (рис. 2.6).
Менее качественно спектры поглощения можно снять и при помощи нерегистрирующих спектрофотометров или фотометров (фотоколориметров), снабженных набором узкополосных светофильтров. В этом случае для получения спектра поглощения, т. е. кривой светопоглощения в координатах А = f (к) или е = = f (К), проводят серию измерений оптической плотности раствора при различных длинах волн в интересующей области спектра. Измерения проводят через 10—20 нм, а найдя границы максимума, промеряют эту область через 1—2 нм. По полученным данным строят кривую. У окрашенных веществ максимум поглощения света в большинстве случаев находится в видимой области спектра, однако он может быть и в ближней ультрафиолетовой области, как, например, у хромата калия (см. рис. 2.6, кривую 1), или в ближней инфракрасной области, как у раствора сульфата меди (кривая 2).
41
F
Рис. 2.5. Электронный спектр поглощения (?-1/2Макс — 7"1/2макс — размытость максимума поглощения).
Рис. 2.6. Спектры поглощения растворов:
/ — Хромата калия; 2 — сульфата меди; 3 роданида кобальта (в ацетоне).
Как уже отмечалось, интенсивность полосы поглощения в электронном спектре характеризуется значением молярного коэффициента светопоглощения при длине волны максимума поглощения данной полосы (емакс). Другим параметром, определяющим интенсивность полосы поглощения, является сила осциллятора [21, 35]:
/«4,6.10-Чакс^*/, <2-17)
Для светопоглощающих соединений, у которых емакс 10®, значение f близко к единице. Из уравнения (2.17) следует, что при полуширине полосы поглощения Avi/2 (ширине полосы на половине ее интенсивности), равной примерно 500 см-1, максимальное значение 8 составляет около 4-105 л-моль-1-см-1.
При одноэлектронном переходе полоса поглощения в электронном спектре характеризуется тремя основными параметрами: максимальным значением молярного коэффициента светопоглощения бма11с, волновым числом поглощаемого излучения vMaK0 (или длиной волны 7на1.с), соответствующим емакс, и эффективной шириной полосы поглощения Avi/a (см. рис. 2.7).
Зависимость е от v для каждого емакс индивидуального компонента обычно
/1 \ выражают уравнением кривой рас-
(7,5емакс-пределения Гаусса:
J\ [ |\ е = емаксехр[-(т —vMaKC)2 1п2/о»] (2 18)
। Т'макс V
_Рис. 2.7. Параметры, характеризующие й полосу поглощения в спектре.
42
Здесь о= 0,5AvIj(]| — полуширина полосы поглощения на половине ее высоты (0,5емакс).
Спектры поглощения таких многоэлектронных систем, как комплексные соединения, имеют сложный характер, и для их расшифровки целесообразно проводить разложение на гауссовы составляющие [35, с. 129]. Но даже в этом случае не всегда удается четко расшифровать полосы поглощения комплексных соединений, поскольку на всегда можно определить ту роль, которую играет центральный ион и лиганд в возникновении того или иного вида электронного спектра [35]. Поэтому для таких сложных молекул предсказать точное положение максимума, и особенно степень поглощения ими света, очень трудно.
При анализе многокомпонентных систем с увеличением значения Av./2 возрастает трудность его проведения из-за сильного перекрывания полос поглощения отдельных компонентов смеси.
Изменение состава и строения поглощающих частиц, а также природы растворителя вызывает соответствующие изменения спектральных свойств поглощающих систем. Эти изменения в электронных спектрах поглощения характеризуются либо увеличением поглощения (гиперхромный эффект), либо его уменьшением (гипсохромный эффект), смещением максимума поглощения в длинноволновую (батохромный сдвиг) или коротковолновую (гипсохромный сдвиг) области спекгра. Но во всех случаях избирательное поглощение света в определенной области спектра, как правило, связано с наличием определенных хромофорных групп. Иногда.в молекуле рядом с хромофором находятся другие активные группы атомов (ауксохромы), которые могут вызывать гиперхромный эффект в спектре поглощения и обусловливать батохромный сдвиг максимума поглощения.
2.3. ВЫБОР СПЕКТРАЛЬНОЙ ОБЛАСТИ ДЛЯ ФОТОМЕТРИЧЕСКИХ ИЗМЕРЕНИЙ
Чувствительность и погрешность фотометрического определения зависят от выбранного интервала длин волн поглощаемого света. Оптимальная спектральная область, в которой проводят фотометрические измерения, определяется спектрами поглощения фотометрируемого комплекса и применяемого реагента. При этом встречаются следующие основные варианты.
1. В рассматриваемой области спектра фотометрический реагент света не поглощает (спектры поглощения не перекрываются).
В этом случае измерение оптической плотности фотометрируемого раствора желательно производить в той области спектра, в которой поглощение света определяемым соединением является максимальным. Это дает возможность провести количественное
43
Рис. 2.8. Сравнение погрешности фотометрического определения при разных длинах волн поглощаемого света:
а — спектры поглощения растворов; б — зависимость Л от С при ^ма(;с 11
определение с наибольшей чувствительностью и меньшей погрешностью.
Рассмотрим влияние результатов измерения оптической плотности раствора при разных длинах волн на чувствительность и погрешность фотометрического определения. Допустим, что рассматриваемый комплекс имеет спектр поглощения, показанный на рис. 2.8, а. Выберем участки спектра, где анализируемое соединение поглощает лучи максимально (при 2. = 550 нм) и минимально (при 2. = 640 нм). Затем, приготовив окрашенные растворы с различными концентрациями так, чтобы Сх < С2 < С3, измерим их оптические плотности при 2ч«акс и ^ыин- Для обеих длин волн построим графическую зависимость оптической плотности от концентрации раствора определяемого вещества (см. рис. 2.8, б). Из рисунка видно, что при изменении концентрации вещества в интервале от Сг до С2 (АС) соответствующее ему изменение оптической плотности АЛ будет гораздо больше при 2.ыакс, чем при ХМШ1. Так как практическая погрешность измерения оптической плотности раствора приблизительно одинакова, то изменение концентрации АС (погрешность определения) будет гораздо больше при ХМ11Н, чем при Дшкс- Угловой коэффициент зависимости А = f (С), характеризующий чувствительность определения, будет значительно выше при 2vMaitc, чем при ^мин- К такому же выводу приводит анализ уравнения основного закона светопог-лощенчя А = fyCl. Продифференцировав это выражение по С, получим
dA Дд
= или = ъ
т. е. с изменением концентрации АС изменение оптической плотности АЛ фотометрируемого раствора тем больше, чем больше
44
Рис. 2.9. Спектр поглощения диэтилдитиокарбамината меди в хлороформе (С8 > >» Сд > Св > С2 > Cj).
молярный коэффициент светопоглощения фотометр и руемого соединения, а .е>. всегда является наибольшим при Хмакс.
В качестве примера можно привести оценку погрешности определения концентрации раствора диэтилдитиокарбамината меди, спектр поглощения которого представлен на рис. 2.9, при X = = 436 нм и X = 540 нм. Зависимость оптической плотности от концентрации раствора диэтилдитиокарбамината меди при этих длинах волн показана на рис. 2.10. Если измерение оптической плотности раствора производить при 436 нм, т. е. при Хмакс, то из рис. 2.10 видно, что погрешность измерения оптической плотности ДД == ±0,01 обусловливает погрешность (ошибку) при определении концентрации раствора меди, равную ±0,01 мкг/мл. При 540 нм та же погрешность в измерении оптической плотности (±0,01) обусловливает ошибку при определении концентрации раствора уже ±0,05 мкг/мл, т. е. ошибка определения концентрации раствора меди при X = 540 нм в 5 раз больше, чем при X = 436 нм.
Измерение оптической плотности раствора в области максимального поглощения лучей позволяет повысить также и чувствительность определения. Поскольку коэффициент чувствительности пропорционален молярному коэффициенту светопоглощения, то наибольшее значение последнего при 2.макс обусловливает и наиболее высокую чувствительность определения (при прочих равных условиях). Значения молярных коэффициентов светопоглощения Диэтилдитиокарбамината меди при X = 436 ими А = 540 нм равны соответственно 12800 и 1600, поэтому коэффициент чувствительности (угловой коэффициент зависимости А от С) определения
45
меди в виде диэтилдитиокарбамината при 436 нм будет в e4se/e640= = 8 раз выше, чем при 540 нм.
2. Спектры поглощения фотометрируемого соединения и применяемого реагента перекрываются (см. рис. 2 15).
Измерения оптической плотности раствора в области максимума поглощения фотометрируемого комплекса в этом случае обычно не позволяют получить хороших результатов, поскольку при этой же длине волны свет поглощает и применяемый фотометрический реагент, концентрация которого значительно превышает концентрацию фотометрируемого комплекса (так как для количественного протекания реакции добавляют избыток реагента). При таких условиях погрешность определения обычно возрастает, а чувствительность — резко падает. Целесообразность использования в этих условиях фотометрических реакций оценивают по их фотометрическим характеристикам: по контрастности реакций и изменению значений молярного коэффициента светопоглощения при комплексообразовании. Контрастность реакций характеризуют интервалом длин волн ДХ между максимумами поглощения фотометрируемого соединения и применяемого реагента. Чем шире ДХ, тем меньше перекрывание полос поглощения в спектрах комплекса и реагента, тем контрастнее применяемая фотометрическая реакция. Эффективность применяемой реакции тем выше, чем больше абсолютное (Де = eMRn—eR) и относительное (еЛ1Кл/ек) изменение молярного коэффициента светопоглощения при комплексообразовании. Таким образом, чем больше значения ДХ и Де, гем ценнее и чувствительнее фотометрическая реакция.
Рнс. 2.10. Зависимость оптической плотности раствора диэтилдитиокарбамината меди от его концентрации при X = 436 нм и Л = 540 нм.
46
Фотометрическое определение при перекрывании спектров комплекса и реагента проводят в области оптимального поглощения лучей, т. е. в том интервале длин волн (или при такой длине волны), где наблюдается максимальная разность Ле комплекса и реагента.
3. В растворе образуются две различные по составу окрашенные формы определяемого элемента с реагентом.
Во избежание больших погрешностей фотометрическое определение анализируемого элемента в этом случае необходимо проводить при длине волны изобестической точки фотометрируемых равновесных форм, где светопоглощение этих форм одинаковое (см. рис. 2.16).
Таким образом, выбор участка спектра при фотометрировании определяется следующими условиями:
1) максимальным (или оптимальным) поглощением лучей проходящего через раствор излучения, обеспечивающим наибольшую чувствительность и воспроизводимость фотометрического определения;
2) высокой чувствительностью приемника излучения (глаза или фотоэлемента) к выбранному интервалу длин волн;
3) соблюдением основного закона светопоглощения при поглощении лучей выбранного участка спектра;
4) воспроизводимостью результатов измерений.
2.4. СВЕТОФИЛЬТРЫ
Для увеличения чувствительности и точности фотометрического определения целесообразно использовать поглощение не смешанного (белого) света, а лишь тех лучей, которые максимально поглощаются фотометрируемым окрашенным раствором. Для того чтобы из всей видимой области спектра выделить лучи опре-
деленных длин волн, на пути световых потоков перед поглощаю-
щими растворами помещают избирательные поглотители света,
называемые светофильтрами Светофильтры пропускают
лучи лишь в определенном интервале длин волн с полушириной
пропускания X. 2 макс — Х:/г макс и практически полностью поглощают лучи других длин волн (рис. 2.11).
Чем уже область максимального пропускания лучей (Х./г ыакг—^;/г макс — размытость максимума пропускания) применяемого светофильтра, тем выше его избирательность к лучам этого ин-
Рис. 2.11. Спектральная характеристика светофильтра (Х1/2Мажс - Х;Лмакс - размытость максимума пропускания).
47
Рис. 2.12. Интерференционный светофильтр-
I — стекло-подложка; 2 — слой серебра; 3 — слой *lgF2. 4 — слой серебра; 5 — защитное стекло.
тервала длин волн. Наиболее эффективные стеклянные узкополосные светофильтры характеризуются размытостью максимума пропускания 20—30 нм.
В качестве светофильтров применяют цветные стекла и пленки, окрашенные жидкости и интерференционные фильтры. Например, из набора № 106 цветного стекла, выпускаемого отечественной промышленностью, можно составить любой интересующий исследователя набор узкополосных светофильтров. Спектры пропускания (или поглощения) стеклянных светофильтров приведены в руководствах к фотоколориметрам и в каталогах цветного стекла *.
В настоящее время выпускают наборы и отдельные интерференционные светофильтры (рис. 2.12) диаметром 40 мм для длин волн 350—800 нм. Светофильтры для длин волн 400—800 нм имеют максимальное пропускание (Т) 25—50% с полушириной пропускания Х1/2 макс —макс порядка 2% от длины волны (8— 16 нм). Наборы интерференционных светофильтров для области спектра 400—800 нм выпускают с интервалом 10, 20 и 40 нм.
Светофильтры для фотометрирования выбирают, исходя из спектра поглощения определяемого вещества так, чтобы спектральная область максимального поглощения лучей окрашенным раствором и область максимального пропускания лучей светофильтром была одной и той же, т. е. максимум поглощения раствора должен соответствовать максимуму пропускания (минимуму поглощения) светофильтра. На рис. 2.13 показаны спектральные характеристики окрашенного раствора (кривая 1) и правильно подобранного к нему светофильтра (кривая 2).
Фотометрическое определение получается тем точнее, чем более узкий участок спектра удается выделить светофильтром. Если светофильтр имеет узкую область максимального пропускания лучей, но она не соответствует области максимального поглощения света фотометрируемым раствором (рис. 2.14), то при работе с таким светофильтром точность определения получается даже меньше, чем при его отсутствии. Иногда, однако, приходится отступать от общего правила подбора светофильтра. Например, если выбранный для анализа реагент R (длина волны максимального поглощения — ZR) имеет окраску и поглощает свет
* Каталоги цветного стекла издаются Государственным оптическим институтом в г. Ленинграде.
48
Рис. 2.14 Спектр поглощения фотометрируемого раствора (?) н не соответствующих ему узкополосных светофильтров (2 и 3).
Рис. 2.13. Спектр поглощения фотометрируемого раствора (/) и соответствующего ему светофильтра (2).
в той же области спектра, что и анализируемое соединение (длина волны максимального поглощения комплекса — 2.к), то измерение оптической плотности раствора производят не в области максимального поглощения лучей окрашенным соединением MR, а в области оптимального поглощения (при Хопт). Эта область соответствует той длине волны (или тому участку спектра), гдедости-гается наибольшая разница (ЛЛ) в оптических плотностях окрашенного соединения и самого реагента (рис. 2.15). В этом случае используют светофильтр с максимальным пропусканием лучей При ^-опт-
Другим примером, требующим специального подхода, является фотометрический анализ раствора, содержащего близкие по со
Рис. 2.15. Перекрывание полос поглощения в спектрах исследуемого соединения (/) и реагента (2).
Рис. 2.16. Спектры поглощения равновесных форм определяемого элемента» I *=» спектр основного соединения; 2 »— спектр равновесной формы.
49
ставу равновесные формы окрашенных соединений, спектры поглощения которых аналогичны, но различаются положением максимумов поглощения. Например, измерения оптической плотности стандартных растворов основного компонента (рис. 2.16, кривая 1) производили в соответствии с общим правилом при При измерении оптической плотности исследуемого раствора при в присутствии некоторого количества его равновесной формы, которая имеет максимум поглощения не при а при Х2 (кривая 2), получаются заниженные значения концентрации определяемого элемента. В этом случае исследуемый и стандартные растворы целесообразно фотометрировать при некоторой наиболее эффективной длине волны Х3, которая соответствует точке пересечения кривых 1 и 2 [57], т. е. при фотометрировании целесообразно применять светофильтр с максимальным пропусканием лучей не в области 2^, а в более эффективной области 2,3. При этой эффективной длине волны, называемой длиной волны изобести-ческой точки, оба окрашенных соединения определяемого элемента имеют одинаковое светопоглощение, поэтому присутствие в исследуемом растворе равновесной формы окрашенного соединения не будет оказывать влияния на результат анализа.
В тех случаях, когда максимум поглощения раствора выходит за пределы области максимальной чувствительности фотоэлемента или отсутствуют спектральные характеристики как для окрашенного раствора, так и для светофильтра, нужный светофильтр подбирают экспериментально. Тот светофильтр, при котором абсолютное значение или разность оптической плотности ДД получается максимальной, является наиболее подходящим для фотометрирования данного окрашенного раствора.
Иногда при подборе светофильтра для фотометрирсвания используют менее точный, но более быстрый прием — выбирают светофильтр по цвету исследуемого раствора.
Набор светофильтров для фотометрирования окрашенных растворов должен удовлетворять следующим требованиям:
а) максимумы пропускания светофильтров должны перемещаться от фильтра к фильтру по спектру, захватывая более или менее одинаковые участки последнего;,
б) светофильтр должен полностью поглощать ультрафиолетовые и инфракрасные лучи;
в) светофильтр должен пропускать лучи узкого интервала длин волн, имея при этом высокое значение коэффициента пропускания.
Глава 3
МЕТРОЛОГИЧЕСКИЕ
И АНАЛИТИЧЕСКИЕ ХАРАКТЕРИСТИКИ
ФОТОМЕТРИЧЕСКОГО АНАЛИЗА.
ВЫБОР ОПТИМАЛЬНЫХ УСЛОВИЙ ФОТОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ
3.1. МЕТРОЛОГИЧЕСКИЕ ХАРАКТЕРИСТИКИ
ФОТОМЕТРИЧЕСКОГО АНАЛИЗА
Фотометрические методы анализа применяют для определения элементов в широких пределах относительных содержаний от 100 до 10“в%. При выборе и описании методов и методик определения содержаний указанного интервала наиболее общий интерес представляют метрологические (правильность, сходимость, воспроизводимость, чувствительность, предел обнаружения, нижняя граница определяемых содержаний) и аналитические (селективность, экспрессность) характеристики, доступность аппаратуры и возможность автоматизации метода анализа.
Область применимости фотометрических методов для определения следов * элементов показана на рис. 3.1, представленном Сенделом [25] в соответствии с номенклатурными рекомендациями ИЮПАК [4] для указанных на рисунке пределов определяемых содержаний и значений аналитических навесок **. В основу классификации положены два признака: значение аналитической навески и содержание определяемого компонента в % или млн. д. (ppm).
По содержанию компонента в аналитической навеске (ось ординат рис. 3.1) различают основные (100—1%), неосновные (1—0,01%) и следовые (<0,01%, <100 ррш) компоненты.
Классификация по значению аналитической навески (ось абсцисс рис. 3.1) может быть рассмотрена по шкалам А и В. По шкале А образцы классифицируют на граммовые (1 — 10 г), дециграммозые (0,1 — 1 г) и т. д. По шкале В в зависимости от аналитической навески виды анализа классифицируют на: макро-
* Иногда содержания в пределах Ы0-4—Ы0~2% (1 — 100 мкг/г) называют следовыми, а ниже 1 • 10~4% (1 мкг/г) — ультраследовыми. В аналитической химии следов элементов наряду с процентным выражением довольно широко распространена шкала содержания частей определяемого компонента на миллион частей основы (млн. д.; ppm) или на миллиард частей основы (ppb), 1 ppm = 1-10-’%; 1 ppb = 1-10-7%.
** Аналитическая навеска — определенная часть пробы, используемая при выполнении единичного определения [1].
51
Деци-грамм
Санти'Мими-Деци-\Санти Микро] грам^грамМ^иллигранн грамм
A
В
Значения аналитических набесок и классификация
Рис. 3.1. Классификация методов анализа по массе пробы и пределам определяемых содержаний [4, 25].
анализ (0,1 г), полумикроанализ (0,1—0,01 г), микроанализ (0,01 — 0,001 г) и т. д.
Заштрихованная на рис. 3.1 часть представляет собой область применимости фотометрических методов анализа следов элементов в координатах: определяемое содержание компонента — значение аналитической навески Пунктирные линии характеризуют абсолютные содержания (г) определяемого компонента как функцию его относительного содержания и значения аналитической навески. Рассмотрение заштрихованной области показывает, что аналитическая навеска при фотометрическом определении следов элементов может изменяться от макро- до микрограммовых значений. Обычно при анализе следов элементов значение аналитической навески не превышает 1—10 г.
52
3.1.1. ЧУВСТВИТЕЛЬНОСТЬ
ФОТОМЕТРИЧЕСКИХ ОПРЕДЕЛЕНИЙ
Понятие «чувствительность определения» часто распространяют [1, 8—10, 21, 25, 58—65] на различные характеристики, применяемые для количественной оценки возможностей определения минимального содержания компонента фотометрическим методом. Как количественная характеристика чувствительность определения характеризуется, в соответствии с номенклатурными правилами ИЮПАК 11,21], коэффициентом чувствительности.
3.1.1.1. Коэффициент чувствительности (sensitivity — S)
Коэффициент чувствительности [1, 21 ] — значение первой производной градуировочной функции при данном определяемом содержании:
(3.1)
В фотометрическом анализе под коэффициентом чувствительности понимают значение первой производной градуировочной функции Л по С при данном определяемом содержании:
(3-2)
Если при выбранной длине волны поглощает свет лишь анализируемое соединение и градуировочная функция (градуировочный график) выражается прямой линией, то, согласно формуле (3.2) и уравнению основного закона светопоглощения, первая производная градуировочной функции равна exZ*. Таким образом, при фотометрических определениях при указанных выше условиях произведение ezZ (тангенс угла наклона, угловой коэффициент градуировочного графика) является коэффициентом чувствительности.
Если же при рабочей длине волны нельзя пренебречь свето-поглощением избытка реагента, участвующего в образовании окрашенного комплекса MuRn, то коэффициент чувствительности определяется не молярным коэффициентом светопоглощения анализируемого соединения ех, а зависит от следующей линейной
* Определение истинных значений молярных коэффициентов светопоглощения описано в разд. 8.2.
53
комбинации: (1/р) (ек — neR), где eR — молярный коэффициент светопоглощения реагента при той же длине волны *.
В литературе используется также понятие «коэффициент аналитической чувствительности» (аналитическая чувствительность) — Sa [64J, который определяется как отношение углового коэффициента градуировочной кривой к стандартному отклонению результатов измерений оптической рлотности раствора:
Sa = ~dC'lSA (33)
Если две методики имеют одинаковую воспроизводимость (сходимость) результатов измерений, то более чувствительной является та, которой соответствует больший угол наклона градуировочной кривой. Если же две методики характеризуются градуировочными кривыми с одинаковым углом наклона, то более точная методика позволяет определять меньшие содержания, т. е. имеет большую аналитическую чувствительность.
Необходимо различать чувствительность метода (методики) анализа и чувствительность фотометрической реакции. Чувствительность фотометрической реакции определяется фотометрическими характеристиками светопоглощающего соединения, а также условиями проведения реакции и фотометрирования.
Чувствительность метода (методики) может отличаться от чувствительности фотометрической реакции, так как она зависит еще и от условий приготовления анализируемой пробы, влйяния мешающих компонентов и нередко определяется вспомогательными операциями по предварительному концентрированию определяемого элемента. Достижение максимальной чувствительности методики фотометрического определения требует соблюдения оптимальных условий при построении градуировочного графика.
3.1.1.2. Условные характеристики чувствительности фотометрического определения
Традиционно при прогнозировании возможностей фотометрического определения следов элементов для быстрой сравнительной оценки чувствительности фотометрических реагентов пользуются
* Оптические плотности анализируемого и холостого растворов, измеренные относительно растворителя при 100%-ном выходе окрашенного комплекса, равны соответственно:
А = I (1/р) е>См + Zep [Cr — (n/u) См]; А2 = ZerCr
Оптическая плотность анализируемого раствора, измеренная относительно холостого опыта, в этом случае равна
А = Al — А2 = Z Ц1/Р) (еь — ntR)[ См
54
следующими теоретически рассчитываемыми характеристиками условной чувствительности *:
Смин — минимальная молярная концентрация элемента в растворе, поддающаяся фотометрическому определению;
т — определяемый минимум;
tn’s — коэффициент Сендела;
а — удельное поглощение — условный коэффициент, широко используемый Марченко [26, 66].
Оценочные расчеты Смин можно провести на основе уравнения (2.3) [26, 60, 62]:
Сдан = “4 мин* (Емакс^макс)
Если при спектрофотометрических измерениях принять Дмин = = 0,001, емакс = 1.105 и I = 1 см, то Смин составит 1.10“8 моль/л. При фотоколориметрических измерениях Дмин « 0,002, значение эффективного молярного коэффициента светопоглощения по меньшей мере в 2 раза меньше емакс. Следовательно, при аналогичных условиях фотометрирования Смин ориентировочно будет в 4 раза больше.
Увеличение толщины слоя до предельного значения (I — 10 см) может позволить снизить Смин на порядок. Однако на практике работают с кюветами толщиной 1—2 см, молярные коэффициенты светопоглощения окрашенных соединений в большинстве случаев не превышают 5-104; кроме того, в ходе выполнения анализа добавляют реактивы, производят разбавление растворов, в результате чего минимальные определяемые концентрации следовых количеств элементов увеличиваются до значений примерно 5.10_? моль/л при спектрофотометрических определениях и до (1—2,5). 1СГ® моль/л при фотоколориметрических определениях. Для элемента с относительной атомной массой 100 минимальные концентрации составят соответственно 0,05 и 0,1—0,3 мкг/мл. Если принять, что оптические плотности исследуемых растворов указанных концентраций измеряют в кювете с I = 2 см, объем которой составляет примерно 10 мл, то общее содержание элемента в этом объеме составит соответственно 0,5 и 1—Змкг. Отсюда следует, что при навеске анализируемой пробы в 1 г обычный спектрофотометрический анализ позволяет определять минимальную массовую долю следов элементов на уровне 5. 1СГ5%, а фото-колориметрический— на уровне (1—3)-1(Г4%.
Таким образом, без применения специальных приемов непосредственно спектрофотометрически возможно определение массовой доли элементов не ниже 5- 10-5%, а фотоколориметрически— не ниже 1. 10~4 % * **. Меньшие содержания находятся
’фактически рассматриваемые в разд. 3.1.1.2 характеристики — это разные варианты формы выражения нижнего предела определяемых содержаний Сн.
** Приведенные значения являются ориентировочными; они могут изменяться в зависимости от конкретных условий. Так, согласно данным [22’, минимальное определяемое значение массовой доли составляет (1—2)-10'6%.
55
ниже чувствительности большинства абсолютных фотометрических методов анализа. На практике эти пределы могут быть снижены до 1. 1СГ®—1- 10“’% за счет увеличения навески пробы, концентрирования и других приемов (см. разд. 3.1.1.3 и 5.1).
Часто, исходя из уравнения (2.3), условную чувствительность фотометрической реакции выражают минимальной концентрацией Смин (мкг/мл) и определяемым минимумом т (мкг) L30, 60, 671:
Смин 103рВЛмин/(е^) (3.4)
т = CMmiV = 103иВЕЛмин (e;.Z) (3.5)
Здесь Лмин — минимальное значение оптической плотности раствора, регистрируемое прибором; Вир — относительная атомная масса и число атомов определяемого элемента, содержащихся в составе молекулы свето.тыло-щающего соединения; V — значение конечного объема фотометрируемого раствора, мл.
Определяемый минимум- характеризует наименьшее массовое содержание (мкг) вещества, которое еще может быть количественно определено по данной методике. Если отношение VH — q принять за «эффективное» сечение кюветы (см2), то уравнение (3.5) можно записать в такой форме:
щ = Лдрщ<7р.В- 103/ех (3.6)
При теоретическом прогнозировании определяемого минимума необходимо учитывать также воспроизводимость результатов, влияние фона и погрешности измерения оптических плотностей, поэтому в уравнения (3.5) и (3.6) необходимо вводить соответствующие поправки, позволяющие объективно оценить значение Лмин- Для учета влияния фона и погрешностей измерения А. Б. Бланк [67] на основе метрологического обоснования предложил принять Амин = 5$л (где — стандартное отклонение, соответствующее оптической плотности Лчин)*. Тогда полученные при этом условии выражения определяемого минимума примут такой вид:
m== 5-10*pBJ|/l-'/(rzZ) (3.7)
т = 5-lO’uBs^g'E^ (3.8)
Во всех вышеприведенных уравнениях при вычислении Смин предполагается, что светопоглощающее соединение, содержащееся в растворе, подчиняется основному закону светопоглощения и что даже при очень малых концентрациях реакция образования окрашенного соединения протекает практически количественно. Поэтому Счин отождествляется с равновесной концентрацией продукта реакции. Однако такое отождествление справедливо только для очень прочных комплексов и в отсутствие конкурирующих равновесий. Если же окрашенный комплекс имеет не очень высо
* В этом случае погрешность определения значения А при числе определений п = 3 и доверительной вероятности Р = 95% не превышает ±50%.
56
кую константу устойчивости, то чувствительность реакции будет определяться не только его минимальной наблюдаемой концентрацией, но и той концентрацией (количеством) определяемого иона, которая останется в растворе не связанной в окрашенное соединение [60].
И. М. Коренман для малопрочных комплексов состава 1 : 1 ввел поправку на диссоциацию комплекса [60], но влияние конкурирующих реакций протонизации фотометрического реагента и комплексообразования определяемого катиона конкурирующими лигандами эта поправка не учитывает. А между тем относительная чувствительность фотометрического определения, достигаемая в присутствии мешающих ионов, может существенно уменьшаться по сравнению с раствором чистой соли. В таких случаях в уравнения (3.4)—(3.8) целесообразно вводить поправку, учитывающую относительную устойчивость фотометрируемого соединения при заданных условиях [68]. Для учета количественного влияния конкурирующих реакций на значения нижних пределов определяемого содержания можно использовать условные константы устойчивости фотометрируемых соединений [42—471.
В реальных условиях, когда вследствие конкурирующего комплексообразования реакция образования окрашенного комплекса с определяемым элементом протекает не до конца, ее чувствительность определяется такой общей (начальной) концентрацией анализируемого иона, при которой возможно образование окрашенного комплекса минимальной наблюдаемой концентрации. В этом случае чувствительность реакции будет определяться суммарной равновесной концентрацией окрашенного комплекса [MRn] и всех ионных форм анализируемого иона [См], не связанного в комплекс:
Смин = [MRn] + [См]
Выражая [См] через условную константу устойчивости, получим
Смии = [MR«] + [MR„]/₽MRn [CRf = [MR„] (1 + 1/₽mr„ [Cr]") откуда
[MRn] = CMIIII₽MRn [CR]n/( 1 + ₽MRn [cr]")
Подставляя равновесную концентрацию комплекса MRn в выражение основного закона светопоглощения и принимая Лмип = = 5s.4, получим уравнения для вычисления минимальной определяемой концентрации и определяемого минимума с учетом влияния сопряженных конкурирующих равновесий:
5s^B-103(l + ₽MRn[CR]n)
5szpBV.103(l + PMRn [Cr]")
(3.10)
®x'₽MRn [CRf
67
Из рассмотрения уравнений (3.9) и (3.10) видно, что влиянием сопряженных равновесий можно пренебречь, если [CR ]" gsl.108, т. е. при достижении избыточной концентрации реагента, равной [CR] (102/рмрЛ1)1//г. При этих условиях уравнения (3.9) и (3.10) становятся тождественными формулам (3.4) и (3.5).
Таким образом, определяемый минимум (мкг) фотометрического определения с учетом влияния различных факторов можно выразить уравнением:
55лИВГ.103(1+Рмр [CRf)
т =----------------------------- (3.11)
1 (®к — ~ "Er) ₽MRn [СRГ
Здесь ек, вад, — молярные коэффициенты светопоглощения комплекса, катиона металла и реагента соответственно.
Для сравнительной оценки условной чувствительности фотометрических реакций в аналитической практике широкое распространение получили коэффициент Сендела ml (мкг/см2) и обратный ему коэффициент а (см2/мкг), предложенный Марченко [26 , 66].
Значительно реже используют условный коэффициент чувствительности Ск (мкг/мл), предложенный Кохом и Кох-Делиц 169], который является частным случаем минимальной определяемой концентрации при Дмин = 0,05 и I = 1 см
С£ = 50|иДел (3.12)
а также показатель поглощения
а = ₽>./( 100В) (3.13)
используемый Хольцбехером и др. [63].
Удельное поглощение а — условный коэффициент, адекватный оптической плотности раствора с концентрацией определяемого элемента 1 мкг/мл при толщине поглощающего слоя 1 см:
я = сх/(103рВ) (3.14)
Типичные значения а для цветных реакций, используемых в неорганическом анализе, составляют 0,1—1,0 см2/мкг.
Л. Б. Гинзбург и Ю. Ю. Лурье [70], а также Сендел 171], отнеся определяемый минимум к эффективному сечению кюветы в 1 см2 и приняв Лмпн = 0,001, из уравнения (3.6) получили простое выражение для условного определяемого минимума (мкг/см2):
= рВ/еА (3.15)
Условная чувствительность по Сенделу определяется числом микрограммов определяемого элемента, превращенного в светопоглощающее соединение, которое в слое раствора с поперечным сечением I см2 показывает оптическую плотность, равную 0,001. 58
Для большинства чувствительных цветных реакций значение т* колеблется от 0,01 до 0,001 мкг/см2. Высокочувствительные цветные реакции с т*. = 0,002-?-0,003 известны, по крайней мере, для 20 элементов (Al, Sb, As, Be, Bi, Cd, Cr, Co, Cu, Ga, Ge, Au, Fe, Pb, Mg, Hg, Pd, Pt, Ru, Tl, Zn). Сравнительно мало встречается реакций, для которых tris < 0,001. В табл. 3.1 в качестве примера приведена условная чувствительность некоторых методик определения меди и никеля.
Удобной для практических расчетов и установления связи между коэффициентом Сендела и удельным поглощением а является форма записи основного закона светопоглощения, в которой Смин выражена в мкг/мл:
Смин ~ ^мин/(я0 (3.16)
Из уравнений (3.15) и (3.16) при Амип = 0,001 следует, что значения коэффициентов ms и а связаны соотношением: а — — и при д = 1
а=Ю-зех/В (3.17)
Сопоставление уравнений (3.15) и (3.16) показывает также, что при Z = 1 см наименьшее значение Смвн, выраженное в млн. д., которое может быть определено прямыми фотометрическими методами, составляет примерно 0,002 ppm.
В обширном обзоре Марченко [26 ] представлены сводные таблицы чувствительных спектрофотометрических методов, рекомендуемых для определения следов элементов с указанием конкретных значений коэффициентов е и а для наиболее интенсивно окрашенных соединений (примеры см. в табл. 3.2). Там же приведены сводные библиографические данные по спектрофотометрическим методам определения следов элементов в воде, воздухе, биологических материалах, растениях, кормах, геологических образцах, сталях, металлах, полупроводниковых материалах и т. д.
Таблица 3.1. Условная чувствительность некоторых фотометрических методик определения меди и никеля [71 ]
Металл Реагент Р астворнтель Длина волны, нм т» . мкг/см2
Си Дитизон Тетрахлорид углерода 533 0,0022
Диэтилдитиокарбаминат натрия Хлороформ 436 0,0040
Салицилальдоксим Амилацетат 344 0,0008
N1 Диметилглиоксим (с окислителем) Вода 445 0,0042
Диэтилдитиокарбаминат натрия Изоамиловый спирт Хлороформ 390 325 0,010 0,0019
59
Таблица 3.2. Чувствительные спектрофотометрические методы, рекомендуемые для определения следовых количеств элементов [26]
Элемент Реагент (среда; метод) Коэффициенты светопоглощения
молярный 8. 10“* удельный 8
а в. 1000
А1 Хромазурол S (pH = 6) 4,9 1,9
Хромазурол S + ЦТА * (pH = 5,8) 13,1 4,9
Ag Дитизон (CC14) 3,1 0,28
Метиленовый голубой (CN-; 1,2-дихлорэтан) 9,9 0,92
В: Дитизон (СС14) 7,9 0,38
Родамин 6G (Вг~; флотация) 15,0 0.70
Cd Дитизои (СС14) 8,8 0,78
Кристаллический фиолетовый (I-: флотация) 13,0 1,1
Со 2-интрозо-1-нафтол (хлороформ) 3,7 0,63
Малахитовый зеленый (SCN',CC14— циклогексан) 8,6 1 5
Си Дитизон (СС14) 4,5 0,71
1,5-Дифенилкарбазид (pH = 9) 15,0 2,4
Fe Батофенантролин (хлороформ + спирт) 2,2 0.40
Родамин В (FeClj; бензол) 9,0 1,60
Ga Родамин В (GaClj; СС14— хлорбензол) 10,0 1,4
Ge Фенилфлуорон (pH = 5) 5,3 0,73
Родамин 6G (ализарин, комплексон; флотация) 29,0 4,0
In Бриллиантовый зеленый (InBrj; бутанол — хлороформ) 9,3 0,81
Бутилродамин В (1пВг7) 11,2 0,97
Mg Титановый желтый (щелочной раствор) 3,6 1,5
Mr ПАН ** (pH = 9,2, хлороформ) 5,8 1,1
Ni Кристаллический фиолетовый (хлороформ) 8,2 1,4
1 Фосфорномолибденовая синь (бутанол) 2,5 0,81
Кристаллический фиолетовый (Мо—Р-кислота; флотация) 27,0 8,8
Pb Дитизон (СС14’ 6,9 0,33
s Метиленовый голубой (СЮу, хлороформ) 3,5 1,1
Sb Бриллиантовый зеленый (SbClj; диизопропиловый эфир) 9,7 0,80
Реакция «усиления» 30,0 2,5
Se Дитизон (СС14) 7,0 0,88
Sn Фенилфлуорон (pH = 1->1,2) 7,7 0,65
Бриллиантовый зеленый (динитропнрокатехин; СС14 — бензол) 17,5 1,5
Si Кремнемолибденовая синь (изоамиловый спирт) 1,7 0,6
Родамин В (Мо—Si-кислота; флотация) 50,0 17,8
Те Висмутол II (хлороформ) 3,6 0,31
Родамин 6G (TeBrg"; флотация) Арсеназо III (6 М НС1) 17,0 1,3
U 12,7 0,50
Хромазурол S (pH = 4,8) 9,9 0,40
W Пирокатехиновый фиолетовый + ЦТА (pH = 1) 8,0 0,43
Кристаллический фиолетовый (SCN-; флотация) 21,0 1,1
Zn Дитизон (СС14) 9,3 1,4
ПАН * (хлороформ) 5,2 0,79
Zr Арсеиазо III (9 М НС1) 12,0 1,3
* Этилродамин В (флотация) ЦТА — цетилтриметиламмоний. ПАН — пирндилазонафтол. 32,0 3,5
60
3.1.2. ИНТЕРВАЛ ОПРЕДЕЛЯЕМЫХ СОДЕРЖАНИЙ
Интервал определяемых содержаний — это предусмотренная данной методикой область значений определяемых содержаний [1]. Интервал определяемых содержаний, в котором соблюдается основной закон светопоглощения, и значения погрешностей определения содержаний элемента на нижней и верхней границах этого интервала являются необходимой составной частью методики фотометрического анализа.
При определении следов элементов метрологическими характеристиками служат предел обнаружения, предел определения и нижняя граница определяемых содержаний, причем последняя является обязательной характеристикой методики количественного определения следов элементов.
3.1.2.1. Предел обнаружения* (limit the detection) и предел определения (limit of determination)
Предел обнаружения (СМЙИ. Р или Смин>ь)**—наименьшее содержание, при котором по данной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью Р или при коэффициенте достоверности k, численное значение которого выбирается в соответствии с уровнем доверительной вероятности [1].
Существуют и другие варианты определения предела обнаружения.
Предел обнаружения представляет собой число, выраженное в единицах концентрации (или количества), характеризующее наименьший концентрационный уровень (или наименьшее количество) элемента, который аналитик может определить как статистически отличный от уровня холостой пробы {73].
Предел обнаружения представляет собой концентрацию Ср (или количество qid, рассчитанную из наименьшего сигнала Хр, который может быть определен с достаточной надежностью для данной аналитической методики [73].
Предел обнаружения элемента — такое наименьшее абсолютное (т, г) или относительное (С, %) его содержание в пробе, при котором аналитический сигнал с заданной статистической достоверностью может быть отличен ст сигнала холостой пробы [76].
Анализ литературных данных, посвященных проблеме оценки пределов обнаружения элементов различными методами, показывает, что расчетные значения этого параметра для одного элемента даже при работе одним методом могут колебаться в пределах порядка и более. Обусловлено это неоднозначностью истолкования статистического термина «предел обнаружения» и отсутствием единого мнения о значении доверительной вероятности, которое должно приниматься при расчетах СЫИН,Р, о методике учета
* Эта метрологическая характеристика является предметом обширных дискуссий [1, 5, 6. 19, 21, 24—26, 64, 72—78].
** Некоторые авторы называют СМИН1 р — относительным пределом обнаружения, а ?мин, Р — абсолютным преде.1 с и обнаружения.
61
сигнала холостого опыта (фона) и методике расчета стандартного отклонения сигнала холостого опыта, которые входят в расчетные уравнения, принятые ИЮПАК 119, 72—74]:
^мин = У ХОЛ 4" Чхол
Уннв УхОЛ = ^хол 18 )
Смин. И»™ СМин,р) = /(^ол) (3-19)
Здгсь уМин — минимальный аналитический сигнал от анализируемой пробы: Ухол — среднее значение сигнала холостого опыта, рассчитываемое из числа пределений nxonJ>20; «й1ол — счапдартьое отклонение сигнала холостого опыта:
"ХОЛ
« Т/г
{.УI ХОЛ ^хол)2/(йхол 1)
.<=1
J'mhij — Ухоп — минимальный разностный аналитический сигнал.
В литературе применяют различные варианты обозначения величин, входящих в уравнения (3.18) и (3 19). ИЮПАК [6, 19] рекомендует использовать обозначения, предложенные Кайзером,
л = х6 + &6 (3.18") и С = f(x) (3.19')
которые в смысловом значении полностью идентичны символам уравнений (3.18), (3.19). В настоящей книге использована совокупность обозначений — с одной стороны, обычно применяемых в отечестве-"но й литературе (например, [58, 75, 76]), а с другой — удобных для наглядного применения математического аппарата главы 9.
В фотометрических методах анализа уравнение (3.18) записывается так:
4.ИВ = Асол + *Хол (3.18")
В общем случае в фотометрических методах анализа стремятся выбирать для работы интервалы содержаний, в которых наблюдается линейная зависимость между значением аналитического сигнала и концентрацией:
у = а 4 Ьх (3.20)
или после замены х и b соответственно на С и S:
V = о + CS (3.21); А = с-|-С5 (3.2Г); C=(A — a)/S (3.21") Простейший вариант расчета СЫ1)И k==3 из уравнений 13 19') и (3.21") в предположении, что погрешности определения параметров a, S незначимы и k = 3, можно проиллюстрировать на примере фотометрического определения Fe (III) с сульфосалициловой кислотой (для и = 50) [64]:
Концентрация Fe (III), — g
мкг/мл А л
0 (холостая проба) .... 0,0125 0,0014
0,07 0 0233 0,0017
1 0,2032 0,0020
Экспериментально установлено, что Fe(IIl) количественно связано в светопоглощающее соединение и уравнение градуировочной кривой^ С = 5,20 (А — — 0,0125) мкг/мл. Расчет предела обнаружения: Амин = Ахол + 3s^X0J1 = = 0,0125 + 3-0,0014 = 0,0167; См„н, k = 5,20 (0,0167 — 0,0125) = 0,02 мкг'мл.
62
Рис. 3.2. Градуировочная кривая зависимости аналитического сигнала у от концентрации С, иллюстрирующая связь А5ухол и предела обнаружения (и Смин, р . ].
При соблюдении линейной зависимости у = f (С) в области очень низких концентраций можно записать:
J/мин ~ Ухоп + Смин, hS (3.22)
откуда
Смин. h — ({/мин — Кхол)/^ (3.23)
Подстановкой уравнения (3.18) в (3.23) получаем выражение:
смин, л = 4xon/s <3-24)
Смысл Смин, k ясен из рис. 3.2.
Следует отметить, что найденное значение СМ1.Н( k будет являться действительно пределом обнаружения, если коэффициент чувствительности S определен правильно и коэффициент а в уравнении (3.21) фактически равен 0 *.
Использование значения k = 3 ** обеспечивает уровень доверительной вероятности 99,86%, при котором уМин (Уход+ + 3Sj,x0Ji) при условии, что случайные погрешности определения сигнала ухол подчиняются закону нормального распределения. Однако распределение значений ухол вблизи предела обнаружения может не следовать закону нормального распределения. В этом случае, согласно неравенству Чебышева [581, вероятность того, что г/1ЛИН (Уход + 3s/;xoJI), окажется равной 89%. Следовательно, для расчетов предела обнаружения нельзя использовать k <Z 3.
Тем не менее численное значение k, которое следует брать для расчета #ыин, явилось предметом дискуссий. В ряде работ предложено значение k — 2, которое соответствует доверительной вероятности Р — 97,7% при законе нормального распределения и Р = 75% при законе распределения случайных погрешностей измерения г/хол, отличном от нормального.
Это привело к противоречивости публикуемых данных по пределам обнаружения элементов. Во избежание недоразумений
* Более распространенный общий случай см. разд. 9.3 и рис. 9.1.
** Разностный аналитический сигнал Д/'Мин = </мин— Ухол = 351/хол ПРИ" нимается за минимально обнаруживаемый.
63
ИЮПАК рекомендует при публикации значений г/мин обязательно указывать значение k, например: (k = 3). Желательно также
указывать значение k и при публикации Смин (k = 3), так как чаще в литературе приведены значения Сыин, fi, а не t/мин-
Немалое значение при сравнении пределов обнаружения элементов, публикуемых различными авторами, имеет выбор оценки стандартного отклонения холостого опыта. Авторы 172 ] провели сопоставление литературных данных по этим вопросам для случаев использования вместо Д^хол, принятого ИЮПАК, значений: ^хол ~~ стандартного отклонения среднего значения $хол; $р = = Ймин/^мин + 5^ХОЛ//7ХОЛ]1^— совокупного стандартного отклонения; sr = $Ухол///хол — относительного стандартного отклонения.
Показано [72], что применение эл вместо Sy приводит к уменьшению Смин, у примерно в 7 раз (при лХ0Л = 3<J) по сравнению с моделью ИЮПАК 16, 73]. Также значительно уменьшается СМ(|Н, й и при использовании Sp В ряде случаев в практике для расчетов применяют значение sr. В этом случае выражение (3.24) может быть преобразовано к виду:
Смин, ft = k (Хол/Ь*хол) (Ухол/5) = Мхол/5 (3.25)**
При расчете СМИВ1 k следует принимать во внимание не только выбор методики расчета погрешности измерения сигнала холостого опыта, но и то обстоятельство, что параметры S и а уравнения (3.21) находят тоже с определенной погрешностью. В связи с этим авторы [71 ] предлагают и анализируют три варианта расчета предела обнаружения Сыин, ь с разной степенью приближения:
1. Приближение, учитывающее лишь погрешность определения сигнала холостого опыта — уравнения (3.24), (3.18) (модель ИЮПАК).
2. Приближение, учитывающее погрешности определения параметров г/хол и S — см. уравнение (3.26).
3. Приближение, учитывающее погрешности определения параметров //хол, S и а — см. уравнения (3.27)—(3.32) и разд. 9.3.
Приближения 2 и 3 позволяют рассчитывать более реальные значения Сипн, ft. В приближении 2 в уравнение (3.24) подставляется доверительный интервал значений коэффициента чувствительности:
CMHH,ft = 4XOJ1/(S±ZP./Ss) <3-26)
* Этот вариант расчета представлен, в частности, в монографии [58].
** Возможности многих методов ограничены значением sr = 0,01 и опубликованные данные с использованием уравнения (3.25) рассчитаны, как правило, при k = 2 CaHHi /г==2 = 0,02 y/S.
64
Здесь tP_f—коэффициент Стьюдента (см.. гл. 9) при доверительной вероятности Р и числе степеней свободы f; ss — стандартное отклонение коэффициента чувствительности S.
Согласно уравнению (3.26) возможны три значения СМ.1Н> k. Когда tPjfss = O, уравнение (3.26) переходит в (3.24). Если S tp, fSs, то значения СМИ11> h, рассчитанные по уравнениям (3.24) и (3.26), будут различаться незначительно. Когда же tP,tss будет сопоставимо со значением S, значение Сыин, ft, рассчитанное по уравнению (3.26), будет значительно больше, чем по уравнению (3.24), и только большее значение должно приниматься за предел обнаружения.
Особое внимание авторы [72 ] обращают на условия выбора коэффициента tPf р Так как коэффициент k — 3 соответствует доверительной вероятности 99,87%, то значение tPtf необходимо брать для доверительной вероятности 99,9% при числе степеней свободы f = п — 2 (для модели линейной регрессии), где п — число экспериментальных точек, использованных для построения градуировочной кривой.
Для расчета предела обнаружения в общем случае (приближение 3), когда свободный член уравнений (3.21), (3.2Г) а 0, выражение (3.21) следует переписать в виде:
C=(z/-a)/S (3.27)
Вклад погрешности измерения каждого из параметров в общую погрешность определения концентрации оценивается из первой производной С по каждому из параметров:
Взяв первые производные и просуммировав слагаемые, можно рассчитать стандартное отклонение $с
,зя>
и после некоторых преобразований:
sc = [ *1 + + (^-)2 3 /s (3-30)
Уравнение (3.30) позволяет рассчитать стандартное отклонение значения С по измеренному значению у и всем остальным рассчитанным параметрам *.
Перепишем уравнение (3.24) в виде:
t-МИН, h = kSg (3.31)
* М :тодику расчета a, S, sy, sa, sg см. в разд. 9.3.
3 М. И. Булатов и др.
65
После некоторых упрощений при щсл да 0 уравнение (3.31) с учетом уравнения (3.30) преобразуется к виду:
Смин, k = k [Я +si+(aSy s^/S (3 32)
Если погрешность измерения коэффициента чувствительности незначительна, уравнение (3.32) упрощается:
Смин.* = *р^о,+ <l‘V (3-33)
Если и $о достаточно мало, уравнение (3.33) преобразуется в (3.24), которое определено ИЮПАК как предел обнаружения.
Анализируя экспериментальные данные, авторы [72 ] показывают, что значения Счпп, к, рассчитанные по модели ИЮПАК и приближению 3 (особенно в случае нелинейных градуировочных кривых), различаются на 1,5—2 порядка. На основании этого делаются следующие выводы
Расчет СМ11Н, к по приближению 2 может быть рекомендован только как оценочный Модель ИЮПАК справедлива только в случае, когда основным источником погрешности является холостой опыт, т. е. когда s£xoJi значительно больше или sj.
Фактически приближение 1 (модель ИЮПАК) в большинстве случаев дает искусственно заниженные значения СЫ1Н) h Приближение 3 является наиболее строгим и дает значения Смин, k, сопоставимые с измеряемыми значениями холостого опыта и с сигналами измеряемых стандартов.
В заключение Лонг и Вайнфорднер [72] считают целесообразной публикацию значений пределов обнаружения, рассчитываемых как по модели ИЮПАК при k = 3(СМИН, так и в приближении 3, которое учитывает суммарный вклад всех составляющих погрешностей (sUxon, sa, ss) аналитических измерений в значение Смин, k-
При оценке пределов обнаружения помимо рассмотренных вопросов необходимо помнить, что значения ушт и СЫИ1> для любого метода анализа имеют смысл только в сочетании с полным описанием методики определения (информации о пробе, ее предварительной подготовке, всех детален аналитической процедуры), так как любое изменение в одной из стадий анализа может сопровождаться изменением значений и Имеется целый
ряд факторов, влияющих на пределы обнаружения. К ним относятся: химические примеси (анализируемые элементы в реактивах, реагентах, растворителях; посторонние компоненты, которые увеличивают значение аналитического сигнала; примеси, попадающие из воздуха, воды, стенок посуды и т. д ), потери анализируемого вещества во время исследования (кажущиеся потери анализируемого вещества из-за разбрызгивания или испарения исследуемой смеси в ходе анализа, потери из-за адсорбции на стенках посуды, при фильтрова-66
нии и т. д.); влияние экспериментальных параметров; прочие помехи.
Особое значение следует придавать постановке корректных холостых опытов и учету влияния примесей в реактивах [21, 67, 75, 76, 79—83] В идеале холостая проба должна быть идентичной реальному анализируемому образцу по составу и физико-химическим свойствам и содержать возможно меньшие количества определяемого элемента. Однако при фотометрических определениях по способу градуировочной кривой холостые пробы обычно не содержат основные компоненты и при проведении холостых определений не поддаются учету погрешности, обусловленные отбором пробы, химической подготовкой образца и специфическим влиянием основы (матрицы) на выбранную цветную реакцию, что может служить причиной ошибочных оценок предела обнаружения, обычно в сторону его необоснованного занижения.
Часто недостаточное внимание уделяется многочисленным погрешностям, источники и значения которых также изменяются с изменением материала пробы и условий анализа.
Приводимые в литературе значения г/МИГ1 (Лмин) и Смин, k (?мин, h) рассчитаны обычно на основании результатов, полученных в оптимальных условиях, или на основании анализа идеальных модельных систем, причем значения k, как правило, не приводятся. Все рассмотренные выше причины резко затрудняют объективную оценку пределов обнаружения элементов разными методами и методиками. В общих случаях пределы обнаружения элементов фотометрическими методами анализа колеблются в интервалах [21 ]:
м iccoBbix долеи Сыин,й 10-2— 10"4 % (sr = 0,014-0,05)
10-4— 10'7 % (sr = 0,054-0,10) относительных содержаний СМИн, fe ’О"3— 10-1 мкг/г массы примеси умив, * 10—104 нг
Рассчитанные значения Сыпн, h (CWHH, л) являются приближенными оценками. Поэтому, как правило, их следует характеризовать числом с одной значащей цифрой.
В связи с рассмотренными выше сложностями и неоднозначностью оценок пределов обнаружения в литературе предложены другие, более надежные, характеристики способности к обнаружению Так, предложена более высокая граница — предел надежного обнаружения сигнала, когда k = 6. В 1980 г. [741 предложен термин — «предел количественного определения» (limit of quantification), который отличается на Ю$ухол от г/хоп. При этом принимается, что содержания, отвечающие аналитическому сигналу 3$йхол с у с 10syxoJ1, соответствуют области обнаружения, а при у ’05Ухол — области определения
Международная комиссия по микрохимической технике и анализу следов [19] пришла к заключению, что вместо «предела обнаружения» в качестве сравнительной характеристики способности аналитических методов определять низкие количества и
3* 67
концентрации элементов следует применять «предел определения» (limit of determination — Lq), характеризующийся более высокой границей. В этом случае используется прежняя расчетная формула (3.18), но значение k берется равным 10 вместо k = 3.
Указанный подход отличается от того, который использовался для оценки предела обнаружения, только более высокой надежностью регистрации полезного аналитического сигнала. Поскольку речь здесь идет об обнаружении элемента, а не о его количественном определении, применение терминов «предел количественного определения» или «предел определения» в этом случае представ-
ляется не вполне удачным.
На основе критерия «предел определения» авторы [19] провели сопоставительный анализ возможностей большой группы
аналитических методов для 83 элементов. По ряду элементов
значения Lq представлены ниже:
Элемент Lq, мкг, мл* Элемент Lq, мкг/мл*
Ag 0,1—0,05 Р 0,03—0,003
Al 0,01 Pb 0,1—0,08
Bi 0,07 S 0,01
Cd 0,04 Sb 0,1—0,08
Co 0,04 Se 0,2—0,08
Cu 0,1—0,03 Si 0,03
Fe 0,1—0,07 Sn 0,07—0,03
Ga 0,04 Те 0,6-0,1
Ge 0,01 Ti 0,1—0,03
In 0,04 и 0,4—0,1
Mg 0,1—0,04 V 0,4—0,04
Mn 0,2—0,002 w 0,2
Ni 0,1—0,02 Zn 0,02
* Значения Lq соответствуют условиям: А — 0,025, погрешность измерения оптической плотности ±0,0025, толщина светопоглощающего слоя I см, объем пробы 1 мл.
3.1.2.2. Нижняя граница определяемых содержаний (lower limit of concentration range)
Область применимости любого метода при определении следов элементов определяется нижней границей определяемых содержаний элемента — Сп*. Здесь, однако, как и в случае предела обнаружения, отсутствует общепринятая методика расчета значений Сн, что связано с некоторой неопределенностью формулировки самого понятия.
Согласно терминологии, рекомендуемой ИЮПАК [1 ], нижняя граница определяемых содержаний — это наименьшее значение определяемого содержания, ограничивающее интервал определяемых содержаний.
* Сн обычно представляет собой массовую долю определяемого компонента в анализируемом веществе (а ие растворе).
68
Традиционно в фотометрических методах анализа теоретические расчеты значений нижней границы определяемых содержаний выполняют, исходя из молярного коэффициента светопоглощения окрашенного раствора и минимального регистрируемого значения оптической тотности. Более строгим является подход [67, 771, при котором нижняя граница определяемых содержаний рассматривается как наименьшее содержание, которое может быть определено с погрешностью, не превосходящей заданную. В качестве такой границы А. Б. Бланк принял содержание, погрешность определения которого не превышает 50%: уравнения (3.7) и (3.8).
Показано, что для линейной градуировочной функции при определенных условиях отношение Сн/С М11Н1 Р = 2, т. е. значение С„ близко к пределу надежного обнаружения. Но чаще всего Са превосходит СНИН1 Р в 3—5 раз [67, 84]. Это подтверждают данные табл. 3.3, в которой приведены полученные авторами 184 ] оценки Сн и СЫПВ1Р для фотоколориметрических методик определения титана, фосфора, кремния, алюминия.
И. А. Блюм [85] указывает, что минимальным содержанием элемента, доступным количественному определению данным методом (нижним пределом определения), принято считать содержание, при котором абсолютная погрешность определения составляет обусловленную долю С, обычно 1/3С, что соответствует srC = — (srC — относительное стандартное отклонение концентрации С):
sch/Ch — ьгСп~1^ (3.34)
Автор рассмотрел воспроизводимость и нижний предел определяемых содержаний методов фотометрического анализа с учетом большого числа факторов. Трактовка отличается от традиционной количественным учетом влияния флуктуаций оптического фона, разделением Сн на составляющие — «аналитическую» Сн (а> и «инструментальную» Сн (н) — и рассмотрением вклада каждой
Таблица 3.3. Оценка нижней границы определяемых содержаний и пределов обнаружения элементов в фотометрических методиках (п = 37, Р = 0,95)
1. Схол — среднее значение результатов холостого опыта; sxoJ1 — стандартное от« клоненне результатов холостого опыта. 2 Сн оценивалось при точности измерения содержания, не превышающей 30% измеряемой величины [см уравнение (3 34)]. 3. ДАПМ.—’ Диантипирил метаи
Элемент Реагент ^ХОЛ’ % *хол» % Смин- Р- % сн. %
Ti ДАПМ 0,005 0,00065 0,002 0,008
Р (NH,)MoO4 0,001 0,00024 0,0008 0,002
Si (NH4;2MoO4 0,004 0,00053 0,002 0,005
Al Хромазурол S 0,006 0,00042 0,001 0,005
69
из них в общую величину Сн. Ниже поиведены соответствующие основные расчетные уравнения (3.35)—(3.38):
сн = ^^тУ -#+ +-F +2so (335)
6м Лм/ ' \ ar < /
Сн = | Сй1а) -г С-(и) (3.36)
36еЙКкС
Си(а) — fRJ( ЬМЛМ
^- = 38—^-
(3.37)
Сн<и) — 4,2Z)Sy/S
(3.38)
+ ^ =
+ в
&гЛ
Д ______
Лр s
Здесь b — безразмерный коэффициент, учитывающий число градуировочных определений п (1 < b < 1Л2; при л -> оо fc -» 1), 10-3iВм’
—истинный молярный коэффициент светоцоглощсния окрашенного соединения MR в области светоиропускания ДА, отнесенный к молю определяемого элемента М; Вп — атомная масса М; eR? = I0~3/Af R — то же для реагента /?;
TVfp — молекулярная масса реагента; ДА — спектральная область пропускания разрешающего устройства фотометра (при спектрофотометрических измерениях — фактическая длина волны А, при которой измеряется оптическая плотность раствора); Rm — отношение концентрации элемента М, связанного в соединение MR, к общей концентрации См (мкг/мл), Дм = 0.54-1; Л'р — отношение концентрации окрашенной формы R к общей концентрации реагента Cr; / — толщина слоя окрашенного раствора (кюветы), см; s—стандартное отклонение*; sc — погрешность нулевого состояния прибора при данной «настройке» (характеристика стабильности «нулевого» состояния фотометра, единицы оптической плот
ности); sr — относительное стандартное отклонение; — стандартное отклонение оптической плотности Др раствора фона (нулевого раствора), выраженное в единицах концентрации элемента М и не зависящее от толщины слоя; S — коэф-
дус = = z г
дС mW 1ГДе
если = 0 (реагент не по-
фициент чувствительности метода, равный 8 = -в общем случае равен разности — meR?‘;
глощает в области ДА), то = емц]-
Анализируя приведенные уравнения, автор обращает внимание на следующее обстоятельство. Общепринятый подход к расчету Сн (см. разд. 3.1.1.2) справедлив, когда Сн (а) = 0, = 0,
т. е. в отсутствие оптического (]она. В этом случае Сп = С„ (и) и из уравнения (3.38) следует, что С„ обратно пропорциональна коэффициенту чувствительности S = еД'Л’м/. Следовательно, оптимальны: реагент и условия аналитического процесса, обеспечивающие наибольшее значение емХКм; характеристика разрешающего устройства фотометра ДА, соответствующая Амаьс соединения MR, максимальная толщина слоя.
При наличии оптического фона вносит свой вклад Сн(а). Аналитическая составляющая, согласно уравнению (3.37), пропорциональна отношению Sa /S. Оптимальны в этом случае: реагент
Расчетные уравнения стандартных отклонений и дисперсий см. в гл. 9.
7о
и условия аналитического процесса, обеспечивающие минимальное отношение (eR?AR)/(e&x£M); характеристика Ы, соответствующая наименьшем!’ значению отношения ErVem*, которое может не совпадать с ^макс-
Таким образом, инструментальная и аналитическая составляющие Сц требуют различных критериев для выбора реагента, условий аналитического процесса и условий измерения. Для обоснованных решений необходимо знать соотношение величин и s0. При анализе результатов экспериментальной оценки погрешностей s0 и при фотометрическом определении В, Th, Ge, Al, Be, Ta, Nb, Те показано [86], что величина ]/2s0 мала по сравнению с ал . Оценка Сн без учета последней была бы в 1,6—30 раз заниженной. Методика определения бериллия с берилловом II при X = 620 нм, соответствующей максимальному отношению ЕрЛ/ем', вместо рекомендуемого в литературе значения X = 640 нм обеспечила уменьшение Сн в 1,4. раза.
В настоящее время нижняя граница определяемых содержаний широко применяется в качестве обязательной метрологической характеристики методик количественного определения следов эле-
Таблица 3.4. Нижняя граница определяемых содержаний отдельных примесей в веществах высокой степени чистоты [22]
Способ определения: КФ — кинетический с фотометрическим окончанием; Ф — фотометрический; ЭФ экстракционно-фотометрический.
Элемент Основа Способ определения; реагент Нижняя граница определяемых содержаний Сн-ю\ %
Fe Кислоты Ф; 1,10-фенантролин 0,1—1
Вода ЭФ; батофенантролин 0,01—0,5
Ag, Zn, Cd Ф; 1,10-фенантролин 4—5
Y2o3 ЭФ, 1- (2-пиридилазо)-2-нафтол 50
TiCl4 Ф; NH4SCN 5
Se ЭФ; 1,10-фенантролин 10
Те ЭФ; роданид 1
Си SnO2 ЭФ; дитизон 50
Те ЭФ; 1,10-фенантролин 1
W Ф; 4-(2-пиридилазорезорцин) 50
Р Ga, AsCl3 Ф; молибдат, аскорбиновая кислота 0,5—2
CHsSiCl3 ЭФ молибдат, N2H4-H2SO4 0,05
О Se Ф; парафуксин 5
Si Ti ЭФ; молибдат, SnCl2 50
В Sn, Si ЭФ; метиленовый голубой, HF 2—10
Те Cd ЭФ; кристаллический фиолетовый, НС1 1
А1 Cu ЭФ 8-оксихинолин 20
Ge NaCi Ф; фенилфлуорон 0.04
As 1 ME Ф; диэтилдитиокарбаминат серебра 0,3
Sn In ЭФ; лючогаллион 5
71
Ментов. Сводные данные значений С„ при определении примесей в веществах высокой степени чистоты приведены в работе [22], откуда следует, что фотометрические методы обеспечивают в основном Св « и-КН %. Типичные значения Сн представлены в табл. 3.4.
3.1.2.3. Пути повышения чувствительности фотометрических реакций, снижения пределов обнаружения и нижней границы определяемых содержаний
Анализ литературных данных показывает, что повышение чувствительности фотометрических реакций (снижение Смин, Р, предела определения и нижней границы определяемых содержаний) развивается по нескольким направлениям:
1. Широкое применен! е экстракционно-фотометрических методов анализа (см. разд. 5.1.2 и 7.4), и не только для анализа следов элементов.
2. Использование реакций «усиления».
3. Применение кинетических методов.
4. Усовершенствование техники измерений, разработка новых типов спектрофотометров и новых вариантов методов абсорбционной спектроскопии.
Наиболее интенсивно окрашенные соединения имеют значения емакс, ^близкие 100 000, и следовательно, дальнейшее увеличение чувствительности на этой основе принципиально ограниченно. Однако значительная часть окрашенных комплексных соединений, используемых в спектрофотометрии, имеет молярные коэффициенты на 1—1,5 порядка ниже предельных значений, и поэтому возможности уменьшения определяемого минимума с помощью новых высокочувствительных реагентов еще не исчерпаны.
Вместе с тем разрабатываются методики, которые позволяют получить значения «эффективных» молярных коэффициентов светопоглощения существенно выше 100 000. Это можно проиллюстрировать на примере определения субмикроколичеств (10~s г) фосфат-иона [87—90].
Принцип метода заключается в применении «усилительной реакции» (amplification reaction), в которой исходный определяемый ион является ответственным за образование нескольких других ионов того же самого или иного вещества. Авторы получали в водном растворе фосфомолибдат-ион (12MoO3-POJ_), который отделяли от избытка молибдата экстрагированием гетерополи-кислоты в бутанол-хлороформ. Зате и фосфомолибдат-ион реэкстра-гировали в аммиачный буферный раствор, где он разрушался, и молибдат-ионы определяли с помощью реактива Киркбрайта и Джойса [90] — 2-амино-4-хлорбензолтиола. Этот реагент образует с молибдат-ионами окрашенное соединение с е = 36 000. Так как каждому фосфат-иону соответствуют 12 молибдат-ионов, то с точки зрения определения фосфат-ионов коэффициент свето-
72
поглощения увеличивается в 12 раз (при А, = 710 нм еалф ~ = 432 000 и ms = 9-10-Б мкг/см2).
Следует отметить, однако, что применимость этого метода ограничена небольшим числом реакций, подобных описанной.
Намного более перспективным является третье направление — каталитические реакции (кинетические методы с фотометрическим определением скорости реакции).
Если следы определяемого иона могут служить катализатором какой-либо реакции [91, 92], продукты которой окрашены, то чувствительность такой «кинетохроматической спектрофотометрии (фотоколориметрии)» может быть повышена во много раз за счет высоких значений (более 100 ООО) «кинетохроматических коэффициентов светопоглощения» [91 ]. Например, реакция между цирконием и индикатором ксиленоловым оранжевым H4Y2“ в солянокислой среде протекает медленно с образованием красного комплекса (2 : 1):
2ZrO2+ + H4Y2' медленно/^ уа+ + 2Н2О
В присутствии некоторых ионов (арсенатов, фторидов, фосфатов и в меньшей степени сульфатов) реакция между ионом металла и реактивом ускоряется и красный комплекс образуется намного быстрее. Можно подобрать условия, при которых неката-лизируемая реакция будет протекать только до ограниченного предела, а катализируемая будет иметь приемлемую скорость, что позволит измерить количественно эффект катализа. При этом соблюдаются следующие соотношения для ионов:
Арсенат : ксиленоловый оранжевый : цирконий 1:4:8
Фторид : ксиленоловый оранжевый : цирконий 1:4:8
Фосфат : ксиленоловый оранжевый : цирконий 1 : 8 : 16
Сульфат • ксиленоловый оранжевый : цирконий 1:2:4
Отсюда видно, что как арсенат-ион, так и фторид-ион катализируют образование четырех хелатных молекул циркония, тогда как фосфат-пон — образование 8 молекул. Микроколичества фосфат-, арсенат-, фторид- и сульфат-ионов образуют с ионом циркония лабильные комплексы, которые могут реагировать с лигандом более быстро, чем устойчивые к замещению полимерные молекулы оксоциркония, обычно существующие в слабокислых растворах. На основе этих кинетохроматических реакций найдены кинето-хроматические коэффициенты светопоглощения для арсенат-иона (200 000), фторид-иона (200 000), фосфат-иона (400 000) и сульфат-иона (40 000).
Описанные системы просты в исполнении, высокоизбирательны и на порядок (до 6-10-8 моль при I = 1 см) чувствительнее, чем любая прямая спектрофотометрическая методика для этих ионов [91 ].
Используя каталитическое действие диэтилдитиокарбамината мед1 на обесцвечивание иод-азидных растворов при А = 420 нм, А. К. Бабко и др. [92] предложили методику определения следов
73
меди (до 0,06 мкг). Максимальное значение молярного коэффициента светопоглощения (^а^с) диэтилдитиокарбамината меди равно 12 800, а условный молярный коэффициент светопоглощения каталитической реакции (е,,) — составляет 3,4 10Б при X = = 420 нм, т. е. ек в 50 раз больше емакс и во столько же раз чувствительность определения меди по иод-азидной реакции больше, чем с диэтилдитиокарбаминатом.
Более подробные сведения по группе кинетических методов анализа можно получить в литературе [93—96].
Значительные перспективы повышения чувствительности и уменьшения Сп открывают достижения в области разработки и выпуска новых моделей спектрофотометров в сочетании с компьютерами, микропроцессорами, данные по которым на 1984 г. обобщены в обзорах [97, 98]. К числу новых высокочувствительных и селективных методов относятся лазерная абсорбционная спектрометрия (laser intracavity absorption spectrometry) [98— 100] и лазерная фото (опто) акустическая спектроскопия (laser-indused photoacoustic spectroscopy) [101, 102].
Лазерная абсорбционная спектрометрия позволяет измерять очень низкие значения оптических плотностей (вплоть до А = — Ы0“3—5-Ю*5) с воспроизводимостью значений А = 2-10_&. Разработан способ определения этим методом следов железа (с помощью ферроцианина — е = 28 600, К = 584 нм) в особочистом SiCl4 в интервале концентраций (0,5—3,0)-Ю-8 г/мл. Фото-акустическая спектроскопия дала возможность снизить предел обнаружения следов кадмия [101] на 2 порядка по сравнению с методами обычной спектрофотометрии.
Правда, указанные методы и модели установок пока не вышли за рамки конструкторских разработок и лабораторных испытаний. Следует отметить, что в ряде случаев на практике оправдывают себя и более простые варианты повышения чувствительности, снижения Сыин> р фотометрических определений. Это достигается уменьшением площади поперечного сечения кювет в сочетании с экстракционным концентрированием [60, 67, 80]. В литературе описано большое число микрокювет различной конструкции [см., например, 22, 60, 67, 80, 103, 104], эффективное сечение q которых на 1—3 порядка меньше, чем у обычных кювет. Так, если q = 10~2 см2, а условная чувствительность реакции = = 0,01 мкг/см2, то т = m't,q = 10~4 мкг = Ю-10 г, т. е. приме-I ение микроспектрофотометрии позволяет определять субмикро-граммовые содержания примесей.
В литературе описаны микроспектрофотометры, созданные на основе микроскопов и монохроматоров, специально предназначенные для кювет малого объема и в большинстве случаев для малых толщин поглощающих слоев. Во многих конструкциях кювету устанавливают на предметном столике микроскопа, в котором на месте окуляра помещены фотоумножитель или фотосопротивление.
74
5
Рис. 3.3. Варианты микрокювет.
а— Вкладыши к кюветам фотоколориметра [61]; б— разборная микрокювета для однолучевых спектрофотометров: 1 — навинчивающаяся крышка из фторопласта; 2— оптическое (или кварцевое) стекло; 3 — корпус из фторопласта [104].
Отечественные фотоколориметры ФЭК-56М, КФК, КФК-2 могут комплектоваться микрокюветами (и = 0,44-0,08 мл, I = = 104-2 мм — см. гл. 6).
А. Б. Бланком 167, 104, 105] предложен ряд приемов и микрокювет (рис. 3.3) для повышения чувствительности фотоколориметрических и спектрофотометрических измерений. Хорошо зарекомендовала себя конструкция разборной микрокюветы (v — — 2,5 мл, I — 50 мм, q — 0,5 см2) (см. рис. 3.3, б) из фторопласта для спектрофотометров типа СФ-16, СФ-26 и др.
Конструктивные особенности фотоколориметров ФЭК-56, ФЭК-56М, КФК, КФК-2 позволяют в 2—5 раз уменьшить отношение объема раствора к толщине поглощающего слоя [67, 105].
В работе [67, 105] двухкратное повышение чувствительности достигалось благодаря тому, что выделенный с помощью диафрагмы узкий пучок света проходил у основания кюветы, заполненной раствором только наполовину. При этом в гнезде держателя кювет помещали отшлифованные прямоугольные пластинки из нержавеющей стали или плексигласа (6,0x23x56 мм), а во входных окошках кюветной камеры устанавливали круглые металлические диафрагмы с центральным отверстием диаметром 3,5 мм. Оптическую плотность измеряли в кюветах с толщиной слоя 2 см, помещенных на подставки у конца кюветодержателя, обращенного к осветителю (в упор). Для заполнения кюветы достаточно 3,4—4 мл раствора.
Поперечное сечение окрашенного раствора можно еще больше уменьшить, используя кюветы, снабженные вкладышами из инертного непрозрачного материала с осевым цилиндрическим отверстием минимального диаметра. Так, вкладыши из фторопласта Ф-4 с диаметром отверстия 9 мм к кюветам с I — 50 мм (рис. 3.3) позволили уменьшить необходимый объем раствора до 3,5—4 мл вместо 20 мл при обычной работе и снизить значение определяемого минимума в 5 раз (от т = 0,37 до т = 0,07 мкг).
Если одним из рассмотренных приемов повышают чувствительность методики фотометрического определения элемента, то при
75
оценке коэффициента чувствительности усовершенствованной методики необходимо использовать градуировочную зависимость (градуировочный график) оптической плотности раствора от массы определяемого элемента — А = f (т).
3.1.3. ВОСПРОИЗВОДИМОСТЬ
ФОТОМЕТРИЧЕСКИХ МЕТОДОВ АНАЛИЗА
3.1.3.1. Воспроизводимость абсолютных фотометрических методов анализа
Сходимость и воспроизводимость * [5S, 82, 106—108] анализа определяются разбросом повторных результатов анализа относительно их среднего значения и обусловливаются наличием случайных погрешностей. Они характеризуются значением стандартного отклонения s (sc) или относительного стандартного отклонения sr (sc. С) — см. гл. 9.1.
В фотометрическом анализе различают два типа случайных погрешностей: инструментальные и аналитические (методические и химические). Обычно полагают, что воспроизводимость абсолютных методов фотометрического анализа характеризуется пределами 2—5%. Однако эти «усредненные» значения могут значительно колебаться в обе стороны в зависимости от содержания определяемого компонента, выбора условий анализа и цветной реакции, характеристик спектрофотометра или фотоколориметра, области измеряемых оптических плотностей, кюветной невоспроизводимое™ и т. д. Ниже рассмотрены основные факторы, определяющие воспроизводимость фотометрического анализа.
1. Случайные погрешности, связанные с пробоотбором, приготовлением анализируемых растворов, измерениями массы, объемов и т. д. (аналитические погрешности).
2. Погрешность холостого опыта. Для учета оптической плотности холостого раствора нередко рекомендуют измерять оптическую плотность исследуемого раствора относительно холостого раствора.
3. Погрешность установки аналитической длины волны, обусловленная несоответствием положения диспергирующей призмы (решетки) и указателя на шкале длин волн.
* Сходимость характеризует рассеяние результатов при фиксированных условиях выполнения эксперимента; воспроизводимость — при варьировании этих условий (ГОСТ 162263—70 Метрология. Термины и определения). В первом приближении можно считать, что стандартное отклонение сходимости в 1,4— 1,5 раза меньше стандартного отклонения внутрплабораторной воспроизводимости [1061. Ввиду наличия такой простой связи между характеристиками сходимости и воспроизводимости в дальнейшем мы говорим о воспроизводимости фотометрических методов анализа (исследованию этой характеристики посвящены работы [8—10, 16, 25, 80, 108—121 ]).
76
4. Погрешность фотометрических измерений, обусловленная диапазоном (областью) измерений аналитического сигнала (А, Т) [16, 25, 108, 109—117].
5. Погрешность абсолютных спектрофотометрических измерений, обусловленная настройкой прибора на нулевое и 100%-ное пропускание, его чувствительностью и нестабильностью работы электронных схем в процессе измерения.
6. Кюветная невоспроизводпмость, связанная с разной толщиной кювет, невоспроизводимостью состояния граней кювет, но самое главное — с невоспроизводимостью положения кювет в кю-ветодержателе. Именно эта составляющая ограничивает воспроизводимость измерений, особенно при низких значениях А [108, 109—117].
Относительный вклад перечисленных факторов в воспроизводимость фотометрических измерений определяется конкретными объектами, условиями анализа и типом спектрофотометра или фотоколориметра.
Некоторые из перечисленных выше факторов могут быть источниками не только случайных, но и систематических погрешностей — например, если данной длине волны соответствует фиксированная по знаку и численному значению погрешность ее установки или если при установке прибора на нулевое пропускание всегда устанавливается близкое к нулю, но не равное ему и практически постоянное пропускание Т.
Основное внимание в литературе уделяется рассмотрению и анализу факторов 4—6 и взаимосвязи воспроизводимости фотометрических измерений со значением оптимальной оптической плотности Аопт.
Погрешность определения содержания, обусловленная погрешностью измерения аналитического сигнала (Л, Т). Оптимальный диапазон измерений аналитического сигнала.
Согласно основному закону светопоглощения, можно записать *:
С = А,а1 (3.39)
Продифференцировав уравнение (3.39) по переменным А и С, получим
dC — dAal (3.40)
где вместо d.C и dA можно подставить соответствующие им стандартные отклонения sc и sA (см. гл. 9).
Из уравнений (3.39) и (3.40) выводится выражение относительной погрешности определения концентрации (srC = sc/Q:
= (З-41)
* При выражении концентрации в мг/мл или мкг/мл. Если а = е, то концентрация выражается в моль/л.
77
Рис. .° 4. Зависимость от носитель! ой погрешности измерения концентрации от оптической плотности раствора (при sr = 0,003)
/ — кривая рассчитана по уравнению Шмидта, 2, 3 — кривые рассчитаны Н. П Ком а рем и В П Самойловым для спектрофотометра СФ-4 и фотоколориме-тра ФЭК-М соответственно [109].
Параллельно можно провести следующие выкладки:
— IgT — аС1 (3.42)
После перехода к натуральным логарифмам производная выражения (3.42) выглядит так:
— 0,434347/7 = a/dC (3.43)
Разделив уравнение (3.43) на (3.42), получим:
dC/C = sc/C = 0,434347/(7’ Ig T) = 0,4343s. /(7 1g T) (3.44)
ИЛИ
ДС/С = 0,4343Д7/(7 1g Т) (3.44')
Из уравнений (3.44) и (3.44') следует, что при заданной абсолютной погрешности Д7 измерения Т значение относительной погрешности определения концентрации АС/С (sc/C)
зависит от абсолютного значения светопропускания (оптической плотности);
проходит через минимум при А = 0,434(7 = 0,368) — рис. 3.4, кривая /;
составляет 1,5—2% *, если А = 0,15-ь 1,0.
Используя уравнения (3.41) и (3.44) и подставляя значения оптической плотности вместо пропускания (Т — 10-л), можно получить широко представленную и анализируемую в литературе теоретическую зависимость
sjC = SA/A = 0,43435у/(1(ГЛЛ) (3.45)
где значение sT определяется из 5—10 параллельных обсчетов пропускания. Эта зависимость в координатах Д($с-100/С) — А представлена на рис. 3.4 (кривая J). Из кривой 1 следует также, что допустимый рабочий интервал оптических плотностей (при
* Более точно: (ДС'С)-100 = 2,00% при А — 0,155 (7 = 0,70) и (ДС/С) X X 100= 2,20% при 4= 1,000 (7=0,10) при Д7 (sT) = 0,005.
Случайные погрешности измерения пропускания на стандартных спектрофотометрах лежат в интервале sr от ±0,002 до ±0,01. Значение 0,005 является типичным.
78
Sj = 0,003), при котором относительная погрешность определения концентрации не превышает удвоенной минимальной погрешности, составляет 0,12—1,2.
Н. П. Комарь, В. П. Самойлов и др. [109, ПО] теоретически и экспериментально показали, что кривая 1 не подтверждается. Обусловлено это следующими обстоятельствами. При выводе уравнений типа (3.44), (3.45) не учитываются:
зависимость sT от Т;
вклад в общую погрешность спектрофотометрического измерения инструментальных погрешностей, обусловленных настройкой прибора на нулевое и 100 %-ное пропускание, чувствительностью прибора, нестабильностью электронных схем, кюветной невос-производпмостью. Эти погрешности в ряде случаев являются определяющими.
На основании результатов исследований авторы [109] сделали два принципиально важных для практики вывода.
1. Широко представленная в литературе кривая 1 рис. 3.4 и расчетные данные по уравнениям (3.44), (3.45) дают заниженную воспроизводимость фотометрирования. Об этом свидетельствуют кривые 2, 3 рис. 3.4, рассчитанные Н. П. Комарем и В. П. Самойловым, согласно которым возможно проводить фотометриро-вание с гораздо лучшей воспроизводимостью: с относительной погрешностью Л определения концентрации, значительно меньшей значения 0,88 %, вытекающего из уравнений (3.44), (3.45).
2. Интервал А оптических плотностей, в котором общая погрешность измерения не превышает удвоенной минимальной, оказался несколько шире указанного кривой Шмидта (рис. 3.4, кривая J) и данными других авторов. Для однолучевых спектрофотометров типа СФ-4 и двухлучевых фотоколориметров типа ФЭК-М этот интервал, в отличие от общепринятого (0,12—1,2), доходит до значений 1,35—1,45. В области малых значений оптических плотностей расширение интервала незначительно.
Авторы [109] обращают внимание на следующее обстоятельство. Из кривой Шмидта следует, что например, при sT = =0,003 (0,3%) имеем Дмин 0,88% и 2ДМНН« 1,76%, т. е. эта кривая указывает, что фотометрирование в интервале А = 0,124-1,2 возможно с погрешностью 2ДМИН« 1,76%. Кривые Кемаря и Самойлова (рис. 3.4, кривые 2 и 3) говорят о возможности фотометрирования с погрешностью Дмин ~ 0,88% в интервале оптических плотностей 0,14—1,9 и с погрешностью 2ДМИН » 1,76% вплоть до значений А л; 2,54-2,6. Таким образом, опубликованные ранее теоретические положения указывали заниженный интервал, где погрешность измерений не превышает удвоенной минимальной, и фактически скрывали возможности спектрофотометрического анализа в области высоких значений оптических плотностей.
Позднее Н. П. Комарь, В. П. Самойлов и др. [’.09, ПО] провели теоретический и тонкий экспериментальный анализ вклада
79
погрешностей чувствительности однолучевых спектрофотометров, нестабильности работы электронных схем и кюветной невоспроизводимое™ на общую относительную погрешность измерения концентрации, впервые с учетом конкретной зависимости sT = f (Т).
Под чувствительностью измерительного прибора понимают отношение линейного или углового смещения указотеля к изменению измеряемой величины, вызвавшему это смещение указателя. Чувствительность спектрофотометра т может быть выражена как отношение числа делений Хи = п2 —Щ, на которое смещается указатель нуль-и негру мент а (миллиамперметра, индикатора), к числу делений шкаты пропускания Д7" = Т2 — при перемещении ее относительно визирной риски, т. е.
m = Д/г \Т = (пг —щ) (Тг — Т,) (3.46)
Зная чувствительность и цену деления шкалы пропускания 0ц = 1 %, можно найти цену деления шкалы нуль-инструмента 0н(%) из уравнения.
= 6П/л (3.47), откуда т = 6И/0Н (3-47')
Размах колебания стрелки нуль-инструмента, выражаемый в миллиметрах деления шкалы или в процентах пропускания, является показателем нестабильности прибора. Нестабильность (невоспропзводимоеть) показаний указателя нуль-инструмента является следствием флуктуаций фото- и темнового токов, обусловленных разного рода источниками — шумами в фотоэлектрической цепи, кюветной невоспроизводимостью, колебаниями накала источника излучения, вибрациями и т. д Нестабильность прибора можно охарактеризовать коэффицие -том нестабильности К, представляющим собой отношение ширины L одного деления шкалы нуль-инструмента к размаху колебания I стрелки.
Согласно [109, ПО] коэффициент нестабильности /С, чувствительность т прибора и стандартное отклонение пропускания sT связаны соотношением:
К = L,I = 0u/(ms?) (3.48), откуда sT = 0а/(тК) (3 48')
Значения sr и К складываются из двух составляющих. При настройке шкалы на Т = 100% невоспропзводимоеть показаний является результатом флуктуаций фото- и темнового токов, обусловленных всеми возможными источниками случайных погрешностей, и достигает максимума. При этом:
Ч = йц('пК1)> где /<! = £'/! (3.49)
При настройке шкалы на Т — 0 (фотоэлемент затемнен) флуктуации фототока исключаются, и невоспроизводимость обусловлена только той группой источников, которые вызывают флуктуации темнового тока. В этом случае флуктуации минимальны и не зависят от значения Т, а
sn = (W(mA'n), где = L//o (3.50)
На основании зависимости sr = f (sn, st) и уравнения погрешности спектрофотометрического измерения концентрации (3.45) авторы [ПО] предлагают расчетное уравнение оценки относительной погрешности (%) измерения концентрации, учитывающее 80
реальные рабочие характеристики спектрофотометров и завися мость s7 = f (Г):
sc _ 0,43v _ о,43 6U Г 1 / 1 1 \ 1
С ТА ТА т L Ко \ Ki Ко / J
(3.51)
Специально разработанная и осуществленная авторами [1101 методика эксперимента позволила разграничить суммарный вклад кюветной невоспроизводимое™ и погрешностей приготовления растворов от погрешностей нестабильности и чувствительности прибора. Результаты представлены кривыми 1—3 рис. 3.5, на основании которых делаются выводы.
1. Кюветная невоспроизводимое™ и погрешность приготовления растворов существенно повышают общую относительную погрешность измерения концентрации (кривая 1 располагается значительно выше кривой 2), особенно в области малых и средних значений оптических плотностей.
2. Кюветная невоспроизводимое™ приводит к смещению области минимальных погрешностей в сторону высоких значений А. Так, у кривой 1 минимальная погрешность Амин находится при Л» 1, ау кривой 2 — при А « 0,454-0,70. Кривая 3 показывает отношение этих погрешностей в исследованном интервале А.
Следовательно, при устранении причин, вызывающих кюветную невоспроизводимое™ и погрешность приготовления растворов, наряду со снижением общей относительной погрешности измерения, область минимальных погрешностей будет смещаться в сторону средних значений оптических плотностей.
3. Область измерений при А < 0,43 является неблагоприятной из-за резко возрастающих значений погрешностей А. В каждом конкретном случае для достижения максимальной воспроизводимости необходимо выяснить оптимальную область измерения с учетом нестабильности и чувствительности прибора.
4. Путями устранения или снижения погрешности спектрофотометрического измерения, обусловленной случайными источни-
81
Рис. 3.6. Зависимость А опт от соотношения коэффициентов нестабильности (кривая характеризует оптимальную область измерений) [109].
ками погрешностей, могут быть, во первых, кардинальное усовершенствование электрических и электрометрических узлов выпускаемых отечественных одколу-чевых спектрофотометров; во-вторых, замена передвижной кюветы неподвижной с проточной подачей растворителя и раствора; в-третьих, снижение погрешностей приготовления рабочих растворов. Обычно погрешность приготовления растворов составляет 0,1—0,2%, и считалось, что она
много меньше инструментальной погрешности измерения спектрофотометра. Однако, как следует из кривой 2 рис. 3.5, эти погрешности соизмеримы при высоком качестве измерительной аппа-
ратуры.
В реальных условиях при работе на спектрофотометрах, в частности на приборах тина СФ-4 с передвижными кюветами, приходится учитывать неизбежность кюветной иевоспроизводи-мости наряду с погрешностями приготовления растворов. Этот учет возможен по методике [109] определения коэффициентов нестабильности 1\п и К^. При наличии таких коэффициентов целесообразно пользоваться номограммой зависимости отношения KJKo от оптической плотности Аопт [109, 1101, по которой определяют оптимальную область измерения. Такая зависимость представлена на рис. 3.6. Она строится решением уравнения (3.52)
и может рассматриваться как единая кривая, указывающая область минимальных погрешностей для всех возможных случаев и приборов:
Ko'Ki = si/so = 1/[ 10Л°пт (2,ЗДОПТ — 1) + 1] (3.52)
Экспериментальное исследование сходимости и воспроизводимости измерений оптической плотности на более совершенных однолучевых спектрофотометрах СФ-26 и СФ-46 (с микропроцессором — см. гл. 6) представлено в работе [116]. Авторами показано, что по метрологическим характеристикам (сходимости и воспроизводимости измерений оптической плотности) эти приборы практически равноценны. Воспроизводимость измерений на обоих приборах лимитируется в основном кюветной погрешностью. Для снижения значений кюветной погрешности могут быть рекомендованы проточные кюветы или промывка и заполнение кювет с помощью шприца (в тех случаях, когда это возможно).
Полная погрешность абсолютного фотометрического анализа. В оценке полной погрешности фотометрического анализа имеется
82
ряд вариантов с различной степенью приближений и с преимущественным акцентом на тех или иных составляющих. Среди них следует выделить работы [80, 85, 86, 117—120], основывающиеся на обобщенной метрологической интерпретации аналитических процессов. Основные вопросы рассмотрены ниже.
В общем виде зависимость между концентрацией раствора и оптической плотностью можно выразить уравнением прямой [117]:
АЦ = Cj -ф ЬХС (3.53)
Здесь ах — отрезок, отсекаемый прямой A/l; Ьх—наклон прямой (коэффициент светопоглощения).
Чтобы найти относительную погрешность определения неизвестной концентрации, следует рассчитать С из уравнения
С = (АЦ — aj/b! (3.54)
и полную абсолютную погрешность, которая равна сумме абсолютных значений всех частных дифференциалов рассматриваемой функции:
дС . дС . дС дС , dC = -^r-dA + -y^-dl -р ——daL ~р - ~d&! (3.55)
дА dl да! 1 йд
Подставляя соответствующие значения частных дифференциалов в уравнение (3.55), получают выражение:
+ <3-“>
Вместо dA, dl, dalt dbx можно подставить значения стандартных отклонений ±5^; ±s;; ±«01; ±5ь1Ф Предполагая, что имеет место наименее благоприятный случай, т. е. что все частные погрешности имеют одинаковый знак (складываются), следует сохранить только положительные значения стандартных отклонений:
1 , А . ! . (А/1) — а! __
SC — bxl Sfl-+ Ч + bi S°i bf Sbi <3’57)
Выражение для расчета относительной погрешности (%) выводится из уравнений (3.54) и (3.57):
~’100= ( A —ad Sa+ I (A —ad) S‘+ A—dd s^ + ~bTs^ 100
(3.58)
Если предположить, что при измерениях отсутствуют систематические погрешности (а± = 0, но s0, Ф 0), то уравнение (3.58) примет следующий вид:
Зл. / S. S, S. lsa \
— .100= ( —+ —+_^+_Л_). 100 (3.59)
Составляющие погрешности в уравнении (3.59) могут быть вычислены в отдельности:
83
а) расчет погрешности sfl/A приведен ранее;
б) относительная погрешность stH может быть определена экспериментально из отклонений толщин кювет I. Если оптическая плотность измеряется во всех случаях в одной кювете, то, по мнению автора [119], погрешность st может быть незначительна;
в) погрешность содержит стандартное отклонение коэффициента светопоглощения Ь^,
г) lsaJA — SaJb^C зависит от концентрации раствора.
Из уравнения (3.60) можно сделать следующие общие заключения для проведения хорошо воспроизводимых спектрофотометрических (фотоколориметрических) измерений.
Погрешность будет меньше, если:
1) пропускание раствора близко к значению 0,386 (А = = 0,4343);
2) меньше значения стандартных отклонений пропускания sr; наклона градуировочной кривой коэффициента $а.;
3) меньше различия между длинами используемых кювет;
4) длиннее используемые кюветы;
5) круче наклон кривой (т. е. больше коэффициент светопоглощения);
6) больше концентрация раствора.
Следует отметить, что некоторые из этих требований, например 1, 4 и 6, противоположны друг другу.
В качестве примера в табл. 3.5 приведены данные расчета максимальной погрешности определения кобальта (II) в виде перхлората спектрофотометрическим методом.
Из приведенных данных видно, что недостаточно считать член sa/A, как это принято, основной относительной погрешностью определения концентрации: в ряде случаев его значение в большой степени зависит от ненадежности параметров и аг.
Таблица 3.5. Погрешности определения кобальта (II)
в виде перхлората классическим спектрофотометрическим методом [117]
X = 511 им, I — 1 см, £73 = 0,0206, sGi = 0,0114.
сСо2+> ол А/1 v.4343s7 SO1 /sot 0/ sc с
ТА °’ t,c д’" (полная погрешность), %
2 0,168 13,57 6,5353 8,95
4 0,334 2,225 3,2676 4,54
6 0,506 1,497 2,1784 3,38
8 0,674 0 1,6338 2,68
10 0.816 1,068 1,3070 2,65
12 1,045 1,424 1,0892 2,28
14 1,194 2,51 0,9336 2,23
Примечен и е jb^ во всех случаях составляет 1,0532%.
84
И. А. Блюм и др. в работе [1191 рассмотрели вопросы повышения воспроизводимости и сокращения трудоемкости массового фотометрического анализа. При этом с учетом многих факторов проанализировано влияние полной погрешности аналитического сигнала и погрешности воспроизведения цветной реакции на погрешность определения концентрации элемента и на полную погрешность фотометрического анализа. В последнем случае учитывались случайная погрешность определения концентрации элемента и погрешность подготовки пробы к определению (погрешности вскрытия, переноса массы определяемого элемента, измерений массы и объемов). Здесь же показано, что в массовом фотометрическом анализе применение общего для партии проб заранее приготовленного реагента, содержащего все необходимые для осуществления цветной реакции компоненты, улучшает воспроизводимость результатов фотометрического анализа и сокращает его трудоемкость.
На практике погрешность определения содержания компонента, как правило, оценивают на основании данных измерений А и уравнения градуировочной кривой, рассчитанной по методу наименьших квадратов (см. гл. 9).
В работе [121 ] показано, что обработка информации с помощью ЭВМ позволяет уменьшить погрешности измерений на спектрофотометрах в УФ-области.
3.1.3.2. Воспроизводимость дифференциального фотометрического анализа
Для определения основных и неосновных компонентов широкое применение получил дифференциальный фотометрический метод анализа (см. разд. 7.3), при котором оптическая плотность исследуемого раствора измеряется не относительно чистого растворителя (или раствора реактивов), а относительно раствора сравнения, содержащего известное количество определяемого компонента. При этом, во-первых, расширяется область рабочих концентраций, в которой соблюдается основной закон светопоглощения; во-вторых, оказывается возможным экспрессное проведение анализа с воспроизводимостью, не уступающей во многих случаях воспроизводимости тнтриметрических и гравиметрических методов анализа.
Имеется несколько вариантов дифференциального фотометрического анализа, сущность, воспроизводимость и правильность которых обобщены в монографиях [122, 123]. Дальнейшее развитие метрологические вопросы дифференциального фотометрического анализа нашли в работах [109, 120, 124—131].
Погрешность определения содержания компонента, обусловленная погрешностью измерения аналитического сигнала (Л, 7). Оптимальный диапазон измерений аналитического сигнала. Наиболее широкое распространение получил метод отношения про
85
пусканий (transmittance ration method), в котором пропускание можно представить как отношение двух пропусканий:
= Л О = ^х/р/Лур
Здесь Txjv = 7’0/р = /0//р, а индексы :<р», «О», и «х» относятся соот-
ветственно к растворителю (или холостой гробе), раствору сравнения и рас гвору определяемого компонента неизвестной концентрации.
Содержание определяемого компонента в растворе сравнения обычно меньше или равно его содержанию в исследуемом растворе (Со < Сх).
Относительную погрешность (%) определения содержания компонента находят из уравнения:
100 ------2------------------s -100
10' х/0 Их/0 + Л0/р) Гх1°
(з.ео)
Функция Fx/0
0,4343 -
—-j---------------- зависит от абсолют-
ю'Лл/0Их/0 + л0/р)
ного значения оптической плотности раствора сравнения Л0/р. Так как последний может быть выбран произвольно, то на графике зависимости Fx/0 = f Их/о) получается ряд кривых, соответствующих выбранному значению АЛ/Р. Некоторые кривые представлены на рис. 3.7, из которого следует:
а) по мере увеличения оптической плотности раствора сравнения по отношению к растворителю АС/р при А 0,4343 на кривых наблюдается исчезновение минимума и функция становится монотонной;
б) по мере увеличения Ло/р уменьшается значение Fx/0 и значение относительной погрешности.
При прочих равных условиях погрешность измерения относительной оптической плотности А' — Ах^ тем меньше, чем больше оптическая плотность раствора сравнения Ао. Для повышения воспроизводимости измерений А' следует стремиться к макси- . мально высокой оптической плотности раствора сравнения и к максимальному сближению значений Ао и А', т. е. содержания Сп и С, компонента в растворах должны быть предельно близки и максимально высеки (но измеримы).
Теоретически наибольшая воспроизводимость определений должна достигаться при Ах о = Ао о = 2.
В ряде случаев определения производят методом полней (двусторонней) дифференциальной фотометрии (см. разд. 7.3), используя раствор сравнения (Со) для определения содержания компонента при С, > Сп и Сх < Со. На рис. 3.8 представлены кривые погрешности двустороннего дифференциального метода для р-яда значений Ао (при S/-)=»]%, sT = 0,01). Номограмма показывает, в каком интервале оптических плотностей (и соответствующем интервале содержаний) в первом приближении можно проводить анализ, сохраняя заданное значение относительной погрешности
86
определения содержания, а также позволяет проводить выбор оптимальных условий фотометрирования растворов.
Последующие работы {109, 124—131], посвященные оптимизации метода отношения пропусканий, показали, что теоретические выводы, вытекающие из уравнения (3.60), на практике не подтверждаются.
Высокая воспроизводимость определений может быть обеспечена только в том случае, если при выборе раствора сравнения (Со, До) учитывается также и чувствительность * измерений фотоколорчметров и спектрофотометров. Этот вопрос явился предметом систематических исследований [127—130] при разработке экспрессных дифференциальных фотометрических методов анализа фосфора и алюминия в минеральных удобрениях, фосфатных растворах, апатитах. Показано |127—130] (рис. 3.9):
1. Метод дифференциального фотометрирования но сравнению с абсолютным не понижает значительно уровень погрешностей Дмпи, а смещает его положение в сторону больших концентраций в пределах соблюдения ochobhoi о закона светопоглощения. Это позволяет повысить воспроизводимость и правильность окончательного результата анализа высоких содержаний определяемого зчемента за счет уменьшения степени разбавления проб.
2. Наибольшая погрешность Фотометрирования наблюдается для концентраций растворов Сх, близких к Со. Поэтому каждому
* Чувствительность фотоколориметра характеризуется отклонением стрелки гальванометра (иг), наблюдаемым при повороте барабана на пт делений шкалы пропускания.
Рис. 3.8. Расчетные кривые погрешности измерения в методе полной (двусторонней) дифференциальной фотометрии [122].
Рис. 3.7 Зависимость функции 0,4343/(10 л*/о (дл/0+ р)] от относи-
тельной оптической плотности Ах^ [122].
87
Рис. 3.9. Зависимость относительной погрешности Д определения концентрации фоаЬора от концентрации раствора сравнения Со и концентрации Р2ОБ при дифференциальном фотометрическом определении фосфора в виде желтой фосфорномолибденованадиевой гете-рополнкислоты [127]:
Ct,, мг Р20ь на 100 мл раствора (абсолютный метод); 1 — 0; 2—1; .3 — 2. « — 4
Примечания1 I. Фотокслориметр ФЭК-56; лампа накаливания; светофильтр N 3 (Аэфф 400 нм) и дополнитечьный светофильтр (кювета I ~ I мм, заполненная раствором 0,02 М KMnOJ, позволивший увеличить крутизну градуировочных кривых; I — 20 мм; F «= 2,2.
2. Д = IOOs^/Д.
значению Со соответствует своя оптимальная рабочая область концентраций АСЖ.
3. С увеличением Со уровень погрешности Амин не снижается, а смещается в сторону больших концентраций; при определенных высоких значениях Со минимальная погрешность Ал1ин возрастает (кривая 3).
4. С ростом Со сужается рабочий интервал концентраций, в котором коэффициент вариации не превышает 2АМИН. Так, из кривой 2 (Со — 1 мг Р2О5 на 100 мл раствора) следует, что этот интервал составляет от 1,7 до 3,7 мг, а при Со = 2 мг Р2ОВ на 100 мл раствора (кривая 3) — примерно 2,5—3,8 мг.
Авторы [127—1301 установили симбатность изменения кривых погрешностей, приведенных на рис. 3.9, и воспроизводимость значений обратного углового коэффициента F = АС/ДА градуировочного графика А' = / (С) при изменении различных условий фотометрирования. Одновременно показано, что выбор оптимальных условий анализа по численному значению и воспроизводимости расчета коэффициента F требует меньше затрат времени, более прост и однозначен (см., например, гл. 11).
Марченко и Рамза [132] для повышения воспроизводимости дифференциальных измерений и экспрессности анализа предложили вместо растворов сравнения применять стеклянные нейтральные светофильтры.
Существенный вклад в теорию оптимизации дифференциального спектрофотометрического метода отношения пропусканий внесли Н. П. Комарь и В. П. Самойлов [109], которые детально теоретически и экспериментально рассмотрели зависимость воспроизводимости от чувствительности и нестабильности работы однолучевых спектрофотометров. Ими предложен ряд уравнений (3.61)—(3.63) для расчета вклада инструментальной погрешности, обусловленной указанными характеристиками спектрофотометров и значениями оптической плотности раствора сравнения, в воспроизводимость дифференциальных измерений;
88
1. Относительная погрешность (%) измерения концентрации (определяемого компонента):
. dCx______________0.43 0п 1 / 1 1 \
х~ сх 7х0(дс/р + дА/С) Ко)
(3.61)
Здесь и ^о/р — соответственно оптическая плотность исследуемого раствора (х) относительно стандартного раствора («0») и стандартного раствора относительно растворителя или холостого опыта (ср»); 0П — цена деления шкалы пропускания, % (см. разд 3.1.3.1); т — чувствительность спектрофотометра; д0 и Ki — коэффициенты нестабильности прибора при настройке шкалы на нулевое (Тх/й — 0) 100%-ное относительное пропускание (7’Л/0= 1), когда на пути светового потока вместо растворителя помещают стандартный раствор с концентрацией Сй.
2. Относительная погрешность (%) измерения оптической плотности Ах/о:
аЛх'О 0,43 6П
Лх/0 д ~ mA Т Лх/0 '' Лх/0 1 х/0
^х/0
(3.62)
3. Минимальная погрешность (%) измерения определяемого содержания:
Дмин = 1(Апт/(тД0) (3.63)
Сопоставляя теоретические уравнения (3.61)—(3.63) и экспериментальные зависимости Ах/Р — Ах/о при разных значениях Ао/Р, Ko/Ki с теоретическими выводами, авторы [109] показали, что для достижения максимальной воспроизводимости в каждом конкретном случае необходимо для выбранных растворов сравнения выяснить Аопг с учетом чувствительности и нестабильности прибора, предварительно определяя коэффициенты нестабильности приемами, описанными в работе [109], и используя их для построения наивероятнейших кривых АЛ/0 — Ах/о или АЛ/Р — АЛ/Р и отыскания Аопт согласно [109].
Развитие ряда вопросов теории и практики дифференциальной фотометрии отражено в работе [131], авторы которой установили зависимость относительной погрешности определения концентрации вещества от оптических плотностей анализируемых растворов и стандартных растворов сравнения в предположении, что стандартные отклонения определения концентрации обоих растворов одного порядка. Авторами оценены оптимальные области соотношения оптических плотностей исследуемых растворов и растворов сравнения для приборов различных типов и марок.
Полная погрешность дифференциального фотометрического анализа (метода отношения пропусканий). Для расчета полной погрешности дифференциального фотометрического анализа, когда
89
Со с Сх и Ах/1 — «! + bjCx, предложено [117, 122] следующее уравнение:
sc
--^-100=. ^х
0,4343
сх—с0 Ч % \
+ ~ с~ sill + ТГ- + ПГ Г100 (3-64) °1^х I
Следует заметить, что второй член правой части уравнения (3.64), содержащий погрешность определения длины кюветы, умножается на фактор (Сх — С^)1СХ. Чем ближе значения Сх и Со друг к другу, тем меньше погрешность, вызванная отклонениями в длине кювет. Если систематически применяют одни и те же кюветы, то погрешность становится незначительной и может не приниматься в расчет.
Из уравнения (3.64) можно также сделать следующие общие заключения по воспроизводимости дифференциальных спектрофотометрических измерений. Погрешность будет меньше, если:
1) больше концентрация раствора сравнения (его относительная оптическая плотность, измеренная по отношению к растворителю, должна быть выше 0,4343);
2) меньше оптическая плотность неизвестного раствора, измеренная по отношению к раствору сравнения, — наиболее благоприятный случай, когда Ах/о = 0, т. е. концентрации неизвестного раствора и раствора сравнения равны;
3) меньше значения стандартных отклонений пропускания sr, наклона градуировочной кривой s*,, коэффициента sC1;
4) меньше расхождение в длинах рабочих кювет;
5) длиннее используемые кюветы;
6) выше концентрации неизвестного раствора;
7) больше наклон градуировочной кривой, т. е. выше коэффициент светопоглощения окрашенного соединения.
Некоторые из этих требований, как и в случае обычной спектрофотометрии, находятся в противоречии. Так, согласно пункту 1, раствор сравнения должен быть как можно более концентрированным. Однако с увеличением концентрации световой сигнал, измеряемый фотоэлементом или фотоумножителем, становится все слабее, и поэтому значение стандартного отклонения пропускания S/- будет возрастать, что противоречит пункту 3.
На практике выбор Со и оптимального интервала значений А' для достижения высокой воспроизводимости требует предварительных исследований [109, 124, 125, 127—131].
Расчет полной погрешности и отдельных составляющих погрешностей дифференциальных спектрофото’ метрических измерений кобальта (14—26 г/л) в виде перхлората, измеренных по отношению к оптимальному раствору сравнения, содержащему 12 г/л Со2+, приведен в табл. 3.6.
90
Таблица 3.6. Погрешности определения кобальта (II)
в виде перхлората методом дифференциальна спектрсфотоиетрнн fl 17] X = 511 нм, / = 1 см.
Сх’ г/л 0.4343ЬГх/( о/ —7^-. % °ТСХ SC С (общая погрешность), %
Ю’ Ах/° (Ах/0 + М/р}
14 0,0432 0,6725 1,15
16 0,0273 0,5885 1,05
18 0,0200 0,5231 0,98
20 0,0159 0 4708 0,92
22 0,0837 0,4279 0,94
24 0,0704 0,3923 0,90
26 0,0448 0,3621 0 84
П р и м е ч а н е. s^jb^ во всех случаях составляет 0,4327%.
Из табл. 3.6 следует, что погрешность, обусловленная членом 1ОО-О,4343«Лх/о/[1СГЛ'":/0 (Ах/0 + Л0/р)], который многие авторы используют как критерий воспроизводимости, существенно уменьшается при применении дифференциальных спектрофотометрических методов. Между тем полная относительная погрешность может оказаться значительно больше из-за погрешностей определения параметров аг и Ьг. Существенное уменьшение экспериментальных погрешностей достигается в том случае, когда для построения градуировочного графика используют большое число точек; например, для интервала, приведенного в табл. 3.6, следует провести около 20 определений.
А. Б. Бланком [120] проведен сопоставительный анализ приближенных выражений для полной относительной погрешности измерения концентрации абсолютным [уравнение (3.65) ] и дифференциальным [уравнение (3.66) ] спектрофсг эмстрическими методами:
Здесь kr — коэффициент пропорциональности между аналитической концентрацией определяемого компонента и равновесной концентрацией окрашенного вещества; k2 — постоянная, характеризующая положительное пли отрица-
91
тельное отклонение данной системы от основного закона светопоглощения;
и п — параметры, характеризующие отклонение функции А = f (С) от линейной; А' = А — Ао и Т' — Т!Тй.
В табл. 3.7 приведены характеристики частных погрешностей уравнений (3.65) и (3.66). Их сопоставление показывает, что не только инструментальная составляющая общей погрешности (№ 1), но и частные погрешности (№Хр 2—4) в случае дифференциального метода ниже, чем при использовании метода непосредственной фотометрии. Частная погрешность (№ 6), обусловленная непостоянством концентрации раствора сравнения, по мнению автора, невелика и в большинстве случаев может не учитываться.
Таким образом, полная погрешность абсолютной спектрофотометрии всегда выше, чем дифференциальной, поэтому применение последней оправдано не только при определении высоких концентраций пробы, но и при использовании медленно образующихся, недостаточно прочных комплексов и в случае низкой воспроизводимости положения кювет при измерениях. При определении низких концентраций компонентов погрешность (№ 5), характеризующая флуктуации общего фона, нередко является определяющей. В этих случаях дифференциальный фотометрический анализ не имеет преимуществ перед абсолютными спектрофотометрическими методами.
Таблица 3.7. Сравнение частных погрешностей методов абсолютной н дифференциальной спектрофотометрии [120]
Колеблющаяся величина Абсолютный метод Дифференциальный метод
1. Оптическая плотность А колеблется за счет инструментального фактора 2. Выход поглощающего соединения (*1, м 3. Окраска поглощающего соединения (е) 4 Толщина поглощающего слоя 5. Общий фон (А'2) 6. Концентрация раствора сравнения (Со) 0,434 „ 10“ Лл т sa-, Ч сп , д Se 8 е sfe, A / «Iй 1^ с? и I X Т | 1 I ° 2 О- - “
V С ) 1 % А Со с
92
3.1.4. ПРАВИЛЬНОСТЬ
ФОТОМЕТРИЧЕСКИХ МЕТОДОВ АНАЛИЗА
Правильность анализа характеризуется значением систематической погрешности [25, 58, 82, 108] *. Результаты анализа правильны, если они не искажены систематической погрешностью, и тем правильнее, чем меньше эта погрешность (см. гл. 9). Доказательство правильности является важной задачей на стадиях разработки, выбора, освоения и эксплуатации методик анализа. Для оценки правильности результатов анализа, в том числе и фотометрического анализа, требуются соответствующие стандартные образцы, стандартные материалы или эталоны строго определенного состава и содержания.
Систематические погрешности подразделяют на положительные, которые приводят к завышению значений аналитического сигнала (Л) и, следовательно, к завышенным значениям определяемых содержаний элемента, и на отрицательные, которые приводят к занижению значений определяемых содержаний элемента. Помимо этого их подразделяют на постоянные (аддитивные), значение которых не связано с абсолютным значением аналитического сигнала, и пропорциональные (мультипликативные), значение которых пропорционально значению аналитического сигнала.
Как и при рассмотрении случайных погрешностей (см. разд. 3.1.3), следует различать инструментальные и аналитические систематические погрешности. Инструментальные систематические погрешности могут быть обусловлены:
а) использованием немонохроматического света;
б) неправильной градуировкой шкалы длин волн спектрофотометра;
в) неправильной градуировкой шкалы пропускания (оптической плотности) прибора;
г) рассеянным излучением (светом), которое может особенно резко сказываться при фотометрировании интенсивно окрашенных растворов (высоких Л) и при длинах волн, экстремальных для данного типа приборов, например при X 220 нм в случае измерений на УФ-спектрофотометрах и Z. = 350-?-400 нм при измерениях на обычных спектрофотометрах. (Подробно этот вопрос рассмотрен в работах [16, 21, 112].)
Для проверки градуировки шкал длин волн и пропускания к настоящему времени предложена серия стандартных материалов сравнения [108, 133—144].
Существенной составляющей, а порой и основной частью систематических погрешностей являются аналитические
* См также ГОСТ 162263—70, ОСТ 6-09-96—79 (Особо чистые вещества для монокристаллов. Обработка результатов анализа).
93
погрешности. Особенно остро этот вопрос стоит при анализе следовых количеств элементов (веществ высокой степени чистоты, микроэлементов и т. д.), когда определяемый элемент, содержащийся в реактивах, или компоненты основы (матрицы), могут приводить к положительным или отрицательным систематическим погрешностям. Причины таких погрешностей и способы их устранения описаны [25]. Приемы, рассмотренные в главах 5 и 7 (разд. 7.2.1 — двухволновая спектрофотометрия и разд. 7.2.2 — производная спектрофотометрия) позволяют во многих случаях ус 1 ранить или резко снизить систематическую погрешность фотометрического анализа. Роль холостого опыта, техника его выполнения приобретают первостепенное значение для достижения правильности результатов анализа следовых количеств элементов и оценки значений Смпн, р и Сн. Этим вопроса™ посвящено значительное число специальных публикаций [21, 79—83, 134, 145].
В широкой практике для установления систематической почетности анализа применяют метод стандартных добавок (см. гл. 9 и [58, 146]) и стандартные образцы (СО) с известным содержанием определяемого элемента.
Следует отметить, что общепринятый прием оценки правильности путем многократнсго анализа стандартного образца и последующего сравнения найденного содержания С с истинным (паспортным) — Сст в определенных случаях может привести к неверным выводам [58]. Полноценная аттестация методики на правильность должна проводиться для нескольких разных содержаний определяемого компонента: по меньшей мере для двух стандартных образцов с меньшим и большим содержанием компонента в сравнении с анализируемыми образцами [58].
3.1.5. СЕЛЕКТИВНОСТЬ
(ПОТОМЕТРИЧЕСКИХ МЕТОДИК АНАЛИЗА
Для оценки селективности фотоме1рической методики анализа обычно исследуют влияние мешающего компонента на значения оптической плотности для трех концентраций стандартных растворов, отвечающих началу, середине и верхнему пределу градуировочной кривой. Соотношения концентраций мешающего и определяемого компонентов (Сь,Р1П/С0Пр) изменяются в широких пределах- от 0,1 до 1000
Строят зависимости А = f (Смгш) при постоянных значениях концентраций определяемого элемента. Предельной минимальной концентрацией мешающего компонента считается концентрация, при которой наблюдается малое, но устойчивое изменение оптической плотности по сравнению с оптической плотностью стандартного раствора в отсутствие примеси. В литературе нет единою мнения о том, какое значение отношения (АДМ)- 100 следует брать в качестве «критического». Разными автопами в качестве критических принимаются значения (ДД/Д)-100 от 1
94
до 10%. Более объективным и рекомендуемым ИЮПАК критерием для оценки мешающего влияния примеси является ее минимальная концентрация, при которой стандартное отклонение оптической плотности раствора, содержащего мешающий компонент, в 2 или 3 раза (2s л или 3$л) превосходит стандартное отклонение метода в отсутствие примеси. Значение рассчитывают статистически из результатов параллельных определений оптической плотности стандартного раствора в отсутствие примеси.
Полученные резучьтаты проверяют также в присутствии двух или более посторонних компонентов, которые содержатся в анализируемых объектах и по отдельнссти не оказывают мешающего влияния, но в совокупности могут влиять на оптическую плотность раствора.
Для устранения мешающего влияния примесей применяют маскирующие реагенты. Эффективное маскирование достигается при отношении условных констант устойчивости комплексов с определяемым и мешающим ионами > 104 и ЕПр™си -*• 0.
Более подробно вопросы селективности и применения маскирующих реагентов рассмотрены в гл 5 и работах [8, 63, 147, 148].
3.2. ВЫБОР ОПТИМАЛЬНЫХ УСЛОВИЙ ФОТОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ
Для достижения высокой воспроизводимости и правильности результатов фотометрического анализа важное значение имеют селективность выбранного реагента и условия проведения фотометрических определений. Некоторые общие рекомендации по основным химическим, оптическим и метрологическим данным, которые необходимы при разработке и выборе фотометрического метода анализа, его оптимизации и представлении материала для публикации, приведены в литературе [8—10, 148—154].
В общем случае методика фотометрического определения должна сопровождаться сведениями о характеристиках применяемых реагентов, метрологических характеристиках полученных результатов и об оптимальных условиях проведения фотометрической реакции.
3.2.1. ВЫБОР РЕАГЕНТА
Реагент для фотометрического определения выбирают, прежде всего, исходя из специфичности взаимодействия анализируемого вещества с определенными аналитическими группами органических реагентов. Например, для никеля специфическими реагентами являются органические вещества, содержащие оксимную группу=N—ОН, для кобальта — реагенты, содержащие в ортоположении группы — N=O и —ОН, для меди—реагенты, содержащие тио- и аминогруппы одновременно и т. д. Необходимо
95
также, чтобы окрашенные соединения удовлетворяли требованиям устойчивости и постоянства состава.
Для оценки специфических реагентов используют критерии, сформулированные А. К- Бабко [351. Лучшим реагентом при прочих равных условиях считают такой, который при образовании окрашенного соединения обеспечивает:
1. Наибольшее смещение максимума поглощения ДА — Ак — — AR (где Ак и AR—длины волн максимального поглощения комплекса и реагента), характеризующее контрастность реакции. Контрастность фотометрической реакции считается достаточно высокой, если обеспечивается ДА = Ак — AR 100 нм.
2. Наибольшие абсолютное и относительное изменения молярного коэффициента светопоглощения: Де = ек — eR и Д = = B|;/eR.
В тех случаях, когда молярные коэффициенты светопоглощения комплекса и реагента неизвестны, реагенты выбирают по наибольшей разности между суммарной оптической плотностью раствора Агч и оптической плотностью реагента: ДА = Асм — AR.
3. Наибольшую разность в значениях pH при образовании окрашенных форм комплекса и реагента: ДрН = pH,. — pHR.
4. Наибольший интервал значений pH, в котором соблюдается постоянство оптической плотности раствора.
Практическим критерием чувствительности реагента служит угол наклона прямой, характеризующей зависимость оптической плотности (или разности оптических плотностей) от концентрации окрашенного вещества. Графическая зависимость определяется при длине волны, где значение А (или ДА) является максимальным. Чем больше угол наклона (или тангенс угла наклона) этой прямой, тем чувствительнее реагент.
Аналогично по тем же критериям можно оценивать селективные и групповые фотометрические реагенты, но их практическое применение для определения отдельных элементов возможно только в отсутствие мешающих компонентов либо в специальных условиях, при которых мешающее влияние сопутствующих элементов проявляется в незначительной степени.
В тех случаях, когда фотометрическая реакция характеризуется невысокой контрастностью и при выбранной длине волны наблюдается светопоглощение не только анализируемого комплекса, но и фотометрического реагента, находят разность оптических плотностей анализируемого комплекса и чистого реагента при той же концентрации, что и в анализируемом растворе.
Однако эту разность оптической плотности можно отождествлять с оптической плотностью раствора светопоглощающего комплекса только в тех случаях, когда светопоглощение комплекса и реагента обусловлено разными хромофорными группами, что на практике встречается сравнительно редко. Если светопоглощение комплекса и реагента обусловлено одной и той же функциональной группой, то этот прием можно использовать только 96
в условиях большого избытка реагента, когда концентрацией реагента, затраченной на комплексообразование, можно пренебречь. Если концентрации реагента и определяемого иона близки, то, как справедливо отмечают М. 3. Ямпольский [591 и С. Б. Саввин [155], измерение оптической плотности раствора относительно раствора реагента той же концентрации может привести к серьезным погрешностям, особенно при определении состава соединений спектрофотометрическими методами. Для определения АД, обусловленной поглощением анализируемого комплекса, из суммарной оптической плотности раствора смеси комплекса (Дсм) необходимо вычитать оптическую плотность только той части реагента, которая осталась не связанной в комплекс (АД = = Дем - ДГ).з
Значение Дц36 непосредственно измерить невозможно, но его находят косвенным путем. В работе [155] для этого строят вспомогательную графическую зависимость оптической плотности (Д') раствора реагента (той же общей концентрации, что и анализируемый раствор) от pH, а затем, измерив оптическую плотность комплекса в условиях полного связывания реагента (Дмш^), вычисляют оптическую плотность, обусловленную поглощением только анализируемого комплекса:
// п А' \
ДД=(Дсы-Д')/р——- (3.67)
I Члмип /
Здесь q = Cr/Cm — соотношение исходных концентраций реагента и металла в растворе.
