/








































































































































Text
АКАДЕМИЯ НАУК СССР
ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ
ИНСТУТИТ НЕФТЕХИМИЧЕСКОГО СИНТЕЗА ИМ. А.В. ТОПЧНЙА
Е. И. БАГРИ Й
Адамантаны получение, свойства, применение
Ответственный редактор доктор химических наук Е.Н. КАРАУЛОВА
МОСКВА ’’НАУКА” 1989
УДК 547.518
Адамантаны: Получение, свойства, применение / Е.И. Багрий. — М.: Наука, 1989.264 с. - ISBN 5-02-001382-х
В монографии рассмотрены получение, физико-химические и химические свойства, а также перспективы практического использования адамантанов, алкиладамалтанов и полициклических адамантаноидных углеводородов - наиболее изученных представителей углеводородов каркасного строения. Разработаны эффективные синтетические методы получения адамантана и алкиладамантанов, делающие эти соединения вполне доступными.
Книга предназначена для специалистов в области органической химии, а также для аспирантов и студентов вузов.
Табл. 40. Ил. 26. Библиогр.: 1235 назв.
Рецензенты:
профессор С.Л. Давыдова профессор Ал А. Петров
Редактор МЛ франк
Adamantanes: Production, properties, application / E.I. Bagrii. - M.: Nauka, 1989,000c.-ISBN 5-02-001382-x
The problems of production, physical and chemical properties and the perspectives of using dadmantane, alkyladamantanes and polycyclic adamantanoid hydrocarbons are considered m the book. These compounds are the most studied hydrocarbons offramework-tipe structure, their chemistry now is intensively developed. The effective synthetic methods of production of adamantane and alkyladamantanes have been developed.
The book is being recommended to the students of advanced courses of chemical and petrochemical profile institutes, research workers, postgraduates and engineering workers.
Tabl. 40. 11.26. Bibliogr.: 1235 ref.
1705000000-472 Б-----------------148-89, kh.2
055 (02)-89
© Е.И. Багрий, 1989
ISBN 5-02-001382-x
Предисловие
Решениями XXVII съезда КПСС, последующих пленумов ЦК КПСС предусматривается интенсивное развитие отраслей малотоннажной химии - получение химических веществ, способных в небольших количествах давать значительный народнохозяйственный эффект. В полной мере это относится к синтезу лекарственных препаратов, средн которых в последние годы успешное применение находят производные адамантана и других углеводородов каркасного строения.
К настоящему времени сотни производных адамантана уже испытаны на биологическую активность и подавляющее большинство из них показало положительный результат. Наиболее широко и всесторонне исследованы азотсодержащие производные адамантана. Хлоргидрат 1-аминоадамантана (препарат ’’мидантан”, ’’симметрел”) проявил широкий спектр противовирусной активности и активности против болезни Паркинсона. Этот препарат спас многие человеческие жизни во время эпидемии гонконгского гриппа в начале 70-х годов. В СССР в качестве лекарственного препарата используется хлоргидрат 1-амиио-(1-адамаитил)этаиа (ремантадин), обладающий высокой активностью в отношении вирусов гриппа серотипа А.
Другая перспективная область применения производных адамантана — получение полимерных материалов. Высокомолекулярные соединения содержащие циклы адамантана, характеризуются высокими значениями объемного модуля; они обладают большой твердостью, термостойкостью, высокими значениями температуры стеклования, устойчивостью по отношению к гидролизу, малой усадкой при формовании. Так, например, полиамид на основе адамантан-1,3-дикарбоиовой кислоты и л<-фенилендиамина имеет т.разм. 294°С; при нагревании его до 380° С не наблюдается потери массы. Волокна, полученные на основе этого полимера, не обнаруживали изменений при их выдерживании 50 ч при 200° С.
* В области теоретической химии исследования каркасных углеводородов и их производных позволяют расширить наши знания о природе активности и устойчивости карбкатионов, установить корреляции между их структурой и реакционной способностью, изучить механизмы реакций ионного и радикального замещения у насыщенного атома углерода, развить расчетные методы по определению энергии напряжения в углеводородах, изучить ориентационные процессы в молекулярных кристаллах.
Адамантан впервые был получен С. ЛандойиВ.Махачеком в 1933 г. при исследовании нефти Годонинского месторождения (Чехословакия). Впоследствии были разработаны синтетические методы получения адамантана путем изомеризации гидрированного димера циклопентадиена (П.Р. Шпейер, 1956 г.; А.Ф. Платэ с сотр., 1961 г.), а также получения алкиладаманта-нов изомеризацией трициклических пергидроаро магических углеводородов
3
над галогенидами алюминия (А. Шнайдер и соавт., 1964 г.) и над алюмо-окисными и алюмосиликатными катализаторами (Е.И. Багрий, П.И. Санин, 1968 г.).
В начальный период развитие экспериментальной и прикладной химии адамантана сдерживалось малой доступностью исходных соединений. Теперь адамантан и алкиладамантаны могут быть получены в достаточном количестве. Их получение базируется на дешевом и доступном углеводородном сырье — жидких продуктах и отходах переработки нефти и каменного угля, содержащих циклопентадиен, аценафтен, флуорен и другие непредельные и ароматические углеводороды. Практически все они после гидрирования и изомеризации могут быть превращены в углеводороды ряда адамантана. При изомеризации трициклических пергидроароматических углеводородов, в частности, образуется, как правило, смесь ал кил адамантанов. Поиск путей их рационального и эффективного использования представляет собой важную научно-техническую и народнохозяйственную задачу, являющуюся частью общей проблемы рационального использования углеводородов газа, нефти и угля.
В отличие от адамантана, имеющего высокую температуру плавления (269° С), алкиладамантаны представляют собой жидкости с низкими температурами застывания (для 1 Д-днметиладамантана —60°С), обладающие повышенной термической и антиокислительной стабильностью. Вместе с тем адамантан и алкиладамантаны проявляют повышенную по сравнению с другими насыщенными углеводородами реакционную способность, что позволяет их широко использовать в органическом синтезе. Некоторые реакции, например реакция ноииого бромирования адамантана, уже прн комнатной температуре протекают с высокой селективностью.
Важно отметить, что адамантан н алкиладамантаны содержатся в нефтях. Как показали исследователи, они образовались в результате каталитических превращений других углеводородов. Изучение закономерностей и особенностей изомерного состава и распределения алкиладамантанов в нефтях различных месторождений представляется важным для выяснения ряда вопросов генезиса, развития н миграции нефтей.
Задачу настоящей монографии автор видит прежде всего в приобщении широкого круга нефтехимиков и химиков-органиков к достижениям в области химии углеводородов каркасного строения, к появившимся широким возможностям их получения и практического использования. Химия адамантана наглядно демонстрирует возросшие возможности современной синтетической органической химии в отношении функционализации насыщенных углеводородов, в особенности полициклических насыщенных углеводородов, — перспективных соединений для основного органического н нефтехимического синтеза.
В основу монографии положены результаты исследований, проводившихся в течение ряда лет автором н сотрудниками лаборатории химии нефти Института нефтехимического синтеза нм. А.В. Топчиева АН СССР.
Автор признателен Т.Н. Долгополовой, Т.Ю. Фрид и Т.С. Бобруйской за помощь при подготовке рукописи к печати.
Автор заранее благодарен читателям, проявившим интерес к книге н приславшим свои замечания, отзывы н пожелания.
4
Глава I
Особенности строения и свойств адамантанов
1 Л. НОМЕНКЛАТУРА АДАМАНТАНА И ЕГО ПРОИЗВОДНЫХ
Среди каркасных углеводородов к настоящему времени наиболее изучены углеводороды ряда адамантана. К ним относят те углеводороды каркасного строения, которые содержат по меньшей мере один фрагмент адамантана. Адамантан - трицикло [3,3,1,13,7 ] декан - представляет собой трициклический насыщенный мостиковый углеводород состава CioHi6> состоящий из трех циклогексановых колец в конформации' ’’кресло”. Атомы углерода в молекуле адамантана расположены в такой же последовательности, как в кристаллической решетке алмаза (рнс. 1.1). Свое название адамантан получил от а5ада£с (непобедимый — греческое название алмаза), в связи с чем в литературе можно встретить для адамантаноидных углеводородов название ’’углеводороды алмазоподобного строения”.
Обычно используют такие изображения структурной формулы молекулы адамантана:
Приведенная нумерация атомов н название - трнцикло [3,3,1,13,7].декан — соответствуют правилу А -32 номенклатуры ИЮПАК [1,2]. В отсутствие заместителей четыре третичных (узловых) атома углерода (в положениях 1, 3, 5,7) эквивалентны. Также эквивалентны шесть вторичных (мостиковых) атомов углеводорода (в положениях 2, 4, 6, 8, 9, 10). При наличии одного заместителя возникают дна структурных изомера, например 1- и 2-пентиладамантаны. Если заместитель более сложный, название производного адамантана обычно строят с использованием наименования радикала — 1- или 2-адамантил, например 2-(1-адамантил)-2,6-днметилгеп-тан
СН, I
Н,С-С —(СПД-СН-СН,
В днзамещенных производных адамантана с одним мостиковым заместителем пространственная ориентация мостикового заместителя может быть
5
Рис. 1,1 Фрагмент кристаллической решетки алмаза
аксиальной (а) или экваториальной (е) в зависимости от расположения заместителя по отношению к плоскости общего для обоих заместителей циклогексанового кольца [3] или ее можно обозначить как цис- и транс-[4]:
СП3
1,4я -диметиладамантан {цис-)
Аналогично могуть быть обозначены и производные с несколькими мостиковыми заместителями, находящимися в одном циклогексановом кольце.
Для полнзамещенных производных адамантана с несколькими мостиковыми заместителями в разных циклогексановых кольцах каркаса в настоящее время не существует общепринятой номенклатуры. Трудность достоит в том, что в связи с особенностями строения молекулы, ее высокой симметрией углеродные атомы могут быть пронумерованы несколькими равноценными способами. Понятие аксиального или экваториального заместителя не может быть использовано, так как замещенная группа, будучи аксиальной в одном циклогексановом кольце молекулы, в то же время является экваториальной по отношению к другому, смежному циклогексановому кольцу.
Снатцке и Марквардннг [3] предприняли попытку расширить Л/.S’-номенклатуру применительно к мостиковым производным адамантана, однако использование предложенных ими правил затруднительно при наличии в соединении нескольких одинаковых мостиковых заместителей. Для обозначения таких производных адамантана могут быть рекомендованы предложенные автором работы [4] правила, вытекающие из родственной связи молекулы адамантана и бицикло [3,3,1] нонана. Задача сво-
6
дится к определению рационального фрагмента бицикло [3,3,1]нонана,
входящего в структуру производных адамантана.
1. В случае отсутствия узловых заместителей нумерацию атомов углерода проводит с учетом предпочтительности заместителя таким образом, чтобы более предпочтительный мостиковый заместитель имел меньший номер, а сумма номеров углеродных атомов была минимальной. При обозначении алкиладамантанов меньший номер получает более простой заместитель. Так, номера увеличиваются в ряду: СН3, C2Hs, н-С3Н7, «зо-С3Н7, н-С4Н9, вгор-С4Н9, ЫЗ0-С4Н9, трет-С4Н9, н-С5Нц, СН2—СН, сн3-сн=сн.
2. Прн наличии одного узлового заместителя ему придается номер 1, нумерация других атомов углерода ядра при этом производится с соблюдением положений пункта 1.
3. При наличии нескольких узловых алкильных заместителей номер 1 получает узловой заместитель, более предпочтительный согласно правилам ИЮПАК.
4. Атомы углерода, получившие, согласно приведенным выше правилам, номера 1—9, составляют рациональный фрагмент бицикло [3,3,1] нонана данного производного адамантана, прн этом положения мостиковых заместителей углеродных атомов 2, 4, 6 и 8 определяются как экзо- или эндо- в зависимости от того, направлен ли заместитель соответственно вверх или вниз по отношению к плоскости рационального фрагмента бицикло [3,3,1] нонана; у атома 10 — как цис- или транс- по отношению к атому 1, а у 9 - как син- или анти- в зависимости от того, направлен ли он вправо или влево относительно заместителя 1.
Таким образом, например, углеводород (1) получает название ”1-изо-бутил-экзо-2-эядо-6-экзо-8-сии-9-цмс-10-пентаметиладамантан.
СИ, СНг —СН—он.
Иная система обозначения полизамещенных производных адамантана предложена Гала [5].
Положение 1 обозначается как г-1 илн г-1Нв зависимости от того, имеется ли узловой заместитель или заместитель отсутствует. Заместите-
7
лн в а-положеииях следует обозначать как Р (плюс) или М (минус) в зависимости от того, направлены лн они по часовой стрелке или против нее при рассмотрении со стороны 1 (г). Заместители в ^положениях обозначаются как цис- (с) или транс- (т) по отношению к г-1. Так, например, приведенное выше соединение (2) получает название ”Р-2д-4-диэтил, г- 1,с-6,М-8-триметиладамаитаи”.... Атомы углерода адамантанового ядра необходимо нумеровать в соответствии с правилами, принятыми для полициклических систем (правилоА— 32). Прн возможных альтернативных вариантах предпочтение необходимо отдавать тому из них, где ближайший по ходу мостиковый заместитель приобретает направление М. Заместители предложено располагать в алфавитном порядке.
В конденсированных биядерных структурах два ядра адамантана могут иметь 1,2,3 или 6 общих атомов углерода. Согласно правилам, предложенным Шпейером, эта группа соединений получила название {«] диадамантанов, где п - число общих атомов углерода. Первым углеводородом этого ряда является (6] диадамантан, тривиальное название ’’диамантан” (конгрессам соединение (3))-
5
,3
2
Соответственно приведенные ниже структуры являются [3]-и [2] диадамантанами.
Для конденсированных полициклических углеводородов ряда адамантана используют также названия в соответствии с правилами номенклатуры ИЮПАК, однако для некоторых нз них чаще применяют тривиальные названия. Это, например, пентацикло [7,3,1,14,12,02,7,061] тетрадекан (конгрессан, диамантан — соединение (3)) [6]; гептацикло [ 7,7,1,13 ’15, О1 2,02 ,04’13,06’11 ] октадекан (триамантан - соединение (4) ) [7,8] и ДР-
Для диамантана возможны монозамешенные; производные трех типов: апикальные (вершинные) (положения 4 н 9), медиальные (1, 2, 7, 6, 11 н 12) н мостиковые.
Углеводороды адамантан, диамантан, трнамантан, тетрамантан нт.д. составляют гомологический ряд углеводородов алмазоподобного строения (другие названия — полиманганы, адамантаноидные углеводороды) общей формулы С4п +6Н4П+ 12 [9]. Каждый последующий член ряда отличается от предыдущего наличием дополнительной группы трет-бутильного строения состава С4 Н4. Подобно тому как этан можно представить в виде соединения двух тетраэдрических фрагментов метана, диамантан является соединением диух тетраэдрических ячеек адамантана. В аналогичном соотноше-
нии находятся пропан н триамантан, бутан н тетрамантан (5) н т.д. Подобно бутану тетрамантан (5) представлен несколькими изомерными формами. Они соотносятся между собой как изобутан (а), анти- (б) н гош- (в) формы я-бу тана [10].
И
I
нЛсн, 5
(5)
1.2. СТЕРЕОХИМИЯ АДАМАНТАНОВ
Молекула адамантана обладает высокой степенью симметрии. Некоторые элементы симметрии структуры адамантана сохраняются прн введении одного или нескольких заместителей в узловые положения ядра. Так, прн наличии одного узлового заместителя [соедвнение (6)] сохраняется плоскость симметрии через углеродные атомы С(1), С(2), С(3), С(6) н ось симметрии третьего порядка (45), проходящая через атом, содержащий заместитель и центр противостоящего циклогексанового кольца. Прн наличии двух одинаковых узловых заместителей [соединение (7)] имеются две плоскости симметрии. Одна из них проходит через атомы, содержащие заместители, н атом С (6), другая — через мостиковый атом углерода С (2), находящийся между замещенными атомами углерода ядра, и атомы С (5 ) иС(7).
Б-ч> Б /
(6) (7)
Имеется также ось второго порядка ВГ.
Плоскость симметрии имеется также при наличии в адамантане трех одинаковых узловых заместителей (у атомов 1, 3, 5). Тетразамещениый в уз-
новых положениях адамантан вновь приобретает высокую степень симметрии молекулы, например для 1,3,5,7-тетраметил адамантана порядок симметрии — 12.
Производные адамантана, содержащие только узловые заместители, из-за отсутствия в ннх асимметрических атомов углерода (хиральных центров) не имеют пространственных изомеров. В то же время такие соединения могут обладать оптической активностью, если в молекуле адамантана имеются четыре разных заместителя. Центр молекулы таких производных играет роль гипотетического атома углерода, валентности которого направлены к узловым заместителям, находящимся в вершинах правильного тетраэдра. Молекула становится хиральной вследствие наличия особого типа асимметрии - асимметрии молекулярного тетраэдра [11, 12]. Оптическая активность производных адамантана такого типа была установлена экспериментально. Так, Хамилл и Мак-Кервн получили оптически активную дегидро-абиетиламмониевуюсоль 3-метил-5-бромадамантан-1-карбоновой кислоты [13]. Оптическую активность проявили также [15] 1-амино-3-метил-5-этил-адамантан [14] и ряд производных адамантана общей формулы
ОН,
СН,
ГД/1 |
[н=и)соон; н
5>conh2; п' / ш
»>• WON] 6г (6)
Величина оптического вращения ([а]25) для этих соединений оказалась весьма незначительной: —0,85 ± 0,11; —0,61 ±0,09; —0,81 ±0,27 для соединений а, б, в соответственно. Эти величины значительно ниже чем, например, для формального аналога этих соединений ot-бромпропионитрила (8) ([ок]2 5 = -19,3 ±3,8°) [16] вследствие большего удаления заместителей от центра молекулы.
Оптическая активность проявляется также в ряде других производных адамантана. Синтезированы и разделены на стереоизомерные формы некоторые производные 1,2-диадамантилэтана [17] и 1,2-бис-(З-метил-1-адаман-тил)этана [18]. Так, рацемический 1,2-бнс-(3-метил-5"Карбоксн-1-адаман-тил)этан, в частности, был расщеплен на оптические антиподы дробной кристаллизацией его оптически активного (+М)-(а-фенилэтил)дмида. Оптически активные адамантаноны получены из энантиомеров эндо-бицикло-[33,1] нонен-6-карбоновой-З кислоты [19]. Результаты изучения оптической активности различных производных адамантана приведены также в работах [20—25]. На основе тетраэамещенного производного адамантана синтезирован (-) 1,3,5,7-тетракис [2-(IS, 3S, 57?, 65, 8Я, 1QR)-D3 -трнсго-мокубанил ацетоксиметил] адамантан, являющийсй первым соединением с Г-симметрней с известной конфигурацией [26].
Возможность образования пространственных изомеров появляется у производных адамантана только при наличии в молекуле мостиковых заместителей. С увеличением числа таких заместителей число возможных стереоизомеров возрастает. Вместе с тем вследствие высокой симметрии молекулы адамантана фактическое число образующихся стереоизомеров много меньше, чем теоретически возможное. Например, 2,6-диметилада-10
мантан (9), имеющий два г асимметрических атома углерода, представлен лишь одним пространственным изомером (вместо ожидаемых четырех), так как все четыре теоретически возможные структуры (а, б, в, г) взаимо-превращаемы-
Структуры а н в представляют собой зеркальное изображение соответственно структур биг; структуры виг пегко получить соответственно из б и а поворотом на 180° последних вокруг вертикальной осн, проходящей через атомы 2 и 6.
Общее число возможных изомеров (структурных н геометрических) в ряду алкилпроизводных адамантана рассчитано Корниловым с использованием комбинаторно-математического метода Пона [27]. Результаты расчета представлены в виде следующего ряда:
1 +2х + Их2 + 49х3 +217х4 +858xs + 3314хб +11995х7 + 41921х8 +
+ 141163х9 +462674*10 + ...,
в котором коэффициенты означают число изомеров алкиладамантанов прн количестве атомов углерода в алкильных группах п, равном показателю степени х. Так, для алкиладамантанов состава С12 и Ci содержащих соот-ветствено 2 и 5 атомов углерода в боковой цепи, общее число изомеров 11 н 858 соответственно. Все изомеры алкиладамантанов состава С12 были получены экспериментально [28], дляалкиладамантанов состава С13 их число определено другими независимыми методами [4, 5, 29] -
С увеличением молекулярной массы резко возрастает число возможных структурных н пространственных изомеров полнмантанов: пентамантан — 7, гексамантан — 24, гептамантан — 88 [30].
1.3. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА АДАМАНТАНОВ
1.3.1. ОСНОВНЫЕ ФИЗИКО-ХИМИЧЕСКИЕ ХАРАКТЕРИСТИКИ
Физические свойства адамантана [11,31] приведены в табл. 1.1.
Температура плавления адамантана в запаянном капилляре, 269° С, необычно высока для такого сравнительно низкомолекулярного насыщенного углеводорода. Она обусловлена высокой симметрией жесткой алмазо-подобиой молекулы адамантана (точечная группа симметрии Td). Вместе с тем относительно слабое межмолекулярное взаимодействие в кристаллической решетке приводит к тому, что углеводород легко возгоняется, частично - уже прн комнатной температуре.
Адамантан является наиболее устойчивым нз трициклических углеводородов. Это обусловлено тетраэдрической направленностью связей всех атомов углерода, весьма близкой к теоретической длине С—С-связей, и фик-
11
Таблица LI
Физические свойства адамантана
Показатель Значение
Молекулярная масса Температура плавления Температура кипения Плотность
Показатель преломления
Теплота образования
Теплота сгорания
Удельная теплоемкость Теплота сублимации Упругость паров
Тройная точка
Точка фазового перехода
136,23
26 9° С (в запаянном капилляре)
194,5° (вычислено иаосновании данных ГЖХ)
1,07 г/см3
1,568 ±0,003
138,0 кДж/моль (25° С, твердое состояние)
6019 кДж/моль (25° С, твердое состояние)
189,6 кДж/моль - град (25°С)
59,4 кДж/моль (27°С)
1лР (ммрт.ст.) =50,27 - (8,416/Т)- 4,21111лТ
(Г = 278-443 К)
460° С [27 кбар; объемно-центрированная решетка - гра-недентрированиая решетка — жидкость]
208,62 К; объемно-центрированная решетка—гран сцентрированная решетка—жидкость; ДЯ = 3,4 кДж/моль;
Д5 = 3,27 э.е.
енрованным положением всех атомов ядра. Кристаллическая структура адамантана была изучена методами рентгеноструктурного анализа и дифракции электронов [11,32—40].
Исследования [34—40] показали, что адамантан кристаллизуется в форме гранецентрированной кубической решетки, необычной для органического соединения, пространственной грулнровки T*dF43m с параметром а = 9,426 ± 0,008А и с четырьмя молекулами, приходящимися на одну ячейку. При —65° С наблюдается фазовый переход в объемно-центрированную тетрагональную структуру [41]. Фазовый переход в тетрагональную структуру прн —64,4° С зафиксирован также методом спинового эха [42]. Интересно, что молекулярная реориентация в твердой пластической фазе наблюдается также для некоторых производных адамантана, в частности для 1-хлорадамантана [43].
Методом дифракции электронов найдены следующие параметры молекулярной структуры адамантана [34]:
Высокая устойчивость каркаса адамантана подтверждается тем, что его структура остается практически неизменной даже в адамантилиденадаман-
12
тане, в котором существует сильное стернческое взаимодействие соединенных непосредственно двух объемистых адамантановых ядер [44]. В этом соединении, за исключением углов прн л-связн, валентные углы близки к тетраэдрическому — 109,4°. Средние значения длины освязей С-< лежат в пределах 1,517—1,532 А. Длина тг-связи 1,336 А.
Ранее считалось, что молекула адамантана лишена как углового, так и торсионного напряжения. Вместе с тем величины С-С—С-углов в адамантане несколько отличны от углов в ациклических и алициклических углеводородах, находящихся в свободных от напряжения конформациях. Так, в пропане такой угол равен 112,4° [45], в изобутане - 111,3° [46], в циклогексане - 111,5° [47, 48]. Помимо этого, в молекуле адамантана, подобно тому как это имеется в циклогексане, должны быть ощутимы взаимодействия несвязанных атомов углерода. Расчет энергии напряжения адамантана [49] показал различные результаты в зависимости от применяемого метода (кДж/моль): метод Аллена—Скиннера — 13,8, Татевского — 8,8, Леддера — 1,7. Шлейер н соавт. [50] определили энергию напряжения в адамантане — 27,1 кДж/моль; для циклогексана и т/инс-декалина получены энергии напряжения 5,6 и 7,5 кДж/моль соответственно.
В расчетах [50] были использованы уточненные термодинамические параметры для адамантана прн 25°С: -Д#° (тв) =6018,7; — ДЯ° = 197,0; Д#субл= 59,3 кДж/моль [51] и метод одиночных конформационных инкрементов. В литературе имеются и другие значения [52]: -Д/7£(т) = = 6027,6 и —ДЯо = 188,2 кДж/моль. Данные по термодинамическим свойствам адамантана приведены также в работах [И, 53—57]. Существует мнение [51], что величина энергии напряжения, полученная в работе [50], завышена, так как не учитывались некоторые факторы, обусловленные особенностью строения молекулы адамантана; величина энергии напряжения, вычисленная с учетом этих факторов, составляет 6,7 кДж/моль. Другие методы расчета дают значения величины общего напряжения в адамантане 6,9 [58] и 5,9 кДж/моль [59]. Для расчета энтальпии и энергии напряжения в адамантане и других насыщенных углеводородах были применены методы молекулярной механики, в частности метод молекулярного силового поля [60]. В зависимости от используемой методики расчета для адамантана получены значения Д#£ (газ 25° С) = -135,9 и -141,4 кДж/моль, что согласуется с экспериментальными данными (кДж/моль): Д#° (газ) = —128,1 [33]; —127,8 [52]; —137,8 [51]. Вычисленные методом силового поля значения энергии напряжения адамантана в газовой фазе при 25° С равны 28,7 или 24,8 кДж/моль.
Таким образом, несмотря на некоторые различия в количественной оценке напряжения в адамантане, обусловленные применением различных методов, в качественном отношении получен однозначный результат: молекула адамантана не лищена напряжения. Источником напряжения в адамантане служат угловое напряжение и отталкивание непосредственно не связанных атомов. Было Высказано предположение, что диамантан будет иметь большую энергию напряжения по сравнению с адамантаном [50, 61]. Однако выполненные затем определения теплот образования [33, 52, 62] показали, что эти углеводороды характеризуются примерно одинаковым значением энергии напряжения (~37,7 кДж/моль). Эти данные, однако, были получены на основании теплоты плавления диамантана 30,1 кДж/моль. Пос-
13
ле обнаружения двух эндотермических эффектов в диамантане [63], обусловленных переходом в пластическое кристаллическое состояние при нагревании выше 100° С,тс пл от а плавления оказалась равной 42,3 кДж/моль. На основании этих данных получено значение энергии напряжения в диамантане 49,8 кДж/моль. Расчет методами силового поля [60] показал для диамантана величину энергии напряжения 44,7 и 42,8 кДж/моль (в зависимости от применяемого метода), что превышает энергию напряжения адамантана примерно на 16,7 кДж/моль. Этим же методом были получены для диамантана значения ДЯ® (газ 25 ° С) = -156,2 н—159,4 к Дж/моль, которые находятся в хорошем соответствии с экспериментальной величиной —153,2 кДж/моль.
Введение алкильных групп в ядро адамантана приводит к нарушению симметрии молекулы н увеличению колебательной и вращательной подвижности ее звеньев; температура плавления углеводородов резко снижается (для 1-метиладамантана т.пл. 103°С, для 1-этиладамантана —58°С).Алкил-адамантаны, как правило, представляют собой жидкости с низкими температурами застывания; их плотность находится в пределах 0,90— 0,94 г/см3. Основные физико-химические характеристики алкил-, циклоалкил- и ариладамантанов приведены в следующей главе (табл. 2.10).
Алкил адамантаны определенной молекулярной массы обычно представлены смесями многих изомеров, различающихся как, строением алкильной группы, так и числом и положением заместителей в ядре адамантана. Эти изомеры зачастую весьма мало отличаются друг от друга по своим физико-химическим свойствам, в связи с чем существенно затруднены их анализ и разделение. Обычно для таких целей используется высокоэффективная капиллярная ГЖХ.
В особенности трудно разделить изомерные метилзамещенные адамантаны с различным числом и положением метильных групп. Вместе с тем задачи исследования индивидуального изомерного состава этих углеводородов возникают при изучений природных алкил адамантанов, содержащихся в нефти. Такие задачи решаются обычно методом ГЖХ с применением высокоэффективных капиллярных колонок с неподвижными фазами: скваланом, апиезоном L, SE-30, OV-101. Для идентификации отдельных изомеров используются относительные времена удерживания, порядок элюирования из колонок, индексы Ковача, часто в сочетании с методом хромато-масс-спектроскопии. Эталонные углеводороды, необходимые для идентификации отдельных изомеров, ввиду трудоемкости их синтеза, получают в вяде смесей известного состава путем реакции метиленирования алкил адамантанов более низкой молекулярной массы.
Метиленирование [64, 65] Представляет собой инициируемую УФ-облу-чением реакцию гомолитического присоединения метилена по связи угле-род-водород:
^С-Н + :СН2 -^С-СН3.
Важные особенности данной реакции, позволяющие заранее предсказать состав образующихся продуктов:
1. Присоединение метилена происходит равномерно по каждой С—Н-свя-зи и практически не зависит от степени замещенности реагирующего углеродного атома. Вследствие этого количественное содержание отдельных 14
Таблица 1.2
Состав продуктов метиленирования 1- и 2-метиладамантанов
Номер углеводорода на рис. 1.2 и 13 Алкиладамантан, получаемый в реакции Число затраги-ваемых С—Н-связей* Содержание, мае. %
вычислено найдено
А Б А Б А Б
1 1,3-Диметил- 3 — 16,7 17 —
2 Чыг-1,4-Диметил- 3 1 16,7 5,6 17 6
3 транс-1,4-Диметил- 3 1 16,7 5,6 17 6
4 1,2-Димет ил- 6 2 33,2 11,1 34 13
5 1-Этил- 3 — 16,7 — 15 —
6 2,6-Диметил- - 2 — 11,1
7 эндо,эндо- 2,4-Диметил- - 2 - 11,1 22
8 2,2-Диметил- — 1 - 5,6 — 5
9 экзо,э«<)о-2,4-Диметил- — 4 - 22,2 — 23
10 экзо, экзо- 2,4-Дим ет ил- — 2 - 11,1 — 10
И 2-Этил- — 3 - 16,7 — 15
* Примечание. А - при метиленировании 1-метиладамантана, Б - при метиленирова-иии 2-мет и л адамантана.
изомеров в продуктах метиленирования может быть заранее определено статистическим расчетом на основании числа затрагиваемых в реакции С—Н-связей.
2. Суммарный выход продуктов метиленирования не превышает 2--3%, что позволяет избежать их последующего вовлечения в реакцию.'
3. Конфигурация исходных изомеров во время реакции не изменяется, и в этом смысле реакция метиленирования является стереоспецифической.
Следовательно, применение реакции метиленирования позволяет не только получить все изомеры последующего гомолога углеводорода, но и дает необходимую информацию о строении получаемых стереоизомеров.
Метилеиирование алкиладамантанов проводят в кварцевом реакторе, снабженном рубашкой для охлаждения. Источником УФ-облучения может служить лампа ПРК-2. Реакционная смесь представляет собой обычно раствор диазометана, получаемого разложением иитрозометилмочевины 40%-ным раствором щелочи в исходном углеводороде или в его циклогексановом растворе. Объем смеси 2-3 мл. Об окончания реакции судят по исчезновению характерной желтой окраски; обычно это происходит через 1,5—2 ч.
В качестве примера использования реакции метиленирования для получения эталонной смеси алкиладамантанов состава С12 приведены результаты метиленирования 1- и 2-метиладамантанов, полученные автором (табл. 1.2, рис. 1.2. и 1.3) [4].
В гл. 2 (табл. 2.10) приведены температуры кипения, определенные на основании данных ГЖХ для моно- и полизамещенных алкиладамантанов Сц— С14, содержащих метильные, этильные и другие алкильные группы (см. также [4]). Данные ГЖХ могут быть использованы как для расшифровки состава сложных смесей алкиладамантанов природного и син-
15
:сн2
1,3-ДМА
тра нс-1,4- ДМА
цис-1,4-ДМА
и
Время
Рис. 1.2. Метиленирование 1-метиладамантана ДМА - диметиладамантан» ЭА - этиладамантан
тетического пронсхоэвдения, так и для определения температур кипения углеводородов, к настоящему времени еще не синтезированных. Такая возможность открывается в связи с определенной экспериментально линейной зависимостью относительных времен удерживания и температур кипения в гомологическом ряду алкиладамантанов [4]. Она является частным случаем общей зависимости [66]: г .кип. =X1lgKf + Х2.
Для гомологического ряда алкиладамантанов в принятых условиях [4] были получены коэффициенты Klt К2, равные соответственно 98,8 и-4.
При использовании данных по относительным временам удерживания или индексов Ковача для идентификации алкиладамантанов необходимо, однако, учитывать, что эти параметры и, следовательно, порядок элюирования углеводородов могут изменяться в зависимости от характера неподвижной жидкой фазы, нанесенной на колонку. Об этом свидетельствуют, в частности, результаты работы {67], где приведены хроматографические характеристики примерно 85 производных адамантана, в том числе и алкиладамантаитанов, на капиллярной колонке с жидкой фазой марки SE-30 (5%) в интервале температур 145—190й С.
~1б
:сн2
экзо, экзо-2,4-ДМА
Время
Рис. 1,3, Метилеиирование 2-м етиладам ан тана
Некоторые изомеры алкиладамантанов весьма значительно различаются по временам удерживания и температурам кипения, что позволяет использовать метод препаративной ГЖХ для их выделения в чистом виде. Методом препаративной ГЖХ из соответствующих смесей были выделены и охарактеризованы, в частности, 1,4-диметил- (цис- и транс-), 1,2-диме-тил-, 13,6-триметил-, 1,2,5-триметил-, 1-метил-4-этиладамантаны, изомеры бутиладамантанов и др. [4]. Ариладамантаны могут быть отделены от других ароматических углеводородов с помощью метода адсорбционной элюентной хроматографии на оксиде алюминия при использовании в качестве элюента гексана. Эдхиаы4*©^*етановлено на примере нафтил-, фенаи-
2. Зак. 18sd
17
трип- и антранил адамантанов [68]. Для разделения изомеров углеводородов ряда адамантана используют также гель-хроматографию [69], для производных адамантана - жидкостную хроматографию на углеродных адсорбентах [70].
Физические свойства адамантана, алкиладамантанов и их производных в связи с оригинальной структурой молекулы достаточно широко исследуются. В настоящей книге перечислены некоторые из работ: изучение кинетики роста кристаллов адамантана из паровой фазы Г711. исследование эластических свойств монокристалла адамантана [72], применение метода зонной плавки для получения особо чистого адамантана (количество примесей в промышленном образце адамантана снижается с 0,05 до 0,002%) [72], изучение влияния давления на молекулярное вращение в твердом адамантане [ 74], изучение сжимаемости и определение фазовой диаграммы адамантана при высоких давлениях [75, 76], изучение самодиффуэии 1,3-диметиладамантана, растворенного в гексафторбенэо-ле или полибутадиене, в результате чего были определены свободные объемы 1,3-двметиладамантана [77].
Физические свойства следующих членов гомологического ряда углеводородов алмазоподобного строения изучены в меньшей мере. В ряду: адамантан—двамантаи—триамантан—онги-тетрамантан температура плавления снижается в такой последовательности: 269 -> 237 -> 221 174°С.
В диамантане наблюдаются два фазовых перехода в ориентационно-беспорядочную фазу при 142 и 174°С, в триамантане — температура фазового перехода 155° С, в онги-тетрамантане 159°С [9]. Триамантан в кристаллическом состоянии показывает такую высокую степень молекулярного движения, что представляет собой весьма интересный объект для изучения движений в молекулярных кристаллах. По мере увеличения молекулярной массы углеводородов алмазоподобного строения, т. е. по мере приближения к алмазу длина связей возрастает. Наибольший (111,8°) и наименьший (107,4°) углы находятся у четвертичного С-атома (см. [9] и цитированную там литературу).
13.2. ТЕРМОДИНАМИЧЕСКАЯ УСТОЙЧИВОСТЬ
Изучение термодинамической устойчивости производных . адамантана и диамантана в настоящее время сводится к исследованию их относительной термодинамической устойчивости, методы определения которой подробно изложены в монографиях [65, 78]. Сущность их сводится к тому, что на основании экспериментальных данных по содержанию компонентов в равновесной смеси определяют уровень свободной энергии одного соединения по отношению к уровню энергии другого соединения, близкого по строению. В последние годы такие методы исследования получили широкое распространение для изучения термодинамических параметров многих соединений, и прежде всего изомеров углеводородов. Создав условия для достижения термодинамического равновесия (когда концентрации каждого компонента остается постоянной во времени), определяют константы равновесия и, пользуясь известным уравнением химической термодинамики, определяют относительную константу равно
18
весия и свободную энергию исследуемых соединений.
ДС° ДЯ° Д$°
1пХр -------=---------+ ----- ,
RT RT RT
где Хр — константа равновесия, Д<7°, ДЯ° и Д5°— изменение соответственно свободной энергии, энтальпии и энтропии реакции изомеризации.
Метод равновесной изомеризации позволяет определить, какой изомер является наиболее устойчивым, установить относительную устойчивость других изомеров и количественно определить избыточную свободную энергию менее устойчивых изомеров.
Значения термодинамических параметров соединений могут быть вычислены также на основе вкладов их отдельных фрагментов [65, 78, 79]. Такие расчетные методы дают хорошие результаты для определения термодинамической устойчивости, в частности, в ряду производных адамантана вследствие конформационной жесткости и устойчивости его структуры. Разницу в свободной энергии (Д(7°) соединений определяют на основании вклада энтальпийной и энтропийной составляющих. Энтальпия соединений (ДЯ°) является функцией числа скошенных бутановых взаимодействий (гош-метил-взаимодействий) и степени разветвленности молекулы. При этом принято (на основании экспериментальных данных, полученных для производных циклогексана), что одно гош-метил-взаимодействие повышает энтальпию соединения примерно на 3,8 кДж/моль, а образование нового третичного атома углерода и замена третичного атома углерода четвертичным понижают энтальпию соответственно на 7,5 и 11,3 кДж/моль.
Изменение энтропии соединений (Д<5°) определяют на основании изменения элементов симметрии молекулы как величину, равную 7? In А, где А — порядок симметрии молекулы. В случае наличия в молекуле оптической изомерии энтропия соединения возрастает на величину Я1п2 (для d,/-пары), а при возможности существования конформационных изомеров следует учитывать вклад от энтропии смешения конформеров, равный где N — число конформеров.
Например, изомеризация 2-метиладамантана (2-МА) над AlBrs приводит к равновесной смеси, содержащей 95% 1-метиладамантана (1-МА) и 2% 2-МА [11]. Изменения термодинамических параметров 2-МА в 1-МА следующие: ДО0 = 10,3 ± ± 0,8 кДж/моль; Д//° = -14,1 ± 0,5 кДж/моль; Д5° = -12,5 ± 1,2 Дж/моль • град [80]. Экспериментально полученное значение разности напряжений, создаваемых аксиальной и экваториальной метильными группами, в метиладамантане (жидкая фаза) составляет 10,9 кДж/моль. Расчет по приведенному выше методу дает значения —ДН° = 11,3 кДж/моль, так как в 2-МА имеется два гош -метил-взаимодействия: 7,5 кДж/моль* и образуется четвертичный С-атом в 1-МА из третичного -3,8 кДж/моль. Эти величины при 27° С отвечают содержанию в равновесной смеси 1-МА 97,2%, что находится в хорошем соответствии с экспериментальным значением 98%.
Хорошее соответствие между вычисленными и найденными экспериментально значениями равновесных концентраций компонентов было получено при изомеризации 1-этиладамантана в двметиладамантаны [81]. Расчетные и экспериментальные данные по составу равновесных смесей алкиладамантанов С12~С14 приведены в табл. 1.3.
* Энглер и соавт. [80] считают, что в случае метиладамантанов следует использовать величину 5,4 кДж/моль для одного гош-метил-взаимодействия.
19
Таблица 1.3
Расчет термодинамических параметров и содержание в равновесной смеси (<?%) изомеров*1 алкиладамантанов С13 -С14 при 293К
Алкиладамантан Расчет Эксперимент
-ДЯ°, кДж/моль AS °, Дж/моль - град С, % С, %
С± 3Н2 0
1,3-Диметип- — — 95,6 9 7, О*1
цис-1,4-Диметил- 11,3 5,9 1,8 1,0
транс-1,4-Диметил- 11,3 5,9 1,8 1,0
1,2-Диметип- 15,0 11,8 0,7 0,6
1-Этил- 18,8 5,9 0,1 0,2
CI j Н2 2
1,3,5-Триметип- — — 94,5 97,0
1,3,6-Триметип- 11,3 9,2 2,7 1,8
цис-1,2,5-Триметил- 15,0 15,0 1,1 0,5
транс-1,2,5-Тр иметил- 15,0 15,0 1,1 0,5
1-Метид-3-этил- 18,8 18,4 0,4 0,1
1,2,3-Триметил- 18,8 9,2 0,1 —
1,4,6-Триметил- 22,6 9,2 0,02 —
1-н-Пропил- 30,1 9,2 0,001 -
с14н„
1,3,5,7-Тетр аметил- — — 92,8 —
1,3,5,6-Тетраметип- 15,0 26,3 4,6 —
1,3-Диметип-5-этил- 18,8 29,7 1,4 —
цис-1,2,3,5-Тетраметип- 18,8 20,5 0,5 —
транс- 1,2,3,5-Тетраметил- 18,8 20,5 0,5 —
1,3,6 ,tr Тетр аметил- 18,8 14,6 0,1 —
** Рассматриваются изомеры, содержащие по меньшей мере один заместитель в узло-
вом положении ядра.
Данные автора [29, 82,83j; см. также [65, с. 139} -
Наиболее высокой термодинамической устойчивостью характеризуются алкиладамантаны, содержащие метильные группы в узловых положениях ядра. Наличие этильного радикала, даже в узловом положении, резко снижает устойчивость углеводородов данного ряда. 1-Пропил- и 1-бутиладаман-таны также значительно менее устойчивы, чем соответствующие нм трн-и тетразамещенные изомеры с узловыми метильными группами. В присутствии А1С1з наблюдалось полное превращение пропил- и бутиладамантанов в метиладамантаны. Однако в связи с тем, что изомеризация пропил- и бутиладамантанов протекает в сравнительно жестких условиях (повышенная температура, большое количество катализатора), эти превращения сопровождаются реакциями деструкции, диспропорционирования и пере-алкилирования. Поэтому равновесные смеси изомеризатов получить не удается. Расчетные же данные показывают, что средн лропиладамантанов (в отличие от пропилбеизолов) более устойчивым является 1-н-пропил-20
адамантан. Причина тому — меньшее число гош- метил -взаимодействий в 1-н-пропиладамантане по сравнению с 1-нзопропиладамантаном (ДЯ° = = —7,5 кДж/моль).
Среди изомерных бутиладамантанов, отличающихся строением бутильной группы, термодинамически наиболее устойчивым неожиданно оказался 1-н-бутил адамантан. На основании зависимостей, наблюдаемых для углеводородов парафинового ряда, следовало ожидать, что термодинамически более устойчивыми будут изомеры с разветвленной алкильной группой. Причиной наблюдаемой аномалии является наличие пространственного 1,5-взаимодействия Н-атомов концевых метильных групп 1-изо бутил адамантана (10) с Н-атомамн мостиковых углеродов ядра н значительное число гош-метил-вэаимодействий, свойственных 1-изо- н 1-трет-бут ни адамантанам.
В 1-н-бутиладамантане (11) такое взаимодействие отсутствует. На основании экспериментальных данных разница в энтальпии 1-н-бутил- и 1-изо-бутиладамантана составляет ~ 10,3 кДж/моль [4]. Наличие такого 1,5-взаимодействия атомов водорода следует рассматривать как одну из особенностей строения алкильных производных углеводородов каркасного типа.
С увеличением длины алкильной группы влияние адамантанового ядра на термодинамическую устойчивость изомеров будет уменьшаться. Тем не менее относительно более низкая устойчивость изомеров, содержащих разветвление у ^-углеродного атома алкильной группы, несомненно будет наблюдаться для углеводородов ряда адамантана различной молекулярной массы.
Среди полициклических углеводородов изомеры, содержащие короткие (метильные) заместители в узловых положениях ядра, также термодинамически более устойчивы, чем изомеры с длинными алкильными группами. Так, среди монозамещенных алкилпроизводиых 1,2-бутаноадаман-тана (тетрацикло[7,3,1,17,11,02,7] тетрадекана) устойчивыми являются два изомера — транс- н i^wc-9-метил замещенные (12); их суммарное содержание в равновесии при ЗООК составляет ~ 80% [84]. Из дизаме-щенных производных 1,2-бутаноадамантана наиболее устойчив изомер (13); при ЗООК его равновесное содержание составляет 42%.
5
21
При изомеризации метилдиамантанов в газовой фазе на катализаторе хлорированном оксиде алюминия с нанесенной на него платиной (с последующей обработкой HQ) в интервале 433—585К было установлено [85], что термодинамически наиболее устойчивым является 4-метилди-амантан (16). Следует отметить, что при наличии меиее (по сравнению с СН3-группой) объемистых заместителей (ОН, Вг, С1) более устойчивыми являются, как правило, 1-замещенные диамантаны [86]. Реакция изомеризации метилдиамантанов характеризуется следующими термодинамическими параметрами:
(16) - (14):
(14) ->(15):
(16) ->(15):
ДН 0, кДж/моль
8,9 ± 0,08
2,7 ± 0,04
11,3 ±0,04
Д S °, Дж/моль • град
5,6 ± 0,1
3,3 ± 0,08
8,0 ±0,8
Наиболее низкой энтальпией и, следовательно, самой высокой устойчивостью среди метилдиамантанов обладает изомер (16), метильная группа которого занимает положение, аналогичное положению 1 в адамантане; она экваториальна по отношению к каждому из трех смежных циклогексановых колец. Второй по устойчивости — 1-метилдиамантан (14), в котором СН3-группа уже аксиальна по отношению к одному циклогексановому кольцу (1, 2, 3, 4, 12, 13). Различие между изомерами (16) и (14) такое же, как между экваториальной и аксиальной формами метилцикло-гексана. Наименьшей термодинамической устойчивостью характеризуется 3-метилдиамантан (15). Как и в изомере (14), метильная группа в (15) также создает два гош-метил-взаимодействия, однако она находится у третичного атома углерода ядра; отсутствие четвертичного атома углерода в (15) и приводит к более высокой энтальпии молекулы.
Результаты изомеризации метилдиамантанов в жидкой фазе (катализатор А1Вг3, 25°С) показали, что при равновесии 4-,1- и 3-метилдиамантаны содержатся в соотношении 87 : 10 : 3 [87]. Согласно другим данным, соотношение названных изомеров в зависимости от температуры колеблется в пределах (93 -г 98) : (13 4,7) : (0,7 v 2,4) в интервале 400—
585К [80, 85]. Сради изомеров диметилдиамантанов термодинамическая устойчивость уменьшается в ряду: 4,9-диметил- > 1,4-диметил-, 2,4-диме-тил- > 4,8-диметил- > 3,4-диметил диамантан. При 298К в равновесии содержание 4,9-изомера составляет 613%; 1,4- и 2,4-изомера — 32% (суммарно); 4,8-изомера — 40% и 3,4-изомера — 2,5% [87].
Как уже отмечалось выше, в случае функциональных производных диамантана 4-замещениые изомеры не всегда преобладают в равновесии. Так, концентрация диамантанола-4 в равновесии понижается с 56,0% при 273К др 35,0% при 473К; соответственно концентрация диамантанола-1 возрастает с 44 до 65% [88]. Равновесное содержание 4-хлор диаманта на уменьшается с 62 до 48% при повышении температуры от 210 до 334К [89].
22
Экспериментальные данные по термодинамической устойчивости производных адамантана и диамантана позволили получить для соединений каркасного типа количественную оценку вклада аксиального заместителя в их энтальпию. Известно, что в относительно гибкой молекуле метил циклогексана аксиальная метильная группа повышает энтальпию соединения на 7,1—7,9 кДж/моль [79]. В более жесткой структуре метил-адамантана, в которой циклогексановые кольца уже лишены возможности инверсионных переходов, вклад аксиальной СН3-группы составляет 10,9 кДж/моль. В диамантане эта величина имеет промежуточное значение — 8,9 кДж/моль [80]. Соответствующие вклады аксиальных заместителей в диамантане для групп С1 и ОН составляют 2,8 и 4,6 кДж/моль [88, 89]. Полученные величины используют при разработке программ по расчету термодинамической устойчивости других каркасных углеводородов .
Триамантан характеризуется следующими значениями термодинамических параметров [60]: ДЯ£ (газ, 25°С) = —186,0 кДж/моль, энергия напряжения Е = 56,2 кДж/моль. Из четырех возможных изомеров метил-триамантана с заместителем в узловом положении термодинамически более устойчив 9-метилтриамантан (17); при изомеризации 2-метилтри-амантана (18) он преобладает в равновесной смеси [90].
Расчет показывает, что для тетрамантанов термодинамическая устойчивость изомеров понижается в ряду: изотетрамантаи (5а) > янтп-тетра-мантан (56) > гош-тетрамантан (5в). Различие в энергии между изо- и антм-формой составляет ~ 8,4 кДж/моль [9].
Термодинамическая устойчивость изомерных каркасных углеводородов зависит также от размера заместителя. При наличии весьма объемистых адамантильных заместителей в ядре адамантана наиболее устойчивыми оказываются изомеры, замещенные в положении 2, т. е. в мостиковом положении ядра. Так, в равновесной смеси диадамантанов в соотношении 25,4 : 69,8 : 4,8 содержатся соответственно изомеры: 1,1-ди-адамантан (19); 2,2-диадамантаи (20) и 1,2-диадамантан (21) [91].
Для ряда других углеводородов каркасного строения термодинамические параметры к настоящему времени получены расчетным путем. В табл. 1.4 приведены значения энтальпии образования (ДЯ®) и энергии
23
м Таблица 1.4
Энтальпия образования (дн£) и энергия напряжения (£) некоторых углеводородов каркасного строения, вычисленные методами молекулярного силового поля [60] *
Название Брутто-формула Структурная формула -ДЯ& (газ, 25° С), кДж/моль Е (газ, 25еС), кДж/моль
3 7 Трииикло[3,3,1,0 ’ ]нонан (норадамантан) с„н14 59,4 83,9
3 7 Трицикло[3,3,1,1 ’ ]декан (адамантан) с10н13 135,9 28,7
3 8 Трицикло [4,3,1,0 ’ ] декан (прото адамант ан) С1#Н„ 88,3 76,4
1-Метил адамантан СиН,, 174,8 24,2
2-Мет ил ад ам ант ан 158,6 35,8
1 -трет-Б утиладам антан СиН14 223,3 61,0
2 трет-Б утиладам антан с14н14 211,7 67,9
1,3,5,7-Тетраметиладамантан
293,7
8.0
3 8 Трицикло[4,3,1,1 ’ ]ундекан (гомоадамантан) С.А,
Трицикло[4,4,1,13,8]додекан (1,5-6исгомо-адамантан)
3 9 Трицикло[4,4,1,1 ’ Jдодекан (1,3-бисгомо-адамантан) С12НЗЭ
3 9 Трицикло[5,3,1,1 ’ ]додекан (1,1-бисгомо-адамантан) C13HI0
Т етр апикло (6,3,1,02,6,05,10] додекан (эт аноадамант ан) CiaHls
Тетрацикло[ 5,3,1,12,6,04,9}додекан (ицсан) C„IIU
ю СП 1,1 '-Диадам анти л Cj о H3 q
125,2
107,8
106,7
109,6
106,7
78,2
224,3
61,0
99,9
100,9
98,0
76,2
104,6
89,7
Таблица 1.4 (продолжение)
ю СП
1 2 1 3 1 « 1 s
1,2'-Ди адамантил a Hj д 213,1 96,3
2,2 ’-Диадамантил Ci о Нэ о 222,4 82,3
[1] Диадам антан с,,н2В 181,3 106,6
[2 ] Диадамантан с1вн1в 177,9 93,2
[3} Диадамантан с,7н34 145,8 99,2
[6] Диадамантан (диамантан, контрессан) с14н10 156,2 44,7
1 -Метил д иам антан С1вн„ 182,1 53,0
З-Метипдиам антан
С,,Н.
4 -М етилд иам антан
с,.н.
Триамантан
с1Вн14
’Приведены результаты по методу Энглера.
Таблица 1.5
Сопоставление экспериментальных и расчетных значений термодинамических параметров алкиладамантанов (кДж/моль)
Адамантан, алкил адамантан Д77О (тв.) зксп ДДЯ (тв.) * ДЯ° (субл.) эксп ДДЯ (субл.) |~дн£ (газ) ДАН (газ)
Адамантан 193,5 -2,9 58,5 0,8 135,0 -2,1
1-Метил- 242,4 0,8 67,7 -0,8 174,7 0,0
2-Метил- 224,5 2,5 68,1 -4,6 156,3 -7,5
2,2-Диметил- 256,2 15,9 73,6 -2,9 182,7 -18,8
1,3,5-Триметил- 332,3 0,4 77,7 4,2 254,6 4,6
1,3,5,7-Тетраметил- 378,3 1,7 83,6 5,9 294,7 7,5
* ДДП — ДНрасч — Д^экси.
напряжения (Е) некоторых алкиладамантанов, углеводородов алмазоподобного строения н ближайших циклических гомологов адамантана, вычисленные методами молекулярного силового поля, опубликованные в работе [60]. Для других полициклических углеводородов значения этих параметров существенно выше [60; 92, с. 120—129; 93]. Например, для эндо-тетрагидродициклопентаднена ДЯ° ~ —60,2 кДж/моль [92] энергия напряжения Е = 174,1 кДж/моль [60].
На основании приведенных данных становится понятным, почему трициклические углеводороды различного строения в результате изомеризации превращаются в адамантан и его гомологи (напомним, что для адамантана ДЯо = -135,9 кДж/моль; Е = 28,7 кДж/моль). Аналогично пента-циклические углеводороды образуют производные диамантана, гепта-циклические — трнамантана. Движущей силой этих перегруппировок является снятие внутреннего напряжения в молекуле и уменьшение энтальпии соединений.
Для алкилпроизводных адамантана самым низким значением энтальпии и наименьшим напряжением характеризуется 1,3,5,7-тетраметиладамантан (ДЯо ~ —293,7 кДж/моль; Е = 8,0 кДж/моль). Как будет показано ниже, в условиях протекания реакций перераспределения алкильных групп в алкиладамантанах данный углеводород является наиболее устойчивым конечным продуктом превращения.
Термодинамические параметры алкиладамантанов вычислены также на основе схемы групповой аддитивности с использованием вкладов групп, найденных при изучении алифатических соединений, путем статистического анализа массива достоверных экспериментальных данных на ЭВМ [94]. Сопоставление экспериментальных [95] н расчетных [94] данных показало, что поправку на адамантановый каркас можно принять равной нулю (табл. 1.5).
Получаемые расчетным путем данные по термодинамической устойчивости полицикланов используются для выяснения наиболее вероятных путей н механизмов их превращений.
1.4. СПЕКТРАЛЬНЫЕ ХАРАКТЕРИСТИКИ АДАМАНТАНОВ
1.4.1. ИК-СПЕКТРБ1
Вопросы идентификации углеводородов ряда адамантана, нх структурно-группового или индивидуального анализа зачастую весьма успешно решаются с помощью метода ИК-спектроскопии. Это возможно благодаря ряду исследований ИК-спектров поглощения индивидуальных алкиладамантанов или их смесей, установлению структурно-групповых корреляций в нх строении н спектральных свойствах. К ннм относятся работы по ИК-спектрам поглощения адамантана и алкиладамантанов [96-101], 1- и 1,3-производных адамантана (ИК- и КР-спектры) [102], дейтерированного адамантана и 1-замещенных производных [103], 2-замещениых производных [104], исследования КР-спектров адамантана, диамантана, трнамантана в интервале от 10 К до комнатной температуры [105], изучение спектров адамантана в дальней ИК-области прн 4,6 К [106] и др. В ИК-спектре поглощения незамещенного адамантана ввиду высокой симметрии его мол$-
28
Рис. 1.4. ИК-спектр поглощения адамантана (КВг)
купы (точечная группа 7^) активны лишь трижды вырожденные колебания (Fa), (рис. 1.4, табл. 1.6) [106, 107]. Отнесение колебательных частот выполнено в работе [107],
Введение алкильного заместителя в молекулу адамантана нарушает симметрию молекулы и снимает вырождение ряда колебаний, Это наглядно демонстрирует табл. 1.7, в которой приведены данные ИК-спектров поглощения алкидамантанов с различным типом замещения [101]. При наличии заместителя в положении 1 устраняется, в частности, вырождение полосы 1360 см”1, что приводит к появлению интенсивных полос 1340—1350 см”1. Кроме того, в спектрах 1-замещенных алкиладамантанов наблюдаются полосы: 770, 890, 1020 1040, 1120 см"1 и др. Вместе с тем полосы, отвечающие в основном скелетным колебаниям адамантанового ядра (800, 970,
Таблица 1.6
Полосы поглощения в ИК-спектре адамантана и их отнесение
Частота колебания, см"1 Форма колебания * Частота колебания, см”1 Форма колебания
444 8 (ССС) 1356 8 (НСС), w(CH2)
638 8 (ССС) 1458 8 (НСН)
798 ” (С-С) 2850 v (С-Н) в СН2 -группах
970 р(СН2), и (С-С), 2910 То же
6 (НСС)
1103 6 (НСС) 2930 >5
1312 V (C-C),w(CH2)
* Обозначения колебаний: 8 — деформационное , v — валентное, р, со — деформационные колебания СН2-группы с выходом из плоскости.
29
Таблица 1.7
Полосы поглощения в ИК-спектрах алкиладамантанов О макс-см'1) [101]
Адамантан 1-Метилада-мантан 1-Этил адамантан 1-м-Пропип-адамантан 1-я-Бутип-адамантан
638 сл 710 705 730
740 р 743
765 сл 775 770 сл 780 ш
798 р, с 792 р, с
812 сл 815 р, с 810р, с 812 р
876 890 р 885 880 сл
900
930 сл 915
940 945
970 р, с 980 970 975 р, с 980
1005 с 1002 р, с 990 р 990
1020
1035 1035 р, сл 1040
1070 с 1060
[1080] 1085
1103 р, с f 1102 ( 1100 р, с Г ИООр Г 1100 с
1 1108 [ 1115 р, с ( 1110р, с 11115
1150 сп
1168
1186 ИЗО 1190 1185
1210
1230 сл
1240 1250 1245
1260 1260 1265
1290 1295
1312сл 1318 1320 р 1320р, с 1320
1356 р, с 1342 1350 р, с 1345 р, с 1350 р, с
1365 1363 р, с 1365 р
1380 1383 с 1380 р, с 1382
1300 см-1), а также высокохарактеристическне полосы углеводородов ряда адамантана 1100 и 1360 см-1 присутствуют в спектрах практически всех исследованных гомологов.
Характерной особенностью ИК-спектров 1-алкиладамантанов является расщепление полос 970 и 1100 см-1 вследствие устранения вырождения соответствующих колебаний, В области 1300-1400 см-1 спектров!-замещенных адамантанов наблюдается специфическая последовательность четырех интенсивных полос, из которых наиболее высокочастотная принадлежит деформационным колебаниям 8 (СН2) замещенных групп. Полоса 1360 см-1, характеризующая деформации углов НСС н НСН адамантана, ин в одном из случаев не перекрывается соответствующими полосами алкильных заместителей. Это дает возможность идентифицировать в алкип-адамантаиах концевые метильные группы, включая изопропильные и трет-бутильные [108].
30
Таблица 1.7 (продолжение)
1-Изобутин-адамантан 2-Метил-а даман тан 2-Этипада-мантан 2-Н-Про-пипада-мантан 2-и-Бутил-адамантан 1,2-Диметил -адамантан
700 сл 700
730р 716 с
740 740 744
770 780 770 р 770 дв 770 дв 780 сл
790 805 р 800 р 795 800 р 790 сл
812р 820р 820 дв 820 р
830 сп 855 833 ш
880 сл 890 сл 870 870 сл [ 875 с 870 сп
1 885 с, дв
|910сл 910 сл 900 р 910 с
[920 сл 930 920 дв
940 940 р 940 с л 945 р 950 р, с 940
955 955
978 р 965 р 965 р 965 р 970
970 980 980 980
990 1005 дв 990 1000
1020 сл 1020 сл 1012 1015 р
1030 сл 1030 1030
1045 сл 1050 сл 1045 р, сл 1040 дв
1070 дв, с 1060 1055 1063 с 1070
[1085] 1180 1080
Г1100 с f ИООр 1100 р, с 1105 р> с, 1102 р, с
(1120 11120с 1120 1120 1122 1115 дв, с
1140 сп 1150 сл
1170 1170 1180сл
1190 1203 1195
1212 1215 сл 1220 1220
1250 1240 т 1240 1240
1260 ш 1270 1260
1290 [1295} сл [1300] сл 1300 дв
1320 дв 1315 дв 1315 1330 дв
1350 с, дв 1355 1361 р, с 1352 р 1346
1370 р, с [1367] [1375] 1376 1360
1388 с 1382 с 1380 с 1386р 1380 1380 дв
В ИК-спектрах поглощения 1,3- н 1,3, 5-замещенных алкиладамантанов сохраняются признаки, свойственные 1-замещению в ядре [99, 101]. Необходимо, однако, отметить, что в случае одинаковых заместителей в 1,3,5-пронзводных степень симметрии молекулы возрастает по сравнению с 1-и 1,3-пронзводиыми. Вследствие этого наблюдается меньшее число полос н значительно ослабевает интенсивность полосы 1100 см-1 в связи с уменьшением числа С—Н-связей у третичного атома углерода в ядре.
Заместитель в положении 2, вероятно, оказывает меньшее деформирующее влияние на ядро, чем заместитель у третичного атома углерода. По этой причине в области 1360 см~‘ для 2-замещенных алкиладамантанов харак-
31
Таблица 1. 7 (окончание)
1,3-Диметил-адамантан 1,3-Дня -пропил адамантан 1,3,5-Тримет ил адамантан 1,3,6-Тримет ил адамантан 1,3,5,6-Тетра-мет ила даман-тан
685
725 сл
740 с 745 745
765 сл 765 дв 760 775 дв, сл
800 сл 800 790
820 823 820 сл
850 дв 830 сл
885 870
910 сл 900 вд 910 сл 890 дв 880 дв
[938] 928 р 930 915 905
948 р, с 960 950 f 920 р, с
970 975 с 940 р 1 935 р, с
988 970 дв 972 р
1010 1015 сл 1005 р, дв 1005 р
1018 р
1045 1040 1045 сл ЮЗОр 1025 р
1040 дв
1065 1070 1060
1080 1085 ш 1080 ш 1080 1080 дв
1090р
1120 дв, с 1118 дв, с 1120 дв, сл 1120р ИООр
1150 сл 1140 дв 1140р
1175 р, с 1165 ш 1160 р, с 1170с 1160 р
1220’ 1205
1223
1235 сл 1250
1260 сл 1255 1260 1275 сл 1270
1320 1322 1300 сл 1320 дв 1300
1350р, с 1325
1342 с 1340 1350 1360
1360р, с 1362 с 1360 р, с 1360 1380 дв
1380 р. с 1380 с 1380 с 1380
Примечание. Обозначения полос поглощения: с — сильная, сл — слабая, р —резкая,ш— широкая, дв — двойная; квадратные скобки обозначают точки перегиба; фигурные скобки объединяют плохо разрешенные полосы.
терна одна интенсивная полоса деформационных колебаний углов НСС и НСН адамантана. Как и для 1-замещенных производных, эта область также может быть использована для идентификации концевых метильных групп в 2-ал к ил ада манта нах. Полоса 1100 см'1, имеющая в спектрах 1-замещенных ярко выраженный двойной характер, я спектрах 2-замещенных алкиладамантанов расщепляется значительно меньще. Названные отличия позволяют определять характер замещения в адамантане.
В ИК-спектрах полнзамещенных алкиладамантанов смешанного типа замещения (1,2-, 1,3,6-, 1?3,5,6-) имеются признаки 1-и 2-замещения, в
32
сяязи с чем определение типа замещения по спектру может быть неоднозначным.
Некоторые различия я ИК-спектрах поглощения наблюдаются в спектрах эпимероя 1, 4-диметиладамантана н 1, 2, 5-триметиладамантана в области 700-1300 см”1 (рис. 1.5) [101].
В ряде случаев ИК-спектры поглощения позволяют также определять строение алкильных заместителей в алкила да мантанах. Для этих целей используется область колебаний выше 1360 см-1, а также область маятниковых колебаний СН2-групп 720—780 см-1, в которой отсутствует собственное поглощение адамантанового ядра [108].
Таким образом, методом ИК-спектроскоп ин можно воспользоваться для идентификации индивидуальных алкиладамантанов, в том числе их пространственных изомеров. Прн этом, как правило, могут быть установлены тип замещения и строение алкильных групп. Наряду с этим ИК-спектроскопия применяется как экспресс-метод группового определения алкиладамантанов в природных; (нефтяные фракции) или искусственных смесях углеводородов [101, 109]. Наиболее удобной для этих целей является полоса поглощения 1100 см'1, так как нефтяные фракции с т, кип. 200— 250°С в области данной полосы обладают ровным фоновым поглощением. Необходимо, однако, отметить, что интенсивность полосы 1100 см-1 относительно невелика (е = 12,8—14,5 л/моль - см для 1-замещенных алкиладамантанов),авобласти! 100см-1, имеют полосы поглощения также некоторые другие полициклические углеводороды. В силу отмеченных выше
_______।_______I_______।_______I______।_______1_
900 1100 1300
V,
Рис, 1,5, ИК-спектры эпимеров 1,4-Диметиладамантана
3. Зак. 1856
33
факторов, как было установлено с помощью эталонных смесей, нижний предел чувствительности метода составляет ~5 мае. % алкиладамантанов в расчете на исследуемую нафтено-парафинов ую фракцию [101]. Отмеченная в работе [109] чувствительность метода 0,5% адамантановых углеводородов на нэопарафино-нафтеновую фракцию, вероятно, завышена. ИК-спектры поглощения адамантановых углеводородов в целом характеризуются теми же особенностями, что и спектры адамантана и алкиладамантанов. Например, ИК-спектр поглощения антц-тетрамантана весьма прост [9]. Кроме полос поглощения при 2900 см"1, относящихся к деформационным колебаниям СН- н СН2-связей, имеется всего 6 узких полос слабой интенсивности.
1.4.2. МАСС-СПЕКТРЫ
Метод масс-спектрометрии, основанный на использовании фрагментации органических соединений под действием электронного удара, является одним из наиболее эффективных методов исследования строения углеводородов и состава сложных углеводородных смесей [110]. Наряду с определением молекулярной массы соединения результаты этого метода позволяют получить информацию, необходимую для отнесения углеводородов к определенному типу, и на основе особенностей фрагментации сделать важные выводы о^их строении. Эффективность метода масс-спектрометрии для исследования сложных углеводородных смесей сильно возросла в связи с разработкой его современного варианта — хромато-масс-спектрометрии, сочетающего высокую разделительную способность капиллярной ГЖХ и информативность масс-спектрометрии.
Масс-спектры углеводородов каркаагого строения, как правило, весьма характеристичны.
Фрагментация адамантана под воздействием электронного удара в силу высокой симметрии и устойчивости его молекулы напоминает распад ароматических углеводородов, Основной пик масс-спектра адамантана представляет собой молекулярный нон СюН16* (m/z 136). Остальные ионы образуются в результате его распада или распада возникших ранее фрагментов. При этом был определен ряд мета стабильных переходов, объясняющих образование ионов cm/z 93,80,79, 67, 41, 39 [111, 112].
Масс-спектры 1 - и 2-монозамещенных алкиладамантанов [99, 111, 113-115] характеризуются значительно меньшей по сравнению с масс-
Рис, 1.6, Зависимость интенсивностей пиков молекулярных ионов (а) и адамантил-нонов (б) в масс-спектрах 1 -моиоалкиладамантанов от числа атомов углерода в молекуле
34
спектром адамантана интенсивностью молекулярного нона. Основные ионы в спектрах возникают в результате отщепления углеводородного радикала oj молекулярного иона вследствие гомолитического расщепления связи Ad-R. В ряду 1-алкилпроизводиых адамантана устойчивость молекулярного нона к электронному удару снижается по мере увеличения длины алкильной группы (например,с 1,65%от полного ионного тока у 1-этипада-мантана до 0,8% для 1-гепт ил адамантана при энергии ионизации 70 эВ (рис. 1.6). Стабильность молекулярных ионов 2-моноалкиладамантанов по сравнению с 1-замещенными выше, одиако общая тенденция изменения этой величины с возрастанием длины алкильного заместителя сохраняется. Например, для 2-этил- и 2-н -пропил ада манта но в стабильность молекулярного иона составляет 6,5 и 5% соответствнио.
Основным направлением распада молекулярных нонов 1- и 2-моноалкиладамантанов является элиминирование алкильной группы согласно следующей схеме:
m/z 135
Образование адамантил-ионов (m/z 135) является характерной особенностью масс-спектров всех 1 - и 2-моноалк ил адаманта нов. Вероятность образования ада мант ил -ионов возрастает с увеличением длины алкильного заместителя (рнс. 1.6). При этом строение алкильных групп практически не влияет на распределение интенсивностей и характер масс-спектра таких алкиладамантанов.
Дальнейшая фрагментация адамантил-иона приводит к следующим ионам в масс-спектре [111]:
На рис. 1.7, и 1.8 приведено распределение интенсивностей ионов в масс-спектрах I- и 2-моноалкипадамантанов (% от полного ионного тока). Видно, что максимум интенсивности среди других ионов приходится на область С7~Сб для всех изученных углеводородов. Это объясняется протеканием ряда общих метастабильных переходов [115]:
m/z
85,0 C\(jCi+s —k + C,H4
64,2 CjoHjs —> cth; + C,H6
46,2 Q о Hi j cX + C4HB
58,2 с.нл —> c6h; + с,н.
35
Рис. 1,7. Распределение интенсивностей ионов в масс-спектрах 1 -моноалкиладаман-танов
1 — 1- метил адамантан; 2 — 1-этиладамантан; 3 - 1-я-пропиладамантан; - 1-н-бутил а даман тан; 5 — 1-пентиладамантам; б — 1-гексиладамантан; 7— 1-гептнладаман-тан
Следует отметить, что в масс-спектрах 1- и 2-монозамещенных алкиладамантанов отсутствуют различия, найденные для 1 - и 2-замещениых производных, содержащих функциональные группы (ОН, СООН, Cl, CONH2) [116], и состоящие в том, что основное направление распада мостиковых производных адамантана (т.е. 2-замещенных) связано с отщеплением заместителя в виде НХ. В случае узловых (1-замещеиие) функциональных производных адамантана имеет место элиминирование заместителя X:. Тем не менее метод масс-спектрометрии может бьпь использован для дифференцированного определения 1- и 2-монозамещенных алкида мантано в. Такая возможность появляется вследствие существенных количественных различий в их масс-спектрах.
Как уже отмечалось, масс-спектры 1-и 2-алкиладамантанов существенно различаются по стабильности их молекулярных ионов. Отражением данного различия является изменение соотношения интенсивностей пиков молекулярного иона (М)+ и иона (М- алкил)+, что, как правило, является вполне надежным критерием определения положения заместителя (табл.
35
60
Рис, 1 .Л Распределение интенсивностей ионов в масс-спектрах 2-мояоалкилацаман-танов
1) 2-метиладамантан, 2) 2-этиладамантан; 5) 2-л-пропиладамантан
1.8). Характерно, что различное соотношение I (М)+ // (М-алкил)+ для 1- и 2-алкиладамантаиов наблюдается также в случае дн- и триметил-адамантанов даже если у мостикового С-атома ядра находится только одна метильная группа.
Характерные особенности проявляют в масс-спектрах полиметилзаме-щениые адамантаны с различным положением метильных групп. Углеводороды данного типа характеризуются высокой устойчивостью к электронному удару (вспомним, что полиметил замещенные алкил адамантаны термодинамически также наиболее устойчивы). Распад молекулярного иона ди-, три- и тетраметиладамантанов вначале происходит путем элиминирования одной метильной группы, что отвечает максимуму на кривой распределения интенсивностей в масс-спектрах (рис. 1.9). Таким образом, в масс-спектрах этой группы алкиладамантанов максимальным по интенсивности пиком в масс-спектре является не адамантил-ион (m/z = 135), аегогомолог (m/z 135 + «14, где л = 1^3). Вероятность же образования адамаитил-иона (m/z 135) здесь невелика (1—1,5%) и мало меняется с увеличением числа метильных групп в молекуле. Как уже отмечалось, здесь также наблюдаются количественные различия в соотношении иоиов с массой (М)+ и ионов с массой (М — алкил)+ для углеводородов, содержащих мостиковый заместитель и ие содержащих его.
37
Таблица 1.8
Соотношение интенсивностей ионов 7(M)+/Z(M-алкил)+ в масс-спектрах алкиладамантанов
Алкил Положение алкилэ в ядре 7Г(МУД(М-алкип)+, %
Метил 1 10
2 72
Этил 1 4
2 28
к-Пропил 1 3
2 14
Диметил 1,3 8
1, 2 13
1,4 14
Триметил 1, 3,5 6
1, 2, 5 10
1, 3, 6 14
С учетом метастабильных переходов, наблюдаемых для адамантана и его некоторых производных в условиях масс-спек трометрин [111] и анализа масс-спектров,полученныхвработе [114], наиболее вероятные пути распада полиметиладамантанов рассмотрены на примере 1,2,5-триметиладамаи-тана,
Я13 It ~сн? HjC'-irj, - он, т/г". 178 r<YH t fl hjc4->^ch2 h3c-^> 153 93 * A A - - - •> 1 1 ф H3CHJC-e45>>e^CH)! M'- >63 io?
38
Рис. 1,9. Распределение интенсивностей ионов в масс-спектрах полиметил адамантанов ,
7) 1,3-диметиладамантан, 2) 1,3,5-триметиладамантан, 3) 1,3,5,7-тетраметиладаман-тан, 4) 1,2-диметипадамантан, 5) 1,4-диметиладамантан, 6) 1,3,4-триметиладамаитан
Полизамещенные алкил адамантаны, содержащие алкильные группы различной величины, также характеризуются рядом особенностей. Эти соединения проявляют весьма низкую устойчивость к электронному удару. Максимальные пики в нх масс-спектрах обусловлены отрывом наиболее длинных радикалов и образованием соответствующих гомологов адамдн-тил-ионов, например;
Как и для монозамещенных алкиладамантанов, увеличение размера алкильной группы здесь также способствует более легкому ее элнмнниро-
39
ванию. Например, интенсивности соответствующих фрагментов гомологов адамантил-ионов (С12) для 1,3-диметил-5-этиладамантана и 1,3-ди-метил-5-гексиладамантана составляют 37 и 53% соответственно. Вместе с тем в углеводородах данного типа имеется некоторая вероятность элиминирования метильной группы (М-СН3)+. Эта вероятность возрастает по мере уменьшения дливы наибольшего заместителя (например, при переходе от 1,3-диметил-5-про пиладамантана к 1,3-диметил-5-этиладамантану).
Для алкиладамантанов, содержащих несколько одинаковых алкильных групп, отличных от метильных, преобладающим направлением распада яв-
Таким образом, для алкиладамантанов характерна общая закономерность их фрагментации под действием электронного удара — элиминирование одного из заместителей (обычно наибольшего) и образование относительно стабильного адамантил-иона или его гомологов. Такой процесс распада является специфическим для углеводородов ряда адамантана, он реализуется независимо от числа и характера алкильных групп, последнее позволяет идентифицировать эти углеводороды в смесях. Кроме того, ряд закономерностей и особенностей молекулярного распада, таких, как стабильность молекулярных ионов, относительная активность адамантил-ионов, распределение интенсивностей осколочных ионов в масс-спектрах
алкиладамантанов различного строения, позволяет использовать метод
масс-спектрометрии для определения типа замещения в ядре, количества заместителей и частично их строения.
Масс-спектры диадамантилов и диадамантилэтанов показывают, что фрагментация бнядерных углеводородов ряда адамантана этого типа протекает по следующим направлениям [117] :
пф 148 А = СН2'1+
| — CH3Ad Н3С-А-А-СНэ1+~ -—*4 m/z 283
1 — СН3 А
m/z 149
-с«н5{ | -С3Н4
m/z: 93 I ~сэн*
107 123
т[г 79 ] -с,нв -с3н. х -с2н, m/z 93 *--Ad ------> m/z 107
m/z 135
— CH2 —CH2 —Ad
Ad—CH2 - CH2 - Ad
А-№СН2П+ m/z 162 (Ad = C10H15; A=C10Hl4).
40
В масс-спектрах 1-ариладамантанов (фенил- или нафтил-) наиболее интенсивными ионами являются молекулярные иоиы, затем фрагментные адамантил-ионы с m/z 135 и ионы, соответствующие М+ -С4Н9 [118,119]. Одновременное присутствие в масс-спектре интенсивного пика молекулярного иона и адамантил-иона является характерной особенностью распада 1-ариладамантанов. Расщепление молекулярного иона с образованием фрагментов М+— СЭН7 и М+— СзН5Аг протекает для этих углеводородов незначительно.
Масс-спектры тетра- и пентациклических углеводородов ряда адамантана (2,4-этано-, 1,2-пропано-, 1,2-бутаноадамантан и др.) характеризуются максимальным молекулярным ноиом [120—122]. При наличии же алкильных заместителей, например для метил адамантанов [123] или метил-1,2-бу-таноадамантанов [84, 124], максимальными являются фрагментные ноны М—алкил + .
Последующие члены гомологического ряда углеводородов алмазоподобного строения, такие, как триамантан,днти-тетрамантан, проявляют под воздействием электронного удара даже меньшую фрагментацию, чем адамантан н диамантан — максимальными пиками в их масс-спектрах также являются молекулярные ионы [9].
Весьма характеристичными являются масс-спектры 1- и 2-замещенных функциональных производных адамантана. Для 1-замещенных производных характерно отщепление заместителя в виде радикала X ’ в тех случаях, когда заместитель обладает электроноакцепторными свойствами (NO2, Cl, Вг, СООН); при наличии же электронодонорного заместителя (С6Н5, ОН, NH2) во всех главных фрагментах масс-спектра заместитель сохраняется [111]. По-иному ведут себя под воздействием электронного удара 2-замещенные функциональные производные адамантана. Основное направление распада нх молекулярных ионов связано с отщепление заместителя X в виде молекулы НХ. Вероятно, такое поведение 2-замещенных производных связано с аксиальной ориентацией заместителя X, в результате чего он сближен с водородными атомами в положениях 4 и 6 того же кольца, что способствует 1,3-элнминированию. Косвенно эти результаты подтверждают возможность 2,4-алкильных и метильных перемещений в производных адамантана, которые будут рассмотрены ниже.
Вместе с тем некоторые 2-замещенные производные также разлагаются с отщеплением X, причем направление распада молекулярного иона таких производных адамантана зависит от относительной стабильности Х-радика-ла (табл. 9.1) [125].
Так, 1- и 2-адамантантиолы имеют практически идентичные спектры — в обоих случаях элиминирование H2S представляет собой весьма незначительный процесс, а фрагментация молекулярных ионов у 2-гидрокси-или 2-аминоадамантанов отлична от фрагментации соответствующих 1-замещенных производных (в случае 2-замещенных отщепление Н2Оили NH3 доминирует) [126].
Важные закономерности наблюдаются при фрагментации производных, содержащих обе — алкильную н функциональную — группы [127, 128]. Здесь имеются два конкурирующих направления фрагментации молекулярного нона: М+ (М—А1к)+ иМ+‘ -> (М—R\). С увеличением длины
алкильной группы, как н в случае незамещенных алкиладамантанов, элнми-
41
Таблица 1.9
Зависимость пути фрагментации молекулярного иона 2-замещенных производных адамантана от характера заместителя [125]
Заместитель Отщепление НХ/ отщепление X Заместитель Отщепление НХ/ отщепление X
ОН OQ | .. OCOCF3 8,0
NH3 290 а 6,5
F 86 Вт 0,016
ососн3 44 I 0,004
пирование алкильного радикала облегчается и доля соответствующего процесса фрагментации возрастает.
