/














































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Text
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
Республиканское унитарное предприятие
«Центр экспертиз и испытаний в здравоохранении»
ГОСУДАРСТВЕННАЯ
ФАРМАКОПЕЯ
РЕСПУБЛИКИ БЕЛАРУСЬ
Разработана на основе Европейской Фармакопеи
(ГФ РБ II)
В двух томах
Том 2
Контроль качества субстанций
для фармацевтического использования
и лекарственного растительного сырья
Введено в действие с 1 июля 2016 года
приказом Министерства здравоохранения Республики Беларусь
от 31.03.2016 года № 270
Молодечно
Типография «Победа»
УДК 615.11 (476)(083.7)
ББК 52.8(4Беи)
Г72
Издание выходит с 2012 г.
Под общей редакцией С.И. Марченко
Государственная фармакопея Республики Беларусь : (ГФ РБ II):
Г 72 разработана на основе Европейской Фармакопеи. В 2 т. Т. 2. Контроль
качества субстанций для фармацевтического использования и
лекарственного растительного сырья / М-во здравоохр. Респ. Беларусь,
УП «Центр экспертиз и испытаний в здравоохранении» ; под общ. ред.
С.И. Марченко. — Молодечно : Типография «Победа», 2016. — 1368 с.
15В1Ч 978-985-6967-31-6
Второй том ГФ РБ II содержит обязательные стандарты и положения,
регламентирующие качество субстанций для фармацевтического использования и
лекарственного растительного сырья. Кроме этого приведены новые общие
фармакопейные статьи и тексты, а также новая редакция некоторых статей первого тома
ГФ РБ II.
Государственная фармакопея Республика Беларусь основана на современных
достижениях медицины, фармации, химии и других смежных наук. Книга предназначена
для специалистов, занимающихся разработкой, производством, контролем качества,
хранением и реализацией лекарственных средств.
УДК 615.11 (476)(083.7)
ББК 52.8(4Беи)
15ВМ 978-985-6967-31-6 (т. 2)
978-985-6967-19-4
©УП «Центр экспертиз и испытаний в здравоохранении», 2016
© Оформление.Типография «Победа», 2016
I
СОДЕРЖАНИЕ
з
Введение 19
1. Общие сведения 21
2. Методы анализа 33
2.2. Физические и физико-химические методы 33
2.2.20. Потенциометрическое титрование 33
2.2.29. Жидкостная хроматография 33
2.2.32. Потеря в массе при высушивании 35
2.2.61. Определение характеристик кристаллических твердых веществ с помощью
микрокалориметрии и калориметрии растворения 36
2.2.64. Спектрометрия ядерного магнитного резонанса для идентификации пептидов 39
2.2.65. Вольтаметрическое титрование 40
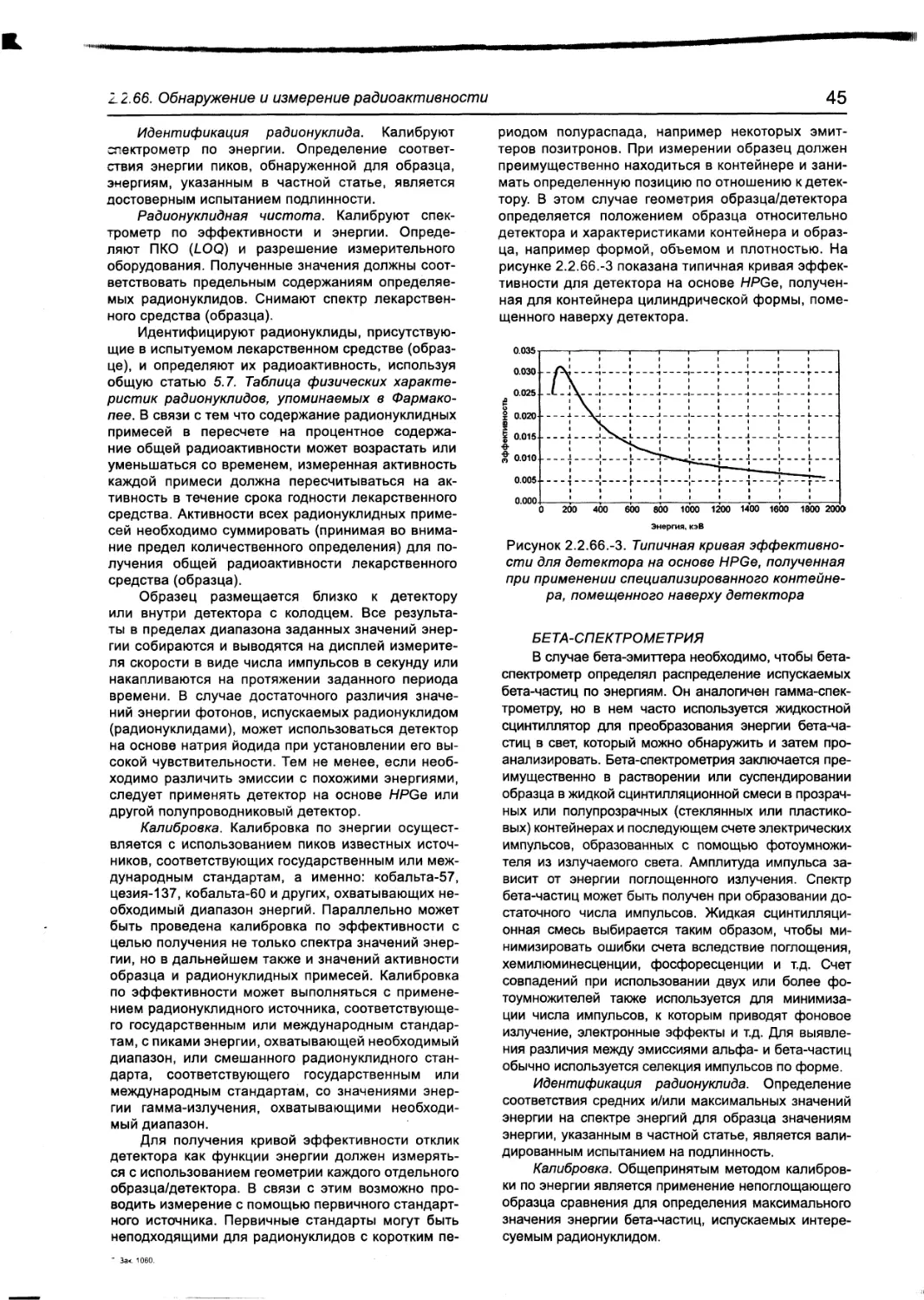
2.2.66. Обнаружение и измерение радиоактивности 40
#2.2.90. Титриметрические методы анализа 48
2.3. Подлинность (идентификация) 50
2.3.1. Реакции подлинности (идентификации) на ионы и функциональные группы 50
2.4. Испытания на предельное содержание примесей 55
2.4.20. Определение остаточных количеств металлических катализаторов
или металлсодержащих реактивов 55
2.4.27. Тяжелые металлы в лекарственном растительном сырье и продуктах из
лекарственного растительного сырья 59
2.5. Методы количественного определения 61
2.5.1. Кислотное число 61
2.5.12. Вода: полумикрометод 62
2.5.40. Метил-, этил- и изопропилтолуолсульфонат в фармацевтических субстанциях 63
2.5.41. Метил-, этил- и изопропилбензолсульфонат в фармацевтических субстанциях 64
2.6. Биологические испытания 65
2.6.31. Микробиологические испытания лекарственных средств растительного
происхождения для внутреннего применения и экстрактов, использующихся для их
приготовления 65
2.8. Методы фармакогнозии 69
2.8.2. Примеси 69
2.8.21. Испытание лекарственного растительного сырья
на наличие аристолохиевых кислот 69
2.9. Фармацевтико-технологические испытания 71
2.9.10. Содержание этанола 71
2.9.11. Испытание на содержание метанола и 2-пропанола 74
2.9.25. Высвобождение действующего вещества из лекарственных жевательных резинок 76
2.9.39. Взаимодействия «вода - твердое вещество»: построение изотерм сорбции-
десорбции и определение активности воды 80
2.9.47. Подтверждение однородности дозированных единиц с использованием большого
количества образцов 84
4. Реактивы 87
4.1.1. Реактивы 87
4.1.3. Буферные растворы 93
4.2. Реактивы, титрованные растворы для объемного анализа 93
4.2.2. Титрованные растворы 93
5. Общие тексты 95
5.4. Остаточные количества органических растворителей 95
5.12. Стандартные образцы ЮЗ
5.15. Функционально-обусловленные характеристики вспомогательных веществ -108
5.20. Остаточные количества металлических катализаторов или металлсодержащих реактивов 110
#5.50. Критерии чистоты пищевых красителей 116
- 3». -060.
4
Государственная фармакопея Республики Беларусь
Общие статьи 147
Радиоактивные фармацевтические препараты 147
Дозированные лекарственные формы 153
Лекарственные средства для парентерального применения 153
Частные фармакопейные статьи
на субстанции для фармацевтического использования 157
Аденозин 157
Адипиновая кислота 159
Адреналина тартрат 160
Азитромицин 162
Азота закись 165
Азотная кислота 166
Апбендазол 167
Аплантоин 168
Алюминия оксид гидратированный 169
Алюминия фосфат гидратированный 170
Алюминия хлорид гексагидрат 171
Амантадина гидрохлорид 172
Амброксола гидрохлорид 174
Амикацин 176
Амикацина сульфат 178
#Аминалон 182
Аминокапроновая кислота 183
Амиодарона гидрохлорид 184
Амитриптилина гидрохлорид 187
Амлодипина бесилат 189
Аммиака раствор концентрированный 191
Аммония хлорид 192
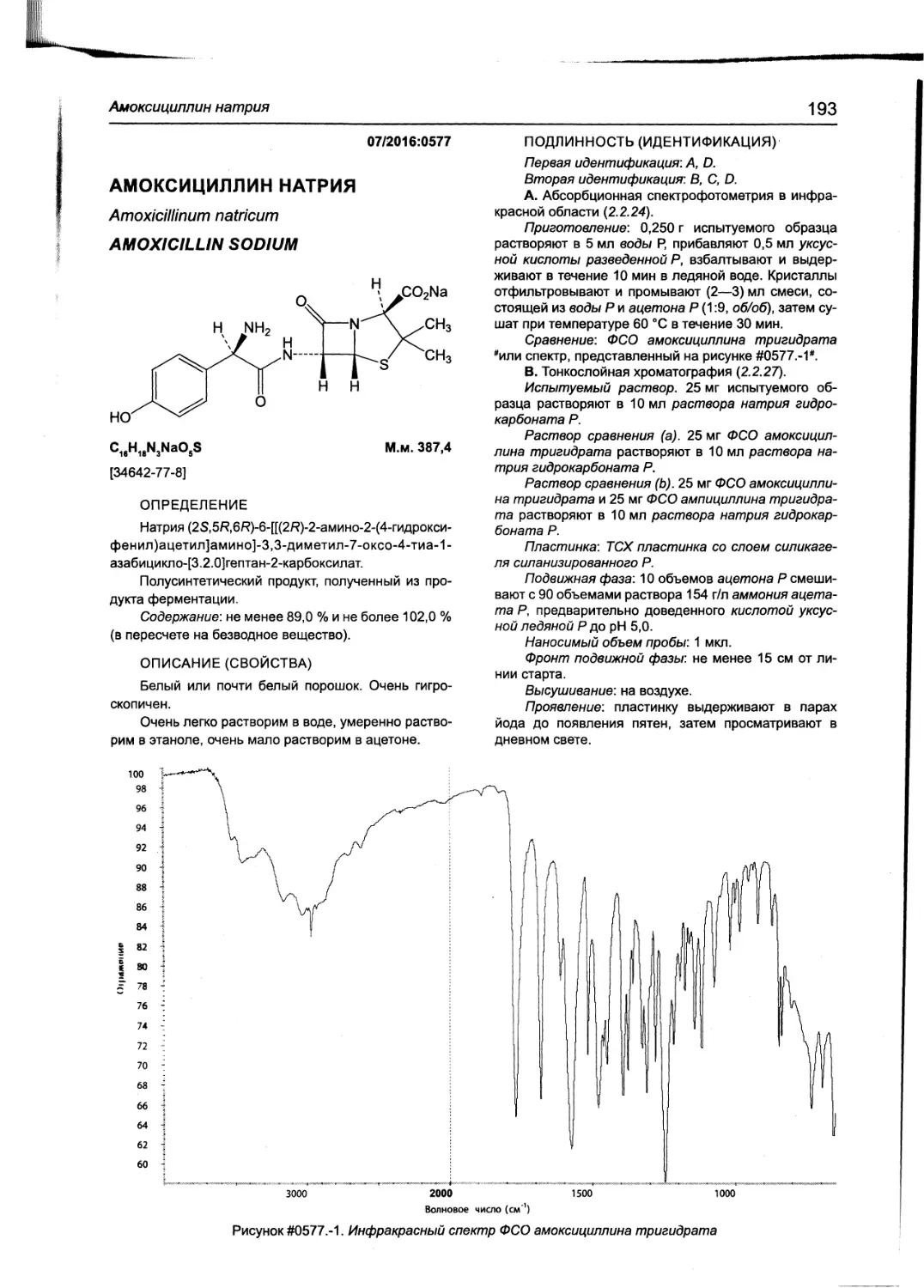
Амоксициллин натрия 193
Амоксициллин тригидрат 196
Ампициллин натрия 199
Ампициллин тригидрат 202
Амфотерицин В 205
Анастрозол 208
Анисовое масло 210
Апельсиновое масло 212
Аргинин 213
Аргинина аспартат 215
Аргинина гидрохлорид 216
Артикаина гидрохлорид 219
Аскорбиновая кислота 221
Аспарагиновая кислота 223
Аспартам 224
Атенолол 226
Аторвастатин кальция тригидрат 228
Атропина сульфат 231
Ацетилсалициловая кислота 233
Ацетил цистеин 235
Ацетон 237
Ацикловир 238
Бария сульфат 241
Беклометазона дипропионат безводный 242
Беклометазона дипропионат моногидрат 245
#Бендазола гидрохлорид 247
Бензалкония хлорид 248
Бензилбензоат 250
Бензиловый спирт 251
Бензилпенициллин натрия 253
Бензойная кислота 256
5
Бензокаин 256
Бензэтония хлорид 258
Бентонит 259
Бетагистина дигидрохлорид 260
Бетагистина мезилат 261
Бетаксолола гидрохлорид 263
Бетаметазона валериат 265
Бетаметазона дипропионат 267
Бисакодил 270
Бисопролола фумарат 272
Бифоназол 275
Борная кислота 277
Бромгексина гидрохлорид 277
Бутилгидроксианизол 279
#Вазелин 280
Валсартан 281
Валин 283
Ванилин 285
Ванкомицина гидрохлорид 286
Варфарин натрия 288
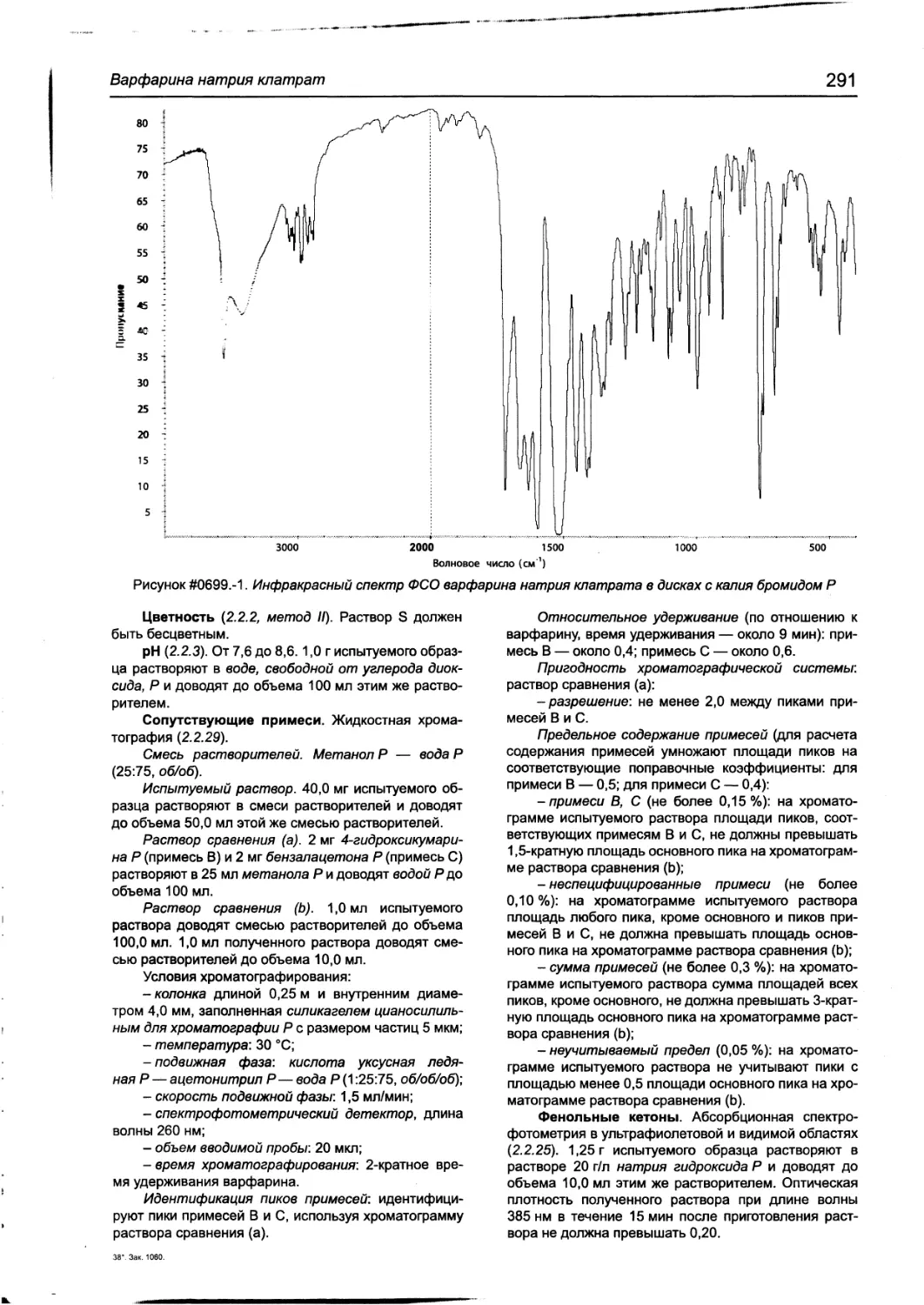
Варфарина натрия клатрат 290
Верапамила гидрохлорид 292
Винная кислота 295
Винорелбина тартрат 296
Винпоцетин 299
Висмута нитрат основной, тяжелый 301
Висмута субгаллат 302
Вода высокоочищенная 303
Вода для инъекций 305
Вода очищенная 309
Водорода пероксида 3 % раствор 311
Водорода пероксида 30 % раствор 312
Воск пчелиный белый 313
Воск пчелиный желтый 313
Галактоза 314
Галоперидол 315
Гвайфенезин 317
Гвоздичное масло 319
Гексаметилентетрамин 320
Гентамицина сульфат 321
Гепарин натрия 323
Гидрокортизон 326
Гидрокортизона ацетат 330
Гидроксикарбамид 332
Гидроксипропилцеллюлоза 334
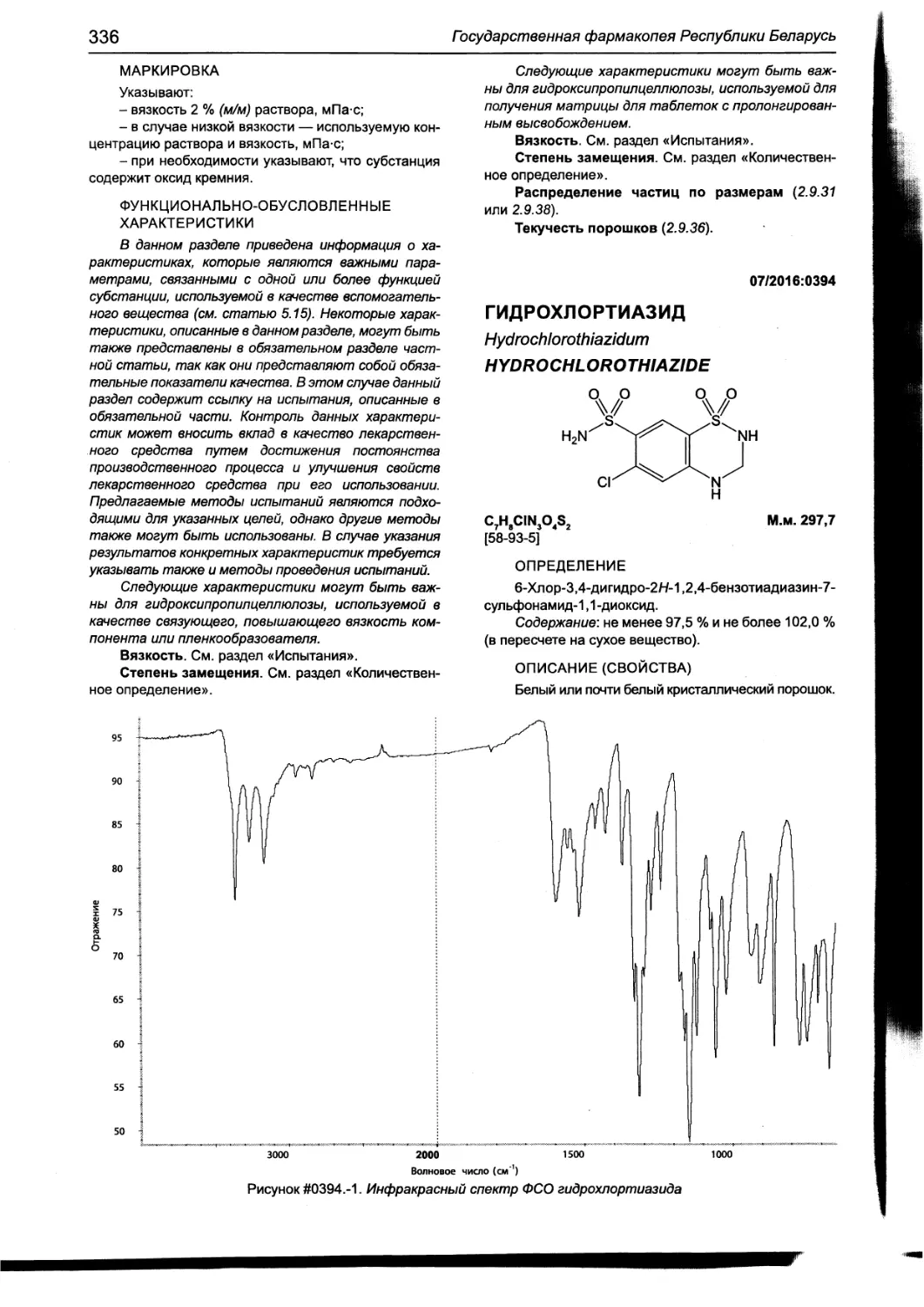
Гидрохлортиазид 336
Гиосциамина сульфат 338
Гиосцина гидробромид 340
Гипромеллоза 342
Гистамина дигидрохлорид 344
Гистидин 345
Гистидина гидрохлорид моногидрат 347
Глибенкламид 348
Гликлазид 351
Глимепирид 353
Глицерин 355
Глицерин 85 % 358
Гпицерина моностеарат 40-55 360
Глицин 361
Глутаминовая кислота 364
Глюкоза безводная 365
Глюкоза моногидрат 367
2. Зак. 1060.