3.2.2. ОПРЕДЕЛЕНИЕ ОПТИМАЛЬНЫХ УСЛОВИЙ ФОТОМЕТРИЧЕСКОГО АНАЛИЗА
При выборе оптимальных условий проведения фотометрических определений в аналитической практике используют два методологических подхода-
первый — преимущественно при разработке методов анализа с применением новых реагентов — включает подробное исследование цветной реакции с определением оптических и термодинамических характеристик образующегося светопоглощающего соединения;
второй — наиболее широко распространенный при использовании уже известных реагентов — не включает ни изучение стехиометрии фотометрической реакции, ни определение термодинамических характеристик светопоглощающего соединения.
При разработке методики с применением новых фотометрических реагентов, прежде всего, устанавливают благоприятные условия образования светопоглощающего комплекса и определяют оптические характеристики растворов комплекса и реагента (спектры поглощения, Хыакс, молярные коэффициенты светопо-
4 М. И. Булатов и др. ,97
глощения при Хмакг). С этой целью исследуют влияние pH раствора, избытка реагента, времени, температуры и последовательности добавления реактивов на светопоглошение фотометрируемого раствора. Затем находят оптимальные значения навески пробы и конечного объема фотометрируемого раствора, пределы концентраций, где соблюдается линейная зависимость А = f (С) при фотометрических определениях, устанавливают стехиометрический состав образующегося светопоглощающего соединения и его константу устойчивости. Для экспериментального определения термодинамических и оптических характеристик светопоглощающих соединений широко применяют методы, изложенные в гл. 8.
Указанные выше зависимости и количественные характеристики служат основой для прогнозирования * оптимальных условий и разработки методики фотометрического определения веществ.
Установление оптимальных условий при фотометрических определениях можно проводить в такой последовательности.
1. Определение области (или длины волны) максимального (оптимального) поглощения света раствором исследуемого соединения.
В диапазоне длин волн 210—750 нм или 360—750 нм с помощью спектрофотометров снимают спектры поглощения растворов определяемого компонента с реагентом при соотношениях CR/CM, равных 1, 10 и 0,1 при различных значениях pH, несколько превышающих pKQ фотометрического реагента. Оптическую плотность растворов измеряют относительно раствсра реагента или чистого растворителя; в последнем случае снимают спектр и раствора реагента. По спектрам поглощения растворов находят длину волны или участок спектра, где наблюдается максимальное значение оптической плотности Лмакс (или разнести оптических плотностей ДЛмакс—в случае светопоглощающего реагента). Все последующие измерения оптической плотности растворов производят при найденном значении длины волны максимального (оптимального) светопоглощения.
Количественный выход светопоглощающего соединения может гарантироваться лишь в присутствии достаточнсго избытка реагента, который обычно превосходит в 2—5 раз оптимальный избыток для верхнего предела градуировочной кривой. Необходимый избыток реагента зависит от относительной устойчивости фотометрируемого комплекса и мешающего влияния сопутствующих компонентов. Его рассчитывают теоретически или определяют экспериментально з предварительных опытах (см разд. 1.4, 8.1.3,
* При наличии совокупных данных для рассматриваемых систем при оптимизации фотометрического анализа успешно применяют планирование экспериментов, прогнозирование оптимальных условий и обработку результатов с использованием ЭВМ [149—153].
98
Рис. 3.10. Зависимость оптической плотности раствора (или разности оптических плотностей ДЛ) от pH.
Рис. 3.11. Графическое определение оптимальной концентрации реагента:
7 — при образовании устойчивого (прочного) окрашенного соединения; 2 — при образовании малопрочного окрашенного соединения.
8.1.4). Если раствор реагента сам поглощает свет в условиях фотометрирования, то значительный избыток реагента крайне нежелателен из-за высокой оптической плотности раствора холостой пробы.
2. Определение оптимального значения pH раствора.
При установлении области pH раствора, наиболее благоприятной для образования светопоглощающего соединения, измеряют оптическую плотность исследуемого раствора при разных значениях pH, начиная с pH < рАа фотометрического реагента, и строят графическую зависимость А (АЛ) = f (pH) (рис. 3.10). По графику определяют интервал оптимальных значений pH раствора АрН = рН( — рН.г, где наблюдается наибольшее и практически постоянное значение оптической плотности раствора.
Если окрашенное соединение извлекается неводным растворителем, то необходимо определить интервал значений pH максимальной степени экстракции анализируемого соединения. При неизвестной оптимальной концентрации избыток реагента в этих опытах берется 5—10-кратным по отношению к количеству определяемого вещества.
3. Определение количества реагента, необходимого для полного связывания анализируемого иона в окрашенное соединение. Требуемое количество реагента рассчитывают теоретически (см. разд. 1.4) или определяют экспериментально по максимальному выходу продукта реакции, т. е. по максимальному светопоглощению. Для этого приготавливают серию растворов с постоянным содержанием определяемого иона *, но с различным и все увеличивающимся содержанием реагента. Затем измеряют оптические плотности растворов и строят график зависимости А (или АА) от концентрации реагента Cr (рис. 3.11).
Для этих опытов желательно брать соли кислот, анионы которых не вызывают дополнительного комплексообразования.
4* <Ю
При образовании устойчивого окрашенного соединения на кривой А (АЛ) = f (CR) наблюдается резкий излом в так называемой «точке насыщения», который и определяет минимальное количество реагента, необходимое для максимального выхода продукта реакции (кривая 1). Оптимальное количество реагента в этом случае должно превышать стехиометрическое на 30—50%. При образовании малопрочного комплекса кривая «насыщения» не имеет резкого излома (кривая 2). В этом стучае необходимо установить наименьшее количество реагента, при котором практически прекращается увеличение А (или АЛ в случае окрашенною реагента). Это количество и принимается за оптимальное.
4. Определение* оптимальной навески и конечного объема фотометрируемого раствора [156]. Обозначив наименьшее содержание определяемого иона М в анализируемом образце через р (%), навеску образца — q (г), а конечный объем раствора — V (мл), получим следующее выражение для наименьшей концентрации определяемого иона М (г/л):
смин = ЯР 1000/(100V) = 10<zp/V (3.68)
Если окрашенное соединение образуется по реакции тМ + 4- nR M^(Rn и реагент тоже окрашен, то наименьшая концентрация (г/л) определяемого иона, согласно основному закону светопоглощения, выражается уравнением:
Смин — ^мкнт^м/{1 (f к — Иеи)} (3.69)
Здесь Лмин — наименьшее значение оптической плотности, измеренное относительно раствора реагента; т и п — стехиометрические коэффициенты, характеризующее состав комплекса МтРп\ Вм — относительная атомная масса определяемого иона М.
Решая уравнения (3.68) и (3.69) относительно неизвестных q и V, получим:
Я, V = Аминт£м/{10р/ (ек — neR)} (3.70)
Вычислив правую часть равенства (3.70) при оптимальном значении оптической плотности Лопт, выбирают разумные значения для q и V. При очень малых отношениях q/V (<10-3) нужные значения навески и объема получают разбавлением раствора пробы в калиброванной посуде. Если отношение q/V больше (^0,1), то чувствительность метода не обеспечивает определение столь малых значений процентного содержания определяемого иона. В этом случае следует либо применить предварительное концентрирование определяемого иона, либо сменить метод анализа.
* Определение носит приближенный характер. Более точный расчет с использованием приемов математической статистики описай А. Б. Бланком [67].
100
5. Выяснение зависимости светопоглощения раствора от времени и температуры, фотометрический анализ обычно проводят при одинаковой комнатной температуре, так как изменение температуры раствора во многих случаях вызывает соответствующее изменение светопоглощения. Если даже небольшое изменение температуры (на 2—3 °C) приводит к значительному изменению оптической плотности фотометрируемого раствора, то измерения последней производят после предварительного термостатирования.
Для выяснения устойчивости светопоглощения фотометрируемого соединения во времени измеряют оптическую плотность раствора через некоторые промежутки времени и строят график зависимости оптической плотности от времени. Для последующей работы выбирают такой интервал времени, в течение которого максимальное значение оптической плотности раствора сохраняется неизменным или меняется незначительно. Кроме того, на протяжении всех измерений строго выдерживают время между измерением оптической плотности и приготовлением фотометрируемого раствора.
3.2.3. ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ ФОТОМЕТРИЧЕСКИХ ОПРЕДЕЛЕНИЙ
В 1978 г. [71 ИЮПАК опубликовал рекомендации по представлению материалов при печатании статей по фотометрическим методам анализа растворов. В развернутом виде они представлены в литературе 18—10, 148—150]. В работе 18] разработана схема для систематического изучения аналитических реакций, применяемых в фотометрическом анализе. Эта схема в виде взаимосвязанных таблиц сочетает в себе представление в компактном виде рекомендаций ИЮПАК и объективную оценку реагентов, применяемых для фотометрических определений.
При публикации методик фотометрического определения веществ рекомендуется [7 ] представлять:
1. Стехиометрический состав светопоглощающего комплекса, значение константы устойчивости и условия ее определения (pH, ионная сила, температура, состав буферного раствора).
2. Спектры поглощения растворов светопоглощающего соединения и реагента, аналитическую длину волны и обоснование ее выбора, указание на проверку правильности шкалы длин волн.
3. Сведения о влиянии pH раствора при выбранной длине волны (или участке спектра) с указанием буферной системы и ионной силы раствора.
4. Сведения о влиянии относительного содержания избытка фотометрического реагента на выход светопоглощающего соединения с указанием отношения CR/CM, отвечающего максимальному значению оптической плотности раствора при постоянной концентрации См.
101
5. Сведения об устойчивости светопоглощающего соединения во времени при выбранной для фотометрирования длине волны и операциях, необходимых для достижения максимального и постоянного значения оптической плотности раствора (например, выдержка в темноте, отсутствие растворенного кислорода и т. п.).
6. Сведения о влиянии порядка добавления реактивов на скорость реакции и выход светопоглощающего соединения с указанием времени достижения максимальной и постоянной оптической плотности раствора при измерениях относительно раствора холостой пробы.
7. Сведения о влиянии температуры на скорость образования и выход светопоглощающего соединения с указанием устойчивости растворов при изменении температуры в интервале 15—30 °C; рекомендуемая температура для пре ведения определений.
8. Сведения о влиянии мешающих компонентов и рекомендуемые способы устранения их влияния.
9. Основные метрологические характеристики с указанием методик построения, статистической обработки и проверки правильности градуировочной кривой:
а) коэффициент чувствительности (см. разд. 3.1.1.1 и 9.3);
б) характеристика воспроизводимости результатов (относительное стандартное отклонение), получаемых по полной аналитической методике в присутствии необходимых маскирующих реагентов или после предварительного отделения (см. разд. 3.1.3 и 9.3);
в) предел обнаружения (или предел определения) (см. разд. 3.1.2.1 и 9.3);
г) правильность результатов, подтверждаемая анализом стандартных образцов или другими независимыми методами (см. разд. 3.1.4 и 9.3);
д) нижняя граница определяемых содержаний (см. разд. 3.1.1.2, 3.1.2.2 и 9.3).
10. Селективность методики определения, оцениваемая отношением концентраций мешающего компонента к определяемому (Смещ/Соир), при котором наблюдается определенное устойчивое отклонение оптической плотности раствора (ДЛ- 100/Л) (см. разд. 3.1.5 и гл. 5).
11. Характеристики реагентов:
а) чистота и устойчивость применяемого фотометрического реагента; рекомендуемые способы очистки реагента;
б) содержание основного вещества и примесей;
в) растворитель для реагента и обоснование его выбора;
г) чистота и устойчивость других применяемых реактивов.
Ниже в компактном виде приведена форма представления необходимых данных и результатов фотометрических определений в соответствии с рекомендациями ИЮПАК [7].
102
Характеристики свойств реагента *
реагент (рациональное название) Растворитель (среда, устойчивость в растворителе) Марка (чистота реагента, необходимые дополнительные операции по очистке реагента)
Характеристики реакции
Спектральные характеристики (спектр поглощения чистого реагента и соединения определяемого элемента с реагентом; молярные коэффициенты светопоглощения при аналитической длине волны)
Значение pH Буферная система Содержание реагента Ионная сила раствора Устойчивость окрашенного соединения Экстр аги-руемость
Характеристики методики
Область рабочих концентраций
Уравнение градуировочного графика
Коэффициент чувствительности (при / = 1 см) нлн условные характеристики е^, л/(моль- см) и а, мл/(мкг- см) и др.
Селективность, методы устранения мешающих иоиов
Воспроизводимость,** правильность
Границы; ^мин, Р»
* Форма таблиц взята из работ [8, 9] и несколько скорректирована с учетом рекомендаций ИЮПАК
** Общие рекомендации по метрологической оценке результатов анализа см. в разд. 9.6.
3.2.4. СРАВНЕНИЕ И ВЫБОР
ФОТОМЕТРИЧЕСКИХ МЕТОДИК ОПРЕДЕЛЕНИЯ
Для выбора методики фотометрического определения используют различные критерии: рабочий интервал определяемых концентраций, природу матрицы анализируемого образца, чувствительность, воспроизводимость, селективность, практически определяемый предел определения. Обычно при выборе методики отдают предпочтение основному определяющему фактору (критерию). Например, нижней границе определяемых содержаний при определении следов элементов, селективности — при анализе сложных многокомпонентных систем и т. д. При отсутствии (незначимости) систематических погрешностей сравниваемых методик объективной оценкой предпочтительности одной из них при прочих равных условиях является значение стандартного отклонения результатов (см. разд. 9.5).
103
Г лава 4
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
И РАСТВОРИТЕЛИ
И ИХ ПРИМЕНЕНИЕ В ЭКСТРАКЦИИ
4.1. ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
Органические реагенты давно вошли в практику аналитической химии для обнаружения и определения неорганических веществ. Исследованы и рекомендованы для аналитического применения сотни самых разнообразных органических соединений. В настоящее время применение органических реагентов в анализе неорганических веществ является одним из важнейших и стремительно развивающихся направлений аналитической химии. Этой проблеме посвящен ряд монографий отечественных и зарубежных авторов 138, 49, 63, 66, 155, 157—164]. Научный Совет АН СССР по аналитической химии и ГЕОХИ АН СССР начали издание многотомной серии монографий «Аналитические реагенты», в которых будут обобщены и систематизированы сведения о наиболее важных органических реагентах. В этой серии уже вышли монографии «Триоксифлуороны» [158], «Оксимы» [159], «Оксихинолин» [160] «Гетероциклические азотсодержащие азосоединения» [161 ]. На каждый элемент составляется рациональный ассортимент аналитических реагентов. В монографии 3. Хольцбехера и др. [63] объединены основные аспекты теории и практики применения органических реагентов в неорганическом анализе. Обширные сведения о применении органических реагентов в виде разнолигандных комплексов содержатся в монографии А. Т. Пилипенко и М. М. Тананайко [38]. Значительная часть монографии Сендела и Ониши [25] также посвящена органическим реагентам и их применению в фотометрических методах анализа следов элементов. Наиболее полные сведения об органических реагентах, применяемых в неорганическом анализе, можно найти в справочниках [165, 166], в которых описано более 5000 реагентов.
Органические реагенты подразделяют на специфические, избирательные и групповые. К избирательным реагентам относят такие, которые реагируют лишь с определенной группой металлов или образуемые комплексы которых с различными металлами отличаются особыми физическими или химическими свойствами. Избирательный реагент называют специфическим, если он взаимодействует только с одним ионом (элементом). Подавляющее большинство органических реагентов является групповыми, которые взаимодействуют со многими элементами периодической системы.
104
Повышения избирательности фотометрических определений можно достигать различными путями. Радикальным способом является направленный синтез избирательных органических реагентов. Однако на практике из-за отсутствия общей теории синтеза избирательных реагентов реализовать этот способ для большинства элементов пока еще не удалось. Поэтому повышения избирательности фотометрических определений добиваются либо созданием ассортимента реагентов, взаимно дополняющих друг друга по избирательности [155], либо применением групповых реагентов в сочетании с маскирующими комплексантами и тщательным выбором условий их аналитического применения. Преимущество применения в фотометрическом анализе ограниченного числа групповых реагентов состоит не только в техническом упрощении анализа при определении многих элементов, но и в том, что благодаря их глубокому и всестороннему изучению можно выбрать наиболее обоснованную и рациональную схему определения каждого элемента даже в сложных объектах [155].
Перспективными в этом отношении являются экстракционнофотометрические методы, особенно с применением разнолигандных комплексов.
Среди важнейших групповых органических реагентов, которые получили широкое распространение для определения и индивидуальных элементов, следует назвать такие, как дитизон, 8-ок-сихинолин, диэтилдитиокарбаминат натрия и свинца, пиридилазонафтол, пиридилазорезорцин, ксиленоловый оранжевый, арсеназо III, метилтимоловый синий, сульфохлорфенол С, дианти-пирилметан, оксифлуороны и некоторые другие. Применение некоторых из этих групповых органических реагентов рассмотрено ниже. Примеры прогнозирования оптимальных условий аналитического применения групповых органических реагентов для экстракционно-фотометрического определения отдельных элементов или их разделенья приведены в гл. 10.
В фотометрическом анализе применяют водные, неводные и смешанные растворы органических реагентов [167]. Для приготовления неводных растворов органических реагентов используют как полярные, так и неполярные органические растворители в зависимости от химической природы реагента и степени гидрофобности образуемого им комплексного соединения.
Дитизон (дифенилтиокарбазон) .NH-NH—/ \
S=C й .
\ , является одним из наиболее важных фо-
N =N —у
Тометрических реагентов. Он обладает слабыми кислотными свойствами и в воде при pH <7 не растворяется. В щелочных растворах дитизон находится в форме аниона HDJ и имеет оранжевый
105
Рис 4 1. Распределение дитизона в зависимости от pH водной фазы: 1 — CCI,; г — CHCI
Рис. 4.2. Распределение диэ тилдитиокарбаминовой кислоты (НДДК) в зависимости от pH водной фазы.
цвет. Растворы дитизона в полярных и неполярных органических растворителях имеют зеленую окраску различного оттенка.
Распределение дитизона между водной фазой и органическими растворителями зависит от pH среды (рис. 4.1). В ка°естве растворителей дитизона используют преимущественно хлороформ и тетрахлорид углерода. Растворимость дитизона в хлороформе (0,04 моль/л) выше, чем в тетрахлорчде углерода (0,003 моль/л), но последний менее летуч, чем хлороформ, имеет большую плотность, способствующую более быстрому разделению фаз после встряхивания с водным раствором. Тетрахлорид углерода менее растворим в воде (0,08 %), чем хлороформ (0,8%), но токсичен. Растворы дитизона (0,01 %) в тетрахлориде углерода можно хранить в течение нескольких недель.
о. Q
Пиридилазонафтол (ПАН) N=N—обра-
I
ОН
зует водные растворы в кислых (pH < 2) и щелочных (pH > 11) средах. При pH = 3-?11 в воде образует коллоидные растворы. Реагент ПАН обычно применяют в виде раствора в метиловом или
этиловом спирте.
Диэтилдитио к арба минат натрия (Na-ДДК)
N—С — групповой хелатообразующий реагент, хорошо
QHZ xSNa растворимый в воде.
Диэтилдитиокарбаминовая кислота в кислых растворах разлагается на диэтиламин и сероуглерод, поэтому при хранении растворы ha-ДДК слегка подщелачивают (примерно до pH = 9). Распределение реагента между водной и органической фазами зависит от pH среды (рис. 4.2). При pH < 4 реагент в виде кис
106
лоты полностью переходит в органическую фазу, а при pH 8 практически количественно остается в водном слое.
Диэтилдитиокарбаминат свинца в хлороформе [РЬ (ДДК)2 в СНС13 ] является избирательным реагентом на Си (II), Hg (II), Ag (I), Bi (III) и T1 (III).
8-0 кси хинолин (оксин)
|| — групповой хела-
ОН тообразующий реагент, обладающий амфотерными свойствами. В щелочных растворах он находится в виде оксихинолинат-аниона, а в кислых — в виде катиона оксихинолиния.
8-Оксихинолин растворяется в различных полярных и неполярных органических растворителях. Распределение реагента между водной и несмешивающейся органической фазами зависит от pH среды (рис. 4.3).
СН3—C=NOH
Диметилглиоксим | — широко извест-
СН3—C=NOH F
ный реагент, представляет собой бесцветные кристаллы, малорастворимые в воде. Реагент обладает слабыми кислотными свойствами, растворяется в растворах щелочей при pH 12, а также в этиловом спирте, диэтиловом эфире.
Применение смешанных водно-спиртовых растворов позволяет значительно повысить устойчивость многих фотометрических реагентов. Так, реагенты трифенилметанового ряда (ксиленоловый оранжевый, хромазурол S и др.) сравнительно хорошо растворяются в воде, однако, часто используют их водно-этанольные растворы, так как добавление спирта значительно увеличивает
срок их хранения.
Многие комплексные соединения, малорастворимые в воде из-за их неполярного характера, хорошо растворяются в несмешиваю-щихся с водой неполярных органических растворителях, образуя истинные растворы. Основную группу таких соединений составляют внутрикомплексные (хелатные) соединения катионов металлов с различными хелатообразующими органическими реагентами. Среди этих соединений следует назвать дитизонаты, диэтилдитио-
Рис. 4.3. Распределение 8-оксихинолина в зависимости от pH водной фазы.
107
карбаминаты, оксихинолинаты, купферронаты, нитрозонафтьлаты, пиридилазонафтолаты, для растворения которых чаще всего используют хлороформ или тетрахлорид углерода.
Элементы, взаимодействующие с дитизоном, представлены в табл. 4.1 [66]. В кислой среде в избытке реагента все указанные элементы образуют с анионом HDz первичные дитизонаты (кето-дитизонаты). Некоторые из этих элементов в щелочной среде и при недостатке реагента образуют с анионом Dg* вторичные дитизонаты (енольные дитизонаты). В фотометрическом анализе чаще используют первичные дитизонаты, которые интенсивнее окрашены и более растворимы в органических растворителях, чем вторичные.
Растворимость первичных дитизонатов металлов в хлороформе, как и самого дитизона, примерно на порядок выше, чем в тетрахлориде углерода, и составляет 10-3 моль/л. Образование дитизонатов отдельных металлов и их переход в органическую фазу зависят от pH водного раствора и константы устойчивости дити-зоната.
Диэтилдитиокарбаминаты металлов плохо растворяются в воде, но хорошо растворимы в таких органических растворителях, как хлороформ, тетрахлорид углерода, эфир, ацетон и другие. Плохо растворяются даже в органических растворителях диэтилдитиокарбаминаты Nb, Ru, Rh, Os, Ir и Pt. Вместе с тем, как сообщают Боде, Туше и Вархаузен [168 ], ряд металлов образуют группу водорастворимых карбаминатов с производными диэтилдитиокар-баминовой кислоты.
Окраска растворов диэтилдитиокарбаминатов металлов в органических растворителях не интенсивна, поэтому применение их в фотометрическом анализе ограничено методами, не трубующими высокой чувствительности определения. Элементы, реагирующие с диэтилдитиокарбаминатом натрия, представлены в табл. 4.2 [66 ].
8-оксихинолин реагирует с катионами металлов, образуя внут-рикомплексные соединения, как правило, хорошо растворимые
Таблица 4.1. Элементы, реагирующие с дитизоном
Окрашенные дитизонаты элементов обведены жирной чертой.
108
Таблица 4.2. Элементы, реагирующие с диэтилдитиокарбамииатом натрия
Штриховкой отмечены элементы, образующие окрашенные соединения.
в неполярных органических растворителях. В табл. 4.3 [66] элементы, образующие оксихинолинаты, взяты в рамку. Условия образования оксихинолинатов, как и других хелатных соединений, зависят от значения pH водного раствора и константы устойчивости оксихинолината металла.
В кислой среде 8-оксихинолин с некоторыми металлами (Fe, Мо, Сг и др.) образует растворимые в воде катионные комплексы, которые можно использовать для фотометрического определения этих металлов [66].
В табл. 4.4 в рамку заключены те металлы, которые образуют окрашенные соединения с пиридилазонафтолом [66, 169]. Значительная часть из них образует электронейтральные внутрикомп-лексные соединения, растворимые в неполярных органических растворителях.
Окрашенные растворы большинства из этих соединений используют для экстракционно-фотометрического определения металлов [38, 66, 167, 170—172]. Присутствие полярных растворителей (спирт, диоксан) в водном растворе препятствует образованию комплексов металлов с реагентом ПАН.
Условия образования хелатных соединений металлов с различными органическими реагентами и возможности их фотометрического определения рассмотрены в монографии [167].
Таблица 4.3. Металлы, образующие 8-оксихинолинаты, которые можно экстрагировать неполярными растворителями
Al Si Р
Ga Ge As
In Sn Sb
TI Tb Bi
о
s
Se
Те
Po
F cT
Br
I
He Ne Ar Kr Xe Bn
В С N
00
1.09
Таблица 4.4. Металлы, образующие окрашенные комплексы с 1-(2-пиридилазо)-нафтолом-2
4.2. ОРГАНИЧЕСКИЕ РАСТВОРИТЕЛИ
Органические растворители в фотометрическом анализе применяют для увеличения растворимости некоторых реагентов и окрашенных соединений, для повышения чувствительности и точности фотометрических определений, а также для концентрирования микроколичеств веществ, подлежащих количественному определению. Очень широкое распространение получили органические растворители для целей разделения анализируемых ионов и непосредственного фотометрического анализа в органической фазе.
Для разделения ионов применяют неполярные, т. е. не смешивающиеся с водой, органические растворители (бензол, хлороформ, тетрахлорид углерода и др ), с помощью которых комплексные соединения экстрагируются (извлекаются) из водной фазы в слой органического растворителя
Полярные органические растворители, хорошо смешивающиеся с водой (ацетон, диоксан, этиловый спирт и др.), обычно используют для повышения относительной устойчивости окрашенных соединений, а следовательно, для повышения чувствительности и точности фотометрических определений.
Влияние растворителей, содержащих гидрофильные группы (ацетон, диоксан, низшие спирты), на растворимость некоторых комплексов в водных системах легко понять, если учесть способность этих групп вести себя в качестве доноров электронов и замещать молекулы воды в координационной сфере комплексов. Этим, по-видимому, и обьясняется повышение растворимости некоторых соединений, например диалкилдитиокарбаминатов меди в водно-этанольных растворах, так как молекула растворителя имеет большее химическое сходство с периферийными частями комплекса, чем молекула воды [49].
С другой стороны, добавление полярного растворителя к воде уменьшает диэлектрическую проницаемость среды и способствует относительному упрочнению растворенных комплексов. Поэтому НО
хорошо смешивающиеся с водой органические растворители используют главным образом для фотометрического определения малоустойчивых окрашенных соединений, которые в водных растворах в значительной степени диссоциированы на ионы.
Неводные растворители уменьшают степень диссоциации окрашенных соединений и создают благоприятные условия для использования малопрочных соединений в фотометрическом анализе. Чувствительность и точность фотометрических определений в полярных растворителях, как правило, повышается по сравнению с водными растворами, где значительная часть определяемого иона остается не связанной в окрашенное соединение. Наиболее удобен для этой цели ацетон, который смешивается с водой в любых соотношениях. Диссоциация большинства электролитов в ацетоне очень сильно уменьшается. Например, фотометрическое определение малоустойчивого синего роданидного комплекса кобальта обычно производят в среде 50%-ного ацетона, так как в водной среде это определение практически провести невозможно. Применение 90%-ного этилового спирта повышает устойчивость роданидного комплекса железа в 250 раз. Прибавление ацетона или этилового спирта оказывается полезным для определения и некоторых других металлов в виде роданидных комплексов [173].
При спектрофотометрических определениях в ультрафиолетовой области спектра необходимо учитывать: возможность присутствия посторонних примесей в растворе, которые будут поглощать свет и мешать анализу; влияние растворителей на характер спектра растворенного вещества; поглощение света самими растворителями. Ниже приведены некоторые растворители, используемые в спектрофотометрии:
Растворитель Нижний предел пропускания света в ультрафиолетовой области, нм * 1 Растворитель Нижний предел пропускания света в ультрафиолетовой области, нм *
Амилацетат 260 Изооктан 210
Ацетон 330 Изопропиловый спирт 210
Aiетонитрил 212 Метиловый спирт 210
Бензол 280 Метилпиклогексан 210
Бутанол 220 Пиридин 300
Бутилацетат 260 Серная кислота (96%) 210
Вода 210 Тетрахлорэтилен 295
Гексан 210 Тол 5 ол 285
Гептан 210 Хлороформ 240
Глицерин 230 Циклогексан 210
п- Диоксан 220 Тетрахлорид углерода 260
Дихлорметан 233 Этилацетат 260
1 2- Дихлорэтан 235 Этиловый спирт 220
Диэтиловый эфир 220
* При пользовании кюветой длиною 1 см.
111
4.3. ЭКСТРАКЦИЯ ОКРАШЕННЫХ СОЕДИНЕНИИ
Экстракция комплексных соединений органическими растворителями является одним из наиболее распространенных методов разделения анализируемых элементов и широко используется в экстракционно-фотометрических методах для определения веществ непосредственно в органической фазе.
Во многих случаях экстракция позволяет значительно повысить чувствительность фотометрических определений, так как дает возможность сконцентрировать малые количества анализируемого вещества в органической фазе. Теории экстракции и ее практическому применению в аналитической химии посвящено большое число работ. Наиболее полно эти вопросы рассмотрены в монографиях [172, 174—180].
Большое влияние на распределение металлов в экстракционных системах оказывает комплексообразование в водной фазе. Вводя в водную фазу различные комплексующие и маскирующие реагенты, можно сознательно управлять распределением веществ и добиться их эффективного разделения.
В общем случае описание количественного распределения вещества в экстракционной системе сравнительно сложно [167], так как требуется знание состава и устойчивости всех комплексов, образующихся как в водной, так и в органической фазах. Однако очень часто, особенно при экстракции хелатов неполярными растворителями, задача существенно упрощается, ибо во многих случаях в широком интервале концентраций образуется единственный экстрагируемый комплекс, а экстракционный реагент практически полностью находится в органической фазе.
4.3.1. КОЛИЧЕСТВЕННЫЕ ХАРАКТЕРИСТИКИ ЭКСТРАКЦИОННЫХ РАВНОВЕСИЙ
Для количественной оценки экстракционного равновесия используют следующие характеристики* [И].
Кп т lMRn]0/[MRn] — константа распределения — отношение концентрации экстрагируемой формы вещества (MRn) в органической фазе к его концентрации в той же самой форме в водной фазе в условиях равновесия. Для каждого несме-шивающегося с водой органического растворителя константа распределения является величиной постоянной, при нормальном давлении зависит только от температуры и ионной силы раствора. Если обе жидкие фазы являются насыщенными относительно твер
* Здесь и далее приведены концентрационные константы, которые от термодинамических (K°D, К^кс и ДР-) отличаются множителем, содержащим коэффициенты активности. 1'
Обозначения, отвечающие равновесным концентрациям в органической фазе, отмечены индексом «о», в отличие от обозначений, относящихся к водной фазе.
112
дой фазы и равновесие достигнуто, то константа распределения равна отношению растворимостей распределяющегося вещества в органической и водной фазах:
AD = S0-S (4.1)
Dc = 1о/ [См ] — коэффициент распределе-
ния * (отношение распределения, коэффициент экстракции) — отношение общей аналитической концентрации вещества в органической фазе к его общей аналитической концентрации в водной фазе в условиях равновесия. Поскольку общая аналитическая концентрация является суммой различных ионных форм, соотношение между которыми в водной фазе зависит от pH и концентрации комплексующих реагентов, коэффициент распределения нс является постоянной величиной, а зависит от условий эксперимента и константы распределения.
£)m = QOi'Q — массовый коэ ф ф ициент распределения — отношение количества распределяемого вещеова (ммоль) в органической фазе к количеству этого же вещества в водной фазе в условиях равновесия.
Из сопоставления выражений Dc и D,„ нетрудно заметить, что
Dc = DmW/V и Dm = DcV/W (4.2)
Здесь И и U7 — объемы водной и органической фаз.
Если, как это часто бывает при экстракции, V/W = г ~ 1, то величины Dc и Dm становятся тождественными. При оценке полноты экстракции из выражения массового коэффициента распределения можно найти мольную долю (или процент) вещества, оставшегося в водной фазе после однократной экстракции:
а= (С?исх — C?o)/(Qhcx) = QKQo + Q) = 1 (Р,п 4- 1) (4.3)
При d-кратной экстракции
a=(l + Dm)-d (4.4)
Вычитая из единицы часть вещества, оставшуюся после экстракции (а), получим степень (фактор) извлечения-
Е = Qo 'Qncx = Dc, (DC + W/V) = Dm '(1 4 Dm) (4.5)
Степень извлечения представляет собой долю или процент от исходного количества вещества в объеме W водной фазы, экстрагируемую объемом Е органической фазы при заданных условиях. Если водный раствор обрабатывают d последовательными порциями экстрагента и каждый раз отношение объемов органической и водной фаз V/W = г, то степень извлечения составляет:
Hd=l ИГ=1 + (4.6)
£ = D/(14-D) (4.7)
* По номенклатурным правилам ИЮПАК [И] к этому коэффициенту можно добавлять термин «концентрационный» и нижний индекс «С», когда возникает Двусмысленность или неясность.
1В
Степень извлечения зависит от тех же факторов, что и коэффициент распределения, и кроме того, от отношения объемов органической и водной фаз.
= (<2м1о/ЗмПоу(<2м1псх/СмПисх) -фактор обогащения — отношение количеств двух разделяемых веществ в фазе экстрагента, отнесенное к исходному отношению их количеств до разделения. Фактор обогащения представляет собою коэффициент, на который нужно умножить исходное отношение количеств двух разделяемых веществ, чтобы получить отношение их количеств в органической фазе после разделения. Следовательно
= £м1/^м1Т (4-8)
5мьмп = 1 - (1 + /{1 - (’ + '%!)'"} (4-9)
При d = 1 и г = 1
SMj/Mir = (! + DMn)/{DMn (! + £%,)} (41°)
•Smi/Мц = — фактор разделения —
отношение коэффициентов распределения двух разделяемых элементов Mj и Мп.
Кекс — константа экстракции — константа равновесия реакции экстракции.
pHj/2 — значение pH раствора, при котором 50% исходного количества вещества экстрагируется в органическую фазу (D = 1).
Коэффициент распределения является наиболее распространенной характеристикой экстракционных процессов, особенно в условиях конкурирующих равновесий в водной фазе. Эта характеристика, зависящая от условий проведения экстракции и взаимосвязанная с константами распределения и экстракции, позволяет получить объективную количественную информацию в реальных условиях анализа. Во многих случаях коэффициенты распределения определяют экспериментально, однако при определенных условиях их можно прогнозировать и теоретически. Для этого необходимо знать состав экстрагирующихся соединений, их константы распределения и экстракции, а также иметь сведения о конкурирующих реакциях и образующихся соединениях в водном растворе.
4.3.2. РАСПРЕДЕЛЕНИЕ ЭКСТРАКЦИОННОГО РЕАГЕНТА
В качестве экстракционного реагента обычно используют многоосновные органические кислоты HmR, анионы которых с катионом М образуют экстрагируемые комплексы. Распределение экстракционного реагента между водной и органической фазами зависит от pH раствора, так как в органический растворитель переходят не все существующие в водной фазе формы экстракционного
114
реагента, а только некоторые из них. Долевое же распределение последних в водном растворе зависит от значения pH. В зависимости от pH водного раствора изменяется соотношение ионных форм, а следовательно, и распределение экстракционного реагента. Если экстракционный реагент HmR образует экстрагирующиеся формы * Hm+1R, HmR, Hm_jR, ..., R, то его коэффициент распределения выражается уравнением:
г) fHm+iR]o + [HmR]o + • • + [R]o _ [Cr]o
R lHm+1R] + [HmR] +••• + [Rj - [CR] (4.11)
Обозначив через , KD , ..., Kb константы распре-
деления экстрагирующихся форм и введя для них значения мольных долей aHfR — [H,R]/[Cr], уравнение (4.11) преобразуем в более простое выражение:
DR = .RaH ,R + ^DH RaH R 4 Ь ^DRaR = 11 I til tit
m+l
— ^DH.RaH.R (4-12)
0 1 1
/m+l
Здесь aH R = P(H [H3O+]‘/ pj' [HjO'1]1— мольная доля протонного ' I и
комплекса HjR от всех форм экстракционного реагента в водной фазе, включая и протонированные частицы Hm^jR; рф — полная концентрационная константа устойчивости протонного комплекса H,R.
Равновесную концентрацию молекулярной формы HmR экстракционного реагента в органической фазе можно найти из совместного решения уравнений (4.11) и (4.12), приняв отношение V/W = г. Исходное количество реагента, содержащееся в одной из фаз, после распределения между двумя фазами составит:
Сксх = [Cr]o V + [Cr] W = [Cr] (DV + №) (4.13)
Если реагент до экстракции был в водной фазе, то из уравнения (4.13) получим:
[Cr] = Сисх/И7 = [Cr] (1 + rD) или [Cr] = [Cr]/(1 + rD) (4.14)
Здесь [Cr]—общая избыточная концентрация экстракционного реагента в исходном растворе до распределения.
Когда применяется раствор реагента в органическом растворителе, его равновесная концентрация в водной фазе составит:
[Cr] = [Cr] о(1 + rD) (4.15)
Согласно уравнению (4.12) получим:
[ЧЛ = [Cr] aHmR^DHmR = [Cr] aHmR^DHmR/(1 + rD) (4.16)
* Знаки зарядов опущены.
115
или
[HmR]o — [Cr] ttHmRr^DHmR/(l + (4.17)
В тех случаях, когда заряженные формы экстракционного реагента не экстрагируются (Kd„ d = 0), можно пользоваться
I Hm+1^ J более простыми выражениями:
1НЛ, = [CrP (’ +гР) и LHmR]o = [Ср]гР/(1 +гР) (4.18)
Из уравнений (4.14) и (4.15) можно найти равновесную концентрацию аниона реагента в водной фазе
fRm'] = [CR]“Rm-/(1+rn) (4.19)
или когда реагент находится в органическом растворителе:
[R-] = [CR]aRm_r,(l+rD) (4.20)
При равном объеме фаз (г = 1) уравнения (4.13)—(4.18) упрощаются, а уравнения (4.16) и (4.19) становятся тождественными уравнениям (4.17) и (4.20) соответственно:
[HmR]o = [C'R]DR/(l+PR) (4.21)
(4.22)
Если общая аналитическая концентрация реагента значительно превышает концентрацию экстрагируемого металла (CR См), то при вычислениях в первом приближении можно принять, что [C'R ] л; CR. Когда концентрация металла в водной фазе соизмерима с концентрацией реагента, их равновесные концентрации при г = 1 могут быть вычислены совместным решением уравнений материального баланса:
(n \
S < 1СкГ 4+ «₽* 1CrP
(N p \
1 + Г ₽Г [Cr]' «r + S Pj- IL]‘ + Kdk^ [CRf a" (4.23) 1 1 /
Здесь и P^ — полные концентрационные константы устойчивости комплексов MR; и МЬ;; [L] — равновесная концентрация конкурирующего ком" плекса (маскирующего реагента) в водной фазе.
4.3.3. РАСПРЕДЕЛЕНИЕ МЕТАЛЛОВ
Распределение металлов, как и экстракционного реагента, между двумя несмешивающимися жидкостями происходит в соответствии с законом распределения Нернста, согласно которому отношение активностей или (при постоянной ионной силе) равновес-
ие
пых концентраций определенной формы вещества, распределяющегося между водным раствором и несмешивающнмся органическим растворителем, при постоянной температуре является величиной постоянной. Допустим, что катион М экстрагируется из водного раствора органическим растворителем в виде комплексных соединений MR, MR», ..., MR, с анионом экстракционного реа!ента HmR, который тоже переходит в органическую фазу в виде соединений HR, H2R, ..., H,R. В водном растворе происходит ступенчатое комплексообразование соединений MR, MR2, ..., MRn. Кроме того, экстрагируемые катионы М участвуют в реакциях конкурирующего комплексообразования с ОН-ионами и маскирующим реагентом L, образуя равновесные системы М (ОН);-и MLP. Тогда выражение для коэффициента распределения можно записать в такой форме-
Dm =
([MR] + [MR2] + + [MRn])o
[M] + V (MRf] + £ [M(OH),-]+ £ [MLi] 1 1 1
[См]о [См]
(4-24)
Выражая равновесные концентрации частиц в уравнении (4 24) через их полные константы устойчивости |3^, pf, .... —для
частиц MR,; |3°н, Р?н, .... Р°н—для частиц М (ОН);; Р?, Р?, Рр —для частиц MLf и учитывая распределение экстрагирующихся соединений, получим ,
₽i [R1 + ₽* (R’2^MP + • • + ₽«
Dm -----------7l-------------------------------- (4 25)
i + JX iRi' + S₽?H + E ₽hLi’
1 1
или в более удобной форме *
= Kr>MRaMR Т KDMRiaKR2 Н “ KDMRnaMRn =
— X^Dmr aMR; (4 26)
Здесь aMR = р* [R]1 /( 1 + £ ₽? [Rf + £ ррн [OH-f + £ ₽b(L]' ) ‘ / \ 1 1 1 /
относительное содержание (мольная доля) экстрагируемой формы от суммарного содержания всех форм экстрагируемого элемента в водном растворе в условия^ равновесия; [R] и [L] — равновесные концентрации стехиометрических форм экстракционного и маскирующего реагентов в водном растворе.
Мольная доля aMR_, а следовательно, и коэффициент распределения £>м зависят от pH и равновесных концентраций [R]
* Если экстрагируемые частицы MR; в органической фазе участвуют в побочных равновесиях и [^Ri]o/[DMRt.]o = aMR,- (0)' то в уравнении (4.26) множитель «МР> необходимо заменить множителем aMR./aftiRi( ,•
117
и IL] в водном растворе. Если исходная аналитическая концентрация экстракционного реагента до распределения значительно превышает аналитическую концентрацию металла, т. е. когда CR См или гС% (о) См, то ее можно принять за равновесную и, учитывая распределение между фазами, по уравнениям (4.19) или (4.20) вычислить необходимую для расчетов равновесную концентрацию стехиометрической формы (аниона) экстракционного реагента в водном растворе. При небольшом избытке реагента необходимо учитывать его расход на образование экстрагируемого комплекса, и тогда [CR ] л; CR — пСм или [CR ] « CR (о) — nC^lr.
В большинстве случаев экстрагируются только некоторые формы комплексов и реагента (обычно незаряженные частицы), а в водном растворе протекают побочные реакции не более двух типов. Поэтому уравнения (4.24) и (4.25) значительно упрощаются. Но при любых условиях, как это следует из уравнений (4.25) и (4.26), коэффициент распределения Е>м всегда будет меньше значения константы распределения наиболее экстрагирующейся формы комплекса MR,. Лишь при самых благоприятных условиях, когда в водном растворе образуется только экстрагируемая форма комплекса, коэффициент распределения может достигать значения константы распределения *. Чтобы обеспечить наибольшее значение коэффициента распределения, необходимо выбрать такие значения pH раствора, концентраций экстракционного и маскирующего реагентов, которые позволяли бы достичь наибольшего выхода экстрагирующегося комплекса (наибольшего значения мольной доли aMR. экстрагирующегося комплекса). При ступенчатом комплексообразовании, когда экстрагируется промежуточный комплекс MRf, наибольшее его извлечение из водного раствора при прочих равных условиях достигается при оптимальной концентрации реагента [R] = (/<,/<,+1)_1/2> обеспечивающей максимальный выход экстрагирующейся формы комплекса MRf (1 < i < п — 1).
Растворимость в органическом растворителе, а следовательно, и экстрагируемость можно увеличить путем образования смешанных комплексов, когда во внутреннюю координационную сферу металла входят молекулы незаряженных органических оснований типа пиридина. При образовании смешанных комплексов с лигандами, концентрации которых можно сознательно изменять, значение коэффициента распределения экстрагирующегося комплекса MRnLg удобно прогнозировать с помощью уравнения, учитывающего все равновесия в водной фазе:
ГГ 1 ₽ЛМ8 L [R]"[L]9
Гу [bMJo __ /д Q7V
М = ---------------1----------- (-27)
Z ,[RHL]l+ 2Хн[ОН-]‘
1=0 '=0 1
* Если экстрагируемые частицы в органической фазе участвуют в побочных реакциях, вследствие которых образуются новые устойчивые соединения, то достигаемый в этом случае коэффициент распределения может превысить константу распределения.
118
Здесь п, q — стехиометрические коэффициенты экстрагирующегося смешанного комплекса MRnLg; 0gn — константа устойчивости экстрагируемого комплекса; Pi, j—константы устойчивости комплексов, образующихся в водном растворе.
Конечная концентрация СЕОВ извлекаемого элемента, остающегося в водном растворе после экстрагирования, зависит от коэффициента распределения и соотношения объема водной и органической фаз:
Скон = СНач {Wl{W + VPM)}d (4.28)
Необходимое число d последовательных экстракций, обеспечивающих заданную полноту извлечения экстрагируемого элемента, можно найти с помощью уравнений (4.4), (4.6) или (4.28):
d = (1g Снач - 1g CK0H)/{lg (W + VDm) — 1g №} (4.29)
4.3.4. ЭНСТРАНЦИЯ ХЕЛАТОВ
Хелатными соединениями называют циклические («клешневидные») комплексные соединения ионов металлов с органическими реагентами, образующиеся путем присоединения к иону металла не менее двух донорных атомов молекулы ре агента Такие соединения чаще всего получаются вследствие одновременного замещения протонов в молекуле реагента и образования координационной связи с другими донорными атомами. Хелатные соединения могут быть нейтральными или заряженными, а их внутренняя координационная сфера либо полностью занята анионами (молекулами) реагента, либо включает еще и другие донорноактивные вещества, в их внутренней координационной сфере молекулы воды могут замещаться незаряженными молекулами реагента.
Большинство хелатных соединений хорошо экстрагируется неполярными органическими растворителями либо в виде незаряженных молекул (внутренних * комплексных солей), либо в виде ионных ассоциатов, представляющих собой продукт ассоциации заряженного хелата с другими ионами. Хелатообразующие реагенты, как правило, нс избирательны, поэтому выбор условий экстракции имеет большое значение. Основными приемами, которые применяются для обеспечения условий успешною выделения или разделения, являются изменение pH раствора и введение маскирующих реагентов.
4.3.4.1. Экстракция внутрикомплексных соединений (незаряженных хелатов)
Экстракция катиона М”+ экстракционным реагентом HR в виде внутри комплексного соединения MRn, растворимого в органическом растворителе, описывается реакцией **
Mn+ + nHRo MRno + «Н+ (4.30)
* По мнению А. А. Гринберга [181], Н. Н. Желиговской и И. И. Черняева [182], к внутр и комплексным соединениям следует относить только незаряженные внутренние комплексные соли — неэлектролиты, хотя в литературе по экстракции внутрикомплексные соединения часто отождествляют с хелат-ньми 1761’ ВКЛ1°чая в них и заряженные формы, как это предложено Леем
** Если в состав экстрагируемого соединения входит q незаряженных молекул реагента HR, то в знаменателе уравнения (4.31) появляется множитель IHR]’.
119
которая характеризуется константой равновесия
«экс = [MR„]0 [H+]n/([M"+] [HR]") (4.31)
Если правую часть уравнения (4.31) выразить через константу устойчивости экстрагируемого комплекса MRn, константу ионизации экстракционного реагента K<?HR и константы распределения KoMR и ^dhr’ то ПОЛУЧИМ новое уравнение, связывающее эти характеристики с КЭкс:
/<SHC = PMR/oMRn/(KoHR/^HR)" (4.32)
Как видно из уравнения (4.32), значение константы экстракции находится в прямой зависимости от константы устойчивости и константы распределения экстрагируемого хелата MRn и в обратной зависимости от константы протонирования Khr и константы распределения Kdhr экстракционного реагента. Если известны ЛомР н Adhr для сравниваемых органических растворителей, то по значению КВ1!С для одного растворителя можно теоретически рассчитать значение Кэкс для другого. Коэффициент распределения £)м при экстракции единственного комплекса MRn определяется соотношением:
DK = [MR„]O/([M"+] + [MRn]) = KoMRn/(l + [M"+]/[MRn]) (4.33)
Вводя константу экстракции КВ1!С в уравнение (4.33), получим зависимость величин £>м и /Свкс:
1/*>М = + [н+]"/(Яэкс [HR]")
(4.34)
Во многих случаях, когда DK , уравнение (4.34) можно
упростить, исключив из его правой части слагаемое
Тогда получим
*
MRn
Ям = КЭкс[НИ]"/[н+1" (4.35)
или в логарифмической форме:
lg^M= 1g Кэкс + «pH + п lg [HR]o 4- п 1g/н+ (4.36)*
Из уравнения (4.36) видно, что при тех условиях, где справедливо это уравнение (KoMR у^м 50), при постоянной ионной силе раствора и неизменной концентрации [Н+ ] или [HR 1о между lg DK и lg [HR ]0 (или pH) существует линейная зависимость. В этой области повышение pH на единицу увеличивает коэффициент распределения в 1О'! раз для «-зарядного катиона металла, образующего экстрагируемый комплекс MRn. Коэффициент рас
* На практике последним слагаемым (коэффициентом активности) этого уравнения часто пренебрегают.
120
пределения внутрикомплексного соединения зависит главным образом от константы экстракции, а также от концентрации хелатообразующего реагента и pH водного раствора. В идеальном случае, как это следует из уравнения (4.36), для данного растворителя при постоянной концентрации реагента в органической фазе и неизменной ионной силе водного раствора наклон зависимости 1g Z)M от pH равен п, а сама линия, выражающая эту зависимость, пересекает ось абсцисс (lg £>м = 0) при значении pH, равном pHi/2:
pHJ/£ = - (1/n) 1g Кэкс - lg [HR]O - lg fH+ (4.37)
Отрезок, отсекаемый на оси ординат этой прямой, в логарифмических единицах численно равен свободному члену уравнения (4.36): 1g Л8КС + п lg ([HR ]ofH+)- Исследование логарифмических зависимостей lg£>M = f(pH) или lg £>м = f (lg [HR ]0) наряду с другими методами [176] широко используется для определения состава экстрагируемого комплекса и количественных характеристик экстракционного равновесия.
Если водный раствор экстракционного реагента при проведении экстракции добавляется непосредственно в водную фазу, где образуется малорастворимое соединение MRn, то константа экстракции будет выражаться соотношением:
Кэкс = [MRn]0/([M"+] [R-]") = ₽MRn^oMRn « So/^sMRn (4.38) Здесь So — растворимость хелата MRn в органическом растворителе. Взаимосвязь величин Z)M и Кэкс примет несколько отличное от уравнения (4.34) выражение
1/DM = i/KoMRn + 1/(КЭЙС [R-1”) (4.39)
которое при £>m//(Omr <: 1/50 упрощается и принимает такой вид: Dm=K0„c[R-]" (4.40)
Здесь Z)M так же, как и в уравнении (4.35), зависит от Кэкс, pH и аналитической концентрации реагента CR в водной фазе (поскольку [R- ] зависит от pH и [CR ]). Правда, значения Какс в литературе обычно не приводятся, поскольку в конечном счете уравнения (4.39) и (4.40) тождественны уравнениям (4.33) и (4.34). Однако рассмотренные выше количественные зависимости экстракционных равновесий, представленные уравнениями (4.30)—(4.40), справедливы только в том случае, когда ни один из компонентов равновесия не участвует в побочных реакциях с другими веществами, что в реальных условиях встречается чрезвычайно редко. Наиболее часто встречающимися побочными равновесиями в водном растворе являются равновесия протонизации экстракционного реагента, гидроксокомплексообразования экстрагируемого катиона металла и образование низших (положительно заряженных) комплексов с экстракционным реагентом. Особенно
121
Рис. 4.4. Зависимссть lg DM от pH водной фазы при экстракции хелатного соединения MR„.
сильное влияние может оказывать присутствие постороннего комплек-санта (маскирующего реагента) или неудачно выбранного буферного раствора, обладающего резко выраженными комплексующими свойствами.
Даже в отсутствие госторонних конкурирующих комплексан-тов максимальное извлечение экстрагируемого металла наблюдается в определенной ограниченной области pH раств.ора. Типичная зависимость 1g от pH раствора при экстракции хелатов металлов показана на рис. 4.4, из которого видно, что Ом увеличивается по мере повышения pH до некоторого определенного интервала, где он достигает максимального значения, а затем начинает падать вследствие уменьшения выхода электронейтраль-ного хелата и образования анионных комплексов. Область pH, где значение 1g £>м максимально и не зависит от pH (Д£>м/ДрН = = 0), характеризуется максимальным выходом внутрикомплекс-ного соединения MRn. При этих условиях значение Ом может достигать Кл.,.. , если в водном растворе распределяемый катион металла практически полностью находится в экстрагируемой форме MRn. Чаще, однако, KD не достигается, а постоянное значение Z)M обеспечивается адекватностью (уравновешиванием) влияния уменьшения концентрации ионов Н+ и уменьшения условной константы экстракции из-за гидроксокомплексообразова-ния катиона в водной фазе.
При изменении этого оптимального значения pH раствора выход экстрагируемого хелатного комплекса } меньшается и коэффициент распределения падает.
В присутствии посторонних комплексантов плато на кривой распределения, как правило, становится значительно короче, а иногда и вообще отсутствует, переходя в максимум. Если в водном растворе в значительных количествах образуются неэкстра-гируемые комплексы экстрагируемого металла с посторонним комплексантом, то наибольшее значение Z)M, достигаемое в этих условиях, значительно меньше, чем в отсутствие постороннего комплексанта. Поэтому для количественной оценки экстракционных равновесий в реальных условиях целесообразно использовать условные константы экстракции X,KC, которые являются производными концентрационных констант и одновременно учитывают влияние побочных равновесий, существующих в экстракционных системах. Равновесия, сосуществую
122
щие при экстракции хелата MRn в органическую фазу, можно изобразить следующей схемой:
HR органическая фаза MRn
KnHP vi It KnMRn
HR, НгО <=> (R~, h+); MRn «=> (M"+, £ MRf, £MLf, £М(ОН)г, R“) водная фаза
Следовательно, если в водном растворе существуют побочные равновесия, то, как это видно из представленной схемы, выраже цие условной константы экстракции хелата MR„ органическим растворителем примет такой вид *:
Сс = [MR„]0[h+P/([Cm] [HR]") = Кэксам (4.41) Здесь
I/ п—1 / р \
ам = [Л1"+1/[СЙ] = 1Д 1 + £ ₽? [R’f + £ ₽?Н [°Н’Г + £ ₽• [L]1’ ) — мольная доля незакомплексованного катиона М от суммарного содержания всех неэкстрагпруемых его форм в водном растворе.
Коэффициент распределения £>м при экстракции единственного комплекса MRn определяется соотношением:
/ / п—1 i р \
DK = [MRn]J [MRn] + £ [MRf] + £ [M (OH)£] + £ [ML,-] =
I \ 0 1 1 /
= ]MR„]()/([MR„] + = KoMRn/(i + (Cm]/[MR„1). (4.42)
Вводя условную константу экстракции Л”кс в уравнение (4.42), получим взаимосвязь величин DK и Кэкс-
= l/KoMRn + (Н+]"/(К'экс [HR]") (4.43)
В тех случаях, когда Kd^/Dm 50, уравнение (4.43) упрощается
dm = ^Kc[HR1o/Ih+]" (4-44)
или в логарифмической форме:
1g Г>м = >g К;кс + n pH + и lg [HR]о + п 1g /н+ (4.45)
Из уравнения (4.45) следует, что при Z)M при постоянной равновесной концентрации экстракционного реагента в органической фазе между 1g £>м и pH раствора существует линейная зависимость при неизменном состоянии побочных равновесий и ионной силы в водной фазе. Если при изменении pH раствора состояние равновесий конкурирующего комплексообразования изменяется и оказывает существенное влияние на условную константу экстракции Лэке, то линейная зависимость будет наблюдаться только в координатах 1g (DM/ccM) = f (pH).
* Если экстрагируемая частица MRn в органической фазе участвует в побочных равновесиях и [MRn]o/[CM]0 = ам (о), то К'экс = (о).
123
Процесс экстракции и экстракционные реагенты часто характеризуют величиной pHi/2 (рН50) — значением pH водного раствора, при котором экстрагируется 50% вещества при равных объемах равновесных фаз (DM = 1). При этих условиях из уравнения (4.45) находим:
рН1/а = - (1/„) 1g К;кс - lg [HR]0 - lg fH+ (4.46)
Рассмотрение уравнений (4.45) и (4.46) показывает, что с увеличением К'экс и pMR значение коэффициента распределения возрастает, a pHi/2—уменьшается. При высоких значениях Юкс, находящейся в прямой зависимости от |3MR и /CoMR , значение рН1(2 может быть очень низким и это благоприятно сказывается на экстракции многозарядных катионов, так как с уменьшением pH водной фазы повышается селективность разделения. В том же направлении оказывает влияние уменьшение констант протонирования и распределения экстракционного реагента. Коэффициент распределения Очи, следовательно, степень извлечения Е будут повышаться, если в органической фазе будет расти равновесная концентрация экстракционного реагента ([HR]0). Изменяя степень протекания побочных конкурирующих реакций в водной фазе путем введения различных комплексующих и маскирующих реагентов и учитывая взаимное влияние экстракционных характеристик, с помощью уравнений (4.41)—(4.46) можно прогнозировать изменение коэффициента распределения DM в нужном направлении, обеспечивая полноту извлечения или маскирования ионов металлов. Таким путем с помощью условных констант экстракции можно сознательно управлять распределением веществ и прогнозировать условия, необходимые для их эффективного разделения *.
4.3.4.2. Экстракция координационно ненасыщенных и заряженных хелатов
Координационно ненасыщенные хелаты. Хелатные соединения, содержащие в своем соыаве координационно связанные молекулы воды, условно называют «координационно ненасыщенными» [176] В таких соединениях в процессе экстрагирования молекулы воды замещаются молекулами полярного naiтворнтеля или нейтралнными молекутачи экстракционного pear енга, например ** Со(О\)2(НОх)2, Cd(O\)2(HOx)2, Г\т (Ох)2(НОх)2, Sc(Ox)2HOx, UO2A2HA и др. Если при экстракции неполярными растворителями координационно связанная вода в молекуле нейтрального хелата остается незамещенной (или не блокированной) коордипа-
* При этом следует иметь в виду, что описанные выше критерии справедливы, только в условиях достигнутого равновесия К сожа тению, в ряде случаев, когда экстракция протекает очень медленно, возможны различные отклонения от тер-модинам гческих прогнозов. Качественное влияние различных факторов на скорость достижения экстракционного равновесия можно оценить с помощью предложенного Ирвингом [167] уравнения. rMR = k [М] [R ]" = k [М] X х 1[hR]o/(KDhr lH+]^R)r.
** НОх — 8-оксихинолин, НА — ацетилацетон.
124
ционно активными молекулами какого-либо органофильного вещества, то такие (хелаты, как правило, почти не экстрагируются. Поэтому при экстракции координационно ненасыщенных хелатов результаты экстрагирования при прочих равных условиях могут существенно зависеть также от присутствияросторонних координационно активных веществ, природы растворителя и природы экстракционного реагента.
Равновесия, возникающие при экстракции хелатов, содержащих в своем составе нейтральные молекулы экстракционного реагента, в основном аналогичны тем, которые имеют место и при экстракции насыщенных внутри комплексных соединений, но при расчетах равновесий, безусловно, необходимо учитывать состав экстрагируемого соединения. Так, если образуется комплекс MRn (HR)g по реакции
м"+ + (л + q) HR0 <=> [MR„ (HR) + пН+
то выражение условной константы экстракции примет такой вид:
С = Л (HR)J0 [н+]'7([с;] [HR]"+?) = Аэксам (4.47)
Зависимость между коэффициентом распределения Z)M и К.'экс
определится выражением:
1/DM = 1/KDm + [h+F/(k;rc [HR]g+9 (4.48)
При 50
DM=«;KC[HR]0"+7[H+]'1 (4.49)
или в логарифмической форме:
Ig^M^ |g«3Kc + '1PH + (n + '7),g[HR]o + nlgfH+ (4.50)
pHi^ в этом случае будет определяться уравнением рН1/2==—(1/n) 1gк;кс —(1+<?/п) lg[HR]o— ]g/H+ (4.51)
Из уравнений (4.47)—(4.51) видно, что при изменении равновесной концентрации экстракционного реагента в органической фазе величины Кэкс, Пм и рН>/2 при экстракции соединений типа MRn(HR)g будут изменяться более резко при прочих равных условиях по сравнению с теми же экстракционными характеристиками для внутри комплексных соединений MRn (когда Z)M < Лдм).
Заряженные хелаты. Образование заряженных хелатов катионного или анионного типов в аналитической практике встречается часто. Такне заряженные комплексы органическими растворителями экстрагируются, как правило, только в виде нейтральных тройных комплексных соединении или ионных ассоциатов с присутствующими в водном растворе анионами нлн катионами соответственно. Анионные хелаты хорошо экстрагируются органическими растворителями в виде ионных ассоциатов с кр1пными гидрофобными органическими катионами. Этот прием экстракции анионных комплексов получил довольно широкое распространение [176]. Некоторые общие вопросы экстракции анионных хелатов рассмотрены В. И. Кузнецовым [183]. Экстракция хелатов катионного типа изучена недостаточно, хотя ее значение в аналитической практике и особенно в экстракционно-фотометрическом анализе представляется не менее важным, чем экстракция анионных хелатов, В работах Ю, А. Золотова с сотрудниками [176, 184]
125
охарактеризованы условия образования и экстракции хелатов катионного типа. Главным нз условий извлечения таких комплексов в органическую фазу авторы указанных работ считают присутствие крупных гидрофобных анионов, образующих с положительно заряженным хелатом экстрагирующуюся ионную пару [176, 183, 184], 'отя в других работах [185—190] показано, что некоторые катионные хелаты хорошо экстрагируются неполярными органическими растворителями и в присутствии таких простых анионов, как СГ, Вг~, Г, SCN~, NO^, CHSCCO~ и др Возможно, это связано с тем, что в последнем случае экстрагируются не ионные ассоциаты, а нейтральные тройные комплексы Однако в обоих случаях экстракционные характеристики в значительной степени зависят от химической природы аниона-партнера и даже от его концентрации [176, 185]. Поэтому теоретическое прогнозирование оптимальных условий выделения или разделения таких соединений можно осуществить только при наличии надежных сведений о константах экстракции, которые найдены экспериментально при условиях, близких к прогнозируемым по составу солевого фона. Если таких данны> в литературе найти не удается, то их необходимо получать экспериментально
В общем виде равновесие экстракции смешанного нейтрального комплекса (или ассоциата) можно представить таким уравнением:
М2+ + (n + q) H,RO + у НгО 4- тА~ <-=>
<=* [М (H^R), ( Н ,R), (ОН), Ат^-пх~У-т + (пх + у) н+ (4.52)
Методом сдвига равновесия из логарифмических зависимостей 1g Ом = f (pH), lg DM = f (lg IHjR ]o) и lg (DM/aM) = f (lg [CA 1) в области, где KD /1)^ 50, находят угловые коэффициенты
линейных участков этих зависимостей: а = пх -\- у, b — п + q и с = т. Если а = 1, то состав экстрагируемого комплекса и стехиометрические коэффициенты становятся известными [185, 189]: п — 1; х = 1; у = 0; q = b — 1; т = с. При а > 1 для определения всех стехиометрических коэффициентов необходимо использовать дополнительные независимые методы определения состава экстрагируемого соединения, которые изложены в литературе [43, 48, 56, 176]. При известном составе экстрагируемого соединения методом сдвига равновесия по любой из логарифмических зависимостей нетрудно найти величины lg KjKC и 1g Ла1;г, которые можно использовать в дальнейшем для прогнозирования условий, необходимых для проведения экстракции. Однако в большинстве случаев общее уравнение (4.52) сводится к более простому:
Мг+ + (п + q) HRO + mA- =₽=> [MR„ (HR), Amf-m~n + «Н+ (4.53)
Условная константа экстракции принимает такой вид:
*жс = [MR„ (HR), Ат1 и [Н+Г/([СМ] [HR]"+’ [СА Г) = Яэксама£ (4.54) Взаимосвязь величин DM и /ф,кс выражается соотношением:
1/Ом = 1/ЯОм + 1Н+Р/(К;КС [HR]"+* [С А]т) (4.55)
В тех случаях, когда 5> 50, уравнение (4.55) упро-
щается
Ом = к;кс [HR]o+9 [СаГДн+Р (4.56)
126
или в логарифмической форме:
lgDM= 'ёК;кс + (п + q) lg [HR]O + т lg [СА] + п pH -f- п lgfH+ (4 57)
Из этого уравнения при 1g Ом = 0 можно найти рН>/2:
рНх/> = —(1/л) lgX;KC — (1 + <?/«) lg [HR]0- (mln) lg [CA] — lg/H+ (4 58)
4.3.4.3. Равновесие обмена при экстракции хелатов
Обменные экстракционные реакции в аналитической практике используют главным образом для повышения избирательности экстрагирования или определения [176]. В качестве экстракционных реагентов часто применяют растворы хелатных соединений в органических растворителях, например диэтилдитиокар-баминат свинца в хлороформе [167, 176], некоторые дитизонаты [191] дитиофосфаты [192], 8-меркаптохинолинаты [193] и др. При перемешивании фаз определяемый (или выделяемый) ион М?+, находящийся в водном растворе, вытесняет ион Мп+ в молекуле хелатного соединения MnRm и переходит в органическую фазу в виде соединения М^п:
+ nMj,Rm(oj <—> mM[Rn(o)-|-пМц+ (4.59)
Условная константа равновесия этой реакции обмена выражается соотношением *:
= [M.Rn]” [СМп]7(1МпктК ) = я;™(4.60)
Поскольку /<8кс = Уравнение (4.60)
можно записать и в такой форме:
^об = (4’61)
Таким образом, полнота обмена зависит от соотношения условных констант экстракции индивидуальных хелатных соединений, от их констант устойчивости и констант распределения. Условия хорошего обмена качественно можно выразить такими неравенствами:
К™ » *экс (4.62)
или
(₽MiR/DMlRnaMi)m (ЧлЛомиктампГ (4’63)
Если обменивающиеся ионы имеют равный заряд и состав экстрагируемых соединений одинаков, уравнения (4.60)—(4.63) упрощаются
коб=я;КС1/к;КСп С1.64)
* Выражение (4.60) является справедливым и в том случае, когда в реакциях обмена участвуют смешанные комплексы типа MRA или MRn (HR)g.
127
и условия хорошего обмена:
» ^СИ 0.65)
₽MTRnKD!A R “мЛРМцй/ом R “Mlr (4.66)
X lb 1 IX lit li-
Однако при оценке конкретных возможностей применения реакций обмена целесообразнее руководствоваться количественными критериями, которые нетрудно получить из анализа выражения условной константы обменного равновесия (4.60). Если реакция обмена (4.59) протекает при отношении Смпит (0)/Смт = g, то при количественном обмене Мх на Мп в условиях равновесия соотношения концентраций будут следующими: [MnRm]o = Cmhr ,— — (пт) [MjRn ]0; [Смп] » (п/т) CMl; [MnRTO]„/[CMn| = (mgr— — n)'rn; IMrRn ]o 0,99CMi/t и [Смг] < 0,01СЧ][. Вводя эти условия в выражение условной константы обменного равновесия (4.60), получим:
^экс/^эксц^ пп- 102m/{rm-" (mgr-n)n\ (4.67)
Даже при большой разнице в величинах KiKCj и /GKC необходимо, чтобы gr > 1. При т =- п = 1 и минимальном избытке реагента-хелата в 10% (gr = 1,1) для полного протекания реакции об лена необходимо, чтобы обеспечивалось соотношение Xskcj/Xjkcjj Ю3 (при равном объеме фаз). Если gr = 2, то при тех же условиях необходимо, чтобы Лэкс1/Кэкс11 1 • Ю2 и т. д. Если константы экстракции обменивающихся ионов близки по величине, то необходимая разница в их условных константах экстракции создается путем избирательного или преимущественного закомплексовывания иона Л4П в водном растворе.
Выражение условной константы обменного равновесия можно использовать также для выяснения возможного обмена при заданных значениях и /С',КСп. Пусть в результате экстракционной реакции ион Мх обменивается на Мп и переходит в органическую фазу на Z %, тогда из уравнения (4.60) следует, что
iz'm 1 гу-'п
* эКСу/^ЭкСц
= Zm-'nrn~mnf7{(100 — Z)m (lOOmgr — т)п\ (4.68)
Решая это уравнение при заданных значениях /(экер r> g> можно прогнозировать степень обмена в реальных условиях. Приняв в уравнении (4.68) Z < 1 %, находим необходимое условие, при котором обменная реакция практически не протекает (при однократной экстракции):
102(m+n) (т!п)п\ (4.69)
При т = п = 1 и г = 1 обменная реакция (4.59) практически не протекает, если /Ckcj/ZGkcii < 10-4/g. Если проводят d последо-128
вательных экстракций, то Z отождествляют с Е и для установления необходимых соотношений между K'3KCl и /<;кс используют уравнение (4.68).
Использование уравнений (4.67)—(4.69) при количественном описании обменных экстракционных равновесий возможно только в том случае, если в процессе экстракции весь реагент остается в органической фазе в виде хелата. Пороговое значение pH, начиная с которого систему можно считать обменной, рассчитывают по уравнению [194]:
,, 1 , CRaMnR 1 , / VCRKMnR \
pH - — lg п — 1g (сМп ) -
~ 1g ^эксц lg (1 — аМцД) Cr (4 70)
Здесь Cr — общая концентрация реагента в системе, моль/л; aMnR — доля реагента, связанного в комплекс MjjR, которая принимается равной 0,99 или 0,999; Смц — общая концентрация металла Мц в системе, моль/л; КЭВС11 — константа экстракции комплекса Мп-
Если в уравнении (4.70) константу экстракции /<эксп заменить ее условным значением ЛХ1(Сп, то это уравнение, по-видимому, можно использовать для определения порогового значения pH обменной экстракции и в присутствии конкурирующих комплек-сантов.
4.3.4.4. Экстракционное разделение хелатов
Возможность и эффективность экстракционного разделения элементов основана на различии коэффициентов распределения и условных констант экстракции разделяемых хелатов. Для количественной оценки эффективности разделения применяют две характеристики: фактор разделения S и фактор обогащения S'. Фактор разделения ионов и Мп, экстрагируемых в виде соединений M1R„. и MnRn--, определяется отношением их коэффициентов распределения [176]:
s ~ £’м1/°м11 (4-71)
Если больший из коэффициентов распределения, например DKl значительно меньше то уравнение (4.71) можно выразить через условные константы экстракции:
sMl/Mn=k;KCMi [HRi"'-'i7(^;KcMn [н+]"'-"э (4.72,
При одинаковом составе экстрагируемых комплексов (п! = ~ п" — п) фактор разделения определяется отношением условных констант экстракции разделяемых элементов;
5МХ/Мц = /(эксМ1//<эксМп (4-73)
5 М. И. Булатов и др.
129
При разделении ионов Мт и Мп соотношение коэффициентов распределения должно обеспечивать переход в органическую фазу определяемого иона не менее чем на 99%, в то время как концентрация отделяемого иона Мп в органической фазе не должна превышать 1 % от концентрации иона Мх. Следовательно, при однократной экстракции
DK1 = [CMllo/[CMl] > 0,99CMl/(0,0ICM/) = Ю2/г (4.74) ^Мц = [Смц]о/[Смц] 0,01Cmj/{(Смц O.OICmj) —
= IO-VRCmh/Cmj - 10-2) r} (4.75)
Разделив почленно уравнения (4.74) и (4.75), получим:
Smj/Мп = > 1 104 (Смп/Смт - °>01) (4.76)
При См /См 5> 0,1 уравнение (4.76) можно упростить и использовать в таком виде:
1 104 (4.77)
Однако это соотношение является необходимым, нс недостаточным, так как соблюдение неравенства (4.77) не гарантирует степень извлечения 99%, а Емп < 1% Поэтому в качестве дополнительного условия необходимо, чтобы степень извлечения отделяемого иона Мп не превышала бы 1 % от количества иона Мх, извлеченного в органический слой не менее чем на 99%. Это дополнительное условие обеспечивается соблюдением неравенств (4.74) и (4.75). В тех случаях, когда при экстракционном разделении применяют d последовательных экстракций, необходимые значения DMl и Z)MjI определяют из уравнений (4.4) и (4.6).
Согласно номенклатурным рекомендациям ИЮПАК 111), под фактором обогащения S' понимают отношение количеств двух разделяемых веществ в фазе экстрагента, отнесенное к исходному отношению их количеств до разделения:
Smi/mii = (^mI(o)/^mikoJ/(^mi /%г ) = £|уч/£мп <4'78) 1/ 11 \ ПО) НИ))/' х *-ИСХ* A^0CXz A' Al
Фактор обогащения представляет собой коэффициент, на который нужно умножить исходное отношение количеств двух разделяемых веществ, чтобы пол) чить отношение их количеств в органической фазе после разделения.
Выражая фактор обогащения через степень извлечения разделяемых хелатов, из уравнения (4.78) получим
SMi/Mn = % (> + г£)мп)/ |°мп (* + ^mJ} I4-79) а при проведении d последовательных экстракций
5мг/мп = {1 - (1 + / {1 - (1+ <4-8°)
В частном случае, когда d = г = 1
«Ц/Мд - % С + оИ11)/(°„п ( + D»I)| <, !|)
130
Если для разделения элементов Mt и Мп используют экстракционные реакции обмена с реагентом-хелатом MRm, то фактор обогащения можно выразить как отношение степеней обмена разделяемых элементов:
(4-82)
Количественное разделение ионов Mj и Мп при однократном применении экстракционной реакции обмена с хелатом MRm, как это следует из уравнений (4.67) и (4.69), возможно, если
/С" Сс""' /К'"1 > 102(2m+n">gn'7n'n'n'm'I7{n"n" (mgr-п)п'} (4.83) эксМг ЭКСМ / эксМп ° ’ 1 ’
а при одинаковом составе разделяемых хелатов (п' = п" — п) (/(' с /Кис \m>(gr)n-^m+n',mnl(mgr-n)n (4.84) \ эксМг' Мп)
Для частного случая, когда т — п = 1
^эксМ1/^эксМ11 104gr/(gr 1) (4.85)
При этом абсолютные значения условных констант экстракции должны удовлетворять уравнениям:
^ксМ1/^ксм^ 102m/{rm'n' (mgr-n'H (4.86)
(mln)n''\ (4.87)
При проведении d последовательных экстракций, обеспечивающих достижение заданных ZMj и ZMn, необходимые соотношения между величинами /Сэксм» A%kcMi> A7kcMii можно найти с помощью уравнения (4.68). Для этого в правую часть неравенства (4.68) необходимо подставить при заданном г значения ZMl и 2мп, полученные при однократной экстракции.
Г лава 5
РАЗДЕЛЕНИЕ, КОНЦЕНТРИРОВАНИЕ
И УСТРАНЕНИЕ МЕШАЮЩЕГО ВЛИЯНИЯ ПОСТОРОННИХ КОМПОНЕНТОВ
После растворения анализируемого образца в растворе, как правило, содержится значительное количество посторонних веществ, которые нередко оказывают мешающее влияние при фотометрических определениях интересующих компонентов. Устранение мешающего влияния посторонних веществ (ионов) осуществляют различными способами:
5*
131
1) разделением или концентрированием определяемых и мешающих компонентов;
2) химическими методами без отделения;
3) специальными приемами фотометрических измерений.
5.1. МЕТОДЫ РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Б.1.1. ОБЩИЕ ПОЛОЖЕНИЯ
Фотометрическому анализу часто предшествуют операции разделения, а при определении следовых количеств элементов и их концентрирование.
Разделение * — это операция (процесс), в результате которой компоненты, составляющие смесь, отделяются один от другого, причем их концентрации могут отличаться или быть близки друг к другу.
Концентрирование — это операция (процесс), в результате которой повышается отношение концентрации или количества микрокомпонентов к концентрации или количеству макрокомпонента (матрице). В этих случаях концентрации разделяемых компонентов резко отличны.
Различают индивидуальное (селективное) и групповое концентрирование (одновременное выделение нескольких микроэлементов).
Индивидуальное концентрирование — процесс, в результате которого из образца выделяется один или последовательно несколько микрокомпонентов Именно этот вид концентрирования стараются применять в фотометрическом анализе, поскольку фотометрические методы чаще применяют для определения содержания одного компонента.
Различают также абсолютное и относительное концентрирование.
При абсолютном концентрировании микрокомпоненты переходят из большой массы образца в малую', при этом повышается концентрация микрокомпонента (например, при упаривании анализируемых проб воды, минеральных кислот). При относительном концентрировании увеличивается соотношение между микро- и мешающими макрокомпонентами (растворитель к последним не относится). Часто главной целью относительного концентрирования является замена неподходящей для анализа матрицы (макрокомпонента) более подходящей средой.
Для описания любого метода концентрирования применяют три следующие количественные характеристики.
Терминология и обозначения взяты из работ 114, 29, 195].
132
Степень извлечения /? — безразмерная величина, показывающая долю от абсолютного количества микроэлемента, сосредоточенную в концентрате: R — qKlqD^ где qv и <7пр — соответственно абсолютные количества микроэлемента в концентрате и пробе. Чаще степень извлечения выражают в процентах.
Коэффициент концентрирования К — показывает, во сколько раз изменилось отношение абсолютных количеств микроэлемента и матрицы в концентрате относительно этого же отношения в исходной пробе; К = qKiQ„ : qai)/Qap = = q«Qn[J <7пР(?к. где Qu и Qnp — соответственно абсолютные количества матрицы в концентрате и пробе. Так как qnp Qnp и qK
Q1;, то Qnp и QK обычно принимают соответственно равными общей массе пробы и концентрата. Если степень извлечения равна 1 (R = 100%), то уравнение упрощается и К = Qnp/QK.
Коэффициент разделения S — величина, обратная коэффициенту концентрирования: S = QJqB : Qnp/?np — MR-
Концентрирование микрокомпонентов может быть осуществлено двумя способами: или их непосредственным выделением, или косвенно — путем отделения матрицы.
Важность операции концентрирования не ограничивается простым увеличением содержания микрокомпонента в концентрате. Помимо этого концентрированием достигается совокупность следующих целей:
1. Как правило, снижается на 2—3 порядка относительный предел обнаружения, а в ряде случаев — и абсолютный предел обнаружения.
2. При определенных условиях улучшаются правильность и воспроизводимость определений за счет устранения мешающих компонентов. Однако при неблагоприятных условиях — частичной потере микрокомпонентов или загрязнениях — возможен и противоположный эффект.
3. Удаление анализируемых матриц разного состава и переведение микрокомпонентов в унифицированную матрицу одного состава (например, в водный раствор) позволяет снять очень важную практическую проблему обеспечения методик анализа стандартными образцами. В этом случае в качестве стандартных образцов могут быть использованы растворы различных элементов.
4. Существенно снижается погрешность, обусловленная него-могенностью распределения микрокомпонента в массе анализируемого вещества.
Обширные экспериментальные результаты последних лет по методам концентрирования и разделения обобщены и представлены в монографиях Ю. А. Золотова и Н. М. Кузьмина [195, 1961, других авторов [198—200 ]. Как следует из этих работ, и для разделения и для концентрирования обычно используют одни и те же методы. Наиболее распространенными из них являются:
1. Экстракция (включая экстракционную хроматографию).
133
2. Сорбционные методы (сорбция, ионообменная и хелатная хроматография).
3. Осаждение и соосаждение.
Б.1.2. ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Экстракция— метод, основанный на распределении растворенного вещества между двумя несмешивающимися жидкими фазами (14, 29, 167, 172, 176, 177, 183, 184, 195—208]. Обычно в практике применяют системы, в которых одной фазой является водный раствор, а второй — органический растворитель.
Для эффективного разделения, как видно из уравнения S — — Dr!D2 (см. разд. 4.3.4.4), необходимо, чтобы коэффициенты распределения определяемого и мешающих веществ различались в достаточной степени. В тех случаях, когда один из коэффициентов распределения очень мал, а другой велик, разделение достигается легко и быстро. Если же фактор разделения велик и меньший из коэффициентов распределения также велик, происходит экстракция обоих компонентов, т. е. в этом случае необходимо тем или иным способом подавить экстракцию нежелательного элемента. Экстракция позволяет селективно разделять не только большие количества различных элементов, но и отделять следы одних элементов от макроколичеств других и концентрировать их в малом объеме органического растворителя, не смешивающегося с водой. Известны случаи извлечения десятых и сотых долей микрограмма вещества (10-7—1(Н г) из 1—2 л водного раствора 5—10 мл органического растворителя.
В настоящее время интенсивно развиваются экстракционнофотометрические методы, которые позволяют объединить этапы разделения и непосредственного фотометрического определения элементов в одну операцию, так как многие ионы металлов образуют окрашенные соединения, которые могут быть извлечены соответствующими растворителями.
Часто измеряют оптическую плотность (пропускание) бесцветного экстракта в ультрафиолетовой области спектра (185—400 нм) или добавляют новый реагент, образующий окрашенное соединение непосредственно в экстракте В случае необходимости используют реэкстракцию, т. е. переводят тем или другим способом определяемый компонент из органической фазы снова в водный раствор и после соответствующей обработки фотометрируют. В ряде случаев чувствительность определения может быть повышена во много раз, если экстрагировать не окрашенное соединение следов определяемых элементов, а продукт цветной реакции, катализируемой этими элементами.
Все экстракционные приемы по извлечению отделяемого (определяемого) иона из водной фазы в органическую сводятся к тому, что его переводят в такое состояние, в котором он легко переходит в органическую фазу.
134
Исходя из типа соединения, переходящего в органическую фазу, выделяют следующие широко применяемые экстракционные системы.
Комплексные металлокислоты общей формулы НГ_5МХР, где разность р — q обычно равна 1 или 2, например: HGaBr4, HFeCl4, HInBr4, H2CdI4 и т. д. Растворителями служат кетоны, спирты, эфиры Экстрагирование ионов в виде металлокислот применяют как для разделения больших содержаний ряда элементов, так и для отделения определяемых микрокомпонентов. Часто комплексные металлокислоты используют для концентрирования микрокомпонентов путем экстракции матрицы, например при анализе металлов высокой степени чистоты и сплавов: Ga, In, InAs, InSb, Cd, Sb и т. д.
Хелаты (внутрикомплексные соединения) металлов с дитизоном, 8-оксихинолином, купфероном, диэтил-дитиокарбаминатом натрия, оксимами, 8-меркаптохинолином, пир-ролидиндитиокарбаминатом аммония, ацилпиразолонами, 1-(2-пиридилазо)-2-нафтолом и т. д. Вышеперечисленные реагенты являются групповыми и применяются для отделения, разделения небольших количеств элементов и микроэлементов. Изменяя pH исходных растворов, добавляя другие комплексанты, используя различия значений констант устойчивости внутри-комплексных соединений, успешно производят и индивидуальное концентрирование определяемых микрокомпонентов. Для растворения внутрикомплексных соединений и извлечения их из водной фазы чаще всего применяют хлороформ и тетрахлорид углерода.
Экстракционные характеристики большинства хелатных соединений могут быть охарактеризованы уравнением (4.34).
Из уравнения (4.34) видно, что извлечение металла зависит от ряда констант, от концентрации ионов водорода в водной фазе и реагента в органической фазе, но не зависит от концентрации металла. Изменение концентрации реагента в органической фазе в 10 раз будет так же эффективно, как соответствующее изменение pH водной фазы на единицу, т. е. использование высокой концентрации реагента весьма полезно при экстракции металлов, которые легко гидролизуются. Однако в тех случаях, когда внутрикомплексное соединение определяют спектрофотометрически или реагент имеет значительный молярный коэффициент светопоглощения при длине волны, используемой для измерений, большой избыток реагента нежелателен.
Внутрикомплексные соединения многих металлов интенсивно окрашены и имеют высокие значения молярных коэффициентов светопоглощения в органических растворителях (до 1-Ю5). Это обстоятельство позволило разработать большое число экстракционно-фотометрических методик определения следовых содержаний (до 1-10~6 %) различных элементов, включая определе-
135
ние нескольких элементов из одной навески путем последовательного извлечения в виде хелатов [205].
Установлены так называемые обменные ряды для 8-меркап-тохинолинатов [196, 201], 8-оксихинолинатов [167].
Fe (III) > Ga > Си (II) > Sc > In > Hg (II) > Al
купферонатов [167]
Mo(VI) > Fe (III) > Ga > Си (II) > In > Al диэтилдитиокарбаминатов (при экстракции в определенных условиях) [196, 201]:
Hg, Ag, Pd, Си, Tl, Bi, Pb, Fe, Co, Cd, Ti, Ni, Zn, In, Sb, Те, Mn
В приведенных рядах каждый предыдущий элемент вытесняет последующие из их комплексов. Обменные реакции в ряду диэтилдитиокарбаминатов широко использ) ют для экстракционного разделения, например для разделения ряда элементов с помощью диэтилдитиокарбамината свинца (а также Cd, Zn, Bi и др.) с последующим фотометрическим определением этих элементов [204].
Интересны практические приложения обменных реакций в ряду диэтилдитиокарбаминатов металлов, предложенные А. Б. Бланком с сотр. [204, 205] для группового концентрирования и фотометрического определения суммы тяжелых металлов в галогенидах щелочных металлов высокой степени чистоты. Так, определялась сумма примесей металлов (Bi, Cd, Си, Fe, Mn, Pb, Zn) путем концентрирования их из водного раствора в виде диэтилдитиокарбаминатов в хлороформе и последующего контактирования экстракта с водным раствором меди (II). Ионы меди (11) при описанных условиях вытесняли вышеуказанные металлы, и по оптической плотности окрашенного в желтый цвет хлороформного экстракта диэтилдитиокарбамината меди определялось суммарное содержание тяжелых металлов — до 5-10-6 % (ат.).
Гетерополисоединения, экстрагируемые кислородсодержащими растворителями, применяют для разделения и концентрирования Р, Si, Ge, As, V, W, Mo и некоторых других элементов [206]. Предложено значительное число схем как последовательного разделения комбинаций вышеуказанных элементов, так и селективного экстрагирования одной из гетерополикислот в присутствии других. Экстракционно-фотометрическое определение проводят, измеряя оптическую плотность экстрактов самих гетерополикислот, окрашенных в желтый цвет, либо экстрактов восстановленных форм гетерополикислот, окрашенных в синий цвет. По таким методикам определяют, например, содержание Si, Р в интервалах КН—КН %.
136
Тройные комплексы: металл — галогенид (роданид или другой электроотрицательный лиганд) — органическое основание (типа пиридина, антипирина или основного органического красителя). В качестве основных красителей чаще всего применяют трифенилметановые (малахитовый зеленый, метиловый фиолетовый, кристаллический фиолетовый, бриллиантовый зеленый) и родаминовые красители.
Экстракцию при помощи перечисленных реагентов главным образом используют в экстракционно-фотометрических методах определения Ga3+, In3+, Sn2+, Sb5+, Ge2+, Zn2+, Tl3+, Te4+, Co2*, AuJ+, находящихся в водном растворе в виде [SbCl6]-, [GaCl4]~, [Т1С14]_, [1пВг4]~, [SnCl4]2- и т. д. Эта группа соединений экстрагируется обычно бензолом или толуолом.
Легкоплавкие органические экстрагенты. Для разделения и концентрирования широко применяют также легкоплавкие органические экстрагенты. В этих случаях исследуемый раствор и реагент помещают в стеклянной посуде на горячую водяною баню и производят экстракцию. При последующем охлаждении экстракт затвердевает и водная фаза отделяется простым сливанием. Процесс характеризуется высокой степенью разделения.
Легкоплавкие экстрагенты применяют в двух вариантах: либо в виде непосредственного расплава органического реагента, либо в виде расплава экстрагента в легкоплавком соединении, в качестве которого наиболее часто используют парафин (tnn — = 50 °C), дифенил (/Ш1 — 70 °C), нафталин (/ЕЛ = 80 СС), а также их смеси.
В настоящее время наибольший интерес среди легкоплавких экстрагентов вызывают хелатообразующие реагенты (в первую очередь 8-оксихинолин, салицилальдоксим). Хорошо изучено экстракционное поведение систем: 8-оксихинолин — М [Zr, Hf, Nb, Та, Си, Ag, Au, Zn, Al, Ga, Zn, Pb, Co, Bi, Cr, Mn, Fe, Ni, Au (III)], салицилальдоксим — M (Cu, Ag, Au, Zr, Al, Ga, Zn, Pb, Bi, Cr, Mn, Fe), 8-эксихинолин + нафталин — M [Zn, Mg, Pb, Mo, In, Cr (III), Rh (III)], диэтилдитиокарбаминат натрия + щафталин — M (Cu, Ni, Bi, Pb, Zn, Cd).
Конечное определение элементов в ряде случаев осуществляют экстракционно-фотометрическим методом. С этой целью затвердевший экстрагент растворяют в хлороформе, диметилформамиде или каком-либо ином растворителе и измеряют оптическую плотность полученного раствора. Так, описан [208] экстракционнофотометрический метод определения микрокомпонентов Pb, Zn, Cd, основанный на количественной экстракции в расплавленный нафталин их диэтилдитиокарбаминатов, растворении затвердевшего нафталина в хлороформе, замещении определяемого металла на Си и фотометрировании комплекса Cu (DDK)2 при X — 440 нм. Некоторые обобщенные сведения по применению органических экстрагентов приведены в работах [195, 2071.
137
6.1.3. МЕТОДЫ ОСАЖДЕНИЯ И СООСАЖДЕНИЯ
При разделении определяемого и мешающих ионов осаждением, выбирают, как правило, такой осадитель, при помощи которого определяемый ион выделяется в осадок, а основной компонент анализируемого образца остается в растворе [195, 198—200, 209—211]. Если осадок при этом будет несколько загрязнен, то его очищают переосаждением. Определяемый ион в виде малорастворимого соединения отфильтровывают, промывают и затем снова переводят в раствор. Методы разделения, при которых в осадок переходит посторонний компонент, нежелательны и применяются лишь при отсутствии другого пути, так как из-за соосаждения могут происходить значительные потери определяемого иона.
При очень малых концентрациях определяемого вещества отделение его затруднено, поскольку произведение концентрации ионов не достигает произведения растворимости (Ks) малорастворимого соединения и осадок не выпадает. Поэтому проводят соосаждение определяемого микроэлемента с соответствующим носителем (коллектором). В качестве коллекторов обычно применяют малорастворимые гидроксиды, сульфиды, фосфаты, карбонаты и сульфаты многих металлов, которые захватывают микрокомпонент в момент осаждения.
Соосаждение микропримесей из раствора представляет собой сложное явление и в зависимости от характеристик компонентов и условий эксперимента может протекать по различным механизмам: путем образования изоморфных или аномальных смешанных кристаллов, за счет образования твердых растворов различных типов, в результате адсорбции на поверхности коллектора или обмена ионов микрокомпоьента с ионами осадка макрокомгюнента. Так или иначе микрокомпоненг, рассеянный ранее в большом объеме раствора, после соосаждения находится в небольшом количестве осадка. Последний растворяют в малом объеме подходящего растворителя и анализируют. Если, например, первоначальный объем раствора равен 1000 мл и полученный осадок, содержащий почти все количество микрокомпонента, затем растворен в 0,5 мл кислоты, то концентрация микрокомпонента в этом растворе примерно в 2000 раз больше, чем в исходном .
Известны случаи концентрирования соосаждением в 10— 20 тыс. раз и более.
Применяя метод соосаждения, можно выделить и обнаружить микрокомпонент при концентрациях до Ю-10—10~12 моль/л.
Среди различных способов выделения определяемых микрокомпонентов путем соосаждения с гидроксидами и сульфидами наиболее широкое распространение получили [14, 209, 210, 222, 225-2271:
138
1) концентрирование путем осаждения вещества, образующего с избытком осадителя коллектор (носитель) для микрокомпонента;
2) концентрирование осаждением части макрокомпонента.
В первом случае для соосаждения следов металла, образующего малорастворимый гидроксид или сульфид, в качестве коллектора, как правило, может быть применен гидроксид (сульфид) любого другого металла. Таким способом количественно соосажда-ются следы металлов (например Ni2+, Со2+, Cd2+, Cu2+, Ag+, Ап3+, Sn2+, Pb2+, Мп2+ и др.) с гидроксидом железа, висмута, магния; микроколичества титана (1 мкг/л) — с гидроксидом алюминия; микропримеси меди — с сульфидами свинца, кадмия, олова, g гидроксидом магния; примеси цинка — с сульфидами кадмия и т. д.
Концентрирование микрокомпонента путем частичного осаждения макрокомпонента в виде гидроксида (сульфида) наблюдается только в том случае, когда микрокомпонент образует с осадителем менее растворимое соединение, чем макрокомпонент. Механизм соосаждения, по-видимому, можно объяснить ионообменными реакциями, протекающими между ионами микро-и макрокомпонента в процессе образования осадка:
Л\А 4“ Мд
осадок ион коллектора микро-компо-
нента
м2а осадок микро-компо-иеита на
коллекторе
+ Мх
ион макро-компонента
Путем частичного осаждения цинка из раствора его соли щелочью соосаждают микропримесь меди (до 1 -10-4%) и определяют ее затем фотоколориметрически. Для определения меди (до 1 • 10-8 %) в солях кадмия осаждают небольшую часть кадмия в виде сульфида и в осадке определяют медь фотометрически. Аналогично поступают при определении меди (до Ы0-Б%) в ацетате и нитрате свинца.
Для выделения микроколичеств (0,01—20 мкг) различных элементов из растворов используют ионообменные реакции, протекающие с введенными в раствор готовыми осадками. Наиболее удобной для этой цели является фильтровальная бумага, пропитанная малорастворимым веществом. При фильтровании растворов обеспечивается очень хороший контакт тонкодисперсного осадка, нанесенного на поверхность фильтра, с раствором. В результате ионообменной реакции, например
CdS + Cu2+ «=> CuS-f-Cd2+ осадок мнкро- осадок микроколлектора компонент компонента на
коллекторе
(^Cscas = 4-10-28; A,4CuS = 9-10-45) микрокомпонент будет поглощаться из раствора. Таким способом можно отделить 0,0001 % меди от никеля или свинца, выделить следы цинка из очень раз-
139
бавленных растворов, применяя в качестве коллектора Cd(OH)s и т. д.
В других случаях с успехом используют органические соосадители, которые имеют ряд преимуществ перед неорганическими коллекторами [211]:
1) легко удаляются из отфильтрованных осадков простым озолением; 2) обладают высокой избирательностью и эффективностью; способны извлекать из чрезвычайно разбавленных растворов одни элементы, не захватывая другие, имеющие большие концентрации.
Принцип действия ряда таких соосадителей заключается в том, что к водному раствору, содержащему удаляемый ион (например, Ni2+), сначала добавляют органический реагент (8-оксихинолин), дающий малорастворимое соединение с неорганическим ионом, а затем спиртовой (или другой) раствор органического соосадителя (р-нафтола), плохо растворимого в воде. При разбавлении спиртового раствора соосаднтель (р-нафтол) выпадает в осадок и может очень полно увлечь с собой соосаждаемое соединение (оксихинолинат никеля). Этим способом можно удалить из раствора никель при разбавлении 1 : 5-Ю8.
Иногда соосаждаемый ион, находясь в виде комплексного аниона, например [Zn(SCN)4 ]2_, образует малорастворимые соли с рядом основных красителей (например, с метиловым фиолетовым) Роданид-ион также дает малорастворимое соединение с метиловым фиолетовым, что позволяет соосаждать цинк из еще более разбавленных растворов. Таким способом удается полностью удалить 1 мкг цинка из 100 мл раствора.
Для фотометрического анализа перспективны бесцветные соосадители. При этом в раствор вводят реагент, образующий с концентрируемым элементом окрашенное соединение, а затем — необходимые количества ингредиентов органического соосадителя. Осадок отфильтровывают, растворяют в подходящем органическом растворителе и фотометрируют.
Б.1.4. СОРБЦИОННЫЕ МЕТОДЫ
Широкое применение для разделения и концентрирования элементов получили адсорбционные и хемосорбционные (особенно ионный обмен и комплексообразование, включая использование хелатных смол) методы [195, 198—200, 212—217].
При ионообменном разделении элементов используют различие в зарядах или объемах ионов, степени их гидратации или протолиза; способности к образованию комплексных соединений с растворителем (элюентом) и изменение этих свойств в зависимости от pH среды и природы ионообменника.
Широко применяют способы разделения ионов, основанные на различии: полярности ионов, устойчивости комплексных соеди-140
нений разделяемых ионов с растворителем (элюентом), степени протолиза комплексных ионов, размеров одинаково заряженных ионов.
При концентрировании с помощью ионообменников можно извлекать или матрицу, или микроэлементы. Удобно, если элемент матрицы находится в анионной форме, а примени — в катионной, или наоборот (первый вариант часто более доступен и предпочтителен). В этом случае возможны два варианта процесса концентрирования: элемент матрицы сорбируется на анионите, а примеси переходят в элюент; примеси сорбируются на катионите, а основной элемент элюируется. После отмывки колонки водой ионит озоляется или микропримеси десорбируются с ионита соответствующим элюентом. В практике отдают предпочтение именно концентрированию микропримесей на ионите. Например, при анализе высокочистого олова его растворяют в смеси кислот HF и HNO3, переводят в комплекс SnF^*", не сорбирующийся катионитом; примеси Cu2+, Pb2+, Са2+, Bi3+, Mn2+, Ni?+, Со2+, Сг3+ поглощаются катионитом, который в дальнейшем может быть озолен.
Процесс концентрирования проводят обычно в динамических условиях, пропуская растворы через микроколонки с ионитом, содержание которого может колебаться от нескольких десятков до нескольких сот миллигра имев.
Для концентрирования следовых количеств элементов в последние годы широкое применение получили хелатные смолы и целлюлозные сорбенты, в состав которых введены функциональноаналитические хелатные группы [195, 214—217]. В результате обеспечиваются простота, экспрессность выполнения процесса, высокая избирательность и эффективность при концентрировании элементов из растворов сложных составов. Так, предложен способ концентрирования микроэлементов Cd, Со, Сг, Си, Fe, Hg, Мп, Mo, Ni, Pb, U, V, Zn при анализе вод с использованием модифицированной целлюлозы, содержащей 4-(2-пиридилазо)-резорцин [216]. На основе целлюлозных волокон, содержащих группы арсеназо, разработан способ селективного извлечения микроколичеств U из морских вод с его спектрофотометрическим определением.
В аналитической практике наряду с рассмотренными применяют целый ряд и других методов разделения и концентрирования (дистилляцию, в том числе различные варианты отгонки, сублимацию, электрохимические методы, различные виды кристаллизации и т. д.), которые, однако, реже сочетаются с фотометрическими методами анализа. К числу таких удачных сочетаний следует отнести анализ чистых веществ с применением кристаллизационного концентрирования [204].
6.1.6. КРАТНОЕ СОПОСТАВЛЕНИЕ МЕТОДОВ КОНЦЕНТРИРОВАНИЯ
Выбор конкретного оптимального метода и способа (удаления матрицы или выделения микроэлементов) концентрирования во многих случаях является непростой комплексной задачей. Химик должен принимать во внимание природу матрицы и микроэлементов, возможности и особенности дальнейшего метода анализа, метрологические параметры и многие другие факторы.
Количественные характеристики операций концентрирования, коэффициент концентрирования, степень извлечения, коэффициент разделения, коэффициент распределения, воспроизводимость — относятся к числу первостепенных.
Наиболее высокие значения одного из основных параметров — коэффициента концентрирования К — 103 достигаются при концентрировании ионообменными методами, соосаждением с коллекторами, зонной плавкой, частичным растворением матрицы; в экстракционных методах значения коэффициентов концентрирования на 1—1,5 порядка ниже.
Существенным моментом при концентрировании являются результаты холостого опыта, которые определяют во многом реальные предел обнаружения и нижнюю границу определяемых содержаний. Результат холостой пробы зависит от содержания примесей (в первую очередь анализируемых) в применяемых реактивах, реагентах и от внесенного количества последних. Ионообменное концентрирование характеризуется низким уровнем холосто! о опыта. Промышленные иониты имеют зольность до 4 % и содержат 10-1—10~2 % железа и 10~2—10~3 % других примесных металлов. Однако несложные специальные приемы позволяют снизить содержание этих примесей на 3—4 порядка [213]. Органические реагенты, применяемые в экстракционном концентрировании, как правило, содержат примеси на уровне 10-1—10_3 %, т. е. дают в ряде случаев относительно высокий уровень холостой пробы Соосаждение во многих случаях тоже связано с высоким уровнем холостой пробы.
Исходя из рассмотрения метрологических характеристик и результатов холостого опыта, можно было бы ожидать предпочтительность применения сорбционного концентрирования по сравнению с другими методами. Однако на практике учитывается совокупность и метрологических характеристик, и результатов холостого опыта, и таких факторов, как простота, доступность, универсальность, экспрессность. Все вместе взятое выдвинуло на первое место (особенно в фотометрических методах анализа) экстракционные методы концентрирования.
Развитие экстракционных методов в сочетании с фотометрическим окончанием количественного определения элементов привело к созданию экспрессного экстракционно-фотометрического метода анализа. Согласно статистическим оценкам (по публи
142
кациям на 1978 г.) второе и третье места по практической применимости делили сорбционные и дистилляционные (с учетом различных вариантов отгонки) методы. Число публикаций по совокупности остальных методов, включая и концентрирование со-осаждением, не превышало 13 % от общего числа публикаций по методам концентрирования.
Что касается выбора способа концентрирования, то в общем случае при простой матрице (один-два элемента) легче удалять матрицу. Если основа является многоэлементной (минералы, сплавы, почвы), то предпочитают выделять микроэлементы. Выбор зависит также и от метода концентрирования. В обобщенном виде эти данные представлены в монографиях 1195, 196], в работах последних лет по концентрированию микрокомпонентов в металлах [29], природных водах [214], различных объектах [197, 218, 219].
Б.2. УСТРАНЕНИЕ ВЛИЯНИЯ
ПОСТОРОННИХ ИОНОВ
ХИМИЧЕСКИМИ МЕТОДАМИ БЕЗ ИХ ОТДЕЛЕНИЯ
Б.2.1. МАСКИРОВАНИЕ МЕШАЮЩИХ ИОНОВ
Устранение мешающего влияния посторонних ионов с помощью реакций комплексообразования сводится в основном либо к связыванию посторонних ионов в бесцветный комплекс, либо к созданию определенной концентрации реагента и pH раствора, обеспечивающих полный перевод в окрашенное соединение определяемого иона и в минимальной степени мешающего иона.
Связывание посторонних ионов маскирующим реагентом в бесцветный комплекс. Маскирующий реагент выбирают таким образом, чтобы при его введении окрашенный комплекс M'Rn. постороннего иона М' разрушался, а окрашенное соединение MRn определяемого иона М практически оставалось бы без изменений. Для этого необходимо, чтобы относительная прочность комплекса постороннего иона с маскирующим реагентом была бы выше, чем с фотометрическим реагентом. С другой стороны, прочность комплекса MRn с фотометрическим реагентом должна быть также значительно выше, чем с маскирующим реагентом (если ион М взаимодействует с маскирующим реагентом).
Для связывания ионов металлов в бесцветные комплексы используют фосфорную, лимонную, винную и некоторые другие органические кислоты. Наиболее распространенные маскирующие реагенты приведены в табл. 5.1.
Ориентировочный выбор маскирующего реагента для преимущественного (или избирательного) маскирования мешающих ионов можно сделать путем сопоставления «-коэффициентов (или функции закомплексованности). Однако для успешного прове-
143
Таблица 5.1. Важнейшие маскирующие реагенты [66, 176]
Элемент
Маскирующий реагент
Элемент
Маскирующий реагент
Ag
Al
As
Au
В
Ba
Be
Bl
Ca
Cd
Ce
Co
Cr (III)
Cu
F
Fe (Ш)
Fe (II) Ga
Ge
Hf
Hg
In
Ir
Br-, I-, C1-, CN-, TM, NH3, SA"
F~, C2OI-, Na-ЭДТА, НТЭ, ДМП, СФСК, ТЭА, OH-
ДМП, S2-, OH-
CN-, C1-, Br-, S2Oy
F-, оксикислоты, гликоли
Na-ЭДТА, HTA, ОЭГ, цитрат, тартрат, SO|~
F-, цитрат, тартрат, СФСК
Цитрат, тартрат, ТМ,
Na-ЭДТА, НТА, ОЭГ,
НТЭ, С1-, I-, р,оа-
Na-ЭДТА, НТА, "ОЭГ, ци трат, тартрат, Р,О1~
Na-ЭДТА, НТА, ОЭГ, цитрат, тартрат, CN-, 1~, SCN-, SA-
Na-ЭДТА, НТА, ОЭГ, цитрат, тартрат, тайрон
Na-ЭДТА, НТА, ОЭГ, ЭДА, ГА, ДМП, ТА, NH3, NOT, SCN-, CN-, SA", Н2О2
Na ЭДТА, НТА, НТЭ, цитрат, тартрат, CHjCOO-, С А"
Na-ЭДТА, НТА, НТЭ, ГА, ДДК, ТА, ТГК, ДМП, ОЭГ, ТМ, цитрат, тартрат, NH3, I-, CN-, S2OT, S2-
Н3ВО3, Al, Be, Zr, Ti, Nb Na-ЭДТА, HT^, ОЭГ, ДДК, ДМП, АК, ТМ, НТЭ, ТГК, ТЭА, цитрат, тартрат, тайрон, F-, C2Oj-, POI-, P2O<7-, SCN-, S2-
Ф, CN-, S2O2-
Na ЭДТА, тартрат, цитрат, C2O?
F-, CA-
Na-ЭДТА, НТА, НТЭ, тартрат, цитрат, F-, H»O2> SO( РОГ, Р2ОГ, CA-
Na-ЭДТА, НТА, ОЭГ, НТЭ, ГА, ТА, тартрат, цитрат, so-;-, И, CN-, С1-, \'О-
Na-ЭДТА, цитрат, СА~, CITAI, тартрат, цитрат, NH3,
С1-, SCN-
Mg
Мп
Мо
Nb
Ni
Os Pb
Pd
Pt
Rh Ru Sb
Sc Se
Sn
Sr
Ta
Те
Th
Ti
Tl
U
V
W
Na-ЭДТА, НТА, ОЭГ, тар трат, цитрат, ОД-
Cp;-, P2O|-, Na-ЭДТА, ТЭА, НТА, ОЭГ, НТЭ, ДМП, цитрат, тартрат, F-
CNS-/CA’, Na-ЭДТА, HTA, тайрон, цитрат, H2O2, тартрат, С1-, F-
F~, СА-, НА. ОН", тайрон, цитрат, тартрат
НТА, Na-ЭДТА, ТА, ГА, цитрат, тартрат, CN-, SCN"
ТМ, С1-, CN-, SCN-
Цитрат, тартрат, Na-ЭДТА, ацетат, НТА, ОЭГ, S.O|"> I-, СН.СОО-, sor
Na-ЭДТА, НТА, ОЭГ, цитрат, тартрат, NH3, CN-, 1_, NO», SCN-, S ОГ
CN-, I-, С1-, NOt, SCN-, NH3, SA". СА"
ТМ, цитрат, тартрат, С1-
ТМ, CN-, Cl-
ДМП, цитрат, тартрат, I-, S2-, F-, OH-
Na-ЭДТА, цитрат, тартрат
ДМБ, цитрат, тартрат, S2-, SOT
ДМП, НТЭ, цитрат, тартрат, F-, I-, ОН-, С А"
Na-ЭДТА, НТА, ОЭГ, цитрат, sor
Цитрат, тартрат, F-, С А", он-
АК, цитрат, тартрат, 1-, F-, SO:!" S2™
Na ЭДТА, НТА, ОЭГ, НТЭ, ацетат, цитрат, тартрат, F-, С Л2- СЛ2-
Na-ЭДТА, НТА, НТЭ, тай-рои, тартрат, цитрат, СФСК, Н2О2, F-, SO’-", OH-
Na-ЭДТА, НТА, НТЭ, тартрат, цитрат, С1-, СА~> CN-
Тартрат, цитрат, F“, СО|-, С А-, НА
Na ЭДТА, НТЭ, тайрон, CN-, н2о2, F-, СА"
Тайрон, тартрат, цитрат, F-, С1-, НА, СА”, SCN-
144
Продолжение табл. 5.1
Элемент
Маскируюций реагент
Элемент
Маскирующий реагент
Zn
Na-ЭД ТА, НТА, ОЭГ, ДМП, ГА, ТА, тартрат, цитрат, гликоколи, CN~, SCN-, NH3, ОН-
Na-ЭДТА, НТА, ОЭГ, НТЭ, тартрат, цитрат, F-, SO|~, С2О1-, Н2О2, р2ог, poj-
Примечание. Принятые сокращения: Na-ЭДТА — этилендиамннтетраацетаг натрия, НТА — нитрнлотриацетат, НТЭ — 2,2'.2" нитрилотриэтанол, ТЭА — триэтаноламин; ДМП — 2,3-димеркапто-1-пропанол; ОЭГ — N, N-(2 оксиэтил)-глицин; ЭДА — этилендиамии. ТА — тетрамин; ГА — гексамин; ТМ — тиомочевина; АК — аскорбиновая кислота; ДДК — диэтилдитиокарбаминат; ТГК — тиогликолевая кислота; ДМБ — диа-мннобензидин; СФСК — сульфосалициловая кислота; Ф — 1,10-фенантролин-
дения маскирования мешающего иона М' необходим количественный подход к установлению избыточной концентрации фотометрического и маскирующего реагентов. В самом деле, концентрации маскирующего реагента L и фотометрического реагента R должны быть такими, чтобы определяемый ион М практически полностью был бы связан в светопоглощающий комплекс MRn, в то время как мешающий ион М' не должен связываться фотометрическим реагентом в комплекс M'Rn-. Если принять, что комплексы определяемого и мешающего ионов обладают близкими по значениям молярными коэффициентами светопоглощения, то мешающее действие постороннего иона М' можно не учитывать, когда [MRn] is 0,99CM, a [M'R„'l < 0,01См, т. е.
₽mr„ = [Cr]") = 0,99CM/(0,01CM fCRf) = 1 -102/[CRf (5.1)
Рмщ„. = [мХг]/([См,] [CRf ) = 0,01CM/{(CM. -0.01CM) [CRf'} =
= 0,01/{(CM,/CM-0,01)[CRf'} (5.2)
Здесь См и CM, — исходные концентрации (ориентировочные) определяемого и мешающего ионов; [Ср] — суммарная равновесная концентрация всех форм реагента, не связанного в комплекс с определяемым и мешающим ионами.
Разделив почленно одно на другое равенства (5.1) и (5.2), получим соотношение между условными константами устойчивости комплексов определяемого и мешающего ионов, при которых возможно фотометрическое определение иона М в присутствии мешающего иона М':
₽mr„
R—1 Ю4 [CR]" (См,/См - 0,01) (5.3)
Учитывая, что в большинстве случаев комплексы определяемого и мешающего ионов с фотометрическим реагентом имеют одинаковый состав (п — п) и отношение См-/См, как правило,
145
не менее 0,1, неравенство (5.3) можно преобразовать в более простое соотношение:
₽ЖпСм/(₽М'В„Аг) 1 • 1оМ (5.4)
Соблюдение условий (5.3) и (5.4) позволяет проводить фото-метрирование определяемого иона М в присутствии мешающего иона М'. Если величины eMR(i, ем-рп,, См и См- неодинаковы, то для теоретического прогнозирования условий селективного фотометрирования следует использовать общее уравнение (5.5),
полученное при Лми„/Лм'рп, 1 -Ю2, [MR„ ] = См — (См 1, lM'R„- ] = СМ' — [См-1:
eMR Рми См(! + Рм-Rn- I^r]" )/{еМ-К„,Рм-Кп,См' х НИХ Ч Z ( Ч Ч
*('+₽MR7l[cRr)pl Ю2 (5.5)
В тех случаях, когда условия (5.1) и (5.2) соблюдаются, т. е. 1 + ₽mr„ [CR ]" 1-102, а 1 + ₽M-Rn. [Cr J”' да 1, уравне-
ние (5.5) упрощается:
емнпРмнпСм/(ем'Рп.Рм-рп,^М')1 Ю1 (5-6)
Создание определенной концентрации реагента и pH раствора. Если определяемый ион М образует относительно более прочное окрашенное соединение по сравнению с мешающим ионом М', то можно создать ограниченную концентрацию реагента, которая достаточна для образования окрашенного соединения MRn, но мала для образования окрашенного соединения M'R„-. Необходимую для проведения анализа концентрацию фотометрического реагента можно найти из уравнений (5.1) и (5.2). Равновесная концентрация реагента, при которой ион М' практически не будет мешать определению, как это следует из уравнения (5.2), не должна превышать величины, равной
[Ск]^(10-2См/Рж,КпСм.)1/'’' (5.7)
Но, с другой стороны, [Cr] должна обеспечивать практически полный переход определяемого иона в светопоглощающий комплекс MRn:
[Ск]^(1-107РмКп)1/'1 <5-8)
В тех случаях, когда неравенство (5.7) невозможно удовлетворить без нарушения неравенства (5.8), изменением pH раствора или введением избирательного маскирующего реагента * заком-плексовывают мешающий ион М' до такой степени, пока Рм-кп.
* Комплекс мешающего иона с маскирующим реагентом не должен поглощать света при выбранных условиях фотометрирования.
146
при выбранном значении [CR] не будет превышать величину 1 • 10-2/(CR к1'. Если принять наиболее часто употребляемую концентрацию фотометрического реагента — примерно 1-10-3 моль/л, то условная константа устойчивости мешающего иона с фотометрическим реагентом должна обеспечивать условие:
Pm-r^IO3"'-2 (5.9)
Использование теоретических критериев, выраженных уравнениями (5.3) и (5.4), как уже отмечалось, справедливо только при одинаковых значениях молярных коэффициентов светопоглощения комплексов MR„ и M'R^. Если величины eMRn и eW'Rn, неодинаковы, то для теоретического прогнозирования следует использовать более общее неравенство (5.6).
Способ «буферного» иона — в раствор определяемого и мешающего ионов вводят третий катион, образующий с фотометрическим реагентом бесцветный комплекс, менее прочный, чем комплекс определяемого иона, но более прочный, чем комплекс мешающего иона. Тогда при достаточном избытке буферного иона реагент будет взаимодействовать, прежде всего, с определяемым ионом, избыток реагента будет связываться в бесцветный комплекс с введенным буферным ионом, а препятствующие! анализу ион вообще не будет вступать в реакцию с фотометрическим реагентом. Так определяют, например, ионы железа сульфосалицилатным методом в присутствии ионов меди, когда в качестве буферного иона применен алюминий.
Основным условием применения этого способа является значительно большая относительная прочность комплекса определяемого иона по сравнению с комплексами посторонних ионов. Количественную зависимость между концентрациями и условными константами устойчивости комплексов определяемого, мешающего и буферного ионов можно найти с помощью уравнений (5.4)—(5.9).
Если посторонние ионы образуют с реагентом бесцветные комплексы, то определяемый ион в окрашенное соединение может связываться не полностью, так как значительная часть реагента затрачивается на реакцию с посторонними ионами. Устранение влияния посторонних ионов в этом случае осуществляется введением в раствор соответствующего избытка фотометрического реагента, достаточного для полного связывания в комплексные соединения как определяемого, так п постороннего ионов.
Если мешающий ион М' образует с реагентом R относительно менее прочный комплекс, чем комплекс MRn, то полного связывания мешающих ионов в бесцветный комплекс добиваться не обязательно. В этом случае при [Cr ] = (102/Рмип)1/п определяемый ион М практически полностью переходит в комплекс MRn
147
и одновременно оказывается закомплексованным и мешающий ион М' в степени, равной величине
/П (5.10)
Общая концентрация реагента, необходимая для проведения анализа, будет слагаться из следующих составляющих:
Cr = пСм + (107pMRn)I/n + «Av (5.П)
Б.2.2. ИЗМЕНЕНИЕ СТЕПЕНИ ОКИСЛЕНИЯ
МЕШАЮЩЕГО ИЛИ ОПРЕДЕЛЯЕМОГО ИОНА
Посторонний ион, образующий с реагентом окрашенное соединение, переводят в такое состояние, в которо.м он не взаимодействует с реагентом или не образует с ни.м окрашенного соединения и не Ихмеет собственной окраски. Например, при определении молибдена роданидным методом в присутствии железа (III) влияние последнего устраняют восстановлением его до железа (II), применяя не очень сильный восстановитель. При определении никеля с диметил глиоксимом в присутствии железа (II) влияние последнего устраняют, наоборот, окислением его до железа (III). Выбор соответствующего окислителя или восстановителя определяется соотношением условных стандартных потенциалов определяемого и мешающего ионов [42, 44, 451. Окислители или восстановители подбирают таким образом, чтобы их способность была достаточной для изменения степени окисления мешающего иона и недостаточной для изменения степени окисления определяемого иона. В некоторых случаях изменяют состояние окисления и определяемого иона.
5.3. УСТРАНЕНИЕ ВЛИЯНИЯ
МЕШАЮЩИХ ВЕЩЕСТВ
СПЕЦИАЛЬНЫМИ ПРИЕМАМИ ФОТОМЕТРИЧЕСКИХ ИЗМЕРЕНИЙ
6.3.1. ФОТОМЕТРИРОВАНИЕ РАСТВОРОВ
В УЗКОМ ИНТЕРВАЛЕ СПЕКТРА
Влияние мешающих веществ с помощью этого приема измерения оптической плотности раствора можно устранить только в тех случаях, когда спектры поглощения окрашенных соединений определяемого и мешающего ионов известны и их максимумы поглощения находятся при различных длинах волн. При этом в спектральной области максимального поглощения света окрашенным раствором определяемого иона светопоглощение мешающего иона должно быть ничтожно мало или вообще отсутствовать (рис. 5.1). Можно определить, например, окрашенное ве-
148
Рис. 5.1. Спектры поглощения окрашенных соединений определяемого (А) и меняющего (В) компонентов.
щество А, раствор которого максимально поглощает лучи в области 440—450 нм, в присутствии мешающего вещества В, у которого максимальное поглощение лучей наблюдается в области 580—600 нм. Для этой цели можно
использовать узкополосный све-
тофильтр с областью пропускания 430—450 нм или измерения проводить с помощью спектрофотометров при 450 нм. Измеряя оптическую плотность в этом интервале длин волн окрашенной смеси компонентов А и В, практически измеряют оптическую плотность лишь вещества А, так как вещество В лучи указан-
ных длин волн не поглощает.
6.3.2. ИЗМЕРЕНИЕ ОПТИЧЕСКИХ ПЛОТНОСТЕЙ
ПРИ ДВУХ ДЛИНАХ ВОЛН
В этом приеме выбирают две длины волны и таким образом, чтобы разность оптических плотностей А?., и А^2 или молярных коэффициентов светопоглощения eZl и е;2 для мешающих компонентов была бы минимальной (или вообще равна нулю), а для определяемого элемента — достаточно большой. Тогда согласно правилу аддитивности можно записать:
= (ехХ,Сх + е2Х,С2 --+ еп?.1с'н) 1 <5-12)
Ах2 = (ежХ2^Ж + '2X2^2 + • • • + ^иХг^п) I (5.13)
Здесь индекс х относится к определяемому элементу, а остальные — к мешающим компонентам.
Вычитая почленно одно уравнение из другого, получим новое уравнение
Axj — ^Х2 = б’ж (СжХх — еа.Х2) + ^2 (М.! — егХ2) ‘ •1 + (enXj — ®пХг)
(5.14)
в котором все разности кроме первой, стремятся
к нулю, и следовательно, в конечной форме уравнение (5.14) принимает такой вид:
= А А = 1СХ (e^Xj — ЕхХа) = (5.15)
т. е. разность оптических плотностей при выбранных длинах волн прямо пропорциональна концентрации определяемого элемента. Этот прием требует обязательного соблюдения основного закона светопоглощения и правила аддитивности.
149
"Вакой прием использовали при определении Nd в Рг и Sm и Ег в Тт и Но с помощью реагента 7-йод-8-оксихинолин-5-сульфокислоты 1220]. В первом случае определение неодима проводили по разности оптических плотностей при 581 и 590 нм, а во втором — при 519 и 525 нм. С помощью этого же приема определяли Ti(IV) в присутствии Fe(III), используя разность оптических плотностей растворов их салицилатных комплексов при 360 и 570 нм [221 ]. Аналогично определяли Cu(II) в присутствии Fe(III) [222].
б.З.З. ФОТОМЕТРИЧЕСКИЕ ПРИЕМЫ
ПРИ ЛИНЕЙНОЙ ЗАВИСИМОСТИ СВЕТОПОГЛОЩЕНИЯ МЕШАЮЩИХ КОМПОНЕНТОВ ОТ ДЛИНЫ ВОЛНЫ
При количественном определении анализируемого вещества в присутствии мешающих компонентов, молярные коэффициенты светопоглощения которых изменяются в линейной зависимости от длины волны на выбранном участке спектра, используют совместное решение линейных уравнений при трех длинах волн:
= (W« + Pi^Ci) I (5.16)
42 = (^c«W0' (5.17)
= (M.3Cx + 1 iXgCi) I (5.18)
Здесь индекс x, как и ранее, относится к определяемому элементу, a i— к мешающим
В литературе описано большое число адекватных приемов [108], различающихся в основном условиями выбора аналитических длин волн, но имеющих одну и ту же основу.