Силилариладамантаны типа Ad—С6Н4 — СН2Х, где X = (СН3)2 SiOCH3, СНЭ (СН3 О) SiCH2 Cl, (CH3O)2SiCH3 и др., под действием электронного Удара претерпевают распад по направлениям а и б с образованием соответствующих фрагментиых ионов [129].
б а
Ad4c6H4- СН2уХ;
। 1
б а
X-C6H44Ad-С6Нд4х
> ।
Фрагментация других ди функциональных производных адамантана в целом также подчиняется установленным ранее закономерностям [130].
Таким образом, углеводороды ряда адамантана различного строения и их производные дают вполне характеристичные масс-спектры, вследствие чего метод масс-спектрометрии является весьма эффективным методом исследования соединений данного класса.
1Д.З. ЯМР-СПЕКТРЫ
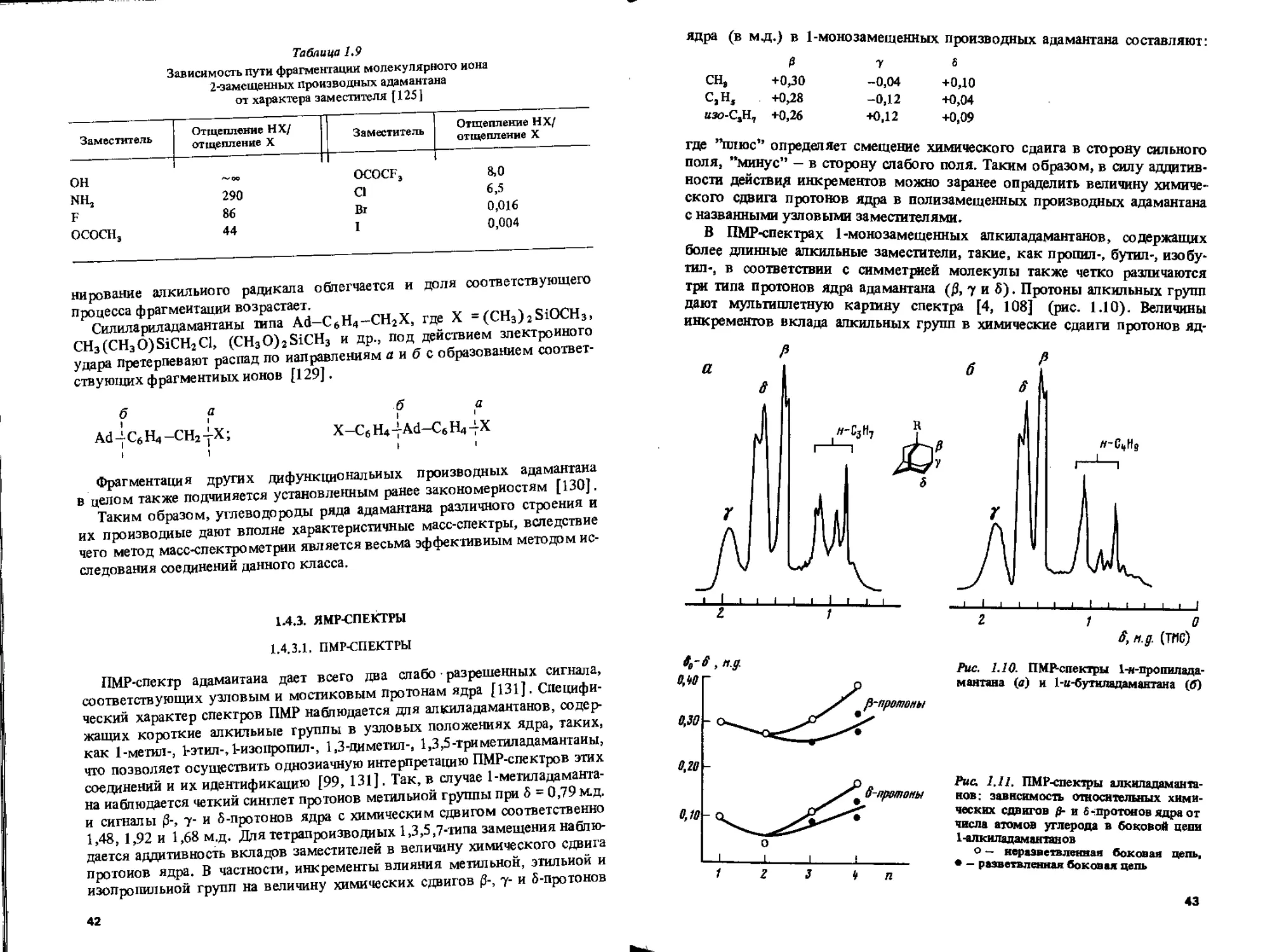
1.4.3.1. ПМР-СПЕКТРЫ
ПМР-спектр адамаитаиа дает всего два слабо разрешенных сигнала, соответствующих узловым и мостиковым протонам ядра [131]. Специфический характер спектров ПМР наблюдается для алкиладамантанов, содержащих короткие алкильные группы в узловых положениях ядра, таких, как 1-метил-, 1-этил-, 1-изопропил-, 1,3-диметил-, 1,3,5-триметиладамантаиы, что позволяет осуществить однозначную интерпретацию ПМР-спектров этих соединений и их идентификацию [99, 131]. Так, в случае 1-метиладамантана наблюдается четкий синглет протонов метильной группы при 5 = 0,79 м.д. и сигналы 0-, у- и 5-протонов ядра с химическим сдвигом соответственно 1,48, 1,92 и 1,68 м.д. Для те трап рои зв о ди ых 1,3,5,7-типа замещения наблюдается аддитивность вкладов заместителей в величину химического сдвига протонов ядра. В частности, инкременты влияния метильной, этильной и изопропильной групп на величину химических сдвигов (3-, у- и 5-протонов
42
ядра (в мд.) в 1-монозамещенных производных адамантана составляют*.
д 7 6
СНЭ +0,30 -0,04 +0,10
с,н5 +0,28 -0,12 +0,04
«яо-СаН7 +0,26 +0,12 +0,09
где ’’плюс” определяет смещение химического сдаига в сторону сильного поля, ’’минус” — в сторону слабого поля. Таким образом, в силу аддитивности действие инкрементов можно заранее опрадалить величину химического сдвига протонов ядра в полизамещенных производных адамантана с названными узловыми заместителями.
В ПМР-спектрах 1-монозамешенных алкиладамантанов, содержащих более длинные алкильные заместители, такие, как пропил-, бутил-, изо бутил-, в соответствии с симметрией молекулы также четко различаются три типа протонов ядра адамантана (3, 7 и 6). Протоны алкильных групп дают мультиплетную картину спектра [4, 108] (рис. 1.10). Величины инкрементов вклада алкильных групп в химические сдаиги протонов яд-
Рис. 1.10. ПМР-спектры 1-я-пропилада-маятанэ (а) и 1-и-бутмладамантана (б)
Рис. 1.11. ПМР-спектры алкиладамантанов; зависимость относительных химических сдвигов р- и 6-протонов ядра от числа атомов углерода в боковой цепи 1-алкиладамантанов
о — неразветвленная боковая цепь, • — разветвленная боковая цепь
43
ра, как правило, того же порядка, что и в случае заместителей меиьшей молекулярной массы. Наблюдается лишь незначительное смещение химического сдвига 3-протонов по мере увеличения длины алкильной группы (от +0,04 мд. для С2Н5 до +0,17 мд. для я-С4Н9. Кроме того, в отличие от других заместителей лишь я-бутильиая группа несколько смещает в сильное поле сигнал у-йротонов ядра. В основе этих явлений лежат, вероятно, пространственные эффекты.
Зависимость изменения величины химического сдвига 0- и 5-протоиов адамантанового ядра от характера алкильной группы (рис. 1.11) показывает, что более сильное влияние оказывает иеразветвленная боковая цепь.
Для углеводородов, содержащих несколько длинных алкильных групп, так»® выполняется правило аддитивности влияния алкильных заместителей на величины химических сдвигов протонов ядра. Это показано в табл. 1.10, где приведены ПМР-спектры 1,3-ди-н-пропиладамантана, вычисленные на основании найденных ранее инкрементов и полученные экспериментально.
В отличие от ПМР-спектров производных адамантана с узловыми заместителями ПМР-спектры мостиковых алкил замещенных адамантанов являются менее характеристичными; даже для простейшего соединения - 2-ме-тиладамантаиа - отсутствует однозначное отнесение сигналов к пяти типам протонов ядра.
Более сложную картину ПМР-спектра с набором множества неразрешенных сигналов протонов ядра образуют поли замещенные алкилпроизвод-иые адамантана, содержащие мостиковые заместители [4]. Единственными хорошо разрешенными сигналами здесь являются синглеты узловых метильных групп в области 0,72—0,88 м.д. и дублеты мостиковых метильных групп в области 0,94-1,04 мд.Расщепление сигналов мостиковыхСНз-групп обусловлено спии-спиновым взаимодействием с незамещенным метиловым протоном, константы спин-спинового взаимодействия находятся в пределах 6,0—7,1 мд.
Данные ПМР-спектров ряда полизамещенных алкиладамантанов с мостиковыми заместителями приведены в табл. 1.11. Весьма существенно, что резонансные сигналы протонов как мостиковых, так и узловых метильных групп различны для разных пространственных изомеров. Как известно, в случае 1-монозамещенных алкиладамантанов, хотя и имеются протоны двух (цис- и транс-) типов в положении 4 (5-протоиы), в спектре ПМР
Таблица 1.10
Химические сдвиги протонов ядра 1,3-дн-н-пропиладамантана
Протоны* S, м.д.
расчет эксперимент
ДО 1,16 1,15
ДО 1,36 1,37
68 1,56 1,56
77 1,94 1,96
•Для незамещенного адамантана сигнал метинового протона наблюдается при 6 = = 1,88 мд., протона метиленовой группы — при 6=1,78 м.д.
44
Таблица 1.11 Химические сдвиги протонов метильных групп полизамещениых алкиладамантанов [4]
Алкиладамантан еСН3. м.д- (ТМС) 7сн3, Н’ 141
для положений 1 (3, 5) для положений 2(4,6)
1-Метил- 0,79
2-Метип- — 1,03 7,0
1,2-Диметил- 0,72 0,94 7,1
цис-1,4-Диметил- 0,75 0,98 6,5
транс-1,4-Диметил- 0,77 1,03 6,3
1,3-Диметил- 0,78 — —
ци с-1 -Метил -4-этил- 0,73 — —
транс-1 -Метил-4-этил- 0,77 — —
1 -Этип-З-метил- 0,77 — —
цис-1,2,5-Триметил- 1 0,77 и 0,72 0,88 6,6
трдяс-1,2,5-Триметил- J 0,92 6,0
1,3,6-Триметил- 0,78 и 0,76 0,98 7,0
1,3,5-Триметил- 0,80 — —
наблюдается только их общий сигнал [131]. В случае, например, 1,4-диме-тиладамантана сигналы протонов метильных групп уже различаются на 0,02 и 0,05 мд. соответственно для узловых и мостиков ыхСН3-групп (рис. 1.12). Аналогичное явление имеет место для эпимеров 1-метил-4-этил-, 1,3,6- и 1,2,5-триметиладамаитанов. При этом наблюдается следующая зависимость: чем ближе расположены друг к другу алкильные группы, тем меньше значения величины химического сдвига 5 их протонов (если сравнивать 1,2-и 1,4-диметиладамантаиы). Это позволяет использовать метод ПМР для установления (или подтверждения) пространственной ориентации заместителей в мостиковых производных адамантана [4, 132], что обычно затруднено в связи с одинаковой термодинамической устойчивостью эпимеров.
Таким образом, спектры ПМР алкиладамантанов, несмотря на трудности, возникающие лри их полной интерпретации, позволяют получать весьма важную информацию о характере замещения в адамантанах для узловых замещенных и сделать отнесения конфигурации геометрических изомеров для мостиковых замещенных алкиладамантанов.
Следует отметить, что зависимости между строением и ПМР-спектра ми углеводородов ряда адамантана сохраняются также для их функциональных производных. Так, в ПМР-спектрах высококипящих адамаитиларомагических соединений [133] и галогеизамещенныхадамантанов, содержащих функциональные группы в мостиковых положениях [134], также наблюдается аддитивность химических сдвигов протонов ядра. Конфигурация пространственных изомеров 2,4-дигалогеизамещенных адамантанов, как и в случае углеводородов, может быть установлена с помощью их ПМР-спектров [135].
Эффекты алкильных групп во фторпроизводных адамантана наблюдались в ЯМР-спектрах I9F [136].
45
Рис. 1.12. ИМР-спектры 1,4-диметил адамантан а
а — ниже кипящий изомер (цис-), б — вышекипящий изомер (транс-) 85% + ниже-кипящий изомер (цис-) 15%; I — сигнал протонов СН3-группы в положении 1, 2 — в положении 4, 3 — сигналы протонов адамантанового ядра
б
Спектры ПМР производных адамантана с кислородсодержащими функциональными группами исследуют обычно в присутствии реагентов сдвига, например трисдипивалоилметаната европия [137—139], 2,2,6,6-тетрамегил-3,5-гептан дионата гадолиния [140, 141]. В этих случаях также удается однозначно определить строение изомеров как производных адамантана, так и родственных соединений, например 1- и 3-гидроксигомоадаманга-нов [142]. ЯМР 2Н-анализ диастереомерных 4-<7-2-гидроксиадамантанов имеется в работе [143].
1.4.З.2. ЯМР-СПЕКТРЫ НА ЯДРАХ 1ЭС
Одним из наиболее информативных и эффективных методов исследования строения углеводородов ряда адамантана, как и углеводородов других типов, является метод ЯМР 13С. Применение полной гетероядерной развязки от протонов и фурье-преобразования позволяет не только получить сигналы всех атомов углерода в молекуле, но и осуществить отнесение пиков к первичным, вторичным, третичным и четвертичным атомам углерода. Возможность такого отнесения вытекает из того факта, что мультиплетно сть пиков (М) в этих спектрах определяется числом протонов (л), присоединенных к данному атому углерода, по формуле М = п + 1. В качестве примера на рис. 1.13 приведены ЯМР 13С-спектр 1-изо бутил адамантана (спектрометр INM—PFT—100, частота 25,15 МГц). В условиях ненастроенного двойного гетероядерно го резонанса (5) в соответствии с отмеченной выше закономерностью сигналы первичных атомов углерода образуют квадруплеты, вторичных — триплеты, третичных — дублеты и четвертич-46
Рис. 1.13.
роядеряой развязки от протонов (а) и ненастроенного двойного гетероядериого резонанса (б)
ных — синглеты. Общее число пиков в спектре, полученном в условиях полной гетероядерной развязки от протонов (л), равно суммарному числу неэквивалентных углеродных атомов адамантанового ядра (а, Д 7, 8) и алкильной группы (а, л'). Отнесение всех сигналов в спектре осу-
ществляется в результате учета интенсивности сигналов данных, полученных в условиях ненастроенного двойного резонанса, а также учета влияния алкильных заместителей в соответствии с правилом аддитивности [144].
В случае как ПМР-, так и ЯМР 1ЭС-спектров наименее сложными являются спектры 1-монозамещенных алкиладамантанов. Данные спектров ЯМР 13С ряда 1-алкиладамантанов и определенные из них аддитивные вклады алкильных групп приведены в табл. 1.12. Аддитивные вклады заместителей показывают характер и величину смещения сигналов по отношению к соответствующим атомам углерода незамещенного адамантана.
47
Таблица 1.12
Химические сдвиги 1 3С (йтмс) и аддитивные вклады алкильных заместителей (AS) в спектрах ЯМР 1 3С 1-алкиладамантанов
б1 S
и
д о
!
5
2
V н
2
е и
й
-снз 39,3 34,0 31,6 39,7 28,3 28,7 38,9 - - 19,1 -1,3 -5,4 5,4 -1,7 0,3 -0,1 -0,9
-СН2-СНЭ 47,8 33,0 33,0 40,7 29,7 30,0 39,9 26,9 - 14,1 -9,8 -4,4 5,0 -2,7 -1,1 -1,4 -1,9
-СН2 -СН2 -СН3 44,4 32,0 32,0 39,6 28,3 28,5 35,3 38,8 20,9 14,7 -6,4 -3,4 6,0 -1,6 -0,3 0,1 2,7
Как видно из приведенных в табл. 1.12 данных, изменение величины алкильной группы в общем мало влияет на значение химических сдвигов атомов углерода каркаса. Как правило, большее влияние испытывают а- и 0-углероды. Характерно, что независимо от своей длины алкильная группа вызывает смещение сигналов а-, 3- и у-углеродов ядра в слабое поле.
Важной особенностью ЯМР 1ЭС-спектров алкиладамантанов является неожиданно сильное экранирующее действие адамантанового ядра на а- углеродный атом боковой цепи; сигналы а-угле родов боковой группы находятся в области спектра, соответствующей наиболее слабому полю (5 = 31-г54 м.д., стандарт ТМС).
Интересно проследить также за изменением величины химического сдвига атома углерода метильной группы по мере ее удаления от ядра адамантана. В «-положении углерод СН3-группы резонирует при 8 =31,3 м.д., в 0-положении наблюдается значительный сдвиг сигнала в сильное поле (8=7,1 м.д.) и, наконец, метильная группа, отделенная от ядра двумя н более метиленовыми звеньями (например, в случае 1-пропил- и 1-бутилада-мантанов), дает в ЯМР 13С-спектре сигнал при 8 = 15-М4 мд. Иными словами, наблюдается эффект чередования, особенно четко проявляющийся в непосредственной близости к ядру.
Известно, что величина химического сдвига в ЯМР 13С-спектре соединений определяется главным образом значением электронной плотности на атоме углерода. Следовательно, ядро адамантана как бы ’’поглощает” существенную долю электронной плотности а’-атома углерода. Аналогичный эффект наблюдается и в ПМР-спектре адамантана [11], где у-атомы водорода более экранированы, чем 0-атомы: смещение электронной плотности внутрь полости ядра адамантана (а), вероятно, обусловлено перекрыванием тыльных сторон орбиталей узловых атомов углерода - так называемый ’’эффект клетки” [11].
Альтернативное смещение электронной плотности а-атомов углерода может происходить по трем о-связям (б). Однако против такого смещения свидетельствует, вероятно, смещение (также в слабое поле) резонансных сигналов 0-углеродов ядра 1-алкилзамещенных адамантанов по сравнению с незамещенным адамантаном (табл. 1.13), т.е. 0-углеродиые атомы, так же как и а'-углеродные атомы, обеднены электронной плотностью.
Наличие ’’эффекта клетки” косвенно подтверждается тем, что для 2-замещениых алкиладамантанов эффект экранирования а’-а тома углерода практически отсутствует (табл. 1.13). В 2-метиладамантане, например, химический сдвиг углерода метильной группы (8 = 19,1 мд.) лишь незначительно превышает значение для удаленной метильной группы (8 = 14,1 и 14,7 в 2-этиладамантане и 2-н-пропиладамаитане соответственно).
Разветвление алкильной группы способствует более значительному смещению сигнала а’-атома углерода и углеродов концевых метильных групп
4. Зак.
49
в сторону слабого поля. Так, например, в 1-изо бутил адамантане, где эффект смещения электронной плотности к ядру усиливается электроиодо-иориым влиянием метильных групп, наблюдается наибольшее смещение сигналов а-СН2-группы, углерод метиленовой группы дает резонансный сигнал в области 5 =54,4 мд. Значительный сдвиг в область слабого поля испытывает также меги новый углерод 1-изопропил адамантана (5 = 38 м.д. по сравнению с 22,8 для 1-изобутиладамантана).
Интересные.в этом отношении данные можно получить из рассмотрения масс-спектров 1-алкениладамаитаиов [146]. Наличие в a-положении боковой группы л-связи значительно стабилизирует молекулярный ион и практически исключает разрыв Сад—Сщлс-связи; именно такой разрыв является доминирующим в случае насыщенных аналогов — 1-алкиладамантанов. Этот результат может быть интерпретирован как свидетельство взаимодействия электронов я-связи и клетки ядра адамантана.
Наблюдаемые в спектрах ЯМР В * * * * 13С эффекты смещения электронной плотности а-углерода боковой группы в сторону ядра оказываются весьма полезными для понимания особенностей реакционной способности углеводородов ряда адамантана. В частности, рассматриваемый ниже (гл. 3) необычный результат бромирования 1 -изо пропил адамантана, когда замещение идет в ядро, а не в боковую цепь, становится понятным, если учесть, что смещение электронной плотности метилов ого а-атома углерода затрудняет элиминирование гидрид-иона из катиона А и образование алкиладамантил-к а тио и а типа Б.
В спектрах ЯМР 1ЭС попизамещениых алкиладамантанов для соедине-
ний, сохранивших некоторые элементы симметрии незамещенного адамантана, картина спектра несложная, и все сигналы могут быть однозначно отне-
сены к соответствующим атомам углерода. Так, 1,3,5-триметиладамаитаи дает пять сигналов — четыре принадлежат четырем типам неэквивалентных
углеродов ядра; для 1,3,5,7-тетраметил адамантана — только три сигнала
(табл. 1.14). Ал кил адамантаны, содержащие мостиковые метильные группы, обладают более сложным ЯМР 1ЭС-спектром (табл. 1.14). Из особенностей ЯМР 13С-спектров этой группы соединений следует отметить некоторое смещение в сильное поле резонансного сигнала углерода мостиковой метильной группы для тех соединений, у которых две метильные группы находятся в положении 1,2 (1,2-диметил-,1,2,5-триметиладамантаиы).Углерод узловой метильной группы менее чувствителен. В то же время для углеродов СН3-группы, расположенных в положении 1,4, пространственная ориентация мостиковой СН3-группы практически не влияет на химический сдвиг углерода (в отличие от химических сдвигов протонов в спектре ПМР).
В целом результаты дают хорошее совпадение вычисленных и найденных величин, показывая, что в ЯМР 13С-спектрах величины химических сдвигов углеродов адамантанового ядра являются аддитивными величинами, отра-
50
Таблица 1.14
Химические сдвиги атомов углерода в спектрах ЯМР1 * С полизамешенных алкиладамантанов* [147]
жающими влияние заместителей. Вместе с тем наблюдается ряд отклонений от правила аддитивности, возможные причины которых рассматриваются в работах [147,148].
Аддитивность величин химических сдвигов наблюдается в спектрах ЯМР 13С 1,3,5,7-тетра-и 1,2,3^,7-пентаметил-2-адамантил-катионов [149] и трн-амантана [150,151].
Большие возможности открывает применение метода ЯМР 1ЭС-спектро-скопии для исследования функциональных производных адамантана. Как при исследовании алкиладамантанов, здесь также можно определить строение соединений и геометрическую конфигурацию изомеров. В ЯМР 13С-спек-трах гидрокси- и дигидроксипроизводиых, например, сохраняется правило аддитивности величин химических сдвигов [152]. Важная информация получена прн исследовании спектров ЯМР 13С 2,6-дизамещенных [153] н 2,4-дизамещенных [154] производных адамантана с различным пространственным расположением заместителей. Следует отметить, что при изучении спектров 4-замещенных адамантанов для некоторых атомов углерода величины химических сдвигов не могли быть предсказаны согласно правилу аддитивности [155]. Вероятная причина этого — взаимодействие непосредственно не связанных атомов углерода по освязям или через пространство.
В ряде случаев для исследования строения 2-гидроксиадамантана и 2-ада-мантантиола методом ЯМР 13С используются лантаноидные реагенты сдвига [156]. Спектры дейтерированных 2-адамантанов приведены в работе [157].
Интересные эффекты наблюдались в спектрах ЯМР 13С 2-фторзамещен-ных адамантанов [158, 159]. В частности, константы расщепления простой С—F-связн отражают внутримолекулярное взаимодействие заместителей, расположенных друг по отношению к другу в виде буквы W, например:
Г
Результаты этих исследований позволяют глубже понять механизмы передачи эффектов заместителей и могут быть использованы для решения стереохимических задач. Информация по спектрам ЯМР 13С 1-фторадаман-танов содержится в работе [160].
Таким образом, вследствие высокой информативности н возможности наблюдения и интерпретации эффектов внутримолекулярного взаимодействия метод ЯМР 13С является весьма эффективным методом установления строения и исследования углеводородов ряда адамантана и нх производных.
Приведем еще несколько примеров. С помощью этого метода была однозначно установлена структура 1,2-пропаноадамантана [122]. Наряду с методами УФ-, НК- и масс-спектроскопии метод ЯМР 13 С с успехом использован для установления строения ряда замещенных нафталинпроизводных адамантана [161], при этом решающая информация, позволяющая доказать структуру синтезированных соединений, была получена благодаря использованию аддитивных закономерностей химических сдвигов ядер13С.
52
Метод ЯМР 1ЭС может быть использован также для конформационного анализа. Это продемонстрировано, в частности, на примере эпимеров 1,4-диме-тил адамантана 1147], стереоизомерных 2,5-дизамещенных производных адамантана и их смесей [162], диамантанкарбоновой кислоты и спирта (с помощью констант спии-спинового взаимодействия) [163].
Из других спектральных методов исследования углеводородов ряда адамантана и их производных можно отметить методы фотоэлектронной спектроскопии [164—166] и электронного спинового резонанса (для исследования 2-адамантил-радикалов) [167].
ЛИТЕРАТУРА
1. Номенклатурные правила ИЮПАК по химии. Т. 2. Органическая химия. М.: ВИНИТИ, 1979. 896 с.
2. StetterH. // Angew. Chem. 1954. Bd. 66, N 13/14. S. 217.
3. Snatzke G., Marquarding D. // Chem. Ber. 1967. Bd. 100, N 5. S. 1710.
4. Багрий Е.И. Исследование углеводородов ряда адамантана: Дис.... д-ра хим. наук. М„ 1977. 385 с,
5. HaJa S. //Chem. listy. 1977. Sv. 71, N 1. S. 18-63.
6. Yogi O., Anderson B.C., Simons DJf. // Tetrahedron Lett. 1966. N 4. P. 415-418.
7. WflZiams V.Z., Schley er P.R., Gleicher G.J., Rodewald L.B // J. Amer. Chem. Soc. 1966. Vol. 88. N4, P. 3862-3863.
8. Bums W., McKervey MA., Rooney J.J.// J. Chem. Soc. Chem. Commun. 1975. N 24. P. 965-966.
9. McKervey MA. //Tetrahedron. 1980. VoL 36,N 8. P. 971-992.
lO. Schleyer P.R., Osawa E., Drew M.G.B. // J. Amer. Chem. Soc. 1968. Vol. 90, N 18. P. 5034-5036.
11. Fort R.S., Schleyer P.R.// Chem. Rev. 1964. VoL 64, N 3, P. 277-300.
12. Степанов Ф.Н., Баклан В.Ф. // Жури, орган, химии. 1966. Т. 2, № 9. С, 1635-1638.
13. ЦатШ Н., McKervey МА. // Chem. Commun. D. 1969. N 15. Р. 864.
14. Cervinka О., Kriz О., Hala S„ Landa S. // Ztschr. Chem. 1971. Bd. 11, N 10. S. 382.
15. Applequist J., Rivers P., Applequist D.E, // J. Amer. Chem. Soc. 1969. VoL 91, N 21. P. 5705-5711.
16. Рату Sturtevant J.M. //Ibid. 1941. VoL 63,N 10.P. 2679-2680.
17. Баклан В.Ф., Даниленко Г.И., Ралко ВА., Смирнова НА. // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1971. № 8. С. 16-18; Chem. Abstr. 1973. VoL 78. 57848 х.
18, Степанов Ф.Н., Смирнова НА., Баклан В.Ф., Юрченко А.Г. // Журн, орган, химии. 1974. Т. 10, №2. С. 234-239.
19. Numan Н, Troostwijk С.В,, Wieringa J.H., WynbergН. //TetrahedronLett. 1977. N 20. P. 1761-1764.
20. Prince R. Preparation and resolution of an optically active adamantane: Ph.D. Dis. Urbana, 1968. 103 p.; Chem. Ab str. 1970. VoL 72. 110878m.
21. Sing Y.L., Numan H„ Wynherg H., Djerassi C. // J. Amer. Chem. Soc. 1979. VoL 101, N 18. P. 5155-5158.
22. Briggs W.S, Suchy M„ Dierassi C. // Tetrahedron Lett. 1968. N 9. P. 1097-1101.
23. Snatzka G., Eckhardt G. // Chem. Ber. 1968. Bd. 101, N 6. S. 2010-2027.
24. Snatzka G., Ehrig B., Klein H. // Tetrahedron. 1969. VoL 25, N 23. P. 5601-5609.
25. Snatzka G., Eckhardt G. // Ibid. 1970. VoL 26, N 4. P. 1143-1155.
2f>.Nakazaki M., Naemura K. // J. Chem. Soc. Chem. Commun. 1980. N 19. P. 911-912.
27. КорниловМ.Ю. // Журн. структур, химии. 1975. Т. 16, № 3. С. 495-498.
28, Долгополова Т.Н. Изомерные превращения би- и трициклических углеводородов над катализаторами на основе окиси алюминия: Дис.... канд. хим. наук, М., 1974. 157 с.
29, Багрий Е.И., Фрид Т.Ю., Санин П.И. //• Нефтехимия, 1970. Т. 10, № 4, С. 480-488.
30. Balaban А.Т., Schleyer P.R. //Tetrahedron. 1978. VoL 34,N24.P. 3599-3609.
53
31. Сасаки Т. //Сэкию гаккайси. 1970. Т. 13, № 10. С 801-805; ВИНИТИ № 90838/1, 1971.
32. Donohue J., Goodman S.H. // Acta crystallogr. 1967. Vol 22, N 3. P. 352-354.
33. Boyd R.H., Sanwal Shjf., Shary-Tehrany Sh., McNally D. // J. Phys, Chem. 1971. VoL 75.N9.P. 1264-1271.
34 Hargittai I., HedbargK. // Chem. Commun. D. 1971. N 22.P. 1499-1500.
35. Giacomello G., Illuminati G. // Gazz. chim. itaL 1945. VoL 75. P. 246-256.
36. Giacomello G., Illuminati G. // Ric. sd. 1945. VoL 15. P. 559; Chem. Abstr. 1946. VoL 40. 692 91.
37 .Nowacki // Helv.chim. acta. 1945. Vol. 28. P. 1233-1242; Chem. Abstr. 1946. VoL 40. 20543.
38. №warcA/ Ж Hedberg K.W. // J. Amer. Chem. Soc. 1948. Vol. 70,N 4. P. 1497-1500. 39МИеи RA., Kraut J., Traylor T.G. //J. Phys. Chem. 1967. VoL 71, N 2. P, 2379-2380; Chem. Abstr. 1967. VoL 67. 111531b.
40. ReynoldsP.A. // Acta crystallogr. A. 1978. VoL 34, N 2. P. 242-249.
41. Nardman C.E., Schmitkons D. // Acta crystallogr. 1965. VoL 18, N 4. P. 765-767. 41. Resing HA. //J. Chem. Phys. 1965. VoL 43, N 5. P. 1828-1829.
43. Bee M., Amoureux J.P. // MoL Phys. 1983. VoL 48, N 1. P. 63-79; РЖХим. 1983. 14Б767.
44. Swen-Walstra S.C., Wisser G. // Chem. Commun. D. 1971. N 2. P. 82-84.
45. Lide D.R(Jr.) //J. Chem. Phys. 1960. VoL 33,N5.P. 1514-1518.
46. Lide D.R. (Jr.) Ц Ibid. P. 1519-1522.
41. Atkinson VA. // Acta chem. scand. 1961. VoL 15, N 3. P. 599-606.
48. Davis M., Hassel O. // Ibid. 1963. VoL 17, N 4. P. 1181.
49. Bratton W.K., Szilard J., Cupas CA. // J. Org. Chem. 1967. VoL 32, N 6. P. 2019-2021.
50. Schleyer РД., Williams J.E., Blanchard K.R // J. Amer. Chem. Soc. 1970. VoL 92, N 8. P. 2377-2386.
51. Mansson M., Rapport N., WestrumE.F. //Ibid. N 25. P. 7296-7299.
52. Butler RS., Carson A.S., Laye P.G., Steele W.V. // J. Chem, Thermodyn. 1971. VoL 3, N2.P. 277-280.
53. Landa S., Machacek V. // ColL CzechosL Chem. Commun. 1933. VoL 5, N 1. P. 1-5. 54. Chang S.S., Westrum E.F. // J. Phys. Chem. 1960. VoL 64, N 10. P. 1547-1551.
55. Westrum E.F. // Phys, and Chem. Solids. 1961. VoL 18. P. 83-85; Chem. Abstr. 1961. VoL 55. 15115b.
56. Мирскап K.B. // Кристаллография. 1963. T. 8, № 2. C. 225-228,
57. Fort R.C Adamantane: The chemistry of diamond molecules. N.Y.: Dekker, 1976. 385 p.
58. Gleicher G.L., Schleyer P.R. // J. Amer. Chem. Soc. 1967. VoL 89, N 3. P. 582—593. 5$.AlHnger N.L., Hirsch J А., Шег MA. et al // Ibid. 1968. VoL 90, N 5. P. 1199-1210. 60. Engler E.M., Andose JJ>., Schleyer p.R // Ibid. 1973, VoL 95, N 24. P. 8005-8025. Gl.AHingerN.L., Tribble M.T.,MSler MA., WertzD.H. //Ibid. 1971. VoL 93.P. 1637-1649. 62. Carson A.S., Laye P.G., Steele W.V. et al // J. Chem. Thermodyn. 1971. VoL 3, N 6. . P. 915-918.
63. Clack T„ Johnston D.E., Mackie H. et aL // J. Chem. Soc. Chem. Commun. 1972. N 18. P. 1042-1043.
64. Doering W.E., Buttery R.G., Laughlin RG.t Chaudhury N. // J. Amer. Chem. Soc. 1956. VoL 78.N13. P. 3224.
65. Петров АлА. Химия иафтеиов. М.: Наука, 1971. 388 с.
66. Smith В., Ohlson R, Larson G. // Acta chem. scand. 1963. VoL 17, N 2. P. 436-454. 67 .Burkhard J., Vais J., Vodicka L.t Landa S. // J. Chromatogr. 1969. VoL 42, N2.P. 207-218.
68. Ушараули ЭА., Картава JIM., Схиртладзе H.H., Мчедлишвили НДж. // Сообщ. АНГССР. 1977. Т. 88, № 1. С. 93-96; РЖХим. 1978. 13Г229.
69. Hale S., Pecka K.t Kremanova В. // ColL CzechosL Chem. Commun. 1974. VbL 39, N 12. P. 3649-3659.
70. Л1ИОИГ D., Cohn H., Guiochen G. // J. Chromatogr. 1982. VoL 234, N 1. P. 1-11.
71. Lubatkin S.D., Dunning W.J. // J. Cryst. Growth. 1978. VoL 43, N l.P. 77-80; Chem. Abstr. 1978. VoL 88. 113486y.
72. Drabble J.R, Husain AH. // J. Phys. C: Solid State Phys. 1980. VoL 13, N 8. P. 1377-1380; Chem. Abstr. 1980. VoL 93.58538t.
54
73. Filby W.G., Gdnther К. //Ub.Pract. 1973. VoL 22, N 9. P. 570; РЖХим. 1974.5Ж183.
74. Liu N.J., Jonas J. //Chem. Phys. Utt. 1972. VoL 14, N 5. P. 555-558.
75. Pistorius C.W.F.T., Snyman H.C. // Ztschr. phys. Chem. N.F. 1964. Bd. 43, N 516. S. 278-281.
76. Геншафт Ю.С., Ларионов Л.В. // Жури.физ.химин. 1969. T. 43, № 3. С. 625-629.
77. Meerwall Е„ Antwerp R.V. ff Macromolecules. 1982. VoL 15, N 4. P. 1115-1119.
78. Илиел Э., Аллинжер H„ Энжиал С., Моррисон Г. Конформационный анализ. М,: Мир, 1969. 592 с.
79. Илиел Э. Стереохимия соединений углерода. М,: Мир, 1965.460 с.
80. Engler Е.М., Blanchard K.R., Schleyer P.R. ff J. Chem. Soc. Chem. Commun. 1972. N22.P. 1210-1212.
81. Schneider A., Warren R W„ Janovski E.J. // J. Org. Chem. 1966. VoL 31, N 5. P. 1617-1625.
82. БагрийЕ.И., Долгополова Т.Н., Санин П.И. // Нефтехимия. 1969. Т. 9, № 5. С. 666-671.
83. Багрий Е.И., Сакин П.И. ff Там же. 1970. Т, 10, № 5. С. 795-799.
84. Берман С.С., Денисов Ю.В., Петров Ал А. ff Там же. 1974. Т. 14, № 3. С 341-348.
85. Hamilton C.R, Johnston D.E., McKervey МА., Rooney J J. // J. Chem, Soc. Chem. Commun. 1972. N 22. P. 1209—1210.
86. Gund TM., Nomura M„ Schleyer p.R. // J. Org. Chem. 1974. VoL 39, N 20. P. 2987-2994.
87. Берман C.C., Петров Ал А. ff Нефтехимия. 1979. Т 19, № 1. С. 17-21.
88. Johnston D.E., McKervey МА., Rooney J J. ff J. Chem. Soc. Chem. Commun. 1972. Nl.P. 29-30.
89. McKervey MA., Johnston D.E., Rooney J.J. ff Tetrahedron Utt. 1972. N 14. P. 1547-1550.
90. Hollo wood F., Karim A, McKervey MA et aL // J. Chem. Soc. Chem. Commun. 1978. N7. P. 306-308.
91. Slutsky J., Engler EM., Schleyer P.R ff Ibid. 1973. N 18. P. 685-685.
92. Общая органическая химия: В 12 т. / Пер. с англ, подред. Н.К. Кочеткова. М.: Химия, 1981. Т. 1.735 с.
93. Godleski SA., Schleyer p.R.} Osawa E. et al // J. Org. Chem. 1976. VoL 41, N 15. P. 2596-2605.
94. Кизик A.H., Лебедев В.П., Лебедев ЮА. // Науч. конф, по химии орган, полиэдра-иов: Тез.докл. Волгоград, 1981. С. 21.
95. Steele W.V., Watt/. ff J. Chem.Thermodyn. 1977. VoL 9,N9.P. 843-849.
96. Mecke R, Spiesecke H. ff Chem. Ber. 1955. Bd. 88, N4. S. 1997-2002.
97. Landa 8., Kamycek Z. // ColL CzechoL Chem. Commun. 1959. VoL 24, N 12. P. 4004-4009.
93. Koch H., Franken J. //Chem. Ber. 1963. Bd. 96, N 1. S. 215-219.
99. Warren R. W., Schneider A., Janovski E.J. ff AppL Spectrosc. 1968. VoL 22, N 2. P. 115 -120.
100. Warren R.W., Schneider A., Janovski E.J. ff Ibid. N l.P. 59-63.
101. Шишкина МВ., Багрий Е.И., Долгополова Т.Н. идр. // Нефтехимия. 1981, Т. 21, № 1. С. 12-19.
102. Оленева Г.И., Краюшкин ММ., Севостьянова В.В. и др. Колебательные спектры моно- и дизамешенных адамантана, М., 1969. 16 с. Дел. в ВИНИТИ 15.09.1969, № 1023-69; РЖХим. 1970. 45259 Ден.
103. Broxton T.J., Deady L.W., Kendall М, Topsom R.L. ff AppL Spectrosc. 1971. VoL 25, N6.P. 600-604; Chem. Abstr. 1972. VoL 76.52353f.
104. Гришина Е.И., Александров ИД, Слободин ЯМ., Костиков РД. ff Журн. прикл. спектроскопии. 1980. Т. 32, №4. С. 664-668.
lOS. Jerafcins Т.Е., Lewis J. ff Spectrochim. acta. A. 1980. VoL 36, N 3. P. 259-264; РЖХим. 1980. 20Б 252.
106. Bertie J.E., Francis B.F., Jacobs SM. ff J. Chem. Phys. 1981. VoL 74, N 11. P. 6522-6523.
107. Snyder R.G., Schachtschneider JJH. ff Spectrochim. acta. 1965. VoL 21, N 1. P. 169-195; Chem. Abstr. 1965. VoL 62. 3536b.
108. Фрид Т.Ю., Багрий Е.И., Санин П.И. и др. // Нефтехимия. 1972. Т. 12, № 4. С. SUSP.
55
109. Куклинский А.Я., Пушкина Р.Я. Ц Там же, 1974. Т. 14, №6. С. 811-816.
ПО. Полякова АЛ., Хмельницкий Р.А. Масс-спектрометрия в органической химии. Л.: Химия, 197 2. 36? с.
lll. Ddlejsek Z., Hala S., Bonus V„ Lande S. // ColL CzechosL Chem. Commun. 1966. Vol. 31,N2.P. 435-449.
112. Митера И., Захарж П, Буркхард И., Водичка Л. // Sb. VSCHT Praze D. 1976. Sv. 32. S. 79-125; Chem. Abstr. 1976. Vol. 85. 7691ly.
113. Slobodin Ya3f., Kovyazin VE., Oshyeva N.A et al. 11 Ibid. 1974. Sv. 30. S. 73-77; Chem. Abstr. 1975. VoL 83. 78693m.
114. Полякова АЛ., Храмова Э.В., Багрий Е.И. и др. // Нефтехимия. 1973. Т. 13, № 1. С. 9-16.
115. Федорова М.С., Денисов Ю.В., Черняк Н.Я. // Там же. № 5. С. 631-634.
116. Карпенко Н.Ф., Чижов О.С., Новиков С.С. и др. // Жури, орган, химии. 1971. Т. 7, №2. С. 416.
117. Карпенко НФ,, Чижов О.С., Новиков С.С. и др. // Изв. АН СССР. Сер. хим. 1971. №2. С. 439-441.
118. Шарбатян ПЛ., Терентьев П.Б., Ковалев В.В., Шоково ЭЛ. // Журн.орган.химии. 1980. Т. 16, № 2. С. 308-314.
119. Scharbatjan РЛ., Terentjew Р.В., Kowaljew W. К, SchokowaE.A. // Sb. VSCHT. Praze D. 1980. Sv. S. 103-111; РЖХим. 1984. 1Б82.
120. Cupas C., Schleyer P.R., Trecker DJ. // J. Amer. Shem. Soc. 1965. VoL 87, N 4. P. 917-918.
121. Яд1и S., Landa S., Hanus V. // Angew. Chem. 1966. Bd. 78, N 23. S. 1060-1061.
122. Бгрман CC., Денисов Ю.В., Мураховская A.C. и др. // Нефтехимия. 1974. Т. 14, № 1. С. 3-8.
123. KurasM., Hala S. // J. Chromatogr. 1970. VoL 51, N 1. P. 45-57.
124. Дягрий Е.И., Фрид Т.Ю., Санин ПИ. // Нефтехимия, 1980. Т. 20, № 6. С. 812-818.
125. Greene R.L., Kieschick И7. A, WahlG.H. // Tetrahedron Lett. 1971. N 47. P. 4577-4579.
126. GreldenusJ.W. // Canad. J. Chem. 1971. VoL 49, N 19. P. 3210:3215.
12"} . Карпенко Н.Ф., Чижов O.C., Новиков С.С. и др. // Изв. АН СССР. Сер.хим. 1972. №2. С. 337-341.
128. Чижов О.С., Новиков С.С., Карпенко НФ, и др. // Там же. № 5. С. 1028-1030. 129. Карпенко Н.Ф., Чижов О.С., Попов Ю.В. и др. // Там же. 1982. № 3. С. 707-708. 130. Чижов О.С., Новиков С.С., Карпенко Н.Ф., Юрченко А.Г. // Там же. 1972. № 7.
С. 1510-1513.
131. Fort R.C., Schleyer P.R. // J. Org. Chem. 1965. VoL 30, N 3. P. 789-796.
132. Багрий Е.И., Федоров ЛЛ„ Какабеков Г.Г. и др. Ц Докл. АН СССР. 1971. Т. 199, № 2. С. 342-345.
133. Podehradska Z, HajekM., Hala S. // Coll. CzechosL Chem. Commun. 1978. VoL 43, N 1. P. 250-256.
134. Deursen F.W., KorverP.K. // Tetrahedron Lett. 1967. N 40. P. 3923-3928.
135. Deursen F.W., Udding A.C. // Rec. trav. chim. 1968. VoL 87, N 11. P. 1243-1248.
136. Wahl G.H., Peterson M.R. // J. Amer. Chem. Soc. 1970. VoL 92, N 24. P. 7238-7239.
137. Юрченко А.Г., Исаев CJL // Жури, органдимии. 1971. T. 7, № 12. С 2628-2629.
138. Hajek М., Vodicka L., Ksandr Z., Lande S. // Tetrahedron Lett. 1972. N 40. P. 4103-4106.
139. Ammon R., Fischer R.D. // Angew. Chem. 1972. Bd. 84, N 16. S. 737-755.
IMS. Hajek M., Mohyla L, Ksandr Z., Vodicka L. // ColL CzechosL Chem. Commun. 1976.
VoL 41, N 12. P. 3591-3599.
IM.Mohyla L, Ksandr Z., HajekM., Vodicka L. // Ibid. VoL 39, N 10. P. 2935-2942.
142. Юрченко А.Г., Новикова М.И„ Исаева С.С. // Жури, орган, химии. 1973. Т. 9, №12. С. 2623-2624.
143. Nordlander J.E., Haky J.E. // J. Org. Chem. 1980. VoL 45, N 23. P. 4780-4782.
IM. Мураховская A.C., Степанянц А.У., Багрий Е.И. и Др. // Нефтехимия. 1974. Т. 14, №2. С. 220-227.
145. Pehk Т., Lippmaa Е., Sevostyanova V. V. et aL // Org. Magn. Reson. 1971. VoL 3, N 6. P. 783-790.
146. Заикин В.Г., Соловьев B.H., Сметанин В.И. и др. // Изв. АН СССР. Сер. хим. 1975. №7. С 1633-1636.
147. Мураховская А.С., Степанянц А. У., Багрий Е.И. и др. // Там же. № 6. С. 1304-1310.
55
148. Мураховская А,С., Степанянц А.У., Зимина К.И. //Там же. 1977. № 5. С. 1072-1076.
149. Schleyer P.R., Lenoir D., Mison P. et aL // J. Amer. Chem. Soc. 1980. VoL 102, N 2. P. 683-691.
150. Dheu M.L., Gagnaire D., Duddeck H. et al // J. Chem. Soc. Perkin Trans. Part II. 1979. N3.P. 357-359.
151. Duddekc H., HoUowood F., Karim A., McKervey MA. 11 Ibid. P. 360-365.
152. Schraml J., Jancke H., Engelhardt G. et al // Coll. CzechosL Chem. Commun. 1979.
VoL 44, N 7. P. 2230-2237.
153. Majerski Z., Vtnkovic K, Meic Z. // Org. Magn.Reson. 1981. Vol. 77, N 3. P. 169-171.
154. Duddeck Я. //Tetrahedron. 19?8.Vol. 34,N2. P. 247-251.
155. Duddeck H., Wolff P. //Org. Magn. Reson. 1977. VoL 9, N 9. P. 528-532.
156. Duddeck H., Dietrich К //Tetrahedron Utt. 1975.N 33.P. 2925-2928.
157. Duddeck H.t Islam M.R. // Org. Magn. Reson. 1983. VoL 21, N 2. P. 140-142.
15%. Duddeck H.t Feuerhelm H.-T. // Tetrahedron. 1980. VoL 36, N 20/21. P. 3009-3015.
159. Duddeck H, Islam M.R. // Ibid. 1981. VoL 37, N 6. P. 1193-1197.
160. Delia E.W., CotsarisE., Hine P.T. // J. Amer. Chem. Soc. 1981. VoL 103, N 14. P. 4131 — 4135.
161. Лузиков Ю.Н., Ковалев B.B., Шокова ЗА. //Нефтехимия. 1978. Т. 18, №3. С. 345-349.
162. Пехк Т.И., Липпмаа Э.Т., Лаврова Л.Н. и др. // Журн.орган. химии. 1978. Т. 14, №8. С. 1634-1640.
163. Marshall J.L., Canada E.D.// J. Org. Chem. 1980. VoL 45, N 15. P. 3123-3125.
164. Morteescu G.D. // Tetrahedron Utt 1972. N 52. P. 5285-5288.
165. Worley S.D., Mateescu G.D., McFarland C.K et aL // J. Amer. Chem. Soc. 1973. VoL 95, N23. P. 7580-7586.
166. Schmidt V. 11 Tetrahedron. 1973. VoL 29, N 14. P. 2129-2134.
167. Waltman RJ., LingA.C.t Bargon J. // J. Phys. Chem. 1982. VoL 86, N 2. P. 325-326.
Глава 2
Методы получения углеводородов ряда адамантана
2.1. ВЫДЕЛЕНИЕ И ИССЛЕДОВАНИЕ АДАМАНТАНОВ НЕФТЯНОГО ПРОИСХОЖДЕНИЯ
В настоящее время единственным природным продуктом, содержащим адамантан и его гомологи, является нефть. Адамантан был обнаружен в нефти раньше, чем он был синтезирован в лаборатории.
Возможность существования декатерпена состава СюН1б, имеющего алмазоподобную структуру, предположил еще в 1924 г. Деккер [1]. Ланда и Махачек в 1932 г. при исследовании состава высок онафтенистых нефтей Годонииского месторождения (ЧССР) выделили углеводород с необычайно высокой температурой плавления (269° С в запаянном капилляре) состава С ц)Н1б, имевший характерный камфорный запах [2—5]. На основании совокупности физических и химических свойств этому углеводороду было приписано строение предсказанного в работе [1] декатерпена и дано название ’’адамантан”. Впоследствии адамантан был найден также в нефтях других месторождений [6—18].
Методы идентификации адамантана в нефтях и его выделения основаны на его необычных для углеводородов данной молекулярной массы свойствах: высокой температуре плавления, летучести, малой растворимости, а также способности образовывать устойчивые аддукты с тиокарбамидом. Весьма характеристичны также ИК-, масс-, ЯМР-спектры адамантана и алкиладамантанов.
Выделение адамантана из нефти Годонииского месторождения, не имеющей бензиновых фракций, осуществляется путем однократной обработки тиокарбамидом дистиллатов, отогнанных из нефти с водяным паром. При охлаждении полученного тиокарбамидиого экстракта до — 50° С адамантан выкристаллизовывается и легко отделяется фильтрованием [9]. Так выделяется ~75% адамантана, присутствующего в нефти.
Если в нефти имеются легкие фракции и содержание адамантана небольшое, то обработку дистиллата тиокарбидом повторяют, используя небольшое количество тиокарбамида, и получают высокоселективные экстракты. Дальнейшее количественное выделение адамантана может быть осуществлено также методами препаративной ГЖХ [16]. Для выделения адамантана из нефти может быть также использован метод азеотропной перегонки циклопарафинового концентрата с три (лерфторбутил)амином [10].
Выделение адамантана из парафинистых нефтей требует более эффективных методов его концентрирования, таких, как термодаффузия и препаративная ГЖХ [15]. Как показали исследования [19], наилучшие результаты при выделении адамантана дает метод, сочетающий перегонку дистиллата (с перегретым водяным паром) с последующим выделением путем препаративной ГЖХ [19].
58
Таблица 2,1
Содержание адамантана в нефтях Бакинского месторождения
Нефть Глубина залегания, м Содержание углеводородов (по данным [21 ]), %* Содержание адамантана, в расчете на нефть, мас.%
алканы циклоалканы ароматические
Надкир макинской свиты Балахано-Сабунчино-Рома-нинского месторождения 710 3,7 88,7 7,6 0,0013
Кирмакинской свиты Бала-хано-Сабунчино-Ром анин-ского месторождения 1302 7,6 77,0 15,0 0,0010
Подкирмакинской свиты Сураханского месторождения *См. 121,с.105, 195, 197]. 2324 45i0 39,0 16,0 0,0004
Количество адамантана в различных нефтях находится в прямой зависимости от химической природы нефти. Наиболее высоким содержанием адамантана характеризуются нефти нафтенового типа. Напротив, парафинистые иефти содержат адамантан в значительно меньших количествах. В табл. 2.1 приведено содержание адамантана в трех индивидуальных нефтях Бакинского месторождения, определенное одинаковым методом [16, 20]. Видно, что с уменьшением общего содержания нафтеновых углеводородов снижается и содержание в нефтях адамантана. Вероятно, это свидетельствует о близкой для этих нефтей степени превращения полициклических углеводородов нефти в адамантан и его гомологи.
В нафтенистой нефти Годонииского месторождения содержится 0,02-0,03% адамантана [6, 9, И], в богатой полициклическими углеводородами нефти месторождения Мартыши (СССР) - 6,6 10"3% [22], в нефти месторождения Понка (США) — 4 • 10-4% [10], в парафинистой нефти Ромаш-кинского месторождения — 1 10~4% [15], в нефти Зыбза—Глубокий Яр — 5 10"3% [23].
В 1962 г. в нефти впервые были обнаружены алкильные производные адамантана. В термодиффузионных фракциях нефти методом ГЖХ с применением синтезированных эталонов были идентифицированы 2-метил-, 1-метил- и 1 -этиладамантаны [13,14]. Эти соединения, подобно адамантану, образуют аддукты с тиокарбамидом [24]. Некоторые алкиладамантаны, главным образом метиладамантаны, были вскоре после этого идеи: ифи-цированы в тиокарбамидных экстрактах бензиновой фракции нефти Ьала-ханского месторождения (АзССР) [25], а также во фракциях, полученных методами гермодиффузионного разделения и равновесной изомеризации нефти Грязевая Сопка (АзССР) [26]. 1-Метиладамантан обнаружен также в нефтях Грузин [27].
Способность образовывать аддукты с тиокарбамидом проявляют только те алкиладамантаны, поперечное сечение молекулы которых составляет ~6,0—6,5 А (табл. 2.2) [28]. Эти размеры близки к геометрическим параметрам сечения канала, создаваемого молекулами тиокарбамида прн
59
Таблица 2.2
Способность алкиладамантанов к образованию аддуктов с тиокарбамидом*
Адамантан, алкиладамантан Температура кипения, С Образование аддукта Ориентировочные размеры поперечного сечения молекулы, А
Адамантан 195 + (0,5) 6,1 6,5
1-Этил- 223 + (5) 6,1 6,5
2-Этил- 227,5 + (20) 6,1 6,5
1-н-Пропил- 242 + 6,1 6,5
1,2-Диметил- 219 + 6,1 6,5
1,3-Диметил- 201,5 — 6,1 7,5
цис-1,4-Диметил- 212 — 6,1 7,5
транс-1,4-Диметил- 213 + (14) 6,1 6,5
1-Мсти л-3-эти л- 226 — 6,1 7,5
цис-1-Метил-4-этил- 230 — 6,5 7,5
транс- 1-Метил-4-этил- 231,5 + 6,1 6,5
цис-1 -Этил-4 -метил- 237 — 6,5 7,5
транс-1 -Этил-4-метил- 240 + 6,1 6,5
цис-1,2,5-Триметил- 221 — 6,5 7,5
транс-1,2,5 -Триметил- 222 ' — 6,5 7,5
1,3,5-Триметил- 205 — 6,5 7,5
1,3,6-Триметил- 216,5 — 6,1 7,5
*Плюс — аддукт образуется; минус — аддукт не образуется. В скобках указана равновесная концентрация углеводорода при образовании аддукта с тиокарбамидом ~ величина, обратно пропорциональная устойчивости аддукта [28].
аддуктообразовании [29]. Следовательно, метод аддуктообразования может быть использован только для обнаружения в нефти отдельных алкиладамантанов вполне определенного строения1.
Среди методов выделения из нефти углеводородов ряда адамантана важное место занимает метод термодиффузиоииого разделения. В основе метода лежит процесс разделения углеводородов по степени цикличности [30—32], В табл. 2.3 приведены результаты термодиффузиоииого разделения эталонной смеси алкиладамантанов, декагидроаценафтена и изоалка-иов—циклоалкаиов. Видно, что трициклические углеводороды (алкил-адамаитаиыи декагидроаценафтены) практически полностью концентрируются в нижних (по высоте колонок для термодиффузии) фракциях (87—94%). Метод термодиффузии был, в частности, успешно применен для концентрирования би- и трициклических углеводородов нефти месторождения Грязевая Сопка [26].
Метод термодиффузии позволяет достаточно полно отделить алкилада-маитаны и другие трициклические углеводороды от моно- и бициклоалканов и алифатических углеводородов. Вместе с тем из-за сложности изомерного состава полициклических насыщенных углеводородов нефти ицеи-
1 С использованием аддуктообразования с тиокарбамидом в иракских нефтях идентифицированы: адамантан, 1-метил-, 2-метил- и 1-этил-2-этиладамантаны, гомоадамантан (Analyst. 1986. N 5. Р. 575—576). — Примеч. ред.
60
Таблица 2.3 Термодиффузионное разделение алкиладамантанов, декагидроаценафтена и изопарафино-циклопарафиновых углеводородов [33]
61
тификация отдельных изомеров затруднена даже в относительно узких термодиффузионных фракциях. Дальнейшее упрощение состава полициклоалкановых нефтяных фракций, содержащих углеводороды ряда адамантана, достигается применением метода дегидрокрекинга. Такая возможность вытекает из высокой термической стабильности низших алкил-адамантаиов, их неспособности к дегидрированию с образованием тг-связи в цикле и незначительному дегидрированию по алкильной группе. Дегидрокрекинг концентратов нефтяных фракций обычно проводят в атмосфере водорода, в присутствии Pt- или Pd-содержащих катализаторов, неспособных вызывать структурную изомеризацию алкиладамантанов [34, 35]. В условиях дегидрокрекинга (410-430°С, 1 атм Н2, Pt на А12О3) конденсированные полициклические углеводороды (пергидроаценафтеи, пергидро флуорен, декалин и др.) нацело превращаются в ароматические углеводороды и легко отделяются адсорбционным хроматографическим методом. Алкиладамантаны с короткими алкильными группами, в том числе пропил адамантаны, в этих условиях практически не изменяются.
Применение метода дегидрокрекинга позволяет исследовать низшие алкиладамантаны (состава Сц^-С^) ряда нефтей. Для группового определения углеводородов ряда адамантана в нефтяных дистиллатиых фракциях предложено использовать также метод ИК-спектроскопии [36] (следует заметить, что чувствительность метода авторами заметно завышена) и сочетание методов ГЖХ и масс-спектрометрии [37].
2.1.1. ИЗОМЕРНЫЙ СОСТАВ И СТРОЕНИЕ
Экспериментальные данные о детальном составе алкиладамантанов нефти немногочисленны. Они ограничены в основном результатами исследований нефтей Балаханскрго месторождения, месторождений Центральное Сабо (о-в Сахалин), Грязевая Сопка, Аиастасьевско-Троицкого (Краснодарский край). Азербайджанские нефти характеризуются высоким содержанием циклических углеводородов и относятся к нефтям нафтенового типа [21,с. 105, 195,197].
Из нефтей адамантаны выделяли следующим путем: фракцию 200-225° С после предварительного отделения ароматических углеводородов подвергали термо-диффузионному разделению (колонка высотой 110 см, зазор 0,3 мм, температура горячей стенки 100-102° С, температура воды 15° С, продолжительность 100 ч). После отделения изоалкано-циклоалкановой части (2,44% в расчете на нефть) из нижней части колонки получали фракции, обогащенные трициклическими углеводородами. Три нижние фракции подвергали дегидрокрекингу пропусканием 3,7 г углеводородов над 50 см3 катализатора АП-56 при 420° С с объемной скоростью 0,1 ч"1, Продукты реакции разделили на силикагеле марки АСМ, получили 0,01 г (0,25%) продукта, представляющего собой практически чистую смесь нефтяных алкиладамантанов (рис. 2,1, табл. 2.4) [21].
Для нефтей с высоким содержанием циклоалканов применение трудоемкого метода термодиффузии необязательно, дегидрокрекингу непосредственно можно подвергать фракцию изопарафино-циклопарафиновых углеводородов. В этом случае, одиако, реакцию дегидрокрекинга необходимо проводить повторно и выход концентрата алкиладамантанов существенно ниже, чем при использо гании гермодиффузии.
Данные табл. 2.4 показывают, что в нефтях содержится значительное число изомерных алкиладамадтаиов состава Cj, -Сг 4 (идентифицированы 62
Рис. 2.1. Хроматограмма концентрата алкиладамантанов, выделенного из фракции с т.кип. 200-225° С балаханской нефти
Колонка: I = 70 м, неподвижная жидкая фаза сквалан, 135° С, газ-носитель азот. 7) Адамантан, 2) 1-мети л ада мант ан, 3) 1,3-циметиладамантан, 4) 1,3,5-триметипада-мантан, 5) 1,3,5,7-тетраметиладамантан, 6) 2-метиладамантан, 7) 1,4-диметиладаман-тан (цис- и транс-), 8) 1,3,6-триметил адамантан, 9) 1,2 диметил а дам ан тан, 10) 1,2,5-триметиладамантан (цис- и транс-), 11) 1 -этиладамантан, 12) 1,3,5,6-тетраметилада-мантан, 13) 1-мегил-3-эти ладамаитан, 14) 2,4-диметиладамантан, 15) 2-этиладаман-тан, 16) 1,3-диметил-5-этнладамантвн, 17) I -мегил-4-этиладамантан, 18) 1,2,3,5-тетра-метиладамантан, 19) 1,2,6-триметиладамантан, 20) 1,2,4-трнметиладамантан
24 изомера). Общее содержание алкиладамантанов в расчете на нефть 0,025 мас.% для нефти Балаханского месторождения и 0,005 мас.% для нефти Сахалинского месторождения, что более чем в 10 раз превышает содержание адамантана,
Алкиладамантаны нефти представляют собой метил- и этил адамантаны. Наибольшее относительное содержание приходится на долю полиметил замещенных алкиладамантанов. Важной особенностью изомерного состава смесей алкиладамантанов, наблюдающейся для всех исследованных нефтей, является наличие наряду с наиболее устойчивыми соединениями (заместители у узловых атомов углерода ядра) также менее устойчивых изомеров (с мостиковыми заместителями). Сравнивая результаты исследования различных нефтей, можно видеть, что, хотя общее содержание алкиладамантанов различно, соотношения отдельных изомеров весьма близки. Наличие общности в распределении изомеров алкиладамантанов в нефтях месторождений регионов, весьма отдаленных друг от друга, свидетельствует о наличии общего пути образования углеводородов ряда адамантана.
63
Таблица 2.4
Алкиладамантаны нефтяного происхождения
Алкиладамантан Содержание в нефти, мае. %
на нефть (с • 10*) на сумму изомеров алкиладамантанов в продуктах изомеризации ПГАУ • в равновесной смеси
Балаханская Центральное Сабо Балаханская [33] Центральное Сабо [33] Анастасьев-ская [35] Грязевая Сопка ] 35]
Cj 1 Hj в 1-Метил- 11 2,5 55,0 52,0 23,0 12,8 98,0
2-Метил- 8 2 45,0 48,0 77,0 87,2 - 2,0
Cj з на 1,3‘ДимеТил* 25 4 33,0 23,0 15,4 12,0 40,0 92,5
1,4-Диметил- (цис- + 21 5 26,0 31,0 30,8 25,8 27,0 6,0
+ транс-) 1,2-Диметил- 15 3 18,0 20,0 22,6 30,2 9,0 1,4
1-Этил- 15 2 18,0 14,0 13,3 15,8 15,0 0,1
2-Этил- 6 1,5 5,0 12,0 17,9 16,2 9,0 —
• Зак. 1856
С, э Я,2.
1,3,5-Триметил- 19 2,5 21,5 14,0 8,4 4,4 28,0 92,5
1,3,6-Триметил- 11 2 14,0 12,0 16,0 11,8 25,0 3
1,2,5-Триметил- (цис- + + транс-) 23 6 28,5 29,0 44,6 49,4 21,0 3
1-Метил-З-этил- 21 4,5 26,0 21,0 31,0 34,4 19,0 1,5
1-Метил-4-этип- (ццс- + + транс-) 4 3 6,0 12,0 - - 5,0
1,2,6-ТримеТил- 1 0,5 1,0 2,0 1,0
1,2,4-Триметил- 2 4 3,0 10,0 — — 1,0 —
с1#н14
1,3,5,7-Тетрамети л- 4 0,5 13,0 5,0 — 12,0 92,8
1,3,5,6 -Тетраметил- 13 3 10,0 40,0 — 33,0 4,6
1,3-Диметил-5-этил- 11 3 36,5 40,0 — 46,0 1,4
1,2,3,5-Тетраметил- (цис- + + транс-) 3 1 10,5 15,0 - - 9,0 1,0
* ПГАУ - пергидроароматические углеводороды. Изомеризация проводилась на катализаторе Н, SO4 /А13 Оэ при 200° С.
Изомерами метил- и этиладамантанов не ограничивается ряд углеводородов типа адамантана в нефти. Семейство этих соединений дополняют тетра- и пентациклические углеводороды адамантаиоидного типа. К настоящему времени из нефти Годонинского месторождения выделены 2,4-эта-иоадамаитан (тетрацикло [6,3,1,0 2>6,05>1 °] додекан, 1), R = Н, идиаман-таи (коигрессаи, пеитацикло [7,3,l,l,4’12,02’7,06fl Чтетрадекаи, 2), R = Н [38].
(?) (J) (Ч) (5)
Углеводороды (1) и (2) находятся во фракции с т.кип. 200—300 С; они выделены [38] сочетанием методов термо диффузии и аддуктообразования с тио карбамидом. Позднее [39] из этой же нефти были выделены еще два тетрациклических углеводорода состава С13Й2О. Предполагается, что это метильный гомолог 2,4-этаноадамаитана (1), R = СН3, и 2,4-пропа-ноадамантан (тетрацикло[7,3,1,02,7,06»1 Чтридекан, 3). 2,4-Этаноадаман-таи (1),R = H, его метильный гомолог (1), R = СН3,и 1,2-пропаноадаман-тан (теграцикло[6,3,1,16’1О,02’б]тридекаи, 4) найдены также в нефти Грязевая Сопка [40] методом хромато-масс-спектрометрии. Некоторые тетра- и пеитациклическне углеводороды ряда адамантана, в том числе 1,2-пропаио- (4), 1,2-бутаноадамантаи (5), а также диамантан и 4-метил-диамаитаи (2), R = Н, СН3, найдены в отечественных (м. Нафталан и Русское) нефтях [41, 42]. В иранской нефти открыт гетероциклический аналог адамантана — тиаадамантан [43, 44], Другие углеводороды каркасного строения, в том числе весьма близкие к адамантану, в природе ие обнаружены.
Исследования углеводородов ряда адамантана в нефти представляют существенный интерес для выяснения весьма важных вопросов происхождения и взаимных превращений углеводородов нефти.
2.1.2. ПРОИСХОЖДЕНИЕ УГЛЕВОДОРОДОВ РЯДА АДАМАНТАНА В НЕФТИ
Со времени открытия адамантана вопрос о его происхождении в нефти интересовал многих исследователей. После разработки Шлейером [45] синтетического метода получения адамантана путем изомеризации гидрированного димера циклопентадиена в присутствии кислот Льюнса Ланда с сотр. [46] высказал предположение, что источником адамантана в нефти служат циклические углеводороды, прн этом природные глниы и алюмосиликаты могут играть роль катализаторов их превращения в адамантан. Каталитическая активность природных глин, вызывающих реакции полимеризации, диспропорционирования водорода и изомеризации ненасыщенных углеводородов при температурах ~~200°С, хорошо известна. Возможная роль этих каталитических систем в процессах катализа углеводородов нефти различных типов отмечалась в работах Фроста и Богомолова [47, 66
48]. Однако сначала применение таких систем, как порода нефтеносного пласта Годонинского месторождения й алюмосиликатные катализаторы, для обработки нефтяных фракций при 190-230° С ие дало положительных результатов. Дополнительное количество адамантана образовалось только при контакте нефтяных фракций с хлористым алюминием [46]. Платэ с соавт. [49] вскоре показали, что изомеризация тетрагидродициклопеита-диеиа в адамантан на алюмосиликатных катализаторах все-таки происходит, ио при более высоких температурах (350—475° С), с выходом адамантана 6-13%. Предположение, что адамантан имеет абиогенное происхождение в нефти высказал также Робинсон [50].
Результаты, полученные автором этой книги с сотр. при изучении изомерных превращений трициклоалканов над алюмоокисными н алюмосиликатными катализаторами (подразд. 2.2.2), и данные, полученные при исследовании изомерного состава алкиладамантанов нефти (подразд. 2.1.2), позволили более глубоко изучить вопрос о возможных путях образования углеводородов ряда адамантана в нефти. В 1967 г. было установлено, что превращения гидрированных ароматических трициклических углеводородов в алкил адамантаны легко протекают иад оксидом алюминия, пропитанным разбавленными растворами некоторых минеральных кислот (в особенности серной кислоты), и над аморфными алюмосиликатными катализаторами при относительно умеренных температурах (180—220°) [51, 52]. Поскольку эти условия сопоставимы с природными условиями залегания и развития нефтей [53, с. 35], то такую реакцию можно рассматривать как модель процесса образования алкиладамантанов в природных условиях. Иными словами, углеводороды ряда адамантана могли образоваться в нефти в результате изомеризации других полициклоалканов. В этом случае особенности изомерных превращений трицикло алканов иад твердыми катализаторами кислотного типа, полученные на модельных смесях, должны найти свое отражение в реальной картине, наблюдаемой в нефтях. Рассмотрим некоторые из них.
Из данных табл. 2.4 видно, что в нефти имеются те же изомеры алкиладамантанов, которые образуются при контакте гидрированных трициклических ароматических углеводородов (пергидроацеиафтеиа, пергидрофлуорена, пергидроантрацена) с катализатором иа основе оксида алюминия прн 200—220° С. В ряду изомеров с одинаковой молекулярной массой соотношения нефтяных алкиладамантанов далеки от равновесных н достаточно хорошо соответствуют их относительному содержанию в неравновесных (промежуточных) изомеризатах; последние характеризуются значительным содержанием менее устойчивых изомеров, содержащих заместители у вторичных атомов углерода идра.
Как будет показано ниже (см. гл. 3), при контакте с алюмоокисными катализаторами алкил адамантаны претерпевают изомерные превращения. Эти превращения управляются термодинамическими факторами и протекают в направлении образования термодинамически более устойчивых изомеров — соединений, содержащих алкильные группы в узловых положениях. Образование последних, т.е. 1-метил- (1-МА), 1,3-диметил-(1,3-ДМА), 1,3,5-триметнл- (1,3,5-ТМА) и 1,3,5,7-тетраметиладамантанов (1,3,5,7-ТеМА), является наиболее медленной стадией изомеризации. Эта особенность изомерных превращений, вероятно, и определяет относи
67
тельно небольшое содержание таких изомеров в нефтях. В этом случае относительное содержание менее устойчивых и более устойчивых трициклоалканов, в частности изомеров алкиладамантанов, может служить критерием оценки степени превращения нефтей на природных алюмооксидных и алюмосиликатных катализаторах.
Другой важный факт, подтверждающий образование углеводородов ряда адмантана в нефтях путем изомеризации, состоит в следующем. Автором [34] было установлено экспериментально, что с увеличением числа замещенных третичных атомов углерода в молекуле алкиладамантанов существенно уменьшается скорость их изомеризации. Отсюда вытекает, что при одном и том же времени контакта степень превращения менее устойчивых изомеров алкиладамантанов в более устойчивые должна быть различной для углеводородов различной молекулярной массы. В ряду алкиладамантанов С12-С14 степень превращения природных алкиладамантанов должна уменьшаться. Найденные экспериментально в нефтях соотношения менее устойчивых изомеров алкиладамантанов и наиболее устойчивого изомера хорошо согласуются с таким выводом для всех исследованных нефтей. Иными словами, для каждой нефти справедливо соотношение
ЬЭА < 1-М-З-ЭА 1,3-ДМ-5-ЭА
1,3-ДМА 1,3,5-ТМА 1,3,5,7-ТеМА ’
где 1-ЭА - 1-этиладамантан; 1,3-ДМА - 1,3-диметиладамантан и т.д.
Соотношения этих величин, полученные экспериментально, равны соответственно
в нефти 0,5 "г 0,6 < 1,2 т-1,6 < 2,8 -г 8,0
в промежуточном изомеризате 0,4 -г 0,6 < 0,7 + 1,3 < 2,8 -г 5,2
Необходимо отметить, однако, что имеются некоторые количественные различия в содержании отдельных изомеров в эталонных изомеризатах пергидроароматических углеводородов и нефтях. Так, например, в нефтях найдено более высокое содержание 1,2-ДМА. Это несоответствие может быть устранено, если использовать другой катализатор - аморфный алюмосиликат.
Еще одним показателем общности состава смесей алкиладамантанов нефти и искусственных изомеризатов является наличие незначительных количеств термодинамически малоустойчивых изомеров с мостиковыми заместителями, как то: 1,4-ДМА, 1,3,6-, 1,2,4- и 1,2,6-ТМА, 1,2,3,5-ТеМА и др. Общим для обеих смесей является также отсутствие пропиладаманта-нов.
Приведенные выше данные позволяют с достаточной степенью достоверности утверждать, что углеводороды ряда адамантана образовались в нефти по реакции изомеризации содержащихся в нефтях полициклических насыщенных углеводородов при контакте последних с природными алюмосиликатными и алюмоокисными катализаторами при 150-220°С. Содержание и соотношение изомеров алкиладамантанов нефти определяется природой катализатора, условиями и продолжительностью контакта с катализатором, т.е. кинетическими факторами.
68
Следует отметить, что для подтверждения этого вывода требуются дальнейшие исследования нефтей различных месторождений: определение содержания в них углеводородов ряда адамантана с учетом геохимических факторов — условий залегания, путей миграции и каталитических свойств сопутствующих пород.
Возможными предшественниками алкиладамантанов в нефти, по всей вероятности, являются конденсированные трициклические углеводороды — фрагменты, возникающие при деструкции реликтовых соединений нефти стеранов, тритерпанов и т.п.
* деструкция изомеризация
1 >> I — --—
(6) (7)
Холестан (6) и другие стераны являются реликтовыми соединениями нефти, т.е. соединениями, сохранившими основные черты строения исходных биологических структур - стероидов. В процессе созревания нефти эти соединения подвергаются термическим, термокаталитнческим и биологическим воздействиям, в результате чего могут образоваться более низкомолекулярные углеводороды, в частности трициклоалканы конденсированного типа (7). Превращения последних в алкиладамантаны, как уже отмечалось, протекают при контакте с природными глинами, обладающими слабыми кислотными свойствами, - алюмосиликатными и алюмо-окисными катализаторами. Заключительный этап превращения установлен н изучен экспериментально. Подробно изомеризация трициклических пергидроароматнческих углеводородов в алкиладамантаны рассмотрена в следующем разделе.
Менее ясны возможные источники образования тетра- и пентацикли-ческих адамантаноидных углеводородов нефти. В настоящее время известен ряд мостиковых углеводородов, которые в присутствии катализаторов кислотного типа образуют этаноадамантан, диамантан и другие подобные углеводороды, однако все они достаточно напряжены, в нефти не содержатся и получаются только синтетическими методами. Так, при контакте с галогенидами алюминия соединение (8) образует 2,4-этаноадамантан (О [54]; димер норборнена (9) образует диамантан (2) [55]; углеводород (10) - 1,2-пропаноадамантан (3) [56].
69
Вполне вероятно, что превращения в тетра- и пентациклнческие адаман-таноидиые углеводороды претерпевают не только мостиковые углеводороды, перечисленные выше, но и конденсированные тетра- и пентацикли-ческие углеводороды, происходящие от стеранов и трнтерпанов; экспериментальные данные по данному вопросу отсутствуют.
2.2. ПОЛУЧЕНИЕ АДАМАНТАНОВ КАТАЛИТИЧЕСКИМИ МЕТОДАМИ
Реакция изомеризации' наиболее часто используется для препаративного получения углеводородов ряда адамантана и служит основой промышленных процессов их получения. Существующее в ряде стран производство адамантана основано на использовании в качестве исходного вещества гидрированного димера циклопентадне на - тетрагидродициклопентадиена (ТГДЦПД) и различных катализаторов кислотного типа.
Экспериментально установлено, что структура адамантана как наиболее устойчивая образуется при изомеризации практически всех известных трициклических углеводородов. Необходимым условием таких превращений является образование в качестве промежуточных высокореакционноспособных частиц карбкатионов (в присутствии катализаторов). Роль инициаторов н катализаторов реакции изомеризации циклоалканов обычно выполняют кислоты Льюиса, сильные протонные кислоты, а также твердые катализаторы кислотного типа на основе оксида алюминия или алюмосиликатов.
2.2.1. ЖИДКОФАЗНАЯ ИЗОМЕРИЗАЦИЯ ПОД ДЕЙСТВИЕМ КИСЛОТ ЛЬЮИСА
Жидкофазная изомеризация в адамантан впервые наблюдалась Шпейером в 1957 г. при изучении превращений эндо-ТГДЦПД (трицикло[5,2,1,02>6] декана) (11) в его экзо-изомер (12) [45].
Реакция представляет значительный практический интерес, так как дициклоп ентадиен доступное вещество и легко гидрируется [56]. В зависимости от применяемого катализатора выходы адамантана изменяются в широком интервале (табл. 2.5). Этим же путем может быть получен сполна дейтерированный адамантан [70]. Метилтетрагидроднциклопентадиен (13), а также гидрированный димер норборнена и изопрена (14) в результате изомеризации образуют метиладамантаны [71—73].
\ А1С13
// /\ —[( + г(
(13) GH3 92„3 % 7,7%
70
При контакте алкил производных ТГДЦПД, содержащих одну или две метильные группы, с катализаторами А1С13-НС1, А1Вг3-НВг или BF3-HF наряду с 1-МА или 1,3-ДМА образуется до 36% метиладамантаиов с заместителями в мостиковых положениях ядра [74]. Характерно, что наличие алкильных групп существенно увеличивает выход конечных продуктов изомеризации. Высокий выход алкиладамантанов попудают при изомеризации (над галогенидами алюминия или комплексами на их основе) трициклических пергидроаромагических углеводородов состава Cj2-C14: пергидроаценафтена (15), пергидрофлуорена (16), пергидроантрацена (17) и других углеводородов [75—77].
Следует отметить, что впервые изомеризацию пергидроантрацена и пергидрофенантрена над А1С13 при 100° С изучали еще в 1938 г. [78], однако продукты превращения - лергидрофеналены (18) и алкиладамантаны не были идентифицированы.
Исходные пергидроароматические углеводороды получают гидрированием соответствующих ароматических углеводородов под давлением водорода на Ni Ренея или других гидрирующих катализаторах.
В качестве катализаторов обычно используют кислоты Льюиса, в частности жидкие каталитические комплексы, получаемые пропусканием НВт через смесь алюми-нийбромида и диметилгексанов (например, 10 г А1Вг3, 16 мл смеси диметилгекса-нов и газообразный НВг при 0° С)продолжительность реакции изомеризации несколько часов.
71
Таблица 2.5
Результаты жидкофазной изомеризации гидрированного димера циклопентадивна (ТГДЦПД) в адамантан
Условия реакции Выход адамантана, % Литература
А1С1з 15-20 [57,58]
А1СЦ.НС1, 40 атм Н3, 120°С 40 [59]
BF3, HF 30 [60]
BFj,HF, 50°С, 23 атм 73 [61]
А1С1Э, Н3О 43,2 [62]
А1С1Э, НС1, СН3 ОН, 70-120°С 35-45 [63]
А1С13, НС1, грег-С4Н9С1 40 [64,65]
А1Вг3, грет-С4 Н9 Вг 25-30 [66]
А1С13, НВг 15 [67]
А1С1а 15 [68]
SbFj.HF, ]00°С, 5 ч 47,2 [69]
Наиболее легко изомеризуется пергидроаценафтен (15); через 78 мин при 0°С продукты реакции содержат: 1-ЭА — 72,4%, 1,3-ДМА (20) — 1,2% и неидентифицированных промежуточных продуктов 27%. Последующее перемешивание реакционной смеси прн 25—28° С в течение 143 мин приводит к получению 96,6% 1,3-ДМА (20) и 3,4% неидентифицированных соединений (в настоящее время установлено, что это в основном диметнлада-мантаны, содержащие одну СН3-группу в мостиковом положении ядра). Изомеризация (16) и (17) в алкиладамантаны протекает медленнее, прежде всего, в связи с образованием в качестве промежуточных соединений устойчивых пергидрофеналенов (18), (21).