6 Гэсударственная фармакопея Республики Беларусь
Глюкозамина гидрохлорид 369
Глюкозамина сульфат натрия хлорид 371
#Деготь березовый 373
Дексаметазона натрия фосфат 374
Декспантенол 377
Декстран 40 для инъекций 379
Декстран 60 для инъекций 380
Декстран 70 для инъекций 382
Декстрин 383
Декстрометорфана гидробромид 384
Диазепам 386
Дигоксин 388
Дикалия фосфат 390
Диклофенак натрия 391
Диметил сульфоксид 394
Динатрия фосфат дигидрат : 395
Динатрия фосфат додекагидрат 396
Динатрия эдетат 396
Дифенгидрамина гидрохлорид 398
#Диэтаноламин 399
Доксиламина гидросукцинат 400
Доксициклина гиклат 401
Доксорубицина гидрохлорид 404
Докузат натрия 405
Допамина гидрохлорид 407
Доцетаксел безводный 408
Доцетаксел тригидрат 410
Дроперидол 413
#Дротаверина гидрохлорид 415
Дутастерид 417
Желатин 419
Железа (II) сульфат высушенный 424
Железа сульфат гептагидрат 425
Железа хлорид гексагидрат 426
Жир твердый 427
Зопиклон 427
Ибупрофен 429
Идоксуридин 432
Изолейцин 434
Изониазид 436
Изопропилмиристат 437
Изопропиловый спирт 438
Изосорбида динитрат разведенный 440
Изосорбида мононитрат разведенный 442
Имипенем моногидрат 445
Индапамид 446
Индометацин 449
Инсулин аспарт 451
Инсулин бычий 453
Инсулин двухфазовый для инъекций 456
Инсулин изофан двухфазовый для инъекций 456
Инсулин изофан для инъекций 457
Инсулин лизпро 457
Инсулин растворимый для инъекций 460
Инсулин свиной 460
Инсулин человеческий 463
Инсулина инъекционные лекарственные средства 466
Инсулина цинка суспензия (аморфная) для инъекций 469
Инсулина цинка суспензия (кристаллическая) для инъекций 470
Интерферона альфа-2 концентрированный раствор 470
Итраконазол 474
Ихтиол 476
7
Йод 477
#Какао масло 477
Калия ацетат 478
Калия бромид 478
Калия гидроаспартат гемигидрат 480
Калия гидроксид 481
Калия дигидрофосфат 481
Калия йодид 482
Калия клавуланат 483
Калия клавуланат разведенный . 485
Калия метабисульфит 487
Калия перманганат 488
Калия сорбат 488
Калия хлорид 489
Калия цитрат 490
Кальция глицерофосфат 491
Кальция глюконат 492
Кальция глюконат безводный 493
Кальция глюконат для инъекций 494
Кальция карбонат 495
Кальция лактат безводный 496
Кальция лактат моногидрат 497
Кальция лактат пентагидрат 498
Кальция лактат тригидрат 499
Кальция стеарат 499
Кальция фолинат 501
Кальция хлорид гексагидрат 504
Кальция хлорид дигидрат 505
О-Камфора 506
Камфора рацемическая 507
Каолин тяжелый 508
#Капсулы твердые желатиновые 509
Каптоприл 511
Карбоплатин 514
Карведилол 516
Картофельный крахмал 518
Касторовое масло нерафинированное 519
Касторовое масло рафинированное 520
Кетоконазол 521
Кетопрофен 523
Кеторолак трометамин 526
Кетотифена гидрофумарат 528
#Кислород газообразный 530
Кладрибин 532
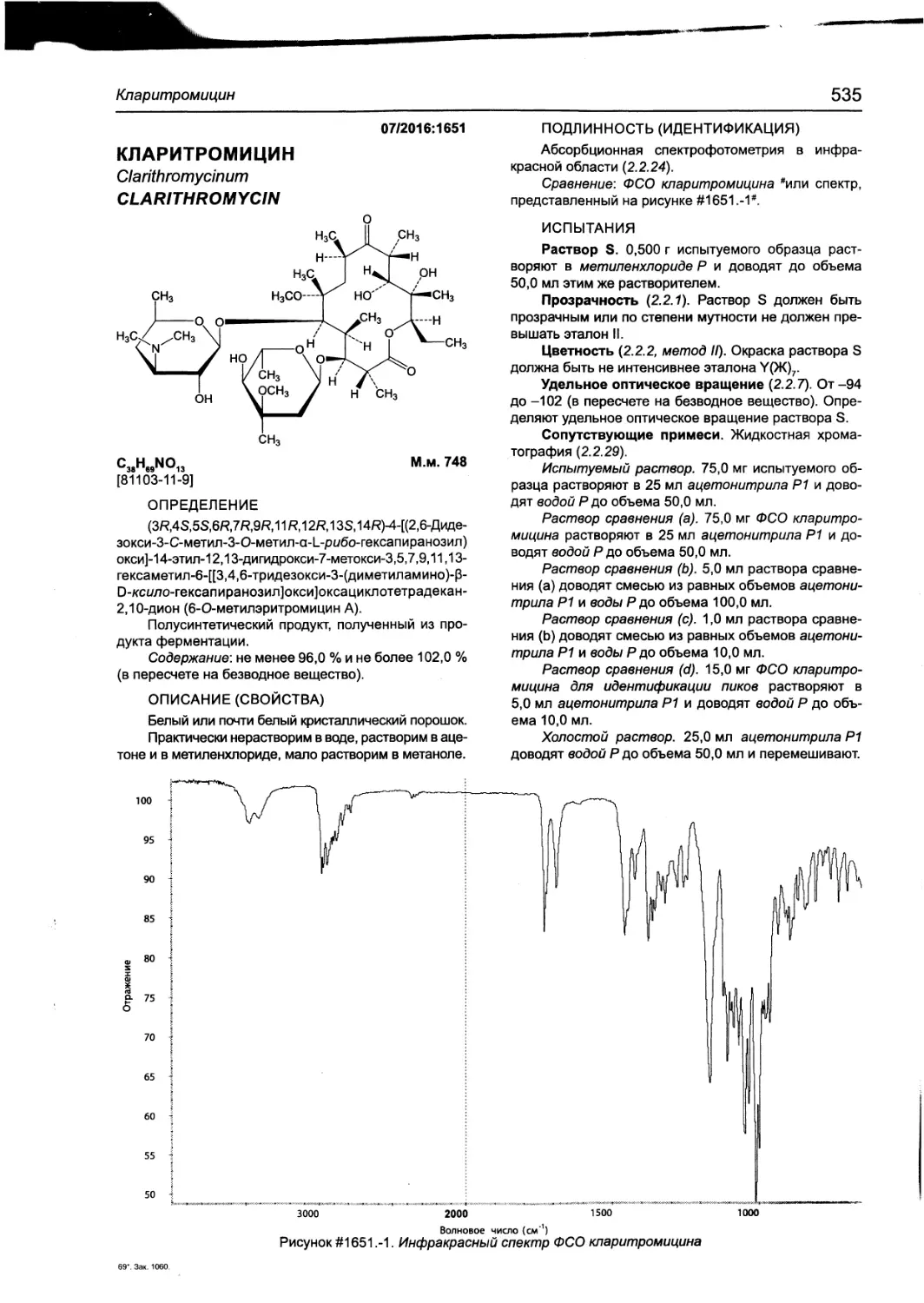
Кларитромицин 535
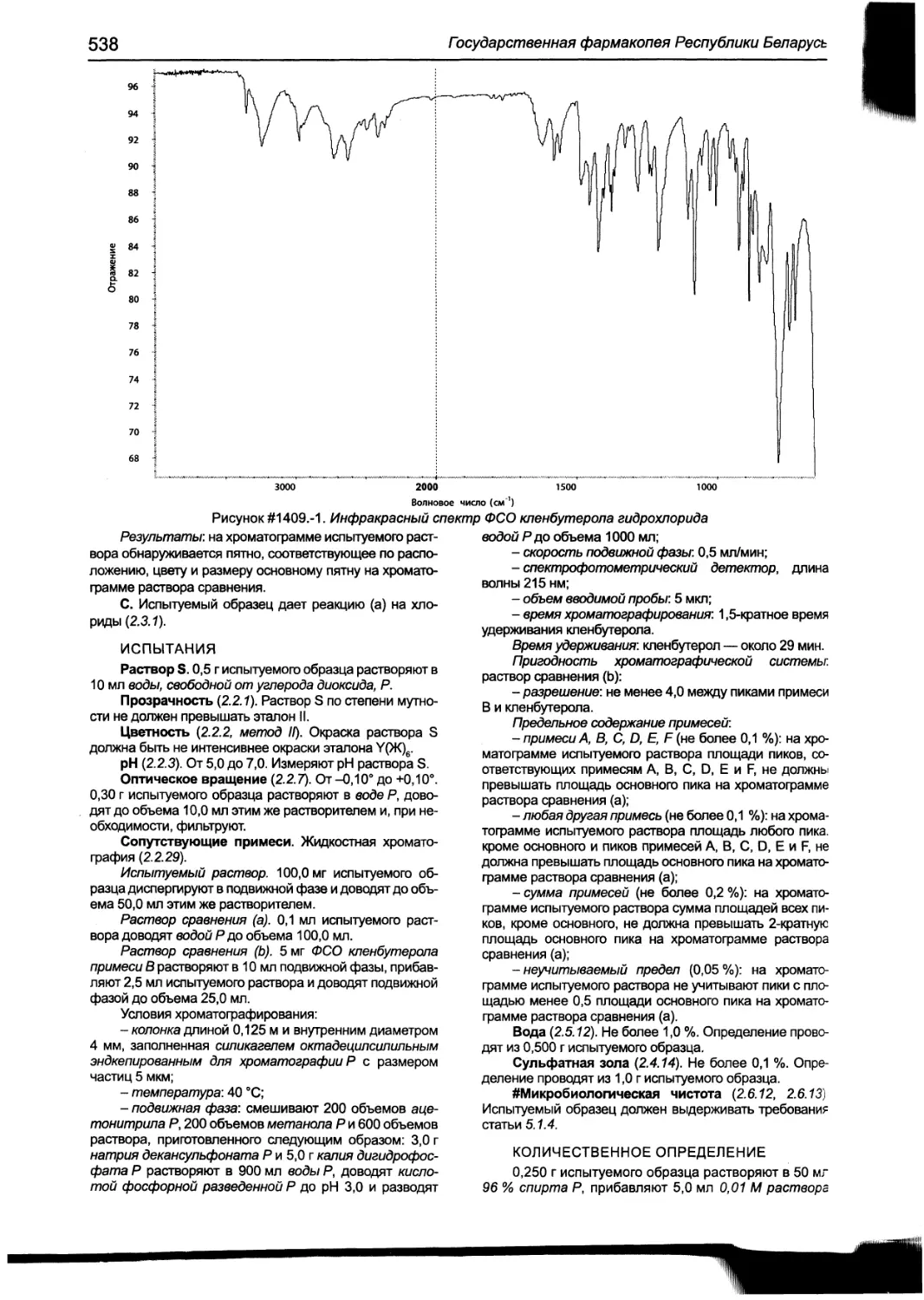
Кленбутерола гидрохлорид 537
Клиндамицина фосфат 539
Клозапин 542
Клонидина гидрохлорид 543
Клопидогреля гидросульфат 545
Клотримазол 548
Кодеин 550
Кодеина фосфат гемигидрат 552
Кодеина фосфат сесквигидрат 555
Кокосовое масло рафинированное 557
Коповидон 558
Кофеин 560
Кремния диоксид коллоидный безводный 562
Кремния диоксид коллоидный гидратированный 563
Кроскармеллоза натрия 564
Кросповидон 566
Ксантановая камедь 568
8 Гэсударственная фармакопея Республики Беларусь
Ксилометазолина гидрохлорид 570
Кукурузное масло рафинированное .. 572
Кукурузный крахмал 572
Лавандовое масло 573
Лактоза безводная 575
Лактоза моногидрат ! 576
Лактулоза 578
Ламотриджин 580
Ланолин 583
Ланолин водный 587
Ланолин гидрогенизированный 588
Лансопразол 589
Леводопа 591
Левоментол 592
Левотироксин натрия 594
Лейцин 596
Лидокаина гидрохлорид 599
Лизина гидрохлорид 601
Лизиноприл дигидрат 603
Лимонная кислота безводная 605
Лимонная кислота моногидрат 606
Лимонное масло 607
Линкомицина гидрохлорид 609
Ловастатин 611
Лоперамида гидрохлорид 614
Лоперамида оксид моногидрат 617
Лоратадин 618
Магния аспартат дигидрат 621
Магния ацетат тетрагидрат 622
Магния гидроксид 623
Магния карбонат основной, легкий 624
Магния карбонат основной, тяжелый 625
Магния оксид легкий 626
Магния оксид тяжелый 627
Магния стеарат 628
Магния сульфат гептагидрат 631
Магния хлорид 4,5-гидрат 631
Магния хлорид гексагидрат 632
Магния цитрат безводный 633
Макрогола цетостеариловый эфир 634
Макроголглицерина гидроксистеарат 635
Макроголы.... 636
Маннит (маннитол) 638
Марганца глюконат 641
Мебендазол ■■■• 642
Меди сульфат безводный 644
Меди сульфат пентагидрат 644
Мекпозина дигидрохлорид 645
Мелоксикам 647
Мельдоний дигидрат 649
Менадион 651
#Менадиона натрия бисульфит 651
Ментол рацемический 653
Меропенем тригидрат 654
Меркаптопурин 656
Метакрезол 656
Метакриловой кислоты и этилакрилата сополимер (1:1) 658
Метакриловой кислоты и этилакрилата сополимер (1:1), 30 % дисперсия 659
Метамизол натрия моногидрат 661
Метилпарагидроксибензоат 663
Метилтиониния хлорид 665
Метилцеллюлоза 667
9
ОЬМетионин 669
Метионин 670
Метоклопрамида гидрохлорид 672
Метопролола тартрат 674
Метотрексат 676
Метронидазол 679
Метронидазола бензоат 681
Метформина гидрохлорид 682
Миконазола нитрат 685
Моксифлоксацина гидрохлорид 687
Моксонидин 689
Молочная кислота 691
(З)-Молочная кислота 691
Морфина гидрохлорид 692
Морфина сульфат 694
Мочевина 696
#Муравьиная кислота 698
#Муравьиная кислота безводная 699
#Мыло зеленое (мыло калийное) 699
#Мыло хозяйственное твердое 700
Мяты перечной масло 702
Напроксен 703
Натрия аминосалицилат дигидрат 706
Натрия ацетат тригидрат 708
Натрия бензоат 709
Натрия бромид 710
Натрия гиалуронат 711
Натрия гидрокарбонат 714
Натрия гидроксид 715
Натрия дигидрофосфат дигидрат 715
Натрия йодид 716
Натрия каприлат 717
Натрия карбонат безводный 718
Натрия карбонат декагидрат 718
Натрия карбонат моногидрат 719
Натрия крахмалгликолят (тип А) 720
Натрия крахмалгликолят (тип В) 721
Натрия крахмалгликолят (тип С) 722
Натрия (З)-лактата раствор 723
Натрия лактата раствор 724
Натрия лаурилсульфат 725
Натрия метабисульфит 726
Натрия метилпарагидроксибензоат 727
Натрия пикосульфат 729
Натрия пропилпарагидроксибензоат 731
Натрия салицилат 733
Натрия сульфат безводный 735
Натрия сульфат декагидрат 735
Натрия сульфит безводный 736
Натрия сульфит гептагидрат 737
Натрия тетраборат 738
Натрия тиосульфат 738
Натрия фторид 739
Натрия хлорид 740
Натрия цетостеарилсульфат 741
Натрия цитрат 743
Натрия этилпарагидроксибензоат 744
Нафазолина гидрохлорид 745
Нафазолина нитрат 747
Никотинамид 749
Никотиновая кислота.. 751
Нимесулид 753
3. Зак. 1060.
Ю Гэсударственная фармакопея Республики Беларусь
Нистатин 755
Нитрофурал 757
Нитрофурантоин 758
Нифедипин 759
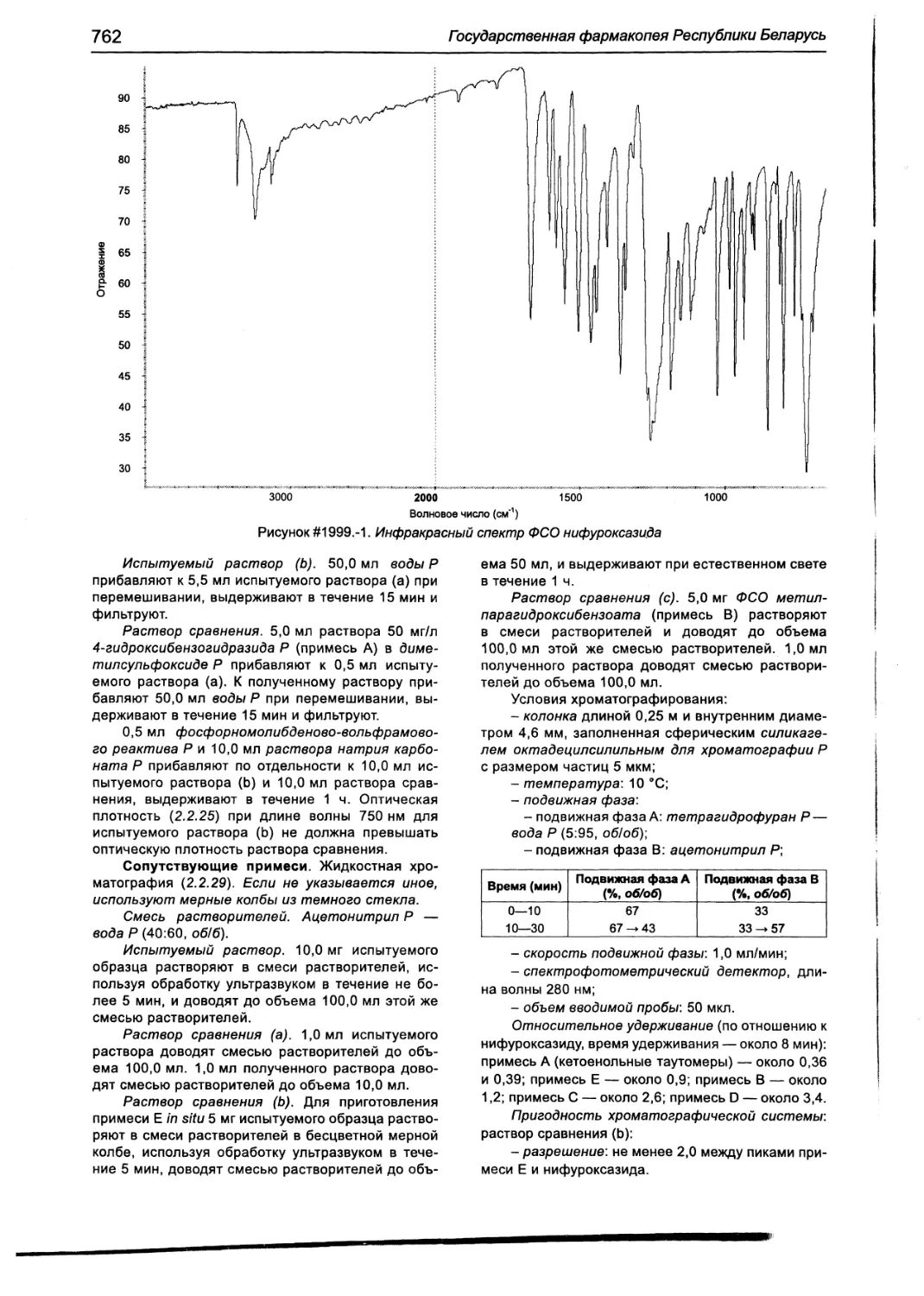
Нифуроксазид 761
Носкапина гидрохлорид 763
Оксалиплатин 765
Оксациллин натрия моногидрат 768
Оксиметазолина гидрохлорид 771
Окситоцин 773
Окситоцина раствор концентрированный 774
Октилдодеканол 775
Омега-З-кислоты этиловые эфиры 60 776
Омега-З-кислоты этиловые эфиры 90 778
Ондансетрона гидрохлорид дигидрат 780
Осельтамивира фосфат 783
Офлоксацин 785
Пакпитаксел 787
Панкреатина порошок 791
Папаверина гидрохлорид 794
Парафин жидкий 796
Парафин мягкий белый 798
Парафин мягкий желтый 799
Парафин твердый 799
Парацетамол 800
Пентоксифиллин 802
Пепсина порошок ; 805
Периндоприл трет-бутиламин 806
Пилокарпина гидрохлорид 810
Пиперазина адипинат 812
Пиперазин гидрат 813
Пирантела эмбонат 814
Пирацетам 816
Пиридоксина гидрохлорид 817
Плазма человеческая для фракционирования 820
Повидон 821
Повидон-йод 825
Подсолнечное масло рафинированное 826
Поливиниловый спирт 826
Полисорбат 20 827
Полисорбат 40 828
Полисорбат 60 829
Полисорбат 80 830
Прамипексола дигидрохлорид моногидрат 832
Преднизолон 834
Прокаина гидрохлорид 836
Прокаинамида гидрохлорид 838
Пролин 839
Прометазина гидрохлорид 841
Пропиленгликоль 843
Пропилпарагидроксибензоат 844
#Прополис 846
Пропранолола гидрохлорид 847
Протамина сульфат 848
Псевдоэфедрина гидрохлорид 850
Пшеничный крахмал 852
Рамиприл 852
Ранитидина гидрохлорид 855
Резорцин 857
Рибавирин 858
Рибофлавин 860
Рибофлавина натрия фосфат 862
11
Рисовый крахмал 864
Рисперидон 865
Рифабутин 867
Рифампицин 869
Рокситромицин 871
Ртути хлорид 874
Рутозид тригидрат 874
Сабаля мелкопильчатого экстракт 876
Салициловая кислота 879
Сальбутамола сульфат 881
Сахарин натрий 884
Сахароза 886
Севофлуран 888
Сера для наружного применения 890
Серебра нитрат 891
#Серебра протеинат 891
Серебро коллоидное для наружного применения 892
Серин 893
Серная кислота . 895
Сертаконазола нитрат 896
Силденафила цитрат 897
Симвастатин 899
Соевое масло гидрогенизированное 902
Соевое масло рафинированное 902
Сорбиновая кислота 903
Сорбитол (сорбит) 904
Сорбитола раствор кристаллизующийся (сорбита раствор кристаллизующийся) 906
Сорбитола раствор некристаллизующийся (сорбита раствор некристаллизующийся) 907
Сосны обыкновенной масло 908
Спирамицин 909
Спиронолактон 912
Стеариновый спирт 915
Стеариновая кислота 915
Стрептомицина сульфат 916
Суксаметония хлорид 918
Сульпирид 919
Сультамициллина тозилат дигидрат 921
Сульфагуанидин 924
Сульфаметоксазол 926
Сульфаниламид 928
Сульфацетамид натрия 929
Тадалафил 931
Тальк.. 934
Таниновая кислота 937
#Таурин 937
Тейкопланин 939
Теобромин 942
Теофиллин 943
Теофиллин-этилендиамин 945
Теофиллин-этилендиамин гидрат 947
#Теофиллин-этилендиамин для инъекций 949
Тербинафина гидрохлорид 951
#Терпентинное масло 953
Терпентинное масло, тип Ртиз р‘таз1ег 953
Тестостерон 954
Тестостерона пропионат 956
Тетракаина гидрохлорид 958
Тетрациклин 960
Тетризолина гидрохлорид 961
Тиамина гидрохлорид 963
Тимолола малеат 965
Тирозин 968
3’. Зак. 1060.