Метод Бейнса и Эби [108 I. Если изменение молярного коэффициента светопоглощения мешающего компонента i линейно связано с изменением длины волны
егХ = ° + М (5.19)
то комбинацией этой зависимости для трех аналитических длин волн Х2 и Хц получают уравнение:
(!Цй — ''Х2)/(11Лз — eiAi)= (^3 — лг)/(^з ^1) (5.20)
Обозначим
(Z3 — ^2)/(^з — ^i) = р (5.21)
п подставим р в уравнение (5.20), тогда
«’ixa-РЧА1 —(1 — Р)чх3 = 0 (5.22)
Затем умножим уравнение (5.17) на р, а уравнение (5.18) — на (1 — р) и вычтем их почленно из уравнения (5.16). Тогда после преобразования получим:
— (1 — Р) А3 = Сх [ехАа — рьжА1 — (1 — р) вхАз] I +
+ Cj |егАа —регА1—(1—р)е1Аз] Z (5.23)
150
Но согласно равенству (5.22), слагаемое во второй квадратной скобке уравнения (5.23) равно нулю, поэтому получим:
Сх = [Д;,2 — рАК1 — (1 — р) ДХз]/{[®хЛ2 — ₽£хХ1 — (1 — Р) ®хл3] Ц (5.24)
Уравнения (5.23) и (5.24) справедливы для любого выбора грех длин волн, при которых наблюдается линейная зависимость оптической плотности мешающих компонентов от длины волны.
Метод Брайса—Швайна [108]. В этом приеме выбирают три аналитические длины волны Х2 и Х3, равноотстоящие друг от друга. При таком условии уравнение (5.21) упрощается и р = 0,5. Тогда уравнение (5.24) принимает более простой вид:
Сх= (2^Х2 ^Х3)/[(2ехЛ2 ®кХ3) П (5.25)
Знаменатель этого уравнения можно найти по оптическим плотностям стандартного раствора определяемого элемента с известной концентрацией Сст:
(2ехЛ2 —®xZj — ®*Х3) I = (^ДстЛ2 ~ Д^ст/бст (5.26)
Если таким же образом обозначить и числитель уравнения (5.25)
2Д/2-ДХ1-ДХз = ДД (5.27)
то конечная форма расчетного уравнения приобретает совсем простое выражение:
Сх = СС1 (ДД/ДДСТ) (5.28)
Возможность применения метода Брайса — Швайна, как и допущение о линейной зависимости светопоглощения от длины волны, проверяют простым приемом [103]. Выбирают вместо и Х3 две другие длины волны и ХБ, тоже равноотстоящие от Л2, и находят для них функцию:
ДД' = 2ДХ2-Д?14-ДЛ5 (5.29)
Если лежащие в основе метода допущения справедливы, то соблюдается тождество
ДД/ДД = ДДС1/ДД„ (5.30)
которое подтверждает линейный характер изменения светопоглощения мешающего компонента в интервале длин волн Х4 — Х6.
При выборе аналитических длин волн следует учитывать две противоположные тенденции. Чем меньше расстояние между аналитическими длинами волн, тем вероятнее и точнее выполнение основного условия применимости метода — линейной зависимости светопоглощения мешающего компонента в используемом интервале спектра. Но с другой стороны, чем меньше расстояние между выбранными длинами волн, тем меньше разница в значе-
151
ниях экспериментально измеряемых оптических плотностей (вследствие этого происходит так называемая «потеря точности при вычитании») и тем больше погрешность в расчете концентрации по уравнению (5.28).
Метод Зиньковской и Киреева [223]. Если для поглощения мешающих компонентов справедливо уравнение (5.19), то для любых длин волн Х7 и Х2, симметричных относительно длины волны X, сумма молярных коэффициентов светопоглощения мешающих компонентов постоянна:
+ ei?.2 = 2а t (Хх -f- Х2) — 2а -f- 2bk = q (5.31)
Сумма оптических плотностей раствора смеси при этих же длинах волн будет равна:
Д?! + Д?.2 — Сх (е*Л1 + ех?.2) I + tfCi (5.32)
Для нескольких пар длин волн, симметричных относительно X, уравнение (5.32) является уравнением прямой с тангенсом угла наклона, равным Сх. Если значения и е^2 неизвестны, то их можно заменить соответствующими значениями оптических плотностей стандартного раствора определяемого компонента с концентрацией Сст. Тогда уравнение (5.32) примет окончательное выражение:
= (Сж/Сст) 0сгЛ1"Ь ^стХа) "Ь (5.33)
Значение Сх можно найти как аналитическим, так и графическим методом из зависимости + А^2 — f(ACT^t + ЛСтл2).
Второй вариант этого метода основан на постоянстве разности молярных коэффициентов светопоглощения мешающего компонента для любых пар длин волн, находящихся на равном расстоянии ДХ друг от друга:
егЛ1 — eiZ2 = ь (Х1 — Х2) = MX (5.34)
Разность оптических плотностей раствора анализируемой смеси при этих длинах волн описывается уравнением прямой-с тангенсом угла наклона, равным Сх
Длх — Дл2 = Сл (ел-^! — е*л2) I + bl (5.35)
или после замены разности ехл, — ехл2 на соответствующую ей разность оптических плотностей Лстх, —Лстх2 Для стандартного раствора определяемого элемента с концентрацией Сст — уравнением прямой с тангенсом угла наклона СЛ/Сст:
— ^л2 — (Сх/Сст) (ДстЛх — ЛстЛг) + (5.36)
Здесь Сх находят так же, как и в предыдущем случае: аналитическим путем или графическим из зависимости — А^г = 152
= f(ACTKt—Лст12). Для уменьшения погрешностей в определении неизвестной концентрации Сх необходимо использовать не менее 4—5 пар длин волн и параметры линейной зависимости рассчитывать методом наименьших квадратов.
6.4. СПОСОБЫ УЧЕТА ПОГЛОЩЕНИЯ ФОНА
6.4.1. ОПРЕДЕЛЕНИЕ МИКРОПРИМЕСЕЙ
В АНАЛИЗИРУЕМЫХ ОБРАЗЦАХ И В ХОЛОСТОЙ ПРОБЕ [81]
Определяемый минимум и точность фотометрического определения тесно связаны с погрешностями аналитического метода, которые, как правило, возрастают с увеличением содержания определяемых микропримесей в применяемых реагентах. Проблема учета содержания примесей в реагентах и снижение их содержания в холостой пробе является одной из важнейших при определении следов элементов. Как показал А. Б. Бланк [81 ], присутствие определяемой примеси в реагентах не вносит каких-либо систематических погрешностей только при строгом соблюдении основного закона светопоглощения. Поэтому прямые измерения оптической плотности анализируемых растворов относительно раствора холостой пробы допустимы только при строгой прямолинейности * градуировочных графиков А — f (С), проходящих через начало координат.
Если градуировочный график А — f (С) оказывается не прямолинейным (что бывает в большинстве случаев при поглощении полихроматического света), то измерения оптической плотности раствора относительно холостой пробы приводят к большим систематическим погрешностям, и только в том случае, когда реагенты практически не содержат определяемой микропримеси, способ прямых отсчетов дает удовлетворительные результаты.
При наличии определяемой микропримеси в реагентах для получения правильных результатов измерения оптической плотности исследуемого и холостого растворов нужно производить последовательно относительно чистого растворителя или раствора реактива строго постоянной концентрации. Определяемое количество микропримеси находят по разности его общего содержания и содержания в холостой пробе.
Измерив оптические плотности анализируемого раствора и холостой пробы (Аан и Ао), по градуировочному графику (рис. 5.2) находят соответствующие им количества определяемой микропримеси 9ан и q0, а по их разности — искомое количество микропримеси: qx = qau — q0.
* Прямолинейность градуировочного графика проверяют математически по уравнению прямой линии методом наименьших квадратов (см. гл. 9).
153
Рис. 5 2. Градуировочный график для раздельного определения количества микропримеси в анализируемом растворе (даи) и в холостой пробе (q0).
Рис. 5.3. Графическое определение оптической плотности анализируемого раствора (Лан) в присутствии смолообразных веществ и нефильтруюшейся взвеси.
— длина волны максимального поглощения света анализируемым раствором; ?-4 — длина волны начала полосы поглощения; — длина волны конца полосы поглощения; Хз — длина волны вне полосы поглощения.
При последовательных отсчетах относительная погрешность определения сохраняется одинаковой вплоть до 1,5-кратного содержания примеси в реагентах по сравнению с определяемой величиной.
6.4.2. АНАЛИЗ В ПРИСУТСТВИИ СМОЛООБРАЗНЫХ ВЕЩЕСТВ ИЛИ НЕФИЛЬТРУЮЩЕЙСЯ ВЗВЕСИ [224]
Присутствие в анализируемых растворах смолообразных веществ I ли тонкодисперсной нефильтрующейся взвеси вызывает существенные затруднения при фотометрических определениях вследствие того, что избирательное поглощение света определяемым веществом маскируется неизбирательным поглощением этих примесей. Однако, если светопоглощение смолообразных веществ пли взвеси в аналитических точках остается постоянным и его удается измерить, то эти примеси можно рассматривать как компоненты анализируемой смеси и соответствующим образом учитывать их светопоглощение. Для учета светопоглощения пользуются различными приемами.
Метод базисных линий*. Сущность метода базисных линий состоит в учете влияния неизбирательного поглощения путем проведения линии нулевой оптической плотности или нулевого поглощения. В качестве такой линии принимают прямую, соединяющую точки видимого начала и конца полосы поглощения на спектральной кривой анализируемого вещества (рис. 5.3, прямая Л2^4). При измерении оптической плотности при за
* Этот прием иногда называют методом «гетерохроматической интерполяции».
154
оптическую плотность раствора определяемого вещества Лан принимают отрезок AAlt отсекаемый базисной линией на вертикали А ]&!
Благодаря простоте метод получил широкое распространение. Однако он имеет и недостатки. Использование метода базисных линий требует проведения измерений во многих точках или записи кривой всего максимума поглощения, а при наложении спектров поглощения двух анализируемых компонентов метод практически неприменим. При нечетко выраженном максимуме метод базисных линий недостаточно чувствителен, дает малый наклон градуировочных графиков А = f (С), а при положении аналитической точки вне максимума поглощения — непригоден.
Метод гетерохроматической экстраполяции [225]. В этом методе оптическая плотность Ао, обусловленная смолообразными веществами (отрезок BZj на рис. 5.3), определяется в аналитической точке путем линейной экстраполяции по значениям оптических плотностей А2 и А3, измеренным при длинах волн Х2 и Л3 вне полосы поглощения.
Значение оптической плотности Лан раствора определяемого вещества (отрезок связано с измеренным значением оптической плотности Аг следующим соотношением: Лан = — Ао.
Из подобия треугольников ОЛ2Л3 и СВЛ3 вытекает, что
DA2:CB = DAs:CAa
откуда
СВ — DA2CAa/DAa = [(Х3 — Xj)/(Z3 — Х2)] (А2 Л3)
Следовательно
Ло = Д3 + (^2 — ^з) [(Х3 — М/(Х3 — Х2)] (5.37)
лап = — лз — (^г — Лз) [(*з — МЖ — Х2)] (5.38)
Если наклон прямой линии Д2Л3 мал Иг ~ -^з)» то> пренебрегая разницей в поглощении примесей при Xt и Х2, значение оптической плотности Лан находят как разность между Лх и А2 (Аав rz
Л - Л).
Метод гетерохроматической экстраполяции позволяет изменять длину волны аналитической точки и даже проводить анализ в аналитической точке, лежащей вне максимума поглощения. При измерении оптической плотности при длине волны максимального поглощения наклон градуировочного графика А = = f (С) получается максимальным. Измерения оптической плотности производят только в трех определенных точках (при Хг, Х2 и Х3).
Недостатком метода является обязательное отсутствие поглощения индивидуальных (исходных) веществ на используемом участке спектра.
155
Г лава 6
АППАРАТУРА И ТЕХНИКА ФОТОМЕТРИЧЕСКИХ ИЗМЕРЕНИЙ
Фотометрические методы определения концентрации растворов основаны на сравнении поглощения или пропускания света стандартными и исследуемыми растворами. Степень поглощения света фотометрируемым раствором измеряют с помощью специальных оптических приборов — фотоколориметров и спектрофотометров, в которых световая энергия, проходящая через фото-метрируемый раствор, с помощью фотоэлементов преобразуется в электрическую. Согласно законам фотоэффекта, сила возникающего фототока прямо пропорциональна интенсивности света, падающего на фотоэлемент. Следовательно, отношение интенсивностей световых потоков, содержащееся в выражении основного закона светопоглощения, может быть заменено равным ему отношением фототоков. Это и используют в фотометрическом анализе, где фактически измеряют не светопоглощение растворов, а значения возникающих фототоков.
При работе на приборах с фотоэлементами следует иметь в виду следующие обстоятельства, влияющие на точность и воспроизводимость получаемых результатов.
1 Спектральная и интегральная чувствительность фотоэлемента со временем уменьшается, и при большой эксплуатационной нагрузке «старение» фотоэлемента вызывает необходимость его замены.
2. Для фотоэлементов характерно явление «утомление», т. е. уменьшение силы фототока при длительном непрерывном освещении фотоэлемента достаточно ярким светом. Поэтому для получения воспроизводимых результатов во вр₽мя работы для фотоэлементов необходим «отдых», т. е. временное прекращение его облучения путем перекрывания световых потоков защитной шторкой. Обычно при работе с фотоэлементами создают постоянную периодичность его облучения и «отдыха».
3. Чувствительность фотоэлемента бывает неодинаковая по всей его поверхности, поэтому для получения воспроизводимых и точных результатов необходимо так поставить осветитель, чтобы при параллельных измерениях всегда освещался один и тот же участок поверхности фотоэлемента, причем площадь этого участка не должна быть меньше 0,8 см2. Иногда равномерная освещенность фотоэлементов достигается использованием матовых рассеивателей.
4. Для того, чтобы наблюдалась линейная зависимость между силой фототока во внешней цепи фотоэлемента и интенсивностью падающего на него светового потока, сопротивление гальвано-156
метра должно быть по возможности малым и не должно превышать внутреннего сопротивления фотоэлемента.
5. Для получения хорошо воспроизводимых и надежных результатов при выборе эффективной длины волны (светофильтра) необходимо учитывать не только спектральные характеристики поглощения фотометрируемого раствора и применяемого светофильтра, но и характеристику спектральной чувствительности используемого фотоэлемента. Изменение спектральной чувствительности фотоэлементов может обусловливать соответствующее изменение эффективной длины волны (светофильтра) при фото-метрировании растворов по одной и той же методике и даже на приборах одного и того же типа.
6.1. ОБЩИЕ ЗАМЕЧАНИЯ
ПРИ РАБОТЕ НА ФОТОНОЛОРИМЕТРАХ
Надежность результатов измерений при работе на фотоколориметрах и фотометрах обеспечивается, в первую очередь, правильной установкой (юстировкой) и эксплуатацией приборов. Поэтому приступать к измерениям можно только после тщательного ознакомления с устройством прибора и правилами его эксплуатации. Измерения на фотоэлектрических приборах можно начинать через 15—20 мин после включения прибора для того, чтобы установился режим накала лампы осветителя.
Большое значение для получения правильных результатов имеет чистота кювет. Кюветы всегда должны быть тщательно вымыты; желательно хранить их заполненными дистиллированной водой. Брать кюветы при измерениях можно только за боковые стенки, через которые не проходит поглощаемый световой поток.
Растворы сравнения (нулевые растворы). Измерение оптической плотности стандартного и исследуемого окрашенных растворов всегда производят по отношению к раствору сравнения (нулевому раствору). В качестве раствора сравнения можно использовать аликвотную часть исследуемого раствора, содержащего все добавляемые компоненты, кроме реагента, образующего с определяемым ионом окрашенное соединение. В том случае, когда сам реагент имеет окраску, раствор сравнения приготавливают следующим образом: к небольшому количеству дистиллированной воды прибавляют реагент и все компоненты (кроме определяемого) в тех же количествах, что и при приготовлении окрашенных растворов определяемого вещества. Затем раствор доводят водой до требуемого объема и перемешивают. Если добавляемый реагент и все остальные компоненты раствора сравнения бесцветны и, следовательно, не поглощают лучей в видимой области спектра, то в качестве раствора сравнения можно использовать дистиллированную воду.
157
При небольшом избытке реагента оптические плотности растворов окрашенного комплекса и чистого реагента целесообразнее измерять отдельно по отношению к чистому растворителю и затем косвенным приемом определить оптическую плотность ДЛ обусловленную поглощением только анализируемого комплекса (см разд. 3.2.1).
Поправка на холостой опыт. Выделение определяемых компонентов из разбавленных растворов и отделение их от мешающих элементов при анализе веществ высокой степени чистоты производят обычно химическим путем с помощью различных реактивов, посуды, аппаратуры. Хотя для этих целей, как правило, применяют специально очищенные реактивы и дважды перегнанную воду, все же они могут содержать определяемую примесь, а стеклянная и кварцевая аппаратура тоже частично растворяется под действием применяемых кислот, щелочей и т. д. * Поэтому при фотометрических определениях микропримесей элементов всегда проводят холостой опыт, т. е. проделывают все те же операции с реактивами только без анализируемого вещества. Обычно в полученном растворе почти всегда обнаруживают какое-то количество искомого вещества. Эту поправку на холостой опыт вычитают из полученного результата определения. Для достижения более высокой чувствительности (при прочих равных условиях) необходимо, чтобы поправка на холостой опыт была бы значительно меньше определяемого количества примеси.
При количественной оценке предела обнаружения большое значение имеет корректная постановка холостого опыта. Обычно холостой опыт выполняют без анализируемого образца со всеми добавленными реактивами, проводя их через все стадии анализа, предусмотренные методикой. Однако полученные таким образом результаты могут оказаться не всегда корректными, поскольку остатки матриц в растворе пробы могут оказывать влияние на результаты опредечения микроэлементов, в то время как в растворе холостого опыта, не содержащего растворенной пробы, подобных влияний нет Ц95]. Для более строгого учета влияния солевого фона растворенной пробы следует использовать методику проведения холостого опыта с применением двух разных навесок пробы [81 ] либо с введением в раствор холостого опыта какой-то части анализируемой пробы, добавляемой в фотометри-руемый раствор [79 J. В последнем случае находят содержание определяемого микрокомпонента в фотометрируемом (анализируемом) и холостом растворах, а затем рассчитывают фактическое содержание определяемого элемента в холостой пробе по уравнениям:
а = С + X; Ь = гС + Х
С = (а — £>)/(! — г); Х = а — С = Ъ — гС
* В последнее время вместо стеклянной и кварцевой посуды широко применяют посуду из полиэтилена и фторопласта, которая не разрушается многими щелочами и кислотами.
158
Здесь а и b — найденные общие содержания (мкг) определяемого элемента в исследуемом и холостом растворах; С — содержание определяемого элемента в анализируемом растворе, мкг; X — фактическое содержание определяемого элемента в холостой пробе (опыте), мкг; г — аликвотная часть анализируемого раствора, добавленная в раствор холостой пробы.
6.2. ФОТОКОЛОРИМЕТРЫ
6.2.1. КОЛОРИМЕТРЫ ФОТОЭЛЕКТРИЧЕСКИЕ КФК, ФЭК-Б6М
Назначение. Технические данные. Колориметры фотоэлектрические типа КФК, ФЭК-56М, ФЭК-56 предназначены для измерения пропускания или оптической плотности растворов в диапазоне 315—630 нм и определения концентрации веществ в растворе фотометрическими методами. Приборы позволяют также производить относительные измерения интенсивности рассеяния взвесей, эмульсий и коллоидных растворов в проходящем свете. Приборы ФЭК-56М, ФЭК-56 могут комплектоваться дополнительным титровальным приспособлением ТПР, которое позволяет проводить фотометрическое титрование.
Все рассматриваемые приборы обеспечивают измерение пропускания от 100 до 5 % (А = 04-1,3). Участок шкалы пропускания от 5 до 0,1 % (А — 1,34-3) служит для ориентировочных измерений. Абсолютная погрешность прибора при измерении пропускания не превышает Т = 1%. Среднее квадратичное отклонение определения пропускания по результатам 10 измерений не превышает sr = 0,3% * (0,003).
Таблица 6.1. Характеристики светофильтров приборов КФК, ФЭК-56М, ФЭК-56
Номер иа рукоятке Маркировка светофильтра Длина волны, соответствующая максимуму пропускания, им Полуширина ПОЛОСЫ пропускания, нм
1 1 315+5 35±15
2 2 364+5 25±10
3 3 400+5 45±10
4 4 440+5 40+15
5 5 490±10 35+10
6 6 540+10 25±10
7 7 572+10 30+10
8 8 590±!0
9 9 630±10
Примечание. Светофильтр № 9 служит для ориентировочных измерений.
Для светофильтров № 8 и № 9 указана предельная длина волны ХПр от максимума.
* Строго значение этой характеристики зависит от абсолютного значения Т (см. разд. 3.1.3).
159
Рис. 6.1 Спектральные характеристики светофильтров в фотоколориметрах КФК, ФЭК56М, ФЭК -56:
J—9 — номера светофильтров.
В качестве источника света в приборе КФК используют лампу накаливания КГМ 6,3-15 (6,3 В, 15 Вт), с которой возможна работа в диапазоне длин волн 315—630 нм. В приборах ФЭК-56, ФЭК-56М
применяют лампу накаливания РН-35 (8 В, 35 Вт) и ртутнокварцевую лампу ДРК-120 сверхвысокого давления мощностью
120 Вт, обеспечивающие возможность работы в диапазоне 315— 630 нм. Все приборы снабжены набором узкополосных свето-
фильтров, спектральные характеристики которых представлены на рис. 6.1 и в табл. 6.1.
Оптическая схема и общий вид фотсколориметров. Фотоколориметры КФК, ФЭК-56М, ФЭК-56 имеют общую оптическую схему, представленную на рис. 6.2. Световой поток от источника света /, пройдя через светофильтр 2, попадает на призму 3, которая делит поток на два: левый и правый. Далее параллельные потоки идут через кюветы 4—4 или 4—4', диафрагмы 5, 6 и попадают на фотоэлементы 7, включенные по дифференциальной
схеме через усилитель постоянного тока на микроамперметр. В правый световой поток можно последовательно вводить кювету 4 с растворителем (или раствором сравнения) или кювету 4'
Рис. 6.2. Оптическая схема фотоколориметров КФК, ФЭК-56М, ФЭК-56:
1 — источник света; 2 — светофильтр; 3 — призма; 4 — кювета с растворителем или раствором сравнения; 4' — кювета с исследуемым раствором; 5 — раздвижная диафрагма; 6 — компенсационная диафрагма; 7 — фотоэлементы.
160
Рис. 6.3. Внешний вид колориметра фотоэлектрического концентрационного КФК:
1 — микроамперметр; 2 — крышка кюветного отделения; 3 — ручка шторки для перекрывания световых потоков; 4, 9 — рукоятка регулировки чувствительности прибора; 5 — рукоятка перемещения кювет (4, 4' на рис, 6.2) в правом световом потоке; 6 — рукоятка измерительной диафрагмы (5 на рис. 6.2); 7 — барабан со шкалой Т (черная) и А (красная); 8 — рукоятка компенсирующей диафрагмы; 10 — рукоятка установки стрелки микроамперметра на «О»; 11 — рукоятка перемещения светофильтров.
с исследуемым раствором. Раздвижная диафрагма 5, расположенная в правом потоке света, при вращении связанного с ней барабана изменяет значение светового потока, падающего на правый фотоэлемент. Правый барабан является измерительным, левый — компенсационным.
Внешний вид фотоколориметра КФК представлен на рис. 6.3. В отличие от этого прибора предшествующая модель — ФЭК-56М снабжена одной рукояткой регулировки чувствительности 9, а более ранняя модель — ФЭК-56 — индикаторной лампой вместо микроамперметра 1. Однако в последнем случае для регистрации уравнивания интенсивности левого и правого световых потоков возможно применение выносного микроамперметра вместо индикаторной лампы.
Порядок работы. Общие указания-.
1. Методика определения концентрации вещества как в окрашенных, так и в мутных растворах одна и та же. Поэтому дальнейшее описание техники измерений (Т или Л) является общим как для фотоколориметрических, так и турбидиметрических определений.
2. Измерения на приборе можно проводить спустя 15—20 мин после включения блока питания и лампы накаливания, когда наступает стабильный режим ее работы. Ртутную лампу включают за 10—15 мин до начала измерения при условии 15—20-минутного прогрева блока питания и лампы накаливания.
6 М- И _ Булатов и др. 161
3. Нельзя оставлять без надобности включенной ртутную лампу прибора, так как это сокращает срок ес службы и, кроме того, лампа разогревает светофильтры прибора, что нежелательно. При возникновении перерыва в работе на время больше 20 мин ртутная лампа должна выключаться.
4. Иногда при работе с некоторыми светофильтрами (приборы ФЭК-56М, ФЭК-56) поступающий на фотоэлементы световой поток оказывается чрезмерно высоким, что приводит к нестабильности работы прибора. Это проявляется в колебании стрелки микроамперметра. В таких случаях необходимо уменьшить чувствительность схемы фотоколориметра, повернув рукоятку чувствительности по часовой стрелке; либо, если нестабильность остается высокой, установить в поток лучей поглотители, прикладываемые к прибору. Поглотители устанавливают в световые окна в кюветном отделении.
Измерение пропускания или оптической плотности раствора. Измерения производят при закрытой крышке кюветного отделения. Прежде всего устанавливают «электрический нуль» прибора. Для этого с помощью ручки 3 (см. рис. 6.3) перекрывают световые потоки шторкой. Рукояткой 10 устанавливают стрелку микроамперметра на «0», после чего открывают шторку. С помощью рукоятки 11 вводят в световой поток выбранный светофильтр. Все измерения производят при чувствительности * электросхемы 1—3 деления микроамперметра при раскрытии измерительной диафрагмы рукояткой 6 на 1 % пропускания. Указанную чувствительность прибора устанавливают вращением рукояток 4** и 9 — на приборе КФК и рукоятки 9 — на приборе ФЭК-56М.
В левом световом потоке на все время измерений устанавливают кювету с растворителем (или раствором сравнения, «холостым» раствором). Если растворитель не окрашен, рекомендуется в левый поток ставить кювету с дистиллированной водой для того, чтобы исключить возможность разогревания левого фотоэлемента теплом светового потока. В правый поток света помещают кювету с исследуемым раствором. Правый барабан 7 вращением рукоятки 6 устанавливают на отсчет 100 по шкале пропускания. Вращением левого барабана (рукоятки 8) добиваются установки стрелки микроамперметра на «0». Если левым барабаном установить «0» не удается, то в правый световой поток (в световое окно) следует установить ослабитель «1» или «2» из комплекта прибора. Затем поворотом рукоятки 5 в правом потоке кювету с раствором заменяют кюветой с растворителем (или раствором сравнения). При этом происходит смещение стрелки
* Чувствительность прибора определиют числом делений по шкале микроамперметра, на которое отклонится стрелка при раскрытии измерительной диафрагмы на 1% пропускания.
** Рекомендуемое положение рукоятки 4 дли светофильтров №№ 1, 2, 8 и 9 — «4» или «5», для остальных — «2» или «3».
162
микроамперметра, установленной на «О». Вращением правого измерительного барабана добиваются первоначального нулевого положения стрелки и производят отсчет пропускания (оптической плотности) исследуемого раствора по шкале правого барабана 7.
В некоторых случаях, особенно при изучении кинетических зависимостей, используют и другую методику измерений. Сначала в оба потока света помещают кюветы с чистым растворителем (или «холостым» раствором), вращением рукоятки 6 правый барабан 7 устанавливают на отсчет 100 по шкале пропускания и вращением левого компенсационного барабана (рукоятка 8) устанавливают стрелку амперметра на «0». Затем в левый кювето-держатель помещают кювету с анализируемым раствором и вращением правого измерительного барабана стрелку микроамперметра вновь устанавливают на «0». Отсчет показаний оптической плотности берут по шкале правого барабана.
Для исключения случайных промахов, которые могут возникнуть в процессе измерения, рекомендуется не ограничиваться одним измерением. При измерениях барабан измерительной диафрагмы следует подводить к индексу Т (А) с одной стороны для исключения влияния люфта в механизме.
При определении концентрации вещества в растворе рекомендуется соблюдать следующую последовательность в работе.
а. Выбор светофильтра — если спектр поглощения анализируемого раствора не известен, то его приближенный вид определяют следующим образом. Заполняют кювету исследуемым раствором и измеряют его оптическую плотность, последовательно используя все светофильтры. По полученным данным строят кривую А = f (X); выбирают область спектра, где оптическая плотность, во-первых, имеет максимальное значение и, во-вторых, мало изменяется с изменением длины волны. Выбирают такой светофильтр, у которого область максимального пропускания соответствует отмеченному выше участку спектра поглощения исследуемого раствора. Если эти условия выполняются для нескольких светофильтров, то выбирают тот из них, для которого чувств! тельность фотоэлемента выше (см. также гл. 2). Светофильтр можно выбирать также по наибольшему значению измеренной оптической плотности раствора.
б. Выбор кюветы — определяется оптимальным диапазоном измеряемых оптических плотностей (см. разд. 3.1.3). Приборы ФЭК-56М, КФК комплектуются наборами кювет:
Рабочая длина кюветы, мм 50 30 20 10 5 3 1
Объем кюветы, мл 20 14 9 5 2,3 1,4 0,5
Помимо этого, по дополнительному требованию заказчиков прибор КФК может комплектоваться для микроанализа держа
6* 163
телем и комплектом микрокювет:
Рабочая длина микрокюве- 10 б 3 2
ты, мм
Объем кюветы, мл 0,40 0,20 0,12 0,08
в. Выбор методики определения концентрации — см. разд.
7.1.1; 7.1.2.3; 7.3.
6.2.2. КОЛОРИМЕТР ФОТОЭЛЕКТРИЧЕСКИЙ КОНЦЕНТРАЦИОННЫЙ КФК-2
Назначение. Технические данные. Однолучевой фотоколориметр КФК-2 предназначен для измерения пропускания, оптической плотности и концентрации окрашенных растворов, рассеивающих взвесей, эмульсий и коллоидных растворов в области спектра 315—980 нм. Пределы измерения пропускания 100—5% (Д — = 0-1-1,3). Основная абсолютная погрешность измерения пропускания 1%. Характеристики светофильтров представлены в табл. 6.2.
Прибор КФК-2 может комплектоваться широким набором прямоугольных кювет:
Набор № 2 — основной
Рабочая длина кюветы, мм 50 30 20 10 5
Объем, мл 20 14 9 5 2,3
Набор № 1
Рабочая длина кюветы, мм 20 10 5 3 1
Набор № 3
Рабочая длина кюветы, мм 100 50 30 20
Мнкрокюветы
Рабочая длина микрокюветы, мм 10 5 3 2
Объем, мл 0,40 0,02 0,12 0,08
Примечание Наборы 1,3 и микрокюветы поставляются по желанию Заказчика
Таблица 6.2. Спектральные характеристики светофильтров к фотоколориметру КФК-2
Маркировка на .писке Маркировка светофильтра Длина волны, соответствующая максимуму пропускания, нм Полуширина полосы пропускания, нм
1 315 315+5 35+15
2 364 364+5 25+10
3 400 400+5 45+10
4 440 440+10 40+15
5 490 490± 10 35±10
6 540 540+10 25+10
7 590 590+10 25±10
8 670 670+5 20+5
9 750 750+5 20±5
10 870 870+5 25+5
11 980 980±5 25±5
164
Рис. 6.4 Принципиальная оптическая схема фотоколориметра КФК-2:
1 — источник света; 2 — теплозащитный светофильтр 3 — нейтральный светофильтр^ 4 — цветной светофильтр; 5 — кювета с исследуемым раствором или раствором сравнения;
6 — пластина, которая делит световой поток на два потока. 7 — фотодиод, 8 — фотоэлемент.
Оптическая схема и общий вид прибора КФК-2. Принципиальная оптическая схема и общий вид фотоколориметра КФК-2 представлены на рис. 6.4 и 6.5. Свет от галогенной малогабаритной лампы (КГЛ1 6,3-15) 1 проходит последовательно через систему линз, теплозащитный 2, нейтральный 3, выбранный цветной 4 светофильтры, кювету 5 с раствором сравнения или с исследуемым раствором, попадает на пластину 6, которая делит световой поток на два: 10% света направляется на фотодиод (ФД-7К) 7 (при измерениях в области спектра 590—980 нм) и 90% — на фотоэлемент (Ф-26) 8 (при измерениях в области спектра 315—540 нм).
Общий вид прибора КФК-2 представлен на рис. 6.5. В качестве регистрирующего прибора применяют микроамперметр 7 типа М-907, оцифрованный в микроамперах и имеющий шкалу 0— —100 дел, соответствующую шкале пропускания Т, или Л1-907-10 со шкалой, оцифрованной в делениях пропускания и оптической плотности. На задней стенке крышки микроамперметра имеются гнезда для подключения цифрового вольтметра с пределом измерения 0,1 В.
Последовательность операций при определении концентрации вещества в растворе (выбор светофильтра, кювет, способа измерений) на приборе КФК-2 такая же, как и на фотоколориметре КФК.
Рис. 6.5. Общий вид прибора КФК-2:
J — осветитель, 2 — рукоятка ввода цветных светофильтров, 3 —- кюветное отделение; 4 — рукоятка перемещения кювет с раствором сравнения и исследуемым раствором; 5 — рукоятка (ввода фотоприем-ников в световой поток) «Чувствительность», б рукоятка настройки прибора на 100%-ное пропускание; 7 —- микроамперметр.
165
Таблица 6.3. Набор кювет и измерительных приставок к прибору Spekol 10
Обозначения кювет Рабочая длина кюветы, см Объем раствора (в мл) иа 1 см толщины слоя Тип измерительной приставки
Малая кювета 0,1; 0,2; 0,5; 0,1; 1,2 ЕК 1
0,2; 0,5; 1; 2; 0,8 ЕК 5
3; 5
Микрокювета 1; 2; 3 0,1 EK Ti
6.2.8. МНОГОЦЕЛЕВОЙ ФОТОМЕТР Spekol 10
Фирма Carl Zeiss lena (ГДР) выпускает многоцелевой однолучевой фотометр Spekol 10 с различными типами приставок. Прибор позволяет производить измерения пропускания или оптической плотности в диапазоне 340—850 нм. В качестве диспергирующего устройства используют прецизионную дифракционную решетку с 651 линиями на 1 мм; цена деления шкалы барабана длин волн 1 нм. Для измерения оптической плотности фотометр снабжен приставками и наборами кювет (табл. 6.3). Для проведения титрований растворов на основе измерений оптической плотности (фотометрическое титрование — см. разд. 7.6), помутнения или флуоресценции прибор комплектуется титровальной приставкой типа Ti, снабженной кюветой (/ = 2 см) с наименьшим титруемым объемом 15 мл.
6.3. СПЕКТРОФОТОМЕТРЫ
6.3.1. СПЕКТРОФОТОМЕТРЫ СФ-26, СФ-16
Назначение. Технические данные. Однолучевые спектрофотометры СФ-26 и СФ-16 предназначены для измерения пропускания и оптической плотности растворов и твердых веществ в диапазоне 186—1100 нм.
Некоторые технические характеристики прибора СФ-26:
Диапазон измерений пропускания
Растяжка участков шкалы пропускания на всю шкалу: от любого целого числа десятков процентов в области от 0 до 10%
Стандартное отклонение пропускания (при п = 10) sj-*, не более: по шкале стрелочного прибора 0—110% по шкалам-растяжкам на стрелочном приборе по табло цифрового вольтметра
Абсолютное значение рассеянного излучения при длине волны 200 нм, не более
3—100%
0,25% (0,0025)
0,1% (0,001)
0,1% (0,001)
1%
* Строго Sj- зависит также от абсолютного значения Т (см. разд. 3.1.3),
166
Рис. 6.6. Оптическая схема однолучевого спектрофотометра:
1 — источник света; 2 — зеркальный конденсор; 3 — веркало; 4 — входная щель;
5, 12 — защитные пластинки; 6 — зеркальный объектив; 7 — кварцевая призма;
8 — выходная щель; 9 — кварцевая линза;
10 — фильтры; 11 — кювета с исследуемым нли стандартным раствором; 13 — фотоэлемент.
Спектрофотометр СФ-26 поставляется в двух вариантах комплектации — основном и дополнительном, включающем цифровой вольтметр Щ 1312, который предназначен вместо стрелочного прибора для более объективного измерения пропускания (оптической плотности).
Оптическая схема. В основе отечественных однолучевых спектрофотометров, начиная с СФ-4 по СФ-26, лежит общая принципиальная оптическая схема (рис. 6.6) (за исключением позиций 9—13 — для СФ-26). Свет от источника 1 попадает на зеркальный конденсор 2, затем на плоское зеркало 3. Зеркало отклоняет поток лучей на 90° и направляет его в щель 4 (автоколлима-ционного монохроматора с 30°-ной призмой), защищенную пластинкой 5. Свет, прошедший через щель, попадает далее на диспергирующую призму 7, разлагающую его в спектр; диспергированный поток направляется обратно на объектив, который фокусирует лучи в щель 8. Призма соединена с помощью специального механизма со шкалой длин волн. Поворачивая призму вращением соответствующей рукоятки на выходе монохроматора, получают монохроматический поток света заданной длины волны, который, пройдя щель 8, кварцевую линзу 9, фильтр 10, поглощающий рассеянный свет, эталон (или образец) 11 и защитную
Рис. 6.7. Внешний вид спектрофотометра СФ-26:
1 — монохроматор; 2 — шкала длин волн; 3 — измерительный прибор; 4 — осветитель с источником излучения и стабилизатором; 5 — кюветное отделение; 6 — рукоятка перемещения каретки с кюветами; 7 — камера с фотоприемниками и усилителем; 8 — рукоятка переключения фотоэлементов; 9 — рукоятка установки чувствительности? 10 — рукоятка установки на «0»; 11 —рукоятка шторкн; 12 — рукоятка раскрытия вход-* ной и выходной щелей (щели открываются в пределах 0,01—2 мм); 13 ® рукоятка sot< счет»; 14 = рукоятка компенсации; 15 — рукоятка шкалы длин волн.
167
пластинку 12, попадает на светочувствительный слой фотоэлемента 13.
В случае прибора СФ-26 после линзы 9 свет проходит через эталон (или образец), линзу и с помощью поворотного зеркала собирается на светочувствительном слое одного из фотоэлементов: сурьмяно-цезиевого (для измерений в области длин волн 186—650 нм) или кислородно-цезиевого (600—1100 нм). Источниками сплошного излучения, обеспечивающими широкий диапазон работы прибора, служат дейтериевая лампа (в области длин волн 186—350 нм) и лампа накаливания (320—1100 нм).
Устройство прибора СФ-26 и принцип измерений. Внешний вид прибора СФ-26 представлен на рис. 6.7. Измерение пропускания (оптической плотности) исследуемого объекта производят относительно эталона, пропускание которого принимается за 100%, а оптическая плотность — равной нулю. Прибор СФ-26 может комплектоваться приставкой ПДО-5, позволяющей снимать спектры диффузного отражения твердых образцов.
6.3.2. СПЕКТРОФОТОМЕТР СФ-46
Назначение. Технические данные. Однолучевой спектрофотометр СФ-46 со встроенной микропроцессорной системой предназначен для измерения пропускания, оптической плотности жидких и твердых веществ в области 190—1100 нм. Диспергирующим элементом служит дифракционная решетка с переменным шагом и криволинейным штрихом.
Некоторые технические характеристики прибора:
Диапазон измерений пропускания
Абсолютная погрешность измерения Т, не более
Стандартное отклонение протекания, не более
Абсолютное значение рассеянного излучения при 200 нм, не белее
3—100% 1% 0,1% (0,001) 0,2%
Источники и приемники излучения те же, что и в приборе ’ СФ-26.
Устройство прибора СФ-46 и
Рнс. 6.8. Блок-схема спектрофотометра СФ-46.
168
Рис. 6.9. Внешний вид спектрофотометра СФ-46:
1 — монохроматор; 2 -* микропроцессорная система; 3 — кюветное отделение; 4 осветители; 5 — камера с фотоприемниками н усилителями; 6 — рукоятка вращения дифракционной решетки; 7 — шкала длин волн.
В спектрофотометре обеспечены следующие режимы работы! измерение пропускания Т, оптической плотности А, концентра* ции С, скорости изменения оптической плотности ЛЛ/Дт. Принцип измерений — общий для всех однолучевых спектрофотометров.
6.3.3. СПЕКТРОФОТОМЕТРЫ СФ-Ю, СФ-14, СФ-18
Назначение и принцип действия. Регистрирующие двухлучевые спектрофотометры СФ-10, СФ-14, СФ-18 предназначены для измерения пропускания (оптической плотности) прозрачных и мутных сред и коэффициентов диффузного отражения твердых и порошкообразных веществ в видимой области спектра. Спектрофотометры состоят из осветителя, двойного призменного монохроматора, фотометра поляризационного типа, приемно-усилительной части и записывающего механизма.
Внешний вид спектрофотометра СФ-14 приведен на рис. 6.10. На плате 5 смонтирована кюветная камера 4 для установки прозрачных образцов, измеряемых на пропускание, и интегрирующий шар 8.
Для измерения коэффициентов пропускания жидкостей имеется набор парных кювет с толщиной слоя 5, 10, 20 и 50 мм, Твердые прозрачные образцы и кюветы с жидкостью помещают в специальные держатели, выполненные в виде тисков, и зажимают винтом. Держатель с исследуемым раствором вставляют в направляющую правой части (по ходу лучей) кюветного отделения, а стандартный раствор помещают слева.
Порошки, измеряемые на отражение, помещают в кювету 7, расположенную на месте правого по ходу лучей эталона. Для исключения зеркальной составляющей отражения имеются кар-
169
Рис. 6.10. Внешний вид спектрофотометра СФ-14:
1 — осветитель с кожухом; 2 — барабан с бланком для записи спектров! 3 -₽ перо; 4 — кюветная камера; 5 — плата, на которой смонтирована кюветная камера; 6 — рукоятки, регулирующие положение экранов, предотвращающих попадание света иа фотоэлемент; 7 — кювета для порошков, измеряемых на отражение; 8 — интегрирующий шар; 9 — карманы для исключения зеркальной составляющей отражения; 10 регулятор скорости развертки спектра.
маны 9, которые можно установить в боковые отверстия шара вместо заглушек.
Внутри шара находятся два экрана, предназначенные длв предотвращения попадания света от образца непосредственно на фотоэлемент при измерении абсолютных значений коэффициентов диффузного отражения — нижний экран — и пропускания — верхний экран (по методу Тейлора). Для включения экранов необходимо оттянуть рукоятку 6.
Измерение абсолютных значений коэффициентов диффузного отражения или пропускания сводится к следующему.
Вначале записывают линию 100%-ного пропускания при нейтральном положении экранов относительно световых потоков; при этом рукоятки 6 вдвинуты в шар. Затем вместо эталона к окну шара в рабочий поток помещают образец, измеряемый на отражение. Оттянув нижнюю рукоятку 6 до упора, экран ставят в положение, при котором прямой свет от образца не попадает на фотоэлемент. Последующая запись дает значение абсолютного коэффициента диффузного отражения образца по спектру.
При измерении абсолютных значений коэффициентов диффузного отражения образец помещают в правое по ходу лучей входное окно шара, с помощью рукоятки 6 устанавливают верхний экран, нижний экран при этом должен занимать нейтральное положение относительно световых потоков.
Результаты всех измерений автоматически записываются на специальном бланке 2, имеющем вид сетки, по оси абсцисс кото-170
рой нанесена равномерная шкала длин волн от 400 до 750 нм с ценой деления 2 нм, а по оси ординат — равномерная шкала оптической плотности от 0 до 2,5 с ценой деления 0,0125 и пропускания (отражения) от 0 до 100% с ценой деления 0,5%.
Спектрофотометр СФ-14 имеет два диапазона измерения пропускания (0—100% и 0—10%) и два диапазона измерения оптической плотности (0—2,5 и 0—1,0), что позволяет повысить чувствительность и точность замеров.
Продолжительность записи одного спектра составляет 2— 4,5 мин.
Оптическая схема. Оптическая система приборов СФ-10, СФ-14 состоит из двух частей — спектральной (двойного монохроматора) и фотометрической.
Нить лампы 1 (рис. 6.11) проектируется конденсором 2 через входную щель 3 в плоскости объектива 4 коллиматора. Входная щель расположена в фокальной плоскости объектива. Выходящий из него параллельный поток света проходит диспергирующую призму 5 и разлагается в спектр. Объектив 6 первого монохроматора дает спектральное изображение входной щели в плоскости средней щели по линии А—А. Средняя щель двойного монохроматора, образованная зеркалом 7 и ножом 8, вырезает участок спектра, который проходит во второй монохроматор и проектируется в плоскости выходной щели 9.
Рис. 6.11. Оптическая схема спектрофотометра СФ-14:
/ — источник света; 2 ₽ конденсор; 3 входная щель; 4 — объектив коллиматора; 5 диспергирующая призма, разлагающая свет в спектр; 6 — объектив первого монохроматора; 7 — зеркало; 8 — нож; 9 — выходная щель; 10 *- линза; 11 « двоякопре-шомляющая призма Рошона; 12 диафрагма; 13 — призма Волластона; 14 линза; 35 — полулинза; 16 — барабан прерывателя; 17 —• призмaf отклоняющая оба потока на 90°; 18 интегрирующий шар; 19 фотоэлемент.
171
При выходе из монохроматора поток света попадает в фотометрическую часть прибора. Сначала поток проходит через линзу 10 и двоякопреломляющую призму Рошона 11. Первая дает изображение объектива выходного коллиматора вблизи диафрагмы 12, вторая разделяет это изображение на два, поляризованных во взаимно перпендикулярных плоскостях: одно, симметричное оси, проходит через призму Волластона 13 и линзу 14, другое, смещенное, срезается диафрагмой 12 Линза 14 дает изображение выходной щели в плоскости полулинз 15 Вследствие двойного лучепреломления призмы Волластона в плоскости полулинз получаются два изображения выходной щели. Пройдя полулинзы 15, установленные внутри барабана прерывателя 16, оба потока отклоняются на 90° призмой 17, проходят через входные окна шара 18 и попадают на окна, к которым прижимаются образец и эталон (в случае измерения коэффициентов отражения) или два эталона (в случае измерения коэффициентов пропускания). Свет, отраженный от образца и эталона, суммируется шаром и попадает на фотоэлемент 19, расположенный за выходным окном шара. Фототок, возникающий под влиянием суммарного светового потока, передается через усилитель на кинематическую систему прибора, и значение оптической плотности (пропускания) автоматически фиксируется на бумажном бланке.
Часть II
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Глава 7
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ КОЛИЧЕСТВЕННОГО АНАЛИЗА
7.1. АБСОЛЮТНЫЕ ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ВЕЩЕСТВ
В ОТСУТСТВИЕ МЕШАЮЩИХ КОМПОНЕНТОВ
7.1.1. МЕТОДЫ ОПРЕДЕЛЕНИЯ ОДНОГО ВЕЩЕСТВА
7.1.1.1. Метод сравнения оптических плотностей стандартного и исследуемого окрашенных растворов
Для определения концентрации вещества берут аликвотную часть исследуемого раствора, приготавливают из нее окрашенный раствор для фотометра рова ни я и измеряют его оптическую плотность. Затем аналогично исследуемому раствору приготавливают два-три стандартных окрашенных раствора определяемого вещества известной концентрации и измеряют их оптические плотности при той же толщине слоя (в тех же кюветах). Сравнивая значения оптических плотностей исследуемого и стандартного растворов, находят неизвестную концентрацию определяемого вещества.
Вс избежание больших погрешностей, концентрации исследуемого и стандартных растворов должны приготавливаться почти одинаковыми, что обеспечивается получением достаточно близких значений оптических плотностей сравниваемых растворов. Поэтому сначала измеряют оптическую плотность исследуемого раствора и лишь после этого подбирают концентрации стандартных растворов так, чтобы получить значения их оптических плотностей, близкие к значению исследуемого раствора. Для каждой пробы исследуемого раствора целесообразно приготовить два-три стандартных раствора с тем, чтобы определить среднее значение неизвестной концентрации определяемого вещества.
Значения оптических плотностей сравниваемых растворов будут равны:
для исследуемого раствора
для стандартного раствора
АС1 = efficjlcB
173
Разделив одно выражение на другое, получим: ^хМсг = ст^ст)
Так как измерения оптических плотностей стандартного и исследуемого растворов производили в одной и той же кювете, то 1Х = /ст; молярный коэффициент светопоглощения ех является постоянным для данного окрашенного вещества. Следовательно, приведенное выше равенство можно упростить:
~ CxICct откуда
= СстДх/^СТ (7«1)
Рассчитав неизвестную концентрацию Сх (мг/мл), с учетом разбавления растворов находят содержание в растворе определяемого вещества (qx, мг):
Qx — хР общ/Р 1
Здесь Рж — объем окрашенного исследуемого раствора, мл; Pf — объем аликвотной части исследуемого раствора, взятой для приготовления окрашенного раствора, мл; РОбщ — общий объем исследуемого раствора, мл.
Метод сравнения применяется при однократных анализах и требует обязательного соблюдения основного закона светопогло-П'.ения.
Существует и другой способ определения неизвестной концентрации Сх. приготавливают два стандартных раствора с концентрациями Сх и С2 так, чтобы оптическая плотность первого из них Лх была бы меньше оптической плотности Ах исследуемого раствора, а оптическая плотность Ай второго стандартного раствора была бы, наоборот, больше, чем Ах.
Неизвестную концентрацию исследуемого раствора рассчитывают по формуле:
Сх = Ci + (С2 - CJ (Ах - ЛХ)/(Л2 - A J (7.2)
Если значения концентраций (или значения оптических плотностей) исследуемого и стандартных растворов достаточно близки, то этот способ более точен.
7.1.1.2. Метод определения по среднему значению молярного коэффициента светопоглощения
Метод определения концентрации вещества по среднему значению молярного коэффициента светопоглощения является разновидностью метода сравнения, только в данном случае нужно непосредственно рассчитывать значение молярного коэффициента светопоглощения и по его значению находить неизвестную концентрацию исследуемого окрашенного раствора. Приготавливают исследуемый и стандартные окрашенные растворы и измеряют значения их оптических плотностей аналогично тому, как это производят при определении по методу сравнения. Поданным,
174
полученным для стандартных растворов, рассчитывают среднее значение молярного коэффициента светопоглощения:
ё= >4CT/(CCTZCT)
Зная значения оптической плотности исследуемого окрашенного раствора и молярного коэффициента светопоглощения, находят неизвестную концентрацию (Ск, моль/л) исследуемого окрашенного раствора и общее содержание в растворе определяемого вещества (qx, мг):
Ся = Лх/ё/я и дх =
Здесь М — молекулярная масса определяемого вещества (иона).
Измерения оптической плотности стандартного и исследуемого растворов можно производить как при одинаковой толщине слоя I (в одинаковых кюветах), так и при разной его толщине (в разных кюветах). Метод требует обязательного соблюдения основного закона светопоглощения и применяется сравнительно редко.
7.1.1.3. Метод градуировочного графина
Для определения содержания вещества методом градуировочного графика при выбранных оптимальных условиях (см. раЗд. 3.2) готовят серию из 5—8 стандартных растворов разных концентраций (не менее 3 параллельных растворов для каждой точки).
При выборе интервала концентраций стандартных растворов руководствуются следующими положениями:
а) он должен охватывать область возможных изменений концентраций исследуемого раствора; желательно, чтобы оптическая плотность исследуемого раствора соответствовала примерно середине градуировочной кривой (см. разд. 3.3, 9.3.1.2);
б) желательно, чтобы в этом интервале концентраций при выбранных толщине кюветы (/) и аналитической длине волны X * (в большинстве случаев X = Хмакс светопоглощающего Соедине-йия) соблюдался основной закон светопоглощения, т. е. график А — f (С) был прямолинейным;
в) интервал рабочих значений А, соответствующий интервалу стандартных растворов, должен обеспечивать максимальную воспроизводимость результатов измерений (минимальное отношение sAM), т. е. А х 0,144-1,9 (см. разд. 3.1.3.1) **. Однако следует
* При фотоколориметрических определениях выбор светофильтра (значения X = ХЭфф) в общем случае определяется не только видом кривых спектров поглощения окрашенного раствора и светофильтра, но и спектральной чувствительностью фотоэлемента.
** Необходимо учитывать, что общая погрешность фотометрического анализа определяется совокупностью слагаемых, из которых в ряде случаев определяющую роль могут играть погрешность невоспроизводимости положения кювет («кюветная» погрешность) в кюветодержателе и погрешность измерения объемов стандартных и исследуемых растворов при работе с неоткалиброванными мерными колбами, пипетками, бюретками [82, ПО, 116].
175
иметь в виду, что на практике при значениях А 1,14-1,3обычно I наблюдается уже нелинейный характер зависимости А = f (С).
При совокупности перечисленных условий измеряют оптические плотности стандартных растворов относительно растворителя и строят график зависимости А — [(C) *. Полученная кривая (рис. 7.1) называется градуировочной (градуировочным графиком). Периодически (раз в неделю или реже) ее проверяют по двум-трем свежеприготовленным стандартным растворам.
Определив оптическую плотность раствора Ах, находят ее значение на оси ординат, а затем на оси абсцисс — соответствующее ей значение концентрации Сх. Содержание вещества qx (мг) в исследуемом растворе определяют по формуле:
Qx =
Этот метод применяют при многократном фотометрировании однотипных по химическому составу растворов, при выполнении серийных фотометрических анализов. Он дает хорошие результаты при соблюдении основного закона светопоглощения.
В отличие от других фотометрических методов, метод градуировочного графика позволяет определять концентрацию окрашенных растворов даже в тех случаях, когда основной закон светопоглощения не собтюдается. Для построения градуировочной кривой в этих случаях приготавливают значительно большее число стандартных растворов, отличающихся друг от друга по концентрации не более чем на 10 %. Такой градуировочный график, имеющий на полотом участке угол наклона не менее 15°, все же позволяет проводить фотометрические определения, несмотря на то, что между концентрацией раствора и его оптической плотностью нет прямолинейной зависимости. Воспроизводимость определений при этом ниже, чем в случае линейной зависимости A=f(O.
* В общем случае, когда свет поглощается не только определяемым соединением, но и реагентом, экспериментальные данные измерений оптической плотности могут быть представлены следующими вариантами:
1 Оптические п тспиости стандартных растворов и раствора реагента (холостого опыта, содержащего определенный объем добавляемого реагента) измеряют относительно растворителя. Определяют разности значений ДД = А —Дк (где А 11 Др — средние значения при разных концентрациях) и строят график или рассчитывают уравнение зависимости ДД = f (С) (см. разд. 9.3). Этот подход наиболее применим для неустойчивых систем или реагентов и систем, содержащих примеси.
2. Оптическую плотность стандартных и исследуемых растворов измеряют относительно раствора реагента в качестве холостого опыта, Д = f (С). Этот вариант применяют нередко, когда раствор холостого опыта имеет высокое значение оптической плотности.
3. Оптическую плотность растворов измеряют относительно растворителя. Результат холостой пробы и его погрешность рассчитывают методом линейного регрессионного анализа (см. разд. 9.3).
176
Рис. 7.1. Градуировочный график.
Несмотря на простоту и удобство, практическое использование градуировочных графиков в ряде случаев вносит дополнительную по-грен1ность при определении концентрации растворов как за счет субъективного построения графической зависимости, так и за счет несоответст-
вия графических (масштабных) погрешностей и погрешностей измерений оптических плотностей. Поэтому для получения более объективных результатов анализа часто пользуются одной из нижеприведенных аналитических зависимостей [см. уравнения (7.3)—(7.5)], которые рассчитывают по экспериментальным данным методом регрессионного анализа (см. разд. 9.3).
Согласно основному закону светопоглощения графическая зависимость А — f (С) выражается прямой линией, проходящей через начало координат:
А = ЬС (7.3)
Здесь Ъ — угловой коэффициент линейного градуировочного графика.
Если в тех же условиях при фотометрировании растворов допускается какая-то систематическая погрешность (например, за счет содержания определяемого элемента в используемых реактивах), то графическая зависимость А — f (С) так же выражается прямой линией, но не проходящей через начало координат. В этом случае в уравнении прямой появляется второй коэффициент (свободный член):
А = а + ЬС (7.4)
Если отклонений от основного закона светопоглощения избежать не удается и графическая зависимость А = f (С) становится » нелинейной, то во многих случаях такую зависимость с достаточной степенью приближения можно аппроксимировать квадратичным уравнением параболы, проходящей через начало координат:
А = аС + ЬС3 (7.5)
Здесь а и b — коэффициенты (параметры) в уравнении параболы.
При построении градуировочного графика различают следующие варианты:
график для чистых стандартных растворов, построенный при оптимальных условиях. Такие графики следует с осторожностью использовать для определений неизвестных концентраций в растворах, содержащих мешающие ионы, или в образцах различных матриц;
177
график, построенный в присутствии отдельных мешающих компонентов матрицы, влияние которых достаточно подробно изучено;
грасЬик, построенный по стандартным растворам, содержащим все элементы анализируемых объектов.
7.1.1.4. Метод добавок
Метод добавок представляет собой разновидность метода сравнения Определение концентрации раствора этим методом основано на сравнении оптической плотности исследуемого раствора и того же раствора с добавкой известного количества определяемого вещества Метод добавок обычно применяю: для упрощения работ! , для устранения мешающего влияния посторонних примесей. в ряде случаев для оценки правильности методики фотометрического определения. .Этот метод позволяет создать одинаковые условия для Фотометрирования исследуемого и стандартного (с добавкой) окрашенных растворов, поэтому его целесообразно применять для определения малых количеств различных элементов в присутствии больших количеств посторонних веществ при анализах солевых растворов. Метод добавок требует обязательного соблюдения основного закона светопоглощения; его возможности и ограничения рассмотрены в работе 1146].
Неизвестную концентрацию находят расчетным или графическим способами.
Расчет неизвестной концентрации по методу сравнения< При соблюдении основного закона светопоглощения и постоянной толщине слоя отношение оптических плотностей исследуемого раствора и исследуемого раствора с добавкой будет равно отношению их концентраций
4х/Ах+а = СХ/(СХ + Са) откуда
Сх — Са,Ах/(Ах+а 4Х) (7.6)
Здесь Ах — оптическая плотность исследуемого раствора; Ах+а — оптическая плотность исследуемого раствора с добавкой; Сх — неизвестная концентрация определяемого вещества в исследуемом окрашенном растворе; Са — концентрация добавки в исследуемом растворе (из расчета только добавленного количества). С концентрацией раствора добавки СдОб она связана соотношением: С а = Сдоб^доб/^к+а-
Учитывая разбавление исследуемого раствора и выражая концентрацию добавки в нем Са через Сдоб, находим содержание (qx, мг) определяемого вещества в растворе:
Ях = ^х^хСдоб^доб^общ/К^х+а ^х) ViVx+a]
Здесь Vx — объем окрашенного исследуемого растзопа без добавки, мл; Гдоб “* объем раствора добавки, мл; УОбщ—общий объем исследуемого раствора, мл; Vx — объем аликвотной части исследуемого раствора, взятой для приготовления окрашенного раствора, мл; Vx+a — объем окрашенного исследуемого раствора с добавкой, мл.
178
Если исследуемый окрашенный раствор и раствор с добавкой приготавливают в одинаковых мерных колбах, то их объемы одинаковы, следовательно
_ ЛжСдобЕдоб v Удоб^кЕобщ ,7 7,
Чх - Гобщ или
Здесь удОб — содержание в растворе добавленного вещества *, мг.
Определение неизвестной концентрации графическим способом. При определении неизвестной концентрации графическим способом (рис. 7.2) на оси ординат откладывают значение оптической плотности исследуемого раствора Ах, а на оси абсцисс из точек Са, и Са2, отвечающих концентрациям добавленного вещества в растворе, восстанавливают перпендикуляры. На этих перпендикулярах откладывают соответствующие значения оптической плотности Ах+а1 и Ах+аа растворов с добавками и а^. Через полученные три точки Ах, Ах^а, и Ах+аа проводят прямую линию до пересечения ее с продолжением оси абсцисс в точке Сх. Абсолютное значение отрезка 0Сх выражает неизвестную концентрацию исследуемого раствора.
Содержание в растворе определяемого вещества (мг) рассчитывают с учетом разбавления:
Чх — бкЕхУобщ/Е1
К сожалению, при большом солевом фоне, особенно когда примеси взаимодействуют с реактивом, метод добавок часто приводит к получению завышенных результатов из-за нарушения прямолинейной зависимости А от С, которое не учитывается ни расчетным уравнением (7.6), ни графически.
Для учета влияния примеси при определении методом добавок предложено несколько вариантов. По мнению Фукишима и др. [226], нелинейную зависимость между концентрацией и опти
* Добавки следует брать в таких количествах, чтобы не происходило «потери точности при вычитании»; минимальная разность Ах+а — Ах должна быть не менее 0,1.
Рис. 7.2. Графическое определение концентрации раствора методом добавок.
Рис. 7.3. Спрямление нелинейной зависимости Л от С.
179
ческой плотностью следует выражать аналитически уравнением наиболее подходящей аппроксимирующей функции, на основании которой и производить количественные расчеты. Однако этот путь очень сложен.
А. Шугар и Ю. Шугар [227 ] при нелинейной зависимости А от С предложили способ расчета, основанный на спрямлении кривой этой зависимости в очень узком интервале концентраций (рис. 7.3).
Если оптические плотности исследуемого раствора Ах, разбавленного в п раз исследуемого раствора Ап и исследуемого раствора с добавкой Аа+0 находятся на криволинейном участке а—с, то спрямив этот участок, мощно рассчитать неизвестную концентрацию Сх по формуле:
f Аж Ап п
Л ° /1х+а —/1Х ‘/1—1
(7.8)
По мере сближения величин Ах, Ап и Аэ;+а погрешность за счет спрямления кривой уменьшается. Однако, как показал Л. П. Адамович [228] даже в самых благоприятных условиях, когда добавка вносится в разбавленный в п раз исследуемый раствор и концентрация рассчитывается по формуле
АХ Ап П
— . • _____________
пп+а ‘‘п п 1
(7.9)
погрешность за счет спрямления кривой принципиально не исключается, а лишь достигает своего минимального значения.
Для исключения погрешностей из-за присутствия взаимодействующей с реактивом примеси, природа которой известна, можно рекомендовать наиболее простой и надежный способ, предложенный Адамовичем [228].
Анализ проводят следующим образом:
1. С двумя разными светофильтрами измеряют значения оптических плотностей исследуемого раствора с примесью А'х и Ах.
2. С теми же светофильтрами измеряют оптические плотности исследуемого раствора с добавкой определяемого вещества А'х+а и А'х+а.
3. С теми же светофильтрами измеряют оптические плотности исследуемого раствора с добавкой некоторого количества примеси Ах+пр И Ах+пр*
Неизвестную концентрацию Сх рассчитывают по формуле:
р С a (AxAe-j-np Ax-j-npAx)
А* 0x4-0 Ах+ьр) + Ах_|_а 0х4-пр Ах) + Ах^пр (Ах ^хч-с)
(7-10)
Если возникают опасения относительно «потери точности при вычитании», то опыт повторяют, изменив значения добавок определяемого элемента или примеси.
180
При неудачном выборе светофильтров, когда относительное изменение молярных коэффициентов светопоглощения определяемого вещества и примеси одинаково
_ "Г и пИ Л*+а ~ А* _ Л*+пр ~ А'х
в* епр Ах+а Ах Ах+пр А'х
расчет положительных результатов не дает. Если замена светофильтров вновь не позволяет получил положительных результатов, то это указывает на чрезвычайное сходство кривых светопоглощения соединений реагента как с определяемым веществом, так и с примесью. В этом случае способ Адамовича не применим. Формулы (7.8) и (7.9) можно использовать также для расчета концентрации Сх в тех случаях, когда в определяемом интервале концентраций зависимость А = f (С) выражается прямой линией, не проходящей через начало координат.
7.1.2. АНАЛИЗ ДВУХ- И МНОГОКОМПОНЕНТНЫХ ОКРАШЕННЫХ СИСТЕМ
Спектрофотометрический анализ двух- и многокомпонентных окрашенных смесей (особенно анализ органических соединений) развивается в различных направлениях:
1. Применение математических методов обработки информации по спектрам поглощения с широким использованием микро-ЭВМ. Специальные вопросы количественного анализа многокомпонентных смесей (качественные и количественные критерии для выбора оптимальных аналитических позиций при анализе; метоцы планирования эксперимента для выбора и уточнений значений аналитических длин волн; применение ЭВМ в анализе смесей; вопросы правильности и воспроизводимости) разрабатываются преимущественно в приложении к многокомпонентным смесям органических соединений 1229—234].
2. Создание аппаратуры и методов двухволновой и производной спектрофотометрии.
3. Разработка приемов, упрощающих определение концентраций компонентов в многокомпонентных системах.
Ниже изложены общие положения и типичные варианты при анализе двух- и многокомпонентных систем (в приложении к анализу неорганических веществ):
Оптическая плотность любой смеси, содержащей ограниченное число окрашенных компонентов, не взаимодействующих химически друг с другом, равна сумме оптических плотностей компонентов смеси при той же длине волны
Ак — а1К + + °3>. + • • • + ОпХ (7-11)
причем каждая из парциальных плотностей равна:
0^ = 6^/ (7.12)
181
Здесь е;>—молярный коэффициент светопоглощения вещества i при длине волны X; I — толщина поглощающего слоя, см; С, — концентрация поглощающего вещества, моль/л.
Если система содержит п окрашенных веществ, то проводят п независимых измерений оптической плотности при п различных длинах волн Х1, Х2, В результате получается система
линейных уравнений:
АЬ, = 8IXIC1Z + 82X1C2Z + ЕЫ,С31 4--fnb,Cnl
АЛ2 = е1Ь„С11 + 82XaC2Z + 83X2C3Z + • • • + 8nX2CnZ
^хп = + e2Xn^2Z + sxnCsl 4- • • • 4- enxnCnZ
Значения A^t определяют в процессе анализа, значения е,-^ находят предварительно, а толщина I постоянна и равна длине кюветы.
В многокомпонентном спектрофотометрическом анализе, как и обычно, измерения оптических плотностей следует проводить относительно раствора сравнения, содержащего все используемые реагенты, для уменьшения систематических погрешностей, обусловленных наличием примесей в самих реагентах.
7.1.2.1. Спектрофотометрический анализ трехкомпонентных систем
Измерив оптические плотности анализируемой смеси при трех длинах волн, составляют систему линейных уравнений
^Х, = 81X2^1Z + 82X,^2Z + 83X,Z'3Z
A't.2 — 8lXa^lZ + 82X2^2Z + 83x/-3Z ^Xa = 8lXa^lZ + 82Xa^2Z + 83X,^3Z
(7.U)
которую решают с помощью определителей н формул Крамера [235]:
А = 81Х182Х,8ЗХ1 81Ха82Ла8ЗХ2 81Ха82Ха8ЗХа = 81Х,82Х28ЗХа + 82Х,8ЗХ281ХЯ + 4" 8ЗХ,81Ха82Ха 8ЗХ,е2Х2е1Хз ~ е1Х,еЗХае2Ха ^Ли/зХ,
А А ~ 82Хаезха Л - = ~ (^Х,82Х28ЗХа 4" ^Ха82Х,8ЗХа 4-,
Л СО « <4 сч сч w со 1- 4 II и Ч 4- ^х.^х/зх, — ^Ха8ЗХ,82Х2 — ^Х,8ЗХас2Х, — ЛХае2Х1еЗХа)
182
Л?1 i
eix, ~Т~ = ~Г (лл.«е1Х1езх. + ^х^зх/хх, +
Ак
е1^г -f- 8зхг + лх,8зхМх, — лх,8зх181х5 ~ лм
е1Л. ~Г 8ЗХ, — ^^ix&x. — лх1е1х18зх,}
лх, S1X182X, 1 1 — (^.^х/гх. + лх1®1х,82х1
Ас„ ~ лх8 81Ха?2Ха 1 + лх181х,8гх, ~ лх181х,82х,
лКз 81Х,82Х» ~j~ — ЛХ8е1Х1Е2Х, — ЛХ,81Х182Х1)
Cl . r AC, = ~л“’ С*~~Г
Если молярные коэффициенты светопоглощения определяемых компонентов при выбранных длинах волн известны, то получают уравнения с эмпирическими коэффициентами а, b и с, с помощью которых по значениям оптической плотности смеси, измеренной при выбранных длинах волн, рассчитывают неизвестные концентрации каждого из компонентов в отдельности;
Ci = аЛХ1 + + сА^
с2 = + ь1Ак„ + с1Лхл
С3 = a2AKt + Ь2Акг + с2Лк
(7.15)
Типовыми примерами могут служить совокупное спектрофото-
метрическое определение золота, платиновых и цветных металлов в виде бромидных комплексов [236] или спектрофотометрическое определение меди, кобальта и никеля при их совместном присутствии в виде диэтилдитиокарбаминатов [237].
Осажденные из водных растворов диэтилдитиокарбаминаты меди, кобальта и никеля хорошо экстрагируются хлороформом и тетрахлоридом углерода. Спектры поглощения их в хлороформе показаны на рис. 7.4. Максимумы поглощения диэтилдитиокарбаминатом меди наблюдаются при 300 и 436 нм. Ди-этилдитиокарбаминат кобальта имеет
Рис. 7.4. Спектры логчощення диэтилдитио-карбамииатов меди (/), кобальта (2) и никеля (3) в тетрахлориде углерода.
183
два пика: один при 367 нм и второй при 321 нм. У диэтилдитиокарбамината никеля один максимум поглощения находится при 328 нм, а второй при 393 нм. Наиболее воспроизводимые результаты при совместном определении трех элементов наблюдаются при следующих длинах волн: 436, 367 и 328 нм *.
Если производят совместное определение всех трех катионов, то оптическую плотность исследуемого раствора измеряют при 436, 367 и 328 нм и содержание меди, кобальта и никеля рассчитывают из системы уравнений (7.14).
Решение системы уравнений (7.15) с учетом конкретных значений молярных коэффициентов светопоглощения при выбранных длинах волн и I = 1 см дает выражения для расчета концентрации каждого компонента:
Сси’Ю5 — 7,93Д43б— 1,ОЗЛ387— 0,27Л328
б*Со 105 — 8,48Лзв7 — 0,91 Л 328 — 0,67Д438
CN, 106 = 3,42Д 328 — 5,19Д387 — 0,08Д436
(7.16)
7.1.2.2. Спектрофютометрический анализ двухкомпонентных систем
При спектрофотометрических определениях возможны следующие варианты.
Кривые светопоглощения обоих веществ перекрываются по всему спектру. Концентрации Сг и С2 раствора находят при двух длинах волн по уравнениям:
Ак, = £1Х,СН + ®2A1C’2Z
ЛХ2 = eU1ClZ + e2X2C2Z
Значения молярных коэффициентов светопоглощения либо берут из таблиц, либо (чаще) определяют экспериментально следующим образом. Приготавливают стандартный раствор исследуемого вещества 1 и измеряют его оптические плотности при тех длинах волн, при которых будут вести спектрофотометрирование смеси обоих компонентов. Затем, зная Ак_, Сст (моль/л) и I (см), по уравнению Аа. — С\1 рассчитывают ** значение молярного
коэффициента светопоглощения eiX для компонента 1 при и Л2.
* Кроме указанных металлов с днэтилдитиокарбаминатом натрия в водных растворах образуют осадки многие другие металлы, например Fe3+, Fe2+, Bis+, Cue+, Zn2+, Hg2+, Sn2+ и т. д.; карбаминаты этих металлов также переходят в слой хлороформа и могут мешать определению Ni2+, Co^.Cu2*.
Значения молярных коэффициентов светопоглощения.
При 436 нм
» 367 нм
» 328 нм
еСи (ДДК)2
12 850
1 260
2 230
еСо (ДДК)2
2 260
14 340
21 820
eNi (ДДЮл
1 720
3 910
35 210
* * Надежные данные получаются только для однотипных по составу анали-
зируемых соединений приблизительно одинаковой прочности.
184
Аналогичным образом определяют значения e22lj для компонента 2 при и Х2. Полученные значения коэффициента светопоглощения подставляют в систему уравнений (7.17) и решают ее относительно двух неизвестных и С2:
ci = ^е2Х2—j1 еА~7^ у/(е1х,е2Лг — e2A,eix2) (7.17)
I t ^2 г )/Йц iS2A2 81Л2®2А.,)
Чтобы относительная погрешность ДС7С была наименьшей, значения Afl и А-,2 должны лежать в интервале 0,3—1, а отношения и e2x2/eix2 должны быть максимальными.
Примерами могут служить определение кобальта и меди (кривые 1, 2 рис. 7.4; = 367 и Х2 = 436 нм); никеля и меди (кривые 1, 3; Хг = 328 и А2 = 436 нм); никеля и кобальта (кривые 2, 3;
= 328 и Х2 = 367 нм) в соответствующих двухкомпонентных системах.
Кривые светопоглощения обоих веществ перекрываются, но имеется участок спектра, где светопоглощением одного из них можно пренебречь. Если имеется участок, в котором поглощает лишь один компонент (например, когда е1>2 = 0) *, система уравнений упрощается и концентрация каждого из компонентов определяется по соотношениям:
_ / Т1д.2 \ /
Cj = j 8 27., I j /(®1Л,®2А.2)> ^2= ^X2/(e2X20 '8)
Например, совместное определение кобальта и никеля при помощи 8-оксихинолина основано на использовании области спектра, в которой поглощает свет только 8-оксихинолинат кобальта [238]. Кобальт 8-оксихинолинатный комплекс в смеси 1 М НС1 — ацетон максимально поглощает свет при длинах волн 365 и 700 нм, в то время как никель-8-оксихинолинатный комплекс имеет пик только при 365 нм (рис. 7.5). Благодаря этому можно провести совместное определение Со2+ и Ni2+ с помощью 8-оксихинолина.
Расчет концентрации кобальта и никеля с учетом значений молярных коэффициентов светопоглощения
«Со <е’> eNi «и
При 363 нм 3529 3228
» 700 нм 428,9 —
производят по уравнению (7.18) или по формулам:
С' = Л7О0/428,9; С" = (ЛМ6 - г’жьС')1<^ = (ЛзеБ- 3529С')/3228
Здесь С — концентрация Co(C8HeON)2-2H2O, моль/л; С" — концентрация Ni(C8HeON)2-H2O, моль/л.
* См. также примеры в разд. 7.1.2.3.
185
Рис. 7.5. Спектры поглощения оксихинолинатов кобальта (/) и никеля (2) в смеси 17И НС1 — ацетон.
Рис. 7.6. Спектры поглощения двух компонентов различного цвета: с и b — области максимального пропускания лучей светофильтрами а и Ъ
Зная С и С", можно вычислить содержание никеля и кобальта в исследуемом объеме.
Определение кобальта и никеля можно провести и методом градуировочного графика, если предварительно построить градуировочные кривые при 365 и 700 нм.
Кривые светопоглощения обоих веществ не перекрываются (рис. 7.6). Расчет ведут по уравнениям:
= и С2 = ^Х,/(е2А,/)
7.1.2.3. Фотоколориметрический анализ двухкомпонентных систем
При фотоколориметрическом анализе двухкомпонентных окрашенных растворов возможны варианты, описанные в разд. 7.1.2.2.
Спектры поглощения определяемых компонентов не накладываются друг на друга, т. е. полосы поглощения в спектрах определяемых компонентов А и В так разграничены между собой, что при фотометрировании смеси с каждым из светофильтров поглощением другого (второго) компонента можно пренебречь (см. рис. 7.6).
Экспериментально или теоретически подбирают светофильтры, один из которых пропускает лучи, поглощаемые в основном только одним из определяемых веществ, в то время как другой светофильтр пропускает лучи, поглощаемые главным образом вторым компонентом.
Фотометрическое определение обоих компонентов, присутствующих в смеси, легко осуществляется любым из описанных выше методов анализа, например методом сравнения. Для этого
186
Рис. 7.7. Спектры поглощения двух компонентов, частично накладавающиеся друг на друга.
а и b - то же, что на рис. 7.6.
приготавливают стандартные растворы определяемых компонентов А и В и измеряют их оптические плотности соответственно со светофильтрами а и Ь. Затем с теми же светофильтрами измеряют оптическую плотность окрашенной смеси двух компонентов. Сравнивая получен-
ные значения оптических плотностей окрашенной смеси и стан-
дартных растворов, определяют неизвестные концентрации обоих
компонентов:
^хА ^стА^х (а)/^ст А (а) и ^"хВ ^ст В^х (Ь)Мст В (6)
Здесь СстА и Сств—концентрации стандартных растворов компонентов А и В; Ах (О) и А ст а (а) — оптические плотности окрашенной смеси и стандартного раствора компонента А с применением светофильтра а; Ах и Лст в — Оптические плотности окрашенной смеси и стандартного раствора компонента В с применением светофильтра Ь.
Спектры поглощения определяемых компонентов частично накладываются друг на друга. В этом случае при фотометрирова-нии с разными светофильтрами можно пренебречь светопоглоще-нием лишь одного из компонентов окрашенной смеси (рис. 7.7). Для этого подбирают такой светофильтр, при котором в области максимального поглощения лучей первым компонентом свето-поглощением второго компонента можно пренебречь. При измерении оптической плотности окрашенной смеси с другим светофильтром в области максимального поглощения лучей вторым компонентом всегда наблюдается суммарное поглощение света обоими компонентами. В этом случае светопоглощением первого компонента пренебрегать нельзя.
Пусть имеется смесь растворов окрашенных компонентов А и В, которые фотометрируются со светофильтрами а и Ь, причем с применением светофильтра а светопоглощением компонента В можно пренебречь. Приготавливают серии стандартных растворов компонентов А и В и измеряют их оптические плотности: растворов компонента А со светофильтрами а и Ь, а растворов компонента В — только со светофильтром Ь. По полученным данным для каждого из компонентов строят градуировочные кривые (рис. 7.8, 7.9). Затем измеряют оптическую плотность окрашенной смеси компонентов А и В со светофильтром а и по измеренному значению Ах (0) и градуировочной кривой для чистого компонента А (кривая а, рис. 7.8) сразу же находят неизвестную концентрацию Сж(а) компонента А в исследуемом растворе.
167
Одновременно при помощи кривой Ъ на том >ке графике определяют оптическую плотность раствора компонента А со светофильтром Ь. После этого измеряют оптическую плотность исследуемой смеси компонентов А и В со светофильтром Ь. Измеренное значение оптической плотности АЛ(*) смеси является суммарной величиной, состоящей из оптической плотности Ав (ь> компонента В и оптической плотности Ад (г>) компонента А. Но так как Ад (Ь) нам известна из рис. 7.8, то по разности величин АХ(Ь) и Ад ((,) находим Ав(Ь)- Ав <г>) = Ах —Ад (ь).
По найденному значению оптической плотности Ав и кривой b (рис. 7.9) определяют неизвестную концентрацию СсВ компонента В в исследуемом растворе.
Таким способом определяют, например, концентрацию ионов Мп (VII) и Cr (VI) в смеси растворов КМпО4 и К2Сг2О7; титана и ванадия.
В первом случае измерения оптической плотности растворов производят при X iv 550 нм (зеленый светофильтр), когда свет поглощает лишь один перманганат-ион, и при X 430 нм (синий светофильтр), когда наблюдается суммарное светопоглощение обоими окрашенными ионами.
По стандартным растворам перманганат- и бихромат-ионов строят градуировочные графики А = f (С) с синим светофильтром (X » 430 нм) (рис. 7.10, кривые 1, 2), а для растворов пер-манганат-иона — с зеленым светофильтром (к 550 нм) (рис. 7.10, кривая 3). По значению оптической плотности исследуемой смеси, измеренному в области 550 нм, и при помощи кривой 3 сразу же определяют неизвестную концентрацию марганца в исследуемом растворе. Одновременно при помощи кривой 2 определяют оптическую плотность раствора перманганат-иона при 430 нм. Затем по разности оптических плотностей исследуемой смеси и раствора перманганат-иона при 430 нм (ААХ t430> = Ах l4i0) — Ам„ изо») по кривой 1 находят неизвестную концентрацию ионов хрома в исследуемой смеси.
Аналогичным образом проводят определение титана и ванадия при их совместном присутствии по спектрам поглощения пер-
Рис. 7.8. Градуировочный график для определения концентрации компонента А.
Рис. 7.9 Градуировочный график для определения концентрации компонента В. 188
Рис. 7.10. Градуировочный график для определения концентраций хрома и марганца при их совместном присутствии:
1 — Для определения хрома при 430 нм, 2 — для определения оптической плотности раствора пермангаиат-иоиа при 430 им; 3 для определения марганца при 550 нм.
Рис. 7.11. Градуировочный график для определения концентраций ванадия и титана при их совместном присутствии:
J — для определения ванадия при 619 им; 2 — для определения оптической плотности раствора ванадия при 400 нм; 3 — для определения титана прн 400 им.
оксидных комплексов [TiO(H2O2) ]2+ и [VO(H2O2) ]2+* [239]. Градуировочные графики (рис. 7.11) строят по измеренным Значениям оптических плотностей стандартных растворов ванадия при 619 нм (оранжевый или красный светофильтр) и при 400 нм (фиолетовый или синий светофильтр) (кривые 1, 2) и титана — при 400 нм (фиолетовый или синий светофильтр) (кривая 5).
По значению оптической плотности исследуемой смеси, измеренному при 619 нм, и при помощи кривой 1 сразу же определяют неизвестную концентрацию ванадия. Одновременно при помощи кривой 2 определяют оптическую плотность раствора ванадия при 400 нм. Затем по разности оптических плотностей исследуемой смеси и раствора ванадия при 400 нм (ДЛХ(4оо) = = Л(Л) аде —Лу (4оо)) по кривой 3 находят неизвестную концентрацию титана.
Спектры поглощения определяемых компонентов накладываются друг на друга на протяжении всей видимой области спектра. В этом случае нельзя выбрать никаких участков видимой области спектра, где можно было бы пренебречь светопоглощением одного из компонентов. Поэтому количественное определение компонентов проводят при помощи спектрофотометров (см. разд. 7.1.2 2), так как этот анализ с фотоколориметрами практически осуществить невозможно.
* При X = 619 нм свет поглощает практически лишь раствор комплексного катиона ванадия, а при 400 нм — оба катиона.
189.
7.1.2.4. Некоторые приемы, упрощающие анализ двух- и многокомпонентных систем
Модели упрощения многокомпонентного анализа основаны на: 1) независимом определении суммарной концентрации компонентов смеси, в частности на использовании точки пересечения спектров поглощения компонентов — модель 1;
2) выборе длин волн с одинаковыми молярными коэффициентами светопоглощения у одного или нескольких комплексов (с использованием «сечения» спектра) — модель 2;
3) использовании разностей суммарных оптических плотностей при маскировании, экстрагировании или изменении состава отдельных комплексов — модель 3;
4) спектрофотометрическом определении двух равновесных форм при их совместном присутствии с использованием смещения равновесия в анализируемой системе — модель 4;
5) определении концентраций таутомерных (равновесных) форм соединений — модель 5.
Модели 1—3 использованы авторами [240, 241 ] при анализе смесей ионов Zn, Mn, Ni; Ni, Си, Fe; Ni, Fe, Си, Mn, Zn с 1-(2-пи-риди тазо)-2-нафтолом.
Модель 1 (рис. 7.12). Спектры поглощения компонентов 1 и 2 двухкомпонентной смеси могут иметь общую точку пересечения, например точку 0 пересечения кривых 1 и 2 рис. 7.12 (аналогичную точку соответственно при пересечении кривых 1 и 3 или 2 и 3). В этом случае eix, = егх,- Система уравнений (7.16) упрощается, и при I = 1 см она будет иметь вид
Ак, = е1М (С1 + С2)
Ак2 = е1Х2С1 + 82Л1С2 откуда:
С1 = Их, — — е2Х2)
С2 = (ЛХ2 — |4Х1еЩ2/е1Х1)/(е2Л2 — е1Х2)
Длину волны Х2 выбирают в области наибольших различий в спектрах поглощения (при определении Мп и Zn длина волны,
При которой EMnRj = £znK2, соста-вляет 560 нм).
Модель 2. При анализе двухкомпонентных систем удобно измерять оптическую плотность при длинах волн с одинаковыми молярными
Рис. 7.12. Спектры поглощения хлороформных растворов пиридилазонафтолатсв цинка (7), марганца (2), никеля (3) и возможные «сечения» по спектрам [241].
190
коэффициентами светопоглощения одного из компонентов: точки А, В-, А', В' сечений АВ, А'В' (кривые 1, 2 рис. 7.12). В случае «сечения» АВ по полосе поглощения второго компонента в точках А, В e2J,1 = е2?.г и решение системы уравнений (7.16) при I — 1 см дает значение:
с = 0Х., Т ^/.2)/(eiXj — е1/.г) (7-21)
При использовании точек А' и В' «сечения» А'В' и при I = = 1 см по аналогии имее и:
С2= И к, е2?,2) (7.22)
Так по «сечению» спектра MnR2 (Х2 = 550 нм, к2 = 565 нм) определяли содержание Zn, а по «сечению» спектра ZnRa (\ = = 548 нм, Х2 = 560 нм) — содержание Мп.
Если спектр поглощения одного из компонентов имеет два максимума, то можно измерять оптическую плотность при 3-или 4-х точках пересечения («сечение» CD, кривая 3 рис. 7.12), понижая размер расчетной матрицы в трех- и четырех-компонент-ных системах.
Модель 3. Примерами этой модели могут служить трех- и пятикомпонентная смеси пиридилазонафталатных комплексов металлов никеля, железа, меди (в первом случае) и никеля, железа, меди, марганца, цинка (во втором случае).
Пиридилазонафтол (HR) образует экстрагируемые комплексы с никелем, медью, железом. При pH = 3-?-4 в хлороформ экстрагируются комплексы NiR2, FeR2, CuRCl. Если экстракт встряхнуть с водным буферным раствором (pH = 9), то состав и окраска комплексов NiRa и FeR2 не изменяется, a CuRCl перейдет в CuRa (CuRCl 4- HR = CuR2 + HC1). Вычитая из суммарной оптической плотности полученного хлороформного экстракта Аа (при к 580 нм) оптическую плотность раствора до промывки, находят концентрацию меди:
CcuRa = CcuRCl = (-4 2 — A)/(eCuR2 — ecuRCi)
В случае анализа пятикомпонентной системы сначала из щелочного раствора (pH = 9-j-10) экстрагируются в хлороформ все комплексы металлов Ni, Fe, Си, Мп, Zn с 1-(2-пиридилазо)-2-нафтолом (экстракт 1). Обрабатывая часть органической фазы буферным раствором с тем же значением pH, но с добавкой сульфида натрия (экстракт 2) и измеряя оптическую плотность экстракта 1 относительно экстракта 2, можно определить содержание меди (при X = 580 нм). Затем часть хлороформного экстракта, не содержащего меди, промывают буферным раствором с pH — 3-?-3,5 (экстракт 3). При этом из органической фазы реэкстрагируются комплексы цинка и марганца. Содержание цинка и марганца рассчитывают по результатам измерения оптической плотности экстракта 2 относительно экстракта 3; никель и железо, оставшиеся в экстракте 3, определяют по измерениям
191
оптической плотности при X = 550 нм (Ni) и к = 560 нм (Fe) относительно холостого опыта.
Модель 4 1242]. Если кривые светопоглощения равновесных форм А и В близки по внешнему виду, полосы поглощения их накладываются. При этом на любом участке спектра светопоглощение одной из форм, например А, всегда больше (ел > ев). В этом случае обычный спектрофотометрический анализ двухкомпонентной системы оказывается малоэффективным, особенно, когда эталонирование затруднено, т. е. определение общей концентрации равновесных форм представляет определенную трудность. Анализ такой системы можно осуществить на основе совместного решения уравнений основного закона светопоглощения и закона сохранения массы веществ с использованием крайних состояний равновесной системы.
Решая систему уравнений
^см = еАСА1 + евсв1 1
С = С а -|- С в J
относительно неизвестных СА и Св и полагая при этом I = 1 см и С — Аа/иа — Ав/ев, получаем расчетные формулы:
СА = Исм8А ~ ^Лев)/[еД (ел — ев)] (7.23)
св = Ид-ЛСм)/(ел-ев) <7-24)
Здесь А см — оптическая плотность исследуемой смеси компонентов Л и Л; А а и Ав — оптические плотности той же смеси в условиях смещенного равновесия при СА1СВ 99 и СВСА 99 соответственно.
При отсутствии значений молярных коэффициентов светопоглощения или при анализе с помощью фотоколориметров (фотометров) отношение еА/ев может быть заменено равным ему отношением Аа/Ав. Вводя эти изменения в формулы (7.23), (7.24) и применяя их для определения относительного содержания компонентов А и В в смеси, получим:
Са/С — (Л см — Лв) (А 4 — А в) и Св/С = (Лд — Лсм)(Лд — Лв)
Все значения оптических плотностей (Асм, Аа и Ав) измеряют при одной и той же длине волны света. При постоянной суммарной концентрации обеих равновесных форм анализ можно проводить с помощью градуировочного графика. Метод требует обязательного соблюдения основного закона светопоглощения и выбора участка спектра, где разница в светопоглощении определяемых компонентов является наибольшей.
Модель 5. При наличии двухкомпонентных систем иногда невозможно получить раствор, содержащий только один компонент (например, при таутомерных превращениях). При этом могут быть неизвестны как спектры поглощения компонентов, так и соотношение между их концентрациями. Графический способ определения молярных коэффициентов светопоглощения и концентрации компонентов в подобных смесях предложен Камин-
192
Рис. 7.13. Графическое определение молярных коэффициентов светопоглощения таутомерных форм соединения
ским, Берштейном и Гинзбургом [2431. Этот способ применим только в тех случаях, когда полосы поглощения анализируемых компонентов не накладываются
друг на друга.
Сущность метода состоит в следующем. Предположим, что спектры поглощения таутомерных форм Л и В не накладываются друг на друга (см. рис. 7.6). При поглощает форма А, а при — форма В, но в условиях опыта невозможно получить соединение, содержащее только одну из таутомерных форм. Всегда обе формы присутствуют совместно. Для дифференцированного определения концентрации каждой из равновесных форм приготавливают несколько растворов с постоянной суммарной концентрацией С форм А и В, но с различным их относительным содержанием. Затем измеряют оптические плотности этих смесей при и Х2 и строят по ним график в координатах Л;., — (рис. 7.13). Через полученные точки проводят прямую и экстраполируют ее в обе стороны до пересечения с осями координат в точках Аа и Ав. Затем определяют молярные коэффициенты светопоглощения
^ = Ал/(С/) и 8В = ЛВ/(С/)
и концентрации каждой из форм:
са = а/Лка1) и св = АкЛев[)
7.2. АБСОЛЮТНЫЕ ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА В ПРИСУТСТВИИ МЕШАЮЩИХ КОМПОНЕНТОВ
7.2.1. ДВУХВОЛНОВАЯ СПЕКТРОФОТОМЕТРИЯ (DUAL-\VAVELE]\GTH SPECTROPHOTOMETRY)
Двухволновая спектрофотометрия является одним из методов повышения селективности, а в ряде случаев и чувствительности фотометрического анализа [244—249]. Интересные практические разработки на двухволновых спектрофотометрах типа Hitachi-356 в приложении к анализу неорганических веществ выполнены японскими исследователями [245, 248], а вопросы правильности и воспроизводимости двухволновой спектрофотометрии подробно рассмотрены в работах [246, 247].
Селективность достигается техникой измерения в присутствии окрашенных реагентов и особенно в дисперсных (светорассеиваю-
7 М. И. Булатов и др. 193
Рис. 7.14. Спектры поглощения!
1 — мешающего компонента; 2 — определяемого компонента; 3— смеси обоих компонент* тов-
щих) системах, которые либо затрудняют, либо не позволяют проводить определения обычными методами.
В общем случае при фотоме-
трировании дисперсных систем наблюдаемые при двух длинах волн и Х2 оптические плотности А'}1 и Л£2 раствора связаны с фактическими оптическими плотностями АК1 и Л?,2 и светорассеянием £>Х1 и D>2 соотношениями:
Aki = + Dkt
Ai,=AK, + Dx2
Длины волн при этом выбирают такими, чтобы — D}^. Тогда ДА = A^t — А}2 не зависит от эффектов светорассеяния. При соблюдении основного закона светопоглощения:
Д^ = ^х1-^х2 = (ех,-ех2)с/ (7-25)
Если раствор содержит определяемое окрашенное соединение и окрашенный реагент или два окрашенных соединения, спектры которых перекрываются (рис. 7.14), то в этом случае в спектре реагента (или мешающего окрашенного соединения) выбирают такие значения и Х2, при которых оптические плотности А^ « » Оптические же плотности, обусловленные изменением концентрации определяемого вещества при одной из длин волн, должны заметно изменяться (на рис. 7.14—Xj). При этих условиях и при соблюдении основного закона светопоглощения
Д^ = ^Х2 = (Г?., 6Х2) Cl ~ Де<Х,—Х2)^ (7.25')
т, е. разность оптических плотностей при выбранных длинах волн прямо пропорциональна концентрации искомого элемента.
Иногда уравнения (7.25) и (7.25') записывают в несколько ином виде, выражая оптические плотности через интенсивности падающих (/ох,. /ох2) и прошедших (IK1, Z>,2) световых потоков:
АЛ = -lg (/Xi//Xs) + lg (l0K1/l0K1) (7.26)
Обозначая отношение Zox,/Zox2 = К, где К — константа для конкретной пары длин волн, получим:
ДА = -lg (Z^2) + 1g К (7.26')
Считается, что в большинстве случаев в выпускаемых промышленностью двухволновых спектрофотометрах К = 1, и тогда:
ДА = -18(/х,//х2) (7.27)
194
Принципиальная блок-схема двухволнового спектрофотометра представлена на рис. 7.15.
Из рис. 7.15 следует, что в двухволновой спектрофотометрии, по существу, исключается «кюветная» погрешность, которая вносит существенный вклад в общую погрешность при измерениях оптической плотности (см. разд. 3.1.3). Помимо этого и другие конструктивные особенности двухволновых спектрофотометров позволяют в ряде случаев повысить правильность и воспроизво» димость измерений по сравнению с «одноволновой» (обычной) спектрофотометрией.
Авторы [245] на примере определения Со * (1—4 мкг/л) с использованием его комплексного соединения с 4-(3,5-дихлор-2-пиридилазо)-1,3-диаминобензолом (R) демонстрируют возможности двухволновой спектрофотометрии как высокочувствительного спектрофотометрического метода анализа неорганических веществ (рис. 7.16—7.18).
В методах обычной «одноволновой» спектрофотометрии оптическая плотность раствора измеряется при Хмакс по отношению к воде или реагенту. В двухволновой спектрофотометрии соответствует максимуму спектра поглощения хелата, а ?.2 — максимуму спектра поглощения реагента. При этом желательно, чтобы образующийся комплекс был как можно более прочным, а свето-поглощение реагента при длине волны, соответствующей максимуму светопоглощения комплекса, было бы минимальным.
При определении Со в виде хелата с реагентом R = 588 нм и Х2 = 425 нм соответствовали максимумам светопоглощения хелата и реагента соответственно.
Для интервала содержаний кобальта в стандартных растворах, по которым строится градуировочный график, выбирается некоторое постоянное содержание реагента R. При этих условиях значения уменьшающейся оптической плотности реагента, обусловленные его убылью при реакции образования хелата, складываются аддитивно со значениями возрастающей оптической плотности образующегося хелата (см. рис. 7.17, 7.18): АЛ == = Лх, — (—AJ — Л?., + ЛхЕ.
Метод двухволновой спектрофотометрии позволяет увеличить эффективный («кажущийся») молярный коэффициент светопогло-
* Следует отметить, что методика демонстрируется на чистых синтетических образцах кобальта.
П К Ф
Рис. 7.15. Принципиальная блок-схема двухлучевого спектрофотометра [245]:
И — источник света; М, и М, -- монохроматоры: П — прерыватель светового потока; К =• кювета с раствором; Ф — фотоумножитель.
7*
195
Рис. 7.16. Спектры поглощения реагента (R) и его комплекса с кобальтом в ЗЛ1 растворе НС1 (Ср = = 1,9-10*5 моль/л) [245]
1 — 5 — концентрации ионов кобальта от 1 10”® до 5 X X Ю“® моль/л соответственно.
Рис. 7.17. Дифференциальные спектры поглощения комплекса кобальта с реагентом R [245]:
ЛЛ = — (-л^2) =
Рис .7.18. Сравнительные градуировочные графики методов двухволновой (5) и обычной (/, 2) спектрофотометрии [245 ].
к, нм: 1 — 425; 2— 588; S — 588—425.
щения хелата Сое Rc 1,35’10® до 1,75-10® и повысить чувствительность определения Со путем значительного увеличения угла наклона градуировочного графика (см. рис. 7.18).
В работе А. И. Лазарева [249] рассмотрен более общий случай выбора эффективных (оптимальных) значений Zj и с целью повышения чувствительности метода, когда спектры поглощения окрашенного комплекса и реагента перекрываются во всем диапазоне длин волн. Автор на примере определения микроконцентраций германия в виде его комплекса с фенилфлуороном (^хэфф = 520 нм, ^2Эфф = 450 нм) продемонстрировал:
во-первых, принципиальную возможность использования двухлучевых фотоколориметров типа ФЭК-60 и однолучевых спектрофотометров типа СФ-4, -4А, -16, -26 и других для анализа методом двухволновой спектрофотометрии;
во-вторых, значительное повышение чувствительности такого метода за счет возрастания эффективного молярного коэффициента светопоглощения eS2O,45o — 7,36 10* (из оценки по градуировочному графику, построенному на фотоколориметре ФЭК-60 при I — 0,5 см) против е520 = 4,94-104.
Суммарный эффективный молярный коэффициент светопоглощения по результатам определений на СФ-16 составлял 5,96-Ю4, т. е. был ненамного выше, чем в методах «однолучевой» спектрофотометрии. Судя по всему, из-за конструктивных особенностей однолучевых спектрофотометров навряд ли можно надеяться на 196
эффективное использование их для повышения чувствительности определений методом двухволновой спектрофотометрии.
Анализируя опубликованные данные по развитию и применению двухволновой спектрофотометрии, следует отметить, что в анализе неорганических веществ широкого применения этот метод не нашел, хотя на ряде примеров и в теоретических работах продемонстрированы его возможности по увеличению селективности, чувствительности, правильности и воспроизводимости определений по сравнению с «однолучевой» спектрофотометрией.
7.2.2. ПРОИЗВОДНАЯ СПЕКТРОФОТОМЕТРИЯ
В основе производной спектрофотометрии 1244, 250—257] лежит изменение формы спектральной кривой при ее дифференцировании по X (X—длина волны или волновое число) (рис. 7.19) [250, 251 ]. Экстремумы первой производной соответствуют точкам перегиба исходной полосы поглощения, а нулевая точка — ее максимуму. Центральный пик второй производной подобен контуру исходной полосы, но значительно сужен.
Количественная производная спектрофотометрия основана на линейности дифференцирования [250, 251]. При выполнении основного закона светопоглощения п-я производная оптической плотности
dnA Cldne
dXn ~ dXn (7,28)
При дифференцировании во времени с помощью аналоговых d11 v^dnE / dx
устройств с учетом [у = ---скорость ска-
нирования) получим:
dnA Clv'We,
Следовательно, при v = const производная пропорциональна концентрации.
Если справедливо условие аддитивности оптических плотностей компонентов многокомпонентной смеси, то оно справедливо и для производных. Тогда при нали-чии мешающих компонентов М{, спектры /г\акс
поглощения которых перекрывают спектр / \ поглощения определяемого компонента М, \
для первой производной по длине волны \
с\ „
Рис. 7.19. Лоренцева кривая (а) (спектр поглощения) и ее первая (б) и вторая (в) производные [2о0].
197
Рис. 7.20. Дифференцированные спектры определяемого (/) и мешающего (2) компонентов [250 ]:
а — первые производные; б — вторые производные.
можно записать выражение
dA ^м^ем dk ~ А
CM.WeM.
i=l 1 '
(7.30)
В случае двухкомгонентной смеси: dA смгйем , d). ~ d). + dX
Когда область дифференцирования совпадает с максимумом поглощения мешающего компонента, то ——► 0 и на оси X появляется точка нулевого вклада (Хо) (рис. 7.20, а) мешающего компонента; подавляется также фон.
т-> / , d2A
Вторая производная (функция 2~, названная «заострением») тоже обладает двумя важными свойствами. На оси X (к, v) появляются точки (в случае двухкомпонентной смеси — точка) нулевого вклада мешающих компонентов, лежащие между центральным пиком и сателлитом (рис. 7.20, б',, и подавляется фон, медленно меняющийся по сравнению с полосой поглощения [2521.
Описан [244] способ определения в водных растворах эрбия в присутствии церия методом первой производной. Представленные на рис. 7.21 дифференцированные спектры смеси солей эрбия и церия наглядно демонстрируют усиление контрастности этих спектров по сравнению с обычными и возможности селективного определения одного компонента (эрбия) в присутствии высоких содержаний другого (церия).
Предложен [255, 256] вариант метода первой производной (автоматический спектрофотометр и методика) для селективного определения U (VI) в присутствии Th, Zr или Fe (III) с использованием в качестве реагента арсеназо III.
198
A
чЫ--->—Cr
400350
-----------(Н--1---.
400350
Л, нм
Рис. 7.21. Спектры поглощения растворов соли эрбия разных концентраций (а), соответствующие им первые производные (б) и градуировочный график (в) [244].
Примечания. /. Значения ДЛ определены по разности между максимумом и минимумом первых производных спектров поглощения при Z 379 нм. 2. Для каждой кривой приведен участок спектра 350—400 нм. 3. Содержание Се постоянно и равно 2000 млн. д.
Достоинства применения вторых производных спектров для селективного определения U (IV) в присутствии Fe (II) наглядно продемонстрированы в работе [257]. На рис. 7.22 приведены спектры поглощения оксиэтилидендифосфоновых (ОЭДФ) комплексов U (IV) (кривая 7) и Fe (II) (кривая 2) и вторые производные спектров поглощения. Характер кривых 1 и 2 таков, что
42 38 J4 30 42 38 34 30
V- 10~}см-'
Рис. 7.22. Спектры погчощения оксиэтилидендифосфоновых (ОЭДФ) комплексов урана (IV) (/), железа (II) (2), спектр поглощения их смеси (5) и вторые производные спектров поглощения комплексов с ОЭДФ в отсутствие (4) и в присутствии железа (II) (4'):
Су цу) = 0,06 мг/мл; Сре yjj = 0,25 нг/мл; 1= 5 мы [257 ],
199
определение U (IV) оказывается невозможным даже при соотношении металлов 1:1. Интенсивности же вторых производных спектров комплекса U (IV) с ОЭДФ как в отсутствие ионов Fe (II) (кривая 4), так и в их присутствии (кривая 4') практически одинаковы. При выбранных условиях регистрации спектров градуировочный график для определения урана был линеен в области концентраций U (IV) 0—40 мкг/мл (X = 269 нм, I = 5 см). Предлагаемый метод успешно опробован на искусственных смесях U (IV) и Fe (II), содержание которых в анализируемой пробе (10 мл) составляло 30—60 мкг и 0—3 мг соо!ветственно. Производные спектры регистрировали в области 45 000—28 500 см-1.
Таким образом, использование дифференцированных спектров (особенно второй производной) позволяет:
добиться значительного усиления контрастности дифференцированных кривых по сравнению с обычными даже при небольших изменениях монотонности спектра, в результате чего облегчается анализ;
проводить анализ многокомпонентных систем;
значительно повысить селективность определений ионов в смесях сложного состава и в дисперсных системах;
подавить фоновый сигнал и тем самым исключить или уменьшить систематические погрешности, обусловленные неучитываемым фоном.
7.3. ДИФФЕРЕНЦИАЛЬНЫЕ
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Дифференциальный метод применяют для повышения воспроизводимости результатов анализа (см. разд. 3.1.3.2) при определении больших количеств веществ, а также для устранения мешающего влияния посторонних компонентов и исключения поглощения реактива. Этот метод, в отличие от других, можно применять еще и в тех случаях, когда из-за большой концентрации растворенного вещества нарушается основной закон светопоглощения или когда значения оптических плотностей окрашенных растворов выходят за пределы шкалы прибора, а дальнейшее разбавление анализируемого раствора нежелательно.
Теоретические основы дифференциальной фотометрии и результаты экспериментальных исследований до 1970 г. обобщены в монографиях [122, 123]. Сущность метода состоит в том, что оптические плотности исследуемого и стандартного окрашенных растворов измеряются не по отношению к чистому растворителю с нулевым поглощением, а по отношению к окрашенному раствору определяемого элемента с концентрацией Со, близкой к концентрации исследуемого раствора.
Относительная степень пропускания света исследуемым окрашенным раствором Г, измеренная по отношению к раствору
200
сравнения со светопропусканием Ц определяется разницей их концентраций ДС и математически выражается уравнением!
1'11 = /о-10_£Сж//(7о10~еСо/) = 10е 1 = ю~еЛС/
На рис. 7.23 показана зависимость относительной степени пропускания 77/ от оптической плотности раствора сравнения Ао для различных отношений концентраций сравниваемых раство* ров (а = CxiC0).
Как видно из рис. 7.23, с возрастанием Ао можно анализировать растворы с меньшим значением а (т. е. с меньшей разницей ДС), так как чувствительность относительного светопропуска-ния при одном и том же значении а повышается (отношение ГН уменьшается). Следовательно, в работе целесообразно использовать раствор сравнения (нулевой раствор), имеющий по возможности большее собственное поглощение. Однако это не всегда можно осуществить на практике, так как использование раствора сравнения с большим поглощением требует мощного источника освещения, высокой чувствительности фотоэлемента и его усилительной схемы, что не всегда предусматривается в конструкциях приборов.
Относительная погрешность определения концентрации дифференциальным методом уменьшается с увеличением концентрации Со раствора сравнения и теоретически * должна быть наименьшей, когда светопоглощение или оптическая плотность исследуемого раствора и раствора сравнения почти одинаковы (Сж » Со и 1'П « 1). При оптимальных условиях, когда отклонения от основного закона светопоглощения ничтожно малы, воспроизводимость дифференциального метода не уступает воспроизводимости классических титриметрических методов. Область концентраций, где соблюдается основной закон светопоглощения, определяется обычным путем по данным измерений оптических плотностей стандартных растворов по отношению к раствору с наименьшей концентрацией в этом ряду.
Показано, что в случае невыполнения основного закона светопоглощения погрешность при дифференциальных измерениях будет минимальной, когда в качестве раствора сравнения используют раствор, для которого величина произведения (ДЛ/ДС) Со имеет наибольшее значение (рис. 7.24).
Практически для определения оптимальной концентрации раствора сравнения Со поступают следующим образом. В области концентраций, где не наблюдается значительных отклонении от основного закона светопоглощения, приготавливают несколько стандартных растворов с такой разностью концентраций ДС, чтобы соответствующие им разности ДЛ были равны 0,3—0,4. Затем измеряют относительные оптические плотности каждого последующего раствора по отношению к предыдущему и рассчи-
* На практике по ряду причин это не всегда соблюдается (см. разд. 3.1.3.2).
201
Рис. 7.23. Зависимость отношения интенсивностей двух световых потоков от оптической плотности раствора сравнения при различных отношениях концентраций поглощающих растворов.
Рис. 7.24. Графическое определение оптимальной концентрации раствора сравнения Со в условиях несоблюдения основного закона светопоглощения:
6' = АА/АС — тангенс угла наклона касательной к кривой зависимости А от С в точке, соответствующей концентрации Со.
тывают величину f = (ДЛ/ДС) Со. (где COj — концентрация раствора, который используется в каждом t-м измерении в качестве раствора сравнения).
Тот раствор, для которого значение f получается наибольшим, и используется в качестве раствора сравнения, так как в этом случае достигается наибольшая чувствительность и воспроизводимость определения. Однако следует иметь в виду, что в фотометрическом анализе увеличение концентрации раствора сравнения Со не всегда приводит к повышению воспроизводимости определения, главным образом из-за возникающих отклонений от основного закона светопоглощения вследствие немонохроматич-ности поглощаемого света. Поэтому при выборе оптимальных условий дифференциальных измерений следует, прежде всего, найти ту предельную концентрацию раствора сравнения, при которой обеспечивается прохождение через поглощаемый раствор достаточного количества света и используемый прибор устанавливается «на нуль». При работе на регистрирующих спектрофотометрах при дифференциальных измерениях перо должно перемещаться с обычной для прибора скоростью и значение максимума поглощения или оптической плотности не должно зависеть от усиления. В противном случае необходимо уменьшить либо толщину поглощающего слоя, либо концентрацию раствора сравнения.
7.3.1. СПОСОБЫ ОПРЕДЕЛЕНИЯ ОДНОГО КОМПОНЕНТА
Дифференциальный метод в зависимости от способов измерения относительной оптической плотности исследуемого раствора и расчета его концентрации может иметь несколько вариантов.
202
Концентрация раствора сравнения меньше концентрации исследуемого раствора (Со < Сж). Измеренная экспериментально относительная оптическая плотность А' представляет собой разность оптических плотностей фотометрируемого раствора и раствора сравнения:
Ах = Ах = 6 (рх Со) I
^ст = Л0 = 6 (^ст Со)
Концентрацию исследуемого раствора определяют или при помощи градуировочной кривой, или расчетным способом. Для построения кривой в области возможных концентраций исследуемого раствора приготавливают серию стандартных растворов с концентрациями Съ С2, .... Ct, ..., Сп (Сп > Ct ... > С2 > > Ci), и измеряют их оптические плотности по отношению к окрашенному раствору сравнения с концентрацией Со. По полученным данным строят градуировочную кривую (рис. 7.25, а), принимая за начало отсчета концентрацию раствора сравнения Со. Измерив относительную оптическую плотность исследуемого раствора А’х, по градуировочному графику определяют его неизвестную концентрацию Сх.
При расчетном способе определения концентрации исследуемого раствора учитывают, что отношение значений относительных оптических плотностей исследуемого и стандартного растворов соответствует отношению разности между концентрациями этих растворов и раствора сравнения, т. е.
Ах/Аст = (Сх С0)7(Сст — Со) откуда
Сх^ + Ах^-СМА'™ (7.32)
или
CX = CO + FA'X (7.32')
Отношение разности концентраций стандартного раствора и раствора сравнения к относительной оптической плотности стандартного раствора (Сст—С0)/Лст (обратный угловой коэффи
Рис. 7.25. Градуировочный график дифференциальным методом:
° со < Сх (ст)- 6 Со > СХ (ст)-
для определения концентрации раствора
203
циент градуировочного графика) называют фактором пересчета F. В одной серии измерений для определенного интервала концентраций исследуемого раствора F является постоянной величиной.
Рассмотренный вариант наиболее широко применяют для экспрессного определения содержания основных компонентов с воспроизводимостью, не уступающей классическим методам анализа. Он хорошо зарекомендовал себя, в частности, при определении фосфора и кремния в различных объектах (табл. 7.1).
При анализе источников систематических погрешностей при дифференциальных фотоколориметрических определениях Р2О5, и в частности в методике ГОСТ 23999—80, авторы [266] выделяют погрешности численной аппроксимации градуировочной зависимости. Для уменьшения влияния нелинейности градуировочного графика на определение концентрации Р2О5 (CJ они рекомендуют применять для расчета «метод ограничивающих растворов» («метод окаймления»):
= Сдт j -|- [(Лж Лст i) (ЛСТ 2 Лег 1)] (Ссг 2 Сст г) (7.33)
Здесь Сеть Сст2 и Лсть ЛСТ2—концентрации и оптические плотности двух ближайших стандартных растворов, причем Л СТ1 < Лж < Л ст г-
Концентрация раствора сравнения больше, чем концентрация исследуемого раствора (Со > Сх). В этом варианте применяют обратный порядок измерений: анализируемые растворы условно принимают за растворы сравнения и по отношению к ним измеряют оптическую плотность раствора сравнения. Максимальная концентрация Сх исследуемого раствора ограничивается известной концентрацией Со раствора сравнения. Концентрацию исследуемого раствора определяют аналогично описанному выше варианту с той только разницей, что здесь относительная оптическая плотность исследуемого раствора равна разности между значениями оптической плотности раствора сравнения и исследуемого раствора:
Концентрацию Сх исследуемого раствора рассчитывают по формуле
сх = с0-гл; (7.34)
(где F = (Со — Сст)/Лст, а Д„ = До — ДСт) или определяют при помощи градуировочной кривой.
Для построения градуировочной кривой приготавливают стандартные растворы с концентрациями С±, С2, С3, ..., Сп, меньшими, чем концентрация Со. По оси абсцисс откладывают значения разности ДС между концентрациями раствора сравнения и стандартных растворов (либо уменьшающиеся абсолютные значения концентраций стандартных растворов, беря за начало отсчета значение Со), а по оси ординат — соответствующие им значения относительной оптической плотности (рис. 7.25, 6).
604
Таблица 7.1. Определение кремния и фосфора в различных объектах дифференциальным фотометрическим методом
Гетерополи кислота (ГПК), в виде которой определяется элемент; окраска раствора Анализируемые объекты Интервал определяемых содержаний, % *; воспроизводимость (sr£ ИЛЕ А С/С) 1, им; L, мм Интервал концентраций ** граду-нров очного графика; Се Примечание Лите** рату-ра
Восстановленная форма фосфорносурь-мяномолнбденовой ГПК; синяя Фосфаты редкоземельных элементов; силикофос-фатные стекла 20- 75 Красный светофильтр; Z = 50 (ФЭК-м). На спектрофотометре h = 882 (1—25). 10" 7 г Р2ОБ в 1 мл Со= 8-КГ? г Р2О5 в 1 мл ФЭК-М Кремний не мешает определению до отношения SiOa/P2O6 = 200/1 [258]
Молибденовая ГПК; желтая Припои Си — Р; Ni—Р; полупроводники ZnP2 и др. 5—48 ^•эфф~ 405 Z= 10 0,07—0,1 мг Р Со = 0,04 мг Р ФЭК-56 [259]
Фосфорнованадиевая ГПК; желтая Апатитсодержащие руды и продукты их обогащения 0,04—20 (классический метод); >20 — дифференциальный метод Л= 420 1. Недостаток: фото-метрироваиие растворов через час после приготовления 2. Мешают Si, As, W, Zr, Nb, Ta, Fe — при высоких концентрациях [260]
Восстановленная форма фосфорномолиб-деновой ГПК; синяя Апатиты, фосфориты, минеральные удобрения 20—60 Светофильтр № 8 (^эфф = 590) 2,0—4,0 мг Р2ОБ Со — 1,0 мг Р2ОБ 4,0—7,0 мг Р2ОБ Со = 2,0 мг Р2ОБ ФЭК-56 [1301
Гетерополикислота (ГПК), в виде которой определяется элемент; окраска раствора Анализируемые объекты Интервал определяемых содержаний, % *; воспроизводимость (sr(j или ДС/С)
Восстановленная форма ГПК фосфопно-молибденовой ГПК; синяя Фосфаты
Фосфорнованаднево-молнбденовая ГПК; желтая Фосфаты, минеральные удобрения 20—60
Кремнемолибденовая ГПК; желтая Горные породы При 67% Src ~ 0,005
Германий, теллур, молибденсодержащие сплавы 1—37% Si; при 15—16% src SC 0,006
Восстановленная форма кремнемолибденовой ГПК; синяя Силикаты, минеральное сырье Сварочные флюсы 10—90; SrC = 0,006 45—15; ДС/С = 0,54-4-0,7%
* Приведено содержание (%) соответственно Р2ОБ и SiO2.
** Для V = 100 мл.
Продолжение
X, им; 1, мм Интервал концентраций ** градуировочного графика; Сц Примечание Литература
^э<ЬФ ~ 595, 0,5—2,5 мг Р2О6 ФЭК-56 [261]
720 Со = 3,35 мг Р2О6 (вместо раствора сравнения использовано кобальтовое стекло, имеющее «фиктивную» концентрацию 3,35 мг Р2О6 [127]
Светофильтры № 3 или № 4 (Кэфф ~ 400—--=-440 нм) 1,0—5,0 мг Р2О5 Со — 1,0 МГ Р2^5
400 Определение в присутствии РП^~ и других мешающих ионов) [262]
Светофильтр 1,1—1,5 мг ФЭК-56; src в 4—5 [263]
We 2 (^-эфф = 370); / = 10 Со = 1,0 мг Si раз меньше, чем при классическом фотометр и ческом анализе [264]
X = 640; / = 5 0,2—2,0 мг SiO2 Со = 0,8 мг SiO2 СФ-4А
Л = 830; 1 = 10 30—220 мкг SiO2 Со = 236 мкг SiO2 СФ-16 [265]
В этом варианте при ДС = 0 концентрации раствора сравнения и исследуемого раствора будут одинаковы (Сж = Со), а при максимальной разности концентраций (ДС = Со) концентрация исследуемого раствора равна нулю (Сх — 0).
Двухстороннее (полное) дифференцирование. Двухсторонним дифференцированием называют сочетание прямого (Со < С) и обратного (Со £> С) порядка измерений при фотометрических (спектрофотометрических) определениях веществ.
В дифференциальной фотометрии для уменьшения погрешности определения используют раствор сравнения с оптимальным значением оптической плотности Ао. Однако получающийся при этом интервал концентрации от Со до Смакс (Со < С), в котором соблюдается основной закон светопоглощения, обычно бывает узок. Возможности метода значительно расширяются при объединении прямого и обратного дифференцирования при измерениях. Двухстороннее дифференцирование при использовании одного и того же
раствора сравнения позволяет увеличить интервал анализируемых концентраций примерно в 2 раза при неизменной воспроизводи-
мости результатов анализа.
При фотометрировании исследуемых и стандартных растворов, концентрации которых больше, чем концентрация раствора сравнения (Со < С), значения относительной оптической плотности берут, как обычно, со знаком плюс. Если концентрация фотометрируемых растворов меньше, чем концентрация раствора сравнения (С < Со), то полученные значения относительной оптической плотности берут со знаком минус.
В последнем случае применяют обратный порядок измерений: анализируемые растворы условно принимают за растворы сравнения и по отношению к ним измеряют оптическую плотность раствора сравнения. Полученные значения относительной оптической плотности принимают знак минус.
Для построения градуировочной кривой приготавливают не-
сколько стандартных растворов с концентрациями определяемого
вещества меньшими, чем в растворе сравнения, и столько же
стандартных растворов с концентрациями большими, чем в растворе сравнения. По полученным данным строят градуировочную кривую (рис. 7.26), при помощи которой и определяют неизвестную концентрацию исследуемого раствора. Неизвестную кон-
Рис. 7.26. Градуировочный график для определения концентрации раствора методом полного (двухстороннего) дифференцирования:
Л*1— относительная оптическая плотность исследуемого раствора, измеренная по отношению к раствору сравнения (Лд-1 > Л q); Л — относительная оптическая плотность раствора сравнения, измеренная по отношению к исследуемому раствору (Л х* < Л
207
центрацию исследуемого раствора можно определить также и расчетным путем по формуле:
С* — £q 4* FAX
При двухстороннем дифференцировании большое значение имеет качество кювет. Если наблюдаются даже незначительные различия в кюветах (длина оптического пути, состояние рабочих поверхностей), то на градуировочном графике в точке С = Со будет происходить изменение угла наклона графика. В этом случае целесообразнее пользоваться расчетным способом, вычисляя значения F для обеих половин графика; Fv — для положительных значений относительной оптической плотности и F2 — для отрицательных значений.
Расчет неизвестной концентрации Сх производят по формулам:
6"^ = С. + Ли С = С- — ГуА'х л (j I л л и & л
Дифференциальный метод добавок [2671. Дифференциальный метод добавок основан на сочетании дифференциальной фотометрии с методом добавок. Его применяют при дифференциальных измерениях в присутствии мешающих компонентов, когда требуется создать одинковый солевой состав в исследуемом растворе и растворе сравнения. Необходимым условием применения этого метода является строгое соблюдение основного закона светопоглощения.
Сущность метода состоит в следующем. Приготавливают исследуемый окрашенный раствор, разбавленный в k раз (k £> 1), исследуемый окрашенный раствор и исследуемый окрашенный раствор с добавкой а определяемого вещества. Затем измеряют относительные оптические плотности неразбавленного исследуемого раствора Ах и раствора с добавкой Ах+а по отношению к разбавленному исследуемому раствору. Неизвестную концентрацию Сх определяемого вещества рассчитывают по формуле:
Л' с — с ______х ----
лх+а 71 х
(7.35)
fe— 1
Воспроизводимость результатов зависит от кратности разбавления k и значения добавки а. Рез}льтаты получаются хорошими, когда берут минимальную добавку и используют наименьшую кратность разбавления исследуемого раствора. При этом следует учесть, что выбранные значения добавки и кратности разбавления не должны сопровождаться «потерей точности результатов при вычитании».