Доступность исходных соединений (соответствующие ароматические углеводороды в значительных количествах легко выделяются из жидких продуктов промышленных процессов коксования каменного угля и пиролиза нефтяных фракций), высокий выход конечных продуктов изомеризации вызвали большой интерес к методу изомеризации, о чем свидетельствует множество полученных патентов, например [79—87]. В патенте [88] предложено использовать в качестве катализатора А1Вг3 с добавкой 1-бром-адамантана и вести реакцию изомеризации прн 30-170° С и 1060— 260 мм рт. ст.; выход 1,3-ДМА из (15) составляет 89% и 1,3,5-ТМА (22) нз (16) - 74%. В качестве инициирующей добавки используют также трет- бутил бромид [66]. Высокоактивным оказался катализатор, полученный на основе А1С1Э и НВг [89]. Галогениды других металлов (ZnCl2, CdCl2, SnCU, SbCl3, SbCls) значительно уступают ему в каталитической активности при изомеризации пергидроароматическнх углеводородов [90].
Исследования по разработке методов получения адамантанов продолжаются. Недавно предложен [90а] препаративный метод получения 1,3-ДМА из промышленного ракетного топлива, состоящего в основном нз различных изомеров димера тетрагидрометилциклопентадиена. Эту смесь подвергают изомеризации под действием смеси бромистого алю
72
миния с трет-бутилбромидом в дихлорэтане, выход целевого продукта 50%, чистота 90%. Следует отметить, что выход 1,3-ДМА здесь ниже, чем при изомеризации пергидроаценафтена (15) [88],
В качестве исходных веществ для получения смеси алкиладамантанов может быть использована фракция трициклических углеводородов с т.кип. 145-217°С (13 мм рт.ст.), выделенная из пиролизного масла производства этилена. Гидрирование и последующая изомеризация этой фракции над А1С13 ПРИ 100 С приводит к алкиладамантанам Сц—Ci4, выход 20—40% [91, 92]. Пригодны также трициклические углеводороды, образующиеся с высоким выходом в результате превращений цикло до декатри-енов при их нагревании с триэтил- или трииэобутилалюмнннем [93].
Перегруппировка в адамантан н углеводороды ряда адамантана вызвала такой большой интерес исследователей, что были изучены многочисленные малодоступные полициклические углеводороды. Образование адамантана было установлено, например, при изомеризации трицикло [5,2,1,04-1 °]декана (23) [94], твистана (24) [95], протоадамантана (25) [96,97].
1?Ч)
Pd, 200-3004
Pt, 160-3004
(25)
Выход адамантана в этих реакциях существенно выше, чем при изомеризации ТГДЩ1Д. Структурная изомеризация соединения (25) имеет место также над платиновыми катализаторами [96, 97].
Гомоадамантен претерпевает быструю гидронзомеризацию в 2-метилада-мантан с выходом 76-84% [98], при этом практически не образуется более устойчивый 1-МА. Это обусловлено, вероятно, значительным различием
73
в скоростях реакций гидроизомеризации гомоадамантена и дальнейшего превращения 2-М А и 1-МА, т.е. кинетическими факторами.
А1Ьга, CS2
25°С, 2 мин
В отсутствие кислоты Льюиса (А1Вг3) 4-гомоадамантантил-катион наряду с 2-МА образует 4-гомоизотвистаи (26) в качестве основного продукта и гомо адамантан (27) [99 J.
Н2&Оч(конц.)
25°С,5-10 мин
(26) Ч5-50Х
25% (27)25%
В зависимости от исходного соединения, используемого для генерирования 4-гомоадамантил-катиоиа, суммарный выход продуктов превращения составляет 40% (из 4-гомоадамаитаиола) или 20% (из 4-гомоадамаита-на). С увеличением времени реакции состав продуктов меняется в связи с тем, что соединения (26), (27) и 2-МА способны изомеризоваться в 1-МА. При этом изомеризация 2-МА протекает, по-видимому, значительно медленнее, чем превращения (26) и (27), так как концентрация 2-МА (29% через 24 ч) все еще далека от равновесной (2%). С таким выводом согласуются результаты изомеризации гомоизотвистана (28); с выходом 85% он образует 1-МА за 10 ч [100],
СП,
Д1СЦ, СНгС12 20’С,10ч
(26)
Изомеризация трицикло [5 О2-6] ундекана (29), получаемого гидрированием димера циклогексадиена-1,3 и циклопентадиеиа, приводит к 1-МА (92%) [101,102].
А1Ха,СНгС1г_ 4 1 “ м Д
При контакте с 95%-ной Н2 S04 в метил цикл о гексане спирт (30) легко образует 2-МА [103],
(30)
74
Дигидроцедрен (31) служит источником относительно малодоступного 1,3,5-три мети л-7-этил адамантана (32) [104].
HjC
В углеводороды ряда адамантана изомеризуются также гомоадаманта-ны [105] и другие трициклические углеводороды различного строения [Ю6].
Скорость изомеризации в адамантан и алкиладамантаны существенно зависит от строения исходного углеводорода. Дня углеводородов ряда трицикло [5,2,1,02,6]декана относительная константа скорости изомеризации лежит в пределах 0,007-20, для трицикло [6,2,1,02,7 ]ундеканов и триццкло[53,1,02'6]ундеканов — в пределах 8—25, для названных выше пергидроароматических углеводородов — 0,001—1 [32, с. 245]. Скорость реакции иногда резко возрастает при наличии метильных и в особенности этильных заместителей.
Помимо трициклических углеводородов, источником адамантанов могут служить также би- и моноциклоалканы и даже углеводороды алифатического ряда или их функциональные производные; для таких превращений предложен специальный термин — ’’адамантизация” [104] (по аналогии с термином ’’ароматизация”). Естественно, что для такого рода превращений необходимы более жесткие условия (большие количества галогенидов алюминия, более высокая температура, продолжительность реакции) и выход продуктов существенно ниже, чем в случае трициклических углеводородов. Например, при контакте с галогенидами алюминия при 110—130 С в течение 2-5 дней смеси алкиладамантанов были получены из хол есте рола, холе стана, абиетиновой кислоты, циклогексана, сквалана, сквалена и других соединений; выход 2—45% в зависимости от исходного соединения [107]. В небольших количествах алкиладамантаны образуются также из циклопентана [108],циклогексана [109],триметилциклогекса-на [ПО], дициклопентила и д нцикл о гексила [111]; выход 5-10%. Если исходить из галогенпроизводиых, то реакции протекает и при более низких температурах. Так, смеси алкиладамантанов, вероятно, с весьма незначительным выходом образуются при контакте с А1С1з хлор- и брбмциклогек-санов или фенилциклоалканов [112, 113], а также циклододекана, полу-щемого из бутадиена [114],
Состав образующихся таким образом смесей алкиладамантанов достаточно сложен. Так, например, при контакте циклопентана с AlBr^ с добавкой трег-€4Н9Вт при 140-160°С с выходом 10 мае. % получают фракцию адамантанов следующего состава (%): адамантан - 0,5; 1-МА - 0,9; 1,3-ДМА - 13; 13Д-ТМА-22; 1,3,5,7-ТеМА - 14; 1-ЭА-З^; 1-М-З-ЭА-25,5; 1,3-ДМ-5-ЭА —10; 1,3,5-ТМ-7-ЭА - 10 [108].
Методы, основанные на реакции дегидроциклизации, в силу малой селективности и невысоких выходов, вероятно, не приобретут препаратив
75
но го значения. Вместе в тем, они представляют определенный теоретический интерес для понимания механизма процесса ’’адамантизации”. Дегидроциклизация проходит ступенчато с образованием в качестве промежуточных соединений бициклических углеводородов ряда декалина [109, 111] . Дополнительное подтверждение этому — увеличение выхода алкиладамантанов при использовании в качестве исходных соединений декалина и метил декалинов. При контакте AICI3 -комплекса с декалином, метил декалинами и гидринданом при 75°С в течение 60 ч выход смеси адамантанов достигает 30% [115]. Катализатором ’’адамантизации” декалина является также хлорид галлия [116]. Образование в результате ’’адамантизации” преимущественно полиметилэамещеиных в узловых положениях алкиладамантанов, количество которых значительно превышает содержание незамещенного адамантана, объясняется тем, что в условиях реакции активно протекают алкилирование адамантана фрагментами алкильных групп [117], а также реакции изомеризации и перераспределения (диспропорционирования) алкильных, в особенности метильных, групп в алкилада-мантанах [20], что приводит в итоге к термодинамически наиболее устойчивым соединениям. Кроме того, уменьшение числа или полное отсутствие реакционноспособных третичных С-атомов (например, 1,3,5,7-ТеМА) делает эти соединения в условиях реакции наиболее устойчивыми также кинетически.
В продуктах ’’адамантизации” наряду с алкиладамантанами содержатся различные углеводороды, что является следствием протекания реакции деструкции, переалкилирования и перераспределения водорода.
Насыщенные полициклические углеводороды, содержащие больше трех циклов, при контакте с кислотами Льюиса образуют углеводороды адамантаноидного строения. Тетрацикло [6,4,0,045,0s ] додекан (33) при обработке комплексом иа основе А1Вг3 _в CS2 при 0—25°С образует 2,4-этаноадамантан (*40—90%) [54]. К такому же результату приводит превращение пеитацикло [6,4,0,02,1О>03'7,0s'9 ] додекана (8) [54] иицеана (34) [118]. Вместе с тем 2,4-этаноадамаитан не удалось получить из соответствующего тетрациклического углеводорода (35), а также из (36) [56].
ГЗЧ)
76
2,4-Этаноадамантаи, несмотря иа наличие в его молекуле пяти- и семичленного циклов, является весьма устойчивым углеводородом. Его энергия напряжения (Ё) составляет всего 76,2 кДж/моль, в то время как для (33) и (34) энергия напряжения составляет 162,5 и 94,9 кДж/моль соответственно [54].
Метилпроизводиые 2,4-этаноадамантана образуются в результате перегруппировки метилтетрацикло алканов (37) и (38), при этом изомерные 1-, 2- и 8-метилэтаноадамаитаны образуются в соотношении 5:3:2 [1191.
(Зв)
Метилциклоалканы (39) и (40), подобно (35), не способны образовывать структуру 2,4-этаноадамантана. Вероятно, механизм перестройки молекулы этих углеводородов требует непременного разрыва одного из имеющихся циклов.
Установлено, что метилэтаноаддмантаны в определенных условиях претерпевают обратимую изомеризацию в 1,2- и 2,4-проп ано адамантаны (1,2- и 2,4-триметилеиадамантаны) [119]. К сожалению, ввиду значительной деструкции углеводородов, приводящей к образованию трициклических углеводородов (алкиладамантанов), состояние равновесия (и, следовательно, относительную термодинамическую устойчивость) этих двух групп углеводородов ряда адамантана экспериментально установить не удалось.
При изомеризации тетрациклических мостиковых углеводородов определенного строения в качестве основных продуктов образуются также 12 -ал кано адамантаны. 1,2-Пропаноадамантан (тетрацик-ло[6Д1,16’10,02,6]тридекан, 4) и 1,2-бутаноадамантан (тетрацик-
ло [7,3,1,171,О2’7]тетрадекан, 5) образуются, в частности, при контакте с А1Вг3 при комнатной температуре соответственно 4-метилтетрацик-ло [6,2,1,13 ’б ,0?,7] додекана (10) и тетрацикло [8,3,0,12 ’9,03’8 ] тетрадекана (41) [56].
77
AlBr}, 2Q°C
(41)
(5)
Изомеризация тетрациклоалкана (42), полученного гидрированием аддукта иорбориена и циклогексадиена, приводит к образованию наряду с 1,2-пропаноадамантаном (4) его 2,4-изомера [119]. Соотношение этих углеводородов (97:3) отражает большую термодинамическую устойчивость 1,2-изомера.
Благодаря этому превращению 1 ^-пропаноадамантан становится впопие доступным тетрациклическим углеводородом ряда адамантана.
Приведенные ниже тетрацикло алканы состава С15Й24 и С1бН2б, как конденсированные, так и мостиковые, в результате изомеризации (А1Вг3, трег-СдЬ^Вг, 20°С) образуют соответственно метил- (43) и диметил-(44) -1,2-бутаноадамантаны [120].
В обоих случаях в
метильные группы в других положениях циклической структуры 1,2-бута-иоадамантана, однако преобладают термодинамически наиболее устойчивые изомеры (43) и (44).
Рассмотренные выше примеры указывают иа ряд особенностей изомеризации полициклоалканов в углеводороды каркасного строения.
1. Важную роль в процессах изомеризации полициклоалканов в углеводороды ряда адамантана играет наличие алкильных групп в молекуле исходного углеводорода. Так, не содержащий алкильных групп углеводород (36) не способен изомеризоваться, в то время как его метильный (10) и изопропильный (45) гомологи легко образуют соответствующие алканоадамантаны. Алкильные группы могут образовываться в результате изомеризации (например, в случае превращений пергидроароматических
76
углеводородов), а также предоставлять атомы углерода, необходимые для образования циклов [изомеризация соединений (10) и (45)].
2. При изомеризации тетрациклоалканов 1,2-алканоадамантаны образуются предпочтительнее чем 2,4-алканоадамаитаны [изомеризация углеводорода (42) ]. Это обстоятельство может быть дополнительным фактором, затрудняющим изомеризацию соединения (36) - ведь 1,2-этаноада-мантан, который мог бы здесь образоваться, вероятно, крайне напряжен.
3. В процессе изомеризации полициклоалканов весьма вероятен гидрогенолиз одного из циклов (вероятно, наиболее напряженного в переходном состоянии) и образование каркасных углеводородов меньшей по сравнению с исходными цикличности. Так, например, при изомеризации трициклического эндо-ТГДЦПД в адамантан выход бициклического углеводорода — транс-декалина — может достигать 87% [121].
Пентациклические насыщенные углеводороды при изомеризации образуют диамантан (следующий член гомологического ряда углеводородов алмазоподобного строения состава С]4Н2о) или его алкильные производные. Пентацикло[8^,1,14,7,02,9,08,3]тетрадекан (46) был первым углеводородом, из которого методом изомеризации был получен диамантан [55, 56,121,122].
(46)
Низкий выход днамантана (1%) вызван разрушением одного из циклов: это подтверждается тем, что одновременно образуется тетрациклический 1,2-бутаиоадамантаи с выходом до 40%.
1-Три метил сил ил адамантаны получают, используя изомеризацию трициклических углеводородов с три метил силильным заместителем в присутствии АИЗгз [ 122а].
Целый ряд полициклических мостиковых углеводородов (47)—(57), полученных иа основе иорбориена по реакции Дильса—Альдера с различными циклическими диенами, также способен перегруппироваться в диамантан
Выход диамантана существенно зависит от строения исходного углеводорода, применяемого катализатора, условий проведения реакции и колеблется от 10 до 90% [121]. Углеводороды (48) и (49) при контакте с А1Вг3 или с предварительно полученными на его основе комплексами низкомолекулярных углеводородов образуют диамаитаи, выход 20—25%. Использование гетерогенного катализатора платины, нанесенной на хлорированный А12Оз,повышает выход диамантана до 45% [123—125]. Изомеризация (52) и (53) в присутствии комплекса А1Вг3 приводит к диамантану [126,127] -
другие
Продукты
80
В качестве побочных продуктов образуются тетра- и трициклические углеводороды ряда адамантана, в том числе 1,2-бутаноадамантан, 1,3,5,7-тетраметиладамантан и др.
Наиболее пригодным для препаративного получения диамантана считается (4+4) -димер норборнадиена (54), тривиальное название - бинор-С [128 -131 ].
СоВгг *2РРЬ3,
(54)
(55),(56)
AlBr3
BF3 • OEt£
, Pt
200 °C,305 атом
г
Гидрирование бииора-С (54) приводит к раскрытию циклопропановых колец и образованию пентациклических углеводородов (55) и (56). Оно протекает с практически количественным выходом при 200° С и 305 атм Н2 в присутствии Pt-катализатора [129] или PtO2 в ледяной СН3СООН [121, 126, 127]. Изомеризация (55) и (56) над А1Вг3 или его комплексом с низкомолекулярными углеводородами приводит к диамантану с выходом 60—70% [126, 127]. Выход 70—90% диамантана был получен при использовании катализатора А1С13 в СН2С12 [123, 124]. грдяс-Пента-цикло[8,2,1,12,5,03,7,08’12 ]тетрадекан (57) образует диамантан с выходом 84% [121]. Выход диамантана в расчете на коммерческий норборна-диен превышает 50%.
В качестве исходных углеводородов для получения диамантана недавно предложено использовать также гидрированные димеры цикл о гептатри-ена [132]. В присутствии катализатора (С2Н5)2 А1С1—Т1С14 димеризация циклогептатриена происходит через стадии [ттбх + тт2$]- и [n4s + п2$] -присоединения. Образуются пентацикло[8,4,0,03,7,04’14,06,1 1 ]тетрадекадиен-8,12 (58) и лентацикло[7,5,0,02,8,05'14?07,11 ]тетрадекадиен-3,12 (59).
практически количественно, изомеризация образующегося при этом насыщенного углеводорода в диамантан сопровождается лишь весьма незначительным образованием полимерных продуктов.
Метилдиамантаны получают изомеризацией пентациклических углеводородов (60) или (61) [133] и го мо диаман тана (62) [134].
6. Зак. 1856
81
А1Вг3) кипячение ь циклогексане
(62)
При изомеризации соединения (60), помимо термодинамически наиболее устойчивого 4-метилдиамантана (63) (65%), образуются его изомеры — 1-метил- (64) и 3-метил диамантан (65) в количествах 10 и 6% соответственно, а также диамантан (5%) и различные алкиладамантаны (18%), Эти же углеводороды образуются при изомеризации гидрированного тримера циклопентадиеиа (66) [ 135,136].
Гидрированный димер 2-метилнорборнеиа (67) [133], а также гидрированный аддукт дициклопеитадиеиа и 1,3-циклогексадиеиа (68) [135] при контакте с кислотами Льюиса образуют диметилдиамантаны; наряду с наиболее устойчивым 4,9-диметил диамантаиом (69) в реакционной смеси содержатся меиее устойчивые 1,4-, 2,4-, 4,8-и 3,4-диметилдиамаитаны.
При изомеризации полицикланов в углеводороды ряда адамантана нередко имеет место ситуация, когда в молекуле исходного углеводорода число атомов углерода недостаточно для образования очередной ячейки
82
адамантана. В простейшем случае, например при изомеризации трициклоал-каиа (70), образуется иорадамаитаи (71) [137].
H0Ag;50°c * / 25°С;О,5т
(70) 69% ' <71) 75%
Тетрациклические углеводороды при такой нехватке атомов углерода изомеризуются в весьма устойчивые изомеры этаноиорадамантана [138]. Так, тетрацикло [6,3,0,02,6,0s ,9] ундекан (72), иорицеаи (73) и метанотвис-тан (74) при контакте с А1Вг3 образуют смесь из 97% 2,4-этаиоиорадамаита-на (75) и 3% 2,8-этаноиорадамантана (76).
При этом норицеан (73) образуется в качестве промежуточного продукта при изомеризации (74). Этаноиорадамантаны (75) и (76) являются наиболее устойчивыми изомерами из 2486 возможных тетрациклических углеводородов общей формулы Ci i Hj 6.
Образование адамантанов с этановым мостиком (с пятичленным циклом) весьма характерно также для изомеризации бопее сложных полициклических углеводородов. Так, превращение изомеров димера гекса-гидроциклооктатетраеиа (77) над А1Вг3 приводит к двум изомерам эта-нодиамантана — (78) и (79) [139].
83
Другой структурный димер циклооктатетраена после гидрирования превращается в соединение (80), которое изомеризуется в бисэтанобис-нордиамантан (81) и соединение (82) [139].
Несмотря на то что изомеризация сложных полициклических структур протекает с трудом, в ряде случаев этим методом можно получить углеводороды каркасного типа регулярного строения, например триамаитан (83). Его предшественниками могут быть непредельный углеводород (84) [140] и тептациклические изомерные углеводороды (85) и (86). Последние получают из бинора-С (54) и бутадиена-1,3 [141].
В4
Бинор-С в газовой фазе над Pt-катализатором или в жидкой фазе в присутствии AgC104 превращается в два димера норборнадиена — (87) и (88), которые образуют аддукты (85) и (86) с бутадиеном-1,4; гидрирование и последующая изомеризация этих аддуктов приводят к триаман-тану. Использование в качестве диена изопрена на стадии образования аддуктов позволяет получить олефины структуры (85) и (86) с метильными группами; дальнейшими превращениями аналогично получают смесь метилтриамантанов с преобладающим содержанием термодинамически бопее устойчивого 9-метилтриамаитаиа (89) [142].
СН3
сн,
(89)
Существенно улучшен метод синтеза каркасных углеводородов из полициклических предшественников за счет проведения изомеризации в присутствии сверхсильных кислот [142а]. эндо- и экзо-Триметиленнорбориан количественно превращается в адамантан в присутствии тристрифлата бора B(OSO2CF3)3. Этот же катализатор, а также сопряженные сверхкислоты
CF3SO3H -SbF5 и CF3SO3H-B(OSO2CF3)3
позволяют получить из тетрагидробинора-С диамантан с выходом 99%, а из гептациклоокта деканов — триамаитан с выходом 70%.
диамантом,
Время изомеризации заметно уменьшается при действии ультразвука, а также в присутствии промоторов — добавок 1-хлор- или 1-фторадаман-
триамаитан.
танов.
Попытка получения тетрамантаиа (91) методом изомеризации была безуспешной [143]. В качестве исходного соединения использовали гидрированный аддукт содимера циклооктатетраена с циклогексадиеиом (90). В результате изомеризации иад А1Вг3 был получен бастардан (92).
CSg 100eCf 2-9ч
(90)
85
(90
Бастардан содержит в своем каркасе напряженный иорборнаиовый фрагмент, и в этом связи его неспособность к дальнейшей изомеризации непонятна. Вероятно, возникают кинетические затруднения.
Не привела к успеху также попытка получения тетрамаитана (91) изомеризацией иоиациклического углеводорода (93), полученного из продукта превращения бинора-С [142].
Углеводород (93) изомеризуется только до образования структуры трнамантана с фрагментом С5. На этой стадии образуется неизвестная структура, препятствующая, дальнейшей изомеризации в тетрамантан.
85
2.2.2. ГАЗОФАЗНАЯ ИЗОМЕРИЗАЦИЯ В УСЛОВИЯХ ГЕТЕРОГЕННОГО КАТАЛИЗА
Прежде чем рассматривать реакции газофазной изомеризации уместно напомнить, что катализаторы типа галогенидов алюминия обладают рядом недостатков, в особенности существенных при использовании их в промышленности. К ним относятся: повышенная коррозионная активность, нестабильность, невозможность регенерации, образование в процессе реакции значительных количеств смолы. Это обстоятельство служило стимулом для исследования изомерных превращений полициклоалканов с использованием стабильных гетерогенных катализаторов кислотного типа, получаемых, как правило, на основе оксидов металлов.
Платз с сотр. [49] первыми показали, что трицикло[5,2,1,02’6] декан (ТГЛТТП.П) при контакте с аморфным алюмосиликатным катализатором при 400-475° С образует сложную смесь продуктов, содержащую 6—13% адамантана, В табл, 2.6 приведена сравнительная оценка активности катализаторов на основе оксида алюминия в реакции изомеризации пергидроаценафтена в алкиладамантаны [20, 51, 52]. Оптимальная температура реакции в случае катализатора H2SO4/A12O3 составляет 220—240°С, а для алюмосиликатных и цеолитсодержащих катализаторов (ЦЕОКАР и АШНЦ) — 280—320° С. Препаративный выход алкиладамантанов достигает 60-70%.
Таблица 2,6
Превращения пергидроаценафтена над алюмоокисными катализаторами*1
Катализатор Температура, 'с Время контакта, мин Степень превращения по реакции
изомеризации дегидрирования деструкции
Оксид алюминия, обработанный: H,SO4 (2N) 210 0,01 97 3
HaSO4 (2N) + 260 1 — 90
+ Pt (0,5%) HF (1%) 235 10 90 10
HC1(1O%) 350 20 — — 10
H3PO4 (10%) 350 20 — — 10
Алюмосиликатный 190 0,01 65 — 15
катализатор крекинга ЦЕОКАР-1 290 0,01 80 — 15
АШНЦ-3 310 0,03 70 — 20
АП-56*’ 360 0,1 98 —*
Окнснохромовый 380 20 — 95 5
Кобальт-алюмомолиб- 380 15 — 95 5
ценовый
** Реакции проводили в микрореакторе.
*3 Реакцию проводили в атмосфере водорода.
87
Таблица 2.7
Состав продуктов изомеризации пергидроаценафтена над HjSO^/Al) Оэ (200-210° С, 0,1 ч“*, ПГАН: катализатор = 1:5)
Продукты реакции Т. кип., ° С Содержание (мае. %) в изомеризатах
1 । 2 1 3
1,3-Ди метил ад а манта и 201,5 45 67 80
, циС‘1,4-Диметиладамантан 212 14 8 4
транс-1,4-Диметиладамантан 213 13 8 4
1,2-Диметиладамантан 219 9 5 3
1-Эти л адамантан 223 12 9 7
2-Этиладамантаи 227,5 4 3 2
^идентифицированные соединения — 3 — —
В лабораторных условиях изомеризацию трициклических пергидроароматическнх углеводородов в алкиладамантаны над алюмооксидным катализатором обычно проводят на установке проточного типа со стационарным слоем катализатора в атмосфере воздуха, инертного газа или водорода (при повышенном давлении). Катализатор H1SO4/A1JO3 готовят путем выдерживания у-А12О3 в 2-3 N растворе HaSO4 в течение 48 ч с последующей промывкой и сушкой прн 120° С. Перед опытом катализатор "тренируют” в атмосфере сухого воздуха или азота до рабочей температуры (220-250°С;)3* Катализаторы ЦЕОКАР и АШНЦ дают лучшие результаты при предварительной тренировке в токе влажного воздуха.
Исходные пергидроароматнческие углеводороды получают гидрированием соответствующих ароматических углеводородов (аценафтена, флуорена) над Ni Ренея при 200“ С и 150 атм Н2 или над другими катализаторами гидрирования. Гидрированные углеводороды пропускают через слой катализатора изомеризации с объемной скоростью 0,1 л на 1 л катализатора в час; объемное соотношение сырье: катализатор = 1:10 при зернистости AljOa 0,5-1 мм. Катализатор регенерируют прокаливанием в токе сухого воздуха при 550-6001 С до полного выжигания продуктов уплотнения.
Типичный состав продуктов изомеризации пергидроаценафтена над На8О4/А1аОэ-каталнзатором приведен в табл, 2.7.
Изомеризаты 1, 2 и 3 получены пропусканием через спой свежего катализатора соответственно пергидроаценафтена, изомеризата 1 и изомеризата 2. Выход изомеризатов 1, 2 и 3 составляет соответственно 90, 82 и 75% в расчете на исходный пергидроаценафтен.
Характерной особенностью изомеризации полициклоалканов в условиях гетерогенного катализа являетси многокомпонентный изомерный состав смеси получаемых алкиладамантанов; в отлнчие от изомеризации над комплексами галогенидов алюминия здесь образуются в значительных количествах изомеры с мостиковым положением алкильных групп.
Прн изомеризации пергидрофлуорена получают (аналогично реакции с пергидроацеиафтеном) изомеризаты 1, 2 и 3 с выходом 65, 50 и 40% соответственно (табл. 2.8), содержащие весьма устойчивый промежуточный продукт реакции — транс-пергидрофенален. Общая сумма изомеров алкиладамантанов, содержащих алкильные группы в мостиковых положениях, в данном случае превышает 60% (в изомеризате 1).
1Попадание в нагретый катализатор вместе с органическим веществом воды приводит к его разложению.
88
Таблица 2.8
Состав продуктов изомеризации пергидрофпуорена над HjSO^/AIjOj (220°С)
Продукты реакции Т. кип., ° С Содержание (мас.%) в изомератах
1 2 3
1,3,5-Триметиладамантан 205 6 18 50
1,3,6-Триметил адамантан 215,5 14 12 6
цие-1, 2,5 -Три метил адамантан 221 10 8 4
транс-1,2,5-Трнметиладамантан 222 11 8 4
1 -Метил-3-этиладамантан 226 14 25 27
дис-1 -Метил-4-этиладамантан 230 5 3 2
транс-1 -Метил-4-этиладамантан 231,5 4 3 2
1,2,6-Трнметил адамантан 233 2 1 —
1,2,2-Триметиладамантан 233,5 2 1 —
траноПергидрофенален — 25 15 1
Невдентифицированные соединения — 7 6 4
Таблица 2.9
Состав продуктов изомеризации пергидроантрацена иад HaSO4/AlaO3 (220°С)
Пр одукты реакции Т. кип. ° С Содержание (мае изомеризате 1 । 2 %) в 3
1,3,5,7-Тетра метил адамантан 208,5 2 3 9
1,3,5,6-Тетра метил адамантан 224,5 15 15 25
1,3-Диме тил-5-этиладамантан 228,5 11 15 35
Другие алкиладамантаны С\ 4 — 42 42 21
Метилпергидрофеналены — 14 18 4
Пергидроантрацен — 16 7 6
При изомеризации пергидроантрацена (табл. 2.9) выход изомеризатов 1,2 и 3 составляет 50, 42 и 35% соответственно. Кинетически крайне затруднено образование термодинамически наиболее устойчивого изомера -1,3,5,7-ТеМ А.
Промышленные цеолитсодержащие катализаторы типа ЦЕОКАР и АШНЦ менее активны в реакции изомеризации в адамантаны по сравнению с катализатором H2SO4/A12O3, ио превосходят его по механической прочности и стабильности. Эти катализаторы представляются более перспективными для использования в промышленности. Катализаторы типа ЦЕОКАР содержат ~20% цеолита типа Y в лантановой форме или в виде смеси редкоземельных элементов (ЦЕОКАР-1). А1ПНЦ-3 содержит ~ 18% цеолита-типа Y в водородной форме.
89
Показатели процесса изомеризации ПГАН над этими катализаторами следующие: температура 260~290°С, объемное соотношение сырье : катализатор = 1:4, выход изомеризата 87 мас.%; состав продуктов реакции (мас.%); 1,3-ДМА - 13,3%, 1,4-ДМА - 20,7%, 1,2-ДМА - 8,9%, 1-ЭА - 14,5%, 2-ЭА - Ц,2%. Общее содержание алкиладамантанов в изомеризате 68,6% при степени превращения пергидроаценафтена 94%.
Катализаторы типа ЦЕОКАР и АШНЦ активны также в реакции изомеризации тетрагидродициклопента диена в адамантан; максимальный выход адамантана составляет 34% [144]. Цеолитный катализатор, содержащий нанесенные металлы (Pt 0,75; Re 0,25; Со 3 мас.%), позволяет получить адамантан с выходом 40—52% при селективности 35% [145].
Катализатор регенерируют в реакторе нагреванием его при 220° С 3-5 ч в атмосфере водорода в отсутствие изомеризуемого углеводорода и НС1.
Нанесение некоторых металлов (Ni, Со, Pd) в 1,5 раза повышает активность аморфных алюмосиликатных катализаторов в реакции изомеризации пергидроаценафтеиа в алкиладамантаны [146].
Катализаторы типа палладий на силикагеле при 200° С в атмосфере водорода активны при изомеризации в адамантан и алкиладамантаны также некоторых мостиковых углеводородов [97, 147]. Так, протоадамантан [32] с высокой селективностью изомеризуется над этим катализатором в адамантан с выходом 3,75 и 99% адамантана соответственно при 152, 240 и 300° С.
Изомеризация 2,3-тетраметиленнорборнана в газовой фазе над платиновым, нанесенным на хлорированный А12О3, катализатором приводит к смеси 1- и 2метиладамантанов. При большой скорости газового потока может быть получен практически чистый менее устойчивый изомер 2-метил-адамаитаи [72, 142]. Как уже отмечалось выше (с. 80), этот катализатор весьма активен также в изомеризации некоторых пеитациклических углеводородов в диадамантаи [123—125].
Опубликованы данные по активности различных цеолитных катализаторов в реакциях изомеризации полициклоалкаиов в адамантаны [148]. В 1976 г. специалисты фирмы ’’Идэмицу косан” разработали и предложили промышленный вариант процесса получения адамантана, 1-МА и 1,3-ДМА с использованием катализаторов изомеризации, полученных нанесением различных металлов на цеолит с применением ионообменииков [149]. В заявке [149а] представлен также способ получения адамантанов изомеризацией трициклических углеводородов в атмосфере водорода или хлористого водорода на цеолитном катализаторе, приготовленном с применением ионообменииков н содержащем Pt, Ni, Со или Re. Этот способ близок к предложенному ранее способу [149].
Как показали исследования, для протекания реакции изомеризации полициклоалканов необходимо наличие на поверхности катализатора протонных кислотных центров [150—154; 155, с. 170].
Подводя итоги рассмотрения поДразд. 2.2.1 и 2.2.2, нужно признать, что метод жидкофазной изомеризации является эффективным, а зачастую и единственным методом получения каркасных углеводородов, в особенности углеводородов алмазоподобного строения, имеющих регулярную адамантаноидную структуру. Важную роль играет Правильный выбор исходного углеводорода, он должен иметь более напряженную структуру, чем конечный продукт изомеризации. Мак-Кервей [142] считает, что это 90
напряжение, однако, не должно быть слишком большим, так как в таких случаях (например, при наличии трехчленных циклов) активно будут протекать реакции деструкции. Необходимо учитывать также и кинетические затруднения, такие, как возможность образования ионов карбо-иия и их устойчивость.
Реакции газофазной изомеризации полициклоалкаиов различного строения в углеводороды ряда адамантана успешно идут также в присутствии твердых алюмо окисных., цеолитсодержащих и цеолитных катализаторов в условиях гетерогенного катализа. Это создает благоприятные предпосылки для осуществления миогоюннажного промышленного производства адамантана и алкиладамантанов иа основе циклопентадиена, аценафтена, флуорена и других продуктов и отходов коксохимического и пиролизного производства. Работы в этом направлении активно развиваются промышленными фирмами, и можно полагать, что адамантан и его гомологи вскоре станут вполне доступными миоготоннажными исходными веществами для получения смазочных сред, полимерных материалов с улучшенными технико-эксплуатационными характеристиками, биологически активных соединений, лекарственных препаратов и других ценных народнохозяйственных продуктов.
2.3. ПОЛУЧЕНИЕ АДАМАНТАНОВ СИНТЕТИЧЕСКИМИ МЕТОДАМИ
Рассмотренные выше каталитические методы изомеризации полициклоалкаиов являются весьма эффективными методами получения углеводородов ряда адамантанов. Многие из иих приобрели препаративное значение, а процесс получения адамантана изомеризацией ТГДЦПД реализован в промышленном масштабе.
Однако по мере повышения молекулярной массы и увеличения числа циклов исходного углеводорода перегруппировка в адамантаноидные углеводороды регулярного строения затрудняется. В ряде случаев методы изомеризации не дают желаемого результата. Так, с их помощью невозможно получить 2-замещенные алкил- и ариладамаитаны. Кроме того, продукты реакции, как правило, состоят из смеси нескольких изомеров, и требуется их разделение.
В этой связи широкое развитие приобрели синтетические методы получения углеводородов ряда адамантана, основанные иа применении функциональных производных адамантана в качестве исходных веществ, а также методы циклизации — построения структуры адамантана исходи из алифатических, моио- и бициклических соединений. Синтезы иа основе функциональных производных широко используются для получения индивидуальных алкил-, циклоалкил- и ариладамаитаиов. Методы циклизации используют обычно при синтезах полифункциоиальных производных адамантана, углеводородов адамаитаиоидиого строения и их производных.
В синтезах углеводородов ряда адамантана из их функциональных производных используют традиционные методы воздействия иа функциональные группы (реакции Вюрца, Гриньяра, Кижиера, Клемеисеиа и др.). Эти реакции протекают с производными адамантана, как правило, без каких-либо осложнений, за исключением случаев, когда возникают существенные пространственные затруднения.
Широко используются также реакции алкилирования с применением различных алкилирующих агентов при воздействии термических, радиационных и каталитических факторов. Реакции алкилирования адамантана и алкиладамантанов имеют самостоятельное значение как один из важных разделов химии этих соединений и будут рассмотрены отдельно.
2.3.1. СИНТЕЗ АДАМАНТАНОВ, ЗАМЕЩЕННЫХ В УЗЛОВЫХ ПОЛОЖЕНИЯХ
Одним нз первых успешных синтезов 1-метиладамантана был многостадийный синтез на основе 1-бромадамантана [156].
Н,О, ТГФ
1-AdBr --------------> 1-AdOH -► l-AdCOOH-> 1-AdCOCl •*
СаСО3, AgNO3
ТГФ
•* 1 - AdCOOC2 Hs •* 1 - AdCH2 OH •* 1 - AdCH2 OSO2 C6 H5-> 1-AdC H3
25%
(1-Ad- l-адамантил).
Позднее был найден другой путь [157]:
АсОН, нх
l-AdCONH2 ->l-AdCH2NH2 ------------>
160° с
1 Р 6 Н в fl
->l-AdCH2X ----------► l-AdCH3 (X=Cl,Br).
CHjOH
Этот же углеводород образуется также прн гидрировании в автоклаве 1-адамантанкарбоновой кислоты (катализатор MoS2, 360°С, 100 атм Н2) [158], при взаимодействии 1-бромадамантана с CH3MgBr (эфирный раствор, 100° С, выход 83%) [159]. В условиях последней работы взаимодействие 1-AdBr с C2HsMgBr приводит к 1-этиладамантану (выход 39%).
Метод [160], приведенный ниже, можно рассматривать как общий метод синтеза алкиладамантанов, попизамещенных в узловых положениях, Он позволяет путем постепенного наращивания углеводородной цепи получить алкиладамантаны с различной длиной алкильных групп нормального строения.
вг3 нсоон
l-AdCH3 -----► 1,3-А(СН3)Вг -------> 1,3-А(СН3)СООН->
HiSO4
1 .СдНдОН НВг
2 LiAIH * 1,3-А(СН3)СН2ОН---------> 1,3-А(СН3)СН2 Вт--+
' Ь АсОН
СО,
-> l,3-A(CH3)CH2MgBr-----> 1,3-А(СН3)СН2СООН—>
1. С2 Н« ОН НВг Mg
----------* 1,3-А(СН3)СН2СН2ОН-----► 1,3-(СН3)СН2СН2Вт—=—►
2. ЫА1Н4 АсОН
Н, О
+ 1,3-А(СН3)СН2СН2MgBr —---► 1,3-А(СН3)СН2СН3
Ь------1,3 -А(СН3 )СН2 СН2 СООН и т.д.
(1,3-А — 1,3-адамантилен).
92
Поли замещенные метил адамантаны можно получить из полифункцио-нальных адамантаикарбоновых кислот действием SOC12 в СН3ОН и восстановлением полученных эфиров NaAlH4 [161, 162]. 1,3,5,7-Тетраметил-адамантан, например, был получен этим методом с выходом 61% [162]. Позднее был предложен метод одностадийного восстановления метилза-мещенных адамантанкарбоновых кислот в углеводороды с помощью алюмогидрида лития [163].
1-Этил- и 1-пропиладамантаны могут быть синтезированы по реакции Вюрца—Внттига исходя из 1-бромадамантана и соответствующих галоге-налкилов. Реакцию ведут прн комнатной температуре в абсолютном эфире над металлическим натрием [164]. При 200° С в присутствии натрия 1-бромадамантан реагирует с бензолом с образованием 1-фениладамантана, последний гидрируют в 1-циклогексиладамантан,
Нагревание 1-бромадамантана с толуолом приводит к n-(l-Ad)C6H4CH3 [165]. Реакция 1-AdBr с нафталином протекает при 80-120° С в присутствии ZnCl2 с образованием 2-(1-адамантил) нафталина [166—168]. 1- и 2- (1-адамантил) нафталиныполучаюттакже взаимодействием 1-AdBr соответственно с 1- и 2-литИЙиафталином [167].
При взаимодействии алкиллитиевых соединений с адамантанкарбоновыми кислотами образуются соответствующие адамантилалкилкетоны, которые затем легко могут быть переведены в углеводороды, например, восстановлением по Клеменсену [169].
RLi RLi
1-AdCOOH -----► 1-AdCOOU ------> l-AdCCRXOLi^ + 1-AdCOR •*
| i.AdCH2 R.
$ C3 Hs OH-HC1
г
й Этим методом получены 1-этил-, 1-н-бутил- и 1-трет-бутиладамантаны. $ В качестве побочных продуктов в реакции 1-AdCOOH с RLi образуются спирты: 2-(1-адамантил)пропанол-2, 3-(1-адамантил)пентанол-3 и 4-(1-адамантил) гептанол-4, которые в результате дегидратации и гидрирова-ния дают соответственно 2-(1-адамантил)пропен и 2-(1-адамантил)пропан;
.£ 3-(1-адамаитил)пеитен-2 и 3-(1-адамантил)пентан,4-(1-адамантил)гептен-3 и 4-(1-адамантил)гептан.
1-Изопропиладамантан получают гидрированием соответствующего непре-5 дельного углеводорода — 2-(1-адамантил) пропена [170]. Олефии легко получить дегидратацией спирта, образующегося при взаимодействии метил (1-адамантил)кетона с реактивом Гриньяра СН3MgBr.
В реакции Гриньяра могут быть использованы эфиры адамантанкарбоновых кислот. При применении соответствующих реактивов Гриньяра с последующей дегидратацией спиртов и гидрированием трициклоалкенов / были синтезированы 1-изопропил-, 1-е гор-бутил а дам ан тан, 2-(1-адамантил)-£ пентан и 3-(1-адамантил) пентан [14], 1-трет-бутиладамантан (выход 22%) I [169], 1-метил-З-изопропиладамантан (выход 75%) [171], 1,3,5-триметил-1 7-изопропиладамантан [172], 1-изобутиладамантан [173] н др. [174].
93
Мегила да мантаны [175], этиладамаитаи [176], изопропиладамантан [170] и др. получают с использованием реакции карбоксилирования адамантана или бромадамаитаиа с последующим получением и восстановлением кетонов или сложных эфиров кислот, например, по схеме
1-AdBr •* l-AdCOOH-> l-AdCOCl-> l-AdCOCH(CO2C2H5)2
• * l-AdCOCH2CO2C2H5 •* l-AdCOCH3 •* l-AdC(OH) (CH3)2 •*
• * l-AdC(CH3)=CH2 •* 1 -AdCH(CH3)2.
В случае использования в реакции Гриньяра сложных эфиров выход конечных продуктов обычно выше, чем при реакции Гриньяра с кетонами.
При действии реактива Гриньяра (RMgBr, где R= СН3, С2Н5, СН2СбН5) на замещенные в узловых положениях адамаитилбромиды получены соответствующие метил-, этил- н бензнладамантаны с выходом более 95% [159, 177].
Оригинальный метод синтеза 1-бутиладамантанов предложен в работе [178]:
I-AdBr + О
S S S
Нг Ni Ренея
1-да(снг)3снэ
Нг Ni Ренея ''
надену снгсн3
трет-Бутил адамантан легко получить циклопропанированием 1-изопро-пениладамантана и последующим гидрогенолизом [161,179, 180].
.СН2 1-Ad-C^
^СН3
(С3Н()аО, Zn/Cu или
С Г’ i 11 > (*“i Нs
-* 1-Ad
• * 1-Ad—С(СН3)3
96%
94
Реакцией циклопропанирования [действием СН212 h(C2H5)2Zd] был получен также простейший спироадамантан — спиро [адамантан-2,1-циклопропан] [181].
Препаративным методом получения смесей моио- и полизамещенных бутиладамантанов является метод деструктивного алкилирования адамантана и низших алкиладамантанов изооктаном [182].
Алкиладамантаны с длинными алкильными группами могут быть получены в одну стадию путем взаимодействия 1 -бромадамантана с соответствующими алкиллитиевыми производными, для 1-н-бутил- и 1-н-гексилада-маитанов выходы соответственно 90 и 50% [183]. Несмотря иа использование малоустойчивых алкиллитиевых соединений, эта реакция представляет препаративный интерес, так как в этом случае не происходит изомеризация алкильных групп и таким способом можно получать индивидуальные алк иладамантаны.
2.3.2. СИНТЕЗ АДАМАНТАНОВ, ЗАМЕЩЕННЫХ В МОСТИКОВЫХ ПОЛОЖЕНИЯХ
Прямой синтез производных адамантана, замещенных в мостиковых положениях, затруднен вследствие низкой реакционной способности мостиковых атомов углерода адамантанового ядра. Большинство синтезированных к настоящему времени 2-алкиладамантанов получены на основе ада-мантаноиа. Успешному использованию адамантанона для синтезов способствовала разработка одностадийного препаративного метода его получения путем обработки адамантана концентрированной сериой кислотой [184]. 2-Метиладамантан получен исходи из адамантанона по схеме [185]
— Н(0
И? .
Необходимо отметить, что взаимодействие адамантанона с реактивами Гриньяра протекает количественно только в случае метилмагнийбромида. Применение других алкилмагиийгалогенидов приводит к значительному восстановлению кето группы в гидроксильную группу и образованию 2-гид-роксиадамантана.По этой причине для синтеза высших 2-алкиладамантанов используют алкиллитий. С помощью алкиллитиевых соединений RLi (R = = C2HS; h-C3H7; н-С4Н9; ИЗ0-С4Н9) синтезированы 2-этил идеи-, 2-этил-, 2-пропил идеи-, 2-пропил-, 2-бутилиден-, 2-н-бутил-, 2-изобутилнден-и 2-изобу-тиладамантаны [186, 187]; можно исходить также из 2-адамаитиллития н соответствующих производных алифатических соединений [188].
Синтез 2-изопропил адамантана, однако, не удается осуществить даже с помощью 2-нзопропилиития, вероятно, вследствие пространственных затруднений. Углеводород синтезирован исходи из адамантаи-2-карбоновой кислоты [189].
CH, N. CH, Mgl (СН-СОК о
2-AdCO2 Н —2—±> 2-AdCO2 СН3 —------> 2-AdC(CH3)2 ОН-------—>
Н1
2-Ad=C(CH3)2+2-AdC(CH3)=CH2-----> 2-AdCH(CH3)2-
95
Разработан также ряд других методов получения 2-алкиладамантанов. 2-Метил адамантан, н частности, образуется (с выходом 80%) в результате превращения гомоадамантена [98]. Прн использовании этой реакции необходимо иметь в виду, что в принятых условиях продукт реакции способен превращаться в более устойчивый изомер — 1-метиладамантан.
Подобно синтезу соответствующих 1-алкиладамантанон, 2-н-бутил-и 2-вгор-бутил адамантаны получают с выходом 40% при взаимодействии 2-бремадамантаиа с тиофеном с последующим гидрогенолизом полученного продукта [178]. 2-трет’-Бутиладамантан образуется при гидрогенолизе соответствующего ал к ил адамантана, содержащего в боковой цепи циклопропановое кольцо [180].
Метод синтеза адамантанов, меченных 14 С, описан в работе [190]. Получены, в частности, адамантан-2-14 С и 1-метиладамантан-2- или -4-14 С.
гем-Замещенные производные, например 2,2-диметиладамантан, получают при гидрогенолизе соответствующих спироциклопропановых углеводородов [180].
Данный метод является общим методом синтеза спиро [адамантан-2,1’-циклопропанов] и может быть использован для получения 2-метил-2-алкиладамантанов с различной длиной алкильной группы. Другой метод синтеза 2,2-диметиладамантана основан на перегруппировке третичного диметилбн-цикло [3,3,1] нонен-2-ола-7, происходящей при карбоксилировании последнего по Коху-Хаафу [191].
Широкие синтетические возможности для получения спнро [адамантан-2,Г-циклопропанов] открывает реакция присоединения олефинов к 2-ада-мантилкарбену [192].
R„ Rt К»
а) СНЭ сн3 сн3 снэ г) СНЭ снэ н сн.
б) сн3 н сн3 н д) с3н5 н н н
в) СН3 снэ н н
В силу высокой реакционной способности карбена строение олефина, вероятно, не будет существенно влиять на выход конечных продуктов,
96
и в зависимости от используемого Непредельного углеводорода по данной реакции можно будет получать самые различные циклопропановые производные адамантана, и затем, после гидрогенолиза трехчленных циклов, — соответствующие труднодоступные 2-алкил- и 2-циклоалкиладамантаны.
Синтез 2,6-диалкил адамантанов основан на использовании адамантан-диона-2,6 [193]. Характерно, что первая карбонильная группа вступает во взаимодействие с реактивом Гриньяра значительно легче, чем вторая.
По данному методу получены 2-метил-б-этил- и 2-метил-б-пропиладаман-таны и соответствующие им алкилидены.
2.3.3. СИНТЕЗ АДАМАНТАНОВ СМЕШАННОГО СТРОЕНИЯ
Синтез ал кил адамантан он смешанного строения, т.е. содержащих наряду с мостиковыми и узловые заместители (1,2- ; 1,4-замещение), может быть осущестнлен исходи из соответствующих функциональных производных. Так, исходи из 2-метил-1-адамантанкарбоновой кислоты и смеси эпимеров 4-метил-1-адамантанкарбоиовой кислоты, образующихся в реакции Коха Хаафа из 2-метил-2-гидроксиадамантана, путем восстановления кислот до соответствующих спиртов, последующего действия SOBr2 и восстановления полученных бромидов получают 1,2- н 1,4-диметиладамантаны [194]. По близкой схеме с использованием алкиллитиевых соединений можно синтезировать также другие алкиладамантаны смешанного типа, в частности 1-этил-2-метил-, 1-пропил-2-метил-, 1-изопропенил-2-метил-, 1-изопропил-2-метиладамантаны и др. [195].
7. Зак.1856 97
В реакцию Гриньяра с адамантаном вступают также а рил магний бромиды (нафтил-, фенантрил магний бромиды), что позволяет получать полициклические арияадамантаны, как, например, 2- (а-иафтил) - и 2-(9-фенант-рип)адамантаны [196].
Би- и полиядерные углеводороды ряда адамантана — 1,2-диадамаитан и его 2,4-дегидропроизводные получают олигомеризацией 2,4-дегидроада-
Обработкой 1,2-дииодадамантана и-бутиллитием прн 0°С получают димеры адамантана - 1,2*; 2,Г- и 1,12,2’-адамантиленадамантаны (выход 98%) [198-200].
Н НН
или
Исходы из соответствующего кетона синтезирован также спиро [адамантан-2,2'-адамантан] [201].
88
2.3.4. СИНТЕЗ ПОЛИЗАМЕЩЕННЫХ И ПОЛИЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ РЯДА АДАМАНТАНА
В синтезах углеводородов, рассмотренных в этом разделе, важную роль играют реакции циклизации. Метод циклизации был первым синтетическим методом, позволившим построить каркас молекулы адамантана. Исходя из бициклического сложного дикетоэфира 4,8-диоксо-3,7-дикарбме-токсибицикло [3,3,1] ионана Прелог и Зейферт [202] впервые осуществили синтез адамантана.
NaOCHj
2. ОН"
Исходный эфир можно легко получить из формальдегида и метил мало -иата с последующим частичным декарбоксилированием. Низкий выход углеводорода на последней стадии декарбоксилирования снижает препаративную ценность метода. Несмотря на это, метод представляет существенный интерес для получения труднодоступных полизамещенных функциональных производных адамантана. Так, например, исходя из эфира Мейер-вейна легко получили полизамещенный дик его и [203], синтез которого из адамантана представляется весьма затруднительным.
2,6-Дикетон адамантана, содержащий дае сложноэфирные группы, можно также получить конденсацией соответствующего производного циклогексанона и пирролидина [204,205].
(Л=согсд; г=н, согсгн,).
88
Незамещенный адамантандион-2,6, являющийся удобным исходным веществом для синтеза 2,6-диалкил адамантанов, получают восстановлением сложноэфирных групп R и Rf [206], а также исходя из бицикло [3,3,1 ]но-нандиона-2,6 через соответствующий дипирролидин [207].
На основе адамантандиона-2,6 с использованием реакции Гриньяра синтезирован ряд 2,6-дизамещенных углеводородов ряда адамантана, в том числе 2,6-диметил- и 2,6-диэтиладамантаны [207], 2-метил-6-этил-и 2-метил-6-пропила дамантаны [193].
Образование структуры адамантана из производных бицикло [3,3,1] нонана достигается в результате циклизации 3-метилен бицикло [3,3,1 ]ноиа-нона-7 [203] либо 3,7-диметиленбмцикло[3,3,1]нонана при действии электрофильных реагентов [208-211].
(X=Cl, Вс, I, ОН, ОСН,, NHCOCH,, я-С$НЧОСН,).
Присоединение электрофильных реагентов к 3-мет илен-7-этил и денби-цикло [3,3,1] нонану позволяет получать 1,2-дизамещенные алкилада-
мантаны [212] через 1,2,3-тризамещенные адамантаны [213, с. 119].
100
В реакцию циклизации вступают также некоторые функциональные производные би цикл о [3,3,1] нонана, такие, как 3-гидроксиметип бицикло [3,3,1] ионанол-7 и 3-гидроксиметилбицикло [3,3,1] нонен-2; при этом образуется смесь адамантанов [214], например:
17’4 39% 26%
Характерной особенностью реакции циклизации производных бицикло-13,3,1] нонана является то, что адамантановая система возникает в результате внутримолекулярной циклизации, при этом метиленовый мостик образуется за счет атома углерода гидроксиметильной группы исходного соединения.
Образование структуры адамантана замыканием молекулы бицикло-[3,3,1 ] нонанов имеет место также под действием инициаторов радикальных процессов. Так, фото инициированная циклизация 3,7-диметиленби-цикло [3,3,1] нонана приводит к образованию напряженного тетрацикло-[4,3,1,14’8,01>4] ундекана (этанонорадамантана), который при взаимодействии с бромом или другими реагентами образует производные адамантана [215].
Раскрытие циклобутанового кольца обусловлено, вероятно, напряжением в норадамантановой структуре этанонорадамантана, так как сам циклобутан при обычных условиях с бромом не реагирует [216, с. 544].
Свободнорадикальное присоединение четыреххлористого или четырех-бромисгого углерода к 3,7-диметиленбицикло [3,3,1] нонану также приводит к производным адамантана и нораддмантана [217, 218].
101
Направление циклизации диена в данном случае зависит как от температуры реакционной среды, так и от легкости гомолиза присоединяемых попит алогенмета но в, Увеличение температуры и использование менее реакционноспособного галогенметана способствует образованию структуры адамантана.
В то же время замкнуть адамантановый цикл на основе 3,7-диметилен-бицикло [3,3,1 ]ионана с помощью дифтор- или дифенилкарбенов, гене-
рируемых фотохимически, не удалось; продуктами реакции были моно и диспиро [бицикло [3,3,1 ]нонан-3,Г-(2',2 -дифенилциклопропан)] [219].
Для синтеза тетрациклических углеводородов ряда адамантана, например 1,2-бутаноадамантана, предложено использовать общий метод расширения циклов путем окисления по месту двойных связей производными трехвалентного таллия [220].
Простейший 2,4-тетрациклический углеводород адамантаноидного строения - 2,4-метаноадамантан - синтезирован Сасаки н соавт. исходя из эта-ноадаманта нона-3 [221].
1G2
2,4-Метаноадамантан представляет собой напряженный углеводород вследствие наличия циклобутанового кольца, а также циклогексанового кольца, фиксированного в конформации ванна. Сужение циклопентанового кольца исходного кетона достигается путем диазотирования н последующего фотолиза полученного карбена. Следует отметить, что попытка получить метаноадамантан по кратчайшему пути — внедрению карбена по связи С (4) —Н — не удалась,
вероятно, вследствие удаленности реакционного центра от С(4)—Н-связи. Следующий гомолог в ряду 2,4-дизамещенных тетрациклических углеводородов ряда адамантана — 2,4-этаноадамантан, как уже отмечалось выше, содержится в нефти и может быть получен также путем изомеризации те-тра1щкло[6}4,0,04’12,05’9]додекана. При синтетическом получении 2,4-эта-ноадамантана путь внедрения карбена по связи С(4)-Н привел к успеху [222]:
Пента циклический углеводород регулярного адамантаноидного строения - диамантан, также содержащийся в нефти, может быть получен и синтетическим путем. Следует отметить, что синтетические методы, основанные на использовании реакции циклизации доя синтезов высших адаманта-ноидных углеводородов, имеют важное значение. Это связано с тем, что каталитические методы изомеризации здесь встречают серьезные затруднения н уже, например, доя получения тетрамантана не могут быть использованы. В качестве исходных соединений доя синтезов высших адамантаноид-ных углеводородов пригодны соответствующие низшие члены гомологических рядов. В результате синтеза могут образоваться либо два, либо
103
сразу четыре новых цикла. Такой общий синтетический подход может быть продемонстрирован на примере синтеза диамантана исходя из адамантана, точнее, - нз адамантанона [222, 223].
1.(СН3)г5(а)=СНг
2.(СгН5)гйЫ3
3. СгО3
Си 501, сн3с6н5
о
Синтезы, как правило, много стадийны, однако достоинство методов состоит в том, что они являются общими и могут в принципе быть использованы для получения полиэдранов различной молекулярной массы. Так, из диамантан-1-карбоновой кислоты по приведенной ниже схеме может быть получен триамантан [140],
В этом синтезе первый новый цикл образуется за счет внедрения карбена при разложении диазокетона. Образование нового цикла на последней стадии синтеза происходит за счет реакций протонироваиия олефина, расши
104
рения пятичленного цикла образовавшегося карбкатиона' и дегидроциклизации, Эти реакции протекают в паровой фазе при 430° С на катализаторе 2% Pt на силикате с выходом конечного продукта 21%.
Аналогично, исходя из диамантан-1,6-дикарбоио вой кислоты, получают соответствующий бисдиазокетои, последний может быть превращен в анти-тетрамаитаи через стадии образования бисциклопеитаноиа (разложением на медном катализаторе), диола (получаемого в результате действия этилмаг-нийбромида) и диена по приведенной ниже схеме [142, 224].
Выход анru-тетрамантана на последней стадии составляет 10%, Предполагается, что, используя этот общий метод, можно будет синтезировать бопее высокомолекулярные углеводороды регулярного алмазо
105
подобного строения, Так, например, синтез гексамантана Мак-Кервей предполагает осуществить по следующей схеме [142] :
Развитие данного метода в конечном счете должно привести к синтезу полиядерных алмазоподобных макромолекул, обладающих не только строением алмаза, но и близкими ему физическими свойствами, Путь этот чрезвычайно сложный н длинный, но вместе с тем весьма интересный. Свойства получаемых соединений и области их применения предсказать заранее трудно.
Многие полиэдрами нерегулярного, но близкого к адамантану строения (так называемые родственные соединения) также получают синтетическим путем. Ниже приведены методы синтеза лишь некоторых циклических гомологов углеводородов ряда адамантана. Синтез норадамантана, например, осуществлен исходя из 1,5-дибромадамантандиона-2,6 [225].
о О
о
9-Гомонорадамантан — трицикло [3,3,2,03’7] декан (А) - один из 18 вполне устойчивых трициклических изомеров адамантана синтезирован двумя независимыми путями: исходя из 9-норадамаитанона методом гомологизации с помощью диазометана и из 10-гомопротоадамантандиона-4,5 [226].
105
Таблица 2.10
Алкил-, циклоалкил- и ариладамантаны
Углеводороды Т.кип., * С/760 мм рт.ст. Т.пл.,°С ао nD Литература
1 2 3 4 5
Монозамещенные:
1 -Метиладаманган 195 102 [32,72]
1-Метил-(2-1 * С; 4-1 *С) -адамантан — — — [190]
1 -Этил ад ам ант ан 219; 223* -60 1,4931 0,9350) [32, 76, 229]
1-н -Пропи ладам ант ан 251; 242* -1 1,4962 [14,229]
1 -Изопропиладамаи-тан 239-240 -20 1,4960 [14, 170, 174]
1 -н-Бутил адамант ан 254* 1,4907 [229]
1-Изобутиладамантан 248* 100—102/3 мм 1,4890 [229]
1 -втор- Бутилад аман-тан 263* -44 1,4870 1,5010 [14,229]
1-iper-Бутил адамант ан — 111-113 — [179]
2-(1-Адамантил) пентан — — 1,4278 [14]
3-(1-Адамангил)пентан 133-134/8мм -23 1,5000 [14,169]
1 -н-Гексипад ам ан-тан - 282 — — [183]
1 -Октиладамантаны — — [230, 231]
1-Цикпогек сил адамант ан — 105 — [14,8, 164]
1 -Фен ил адамант ан — 82 [14, 164, 232]
1- (4'-Метилфен ил) -адамантан — 100 — [165]
1-(3'-Метилфенил) -адамантан Возгоняется при 100--110/3 мм 77 — [165]
1-(1 -Метилциклопропил) адамантан 65-70/0,005 63-65 [161, 179,181]
1 -Бензиладамантан — 42-43 — [159]
144 -Метилфенил) метил адамантан — — [234]
Метилфеиил (1-ада-мантил) метан 182-5/12 мм — — [232]
Метил (4' -метилфенил) (1-адамантил) -метан 200-205/12 мм — — [232]
1-ОНафтил)-адамантан — 127-129 — [166, 167 ]
1-(«-Нафт ил)-адамантан — 199-201 — [166,167]
1, Г-Диад ам ант ил — 296 — (160, 233, 235]
1,2’ -Диадам ант ил — 266-8 — [197, 236]
2.2'-Диадамангил - — — [188, 236]
107
Таблица 2.10 (продолжение)
1 2 3 4 5
Диадам ант ил алканьт - - - [39, 160, 237, 238]
2-М етиладам антан 210* 146,0- -149,8 — [24, 32, 239]
2-Этил адамантан 229;227,5* -24 1,5025 [32, 186, 239]
2-м-Пропмладаманган 246 -31 1,4984 [32, 186, 239]
2-н- Бут иладам антан 261,5 -24,5 1,4965 [32,186]
2-Изобутиладам антан 255 -62 1,4947 [32,186]
2-трет- Бут ил адамантан 258 — — [180]
2-Цикло! юнти ладам антан — — 1,5197 [240, 241]
2-Фениладамантан — 28-30 — [242]
2-(а-Нафтил) адамантан — 176-8 — [196]
2- (9'-Фенантрид) -адамантан — 328-330 — [196]
2-Цикпогексиладамант ан Дизам ещенные: 109/0,8 мм [243]
1,3 -Дим егил адамант ан 204; 201,5* -30 1,4783 (₽4°0,9016) [32,76, 239, 244]
цис-1,4-Дим егнл-адам антан 212* 1,4850 [229]
трйгнс-1,4-Диме-тиладамантан 213* — 1,4840 [229]
1,2-Диметилада-мантан 219* 106 — [195, 239]
2,6-Диметил-адам антан 225 -11,6 1,5018 [207, 239]
2,4-Диметилада-мантаны 224,5-226,5* — [239]
2,6- Диэтил адамантан 244 -8,8 1,4987 [207]
1,3-Ди-н-пр опилада-м антан 275* — 1,4882 [229]
1,3-Диизопропил-адам антан 294 ниже -70 — [14]
1,3-Дифенил-адамантан — 94 [245]
1,3-бис (4'-Метип-феиил) адамантан 157 — [245]
1-М етил-3-этилада-мантаи 226* — 1,4850 [229]
1-Этил-2-метил-адамантан 259,5/ 745 мм -38т -39 1,5023 [195]
1-Метил-2-этил-адамантан 236* — — [239]
цис-1 -Метил-4- 230 — 1,4880 [239]
эгиладам антан
108
Таблица 2.10 (продолжение)
1 2 3 4 5
1 трлн с-1-Мети л4- этиладамантан 231,5* - 1,4880 [239]
цис-1-Этил4-метил адамант ан 237* — — [239]
транс- 1-Этил4-ме-тиладамантан 240* — ]239]
1-Пропил-2-ме-тиладаманган 271/745 мм -68,2: + -69,7 1,4963 [195]
1 -Из опропил-2-ме-тиладаманган 263/753 мм -66,5 + -г-65,0 1,5051 [195]
2 -Метил-6-этил- | адамантан 237 -12,5 + + -11,5 1,4873 [193]
2-Мегил-б-про- | пиладамантан 255 -65-г -Г-64 1,4973 [193]
1-Метил-3-(4'-ме- S типфенил) ада- 1 мантан — 60+61 — [232]
В 1,3-Полиада- Ц манганы — — - [246]
Ц 1,4-Попиадаман- таны Ж Тризамещеиные-. — — [197]
"Й 1,3,5-Триметил- X адамантан 205* - 1,4751 [ 32, 163, 239]
* 1,3,6-Триметил- 1 адамантан 215,5* — 1,4780 [229, 239]
1,2,5-Триметил- 51 адамантан 221* — 1,4830 [229, 239]
1,2,3-Триметил-адамантан 228,5* — - [239]
1,2,4-Триметил-адамантаны 233-235* — — [239]
1,2,6-Триметил-адамантаны 227-238* — — [239]
1,4,6-Триметил-адамантаны 226- 230* — — [239]
1,4,4-Тримет ила да-мантан 236* — — [239]
1,2,2-Триметил-а да мантан 242* — — [239]
1,2,8-Грим стил адамантаны 239-242* — — [239]
1,3,5-Трифенил-адамантан — 230-233 - [247]
1,3-Диметил-5-эт иладам антан 228* — 1,4805 [76,229]
1,3-Диметил-5-изобут кладам антан 261,5 — 1,4798 [248]
1,3-Диметил-5-и -бутил адамантан 266* — — [248]
109
Примечание. Адамантаны, физико-химические константы которых не приведены, идентифицированы другими методами,
* Температура кипения определена на основании хроматографических времен удержи-
Таблица 2.10 (окончание'
Таблица 2.11
Полициклические углеводороды ряда адамантана
Структурная формула Название Брутто-формула TjUL, °C | Литература
Тетрациклические: £ д g 2,4- Метаноадамантан; тетрацикло [5,3,1,0 • ,0 ’ ] -ундекан СцН1( 206-208 [221]
2,4-Этаноадамантан; тетрацикло[6,3,1,02,6,05,10]-додекан Ci jHi s 174-176 [38,54,223,258]
Г| Т Спиро [адамантан-2,1 '-циклопропан] С1аН|Ш 113-114 [180,181,256]
Спиро [ адамантан-2,1 '-циклобутан] Cj э о - [257,258]
Спиро [адамантаи-2,1 ’-циклогексан] Cl 3^24 - [241]
кА 1 2,4-Пропаноадамантан; тетрацикло [7,3,1,02,7,06,11 тридекан !- c13Ha0 102-105 [40, 119,258]
Таблица 2,11 (продолжение)
Структурная формула Название Б p у т то-фо рм у л a Т.пл., °C Литература
1,2-Пропаноадамантан; тетрацикло-[6,3,1,13’10,03,7] тридекан Cx aHj 0 45-51 [56,119,258]
1,2-Бутаноадамантан; тетрацикло-[7,3,1,13 ’1 \о3,8] тетрадекан C14Ha, - [56,119,220,258]
V 9-М етилтетрацикл о [ 7,3,1,0 2,7,17 ’11 ] -тетрадекан CtfH24 т.кип, 280—284 [120]
СК Диметилтетрацикло [7,3,1,02,7,17,11 ]-тетрадеканы в Иде т.кип, 281 [120]
CH, Спиро [адамантан-2, Г-алкилциклопропаны] [192]
ЛзуЯ, fTn 1) Rj.-Ra =R, =R4 =CHs 2) R3 -R, =CH1;R1 = R4 =H 3)R! = Ra =CHa;Ra =R4 =H 4)R, =R4 =CH3; Rj =R3 =H 5)Ri =CeH,;Ra =R3 =R4 =H P P P P P Я к я и к » к» м м ►> н »
8. Зак. 1856
Этил-1,2-пропаноадамантан; этилтетрацикло-[6,3,1,13,1 °,О3,7] тридекан
1-, 2- и 8-Метил-2,4-этаноадамантаны
Пентациклические:
Диамантан; конгрессан; пентацикло- С14Н20
[ 7,9,1,14,^2,02,7,08,11 ] тетрадекан
Метилдиамантаны
Н
З-Метилдиамантан
[241]
[119]
236-237 [42, 118, 121,
122,259]
[42,55,133, 135,260]
117-118 [133]
4-Метилдиамантан
С13Наа 91,6-94,4 [133]
Диметилдиамантаны
— (135]
Таблица 2.11 (окончание)
114 ® 115
Структурная формула Название Брутто-формула Т.пл., °C Литература
Гексациклическне:
3,5-Этанодиамантан; гексацикло- С1ЛНаа
[10,3,1,02,1 °, О3,7,06’15,09’14] гексадекан
1,3-Этанодиамантан; гексацикло- С16Наа
[7,6,1,01,12 Д2,7,04,13,06,11 ] гексадекан
[2] Диадамантан; гексацикло- CieHJ4
[9,3,1,12’6,14,8,19,13,01’8] октадекан
[11 Диадамантан; спиро [адаман-тан-2>2'-адамантан]
Спиро [адамантан-2,2’-гомоадамантан ]
Гептациклические:
с1#н3,
110-111,5 [39,261,262]
206,5-208 [261]
194-195,2 [259]
252,3-255,1 [201,259]
200 [201]
Триамаитан; гептацикло- С1вНа4 221
[7,7,1,13,15,01,12,02’7,04,13,06,11] октадекан
[ 106, 140, 141, 201]
1,2’; 2,1’-Адам антилен адамантан; гептацикло-[11,3,1,13,7,15’9Д11,15»01’1®, О2’9] Эйкозан
1, Г; 2,2'-Адам антилен адамантан; гептацикло-[11,3,1,12,6,14,8,111’15,01’1О,02,9]эЙкозан
ан ги-Те трам ан тан
С н
Нон ациклические:
с„н„
Б астар дан; нонацикло- Ci
[11,7,1,12’18,03’16,04’13,05’10,06’14,07’11,015*20]-
докозан
152-153 [198,199,200]
152-153 [198,199,200]
173,5-174 [25,143,224,
263]
144,5-146,5 [143]
1, 3-Бис го мо адамантан (Б) получен при взаимодействии 4-гомоадаман-танона-2 с KCN и (NH4)2CO3 в 50%-ном спирте [227].
Синтез одного из наиболее интересных тетрациклоалканов состава С12Н18, имеющего более высокую, чем адамантан, температуру плавления (328, 5° С), — идеала осуществлен исходя из циклопентенилднгидротропо-нов [118].
Как уже отмечалось ранее, при контакте с кислотами Льюиса ицеан изомеризуется в 2,4-этаноадамантан. Исходя из дибромгомоадамантана действием гексаметилфосфортриамида при 180° С недавно получены норицеан и 3,5-дегидронорицеан [228].
Сведения об углеводородах ряда адамантана обобщены в табл. 2.10 и 2.11.
ЛИТЕРАТУРА
Decker Н. Ц Angew. Chem. 1924. Bd. 37, N 41. S. 795.
2. Landa S., Machacek V. 11 Coll. CzechosL Chem. Commun. 1933. VoL 5, N 1. P. 1-5.
3. Landa S., Machacek V., Mzourek J. // Chem.listy. 1933. Sv. 27. S. 415-418.
4. Landa S., Machacek K, Mzourek J., Landova M. // Chem. and Industr. 1933. Spec. N. P. 506-510; Chem. Abstr. 1933. Vol. 27. 5949.
5. Landa S., Machacek V., Mzourek J. Ц Petrol. J. 1934. Vol. 30, N 16. P, 1-5; Chem. Abstr. 1934. Vol. 28,4873*.
6. Landa S., Kriebel S„ Knobloch E. Ц Chem.listy. 1954. Sv. 48, N 1. S. 61-64.
7. Crozier A. 11 Rev. Inst. ft. petroL 1956. Vol. 11, N 10. P. 1232-1268.
8. Landa S„ Hala S. Ц Chem. Usty. 1957. Sv. 51, N 12. S. 2325-2329.
9. Landa S., Hala S. 11 Erdol und Kohle-Erdgas-Petrochem. 1958. Bd. 11, N 10. S. 698-700.
10. Mod* B.J., Shamaiengar M., Krouskop N.C., Rossini F.D. 11 AnaL Chem. 1959, Vol. 31, N 12. P. 2082-2083.
11. Landa S. 11 Ropa a uhlie. 1959. Vol. 1, N 9.P. 5-7; Chem. Abstr. 1962. Vol. 57. 6206a.
12. Landa S., Kamy&k Z„ KamyUkova J. // Ropa a uhlie. 1961. Vol. 3. P. 261—262; Chem. Abstr. 1962. Vol. 56. 75711.
13. Landa S. Ц Acta chim. Acad. sci. hung. 1962. VoL 31, N 1/3. P. 123—136; Chem. Abstr. 1963.Vol.58. 3326h.
14. Landa S. Ц Curr. Sci. (Ind.), 1963. Vol. 32, N 11. P. 485-489.
15. Лид С, Кураш M., Ланда C. 11 Нефтехимия. 1966. T. 6, № 1. C. 3—8.
16. Багрий Е.И., Амосова Е.И., Санин П.И. // Там же. № 5- С. 665-670.
17- Слободин Я.М., Ковязин В£., Мотовилов П.И., Васильева В.Ф. Ц Там же. 1969. Т. 9. №6. С. 921-924.
18. Ушараули Э.А., Топурия Э.Н. // Сообщ. АН ГССР. 1973. Т. 72, № 2. С. 361-364; РЖХим. 1974. 1ОП237.
19. Топурия ЭЛ. И Сообщ. АН ГССР. 1977. Т. 85, № 2. С. 363-365; РЖХим. 1977. 16П297.
20. Багрий Е.И. Исследование углеводородов ряда адамантана; Дис. ... д-ра хим. наук. М., 1977. 385 с.
21. Ашудюв Г.Г. Азербайджанские нефти. Баку: Изд-во АН АзССР, 1961. 555 с.
22. Куклинский А.Я., Пушкина Р.А., Караев Я. Ц Изв. АН ТССР. Сер. физ.-техн„ хим. и геол. наук. 1977. № 5. С. 119-121.
23. Бондаренко М.Ф., Варфоломеев Д.Ф., Паис М.А., Сколдинов А.П // Тр. НИИ нефтехим. пр-ва. 1969. Вып. 1. С. 217-220; РЖХим. 1969. 22П278.
24. Hala S„ Landa S. Ц Erdol und Kohle-Erdgas-Petrochem. 1966. Bd. 19, N 10. S. 727-729.
25. Багрий Е.И., Санин П.И. // Газоконденсаты и нефти. Ашхабад; Ыпым, 1968. С. 110-115.
26. Солодков В.К., Михновская А.А., Смирнов Б.А., Петров Ал.А. // Нефтехимия. 1969. Т. 9, У 4. С. 491-499.
116
27. Меликадзе Л'Д., Ушараули ЭЛ., Дзамукашвили АЛ., Мачабали МЛ. // Сообщ. АН ГССР. 1972. Т. 68, № 1. С. 81-84; РЖХим. 1973. 2П75.
28. Алкабекое Г.Г., Багрий Е.И, Санин П.И. // Нефтехимия. 1972. Т. 12, № 2. С. 290-297.
29. Schiessler R.W., Flitter D. Ц J. Amer. Chem. Soc. 1952. Vol. 74, N 7. P. 1720-1723.
30. Kramers H., BroederJ.J. 11 Anal. chim. acta. 1948. Vol. 2, N 7. P. 687-692.
31. Jones A.L., Milberger E.C. // Industr. and Eng. Chem. 1953. VoL 45, N 12. P. 2689-2696.
32. Петров АлЛ. Химия нафтенов. М.: Наука, 1971. 388 с.
33- Санин П.И, Багрий Е.И, Цицугина Н.Н. и др. // Нефтехимия. 1974. Т. 14, № 3. С. 333-340.
34. Багрий Е.И., Амосова Е.И, Санин П.И. // Там же. 1971. Т. 11, № 5. С. 639-647.
35. Якубсон 3.В., Арефьев О.А., Петров АлЛ. Ц Там же. Т. 13, № 3. С. 345-351.
36. Куклинский АJL, ПушкинаРЛ. // Там же. 1971. Т. 14, №6. С. 811-816.
37. Arpino Р„ Schmitter JJtf., Selves J.L. // Rev. Inst. fr. petrol. 1978. Vol. 33, N 3. P. 467-483; РЖХим. 1978. 24Г290.
38. Hala S., Landa S.t Harms V. Ц Angew. Chem. 1966. Bd. 78, N 21/23. S. 1060-1061.
39. Weidenhoffer Z„ Hala S. 11 Sb. VSCHT Praze D. 1971. Sv. 22. S. 5-84; Chem. Abstr. 1972. Vol. 76.342И,
40. Воровьева H.C., Земскова 3JG, Петров АлЛ. I/ Нефтехимия. 1979. Т. 19, № 1. С. 3-6.
41. Мусаев ИЛ., Заикин В.Г., Курашова Э.Х. и др. Ц Там же. 1982. Т. 22, № 1. С. 175-179.
42. Мусаев ИЛ., Багрий Е.И, Курашова Э.Х. и др. Ц Там же. 1983. Т. 23, № 5- С. 600.
43. Birch S.F., Cullum T.V., Dean KA., Denyer R.L. Ц Nature. 1952. Vol. 170, N 4237. P. 629-630.
44. Birch S.F„ Cullum T. V., Dean R.A., Denyer R.L. // Industr. and Eng. Chem. 1955. Vol.47, N 2.P. 240-249.
45. Schley er P.R. 11 J. Amer. Chem. Soc. 1957. Vol. 79, N 12. P. 3292.
46. Landa S., Kamy^ek Z„ Kamyikova J. // Erdol und Kohle-Erdgas-Petrochem. 1961. Bd. 14, Nil. S. 904-905.
47-Фрост A.B. // Успехи химии. 1945. Т. 14, № 6. С. 501-509.
^.Богомолов А.И Ц Тр. Всесоюэ. нефт. геол.-развед. ин-т. 1964. Вып, 227. С. 10-20; РЖХим. 1964. 19Е106.
49. Платэ А.Ф., Никитина З.К., Бурцева ТЛ. Ц Нефтехимия. 1961. Т. 1, № 5. С. 599— 603.
50. Robinson RJI Nature. 1966. Vol. 212, N 5068. Р. 1291-1295.
51. A.c. 239302 СССР, МКИС 07 С 15/20. Способ получения гомоголов адамантана / Е.И. Багрий, П.И. Санин// Б.И. 1971. № 25. С. 241.
52. Багрий Е.И., Санин П.И, Долгополова Т.Н. // Нефтехимия. 1969. Т. 9, № 3. С. 353-358.
53. Fort R.C. Adamantane: The chemistry of diamond molecules. N.Y,: Dekker, 1976. 385 p.
54. Farcasiu D., Wiskott E., Osawa E. et al. // J. Amer. Chem. Soc. 1974. Vol. 96, N 14. P.4669-4671. v
55. Hala S„ Novak J., Landa S. // Sb. VSCHT Praze D. 1969. Sv. 19. S. 19-37; Chem. Abstr. 1971. Vol. 74. 99508j.
56,Берман C.C., Денисов Ю.В., Мураховская A.C и др. // Нефтехимия. 1974. Т. 14, № 1. С. 3-8.
57- Schleyer P.R., Donaldson М.М. // J. Amer. Chem. Soc. 1960. Vol. 82, N 17. P. 4645-4651.
58. Schleyer P.R., Donaldson M.M., Nicholas R.D., Cupas С. 11 Org. Synth. 1962. Vol. 42. P. 8-11.
59. Koch H„ Franken J. 11 Brennst. Chem. 1961. Bd. 42, N 3. S. 90.
60. Stetter H 11 Angew. Chem. 1962. Bd. 74, N 11. S. 361-374-
61. Пат. 5538935 Япония, МКИа С 01 С 13/615, 5/22. Получение адамантана / Т. Такахаси, С. Фудзияма // РЖХим. 1981. 10Н94П.
62. Пат. 1431816 Франция, МКИ2 С 07 С. Adamantane / R. Achard. // Chem. Abstr. 1966. Vol. 65. 15250b.
117
63. Пат. 324274 США. Adamantane process / Н.Е. Cupery // Chem. Absti. 1967. Vol. 66. 37512g.
64. Пат. 135674 ЧССР, МКИ1 С 07 С. Adamantane / S. Hala, M. Kuras, S. Landa, Z. Ti-chy // Chem. Abstr. 1971. Vol. 74.124987z.
65. Kuras M„ Hala S., Landa S. // Sb. VSCHT Piaze D. 1971- Sv. 22. S. 95-104; Chem. Abstr. 1972. Vol. 76. 59046».
66. Robinson Tarrat H.J.F. // Tetrahedron Lett. 1968. N 1. P. 5-7-
67. Заявка 7511395 Нидерланды, МКИ3 С 07 С 13/54. Адамантан // Chem. Abstr. 1978. VoL 88. 6433g.