12 Государственная фармакопея Республики Беларусь
Титана диоксид 970
Тозилхлорамид натрия 971
а-Токоферилацетат 972
а-Токоферол 974
ЯКЯ-а-Токоферилацетат 976
Толбутамид 978
Толнафтат 979
Трагакант 981
Трамадола гидрохлорид 983
Треонин 984
Триамцинолона ацетонид 987
Трибенозид 990
Триметазидина дигидрохлорид 991
Триметоприм 993
Триптофан 996
Троксерутин 1000
Троламин 1002
Трометамол 1004
Трописетрона гидрохлорид 1005
Углерода диоксид 1007
Уголь активированный 1009
Уксусная кислота ледяная 1010
Фамотидин 1011
Феназон 1013
Фенилаланин 1015
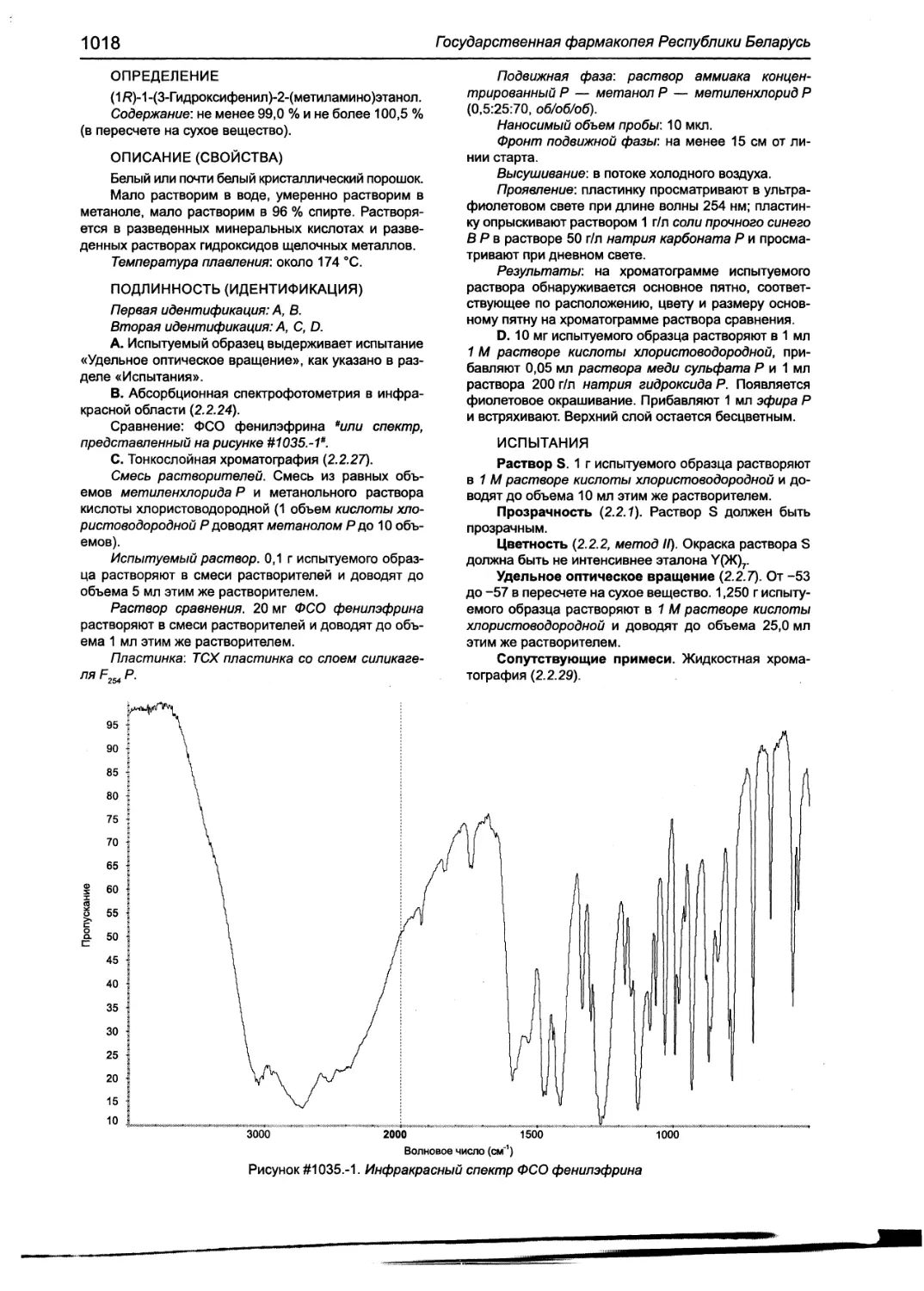
Фенилэфрин 1017
Фенилэфрина гидрохлорид 1020
Фенирамина малеат 1022
Фенобарбитал 1024
Феноксиметилпенициллин 1026
Фенол 1028
Фенолфталеин 1028
Фентанил 1029
Фибриноген человеческий 1031
Флударабина фосфат 1032
Флуконазол 1035
Флуоцинолона ацетонид 1037
Флутамид 1040
Фолиевая кислота 1041
Формальдегида 35 % раствор 1043
Фосфорная кислота концентрированная 1044
Фосфорная кислота разведенная 1045
Фрамицетина сульфат 1045
Фруктоза 1047
Фторурацил 1048
#Фуразолидон 1050
Фуросемид 1051
Химотрипсин 1053
Хинидина сульфат Ю55
Хинина гидрохлорид 1057
Хинина сульфат 1058
Хлоралгидрат 1060
Хлорамфеникол 1061
Хлорамфеникола пальмитат 1062
Хлорбутанол безводный 1064
Хлорбутанол гемигидрат 1064
Хлоргексидина диацетат 1065
Хлоргексидина диглюконата раствор 1067
Хлористоводородная кислота концентрированная 1071
Хлористоводородная кислота разведенная 1072
Хлоркрезол 1072
#Хлороформ 1073
13
Хпорталидон Ю74
Хлорфенамина малеат 1076
Холекальциферол 1078
Холестерин 1079
Хондроитина натрия сульфат 1081
Целекоксиб 1083
Целлюлоза микрокристаллическая 1085
Цетиловый спирт 1088
Цетил пал ьмитат 1089
Цетилпиридиния хлорид 1090
Цетиризина дигидрохлорид 1091
Цетостеариловый спирт 1093
Цетостеариловый спирт (тип А) эмульсионный 1094
Цетостеариловый спирт (тип В) эмульсионный 1096
Цетримид Ю97
Цефазолин натрия 1098
Цефаклор 1101
Цефалексин моногидрат 1103
Цефепима дигидрохлорид моногидрат 1106
Цефоперазон натрия 1108
Цефотаксим натрия 1111
Цефтазидим пентагидрат 1113
Цефтазидим пентагидрат с натрия карбонатом для инъекций 1116
Цефтриаксон натрия 1119
Цианокобаламин 1121
Циклофосфамид 1122
Циластатин натрия 1123
Цинка ацетат дигидрат 1126
Цинка глюконат 1127
Цинка оксид 1128
Цинка сульфат гексагидрат 1128
Цинка сульфат гептагидрат 1129
Цинка сульфат моногидрат 1129
Цинка хлорид 1130
Циннаризин 1130
Ципрофлоксацин 1132
Ципрофлоксацина гидрохлорид 1134
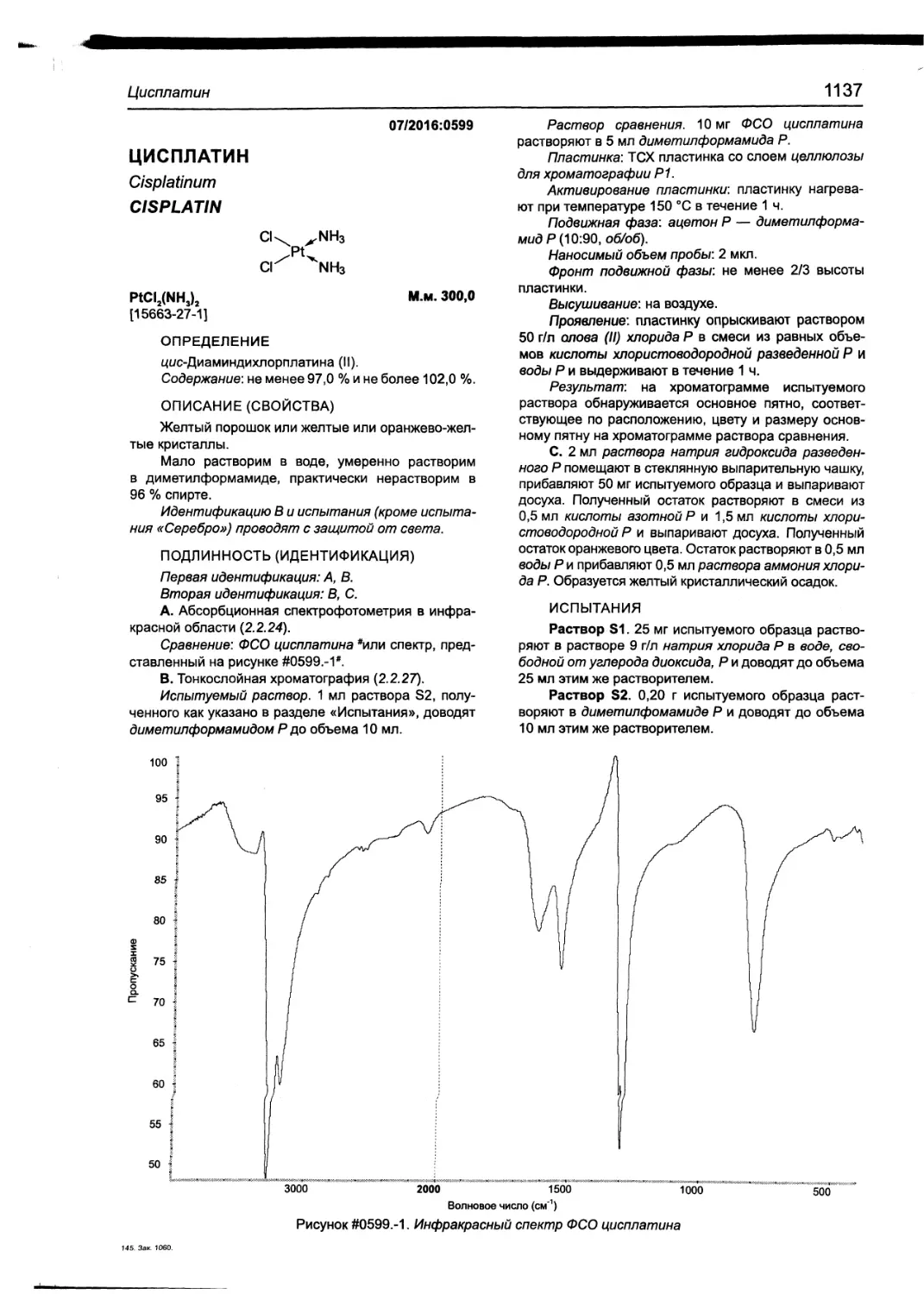
Цисплатин 1137
Цистеина гидрохлорид моногидрат 1139
Цитарабин 1141
Чайного дерева масло 1142
Шалфея мускатного масло 1143
Шеллак 1144
Эвкалиптовое масло 1146
Эметина гидрохлорид пентагидрат 1147
#Эмульгатор № 1 1148
Эналаприла малеат 1150
Эргокальциферол 1152
Эритритол 1154
Эритромицин 1156
Эритромицина эстолат 1159
Этакридина лактат моногидрат 1162
Этанол безводный 1163
Этил морфина гидрохлорид 1165
#Этиловый спирт 95 %, 90 %, 80 %, 70 %, 60 %, 40 % 1167
Этиловый спирт 96 % 1167
Этил парагидроксибензоат 1172
Эфедрина гидрохлорид 1173
Эфедрина гидрохлорид рацемический 1175
Эфир 1177
Эфир анестезирующий 1177
4. Зак. 1060.
14
Гэсударственная фармакопея Республики Беларусь
Частные фармакопейные статьи
на лекарственное растительное сырье.
Аира корневища
Алтея корни
Аралии маньчжурской корни
Арники цветки
Багульника болотного побеги
Бадана корневища
Барбариса обыкновенного корни
Барбариса обыкновенного листья
Бегонии листья
Белены черной листья
Белладонны листья (красавки листья)
® Березы листья
Березы почки
Бессмертника песчаного цветки
@> Боярышника листья и цветки
Боярышника листья
Боярышника плоды
Боярышника цветки
Брусники листья
Бузины черной цветки
Валерианы корневища с корнями
Василька синего цветки
Вахты трехлистной листья
® Гинкго листья
® Горечавки корни
Горицвета весеннего трава
Горца змеиного корневища (змеевика корневища)
Горца перечного трава (водяного перца трава)
Горца почечуйного трава
Горца птичьего трава (спорыша трава)
Девясила корневища и корни
Девясила цветки
Донника трава
Дуба кора
Дурмана листья
Душицы трава
® Дягиля корни
Женьшеня корни .
Жостера слабительного плоды
Зверобоя трава
Земляники лесной листья
Земляники лесной плоды
Золототысячника трава
Ивы кора
Имбиря корневища
® Исландского мха слоевища
Календулы цветки (ноготков цветки)
Калины кора
Каштана конского семена
Кориандра плоды
Крапивы листья
Кровохлебки корни
Крушины кора
Кукурузы столбики с рыльцами
@> Лабазника вязолистного трава
Лабазника вязолистного цветки
® Лаванды цветки
Ламинарии слоевища (морская капуста)
Ландыша листья
Ландыша трава
1179
...1179
...1180
...1180
...1181
...1184
...1185
...1185
...1186
...1187
...1188
...1189
...1191
...1193
...1193
...1194
...1196
...1197
...1199
.. 1200
.. 1202
..1204
.. 1207
.. 1207
.. 1208
.. 1210
...1211
..1212
..1213
..1214
..1214
..1216
..1217
..1218
.. 1220
..1221
..1223
..1224
.. 1226
..1228
..1228
..1230
..1231
..1231
.. 1233
..1234
..1235
..1236
.. 1238
..1238
..1240
.. 1241
..1243
.. 1244
..1246
..1246
..1248
..1249
..1251
..1252
..1252
15
Ландыша цветки 1253
Лапчатки белой трава 1254
Лапчатки корневища 1255
Левзеи сафлоровидной корневища с корнями 1256
Левзеи сафлоровидной листья 1257
® Лимонника плоды 1258
Лимонника семена 1260
Липы цветки 1260
Льна семена 1262
® Любистка корни 1262
Маклейи листья 1264
Малины листья 1265
Малины плоды 1266
Марены корневища и корни 1266
Мать-и-мачехи листья 1268
® Мелиссы листья 1269
Мелиссы трава 1270
Многоколосника морщинистого трава (лофанта трава) 1271
® Можжевельника плоды 1272
Мяты перечной листья 1273
Наперстянки пурпурной листья 1275
Облепихи плоды свежие 1276
Одуванчика лекарственного корни 1277
Ольхи серой листья 1278
Ольхи соплодия 1280
Ольхи черной листья 1280
Ортосифона тычиночного листья (почечного чая листья) 1283
Пассифлоры трава 1284
Пастушьей сумки трава 1285
® Первоцвета корни 1286
Пижмы цветки 1287
Пиона уклоняющегося корневища и корни 1288
Пиона уклоняющегося трава 1289
Подорожника большого листья 1290
® Подорожника ланцетного листья 1291
® Полыни горькой трава 1292
Пустырника листья 1293
Пустырника трава 1294
Расторопши плоды 1296
Ревеня корни 1298
® Репешка трава 1300
Родиолы розовой корневища и корни 1301
Ромашки цветки 1302
Рудбекии шершавой цветки 1303
Рябины плоды 1305
Сабельника болотного корневища с корнями 1307
® Сенны листья 1308
Сенны листья с плодами 1309
® Сенны остролистной плоды 1311
® Сенны узколистной плоды 1312
Синюхи корневища с корнями 1314
Солодки корень (лакричный корень) 1315
Сосны почки 1317
Сушеницы топяной трава 1317
Термопсиса ланцетного трава 1318
® Тимьяна трава 1319
Тмина плоды 1321
Толокнянки листья 1322
Тыквы семена 1324
Тысячелистника трава 1324
Укропа пахучего плоды 1325
Фасоли створки 1326
4*. Зак. 1060.
16
Гэсударственная фармакопея Республики Беларусь
Фенхеля горького плоды 1327
@> Фенхеля сладкого плоды 1328
Фиалки трава 1329
Фукус 1331
Хвоща полевого трава 1332
Хмеля шишки 1334
Чабреца блошиного трава 1335
Чабреца ползучего трава 1337
Чага (черный березовый гриб) 1338
Череды трава 1339
Черемухи плоды 1340
® Черники плоды свежие 1341
® Черники плоды сухие 1342
@> Чеснока порошок 1342
Чистотела трава 1343
Шалфея листья 1345
Шиповника плоды 1346
Эвкалипта прутовидного листья 1347
(§•> Эвкалипта шаровидного листья 1348
Элеутеррококка корневища и корни 1349
Эхинацеи пурпурной трава 1351
Указатель общих фармакопейных статей 1355
Алфавитный указатель частных фармакопейных статей
на субстанции для фармацевтического использования 1361
Алфавитный указатель частных фармакопейных статей
на лекарственное растительное сырье 1366
17
Редакционный совет
Государственной фармакопеи Республики Беларусь II, том 2
Марченко С.И. — председатель редакционного совета
Стреха И.С. — заместитель председателя, координатор
Члены редакционного совета
Бузук Г.Н., д.ф.н.
Гурина Н.С., д.б.н.
Заневская Ю.В., к.х.н.
Кузьмичева НА, к.б.н.
Кравец М.М.
Марченко С.И.
Моисеев Д.В., к.ф.н.
Рафалович Л.И.
Реутская Л.А.
Стреха И.С.
Тимошина В.В.
Хишова О.М., д.ф.н.
Список авторов
Александрова Н.В.
Алексеев Н.А., к.ф.н.
Бернович Л.С.
Бузук А. Г.
Бузук Г.Н., д.ф.н.
Вернигорова М.Н.
Германенко Е.В.
Глушко Т.В.
Голик Г.И.
Голяк Ю.А.
Гурина Н.С., д.б.н.
Дергачева Ж.М.
Дорошкевич Н.А., к.б.н.
Дубашинская Н.В.
Дунец Л.Н.
Ершик О.А., к.ф.н.
Жулина Н.В.
Заневская Ю.В., к.х.н.
Зенько Л. А.
Казаровец Я.С.
Карусевич А.А.