Определив одним из описанных выше способов концентрацию исследуемого раствора, рассчитывают общее содержание определяемого вещества в анализируемой пробе. Учитывая разбавление исследуемого раствора, находят содержание определяемого вещества в растворе (дх, мг):
Чх —
£С8
Дифференциальный метод применяют при анализах различных объектов на содержание основных компонентов. Относительная погрешность определения составляет 0,2—1,0%,
7.3.2. ДИФФЕРЕНЦИАЛЬНАЯ СПЕКТРОФОТОМЕТРИЯ МНОГОКОМПОНЕНТНЫХ СИСТЕМ
Для дифференциальной спектрофотометрии многокомпонентных систем может быть применен метод относительного пропускания.
Измерения двухкомпонентных систем проводят при двух длинах волн — Xi и Х2, соответствующих максимумам поглощения веществами 1 и 2. Оптическую плотность неизвестного раствора измеряют по отношению к раствору сравнения, содержащему вещество 1 (при известной концентрации раствора сравнения C,J Ах/Г1, но не содержащему вещество 2. При длине волны Х2 оптическая плотность Ах/Г2 должна измеряться по отношению к раствору, содержащему известную концентрацию СГ2.
Предполагая, что толщина слоя раствора / = 1 см, и зная (или измерив) коэффициенты светопоглощения вещества 1 при М и Х2 (feix, и *ы2) и вещества 2 при Xi и X, (/г2?„ и *2?.2), неизвестные концентрации CV1 и СХ2 (г/л) веществ 1 и 2 можно рассчитать из следующих выражений:
1 / *21, „ , *21,
*111 Х/Г- *11, *11,*21,
1 _ feg?4 fe|>z *11, *21,
(7.36)
Если условия выбраны так, что обе относительные оптические плотности Лх/r, и Ах/,2 равны или близки к нулю, то для расчетов СХ1 и СХг достаточно знать отношения коэффициентов светопоглощения Y' = foi./fox, и У" = &ii2/Z?2is, которые могут быть легко найдены экспериментально. Погрешность определения концентрации в этом случае рассчитывается сложно и здесь не приводится .
Практические требования для получения минимальной погрешности сводятся к следующему:
а) концентрации стандартных растворов сравнения СГ1 и СГ1 должны быть как можно больше;
б) измеряемые оптические плотности Ax/rt и А"х/Гг должны быть близки к нулю и не должны превышать значений 0,2—0,3;
в) отношение коэффициентов светопоглощения должно быть вычислено точно.
209
При использовании этого метода достигается высокая воспроизводимость и относительная погрешность определения концентраций может быть уменьшена до 0,2—0,5% (отн.).
7.4. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД
Экстракционно-фотометрический метод [38, 66, 118, 161, 167, 176, 268—275] основан на сочетании экстракции определяемого вещества с его последующим фо -ометрическим определением. Этот метод применяют при анализе сложных смесей, когда нужно определить малые количества одних веществ в присутствии больших количеств других, при определении примесей в присутствии основных компонентов, а также в тех случаях, когда непосредственное определение интересующего элемента в смеси связано с большими трудностями. При экстракции малых количеств примесей происходит не только их выделение, но и концентрирование. Поэтому экстракционно-фотометрический метод приобретает особо важное значение в связи с определением малых количеств примесей в веществах высокой степени чистоты, широко применяемых в атомной и полупроводниковой технике. Экстракционно-фотометрические методы анализа являются высокочувствительными методами, они быстро развиваются и очень перспективны.
Экстракция разнолигандных комплексов — одно из наиболее интенсивно развивающихся направлений в аналитической химии, при этом разнолигапдные комплексы используют для прямого определения не только ионов металлов-комплексообразователей, но и анионов-реагентов (лигандов) [38, 273]. Разнообразие лигандов при образовании смешанных экстрагирующихся комплексов значительно расширяет возможности в повышении чувствительности и избирательности экстракционно-фотометрических методов анализа.
В экстракционно-фотометрических методах для определения металлов применяют экстракционные системы различных типов, выбор которых зависит от химической природы определяемого компонента, состава растворенных веществ и условий проведения экстракции.
7.4.1. НАИБОЛЕЕ РАСПРОСТРАНЕННЫЕ
ЭКСТРАКЦИОННЫЕ СИСТЕМЫ
1. Комплексные металлокислоты (галоге-нидные и псевдогалогенидные ацидокомплексы) общей формулы Нр_2МАр {р — лигадное число, г — заряд иона-комплексообра-зователя). Эти соединения экстрагируются активными растворителями, способными протонироваться в кислой среде (кетоны, спирты, эфиры). Такие системы применяют для экстракционнофотометрического определения Fe (III), Со (II), Cu (II), Мп (II),
210
Ni, Al, Ga, In, TI, содержащихся как в микроколичествах, так и в виде основных компонентов.
2. Гетерополисоединения. Различного состава гетерополисоединения, экстрагируемые кислородсодержащими растворителями, используют для экстракционно-фотометрического определения As, Ge, Mo, W, V, Si, Р и некоторых других элементов.
3. Хелатные комплексы. Хелаты, образуемые ионами металлов с органическими реагентами, способными координироваться одновременно не менее чем двумя донорными атомами (обычно атомами О, N и S), составляют одну из самых обширных групп комплексных соединений, применяемых в экстракционно-фотометрических методах анализа. Наиболее распространенные органические реагенты, применяемые для экстракции хелатов металлов, описаны в монографии [167], а количественные зависимости распределения хелатов между водной и органической фазами рассмотрены в гл. 4.
4. Разнолигандные комплексы. Для аналитических целей наибольший интерес представляют разнолигандные комплексы-аддукты, в состав которых входят хелатирующие и нейтральные лиганды. Образование таких смешанных комплексов, как правило, сопровождается явлением синергизма — повышением степени извлечения металла в органическую фазу за счет присоединения нейтрального лиганда к хелату извлекаемого металла. При синергетической экстракции коэффициент распределения разнолигандного комплекса значительно превышает коэффициенты распределения однороднолигандных комплексов этого металла с каждым из лигандов в отдельности. Для количественного определения элементов применяют следующие типы разнолигандных компонентов.
a) MR,R,' — комплексы, образованные хромофорным хелатирующим органическим реагентом (дитизон, пирокатехиновый фиолетовый, хромазурол S, эриохромцианин R и другие) и органическим основанием (1,10-фенантролин, а,а'-дипиридил, диан-типирилметан).
б) МЕ;Аг — комплексы со смешанной координационной сферой, образованной амином R (пиридин, 1,10-фенантролин, а, а'-дипиридил, диантипирилметан) и анионом А минеральных и органических кислот.
в) MRjR, — комплексы с двумя электроотрицательными реагентами, один из которых обычно является хелатообразующим (пиридилазонафтол +SCN-, С1", СН3СОО", пиридилазорезорцин капроновая кислота, ксиленоловый оранжевый + молочная кислота и др.).
г) [MR;] RJ, [MR;] (НАД (гдеМ — ион металла; R—электроотрицательный хелатирующий лиганд; R' —электроотрицательный анион кислоты; А — органическое основание) — комплексы типа ионных ассоциатов (ионных пар).
211
Во всех случаях ион металла входит в состав комплексного катиона или аниона, а противоионом может быть анион органической или минеральной кислоты, амин, основной краситель, «ониевый» катион.
Ионные ассоциаты по существу представляют собой сочетание внутри- и внешнесферных комплексов и обладают свойствами разнолигандных комплексных соединений, в которых полнее проявляются индивидуальные свойства ионов металлов. Это дает возможность повышать избирательность экстракционно-фотометрического определения металлов. При экстракции с использованием «ониевых» солей (солей тетрафениларсония, тетрафенилфосфония, тетрафенилсульфония, тетрабутиламмония, цетилпиридиния, ди-фенилгуанидиния и др.) эффективность экстракции увеличивается еще и за счет того, что катионы многих из них одновременно являются коллоидными поверхностно-активными веществами (ПАВ.) В процессе мицеллообразования катионных ПАВ на их поверхности происходит адсорбция анионов металлохромных реагентов, это повышает их концентрацию и способствует образованию более координационно-насыщенных металлокомплексных хелатов [268]. Кислотные свойства адсорбированных реагентов при этом повышаются, а значения pH образования разнолигандных комплексов смещаются в более кислую область, что особенно важно для ионов металлов, реагирующих в кислых средах (Т., Zr, Mo, W, Sn, Fe). Таким образом, применение катионных ПАВ повышает избирательность экстракционно-фотометрического определения и значительно повышает чувствительность и контрастность реакций ионов металлов с хромофорными реагентами [269—2711.
д) Разнолигандные соединения окрашенных металлокомпле-ксов с основными красителями. К этой группе ионных ассоциатов относят разнолигандные комплексы, содержащие две хромофорные группы — окрашенный металлокомплекс и основный краситель (родамин 6Ж, родамин С, кристаллический фиолетовый и др.).
Применение ионных ассоциатов с двумя хромофорными группами является одним из эффективных способов повышения чувствительности экстракционно-фотометрических определений [381. Высокая чувствительность таких методов объясняется большими значениями молярных коэффициентов светопоглощения основных красителей — до 1 • 105. Если в состав экстрагируемого соединения на один атом определяемого элемента входит п молекул основного красителя, то молярный коэффициент светопоглощения в пересчете на определяемый элемент может достигать величины порядка п-105. Высокая эффективность в повышении чувствительности достигается при сочетании многозарядных комплексных анионов с катионами основных красителей. Например, многозарядные анионы гетер ополи кислот взаимодействуют с основными красителями с образованием ионных ассоциатов с молярными коэффициентами светопоглощения до ЫС®.
212
В ряде случаев образующийся осадок гетерополианиона с основным красителем сначала флотируют подходящим органическим растворителем, а затем растворяют в ацетоне и фотометри-руют стехиометрическое количество «освободившегося» основного красителя. Примеры косвенного фотометрического определения веществ, основанного на использовании сильно окрашенных красителей, рассмотрены в монографии [33]. Таким способом, например, можно определять нанограммовые содержания (1-10_® г) Ni, Си, Fe, Со, и Pd, используя хелатные соединения бора [274]. При взаимодействии внутрнкомплексных соединений определяемых металлов с органическими соединениями бора образуются ионные ассоциаты, хорошо экстрагируемые органическими растворителями. После разрушения таких сложных ассоциатов определяют стехиометрическое (удвоенное по отношению к определяемому иону металла) количество бора по очень чувствительной реакции с куркумином. Например, при определении никеля путем взаимодействия диметилглиоксимина никеля и тетрафенил-бората натрия молярный коэффициент светопоглощения в пересчете на никель достигает значения 3,6-105 [274]. Аналогичные определения выполняют при помощи кристаллического фиолетового, родамина Б [275] и некоторых других красителей [38].
При экстракционно-фотометрическом определении с использованием основных красителей реакцию проводят таким образом, чтобы определяемый элемент входил в состав аниона, а краситель — в состав катиона. Образующееся окрашенное соединение (RKpac)n Апэл экстрагируется органическими растворителями и либо непосредственно фэтометрнруется экстрагируемый ионный ассоциат, либо определяется стехиометрическое количество основного красителя после разрушения экстрагируемого комплекса. Молярный коэффициент светопоглощения в пересчете на моль определяемого элемента можно выразить следующим образом:
eMR = 6R/i/7100 (7.37)
Здесь eR — молярный коэффициент светопоглощения фотометрируемого красителя (или его соединений); п — стехиометрический коэффициент, показывающий число молекул красителя, связанное в экстракте с одним атомом определяемого элемента; г — степень извлечения окрашенною соединения определяемого элемента экстрагентом.
Чувствительность определения тем выше, чем больше eR, г и и.
При фотометрировании экстрактов обычно используют метод градуировочного графика или метод сравнения.
При всем многообразии экстракционных систем, применяющихся в экстракционно-фотометрических методах анализа, экстракция ионов металлов Хелатообразующими органическими реагентами либо в виде координационно-насыщенных хелатов, либо в виде разнолигандных комплексов получила наиболее широкое £13
распространение. Количественное распределение веществ и рассмотрение равновесий, сосуществующих при экстракции ионов металлов хелатообразующими реагентами, изложены в гл. 4. Конкретные примеры теоретических обоснований в выборе оптимальных условий экстракционного выделения и разделения хелатов рассмотрены в гл. 10.
7.4.2. УСЛОВИЯ СЕЛЕКТИВНОГО ОПРЕДЕЛЕНИЯ
При определении иона Мх в присутствии мешающего иона Мп, когда они оба образуют с реагентом экстрагируемые окрашенные комплексы MTRn и MnRn с перекрывающимися спектрами поглощения, правильные результаты получают только в том случае, если оптическая плотность экстракта мешающего иона Мц (Ап) не превышает 1 % от оптической плотности экстракта определяемого иона Mj (Aj):
Л j/Лп = ei [MiRn]0/(en [MnRn]0) >1-102 (7.38)
Концентрации комплексов определяемого и мешающего ионов в органической фазе, как это следует из уравнения (4.5), связаны с их коэффициентами распределения выражениями:
[MxRnlo = CiEjUZ/HOOV) = CjA/frZh 4- 1) (7.39)
[MiiRn)o == СцОцДгРц + 1) (7.40)
Здесь Cj, Сп — аналитические концентрации ионов ЛД и Л-1 ц в водной фазе до распределения.
Выразив равновесные концентрации комплексов в уравнении (7.38) через равные им значения в уравнениях (7.39) и (7.40), получим следующее соотношение:
ejCiDi (rDn + l)/[enCnD„ (rDx + 1)] > 1(Я (7.41)
Неравенство (7.41) устанавливает необходимые соотношения между коэффициентами распределения определяемого и мешающего ионов и их общими концентрациями для обеспечения селективного экстракционно-фотометрического определения анализируемого иона. Если гО[ 1, a rDu 1, то неравенство (7.41) упрощается и принимает такой вид:
eiCj/fenCi 1£>пг) > 10» (7.42)
Для случая, когда Сг = Сп и ej та еп, неравенство (7.42) становится еще более простым:
rDn «S 0,01 (7.43)
Таким образом, соблюдение неравенств (7.41)—(7.43) является основным критерием при прогнозировании условий селективного экстракционно-фотометрического определения анализируемого иона Mj в присутствии мешающего иона Мп.
214
7.5. СПЕКТРОФОТОМЕТРИЧЕСКИЙ АНАЛИЗ ПО СПЕКТРАМ ОТРАЖЕНИЯ
В этом методе, применяемом для анализа твердых непрозрачных сред, о поглощении света частицами судят по диффузному отражению света рассеивающими частицами. Для рассеивающих объектов, у которых направление света внутри слоя многократно изменяется вследствие многочисленных отражений и преломлений твердыми частицами, основной закон светопоглощения не применим [276]. В этом случае световой поток ослабляется как за счет поглощения света частицами исследуемого вещества, так и за счет рассеяния, поэтому истинное поглощение света оказывается замаскированным рассеянием. Тем не менее, при анализе спектральная кривая отражения рассеивающими объектами обычно отождествляется с кривой истинного поглощения анализируемого вещества, и количество вещества в твердой фазе определяют по минимуму отражения аналогично фотометрическому анализу растворов.
Количественное определение основано на сравнении степени отражения света исследуемой и стандартной рассеивающими средами при длине волны максимального поглощения света анализируемым веществом.
Спектры отражения исследуемого образца и эталона снимают при одинаковых условиях (однородность, дисперсность, толщина слоя и т. п.) относительно той же рассеивающей среды, но не содержащей определяемого вещества. Реагент для анализа выбирают таким образом, чтобы обеспечивалась наибольшая разница в коэффициентах отражения определяемого вещества и самого реагента (прн длине волны минимума отражения определяемого вещества). Прн помощи спектрофотометров СФ-10, СФ-14 и СФ-18 можно проводить качественный и количественный анализ по спектрам отражения в видимой области спектра.
Анализ по спектрам отражения проводят при количественных определениях адсорбированных веществ, полученных после хроматографического или электрофоретического разделения на бумаге [302].
В качестве примера рассмотрим определение лантана на хроматограмме при помощи метилового тимолового синего [278].
Определение лантана на бумажной хроматограмме основано на различии в положении минимума отражения метилового тимолового синего и его комплекса с лантаном.
Приготавливают полоски хроматографической бумаги с окрашенными зонами метилового тимолового синего с лантаном и на спектрофотометрах СФ-10 или СФ-14 снимают спектры отражения цветных зон относительно хроматографической бумаги с зоной чистого реактива той же концентрации. Для получения однородной окраски пятен хроматографическую бумагу погружают в раствор реагента в растгорителе, не обладающем хроматографическим эффектом. Построив градуировочный график «коэффициент отражения — концентрация» по цветным зонам с известным содержанием лантана (рис. 7.27), определяют неизвестную концентрацию лантана на исследуемой хроматограмме.
215
Рис. 7.27. Градуировочный график для определения концентрации лантаНа rto значению коэффициента отражения
На полоску хроматографической бумаги наносят микропипеткой 0,05 мл рас-створа и трата лантана, имеющего pH » 5, и высушивают на воздухе. Затем бумагу погружают на 5 с в 0,001 М водно-спиртовой раствор реактива метилового синего, высушивают на воздухе в течение 30 мин и снимают спектр отражения. Для построения градуировочного графика на полоски хроматографической бумаги наносят микропипеткой по 0,05 мл растворов нитрата лантана, содержащих 60, 90, 120, 150 и 180 мкг лантана и имеющих pH а; 5 Бумагу высушивают на воздухе и затем каждую полоску в отдельности погружают на 5 с в 0,001 М водно-спиртовый раствор метилового тимолового синего. После высушивания на воздухе в течение 30 мин определяют коэффициенты отражения окрашенных зон при Л = 600 нм и строят график «коэффициент отражения — концентрация лантана».
Эталоном сравнения служит хроматографическая бумага, погруженная на 5 с в раствор метилового тимолового синего той же концентрации и высушенная в течение 30 мин на воздухе.
Определив коэффициент отражения исследуемой хроматограммы при 600 нм, при помощи градуировочного графика находят неизвестное содержание лантана в исследуемой пробе.
7.6. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Спектрофотометрическое титрование — группа методов титри-метрического анализа, в которых конечная точка титрования определяется по изменению оптической плотности раствора. При этом используют все реакции, встречающиеся в титриметрии: кислотно-основные, осаждения, окислительно-восстановительные и особенно широко — комплексообразования (в первую очередь с различными комплексонами)*.
Спектрофотометрическое титрование имеет следующие достоинства:
1. Позволяет быстро, престо и с высокой воспроизводимостью результатов проводить анализ. Если объем рабочего раствора (титранта) измерен с достаточной степенью точности, то ошибка титрования зависит только от погрешности определения концентрации титранта.
2. Можно титровать с высокой воспроизводимостью очень разбавленные растворы (С < Ю-6 Л1), а также сильноокрашенные и даже мутные растворы. Абсолютные содержания веществ, определяемых этим методом, лежат в пределах I • 10-1—П10-8 г.
* Вопросам теории, практики и аппаратурного оформления метода посвящена литература 1279—322].
£16
3. Его можно проводить во многих случаях, когда изменения окраски раствора плохо различаются глазом. Применение фотоэлементов, фотосопротивлений, фотодиодов и фотоумножителей в качестве приемников света, прошедшего через исследуемый раствор, позволяет получать объективные данные и проводить титрование не только окрашенных, но и «бесцветных» для глаза растворов, поглощающих излучение в ультрафиолетовой и ближней инфракрасной областях спектра, что значительно расширяет возможности титрометрического определения многих элементов.
4. Легко может быть автоматизировано.
Ниже приведена принципиальная блок-схема установки для спектрофотометрического титрования:
7.6.1. СУЩНОСТЬ МЕТОДА И КРИВЫЕ
СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Метод спектрофотометрического титрования основан на последовательном измерении оптической плотности раствора в процессе титрования при заранее заданной длине волны. Если анализируемый раствор подчиняется основному закону светопоглощения и относительная устойчивость образующегося при титровании комплекса достаточно высокая (РмяпСм 102 ("+1)№), то кривые спектрофотометрического титрования выражаются двумя пересекающимися прямыми линиями (рис. 7.28 кривая 1). Для определения эквивалентного объема титранта в этом случае строят кривую титрования в координатах А = f (V) (где V — объем рабочего раствора реагента-титранта, мл) и по точке излома на кривой титрования находят эквивалентный объем титранта. На воспроизводимость результатов титрования оказывает влияние значение угла, образованного двумя линейными участками кривой по обе стороны от точки эквивалентности.
Если относительная устойчивость образующегося при титровании комплекса невысокая, то в равновесии с комплексом всегда будут находиться сравнительно большие количества исходных ком-
217
Рис. 7.28. Кривые спектрофотометрического титрования:
1 — при образовании высокопрочного комплекса; 2 — при образовании малоустойчивого комплекса; х == — титровальная Доля; Аэ —
оптическая плотность раствора в точке эквивалентности; ДЛЭ — отклонение оптической плотности от ее значения в точке эквивалентности при образовании малопрочного комплекса; ЛЛх/ —• отклонение оптической плотности при х = 0,5,
понентов и кривая титрования в области точки эквивалентности вместо резкого излома будет иметь плавный переход от одной ветви к другой (рис. 7.28, кривая 2). При таких условиях конечная точка титрования может быть определена графически экстраполяцией линейных участков кривой до их взаимного пересечения (см. рис. 7.28). Из рассмотрения неравенства (1.15) видно, что если условная константа устойчивости комплекса MRn в условиях титрования достигает значения не менее чем пЮ7—и108, то скругление кривой титрования около точки эквивалентности будет незначительным.
Ошибка спектрофотометрического титрования слагается из следующих составляющих:
а) «химических» ошибок, обусловленных неколичественным связыванием титруемого нона в светопоглощающее соединение в конечной точке титрования;
б) погрешностей измерения светопоглощения на используемых оптических приборах;
в) погрешностей измерения объемов титранта.
Спектрофотометрическое титрование можно проводить как с индикатором, так и без него. Если хоть один из компонентов реакции, используемой при титровании, обладает избирательным светопоглощением, то титрование можно проводить без индикатора. В тех случаях, когда все компоненты реакции оказываются индифферентными к светопоглощению на выбранном участке цпекгра, применяют специальные индикаторы—дополнительные реагенты, которые могут сами поглощать свет на выбранном участке спектра или образуют светопоглощающие соединения с одним из компонентов реакции, использующейся при фотометрическом титровании.
7.6.2. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
БЕЗ ИНДИКАТОРА
Когда в точке эквивалентности наступает резкое изменение поглощения света раствором, возможно спектрофотометрическое титрование без индикатора. Если молярные коэффициенты светопоглощения определяемого вещества, реагента и продукта
218
реакции обозначить соответственно через еА, еБ, еАБ, то ход кривой титрования (изменение оптической плотности до и после точки эквивалентности) зависит в общем случае от разницы Де = = еАБ — еБ — еА. При титровании одного компонента чаще всего встречаются случаи, изображенные на рис. 7.29.
а. Определяемое вещество А и конечный продукт АВ не поглощают излучения, а реагент В поглощает его (рис. 7.29, а). Например, при титровании железа (II) перманганатом калия при X = 550 нм. Оптическая плотность раствора начинает увеличиваться после точки эквивалентности в результате поглощения света избытком КМпО4.
б. Конечный продукт АВ поглощает излучение, а вещества А и В не поглощают его. Так, если исследуемый раствор, содержащий малые количества Си2+, титруется стандартным раствором ЭДТА, то при X = 260 нм значительно поглощает излучение только продукт реакции (комплекс Cu-ЭДТА) и получается кривая, изображенная на рис. 7.29, б. Подобная же кривая получается при титровании железа (II) кобальтом (III) при к = 360 нм.
в. Определяемое вещество А поглощает излучение; добавляемый реагент В и конечный продукт АВ не поглощают его (рис. 7.29, в). Например, при титровании бихромат-иона железом (Ц) или мышьяком (III) при X = 350 нм.
г. Определяемое вещество А и реагент В поглощают излучение; продукт реакции АВ его не поглощает. Светопоглощение раствора уменьшается по мере того, как испытуемое вещество А вступает в реакцию и возрастает с увеличением избытка «окрашенного» реагента В (рис. 7.29, г).
Рис. 7.29. Кривые спектрофотометрического (фотометрического) титрования.
219
д. Продукт реакции АВ и реагент В поглощают излучение; определяемое вещество А его не поглощает (рис. 7.29, д). При этом могут наблюдаться два случая:
1) реагент В поглощает излучение в большей степени, чем продукт реакции АВ. В процессе титрования светопоглощение увеличивается до наступления момента эквивалентности, так как накапливается «окрашенный» продукт реакции АВ. После достижения точки эквивалентности светопоглощение вновь возрастает после добавления избытка «окрашенного» реагента В (кривая /);
2) продукт реакции АВ поглощает излучение больше, чем реагент В. Светопоглощение увеличивается по мере накопления в растворе «окрашенного» продукта реакции АВ. После точки эквивалентности излучение поглощает избыток реагента В; поглощение излечения реагентом В меньше, чем продуктом реакции АВ (кривая 2).
е. Определяемое вещество А и продукт реакции АВ поглощают излучение; реагент В его не поглощает (рис. 7.29, е). Здесь также следует резличать два случая:
1) определяемое вещество А сильнее
поглощает излучение, чем продукт реакции АВ. Светопоглощение раствора падает по мере уменьшения концентрации определяемого вещества А и становится постоянным после достижения точки эквивалентности (кривая /);
2) продукт реакции АВ поглощает излучение больше, чем определяемое вещество А. Светопоглощение раствора увеличивается до наступления момента эквивалентности (кривая 2).
ж. Излучение поглощают все три компонента, определяемое вещество А, реагент В и продукт реакции АВ. Следует различать три случая (рис. 7.29, ж):
1) определяемое вещество А и продукт АВ эквивалентно поглощают излучение, поэтому до наступления момента эквивалентности светопоглощение раствора остается постоянным и увеличивается только после добавления избытка реагента В (кривая /);
2) определяемое вещество А сильнее поглощает излучение, чем продукт реакции АВ; светопоглощение раствора уменьшается до момента эквивалентности (кривая 2);
3) продукт реакции АВ сильнее поглощает излучение, чем определяемое вещество А. Светопоглощение раствора увеличивается до точки эквивалентности (кривая 3).
220
Рис. 7.30. Спектры поглощения аква-1 с ЭДТА.
Рис. 7.31. Кривая спектрофотометрического титрования ионов висмута и свинца при их совместном присутствии.
В двух последних случаях, как и в первом, светопоглощение раствора после точки эквивалентности определяется избытком реагента В.
Примерами спектрофотометрического титрования без индикатора могут служить определения: бихромат-ионов железом (II) или мышьяком (III) при X — 350 нм; урана (IV) и железа (II) церием (IV) при X = 360 нм; висмута, цинка( кадмия диэтил-дитиокарбаминатом натрия; магния, меди — турбидиметрически 8-оксихинолином в 50%-ном этиловом спирте; магния, кальция, цинка, кадмия — раствором ЭДТА при X, равной 222 и 228 нм.
Вилхайт и Ундервуд [315] предложили интересную методику спектрофотометрического титрования «бесцветной» смеси Bi3+ и РЬ2+ в ультрафиолетовой области. Известно, что Bi3+ и РЬ2+ и их комплексы с ЭДТА бесцветны. Однако они хорошо поглощают излучение в ультрафиолетовой области (рис. 7.30). Пики кривых поглощения Bi3+ и РЬ2+ могут несколько смещаться в зависимости от присутствия тех или других анионов из-за образования комплексных ионов. Спектры же поглощения Bi3+ и РЬ2+ с ЭДТА при pH = 2 не зависят от наличия посторонних ионов. Максимумы поглощения для РЬ-ЭДТА — при 240 нм, а для Bi-ЭДТА — при 265 нм. Так как Bi3+ дает значительно более устойчивый комплекс с ЭДТА, чем РЬ2+, то можно производить одновременное количественное определение каждого из ионов. Титрование ведут при X = 240 нм, т. е. при Хмак0 для Pb-ЭДТА. При этом в первую очередь Bi3+ реагирует с ЭДТА (рис. 7.31). Оптическая плотность раствора увеличивается медленно, по мере образования комплекса Bi-ЭДТА, так как данный комплекс при этой длине волны поглощает излучение несколько сильнее, чем сам катион Bi3+ (см. рис. 7.30). После того, как практически весь Bi3+ прореагирует, начинает образовываться менее стойкий комплекс РЬ-ЭДТА
221
ц оптическая плотность раствора изменяется более резко, так как Pb-ЭДТА при X = 240 нм поглощает излучение значительно сильнее, чем Bi-ЭДТА. Наконец, за точкой эквивалентности для свинца оптическая плотность не изменяется, так как ЭДТА не поглощает излучения с X = 240 нм.
При выполнении спектрофотометрического титрования важно знать, с какой точностью может быть определена конечная точка титрования и насколько разбавленные растворы (при заданной точности определения) можно титровать.
Рассмотрим простейший случай, когда образуется светопоглощающий комплекс состава 1 : 1 (см. рис. 7.28). Так как реагент R в процессе титрования в комплекс связывается не полностью и некоторая его часть остается в равновесии, то измеряемая в действительности оптическая плотность раствора, обусловливаемая равновесной концентрацией комплекса MR, будет несколько меньше, чем соответствующее ей значение на идеальной кривой титрования. Относительное отклонение оптической плотности раствора от теоретического значения будет увеличиваться по мере приближения к точке эквивалентности. Степень комплексообразования (выход комплекса) после точки эквивалентности в условиях избытка реагента, напротив, увеличивается по мере удаления от нее. Отклонение оптической плотности раствора от теоретического значения на идеальной кривой титрования соответственно уменьшается по мере увеличения отношения CR/CM. Поэтому эквивалентный объем, определяемый экстраполяцией прямых участков титрования (рис. 7.28, кривая 2), будет несколько завышен по сравнению с его точным теоретическим значением (рис. 7.28, кривая J).
К аналогичным выводам приводит рассмотрение и других видов кривых титрования с той лишь разницей, что в одних случаях титровальная доля увеличивается на Ах, а в других, наоборот, уменьшается. Из выражения условной константы устойчивости комплекса при х — 0,5 получим, что [CR ]х=о,5 = [MR ]/([См ] X X Pmr) ~ 1/pMR и Ах = [CR]/CR tv 1/(0,5CmPmr)-
Следовательно, относительная погрешность определения эквивалентного объема, обусловленная неколичественным протеканием реакции комплексообразования, будет тождественна относительному содержанию незакомлексованного реагента вблизи точки эквивалентности
4>kb=V[Cm₽mr(1-4] (7-44)
причем с увеличением значений х, при которых спрямляется зависимость А = f (х), относительная погрешность Ахэкв возрастает.
Чтобы получить кривую титрования с резким изломом, необходимо обеспечить в точке эквивалентности закомплексованность реагента не менее чем на 99%, что, согласно уравнению (7.44), 222
достигается при Cm₽'mr 1-Ю4 или при См = 1.10-4 моль/л, 0mr Ь Ю8. Однако при титровании часто используют комплексные соединения и с меньшими значениями Pmr- Если йрямую начальной ветви кривой титрования провести по точкам от х =* — О до х = 0,5 (что чаще всего осуществляется на практике), то для обеспечения того же значения Ахэкв — не более чем ±1% от хэкв = 1 — необходимо, чтобы при х = 0,5 Cm₽mr jgs 2-102. Отсюда минимальное значение Pmr при См = 1- 10-5 моль/л составит 2.107, при См = 1- Ю-4 моль/л — 2- 10е и т. д.
Таким образом, даже если прямую начальной ветви кривой титрования проводить по начальным точкам от х = 0 до х = = 0,1 при См ~ 1-10-4 моль/л, значение PmR л: 1. 10е следует рассматривать как минимальное для обеспечения «химической» ошибки титрования не более 1%.
Для определения погрешности измерения светопоглощения рассмотрим уравнение (2.3).
Если оптическую плотность выразить через светопропуска-ние, то А = —IgT.
Для измерения угла между двумя пересекающимися линиями в точке эквивалентности (рис. 7.29, а—е) можно принять, что;
ЛА/ДС = tel (7.45)
При рассмотрении вопросов, касающихся точности определений, часто бывает целесообразно пользоваться значениями АТ вместо АЛ, так как, во-первых, шкала пропускания равномерная и, во-вторых, чувствительность спектрофотометра по шкале пропускания не зависит от Т. Соотношения между этими двумя величинами равны
ДЛ = ДТ/(2,ЗГ) (7.46)
(при условии, что количественные изменения очень малы). Из уравнений (7.45) и (7.46) следует, что
ЛС/ЛТ = 1/(2,ЗТ tel) (7.47)
Этим уравнением можно пользоваться для вычисления ошибки титрования и для определения допустимой степени разбавления раствора. Предположим, что светопропускание измеряется с погрешностью ±0,2%, т. е. что АТ = 0,002, и в точке эквивалентности оно равно приблизительно 0,85. Уравнение (7.47) дает соответственно:
ДС « 10'3/(Де0
Здесь ДС — концентрация вещества, которая соответствует наименьшему измеряемому значению пропускания.
Если, например, Ае = 103 и I — 1 см, то С — 10”®. При титровании раствора с концентрацией С = 10"2 М погрешность составляет;
ДС-100/С= 10“в-100/10-2 = 0,01% (отн.)
£23
Аналогичным путем можно рассчитать допустимую степень разбавления. Если, например, требуется погрешность определения <р = ДС/С = 0,1%, это означает, что АС не должно превышать:
АС = фС/100 = 10~»С
Следовательно, для приведенного выше примера:
ДС = 10-зС = 10-з/(Ле/) и С» 1 (Ле/)
Расчет ошибки титрования для случая, когда излучение поглощает только одно из веществ — А, В или АВ, — может быть выполнен также по уравнению
ср = ДЛ (еС/) (7.48)*
предложенному К. Б. Яцимирским и И. И. Алексеевой [297]. Из уравнения (7.48) видно, что ошибка титрования уменьшается с уменьшением погрешности в измерении оптической плотности, с увеличением концентрации поглощающего излучение раствора С, толидины поглощающего слоя / и значения молярного коэффициента светопоглощения е. Уравнение (7.48) может быть использовано также для расчета минимальной допустимой концентрации С при заданной ошибке титрования:
С» Л л р/ф)
7.6.3. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
С ИНДИКАТОРОМ
Если все компоненты реакции (исходные компоненты и образующийся комплекс), используемой в фотометрическом титровании, на выбранном участке спектра света не поглощают, то титрование проводят в присутствии индикатора, который образует светопоглощающий комплекс с одним из компонентов (чаще с катионом металла) основной реакции. Сам индикатор при этом либо не поглощает света при выбранной длине волны, либо поглощает его незначительно**. Изменение концентрации «свободного» и «связанного» индикатора в процессе титрования является основной теоретической предпосылкой определения точки эквивалентности. Фотометрическое титрование с индикатором имеет много общего с визуальным титрованием В ряде случаев даже кривые фотометрического титрования совершенно аналогичны кривым визуального титрования (см. рис. 7.29,з, и).
При визуальном комплексометрическом титровании с индикатором условия титрования и достигаемую воспроизводимость, как известно [42, 47, 290], можно прогнозировать с помощью номограммы Рингбома (рис. 7.32). Для теоретической оценки возможностей комплексометрического титрования номограмма
* В общем случае в уравнение (7.48) следует подставлять значение Де = = еав - еа ~ ев вмест0 Е-
** Иногда титрование проводят при такой длине волны, где индикатор обладает большим светопоглощением, а комплекс поглощает незначительно.
224
Рис. 7.32. Номограмма Рингбома:
А — минимальная теоретическая ошибка титрования, %: С^\ и ft^R ориентировочная концентрация и условная константа устойчивости комплекса определяемого металла- у — коэффициент разбавления раствора при титровании.
Рипгболга очень удобна, позволяет быстро оценивать достигаемую теоретическую воспроизводимость титрования, выбрать нужные условия (pH раствора, состав буферного раствора, концентрацию дополнительного комплек-санта и т. п.), при которых условная константа устойчивости комплекса (0mr) обеспечивает титрование с заданной воспроизводимостью.
Теоретическая ошибка титрования (А, %) определяется константой Pmr. началь
ной концентрацией титруе- ^(Смр'мн/р
мого иона См и разностью
значений рМ* в конечной точке титрования (рМпег) и в точке эквивалентности (рААэ):
дРм = рмпср - Рмэ = ig рМ11Лп - Рмэ
Здесь р М1и — концентрационная константа устойчивости металлокомплекса с индикатором; а1п — доля стехиометрической формы индикатора от его общей равновесной концентрации, не связанной с титруемым катионом.
При фотометрическом титровании эти факторы также имеют очень важное значение, но для обеспечения той же воспроизводимости условия могут быть менее строгими, чем при визуальном титровании, поскольку интервал значений рМ для определения конечной точки титрования значительно шире. Если для визуального титрования можно использовать только такие индикаторы, которые обеспечивают изменение окраски раствора в области скачка на кривой титрования, то в фотометрическом титровании применяют разнообразные индикаторы, в том числе такие, переход окраски которых выходит за пределы скачка на кривой титрования и наблюдается или раньше (рис. 7.33, а) или позже
* рЧ=—lg [MJ, где [М] — концентрация катиона, определяемого комплексометрически
8 М И Булатов и др.
225
а Рис. 7 33. Кривые спектрофотометрического титрования:
—।---------------- а и е — асимметричные кривые с резким изломом; б — сим*
1 метрично закругленная кривая.
|Макс у а макс
’мт Л т
________-------- (рис. 7.33, с) точки эквивалентности. Ширина макс V интервала значений рМ для определения ко-Т'мш | С нечной точки титрования зависит от чувстви-__________________________________ тельности используемого оптического прибора. -----------------6 Рассмотрим случай изменения окраски при I s' образовании простейшего комплекса состава ХСакС 1’ (рис. 7.33). Фотометрическое определение п /\Л точки эквивалентности возможно во всех трех — —случаях. В случае на рис. 7.33, б имеем сим-V~ метрично закругленную кривую титрования, в случаях на рис. 7.33, а, б, наоборот, асимметричные кривые с резким изломом, которые четко могут воспроизводиться современными фотометрическими приборами.
Точка эквивалентности при титровании находится в пределах перехода окраски индикатора. Допустим, что при выбранной длине волны поглощает свет как комплекс металла с индикатором (Mln), так и реакционная форма самого индикатора (In). Тогда, согласно основному закону светопоглощения, можно записать, что
Ап = Егп [In] I; АГС = Eincinz <7-49)
^мш = [Mln] /; = Win' <7'5°)
Здесь С1п — общая концентрация индикатора в растворе; ein и еМш—молярные коэффициенты светопоглощения индикатора и его комплекса с катионом М; ХГпКС — наибольшее значение оптической плотности раствора индикатора в условиях избытка реагента-титранта; Дм!™ — наибольшее значение оптической плотности раствора металлокомплекса с индикатором в условиях избытка титруемого катиона металла.
Комбинируя уравнения (7.49) и (7.50), получим:
^м,п^ГпК7(^т^ма1пС) = [М] ₽М1п (7.51)
Оптическая плотность в процессе титрования будет слагаться из двух составляющих:
л = Ап+^мш <7-52)
Из уравнений (7.51) и (7.52) найдем, что в точке эквивалентности оптическая плотность раствора будет равна:
А = (Ап/<кс) (АТС + Аппс [М]э PMItl) (7.53)
226
Но учитывая, что
л"пК7л1п = + [In])/[In] = 1 + [М] ₽м1п (7.54)
получим окончательное уравнение для вычисления оптической плотности раствора в точке эквивалентности:
= (ЛГпакс + А^ [М]э ₽м111)/(1 + [М]э ₽М1п) (7.55)
Значения А”пКС и Дм1пСв уравнении (7.55) легко измеряются экспериментально, а [М]э нетрудно вычислить из выражения условной константы устойчивости комплекса MR:
[м1е = «м <7-56)
Здесь Cr — исходная концентрация реагента; ам — мольная доля незакомплексованного посторонними комплексантами титруемого катиона в условиях титрования.
С помощью уравнения (7.55) можно найти эквивалентный объем титранта с достаточно высокой степенью точности, правда, это оказывается возможным лишь при очень точном измерении объемов титранта в процессе титрования.
При титровании очень разбавленных растворов необходимо вычислить концентрацию комплекса Mln в точке эквивалентности
[М1п]8 = С1п (Д# - А Гпакс)/(ЛХС - <КС) (7-57)
и найти поправку к полученному эквивалентному объему титранта
ДЕэ=[М1п]эК/Ск (7.58)
Здесь V — общий объем раствора в точке эквивалентности; CR — молярная концентрация раствора титранта.
Если при титровании используют очень низкие концентрации индикатора (CIn < 1- 10-Б моль/л), то поправка ДЕЭ часто оказывается пренебрежимо малой.
Чувствительность измерения светопоглощения прибором обычно характеризуют величиной отношения ДрМ/ДТ. Согласно Рингбому [42]
ДрМ/ДГ = 0,188 (14- [М] РМ1П)/[(Л - А “акс) Т [М] ₽М1п] (7.59)
Вычислив по уравнению (7.59) значение ДрМ при надежно фиксиру мой прибором разнице пропускания ДТ и зная См и ₽mr, по номограмме Рингбома (рис. 7.32) находят ошибку титрования (Д, %).
Для достаточно прочных комплексов вычисление эквивалентного объема титранта с помощью уравнения (7.55) и графической экстраполяции показывает, как правило, хорошую сходимость результатов. Если эквивалентный объем титранта определяется графической экстраполяцией линейных участков кривой титрования, то концентрацию комплекса Mln в точке пересечения прямых (Дизл) удобнее вычислять по уравнению (7.60), аналогичному
8* 227
уравнению (7.57), но не содержащему неизвестных констант устойчивости:
[М1п]изл = С1п (Л„зл - <“)/№? - <кс) (7.60)
Точка эквивалентности при титровании находится вне видимого интервала перехода индикатора. Этот случай часто наблюдается при титровании очень разбавленных растворов. Конечную точку титрования определяют графической экстраполяцией, и абсолютная величина оптической плотности раствора при этом не имеет
существенного значения.
Если относительная устойчивость металлокомплекса титруе
мого катиона сравнительно невысокая, индикатор характеризуется ранней чувствительностью и изменение окраски раствора наблюдается до точки излома на кривой титрования (см. рис. 7.33, а). Поправка на индикатор в этом случае вообще не требуется, так как в точке излома практически ве ь индикатор находится в незакомплексованном состоянии.
Фортуин и др. [285] показали, что при раннем переходе индикатора на кривой титрования наблюдается точка излома, если ₽ми/рм1п 3s 1- W4. Если точка излома наблюдается нечетко, то эквивалентный объем титранта находят экстраполяцией, как и при средней чувствительности индикатора (см. рис. 7.33, б), учитывая при этом индикаторную поправку по уравнению (7.60). При позднем переходе индикатора, вследствие высокой относительной устойчивости комплекса Mln, изменение окраски раствора происхо
дит после точки эквивалентности, поэтому к эквивалентному объему, найденному с помощью уравнения (7.55) или графической экстраполяцией, всегда необходимо вносить индикаторною поправку по уравнениям (7.57), (7 58) или (7.60).
Хигуши, Рем и Бернштейн [303] предложили оригин. льный способ определения конечной точки титрования, когда светопо-глощением обладает применяемый индикатор. Если кривую титрования представить как функцию [HIn]/[In] = / (VR) или, что то же самое, (Л“пКС — А)/А = / (VR), то вблизи точки экви-
валентности эта функция становится линейной и экстраполяция прямой линии до пересечения с осью объемов позволяет найти эквивалентный объем титранта (рис. 7.34). Если при этом окажется, что Рм1п/₽мк 0,05, то по оси абсцисс вместо объемов |/R целесообразнее откладывать обратные им величины (1/VR). Однако
Рис. 7.34. Кривая спектрофотометрического титрования с линейным участком.
228
этот метод не предназначен для титрований очень разбавленных растворов, когда концентрация индикатора такого же порядка, что и концентрация титруемого иона.
При титровании разбавленных растворов для оценки чувствительности можно использовать уравнение (7.47), применяемое при фотометрическом титровании без индикатора. Значение рЛ4 в конечной точке титрования можно найти по уравнению [42]:
Рмизл = 'g ₽Min ± lg [2.37 - АГпакс)/Д7] (7.61)
Уравнение (7.61) справедливо только в том случае, если конечная точка титрования находится на закругленной части кривой титрования, когда рА1ичл не слишком отличается от рМэ.
При раннем переходе индикатора второе слагаемое в уравнении (7.61) прибавляют, а при позднем переходе — вычитают.
Если индикатор образует с титруемым катионом комплекс состава Mln,, то значения рМизл можно найти [42] по уравнениям (7.62) при раннем переходе индикатора и (7.63) при позднем его переходе:
рМИзл = ’g + lg 2CIn ± lg [2,37 - Л"пакс)/Д7] (7.62)
Рмизл = 'g ₽Min «In + lg 2CIn - lg [2,37 (A-- - А“акс)/ Д7] (7.63)
Определив эквивалентный объем по излому кривой титрования (включая и метод экстраполяции) и вычислив по уравнениям (7.61)—(7.63) рМизл, с помощью номограммы Рингбома можно оценить достигаемую точность титрования. При этом следует иметь в виду, что изменение светопоглощения АТ обеспечивает надежное фиксирование конца титрования в интервале ДрМ = ±0,3 независимо от чувствительности прибора и индикатора. Следовательно, если pMj — рМига > 0,3, то к найденному объему титранта в конце титрования необходимо внести индикаторную поправку с помощью уравнения (7.60).
Если индикатор, применяемый при титровании, сам не обладает светопоглощением, то в таких случаях кривые титрования аналогичны рис. 7.29, а—в. Например, для титрования малых количеств Th4+ раствором ЭДТА в качестве индикатора используют соли меди (II). Комплекс Си2+ с ЭДТА сильно поглощает излучение при X = 260 нм, в то время как остальные компоненты незначительно поглощают излучение этой длины волны. Устойчивость же соединения Th4+ с ЭДТА значительно больше устойчивости комплекса Сп2+ с ЭДТА. Поэтому светопоглощение остается постоянным до тех пор, пока не будет оттитрован весь Th4+, и только после этого титрант дает соединение с медью и оптическая плотность раствора начинает расти по мере добавления избытка ЭДТА (рис. 7.29, а).
Кривые, изображенные на рис. 7.29, б, в, могут получаться если излучение будут поглощать только соединения АВ In или Ain. Например, при титриметрическом определении содержания
229
Fe3+ в исследуемом растворе обычно его предварительно восстанавливают до Fe2+, а затем титруюг перманганатом калия. Методом спектрофотометрического титрования это определение можно провести значительно быстрее без предварительного восстановления железа (III). Салициловая кислота и Fe3+ образуют интенсивно окрашенный комплекс, максимум поглощения которого находится примерно при X = 525 нм. Этот комплекс при pH ж 2,4 значительно менее устойчив, чем комплекс Fe3+ с ЭДТА. Поэтому по мере титрования салицилата железа раствором ЭДТА наблюдается постепенное обесцвечивание раствора, которое в точке эквивалентности полностью заканчивается. При визуальном титровании это изменение наблюдается недостаточно ясно, зато при спектофото-метрическом титровании конечная точка определяется совершенно четко при X = 525 нм (рис. 7.29, в).
Аналогичным образом можно провести титрование Bi3+ раствором ЭДТА в присутствии тиомочевины как индикатора при & = 400 нм; титрование меди (II) раствором ЭДТА, используя в качестве индикатора NH4OH, проводят при X = 630 нм. Комплекс [Cu (NH3)4]2+, сильно поглощающий свет при X = 630 нм, по мере титрования разрушается с образованием более устойчивого комплекса меди с ЭДТА и др.
Во всех перечисленных случаях точка эквивалентности соответствует точке пересечения двух прямых линий, аналогичных кривым амперометрического или кондуктометрического титрования.
В противоположность этому кривые, показанные на рис. 7.29, з, и, аналогичны кривым потенциометрического титрования и являются типичными случаями, когда в точке эквивалентности небольшой избыток рабочего раствора вызывает резкое изменение pH, рМ или окислительно-восстановительного потенциала системы. Индикатор при этом меняет свою структуру, что сопровождается резким изменением окраски раствора, а следовательно, и светопоглощения раствора. Так, для кривой рис. 7.29, з содержание определенной формы индикатора с максимальным поглощением при выбранной длине волны резко возрастает в точке эквивалентности, давая резкое увеличение поглощения, а для кривой на рис. 7.29, и — наоборот. Эти резкие переходы окраски можно хорошо заметить и простым глазом, если концентрация определяемого вещества достаточно велика и полосы поглощения находятся в видимой области спектра, как в случае классических визуальных индикаторов. Однако при титровании микрограммовых содержаний веществ часто не происходит резкого изменения окраски в точке эквивалентности (индикатор постепенно изменяет свою окраску в ходе титрования, особенно при комплехсонометрическом титровании). В этих случаях спектрофотометрическое титрование позволяет получать более четкие кривые титрования и достаточно высокую воспроизводимость.
В ряде случаев при титровании с индикаторами, когда известны состав и константы устойчивости комплексов титруемого иона 230
как с реагентом-титрантом, так и с индикатором, значение оптической плотности раствора в точке эквивалентности можно теоретически рассчитать по уравнению (7.55) и по экспериментальной кривой титрования найти эквивалентный объем.
Вопросы теории и практики фотометрического мпкротитрования с применением комплексонов освещены в ряде работ [42, 282—289, 292, 295, 313, 314]; Cd, Zn — [286, 292]; Ni — [288]; Ca, Mg — [292, 302[.
При титровании разбавленных слабых кислот (оснований) в водных и неводных средах не удается четко определить точку эквивалентности. Для повышения воспроизводимости фотометрического титрования таких систем предложены специальные графические методы [303—308 [. Относительные ошибки титрования в случае применения окрашенных нндькаторов вычисляются значительно сложнее.
7.6.4. ФАКТОРЫ, ВЛИЯЮЩИЕ НА ВОСПРОИЗВОДИМОСТЬ
И ПРАВИЛЬНОСТЬ СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Погрешность разбавления. Если к титруемому раствору добавлен относительно большой объем рабочего раствора, то необходимо внести поправку на разбавление. Для этого умножают найденные значения оптической плотности раствора на фактор:
(И + v)!V
Здесь V — объем в начале титрования; v — объем добавленного рабочего раствора.
Если не внести эту поправку, то могут возникнуть «неожиданные» погрешности особенно для кривых, аналогичных рис. 7.29, б. Без поправки на объем получается пунктирная кривая и точка эквивалентности может быть определена неверно. Поправки на объем важны также для кривых рис. 7.29, г, д, и, несколько менее для кривой рис. 7.29, в. Для кривых титрования, подобных рис. 7.29, а, поправки важны только после точки эквивалентности, так как вплоть до нее титруемый раствор остается бесцветным.
Чтобы свести погрешность разбавления к минимуму, рекомендуется использовать по возможности концентрированный рабочий раствор (титрант), объем которого обычно измеряют полумикробюреткой (v = 5ч-10 мл). Когда разбавление не превышает нескольких процентов, прямолинейность обеих кривых титрования сохраняется и погрешностью разбавления можно пренебречь.
Характеристики рабочих ячеек. Для титрования в области длин волн более 350 нм годятся обычные стаканы из пирексового стекла; никаких особых оптических поверхностей высокого качества не требуется. Однако очень важно, чтобы стакан был защищен от рассеянного света и надежно зафиксирован в течение всего времени титрования, так как даже незначительный поворот или боковое освещение может заметно изменить оптические характеристики раствора. Объем анализируемого раствора составляет обычно 100—200 мл.
Для титрования в ультрафиолетовой области спектра применяют кварцевые кюветы или кюветы из боросиликатного стекла
231
[для ближней ультрафиолетовой области). В литературе [302— 308] описаны различные конструкции кювет, предназначенных Для определения как миллиграммовых, так и субмикрограммо-Ьых содержаний элементов.
Рабочий раствор подают в кювету из полумикробюретки по 0,3—0,5 мл в процессе титрования и по 0,1 мл (или по каплям) вблизи точки эквивалентности. Кончик бюретки, помещенный в стакан вблизи поверхности исследуемого раствора, вместе с последним защищен от рассеянного света и от глаза экспериментатора, поэтому не может быть гарантии, что последняя капля каждой порции рабочего раствора не остается на конце бюретки (некоторые исследователи снабжают кончик бюретки капилляром, который погружают в исследуемый раствор. Перемешивание раствора в ходе титрования может осуществляться током газообразного СО2, N2, механической или магнитной мешалками. Если перемешивание производится механической мешалкой, то необходимо, чтобы она не препятствовала прохождению света через раствор.
Работая с чувствительными к температурам системами, следует помнить, что механическая и магнитная мешалки могут дать значительное повышение температуры, которое скажется на измерении оптической плотности раствора.
Выбор рабочей длины волны. Рабочую длину волны выбирают с учетом двух обстоятельств:
1. Необходимо, чтобы по мере возможности были исключены помехи со стороны других окрашенных веществ.
2. Из уравнений (7.47) и (7.48) видно, что ошибка титрования <р зависит как от С, так и от AT1, Т и АЛ. Она будет тем меньше, чем больше значение молярного коэффициента светопоглощения е. Но растворы, титруемые при Хмакс, даже при небольших концентрациях, но значительных толщинах кювет будут сильно поглощать свет, что может привести к серьезным фотометрическим погрешностям при измерении Т и А как за счет того, что при высоких А (низких Т) воспроизводимость их определения уменьшается, так и за счет отклонений от уравнения (2.3). Поэтому желательно экспериментально выбрать такую длину волны, при которой е был бы достаточно велик, но изменение поглощения (пропускания) во время титрования происходило бы в удобных для измерения пределах.
При работе на фотоэлектрических титрометрах уменьшения значений оптической плотности сильноокрашенных веществ добиваются подбором подходящего светофильтра, уменьшением диаметра светового пучка, проходящего через исследуемый раствор (диафрагмированием светового пучка). Иногда уменьшают толщину I фотометрируемого слоя.
Аппаратура. Для спектрофотометрического (фотометрического) титрования могут быть использованы как специальные титраторы, выпускаемые промышленностью, так и обычные спектрофотоме
232
тры и фотоколориметры [298, 309, 310, 318—322]. Для автоматического титрования применяют различные модели зарубежных спектрофотометров: Beckman Model В, Сагу-14, Spectromom 360, Beckman Model DU, Unicam SP-600.
7.6. Б. РАСЧЕТ КОЛИЧЕСТВА ВЕЩЕСТВА
ПО ДАННЫМ СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Количество вещества, определяемое спектрофотометрическим титрованием, равно количеству реагента, затраченному на титрование до точки эквивалентности. Тогда масса (мг) вещества
9 = УЭМВ
Здесь М — молярность раствора реагента; В — атомная масса определяемого элемента.
Расчет эквивалентного объема (Va, мл) рабочего раствора реагента (титранта) зависит от формы кривой спектрофотометрического титрования А = f (V). При соблюдении основного закона светопоглощения кривая титрования выражается двумя прямыми, точка пересечения которых соответствует точке эквивалентности. Абсцисса этой точки равна vs. Значение vs рассчитывают, решая систему уравнений двух прямых относительно общей точки пересечения:
А — brv
А = а2 + b2v
Здесь щ, й2. ^1. Ь2 — параметры jравнений прямых, рассчитанные по данным титрования методом наименьших квадратов; v — объем добавленного рабочего раствора реагента, мл.
Все значения оптических плотностей, используемые в расчетах, должны содержать поправку* на разбавление раствора при титровании, т. е.
А = Лизм(к + и)'к
Здесь V — первоначальный объем титруемого раствора, мл.
Во многих случаях, хотя и с меньшей точностью, можно использовать расчетные формулы, полученные из геометрического подобия составных частей кривых титрования.
Титрование одного компонента без индикатора. В практике спектрофотометрического титрования одного компонента без индикатора используют две основные реакции:
А + В «=> АВ; А + В « > А' + В'
Здесь А — определяемый ион; В — реагент; АВ, В' и А' — продукты реакции.
* Если обусловленная разбавлением раствора при титровании поправка в определении количества (концентрации) вещества несоизмеримо мала по сравнению с погрешностью определения, то разбавлением раствора в расчетах можно пренебречь.
233
При титровании одного компонента в соответствии с основными формами кривых титрования можно выделить следующие варианты:
а. Излучение поглощают (рис. 7.35, а): 1) В; 2) А, В и АВ, но еА = еАВ; 3) А, В и В', но еА = ев.; 4) А, А' и В, но еА = = еА'; 5) А, В, А', В', но еА = еА' + еВ'-
Уэ = V' (7.64)
Здесь и' и у" — последовательно добавленные объемы рабочего раствора после точки эквивалентности v">v' > Vo); А' и А" — оптические плотности после точки эквивалентности, соответствующие V' и V" (А" > А' > Ло); Ао — постоянное значение оптической плотности раствора, измеряемой до точки эквивалентности.
б. Излучение поглощают (рис. 7.35, б): 1) АВ; 2) А и АВ, но еАВ > еА; 3) А' или В'; 4) А и А', но еА- > еА; 5) А и В', но ев, > > »А-
уэ = (7 65)
Л2 — Al
Здесь щ и и2 — последовательно добавленные объемы реагента до точки эквивалентности; А± и Л2— оптические плогносги до точки эквивалентности, соответствующие щ и у2; Ав — постоянное значение оптической плотности раствора, измеряемой после точки эквивалентности.
в. Излучение поглощают (рис. 7.35, в): 1) А; 2) А и АВ, но еа > еАв! 3) А и А', но еА > еА,; 4) А и В', но ев> ев,; 5) А, А' и В', но еА > еА- -[- еВ'.
v2Al — vlA2 — Ab(v2 — vl) „
г) Излучение поглощают (рис. 7.35, г): 1) В и АВ, причем ев ¥= еАВ; 2) В и АВ, но еАВ > еА и ев ф еАВ — еА; 3) А'
и В; 4) В и В'; 5) А, А' и В, но еА < еА- и ев еА- — еА; 6) А,
В и В', но еА < ев. и ев Ф ев, — еА; 7) А, А', В и В', но еА <
< еА- + ев< и ев еА' + еВ' — еА.
У = (оа — V1) (A'tf — А"у') + (у" — у') (У1А2 — v2AL) _
3 (v"-v’){A2-Al)-(y2-vl)(A''-A')
д) Излучение поглощают (рис. 7.35, д): 1) А и В; 2) А, В и АВ, но > еАВ; 3) А, А' и В, но еА < еА-; 4) А, В и Е', но еА > ев< 5) А, А', В и В', но еА > еА- + ев,.
v _ (у" — И (Mt — щД2) + (v2 — nJ (у'А" — и"Л')
8 (А''-А')(у2-у1)+(А1-А2)(у"-у1)
Одновременное титрование двух компонентов* при их совместном присутствии. При титровании по реакции
+ А2 В < > АгВ А2В практическое значение имеют лишь два основных случая.
* Определение возможно только в том случае, когда компоненты Ах и А2 или AiB и А2В резко различаются по своим свойствам (константы устойчивости, окислительный потенциал и т. п.).
234
A
Рис. 7.35. Кривые титрования одного компонента: а—д — варианты.
а. Излучение поглощают компоненты Ах и А2 (рис. 7.36, а), причем еА1 =£= еАя.
еА, <еА2; еА,>еАа
V — ~ М ~~ ~~ V'^ ~ П АЩ
зА*~ (^_иЭ(Л2_Л1)_(^__Ц1)(Л''_Л') <• '
_ иМ'-г'Л"-Л(1(^-Н ,770)
ПэАЩ-А,- Д' —Л" (/- ’
б. Излучение поглощают продукты реакции А2В и А2В (рис. 7.36, б), причем eAlB Ф еАгВ.
® А,В > fA2B‘, еА,В<еА2В
v fa — t>i) (А'у" — AV} 4- (и" — у') (У1А2 — и2Л1)
эА*в (г,»_ц')(л2_л1)_(Ц2_О1)(л''_л-) < >
v'A"-v"A- +Ав(у"-у') vsA,B-J-A2B--------Д'
Титрование одного компонента в присутствии индикатора. При титровании по реакции А + В АВ в присутствии индикатора In форма кривых титрования различается в зависимости от того, с каким продуктом — А, В или АВ — индикатор образует соединение, поглощающее свет при выбранной длине волны (компоненты реакции А, В и АВ обычно света не поглощают при выбранной длине волны)-
1. Если индикатор образует окрашенное соединение с определяемым компонентом А, то при прямом титровании форма кривой
235
Рис. 7.36. Кривые титрования двух компонентов: а и б — варианты.
и расчет эквивалентного объема рабочего раствора такие же, как и в варианте в (см. рис. 7.35).
2. Когда индикатор образует окрашенное соединение с реагентом В, форма кривой титрования и рачет эквивалентного объема раствора реагента аналогичны варианту а (рис. 7.35).
3. Если индикатор образует окрашенное соединение с продуктом реакции АВ, то расчет эквивалентного объема рабочего раствора производят по формуле (7.65) так же, как и в варианте б (рис. 7.35).
Учитывая, однако, что концентрация индикатора в большинстве случаев бывает несоизмеримо мала по сравнению с общей концентрацией определяемого компонента, плавного изменения оптической плотности раствора в процессе титрования не наблюдается, а в области точки эквивалентности происходит резкий скачок величины А, как это показано на кривых з и и (см. рис. 7.29). Поэтому эквивалентный объем рабочего раствора следует находить по дифференциальной кривой АЛ/А У = f (v) аналогично потенциометрическому титрованию. Абсцисса максимума или минимума дифференциальной кривой АЛ/А У = / (у) соответствует эквивалентному объему рабочего раствора реагента (здесь А У — разность между последовательными порциями добавленного объема рабочего раствора; АЛ — соответствующая им разность оптических плотностей).
7.6.6. ПРИМЕРЫ И ЗАДАЧИ
Пример 1. Неизвестное количество Fe (III) в присутствии некоторого избытка салициловой кислоты тируют при К = 525 нм 0,01 М раствором ЭДТА (Y) при толщине слоя / = 3 см до постоянного значения оптической плотности До = 0,25; значения оптических плотностей раствора до точки эквивалентности соответствующие добавленным объемам рабочего раствора Oj = 2,5 см3 и v2 = = 3,5 см3, равны соответственно 0,75 и 0,50. Первоначальный объем V титруемого раствора составляет 15 см3. Максимум светопоглощения салицилатом 236
железа наблюдается при 525 нм; светопоглощения ЭДТА и образующимся комплексонатом железа (III) при этой длине волны не наблюдается, Определить содержание железа (III) в растворе.
Решение. qFe = /ИуУ8уВре. Значение Ув рассчитывают по уравнению (7 66):
а) без учета разбавления раствора
Уа= [3,5-0,75 —2,5-0,5 — 0,25 (3,5 — 2,5)1/(0,75 — 0,50) = 4,5 см3 qFe= 0,01 -4,5-55,85 = 2,52 мг
б) с учетом разбавления раствора
А/ = АиЗМ (У + vt)'V; Aj = 0,75-17,5/15 = 0,875
А2 = 0,5-18,5/15 = 0,617; Ао = 0,25-19,5/15 = 0,325
Уа = [3,5-0.875 — 2,5-0,617 — 0,325 (3,5 — 2,5)1/(0,875 — 0,617) = 4,6 см3 qFe = 0,01 -4,6 -55,85 = 2,56 мг
Пример 2. Сульфат Fe(II) в объеме 25 см3 титруют 0,01 М раствором сульфата церия(1У) при Z = 310 нм и I = 3 см. Значения оптических плотностей раствора до точки эквивалентности, соответствующие объемам добавленного рабочего раствора v± — 2,5 см3 и v2 = 4,0 см3, равны соответственно 0,25 и 0,45. Оптические плотности раствора после точки эквивалентности, соответствующие добавленным объемам рабочего раствора v' = 4,5 см3 и v" — 4,6 см3, равны соответственно А' = 0,75 и А" ~ 1,55. При Л= 310 нм излучение поглощают Fe2(SO4)3 (е = 2,2-103) и Ce(SO4)2 (е = 7,9-10э). Рассчитать содержание Fe в растворе.
Решение. <?Fe = ^ce^sCe^Fe’ Значение Va рассчитывают по уравнению (7.67) с учетом разбавления:
Лг = Лизм(У + ^)/У; Ах = 0,25-27,5/25 = 0,275; /2 = 0,45-29,25 = 0,522
А' = 0,75-29,5/25 = 0,885; А" = 1,55-29,6/25 = 1,835
(4 — 2,5) (0,885-4,6 — 1,835-4,5) + (4,6 — 4,5) (2,5-0,522 — 4-0,275) (4,6 —4,5) (0,522 —0,275) —(4 —2,5) (1,835 —0,885)
= 4,48 см3
<?Fe = 0,01 -4,48-55,85 = 2,50 мг
Пример 3. Раствор объемом 25 см3 (о), содержащий смесь солей РЬ(П) и Bi(III), титруют при Л = 240 нм 0,01 М раствором ЭДТА (У). Комплексонат Bi(III) более прочен, чем Pb(II), поэтому на кривой титрования получаются две точки излома, отвечающие точкам эквивалентности соответственно для Bi и РЬ. Из всех компонентов излучение при Л «= 240 нм поглощают только комплексона™ Bi н РЬ; еРЬ-эдтА >еBi-эдтА-
Рассчитать содержания Bi и РЬ в растворе если известно, что значения оптических плотностей Af и А2 до первой точки эквивалентности, соответствующие добавленным объемам рабочего раствора = 1 см3 н и? = 1,8 см3, равны соответственно 0,15 н 0,25. Значения А' и А" после первой точки эквивалентности, соответствующие объемам рабочего раствора v' — 3,0 н и" = 3,5 см3, равны соответственно 0,75 и 1,00. Постоянное значение оптической плотности Ао, достигаемое в конце титрования, равно 1,20.
237
Решение. <7BI = AfyVe BlfiBP <7Pb — Л1у (Уэ Bi+Pb — Ve Bi) BPb-Значения Ve В1 и Vg Bi+Pb рассчитывают по формулам (7.71) и (7.72) с учетом разбавления:
Лг = Аизм(Р + пг)/У
У a BI =
(1,8-1,0) (о,75-^р-------!^^)+(3,5-3) (ПО,25-26,8/25-!,8-0,15-26/25)
(3,5 — 3) (0,25-26; 8 25 — 0,15-26 25) — (1,8 — 1) (1 -28,5/25 — 0,75-28/25) .
= 2,11 см3
3-1-28,5/25 —3,5-0,75-28/25+ 1,2-28,9(3,5 —3)/25 _
Bi+Pb — 1-28,5/25 — 0,75-28,25 - З.У2 см
qbi = 0,01-2,11-208,98 = 4,41 мг; ?pfj = 0,01 (3,92 — 2,11) 207,2 = 3,76 мг
Задача 1. Неизвестное количество Pb (II) в объеме 15 см3 в присутствии некоторого избытка 4-(2-пиридилазо)-резорцинола (ПАР) титруют 0,001 М раствором Na-ЭДТА при pH = 9,5 и /. = 516 нм до постоянного значения оптической плотности До = 0,15. Значения оптических плотностей, соответствующие добавленным объемам рабочего раствора = 2,5 см3 и и2 = 3,5 см8, равны соответственно Ах = 0,65 и А2 = 0,25.
Рассчитать содержание свинца в растворе: а) без учета разбавления; б) с учетом разбавления раствора при титровании. При 516 нм излучение поглощает только комплекс свинца с ПАР.
Ответ: дрь — 0,774 мг; б) дрь = 0,781 мг.
Задача 2. Неизвестное количество оксалата натрия в объеме 20 см3 титруют 0,01 М раствором Ce(SO4)2 при Z = 365 нм. Значения оптических плотностей при добавлении рабочего раствора объемами = 2,0 см3 и v2 = 3,5 см3 равны соответственно Аг = 0,09 и А2 = 0,07, после точки эквивалентности при добавлении объемов v' = 4,8 см3 и v" = 5,0 см3 — соответственно А' = 0,35 и А" — = 0,75.
Рассчитать содержание оксалат-иона в растворе, учитывая разбавление раствора при титровании. При 1 = 365 нм излучение поглощает только Ce(SO4)2.
Ответ: о„ _.2_ = 2,05 мг.
Задача 3. Неизвестное количество SnCl2 в объеме 18 см3 титруют 0,01 /И раствором Ce(SO4)2 при 1 = 320 нм. Значения оптических плотностей раствора до точки эквивалентности, соответствующие добавленным объемам рабочего раствора vL = 1,25 см3 и v2 — 2,50 см3, равны соответственно Ах = 0,35 и А2 = = 0,20, после точки эквивалентности при добавлении объемов о' = 3,50 см3 и v" = 3,70 см3 — соответственно А'= 0,30 и А"= 1,10.
Рассчитать содержание олова в растворе, учитывая разбавление раствора при титровании. Прн 1 = 320 нм излучение поглощают SnCl2 и Ce(SO4)2.
Ответ: r/Sn = 3,43 мг.
Задача 4. К неизвестному количеству Hg(NO3)2 в объеме 15 см3 добавлено 5 см3 0,01 М раствора ЭДТА, избыток которого титруют при Л = 578 нм в присутствии 5—7 капель 0,5% раствора ксиленоловою оранжевого (при pH = 6) 5-10-4 Л1 раствором Pb(NO3)2. Значения оптических плотностей при добавлении раствора соли свинца объемами — 2,0 см3 и v2 = 4,50 см3 составляли соответственно Аг— 0,20 и А2 = 0,18, после точки эквивалентности при добавлении объемов и' — 5,0 см3 и и" = 5,50 см3 — соответственно А' = 0,45 и А" = 0,75.
Рассчитать содержание ртути в исследуемом растворе, учитывая разбавление раствора при титровании. При 1 = 578 нм излучение поглощает практически только комплекс свинца с ксиленоловым оранжевым.
Ответ: ?Hg = 9,57 мг.
238
Задача 5. Смесь солей Ва и Pb(II) в объеме 20 см3 титруют при Л = 240 нм 0,01 М раствором ЭДТА до постоянного значения оптической плотности Ао = = 1,25. Значения оптических плотностей, соответствующие объемам добавленногс раствора ЭДТА Vf = 0,45 см3 и v2 = 1,35 см3 (до первой точки эквивалентности), v' == 2,50 см3 и tr = 3,50 см3 (после первой точки эквивалентности), равны соответственно: А1= 0,30; А2 = 0,90; А' = 1,15; А" = 1,20.
Рассчитать содержания Ва и РЬ в раствора с учетом разбавления раствора при титровании. При Л = 240 нм излучение поглощают только комплексонаты Ва и РЬ (8рЬ_эдтА = 88^0; еВа_ЭдТА = 1500).
Ответ: ?Ва = 4,14 мг; ?рь = 3,42 мг.
Глава 8
СПЕКТРОФОТОМЕТРИЧЕСКИЙ АНАЛИЗ РАСТВОРОВ ОКРАШЕННЫХ СОЕДИНЕНИЙ
Для полной характеристики окрашенного * комплексного соединения, используемого в фотометрическом анализе, необходимо знать:
уравнение химической реакции образования комплексного соединения;
состав комплекса;
константу равновесия реакции образования комплекса;
константу устойчивости (или константу нестойкости) комплексного соединения;
молярный коэффициент светопоглощения комплекса;
значения pH раствора, при которых достигается наибольшая степень образования окрашенного комплекса.
Несмотря на большое число работ по исследованию комплексных соединений, применяющихся в фотометрическом анализе, химизм многих фотометрических реакций до настоящего времени остается неясным, а полностью изученных реакций, по-видимому, вообще не существует [323]. Поэтому при выполнении фотометрического анализа часто приходится использовать спектрофотометрические методы для исследования как самой фотометрической реакции, так и образующегося окрашенного соединения. Спектрофотометрический анализ растворов окрашенных соединений основан на совместном использовании закона действующих масс и основного закона светопоглощения; он включает в себя определение состава, прочности и оптических~характеристик окрашенных соединений.
* В общем случае соединение может быть и бесцветным, но должно обязательно обладать избирательным поглощением света в ультрафиолетовой или инфракрасной областях спектра.
239
В литературе описано большое число различных спектрофотометрических методов анализа комплексных соединений [41, 48, 56, 323—333]. Наиболее распространенным методом определения состава комплексных соединений является метод Остромыслея-ского [326]. Метод Комаря [324, 325], пожалуй, является основным методом определения точных значений молярных коэффициентов светопоглощения и констант равновесйя'фотометрических реакций. При ступенчатом комплексообразовании для анализа образующихся соединений обычно используют функцию образования Бьеррума [326, 327] или метод Яцимирского [41, 328]. Янсен [326, 329], Ромен и Коллете [326, 330] описывают методы анализа комплексных соединений, образованных слабыми кислотами, не требующие специального выбора длины волны поглощаемого света. Наиболее надежным и общим спектрофотометрическим методом определения состава и констант образования смешанных комплексных соединений является метод Ньюмена и Хьюма [326, 331]. Ниже кратко рассмотрены наиболее распространенные методы спектрофотометрического анализа окрашенных соединений. Более подробно и полно эти методы анализа изложены в специальных монографиях Бабко [56], Яцимирского и Васильева [328], Ф. и X. Россотти [332], Шлефера [329], Бека [48], в работах Комаря [333] и других авторов [326].
8.1. ОПРЕДЕЛЕНИЕ СОСТАВА КОМПЛЕКСНОГО СОЕДИНЕНИЯ
Анализ состава образующихся в растворе комплексных соединений удобно проводить с помощью диаграмм состояния, которые особенно полезны в методе изомолярных серий. На рис. 8.1 изображена диаграмма состояния тройной системы, вершины которой соответствуют 100%-ному содержанию каждого из компонентов, а точки внутри треугольника — растворам с определенным соотношением компонентов системы Параллельные разрезы диаграммы (линии, параллельные одной из сторон треугольника MSR) отвечают серии растворов с постоянной концентрацией компонента в вершине треугольной диаграммы и постоянной суммарной концентрацией двух других компонентов. Разрез т —г соответствует серии растворов, содержащих одинаковое количество растворителя S; в растворах этой серии сумма концентраций компонентов М и R является величиной постоянной. Разрез s—г соответствует серии
Рис. 8.1. Диаграмма состояния трехкомпонентной системы.
М— комплексообразователь; R — фотометрический реагент; S — растворитель.
S
240
растворов с постоянной концентрацией комплексообразователя М и переменной концентрацией реагента R, а разрез т —s — постоянной концентрации реагента-лиганда R и переменной — комплексообразователя М.
Параллельные разрезы диаграммы состояния системы дополняют друг друга, но важнейшим из них, безусловно, является разрез s — г, позволяющий обнаружить области существования образующихся моноядерных комплексов. Во всех спектрофотометрических методах определения состава светопоглощающих соединений используют тот или иной разрез диаграммы состояния.
8.1.1. МЕТОД НЕПРЕРЫВНЫХ ИЗМЕНЕНИЙ (МЕТОД ИЗОМОЛЯРНЫХ СЕРИЙ)*
Метод предложен И. И. Остромысленским в 1910 г. [3261. Позднее П Жоб [3261 уточнил выводы Остромысленского, а Восберг и Купер [326] повторили вывод Жоба для простейшего случая: М + R s* MR. В СССР метод изомолярных серий получил широкое распространение благодаря работам А. К. Бабко с сотрудниками [56, 334—337].
Метод основан на определении отношения изомолярных концентраций реагирующих веществ, отвечающего максимальному выходу образующегося комплексного соединения MmRn. Кривая зависимости выхода комплекса от состава раствора характеризуется экстремальной точкой (рис. 8.2). Такая точка отвечает максимально возможной концентрацией комплекса MmRn, образующегося по реакции тМ. + nR MmRn, а ее положение (абсцисса) однозначно связано со стехиометрическими коэффициентами /пип:
Хмаьс = CR/(CM + CR) = n/(m + п)
Здесь См и Cr — начальные концентрации реагирующих компонентов Ми R.
Для выполнения анализа приготавливают растворы обоих компонентов одинаковой молярной концентрации и смешивают их в антибатных соотношениях (чаще всего от 1:9 до 9:1), сохраняя неизменным общий объем раствора (VM + Vr = V = const). При этом суммарное число молей обоих компонентов в общем объеме смеси всегда остается постоянным (См + CR ~ С — — const).
Измерение оптической плотности проводят при постоянных значениях ионной силы и pH растворов. Буферный раствор для поддержания постоянного значения pH среды подбирают так, чтобы между компонентами изомолярной серии и буферной смеси комплексообразование отсутствовало.
* Серия растворов с постоянной суммарной концентрацией реагирующих компонентов М и R, нос различным отношением их концентраций Cr/Cm, называется изомолярной.
241
концентраций или объемов
Рис. 8.2. Зависимость А (ДД) или концентрации комплекса от состава изомоляр-ного раствора.
Измерив оптические плотности приготовленных растворов изомолярной серии, строят график зависимости А от соотношения компонентов изомолярной серии
л = /(Си/см); ^ = /(rR/vM)
или
А = f [CR/(CM + CR)]
(см. рис. 8.2) и определяют положение максимума поглощения на изомолярной кривой. Максимальным светопоглощением обладает такой раствор, в котором содержание образующегося комплексного соединения является наибольшим. Поэтому объемное соотношение компонентов изомолярной серии, отвечающее максимуму поглощения, соответствует стехиометрическому соотношению реагирующих веществ.
Если максимум поглощения на изомолярной кривой нечеткий, то его положение определяют экстраполяционным приемом: через начальные точки обеих ветвей кривой проводят прямые линии, продолжая их до взаимного пересечения. Экстраполяционная точка пересечения прямых соответствует экстремальной точке на изомолярной кривой.
В общем случае, когда при выбранной длине волны А, свет поглощают наряду с комплексным соединением MmRn и исходные компоненты М и R, строят графическую зависимость между отклонением оптической плотности от аддитивности АЛ = Лсм — — Лм — Лк (где Лсм — суммарная оптическая плотность всех компонентов раствора; Лм и Ли — оптические плотности растворов компонентов М и -R при концентрациях, равных концентрациям в исследуемой изомолярной смеси*) и составом изомолярной смеси.
Если на графике Л = f [CR/(CM + CR) I положения максимумов совпадают для различных концентраций анализируемых изомолярных серий, то это свидетельствует о постоянстве состава комплексного соединения.
К сожалению, метод изомолярных серий не является универсальным. Он применим только при следующих условиях [323 J:
1) химическая реакция между реагирующими веществами протекает строго по рассматриваемому уравнению и не осложняется
* Суммарная величина Дм+Дк может быть вычислена теоретически, если известны молярные коэффициенты светопоглощения компонентов: Дм4~ + Дк = емСм/ 4- eRCRZ.
242
никакими побочными процессами (протолиз, ассоциация и т. п.);
2) в системе образуется только одно комплексное соединение;
3) ионная сила растворов изомолярной серии сохраняется постоянной.
Однако и при этих условиях метод изомолярных серий как графический способ определения состава неодинаково эффективен, поскольку внешняя форма кривой зависит от прочности образующегося комплекса, концентрации реагирующих компонентов и значений стехиометрических коэффициентов [338].
При исследовании очень прочных комплексов изомолярная кривая вырождается в две пересекающиеся прямые, и положение абсциссы максимума на таких кривых не зависит ни от константы образования комплекса, ни от начальных концентраций реагирующих компонентов. Определение максимума на изомолярной кривой значительно затрудняется при образовании малопрочных комплексных соединений, вследствие его размытости.
Другим серьезным затруднением в определении максимума на кривой является образование высококоординированных соединений типа MRn (где п 4) или многоядерных соединений с дробным отношением стехиометрических коэффициентов. При образовании таких соединений максимумы на кривой сильно смещаются к краям изомолярной диаграммы и становятся малоразличимы. Поэтому наложение небольших экспериментальных погрешностей может серьезно исказить результаты анализа. В этих случаях, когда графическое определение экстремальной точки на изомолярной диаграмме становится совершенно ненадежным, либо используют прием Л. П. Адамовича [339] (прием параллельных разрезов изомолярной кривой), либо определяют положение максимума аналитическим методом [323]. В последнем случае вычисляют уравнения обеих ветвей изомолярной кривой, отбрасывая кажущуюся экстремальную точку, решают их совместно и находят искомое соотношение объемов.
Для получения надежных результатов рекомендуется готовить несколько изомолярных серий с различными суммарными концентрациями* и измерять светопоглощение при различных длинах волн. Опыты проводят при соблюдении необходимых мер предосторожности, и полученные результаты подвергают последующему статистическому контролю.
Состав соединения можно считать установленным в следующих случаях:
а) найденные стехиометрические соотношения почти точно удовлетворяют целочисленным коэффициентам;
* Смещение максимума на изомолярной диаграмме при различных исходных концентрациях изомолярной серии указывает на наличие побочных процессов или на образование нескольких комплексных соединений.
243
б) эти соотношения подтверждаются свойствами ожидаемого соединения;
в) последующие определения константы равновесия подтверждают образование предполагаемого продукта реакции.
Глубокое теоретическое рассмотрение метода изомолярных серий и возможностей его применения к исследованию разных типов реакций проведено Н. П. Комарем [333, 340]. Автор показал, что при осложнении рассматриваемых равновесий различными побочными процессами метод изомолярных серий дает возможность установить лишь стехиометрические коэффициенты в уравнении реакции, а не стехиометрические индексы в формуле образующегося комплексного соединения. Кроме того, метод содержит некоторую неопределенность, которая иногда не позволяет однозначно решить вопрос о составе продукта реакции даже при правильно установленных стехиометрических коэффициентах, например для реакций типа:
MR + Rj <=> MR! + R и MR + R! <=> MRRX или
M R <=£ MR и nM + nR <=> (MR)n
Однако, несмотря на некоторые ограничения, метод изомолярных серий в ряде случаев успешно применяют для определения состава экстрагируемого комплексного соединения [341—343]. Н. П. Комарь доказал, что этим методом можно определить состав экстрагируемого комплекса и в более сложных случаях, когда вместе с комплексом экстрагируется светопоглощающий реагент, ассоциирующий в неводном растворителе, или когда в водной фазе протекают процессы ступенчатого комплексообразования [344 ].
8.1.2. МЕТОД СДВИГА РАВНОВЕСИЯ
Метод применяют для исследования состава моноядерных комплексных соединений. Из выражения условной константы устойчивости комплекса MRn следует, что при постоянной концентрации компонента М относительное изменение степени закомплексованности иона М (сдвиг равновесия) находится в прямой зависимости от равновесной концентрации второго компонента:
lgT§r=nlg[CR1 (81)
Полагая пропорциональной зависимость между изменением степени закомплексованности и изменением оптической плотности раствора, это уравнение прямой без упрощающих допущений можно записать в такой форме
Л
’б -дпр_л = «Jg [Cr] (8.2)
244
или при наличии поглощения реагирующих компонентов
АЛ
= <8-3>
Здесь Апр(ДДпр)— предельное значение оптической плотности (или разности оптических плотностей), достигаемое в опытах в условиях (насыщения».
Для определения стехиометрического коэффициента п по данным измерения А и АД растворов с постоянной концентрацией компонента М и переменной концентрацией компонента R строят график в координатах 1g -дд-----------lg 1R 1. п0 наклону кото-
рого и определяют значение п (п = tg а, где а — угол наклона прямой).
Если опыты проводить при постоянной концентрации компонента М в условиях большого избытка переменной концентрации реагента R (CR См), когда разницей между общей и равновесной концентрацией реагента можно пренебречь, то график для определения п можно строить в координатах 1g -дд-~—уд lg Cr-Этот метод применяют также для определения состава одноядерного комплекса при ступенчатом комплексообразовании.
Ниже рассмотрены в качестве самостоятельных методов различные варианты метода сдвига равновесия.
8.1.3. МЕТОД МОЛЯРНЫХ ОТНОШЕНИЙ
(МЕТОД «НАСЫЩЕНИЯ»)
Метод молярных отношений [56, 345] является наиболее общим приемом исследования прочных комплексов. Сущность метода заключается в установлении зависимости А (или АД) * от концентрации одного из компонентов при постоянной концентрации второго компонента и наоборот. Зависимость А (АД) от CR/CM при постоянной концентрации См (кривая насыщения) изображена на рис. 8.3. Точка излома на кривой отвечает отношению стехиометрических коэффициентов, которое равно отношению концентраций реагирующих компонентов в точке хт э — абсциссе точки эквивалентности. Если точка излома на кривой насыщения наблюдается нечетко, ее определяют экстраполяцией прямолинейных участков кривой до взаимного пересечения (см. рис. 8.3).
При исследовании простейших реакций типа
М + nR MRn или mM + R A4mR стехиометрические коэффициенты можно определять и аналитическим путем.
1. Изучая зависимость оптической плотности раствора от концентрации реагента R при постоянной концентрации компонента М (См = const), находят предельное значение А (ДПр). Затем
* ДА = Асм — Ам — Ar.
245
Рис. 8.3. Кривая насыщения:
1 — для прочного комплекса; 2 — для малопрочного (неустойчиво! о) комплекса.
аналогично определяют предельное значение А (Л,',р) при постоянной концентрации реагента R (CR = const). В этом случае насыщение наступает,
очевидно, при концентрации комплекса в п раз меньшей (или, наоборот, в т раз большей при образовании комплекса MmR), чем в предыдущем случае.
Искомые стехиометрические коэффициенты п или т определяют из отношения кажущихся молярных коэффициентов све-
топоглощения
__ 8 ___ ^1|р/(Сд-/) .
“ 8' “ ’
"pV (8.4)
п г' Л'пр/(СЯ1)
8 Лцр/(бмО
или, когда измерения оптической плотности производят при одинаковой толщине слоя /:
п = ЛпрСи/(ЛпрСм); т = ЛпрСм/(ЛпрСн)
Здесь е и е' — кажущиеся молярные коэффициенты светопоглощения при См = const и Cr = const соответственно.
2. Когда кривая насыщения выражается двумя пересекающимися прямыми, искомое соотношение CR/CM, отвечающее точке насыщения, находят решением системы уравнений [346]:
Здесь b — угловой ксвффициент прямой, проходящей через начало координат; а — наибольшее значение оптической плотности в условиях полного насыщения (Лпр); х — переменная концентрация реагента (или Cr/Cm).
В условиях недостигнутого насыщения задаются величиной х = CR/CM и, измеряя оптическую плотность полученных растворов, находят значение углового коэффициента kyr:
^уг = (Аг — '41)/(*'2 — *1)
Затем определяют оптическую плотность в условиях полного насыщения Лпр и находят искомое стехиометрическое отношение реагирующих компонентов:
хт. э = 4пр/&уг
Этот аналитический прием можно применять в условиях, когда поглощают свет все три компонента системы. В этом случае в уравнения вместо А подставляют разность оптических плотностей АЛ. 246
При использовании метода молярных отношений следует иметь в виду, что на кривых насыщения АЛ = f (С) точка излома определяется, как правило, менее четко по сравнению с графиками А = = f (С); причем, чем меньше прочность образующегося комплекса, тем больше происходит «сглаживание» излома на кривой насыщения [347]. В случае линейной зависимости оптической плотности раствора от концентрации одного из исходных компонентов аналитический прием вообще неприменим [348].
8.1.4. ТИТРИМЕТРИЧЕСКИЙ МЕТОД
Титриметрический метод [349] применяют для определения состава простейших одноядерных комплексных соединений. Кривая спектрофотометрического титрования (титриметрическая кривая) является простой модификацией диаграммы «состав — оптическая плотность» и по физическому смыслу аналогична кривой насыщения.
Приготавливают эквимолярные растворы реагирующих компонентов М и R. Взяв определенный объем вещества М (Ем\, из полумикробюретки титруют его раствором реагента R, одновременно измеряя оптическую плотность раствора. Затем на кривой титрования находят точку излома, отвечающую эквивалентности взаимодействия реагирующих компонентов (рис. 8.4), и измеряют объем реагента VR, затраченный для достижения точки эквивалентности (т. э.). Если реакция протекает согласно простейшей схеме
М + nR <=> MRn
то отношение объемов реагирующих вещест в в точке эквивалентности соответствует стехиометрическому отношению реагирующих компонентов:
п = Vr/Vm
(8-6)
Титриметрический метод применим для реакций комплексообразования, имеющих константы равновесия не менее 100, а также при условии, что светопоглощение реагирующих компонентов либо вообще отсутствует, либо одинаково, но меньше поглощения образующегося комплекса, либо одинаково для всех трех компонентов системы. Метод очень удобен для исследования продуктов реакций комплексообразования, протекающих с большой скоростью.
Рис. 8.4. Кривая спектрофотометрического титрования (Vr — добавленный объем реагента-титранта).
247
8.1.В. МЕТОД ИЗОБЕСТИЧЕСКОЙ ТОЧКИ
Метод применяют [350] для определения состава прочных моно-ядерных комплексных и хелатных соединений, когда поглощают все компоненты раствора.
Если прочный комплекс образуется по реакции
М + nR MRn
и поглощают все три компонента, то спектры поглощения изомолярной серии растворов имеют две изобестические точки при „—> - п Ср _ п
> п у и Cm + Cr п'+’Т* в КОТОРЬ1Х суммарное поглощение всех компонентов раствора не зависит от их концентрации в смеси и равно постоянной величине (А = Ак + Лм + AR = const; АЛ =0; Ае = ек — 8М — neR = 0). Однако системы, в которых поглощают все три компонента раствора, встречаются сравнительно редко; обычно поглощают только два компонента: окрашенный комплекс и реагент.
Если комплексообразователь М не поглощает свет (ем = 0), то спектры поглощения изомолярных растворов имеют только одну изобестическую точку при ~с > nJ-Т (Рис-
Состав изомолярного раствора, кривая поглощения которого проходит через изобестическую точку и имеет наибольшее значение максимума поглощения образующегося комплекса, отвечает составу комплекса.
Зависимость оптической плотности от состава раствора изомолярной серии при длине волны изобестической точки выражается двумя прямыми линиями (рис. 8.6), точка пересечения которых определяет состав комплекса. Абсцисса точки пересечения:
<°r
См + Cr
п
п г 1
(8 7)
Сослав изомолярного раствора, отвечающий точке пересечения, соответствует составу, для которого абсорбционная кривая А — = / (л) имеет наибольшее значение максимума поглощения образующегося комплекса. При исследовании высокопрочных комплексов пересекающиеся прямые А = f с ) строить не обязательно, так как из спектров поглощения изомолярных растворов видно, при каком составе раствора изобестическая точка перестает существовать — этот состав раствора и отвечает составу комплекса.
Если комплекс не очень прочный, то изобестическая точка наблюдается нечетко и определение состава по кривым светопоглощения становится менее надежным.
На графике зависимости оптической плотности от состава изомолярного раствора при длине волны изобестической точки 248
Рис. 8.5. Кривые поглощения изомолярной серии растворов»
В процентах выражена мольная доля одного из компонентов изомолярной серии, максимальный выход комплекса отвечает отношению реагирующих компонентов 1 : 2.
Рис. 8 6. Зависимость А от состава и зомол яркого раствора
наблюдается плавный переход от одного прямолинейного участка к другому. В этом случае точка пересечения, определяющая состав комплекса, находится экстраполяцией прямолинейных участков до взаимного пересечения.
8.1.6. МЕТОД ОТНОШЕНИЯ НАКЛОНОВ
(МЕТОД ГАРВЕЯ-МЕННИНГА)
Метод Гарвея и Меннинга [351 ] применяют для исследования различных реакций, в результате которых образуется один достаточно прочный комплекс. Чаще всего такие реакции происходят между двумя бесцветными компонентами М и R и сопровождаются образованием окрашенного продукта MmRn:
тМ + nR <=> MmRn
Опыты ставят двумя сериями. В первой серии начальная концентрация реагента CR сохраняется постоянной и значительно большей, чем концентрация компонента М, которая берется различной: CR См.
Из-за большого избытка реагента R можно считать, что компонент М будет целиком переходить в комплекс MmRn, концентрация которого составит Ск!т. Измеряя оптическою плотность для каждой концентрации См, получают графическую зависимость (рис. 8.7)
A = kjC^/rn
249
Рис. 8.7. Графическое определение отношения стехиометрических коэффициентов т : п методом отношения наклонов (на рисунке т : п = 1).
(где ky — коэффициент пропорциональности) и находят угловой коэффициент этой прямой: by = kjm.
Во второй серии опытов, сохраняя постоянной концентрацию компонента М
(См) и изменяя концентрацию реагента R, при аналогичных
условиях получают графическую зависимость
Л 2 = k^CeJn
и находят угловой коэффициент прямой b2 = kjti.
Применяя основной закон светопоглощения к выражениям оптической плотности Ау и /2, получим уравнения:
Л = kiCt^m = bjCfA == zlCwJrn
(8.8)
Л2 = kiP^Jn = 62CR = eZCp/n
Отсюда
by = zllm', b2 = e//n
Разделив одно равенство на другое, получим отношение угловых коэффициентов, которое равно отношению стехиометрических коэффициентов в формуле комплекса:
Ьу/Ь2 — п/т (8.9)
Детальный анализ спектрофотометрического метода Гарвея и Меннинга проведен Л. П. Адамовичем [352], который установил, что этот прием можно использовать для анализа систем, где поглощают все компоненты при выбранной длине волны. В этом случае отдельно измеряют оптическую плотность реагирующих компонентов и всей смеси. По данным отклонения оптической плотности от аддитивности строят графики зависимости ДА от См (при CR = const) и CR (при См = const) и определяют отношение угловых коэффициентов полученных прямых.
Погрешность метода тем больше, чем меньше прочность комплекса. Для анализа малопрочных комплексов, особенно при ступенчатом комплексообразовании, метод отношения наклонов вообще неприменим.
8.1.7. МЕТОД ПРЯМОЙ ЛИНИИ (МЕТОД АСМУСА)
Метод применяют для определения состава малопрочного моно-ядерного комплекса при отсутствии поглощения исходными компонентами. Применяя закон действующих масс и основной закон светопоглощения к реакции комплексообразования
М + nR MRn
250
Асмус путем несложных преобразований получил зависимость [353]:
1 А'В 1 В
= (8.10)
К л
А А' СМг7Й (8.П)
тА 1 ц_ V(1+AV£/W)
Здесь А' и В — постоянные для данной серии опытов; К — константа нестойкости комплексного соединения; п — стехиометрическое отношение компонентов в составе комплекса; Vr — объем реагента R, добавляемый к постоянному объему Ум компонента М; См и Cr — исходные концентрации компонентов М и R; V — постоянный общий объем анализируемых растворов; тА— модуль оптической плотности.
Для определения стехиометрического отношения реагирующих компонентов применяют графический прием (рис. 8.8). Если nd оси ординат откладывать величину 1/Vr, а по оси абсцисс — соответствующее ей значение 1/тА и задавать различные значения п, то в общем случае получим набор кривых в координатах 1/Vr— — \1тА. Эта зависимость только для истинного значения п будет выражаться прямой линией.
Практически к постоянному объему исходного раствора компонента М (VM) с концентрацией См прибавляют различный объемы раствора реагента (VR) с концентрацией CR и доводят общий объем приготовленного раствора до постоянного значения V. Затем измеряют оптические плотности растворов, определяют значения тА = АН и строят графические зависимости 1/Vr от 1/тА для различных задаваемых значений п (где п — целое число). Удовлетворяя требованию прямолинейности этой зависимости, находят искомый коэффициент п.
Выгодными преимуществами метода Асмуса являются:
1) возможность правильного определения состава комплекса в условиях, когда концентрации исходных растворов реагирующих компонентов определены неточно и даже вообще неизвестны;
2) возможность работы с загрязненными препаратами (при условии, что сами примеси не образуют комплексов с компонентами основной реакции);
3) простота выполнения.
Клаузен и Лангмюр [354 ] использовали метод Асмуса для анализа многоядерного комплекса типа MmRn.
Рис. 8.8. Графическое определение стехиометрического коэффициента п методом Асмуса (при п = = 2 функция прямолинейна).
251
8.1.8. ОГРАНИЧЕННО-ЛОГАРИФМИЧЕСКИЙ МЕТОД (МЕТОД БЕНТА—ФРЕНЧА)
Метод применяют [355—357] для определения состава малопрочных моноядерных комплексных соединений. Он основан на установлении логарифмической зависимости оптической плотности раствора от концентрации одного из компонентов (рис. 8.9). Применяя закон действующих масс к простейшей реакции комплексообразования
М + nR <=> MRn
находят логарифмическую зависимость между концентрацией комплекса MRn и переменной концентрацией одного из компонентов, например CR:
lg₽K='g’iMW: n,g[R] = lg[MR'll-,gpi:[Ml (8J2)
Здесь Рк — константа устойчивости комплекса.
При постоянной концентрации См эта зависимость выражается прямой линией, угловой коэффициент которой равен искомому стехиометрическому коэффициенту п. Если оптическая плотность раствора пропорциональна концентрации образующегося комплекса, то стехиометрический коэффициент п находят по наклону прямой логарифмической зависимости оптической плотности от переменной концентрации одного из реагирующих компонентов.
Для этого смешивают растворы реагирующих компонентов таким образом, чтобы начальная концентрация первого компонента См была постоянной, а концентрация второго CR непрерывно возрастала; общий объем смеси при этом должен оставаться неизменным. Затем, измерив оптические плотности приготовленных растворов, строят график логарифмической зависимости оптической плотности от концентрации компонента R (см. рис. 8.9): —lg Л = f(—lg Cr). В первом приближении угловой коэффициент этой прямой b принимают численно равным стехиометрическому отношению реагирующих веществ п. Этот прием, не учитывающий концентрацию реагирующих компонентов в составе комплекса MR„, можно применять при условии, что концентра-
ция компонентов во всех опытах значитрльно превосходит кон-
центрацию образующегося комплекса Ск, т. е. CR пСк и . См Ск.
Исследуя применение метода ограниченного логарифмирования, В. Н. Толмачев [356] рекомендует следующие наиболее благоприятные условия, при
Рис. 8.9. Логарифмическая зависимость оптиче-
——*• ской плотности раствора от концентрации ре-*‘9 ‘'R агента.
252
которых наклон прямой b будет близок к истинному значению п:
1) малое значение константы образования комплексного соединения;
2) небольшое абсолютное значение постоянной начальной концентрации компонента М — тем меньше, чем больше значение константы устойчивости комплекса (Зк;
3) незначительный избыток реагента R по сравнению с компонентом М.
Если при реакции происходит выделение ионов водорода, то с целью снижения выхода комплекса анализ необходимо проводить при низких значениях pH раствора.
По данным Адамовича [357] этот метод можно применять, когда кроме комплекса один из реагирующих компонентов также поглощает свет при выбранной длине волны X. Для этого при аналогичных условиях строят график логарифмической зависимости разности оптической плотности (АД = Асы — Ам или АД = = Асм — Ак) от переменной концентрации второго компонента. По наклону прямой находят стехиометрический коэффициент п.
При анализе этим методом иногда применяют аналитический прием: приготавливают два раствора с постоянной концентрацией компонента М (См) и разными концентрациями реагента (Cr и Cr) и измеряют оптические плотности этих растворов (А' и А"). Предварительно измеряют оптические плотности раствора компонента М при концентрации См (Ам) или растворов реагента R с концентрациями Cr и Cr (Ar и Ar). Искомый коэффициент п рассчитывают по формуле *:
, ДД' /. CR , Д' — АК I. CR , a' — ar/, cr „ „ n~ g ЛА" g C" g А" — Дм / g Cr ~ g А" — Д'Д / g Cr' (8J3)
Концентрации реагента Cr и Cr должны быть малы и очень близки. Однако концентрации компонентов должны обеспечивать достаточную разность значений АА во избежание «потери точности при вычитании».
8.1.9. МЕТОД ОТНОСИТЕЛЬНОГО ВЫХОДА
(МЕТОД СТАРИКА И БАРБАНЕЛЯ)
Метод [358] основан на использовании уравнения алгебраической суммы стехиометрических коэффициентов реакции, которое характеризует состав равновесной смеси в точке максимального относительного выхода (максимального отношения концентрации продукта реакции к переменной начальной концентрации одного
* Этот расчет, по-видимому, можно использовать, когда поглощают все три компонента системы. Тогда при вычислении ДД от значения Дсы отнимают суммарную оптическую плотность чистых растворов обоих компонентов: ДД = = ^см — — Дк-
253
из реагирующих веществ). Этим методом можно определять состав комплексных соединений, образующихся по любому стехиометрическому уравнению. Для реакции комплексообразования
mM + «R <=*: M,nRn
при постоянной концентрации компонента М и переменной концентрации компонента R уравнение Барбанеля принимает вид:
с — См п — 1 к т т п — 1
(8.14)
Анализ проводят аналитическим методом в сочетании с графическим построением кривой относительного выхода. Приготавливают две серии растворов, в одной из которых изменяется концентрация реагента R при постоянной концентрации компонента М, а в другой, наоборот, постоянной остается концентрация компонента R. Затем измеряют оптические плотности приготовленных растворов чистых компонентов М и R тех же концентраций и определяют отклонение оптической плотности от аддитивности (ДА) *. После этого находят ДАир — максимальные значения ДА, соответствующие предельным значениям концентрации образующегося комплекса:
Cjt. пр — Сд|/Г71 или Ск. пр = Cr /п
По полученным данным строят кривые относительного выхода в координатах CK/CR—Ск/Ск. пр или AA/CR—ДА/ДАпр при См »= const и в координатах Ск/См—Ск/Ск. пр или ДА/СМ — ДА/ДАцр при CR = const.
Определив абсциссы, отвечающие максимумам на кривых для обеих серий опытов, рассчитывают стехиометрические коэффи циенты тип:
Ск
Ск. пр
с,.
бк. пр
Для определения стехиометрических коэффициентов одноядерных комплексов типа MmR или MRn используют кривые относительного выхода лишь для одной из серий опытов. Так, при определении состава комплекса MRrt строят экспериментальную кривую относительного выхода в координатах ДА/Cr—ДА/ДАпр при См = const и, определив на кривой абсциссу максимума, рассчитывают стехиометрический коэффициент п по уравнению:
»= 1 "дл Мц" (при ДЛ'бк = макс) (8.16)
п — 1
। __j (при См = const и ДДСв— макс)
(8.15)
т — I
п _ 1 (при Cr = const и ДЛ/СМ = макс)
\ длир)
/ АЛ \ = \ Л^ир )
* Если при выбранной длине волны X ни один из компонентов системы, кроме комплекса, света не поглощает, то ДЛ = Дсм.
254
Рис. 8.10. Кривые относительного выхода, построенные для произвольных комбинаций тип при постоянной концентрации компонента М (С,ц = const).
' При анализе комплекса MmR стехиометрический коэффициент т определяют аналогично, используя кривую относительного выхода ЛЛ/СМ = f (ДЛ/ДЛПр) при CR = const:
т = 1 —ДЛ/ДЛпр (при ДЛ/См=макс)
(8.17)
Отсутствие максимума на кривой относительного выхода для любой из серий опытов указывает на то, что стехиометрический
коэффициент компонента переменной концентрации равен единице (M2R и MR, рис. 8.10). Стехиометрический коэффициент второго компонента в этом случае определяется, как указывалось выше, с использованием кривой относительного выхода при переменной концентрации этого же компонента. Если кривая относительного выхода выражается прямой линией, то стехиометрические коэффициенты, как правило, одинаковы и равны единице (m = п = 1).
Метод относительного выхода имеет большие возможности и, по-видимому, очень перспективен. В отличие от метода изомолярных серий он позволяет определять не отношения стехиометрических коэффициентов, а их абсолютные значения. К другим достоинствам этого метода относятся:
1) применимость к -любым стехиометрическим реакциям;
2) отсутствие каких-либо ограничений и допущений, связанных с прочностью комплекса;
3) отсутствие ограничений в выборе интервала концентраций;
4) возможность установления состава комплекса при отсутствии данных о концентрациях вещества в исходных растворах, так как достаточно лишь поддерживать постоянство начальной концентрации одного вещества и знать относительную концентрацию другого вещества в каждом из растворов серии.
8.2. ОПРЕДЕЛЕНИЕ СОСТАВА,
ПРОЧНОСТИ И МОЛЯРНЫХ КОЭФФИЦИЕНТОВ СВЕТОПОГЛОЩЕНИЯ ОКРАШЕННЫХ СОЕДИНЕНИЙ
Значения молярных коэффициентов свеюпоглощения, вычисленные по опытным данным 8 = не всегда соответствуют истинным. Чаще всего это лишь среднее значение молярного коэф
255
фициента светопоглощения, которое может изменяться при изменении концентрации реагирующих веществ вследствие побочных процессов (изменение степени диссоциации комплекса, ступенчатое комплексообразование, полимеризация и др.).
При наличии побочных процессов основной закон светопоглощения, как правило, нарушается и при изменении концентрации окрашенных растворов максимумы поглощения i.a спектральных кривых е = f (X) смещаются (рис. 8.11). Однако этого не происходит, когда молярные коэффициенты светопоглощения всех компонентов раствора одинаковы. При этом значение среднего молярного коэффициента светопоглощения ё сохраняется неизменным в широком интервале концентраций.
Постоянство значений е для растворов с различной концентрацией как при наличии побочных процессов, так и при их отсутствии не является доказательством их истинности, так как при таком определении не учитывается состав поглощающего соединения. Значения молярных коэффициентов светопоглощения в таких случаях называют кажущимися. Истинные значения е определяют специальными спектрофотометрическими приемами .
Рассмотренные ниже спектрофотометрические методы позволяют также определять условные и концентрационные константы устойчивости светопоглощающих соединений. Если в методе используют выражение условной константы устойчивости, когда в экспериментальной или расчетной зависимости равновесную концентрацию исходных компонентов [См]и 1CR ] отождествляют с разностью между общей их концентрацией и концентрацией комплекса ([СМ1 = СМ— Ск и [CR] ~- С^ — пСя), то найденное значение константы устойчивости всегда является условным и может изменяться в зависимости от условий проведения эксперимента (pH, состав буферного раствора и т. п.).
Если в методе используют выражение концентрационной константы устойчивости или константы равновесия, то в рассматриваемой зависимости необходимо учитывать равновесную концентрацию только стехиометрических форм компонентов. Следовательно, при определении концентрационных констант недопустимо отождествление равновесных концентраций компонентов с разностью между их общими концентрациями и концентрацией в комплексе. В этом случае необхо-£" /'Т'^х4 димо вводить поправку на относитель-
///I уЛ'/с' ное содержание стехиометрической
/,'/ ' И\\/С2 формы компонента в условиях прове-
JJ [ Рис. 8.11. Смещение максимума поглощения
, Jr | на кривых е = f (X):
у ' С, —С, — различные концентрации светопоглощаю-
1 щего соединения.
256
дения эксперимента). Так, [М] = (См — Ск) <zM; [R ] = (CR — — пС,.) aR и т. д., где а — мольная доля стехиометрической формы компонента.
Я2.1. МЕТОД ЯЦИМИРСКОГО
Метод [41 ] основан на совместном использовании закона действующих масс и основного закона светопоглощения в сочетании с правилом аддитивности оптической плотности. Метод применим к системам со ступенчатым комплексообразованием, в которых свет поглощают все компоненты, кроме лиганда-реагента.
Согласно правилу аддитивности, оптическая плотность раствора такой системы определяется уравнением:
А = ем [М] I -г Ч [MR] I + е2 [MR2] I + • • • + % [MRJ I (8.18)
Среднее значение молярного коэффициента светопоглощения будет равно:
_ _ А _ ем + еА [R1 + e2$-i [R12 + • • • + enPn [R]n i+₽1 [R] + ₽2 [R]2+...+pn [Rp (819)
После вычитания из обеих частей уравнения (8.19) значения ем получим:
= ApiPi [R1 Ае'зРз [R]2 Ч- • • • 4 AfnPn [Rp
1 + ₽i[R] + Ра[R]2 + •••+₽n[R]n ’
Для решения уравнения (8.20) и определения неизвестных значений (3, и Дег К. Б. Яцимирский разработал математический прием с использованием вспомогательных функций и экстраполяции их на нулевое значение. Вводя вспомогательную функцию ft = Ae/[R], из уравнения (8.20) получим:
. = ДедР1 + Af A [R] )-••• + Лепрл [R]"-1
1 1 + ₽i [R] + Рг[R]2 + •” + ₽n[R]" J
Экстраполяция этой функции на нулевое значение концентрации лиганда дает
= = Д’! Pj
[R]-*o
Такую экстраполяцию проще осуществить графическим методом, откладывая по оси абсцисс равновесную концентрацию лиганда; а по оси ординат — значение функции Д. Экстраполируя эту зависимость до пересечения с осью ординат, находят отрезок, численно равный Де^. При дифференцировании функции и экстраполяции ее производной на нулевую концентрацию лиганда получаем:
lim тля = й2 = Ае2р2 + ДеД2
[R]-и “ iRJ
9 М. И. Булатов и др.
257
Значение аг можно получить построением новой вспомогательной функции /2:
/2 = (fi — °i)/[RJ (8.22)
Функция (8.22) при экстраполяции на нулевую концентрацию лиганда принимает следующее значение:
lim /2 = а2 = Де2В, — Ле. Р?
[Rl^o
Аналогично строится третья вспомогательная функция f3 = — (/2 — o2)/[R ]. При экстраполяции /3 на нулевую концентрацию лиганда получаем:
Игл /3 = щ = ДеаВ3 —Де.В?
[R]-*o
Совершенно аналогично могут быть построены и последующие вспомогательные функции f4, f?,, f6 и т. д.
A = (A-i-Ci_i)/[R] (8.23)
Экстраполяция такой функции на нулевую концентрацию лиганда дает:
lim /z = a( = ДеД—Де^ [RJ-o
Однако общее число экстраполяционных уравнений оказывается в два раза меньше, чем число подлежащих определению значений р и Де. Поэтому вводят новую переменную у, связанную обратной зависимостью с концентрацией лиганда:
j/=l/|R] (8.24)
После деления числителя и знаменателя уравнения (8.20) на [R ]" и учета соотношения (8.24) получим:
дё = AFflPn ~Ь • • • + Де1Р1!/п 1 ,g 25ч
Рп + Рп-Ц/ + • • • + Pi!/*1 1 +
Экстраполяция Де на нулевое значение у дает:
Игл Де = Ьг = Деп (8.26)
J/-0
Дифференцирование Де по у и экстраполяция производной на нулевое значение у приводит к пределу:
*'т = <Деп-1 — Деп) Pn-1/Pn
j/->o “У
Такой же результат можно получить и при построении графической функции ф!
Ф1 = (Де — Ьг)!у (8.27)
и ее экстраполяции на нулевое значение у\
lim <рх = Ь2 = (Деп_1 — Деп) Pn-1/Pn
H-t-0
258
Аналогично строят функции <р2, <р3 ... <рп_! и экстраполируют на нулевое значение у. Комбинируя значения, полученные экстраполяцией функций Де, <рх, <р2 ... <рп-1, с результатами экстраполяции функций flt f2, f3 ... fn, можно определить все неизвестные коэффициенты рг и Ае, в уравнении (8.20). В простейшем случае, когда в растворе образуются только комплексы MR и MR2, по вышеописанной схеме получаем:
= Деф 1; а > = Деф.2 — Деф'(
fci = Деа; = (Де! — Де2) ₽!/₽а
Из этих данных находим константы устойчивости комплексов и их оптические характеристики:
.аф1 —д26а . о ат + аА
Р1 дф2 + bl ’ Р2 afa + bl
Дв!= +
arbt — агЬ2
Метод пригоден для исследования малопрочных и умеренно прочных комплексов, когда не возникает трудностей в вычислении равновесной концентрации лиганда. В этих условиях общая концентрация лиганда обычно отождествляется с равновесной Г41 1.
8.2.2. ИСПОЛЬЗОВАНИЕ ИЗОМОЛЯРНОЙ ДИАГРАММЫ
И КРИВОЙ НАСЫЩЕНИЯ
Молярные коэффициенты светопоглощения ек и условные константы устойчивости Рй комплексов можно рассчитать по данным изомолярной кривой или кривой насыщения.
а. Условная константа устойчивости простейшего комплекса MR для любых двух точек изомолярной кривой определяется выражением:
(Cr-c;)(Cm-c;) (c'r-ck)(c;-q
Здесь С^, и Ср, Ср — начальные концентрации компонентов М и R в двух точках; С^ и С" — равновесные концентрации комплекса.
Если при выбранной длине волны поглощает только комплекс
MR, то при I = 1 см и Ск = А/ек получим:
___________^''"ь_______ _______________Л7е«___________ го oq\
[c(l-x')—dLJ(cx'--dL) [с(1-х“)-^-]
Здесь xl = С^/С; С = См + CR — суммарная концентрация компонентов в изомолярной серии.
9* 259
Решаем уравнение (8.29) относительно неизвестного значения 8К*:
е - 1 1/- Л'(А"У-А"(А'У -
к с V А"[х' — (х')2| + Д' [(х")а — х"] 1 ;
Рассчитав ек, определяют концентрацию комплекса Ск для любого изомолярного раствора и по уравнению (8.28) находят значение
б. Если состав комплекса MmRn и предельная оптическая плотность АЛпр могут быть определены непосредственно из кривой насыщения, по этим данным можно рассчитать и значения еи и рй:
= п ААц-р/(1Сц) (8.31)
Здесь п — стехиометрический коэффициент Cr — концентрация компонента R, отвечающая предельному значению Д Дпр при См = const.
Концентрацию комплексной формы Ск рассчитывают из выражения
ск = л//[/(ек-/тем-«Ек)]
причем при выбранной длине волны могут поглощать все компоненты системы.
На основании полученных данных рассчитывают
Р'к = СК/[(СМ - ™ск)т (CR - пск)"]
в. Бабко [56] показал, что для систем с малопрочным одноядерным комплексом MRn приближенные значения ек и Рй можно рассчитать по данным измерения оптических плотностей двух растворов при одинаковой концентрации комплексообразователя М. Применяя закон действующих масс к реакции образования комплекса в условиях, когда CR Ск и (CR — пСк) лг CR, получим выражение условной константы устойчивости комплекса:
₽к = Ск/[(См-Си)С£1 (8.32)
Сопоставляя данные двух растворов с одинаковой концентрацией См и полагая при этом, что А = екСк1, получаем выражения для расчета ек и Си:
1
1 |См[(с'кУ-^"М')(СкП
(8.33)
, = Д' = См[(с;у-(л7дЖ)Ч
к (д7д')[(с'Еу-(ск)"]
* В случае окрашенного реагента для расчетов используют значения разности оптических плотностей ДД' и ДД" и соответственно получают разность молярных коэффициентов светопоглощения комплекса и реагента Де = ек — neR при выбранной длине волны.
260
Рассчитав Ск, по уравнению (8.32) вычисляют значение условной константы устойчивости комплекса Это'- способ расчета можно применять, когда в системе поглощают не только комплекс MRn, но и исходные компоненты М и R. В этом случае вместо значений А', А" и ек используют соответствующие им значения разности оптических плотностей Л/Г, ЛЛ" и молярных коэффициентов светопоглощения Ле = ек — ем — neR.
8.2.3, МЕТОД КОМАРЯ
Этот универсальный метод точного определения молярных коэффициентов светопоглощения и констант равновесия реакций комплексообразования * основан на решении уравнения с двумя неизвестными для двух или более опытов. Метод предполагает знание типа реакции или состава комплекса, установленных независимым путем. Применяется для исследования реакций типа:
Мп+ + «HR «=> MRn + /?Н+ или Мл+ + nR~ <=> MRn
Аналитический вариант метода. Приготавливают ряд растворов с постоянной концентрацией ионов водорода Сн и стехиометрическим отношением реагирующих компонентов CHR/CM = = п. Затем измеряют оптические плотности полученных растворов при выбранной длине волны X. Применяя закон действующих масс к приведенным выше реакциям комплексообразования, находят выражение констант равновесия Лр:
Кр = ^Жм-Ск)П+‘<1 <8'35)**
Оптическая плотность раствора, когда при выбранной длине волны X поглощает только комплекс и один из реагирующих компонентов, например реагент HR-, будет выражаться уравнением:
Л=п(См-Сн)8к/ + е1(Ск/ (8.36)
Подставив значение Ск из уравнения (8.36) в (8.35) для I- и j-го значений концентрации компонента М и разделив полученные выражения одно на другое, получим:
1
Cil^-At lAi-nfRlCi\n+i /OQ,V
С;1ьк-А, -[Aj-r^lCj )
Полагая, что >
* Метод Комаря можно успешно применять при исследовании диссоциации слабых электролитов (индикаторов, кислот и т. п.) [333].
** В выражении константы равновесия для реакций, протекающих без выделения ионов водорода, множитель Сн отсутствует.
261
получаем формулу для вычисления молярного коэффициента светопоглощения комплекса:
*к = -р (+ В -£\1Г АВ} ) (8’38)
Если удается так выбрать длину волны X, что кроме комплекса не поглощает ни один из реагирующих компонентов, то выражение для В упрощается и принимает вид:
1
В = (Л/М>)Л+1
Измерения оптических плотностей для i- и /-го значений концентраций следует проводить в строго одинаковых условиях (pH раствора, длина волны, толщина слоя, температура, ионная сила). Таким образом, из данных любой пары растворов определяется ек, а затем и значение константы равновесия Др. Концентрацию комплекса Ск вычисляют по рассчитанному значению ек из уравнения (8.36).
Однако при вычислении ек и Лр может произойти «потеря точности при вычитании», особенно в случае очень прочных комплексов. Для предотвращения «потери точности» используют разные отношения С^Сj = b, разбавляют раствор (в пределах сохранения е„) или уменьшают выход комплекса понижением pH среды, когда реактив является слабой кислотой. Если определяются характеристики малопрочных комплексов, то сразу же можно получить удовлетворительные результаты.
Для получения надежных данных, когда кроме комплекса свет поглощает и реагент HR, аналитический вариант метода Комаря следует использовать при таких условиях, когда pH «С рКни, так как в уравнениях (8.35)—(8.38) равновесная концентрация кислотной формы реагента [HR ] отождествляется с разностью CR— пСк, а равновесная концентрация [Мп+] — с разностью См — Ск. В действительности же [HR ] = = (CR — nCK) aHR. a lMn+ J = (CM — Ск) ам (где aHR и aM — мольные доли компонентов HR и Мл+), поэтому при произвольном выборе pH раствора для проведения эксперимента в уравнения (8.35)—(8.38) необходимо вводить коэффициент aHR (коэффициент ам можно не вводить, так как используется не абсолютное значение концентрации [Мл+ ], а их отношение).
Тогда уравнения (8.36) и (8 37) примут вид:
Л = n (См Ск) eHRaHR/ + (8.39)
и
1
CfiKl~Aj _ / \ n+l ,я 4fn
Cflv.1 Aj \ А. — neHKaHRlCj /
Если метод Комаря применять при pH < p/CHR — 2, то значение aHR можно принять равным 1 (с погрешностью, не 262
превышающей 1%) и вместо уравнений (8.39), (8.40) использовать вышеприведенные уравнения (8.36), (8.37).
Аналогичные ограничения в условиях использования аналитического варианта метода Комаря возникают и в случае поглощения анионной формой реагента R'. Здесь также при произвольном выборе pH раствора в уравнения (8.36) и (8.37) необходимо ввести поправку на относительное содержание поглощающей формы реагента:
/t = n(CM-Ct!)eRaRZ + eKCKZ (8.41)
и
1
C^Kl -At At~ MffrlCt \ n+1
CfKl -A; ~ \ A3 - neRaRtC} )
Если эксперимент проводить при pH р/Снв + 2, то с погрешностью не более 1% можно принять, что aR 1, и для расчетов использовать уравнения (8.36) и (8.37). Еще более простым может оказаться случай, когда при выбранной длине волны кислотная форма реагента HR света не поглощает и при pH с < рЛня — 2 выход комплекса достаточен, тогда при eR < еи светопоглощением реагента вообще можно пренебречь.
В общем случае, когда при выбранной длине волны свет поглощает кроме исследуемого комплекса как кислотная, так и солевая формы реагента HR, использование приближенных уравнений (8.36) и (8.37) допустимо только при pH < рКня — 2 и eR < ек.
В противном случае необходимо применять более точные уравнения:
Л = nZ (См — ск) (eHRaHR + eRaR) + екСк и
CjeKl — Aj _ At ~ n^i (енякня енкк) CjEKZ Aj A. — nlCj (eHRaHR + eRaR) _
Z (8.43)
4т
(8.44)
Если значения eHR и eR неизвестны, то иногда используют условные молярные коэффициенты светопоглощения реагента (eR), определенные предварительно в таких же условиях, в которых проводят исследование светопоглощающего комплекса. Тогда во всех рассмотренных случаях для определения еи и Си, хотя и с большими погрешностями, можно использовать уравнения (8.36)—(8.38). Но при вычислении константы равновесия а-коэффициенты следует обязательно учитывать:
снСк
Кр=(С — С]^Л~ап~ (8Л5)
(СМ Ск) ” aMaHR
Графический вариант метода. Графическая интерпретация метода Комаря впервые предложена В. Н. Толмачевым 1325).