68. Addison A.f Rachal K. // J. Chem.Educ. 1969. Vol. 46, N 9.P. 612.
69. Olah J.A., Olah G.A. // Synthesis. 1973. N 8. P. 488.
7Q. Luke M.O., Atkinson J.G. //Tetrahedron Lett. 1971. N 2.P. 117-120.
71. Fort R.C., Schleyer P.R. // Chem. Rev. 1964. Vol. 64, N 3. P. 277-300.
22. Schleyer P.R., Nicholas R.D. // Tetrahedron Lett. 1961. N 9. P. 305-309.
73. Cash D.t Wilder P. И Ibid. 1966. N 52. P. 6445-6451.
74. Пат. 3356751 США, МКИ’ С 07 C. Preparation of methyladamantane and dimethyladamantanes / A. Schneider // РЖХнм. 1975.116Н115П.
75. Пат. 3128316 США. Adamantanes / A. Schneider // Chem. Abstr. 1964. Vol. 61.4244a.
76. Schneider A., Warren Л.К, Janovski E.J, //J. Amer. Chem. Soc. 1964. Vol. 86, N23. P. 5365-5367.
77- Schneider A., Warren R.W., Janovski E.J. // J. Org. Chem. 1966. Vol. 31,N 5. P. 1617 — 1625.
7%. Прокопец Е.И. //Журн. прикл. химии. 1938. Т. 11, № 10/11. С. 1471-1474.
79. Пат. 1266299 ФРГ, МКИ’ С 07 С. Adamantanes / A. Schneider // Chem. Abstr. 1964. VoL 61.4244a.
80. Заявка 639878 Бельгия, МКИ1 С 07 С. Adamantanes / A. Schneider // Ibid. 4244а.
81. Заявка 300223 Нидерланды, МКИ1 С 07 С. Adamantanes / Sun oil со // Ibid. 1966. Vol. 64.6528e.
82. Заявка 658783 Бельгия, МКИ3 С 07 С. Ethyl substituted alkyladamantanes / E.J. Ja-noski, R.E. Moore // Ibid. II 104h.
83. Пат. 1393283 Франция, МКИ’ С 07 С. Condensed polynuclear compounds / Sun oil co // Ibid. 1965. Vol. 63.2910a.
84. Заявка 6609427 Нидерланды, МКИ’ С 07 С- Polymethyladamantanes / Sun oil co // Ibid. 1967. Vol. 67.90463р.
85. Пат. 3336405 США, МКИ3 С 07 С. Alkyladamantanes having one ethyl substituent / A. Schneider, O. Hills, E.J. Janoski, R.W. McGinnis // Ibid. 108330e.
86. Пат. 3275700 США, МКИ3 С 07 С. Ethyl substituted alkyladamantanes /EJ. Janoski, R.E. Moore // Ibid. 1966. VoL 64.11104h.
87. Пат. 1068518 Великобритания, МКИ’СО 7 C. Isomerization of tricyclic saturated alkanes to alkyl-substituted adamantanes / A. Schneider //Ibid. 1967. VoL 67. 81859v.
88. Пат. 1802055 ФРГ, МКИ3 С 07 С. Substituted adamantanes / К. Bott // Ibid. 1970. VoL 73. 1431 Iw.
89. Пат, 169266 ЧССР, МКИ1 С 07 13/28. Alkyl adamantanes / J. Burkhard, J. Janku, L. Vodi&e, J. Nostecky // Ibid. 1978. VoL 88. 104787v.
90. Landa S.,MandikL. fl Sb. VSCHT Praze D. 1973. Sv. 29. S. 111-124; Chem. Abstr. 1976. VoL 84.105070k.
90aJ3oofey TJ„ Rowe W. // Org. Prepr. Proc. Intern. 1988. Vol. 20, N 3. P. 293-295.
91. Пат 143879 ЧССР, МКИ3 С 07 С 13/28. Zpiisob vyroby aikyladamantanove frakoe / J. Mostecky, M. PopL S. Hala, M. Kuras // Chem. Abstr. 1972. VoL 77. 126153k.
92. Hala S., Kura# M., Popl M., Mostecki J. // Erdbl und Erdgas-Kohle-Petrochem. 1971. Bd. 24, N 2. S. 85-88.
93. Толстиков Г.А., Юрьев В.П., Салимгареева И.М., Кучин A.R fl Изв. АН СССР. Сер. хим. 1974. № 7. С. 1631-1632.
94. Baguette Т.Л., Meehan G.V., Marshall S.J. If I. Amer. Chem. Soc. 1969. VoL 91, N 24. P. 6779-6784.
95. Whitlock HW., SiefkenM.W. //Ibid. 1968. VoL 90, N 18. P. 4929-4939.
96- Lenok D.t Schleyer P.R. //Chem. Commun. D. 1970, N 15. P. 941—942.
97. McKervey M.A.„ Rooney JJ., Samman NG, ff J. CataL 1973. VoL 30, N 2. P. 330-331.
98. Majerski Z.t Mlinaric K, ff J. Chem. Soc. Chem. Commun. 1972. N 18. P. 1030-1031.
118
99. Mdjerski K.M., Mdjerski Z. // Tetrahedron Lett. 197 3. N 49. P. 4915-4918.
100. Пат. 77118455 Япония, МКИ1 С 07 С 13/54. 1-Метиладамантан / Y. Inamoto, К. Ajgami, Y. Fujikura, К. Tsuchihashi // Chem. Abstr. 1978. Vol. 88. 104786u.
101. Пат. 5522451 Япония, МКИ1 С 07 С 13/615, С 07 С 5/29, Получение 1-метилада-мантана /N. Takaishi, Y. Inamoto, S. Dobashj // РЖХим, 1981. 12Н93П.
102. Пат. 5522452 Япония, МКИ1 С 07 С 13/615, С 07 С 5/29. Получение 1-мегилада-мантана / N. Takaishi, Y. Inamoto, S. Dobashi // Там же. 12Н94П.
103. Пат. 78845 Япония, МКИ1 С 07 С 13/54. 2-Метиладамантан / К. Sogami, Н. Reda, Y. Inamoto, Y. Fujikura; N. Takaishi // Chem. Abstr. 1979. VoL 90. 22437h.
104. Schleyer P.R., Gteicher GJ., Cupas C.A. // J. Org. Chem. 1966. Vol. 31, N 6. P. 2014-2015.
105. Арефьев O.A., Воробьева H.C., Епишев В.И., Петров Ал.А. // Нефтехимия. 1972. Т.‘ 12, №4. С. 488-494.
106. Williams V.Z., Schleyer P.R., Glekher G.J., Rodewahl L.B. // J. Amer. Chem. Soc. 1966. Vol 88, N 16. P- 3862-3863.
107. Nomura M., Schlayer P.R., Arz A. A. // Ibid. 1967- VoL 89, N 14. P. 3657-3659. : 108. Гаврилов Б.Г, Лукша В.Г., Тропинова Л.Н. //Журн. орган, химии. 1976. Т. 12,
№ 7. С-1599-1600.
109. Гаврилов Б.Г, Лукша В.Г, Слободин Я.М., Ковязин В.Е. // Тйм же. 1975. Т. 11, №3. С. 597-600.
110. Гаврилов Б.Г, Лукша В.Г., Мартюшин С.В. и др. // Журн, прикл. химии. 1976.
Т. 49, №8. С 1817-1819.
111. Гаврилов Б.Г, Лукша В.Г, Аверьянов С.Ф. // Там же. 1978. Т. 51, № 3. С- 671— ; 673.
112. Тахистов В.В., Ковязин В.Е. // Науч. конф, по химии орган, полнэдранов: Тез. докл, Волгоград, 1981. С. 9.
113- Ковязин В.Е., Тахистов В. В. // Там же. С- 11.
114. Гаврилов Б.Г, Кдсниковская О.С, Николаева Т.Н. // Журн, прикл. химии. 1984. Т. 57, №2. С. 469-471. *
115. Landa S., Mandik L. // Sb. VSCHT Praze D. 1973. Sv. 29. S. 99-100; Chem. Abstt. 1976. Vol. 84. 58738x.
116. Ковязин B.E., Слободин Я.М., Темянко М.Б. // Нефтехимия. 1980. Т. 20, № 1. С 64-70.
117. Багрий Е.И., Фрид Т.Ю., Санин НИ. // Изв, АН СССР. Сер. хим. 1970. № 2. С. 498.
118. Cupas Ch.A., Hodakowski L. // J. Amer. Chem, Soc. 1974. VoL 96, N 14. P. 4668-4669.
119. Osawa E., Tahara Y.r Ezuka T. et al. // J. Org. Chem. 1982. VoL 47, N 10. P. 1923-
' 1932.
; 120. Берман C.C, Денисов Ю.В., Петров Ал.А. // Нефтехимия, 1974. Т. 14, № 3. С. 341 —
348.
121. Gund Т.М., Osawa Е., Williams V.Z., Schleyer P.R. // J. Org. Chem. 1974. VoL 39, £ N 20. P. 2979-2987-
122. Cupas C, Schleyer P.R., Trecker DJ. // J. Amer. Chem. Soc. 1965. VoL 87, N 4.
; P. 917-918.
1 122a. Салимгареева И.М., Юрьев В.П., Рафиков C.P. // 6-я Всесоюз, конф, по химии
В и применению кремнийорган. соед.: Тез, докл. Рига, 1986. С. 215—216; РЖХим. I 1986. 17Ж359.
К 123. Couctney Т., Johnston D.E., McKervey М.А., Rooney J.J. //J. Chem. Soc. Perkin Trans. I Part 1.1972. N 21. P. 2691-2696.
й 124. Faulkner D.t Glendinning /L4., Jhnston- D.E., McKervey M.A. // Tetrahedron Lett.
I 197 l.N 20. P. 1671-1674.
| 125. McKervey M.A., Johnston D.E., Rooney J J. //Ibid. 1972. N 16-P. 1547-1550.
126. Gund TJf.r Williams V.Z., Osawa E„ Schleyer P.R, // Ibid. 1970. N 44. P. 3877-£ 3880.
£ 127. Gund T.M., Thielecke W., Schleyer P.R. Ц Org. Synth. 197 3. Vol. 5 3. P. 30- 34.
£ 128. Schrauzer G.N. // Advances in catalysis and related subjects / Ed. D.D. Eley et al. N.Y.:
I Acad, press, 1968. Vol. 18. P, 37 3-396.
t 129. Schrauzer G.N., Bastian B.N., Fosselius G.A. //J. Amer. Chem. Soc. 1966. VoL 88, Ь N21.P.4890-4894.
I; 130. Scharf H. D., Weisgerber G., Hover H. //Tetrahedron Lett. 1967-N 43. P. 4227-4229.
f 131.Schrauzer G.N., Ho R.K.Y., Schlesinger G. //Ibid. 1970. N 8. P. 543-545.
119
132, Turecek F., Hanus V., Sedmera P. et aL // Coll. Czechosl. Chem. Commun. 1981
Vol. 46, N 6. P, 1474- 1485.
133. Gund T.M., Nomura M., Schleyer P.R. // J. Org. Chem. 1974. Vol. 39, N 20. P. 2987-2994.
134. Tabushi Aoyama Y., TakahashiN. et aL // Tetrahedron Lett. 1973. N 2. P. 107-110.
1 35. Берман C.C., Петров Ал.A. // Нефтехимия. 1979. Т. 19, № 1. С. 17-21.
136. Берман С.С, Петров Ал.А. // Науч.конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 20. |
137. Schleyer P.R., Wiskott Е, // Tetrahedron Lett. 1967. N 30. P. 2845-2850.
138. Godleskl SA., Schleyer P.R., Osawa E. et al. // J. Org. Chem. 1976. VoL 41, N 15.
P. 2596-2605.
139. Osawa E., Furusaki A., Hashiba N. et aL // Ibid. 1980. VoL 45, N 15. P. 2985-2995.
140. Burns W., McKervey MA„ Rooney J.J, // J. Chem. Soc. Chem. Commun. 1975. N 24.
P. 965—966. I
141. Hamilton R., McKervey M.A., Rooney JJ., Malone IT. // Jbid. 1976.N 24.P. 1027-1028.
142. McKervey MA. //Tetrahedron. 1980. Vol. 36,N 8.P. 971-992.
142z.Farooq 0., Farnia S.M., Stephenson M., Olah GA. 11 J. Org. Chem. 1988. Vol. 53, N12.P. 2840-2843. ?
143. Schleyer P.R., Osawa E., Drew M.G.B. // J. Amer. Chem. Soc. 1968. Vol. 90, N 18.
P. 5034-5036.
144. Масленникова Г.А., Соловьев B.H., Крупцов Б.К, Багрий Е.И. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 10. ।
145. Пат, 7752888 Япония, МКИ3 В 01 J 29/38. Regeneration of zeolite isomerization catalysts / H. lida, K. Kawamura, M. Sugimoto, H. Ichikawa, N. Komori, H. Sugahara, K. Honna // Chem. Abstr. 1978. Vol. 88. 62058a.
146. Ланда С., Подроужкова В //Нефтехимия, 1974. T. 14, N® 4. С. 551-560.
147. Quinn НА., Graham J.H., McKervey MA., Rooney J J. // J. CataL 1972. Vol. 26, N 3.P. 333-337. i
148. Honna K, Sugimoto M., Shimizu N., Kurisaki К. Ц Chem. Lett. 1986. N 3. P. 315-318. :
149. Пат. 4022845 США, МКИа С 07 С 5/24. Process for producing alkyl adamantanes / K. Honna, N. Shimizu, K. Kurisaki // РЖХим. 1978. 4H116П.
149a. Заявка 6024633 Япония, МКИ С 07 С 1 3/615,С 07 С5/22. Получение адамантана/ X. Ида, К. Хонна //РЖХим, 1986. 22Н84П.
150. Соловьев ВН„ Багрий Е.И, Санин П.И. // Изв. АН СССР. Сер. хим. 1979. N® 6.
С. 1198-1201.
151. Parry Е.Р. //J. CataL 1963. Vol. 2, N 5. Р. 371-379. i
152. Соловьев ВН., Багрий Е.И, Маркова Е.И., Санин П.И. // Нефтехимия. 1974. >
Т. 14, N® 4. С. 561-567.
153. Ола ГА. // Успехи химии. 1975. Т. 44, N® 5. С. 793-867.
154. Миначев Х.М., Гаранин В.И., Харламов В.В. // Изв. АН СССР. Сер. хим. 1970.
№4. С. 835 - 84 0. ‘
155. Жермен Дж. Каталитические превращения углеводородов. М.: Мир, 1972. 308 с. ;
156. Stetter Н., Schwarz М., Hirschhorn А. // Chem. Ber. 1959. Bd. 92, N 7. S. 1629-1635. 1 ,
157. Stetter H., Goebel P. //Ibid. 1963. Bd. 96. N 2. S. 550-555.
158. KuraSM., Hala S., Landa S. // Sb. vScHT Praze D. 1971. Sv. 22. S. 131-1 37; РЖХим, 1972. 5Ж236.
159. Osawa E., Majerski Z., Schleyer P.R. 11 J. Org. Chem. 197 1. Vol. 36, N l.P. 205-207.
160. Степанов Ф.Н„ Баклан В.Ф. // Журн. общ. химии. 1964. Т. 34, N® 2. С. 579—584.
161. Landa S., KamyXek Z. // Coll. Czechosl. Chem. Commun. 1956. VoL 21, N 3.P. 772-773.
162. Landa S, Kamycek Z. // Ibid. 1959. Vol. 24, N 12.P. 4004-4009.
163. Koch H, Franken J. //Chem. Ber. 1963. Bd. 96, N l.S.213-219.
164. Lande S., Hala S. // Coll. Czechosl. Chem. Commun. 1959. Vol. 24, N 1. P. 93-
98.
165. Stetter H., Weber J„ Wulff C. // Chem. Ber. 1964. Bd. 97, N 12. S. 3488-3492.
166. A.c. 376347 СССР, МКИ С 07 15/24. Способ получения 2- (адамаитил-1)-нафталина/ Ф.Н. Степанов, С.Г. Рыклис // Б.И. 1973. N® 1 7. С. 70.
167. Рарог Б.Г., Топчий В.А., Юрченко А.Г. // Журн. орган, химии. 1981. Т. 17, № 3.
С. 553-555.
120
J
168. Меликадзе Л.Д., Схиртладзе Н.Н., Дзамукашвили А.А., Гецадзе М.П. // Изв. АН ГССР. Сер. хим. 1975. Т. 1,№ 2. С. 132-137; РЖХим. 1976. 7Ж199.
169. Ланда С, Буркхард И., Вайс И. // Нефтехимия. 1968. Т. 8, N® 3. С. 323- 325.
170. Grob С.А., Schwarz W., Fischer HP. // Helv. chim. acta. 1964, Vol. 47, N 6. P. 1385— 1401.
111. Raber DJ„ Fort R.C., Wiskott £. et al, // Tetrahedron. 1971. Vol. 27, N 1. P. 3-18.
172. Степанов Ф.Н., Юрчепко А.Г., Мурзинова З.Н. и др. // Журн. орган, химии. 1972. Т. 8,№ 9. С. 1837-1841.
173. Степанов Ф.Н., Сидорова Л.И., Довгань Н.П. // Там же. № 11. С. 2338-2342.
174. А.с. 304248 СССР, МКИ С 07 С 13/54, С 07 С 3/00. Способ получения 1-изоалкил-адамантанов / Я.Ю. Полис, Б.П. Рагу ель// Б.И. 1971. № 17. С. 84.
175. Лерман Б.М., Арефьева З.Я., Кузырева А.Р. и др. // Изв. АН СССР. Сер. хим. 1972. №8. С. 1815-1819.
1Т6.Hala S., Landa S. // Coll. Czechosl Chem. Commun. 1960. Vol. 25, N 10. P. 2692-2694.
177. Довгань Н.Л., Мешкова C.K., Лихотворик И.Р, // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1980. N® 17. С. 14-18; РЖХим. 1981. 12Ж117.
178, Hoek W., Strafing Z, Wynberg Н. // Rec. trav. chim. 1966. VoL 85, N 9/10. P. 1045-1053.
179. Fischer ИСР,, Grob CA„ Katayama H. // Helv. chim. acta. 1976. VoL 59, N 6. P. 1953— 1962.
180. Woodworth C.W., Buss И, Schleyer P.R. // Chem. Commun. 1968. N 10. P, 569-570.
181. Vais Z, Burkhard J., Landa S, // Ztschr. Chem. 1968. Bd. 8, N 8. S. 303-304.
182, Багрий Е.И,, Фрид Т.Ю., Санин П.И. // Нефтехимия. 1973. Т, 13. N® 6. С. 800—807.
183. Spengler G.r Wunsch F. // Erdol und Kohle-Erdgas-Petrochem. 1962. Bd, 15, N 9. S. 702-707.
184. Geluk H.W., Schlatmann J.L.M.A, // Chem. Commun. 1967. N 9. P. 426.
185. Schleyer P.R,, Nicholas R.D. // J. Amer. Chem. Soc. 1961. Vol 83, N 1. P. 182-187.
186. Landa S., Vais J., Burkhard Z // ColL Czechosl. Chem. Commun. 1967. VoL 32, N 2. P, 570-575.
187. Applequist D.E., Kaplan L. // J. Amer. Chem. Soc. 1965. Vol. 87, N 10. P. 2194-2200.
188. Wieringa J.H., Wynberg H., Strafing J. // Synth. Commun. 1971. VoL 1, N 1. P. 7-10; РЖХим. 1971. 22Ж283.
189. Burkhard Z, Vais J., Landa К Ц Ztschr. Chem. 1969. Bd. 9, N 1. S. 29-30.
190. Liggero S.H., Majerski Z., Schleyer PR. et aL // J. Label Compounds. 1971. VoL 7, N l.P. 3-10; Chem. Abstr. 1971. Vol. 74.141076u.
191. McKervey M.A., Faulkner D., Hamill H. // Tetrahedron Lett. 1970. N 23. P. 1971 -1974.
192. Moss R.A., Chang MJ. // Ibid. 1981. VoL 22, N 38. P. 3749-3752.
193. /anfai Z, Landa S. Ц ColL Czechosl Chem. Commun. 1970. VoL 35, N 11. P. 3481-3483.
194. Vais J., Burkhard J., Landa S. // Ztschr. Chem. 1969. Bd. 9, N 7. S. 268-269.
195. Landa S., Hlavaty Z // ColL Czechosl Chem. Commun. 1971. Vol. 36, N 8. P. 3059-3065.
196. Схиртладзе H.H., Меликадзе ЛД., Табашидзе НИ. и др. // АН ГССР. Сер. хим. 1981. Т. 7, № 1. С. 17-22; РЖХим. 1982. 1Ж108.
197. StoresundHJ. //Tetrahedron Lett. 1971. N 42. Р. 3911-3914.
198, Grant D., McKervey M.A., Rooney J J. et al // J. Chem. Soc. Chem. Commun. 1972. N21. P. 1186-1187.
199. Lenoir D. // Tetrahedron Lett. 1972. N 40, P. 4049-4052.
200. Bums W., McKervey M.A. // Ibid. 1974. N 21. P. 858-859.
201. Boelema E,, Strafing Z, Wynberg H. // Tetrahedron Lett. 1972. N 12. P. 1175-1177.
202. Prelog V.,SciwerthR. // Chem. Ber. 1941. Bd. 74, N 10. S. 1644-1648.
203. Stetter H, Gartner Z, Таске P. //Ibid. 1965. Bd. 98, N 12. S. 3888-3891,
204. Stetter H., Thomas HG. Ц Angew. Chem. 1967, Bd. 79, Nll.S. 529-530.
205. Stefter H, Thomas HG. //Ghem.Ber. 1968. Bd. 101, N 3. S. 1115-1119.
206. Stetter H., ThomasHG. // Ibid. 1970. Bd. 103, N3. S. 863-867.
207. Janku Z, Landa S. // ColL Czechosl Chem. Commun. 1970. Vol. 35, N 1. P. 375-377.
208. Stetter H., Gartner Z, Таске P. // Angew. Chem. 1965. Bd. 77, N 4. S, 171.
209. Stetter H„ Gartner J. // Chem. Ber. 1966. Bd. 99, N 3. S. 925-929.
121
210. Stepanov F.N., Suchowerchow W.D. // Angew. Chem. Bd. 79, N 19. S. 860.
211. Степанов Ф.Н., Красуцкий П.А., Руданок Л.И. // Журн. орган, химии. 1972. Т. 8, № 12. С. 2616.
212, Юрченко А.Г., Мурзинова З.Н., Степанов Ф.И. // Там же. №11. С. 2332-2338.
213. Юрченко А.Г. Исследование взаимных превращений производных адамантана и бицикло [3,3,11 нонана: Дис* — Д"Ра химлаук. Киев, 1974. 334 с.
214. Udding А.С., Wynberg Н, Strating J. Ц Tetrahedron Lett. 1968. N 55. P. 5719-5722.
215. Юрченко А.Г., Ворощенко A.T., Степанов Ф.Н // Журн. орган, химии. 1970. Т. 6, № 1. С. 189-190.
216. Fieser L.F., Fieser М. Advanced organic chemistry. N.Y.: Reinhold, 1961. 1158 p.
217. Yurchenko A.G.t Zostm LA.t Dovgan’ NL., Verpovsky NS. // Tetrahedron Lett. 1976. N 52. P. 4843-4846.
218. Довгань Н.Л., Лихотворик И.Р., Юрченко А.Г. // Науч.конф. по химии орган, по-лиэдранов: Тез.докл. Волгоград, 1981. С. 43.
219. Krasutsky РА, Jones М. // J. Org. Chem. 1980. Vol 45, N 12. P. 2425-2429.
220. Farcasiu D., Schleyer P.R.t Ledlie D.B. // Ibid. 1973. VoL 38, N 20. P. 3455-3459.
221. Sasaki T.t Eguchi Sh., Hirako Y. // Ibid. 1977. Vol 42, N 18. P. 2981-2985.
222. FarcasiuD. // Synthesis. 19?2.N ll.P. 615-616.
223. Farcasiu D., Bohm H.t Schleyer P.R. // J. Org. Chem. 1977. Vol. 42, N 1. P. 96-102.
224. Bums K, McKervey MA., Mitchell T.R.B., Roony J.J. // J. Amer. Chem. Soc. 1978. VoL 100, N 3. P. 906—911.
225. Vogt B.R., Hoover J.R.E, // Tetrahedron Lett. 1967. N 30. P. 2841-2843.
226. Majerski Z., Dflgas &, Vinkovtf V. // J. Org. Chem. 1979. VoL 44, N 23. P. 4064-4069.
227. Sasaki T., Eguchi Sh., Hattori S. // Tetrahedron. 1978. VoL 34, N 1. P. 67—71.
22%. Katsushima T, YamaguchiR., KawanisiM. // Bull Chem. Soc. Jap. 1982. VoL 55, N 10. P. 3245-3247.
229. Полякова A.A., Храмова Э.В., Багрий Е.И. и др. // Нефтехимия, 1974.Т. 13,№1. С. 9-16.
230. Багрий ЕЛ., Фрид Т.Ю., Соловьев В.Н. и др. // Кинетика и катализ. 1977. Т. 18, №6. С. 1604.
231. Фрид Т.Ю., Соловьев В.Н., Заикин В.Г. и др. // Нефтехимии. 1978. Т. 18, № 1. С. 39—43.
232. Зосим Л.А., Юрченко А.Г. // Жури, орган, химии. 1980. Т. 16, № 4. С. 887-888.
233. Lansbury Р.Т., Sidler J.D. // Chem. Commun. 1965. N 16. P. 373.
234. Margostan D., SpeierJ., Kovacic P. // J. Org.Chem. 1981. VoL 46, N 7. P. 1346-1350.
235. Reinhardt HF. // Ibid. 1962. VoL 27, N 9. P. 3258-3261.
236. Slutsky J.t Engler E.M., Schleyer P.R. // J. Chem. Soc. Chem. Commun. 1973. N 18. P. 685-686.
237. Пат. 2041275 ФРГ, МКИа С 07 DC. Dimerization of alkyl adamantanes / R.E. Moore, A. Schneider // Chem. Abstr. 1971. VoL 75. 76268и.
238. Степанов Ф.Н., Смирнова НА., Бакланов В.Ф., Юрченко А.Г. // Журн,орган.химии. 1974. Т. 10,№ 2. С. 234-239.
239. Багрий Е.И., Фрид Т.Ю., Санин П.И. // Нефтехимия. 1970. Т. 10,№4. С. 480—488.
240. Landa S., Burkhard J., Vais J. // Ztschr.Chem. 1967. Bd. 7, N 10. S. 388-389.
241. Багрий Е.И., Фрид TJ0.t Саннн П.И. // Нефтехимия. 1980. T. 20, № 6. С. 812-818.
242. Udding А.С., Strating J.t Wynberg Н // Tetrahedron Lett. 1968. N 11. P. 1345-1350.
243. Исаев СД., Юрченко А.Г, Степанов ФМ. и др. // Журн.орган, химии. 1973. Т. 9, №4. С. 724-727.
244. Степанов Ф.Н., Столяров З.С. // Там же. 1969. Т. 5,№ 1. С. 91-92.
245. Новиков С.С., Радченко С.С., Новаков ИА. и др. // Функциональные органические соединения и полимеры. Волгоград, 1977. С. 6-9; РЖХим. 1977. 22Ж53.
246. Reinhardt H.F. // J. Polym, Sci. В. 1964. VoL 2, N 5. P. 567-568.
247. Newman H. // Synthesis. 1972. N 12. P. 692-693.
248. Сагинаев A.T., Багрий EM // Нефтехимия. 1982. T. 22, № 6. С. 729-734.
249. Пат. 3419631 США, МКИ С 07 С. 1-(a-Mesityl)-3,5-diinenthyladamantane / J.J. Ven-rooy // Chem. Abstr. 1969. VoL 70. 67841р.
250. Карпенко НФ., Чижов О.С., Новиков С.С. и др. // Изв. АН СССР. Сер.хим. 1971. №2. С. 439-441.
25\. Hala Pecka К., Kremanova В. // ColL CzechosL Chem. Commun. 1974. VoL 39, N 12. P. 3649-3659.
122
252. Степанов Ф.Н., Даниленко ГМ., Диколенко ЕМ., Новикова ММ. // Веста. Киев, политехи, ин-та. Хим. машиностроение и технология. 1969. №6. С. 59-64; РЖХим. 1970. 11Ж235.
253. Stetter Н, Krause М. // Tetrahedron Lett. 1967. N 19. Р. 1841-1843.
254. Pincock R.E., Schmidt J., Scott W.B., Torupka EJ. // Canad. J. Chem. 1972. Vol 50, N24. P. 3958-3964.
255. Пат. 3649702 США, МКИ3 С 07 С. Polymerizable adamantane derivatives/КЕ. Pincock, E. Torupka, W.B. Scott // Chem. Abstr. 1972. Vol 77. 6006t,
256. Liao C.C., De MayoP. //Chem. Commun. D. 1971. N 23. P. 1525-1526.
357.Engler E.M., Schleyer P.R. // AHcycle compounds. L.etc., 1973. P. 239-317; РЖХим. 1974. 15Ж188.
258. Hala S // Chem. tisty. 1977. Sv. 71, N 1. S. 18-63.
259. Graham W.D., Schleyer P.R., Hagaman E.W., WenkertE. // J. Amer. Chem. Soc. 1973.
VoL 95, N 17. P. 5785-5786.
260. Kura$M„ Hala S. Ц J. Chromatogr. 1970. VoL 51, N 1. P. 45-57.
261. Rao S.T., SundaralingamM., OstrwaE. etaL // Chem. Commun. D. 1970.N 14.P.861-862.
262. Rao ST., SundaraUngam M. // Acta aystallogr. B. 1972. VoL 28, N 3. P. 694-699.
263. Bums W., Mitchell T.R.B., McKervey M.A. // J. Chem. Soc. Chem, Commun. 1976. N 21. P. 893-895.
Глава 3
Химические свойства адамантанов
3.1. РЕАКЦИОННАЯ СПОСОБНОСТЬ АТОМОВ УГЛЕРОДА В МОЛЕКУЛЕ АДАМАНТАНА
Известно, что насыщенные углеводороды характеризуются меньшей по сравнению с ненасыщенными или ароматическими углеводородами реакционной способностью. Не так давно их считали ’’химическими мертвецами”. Это обусловлено предельным характером всех С -С-св язей, образуемых зр3-гибридизованными атомами углерода. Насыщенные углеводороды каркасного строения также содержат только освязи, однако такие особенности их строения, как наличие нескольких третичных атомов углерода, чередующихся с метиленовыми мостиками, и объемистой структуры типа клетки заметно увеличивают реакционную способность этих соединений, в особенности в реакциях ионного типа.
3.1.1. КАРБЕНИЙ-ИОННЫЕ РЕАКЦИИ
Высокую реакционную способность и селективность углеводороды ряда адамантана проявляют в реакциях ионного типа, протекающих с образованием карбкатионов в качестве промежуточных соединений. Способность к образованию карбкатиона является лимитирующей стадией, она и определяет, как правило, скорость реакции.
Легко образуется и относительно устойчив по сравнению с другими карбкатионами третичный 1-адамантил-катион. Он стабилизируется как электронодонорным эффектом трех соседних метиленовых групп, так и, вероятно, ’’эффектом клетки”, возникающим в результате перекрывания тыльных сторон орбиталей четырех третичных С-атомов внутри каркаса.
Современные спектральные методы позволяют наблюдать сравнительно устойчивые карбкатионы. ЯМР-спектр 1-адамантил-катиона был получен для вещества, образующегося при действии на фторадамантан ляти-фтористой сурьмы [1], а также при растворении
адамантана и диамантана в сверхсильной кислоте (смеси FSO3H и SbFs [2]. 1-Адамантил-катион легко генерируется из 1-бромадамантана в смеси конц. Н2 SO4 н 50%-ной HNO3 [3], а также при действии других электрофильных реагентов и катализаторов.
124
Таблица 3.1
Способность производных насыщенных углеводородов к сольволизу при 25° С [4-6]
Соединение Константа скорости сольволиза ди*, кДж моль AS, э.а.
80%и-ныйС2 HjOH
1 -Б ромадамантан 4,38 • 10"7 94,5 -12,0
трег-Буталбромид 3,58 10"* 89,9 -2,3
1-Бромбицикло[ 2,2,2] октан 8,68- 10-11 110,4 -16,0
1-Бромбицикло [ 2,2,1 ] гептан 7,0 10-16 - -
сн3соон
1 -Адамантилтозилат 5,68 • 10"* — —
2-Адамантилтозилат 3,25 - 10"’ 30,0 +3,2
Относительная устойчивость 1-адамантил-катиона по сравнению с катионами таких насыщенных полициклических углеводородов, как гомоадамантан, бицикло [2,2,2,1октан, бицикло [3,2,1] нонан, бицикло [2,2,1] гептан, бицикло [3,3,1] нонан, а также в сравнении с трег-бутил-кагионом определена на основании изучения скорости сольволиза их бромидов и тозилатов [4—6]. Полученные экспериментальные данные хорошо объясняются на о снове концепции о предпочтительности планарной конфигурации катионов (табл. 3.1). Разница в скоростях сольволиза 1- и 2-адамантилтозилатов (КГ5) указывает на значительно большую устойчивость 1-адамантил-ка-тиона по сравнению с вторичным 2-адамантил-катионом, чем объясняется селективное протекание реакций ионного типа по узловым атомам углерода ядра.
Из сравнения данных табл. 3.1 видно также, что скорость сольволиза трет-бутилбромида примерно на 103 выше, чем таковая 1-адамантилбро-мида; последнее объясняется тем обстоятельством, что 1-адамантил-катион не может иметь плоской конфигурации. Рассчитано, что для образования ’’уплощенного” катиона, у которого все углы при С-атоме были бы равны 120°, требуется затрата энергии в 50,2 кДж/моль [7]. При углах, соответствующих углам правильного тетраэдра (109,5°), карбкатион будет иметь избыточную энергию в 24,2 кДж/моль. Минимумом энергии 1-адамантил -катион будет обладать, если величина углов у С (1) атома будет промежуточной и равной 113,5° [6]. Для получения катиона такой геометрии требуется 14,6 кДж/моль.
В случае диамантана, где возможно образование уже нескольких неэквивалентных катионов с третичными атомами углерода, легче образуются ’’медиальные” (срединные) катионы (положения 1 и 2) [8]. Эти результаты, однако, не следует отождествлять с данными по устойчивости соответствующих производных. В силу действия пространственных факторов более устойчивыми, напротив, являются ’’апикальные’’ (вершинные) производные (положение 4).
125
ЬЬГ5—SOgClF
Разница в энтальпии 1- и 4-бромдиамантана составляет 2,6 кДж/моль [9, Ю].
В триамантане наиболее стабильный карбкатион образуется в положении 2 [8]. Это подтверждается результатами бромирования триамантана
15
[11], где первоначально образуются 2-замещенные производные. Однако в положении 2 триамантана заместитель испытывает четыре неблагоприятных 1,3-взаимодействия с сии-аксиальными атомами водорода, находящимися в положениях 8, 17, 16и 18. Это существенно снижает термодинамическую устойчивость 2-производных триамантана, вследствие чего они затем изомеризуются в 9-, 3- и 4-замещенные производные.
Образование 2-карбкатиона триамантана наблюдалось также в сверхкислых средах [11]. Повышенная устойчивость этого катиона, вероятно, связана с тем, что атом углерода С (2) соединен непосредственно с тремя третичными атомами углерода, оказывающими дополнительный индуктивный эффект сопряжения.
Вполне устойчив также 3-гомоадамантил-катион, полученный в сверхсильной кислоте при -78°С и изученный с помощью спектроскопии ПМР
1
и ЯМР 13С [12]. Спектры указывают на классическую природу 3-гомоада-мантил-катиона, являющегося трехвалентным карбкатионом. Найдено, что его ’’клеточный эффект” незначителен по сравнению с эффектом в 1 -адамантил-катионе.
В настоящее время получены и исследованы также некоторые дикатионы углеводородов ряда адамантана. Их образование наблюдалось при низких температурах в сверхсильных кислотах [13].
5(13С) 301,0 м.д.
126
Дикатионы типа (1) устойчивы только тогда, когда они содержат делокализующие заместители, такие, как циклопропил или фенил.
Из результатов работ по данной тематике в последние годы необходимо отметить прежде всего синтез Шлейером с сотр. [13а] чрезвычайно интересного нового дикатиона ряда адамантана — 1,3-дегидро-5,7-адамантандиила. Синтез был предпринят после расчета, показавшего, что такой дикатион должен быть стабильнее 1-адамантил-катиона на 198 кДж/моль.
Из 1,3,5,7,-тетраиодадамантана действием двухфтористой ртути был получен 1,3,5-трифтор-7-иодадамантан, а из него с и-бутиллитием — 1,3-де-гидр о-5,7-двфторадамантан. Последний прибавили к раствору SbFs в S0C1F и получили дикатион, который обладал необычной стабильностью — выдерживал нагревание от —80° С (температура синтеза) до 0°С.
В этом дикатионе два электрона С (1)-С (3)-связи делокализованы по всем узловым атомам, углы С—С—С-связей деформированы до 88° (строение установлено на основании спектров ЯМР, данные которых согласуются с квантовомеханическими расчетами).
В силу рассмотренных особенностей строения углеводороды ряда адамантана и их производные являются весьма перспективными объектами исследования для химии дикатионов — новой активно развивающейся области химии.
Как уже отмечалось выше, ионные реакции адамантана и его гомологов протекают легко и с высокой селективностью вследствие легкого образования соответствующих третичных карбкатионов. Указанием на ионный характер реакции обычно является предпочтительное образование производных с заместителями в узловых положениях или с заместителем у других третичных атомов углерода. Как будет показано ниже, в реакциях с участием радикалов такой селективности не наблюдается. Из реакций ионного типа наиболее изученными являются реакции галогенирования, они будут рассмотрены в разд. 3.4.
При реакции ионного гидрирования адамантан играет роль донора гид-рид-ионов [14]. В качестве акцепторов гидрид-иона были исследованы 1-метилциклогексен-1, трифенилкарбииол, циклогексанои, адамантанон. Максимальная конверсия 1-метилциклогексена-1 составляет 25%.
Н®(ИЗ CFjCOOHl
Ij ---------------2-------
Add
127
Реакцию проводят при 40-90° С в малополяриом (м-гексан) или полярном (CS2, СНС1Э, СН2 С13 ) растворителе или вовсе без растворителя.
Повышенная устойчивость трег-адамантил-катионов позволяет получать на их основе устойчивые соли (фторантимонаты) типа Ad+ SbF6“, подобно тому как это имеет место в случае трег-бутил- и трициклопропил карбкатионов [15]. Для получения таких солей используют 1-галогеиадамантаны (Hal = F, Cl, Вт) и избыток SbF5 в 1,1,2-трихлорфторэтане, выходы солей 96%.
Стабильность солей адамантил-катиона выше, чем аналогичных солей трет-бутил- и трициклопропил карбкатионов, что весьма важно для их применения в качестве алкилирующих агентов и катионных катализаторов полимеризации. Высокая устойчивость третичных адамантил-катионов играет также важную роль в реакциях гидридного переноса, протекающих при окислении адамантана серной кислотой, реакциях диспропорционирования гидрокси- и галогенпро из водных адамантана, карбоксилирования и других, рассмотренных ниже.
3.1.2. СВОБОДНОРАДИКАЛЬНЫЕ РЕАКЦИИ
Углеводороды ряда адамантана весьма активны также в реакциях радикального типа. Относительная устойчивость и реакционная способность 1-адамантил-радикала по сравнению с радикалами других мостиковых углеводородов детально исследовалась на примере реакции декарбонилирования альдегидов [16].
RCO
R + СО RH, RC1 I__________f
н*
—+ RCOC1
СС14
Так как к2 (константа скорости захвата радикала RCO растворителем) малочувствительна к природе радикала R, то устойчивость свободных радикалов оценивалась соотношением к\ /кг (табл. 3.2) -
Порядок относительной устойчивости радикалов прн сравнении 1-ада-маитил- и трег-бутил-радикалов (согласно данным табл. 3.2) обратен порядку относительной устойчивости соответствующих катионов (см. данные по сольвол из у, табл. 3.1) — 1 -а дамантильный радикал примерно на 4,18 кДж/моль более устойчив, чем трег-бутильный радикал [1]. Однако данные, полученные диумя методами при разложении пероксидных эфиров, свидетельствуют о том, что 1-адамантильный радикал менее устойчив, чем 128
Таблица 3.2
Относительная устойчивость углеводородных радикалов
Углеводородный радикал *»/*»
(СН3)ЭС- 1,00
1-Ad 2,5
1 -бицикло [ 2,2,2] октил 1,2
1-бицикло[2,2,1 ]гептил 0,007
трег-бутильный [17, 18]. К аналогичному выводу приводят результаты работы [19], где из данных по распаду азосоединении следует, что 1-адамантильный радикал менее стабилен, чем ациклические третичные радикалы. Изучение реакционной способности 1-адамантил-радикала, образующегося при фотолизе соответствующего азосоединения, в различных растворителях показало [20], что этот радикал, подобно 1-бицикло [2,2,2] октильному радикалу, легко отрывает водород от насыщенных углеводородов, а его присоединение к бензольному кольцу происходит значительно легче, чем присоединение трег-бутильного радикала. 1-Адамантильный радикал проявляет высокую селективность в отношении водорода, находящегося в ароматическом кольце; присоединение такого водорода протекает в 25 раз быстрее, чем присоединение водорода циклогексанового кольца.
Не менее важным н интересным в теоретическом отношении является вопрос о сравнительной устойчивости и реакционной способности 1- и 2-ада-м ант ильных радикалов. Данные, полученные при изучении реакции свободнорадикального галогенирования адамантана, позволили сделать вывод [21, 22], что в реакциях радикального типа 1-адамантильный радикал образуется легче, чем 2-адамантильный. Вместе с тем 1-адамантильный радикал менее стабилен, что приводит к предпочтительному образованию 1-замещенных производных (соотношение 1- и 2-замещенных равно 4:1). 1-Адамантильный радикал предпочтительно отрывает хлор в хлорсодержащих растворителях [22], а при радикальном хлоркарбонилированни адамантана 1- и 2-изомеры образуются в соотношении 55 : 45 [23]. Атомарный бром, генерируемый фотохимически из N-бромсукцинимида, при действии на адамантан в растворе СС14 дает смесь 1- и 2-замещенных Производных [24, 25]. В то же время реакция прямого фотооксимнровання адамантана смесью хлора и монооксида азота, протекающая, вероятно, также по радикальному механизму, приводит к образованию только 2-замещенных производных [26]. Аналогичный результат был получен в реакции окислительного фосфорилирования адамантана [27].
Причиной такого различия в протекании реакций радикального типа с участием адамантана является, вероятно, не только различная скорость образования, стабильность н реакционная способность радикалов, но и взаимная изомеризация 1- и 2-адамантил-радикалов посредством межмолекулярного обмена атомом водорода; вследствие образования стабильных конечных продуктов равновесие может постоянно смещаться в сторону образования одного из них. В связи с этим интересны результаты по облучению адаман-9.Зак.1856 129
тана (60Со, у-облучение, 0,5 Мрад) при 77 К [28]. С помощью ЭПР-метода было установлено, что образуются оба 1- и 2-ацамантильных радикала. При 77 К преобладает 2-адамантил-радикал, в то время как при 298 К происходит частичная изомеризация радикалов и соотношение 1- и 2-адамантил-радикалов изменяется до 4 : 1. Из работы [29] следует, что 1-адамантил-радикал заметно более долгоживущ, чем 2-адамантил-радикал. Незамещенный 2-адамантил-радик ал по своей конфигурации весьма близок к планарному. При введении заместителя, например триметил силокснгруппы, в положение 2 радикал далек от планарного н легко инвертирует [30].
Адамантан, как и другие насыщенные циклические углеводороды, активен в реакциях свободнорадикального отщепления водорода при действии СС1з [31], в реакциях внутримолекулярной циклизации, протекающих через промежуточные бирадикалы, например при фотолизе 2-адамантилтри-фторацетата [32], а также в реакциях замещения и фрагментации, протекающих с участием катион-радикалов [33]. В реакции свободнорадикального замещения легко вступают также высшие адамантаноидные углеводороды, в частности триамаитан [11]. Изучены реакции с участием 1- и 2-адамантилкарбенов [34,35].
3.1.3. ВЗАИМНОЕ ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ
Вследствие особенностей строения молекула адамантана представляет большой интерес в качестве объекта для теоретических и экспериментальных исследований пространственных и индукционных эффектов в насыщенных соединениях. Одна из наиболее интересных проблем — выяснение влияния заместителей на реакционную способность производных каркасных углеводородов.
1-Адамантильная группа оказывает более значительный индукционный эффект по сравнению с грет-бутильной группой. Это следует из результатов изучения дипольных моментов ряда 1-замещенных производных адамантана (С1, Вт, ОН, NH2, СН2 С1 и др.) [36]. При соответствующей корреляции длины диполя наблюдается линейная зависимость между дипольными моментами производных адамантана и о*-константами Тафта. Повышенный по сравнению с другими алкильными группами положительный индукционный эффект проявляет адамантильная группа также в 2-замещенных производных; последние также характеризуются повышенными дипольными моментами [37].
С помощью метода ядерного квадрупольного резонанса (ЯКР) в замещенных в ядре алкил адамантанах (35С1) была найдена значительная электронная плотность на атоме хлора, т.е. сравнительно большая ионность связи С—С1, что подтверждает значительный злектронодонорный эффект адамантанового ядра, содержащего алкильные заместители [38].
Вопрос об индукционном эффекте алкильных групп, находящихся в ядре адамантана, остается неясным. З-Алкилзамешениые 1-бромадамантана показывают меньшую скорость сольволиза по сравнению с незамещенным 1 -бром адамантан ом, из чего можно было бы сделать вывод об электроноакцепторном эффекте алкильных групп [39]. Однако, возможно, это является результатом стерическлх препятствий, создаваемых алкильными 130
группами; пока, вероятно, следует воздержаться от вывода о наличии индукционного эффекта [40], Интересные в этом отношении результаты были получены при исследовании потенциалов ионизации некоторых алкиладамантанов с помощью фотоэлектронных спектров. Установлено, что алкильные заместители оказывают лишь весьма незначительное влияние на значение первых потенциалов ионизация (табл. 3.3).
В то же время потенциалы ионизация существенно изменяются в парах адамантан-адамантанон (8,76) и адамантан-диамантан (8,93).
Для прогнозирования величины реакционной способности алкиладамантанов, других каркасных углеводородов н их функциональных производных важно выяснить механизм передачи взаимного влияния заместителей. Адамантановое ядро представляет собой жесткую трициклическую систему, построенную нз простых С—С-связей, вследствие чего взаимное влияние заместителей должно передаваться по индукционному механизму. На примере 1-С1-3-Х-зам еще иных производных адамантана (X = мзо-С3Н7, СН3, Н, СН2Вг, СООН, С0С1) такой механизм был подтвержден на основании спектров ядерного квадрупольного резонанса этих соединений при 77К [38, 43]. Была найдена четкая корреляция н линейная зависимость частот спектров ЯКР 3/С1 исследованных соединений с константами о* заместителя X: р = 31,337+0,192 Эта корреляция была одинаково хорошей как в случае злектроноакцепторных, так и в случае злектронодопорных заместителей. Коэффициент проводимости электронного влияния 1,3-за-местнтелей в молекуле адамантана соответствует числу освязей (четыре), разделяющих заместители. Проводимость индукционного влияния через неподвижную систему, состоящую из трех углеродных атомов каркаса, оказалась несколько выше, чем через ациклическую цепь (через СН2-группы). Доля индукционного влияния, передаваемого через пространство, оценена в 23% от величины влияния по связям цикла [44].
Для производных, содержащих один мостиковый заместитель (1,4-ди-замещенне), взаимное индукционное влияние заместителей (положение 4) на реакционный центр (положение 1) передается либо по освязям (для цис-положения), либо полем (транс-положенне заместителей) [45]. В случае 1,2-дизамещенных производных индукционное влияние также передается по о-связям [46].
Как уже отмечалось, на реакционную способность углеводородов ряда адамантана существенное влияние оказывают стерическне эффекты. Нали-
Таблица 3.3
Потенциалы ионизации мостиковых углеводородов [41,42]
Углеводород 1 Потенциал ионизации, эВ
Адамантан 1 9,25
1 -Метиладамантан 9,24
2-Мети ладам антан 9,24
1,3,5,7-Тетраметил адамантан 9,23
Диамантан 8,93
Бицикло [ 2,2,11гептан 9,80
131
чие объемистого ядра затрудняет протекание химических реакций. Это рассмотрено в разд. 4.2. Здесь лишь отметим, что стерическнн эффект наглядно проявляется при комплексообразовании адамантилпирогаллола с гига-hom(IV) [47]. Комплексообразующая способность адамантилпронзводного пирогаллола значительно ниже, чем у незамещенного пирогаллола.
3.14. КОЛИЧЕСТВЕННЫЕ ДАННЫЕ ПО РЕАКЦИОННОЙ СПОСОБНОСТИ
Для количественной оценки реакционной способности адамантана и близких по строению углеводородов были использованы расчетные методы. Методом ЛКАО МО рассчитаны величины делокализуемое™ Ф^, Ф? положений 2, 3 и 4 молекулы адамантана и 1-R-замещенных адамантанов (R = СН3, ОСН3, СООСН3, CN) при отщеплении Н+ н И' [48]. Расчет, в частности, подтвердил экспериментальные данные, что наибольшей активностью обладают узловые атомы водорода. Методы квантовой механики дали весьма ценную информацию о величине реакционной способности также других мостиковых соединений [49, 50], и в этом случае расчетные данные хорошо согласуются с экспериментальными. Последние для углеводородов ряда адамантана базируются главным образом на результатах исследования реакции сольволиза галогенпроизводных, в частности бромпро-нзводных. Реакция замещения брома в 1-бромзамещенных производных бромадамантанов протекает исключительно по моно молекулярно му механизму [51]. На скорость сольволиза бромидов существенное влияние оказывают заместители, содержащиеся в других узловых положениях ядра [40, 52—54]. Электроноакцепторные группы, снижая устойчивость адаман-тил-катиона, тем самым понижают скорость реакции сольволиза. К аналогичному эффекту приводит н введение метильной группы [55], хотя последняя, как известно, характеризуется электронодонорными свойствами; такое аномальное поведение метильной группы пока не нашло объяснения. Сопоставление констант скорости сольволиза 3-замещенных 1-бромадамантанов с о*-константами заместителей показало весьма высокий коэффициент корреляции (г =0,996) [40]. Корреляция логарифмов констант скоростей реакции сольволиза с рКа соответствующих адамантанкарбоновых кислот также достаточно высокая (г =0,986) [52]. Коэффициенты корреляции от 0,947 до 0,999 были получены также между потенциалами полуволн 614 н (д -константами заместителей при изучении полярографического поведения серий производных 1-бромадамантана н 1-бромметиладамантана [44].
К важным результатам привели исследования сольволиза других производных адамантанов, в частности хлоридов н n-толуолсульфонатов. Изучение кинетики сольволиза 1-адамантилхлорида в уксусной, муравьиной, трифторуксусной кислотах, а также в некоторых бинарных средах показало, что 5^1-ре акции 1 -адамантилгалогештроизводных протекают быстрее, чем такие реакции аналогичных трет-бутильных производных. Иными словами, в реакции RX + R + R+ + R* X предпочтение по стабильности отдается адамантил-катиону [56].
Механизм реакции сольволиза, вероятно, различен в зависимости от условий. Данные, полученные при сольволизе 1-адамантилтозилата в ацего-132
нитриле, например, позволили предположить, что лимитирующей стадиен реакции является превращение первоначально образующейся контактной ионной пары субстрата в сольватно-раздельную ионную пару [57].
При изучении сольволиза дейтерированных производных типа
(?) (3)
(X=0Ts,Br)
В (Ч)
наблюдались эффекты сопряжения в 1-адамантил-катионе [58], Проведение реакции в 80%-ном CF3CH2OH показало отсутствие |3-эффекта дейтерия при сопьвопнзе 2,2-дейтерированного соединения (2) по сравнению с сольволизом 1-бромадамантана. Этот факт объясняется взаимной компенсацией действия даух противоположных эффектов — сверхсопряжения даух атомов дейтерия у С(2)-атома ядра (двугранный угол 60°) с рюрби-талью катиона и конформационно-независимого индуктивного эффекта, обусловленного несколько большим электроноакцепторным влиянием D в сравнении с Н. Однако при сольволизе соединений (3) н (4) все же наблюдается заметный кинетический положительный изотопный эффект у-атомов дейтерия (положения 3, 5 и 7), что, вероятно, является результатом взаимодействия псевдо-п-орбиталей С(3, 5, 7)—H(D) с тыльной стороной свободной р-орбиталн С (1)-атома. Как уже отмечалось выше, симметрия молекулы такое взаимодействие разрешает.
Передача взаимного влияния заместителей на скорость сольволиза наблюдается в реакции гидролиза моно- и дибромпронзводных гексафтор-пропил адамантана [59]. Введение в ядро второго атома брома приводит к значительному уменьшению константы скорости реакции вследствие его заметного электроноакцепторного эффекта.
Значительный теоретический интерес представляет изучение реакций сольволиза 2-адамантильных производных, в которых рассмотренные выше эффекты передачи влияния заместителей через ядро (’’эффекты клетки”) не должны проявляться. Сольволиз 2-адамантилъных н 2-метил-2-адаман-тильных производных протекает через образование классических карбкатионов [60]. Однако имеются случаи, когда результаты указывают на образование ”неклассического” (мостикового) карбкатиона. Так, при сольволизе 2-замещенных соединений типа (5) для соединений, содержащих заместитель также в узловом положении,
fR=cn,; x=Br,OTs),
скорость реакции замещенных соединений при 25° С примерно в 35 раз выше скорости реакции сопьволнза незамещенных соединений (R - Н)
133
[61]. Вероятно, при сольволизе образуется неклассический карбкатион (6), который стабилизируется вследствие электроиодонориого влияния метильной группы. Возможность образования иеклассического карбкатио-на при сольволизе 2-замещеииых производных адамантана рассматривается таки® в работе [62]. Из-за стерических факторов реакция сольволиза 2-а да мантил производных (бромида и n-толуол сульфоната) в 80%-иом спирте и СН3СООН при 25° С протекают по механизму (через карбкатион), свойственному сольволизу субстратов с карбкатионом при третичном, но не вторичном атоме углерода [63, 64]. Поведение исследованных соединений резко отличается от поведения других производных углеводородов, например, содержащих изопропильную группу. Такое различие обусловлено нуклеофильным участием растворителя в сольволизе производных изопропильного ряда; в сольволизе 2-адамантилпроизводных влияние растворителя исключается из-за наличия объемистого ядра адамантана. При сольволизе 2-адамаитиларенсульфонатов наблюдается образование ионных пар [65]. Образование как "классических”, так и ’’неклассических” полиметил-2-а дамантил-катионов имеет место при сбльволизе пространственно-затрудненных 2-адамантилпроизводиых [66, 67], в частности 1 -алкил-2-адамантилтозилатов, а также при сольволизе 4-протоадамантил-и 4-метил-4-протоадамантилпроизводиых [68—70].
Исследование кинетических закономерностей реакции сольволиза 1-замещенных 2-адамантилсульфонатов (заместители: СН3, СО2СН3, CN) показало, что имеет место линейная зависимость констант скорости сольволиза от коистаит Гаммета—Тафта о*; при 25°C наклон прямой /3 = 4,09. Реакцию изучали в интервале температур 0—120°С в 50%-иом водном этаноле [71].
Производные тетра- и пентациклических углеводородов ряда адамантана характеризуются более высокой по сравнению с соответствующими производными адамантана реакционной способностью в реакции сольволиза. Если скорость сольволиза 1-бромадамантана принять за единицу, то скорость сольволиза 1- и 6-бром-2,4-зтаноадамантанов составляет соответственно 2,6 и 13 (в 80%-иом этаноле) [72].
Бромпроизводиые днамантана вступают в реакцию сольволиза с различной скоростью в зависимости от числа атомов брома в молекуле. Сольволиз моиобромидов протекает легко, а гидролиз дибромпроизводиых требует большего времени и более высокой температуры. Это происходит при гидролизе под действием НС1 [9], смеси К2СО3 и AgNO3 или водного спирта и ацетона [8, 73]. Согласно данным работы [8], скорость сольволиза медиального 1-бромдаамантана в 8 раз превышает скорость сольволиза 1-бромадамантана; для апикального же изомера — 4-бромдиамантаиа — она в 3 раза ниже. Ввиду того что 1- и 4-бромдиамаитил-катионы, вероятно, более напряжены, чем 1-адамантил-катиои, можно было ожидать меньшей скорости сольволиза обоих изомеров бромдаамаитаиов. Вероятно, скорость сольволиза повышается за счет большего напряжения, вызываемого уходящей группой; важную роль играет также некоторое повыще-иие устойчивости 1-днамантил-катиона благодаря наличию даух соседних тртичных атомов углерода (/3-разветвление). Скорость сольволиза вторя шых 3-даамантилтозилатов в 3,5 раза выше, чем аналогичных 2-адамантил то зил ато в [73].
134
При проведении гидролиза полизамещенных бромдиамантанов в конц. HNO3 (65%-иой) для разных по степени замещения бромидов требуется различная температура: для моно бромида 50—60° С, да бромида 70-80° С, трибромида 80—90° С, а тетрабромид вообще не подвергается гидролизу даже после кипячения в HNO3 в течение 1 ч [74]. Сольволитическая активность некоторых циклических аналогов адамантана, например 1-гомоада-маигилкарбннилтозилата и производных твистана (трицикло [4,4,03,8] декана), рассмотрена в работах [75, 76].
3.2. ТЕРМИЧЕСКИЕ И КАТАЛИТИЧЕСКИЕ ПРЕВРАЩЕНИЯ АДАМАНТАНОВ
3.2.1. ПРЕВРАЩЕНИЯ АДАМАНТАНОВ ПОД ДЕЙСТВИЕМ ПОВЫШЕННОЙ ТЕМПЕРАТУРЫ И МЕТАЛЛСОДЕРЖАЩИХ КАТАЛИЗАТОРОВ
Адамантан - наиболее устойчивый из алициклических углеводородов. Его термический распад начинается при 660—675°С и приводят к образованию сложной смеси газообразных, жидких и твердых продуктов [77]. В присутствии катализаторов температура термолиза снижается до 550°С над алюмосиликатным катализатором и до 570° С — над алюмо хромовым [78].
Радиационное облучение адамантана при относительно низких температурах приводит первоначально к выделению водорода. Эффективная энергия активации реакции образования водорода в интервале —196-Н-240°С изменяется от 0,2 до 3,8 кДж/моль [79]. При температуре выше 160°С наблюдается снижение выхода водорода и начало радиационно-термического крекинга адамантана вследствие разрыва С—С-связей [79—81]. Образуются как низкомолекулярные, так и более высокомолекулярные, чем адамантан, вещества - продукты алкилирования и конденсации. В этих процессах промежуточными частицами являются радикалы. Радиационно-химический выход радикалов адамантана при его у-облучении от источника 60Со при 74 К составляет 3,2 молек./100 эВ, для 1-и-пропиладамантана -1,6 [82].
Температуру разложения адамантана резко снижают также сильные электрофильные катализаторы, например галогениды алюминия. Награва-ние адамантана с А1Вг3 уже при 100° С приводит к образованию водорода, газообразных углеводородов и смеси алкиладамантанов и ароматических углеводородов, в том числе состава С8Ню, СюН14, СцН16 [83, 84].
Состав продуктов разложения адамантана в присутствии металлсодержащих катализаторов гидрирования существенно зависит от природы катализатора. Алюмоокисиые катализаторы, содержание Сг и Pt, вызывают фрагментацию с образованием газообразных парафинов, бензола, алкилбензо-лов, гидриидаиа и нафталина [85]. На катализаторах Pt, Ст и Pd, нанесенных на активированный уголь, образуются те же продукты, кроме нафталина. В присутствии никелевых катализаторов [Ni (50%) /кизельгур, Ni(20%)/A12O3, №-(50%УА12Оз] образуются исключительно газообразные углеводороды.
135
Алкиладамантаны в условиях термического и термокаталитического разложения, как правило, менее устойчивы, чем адамантан. Наиболее легко протекающий процесс здесь - деалкилирование. Однако, как будет показано ниже, в условиях реакции деструктивного алкилирования (катализаторы AlHai3. температура выше 50°С) адамаитаи значительно менее устойчив, чем 1,3,5,7-теграметиладамантан, у которого нет незамещенных третичных наиболее реакционноспособных атомов углерода.
Каталитическое деалкилирование алкиладамантанов имеет место при нагревании в присутствии металлических катализаторов. Реакции протекает при 300—800 °C [86] над металлами VII и VIII групп таблицы ДМ. Менделеева [86], над металлами VI6 или VIII группы или оксидами металлов, пропитанными гидроксидами щелочных металлов при 500—800 ° С и давлении 68 атм [87].
В присутствии металлсодержащих катализаторов в атмосфере водорода полизамещеиные алкиладамантаны образуют адамантан и алкиладамантаны меньшей молекулярной массы. Максимальный выход адамантана на катализаторе Ni/Al2O3 (37% Ni) из 1,3-ди метил адамантана составляет 62,5%. При гидродеалкилировании 1 -этиладамантана основными продуктами являются адамантан и 1-метиладамантан (соотношение 81,2:18,8); 13-ДЙ' метил-5-этиладамантана — 1,3-диметил-, 1-метиладамантаны и адамаитаи (0,9:22,5:76,5); при деалкилировании 1,3,5,7-тетраметиладамантана — 1,3,5-трнметил-, 1,3-диметил-, 1-метиладамантаны и адамантан (1,2:8,2:24,1 :66,5) [88].
Деалкилирование алкиладамантанов протекает по консекутивной схеме с последовательным отщеплением метальных групп. Зависимость состава
Рис. 3.1. Деалкирование 1,3,5-триметиладамантайа иа никель-алюмохромовом катализаторе
Импульсный микрокаталитический метод, Н2, время контакта 0,2 с, 1) 1,3,5-Три-мети л адамантан, 2) 1,3-диметиладамантан, 3) 1-метиладамантан, 4) адамантан, 5) газообразные продукты
136
I продуктов деалкилирования 13,5-триметиладамаитана от температуры | реакции представлена на рис • 3.1.
к Отщепление заместителя — основное направление распада ариладаманта-1 нов под действием каталитических глин при 400—450°C. В результате | деструкции 1-(1-нафтил)-, 1-(7-(2-метил) нафтил)*, 1-(л-дифенил)-, | 1-(2-флуоренил) *, 1-(2*антрил) - и 1-(2-фенантрил)адамантанов образуют-
| ся адамантан и соответствующие ароматические углеводороды [89].
I При термокаталитических превращениях тетра- и пентациклических f углеводородов ряда адамантана доминирующим направлением их распада ? также является расщепление с образованием производных адамантана S или диамантана, те. с сохранением устойчивого ядра молекулы. Так, например, гидрокрекинг 1,2-бутаноадамаитана приводит к смеси 1- и 4 2-бутиладамантанов [90]; метиладамантаны в условиях каталитического
гидрогенолиза легко отщепляют метильные группы [91]. Последняя реак-'* ция изучалась на примере метиладамантанов, метилдиамантаиов и 2влетал триамантана в газовой фазе в атмосфере водорода при 150—250 °C над N1/A12O3. Интересно, что адамантаноидные углеводороды также вполне | устойчивы к каталитическому расщеплению в атмосфере водорода; их пол-ная деструкция ДР метана происходит при относительно высоких Темпе-S' ратурах. Так, 2-метилтриамантан последовательно превращается в трнаман-£ тан, диамантан, адамантан и метан при 280 °C.
I В УСЛОВИЯХ ЖИДКОФАЗНОГО КИСЛОТНОГО КАТАЛИЗА
В условиях кислотного катализа, когда возможно образование адаман-f тил-катионов, деструкции углеводородов ряда адамантана предшествует нх пространственная и структурная изомеризация. Изучению процессов изомерных превращений в химии углеводородов ряда адамантана уделяет-< ся значительное внимание, так как вследствие специфического строения молекулы адамантана, содержащей жестко фиксированные циклогексановые кольца (неспособные дегидрироваться и лишенные инверсионных л переходов), представляется редкая возможность изучения таких перегруп-S пировок производных циклогексана, наблюдение и точная интерпретация которых в случаях подвижных инверсионных систем затруднительна или > невозможна.
Одной из наиболее известных перегруппировок в ряду алкиладамантанов является изомеризация 1-этиладамантана в 1,3-диметиладамантан при контакте с галогенидами алюминия [92].
СП,
Д1На1,,ЭО°С
снгсн,
СП,
Аналогично 1-метил-З-этил адамантан изомеризуется в 1,3,5-гриметил-адамантан, а 13 -метил-5 -этиладамантан образует 1,3,5,7-тетраметиладамантан [93]. Направление превращений этиладамантаиов контролируется термо динамическими факторами и обусловлено большей термодинами
137
ческой устойчивостью таких полиметил адамантанов по сравнению с этил-адамантанами. Кинетически эта перегруппировка является самым медленным этапом в цепи превращения трициклических насыщенных углеводородов. Относительные скорости изомеризации 1-этил-, 1-метил-3-этил- и 1,3-днметил-5-этил адамантанов (по отношению к скорости изомеризации этил циклогексана, принятой за единицу) составляют 0,02, 0,01 и 0,005 соответственно [94, с. 242-245].
По мере увеличения длины алкильной группы изомеризация алкиладамантанов затрудняется. Так, значительная степень превращения 1-н-про-пиладамантана достигается только при 70 °C и большом мольном соотношении катализатор-.субстрат (1:1); в этих условиях изомеризация уже осложняется протеканием побочных реакций деалкилирования и межмолекулярного перераспределения алкильных групп. Из 1-изобутилада-мантаиа (60 °C, 0,5 ч, мольное соотношение алкиладамантан : А1С13 =1:1, конверсия 98%) образуется смесь, содержащая ~50% адамантана, 18% 1-м-бутила дамантана и всего 7,5% 1,3 -диметил -5 -эти л адамантана. Таким образом, деалкилирование и изомеризация боковой цепи становятся основными направлениями превращений. Рис. 32 показывает, что обе реакции протекают параллельно.
Увеличение степени деструкции алкиладамантанов при их контакте с галогенидами алюминия по мере возрастания длины алкильной группы обусловлено, вероятно, понижением устойчивости промежуточного не
Рис. 3.2. Превращения 1-изобутиладамантана
Катализатор А1Втэ и НВг,раствор в гексане, 20° С Зависимость содержания адамантана ()) и 1-н-бутил адамантана (2) от времени реакции
Рис. 3.3. Изменение относительного содержания метиладамантанов в реакционной смеси, полученной при контакте адамантана с 2,3,5-триметилгексаном
Катализатор А1Вг3, 160°С. 1) Адамантан, 2) I метиладамаятая, 3) 1,3-диметил адамантан, 4) 1,3,5-триметиладамаятан, 5) 1,3,5,7-тетраметиладамантан
138
классического карбкатиона
При наличии длинной боковой цепи происходит одновременный разрыв связей С(1)—С(3) и С(2)—С(3). Причиной этого может являться уменьшение электро но до норного эффекта заместителя, влияющего на устойчивость промежуточного карбкатиона, а также увеличение числа колебательных и вращательных степеней свободы заместителя. Стабилизация катиона может приводить также к образованию в качестве интермедиата замещенной структуры гомоадамантана, являющемуся начальным этапом дальнейшей структурной изомеризации.
Алкиладамантаны, содержащие длинные алкильные группы, при контакте с галогенидами алюминия испытывают также другой тип фрагментации, обнаруженный Казанским с сотр. [95]. Наблюдалось отщепление метильных, этильных и пропильных звеньев от длинных алкильных групп. Продукты превращения 1-бутиладамантанов, например, содержали 1-метил-, 1-этил- и 1-пропиладамантаны и продукты их структурной изомеризации. При данном типе фрагментации с несколько большей вероятностью реализуется расщепление С—С-связи, расположенной в 0-положении по отношению к ядру, т.е. образование метиладамантанов.
При температурах выше 100 °C при действии АНа1з активно протекает реакция перераспределения метильных групп.
Если время реакции достаточно велико, то постепенно равновесие смещается в сторону сполна замещенного (в узловых положениях) углеводорода — 1,3,5,7-те траме тил адаманта на. Дополнительным источником метильных групп могут служить разветвленные алканы (если реакцию проводят в их среде); в этом случае протекает реакция деструктивного алкилирования адамантана н алкиладамантанов. Изменение относительного содержания метиладамантанов при контакте с А1Вгз продуктов взаимодействия адамантана с 2,3,5-триметилгексаном приведено на рис. 33.
Протекание реакции перераспределения метильных групп является наиболее вероятной причиной образования полизамещенных метиладаман-танов, ио не самого адамантана в процессе так называемой ’’адамаитиза-ции” (см. гл. 2). Уместно напомнить, что ее сущность заключается в том, что при контакте с галогенидами алюминия в достаточно жестких условиях самые различные углеводороды или их производные способны образовывать (правда, в относительно небольших количествах) соединения со структурой адамантана [96]. Если адамантан и образуется на определенном
139
этапе этих превращений, то протекающие затем реакции алкилирования, изомеризации, деструкции и перераспределения заместителей приводят к поли замещенным метиладамантанам.
Таким образом, в условиях кислотного катализа углеводороды ряда адамантана претерпевают сложный комплекс химических превращений, в основе которых лежат реакции изомеризации, алкилирования—деалкилирования, фрагментации и перераспределения алкильных групп, что приводит к образованию сложной смеси продуктов реакции. Несмотря на это, путем подбора исходного субстрата и условий реакции можно обеспечить протекание превращений по желательному направлению и получение требующихся продуктов.
Из перечисленных выше превращений углеводородов ряда адамантана к настоящему времени наиболее глубоко изучены реакции изомеризации и алкилирования. Эти реакции представляют значительный теоретический интерес главным образом для выяснения лежащих в их основе перегруппировок алициклических катионов, а также большой практический интерес — с их помощью могут быть получены важные в практическом отношении углеводороды.
Среди перегруппировок, свойственных адамантил-катионам, наблюдаются как перегруппировки, присущие карбкатионам других алициклических и алифатических углеводородов, так и специфические перегруппировки, возможность протекания которых обусловлена особенностями строения ядра адамантана. К первым относятся прежде всего перегруппировки, вызывающие изменение боковой цепи (например, изомеризация 1 -изо-бутиладамантана в 1 -н-бутиладамантан), и перегруппировки, приводящие к увеличению числа заместителей в ядре адамантана. Последние протекают по пути расширения шестичленного цикла и образования промежуточной неустойчивой структуры гомо адамантил-катиона [94], например изомеризация 1-этиладамантана в 1,3-диметиладамантан:
Несмотря на то что в промежуточных продуктах изомеризации 1-этиладамантана структура гомо адамантана (вследствие высокой скорости изомеризации карбкатиона) не обнаружена, такой ход реакции не вызывает сомнения по двум соображениям. Во-первых, расширение шестичленного цикла за счет а-метиленовой группы боковой цепи легко протекает в ряду алкилциклогексанов и, во-вторых, образование структуры гомоадамантаиа наблюдается в целом ряде химических превращений различных производных адамантана [97], например в случае превращения 1 -гидроксиметиладамантана в 1 -гидроксигомоадамантан:
Структура гомоадамантана в условиях кислотного катализа легко изомеризуется в адамантановую. Так, например, если 1- и 3-гидрокси-
140
"’’лк
W гомоадамантаны нагревать с 75%-ной H2SO4 ДР 70 °C, то образуются roll моадамантан, 1-и 2-метиладамантаны и 1 -гидроксиметиладамантан [98]
Изомеризация алкильной группы в алкил адамантанах, по всей вероят-$ ности, также протекает аналогично изомеризации других насыщенных $ углеводородов, а именно за счет 1,2-гидридных и алкильных сдвигов $ в соответствующих карбкатионах. Большой экспериментальный материал по этим вопросам обобщен в монографиях Ал .А. Петрова [94,99].
К перегруппировкам, которые не встречаются при изомеризации насыщенных углеводородов, отличных от адамантановых, относятся: скелетная ’’вырожденная” перегруппировка адамантанового ядра и 2,4-гид-ридные и алкильные сдвиги.
Вследствие неблагоприятного пространственного расположения свободной орбитали карбкатионного центра и соседней мигрирующей алкильной группы или атома водорода (угол равен 60 и 90°) 1 ^-сдвиги в адамантане сильно затруднены [100].
По этой причине 1 ^-перемещения в жесткой структуре адамантана протекают либо по межмолекулярному механизму, либо по механизму вырожденной изомеризации адамантанового скелета*. Первый путь наглядно демонстрируется реакцией карбоксилирования по Коху—Хаафу (см. ниже). Так, при карбоксилировании 2-гидрокснадамантана (НСООН, H2SO4) выход 1-адамантанкарбоновой кислоты — продукта, образующегося в результате 1,2-гидридного перемещения в катионе, — зависит от объема используемой 96%-ной H2SO4 [101] (табл. 3.4). Разбавление реакционной среды серной кислотой сильно уменьшает вероятность встречи 2-адамантиллсатионов, вследствие чего вероятность межмолекулярных переходов падает. При больших объемах H2SO4 основным продуктом реакции является 2-адамантанкарбоновая кислота, т.е. 1,2-гидридные перемещения не реализуются. Аналогичным образом ведут себя 2-гид-рок си-2-метиладамантан и другие адамантанолы. В реакции Коха-Хаафа 2-метиладамантанол-2 прн более высокой температуре претерпевает изомеризацию в З-гидрокси-1-метиладамантан, т.е. параллельно идет перемещение также метильной группы в узловое положение.
Протекание вырожденной скелетной перегруппировки установлено в адамантане с помощью меченого 14С в положении 2 [102] , а в 2-метил-
*Под вырожденной понимают такую перестройку молекулы, когда, несмотря на перемещение связей или заместителей, структура молекулы остается неизменной.
141
Таблица ЗА
Зависимость выхода 1-адамантанкарбоновой кислоты от количества серной кислоты при реакции Коха—Хаафа с 2-гидр окси адамантаном
Количество 96%-ной Н , SO4, мл j" Выход 1 -AdCOOH,%
30 100
52 35
300 14
1000 0,5
адамантане — в результате изучения изомеризации меченого 2-метиладамантана, в котором метильная группа находится у 14С-атома в положении 2 [100]. Действие А1Вг3 в CS2 на адамантан, меченный 2-14С, привело к распределению метки по всем статистически возможным положениям ядра. Степень распределения метки при 25 °C за 14 цн. составила 1,9%, апри 110°Сза8ч —78,4% [102].
Аналогично протекает вырожденная перегруппировка в меченом 2-м тиладамаитане [ 100].
Таким образом, несмотря иа то что метильная группа 2-метиладамантана "мигрировала” в узловое положение ядра, те. образовался 1-метиладамантаи, она оказалась присоединенной к тому же самому атому углерода, что в исходном 2-метиладамантане.
142
Исключительно высокий потенциальный барьер 1,2-гидридного пере мещения в 1-адамантильном катионе установлен экспериментально методом ЯМР с помощью дейтерированного производного адамантана [103]. 3,5,7-</3-Адамантил-катион был получен из соответствующего хлорида при взаимодействиям последнего со сверхсильной кислотой (SbF 3, SOjClF) при —100 QC в вакууме. При нагревании катиона до 105 °C в течение 13 ч не происходило заметной миграции дейтерия из 7-положения, что можно было бы ожидать в результате двух последовательных 1,2-гид-ридных сдвигов.
Н
D D
Константа скорости 1,2-сдаига была меньше 2 • 10-5 с-1, и энергия активации больше 121,2-127,5 кДж/моль.
Методом спектроскопии ЯМР 13С и ПМР ХН при изучении серии 2-ада-мантил-катионов (2-алкил-, 2-фенил-и 2-галогензамещениых) установлено интересное различие в поведении вторичных и третичных 2-адамантил-катионов в среде сверхсильных кислот при —78 °C [104]. В то время как вторичные катионы претерпевают быструю внутримолекулярную скелетную перегруппировку в узловой 1-адамантильный катион, скелетная перегруппировка третичных (т.е. замещенных в положении 2) 2-адамантиль-ных катионов в этих условиях ие происходит. В случае 2-фенил-2-адяман-тил-катионов можно было ожидать наличия сопряжения между я-системой бензольного кольца и 2р-орбиталью при карбениевом центре, однако найдено,что такое сопряжение незначительно.
1 ^2-Гидридные перемещения ие имеют места при сольволизе 1-адамантил-и 2-адамантил-л-толуолсульфонатов [105]; в то же время в 2-грег-бутил-2-адамантил-катионе четко наблюдаются обратимые перемещения метильных групп [106].
Существенное различие в химических свойствах вторичных и третичных катионов установлено при изучении 1,1-диадамантилметил-катиоиов методами спектроскопии ПМР и ЯМР 13С [107]. Катионы получают из соответствующих спиртов или галоген алкилов в среде сверхсильиых кислот.
X
Третичные катионы (R - Aik), за исключением трет-бутил-1,1-диамантил-метил-катиона (R = трег-С^Нд), при низких температурах вполне устойчивы, в то время как вторичные катионы (R = Н) претерпевают перегруппировку с расширением цикла в 4-(1-адамантил)-3-гомоадамантил-ка-тион (7).
143
(7)
Катион (7) даже прн весьма низкой температуре (~ —140 °C) претерпевает быструю перегруппировку Вагнера—Мее рв ей на с образованием набора шести эквивалентных катионов.
1,2- и 1,3-Алкнльные перемещения лежат в основе изомерных превращений 2-циклопе я тил адамантил-катионов [108].
(На схеме изображены лишь наиболее устойчивые промежуточные продукты.)
Конечными продуктами изомеризации 2-цикл опешил адаманта на над А1Вт3 являются термодинамически наиболее устойчивые тетрациклоалканы — эпимеры 1,2-бутаноадамантана. В качестве промежуточного продукта образуется спиро [ адамантан-2,1'-циклогексан] (образование его удается зафиксировать, вероятно, вследствие ингибирования 1 Д-алкильного сдвига в адамантане, необходимого для дальнейшей перегруппировки соответствующего катиона).
Ряд интересных перегруппировок протекает в 2,4-дегидроадамантил-ка-тиоиах. 8,9-Дегидро-2-адаман тил-катион испытывает вырожденную скелетную перегруппировку прн -120° С [109,110].
Введение электронодонорного заместителя прн катионном центре С (2) существенно увеличивает энергетический барьер перегруппировки. Так,
144
катионы типа
(Л=СМ3, СБН5, иякло-СэН5)
уже вполне устойчивы в данных условиях. В интервале — 95^-10° С не изменяется также 1,2-диметил-8,9-дегидро-2-адамантил-катион. По-иному ведет себя изомерный 2,8-диметил-8,9-дегидро-2-адамантил-катион (8) [111]. При температуре -128°С он достаточно устойчив, ио при повышении температуры до -26° С претерпевает вырожденную циклопропилкарби-нильную перегруппировку, которая протекает либо через З^-днметил-2,5-дегидро-4-протоадамантил-катиои (9), либо через 2,3-днметил-2,5-дегид-ро-4-протоадамантил-катиои (10) и 8,9-днметил-8,9-дегидро-2-адамантил-ка-тион (И).
4-Фенил-2,5-дегидро-4-протоадамантил-катион (12) не изменяется при — 135 ° С, ио при нагревании до 40° С претерпевает трехкратную вырожденную перегруппировку, протекающую, вероятно, через образование 1-фенил-8,9-дегидро-2-адамантил-катиона (13).
(12)
Энергетический барьер изомеризации (8)^(9) или (10)->(11) равен ДС = 30,9±2,1 кДж/моль, для (12)-*(13) — 28,8 ±2,1 кДж/моль [111].