Коноплева М.М., к.ф.н.
Корожан Н.В.
Коцур О.М.
Кузьмич А.Б., к.б.н.
Кузьмичева Н.А., к.б.н.
Кукалевич Т.В.
Кухарева Л.В., к.б.н.
Линкевич В.Г.
Лукашов Р.И.
Марченко С.И.
Медяков М.М.
Моисеев Д.В., к.ф.н.
Мушкина О.В.
Пленина Л.В., к.б.н.
Погирницкая А.В.
Погоцкая А.А.
Подолинская О.Г.
Потапнев М.П., д.м.н.
Прохорова М.В.
Рафалович Л.И.
Родионова Р.А., к.ф.н.
Родионова Т.В.
Руткевич Е.Б.
Стреха И.С.
Су гакова А. В.
Телегина Т.В.
Трикашова Л.В.
Фомичева Н.В.
Хишова О.М., д.ф.н.
Холина Н.А.
Цаприлова С.В.
Цвилик Г. Л.
Шеряков А.А., к.ф.н.
Шимко О.М.
Шуть Н.В.
Юрченко РА.
5. Зак. 1060.
18
Гзсударственная фармакопея Республики Беларусь
Список организаций, учреждений и предприятий
Республики Беларусь, принимавших участие в разработке
Государственной фармакопеи Республики Беларусь II, том 2
РУП «Центр экспертиз и испытаний в здравоохранении»
УО «Белорусский государственный медицинский университет»
УО «Витебский государственный медицинский университет»
РУП «Белмедпрепараты»
ОАО «Борисовский завод медицинских препаратов»
УП «Минскинтеркапс»
ООО «Фармтехнология»
ГУ «Республиканский научно-практический центр трансфузиологии и медицинских
биотехнологий»
19
ВВЕДЕНИЕ
Во втором издании тома 2 Государственной фар¬
макопеи Республики Беларусь (ГФ РБ II, том 2) приве¬
дены частные фармакопейные статьи на субстанции
для фармацевтического использования и лекарствен¬
ное растительное сырье. Кроме этого приведены но¬
вые разделы, общие фармакопейные статьи и тексты,
а также новая редакция некоторых статей второго
издания тома 1 Государственной фармакопеи Респу¬
блики Беларусь (ГФ РБ II, том 1).
Новые частные фармакопейные статьи на суб¬
станции для фармацевтического использования:
Азота закись (0416)
Азотная кислота (1549)
Албендазол (1386)
Амикацин (1289)
Амикацина сульфат (1290)
Амоксициллин натрия (0577)
Амфотерицин В (1292)
Анастрозол (2406)
Аргинин (0806)
Аторвастатин кальция тригидрат (2191)
Бария сульфат (0010)
Беклометазона дипропионат безводный (0654)
Беклометазона дипропионат моногидрат (1709)
Бентонит (0467)
Бетагистина дигидрохлорид (1665)
Бетагистина мезилат (1071)
Бетаксолола гидрохлорид (1072)
Валсартан (2423)
Винорелбина тартрат (2107)
Висмута субгаллат (1493)
Галактоза (1215)
Галоперидол (0616)
Гэнтамицина сульфат (0331)
Гиосциамина сульфат (0501)
Гиосцина гидробромид (0106)
Гипромеллоза (0348)
Гистамина дигидрохлорид (0143)
Гистидин (0911)
Гистидина гидрохлорид моногидрат (0910)
Гпимепирид (2223)
Гпюкозамина сульфат натрия хлорид (2447)
Динатрия фосфат додекагидрат (0602)
Доксиламина гидросукцинат (1589)
Докузат натрия (1418)
Допамина гидрохлорид (0664)
Доцетаксел безводный (2593)
Доцетаксел тригидрат (2449)
Дутастерид (2641)
Железа (II) сульфат высушенный (2340)
Зопиклон (1060)
Идоксуридин (0669)
Имипенем моногидрат (1226)
Инсулин аспарт (2084)
Инсулин бычий (1637)
Инсулин двухфазовый для инъекций (0831)
Инсулин изофан двухфазовый для инъекций (0832)
Инсулин изофан для инъекций (0833)
Инсулин лизпро (2085)
Инсулин растворимый для инъекций (0834)
Инсулина инъекционные лекарственные сред¬
ства (0854)
Инсулина цинка суспензия (аморфная) для инъ¬
екций (0835)
Инсулина цинка суспензия (кристаллическая)
для инъекций (0836)
Интерферона альфа-2 концентрированный раст¬
вор (1110)
Итраконазол (1335)
Калия гидроксид (0840)
Калия клавуланат (1140)
Калия клавуланат разведенный (1653)
Калия цитрат (0400)
Карбоплатин (1081)
Касторовое масло рафинированное (2367)
Кислород газообразный (РБ0001)
Клозапин (1191)
Клопидогреля гидросульфат (2531)
Лактулоза (1230)
Ламотриджин (1756)
Лансопразол (2219)
Леводопа (0038)
Магния ацетат тетрагидрат (2035)
Макрогола цетостеариловый эфир (1123)
Марганца глюконат (2162)
Мебендазол (0845)
Меклозина дигидрохлорид (0622)
Мелоксикам (2373)
Мельдоний дигидрат (2624)
Менадион (0507)
Меропенем тригидрат (2234)
Метионин (1027)
Метотрексат (0560)
Метронидазола бензоат (0934)
Моксифлоксацина гидрохлорид (2254)
Моксонидин (1758)
Молочная кислота (0458)
(8)-Молочная кислота (1771)
Натрия гиалуронат (1472)
Натрия йодид (0196)
Натрия метилпарагидроксибензоат (1262)
Натрия пропилпарагидроксибензоат (1263)
Натрия фторид (0514)
Натрия цетостеарилсульфат (0847)
Натрия этилпарагидроксибензоат (2134)
Нимесулид (1548)
Нифуроксазид (1999)
Носкапина гидрохлорид (0515)
Оксалиплатин (2017)
Окситоцин (0780)
Окситоцина раствор концентрированный (0779)
Омега-З-кислоты этиловые эфиры 60 (2063)
Омега-З-кислоты этиловые эфиры 90 (1250)
Осельтамивира фосфат (2422)
Панкреатина порошок (0350)
Пентоксифиллин (0851)
Пепсина порошок (0682)
Пиперазин гидрат (0425)
Прамипексола дигидрохлорид моногидрат (2416)
Протамина сульфат (0569)
Рамиприл (1368)
Рибавирин (2109)
Рисперидон (1559)
Ртути хлорид (0120)
Сабаля мелкопильчатого экстракт (2579)
Севофлуран (2269)
Серебра нитрат (0009)
Серин (0788)
5*. Зак. 1060.
20
Гэсударственная фармакопея Республики Беларусь
Сертаконазола нитрат (1148)
Спирамицин (0293)
Стрептомицина сульфат (0053)
Суксаметония хлорид (0248)
Сульпирид (1045)
Тадалафил (2606)
Тейкопланин (2358)
Теобромин (0298)
Теофиллин (0299)
Терпентинное масло, тип Р/пиз ртаз1ег (1627)
Тестостерон (1373)
Тестостерона пропионат (0297)
Тирозин (1161)
Тозилхлорамид натрия (0381)
Толбутамид (0304)
Трагакант (0532)
Трамадола гидрохлорид (1681)
Трометамол (1053)
Углерода диоксид (0375)
Уголь активированный (0313)
Феназон (0421)
Фенилаланин (0782)
Феноксиметилпенициллин (0148)
Фибриноген человеческий (0024)
Фруктоза (0188)
Фторурацил (0611)
Хинидина сульфат (0017)
Хинина гидрохлорид (0018)
Хинина сульфат (0019)
Хлорбутанол безводный (0382)
Хлорбутанол гемигидрат (0383)
Хлоргексидина диацетат (0657)
Холекальциферол (0072)
Холестерин (0993)
Хондроитина натрия сульфат (2064)
Целекоксиб (2591)
Целтилпиридиния хлорид (0379)
Цетримид (0378)
Цефепима дигидрохлорид моногидрат (2126)
Цефтазидим пентагидрат (1405)
Цефтазидим пентагидрат с натрия карбона¬
том для инъекций (2344)
Цианокобаламин (0547)
Циластатин натрия (1408)
Цинка глюконат (2164)
Цитарабин (0760)
Эметина гидрохлорид пентагидрат (0081)
Эритриол (1803)
Этанол безводный (1318)
Этилпарагидроксибензоат (0900)
Новые частные фармакопейные статьи на ле¬
карственное растительное сырье:
Каштана конского семена (РБ0008)
Лимонника плоды (2428)
Малины листья (РБ0007)
Подорожника ланцетного листья (1884)
Рудбекии шершавой цветки (РБ0099)
Чабреца блошиного трава (РБ0100)
Чеснока порошок (1216)
Новые общие фармакопейные статьи и тек¬
сты:
2.2.61. Определение характеристик кристалли¬
ческих твердых веществ с помощью микрокалори¬
метрии и калориметрии растворения
2.2.64. Спектрометрия ядерного магнитного
резонанса для идентификации пептидов
2.2.65. Вольтаметрическое титрование
2.2.66. Обнаружение и измерение радиоактив¬
ности
#2.2.90. Титриметрические методы анализа
2.4.20. Определение остаточных количеств ме¬
таллических катализаторов или металлсодержа¬
щих реактивов
2.5.40. Метил-, этил- и изопропилтолуолсуль-
фонат в фармацевтических субстанциях
2.5.41. Метил-, этил- и изопропилбензолсуль-
фонат в фармацевтических субстанциях
2.8.21. Испытание лекарственного раститель¬
ного сырья на наличие аристолохиевых кислот
2.9.39. Взаимодействия «вода - твердое веще¬
ство»: построение изотерм сорбции-десорбции и
определение активности воды
2.9.47. Подтверждение однородности дозиро¬
ванных единиц с использованием большого количе¬
ства образцов
5.20. Остаточные количества металлических
катализаторов или металлсодержащих реактивов
#5.50. Критерии чистоты пищевых красителей
В новой редакции приведены следующие
разделы и статьи:
1. Общие сведения
2.2.20. Потенциометрическое титрование
2.2.29. Жидкостная хроматография
2.2.32. Потеря в массе при высушивании
2.3.1. Реакции подлинности (идентификации)
на ионы и функциональные группы
2.4.27. Тяжелые металлы в лекарственном рас¬
тительном сырье и продуктах из лекарственного
растительного сырья
2.5.1. Кислотное число
2.5.12. Вода: полумикрометод
2.6.31. Микробиологические испытания лекар¬
ственных средств растительного происхождения
для внутреннего применения и экстрактов, исполь¬
зующихся для их приготовления
2.8.2. Примеси
2.9.10. Содержание этанола
2.9.11. Испытание на содержание метанола и
2-пропанола
2.9.25. Высвобождение действующего веще¬
ства из лекарственных жевательных резинок
5.4. Остаточные количества органических
растворителей
5.12. Стандартные образцы
5.15. Функционально-обусловленные характери¬
стики вспомогательных веществ
Радиоактивные фармацевтические препараты
(0125)
Лекарственные средства для парентерального
применения (0520)
Приведены дополнения к статьям, опублико¬
ванным в ГФ РБ II, том 1:
4.1.1. Реактивы
4.1.3. Буферные растворы
4.2.2. Титрованные растворы
1. Общие сведения
21
07/2016:10000
1. ОБЩИЕ СВЕДЕНИЯ
1.1. ОБЩИЕ ПОЛОЖЕНИЯ
Положения главы 1. Общие сведения распро¬
страняются на все общие статьи, частные статьи и
другие тексты Государственной фармакопеи Респу¬
блики Беларусь.
В текстах Государственной фармакопеи Респу¬
блики Беларусь слово «Фармакопея» без уточнений
подразумевает Государственную фармакопею Рес¬
публики Беларусь. Наряду с этим для обозначения
Государственной фармакопеи Республики Беларусь
может быть использовано также официальное сокра¬
щение ГФ РБ.
Частные и общие статьи Фармакопеи гармони¬
зированы с Европейской Фармакопеей и построены в
следующем формате:
- адаптированный перевод соответствующего
материала Европейской Фармакопеи;
- национальные общие и частные статьи, допол¬
нительные испытания, информационные и иные ма¬
териалы отмечены значком «#» перед названием гла¬
вы, статьи, раздела, таблицы, рисунка или пункта, на
которые он распространяется; текст, отличающийся
от текста Европейской Фармакопеи, отмечается меж¬
ду двумя значками «#», например контрольный опыт#;
- в разделе «Частные фармакопейные статьи на
лекарственное растительное сырье» статьи на лекар¬
ственное растительное сырье или отдельные испы¬
тания и нормы, соответствующие требованиям Евро¬
пейской Фармакопеи, обозначены значком «сеф)».#
Ссылка в материалах Фармакопеи на какую-либо
статью и/или ее раздел означает, что продукт соот¬
ветствует требованиям этой статьи. Название статьи,
на которую дается ссылка, и/или ее номер выделены
курсивом.
#Дата введения в действие (месяц/год) и номер
статьи (четырехзначное или пятизначное число; РБ
перед номером статьи обозначает статью, не описан¬
ную в Европейской Фармакопее) указываются над ее
заголовком. Например, 07/2016:2624 соответствует
частной статье Мельдоний дигидрат (2624), которая
вступит в действие 1 июля 2016 года, а 01/2013:20101
соответствует общей статье 2.1.1. Каплемер, которая
действует с 1 января 2013 года.#
Лекарственное средство должно соответствовать
требованиям Фармакопеи на протяжении срока годно¬
сти. Компетентный уполномоченный орган может при¬
нять решение о необходимости установления отдель¬
ного срока годности и/или требования спецификации
для лекарственного средства во вскрытом контей¬
нере. Объект любой частной фармакопейной статьи
должен соответствовать установленным требованиям
в течение всего периода его использования. Срок год¬
ности и дата, от которой он должен отсчитываться, со¬
гласовываются компетентным уполномоченным орга¬
ном на основании экспериментальных исследований
по стабильности данного готового продукта.
Требования частной статьи являются обязатель¬
ными, если нет специальных указаний в разделе «Об¬
щие сведения» или в данной частной фармакопейной
статье. Общие статьи становятся обязательными, ког¬
да на них приводится ссылка в той или иной частной
или общей фармакопейной статье, если только спе¬
циально не указано, что ссылка приводится исключи¬
тельно как информация или рекомендация.
Действующие вещества (фармацевтические суб¬
станции), вспомогательные вещества, лекарственные
средства и другие продукты, описываемые в частных
статьях Фармакопеи, предназначены для использова¬
ния в медицине.
Качество продукта является фармакопейным
только при его соответствии всем требованиям фар¬
макопейной статьи. Это условие не подразумевает
необходимость выполнения производителем всех
испытаний, описанных в статье, при оценке соответ¬
ствия требованиям Фармакопеи при выпуске в об¬
ращение. Для подтверждения соответствия продукта
требованиям Фармакопеи производитель может ис¬
пользовать данные, полученные, например, в ходе
валидации производственного процесса и внутрипро¬
изводственного контроля. Таким образом, требование
соответствия продукта Фармакопее может выполнять¬
ся и при выходном контроле в виде выпуска по пара¬
метрам (рагатеМс ге1еазе) при наличии разрешения
компетентного уполномоченного органа.
Фармацевтические субстанции, вспомогатель¬
ные вещества и другие продукты, на которые рас¬
пространяются требования Фармакопеи, могут иметь
различные параметры качества в зависимости от
цели использования. Если на этот счет нет указаний
в соответствующей частной фармакопейной статье,
ее требования распространяются на продукт незави¬
симо от целей его применения. В некоторых случаях,
в частности в случае вспомогательных веществ, для
информации частная статья может быть дополнена
списком функционально-обусловленных характери¬
стик, которые являются важными для использования
данного вещества. Также в информационных целях
могут быть приведены методики контроля одной или
нескольких таких характеристик.
Системы качества. Стандарты качества, уста¬
новленные в фармакопейных статьях, применимы к
продукту только при условии его производства в рам¬
ках соответствующей системы обеспечения качества.
Общие статьи. Субстанции и лекарственные
средства, описанные в частных фармакопейных ста¬
тьях, также должны выдерживать требования соот¬
ветствующих подходящих общих фармакопейных
статей. В частных фармакопейных статьях обычно не
указывают перекрестные ссылки на общие фармако¬
пейные статьи.
Действие общих фармакопейных статей рас¬
пространяется на все субстанции и лекарственные
средства, указанные в разделе «Определение» об¬
щей фармакопейной статьи, за исключением случа¬
ев, когда вводная часть ограничивает ее применение,
например только для субстанций и лекарственных
средств, описанных в этой фармакопейной статье.
Требования фармакопейных статей на лекар¬
ственные формы распространяются на все лекар¬
ственные средства, изготовленные в виде этой ле¬
карственной формы. Для конкретного лекарственного
средства требования соответствующей фармакопей¬
ной статьи не обязательно являются исчерпываю¬
щими, и компетентный уполномоченный орган может
ввести дополнительные требования помимо указан¬
ных в статье.
Общие и частные фармакопейные статьи явля¬
ются взаимодополняемыми. Если требования общей
фармакопейной статьи неприменимы к определенно¬
му лекарственному средству, это однозначно указы¬
вается в частной фармакопейной статье.
22
Гэсударственная фармакопея Республики Беларусь
Валидация фармакопейных методик. Методи¬
ки, приведенные в частных и общих фармакопейных
статьях, были валидированы в соответствии с обще¬
принятой научной практикой и современными реко¬
мендациями по валидации аналитических методик.
Если иное не указано в частной или общей фармако¬
пейной статье, валидация таких методик не требуется.
Испытания и методики определения, приведен¬
ные в Фармакопее, являются официальными мето¬
диками, однако по согласованию с компетентными
уполномоченными органами могут использоваться и
другие методики, при условии, что они дают результа¬
ты, соответствующие фармакопейным методикам. В
случае сомнений или разногласий решающей являет¬
ся фармакопейная методика/
Общепринятые термины. Термин «компетент¬
ный уполномоченный орган» означает Министерство
здравоохранения Республики Беларусь или Респу¬
бликанское унитарное предприятие «Центр экспертиз
и испытаний в здравоохранении» в соответствии с его
компетенцией/
Выражение «если нет других указаний в частной
статье» #(под частной статьей подразумевается част¬
ная фармакопейная статья Фармакопеи или норма¬
тивный документ по контролю качества (НД или ФСП),
согласованный уполномоченным органом)# означает,
что требования общей фармакопейной статьи долж¬
ны быть выполнены, за исключением случаев, когда
компетентный уполномоченный орган согласовал (ут¬
вердил) изменения или исключения для таких требо¬
ваний, что указывается в частной статье.
Положения со словом «следует» носят рекомен¬
дательный или информационный характер.
В некоторых общих и частных статьях Фармако¬
пеи при описании реактива, микроорганизма, методи¬
ки и т.д. используется термин «подходящий». Если при
этом критерии их пригодности не сформулированы, то
пригодность конкретных реактивов, методик и т.д., ис¬
пользуемых в частной статье, должна быть обоснова¬
на перед компетентным уполномоченным органом.
Лекарственное средство. #Вещество или ком¬
бинация нескольких веществ природного, синтетиче¬
ского или биотехнологического происхождения, об¬
ладающие фармакологической активностью и в опре¬
деленной лекарственной форме применяемые для
медицинской профилактики, диагностики, лечения и
медицинской реабилитации пациентов, предотвраще¬
ния беременности путем внутреннего или внешнего
применения/
Лекарственное средство растительного про¬
исхождения. Лекарственное средство, содержащее
в качестве действующего вещества (действующих
веществ) только лекарственное растительное сырье
или продукты из лекарственного растительного сы¬
рья или лекарственное растительное сырье в ком¬
бинации с продуктами из лекарственного раститель¬
ного сырья.
#Продукт из лекарственного растительного
сырья. Однородный продукт, полученный из лекар¬
ственного растительного сырья путем экстракции,
перегонки, отжима, фрагментирования, очистки,
концентрирования или ферментации. Продукты из
лекарственного растительного сырья включают, на¬
пример, экстракты, эфирные масла, соки, экссудаты
и лекарственное растительное сырье, измельченное
для соответствующего применения, например приго¬
товления настоев, чаев, отваров, получения сборов,
капсулирования или таблетирования. Продукт из ле¬
карственного растительного сырья может представ¬
лять собой фармацевтическую субстанцию, промежу¬
точный продукт или лекарственное средство.
#Лекарственное растительное сырье. Исполь¬
зуемые для промышленного производства, аптечного
изготовления лекарственных средств цельные лекар¬
ственные растения или части лекарственных расте¬
ний, на которые имеются соответствующие фармако¬
пейные статьи.
Фармацевтическая субстанция (действующее
вещество). #Вещество или комбинация нескольких
веществ природного, синтетического или биотехно¬
логического происхождения, обладающие фарма¬
кологической активностью, используемые для про¬
мышленного производства, аптечного изготовления
лекарственных средств/ Такие вещества предназна¬
чены для оказания фармакологического или иного не¬
посредственного действия при диагностике, лечении
и профилактике заболеваний или для воздействия на
органы и функции организма.
Вспомогательное вещество. #Вещество или
комбинация нескольких веществ, не обладающие
фармакологической активностью и используемые в
процессе промышленного производства, аптечного
изготовления лекарственного средства для придания
ему определенной лекарственной формы/ Напри¬
мер, вспомогательными веществами являются стаби¬
лизаторы, антимикробные консерванты, антиоксидан¬
ты, растворители, адъюванты.
Субстанции для фармацевтического использо¬
вания. Термин распространяется на фармацевтиче¬
ские субстанции и вспомогательные вещества.
1.2. ДРУГИЕ ПОЛОЖЕНИЯ,
РАСПРОСТРАНЯЮЩИЕСЯ НА ОБЩИЕ И
ЧАСТНЫЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ
Количество вещества. При описании количе¬
ственного определения или испытания с численно за¬
данными пределами количество вещества, необходи¬
мое для проведения испытания, может отклоняться в
пределах ±10 % от указанного количества. Необходи¬
мо взять точную навеску испытуемого вещества (или
отмерить его каким-либо другим способом) и все вы¬
числения производить для этого точного количества.