263
Преобразовав выражение константы равновесия, Толмачев вывел уравнение:
v + <8'46)
у Ап
Здесь е' — кажущийся молярный коэффициент светопоглощения; Q — в условиях опытов постоянная величина:
Q =
ппекКр
1 п+1
В координатах 1/е' — 1/ у Ап уравнение Толмачева описывает прямую линию, отсекающую на оси ординат отрезок, равный 1/ек. При определении ек и Кр, как и в аналитическом варианте, измеряют оптические плотности растворов с различными концентрациями реагирующих компонентов, но при постоянном стехиометрически требуемом соотношении. По данным измерений оптической плотности растворов строят график в координатах Cl/А — 1/ Ап, по которому находят * сначала ен, а затем, определив угловой коэффициент прямой (b = Q), рассчитывают константу равновесия по уравнению:
______/ Сц1 \п 1
1 ~ 1 ен ь«+1
(8.47)
8.2.4. МЕТОД АДАМОВИЧА
Метод [359] основан на использовании кривой насыщения и применяется для анализа достаточно прочных комплексов. Сущность метода заключается в выражении опытной кривой насыщения соответствующим аппроксимирующим уравнением, решение которого позволяет определить состав, прочность и молярный коэффициент светопоглощения комплексного соединения.
Рекомендуется следующий порядок работы.
1. Приготавливают серию растворов, в которых концентрация одного из компонентов, например реагента CR, остается неизменной, а концентрация второго компонента См увеличивается от опыта к опыту. Затем, измерив оптическую плотность приготовленных растворов или отклонение ее от аддитивности АЛ, строят кривую насыщения в координатах А (ЛА) — CM/CR. Опыты проводят при постоянном значении pH раствора, обеспечивающем максимальный переход компонента М в комплексное соединение MR„.
* Для получения более точных данных следует рассчитать уравнение прямой по способу наименьших квадратов. Из свободного члена уравнения находят величину ек, а по угловому коэффициенту рассчитывают Кр.
264
2. Полученную кривую насыщения выражают уравнением гиперболы второго порядка, проходящей через начало координат
ау*— 2(Ь-\-х)у— 2сх = 0, где 4/=Л(ДЛ); х = См (8.48)
Способом наименьших квадратов рассчитывают параметры а, b и с уравнения гиперболы.
с Е J/4 — 2Ь £ У3 + 2с *У3 — 2 U ху* = О
Y^ + ZcYxy-2 = °
с £ ху* — 2Ь £ ху + 2с £ х2 — 2 £ х^у = О
Найдя параметры гиперболы, вычисляют для всех значений х = См величины у~А (АЛ) и сопоставляют рассчитанные величины с опытными. Если расхождение между ними укладывается в пределы точности прибора, то выражение опытной кривой насыщения уравнением гиперболы можно считать обоснованным.
3. Использование параметров уравнения гиперболы для характеристики комплекса.
а. Если А или АЛ линейно связана с концентрацией образующегося комплекса MR^, то параметр Ь уравнения гиперболы можно использовать для определения стехиометрического коэффициента п в комплексе MRn. При достаточно высокой прочности комплекса значение параметра b практически равно концентрации реагента R, стехиометрически связанной с постоянной концентрацией компонента М. Поэтому
п — Cr'Cm = Ск/Ь
Критерием законности применения этого приема является неравенство:
К/С'[> 0,02
Здесь К — константа нестойкости комплекса.
Если оно не выполняется, то параметр Ъ нельзя использовать для определения состава комплекса.
б. При помощи значения параметра с, равного предельному значению оптической плотности Лпр (или АЛпр), находят молярный коэффициент светопоглощения комплекса ек (или Ае);
= Лпр/г/((Сц) = сп/(1Сц) (8.49)
Важным преимуществом этого приема является возможность расчета ек (или Ае) в тех случаях, когда предельное значение оптической плотности Лпр (или ААпр) в условиях опыта не достигается.
265
4. Определение константы нестойкости комплекса. Для каждого опыта серии находят равновесную концентрацию образовавшегося комплекса *:
CK=A4/[Z(eK-eM-neR)]
Затем для каждого опыта подсчитывают значение константы нестойкости:
= (См Ск) (CR яСк)« aMaR/CK
Стабильность значения К подтверждает правильность определения стехиометрического коэффициента п. Метод применяют для исследования, главным образом, моноядерных комплексных соединений. В случае образования окрашенного комплекса его можно использовать также в условиях полимеризации бесцветного компонента М. Однако область применения этого метода ограничена системами, где образуются достаточно прочные комплексы (со степенью связанности не менее 98%) и отсутствует ступенчатое комплексообразование.
8.2.5. МЕТОД ПЕРЕСЕЧЕНИЯ КРИВЫХ
Метод [360] используют для определения состава и прочности моноядерных комплексных соединений, когда имеется возможность определения равновесной концентрации образующегося комплекса MRn.
Приготавливают несколько растворов компонента М с различным содержанием реагента R (Cr,, Cr2 и т. д.) и определяют равновесную концентрацию комплекса MRn:
Ск = См (А/АПр) (8.50)
Здесь Апр— предельное значение оптической плотности раствора в условиях насыщения при См = const; А — измеренное значение оптической плотности ** при См = const.
Затем, задаваясь различными значениями п = 1, 2, 3, рассчитывают значения констант устойчивости комплекса для любых двух растворов:
₽к = С’к/|.(С’м Q) (CR — «Ск)" «m«r]
По полученным данным строят график зависимости 1g 0К от п для обоих растворов (рис. 8.12). Если состав комплекса в обоих растворах один и тот же, кривые 3* пересекаются в некоторой
* Если удается выбрать такую длину волны поглощаемого света, при которой ем — cr = 0, то вместо ДА подставляют измеренное значение оптической плотности А.
** Если при выбранной длине волны кроме комплекса поглощают также и исходные компоненты М и R, то вместо значений А нужно использовать ЛА = — Асм ~ Ам — Ar.
s* Зависимость 1g рк от п может быть и не прямолинейной, но все кривые пересекутся в одной точке.
266
Рис. 8.12. Кривые зависимости 1g ₽к от п, рассчитанные при п= 1,2,3 и См = = const.
Рис. 8.13. Кривые зависимости 1g и 1g (₽^/₽") от п, рассчитанные при п = = 1, 2, 3 и См = const.
точке, проекция которой на ось абсцисс покажет истинное значение стехиометрического коэффициента п, а на ось ординат — значение 1g рк данного комплекса. Однако при небольшой разнице равновесных концентраций комплекса в приготовленных растворах кривые пересекаются под очень острым углом и определение значений п и рк становится ненадежным. В этом случае для повышения воспроизводимости результатов определения строят график зависимости 1g Рк и 1g Рк/Р'к от п р« и Рк‘ — расчетные константы устойчивости комплекса для первого и второго растворов), задаваясь различными значениями п (рис. 8.13). Кривая зависимости 1g Р,'/Рк — f (ri) (кривая 2) пересекает ось абсцисс в точке, которая соответствует искомому значению стехиометрического коэффициента п. Расстояние по вертикали от этой точки до кривой 1g Рк = f (п) (кривая /) равно 1g рк исследуемого комплексного соединения.
Метод пересечения кривых дает надежные результаты, если состав комплекса в исследуемых растворах остается постоянным. Этот метод, по-видимому, можно использовать и при ступенчатом комплексообразовании, когда в анализируемых растворах доминирует комплекс одинакового состава.
8.2.6. МЕТОД РАЗБАВЛЕНИЯ (МЕТОД БАБКО)
Метод (35, 56] применяют для определения констант устойчивости умеренно прочных комплексов.
Приготавливают раствор со стехиометрическим соотношением концентраций компонентов М и R и разбавляют его растворителем в q раз. При этом степень диссоциации комплекса увеличивается и определяется выражением:
б=д/(1/7—1) (8.51)
Здесь Д — отклонение от основного закона светопоглощения вследствие диссоциации комплекса
Д = (Л — qAq)/A (8.52)
267
А и Ад — оптические плотности исходного и разбавленного растворов.
Если образуется простейший комплекс типа MR, то (Зк будет равно:
₽к = (1-б)/(й2См) (8.53)
Метод можно применять для оценки констант устойчивости малопрочных комплексов, образующихся при значительном избытке реагента. В этом случае следует разбавлять раствор, содержащий р-кратный избыток реагента R. При разбавлении исходного раствора в q раз
Ъд = 6 и Л = 6 (<? — 1)
Здесь f>g — степень диссоциации комплекса в разбавленном растворе.
При б « 1 и р 1 получим:
₽к = (1-б)/(брСм) (8.54)
Однако предложенный А. К. Бабко метод разбавления позволяет получить надежные данные только при таких условиях, когда влияние сопряженных равновесий, протекающих в водном растворе, незначительное. Метод разбавления основан на использовании уравнений (2.4) и (2.5), поэтому ограничения, относящиеся к этим уравнениям, в полной мере распространяются и на метод разбавления. При наличии в водном растворе сопряженных равновесий, накладывающихся на равновесие образования светопоглощающего соединения, метод разбавления можно использовать для определения условной константы устойчивости исследуемого комплекса. При этом pH и солевой фон исходного и разбавленного растворов должны быть одинаковыми. При стехиометрическом соотношении катиона металла и реагента из уравнения (2.11) можно получить зависимость между Рмкп и Д:
/„l/(n+D _ 1 \м-1 /
₽«% = (—та—) /(«"") <8-55>
Вычислив Д по экспериментальным данным согласно уравнению (2.6), при заданном солевом фоне и pH раствора по уравнению (8.55) можно рассчитать условную константу устойчивости светопоглощающего комплекса.
При р-кратном избытке реагента уравнение для расчета условной константы устойчивости комплекса получаем из уравнения (2.15):
₽мип = 100 № - 1)/[А (Р«СМ)"] (8.56)
Для получения правильных результатов необходимо обеспечить одинаковое значение условной константы устойчивости [<мчп в исходном и разбавленном растворах, поэтому при разбавлении раствора следует строго соблюдать идентичность количественного состава солевого фона (концентрация конкурирующих комплексан-тов, pH раствора) в обоих растворах.
268
Если состав солевого фона в исходном и разбавленном растворах все же оказывается неодинаковым, то преобразованием уравнений (2.11) и (2.15) можно получить новые расчетные уравнения (8.57) и (8.58), позволяющие по экспериментальным данным вычислить концентрационную константу устойчивости исследуемого комплекса в отсутствие избытка реагента
]. jq2 (п4-1) /п+1 Г п+1 Г [ \ п+1
₽MR" = \]/ (ама^ “ V (ама")0 J
и при р-кратном избытке реагента:
1 • Ю2 [<7п («m«r)0 ~ («m«r) J Д (ама" )о (амад)в (р/гСм)"
(8.57)
(8.58)
Здесь («м«4о и («ман)у — коэффициенты, учитывающие влияние сопряженных равновесий в исходном и разбавленном в q раз растворах.
8.2.7. СПОСОБ КЛОТЦА
Способ 1361] применим для определения значений ек и 0к умеренно прочных комплексов в условиях, когда поглощают все компоненты системы. Этот способ требует предварительного определения состава комплекса и молярных коэффициентов светопоглощения исходных веществ.
Для реакции mM + nR MmRn суммарная оптическая плотность аддитивно складывается из составляющих: Лсм = Ак 4-4- Лм 4- 4R. Если Ск пропорциональна А, то при постоянной концентрации CR и переменной См, когда См CR, степень комплексообразования
z = 0см ^м f r^rO/I^V (ек т?м пеи)1
Условная константа устойчивости комплекса
о,____________Ск________________________kCr__________
к“ (См-тСк)т (Cr-яСн)'1 ~ (Cm-wkCr)”1 (Cr — nzCR)n =
(См — mztCR)/fi (1 nx)”CR
Значение е„ определяется из экспериментальных данных при CR = const в условиях большого избытка См (См CR):
Еи ~ 0 см ^m^mM^rO
Если при выбранной длине волны ем = 0, то
Ек = 4cm/(CrZ)
Для определения е„ приготавливают серию растворов с постоянной концентрацией реагента R и переменной концентрацией компонента М в условиях, близких к насыщению (См CR); измеряют оптические плотности полученных растворов и строят
269
Рис. 8.14. Определение оптической плотности раствора графической экстраполяцией иа бесконечно большую концентрацию непоглощающего иона-комплек-сообразователя М.
график зависимости А (АЛ) * = f (1/См) (рис. 8.14). Экстраполяцией прямолинейного участка кривой до пересечения с осью ординат получают искомое значение А (или АЛ), из которого и рассчитывают значение ек (или Ае). Определив ек, рассчитывают х, а затем и Рк.
8.2.8. ГРАФИЧЕСКИЕ ПРИЕМЫ
Определение еи и рк на основе экспериментального определения равновесной концентрации реагента** R [326, 362]. Применяя закон действующих масс и основной закон светопоглощения к реакции образования моноядерного комплекса MRn, без упрощающих допущений получают уравнение8*:
ес₽ = (£м + еЛ lR]")/(1 + ₽к [RD (8-59>
Здесь еСр = — средний молярный коэффициент светопоглощения.
Уравнение (8.59), представленное в виде
(8cP-^)/[R]" = ^k-Mcp (8-60)
выражает прямую линию с тангенсом угла наклона, равным Рн, которая отсекает на оси ординат величину е„Р„.
Для построения графической зависимости (еср— eM)/[R]re от еср приготавливают несколько растворов с известным содержанием компонентов М и R. По измеренному значению оптической плотности раствора вычисляют еср = Л/(СМ/); равновесную концентрацию реагента определяют экспериментально. Этот прием требует предварительного знания стехиометрического коэффициента п.
Прием Франка—Освальда [326, 363]. Этим приемом пользуются для определения ек и Рк простейших комплексов типа MR в условиях избытка реагента. Согласно закону действующих масс
Ck = ₽k(cm-Ck)(Cr-Ck)
Пренебрегая после преобразования значением С|, имеем:
Ск = CMCR/(l/₽; + CR + См) (8.61)
* ДА = ДРм Дм*
** Равновесную концентрацию реагента можно найти из данных рН-метрии, если реагент является анионом слабой кислоты.
Предполагается, что ер = 0.
270
Применяя основной закон светопоглощения Ск — АА/(1 Де) (где Де = ек — ем — eR) при I = 1 см, получим;
_£mCr_ _ 1 См + CR
АЛ ₽;Ае "г Де
(8.62)
Уравнение (8.62) выражает прямую линию с тангенсом угла наклона, равным 1/Де, которая на оси ординат отсекает отрезок, численно равный 1/р« Де.
Приготавливают серию растворов с переменной концентрацией обоих компонентов, но так, чтобы суммарная концентрация См + CR не оставалась постоянной. Измерив оптические плотности растворов, устанавливают графическую зависимость в координатах (СкСм/ДЛ) — (См + CR) и определяют значения Дек и р;.
Прием Шварценбаха [364]. Этим приемом пользуются для определения ек и рй умеренно прочных комплексов типа MR в условиях избытка реагента и отсутствия поглощения реагирующими компонентами.
Если ем = eR = О и Z = 1 см, то выражение условной константы устойчивости Р^ принимает вид *:
₽к = -Г-/(см—r-)(cR-T")
ИЛИ
dr = TT+ ^K(Cr-a/8k) (8-63)
Если CR Ск и CR — А/ек CR, то уравнение (8.63) выражает прямую линию СмМ = / (1/CR), которая отсекает на оси ординат отрезок, приближенно равный 1/ек.
Определив приближенно 1/ек, рассчитывают разность CR — — Л/ек и строят прямую в координатах СмМ — 1/(CR — Л/ев). После этого более точно находят значение 1/ек, а по наклону прямой судят о величине 1/(екРк).
8.2.9. МЕТАЛЛ-ИНДИКАТОРНЫЙ МЕТОД БАБКО
Метод [365] является приближенным, но очень быстрым. Применяется для определения относительной прочности простейших однотипных комплексов различных металлов. Он основан на распределении некоторого количества вспомогательного вещества (иона) между изучаемым металлом и металлом-индикатором.
В качестве индикаторных систем используют комплексные соединения металла-индикатора МиндН, светопоглощение которых обратимо изменяется в зависимости от концентрации в растворе
* Если 8r#=0, то строят графическую зависимость из которой определяют значения Де и 0'.
См — ; /____1
ДД \Cr — ДД/Де
271
вспомогательного вещества R', образующего комплексные соединения как с катионом металла-индикатора, так и с катионами исследуемых металлов. К индикаторной системе добавляют известное количество вещества R', являющегося общим для изучаемых металлов, и измеряют оптическую плотность раствора. Затем добавляют контролируемые количества катионов исследуемых металлов до достижения одинаковых значений оптических плотностей, приближающихся к значению оптической плотности индикаторной системы в отсутствие общего адденда R'. Концентрация катионов металлов должна быть значительно больше, чем концентрация вещества R' (См Сщ).
Зная начальные концентрации вещества R' и добавляемых катионов (Мп М2 и т. д.), вычисляют их равновесные концентрации:
plj] == CMj — CR,; [М2] = СМа — CR, и т. д.
Рассчитав равновесные концентрации добавленных катионов и зная условную константу нестойкости (или константу устойчивости) одного из соединений MR', можно найти условные константы нестойкости любого из комплексов этой системы:
[MJ : 1мг]! [мз]: • • ! Iм,] = : Km3R' ’ • • • : ^мгК'
Если в качестве индикаторной системы использовать слабую кислоту HR, анион R- которой является общим аддендом для изучаемых комплексов металлов, то прочность комплексов можно определить аналогично, зная концентрацию аниона R" и константу диссоциации слабой кислоты HR. Такую индикаторную систем}' можно использовать при низком значении pH среды, koi да pH < р/Сню
Металл-индикаторный метод пригоден для изучения лишь простейших комплексов и сильно ограничен различными побочными реакциями, возникающими в системе.
Результаты исследования становятся более надежными, когда комплексы индикаторного и исследуемого металлов не очень прочные, причем значение Дм R' должно быть в 10—1000 раз меньше, чем Дм.ц'.
8.3. ОПРЕДЕЛЕНИЕ ЗАРЯДА КОМПЛЕКСНОГО ИОНА
Определение заряда комплекса, образованного ионами сильных электролитов. 1. Для определения заряда комплексного иона, образованного по реакции
Mx+ + nRy- <=> [MRn]*-п« используют значение стехиометрического коэффициента, найденного по одному из вышеописанных методов определения состава. 272
Если опыты проводить при постоянной концентрации металла См и при большом избытке реагента (CR Ск), то концентрация комплекса Ск будет определяться его условной константой нестойкости К' и переменной концентрацией реагента CR:
Ск = (См — Св) (CR — пСи)п/К' (8.64)
Поскольку CR Ск, можно принять, что (CR — nCK) CR. Концентрация комплекса Ск = Ск-А/Авр (где А!Авр— отношение измеренного значения оптической плотности к предельной в условиях насыщения при той же концентрации металла См).
Применяя эти упрощения к уравнению (8.64), получим соотношение
MA.₽-7i) = ^/K' которое после логарифмирования превращается в уравнение прямой:
Л
lg ~А---Г = « Cr — 1g К' (8.65)
Тангенс угла наклона этой прямой равен искомому коэффициенту п. Для практического определения п в условиях избытка реагента строят график в координатах 1g ------------lg CR, по
которому и определяют значение п. Можно строить также график в координатах 1g Ск — lg CR, если комплекс количественно экстрагируется из смеси и его концентрация легко определяется. Полученная прямая отсекает на оси ординат величину, численно равную 1g К’ комплекса.
Определение заряда комплекса, образованного слабой кислотой. 1. Если взаимодействие слабой кислоты (реагента) с ионами металлов протекает по уравнению
M₽+ + HmR <=* [МН,n_nR]<₽-"’++ пн+
то константа равновесия выражается соотношением:
С [н+р
Р (СМ — Ск) (CR ~ Ск) aMaHm R
Здесь Ск — равновесная концентрация комплекса [MH,n_nR ](₽-п,+; См — начальная концентрация ионов металла М₽ ; Cr — начальная концентрация слабой кислоты (реагента) HmR.
Концентрация комплекса Ск может быть найдена из соотношения Ск = См- А/Апр (где Авр — предельное значение оптической плотности раствора, когда ионы металла при концентрации См полностью переведены в комплекс; А — оптическая плотность раствора, измеренная в условиях опыта при концентрации металл-иона, равной См)- Заменив Ск на равное ей значение См-^/^пр и используя в опытах эквимолярные растворы
273
соли металла и реагента, получим следующее выражение кон-; станты равновесия:
ЛСМ [Н+]П
К = _________ ЛЛдр[Н+Г
Р aMaHmR^np(CM СМ^^вр)2 СмИпр~ ^)2aMaHrftR
Обозначив —т-з ~4jn?--------- = В и прологарифмировав
это выражение, получим: lg КР = 1g В + п lg [Н+] = 1g В — — п pH. Решая это уравнение относительно неизвестной п для двух опытов, получим [3461:
n = (lgB1--lgBI!)/(pHi-pHK) (8.67)
Для практического определения значения п готовят два раствора эквимолярных концентраций иона металла и реагента (См = CR) и измеряют их оптическую плотность при разных значениях pH раствора. Предварительно определяют предельное значение оптической плотности раствора Лпр той же концентрации См при высоком значении pH. По полученным данным рассчитывают п.
Величины п и /Ср можно определить также и графическим путем, построив прямую в координатах 1g В — pH.
2. Если комплексное соединение образуется по той же схеме, а концентрация реагента значительно больше концентрации комплекса (CR Ск), то для определения заряда комплексного иона, по-видимому, можно использовать упрощенный графический прием.
В выражении константы равновесия
Ск[н+Г
Р (СМ Ск) (CR пСк) aMaHwR
вместо равновесной концентрации реагента используют его начальную концентрацию CR, так как CR Ск. При постоянных начальных концентрациях ионов металла и реагента, равных соответственно См и CR, концентрация комплекса будет зависеть от концентрации ионов Н+. Выразив Ск через равное ей значение См-Л/Лцр и прологарифмировав выражение константы равновесия, получим уравнение прямой в координатах 1g -,-—j —pH:
^пр -
Л
,g лпр —л~ = - ,g KPCRaM“HmR + "РН <8’68)
Тангенс угла наклона этой прямой равен п.
Для построения графика приготавливают серию растворов с постоянными значениями концентраций См и CR при различных pH, но так, чтобы CR Сн. Затем измеряют А и Лпр при различных pH и стр ят график зависимости 1g-з—-—з- от pH.
пр
По углу наклона этой прямой (tg а) определяют искомое зна
274
чение п. Полученная прямая отсекает отрезок на оси ординат, численно равный величине 1g КрСрамантп.
3. В тех случаях, когда при выбранной длине волны наблюдается поглощение света как окрашенным соединением, так и самим реагентом, применяют графический прием Астахова [366, 367]: приготавливают серию растворов эквимолярной концентрации реагирующих компонентов, но с различными значениями pH. Затем измеряют оптические плотности приготовленных растворов Ах и растворов реагента AR при тех же значениях концентраций и pH. По полученным данным строят график зависимости 1g -------рту от pH (где Апр — предельное
(^пр — ™x)
значение оптической плотности раствора, достигаемое в опытах при высоком значении pH). Угловой коэффициент (Ь) построенного графика численно равен значению п.
Если реагент имеет собственную окраску, но известен молярный коэффициент светопоглощения комплекса ек, то для определения числа замещаемых ионов водорода в молекуле реагента можно использовать графический прием Чудинова [368]. Для этого строят график зависимости в координатах
, (екС/ — Лк) _ „
Ig (е«С/-Лж)а Р
для растворов эквимолярных концентраций реагирующих компонентов (/ — толщина слоя раствора, см). Угловой коэффициент (Ь) полученной прямой численно равен числу п замещенных ионов водорода. На оси ординат эта прямая отсекает отрезок, численно равный величине 1g АрСам«н к (где С — концентрация раствора эквимолярной серии).
Если известна константа диссоциации реагента, то можно рассчитать константу нестойкости комплекса:
Kmr = Khr/^p
8.4. ИССЛЕДОВАНИЕ СМЕШАННЫХ (РАЗНОЛИГАНДНЫХ) КОМПЛЕКСОВ
Система, содержащая комплексообразователь М, лиганды R и R' и растворитель S, графически может быть представлена в виде тетраэдра состава [38], основанием которого служит треугольник MRR', а вершиной — растворитель S (рис. 8.15). Каждое сечение тетраэдра, параллельное плоскости треугольника MRR', соответствует тройной изомолярной серии. Любая точка на стороне MR или MR' треугольника отвечает только бинарному соединению. Если в системе образуется тройное (смешанное) соединение, то ему соответствует некоторая точка внутри тре-
275
Рис. 8.15. Тетраэдр состава:
М — комплексосбразователь, R и R' — лигаиды; S — растворитель, M1RJR'— тройная изомолярная серия.
Рис. 8.16. Диаграмма состояния трехкомпонентной системы М—R—R'.
Состав комплекса MR2R' [38].
угольника MRR'. Углы треугольника (рис. 8.16) соответствуют растворам с одинаковой концентрацией всех трех компонентов: М, R и R'. Для получения сведений о составе образующихся комплексов применяют способ построения и интерпретации изохром * в тройной изомолярной диаграмме, предложенный А. К- Бабко [56 J.
На основании полученных диаграмм путем интерполяции находят точки для различных составов с одинаковой оптической плотностью. Соединив эти точки линией, получают изохрому (рис. 8.17). Примеры составления таких диаграмм и интерпретация изохром приведены в литературе [38 , 41, 56].
8.4.1. МЕТОД ИЗОМОЛЯРНЫХ СЕРИЙ
Метод применяют для определения состава разнолигандного комплекса, когда в системе образуется только один такой комплекс. На практике используют два варианта метода.
1. Изменяются молярные отношения металла и одного из лигандов при постоянной их суммарной концентрации; значительно более высокая концентрация второго лиганда при этом сохраняется постоянной во всех растворах изомолярной серии. Метод аналогичен ранее описанному методу определения состава однороднолигандных комплексов. Следует, однако, иметь в виду, что в условиях большого избытка третьего компонента состав разнолигандного комплекса может отличаться от его состава при эквимолярных количествах всех трех компонентов [38].
* Из о хрома—линия, соединяющая в треугольнике состава MRR' точки с одинаковой оптической плотностью (или одинаковой разностью ДД).
276
Рис. 8.17. Диаграмма состояния трехкомпонентной системы (с изохромами) ион меди — хинолин — салицилат-ион [38].
Состав комплекса 1:2: 2. СНСЦ, 0,05 М растворы, X — 670 нм.
2. Изменяются молярные соотношения всех трех компонентов при соизмеримой их концентрации. Для исследования состава в условиях примерно одинаковой концентрации компонентов
используют метод тройной диаграммы [38, 41J. Обязательным условием при этом является постоянство суммарной концентрации всех трех компонентов. Для большей надежности измерения оптической плотности проводят при различных длинах волн, сохраняя неизменными все прочие условия (pH, ионная сила и т. п.). Анализ изохром в тройной диаграмме целесообразно дополнять изучением наиболее характерных разрезов тетраэдра состава. Так, при образовании разнолигандного комплекса MR;R[-более точные данные о составе комплекса можно получить при ;пализе разрезов MR,-—R' или MR[—R. Этот вариант метода целесообразно использовать в тех случаях, когда прочность связи центрального атома с обоими лигандами примерно одинаковая.
Метод изомолярных серий удобнее использовать в экстракционном варианте, когда в органическую фазу экстрагируется только разнолигандный комплекс, а однороднолигандные комплексы не экстрагируются.
84.2. МЕТОД СДВИГА РАВНОВЕСИЯ
Метод применяют для определения состава разнолигандного комплекса в условиях, когда изменяется концентрация только одного из компонентов, а концентрации двух других компонентов постоянны (pH и ионная сила тоже постоянны). Можно использовать различные варианты этого метода, которые применяют при исследовании однороднолигандных комплексов. Так, по кривым насыщения можно найти число молей лиганда при образовании смешанного комплекса по схемам:
MR;-HR' <=> mr;r: (8.69)
MR;+/R <=> MR,R: (8.70)
Применение обычного варианта метода сдвига равновесия (см. разд. 8.1.2) к уравнениям (8.69) и (8.70) позволяет определить стехиометрические коэффициенты j и i. Первый из них определяется [MR.R.'i
как тангенс угла наклона зависимости lg r )C. ' — от lg lR L
277
[MRR'l
а второй — из зависимости 1g j = f (lg [R' ]). Отношение концентраций [MRyRJ ]/[MRj] \ [MRjR; ]/ [MR(- ] в соответствии с основным законом светопоглощения можно заменить адекватным отношением оптических плотностей: Л/(Лпр — А), где А — оптическая плотность анализируемого раствора; Лпр — предельное значение оптической плотности раствора, достигаемое в условиях «насыщения». Равновесная концентрация реагента определяется по разности: [R] = CR — jCK или [R'] = = CR- — iCK. Если CR.CR-»CK, то [R]»Cr, [R'1«Cr-. Аналогично можно использовать и другие варианты метода сдвига равновесия (методы Асмуса, Гарвея и Меннинга, Старика и Барбанеля и др.).
8.4.3. МЕТОД НЬЮМЕНА И ХЬЮМА
Метод [326, 331 ] применяют для исследования смешанных комплексов MRjRz в условиях, когда образуется не более шести комплексных форм, причем две крайние из них являются однороднолигандными с каждым из лигандов в отдельности (MRm и MR„). Метод применим при условии, что координационная емкость центрального иона (атома) М остается неизменной, а координационные числа лигандов R и R' могут быть как одинаковыми, так и различными. Применение метода требует соблюдения таких условий, при которых диссоциация обоих однороднолигандных комплексов практически подавлена. В основе метода Ньюмена и Хьюма лежат реакции обмена лигандов при образовании разнолигандных комплексов:
MR; + sR (8.71)
mr;_?_prs+w + pr' mr;_,R54 ^R (8.72)
Здесь s, п, q, р, w — стехиометрические коэффициенты.
Различие стехиометрических коэффициентов в реакциях (8.71), (8.72) указывает на то, что лиганды R и R' обладают различной координационной емкостью.
Согласно закону действующих масс константы равновесий (8.71), (8.72), характеризующие прочность образующихся разнолигандных комплексов, будут равны:
кп = [mr;j ir]7[mr;_,rs] irf м
Kn-q = [MR;_,RS] [R]7'[MR;_,_pRs+J [R'p (8.74)
Если все три комплексные формы MRn, MR^_, Rs и MR^-f-pRs+tti поглощают свет при выбранной длине волны X, то согласно правилу аддитивности для раствора, содержащего все указанные комплексы, получим (при I = 1 см):
А = гп Р^л] + еп—q [MRn-^Rs] + en—q—p [MRn—q—pR 54-01] (8.75)
278
Здесь en, en_g, en_g_p — молярные коэффициенты поглощения соединений MR„, MR„_gRs и MRg_g_pRs+tt, соответственно.
Значение еп следует определить заранее (из отдельных опытов) в растворе, содержащем комплексообразователь М и избыток лиганда R':
®„ = 71O/C^ = 71o/[MR;] (8.76)
Здесь Д° и — оптическая плотность и концентрация компонента М в отдельном опыте.
В присутствии избытка лигандов R и R' (Cr, Cr- См) условие материального баланса для комплексообразователя М можно выразить уравнением:
См = [mr;] + [MR;_gRs] + [mr;_?_prs+№] (8.77)
При достаточно большом избытке обоих лигандов, когда концентрация не связанного в комплекс металла очень мала, ею можно пренебречь, поэтому она и не учитывается в уравнении (8.77). При этих условиях можно принять также, что [R] « CR и [R'] ж Cr'. Решая совместно уравнения (8.73)—(8.76) и учитывая условие материального баланса (8.77), получим значения равновесных концентраций разнолигандных комплексов:
[MR,',. eRs] =______________А-гп_д_рСм__________________
*n-q + (л°/см) kmcsr - еп_ч_р [1 + (ад./с*)]
rMR. р х - См - А + (*Л'/<щ)]
I q—p's-J-w)
KnC^,/CsR-en^_p [1 + (K„C’,/CSR)] (8.79)
Подставив затем в выражение (8.74) значения концентраций комплексов из уравнений (8.78) и (8.79), получим:
Л = *„-,{[*„ (AnCK/C°K-A) (Cl^)-A+en_qCK] (С^/С-)}+е„_д_рСм
(8.80)
На основе уравнения (8.80) можно определить константу равновесия реакции (8.72) и молярный коэффициент светопоглощения разнолигандного комплекса MRn_9_pRs+te, путем построения графической зависимости А от выражения в фигурных скобках. Эта зависимость, если правильно заданы значения р и w, выражается прямой линией, у которой b = Kn-q и которая на оси ординат (Д) отсекает отрезок, численно равный en_g_pCM. Однако, прежде чем строить указанную графическую зависимость, необходимо найти значения Кп и en_g первого разнолигандного комплекса MR„_CRS. Эти неизвестные величины нахсдят на основе уравнения, аналогичного уравнению (8.80), но полученного при условиях, когда в растворе существуют только две комплексные формы MRn и MRA-<zRs:
А = Кп [(А°СМ - А) (С*,/(-)] + Е„_дСм
(8.81)
279
Значения См, CRi, Cr, известны, А и А° — определяют экспериментально, выбрав длину волны, при которой поглощают только два комплекса: MR„ и MRA-9RS. Построив графическую зависимость А от выражения в квадратных скобках уравнения (8.81) при разных значениях q и s, выбираем такие значения этих коэффициентов, при которых зависимость получается прямолинейной. Тангенс угла наклона этой прямой будет соответствовать значению К.п\ на оси ординат эта прямая отсекает отрезок, численно равный значению en_QCM.
Если применяют лиганды R и R' с одинаковой координационной емкостью (q = s и w = р), то равновесия (8.69), (8.70) принимают такой вид:
mr;_,r, + </R' mr; + <?r (8.82)
mr;_,_pr,+p + ₽R' <=+ mr;_^ + pr <8.83)
Тогда уравнения (8.80) и (8.81) упрощаются
* = Kn-q {[*„ (А^/С^-А) (CR./CR)<- A +en_QCM] (CR,/CR)₽} +
(8.84)
Л = К„ [(Л°СМ/С«Ч - Л) (CR./CR)q + ел_рсм (8.85)
и их можно использовать для определения состава, констант устойчивости и оптических характеристик разнолигандных комплексов, как это описано выше.
8.5. ОПРЕДЕЛЕНИЕ КОНСТАНТ ДИССОЦИАЦИИ ОРГАНИЧЕСКИХ РЕАГЕНТОВ
Методы определения констант диссоциации органических реагентов основаны на совместном решении уравнений основного закона светопоглощения и закона действующих масс.
Большинство методов позволяет рассчитать лишь кажущуюся константу диссоциации реагента, так как она зависит от ионной силы раствора и только при бесконечном разбавлении становится равной истинной величине. Для определения истинного значения константы диссоциации либо дополнительно определяют коэффициенты активности составляющих протолитического равновесия, либо, получив ряд кажущихся значений констант диссоциации при различной ионной силе раствора, применяют графическую экстраполяцию на нулевую ионную силу.
Если органический реагент диссоциирует как одноосновная кислота
HR а~> H+ + R-
то суммарное значение оптической плотности раствора Лсм аддитивно складывается из поглощения обеими формами реагента:
Лсм = fhrc/ 0 ~ *) + гяс1х (8-88)
280
Здесь ер и 6hr — моляриь,^ коэффициенты светопоглощения солевой и кислотной форм реагента; х и (1 — х) — мольные доли этих форм реагента; С — общая молярная концентрация реагента.
В зависимости от способа совместного решения уравнения (8.86) и уравнения закона действующих масс различают несколько вариантов определения кажущейся константы диссоциации реагента :
KaHR = IH'l lR~WHR]
Расчетный способ [369, 370]. Подставив в выражение константы диссоциации реагента величины С, х и (1 —х), получим:
_ [Н+] Сх _ Kohr _ [Н+]
с (1-Х) ИЛИ [H+] + XoHr’ ° ’ } [Н+] + KaHR
Подставляя найденные концентрации кислотной и солевой форм реагента в уравнение основного закона светопоглощения (8.86), получим выражение:
д ___.. 1С [Н+]_________I р /с ^chr
см - eHRiL [H+J + KOHR + [Н+] + КоНр
(8.87)
Здесь fiirZC = Дни и eRZC = Др — оптические плотности раствора реагента соответственно в кислотной и солевой формах при выбранной длине волны, концентрации реагента С и толщине поглощающего слоя I.
Решая уравнение (8.87) относительно неизвестной KiHR, получают выражение:
K«hr = <лсм Лни) [Н*]/(Дr Дем) (8.88)
Величины t4Hr и >4r измеряют при выбранной длине волны в условиях, когда реагент полностью находится в кислотной и солевой формах соответственно. Измерив значение оптической плотности смеси равновесных форм реагента и определив pH раствора, рассчитывают константу диссоциации.
Для практического определения константы диссоциации готовят ряд растворов реагента, сохраняя постоянной его общую концентрацию и изменяя значение pH в широких пределах. Снимают спектры поглощения приготовленных растворов, используя кювету с постоянной толщиной слоя. Для расчетов применяют ту область длин волн, в которой наблюдается наибольшее различие в величинах Лсм, >4HR и /R.
Графический способ. Графический способ основан на определении pH среды, отвечающей 50%-ной диссоциации реагента, когда ЛаНц = 1Н+ 1- Эта точка соответствует полусумме значений оптических плотностей кислотной и солевой форм реагента при одинаковой концентрации (рис. 8.18)
•^см = (71hr + Др)/2
XaHR = Исм— ^hr) [Н+]/(Дц —Дсм) = (Дц — Дни) [Н+]/(Дц — Дни) = [Н+]
(8.89) откуда рЛа = pH.
281
Рис. 8.18. Графическое определение рКа кислоты по кривой зависимости А от pH.
Для графического определения кажущейся константы диссоциации реагента снимают спектры поглощения нескольких растворов при разных pH и определяют длину волны, при которой происходит наибольшее изменение оптической плотности при диссо
циации реагента.
Затем измеряют оптическую плотность раствора реагента при выбранной длине волны, изменяя pH таким образом, чтобы реагент
при этом полностью переходил из кислотной формы в солевую (или наоборот). Построив график зависимости А от pH, находят точку на кривой, отвечающую полусумме крайних значений: А см = 2/(AHR + AR). Абсцисса этой точки и соответствует значению pAa.
Упрощенным вариантом этого способа является графическое
определение константы диссоциации по кривым спектрофотометрического титрования. Графический способ можно применить и к многоосновным кислотам при условии достаточно большого различия в значениях молярных коэффициентов светопоглощения и констант диссоциации всех равновесных форм многоосновной
кислоты.
Определение Аонк реагента по значению молярного коэффициента светопоглощения его солевой формы eR [371 ]. Приготавливают несколько растворов реагента одинаковой концентрации CR с различными значениями pH и измеряют оптическую плотность при длине волны максимального светопоглощения солевой формы реагента. Значение кажущейся константы диссоциации реагента определяют по формуле:
— 1н+]л I [eRZ (cr е/
(8.90)
Метод Комаря [372]. Метод Комаря является универсальным. В отличие от других методов его можно применять и в тех случаях, когда практически невозможно получить точные данные о поглощении кислотной и солевой форм реагента из-за наложения их спектров поглощения. Метод не требует предварительного знания значений молярных коэффициентов светопоглощения (или соответствующих им значений оптических плотностей) кислотной и солевой форм реагента в области их максимального светопоглощения.
Приготавливают несколько растворов с постоянной концентрацией реагента CR и различными значениями pH и измеряют их оптические плотности при выбранной длине волны. Если реагент 282
диссоциирует как одноосновная кислота HR Н+ + R”, то его константа диссоциации определяется соотношением:
К =С хНС — х) (8.91)
°HR н /
Здесь Сн — концентрация ионов водорода.
Согласно основному закону светопоглощения, суммарная оптическая плотность равновесных форм реагента равна:
А = eHR/ (С — х) + eR/x (8.92)
Решая совместно уравнения (8.91) и (8.92) относительно х, получим уравнение
ЛК — К есС1 = сн8н„с/ — С„А (8.93)
в котором три неизвестных: KoHR, eHR и eR.
Применяя уравнение (8.93) к данным трех опытов, получают систему 3-х уравнений с тремя неизвестными, решение которой позволяет определить каждую из неизвестных величин отдельно:
_ (Сн ~ Сн) (Л'СН ~ ЛПС£) - (СЬ - С”н) (A«Cj, - А*С* )
“HR (Л. _ ЛА) _ Сп^ Ап) (С>н _ СЛ )
1 (Л* — Ak) (А‘С1Н — АпСЪ) — (Ас — Dn) (Л‘Сц — AkC^)
ЁнК = -сГ (л < _ ЛЛ) (С^ _ с«н) _ (Л* _ лп) _ Ckj
(8.94)
(8.95)
А‘А*Спн (С‘н - CkH) + AftA"Cj, (С*н - С" ) - А‘АпС^ (С‘н - С£) С' [(с^ - с*) (А‘С‘Н - А"С"Н) - (с£, - С^) (Л‘с£, - л*с*)1
Здесь I, п, k — номера опытов.
Если можно выбрать такой участок спектра, где поглощает только одна из равновесных форм реагента, то расчеты упрощаются. Допустим, что при выбранной длине волны поглощает только солевая форма реагента R, тогда
?HR = 0 и A = eRxl нли x=A/(eR/)
Подставив полученное выражение концентрации х в уравнение константы равновесия (диссоциации), получим уравнение с двумя неизвестными — KaHR и er:
к _ cHA/(eRl) ни С —A/(eR/)
(8.97)
Решая это уравнение по данным двух опытов, получим простые расчетные формулы для KaHR и eR:
КанК = (Л‘С^-^)/(АЛ-Д1')
eR = A‘Ak (С^- CkH)/[Cl (А‘С‘Н - AftC‘)] (8.98)
283
Метод Комаря можно использовать также для спектрофотометрического определения pH раствора. Зная KOhr, eR и начальную концентрацию реагента CR, по измеренному значению А можно вычислить равновесную концентрацию Н+.
Спектрофотометрическое определение констант диссоциации двухосновных кислот представляет собой более сложную задачу и является предметом специального рассмотрения в работе [370].
8.6. РАСЧЕТ СОСТАВА, КОНСТАНТ УСТОЙЧИВОСТИ, КОНСТАНТ ДИССОЦИАЦИИ И МОЛЯРНЫХ КОЭФФИЦИЕНТОВ СВЕТОПОГЛОЩЕНИЯ ФОТОМЕТРИРУЕМЫХ СОЕДИНЕНИЙ
8.6.1. ПРИМЕРЫ И ЗАДАЧИ
Пример 1. Рассчитать состав комплекса 14 с ксиленоловым оранжевым методом молярных отношений по следующим данным
Cr-105, моль/л 0,12 0,16 0,20 0,40 0,60 0,80 1,00 1,20 1,48 1,80
А 0,040 0.050 0 065 0,130 0 200 0,270 0,315 0,360 0,365 0,370
Постоянная концентрация никеля 1-10~5 моль/л.
Решение. Эквивалентная концентрация реагента соответствует точке излома кривой насыщения, которая определяется как точка пересечения пря-мочинейных участков обеих ветвей кривой насыщения. Точку излома кривой насыщения рассчитывают решением системы уравнений обеих прямых относи-течьно общей точки
А = а + 6Cr
A = d
откуда CR = (d — a)ib Две точки, предшествующие прямолинейному участку А = d, отбрасывают, полагая, что они не лежат ни на одной из прямых. Сред-п
нее значение величины
АПр = 0,365. Параметры а н b уравнения
J
А = а + ЪСц рассчитывают по первым пяти точкам методом наименьших ква-
дратов
£ с] 2 Ъ 1
6 10"11 0,485— 14,8-Ю'в 1П7,8 10~8
5 60 10'12--218,5 10-12 “'
« £ CtAi— £ С( JIАг 5-197,8 IO”8 — 14,8-10“8-0,485
" L —(1j C<)2 5 60 10 12 — 218,5-10~12 “
_ d — a 0,365 + 0,002 , , ln . , . Cr l.l-lO’8
Cr b 3,33.104 1,1-10 - моль/л, «-Cni uq-s - 1.1
т. e. состав анализируемого соединения 1 : 1 (NiR).
284
Пример 2. Рассчитать отношение стехиометрических коэффициентов, характеризующих состав комплекса MmRn, методом отношения наклонов по следующим данным:
См- 10е (при Cr = const), моль/л 2,2 4,3 6,5 8,6
А, 0,103 0,215 0,316 0,407
CR.106 (при См= const), моль/л 0,43 0,86 1,30 1 71
Аг 0,091 0,196 0,296 0Д88
Постоянные концентрации компонентов М и R, условия приготовления растворов и измерения оптических плотностей для обеих серий опытов одинаковые.
Решение. Лх= zlC^Jm — bjC^; А2 = elC#/n — 62Cr; Ь2/Ь2 ~ п/т.
По приведенным данным методом наименьших квадратов рассчитывают угловые коэффициенты (Ь), характеризующие прямолинейную зависимость Ах от См н А2 от Ср:
п Г CtAi- J] С; £ At
4-0,67-10~® — 2,16-10"®•1,04
4 1,39-10-10 — 4,66-iO-10 ’
4-1,257.10-®— 4,30-10"®-0,971
bi~ 4-5,54-IO"10 — 18,49-IO"10 ’ 10
n 4,78-lQ4 _
m b2 2,34-104
Следовательно, отношение компонентов M : R в составе комплекса равно 1 : 2.
Пример 3. Для определения истинного значения молярного коэффициента светопоглощения комплекса никеля с ксиленоловым оранжевым состава 1 : 1 используют метод Клотца. Значения оптических плотностей растворов с постоянной концентрацией реагента 9,9-10"» моль/л, измеренные прн к — 584 нм в кювете с толщиной слоя 1 см, изменялись в зависимости от концентрации никеля следующим образом:
CNi, моль'л 9,9-10-® 1,1.10-® 1,37.10-^ 1,79-10-6 2,0-10-5
А 0,320 0,330 0,335 0,340 0,345
Рассчитать по этим данным истинное значение молярного коэффициента светопоглощения комплекса.
Решение. Составляют зависимость А от 1/СКм:
А 0,320 0,330 0,335 0,340 0,345
(1/CN1). IO"4 10,1 9,1 7,3 5,6 5,0
Экстраполяцией на бесконечно большую концентрацию никеля (l/C^j » 0) находят значение оптической плотности (А8КСтр), из которого и рассчитывают молярный коэффициент светопоглощения.
При Cni Cr зависимость А — [ (1/Cni) выражается прямой линией, которая отсекает на оси ординат отрезок, численно равный Аэкстр. Для его определения решают уравнение А = а+ b/C^i и по методу наименьших квадратов рассчитывают параметр а = Аэкстр:
в £(1/^1! А'-УКу^.Х-ЕИ/М»
« S(VCNi)?-[S(l,CNi)l2 2,94-Ю10-1,67 — 3,71-10®. 1,23-10® _ _ 5-2,94-1010— 13,76-101°
285
Затем рассчитывают значение Enir:
eNiR = ^bctp'W) = 0,37/(9,9. Ю-e-1) = 3,74- 10*
Пример 4. Рассчитать методом Толмачева — Комаря молярный коэффициент светопоглощения и константу устойчивости сульфосалицилатного комплекса железа (III), образующегося по реакции:
Fe3+ + 3H2R- =г± [FeR3 ]в- + 6Н+
Зависимость между концентрацией железа и оптической плотностью растворов, измеренной при X = 430 нм в кювете с толщиной слоя 2 см, характеризуется следующими данными:
Сре. 10®, моль/л 2,30
А 0,08
pH растворов равен 8,0; константы лоты Кп = 1,4-10-» н К„ = 1,8-10-12.
С2 °5
4,00 6,60
0,18 0,36
диссоциации
8,00 10,00 12,00 16,00
0,50 0,64 0,80 1,10
сульфосалициловой кнс-
Решение.
± = _L+C_!_ 8
hl 1
’ РК К*
В данном примере п = 3; h = 1-Ю'8 моль/л; I — 2 см; К„ = К„ .
По данным зависимости (Cl/A)i от (1/A3^4)j, методом наименьших квадратов рассчитывают значения 1/ек и Q, по которым вычисляют ек и рк:
ек
Кр =
1
ек
68,05-254,00-10'®— 17,55.773,90-10-6 „
2,2-10-*
7-68,05 — 308,0
7-773,9-10-®— 17,55-254,0-IO-5
7-68,05 — 308,0
5,66-10 «
ек= 1/(2,2- IO'*) = 4,55 10»
Кр = (1 10-8-2/3)» {1/[4,55-10' (5,66-10~6)4} = 6,37- 1(ГИ
₽к = 6,37-10~12/( 1,4- IO’» -1,8- 10“12)3 = 3,98-10»2
Пример 5. Рассчитать методом Комаря кажущуюся константу диссоциации реагента HR и значения молярных коэффициентов светопоглощения обеих форм реагента по следующим данным:
Номер опыта
pH А
С k п
2,96 7,60 9,80
0,017 0,516 0,705
Концентрация реагента во всех растворах одинакова — Chr — 2,14-10_₽ М.
Оптическая плотность растворов измерена в кювете с толщиной слоя 1 см.
П
2
286
Решение. h= Сн+ моль/л; I = 1 см; CHR = С;
„ ___(fit hk) {Aihi Anhn) {hi hn) (Л tht — Лййй)
Л°- (Лг-Лй)(Л{-Лп)-(Лг-Л„)(Лг-Лй)
(1,1-IO-8 — 2,51 • IO"8) (0,017-1,1 • 10-3 — 0,705-1,58-10'“) —
— (1,Ы0-3— 1,58-10-“) (0,017-1,1-Ю'8 — 0,516-2,51-10-8)
= (0,017 —0,516) (1,1-IO'3—1,58-10'“) — -6,79-10
— (0,017 — 0,705) (1,1- IO'3 — 2,51 • IO’8)
„ _ 1 (Л; Лй) (Ajhi Anhn) (Л; Лп) {Ajhj A^hk) ______________1_____
HR- C7 (Ai-Ah)(hi-hn)-{Ai-An)(hl-hh) ~ 2,14-10'5-1 x
(1,7-10-2 — 0,516) (0,017-1,1 IO'3 — 0,705-1,58-IO'10) —
— (0,017 — 0,705)(0,017-1,1•IO'3 — 0,516-2,51 - IO'8)
X (0,017 — 0,516) (1,1 -IO"3— 1,58-10-“)— “ 792
— (0,017 — 0,705) (1,1-10-3—2,51- IO'8)
„ Л{Лййп (h; hjj) AfrAnht (h^ hn) AjAnhk (hj hn) ____________
~ Cl [(hi - hh) (Aihi - Anhn) - (йг - hn) (Aihi - ЛйАй)] ~
0,017-0,516-1,58- IO'10 (1,1 - IO'3 _ 2,51.Ю'8) +
+ 0,516-0,705-1,1 - IO’3 (2,51 • IO'3 -1,58-10'“) —
— 0,017-0,705-2,51-IO'8 (1,1 • IO'3 — 1,58.10"“)
2,14-10-3-l [(1,1-10-3 —2.51-10-8) (0,017-1,1-10'3 —0,705-1,58-10-“)— =
-(1,1- IO'3 — 1,58-10-“) (0,017-1,1 • IO'3 — 0,516-2,51 • 10 е)]
= 3,2-10'’
Пример 6. Рассчитать кажущуюся константу диссоциации фотометрического реагента HR и молярный коэффициент светопоглощения его солевой формы в условиях, когда молекулярная форма HR света не поглощает. Концентрация реагента С = 2,00-10'4 М. Значения оптической плотности растворов, измеренные в кювете с толщиной слоя 1 см, при pH 7,32 и 7,75 равны соответственно 0,785 и 0,996.
Решение.
= Ajhj-Ahhh = 0,785-4,78-10-з-0,996-1,78-Ю-з = g 3g jQ_8
° Ak — Ai 0,996 — 0,785 ’
.______!_ AjAk(hi-hk) __
R Cl ’ Aihi — Ahhh
0,785-0,996 (4,78-IO'8—1,78-IO'8) , ,
“ 2-10'4 (0,785-4,78-IO'8 — 0,996-1,78-10~8) ’ b’1U
Пример 7. Рассчитать кажущуюся константу диссоциации фотометрического реагента HR по следующим данным. Оптическая плотность раствора реагента при pH = 7,33 равна 0,442. Оптические плотности тех же растворов, когда реагент находится полностью в солевой н кислотной формах, равны соответственно 0,705 и 0,017.
Решение.
« - ih*i - •4-67- '«’=’-ss- >»-•
Задача 1. Рассчитать состав хелатного соединения Cu(II) с диэтилдитиокар-баминатом натрия (ДДК) методом молярных отношений по следующим данным:
СнаДДК"10®, °-85 *.7’ 2-56 3,42 4.28 5-14 6>42 8>56 10-70 12>84 моль/л
А 0,091 0,196 0,296 0,388 0,458 0,502 0,520 0,525 0,531 0,533
287
Постоянная концентрация меди в растворах равна 2,2-Кг6 моль/л.
Ответ: Си(ДДК)2, т. е. Си : ДДК равно 1 : 2.
Задача 2. Рассчитать состав хелатного соединения РЬ(П) с пиридилазорезорцином (ПАР) методом молярных отношений по следующим данным:
Спар- Ю5, моль'л 0,85 1,70 2,50 3,40 4,30 5,20 6,40 8,60 10,70 12,80
А 0,18 0,39 0,59 0,78 0,92 1,00 1,04 1,05 1,06 1,04
Постоянная концентрацья свинца в растворах равна 4,4-10-5 моль/л.
Ответ: PbR, т. е. РЬ : ПАР равно 1 : 1.
Задача 3. Рассчитать состав комплекса Zn с ксиленоловым оранжевым методом отношения наклонов по следующим данным:
Czn-Ю5 (при Cr = const), моль/л 0,86 1,72 2,60 3,42
А 0,205 0,430 0,630 0 810
Ср.106 (при CZn = const), моль'л 0,86 1,72 2,60 3,42
А 0,182 0,388 0,592 0,776
Постоянные концентрации компонентов н условия проведения опытов для обеих серий одинаковые.
Ответ: ZnR (1 : 1).
Задача 4. Рассчитать состав комплекса Fe (II) с 1-нитрозо-2-нафтолом методом отношения наклонов по следующим данным:
Сре- 10е (при Со = const), моль/л 1,4 2,9 4,3 5,7
А 0,10 0,22 0,32 0.41
Ср. Ю6 (при Сре = const), моль/л 0,43 0,86 1,30 1,71
А 0,095 0,196 0,296 0,390
Ответ: FeR3 (1 : 3).
Задача 5. Для определения истинного значения молярного коэффициента светопоглощения комплекса свинца с пиридилазорезорцином (ПАР) состава PbR применяют метод Клотца. Значения оптических плотностей растворов с постоянной концентрацией реагента 2,0-10-5 моль/л, измеренные в кювете с толщиной слоя 1 см, изменялись линейно в соответствии с изменением концентрации свинца следующим образом:
Срь, моль/л 5-10-6 1-Ю"6 1,5.10“Б 2.10-6 3-10~6
А 0,55 0,70 0,75 0,77 0,80
Рассчитать по приведенным данным истинное значение молярного коэффициента светопоглощения комплекса свинца с ПАР.
Ответ: = 4,18-104.
Задача 6. Рассчитать методом Толмачева — Комаря молярный коэффициент светопоглощения и константу устойчивости диэтилдитиокарбамината меди(П), образующегося по реакции Си2+ + 2ДДК Си(ДДК)2, используя следующие данные
С1Ц.1О5, моль/л 1,15 2,00 3,30 4,00 5,00 6 00 8,00
А 0.04 0,09 0.18 0.25 0,32 0.40 0 5
Оптическая плотность растворов измерена при X = 460 нм в кювете с толщиной слоя 3 см.
Ответ: ек = 2,95-103; ₽к = 3,33-10».
Задача 7. Рассчитать методом Толмачева — Комаря молярный коэффициент светопоглощения и константу устойчивости ннтрозонафтолата ксбальта (III) по следующим данным:
ССо. 10е, моль/л 2,30 4,00 6,60 8,00 10,00 12,00 16,00
А 0,08 0,18 0,36 0,50 0,64 0,80 1,10
Питрозонафтолат кобальта (III) образуется по реакции: Со3+ + 3HR 4^CoR3+3H+. Оптические плотности растворов измерены при X = 360 нм, pH = 6 в кювете с толщиной слоя 2 см. Константа диссоциации 2-нитрозо-1-нафтола равна 6-Ю-8.
Ответ: ек = 4,55-104; ₽к = 2,95-1018.
288
Задача 8. Рассчитать методом Комаря кажущуюся константу диссоциации (первой ступени) и молярные коэффициенты светопоглощения обеих форм ди-метилглиоксима (H2R н HR-) по следующи! [ данным:
Номер опыта i k п
pH 2,00 10,00 12,00
А 0,10 0.20 1,00
Оптические плотности 5-10“4 М раствора диметилглиоксима измерены при к = 250 нм в кювете с толщиной слоя 1 см.
Ответ: Ка = 2,38- IO-11; eHR = 200; eHR_ = 1,05-103.
Задача 9. Рассчитать методом Комаря кажущуюся константу диссоциации ксиленолового оранжевого (по четвертой ступени) и молярные коэффициенты светопоглощения обеих форм реагента (H3R3- и H2R4-) при к = 5b0 нм. Зависимость оптической плотности 2,14-10-5 М растворов реагента от pH следующая:
pH 2.96 5,80 7,33
А 0,019 0,085 0,472
Оптические плотности измерены в кювете с толщиной слоя 1 см. Ответ: KaHR3_ = 2,1-10-7; e4jR3_ = 8,8-Ю2; eHoR1-= 2,7-104. Задача 10. Рассчитать методом Комаря кажущуюся константу диссоциации 2-нитрозо-1-нафтола и молярный коэффициент светопоглощения солевой формы реагента по данным фотометрирования при к — 440 нм, где светопоглощением молекулярной формы реагента можно пренебречь. Значения оптической плотности 2,5 X 10-5 М раствора реагента, измеренные в кювете с толщиной слоя 3 см, прн pH 7,3 и 9,8 равны соответственно 0,44 в 0,80.
Ответ: Ка = 5,7-10-8; е= 1,06-104.
Задача 11. Рассчитать кажущуюся константу диссоциации водорода оксим-нон группы нитрозо-К-соли по следующим данным. Оптическая плотность раствора реагента при pH = 7,2 равна 0,44. Оптические плотности раствора реагента, находящегося полностью в молекулярной и солевой формах, равны соответственно 0,02 и 0,79.
Ответ: Ка — 9,5-10-8.
8.6.2. ПРАКТИЧЕСКИЕ РАБОТЫ
В зависимости от особенностей образования окрашенного соединения для его исследования используют наиболее целесообразные методы.
В качестве примера приведем несколько практических работ, иллюстрирующих применение отдельных методов к исследованию окрашенных соединений. Предлагаемые практические работы будут также полезны для приобретения навыков спектрофотометрических исследований.
Работа 1. Определение состава соединения железа(Н) с 1-нитрозо-2-нафтолом методом изомолярных серий
Выполнение работы. Приготавливают эквимолярные растворы сульфата железа(Ш) и 1-нитрозо-2-нафтола концентрацией 5,0- 1(Г4 М. Затем смешивают эти растворы в соотношениях от 10 до 90% так, чтобы общий объем оставался постоянным — 10 мл. Приготовленные растворы изомолярной серии фотометри-руют в интервале длин волн 600—800 нм, и по экстремальной точке на изомолярных диаграммах определяют состав нитрозонафтолата железа(П).
10 М. И. Булатов и др.
289
Приготовление каждого раствора изомолярной серии для фото-метрирования производят следующим образом. В делительную воронку на 100 мл вводят 5 мл 1% раствора винной кислоты, v мл раствора сульфата железа(Ш), 5 мл 10% раствора аскорбиновой кислоты, разбавленный раствор аммиака (1 : 10)дорН=7,5 (объем аммиака определяйся заранее в параллельной пробе нейтрализацией по феноловому красному) и (10 — п) мл водно-спиртового раствора 1-нитрозо-2-нафтола. Через 30 мин прибавляют 25 мл изоамилового спирта и извлекают питрозонафтолат железа (II) в органическую фазу при интенсивном встряхивании в течение 3 мин. Полученный экстракт фильтруют через комочек ваты (для удаления взвешенных капель воды) в мерную колбу на 25 мл и в случае необходимости растворителем доводят объем до метки. Оптическую плотность экстракта измеряют на спектрофотометре при длинах волн 640 и 700 нм в кювете с толщиной слоя 2 см относительно экстракта холостой пробы.
По данным измерений оптической плотности строят изомоляр-ные диаграммы «оптическая плотность — состав изомолярного раствора» при 640 и 700 нм и определяют стехиометрические коэффициенты реакции железа (II) с 1-нитрозо-2-нафтолом.
По литературным данным [67, 373], состав комплекса нитрозо-нафтолата железа (II) отвечает формуле FeR3.
Реактивы
1-Нитрозо-2-нафтол, 0,0005 М раствор.
Fe2(SO4)3, 0,0005 7И раствор.
Винная кислота, ч. д. а., 10% раствор.
Аскорбиновая кислота, ч. д. а., 10 % раствор. NH3, раствор (1 : 20).
Изоамиловый спирт, ч. д. а.
Работа 2. Определение константы кислотной диссоциации нитрозо- R-соли
В нитрозо-R-соли водород оксимной группы способен к диссоциации:
N—ОН NO-
ll О || О
NaO3S—C3X^“SO3Na NaO3S + Н*
Максимум поглощения недиссоциированной формы находится при 260—265 нм, диссоциированной — при 420—430 нм. Поэтому для измерений светопоглощения можно использовать спектрофотометры СФ-16, СФ-26 или СФ-14.
Вылолнение работы. Готовят серию растворов реагента постоянной концентрации (5-10“4 М) в интервале значений pH — = 34-12.
Постоянство ионной силы растворов (/ 0,5) поддерживается
добавлением буферного раствора и перхлората натрия так, чтобы 290
суммарное значение произведений C,Z? для всех компонентов раствора оставалось постоянным.
Приготовленные в мерных колбах на 50 мл растворы фотоме-трируют при длине волны максимального поглощения одной из форм, и по найденным значениям оптических плотностей рассчитывают константу диссоциации нитрозо-И-соли (см. разд. 8.5.1).
По литературным данным [374], Ка водорода оксимной группы нитрозо-И-соли равна l-10~z.
Реактивы
Нитрозо-Ъ-соль, 0,0005 М раствор.
Ацетатные буферные растворы (pH = 34-7).
4>осфаи1ные буферные растворы (pH = 74-Ь).
Тетраборат натрия, 0,005 Л! раствор (pH = 9,24).
Боратные буферные растворы (pH = 94-11).
NaOH, 0,1 М раствор.
Работа 3. Исследование реакции ионов цинка с ксиленоловым оранжевым [375]
Трифенилметановый краситель ксиленоловый оранжевый (КО) является шестиосновной кислотой H6R, которая при pH = 24-5 находится преимущественно в форме HaR3“. Эта форма реагента характеризуется наличием максимума поглощения при 440 нм; при pH > 5, вследствие частичного перехода реагента в форму H2R4~, появляется второй максимум поглощения при 580 нм. Комплекс цинка с КО имеет максимум поглощения при 570 нм (рис 8.19), но при pH < 4,5, вследствие малой степени комплексообразования, светопоглощение сравнительно невелико (кривая 3). При pH > 5 значение максимума поглощения комплекса при 570 нм резко возрастает (кривые 4—6), однако, светопоглощение самим реагентом при этой длине волны также увеличивается.
Пользуясь спектрофотометрическими методами, нужно установить оптимальные условия образования окрашенного соединения, а также определить состав образующегося комплекса, его устойчивость и молярный коэффициент светопоглощения.
Рис. 8.19. Спектры поглощения растворов КО и его комплекса с цинком при различных pH: 1, 2 — реагент КО при pH = 4.85 и pH = 5,96; 3, 4, 5, 6 — комплекс цинка с КО при pH, равном 4,85; 5,57;
5,96; 6,55 соответственно.
10*
291
Рис. 8.20. Зависимость оптической плотности раствора от pH:
1 — раствор комплекса и реагента; 2 —раствор [реагента; 3 —разность оптических плотностей комплекса и реагента.
Выполнение работы. Определение оптимального значения pH раствора. Приготовив несколько растворов комплекса и реагента одинаковой концентрации, но с различным значением pH, измеряют их оптические плотности относительно раствора сравнения при 570 нм.
Зависимость оптической плотности растворов комплекса и реагента от pH показана на рис. 8.20. Максимальная разность оптической плотности комплекса и самого реагента наблюдается в интервале pH = 5.7ч-6,0, поэтому для последующих опытов используют ацетатный буферный раствор с pH = 5,8.
Исследование состава комплекса цинка с КО. Для исследования состава комплекса цинка с КО применяют различные методы. Сначала приготавливают и анализируют изомолярную серию растворов. На рис. 8.21 показаны спектры поглощения изомолярной серии растворов при общей (суммарной) концентрации компонентов 5,36- 10-Б моль/л.
.Из сравнения спектральных кривых видно, что при отношении концентраций CKQfCZn 1,5 наблюдается значительное поглощение света при 440 нм. Это свидетельствует о наличии больших количеств реагента в свободном состоянии. Максимальный выход комплекса цинка с КО (метод изобестической точки, см. разд. 8.1.5) наблюдается при отношении концентраций CZn/CKo = = 1 (кривая 5). Такое же отношение концентраций цинка и КО отвечает экстремальной точке на изомолярных диаграммах (рис. 8.22), построенных для разных изомолярных концентраций при 570 нм. Из этих данных следует, что ионы цинка реагируют с КО в отно-292
Рис. 8.21 Спектры поглощения растворов изомолярной серии:
1—9 — растворы с соотношением Czn/(Cgn + ^КО) от 0,1 до 0.9 соответственно (Czn + + = 5,36-10-6 моль/л, pH = 5,8, I = 2,09 см).
Gco/^ko+ Сгп)/моль/л
Рис. 8.22. Изомолярная диаграмма:
1 — CZv + ~ Ю~* моль/л; 2
X = 570 нм; I = 2,09 см).
4- СКО = 5,36.10~6 моль/л (pH = 5.8:
Рис. 8.23. Определение состава комплекса по методу Асмуса (С2п = ЫО'^моль т; pH = 5,8; / = 2,09 см).
293
Таблица 8.1. Данные для определения состава комплекса по методу Асмуса
А ико, мл 1 1 тА~ А (Я’-- (Я'“-
0,060 0,072 34,900 1,390 1,930 2,670 3,720
0,087 0,108 24,100 0,925 0,855 0,790 0,730
0,113 0,144 18,500 0,695 0,482 0,335 0,230
0,144 0,180 14,500 0,555 0,310 0,170 0,096
0,163 0,216 12,800 0,463 0,210 0,098 0,044
0,193 0,252 10,800 0 397 0,160 0,062 0,026
0,216 0,288 9,700 0,347 0,120 0,041 0,014
0,238 0,324 8,800 0,309 0,095 0,029 0,009
0,266 0,360 7,850 0,278 0,077 0,021 0,006
0,290 0,396 7,200 0,253 0,064 0,016 0,004
0,350 0,500 6,000 0,200 0,040 0,008 0,002
шении 1:1. Однако, как показал Н. П. Комарь [340], отношение стехиометрических коэффициентов реакции, определяемое методом изомолярных серий, часто не соответствует индексам в формуле образующегося комплексного соединения. Поэтому для окончательного выяснения состава комплекса необходимо применять дополнительные спектрофотометрические ме-
тоды.
В табл. 8.1 представлены данные для определения состава комплекса по методу прямой линии Асмуса ([3531; см. также разд. 8.1.7). Опыты проводят при постоянной концентрации ионов цинка 1- 10~Б моль/л с различными объемами реагента КО концентрации НО'3 моль/л при pH = 5,8. Общий объем фотометрируемого раствора составляет 50 мл, толщина слоя I = = 2 см.
Построенные по данным табл. 8.1 кривые зависимости (l/VR)n = — f для значений п — 1, 2, 3 и 4 показаны на рис. 8.23.
Из сопоставления кривых видно, что только п = 1 удовлетворяет
0
J__।_I_1__1_1_1_1__1—j
0,4 0,8 V 1,6 2,0 С-Ю^ МОЛЬ/Л
требованию прямолинейности функции Асмуса.
Для определения состава комплекса по методу отношения наклонов ([351 ]; см. также разд. 8.1.6) измеряют оптические плотности двух серий растворов с постоянной концентрацией ионов Zn и КО, равной 2,14- 10-Б моль/л при пе-
Рис. 8.24. Определение состава комплекса ме-
тодом отношения наклонов:
1 — при постоянной концентрации циика 2rI4 X X 10“6 моль/л; 2 — при постоянной концентрации КО 2,14-10“* моль/л (Л = 570 нм; pH = 5,8;
I = 1,0 см).
294
Рис. 8.25. Определение состава комплекса методом сдвига равновесия (1 = = 570 нм; pH = 5,8; 1= 1,0 см).
ременной концентрации второго компонента. .Отношение стехиометрических коэффициентов цинка и КО в составе комплекса, получаемое методом отношения наклонов (рис. 8.24), равно 1 ! 1.
Исследование состава комплекса методом сдвига равновесия ([376]; см. также разд. 8.1.2) (рис. 8.25) показывает, что влияние одинакового избытка реагента и ионов цинка (при постоянной и одинаковой концентрации второго компонента) на сдвиг равновесия реакции сказывается совершенно идентично, а это возможно только в случае образования комплекса с отношением стехиометрических коэффициентов, равным 1. Отношение стехиометрических коэффициентов в составе комплекса, определенное из кривой насыщения (рис. 8.26), также равно 1.
Таким образом, результаты различных спектрофотометрических методов показывают, что при взаимодействии ионов цинка с КО при pH = 4-4-6 образуется простейший комплекс состава 1 : 1.
Для определения количества ионов водорода, замещаемых ионами цинка в молекуле реагента, используем графический прием Астахова ([366, 367 ]; см. также разд. 8.3.2). Как видно из рис. 8.27, прямая зависимости lg ,дХ~ / (pH) имеет угловой Мпр —
коэффициент, равный 1. Следовательно, Zn2+ при взаимодействии с КО замещает только один Н+, образуя анионный комплекс ZnH2R2-.
Определение молярного коэффициента светопоглощения^^ и константы устойчивости комплекса. Решая совместно уравнения закона действующих масс и основного закона светопоглощения для реакции
Zn2+4-H3Rs- ZnH2R2--bH+
когда кроме комплекса при выбранной длине волны (570 нм) поглощает и сам реагент, без упрощающих допущений получаем за-
295
Рис. 8.26. Кривая насыщения (С2п = 2,14-10*’ моль/л; X = 570 нм, I — 1,0 см).
Рис. 8.27. Графическое определение числа замещаемых протонов в молекуле реагента по методу Астахова.
висимость (при постоянной концентрации Zn2+ и толщине поглощающего слоя I = 1 см):
cz, ,/ла = 1/де+ 1 I [д.д; (ско--
Здесь Де — разность молярных коэффициентов светопоглощения комплекса и реагента; Cj<o — переменная начальная концентрация реагента; ДА — получаемая разность оптических плогноией растворов комплекса и чистого реагента.
Эта зависимость, построенная в координатах CZa/&A — — ---выражается прямой линией, которая на оси
ординат отсекает отрезок, равный l/Де, а угловой коэффициент прямой равен 1/Дерк. Для построения * такой прямой сначала проводят различные приближенные расчеты равновесной концентрации реагента, затем по приближенным графикам (рис. 8.28, а) получают приближенные значения Де. Используя найденные значения Де, уточняют равновесную концентрацию реагента и получают более точную графическую зависимость (рис. 8.28, б), из которой находят величины Де и Рй-
Приближенный расчет равновесной концентрации реагента в условиях избытка его общей концентрации производят:
1) по методу Шварценбаха ([364]; см. также разд. 8.2.8), пренебрегая равновесной концентрацией образующегося комплекса;
2) по методу Бабко, учитывая равновесную концентрацию комплекса ([56]; см. также разд. 8.2.8):
с'к — CZn ( ско
ЛА"
ДА'
Хд7- (Ско — ско)]
* Графическую зависимость строить не обязательно. Параметры прямой можно рассчитать по опытным данным методом наименьших квадратов (гл. 9).
295
Рис 8.28. Графическое определение Де и р' методом последовательного приближения:
а — приближенная зависимость; б — зависимость по уточненным данным: / « по методу Шварценбаха; 2 — по методу Бабко; 3 — по данным кривой насыщения (h — 570 нм? pH = 5,8; €?2п = 2,14 10-ь моль/л, I == 1,0 см).
3) по данным кривой насыщения, учитывая равновесную концентрацию комплекса (см. разд. 8.1.3):
Сц — Czn ДЛ/ДЛПр
Рассчитанные по опытным данным приближенные значения 1/(Ско — Ск) и соответствующие им значения С2п/ДЛ приведены в табл. 8.2. По данным таблицы методом наименьших квадратов рассчитывают приближенное значение Де:
Гю методу Шварценбаха
По методу Бабко
По данным кривой насыщения
2,53.104
2,53.104
2,525-104
Таблица 8.2. Данные приближенных расчетов
АЛ ско-10Б-моль/л ско —-10» АЛ 1 го-» ско-с„
по Шварцен-баху по Бабко по кривой насыщения
0,458 2,14 1,0 4,67 3,450
0,502 2.57 1,2 4,27 0.389 1,490 1,820
0,520 3,21 1,5 4,12 0,311 0,850 0,900
0,525 4,28 2,0 4,07 0,233 0,440 0,463
0,531 5.35 2.5 4,03 0,187 — 0,311
297
По найденным значениям Де более точно рассчитывают значе-Ск0--------л~)> соответствующие тем же значениям С2п/ДЛ.
Уточненные данные приведены в табл. 8.3. По данным таблицы снова рассчитывают параметры прямолинейной зависимости С2п/ДЛ — 1|(Ско-----мет°Д°м наименьших квадратов:
2 [ско — ДЛ/Де
И 1
2 [б?ко —ЛЛ/Дв
1
1
Ско — ДЛ/Де]г ^2(сцо — ДЛ/Де
Рассчитав величины Де и Рй определяют их интервальные значения (см. расчет градуировочного графика, гл. 9).
По данным расчетов получают:
Де. 10"4 Р'.10~в
К
По методу Шварценбаха 2,54±0,03
По методу Бабко 2,54±0,03
По данным кривой насыще- 2,52±0,04 ния
1,7±0,17
1,7±0,17
1,7±0,22
Таблица 8.3. Уточненные данные
ДЛ ско- 1О’> моль/л —Zn...IQB дл ско 5 io~‘ CR0 — Д Л/Дб
по Шварцен-баху по Бабко по кривой насыщения
0,458 2,14 4,67 1,00 3,030 3,030 3,070
0,502 2,57 4,27 1,20 1,690 1,690 1,720
0,520 3,21 4,12 1,50 0,860 0,860 0,869
0,525 4,28 4,07 2,00 0,450 0,450 0,454
0,531 5,35 4,03 2,50 0,807 0,307 0,307
298
Значение Де представляет собой аддитивную разность молярных коэффициентов светопоглощения комплекса ек и самого реагента еКо при 570 нм. Однако вследствие того, что реагент является многоосновной кислотой, которая при pH = 5,8 находится преимущественно в форме H3R3- и частично в форме H2R4“, суммарное значение ек0 определяется молярными коэффициентами светопоглощения обеих форм реагента при этих условиях.
Определение молярных коэффициентов светопоглощения равновесных форм реагента при 570 нм. Молярные коэффициенты светопоглощения е'Ко (форма 7/3R3-) и е"к0 (форма H2R4-) определяют по методу Комаря ([3721; см. также разд. 8.2.3). Приготавливают несколько растворов реагента КО с постоянной концентрацией 2,14-Ю'6 моль/л и различными значениями pH от 2,96 до 9,80 и измеряют их оптические плотности при 570 нм в кюветах G толщиной слоя 1 см. По полученным данным рассчитывают величины Еко, еко и KaHaRa_ (где KaHaR3_ — константа диссоциации реагента, равная [Н+ ] [H2R4-/ IH3R3- ]).
Результаты расчетов приведены в табл. 8.4.
Исходя из суммарного светопоглощения комплекса и обеих форм реагента, получают равенство:
Л = екС J + е'к0 (Ск0 — Ск) “+Сн +
+ еко (Ско - Ск) KeH>R,_ + CH-
Здееь Сн — концентрация Н+ в растворе.
Отсюда разность молярных коэффициентов светопоглощения комплекса и реагента равна
. , СН ДаН№
к ®ко Koh,r-. + Ch ко K«h,r»- + Ch
Таблица 8.4. Значения молярных коэффициентов светопоглощения (®КО> ®ко) и ^“h.r»- реагента К0
Концентрация КО — 2,14.10"’ моль/л', К = 570 нм.
pH А ЕК0 еко К„ “h3r3-
2,96 0,017 713 2,86-104 9,09-10-8
4,84 0,033 716 3,19-104 7,70-10-8
5,80 0,082 792 3,18-104 5,88-IO*8
7.33 0,442 791 3,11-104 10,40.10-8
7,60 0,516 795 2,68.104 6,79-10-8
9,80 0,705 790 3,31-104 3,04.104 8,02.10-8 6,01.10-’ 8,91.10-8
Среднее Значение 760 ЗЛО4 7,8-10-8
299
откуда
е“ ” Де + ®к0 KaHsR3- + CH + 8к0 KflH3R3- + CH =
_ 9 „ i™ , 7т 1,58-10~6 3-10«-7,8-10-8’ _
2,53-10 -1-760 7>8.ю-ь-|-1,58-10'6 + 7,8 10’в-j-1,58 Ю”» 2’74' 0
Примечание. Все опытные данные получены при анализе растворов, содержащих 3% этилового спирта при постоянной ионной силе раствора (/ = == 0,04), на спектрофотометре СФ-14 с использованием шкалы повышенной чувствительности (dj.
Реактивы
Кеш ноюеый оранжевый, 0,001 М раствор.
Ацетат цинка, 0,001 Л1 раствор.
Ацетатные буферные растворы (pH = 2,94-7,6).
Боратные буферные растворы (pH =94-10).
Часть HI
РАСЧЕТЫ И ВЫЧИСЛЕНИЯ
В ФОТОМЕТРИЧЕСКОМ АНАЛИЗЕ
Глава 9
КРАТКИЕ СВЕДЕНИЯ
ПО МАТЕМАТИЧЕСКОЙ ОБРАБОТКЕ
ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ
Каждый результат анализа по тем или иным причинам имеет погрешность определения. Исследователь всегда получает лишь приближенное значение определяемой величины. Поэтому завершающей стадией количественного анализа химического состава вещества любым методом является статистическая обработка результатов измерений, которая включает расчет результатов анализа, контроль воспроизводимости (сходимости) и правильности результатов [9, 10, 15, 58, 82, 107, 377—404].*
Воспроизводимость ** зависит от случайных погрешностей. Чем больше их значение, тем менее «точный» анализ. Правильность ** результатов определяется систематическими погрешностями. Метод или методика анализа дают лишь тогда правильный результат, когда он свободен от систематических погрешностей. Статистическая обработка результатов измерений позволяет оценить, свести к минимуму систематические и случайные погрешности результатов измерений.
Ниже приведены некоторые приемы современной математической статистики, позволяющие:
1. Рассчитать основные метрологические характеристики методики анализа (оценить воспроизводимость и правильность полученных данных, отбросив результаты, содержащие промахи);
* См. также: 1. Отраслевой стандарт. Особо чистые вещества для монокристаллов. Обработка результатов анализа ОСТ 6-09-96-79.
2. ГОСТ 16263—70. Государственная система обеспечения единства измерений. Метрология. Термины и определения.
3. Стандарт ИСО 2602—73. Статистическая интерпретация результатов испы- -танин. Оценка математического ожидания. Доверительный интервал.
4. ГОСТ 11.004—74. Прикладная статистика. Правила определения оценок и доверительных границ для параметров нормального распределения.
** Подробно о воспроизводимости и правильности фотометрических методов анализа см. разд. 3.1.3, 3.1.4 и литературу (15, 21, 25, 58, 80, 82, 109, 110, 118, 119].
301
2. Определить методом регрессионного анализа вид функциональной зависимости А — f (С); рассчитать метрологические характеристики градуировочного графика и результатов анализа;
3. Объективно сравнить две методики анализа одного объекта или результаты его анализа, полученные в разных лабораториях, и выбрать лучшие из них;
4. Представить результаты статистической обработки в виде компактных табличных данных, позволяющих оценить воспроизводимость и правильность полученных результатов;
5. Рассчитать определяемый минимум цветной реакции.
9.1. РАСЧЕТ ОСНОВНЫХ
МЕТРОЛОГИЧЕСКИХ ХАРАКТЕРИСТИК РЕЗУЛЬТАТОВ ПРЯМЫХ (НЕПОСРЕДСТВЕННЫХ) РАВНОТОЧНЫХ ИЗМЕРЕНИЙ
В химическом анализе содержание вещества в пробе устанавливают, как правило, по небольшому числу параллельных определений (п 3). Для расчета погрешностей определений в этом случае пользуются методами современной математической статистики, разработанной для малого числа определений. Полученные результаты рассматривают как случайную (малую) выборку из некоторой гипотетической генеральной совокупности, состоящей из всех мыслимых в данных условиях наблюдений. Соответственно различают выборочные параметры (параметры малой выборки) случайной величины, которые зависят от числа наблюдений, и параметры генеральной совокупности *, не зависящие от числа наблюдений.
Все измерения в метрологии делят на прямые и косвенные.
При прямых непосредственных измерениях числовое значение измеряемой величины х сразу получается из показаний прибора, при помощи которого выполняется данное измерение, например значение оптической плотности или пропускания по шкале оптической плотности (пропускания).
Результат каждого прямого измерения включает случайную погрешность, которая зависит от большого числа случайных факторов.
Если отклонения, вызванные случайными факторами, сравнимы по абсолютному значению с чувствительностью прибора, то они обнаруживаются приборами, и при п измерениях одной и той же величины получаются результаты хъ х2, ..., xif хп, которые могут отличаться друг от друга в пределах чувствительности данных измерений.
* Для практических целей можно считать, что при числе измерении п 20ч-30 значения стандартного отклонения генеральной совокупности (о) — основного параметра — и стандартного отклонения малой выборки (з) близки (з ~ о).
302
Статистическую обработку результатов измерений малой выборки (и), оценку их воспроизводимости и правильности проводят по следующей схеме.
9.1.1. ОЦЕНКА ВОСПРОИЗВОДИМОСТИ
РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ*
Среднее выборки. Пусть xlt х2, ..., хп обозначают п результатов измерений величины, истинное значение которой р. Предполагается, что все измерения проделаны одним методом и с одинаковой тщательностью. Такие измерения называют равноточными.
В теории ошибок доказывается, что при условии выполнения нормального закона (закона распределения Гаусса) при п измерениях одинаковой точности среднее арифметическое из результатов, полученных при всех измерениях, является наиболее вероятным и наилучшим значением измеряемой величины:
п
х = Xj + х2 + • • • + Xi -J- • • + хп = Xi (9.1)
i=i
Это среднее значение принимают за приближенное и пишут р « х.
Единичное отклонение — отклонение отдельного измерения от среднего арифметического:
ег — Х{ — X (9.2)
Алгебраическая сумма единичных отклонений равна нулю:
S 8i = О
1=1
Дисперсия, стандартное отклонение, относительное стандартное отклонение, коэффициент вариации. Рассеяние результатов измерений относительно среднего значения принято характеризовать дисперсией sa
s2= £ (хг-х)2/(н-1) (9.3)
i=i
или стандартным отклонением (средним квадратическим отклонением — СКО) — s
S = ~ ~ (9-4)
* Здесь предполагается, что выборка имеет приближенно нормальный закон распределения. Отклонения от гауссовского распределения могут иметь место, особенно при определении следовых содержаний компонентов. При этом возможно логарифмическое нормальное распределение. При необходимости следует осуществить проверку нормальности распределения (см. например, г58, 82, 380, 386, 388]).
303
которое обычно приводят при представлении результатов измерений (анализа) * и которым характеризуют их воспроизводимость.
Стандартное отклонение, деленное на среднее выборки, называют относительным стандартным отклонением
sr = s/x (9.5)
а это же выражение, умноженное на 100 %, — коэффициентом вариации v (термин, не рекомендуемый в литературе по аналитической химии).
В общем случае метод (методика) анализа оптимален в той области содержаний, в которой и абсолютное (s), и относительное (sr) стандартное отклонения имеют минимальные значения (см. разд. 3.1.3).
Дисперсия среднего арифметического (выборки), стандартное отклонение среднего арифметического (выборки). При оценке воспроизводимости полученных результатов вычисляют также дисперсию среднего арифметического
sx= X, (xi ~ *УИп (п — (9 6>
и стандартное отклонение среднего арифметического (СКО среднего арифметического):
sjf=s/K«=|/r(х, — х)2 * */[я (я—!)] (9.7)
9.1.2. ОПРЕДЕЛЕНИЕ И ИСКЛЮЧЕНИЕ
ГРУБЫХ ПОГРЕШНОСТЕЙ (ПРОМАХОВ)
В литературе приведены различные методы оценки и исключения грубых погрешностей. Рассмотрим два наиболее простых для практического использования метода.
* Химический анализ миогостаднен. Поэтому воспроизводимость результатов измерений аналитического сигнала (s) не совпадает с воспроизводимостью методики анализа в целом (sM) (обычно в литературе применяют один индекс—з).
В общем случае sM > s, так как значение s,,, складывается из совокупности по-
грешностей отбора пробы (so. п), погрешностей подготовительных операций пробы перед измерениями (sn о) и погрешностей измерения (sH) [107] (см. также разд. 3.1.3)
Sm п + sn о + sh
Слагаемые могут быть оценены методом дисперсноннот'о анализа (см., например, [58]). Значение sMследует вычислять для п самостоятельных параллельных проб анализируемого вещества, прошедших все этапы анализа. При подсчете значений s всегда используют неокругленные результаты измерения (анализа) с ненадежным последним знаком после запятой [82].
304
Таблица 9.1. Численные значения для Q (Р, гц) [82]
п1 Р = 0,90 Р = 0,95 Р == 0,99
3 0,89 0,94 0,99
4 0.68 0,77 0 89
5 0,56 0,64 0,76
6 0,48 0,56 0,70
7 0,43 0,51 0.64
8 0,40 0 48 0,58
Исключение грубых погрешностей по Q-критерию. При малых выборках с числом измерений п < 10 определение грубых погрешностей лучше оценивать при помощи размаха варьирования по Q-критерию [82]. Для этого составляют отношение:
Q = I Ч — х21/7? (9 8)
Здесь х( — подозрительно выделяющийся результат определения (измерения), х2 — результат единичного определения ближайший по значению к Xf, 7? — размах варьирования *, R — хмакс — хмин — разница между наибольшим и наименьшим значением ряда измерений.
Вычисленное значение Q сопоставляют с табличным значением Q (Р **, nt) (табл'. 9.1). Наличие грубой погрешности доказано, если Q > Q (Р, tii).
Исключение грубых погрешностей методом вычисления максимального относительного отклонения [378, 380, 388]. Статистический критерий обнаружений грубых погрешностей основан на предположении, что выборка взята из генеральной совокупности, распределенной нормально. Это позволяет использовать распределение наибольшего по абсолютному значению нормированного отклонения:
tT = макс | Xi — х |'s (9.9)
Здесь /т — теоретическое значение квантиля распределения статистики.
* При малой выборке (п < 10) размах варьирования служит также одной из характеристик рассеяния результатов измерений (подробно см. [82]).
** Р — доверительная вероятность — доля случаев, в которых среднее (арифметическое) при данном числе определений будет лежать в определенных пределах. Доверительная вероятность Р связана с двусторонней — верхней и нижней — границей разброса среднего значения выборки. В аналитической химии, как правило, пользуются доверительной вероятностью Р — 0,95 и значительно реже Р — 0,90 или Р = 0,99.
Часто устанавливают одностороннее требование, например, чтобы Значение х не превышало некоторой верхней границы. В этом случае говорят об односторонней границе с соответствующей вероятностью Р. Между Р и Р существует соотношение Р = 0,5-|-Р/2. Подробнее см. [82].
305
Таблица 9.2. [388]. Квантили распределения максимального отклонения, /т = макс | х, — х \[s
п Уровни значимости р п Уровни значимости р
0,1 о.иб 0,025 0,01 0,1 0,05 0,025 0,01
3 1,41 1,41 1,41 1,41 15 2,33 2,49 2,64 2,80
4 1,65 1,69 1,71 1,72 16 2,35 2,52 2,67 2,84
5 1,79 1,87 1,92 1,96 17 2,38 2,55 2,70 2,87
6 1,89 2,00 2,07 2,13 18 2,40 2,58 2,73 2,90
7 1,97 2,09 2,18 2,27 19 2,43 2,60 2,75 2,92
8 2,04 2,17 2,27 2,37 20 2,45 2,62 2,78 2,96
9 2,10 2,24 2,35 2,46 21 2,47 2,64 2,80 2,98
10 2,15 2,29 2,41 2,54 22 2,49 2,66 2,82 3,01
11 2,19 2,34 2,47 2,61 23 2,50 2,68 2,84 3,03
12 2,23 2,39 2,52 2,66 24 2,52 2,70 2,86 3,05
13 2,26 2,43 2,56 2,71 25 2,54 2,72 2,88 3,07
14 2,30 2,46 5,60 2,76
Для уровней значимости * р = 0,10; 0,05; 0,01 или доверительной вероятности 1 — р = 0,90; 0,95; 0,99 и п 25 значения /т приведены в табл. 9.2 **. На практике обычно используют уровень значимости р — 0,05 (результат получается с 95%-ной доверительной вероятностью).
Для того, чтобы в группе из п наблюдений х2, .... хп отбросить результат хмавс (или хмин), надо:
а) вычислить дробь /макс = |хмакс — x|/s;
б) по табл. 9.2 найти теоретическое значение /т в зависимости от и и выбранного уровня значимости р;
в) сравнить рассчитанное по п. «а» значение (макс с tT. Если /макс > ^т> т0 результат хмакс следует отбросить как промах.
Процедуру исключения промахов можно повторить и для следующего по абсолютному значению максимального относительного отклонения, но предварительно необходимо пересчитать х и s для выборки п — 1.
9.1.3. ОЦЕНКА ПРАВИЛЬНОСТИ
РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ(ОПРЕДЕЛЕНИЙ)
После того как осуществлена проверка грубых погрешностей (в случае подозрительных результатов измерений) и их исключение производят оценку доверительного интервала (Ах) для сред
* Уровень значимости р = (1 — Р) — максимальная вероятность того, что погрешность гревзойдет некое предельное (критическое) значение ±ДхКр, т. е. такое значение, что появление этой погрешности можно рас сматривать как следствие значимой (неслучайной) причины [58].
В разных литературных источниках уровень значимости обозначается по-разному Р, а, р
** В развернутом виде для п 50 соответствующая таблица дана в работе [380]. Там же приведена методика определения и исключения грубых погрешностей и при п > 50.
306
него значения х, интервальных значений х ± Ах и при необходимости — оценку правильности результатов.
Доверительный интервал (Ах). Если воспроизводимость результатов измерений (методику анализа) характеризуют стандартным отклонением, то сами результаты измерений (определений) характеризуют доверительным интервалом среднего значения Ах, который рассчитывают по формуле:
Дх = T(Pi f)s/^п (9.10)*
Здесь tp f— квантиль распределения Стьюдента при числе степеней свободы f =- — 1 и двусторонней доверительной вероятности Р (значения tp, f см. в табл. 9.3).
Обычно для расчетов доверительного интервала пользуются значением Р = 0,95; иногда достаточно Р — 0,90, но при ответственных измерениях требуется более высокая надежность (Р = = 0,99).
Необходимо отметить, что, если при отработке методики выполняют п параллельных определений, а методика анализа в дальнейшем предусматривает выдачу результатов из т параллельных определений (обычно п 10, т — 2-?-3), то доверительный интервал для рядовых анализов следует рассчитывать по формуле Ах = £p,ys/-j/m [389], а не по формуле Ах = tPitsl^n (где s — стандартное отклонение для выборки из п опытов). В противном случае значение Дх рядового анализа окажется слишком заниженным.
Коэффициент tPtf показывает, во сколько раз разность между истинным и средним результатами больше стандартного отклонения среднего результата:
tp, f = I 8 — •* l/sx = If* —*l^«/s (9.И)
Интервальные значения измеряемой величины. В общем случае интервальные значения изменяемой величины при выбранной доверительной вероятности определяются выражениями:
х — Дх<Ц<х-}-Дх (9.12)**
* — \р, f)s* <!*<* + t(P, f)sx (9.13)
х — t(P' ^s/Vn <р<х-М(Р> (9.14)
* Доверительный интервал может быть задан как абсолютной погрешностью с представлением в тех единицах, в которых выражается результат анализа, так н относительной погрешностью, выраженной в процентах от результата анализа. ЮОДх/х [% (отн.)] или ЮОДх/р [% (отн.)] (при наличии стандартных или эталонных образцов).
** При оформлении окончательного результата придерживаются следующего правила: погрешность должна иметь одну—две значащие цифры, а число, выражающее среднее значение измеряемой величины, должно оканчиваться разрядом, которым начинается погрешность, т. е. значения среднего результата и доверительного интервала должны быть выражены числами с одинаковым числом знаков после запятой; их положено округлять в одинаковой степени. Правила округления чисел см., например, в работах [15, 82, 380].
307
Таблица 9.3 [82]. Значения квантиля ^-распределения в зависимости от вероятности Р (двусторонняя постановка задачи) и Р (односторонняя постановка задачи) и степени свободы f
р
0,75 0,90 0,95 0,98 0,99
t
р
0,875 0,95 0.975 0,99 0,995
1 2,41 6,31 12,7 31,82 63,7
2 1,60 2,92 4,30 6,97 9,92
3 1,42 2,35 3J8 4,54 5,84
4 1,34 2,13 2,78 3,75 4,60
5 1,30 2,01 2,57 3,37 4,03
6 1,27 1,94 2,45 3,14 3,71
7 1,25 1,89 2,36 3,00 3,50
8 1,24 1,86 2,31 2,90 3,36
9 1,23 1,83 2,26 2,82 3,25
10 1,22 1,81 2,23 2,76 3,17
11 1,21 1,80 2,20 2,72 3,11
12 1,21 1,78 2,18 2,68 3,05
13 1,20 1,77 2,16 2,65 3,01
14 1,20 1,76 2,14 2,62 2,98
15 1,20 1,75 2,13 2,60 2,95
16 1,19 1,75 2,12 2,58 2,92
17 1,19 1,74 2,11 2,57 2,90
18 1,19 1,73 2,10 2,55 2,88
19 1,19 1,73 2,09 2,54 2,86
20 1,18 1,73 2,09 2,53 2,85
21 1,18 1,72 2,08 2,52 2,83
22 1,18 1,72 2,07 2,51 2,82
23 1,18 1,71 2,07 2,50 2,81
24 1,18 1,71 2,06 2,49 2,80
25 1,18 1,71 2,06 2,49 2,79
26 1,18 1,71 2,06 2,48 2,78
28 1,17 1,70 2,05 2,47 2,76
30 1,17 1,70 2,04 2,46 2,75
СО 1,15 1,64 1,96 2,33 2,58
Доверительный интервал ограничивает область, внутри которой, при отсутствии систематических погрешностей, находится истинное значение измеряемой величины [см. уравнение (9.12)] с заранее заданной доверительной вероятностью Р.
Из уравнений (9.10) и (9.14) следует, что значение доверительного интервала зависит от размера выборки, т. е. от числа проведенных опытов: е уменьшением числа измерений п увеличивается доверительный интервал (при той же доверительной вероятности) или при заданном доверительном интервале уменьшается надежность измерений.
308
Пользуясь соотношениями (9.10) и (9.12) и данными табл. 9.3, можно рассчитывать доверительные интервалы при выбранных значениях Р или, наоборот, задавшись определенным доверительным интервалом, можно рассчитать tPif и по табл. 9 3 оценить его надежношь. Кроме того, на основании формулы (9.11) и табл. 9.3 можно установить число параллельных определений, необходимых для того, чтобы средний результат имел доверительный интервал не ниже заданного.
Значимость систематической погрешности. Значение систематической погрешности характеризует меру правильности резуль-таюв определений (анализа). О значимости систематической погрешности, т. е. о правильности результата анализа, судят в зависимости от того, попадает ли истинное значение определяемой величины в установленный доверительный интервал или находится вне его. Если | х — р | > Ах, то можно говорить о значимой систематиче кой погрешности Е, интервальное значение которой заключено в пределах:
х — р — Ах<Е<х —р-)-Дх (9.15)
В этом случае необходимо выяснить причину появления систематической погрешности.
Следует отметить, что задача освобождения результатов измерений от систематических погрешностей требует глубокого анализа всей совокупности данных измерений. Наиболее вероятными источниками систематических погрешностей фотометрических измерений могут служить: недостаточная представительность состава отобранной аналитической навески, погрешности в подготовке аналитической навески к фотометрическим измерениям, погрешности градуировки весов, мерной посуды, шкал спектрофотометров (фотоколоримегров), несоответствие составов анализируемых растворов и стандартных растворов, по которым строились градуировочные графики A =f(C) и т. д. ([21, 25, 58, 82, 3801, см. также разд. 3.1.4, 3.2.2). Правильность результатов определений может контролироваться различными способами и, в первую очередь, путем сопоставления следующих величин:
а) результата анализа стандартного образца (СО) с аттестованной величиной содержания определяемого компонента;
б) найденного содержания добавки в анализируемый раствор определяемого компонента;
в) результатов анализа одного и того же образца данного вещества, полученных в разных лабораториях;
г) результатов анализа одного и того же образца данного вещества при помощи методики, используемой для контроля продукции, и арбитражной методики анализа *.
* Предполагается, что погрешности аттестации СО и априорных оценок введенных добавок пренебрежимо малы по сравнению с погрешностями аналитических определений.
309
При сопоставлении величин по п. «а—г» правильность результатов аналитических определений считают удовлетворительной, если выполняется одно из следующих неравенств:
11 — а;х | < 2sr/|^п (9.16)
11 — sa/x । < 2 + S2 (а)/па (9.17)
Здесь а — аттестованное значение содержания определяемого компонента в стандартном образце или известное содержание добавки согласно п. «б»- х — среднее арифметическое результатов п параллельных определений при анализе образца по п. «-а—г» в лаборатории; ха — вреднее арифметическое результатов па параллельных определений при анализе образца по п. «С» в арбитражной лаборатории или при помощи арбитражной методики анализа; sr (а) — относительное стандартное отклонение единичного результата, полученное в арбитражной лаборатории или для арбитражной методики анализа.
В отсутствие стандартных образцов правильность результатов определений может быть оценена путем постановки специальных опытов по «схеме удвоения» в сочетании с введением добавок [58].
Для обнаружения и исключения систематических погрешностей широко применяют также регрессионный и корреляционный анализы (см., например, [58, 82, 380, 3881).
В монографии [25] детально проанализированы источники и причины систематических погрешностей в фотометрических методах анализа.
Для полной характеристики методики на правильность необходима оценка систематических погрешностей вблизи нижней и верхней границ интервала Св — Св.
9.2. ПОГРЕШНОСТИ КОСВЕННЫХ ИЗМЕРЕНИЙ
Косвенным называют измерение, при котором сначала проводят прямые измерения некоторых величин (хь х2, х3, ..., xt, ..., хв), а затем по определенным формулам, связывающим эти величины с измеряемой величиной у, вычисляют ее значение, т. е. у является функцией непосредственно измеренных величин:
У = i (Л. *г. .... xit ..., хп) (9.18)
Каждая из величин хг, х2, ..., xt, ..., хп имеет свою погрешность и в зависимости от формулы, по которой вычисляют результат анализа, эти погрешности различно влияют на погрешность результата.
При оценке воспроизводимости (сходимости) косвенных измерений применяют следующие формулы.
Среднее значение косвенно определяемой величины получают подстановкой в расчетную формулу средних арифметических значений непосредственно измеренных величин: у = f (хъ х.г, xit ..., хп) и считают у ха (где а — истинное значение косвенно определяемой величины).
310
,2 , , ( Of
'*i+ + \д^
частная производная функции f (xj, х2, xt.
si
s2 (9.19) п
• *п) ПО *1 —
— дисперсия прямо измеренной величины х.,
накопления ошибок. Поль-
Дисперсию косвенно измеряемой величины вычисляют по формуле:
Здесь. -gy -прямо измеренной величине;
Формулу (9.19) называют законом или законом распределения зуясь формулой (9.19), можно оценить погрешность косвенного измерения, если известны погрешности прямых измерений.
Иногда при обработке результатов косвенных измерений ргра-ничиваются только расчетом дисперсии Sy или дают расчеты так называемых предельных абсолютной и относительной ошибок, вычислив предварительно предельные абсолютные ошибки прямых измерений по формуле: X = Зяд.
Доверительный интервал косвенно измеренной величины &У можно получить из выражения:
&y = tp, f^=
V п значения tPti находят из табл. 9.3.
Интервальные значения косвенно измеряемой величины ф
(9.20)
Пример. Для определения концентрации железа в исследуемом pjcfeogo (Сх) применили фотоколориметритеский метод сравнения, и из трех измерении бьрш получены следующие средние значения оптической плотности исследуемого и стандартного растворов: Ах = 0,216; Лст= 0,148.
Выборочные дисперсии соответственно были равны: = 4- 10~е; 5лст =
= 4-10'8; концентрация стандартного раствора Сст = 1,20 мкг/мл (для простоты расчетов Сст примем за точное чясло).
Определяем среднее и интервальные значения Сх с доверительной вероятностью Р — 0,95
Сх « Сх = С^АХ/Аст = 1,20-0,216/0,148
Д (СстЛх/Лст)
где
S2
Lx
дА-
2
«л +
X
д (СстАкМст) дЛСт
1.75 мкг/мл
2
®л Лст
д (Cct^xZ-^ct) Сст • (СстЛхМст) Сст
&Ах Лет * дА™ Лет
Подставив эти выражения в приведенную выше формулу, получим:
С
^СТ у nCTSJ
'X
ст
sc
1,20-2-Ю-з /219-10'4 + 467-1 (Г4 _ = 219-IO"4 -U.U24
При Р = 0,95 f = п — 1 = 2 и ДС = 4,30-0,029,1,73 = 0,072 получим Сх = (1,75 ± 0,07) мкг/мл.
311
9.3. РЕГРЕССИОННЫМ АНАЛИЗ
ПРИ РАСЧЕТЕ ГРАДУИРОВОЧНЫХ ГРАФИКОВ
И МЕТРОЛОГИЧЕСКИХ ХАРАКТЕРИСТИК
РЕЗУЛЬТАТОВ АНАЛИЗА
В разд. 7.1.1 рассмотрена методика построения градуировочного графика при выбранных оптимальных условиях фотометрирова-ния (см. разд. 3.2). Однако описанное представление графической зависимости А = f (С) имеет два существенных недостатка. Во-первых, такой вариант построения градуировочной кривой носит субъективный характер: прямая не лучшим образом будет проходить через экспериментальные точки (г/г, хг) и это может привести к большим погрешностям конечного результата. Во-вторых, если определять более чем две значащие цифры, то график должен быть очень большим.
В связи с этим более объективным и правильным является установление математической зависимости А = / (С), которую находят методом регрессионного анализа (см., например, [9, 10, 15, 82, 380, 388, 390—3931).* В фотометрических методах она выражается обычно линейной и, значительно реже, параболической зависимостью. Все расчеты при этом могут быстро выполняться на программируемых микрокалькуляторах типа «Электроника» БЗ-21, БЗ-34, МК-54 и мини-ЭВМ (см., например, [394—401 ]).
9.3.1. РАСЧЕТ УРАВНЕНИЯ
ЛИНЕЙНОГО ГРАДУИРОВОЧНОГО ГРАФИКА,
ЕГО МЕТРОЛОГИЧЕСКИХ ХАРАКТЕРИСТИК
И МЕТРОЛОГИЧЕСКИХ ХАРАКТЕРИСТИК
РЕЗУЛЬТАТОВ АНАЛИЗА
9.3.1.1. Вычисление метрологических характеристик линейного графика
Вычисление параметров а и Ь. В общем случае линейная зависимость выражается уравнением (3.20) **:
у = а + Ьх
* Особо следует отметить монографии В. В. Налимова [377], К. Доерфеля [82] и Е. Н. Львовского [388].
** Уравнение (3.20) называют линейной регрессией; с — свободным членом; Ь — угловым коэффициентом, коэффициентом регрессии. При фотометрическом анализе а — оптическая плотность холостой пробы, Ь — в простейшем случае (разд. 3.1.1.1 и уравнения (3.21)—(3.21") характеризует собой коэффициент чувствительности 8, равный е (моль-1-л см-1) или е' = е В (г^1-л-см"1), где В — атомная масса.
Существуют две модели регрессии [388]. Условно модель у — ау, ж+ Ьу< Хх можно назвать прямой регрессией, а модель х = ск> е -f- Ьх, уу — обратной регрессией.
312
Значения параметров а и b вычисляют методом регрессионного анализа.
Если имеется п взаимосвязанных пар значений (хг, у^, то можно записать;
— а +
Ui = а + bxt
Уп = й + Ьхп
Здесь п — число измерений, х, — известное содержание (концентрация) определяемого компонента в j-м стандартном растворе; yi — результат прямых измерений аналитического сигнала (оптической плотности) i-ro стандартного раствора.
Непременным при построении градуировочного графика является условие, чтобы погрешности определения значений х были существенно (намного) меньше погрешностей измерения у *.
В левой части системы уравнений (9.22) находятся измеренные значения yt, а в правой — вычисленные значения Yt = а 4~ Ьх,. Разность между обеими величинами дает погрешность. Аналитически задача метода наименьших квадратов может быть выражена в следующей форме [82, 380, 388, 390—393]:
SQ= (yt—Yt) = V1, [yi—(а + йх,)]мин (9.23)
1 = 1 -1
Если yt — (а + bxi) — 6г, тогда
« = 1.....п (9.23')**
1=1
Следовательно, задача линейного регрессионного анализа (метода наименьших квадратов) состоит в том, чтобы сумма квадратов отклонений SQ экспериментальных точек (х;, yt) вдоль ординаты от прсвсденной прямой была минимальной.
Для того, чтобы найти параметры а и Ь, удовлетворяющие минимуму SQ, берут частные производные выражения (9.23) относительно а, затем относительно Ь, полученные выражения приравнивают нулю и, решая уравнения, находят;
П — S X; S f/j ”£д-(2>1)2
(9-24)
* Для этого в перзую очередь необходимо выбирать соответствующие аналитические навески и мерную хорошо отградуированную посуду, обеспечивающую малые погрешности отбираемых для анализа аликвотных объемов [82].
л
** В дальнейшем с целью упрощения символ суммы У будем обозна-=1
чать просто у.
ЗГЗ
или
V S Xi 2 Di ь_ "_____2- = (9.2,-)
^i-nx2
S *i , £j уi
где x = г- и В = — .
n n
a = (S M — * 2j xi)7n (9.25)*
или в несколько ином варианте;
S *? S si - Е xi S xi«i S *? S yt - S xi S xi9i ,9 254
Константы а и b являются случайными величинами, ввиду чего необходима оценка их доверительных интервалов.
Дисперсии констант а и Ь. Дисперсии констант а и b вычисляют по уравнениям
2(xf —х)2 п£х2.— (£х4)2
(9.26)
2 _ sb v 2
“ n%(xt-xp n L 1
(9.27)
co степенями свободы f = n — 2.
Дисперсию syx, характеризующую рассеяние результатов относительно прямой, вычисляют по формуле
slx= £(^-Гг)2/(«-2) (9.28)
со степенями свободы f = п — 2. Однако, если для каждой из п проб проводят по kj параллельных измерений, так что имеется nkj = п' результатов измерений, то с уравнением (9.28) будет связано не f = п — 2, a f = ti — 2, которое и будет в знаменателе.
Сумму квадратов в уравнении (9.28) удобнее определять, пользуясь следующим выражением:
SQ = '^i(yt—Vi)2=s2yx(n — 2)= yiyj — aYlyi~b^ixly{ (9.29)
При проведении вычислений по формуле (9.29) все расчеты необходимо выполнять при достаточно большом числе знаков после запятой, так как искомую сумму квадратов часто ищут для весьма близких значений. Поэтому на данном этапе даже незначительные погрешности в вычислении могут привести к большим погрешностям.
* Если а мало отличается от 0, то уравнение может иметь вид у — Ь'х. Проверку константы а см. разд. 9.3.2.
314
Анализ уравнения (9.26) показывает, что для константы Ь дисперсия тем меньше, чем дальше значение хг лежит от его среднего значения х, т. е. чем шире интервал содержаний (концентраций), выбранный для построения градуировочной кривой.
Доверительный интервал для значений констант а и Ь. Доверительный интервал для а и Ь рассчитывают по уравнениям:
ЛЬ = sbtp,} (9.30)
Да = satPt f (9.31)
при f = п — 2 или f = п’ — 2 (см. выше). Зная Afe и Ас, определяют число необходимых знаков после запятой для b и а.
Доверительный интервал вычисляемых значений зависимой переменной Yk. Полученную функцию у — а + Ьх можно использовать для расчета Yk и АУЛ для одного заданного значения xk. Доверительный интервал для вычисленного значения Yh рассчитывают по формуле:
(Xk — х)2 ~| _ J
n {Xk — х)2 ]
(9.32)
Из уравнения (9.32) следует, что доверительный интервал зависит от разности (xh — х) и будет тем больше, чем дальше xh лежит от среднего значения х (см. рис. 9.1). Поэтому экстраполяция даже при наличии линейной связи может сопровождаться очень большими погрешностями.
9.3.1.2. Вычисление метрологических характеристик результатов анализа:
^ан» ®Лан’ Д^ан’ ^мин, Р
После того как определена функциональная зависимость у = = а -|- Ьх и рассчитаны значения а, b, Ап, Afe, по данным измерений аналитического сигнала у (оптическая плотность А) анализируемых проб рассчитывают метрологические характеристики результата анализа.
Среднее значение результата анализа (хан). Проба определяемого компонента с неизвестным содержанием [оно должно быть в интервале содержаний, для которых вычислена функциональная зависимость у = f (х) J анализируется т раз (обычно т^З). При этом получают т значений аналитического сигнала уаП1, У ин,» ..., рассчитывают среднее уан, а по уравнению
Хан = (Уан — а)/Ь (9.33)
* В случае подозрительно выделяющегося результата yaRi осуществляют его проверку по Q- или /^-критерию (см. разд. 9.1).
315
находят среднее значение содержания компонента в анализируемой пробе.
При фотометрическом анализе уравнение (9.33) преобразуется к виду:
Сан= (^ан (9.33')
Стандартное отклонение результата анализа. В случае градуировочного графика вида у = а + Ьх погрешность метода анализа состоит из трех частных погрешностей, обусловленных погрешностями определения констант а, b и значения уаа. Эти три погрешности суммируются по закону распределения погрешностей *. Если каждый из п стандартных растворов анализируется без повторений, а проба с неизвестным содержанием — т раз и для этих т проб среднее значение ула, то:
1 , /3 11 п(-/ап —У)-
b V “х[т п
(9 34)
с f = п — 2 **.
Анализ уравнения (9.34) показывает, что стандартное отклонение результата анализа 5Хан тем меньше, чем:
а) круче I радуировочный график (чем больше коэффициент регрессии), т. е. чем больше значение е и толщина кюветы /;
б) больше число измерений п при построении градуировочного графика. В общем случае п должно быть не менее 5. При фотометрических определениях погрешность измерения у (оптической плотности А) зависит еще и от абсолютного значения оптической плотности Ввиду этого в области высоких значений погрешностей sr (или sx) (см. разд 3.1 3) следует предусмотреть дополнительные стандартные растворы;
в) меньше разность значений у,,,— у. Она будет наименьшей, кшда уа, = у При больших различиях уан и у значения s, возрастают. Поэтому интервал содержаний (концентраций), для которого строится градуировочный график, не должен быть сильно растянут.
Доверительный интервал результата анализа Дхан вычисляют, как обычно, по формуле
ЛАш = «л zp. f (9 35)
Аан
но при f = п — 2, а не при f — т — 1.
* См. уравнение (9.19).
** Если проводят параллельные измерения для каждого из п стандартных растворов и число параллельных измерений равно kr то в уравнении вместо п берется п' = nk} и f = п' — 2.
316
Рис. 9.1. Доверительные границы концентраций и предел обнаружения, рассчитываемые по уравнению линейной регрессии у = а + 4- Ьх.
Границы доверительного интервала результата анализа находят по формуле:
*ан ± Л-Хан (9.36)
На рис. 9.1 приведены границы доверительного интервала в зависимости от содержания (концентрации) х. Они задаются двумя ветвями гиперболы, расположенными
линии регрессии, и убедительно демонстрируют возрастание доверительного интервала Дх по мере увеличения разности уап — у, т. е. по мере удаленности значений хан от средней точкц с координатами X, у.
Предел обнаружения (хи р) * [82]. Ранее (см. разд. 3.1.2.1) указывалось, что предел обнаружения определяется значением холостого опыта и его случайными отклонениями. Имея уравнение регрессии, значение у для холостого опыта (г/мпв) можно получить, экстраполируя прямую к значению концентрации х = 0. В этом случае
#мин — а + *Р, fsa (9.37)
При переходе от г/мин к хмин р- необходимо принимать во внимание доверительный интервал линии регрессии (см. рис. 9.1). Так, прямую уыкн = а + t-p fsa следовало бы провести до пересечения с нижней ветвью гиперболы и таким образом оценить хмин р**- Однако на практике этот расчет труден, и поэтому вместо гиперболы приближенно используют соответствующую асимптоту. Уравнение асимптоты:
у = а + Ьх + tp, fsb (х — х)
(9.38)
Подставляя найденное из уравнения (9.37), получаем:
*мин, Р ^мии, Р
tp, г К + xsfc] Й + /р_ fSb
(9.39)
Пз уравнения (9.39) следует, что на значение хмни р абсолютное значение холостого опыта не оказывает влияния. Значе
* Другие варианты расчета предела обнаружения см. разд. 3.1.2.1.
** Получающееся значение абсциссы представляет — с вероятностью Р (односторонняя постановка задачи).
317
ния хнии р определяется значениями случайных погрешностей sa, sb, наклоном градуировочной прямой и средним значением х. Число стандартных растворов мало влияет на хнин р, но оно должно быть более 12; доверительные границы в большинстве случаев строят для Р — 0,99.
9.3.1.3. Пример расчета (82]
Для построения градуировочного графика при фотометрическом определении бензола в УФ-области измерены оптические плотности семи стандартных растворов. Полученные результаты приведены в табл. 9.4 Далее проанализированы две серии двух растворов неизвестной концентрации бензола в спирте (по три параллельных _пробы). По экспериментальным данным рассчитаны средние значения уап (Лан) = 1 >53 (для первой серии) 'и уяп (Лан) = 0,93 (для второй серии). Рассчитать значения хан и Лхан и предел обнаружения хмин р.
Расчет характеристик линейного графика. Вычистечие коэффициентов регрессии удобно проводить в табличной ферме, где приводятся результаты расчетов сумм ^Хр ^ур и сРед;1их значений у и х.
x = ^i-=l,53; 0 = 2^-= 0,951; (2 xt)2 = 114,49. п п ь/
Проверку правильности вычислений осуществляют по уравнению?
£(ч+&)2 = £ 4-2 Г -'^1 + Е И <9-
Значения сумм из табл. 9.4 подставляют в уравнение (9.40). При условии правильности вычислений значения сумм левой и правой частей уравнения будут равны и можно будет приступить к расчету констант а и b по уравнениям (9.24) и (9.25) или (9.24') и (9.25')г подставляя в них найденные значения для сумм из табл. 9.4.
Т а б л и ц а 9.4. Вычисление коэффициентов регрессии
Номер по пор. Концентрация г/л Оптическая плотность »i (Al) Х1 у1 х1«1 Xi+Vl
1 0,2 0,20 0,04 0,0400 0,040 0,40 0,1600
2 0,5 0,37 0,25 0,1369 0,185 0,87 0,7569
3 1,0 0,64 1,00 0,4096 0,640 1,64 2,6896
4 1,5 0,93 2,25 0,8649 1,395 2,43 5,9049
5 2,0 1,22 4,00 1,4884 2,440 3,22 10,3684
6 2,5 1,50 6,25 2,2500 3,750 4,00 16,0000
7 3,0 1,80 9,00 3,2400 5,400 4,80 23,0400
Е= 7 10,7 х = 1,53 6,66 у = 0,951 22,79 8,4298 13,850 17,30 58,9198
Примечание. согласно уравнению (9.40) и табличным данным: 58,9198 = 22,79 + 2. 13,850 + 8,4298; 58,9198 = 58,9198, т. е. вычисления в табл. 9.4 выполнены правильно.
318
Из уравнений (9.24), (9.25) * вычисляем:
Ь = (7-13,850— 10,7-6,66)/(7-22,79 — 114,49) = 0,5700337
а = (6,66 — 0,570337-10,7)/7 = 0,079628
По уравнению (9.29) рассчитываем сумму квадратов для дисперсии, характеризующей отклонение измеренных значений у-{ относительно вычисленных У,:
2j ({/; —У/)2 = 8,429800 —0,079628-6,66 —0,570337-13,850 = 0,000311
4* = 0,00031/5 = 0,0000622
Из уравнений (9.26) и (9.27) находим дисперсии для констант Ь и ал
s2b = 7-0,0000622/(7-22,79 — 114,49) = 0,00000967;
st = 0,0031 (с f = 5)
s2 = 0,0000622-22,79/(7-22,79— 114,49) = 0,00003147;
sa = 0,00561 (c f = 5)
При P = 0,95 tp, f = 2,57 (см. табл. 9.3). Отсюда Д6 = Sbtp, f = 0,00799 и Да = satPf [ = 0,014. Следовательно, при Р = 0,95: b = 0,570 ± 0,008; а = 0,079 ± 0,014.
Расчет характеристик результатов анализа. Для серии растворов с £/ан (^ан)= = 1,53 согласно уравнениям (9.33)—(9.35) вычисляем среднее значение результата анализа:
*ан = (1 >533 — 0,070)/0,570 = 2,55 г/л
При Р = 0,95 и f = 5 доверительный интервал для хап равен!
Дх . 2’57 l/o 0000622 Г 1 ! 1 ' 7(1.533-6,66/7)2 ]_
ан 0,570 V 0,0000622 £ g + ? + 0 570а (7.22,79 — Ц4,49) J
= 0,028 г/л
Следовательно, граница доверительного интервала при Р — 0,95 равна 2,55 ± 0,03 г/л.
Аналс~ичные расчеты для второй серии растворов с уан (Аав) = 0,93 дают значение хав = 1,39 г/л. Так как в этом случае уан — у, то третий член под корнем уравнения (9.34) мал. При Р — 0,95 абсолютная погрешность анализа становится менвше (tixaH ~ 0,02 г/л), и граница доверительного интервала равна 1,39 ± 0,02 г/л.
Расчет умнн и предела обнаружения хмин р (Смин> р) проводят согласно уравнениям (9.37) и (9.38) для Р = 0,99:
Умни = 0,079 + 3,37-0,0056 = 0,098
*мнн. р = 3,37 (0,0056— 10,7/7-0,0031)/(0,570 + 3,37-0,0031) = 0,060 г/л
* Программы расчетов констант а, Ь на микрокалькуляторах «Электроника» БЗ-21, БЗ-34 [394—401].
319
9.3.2. ПРОВЕРКА ЗНАЧИМОСТИ КОНСТАНТЫ а
В ряде случаев значение а настолько мало, что этим членом уравнения прямой можно пренебречь. При зависимости у = Ьх вы-
числения значительно упрощаются. Тогда: *= (9-41)
sh= = (9 42)
SQ' = LGg—= (9 43)
sl = Aixi 2 ч и =n -') <9 44>
Afc = SbfP, f = {P. I ^syx! E x\ (9 45)
В ситуации, когда значение а уравнения у = а + Ьх мало отличается от нуля и возникает предположение, что уравнение может иметь вид у = Ь’х, осуществляют проверку константы а по следующей схеме. Вычисляют сумму квадратов SQ [уравнение (9 29) ] и SQ' [уравнение (9.43) ] соответственно с f — п — 2 и
Таблица 9.5. Значения F-распределения в зависимости от числа степеней свободы fr и /2 Для Р = 0,95 [82] *
ft 1 2 3 4 6 8 10 12 20
1 161 200 216 225 234 239 242 244 248
2 18,51 19,00 19,16 19,25 19,33 19,37 19,39 19,41 19,44
3 10,13 9,55 9,28 9,12 8,94 8,84 8,78 8,74 8,66
4 7,71 6,94 6,59 6,39 6,16 6,04 5,96 5,91 5,80
5 6,61 5,79 5,41 5,19 4,95 4,82 4,74 4,68 4,56
6 5,99 5,14 4,76 4,53 4,28 4,15 4,06 4,00 3.87
7 5,59 4,74 4,35 4,12 3,87 3,73 3,63 3,57 3,44
8 5,32 4,46 4,07 3,84 3,58 3,44 3,34 3,28 3,15
9 5,12 4.26 3,86 3,63 3,37 3,23 3,13 3.07 2,93
10 4,96 4,10 3,71 3,48 3,22 3,07 2,97 2,91 2,77
11 4,84 3,98 3,59 3,36 3,09 2,95 2,86 2,79 2,65
12 4,75 3,88 3,49 3,26 3,00 2.85 2.76 2,69 2,54
13 4,67 3,80 341 3,18 2,92 2,77 2,67 2,60 2,46
14 4,60 3,74 3,34 3,11 2.85 2,70 2,60 2,53 2,39
15 4.54 3,68 3,29 3,06 2,79 2 64 2,55 2,48 2,33
16 4,49 3,63 3,24 3,01 2,74 2,59 2,49 2,42 2,28
17 4,45 3,59 3,20 2,96 2,70 2,55 2,45 2,38 2,23
18 4,41 3,55 3,16 2,93 2,66 2,51 2,41 2,34 2,19
19 4,40 3,52 3,13 2,90 2,63 2,48 2.38 2,31 2,15
20 4,35 3,49 3,10 2,87 2,60 2,45 2,35 2,28 2,12
* При flt f2 > 20 и для промежуточных значений ft и /г значения F можно найти в приложении монографии [82] Там же приведена таблица значений F для Р = 0,00
320
f — п — 1 степенями свободы. При этом всегда SQ' > SQ. Затем находят соотношение:
F = (SQ'-SQ)/s2bX (9.46)
Здесь s%x — дисперсия, вычисленная по уравнению (9.28).