Интересный каскад перегруппировок претерпевает 2-трет-бутил-2-ада-мантил-катион (14) [106, 112], схема 1. Катион изучен с помощью методов спектроскопии ПМР и ЯМР 13 С. Эго основная промежуточная частица, образующаяся при действии на 2-трет-бутила даман танол-2 (15) суперкислоты FSO3H—SbFs — SO2C1F в интервале температур — 784—20°С. Этот же катион образуется также при действии кислоты на 2-изопропенил-2-метиладамантан (16) и на спиро [адамантаи-2,1'-(2,^’-днметилциклопро- 10
10. Зак. 1856 1ДБ
Схема 1
СН3
Н3С- С” си3
(14)
СМ,
-н®
„ zCH = н® Z-AJ
СН3 (ГО) CHj
м^-с-сНз^£_^сн-сн-сиэ
® н (Z1)
пан)] (17). С помощью дейтерирования установлена быстрая взан-мопревращаемость катионов (14) и (18) в кислой среде. В присутствии восстановителя (HI или три-н-гексилсилаи) соединения (15) и (16) образуют один и тот же продукт — 2-трет-бутиладаманган в качестве основного продукта. В среде (С2Н5)2О, СН3С12 или СНС13, насыщенных хлористым водородом, при 100° С алкен (16) образует до 5% изомерного соединения (17). Это же соединение (17), вероятно, образуется как промежуточное в результате 1,2-гидридного перемещения нз катиона (14) через неклассический катион (19). Дальнейшие превращения соединения (17) протекают через стадию протонирования трехчленного цикла и приводят в итоге к образованию углеводородов (20) (30—40%) и (21) (60- 70%). В небольших количествах (до 10%) образуется 2-метиленадамантан, вероятно, вследствие фрагментации катиона (18).
146
С помощью дейтериевой метки установлено также наличие 1,3-гидрид-ного сдвига, обусловливающего взаимное превращение соединений (20) и (21) через соответствующие катионы [112]. Характерные превращения, также сопровождающиеся частичной фрагментацией, испытывает первичный 1-адамантил-трет-бутильный катион (22) [ИЗ].
СН3
I '
1-Ad—С—СН2ОН
I
(23) СН3
СН3
1-Ad—С—CHj (22) СН3
1-Ad- СН- СН2- СН3
СН3 (24)
перегруппировку bAd_J_CH СН 80% |
СН3 (26)
1-AdH
1-Ad—СН2 —С—СН3
(27) СН3
фН®
1-Ad—СН2—СН—СН3 I
(25) СН3
Смесь продуктов, приведенная на схеме, получена при —40°С в течение H2SO4- Основным продуктом перегруппировки катиона является 2-(1-адамантил)бутан (24), что свидетельствует об 1,2-перемещении метильной группы. Наличие в продуктах реакции адамантана и 1-гидроксиадамантана свидетельствует о частичной фрагментации катиона (22). Рекомбинация фрагментной пары (1-адамантил-катиона и изобутилена) приводит к образованию 1-изобутиладамантана (25). Альтернативный путь образования (25) - возможное взаимное превращение катионов (26) и (27) вследствие одного 1,2-гидридвого и 1,2-метильного перемещений. Несмотря
-н®
1-Ad-CH2—СН—СН3 -2-
СН3 е * -Н
1-Ad—СН2—СН—СН2
I СН,
• ^СН3
* 1-Ad-CH2-C
^СН3
~1Д-Н
-----► 1-Ad—СН2—СН—СН2—СН3
1-Ad—СН2 — СН2—СН2—СН3
147
на то что для такой перегруппировки требуется образование менее устойчивого вторичного карбкатиона (из более устойчивого третичного), этот путь представляется предпочтительнее, нежели бимолекулярный процесс рекомбинации фрагментной пары. Известно, например, что 1-изобутилада-мантан легко превращается в 1-н-бутиладамантан, хотя для этого требуется образование еще менее устойчивого первичного катиона [114].
Широко исследованы также превращения различных функциональных производных адамантана, такие, как гофманское расщепление [115], перегруппировки Курциуса и Лоссеня [116], Бекмана [117], Вольфа [118] н др. Подробное рассмотрение этих перегруппировок выходит за рамки настоящей монографии, посвященной главным образом химии углеводородов (могут быть рекомендованы монография [119] н обзор [120]). Уместно рассмотреть здесь лишь превращения производных, имеющих близкую аналогию с превращениями углеводородов. Например, в присутствии галогенидов алюминия происходят миграция н перераспределение галогена в галогенпронзводных адамантана [121], подобно перераспределению метильных групп в метиладамантанах (содержание производных в смеси указано в процентах).
Х= С1‘. о,3 6,4 1,8 85,6 5,9
Х=Вг: 3,9 16,0 0,3 63,0 16,8
Смесь продуктов, приведенная на схеме, получена прн —40° С в течение 3 ч. Образование значительного количества производных, содержащих меньшее н большее число атомов галогена в молекуле по сравнению с исходным соединением, свидетельствует о значительном участии межмолекулярного механизма перемещений галогена н гидрида. Вместе с тем преобладающим является, вероятно, внутримолекулярный механизм, так как днзамещенные производные содержатся в наибольшей концентрации.
В заключение отметим еще два типа перегруппировок, наблюдаемых для функциональных производных адамантана. Эго 1,3-гидрндные сдвиги между положениями 2 н 4, которыми объясняют [122] превращения днадамантилпрон зво дных
50% fySOii
1оо°С
н частичная фрагментация, например, дибромпронзводных адамантана с образованием структуры бисметиленбицикло [3,3,1 ].нонана [123]
148
Вопросы частичной фрагментации производных адамантана и последующей циклизации получаемых продуктов всесторонне изучены в работах Юрченко, см., например, [124]. Эти реакции представляют большой практический интерес для синтетической химии адамантана, так как на основе получаемых производных метиленбицикло [3,3,1] нонана путем циклизации могут быть легко получены самые разнообразные полизамещенные производные адамантана.
Перегруппировку, обусловленную частичной фрагментацией адамантанового скелета, наблюдали также при изучении реакции карбоксилирования 2-(1-адамантил)пропанола-2 по Коху-Хаафу (96%-ная H2SO4, НСООН) [125].
В результате этой перегруппировки прн карбоксилировании спирта (28) образуется не ожидаемая 2-нзопропил-1-адамантанкарбоиовая, а 3-нзо-пропил-1-адамантанкарбоновая кислота. Впервые частичная фрагментация производных адамантана наблюдалась на примере гидроксиадамантана [126].
Для производных адамантана известна перегруппировка с образованием структуры протоадамантана (напомним, что такая перестройка структуры адамантана является первым этапом вырожденной скелетной перегруппировки) . Так, прн сольволизе некоторых 1-К-2-И’-адамантил-2-суль-фонатов (где R, R* = Н; СН3, Н; СО2СН3, Н; CN, Н, а сульфонатная группа - OSO2CH2CF3 или OSO2C6H4~СН3-л) в 50%-ном водном этаноле наряду с ожидаемыми продуктами сольволиза адамантанолами образуются также протоадамантанолы [71 ].
149
32.3. ПРЕВРАЩЕНИЯ АЛКИЛАДАМАНТАНОВ В УСЛОВИЯХ ГАЗОФАЗНОГО ГЕТЕРОГЕННОГО КИСЛОТНОГО КАТАЛИЗА
Эффективными катализаторами превращений алкиладамантанов, помимо галогенидов алюминия, являются активированный оксид алюминия, алюмосиликатные и цеолит содержащие катализаторы. Использование этих катализаторов, в частности у-о к сида алюминия, активированного серной кислотой, позволило применить импульсный микрокаталитический метод для изучения кинетики и механизма изомерных превращений алкиладамантанов различного строения [127]. Над перечисленными выше катализаторами изомеризация алкиладамантанов состава Сц-С14 протекает в интервале 180—300° С.
Реакцию проводят в микрореакторе, встроенном в газовую линию капиллярного хроматографа с пламенно-ионизационным детектором, что позволяет с высокой точностью определять состав продуктов на самых ранних этапах превращения.
Реакция газофазной изомеризации алкиладамантанов хорошо описывается уравнением для необратимых реакций первого порядка [128]
Fo 1
кК = ---— in------,
213mR 1 -у
где к - константа скорости реакции; К — константа адсорбционного равновесия; Fo — скорость газа носителя, приведенная к нормальным условиям; у - степень превращения; т — навеска катализатора.
Реакция протекает в кинетической области в интервале 180—200°С при использовании катализатора с размером зерен 0,5—1 мм; энергия активации составляет 62,7 ± 2,1 кДж/моль.
В табл. 3.5 приведены значения констант скорости изомеризации алкиладамантанов. Зависимость логарифма констант скорости от числа незаме-
ГвДлица 3.5
Изомеризация алкиладамантанов состава С2, - 4
(190° С, F9 = 3,9 мл/с, т = 4 г)
Исходный алкилада-мантан а* nD Степень превращения, мас.% Продукты деструкции, мас.% Константа скорости к, ммоль/г - атм • с
1,4-Диметил- 1,4835 64 5,6 0,05
2,2-Диметил- — 75 7 0,06
1-Этил- 1,4930 51 4 0,03
2-Этил- 1,5015 87 3 0,09
1-Метип-З-этил- 1,4850 55*1 12 0,02
2-я-Пропил- 1,4964 96 17*э 0,14-
1,3,5,7-Тетраметил- 1,4802 15 6 0,01
(82%) + 1,3-ди метил -5-этил-(18%)
** т = 8,3г; *2втом числе 4,5% адамантана.
150
щенных третичных углеродных атомов в ядре адамантана, т.с. атомов, наиболее легко образующих устойчивые третичные ада мант ил-кат ионы, представлена на рис. 3.4. Наблюдаемая корреляция свидетельствует о том, что лимитирующей стадией реакции изомеризации алкиладамантанов в принятых условиях является образование третичного адамантил-катиона. Последнее обстоятельство значительно ограничивает возможности кинетического исследования других стадий реакции изомеризации в условиях газофазного гетерогенного катализа, в том числе определения скорости различных перегруппировок, лежащих в основе этих превращений. Основную информацию о протекающих при этом перегруппировках получают при изучении ранних этапов изомеризации на основании исследования первичных продуктов реакции-
1,3-Диметил- н 1,3,5-три метиладамантаны, как наиболее устойчивые изомеры соответственно в ряду Ci2- и С13-алкиладамантанов изомеризуются лишь в незначительной степени (рис. 35).
1,3-Диметил адамантан (1,3-ДМА) превращается в 1,4-и 1,2-диметилада-мантаны. 1,4-Изомер представлен в продуктах изомеризации двумя геометрическими изомерами, образующимися в равном количестве. Суммар-
Рис. 3.4. Зависимость логарифма константы скорости реакции изомеризации алкиладамантанов от числа третичных атомов углерода в молекуле
Катализатор На SO4 /А1а Оэ, 190° С
Рис. 3.5. Изомеризация 1,3-димстил- (а) и 1,3,5-триметиладамантанов (б)
Катализатор HjSO^/AljO-, 220°С (д) и 240° С (б). Продукты реакций: У) 1,4-диметнладаман-тан, 2) 1,2-диметил адамант ан, 5) 1,3,б-триметил-адамантам, 4) 1,2,5-гриметиладамантан. у — степень превращения исходного углеводорода, с — содержание изомеров в продуктах реакции
151
ная концентрация эпимеров 1,4-изомера выше, чем 1,2-иэомера. Помимо термодинамических факторов (1,2-диметипадамантан менее устойчив, чем 1,4-изомер), здесь, вероятно, играют роль и кинетические факторы, связанные с меньшей вероятностью образования катиона в «-положении к метильной группе. Изомеризацию 1,3-ДМА можно описать с помощью 1,2-перемещений метильных групп.
Аналогичной схемой может быть объяснение образование продуктов изомеризации 1,3,5-триметиладамантана (1,3,5-ТМА). Интересной особенностью изомеризации 1,3,5-ТМА является то, что содержание 1,3,6-ТМА всегда существенно выше, чем содержание 1,2,5-изомера. Это обусловлено первоначальным образованием третичного адамантил-катиона; последующие 1,2-гидридное и алкильное перемещения в положения, отмеченные звездочками, обязательно приводят к образованию 1,3,6-изомера.
Результаты изомеризации 13-ДМ А н 1,3,5-ТМА можно объяснить не только 1,2-гидридным и алкильным перемещением, но и тем, что имеет место скелетная вырожденная перегруппировка, рассмотренная ранее, например:
152
Рис. 3.6. Изомеризация 1,2-диметил-адамантана
Продукты реакции: 1) 1,4-диме-тиладамантан, 2) 1,3-диметиладаман-там
Рис. 3.7. Изомеризация 1,2,5-триме-тиладамаитанов
Продукты реакции'- 2) 1,3,6-три-метиладамантан (ТМА), 2а) цис-1,2,5-триметил адамантан, 26) транс-1,2,5-триметиладамантан, 3) 1,3,5-три-метил адамантан, 4) 1-метип-3-этил-
адамантан
Тем не менее можно предположить, что в газовой фазе протекают гид-рндные и алкильные перемещения несмотря на то что они в значительной степени затруднены. Это следует из приведенных ниже результатов изомеризации других алкиладамантанов. Так, 2,4-алкильные перемещения, в частности, протекают при изомеризации 1,2-диметил- и 1,2,5-триметил-адамантанов (рис. 3.6 и 3.7). В продуктах изомеризации 1,2-диметилада-мантана преобладает 1,4-изомер, в том числе и на самых ранних этапах.
153
Значит, он образуется непосредственно из 1,2-изомера, минуя стадию образования 1,3-нзомера, вероятно, по одному из следующих путей:
Аналогично в продуктах изомеризации 1,2,5-ТМЛ преобладает содержание 1,3,6-ТМА над 1,3,5-ТМА.
В случае изомеризации как цис-, так и транс-1 ,2,5-триметиладамаитана (рис. 3.7) иа начальных этапах изомеризации наиболее легко протекает геометрическая изомеризация (эпимеризация). Этот вывод следует также из результатов изомеризации индивидуальных эпимеров 1,4-диметилада-мантана (рис.3.8).
Реализация 2,4-метильиого сдвига при изомеризации алкиладамантанов в особенности наглядно иллюстрируется результатами изомеризации 1,3,6-трнметиладамантана (рис. 3.9). В данном случае 1,2-сдвиг метильной группы или вырожденная скелетная перегруппировка могут привести только к 1,3,5-изомеру, т.е. изомеру, термодинамически наиболее устойчивому, способному, как это отмечалось выше, к дальнейшей изомеризации лишь в весьма незначительной степени (~3%) (рис. 3.4). Иными словами, узловое положение 5 играет роль своеобразной ловушки, попав в которую метильная группа лишается способности к дальнейшему перемещению. В то же время эксперимент показывает, что на начальном этапе изомеризации 1,2,5-ТМА даже преобладает над 1,3,5-ТМА, т.е. образуется непосредственно из 1,3,6-ТМА посредством 2,4-метильного перемещения.
Можно было бы предположить, что реализацию 2,1-алкильного перемещения в алкиладамантил-катноне в условиях гетерогенного катализа удастся наблюдать при изомеризации 2,2-диметиладамаитана. Действительно, если такая изомеризация имеет место и продуктами реакции будут диметиладамантаны с различным положением метильных групп, то превращение 2,2-ДМА можно было бы описать только следующей схемой:
154
Рис, 3,9. Изомеризация цис- (а) и транс-1,4-димсти ладам ан танов (б)
Продукты реакции: 2) транс-1,4-диметил-адам антан, Г) гдас-1,4-дим етнладамантан, 2) 1,2-диметиладамантан, 3) 1,3-диметил-адам антан
Рис. 3.9. Изомеризация 1,3,6-триметилада-мантана
Продукты реакции: 1) 1,3,5-триметил-адамантан, 2) 1,2,5-триметиладамантан (цис-+ транс-), 3) 1-метип-З-этиладамантан
На первый взгляд эксперимент, результаты которого представлены на рнс. 3.10, подтверждает такое протекание реакции. Действительно, преобладающим продутом здесь является 1,2-диметиладамантан (1,2-ДМА). Тем не менее этих результатов недостаточно для подтверждения прямого 2,1-перемещения (скелетная изомеризация не может объяснить наблюдаемые экспериментальные результаты), ибо возможен также протоадамантановый путь изомеризации.
С учетом ингибирования 2,1-алкильного перемещения протоадамантаио-вый путь представляется более вероятным.
Изомеризация алкиладамантанов, содержащих алкильные группы, отличные от метильных, приводит к более сложному составу продуктов реакции. Зно видно из рис. 3.11 и 3.12, на которых представлены результа-
155
Рис, 3.10. Изомеризация 2,2-диметил-адамантана
Продукты реакции: 1) 1,2-диметил адамантан, 2) 1,4-диметиладамантан (цис- + транс-), 3) 1,‘З-диметилада-мантам
Рис. 3.11. Изомеризация 1- (с) и 2-этиладамантанов (б)
Продукты реакции: 1}. 1,4-диме-тиладамантан, 2) 1,2-диметиладаман-тан, ?) 1,3-диметиладамантан, 4) 2-эти л адамантан, 5) 1 -этиладамантан
Рис. 3.12. Изомеризация 1-ы-пропил-адамантана
Продукты реакции: 1) адамантан, 2) 1-метил-4-этиладамантан, 3) 2-н-пропиладамантан, 4) 1 -этил-4-метил-адамантаи, 5) 1 -метил-2-этиладаман-тан, б) i-метил-З-этиладамантан, 7) 1,3,5-три мети л адамантан, Л) 1,2,5-три-метиладамантан, 9) 1,3,6-триметил-а дам ан тан
W 20 30 0 20 90 SO yt %
% Рис. 3.11
о 20 90 , SO 0 20 90 SO у, %
156
Рис. 3.12
ты изомеризации этил- и пропиладамантанов. В начальный период изомеризации 1-этиладамантана (1-ЭА) в продуктах реакции преобладает 2-этил-адамантан (2-ЭА), образующийся по механизму скелетной изомеризации. Аналогично из 2-этиладамантана первым образуется 1-ЭА. Кривая образования 2-ЭА имеет экстремальный характер - углеводород претерпевает дальнейшую изомеризацию, причем скорость его изомеризации выше, чем скорость изомеризации 1-ЭА. Механизм изомеризации этил- и пропиладамантанов соответственно в циметил-и триметиладамантаны, вероятно,включает протекание рассмотренной выше перегруппировки — расширение циклогексанового кольца за счет метиленовой группы боковой цепи и образование структуры алкилгомоадамантил-катиона. Общая схема изомеризации 1-ЭА может быть представлена так:
Таким образом, в условиях гетерогенного катализа над катализаторами на основе оксида алюминия (например, H2SO4/A12O3) при 190—230°С активно протекают изомерные превращения алкиладамантанов. Преобладающие направления изомеризация — увеличение числа алкильных групп, уменьшение величины заместителей н образование метил адаманта нов с метильными группами в узловых положениях. Для пропиладамантанов наблюдается также частичное отщепление заместителя. Превращения алкиладамантанов с более длинными, чем пропильная, алкильными группами протекают исключительно в направлении гидродеалкилирования.
3.3. АЛКИЛИРОВАНИЕ
Реакции алкилирования в химия адамантана можно условно разделять иа два типа: реакции, где адамантан или его производные подвергаются действию олефинов, галогеналкилов, спиртов или других алкилирующих агентов, и реакции, где адамантаны сами являются алкилирующими агентами, например при взаимодействии с ароматическими соед инениями. Реакции
157
алкилирования могут протекать как по ионному, так и по радикальному механизму.
Ионное алкилирование адамантанов идет при действии катализаторов, обеспечивающих образование адамантил-катионов как основных промежуточных частиц. Обычно такими катализаторами являются сильные протонные кислоты (H2SO4, HF, CF3COOH) или кислоты Льюиса (А1На13). Протонные кислоты в особенности активны при алкилировании непредельными соединениями или спиртами. Электрофильные катализаторы типа галогенидов алюминия позволяют осуществить алкилирование адамантанов даже такими относительно мал о реакционными соединениями, как алканы. При повышенной температуре в реакциях алкилирования адамантанов активны также твердые кислотные катализаторы на основе оксида алюминия и цеолитные катализаторы.
Характерной особенностью протекания реакций ионного алкилирования углеводородов ряда адамантана является высокая селективность образования продуктов, замещенных в узловом положении ядра. Это обусловлено значительно большей устойчивостью и большей легкостью образования третичных (узловых) адамантил-катионов по сравнению со вторичными (мостиковыми). Вместе с тем в реакционной смеси, как правило, присутствуют продукты протекания побочных реакций олигомеризации олефинов н изомеризации алкильных групп.
Образованию третичных адамантил-катионов способствует наличие в узловом положении ядра адамантана функциональной группы (Вг, С1, ОН), вследствие чего в ряде случаев реакции ионного алкилирования протекают значительно легче, если в качестве исходных соединений используют галоген-или гидроксипроизводные адамантанов.
Реакции радикального алкилирования адамантанов протекают при повышенной температуре (>290°С), прн 7-облученин или прн действии инициаторов радикальных процессов. В этих реакциях затрагиваются как третичные, так и вторичные атомы углерода ядра в результате чего образуются, как правило, сложные смесн продуктов реакции различного строения.
33.1. ИОННОЕ АЛКИЛИРОВАНИЕ
Адамантан реагирует с олефинами (этиленом, пропиленом, изобутиленом) в присутствии галогенидов алюминия при температурах ниже 0°С; даже в мягких условиях (-10° С) в смесн продуктов реакции содержится не менее 20 алкиладамантанов, имеющих различную длину н строение алкильных групп [95].
В случае алкилирования 1-бромадамантанов прн более низкой температуре селективность повышается. Так, например, из 1-бром-З^-диметил-адамантана при взаимодействии с этиленом (А1Вг3, —30°С) выход 1,3-диметил-5,7-диэтиладамантана составляет 85% [129]. Хороший выход продуктов реакции и высокая селективность получаются при алкилировании адамантана ц> клоолефииами (цикл о пентеном, З-мешлциклопен-теном или цикл отеке еном) в присутствии углеводородного комплекса А1Вг3 [130]. С высоким выходом получается 1-циклогексиладамантан прн взаимодействии адамантана ,н 1-гидроксиадамантана с циклогексеном
158
в CFgCOOH [131]. Адамантан н алкиладамантаны также легко вступают в реакцию с алкенилхлорсил анами [132].
(r=CjHs,r'=r"=m; R=r'=ch3,r"=h; R=R'=R"=CHj;Hai=ci,Br; я=о,п.
В этом случае лучшие выходы (80%) получены при использовании винилтрихлорсилана, наиболее активным катализатором является А1С1з; при использовании А1Вг3 выход целевых продуктов снижается до 30—50%. В случае адамантана, 1- и 13-алкиладамантанов наблюдается также образование 5,7-днсилил замещенных производных. На основе этих соединении могут быть получены алкокси производные кремиийорганическнх мономеров [133].
Легкое образование третичных карбкатионов адамантана использовано в других реакциях ионного алкилирования [133а, 1336]. Так, сложная смесь моно- н полиалкиладамантанов различного строения образуется уже при комнатной температуре из адамантана и гексана нли октана в присутствии катализатора СН3СОВг - 2А1Втэ [ 1336]. По существу, здесь имеет место алкилирование адамантана олефинами, так как этот катализатор в данных условиях способен крекировать алканы с образованием алкенов.
Алкилирование адамантана и алкиладамантанов олефинами осуществляется также в присутствии твердых алюмоокнсных катализаторов прн повышенной температуре [134]. Используют оксид алюминия, активированный серной кислотой, а также алюмосиликатные н цеолитсодержащие катализаторы типа АШНЦ, ЦЕОКАР, т.е. те же катализаторы, которые активны и в реакциях изомеризации алкиладамантанов.
Катализатор H2SO4/A12O3 активен при 190—200°С, выход продуктов алкилирования (алкиладамантанов) при использовании адамантана и олефинов С3—С7 составляет 70—80%. В случае катализатора ЦЕОКАР температуру реакции следует повысить до 280—290°С. Образуются, как правило, смеси алкиладамантанов; преобладают те из них, в которых алкильная группа содержит то же число атомов углерода, что и исходный олефин. В некоторых случаях, например при алкилировании пропиленом, наряду с алкиладамантанами образуются ненасыщенные апкениладамантаны, их содержание может достигать 29% [127], табл. 3.6. В случае 1,3-днметил-адамантана ненасыщенные продукты образуются также на цеолнтсодержащих катализаторах. С увеличением молекулярной массы олефинов выход таких производных падает; при взаимодействии пентена-1 н гексена-1 с адамантаном они полностью отсутствуют.
159
Таблица 3.6
Состав продуктов алкилирования адамантана пропиленом над алюмоокисными катализаторами (Зч, адамантан:пропилен = 1:1, моли)
Адамантан, алкиладамантаны Содержание в реакционной смеси, мае.%
алюмос ил нк ат типа Г удри, 28(У С ЦЕОКАР-2, 280“ С H2SO4/A12 о3, 190° С
Адам антан 48 41 53
1-Пропенил- — 6 29
1-я-Пропил- 28 28 6
2-я-Пропил- 3 2 —
1-Изопропил- 7,5 7,5 2
1,3 -Дипропенил- — — 4
Пропилпропенил- и дипропил- 12 14 5,5
1-Гексил- 1,5 1,5 0,5
Алкилирование адамантана и низших алкиладамантанов (1-этил-, 1,3-диметил-, 1,3-диметил-5-эти л адамантанов) олефинами, а также алифати ческими (С2 -С5) и насыщенными циклическими спиртами (такими, как циклопентанол и циклогексанол) имеет место при контакте с 96%-иой H2SO4 или 90—100%-ной HF при -20°^100°С [135], Образуются соответствующие алкил- и циклоалкиладамаитаны, содержащие от одной до четырех (в случае адамантана) алкильных групп. При алкилировании изомерными бутиловыми спиртами выход продуктов реакции невелик и образуются алкиладамантаны, представляющие собой изомеры, отличающиеся строением боковой цепи [136].
Введение этильной группы в узловое положение ядра адамантана достигается взаимодействием углеводородов с эфиратом трифторида бора BF3 Et2OBH2SO4 [132].
Галогеналкилы малоэффективны в качестве алкилирующих агентов; реакции адамантанов в этих случаях протекают неселективно и зачастую сопровождаются реакциями галогенирования, например [137]:
При алкилировании адамантанов иад алюмоокисными и цеолитсодержащими катализаторами легче всего реакция протекает при взаимодействии с олефинами, труднее - со спиртами и галоген алкилами [138], Реакционная способность 1,3-днметиладамантана ниже, чем адамантана. Увеличение
160
соотношения олефин : адамантан способствует увеличению выхода ди- и полизамещенных продуктов.
В качестве алкилирующих агентов в реакциях с углеводородами ряда адамантана могут быть с большим успехом использованы и алканы. Реакция алкилирования адамантана или низших алкиладамантанов алкаиами состава С3—С2о протекает в H2SO4 (96-105%-иой) при 10-75°С [139, 140] с образованием наряду с алкил адамантанами также диадамантил алканов, Алкилирование алканами С2-С5 имеет место при — 20^100°С при контакте с HF [135]. Сложные смеси диадамаитилалканов с выходом 40-70% образуются при ~ 100° С иад 94-102%-иой H2SO4 [141].
При контакте адамантана или низших алкиладамантанов с алкаиами в присутствии галогенидов алюминия реакция протекает по механизму деструктивного алкилирования. Алкай первоначально претерпевает деструкцию с образованием более низкомолекулярных фрагментов, которые затем участвуют в реакции алкилирования. Реакция была впервые осуществлена автором с сотр. [114, 142-144]. Она в особенности легко идет с алкаиами разветвленного строения, в частности с изооктанами при 50—60°С с выходом алкиладамантанов 90—80% от теорет. При реакции с изооктаном образуются преимущественно бутиладамаитаны (моио-и полизамещенные).
(R=HJ C„H2w+j при *аЧ;5).
В отличие от деструктивного алкилирования ароматических углеводородов деструктивное алкилирование углеводородов ряда адамантана идет по механизму нуклеофильного присоединения. Это и определяет состав образующихся продуктов. Так, при взаимодействии бензола с изооктаном образуется главным образом трет-бутил бензол, в реакции же изооктана с адамантаном основной продукт — изо бутил адамант ан.
Использование других алканов приводит к более сложной смеси продуктов. Проведение реакции иа алюмоокисных и цеолитсодержащих катализаторах требует более высокой температуры (100-350° С); в этих условиях наблюдается также образование наряду с бутил адамантанами октил адамантанов [145, 146].
Нагревание адамантана при 100°С [83] или алкиладамантанов при 120—190°С с А1На1з приводит к образованию сложной смеси полиалкил-, главным образом полиметил замешенных алкиладамантанов, вследствие протекания реакций переалкилирования (перераспределения алкильных групп) [ 147]. Реакция адамантана с алканами исследовалась и в бескислородной среде иад А1С13 при 75°С [ 148].
Реакция деструктивного алкилирования адамантана изооктаном может быть использована как препаративный метод получения смеси бутилада-м ант ано в (полибутиладамантаиовых смазочных масел). В реакцию вступают также диадамантил, диамантан [149] и др.
11, Зак. 1856
161
В качестве источника метильных групп при получении из адамантана или алкиладамантанов полиметилзамещенных алкиладамантанов может быть использован гетрам етилсилан.
Реакция протекает с количественным выходом в присутствии А1На13 и характеризуется высокой селективностью в отношении образования метил* адамантанов, замещенных в узловых положениях [150, 151]. При 20°С над А1С1з из адамантана за 6 ч может быть получен практически чистый 1-метиладамантан с выходом 25%. Повышение температуры до 65°С при* водит к образованию главным образом тетра- и пент аз вмещенных алкиладамантанов, в том числе 1,3>5-триметил-7-этиладамантана. В реакцию ионного метилирования вступают также галогеипроизводные адамаитаиа [152]. Эта интересиан реакция, впервые предложенная для метилциклогексана [153], является общей для углеводородов, имеющих третичный атом углерода.
В реакциях ионного алкилирования широко используются некоторые производные адамантана и алкиладамантанов (заместители: Вг, С1, ОН) [129, 154], проявляющие большую активность, чем сами углеводороды. К настоящему времени известны лишь отдельные случаи применения этих реакций для получения алкиладамантанов, например взаимодействие 1-б ром-3,5-ди метил адамантана с этиленом [129].
ОН, он,
В большинстве случаев функциональные производные адамантанов используются сами как алкилирующие, точнее, циклоалкилирующие агенты в реакциях с ароматическими углеводородами или их производными.
В реакцию Фриделя—Крафтса (катализатор А1С13) с бензолом легко вступает 1-6 рома да мант ан с образованием смеси замешенных в узловых положениях моио-, ди-, три- и тетрафениладамаитанов [155, 156]. При взаимодействии бензола, толуола, о-ксилола с 1-бром- и 1,3-ди бромада-мантаном (катализаторы; А1С1з, T1CI4, FeCl3, ZnCl2) образуются 1-фенил-, 1,3-дифеиил-, 1-(4-метилфенил)-, 1,3-бис (4*-метилфенил)-, 1-(3,4-ди-метилфенил)- и 1,3-бис (3*,4-диметилфенил) адамантаны [157]. Направление замещения в бензольном ядре определяется в этих реакциях как ориентирующим влиянием заместителей бензольного ядра, так и стерическими факторами, обусловленными наличием объемистого адамантанового цикла.
Единственным продуктом реакции 1-бромадамантана (1-AdBr) с нафталином (60-140°С, FeCl3) является монозамещенный 2-(1-адамантил) наф-162
тални [158]; не исключено, что обреется и 1-(1'-адамантил) нафталин, который в условиях реакции изомеризуется в 2-замещенный изомер Реакции 1-AdBr с тиофеном приводит к образованию двух изомерных продуктов с различным положением 1-адамантила в тиофеновом кольце, а при реакции с N-ацетил-!-аминонафталином 1-адамантил замещает водород в положении 4. Взаимодействие 1-AdBr с бифенилом или фениловым эфиром (C6Hs )2О в присутствии ZnCl2 при 250—260°С приводит к образованию олигомеров линейного строения (выход 48-59%) [ 159]
Нагревание 1,3-ди бром-5,7-диметил адамантана с избытком фенола при 180°С в течение 5-6 ч приводит к получению 1,3-бис (л-гидроксифе-нил) -5,7 -диметиладамантана с выходом 85%. Поликарбонат, получаемый иа основе этого диола, термически более устойчив, чем получаемый из бисфенола А [160].
Проведение реакции 1-AdBr с аминами требует достаточно жестких условий (170—180°С для первичных аминов и 200-210° С для вторичных) [161]. Это обусловлено тем, что амины сильно затрудняют необходимую поляризацию связи в 1-AdBr. Реакция циклоалкилирования ароматических аминов идет как по аминогруппе, так и в бензольное кольцо.
В условиях реакции Фриделя-Крафтса 1-хлорадамантан взаимодействуете реагентами, содержащими триметилсилилаллвльную группу RSi(СН3)3 (где R: СН2 =СН—СН2; (СН2)4СН=С=О; СбН3СН2 и др.), с образованием соответствующих производных, замещенных в узловом положении производных адамантана [162]. Реакция протекает также с алкинил-, арил- и г етероарил триметил силанами.
Весьма активно в реакцию циклоалкилирования вступают адамантанолы. С олефинами реакция протекает в среде коиц. H2SO4 [163] или CF3COOH [164]. При обработке 1-гидроксиадамантана в серной кислоте гептенами образуется сложная смесь углеводородов, содержащая наряду с гептил-адамантанами также дигептил- и тетрагептиладамантаны, а также диаман-тилгептаны и адамантилгептиладамантилгептаны. Реакция может быть использована для получения технической смеси высококипящих алкиладамантанов. В трифторуксусиой кислоте основными продуктами являются гидроксиадамантаны или их смеси с алкенил ада г ан танами.
1 -AdOH + CH2=CHR—1-AdCH(OR)CH2 R +
+ l-Ad(CH2)4C(OH)R1 R2 + l-AdC3Hs.
Реакция может быть использована как препаративный метод получения гидроксиалкиладамантанов. Выход и состав продуктов реакции зависит от природы олефинов, соотношения реагентов и условий реакции. При взаимодействии 1 -гидрокснадамантана с пропиленом доля вторичного спирта l-AdCH(OH)C2H5 в продуктах реакции достигает 94% (55°С и мольное соотношение 1-гидроксиадамантан :пропилен: трифторуксусная кислота = 1:5:8,5), При реакции 1-гидроксиадамантана с гексадиеиом-1,5
163
имеет место его избирательное взаимодействие с одной двойной связью диена; образуются в основном углеводороды.
Гидроксипроизводные адамантана, в том числе содержащие гидроксильную группу в боковой цепи, реагируют с ароматическими углеводородами в присутствии фосфорного ангидрида с образованием арилалкиладаман-танов [165].
46-73%
В случае гидроксибромпроизводних реакция идет по гидроксильной группе и образуются бромзамещенные адамантил ароматические соединения, содержащие галоген в адамантановом ядре. В связи с тем, что прямое галогенирование арил адамантанов обычно приводит к замещению в бензольном ядре, эта реакция является важным методом получения таких галогензамещенных алкиладамантанов.
В ряде случаев реакции алкилирования приводят к образованию полимерных продуктов. Так, при взаимодействии алкиладамантанов с дн-трет-алкилбензолами (А1На1з, О—25°С) образуется твердый полимер с молекулярной массой 2300, плавящийся при 250°С [166]. Исходные соединения для получения термостойких полимерных материалов получают при взаимодействии 13-дихло радам ант ана или адамантанона с фенолом или метилфенолами (в присутствии НС1 или тиоуксусной кислоты) [167].
В реакцию с бензолом или толуолом (А1С1з, 80°С) вступает 3-азидо-гомоадамантан с образованием (с выходом 91—97%) 1 -гидроксиадамантил-аренов (l-R-адамантанол, где R = СН2С6Н5; СН2С6Н4СН3) [168]. При алкилировании адамантана винилтрихлоргерманием [169] образуются 1-и 2-(со-трихлоргермил) алкиладамантаны.
33.2. РАДИКАЛЬНОЕ АЛКИЛИРОВАНИЕ
Термическое алкилирование адамантана олефинами протекает при 270— 450° С и 30 атм по цепному механизму [170]. В отличие от ионного алкилирования скорость образования соответствующих алкиладамантанов уменьшается в раду: этилен > пропилен > изобутилен. Основными продуктами реакции в таких условиях являются 1-моноалкиладамантаны, выход которых возрастает с увеличением давления.
Свободнорадикальное алкилирование адамантана и алкиладамантанов олефинами н цикло олефинами (С2 -С7, циклопентен, циклогексен) имеет место в присутствии инициаторов образования свободных радикалов, таких, как пероксид водорода, надсульфат калия, надсульфат аммония, пероксиды бензоила, ацетила, лаурила, дназоаминобензол, нитрозоацил-аллиламин; использовалось также инициирование у-изл учением [171, 172]. В реакции адамантана с этиленом образуются наряду с этиладаманта-ном также бутил- и гексиладамантаны; выход последних возрастает с увеличением давления (от 5 до 20 атм). Конверсия адамантана мало зави
164
сит от давления. Мольный выход продуктов реакции зависит от используемого олефина и составляет от 89% (для этилена) до 5% (для циклогексена) .
В присутствии пероксидных соединений имеет место свободнорадикальное присоединение адамантана к винил- и алл ил ацетатам; реакция протекает в среде нейтрального растворителя (декана) [173].
AdH £д^сн(СНа )н ососн?, 1-AdCH2 СНз (СНг 0С0СНз +
+ l-Ad-[—СН2 (СН2 )„ СН-] т -СН2СН2 (СН2 )„ ОСОСН3
ОС ОС И з
(и = 0,1 = 3-? Ю).
По эффективности инициирования реакции пероксидные соединения располагаются в следующий ряд: пероксиды алкилов > перэфиры > пероксиды ацилов. Из пероксидов алкилов наиболее эффективным инициатором является 4-метил-4-трет-бутилпероксипентанон-2. Побочную реакцию тег^меризации можно частично подавить путем изменения соотношения адамантан:непредельный эфир (от 1:2 до 1:4). Алкилирование адамантана фторолефинами можно осуществить при инициировании у-излучением. Реакция адамантана с тетрафторэтиленом в этих условиях происходит по механизму реакции теломеризации [174]. Легко протекает реакция радиационного алкилирования адамантана гексафторпропиленом с образованием моно- и дифторалкиладамантанов [175, 176]. Изучение зависимости скорости радиационного инициирования от состава исходной реакционной смеси показало существенное влияние молекулярных комплексов, образуемых реагентами [177, 178]. Реакция радиационного алкилирования адамантана гексафторпропиленом лежит в основе препаративного метода получения 1 - (1 -адамантил) -1,1Д ,3 Д,3-гексафторпропана [179].
Рассмотрение реакций алкилирования адамантанов и некоторых их функциональных производных (содержащих группы Br, С1, ОН) приводит к заключению, что они весьма активно вступают в эти реакции. В результате, как правило, образуются смеси ал кил замещенных и других производных адамантана. Они, вероятно, смогут найти применение в виде техничес ких смесей для получения смазочных материалов, термостабильных жидкостей и в полимерных композициях. Использование современных методов разделения позволяет получать из таких смесей также индивидуальные соединения, в частности высшие алкиладамантаны, фто рал кил адамантаны и др. Перспективным представляется использование этих соединений в промышленности в качестве исходных в процессах, основанных на реакции алкилирования не самого адамантана, а низших алкиладамантанов (например, 1,3-ди метил адамантана). Они в отличие от адамантана — жидкости и, следовательно, более технологичны.
165
3.4. ГАЛОГЕНИРОВАНИЕ
Реакции галогенирования — одни из первых химических реакций, позволившие осуществить успешную функционализацию адамантана. Многочисленные исследования проводились главным образом с незамещенным адамантаном.
Бромирование углеводородов ряда адамантана и их производных молекулярным бромом в жидкой фазе представляет собой ионный* процесс, катализируемый кислотами Льюиса и нечувствительный к инициаторам радикального типа (180, 181]. Ввиду значительно большей по сравнению со вторичным карбкатионом устойчивости третичного карбкатиона замещение бромом идет в узловые положения ядра адамантана с высокой селективностью [180, 182—184]. и
1,3,5-Трибромадамантан высокой степени чистоты получают с количественным выходом, бронируя адамантан бромом (со следами влаги) в присутствии восстановленного железа [184а].
Бромалкильные производные адамантана синтезированы нагреванием гидроксиалкиладамантанов с 47,5%-ной НВг [1846].
Применение катализаторов типа Фриделя—Крафтса (А1Вг3, ВВг3) позволяет повысить степень галогенирования и последовательно заместить иа бром от деух до четырех атомов водорода узловых атомов углерода ядра; из адамантана получают в итоге 1,3,5,7-тетрабромадамантан [185, 186]. Согласно Юрченко с сотр. [187], механизм реакции бромирования адамантана и алкиладамантанов жидким бромом включает гетеролиз связи С—Н в узловом положений ядер в циклическом комплексе (29) субстрата с одной или двумя поляризованными молекулами брома.
(29) (R, ,Лг,й,=М,СНэ, СгН5).
Энергия активации реакции бромирования составляет Е = 26,3 кДж/ /моль, энтропия активации - —40,5 з.е. Реакции бромирования протекает, вероятно, по механизму нуклеофильного замещения [188]. Подтверждением этому служат результаты галогенирования адамантана монобромидом иода, молекула которого поляризована [189]. При этом образуются 1-6 юмадамантан и 1,3-дибромадамантаи. В случае же электрофильного замещения следовало бы ожидать образования иодзамещениых адамантана, так как электрофильной частью реагента является атом иода. В литера
166
туре, однако, есть представления о бромировании адамантана как о процессе электрофильного замещения [190].
Обычно алкиладамантаны бронируются, как и адамантан, по узловым атомам ядра. Вместе с тем имеются случаи ’’аномального” протекания реакции бромирования. Это относится прежде всего к 2-замещенным алкиладамантанам. Например, бромирование 2-метиладамантана протекает по сложной схеме [191]:
Следует подчеркнуть, что это одна из немногочисленных известных к настоящему времени реакций алкиладамантанов, протекающих с участием алкильных групп.
При бромировании 1-изопропиладамантана следовало ожидать вхождения атома брома в изопропильную группу - с участием третичного атома углерода боковой цепи - вследствие того, что соответствующий карбкатион более устойчив, чем трег-адамантил-катион (первый может принять плоскую конфигурацию у реакционного центра). Эксперимент, однако, показал, что реакция вначале протекает главным образом по узловому положению ядра и лишь затем в реакцию вовлекается изопропильная
167
группа [188, 192]. Согласно данным работы [192], продукт бромирования по изопропильной группе составляет всего 10% от суммы других продуктов реакции.
Напомним, что при сольволизе бромидов алкиладамантанов реакция протекает значительно быстрее в тех случаях, когда бром находится в изопропильной группе, а не в ядре адамантана, что согласуется с относительной устойчивостью карбкатионов. Степанов с сотр. [188] высказал предположение, что в каждом случае имеется своя лимитирующая стадия реакции: прн сольволизе это образование комплекса типа (29).
В реакции бромирования алкиладамантанов отчетливо проявляется индукционное влияние ядра. Известно, что изобутан или 2-метилгептан не способны бронироваться в среде жидкого брома. Производное этих углеводородов, в котором метильная (или пентильная) группа заменена на адамантильный радикал — 1,3,5-триметил-7-изопропиладамантан, в среде жидкого брома бронируется по изопропильной группе с образованием 2-(3,5,7-триметил-1-адамантил)-1,3,3-трибромпропена [193].
СМ, I 1 Вг« и3с-(смд—см ;
сн3
Достаточно, чтобы между адамантильным радикалом и изопропильной группой находилась хотя бы одна метиленовая группа (1,3,5-триметил-7-изобутиладамантан), как углеводород снова теряет способность вступать в реакцию с жидким бромом в принятых условиях [193]. Таким образом, введение полиэдрического ядра в молекулу алканов может служить одним из путей повышения их реакционной способности.
С образованием непредельных полизамещенных бромидов вступают в реакцию бромирования также некоторые тетрациклические углеводороды ряда адамантана [194], например:
Вг
В 2,4-этаноадамантане различную реакционную способность проявляет каждый из четырех третичных атомов углерода. В присутствии бромистого алюминия и бутилбромида углеводород с высокой селективностью образует 1-бромэтаноадамантан [72].
168
Br
Br 967, 4%
Диамантан при взаимодействии с молекулярным бромом может образовывать в зависимости от условий реакции различные продукты ’’нормального” бромирования. При комнатной температуре образуется преимущественно 1-бромдиамантан (1-диамантил-катион является наиболее стабильным среди других диамантил-катионов). Нагревание реакционной смеси приводит к дибромидам. Действие зрет-СдЩВг и Вг2 (катализатор А1Вг3) при низкой температуре приводит к смеси монобромидов — 1- и 4-бром-диамантанов. Более жесткие условия способствуют образованию три- и тетрабромидов. Бромпроизводные диамантана, содержащие атом брома в узловых положениях ядра диамантана, ио термодинамической устойчивости близки, поэтому в равновесной смеси они представлены несколькими изомерами в примерно одинаковых количествах.
Триамантан в реакции бромирования проявляет более высокую реакционную способность, чем адамантан и диамантан. При взаимодействии с бромом в зависимости от условий образуются с достаточно высокой селективностью (>70%) 2- или 3-бромтриамантаны [11].
Легко — уже при комнатной температуре - вступает в реакцию бромирования бнсгомоадамантан. Наличие семичленного цикла в молекуле этого углеводорода способствует повышению стабильности третичного карбкатиона по сравнению с третичным адамантил-катионом.
Как уже отмечалось, легкость протекания реакции бромирования адамантана и алкиладамантанов зависит как от устойчивости карбкатиона, так и от степени поляризации связи Сад—Н. Вследствие передающихся по связям и через пространство индуктивных эффектов введение брома в узловое положение ядра должно облегчаться прн наличии в других узловых положениях электронодонорных заместителей и затрудняться при наличии электроноакцепторных групп. Это подтверждают данные работ [4, 186, 191, 195—197]. Так, адамантан реагирует с бромом даже при комнатной температуре [185]. Введение же второго атома брома, т.е. бромирование 1-бромадамантана, идет только в присутствии катализатора — кислоты Льюиса [186]. Аналогичный ингибирующий эффект при реакции бромирования ядра адамантана оказывают также такие электроноакцелторные группы,кактрифторметильная [198,199],гексафторпропильная [200,201], гидрокси метильная [202], находящиеся в других узловых положениях ядра адамантана. Для бромирования применяют сильные электрофильные катализаторы: А1С13, А1Вт3, BF3, ВВг3, FeCl3, FeBr3, Fe.
CFZCHFCF5
Br2,Fe или FeBr3 кипячение, 5ч, 86,5 7.
169
CFgCMFCFj
В качестве общего метода селективного введения одного атома галогена в узловое положение адамантанов, содержащих заместитель в другом узловом положении, авторы работы [203] предлагают использовать де протонирование под действием трет-бутилового спирта.
В отличие от ионного галогенирования адамантана, протекающего селективно по узловым атомам ядра, свободнорадикальное галогенирование самого углеводорода илн его производных приводит к смеси продуктов, состоящих из 1- н 2-замещенных производных. При действии на адамантан атомарного брома, генерируемого фотохимически нз N-бромсукцинимида (СС14,40°С), образуется сложная смесь продуктов вследствие протекания конкурирующего замещения по узловым и мостиковым положениям бромом н хлором (из СО4) [24]. Облучение 1-фтор-, 1-хлор- или 1-бромадамантана в галогенированных растворителях также приводит к продуктам с замещением у третичных и вторичных атомов углерода ядра [204]. Наряду с этим протекают реакции обмена галогена, доля которых зависит от природы исходного соединения и применяемого растворителя. Так, в СС14 протекает в основном галогенирование, в СВгС13 наблюдается некоторая селективность галогенирования по узловым атомам углерода, 1-иодадамантан образует вначале исключительно продукты обмена галогена.
Реакции хлорирования и фторирования адамантана, алкиладамантанов или их производных также протекаютпо ионному (в присутствии активаторов образования адамантилкарбкатионов) или же по радикальному механизму (см. табл. 3.7).
Ионное хлорирование адамантана, 1,3-диметил- и 1,3,5-триметиладамаи-танов с помощью хлорсульфо новой кислоты [205,206] приводит к различной степени замещения в узловые положения в зависимости от условий. В адамантане в мягких условиях замещается на хлор от одного до трех узловых атомов водорода в зависимости от продолжительности реакции. Для алкиладамантанов образуются соответственно моио- или дихлор-производиые. Прн этом важно отметить, что замещение происходит ступенчато, что позволяет получать с хорошим выходом как промежуточные, так и конечные продукты.
В присутствии H2SO4 можно хлорировать в мягких условиях 1-этил-адамантан; изомеризация его в диметиладамантан в этих условиях не происходит [207].
Метод хлорирования с помощью хлорангидридов кислот [208] позволяет с достаточной селективностью получать из адамантана монозамещен-ный 1-хлорадамантан. Механизм реакции включает гидридный перенос от адамантана к ацил-катиону, о чем свидетельствует образование, например, 1-формилциклогексана (при использовании цикло-С6 Hi t COCI).
В различных растворителях взаимодействие адамантана с FeCl3 н SbCl5 приводит главным образом к 1- и 2-хлорадамантаиам (наряду с некото-170
рым количеством 1,3-дихлорадамантана) [209, 210], что указывает на существенный вклад радикальных реакций.
AdH + 2FeCl3 -> AdCl + 2FeCl2 + HC1,
AdH + SbCls -+ AdCl + SbCl3 + HC1.
В зависимости от условий реакции соотношение 1- и 2-хлорадаманта-нов колеблется в интервале от 7:1 до 42:1 прн хлорировании с помощью H-C4H9CI; когда же имеет место чисто ионный процесс, это соотношение составляет не менее 1000. С применением системы SbCl5—А1С13 доля ионного хлорирования существенно возрастает.
Реакция хлорирования широко используется для препаративного получения хлорпроизводиых адамантана и алкиладамантанов. Наряду с рассмотренными выше случаями для этой цели могут быть использованы также другие пути. К 1-хлорадамантану приводит, например, расщепление 1-адамантил (аминоалкил) сульфидов при их нагревании с конц.НО [211].
Хлор метил производные адамантана (и норадамантана) с трихлорметильной группой образуются при свободнорадикальном присоединении СС14 к 3,7-диметиленбицикло[3,3,1] нонану [212].
Используют также хлорирование адамантана с помощью азотсодержащих катион-радикалов [213], хлорирование при действии солей серебра в четыреххлористом углероде [214], взаимодействие 4-гомоадамантана с дихлоркарбеном [215].
Реакции фторирования углеводородов ряда адамантана требуют более жестких условий. Реакция адамантана с четырехфтористой серой в безводном HF протекает при 60-200° С с образованием фторзамещенных производных [216]. При пропускании паров адамантана или алкиладамантанов в токе азота над CoF3 при 350° С образуются продукты исчерпывающего фторирования [217]. В 1,3-диметил-5-этиладамаитайе удается заместить на фтор до 90% атомов водорода; при этом образуется продукт состава Ci4F2JH3, представляющий собой жидкость с т. кип. 185—210° С [218].
Для получения фторпроизводных адамантана и алкиладамантанов часто используются функциональные производные; при этом наряду с замещением по функциональной группе нередко происходит замещение в ядре. Так, 1-фторадамантан образуется, например, при действии иа адамантанол-1 комплекса BF3 • Н3РО4 [219]. Действием SF4 на 1-адамантанкарбоиовую кислоту могут быть получены замещенные в узловом положении трифтор-метил производные адамантана [198, 220]. Фторадамантаны получают также при непосредственном воздействии элементного фтора на галогенадамаи-таны [221]. Перфторированный 1,3-диметиладамантан получают через промежуточный бис (трифторметил) адамантан, образующийся при фторировании адамантандикарбоновой кислоты с помощью SF4 при 110е С, выход 80% [222]. Бис (трифторметил) адамантан подвергают действию
171
CoF3 сначала при 250 -300° С, полученный продукт обрабатывают повторно при 275—280°С, получают перфтор-1,3-Диметипадамаитаи 75—90%-ной чистоты.
Исчерпывающее фторирование 1-аминоадамантана происходит при действии фтора [223].
I'JHe, 20° С
1-Ci0H15NH2 —------------► 1-C10Fi5NF2.
6,3%
Другие амииопроизводные адамантана в реакции с SF4 в HF в зависимости от положения аминогруппы образуют как фторсодержащие амино-адамаитаны, так и продукты дезаминирования [224].
Из 1-аминоадамантана в этих условиях образуется смесь 1,3,5-трифтор-и 1,3,5,7-тетрафторадамаитаиов. 1-Амииометил адамантан превращается в смесь 1 -аминометил-3,5-дифторадамантана и нитрила 3,5-дифтор-1-адамаи-танкарбоиовой кислоты.
При действии SF4 на 2-замещеииые карбонипьиые или карбоксильные производные адамантана образуются трифторметил-и фтор(трифторметил)-производные [225].
SF4lHF
Г
Действие SF4 на адамаитаиои приводит без HF к 2,2-дифторадаман-таиу [226], а в присутствии HF — к 2,2,5,7-тетрафторадамантану. Из диметилового эфира адамаитаи-2,6-диоксо-1,5-дикарбоиовой кислоты и SF4 получают диметиловый эфир 2,2,6,6-тетрафтор-! ,5-адамаитаидикарбоиовой кислоты. В присутствии HF удается заместить все функциональные группы в 2,6-диоксо-1,ЗД7-тетракарбоновой кислоте.
172
При фторировании с помощью четырехфтористой серы из соответствующих триметилсилоксиадамаитаиов образуются 1- и 2-фторадамантаны, а из тримета лсилокснгомоадамантана или из триметил си л окси мета л адамантана - 3-фторгомоадамаитаи [227].
SF4
CHzOSi(CH3)3
SF4
OSi(CHj)3
Для получения трифторметилпроизводных адамаитаиа используют также видоизмененную реакцию карбоксилирования по Коху—Хаафу [228]. При действии иа адамантан SF4, HF и НСООН в СН2С12 (20°С, 50 ч) образуется 1-(трифторметил) адамаитаи с выходом 75-80%, а в качестве побочного продукта — 1-фторадамаитаи. Аналогично протекает реакция с 2,2-дифто радамантаиом.
SF4, HF, НСООН, 20°С
F
F
(X=M,F).
СГ
Из рассмотренного выше следует, что SF4 в сочетании с HF. является весьма сильным электрофильным реагентом, способным генерировать адамантильный катион уже при 20° С. Затем происходит карбонилирование катиона с последующим фторированием карбонильной группы.
При фторировании 2-фтор- и 2-гидроксиадамантан-2-карбоновых кислот с помощью SF4 образуются производные, содержащие трифторметильную группу не в мостиковом (как при фторировании незамещенной 2-карбоновой кислоты), а в узловом положении ядра. Вероятно, пространственные факторы способствуют протеканию 1,2-гидридных межмолекулярных перемещений.
Как уже отмечалось, для получения фторалкильных производных адамантана также широко используется реакция радикального фторалкили-роваиия, например, гексафторпропилеиом [175]. Реакция ионного фтор-алкилирования фторэтиленом [SbF5, (СН3)3СС1] приводит к образованию до 40% 1,3,5-трис (пеитафторэтал) адамаитаиа [229]. В табл. 3.7 рассмотрены также другие случаи галогенирования углеводородов ряда адамантана.
Галогеизамещеиные углеводороды ряда адамаитаиа служат исходными соединениями для получения разнообразных функциональных производных. Реакционная способность галогеипроизводных адамантанов в этих реакциях, как правило, превышает таковую для других насыщенных углеводородов, за исключением случаев, когда решающую роль играют пространственные затруднения. Это обусловлено большей поляризацией связей Сад—Hal (в силу индуктивного эффекта адамантанового ядра) и легкостью образования адамантил-катионов, в особенности трег-ада-мантил-катионов.
Бромпроизводные адамантана легко вступают в реакцию сольволиза, которая подробно рассматривалась ранее. Эта реакция может быть использована для препаративного получения гидроксипроизводных адамаи-
173
Таблица 3,7
Реакции галогенирования адамантанов
Исходное соединение Реагент, катализатор, условия Продукты реакции, выход, % Литература
1 2 3 4
AdH Вг3, кипячение Вг3, ВВгэ, А1а Вт*, кипячение Вга, А1Вг3, кипячение 1-Бромадамантан (1-AdBr) 1-AdBr; 1,3-Дибромадаман-ган (1,3-АВг3) 1,3,5-Трибромадамантан (АВг3) (180, 182, 184,186] [186,261, 262,263] [186,263, 264]
1,3,5 -Трибромадаман-тан (АВгэ) Вг3, А1Вг3 ,140° С 1,3,5,7-Тетрабромадаман- тан (АВг4) [264]
AdH или 1-AdBr Вг3 , А1Вг3, кипячение 1,3-АВг3,70-80 (265]
AdH Вг3, комнатная температура, 2 ч 1-AdBr, 91 [266]
AdH илн 1-AdBr Br3, Fe, Н3 О, комнатная температура, 1ч 1,3-АВг3,97 [267]
AdH Вг3, пиридин Вг3, HNO3 (65%), 80°С 1-AdBr, 89 1-AdBr, 0,4 l-AdONO3, 6,6 1-AdOH, 92,7 [268] [269]
экзо- или зндо-Трн-метиленнорборнан Вг3, А1Вгэ 1,3,5-и 1,3,6-Трибромада-мантаны (АВг3) [270]
Поли-1 -адамантилок-сид Н (OR), ОН (R = = 1,3-адамантенил) Вг3, кипячение 1,3-АВг3,73,5 [271]
2-AdCl Вг3, кипячение, 9 ч 1,3,5,7-Тетрабромадам ан-ган (АВг4) [272]
2,4-Дегидроадамантан или протоада-м антен Вг3 2,4-АВг3 (273,274]
1 -Метиладам антан Вг3, кипячение L -Бром-3 -метил адамантан [275]
1 -Бромметиладам антан Вг3, кипячение 1 -Бромметил-3 -бромада-мантан [195]
1 -Гидроксиметил-адамантан Вг3, кипячение 1-Гидроксиметил-3-бром- _ адамантан [195]
1 -Метил-3 -зтилада-мантан Вг3, СС1„, кипячение 1-Бром-3-метил-5-этил-адамантан, 90 [195]
2-Метиладамантан Bra, HNO3 (65%), 0°С, 0,3 ч Вг3, кипячение 1-Бром-2-метил- и 1-бром-4 -метиладамантаны 2-Дибромметилен-4-бром- [196] [191]
адамантан
174
Таблица 3.7 (продолжение)
1 1 2 3 4
1,3,5-Триметиладаман- Вг3, грег-С4Н?ОН, 1 -Бром-3,5,7-триметилада- [203]
тан H3SO4 (конц.) мантан
1 -Изопропиладаман-тан Вг3, кипячение 1-Бром-З-изопропилада-мантан Вначале: 1-бром-3-нзопро-пиладамантан, затем: 1,3-дибром-2- (З-бром-1-адаман тил)пропен 1,3,3-трибром- 2- (1-адамантил) пропен 1,3,3-трибром-2-(3-бром- 1 -адамантил) пропан 1,3 -дибром-5-изопропил-адамантан [188] [192]
2-(1-адамантил)- 2-бромпропен Вг3, кипячение 1,3,3-Трибром-2- (1-адамантил) пропен [188]
1 -трег-Бутиладаман-тан Вт2,70°С, 2ч 1 -Бром-З-трег-бутилада-мантан, 85 [276]
1,1-Диадам анти л Вг2, кипячение 3,3 -Дибром-1,1 -диадамантил [237]
1,2-Бнс (3-метилада-мантил)этан Вг2 ,кипячение, 4 ч 1,2-Бис (З-бром-5-ме- тил)адамантилзтан, 98 [277]
1 -Фениладам антан Вг2, 0°С, 4 ч 1 - (4-Бромфеннл) адамантан [278]
Вг2,25°С, 4ч 1 - (3,4-Дибром фенил) адамантан [278]
1 -Фениладам антан Вг3,20°С, 3,5 ч 1- (2,4-Дибромфенил) адамантан, 47 [276]
2,4-Этаноадамантан Вг3, С^Н, Br, А1Вгв 1- н 6-Бромэтан садам антаны (96 :4) [72]
Диамантан Вг3,25°С 1 -Бромдиамантан [8]
Вг2, кипячение 1,6-н 1,4-Дибромд намантаны (8J
трет-С4 Н9 Вг, А1Вт3, о°с 1- и 4-Бромдиамантаны [8]
Вг2, А1Вг3 (следы) 1,6-, 1,4- н4,9-Дибромди-амантаны [8]
Вга, А1Вг3 (избыток) , кипячение 1,4,9-Три-и 1,4,6,9-тетра-бромдиамантаны [8]
3, 5-Дегидродиаман- N-Бромсукцияимид, 3,5-Б ром гидрины диаман- [279]
тан 20° С, растворитель дмсо тана, 72
175
Таблица 3.7 (продолжение)
1 2 3 4
Триамантан Вг2 Вт2, тетрабензоат свинца 2-Б ромтр иамантан З-Бромтриамантан [11] [И]
Бисгомоадамантан Вг2,25°С (трицикло [4,4,1,1 1,3-Бисбромметил адамантан, 91 [280]
додекан)
Бисгомоадамантан (трицикло [ 4,4,1,13> *|- Вг2,25° С Бромпроиэводные адамантана и гомоадамантана [280]
додекан)
Протоадамантан Вг2,80° С, 3 ч 6-Бромпротоадамантан [50,281]
Адамантанон-1 Вг5,20° С, грет-С4 Но Вг Вг2,55°С НВг, NH4 ОН, НС! 1-Бромадамантанон-4, 25 1-Бромадамантанон-4, 25 2 -Б ромадамантанон-4 [282] [282] [283]
AdH, 1,3-диметилада-м ант ан, 1,3,5-триме-тиладамантан C1SO2 ОН 1-Моно-, 1,3-ди- или 1,3,5-трихлорадам антаны, 60-92 [205,206]
1,З-Диметиладаман-тан, адамантан и другие алкиладамантаны нг SO4 (олеум), NaCl 1-Хлор-3,5-диметил- и 1,3 -дих л ор-5,7 -диметил-адамантаны [284]
AdH А1С13, инертный растворитель Гексахлор- и додека-хлорадамантаны [285,286]
1 -Этиладамантан Н2 SO4 (96%), трет-С4Н4С1, 0°С, 1,3 ч 1-Хлор-З-этил-, 1,3-ди-хлор-5-этил- и 1,3 >5 -трихлор-7 -этиладамантаны (98,2 : 1,4 : 0,4) [207]
AdH СС14 , А1С1,, 20°С SOC12 , А1С1Э,-15°С RCOC1 (R «СН3; цик ло-Св Н21, н-С3 Н7; дэо-Сз Н7), А1С13 СС14, А1С13, кипячение 1-Хлор-и 1,3-дихлорада-мантаны (15 : 85), 84 1,3,5-Трихлор-и 1,3-ди-хлорадамантаны (72 : 12) 1-Хлорадамантан при избытке реагента, а также 1,3 -дихлор адам антан 1,3,5,7-Тетрахлорадаман-тан [287,288] [287,288] [208] [251]
AdH или алкилада- СНС13, Na ОН, Дихлорадамантаны и [289]
м антаны нагрев а лк и ладам антаны
AdH ГеС13, SbCl5, растворитель или SbCl3 -AlCla a2,Av 1- и 2-Хлорадамантаны [(7:1)- (42:1)], 1,3-ди-хлорадамантан, 2-72 1-Хлор- + 2-хлор- + дихлорадамантаны [209,210] [290]
176
Таблица 3.7 (продолжение)
I 2 3 4
SF4, HF, 60-200°С Фторзамещенные производные адамантана [216]
AdH или алкиладамантаны CoF3,N2, 350°С Перфторпроизв одные адамантана и алкиладамантанов [217]
1,3-Дим етил-5-эти лада- CoF3, N5,35tf* С мантан Продукт состава CI4F51H3 (т.кип. 185-210°) [218]
1-AdOH bf3,h3po4 1-Фторадамантан [219]
1-AdCOOH bf3,h3po4 Трифторметилпроизвод-ные адамантана [198,220]
Галогенадамантаны Fa SF4, 85—140°C Фторадамантаны 1-Фторадамантан, 61-84 [221] [254]
1,3-Адамантандикарбоновая кислота sf4, ncTc 1,3-Бис (трифторметил) адамантан, 80 [222]
1,3-Бис (трифторметил) адамантан CoF3,250-300° C Перфтор-1,3-диметил-адамантан [222]
l-AdNH2 F3 , He, 20°C Перфторамнноадамантан, [223] 6,3
1,3,5-Трифтор- и 113,5,7-тет- [224] рафтор адамантаны
1 - Амином ети лада- SF4, HF 1-Аминометип-3,5-дифтор- [224]
мантан адамантан + нитрил 3,5-ди-фтор-1 -адамантанкарб оио-вой кислоты
2-Диметиламино- LSF4,HF; 2-Диметиламин о-5,7-ди- [224]
адамантан 2. HC1 фторадаманган
2-AdCOOH sf4 2-Трифторметил-+ 1-фтор-4-трифторметиладаманта- ны [225]
sf4,hf 1,3-Дифтор-6-трифтор-метил адамант ан [225]
Адамантанов sf4 2,2-Дифторадамантан [226]
SF4, HF 2,2,5,7-Тетрафторадаман-тан [226]
Диметиловый эфир адамантан-2,6 -диоксо- 1,5-дикарбоиовой кислоты sf4 Диметиловый эфир 2,2,6,6-тетрафтор адамантан-1,5-дикарбоновой кислоты [226]
2,6-Диои-1,3,5,7-адамантантетракарбоновая кислота SF4, HF 2,2,6,6-Тетрафтор-1,3,5,7-трифторметиладамантан [226]
1- или 2-Триметил-силоксиадамантаны ' Tqiz t яса sf4 1- и 2-Фторадамантаны [227] 177
12. Зак. 1856
Таблица 3.7 (окончание)
1 2 | 3 4
AdH SF4, HF, НСООН,. CHaCIj, 20°С, 50ч 1 -Трнфторметиладаман-тан (75-80) + 1-фторада-мантан [228J
2,2-Ди фтор адамантан SF4,HF, НСООН, СНаСТ,, 20°С, 50ч 1-Трифторметил-2,2-дн-фторадамантан [228]
Эфиры нхлорангид- (CHa)8SiI, Na,20°C 1-AdI, 70-86 [292]
риды 1-адамантанкарбоновой кислоты (например, 1-AdCOOCHa )
Примечание. Здесь и далее Ad — одновалентный адамантильный заместитель, А — дн-и поливалентный адамантильный заместитель.
тана. Например, 1-бромадамантаи (1-AdBr) при кипячении в НС1 образует 1-гидроксиадамантан с выходом до 90% [230].
1-Бромадамантан, подобно галогеипроизводным других насыщенных углеводородов, реагирует с магнием в условиях реакции Гриньяра с образованием 1 -адамантилмагнийбромида с выходом до 64% [231]. При действии на 1-AdBr метиллития образуется 1,1-диамантил. Бромпроизводные адамантана, содержащие галоген в боковой цепи, также легко реагируют с магнием, образуя адамантилалкилмагнийбромидьг, т.е, реакции протекает аналогично таковой для алифатических бромидов.
Изучена реакционная способность адамантилмагнийбромидов [231 ] с карбонильными соединениями и с хлоридами трехвалентного фосфора [231а, 2316]. 1- и 2-AdMgBr взаимодействуют с карбонильными соединениями по гомолитическому механизму. При этом реагент Гриньяра либо присоединяется по карбонильной группе, либо превращается в адамантан, причем водород отщепляется от карбонильного соединения. При взаимодействии адамантилсодержащих реагентов Гриньяра с РС13 на 1-Ad замещается не более двух атомов хлора вследствие пространственных затруднений, возрастающих в ряду: l-AdCH2 < 2-Ad < 1-Ad.
1'Литинадамантаи можно получить, действуя на 1-иодадамантан трет-бутиллитием [232]. 1- и 2-Литийадамантаны, а также 1-литнйдиамантан и литийпроизводиые других каркасных углеводородов (твистана, норадамантана) получают из соответствующих галогенидов в пентане при 35°С или в эфире при —45°С [233]. В кипящем пентане образуется также 1-литий-3,5,7-триметиладамантан. Эти Соединения неустойчивы и при 20° С в эфире или тетрагидрофуране разлагаются. В среде пентана, однако, они вступают в реакцию с кетонами, неспособными к енолизацни (адаманта-нон, бензофенон, гекса метилацето н), с образованием соответствующих продуктов конденсации; при взаимодействии с СО2 1-литийадамантан образует 1-AdCOOH.
176
Аналогично действием 1-литийадамантана на 2-иодадамантан получают 2-адамантиллитий [234]. Реакция различных галогенадамантанов с литием изучалась в работе [235].
В реакции Гриньяра 1 -бромалкиладамантаны образуют продукты рекомбинации [236]. К таким же продуктам приводит и реакция Вюрца. При взаимодействии бифункциональных производных (например, 3,3-дибром-1,1 -диадамантила) с натрием образуются полимерные продукты — полиадамантаны [237].
Подобно алкиладамантанам, галогенадамантаны с различным положением атома галогена в ядре под действием галогенидов алюминия претерпевают изомерные превращения с образованием более устойчивых изомеров, содержащих атомы галогена в узловых положениях. Например, 2,2-дибром- и 2,2-дихлорадамантаны превращаются соответственно в 1,3-дибром- и 1,3-дихлорадамантаны [238, 121], При нагревании в присутствии AlHal3 (Hal = Cl, В г) перенос галогена в галогенадамантанах идет по межмолекул яркому механизму. Так, 1-AdBr при нагревании в растворе н-гексана образует 17% AdH и 7% 1,3-дибром адамантана (время реакции 18 ч) [239]. Реакция протекает также в присутствии FeCl3 и конц. H2SO4.
Бромиды углеводородов ряда адамантана легко вступают в реакцию карбоксилирования по Коху—Хаафу с образованием соответствующих карбоновых кислот. При взаимодействии 1-AdBr с нитрилами (реакция Риттера) с высокими выходами образуются N-1-адамантиламиды [240].
1-AdBr 1-Acf® HSO? 1-AdN = CRHSO^^^-
-* 1-AdNHCOR,
При действии азида натрия 1-AdBr легко образует 1-азидоадамантаиы [24].
дмсо
1-AdBr + NaN3 ------► l-AdN3 + NaBr.
При взаимодействии 1-AdBr с СНС1=СС12 в условиях, необходимых для образования адамантил-катиоиа, получается 1-адамантил хлоруксус-иая кислота [242].
Как уже отмечалось, бромиды адамантана весьма активны в реакции алкилирования олефинами. Они также вступают во взаимодействие с ароматическими углеводородами. Например, реакция 1-AdBr с бензолом протекает легко и позволяет получать в зависимости от соотношения реагентов либо адамантан, содержащий несколько фенильных групп, либо бензол, содержащий до трех адамантильных остатков [155]. Из бромада-мантана и ферроцена в присутствии А1С13 можно получить моно- и 1,1*-диадамантилферроцены [155а]. Активность галогена в различных реакциях галогенадамантанов была изучена в работе [54].
Галогенпроизводиые углеводородов ряда адамантана вступают также в некоторые специфические реакции; к таким реакциям относится, в частности, реакция фрагментации. Дибромпроизводиые адамантана (например, 1 -бром-3-бромметиладамантан) при нагревании с цинковой пылью (150°С, ДМФА, Na2CO3 и Nal) образуют с высокой селективностью продукты частичной фрагментации — производные бицикло [3,3,1] нонана,
179
например (30) [123, 243], фотоизомеризация которого приводит к тетра-
ОС)
циклическому углеводороду, содержащему четырехчлеииый цикл [244]. 3,7-Диметилеибицикло [3,3,1] нонан (30) представляет собой весьма ценный продукт для синтеза (путем последующего замыкания) различных производных адамантана, недоступных другими методами [124].
При фрагментации тетрабромпроизводных адамантана продуктом реакции является симметричный тетраметиленциклооктан [245].
1,3-Дибром- и 1,3,5,7-тетрабромадамаитаиы, 1,5-дибром- и 1,3,5,7-тетра-бромадамаитаидионы-2,6 изменяются под действием коиц.Н^ЭОд, при
этом ди- и тетрабромадамаитаиы гидролизуются до соответствующих гидроксипроизводных, а ди- и тетрабромадамантандиоиы претерпевают фрагментацию адамантанового ядра [246]. Если реакцию 1,3,5,7-тетра-бромадамантана с H2SO4 проводить в присутствии Ag2SO4, то наряду с 1,3,5,7-тетрагидроксиадамаитаиом образуется продукт частичной фрагментации — 1,5-дигидро к си-7-метилеи би цикло [3,3,1] иоианон-3 [247].
1-NqOH,-Вг0
Z.RM, -R®
180
Реакция 1,3,5,7-тетрабромадамаитаиа со щелочью приводит к образованию 7-метилеибицикло [3,3*1 ]иоианоиа-3 [248].
Частичную фрагментацию претерпевают и пентациклические углеводороды ряда адамантана. Кипячение 1,6-дибромадамантаиа с активированной цинковой пылью в ДМФА приводит к тетрацикло [7,3,1,14>12,02'7] тетра-декадиеиу-6,11 (выход 83%); последний можно количественно превратить в диамантан при обработке Н2 SO4 в метилциклогексаие при О С, а при взаимодействии с 48%-иой НВг в гексаие при 0°С - в смесь 1-бромдиамаи-таиа (45%-) и бромпротодиамантана (55%) [249].
При действии литийалюмогидрида, содержащего каталитические количества хлоридов переходных металлов (FeCl2, СоС12, TiCl2), галогеиада-мантаны восстанавливаются до соответствующих углеводородов [250]. Этим методом, например, из 1,3,5,7-тетрахлорадамаитана может быть получен 1,3,5,7-тетрадейтероадамаитаи [251].
Галогенадамантаны под действием галогеипроизводных кислот легко вступают в реакции обмена атомов галогена [54].
MCI HF
' 1-AdGl
I НВг НС1
HF
1-AdBr 2- 1-AdF
НВг
Бром замещается на хлор также в функциональных производных, например в З-бром-1-адамантилкарбоиовой кислоте [252]. 2-Хлор- и 2,2-ди* хлорадамантаиы при действии жидкого брома образуют три- и тетрабром-адамаитаны [253], при этом получаются также 2,4-дибромадамантаны, содержащие значительные количества диаксиальиого изомера, а также 1,3,6-трибромадамантай.
Опубликован патент иа способ получения 1-хлорадамаитаиа с выходом 94% последовательной обработкой адамаитаиа смесью брома скоиц.НМО3, водио-спиртовой щелочью и, наконец, конц. НС1 [153а].
Благодаря способности трициклоалканов к изомеризации в адамантаны в присутствии кислот Льюиса удалось синтезировать из таких циклоалканов и непредельных хлорсиланов в присутствии смеси А1С13 и А1Вг3 крем-иийорганические мономеры с алкиладамаитильиым заместителем — алкил-хлорсилил ал кил адамантаны [2536],
1-Галогенадамантаны (Hal = Cl, Br, I) взаимодействуют c SF4 в сравнительно мягких условиях (85—140°С, 3—8 ч), образуя 1-фторадамаитаи с выходом 61—84% [254]. 1-Фторадамантан при нагревании с безводным Вг2 дает 1-AdBr (80—90%). При действии иа полизамещеиные фторпроиз-водные адамантана ВВг3 при 50—60°С на бром замещаются только наиболее подвижные (узловые) атомы фтора [199].
181
Вместе с тем добавление каталитических количеств безводного А1Вг3 позволяет провести обмен атомов фтора на атомы брома и в трифторме-тильиой группе [255]. Тетрафторадамаитаиы с геминальными атомами фтора реагируют с ВВг3, образуя соответствующие геминальные тетрабромиды; последние прн действии трифторацетата серебра и последующего щелочного гидролиза превращаются в бромзамещеииые адамантаны [256].
1. CFjCOOAg g.OMQ, Нг0
Облучение 1 -галогеиадамантанов (Hal = F, Cl, Вт) в галогенированных растворителях вызывает свободнорадикальное галогенирование ядра адамаитаиа без обмена атома галогена [257]. Реакции с СС14, например, приводят к продуктам замещения у обоих третичных и вторичных атомов углерода ядра. Продукты, молекулы которых содержат атомы брома в узловых положениях, преобладают, если в качестве растворителя используют ВгСС1э, Облучение 1-AdI в эфире приводит первоначально только к обмену галогена.
Фторадамантаиы, содержащие атомы фтора в боковой цепи, дегидро-фторируются с образованием фторалкениладамантанов. Так, при нагревании в спиртовом растворе щелочи моио- и ди (гексафторпропил) адамантаны образуют соответствующие моио- н ди (перфторпропенил) адамантаны [258,259].
CF = CFCf3
ЫаОН/EtQH, BZ-a5°C
-2HF
CF = CFCF3
Некоторые реакции галогенадамаитаиов протекают с расширением одного из шестичленных циклов ядра адамантана. Так, взаимодействие 1-AdBr с пропаргиловым спиртом под действием 98%-ной H2SO4 приводит к образованию гомоадамантилметилкетона [260]. Примеры реакций галогенирования адамантана, низших алкиладамантанов и их производных приведены в табл. 3.7.
3.5. ПОЛУЧЕНИЕ КИСЛОРОДСОДЕРЖАЩИХ ПРОИЗВОДНЫХ АДАМАНТАНОВ
3.5.1. ОКИСЛЕНИЕ АДАМАНТАНОВ
Реакции окисления углеводородов ряда адамантана лежат в основе важных препаративных методов получения нх кислородсодержащих производных, главным образом гидроксипронзводных.
Исследование окисления адамантана и алкиладамантанов началось как только появились методы синтеза этих углеводородов. В качестве окне-
182
Таблица 3.8
Окисление адамантанов
Исходное вещество Окислитель, катализатор, условия Продукты реакции (выход, %) Литература
Адамантан или алкиладамантаны СгОэ,120°С Моногидроксипроизвод-ные [292]
Адамантан Воздух, 60 атм, 140° С, iper-Cj Нв ООН, (СНэСОО)3Со, снэсоон Смесь продуктов (42-90), 1-гидроксиадамант ан (1-AdOH) (3-46 ),ада-адамантанон (3—22 ) [293[
1 -Фени ладамантан СгО,, снэ соон, (СН3СО)2О, 0° с 1-Фен ил-3-гидроксиада-мантан (51) [276]
Адамантан или алкиладамантаны сн3 СООН, Н2СгО4 (хром ; углеводород = 3:1) 1,3-Дигвдроксипроизвод-ные [2941
Адамантан ОЭ,-78°С Продукты окисления [295]
1,3-Диметил-5-этил-адамантан СгОэ,СН3СООН (водн.) 1 -Гидрокси-3,5-диметил-7-этиладамантан (22) [296]
.Адамантан или 1-гидро- Н2 SO4 (коиц.), ксиадамантан нагрев Адамантаном (66- 84) [297, 230]
1-Метиладамантан H2SO4 (конп>), 80°С 5-Метил-2-адамантанои [298[ (78 ), 1-метил-2-адамантаг ион (10 ), 1-гидрокси-3-метиладамантан (5 ), адамантаном (5 )
лителей применяли кислород воздуха, озон, серную или хромовую кислоту и др. Реакцию обычно проводили в присутствии катализаторов (металлоорганические соединения Со, Мп, Fe), в среде растворителей (уксусная кислота или уксусный ангидрид) прн нагревании (табл. 3.8), Способность углеводородов ряда адамантана к окислению выше, чем у разветвленных алканов.
Вследствие значительной роли радикальных путей окисления прн этом обычно образуются смеси 1- и 2-замещеиных производных. Для адамантана, например, образуются три первоначальных продукта окисления:
Ой
К моиогидроксипроизводным адамантана и алкиладамантанов приводит окисление хромовым ангидридом при 120—125°С [292], Окисление адамантана воздухом в автоклаве при 60 атм и 140° С в присутствии пе-
183
роксида трет-бутила, ацетата кобальта и СН3СООН также приводит к смеси веществ, содержащей 1-гидроксиадамантан, адамантанон и непрореагировавший адамантан [293]. Лучшие выходы кетона получают при окислении в растворах бензола или гексана.
Окисление ариладамантанов в отличие от галогенирования протекает по адамантильиому кольцу с образованием 1-арил-З-гидроксиадамантана [276]. Окисление адамантана и алкиладамантанов смесью хромовой и уксусной кислот приводит к 1,3-диолам [294].
Рассмотренные выше методы окисления отличаются весьма низкой селективностью в отношении образования отдельных соединений. В этой связи важное значение приобрела реакция адамантанов или их производных с серной кислотой, позволившая существенно повысить выход кетона (Гелук и Шлатман) [230, 297]. Например, при взаимодействии адамантана или 1-гидроксиадамантана с конц.Н2£Ю4 максимальный выход ада-мантаноиа составляет 84%. Такой результат реакции обусловлен активным протеканием процессов гидридного переноса.
Роль акцепторов гидрид-ионов при межмолекулярном гидридиом переносе играют адамаитил-катионы. Это подтверждается тем, что 1- и 2-гид-роксиадамаитаны в реакциях с участием адамаитильных катионов образуют 1,3- и 1,4-дигидроксиадамантаны [299]. 2-Гидрокси-2-метиладамантан в коиц.Н2ЗО4 при 0—50° С претерпевает изомеризацию, сопровождающуюся межмолекулярными гидридными переходами; первоначально образующийся 2-метил-2-адамантил-катион быстро превращается в смесь всех возможных третичных катионов [300].