Если пределы испытания заданы не численно,
а определяются путем сравнения со стандартом при
тех же условиях, для испытания берут указанное ко¬
личество вещества. Реактивы всегда берут в указан¬
ных количествах.
Количества вещества взвешивают или отмерива¬
ют с точностью, соответствующей указанной степени
прецизионности. Точность взвешивания должна быть
±5 единиц после последней указанной цифры (напри¬
мер, навеска 0,25 г может находиться в пределах от
0,245 г до 0,255 г). Объемы отмеривают следующим
образом: если после десятичной запятой стоит ноль
или число, заканчивающееся на ноль (например,
10,0 мл или 0,50 мл), требуемый объем отмеривают
с помощью пипетки, мерной колбы или бюретки. В
остальных случаях можно использовать градуирован¬
ный мерный цилиндр или градуированную пипетку.
Микролитры отмеривают с помощью микропипетки
или микрошприца.
Тем не менее в некоторых случаях точность, с
которой указаны количества вещества, не совпадает
с количеством значащих цифр, указанных в заданных
1. Общие сведения
23
числовых пределах. В таких случаях взвешивания и
отмеривания проводят с существенно более высокой
точностью.
Оборудование и аналитические операции.
Стеклянная мерная посуда должна соответствовать
требованиям класса А Международного стандарта,
выпущенного Международной организацией по стан¬
дартизации (180), #или 1-го класса точности, соот¬
ветствующая национальному стандарту Республики
Беларусь*.
Аналитические операции, если нет других указа¬
ний, осуществляют при температуре от 15 °С до 25 °С.
Сравнительные испытания, если нет других
указаний, проводят с использованием идентичных
пробирок из бесцветного прозрачного нейтрального
стекла с плоским основанием и внутренним диаме¬
тром 16 мм, так как указываемые объемы жидкостей
рассчитаны для этого диаметра; однако, при условии
корректировки объемов, могут быть использованы
также пробирки с большим внутренним диаметром
(2.1.5). Сравнивают равные объемы жидкостей вдоль
вертикальной оси пробирки на белом (или, при необ¬
ходимости, на черном) фоне. Испытания проводят в
рассеянном свете.
Если для проведения испытания или количе¬
ственного определения требуется использовать раст¬
воритель с растворенным в нем индикатором и при
этом не предусмотрен контрольный опыт, этот раст¬
воритель предварительно нейтрализуют по этому ин¬
дикатору.
#Под контрольным (холостым) опытом подраз¬
умевают определение, проводимое с теми же количе¬
ствами реактивов и в тех же условиях, но без испыту¬
емого образца*
Водяная баня. Если не указана вода с другой
температурой, то подразумевается баня с кипящей
водой. Можно использовать и другие способы нагре¬
вания, если они гарантированно обеспечивают тем¬
пературу, близкую, но не превосходящую 100 °С (или
другую указанную температуру).
#Ледяная баня. Если не указана другая тем¬
пература, то подразумевается баня с температурой
около О °С. Если необходимо охлаждение до более
низкой температуры, применяют смесь льда с некото¬
рыми электролитами (солями, кислотами).
Высушивание и прокаливание до постоян¬
ной массы. Результаты двух последних взвешиваний
должны отличаться не более чем на 0,5 мг; интервал
времени между двумя взвешиваниями определяется
свойствами и количеством высушиваемого/прокали-
ваемого остатка.
В тех случаях, когда требуется высушивание «в
эксикаторе» или «в вакууме», оно осуществляется
в соответствии с условиями, описанными в статье
2.2.32. Потеря в массе при высушивании.
Реактивы. Надежность результатов, получаемых
с помощью описанных в фармакопейных статьях ана¬
литических операций, зависит, в частности, от каче¬
ства используемых реактивов. Реактивы описаны в
главе 4. Реактивы. Подразумеваемая степень чисто¬
ты — не ниже квалификации «аналитической чисто¬
ты» (апа!уИса1 дгаде) #или квалификации «чистый для
анализа» (ч.д.а.)#. Для некоторых реактивов включе¬
ны испытания для определения пригодности.
Растворители. Если для растворов не указан
растворитель, то подразумевают водные растворы.
Для проведения описанных в статьях Фармако¬
пеи аналитических операций и для приготовления
реактивов используют воду, соответствующую тре¬
бованиям частной статьи Вода очищенная (0008), за
исключением случаев, когда требования по содержа¬
нию бактериальных эндотоксинов (Вода очищенная т
Ьи1к) или по микробиологической чистоте (Вода очи¬
щенная в контейнерах) не являются существенными.
Под термином «вода дистиллированная» понимают
воду очищенную, полученную путем дистилляции.
Термин «этанол» без уточнений означает абсо¬
лютный этиловый спирт. Термин «спирт» без уточ¬
нений означает 96 % (об/об) этанол. Другие степени
разбавления обозначаются термином «этанол» или
«спирт» с указанием содержания этанола (С2НеО) в
объемных процентах.
*Термин «эфир» без уточнений означает диэти-
ловый эфир.#
Способы выражения концентрации. Выраже¬
ние «%» может иметь одно из трех значений:
- массовый процент (м/м) — число граммов ве¬
щества в 100 граммах конечного продукта;
- объемный процент (об/об) — число миллили¬
тров вещества в 100 миллилитрах конечного продукта;
- #массо-объемный процент (м/об) — число грам¬
мов вещества в 100 миллилитрах конечного продукта*
Если не указано иного, обозначение «ррт» (ча¬
стей на миллион) подразумевает массовое соотноше¬
ние.
*Если указано, что при приготовлении смеси раст¬
ворителей их берут в соотношении (а:Ь), то имеется в
виду соотношение объемов. Например, соотношение
гексан — бензол (1:3) означает, что смешивают 1 объ¬
ем гексана с 3 объемами бензола.#
Температура. Кроме конкретного указания тем¬
пературы при проведении испытаний используют так¬
же следующие термины:
- глубокое охлаждение (ниже -15 °С);
- в холодильнике (от 2 °С до 8 °С);
- в холодном или прохладном месте (от 8 °С до
15 °С);
- при комнатной температуре (от 15 °С до 25 °С).
#Кроме терминов, приведенных выше, могут ис¬
пользоваться также следующие термины:
- теплый (от 40 °С до 50 °С);
- горячий (от 80 °С до 90 °С);
- температура «водяной бани» (от 98 °С до
100 °С);
- температура «ледяной бани» (около 0 °С).#
1.3. ОБЩИЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ
Контейнеры. Материалы, используемые для
контейнеров, описаны в главе 3.1. Для материалов,
используемых для производства контейнеров, осо¬
бенно для полимерных материалов, используют об¬
щие названия, каждое из которых охватывает ряд
материалов, отличающихся как свойствами основ¬
ного компонента, так и используемыми добавками.
Испытания и пределы нормирования зависят от кон¬
кретного состава материала и, таким образом, приме¬
нимы только при условии, что материал соответствует
вводной части его спецификации. По согласованию с
компетентным уполномоченным органом могут ис¬
пользоваться материалы других составов, а также ис¬
пытания для них.
Спецификации на контейнеры, включенные в
главу 3.2, разрабатывались для всех контейнеров ука¬
занной категории. Однако, учитывая большое разно-
24
Гэсударственная фармакопея Республики Беларусь
образие существующих контейнеров и возможность
появления новых контейнеров, публикация специ¬
фикации не исключает возможности использования
контейнеров, соответствующих другой спецификации,
если это обосновано и согласовано с компетентным
уполномоченным органом.
В статьях Фармакопеи могут даваться ссылки на
определения и спецификации контейнеров, приве¬
денные в главе 3.2. Контейнеры. В разделах «Опре¬
деление» или «Производство» общих статей на ле¬
карственные формы может содержаться требование
по использованию определенного типа контейнера. В
разделе «Хранение» некоторых статей может указы¬
ваться тип рекомендуемого контейнера.
1.4. ЧАСТНЫЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ
НАЗВАНИЯ
#Кроме названий на русском языке приводятся
также английское и латинское названия/
ОТНОСИТЕЛЬНЫЕ АТОМНЫЕ И
МОЛЕКУЛЯРНЫЕ МАССЫ
Относительную атомную массу (А.м.) или отно¬
сительную молекулярную массу (М.м.) указывают где
применимо в начале частной фармакопейной статьи.
Относительную атомную массу, относительную моле¬
кулярную массу, молекулярную формулу и графиче¬
скую формулу приводят как информационный мате¬
риал.
РЕГИСТРАЦИОННЫЕ НОМЕРА САЗ
Регистрационные номера Химической рефера¬
тивной службы САЗ (СЬетюа! АЬ$1гас1з Земсе) могут
включаться для информации в частные фармакопей¬
ные статьи, где это применимо, с целью предостав¬
ления пользователю удобного доступа к полезной
информации. Регистрационный номер САЗ® (САЗ
Ред181гу ИитЬег®) является зарегистрированной тор¬
говой маркой Американского химического общества
(Атепсап СПетюа! Зо^у).
ОПРЕДЕЛЕНИЕ
В разделе «Определение», идущем после назва¬
ния частной статьи, приводится официальное опреде¬
ление субстанции, лекарственного средства или ино¬
го продукта, являющегося предметом частной статьи.
Пределы содержания. Если указаны пределы
содержания, то это пределы, полученные с использо¬
ванием метода, указанного в разделе «Количествен¬
ное определение».
Лекарственное растительное сырье. В част¬
ных статьях на лекарственное растительное сырье
в разделе «Определение» указывают на предмет
частной статьи. Это может быть, например, лекар¬
ственное растительное сырье в исходном виде или
лекарственное растительное сырье, измельченное в
порошок. Если частная статья распространяется на
несколько вариантов, например на оба из указанных,
то это оговаривается в разделе «Определение».
ПРОИЗВОДСТВО
Информация в разделе «Производство» призва¬
на привлечь внимание к некоторым важным аспектам
процесса производства и не обязательно является
исчерпывающей. Содержащиеся в ней инструкции
адресованы производителю. Они могут относиться,
например, к материалам, к процессу производства,
к его валидации и контролю, к постадийному контро¬
лю, а также к испытаниям, которые производитель
должен проводить перед выпуском для каждой серии
продукта или для выбранных серий. Эти положения
не обязательно должны быть подтверждены посред¬
ством анализа конечного продукта. Компетентным
уполномоченным органом может быть установлено,
что приведенные выше аспекты были выполнены.
Такое заключение может быть сделано на основании
проверки полученных от производителя данных, или
при инспектировании производства или при испыта¬
нии соответствующих образцов.
Отсутствие раздела «Производство» не означа¬
ет, что аспекты производственного процесса, отме¬
ченные выше, не требуют внимания.
Выбор вакцинного штамма, выбор состава
вакцины. В разделе «Производство» частной фар¬
макопейной статьи на вакцину могут быть указаны
характеристики вакцинного штамма или состав. Если
иное не указано, описываемые в этом разделе мето¬
дики испытаний для подтверждения этих параметров
приводятся для информации в качестве примера. В
случае разрешения компетентным уполномоченным
органом могут использоваться иные методики испы¬
таний без проведения их валидации в сравнении с
методиками, приведенными в статье.
ВОЗМОЖНОСТЬ ФАЛЬСИФИКАЦИИ
В связи с увеличением мошеннической активно¬
сти и случаев фальсификации осведомленность может
помочь пользователям Фармакопеи в обнаружении
фальсификатов (т.е. фармацевтических субстанций,
вспомогательных веществ, промежуточных продуктов,
нефасованных продуктов и лекарственных средств).
С учетом этого в раздел «Возможность фальси¬
фицирования» частных статей на субстанции, для
которых встречались случаи фальсифицирования
или у которых присутствует риск наличия умышленно
добавленных примесей, могут быть включены мето¬
дика обнаружения возможных фальсификатов и со¬
ответствующие пределы вместе с напоминанием, что
все стадии производства и снабжения должны быть
частью надлежащей системы качества. Частота ис¬
пытаний, проводимых производителем или потреби¬
телем (например, производителем промежуточных
продуктов, нефасованных продуктов и лекарственных
средств, где применимо), зависит от оценки риска,
принимая во внимание национальные требования и
уровень знаний всей цепочки поставок.
Данный раздел устанавливает требования ко
всей цепочке поставок в целом, от производителей
до потребителей (например, производителей проме¬
жуточных продуктов, нерасфасованных продуктов и
лекарственных средств, где применимо). Отсутствие
данного раздела не предполагает того, что не должно
уделяться внимание моментам, упомянутым выше.
ОПИСАНИЕ (СВОЙСТВА)
Информация, приведенная в этом разделе, не
должна рассматриваться как прямое указание и не
является требованием.
#В данном разделе может быть указано о необ¬
ходимости измерения размера испытуемого образца
(лекарственного средства или лекарственного расти¬
тельного сырья). В таком случае определения прово¬
дятся с использованием подходящего измерительного
оборудования (линейки, штангенциркуля, микрометра
и др.) с требуемой точностью/
Растворимость. #Под «растворимостью» под¬
разумевают свойство вещества растворяться в раз-
1. Общие сведения
25
личных растворителях, принятых в ГФ РБ. Показатели
растворимости вещества в различных растворителях
приводятся в частных фармакопейных статьях.#
Для обозначения растворимости в разделе «Опи¬
сание (Свойства)» используют описательные терми¬
ны, которые в температурном интервале от 15 °С до
25 °С имеют следующие значения:
Термин
Примерное количество
растворителя (мл), необ¬
ходимое для растворения
1 г вещества
Очень легко растворим
менее
1
Легко растворим
от
1
ДО
10
Растворим
от
10
до
30
Умеренно растворим
от
30
ДО
100
Мало растворим
от
100
до
1000
Очень мало растворим
от
1000
до
10 000
Практически нерастворим
более
10 000
Термин «частично растворим» используют для
характеристики смесей, у которых растворимы только
некоторые компоненты. Термин «смешивается с ...»
используют для характеристики жидкостей, смешива¬
ющихся с указанным растворителем во всех соотно¬
шениях.
#Примечание: для веществ, образующих при рас¬
творении мутные растворы, соответствующее указа¬
ние должно быть приведено в частной фармакопей¬
ной статье. Если указано, что субстанция растворима
в жирных маслах, то имеется в виду, что она раство¬
рима в любом масле, относящемся к классу жирных
масел.#
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Область применения. Приводимые в этом
разделе испытания не рассчитаны на полное под¬
тверждение химической структуры или состава
продукта. Они предназначены для подтверждения
с приемлемой степенью достоверности того, что
продукт соответствует информации, приведенной
на этикетке.
Первая и вторая идентификации. В некоторых
частных статьях есть подразделы «Первая идентифи¬
кация» и «Вторая идентификация». Испытания, опи¬
санные в подразделе «Первая идентификация», мо¬
гут быть использованы во всех случаях. Испытания,
описанные в подразделе «Вторая идентификация»,
могут быть использованы в аптеках, если есть дока¬
зательства того, что данная серия субстанции была
сертифицирована на соответствие всем другим тре¬
бованиям частной фармакопейной статьи.
#Для лекарственного растительного сырья могут
использоваться испытания, описанные в подразделе
«Вторая идентификация», если есть доказательства
того, что данная серия сырья была сертифицирована
на соответствие всем требованиям частной фармако¬
пейной статьи/
В некоторых частных статьях для первой иден¬
тификации приводятся два или более вида испыта¬
ний, которые являются эквивалентными и могут ис¬
пользоваться независимо друг от друга. Один или
более из этих рядов обычно содержат перекрестные
ссылки на испытания, приведенные в разделе «Ис¬
пытания» частной статьи. Это может быть исполь¬
зовано для облегчения работы аналитика, прово¬
дящего идентификацию и предписанные испытания.
Например, один ряд испытаний для идентификации
имеет ссылку на определение энантиомерной чисто¬
ты, в то время как в другом ряде проводится опре¬
деление удельного вращения: оба ряда преследуют
одну и ту же цель — подтвердить присутствие необ¬
ходимого энантиомера.
Порошкообразное лекарственное раститель¬
ное сырье. Частные статьи на лекарственное рас¬
тительное сырье могут содержать схематические
рисунки порошкообразного лекарственного средства.
Эти рисунки дополняют описание, приведенное в со¬
ответствующем испытании на подлинность.
ИСПЫТАНИЯ И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
Область применения. Эти требования не рас¬
считаны на охват всех возможных примесей. В част¬
ности, если примесь не определяется с помощью опи¬
санных испытаний, а здравый смысл и надлежащая
производственная практика не допускают ее присут¬
ствия, не следует делать вывод, что она допустима.
См. также ниже раздел «Примеси».
#Если в испытаниях с использованием хромато¬
графических методов после указания вводимого или
наносимого объема раствора в микролитрах в скобках
указывается количество вещества в микрограммах, то
имеется в виду приблизительное количество.
Если указано, что испытание проводят «в защи¬
щенном от света месте», то это означает, что следует
принять меры, исключающие попадание прямого сол¬
нечного света, любого другого яркого света, а также
исключить попадание ультрафиолетового света, на¬
пример, путем использования посуды из специально¬
го стекла, работы в затемненной комнате и т.д.#
Расчеты. Если при проведении вычислений тре¬
буется выполнить пересчет на сухое вещество или
безводное вещество или оговорено какое-либо дру¬
гое условие, то потерю в массе при высушивании,
содержание воды или иной показатель определяют с
помощью метода, описанного в частной фармакопей¬
ной статье. Слова «в пересчете на сухое вещество»
или «в пересчете на безводное вещество» и другие
указывают после результата.
Пределы. Указываемые пределы основыва¬
ются на результатах, полученных в рамках обычной
аналитической практики; в них уже учтены обычные
аналитические погрешности, допустимый разброс при
производстве и приготовлении, а также ухудшение
качества в процессе хранения в пределах, которые
считаются приемлемыми. При определении соответ¬
ствия продукта требованиям частной фармакопейной
статьи к указанным пределам не должны добавляться
никакие дополнительные допуски.
Результат, полученный в испытании, округляют
до указанного в пределе количества значащих цифр
(если нет других указаний). Пределы, независимо от
того, выражены они в процентах или в абсолютных
значениях, считаются значимыми до последнего ука¬
занного знака (например, число 140 обозначает три
значимых знака). При этом последнюю цифру увели¬
чивают на единицу, если цифра, отбрасываемая при
округлении, больше или равна пяти. Если цифра, от¬
брасываемая при округлении, меньше пяти, послед¬
нюю цифру оставляют неизменной.
Определение допустимого предела приме¬
сей. Для сравнительных испытаний примерное до¬
пустимое содержание примеси или суммы примесей
может быть указано в скобках только для информа¬
ции. Решение об одобрении или отклонении принима¬
ют на основании соответствия либо несоответствия
26
Гэсударственная фармакопея Республики Беларусь
приведенному в статье испытанию. Если для данной
примеси не указано использование стандартного об¬
разца, ее содержание может быть выражено исходя
из номинальной концентрации вещества, используе¬
мого для приготовления указанного в частной фарма¬
копейной статье раствора сравнения (если нет других
указаний).
Лекарственное растительное сырье. Для ле¬
карственного растительного сырья сульфатную золу,
общую золу, растворимые в воде вещества, раство¬
римые в спирте вещества, содержание воды, содер¬
жание эфирных масел и содержание действующих
веществ рассчитывают в пересчете на лекарствен¬
ное средство, которое не было специально высушено
(если нет других указаний в частной фармакопейной
статье).
Эквиваленты (титры). В тех случаях, когда
приводят эквивалент, его дают с таким количеством
значащих цифр, которое требуется в данной частной
статьи.
Питательные среды. Питательные среды, опи¬
санные в частных и общих статьях, являются удовлет¬
ворительными для использования их по предназначе¬
нию. Однако из-за непостоянного качества компонен¬
тов среды, особенно биологического происхождения,
может понадобиться коррекция концентраций некото¬
рых ингредиентов, например:
- пептонов, мясных или дрожжевых экстрактов с
учетом их питательных свойств;
- буферных веществ;
- желчных солей, желчного экстракта, дезоксихо-
лата и красящих веществ в зависимости от их селек¬
тивных свойств;
- антибиотиков в зависимости от их активности.
ХРАНЕНИЕ
Информация и рекомендации, приводимые в
разделе «Хранение», не являются исчерпывающи¬
ми фармакопейными требованиями, и компетентные
уполномоченные органы могут указывать конкретные
условия хранения, обязательные для исполнения.
Описанные в Фармакопее продукты следует
хранить таким образом, чтобы предотвратить их за¬
грязнение и, по возможности, разложение. Если ре¬
комендуются особые условия хранения, включая тип
контейнера (см. раздел 1.3. Общие фармакопейные
статьи) и температурные пределы, эти рекомендации
приводятся в частной статье.
Ниже разъясняются термины, используемые в
частных фармакопейных статьях в разделе «Хране¬
ние».
«В воздухонепроницаемом контейнере» обозна¬
чает, что лекарственное средство должно храниться в
воздухонепроницаемом контейнере (3.2). При вскры¬
тии контейнера во влажной атмосфере принимают
меры предосторожности. При необходимости низ¬
кая влажность содержимого может поддерживаться
с помощью осушающего вещества, помещаемого в
контейнер, при условии отсутствия прямого контакта
между ним и лекарственным средством. #При указа¬
нии «защищать от влаги» относительная влажность
в условиях хранения должна быть не более 60 %.#
«Б защищенном от света месте» обознача¬
ет, что лекарственное средство должно храниться в
контейнере, изготовленном из материала, в достаточ¬
ной степени поглощающего свет, способный вызвать
фотохимические превращения (актиничный свет); или
контейнер должен быть помещен во внешний контей¬
нер, обеспечивающий такую защиту; или лекарствен¬
ное средство должно храниться в месте, исключаю¬
щем возможность попадания такого света.