Сравнивают значения F и F (Р, Д = 1, f2 — п — 2) (табл. 9.5). Если F < F (Р, /2), то имеются основания для перехода к урав-
нению у = Ь'х.
Пример. На основании данных фотометрических определений рассчитано уравнение градуировочного графика у = а + Ьх. Требуется проверить, возможно ли более простое уравнение вида у — Ь' х.
Имеются следующие данные;
«/= 0,101 + 115,567* у =119,625*
SQ = s2 (п — 2) = 0,4107 SQ' = (s')2 (п — 1) = 0,4682
Результаты представляем по схеме:
•SQ f
«/= Ь'х 0,4682 18
у — а + Ьх 0,4107 17 0,0242
Разность 0,0575 1
F = 0,0575'0,0242 = 2,38 Из данных табл. 9.5 получаем F (Р = 0,95.
= 1, f2 = 17) = 4,45. Следовательно, между обеими дисперсиями [syJr = = 0,0242, вычисленной по уравнению (9 28) и s^x — 0,0260 — по уравнению (9.42)] нет значимой разницы, поэтому переход к уравнению у = Ь'х допустим.
9.3.3. ПРОВЕРКА ГИПОТЕЗЫ ЛИНЕЙНОСТИ ГРАДУИРОВОЧНОГО ГРАФИКА {82, 380, 388, 390]
Линейная зависимость у от х при построении градуировочных графиков, используемых в аналитических методах, нередко нарушается по тем или иным причинам. Поэтому следует проверять гипотезу линейности
Эксперименты должны планироваться так, чтобы для каждого стандарта было выполнено т параллельных определений. Гипотеза линейности проверяется путем сравнения дисперсии (s2), обусловленной рассеянием средних значений (#г) относительно линии регрессии с дисперсией (5вОспР), обусловленной погрешностями воспроизводимости при параллельных определениях. Составляют отношение
__ Дисперсия разброса средних значений
Дисперсия разброса внутри параллельных определений
= ~~ (9.47)
С“
воспр
которое сравнивают со значением Гтаел, найденным для Д == = п — 2 (для числителя) и f2 = п (т — 1) (для знаменателя) из табл. 9.5.
11 М. И Булатов и др.
321
Значение s2 находят из уравнения;
п
s2-=-^^(yi-Yi}2 <9-48)
Здесь у — среднее значение для г-й серии, состоящей из т определений} Yj — вычисленное значение для каждот о отдельного опыта i-й серии.
Дисперсию для разброса «внутри параллельных определений» определяют по формуле:
У (уц—У1)2 л = ________
воспр г т—1
У (.УЦ Si)2 У (Уг) Sr)2 У У (Уи Si)2
+ 4-^?-m--T- +-+VJ=1/n-l - (9Л9)
tn
Здесь у (уц — Si)2Hm — 1) = si — Дисперсия i-it серии; т — число опре-делений в каждой серии; г — число серий; yAj, y2j, , yij Ут] — /-е определение соответственно в 1, 2, i, г сериях; St, S& Sb ••• Sr — средние значения соответственно в 1, 2, ..., I, .... г сериях
Если есть основания сомневаться в однородности дисперсий 2 2 2 2
Si, S2, .... Si. .... sr. полученных при анализе отдельных стандартов и применяющихся в формуле (9.49) для расчета ^воспр, то предварительно проверяют гипотезу однородности этих дисперсий.
Если Етабл > F, то дисперсионное отношение незначимо, т. е. рассеяние точек относительно линии регрессии определяется только погрешностью воспроизводимости и, следовательно, градуировочный график имеет линейный характер. Когда же БтаСл < <Z F, то различие между дисперсиями № и slocnp не является случайным и гипотеза линейности графика должна быть отброшена.
Нарушение линейности обусловливается различными причинами. Так, при фотоколориметрических определениях линейная зависимость между оптической плотностью растворов и концентрацией определяемого элемента в ряде случаев не выполняется. Эти отклонения бывают обусловлены рядом причин, связанных с состоянием вещества в растворе, а чаще с недостаточной монохроматичностью светового потока при использовании фотоколориметров с относительно широкими полосами пропускания света.
Градуировочные графики в общем случае отличаются от прямых, поскольку по отношению к полихроматическому световому потоку окрашенное вещество является светофильтром, неодинаково поглощающим проходящий через него свет различных длин волн. А. Б. Бланк [80] показал, что пренебрежение этим фактом при определении микроколичеств элементов приводит к значительным систематическим погрешностям.
322
Часто в этих случаях характер расположения точек на кривой указывает на то, что градуировочный график лучше описывается уравнением параболы, проходящей через начало координат!
у=ах-\-Ьх2 (9.50)*
Л = аС + ЬС2 (9.51)
Для вычисления параметров а и b параболы применяют формулы;
(и п п п \ I Г п п / п VI
SЕ А- Е A S / Е A S A- S
4=1 4=1 4=1 4=1 / / 1.4=1 4=1 \4=1 / J
(9.52)
(п п п п \ I Г п п / п \21
4=1 4=1 4=1 4=1 / / L 4=1 4=1 \4=1 / J
(9.53)
Дисперсии параметров а и b вычисляют из уравнений;
п I Г п л / п \21
6« = Ах Ё А / S А Ё х4 - ( Ё *4 ) <9- 54)
4=1 / L4=l 4=1 \4=1 / J
n / Г п п / п \21
(9.55)
z=i / L»=i »=i м=1 / J
п
Здесь s=x = J] (yt — Y,)2/(я — 2); (y{ — Yt) или (A{— A) — разность t’s=l
между экспериментально найденным значением t/j (Ag) в 4-м опыте и значением Vi (Л), вычисленным по уравнению (9.51).
В тех случаях, когда градуировочный график искривляется, расчет по уравнению (9.20) дает прямую с относительно большим свободным членом, и при малых концентрациях точки, найденные опытным путем и вычисленные по уравнению, особенно заметно отличаются [80].
Уравнение параболы (9.50) в этом случае значительно лучше описывает зависимость оптической плотности (А) от концентрации (С), что иллюстрируется данными, приведенными в табл. 9.6.
Если для расчетов пользуются не уравнением, а построенным градуировочным графиком, то следует принимать во внимание;
1) графики, которые служат измерительным целям, необходимо
* Алгоритм и укрупненная блок-схема алгоритма расчета на ЭВМ оптимальной формы связи между двумя переменными физическими величинами, включающие вычисление констант а, Ь, проверку значимости константы о, проверку гипотезы линейности связи (г/;, хг-), выбор оптимальной формы связи, представлены в монет рафии Е Н. Львовского [388 ]. Разработана также программа для ЭВМ ЕС 1020, проверяющая 19 различных форм связи, включая линейную и квадратичную формы (Негура И. Г. — В кн.: Повышение качества и эффективности бетонных конструкций и изделий на базе применения передовой технологии. Кишинев, 1978).
1И
323
Таблица 9.6. Рассчитанные и найденные из опыта значения оптической плотности при построении градуировочного графика для определения меди с диэтилдитиокарбаминатом свинца [80]
Линейное уравнение у ~ 1,725- 10-2 + 2,303-10-2 х; параболическое уравнение у = 2,706- IO"* X — 1,771- 10-4х2.
Концентрация Х-, МКГ/мл Число опытов п А (измеренная) А (рассчитанная по линейному у равнению) А (рассчитанная по параболическому уравнению)
1,0 1 0,027 0,040 0,027
2,5 4 0,067 0,075 0,067
5,0 4 0,136 0,132 0,131
7,5 4 0,194 0,190 0,193
10.0 4 0,252 0,248 0,253
12,5 4 0,309 0,3('5 0,311
15,0 3 0,363 0,363 0,366
вычерчивать с большой точностью и в достаточно большом масштабе (во mhoihx случаях примерно 20x25 см или 25x30 см);
2) если данные разбиты на небольшие группы, то примерно половина точек каждой группы должна лежать но одну сторону кривой, а половина — по другую;
3) если необходимо, чтобы воспроизводимость отсчетов по графику отвечала воспроизводимости измерений, то наименьшее расстояние, которое можно отсчитать на графике, должно соответствовать погрешности измерений. Так, если погрешность отсчетов достигает ±0,5 мм, то абсолютную погрешность измерений и следует принять равной этому значению (в масштабе). Однако рассмотренное требование далеко не во всех случаях можно выполнить на практике и часто приходится от него отступать обычно в сторону уменьшения масштаба. Следовательно, отсчеты по графику могут быть менее воспроизводимы, чем измерения, для которых он составлен.
9.4. РАСЧЕТ ОПРЕДЕЛЯЕМОГО МИНИМУМА ЦВЕТНОЙ РЕАКЦИИ [67, 80]
При расчете определяемого минимума т (мкг)
т = AqjiB- 103/е*
следует исходить из того, что значение А в этом уравнении определяется погрешностью анализа, которая характеризуется при помощи стандартного отклонения. Так как стандартное отклонение зависит от количества определяемого вещества [671, то целесообразно оценить его величину для значения оптической плотности А, соответствующего определяемому минимуму т.
* См. разд. 3.1.1.2.
324
Для этого следует воспользоваться экспериментально найденной зависимостью sA — f (Д) (в форме графика или эмпирического уравнения). Мерой определяемого минимума можно считать, по-видимому, такое значение А, для которого абсолютная погрешность при заданном числе опытов и достаточно большой доверительной вероятности Р не превысит половины определяемой величины. Вычисляя доверительный интервал ДД по формуле kA=tptfSAlif п, получим для Р — 0,95 и числа опытов п = 3 значение ДД = = 2,5sa, а Дмин = 5$л. Значение находят путем аналитического (если это возможно) или графического решения системы уравнений!
IА = ч
(9.56)
Этот способ применяли для расчета определяемого минимума меди при анализе иодида натрия особой чистоты [67, 80]. Различное содержание меди в иодиде натрия получали, смешивая в необходимых соотношениях раствор препарата с содержанием меди 8-10“5% с раствором препарата, практически полностью очищенным от меди; масса Nal в каждом опыте составляла 10 г. Определение меди в Nal проводили фотометрированием хлороформных экстрактов диэтилдитиокарбамината меди [205]. Оптическую плотность исследуемых растворов измеряли относительно раствора холостой пробы (не содержащей иодида натрия). Использованные в расчетах значения оптической плотности представляли средние арифметические величины из трех отсчетов на приборе, исправленные на собственное поглощение кювет.
В табл. 9.7 приведен расчет стандартного отклонения sA для одной серии опытов [205].
2Лг — 3951 -10~4; А = 394.10~4; S (Лг — А)2 = 5443-10"8. Число опытов п = 10.
Дисперсия sA = 5443-10 8/9 = 6,05-10 6.
Стандартное отклонение = т/6,05-10-6 — 2,46-10~3.
Таблица 9.7. Расчет стандартного отклонения для одной серии опытов
Номер опыта Оптическая плотность Лг10* (Л—Л) х х Ю' (Лг-лр< хю8 Номер опыта Оптическая плотность Аг 104 (Л—Л)Х ХЮ4 ХЮ»
1 430 35 1225 6 397 2 4
2 360 —35 1225 7 407 12 144
3 383 — 12 144 8 383 — 12 144
4 397 2 4 9 357 —38 1444
5 417 22 484 10 420 25 625
325
Таблица 9.8. Стандартные отклонения при разных значениях А (число опытов в каждой серии п = 10)
Номер серим Л. 10 s„.10’ Аг. 10г Asz. 10‘
1 1,868 4,34 3,489 8,007
2 1,022 3,42 1,045 3,495
3 0,055 2,62 0,300 1,436
4 0,395 2,46 0,156 0,097
5 0,222 1,92 0,049 0,426
6 0,083 1,79 0,007 0 149
Значения стандартных отклонений для шести серий приведены в табл. 9.8.
Зависимость = f (Л) оказалась в данном случае линейной. Коэффициенты прямой sz = а ф- ЬА рассчитывали по табличным данным (табл. 9.8) методом наименьших квадратов:
«U^sa—
-
Уравнение прямой имело вид:
= 1,761 IO’3 + 1,446 - 10-=Л
Аналитическое или графическое (рис. 9,2) решение системы уравнений
I sA = 1,761-10"3 -Ь 1,446-10’М
(sz = А/5
дало значения: 5/
При расчете т вечных графиков
Sfi-Ю3
А
= 1,9.10-3; Амив = 9,5.10"3.
пользовались уравнением одного из градуиро-для определения меди на фотоколориметре
ФЭК-М:
А = 2,706 10-2т -1,771- 10"4та
Подстановка найденного значения АМ11Нв это уравнение дала т = 0,35 мкг меди.
Рис. 9.2. Определяемый минимум меди:
J — прямая = / (А) рассчитана методом наименьших квадратов; 2 — прямая получена по уравнению А = 5sy]e Точка пересечения прямых 1 и 2 (s^ = 1,9.10-а, А = 0,01) соответствует определяемому минимуму.
826
Условная чувствительность реакции = 5-10“2 мкг/см2 рассчитана по уравнению
= Лр.В-1б3/е
при в = 12,8-103 (см. разд. 3.1.12). Это значение чувствительности при 9 = 5 см2 соответствует значению определяемого минимума 0,25 мкг.
Таким образом, при тщательной аккуратной работе фактически достигнутое значение определяемого минимума лишь немного выше соответствующего значения, рассчитанного теоретически.
9.Б. СОПОСТАВИТЕЛЬНАЯ ОЦЕНКА ВОСПРОИЗВОДИМОСТИ ДВУХ ФОТОМЕТРИЧЕСКИХ МЕТОДИК АНАЛИЗА [82]
Нередко перед химиком возникает вопрос объективного выбора (или сопоставления) различных фотометрических методов (методик) анализа одного элемента. Решение этой задачи определяется во многом поставленными целями, среди которых, как правило, на одном из первых мест стоит «высокая воспроизводимость». Ответ на этот вопрос может быть получен путем сравнения стандартных отклонений двух методик (по Е-критерию).
Пусть необходимо сравнить воспроизводимость двух методик или результатов, полученных по одной методике в разных условиях. Примем, что первая методика имеет большее стандартное отклонение — su а вторая — меньшее s2; число степеней свободы в первом случае пусть будет Д, а во втором — f2*. Требуется определить, обусловлена ли разница значений sx и s2 случайными колебаниями или второй метод обладает лучшей воспроизводимостью. Для этого сравнивают дисперсии обоих методов по F-критерию. Рассчитывают значение
F = s2/s2 (9.57)
и сравнивают его со значениями табл. 9.5. Если F <Z F (Р, flt /2), то расхождения Sj и s2 незначимы и, следовательно, обе методики обладают одинаковой воспроизводимостью. Если же F > F (Р, fit /г), то расхождения Sj и s2 значимы и при прочих равных условиях следует отдать предпочтение второй методике (с s2), даже если она обладает и небольшой систематической погрешностью. Несмотря на небольшой систематический сдвиг результатов, при этом можно получить значение более близкое к истинному содержанию, чем при применении «более правильного» метода, но дающего большую случайную погрешность.
* Если значения s получены методом регрессионного анализа и коэффициент а линейного градуировочного графика значим, то / = п — 2; в других случаях f = п — 1.
827 1
9.6. ОБЩИЕ РЕКОМЕНДАЦИИ ПО МЕТРОЛОГИЧЕСКОЙ ОЦЕНКЕ РЕЗУЛЬТАТОВ АНАЛИЗА [402-4041
Вопросы, касающиеся формы представления результатов фотометрических опредетений, рассмотрены в разд. 3.2.3. Однако в связи с особым вниманием к оценке правильности результатов анализа Журнал аналитической химии [402] предлагает авторам и исследователям пользоваться унифицированными таблицами для представления данных о воспроизводимости результатов анализа и правильности предлагаемых методик * с минимальным необходимым набором метрологических оценок экспериментальных данных.
Воспроизводимость результатов анализа
Объект Определяемый
анализа компонент
Примечав не. Здесь и в последующих таблицах: т — навеска пробы для анализа (мкг, гит. д.); с — результат анализа как среднее выборки из и определений (%, г/л, моль/л, гит. д.); s — стандартное отклонение (%, г/л, моль/л, гит. д.); sr —относительное стандартное отклонение; с ± б — доверительный интервал величины с (%; г/л, моль/л, гит. д.), где б « tp, js/Уи; Р — доверительная вероятность.
Правильность результатов анализа. Проверка по стандартным образцам (СО)*
Название и номер СО Аттестованное содержание определяемого компонента, % т п с S(M с ± б (Р =...)
* Следует обратить особое внимание на то обстоятельство, что ввиду двух составляющих систематических погрешностей (пропорциональной и постоянной) полноценная аттестация методики анализа на правильность должна проводиться для нескольких разных содержаний определяемого компонента, т. е. по меньшей мере для двух СО с меньшим и большим содержанием компонента в сравнении с анализируемыми образцами. Аналогичным образом оценку пропорциональной погрешности следует проводить минимум для двух уровней добавок [58 ].
Правильность результатов анализа. Проверка со поставлен и e^i результатов, полученных по разработанной методике, с результатами, полученными другим методом
* Возможно также объединение данных, характеризующих вопроизводи-мость и правильность в одну таблицу.
328
Правильность результатов анализа. Проверка методом стандартных добавок
Объект анализа Определяемый компонент т Введено, %, г/л н т. д. п 6 8
Примечания: 1. Де сс доб. ^без доб.
2. Величину s при вычислении Дс находят как корень квадратный нз суммы квадратов стандартных отклонений значений s-*c д0^ н §2без доб. Эту величину принимают во внимание при вычислении значения (Дс ± бд^).
3. Дня надежной проверки правильности результатов введенная стандартная добавка должна увеличивать аналитический сигнал определяемого компонента в 2—3 раза.
Правильность результатов анализа. Проверка систематических погрешностей (и опенка селективности методики)
Определяемый компонент Концентрация определяемого компонента, %» г,л, моль/л и т. д. Концентрация сопутствующего компонента, %, Г/л, моль/л н т. д. Варьируемые факторы п С S с ±С (Р =
t pH и т. д.
1 2 3 4 5 6 7 8 9 10
Примечание. Графа 3 может быть заменена графой «Соотношение концентраций определяемого н сопутствующих компонентов», например, 1 : 10; 5 : 7 и т. д.
Важной метрологической характеристикой является интервал содержаний данного компонента, определяемых по предложенной методике. При анализе следовых содержаний нижней границей этого интервала часто считают то минимальное содержание, которое можно определить с относительным стандартным отклонением sr — 0,33. Критерий верхней границы определяемых содержаний компонента зависит от назначения методики.
Выше приведены общие рекомендации по представлению метрологических характеристик результатов определений. Необходимо также помнить и учитывать, что основные метрологические характеристики методики анализа—воспроизводимость (см. также разд. 9.1.1), правильность, нижняя граница определяемых содержаний, предел обнаружения (или предел определения) — в большой степени определяются метрологическими характеристиками используемых приборов (классом их точности), классом точности мерной посуды и весов [58, 82], качеством реагентов и реактивов, условиями проведения определения (см. разд. 3.2), природой анализируемого объекта и т. д. Поэтому перечисленные характеристики также следует отражать при описании методики.
329
Глава 10
ПРИМЕРЫ РАСЧЕТОВ И ОБОСНОВАНИЙ В ФОТОМЕТРИЧЕСКОМ АНАЛИЗЕ
Расчеты в фотометрическом анализе в большинстве случаев выполняют с использованием основного закона светопоглощения, закона действующих масс и закона сохранения массы вещества. Обстоятельные расчеты, связанные с различными вопросами фотометрического анализа и аналитического определения веществ, описаны в монографии Н. П. Комаря [405] и книге Л. П. Адамовича [404]. В монографии Батлера [406], посвященной математическому описанию ионных равновесий, изложены методика составления и способы решения системы уравнений. В предлагаемом ниже материале приведены некоторые приемы и способы расчета ионных равновесий., основанные на применении вышеназванных химических законов с использованием условных констант. Там же даны различные примеры приближенных расчетов, которые, несмотря на упрощающие допущения, позволяют получать вполне удовлетворительные результаты.
10.1. РАСЧЕТ ОТКЛОНЕНИЙ ОТ ЗАКОНА БЕРА ПРИ РАЗБАВЛЕНИИ ОКРАШЕННЫХ РАСТВОРОВ
Пример 1. Вычислить отклонение от закона Бера при разбавлении в 10 раз 5-10~4/И раствора окрашенного комплекса Pb (IP с ксиленоловым оранжевым при pH = 5,5 в ацетатном буферном растворе при равновесной концентрации ацетат-ионов 0,1 моль/л в условиях 10- и 100-кратного избытка реагента: а) при разбавлении буферным раствором исходного состава; б) при разбавлении дистиллированной водой.
Комплекс имеет состав PbH2R2~ и константу устойчивости Рк=3,3-108.
Решение. При pH = 5,5 и [СН3СОО~ ] = 0,1 моль/л вычисляем значение 0[t;
/6
£ Р.Н [Н+]‘ = 0,11;
о
= S РНсн3СОО-]'= 1,2. ю4; о
р; = з.з- ю8-о,11/1,2- ю4 = з-ю3.
При 100-кратном избытке реагента неравенство (2.13) соблюдается, и поэтому для вычисления отклонения от закона Бера можно использовать приближенное уравнение (2.15).
В условиях 10-кратного избытка реагента P’(Cpb =3.103.5.10'4 = 1,5 вначительно меньше, чем 100/[п" (р— 1)"]= 11,1, и для вычисления отклонения от закона Бера необходимо по уравнению (2.12) отдельно найти степень 330
диссоциации комплекса в каждом из растворов, а затем го общему уравнению (2.6) рг .считать значение отклонения А (%).
а. При р = Ю, решая уравнение Р’ = (1 — б)/[б (10 + 6) 6РЬ] для негодного и разбавленного в 10 ра® растворов, находим 60 = 0,07 и 6д == 0,40. Еатем вычисляем значение А
А = 100 (6? — 6<,)/( 1 — 6„) = 100 (0,40 — 0,07)/(1—0,07) = 35,5 %
При р — 100 ио уравнению (2.15) яолучим
а=юо (<?%- ₽;)/[₽;₽; (рпсрь)п]=
= 100 (10-3-103 — 3 103)/((3 103)2 100-5-10'4] = 6 %
б. При разбавлении в 10 pas водой концентрация ацетат-ионов уменьшается до 0,01 моль/л, чю приводит к увеличению условной конетани- устойчивости окрашенного комплекса.
При pH = 5.5 и [СН,СОО~] = 0,01 моль/л «Нац*- = 0,11, 1/арь,+= 11 и ₽„= 3,3-108-0,11/11 = 3,3- 10е.
При р = 10, решая, как и в е лучае «а», уравнение (2.12) для исходного и разбавленного растворов, находим во — 0,07 и од = 6-10~4, затем по уравнению (2.6) определяем
А= 100 (6-10'4— 7 10-2)/(1 — 7-Ю'2) = — 7,5
Ери р = 100 по приближенному уравнению (2.15) получим!
А = 100 (10-3-103 — 3,3-108)/(3 10»-3,3- 10е-100-5-КГ4) = — 0,66 %
Как показывает расчет, при разбавлении водой происходит относительное упрочнение комплекса Pb (II) вследствие значительного уменьшения влияния сопряженного комплексообразования ионов Pb (II) аце, ат-иоиами.
Пример 2. Определить относительное отклонение от закона Бера при разбавлении 0,2 М окрашенного раствора роданида железа FeSC№+ дистиллированной водой в 20 раз, если известно, что pH раствора равен 1 (0,1 М НС1), констаига устойчивости рк = 1 • 103.
Решение. При pH ® 1: aSCN~= t = °’58' 1/°^+ = 1 +
4 2
+ S Рк [СП* 4- S (OH“"]f = 5,2; = Ь ЮМ,58/5,2 = 1,14-102.
1 1
При разбавлении 0,1 М раствора НС1 водой в 20 раз можно принять, что [H+J = -^-= 5-10-3 моль/л. Тогда aSCN_=l; 1/Оре,+ = 2,6; = 1 X
X 103/2,6 — 3,8-102.
При стехиометрическом соотношении Fe (III) и CNS- как в исходном, так и разбавленном растворах неравенство (2.8) не соблюдается, поэтому по уравнению (2.7) необходимо найти долю не связанных в окрашенный комплекс ионных форм Fe (III) н по уравнению (2.6) — аначение А (%):
6o = O,21; — 0,40; А= 100 = 24 %
10.2. РАСЧЕТ РАВНОВЕСНЫХ КОНЦЕНТРАЦИЙ КОМПЛЕКСНЫХ ИОНОВ
Равновесные концентрации ионов и долевое распределение частиц рассчитывают с помощью функций образования, распределения и закомплексованности [43, 47, 48]. Расчет равновесных концентраций моноядерных комплексов по исходным аналитическим
331
концентрациям не вызывает особых затруднений, если известны константы всех сосуществующих равновесий. Правильность выполненного расчета можно легко проверить соблюдением материального баланса по концентрации комплексанта (лиганда), аналитическая концентрация которого, как правило, всегда известна. Некоторые затруднения могут встретиться, когда в рассматриваемых системах в заметных количествах существуют поли-ядерные комплексы типа MmLn. Функция закомплексованности в таких случаях выражается уравнением *:
р
= [СМ]/{Л1] = У [Lf + [L]" (10.1)
Решая это уравнение приближенно при заданных значениях [L J и [См], можно найти а-доли (и концентрации) любой из равновесных частиц ML/ и полиядерного комплекса MmLn. При проведении анализа присутствие полиядерного комплекса определяемого иона с посторонним комплексантом является нежелательным из-за осложнения применяемой фотометрической реакции, поэтому для фотометрпрования выбирают такие условия, при которых равновесная концентрация полиядерного комплекса с посторонними комплексантами несоизмеримо мала по сравнению с концентрацией моноядерпых частиц.
Из уравнения (10.1) следует, что образованием полиядерных частиц можно пренебречь, если
и 1МГ-1 £ pf [L]1 4 т [Lfp„m ) 110-2
I \ V /
(Ю2)
В тех случаях, когда концентрация [MmLn] не превышает 1% сп концентрации всех равновесных частиц, уравнение (10.2) упрощают, сопоставляя концентрацию полиядерного комплекса М„,ЕП с суммарной концентрацией только моноядерных комплексов:
т [MmLn] <0,01 1М£/] (10 3)
ИЛИ
1М) <
0,01 % pj- IL]")
о
(10.4)
(Р \ /
2+ lgOT-lg У} pj- [L)‘ -]_igpnm+nlg (L) / (m-1) (10 5)
Соблюдение неравенств (10.3)—(10.5) позволяет создавать условия, практически исключающие образование полиядерных ча
* Если в растворе сосуществуют полиядерные комплексы различного состава, то в уравнении материального баланса, а следовательно, и в уравнении (10.1) их необходимо учитывать.
332
стиц при любых концентрациях лиганда L. Если неравенства (10.3)—(10.5) не соблюдаются, то концентрацию полиядерных частиц можно уменьшить изменением концентрации лиганда L, что часто используют для предотвращения образования полиядерных гидроксокомплексов. Образование полиядерных комплексов при неизменной концентрации [М ] практически можно не учитывать, если
m [MJ"1-1 / j] pL [L]1 1 - IO'2 (10.6)
/ о
Пример 1. Рассчитать в общем виде равновесные концентрации комплексов MR, MR2....MRn при равновесных концентрациях реагента [Cr], конку-
рирующего комплексанта [Сц] и заданном значении pH.
Сбщая концентрация катиона М равна См- Полные константы устойчивости комплексов равны соответственно: для реагента RH? — 0J1, Р^1, ..., 0^*. ддЯ комплексанта LH? Pf1, Р|*,..., Р^; для комплекса МЦ—Р}“, 02.... 0^;для
комплекса М (ОН);- — 0®н, Р°Н, ..., Р°Н; для комплекса MRn — 0Г 02.0п,
Решение. При заданном pH по уравнению (1.6) рассчитываем а-доли депротонированных форм фотометрического реагента R и конкурирующего комплексанта L' о е
i/^ZW
0 и
Затем вычисляем равновесные концентрации стехиометрических форм фотометрического и комплексующего реагентов
[R] == [CR] aR и [L] = [CL] aL
Мольную a-долю комплексов MR, рассчитываем, учитывая все сопряженные равновесия, в которых участвуют катионы М:
a ₽1[Ю <10 71
aMR— п „ 1 110-О
14- Z Pi IR]’ 4- Z [L]‘ -J- Z [°н-]г i i i
a MR]2 <10 81
KMR,~ ' n p i (10-Ь1
1 4- Z ₽« 4- £ & + S 1 1 1
a ₽"[R]” (10 91
°MR„ 1 p j
1 4- Z MR1‘ 4- Z ₽HLll 4- Z ₽°H 1 1 1
n p i
l/aM = Z ₽» 1R1‘ 4- Z 1Ч‘ 4- Z P?H (10.10) 0 1 1
Равновесную концентрацию каждого компонента находим умножением a-доли этого компонента на общую концентрацию в растворе катиона М;
[MR] = CMaMR; [MR2] =
[MRnJ = CMaMR„; = СмаМ
333
П шмер 2. Рассчитать равновесные концентрации роданидных комплексов Со (II) при равновесной концентрации [CscnJ = 0-17 моль/л и pH = 1. Общая концентрация Со (II) в растворе составляет ЫО'2 моль/л. Полные константы устойчивости для роданидных комплексов Со (II) равны: Pi = 1 • Ю3, Р2 = !• Ю3, Рз = 2-102 и Р4 = 1,8-Ю2. Константа ионизации HSCN равна 1,4-10"1.
Решение. Используя уравнении Л0.7)—(10.10;, находим при l/asCN-=1 4- 10-Л/0,14 = 1,7; [SCN'J = CSCNaSCN_ = 0,17/1,7 = 0,1 моль/л 1 • 103-0,1
aCoSCN+ - 1 + ю3-0,1 + 103-10'2 +2-Ю2-10'3 4- l.e-iCH-lO"4 “ = 1 • 102/1,11 • 10я = 0,90
103-10-® ппп 2-102-10'3 , о„
а~ /сгкп = i n — 0,09 ; аг ,сгкч_ = . .г.лГ = 1,98-10 8 Со (SCN)2 1»11 дог с© (sci\)3 1ДЫ02
1,8 102-10~4 , д ш .
“Со (SCN)2' ~ 1,11-Ю2 “ 1,610 ’
aCo2+= 1/1.Н •102 = 9,0-Ю-з
[Со SCN+J = 1 10'2-0,9 = 9,0-10'3 моль/л
[Со (SCN)2J = 1 10'2-9-10~2 — 9,0 10'4 моль/л
[Со (SCN)j] = 1 • 10'2-1,98 10'3= 1,98-10~Б моль/л
[Co(SCN)('] = 110'2-1,6-10-4= 1,6-10-® моль/л
[Со2+] = 1 • 10'2-9- Ю'3 = 9,0-10“^ моль/л
Пример 3. В 1 • 10~3 М растворе FeCl3 рассчитать равновесную концентрацию роданидного комплекса Fe (III) состава 1 : 2 при pH = 3 и равновесных концентрациях роданида калия и хлорида натрия 0,1 моль/л. Как изменится концентрация комплекса в присутствии 0,1 моль/л K2SO4?
Полные константы устойчивости равны: для Fe(SCN)?~z —Pj ~ 1,07 X X 10е; р2 = 2,14-104, рз = 4,27-104, Р4 = 3,39-104, Р5 = 1,7-104, Рв = 1,7.103; для FeCl3'1 — Pj = 28,2, Р2 = 1,26-102, Р3 = 12,6, Р4 = 0,14; для Fe(OH)|~f — — р, = 1-1011, р2 = 5-1021; для Fe(SO4)?'2Z - Pt = 1,51 -104, Р2 = 2,51-107; для HSCN — Р = 7,15; для H2SO4 — Pi = 83,4.
Решение. Используя уравнения (10.7)—(10.10), находим при pH = 31 1/aNCS-= 1 + 7,15.10'3= 1; 1/аь02- = 1 + 83,4-10'3= 1,08
2
£ РНОН"Г= 110и-10_11 + 5 1021-10’22= 1,5 1
4
У, pf [СГ]г = 28,2-0,1 + 1,26-102-10-2 + 12,6-10“3 + 0,14-10-4 = 4,1 1
6
У Pi [SCN-]' = 1 + 1,07-103-0,1 +2,14-104-I0'2 + о
+ 4,27- IO4-10'3 + 3,39- IO4-10'4 + 1,7-104-10'$ + 1,7-103-10'® = 3,68-102 aFe (SCN)J = 2,14- Ю4-10'2/(3,68.102 + 4,1 + 1,5) = 5,74-10'4 (57,4 %)
[Fe (SCN)t[ = 1 • 10's-5,74-10'1 = 5,74-10'4 моль/л
334
В присутствии K2SO4i
2
Е Pi ISOl-]£ = 1.51 • 10*-0,1/1,08 + 2,51 • 107/(0,1/1,08)2 = 2,16-105 1
aFe(sCN)+ = 2’14’I°4'10‘2/(3-68-102 + 4,1 + 1.5 + 2,1б-106)= 1-Ю-з
[Fe (SCN)JJ = 1 • IO-3-1 • IO'3 = 1 IO"3 моль/л
Таким образом, в присутствии 0,1 моль/л K2SO4 окрашенный комплекс роданида Fe (III) практически не образуется (0,1 %).
Пример 4. Рассчитать и построить диаграмму, характеризующую состояние равновесия системы железо (II1) — сульфосалициловая кислота в зависимости от pH раствора и равновесной концентрации сульфосалициловой кислоты. Данные о составе и прочности образующихся в этой системе комплексов приведены ниже.
Константы устойчивости равны: для H2SSar — Р]4 = 4- 10u, Р^1 = 1,8-1014; для FeSSal+3f — Pj = 2,5-1014, P2 = 1,8-1025, P3 = 1,8-1032; для Fe(OH)f-‘‘ — _рон=1.10П1 pOH = 5.102i.
Решение. Для каждого рассматриваемого значения pH раствора вычисляем
2 2
i/«ssai = S pF [h+]z* Е р°н [°н“И о о
ttFe (SSallj =
Pt [CSSal ]taSSal_ 3_____________________________2
EP.[cssai]£4sal+EP?H[0H"r 1 о
при различных концентрациях [CssaiL изменяющихся от 10~4 до I моль/л. 2 2
Например, прн pH = 2: l/aSSal = Е PF [Н+Р = 2,2-1010; У Р°Н [ОН ]‘ = о о
= 1,1.
При [CSSa[ ] = 1 моль/л
«FeSSa! = [1.5-Ю14-1;(2,2-1О10)]
3
2 Рг1/(2,2 .101оу + 1,1] = 0,23 1
aFe(SSal)a “
= 1,8 -1025-1/(2,2 -IO10)2
з
Е Pi • 1/(2,2.1О10)‘ + 1,1
1
= 0,77
aFe <SSaI)a —
= 1,8-1032-1/(2,2- Ю10)3
3
Е Рг1/(2,2. 1О10У + 1,1
1
= 3,5-КГ4
aFe+Fe (ОН), — 1.1
3
Е Рг1/(2,2.1О1о)‘+1.1
1
= 2,3-10“5
333
Рис. 10.1. Диаграмма распределения (относительного содержания) комплексных ионов:
с — аЛ — а з. о j; сх — ар.сся1; б — схЛ — и ч-l а„ — а в-'
0 Fe+4-Fe(OH)? 1 1 hebSal a Fe(SSal)| 3 Fe (SSal)f *
[CSSai], моль/л; / — 1,0; 2 — 0,1; 3 — 0,01; 4 — 1.10"’; 5 — 1.10~<.
Здесь ciFe+Fe (OH); — суммарная а-доля Fe (III), находящегося в равновесии в виде акваиона и гидроксокомплексов.
Аналогично вычисляют а-дрли сульфосалицилатных комплексов при других значениях концентрации реагента н pH.
Построенная по этим данным диаграмма показана на рнс. 10.1.
Расчет показывает, что ни при каких условиях сульфосалицилатные комплексы состава 1 : 1 и 1 : 2 не достигают 99% Максимальные a-доли для комплексов состава 1 : 1 и 1 : 2 составляют соответственно 0.96 и 0,98. Образование комплекса состава 1 :3 наблюдается в зависимости от равновесной концен-т[ ации реатента при pH выше 4, но полнее связывание Fe (III) в этот комплекс может произойти только при pH выше 7.
10.3. РАСЧЕТ ОПТИМАЛЬНОГО ЗНАЧЕНИЯ pH ПРИ ФОТОМЕТРИЧЕСКИХ ОПРЕДЕЛЕНИЯХ
При взаимодействии гидролизующихся металл-ионов с реагентами слабокислотного характера выход продукта реакции зависит от pH раствора. Повышение pH приводит к увеличению концентрации аниона реагента, но из-за увеличения гидролиза уменьшается концентрация металл-иона. Снижение pH подавляет гидролиз, по увеличивает протонизацию реагента. Существует некоторое оптимальное значение pH раствора, при котором наблюдается максимальный выход продукта реакции (см. разд. 1.5).
Пример I. Рассчитать оптимальное значение pH раствора для реакции Ве2+ с альбероиом (H3R_), в результате которой образуется окрашенное соединение состава BeH2R (1 : 1).
Константы устойчивости гидроксокомплексов Ве2+ : Р^н = 2-108, Р®н = = 4-1015, PgiH = 2-1015, Р^и = 1' I015. Соогв₽тствующне константы протонирования аниона реагента: Р}1 = 3,16-1011, Р*1 = 2,5-1016, р.^ = 8-1018.
Решение. Решая уравнение (1.36) при п = 1, q — 3 и j = 4, получим:
3,2 • 103 (Н+р -1,5- 10“5[Н+}Б -5,1- 10-и [н+р _ 2,2• 10'18 щ+р _ — 2,5- IO'24 [н+р — 1,9. Ю"38 [Н+] — 1,3-10-* = 0 (10.11)
336
Решая уравнение (10.11) методом итераций, находим, что оно превращается в тождество при [Н+]=2-10-4 моль/л. Следовательно, рН0Пт — 3,7. Полученное значение pH хорошо согласуется с установленным экспериментально (pH = 4,5) [407].
Пример 2. Найти оптимальное значение pH для реакции железа (II) с 1-ни-трозо-2-нафтолом (HR). По литературным данным [67 ], в результате этой реакции образуется соединение состава 1 : 3.
Константа протонирования аниона реагента составляет 5-107. Константы устойчивости гидроксокомплексов Fe (II) равны: = 3.6-105, РРН = 5,9 X
X 109, Р2Н = 4,7-109, ₽РН = 3,6-108.
Решение. Решая уравнение (1.33) при п = 3, j = 4 и q = 1, получим:
1,5• 10s [Н+]5 + 0,36 [Н+]4 — 3,57- Ю'8 ]Н+]3 —1,18- 10’1в [Н+]2 —
— 1,41-IO'32 [Н+] - 1,44-10“47 = 0 (10.12)
Решая приведенное уравнение методом итераций, находим, что оно превращается в тождество при [Н+]= 4-10-9 моль/л. Найденное значение pH раствора, равное 8,4, хорошо согласуется с экспериментально установленным значением pH для этого определения равным 7,5 [408].
Пример 3. При фотометрическом определении Fe (III) применяемый реагент очень медленно взаимодействует с полнядерным гидроксокомплексом Fe2(OH)^+. Установить, при каких условиях концентрация полиядерного гидроксокомплекса Fe (III) будет несоизмеримо мала (^1%) по сравнению с моноядерными.
Решение. Запишем функцию закомплексованности для рассматриваемого случая:
CFe/[Fe3+] ее l/aFe3+ =
= 1 + 1011 [ОН'] + 5-1021 [ОН"]2 + 2-1,26-1025 [Fe3+] [ОН"]2
Затем в соответствии с заданием введем ограничивающее условие, а именно:
2-1,26-1025 [Fe3+] [OH-]2/CFe < 1 • 10~2 (10.13)
Комбинируя уравнение (10.13) с функцией закомплексованности, получим приведенное уравнение-
[ОН-]2 + 2- 10-22(10u ± 5- 1013;/Cj~) [ОН-] + 2-10’22 = 0 (10.14)
Это уравнение может иметь положительные действительные корни только при отрицательном значении линейного члена, т. е. когда 5-1013 КCFe 1- 10'1 или CFe > 4- IO'9 моль/л. Следовательно, при CFe 4-10-6 моль/л полиядерные частицы Fe2(OH)I+ практически не образуются ни при каких значениях pH раствора. Если аналитическая концентрация Fe (III) превышает 4-10~6 моль/л, то область pH, где образованием частиц Fe2(OH)J+ можно пренебречь, нетрудно найти решением уравнения (10.14)
[ОН-]= — 10"22(10i1 — 5-1013 j /-С^7) ±
± IO'14 j/10-22(101i±510131' С^)2 —2 (10.15)
Так при Сре— 1-10-2 моль/л из уравнения (10.15) получаем: [OH-Jj — = 1-10"9 моль/л и [ОН-]2 = 2-10-13 моль, л Следовательно, при Сре = 1 X X I0-2 моль/л образованием частиц Fe2(OH)4+ можно пренебречь при pH Z>5 и pH 1,3, если Сре — 1 • 10 4 моль л — при pH > 4 и pH 2,4 и т. д.
Пример 4. Комплексонат Fe (111) интенсивно поглощает свет с/. = 260 нм (1g е = 3,9) [409] Определить оптимальный интервал pH раствора, где образованием смешанных комплексонатов FeHY и FcOHY2- можно пренебречь, Константы образования lg KFeHY = 1-4 и lg Kf^qhy = 6.5 [42]. Общая концентрация Fe (111) примерно 5-10—моль/л.
337
Решение. Применяя уравнения (1.37) и (1.38) к рассматриваемым равновесиям, находим pHj > lg KpeHY + 2 = 3,4; рН2 12 — lg ^eOHY = = 5,5. При pH = 3,4 и pH = 5,5 значения lg Равны соответственно 14,3 и 14,7. При этих условиях КРеСРе составляет IO14-3- 10~4-3 = 1010 и 10'°’4, что значительно превышает значение 104, необходимое для полного Связывания Fe(lII) в комплексонат согласно неравенству (1.15). Следовательно, рНоцф — 3,4—5,5.
Пример 5. Найти опти лальное значение pH раствора при фотометрическом определении Fe (III) в виде сульфосалицилатного комплекса [Fe(SSal)3]6- в условиях 100-кратного избытка реагента. Общая концентрация Fe (III) в исследуемом растворе составляет 1 • 10“4 моль/л.
Константы устойчивости для Fe(SSal)3-31 : р1 = НО15, р2 = 5,7-1025, Р3= 4-1032; для Ре(ОН)?"‘; = 1-Ю1*, Р°н = 5-Ю21; для H2SSaF: Pf =
= 4-1011, Й1 — 1,8-1014. =3,2-10’38
2 sFe (OH)3
Решение. Оптимальное значение pH раствора, при котором достигается максимальный выход сульфосалицилатного комплекса Fe (III), находим по уравнению (1 36) при п = 3, т = 2, j = 2 и q = 0:
(tf'X [Нзо+]-2 + 2р°н^ [Нзо+]-3) (1 + pH [Нзо+] +
+ ₽2Н 1нзо+]2) - 3 0 + ₽РЧЛН3О+] +
+ ₽2°Х/[Н3о+]2) (pH + 2рН [Нзо+]) = 0
Решая это уравнение методом последовательных приближений, находим, что оно практически превращается в тождество при [Н3О+ ] = 5-10-12 моль/л, следовательно, рН0ПТ = 11,3. По приближенному уравнению (1.33) получаем рНопт = И,8 *. Более широкий интервал значений pH, где возможно надежнее фотометрирование окрашенного комплекса, можно получить из рассмотрения графической зависимости lg Рре <ssai)3 = f (PH). При вычислении значений ₽pe(Ssai) a-коэффициенты должны учитывать сопряженные реакции гидро-ксокомплексообразования Fe (III) и протонизации сульфосалицилат-ионов. Например, при pH = 9 получим l/«pes+ = 1+ 1011-10~5 + 5-1021-10-10 = = 5-1011; l/aSSap- =1 + 4-1011-10"» + 1,8-1014-10~2« = 4-102; ₽ре {SSal)> = = 4-1032/[5-1011 (4-102)3] = 1,25-1013.
Производя аналогичные вычисления в интервале pH от 7 до 13, получим графическую зависимость lg Р' = f (pH), представленную на рис. 10.2. Одновременно необходимо установить pH раствора, при котором достигается условное произведение растворимости Ks осадка Fe (ОН;3 и фотометрическое определение Fe(IIl) практически становится невозможным. При вычислении = ^s/ape>+ коэффициент aFe3+ должен тесину учитывать конкурирующее комплексообразование
* При этих значениях pH достигается максимальное развитие окраски раствора [162, с. 301].
Рис. 10.2. Зависимость условной константы устойчивости сульфосалицилатного комплекса Fe (III) И ₽^Fe(OH), ОТ РН:
1 - 1к ₽'; 2 - з - р <ип).
338
не только ОН-ионов, но и сульфосалицилат-ионов. Тчк, при pH =9 И ICssad = lOOC’Fe = 3-10-2 моль/л: [SSaF- ] = 3-10-2/(4-102) = 7,5X X 10-6 моль/л; l/aFe8+= 5-10u+ ЫОЧ.7,5-10-6 + 5,7-1026 (7.5-10-6)2 + + 4-1032 (7,5-10-6)3= 1,7-1020; K's = 3,2- IO-31*- 1,7-1020= 5,4-10-».
Ионное произведение 'ИП) при этих условиях составит: [Сре] [ОН_]3 = = 10** (10*6)3 = 10-18. Выполнив аналогичные вычисления во всем интервале вначений pH от 7 до 13, получим (см. также рис. 10.2):
pH 7 8 9 10 11 12 13
106 12-1 13,1 14-! 14-8 14.5 12,8 рД' 23,6 20,2 17,2 14,2 11,5 9,7 9,4
6Fe (ОН)3
р (ИП)Ре (ОН)> 25,0 22,0 19,0 16,0 13,0 10,0 7,0
paSSal3- 4,8 3,6 2,6 1,6 0,7 0,1 0
Из этих данных следует, что оптимальный интервал pH для фотометрического определения Fe (III) в виде [Fe(SSal).,j6~ находится в пределах от 10 до 12 (pHL„~= 11,3), где В' комплекса достаточно высока и /Се не дости-
' опт 1 лРе(ОН)3
гается При рН>12 фотометрирование проводить нецелесообразно, так как выход комплекса уменьшается, а вероятность выпадения осадка Fe(OH)3 увеличивается и при pH около 12,3 ионное произведение превышает значение I('s осадка Fe(OH)g. Нижняя граница pH раствора определяется таким значением, где Fe (III) еще количественно связывается в комплекс Fe(SSaJ)§~. При = 3-10-2 моль/л
минимальное значение pH раствора определяется соблюдением неравенства рз [cssai J3 “ssal-/ (₽2 [Cssai]2 “ssal-) > 1 ’1С|2> откУДа следует, что 1/ctssal3- 4‘ Ю32’3-10-2/(5,7-1026-102) = 2,1-103. Сопоставляя найденное значение рре jSSal) и й^аГ1- с приведенными выше данными, видим, что с досточкой точностью фотометрирование комплекса можно проводить в широком интервале pH — от 8,3 до 12.
10.4. РАСЧЕТ КОНЦЕНТРАЦИИ РЕАГЕНТА, ОБЕСПЕЧИВАЮЩЕЙ НЕОБХОДИМУЮ ПОЛНОТУ СВЯЗЫВАНИЯ ОПРЕДЕЛЯЕМОГО ЭЛЕМЕНТА
При взаимодействии гидролизующегося иона металла М с фотометрическим реагентом HgR, являющимся слабой кислотой, в присутствии конкурирующих комплексантов L степень связанности определяемого элемента в окрашенный комплекс и необходимую для этого концентрацию реагента рассчитывают из выражения условной константы устойчивости исследуемого комплекса MRn.
Пример 1. Вычислить в общем виде концентрацию фотометрического реагента HgR, необходимую для связывания определяемого иона М в окрашенный комплекс AIRn на 99,5%. Общая концентрация иона М равна С.
Решение. При связывании определяемого иона в комплекс MRn на 99,5% [MRn] = 0,9950, а [Сод] = 0,0050. Из выражения условной константы устойчивости находим, что [MRn]/[CM] = Рд; [CR]". по условию [MRn ]/[Сд11 = 0,9950/(0,0050) = 2-102. Следовательно, определяемый ион М будет связан в окрашенный комплекс MRn в том случае, когда P^R [CR]n > 2-102. П
339
Общая концентрация фотометрического реагента Cr (моль/л) будет равна С"мме его равновесной и координационно связанной концентраций: Cr = = (2-102/₽MRn)'/"+«C
Пример 2. Вычислить в общем виде концентрацию фотометрического реагента H^R, обеспечивающую за счет связывания определяемого иона М в окрашенный комплекс MRn понижение его равновесной концентрации, не связанной в комплекс, до 1-10~3 ст первоначального значения. Общая концентрация определяемого иона М равна С.
Решение. Определяемый ион М необходимо г,:.лзать в окрашенный комплекс MRn и в такой степени, чтобы [См] 1- 10~3С. При этих условиях [MRJ = С— Ы0-3С= С Н — Ы0-3) мольл. [MRn)/1См] = С (1 — 1 X X 10—3)/(1- 10"3С) = 1-103. Из выражения условной koi статы устойчивости комплекса MRn найдем равновесную концентрацию [Cr], обеспечивающую заданную степень связанности определяемого иона в окрашенный комплекс: [CR] (1- 1О3/РдШ )|/п. Общая концентрация фотометрического реагента CR (мель/л; будет сла!аться из ею равновесной и координационно связанной концентраций: CR = пС Ч- (1 • 103/pMR )1/п.
Пример 3. Вычислить концентрацию альберона (H3R~), обеспечивающую связывание Be24 в окрашенный комплекс состава BcIL R не менее чем на 99,9%. Фотометрическая реакция между Ве2+ и реагентом проводится при pH = 4,5 и концентрации Ве2+, равной 1 10~4 моль/л, в отсутствие других катионов, реагирующих с ал: бероиом
Константы устойчивости: для BeH2R — ₽ = 2,34-108; для Н ,R_ — Р[1== = 3,16-1011, = 2,5-10;с, fi*'= 8-101к; для Be(OH)?~'— f^H = 2-108,
fOH = 4. 1015 рОН = 2.1015 рон = ь i015
Решение. При pH = 4,5 ([Н ] — 3,2-10~5 моль/л) имеем: ан R!_ = / 3 1
= Р2Н [Н+]2/ £ Р,Н ШЧ: = 0,72; 1 аВе2+ = £ р?н [ОН*У « Г, ₽'BeHjR = / о о
= 2,34-108-0,72 = 1,68-10s.
По условию. [ВеН R ]'[Све] = 0,999С/(0,001С) = Ь103. Тогда: [Cr] 1 X X 103/pBeH,R = 1 • 10=7(1,68- 10s) = 6- 10-е Моль/л; CR = CBl + [CR] = = 1,1-Ю-4 моль/л.
По опытным данным [407] количество альберона, добавляемое при определении бериллия в этих условиях, составляет 1 мл 0,1% рас.вора в объеме 10 мл, что соответствует концентрации Cr = 1,8-10"' мольл.
Пример 4. Вычислить концентрацию ксиленолового оранжевого (H6R), обеспечивающую:
а) связывание Zn2+ в 1-10“ ' А1 водном растворе ZnCl2 до равновесной концентрации [CZn], равной 1-10-® моль/л при pH = 5,8;
б) связывание Zn2'’ в скрашенный комплекс ZnH2R2~ при pH = 5,8 на 99,9%.
Посторонние катионы, взаимодействующие с ксиленоловым оранжевым, отсутствуют. Константа устойчивости комплекса ZnH2R2~ равна 8,5-106.
Решение. При pH = 5,8 ([Н+ ] — 1.6-10-6 моль/л) имеем-
/ 6 4
«h2r<- = ₽2Н [НЧ2/ Ё ₽" 1н+11' = °’2: VaZn!+ = Ё ₽°Н Юи-]' = 1;
/ о о
₽ZnH2R = 8-5- 10S-0.2 = 1,7-10».
а. По условию, [ZnH2R2-]/[Cz„] = (1-10~4 — 1-10~G)/(l-10~°; = 1-102. Следовательно, PZnH Rs- [CR ] = ЫО2, откуда равновесная концентрация [CR] = 1' 102/pZnH R’-= Ь 102/(l,7-106) = 6-1О~6 моль/л. Общая концентрация реагента: Cr"= CZn+ [Cr]= 1-10~4+ 0,6-10-4= 1,6-10—4 моль/л.
340
б. По условию, [ZitHR2* J/[Czn] — 0,999/0,001 1 • 103. Равновесная кон-
центрация реагента, обеспечивающая связывание в комплекс на 99,9%, будет равна: [Cr] = 1-108/(1,7-106) — 6-10*4 моль/л. Общая концентрация реагента; Cr = I- Ю-4-Ь 6-10“4= 7-Ю*4 моль/л.
10.5. УСТАНОВЛЕНИЕ ВОЗМОЖНОСТИ ПРИМЕНЕНИЯ МАСКИРУЮЩИХ РЕАГЕНТОВ И АНАЛИЗА В ПРИСУТСТВИИ МЕШАЮЩИХ ИОНОВ
Пример 1. Около 1- Ю*5 моль/л Fe (III) после предварительного восстановления предполагают определять фотометрически с 1,10-фенантролином^НР+) в присут ствии примерно 1-Ю"4 моль/л Си (II), для устранения влияния которой используют маскирование ЭДТА. Какую следует создать концентрацию ЭДТА, чтобы устранить мешающее влияние Си (II) и не разрушить окрашенное соединение Fe(II, с реагентом?
Определение проводят при pH = 7 и общей концентрации реагента Cr = = 1-Ю"3 моль/л. Константы устойчивости соединений равны: k fpeR = 5,86, >g PFeRe = "-I, lg PFeRa = 21,3; lg PCuR = 8,82, lg PCuRf = 15,44, lg PCuRa = = 20,3; lg K^R = 5,0. Значения eFeR и eC(jRa приблизительно одинаковые.
Решение. При pH = 7 aR = 1, ау4_ = IO-8-4, aC(j2+ = 1, PFeR = = IO21-8, fcUR ~ Ю20'3- Согласно уравнению (5.4) мешающее влияние Си .11' будет устранено, если Рнер.,/^СиР. Ы0- и, кроме то’’©, как это это следует из неравенства (5.2), С или PcuR3 1-Ю6. Для того
чтобы в такой степени уменьшить P^uR , необходимо ввести избыток ЭДТА, обеспечивающий закомплексованность Си (II): l/aClj2+= 1+ 1018>8 [CY ] X X Ю-8"4 = 102°’8/10в= 10м.8; [Су] > 10-1.1 = 0,08 моль/л.
При этой концентрации ЭДТА l/aFe2+= 1+ 1014> 8-10-1.4-10-8>4 = = 109>8; fFeR = Ю21>8-10*®’8 = 1011’6. Учитывая, что при маскировании Си (II) необходимо обеспечить количественное образование комплекса FeR3> проверяем соблюдение условия (5.1): PFeRs [CR ]s = 10u> 10~® = 102-6> > 102. Максимально допустимая концентрация ЭДТА будет определяться соблюдением неравенства (5.1). Из этого условия следует, что минимальное значение PFeR должно составлять 1011, следовательно, максимальная закомплексованность Feyll): 1 aFe!+= 1 + J014’4[CY]-10-3’4= 1021’3/1014= Ю10-8 или [CY ] IO-0-8 = 0,25 моль.'л.
Таким образом, введя в раствор 0,1—0,2 моль/л ЭДТА при заданных условиях можно фотометрически определять Fe (II) в присутствии Си (II) при pH = 7.
Пример 2. При фотометрическом определении Ве2+ с альбероном (хромазу ролом S) в присутствии примесей Fe (111), Си (II) и А184 последние предлатают маскировать раствором ЭДТА. Нужно установить максимальную допустимую концентрацию маскирующего реагента и оценить достижимую при этом полноту маскирования мешающих примесей Фотометрическое определение Be2* проводят при pH = 4,5, концентрации Be2 , равной примерно 1-10*4 моль/л и общей концентрации реагента H3R* 1,1-10*8 моль/л Альберон с присутствующими катионами образует комплексные соединения состава MH2R с константами устойчивости PBeHaR = 1,6ч ,d, PAiH2r+= 7,9-1012, PpeHaR+= 4-1042 и PCijHi!r = = 2,5-10®.
Решение. При pH = 4,5 имеем:
aHY=-
= 1,78-IO'2; aY<_
/4
2 рГ [н3°+У =2’57-10~8;
0
341
/3
2₽Г [Н3О+]г=0,71. о
Чтобы обеспечить количественное связывание Ве2+ в окрашенный комплекс, допустима такая концентрация ЭДТА, при которой Рцен r 1-10а или при [CR ] = CR — Cge = 1 • 10"s моль/л PBeH2R Но РвеП2р “
= 1,6-1010 аВе2+-0,71, следовательно, 1/аВег+ С 1,1-10». При заданных условиях:
4
^аВе1+= ^BeY [CY ] aY‘- ^ВеНY [CY1 aHY’“ + S ₽?H 1* =
1
= 1+ 51,4 [Су] + 1,19-10’ [Су] 1,1-10», откуда [Су]< 1,1-Ю»/(1,19-10’)= 9-Ю-3 моль/л.
При такой избыточной концентрации ЭДТА обеспечивается полнота образования окрашенного комплекса бериллия с альбероном, ио обеспечивается ли полнота маскирования мешающих примесей? Для простоты примем, что общие концентрации всех ионов и молярные коэффициенты светопоглощения соединений MH2R приблизительно одинаковые. Тогда, согласво уравнению (5.6), минимальная разница в условных константах устойчивости комплексов бериллия и мешающих ионов должна составлять четыре порядка, а для полного маскирования Cu, Fe и А1 необходимо соблюдение неравенства (5.2) для суммы всех комплексов. При заданных условиях, т. е. при [Су ] = 9-10"3 моль/л и pH = 4,5, имеем: 1/аА13+ = 2,9- 10е, l/aFe„+ = 2,9-1015, 1/аСц2+= 1,5-10®, Paih2r = = 1,9- 10е, PpeH.R “ Ь Pcuh2r ** 1’2’
Сопоставление условных констант устойчивости показывает, что Fe (111) и Си (11) полностью маскируются и для этих ионов избыточная концентрация ЭДТА для обеспечения их полного маскирования и соблюдения условия (5.6) может бть уменьшена до 3-10-3 моль/л. Ионы АР при заданных условиях не маскируются ЭДТА, и с термодинамических позиций определение Ве2+ в присутствии А13+ невозможно. Однако из опыта известно [407 ], что при концентрации ЭДТА 6-Ю-3 моль/л ионы Ве2+ можно определять в присутствии всех указанных катионов. По-видимому, происходит «кинетическое» маскирование А13+ и переход последнею в комплекс с реагентом идет настолько медленно, что не возникает помех для фотометрирования бериллия.
Правда, следует иметь в виду, что комплексонаты железа (III) и меди (II) окрашены и могут создавать определенные помехи. Комплекс бериллия поглощает при 570 нм (е — 2,1-104). Комплексонат меди максимально поглощает при 735 нм (есиу = 1,5-102', и определению Be практически не мешает. Комплексонаг железа (ПР интенсивно поглощает при 450 нм (eFey = 45) и может создавать помехи при определении Be. При 570 нм eFey 45, поэтому допустимые в пределах погрешностей (±1 %) количества Fe (III) можно найти из соотношения: ABeHsR/AFeY = еВеН2КСВе/(ереУСре) > 1-102, откуда CFe/CBe = = 2,1-104/(45-102)s^5. Если содержание Fe (III) превышает пятикратное по отношению к Be, то в холостой раствор необходимо вводить соответствующие количества комплексоната железа (Ш).
Пример 3. Установить возможность применения в качестве маскирующих веществ для Fe (III) при фотометрическом определении Со (II) роданидным методом (при pH = 2): а) оксалат-ионов; б) фторид-ионов.
Полные константы устойчивости комплексов равны: для Co(SCN); — Pi = = 1-103, Р2= ЫО3, Р3= 2-102, Р4 = 1,6-102; для Fe(SCN)^1' — см. пример 3 в разд. 10.2; для Со(С2О4)2-2' — = 5-104, Р2 = 5-106, Р3 = 5-109; для
Fe(C2O4)?-2iг — Р, = 2,5-1°9, р2= 1,6-1016, Р3 = 1,6-1О20; для HSCN —Р =
342
= 7,15; для ВДА — hi= 2-10'*, Р2 = 3,5-10®; для HF — Р = 1,5-10s; для FeF3-'-p1= 1,1-Ю6; р2 = 5,5- Ю10, р3 = 5,5-1013, р4=5,5-1015, ps « = 1,16- 10м», рв= 1,26-1010.
Решение. При pH = 2 имеем: l/aSCN_ = 1 + 7,15-10“2 •= 1,07; 1/а „ = 1+2-10М0-2 + 3,5.105-10-4= 2,36-102; 1/а = 1+1,5- 10s-10~2 =
С2О4 F~
= 16.
Из сопоставления констант устойчивости роданидных комплексов Со (JI) видно, что для фотометрирования целесообразно использовать комплекс CoSCb(+. Доля иесвязанного в комплекс Со (П) не будет превышать 1% при 1/(РЛСчгм] X X aSCN-) С 0.01 или [SCN]>l,07/(10'2-103)= 0,11 моль/л. При такой равновесной концентрации доля комплекса Co(SCN)2 составит: aCo(SCN)2 = 1 X X 10s (0,1I)2/ £ Рг (О-11) = О.1. т. е. 10%-
/ о
Если принять, что фотометрирование Со (11) допустимо при соизмеримых концентрациях роданидных комплексов разного состава (в противном случае задача не решается даже в отсутствие ионов железа), то для определения Со (II) роданидным методом в присутствии Fe (III) необходимо, чтобы при заданных условиях соблюдались неравенства:
[cscn] PcoSCN + > 1 • 102 и
[CsCN]₽FeSC№+
а. В присутствии оксалат-ионов
PcoSCN+aCoI+aSCN- [CSCn1 ’ Ю2 ИЛИ
1/аСо’+ < 1,07-10™1 = 9’3(CSCN]
Предполагая, что при маскировании Fe (III) оксалат-ионами Со (IJ) тоже будет связываться в простейший комплекс СоС2О4, найдем предельно допустимую концентрацию [CCsOJ, при которой возможен полный переход Со (П) в окрашенный роданидный комплекс. В присутствии ионов С2О|_
1/аСоЕ+ = 1 + Рх [CCxOJ «c2oi- ~ Pi 1ССгОJ/(2.36-102) С 9-3[Cscn] откуда
[GCloJ < 2,36-103-9,3 [Cscn]/(5• 10*) = 4,4- IO’2 [CSCN]
при тех же условиях PFeSCN2+aFe3+aSCN~ [^sCn] 1-10-2 ИЛИ
.. ’ -е sCN!+aSCN-[CSCn1 _ 1,07’,03(CSCn1 . >r- 1
*/“Fe»+ — 10-2 1,07. IO"2 ~ 1uLgSCn1
Полагая, что Fe (III) необходимо закомплексовать хотя бы в простейший комплекс FeCaOf, найдем минимальное значение концентрации оксалат-ионов:
l/aFes+ < 1 + Pi [CC2OJ =
= 2,5- 10s [CCjOJ/(2,36-102) i. 105 [Cscn] откуда
[CctoJ > I2-36’ ’О2’ ’ • Ю=/<2,5-10»)] [CSCN] = 9,4.10-s [CSCN]
Следовательно, при 9,4-10-3 [CSCN] [C^oJ 4,4-10“2 [CSCN] Fe (III) будет практически полностью связано в бесцветный оксалатный комплекс, в то время как Со (II) будет находиться в виде окрашенного комплекса с роданид-ионами. При этом минимальная концентрация [CscnI. как это было показано выше, ие может быть менее 0,11 моль/л.
843
Например, если фотометрическое определение Со (II) в присутствии Fe (III) проводить при pH = 2 и равновесных концентрациях [CscnI = 0,75 моль/л и [Сс о ] = 2,36- IO"2 моль/л, то относительное содержание комплексных ионов (в условиях равновесия), рассчитанное по формуле (10.9), будет следующим:
аСо SCN+ = о,54: аСо (SCNh = 0>38’
аСо (SCN)~ = 0,05, аСо (scn)2- = 0>03
асос2о4 = 3,9-10 3; аСо (с2о4)2- — 3,9• 10~5; аСо (с2о4)|~ = 3,9- 10-в
aFeC О4 = 7’8'10 4; aFe(C2O4)~ = 5>0-10 aFe (С2О4)3- = Ю 1
“Fa^CN2* = 0’2’ 10 5> aFe (SCN)^ = 3,3-10 5;
aFe (SCN), = 4’5’ Ю"5! aFe (SCN)~ ~ 2>5' Ю
aFe (SCN)2~-|-Fe (SCN)g" = °>9’ Ю 5
При выбранных равновесных концентрациях роданид- и оксалат-ионов Со (II) на 99,5% будет находиться в виде окрашенных комплексов с роданид-ионами, a Fe (III) на 99,98% — в виде бесцветных комплексов с оксалат-ионами. Общие концентрации C-CN и Ссс^ (моль'л), которые необходимо создать в растворе, учитывая затраты и на комплексообразование, будут равны:
^scn = PscnI + 1’О^со = О’7® + 1'О^со
Сс2о4 = [Сс2о J + 2-5CFe = 2,36-10-2 + г>5Сре
Таким образом, при фотометрическом определении Со (II) роданидным методом при заданных условиях оксалат-ионы можно использовать для маскирования Fe (III). При этом необходимо, чтобы [CscnI >0,11 моль/л и 9,4- 10~3Х х [cscn] С [СС1 о ] гС 4,4-10-2 [CGCN]. Кроме того, для надежного обеспечения соотношения концентраций фотометрического и маскирующего реагентов в требуемых пределах необходимо, чтобы равновесные концентрации этих реагентов значительно превышали концентрации определяемого и маскируемого катионов соответственно.
б. В присутствии фторид-ионов для полного связывания Со (II) в окрашенный роданидный комплекс, как и в вышерассмотренном случае, необходимо, чтобы PCo<.CN+ [QscnI ЫО2. Поскольку Со (II) с фторид-ионами образует очень неустойчивые комплексы, в первом приближении можно принять, что в присутствии фторид-ионов «со2+ 1- Тогда полный перевод Со (II) в окра-
шенный комплекс бу дет достигаться при Cscn > 1’ Ю2-1,07/(1-103) =0,11 моль/л.
При тех же условиях для Fe (III) необходимо, чтобы PFesc№+[^scn]
1-10~2 или l/aFe.+ 1,07-103 [Cscn]/(1,07- 10-2) = 1-105 [CSCN].
Полагая при маскировании Fe (III) связывание его в простейший комплекс FeF2+, получим неравенство, обеспечивающее маскирование:
l/«Fe,+ — 1 + Pi [CF] aF_ > 1 Ю5 [CSCN]
[CF] >1-10* [CscrJ -16/(1,1 • 10«) = 1,45 [CSCN]
Следовательно, маскирование Fe (III) фторид-ионами можно осуществить проще, чем оксалат-ионами. Для маскирования Fe (III) при pH = 2 необходимо, чтобы равновесная концентрация фторида аммония в 1,5 раза превышала равновесную концентрацию роданида аммония. Но при этом минимальное значение [CscnI должно составлять не менее 0,11 моль/л. Общие концентрации Cscn и CF должны удовлетворять уравнениям:
CSCN = [^SCnI + ЙССо> CF = 1 >5 [CSCn1 + ” CFe
344
Здесь пип' — среднее координационное число для роданидных комплексов Со (II) и фторидных комплексов Fe (III) при выбранных равновесных концентрациях [CScn] и [Cf],
Пример 4. Выяснить возможность маскирования Ag+ при рН= 9,5: а) суль-фит-ионами; б) аммиаком. Оценить эффективность маскирования Ag+ этими веществами и установить, целесообразно ли добавление аммиака к су льфит-иону для увеличения эффективности маскирования Ag*. Какова должна быть концентрация сульфит-ионов и аммиака для обеспечения полного маскирования 0,1 моль/л Ag ?
Решение. Поль се маскирование ионов серебра можно считать достигнутым, если [Ag+ ] = [CAg]aAg+ < 1- 10_® моль/л или l/aAg+ > 1- 10е [CAg] При [CAg ] = 0,1 моль/л l/aAg+5г ЫО7. Следовательно, если при избирательном комплексообразовании Ag(I) l/«Ag+ превысит ЫО7, то маскирование Ag (I) можно считать достигнутым. Мольная доля гидроксокомплексов Ag (I), з
равная J? Р?н [ОН-]1 = 6,3-10-3, несоизмеримо мала и в расчетах, естественно, 1
не учитывается.
2
а. При использовании сульфит-ионов при pH = 9,5. 1/а£О.- = J] Pj1 X о
з
X [Н+]1' = 1 + 1,6-107-13,16- Ю-10 + 9,35-108-10-19 = 1; l/aAg+ = £ ₽( X о
X [CSOs]4oF= 1 + 2>5-105[СЬОз]+6,3.108[С£Оз]2+ 1- 109[CSOs]3.
По условию, 2,5-105 [CSOJ + 6,3- 10е [CSOJ2 + ЫО" [CSOa]3 > ЫО7. Полученное неравенство сравнительно легко можно решить относительно неизвестной |CSO ] методом последовательных приближений.
При [CSOj] = 0,1 моль/л
2,5 104 + 6,3-10в+ ЫО6 = 7,33-106 < ЫО7
При [C,;OJ=0,12 моль/л
2,5-105-0,12 + 6,3-10® (0,12)г + 1.1о» (0,12)3 ~М0’
Следоватепьно, при [SOF ] 5= 0,12 моль/л ионы Ag+ будут замаскированы сульфит-ионами Общая концентрация сульфит-ионов в растворе будет слагаться из их равновесной концентрации и координационно связанной с ионами серебра:
CSO8 = [S03 ] + пС Ag 3
Здесь ii = 1ал- <so3). — среднее координационное число сульфит-ионов в соединениях с ионами Ag.
При [SO(~1 = 0,12 моль/л
п = [2,5-105-0,12 + 2-6,3-10® (0,12)3 + 3-10* (0,12)3]/(1 • 107) =
= 2-0,84 + 3-0,16 = 2,2
.Минимальное (и достаточное) значение общей концентрации сульфит-ионсв, необходимой для маскирования 0,1 моль/л Ag (I), будет равно: Cso =0,12 + + 2,2-0,1=0,34 мольл.
б. при использовании NH3 при pH = 9,5: 1/«nH = 1+ 1,8-10s-3,16 X X 10-10= 1,57; l/aAg+ = 1 + 2,14-10® [CNHJ/1,57 + 1?74-107 ([CNH>]/1,57)2= = 1,4-10’ [CNHJ+ 7,Ы0в
345
По условию, 1,4-103 [CNHa] + 7,1-10е [CNHJ2 1-107. В полученном квадратном уравнении линейным членом пренебрегаем и находим:
K'NHjS’l/ Ю7/(7,1 -106) = 1,2 моль/л
При [CNHJ — 1,2 моль/л
_ 2,14-103-1,2/1,57 + 2-1,74-Ю7 (1,2/1,57)2
П~ 2,14-103-1,2/1,57+ 1,74-107 (1,2/1,57)2 — 2
Общая минимальная концентрация NH3 в растворе, необходимая для маскирования 0,1 моль/л Ag (I), составит. CNHj = [Сын,] + 2CAg = 1,2 + + 2-0,1 = 1,4 моль л. Следовательно, при концентрации NH3 не менее чем 1,4 моль/л ионы Ag (1) можно замаскировать и аммиаком. Но эффективность маскирования аммиаком меньше, чем сульфит-ионами, так как для достижения практически одинакового результата равновесная концентрация аммиака должна быть в 10 раз больше, чем концентрация сульфит-ионов. Тем не менее добавление NH3 к ионам SOj” следует признать целесообразным, поскольку NH3 создает необходимую для pH = 9,5 щелочную среду, и кроме того, прн концентрации [CNHj] 105/(7,1- 10е) =0,12 моль/л эффективность маскирования суль-
фит-ионами будет повышаться за счет дополнительного комплексообразования Ag (I) аммиаком.
10.6. ОБОСНОВАНИЕ ВОЗМОЖНОСТИ КОСВЕННОГО ОПРЕДЕЛЕНИЯ
Пример 1. Установить на основании расчета возможность косвенного фотометрического определения 1-10 4 моль/л Fe (III) при pH = 4 в присутствии 2-10~4 моль/л комплексоната меди, реагента PAN и 1 • 10“2 моль/л С1_ *.
Решение. Для обеспечения возможности косвенного определения Fe (III) необходимо соблюдение следующих условий.
I. Количественное замещение Cu (II) в комплексе с ЭДТА ионами Fe (III).
При pH = 4
*FeY = Ю25,1«у / S Р?Н [ОН']'' + £ 0? [Cl"]' + J] ₽« [R-]f =
I \ 0 1 1 /
/ / 2 \
= io14’7-101-8 / Ю'-Ч Ё [R-]£);
Kcuy = 1018,8«y / ( £ ₽?H [OH-]‘ + РГ [R-] = Ю6-4 I \ о /
К сожалению, из-за отсутствия данных о константах устойчивости комплексов Fe (III) с PAN невозможно рассчитать точное значение KpeY- Однако, если принять, что Cu (II) образует с PAN более устойчивый комплекс, то в первом приближении ДреУ/^СиУ Ю12'7-614 = 106'3. Если для обоих комплексов FeY- и CuY2- закомплексованность Fe (III) и Cu (II) реагентом PAN вообще не учитывать, то ApeY/AcuY = 10и’7-10,2 = 104,Б. Поскольку протекание реакции замещения рассматривается при Ссиу » 2Сре, то согласно уравнению (1.24) Cu (II) в комплексе с ЭДТА замещается ионами Fe (III) количественно.
* Присутствие С1” необходимо для образования окрашенных соединений FeR2A, CuRA в органическом растворителе [185, 411].
346
2. Ничтожно малое вытеснение Си (II) из комплекса с ЭДТА реагентом PAN При pH = 4PcuR= 1016'10-8,2 = 10''8; PcuY = 10,8’s-8-e = 10Ю.2’ Поскольку при CCuY/CFe « 2 значение CR/CFe ~ 2Pcur/₽Cuy ~ 10-2,4 <10~2э то, согласно уравнению (1.27), при заданных условиях реагент может вытеснить Си (II) из комплекса с ЭДТА не более чем на 1%. Более точный расчет равновесия CuY2- + R- CuR+ + Y4- показывает, что концентрация комплекса CuR+, образующегося по этой реакции, не превышает 0,4% от концентрации определяемого иона.
3. Количественный переход ионов Си (II), вытесненных из комплекса с ЭДТА ионами Fe (III), в фотометр ируемый комплекс CuR+.
При заданных условиях PFuR I^r] ~ Ю7,8-10-4 — 10я'8 >> 102, следовательно, стехиометрически замещаемые ионы Си (II) будут более чем на 99,9% переведены в фотометрируемое соединение.
Таким образом, все условия, необходимые для проведения косвенного фотометрического определения Fe (III) с помощью комплексоната меди и реагента PAN, соблюдаются, что подтверждается и экспериментально [410].
10.7. ПРИМЕРЫ РАСЧЕТОВ И ОБОСНОВАНИЙ ПРИ ЭКСТРАКЦИОННОМ ВЫДЕЛЕНИИ
И РАЗДЕЛЕНИИ ЭЛЕМЕНТОВ
Пример 1. При добавлении 20 мл 1'10“яЛ1 раствора экстракционного реагента HR в органическом растворителе к 100 мл Ы0*5 М раствора соли металла М при pH = 5 экстрагируется 33% металла в виде MR2. Какова была бы степень извлечения, если бы экстракцию проводили при pH = 6 и 50 мл 5-10-4 Л1 раствора реагента в отсутствие конкурирующих реакций в водном растворе? Каково будет значение pHt/ ?
Решение. Сначала находим коэффициент распределения:
п Ек 117 33 •100 о с
м 100 — Ем' V (100 — 33)20 ’
Затем необходимо найти условную константу экстракции. Для вычисления Кзкс воспользуемся приближенной формулой (4.44):
К'зкс = [h+]2/[HR ]| = 2,5 (Ю-^/Ю-3)2 = 2,5- Ю“4
Достоверность полученного значения К'экс проверяем по уравнению (4.43): j________________________1 , [нч2 .
Dm ^mr/a;kc[HR]2’
1 1 (IO-5)2
2,5“ ADMRs + 2,5-10-4 (10-3)2
Проверка показывает, что при заданных условиях величина 1/ЛДмк несоизмеримо мала по сравнению с 1/£>м, следовательно, £>м ^£>MR, и исполь*
зование приближенной формулы (4.44) можно считать обоснованным. По этой же формуле вычисляем коэффициент распределения при pH = 6 и [HR ]о = = 5-10-4 моль/л
Dm = *;KC [HR]2/[H+]2 = 2,5- IO"4 (5-10-4/Ю-6)2 = 62,5
а затем
Дм = Ю0/[1 4- 100/(50-62,5)] = 96,9 %
347
По уравнению (4.46) нетрудно вычислить рН,/г при {HR]о = 5-10*4 моль/л.’ рН,/г = — */г 1g 2,5 • 10~4 — 1g 5 • КГ4 = 5,1
Пример 2. При экстракционно-фотометрическом определении Си из 100 мл 1 М раствора НС!, содержащею 1-10*’ моль/л Cu2+, Си (II) экстра!провали 10 мл 4-10-4 М раствора дитизона (HR) в CCis. Определить степень извлечения меди, если известно, что состав экстрагируемого комплекса CuR2 и Кпк = = 3-10~5, KoHR = 1.1-104; РСиКг = Ю22 и /CDcuR2 = 7-104 Сопряженные равновесия в водной фазе не учитывать.
Решение. Прежде всего по уравнению (4.32) вычисляем KdEC KJKC = = 5-1022-,7-104 [3- 10-ь/< 1,1 • 104)f = 2,6-1010, К;кс = Лэкс, так как ам = 1.
При KOj]R = 1,1-10' и pH С pKhr можно принять, что [HR ]0 = Chr — — 4-10~4 моль/л.
Затем по формуле (4.35) находим приближенное значение Оси, т- е- /^Си = = 2.6-1010 (4-10-4)2 = 4,16-103 и
£Си = 100/11 + 100/(10-4,16.103)] = 99,77 %
Если использовать для вычисления £>си более точное уравнение <4.34), то получим.
1 — * л________________!_________— 2 54 10*4
Оси 7-104^ 2,6-1010 (4-10*4)2
Оси = 1/(2,54- IO’4) = 3,93-103
ЕСи = 100/11 + 100/(10-3,93.103)] = 99,74 %
Различие в значения Еси незначительное, так как Ocu <S KdCuR > и ис" пользование приближенного уравнения (4.35) вполне оправданно.
Пример 3. Торий экстрагируют из Ы0~4 М водного раствора ацетилацето-ном (HR) в бензоле. Вычислить степень извлечения Th из раствора при рН= 6, если известно, что Th экстрагируем в виде комплекса ThR и конечная равновесная концентрация ацетилацетона в бензоле составляет 1 • 10~3 моль/л При расчете учесть ступенчатость комплексообразования в водном растворе.
Константы устойчивости ацетилацетонатов гория; f1=7-107, = 3,8 X
X 1015, р8 = 7,2-1021, = 7,2-102®. Соотношение объемов фаз IV7V — 10;
*WThR. = 315; KaHR = 1’17’ 1О’“: ^HR = 5195
Решение. Зная величины [HR ]0 и KoHR, найдем общее количество реагента и его суммарную равновесную концентрацию в водной фазе;
р 100 100 ЧТ ч
j + uz/(r>£-R) 1 + Ю/5,95 7’ /о
[HR]nV 10"3V „ сс ,
9hR~ £/100 “ 0,373 ~2’68' ° V|M0JIbl
2 68.10~3V____ Щ-зр
[CR]W = --—- = 1,68.10-3.0,1 = 1,68-10"4 моль/л
Вычислив при pH = 6 мольную долю aR аниона реагента, находим его равновесную концентрацию в водном растворе:
aR = 1/(1 + 10-3/1,17-10-3) = 1,17- 10-а
[RJW= [CR]w,aR= 1,68-10-4-1,17-10'3= 1,97-10-’ моль/л
348
Теперь можно вычислить мольную долю экстрагируемого комплекса ThR< при pH = 6 и [R-1 = 1,97-10-7 моль/л, а затем найти величины Djh и £ц,:
aThR4 — (~j — —
S ₽£ ?=о
7,2-1028 (1,97-10'7)4
= 1 + 7-10’- 1,97-Ю'7 + 3,8-101= (1,97-10-’)2+ 7,2-1021 (1,97- Ю’7)3 + = + 7,2- 102в (1,97-Ю-’)4
= 4,9-10-3
°ть = = 3,5'4>9-10’3 = ’>56
£Т11 = Ю0/(1 + 10/1,56) = 13,5 %
Пример 4. При экстракции из 1О~6 М раствора Pb JI) 0,25 М раствором теноилтрифторацетона (HR) в бензоле pHt/ составляет 3,2, а константа экстракции /<ЭцС = 7-10"6. Найти константу распределения и константу устойчивости экстрагируемого комплекса PbR2, если известно, что ^oHR= 7-10“7 и Adhr = = 40.
Какова степень извлечения РЬ при pH = 6 и Vi IV' = 0,1? Сколько последовательных экстракций необходимо для 99%-ного извлечения свинца?
Решение. Сначала необходимо найти коэффициент распределения Dr и равновесную концентрацию реагента в органической фазе:
°R = ^£>hr“hr!
0Н[Н+] 6,3-10-4/7-10"7
aHR “ j + pH [Н+] - 1 + 6,3-10-4/7-10-- ~ 1
OR = 40*1 =40
[HR]0 = Dr [Cr]/(1 + Dr) = 40-0,25/(1 + 40) = 0,244 моль/л
Затем вычисляем значение KbpbR учитывая, что при pH = 3,2 гидроксо-комплексообразование Pb (II) протекает в незначительной степени, арь = 1 и ^экс ^<жс’
___1 _ _J______[НТ _ i _ (6,3-ю-т
«ВРИ, Лэке fHR]o 7-10"® (0.244)2
£ppbR2=1/(4,5-1()-2)=22,2
о_______________Kjkc_________________10 6 = । । «а
₽PbR* - ^opbR.^W^HR)2 22,2(7-10-7/40)4
При pH - 6 и r = 0,1
l-10-e/(7-10-7)
KHR = 1 + Ю~8/(7 -10~7) - °’59: ^ = 40-0,59 = 23,6
[HR]0 = 0,25-23,6-0,1/3,36 = 0,12 моль/л
П^ = -2^2+7-7тДда = 4-5-,0-2= ^ = 22,2
р 100 100 fit) 07
pb “ 1 + r/(VDPb) “ 1 + 10/22,2 z0
349
Определение числа последовательных экстракций, необходимых для 99%-ного извлечения свинца в органический слой, производим по формуле (4.29):
j _ 1g ^вач — 1g 5-кон_1g Ю 5— lg I0~7__ .
lg (1+ PpbV/Г) lg (1 + 22,2-0,1)“’
Одиако, если учитывать гидроксокомплексообразование Pb (II), особенно образование полиядерных гидроксокомплексов: РЬ3(ОН)|+, РЬ4(ОНД+, РЬ6(ОН)|+, то получим следующие результаты.
Запишем функцию закомплексованности Pb (II) в общем виде;
з
Срь/[ро2+]=2 0°Н [°н’]' + о
+ 3 [РЬ2+]210Н-]*р43 + 4 [Pb2+]3 [OH"J4p44 4- 6 [Pb2+]?[OH"]8pge (10.16)
Подставляя в уравнение (10.16) численные значения CPb = 1-10"6 моль/л и констант образования гидроксокомплексов свинца (1g fj43 = 32,6; lg fJ44 = = 35,1, lg Pge = 99,3), получим приведенное уравнение
10~6/[РЬ2+] = 1,75 + [Pb2+J21 033-i [ОН']4 + |Pb2+]3103^ [ОН']4 -J-
+ [РЬ2+[61О100-2 [ОН']8 (10.17)
Которе-, превращается в тождество при концентрации [ОН"]=1-10“8 моль/л (pH = 6) и [Pb2+] = IO*6-87 моль/л. Отсюда находим, что арЬ2+ = = IO"6'87/10"5 = IO’1-87. Тогда Я;кс = 7-10“6-10"1'87 = 10"8'78
1 1 , 10*12
------ =________। = 7 он. 1 п~2 НРЬ 22,2________0,122-IO-8-78---’
Орь = 12,6; £рь = 55 %; d = 6
Пример 5. Определить оптимальный интервал pH раствора для экстракции Zn тетрахлоридом углерода в виде дитизоната Zn(HR)2 при экстракционном разделении Zn и Pb (И) в 1 М растворе ацетата натрия. В процессе экстракции во всем интервале pH равновесная концентрация [H2R]0 = 1-10~4 моль/л поддерживается постоянной. 1g АоНгц = 4,04; 1g КоРЪ = 4,0; lgAozn=4,0; lg ^аксг , = 2,7; 1g /СЭксрь = 0,75; lg fz , (HR)S = 15,83; lg PPb(HR)2 = = 13,83; lg 12,0; Igp^ r = 16,5. Экстракцию проводят при г =0,1 из
водного раствора, содержащего 1 • 10“5 моль/л Zne+ и 1-10—4 молы л РЬ2+
Решение. При экстракционном распределении Zn2+ и РЬ2+ необходимо создать такие условия, чтобы из водного раствора в органическую фазу переходил бы только Zn, в то время как РЬ оставался бы практически полностью в водном растворе. Для количественной оценки фактора разделения S используем уравнения (4.73)—(4.77). Необходимые соотношения между коэффициентами распределения дитизонатов цинка и свинца, как это следует из этих уравнений, должны обеспечивать соблюдение неравенства: lg DZa — 1g Орь > 5} lg ^Zn > 3; 1g Орь —2.
С изменением pH раствора изменяется равновесная концентрация ацетат-ионов, а следовательно, значения условных констант экстракции и коэффициентов распределения. Ниже в зависимости от pH раствора приведены значения а-коэффициентов;
Рн, 1g 1/аАс-lg l/«zn«+ 1g 1/«рь»+
1 2 3
3,76 2,76 1,76
0 0 0,15
0,02 0,2 1,72
4 5 6
0,83 0,19 0,02
0,83 1,82 2,11
5,20 7,72 8,41
7 8 9
0 0 0
2,11 2,17 2,22
8,50 8,50 8,50
850
По этим данным вычисляем в том же интервале pH следующие значения:
pH 1 2 3 4 5 6 7 8 9
^SKCZtl 2,70 2,70 2,56 1,88 0,89 0,58 0,53 0,53 0,50
1g ^эксрь 0,72 0,54 —0,97 —4,46 —6,98 —7,66 —7,75 —7,75 —7,75
lg OZn —3,3 - -1,3 0,56 1,88 2,89 3,90 4,00 4,00 4,00
lg Dpb —5,28 -3,46 —2 93 —4,46 —5,00 —3,66 — 1,75 0,25 2,25
IgS 1,98 2,16 3,52 6,34 7,89 7,56 5,75 3,75 1,75
Из представленной зависимости lg S от pH (см. также рис. 10 3) видно, что по значению фактора разделения для избирательной экстракпии дитизоната цинка можно использовать интервал pH от 3,3 до 7,3, так как в этом интервале соблюдается необходимое по условию требование lg S ^5. Максимальное значение S достигается в более узком интервале pH — от 5 до 5,5. Однако при pH > 6,8 коэффициент распределения Орь превышает значение, допустимое по требованиям разделения. С другой стороны, минимальное значение Р/п. обеспечивающее полноту извлечения дитизонта цинка, достигается только при pH 5,2. Следовательно, интервал pH, где удовлетворяются требования допу-с'-имых значений S, DZB и Dp;,, соответствует области 5,2—6,8. Оптимальный же интервал отвечает области максимума на кривой lg S = f (pH), т. е. 5,5— 6,0. На рис. 10.3 область pH зодного раствора, где целесообразно проводить экстракционное разделение дитизонатов цинка и свинца, заштрихована. В этой области lg S 5? 6.
д lg Dzn = lg (DZn/DznM1IH) > 0
и Д lg DPb = lg (£)рь/ОрЬма1,с) < 0
Пример 6. Найти интервал pH раствора, в котором обеспечивается количественное извлечение (Е 99,9%) оксихинолината железа (III) из 100мл 2-10*5 М водного раствора, не содержащего конкурирующих komhj ексаитов, тремя последовательными экстракциями по 10 мл 0,01 М раствора 8-оксихинолина в хлороформе. Какой можно использовать интервал pH, если экстракцию проводить при тех же условиях из водного раствора, содержащего 0,01 моль/л ЭДТА?
По данным работы [176], оксихинолинат железа (III) экстрагируется хлороформом только в виде незаряженного хелата FeR3, а реагент — только в молекулярной форме HR.
1g = 2,6; lg P«R = 9,9; lg ₽«jR+ = 14 9; lg ₽FeR2+ = 12,7; lg ₽FeR+ = = 25,6; lg ₽FeRs = 37,0; lgK3KC = 4,ll.
Решение. Используя уравнение (4.5), найдем минимальное значение коэфф, циента распределения Dpe, обеспечивающее степень извлечения Е 5? 99% при трех последовательных экстракциях 3 1g (100 + lODp^ — 31g 100== = lg 2- 1(Ге — ig 2- IO-8, откуда DFe 90. По формуле (4.32) находим-Ki>FeRs = 10'*"61- П° литературным данным [167] Fe (III) экстрагируется в интервале pH = 2ч-10. Так, при pH = 2 : lg l/ccR_ = 10,9; lg«HR = —3,0; OR = 0,4; [HR]o= IO-"”42 моль/л; [R“] = 10~13,в моль/л; lg l/«Fe3+ = 0,07; lg«Fea+aR- = -32,77; lg K'SKC = 4,04; OFe = 0,6.
Производя аналогичные вычисления н при других значениях pH, получим следующую зависимость;
рн 2 3 4 5
lg «Fe3+ —0,07 — 1,64 —4,4 —6,6
lg «R- — 10,9 —8,9 —6,94 —5,2
1g «HR , —3,0 —2,0 — 1,04 —0,3
lg «Fe’+aR- —32,77 —28,34 —25,22 —22,2
1g [HR]0 —3,42 —2,54 —2,1 —2,02
lg [R-] — 13,9 — 12,0 — 10,6 —9,52
lg^K0 4,04 2,47 - 0,29 —2,49
lg DFe —0,22 3,78 4,54 4,61
351
pH lg ctfe3+ ’gaR-Ig«HR lg <^fey4'^R-1g [HR |0 lg 1R-] lg ^экс 1g Dpe
6 7 8 9 10
— 8,58 — 10.6 — 12,6 — 14,6 — 16.96
—3,94 —2,9 — 1,9 —0,95 —0,25
—0,04 0 0 —0,05 —0,35
—20,4 — 19,3 — 18,3 — 17,45 — 17,71
—2,01 —2,0 —2,0 —2,01 — 2,02
— 8,51 —7,5 —6,5 —5,51 —4,52
—4,47 —6,49 —8,49 — 10,49 — 12,85
4,61 4,61 4,61 4.61 4.61
Из расчетных данных видно, что выход комплекса FeR3 достигает максимума при pH около 9, но на распределении это практически не сказывается, поскольку уже при pH = 5 в водном растворе количественно доминируют экстрагируемые комплексы и коэффициент распределения достигает константы распределения Поэтому в широкой области pH от 5 до 10 максимальное значение Dpe определяется константой KoFe (рис. 10.4) и сохраняется неизменным Однако при pH 7 достигается произведение растворимости осадка гидроксида железа (HI), и при проведении экстракции при более высоких значениях pH необходимо выяснить возможность сопряженного осаждения Fe (III) в виде Fe(OH)3 совместно с основным осадком FeR3, переходящим в органическую фазу. Условная константа обменного равновесия FeRa-|- 301Г Fc(OI I)., Д 3R~
= /к; =
6Ре(ОН)з / sFeR,
= ^sFe (ОН)заре’+ (OH)aR-/(\sFeR4aFes+ (R>) =
= 10 ’'•5CZpe3+ (oH)aR-/(l° W’’aFe’+(R))
котя и возрастает с увеличением pH раствора, тем не менее даже при самом высоком значении pH выбранного интервала, т. е. при pH = 10, не превышает 1 10~*. Если учесть, что частицы FeRs количественно переходят в органическую фазу, то можно утверждать, что во всем интервале pH от 2 до 10 сопряженною осадка Fe(OH)3 не образуется и для проведения экстракции при заданных условиях можно использовать широкий интервал pH от 3 до 10.
В присутствии 0.01 моль'л ЭДТА значительно увеличивается сопряженное комплексообразование Fe (III) в водном растворе, значения К,кс и будет существенно отличаться. Так, при pH = 2 1g = 13,8; lg 1 aFe4+ =
= 9,3; lg К;кс = —5,19, DFe= 10~‘'-
Производя аналогичные вычисления и при других значениях pH, получим зависимость lg Dpe or pH, представленную на рис 10.5. Из рассмотрения этой
зависимости видно, что экстракцию окспхинолината железа (III) хлороформом
из 0,01 М раствора ЭДТА можно осуществить только в щелочной среде (pH, , » 7,2). Для решения поставленной задачи пригодно значение pH 8,5.
Пцимер 7. 5 мл 4-10-4 А1 раствора диэтилдитиокарбамината никеля (NiR?) в тетрахлориде углерода перемешивают с 50 мл 2- 10~° М водного раствора Cu (II) при pH = 5 Будут ли ионы Сц-+ вытеснять ионы NF+ из соединения NiR2 в органической фазе? Какова бу чет
Рис. 10.3. Зависимость фактора разделения Szn/РЬ от PH в°Дн°й фазы.
352
Рис. 10.4. Зависимость lgD?e от pH.
Рис. 10.5. Зависимость lgDpe от pH в 0,01 М растворе ЭДТА.
полнота протекания реакции обмена? При какой концентрации NiRa в органической фазе реакция обмена пройдет не менее чем на 99% при однократном экстрагировании? Изменятся ли результаты, если в водном растворе будет содержаться 0,1 моль/л ионов SCN"?
1g КэксСи = 13.7, 1g Кэкс№ = 11.6. Для Си (SCN)’"*: 1g ₽, = 2,3, lg ₽а= = 3,65, lg p3 = 5,2; lg ₽4 = 6,5. Для Ni(SCN)2-*: lg = 1,2, lg p2 = 1,64, lg Рз = 1.8.
Решение. По уравнению (4.70) находим, что пороговое значение рНП0р = = 5. При pH = 5 «си”-(ОН)= 1 и aNi*+ (ОН) = 1- В отсутствие посторонних комплексантов К;кССц = Ка„ССи и = K8K0N| Поскольку
К' > . ноны меди будут вытеснять ионы Ni?+ из диэтилдитиокарба-
мината в СС14 при перемешивании фаз, что наблюдается и экспериментально 1167].
Чтобы определить, в какой степени пройдет реакция обмена при заданных условиях, воспользуемся уравнением (4.68): g= 4-1(Г4/(2 • 10“®) = 20; 1013,7/1011,e = Z2/[(100— Z) (200 — Z); откуда 7=99,2%. Практически количественное протекание реакции обмена подтверждается также соблюдением неравенства (4.67): (1013,7/1011,в2) > 104.
4
В присутствии 0,1 моль/л ионов SCN“ : l/aCu,+ = J] ₽j [SCN-]* = IO2,7; о
з
l/aNil+ = £ Pf [SCN"]* = 10°’«; lg 7^ = 13.70 - 2,7 = 11,0; lg = — 11,6 — 0,5= 11,1. Поскольку значения Кэкс соединений CuRa и NiRa приблизительно одинаковы, реакция обмена протекать будет, но не количественно. При заданных условиях 10~0,1 = Z2/[(100— Z) (200 — Z), откуда Z=63%.
Для практически полного извлечения Си (II) из водного раствора необходимо последовательно экстрагировать новыми порциями NiRa в СО,. Отождествив величины Z и Е (%), по формуле (4.5) найдем условную величину Dcu-63 = 100/(1 + 10/Dcu); Оси = 17. Воспользовавшись уравнением (4.6), находим необходимое число последовательных экстракций: d = (1g 2- IO"6 — lg 2 X X 10-7)/[lg (50 + 17-5) — lg50] = 5. Если концентрацию NiRa в CC14 повысить до 2-10"3 моль/л, то число последовательных экстракций можно сократить до трех.
Пример 8. Каких можно ожидать результатов в условиях экстрагирования, описанных в примере 7, если в качестве экстрагента использовать 4- IO'4 М
12 М. И. Булатов и др. . 353
раствор диэтилдитиокарбамината свинца в тетрахлориде углерода? 1g КЭКсрЬ = = 7,77.
Решение. По уравнению (4 70) находим, что рНпор = 5. При pH = = 5аСи2+ (ОН) — 1; аРЬ2+ (ОН) = 1; ^экс — ^экс: ^эксСи ^ЭКСРЬ’ поэт°му условия обмена благоприятные. Согласно уравнению (4.67)
к;КСси / к;КСрь=io13-7-7-77» w2/(gr _ и=ю2
Следовательно, обмен ионов Си2+ на ионы РЬ2+ протекает количественно за одну экстракцию. В присутствии 0,1 моль/л ионов SCN- : 1/аСц!+ = Ю217; 6
1/aPb2+ = S PjSCN-f = 10°-77; 1g к;ксси= 11,0; 1g <КГрь = 7,0;
^эксСи/^эксрь = Ю4 > Ю2- Следовательно, результаты экстрагирования не изменятся и в присутствии 0,1 моль/л ионов SCN- в водном растворе. Полученные результаты прогнозирования хорошо согласуются с широким использованием [167] экстрагента PbR2 в СС14 для избирательного извлечения Си (П).
Пример 9. При какой концентрации ЭДТА в водном растворе при pH = 4 ионы Hg (И) из диэтилдитиокарбамината ртути HgR2 в СС14 можно количественно заместить ионами Ag+, содержание которых в водном растворе составляет 2-1(ГБ моль/л. Концентрация HgR2 в органической фазе Ы(Г® моль/л. Соотношение объемов фаз г= 0,1; 1g КЭксА„ = 4,9; 1g K8KcHg = 31,9.
Решение. Согласно уравнению (4.70), рНпор = 4. При pH = 4 'g 1 /aAg+ (ОН) = °> ]g ^aHg2+ (ОН) = 1 ’9; 1g /<эксА8 = 11 'g ^экс Hg = = 31,9 — 1,9=30,0 Согласно уравнению (4.60), значение = _ 1О2.ц,9/1озо= ip-6.2, т е реакция обмена без введения комплексующих Hg (II) веществ невозможна. Для того чтобы реакция обмена могла протекать количественно, необходимо, чтобы -^эксА /^эксНа > (Ю0)2/[(2-5О X X 0,1 - 1)0,1]= 10* или ^KcAgaAg+ (Y)/(^3KCHgaHg2+(Y)) > 104 или ^Ag+ (Y)/^Hg2+ (Y) Ю12*1. При pH = 4: lg l/cty*- — 8,6; lg K^gy ~ 21,8;
lg KAgY = 7,3; (1 + 102ll8CY- 10-e-b)/(l + 10,,3CY- IO-816)2 > 1012-1, откуда Су 10-111 моль/л.
Если реакцию обмена проводить в присутствии 0,1 моль/л ЭДТА и концентрации HgR2 в СС144-10-3 моль/л, то при этих условиях lg l/aAg+= 0,1; lg ^«Hg2-*-^ 12’!; lg ^3KcAg= 11 >8; !g 7<;KcHg = 19.8- Согласно уравнению (4.68), 1023<6/101S’8 * * = JM0/[(10O — Z)2 (4000 — Z)], откуда Z=99,4%. Следовательно, реакцию обмена можно завершить количественно при однократной экстракции.
Г лава 11
ПРАКТИЧЕСКИЕ РАБОТЫ И ЗАДАЧИ
В главе приведены практические работы, иллюстрирующие при-
менение описанных выше фотометрических методов для опреде-
ления различных веществ в конкретных объектах или в модель-
ных растворах, имитирующих эти объекты.
354
ОПРЕДЕЛЕНИЕ МЕТОДОМ СРАВНЕНИЯ
Работа 1. Определение микроколичеств железа при помощи о-фенантролина
Определение основано на образовании оранжево-красного цвета комплексного иона
Приготавливают исследуемый и стандартные окрашенные растворы Fe2+ с о-фенантролином и измеряют нх оптические плотности по отношению к раствору сравнения на фотоколориметре с синим светофильтром. Неизвестную концентрацию ионов железа определяют методом сравнения по следующим формулам;
Сд; = ^СТ^к/^СТ ИЛИ Cg; = Cj = (Сп-С2) (Их- А1)/(А2- ^1)
Здесь Ci и С2 — концентрации стандартных растворов железа; Af и Л2 — оптические плотности этих растворов (причем Сг < Сх < С2 и < Ая < Д2).
В интервале концентраций железа 2—40 мкг/мл соблюдается прямолинейная зависимость между оптической плотностью и концентрацией раствора.
Выполнение работы. В мерную колбу на 25 мл помещают 20 мл раствора, содержащего 0,05—1,00 мг железа, добавляют 0,5 мл 10% раствора солянокислого гидроксиламина. В другой колбе с помощью универсальной индикаторной бумаги определяют объем ацетата натрия, необходимый для того, чтобы довести значение pH до 4 — 6 в 20 мл исследуемого раствора. Найденный объем ацетата натрия добавляют к анализируемому раствору, прибавляют 1 мл 0,5% раствора о-фенантролина и доводят объем водой до 25 мл. Через 10 мин раствор фотометрируют на фотоколориметре с синим светофильтром в кювете с толщиной слоя 5 см. Одновременно в мерных колбах на 25 мл аналогично приготавливают два стандартных раствора так, чтобы оптическая плотность одного из них была немного меньше оптической плотности исследуемого раствора, а оптическая плотность другого, наоборот, немного больше.
Раствором сравнения служит раствор, содержащий все реактивы, кроме соединений железа
Примечание. Алюминий, медь, хлориды и сульфаты при концентрациях до 10 мг/л определению железа ие мешают.
Реактивы
Солянокислый гидроксиламин, 10% раствор.
Ацетат натрия, 0,2 М раствор.
о-Фенантролин, 0,5% водный раствор моногидрата (растворяют в воде при нагревании).
Стандартный раствор соли железа, содержащий 0,1 мг/мл железа.
ОПРЕДЕЛЕНИЕ МЕТОДОМ ГРАДУИРОВОЧНОГО ГРАФИКА
Работа 1. Определение алюминия в медно-цинковых сплавах [4121.
Определение основано на образовании окрашенного соединения алюминия с реагентом эрнохромцнанинэм R.
12* 355
Выполнение работы. Помещают 0,1 г латуни в коническую колбу на 50 мл, добавляют 2 мл HNO3 и растворяют при умеренном нагревании. Затем приливают 40—50 мл дистиллированной воды и переносят жидкость в мерную колбу на 100 мл, добавляют 8 мл НС1, доводят объем до метки дистиллированной водой и тщательно перемешивают. Отбирают аликвотную часть 2,5 мл в мерную колбу на 100 мл, прибавляют 40 мл воды, 0,1 мл 4% раствора аскорбиновой кислоты, 0,25 мл 15% раствора тиосульфата натрия и взбалтывают. Затем добавляют 2,5 мл 0,075% раствора эрио-хромцианина, 20 мл 40% раствора ацетата натрия, доводят дистиллированной водой до метки и тщательно перемешивают. Через 2 мин измеряют оптическую плотность на фотоколориметре с зеленым светофильтром в кювете с толщиной слоя 2 см относительно раствора сравнения.
Для приготовления раствора сравнения берут навеску 0,1 г латуни, не содержащей алюминия, прибавляют те же количества реактивов, что и при приготовлении исследуемого раствора.
Градуировочную кривую строят по данным, полученным для стандартных растворов образцов латуни, не содержащей алюминия, к навескам которых добавляют соответствующее количество стандартного раствора алюминия; градуировочную кривую можно строить также по разным навескам стандартного образца латуни с известным содержанием алюминия.
Примечание. Цинк, свинец, никель, олово и марганец в тех количествах, в которых они находятся в медно-цинковых сплавах, определению алюминия не мешают. Влияние ионов железа устраняют введением в раствор аскорбиновой кислоты, которая восстанавливает ионы Fe3+до Fe2+, образующих с эрио-хромцианином бесцветный комплекс; влияние ионов меди устраняют добавлением тиосульфата натрия, образующего бесцветный тиосульфатный комплекс. Анализ выполняют за 12—15 мин с погрешностью, не превышающей ±3% (отн.).
Реактивы
HNO3 (плотность 1,40).
НС1 (1 : 1).
Аскорбиновая кислота, 4% раствор.
Тиосульфат натрия, 15% раствор.
Ацетат натрия, 40% раствор.
Эриохромцианин R, 0,075% раствор. В колбу на 200 мл помещают 0,15 г реактива, растворяют в 50 мл дистиллированной воды, прибавляют 5 г NaCl, 5 г NaNO3, 0,4 мл HNO3 (плотность 1,40), разбавляют водой до метки и перемешивают.
Стандартный раствор нитрата алюминия, содержащий 25 мкг алюминия в 1 мл. Навеску 0, 3475 г х. ч. А1('Ю3)3-9Н2О растворяют в 50 мл дистиллированной воды, подкисляют 2—3 мл HNO3 (плотность 1,40) и доводят объем водой до 1 л.
Работа 2. Определение железа (111) с помощью сульфосалициловой кислоты
Железо (III) с сульфосалициловой (2-гидроксн-5-сульфобензойной) кислотой п-он __________________________Х J—соон
356
образует различные по составу комплексы [66, 71 ] в зависимости от кислотности раствора. В кислой среде в интервале pH = 1,8ч-2,5 образуется комплексный катион моносульфосалицнлата железа FeSSal красно-фиолетового цвета (Хмакс = = 510 им, е — 1800, Pi = 1,1-Ю14), при pH = 4,0-^8,0 доминирует комплексный аиион дисульфосалицилата железа Fe(SSai)^", а в интервале pH = 8,0-4--s-11,5 образуется трисульфосалицилат железа Fe(SSal)!j- желтого цвета (Хмакс = = 416 нм, е= 5800, Рз = 1,25-10м). Сульфосалицилатные комплексы железа используют для дифференцированного определения железа (III) и железа (II) при их совместном присутствии. Железо (III) определяют в кислой среде в виде моносульфосалицилатниго комплекса, а в щелочной среде в виде трисульфосалицилата определяют суммарное содержание железа (III) и железа (II).
Трисульфосалицилатный комплекс железа (III) достаточно устойчив и позволяет проводить определение железа в присутствии ацетат-, борат-, фосфат и фторид-ионов. Ниже приведена методика определения железа (III) в виде три-сульфосалицилатного комплекса методом градуировочного графика.
Выполнение работы. К исследуемому раствору, содержащему от 0,1 до 0,6 мг железа, в мерной колбе на 100 мл добавляют 10 мл 10% раствора сульфосалипиловой кислоты, перемешивают и затем добавляют 10 мл 10% раствора аммиака, доводят дистиллированной водой до метки, перемешивают и через 5 мин фото-метрируют на фотоколориметре или спектрофотометре при 416 нм относительно раствора холостой пробы, содержащей все добавляемые реактивы, кроме железа. Массовое содержание (мкг) железа находят по уравнению градуировочного графика, рассчитанному методом наименьших квадратов (см. разд. 9.3) по результатам фотометрирования стандартных растворов соли железа:
<7х = (Л— а)/Ь
Здесь А — среднее значение оптической плотности исследуемого раствора; а и Ь — параметры уравнения линейного градуировочного графика (Ь — угловой коэффициент).
Для приготовления стандартных растворов в мерные колбы вместимостью 100 мл вносят 1, 2, 3, 4 и 5 мл основного стандартного раствора соли железа, увеличивая содержание железа в каждом стандартном растворе на 0,06 мг, добавляют по 10 мл раствора сульфосалициловой кислоты, по 10 мл раствора аммиака, доводят дистиллированной водой до метки и перемешивают. Оптическую плотность приготовленных стандартных растворов измеряют при 416 нм (фиолетовый или синий светофильтр) относительно раствора холостой пробы. По полученным данным строят линейный градуировочный график и рассчитывают его параметры.
Реактивы
Сульфосалициловая кислота, 10% раствор.
Аммиак, 10% раствор.
Стандартный раствор соли железа, содержащий 0,06 мг Fe в I мл. Навеску 0,5190 г х. ч. невыветреиных квасцов (Nh4)2Fe2(SO4)4-24H2O растворяют в воде, подкисляют 10 мл серной кислоты (1 : 1) и доводят объем водой до 1 л.
Работа 3. Определение висмута в олове
Определение [413] основано на образовании окрашенного комплексного катиона висмута с тиомочевиной Bi(NH2CSNH2)g+ в кислой среде.
357
Приготавливают исследуемый и стандартные окрашенные растворы, содержащие комплексные катионы висмута с тиомочевиной, и измеряют нх оптические плотности на фотоколорнметре или спектрофотометре прн 460—470 нм относительно раствора олова без тиомочевины (раствор сравнения). По данным, полученным для стандартных растворов, строят градуировочную кривую и с ее помощью определяют неизвестную концентрацию висмута в исследуемом растворе.
Выполнение работьь 1 г олова растворяют в 5 мл царской водки в мерной колбе на 50 мл при слабом нагревании. Раствор охлаждают, прибавляют 15 мл HNO3, 10 мл 10% раствора тиомочевины, доводят дистиллированной водой до метки, перемешивают и спустя 15 мин фотометрируют на приборе ФЭК-56М с синим светофильтром в кювете с толщиной слоя 5 см.
Все рактивы, после их приготовления, фильтруют через плотный бумажный фильтр. Раствор тиомочевины хранят в склянке из темного стекла.
Для построения градуировочной кривой приготавливают стандартные растворы олова, не содержащего висмута. К этим растворам, помещенным в мерные колбы на 50 мл, добавляют различные количества стандартного раствора висмута, приливают те же количества реактивов, что и к исследуемому раствору, объем доводят водой до метки, перемешивают и фотометрируют относительно раствора сравнения.
Примечание. Небольшие количества сурьмы, меди и железа, содержащееся в олове, определению висмута ие мешают.
Реактивы
НС1, конц.
HNOS, конц.
HNOS (2 : 13).
Тиомочевина, 10% раствор.
Стандартный раствор висмута, содержащий 0,1 мг висмута в 1 мл. Для приготовления стандартного раствора берут 0,1 г металлического висмута, обрабатывают 20 мл HNOS (2 : 1), добавляют 10 мл H2SO4 (1 : 1) и выпаривают до выделения белых паров. Раствор охлаждают, добавляют 100 мл дистиллированной воды, переносят в мерную колбу на 1 л и разбавляют до метки серной кислотой (1 :9).
Работа 4. Определение молибдена в сталях и сплавах [414]
Определение основано на образовании окрашенного роданидного комплекса [Mo(SCN)6]. В качестве восстановителя используют аскорбиновую кислоту.
Выполнение работы. Определение молибдена в хромистой стали. Навеску хромистой стали 0,1 г обрабатывают при нагревании 10 мл 20% раствора H2SO4; нерастворившиеся карбиды окисляют 1 мл НЫО8, нагревают до начала выделения паров SOs, охлаждают, добавляют 50 мл воды и снова нагревают до растворения солей. Затем раствор переносят в мерную колбу на 250 мл, доводят до метки водой и перемешивают.
Аликвотную часть раствора, соответствующую оптической плотности A tv 0,43, вносят в колбу на 25 мл, приливают 4 мл HaSO4, дистиллированной воды до 15 мл и охлаждают. Затем 358
вносят 4 мл 4 М раствора KSCN, 5 мл 10% раствора аскорбиновой кислоты, доводят до метки водой и тщательно перемешивают. Фотометрируют через 20 мин в кювете с толщиной слоя 3 см, применяя светофильтр с областью светопропускания около 420— 440 нм. Раствором сравнения служит раствор, содержащий все реактивы, кроме роданид-иона.
Градуировочную кривую строят по растворам стандартных образцов стали, содержащих известное количество молибдена. Растворение стандартных образцов стали и приготовление растворов для фотометрирования производят аналогично тому, как приготавливают и фотометрируют исследуемый раствор.
Определение молибдена в вольфрамовых сталях. Навеску 0,1 г вольфрамовой стали обрабатывают 10 мл 20% раствора НС1, карбиды разрушают азотной кислотой, выпаривают, добавляют 10 мл 50% раствора лимонной кислоты и 40 мл дистиллированной воды или 4 г щавелевой кислоты и 50 мл воды и осторожно нагревают до растворения вольфрамовой кислоты. Затем раствор переносят в мерную колбу на 250 мл, доводят до метки дистиллированной водой и перемешивают. Аликвотную часть раствора, соответствующую оптической плотности А » 0,43, вносят в колбу на 25 мл, прибавляют все реактивы и определение ведут так же, как и при анализе хромистой стали.
ПР и меч а н и е. Применение аскорбиновой или лимонной кислоты позволяет определить молибден в присутствии вольфрама, ванадия, хрома, никеля и кобальта. Присутствие ионов NO3 до 0,15 моль/л также не мешает определению.
Реактивы
H2SO4, 20% раствор.
HNOS, конц.
H2SO4 (1 : 1)
KSCN, 4 М раствор.
Аскорбиновая кислота, 10% раствор (10 г аскорбиновой кислоты, 2 мл 0,1 М раствора ЭДТА, 10 капель муравьиной кислоты, 90 мл воды).
Лимонная кислота, 50% раствор.
Щавелевая кислота, х. ч.
Стандартные образцы сталей, содержащие 0,5—2% молибдена.
Работа 5. Определение циркония в сплавах [415]
Определение основано на образовании окрашенного соединения циркония с реагентом арсеназо III в кислой среде.
Выполнение работы. Навеску сплава 0,1—0,2 г растворяют либо в соляной (сплавы на алюминиевой, титановой основе и др.), либо в азотной (сплавы на медной основе) кислотах. При анализе сплава на титановой основе, после растворения навески в соляной кислоте, окисляют титан пероксидом водорода и кипятят раствор до обесцвечивания. Раствор переводят в мерную колбу, доводя кислотность до 2 моль/л. Для фотометрирования отбирают аликвотную часть раствора, содержащую 5—25 мкг циркония, разбавляют до 10 мл 2 Л! НС1 и нагревают до кипения.
359
При анализе сплавов на основе железа добавляют солянокислый гидроксиламин до уничтожения в растворе желтой окраски Fe3*. После охлаждения переводят раствор в мерную колбу на 50 мл, приливают 3 мл 1 % раствора желатины, 1 мл 1 % раствора арсеназо III, доводят объем до метки 2 М НС1, перемешивают и измеряют оптическую плотность по отношению к раствору сравнения, содержащему те же количества желатины и арсеназо III в 50 мл 2 М НС1. При подготовке раствора сравнения для анализа сплавов на основе меди в мерную колбу дополнительно вводят солянокислый раствор соли меди (2 М НС1) в количестве, отвечающем содержанию меди в аликвотной части исследуемого раствора, взятого для фотометрирования. Измерения оптической плотности проводят в кювете с толщиной слоя 2 см.
Градуировочную кривую строят по растворам стандартных образцов сплавов с известным содержанием циркония. Приготовление стандартных растворов для фотометрирования производят аналогично исследуемому раствору.
Методика применима для определения циркония в различных сплавах на основе титана, железа, меди, ванадия, алюминия, магния и др.
Примечание. В кислой среде (2 М НС1) алюминий, магний, железо титан (до 10 иг), медь (до 10 мг), сульфат-иои (до 100 мг) определению ие мешают.
Реактива
НО, конц.
НО, 2 М раствор.
HNOj» конц.
Гидролси.. ашя ‘юлвяокислый, к. ч.
Желатина, ! % раствор
Стандартный раствор соли меди, содержащий 0,5 мг меди в 1 мл. Навеску 1,3401 г н. ч. CuCL--2HjO растворяют в дистиллированной воде, добавляют 166 мл конц. НС (плотность 1,19) и разбавляют раствор до 1 л.
Арсеназо III, 1% раствор, 10 мг арсеназо III растворяют при нагревании в 50— 60 мл воды, добавляют 15 мл НО (1 : 5) и разбавляют раствор водой до 100 мл.
ОПРЕДЕЛЕНИЕ МЕТОДОМ ДОБАВОК
Работа I. Определение титана в легированных сталях [416]
Определение основано на образовании в кислой среде окрашенного в желтый цвет комплексного катиона титана с пероксидом водорода [TiO(H,O2) ]s+.
Анализируемый образец стали переводят в раствор и приготавливают из него раствор сравнения, исследуемый раствор и исследуемый раствор с добавкой а определенного количества титана. Измерив значения оптических плотностей исследуемого раствора Ая и того же раствора с добавкой Ах+а относительно раствора сравнения, неизвестное содержание титана в аликвотной части исследуемого раствора определяют по формуле:
4ti = °^х/(^х+в ^х)
Здесь а — масса титана, добавленная к аликвотной части исследуемого раствора образца, мг.
Наибольшая воспроизводимость результатов при определении содержания гитана достигается при = 2АЯ = 0,43.
360
Выполнение работы. Навеску стали 0,25 г обрабатывают при нагревании 50 мл H2SO4 (1 : 4). После растворения по каплям прибавляют азотную кислоту до прекращения вспенивания. Кипятят до удаления оксидов азота, охлаждают, переносят в мерную колбу на 100 мл, разбавляют ее содержимое дистиллированной водой до метки и перемешивают. Отбирают три аликвотные части раствора по 25 мл в колбы на 50 мл. Для приготовления раствора с добавкой к одной из аликвотных частей добавляют точно отмеренный объем сульфата титана (0,05—1,00 мг титана), 2 мл НгОа и 3 мл ортофосфорной кислоты для связывания Fe3+ в бесцветный комплекс. Ко второй аликвотной части добавляют 2 мл НгО2 и 3 мл Н3РО4 (исследуемый раствор). В третью колбу добавляют только 3 мл НЯРО4 (раствор сравнения). Содержимое колб разбавляют дистиллированной водой до метки, перемешивают и фотометрируют на фотоколориметре с синим светофильтром в кюветах с толщиной слоя 3 см относительно раствора сравнения.
Методика применима для определения 0,1—0,4% титана в образцах сталей, содержащих до 70% Fe, 25% Ni и 5% Си.
Реактивы
HaSO4 (1 : 4), х. ч.
Н3РО4 (плотность 1,70), х. ч.
HNOS (плотность 1,40), х. ч.
НаОа, 3% раствор.
Стандартный раствор Ti(SO4)s, содержащий 0,05 мг титана в 1 мл. Навеску 0,0834 г х. ч. TiO2 обрабатывают смесью серной (1:3) н плавиковой кислот при нагревании до полного растворения диоксида титана и выпаривают большой избыток H2SO4 до выделения белых паров SO3, для удаления фторида водорода. Затем раствор охлаждают, разбавляют водой и выпаривание повторяют. К полученному раствору добавляют 100 мл дистиллированной воды н 5% раствором HtSO4 доводят объем до 1 л.
ОПРЕДЕЛЕНИЕ ДИФФЕРЕНЦИАЛЬНЫМ МЕТОДОМ
Работа 1. Определение титана в ильменитовых концентратах [417]
Определение основано на образовании в кислой среде окрашенного комплексного катиона тнгана с пероксидом водорода [ТЮ(Н2О2)Р+.
Приготавливают раствор сравнения — окрашенный раствор пероксидного соединения титана определенной концентрации Со. Затем приготавливают исследуемый и стандартные окрашенные растворы этого же соединения титана н измеряют их относительные оптические плотности по отношению к раствору сравнения. Концентрацию титана в исследуемом растворе определяют либо при помощи градуировочной кривой, построенной по стандартным растворам (рис. 11.1), либо расчетным способом по формуле (7.32).
Концентрации исследуемого и стандартных растворов должны быть больше концентрации раствора сравнения.
Из рнс. 11.1 видно, что в интервале концентраций 12—18 мг титана в 100 мл раствора соблюдается прямолинейная зависимость между оптической плотностью н концентрацией раствора.
В качестве раствора сравнения используют раствор, содержащий 12 мг титана в 100 мл.
Мешающее действие Fe* устр 1ияют добавлением фосфорной кислоты.