При кипячении экви мольных количеств адамантана с 96—98%-ной Н2 SO4 в трифторуксусном ангидриде с количественным выходом образуется смесь, состоящая из 80% 1-AdOH и 15% 2-AdOH [301а]. С образованием такой же смеси, но менее селективно и с худшими выходами протекает окисление адамантана кислородом воздуха в присутствии различных инициаторов радикального окисления [3016, 301в1.
Окисление адамаитаиа в двухфазной системе водным раствором, содержащим Сг2О,~, H2SO4 и (C4H9)4NBr, приводит при облучении светом ртутной лампы мощностью 500 Вт к смеси 1-гидроксиадамантана и адаман-танона-1 в соотношении 7:1с общим выходом 52% [301г]. При фото-окислеиии адамантана стехиометрическим количеством (NH4)2 [CeNO3]6 184
в ацетонитриле образуются 1-адамаитилнитрат и немного 2-адамаитил-иитрата, а также продукты их гидролиза — N-(адамантилацетамиды). В случае, если положение 1 ядра адамантана занято объемистым трет-бу-тильным заместителем, окисление в этих условиях направляется в положение 3 [305]. Фотоокислеиие AdH можно проводить, используя (NHJ2 [CeNO3]6 в качестве катализатора (в присутствии HNO3), а в качестве окислителя — кислород воздуха. В этом случае образуется главным образом адамантилпероксид, а после его разложения - 1-гидроксиадамантан с селективностью 90%, самой высокой среди реакции автоокисления адамантана [301д].
Аналогично адамаитаиу реагируют с H2SO4 и алкиладамантаны. 1-Ме-тиладамантан или его производные (заместители: Вт или ОН) образуют смесь продуктов, например [298]:
О
7В%
Окисление 1,3-диметиладамантана кислородом воздуха при 80—100° С осуществлено над платииосодержащим катализатором, получена смесь, в основном состоящая из 65-70% 1-гидрокси-3,5-диметиладамантаиа и 10—15% 1,3-дигидрокси-5,7-диметиладамантана; степень превращения исходного углеводорода 85% [301е].
В результате окисления 1-гидрокси-З-метиладамантана или 2-гидрокси-2-метиладамантана серной кислотой в качестве основного продукта также образуется 5-метиладамантанои-2 [301].
На протекание реакции окисления в серной кислоте существенное влияние оказывают концентрация кислоты и условия реакции-
Адамантан или 1-гидроксиадамантан при нагревании в коиц.Н28О4 образуют адамантанон в качестве основного продукта. В 20%-ном олеуме образуются главным образом 1,4-, 2,6- и 1,3-дигидроксиадамантаны [302]. Если реакцию вести при 16—35° С в среде 18—25%-ного олеума, то образуется преимущественно -1,4-дигидроксиадамантан [303]. Образующиеся диолы действием СгО3 могут быть переведены в кетоны [304]. При окислении адамантана хромовой кислотой из продуктов реакции можно выделить адамантандиои-2,6 [302]. Дымящая серная кислота при 8—25° С окисляет алкиладамантаны по свободным третичным атомам углерода с образованием главным образом третичных спиртов. 1,3-Диметиладамантан образует 1-гидрокси-3,5-диметил адамантан, а 1,3-диметил-5-этиладамаи-тан — 1-гидрокси-3,5-диметил-7-этиладамантан с выходом 96-98% [305]. Обработка 1,3-диметиладамантаиа 100%-йой H2SO4 в течение 6 ч при 25°С приводит к сложной смеси продуктов, в которой наряду с гидроксидиме-тиладамантанами содержатся 1,1-бис(3,3’,5,5-тетраметил)адамантан, эфир 1,3-диметиладамантана и кетоны [306]. 2-Метиладамантан также реагирует с конц.Н2 S04 с образованием продуктов окисления [307].
185
Исследование реакции адамантана с Н2 S04 привело к разработке простого препаративного метода получения адамантанона [308] — весьма важного полупродукта прн синтезе 2-замещенных производных адамаитаиа.
Адамантан перемешивают 2 ч в 98%-ной H2SO4 при 70-80° С, поднимают температуру до 82° С и после полного растворения адамантана перемешивают еще 1 ч. Адамантанон выделяют перегонкой с водяным паром, выход 52-54%. Дополнительную очистку продукта проводят хроматографией иа Als Оэ или обработкой 20%-ным олеумом. Основная побочная реакция - образование 1,3-дигидроксиадамантана и полиадамаитилового эфира из образующегося вначале 1-дигидроксиадамантана.
В сверхсильных кислотах, иапрнмер в смесях FSO2OH— SbFs (1 : 1) и HF—SbFs, адамантан легко генерирует третичный адамантил-катнон; последующая обработка реакционной смеси водой приводит к 1-гидроксиадамантану [2]. Аналогично протекает реакции адамантана и алкиладамантанов в неочищенной фторсульфоновой кислоте [309].
(М'=н,сн3).
Важно отметить, что чистая фторсульфоиовая кислота, полученная повторной перегонкой, не растворяет углеводороды ряда адамантана и инертна по отношению к ним при комнатной температуре. Для появления активности необходима добавка HF. Это напоминает катализ с помощью соединений А1На1з (Hal = Cl, Вг), которые в чистом виде также неактивны в реакциях насыщенных углеводородов.
При действии на адамантан или его функциональные производные (заместители: F, С1, Вт, СНаОН и др.) азотной кислоты образуются нитрат 1-гидроксиадамантана, динитрат 1,3-дигидроксиадамаитана или нитраты 3-замещенных 1-гидроксиадамаитанов [246]. Эти интраты способны легко генерировать адамантил-катиоиы, легко вступают в реакции Коха—Хаафа, Риттера, Фриделя—Крафтса, вследствие чего являются ценными веществами для получения различных производных на основе углеводородов ряда адамантана.
Полициклические углеводороды ряда адамантана также вступают в реакцию с серной кислотой. Диамаитаи образует 3-диамантанои в качестве основного продукта [9, 310—312]. Обработка триамантана 96%-иой HaSO4 приводит к образованию триамантаиоиа-8, триамантандиоиа-8,16 и 15-гид-рокситрнамантаноиа-8 [313].
Н; S04 , нагревание
a)A, = O,Rf=H,ll,=Hf;
«А<=0, R2=tt,R,= f=0); Ь>Я, = О. Вг=ОН. и,=нг.
Как и другие насыщенные углеводороды, адамантан окисляется молекулярным кислородом с образованием 1- и 2-гидроксиадамантанов и 186
адамантанона в присутствии комплексного железного катализатора (31) [314].
aCH=N1, = CH.
\/ Y] о
О----Fe---
I J2 (31)
Характерной особенностью этой реакции является более высокая селективность образования вторичных (мостиковых) продуктов окисления; соотношение 2- и 1-гидроксиадамантанов составляет 3 : 1, в то время как при окислении в отсутствие катализатора оио равно 1 : 1 [315],
При окислении воздухом (подаваемым с небольшой скоростью) в присутствии катализатора Жиф-системы: кластер железа, металлический цинк—пиридин, водная СН3СООН (Zn, [Fe11, Ре|пО(ОАс)бРугз]2Руг) при 40°С достигается практически полная селективность в отношении окисления вторичных атомов углерода ядра адамантана — соотношение образующихся 2- и 1-замещевдых производных [ (2-гидрокснадамантан + адаманта-нои) ; 1-гидроксиадамантан] составляет от 10 : 1 до 24,8 : 1 [316] (общий выход продуктов реакции 16—6%).
Соли свинца также катализируют окисление углеводородов ряда адамантана; образуются замещеииые в узловых положениях гидроксипроизводные. Окисление адамантана, алкиладамантанов, диамантана можно проводить, в частности, в присутствии тет^аацетата свинца в среде три-фторуксусиая кислота—дихлорметан при 20 С в присутствии ноиов хлора [317]. С высоким выходом образуются трнфторацетаты спиртов, которые после гидролиза дают соответствующие спирты, выход 1-гидроксиадамантана 88%. Из диамантаиа образуется смесь 1- и 4-гидроксиднамантаиов.
Электрохимическое окисление адамантана в ацетонитриле (Pt-электроды) приводит к образованию N-(l-адамантил) ацетонитрил-иоиов; в присутствии воды образуется 1-адамантилацетамид [318, 319]. Процесс, вероятно, включает прямое окисление адамантана. 1-Замещенные производные адамантана (заместители; Вт, СН3СО, СН2ОН) также претерпевают расщепление, иа аноде с образованием 1-адамантилацетамида, а производные с заместителями Cl, F, СН2СООН, CN ие расщепляются и образуют 3-за-мещеиные 1-ацетамидоадамаитаиы, в таких случаях реакция протекает по связи С—Н свободного узлового атома ядра. В результате электрохимического окисления в трифторуксусиой кислоте образуется 1,3-дигидрокси-адамантан, выход 70% [320], а в ацетонитриле можно получить N,N'-(1,3-дигидро к сиадамантил) бисацетамид, выход 58%.
Изучение кинетики и механизма окисления адамантана кислородом (растворитель хлорбензол, инициатор азодиизобутироииТрил) показало, что (как в случае других насыщенных углеводородов) первичным актом окисления является образование трет-гидро пероксида [321]. В растворах нормальных алканов скорость окисления адамантана значительно ниже, чем в хлорбензоле. Небольшие добавки гидропероксида кумила и трет-бутила приводят к прекращению окисления. Уменьшение скорости окисления имеет место при наличии электроиоакцепториого заместителя в ядре
187
адамантана: 1-AdBr окисляется медленнее адамантана. Добавление небольших количеств 1-AdBr к кумолу в растворе хлорбензола приводит к возрастанию скорости поглощения кислорода по сравнению со скоростями, наблюдаемыми при окислении индивидуальных веществ; энергия активации реакции окисления адамантана и 1-AdBr равна соответственно ПО и 118 кДж/моль.
Аналогично протекает жидкофазное окисление адамантана в ацетонитриле и бензонитриле [322]. Реакционная способность адамантана (на одну третичную С Н-связь) по отношению к пероксирадикалам близка к таковой у изопропилбензола и декалина и в 3-10 раз превышает реакционную способность разветвленных алканов [323].
Окисление (гексафторпропил) адамантана смесью азотной и серной кислот приводит к получению соответствующего моногидроксипроизводного [59].
Первичные продукты окисления адамантана или алкиладамантанов могут подвергаться дальнейшему окислению с образованием полифункционал ьных производных, окисление адамантанона приводит, например, к 5-гидрокси- и 5-ацетоксиадамаитаиону-2 [324].
(Х = ОН)ОСОСН3).
Реакция окисления адамантанов иногда приводит к расширению цикла. Так, 1-дихлорметиладамантан лри нагревании в фосфорной кислоте образует 4-гомоадамантаион [289].
3.5.2. КАРБОКСИЛИРОВАНИЕ АДАМАНТАНОВ
Для синтеза карбоновых кислот адамантанового ряда чаще всего используют реакцию карбоксилирования. Кох и Хааф впервые в 1960 г. осуществили таким путем прямой синтез 1-адамантанкарбоновой кислоты (1-AdCOOH) [325], Реакцию проводят в среде конц. H2SO4 или олеума, что обеспечивает образование адамаитил-катионов. В качестве исходных соединений используют либо углеводороды, имеющие свободные третичные углеродные атомы, либо их простейшие функциональные производные (заместители: Вг, ОН, ONO2).
Целевой продукт 1-AdCOOH с выходом до 80% получили при обработке адамантана в растворе циклогексана и 96%-ной H2SO4 при 17-19° С муравьиной кислотой с добавлением трет-С4 Н9 ОН. Реакцию карбоксилирования проводят также, прибавляя НСООН к суспензии адамантана в 5%-ном олеуме при 0-20° С [326] или в 15-20%-ном олеуме в среде СНа С12 [327].
Нг50ч(олеум)
НСООН,20°С
СООН
188
Если реакцию карбоксилирования адамантана вести в смеси 94%-ной H2SO4, 60%-ного олеума и 56%-иой HNO3, то наряду с 1-AdCOOH можно получить также 1,3-адамаитаидикарбоновую кислоту [1,3-А(СООН)2] [328]. Суммарный выход этих кислот достигает 94%; соотношение 1-AdCOOH : 1,3-А (СООН) 2 = (5 4- 10) : (95 4-90). 1,3-Адамантандикарбоновая кислота образуется также при последующем карбоксилировании 1-AdCOOH в среде олеума и HNO3 [329]. Аналогично реагирует 3,3'-ди-амантил-1 Д'-дикарбоиовая кислота - образуется 5,5 '-диамантил-Ь^ЗЗ'-тётракарбоновая кислота. 1,3-А(СООН)2 образуется также из адамантана при действии газообразного СО при 90—160° С под давлением 6-150 атм в присутствии 10-20 вес.ч. 90-103%-ной H2SO4 [330],
Скорость карбоксилирования и выход продуктов реакции зависят от легкости образования адамантил-катионов. Вследствие этого функциональные производные адамантана и алкиладамантанов вступают в реакцию легче, чем адамантановые углеводороды, так, например, Карбоксилирование 1-бром-3-метил-5-этиладамантана в условиях реакции Коха-Хаафа приводит к 3-метил-5-этил-1-адамантанкарбоновой кислоте с выходом 92% [195]. В качестве доступных исходных веществ в реакции карбоксилирования предложено использовать также адамантанолы и нитраты адамаитанолов [331]. Возможность использования функциональных производных в реакции Коха—Хаафа особенно важна в тех случаях, когда соответствующие адамантил-катионы непосредственно получить из углеводородов затруднительно или невозможно (например, при получении 2-ада-мантанкарбоиовых кислот). Для получения 2-AdCOOH можно использовать 2-AdOH. Необходимо помнить, что при карбоксилировании 2-AdOH состав продуктов реакции существенно зависит от соотношения реагентов. В обычных условиях проведения реакции вследствие интенсивно протекающих межмолекуляриых гидридиых переходов образуется 1-AdCOOH, а ие ожидаемая 2-AdCOOH. Для получения 2-AdCOOH в качестве главного продукта необходимо сильное разбавление кислот в реакционной смеси — с целью уменьшить относительную концентрацию спирта и свести к минимуму межмолекулярные гидридные переходы [332]. 2-AdCOOH высокой степени чистоты (98-99%) получают при мольном соотношении 2-AdOH : НСООН : Н2 SO4 = 1 : 100 : 300 [333].
Карбоксилирование 2-гидрокси-2-метиладамантана также приводит к различным продуктам в зависимости от условий реакции [334, 335]. При 0°С и обычно применяемых условиях в этой реакции основным продуктом является 2-метил-1-адамантанкарбоиовая кислота, т.е. протекание межмолекулярных гидридных переходов приводит к изомеризации первоначального катиона и в результате к вхождению карбоксильной группы в узловые положения ядра.
При 50° С и тех же концентрациях реагентов основным продуктом реакции является 3-метил-1-адамантанкарбоновая кислота (’’миграцию” в узловое положение претерпевает и метильная группа). Наконец, при высоком разбавлении реакционной смеси образуется ’’нормальный” продукт реакции — 2-метил-2-адамантанкарбоновая кислота. Аналогично протекает карбоксилирование гидроксиадамантанов с гидроксильной группой в алкильной цепи. 1-Адамантилнзопропанол в 96%-ной H2SO4 при обычном соотношении реагентов образует не ожидаемую 2-изопро-
189
пил-1-адамантановую кислоту (32), а продукт перегруппировки -З-нзопропил-1-адамантанкарбоновую кислоту (33) [125, 336]. Таким образом, равновесие между катионами (34) и (35) в обычных условиях сдвинуто в сторону более реакционноспособного катиона (35) (Лл >^г):
(ЗП
(34)
со,он~
Получить карбоновую кислоту (32) можно только при сильном разбавлении реакционной смеси серной кислотой, когда из-за малой вероятности встречи катиона (34) с молекулой исходного спирта межмолекулярные гидридные переходы сведены к минимуму. Аналогично протекает реакция с 2-(3-бром-1-адамантил)пропанолом [125].
В случае гидроксналкиладамантанов протекание реакции карбоксилирования зависит от положения гидроксигруппы по отношению к ядру адамантана. Карбоксилирование l-AdCH2OH протекает с расширением шестичленного цикла за счет метиленового звена боковой цепи и приводит к двум изомерам 1-гомоадамантан- и 3-гомоадамантанкарбоновым кислотам [337]. В результате карбоксилирования 2-(1-адамантил) этанола, в котором гидроксигруппа более удалена от ядра адамантана, образуется 3-этил-1-адамантанкарбоновая кислота (вследствие межмолекулярных гндрид-ных переходов) и 1-адамантанкарбоновая кислота (вследствие фрагментации 1-адамантилэтильного катиона) [335, 337].
Диамантан и его производные также вступают в реакцию карбоксилирования. 1-Диамантил-катион, образующийся в условиях реакции, превращается в 1-диамантанкарбоновую кислоту [310, 311], 3-Гидроксиадаман-тан при сильном разбавлении реакционной среды подобно 2-гидроксиада-мантану образует З-диаь антанкарбоновую кислоту, а в обычных условиях — только продукт перегруппировки — 1-диамантанкарбоновую кислоту' [312].
190
M2SO4,HCDOH
с разбавлением МООС
сооп
1- н 4-Бромдиамантаны в реакции карбоксилирования также образуют соответствующие 1- и 4-диамантан карбоновые кислоты [338], На основе монобромдиамантанкарбоновых кислот путем их бромирования и после-» дующего карбоксилирования могут быть получены дикарбоновые кис-поты диамантана, содержащие карбоксильные группы у третичных С-ато-мов [339].
В реакцию карбоксилирования вступают также те производные адамантана, которые содержат электроноакцепторные заместители, не отщепляющиеся в процессе реакции (l-AdCF3, l-AdCH2CHFCF3). В связи с тем что такие группы существенно затрудняют образование iper-адамантил-катионов, нужны более жесткие условия реакции, например использование смесн олеума и азотной кислоты [340]. 1-(1,1,2,3,3,3-Гексафторпропил)-3-бромадамантан также менее активен в реакции, чем 1-бромадамантан, но прн действии олеума с добавкой азотной кислоты и в этом случае могут быть получены хорошие выходы (до 70%) соответствующих карбоновых кислот [341].
При наличии диух гексафторпропильных групп в ядре реакция карбоксилирования замедляется настолько, что даже в HNO3 выход продукта карбоксилирования не превышает 6% (в таких случаях для получения полифторалкилированных адамантанкарбоновых кислот лучше использовать радикальное алкилирование адамантанкарбоновых кислот перфтор-олефинами).
В реакцию карбоксилирования по Коху—Хаафу вступают также производные гомоадамантанов; прн этом образуются 1- н 3-гомоадамантановые кислоты [342]. Ввиду большей по сравнению с iper-адамантильным катионом устойчивости iper-гомоадамантильного катиона реакция должна протекать легче, однако возникает возможность перегруппировки в адамантан. Другой изомер адамантана — триметиленнорборнан, третичный катион которого меиее плоский н менее устойчивый, чем трег-адамантильный катион, в реакцию карбоксилирования вступает значительно труднее адамантана [343].
191
ЗЛ.З. ДРУГИЕ МЕТОДЫ ПОЛУЧЕНИЯ
Генерирование 1-адамантил-катиона из адамантана происходит также под действием CF3COOH и грет-С4Н9ОН. На этом осиоваи одностадийный способ получения 1-гидроксиадамантаиа (выход до 65%) [344]. Реакцию проводят в среде СН2С12, в качестве побочных продуктов образуются алкениладамантаны. Взаимодействие адмантана с винилиденхлоридом в присутствии трет-С4Н9ОН и H2SO4 [345] или в смеси H2SO4 и HNO3 [328, 346] приводит к адамантанмоио- или-диуксусиой кислоте (в зависимости от соотношения реагентов).
Рид гидроксипроизводных адамантана может быть получен с использованием реакции циклизации. Помимо тех случаев, которые были описаны ранее (в синтезах углеводородов, гл. 2), следует рассмотреть синтез 2-гидроксизамещенных производных из соответствующих 1-замещенных через производные бицикло [3,3,1 ]иоиана и протоадамантаиа [347]. Так, 1-гидрокси-3-метиладамантан с иодом в присутствии РЬ(ОАс)4 в бензоле образует неустойчивый гипоиодид, распад которого с разрывом С-С-связи приводит к 1 -метил-7-иодметилбицикло [3,3,1 ] ноианону-3; из него после обработки спиртовым раствором КОН получают смесь 6- и 8-метил-4-ок со-протоадамаитаиов. Эти соединения подвергают действию реактива Гриньяра и проводят изомеризацию полученных продуктов, иалример:
Оптически активный кетой — 4,4-диметиладамантадои-2 — получают путем циклизации эндо-бицикло [3,3,1 ] иои-6-ен-З-карбоиовой кислоты [348]. К замещенным дикетоиам приводит кислотио-каталитическое ацилирование бицикло [3,3,1 ]ионандиона-2,6 и его 9-тиа-2- и 9-оксо-З-ана-логов с помощью (СН3СО)2О [349].
Хлорангидриды карбоновых кислот получают хлоркарбоиилированнем адамантана и его производных [23,350].
(0001)?, пероксид бенаоилй
C6HsCl, во°с
(И = СНЭ ,СНЭО, М, СОгСМэ, CN).
2-Замещенные (гидрокси-, метокси-, бром-, хлор-, ннтро-)-1-адамантан -карбоиовые кислоты получают исходя из адамантаноиа через стадии образования гомоадамаитандиона н 2-оксо-1-адамантанкарбоиовой кислоты [351]. З-Гндроксиадамантаи-1-карбоновая кислота может быть получена кз 1-адамантанкарбоиовой кислоты через соответствующий бромид [352], Ряд методов синтеза кислородсодержащих производных основан на использовании реакции алкилирования адамантана или алкиладамантанов ацетиленом. Образующиеся 1-адамантилацетилены при обработке их 192
90%-иой H2S04 превращаются в смесь 1-адамантил кетонов и гомо адамантанов [353]. Реакция протекает через промежуточные адамантилвинил-
катиоиы; оии образуются как путем протоиирования 1-адамантилацетиленов, так и в результате присоединения 1-адамантил-катионов к ацетилену (более подробно образование и превращения виниладамантил-катионов
рассмотрены в непредельных производных адамантана, гл. 4).
Если источником адамантил-катиоиов служит 1-гидроксиадамантан, то в результате взаимодействия его с 90—95%-иой H2SO4 в атмосфере ацетилена (3-6 мин) образуется смесь 1-адамантилацетальдегида, 1-ацетил-адамантана и З-метилгомоадамантанона-4 [354]. Аналогично реагирует с ацетиленом в присутствии конц. H2SO4 при 5°С 1-гидрокси-3,5,7-триме-тиладамантан — образуются триметиладамантилацетальдегид (73%) и триметилгомоадамаитанои (24%) [355].
Известны пути синтеза адамантилсодержащих альдегидов типа 1-AdCHO, l-AdCH2CHO, l-Ad(CH2)2CHO [356], 2-адамантанкарбоновой и 2-адамаи-тануксусной кислот исходя из адамантаноиа [357] и из 1,4-дизамещенных кислородсодержащих соединений (оксо- и гидроксикислот и их эфиров) ряда адамантана [354], например:
Гидроксикислоты ряда диамантана получают путем окисления моно-бромдиамантановых кислот КМпО4 [339], гидрокситриамантаны - гидролизом соответствующих бромидов [11].
Алкиладамантилкетоиы легко получают по реакции Гриньяра 1-AdCOO или l-AdCH2COCl с RMgBr (R = СН3, С2Н5 и др.) в эфире в присутствии СН2С12 [358].
Для кислородсодержащих производных адамантанов в общем характерны те же химические превращения, что и для циклогексанов или алканов. Вместе с тем наличие объемистого адамантанового ядра, обладающего значительным индуктивным эффектом, и системы циклогексановых колец с сочленением мостикового типа в составе этого ядра оказывают влияние иа характер протекания некоторых реакции и строение образующихся при этом продуктов.
Одной нз характерных реакций кислородсодержащих адамантанов является расширение шестичленного цикла и образование структуры гомо-
13.Зак.18S6 193
адамантана. Впервые перегруппировка производных адамантана в производные гомоадамантана была обнаружена Штеттером с сотр. при карбоксилировании 1-гидроксиметила дамантана по Коху—Хаафу; реакция приводит к смеси изомерных 1- и З-гомоадамантаикарбоновых кислот [180, 359, 360], причем первая преобладает (80%). Механизм реакции включает перегруппировку первичного неустойчивого метил енадамантиль-ного катиона в более устойчивый третичный гомоадамантильнйй катион.
соон
Расширение шестичленного цикла в структуре адамантана имеет место также прн электролизе натриевой соли 1-адамантануксусной кислоты [361], при взаимодействии 1-гидроксиметиладамантана с трифенил фосфитом и бромом [362] (в последнем случае образуются 3-бромгомоадамантан и 1-бромметиладамантан в соотношении 8:1, выход 40%), а также при взаимодействии адамантанона с диазометаном или цианистоводородной кислотой [363, 364] ив ряде других реакций.
1 -Гнпроксня даман таны легко вступают в реакцию Риттера с различными нитрилами с образованием соответствующих амидов, катализатором и растворителем служит трифторуксусная кислота [365]. С использованием реакции Риттера может быть получен лекарственный препарат мядантан -гидрохлорид 1-аминоадамантана.
Подобно алифатическим спиртам, 1-алкиладамантанолы при окислении образуют альдегиды, причем в зависимости от растворителя окисление тетраацетатом свинца протекает по-раэиому [366, 367]. Прн окислении 1-гидрокснметиладамантана в пиридине образуется 1-адамантилкарбаль-дегид; реакция протекает по гетеролитическому механизму. В среде бензола же имеет место гомолитическое расщепление спирта; образуется первоначально 1-ада мантия-радик ал, который затем дает 1-ацетокси- и 1-фенил-адамантаны. 2-Гидроксиадамантан во всех исследованных средах окисляется по ионному механизму, и в качестве основного продукта образуется ада-мантанон.
В результате окисления (2-адамантил) ал канонов-1 тетраацетатом свинца в зависимости от природы растворителя образуются либо соответствующие альдегиды (36) (в пиридине), либо циклические эфиры (37) и (38) (в бензоле).
(36) (37) (38)
194
Если гидроксильная группа адамантанола соединена непосредственно с ядром, то продуктами реакции являются кетоны протоадамантана [50].
СМЭ
Под действием комплекса BF3 Н3РО4 1-гидроксиадамантан образует смесь трех продуктов — адамантана, адамантанона и вещества неустановленного строения, а 2-гидроксиадамантан — смесь только адамантана и адамантанона. В присутствии чистой ортофосфорной кислоты (100 или 87%-ной) оба спирта превращаются только в адамантан [368]. Подобные реакции адамантанолов протекают также при действии уксусной и трифторуксусной кислот н комплекса BF3 • 2СН3СООН [369], а также в присутствии А1Вгэ и SnCl4 [370].
Кетоны - производные углеводородов ряда адамантана - также вступают во все реакции, свойственные соединениям с карбонильной группой, однако прн этом существенное влияние может оказывать строение кетона. Например, для кетонов типа AdCH2COR и (3-RAd)—COR* в реакции оксн-мнровання в случае нахождения между адамантилом н карбонилом метиленового звена реакционная способность карбонильной группы уменьшается, по мере увеличения размеров заместителя скорость реакции оксн-мнрования также падает [371, 372]. Как н другие окснмы, окснм адамантанона претерпевает перегруппировку Бекмана [373], сам адамантанов, однако, не подвергается енолнзации в условиях, вызывающих енолизацию других циклических кетонов, в частности камфенилона [374, 375].
С реактивом Гриньяра кетоны углеводородов ряда адамантана легко образуют третичные спирты. Как уже отмечалось, реакция широко используется для получения 2-замещенных производных, в том числе 2-алкиладамантанов, но вследствие пространственных затруднений в адамантан-дионе-2,4 реагирует только одна карбонильная группа.
Кетоны легко могут быть переведены в серосодержащие производные [376].
Адамаитантнон способен к димеризации н трнмеризацин с образованием слнропронзводных [377].
195
В сверхсильиых кислотах (HSO3F и SbFs) адамантанон существует в оксоииевой форме [ 378]
Замещенные адамантаноиы широко используются при изучении циркулярного дихроизма (см., например, (379]).
Окисление адамантаноиов (окислитель - СтО3, растворители -СН3СООН, СР3СООН)при комнатной температуре приводит к гидроксике-тоиам, например из адамантаноиа образуется 5-гидрокси-2-адамаитанон (380], гидроксикетоиы легко восстанавливаются действием LiAlH4 в соответствующие диолы (381]. Восстановлением кетонов по Кижнеру— Вольфу можно получить углеводороды, например из 8,9-дегидроадаман-таноиа-2 — 2,4-дегидро адамантан [382].
При действии иа адамантанон йодистого триметилсульфоксоиия образуется устойчивый 2-эпоксиметиленадамантан; из последнего получают 2-адамантан карбоновую кислоту [383].
Весьма интересны и пока мало изучены пероксиды углеводородов ряда адамантана и их производных. трет-Бутилпероксиэфир 1-адамантан-карбоновой кислоты, например, претерпевает распад с образованием ада-мантильного радикала по схеме [384]:
О
//
1-AdC—О—OC(CH3)3-----------> 1-Ad + СО2 + (СН3)3С0‘.
Пероксид адамантилиденадамантаиа вследствие сильного пространственного экранирования весьма устойчив [385].
В ряду 1-адамантанкарбоиовых кислот в условиях кислотного катализа протекает межмолекуляриый обмен карбоксильными группами. При нагревании 1-AdCOOH в конц. П2 SO4 при 60-80° С в течение 7 ч образуется адамантан (5%), 1-гидроксиадамантан (5,3%), адамантанон (2,9%) и 1,3-адамантандикарбоиовая кислота (10,7%)- Из З-гидрокси-1-адаман-таикарбоиовой кислоты образуются 1,3-адамантандикарбоиовая кислота и 1,3-дигидроксиадамантан [386].
Как уже отмечалось, введение в узловые положения адамантана электро-иоакцепторных групп ингибирует образование третичных адамантил-катио-иов с зарядом у незамещенных атомов углерода ядра и снижает реакционную способность соединений. Для протекания реакций ионного типа в этих случаях требуются более жесткие условия. Так, например, взаимодействие 1-AdCOOH с CH3CN, приводящее к образованию 1-ацетамино-З-адамантан-карбоиовой кислоты (выход 77%) протекает лишь в среде смеси H2SO4 и HNO3 [387]. 1-AdCOOH и l-AdCH2COOH окисляются до соответствую-
196
щих З-гндрокси-1-адамаитаикарбоиовых кислот лишь 98%-иой HNO3 [388].
Хлораигидриды 1-адамантанкарбоиовых кислот при декарбоиилирова-иии превращаются в 1-хлорадамантаиы [389].
Весьма интересными в теоретическом и прикладном отношении являются реакции внутримолекулярной циклизации кислородсодержащих производных адамантана, протекающие с участием мостиковых углеродных атомов ядра адамантана. Размыкание цикла в образующихся при этом соединениях позволяет получать таким методом труднодоступные иным путем 1,2-дизамещенные производные адамантана [390, 391].
Н0С1
Реакция производных 1-адамантанкарбоновой кислоты с алкинами приводит к образованию производных циклопеитаноадамаитаиа [393, 394]. Предполагается, что циклизация предшествует 1,5-гидридиый сдвиг.
Некоторые кислородсодержащие производные адамантана (адамантанон, гидроксикетоны и даже 1-AdOH) образуют весьма устойчивые комплексы при их нагревании с 65% HNO3,60°С [395]. Аналогичные, ио менее стабильные комплексы адамантаиои образует также с серной, хлорной и пикриновой кислотами. ИК- и ЯМР-спектры комплексов указывают на наличие водородной связи. 1- и 2-Гидроксиадамаитаны и гидрокси-метиладамантаны образуют также комплексные соединения с участием иоиов переходных металлов (Ti, V, Nb, Сг, Со, Мп и др.) [396].
197
3.6. ПОЛУЧЕНИЕ АЗОТСОДЕРЖАЩИХ ПРОИЗВОДНЫХ АДАМАНТАНОВ
Азотсодержащие производные углеводородов ряда адамантана получают чаще всего обычными методами, основанными на использовании соединений, уже содержащих такие функциональные группы, как бром, хлор, гидрокси. Наряду с этим разработаны методы одностадийного получения азотсодержащих производных адамаитаиа исхода из углеводородов. Одним из таких методов получения аминов ряда адамантана является взаимодействие адамантана или алкиладамантанов с NC13 в присутствии AICI3 в СН2С12 при 0—15° С. Образуются 1-аминоадамантан (l-AdNH2) или соответствующие 1-амииопроизводные алкиладамантанов — метил-, диметил- или триметил адамантанов, выход 60-75% [397, 398]. Выход продуктов может быть несколько повышен при использовании в качестве катализатора системы AlBr-jper-C^^Bf. К числу таких методов относится также реакция с нитрилами. Адамантан или низшие алкиладамантаны реагируют с нитрилами в присутствии грет-бутилового спирта (выход 1-формиламиноадамаитана 76%) [399] под действием брома в 95%-ной H2SO4 [этим путем с ацетонитрилом получают N-(l-адамантил) ацетамид] [400] или в Присутствии нитрозония гексафторфосфата [401]:
1 = AdH1 - AdNHCOR (R = СН3, С2 Н5, С3 Н7, С6 Н5, Сб Н5 СН2 ).
NOPF(
В реакцию Риттера активно вступают также 1-бромадамантаны [240].
1 -AdBr -AdeHSO® ~*l-AdN=CRHSoT -AdNHCOR.
1-Бромадамантаны образуют аминоадамантаны и при прямом аминировании (нагреванием с аминами) [402] или при взаимодействии с мочевиной, 180—240°С [403]. При обработке 1-бромметиладамантаиа формамидом образуется 3-аминогомоадамаитаи [404]. 1,3,5,7-Тетрааминоадаман-тан получают восстановлением 1,3,5,7-тетранитроадамантаиа [405], 1,3-аминоадамантаи — соответственно через 13-динитроадамаиган, образующийся непосредственно при нитровании адамантана [406],
Прямое фотооксимирование адамантана смесью оксида азота и хлора в CCI4 приводит к оксиму адамантанона [36].
С1, + no
AdH -------> Ad=NOH.
CCi«
Аналогично реакция, вероятно, будет протекать в случае низших алкил-адамаитанов. Оксим адамантанона претерпевает бекманскую перегруппировку в тиоиилхлориде с образованием 4-азагомоадамантанона (85%) [117, 407]; перегруппировка протекает и фотохимически при фотолизе оксима в СН3СООН в атмосфере азота при комнатной температуре [408]. ф
СН3 СН3 С-СН3
Ad - Ad - (j-> Ad - N: H-^°; > AdNHCOCH3.
NOH
198
Интересный метод получения аминопроизводных адамантана предложен в работах [409, 410]. Если термолиз этилазидоформиата проводить в адамантане, то реакция протекает с образованием 1- и 2-замещениых производных адамантана (выход 23%) в соотношении 6 : 1.
C-H.OCON,
AdH l-AdNHCOOCa Н5 + 2-AdNHCOOC2 Н5.
При взаимодействии NO2 с адамантаном в CCI4, индуцированном излучением лазера на аргоне (в видимой области), основными продуктами реакции являются 1-иитроадамантан и 1-адамантнлиитрит, образующиеся в примерно равных количествах [410а].
Из других азотсодержащих производных адамантана доступны нитраты адамантанопов. Они легко образуются при взаимодействии адамантана или алкиладамантанов с 90-95 %• ной HNO3 или смесью конц. HNO3 и СН3СООН (например, при 0—30°С за 40—45 мин из адамантана образуется нитрат 1-гидроксиадамантаиа с выходом 45%; наряду с основным продуктом в смеси присутствуют в небольшом количестве 1-AdOH и 1,3-дигид-роксиадамантан) [411, 412]. Наиболее высокий выход нитратов получают при обработке углеводородов смесью конц. HNO3 (d =1,5) и уксусного ангидрида (выход 65—90%). Нитраты спиртов реакционноспособны и позволяют получать с выходом 56—90% гамму функциональных производных [413] , например:
a)R, = CfH,, R,= R,=fi; 6)R,=Rz=Ct1,, R,=fi; S)RI=R2=R,= CH1).
Реагентом — катализатором реакции нитрования обычно служит коиц. H2SO4 (10—40-кратный избыток). В аналогичных условиях проводят реакцию и с нитратами незамещенных адамантанопов [331, 414, 415]. N-Ацил-амииоалкиладамантаны могут быть также получены исходя из соответствующих бром- и гидроксипроизводиых [416,417].
Ар ил адамантаны в условиях реакций нитрования (с помощью HNO3), ацилирования [реагенты: CHjCOCl и А1С1э ] и хлорметилирования [реа
199
генты: (СН2О)Х, НС1, H2SO4] претерпевают замещение в ароматическое кольцо. Так, 6-(1-адамантил) тетралин образует соответственно 8-нитро-, 8-ацетил- и 8-хлорметилпроизводные [418].
Циклические азотсодержащие 2-замещенные адамантаны получают на основе адамантанона. Например, при действии гидроксиламин-О-супьфо-кислоты (ГАСК) и аммиака на 5-замещенные адаманганоны-2 образуются соответствующие диазнридины; при окислении хромовым ангидридом они превращаются в диазирины [419]
(R = ОИ, 01, Вг, СО ОН, СООСП,, CON Мг), а при действии гидразина образуют дизамещенные гидразины, из которых получают диспиро [адамантан-2,2'-тиадиазоп-5',2"-адамантаны] [420].
Из других реакций углеводородов ряда адамантана, затрагивающих Сад—Х-связи н приводящих к азотсодержащим производным адамантанов, следует отметить взаимодействие 1-AdBr с алифатическими амино-спиртамн H2N(CH2)nOH, происходящее с образованием амнноалкокси-адамантанов [421]; фторирование 1-аминоадамантана [реакция идет при фотооблучении прн —78° С и осуществляется с помощью фторокситрнфтор-метана CF3OF] с образованием смеси 1-амино-3- и 1-амино-4-фторадаман-танов [422]; перегруппировку Курциуса ацилазидов адамантана, приводящую к образованию изоцианатов [424]; н, наконец, электрохимическое адамантилнрование N-гетероцикпов [423].
Методы синтеза азотсодержащих производных днамантана рассмотрены также в работах [9, 73,310,311,425].
Непосредственным воздействием на адамантановые углеводороды могут быть получены их производные, содержащие фосфор. Следует отметить, однако, что прямое окислительное фосфорилирование адамантана протекает медленно и с небольшим выходом. Образуются главным образом 2-замещенные фосфоновые производные [426]. Адамантанфосфорные кислоты, их хпорангидриды, адамантилдихпорфосфиты, триадамантилфос-фиты н другие фосфорсодержащие производные получают действием РС1з на 1-AdBr [427] или фосфорилированием 1- и 2-гидроксиадамантана хлорангидридами кислот трехвалентного фосфора [428]. Подобно другим электроноакцепторным заместителям, фосфорильная группа оказывает пассивирующее действие на реакционную способность узловых положений идра адамантана [429].
200
Серосодержащие производные адамантана - тиопы, сульфиды, дисульфиды - получают нз таких функциональных производных адамантана, как спирты и бромиды, известными методами [377,430].
По данным работы [431], реакция сульфирования 1-(1-адамантил) -алканолов-1 серной кислотой в трифтор уксусном ангидриде (или в смеси уксусного ангидрида с СН2С12) сопровождается адамантан-гомоадаманта-новой перегруппировкой — образуются у-сультоны 1-(3-гидрокен-4-гомо-адамантил ) -1-алкилсульфокнслот.
1-Триметилсилиладамантаны получают, используя изомеризацию в присутствии А1Вг3 трициклических углеводородов с трнметилсилильным заместителем [432], например:
Реакции дихлорметилсипильной группы алкил (силилэтил) адамантанов X(CH2)2SiMeCl2, где X = 1-адамантил илн 3,5-диметил-1-адамантил, изучены в работе [433].
ЛИТЕРАТУРА
1. Schleyer P.R., Fort R.C., Watts W.E. et ah // J. Amer. Chem. Soc. 1964. Vol. 86, N 19. P. 4195-4197.
2. Olah J.A., Lucas J. /} Ibid. )968. Vol, 90, N 4. P. 933-938.
3. Бутенко J1JL, Хардин АЛ. // Журн. орган, химии. 1973. Т. 8, № 4. С. 846.
4. Fort R.C., Schleyer P.R. Ц Chem. Rev. 1964. Vol. 64, N 3. P. 277-300.
5. Gleicher GJ., Schleyer P.R. Ц J. Amer. Chem. Soc. 1967. Vo). 89, N 3. P. 582-593.
6. Schleyer P.R., Nicholas R.D. jI Ibid. 1961. Vo). 83, N 12. P. 2700-2707.
J.SchoUkopf U. // Angew, Chem. 1960. Bd. 72, N 5. S. 147-159.
201
8. Gund T.M., Schleyer P.R., Unruh G.D., Gleicher GJ. // J. Org. Chem. 1974. Vol. 39, N 20. P. 2995- 3003.
9. Courtney T„ Johnston D.E., McKervey M.A., Rooney J J. 11 J. Chem. Soc. Perkin Trans. Part 1.1972. N 21. P. 2691-2696.
10. Hamilton C.R., Johnston D.E., NcKervey M.A., Rooney J.J. // J. Chem. Soc. Chem. Commun. 1972.H. 22.P. 1209-1210.
Il. Hollowood F.t Karim A., McKervey M. A. et al. // Ibid. 1978. N 7. P. 306-308.
12. Olah G.A., Liang G. // J. Amer. Chem. Soc. 1973. VoL 95, N 1. P. 194-197.
13. Prakas/i G.K.S., Rawdach T.N., Olah G.A. // Angew. Chem. 1983. Bd.95,N5. S. 356-367.
13a. Schleyer P.R., Bremer M., SchotzK.et aL // Chem. and Industry. 1987. Vol. 65, N 16. P. 60-61.
14. Шоково Э.А., Клоп В.В. // Нефтехимия. 1975. T. 15, № 1. С. 82-87.
15. Olah G.A., Svoboda J J, KuA.T. // Synthesis. 1973. N 8. P. 492-493.
16. Applequist D., Kaplan L. // J. Amer. Chem. Soc. 1965. VoL 87, N 10. P. 2194-2200.
17. Chick W.H., Ong S.H. // Chem. Commun. D. 1969. N 5. P. 216-217.
18. Lorand J.P., Chodroff S.D., Wallace R.W. // J. Amer. Chem. Soc. 1968. Vol 90, N 19. P. 5266-5267.
19. РюхардтХ. // Успехи химии. 1978. T. 47, № 11. С. 2014-2043.
20. Engel P.S., Chae W.-K., Baughman Sh.A. et al. // J. Amer. Chem. Soc. 1983. Vol 105, N15.P. 5030-5034.
21. Tabushil, HamuroJ., OdaR. // J. Chem. Soc. Jap. 1968. VoL 89, N 8. P. 789-794.
22. Tabushi L, Hamuro J., Oda R. // J. Amer. Chem. Soc. 1967. Vol. 89, N 26. P. 7127-7129.
23. Tabushil, HamuroJ, Oda R. // Org. Chem. 1968. Vol. 33, N 5.P. 2108-2109.
24. Gleicher G.J., Jackson J.L., Owens P.H., Unruh JD. // Tetrahedron Lett. 1969. N 10. P. 833-836.
25. Muller E.t Trensc U. // Ibid. 1967. N 49. P. 4979-4982.
26. Muller E.. Fiedler G. // Chem. Ber. 1965. Bd. 98, N U.S. 3493-3496.
27. Шепелева E.C., Олейник ДМ., Багрий EJf., Санин П.И. // Тр. V конф, по химии и применению фосфорорган. соединений. М.: Наука, 1974. С. 369-373; РЖХим. 1975. 12Ж405.
ZS.GeeD.R., FabesL., WanjK.S. //Chem. Phys. Lett. 1970. Vol. 7, N 2. P. 311-313.
29. Tabushi I, Aoyama Y„ Koto Sh. et al // J. Amer. Chem. Soc. 1972. Vol. 94, N 4. P. 1177-1183.
ЗО .Оа M., Watanabe M., Ichinose M., Sakurai H. // Ibid. 1982. VoL 104, N 13. P. 37 62-3764.
31. Koch V.R., Gleicher GJ. // Ibid. 1971. Vol 93, N 7. P. 1651-1661.
32. Marron N.A., GanoJ.E. // Ibid. 1976. Vol 98, N 15. P. 4653-4655.
33. Edwards G.J., Jones S.R., Mellor J.M. // J. Chem. Soc. Chem. Commun. 1975. N 20. P. 816-817-
34. Sasaki T, Eguchi S., Ruy 1.Н., Hirako Y. Ц Tetrahedron Lett. 1974. N 23. P. 2011-2014.
35. Sasaki T„ Eguchi S, Hirako Y. Ц J. Org. Chem. 1977. Vol. 42, N 18. P. 2981-2985.
Зб. ЛГалгейкд И.Б., Янковская И.С., Полис Я.Ю. // Журн. общ. химии. 1971. Т. 41, №7-С. 1633-1635.
37. Янковская И.С., Мажейка И.Б., Крускоп ДК., Полис Я.Ю. // Там же. 1973. Т. 43, №3. С. 490-492.
38. Воронков М.Г., Фешин ВЛ., Полис Я.Ю. // Теорет. и эксперим. химия. 1971. Т. 7, №4, С. 555-558.
39. Fort R.C., Schleyer P.R. // J. Amer. Chem. Soc. 1964. Vol. 86, N 19. P. 4194-4195.
40. Schleyer P.R., Woodworth C. W. 11 Ibid. 1968. Vol. 90, N 23. P. 6528-6530.
41. BoderN., DewarM.J.S., Worley S.D. // Ibid. 1970. Vol. 92, N 1. P. 19-24. ’
42. Broxton TJ., Capper G„ Deady L. IP. at aL // J. Chem. Soc. Perkin Trans. Part II. 1972. N9.P. 1237-1240.
43. Воронков М.Г., Фешин ВЛ. // Теорет. и эксперим. химия. 1971. Т. 7, № 4. С. 444-453.
44. Лейбзон В.Н., Майрановский С.Г., Краюшкин ММ. и др. // Изв. АН СССР. Сер. хим. 1971. №4. С. 764-769. .
202
* 45. Лантвоев ВМ. // Журн. орган, химии. 1977. Т. 13, № 1. С. 88-92.
46. Лантвоев В М. // Там же. 1976. Т. 12,№ 12. С. 2516-1523.
47- Степанова Г.Ю., Блейхер Я.И. Ц Укр. хим. журн. 1973. Т. 39, № 11. С. 1176-1179. { 48. Fujimoto Н., Kitagawa Y„ Hao Н, Fukui К. // Bull. Chem. Soc. Jap. 1970. VoL 43,
; Nl.P. 52-56.
49. Bingham R.C., Schleyer P.R. // J. Amer. Chem. Soc. 1971. Vol. 93, N 13. P. 3189-; 3199.
50. Калт A., McKervey М.Д., Engler E.M., Schleyer P.R. // Tetrahedron Lett. 1971. ! N43.P. 3987-3990.
51. Степанов Ф.Н., Даниленко ГМ. // Журн. орган, химии. 1966. Т 2, № 9. С. 1630-; 1632.
52. Краюшкин ММ., Севостьянова ВЛ., Даниленко ГМ. // Изв. АН СССР. Сер. хим. 1969. №12. С. 2844-2845.
53. Даниленко ГМ., Краюшкин ММ., Севостьянова ВВ. // Там же. 1970. № 2. С. 444-‘ 445.
54. Степанов Ф.Н., Сребродолъский ЮМ. // Журн. орган, химии. 1966. Т. 2, № 9.
i С. 1633-1634.
? 55. Perkins R., ПЬЪ Р., Reid К., Doman /. // Chem. and Ind. 1981.N 16. P. 571-572.
56.Bentley T.W., Carter G.E. // J. Amer. Chem. Soc. 1982. VoL 104, N 21. P. 5741-5747.
! 57.Пономарева Э.А., Тарасенко ПВ., Юрченко А.Г., Дворко ГФ. // Журн. орган,
химии. 1983. ТЛ9, №у3. С. 548-561.
* S8.Surtfco D.E., Hfrsl-Starcevic S., Pollack S.K., Hehre W.J. Ц J. Amer. Chem. Soc. 1979. [ VoL 101, N 21. P. 6163-6170.
59. Назарова МЛ., Подхалюзин A.T. // Журн. орган, химии. 1981. Т. 17. № 1. С. 128-j. 132.
! 60. Bone JA., Pritt J.R., Whiting M.C. 11 J. Chem. Soc. Perkin Trans. Part II. 1975. N 13.
P. 1447-1452.
( 61. Lenoir D., Schkeyer P.R. Ц Angew. Chem. 1971. Bd. 83, N 22. S. 918.
; 62. Bone J A., WhitbagM.C. // Chem. Commun. D. 1970. N2.P. 115-116.
63. Fry J.L., Lancelot CJ., Lam L.KM. et al. // J. Amer. Chem. Soc. 1970. VoL 92, N 8. ? P. 2538-2540.
64. Fry J.L., Harris JM., Bingham R.C, SchleyerP.R. //Ibid. P. 2540-2542.
65. Harris JM„ Jagan J.F., Waldon FA., Clerk D.C. // Tetrahedron Lett. 1972. N 30. P. 3023-3026.
I 66. Faulkner D., McKerwey MA., Lenoir D. et al. // Ibid. 1973. N 9. P. 705-708.
i 61. Lenoir D. //Chem. Ber. 1973. Bd. 106, N 7. S. 2366-2378.
’ 68. Lenoir D„ Misow P„ Hyson E. et aL // J. Amer. Chem. Soc. 1974. VoL 96, N 7.
P.2157-2164.
69. Lenoir D., Hall R.E., Schleyer P.R. //Ibid. P. 2138-2148.
70. Lenoir D., Raber DJ., Schleyer PM. // Ibid. P. 2149-2156. > 71. FdrcasiuD. // Ibid. 1976. VoL 98, N 17. P. 5301-5305.
72. Osawa E., Engler EM., Godleski SA. et aL // J. Org. Chem. 1980. VoL 45, N 6. P. 984-991.
73. Gund TM., Nomura M„ Schleyer P.R. // Ibid. 1974. VoL 39, N 20. P. 2987-2994. ' 14. Janku J., Burkhard J., Vodicka Z. Ц Ztsdur. Chem. 1981. Bd. 21, N 9. S. 325-326.
г 75. Новикова ММ., Юрченко А.Г. // Весгн. Киев. политехи, ии-та. Хим. машинострое-' ние и технология. 1980. № 17. С. 8-10; РЖХим. 1981. 12Ж115.
76. Bingham R.C., Schleyer P.R., Lambert Y., DeslongchampsP.//Canad.J.Chem.. 1970. VoL 48, N23. P. 3739-3741.
177. Казанский БА., Шокова ЭА., Коростелева ТВ. // Изв. АН СССР. Сер. хим. 1968. № И. С. 2640-2642.
78. Казанский БА., Шокова ЭА., Коростелева ТВ. // Там же. С. 2642-2645.
19. Антонова ЕА., Демина Н.Г., Пичужкнн ВЛ., Сараева ВВ. // Химия высоких энергий. 1974. Т. 8, № 1. С. 49-51.
80. Podkhalyuzin А.Т., Vereshchinskii I. V. // Proc. 1П Tihany symp. radiat. chem. Budapest, 1971. VoL 1. P. 115-121; Chem. Abstr. 1973. VoL 78.110179h.
t 81. Зеделашвили EM., Шаншиашвили М.И., Ершов Б.Г. // Сообщ. АН ГССР. 1974. Т. 75, № 3. С. 605-608; РЖХим. 1975. 5Ж94.
203
82. Викулин ВВ., Клиншпонт Э.Р., Подхалюзин А.Т. // Химия высоких энергий. 1978. Т. 12, № 1.С. 85-86.
83. Слободин Я.М., Ковязин В.Е., Фролова Р.И. и рр. // Журн. орган, химии. 1975. Т.Н, №10. С. 2218.
84. Слободин ЯМ., Ковязин В.Е., Варенье Г.В. и др. // Там же. 1978. Т. 14, № 3. С. 542-544.
$5. Казанский БА., Шокова ЗА., Коростелева ТВ. // Докл. АН СССР- 1969. Т. 187, №5. С. 1056-1059. f
86. Пат. 3418387 США, МКИ2 С07 С. Process for production of adamantane I S. Hala, S. Landa, Z.Weidenhoffer// Chem. Abstr.. 1968. Vol. 69.106010t.
87. Пат. 3489817 США, МКИ2 С07 С 3/58. Hydrodealkylation of alkyladamantanes / E.C. Capaldi, H. Bromall, L.N. Leum, M. Shalit, H. Shalit, D. Hill // Chem. Abstr. 1970. VoL 72.89901b.
88. Weidenhoffer Z.r Hala S., Landa S. // Sb. VSCHT Praze D. 1971. Sv. 22. S. 85-94; Chem. Abstr. 1972.Vol.76.72091x.
89. Меликадзе ЛЛ., Уша pay л и ЗА., Кортави ЛМ. // Сообщ. АН ГССР. 1976. Т, 84, № 2. С. 385-388; РЖХим. 1977. 12Б1177.
90. Берман С.С., Денисов Ю.В., Мураховская А.С. и др. // Нефтехимия. 1974. Т. 14, № 1. С. 3-8.
91. Grubmuller Р., Schleyer P.R., McKervey МА. // Tetrahedron Lett. 1979. N 2. Р. 181-184.
92. Hala S.. Landa S. // Coll. CzechosL Chem. Commun. 1964. Vol. 29, N 5. P. 1319-1322.
93. Schneider A., Warren R.W., Sanovsky EJ. 11 J. Org. Chem. 1966. Vol. 31, N 5. P. 1617-1625.
94. Петров АлА. Химия нафтенов. М.: Наука, 1971- 388 с.
95. Казанский БА.Г Шокова ЗА., Коростелева ТВ. // Докл. АН СССР. 1970. Т. 191, №4. С. 831-834.
96.N omura М., Schleyer P.R., Azz А. А. // J. Amer. Chem. Soc. 1967. Vol. 89, N 14. P. 3657-3659.
97. Sasaki T., Eguchi S., Тоги T. // Tetrahedron Lett. 1968. N 38. P. 4135-4138.
98. Janjatovic J., SkareD., MajerskiZ. // Org. Chem. 1974. Vol. 39, N 5. P. 651-654.
99. Петров Ал.А. Каталитическая изомеризация углеводородов. М.: Изд-во АН СССР, I960. 215 с.
100. Majerski Z„ Schleyer P.R., Wolf А. Р. // J. Amer. Chem. Soc. 1970. Vol. 92, N 19. P. 5731-5733.
101. Schleyer P.R., Lam L.K.M, Raber D. J. et al. // Ibid. N 17. P. 5246-5247.
102. Majerski Z., Liggero S.H., Schleyer P.R., WolfA.P. // Chem. Commun. D. 1970. N 23. P. 1596-1597.
103. Vogel P.r Saunders M., Thielecke IP., Schleyer P.R. 11 Tetrahedron Lett. 1971. N 18. P. 1429-1430.
104. Oleh G.A, Liang G., Mateescu G.D. // J. Org. Chem. 1974. VoL 39, N 25. P. 3750-3754.
105. Sinnott M.L., Whiting MC. //J. Chem. Soc. Perkin Trans. Part II. 1975. N 13. P. 1446-1447.
106. 7^yZ£., Saba SA. Ц Tetrahedron Lett. 1982. Vol. 23, N 17. P. 1743-1746.
107. Oleh G.A., Prakash G.K.S., Liang G. et aL // J. Org. Chem. 1982. Vol. 47, N 6. P. 1040-1047.
108. Faapuu ЕЙ, Фрид Т.Ю., Санин П.И. Ц Нефтехимия. 1980. Т. 20. №6. С. 812-818.
109. Leone R.E., Schleyer P.R. // Angew. Chem. 1970. Bd. 82, N 22. S. 889-919.
110. OlahG.A., Liang G., Babiak K.A. et al. // J. Amer. Chem. Soc. 1978. Vol. 100, N 5, P. 1494-1500.
lll. iMtawy R.K., Ford T.M., Prakash G.K.S., OlahG.A. // Ibid. 1980. Vol. 102, N 6. P. 1865-1868.
112. SabaS.A., Fry J.L. // Ibid. 1983. Vol. 105, N 3. P. 533-537.
113. Федоренко A.B., Довгань Н.Л. // Вести, Киев, политеха, ин-та. Хим. машиностроение и технология. 19 83. № 20. С. 11-15.
114. Багрий Е.И, Фрид Т.Ю., Санин П.И. Ц Нефтехимия. 1978. Т. 13, №6. С, 800-807-115. Stetter И, Wulff С. // Chem. Вег. 1962. Bd. 95, N 9. S. 2302-2304.
204
116. Степанов Ф.Н., Столяров З.Е. Ц Журн. орган, химии. 1970. Т. 6, № 6. С. 1186-1189.
117. Степанова Г.Ю., Диколенко В.М., Диколенко ЕЙ, Джетуа Ф. Ц Там же. 1974. Т. 10, №7. С. 1455-1457-
118. Majerski Z., Redwanly C.S. // J. Chem. Soc. Chem. Commun. 1972. N 11. P. 694-695.
119. Fort R.C. Adamantane: The chemistry of diamond molecules. N.Y.: Dekker, 1976. 385 p.
120. Севостьянова B.B., Краюшкин ММ., Юрченко А.Г. // Успехи химии. 1970. Т. 39, № 10. С. 1721-1753.
121. Yang К.Н, Kimoto К., Kawanisi М. // Bull. Chem. Soc. Jap. 1972. Vol 45, N 7. P. 2217-2219.
122. Boelema E, Wieringa EH., Wynberg H, Strafing /. // Tetrahedron Lett. 1973. N 26. P. 2377-2380.
123. Stepanov F.N., Suchowerchow W.D. // Angew. Chem. 1967. Bd. 79, N 8. S. 860.
124. Юрченко А.Г. Исследование взаимных превращений производных адамантана и бицикло [3,3,1] нонана: Дис.... д-ра хим. наук. Киев, 1974. 333 с.
125. Raber D.E, Fort R. С, Wiskott Е. et al. // Tetrahedron. 1971. Vol. 27, N 1. P. 3-18.
126. Stetter H, Tillmann V. // Chem. Ber. 1972. Bd. 105, N 2. S. 735-739.
127. Багрий Е.И. Исследование углеводородов ряда адамантана: Дис.... д-ра хим. наук. М., 1977. 385 с.
128. Basset D.W., Hagood HW. // J. Phys. Chem. 1960. Vol. 64, N 6. P. 769-773.
129. Пат. 3655782 США, МКИ1 С 07 С. Каталитическое диалкилирование галоидада-мантанов = Catalytic dialkylation of haloadamantanes/ E.R. Moore // Chem. Abstr. 1972. Vol. 77. 5O53u.
130. Шокова Э.А., Кнопова С.И., Казанский Б.А. // Нефтехимия. 1972. Т. 12, № 3. С. 314-317-
131. Шокова Э.А., КлопВ.В. // Там же; 1975. Т. 15, № 2. С. 206-211.
132. Федотов НС., Болосова Е.В., Эверт Г.Е. н др. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 106.
133. Федотов НС., Болосова Е.В., Эверт Г.Е. и др. // Там же. С. 107.
133а . Болестова Г.И, Парнес З.Н., Вольпин М.Е. // II Всесоюэ. симпоз. по гомоген. катализу; Тез. докл. Черноголовка, 1988. С.12.
1336. Ахрем ИС., Орлинков А.В., Витт С.В» н др. // Докл. АН СССР. 1986. Т. 288, №1.С. 130-134.
134. Багрий Е.И, Соловьев В.Н, Заикин В.Г., Санин ПИ. // Нефтехимия. 1975. Т. 15, № 1.С. 88-94.
135. Пат. 3382288 США Alkylated adamantanes / A. Schneider // Chem. Abstr. 1968. Vol. 69. 3S584v.
136. Фрид Т.Ю., Багрий Е.И, Кошевник А.Ю. н др. // Нефтехимия. 1972. Т. 12, №4. С. 511-517-
137. Пат. 1570330 Франция, МКИ1 С 07 С. Adamantane nuclei joined by short polymethylene chains / A. Schneider // Chem. Abstr. 1970. VoL 72. 89899g.
138. Соловьев B.H., Багрий Е.И., Санин П.И. // Нефтехимия. 1974. Т. 14, №6. С. 817-821.
139. Пат. 3646233 США МКИ1 С 07 С 3/54. Reaction of paraffins with adamantane compounds j R.E. Moore j/ РЖХим. 1972. 24П347П.
140. Пат 2055732 ФРГ, МКИ1 C 07 C. Alkyl adamantanes / l.N. Duling, F.P. Glazier, D.S. Gates, R.E. Moore // Chem. Abstr. 1971. Vol. 75, 109956г.
141. Пат. 4008251 США, МКИ1 С 07 D 313/14, 311/80. Reation of alkyladamantane compounds to form products having two linked adamantane nuclei / R.E. Moore, A. Schneider // РЖХим. 1977. 20П225П.
142. A.c. 302008 СССР, МКИ С 07 С 23/39, 5/24. Способ получения алкилированных адамантанов / Е.И. Багрий, П.И. Санин, Т.Ю. Фрид // Б.И. 1972. № 28. С. 193.
143. Багрий Е.И., Фрид Т.Ю., Санин ПИ // Изв. АН СССР. Сер. хим. 1970. № 2. С. 498.
144. Сагинаев А.Т., Багрий Е.И. // Нефтехимия. 1982. Т. 22, №6. С. 729-734.
145. Фрид Т.Ю., Соловьев В.Н, Заикин В.Г. и др. // Там же. 1978, Т. 18, № 1. С. 39-43.
146. Багрий Е.И., Фрид Т.Ю., Соловьев В.Н и др. // Кинетика и катализ. 1977. Т. 18. №6. С. 1604.
205
147. А-с. 287933 СССР, МКИ С 07 С 23/39, 5/24. Способ получения полиметипада-мантанов / Е.И. Багрий, П.И. Санин // Б.И. 4970. № 36. С. 28.
148. А-с. 376346 СССР, МКИ С 07 С 13/62. Способ получения смеси метиладаманта-нов/ А.Т. Подхалюзин,Ю.П.Щелков //Б.И. 1973.№ 17.С. 70.
149. Гала С, Кураш М„ ЗахарджП. //Sb. VSCHT Proze D. 1976. Sv. 33. S. 305-321;
150. A.C. 915407 СССР, МКИ С 07 С 13/615. Способ получения смесн моно- н поли-метиладамантанов / Е-И. Багрий, Т.Н. Долгопопова, П.И. Санин // Б.И. 1985. №. 2. С. 207.
151. Долесговд RH., Парне с З.Н., Курсанов Д.Н. // Журн. орган, химии. 1983. Т. 19, № 2. С. 339-343.
152. Болестова Г.И., Парнес З.Н., Латыпова Ф.М., Курсанов Д.Н, // Там же. 1981. Т. 17, № 7. С. 1357-1363.
153. Курсанов ДЛ„ Болестова Г.И., Катаев ВЛ., Парнес З.Н. // Изв. АН СССР. Сер. хим. 1979. №8. С. 1919-1920.
154. Пат. 3437701 США, МКИ3 С 07 С. Алкиладамантаны = Alkyladamantanes / Е.С. Capaldi//Chem. Abstr. 1969. Vol. 70. 114713k.
155. StetterH., Weber J., Wulff C. //Chem. Ber. 1964. Bd. 97, N 12. S. 3488-3492.
155a. Кравцов Д.Н., Ильчевская ВД., Петровский П.В., Гореликова И.И. // Изв. АН СССР. Сер. хим. 1988. №5. С. 1130-1133.
156. Newman Н. // Synthesis. 1972. N 12. Р. 692- 693.
157.Н овиков С.С., Радченко С.С., Новаков ИЛ.идр. // Функциональные органические соединения н полимеры. Волгоград, 1977- С. 6-9; РЖХим. 1977. 22Ж53.
158. Юрченко А.Г., Рарог Б.Г, Хоткевич А.Б., Топчий ВЛ. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 84.
159. Козлов О.Ф., Юрченко А.Г. // Вести. Киев, политехи, ии-та. Хим. машиностроение н технология. 1977. № 14. С. 3-5.
160. Пат. 3594427 США, МКИ3 С 07 С. Adamantanebisphenols / R.E. Moore // Chem. Abstr. 1971. VoL 75. 118780f.
161. Степанов Ф.Н., Столяров З.Г. // Журн. орган, химии. 1969. Т. 5, № 3. С. 537-541.
162. Sasaki Т., UsukiA., OhnoM. //J. Org. Chem. 1980. VoL 45, N 18. P. 3559-4564.
163. Vodicka L., Burkhard J„ Janku J. // Coll. CzechosL Chem. Common. 1980. VoL 45, N3.P. 835-842.
164.К овальB.B., ШоковаЭ.Л II Журн. орган, химии. 1981. Т. 17, № 1. С 109-116.
165. Зосим Я.А., Юрченко А.Г. //Там же. Т. 16, № 4. С. 887—888.
166. Пат. 3563919 США, МКИ3 С 08 G 33/00. Copolymers of adamantane and benzene / A. Schneider // РЖХим. 1971. 20С284П.
167. Лерман Б.М., Арефьева З.Я., Галин Ф.З., Толстиков Г.А АН СССР. Сер.
хим. 1984. № 3. С 720-721.
168. Margosiaп О., SpeierJ., Kovacik Р. // J. Org. Chem, 1981. Vol. 46, N 7. P. 1346-1350.
169. Гар Т.К, Чернышева O.H., Колесникова E.M. н др. // Журн. общ. химии. 1983. Т. 53, №3. С. 574-577.
170. Подхалюзин А.Т., Викулин В.В. // Нефтехимия. 1974. Т. 14, №6. С. 822-827.
171. Tabushil., FUkunishi К. //J.Org. Chem. 1974. VoL 39.N25. P. 3748-4750.
172. Подхалюзин A.T., Викулин ВД., Ворощинский И.В. // Докл. АН СССР. 1975. Т. 22, №2. С. 381-383.
173. Брель А.К., Рахимов А.И. Соловьева ВЛ. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 66.
174. Морозов В.А,, Алимирзоев ФЛ., Степанянц А.У., Подхалюзин А.Т. // Химия высоких энергий. 1980. Т. 14, № 5. С. 392-399.
175. Подхалюзин А.Т., Морозов ВЛ., Янкелевич А.З. // Докл. АН СССР. 1976. Т. 228, №3, С 609-612,
176.М орозов ВЛ., Подхалюзин А.Т. // Химия высоких энергий. 1979. Т. 13, № 1. С. 27-33.
177. Л^муэктое В.А., Бегишев И.Р., ПодхалхЯзин А.Т. н др. // Там же. 1977. Т. И, №4. С. 309-315.
178. Пдлузктов В.А., Бегншев И.Р., Подхалюзин А.Т. н др. // Докл. АН СССР. 1976. Т. 228, № 2. С 401-404.
179. Подхалюзин А.Т., Морозов ВЛ., Янкелевич А.З. // Там же. С. 609-612.
18й StetterH., Schwarz Н., Hirschham А. // Chem. Ber. 1959. Bd. 92, N 7. S. 1629-1635.
206
181. Stetter ft, Wulff С/j Angew.Chem. 1960. Bd.72,N U.S. 351.
182. Landa S., Kriebei S.t Knobloch К // Chem. hsty. 1954. Sv. 48, N 1. S. 61-64.
183. Landa S., Kamycek Z. Ц CoB. CzechosLChem. Commun. 1959. Vol. 24, N 4. P. 1320-1326.
184, Stetter H„ Schwarz M„ Hirschhom A. // Angew. Chem. 1959. Bd. 71, N 13. S. 429-430.
184a . Делимарский P.E., Родионов ВЛ., Юрченко АЛ. // Укр. хим. журн, 1988. Т. 54, №4. С. 437-8.
1846, Довгань Н.Л., Лихотворик НЛ., Машковский В.Н. // Вести. Киев, политехи, ин-та. 1988. № 25. С. 9-10.
185. StetterH., Mayer J., Schwarz M„ Wulff C //Chem. Вет. i960. Bd, 93, N l.S. 226.
186. Stetter ft, Wulff C //Ibid. N 6. S. 1366-1371.
187.Ю рченко А.Г., Баклан В.Ф., Дьяковская B.M., Кулик НИ //Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 35.
188. Степанов Ф.Н, Юрченко АЛ., Мурзинова ЗЛ. н др. // Журн. орган, химии. 1972. Т. 8. №9. С. 1837-1841.
189. Юрченко АЛ., Дьяковская В.М„ Баклан В.Ф. // Там же. 1984. Т. 20. №10. С. 2239-2240.
190. Ола ГЛ. // Успехи химии. 1975, Т. 44, № 5. С. 793—867.
191. Alford J.R., Grant D., McKervey MA. //J. Chem. Soc. C. 1971. N 5. P. 880-883.
192. Рагу ель Б.И, Полис Я.Ю., Шатц ВД., Гаварс М.П. // Науч. конф, по химии орган, поннэдранов: Тез. докл. Волгоград, 1981. С. 87.
193. Баклан В.Ф., Фесенко Т.Е., Дьяковская В.М., Кравченко ЛЛ. // Там же. С. 40.
194. // Tetrahedron Lett. 1975. N 49. P. 4403-4404.
195. Степанов Ф.Н., Баклан В.Ф., Исаев СД. // Жури, орган, химии. 1965. Т. 1, № 2. С. 280-283.
196. Landa S., Hlavaty /. // Ztschr. Chem. 1971. Bd. 11, N 3. S. 110.
197. Schleyer PR.. Buss V. //J. Amer. Chem. Soc. 1969. Vol. 91, N 21. P. 5880-5882.
198. Александров A.M., Даниленко Г.И., Ягупольский Л.М. // Журн. орган, химии. 1973. Т. 9, №5. С. 951-953.
199. Сорочинский А.Е., Александров А.М., Кухарь В.П., Краснощек А.П. // Там же.
1979. Т. 15, № 2. С. 445-446.
200. Назарова М.П., Алимирзоев Ф.А., Степанянц А.У., Подхалюзин А.Т. // Там же.
1980. Т. 16, №9. С. 1851-1857.
201. Назарова М.П., Подхалюзин А.Т. // Кинетиканкатализ. 1980. Т. 21, № 5. С. 1355.
202. Ас. 316680 СССР, МКИ С 07 С 23/20. Способ получения 1-бром-З-бромметилада-мантана / Ф.Н. Степанов, Л.А. Зосим // Б.И. 1971. № 30. С. 77.
203. Inamoto J., Kadono Т„ Takaishi N. // Synth. Commun. 1973. Vol. 3, N 2. P. 147-151; РЖХим. 1974. 2Ж158.
204. Perkins R.R., Pincock R.E. // Canad. J. Chem. 1978. Vol. 56, N 9. P. 1269-1272; РЖХим. 1978, 20Б1431.
205. Лерман Б.М., Арефьева З.Я., Кузыев А.Р., Толстиков Г.А. // Изв. АН СССР. Сер. хим. 1971. №4. С. 894.
206. Лерман Б.М., Арефьева З.Я., Кузыев А.Р. и др. // Там же. 1972. № 8. С. 1815-1819.
207. Пат. 1913998 ФРГ, МКИ3 С 07 С. Monogelogenation of alkyladamantane / A. Schneider // Chem. Abstr. 1970. Vol. 72. 21405n.
208. Tabushi I., HamuroJ., Oda R. // J. Chem. Soc. Jap. 1968. VoL 89, N 8. P. 794-797.
209. Kovacick P., ChangJ.-H.C. //Chem. Commun. D, 1970.N21.P. 1460-1461.
210. KovacickP., Chang L-HC //J. Org. Chem. 1971. Vol. 36, N 21. P. 3138-3145.
211. Khullar K.K., Bauer L. // Ibid. N 20. P. 3038-3040.
212. Yurchenko A.G., Zocim L.A., Dovgan' N.L., Verpovsky N.S. //Tetrahedron Lett. 1976. N 52. P. 4843-4846.
213. Smith СИ, BuUups W.E. //J. Amer. Chem. Soc. 1974. Vol. 96, N 13. P. 4307-4311.
214. Schwartz A.L., Moon S. // J. Org. Chem. 1975. Vol. 40, N 7. P. 865-869.
215. Sasaki T.r Eguchi Sh., Mizutani M. // Org. Prep. Proc. Intern. 1974. VoL 6, N 2. P. 57-62.
216. A.c. 598859 СССР. МКИ С 07 С 23/40, 23/18. Способ получения фторозамещенных адамантана / АП. Хардин, АД. Попов, П.А. Протопопов // Б.И. 1978. № 11. С. 69.
207
217. Пат, 3641167 США, МКИ3 С 07 С. Highly fluorinated alkyladamantanes / R.E. Moore, J. Janovski // Chem. Abstr. 1972. Vol. 76. 126483x.
218. Пат. 4041086 США, МКИ2 С 07 С 23/18. Process of manufacture of fluorinated alkyladamantanes / R.E. Moore, E.J. Janovski// РЖХим. 1978. 7284П.
219. Шокова Э.А, Кнопова С. И., Нгуен Данг Къен, Казанский Б.А. // Изв. АН СССР. Сер. хим. 1973. № 6. С. 1429-1430.
220. Пат. 3714273. США. МКИ2 С 07 С. Fluorine-containing adamantanes and bicyclo [2,2,2] octanes / C.W.Tullock // Chem. Abstr. 1973. Vol. 78. 110713c.
221. Rozen S, BrandM. // J. Org. Chem. 1981. Vol. 46, N 4. P. 733-736.
222. Пат. 2808112 ФРГ, МКИ2 С 07 С 23/20. Perfluorination of cyclic hydrocarbons / R.E. Moore // Chem. Abstr. 1979. Vol. 90. 22436g.
223. Robertson G., Liu E.K.S., Lagow R.J. // J. Org. Chem. 1978. Vol. 43, N 26. P. 4981-4983.
224. Сорочинский A.E., Александров A.M., Кухарь В.П. и др. // Журн. орган, химии. 1979. Т. 15, № 12. С. 2480-2484.
225. Александров А.М., Сорочинский А.Е, Краснощек А.П. // Там же. № 2. С. 336-342.
226. Александров А.М., Кухарь В.П., Даниленко Г.И., Краснощек А.П. // Там же. 1977-Т. 13, №8. С. 1629-1634.
227. Сорочинский А.Е., Александров А.М., Гамалея В.Ф., Кухарь В.П. // Там же. 1981. Т. 17, №8. С. 1642-1648.
228. Сорочинский А.Е„ Александров А.М., Кухарь В.П. Ц Там же. 1982. Т. 18, № 1. С. 228-229.
229. Беленький Г.Г.Г Савичева Г.И., Герман Л.С. // Изв. АН СССР. Сер. хим. 1978. №6. С. 1433-1434.
230 Geluk H.W., Schlatmann J.L.M.A. // Tetrahedron. 1968. Vol. 24, N 15. P. 5361-5368.
231.Ю рченко А.Г, Федоренко T.B., Родионов В.Н. // Журн. орган, химии. 1985. Т. 21, №8. С. 1637-1677.
231а. Юрченко А.Г., Федоренко Т.В. // Там же. 1987. Т. 23, № 5. С. 970-976.
2316. Федоренко Т.В. Магнийорганическне производные ряда адамантана: Автореф. дис.... канд. хим. наук. Киев. 1988. 28 с.
232. Wieringa J.H., Strating Z. Wynberg Н. // Synth. Commun. 1972, Vol. 2, N 4. P. 191-195; Chem. Abstr. 1972. VoL 77.126727a,
233. Molle G., Briand S., Bauer P., Dubois J.E. // Tetrahedron. 1984. Vol. 40, N 24. P. 5113— 5119.
234. Wieringa J.H., Wynberg H„ Strating J. // Synth, Commun. 1971. Vol. 1, N 1. P. 7-10; РЖХим. 1971. 22Ж283.
235. Но Б.И., Сон B.B., Белякова T.B., Куликова Н.И. // Журн общ. химии. 1982. Т. 52, №9. С. 2139-2140.
236. Гуц С.С., Суховерхое В.Д. // Вести. Киев, политехи, ии-та. Хим. машиностроение и технология. 1969. №6. С. 80-83; РЖХим. 1970. 10Ж221.
237. Reinhardt H.F. // J. Polym. Sci. В. 1964. Vol. 2,N5.P. 567-568.
238. McKervey M.A., Grant D., Hamill H // Tetrahedron Lett. 1970. N 23. P. 1975-1977.
239. Степанов ФЛ., Даниленко Г.И., Бузаш B.M., Дайси К // Журн. орган, химии. 1969. Т. 5, № 12. С. 2187-2189.
240. Sasaki Т, EguchiS, Тоги Т. // Bull. Chem. Soc. Jap. 1968. Vol. 41, N 1. P. 236-238.
241. Sasaki T, EguchiS, Тоги T. // Ibid. Vol 42. N 12. P. 3613.
242. Janku Z, Burkhard Z, Landa S. // Sb. VSCHT Praze D. 1973. Sv. 29. S. 135-144; РЖХим. 1976. 10Ж175.
243. Степанов Ф.Н., Суховерхое ВД. // Анилино-красочная гфомышлеииость. М., 1970. Вып. 1. С. 38-43; РЖХим. 1971. 12Ж08.
244. Юрченко А.Г., Ворощенко А.Т., Степанов Ф.Н. // Жури, орган, химии. 1970. Т. 6, № 1. С. 189-190.
245.С тепанов Ф.Н., Суховерхое ВД., Баклан В.Ф., Юрченко А.Г. // Там же. №4. С. 884-885.
246. Моисеев И.К // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 34.
ТА!. Моисеев И.К, Беляев П.Г., Тартаковский В.А. // Изв. АН СССР. Сер. хим. 1973. №1. С. 231.
208
248. Юрченко А.Г., Исаев CJJ., Мурзинова З.Н., Черняховская Е.Е. // Журн. орган, химии. 1972. Т. 8, №12. С. 2617.
249. Gund Т.М., Schleyer P.R. // Tetrahedron Lett. 1973. N 22. P. 1959-1962.
250. Ashby EC, Lin J J. (( J. Org. Chem. 1978. Vol. 43, N 6. P. 1263-1265.
251. Леей R.D., Badger R.C // Synthesis. 1979. N 7. P. 529.
252. Фридман А.Л., Колобов H.A., Петухов C.A. // Журн. орган, химии. 1975. Т. И, №3. С. 660-661.
253. Симоненко Л.С., Котляров АЛ., Новиков С.С. Ц Изв. АН СССР. Сер. хим. 1973. №1.С. 49-53.
253а. А.с. 216575 ЧССР, МКИ С 07 С 23/38. Способ получения 1-хлорадамантана / J. Belusa, V. Hruskova, V. Kysilka, Р. Macoun, J. Vacek, A. Hanak O. Pospichal // РЖХим. 1985. 8Н101П.
2536. Федотов H.C., Гришина E.B., Багрий Е.И. Ц VI Всесоюз. конф, по химии н применению кремнийорган. соединений: Тез. докл. Рига, 1986. С. 22- 24.
254. Хардин А.П., Попов А.Д., Протопопов П.А. // Журн. орган, химии. 1975. Т. 11, № 9. С. 1982.
255. Сорочинский А.Е., Александров А.М., Кухарь В.П. // Там же. 1979. Т. 15, №7. С. 1555-1556.
256. Сорочинский А.Е., Тарасевич А.С., Александров А.М., Кухарь В.П // Там же. 1981. Т. 17, №11. С. 2339-2343.
257. Perkins R.R., Pincock R.E. // Tetrahedron Lett. 1975. N И. P. 943-946.
25Ъ.Подхалюзин A.T., Назарова М.П //Докл. АН СССР. 1976. Т. 229, № 1. С. 105-107.
259. Подхалюзин А.Т., Назарова М.П., Янкелевич А.З. // Журн. орган, химии. 1976. Т. 12, №4. С. 910.
260. Chakrabarti J.K., Todd А. // Chem. Commun. D. 171. N И. Р. 556-557.
261. Baughman G.L. //J. Org. Chem. 1964. Vol. 29, N 1. P. 238-240.
262. Talaty E.R., Cancienne A.E., Dupuy A.E. // J. Chem. Soc. C. 1968. N 15. P. 1902-1903.
263. Хардин А.П, Новаков И.А., Радченко С.С. // Журн. орган, химии. 1973. Т. 9, № 2. С. 429.