#«Температура». Лекарственные средства, суб¬
станции для фармацевтического использования, ле¬
карственное растительное сырье должны храниться в
соответствии с требованиями, указанными в таблице
МАРКИРОВКА
Маркировка регламентируется компетентным
уполномоченным органом с изданием соответствую¬
щего нормативного правового акта. Таким образом,
информация в разделе «Маркировка» не претендует
на полноту. Она ориентирована прежде всего на фар¬
макопейные цели, и обязательными являются только
те положения, которые необходимы для подтверж¬
дения соответствия продукта статье. Вся остальная
Таблица #1.4.-1
Условия хранения лекарственного
средства
Температурные пределы, указывае¬
мые на упаковке
Дополнительное указание
(при необходимости)
Лекарственное средство не требует
особых условий хранения
Нет
«Не охлаждать» или «Не замораживать»
Лекарственное средство требует усло¬
вий хранения при температуре не выше
30 °С (температура хранения от 2 °С до
30 °С)
«Хранить при температуре не выше
30 °С» или «Хранить при температуре
ниже 30 °С»
«Не охлаждать» или «Не замораживать»
Лекарственное средство требует усло¬
вий хранения при температуре не выше
25 °С (температура хранения от 2 °С до
25 °С)
«Хранить при температуре не выше
25 °С» или «Хранить при температуре
ниже 25 °С»
«Не охлаждать» или «Не замораживать»
Лекарственное средство требует хране¬
ния при комнатной температуре
«Хранить при температуре от 15 °С до
25 °С»
«Не охлаждать» или «Не замораживать»
Лекарственное средство требует хране¬
ния в холодном или прохладном месте
«Хранить при температуре от 8 °С до
15 °С»
«Не замораживать»
Лекарственное средство требует хране¬
ния в холодильнике
«Хранить при температуре от 2 °С до
8 °С»
«Не замораживать»
Лекарственное средство требует хране¬
ния при температуре ниже 0 °С
«Хранить в морозильной камере» или
«Хранить и транспортировать в моро¬
зильной камере»
Примечания:
- при дополнительном указании «Не охлаждать» не допускается подвергать лекарственное средство воздействию температуры ниже 8 °С;
- допускается кратковременное изменение условий хранения в процессе местной транспортировки (в пределах одного населенного
пункта) для лекарственных средств, не имеющих дополнительное указание по хранению «Не охлаждать» или «Не замораживать».
1. Общие сведения
27
информация носит рекомендательный характер. В
тех случаях, когда в Фармакопее употребляется тер¬
мин «этикетка», соответствующая информация может
быть размещена на контейнере, на упаковке, на лист¬
ке-вкладыше или в сертификате анализа, сопрово¬
ждающем лекарственное средство, в зависимости от
решения компетентного уполномоченного органа.
ПРЕДОСТЕРЕЖЕНИЯ
Описываемые в Фармакопее продукты и реак¬
тивы могут оказаться опасными для здоровья, если
не предпринять необходимые меры. Во всех случа¬
ях следует придерживаться принципов Надлежащей
лабораторной практики (НЛП, 0/.Р), а также соответ¬
ствующих положений техники безопасности. В неко¬
торые статьи включены специальные указания о не¬
обходимых мерах предосторожности, но отсутствие
таких указаний не следует трактовать как отсутствие
всякого риска.
ПРИМЕСИ
В частной фармакопейной статье может быть
приведен перечень всех известных и потенциальных
примесей, для которых показано, что они контролиру¬
ются испытаниями. См. также статью 5.10. Контроль
примесей в субстанциях для фармацевтического
использования. Примеси обозначают буквой или бук¬
вами латинского алфавита. Если какая-либо буква
пропущена, это означает, что примесь, обозначенная
с помощью нее, исключена из списка примесей при
разработке частной статьи до публикации либо при
пересмотре этой статьи.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ ВСПОМОГАТЕЛЬНЫХ
ВЕЩЕСТВ
Частные статьи на вспомогательные вещества
могут содержать раздел с перечнем функционально-
обусловленных характеристик. Параметры, любые
методики определения и нормы, приведенные в этом
разделе, не являются обязательными требованиями;
тем не менее они могут быть важны для использова¬
ния вспомогательных веществ и приведены для ин¬
формации (см. также раздел 1.1. Общие положения).
СТАНДАРТНЫЕ ОБРАЗЦЫ
Некоторые частные статьи предусматривают ис¬
пользование стандартных образцов (фармакопейных
стандартных образцов, биологических стандартных
препаратов, эталонных спектров). См. также статью
5.12. Стандартные образцы. 3ти стандартные об¬
разцы являются официальными в случае арбитража.
1.5. СОКРАЩЕНИЯ И ОБОЗНАЧЕНИЯ
А Оптическая плотность
А]^ Удельный показатель поглощения
А.м. Относительная атомная масса
[а]*° Удельное оптическое вращение
БСП Биологический стандартный препарат
ФСО Фармакопейный стандартный образец
Относительная плотность
А Длина волны
МЕ
Международные единицы
М
Молярность
М.м.
Относительная молекулярная масса
п20
Показатель преломления
РН. Еиг. и
Единица действия Европейской Фармакопеи
РРЬ
Частей на миллиард (микрограммов на кило¬
грамм)
РРт
Частей на миллион (миллиграммов на кило¬
грамм)
Р
Вещество или раствор, указанный в главе 4.
Реактивы
Фактор подвижности (статья 2.2.46)
К*
Используется в хроматографии для обозна¬
чения пути, пройденного испытуемым веще¬
ством, к пути, пройденному стандартным об¬
разцом
РО
Исходное стандартное вещество для уста¬
новки титра титрованных растворов в объ¬
емном анализе (статья 4.2.1)
Аббревиатуры, используемые в частных
фармакопейных статьях на иммуноглобулины,
сыворотки и вакцины
^50
Медианная летальная доза. Статистиче¬
ски определенное количество вещества, ко¬
торое при соответствующем применении
может вызвать смерть 50 % испытуемых жи¬
вотных за исследуемый период
мю
Минимальная летальная доза
1-+/10 <Зозе
Наименьшее количество токсина, которое
в смеси с 0,1 МЕ антитоксина при обычном
пути введения способно вызвать гибель ла¬
бораторных животных в экспериментальных
условиях за исследуемый период
Ь+ с1озе
Наименьшее количество токсина, которое в
смеси с 1 МЕ антитоксина при обычном пути
введения способно вызвать гибель лабора¬
торных животных в экспериментальных ус¬
ловиях за исследуемый период
1г/100 с!озе
Наименьшее количество токсина, которое
в смеси с 0,01 МЕ антитоксина при внутри-
кожном введении способно вызвать харак¬
терные реакции в месте введения у лабо¬
раторных животных в экспериментальных
условиях за исследуемый период
1_р/10 <Зозе
Наименьшее количество токсина, которое
в смеси с 0,1 МЕ антитоксина при внутри-
кожном введении способно вызвать харак¬
терные реакции в месте введения у лабо¬
раторных животных в экспериментальных
условиях за исследуемый период
1.0/10 <Зозе
Наименьшее количество токсина, которое в
смеси с 0,1 МЕ антитоксина и обычном пути
введения неспособно вызвать токсическую
реакцию у лабораторных животных в экс¬
периментальных условиях за исследуемый
период
28
Гэсударственная фармакопея Республики Беларусь
и дозе
Количество токсина или токсиноподобного
вещества, которое способно связать 1 МЕ
антитоксина за кратчайшее время
1ЧС1МВ
Национальная коллекция промышленных и
морских бактерий
Л/а#ола/ СоНесНоп о7 ХпбизХпа1 апб Маппе
со ю„
Медианная инфицирующая доза культуры
клеток. Статистически определенная доза
вирусных частиц, которая при внесении в
ВасХепа Ш
23 5^ МасЬаг ^пVе
АЬегбееп АВ2 1 РУ, СгеаХ ВгНат
клеточную культуру способна инфицировать
50 % клеток
ЫСРР
Национальная коллекция патогенных грибов
ЫаИопа1 СоНесНоп оХ РаНюдеп/д Рипд/
Медианная инфицирующая доза яиц. Ста¬
тистически определенная доза вирусных
частиц, которая при внесении в оплодотво¬
ренные яйца способна инфицировать 50 %
1-опбоп Зс1юо1 оТ Нуд/епе апб Тгорюа1 МебР
сте
Керре1 ЗХгееХ
1_опбоп УУС1Е7НТ, СгеаХ ВгНат
ю»
эмбрионов
(ЧСТС
Национальная коллекция типовых культур
Медианная инфицирующая доза. Статисти¬
чески определенная доза вирусных частиц,
которая при введении в организм животных
способна инфицировать 50 % особей
1ЧСУС
Л/аГ/ола/ СоНесНоп оХ Туре СиНигез СепХга1
РиЬНс НеаНН Х.аЬогаХогу СоНпба1е Ауепие
1-Опбоп Л/И/9 5НТ, СгеаХ ВгНат
Р°50
Медианная защитная доза. Статистически
определенная доза вакцины, которая в экс¬
периментальных условиях способна защи¬
тить 50 % животных от инфицирующей дозы
микроорганизмов или токсинов, в отношении
которых эта вакцина активна
Национальная коллекция дрожжевых
культур
А/а^/ола/ СоНесНоп оХ УеазХ СиНигез
АРСР Рооб РезеагсЬ ХпзИХиХе
Со1пеу \-апе
Мопл/юН ИРЛ 10А, СгеаХ ВгНат
т
о
а
Статистически определенная доза вакцины,
которая в экспериментальных условиях спо¬
собна индуцировать выработку антител к со¬
ответствующим антигенам вакцины у 50 %
животных
1Ч1ТЕ
Центр биологических ресурсов
Вю1од1са1 Резоигсез СепХег
йерагХтепХ оХ В'юХесЬпоХоду
МаИопа! /пзНХиХе оХ ТесХтоХоду апб ЕVаIиаX^оп
2-5-8 КагизакатаХагу, К/'загаги-зЫ, СЫЬа,
РР11
Оспинообразующие единицы или бляшкоо¬
бразующее число
292-0818
Зарап
5РР
Свободный от специфических патогенов
3.3.1.
Государственный институт сывороток
ЗХаХепз Зегит 1пзХНиХ
Коллекции микроорганизмов
80 Атадег Вои^агб, СорепНадеп, йептагк
АТСС
Американская коллекция типовых культур
Атепсап Туре СиНиге СоНесНоп
10801 ШЫегзНу ВоиХеуагс1
Мапаззаз, \ЛгдЫа 20110-2209, 113А
#Украинская коллекция микроорганизмов
Институт микробиологии им. Д.К. Заболотно¬
го НАН Украины
01000, Киев, ул. Заболотного, 154, Украина
С.1.Р.
Коллекция Пастеровского института
(штаммы бактерий)
СоНесНоп бе ВасХёпез ПпзХННиХе РазХеиг
В.Р. 52, 25 иге би ЭосХеигРоих
75724 Рапз Себех 15, Ргапсе
#Коллекция штаммов Российского музея па¬
тогенных бактерий
Государственный институт стандартизации и
контроля медицинских биологических препа¬
ратов им. Л.А. Тарасевича
121002, Москва, Сивцев-Вражек, 41, Россий-
1М1
Международный институт микологии
1п(егпаНопа1 Мусо1одюа1ХпзХНиХе
ВакеЬат 1-апе
Зиггеу Ш20 9ТУ, Сгеа1 ВпХа1п
ская Федерация
#Белорусская коллекция непатогенных ми¬
кроорганизмов (научная коллекция непа¬
тогенных типовых и промышленно ценных
1.Р.
Национальная коллекция культур
микроорганизмов
СоПесХюп МаХюпаХе бе СиНиге бе Мюгоогдап-
/зтез (С.Л/.С.М.)
1пзХННиХе РазХеиг
25, гие б и йосХеиг Роих
75724 Рапз Себех 15, Ргапсе
микроорганизмов института микробиологии
НАН Беларуси)
ГНУ «Институт микробиологии» НАН Бела¬
руси
220141, Минск, ул. Купревича, 2, Республика
Беларусь
1.6. ЕДИНИЦЫ МЕЖДУНАРОДНОЙ СИСТЕМЫ (СИ), ИСПОЛЬЗУЕМЫЕ В ФАРМАКОПЕЙНЫХ СТАТЬЯХ,
И ИХ СООТВЕТСТВИЕ ДРУГИМ ЕДИНИЦАМ
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ (СИ)
Международная система единиц состоит из трех классов единиц физических величин, а именно: основные
единицы, производные единицы и вспомогательные единицы1. Основные единицы и их определения приведены в
таблице 1.6.-1.
Производные единицы могут быть образованы путем комбинирования основных единиц согласно алгебраиче¬
ским взаимоотношениям, связывающим соответствующие количества. Некоторые из таких производных величин
имеют свои собственные названия и обозначения. Единицы СИ, используемые в Фармакопее, приведены в табли¬
це 1.6.-2.
1 Определения единиц, используемых в Международной системе, даны в буклете /_е ЗузХёте 1п1егпаНопа1 йУпНёз (31), опубли¬
кованном Вигеаи 1пХетаНопа1 без Ро/бз еХ Мезигез, РэуШоп бе ВгеХеиН, Г-92310 Зёугез.
1. Общие сведения
29
Таблица 1.6.-1
Основные единицы СИ
Величина
Единица
Определение
Наименование
Символ,
принятый в
Фармакопее
#Размер-
ность**
Наименова¬
ние
Символ
Длина
/
метр
м
Один метр представляет собой длину
пути, который проходит свет в вакууме за
1/299 792 458 долю секунды
Масса
т
м
килограмм
кг
Один килограмм равен массе международного
прототипа килограмма
Время
г
т
секунда
с
Одна секунда представляет собой продол¬
жительность 9192 631 770 периодов излуче¬
ния, соответствующих переходу между двумя
сверхтонкими уровнями основного состояния
атома цезия-133
Электрический ток
1
1
ампер
А
Один ампер представляет собой силу неиз-
меняющегося тока, который при прохождении
по двум прямолинейным параллельным про¬
водникам бесконечной длины и пренебрежи¬
мо малой площади кругового поперечного се¬
чения, расположенным на расстоянии 1 метр
один от другого в вакууме, вызывал бы силу
взаимодействия, равную 2*107 ньютона на
один метр длины
Термодина¬
мическая
(#абсолютная#)
температура
Т
О
кельвин
К
Один кельвин представляет собой 1/273,16
часть термодинамической температуры трой¬
ной точки воды
Количество веще¬
ства
п
N
моль
моль
Один моль представляет собой количество
вещества системы, содержащей столько же
структурных элементов, сколько атомов со¬
держится в 0,012 кг углерода-12*
Сила света
1у
кандела
кд
Кандела представляет собой силу света ис¬
точника, испускающего в данном направле¬
нии монохроматическое излучение частотой
540-1012 герц, энергетическая сила которого
в этом направлении составляет 1/683 ватт на
один стерадиан
(*) При выражении количества вещества в молях следует указывать, к чему они относятся - например, атомы, молекулы, ионы,
электроны, иные частицы или определенные группы таких объектов.
(**) Размерность приведена в соответствии с техническим регламентом «Единицы измерений, допущенные к применению на тер¬
ритории Республики Беларусь» (ТР 2007/003/ВУ).
Таблица 1.6.-2
Единицы СИ, используемые в Фармакопее, и их соответствие другим единицам
Величина
Единица
Преобразование иных единиц
в единицы СИ
Наименование
Символ,
приня¬
тый в
Фарма¬
копее
#Раз-
мер-
ность*
Наиме¬
нование
Символ
Выраже¬
ние в ос¬
новных
единицах
Выра¬
жение в
иных еди¬
ницах СИ
Волновое число
V
-
единица
на метр
1/м
м-1
Длина волны
Л
-
микро¬
метр
нанометр
мкм
нм
Ю*6 м
10-9м
Площадь
А, 8
-
метр ква¬
дратный
м2
м2
Объем
V
-
метр ку¬
бический
м3
м3
1 мл = 1 см3 = 10*6 м3
Частота
V
т1
герц
Гц
си
Плотность
Р
-
кило¬
грамм на
метр ку¬
бический
кг/м3
КГ-М'3
1 г/мл = 1 г/см3 = 103 кг-М'3
Скорость
V
-
метр в
секунду
м/с
М-С’1
Сила
Р
1.МТ2
ньютон
н
М‘КГС'2
1 дин = 1 г-см-с'2 = 10'5 Н
1 кгс = 9,80665 Н
Давление
Р
12МТ2
паскаль
Па
М'1-КГ-С‘2
Нм2
1 дин/см2 = 10-1 Па = 10'1 Н-м*2
1 атм = 101 325 Па = 101,325 кПа
1 бар = 105 Па = 0,1 МПа
1 мм рт. ст. = 133,322387 Па
1 торр = 133,322368 Па
1 фунт на квадратный дюйм (р$/) =
6,894757 кПа
30
Государственная фармакопея Республики Беларусь
Величина
Единица
Преобразование иных единиц
в единицы СИ
Наименование
Символ,
приня¬
тый в
Фарма¬
копее
#Раз-
мер-
ность*
Наиме¬
нование
Символ
Выраже¬
ние в ос¬
новных
единицах
Выра¬
жение в
иных еди¬
ницах СИ
Динамическая вяз¬
кость
п
-
паскаль-
секунда
Пас
М'1КРС'1
Нем2
1 пз = 10*1 Пас=10-1 Нем2
1 спз = 1 мПа-с
Кинематическая
вязкость
V
-
метр ква¬
дратный
в секунду
м2/с
М2С'1
Пасм3кг1
Н-м-с-кг1
1 Ст = 1 см2 с'1 = 10^ м2 С'1
Энергия
м
имт2
джоуль
Дж
М2КГС'2
Нм
1 эрг = 1 см2-гс2 = 1 дин-см = 10'7 Дж
1 кал = 4,1868 Дж
Мощность
Поток излучения
р
имт-3
ватт
Вт
М2*КГС'3
Н-м-с*1
Дж-с1
1 эрг/с = 1 дин см с'1 = 10'7 Вт =
10'7 Н-м-с1 = 10'7Дж с'1
Поглощенная доза
ионизирующего из¬
лучения
О
12Т2
грей
Гр
М2С'2
Дж-кг1
1 рад = 10"2 Гр
Электрический по¬
тенциал, электро¬
движущая сила
и
имтч*
вольт
в
м2кг-с*3А'1
ВтА-1
Электрическое со¬
противление
к
имтч2
ом
Ом
м2кгс‘3А'2
В А1
Количество элек¬
тричества
0
Т1
кулон
Кл
Ас
Радиоактивность
вещества
А
т1
бекке-
рель
Бк
с1
1 Ки = 37-109 Бк = 37-109 с-1
Концентрация (ко¬
личества веще¬
ства), молярная
концентрация
с
-
моль на
метр ку¬
бический
моль/м3
МОЛЬ’М'3
1 моль/л = 1 М = 1 моль/дм3 =
103 моль-м 3
Массовая концен¬
трация
Р
-
кило¬
грамм на
метр ку¬
бический
кг/м3
кг-м3
1 г/л = 1 г/дм3 = 1 кг-м'3
(*) Размерность приведена в соответствии с техническим регламентом «Единицы измерений, допущенные к применению на
территории Республики Беларусь» (ТР 2007/003/ВУ).
Некоторые важные и широко используемые единицы, не входящие в Международную систему СИ, приведе¬
ны в таблице 1.6.-3.
Таблица 1.6.-3
Единицы, используемые наряду с Международной системой единиц
Величина
Единица
Значение в единицах СИ
Наименование
Символ
Время
минута
мин
1 мин = 60 с
час
ч
1 ч = 60 мин = 3600 с
сутки
сут
1 сут = 24 ч = 86 400 с
Плоский угол
градус
о
1° = (тг/180) рад
Объем
литр
л
1 л = 1 дм3 = 10'3 м3
Масса
тонна
т
1 т = 103 кг
Частота вращения
оборот в минуту
об/мин
1 об/мин = (1/60) С'1
Множительные приставки для образования десятичных дольных и кратных единиц приведены
в таблице 1.6.-4.
Таблица 1.6.-4
Множители и приставки для образования десятичных кратных и дольных единиц
Множитель
Приставка
Обозначение
Множитель
Приставка
Обозначение
1018
экса
Э
10-1
деци
Д
1015
пета
П
ю-2
санти
с
1012
тера
Т
10'3
милли
м
109
гига
Г
Ю-е
микро
мк
10®
мега
м
10'9
нано
н
103
кило
к
10-12
пико
п
ю2
гекто
г
10'15
фемто
ф
101
дека
да
Ю'18
атто
а
1. Общие сведения
31
ПРИМЕЧАНИЯ
1. В Фармакопее используется температура по
Цельсию (символ {). Температура по Цельсию опреде¬
ляется согласно формуле:
1 = Т-Т
где:
Г0- 273,15 К.
Температура по Цельсию выражается в граду¬
сах Цельсия (символ °С). Один градус Цельсия равен
одному кельвину.
2. Практические выражения для концентраций
определены в Общих положениях.
3. Радиан представляет собой плоский угол, вы¬
резающий на окружности дугу, равную по длине ра¬
диусу.
4. Условия центрифугирования определяют¬
ся центробежным ускорением по отношению к уско¬
рению свободного падения (д), которое принимается
равным
д = 9,80665 м с'2.
5. Некоторые величины используются без раз¬
мерности, как, например, относительная плотность
(2.2.5), оптическая плотность (2.2.25), удельный по¬
казатель поглощения (2.2.25) и показатель преломле¬
ния (2.2.5).
6. Микрокатал определяется как энзиматическая
активность, которая при указанных условиях приво¬
дит к превращению (например, к гидролизу) 1 микро¬
моль субстрата за одну секунду.
#1.7. ОТБОР ПРОБ
ОСНОВНЫЕ ПОНЯТИЯ
Следующие понятия даны для применения в
рамках данного раздела.
Выборка — количество штучной продукции, ото¬
бранной из контролируемой серии (партии).
Гэтовое лекарственное средство (продукт) —
лекарственное средство (продукт), прошедшее все
стадии технологического процесса, включая марки¬
ровку, упаковку, лабораторный контроль.