361
Рис. И.!. Градуировочный график для определения концентрации раствора титана.
Концентрация титана в растворе сравнения 12 мг в 100 мл.
Измерения оптической плотности проводят на спектрофотометре СФ-16 при длине волны 390 нм. (Можно применять также и фотоколориметр ФЭК-56М). Погрешность определения не превышает 0,5% (отн.).
Выполнение работы. Навеску 0,5 г х. ч. ТЮ2 помещают в коническую колбу, прибавляют 12,5 г (NH4)2SO4 и 27,5 мл H2SO4 (плотность 1,12) и нагревают до полного растворения. После охлаждения раствор переводят в мерную колбу на 250 мл, доводят водой до метки и перемешивают. 1 мл полученного рабочего раствора содержит 1,2 мг титана.
Для приготовления раствора сравнения в мерную колбу на 100 мл помещают 10 мл рабочего раствора титана, прибавляют 20 мл Н3РО4, 50 мл 20% раствора H2SO4 и 5 мл 6% Н2О2 доводят объем до метки тем же раствором H2SO4 и тщательно перемешивают. Этот раствор, содержащий 12 мг титана в 100 мл, используют в качестве раствора сравнения.
Стандартные растворы титана приготавливают следующим образом: в мерные колбы на 100 мл помещают 12, 14 и 15 мл рабочего раствора титана и те же количества реактивов, что и при приготовлении раствора сравнения. После доведения объемов до метки и перемешивания растворы фотометрируют в кюветах с толщиной слоя 1 см.
Для определения титана в исследуемом концентрате берут навеску 0,3 г, добавляют несколько капель концентрированной H2SO4 и сплавляют ее при 800—900 °C в платиновом тигле с 5 г пиросульфата калия до получения прозрачного плава. Затем при нагревании выщелачивают плав 50 мл H2SO4 (плотность 1,12), содержащей 1 мл 6% раствора Н2О2. Раствор охлаждают, переводят в мерную колбу на 100 мл, доводят тем же раствором H2SO4 до метки и перемешивают, отбирают 15 мл в мерную колбу на 100 мл и приливают все реактивы в тех же количествах, что и при фотометрировании стандартных растворов.
Относительную оптическую плотность исследуемого раствора измеряют по отношению к раствору сравнения, содержащему 12 мг титана в 100 мл.
Содержание титана в отобранной аликвотной части раствора вычисляют либо по формуле (7.32), либо по градуировочной кривой зависимости оптической плотности от концентрации.
Если анализируемый продукт содержит малые количества железа, методика определения титана остается той же, но исключается добавление Н8РО4.
362
Реактивы
TiO2, х ч. (NH4)2SO4, х. ч.
H2SO4 (плотность 1,12 и 1,84).
HSPO4 (плотность 1,70).
H2SO4, 20% раствор.
Н2О2, 6% раствор.
Пиросульфат калия, х. ч.
Работа 2. Определение железа в продуктах титанового и магниевого производств {418]
Определение основано на образовании окрашенного комплексного катиоиа двухвалентного железа с о-фенантролнном.
Приготавливают раствор сравнения — окрашенный раствор соединения железа с о-фенантролином. Затем приготавливают исследуемый н стандартные окрашенные растворы этого же соединения железа с о-фенантролином и измеряют нх относительные оптические плотности по отношению к раствору сравнения. Неизвестную концентрацию железа определяют по градуировочной кривой, построенной по стандартным растворам (рис. 11.2), или расчетным способом по формуле (7.32).
Измерения оптической плотности производят на спектрофотометрах СФ-14 нлн СФ-16 прн 516 нм в кюветах с толщиной слоя 1 см. Для этих целей можно применять также фотоколориметры с узкополосным светофильтром, имеющим область пропускания 510—520 нм.
Применение спектрофотометра и определение неизвестной концентрации ионов железа прн помощи градуировочной кривой позволяют получить результаты с погрешностью, не превышающей ±0,5% (отн.).
В связи с тем, что в анализируемых образцах содержится различное количество ионов железа, целесообразно иметь два раствора сравнения, содержащих 0,5 мг и 1,0 мг железа в 100 мл.
Выполнение работы. Навеску образца 0,5 г обрабатывают при нагревании 30 мл раствора HCI. Раствор фильтруют через беззольный фильтр «с белой лентой» с добавлением бумажной массы и промывают горячей водой Фильтр с осадком озоляют в платиновой чашке и сплавляют со смесью 3 г Na2CO3 и 1,5 г Na2B4O7. Выщелачивают водой при добавлении 300 мл раствора HCI. Раствор объединяют с первоначальным фильтратом и объем его доводят в мерной колбе до 500 мл. Отбирают аликвотную часть раствора 5 мл в мерную колбу на 100 мл, прибавляют 10 мл 5% раствора тартрата натрия, 10 мл 2% свежеприготовленного
раствора солянокислого гидроксиламина, 10 мл 25% раствора ацетата натрия, 10 мл 0,2% раствора о-фенантролина и доводят объем раствора водой до метки.
Одновременно готовят два раствора сравнения, для чего в две
Рис. 11.2. Градуировочный график для определения концентрации раствора железа: / — концентрация железа в растворе сравнения 0.5 мг в 100 мл: 2 — концентрация железа в растворе сравнения 1.0 мг в 100 мл.
0,5 1,0 1,5 2,0 2,5 3,0 С,мг на 100 мп
363
Рнс. 11.3. Градуировочный график для определения концентрации раствора никеля.
Концентрация никеля в растворе сравнения 3,0 иге 100 мл.
мерные колбы на 100 мл помещают соответственно 5 и 10 мл стандартного раствора соли железа и все те же реактивы, которые используют для приготовления исследуемого раствора. Через 30 мин изме
ряют оптическую плотность исследуемого раствора на спектрофотометре СФ-14 (или СФ-16) в кюветах с толщиной слоя 1 см при длине волны 516 нм по отношению к тому раствору сравнения, с которым получается минимальная оптическая плотность.
Если в исследуемом образце имеется от 10 до 20% железа, для построения градуировочной кривой берут стандартные растворы, содержащие 6, 7, ..., 10 мл железа, а от 20 до 30% — 11, 12, ..., 15 мл железа. В первом случае в качестве раствора сравнения используют раствор, содержащий 0,5 мг железа в 100 мл, а во втором — 1,0 мг железа в 100 мл. При содержании железа от 5 до 10% необходимо брать аликвотную часть раствора — 10 мл.
Реактивы
НС1 (1 : 1), ч. д. а.
Na2COs, х. ч.
Na2B4O7, х. ч.
Тартрат натрия, 5% раствор.
Гидроксиламин солянокислый, 2% раствор.
Ацетат натрия, 25% раствор.
о-Фенантролин, 0,2% раствор.
Стандартный раствор соли железа, содержащий 0,1 мг железа в I мл. Навеску 0,7238 г х. ч. Fe(NOs)s-9H2O растворяют в дистиллированной воде, добавляют 2—3 мл HNOS (плотность 1,40) и доводят объем до 1 л.
Работа 3. Определение никеля в стали [419]
Определение основано на образовании в щелочной среде окрашенного соединения никеля с диметилглноксимом в присутствии окислителя.
Приготавливают раствор сравнения — окрашенный раствор диметилглиокси-мнна никеля определенной концентрации Со, затем приготавливают исследуемый н стандартные окрашенные растворы н измеряют их относительные оптические плотности. Концентрацию ионов никеля в исследуемом растворе определяют при помощи градуировочной кривой (рнс. 11.3), построенной по стандартным растворам, содержащим никель. Концентрация стандартных растворов меньше концентрации раствора сравнения (Сет < Со).
Примечание. Определение никеля можно проводить в присутствии Fe?*, Мп2*, Си2* н Сг2*. Влияние Fe2* устраняют связыванием его в устойчивый виннокислый комплекс; Мп2* даже прн трехкратном избытке по отношению к нонам никеля определению не мешает. Мешающее действие ионов меди не сказывается прн содержании до 20% (по отношению к количеству никеля), а влияние ионов хрома устраняют добавлением Ст2* в раствор сравнения и стандартные растворы.
364
Выполнение работы. Градуировочную кривую строят по стандартным растворам, для чего в мерные колбы на 100 мл помещают 3--30 мл стандартного раствора никеля концентрации 0,1 мг/мл. В каждую колбу добавляют 4 мл раствора Cr(NO3)3 (с содержанием хрома 2 мг/мл), 12 мл раствора Fe(NO3)3 (с содержанием железа 1 мг/мл), 5 мл 20% раствора сегнетовой соли, 10 мл 10% раствора (NH4)2S2O8, 15 мл 10% раствора NaOH и 5 мл 1% щелочного раствора диметилглиоксима. Содержимое колб перемешивают, через 1 ч доводят водой до метки и снова перемешивают.
Раствор сравнения, содержащий 30 мл стандартного раствора никеля и все остальные реактивы, фотометрируют с сине-зеленым светофильтром в кювете с толщиной слоя 5 см по отношению к стандартному раствору.
Для определения никеля в анализируемом образце стали берут навеску 0,2 г (при содержании Ni от 5 до 12%) или 0,1 г (при содержании Ni более 12%) и обрабатывают 20 мл раствора НС1 (1 : 1), разрушают карбиды и окисляют железо несколькими каплями HNO3 (плотность 1,40). Охлаждают раствор, переносят в мерную колбу на 200 мл, разбавляют водой до метки и перемешивают; 20 мл полученного раствора переносят в мерную колбу на 100 мл и далее поступают так же, как при построении градуировочной кривой.
Методика рекомендуется для определения высоких содержаний никеля в стали (до 26%).
Реактивы
Сегнетова соль, 20% раствор.
(NH4)2S2O6, 10% раствор.
NaOH, 10% раствор.
Диметилглиоксим, 1% щелочной раствор.
Стандартный растворами никеля, содержащий 0,1 мг никеля в 1 мл. Навеску 0,4786 г х. ч. NiSO4-7H2O растворяют в дистиллированной воде, подкисляют раствор 2—3 мл конц. H2SO4 и доводят объем водой до 1 л.
Стандартный раствор нитрата хрома, содержащий 2 мл хрома в 1 мл раствора. Навеску 15,39 г х. ч. Cr(NO3)3-9H2O растворяют в 200 мл дистиллированной воды, добавляют 2—3 мл HNO3 (плотность 1,40) и доводят объем водой до 1 л. Стандартный раствор нитрата железа, содержащий 1 мг железа в 1 мл раствора. Навеску 7,238 г х. ч. Fe(NO3)3-9H2O растворяют в дистиллированной воде, добавляют 2—3 мл HNOS (плотность 1,40) н доводят объем до 1 л.
Работа 4. Определение больших количеств марганца в сталях [420]
Определение основано на поглощении света окрашенным раствором перманганат-иона. Сущность работы заключается в окислении ионов марганца (II) до марганцевой кислоты с последующим измерением оптического плотности полученного раствора относительно раствора сравнения, содержащего перманганатной
Сначала приготавливают раствор сравнения — окрашенный раствор перманганат-иона с известной концентрацией Со (6,62 мг марганца в 100 мл), затем приготавливают исследуемый и стандартные растворы, содержащие перманга-нат-ионы, и по отношению к ним измеряют относительную оптическую плотность раствора сравнения.
365
Рнс. II.4. Градуировочный график для определения концентрации раствора марганца.
Концентрация марганца в растворе сравнения 6.62 мг в 106 мл.
Концентрацию нонов марганца в исследуемом растворе находят прн помощи градуировочной кривой, построенной по стандартным растворам соли марганца. Содеожание нонов марганца в стандартных растворах должно быть меньше, чем в растворе сравнения (Сст< <Со). Оптическую плотность растворов
намеряют на фотоколориметрах КФК или ФЭК-56М с зеленым светофильтром.
Примечание. Соли Сг3+ н Ni2+ не мешают определению марганца; влияние Fe®+ устраняют добавлением Н3РО4.
Выполнение работы. Персульфатный метод. Навеску 0,2 г стали в конической колбе на 250 мл обрабатывают 30 мл раствора HNO3 и кипятят до полного удаления оксидов азота. К горячему раствору добавляют 10 мл 0,01 М раствора AgNO3 и 30 мл 25% раствора (NH4)2S2O8 и нагревают до кипения, после чего выдерживают на горячей бане до начала разложения избытка (NHJaSgOg. Раствор охлаждают, добавляют 1,0 мл раствора Н3РО4, переносят в мерную колбу на 200 мл, доводят водой до метки и перемешивают. Затем измеряют оптическую плотность раствора сравнения, содержащего 6,62 мг марганца в 100 мл и все реактивы, используемые для приготовления исследуемого раствора. Содержание марганца находят по калибровочной кривой (рис. 11.4). Для приготовления стандартных растворов в мерные колбы на 200 мл добавляют 9, 10, 11, ..., 15 мл раствора соли марганца; затем в каждую колбу добавляют по 1,8 мл раствора КаСгаО7, содержащего 3,4 мг хрома в 1 мл, 7 мл раствора соли железа (5 мг железа в 1 мл), 1 мл раствора Н3РО4, 5 мл раствора H2SO4 (1 : 1), разбавляют водой до 200 мл, перемешивают и измеряют оптические плотности.
Перйодатный метод. Навеску 0,1 г стали обрабатывают 10 мл смеси растворов кислот (3 об. ч. конц. HNO3 и 1 об. ч. конц. HCI); после растворения приливают 20 мл H2SO4 (1 : 1) и 10 мл Н3РО4 и нагревают до прекращения выделения паров SO3. Раствор охлаждают, приливают 50 мл воды и 10 мл HNO3 (плотность 1,4), снова нагревают до кипения, прибавляют ~0,5 г КЮ4 и кипятят 2—3 мин для окисления марганца, затем охлаждают, переносят раствор в мерную колбу на 100 мл, доводят водой до метки и перемешивают. По отношению к нему измеряют оптическую плотность раствора сравнения, для приготовления которого в мерную колбу на 100 мл отбирают 30 мл титрованного раствора соли марганца, содержащего 0,44 мг марганца в 1 мл, и все реактивы, используемые для приготовления исследуемого раствора. Для приготовления стандартных растворов отбирают 12, 14, ..., 28 мл
366
титрованного раствора соли марганца (0,44 мг марганца в 1 мл) и приготавливают их для фотометрирования аналогично исследуемому раствору. Раствор КгСг2О7 в стандартные растворы марганца не вводят, так как содержащийся в стали хром перйодатом калия не окисляется и, следовательно, определению марганца не мешает.
Реактивы
НЬЮ3 (плотность 1,12).
AgNO3> 0,01 М раствор. ((NH4)2S2O8, 25% раствор. Н8РО4 (плотность 1,7).
Стандартный раствор MnSO4, содержащий 0,44 мг марганца в 1 мл. Навеску 1,932 г х. ч. MnSO4-5H2O растворяют в 100 мл воды, подкисляют 2—3 мл H2SO4 (1:1) и доводят объем водой до 1 л.
Стандартный раствор К2Сг2О7, содержащий 3,4 мг хрома в 1 мл. Навеску 9,615 г х. ч. К2Сг2О7 растворяют в 200 мл дистиллированной воды, подкисляют 3 мл HNO3 (плотность 1,40) и доводят объем до 1 л.
Стандартами раствор Fe2(SO4)3, содержащий 5 мг железа в 1 мл. Навеску 25,16 г х. ч. Fe2(SO4)3-9H2O растворяют в 200 мл дистиллированной воды, подкисляют 2—3 мл H2SO4 (плотность 1,84) и доводят объем водой до 1 л. H2SO4 (1:1).
HNO3 (плотность 1,40).
НС1 (плотность 1,19).
КЮ4, х. ч.
Работа 5. Фотометрическое определение Р2О6 в фосфорсодержащих удобрениях по молибденовой сини [130, 421]
Определение основано иа реакции восстановления фосфорномолнбденового комплекса до соединения, отвечающего предположительно формуле (МоО2 X X 4МоО8)2-Н3РО4 и окрашенного в синий цвет, и на фотометрировании растворов на фотоколориметрах ФЭК-56, ФЭК-56М, КФК при Лэфф = 590 нм (светофильтр № 8).
Выполнение работы. Для устранения погрешностей, связанных с построением градуировочного графика, концентрацию Р2О5 в фотометрируемых пробах определяют расчетным путем (см. разд. 7.3.1) по уравнению:
Сх = Си + FA ’х
Здесь F = ЬС/А'ст—обратный угловой коэффициент; АС — разность концентраций Р2О5 в стандратном растворе и растворе сравнения (Со); А' — относительная оптическая плотность анализируемого раствора.
Определение обратного углового коэффициента. Готовят серию стандартных растворов, содержащих 1,0—4,0 мг Р2О6 в 100 мл фотометрируемого раствора. Для этого в мерные колбы вместимостью 100 мл отмеряют из бюретки соответственно 5,0; 10,0; 12,5; 15,0; 17,5; 20,0 мл стандартного раствора (0,2 мг Р2ОБ в 1 мл). Затем приливают 2 мл соляной кислоты, дистиллированную воду (примерно до 50 мл), 7 мл раствора восстановителя, 10 мл молибдата аммония, через 10 мин — 20 мл раствора ацетата натрия. Доводят объемы до метки дистиллированной водой и перемешивают. Через 30—40 мин каждый раствор трижды фотометрируют относительно раствора сравнения в кюветах толщиной 1 см. Результаты изме
367
рений вносят в табл. 11.1 и вычисляют среднее значение обратного углового коэффициента. Графическое построение градуировочной кривой — не обязательно.
Дифференциальное фотометрическое определение высоких содержаний Р2ОБ*. При анализе продуктов, содержащих 20—60% Р2ОБ (удобрения, фосфориты, экстракционные кислоты и Др-), берут навеску 2,5 г (д) с погрешностью 0,0002 г, помещают в термостойкий стакан вместимостью 100 мл, прибавляют 50 мл НС1 (1:1) и кипятят 30 мин. После охлаждения содержимое стакана переносят в мерную колбу вместимостью 250 мл, доводят до метки дистиллированной водой и перемешивают. Если раствор прозрачен, то сразу отбирают аликвотный объем (VJ пипеткой на 25 мл. Если же раствор мутный, то его фильтруют, отбрасывая первые порции фильтрата, а из последующего фильтрата отбирают 25 мл (V\). Аликвотный объем (V\) переносят в мерную колбу на 250 мл и разбавляют его дистиллированной водой до метки. Затем из полученного раствора отбирают аликвоту У2 = = 10 мл, содержащую 2—4 мг Р2ОБ, в мерную колбу вместимостью 100 мл, добавляют 2 мл НС1 (1 : 10), 10 мл 10 % раствора борной кислоты (при наличии высоких содержаний фторид-ионов), дистиллированную воду примерно до 50 мл и далее —
Таблица 11.1. Расчет обратного углового коэффициента F градуировочного графика
Со = 1,0 мг PsOi в 100 мл, I — 1 см.
в 100 мл ДС = Сст — Со при измерениях 2iAi Л 3 р.=-*£ « Л'
1 2 3
2,0 2,5 3,0 3,5 4,0 1,0 1,5 2,0 2,5 3,0 ....
Примечание: А' и F — средние значения, вычисленные соответственно из 1—5
результатов 3-х и 5-тн параллельных измерений (F — •/» У Fj).
1=1
* В работе приведены соотношения реактивов, порядок их приливания и условия фотометрирования, обеспечивающие 1130] воспроизводимость результатов, сопоставимую с воспроизводимостью классических методов анализа. При выполнении работы на фотоколориметре КФК-2 или на спектрофотометрах выбор Адфф должен быть произведен как с учетом Хманс спектра поглощения «молибденовой сини», так и с учетом чувствительности этих приборов.
368
все реактивы, как при приготовлении и фотометрировании стандартных растворов.
Одновременно готовят раствор сравнения (Со = 1,0 мг Р2О8 в 100 мл раствора). В случае необходимости проверки значений F готовят стандартный растзор с концентрацией Сст = 2,0 мг Р2О6 в 100 мл (или иной). Через 30—40 мин измеряют оптическую плотность приготовленных растворов относительно Со. Значения А' должны укладываться в пределы 0,3—1,0. В противном случае необходимо изменить разбавление анализируемого раствора.
Концентрацию (Сх, мг на 100 мл раствора) и массовую долю (и, %) Р2О6 в объекте анализа рассчитывают по уравнениям *:
Сх = Со + FA'X; и, = Сх-250-250- 1000)
Примечание. Определение Р2ОЬ по данной методике возможно в при-сутств! и примесей (в пересчете на CaO, Fe2O2, Al2OSt SiO2, SiFj“, F-) при соотношениях содержание прнмеси к содержанию P2Ot не более 25, 15, 15, 4, 22, 15 (без HSBOS) и 70 (в присутствии Н2ВО3) соответственно.
Определение малых содержаний Р2ОЬ абсолютным методом. Определение содержаний 0,1—20% Р2О6 может быть проведено абсолютным методом по градуировочному графику, построенному для интервала концентраций 0,5—2,5 мг Р2О5 на 100 мл раствора. При этом используют все вышеуказанные реактивы в тех же количествах и в том же порядке, что и при дифференциальном методе. В качестве раствора сравнения берут раствор «холостого» опыта, содержащий все добавляемые реагенты.
Реактивы
Соляная кислота (1 : 1).
Молибдат аммония; раствор в серной кислоте. 50 г молибдата аммония (х. ч. нлн ч. д. а.) переносят в стакан вместимостью 700—800 мл н растворяют в 500 мл 5 М раствора серной кислоты. Содержимое стакана переносят в мерную колбу емкостью 1 л, доводят объем дистиллированной водой до метки, раствор перемешивают н фильтруют.
Ацетат натрия. 600 г ацетата натрия (х. ч. нлн ч. д. а.) растворяют в 2 л дистиллированной воды н фильтруют.
Фосфат калия КН2РО4, одпозамещенный (х. ч. нлн ч. д. в.).
Стандартный раствор фосфата калия. Навеску 0,3834 г однозамещенного фосфата калия растворяют в мерной колбе вместимостью 1 л, прибавляют 2 мл концентрированной серной кислоты и доливают дистиллированной водой до метки. 1 мл полученного раствора содержит 0,2 мг Р2О2.
Раствор восстановителя. 5 г кристаллического сульфита натрия вносят в стакан вместимостью 500—600 мл, растворяют в 400 мл дистиллированной воды, добавляют 1 г метола и после растворения вводят 150 г бисульфита натрия (или пиросульфита натрия Na2S2OB). После растворения бисульфита натрия содержимое стакана переносят в мерную колбу вместимостью 500 мл, доводят объем дистиллированной водой до метки, раствор перемешивают и фильтруют.
* Для достижения большей воспроизводимости и правильности может быть рекомендо ано уравнение (7.33) [266].
13 М, И. Булатов др. 369
Работа 6. Фотометрическое определение Р2О6 фосфсрно-молибденованадиевым методом в удобрениях и фосфатных растворах [127]*
Определение основано на образовании фосфорномолибденованадиевой гетерополикислоты (ГПК) желтого цвета и фотометрированин желтых растворов. Фотометрнрование осуществляют в кюветах с толщиной слоя I = 20 мм на фотоколориметрах ФЭК-56, ФЭК-56М, КФК с лампой накаливания при Х3фф = = 400ч-440 нм (светофильтры № 3 или № 4) и дополнительным жидким светофильтром 0,02 М КМпО4. Последний позволяет получать в более широком интервале концентраций и с большим углом наклона линейные градуировочные графики.
Выполнение работы. Определение обратного углового коэффициента. В мерные колбы вместимостью 100 мл вводят из мерной бюретки 5,0; 10,0; 12,5; 15,0; 17,5; 20,0 мл стандартного раствора (0,2 мг Р2О6 в 1 мл), прибавляют дистиллированную воду (примерно до 50 мл), по 30 мл единого реактива, доводят объемы до метки дистиллированной водой и перемешивают. Через 5—10 мин каждый раствор фотометрируют трижды в кювете (I = 20 мм) относительно раствора сравнения (Со = 1 мг Р2О6 в 100 мл). Результаты заносят в таблицу по форме табл. 11.1 и рассчитывают среднее значение F. Значения Fs, полученные для каждого стандартного раствора, не должны отклоняться от F более чем на 1—2% (отн.).
При массовом экспрессном анализе периодически производят проверку значения F по одной точке путем фотометрирования стандартного раствора, содержащего 2,0 мг Р2О6 в 100 мл, относительно раствора сравнения (Со = 1,0 мг Р2О6 в 100 мл). В этом случае F = (Сст — С0)Л4ст = 1,0Мст.
Дифференциальное фотометрическое определение высоких содержаний Р2О6 (20—60%). Навеску (<?) пробы, взятую с погрешностью 0,0002 г, помещают в термостойкий стакан вместимостью 100 мл, прибавляют 50 мл HCI (1 : 1) и кипятят 30 мин. После охлаждения содержимое стакана переносят в мерную колбу на 250 мл и доводят до метки дистиллированной водой **. Аликвотный объем (V) полученного раствора, содержащий от 2 до 4 мг Р2О6, переносят в мерную колбу на 100 мл, разбавляют дистиллированной водой примерно до 50 мл и далее поступают, как при приготовлении серии стандартных растворов. Одновременно готовят стандартные растворы (Со = 1,0 и Сст — 2,0 мг Р2ОБ в 100 мл). Через 5—10 мин проводят измерение оптической плотности приготовленных растворов по отношению к раствору сравне
* В работе воспроизводятся оптимальные условия фотометрирования по данным [127] для получения высокой воспроизводимости результатов, сопоставимой с воспроизводимостью классических методов анализа (см. разд. 3.1.3.2). При работе на других марках и типах приборов для достижеиня аналогичной воспроизводимости следует проводить измерения оптической плотности при максимальной чувствительности прибора.
** Если раствор мутный, то его фильтруют и аликвотную часть отбирают нз фильтра.
370
ния (Со). Значение Ах должно находиться в пределах 0,2—0,9. В противном случае необходимо повторить анализ с другой навеской или другим аликвотным объемом (V 5 мл).
Концентрацию (Сх, мг на 100 мл раствора) и массовую долю (ов, %) Р2О6 рассчитывают по формулам *:
Сх = Со + FA* = 1 + FAx-, u = Сх • 250 • I00/(<?V-1000)
Примечание. Определению не мешают примеси Са, Mg, Fe, Al, F, SIOt, SiF|~ в количествах, ие превышающих массовое содержание Р2ОБ в фото-метрируемой пробе соответствен ио в 57, 45, 5, 13, 9, 3, 250 раз.
Определение малых содержаний (<20%) Р2ОБ абсолютным методом. Определение Р2ОБ по желтой окраске фосфорномоли-бденованадиевой кислоты можно провести быстро и абсолютным методом. В этом случае при вышеприведенных условиях градуировочный график строят для интервала концентраций 0,5—2,5 мг Р2ОБ в 100 мл раствора, для которого соблюдается основной закон светопоглощения.
Реактивы
Соляная кислота (I: I)
Фосфат калия КН2РО4 и стандартный раствор фосфата калия (см. работу 5). Единый реактив. Готовят смешением трех компонентов в объемном соотношении 1 : 1 : 1 в следующей последовательности: HNOS (1 : 1), молибдат аммония (водный 10% раствор) и раствор NH4VO2 (2,5 г/л) с добавлением 20 мл НЬ1О3 (конц.).
Работа 7. Дифференциальный фотометрический метод определения кремния в фосфорсодержащих удобрениях [421]
Определение основано на реакции восстановления кремнемолибденовой кислоты и фотометрнрованнн растворов «молибденовой сини» на фотоколориметрах ФЭК-56, ФЭК-56М, КФК при Х3фф= 590 нм (светофильтр № 8) в кюветах с I = = 30 мм относительно Раствора сравнения (Со = 0,05 мг SiOs в 100 мл раствора).
Выполнение работы. Определение обратного углового коэффициента F. В мерные колбы вместимостью 100 мл из бюретки отбирают 5,0; 10,0; 15,0; 17,5; 20,0; 25,0 мл (0,05—0,25 мг SiO2 в 100 мл) стандартного раствора, доводят дистиллированной водой примерно до 50 мл, прибавляют 50 мл НС1 (1 : 10), 10 мл 5% раствора молибдата аммония, выдерживают 5 мин и добавляют 5 мл винной кислоты для устранения влияния Р2ОБ. Через следующие 5 мин добавляют 10 мл раствора аскорбиновой кислоты для восстанозлэния кремнемолибденовой гетерополикис-лоты и доводят объем раствора до метки дистиллированной водой. Через 40—60 мин трижды измеряют оптическую плотность каждого стандартного раствора относительно раствора сравнения (Со). Полученные данные заносят в таблицу, аналогичную по форме табл. 11.1, и рассчитывают значение обратного углового коэффициента F.
• См. сноску в работе 5.
13*
371
Определение кремния в образцах. Навеску (q) 0,1—0,2 г, взвешенную с погрешностью 0,0002 г, помещают в платиновый тигель, добавляют 3 г плавня (60 г соды и 40 г буры), на кончике шпателя — нитрат калия и помещают тигель в муфельную печь. Доводят температуру до 950 °C и через 15—30 мин обрабатывают HCI (1 : 10). Полученный раствор переносят в мерную колбу вместимостью 500 мл и доводят до метки дистиллированной водой. Аликвотный объем (V) полученного раствора, содержащий 0,1— 0,3 мг SiO2, помещают в мерную колбу вместимостью 100 мл, прибавляют 10 мл буферного раствора, дистиллированную воду примерно до 50 мл. Затем добавляют 10 мл молибдата аммония, через 5 мин — 5 мл винной кислоты, еще через 5—10 мин — 10 мл аскорбиновой кислоты, доводят дистиллированной водой до метки и через 50—60 мин измеряют оптическую плотность окрашенных растворов относительно раствора сравнения (Сп), приготовленного параллельно.
При периодической проверке значения F параллельно с исследуемым раствором и раствором сравнения приготавливают еще один стандартный раствор (Сст = 0,15 или 0,20 мг SiO2 в 100 мл), измеряют его оптическую плотность относительно раствора сравнения и рассчитывают значение F.
Определение массовой доли (со, %) кремния проводят по формуле •:
<п = (С0 + ДЛ^).25/(У9)
Примечание. Иногда буферной емкости добавляемого буферного раствора с pH = 1,15 не хватает. В этих случаях устанавливают pH по индикаторной бумаге «Рнфан> или по рН-метру
Реактивы
Соляная кислота 1:1, 1 : 10.
Карбонат натрия, ч. д. а.
Тетраборат натрия, ч. д. а.
Стандартный раствор. Навеску 3,1330 г кремнефторида калия (х. ч. или ч. Д1 а.) растворяют в дистиллированной воде в мерной колбе вместимостью 1л 10 мл полученного раствора содержат 0,1 мг SiO2.
Молибдат аммония. 50 г молибдата аммония (х. ч. нлн ч. д. а.) растворяют прн нагревании до 50 °C в 400 мл дистиллированной воды в термостойком стакане вместимостью 1 л, переносят в мерную колбу на 1 л, доводят дистиллированной водой до метки и фильтруют.
Винная кислота. 30 г препарата (х. ч.) растворяют в 70 мл дистиллированной воды.
Аскорбиновая кислота. 2,5 г препарата (ч. д. а.) растворяют в 100 мл дистиллированной воды.
Буферный раствор, pH = 1,15.
Работа 8. Дифференциальный фотометрический метод определения алюминия в минеральных удобрениях 1129, 421)
См. сноску в работе 5.
372
Определение основано на образовании окрашенного комплекса алюминия с хромазуролом S (Хманс MR = 549 нм, е=5,9-104) в уксуснокислой среде (pH = 5,7ч-5,8) и фотометрированин полученных растворов. Оптическую плотность измеряют при ^фф = 540 нм (светофильтр № 6) на фотоколорнметрах ФЭК-56, ФЭК-56М, КФК в кюветах (/ = 10 мм) относительно раствора сравнения (Со = 0,018 мг А12О3 в 100 мл раствора) *.
Примечание. В случае высоких содержаний примеси железа (при Fe2Os/Al2Os > 2) его влияние устраняют добавлением аскорбиновой кислсты или 0,02 М раствора ЭДТА, образующих с нонами Fe (III) комплексы, не влияющие на ход определения. Влияние титана устраняют введением фосфорной кислоты, а мешающее действие последней совокупностью приемов: созданием высокого фосфатного фона, увеличением количества добавляемого реагента (хромазурола S) по сравнению с общеизвестными методиками, применением дифференциального метода измерения оптической плотности. Определению не мешают 2500-, 3000-, 2500-, 2-кратные количества фосфат-ионов, кальция, магния, фторид-нонов соответственно.
Выполнение работы. Определение обратного углового коэффициента. Готовят серию стандартных растворов, содержащих 0,04—0,09 мг А12О3 в 100 мл. Для этого в мерные колбы вместимостью 100 мл вводят из бюретки 5,0; 10,0; 15,0; 17,5; 20,0; 25,0 мл стандартного раствора Б, добавляют 5 мл НС1, 0,5 мл раствора Н3РО4 (10 мг/мл), 2 мл 0,1% раствора аскорбиновой кислоты, дистиллированную воду примерно до 50 мл, затем приливают при постоянном перемешивании (как можно более точно) 10 мл 0,1% раствора хромазурола S, 10—20 мл буферного раствора, доводят до метки дистиллированной водой и сразу измеряют оптическую плотность окрашенных растворов относительно раствора сравнения (Со = 0,018 мг А12О3 в 100 мл раствора). Оптическую плотность каждого раствора измеряют трижды, результаты заносят в таблицу, составленную по форме табл. 11.1, и рассчитывают значение F.
Определение высоких содержаний алюминия в сырье (апатитах, фосфоритах, известняках и т. д.). Навеску (у), взятую с погрешностью 0,0002 г, помещают в термостойкий стакан вместимостью 200—250 мл, прибавляют 50 мл НС1 (1 : 1) и 5 мл концентрированной азотной кислоты и кипятят до растворения. Затем содержимое стакана количественно переносят в мерную колбу вместимостью 250 мл и доводят раствор до метки дистиллированной водой. Из полученного раствора отбирают аликвотную часть (V), содержащую 0,04—0,09 мг А12О3, переносят в мерную колбу вмести юстмо 100 мл, прибавляют 1 мл фосфорной кислоты, 2 мл 0,1 % раствора аскорбиновой кислоты, добавляют дистиллированную воду примерно до 50 мл, приливают прн сильном помешивании 10 мл 0,1% раствора хромазурола S и 10—25 мл буферного раствора н дистиллированную воду до метки.
Одновременно готовят раствор сравнения (Со — 0,018 мг А1аО3 в 100 мл раствора), а в случае периодической проверки значе
• При выполнении работы на фотоколориметре КФК-2 н на спектрофотометрах выбор X производят с учетом чувствительности этих приборов для достижении высокой воспроизводимости результатов анализа.
373
ний F — еще стандартный раствор (Сст = 0,24 или 0,072 мг А12О3 в 100 мл). Исследуемый и стандартный растворы фотометрируют относительно раствора сравнения. Измеряемое значение А' должно находиться в пределах 0,2—0,5.
Фотометрирование растворов должно производиться в течение часа после их приготовления.
Массовую долю (со, %) алюминия определяют по формуле:
<в = (С0 + М;)-25/(У<?)
Определение малых содержаний А1аО3 (экстракционные кислоты, известняки и др.). При малых содержаниях А12О3 навеску увеличивают до 5—10 г. После операции растворения и отбора аликвотной части, содержащей 0,04—0,09 мг А1аО3, в колбу вместимостью 100 мл (см. выше) туда же последовательно прибавляют: 1 мл Н3РО4, 2 мл 0,1% аскорбиновой кислоты, дистиллированную воду (примерно до 50 мл), 1—2 капли индикатора тропеолина 00 и водного раствора аммиака или НС1 (в зависимости от среды раствопа) до желтой окраски раствора, 10 мл 0,1 % раствора хромазурола S, 10 мл аце атного буферного раствора и дистиллированна ю воду до метки. Дальнейшие операции проводят, как описано выше.
Примечание. Если аликвотную часть раствора берут в пределах 5—10 мл, то можно просто увеличить объем прибавляемого буферного раствора с 10 до 20 мл н не устанавливать в этом случае pH по тропеолииу.
Определение ведут следующим образом. Аликвотную ч«,ст! раствора (5— 10 мл).помещают в мерную колбу на 100 мл, прибавляют 1 мл Н3РО4, 2 мл аскорбиновой кислоты, дистиллированную воду (примерно до 50 мл), 10 мл 0,1% раствора хромазурола S, 25 мл буферного раствора, доводят дистиллированной водой до метки и фотометрируют относительно окрашенного раствора сравнения (Со). В стандартный раствор вводят такой же объем буферного pacToOja.
Реактивы
Соляная кислота, 0,1 М раствор.
Фосфорная кислота, 60%-ная, реактивная.
Ацетат натрия.
Хромазурол S, 0,1% раствор.
Алюминиевая фольга
Аскорбиновая кислота, 0,1% раствор. ЭДТА, ч. нлн ч. д. а.
Стандартный раствор алюминия. 0,1900 г металлической фольги (предварительно промытой спиртом) помещают в термостойкий стакан на 520 мл и прибавляют 50 мл HCl (I : 1). После растворения всей навески содержимое стакана количественно переносят в мерную колбу на 1 л, прибавляют 50 мл HCl (1 : 1) и доводят до метки дистиллированной водой. Полученный раствор А содержит 0,36 мг А12О3 в 1 мл. Затем 5 мл раствора А переносят мерной пипеткой в мерную колбу на 500 мл. Полученный раствор Б содержит 0,0036 мг А1аЭ3 в 1 мл.
Раствор фосфорной кислоты. Навеску 22 г 60%-ной фосфорной кислоты помещают в мерную колбу на 1 л и доводят дистиллированной водой до метки. Буферный раствор. 272 г ацетата натрия растворяют примерно в 700 мл дистиллированной воды, доводя pH раствора по pH-метру до 5,9 и разбавляют дистиллированной водой до метки в мерной колбе на 1 л; затем полученный раствор фильтруют и используют для работы.
374
Работа 9. Дифференциальный фотометрический метод определения железа в фосфорсодержащих удобрениях [128, 421]
Определение основано на образовании трисульфосалицилата железа при взаимодействии ионов Fe (111) с сульфосалициловой кислотой в щелочной среде и фотометрированни желтых окрашенных растворов. Оптическую плотность измеряют на фитоколориметрах ФЭК-56, ФЭК-56М, КФК при ХЭфф = 400 нм (светофильтр № 3) в кюветах с I = 10 мм относительно раствора сравнения (Со= 0,143 мг Fe2Os в 100 мл).
Примечание. Определению не мешают высокие содержания фосфора, кальция, алюминия, фторид-нонов н кремния.
Выполнение работы. Определение обратного углового коэффициента. Готовят серию стандартных растворов, содержащих 0,6—1,5 мг Fe2O3 в 100 мл раствора. В мерные колбы на 100 мл вводят из бюретки 1,0; 4,0; 6,0; 7,0; 8,0; 10,0 мл типового раствора железа, добавляют 20 мл 20% раствора сульфосалициловой кислоты и водный раствор аммиака до появления желтой окраски, затем прибавляют избыток аммиака 0,5 мл и доводят объем раствора до метки дистиллированной водой. Измеряют оптическую плотность каждого раствора трижды относительно окрашенного раствора сравнения (Со = 0,143 мг Fe2O3 в 100 мл). Полученные результаты заносят в таблицу, аналогичную табл. 11.1, и рассчитывают значение F.
Определение железа в фосфоритах. Навеску (q) фосфорита, взвешенную с погрешностью 0,0002 г, растворяют при нагревании в 50 мл HCI (1 : 1) с добавлением 1 мл концентрированной HNO3, переносят в мерную колбу на 250 мл, доводят до метки дистиллированной водой, отфильтровывают. Из фильтрата берут аликвотную часть (V), содержащую 0,6—1,5 мг Fe2O3, и помещают в мерную колбу на 100 мл; прибавляют 40 мл 20% раствора сульфосалициловой кислоты и водный раствор аммиака (25%-ный) до появления желтой окраски, затем еще избыток аммиака — 0,5 мл, доводят до метки дистиллированной водой.
Одновременно готовят окрашенный раствор сравнения (Со = = 0,143 мг FejOs в 100 мл), а если надо проверить значение F, то окрашенный стандартный раствор (Сст = 0,858 мг Fe2O3 в 100 мл). Оптическую плотность растворов измеряют относительно раствора сравнения. Значение А' должно находиться в пределах 0,3—0,8. Фотометрируемые растворы сохраняют окраску неизменной в течение длительного времени.
Массовую долю (ш, %) железа рассчитывают по формуле:
Щ = (Со + ГЛ;)-25/(1/9)
Примечание. В случае появления осадка прн добавлении раствора аммиака (в присутствии больших количеств кальция и фосфат-нонов) необходимо прибавить еще сульфосалициловой кислоты до полного растворения осадка, затем осторожно прибавить водный раствор аммиака. Если повторение подобной операции не приводит к желаемому результату, то растворы отфильтровывают прямо в кюветы.
375
При высоких содержаниях железа в образцах (А' >0,9) необходимо производить двойное разведение (25/250—25/250), т. е. из колбы на 250 мл следует отобрать 25 мл раствора в другую мерную колбу на 250 мл и уже из этой колбы отобрать аликвотную часть для фотометрирования.
Реактивы
Аммиак, 25% раствор.
Сульфосалициловая кислота, 20% раствор.
Железо-аммонийные квасцы, ч. д. а.
Стандартный раствор железа. Навеску железо-аммонийных квасцов, 0,8634 г (х. ч. нли ч. д. а.) растворяют в стакане вместимостью 500 мл, добавляют 10 мл концентрированной азотной кислоты, стакан накрывают часовым стеклом н кипятят раствор на песчаной бане 2—3 мин. После охлаждения количественно переносят содержимое стакана в мерную колбу :ia 1 л и доводят до метки дистиллированной водой. 1 мл раствора содержит 0,143 мг FejOj.
ОПРЕДЕЛЕНИЕ ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИМ МЕТОДОМ
Работа 1. Прогнозирование условий и методика экстракционно-фотометрического определения никеля в присутствии кадмия, марганца и цинка с помощью 1-(2-пиридилазо)-2-нафтола
Прогнозирование условий экстракционного разделения и селективного экстракцнонно-фтометрнческого определения Ni с ПАН (HR) в присутствии Cd, Мп и Zn проводят на основе критериев, изложенных в разд. 4.3.4.4 и 7.4.2.
Пиридилазонафтолаты никеля, кадмия, марганца и цинка имеют следующие экстракционные и фотометрические характеристики:
Экстрагируемый комплекс ^макс' км •макс"10 * ₽н‘/. 1в Кэке
NiRj 575 5,1 2,1 1,9
CdRa 560 4,9 7,0 —7,9
MnRa 550 5,9 8,5 — 11,0
ZnRg 560 2,9 5,7 —5,3
Из представленных в табл. 11.2 данных видно, что оптимальное значение pH для экстракции пиридилазонафтолата никеля находится в интервале 4,0— 8,0. Основные критерии экстракционного разделения и селективного экстракционно-фотометрического определения Ni выполняются при pH — 4,04-6,0 — для Cd, 4,0 — для Zn и 4,04-7,0 — для Мп. Поэтому ионы никеля можно определять с помощью ПАН в присутствии соизмеримых количеств Cd, Мп и Zn, экстрагируя хелатный комплекс NiRa хлороформом при pH « 4,0. При этом отпадает необходимость предварительного отделения или маскирования Cd, Мп н Zn.
Выполнение работы. К кислому раствору, содержащему до 20 мкг никеля, кадмия, марганца и цинка, добавляют 1 мл 1 М раствора KNO3, 1 мл 0,01 М спиртового раствора реагента ПАН и доводят объем дистиллированной водой до 10 мл. Через 5 мин к раствору с выпавшим осадком добавляют 10 мл хлороформа и встряхивают смесь на механическом встряхивателе колб в течение 5 мин. После расслаивания органическую фазу фильтруют через комочек сухой ваты в кювету с толщиной слоя 1 см, и изме-
376
Таблица 11.2. Результаты расчетов критериев экстракционного разделения и селективного фотоне.рического определения Ni
в присутствии Cd, Мп и Zn при экстракции их пиридилазонафтолатов хлороформом
[НР1о = 0.001 моль/л, [NO3] = 0.1 моль/л, г = 1. Cd, Mn, Zn — М.
Элемент рн КЭкс IgD E. % , CNlPN'l CMPM 1 e ”1 iCNlDNI (1+dm) EMCMPM (* + PN1)
Ni 3,0 1,85 1,85 98,6
4,0 1,79 3,77 99,9 — —
5,0 1,44 5,16 99,9 — —
6,0 0,63 5,17 99,9 — —
7,0 —0,34 5,18 99,9 — -—
8,0 — 1,34 5,18 99,9 — —
Cd 4,0 —8,0 —6,0 10-4 9,77 6,02
5,0 —8,0 —4,0 0,01 9,16 4,02
6,0 —8,0 —2,0 1,0 7,17 2,02
7,0 —8,0 0 50,0 5,18 0,32
8,0 —8,0 2,0 99,1 3,18 0,02
Mn 4,0 — 11,0 —9,0 10-’ 12,77 8,94
5,0 — 11,0 —7,0 10_B 12,16 6,94
6,0 — 11,0 —5,0 io-s 10,17 4,94
7,0 — 11,0 —3,0 0,09 8,18 2,94
8,0 — 11,0 —1,0 9,1 6,18 0,98
Zn 4,0 —5,25 —3,25 5,6-IO-2 7,02 3,27
5,0 —5,25 — 1,25 5,6 6,41 1,29
6,0 —5,25 0,75 84,9 4,42 0,09
7,0 —5,42 2,58 99,7 2,60 0,02
8,0 —5,75 4,25 99,9 0,93 0,02
ряют оптическую плотность раствора при 575 нм относительно экстракта холостого опыта. Исходное значение pH (—4,5) создают добавлением растворов NH3 и HNO3. Значения pH водных растворов после экстракции контролируют с помощью pH-метра, обеспечивая их равновесные значения в интервале 4,0—4,5. Содержание никеля находят по градуировочному графику.
Результаты определения 11,7 мкг Ni в присутствии 11,0 мкг Cd, 11,0 мкг Мп и 13,0 мкг Zn характеризуются высокой надежностью и хорошей воспроизводимостью (при п = 5, Р = 0,95, sr = 0,024, mNl = 11,7 ± 0,3 мкг).
Реактивы
Стандартные растворы нитратов никеля, кадмия, марганца и цинка, содержа-щ.1 2—4 мкг Ni, Cd, Мп и Zn в 1 мл.
/CNOS, х. ч., 1 М раствор.
ПАН, 0,01 М спиртовый раствор.
Аммиак, 10% раствор.
Хлороформ, ч. д. а.
Работа 2. Теоретическое обоснование и методика экстракционно-фотометрического определения меди с помощью диэтилдитиокарбамината свинца РЬ (ДДК)г в хлороформе
Экстракционно-фотометрическое определение медн этим методом основано на реакции вытеснения свинца из его диэтилдитиокарбамннатного комплекса в хлороформе (или тетрахлориде углерода):
РЬ(ДДК)2 + Си3* Си(ДДК)2 + РЬ2*
1(> -Каиссц — 13,70, 1g Кайеру — 7,77
Прогнозирование условий проведения обменной экстракции проводят на основе теоретических критериев, изложенных в разд. 4.3.4.3 и 4.3.4.4.
Предполагая извлекать ионы меди из 50 мл примерно 2- Ю-5 М раствора, принимаем, что минимальный избыток экстрагента должен составить около 100% (Е мл 4-10-1 М раствора диэтилдитиокарбамината свинца в хлороформе). По уравнению (4.70) находим минимальное (пороговое) значение pH водного раствора, при котором возможна обменная реакция (рНп = 5). При pH = 5 аСц1+ = 1, аРЬ*+ 1’ ^эксСи ^эксСи’ ^ЭКСРЬ ^KCPb' Поскольку ^3KCCv
/СэКСрь> условия для обмена ионов Си2* на ионы РЬ2* в процессе экстракции благоприятные.
Согласно уравнению (4.67), минимальное отношение условных констант экстракции 7С,КСс при заданных условиях (т = п = 2, g = 20, г =
= 0,1) должно составлять не менее 4-10s. Действительное же соотношение равно 105,в, т. е. при заданных условиях количественное извлечение ионов меди достигается за одну экстракцию.
Полученные результаты прогнозирования хорошо согласуются с широким использованием экстрагента РЬ(ДДК)2 в хлороформе или тетрахлориде углерода при избирательном извлечении ионов меди из водных растворов.
Выполнение работы. В делительную воронку на 50—100 мл помещают 10—15 мл дистиллированной воды, 10 мл исследуемого раствора и устанавливают значение pH = 5 (по индикаторной бумаге), добавляют 5 мл хлороформного раствора РЬ (ДДК)2 и интенсивно встряхивают в течение 2 мин. Органический слой сливают в мерную колбу вместимостью 25 мл, а к водному раствору добавляют еще дважды по 5 мл экстрагента и снова повторяют экстрагирование. Экстракты соединяют, разбавляют хлороформом до 25 мл и измеряют оптическую плотность полученного раствора при 436 нм в кювете с толщиной слоя 5 см относительно раствора сравнения, приготовленного из 15 мл реактива и разбавленного хлороформом до 25 мл.
Для построения градуировочного графика приготавливают стандартные растворы, содержащие 5, 10, 15, 20, 30 и 40 мкг меди, и проводят с ними те же операции, что и с исследуемым раствором. По полученным значениям оптических плотностей строят градуировочный график, при помощи которого и определяют неизвестное содержание меди в исследуемом растворе.
Примечание. Определению меди по этой методике не мешают большинство катионов металлов. Мешают висмут, ртуть, серебро, металлы платиновой группы, большие содержания ни келя (больше 10 г/л) и кобальта (больше 20 г/л).
Реактивы
Хлороформ, ч. д. а.
РЬ (ДДК)2 — диэтилдитиокарбаминат свинца в хлороформе. К 50—100 мл водного раствора, содержащего —0,1 г ацетата свинца (ч. д. а.) прибавляют 25— 50 мл водного свежеприготовленного раствора, содержащего — 0,1 г диэтилди-
378
тиокарбамииата натрия. Образующийся белый осадок растворяют в 250 мл хлороформа. Хлороформный слой отфильтровывают через сухой фильтр в мерную колбу вместимостью 500 мл, разбавляют хлороформом до метки и тщательно перемешивают. Реактив пригоден для работы в течение длительного времени (3—4 недели, если он защищен от солнечного света).
Стандартный раствор соли меди содержащий 10 мкг меди в 1 мл. Навеску 0,1 г электролитической меди, помещенную в стакан вместимостью 50—100 мл, обрабатывают 3—5 мл HNOS (1: 1), добавляют 20—30 мл дистиллированной воды; полученный раствор переводят в мерную колбу на 1 л и разбавляют водой до метки. Разбавлением приготовленного раствора в 10 раз получают стандартный раствор, содержащий 10 мкг меди в 1 мд.
Работа 3. Определение кобальта в присутствии Fe (111) 171]
Определение основано на образовании окрашенного соединения Со с 2-нитрозо-1-иафтолом и измерении светопоглощения его хлороформного экстракта.
Выполнение работы. Кислый раствор анализируемого образца (до 0,2 мг Со2+), содержащий до 0,5 г Fe (III), обрабатывают 15 мл раствора цитрата натрия, разбавленного 50—75 мл, и доводят pH до 3—4 (по индикаторной бумаге), добавляя 2М раствор НС1 или NaOH. Раствор охлаждают до комнатной температуры, добавляют 10 мл раствора Н2О2 и спустя несколько минут — 2 мл раствора реактива. Выдерживают в течение 30 мин при комнатной температуре, после чего помещают в делительную воронку и энергично встряхивают в течение 1 мин с 25 мл хлороформа. Хлороформный экстракт сливают в мерную колбу на 50 мл и повторяют экстракцию двумя порциями хлороформа объемом по 10 мл. Объединенные экстракты разбавляют хлороформом до 50 мл и помещают раствор в чистую делительную воронку. Добавляют 20 мл 2Л1 раствора НС1 и встряхивают в течение 1 мин. Сливают хлороформный экстракт в другую делительную воронку, куда добавляют 20 мл 2М раствора NaOH и встряхивают в течение 1 мин. Хлороформный слой сливают, и измеряют оптическую плотность прозрачной органической фазы при 530 нм.
Для построения градуировочного графика берут стандартные растворы хлорида кобальта, содержащие 0,5; 10; 20; 40; 80; 100; 150 и 200 мкг Со2+. и анализируют их аналогично исследуемому, проводя опыт через все стадии анализа.
Устойчивая окраска раствора сохраняется не менее 12 ч. Определению не мешают большинство элементов при содержании до 0,1 г.
Реактивы
2-Нитроэо-1-нафтол, 1% раствор. Растворяют 1 г 2-ннтрозо-1-нафтола в 100 мл ледяной уксусной кислоты. Добавляют 1 г активированного угля. Раствор перед употреблением встряхивают н отфильтровывают нужный для работы объем. Цитрат натрия, х. ч., 40% раствор.
НС1, х. ч., 2 М раствор.
NaOH, х. ч., 2 М раствор.
Н2О2, х. ч., 3% раствор. Хлороформ, ч. д. а.
Станда тный раствор хлорида кобальта, содержащий 10 мкг кобальта в I мл.
379
Растворяют в воде 0,0404 г. х. Ч. СоС1а-6Н2О, добавляют 2 мл конц. НС1 и разбавляют водой до 1 л.
Работа 4. Определение’малых количеств хрома с дифенил-карбазидом [423]
Определение основано на образовании окрашенного соединения Cr (VI) с дифеннлкарбазидом и экстракции его комплекса с нафталнн-Р-сульфонатом натрия изоамнловым спиртом.
Окрашенное соединение Cr (VI) с дифеннлкарбазидом является однозарядным катионом, который с анионом — нафталин-р-сульфонатом образует соединение, хорошо экстрагируемое изоамнловым спиртом При умеренных количествах большинство посторонних ионов определению хрома не мешает. Предел определения — 0,001 мкг Cr (VI) в 1 мл исследуемого водного раствора.
Выполнение работы. К 80—100 мл исследуемого раствора, содержащего ионы * СггО^, прибавляют 2 мл ЮМ раствора H2SO4, 0,3 мл 0,1% спиртового раствора дифенилкарбазида и через 5 мин — 50 мл 0,1 М раствора нафталин-р-сульфоната натрия. Объем доводят бидистиллятом до 200 мл и переносят в делительную воронку; туда же прибавляют 10 мл изоамилового спирта. Содержимое воронки энергично встряхивают, и через 5—10 мин органический слой сливают в кювету с толщиной слоя 1 см. Через 5 мин измеряют оптическую плотность экстракта при X — 545 нм (при работе на колориметре используют зеленый светофильтр) относительно экстракта холостой пробы.
Для построения градуировочного графика приготавливают стандартные растворы, содержащие в пересчете на Cr (VI) 1—10 мкг хрома в 1 л. Затем с каждым из стандартных растворов выполняют те же операции, что и при анализе исследуемого раствора. По найденным значениям оптической плотности экстрактов стандартных растворов строят градуировочный график, по которому и определяют неизвестную концентрацию хрома в исследуемом растворе.
Погрешность определения в интервале концентраций хрома 1—10 мкг/л не превышает ±6% (отн.).
Реактивы
H2SO4, х. ч., 10 М раствор.
Дифенилкарбазид, ч. д. а., 01% раствор в этаноле.
Нафталин-$-сульфонат натрия, ч. д. а., 0,1 М водный раствор. Изоамиловый спирт, ч. д. а.
Na2COs, х. ч.
Бромная вода, насыщенная.
Стандартный раствор соли хрома, содержащий 1 мкг Cr (VI) в 1 мл. Навеску 0,0960 г х. ч. хромо-калневых квасцов KCr(SO4)2- 12Н2О растворяют в 100 мл бидистиллята, подкисляют 3 мл H2SO4 (1 : 1) и доводят объем бндистил-лятом до 1 л. Разбавлением полученного раствора в 10 раз приготавливают раствор содержащий 1 мкг Cr (VI) в 1 мл.
Работа 5. Определение следов железа с 1-нитрозо-2-наф-толоп. в веществах высокой степени чистоты [408]
* Если в растворе содержится Cr (III). его окисляют бромной водой.
380
Определение основано на образовании окрашенного соединения Fe (II) с 1-ннтрозо-2-нафтолом и измерении светопоглощения его в растворе изоамилового спирта.
Выполнение работы, а. Определение железа в иодиде натрия «особой» чистоты. Навеску препарата до 10 г помещают в стакан вместимостью 100 мл, растворяют в 25 мл раствора тартрата аммония и переносят в делительную воронку на 250 мл. В другую делительную воронку (холостой опыт) вводят 25 мл тартрата аммония. Далее в каждую из воронок добавляют по 15 мл аскорбиновой кислоты, нейтрализуют раствор аммиаком по феноловому красному (до pH = 7,5), прибавляют по 5 мл 0,1% раствора 1-нитрозо-2-нафтола, разбавляют дистиллированной водой до 150 мл и перемешивают. Через 40—50 мин производят двукратную экстракцию изоамиловым спиртом (по 10 мл), предварительно насыщенным углекислым газом. Объединенный экстракт разбавляют растворителем до 25 мл в мерной колбе и измеряют оптическую плотность на колориметре с красным светофильтром (^афф = 700 нм) в кювете с толщиной слоя 5 см относительно экстракта, полученного одновременно в холостом опыте. Измерения оптической плотности раствора производят не позднее, чем через 1 ч после начала экстракции.
Для приготовления стандартных растворов в делительную воронку вводят 20 мл винной кислоты (0,05 М раствор, содержащий 15—20 капель фенолового красного), отмеренные объемы титрованного раствора сульфата трехвалентного железа и 5 мл аскорбиновой кислоты. После нейтрализации раствора аммиаком по феноловому красному в воронку вводят 5 мл 0,1% раствора реактива и через 40 мин экстрагируют двумя порциями по 10 мл изоамилового спирта. Объединенный экстракт разбавляют растворителем до 25 мл в мерной колбе и измеряют оптическую плотность при тех же условиях, что и исследуемый раствор. Раствором сравнения служит экстракт, полученный одновременно в холостом опыте (последний отличается от анализируемого экстракта лишь заменой раствора сульфата железа равным объемом дистиллированной воды, подкисленной, как и упомянутый раствор, серной кислотой).
Для построения градуировочного графика берут стандартные растворы сульфата трехвалентного железа, содержащие 1—20 мкг железа. Неизвестную концентрацию железа в исследуемом растворе определяют по градуировочному графику. Закон Бера хорошо соблюдается в интервале концентраций 0,5—20 мкг Fe в 25 мл.
б. Определение железа в винной кислоте. Две навески винной кислоты (например, 4,5 и 0,5 г) после растворения в воде переводят в делительные воронки на 250 мл. В каждую из воронок прибавляют по 15 мл аскорбиновой кислоты, несколько капель индикатора фенолового красного и после нейтрализации аммиаком вводят по 5 мл 0,1% раствора 1-нитрозо-2-нафтола. Далее пробы
381
разбавляют водой приблизительно до 150 мл, перемешивают и через 40—50 мин производят двукратную экстракцию изоамиловым спиртом (по 10 мл). Объединенные экстракты для каждой пробы разбавляют растворителем до 25 мл в мерной колбе и перемешивают. Фотометрируют первый экстракт, полученный ив большей навески, относительно второго. Затем определяют содержание железа в 1 мл раствора аммиака. Для этого в несколько стаканов помещают по 200 мл раствора аммиака, выпаривают досуха, остаток растворяют в 20 мл 0,05 Л4 раствора винной кислоты и далее растворы готовят для фотометрирования так же, как и стандартные растворы (см. пункт «а»).
Из л ерив оптические плотности исследуемого раствора и раствора холостой пробы, используют предложенный А. Б. Бланком прием [81 ] учета примесей в реактивах при определении следовых количеств, т. е. по градуировочному графику отдельно находят суммарное содержание железа в пробе и реактивах и только в реактивах. Искомое содержание железа (мкг) в 1 г препарата (р) находят по формуле:
р = (Дх — q W'l/hm
Здесь Дх — содержание железа, отвечающее найденному значению оптической плотности раствора пробы и реактивов, мкг; q — содержание железа в 1 мл раствора аммиака, мкг; ДУ — разность объемов раствора аммиака, пошедшего на нейтрализацию двух навесок винной кислоты, мл; Д/п — разность двух навесок, г.
Примечание. Определению мешают большие количества Си,+ и Ag*.
Реактивы
Стандартный раствор сульфата железа, содержащий 0,1 мг Fe*+ в 1 мл. Навеску 0,4977 г перекристаллизованной, х. ч. соли Мора FeSO4-7H2O окисляют в сернокислой среде медицинским пергидролем и разбавляют водой до 1 л. Разбавлением полученного раствора получают растворы с меньшим содержанием железа. Вода, дважды перегнанная.
Изоамиловый спирт, х. ч. или ч. д. а.
Аскорбиновая кислота (медицинская), 10% раствор. Кислоты растворяют в воде, насыщенной СОа, н хранят в темной склянке.
1-Нитрозо-2-нафтол, 0,1% раствор. Навеску реактива 1 г растворяют в 120 мл 1 М раствора КОН (х. ч.), очищают от примесей металлов встряхиванием с хлороформом, после чего фильтруют н разбавляют водой до 1 л.
Винная кислота, ч. д. а.. 0,05 М раствор.
NH, (I : 20).
Тартрат аммония. Навеску 7,5 г винной кислоты растворяют в небольшом объеме воды, добавляют 2—3 капли фенолового красного и нейтрализуют аммиаком; прибавляют еще 15—13 капель раствора индикатора и разбавляют водой до 1 л. Перед употреблением прибавляют на 100 мл раствора 2 мл 3% раствора тиосульфата натрия и перемешивают.
Работа 6. Последовательное определение следов меди, никеля, железа и марганца в препаратах КС1 и КВг [424]
Определение основано на последовательном экстракционном извлечении из аммиачно-цитратного буферного раствора комплексов: медн с j иэтилдитнокарба-минатом, никеля с димстилглнокснмом, двухвалентного железа с 1-нитрозо-2-нафтолом н марганца с диэтилдитиокарбаминатом.
382
Примечание. Определению меди мешают только ноны Ag+, Hg,+, Bi3+ и Т13+ при соотношениях Си : Me соответственно меньших, чем 1 5оо' 1 : 500, 1:4 и 1 : 10.
Определению железа мешают, кроме меди и никеля, еще Со3+ и Ag+ в соотношении превышающем соответственно 4 : 1 и 10 : 1.
Определению марганца после отделения железа, меди и никеля мешают лишь Со2+ н MoOj — в количествах, превышающих количество марганца соответственно в 10 и 20 раз.
Выполнение работы. Навеску 5 г хлорида или бромида калия растворяют в воде с прибавлением 5 мл аммиачно-цитратного буферного раствора и, если нужно, дополнительно нейтрализуют аммиаком до pH = 7,5 по феноловому красному. Раствор переводят в делительную воронку на 250 мл, разбавляют водой приблизительно до 50 мл и затем последовательно экстрагируют из раствора медь, никель, железо и марганец.
Оптическую плотность измеряют на фотоколориметре с синим светофильтром в кювете с толщиной слоя 5 см относительно экстракта холостой пробы. (При определении железа применяют красный светофильтр.)
Определение меди. Производят двукратную экстракцию меди раствором диэтилдитиокарбамината свинца в хлороформе (по 5 мл), после чего раствор промывают 5 мл хлороформа и присоединяют его к экстракту. После разбавления хлороформом в мерной колбе на 25 мл раствор фильтруют в кювету и измеряют оптическую плотность.
Определение никеля. К раствору после отделения меди прибавляют, если нужно, 1—2 капли аммиака (1 : 20) до появления фиолетовой окраски индикатора (pH = 8.0), 1 мл 1% раствора диметилглиоксима и через 15 мин экстрагируют комплекс никеля с диметилглиоксимом тремя порциями по 5 мл хлороформа. Объединенные экстракты встряхивают с тремя порциями по 5 мл 0,5 М соляной кислоты. К полученному солянокислому раствору, помещенному в мерную колбу на 25 мл, прибавляют 0,2 мл бромной воды, 1 мл 25% раствора аммиака и 0,5 мл раствора диметилглиоксима, разбавляют водой до метки и через 15 мин измеряют оптическую плотность.
Определение железа. К раствору после отделения никеля прибавляют 15 мл 10% раствора аскорбиновой кислоты, осторожно нейтрализуют раствор аммиаком по феноловому красному, приливают 5 мл 0,1% раствора 1-нитрозо-2-нафтола и разбавляют водой примерно до 150 мл. Через 40 мин производят двукратную экстракцию изоамиловым спиртом (по 10 мл) нитрозонафтолата железа. Объединенные экстракты разбавляют в мерной колбе растворителем до 25 мл и измеряют оптическую плотность.
Определение марганца. После отделения железа раствор встряхивают с 10 мл хлороформа для удаления оставшегося изоамилового спирта. Хлороформный слой отбрасывают, вводят в делительную воронку 5 мл 5% раствора диэтилдитиокарбамината натрия и встряхивают с 5 мл хлороформа. Последнюю операцию
383
повторяют еще раз, после чего промывают раствор еще 5 мл хлороформа. Объединенные экстракты после разбавления хлороформом в мерной колбе до 25 мл фильтруют в кювету и через 15 мин после перемешивания измеряют оптическую плотность.
Градуировочные графики для определения меди, железа и марганца строят по стандартным растворам, проводя экстракцию каждого иона металла из аммиачно-цитратного буферного раствора по тем же методикам, что и для исследуемого раствора.
Стандартные растворы для определения никеля приготавливают, прибавляя определенные количества Ni2+ к 15 мл 0,5 М раствора НС1 с последующим введением бромной воды, аммиака и диметилглиоксима.
Градуировочные графики для всех определяемых ионов строят в интер вале концентраций 0,5—20 мкг в 25 мл конечного объема. В этом интервале концентраций хорошо выполняется основной закон светопоглощения. Неизвестные концентрации определяемых примесей Си, Fe, Ni и Мп находят по соответствующим градуировочным графикам. Для внесения поправки в результат определения периодически производят определение железа в аммиаке, для чего 50 мл последнего выпаривают досуха в кварцевом стакане, остаток растворяют в воде, содержащей аммиачно-цитрат-ный буферный раствор, и далее поступают, как при определении железа.
Относительная погрешность при определении 5 мкг Си, Ni, Fe и Мп в анализируемых образцах не превышает 6%.
Реактивы
Раствор диэтилдитиокарбамината свинца в хлороформе. Водный раствор, содержащий 0,2 г диэтилдитиокарбамината натрия, 0,2 г нитрата свинца и 1 г тартрата калия-натрия нейтрализуют аммиаком по феноловому красному н встряхивают с хлороформом. После промывания двумя порциями воды экстракт фильтруют и разбавляют хлороформом до 1 л.
Бромная вода, водный раствор брома, ч. д. а.
Диметилглиоксим, ч. д. а., 1% раствор в этаноле.
Цитрат натрия трехзамещенный, ч. д. а.
Аммиачно-цитратный буферный раствор. К 500 мл 4% раствора трехзамещенного цитрата натрия добавляют 4 мл 0,1% раствора фенолового красного, нейтрализуют аммиаком (1 : 20) по индикатору и разбавляют до 1 л.
Вода, дважды перегнанная.
Хлороформ, ч. д. а.
NHS, 25% заствор.
НО, х. ч., 0,5 М раствор.
Аскорбиновая кислота, 1% раствор.
1-Нитрозо-2-нафтол, ч. д. а., 0,1% раствор, 1 г реактива растворяют в 120мл 1 М раствора КОН и разбавляют до 1 л.
Изоамилосый спирт, ч. д. а.
Дизтилдитиокарбаминат натрия, ч. д. а., 5% раствор.
Стандартный раствор нитрата меди, содержащий 10 мкг медн в 1 мл. Навеску 0,1 г электролитической меди, помещенную в стакан на 50—100 мл, обрабатывают 3—5 мл HNOS (1 1), добавляют 20—30 мл бидистиллята, переводят в мерную колбу н разбавляют водой до метки 10 мл полученного раствора переносят в мерную колбу на 100 мл и разбавляют водой до метки
384
Стандартный раствор сульфата железа, содержащий 10 мкг железа в 1 мл. Навеску 0,7238 г. х. ч. Fe(NO3)2-9H2O растворяют в воде, добавляют 1—2 мл НЧО3 (плотность 1,40) н доводят объем до 1 л. 10 мл полученного раствора переносят в мерную колбу на 100 мл и разбавляют водой до метк !.
Сг. андартный раствор сульфата никеля, содержащий 10 мкг никеля в I мл. Навеску 0,4786 г х. ч. NiSO4-7H2O растворяют в воде, подкисляют 2—3 мл тарнц. H2SO4 и доводят объем водой до 1 л. 10 мл полученного раствора пере-нрсят в мерную колбу на 100 мл и доводят объем водой до метки.
Стандартный раствор хлорида марганца, содержащий 10 мкг марганца в 1 мл. Навеску 0,3597 г х. ч. МпС12-4Н2О растворяют в воде, подкисляют 2—3 мл конц. НС1 и доводят объем до 1 л. 10 мл полученного раствора помещают в мерную колбу на 100 мл и разбавляют водой до метки.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ПРИ ДВУХ ДЛИНАХ ВОЛН
Работа 1. Определение титана (IV) и железа (III) в шихте, содержащей W, Ti, Со, Al, Si [221 ]
Одновременное спектрофотометрическое определение Ti (IV) и Fe (III) в присутствии Со (II), А1 и Si основано на фотометрировании раствора смеси салицилатных комплексов этих металлов при двух длинах волн, где светопоглоще-ние салицилатного комплекса Fe (III) одинаковое.
Из рассмотрения спектров поглощения салицилатных комплексов титана и железа (рис. 11.5) видно, что наиболее целесообразно фотометрирование раствора смеси проводить при 360 и 570 нм. Прн 570 нм светопоглощенне салицилатного комплекса Fe точно такое же, как и прн 360 нм, но светопоглощенне салицилата титана отсутствует. Следовательно, разность оптических плотностей раствора смеси, измеренных прн 360 и 570 нм, тождественна оптической плотности раствора титана прн 360 нм. Затем по градуировочному графику или с помощью его уравнения находят неизвестные содержания Fe (III) Ti (IV).
Салицилатные комплексы Со (II) н А1 в интервале от 360 до 750 нм света не поглощают н определению Ti и Fe не мешают. Салицилатные комплексы Ti (IV) и Fe (III) при pH — 4 образуются практически мгновенно и устойчивы в течение длительного времени.
Выполнение работы. Навеску шихты 0,2 г, содержащую 3— 5% Ti и Fe, растворяют в 30 мл «царской водки» при слабом нагревании, добавляют 5—7 капель концентрированной серной
кислоты и упаривают до влажных солей. Затем прибавляют 20 мл
HCl (1 : 1), кипятят в течение 5—10 мин, добавляют 50 мл дистиллированной воды и охлаждают. После охлаждения отфильтровывают вольфрамовую кислоту, промывая осадок сначала раствором НС1 (1:1), а затем 2% раствором NH4NO3. Фильтрат и промывные воды собирают в мерную колбу на 250 мл, доводят дистиллиро-
Рис. 11.5. Спектры поглощения салицилатных комплексов:
1 — раствор титана; 2 — раствор железа.
ванной водой до метки и используют для определения Ti (IY) и Fe (III) в присутствии Со (II) и А1. Аликвотную часть фильтрата (10 или 20 мл) помещают в мерную колбу на 100 мл, добавляют 10 мл ацетатного буферного раствора (pH = 4) и 2 мл 10% спиртового раствора салициловой кислоты. Затеял добавляют заранее установленный объем 1 М раствора NaOH, необходимый для достижения pH = 4. Раствор в мерной колбе доводят до метки дистиллированной водой, перемешивают и измеряют оптическую плотность при 360 и 570 нм на спектрофотометре СФ-16 в кювете с толщиной слоя 1 см. Неизвестные содержания Ti и Fe находят по уравнениям градуировочных графиков, рассчитанным методом наименьших квадратов по стандартным растворам чистых солей титана и железа.
Основной закон светопоглощения соблюдается при содержании Ti до 0,5 мг в 100 мл.
Реактивы
Салициловая кислота, х. ч., 10% спиртовый раствор.
НС1, х. ч., конц.
HNO3, х. ч., конц.
H2SO4, х. ч., конц.
NH4NO3, х. ч., 2% раствор.
Ацетатный буферный раствор (pH = 4). К 50 мл 1 М раствора NaOH добавляют 284,4 мл 1 М уксусной кислоты и разбавляют дистиллированной водой до 500 мл.
NaOH, х. ч., 1 М раствор.
ЗАДАЧИ
1. Содержание меди в полупроводниковых материалах 1Х Х10-6%. Каким минимальным молярным коэффициентом светопоглощения должно обладать комплексное соединение меди, в виде которого ее определяют спектрофотометрически, если навеска образца не должна превышать 1 г, конечный объем измеряемого раствора не должен быть меньше 5 мл, кювета имеет 1 = 5 см, минимальное значение оптической плотности, при котором возможна погрешность не более 10%, составляет 0,020.
Ответ: емив = 1,29-105.
2. Рассчитать определяемый минимум для фото колориметрического определения железа (III) с сульфосалициловой кислотой в аммиачной среде. Толщина поглощающего слоя Z = 5 см, минимальный объем окрашенного раствора в кювете составляет 15 см3. При измерениях используют синий светофильтр. Среднее значение молярного коэффициента светопоглощения комплекса равно 4000. Минимальная оптическая плотность, измеряемая прибором, Амми = 0,01.
Ответ: mFe = 0,42 мкг.
3. Рассчитать условный коэффициент чувствительности (эффективное сечение кюветы 1 см2) спектрофотометрического определения меди с пиридилазонафтолом при 550 нм. Молярные 386
коэффициенты светопоглощения комплекса и реагента при 550 чм р^вны соответственно 4,5.104 и 4-102. Минимальное значение оптической плотности, регистрируемое прибором, равно 0,005. Состав комплекса 1:1.
Ответ: т3 — 7,1.10-3 мкг/см2.
4. Рассчитать неизвестное содержание железа в 50 см® анализируемого раствора по результатам фотометрирования методом добавок. Оптическая плотность исследуемого окрашенного раствора, приготовленного из 5 см3 анализируемого раствора, равна 0,45; значение оптической плотности такого же исследуемого раствора с добавкой 100 мкг Fe, измеренной в тех же условиях, составляет 0,85.
Ответ: 1,125 мг.
5. Для определения примеси А1 в силикате магния взята навеска последнего 0,2 г; после сплавления с содой и последующей обработки кислотой получено 200 см3 анализируемого раствора. Для приготовления окрашенного раствора алюминия с алюминоном отбирают аликвотную часть объемом 20 см® и разбавляют после добавления реактивов до 50 см3. Оптические плотности исследуемого окрашенного раствора и такого же раствора с добавкой 5 см® 5-10-1 М раствора А1С13, измеренные по отношению к дистиллированной воде в кювете с толщиной слоя 2 см на фотоколориметре, равны соответственно Ах = 0,25 и Ах+а — 0,55.
Рассчитать содержание (%) А1 в безводном силикате магния, если известно, что силикат содержит 5% воды.
Ответ: 0,3%.
6. Приготовлены четыре стандартных раствора соли же-леза(П), содержащих 1,0; 1,05; 1,10 и 1,15 мг железа. Оптические плотности трех последних окрашенных растворов железа с о-фе-нантролином, измеренные относительно первого стандартного окрашенного раствора, соответственно равны: А{ = 0,25; А? — = 0,50; Аз = 0,75. Оптическая плотность исследуемого окрашенного раствора, измеренная при тех же условиях по отношению к первому стандартному раствору, составляет 0,80.
Рассчитать содержание Fe в анализируемом растворе, если известно, что для приготовления окрашенного раствора использовали х/в аликвотную часть.
Ответ: 5,8 мг.
7. Приготовлено 9 стандартных окрашенных ргстворов, содержащих в объеме 50 см3 соответственно 1, 2, 3, 4, 5, 6, 7, 8 и 9 мг Fe. Оптическая плотность пятого раствора (5 мг Fe), измеренная поочередно относительно 1, 2, 3 и 4 растворов, была равна соответственно 1,0; 0,75; 0,50 и 0.25. Оптические плотности 6, 7, 8 и 9 растворов, измеренные в тех же условиях по отношению к 5-му раствору, составляли соответственно 0,25; 0,50; 0,75 и 1,0.
Рассчитать по этим данным уравнение прямой градуировочного графика для двустороннего дифференцирования.
Ответ: А' = 0,25С — 1.25.
387
8. Никель в виде диметилглиоксимина в присутствии окислителя определяют методом двустороннего дифференцирования и рассчитывают по уравнению прямой градуировочного графика А' = 0,50С — 1,20 (С в мг).
Рассчитать содержание никеля в двух исследуемых растворах, если известно, что оптическая плотность первого из них, измеренная относительно нулевого раствора, равна 1,2, а оптическая плотность нулевого раствора, измеренная в тех же условиях относительно второго исследуемого раствора, составляет 1,0.
Ответ: qx = 4,8 мг; q2 = 0,4 мг.
9. При определении методом добавок Ti(IV) в виде переоксид-ного комплекса в присутствии некоторого количества V (V) было приготовлено три раствора: исследуемый, исследуемый раствор с добавкой 200 мкг Ti и исследуемый раствор с добавкой некоторого количества V. Оптические плотности растворов, измеренные на фотоколориметре с синим и зеленым светофильтрами, были следующими:
Асх = 0,50; Л^+Т1 = 0,90; = 0,55
Л3 = 0,25; Л*+Т5 = 0,40; Л3+у=0,35
Рассчитать неизвестное содержание титана в фотометрируе-мом растворе.
Ответ: 230 мкг.
10. Для определения кобальта с нитрозо-Р-солью применяют дифференциальный метод добавок. Для этого из анализируемого раствора объемом 100 см3 приготавливают одинаковые объемы исследуемого раствора, раствора с добавкой и раствора сравнения. К одной из аликвотных частей объемом 10 см3 добавляют 2 см3 2-10-4 М раствора СоС1а и после приготовления окрашенных растворов фотометрируют относительно окрашенного раствора сравнения, приготовленного при тех же условиях из 8 см8 анализируемого раствора. Относительные оптические плотности исследуемого раствора с добавкой и без нее равны соответственно А'х+а — 0,75 и Ах = 0,35.
Рассчитать содержание кобальта в анализируемом растворе. Ответ: 1,03 мг.
11. Рассчитать определяемый минимум ионов никеля с ксиленоловым оранжевым при А = 584 нм, толщине поглощающего слоя Z = 5 см и конечном объеме фотометрируемого раствора V = 50 см3. Избыточная концентрация реагента составляет 5-10-5 моль/л, константа устойчивости комплекса = 2- 10е, состав комплекса 1:1. Наименьшее значение оптической плотности, измеряемое прибором, Лмин = 0,005. Молярный коэффициент светопоглощения комплекса 3,6-10*.
Ответ: 0,10 мкг.
12. Рассчитать определяемый минимум железа(Ш) в виде роданидного комплекса при X = 480 нм. Значение молярного
388
коэффициента светопоглощения равно 7000. Константа нестойкости комплекса [FeSCNl2+ равна 9,4-10-4; избыточная концентрация роданид-иона составляет 5-10"4 моль/л, толщина поглощающего слоя I — 5 см, конечный объем фотометрируемого раствора V — 25 см3. Среднее значение стандартного отклонения вд в интервале оптических плотностей 0,005—0,05 составляет ±2-10"3.
Ответ: 1,15 мкг.
13. Рассчитать определяемый минимум никеля в виде ди-метплглиоксимина в присутствии окислителя, используя при измерениях синий светофильтр. Состав образующегося соединения 1 : 3. Значения стандартных отклонений sA и средние значения оптических плотностей для различных концентраций равны соответственно:
А 0,10 0,18 0,24 0,32 0,40 0,50
sA 3-10"3 9-10-® 1,5-10"? 2,1-10"? 2,5-10"? 3,0-10"?
Среднее значение молярного коэффициента светопоглощения комплекса равно 9000. Минимальный объем раствора составляет 25 см3, толщина слоя I — 5 см. Реагент в этой области спектра излучения не поглощает.
Ответ: mN1 = 1,26 мкг.
14. При определении висмута в сплаве, содержащем до 0,02% Bi, используют фотометрическую реакцию его с тиомочевиной. Рассчитать оптимальные значения навески сплава и конечного объема фотометрируемого раствора, если известно, что молярный коэффициент светопоглощения комплекса висмута с тиомочевиной 3,5-104, состав комплекса 1 : 3, толщина поглощающего слоя I = 5 см, оптимальное значение оптической плотности ДОпт = 0-43. Фотометрирование проводят при X = 322 нм; молярный коэффициент светопоглощения раствора тиомочевины при 322 нм равен 3000.
Ответ: q/V = 3,46-10"3; q = 0,35 г; V = 100 см3.
15. Содержание примеси Ag в полупроводниковом материале (PbS) составляет 1-10"1® атомов/см3. Установить, возможно ли определение серебра с n-диметиламинобензилиденроданином, если известно, что фотометрирование следует проводить при X = = 460 нм в кювете с толщиной слоя 1 см. Молярные коэффициенты светопоглощения при 460 нм комплекса и реагента равны соответственно 20 000 и 900; состав комплекса 1 : 2. Минимальная оптическая плотность, измеряемая прибором, равна 0,01; плотность PbS равна 7,5 г/см3.
Ответ: q/V — 0,25. Определение невозможно.
16. При спектрофотометрическом определении Ni концентрацию последнего рассчитывают по уравнению прямой градуировочного графика CNi — 25,4-10“2 А (мкг/мл) при X = 580 нм. Оптическая плотность, измеренная при Хг = 580 нм в присутствии смолообразных веществ, Ai = 0,95. Значения оптической плот
389
ности раствора вне полосы поглощения при Х2 = 620 нм и = = 680 нм равны соответственно Л2 = 0,18 и А3 = 0,12.
Рассчитать неизвестное содержание никеля в растворе, если известно, что объем фотометрируемого раствора равен 50 мл.
Ответ: 9,25 мкг.
17. Хлорид никеля из водного раствора объемом 100 мл экстрагируют в виде диметилглиоксимина 10 мл хлороформа и разбавляют хлороформом до 25 мл. Из полученного раствора аликвотные части объемом 5 мл фотометрируют методом добавок. Рассчитать неизвестное содержание никеля в растворе с учетом его неполного извлечения, если известно, что оптические плотности хлороформных экстрактов с добавкой 20 мкг никеля и без нее равны соответственно 0,45 и 0,20. Коэффициент распределения диметилглиоксимина никеля в системе хлороформ — вода равен 410.
Ответ: 82,1 мкг.
18. Рассчитать содержание РЬ и Bi в смеси по светопоглоще-нию водных растворов комплексонатов этих металлов при X = = 240 и X — 365 нм. Значения оптической плотности раствора смеси комплексонатов РЬ и Bi, измеренной в кювете с толщиной слоя 3 см, равны соответственно Л240 = 0,87 и Лзе5 = 1,24. Общий объем фотометрируемого раствора составляет 50 мл. Значения молярных коэффициентов светопоглощения комплексонатов равны:
ЕРЬ-ЭДТА 'Bi-ЭДТА
X = 240 нм 8900 2800
X = 365 нм 900 9900
Ответ: <?РЬ = 207,2 мкг; <7Bi = 418 мкг.
19. Рассчитать содержание Си (II), Fe (III) и TI (I) в смеси по светопоглощению водных растворов комплексонатов этих металлов при 240, 250 и 260 нм. Значения оптической плотности
раствора смеси комплексонатов меди, железа и таллия, измеренной в кювете с толщиной слоя 2 см, равны соответственно: Л240 = = 0,44; Л250 = 0,48; Л260 = 0,50. Общий объем фотометрируемого раствора составляет 100 мл. Значения молярных коэффициентов
светопоглощения комплексонатов равны:
X = 240 нм
X = 250 нм
X = 260 нм
ЕСи-ЭДТА
5400
4000
3000
'Fe-ЭДТА
6000 /000
8800
'Т1-ЭДТА
1500
2000
1500
Ответ: qCu = 63,5 мкг; qFe = 111,7 мкг; </Т1 = 613,1 мкг.
20. Из 100 мл водного раствора, содержащего 1 • 10-4 моль/л NiCl2, 1-10-4 моль/л CuCl2, 1 моль/л ЫН3 и 0,1 моль/л цитрата натрия, предполагают экстрагировать Ni в виде диметилглиоксимина тремя последовательными порциями хлороформа по 10 мл, содержащего 4,8.10“4 моль/л диметил глиоксима. Определить интервал pH водного раствора, где никель практически полностью переходит в хлороформный слой, а медь практически полностью
390
остается в водной фазе. Диметилглиоксим переходит в органическую фазу только о молекулярной форме HR.
^„„-0.12; ^„,„,-410: -0.12;
Кэксм = 6-10 *; Лэкссц == 1«7.
Ответ: pH = 9-4-12.
21. Для отделения меди от цинка используют экстракцию 0,1 М раствором бензоилацетона (HR) в бензоле. Найти фактор разделения и оптимальный интервал pH, где обеспечивается практически полное разделение однократной экстракцией при отношении W1V = 10. В водном растворе содержится I- 10-а моль/л ZnCl2 и I-I0-5 моль/л СиС12. Конкурирующие комплексанты отсутствуют. По данным Стары [167], соединения экстрагируются в форме хелатов MR2, а реагент — в молекулярной форме HR.
₽hr = 5-1°8; ^dHr = 1,15'1о3;
Лэксеи= 6,75-10 s; КЭисг„= 1,68- 10"и.
Ответ: S = 4-10е; pH = 4,14-5,4.
22. При экстракции оксихинолината Со (II) из 100 мл 1- 10“Б М водного раствора при pH — 5 возможна совместная экстракция оксихинолината серебра. В водном растворе содержится 1X X 10-4 моль/л соли Ag (I), 0,1 моль/л Na2SO3, и экстракцию проводят 10 мл 0,1 М раствора 8-оксихинолина (HR) в хлороформе. Определить степень извлечения серебра, если известно, что при этих условиях K3KcAg = 1,26-10"6 и экстрагируемый комплекс имеет состав AgR2HR“. При каком значении pH степень извлечения Ag (I) составит не менее 90%? Реагент экстрагируется только в форме HR.
p«R = 5-109; ₽h,r* = 7,7 • 10й;
KdhR = 4,5-10*; KaKCAg = 1,26-10“«.
Ответ: 0,3%; pH 2s 8,8.
23. Свинец предполагают определить по светопоглощению оксихинолинатного комплекса в хлороформе при X. = 400 нм. Найти интервал pH, в котором 3-кратная экстракция по 10 мл 0,1 М раствором 8-оксихинолина в хлороформе обеспечивает практически полное (Е 99%) извлечение Pb (II) из 100 мл 1-10-4 М водного раствора соли свинца, содержащего 0,1 моль/л ацетата натрия. В органический слой переходит молекулярная форма реагента (HR) и хелат PbR2.
₽hr = 5-109; ₽S1R+ = 7,7- 10й; ADhr = 4,5-102; /f3KCpb = = ЫО"8.
Ответ: pH = 8,1—12.
391
24. Вычислить pHi/, и степень извлечения лантана при экстракции оксихинолината лантана из I-10-6 М водного раствора 0,01 М раствором 8-оксихинолина в хлороформе в отсутствие конкурирующих комплексантов. Отношение объемов фаз W/V = = 10. Экстрагируются незаряженные формы реагента и комплекса.
Phr = 5-109; = 7,7- 10й; 7GHR = 4,5-102; pLaRa =
= 8.1016; /CCLaR3 = 3,7-102.
Ответ: рН^, = 7,5; Е — 9,1 %.
25. Из 100 мл водного раствора, содержащего по 1-10~4 моль/л Fe (III) и Со (II), при pH = 5 проводят экстракцию 10 мл 0,1 М раствора 8-оксихинолина (HR) в хлороформе. Определить: а) степень извлечения Fe (III) и Со (II) при этих условиях; б) интервал pH, где можно было бы количественно извлекать Fe (III) при степени извлечения Со (II) не более чем 0,5%; в) интервал pH, обеспечивающий селективное извлечение Со (II) в присутствии Fe (III) двумя последовательными экстракциями по 10 мл экстрагента при использовании в качестве маскирующего реагента 0,1 М раствора пирофосфата натрия. По данным Стары [167], Fe (III) и Со (II) экстрагируются в виде хелатных соединений FeR3 и CoR2(HR)2, а реагент — только в молекулярной форме HR.
7<ЭКСре = 5- IO3; tfDFeR3 = 5,7 - IO3; pFeR2+ = 5-1012; ₽FeR+ = 4 -1025; fW = 1- 1037.
KSKCCo = 1,7. IO"3; AdCoRi (HR)j = 5.102; pCoR+ = 1,2. IO9; PcoR, = 1,6-101’.
= 4,5. IO2; |$R = 5-109; рП2₽+ = 7,7-1014.
Fe (III) с пирофосфат-ионом образует комплекс состава Fe (HPA)!’; Pre (нр2о7)Г = I>6.1022.
Для H4P2O7 — рн = 3,2- 10s; p2H = 4- 10м; p3H = 1,2-10”; P4H = 1,2-1018.
Ответ: а) при pH = 5; EFe — 99,8%; £co = 58,5%; б) при pH = 24-3; £Fe = 99,8%; £co < 0,5%; в) при pH — = 4,54-5,5; £CO^99%; EFe < 1% (0,1 M Na4P2O7).
ПРИЛОЖЕНИЯ
Соотношение между величинами светопропускания и оптической плотности
Пропуска-ние, % Пропускание, %
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0,7 0,8 0.9
Оптическая плотность
0 3,000 2,699 2,523 2,398 2,301 2,222 2,155 2,097 2,046
1 2,000 1,959 1,921 1,886 1,854 1,824 1,796 1,770 1,745 1,721
2 1,699 1,678 1,658 1,638 1,620 1,602 1,585 1,569 1,553 1,538
3 1,523 1,509 1,495 1,481 1,469 1,456 1,444 1,432 1,420 1,409
4 1,398 1,387 1,377 1,367 1,357 1,347 1,337 1,328 1,319 1,310
5 1,301 1,292 1,284 1,276 1,268 1,260 1,252 1,244 1,237 1,229
6 1,222 1,215 1,208 1,201 1,194 1,187 1,180 1,174 1,167 1,161
7 1,155 1,149 1,143 1,137 1,131 1,125 1,119 1,114 1,108 1,102
8 1,097 1,092 1,086 1,081 1,076 1,071 1,066 1,060 1,056 1,051
9 1,046 1,041 1,036 1,032 1,027 1,022 1,018 1,013 1,009 1,004
10 1,000 0,9957 0,9914 0,9872 0,9830 0,9788 0,9747 0,9706 0,9666 0,9626
11 0,9586 0,9547 0,9508 0,9469 0,9431 0,9393 0,9355 0,9318 0,9281 0,9245
12 0,9208 0,9172 0,9136 0,9101 0,9066 0,9031 0,8996 0,8962 0,8928 0,8894
13 0,8861 0,8827 0,8794 0,8761 0,8729 0,8697 0,8665 0,8633 0,8601 0,8570
14 0,8539 0,8508 0,8477 0,8447 0,8416 0,8386 0,8356 0,8327 0,8297 0,8268
15 0,8239 0,8210 0,8182 0,8153 0,8125 0,8097 0,8069 0,8041 0,8013, 0,7986
16 0,7959 0,7932 0,7905 0,7878 0,7852 0,7825 0,7799 0,7773 0,7747 0,7721
17 0,7696 0,7670 0,7645 0,7620 0,7595 0,7570 0,7545 0,7520 0,7496 0,7471
18 0,7447 0,7423 0,7399 0,7375 0,7352 0,7328 0,7305 0,7282 0,7258 0,7235
19 0,7212 0,7190 0,7167 0,7144 0,7122 0,7100 0,7077 0,7055 0,7033 0,7011
20 0,6990 0,6968 0,6946 0,6925 0,6904 0,6882 0,6861 0,6840 0,6819 0,6799
21 0,6778 0,6757 0,6737 0,6716 0,6696 0,6676 0,6655 0,6635 0,6615 0,6596
22 0,6576 0,6556 0,6536 0,6517 0,6498 0,6478 0,6459 0,6440 0,6421 0,6402
23 0,6383 0,6364 0,6345 0,6326 0,6308 0,6289 0,6271 0,6253 0,6234 0,6216
24 0,6198 0,6180 0,6162 0,6144 0,6126 0,6108 0,6091 0,6073 0,6055 0,6038
25 0,6021 0,6003 0,5986 0,5969 0,5952 0,5935 0,5918 0,5901 0,5884 0,5867
26 0,5850 0,5834 0,5817 0,5800 0,5784 0,5768 0,5751 0,5735 0,5719 0,5702
27 0,5686 0,5670 0,5654 0,5638 0,5622 0,5607 0,5591 0,5575 0,5560 0,5544
28 0,5528 0,5513 0,5498 0,5482 0,5467 0,5452 0,5436 0,5421 0,5406 0,5391
29 0,5376 0,5361 0,5346 0,5331 0,5317 0,5302 0,5287 0,5272 0,5258 0,5243
30 0,5229 0,5214 0,5200 0,5186 0,5171 0,5157 0,5143 0,5129 0,5114 0,5100
31 0,5086 0,5072 0,5058 0,5045 1,5031 0,5017 0,5003 0,4989 0,4976 0,4962
32 0,4949 0,4935 0,4921 0,4908 0,4895 0,4881 0,4868 0,4855 0,4841 0,4828
33 0,4815 0,4802 0,4789 0,4776 0,4763 0,4750 0,4737 0,4724 0,4711 0,4698
34 0,4685 0,4672 0,4660 0,4647 0,4634 0,4622 0,4609 0,4597 0,4584 0,4572
35 0,4559 0,4547 0,4535 0,4522 0,4510 0,4498 0,4486 0,4473 0,4461 0,4449
36 0,4437 0,4425 0,4413 0,4401 0,4389 0,4377 0,4365 0,4353 0,4342 0,4330
37 0,4318 0,4306 0,4295 0,4283 0,4271 0,4260| 0,4248 0,4237 0,4225 0,4214
38 0,4202 0,4191 0,4179 0,4168 0,4157 0,4145 0,4134 0,4123 0,4112 0,4101
39 0,4089 0,4078 0,4067 0,4056 0,4045 0,4034 0,4023 0,4012 0,4001 0,3989
40 0,3979 0,3969 0,i п58 0,3947 0,3936 0,3925 0,3915 0,3904 0,3893 0,3883
41 0,3872 0,3862 0,3851 0,3840 0,3830 0,3820 0,3809 0,3799 0,3788 0,3778
42 0,3768 0,3757 0,3747 0,3737 0,3726 0,3716 0,3706 0,3696 0,3686 0,3675
43 0,3665 0,3655 0,3645 0,3635 0,3625 0,3615 0,3605 0,3595 0,3585 0,3575
44 0,3565 0,3556 0,3546 0,3536 0,3526 0,3516 0,3507 0,3497 0,3487 0,3478
45 0,3468 0,3458 0,3449 0,3439 0,3429 0,3420 0,3410 0,3401 0,3391 0,3382
46 0,3372 0,3363 0,3354 0,3344 0,3335 0,3325 0,3316 0,3307 0,3298 0,3288
47 0,3279 0,3270 0,3261 0,3251 0,3242 0,3233 0,3224 0,3215 0,3206 0,3197
48 0,3188 0,3179 0,3170 0,3161 0,3152 0,3143 0,3134 0,3125 0,3116 0,3107
393
%
ФФфСОЮфф1О<ОФОЗООООСрОогаООООООООЧ^1^]~4^1м^'Ч-ЧМОТра>ОТОС»а)05С1®СЛСЛСЛСЛу1рСЛрСЛО1Ф;
ФООЧОСЛ^СЙКЗ — ОФОО'ЧС»СЛ.₽>-С>ЭЮ — 0'OC04CW*"MW - ОФОО'ЧОТС1*-СлЖ> — ОФ05ЧС5б1Ь-СОЮ — ОФ
Пропуска" вне, %
oooooooooooooooooooo— — oo — —K>K>ww1bAuienfflfflS'4a><»to<ooo ^оэмчкза-а-ою&оспоспот'-тюч *03ИЧСй<00Ч0О050>иЧЛСЛ0>ЧС0К>01ф1МО
ЮЫЫМИИИИИЫКМЙЙЙ о о — имлАслстачдаооо оч*-аз<лд-®чсл£Ь?;;£
ооо о оо о ооо оорр ороороооооооо оооооооооооооооо 0ppppop О о о о о о о р О О О О О О О О О О О О — '— ’— “— "— — "— — "— ’— “— '— “— “— "— — ЬЭ Ю "to "to "to ЬЭ “to ЬЭ КЗ "ю to к>ю й и од- - totocoGo.fi^cncnoOiJ'-JOTootoooo — — - к>юы*кспсло-ч|0офдр о> 00 to -j — о> — слослослоспослосл — о, — ч к a i-a о ми t*o ама гпиоа А- оори — мр^сохрсо «осооосооэи^ — *ч сл со — ооо — м>1>^о^даыоа*м^- — — со сл <о со оо & — оюо — джлахсо'оюаио 1*0
ррроооооорро оррр ооооооооооооооооооосооооооооорррорр О О О О О О О О О О Q О О О ООО О О О — "— — — — — — "— — — — — — — WMtOW to Ь? t-sto to to со ор ——ююшсол^.^рслоо-ч'чсоффоо — — юк5со4»*ьелслр'4-^с»ффо — ююсо^слдомроФФО w ч w о — сл о сл о л о о о * 'О сл а сп - от to а> со ф сл — »»]союотюфслк5а5отсоо'^01мооод?^ьэд ф 00 СЛФСОСИ^ФОТСООСООТСЛСЛСЛОТ'ЧсО — ЛьСПСйОО^ьОСвОТСЛСЛСЛ^ФЬООТ—&CnMtOtO*»«4—ОТСО — О — СОМСМО г'а
pppooooppppoppopoppoooooooooooooooooooooooooppppppo О ООО О О О О О О О О О О О О О О О О “ф — “— "— — — — "— — — — "— "— — “— — “— "to to "to "to to "to "to "to м "to “to "to to co 00 — — ккс:ис1АЛС1спос1ччааооа - — до-г.ляччйо'ОО- — M'j-fr*wci4®aoq US- aotno ДО^'ОЛ'Р^Ой'ОЛОСПОа - JCOoS-OOTCOOOTtOOOOT — co СЛ to Ф ОТ *» — Ф $Л CO — ~?4 — Лф^соа— oo сл co — ооо — №л.отфс*з"Чьооослюофффосоотосл — ooocnco^jocooocncotoocno — to 0.3
pppppppppppppppppppppOOOOOOOOOOOOOOOOOOO0 0000pppop0 OOOOOO OOOOQOOO OOOOOO X- X- X-X- X- X- X- X- X- X- X- X- X- X- X- X- X- ND ND ND ND ND ND ND ND ND ND ND ND ЬЭ C0 00*-*-NDNDNDCpWJ^^CnCHOO>J'*«JOOOOOOO*-’—NDNDCo^^CnWO,4,*JOot©00*-*-NDC04*4>-CnO^JQOOOpO NDS>-U10CrO>MJDU03WOOW03WtX>N(CtNOUl“CDN)a^O^N7COtN“-1s]^i-4jJ».t--0(7)W-OOCD^WOg'^^ OTO^tOtnOSW- CONCnUlCnUl^a-tNsJtOslWUDaS-CoMCO^OOCAJCOtN-^COCOONDC} - ОСЛ‘^СЛ‘ЧООСлЭ p
ppоopоооооOppopopop оо ооооооооооооооооооооооооррооор о "о о о "о "о о "о о о о о о о о о о о "о о "о"—"——’— — — — — '— "— ’— ”— "— "— "— "to "to "to "to "to "to "to "to "to "to "to to to CO po — —tototoMM^-jg-cncxippsaJOogootpo — — юьэсосо^слслга'ЧмооФФО — — юсоф">(ьслр'4^}оосос5 to <2 — счоАФм'вмаыиысоисоыспдйоюаюч'ошсл- а>*очсооч5 - oocntoooocnco — ® й o> юоосластмфа^м-000 —wcnootocn—4С0 —э:чччсоос:чм<клгЗ“ — исласоочаасомч^ 0.5
ppp о oppppppp o-^ppoppooooooooooooooooooooooooooopppp^3 0 0 "o 0 0 "p p 0 "p "p "p О О 2 "0 "0 "0 "0 "0 Q 0 '— "— — — "— — — — — — — "— "— X_ „ _ to "to "to "to "to “to "to “to "to to "to "to to CO OO — — — 10юрсо&^>.спу}^р'0~000 00фф0 — — СОГОСОСООХСЛСЛОЧ-ЧОЭООФО — — tOCOCO^CnmiJ^JOOCDg — аосл’о^оом'хмч^чтчачойщшдоо’. - чссосл- чиоаЙЕойслОч^юФЧл^ой^ЯЙ; М — 050СЛ — CO*OC5CnUCnOC»OC*0,*J— О — ООСЛСО — — — tOtS^q — Cl—OOUll^^UlOO— СЗЮФООООО^ООСП 9*0
ppppppppppppppppoppoooooooooooooooooooooooooopppppo p "о "о О О О 'р О "р "р "р “о О "о О "о “о "о "о "о О “— “— “— "— "— — — — — — „ “to “to “to "to "to to "to "to “to “to "to “to to CO T1 — КЙЙЙЙЙЙЧЙ°!^^й°;,“:,£9'ЗгйК1йМ’й'спс'’®“''0’®1ооо - юащ^спочч j-co r;p'C5£t-OCOpCtO^JtO'JtO'JtC'JtO<tO^lC>OCO^COOnOClMCO^OOCOr0OntoCOOntO®Cl-> — ooct^toocomcnco W**J — СЭ — 4CO00->tO— ООО — СОСЛ0О — СЛООГОФ'ЧСЯСЛСЛСПООООхфкеС — un^j^qoco->oco>- — ootocnocn 0.7
poppppppppppp000ppp opoooooooooooooooooooooooooooooo 0'0 00'0 p“p p p p p"o“p“o“o"o o"p'p“o"p“—“to "to "to “to "to “to to "to “to "to “to “to "to w 252T^K^H4'ftistu’Silo2'!iSfflH£o'2“!5totJWq'Cncn004tot»fflOO-‘jwc:^cn00 4i0o рСЛФ^-OOCO^JtO-J — P“P7'PxptO-4gCOCOO^.OOT — ЧЫООЮИСЛ - СОСЛПОФОСООООСЛСО — ф -О СП to Oto^l — ОПОСХСЛЮФ'ЧОСЛСЛСЛООООКЭООСЛООСО - ФФФО — ifiCOtOCoJttoOO — COO — 4^tOIO»|t 4 — ОО 8*0
ООО 000 OOOOOO 00 000 Оо ООО ООООООООООООООООООООООООООООО ооооооооооооооооооооо"—• Х-Х— X- >— — X- "X- X- X- X- X— X- Хл no ьэ м Хл Хэ кэ ьо ьо ьо Ъз •NOJMNltO'JWOsl^ND'-OOO—ND^S>“Cn'X)Cnt-00^>NC0C0^Cna)'-,CD‘-C0WWljjXcD<3M<OCD>^S‘CnCCW!r> 6*0
Продолжение
Логарифмы констант устойчивости комплексонатов некоторых металлов [42] (7 = 0,1; t = 20 °C)
Ион металла ДЦТА ДТП A ЭДТА
jrH amhl Kml vOH MOHL MHL *ML THOWV HO* SS * лмнь 1 d X IO oS *
Ag* 8,7 6,0 7,3 1,9
А1’+ 2,0 17,6 6,4 18,5 2,6 16,1 8,1
Ва2+ 6,7 8,0 5,3 8,8 4,6 7,8 0,3
Ве2+ 8,3 10,8 5,2
В1а+ 31,2 2,8 35,4 2.9 1,4 28,2 3,5
Са2+ 12,5 6,4 10,6 2,0 3,1 10,7 0,6
Cd2* 3,0 19,2 3,9 19,0 3,0 2,9 16,5 2,9
Сс’+ 16,8 20,5 3,0 18,4
Со«+ 2,9 18,9 4,8 19,0 3,5 3,2 16,3 3,1
Со’* 1,3 36,0 6,6
Сг’+ 2,3 23,0 6,6
Си2* 3,1 21,3 5,0 20,5 5,5 3,0 18,8 2,5
Dya+ 19,7 2,2 22,8 2,8 18,3
Ега+ 20,7 2,0 22,7 2,7 19,0
Еиа+ 18,6 2,1 22,4 2,6 17,4
Ее2* 18,2 5,4 16,0 5,0 3,0 2,8 14,3 4,9
Fe»* 29,3 4,7 3,4 27,5 4,1 1,4 25,1 6,5
Gaa+ 22,9 4,4 25,5 2,1 1.7 20,3 8,9
Gda+ 18,8 2,3 22,5 2,6 17,4
Hg2* 3,1 24,3 3,5 3,6 27,0 3,1 21,8 4,9
Hoa* 2,2 22,8 2,7 18,7
Ina+ 1,6 25,0 5,0
Laa+ 2,6 16,3 3,0 19,1 2,0 15,5 2,2
Lua+ 21,5 2,2 22,4 2,5 19,8
Mg2* 10,3 6,9 9,3 3,9 8,7 2,1
Mn2+ 2,8 16,8 4,5 15,5 2,1 3,1 14,0 2,9
Nd3* 17,7 2,4 21,6 4,4 16,6 1,6
Ni2+ 19,4 5,6 20,0 5,4 3,2 18,6 3,3
Pb2* 2,8 19,7 4,5 18,9 3,4 2,9 18,0 4,7
Pr3* 17,3 2,4 21,1 16,4 2,2
Sc3* 2,0 23,1 3,5
Sma+ 18,4 2,2 22,3 2,6 17,1 2,4
Sn2+ 22,1
Sr2* 10,0 5,4 9.7 4,0 8,6
Tba+ 19,5 2,2 22,7 2,6 17,9
Ti3* 21,3
TiO2* 17,3
Tla+ 21,0 460 2,3 22,5 10,4
Tma+ 1,9 22,7 2,6 19,3
Th1* 23,2 6,4 2,1 28,8 5,1 2,0 23,2 7,0
V2* 12,7
v«+ 25,9
VO2* 19,4 18,8
vot 3,6 18,1
Yba+ 21,1 2,3 22,6 2,7 19,5
Y3* 19,2 18,1
UOf* 17,7
Zn2* 3,0 18,7 5,6 18,0 4,4 4,4 16,5 3,0
Zr1* 36,9 29,9 7,8
Н . [MHL] . он [MOHL] . м [M,L]
MHL [ML] [Н*] ‘ MOHL [ML] [ОН~] ' AM,L~ [ML] [М]
Логарифмы условных констант устойчивости комплексонатов некоторых металлов
oH
иБталла a 1 2 3 « 5 6
Ag+ 0,7 1.7 2,8
А13+ 3,0 (2,9) 5.4 (5.4) 7,5 (7.4) 9,6 (7,2) 10,4 (6,6)
Ва2* 1,3 3,0
Be2* 3,3 (3,3) 5,3 (5,3) 6,5 (6,5) 7,4 (7,1) 7,2 (6,9)
Bi3* 5,7 (5,7) 10,2 13,6 14,9 15,1 15,1 14,9
(10,2) (12,7) (13,6) (13,6) (13,4) (13,0)
Са2+ 2,2 4,1 5,9
Cd3* 0,1 3,7 6,0 7,9 9,9 11,7
Сея* 0,4 4,9 7,8 10,0 11,9 13,7
Со3* 0,1 3,6 5,9 7,8 9,7 11,5
Сг3+ 6,0 9,5 12,2 14,0 15,1 15,6
Си2* 2,5 (2,5) 6,0 (6,0) 8,3 (8,3) 10,2 12,2 14,0
(10,2) (12,2) (14.0)
Fe2* 1,4 3,7 5,7 7,7 9,5
Fe3* 2,5 (2,5) 7,2 (7,2) 11,3 13,9 14,7 14,8 14,6
(11,3) (13,8) (14.6) (14.8) (14,6)
Ga3+ 3,1 6,9 9,7 10,1 10,3 10,8
Hg2* 0,9 5,6 9,1 11.1 11,3 11,3 H.l
In3* 7,1 11,4 14,3 16,1 16,5 16,4
La3* 2,1 4,9 7,0 8,9 10,7
Mg2* 2,1 3,9
Mn2+ 1,3 3,6 5,5 7,4 9,2
Nd3* 3,1 5,5 7,6 9,5 11,3
Ni2* 2,5 6,0 8,2 10,1 12,0 13,8
Pb2* 1,6 (1,6) 5,2 (5,2) 7,4 (7,4) 9,4 (9,4) 11,4 13,0
(П.4) (13,0)
Pr3* 2,7 5,6 7,8 9,7 11,5
Sc3* (5.D (9.6) (12,5) (14,6) (15,5) (15,3)
Sm3+ 3,5 5,8 7,9 9,8 11,6
Sr2* 2,0 3,8
Th4* 6,7 (6,7) 10,3 12,6 14,5 15,8 15,5
(10.3) (12,6) (14.3) (15,2) (15.4)
TiO2* 3,6 5,0 6,2 7,2 8,0
Tl3+ 5,5 8,0 8,7 8,8 10,0 10,7
Zn2* 1,6 5,1 7,1 8,3 10,0 11,7
Zr4+ 8,2 10,9 11,1 10,1 8,3 6,3 4,1
lg“Y«- 24,0 18,3 13,8 10,8 8,6 6,6 4,8
lg aHY»- 13,6 9,0 5,5 3,5 2,3 1,3 0,5
* Для А1. В1, Сп, Fe (III), Pb, Sc, Th условные константы устойчивости рассчитаны в скобках указаны значения условных констант, рассчитанные с учетом образования поли условных констант устойчивости длн редкоземельных элементов приведены по данным ра
396
с ЭДТА в отсутствие посторонних комплексантов (7 = 0,1, t = 20—26 °C)
pH
7 9 9 ID П 12 13 14
3,9 5,0 5,9 6,8 7,1 6,8 5,0 2,2
8.5 (6,5) 6,6 (6,1) 4,5 (4,5) 2,4 (2,4)
4,4 5,5 6,4 7,3 7,7 7,8 7,7 7,3
5,7 (5,7) 3,9 (3,9) 2,9 (2,9) 2,5 (2,5)
14,3 13,4 12,3 11,3 10,2 9,2 (5,9) 8,2 (4,7) 7,2 (3,5)
(12.1) (П.О) (9,71 (8,5) (7.1)
7,3 8,4 9,3 10,2 10,6 10,7 10,4 9,7
13,1 14,2 15,0 15,5 14,4 12,0 8,4 4,5
15,1 16,1 17,0 17,1 16,7 13,2 10,3 7,3
12,9 13,9 14,5 14,7 14,0 12,1
15,3 15,0 14,9 14,8 14,2 13,3 12,3 11,3
15,3 15,2 14,2 13,1 11,5 10,1 8,7 (8,7) 6,5 (6,5)
(15,3) (15,0) (14.2) (13.1) (П.5) (Ю.1)
10,9 12,0 12,8 13,2 12,7 11,8 10,8 9,8
14,1 13,7 13,6 14,0 14,3 14,4 14,4 14,4
(14,1) (13,7) (13,6) (14.0) (14.3) (14,4) (14,3) (13,8)
11,1 10,3 8,5 6,3 3,0
10,5 9,6 8,8 8,4 7,7 6,8 5,8 4,8
15,8 14,9 13,9 12,7 10,8 8,1 5,2 2,2
12,1 12,6 13,3 12,6 10,9 8,2 4,9 1,9
5,3 6,4 7,3 8,2 8,5 8,2 7,4
10,6 П.7 12,6 13,4 13,4 12,6 11,6 10,6
12,7 13,7 14,4 14,2 12,6 10,3 7,1 4,1
15,2 16,3 17,1 17,4 16,9
13,8 14,0 13,8 13,6 13,0 11,9 10,2 7,9 (7,9)
(13,8) (14,0) (13,8) (13,6) (13,0) (11,9) (Ю,2)
12,9 13,8 14,3 13,6 11,8 9,4 6,0 3,0
(14,7) (13,5) (12,0) (10,0) (7,9) (5,8) (3,8) (1,8)
13,0 14,0 14,6 14,3 12,7 10,5 7,0 4,0
5,2 6,3 7,2 8,1 8,5 8,6 8,5 8,0
13,6 10,5 9,3 (9,3) 7,2 (7,2) 4,7 (4,7) 1,7 (1,7)
(13,6) (10,5)
8,4 8,5 8,4 8,3 7,7 6,8 5,8 4,8
10,5 9,6 8,6 7,5 5,8 3,9 1,9
13,1 14,2 14,9 13,6 11,3 9,0 6,7 4,0
1,5
3,4 2,3 1.4 0,5 0,1
0,1 0 0,1 0,2 0,8 1,7 2,7 3,7
по данным 1421 для условий Доминирования гидроксндных моноядерных комплексоа; ядерных гидроксокомплексов, прн общих концентрациях металлов 0,001 моль/л. Значения боты 1425].
397
Значения lg 1/вм для некоторых металлов с различными комплексантами (42 j
Концен- pH
Металл Комплексавт трация.