264. Stetter Н, Krause М. // Tetrahedron Lett. 1967. N 19. Р. 1841 — 1843.
265. Gorrie Т.М., Schleyer P.R, // Org. Prep. Proc. Intern. 1971. Vol. 3, N 3. P, 159-162; Chem. Abstr. 1976. Vol. 75. 76247e.
266. Osawa E. // Tetrahedron Lett. 1974. N 1. P. 115-117.
267.Л ихотворник И.Р., Довгань Н.Л., Даниленко Г.И. // Журн. орган, химии. 1977. Т. 13, №4. С. 897.
268. Wolinski Kordonowska В., Plachta D. // Acta polym. pharm. 1977. Vol. 34, N 1. P. 17-22; РЖХим. 1977. 20Ж119.
269. Vodicka L., Jankii Burkhard /. // Coll. CzechosL Chem. Commun. 1978. Vol. 43, N5.P. 1410-1412.
THi.HatnUl H., Karim A,, McKervey M.A. // Tetrahedron. 1971- Vol. 27, N 18. P. 4317-4322.
271. Степанов Ф.Н, Уточка Т.Н. // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1973. № 10. С. 16-17; РЖХим. 1974. 6Ж136.
272. Симоненко Л.С., Котляров А.М., Новиков С.С. // Изв. АН СССР. Сер. хим. 1971-№5. С. 1125.
273. Lenoir D., Schleyer P.R., Cupas С.А.. Heyd W.E. // Chem. Commun. D. 1971. N 1. P. 26-27.
274. Cuddy B.D., Grant D., McKervey M.A. // J. Chem. Soc. C. 1971. N 19. P. 3173-3179.
275. Степанов Ф.Н., Баклан В.Ф. // Журн. общ. химии. 1964. Т, 34, № 2. С. 579-584.
276. Fischer W., Grob С.А., Katayama Н. // Helv. chim. acta. 1976. Vol. 59, N6. P. 1953-1962; РЖХим. 1977. 6Ж121.
277- Баклан В.Ф., Даниленко Г.И., Ралко В.А., Смирнова Н.А. // Вести. Киев, политехи. ин-та. Хим. машиностроение и технология. 1971. № 8. С. 16-18; Chem. Abstr. 1973. Vol. 78. 57848х.
278. Диколенко Е.И, Даниленко Г.И. // Вести. Киев, политехи, ии-та. Хим. машиностроение и технология. 1976. № 13. С. 47—49; РЖХим. 1977- 8Ж113.
279. Farcasiu D.t Schleyer P.R. // Tetrahedron Lett. 1973. N 39. P. 3835-3838.
14.Зак.1856 209
280.Ю рченко А.Г., Новикова М.И., Заикин В.Г. // Журн. орган, химии. 1978. Т. 14, №5. С. 984-989.
281. Karim A., McKervey М.А. // J. Chem. Soc. Perkin Trans. Part. 1.1974. N 21. P. 2475-2479.
282. Klein H, Wiartalla R. // Synth. Commun. 1979. Vol. 9, N 9. P. 825-830; РЖХим 1980. 18Ж123.
283. Vodicka L.t Triska J., Hajek M. // Coll. Czechosl. Chem. Commun. 1980. Vol. 45, N 10. P. 2670-2674.
284. Пат. 3666806 США, МКИ1 С 07 С. Haloadamantanes / R.E. Moore // Chem. Abstr. 1972. Vol. 77.100937U.
285. Пат. 819240 Великобритания. Chlorinated and brominated polycyclic hydrocarbons / W.C. Webber, P.A. Harthoom 11 Chem. Abstr. 1960. Vol. 54.15272d.
286. Пат. 3218355 США. Halogenated adamantane derivatives / M. Paulshock // Chem. Abstr. 1966. Vol. 64. 3381a.
287. Stetter H.t Krause M., Last W.-D. // Angew. Chem. 1968. Bd. 80, N 22. S. 970—971.
288. Stetter H.t Krause M., Last W.-D. // Chem. Ber. 1969. Bd. 102, N 10. S. 3357-3363.
289. Tabushi I., Yoshida Z., Takahashi N. // J. Amer. Chem. Soc. 1970. Vol. 92, N 22, P. 6670-6672.
290. Smith G. W., Williams HD. // J. Org. Chem. 1961. VoL 26, N 7. P. 2207-2212.
291. Olah G.A., NarangS.C // Tetrahedron. 1982. Vol. 38, N 15. P. 2225-2277.
292. Landa S., Vais J., Burkhard J. // Ztschr. Chem. 1967. Bd. 7, N 6. S. 233.
293. Ланда С., Буркхард Й., Вайс Й. Ц Sb. VSCHTPrazc D. 1969. V. 19. S. 5-17.
294. Заявка 6516807 Нидерланды. МКИ1 С 07 С. 1,3-Dihydroxyadamantanes / Sun oil co /I Chem. Abstr. 1966. Vol. 65. 15249e.
295. Whiting M.C., Bolt A.J.N., Parish J.N. // Adv. Chem. Ser. 1968. N 77. P. 4-14; Chem. Abstr. 1969. VoL 70.11205a.
296. Пат. 3450761 США, МКИ1 С 07 С. l-Amlno-3-ethyl-5,7-dimethyladamantane/A-Schnei-der II Chem. Abstr. 1969. Vol 71. 60869r.
297. Gehik H W., Schlatmann J.L.M.A. // Chem. Commun. 1967. N 9. P. 426.
298. Geluk H W., Schlatmann J.L.M.A. // Rec. trav. chim. 1969. VoL 88, N I. P. 13-16.
299. GahikH.W., Schlatmann J.L.M.A. Ц Tetrahedron. 1968. Vol. 24, N 15. P. 5369- 5377.
300. McKervey M.A., Alford J.R., McGarrity J.F., Rea E.J.F. 11 Tetrahedron Lett. 1968. N50.P. 5165-5168.
301. Bone J.A., Pritt J.R., Whitng M.C. // J. Chem. Soc. Perkin Trans. Part I. 1972. N 21. P. 2644-2647.
301a. Ковалев B.B., Федорова О.А., Шокова ЭЛ. Ц Журн. орган, химии. 1987. Т. 23, №2. С. 451-452.
3016. Опейда И.А., Залевскал НМ., Мироненко Н.И. // Укр. хим. журн. 1986. Т. 52, № 8. С. 884-889.
301в. Залевскал Н.М., Мироненко Н.П., Опейда ИЛ. // Нефтехимия. 1986. Т. 26, № 1, С. 99-104.
301г. Шульпин Г.Б., Ледерер П., Мацова Е. // Изв. АН СССР. Сер. хим. 1986. № 11. С. 2638- 2639.
301д. Baciocchi £, Del-Gacco T.t Sebastian: G.V. // Tetrahedron Lett. 1987. VoL 28, N I? P. 1941-1944.
30Ie. Урин А.Б., Багрий Е.И. // Il Всесоюз. симпоз. по гомоген. катализу: Тез. докл. Черноголовка. 1988. С. 74.
302. Schlatmann J.L.M.A. 11 Angew. Chem. 1971. Bd. 83, N 17. S. 732.
303. A.C. 379558 СССР, МКИ С 07 С 35/44. Способ получения 1,4-диоксиадамантана / Ф.Н. Степанов. З.Е Столяров, А.Г. Юрченко // Б.И. 1973. №20. С. 71-72.
304. GalukH.W., Schlatmann JL.M.A. // Rec. trav. chim. 1971. VoL 90, N 6. P. 516-520.
305. Пат. 3646224 США, МКИ1 С 07 С. Conversion of adamantane hydrocarbons to monools/ R.E. Moore // Chem. Abstr. 1972. VoL 76.112794m.
306. Пат. 4043927 США, МКИ1 С 10 M 1/20. Friction of tractive drive containing etheres of adamantanes / J.N. Doling D.S. Gates, F.P. Glazier, R.E. Moore // Ibid. 1977. Vol. 87. 187 080n.
307» Lancto S., Hlavaty J. // ColL Czechosl. Chem. Commun. 1975. VoL 40, N 2. P. 448-451.
308. Galuk H.W., Keizer V.G. // Synth. Commun. 1972. VoL 2, N 4. P„ 102-206; РЖХим. 1973.1Ж197.
210
309. Лерман Б.М., Арефьева ЗЯ» Толстиков ГЛ, // Журн.орган.химии. 1971, Т. 7, №5. С. 1084.
310. Gund Т.М., Nomura М., Williams V.Z. et aL // Tetrahedron Lett. 1970. N 56. P. 4875-4878.
311. Gund T.M., Osawa E., Williams V.Z., Schleyer P.R. // J. Org. Chem. 1974. VoL 39, N20. P. 2979-2987.
312. Janku J., Burkhard J., Vodtfka L. // Ztschr. Chem. 1981. Bd. 21, N 2. S. 67-68.
313. Kafka Z.t Vodtika L., Hajek M. // ColL Czechosl. Chem. Commun. 1983. VoL 48, N 4. P. 1074-1076.
314. Пат. 77122311 Япония, МКИ2 C 07 C 31/02. Selective hydroxylation of saturated hydrocarbons /1. Tabushi, T. Nak^jima, K. Seto 11 Chem. Abstr. 1978. VoL 88.1047 8 St.
315. Tabushi I, Nakajima T., Seto K. // Tetrahedron Lett. 1980. VoL 21, N 26. P. 2565-2568.
316. Barton D.H.R., Boivin J., Ozbalik N., Schwartzentuber KM. // Ibid. 1984. Vol. 25, N38. P. 4219-4222.
317. Jones S.R., Mellor JM. // J. Chem. Soc. Perkin Trans. Part I. 1976. N 23. P. 2576-2581. 31%. Koch V.R., Miller L.L. // J. Amer. Chem. Soc. 1973. Vol. 95, N 26. P. 8631-8637.
319. Koch V.R., Miller L.L. // Tetrahedron Lett. 1973. N 9. P. 693-696.
320.B ewick A., Edwards G.J., Jones S.R., Mellor J.M. // J. Chem. Soc. Perkin Trans. Part I. 1977. N 16. P. 1831-1834.
321. Залевскал HM„ Опейда ИЛ, // Всесоюз.науч.конф, по жидкофаз.окисл. орган, соединений: Тез.докл.Баку, 1979. Ч. 2. С. 31.
322. Залевскал НМ,, Мироненко Н.И., Опейда ИЛ. // Науч.конф, по химии орган.по-лиэдранов; Тез.докл. Волгоград, 1981. С. 86.
323. Тимохип В.И., Симонов МЛ,, Романцевич АМ„ Опейда ИЛ. // Нефтехимия. 1984. Т. 24, № 3. С. 411-414.
324. Morat С., RassatA. // Tetrahedron Lett. 1979. N 45. Р. 4409-4410.
325. Koch H.,HaafW. // Angew. Chem. I960. Bd. 72, N 17. S. 628.
326. A.c. 255242 СССР, МКИ С 07 С 61/12. Способ получения 1-адамантанкарбоновой кислоты / С.Д. Исаев, Ф.Н. Степанов // Б.И. 1969, № 33. С. 21.
327. А.с. 583999 СССР, МКИ С 07 С 61/12. Способ получения 1-адамантанкарбоновой кислоты / Я.Ю. Полис, БД. Рагуель, Б.Ю. Внльне // Б.И. 1977. № 46. С. 54.
328, Хардин А.П, Бутенко Л.Н„ Дербишер В.Е., Косенков РЛ. // Изв. вузов. Химия и хим. технология. 1977. Т. 20, № 4. С. 495-498.
329. Бутенко ЛД, Дербишер В.Е., Хардин А.И., Шрейберт А.И, // Жури, орган л имии. 1973. Т. 9, №4. С. 728-729.
330. Пат. 1353906 Франция, МКИа С 07 С. Adamantanecarboxylic acids / A. Lam о la // Chem. Abstr. 1964. VoL 61. 593g.
331.М оисеев HJC., Дорошенко Р.И. // Журн.органдсимии. 1983. T. 19, № 5. С. 1117-1118.
332. Burkhard J., Vais J., Landa S. // Ztschr. Chem. 1969. Bd. 9, N 1. S. 29-30.
333.J anku J., Burkhard J., Voditka L. // Sb. VSCHT Praze D. 1978. Sv. 39. S. 51-55; РЖХим. 1980. 21Ж103.
334. Vais J., Burkhard J„ Landa S. // Ztschr. Chem. 1969. Bd. 9,N 7. S. 268-269.
335.A lford J.R., Cuddy B.D., Grant D., McKervey МЛ. // J. Chem. Soc. Perkin Trans. Part 1.1972. N 21. P. 2707-2713.
336. Schleyer PX. Ц Angew. Chem. 1969. Bd. 81, N 14. S. 539-540.
337. Юрченко А.Г., ХрнсгичА.И„ Довгань НЛ. // Науч.коиф. по химии орган, пол подрайон: Тез.докл. Волгоград, 1981. С 85.
338. Водичка Л„ Япку Й„ Буркхард Й. // Там же. С 76.
339. VodiUkaL., Janku J., Burkhard J. // ColL Czechosl Chem. Commun. 1983. Vol. 48, N 4. P. 1162-1172.
340. Хардин АЛ., Бутенко Л.И., Понов Протононов ПЛ. // Науч. конф, по химии орган, полиэдранов: Тез.докл. Волгоград, 1981. С. 69.
341. Назарова МЛ„ Морозов ВЛ., Подхалюз ин А.Т, // Жури, орган, химии. 1983. Т. 19, №3. С. 565-571.
342. Longhair Н., Rilchardt Ch. // Chem. Ber. 1974. Bd. 107, N 4. S. 1245-1256.
343. Хардин АЛ., Бутенко ЛЛ„ Радченко С.С. // Научжонф. по химии орган, полн-эдранов: Тездокл. Волгоград, 1981. С. 70.
211
344. Шокова Э^4., Ковалев В.В., Лузиков Ю.Н., Шарбатян ПЛ. // Нефтехимия. 1981 Т. 21, №2. С. 271-273.
345, Inamoto Y., Nakayama Н. // Synth. Commun. 1971. VoL 1, N 2. P. 133-135; Chem Abstr. 1971. VoL 75. 118018р.
346. Новиков C.C., Хардин А.П., Бутенко Л.Н. и др. // Изв. АН СССР. Сер.хим, 1976. № 11. С. 2597-2599.
347. Drivas I.Mison Р. // Tetrahedron lett, 1981. VoL 22, N 7. P. 641-644.
34%. Numan H, Troostwijk C.B., Wieringa J.H, Wynberg H. // Ibid. 1977. N 28. P. 176] -1764.
349,A fcCabe P.H., Nelson C.R., Routedge W. // Tetrahedron. 1977. Vol. 33, N 14. P. 1749-1753.
350. Tabushi I., Okada T., Aoyama Y., Oda R. // Tetrahedron Lett. 1969. N 46. P. 4069-4072.
351. Багал МЛ., Клиндукова T.K., Лантвоев В.И. // Журн.огран.химии. 1975. Т. 11, №8. С. 1645-1648.
352. Фридман АЛ., Сивкова МЛ., Колобов НЛ. // Там же. 1976. Т. 12, № 10. С. 2260-2261.
353. Bott К. //Chem. Commun. D. 1969. N 22. P. 1349-1350.
354. Bott К. Ц J. Liebigs Ann. Chem. 1972. Bd. 766. S. 51-57.
355. Bott К. // Tetrahedron Lett. 1969. N 22. P. 1747-1749.
356. Степанов Ф.Н., Довгань НЛ. // Журн.органлимии. 1968. Т. 4. № 2. С. 277-280.
357. Scharp Wymberg Н., Strafing /. // Rec. trav. chim. 1970. VoL 89, N 1. P. 18-22.
358. Грева ИЛ., Полис Я JO., Лидак MJ0. и др. // Журн. орган, химии. 1981. Т. 17, № 4. С. 778-786.
359. Степанов Ф.Н., Гуц С.С. Ц Изв. АН СССР. Сер.хим. 1970. № 2. С. 430-434.
360. Yurchenko A.G., Stepenov F.N., Isaeva S.S. et al. // Org. Mass Spectrom. 1970. Vol. 3, N Ц. P. 1401-1410;Chem. Abstr. 1971. VoL 74. 31322р.
361. Степанов Ф.Н., Гуц С.С. Ц Журн. орган.химии. 1968. Т. 4, № 11. С. 1933-1936.
362. Степанов Ф.Н, Диколенко Е.И, Мартынова Э.Н. // Там же. 1971. Т. 7, №7. С. 1544-1545.
363. Schleyer P.R., Funke Е., Liggero S.H. // J. Amer. Chem. Soc. 1969. VoL 91.N44. P. 3965-3967.
364. Nordlander J.E., Wu F. Y.-H.,Hndal S.P., Hamilton J.B, // Ibid. P. 3962—3964.
365. Плахотник BM., Ковтун BJ0., Яшунский В.Г. Ц Журн.орган.химии. 1982. Т. 18, №5. С. 1001-1005.
366. Burkhard Z, Janku Z, Landa S. // Co IL Czechosl. Chem. Commun. 1974. VoL 39, N 4. P. 1072-1082.
367. Burkhard J., JankuJ., Kubelka V. et al. 11 Ibid. P. 1083-1090.
368. Шокова ЭЛ., Кнопова СМ., Нгуен Данг Кьен, Казанский БЛ. // Нефтехимия.
1973. Т. 13, №4. С. 585-591.
369. Шокова ЭЛ., Гавричева ОМ. // Там же. 1975. Т. 15, № 3. С. 363-368.
370. Шокова ЭЛ., Кнопова СМ., Казанский БЛ. // Докл. АН СССР. 1973. Т. 212, №2. С. 386-388.
371. Иванова ЛЛ., Полис Я.Ю., Грева ИЯ. и др. // Журн.оргая. химии. 1978. Т. 14, №-9. С. 2013.
372, Иванова Л.П., Полис ЯД)., Грава ИЛ. и др. // Там же, 1981. Т. 17, № 2. С. 325-329,
373. Koraloot J.G., Keizer V.G. // Tetrahedron Lett. 1969. N 40. P. 3517-3520.
374. Nordlander J.E., Jindal SJ>., Kitko D.J. // Chem. Commun, D. 1969. N 19. P. 1136-1137.
375. Stothers J.B., Tan C.T. // J. Chem. Soc. Chem. Commun. 1974. N 18, P. 738-739.
376. Greidanus J.W., Schwalm WJ. Ц Canad. J. Chem. 1969. Vol. 47, N 19. P. 3715-3716.
377. Greidanus J. W. //Ibid. 1970. VoL 48, N 22. P. 3530-3536.
378. Olah GM., Calin M. //J. Amer. Chem. Soc. 1968. Vol. 90, N 4. P. 938-943.
379. Snatzke G,, Eckhardt G. // Tetrahedron. 1970. VoL 25, N4.P. 1143-1155.
380. Vodicka L., Hlavaty J., Sevostjanova V. V. // Sb. VSCHT Praze D. 1978. Sv. 39.
S. 357-368;Chem. Abstr. 1980. VoL 93.185830g.
381. Vodicka L., Hlavaty J. // Coll. Czechosl. Chem. Commun. 1979. Vol. 44, Nil. P. 3296-330.
382, Murray R.K.,Babiak КЛ. ЦЗ. Org. Chem. 1973. VoL 38, N 14. P. 2556-2557.
383, Farcasiu D. // Synthesis. 1972. N 11. P. 615-616.
212
384. Разуваев ГЛ., Богуславская Л.С., Этлис В.С., Бровкина Г.В. //Докл. АН СССР. 1968. Т. 183, №6. С. 1346-1349.
385. Wieringa J.H., Strafing Wynberg И., Adam W. // Tetrahedron Lett. 1972. N 2. P. 169-172.
386. Лантвоев B.H. // Журн.орган.химин. 1977. T. 13, № 11. С. 2451-2452.
387. Новиков С.С., Хардин А.П., Бутенко Л.Н. и др.//Там же. 1980. Т. 16, № 7. С. 1433-1435.
388. Довгань НЛ, Верповский Н.С., Зосим ЛА. // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1977. N 14. С. 9-11; Chem. Abstr. 1978. Vol. 89. 59698z.
389. Столяров З.Е. // Вести.Киев.понитехн.ин-та. Хим.машиностроение и технология. 1979. №16. С. 15-17; РЖХим. 1980. 10Ж124.
390. McKervey МА. //Chem. and Ind. 1967. N 42. P. 1791; РЖХим. 1968. 15Ж137.
391. Curran W. V., Angier RB. // Chem. Commun. 1967. N 11. P. 563.
З92.£ ипл W.H.W., Podmore W.D., SzinaiS.S. //J. Chem. Soc. C. 1968. N 13. P. 1657-1660.
393. Канищев М.И., Смит BA., Щеголев AA., Кэпл P. // Изв, АН СССР. Сер.хим. 1977. №9. С. 2175.
394. Канищев М.И., Смит ВА., Щеголев АА„ Кэпл Р, // Там же. 1978. № 2. С. 511.
395. GelukH.W. // Tetrahedron Lett. 1971. N 47. P. 4473-4476.
396.B ochmann M, Wilkinson G., Young G.B. et al. //J. Chem. Soc. Dalton Trans. 1980. N6.P. 901-910.
397- Kovadc P, Roskos Ph.D. // Tetrahedron Lett. 1968. N 56. P. 5833-5835.
398. Kovadc P., Roskos Ph.D. // J. Amer. Chem. Soc. 1969. VoL 91, N 23. P. 6457-6460.
399. Haaf W. // Angew. Chem. 1961. Bd. 73, N 4. S. 144.
400. OhsukiM., Inamoto Y., Takaishi N. et al. // Synthesis. 1977. N 9. P. 632-633.
401. О&й GA., Gupta B.G. // J. Org. Chem. 1980. Vol. 45, N 15. P. 3532-3533.
402. KrumkalnsE. V., Pfeifer W. //J. Med. Chem. 1968. Vol. 11, N 5. P. 1103.
403. Пат. 1294371 ФРГ, МКИ’ С О? С, А 61 К. 1-Aminoadamantane and its derivatives / E. Gottwald, H. Machoczak // Chem. Abstr. 1969. VoL 71. 38444b.
404. A.c. 254518 СССР. МКИ С 07 С. Способ получения 3-аминогомоадамантана / С.Д. Исаев, П.А. Красуцкий, Ф.Н. Степанов // Б.И. 1969. № 32. С. 25.
405. Sollott G.P., Gilbert Е.Е. // J. Org. Chem. 1980. Vol. 45, N 26. P. 5405-5408.
406. Пат. 3053907 США. Nitroadamantanes / G.W. Snith, H.D. Williams // Chem. Abstr. 1963. VoL 59. 480d.
407. Kovadc P., Field K.W., Wnuk TA. // J. Chem. and Eng. Data. 1971. Vol. 16, N 2. P. 141-142; Chem. Abstr. 1971. VoL 74. 141487d.
408. Sasafci T., Eguchi S.r Тоги T. 11 Chem. Commun. D. 1970. N 19. P. 1239-1420.
409. Breslow D.S., Edwards EJ.r Leone R., Schleyer P.R. // J. Amer. Chem. Soc. 1968. VoL 90, N 25. P. 7097-7102.
410. Casagrande P.r PellaeaniL.r Tardella P.A. 11 J. Org. Chem. 1978. Vol. 43, N 13- P. 2725-2726.
410a.l7msfed M.E., Lin M.C. // Appl. Phys. 1986. VoL 39, № 1. P. 61-63; РЖХим. 1986. 10Б4395.
4ll.A fouceee И.К., Беляев П.Г., Барабанова H.B. и др. // Журн. орган.химии. 1975. Т. II, № 1. С. 214-215.
412. Моисеев И.К., Багрий Е.И., Климочкин Ю.Н и др. // Изв. АН СССР. Сер.хим. 1985. №9. С. 2141-2143.
413. Моисеев ИЛС., Багрий Е.И., Климочкин Ю.Н. и др. // Там же. С. 2144-2146.
414. Моисеев И.К., Дорошенко Р.И. // Журн,орган.хнмии. 1982. Т. 18, № 6. С. 1233-1236.
415. Моисеев И.К., Дорошенко Р.И., Иванова В.И. // Хим.-фармацевт, жури. 1976. Т. 10, №4. С. 32-33.
416. Gerzon К., Tobias D.J., Holmes R.E. etal. // J. Med. Chem. 1967. Vol. 10, N 4, P. 603 — 606; Chem. Abstr. 1967. VoL 67. 7321 Щ.
417. KochH., Franken J, // Chem. Ber. 1963. Bd. 96, N 1. S. 213-219.
418. Ковалев В.Д, Васильева МЛ., Шокова ЭЛ. // Нефтехимия. 1978. Т, 18, № 1. С. 17-22.
419. Юрченко А.Г., Исаев СД., Новоселов Е.Ф, и др, // Науч.конф, по химии, орган, по-лиэдранов; Тез.докл. Волгоград, 1981. С. 97.
420. Adam W., De Lucchi О. // Angew. Chem. 1980. Bd. 92, N 10. S. 815-832,
213
421. Плахотник В.М,, Ковтун В.Ю., Красуцкий ПЛ. и др. // Журн.хнмии. 1981. Т. 17, №7. С. 1447-1450.
422. Kolfonftscft Z, Barash L., Doldouras G.A. Ц J. Amer. Chem. Soc. 1970. VoL 92, N 25. P. 7494-7495.
423. Hess U.t Huhn D.t Lund H. // Acta chem. scand. B. 1980. Vol 34, N 6. P. 413-417.
424. Хардин AJT„ Гуреев Н.Г., Радченко С.С. jj Журн.орган.химии. 1980. T. 16, № 1 С. 60-62.
425. Tabushi I., Koto Sh.t Schleyer P.R.t Gund T.M. // J. Chem. Soc. Chem. Commun. 1974. N15.P. 591.
A2(>.Шепелева E.C., Санин П.И., Олейник Д.М, и др. // Докл. АН СССР. 1972. Т. 203, №3. С. 608-611.
427. Stetter H.t Last W.D. // Chem. Вет. 1969. Bd. 102, N 10. S. 3364-3366.
428.Ю рченко Р.И., Клепа Т.И., Мишак М.И., Тихонов В.П. // Жури,обш.химни. 1980 Т. 50, № 11. С. 2443-2447.
429. Юрченко Р.И., Войцеховская О.М., Пинчук AJf. н др. // Там же. 1982. Т. 52, № 3. С. 558-561.
430. Jorgensen F.S., Snyder J.P. 11 J. Org. Chem. 1980. Vol. 45, N 6. P. 1015-1020.
431. Ковалев B.B., Шокова ЭЛ. // Журн.орган.химии. 1988. Т. 24, № 4. С. 738-742.
432. Салимгареева И.М., Юрьев В.П., Рафиков С.Р. // VI Всесоюэ. конф, по химии и применению кремнийорган. соединений: Тез. докл. Рига, 1986. С. 215-216.
433. Гришина Е.В., Федотов Н.С., Шелудяков ВД. // Журн,обш.химнм. 1988. Т. 58, № 8. С 1949-1953.
Глава 4
Непредельные соединения ряда адамантана
Непредельные углеводороды каркасного строения вследствие их высокой реакционной способности могут быть использованы как исходные вещества для синтеза различных функциональных производных и в качестве мономеров. В практическом отношении большой интерес представляют прежде всего непредельные производные адамантана, ставшие относительно доступными соединениями. Для удобства рассмотрения в данной работе они условно разделены на три основные группы: дегидроадамантаны; соединения, содержащие экзоциклическую двойную связь н соединения с кратной связью в боковой цепи.
4.1. ДЕГИДРОАДАМАНТАНЫ
Теоретически возможны три изомера дегидроадамантана составаС10Н14: 1,2-дегидроадамантан (1), 1,3-дегидроадамантан (2) и 2,4-де гидро адамантан (3).
(3)
1,2-Дегидроадамантан относится к так называемым антибредтовым олефинам [1, с. 290]. Изомеры (2) и (3) лишь условно могут быть отнесены к непредельным производным. Их рассмотрение здесь, однако, целесообразно в связи с тем, что по своим химическим свойствам (прежде всего по высокой реакционной способности) они весьма близки к производным адамантана, содержащим кратную связь.
Возможность существования 1,2дегидроадамантана(грицикло[ЗД1,13’7]-децена-1, адамантена) — важная и интересная проблема химии адамантана. Расчет показал, что адамантен является высоконапряженным олефином, так как для атомов С(1) и С (2) затруднено перекрывание свободных р-орби-талей, находящихся в различных плоскостях (требуется большая деформация жесткого скелета). В обычных условиях зто соединение неустойчиво.
Вместе с тем имеется ряд косвенных доказательств существования адамантена, образующегося в качестве интермедиата во многих реакциях, обычно
215
используемых для получения напряженных олефинов. Так, при бнсдегало-генировании 1-бром-2-иодадамантаиа с помощью и- или втор-бутиллнтня получен углеводород С2 о Н2 8 [2].
Аналогичные димеры получены с выходом 98% прн обработке 1,2-дииод-адамантана н-бутиллитием в пентане при 0°С [3]; прн —80° С в пентане в условиях большого избытка бутадиена, кроме димера (85%), был выделен аддукт бутадиена и адамантена [4,5] .Нагревание смеси 1,2-дннодадаманта-на и днфенилфосфорнокислого Лития в ТГФ и последующее окисление пероксидом водорода также приводит к димеру адамантена [6].
В качестве интермедиата адамантеи образуется также при разложении бис-трег-бутиловок) эфира 1,2-адамантандикарбоновой кислоты в 2,5-ди-метилфуране [7]. Прн этом по реакции Дильса—Альдера получен аддукт адамантена и ди метил фурана.
Де бромирование 1,2-днбромадамантана действием (CH3Si)2Hg в кипящем бензоле также приводит к образованию в качестве промежуточного продукта адамантена, который либо димеризуется, либо прн проведении реакции в присутствии 2,5-днметилфурана дает соответствующий аддукт Дильса—Альдера [8].
Образование адамантена удалось подтвердить спектральными методами. По данным ИК- и ЭПР-спектров в продуктах взаимодействия 1,2-днбром-или 1,2-дииодадамантана с парами Na или К, стабилизированных в аргоновой матрице при 10 К, основным является адамантеи; в небольших количествах образуется также 1-адамантильный радикал [9]. После отогрева матрицы до ~20°С в реакционной смеси методом хромато-масс-спектро-метрнн обнаружены [2+2]-димеры и небольшие количества 1,1-диадаманти-ла. Судя по данным ИК-спектров, адамантеи, скорее, содержит ненапряженную двойную связь, нежели является бирадикалом; возможно, эта диойная связь имеет частично радикальный характер.
Попытка получить адамантеи реакцией фотозлиминирования нз 1-ада-мантилацетона не привела к успеху [10]. Вместе с тем наблюдалось образование адамантена по реакции фотозлиминирования из адамантиловых эфиров (1- и 2-адамантил фенил ацетате в) в метаноле; в обоих случаях после-
216
Таблица 4,1
Устойчивость некоторых олефинов с С-С-связью у основания мостика [13]
Алкен д№ дк°
ккал/мопь
Бицикло [ 2,2,2] октен-1 +42,72 50,6 40,4
Адамантеи +32,26 43,0 39,5
Гомоадамантен-3 + 18,92 35,3 20,2
Бицикло [3,3,2] децен-1 +6,18 25,2 4,7
Бицикло[4,4,1] уидецен-1 -8,95 15,7 -1,5
Примечание. — теплота образования, Е° — энергия напряжения, — разница в энергии напряжения алкена и соответствующего алкана.
дующее ионное присоединение остатка молекулы растворителя к адаманте-ну приводит к одному н тому же продукту - 1-метоксиадамантану [11].
Прн ионизации 2-(1-адамантил) циклогексанона под электронным ударом в высоком вакууме возникает катион-раднкал 1-гидроксициклогексе-нилий и нейтральный углеводород (4) [12].
Ближайший циклический гомолог адамантена — гомоадамантен-3 (5); его энергия напряжения значительно ниже вследствие большего размера цикла, содержащего кратную С-С-связь (табл. 4.1) [13]. Теоретически существование гомоадаманте на-3 было предсказано в работах [14—16]. В чистом виде (5) получен (сконденсирован на пластине из NaClnpn -196°С) пиролизом в вакууме (10-3 мм рт. ст., 375°С) 1-адамантил дназо-метаиа. При нагревании соединения (5) до комнатной температуры образуется димер гомоадамантена-3 [17], описанный также в работах [18, 19]. Пиролиз (5) при 500—520° С приводит к 3-винилнорадамантану (6), 4-мети-ленпротоаддмантану (7) и гомоадаманте ну-4 (8).
217
Симметричный бисгомо адамантан диен может быть получен исходя из адамаитандиона-2,6 по схеме [20] :
СНгЫ|-Ц
LLiAIHq__________
2.CttjC6H40C[-S)CL
1,3- и 2,4-Дегидроадамантаны, представляющие собой уже вполне устойчивые соединения, получают с использованием реакций, обычно применяемых для синтеза циклопро Пановых углеводородов (1,3-элиминирование, внедрение карбена и др.).
1,3-Дегидроадамантан (2) представляет собой напряженную систему с двумя ’’вывернутыми” (inverted) атомами углерода. Молекула 2,4-дегидро-адамантана (3) является менее напряженной; по своим свойствам (3) близок к другим алициклическим углеводородам, содержащим трехчленный цикп.
1,3 Дегидроадамантан (2) впервые был синтезирован действием нагрнй-калиевого сплава на 1,3-дабромадамантан в кипящем гептане; его структура подтверждена химическими превращениями, связанными с раскрытием трехчленного цикла [21]. Аналогично из 1,3-дабромадамантана в эфире, содержащем следы трет-бутилового спирта и (или) гексаметиленфосфамида (ГМФА), была получена смесь (9:1) 1,3-дегидроадамантана и адамантана с выходом 77% [22]. В тех же условиях 1, З-да бро м-5,7-да метил адамантан с выходом 78% образует смесь (1:1) 1,3-дегидро-5,7-даметиладамантана и 1,3-диметил адамантана.
При использовании пития реакция циклизации 1,3-дабромадамантана протекает в ТГФ при комнатной температуре. В этом случае требуется, одаако, присутствие катализатора — алкил арил хлорсилаиа; выход соединения (2) составляет 80% [23]. Обработка 1,3,5-трибромадамантана н-бу-тиллитием при —35°С в эфире, содержащем ГМФА (25:1), приводит к 1,3-дегидро-5-бромадамантану, который при добавлении NaCN превращается в стабильный 1,3-дегидро-5-циаиадамантан [24].
Вг
Образование соединения (2) наблюдается также прн полярографическом восстановлении 1,3-дабромадамантана [25].
Подобно другим алициклическим углеводородам, содержащим напряженный трехчленный цикл, 1,3-дегидроадамантан (2) и его производные
218
обладают высокой реакционной способностью. Связь С(1)—С(3) в (2) значительно ослаблена; ее длина 1,64 А (длина других С-С-связен в адамантане 1,53 А). Вследствие этого реакции 1,3-дегидроадамантана протекают селективно с раскрытием трехчленного цикпа и образованием 1-моно- или 1,3-дизамещенных производных адамантана; иногда происходит также фрагментация адамантанового цикла.
Окисление соединения (2) воздухом при 20° С в гептане приводит к пероксиду, который разлагается со взрывом при 160 С. На воздухе в растворе пеитана при комнатной температуре (2) превращается в смесь сополимеров с кислородом, главным из которых является взрывоопасный поли-пероксиадамаитан(ОС10Н14О)„.При нагревании (13О-16О°С) взапаянной ампуле соединение (2) превращается в белый полимер — 1,3-полиадамантан состава (СюН14)л,нерастворимый в органических растворителях и устойчивый в атмосфере азота при 400° С. Аналогичный поли диметил адамантан образуется из 1,3-дегидро-5,7-диметил адамантана при 90—145° С. В отсутствие кислорода соединение (2) термически устойчиво, период полупревращения при 195°С в растворе н-октана составляет 4ч [21,22].Каталитическое гидрирование соединения (2) приводит к адамантану с выходом 82%; при взаимодействии (2) с бромом или иодом образуется соответственно 1,3-дибром-или 1,3-дииодадамантан.
При обработке соединения (2) иодом в присутствии пиридина получается иодид М-(3-иод-1-адамантил) пиридиния [26].
I
(Z)
Эта реакция является одним из первых примеров сопряженного межмоле-купярного галогенаминирования; реакция протекает и с другими соединениями общей формулы NI^R’R3 [27],
1,3- Дегидроадамантан (2) присоединяет галоген ангидрид бензолсульфо-кислоты в отсутствие катализатора [28].
В условиях каталитического генерирования дихлоркарбена 1,3-дегкдро-адамантан присоединяет 2 экв. карбеиа с образованием спиро [7-дихлор-метиленбицикл о [33,1] ионан-ЗД*- (2*,2*-дихлорциклопропана) ] [29].
0
218
Исследована реакция (2) с арилиодозокарбоксилатами общей формулы Аг(ОСОК)з» где R = Aik, Аг, Основными продуктами реакции, протекающей в инертных растворителях при комнатной температуре, являются иодбеизол и 1,3-диацилоксипроизводные адамантана [30].
1,3- Дегидроадамантаи (2), образующийся при электро восстановлении 1,3-дибромадамантана в ацетонитриле на ртутном электроде, реагирует с ацетонитрилом и содержащейся в нем водой, давая 1-гидрокси-и 1-аце-таминоадамантаиы [31].
Присоединение уксусной кислоты к соединению (2) приводит к 1-ада-маитилацетату [32],
При взаимодействии (2) с малеиновым ангидридом, акрилонитрилом, диметиловым эфиром ацетилендикарбоиовой кислоты образуются олигомерные продукты [33]. Реакцию проводят при комнатной температуре и в отсутствие инициаторов полимеризации; гомополимеризация каждого из этих мономеров протекает в более жестких условиях. Известен ряд других реакций 1,3-дегидроадамантана, протекающих с раскрытием трехчленного цикла н приводящих к получению производных, представляющих интерес в отношении физиологической активности.
(2)
СН,(СО2С2Н5),
100° С, 75% CH,CeH4SOa nh2
80°С, 50% C6HsNHa
100°С, 55% C6H5NHjC1
100°С, 55% Cl-S— С6У,
HSi(R)X3 ; HaPtCl6; Уф
(Х=Н,С1; К=СНз), СеН^С1
1-Ad-CH(CO2C2HS)2 [34]
1-Ad- NHSO2C6H4CH3 [34]
l-AdNHC6Hs [34]
l-AdC6H4NH2 [34]
3-Cl-l-ASC6Fs [351
l-AdSi(R)X2 [36]
2,4Дегидроадамантан (3) (тетрацикло [4,3,1,0 2)4,0 3)8] декан) - весьма устойчивое соединение (тля. 202,5—203,5°С). В чистом виде (3) получен пиролизом сухой литиевой соли n-тозилгидразона адамантанона-2 при 130—140° С (выход 65%) [37] и спиро [адамантан-2,3'-диазирнна] при 320° С (выход 96%) [38].
220
(3)
Гидролизом соответствующих щелочных солей тозилгидразоиов получены производные 2,4-дегидроадамантана, содержащие заместители в узловых положениях [39]. Установлено, что распределение продуктов реакции внутримолекулярного С—Н-виедрения промежуточно образующихся карбенов зависит от природы заместителя и его положения относительно карбенового центра.
Пиролиз 2-адамантилсульфоната при 520° С приводит к соединению (3) и протоадамантену (выходы 57% и 38% соответственно) [40],
УФ-облучение растворов 2-иод- н 2-бромадамантанов приводит к смеси, состоящей из адамантана, соединения (3), протоадамантана и соответственно (в зависимости от используемого растворителя) 2-этоксн- или 2-меток-сиадамантана. Последние соединения образуются в результате нуклеофильной атаки молекул растворителя на промежуточный 2-адамантильный катион; депротоиирование катиона приводит к смеси (3) и протоадамантана [41].
221
Таблица 4.2
2-Замещенные адамантаны, образующиеся при взаимодействии 2,4-дегидроадамантана с электрофильными реагентами [42)
Реагенты Заместитель, X | Выход, %
HI I 24
CHjCOOH, H2SO4 OCOCHj 94
СН3ОН, BFS осн3 80
С6 He , А1С1Э, BF3 CeHs 70
С6 Н5 СН3 , A1.CI,, ВРЭ с6 Н4СН3 50
С6Н5ОСНз, A1C13,BF, св Н4ОСН3 86
Таблица 4.3
2,4-Дизамешенные адамантаны, образующиеся при взаимодействии 2,4-дегидроадамантана с электрофильными реагентами (42)
Реагенты Заместители Выход, %
X 1 Y
С12, СС14 Cl Cl 77
Вг2, СС14 Bi Br 86
Jj,CCl4 1 I 100
С12,СНЭCN, Н2О Cl HNCOCH3 76
Вга, СН3 CN, нз О, AgC104 Bi HNCOCH3 78
Ia,CH3CN, Н2О, AgQO4 I HNCOCH3 81
С11,Н1О, (СаН40)2 Cl OH 71
N-Бромсукцинимид, НаО Br OH 83
С1Н3ОН,Н3О, AgNO3 OH OH 50
СН3ОН, N-бромсукцинимид Br OCH9 23
Hg(CH3COO)3,H3O OH HgOCOCH3 95
noci, снаэ Cl NOH 38
Структура 2,4-дегидроадамантана (3) представляет уникальную возможность синтеза на его основе труднодоступных 2-моно- и 2,4-диза-мещенных производных адамантана путем размыкания трехчленного цикла электрофильными реагентами (табл. 4.2 и 4.3) [42, 43] .С такими реагентами реакции соединения (3) протекают по следующей общей схеме:
(3)
222
Олигомеризация соединения (3) под влиянием А1С13 приводит к димеру и трнмеру; высшие олигомеры в продуктах реакции не обнаружены [44]. Предполагается, что олигомеризация протекает через 1^адамантильный катион по ионному цепному механизму.
Производное 2,4-дегидроадамантана (9) получено фотоизомеризацией [45, 46] или изомеризацией в присутствии кислотного катализатора [47] протоадамантенона-2.
96 %-ная Иг30ч, пентан
(9)
Исходя из иорборнадиена соединение (9) было синтезировано в несколько стадий с суммарным выходом 2% [48] ; по реакции Вольфа— Кинжне-ра оно легко превращается в соединение (3), выход 75% [49].
Наличие карбонильной группы делает возможным получение на основе (9) других 2-замещенных 8,9-дегидроадамантанов.
Ближайший циклический гомолог 2,4-дегидроадамантана — 2,4-мета -ио-2,4-дегидроадамантан (получен при пиролизе сухой натриевой соли то зил гидразона 4-метиленадамантанона-2 с выходом 70% [50]) — относительно устойчив при комнатной температуре, быстро реагирует с кислородом, метанолом и бромом, образуя сложную смесь продуктов. Структура этого пропеллана доказана методом ЯМР13 С [51].
Большой интерес представляет 2,4,6,9-тетрадегидроадамантан (10) — устойчивое соединение с т.шт, 152—154° С, выдерживающее нагревание при 250 С в течение 10 мин без изменения структуры. Углеводород (10) получен при термолизе дилнтиевой соли бисто зил гидразона ада манта ндиона-2,6 [52,53].
223
(Э) (11)
В результате каталитического гидрирования соединения (10) над Pt-катализатором при комнатной температуре и атмосферном давлении в присутствии ТГФ и СН3СООН происходит расщепление только одного циклопропанового кольца с образованием смеси двух СюН14-углеводородов: 2,4-дегидроадамантана (3) и главным образом дегидропротоадамантана (11). Изомеризация соединения (Ю) под действием температуры и катализатора (80°С, AgBF4) не происходит [53].
Подобно 2,4-дегидроадамантану, разложением литиевой соли тозилгид-разона диамантанона при 165—170°С получают 3,5-дегидродиамантан (выход 20%); размыканием трехчленного цикла под действием различных реагентов (например, N-бромсукциннмида - N-БС) на его основе могут быть получены различные 3,5-дизамещенные производные диамантана [43, 54], например:
4.2. АДАМАНТАНЫ
С ЗКЗОЦИКЛИЧЕСКОЙ КРАТНОЙ СВЯЗЬЮ
Вследствие особенностей строения адамантана экзо циклическая тг-связь возможна только в мостиковом положении ядра.
(12)
A = GRR'
r'rc=a=grr'
Соединения типа (12), как правило, получают традиционным методом -дегидратацией соответствующих третичных спиртов — продуктов взаимодействия адамантанона и литийалкилов или реактивов Грииьяра. Таким путем получены, в частности, 2-метиле на даман тан [55, 56], 2-этилиденада-мантан [57, 58], 2-прол ил идеи-, 2-изобутилиден- и 2-бутилиденадамантаны [57, 59].
224
Другим путем получения этих олефинов является сольволиз 2-алкил-адамантил-2-к-нитробензоатов [60].
[К = СНЭ, СгН5, СМгС(СН3)3 г а'=снг,снсн3, СНС(СН3)3С(СН3)г].
Имеются сведения о синтезе ди-трет-бутил метилена дамантаиа путем термолиза соответствующего спиро [адамантан-2,2*-тиадиазол ина] [61].
Бензилиденадамантан получен с 90%-ным выходом дегидратацией 2-бен-зиладамантанола-2 в присутствии десятикратного избытка 85%-ной ортофо-сфорнон кислоты при 120° С [62].
C6HsCH2MgCl А=О ----------------> А-
н3ро4
А-СНС6Н5 .
сн2сбн5
Существенный интерес представляет адамантилиденадамантан (14); он образуется при фотохимическом декарбонилировании димера 2-адамантил-кетона [63]
аЛа-^*а=а
(14)
о
и при дебромированни гем-2-дибромадамантана с помощью цинк-медной пары [64].
Синтезированы также многие метильные производные адамантилиден-адамантана (14) [65 67]
R2 R’^ (14, а-г)
qIR'.r^h; r*,r’=chs', 6)R',Rj = H;R’,R’=CHs; b)R',R2=H; к5, вч=сн3; г) R1 ,R2,R5,R4 = Cti3.
Бнсиепредельные производные адамантана типа (13) получены дегидратацией соответствующих диолов [68].
^0
Н0/АМН--------—*R'=A=R' a)R=CHj, R=CH2 (45Ц ‘
(13) б) R=(yts,R'=CA (907.1
1/2 15. Зак. 1856
225
Целый ряд ненасыщенных углеводородов типа (12), содержащих циклопропановые кольца, получен путем взаимодействия адамантилиденкарбе-на с различными алифатическими моно- и бициклическими олефинами, в том числе норборненом и норборнадиеном [69].
По химическим свойствам непредельные производные адамантана типа (12) и (13) близки к другим алициклическим углеводородам, содержащим изолированную экзоциклическую кратную С=С-связь. Каталитическое гидрирование углеводородов приводит к 2-алкил- и 2, 6-дналкилада-маитаиам; метилен- и этилиденадамантаны при взаимодействии с иоднстым метиленом в присутствии цинк-медной пары или с дназометаном в присутствии бромистой медн дают соответствующие производные циклопропана [70]; метиле на дамантаи присоединяет днхлор- и днбромкарбены с образованием спиро [ 2*,2*-дибром(или 2*,2'-дихлор) адамантан-2,1 '-циклопропанов] [71]. Вместе с тем наличие объемистого адамантильного радикала создает серьезные стерические затруднения, понижающие реакционную способность ОС-связи, а жесткость структуры препятствует проявлению возможных эффектов сопряжения. Так, вследствие затруднения сопряжения между катионным центром и соседней двойной связью в тозилате 2-метиленадамантаиола-1 скорость сольволиза тозилата (15) ниже скорости сольволиза незамещенного (16) в 104 раз, хотя можно было ожидать обратную зависимость [72].
(15) (16)
Активность кратных связей олефинов (12) в реакции с дихлоркарбеном существенно зависит от величины и характера заместителя [73],
А=С
/К' :сс1г
Радикалы R и Rr увеличивают (первичные алкильные группы) или уменьшают (изопропильная и фенильная группы) активность двойной связи в соответствии с проявляемыми ими индуктивными н стерическнми эффектами. Углеводород (12), R = R' = СбН5, не вступает в реакцию с дихлоркарбеном.
Стерические эффекты особенно ярко проявляются в случае адамантилн-денадамантана (14). При галогенировании (14) были выделены его устойчивые комплексы с хлором и бромом [74, 75]. На основании исследований методом ЯМР 13С им была приписана структура двухэлектронных трехцентровых я-комплексов [76].
Уникальное поведение двойной связи соединения (14) делает возможным получение термически стабильных комплексов [77]
226
Взаимодействие SC12 со стерически сильиозатрудненной двойной связью в (14) протекает необычно и в зависимости от условий реакции приводит к образованию соединений, содержащих S-гетероцикпы, а также хлорза-мещенных адамант илиденадамантанов [78-80].
Реакция (14) с МоОС14 или SbCl5 в присутствии кислот Льюса приводит соответственно к гепгахлордио к со димолибдату и гексахлоран! имона-ту адамантилиденадамантанхлорония [81]
2MOOCU + А = А
-----------► [А^-^А]Ф [Мо2О2С17] ®
2SbCl5 + А = А ------------►
[А'^^^А] 9 [SbCl6] ° + SbCl3.
Вследствие значительного экранирования кратной связи присоединение дихлоркарбеиа к (14) происходит в 20 раз медленнее, чем к углеводороду (12),R = R' = H.
Взаимодействие (14) с трифторуксусной кислотой в растворе дихлор-метана в атмосфере кислорода в темноте [82] и реакция с,эО2 при каталитическом действии трис(о,л-дибромфенил)аминийгексахлорантимината в том же растворителе при -78" С [83] приводит к быстрому окислению олефииа с образованием дноксетана в качестве главного продукта.
о—о
I I
А—А 607.
Интересными синтетическими возможностями обладает 2-циклопента-дненилидеиадамаитан (17), легко получаемый из адамантаноиа-2 и циклопентадиена [84]. Гидрирование его приводит к 2-циклопентенил- и 2-циклопе нтил адамантану [84, 85]. Углеводород (17) активно вступает в реакции конденсации с диенофилами, например [86]:
227
4.3. АДАМАНТАНЫ
С КРАТНОЙ СВЯЗЬЮ В БОКОВОЙ ЦЕПИ
Производные адамантана,относящиеся к этой группе, включают соединения, содержащие ненасыщенный заместитель как в узловом, так и в мостиковом положении, а также соединения, имеющие два (одинаковых или разных) заместителя в положениях 1 и 3. Для их синтеза обычно с успехом используют традиционные методы, хотя известны и некоторые исключения. Так, несмотря иа широкое варьирование условий реакции дегидратации 1-(1-гидроксиэтил)адамантана получить 1-виниладамантан (18) не удалось [87]. Неудачной оказалась также попытка синтеза (18) пиролизом четвертичного аммониевого основания - гидроксида 2-(1-адамантил) этил три-метиламмония [88],
1 -Винил ада манта и получают дегидрированием 1-этил адамантана иа катализаторе, содержащем алюминий и оксиды хрома, молибдена, вольфрама [89] или дегидратацией 1-(1-гидроксиэтил)адамантана [90]. С наибольшими выходами (85-95%) (18) получали пиролизом бората и ацетата исходного спирта иад оксидом алюминия при 350—400° С.
Термическое расщепление 2- (1-адамаитил)этил-3-метилксаитогената приводит к соединению (18) с выходом 77% [88].
270° с
Ad(CH2)2OCSCH3 —► AdCH=CH2 + S=C(OH)SCH3. (18)
S (18)
Дегидратацией бората 1-(2-гидроксиэтил) адамантана при 300° С получен (18) с выходом 80% [91]. 1-Виниладамантан является продуктом дебро-мнроваиия (Zn, ДМФА) 1,2-дибром (1-адамантил)-этана [92,93].
Взаимодействие адамантана с ацетиленом под действием 7-излучения б0Со также приводит к соединению (18) (8%) [94]. Метод, очевидно, не будет иметь препаративного значения, но ои интересен как первый пример образования 1-виниладамаитана по радикально-цепному механизму.
Предложен способ получения гомологов 1-виниладаматана взаимодействием 1-пропениладамантанов с этиленом в присутствии алюмо рениевого катализатора (Re2O7 - 10-20%, Ai2O3 - 80—90%) при 20е С и атмосферном давлении [95]. Пропениладамантаны образуются при взаимодействии адамантана и его гомологов с пропиленом иад промышленными алюмо-окисиыми и цеолитсодержащими катализаторами [96]. Если исходить из адамаитаиопа-1, то в качестве катализатора может быть использована также трифторуксусная кислота [97,98].
Независимо от способа получения 1-пропениладамантана (19)одновременно с ним образуется его изомер — 1-ал пил а дамантан (20). При дегидрогалогенировании 1-(2-бромпропил) адамантана в реакционной смеси (19) и (20) содержатся в количествах 80 и 7,4% соответственно [99].
AdCH2CHBrCH3—► AdCH=CHCH3 + AdCH2CH=CH2
(19) (20)
Продукты пиролиза ацетата 1-(1-адамаитил)пропанола-2 содержат 70%
228
(19) и 30% (20) [100]. Вероятно, соединение (19) термодинамически более устойчиво.
1-Алпиладамаитаи получен де бромированием 1,2-дибром-3- (1-адамаи-тил)пропана цинком в кипящем этаиопе с выходом 89% [99], а также реакцией 1-хлорадамаитана с СН2=СНСН2 Si(CH3)3 при участии кислот Льюиса (TiCl4) [101,102].
1-Изо пропенил адамантан (21) синтезирован дегидратацией третичного спирта - 2- (1-адамантил)пропанопа-2 - с помощью уксусного ангидрида [103] или иода [104].
AdC(CH3)2OH—> AdC(CH3)=CH2.
(21)
При попытке синтеза трет-бутил адамантана из 2-(1-адамантил)пропаио-па-2 в качестве основного продукта также получили (21) [105].
SOBr, CH.Mgl
AdC (СН3) 2 ОН----► AdC (СН3) 2 Вг ---*• AdC (СН3) 3
I------------- AdC(CH3)=CH2 -<-----------1
(21)
1-Изопропенил-3,5,7-триметиладамантан (22) синтезирован [106] дегидратацией соответствующего спирта, который был попучеи из метилового эфира 3,5,7-триметил-1-адамаитаикарбоновой кислоты (23).
CH.Mgl -н,о
RC (О) ОСН3 —----> RC (СН3) 2 ОН-----RC (СН3) = СН2
(23) (22)
1-Изопропеиил-2-метиладамантан является продуктом дегидратации 2-(2-метил-1-адамантил)пропанопа-2, протекающей под действием уксусного ангидрида [107].
Для синтеза бутеииладамантанов (24) и (25) был использован 1-(1-ада-мантил)-2-метилпропаиоп-2 [108]. Дегидратация протекает как при нагревании спирта с уксусным ангидридом [107] или с безводной щавелевой кислотой [100], так и в процессе обработки этого спирта бромистым водородом в ледяной уксусной кислоте [100, 108]. Очевидно, превращение спирта протекает двухстадийно — через промежуточное образование третичного карбкатиона (26), который стабилизируется за счет злектронодоиор-иого влияния адамантанового ядра.
НВг, СН.СООН Ф
AdCH2С(СН3)2ОН---------------- AdCH2C(CH3)2 ->
-н®
-> AdCH2C(CH3)=CH2 + AdCH=C(CH3)2
— (24) (25)
^/d+ СН2=С(СН3)2
Дальнейшие превращения иоиа (26) происходят с образованием в качестве электрофильного фрагмента более устойчивого 1-адамантильиого ка-
229
тиоиа или с отщеплением протона и образованием непредельных соединений.
Адамантаны с ненасыщенной боковой цепью в положении 2 ядра практически не изучены. 2-Винил адамантан может быть получен с высоким выходом путем прямого превращения адамантаноиа под воздействием 0-[(триметилсилил)этил]лития [109].
A=O+(CH3)sSiCHfCtt?Li
-78е С
> А
/ОН ЧН/
ЬГ51СНэС00Н(СН1С»,,
7Z7. (CHs,i
/Н ЧН=СНг
100 7.
Адамантаны, содержащие два непредельных заместителя в молекуле, получены аналогичными методами. Так, 1,3-динзопропеииладамаитаи был синтезирован дегидрохлорированием бисхлорпроизводиого углеводорода [110].
1-Аллил-З-вииил- и 1,3-диаллиладамаитаиы получены дегидробромиро-ваннем соответствующих бромпроизводных [99].
Непредельные углеводороды ряда адамантана, содержащие двойную связь в a-положении боковой цепи, являются, как правило, жидкостями с температурами кипения выше 200°С, достаточно устойчивыми при нагревании. Исследование масс-спектров этих соединений показало, что их молекулярные ионы (М+) значительно более устойчивы, чем М+ соответствующих алкиладамантанов (для 1-этил- и 1-виииладамаитаиов интенсивность М+ составляет 4 и 92% соответственно) [111, 112], интенсивность ионов с т/е 135 (или их гомологов), соответствующих элиминированию алкенильных групп, невелика. Эти данные свидетельствуют о большей по сравнению с ал кил адамантанами прочности Сад-Салк-связи в алкеиил-адамаитаиах. Возможно, это результат взаимодействия С=С-связи с электронной плотностью внутри ядра адамантана; последняя может образоваться за счет перекрывания внутри каркаса тыльных сторон орбиталей четырех узловых атомов углерода (предполагаемый ’’эффект клетки”). Такое взаимодействие, одвако, если оно и существует, энергетически незначительно, так как заметного его влияния на химические свойства соединений не наблюдалось. Так, 1-винил- и 1-аллиладамаитаиы проявляют одинаковую реакционную способность при взаимодействии с иодом [113]. Гидрированием этих олефинов с хорошими выходами (80-95%) получены соответствующие алкиладамантаны [88, 100, 108]. 1-Виниладамантаи обычно присоединяет НВт по правилу Марковникова. В определенных условиях, однако, основным продуктом присоединения НВг к 1-виниладамантану
230
является ие 1-бром-1-(1-адамаитил)этаи (27), а 2-бром-1-(1-адамаитил)-этаи [114].
5® s® н®(нвг) ф Ф Вг9
AdCH=CH2-----► AdCH2CH2-----> AdCHCH3—> AdCHBrCH3
[вг9 , <27)
1---► AdCH2CH2Br
73%
Присоединение брома к 1-виниладамантаиу дает 1-(1-адамантил)-1,2-дибромэтан, дегидро бромирование которого приводит к смеси непредельных соединений [114].
с, н.он + кон
AdCH=CH2 +Вг2---> AdCHBrCH2Br—--------*
5 ч, кипячение
->AdCBr=CH2 + TpaHc-AdCH=CHBr +AdC=CH 59% 34% 7%
Осуществлено эпоксидирование 1-виииладамантаиа с помощью гидропероксида трет-бутила в присутствии Мо(СО)6; 1-эпоксиадамантаи запо-лимеризоваи с раскрытием эпоксидного цикла [115].
Введение адамантанового ядра в третичные гидропероксиды должно существенно влиять на их перегруппировку в кислой среде, поскольку отличительной особенностью производных адамантана является большая склонность к образованию .карбкатиона. Так, гидропероксиды, полученные из 1-изопропенил- и 1-изо бутенил адамантанов в 70%-иой H2SO4 в Присутствии циклогексана, образуют сложную смесь продуктов [116].
грег-ROOH н, so. л
AdC (СНз) = СН2-----> AdC (СНЭ) 2 О AdC ® (СН3 ) ОСНЭ
I ; —|
AdCH2CH3 AdH AdCOCH3
AdCH=CH2 AdOH 14%
AdCH(OH)CH3 51% 35%
При взаимодействии с иодистым метиленом 1-изо про пений- и 2-изопро-пеииладамантаны [117] или З-бром-1-изопропеииладамаитан [118] в присутствии цинк-медной пары или с диазометаиом в присутствии бромистой меди образуют соответствующие производные циклопропана; гидрогенолиз последних приводит к алкиладамантанам.
Разнообразные кремнийоргаиические производные адамантана с заместителем в узловом положении, в том числе непредельные, получены путем гидросилилирования 1-винил- и 1-этиниладамаитанов [118а]. Образуются только 0-изомеры (в случае 1 -этиниладамаитаиа — 0-трднс-изомеры); ре-гиоселективность реакции, вероятно, связана с положительным индуктивным эффектом (а* = -0,26) адамантильиой группы.
231
, H.PtCL • н,о,ызо-с3н,он
Ad—СН= СН2 + RR] SiH-------------- ^Ad-CH 2 СН2 SiRR2,
Н
Ad -C=CH+ RR’SiH -> Adot-SiRR’ А I *
н
[a)R - 2-тиенил, R* =СН3; б) R=C6HS,R1= СН3; в) R=C1,R1 = СН3;
г) R=R‘ =С2Н5].
Непредельные производные адамантана активны в реакции 1,3-полирно-го циклоприсоединения N-оксидов нитрилов [113]. Реакционнаи способность исследованных непредельных соединений измениется в ряду:
AdCH=CH2 ^AdCH2CH=CH2 > AdC(CH3) = CH2 > AdC=CH .
Такой порядок снижения реакционной способности можно объяснить частично влиииием стерических факторов.
Ненасыщенные углеводороды ряда адамантана вступают в реакцию полимеризации. Так, на катализаторе Циглера - Патта получен полимер 1-аллил-адамантана с выходом 12,5%. Сополимеризация 1-аллиладамантана с винил циклогекса ном привела к сополимеру с выходом 10% [99]. Изучена кинетика сополимеризации 1-винил-3,5-днметиладамантана с винилацетатом [119].
В безводных растворителих под действием А1ХЭ (X = С1, Вг) протекает катионная полимеризация 1-внниладамантана [120,121]; температура начала распада поли(1-адамантана) 300° С [91].
A1CL
AdCH= СН2----AdCH2 СН2 - [-СН (Ad) СН2 -] п -CH (Ad) СН2 Ci.
Применение других катализаторов типа кислот Льюиса (эфират трехфтористого бора* борфторид триэтил о ксония) не приводит к образованию полимера.
Для непредельных производных адамантана
СН2 = С (СН3) С (О) O(R) AdX; СН2 = СН-0 (О) С (R) AdX
[X = Н,Вт,С(О)ОСНз; R = (СН2)„, и = 0^2]
было установлено, что введение адамантилького радикала в состав мономеров не только не ингибирует их полимеризапию. но и в некоторых случаях приводит к возрастанию полимеризационнй способности. В ряду метакриловых производных адамантана и виниловых эфиров адамантанкарбоновых кислот характер изменения констант полимеризации указывает на то, что по мере удаления адамантильного ядра от двойной связи полимеризацион-ная способность мономера снижается [122—124]. Отличительными свойствами полимеров, полученных иа основе этих производных адамаитаиа, является термическая стойкость, устойчивость к гидролизу и повышенная поверхностная твердость.
232
4.4. АДАМАНТАНЫ С НЕСКОЛЬКИМИ КРАТНЫМИ СВЯЗЯМИ
1.3-Дивинил адамантан получен [124а] восстановлением диэтилового эфира адаманталиден-1,3-диуксусной кислоты под действием LiAlH4, последующим превращением полученного диола в эфир бориой кислоты и пиролизом этого эфира, общий выход 30%.
Изучены также соединения, содержащие в боковой цепи несколько двойных связей. Действием бутиллнтия на спиро [2",4-дибром (или 2'2-дихлор)-aдaмaнтaн-2^l,-циклопропан] получен винилиден адамантан (28а) [71].
X
ащ=н; б)ц=сн5;
(28) b)R=CtH5,X=Cl,Br.
Аналогично синтезированы 1-лропенилиден- (286) и 2-фенилэтннили-деиадамантан (28в) [125]. Дегидратация 2-аллиладамантанола-2 приводит к аллилидеиадамантану (29) [126].
хСН2—СН=СН2 SOC1,, пиридин 12 3
Af ----------*-А= СН-СН=СН2.
он -н»о
(29)
Метилпропенилидеиадамантан (30) получен с использованием в качестве дегидробромирующего агента трет-бутилата калия в диметилсульфоксиде [125].
грег-С.Н9ОК ДМСО I 2
А=С(Вг)СН(СН5)2 ----~-----------*- А=С=С(СН3)2.
(30)
Продукты реакции адамантэнона-2 с 1-фенил-3-хпорпропеном-1 и магнием после дегидратации дают смесь адамантилиденпропенов-2 [125].
1,2-Диадамантилиденэтан получен действием литии на бромметилеиада-маитан в присутствии CuBr.
Li, CuBr
А=СНВг------> А=СН-СН=А.
В реакции дигалокарбенов с алленами ряда адамантана выявлены закономерности в изменении реакционной способности двойных связей в зависимости от величины заместителя, степени замещения и удаления двойных связей от ядра [125].
Реакционная способность ближайшей к ядру двойной связи С(1) = С(2) винилиденадамантана (28а) 2-А=С=СН2 в 12 раз ниже, чем в винилиден-циклогексаие, ив 1,8 раза ниже, чем в 1,1-диметил ал лене. Причина этого заключается в пространственном экранировании ее адамантановым ядром. Активность ближайшей к ядру С(1)=С(2)-связи 1-пропенил иденадаманта-иа (286) по сравнению с соединением (28а) меньше почти в 2 раза, а в ме-
16. Зак. 1856
233
тил про пенилиденадамантане (30) равна нулю. Очевидно, такое снижение реакционной способности объясняется стерическим взаимодействием между карбеном и метильными группами при С(2)-атоме в переходном состоянии. В соединении (28а) подход карбена к плоскости связи С(1)=С(2) одинаково возможен с обеих сторон; в (28б) один из путей подхода заслонен метильной группой, а в соединении (30) метильные заместители с обеих сторон экранируют связь С (1)=С (2).
Реакционная способность концевой связи С(2)=С(3) в аллилиденада-мантаие (29) в 6 раз выше, чем в бутадиене-1,3, и в 20 раз выше реакционной способности двойной связи в метилеиадамантане (12) (R = R’ = Н). Активность связи 2-А=С(1) диена (29) в 36 раз ниже активности связи С(2)=С(3). По-видимому, основной причиной различной активности двойных связей в аллилидеиадамантаие, как и в случае винилиденадамантаиа, является пространственное влияние адамантанового ядра, экранирующего прилегающую к нему связь 2-А=С(1). С другой стороны, снижение реакционной способности связи 2-А=С(1) в аллилиденадамаитане по сравнению с метиленадамантаиом в 1J8 раза свидетельствует о том, что в случае диена (29) действует не только стерический фактор. Благодаря поляризации молекулы донорный эффект адамантанового заместителя передается на концевую двойную связь С(2)=С(3) и активирует ее, в то время как ближняя к циклу связь 2-А=С(1) относительно дезактивируется. Таким образом, адамантановое ядро оказывает сильное пространственное влияние иа примыкающую к нему кратную С=С-связь, экранируя ее и затрудняя к ней подход карбена.
Описаны 1-адамантилзамещеииые аллены — 1-(1-адамаитил)-3-хлорал-леи [127] и 1,3-ди (1-адамантил) алл ей [128].
SOCL
AdCH(OH)-C=CH -------AdCH=C=CHQ,
LiAlH.
AdC=CLi + AdCHO AdC=C-CH(OH)Ad -----------AdCH=C=CHAd.
При нагревании аллены, как правило, полимеризуются; одиако при определенных условиях из 1-(1-адамантил)-3-хлораллеиа можно выделить димерные продукты цикпобутановой структуры. Попытка получить димеры при нагревании 1,3-ди(1-адамаитил)аллена оказалась неудачной; реакционная масса содержала только полимерные продукты.
Алленовые производные адамаитаиа (31) получены на основе алкеии-иов и AdBr (реакция протекает в присутствии металлического лития) [129].
, Li, ТГФ л е . AdBr
RCC-C(R>CH2 -------------RC=C=C(Rr)CH2--------------->
e . H,o ,
-> RC=C=C(R )CH2 Ad —----► RCH=C=C(R )CH2 Ad [a) R = C2 H5, R' = H;
(31)
6) R = C(CH3)3, Rr = H;
в) R=R' = CH3], 234
Действием пр опар гил три мети леи лана на 1-хлорадамаитаи синтезирован 1-адамаитилаллеи [101, 102].
Ada + СН=С-СН2 Si(CH3 )з -+ AdCH=C=CH2.
Обработкой 2,5-дигидрокси-2-этиниладамантаиа тионилхлоридом- получают хлораллеи с сохранением конфигурации [130]. Аллеи (32) можно получить из кетену [131].
0-^=0
* л « А’СЬ *1 1 Z60“C . п л
А=0=0--------А->А=1—А ——-*А=С=А
—сог
(3?)
Подобно непредельным углеводородам алифатического ряда, непредельные производные адамантана легко вступают в реакцию с карбенами с образованием замешенных спиро [адамантанциклопропанов] (выходы до 75%) [132,133].
-у
А У A^-V9 (X=Y=ci; х=Y=Br;
R х X=H,Y=CO2C2t15).
Сопряженные диены и аллены ряда адамантана образуют в этих условиях моио- и диадцукты.
(к'=W, в'=н,сд),
4R'=H,CHS; Вг=Н,СН,|.
2-Фенилэтенилидеиадамантаи в реакцию с дихлоркарбеиом не вступает [133].
Высокой реакционной способностью характеризуются ацетиленовые производные адамантана. 1-Этиниладамаитаи получают взаимодействием AdBt с винилбромидом в присутствии А1Вг3 и последующим дегидрогалогенированием образовавшегося 1,2-дибром-1-(1 -адамантил) этана с помощью КОН в диэтнлеигликоле. 1-Этиниладамантан вступает во многие реакции, свойственные ацетиленовым углеводородам [134].
235
HgO,н, so.
----——AdCOCH3
AdC=CH 'Cu(OAc), h«
------—* AdC=C-C=CAd-------> Ad(CH2 )4 Ad
Ni Ренея
Na, CH,OH
AdOCH -lC°a
AdC(OCH3)=CH2
CaHsMgI, CH3C(O)CHS
—J---> AdOCC(CH3)2OH
C-H.Mgl, C,H.C(O)Ce H.
L2_2-----6 L* AdC=CC(C6 H5 )2 OH
AdOC-COOH
Синтезированы также производные 2-этиниладамантаиа [ 130]:
(X = CltOH).
Предполагается, что реакции 1-этиниладамантаиа с 90%-ной H2SO4 и НВг в ледяной СН3СООН проходят через промежуточно образующиеся адамантилвииильные катионы (33), легко изомеризующиеся в более стабильные катионы (34), Пути превращения катионов (33) и (34) зависят от исходных соединений (33) (AdX + НС=СН или AdC=CH), природы уходящей группы X (Вт или ОН) в AdX, степени замещения в адамантановом ядре. Так, 1-этиниладамантан присоединяет НВг в ледяной СН3СООН, образуя смесь всех возможных продуктов [135, 136].
ф
AdC=CH2
©I (34)
Вг (--_---------
Н Ad L
/ НВг НВг \ НВг
С=С --------* AdCHBrCH2 Вг<--- С=СН2--------
IIх 4 Вт Вг-"'
5® 6® НФ(НВг) в быстро
AdOCH -----> AdCH-CH------------
Вг*
Ad Н Вг AdCH2CHBr2
AdCBr2CH3.
Если в реакцию 1-AdBr с винилбромидом в присутствии А1Вг3 проводить в дихлорэтане, то образуются 1,3-дизамещенные дибромиды, дегидрогалогенирование которых приводит к 1,3-диэтиниладамантану [137]; метилированием последнего получен 1,3-ди(1-пропилил) адамантан.
AIBr, кон.дмсо
1-AdBr + СН2=СНВг-----=—► 1,3-А-(СН2СНВг2)2 ----------->
CHjClj
->1,3=А-(ОСН)2 -С - 1.3-А -(С=С-СН3)2.
Эти 1,3-диалкиниладамантаны с пропином в присутствии А1Вг3 вступают в реакцию co-цикл о димеризации и образуют ст-комплексы [137а].
236
Полимерный (полиядерный) комплекс нерастворим в органических растворителях; по мнению авторов, он представляет интерес как предшественник для получения других полимеров.
Таким образом, химия непредельных производных адамантана наряду с тем общим, что свойственно химии непредельных соединений других алициклических углеводородов, характеризуется рядом особенностей, обусловленных прежде всего наличием объемистого адамантанового ядра. Вследствие жесткости структуры адамантана дегидроадамантаны представляют собой высоконапряженные реакционноспособные углеводороды, что позволяет получать на их основе в одну стадию самые различные моно- н дизамещенные производные адамантана. Реакционная способность соединений, содержащих экзоциклическую кратную связь, заметно ниже из-за пространственного ее экранирования; вместе с тем эти соединения представляют интерес в связи с возможностью получения стабильных комплексных соединений. В производных с кратной связью в ajB-положении боковой цепи С=С-связь испытывает заметное экранирующее и электроиодо-норное влияние адамантильного радикала. Эти соединения также весьма реакционноспособны, вступают в реакции гомо- и в особенности сополимеризации; они могут быть использованы для получения полимерных материалов, а также различньгх функциональных производных адамантана, представляющих интерес с точки зрения их биологической активности. Доступность этих соединений может быть существенно увеличена за счет технических смесей алкеннладамантанов, получаемых окислительным дегидрированием алкиладамантанов [137].
ЛИТЕРАТУРА
1. Иди ел Э.Л Стереохимия соединений углерода. М.: Мир, 1965. 460 с.
2. Lenoir D. // Tetrahedron Lett. 1972. N 40. P. 4049-405 2.
3. Grant D., McKervey M.A., Rooney J. J. et al. // J. Chem. Soc. Chem. Commun. 1972. N 21. P. 1186-1187.
4. Bums K, McKervey M.A. // Ibid. 1974. N 21. P. 858-859.
5. Bums IP., Grant D,, McKervey M.A,, Step G // J. Chem. Soc. Perkin Trans. Part 1. 1976. N 2. P. 234-238.
6. Gillespie D.G.. Walker B.J. Ц Tetrahedron Lett. 1977. N 19. P. 1673-1676.
T Alberts A.H.. Strating J., Wynberg H. 11 Ibid. 1973. N 32. P. 3047-3050.
8. Cadogan Leardini R. // J. Chem. Soc. Chem. Commun. 1979. N 17. P. 783-784.
237
9. Conlin R.I., Miller R.E.. Michl J. // J. Amer. Chem. Soc. 1979. Vol. 101, N 25. P. 7637 7638.
10. Gagosian R.B., Dalton J.C., Turro N.J. // Ibid. 1970. Vol. 92, N 15. P. 4752 -4754.
11. Gano J.E., Eizenberg L. // Ibid. 1973. Vol. 95, N 3. P. 972-974.
12. Schwarz H., Reetz M.T., Maier W.F et al. // Angew. Chem. 1979. Bd, 91, N 12. S. 1019.
13 .Maier W.F., Schleyer P.R. // J. Amer. Chem. Soc. 1981. Vol. 103, N 8. P. 1891-1900.
14. Adams B.L., Kovacic P. Ц Ibid. 1973. Vol. 95, N 24. P. 8206-8207.
15. Adams B.L., Kovacic P. //Ibid. 1974. VoL 96, N 22. P. 7014-7018.
16. Adams B.L., Kovacic P. // J. Org. Chem. 1974. VoL 39, N 21. P. 3090-3094.
17. Martella D.J., JonesM., SchleyerP.R., Maier W.E // J. Amer. Chem. Soc. 1979. VoL 101, N 25. P. 7634-7637.
18. Klebach T.C. Jones M., Kovacic P. // Tetrahedron Lett. 1981. VoL 22, N 6. P. 497-500.
19- Farcasiu M„ Farcasiu D., Conlin R. T. et al. // J. Amer. Chem. Soc. 1973. Vol. 95, N 24. P. 8207-8209.
20. Gerlach H. // Hetv. chim. acta. 1972. VoL 55, N8.P. 2962-2964.
21. Pincock R.E., Torupka EJ. // J. Amer. Chem. Soc. 1969. VoL 91, N 11. P. 4593.
22. Pincock R.E., Schmidt J., Scott W.B., Torupka E.J. // Canad. J. Chem. 1972. Vol. 50, N 24. P. 3958-3964.
23. Но Б.И, Сон B.B., Белякова T.B., Куликова Н.И. 11 Науч. конф, по химии орган, полиэдранов: Тез, докл. Волгоград, 1981. С. 82.
24. Scott W.B., Pincock R.E // J. Amer. Chem. Soc. 1973. Vol. 95, N 6. P. 2040-2041.
25. Лейбзон B.H.t Майрановский С.Г., Краюшкин M.M. и др. // V111 Всесоюз. совещ. по электрохимии орган, соединений: Тез. докл. Рига: Зинатне, 1973. С. 11-12.
26. Соколенко В.А., Когай Б,Е. // Журн. орган, химии. 1976. Т. 12, № 6. С. 1370-1371.
27. Когай Б.Е., Губернаторов В.К., Соколенко В.А. // Там же. 1984. Т. 20, № 12. С. 2554-2558.
28. Кишкань Л.Н., Маркова В.А., Когай Б.Е., Соколенко В.А. // Науч. конф, по химии полиэдранов : Тез. докл. Волгоград, 1976. С. 51.
29- Соколенко В.А., Маркова В.А., Когай Б.Е. // Журн. орган, химии. 1978. Т. 14, №5. С. 1111.
30. Когай Б.Е., Карпицкая ЛГ., Меркушев Е.Б. и др. // Науч. конф, по химии полиэдранов: Тез. докл. Волгоград, 1976. С. 52.
31. Лейбзон В.Н., Мендкович А.С., Климова T,A.t Краюшкин М.М, // Науч. конф, по химии орган, попиэданов: Тез. докл. Волгоград, 1981. С. 81.
32. Wiberg К.В., Соппоп Н.А., Pratt W.E // J. Amer. Chem.Soc. 1979. VoL 101, N 23. P. 6970-6972.
33. Смирнов А.Г., Кочмарева Т.И, Когай E.E. и др. // Науч. коиф. по химии полиэдранов: Тез. докл. Волгоград, 1976. С. 53.
34. Когай Б.Е., Соколенко В.А. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 73.
35. Когай Б.Е, Соколенко В.А., Фурин Г. Г., Якобсон Г.Г. // Там же. С. 74.
36. Но Б.Н., Сон В.В., Ущенко В.П. и др. // Там же. С. 108.
37. Udding А.С., Strafing J.r Wynberg И. // Chem. Commun. 1966. N 18. P. 657-658.
38. Исаев С.Д., Юрченко А.Г., Степанов Ф.Н. и др. // Жури, орган, химии. 1973. Т. 9, № 2. С. 430.
39. Hirsl-Starcevic S., Majerski Z. // J. Org. Chem. 1982. Vol. 47, N 13. P. 2520-2525.
40. Boyd J., Overton K.H. // J. Chem. Soc. Perkin Trans. Part 1. 1972. N 20. P. 2533 — 25 39.
41. Kropp P.J.r Gibson J.R., Snyder J. J., Poindexter G.S. // Tetrahedron Lett. 1978. N 3. P. 207-210.
42. Udding A.C., StratingJ., Wynberg H. // Ibid. 1968. N 11. P. 1345-1350.
43. Farcasiu D., Schleyer P.R. // Ibid. 1973. N 39. P. 3835-3838.
44 Storesund N.J. // Ibid. 1971. N 42. P. 3911-3914.
45 Murray R.K., Morgan T.K., Babiak K.A. 11 J. Org. Chem. 1975. VoL 40, N 8. P. 1079-1083.
46. Murray R.K., Babiak K.A. // Tetrahedron Lett. 1974. N 4. P. 319-322.
47. Karlovic G.r Majerski Z. // J. Org. Chem. 1978. Vol. 43, N 4. P. 746-747.
238
48. Baldwin J.E., Foglesong W.D. // J. Amer. Chem. Soc. 1968. Vol. 90, N 16. P. 4303-4310.
49. Murray R.K., Babiak K.A. Ц J. Org. Chem. 1973. Vol. 38, N 14. P. 2556-2557.
SQ.Mlinaric-Majerski K., Majerski Z. Ц J. Amer. Chem. Soc. 1980. Vol. 102, N 4. P. 1418-1419.
51. Majerski Z., Mlinaric-Majerski K., Meic Z. Ц Tetrahedron Lett. 1980. Vol. 21, N 42. P. 4177-4188.
52. GelukH.W., Boer Th J. Ц Tetrahedron. 1972. Vol. 28, N 12. P. 3351-3359.
53. Gehik H.W., Boer Th. J. //J. Chem. Soc. Chem. Commun. 1972. N 1. P. 3-4.
54. B/wiey F., Faulkner D.r McKervey M.A. // Synth. Commun. 1973. Vol. 3, N 6. P. 435.
55. Schleyer P.R.. Nicholas R.D. Ц J. Amer. Chem. Soc. 1961. VoL 83, N 1. P. 182-187.
56. Kuthan J., Palecek J., Musil L. // Coll. Czechosl. Chem. Commun. 1973. VoL 38, N 11. P. 3491-3495.
57. Landa S., Vais J., Burkhard J. Ц Ibid. 1967. Vol. 32, N 2. P. 570-575.
58. Vais J., Burkhard J., Landa S. Ц Ztschr. Chem. 1969. Bd. 9, N 7. S. 268-269.
59. Багрий Е.И., Сагинаев A.T. // Успехи химии. 1983. T. 52, № 9. С. 1538-1567.