Контейнер — изделие, которое содержит про¬
дукцию или предназначено для ее хранения и нахо¬
дится или может находиться в непосредственном кон¬
такте с ней. Укупорочное средство является частью
контейнера.
Контроль — процедура оценки соответствия пу¬
тем измерений, наблюдений, испытаний или калибро¬
вания соответствующих характеристик.
Контаминация — процесс загрязнения.
Нефасованный продукт (ангро, т Ьи1к) — про¬
дукт, прошедший все стадии технологического про¬
цесса, кроме упаковывания.
Отбор проб — действия по изъятию проб сырья,
материалов, полупродуктов, промежуточной и готовой
продукции для исследования их качества.
Объем выборки — число выборочных единиц в
выборке.
Объединенная проба — проба продукции, со¬
стоящая из нескольких точечных проб, отобранных из
контролируемой партии.
Образец продукции — единица конкретной про¬
дукции, используемая в качестве представителя этой
продукции при исследовании, контроле и оценке.
Промежуточная продукция — частично обрабо¬
танное сырье или продукция, которые должны пройти
последующие технологические операции прежде, чем
стать нерасфасованной продукцией.
Проба — количество нештучной продукции,
отобранной из контролируемой серии (партии) для
принятия решения, состоящее из нескольких точеч¬
ных проб.
Сырье (исходные материалы) — субстанции
для фармацевтического использования, части ле¬
карственных растений или продукты их переработ¬
ки, вспомогательные вещества собственного про¬
изводства или получаемые от внешних поставщи¬
ков, используемые в производстве лекарственных
средств, за исключением маркировочных и упако¬
вочных материалов.
Серия (партия) — количество продукции одно¬
го наименования, полученное в одном технологи¬
ческом цикле или в течение определенного интер¬
вала времени, в одних и тех же условиях и одно¬
временно представленное на контроль. Качество
серии (партии) должно быть удостоверено одним
документом.
Точечная проба — количество продукции, взя¬
тое за один раз из одного места серии (партии) од¬
номоментно.
Упаковка — средство или комплект средств,
обеспечивающие защиту продукции от поврежде¬
ний и потерь, от загрязнения окружающей среды, а
также процесс обращения продукции.
Упаковочная единица — упаковка, содержа¬
щая установленное количество продукции.
Чистая зона — зона, в которой окружающая
среда контролируется на наличие контаминирую-
щих частиц и микроорганизмов, построенная и экс¬
плуатируемая таким образом, чтобы уменьшить их
проникновение и образование контаминантов вну¬
три зоны.
ОБЩИЕ ПРАВИЛА
Если в частных статьях не указано иное, отбор
проб для испытаний должен проводиться в соответ¬
ствии с процедурой отбора.
Процедура отбора проб включает:
- план или схему отбора проб;
- место и время отбора проб;
-извлечение и подготовку проб продукции для
испытаний;
- объем и тип отбора проб;
-параметры окружающей среды при отборе и
подготовке проб для испытаний.
Все оборудование, используемое для отбора
проб, включая измерительное оборудование для
проведения испытаний, связанных с отбором проб,
должно удовлетворять требованиям нормативной до¬
кументации или процедуре отбора проб и работать в
соответствии с данной процедурой или инструкциями
(эксплутационной документацией) на оборудование.
Измерительное оборудование должно пройти в уста¬
новленном порядке поверку или аттестацию.
Персонал, выполняющий отбор проб, должен
владеть процедурой отбора. Документация по проце¬
дуре отбора проб должна находиться в местах отбора
проб и быть доступной для персонала.
В процессе проведения отбора проб необходи¬
мо учитывать факторы, которые должны контроли¬
роваться, чтобы обеспечить достоверность резуль¬
татов испытаний. К ним относятся: показатели, обе¬
спечивающие соответствие контролируемых пока-
32
Гэсударственная фармакопея Республики Беларусь
зателей качества и безопасности проб показателям
объекта испытаний и стабильность этих показате¬
лей в течение периода проведения испытаний; па¬
раметры окружающей среды и других воздействую¬
щих на пробы факторов.
Пробы, прошедшие отбор, должны соответ¬
ствующим образом идентифицироваться с исполь¬
зованием единой маркировки и оформляться ак¬
том отбора или другим документом, включающим
дату, время и место отбора, условия окружающей
среды при отборе, фамилию, имя и отчество лица,
проводившего отбор, и другую необходимую ин¬
формацию.
Если поставка сырья или готовой продукции
состоит из нескольких серий (партий), то каждую
серию (партию) необходимо рассматривать как от¬
дельную в отношении отбора проб и проведения
испытаний.
Во избежание ошибок при отборе проб не допу¬
скается отбор проб одновременно от двух и более
наименований, двух и более серий (партий) сырья
или готовой продукции.
Отбор проб для нерасфасованной продукции
(ангро, или т Ьи!к) должен осуществляться в сте¬
рильные контейнеры. Упаковка должна обеспечи¬
вать пригодность пробы для проведения испытаний.
К отбору от следующей серии (партии) посту¬
пившего сырья или готовой продукции можно при¬
ступать только после выполнения всей процедуры
отбора от предыдущей серии (партии).
Отбор проб следует производить только из не¬
поврежденных укупоренных и упакованных соглас¬
но нормативной документации упаковочных единиц.
До и после проведения испытаний пробы долж¬
ны храниться в отдельном помещении. Условия в
помещении должны обеспечивать сохранность
проб в течение всего срока хранения.
Перед отбором проб необходимо произвести
внешний осмотр упаковочной тары (ящиков, ко¬
робов, барабанов, бутылей и т.д.), определить ее
количество, целостность, наличие пломб, правиль¬
ность маркировки и оформление сопроводительной
документации, а также соответствие тары и упаков¬
ки требованиям спецификации.
Количество упаковочных единиц, отобранных
для отбора, рассчитывают по формуле:
0,а4п ,
где:
п — общее количество упаковочных единиц од¬
ной серии (партии).
Количество вскрытых упаковочных единиц долж¬
но быть не менее 3 и не более 30.
Отбор проб сырья необходимо производить в
отдельном помещении или в складском помещении
таким образом, чтобы предотвратить контамина¬
цию.
Для проведения испытаний лекарственных
средств на соответствие требованиям норматив¬
ной документации проводят многоступенчатый от¬
бор проб. При многоступенчатом отборе пробу об¬
разуют по ступеням и продукцию в каждой ступени
отбирают случайным образом в пропорциональных
количествах из единиц, отобранных в предыдущей
ступени. Число ступеней определяется видом упа¬
ковки.
I ступень: отбор единиц упаковочной тары (ящи¬
ков, коробок, мешков и др.)
II ступень: отбор упаковочных единиц, находя¬
щихся в упаковочной таре (пакетов, флаконов, банок,
бутылок, рулонов и др.)
III ступень: отбор продукции в первичной упаков¬
ке (ампул, флаконов, туб, контурных упаковок и др.)
При отборе проб необходимо принимать меры
предосторожности и выполнять требования безопас¬
ности с учетом токсичности, взрывчатости, огнеопас¬
ности, гигроскопичности и других свойств сырья, а
также меры, направленные на предохранение проб
(образцов) от повреждения и загрязнения во время
работы с ними, к их упаковке, транспортировке, скла¬
дированию и хранению с учетом требований и мето¬
дов последующих испытаний.
При отборе проб лекарственных средств списка
«А», наркотических и психотропных лекарственных
средств следует руководствоваться правилами, ин¬
струкциями и положениями, утвержденными компе¬
тентным уполномоченным органом, и частными ста¬
тьями на эти лекарственные средства.
При отборе проб запрещается принимать пищу,
пить, курить, а также хранить еду, средства для куре¬
ния в специальной одежде или месте отбора проб.
Персонал, занятый отбором проб, должен строго
соблюдать инструкции, регламентирующие состояние
здоровья и требования личной гигиены, носить специ¬
альную обувь.
ОТБОР ПРОБ ИЗ НЕФАСОВАННОЙ
ПРОДУКЦИИ (АНГРО, ИЛИ /Л/ ВШК)
Проба из нефасованной продукции должна пред¬
ставлять собой объединенные точечные пробы, взя¬
тые примерно в равных количествах.
Отбор точечных проб (объединенная проба) не¬
обходимо производить из верхнего, среднего и ниж¬
него слоев каждой упаковочной единицы, убедиться
в однородности сыпучих, вязких и гетерогенных пре¬
паратов.
При отборе проб сыпучих и вязких лекарственных
средств, фармацевтических субстанций и вспомога¬
тельных веществ для предотвращения перекрестной
контаминации точечные пробы необходимо отбирать
чистыми и стерильными пробоотборниками, изготов¬
ленными из материала, не реагирующего с данным
сырьем. Детали пробоотборников, контактирующие
с сырьем, должны быть обработаны специальными
антисептиками, разрешенными к применению компе¬
тентным уполномоченным органом.
В случае если перемешивание жидкости затруд¬
нено (большие емкости), точечные пробы отбирают
без перемешивания из разных слоев.
ОТБОР ПРОБ ГОТОВЫХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ (ГОТОВОЙ ПРОДУКЦИИ)
Отбор проб готовых лекарственных средств (про¬
дукции) должен производиться из ненарушенных за¬
водских упаковочных единиц.
Отбор проб готовых лекарственных средств для
инъекций и глазных лекарственных средств на от¬
сутствие в них механических включений (видимых
частиц) должен производиться в соответствии со ста¬
тьей 2.9.20. Загрязнение механическими включени¬
ями: видимые частицы, если нет других указаний и
разрешений компетентного уполномоченного органа.
2.2.29. Жидкостная хроматография
33
2. МЕТОДЫ АНАЛИЗА
2.2. ФИЗИЧЕСКИЕ И ФИЗИКО¬
ХИМИЧЕСКИЕ МЕТОДЫ
07/2016:20220
2.2.20. ПОТЕНЦИОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
При потенциометрическом титровании (объем¬
ный метод титрования с потенциометрическим опре¬
делением конечной точки титрования) конечную точку
титрования определяют путем измерения разницы
потенциалов между двумя электродами (обычно один
индикаторный электрод и один электрод сравнения
или комбинированный электрод), погруженными в ис¬
пытуемый раствор (#в случае когда ионы из электрода
сравнения мешают титрованию, электрод сравнения
соединяют с испытуемым раствором электролитиче¬
ским мостом#), как функцию количества прибавленно¬
го титранта.
Прибор. Используемый прибор включает мил¬
ливольтметр. Могут использоваться коммерчески до¬
ступные приборы для автоматического титрования,
которые должны эксплуатироваться в соответствии с
инструкциями производителя, с электродами, подхо¬
дящими для конкретного типа титрования.
В зависимости от природы определяемой суб¬
станции подбирают подходящий индикаторный элек¬
трод, который может быть стеклянным или металли¬
ческим (например, платиновым, золотым или сере¬
бряным).
#Индикаторный электрод выбирают так, чтобы
его потенциал закономерно изменялся при протека¬
нии химической реакции между титруемыми ионами
и ионами титранта (таблица #2.2.20.-1). Электродом
сравнения обычно служит каломельный или хлорид-
серебряный электрод/
Для кислотно-основного титрования обычно ис¬
пользуют комбинацию электродов стеклянный - сере¬
бряный - хлоридсеребряный.
Методика. Готовят испытуемый образец, как
указано в частной статье. Титрованный раствор при¬
бавляют подходящими аликвотами, уделяя особое
внимание скорости прибавления и приросту объема
прибавляемого титранта вблизи конечной точки ти¬
трования. Продолжают прибавлять титрованный раст¬
вор сверх предполагаемой конечной точки титрования
для возможности ее четкого определения.
Конечная точка титрования соответствует макси¬
мальному изменению потенциала на кривой титрова¬
ния (график зависимости потенциала от прибавлен¬
ного объема титрованного раствора) и выражается
соответствующим объемом прибавленного титрован¬
ного раствора. Записывая первую или вторую произ¬
водную кривой титрования можно облегчить опреде¬
ление конечной точки титрования.
При потенциометрическом титровании слабых
кислот или оснований с использованием неводных
растворителей перед растворением испытуемого ве¬
щества при необходимости проводят либо холостой
опыт, либо предварительно нейтрализуют смесь
растворителей. При невозможности использования
потенциометрического детектирования смесь раст¬
ворителей может быть предварительно нейтрали¬
зована путем титрования с использованием подхо¬
дящего индикатора. Далее представлено несколько
примеров:
Титрант
Индикатор
Хлорная кислота
Кристаллического
фиолетового раствор Р
Тетрабутиламмония
3 г/л раствор тимолового
гидроксид
синего Р в метаноле Р
Натрия гидроксида
спиртовой раствор
Тимолфталеина раствор Р
07/2016:20229
2.2.29. ЖИДКОСТНАЯ
ХРОМАТОГРАФИЯ
Жидкостная хроматография (ЖХ) представляет
собой метод разделения, основанный на различном
распределении образца между двумя несмешиваю-
щимися фазами, где подвижной фазой является жид¬
кость, которая проходит сквозь неподвижную фазу,
помещенную в колонку.
Жидкостная хроматография основана на меха¬
низмах адсорбции, распределения, ионного обмена,
разделения по размерам молекул (эксклюзии) или
стереохимического взаимодействия.
Таблица # 2.2.20.-1
Электродные системы для потенциометрического титрования
Тип аналитической реакции
Индикаторные электроды
Электроды сравнения
Применение
Кислотоно-основный
Стеклянный
Хлоридсеребряный, кало¬
мельный
Титрование кислот, оснований
и солей
Осаждения
Серебряный, сульфидсере-
бряный
Хлоридсеребряный, кало¬
мельный, стеклянный
Титрование галогенидов, ро-
данидов, цианидов, сульфи¬
дов
Комплексонометрический
Ртутный, ионоселективный
Хлоридсеребряный, кало¬
мельный, стеклянный
Титрование катионов, образу¬
ющих прочные комплексонаты
Окислительно-восстанови¬
тельный
Платиновый
Хлоридсеребряный, кало¬
мельный, стеклянный
Титрование восстановите¬
лей различными окислите¬
лями, например броматом,
дихроматом, перманганатом,
йодом, церием (IV). Титрова¬
ние окислителей различными
восстановителями, например
арсенитом, тиосульфатом, ни¬
тритом
34
Гэсударственная фармакопея Республики Беларусь
Если нет других указаний, вся представленная
ниже информация применима как к стандартной жид¬
костной хроматографии, так и к жидкостной хромато¬
графии, в которой используются колонки с уменьшен¬
ным размером частиц (например, менее 2 мкм).
В последнем случае требуется оборудование, ха¬
рактеризующееся следующим: возможность исполь¬
зования высоких давлений (обычно до 100 МПа, то
есть около 15 000 рз/), уменьшенное внеколоночное
уширение пиков, улучшенные параметры градиент¬
ного смешивания и увеличенная скорость получения
данных системой детектирования.
ПРИБОР
Прибор обычно состоит из системы подачи под¬
вижной фазы, блока ввода пробы (инжектора), хро¬
матографической колонки (может использоваться
устройство контроля температуры колонки), детек¬
тора и регистрирующего устройства (интегрирующе¬
го устройства или самописца). Подвижная фаза из
одной или нескольких емкостей протекает обычно с
постоянной скоростью через колонку, а затем через
детектор.
СИСТЕМА ПОДАЧИ ПОДВИЖНОЙ ФАЗЫ
Система подачи подвижной фазы (насосная уста¬
новка) необходима для поддержания контролируемой
скорости подачи подвижной фазы. Колебания давления
должны быть минимизированы, например, путем про¬
пускания подвижной фазы, находящейся под давлени¬
ем, через устройство, сглаживающее пульсацию дав¬
ления (демпфер). Проводящая система капилляров и
соединительные узлы должны выдерживать давление,
которое развивается при работе насосной установки.
Насосные установки, использующиеся в жидкостной
хроматографии, могут оснащаться дегазаторами.
Системы подачи подвижной фазы, контролиру¬
емые микропроцессором, должны обеспечивать точ¬
ную подачу подвижной фазы постоянного (изократи-
ческое элюирование) или переменного (градиентное
элюирование) состава в соответствии с определенной
программой. В случае градиентного элюирования не¬
обходима насосная установка, подающая растворите¬
ли из нескольких емкостей, а смешивание раствори¬
телей должно проводиться на стороне либо низкого,
либо высокого давления насоса (насосов).
БЛОК ВВОДА ПРОБЫ (ИНЖЕКТОР)
Раствор испытуемого образца вводится в поток
подвижной фазы, поступающей в колонку, при по¬
мощи блока ввода, функционирующего при высоком
давлении. Для ввода пробы используют петлевые
дозаторы фиксированного объема или устройства с
регулируемым объемом, которые могут управляться
вручную либо посредством автосамплера. Частичное
заполнение петлевого дозатора, осуществляющееся
вручную, приводит к снижению точности вводимого
объема пробы.
НЕПОДВИЖНАЯ ФАЗА
В жидкостной хроматографии используется боль¬
шое количество типов неподвижных фаз:
- силикагель, оксид алюминия или пористый гра¬
фит, используемый в нормально-фазовой хромато¬
графии, в которой разделение основано на разнице
в адсорбции и/или массовом распределении (распре¬
делительная хроматография);
- смолы или полимеры, модифицированные кис¬
лотными или основными группами, использующиеся в
ионообменной хроматографии, в которой разделение
основано на конкурирующем взаимодействии'между
разделяемыми ионёми и ионами подвижной фазы;
- пористый силикагель или полимеры, использу¬
ющиеся в эксклюзионной хроматографии, в которой
разделение основано на разнице в размерах молекул,
соответствующих стерической эксклюзии;
-большое количество химически модифициро¬
ванных носителей, изготовленных из полимеров, си¬
ликагеля или пористого графита, использующихся в
нормально-фазовой (абсорбционная хроматография)
и обращенно-фазовой хроматографии, в которых раз¬
деление основано на разделении молекул между под¬
вижной и неподвижной фазой;
-специальные неподвижные фазы, химически
модифицированные, например, производными цел¬
люлозы или амилозы, белками и пептидами, цикло-
декстринами и т.д., использующиеся для разделения
энантиомеров (хроматография на хиральных непод¬
вижных фазах).
Большая часть разделений основана на механиз¬
ме распределения между химически модифицирован¬
ным силикагелем, использующимся в качестве непод¬
вижной фазы, и полярными растворителями, исполь¬
зующимися в качестве подвижной фазы. Поверхность
носителя, например силанольные группы силикагеля,
реагирует с различными силановыми реагентами с
образованием ковалентно связанных силильных про¬
изводных, покрывающих различное число активных
групп на поверхности носителя. Природа модифици¬
рованной фазы является важной характеристикой,
определяющей разделяющие свойства хроматогра¬
фической системы.
Ниже представлены наиболее часто используе-
мые модифицированные фазы:
Октильная - $1-[СН2]7-СН3
С8
Октадецильная
- 31-[СН2]17-СНЭ
С„
Фенильная
- ЗЧСНД.-СД
с6н!
Цианопропильная
- 3|-[СН2]3-СЫ
см
Аминопропильная
- 8|-[СН2]3-1\1Н2
N4,
Диольная
- 3|-[СН2]3-0-СН(0Н)-СН2-0Н
Если производитель не дает другой информации,
находящаяся в колонках обращенно-фазовая непод¬
вижная фаза на основе силикагеля считается стабиль¬
ной в подвижных фазах при значениях рН в диапазоне
от 2,0 до 8,0. Неподвижная фаза, представляющая со¬
бой пористый графит или частицы полимерного мате¬
риала, такого как сополимер стирол-дивинилбензола,
стабильна в более широком диапазоне значений рН.
В некоторых случаях для анализа применяют
нормально-фазовую хроматографию, используя в
качестве неподвижной фазы немодифицированный
силикагель, пористый графит или силикагель, хими¬
чески модифицированный полярными группами (на¬
пример, цианопропильными или диольными).
Размер частиц для большинства неподвижных
фаз, использующихся для аналитического разделе¬
ния, составляет от 2 мкм до 10 мкм. Частицы могут
иметь сферическую или неправильную форму, раз¬
личную пористость и удельную площадь поверхно¬
сти. Эти параметры определяют хроматографические
свойства конкретных неподвижных фаз. В обращен¬
но-фазовой хроматографии дополнительными опре¬
деляющими факторами являются природа неподвиж¬
ной фазы, степень связывания, выраженная в содер¬
жании углерода, а также эндкепирование подвижной
2.2.32. Потеря в массе при высушивании
35
фазы (т.е. силилирование части остаточных сила-
нольных групп). Остаточные силанольные группы мо¬
гут являться причиной размытия тыла пика, особенно
для веществ основного характера.
Кроме пористых частиц могут быть использованы
поверхностно-пористые или монолитные материалы.
Если нет других указаний в частной статье, в
аналитической хроматографии используют колонки,
изготовленные из нержавеющей стали, с различными
длиной и внутренним диаметром (0). Колонки с вну¬
тренним диаметром менее 2 мм часто относят к ми¬
кроколонкам. Температура подвижной фазы и колон¬
ки в течение анализа должна сохраняться постоян¬
ной. Большинство анализов проводится при комнат¬
ной температуре, но для обеспечения оптимальной
эффективности может потребоваться использование
других температур.
ПОДВИЖНАЯ ФАЗА
В нормально-фазовой хроматографии обычно
используют малополярные органические растворите¬
ли. Для получения воспроизводимых результатов нор¬
мально-фазовой хроматографии необходимо строго
контролировать содержание воды в растворителях,
использующихся в подвижной фазе. В обращенно-
фазовой ЖХ используют водные подвижные фазы,
содержащие либо не содержащие органические рас¬
творители.
Компоненты подвижной фазы обычно фильтруют
для удаления частиц с размером более 0,45 мкм (или
0,2 мкм, в случае если неподвижная фаза состоит
из частиц с размером менее 2 мкм, а также если ис¬
пользуются специальные детекторы, например детек¬
тор светорассеяния). При приготовлении многоком¬
понентных подвижных фаз смешивают отмеренные
объемы (или массы) индивидуальных компонентов.