моль/л 0 1 I 2 3 «
Ag Гидроксид
Цианид 0,1 0,8 -2,7 4,7 6,7 8,7
Сульфид 0,01 4,4 5,4 6,4 7,4 8,5
Тиомочевина 0,1 10,1 10,1 10,1 10,1 Ю.1
Аммиак 1,0
0,1
0,01
Трнен 0,1
ЭДТА 0,1 0,1
0,01
Ен 0,1
Ден 0,1
Глицин
Хлорид 0,1 2,8 2,8 2,8 2,8 2,8
Al Гидроксид
Цитрат 0,1 1,8 5,2
Ацетилацетонат 0,01 0,1 0,6 2,2
ЭДТА 0,1 1,8 4,1 6,2
0,01 1,5 3,4 5,5
Фторид 0,1 3,3 6,1 10,0 12,9 14,3
ДЦТА 0,01 0,2 2,8 5,5
Оксалат 0,1 2,4 5,6 8,5 Ю.7
Ва Гидроксид
ДТПА 0,01
ДЦТА 0,01
ЭДТА 0,1
0,01
НТА 0,1
0,01
Цитрат 0,1
Тартрат 0,1 0,1 0,4
Be Гидроксид
Сульфосалицилат 0,1 0,5 2,1 3,7
Ацетилацетонат 0,1 0,1 0,8 2,4
ЭДТА 0,1 0,1
0,01
Цитрат 0,1 0,2 1.1
Bi Гидроксид 0,1 0,5 1.4 2,4
Иодид 0,1 12,8 12,8 12,8 12.8 12,8
Бррмид 0,1 4,5 4,5 4,5 4,5 4,5
Хлорид 0,1 2,7 2,7 2,7 2.7 2,9
Са Гидроксид
ДЦТА 0,01 0,5
ЭДТА 0,1 0,1 1,1
0,01 0,4
ДТПА 0,01 0,1
НТА 0,1 0,1
0,01
Цитрат 0,1 0,3 1,0
Тартрат 0,1 0,2 0,5
Ацетат 0,1
Cd Гидроксид
ДЦТА 0,01 0,1 2,2 4,8 7,1
ДТПА 0,01 0,4 3,1 5,5
398
pH
6 6 1 7 8 1 9 10 11 12 13 1 14
16,7 0,1 0,5 2,3 5.1
10,7 12,7 14,7 18,4 19,0 19,2 19,2 19,2 19,2
9,9 11,8 13,2 13,7 13,7 13,7 13,8 14,2 14,7 14,8
10,1 10,1 10,1 10,1 10,1 10,1 10,1 10,1 10,1 10,1
0,1 0,8 2,6 4.6 6,4 7,2 7,4 7,4 7,4 7,4
0,1 0,8 2,6 4,4 5,2 5,4 5,4 5,4 5,6
0,1 0,8 2,4 3,2 3,4 3,4 3,4 5,1
1,2 3,4 5,2 6,4 6,7 6,7 6,7 6,7
0,5 1,5 2,6 3,7 4,7 5,5 5,9 6,0 6,0 6,0
0,1 1,9 3,0 4,0 4,8 5,2 5,3 5,3 5,5
0,4 1,7 3,5 4,9 5,5 5,7 5,7 5,8
0,3 • 1,8 3,6 4,7 5,1 5,1 5,1 5,4
0,2 1,5 3,4 4,4 4,8 4,8 4,8 5,3
2,8 2,8 2,8 2,8 2,8 2,8 2,8 2,8 2,9 5,1
0,4 1,3 5,3 9,3 13,3 17,3 21,3 25,3 29,3 33,3
8,6 11,3 13,6 15,6 17,6 19,6 21,8 25,3 29,3 33,3
4,3 6,8 9,8 12,5 14,6 17,3 21,3 25,3 29,3 33,3
8,2 10,3 12,5 14,5 16,5 18,3 21,3 25,3 29,3 33,3
7,6 .9.7 11,8 13,9 15,8 17,8 21,3 25,3 29,3 33,3
14,5 14,5 14,5 14,5 14,5 17,7 21,3 25,3 29,3 33,3
7,6 9,4 10,8 12,3 14,3 17,3 21,3 25,3 29,3 33,3
H.5 H.6 11,6 11,6 13,3 17,3 21,3 25,3 29,3 33,3
0,1 0,5
0,3 1,6 3,5 5,1 6,1 6,7 6,8 6,8 6,8
0,2 0,7 1,3 2,2 3,2 4,2 5,1 5,8 6,0 6,0
1,8 3,2 4,2 5,2 6,0 6,2 6,3 6,3 6,3
0,1 1,1 2.4 3,5 4,4 5,3 5,7 5,8 5,8 5,8
0,2 0,8 1,7 2,6 3,3 3,4 3,4 3,4 3,4
0,3 1,0 1,9 2,6 2,8 2,8 2,8 2,8
0,5 1,1 1,4 1,4 1,4 1,4 1,4 1,4 1,4 1,4
0,6 0,6 0,6 0,6 0,6 0,6 0,6 0,6 0,6 0,8
0,1 1,2 3,1
5,6 7,6 9,6 11,6 13,6 15,6 17,4 18,6 18,8 18,8
4,3 6,3 8,3 10,1 11,5 11,9 11,9 11,9 11,9 11,9
1,6 3,3 4,7 5,7 6,7 7,5 7,7 7,8 7,8 7,8
0,8 2,5 3,9 5,0 5,9 6,8 7,2 7,3 7,3 7,3
2,3 3,0 3,3 3,3 3,3 3,3 3,3 3,3 3,3 3,5
3,4 4,4 5,4
12,8 12,8 12,8
4,5 4,8 5,5
3,5 4,4 5,4 0,3 1,0
2,5 4,3 5,6 6,7 7,7 8,7 9,6 10,3 10,5 10,5
3,2 4,7 6,1 7,1 6,4 8,1 8,9 9,1 9,2 9,2 9,2
2,1 3,9 5,3 7,3 8,2 8,6 9,7 8,7 8,7
0,8 1,9 3,4 5,3 6,9 7,9 8,5 8,6 8,6 8,6
0,5 1.3 2,3 3,3 4,2 4,9 5,0 5,0 5,0 5,0
0,2 0,7 1,6 2,6 3,5 4,2 4,4 4,4 4,4 4,4
1,8 2,2 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5
0,7 0.8 0,8 0,8 6,8 0,8 0,8 0,8 0,9 1,2
0,1 0.1 0,1 0,1 0,1 0,1 0,1 0,1 0,3 1,0
0,1 0,5 2,0 4,5 8,1 12,0
9,2 11,0 12,3 13,4 14,4 15,4 16,3 17,0 17,2 17,2
7,6 9.7 11,7 13,6 15,2 16,3 16,9 17,0 17,0 17,0
399
Продолжение
Металл Комплексен? Концентрация, моль/л pH
0 * 2 3 ! «
Cd Цианид 0,1 2,7 4,8 0,1
ЭДТА 0,1 0,3 6,8
0,01 1,8 4,0 5,9
ГЭДТА 0,01 0,1 1,5 3,3
НТА 0,1 0,4 1,9 3,0
0,01 1,2 2,3
Фенантролин 0,01 0,3 1,4 3,5 6,3
Дея 0,1
Триен 0,1 0,5 1,8 4,1
Пиколинат 0,1
Аммиак 1,0
0,1
0,01
Цитрат 0,1 0,1 0,8
Ацетил ацегонат 0,1
ТЭА 0,25
Иодид 0,1 2,5 2,5 2,5 2,5 2,5
Оксалат 0,1 0,2 1,8 2,2
Тартрат 0,1 0,6 1,4
Ацетат 0,1 5,1 0,1
Се (IV) Гидроксид 0,1 0.1 1.2 3,1 7,1
Сульфат 2,5 4,8 6,8 7,4 7,6
Со (II) Гидроксид Фенантролин 0,01 0,4 2,1 4,8 7,8 10,8
ДТПА 0,01 1.0 3,8 6,0
ДЦТА 0,01 1,8 4,4 6.8
ЭДТА 0,1 0,2 2,6 4,7 6,6
0,01 1.7 3,5 5,7
Тетрен 0,1
НТА 0,1 0,8 2,4 3,5
0,01 0,3 1,7 2,8
Ден 0,1
Пиколинат 0,1 0,6 2,0 4,3 7,2
ГЭДТА 0,01 0,3
Триеи 0,1 1.5
Цитрат 0,1 0,5
Сульфосалицилат 0,1
Ацетилацетонат 0,1
Аммиак 1.0
0,1
Оксалат 0,1 0,1 0,5 1,0 2,0 3,2
Тартрат 0,1 0,2 0,8
Си Гидроксид Тетрен 0,1 2,8 6,3 9.4
ЭДТА 0,1 1,9 4,6 6,8 8,7
0,01 1,5 4,2 6,3 8,2
ДЦТА 0,01 1,5 4,4 6,9 9,2
Триен 0,1 0,1 2,6 5,4
Ден 0,1 0,5 3,2
ДТПА 0,01 2,8 5,7 7,8
Фенантролин 0,01 2,1 3,5 5,9 8,8 11,8
Трен ТЭА 0,1 1,1
0,1 0,1
ГЭДТА 0,01 0,1 1,8 3,7 5,1
Сульфосалицилат 0,1 0,2 1,0
400
pH
"1 * 1 6 1 7 1 8 1 9 1 10 1 11 1 12 1 13 14
— 0,7 2,9 6,2 10,1 13,3 14,5 14,9 14,9 14,9 14,9
8,Я 10,5 11,9 12,9 13,9 14,7 14,9 15,0 15,0 15,0
7,9 9,7 11,1 12,2 13,2 14,0 14,4 14,5 14,5 14,5
5,1 7,7 9,1 11,1 12,7 13,5 13,6 13,6 13,6 13,6
4,0 5,3 6,9 8,9 11,7 12,1 12,3 12.3 12.3 12,5
3,3 4,3 5,4 7,0 8,7 10,1 10,5 10,5 10,5 12,0
84 9,3 9.3 9,3 9,3 9,3 9,3 9,3 9,3 12,0
0, 1,3 3,7 5,5 9,1 11,3 11,9 11,9 11,9 12,3
0,4 2,2 4,4 6,5 8,3 9,5 9,8 9,8 9,8 12,0
6,3 7,5 7,8 7,8 7,8 7,8 7,8 7,8 8,3 12,0
0,1 0,5 2,3 5,1 6,7 7,1 7,1 8,1 12,0
0,1 0,5 2,0 1,0 3,6 4,5 8,1 12,0
0,1 0,6 1,4 2,0 4,5 8,1 12,0
2,0 2,7 3,0 3,1 3,5 4,3 5,3 6,3 8,2 12,0
0,1 0,9 2,3 3,6 4,0 4,0 4,6 8,1 12,0
1,9 2,2 2,8 3,7 4,5 8,1 12,0
2,5 2,5 2,5 2,5 2,5 2,5 2,6 4,5 8,1 12,0
2,7 2,7 2,7 2,7 2,7 2,7 2,8 4,5 8,1 12,0
1,8 1,8 1,8 1,8 1,8 1,8 2,2 4,5 8,1 12,0
0,3 0,4 0,5 0,5 0,5 0,7 2,0 4,5 8,1 12,0
9,1 11,1 13,1
9,1 11,1 13,1
0,1 0,4 1,1 2,2 4,2 7,2 10/6
12,9 13,8 13,8 13,8 13,8 13,8 13,8 13,8 13,8 13,8
7,8 9,7 11,7 13,6 15,2 16,3 16,9 17,0 17,0 17,0
8,9 10,7 12,0 13,1 14,1 15,1 16,0 16,7 16,9 16,9
8,6 10,3 11,7 12,7 13,7 14,5 14,7 14,8 14,8 14,8
7,7 9,5 10,9 12,0 13,0 13,8 14,2 14,3 14,3 14,3
1.3 4,6 7,6 10,4 12,7 13,7 14,1 14,1 14,1 14,1
4,5 5,6 7,1 9,0 10,8 12,2 12,4 12,4 12,4 12,4
3,8 4,8 5,9 7,2 8,8 10,2 10,6 10,6 10,6 10,8
0,2 2,1 5,7 9,3 11,5 12,1 12,1 12,1 12,1
9,6 10,8 11,1 11,1 11,1 11,1 11,1 11,1 11,1 11,1
1,9 3,9 5,9 7,9 9,5 10,2 10,3 10,3 10,3 10,6
0,3 2,1 4,5 6,7 8,5 9,7 10,0 10,0 10,0 10,4
2,5 3,1 3,4 3,8 4,5 5,5 6,5 7,5 8,5 10,3
0,1 0,5 1,5 2,8 4,6 6,4 7,6 7,9 10,2
0,4 1,6 3,3 5,1 6,5 6,9 6,9 6,9 7,4 10,2
0,2 1,2 3,7 5,3 5,7 5,8 7,2 10,2
0,2 1,0 1,8 2,9 4,9 7,2 10,2
3,8 3,8 3,8 3,8 3,8 3,8 3,8 4,3 7,2 10,2
1.1 1,1 1,1 1,1 1,1 1,1 2,2 4,2 7,2 10,2
0,2 0,8 1,7 2,7 3,7 4,7 5,7
12,0 14,8 17,6 20,3 22,3 23,3 23,3 23,3 23,3 23,3
10,7 12,5 13,9 15,0 16,0 16 8 17,3 18,0 18,9 19,9
10,2 12,0 13,4 14,5 15,5 16,3 16,8 17,5 18,4 19,4
11,3 13,1 14,4 15,5 16,5 17,5 18,4 19,1 19,3 19,3
8,4 11,3 13,9 16,1 17,9 19,1 19,4 19,4 19,4 19,4
5,7 7,8 9,8 12,9 16,5 18,7 19,3 19,3 19,3 19,3
9,5 11,2 13,2 15,1 16,7 17,8 18,4 18,5 18,5 18,5
13,9 14,8 14,8 14,8 14,8 14,8 14,8 14,8 14,8 11,8
4,1 7,1 10,1 13,0 15,6 17,1 17,7 17,8 17,8 17,8
0,7 1,9 3,7 6,0 8,1 10,1 12,3 15,0 17,9 20,9
6,5 8,5 10,5 12,5 14,1 14,9 15,0 15,0 15,0 15,0
2,0 3,4 5,3 7,3 9,3 11,3 13,1 14,3 14,5 14,5
14 M. И. Булатов и др.
401
Продолжение
Металл Комплекс ант Концентрация, моль/л pH
0 1 2 3 4
Си Цитрат 0,1 0,2 0,8 2,9
НТЛ 0,1 0,3 2,6 4,3 5,5
0,01 0,1 2,0 3,7 4,9
Пиколинат 0,1 2,4 4,4 6,4 8,4 10,4
Аммиак 1,0
0,1
0,01
Ацетилацетопат 0,01 0,3 1,2
Пирофосфат 0,1 4,9 0,1
Оксалат 0,1 0,5 1,1 3,1 6,3
Тартрат 0,1 0,1 1,0 2,5
Ацетат 0,1 0,3
Fe (11) Гидроксид ДТПА 0 01 1,6 3,7
ДЦТА 0,01 0,5 3,4 6,1
ЭДТА 0,1 0,6 2,6 4,6
0,01 0,1 1,8 3,7
Цитрат 0,1 0,7 0,5
НТА 0 1 1,7
0,01 0,3 1,0
Тетрен 0,1 5,4
Пиколинат 0,1 0,1 0,9 2,7
Ден 0,1
Трен 0 1
Трнен 0,1
Ацетилацетонат 0,01
Fe (III) Гидроксид ТЭА 0,1 0,4 1,8
ЭДТА 0,1 3,8 7,0 10,3 13,0 15,2
0 01 3,1 62 9,5 12,3 14,5
Салицилат 0,1 0,9 2,7 5,1 7,3
ДЦТА 0,01 3,2 7,2 11,1 14,5 17,2
ДТПА 0,01 0,6 2,5 6,5 10,5 13,8
Сульфосалицилат 0,1 0,1 1,2 3,2 5,8 8,0
НТЛ 0 1 0,4 3,2 5,8 8,2 10,3
0,01 0,1 2,5 5,2 7,1 8,9
Пирофосфат 0,1 0,4 6,0 10,6 13,8 16,0
Цитрат 0,1 0,3 3,0 6,4 9,5
Оксалат 0,1 2,5 6,0 9,8 12,5 14,6
Фторид 0,1 1,4 3,3 5,7 7,9 8,7
Ацетат 0,1 0,2 1,3 3,5
Роданид 0,1 2,9 2,9 2,9 2,9 2,9
Hg (II) Гидроксид 0,5 1,9
Цианид 0,1 14,3 16,3 18,3 20,3 22,3
ДЦТА 0.01 1,5 4,5 7,4 9,9 12,3
Иодпд 0.1 25,8 25,8 25,8 25,8 25,8
ЭДТА 0,1 2,4 5,4 8,1 10,2 12,1
0,01 1,5 4,5 7,2 9,4 11,2
ДТПА 0,01 0,4 4,2 7,9 10,9 13,4
Триен 0,1 0,6 3,5 6,5 9,4 11,8
Тиомочевина 0,1 22,4 22,4 22,4 22,4 22,4
ГЭДТА 0,01 1,0 3,9 6,6 8,8 10,8
Треи 0,1 2,1 5,1
Ден 0,1 0,4 3,1 6,1 9,0
Аммиак 1,0 0,9 2,7 4,7 6,7
402
pH
"1 • 1 6 1 7 8 1 9 1 10 11 1 12 13 1 14
5,1 6,7 8,0 9,0 10,0 11,0 12,0 13,0 14,0 15,0
6,5 7,5 8,8 10,5 12,3 13,9 14,1 14,3 15,1 16,1
5,9 6,8 7,9 9,1 10,6 12,0 12,6 13,4 14,4 15,4
12,0 12,8 13,0 13,0 13,0 13,0 13,0 13,0 13,0 13,0
oj2 1,2 3,6 7,1 10,6 12,2 12,7 12,7 12,7 12,7
0,2 1,2 3,6 6,7 8,2 8,6 8,6 8,6 8,6
0,2 1,2 3,3 4,5 4,9 4,9 5,1 5,8
2,8 4,7 6,7 8,5 9,9 10,3 10,3 10,3 10,3 10,3
1.1 2,8 4,3 5,9 6,8 7,0 7,0 7,0 7,0 7,0
6,9 6,9 6,9 6,9 6,9 6,9 6,9 6,9 6,9 6,9
3.1 3,2 3,2 3,2 3,2 3,2 3,3 3,8 4,7 5,7
0,9 1,1 1,1 1,1 1,3 1,8 2,7 3,7 4,7 5,7
0,1 0,6 1,5 2,5 3,5 4,5
5.2 6,8 8,7 10,7 12,6 14,3 15,9 17,0 18,0 19,0
8,2 10,0 11,3 12,4 13,4 14,4 15,3 16,0 16,2 16,2
6,6 8,3 9,7 10,7 11,7 12,5 12,7 12,8 12,8 12,8
5,7 7,5 8,9 10,0 11,0 11,8 12,2 12,3 12,3 12,3
2,6 4,2 5,5 6,5 7,5 8,5 9,5 10,5 11,5 12,5
2,7 3,7 4,7 5,7 6,6 7-4 7,9 8,8 9,8 10,8
2,0 3,0 4,0 5,0 5,9 6,7 7,3 8,1 9,1 10,1
1,0 3,9 6,7 9,0 10,0 10,4 10,4 10,4 10,4
7,8 9,0 9,3 9,3 9,3 9,3 9,3 9,3 9,3 9,3
0,3 2,3 5,6 7,8 8,4 8,4 8,4 8,4
0,4 30 5,6 7,1 7,7 7,8 7,8 7,8
1,3 3,5 5,3 6,5 6,8 6,8 6,8 6,8
0,3 1,1 2,3 3,6 4,0 4,0 4,0 4,1 4,6
3,7 5,7 7,7 9,7 11,7 13,7 15,7 17,7 19,7 21,7
24,2 28,2 32,2 36,2 40,2
17,2 18,9 20,4 22,0 24,0 26,4 28,5 30,6 32,6 34,6
16,5 18,3 19,8 21,4 23,3 25,7 28,0 30,1 32,1 34,1
9,3 11,5 14,1 17,0 20,0 23,0 26,0 29,0 31,4 32,3
19,3 21,1 22,4 23,5 24,7 26,3 28,1 29,8 31,0 32,0
16,2 18,2 20,2 22,2 23,9 25,2 26,5 27,6 28,6 29,6
10,1 12 5 15,4 18,4 21,4 24,4 27,1 28,9 29,2 29,2
12,3 14,3 16,3 18,3 20,1 21,5 21,8 22,4 23,3 24,3
10,8 12,8 14,8 16,8 18,6 20,1 20,9 21,8 22,8 23,8
18,0 19,4 20,0
12,0 13,7 15,0 16,0 17,0 18,0 19,0 20,0 21,0 22,0
15,5 15,5 15,5 15,5 15,5 15,5 15,9 17,7 19,7 21,7
8,9 8,9 8,9 9,8 11,7 13,7 15,7 17,7 19,7 21,7
5,2 6,0 7,7 9,7 11,7 13,7 15,7 17,7 19,7 21,7
3,8 5,7 7,7 9,7 11,7 13,7 15,7 17,7 19,7 21,7
3,9 5,9 7,9 9,9 11,9 13,9 15,9 17,9 19,9 21,9
2^,3 26,4 29,2 32,8 35,9 37,1 37,5 37,5 37,5 37,5
14,3 16,1 17,4 18,5 19,5 20,7 21,9 23,6 24,8 25,8
25,8 25,8 25,8 25,8 25,8 25,8 25,8 25,8 25,8 25,8
14,1 15,8 17,2 18,2 19,5 21,0 22,2 23,3 24,3 25,3
13,2 15,0 16,4 17,5 18,8 20,3 21,6 22,7 23,7 24,7
15,6 17,7 19,7 21,6 23,2 24,3 24,9 25,0 25,0 25,0
14,0 16,3 18,8 21,0 22,8 24,0 24,3 24,3 24,3 24,3
22,4 22,4 22,4 22,4 22,4 22,4 22,4 22,4 22,4 22,4
12,8 14 8 16,8 18,8 20,4 21,1 21,2 21,2 21,2 22,0
8,1 11,1 14,1 17,0 19,6 21,2 21,7 21,8 21,8 22,2
11,7 13,6 15,6 17,6 19,4 20,5 20,8 20,8 20,8 21,9
8,7 1 10,7 12,7 14,9 17,6 19,1 19,4 19,4 20,0 21,9
403
Продолжение
Металл Комллексаит Концентрация, моль/л pH
0 1 2 3 4 1
Hg (II) Аммиак 0,1 0,9 2,7 4,7
0,01 0,9 2,7
Фенантролин 0,01 5,0 7,0 9,0 11,0 13,0
Роданид 0,1 16,9 16,9 16,9 16,9 16,9
Пиколинат 0,1 3,0 5,0 7,0 9,0 11,0
In Гидроксид
La Гидроксид
НТА 0,1 0,5 2,3 4,2
0,01 0,2 1,5 2,9
ДЦТА 0,01 1,7 4,2
ЭДТА 0,1 0,8 3,5 5,7
0,01 0,2 2,6 4,8
Mg Ацетнлацетонат 0,01
Гидроксид
ДЦТА 0,01
ДТПА 0 01
ЭДТА 0,1
0,01
Глицин 0,1
НТА 0,1
0,01
Цитрат 0,1 0,2
Тартрат 0,1 0,1 0,3
Мп (II) Гидроксид
ЭДТА 0,1 0,5 2,4 4,3
0,01 0,1 1,6 3,5
Тетрен 0,1
НТА 0.1 0,1 0,5
0,01 0,2
Трен 0,1
Ацетнлацетонат 0,1
Ен 0,1
Оксалат 0,1 0,2 0,6 1,8
Цитрат 0,1 о,1 0,7
Ni Гидроксид
Цианид 0,1 2,5 6,5
Фенантролин 0,01 3,4 6,3 9,3 12,3 15,3
ДТПА 0,01 0,1 2,9 5,8 7,9
Триен 0,1 0,3
ЭДТА 0,1 0,1 2,4 5,1 7,1 9,0
0,01 1,5 4,2 6,3 8,1
Ден 0,1
Тетрен 0,1 0,3
Пиколинат 0,1 0,6 2,2 4,5 7,3 10,3
Трен 0,1
НТА 0,1 1,4 3,1 4,2
0,01 0,7 2,4 3,5
Цитрат 0,1 0,2 0,5 1,7
ГЭДТА 0,01 0,5 1,5
ТЭА 0,1
Ацетнлацетонат 0,1 °,2
Аммиак 1,0
0,1
0,01
Сульфосалнцилат 0,1
404
pH
1 5 1 e 1 7 1 8 I 9 1 10 1 11 1 12 13 1 14
6,7 8,7 10,7 12,7 14,6 15,7 16,2 17,9 19,9 21,9
4,7 6,7 8,7 10,7 12,5 14,0 15,9 17,9 19,9 21,9
14,4 15,0 15,0 15,0 15,0 15,0 15,9 17,9 19,9 21,9
16,9 16,9 16,9 16,9 16,9 16,9 16,9 17,9 19,9 21,9
12,6 13,4 13,6 13,6 13,6 14,1 15,9 17,9 19,9 21,9
0,3 1,0 2,0 3,0 4,0 5,0 6,0 7,0
0,3 1,0 1,9 2,9 3,9
6,1 8,1 10,1 12,1 13,9 15,3 15,5 15,5 15,5 15,5
4,6 6,5 8,5 10,5 12,3 13,7 14,1 14,1 14,1 14,1
6,3 8,1 9,4 10,5 11,5 12,5 13,4 14,1 14,3 14,3
7,7 9,4 10,8 11,8 12,8 13,6 13,8 13 9 13,9 13,9
6,8 8,6 10,0 11,1 12 0 12,9 13,3 13,4 13,4 13,4
0,2 1,0 2,5 4,3 4,8 4,8 4,8 4,8 4.8
0,1 0,5 1,3 2,3
0,4 2,1 3,4 4,5 5,5 6,5 7,4 8,1 8,3 8,3
0,3 1,0 2,3 4,0 5,6 6,6 7,2 7,3 7,3 7,3
0,7 2,4 3,8 4,9 5,8 6,7 7,1 7,2 7,2 7,2
0,4 1,9 3,3 4,4 5,3 6,2 6,6 6,7 6,7 6,7
0,1 1,0 2,6 3,8 4,1 4,1 4,1 4,1
0,1 0,4 1,2 2,2 3,1 3,8 4,0 4,0 4,0 4,0
0,1 0,7 1,6 2,5 3,2 3,4 3,4 3,4 3,4
0,9 1,5 1,8 1,8 1,8 1,8 1,8 1,8 1,9 2,4
0,4 0,4 0,4 0,4 0,4 0,4 0,4 0,7 1,3 2,3
0,1 0,5 1,4 2,4 3,4
6,3 8,0 9,4 10,4 11,4 12,2 12,4 12,4 12,4 12,4
5,4 7,2 8,6 9,7 10,6 11,5 11,9 120 12,0 12,0
1,0 3,6 5,6 6,5 6,6 6,6 6,6 6,6
1,3 2,3 3,3 4,3 5,2 5,9 6,0 6,0 6,0 6,0
0,7 1,6 2,6 3.6 4,5 5,2 5,4 5,4 5,4 5,4
0,3 2,5 4,1 4,7 4,8 4,8 4,8
0,3 1,3 2,9 4.2 4,6 4,6 4,6 4.6 4,6
0,1 1,0 2,2 2,8 3,1 3,2 3.5
2,2 2,2 2,2 2,2 2,2 2,2 2,2 2,2 2,6 3,4
1,5 2,1 2,4 2,4 2,4 2,4 2,4 2,4 2,7 3.4
0,1 0,7 1,6
10,5 14,5 18,5 22,5 25,7 26,9 27,3 27,3 27,3 27,3
17,4 18,3 18,3 18,3 18,3 18,3 18,3 18,3 18,3 18,3
9,4 10,7 12,7 14,6 16,2 17,3 17,9 18,0 18,0 18.0
2,3 4,9 7,5 10,8 14,2 16,6 17,4 17,4 17,4 17,4
10,9 12,6 14,0 15,0 16,0 16,8 17,0 17,1 17,1 17,1
10,0 11,8 13,2 14,3 15,3 16,1 16 5 16,6 16,6 16,6
0,5 2,8 6,5 10,5 14,1 16,3 16,9 16.9 16,9 16,9
3,8 7,1 10,1 12,9 15,2 16,2 16,6 16.6 16,6 16,6
12,7 13,9 14,2 14,2 14,2 14,2 14,2 14,2 14,2 14,2
0,4 3,1 6,0 9,0 11,6 13,1 13,7 13,8 13.8 13,8
5,2 6,5 8,2 10,2 12,0 13,4 13,6 13.6 13,6 13,6
4,5 5,5 6,7 8,3 10,0 11,4 11,8 11.8 11,8 11,8
2,9 3,6 4,4 5,3 6,3 7,3 8,3 9,3 10,3 11,3
2,5 3,8 5,5 7,5 9,1 9,9 10,0 10,0 10,0 10,0
0,3 0,9 1,6 2,2 3,0 4,0 5,1 6,7 9,0
0,9 2,3 4,3 6,4 8,4 8,9 8,9 8,9 8,9 8,9
0,1 0,6 2,8 6,3 8,3 8,8 8,8 8,8 8,8
0,1 0,6 2,5 3,8 4,5 4,5 4,5 4,5
0,1 0,5 1,3 1,8
0,2 0,8 1,8 3,2 5,0 6,8 8,0 8,0 ' 8,0
405
Пр одолжение
Коицен- pH
Металл Комплексант трация, моль/л 0 1 2 1 3 4
Ni Pb Sc Sr Th Zn Оксалат Ацетат Гидроксид ДЦТА ТЭА ДТПА ЭДТА ГЭДТА НТА Цитрат Пиколинат Тартрат Ацетат Гидроксид Гидроксид ДТПА ЭДТА ГЭДТА НТА Цитрат Тартрат Гидроксид ЭДТА Оксалат Ацетилацетонат Фторид Гидроксид ДЦТА ДТПА ЭДТА Тетрен Трен Цианид Ден Триен ГЭДТА НТА Пиколинат Цитрат Аммиак Ацетилацетонат Ен Оксалат Тартрат Ацетат 0,1 0,1 0,01 0,10 0,01 0,1 0,01 0,01 0,1 0,01 0,1 0,1 0,1 0,1 0,01 0,1 0,01 0,01 0,1 0,01 0,1 0,1 0,1 0,01 0,1 0,01 0,1 0,01 0,01 0,1 0,01 0,1 0,1 0,1 0,1 0,1 0,01 0,1 0,01 0,1 0,1 1,0 0,1 0,01 0,01 0,1 0,1 0,1 0,5 0,2 0,1 6,0 0,1 0,2 0,1 1,4 0,6 0,1 4,3 3,8 6,5 8,9 0,3 0,1 0,3 0,6 1,6 2,6 0,8 4,2 3,3 1,9 1,1 1 0 0,5 0,2 8,0 7,5 12,1 0,1 11,7 1,6 1,0 2,8 1,9 0,7 0,2 1,3 1,2 0,2 3,3 5,2 3,6 6,3 5,5 0,5 3,6 3,9 2,6 1,6 1,4 0,1 10,8 10,4 15,7 0,7 14,1 4,2 3,8 4,9 4,0 2,3 1,6 3,3 0,3 2,1 1,4 4,9 7,6 5,8 8,2 7,4 1,8 4,7 4,0 3,7 3,3 2,4 0,6 0,1 0,1 0,1 0,3 0,2 13,0 12,6 18,5 1,9 14,9 6,6 5,9 6,8 6,0 0,5 3,4 2,7 6,0 1,4 3,4 2,4 0,1
Примечание. Принятые условные обозначения: НТ А—ннтрилтри ацетат; ЭДТА — ацетат; ДТПА — диэтилентриаминиеитаацетат; ДЦТА—транс-1,2-ди аминоциклогексантетра тилентриамин; Ен — этнлендиамвн; ТЭА — триэтаноламин; Тетрев — тетраэтилеи пентамин.
406
pH
5 i 6 1 7 8 1 9 1 10 11 1 12 13 14
5,7 5,7 5,7 5,7 5,7 5,7 5,7 5,7 5,7 5,7
0,1 0,2 0,2 0,2 0,3 0,7 1,6
0,1 0,5 1,4 2,7 4,7 7,4 10,4 13,4
9,7 11,5 12,8 13,9 14,9 15,9 16 8 17,5 17,7 17,7
3,0 4,5 7,0 10,0 13,9 17.5
7,6 9,6 11,6 13,5 15,1 16,2 16,8 16,9 16.9 16,9
10,2 12,0 12,4 14,4 15,4 16,2 16,4 16,5 16,5 16,5
9,4 11,2 12,6 13,7 14,7 15,5 15.9 16,0 16,0 16,0
3,0 4,6 6,5 8,5 10,1 10,9 11,0 11,0 11,1 13,4
5,7 6,7 7,7 8,7 9,6 10,3 10,4 10,4 10,7 13,4
5,0 6,0 7,0 8 0 8,9 9,6 9,8 9,8 10,5 13,4
4,2 4,2 4,2 4,2 4,5 5,3 6,3 7,7 10,4 13,4
4,9 5,7 5,9 5,9 5,9 5 9 5,9 7,4 10.4 13,4
2 8 2,8 2,8 2,8 2,8 3.0 4,7 7,4 10.4 13,4
1,2 1,5 1,5 1,5 1,8 2,7 4,7 7,4 10,4 13,4
0,6 2,2 4,2 6,2 8,2 10,2 12,2 14,2 16,2 18,2
0,1 0,6
0,6 2,4 4,3 5,9 7,0 7,6 7,7 7,7 7,7
1,0 2,6 4,0 5,0 6,0 6,8 7,0 7,1 7,1 7,1
0,3 1,8 3,2 4,3 5,2 6,1 6,5 6,6 6,6 6,6
04 2,1 4,4 5,7 6,4 6,5 6,5 6,5 6,5
0,3 1,0 1,9 2,8 3,5 3,6 3,6 3,6 3,6
0,1 0,4 1,2 2,1 2,8 3,0 3,0 3,0 3,0
0,9 1,5 1,8 1,8 1,8 1,8 1,8 1,8 1,8 1,8
0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,6
0,8 1,7 2,7 3,7 4,7 5,7 6,7 7,7 8,7 9,7
15,0 16,7 18,5 20.2 22,2 24,0 25,2 26,3 27,3 28,3
14,6 16,4 18,1 19,9 21,8 23,7 25,1 25 2 27,2 28,2
19,7 19,7 19,7 19,7 19,7 19,7 19,7 19,7 19,7 19,7
3,7 6,2 9,9 13,5 16,3 17,1 17,1 17,1 17,1 17,1
15,0 15,0 15,0 15,0 15,0 15,0 15,0 15,0 15,0 15,0
0,2 2,4 5,4 8,5 11,8 15,5
8,7 10,5 11,8 12,9 13,9 14,9 15,8 16,5 16,7 16,7
7.4 8,9 10,7 12,6 14,2 15,3 15,9 16,0 16,0 16,1
8,8 10,5 11,9 12,9 13,9 14,7 14,9 15,0 15,0 15,6
7,9 9,7 11,1 12,2 13,2 14,0 14,4 14,5 14,5 15,5
1,6 4,9 7,9 10,7 13,0 14,0 14,4 14,4 14,4 15,5
0,3 3,0 6,0 8,9 11,5 13,0 13,6 13,7 13,7 15,5
0,1 3,5 7,5 10,7 12,3 12,7 12,7 12,8 15,5
0,8 2,8 6,1 9,7 11,9 12,5 12,5 12,6 15,5
0,6 3,1 5,6 7,8 9,6 10,8 11,1 11,1 11,9 15,5
2,4 4,4 6,4 8,4 10,0 10,7 10,8 10.8 11,9 15,5
4,4 5.4 6,4 7,4 8,3 9 0 9,1 9,2 11,8 15,5
3,7 4,7 5,7 6,7 7,6 8,3 8,5 8,8 11,8 15,5
8,4 9,6 9,9 9,9 9,9 9,9 9,9 9,9 11,8 15,5
2,6 3,2 3,5 3,8 4,4 5,4 6,4 8,5 11,8 15,5
0,4 3,6 7,1 8,7 9,1 9,2 11,8 15,5
0,4 3,2 4,7 5,6 8,5 11,8 15,5
0,2 1,1 2,5 3,8 4,2 5,4 8,5 11,8 15,5
0,2 0,8 2,3 4,2 5,7 6,3 8,5 11,8 15,5
4,0 4,0 4.0 4,0 4,0 4,0 5,4 8,5 11,8 15,5
2 8 2,8 2,8 2,8 2,8 2,8 5,4 8,5 11,8 15,5
0,5 0,6 0,6 0,6 0,7 2,4 5,4 8,5 Н.8 15,5
этилен ди аминтетраацетат; ГЭДТА — |3,3'-диаминодиэтилглнколевый эфир-N, N, N', N'-тетра-ацетат; Трен — Э,0',0*-трис (амниоэтил) амин; Триеи — триэтилентетрамни; Ден—диэ-
407
S »»- |_ I—1 1—I О 45» W ND — O^CoslCrJUl ^WW - О pH § Значения lg 1/«L для некоторых маскирующих реагентов [42 j ©0
s §a ° ® 53 ® r* m0*3 0 О '—• -4 CD CD CD о ND CD 45» CD СЛ Сл СЛ Ацетат
W х " —“ М S О О ° Д Ю W * СП р 4 р а П ND 'со 00 Q0 со 00 00 00 00 00 го д _ Ацетнлацетонат
•О м — — — — В5Я о О и— KD со О) 00 О М rfb р S s W О О СП W W со со W со Карбонат
н IP О • >—‘ — 4 О О О — ND 45» j4 О Q0 3 S’ О ND ND “4 00 СЛ СЛ СЛ Qjg СЛ СЛ Цитрат
О Hg о о О *• М СО Л СП р Ч 00 р ^Е — Ф» ND ND ND ND ND ND ND ND ND 1 ft — Цианид
' a о a — nd go TOO - - - - - -ок о cc — a о ™P СЛ СП СЛ Фторид
J3 s s ® о О О *- ND CO СЛ Eft CD CO О О W — g и СИ СЛ СЛ О1 Оксалат
5 ® ~ о • — — — — ND -5 © О О »— Ю Со СЛ СТ) 00 О ND СЛ ^4 О У I ND 00 -4 \] 4 О СТ Ъ) СТ) CD о ”*-4 ~-4 s н СЛ СЛ Фосфат
Д fit . - .. _ - я 1 О О О г- ND 4* СТ) 00 ~ 00 S Я '*►— СТ) О СО о CD '*-4 00 4* — 9 s - - Пирофосфат
1з 3 • — . - — 2 -о ООО — ND 4^ СЛ -4 Е S О Ф» ND СО О О О ®н СЛ Фталат
S.s - - - - яд оо-мй>дсло>ч<»<оомд® V ч w — — — — — — — " Оо — о о Салицилат
? н . . . ? ООО — МСО^СЛОЧОоОМ^ Z^> '*— М-Ч Ст) СТ) СТ) СТ) СТ) Ъ) Ъ) СО ND ND • 'Jm _ .. — Сульфосалицилат
zh „ _ _ _ _ a о о J— ND CO 4^- СЛ “4 О — CO СЛ vj co ft f '^-‘^4 0)ОСТ)СТ)00Сл'слСЛСЛСЛСлСЛ d СЛ СЛ “ Щ Сульфид
ш 4 и S’ О О О СО СЛ -4 ft ft О 4х 4х О О О Н д СЛ СЛ 5 Тартрат
U я . _ 5дг о©о— _кэ w 4^ сл сл ао р ко Ч § N Ч Ч «4 N Ч -ч 00 W “ — Глицин
Д Ч -Л . . - . >2 — . «о ООО — ND СО 45». СЛ СТ) 00 О ND 1 “ — Ъ) СЛ СЛ СП сл СП М-4 Со ND ND Имииодиуксус-ная кислота
® 2 — «— Н to О О О ~ ND W Ф». СЛ jH 00 “ 45» §С ND О 00 00 00 00 00 О ’*’4 4^- 45». п>£з _ _ . НТА
»Е к, 3 Н ООО — МСОЛООООрр^ § 5* — СЛ Ф- СО 4» 00 СТ> Ъ> 00 00 W о ЭДТА
2 [ X — — ND ND S3 О о О — ND W 45». СТ) 00 О ND CD О 45». ft"g ND Ъ 00 00 00 О ND О - 00 М - “ ДЦТА
ы 7 — — — — nd И— О О О ND 45» О) ОЭ G ND СЛ СО СО ft М СО СИ СЛ СЛ СЛ СЛ “-4 СТ) СО СО ГЭДТА
to й — . - - - - ,Н S — >— — ND ND g О Р О J- СО СЛ j4 tO J— 4^. 00 GO 00 Д OJ co CO CD 00 CO 00 CH 4*. ДТПА
Химические элементы
Название Химический символ Порядковый номер Относительная атомная масса*) Валентность
Азат N 7 14,0067 3, 5
Актиний Ас 89 {227] 3
Алюминий А1 13 26,9815 3
Америций Ат 95 1243] 2-6
Аргон Аг 18 39,948 0
Астат At 85 1209,9871 ] L 5
Барий Ва 56 137,34 2
Бериллий Be 4 9,01218 2
Берклий Bk 97 [247] 3, 4
Бор В 5 10,811 3
Бром Bi 35 79,904 1. 5
Ванадий V 23 50,942 5, 3, 4
Висмут Bi 83 208,980 3, 5
Водород Н 1 1,00797 1
Вольфрам W 74 183,85 6
Гадолиний Gd 64 157 25 3
Галлий Ga 31 69,72 3
Гафний Hf 72 178,49 4
Гелий He 2 4,0026 0
Германий Ge 32 72,59 4
Гольмий Ho 67 164,930 3
Диспрозий Dy 66 162,50 3
Европий Eu 63 151,96 3, 2
ДСелезо Fe 26 55,847 2, 3
Золото Au 79 196,9665 3, 1
Индий In 49 114,82 3
Иод I 53 126,9044 1. 5, 7
Иридий Ir 77 192,22 3, 4
Иттербий Yb 70 173,04 3, 2
Иттрий Y 39 88,9059 3
Кадмий Cd 48 112,41 2
Калий К 19 39,0983 1
Калифорний Cf 98 [251] 3
Кальций Ca 20 40,08 2
Кислород О 8 15,9994 2
Кобальт Co 27 58,9332 2, 3
Кремний Si 14 28,0855 4
Криптон Kr 36 83,80 0
Ксенон Xe 54 131,30 0
Курчатовий Ku 104 1261]
Кюрий Cm 96 [247] 3, 4
Лантан La 57 138,9055 3
Литий Li 3 6,939 1
Лоуренсий Lr 103 260 3
Лютеций Lu 71 174,967 3
Магний Mg 12 24,305 2
Марганец Mn 25 54,9380 2, 4, 7
Медь Cu 29 63,546 2, 1
Менделевий Md 101 {258 ] 3
Молибден Mo 42 95,94 6, 3, 4
Мышьяк As 33 74,9216 3, 5
Натрий Na 11 22,98977 1
Неодим Nd 60 144,24 3
Неон Ne 10 20,179 0
409
Продолжение
Название Химический символ Порядковый номер Относительная атомная масса *) Валентность
Нептуний Np 93 1237] 4, 5, 3, 6
Никель Ni 28 58,69 2, 3
Нильсборий Ns 105 262
Ниобий Nb 41 92,906 5, 3
Нобелий No 102 1259]
Олово Sn 50 118,71 2, 4
Осмий Os 76 190,2 8, 4
Палладий Pd 46 106,42 2, 4
Платина Pt 78 195,08 2, 4
Плутоний Pu 94 [244] 3, 4, 5, 6
Полоний Po 84 ]208,9824] 2, 4
Празеодим Pr 59 140,907 3, 4
Прометий Pm 61 [144,9128] 3
Протактиний Pa 91 [231 | 5, 4
Радий Ra 88 (226] 2
Радон Rn 86 ]222] 0
Рений Re 75 186,207 7. 6, 4
Родий Rh 45 102,905 3, 2
Ртуть Hg 80 200,59 2, 1
Рубидий Rb 37 85,4678 1
Рутений Ru 44 101,07 4, 8
Самарий Sm 62 150,36 3.2
Свинец Pb 82 207,19 2, 4
Селен Se 34 78,96 2, 4, 6
Сера S 16 32,066 2, 4, 6
Серебро Ag 47 107,868 1
Скандий Sc 21 44,95591 3
Стронций Sr 38 87,62 2
Сурьма Sb 51 121,75 3, 5
Таллий TI 81 204,383 1, з
Тантал Ta 73 180,948 5
Теллур Те 52 127,60 2, 4, 6
Тербий Tb 65 158,9254 3, 4
Технеций Tc 43 [97,907] 7
Титан Ti 22 47,90 4, 3
Торий Th 90 232,038 4
Тулий Tm 69 168,934 3
Углерод C 6 12,01115 4, 2
Урай u 92 238,0289 6, 4, 3, 5
Фермий Fm 100 [257] 3
Фосфор P 15 30,97376 5
Франций Fr 87 [223] 1
Фтор F 9 18,9984 1
Хлор Cl 17 35,453 I, 5, 7, 3
Хром Cr 24 51,996 3, 6, 2
Цезий Cs 55 132,905 1
Церий Ce 58 140,12 3, 4
Цинк Zn 30 65,39 2
Цирконий Zr 40 91,224 4
Эйнштейний Es 99 1252] 3
Эрбий Er 68 167,26 3
* Элементы с порядковыми номерами 43, 61, 84 — 105 не имеют стабильных изото-пов. В квадратных скобках указана относительная масса изотопа с наиболее продолжительным периодом полураспада.
410
БИБЛИОГРАФИЧЕСКИМ СПИСОК
1. ЖАХ, 1975, т. 30, № 10, с. 2058—2063.
2. Compendium of analytical nomenclature, ШРАС — Pergamon Press, 1977.
3. Boef D. G., Nulanicki A.— Pure a. Appl. Chem., 1983, v. 55, № 3, p. 553— 556. Перевод: ЖАХ, 1984, т. 39, № 5, с. 945—947.
4. Sandell Е. В., West Т. S. — Pure a. A ppi. Chem., 1979, v. 51, № 1, р. 43— 47.
5. Kaiser И — Spectrochem. Acta, 1978, v. ВЗЗ, № 9, p. 551—576.
6. Koch О. G. — Anal. Chim. Acta, 1979, v. 107, № 5, p. 373—376.
7. Kirkbright G. F. — Pure a. Appl. Chem., 1978, v. 50, № 3, p. 237—242.
8. Ackermann G., Kothe J., Koch S. —Scripta Fac. Sci. Natur. UJEP Bru-nensis. Chemia 1, 1978, v. 8, № 1, p. 1- -12.
9. Sommer L., Langova M., Kuban V. — Ibid., p. 13—34.
10 Sommer L., Langova M., Kuban V. et. al. — Ibid., Chemia 1—2, 1980, v. 10, N 1—2, p. 71—80.
11. Номенклатурные правила ИЮПАК по химии. М.: ВИНИТИ, 1979. Т. 1, полутом 2. 660 с.
12. Нейман Е. Я-, Каплан Б. Я. — ЖАХ, 1978, т. 33, № 3, с.607—609.
13. Чарыков А. К- — Вести. ЛГУ, 1984, № 4, с. 64—74.
14. Zolotov Yu. А. —Pure a. Appl. Chem., 1979, v. 51, p. 1195— 1211.
15. Петерс Д., Хайес Дж., Хифтье Г. Химическое разделение и измерение. Теория и практика аналитической химии/Пер. с англ, под ред. П. К. Ага-сяна. М.: Химия, 1978. 816 с.
16. Скуг Д., Уэст Д. Основы аналитической химии/Пер. с англ, под ред. Ю. А. Золотова. М.: Мир, 1979, т. 1. 480 с.; т. 2. 438 с.
17. Ляликов Ю. С., Клячко Ю. А. Теоретические основы современного качественного анализа. М.: Химия, 1978. 312 с.
18/ Карпов Ю. А., Алимарин И. П. —ЖАХ, 1979, т. 34, Кв 7, с. 1402—1410.
19. Koch О. G., La Eleur Р. D., Morrison G. Н —Pure a. Appl. Chem., 1982, v. 54, № 8, p. 1565—1577 Перевод: ЖАХ, 1984, т. 39, № 6, с. 1135— 1144.
20. Александрова Г. И., Главин Г. Г., Гузеев Н. Е. и др. — Зав. лаб., 1978, т. 44, № 12, с. 1427—1431.
21. Вайнфорднер Дэю. — В кн.: Спектроскопические методы определения следов элементов/Пер. с англ под ред. О. М. Петрухина, В. В. Недлера. М.: Мир 1979, с. 9—19; 428—477.
22. Назаренко В. А., Флянтикова Г. В. —ЖАХ, 1977, т. 32, Кв 6, с. 1217 — 1236.
23. Пешкова В М., Луфт Б. Д., Громова М. И. Орлов Ю. Ф. — В кн.: Проблемы аналитической химии. М.: Наука, 1977, вып. 4, с. 52—75.
24. Бусев А. И. — Там же, 1976, вып. 3, с. 7—45.
25. Sandell Е В., Hiroshi Onishi. Photometric Determination of Traces of Metals. General Aspects. 4 th Ed. N. Y. — Toronto: J. Wiley, 1978. 1085 p. (Chem. Anal v. 3, Part 1).
26. Marczenko Z. —Crit. Rev. in Analyt. Chem., 1981, v. 11, N 3, p. 195—260.
27. Джог П. — В кн.: Спектроскопические методы определения следов элементов/Пер. с англ под ред. О. М. Петрухина, В. В. Недлера. М.: Мир, 1970, с. 200 — 228.
28. Марченко 3., Минчевский Е. — В кн.: Достижения в аналитической химии. М.: Наука, 1974, с. 164—170.
411
29. Jackwerth E., Mizuike A., Zolotov Yu. A. et al. — Pure a. Appl. Chcm., 1979, v. 51, Ns 5, p. 1195—1211.
30. Пилипенко A. T. — ЖАХ, 1976, т 3, № 2, c. 220—229.
31. Pilipenko A. T. — Wissenchaft Z. Karl Marx—Univ. Leipzig. Math.-Natur-wiss. Rundschau, 1975, Bd. 24, № 4, S. 367—380.
32. Бабко А. К., Пилипенко A. T. Фотометрический анализ. Методы определения неметаллов.-М.: Химия, 1974. 359 с.
33. Boltz D., Howel J. A. Colorimetric Determination of Nonmetals. 2nd. Ed. N. Y. — Toronto: J. Wiley, 1978. 543 p. (Chem. Anal. v. 8).
34. Коренман И. M. Фотометрический анализ. Методы определения органических веществ. М.г Химия, 1970. 334 с.
35. Бабко А. К., Пилипенко А. Т. Фотометрический анализ. Общие сведения и аппаратура. Ж.: Химия, 1968. 388 с.
36. Яцимирский К. Б. Кинетические методы анализа. Л!.. Химия, 1967. 200 с.
37. Крейнгольд С. У Кататиметрия в анализе реактивов и веществ особой чистоты. М.: Химия, 1983. 192 с.
38. Пилипенко А Т., Тананайко М. М. Разнолигандные и разнометальиые комплексы и их применение в аналитической химии, М. Химия, 1983. 224 с.
39. Babko А. К. — Taianta, 1968, v. 15, N 8, р. 721—733.
40. Устойчивость смешанных комплексных соединений в растворах''Под ред. Фридмана Я. Д- Фрунзе: Илым, 1971. 181 с.
41. Яцимирский К. Б. — ЖНХ, 1956, т. 1, № 10, с. 2306—2313.
42. Ringbom A. Complexation in Analytical Chemistry. N. Y J. Wiley, 1963. 395 p
43. Инцсди Я- Применение комплексов в аналитической химии/Пер. с англ. О. М. Петрухина и Б Я- Спивакова. Л!.. Мир, 1979. 376 с.
44. Булатов М И. Примеры теоретических расчетов в химическом анализе. Л.: ЛТИ им. Ленсовета, 1972. 202 с.
45. Лайтинен Г. А., Харрис В. Е. Химический анализ/Пер. с англ, под ред. Ю. А. Клячко. М Химия, 1979. 624 с.
46. Янсон Э. Ю., Путнинь Л- К. Теоретические основы аналитической химии. М.: Высшая школа, 1980. 260 с.
47. Булатов М. И. Расчеты равновесий в аналитической химии. Л.: Химия, 1984. 184 с.
48. Бек М. Химия равновесий реакций комплексообразования/Пер. с англ, под ред. И. Н. Марова. М.: Мир, 1973. 359 с.
49. Перрин Д. Органические аналитические реагеиты'Пер. с англ, под ред. Ю. А. Золотова. М.: Мир, 1967. 407 с.
50. Карлышева К- Ф-, Кошель А. В., Вашунь Л. Ф. — Укр. хим ж., 1972, т. 37, Ns 5, с. 493—497
51. Moehan Е. I. — In Treatise on Analytical Chemistry/Ed. I. M. Kolthoff, P. J. Elving. N.Y.Jntersci. Encyclopedia, 1964. Part 1, v. 5, p. 2754—2804.
52. Boltz D. F., Mellon M. G.— In: Standard Methods of Chemical Analysis/Ed. F. J. Welcher. 6th ed. Princeton e. a.: D. van Nostrand Company Inc., 1966. v. 3, Part A, p. 3—22.
53. International Congress of Pure and Applied Chemistry. Comission on Optical Data Belgium: Liege, Sept. 6—8, 1958.
54. Львов Б. В. — ЖАХ, 1979, т. 34, № 9, с. 1864—1865.
55. Braud1 Е. А. — In: Determination of Organic Structures by Physical Me-thods/Ed. E. A. Braude, F. C. Nachod. N. Y.: Academic Press, 1955, p. 131— 194.
56. Бабко А. К- Физико-химический анализ комплексных соединений в растворах. Киев: Изд. АН УССР, 1955. 328 с.
57. Бабко А. К. — Хим наука и пром., 1959, т. 4, № 2, с. 164— 171.
58. Чарыков А. К. Математическая обработка результатов химического анализа. Л.: Химия, 1984. 168 с.
59. Ямпольский М. 3., Дроздова С. Н. — Уч. зап. Курского пед. ин-та, 1969, . т. 57, вып. 2, с. 88—95.
60. Корецман И. М. Аналитическая химия малых концентраций. М.: Химия, 1937. 168 с.
412
61. Kaiser H. — Pure a. Appl. Chem., 1973, v. 34, № 1, p. 35—60.
62. Мустафин И. С.—ДАН СССР, 1959, т. 126, № 3, с. 579 — 581.
63. Хольцбехер 3., Дивиш Л., Крал М. и др. Органические реагенты в неорганическом анализе/Пер. с чешек. 3. 3. Высоцкого. М.: Мир, 1979. 752 с.
64. Stefanini-Oreiic L., Gradinii. — Farmacentski glasnik, 1978, v. 34, № 2—3, p. 85—104.
65. Каплан Б. Я-—Изв. CO АН СССР. Сер. хим. наук, 1978, № 12/5, с. 27—38.
66. Марченко 3. Фотометрическое определение элементов/Пер. с польск. под ред. А. А. Золотова. М.: Мир, 1971. 501 с.;
Marczenko Z. Spectrophotometric Determination of Elements. Chichester: Horwood — Wiley, 1976.
67. Бланк А. Б. — Ж AX, 1962, t. 17, № 9, c. 1040—1044; Автореф. канд. дисс Харьков: ВНИИ Монокристаллов, 1966, 18 с.; В ки.: Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты. Харьков: ВНИИ А1онокристаллов, 1971, с. 121—135.
68. Булатов М. И. — ЖАХ, 1975, т. 30, № 1, с. 5—8.
69. Koch О. О., Koch-Dedic G. A. Hadbuch der Spurenanalyse. Berlin: Springer— Verlag, 1964. 1232 S.; 2nd Ed., 1974.
70 Гинзбург Л. Б., Лурье Ю. Ю. — Зав лаб., 1948, т. 14, № 5, е. 538—545.
71. Сендел Е. Б. Колориметрические методы определения следов металлов/Пер. с англ, под ред. В. Н. Прусакова, М.: Мир, 1964. 902 с.
72. Wmefordner J. D., LongG. L. — Analyt. Chem., 1983, v. 55, № 7, p. 712A— 724A.
73. Spectrochim. Acta, 1978, v. 33B, № 6, p. 242—285.
74. Analyt. Chem., 1980, v. 52, № 14, p. 2242—2249.
75. Гринзайд E. Л., Зильберштейн X. И., Надежина Л. С., Юфа Б. Я. — ЖАХ, 1977, т. 32, № 12, с. 2106—2112.
76. Зильберштейн X. И. — Изв. СО АН СССР. Сер. хим. наук. 1978, № 9/4, с. 39—48.
77. Бланк А. Б. - ЖАХ, 1979, т. 34, № 1, с. 5—9.
78 Luteany С., Rica I. — Pure a. appl. Chem., 1975, v. 44, № 3, p. §35______553.
79. Кузьмин П. M., Журавлев Г. И., Кувовлев И. А. и др.—ЖАХ, 1969, т. 24, № 3, с. 429—434.
80. Бланк А. Б. — ЖАХ, 1965, т. 20, № 1, с. 3—15; В кн.: Методь. анализа веществ особой чистоты и монокристаллов/Под ред. А. Б. Бланка, А. М. Булгаковой, Ю. Ф. Рыбкина. Харьков1 ВНИИ Монокристаллов, 1971. Ч. I, с. 136—155.
81 Бланк А. Б. — ЖАХ, 1960, т. 15, Ns 3, с. 359—361.
82. Доерф'-ль К- Статистика в аналитической химии. М.: Мир, 1969. 247 с.
83. Murphy Т. J. — U. S. Dep. Commer. Nat. Bur. Stand. Spec. Publ. 2, 1977, № 492, p. 117—147.
84. Федорова С. Ф., Пенкина H. В., Нустрова В. С., Захарова Т. Н. — В kh.s Стандартные образцы в черной металлургии. М.: Металлургия, 1980, № 9, с. 64—67.
85 Блюм И. А. — Зав. лаб., 1977, т. 43, № 12, с. 1441—1449.
86. Блюм И. А. — Зав. лаб., 1978, т. 44, № 6, с. 660—664.
87. Recommedations on use of the term amplification reactions. Pure a. Appl. Chem., 1982, v. 54, № 12, p. 2554—2556.
88. Djurkin V., Kirkbrighi G F., West T. S. — Analyst, 1966, v. 91, № 1079, p. 89—93.
89. Kirkbrighi G. F. — Proc. Soc. Analyt. Chem., 1966, v. 3, № 10, p. 164.
90. Kirkbrighi G. F., Yoe J. H. — Taianta, 1964, v. 11, № 3, p. 415—422.
91 West T. S. — Chem. and Ind., 1966, v. 25, Ns 5, p. 1005—1016.
92. Бабко А. К., МарковаЛ. В., Приходько M. У. —ЖАХ, 1966, т. 21, Ns 8, с. 935—939.
93 Набиванец Б. И., Линник П. И., Калабина Л. В. Кинетические методы анализа природных вод. Киев: Наукова Думка, 1981. 139 с.
94. Ушакова Н. М., Долманова И. Ф. — ЖАХ, 1983, т. 38, Ns 8, с. 1515— 1531.
413
95. Мюллер Г., Отто М., Вернер Г. Каталитические методы в анализе следов элементов/Пер. с нем., М.: Мир, 1983. 195 с.
96. Kopanica М., Stara V. Kinetic Methods in Chemical Analysis. Amsterdam e. a.: Wilson and Wilson, 1983. (Compr. Anal. Chem., v. 18, p. 11—250).
97. Howell J. A., Hargle L. G. — Analyt. Chem., 1982, v. 54, № 5, p. 171R— 188R; 1984, v. 56, № 5, p. 225R—240R.
98. Harris T. D. — Ibid., 1982, v. 54, № 6, p. 741A—750A.
99. Shirk J. S., HarrisT. D., Mitchell J. W. — Ibid., 1980, v. 52, № 11, p 1701— 1705
100. Harris T. D., Mitchell J. W. — Ibid., p. 17C6—1708.
101. Oda Shohei, Sawada T., Nomura M., Kamada H. — Ibid., 1979, v. 51, № 6, p. 686—688.
102. Lahmann Ludewig H. T., Welling H. — Ibid., 1977, v. 49, № 4, p. 549—551.
103. Yarbo S., Flaschka H. A. —Microchem J., 1976, v. 21, № 4, p. 415—423.
104 Бланк А. Б., Чепурная В. Г., Вакуленко В. fl. — В ки.: Монокристаллы и техника. Харьков: ВНИИМонокристаллов, 1972, вып. 7, с. 149—155.
105. Бланк А. Б. — ЖАХ, 1962, т 17, № 8, с. 1015—1017; 1964, т. 19, № 3, с. 363—365.
106. Плинер Ю. Л., Свечникова Е. А., Огурцов В. М. Управление качеством химического анализа в металлургии. М.: Металлургия, 1979, с. 103.
107. Данцер К., Таи Э., Мольх Л. Аналитика. Систематический обзор/ Пер. с нем. Под ред. Ю А. Клячко. М.: Химия, 1981. 280 с.
108. Берштейн И. fl , Каминский Ю. Л. Спектрофотометрический анализ в органической химии. 2-е изд. Л.: Химия, 1986. 200 с.
109. Комарь Н П., Самойлов В. П. а) ЖАХ, 1963, т. 18, № 11, с. 1284—1290; б) там же, 1967, т 22, № 9, с. 1285—1296, в) там же, 1969, т. 24, № 12, г 1801—1808; г) там же, 1975, т. 30, № 3, с. 465—471.
ПО Комарь Н П., Самойлов В. П , Залкинд В. М., Степаненко И. А. — ЖАХ, '982, т. 37, № 2, с. 349 -356
111. Rothman L. D., Grouch S. R., Ingle J. D. —Analyt. Chem., 1975, v. 47, p. 1226—1233.
112. Sharpl M. R — Analyt. Chem., 1984, v. 56, № 2, p. A339, A340, A342, A344, A,348, A350.
113. Youmans H. L., Brown V. H. — Analyt. Chem., 1976, v. 48, № 8, p. 1152— 1155.
114. Sloane H. J., Gallaway \V. S. — Appl Spectroscopy, 1977, v. 31, № 1, p. 25— 30
115. Burnett R. W. — Analyt. Chem., 1973, v. 45, № 2, p. 383—385.
116. Берштейн И. fl , Кабанов В А., Комаров E. В. Экспериментальное изучение погрешиосгей измерения иа спектрофотометре СФ-46. Черкассы: Н14ИТЭХИМ. 323 — ХП 86 Деп.
117. Svehla G„ Pall A., Erdey L. — Та.anta, 1963, v. 10, № 7, p. 719—726.
118. Блюм И. А. Экстракционно-фотометрические методы анализа с применением основных красителей. М.: Наука, 1970, 220 с.
119. Блю» И. А , Овчинникова Л М , Степина Н. И. — Зав. лаб., 1979, т. 45, № 9. с. 788—794.
120. Бланк А. Б. — ЖАХ, 1973, т. 28, № 7, с. 1435—1436.
121. Лагутин В. И. —Метрология, 1981, 8, с. 58—62.
122. Берковский В. Ф., Ганопольский В И. Дифференциальный фотометрический анализ. М.: Химия, 1969 167 с.
123. Беликов В. Г. Дифференциальная фотометрия. Ставрополь: Ставропольское книжное изд-во, 1970. 136 с.
124. Pall A., Svehla G„ Erday L.— Taianta, 1964, v. 11, № 10, p. 1383—1390.
125. Suehla G. — Taianta, 1966, v. 13, № 5, p. 641—666.
126. Кораблев ИВ,— ЖАХ, 1973, т. 28, № 5, с. 837—847.
127. Пантелеем Е. П , Черепанова Э. В. — В кн.: Технология минеральных удобрений/Под ред. М. Е. Позина, В. А. Копылева. Л.: ЛТИ им. Лснсо-вега, 1973, с. 218—233.
128. Пантелеева Е. П., Крупина И. Н. — Зав. лаб., 1972, т. 38, № 11, с. 1323.
414
129. Крупина И. Н. Автореф. канд. дисс., ЛТИ им. Ленсовета, 1978.
130. Пантелеева Е. П., Черепанова Э. В. — Химия в сельском хозяйстве, 1981, т. 9, № 8, с. 55—59.
131. Коковкин-Щербак Н. И., Масленников Ю. Н. —ЖАХ, 1980, т. 35 № 9 с. 1669—1676.
132. Marczenko Z., Ramsza А. Р.—Chem. Anal. (PRL), 1976, v. 21 № 4, p. 805^811.
133. Urino G. A., Gravatt С. C. — Analyt. Chem., 1977, v. 49, № 6, p. 361— 365.
134. Cali J. P., Reed IT. R. The role of NBS Standard reference materials in accurate trace analysis. NBS spec. publ. 422. Washington, 1976.
135. Alvarez R., Rasberry S. D., Uriano G. A. — Analyt. Chem., v. 54, № 12, p. 1226A—1244A.
136. Standards in Absorption Spectrometry Ultraviolet Spectrometry Group/Ed. C. Burgess, A. Knowles. London—N. Y.: Chapman a. Hall, 1981. 142 p.
137. Proceedings of a Workshop Seminar. Gaitherburg, Nov. 19—20, 1975; U. S. Dep. Comm. Nat. Bur. Stand. Spec. Publ., 1977, № 466 p. 1—150.
138. Herington E. F. G., Milazzo G. — Pure a. Appl. Chem., 1977, v. 49, №5, p. 661—674.
139. Almon D. H., Billmeyer F. IF. Jr. — J. Chem. Educ., 1975, v. 52, № 5, p. A281, A282, A284, A290; № 6, p. A315, A318—A321.
140. West M. A., Kemp D. Practical standards for UV absorption and fluorescence spectrophotometry. International Laboratory, 1976, Мау/June, p. 27—39.
141. Milazzo G., Caroil S., Palumbo-Doretti M., Violente N.—Analyt. Chem., 1977, v. 49, № 8, p. 711—717.
142. Burke R. W., Mavrodineanu R. Standard Reference Materials: Certification and Use of Acidic Potassium Dichromate Solutions as an Ultraviolet Absorbance Standard — SRM 935. NBS. August 1977. 48 p.
143. Venable W. H. Jr, Eckerle K. L. Didimium Glass Filter for Calibrating the Wavelenght Scale of Spectrophotometers. — SRM 2009, 2010, 2013, 2014. Washington D. C., 1979, NBS Spec. Publ., p. 260—266.
144. Mavrodineanu R., Baldwin J. R. Standard Reference Materials: Glass Filters as a Standard Reference Material for Spectrophotometry—Selection, Preparation, Certification, USe SRM—930, Sc. Cat. № C 13.10 : 260—51, U. S. Washington, D. C. 107 p.
145. Morrison G. — Pure a. Appl. Chem., 1975, v. 41, № 3, p. 397—403.
146. Klein R., Clifford H. — Amer. Lab., 1977, v. 9, № 7, p. 21—22; 24—27.
147. Чарыков А. К. — ЖАХ, 1984, t. 39, № 9, c. 1708-1714.
148. Klika Z., Sommer L. — Scr. Fac. Sci. Natur. UJEP Brun. Chem. 1978, v. 8, № 2, p. 53—76.
149. Sommer L., Kuban V., Langovd M.—Fresenius Z. Anal. Chem., 1982, Bd. 310, H. 1/2, p. 51—61.
150. Novotny I., Horak J., — Scripta Fac. Sci. Natur. UJEP Prun. Chem., 1977, v. 7, № 2, p. 85—94.
151. Mullerova L., Vrchlabsky M., Havel J. — Scripta Fac. Sci. Nat. Univ. Purk. Brun., 1980, v. 10, № 1—2 (Chemia), p. 13—22.
152. Havel J., Vrchlabsky M., Sekaninova J., Komarek J. — Scripta Fac. Sci. Univ. Brno, 1979, v. 9. (Chemia), p. 51—61.
153. Gruberova A., Suchanek M. — Sb. VSCHT Praze, 1983, H. 18, p. 167—177.
54. Bajo S. — \ualyt. Chim. Acta, 1979, v. 105, № 1, p. 283 — 287.
155. Саввин С. Б. Органические реагенты группы арсеназо III. М.: Атомиздат, 1971. 349 с.
156. Адамович Л. П. — ЖАХ, 1965, т. 20, № 12, с. 1273—1279.
157. Бургер К. Органические реагенты в неорганическом анализе/Пер. с англ. И В. Матвеевой. М.: Мир, 1975. 272 с.
158. Назаренко В. А., Антонович В. П. Триоксифлуорэиы. М.: Наука, 1973.
159. Пешкова В. М., Савостина В. М., Иванова Е. К., Оксимы. М.: Наука, 1978. 236 с.
160. Виноградов А. В., Елинсон С. В. Оксихинолин. М.: Наука, 1979, 328 с.
415
161. Иванов В. М. Гетероциклические азотсодержащие азосоединения. Мл Наука, 1982. 230 с.
162. Умланд Ф., Янсен А., Тириг Д. и др. Комплексные соединении в аналитической химии/ Пер. с нем. О. М. Петрухина. Мл Мир, 1975 531 с.
163. Singh R. В., Garg В. S., Singh R. Р. — Taianta, 1979, v. 26, № 6, р. 425— 144
164. Диантипирилметаи и его гомологи как аналитические реагенты: Уч. зап. Пермского ун-та, № 234, Пермь, 1974. 280 с.
165. Коренман И. М. Органические реагенты в неорганическом анализе: Справочник. Мл Химия, 1980. 448 с.
166. Лазарев А. И. Органические реактивы в анализе металлов: Справочник. М.: Металлургия, 1980. 232 с.
167. Стары И. Экстракция хелатов Пер. с аигл. под ред. Ю. А. Золотова. Мл Мир, 1966. 392 с.
168. Bode И., Tusche К-, Wahrhausen Н. — Z. anal. Chemie, 1962, Bd. 190, Н. 1, S. 48—60.
169. PiiKhel R. — Ibid., 1966, Bd. 221, Teil I, S. 132—142.
170. Shibata Sh. — Anal. Chim. Acta, 1960, v. 23, № 4, p. 367—369; 1961, v. 2t>, № 4, p. 348—359.
171. Betteridge D., Fernando Q., Freiser H. — Analyt. Chem., 1963, v. 35, N 3, p. 294—298.
172. Моррисон Д., Фрейзер Г. Экстракция в аналитической химии/Пер. с англ, под ред. В. М. Вдовенко. Л.: Госхимиздат, 1960. 311 с.
173. Rozycki С — Chemia Analityczna, 1966, v. 11, Z. 3, p. 447—472.
174. De A. K. Khopkar Sh. M., Chalmers. R. A. Solvent Extraction of Metals. London: van Nostrand, 1970. 259 p.
175. Фомин В. В. Химия экстракционных процессов. Мл Атомиздат, 1960. 166 с.
176. Золотов Ю. А. Экстракция внутрикомплексных соединений. Мл Наука, 1968. 313 с.
177. Аналитическая химия и экстракционные процессы/Под ред. А. Т. Пилипенко, Б. И. Набиваица. Киев: Наукова Думка, 1970. 119 с.
178. Комплексообразование и экстракция актинидов и лантаноидов/Под ред. В. М. Вдовенко. Лл Наука, 1974. 161 с.
179. ШевчукИ. А. Экстракция органическими основаниями. Киев Вища школа, 1978, 170 с.
180. Шмидт В. С. Экстракция аминами. At. Атомиздат, 1970, 308 с.
181. Гринберг А. А. Введение в химию комплексных соединений. Лл Химия, 1966. 632 с.
182. Желиговская Н. И., Черняев И. И. Химия комплексных соединений. Мл Высшая школа, 1966. 388 с.
183. Кузнецов В. И. Химические основы экстракционно-фотометрических методов анализа. At.: Госгеолтехиздат, 1963. 42 с.
184. Золотов Ю. А., Петрухин О. М., Кузьмин Н. М. — В кил Химические основы экстракционного метода разделения элементов. М.: Наука, 1966, с. 28—43.
185. Булатов М. И., Муховикова Н. П. — ЖАХ, 1979, т. 34, № 6, с. 1073— 1080.
186. Булатов М. И., Муховикова Н.П. — В кнл III Всесоюзная конференция по аналитической химии: Тезисы докладов. А1ииск: БГУ, 1979. Ч. 1. с. 211 — 213.
187. Zolotov Yu. A., Seryakova I. V., Vorobyeva G. A. — Taianta, 1967, v. 14, p. 737—749.
188. Betteridge D., Fernando Q., Freiser H. — Analjt. Chem., 1963, v. 35, N 3, p. 294—298.
189. Булатов M. И., Муховикова H. П. — Изв. вузов. Химия и хим. технол., 1979, т. 22, № 8, с. 961—964.
190. Galik Q. — Taianta, 1969, v. 16, № 2, р. 201—213.
191. Иванчев Г. Дитизон ч его применение/Пер. с нем. под ред. И. Б. Супру-новича. Мл ИЛ, 1961. 451 с.
192. Handley Т. Н., Dean J. А. — Analyt. Chem., 1962, v. 34, р. 1312—1327.
416
193. Банковский Ю. А., Иевиньш А. Ф., Лиепиня 3. Э. —ЖАХ, 1960 т 15 № 1, с. 4—И.
194. Спиваков Б Я., Золотов Ю. А. —ЖАХ, 1970, т. 25, № 5, с. 616—633.
195. Золотов Ю. А., Кузьмин И. М. Концентрирование микроэлементов. Мл Химия, 1982. 284 с.
196. Золотов Ю. А., Кузьмин Н. М. Экстракционное концентрирование. Мл Химия, 1971. 272 с.
197. Кузьмин Н. М. —Зав. лаб., 1982, т. 48, № 2, с. 11—15.
198. Roland S. Young separation procedure in inogranic analysis. N. Y.: J. Wiley, 1980. 475 p
199. Minczewski J., Chwastowska J., Dybczynski R. Separation and preconcentration methods in inogranic trace analysis. N. Y.: Horwood — J. Wiley, 1982. 543 p.
200. Mizuike A. Enrichment techniques for inogranic trace analysis. Berlin, Heidelberg, N. Y.: Springer—Verlag, 1983. 144 p.
201. Gottschalk G. — Z. anal. Chem., 1963, Bd. 194, H. 5, S. 321—333.
202. BodeH. — Z. anal. Chem., 1954, Bd. 143, H. 1, S. 182—194; 1955, Bd 114, H. 1, S. 165—186.
203. Vanco D. —Chem. Listy, 1983, v. 77, № 1, p. 16—30.
204. Бланк А. Б., Экспериандова Л. П. —Зав. лаб., 1979, т. 45, № 1 с. 7—9.
205. Бланк А. Б., Булгакова А. М., Сизоненко Н. Т. — В ки: Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты. Харьков: Изд. ВНИИ Монокристаллов, 1971. Ч. 2, 65—71; ЖАХ, 1961, т. 16, № 6, с. 715—719.
206. Дорохова Е. ЕЕ, Алимарин И. П. — Успехи химии, 1979, т. 48, № 5, с. 930—956.
207. Лугинин В. А., Коробейникова Е. Г., Церковницкая И. А.—В кн. Проблемы совр. аналит. химии. Л.: ЛГУ им. А Жданова, 1981, № 3, с. 200— 207.
208. Wasey Abdul, Bansal Ray K-, Satake Masatada, Puri Bal K- — Bull. Chem. Soc. Jap., 1984, v 57, № 4, p. 980—983.
209. Чуйко В. T Автореф. докт. дисс. Тернополь. 1962
210. Bruninx Е. — Philips J. Res., 1978, v. 33, № 5—6. р. 264—280.
211. Кузнецов В. И. — В ки.: Труды комиссии по аналитической химии. Мл Наука, 1965, т. 15, с. 279—295.
212. Чащина О. В. Ионообменное концентрирование в анализе микропримесей. Томск Томск, ун-т, 1980. 95 с.
213. Макарова С. Б., Егоров Е. В Ионообменные смолы для получения и анализа особо чистых веществ. М.: НИИТЭХИМ, 1975. 35 с.
214. Мясоедова Г. В., Саввин С. Б. — ЖАХ, 1982, т. 37, № 3, с. 499—519.
215. Мясоедова Г. В., Щербинина И. И., Саввин С. Б. —ЖАХ, 1983, т. 38, № 8, с. 1503—1514.
216. Lieser К- Н., Breuwieser Е., Burba Р. е. а. — Mikrochim. Acta (Wien), 1978, № 1, р. 363—373.
217. Sturgeon R. E., Berman S. S., Desaulniers Russell D. S. — Taianta, 1980, v. 27, № 1, p. 85—94.
218. Марченко 3. — Зав. лаб., 1979, т. 45, № 1, с. 1—7.
219. Bachmann К- — Taianta, 1982. v. 29, № 1 р. 1—26.
220. Романцева Т. И., Громова М. И., Пешкова В. М. — Вести. МГУ. Сер. 11. Химия, 1965, № 6, с. 74—78.
221. Булатова А А., Алесковский В. Б., Булатоь М. И. — Изв. вузов. Химия и хим. технол., 1974, т. 17, № 3, с 345—347.
222. Бардин В. В., Булатов М. И., Муховикова Н. П. — Зав. лаб., 1981, т. 47, № 7, с. 7—9.
223. Зиньковская Г. Т., Киреев М. Н. —• Труды Омского мединститута им. М. И. Калинина, 1969, № 88, с. 122—129.
224. Финкечьштейн А. И. — Зав. лаб., 1961, т. 27, № 8, с. 923—928.
225. Шабадаш А. Н., Пшечицина В. П., Хишева В. М. — В кн.: Материалы X Всесоюзного совещания по спектроскопии. Т. 1. Львов: Львовск. ун-т, 1957. с. 275.
417
226. Fukushima S., Shigemoto M., Kato J., Otazai K- — Alikrochim. Acta, 1957, H. 1, p. 35—70.
227. Шугар А., Шугар Ю. — Изв. ТСХА, 1960, вып. 3 (34), с. 206.
228. Адамович Л. П. — ЖАХ, 1962, т. 17, № 8, с. 912—916.
229. Кац М. Д. — Зав. лаб., 1973, т. 39, №2, с. 160—163.
230. Васильев А. Ф., Арюткина Н. Д. — Зав. лаб., 1977, т. 43, № 11, с. 1330— 1338.
231. Манжелий Л. С. О некоторых вопросах многокомпонентной спектрофотометрии. Черкассы: НИИТЭХИМ, Деп. № 650/75 деп., 1984, 29 с.
232. Пилипенко А. Т., Савранский Л. И., Масько А. Н. —ЖАХ, 1983, т. 38, № 8, с. 1455—1462; 1985, т. 40. № 2, с. 232—236.
233. Sustek J., Schiessl О. —Chem Zvesti, 1975, v. 29, № 1, p. 24—27
234. Conncs K. A., Eboka J. Analyt. Chem., 1979, v. 51, № 8, p. 1262—1266.
235. Бронштейн И. H., Семендяев К. А. Справочник по математике для инженеров и учащихся втузов. М.- Наука, 1965, 608 с.
236. Нестеренко II. И., Иванов В. М. —ЖАХ, 1983, т. 38, № 8, с. 1415—1421.
237. Chilton J. М. — Analyt. Chem., 1953, v. 25, № 8, р. 1274—1279.
238. Nukhedkai A J., Deshpande N. V. — Ibid., 1963, v. 35, Ks 1, p. 47—48.
239. Бабко А. К., Волкова A. M. — Зав. лаб., 1952, т. 18, № 5, с. 518—523.
240. Перьков И. Г., Дрозд А. В. — Вести. Харьк. ун-та, 1983, № 242, с. 33— 37.
241. Перьков И. Г., Шевцов Н. И. — Вести. Харьк. ун-та, 1974, № 115, Химия, вып. 5, с. 80—85.
242. Булатов М. И. В кн.: Краткие сообщения НТК ЛТИ им. Ленсовета. Л.: ЛТИ им. Ленсовета, 1969, с 39.
243. Каминский Ю. А., Берштейн И. Д., Гинзбург О. Ф. —ДАН СССР, 1962, т. 145, Кв 2, с. 330—331.
244. Porro Т. J. — Analyt. Chem., 1972, v. 44, № 4, р. 93А—ЮЗА.
245. Shibata Shozo, Furukawa Masamichi, Honkatza Tabashi — Analyt. Chem. Acta 1975, v. 78, № 2, p. 487—491.
246. Ratzlaff K- L., Natusch D. F. S. — Analyt. Chem., 1977, v. 49, p. 2170— 2176; 1979, v. 51, № 8, p. 1209—1217.
247. Ratzlaff К L., Darus H. — Analyt. Chem., 1979, v. 51, № 2, p.2E6—261.
248 Watanabe H., Ohmori H. — Taianta, 1979, v. 26, №, 10, p. 959—961.
249. Лазарев А. И. — Зав. лаб., 1983, т. 49, № 1, с. 3—7.
250. Дубровкин И. М. — ЖПС, 1983, т. 39, № 6, с. 885—899.
251. Дубровкин И. М., Беликов В. Г. Обзорная инф. НИИ ТЭИ. Общеотрасл. вопр. развития хим. пром-сти, 1981, № 12, 42 с.
252. Дубровкин И. М., Сагадссв Р. С., Соболев А. С. —Зав. лаб., 1978, т. 44, № 6 с. 685—686.
253. O'Haver Т. С. — Analyt. Chem., 1979, v. 51, № 1, р. 91А—100А; Analyt. Proc., 1982, v. 19, № 1, p. 22—28.
254. Cottrell С. T. — Ibid., 1982, v. 19, Ks I, p. 43—45.
255. Кварацхели IO. К., Демин IO B.—ЖАХ, 1983, t. 38, № 8, c. 1427—1433.
256. Демин Ю. В., Кукушкин Г. Р. —ЖАХ, 1981, т. 36, № 10, с. 2054—2058.
257. Перфильев В. А., Мищенко В. Г., Полуэктов Н. С. —ЖАХ, 1984, т. 39, № 12, с. 2132—2133.
258. Пирютко М. М., Бибаина Т. В., Доронина Л. А.—Зав. лаб., 1983, т. 49, Кв 3, с. 27—28.
259. Шкаравский Ю. Ф., Лынчак К. А., Черногоренко В. В. —Зав. лаб., 1983, т. 49, Кв 2, с. 1- -3
260 Василевская А. Е., Горда И. И. — Зав. лаб., 1980, т. 46, Кв 10, с. 888— 889.
261. Кършев Ив., Попов С. — Годишн. Висш. хим.-техиол. ин-т. София, 1977 (1978), т. 23, Кв 1, с. 39—48.
262. Sarkar R. С., Das М. S. — Anal. Chim. Acta, 1982, v. 134, p. 401—405.
263. Бешикдашьян M. T., Васильева М. Г. — ЖАХ, 1981, т. 36, Кв 6, с. 1082— 1084.
264. Филимонова Г. П. — Труды ВНИИ золота и редких металлов, 1977, т. 36, с. 149—155.
418
265. Смирнова Г. М., Столяров К. П., Харцизов А. Д. — В кн.: Физико-химические методы анализа. Горький, 1981, с. 25—27.
266. Бугаевский А. А., Каминская В. А., Кравченко В. А., Науменко В. А. — Вести. Харьк. ун-та, 1983, № 242, с. 37—40.
267. Булатов М. И. — В кн.: Тезисы докладов научно-техн. конф. ЛТИ им. Ленсовета. Л.. Химия, 1964, с. 68.
268. Новоселова М. М., Голинова Е. В., Чернобежский Ю. М., Антонович В. П. — ЖАХ, 1981, т. 36, № 5, с. 914—919.
269. Тананайко М. М., Пилипенко А. Т. — 1977, т. 32, № 3, с. 430—
436.
270. Пилипенко А. Т., Тананайко М. М. — ЖАХ, 1973, № 4, т. 28, с. 745— 778.
271. Чернова Р. К. — ЖАХ, 1977, т. 32, № 8, с. 1477—1477; 1978, т. 33, № 10, с. 1934—1939.
272. Назаренко А. Ю., Тананайко М. М., Тодрадзе Г. А. —ДАН УССР, 1983, Б, № 11, с. 50—52.
273. Булатов М. И., Муховикова Н. П. —Изв. вузов. Сер. хим. и хим. технол., 1980, г. 23, № 11, с. 1368—1370.
274. Umland F., Thiering D. —Z. anal. Chem., 1963, Bd. 197, № 3, p. 151— 160.
275. Блюм И. А., Душина T. К., Семенова Т. В. и др. —Зав. лаб., 1961, т. 27, № G, с. 644—650.
276. Савостьянова М. В. — В кн.: Труды комиссии по аналитической химии, М.: Изд. АН СССР, 1958. Т. 8 (10), с 3—20.
277. Бабко А. К., Вдовенко М. Е., Копа М. В. — Зав. лаб., 1963, т. 29, № 6, с. 645—649.
278. Бабко А. К., Вдовенко М. Е. — Укр. хим. ж., 1966, т. 32, № 2, с. 209— 212.
279. Headridge J. В. Photometric Titrations. Oxford: Pergamon Press, 1961. 131 p.
280. Underwood A. L. — In: Advances in Analytical Chemistry and Instrumen-tation/Ed. Ch. N. Reilley. N Y. e. a.: Interscience Publ. J. Wiley, 1964. v. 3, p. 31—104.
281. Leonard M. A. — In: Comprehensive Analytical Chemistry/Ed. G. Svehla. Amsterdam: Elsevier, 1977. V. 8, p. 207—389.
282. Kotrbi S., Mach V., Riha V. e. a. — Sb. ved pr. VSCHT Pardubice, 1976, v. 35, p. 9—27.
283. Kotrly S., Vachla J. — Collect. Czech. Chem. Communs., 1972, v. 37, № 2, p. 550—559.
284. Kotrly S. — Mikrochim. Acta, 1964, H. 2—4, p. 407—413.
285. Fortuin J. M., Karsten P., Kies H. L. — Anal. Chim. Acta, 1954, v. 10, № 4, p. 356—372.
286 Flaschka H. F., Ganchoff J.—Taianta, 1961, v. 8, № 12, p. 885—891; 1962, v. 9, № 1, p. 76—79.
287 Flaschka H. A., Paschal D. C. — Microchim. J., 1975, v. 20, № 1, p. 70.
288. Flaschka H. F., Sawyer P. — Taianta, 1962, v. 9, № 3, p. 249—263.
289. Ringbom A., Eklund B., Bergman L. — In Proc. Intern. Symp. Microchem. Technol/Ed. Cheronis N. D. N. Y. — London' Interscience publ. 1962, p. 797.
290. Булатов M. И.—Изв. вузов. Химия и хим. технол. 1974, т. 17, № 5, с. 670— 672.
291. Axel J. Anal. Chim. Acta, 1972, v. 61, № 2, p. 285—296.
292. Smit W., Stein H. N. — Anal. Chim. Acta, 1976, v. 83, may, p. 297— 307.
293. Sato H. — Ibid., 1978, v. 96, № 1 p. 215—219.
294. Scott R. Gocde — Anal. Chern., 1977, v. 49, № 9, p. 1408—1413.
295 Kragten J. — Taianta, 1973, v. 20, № 10, p. 937—946.
296. Jongkind ll7., Eelderlnk G. H. В , Verbruggen H. B. et. al. — Z. anal. Chem., 1977, v. 226, № 1—2, p. 76—79.
297. Дцимирский К. Б., Алексеева И. И. — ЖАХ, 1962, т. 17, К» 5, с. 574— 577.
419
298/ Абрамов В. В. — В кнл Контроль в производстве стекла. Иваново, 1975, с. 5—12.
299. Нечипоренко А. П., Калинкин И. П., Вентов Н. Г,, Алесковский В. Б. — ЖПХ, 1970, т. 43, № 11, с. 2558—2561; Изв. вузов. Химия и хнм. технол., 1969, т. 12, № 12, с. 1645—1648.
300 Тихонов В. Н. — ЖАХ, 1972, т. 27, № 8, с. 1485—1491.
301 Кузнецова В. В., Саморукова О. Л. — Зав. лаб., 1979, т. 45, № 2, с. 97— 99
302. Rinehart R. W., Stafford J. Е. — Microchem. J., 1962, v. 6, № 4, p. 567— 574.
303. Higuchi T„ Rehm C., Bernstein C. — Analyt. Chem., 1956, v. 28, № 10, p. 1506—1510.
304. Connors K. A., Higuchi T. — Ibid., 1960, v. 32, № 1, p. 93—96.
305 Bruckenstein S., Nelson D. C. — Ibid., 1961, v. 33, № 3, p. 438— 444.
306. Connors K. A. —Anal Chim. Acta, 1961, v. 25, № 6, p. 509—512.
307. Bruckenstein S., Gracias M. — Analyt. Chem., 1962, v. 34, № 8, p. 975— 980.
308. Higuchi T., Barnstein Ch H., Chassemi H. G., Reree W. E.— Ibid., № 3, p. 400—403.
309. Lalancette R A., KoubekK- G.—Microchem. J., 1972, v. 17, № 1, p. 72—74.
310. Stur J., Marek N. — Hung. Sci. Intsrum., 1973, May, p. 55—58.
311. Underwood A. L., Robertson T. M. —Analyt. Chem., 1963, v. 35, № 11, p. 1761—1762
312. Haartman Ch. — Taianta, 1980, v. 27, № 2, p. 217—221.
313. Bishop E — Analyst, 1978, v. 103, № 1222, p. 93—100.
314. Hulden S.-G., Harju L. —Taianta, 1980, v. 27, № 10, p. 815—817.
315. Wilhiete R. N., Underwood A. L. — Analyt. Chem., 1954, v. 27, № 8, p. 1334—1336.
316. Scaife D. B. — Analyst, 1963, v. 88, № 1049, p. 618—621.
317. AspilaK- T., Chakrabarti C. L., Sastri V. S. — Can J. Spectrosc. 1972, v. 17, № 5, p. 125—127.
318. Столяров К. П. Новый метод микрофотометрического титрования веществ в ультрафиолетовых лучах. Л.: ЛДНТП, 1963. 29 с
319. Мартынов В. И. Приставка для фотометрического титрования, А. с. 983536 СССР. Заявл. 12.02.81, № 3247708/25—26, опубл, в Б. И., 1982, № 47. МКИ G 01 № 31/16.
320. Oort W. J. van, Schalkwijk Р. С., Brandenburg R. A., Griepink В. — Z. anal. Chem., 1975, Bd. 276, № 3, S. 177—180; Ibid., S. 181 — 187.
321. Hanisch G., Kaden Th. A., ZuberbUhler A. D. — Taianta, 1979, v. 26, № 7, p. 563—567
322 Jongkind W., Eelderink G. H. B., Verbruggen H. B. e. a., Z. anal. Chem., 1977, Bd. 286, № 1—2, p. 72—75.
323. Комарь H. П. — Уч. зап. ХГУ, т. 37. Труды хим. ф-та и НИИ химии, 1951, т. 8, с. 37—41.
324. Комарь Н. П. — ЖАХ, 1950, т. 5, № 3, с. 139—144; Уч. зап. ХГУ, т. 37. Труды хим. ф-та и НИИ химии, 1951, т. 8, с. 43—49, 51—56.
325. Толмачев В. Н. — Уч. зап. ХГУ, т. 54. Труды хим. ф-та и НИИ химии, 1954, т 12, с. 99—106; Уч. зап. ХГУ, т.'37. Труды хим. ф-та и НИИ химии, 1951, т. 8, с. 65—69.
326. Колычев В Б., Парамонова В. И. — В кн.: Спектроскопические методы в химии комплексных соединений/Под ред. В. М. Вдовенко. М.: «Химия», 1964, с. 30—73.
327. Бьеррум Л- Образование амминов металлов в водном растворе/Пер. с англ, под ред. И. В. Тананаева. М.: ИЛ, 1961. 308 с.
328. Яцимирский К. Б., Васильев В. П Константы нестойкости комплексных соединений. Мл Изд. АН СССР, 1959. 206 с.
329. Шлефер Г. Л. Комплексообразование в растворах. Методы определенья состава и констант устойчивости комплексных соединений в растворах/Пер. с нем. под ред. А. А. Гринберга. М.—Л.: Химия, 1964. 379 с.
420
330. Romain P., Colleter J. — Bull. Soc. chim. Prance, 1959, № 10, р 155&— 1559.
331. Newman L., Hume D. — J. Am. Chem. Soc., 1957, v. 79, № 17, p. 4571— 4576, 4581—4585.
332. Poccomrnu Ф., Poccommu X. Определение констант устойчивости и других констант равновесия в растворах/Пер. с англ, под ред. Д. И. Рябчикова. М.: Мир, 1965, 564 с.
333. Комарь Н. П. — Уч. зап. ХГУ, т. 37. Труды хим. ф-та и НИИ химии, 1951, т 8, с. 37—41, 43—49, 51—69; Уч. зап. ХГУ, т. 54. Труды хим. ф-та и НИИ химии, 1954, т. 12, с. 5—12, 13—45, 53—75; Уч. зап. ХГУ, т. 71. Труды хим. ф-та и НИИ химии, 1956, т. 14, с. 119—128; Уч зап. ХГУ, т. 133. Труды хим. ф-та и НИИ химии, 1963, т. 19, с. 9—16; ЖАХ, 1950, т. 5, № 3, с. 139—144
334. Бабко А. К. — Зап. Ин-та химии АН УССР, 1946, т. 8, вып. 2, с. 3— 13; ЖОХ, 1945, т. 15, № 9—10, с. 743—757; 1946, т. 16, № 10, с. 1549— 1560, 1947, т. 17, № 4, с. 443—449; № 4, с. 642—648; 1948, т. 18, № 5, с 816—822.
335. Бабко А. К., Кодснская В. С. —ЖОХ, 1947, т. 17, № 6, с. 1080—1087; Заг лаб., 1950, т. 16, № 6, с. 643—648.
336. Бабко А. К., Драко О Ф —ЖОХ, 1949, т. 19, № 10, с. 1809—1815. 337. Бабко А. К-, Васильева Е. В. — ЖАХ, 1947, т. 2, № 3, с. 159—166.
338. Степанов Н. И. — Изв. АН СССР, Сер. хим., 1936, № 2, с. 219—253. 339. Адамович Л. П. — ЖНХ, 1959, т. 4, № 7, с. 1552—1557.
340. Комарь Н. П. — Там же, 1956, т. 1, К» 6, с. 1243—1251.
341 Бабко А. К., Пилипенко А. Т. — ЖАХ, 1947, т. 2, Ns 1, с. 33—42.
342. Пилипенко А. Т — Там же, 1950, т. 5, № 1, с. 14—20
343. Комарь Н. П. — Уч. зап. ХГУ, т. 71. Труды хим. ф-та и НИИ химии,
1956, т. 14, с. 119—128; ЖНХ, 1957, т. 2, Ns 5, с. 1015—1024.
344. Specker Н., Gremer М. — Z. anal. Chem., 1959, Bd. 167, Н. 2, S. 110—114.
345. Толмачев В. Н. — Уч. зап. ХГУ, т. 54. Труды хим. ф-та и НИИ химии, т. 12, с. 83—87.
346. Салихов В. Д., Ямпольский Л1. 3 — ЖАХ, 1965, т. 20, Ns 12, с. 1299— 1305.
347. Комарь И. П., Толмачев В Н., Новик Г. Ю-— Уч. зап. ХГУ, т. 37.
Труды хим. ф-та и НИИ химии, 1951, т. 8, с. 81—86.
348. Толмачев В. И. — Уч. зап. ХГУ, т 54. Труды хим. ф-та и НИИ химии,
1954. т. 12, с. 77—81
349. Толмачев В. Н. — Там же, с. 95—98.
350. Asmus Е., Bull A., Wolhdorf F. - Z. anal. Chem., 1963, Bd. 193, H. 2, S 81—85.
351. Harvey A., Manning D. —J. Am. Chem. Soc., 1950, v. 72, Ns 10, p. 4488—4493
352. Адамович Л. П. — Уч. зап. ХГУ, т. 54. Труды хим. ф-та и НИИ химии, 1954, т. 12, с. 107—111
353. Asmus E.—Z. anal. Chem., 1960, Bd. 178, H. 2, S. 104—116.
354. Klauscn К S., Langmuhr F J. — Anal. chim. acta, 1963, v. 28, Ns 6, p. 501—505.
355. Bent H., French C. — J. Am. Chem. Soc., 1941, v. 63, Ns 2, p. 569—572. 356. Толмачев В. H. — Уч. зап. ХГУ, т. 54. Труды хим. ф-та и НИИ химии, 1954, т. 12, с. 89-91
357. Адамович Л. П. — Там же, с. 131 —135.
358. Барбанель Ю. А. — ЖНХ, 1964, т. 9, № 2, с. 437—446.
359 Адамович Л. П. — Там же, 1960, т. 5, As 4, с. 782—790; 1961, т. 6, №6, с 1267—1271.
360. Шевченко Ф. Д. — Укр. хим. ж., 1965, т. 31, № 2, с. 229—232.
361 Klotz /. М., Loh Ming W. С. — J. Am. Chem. Soc., 1953, v. 7E, № 17, p. 4159—4162.
362. Nach С. P. — J. Phys. Chem., 1960, v. 64, p. 950.
363. Frank H., Oswald R. — J. Am. Chem. Soc., 1947, v. 69, Ns 6, p. 1321— 1325.
421 •
364. Heller J, Schwa, r-enbach G. — Helv chim. acta, 1951, v. 34. № 225, p. 1876—1889.
365. Бабке А. К. — ЖНХ, 1959, t. 4, № 5, c. 1055—1059.
366. Астахов Д. В., Вериникин В. Б., Зимин В. и др. — Там же, 1961, т. 6, № 9, с. 2069—2076.
367. Галактионов Ю. П., Астахов К. В. — Там же, 1963, т. 8, № 4, с. 896— 994.
368. Чудинов Э. Г. — ЖАХ, 1965, т. 20, Л» 8, с. 805—811.
369. Пешкова В. М., Громова М. И. Практическое руководство по спектрофотометрии и колориметрии. М. Изд. МГУ, 1965. 228 с.
370. Никольский Б. П., Пальчевский В. В. — В кн.: Спектроскопические методы в химии комплексных соединений/Под ред. В. М. Вдовенко. М.: 1964, с. 74—101
371. Бусев А. И., Типцова В. Г. —ЖАХ, 1960, т. 15, № 5, с. 573—580.
372. Комарь Н. П. — Уч.-зап. ХГУ, т. 37. Труды хим. ф-та и НИИ химии. 195:, т. 8, с. 57—60.
373. Толмачев В. Н., Подольская Г. Н., Серпухова Л. Н. — ЖНХ, 1957, т. 2, № 9, с. 2073—2077
374. Комарь Н П., Толмачев В. Н. — Уч. зап. ХГУ, т. 37 Труды хим. ф-та и НИИ химии, 1951, т. 8, с. 95—102
375. Булатов М. И — ЖНХ, 1968, i. 13, № 11, с. 3002—3008.
376. Бусс А. И., Чжань Фань — ЖАХ, 1961, т. 16, с. 1308.
377. Налимов В. В. Применение математической статистики при анализе вещества. М. Физматгиз, 1960 431 с.
378. Зайдель А Н. Элементарные оценки ошибок измерений. Изд. 2-е. Мл Наука, 1974. 108 с.
379. Беляков А. А. Метрологическое обеспечение результатов анализа Саи.-хим. методы определения вредных веществ Мл 1981, 51 с.
380. Методы обработки результатов наблюдений при измерениях: Труды метрологических институтов СССР/Под ред К. П Широкова М.-Лл Изд-во Стандартов, 1972. Вып 134 (194). 117 с.
381. Спиридонов В. П., Лопаткин А. А Математическая обработка физико-химических данных. М.. Изд-во МГУ, 1970, 221 с.
382. Снесарев К- А., Зараковская Л. И., Воробьева М. Т. Метрологические основы аналитического контроля химических производств. М.-Л.: Гослес-бумиздат, 1960. 206 с.
383. Поллард Дж. Справочник по вычислительным методам статистики. Мл Финансы и статистика, 1982. 344 с.
384. Исмаилов Ш. Ю. Математическая статистика и обработка результатов измерений. Мл ЛЭТИ, 1977. 60 с.
385. Алексеев Р. И., Коровин Ю. И Руководство по вычислению и обработке результатов количественного анализа. М. Атомиздат, 19Z2. 72 с.
386. Румшинский Л. 3., Смирнов С. Н. Методы обработки результатов экспериментов Мл МИСиС, 1973. 162 с.
387 Мокулов Н А. — Зав лаб., 1977, т. 43, № 12, с. 1457—1464, В кнл Прикладная спектроскопия. Минск, 1974, с 277—290.
388. Львовский Е. Н. Статистические методы построения эмпирических формул. Мл Высшая школа, 1982. 224 с.
389. Бланк А. Б., Бугаевский А А. — ЖАХ, 1969, т. 24, К» 9, с. 1445; 1970, т. 25, № 1, с. 11-17.
390. Пирятин В. Д Обработка результатов экспериментальных измерений по способу наименьших квадратов Харьков: Изд ХГУ, 1962. 216 с.
391 Айвазян С. А. Статистическое исследование зависимостей М-. Металлургия, 1968. 227 с.; Зав лаб. 1964, т. 30, № 7, с. 832—851, там же, Ks 8, с. 973—996.
392. Прикладная статистика: Исследование зависимостей: Справочиик/Под ред. С. А. Айвязяна. Мл Финансы и статистика 1985. 487 с.
393. Mandel Linnig F. J. — Anal. Chem., 1957, v. 29, Ks 5, p. 743—749.
394. Поснов H. H., Поснова M. Ф. Микрокалькуляторы «Электроника БЗ» в учебной лаборатории. Минск: БГУ, 1981. 190 с.
422
395. Алимов В. И. Вычисления на калькуляторе «Электроника БЗ-34». Томск-ТЛИ, 1984. 95 с.
396. Дьяконов В. П. Справочник по расчетам на микрокалькуляторах. М.: Наука, 1985. 224 с.
397. Трохименко Д. К., Любич Ф. Д. Инженерные расчеты на программируемых микрокалькуляторах. Киев: Техшка, 1985. 328 с.; Изв. вузов. Радиоэлектроника, 1984, т. 27, № 11, с. 60—66; № 3, с. 43—50; 1983, т. 26, № 7, с. 50—55; № 6, с. 68—74.
398. Бочков А. Ф. — Химия и жизнь, 1984, № 10, с. 60—63.
399. Славин Г. Программирование на программируемых микрокалькуляторах типа «Электроника БЗ-34». Таллии: Валгус, 1984. 129 с.; Прикладные программы для микрокалькулятора «Электроника БЗ-34» Тарту: Тартуский ун-т, 1983. 88 с.
400. Цветков А. Н. Сглаживание экспериментальных зависимостей с помощью микрокалькулятора «Электроника БЗ-34». М.: ИРЭ АН СССР, 1982. Препринт № 20 (347). 30 с.; Прикладные программы для микро-ЭВМ «Электроника БЗ-21». М.: Финанасы и статистика, 1982. 128 с. Прикладные программы для микрокалькулятора «Электроника БЗ-21». М.: ИРЭ АН СССР, 1979. Препринт № 19 (275). 46 с.
401. Цветков А. Н., Епанечников В. А. Прикладные программы для микрокалькулятора «Электроника БЗ-34». М.: ИРЭ АН СССР, 1982. Препринт № 19 (346). 47 с.; Прикладные программы для микро-ЭВМ «Электроника БЗ-34», «Электроника МК-56», «Электроника МК-44». М.: Финансы и статистика, 1984. 175 с.
402. Нейман Е. Д., Каплан Б. Д. —ЖАХ, 1978, т.ЗЗ, № 3, с. 607.
403. Михайлов В. А. —ЖАХ, 1977, т. 32, Ns 6, с. 1260—1262.
404. Адамович Л. П. Рациональные приемы составления аналитических прописей. Харьков: ХГУ, 1973. 96 с.
405. Комарь Н. П Химическая метрология. Гомогенные ионные равновесия. Харьков: Вища школа, 1983. 208 с.
406. Батлер Дж. Ионные равновесия (математическое описание)/Пер. с англ, под ред. А. А. Пендина. Л.: Химия, 1973, 448 с.
407. Мустафин И. С., Матвеев Л. Н. — Зав. лаб., 1958, т. 24, № 3, с. 259.
408. Бланк А. Б., Булгакова А. М.—ЖАХ, 1960, т. 15, с. 605—609.
409. Столяров К. П- Химический анализ в ультрафиолетовых лучах. Л.: Химия, 1965, 176 с.
410. Inczedy J. — Taianta, 1970, v. 17, р. 1212—1223.
411. Inczedy J., Varga-Puchony Z.—Acta Chim. Hung , 1973, v. 79, p. 63—69.
412. Мальцев В. Ф., Лукьяненко Л. И., Кукуй Д. М. — Зав. лаб., 1961, т. 27, № 7, с. 807—808.
413. Швайгер М. Н., Паклина В. П., Медведева А. С. — Зав. лаб., 1958, т. 24, № 1, с. 16—17.
414. Лазарев А. И., Лазарева В. И. — Зав. лаб., 1958, т. 24, № 7, с. 798.
415. Горюшина В. Г., Романова Е. В., Арчакова Т. А. — Зав. лаб., 1961, т. 27, № 7, с. 795-797.
416. Лазарев А. И., Лазарева В. И. — Зав. лаб., 1958, т. 24, № 2, с. 145.
417 Малютина Т. М., Добкина Б. М. — Зав. лаб., 1961, т. 27, № 6, с. 650—652.
418. Тихонов В Н., Рычкова Н. Д. —ЖАХ, 1963, т. 18, № 9, с. 1131—1133.
419. Барковский В. Ф, Вторыгина И. Н. — Зав. лаб., 1962, т. 28, № 3, с. 275—277.
420. Барковский В. Ф., Вторыгина И. Н. — ЖАХ, 1962, т. 17, Ns 7, с. 865.
421. Анализ фосфорсодержащих продуктов: Метод указания. Л.: ЛТИ, 1982, 28 с.
422. Булатов М. И., Муховикова Н. П. —ЖАХ, 1985, т. 40, Ns 11, с. 1956.
423. Коваленко Е. В. Автореф. каид. дисс., Новочеркасск, НПИ, 1964.
424. Бланк А Б., Булгакова А, М., Сизоненко Н. Т.—ЖАХ, 1961, т. 16, № 6, с. 715—719.
425. Амшсева А. А.—ЖАХ, 1978, т. 33, Ns 6, с. 1054—1061.
предметный указатель
Адамовича метод 264
Аддитивности правило 39
Аппаратура 156 сл.
Асмуса метод 250
Бабко методы 260, 267, 271
Бента—Френча метод 252
Бера закон 32
Бугера—Ламберта—Бера закон 33 отклонения 35, 330
Внутрикомплексные соединения 106 сл., 136
Воспроизводимость
абсолютных фотометрических методов 76
дифференциального фотометрического анализа 85
методик анализа 327 результатов измерений 303
Гарвея—Меннинга метод 249
Градуировочный график 33, 175 определение концентрации 175, 188, 189
расчет параметров 312
Дисперсия 303, 311, 314, 321
Дитизон 105, 106
Дитизонаты 105, 106
Доверительный интервал 307, 311, 315, 316
Диэтилдитиокарбамината 106, 107
Диэтилдитиокарбаминовая кислота 106, 107
Задачи 238, 239, 288, 289, 386—392
Изобестическая точка 50
Изомолярная серия 241
Клотца метод 269
Комаря методы 261, 282
Комплексы металлов
галогенидные 135 сл.
графические приемы исследования 276 сл.
диссоциация 22 сл.
комплексы металлов
методы исследования 240—280 образование, влияние
pH 25
сопряженных равновесий 23—28
разложение во времени 23 разнолигандные 14—20, 135—137 роданидные 15, 16, 22, 140, 144 с анионами кислот 25, 26 смешанные 14—20, 135—137
состав
влияние pH 25—29 определение 240—272, 276—280 расчет 284—287
тройные 14—20, 135—137 устойчивость 14—18
хелатные 105—110, 115—140
экстракция 105—110, 135—140, 210—214, 376—384
Константы
диссоциации 22 сл. концентрационные 14—18 сл.
нестойкости 14
образования 14—18 сл.
определение 255—272, 280—287, 290—300
протонирования 17, 20 сл.
распределения 112 сл. сопропорционирования 14 ступенчатые 17 сл.
термодинамические 14 условные 16—20, 396 устойчивости 14—20, 255, 272 экстракции 114 сл.
Контрастность реакций 46, 96 Концентрация
ионов водорода 20, 25 сл.
маскирующие реагенты 143—146, 341
методы определения 173—230 минимальная определяемая 55—59 определяемая, интервал 61 равновесная 19, 20
фотометрического реагента 30 сл.
экстракционного реагента 114—
116
Коэффициент концентрирования 133 сл. разделения 133 сл.
424
Коэффициент
распределения 4, 113 сл.
светопоглощения молярный 35 методы определения 255—280 расчет 284—287
селективности 94
Сейдела 58
чувствительности 53 сл.
Кривые фотометрического титрования 217—221, 226
Кюветы 74, 163, 164
Маскирование ионов 143—147 Маскирующие реагенты
выбор 144, 398—408
расчет необходимой концентрации 143—147, 341 с л.
Методы
исследования
графические 270 сл.
изобестической точки 243
изомолярных серий 241, 276 металл-индикаторный 271 молярных отношений («насыще-
ния») 245
непрерывных изменений 241 ограниченно-логарифмический
252
относительного выхода 253
отношения наклонов 249
пересечения кривых 266
прямой линий 250
разбавления 267
сдвига равновесия 244, 277
титриметрический 247
определения концеитрацин абсолютные фотометрические 76,
173, 193
анализа двух- и многокомпонентных систем 181
градуировочного графика 175, 355
дифференциальные 85, 200, 361
добавок 208, 360
прямые и косвенные 12 сл.
сравнения 173
экстракционно-фотометрический 210—214
разделения и концентрирования отгонкой 141
осаждения и соосаждения 138
сорбционные 140
экстракционные 134
Навеска пробы
взаимосвязь с пределом определения 51, 52
расчет оптимального значения 100
Нулевой раствор 157
Ньюмена и Хьюма метод 278
Окрашенные соединения малорастворимые 105—НО определение состава 240—290 определение констант устойчивости 255—272, 280—287, 290— 300
экстракция 112 сл.
Оксин (8-оксихинолин) 107 Оксихинолинаты 107
Оптическая плотность 33, 34 влияние разбавления 36 состава соединения 22 температуры 101 зависимость от pH 99 методы измерения 159, 164 погрешность измерений 76—92
Ошибка титрования 4, 218 1-(2-Пиридилазо)-2-нафтол (ПАН), свойства 106
Поглощение света зависимость от концентрации 33 молярный коэффициент 35 полоса, полуширина 42 спектр 40 фотометрическим реагентом 49
Погрешность(и)
грубые, исключение 304 измерения 78, 85 косвенных измерений (определений) 310
определения абсолютная 77, 85, 301
систематическая 77, 93, 309 случайная 76, 302
Посторонние ионы
влияние на определение 143—155 допустимые концентрации 94, 95 устранение влияния 132 маскирование*' 143 осаждением 138 сорбционными методами 140 специальными приемами 148 экстракцией 134
Правильность
результатов 306, 328
фотометрических методов 93
Практические работы исследования фотометрических реакций 289—300
определение концентрации элементов 354—386
Примеры расчетов 236—238, 284—287, 330—354
PH влияние на диссоциацию комплексов 25— 10 образование комплексов 26—30 оптическую плотность 99
425
pH
состав комплексов 25 сл.
выбор 11ри анализе 99
расчет оптт ального значения 27—30, 336—340
Растворители органические 105—111
Растворы сравнения 157
Расчет
градуировоцкого графика 312 констант устойчивости 286—289
условных 16—18, 295—299 констант экстракции 120—123
условных 123, 350—354 концентрации
равновесной 331 сл.
маскирующих реагентов 145, 341 фотометрического реагента
339 сл.
экстракционного реагента 115, 116
коэффициентов
распределения 112 сл светопоглощения 255—271, 284—287
нижней границы определяемых содержаний 69
определяемого минимума 324 оптимального значения pH 336 сл. отклонения от закона Бера 36—39, 330, 331
предела обнаружения 61—67, 317 предела определения 67, 68 состава соединения 284—287
степени извлечения 113 сл., 347— 354
фактора разделения 350—352 эквивалентного объема 233—238
Реагенты органические выбор 95 сл. индикаторные свойства 29 количество 30—31
расчет 339, 340
маскирующие 144, 145, 398—409 распределение 114—116 фотометрические 104—ПО экстракционные 104—ПО, 134—137
Светопоглощенне 31
молярный коэффициент 35
основной закон 33
отклонение 36
Светофильтры 47, 160, 164 Селективность 94
Смеси компонентов, анализ 181 Состав комплексов см. Комплексы Соосаждение с коллектором 138 Спектральная область, выбор 43, 96, 98, 182—192
Спектрофотометрический анализ 181—< 187, 190—200 абсолютный 173—186 дифференциальный 200 многокомпонентных смесей 182— 184
одного компонента 173—181 специальные приемы 149, 150— 152, 191
Спектрофотометрическое титрование 216—232
аппаратура 232
без индикатора 218
расчет количества вещества 233
с индикатором 224
Спектрофотометрия абсолютная 173—182 двухволновая 193 дифференциальная 200, 209 по спектрам отражения 215, 216 производная 197
Спектрофотометры 166, 168, 169
Спектры поглощения 40
Специальные приемы спектрофотометрического анализа 149—155, 190— 192
Стандартное отклонение 62, 69, 76, 86, 303, 304, 316
Старика и Барбанеля метод 253
Титрование см. Спектрофотометрическое титрование
Фотоколориметры 159—165
Фотометр Spekol 10 166 Фотометрический анализ аппаратура 159—172 воспроизводимость см. Воспроизводимость методы измерения оптической плотности 161, 165, 168 методы определения концентрации см. Концентрация общие условия 156 одновременное определение нескольких компонентов 182, 184, 186
оптимальные условия 43, 95 практические работы 355 сл. специальные приемы 149—155, 190—192
Форма представления результатов определения 101, 328
Фотометрическое определение элементов 354—386
алюминия
в медно-цинковых сплавах 355
в минеральных удобрениях 372 висмута в олове 357 железа с о-фенантролином 355
426
Фотометрическое определение элементов
в промышленных материалах 363
в удобрениях 375
с сульфосалиниловой кислотой 356
марганца в стали 365
молибдена в сталях и сплавах 358
никеля в стали 364
титана в промышленных материалах 361
титана и железа в шихте 385
фосфора в удобрениях 367, 370, 371
циркония в сплавах 360
Функция
закомплексованности 17, 332
образования 345, 346
распределения 17, 334—336
Характеристики
аналитические 51, 94
методики 103, 328
метрологические 51 сл.
реакции 103
свойств реагента 103
фотометрические 32—35
экстракционные 112—114
Холостой опыт 153—158
Цветная реакция, определяемый минимум 55, 324
Чувствительность 53
коэффициент 53
Чувствительность
методики 75, 76
реакции 54
фотометрических определений абсолютная 52, 55 определяемый минимум 56, 324 относительная 52, 55 увеличение 72
Экстракционно-фотометрический анализ 210—214
Экстракционно-фотометрическое определение элементов 376—384
кобальта в присутствии железа 379
меди с диэтилдитиокарбаминатом свинца 377
меди, никеля, железа и марганца в препаратах КО и КВ г 382
никеля в присутствии кадмия, марганца и цинка 376
хрома с дифенилкарбазидом 380 Экстракция
количественные характеристики 112 сл.
окрашенных соединений 112 сл„ распределение металла 116 распределения реагента 114 сл. хелатов 119 сл.
обменная 127 сл.
разделение 129 сл.
условия проведения 214, 347, 376
Яцимирского метод 257
ОГЛАВЛЕНИЕ
Предисловие к пятому изданию ......................................... 3
Принятые термины и обозначения........................................ 4
Введение.............................................................. 6
Часть I, ОБЩИЕ ПОЛОЖЕНИЯ И АППАРАТУРА ФОТОМЕТРИЧЕСКОГО АНАЛИЗА................................................... 12
Глава 1. Физико-химические свойства окрашенных соединений. ... 12
1.1. Прочность окрашенных соединений................................. 14
1 1.1. Условные константы устойчивости........ 16
1.1.2. Основные количественные зависимости...................... 18
1.2. Постоянство состава окрашенных соединений....................... 22
1.3. Влияние pH раствора на образование окрашенных соединений ... 25
1.4. Количество фотометрического реагента............................ 30
Глава 2. Оптические свойства окрашенных соединений в растворах. . . 31
2.1. Поглощение света растворами окрашенных соединений.............. 31
2.1.1. Основной закон светопоглощения........................... 33
2.1.2. Молярный коэффициент светопоглощения..................... 35
2.1.3. Отклонения от основного закона светопоглощения........ 35
2.1.4 Правило аддитивности...................................... 39
2.2. Электронные спектры поглощения.................................. 40
2.3. Выбор спектральной области для фотометрических измерений ... 43
2.4. Светофильтры ................................................... 47
Глава 3. Метрологические и аналитические характеристики фотометрического анализа. Выбор оптимальных условий фотометрического определения . ......... ...................................... 51
3.1. Метрологические характеристики фотометрического анализа .... 51
3.1.1. Чувствительность фотометрических определений............. 53
3.1 1.1. Коэффициент чувствительности...................... 53
3.1 1.2. Условные характеристики чувствительности фотометрического определения .................................... 54
3.1.2. Интервал определяемых содержаний......................... 61
3.1.2.1. Предел обнаружения и предел определения.......... 61
3.1.2.2. Нижняя граница определяемых содержаний. ... 68
3.1.2.3. Пути повышения чувствительности фотометрических реакций, снижения пределов обнаружения и нижней границы определяемых содержаний ................................... 72
3.1.3. Воспроизводимость фотометрических методов анализа. ... 76
3.1.3.1. Воспроизводимость абсолютных фотометрических методов анализа.............................................. 76
3.1.3.2. Воспроизводимость дифференциального фотометрического анализа.............................................. 85
428
3.1.4. Правильность фотометрических методов анализа. ..... 93
3.1.5. Селективность фотометрических методик анализа............ 94
3,2. Выбор оптимальных условий фотометрического определения.... 95
3.2.1. Выбор реагента........................................... 95
3.2.2. Определение оптимальных условий фотометрического анализа 97
3.2.3. Представление результатов фотометрических определений 101
3.2.4. Сравнение и выбор фотометрических методик определения 103
Глава 4. Органические реагенты и растворители и их применение в экстракции ... 104
4.1. Органические реагенты........................................... 104
4.2. Органические растворители....................................... ПО
4.3. Экстракция окрашенных соединений................................ 112
4.3.1 Количественные характеристики экстракционных равновесий 112
4.3.2. Распределение экстракционного реагента.................. 114
4.3.3. Распределение металлов ................................. 116
4.3.4. Экстракция хелатов .................................... 119
4.3.4.1. Экстракция внутрикомплексных соединений (незаряженных хелатов) ....................................... 119
4.3.4.2. Экстракция координационно ненасыщенных и заряженных хелатов ......................................... 124
4.3.4.3. Равновесия обмена при экстракции хелатов.... 127
4.3.4.4. Экстракционное разделение хелатов............... 129
Глава Б. Разделение, концентрирование и устранение мешающего влияния посторонних компонентов........................................ 131
5.1. Методы разделения и концентрирования.......................... 132
5.1.1. Общие положения........................................ 132
—>5.1.2. Экстракционные методы.................................. 134
5.1.3. Методы осаждения и соосаждения ........................ 138
5.1.4. Сорбционные методы..................................... 140
5.1.5. Краткое сопоставление методов концентрирования......... 142
5.2. Устранение влияния посторонних ионов химическими методами без их отделения....................................................... 143
5.2.1 Маскирование мешающих ионов............................. 143
5 2 2. Изменение степени окисления мешающего или определяемого иона ......................................................... 148
5.3. Устранение влияния мешающих веществ специальными приемами фотометрических измерений ......................................... 148
5.3.1 Фотометрирование растворов в узком интервале спектра. . . 148
5.3.2. Измерение оптических плотностей при двух длинах волн 149
5.3.3. Фотометрические приемы при линейной зависимости светопо-
глощения мешающих компонентов от длины волны ........ 150
5.4. Способы учета поглощения фона................................. 153
5.4.1. Определение микропримесей в анализируемых образцах и
в холостой пробе.............................................. 153
5.4.2. Анализ в присутствии смолообразных веществ или иефильтру-ющейся взвеси................................................. 154
Глава 6. Аппаратура и техника фотометрических измерений............. 156
6.1. Общие замечания при работе на фотоколориметрах................. 157
6.2. Фотоколориметры ............................................... 159
6.2.1. Колориметры фотоэлектрические КФК, ФЭК-56М............ 159
6.2.2. Колориметр фотоэлектрический концентрационный КФК-2 164
6.2.3. Многоцелевой фотометр Spekol 10......................... 166
•6,3. Спектрофотометры.............................................. 166
6.3.1. Спектрофотометры СФ-26, СФ-16........................... 166
6.3.2. Спектрофотометр СФ-46 .................................. 168
6.3.3. Спектрофотометры СФ-10, СФ-14, СФ-18............... * 169
429
Часть II. ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА .......................... 173
Глава 7. Фотометрические методы количественного анализа............ 173
7.1. Абсолютные фотометрические методы определения веществ в отсутствие мешающих компонентов....................................... 173
7.1.1. Методы определения одного вещества...................... 173
7.1.1.1. Метод сравнения оптических плотностей стандартного и исследуемого окрашенных растворов ..................... 173
7.1 1.2. Метод определения по среднему значению молярного коэффициента светопоглощения ....................... 174
7.1 1.3. Метод градуировочного графика................... 175
7.1.1.4. Метод добавок................................... 178
7.1.2. Анализ двух- и многокомпонентных окрашенных систем. . . 181
7.1.2.1. Спектрофотометрический анализ трехкомпонентных систем 182
7.1.2.2. Спектрофотометрический анализ двухкомпонентных систем .................................................. 184
7.1.2.3. Фотоколориметрический анализ двухкомпонентных систем ........ ... ....................... 186
7.1.2.4. Некоторые приемы, упрощающие анализ двух- и многокомпонентных систем.................................... 190
7.2. Абсолютные фотометрические методы анализа в присутствии мешающих компонентов................................................... 193
7.2.1. Двухволновая спектрофотометрия.......................... 193
7.2.2. Производная спектрофотометрия .......................... 197
7.3. Дифференциальные фотометрические методы анализа............... 200
7.3.1. Способы определения одного компонента................... 202
7.3.2. Дифференциальная спектрофотометрия многокомпонентных систем 209
7.4. Экстракционно-фотометрический метод........................... 210
7.4.1 Наиболее распространенные экстракционные системы. . . . 210
7.4.2. Условия селективного определения ....................... 214
7.5. Спектрофотометрический анализ по спектрам отражения........... 215
7.6. Спектрофотометрическое титрование ............................ 216
7.6.1. Сущность метода и кривые спектрофотометрического титрования . . . . 217
7.6.2. Спектрофотометрическое титрование без индикатора .... 218
7 6.3. Спектрофотометрическое титрование с индикатором..... 224
7.6.4. Факторы, влияющие на воспроизводимость и правильность спектрофотометрического титрования............................ 231
7.6.5 Расчет количества вещества по данным спектрофотометрического титрования .............................................. 233
7.6.6. Примеры и задачи........................................ 236
Глава 8. Спектрофотометрический анализ растворов окрашенных соединений 239
8.1. Определение состава комплексного соединения................... 240
8.1 1. Метод непрерывных изменений (метод изомолярных серий) 241
8.1.2. Метод сдвига равновесия ............................... 244
8.1.3. Метод молярных отношений (метод «насыщения»)............. 245
8.1.4. Титриметрический метод ... ......... 247
8.1.5. Метод изобестической точки............................... 248
8.1.6. Метод отношения наклонов (метод Гарвея—Меннинга). . . 249
8.1.7. Метод прямой линии (метод Асмуса)........................ 250
8.1.8. Ограниченно-логарифмический метод (метод Бента—Френча) 252
8.1.9. Метод относительного выхода (метод Старика и Барбаиеля) 253
430
8.2. Определение состава, прочности и молярных коэффициентов светопоглощения окрашенных соединений .................................. 255
8.2.1. Мэтод Яцимирского ....................................... 257
8.2.2. Использование изомолярной диаграммы и кривой насыщения 259
8.2.3. Метод Комаря....................................... ... 261
8.2.4. Метод Адамовича ......................................... 264
8 2.5. Метод пересечения кривых ............................ 266
8.2.6. Метод разбавления (метод Бабко).......................... 267
8.2.7. Способ Клотца............................................ 269
8.2.8. Графические приемы....................................... 270
8.2.9. Металл-индикаторный метод Бабко......................... 271
8.3. Оределение заряда комплексного иона............................ 272
8.4. Исследование смешанных (разнолигандных) комплексов............. 275
8.4.1. Метод изомолярных серий ................................. 276
8.4.2. Метод сдвига равновесия ................................. 277
8.4.3. Метод Ньюмена и Хьюма.................................... 278
8.5. Определение констант диссоциации органических реагентов .... 280
8.6. Расчет состава, констант устойчивости, констант диссоциации и мо-
лярных коэффициентов светопоглощения фотометрируемых соединений 284
8.6.1. Примеры и задачи......................................... 284
8.6.2. Практические работы...................................... 289
Часть III. РАСЧЕТЫ И ВЫЧИСЛЕНИЯ В ФОТОМЕТРИЧЕСКОМ АНАЛИЗЕ............................................................. 301
Глава 9. Краткие сведения по математической обработке экспериментальных данных............................................... .... 301
9.1. Расчет основных метрологических характеристик результатов прямых (непосредственных) равноточных измерений........................ 302
9.1.1. Оценка воспроизводимости результатов измерений. . . . 303
9.1.2. Определение и исключение грубых погрешностей (промахов) 304
9.1.3. Оценка правильности результатов измерений (определений) 306
9.2. Погрешности косвенных измерений................................ 310
9.3. Регрессионный анализ при расчете градуировочных графиков и метрологических характеристик результатов анализа...................... 312
9.3.1. Расчет уравнения линейного градуировочного графика, его метрологических характеристик и метрологических характеристик результатов анализа .......................................... 312
9.3.1.1. Вычисление метрологических характеристик линейного графика................................................... 312
9.3 1.2. Вычисление метрологических характеристик результатов анализа........................................... 315
9.3.1.3. Пример расчета................................. 318
9.3.2. Проверка значимости константы а.......................... 320
9.3.3. Проверка гипотезы линейности градуировочного графика 321
9.4. Расчет определяемого минимума цветной реакции.................. 324
9.5. Сопоставительная оценка воспроизводимости двух фотометрических методик анализа..................................................... 327
9.6. Общие рекомендации по метрологической оценке результатов анализа 328
Глава 10. Примеры расчетов и обоснований в фотометрическом анализе 330
10.1. Расчет отклонений от закона Бера при разбавлении окрашенных растворов........................................................... 330
10.2. Расчет равновесных концентраций комплексных ионов............. 331
431
10.3 Расчет оптимального значения pH при фотометрических определениях ... ................................................ 336
10.4. Расчет концентрации реагента, обеспечивающей необходимую полноту связывания определяемого элемента ........................... 339
10.5. Установление возможности применения маскирующих реагентов и анализа в присутствии мешающих ионов............................ 341
10 6 Обоснование возможности косвенного определения............. 346
10.7. Примеры расчетов и обоснований при экстракционном выделении и разделении элементов............................................ 347
Глава 11. Практические работы и задачи............................ 354
Приложения ...................................................... 393
Соотношение между величинами светопропускания и оптической плотности 39з Логарифмы констант устойчивости комплексонатов металлов 395
Логарифмы условных констант устойчивости комплексонатов металлов 396
Значения lg 1 «у для некоторых металлов с различными комплексантами 398 Значения 1g 1/аь для некоторых маскирующих реагентов.............. 408
Химические элементы . . 409
Библиографический список ......................................... 411
Предметный указатель.............. 424
ПРАКТИЧЕСКОЕ РУКОВОДСТВО
Михаил Ильич Булатов Игорь Петрович Калинкчн
ПРАКТИЧЕСКОЕ РУКОВОДСТВО ПО ФОТОМЕТРИЧЕСКИМ МЕТОДАМ АНАЛИЗА
Редактор Л. Ф. Травина
Техн, редактор Л. Ю. Коровенка
Корректор М. 3. Басина
Переплет художника Б. Н. Осенчакова
ИБ № 1815
Сдано в набор 18.02.86 Подписано к печати 25.07.86. М-33876.
Формат 60X90Vie. Бумага типографская № 2. Печать высокая.
Гарнитура литературная. Усл. печ. л 27.0. Усл. кр.-отт. 27.0.
Уч-изд. л 28,7. Тираж 15700 экз. Зак. 51- Цена 1 р. 80 к.
Изд № 2715
Издательство «Химия», Ленинградское отделение
191186, г. Ленинград, Д-186, Невский пр., 28
Ленинградская типография № 6 ордена Трудового Красного Знамени Ленинградского объединения «Техническая книга» им. Евгении Соколовой Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
193144, г. Ленинград, ул. Моисеенко, JD.