60. Fty J.L., Engler Е.М., Schleyer P.V.R. Ц J. Amer. Chem. Soc. 1972. Vol. 94, N 13. P. 4628 -4634.
61. Cordt F., Frank R.M., Lenoir D. // Tetrahedron Lett. 1979. N 6. P. 505-508. bl.KeulH. И Chem. Ber. 1975. Bd. 108, N 4. S. 1198-1206.
63. Strafing J., Scharp J., Wynberg И. Ц Rec. trav. chim. 1970. Vol. 89, N 1. P. 23-31.
64. Geluk H.W. И Synthesis. 1970. N 12. P. 652-653.
f>5. Schaap A. P.t Faler G.R. 11 J. Org. Chem. 1973. Vol. 38, N 17. P. 3061-3062.
66. Gill G.B., Hands D. Ц Tetrahedron Lett. 1971. N 2. P. 181-184.
67. Lenoir D., Frank R. Ц Ibid. 1978. N 1. P. 53-56.
68. Janku J., Landa S. Ц Coll. CzechosL Chem. Commun. 1970. Vol. 35, N 1. P. 375-377.
69. Sasaki 71, Eguchi S.r Tanida M. et al. // J. Org. Chem. 1983. Vol 48, N 10. P. 1579-1586.
70. Vais J., Burkhard J.r Landa S. Ц Ztschr. Chem. 1968. Bd. 8,N 8. S. 303-304.
71. Sasaki T., Eguchi S., Hirako Y. // Tetrahedron Lett. 1976. N 7. P. 541-544.
72. Buss V., Gleiter R., Schleyer P.R. Ц J. Amer. Chem. Soc. 1971. VoL 93, N 16. P. 3927-3933.
73. Костиков P.P., Гришина Е.И., Слободин Я.М. Ц Журн. орган, химии. 1979. Т. 15, №2. С. 331-336.
74. Wieringa J.H., Strating J., Wynberg И. Ц Tetrahedron Lett. 1970. N 52. P. 4579-4582.
75. Strating J., Wieringa J.H., Wynberg H. // Chem. Commun. D. 1969. N 16. P. 907-908.
76. Olah G.H., Schilling P.r Westerman P.W., Lin H.C. Ц J. Amer. Chem. Soc. 1974. Vol.96, N 11. P. 3581-3589.
77. Garratt D.G. Ц Tetrahedron Lett. 1978. N 22. P. 1915-1918.
78. Толстиков ГА., Лерман Б,М., Уманская Л.И. Ц IX Междунар. симпоз. по химии орган, соединений серы: Тез. докл. Рига, 1980. С. 98.
79. Tolstikov G.A., Lerman В.М., Umanskaya L.L et al. Ц Tetrahedron Lett. 1980. Vol. 21, N43.P. 4189-4192.
80. Толстиков Г.А„ Лерман Б.М., Уманская Л.И.г Стручков Ю.Т, Ц Изв. АН СССР. Сер. хим. 1982. № 3. С. 661-669.
81. Nugent W.A. Ц 3. Org. Chem. 1980. VoL 45, N 22. P. 4189.
82. ЛАпЬй R., Sakuragi H., Tokumaru К. Ц Tetrahedron Lett. 1984. VoL 25, N 6.P. 665-668.
83. Nelsen S.E, Kapp D.L., Teasley M.E Ц J. Org. Chem. 1984. Vol. 49, N 3. P. 579-580.
84. Landa S, Burkhard J, Vais J. Ц Ztschr. Chem. 1967. Bd. 7, N 10. S. 388-389.
85. Багрий Е.И., Фрид Т.Ю., Санин П.И. // Нефтехимия. 1980. Т. 20, № 6. С. 812-818.
86. Шокова Э.А., Ковалев В.В.Г Платэ А.Ф. Ц Там же. 1977. Т. 17, № 2. С. 252-256.
87. Stetter H.r RauscherE. // Chem. Вег. 1960. Bd. 93, N 9. S. 2054-2057.
88. Степанов Ф.Н., Столяров З.Е. // Журн. орган, химии. 1969. Т. 5, № 1. С. 91-92.
89. Пат. 3255268 США, МКИ3 С 07 С. Vmyladamantanes / G. Suld, R.E. Moore // Chem. Abstr. 1966. VoL 65. 3767a.
90. Пат. 3433844 США, МКИ! С 07 С. Vmyladamantanes / ЕС- Capaldi, L.N. Leum Ц Ibid. 1969. Vol. 71. 3053.
91. Zuanic M.r Majerski Z.r Janovic Z. Ц J. Polym. Sci.: Polym. Lett. Ed. 1981. Vol. 19, N8.P. 387-389.
239
92. Stepanov FJV., Suchoverchov V.D., Yurchenko A. G. // Tetrahedron Lett. 1971. N 36. P. 3357-3358.
ЭЗ. Йосида Дзэнтъити // Катаку. 1972. T. 27, № 7. С. 712-715; РЖХим. 1973.4Н137-
94. Подхалюзин А.Т., Викулин В.В., Верещинский И.В. // Укр. респ. конф, по химии и перспективам применения углеводородов ряда адамантана и родств. соединений: Тез. докл. Киев, 1974. С. 88.
95. А.с. 789472 СССР, МКИ С 07 С 13/54, С 07 С 5/52. Способ получения 1-винил-адамантана или его замещенных / В.Н. Соловьев, Е.И. Багрий, С.М. Носакова, Л.Г. Либеров, П.И. Санин // Б.И, 1980. № 47. С. 92.
96. А.с. 639844 СССР, МКИ С 07 С 13/54. Способ получения Ci э-С, в-алекенил-адамантанов / В.Н. Соловьев, В.Г. Заикин, Е.И. Багрий, НИ. Санин // Б.И. 1978. № 48. С. 81.
97. А.с. 606853 СССР, МКИ С 07 С 35/22. Способ получения 1-(адамантил-1)-пропанола-1 / В.В. Ковалев, Э.А. Шокова, А.Ф. Платэ //Б.И. 1978. № 18. С. 70.
98. Ковалев В.В., Шокова ЗА. // Жури, орган, химии. 1981. Т. 17. № 1. С. 109-116.
99. Пат. 3457318 США, МКИ3 С 07 С. Alkenyl adamantanes and their polymers useful for manufacture of molded articles IEJZ. Capaldi, A±. Borchert // Chem. Abstr. 1969. Vol. 71. 81931V.
100. Степанов Ф.Н, Сидорова Л.И., Довгань НЛ, // Журн. орган, химии. 1972. Т. 8, № 11. С. 2338-2342.
101. Sasaki T.f UsukiA., OhnoM. // Tetrahedron Lett. 1978.N 48. P. 4925—4928.
102. Swett T., Usuki A., Ohno M. // J. Org. Chem. 1980. Vol. 45, N 18. P. 3559-3564.
103. Grob CA., Schwarz W., Fischer HP. // Helv. chim. acta. 1964. Vol. 47, N 6. P. 1385-1401.
104. A.c. 304248 СССР, МКИ С 07 С 13/54, С 07 С 3/00. Способ получения 1-изоалкил-адамантанов / Я.Ю. Полис, Б.П. Рагуель // Б.И. 1971. № 17. С 84.
105. Ланда С., Буркхард Й. // Нефтехимия. 1968. Т. 8, № 3. С. 323—325.
106. Юрченко А.Г. Исследование взаимных превращений производных адамантана и бицикло[33.1] нонана: Дис.... д-ра хим. наук. Киев, 1974. 334 с.
107. Landa S., Hlavaty J. // Coll. Czechosl. Chem. Commun. 1971- Vol. 36, N 8. P. 3059-3065.
108. Степанов Ф.Н, Сидорова Л.И., Довгань НЛ. // Журн. орган, химии. 1973. Т. 9, № 7. С. 1545,
109. Wilson S.R., Shedrinsky А. // J. Org. Chem. 1982. Vol. 47, N 10. P. 1983-1984.
110. Landa S., Kamycek Z. // Coll. Czechosl. Chem. Commun. 1959. Vol. 24, N 4. P. 1320-1326.
Ill, Заикин В.Г., Соловьев B.H, Сметанин В.И. и др. // Изв. АН СССР. Сер. хим. 1975. №7. С. 1633-1636.
112. Полякова АЛ., Храмова Э.В., Багрий Е.И. и др. // Нефтехимия. 1973. Т. 13, № 1. С. 9- 16.
113. Сафонова О А. Реакция 1,3-диполяриого циклоприсоединения N-окисей нитрилов с непредельными производными адамантана: Дне. ... канд. хим. наук. М_, 1975. 125 с.
114. Полис Я.Ю., Грава ИЛ., Лиепиныи Э.Э., Шатц ВД. // Науч. конф, по химии полн-эдранов-. Тез. докл. Волгоград, 1976. С. 24.
115. Пат. 3536732 США, МКИ3 С 07 D. Adamantyl epoxides whose polymers are useful in making way additives, gaskerts and rubberlike compositions / A Ji. Borchert, E.C. Capaldi // Chem. Abstr. 1971. Vol. 74.4282u.
116. Довгань НЛ., Баклан В.Ф., Сидорова Л.И., Христин А.И. // Укр. респ, конф, по химии и перспективам применения углеводородов ряда адамантана и родств. соединений: Тез. докл. Киев, 1974. С. 84.
117. Woodworth C.W., Buss К, Schleyer P.R. // Chem. Commun. 1968. N 10. P. 569-570.
118. Broxton T.L, Capper G., Deady L.W. et al. // J. Chem. Soc. Perkin Trans. Part II. 1972. N9.P. 1237-1240.
ИЗаЛукевиц Э., Стуркович Р.Я., Пудова OA. и др. // Журн. общ. химии. 1985. Т. 55, №3. С. 624-627.
119, Miller R.S. Kinetics of the free radical initiated copolymerization of 1-vinyl-3,5-dimethyladamantane and vinyl acatate and the glass ttansition temperatures of the resultant co-plymers: Diss. N.Y., 1970.505 p.; Chem. Abstr. 1971. Vol. 75.49831j.
240
120. А.с. 478840 СССР, МКИ С 08 F 7/02. Способ получения полнвиннладамантана / Р.А. Зосим, Н.Л. Довгань, А.Г. Юрченко // Б.И. 1975. № 28. С. 58.
121. Довгань НЛ., Зосим ЛЛ., Юрченко А.Г. и др. // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1976. № 13. С. 54-55.
122. Новиков С.С., Хардин А.П., Радченко С.С. // Укр. респ. конф, по химии и перспективам применения углеводородов ряда адамантана и родств. соединений: Тез. докл. Киев, 1974. С. 5.
YlAzJMajerskl Z., Skar D., Vulic L. // Synth. Commun. 1986. Vol. 105, N 9. P. 51; Chem. Abstr. 1986. Vol. 105.78559V.
123. Новиков C.C., Хардин АЛ., Радченко С.С. и др. // Науч. конф, по химии полиэдра-нов: Тез. докл. Волгоград, 1976. С. 63.
124. Новиков С.С., Хардин АЛ, Радченко С.С. и др. // Изв. АН СССР. Сер. хим. 1977. № 12. С. 2765-2767.
125. Гришина Е.Н, Исследование реакции дигал окарбенов с производными адамантана. содержащими углерод-углеродные кратные связи: Дис. ... канд. хим. наук, Л., 1980. 115 с.
126. Bemaert Е., Dannels D., Anteunis М., Verhegge G. // Tetrahedron. 1973. Vol. 29, N 24. P. 4127-4136.
127. Jacobs T.L., Muscio O.J. // Tetrahedron. Lett. 1970. N 55. P. 4829-4832.
128. Jacobs T.L., Kammerer R.C. // J. Amer. Chem. Soc. 1972. Vol. 94, N 20. P. 7190-7191.
129. Романов O.E., Зубрицкий Л.М., Бальян X.B. // Журн. орган, химии. 1977. Т. 13, №2. С. 468.
130. Noble W.L., Chiou D.-M., Matuszynska H, Okaya Y. Ц Tetrahedron Lett. 1977. N 4. P. 3865-3868.
131. Strafing J., Alberts A.H, Wynberg H. //Chem. Commun. D. 1970. N 13. P. 818.
132. Гришина E.H., Костиков P.P., Слободин Я.М. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 79.
133. Костиков Р.Р., Гришина Е.Н., Слободин ЯМ. // Жури, орган, химии. 1984. Т. 20, № 7. С. 1426-1435.
134. StetterH, Goebel Р. // Chem. Вег. 1962. Bd.95, N 4. S. 1039-1042.
135. Полис Я.Ю., Грава ИЛ., Лиепинъш Э.Э., Майц В.Д. // Науч. конф, по химии полиэдранов.- Тез. докл. Волгоград, 1976. С. 23.
136. Bott К. /I J. Chem. Soc. Chem. Commun. 1969. N 22. P. 1349-1350.
137. Сагинаев A.T., Багрий Е.И. // Нефтехимия. 1982. Т. 22, № 6. С. 729-734.
137Л-Вгохterman Q.B., Hogeveen Н, Kingman R.F. // Tetrahedron Lett. 1986. Vol. 27, N 9. P. 1055-1058.
Глава 5 Перспективы и основные направления практического использования углеводородов ряда адамантана и их производных
Высокосимметричная компактная структура молекулы адамантана и других каркасных углеводородов сообщает необычные свойства их производным, в том числе такие, которые обусловливают их использование не только для научных исследований н в технике. Такие соединения применяются для получения медицинских препаратов, высокомолекулярных (полимерных) материалов, синтетических смазочных масел, резин, устойчивых к растворителям.
Естественно, что широкое использование в науке, технике и медицине углеводородов ряда адамантана и их производных станет возможным лишь при условии их доступности и дешевизны. Этн показатели зависят прежде всего от технологии получения и объема производства. В табл. 5.1 показана зависимость стоимости одного из перспективных для практического использования углеводорода -- 1,3-диметиладамантана (13-ДМ А) и его производного — 5,7-диметиладамантандиола-13 (5,7-ДМА-диола-1,3) от объема их производства [1].
Одна нз японских фирм предложила технологическую схему непрерывного процесса получения адамантана. В США фирма ’’Дюпон” производит адамантан для покрытия собственных потребностей в сырье с целью получения лекарственных препаратов; а фирма ”Сан Ойл” осуществила пробный выпуск 13-диметиладамаятана [2]. В Европе адамантан производят
Таблица 5.1
Зависимость стоимости 1,3-диметиладамантана от объема его производства [1 ]
Объем производства, т/год Цена за 1 л, доллары
1,3-ДМА 5,7-ДМА-днол-1,3
230 2,5-5,0 5,0-10,0
460 — 2,5-5,0
4600 0,5-1,0 -
фирмы ’’Силаг хеми” (Швейцария), ’’Филлипс—Днодпер” (Нидерланды) и ’’Марк—Шукардт”(ФРГ). В настоящее время практически весь производимый адамантан, производство которого составляет, вероятно, не более нескольких десятков тонн в год (официальные данные отсутствуют), используется для получения лекарственных препаратов — снмметрела (амантадина), ремантадина и некоторых других.
242
5.1. ПЕРСПЕКТИВЫ ИСПОЛЬЗОВАНИЯ АДАМАНТАНОВ И ИХ ФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
Как показали многочисленные исследования, диапазон возможного использования адамантановых углеводородов и их производных чрезвычайно широк. Онн перспективны для получения на их основе термо-стабильных смазочных материалов, полимеров, а также взрывчатых веществ.
Адамантан может быть использован в качестве основы для получения душистых веществ, адамантилгексанол и 1-(фенилэтокси) адамантан — в качестве душистых веществ; перфторированный адамантан предложен в качестве компонента искусственной крови, различные производные адамантана — возможные антистатики, поверхностно-активные вещества, пластификаторы, инсектициды, бактерициды, замасливатели для волокон нт. д. [2].
На основе адамантана получают алмазоподобные пленки, по своей твердости лишь в 3 раза уступающие алмазу. Они были получены из паров адамантана при наложении днух видов электрического разряда — тлеющего и высокочастотного [2а]. Такие пленки, нанесенные на кварцевую или молибденовую поверхность, увеличивают твердость поверхности соответственно в 1,3 и 2,1 раза. Пероксидные производные адамантана, в частности 1-адамантил-1рет-бутилмонопероксикарбонат, используются в качестве инициаторов блочной полимеризации метилметакрилата [3].
Еще в то время, когда не были разработаны препаративные методы получения алкиладамантанов, эти соединения, характеризующиеся подходящими значениями таких показателей, как плотность и соотношение С и Н, было предложено использовать в качестве углеводородного реактивного топлива [4]. В этой связи были определены их температуры воспламенения (табл. 5.2) [5].
Техническая смесь алкиладамантанов с т. кип. 220—240° С (она была получена изомеризацией гидрированного пиролизного масла и содержала ~ 11% декалина) имела т. всп. 290°С. Для получения углеводородного
Таблица 5.2
Температура самовоспламенения циклоалканов
У глвводор оды 7, °C Углеводороды Т,°С
Моноциклические: Би- и трициклические:
Цкклопентан 412 дицикпопеитил 252
Метил цнкпопентан 341 Дициклогексил 256
Этил циклопентан 290 Гидриндан 296
Циклогексан 294 Декалин 268
Этилцикпсгекеан 275 Пергидр оаценафтен 275
1,2,4-Тр иметилцнк логексан 315 Пергидроантрацен 252
Гексаметилциклогексан 340 Пергидрофпуорен 266
Адамантан 287
1,3-Диметил адамантан 288
1-Этил адамантан 271
243
топлива с максимальной устойчивостью к самовоспламенению к циклоалканам следовало добавлять ароматические фракции. Однако алкиладамантаны как ненапряженные углеводороды имеют недостаточно высокую теплотворную способность, поэтому использование их в качестве реактивных топлив в настоящее время представляется нецелесообразным. В качестве углеводородных топлив для реактивных двигателей более пригодны топлива на основе более напряженных три- и полициклических углеводородов, содержащих норборнановую структуру, например гидрированный димер циклопентаднена [6]. Предлагают также в качестве топлива для ди иг ат ел он и ракет и для применения в сигнальных противопожарных системах 13,5,7-тетранитроксиадамантан, полученный нитрованием 13,5,7-тетрагидрокси- или 133,7-тетраацетоксиадамантана смесью 90%-ной HNO3 и 96%-ной H2S04 в СН2С12 при 0°С [6а].
Для техники значительно больший интерес представляет высокая термическая стабильность алкиладамантанов. Так, при нагревании 13-ДМА при 350°С в течение 107 дней на поверхности нержавеющей стали н алюминия был обнаружен только легкий голубоватый налет [7]. Важно и то, что 13-ДМА характеризуется весьма низкой токсичностью. В этой связи 13'ДМА, как и другие алкиладамантаны с короткими алкильными группами, рекомендовано использовать в качестве рабочих жидкостей в газотурбинных установках. Целесообразность применения его для этих целей подкрепляется еще тем, что для алкиладамантанов характерна большая разница между критической температурой и температурой кипения (435 и 201,5°С, что на 63°С выше чем, например, для додекана — 385 и 214,5°С соответственно).
Цикло алкиладамантаны характеризуются высоким коэффициентом трения. Так, для циклогексил-13*диметиладамантана он равен 0,055 [8]. Таким же коэффициентом трения (0,05) и хорошими вязкостными свойствами обладает смесь полиалкиладамантанов состава С22—С3о (ее получают обработкой низших алкиладамантанов конц. H2SO4) в композиции с гидрированным полнизобутиленом (60%) [9, 10]. Продукт рекомендуют использовать в качестве компонента гидравлической трансмиссионной жидкости с улучшенным коэффициентом сцепления. В этих целях рекомендуют использовать алкиладамантаны также в патентах [11, 12]. Путем алкилирования адамантана, низших алкиладамантанов, диадамантила или диамантана нзооктаном получают бутиладамантановые масла [13]. Масло, основной компонент которого 13^5,7-тетранзобутиладамантан, имело прн 50° С ркин = 467,2 сст, прн 100° С — 30,6 сст, индекс вязкости — 2, т. заст. — 24°С. т. всп. 202°С, плотность при 20°С — 0,9075 г/см3. Масла такого типа рекомендовано использовать в качестве термостойких синтетических смазочных масел или их компонентов, а с учетом их хороших электроизоляционных свойств н низкого индекса вязкости — в качестве кабельного масла.
Весьма перспективны в качестве синтетических смазочных масел сложные эфиры — производные адамантансодержащих спиртов н алифатических кислот [14]. Основная особенность таких эфиров — их повышенная термическая стабильность по сравнению с маслами, применяемыми в настоящее время в промышленности (турбинное масло фирмы ЭССО-15 и ВНИИНП-50-1-4) (табл. 5.3) [15]. Ввиду повышенной термической 244
Таблица 5.3
Термическая стабильность синтетических эфирных масел (%) при400°С [15]
Эфирное масло Время нагревания, ч
1 I- 2 4 6
Турбовинтовое масло фирмы 100 100 100 100
ЭССО-15 Масло ВНИИНП-50-1-4 100 100 100 100
[бис (2-этилгексил) себацинат] Неопентилы ир истат 11 16 38 42
(3,5-Димег ил -7-этил-1-адамантил- 6 12 17 29
метил)лауринат 3,5-Дим етил-1-адам антилметилми- 3 7 24 32
ристат 1-Неопентил алкил адамантил- 2 5 14 21
карбоксилат
Примечание. В процентах указана степень разложения.
стабильности масла на основе адамантандиолов (например, производные 13*Диметил адамантандиола-5,7) предложено использовать в качестве масел для авиационных диигателей [2].
Алкиладамантаны обладают бактерицидным действием, вследствие чего рекомендовано использовать их в качестве антимикробных присадок к смазочным материалам [16]. Диметилбутил-, диметилоктил- н ди-метилциклопентиладамантаны предлагают применять [17, 18] в качестве компонентов эффективных смазочных жидкостей для фрикционных передач. Для этих же целей рекомендуют высоко- или сполна фторированные адамантаны [19]. Онн обладают хорошими диэлектрическими свойствами, высокой устойчивостью к кислотам, щелочам, окислителям н восстановителям, а также негорючи, не вызывают коррозию, имеют высокую плотность. В качестве жидкостей для трансмиссионных н фрикционных систем предложено использовать высшие алкиладамантаны и диадамантилалканы [20,21].
Простые эфиры алкиладамантанов могут служить добавками, повышающими окислительную стабильность и вязкость смазочных масел и трансмиссионных жидкостей [8, Ilf 22—25]. В качестве антиоксидантов и синтетических добавок предложено использовать N-галоформамидо- и бнс-(N-галоформамидо)алкиладамантаны [26].
В качестве добавок к смазочным маслам, повышающих их термическую н окислительную стабильность, рекомендовано использовать днэфиры алкиладамантанолов и дикарбоновых алифатических кислот типа [27]
245
полиалкиленадамантаны [28] — в качестве уплотнительных жидкостей и термостойких связующих.
Среди других перспективных направлений применения производных адамантана следует отметить возможность использования 1- (2Н-гекса-фторпропил)-3-бромадамантана в качестве охлаждающей негорючей жидкости, стойкой к гидролизу [29]; N-метилстеариламида 1-адамантанкарбоновой кислоты — в качестве селективной стационарной жидкой фазы в ГЖХ (рабочий диапазон температур 80—150°С) [30]; (1-адамантил)-1-метил-1-этоксикарбонил ьной группы — в качестве защитной группы в синтезах пептидов [31]. Производные адамантана и некоторых других каркасных соединений служат пластификаторами нитрата целлюлозы [32]; диадамантилхромат является катализатором полимеризации этилена и смеси этилена и пропилена [33].
Многие практически важные направления использования производных адамантана, по-видимому, не публикуются. Известно, однако, что Управление вооружений армии США намеревалось провести испытания нового взрывчатого вещества — 13,5,7-тетранитроадамантана, обладающего высокой стойкостью к ударным нагрузкам и в то же время по эффективности превосходящего ’’классический” тол [34].
В настоящее время активно ведутся работы по расширению применения адамантанов и их производных.
Добавка адамантана, адамантанола, бромадамантана илн ацетиламиноадамантана к сернофосфорному электролиту (смесь H2SO4 и Н3РО4) повышает технологичность процесса электрополирования нержавеющей стали при одновременном улучшении качества обработки [34а, 34б].
Модификация силикагелевой фазы адамантилэтилтрихлорсиланом успешно применяется при анализе с помощью жидкостной хроматографии препаратов от кашля типа эфедрина [34в].
Небольшие добавки (~ 1%) нитрата адамантана к дизельному топливу позволяют увеличить цетановое число дизельного топлива с 50 до 58 единиц [35].
Следует остановиться также иа использовании углеводородов ряда адамантана и их производных для сугубо научных исследований. Так, адамантан служит в качестве матрицы при исследовании методом ЭПР радикалов, образующихся при рентгеновском облучении алифатических кетоиов [36], и в риде других исследований [37—39] для получения и консервирования радикалов.
5.2. НЕКОТОРЫЕ НАПРАВЛЕНИЯ ИСПОЛЬЗОВАНИЯ УГЛЕВОДОРОДОВ РЯДА АДАМАНТАНА
52.1. АДАМАНТАНСОДЕРЖАЩИЕ ПОЛИМЕРНЫЕ МАТЕРИАЛЫ
Весьма перспективны для практического использования полимерные материалы, полученные иа основе производных углеводородов каркасного строения (прежде всего, адамантана и алкиладамантанов); они характеризуются целым комплексом ценных свойств. По этой проблеме опубликованы обстоятельные обзоры [2, 40, 41] и монография [42]. В связи с этим ниже рассмотрены наиболее важные аспекты химии полимеров на 24в
основе адамантанов, в частности вопрос о влиянии полиэдрического углеводородного ядра на свойства полимерных материалов.
Полученные к настоящему времени данные показывают, что введение молекулы адамантана в состав высокомолекулярных соединений существенно улучшает ряд их свойств. Как правило, адамантансодержащие полимеры термостойки и размягчаются при относительно высоких температурах. Они проявляют высокую устойчивость к окислению, гидролизу, воздействию света и растворителей. По этим свойствам адамантансодержащие полимеры превосходят многие известные промышленные полимерные материалы и могут найти применение в различных отраслях техники и народного хозяйства. Один из первых углеводородных полимеров — полиадамантан — был получен путем взаимодействия 33'-дибром-1, Г-ди-адамантана с натрием [43]. Полимер Представляет собой белый порошок,
нерастворимый в органических растворителях и устойчивый к действию конц. НЯ, 20%ного раствора NaOH. При нагревании выше 325° С он постепенно темнеет,но ие плавится до 420°С. Подобные полимеры образуются из 1,3- и 2,4-дегидроадамантанов.
Полиадамантан с метиленовым мостиком в цепи может быть получек катионной полимеризацией 3,7-бисметиленбицикло[3,3,1]нонана [44, 45].
Полимер плавится при температуре выше 340° С и нерастворим в органических растворителях, так же как н полимер, полученный катионной полимеризацией 1-виниладамантана над хлористым алюминием в метиленхло-риде [46].
В патенте [47] сообщается о получении гомополимера 1 -аллиладаманта-на и его сополимера с в ин ил циклогексаном на комплексном катализаторе триизобутил алюминий — четыреххлористый титаи, однако данные о свойствах полимера не приводятся. Плохая растворимость и высокая температура размягчения таких полимеров, вероятно, существенно затрудняют их использование для практических целей.
Сополимеры алкиладамантанов и алкилбензолов характеризуются высокой температурой размягчения (250°С), они образуют защитные пленки и рекомендованы в качестве покрытий с высокой термоокислительной устойчивостью [48].
247
Полимеры иа основе акрилатов, метакрилатов и сложных виниловых эфиров адамантана отличаются высокими температурами стеклования, значительной поверхностной твердостью и высокой вязкостью их растворов. Недостатком их является хрупкость и невысокая механическая прочность, что обусловлено присутствием в макромолекулах объемистого ада-мантильного радикала [42]. Вследствие повышенной вязкости полиакрилаты адамантана используются в качестве загустителей смазочных масел.
Полимеры иа основе производных адамантана устойчивы к действию концентрированных минеральных и органических кислот. Полимеры адамантил ьных производных монометакрилового эфира этиленгликоля в этих кислотах лишь набухают, другие полимеры разлагаются конц. HNO3 только после продолжительного воздействию и медленно деструктурируют-ся в конц. H2SO4. Полиметакрилаты адамантана и гомоадамантана не разрушаются в плавиковой кислоте, а их поливиниловые эфиры не омы-ляются водным и спиртовым растворами щелочей. Поливиниловые эфиры также обладают высокой термостабильностью, начало потери массы при 302~315°С, температура полураспада 360°С. Их алифатический аналог — поливинилацетат — начинает разлагаться уже при 170°С.
Акриловые и метакриловые эфиры адамантанопов сополимеризуются с метилметакрилатом, стиролом, акрилонитрилом и хлористым винилом [49]. Сополимеризация легко протекает как под действием свободнорадикальных и анионных катализаторов, так и УФ-облучения [50], Причем адамантильная группа не ингибирует скорость полимеризации [51]. Свойства сополимеров зависят от их состава, однако во всех случаях сополимеризация с адамантилакрилатами приводит к повышению температуры стеклования и твердости сополимеров.
Полимеры, содержащие остатки амантадина и обладающие способностью к гидролитическому отщеплению молекулы лекарственного вещества, получают следующим образом [51а]: синтезируют винильные производные амантадина и сополимеризуют их с 2-диметиламиноэтилметакрилатом, стиролом или метилметакрилатом. Гидролиз низко- и высокомолекулярных Производных амантадина проводят в воде при 37° С и pH 1—7.
Адамантансодержащие полиэфиры характеризуются высокой устойчивостью к термической и окислительной деструкции, действию УФ-облуче-иия и солнечного света и хорошей гидролитической стойкостью; для них также характерны высокие температуры стеклования и размягчения (табл. 5.4).
По термической и термоокислительной стабильности полиэфиры на основе адамантансодержащих дикарбо новых кислот или диолов находятся на уровне полиарилатов, начало потери массы которых при нагревании на воздухе 350-370°С, однако оии превосходят последние по стойкости к гидролизу.
248
Таблица 5-4
CHj
290 350 370 415
Примечание. В скобках — температура нагрева в атмосфере .
300 360 425 470
Адамантансодержащие полисульфоны и полисульфонаты в отличие от полисульфонатов на основе других углеводородов прочные, жесткие, ио не хрупкие. Они размягчаются при 180°С и обладают высокой окислительной стабильностью [52, 53].
Значительный положительный эффект проявляется при введении ада-мантильных групп в полимерные цепи при получении полиамидов [42].
17. Зак. 1856
249
Наибольший интерес среди полимеров этого типа представляет поли(л4-фениленадамантилен-13 -дикарбоксамид) — продукт конденсации м-феии-лендиамина с хлорангидридом 1,3-адамантандикарбоновой кислоты. Поли-
мер образует термостойкие пленки н волокна с т. разм. 294°С, при выдерживании на воздухе в течение 2 ч при 325° С потеря массы не превышает 0,86% [42, 54]. Полученные на основе этого полиамида волокна обладают рядом других интересных свойств: они аморфны, полученное из них полиамидное волокно отличается высокой термической стабильностью и сохраняет 50% первоначальной прочности при выдерживании в течение 100 ч при 250°С [42,55- 57].
Из мономеров 2-(1-адамантил)- и 2-(1-адамантилметан)-2-оксазолинов получены блокполимеры, а также диблок и триблоксополимеры со вторым
н-с=о
мономером — 2-этил-2-оксазолином [57а]. Блокполимеры высококристал-личны (т. пл. адамантилсодержащего полимера 269° С, адамантилметансодержащего — 320°С), плохо растворимы в обычных органических растворителях. Диблокполимеры обладают обратной эмульгирующей способностью при эмульсионной полимеризации в системе стирол + вода.
Бесцветные прозрачные полимеры с малой усадкой после полимеризации и высоким показателем преломления получают путем полимеризации диаллиловых эфиров адамантан- или 1,3-адамантилидендикарбоно-вых кислот в присутствии пероксидных инициаторов [576].
Большой практический интерес представляют циклоцепные полимеры с адамантиленовыми группами — полннмиды, например:
Исходными мономерами для их получения служат диамины адамантанов
и диангидриды ароматических тетракарбоновых кислот.
Эти полимеры ие уступают ароматическим лолинмидам по термической стабильности и в отличие от последних бесцветны [42, 58, 59]. Они устойчивы к гидролизу, воздействию органических растворителей и активных
250
химических реагентов, размягчаются при нагревании и могут перерабатываться прессованием или поливом из раствора.
Например, полинмид, полученный иа основе 4,4'-диаминодифенилового эфира и диангидрида 1,3-бис (3,4-дикарбоксифенил) адамантана устойчив при действии конц. H2SO4 и 10%-иого NaOH (при выдерживании в этих средах в течение 120 ч при 25°С сохраняется 84—86% первоначальной прочности), в то время как аналогичный полиимид иа основе пиромепли-тового диангидрида в этих условиях нацело разрушается. Повышенная гидролитическая устойчивость адамантан содержащих полиимидов, вероятно, связана с их явно выраженными гидрофобными свойствами и с замедлением диффузии гидролизующего реагента к реакционным центрам полимера [42]. Полиимиды, синтезированные из 1,3-ди (аминометил)-5,7-диметиладамантана и ангидрида пиро меллитовой кислоты, имеют желтоватый оттенок; оии не плавятся при 500° С и совершенно устойчивы при 370° С даже в присутствии кислорода.
Обычно разложение адамантансодержащих полиимидов начинается в пределах 370—400° С. По термоокиспигольной устойчивости эти полими-ды превосходят аналогичные полимеры на основе 1,6-гексаметилендиамина или 1,4-диаминоциклогексана, однако немного уступают ароматическим полиимидам.
Адамантансодержащие полиди (метиленимииы) общей формулы (CH2NRNCH2)n, где R — адамантилен, п = 3 -г 10, применяют в качестве вулканизующих агентов с целью повышения (в 1,3 раза) термостойкости фторсодержащих резин и увеличения срока службы деталей из этих резин, работающих при повышенных температурах [60].
Весьма ценными свойствами обладает адамантансодержащий поли бензоксазол; его получают из хлорангидрида 5,7-диметил-1,3-адамаитандикарбоновой кислоты и 3,3-дигид рок си бенз идина [40, 61].
Полимер бесцветен и растворим в воде. Пленка, полученная из него, имеет Стр = 1,2 МПа, модуль при растяжении Ер = 210 МПа и при нагревании до 500° С теряет в массе только ~10%.
Важный класс полимеров на основе каркасных углеводе родов составляют полиуретаны. Они могут быть, в частности, получены на основе адамантил ен диизоцианатов и полиолов [40, 62]. Такие полимеры служат основой для создания светостойких полимерных материалов, дающих волокно типа ’’спандекс”. Пленки из этого полимера отличаются прозрачностью, низкой оптической плотностью, высокой устойчивостью к УФ-облучению и повышенной гидролитической стойкостью [63—66]. Можно полагать, что использование адамантансодержащих компонентов для конструирования таких сополимеров, как сетчатые и линейные полиуретаны, позволит существенно повысить их светостойкость.
251
Адамантан содержащие поликарбонаты и сополикарбонаты могут быть получены на основе адамантанбисфенопа. Гомополимер адамантанбисфе-иола превосходит некоторые гомополимеры бисфенолов по термическим и оптическим свойствам, но уступает им в прочности на растяжение. Сополикарбонаты из 1,3-бис (4-гидроксифенил) адамантана начинают терять массу при 250° С; их показатель прочности иа разрыв стр = 91 МПа [67,68]. Присутствие адамантильиых группировок в составе поликарбонатов всегда приводит к значительному повышению их теплостойкости и термической стабильности. В зависимости от структуры бисфенола температура размягчения составляет обычно 240—350° С, а температура начала разложения -300- 350° С [69].
Окислением алкениладамантанов получают адамантан со держащие эпоксиды [70, 71] типа
(R, R1, R2) А(СН2 ЬR3 (R, R1, R3 = Н, алкил Cj _ х 0;
О R2 = алкил Ci_! о,
алкенил С2 _ 10; п = 0,1; R + R1 +R2 <20).
Полимеризация этих соединений приводит к полиэпоксидам, предствля-ющим собой вязкие жидкости или твердые вещества. Они перспективны для использования в качестве присадок к смазочным маслам. Полиэпоксиды с в ин ильными или аллильными группами могут вулканизироваться, образуя резиноподобные материалы. По термостойкости эпоксидные смолы на основе адамантана или 1,1-диадамантила превосходят полиэпоксид с изопропилиденовыми группами. Температура размягчения адамантановых эпоксидов 250 и 220°С соответственно, а изопропилиденового — 180°С, начало потери массы у адамантановых эпоксидов 390° С, у изопропилиденового - 340° С [40, 72, 73]. Полимеры, полученные на основе адамантансодержащих эпоксидных соединений типа
характеризуются хорошей устойчивостью к нагреванию [74].
Винил алкиладамантаны, в том числе 1-в ин ил-3,5-диметил адамантан, а также алкиладамантилакрилаты использовали в качестве сомономеров [75—77]. В работе [78] описаны кремний со держащие полимеры на основе адамантана типа
(»= 35тчэ; w=f-rf5).
252
При введении триметил сил ок си-0-адамантил этил сил оксановых звеньев в диметилсилок сановую цепь пов ысипись уров ень межмоп екулярного взаимодействия и потенциал внутреннего вращения звеньев вокруг силоксановой связи, в результате чего возросла жесткость цепи и термическая стабильность олигомеров.
Следует ожидать интересных свойств для полимеров на основе фторал-кениладамантанов; последние стали доступными благодаря разработке методов радиационного алкилирования адамантана и алкиладамантанов гексафторпропиленом и последующего дегидрофторирования [79].
Большой интерес предствляют биологически активные высокомолекулярные соединения на основе каркасных углеводородов. Такие полимеры могут служить основой для создания противовирусных и других лекарственных препаратов пролонгированного действия. Так, например, полимер строения
СО I О I снг I so
I см3
сн3 сн3
— ( —СНг — С-)„- ( — снг — с — )т-I со
о I снг 1 снг I R
[к = - Ж СНэ1г - СНг CON Н - Ad ]
обладает противовирусной активностью [80].
Полимерные комплексы солевого типа, полученные обработкой 1-амино-адамантаном и 1-(2-аминоэтил)адамантаном сополимеров поливинилового спирта или N-винилпирролидона с N-внниламидоянтарной и кротоновой кислотами, менее токсичны, чем хлоргидраты аминопроизводных адамантана, а срок профилактического действия их против вирусных заболеваний в 5 раз больше [40, 81]; на основе этих комплексов разработаны лечебные препараты пролонгированного противовирусного действия.
Таким образом, рассмотренные в этом разделе данные показывают, что введение углеводородного каркаса в молекулы полимера позволяет существенно улучшить многие их свойства. Тем не менее такие полимерные материалы до сих пор промышленностью не выпускаются, вероятно, прежде всего ввиду отсутствия промышленного процесса получения исходных углеводородов; есть основания полагать, что вскоре ситуация заметно улучшится.
253
5.2.2. БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ И ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ
В настоящее время широко исследована физиологическая активность производных углеводородов каркасного строения, и прежде всего производных адамантана; некоторые из них уже используются в качестве лекарственных препаратов. Активность этих соединений вызвана явно выраженной липофильной природой компактного каркасного углеводородного фрагмента, а также, вероятно, абиогенным происхождением большинства этих соединений.
Из производных адамантана обладают физиологической активностью прежде всего азотсодержащие производные, в особенности имеющие аминогруппы. В 1966 г. после обширных клинических исследований фирмой ’’Дюпон” был введен в медицинскую практику гидрохлорид 1-аминоада-мантана (фирменные названия — мидантаи, амантадин, симметреп) [I]. Препарат активно предохраняет от респираторного заболевания, вызванного вирусом типа А2. Механизм действия препарата состоит в том, что он не убивает вирусы, а лишь блокирует их проникновение в клетку, вследствие чего он особенно эффективен для профилактики инаранинх этапах заболевания [82]. Имеется, однако, другая точка зрения на механизм действия препарата, согласно которой аминоадамантан и его производные ’’вмешиваются” в ранние этапы репродукции вирусов, а их ингибирующее действие связано с латентным периодом — началом репликации и синтеза вирус-специфических РНК [83].
Симметреп эффективен и против гриппа у лошадей [84], а также против вируса саркомы Рауса, поражающего птиц, и вируса лейкоза цыплят. У относительно устойчивых к этим заболеваниям цыплят рост опухолей почти полностью прекращается, небольшая опухоль даже иногда регрессирует.
Другим эффективным противовирусным препаратом, используемым в настоящее время, является гидрохлорид 1-( 1-адамантил) этил амина (Du Ponts’remantidine, ремантадин) [85, 86]
NMg-HCl
Противовирусная активность ремантадина характеризуется более широким по сравнению с симметрепом спектром противовирусного действия, меньшей токсичностью и более выраженным терапевтическим эффектом [86, 87].
Высокую противовирусную активность в отношении вирусов, к которым симметреп неактивен, — herpes simplex, strains of rhino viruses — показали производные алкиладамантанов, такие, как 1-гидрокси-3,5-диметил-7-этиладамантан [88] и 1-мето кси-3,5-диметил адамантан [89]; эти соединения также активны и в отношении вирусов А2. В качестве противовирусного препарата предлагают использовать также 1-амино-3^-диметил-7-этилада-мантан [90].
В последние годы противовирусная активность была обнаружена [91] У ряда галоген-, гидрокси- и меркаптопроизводных адамантан со держащих
254
амидов общей формулы I-AdCONHCH2CH2X и l-AdNHCOCH2X, где X = “ ОН, SH, C(S)(NH2b> Hal. Эти соединения активны в отношении вируса гриппа А, вируса классической чумы птиц, штамма В ей бридж и вируса Синдбис; введение нзотиурониевой группы приводит к увеличению как противовирусной активности, так и токсичности.
Против болезни Паркинсона активны хлорангидриды 3,5,7-замещениых 1-аминоадамантанов (заместители: Н, СН3, С2Н5, С3Н7) [92] - Некоторые из них могут быть использованы в качестве антагонистов дофамина. Кура-репо добную активность проявляют некоторые четвертичные аммониевые основания с 2-адамантильным радикалом [93], противовирусную - производные 2-(1-адамантил)имидазола [94], противокаталептическую — производные 1-аминоадамантана и 3,3-диамино-1,1-диадамантила [95], бактериостатическую — адамантанкарбоновые кислоты [96] и фосфаты адаманта-нолов, адамантантиолов и их производных [97], противосудорожную — оксимы адамантил-а-гидроксиметилкетоиов [98], замещенные в положении 3 (заместители: □, Br, I, F, CeH5, NO2, ONO2), адамантанкарбоновые кислоты и их производные (хлорангидриды и сложные эфиры) [99], амиды адамантан- и 1-бромадамантанкарбоновой кислот и- диамид адамантан-2, 6-диоксо-1,3,5,7-тетракарбоновой кислоты [100]. 2-Бром- и 2-хлор-адамантил-1-метиламины проявляют в 2 раза большую противопаркинсо-ническую активность, чем симметреп [101] .Противовирусной активностью обладают также замещенные 1-аминопроизводные адамантана [102], 1 - (4-этиламинофенил )ада мантан [103], N,N*-6hc[ 1-адамантил] формамид [104], а-метил-1-адамантилметиламин [105], М-(1-адамантил)мочевина [106], а также производные гомоадамантана, такие, как 3-аминогомоадамантан [107], 3-гомо-Ас1СН2 NH2, 3-roMO-AdCH(CH3)2NH2, 3-гомо-Ad(CH3)2NH2 [108] и другие производные каркасных углеводородов.
1-N-(Р-Гидроксиэтиламино)метиладамантаны и их соли служат исходными веществами для синтеза радиопротекторов [109], 2-бис(и-заме-щенные) фенил метил енадамантаны являются ингибиторами оплодотворения крыс [110]. Бактерицидную, фунгицидную н гербицидную активность проявляют диалкиламиноалкиловые эфиры адамантантиокарбоновой кислоты [111]. Анестезирующим действием обладают производные типа l-AdCH2OCH2CH(OH)CH2NRR, [112] и 1-адамантиламмоний-^хлор-этилоксамннат [ИЗ], желчегонным — натриевая соль0-(1-адамаитаноил)-пропноновой кислоты [114].
Производные 1-аминоадамантана типа l-AdNHCONHNHCONH2 являются хемо стерилизаторам и [115], замещенные аминоадамантаиа типа 1-AdNHCH2 CHRCXYZ — депрессантами [116].
На основе 1-адамантилглицина и а-(1-адамантил)аланина получены новые биологически активные производные пенициллина [117]. Некоторые адамантановые а-аминокислоты, такие, как 2-аминоадамантан-2-карбо-новая кислота и N-гидроксиадамантаннн, проявляют противоопухолевую активность в отношении саркомы Эрмеха [118].
(£)-Адамантилаланин, введенный в положение 5 молекулы энкефалина, придает последнему резистентность по отношению к таким энзимам, как химотрипсин, нейтральная протеаза, проназа и термолизин, которые легко разрушают энкефалин 11191.
Комплексы марганец (III)—(адамантаноиламинофенил)порфин в конц.
255
H2SO4 экранируют металлопорфирин и замедляют сольволитическую диссоциацию по связи металл—ацидолиганд [120].
2,2,5,7-Тетрабром- н 2,2,5,7-тетрафторадамантаны увеличивают содержание цитохрома Р 450 в микросомах печени мышей и значительно усиливают его ферментативную активность [121].
В настоящее время получен обширный экспериментальный материал по химиотерапии с помощью производных адамантана, гомоадамантана и других углеводородов. Литература по данному вопросу столь обширна, что заслуживает самостоятельного рассмотрения. Читателю, желающему подробно ознакомиться с вопросами синтеза и биологической аткивности производных каркасных углеводородов, можно рекомендовать ряд обзоров и монографий [1, 87,122—133]. В монографии Форта [124], например, приведены свыше 110 производных адамантана, обладающих различной биологической активностью — противосудорожной, анальгетической, цитотоксической, курареподобной, транквилизационной, гипогликемической, противоаритмической и др. 1-Аминоадамантан успешно испытан на 23 вида противовирусной активности, в том числе против злокачественных вирусов и против болезни Паркинсона.
Исследования в области изучения биологической активности производных каркасных углеводородов продолжают успешно развиваться, осуществляется напранленный синтез химиотерапевтически активных соединений этого типа. Основные методические подходы исследований таковы [134,135]:
— использование высокой липофильности и большого объема адаман-тильиой группы или другого каркасного углеводородного радикала, что определяет проницаемость или адсорбцию вещества на клеточных мембранах, с чем обычно связаны такие виды активности, как противовирусная, иммунотропная и др.;
— введение каркасного углеводородного радикала в структуру уже известных физиологически активных веществ, что может приводить к изменению взаимодействия их с соответствующими рецепторами и, следовательно, менять физиологическую реакцию;
— применение циклической каркасной углеводородной структуры в качестве объекта для построения с ее помощью закрепленных конфигураций физиологически активных веществ.
Модификация биологически активных соединений и лекарственных средств адамантанкарбоиовыми кислотами с большой вероятностью вызывают появление антивирусной активности [135].
Как отмечено в разд. 5.2, весьма перспективным направлением использования производных каркасных углеводородов для получения физиологически активных соединений и лекарственных препаратов широкого спектра активности является введение таких фрагментов в полимерные цепи с целью получения препаратов пролонгированного действия. Адамантильный радикал не является единственным углеводородным радикалом, обусловливающим физиологическую активность. Аналогичный эффект уже получен на примере производных гомоадамантана, норадамантана; без сомнения, носителями биологической активности будут и другие углеводороды каркасного строения, н по мере возрастания нх доступности исследования в этой области будут расширяться.
256
* * *
Таким образом, даже из приведенных, далеко не исчерпывающих данных следует, что углеводороды каркасного строения и их производные могут быть с успехом использованы в науке, технике, медицине, сельском хозяйстве и других областях народного хозяйства. Полученный к настоящему времени фактический материал базируется в основном на производных адамантана и алкиладамантанов, ставших уже относительно доступными соединениями.
Необходимо, однако, отметить, что для широкого промышленного использования производных адамантана и других каркасных углеводородов к настоящему времени в нашей стране сырьевая база еще не создана. Хотя исходные вещества для получения адамантана и алкиладамантанов -циклопентадиен, метил циклопентадиен н аценафтен — имеются в достаточном количестве, крупномасштабное производство адамантанов на их основе не организовано. Вместе с тем такого рода промышленные процессы могут быть созданы без особых трудностей на основе типовой технологии риформинга, крекинга и гидрокрекинга.
Производство и применение продуктов на основе каркасных углеводородов, прежде всего в технике, медицине и ветеринарии, приведет к значительному народнохозяйственному эффекту.
ЛИТЕРАТУРА
1. Moore R.E. // Kirk-Othmer Encycl. Chem. Technol. / Ed. A. Standen, 2nd ed. N.Y.: In-tersci. Publ., 1971. Suppl. Vol. P. 1-15.
2. Kurisaki K. // Kagaki to kogyo-Chem. and Chem. Industry. 1978. Vol. 31, N 4. P. 270-273.
2a. Федосеев Д.В., Варшавская И.Г., Варнин В.П. и др. // Журн. физ. химии. 1987. Т. 61, №1. С. 3070-3073.
3. Богуславская Л.С., Этлис В.С., Бровкина Г. В., Разуваев Г. // Журн. орган, химии. 1970. Т. 6, № И. С. 2243-2245.
4. Landa S. // Сил. ScL (Ind.). 1963. Vol 32, N 11. Р. 485-489.
5-Kuras M., Hala S.r Landa S. // Erdol und Kohle-Erdgas-Petrochem. 1971. Bd. 24, N 7. S. 467-471.
6, Пат. 4222800 США, МКИ2 С 06 В 23/00. Izomerization of endo-hexacyclic .olefinic dimer of norbornadiene / H.K. Myers, A. Schneider; РЖХим. 1981. 10П275П.
6a. Пат. 4476060 США, МКИ С 07 С 77/00. 1,3,6,7-Tetranitroxyadamantane / Е. Gie-bert//РЖХим. 1985. 16Н241П.
7- Пат. 3648456 США, МКИ2 С 09 К. Power generation with Rankine-cycle engines using alkylated adamantanes as working fluids / M.F. Bechtold, C.W. Tullock // Chem. Abstr. 1972. Vol. 77. 37O37h.
8. Пат. 3646234 США, МКИ2 С 07 С. Alkyl adamantanes /R.E. Moore // Ibid. 1971. 75. 109956 r.
9. Пат. 4043927 США, МКИ2 С 10 М 1/20. Friction of tractive drives containing etheres of adamantanes / l.N. Doling, D.S. Gates, F.P. Glazier, R.E. Moore // Ibid. 1977. Vol87. 187080П.
10. Пат. 3648531 США, МКИ2 С 09 К. Friction of tractive drive fluids / l.N. Duling, D.S. Gates, R.E. Moore, F.P. Glazier // Ibid. 1972. Vol. 76. 143219h.
11. Пат. 3646233 США, МКИ2 С 07 С 3/54. Reaction of paraffins with adamantane compounds / R.E. Moore // РЖХим. 1972. 24П347П.
12. Пат. 3382288 США. Alkylated adamantanes /А. Schneider // Chem. Abstr. 1968. Vol. 69. 3558v.
257
13. Hala S., Kuras M., Zachar P. // Sb. VSCHT Praze D. 1976. Sv. 33. S. 305-321; РЖХим. 1977. 13П337.
14. Landa S. // Ropa a uhlie. 1975. Vol. 17, N 12. P. 659-669; РЖХим. 1976. 12П294.
15. Podehradska Vodicka L. // Sb. vSCHT Praze D. 1983. Sv. 47. S. 101-122;РЖХим.
1984. 1ОП312.
16. Пат. 522909 Япония, МКИ2 С 07 С 13/54. Получение адамантанов изомеризацией трициклических насыщенных углеводородов / К. Хоииа, Н. Симидзу, К. Курдза-ки // РЖХим. 1978. 10П275П.
17. Пат. 3597358 США, МКИ2 С 09 К 3/00. Traction drive transmission containing adamantane compounds as lubricant I.N. Doling, D.S. Gates, R.E. Moore, F.P. Glazier // РЖХим. 1972. 10П206П.
18. Пат. 3645902 США, МКИ2 С 09 К 3/00. Friction or tractive drive fluid comprising adamantanes / I.N. Doling, F.P. Glazier, D.S. Gates, R.E. Moore // РЖХим. 1972. 23П233П.
19. Пат. 4041086 США, МКИ2 С 07 С 23/18. Process of manufacture of fluorinated alkyl-aclamantanes / R. E. Moore, E.J. Janovski // РЖХим. 1978. 1П284П.
20. Пат. 2055732 ФРГ, МКИ2 С 07 С ЮМ. Alkyl adamantanes / I.M. Duling, F.P. Glazier, D.S. Gates, R.E. Moore // Chem. Abstr. 1971. Vol. 75. 109956г.
21. Пат 4008251 США, МКИ2 С 07 Л 313/14, С 07 Д 311/80. Reaction of alkyladamantane compounds to form products having two linked adamantane nuclei // РЖХим. 1977. 20П225П.
22. Пат. 3843537 США, МКИ2 С 10 М. Blended traction fluid containing cyclic compounds / I.N. Duling, D.S. Gates // Chem. Abstr. 1975. Vol. 82. 46259n.
23. Пат 3928480 США, МКИ2 С 01 C. Alkyl adamantanes / I. Tabushi, К. Fukunishi // Ibid. 1976. Vol. 84.164288k.
24. Пат. 3966624 США, МКИ2 С 10 M 1/20. Blended traction fluid containing hydrogenated polyolefin and an adamantane ether / I.N. Duling, D.S. Gates, F.P. Glazier, R.E. Moore, T.D. Newingham // Ibid. 1977. Vol. 86.142712h.
25. Spengler G., Wlnsch F. // Erdol und Kohle-Erdgas-Petrochem. 1962. Bd. 15, N 9. S. 702-707.
26. Пат. 1576671 Франция, МКИ2 С 07 С. Adamantane derivatives / G.L. Driscoll // Chem. Abstr. 1970. VoL 72. 89900a.
27. Пат. 1113020 Великобритания, МКИ2 C07 C. Adamantane diester lubricants / Sun oil co // Ibid. 1968. VoL 69. 776749w.
28. Пат. 2611964 ФРГ, МКИ2 С 07 С 69/74. Polyadamantyl compounds / P.J.M. Bauer, G.M.P. Molle, J.L. Daza // Ibid. 1977. VoL 86. 72037г.
29. А.с. 755775 СССР, МКИ С 07 С 23/38, С 07 С 17/10. 1-(2Н-гсксафторпропил)-3-бромадамантан - охлаждающая жидкость, проявляющая стойкость к гидролизу и негорючие свойства / А.Т. Подхалюзин, М.П. Назарова // Б.И. 1980. № 30. С. 132.
30. Зосши Л.А. // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1977. № 14. С. 11-12; Chem. Abstr. Vol. 89. 84354s.
31. Kalbacher H„ Voelter // Angew. Chem. 1978. Bd. 90, N 12. S. 998-999.
32. Юдинова A.A., Федосеев B.A., Юрченко А.Г. и др. // Хим. технология. 1980. № 2. С. 28-30.
33. Пат. 2014031 ФРГ, МКИ2 С 08 F. Polymerization catalysts for olefines / R.N. Johnson, F.J. Karol, L.A. Pilato // Chem. Abstr. 1971. Vol. 74, 3977n.
34. Gilbert E.E., Sollott G.P. // Chem. and Eng. News. 1980. Vol. 58, N 4. P. 32.
34a. Федорова E.A., Митрофанов Э.В., Флеров B.H. // Изв. вузов. Химия и хим. технология. 1986. Т 29, № 5. С. 27-29.
346 Федорова Е.А., Митрофанов Э.В., Флеров В.Н. // Журн. прикл. химии. 1985. Т. 58, № 8. С. 1897-1900.
34в. Yang S.S., Gilpin R.K. // J. Cbromatogr. Sci. 1988. Vol. 26, N 8. P. 416-420.
35. Но И.Б., Ущен ко В.IL, Бутенко Л.Н., Румянцев А.Г. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 132.
36. Camaioni D.M., Walter H.F., Jordan J.E., Pratt D.W. // J. Amer. Chem. Soc. 1973. VoL 95, N 24. P. 7978-7992.
37. Wood D.E., Lloyd R.V. Ц J. Chem. Phys. 1970. Vol. 52, N 7. P. 3840-3841.
38. Wood D.E., Lloyd R.V., Pratt D.W. // J. Amer. Chem. Soc. 1970. Vol. 92, N 13. P. 4115-4117.
258
39. Chang J.C., Thomas T.F. //J. Photochem. 1978. Vol. 9, N 2/3. P. 312-314.
40. Хардин А.П., Радченко С.С. // Успехи химии. 1982. Т. 51, № 3. С. 480-506.
4\.Sakurada I. 1. // Kobunshi Како. 1972. Vol. 21, N 7. Р. 382-383; Chem. Abstr. 1972. Vol. 78. 31202g.
42. Хардин А.П., Радченко С.С. Высокомолекулярные соединения на основе поли-эдрановых углеводородов. Волгоград: Политехи, ин-т, 1981. 130 с.
43. Reinhardt H.F. // J. Polym. Sci. В. 1964, Vol. 2, N 5. Р. 567—568.
44. А.с. 521951 СССР, МКИ С 08 83/00. Способ получения полиметиладам антана / А.Г. Юрченко, Л. А. Зосим, З.Н. Мурзинова, П.А. Красуцкий // Б.И. 1976- № 27. С. 24.
45. Довгань Н.Л., Зосим Л.А., Юрченко А.Г. и др. // Вести. Киев, политехи, ин-та. Хим. машиностроение и технология. 1976. № 13. С. 54—56; РЖХим. 1977. 3C385.
46. А. с. 478840 СССР, МКИ С 08 7/02. Способ получения поливин ил адамантана /Л.А. Зосим, Н.Л. Довгань, А.Г. Юрченко // Б.И. 1975. № 28. С. 58.
47. Пат. 3457318 США, МКИ3 С 07 С. Alkenyl adamantanes and their polymers useful for manufacture of molded articles / E.C. Capaldi, A.E. Borchert // Chem. Abstr. 1969. Vol. 71. 81931V.
48. Пат. 3563919 США, МКИ3 С 08 G 33/00. Copolymers of adamantane and benzene / A. Scheider // РЖХим. 1971. 20С284П.
49. Пат. 363962 США, МКИ3 С 08 F 15/16. Adamantane acrylate and methacrylate esters and polymers thereof / I.N. Duling, A. Schneider, R.E. Moore // РЖХим. 1972. 20С302П.
50. Пат. 3533947 США, МКИ3 С 10 М 1/26. Use of adamantane polymers as VI impovers / I.N. Duling, M. Hoagland // РЖХим. 1971. 17П241П.
51. Новиков С.С., Хардин А.П., Радченко С.С. и др. // Изв. АН СССР. Сер. хим. 1977. № 12. С. 2765-2767.
51а. Sugiyama К., Kinoshita К. // Кинки дайгаку когакубу кэикю хококн. 1984. № 18. С. 27-33; РЖХим. 1985. 22С370.
52. Пат. 373 8960 США, МКИ3 С 08 G. Polysulfone polymers from adamantane bisphenols/ R.M. Thompson, I.N. Duling // Chem. Abstr. 1973. Vol. 79. 79506g.
53. Пат. 3738965 США, МКИ3 С 08 G. Polysulfonate from adamantane bisphenols ! R.M. Thompson, I.N. Duling // Ibid. 1973. Vol. 79. 67074b.
54. Пат. 11994481 Великобритания, МКИ3 С 08 G. Adamantane polymers / W. Flavell, B.A. Nodar // Ibid. 1970. Vol 73.46052d.
55. Пат. 1333084 Великобритания, МКИ3 С 08 G. Spinning of adamantane polyamides / E. Dyson, F. Happey, D.E. Montgomery, I.N. Sewell, K. Tregonning // Ibid. 1974. VoL 80. 84565a.
56. Tregonning K., Dyson E. // J. Appl. Polym. Sci. 1972. Vol. 16, N 8. P. 1911-1924.
57. £>ywn EL, Montgomery D.E., Tregonning K. // Polymer. 1972. Vol. 13, N 2. P. 85-88. 57a. Hsieh B.R., Litt M.H. // J. Polym. Sd. A. 1988. Vol. 26, N 9. P. 2501-2515.
576. Пат. 58-23 2382 Япония, МКИ С 08 P 36/02. Способ получения полимеров из производных адамантана / К. Исакава, Ю. Хаякава, Й. Йосида; РЖХим. 1986. 12С543П.
58. Пат. 3814735 США, СКИ3 С 08 G. Potimide polymer of aklyladamantane diamine and polyanhydride / R.M. Thompson // Chem. Abstr. 1974. Vol. 81. 153090b.
59. Пат. 3847872 США, МКИ3 С 08 G 20/32. Water write polyimide polymer prepared from di(aminomethyl)dialkyladainantanes / R.M. Thompson // РЖХим. 1975. 16С296П.
60. A.c. 602497 СССР, МКИ С 07 С 119/08; C08G 12/06- По л и диметилен имины ряда адамантана - вулканизирующие агенты фторполимеров / С.С. Новиков. А.П, Хардин, С.С. Радченко, И.А. Новаков, А.П. Тусеев // Б.И. 1978. № 14. С. 83.
61- Пат. 2330542 ФРГ, МКИ3 С 08 G. Polybenzoxazoles / G.G. Bellman, A.M. Groult, I.N. Arendt // Chem. Abstr. 1974. Vol. 81. 26216.
62. Пат. 3646095 США, МКИ3 С 07 С 19/04. Organic dHsocyanates of alkyladamantanes / R£. Moore // РЖХим. 1973.1Н96П.
63. Пат. 3301 827 США. Fiber-forming, oxidation-resistant polycyclic diamines / L.M. Ehno-re // Chem. Abstr. 1967. Vol. 66. 76848.
64. A.c. 507Jn4 СССР, МКИ С 08 G 18/75. Способ получения полиуретан мочевин в растворе / О.С. Новожилова, В.С. Наумов, Т.К. Хлыстапова, Л.В. Невский, А.П. Хардин, Н.Г. Гуреев //Б.И. 1976-№ 11. С. 77.
259
65. А.с. 647313 СССР, МКИ С 08 G 18/75. Способ получения светостойких полиуретан мочевин в растворе / А.П. Хардин, И.Г. Гуреев, С.С. Радченко, В.В, Першин // Б.И. 1979. № 6. С. 85.
66. Новиков С.С., Хардин А.П., Гуреев Н.Г., Радченко С.С. // Высокомолекуляр. соединения А. 1976. Т. 18, № 3. С. 619-624.
67. Пат. 3516968 США, МКИ1 С 08 G 17/13. Polycarbonates from adamantyl bisphenols/ RJE. Moore, LN. Duling // РЖХим. 1971. 8С292П.
68. Пат. 3516969 США, МКИ2 С 08 G 17/13. Adamantane copolycarbonates / MJE. Hoag-lang// РЖХим. 1971. 8С293П.
69. Папава Г.Ш., Гелашвили H.C., Беридзе Л.А., Цискаришвили П.Д. Ц Сообщ. АН ГССР. 1977. Т. 88, № 3. С. 597-600.
70. Пат, 3536732 США. МКИ3 С 07 D 1/00. Adamantyl epoxides / AJE. Borchert, Е.С. Capaldi// РЖХим. 1971. 14С294П.
71. Пат. 3676375 США, МКИ3 С 08 G 23/02. Adamantyl epoxide polymers / AJE. Borchert, E.C. Capaldi//РЖХим. 1973. 8С375П.
ll.P ezdirtz G.F., Guidotti V. // J. AppL Polym. Sci.: Appl. Polym. Symp. 1973. N 22. P. 101-102.
TS.Burreson B.J., Levine H,H // J. Polym. Sci.: Polym. Chem. Ed. 1973. Vol. 11, N 1. P. 215-224.
14. Строганов В.Ф., Батог A.E., Петько И.П. и др. // Науч. конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С- 122.
75. Miller R.S. Kinetics of the free radical initiated copolymerization of l-vmyl-3,5-dimethyl adamantane and vinyl acetate and glass transition temperatures of the resultant copolymers: Diss. N.Y., 197 0.505 p.; Chem. Abstr. 1971. Vol. 75. 4983 Ij.
76. Пат. 3433844 США, МКИ3 С 07 С. Vinyladamantanes / Е.С. Capalli, L.N. Leum // Chem. Abstr. 1969. Vol. 71. 3053w.
77. Hoagland M.E., Duling LN. // Amer. Chem. Soc,: Div. Petrol. Chem. Prepr. 1970. VoL 15, N 2. P. B85-B95; Chem. Abstr. 1972. Vol. 76. 487h.
78. Лавыгин И.А., Скороходов И.И., Соболевская Л.В. и др. // Пласт, массы. 1984. №12. С. 14-15.
79. Подхалюзин А. Т., Назарова М.П. // Докл. АН СССР. 1976. Т. 229, № 1. С. 105-107.
80. Ringsdorf Н, Ritter Н, Roily Н. //MakromoL Chem. 1976.Bd.177, N 3. S. 741 — 746.
81. Николаев А.Ф., Бондаренко B.M., Шальнова Л.И. //Журн. прикл. химии. 1976. Т. 49, №7. С. 1601-1605.
82. Hoffman С.Е., Neumayer Е.М., Haff R.F., Goldsby R.A. // J. Bacteriol. 1965. Vol. 90, N 3.P. 623-628.
83. Козелецкая K.H, Дубровина Т.Я., Мещерякова И.Е. и др. // Вирусные ингибиторы н механизм их действия / Под ред. В.П. Ложи. Рига: Зинатне, 1977. С. 55-67.
84. Bryans J.T.r Zent Grunert R,R., Roughton D.C. 11 Nature. 1966. Vol. 212, N 5070. P. 1542-1544.
85. Dawkins A.T., Gallager L.R.t Togo К et aL // J. Airier. Med. Assoc. 1968. VoL 203, N13.P. 1095-1099.
86. Индулен M.K, Конель И.А., Рязанцева Г.М. и др. // Изв. АН ЛатвССР. 1972. № 9. С. 98-106.
87. Злыдников Д.М., Романов Ю.А. // Химиопрофилактика и химиотерапия гриппа / Под ред. А.А. Смородинцева, Д.М. Злыдникова. Л.: ВНИИ гриппа, 1972. С. 71-76.
88. Пат. 3450775 США, МКИ3 С 07 С- l-Hydroxy-3^-dimethyl-7-ethyladamantane / A. Schneider // Chem. Abstr. 1969. VoL 71. 69666e.
89. Пат. 3383423 США. Methoxy alkyladamantanes, antiviral agents / R.E. Moore // Ibid. 1968. Vol. 69. 35579x.
90. Пат. 3450761 США, МКИ1 С 07 С. l-Amino-3-ethyl-5,7-dimethyladamantane I A. Schneider // Ibid. 1969. Vol. 71. 60869г.
91. Галегов Г.А., Петрова И.Г., Леонтьева НА. и др. // Науч. конф, по химики Орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 58.
92,Henkel J.G., Напе J.T., Gianutsos G. // J. Med. Chem. 1982. Vol. 25, N 1. P. 51-56.
93. Харкевич ДА., Сколдинов А.П., Климова Н.В. и др. // Фармакология и токсикология. 1981. Т. 44, №6. С. 670-672.
94. Pellicciari R., Fioretti М.С., Cogolli Р., Tieeeo М. // Arzneimittel-FoTsch. 1980. Bd. 30, N 12. S. 2 1 03 -2 1 05.
260
95. Вихляев Ю.И., Ульянова О.В., Воронина Т.А. и др. // Хим.-фармацевт. журн. 1980. Т. 14, № 3. С. 59-63.
96. Даниленко Г.И., Палий Г.К., Мохорт НА., Баклан В.Ф. // Физиологически активные вещества: Респ. межвсд. сб. 1975. № 7. С. 144—147.
97. Bohringer Я, Vogt Я. // Arch. Pharm. 1978. Bd. 311, N 9. S. 721-727.
98. A.c. 584003 СССР, МКИ С 07 С. 131/00. Оксимы адамантил за мешенных а-окси-метилкетонов, проявляющие противосудорожную активность / А.Л. Фридман, B.Q Залесов, М.П, Сивкова, К.В. Долбилкин, И.А. Колобов // Б.И. 1977. № 46. С. 54.
99. Фридман А.Л., Сивкова М.П, Залесов В.С. и др. // Хим.- фармацевт, журн. 1979. Т. 13, N<> 12. С. 24-31.
100. Фридман А.Л., Залесов В.С., Моисеев И.К. и др. // Там же. 1974. Т. 8, № 7. С. 6-8.
101. Chakrabarti J.K., Hotten Т.М., Sutton S., Utpper D,E. //J. Med. Chem. 1976. Vol. 19, N 7. P. 967-969.
102. Arya V.P., Fernandes F, Ghate S.P. et al. // Ind. J. Chem. 1972. VoL 10, N7. P. 686-690.
103. Митин НИ, Лагуткин И.А., Старовойтова B.A. и др. // Физиологически активные вещества: Респ. межвед. сб. 1977. №9. С. 31-35.
104. Kreutzberger A., Schroders Н.-Н. // Tetrahedron Lett. 1971. N 39. Р. 3635-3638.
105. Пат. 1568056 Франция, МКИ1 С 07 D, А 61 К. Antiviral polycyclic a-methyl-methyl-amines / L.D. Brake // Chem. Abstr. 1970. Vol. 72. 100166 p.
106. Пат. 37035 37 США, МКИ3 С 07 С. N-(l-Adamantyl)-ureas / С. Richter, К. Pluss, G. Eeth // Ibid. 1973. Vol. 78. 7157 Ih.
107. Ин дул ен М.К, Канель И.А., Дзегузе Д.Р. и др. // Ингибиторы вирусной активности / Под ред. Э.М. Пландере. Рига: Зинатне, 1972. С. 41-60.
108. AIdrich Р.Е., Hermann Е.С., Meier W.E. et al. // J. Med. Chem. 1971. Vol. 14, N 6. P. 535-543.
109. A.c. 585155 СССР, МКИ С 07 С 91/06. 1-[К-(0-оксиэтиламино) метил] адамантаны или их соли — исходные продукты для синтеза радиопротекторов / В.М. Плахот-ник, А.Г, Юрченко, В.Г. Ящунский //Б.И. 1977. №47. С. 79.
ПО. Пат. 3711556 США, МКИ2 С 07 С. 2-[Bis(p-substituted phenyl)methylene] adaman-tanes/K.T. Lee // Chem. Abstr. 1973. Vol. 78. 71671г.
111. Пат. 3671527 США, МКИ2 С 07 D. Dialk у lam inalkyl esters of adamantanetiocarbo-xylic acids / C.P. Krimmel // Chem. Abstr. 1972. Vol. 77. 113910e.
112. Pedrazzoli A., DalVAsta L., Ferrero E. // Parmaco. Ed. Sci. 1970. VoL 25, N 11. P. 822-829; Chem. Abstr. 1971. VoL 74. 41992w.
113. A.c. 755778 СССР, МКИ С 07 С 87/46. 1-Адамантиламмонйй-/1-хпорэФилоксаминат, проявляющий местноанестезирующую активность / ГЛ. Петюнин,Т.А. Колесникова, И.Е. Ковалев, И.Д. Ионов, В.В. Шайдров // Б.И. 1980. № 3. С.133.
114. Laurie F, Vecchietti V., Bergamaschi М. // Farmaco Ed. Sci. 1967. VoL 22, N 9. P. 681-691; РЖХим. 1968. 16Ж161.
115. Oliver J.E., Stokes J.B. //J. Med. Chem. 1970. VoL 13, N 4. P. 779-780.
116. Пат. 1181520 Великобритания, МКИ3 С 07 С. Depressant substituted 1-aminoada-mantanes/C. Podesva,C. Solomon//Chem. Abstr. 1970-Vol. 72. 110902q.
117. Пат. 3325478 США. a-Amino-l-adamantylmethylpenicillins / Е.С. Hermann, J A. Snyder //Chem. Abstr. 1967. Vol. 67.90798b.
118. Nagasawa H.T., Elberling J.A., Shirota F.N. // J. Pharm. Set 1980. VoL 69, N 9. P. 1022-1025.
119. Do K.Q., Sehwyzer R. //Helv. chim.acta. 1981. VoL 64, N 7. P. 2084—2089.
120. Ломова Т.Н, Березин БД. // V Всесоюз. конф, по координац. и физ. химии порфиринов: Тез. докл. Иваново, 1988. С. 10-11.
121. Шипулина Н.В. // Всесоюз. конф. ’’Цитохром Р 450 и охрана окружающей среды”: Тез. докл. Новосибирск. 1987. С- 119.
122. Полис Я.Ю. // Современные аспекты исследований в обоасти фармации. Рига, 1977. С. 111-112; РЖХим. 1977.20045.
123. Ковалев И.Е. //Хим.-фармацевт. журн. 1977. Т. 11, № 3. С. 19-27.
124. Fort R.C. Adamantane: The chemistry of diamond molecules. N.Y.: Dekker 1976. 385 p.
26t
125. Hoffman C.E. // Annu. Rep. Med. Chem. 1967- P. 116-125; Chem. Abstr. 1969. Vol. 70. 27299m.
126. Mueller R., Auterhoff H. // Mitt. Dt. Pharm. Ges. DDR. 1969. Bd. 39, N 1. S. 1-6; Chem. Abstr. 1969. Vol. 70. 807 99a.
127. &*fe* W. // Dt. med. Woch.-Schr. 1969. Bd. 94, N 16. S. 857-862; Chem. Abstr.
1969. Vol. 70. 11252lx.
128. WishnokJ.S. ff3. Chem. Educ. 1973. Vol. 50,N ll.P. 780-781.
129, Галегов Г.А. // Журн. Всесоюз. хим. о-ва им. Д.И. Менделеева. 197 3. Т. 18, № 2. С. 200-207.
130. Першин Г.Н. // Фармакология и токсикология. 1975. Т. 38, № 5. С. 517-527-
131. Першин Г.Н, Богданова Н.С. Химиотерапия вирусных инфекций. М.: Медицина, 1973. 144 с.
132. Индулен М.К, Балоде-Калныня В. А. // ХХП Науч. сес. по общ. вирусологии: Тез. докл. М.» 1971. Ч. 1. С. 36.
133. Севостьянова В.В., Краюшкин М.М., Юрченко А.Г. ff Успехи химии. 1970. Т. 39, № 10. С. 1721-1753.
134. Сколдинов А.П ff Науч, конф, по химии орган, полиэдранов: Тез. докл. Волгоград, 1981. С. 133.
135. Ковтун В.Ю., Плахотник В.М. ff Хим.-фармацевт, журн. 1987. Т. 28, № 8. С. 931-940-
Оглавление
ПРЕДИСЛОВИЕ.................................................... 3
Глава 1
ОСОБЕННОСТИ СТРОЕНИЯ И СВОЙСТВ АДАМАНТАНОВ ........................... 5
1.1. Номенклатура адамантана и его производных........................ 5
1.2. Стереохимия адамантанов ......................................... 9
1.3. Физико-химические свойства адамантанов.......................... 11
13.1- Основные физико-химические характеристики ................. Ц
1.3.2 . Термодинамическая устойчивость ........................... 18
1.4. Спектральные характеристики адамантанов ........................ 28
1.4.1. ИК-спектры................................................. 28
1.4.2. Масс-спектры............................................... 34
1.43. ЯМР-спектры ............................................... 42
1,4.3.1. ПМР-спектры ,......................................... 42
1.4.3.2,ЯМР-спектрынаядрах ,3С................................. 46
Литература........................................................... 53
Глава 2
МЕТОДЫ ПОЛУЧЕНИЯ УГЛЕВОДОРОДОВ РЯДА АДАМАНТАНА....................... 58
2.1. Выделение и исследование адамантанов нефтяного происхождения.... 58
2.11. Изомерный состав и строение................................ 62
2.1.2 . Происхождение углеводородов ряда адамантана в иефти....... 66
2.2. Получение адамантанов каталитическими методами.................. 70
2.2.1. Жидкофазная изомеризация под действием кислот Льюиса....... 70
2.2.2 Газофазная изомеризация в условиях гетерогенного катализа.. 8?
2.3, Получение адамантанов синтетическими методами................... 91
2.34 .Синтез адамантанов,замешенных в узловых положениях......... 92
2.3.2. Синтез адамантанов, замещенных в мостиковых положениях.... 95
2.3.3. Синтез адамантанов смешанного строения .................... 97
2.3.4. Синтез полизамещенных и полициклических углеводородов ряда адамантана......................................................... 99
Литература........................................................... 116
Глава 3
ХИМИЧЕСКИЕ СВОЙСТВА АДАМАНТАНОВ ..................................... 124
34. Реакционная способность атомов углерода в молекуле адамантана... 124
34.1. Карбений-ионные реакции ..................................... 124
3.1.2. Свободнорадикальные реакции................................. 128
3.1.3. Взаимное влияние заместителей............................... 130
3.1.4. Количественные данные по реакционной способности.......... 132
3-2. Термические и каталитические превращения адамантанов........... 135
324. Превращения адамантанов под действием повышенной температуры и металлсодержащих катализаторов ............................. 135
3.2.2- Превращения адамантанов в условиях жидкофазного кислотного катализа ..................................................... 137
32.3. Превращения алкиладамантанов в условиях газофазного гетерогенного кислотного катализа ..................................... 150
3.3. Алкилирование.................................................. 157
3.3. 1. Ионное алкилирование .................................. 158
3.32. Радикальное алкилирование................................ 164
263
3.4. Галогенирование............................................... 166
3.5. Получение кислородсодержащих производных адамантанов ......... 182
3.5.1. Окисление адамантанов................................... 182
3.5.2. Карбоксилирование адамантанов............................ 188
3.5.3. Другие методы получения ................................. 192
3.6. Получение азотсодержащих производных адамантанов.............. 198
Литература......................................................... 201
Глава 4 НЕПРЕДЕЛЬНЫЕ СОЕДИНЕНИЯ РЯДА АДАМАНТАНА.............................215
4.1. Дегидроадамантаны..............................................215
4.2. Адамантаны с экзоциклической кратной связью.................. 224
4.3. Адамантаны с кратной связью в боковой цепи.....................228
4Д. Адамантаны с несколькими кратными связями.......................233
Литература........................................................ 237
Глава 5 ПЕРСПЕКТИВЫ И ОСНОВНЫЕ НАПРАВЛЕНИЯ ПРАКТИЧЕСКОГО ИСПОЛЬЗОВАНИЯ УГЛЕВОДОРОДОВ РЯДА АДАМАНТАНА И ИХ ПРОИЗВОДНЫХ .............................................................242
5.1. Перспективы использования адамантанов и их функциональных производ-
ных ............................................................243
5,2. Некоторые направления использования углеводородов ряда адамантана . . 246
5 .2.1. Адамантан со держащие полимерные материалы..............246
5 22. Биологически активные соединения н лекарственные препараты .... 254
Литература..........................................................257
Научное издание
БАГРИЙ Евгений Игнатьевич
АДАМАНТАНЫ получение, свойства, применение
Утверждено к печати ордена Трудового Красного Знамени Институтом нефтехимического синтеза им. А.В. Топчиева АН СССР
Художник Э.А. Дорохова. Художественный редактор И.Ю. Нестерова Технические редакторы И.И. Джиоева, Л.Н. Богданова Корректор Р.Г. Ухина
Набор выполнен в издательстве на иаборно-печатающих автоматах
ИБ №40140
Подписано к печати 19,10.89. Т — 14043. Формат 60 X 901 /lt Бумага офсетная К* 1, Гарнитура Пресс-Роман. Печать офсетная Усл.печл. 16,5, Усл.кр.-отт. 16,8. У я.-изд л. 18,9
Тираж 1200 экз. Тнп.эак. 1856, Цена 3 р. 90 к.
Ордена Трудового Красного Знамени издательство ’’Наука”
117864 ГСП-7, Москва В-485, Профсоюзная ул., д. 90
Ордена Трудового Красного Знамени I-я типография издательства "Наука” 199034, Ленинград В-34, 9-я линия, 12
Опечатки и исправления
Стр. Напечатано Должно быть
6 (5 строка св.) 47 (рис. 1.13, а) 147 1,4 Ь- а’СН. Смесь продуктов, приведенная на схеме, получена при -40°С в течение 1,4е- a'CHj Образование катиона имеет место при действии на спирт (23) конц.
Зак. 1856