Кроме этого, растворители могут подаваться индиви¬
дуальными насосами и с помощью дозирующих кла¬
панов смешиваться в необходимых пропорциях. Для
предотвращения образования пузырьков газа и попа¬
дания их на детектор растворители обычно дегазиру¬
ют при помощи барботирования гелием, обработкой
ультразвуком и/или с использованием мембранно-ва¬
куумных модулей, работающих в режиме оп-Нпе.
Растворители для приготовления подвижных
фаз обычно не содержат стабилизаторов, а при ис¬
пользовании ультрафиолетовых детекторов должны
быть прозрачными при длине волны детектирова¬
ния. Растворители и другие компоненты подвижной
фазы должны быть соответствующей степени чисто¬
ты. Корректировку рН при необходимости проводят
для водного компонента подвижной фазы, а не для
смеси. Для предотвращения кристаллизации солей
после проведения хроматографирования с использо¬
ванием буферных или солевых растворов проводят
промывку системы с использованием смеси воды и
небольшого количества органической части подвиж¬
ной фазы (5 % (об/об)).
Подвижные фазы могут содержать другие ком¬
поненты, например контр-ионы для ион-парной хро¬
матографии или хиральные модификаторы для хро¬
матографии, использующей ахиральные неподвиж¬
ные фазы.
ДЕТЕКТОРЫ
Наиболее часто используемыми детекторами
являются спектрофотометры, работающие в уль-
трафиолетовой/видимой ((УУ/\//$) областях, а также
диодно-матричные детекторы. Кроме этого могут
использоваться специальные детекторы: флуорес¬
центные спектрофотометры, дифференциальные
рефрактометры (/?/), электрохимические детекто¬
ры (ЕСО), детекторы заряженных аэрозолей (ОАО),
масс-спектрометры (М3), детекторы радиоактивности
и другие.
МЕТОДИКА
Колонку уравновешивают при указанном составе
и скорости подвижной фазы, а также при комнатной
температуре или температуре, указанной в частной
статье, до получения стабильной базовой линии. Го-
товят испытуемый раствор(ы) и раствор(ы) сравне¬
ния, как указано в частной статье. Растворы не долж¬
ны содержать твердых частиц.
Критерии для проверки пригодности хроматогра¬
фической системы описаны в статье 2.2.46. Хромато¬
графические методы разделения, где, кроме этого,
приводятся интервалы, в пределах которых могут из¬
меняться различные параметры хроматографических
испытаний без принципиального изменения методики
для того, чтобы удовлетворять критериям пригодно¬
сти хроматографической системы.
07/2016:20232
2.2.32. ПОТЕРЯ В МАССЕ
ПРИ ВЫСУШИВАНИИ
Определение потери в массе при высушивании
проводят одним из приведенных способов и выража¬
ют в процентах (м/м).
Методика. Указанное в частной статье количе¬
ство испытуемого образца помещают во взвешенный
бюкс, предварительно высушенный в условиях, опи¬
санных для испытуемого вещества. Вещество сушат
до постоянной массы или в течение времени, указан¬
ного в частной статье, одним из следующих ниже спо¬
собов. Если температурный интервал не указан, то
высушивание проводят при указанной температуре
±2 °С.
a) «в эксикаторе»: высушивание проводят над
фосфора (V) оксидом Р при атмосферном давлении
и комнатной температуре;
b) «в вакууме»: высушивание проводят над
фосфора (V) оксидом Р при давлении от 1,5 кПа до
2,5 кПа и комнатной температуре;
c) «в вакууме в пределах указанного температур¬
ного интервала»: высушивание проводят над фосфо¬
ра (V) оксидом Р при давлении от 1,5 кПа до 2,5 кПа и
температуре, указанной в частной статье;
д) «в пределах указанного температурного ин¬
тервала»: высушивание проводят в сушильном шка¬
фу в температурном интервале, указанном в частной
статье; проверка пригодности прибора может быть
проведена с использованием подходящего серти¬
фицированного стандартного материала (например,
ФСО амоксициллина тригидрата для проверки при¬
годности);
е) «в глубоком вакууме»: высушивание проводят
над фосфора (V) оксидом Р при давлении не более
0,1 кПа и температуре, указанной в частной статье.
Если указаны иные условия, используемая мето¬
дика полностью описывается в частной статье.
36
Государственная фармакопея Республики Беларусь
07/2016:20261
2.2.61. ОПРЕДЕЛЕНИЕ
ХАРАКТЕРИСТИК
КРИСТАЛЛИЧЕСКИХ ТВЕРДЫХ
ВЕЩЕСТВ С ПОМОЩЬЮ
МИКРОКАЛОРИМЕТРИИ
И КАЛОРИМЕТРИИ
РАСТВОРЕНИЯ
В рамках данной главы в качестве твердых
веществ рассматриваются кристаллические ве¬
щества, частично кристаллические вещества и
аморфные вещества.
ВВЕДЕНИЕ. ОБЩЕЕ ПРЕДСТАВЛЕНИЕ
О КРИСТАЛЛИЧНОСТИ
Полностью упорядоченная кристаллическая ре¬
шетка, в которой каждая молекула занимает предпо¬
ложительно известное положение, является идеаль¬
ной, но редко встречающейся, если вообще когда-ли¬
бо бывает возможной. Другим крайним состоянием
является аморфное, находясь в котором твердое ве¬
щество содержит максимально возможную плотность
отклонений (дефектов различных размерностей), что
обусловливает полную потерю дальнего порядка и
присутствие только ближнего, представленного бли¬
жайшим окружением. Реальные кристаллы находятся
между этими двумя крайними состояниями. Положе¬
ние кристалла на шкале, ограниченной этими состоя¬
ниями, называется кристалличностью.
Все реальные кристаллы, даже находясь в чи¬
стом состоянии, обладают некоторыми искажениями
кристаллической решетки или дефектами, которые
увеличивают энергию (энтальпию при условии по¬
стоянного атмосферного давления) и неупорядочен¬
ность (выраженную через энтропию) кристаллической
решетки. Кристалл, имеющий относительно низкую
плотность дефектов, называют высоко кристалличе¬
ским и обладающим высокой кристалличностью. И
наоборот, частицу с относительно высокой плотно¬
стью дефектов называют частично аморфной и обла¬
дающей низкой кристалличностью. В идеальных ус¬
ловиях полностью аморфной частице соответствует
нулевая кристалличность. Аморфные частицы могут
содержать в некоторой степени упорядоченные обла¬
сти, которые могут играть роль ядра кристаллизации;
о подобных так называемых аморфных частицах гово¬
рят, что они обладают низкой, но ограниченной степе¬
нью кристалличности.
На протяжении разработки и последующего про¬
изводства лекарственного средства большое значе¬
ние имеет возможность обнаружить и рассчитать ко¬
личество аморфного вещества в высоко кристалличе¬
ской субстанции.
В реальности порошок вероятно содержит части¬
цы с различной степенью кристалличности, а также
может содержать и частицы с различными размерами
и формой. Чем ниже кристалличность твердого веще¬
ства, тем больше его энтальпия и энтропия. Увеличе¬
ние энтальпии никогда полностью не компенсируется
увеличением энтропии; поэтому свободная энергия
Гиббса, отражающая равновесие между ними, фак¬
тически возрастает. Следовательно, чем меньше
кристалличность вещества (порошка) и больше его
аморфный характер, тем больше его кажущаяся ха¬
рактеристическая растворимость и скорость раство¬
рения, но тем ниже его термодинамическая стабиль¬
ность. В связи с большой значимостью этих свойств
кристалличность также является важным свойством и
требует измерения соответствующим методом.
В следующем разделе кристалличность или со¬
держание аморфных частей порошка измеряются
калориметрическими методами, такими как микро¬
калориметрия или калориметрия растворения, хотя
могут использоваться и другие методы (например,
описанные в общей статье 2.9.33. Определение па¬
раметров кристаллических и частично кристалли¬
ческих твердых веществ при помощи дифракции
рентгеновского излучения на порошке).
Способность вещества кристаллизоваться с
образованием более одного типа кристаллической
решетки называют полиморфизмом. Если вода или
растворитель включены в кристаллическую решетку,
то кристаллы называются гидратами или сольватами.
Они обычно проявляют разные физические свойства,
что связано с разной упаковкой кристалла и/или мо¬
лекулярной структурой и энергией решетки. Для про¬
стоты проведения калориметрических измерений при
определении степени кристалличности, рассматри¬
ваемом в данном случае, делается допущение, что в
исследуемом веществе присутствует только одна кри¬
сталлическая форма. Теория и экспериментальный
метод могут распространяться на полиморфные си¬
стемы при тщательном изучении различий в энталь¬
пии полиморфных веществ.
МЕТОД 1. МИКРОКАЛОРИМЕТРИЯ
(ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ АМОРФНОЙ
ЧАСТИ)
Большинство химических, физических и биоло¬
гических процессов связано с теплообменом. Микро¬
калориметрия является высокочувствительным мето¬
дом мониторинга и расчета экзотермических (выделя¬
ющих теплоту) и эндотермических (поглощающих те¬
плоту) превращений, сопровождающих эти процессы.
Метод позволяет определять скорость и глубину про¬
текания химических реакций, фазовых превращений
или изменений структуры.
С применением микрокалориметрии могут быть
исследованы тепловые процессы, в результате кото¬
рых образуется только фракция напряжения в микро¬
ваттах. Это означает, что различия в температуре
менее 10_6 К должны быть обнаруживаемы. Микро¬
калориметрия обычно использует принцип теплового
потока (тепловое рассеивание), когда теплота, выде¬
ляемая (или поглощаемая) в определенном термо¬
стойком сосуде, перемещается из (в) него с целью по¬
вторного установления теплового равновесия с окру¬
жающей средой. Исключительная термическая ста¬
бильность по отношению к окружающей среде должна
достигаться с помощью теплоотвода или окружающих
электронно-регулируемых устройств.
Тепловая энергия, идущая от активного образца
в реакционном сосуде, обычно направляется через
элементы Пельтье; они выступают в качестве термоэ¬
лектрических генераторов, используя эффект Зеебека
(ЗееЬеск). Тепловая энергия конвертируется в сигнал
напряжения, пропорциональный тепловому потоку.
Результаты представляют обычно в виде количе¬
ственной характеристики тепловой энергии, которая
образуется в единицу времени (Ватт), как функции
времени.
2.2.61. Определение характеристик кристаллических твердых веществ
37
ПРИБОР
Микрокалориметры обычно представлены в виде
парных систем с измерительным сосудом и сосудом
сравнения. Сосуды обычно сделаны из стекла или
нержавеющей стали. Для определенных целей могут
применяться специально сконструированные сосуды,
позволяющие добавлять газ, жидкость или твердое
вещество.
КАЛИБРОВКА
Микрокалориметр калибруется по тепловому по¬
току (энергия в единицу времени) с использованием
откалиброванных внешнего или внутреннего электри¬
ческих тепловых источников или соответствующей
стандартной реакции.
ЧУВСТВИТЕЛЬНОСТЬ
В основе оценки чувствительности микрокало-
риметрического метода может быть положено ис¬
пользование подходящего стандартного образца,
проанализированного соответствующим методом, в
совокупности с определением флуктуационного шума
измерительного прибора.
МЕТОД
Соответствующее количество испытуемого об¬
разца взвешивают в подходящую колбу. Закрывают
колбу тщательно, чтобы избежать испарения раство¬
рителей, и помещают в держатель образца. При не¬
обходимости уравновешивают колбу при температуре
измерения перед помещением ее в положение для
проведения измерения.
Начинают испытание и фиксируют значения ве¬
личины теплового потока, откладывая значения вре¬
мени на оси абсцисс, а теплового потока — на оси ор¬
динат (устанавливают направление экзотермического
и эндотермического теплового потока).
ОБНАРУЖЕНИЕ И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ АМОРФНОЙ
ЧАСТИ В ПОРОШКАХ
Аморфное состояние является метастабильным
по отношению к кристаллическому состоянию, поэтому
может происходить перекристаллизация. Измерение
теплоты перекристаллизации позволяет определять
содержание аморфной части, используя площадь пика
перекристаллизации. Рассчитать содержание аморф¬
ной части образца можно с помощью отношения вы¬
ходных данных микрокалориметра для образца к вы¬
ходным данным для аморфного стандарта. Диапазон
значений содержания аморфной части, охватываемый
данным методом, зависит от конкретного испытуемого
образца; в благоприятных случаях могут быть достиг¬
нуты пределы обнаружения ниже 1 %.
Перекристаллизация может быть инициирована,
если подвергнуть образец воздействию более высо¬
кой относительной влажности или атмосферы, содер¬
жащей органический пар. Образец обычно помещает¬
ся в ампулу, которая также содержит небольшую про¬
бирку с водным насыщенным раствором соли, орга¬
ническим растворителем или смесью растворителей.
Теплота перекристаллизации обычно измеряет¬
ся с использованием образца фиксированной массы,
помещенного в стакан или стальной сосуд. Пробирка,
содержащая насыщенный раствор соли или органиче¬
ский растворитель, выбирается таким образом, чтобы
она была достаточно большой для полного насыще¬
ния атмосферы над образцом. Масса образца и при¬
рода атмосферного пара над образцом выбираются
таким образом, чтобы в процессе перекристаллиза¬
ции наблюдался отчетливый пик, четко отделенный от
исходных тепловых эффектов, вызванных введением
образца.
Условия, при которых происходит переход
аморфной фазы в термодинамически более стабиль¬
ное кристаллическое состояние, оказывают значи¬
тельное влияние на время перекристаллизации. В
частности, физические смеси из чистых аморфных и
чистых кристаллических веществ будут отличаться от
частично кристаллического вещества в проявлении
своих свойств. Такие влияния следует учитывать при
разработке метода.
Типичный отклик для перекристаллизации пре¬
имущественно аморфного вещества показан на ри¬
сунке 2.2.61.-1. Первая часть кривой отображает не¬
сколько конкурирующих процессов, происходящих
одновременно, таких как поглощение водяного пара
аморфными частями порошка и выделение водяного
пара из пробирки. После данного начального отклика
происходит большой экзотермический отклик, вызван¬
ный перекристаллизацией аморфного вещества. При¬
сутствуют также происходящее, но невидимое удале¬
ние избытка воды из перекристаллизованных частей
и ее конденсация. Таким образом, площадь под этим
экзотермическим откликом перекристаллизации про¬
порциональна теплоте перекристаллизации.
Рисунок 2.2.61 .-1. Типичные для микрокалориметри-
ческого измерения выходные данные по мощности
(мкВт) в виде функции времени (часы) : пики разруше¬
ния аморфной структуры (I) и пик кристаллизации (II)
для преимущественно аморфной лактозы при темпе¬
ратуре 25 °С и относительной влажности 75 %
МЕТОД 2. КАЛОРИМЕТРИЯ РАСТВОРЕНИЯ
(ОПРЕДЕЛЕНИЕ КРИСТАЛЛИЧНОСТИ)
Калориметрия растворения устанавливает спосо¬
бы определения энтальпии растворения вещества (то
есть теплоты растворения при постоянном атмосфер¬
ном давлении). Энтальпия растворения определяется
как разность энтальпии вещества, растворенного до
определенной концентрации, и энтальпии исходного
вещества. Для растворения должен использоваться та¬
кой растворитель, чтобы твердое вещество определен¬
ной массы растворилось в течение периода времени,
соответствующего времени отклика калориметра, как
это описано ниже. Энтальпия растворения пропорцио¬
нальна количеству растворяемого твердого вещества.
38
Гэсударственная фармакопея Республики Беларусь
Это количество может приниматься за 1 моль для мо¬
лярной энтальпии или 1 г для удельной энтальпии.
Если вещество имеет достаточную чистоту (опреде¬
ленную с требуемой степенью точности) и известна его
молекулярная масса, то предпочтительной является
молярная энтальпия, в ином случае должна исполь¬
зоваться удельная энтальпия. Энтальпия растворения
слабо зависит от температуры, которая обычно имеет
значение 25,0 °С, и конечной концентрации растворен¬
ного вещества.
Обычно предпочтительнее выражать кристал¬
личность вещества Рс на шкале процентного со¬
держания. Для данного метода необходимо два
стандарта сравнения, а именно: высоко кристалли¬
ческий образец, при условии, что он имеет кристал¬
личность 100% и измеренную энтальпию раство¬
рения АН*, и аморфный образец, допуская, что он
имеет кристалличность 0 % и измеренную энталь¬
пию растворения АН*. Кристалличность твердого
вещества (Рс) в процентах может быть рассчитана с
использованием значения указанных величин и из¬
меренной энтальпии растворения изучаемого твер¬
дого вещества (АН*), по формуле:
рс(%)=юо{ан1 -ан:)/(ан°с-ан:).
Очевидно, что кристалличность, представленная
на шкале процентного содержания, зависит от трех
измеренных величин, а энтальпии растворения могут
быть заменены другими подходящими физическими
величинами, зависящими от кристалличности. Значе¬
ние кристалличности образца в процентах зависит не
только от природы и метода приготовления двух стан¬
дартов сравнения, но также от выбора измеряемой
физической величины. Энтальпия растворения изме¬
ряется с помощью изопериболического жидкостного
калориметра (постоянная оболочка, то есть рубашка)
или изотермического жидкостного калориметра (по¬
стоянная температура). Обычно проводится не менее
3 измерений для каждого образца. Затем рассчитыва¬
ется среднее значение этих величин. Конкретные тре¬
бования будут зависеть от возможностей оборудова¬
ния и необходимой степени точности.
ИЗОПЕРИБОЛИЧЕСКАЯ ЖИДКОСТНАЯ
КАЛОРИМЕТРИЯ
В изопериболическом жидкостном калориметре
теплообмен в течение процесса растворения вы¬
зывает соответствующее изменение температуры в
системе растворитель - растворенное вещество (то
есть раствор). Данное изменение температуры изме¬
ряется температурным датчиком, включенным в элек¬
трическую цепь, который записывает электрический
сигнал, соответствующий изменению температуры.
Обычно такое изменение температуры, представляе¬
мое в электронной форме, измеряется в точно опре¬
деленные интервалы времени для получения данных
температура - время, которые накапливаются, анали¬
зируются с помощью компьютера и затем отобража¬
ются графически. Контрольный опыт без прибавления
раствора твердого вещества к растворителю обычно
показывает заметное изменение наклона графика за¬
висимости температура - время.
Для изопериболических жидкостных калориме¬
тров отклик достаточно быстрый, но должны быть
сделаны поправки на любые потери теплоты на баню
или на поступление теплоты от нее. Следовательно,
если процесс растворения относительно быстрый,
то изопериболические жидкостные калориметры
имеют преимущества перед изотермическими жид¬
костными калориметрами. Для измерения энтальпии
растворения с использованием изопериболических
жидкостных калориметров важным является выбор
растворителя. Природа, масса растворителя и масса
образца должны быть такими, чтобы общий теплооб¬
мен, соответствующий полному растворению твердо¬
го вещества, подошел к завершению в течение 5 мин
при интенсивном перемешивании с постоянной скоро¬
стью вращения, находящейся в пределах интервала
(400—600) об/мин.
Эффективная теплоемкость калориметрической
ячейки и ее содержимого определяется для каждого
калориметрического измерения. Это определение со¬
провождается электрическим нагревом содержимого
калориметрической ячейки. Эффективная теплоем¬
кость определяется в соответствии с одной из двух
последовательностей действий: выполнение первого
определения после разбивания ампулы или выпол¬
нение первого определения перед разбиванием, а
второго — после разбивания и затем усреднение двух
результатов. Точность и надежность электрического
нагрева устанавливаются на основании точности и на¬
дежности вышеупомянутых химических калибровок.
ИЗОТЕРМИЧЕСКАЯ ЖИДКОСТНАЯ
КАЛОРИМЕТРИЯ
В изотермическом (постоянная температура)
жидкостном калориметре изменение теплоты в тече¬
ние процесса растворения компенсируется равным,
но противоположным изменением энергии, таким,
чтобы температура системы растворитель - раство¬
ренное вещество (то есть раствора) оставалась пре¬
имущественно постоянной. Измеряется равное, но
противоположное изменение энергии, которое при
изменении знака на обратный обусловливает энталь¬
пию растворения. Для изотермических калориметров
отклик относительно медленный, но компенсацион¬
ный процесс уменьшает эффекты потери теплоты на
баню или поступления теплоты от нее. Следователь¬
но изотермические жидкостные калориметры имеют
преимущества перед изопериболическими жидкост¬
ными калориметрами в случае, когда процесс раство¬
рения относительно медленный.
КАЛИБРОВКА ЖИДКОСТНЫХ
КАЛОРИМЕТРОВ
Для обеспечения точности калориметра химиче¬
ские калибровки должны выполняться на постоянной
основе. Для эндотермического процесса растворения
калибровка калориметра выполняется посредством
измерения теплоты, поглощенной в течение процес¬
са растворения калия хлорида в дистиллированной
воде при температуре 298,15 К (25,0 °С). Установ¬
ленное изменение энтальпии в данном эндотермиче¬
ском процессе равно 235,5 Дж/г (17,56 кДж/моль). Для
экзотермического процесса растворения проверка
калориметра осуществляется измерением теплоты,
выделенной в течение растворения 5 г/л тромета-
мина ((трис(гидроксиметил)аминометана, ТНАМ) в
0,1 моль/л водном растворе хлористоводородной кис¬
лоты при температуре 298,15 К (25,0 °С). Установлен¬
ная теплота для вышеупомянутого процесса равна
-246,0 Дж/г (-29,80 кДж/моль).
РАБОТА С ОБРАЗЦАМИ
Химическая и физическая стабильность твер¬
дых веществ может уменьшаться с уменьшением
кристалличности. В частности, твердые вещества с
2.2.64. Спектрометрия ядерного магнитного резонанса для идентификации пептидов
39
низкой кристалличностью, особенно аморфные твер¬
дые вещества, стремятся сорбировать пары воды из
атмосферы, приводя к кристаллизации и соответству¬
ющему увеличению кристалличности. В связи с этим
перед проведением определения кристалличности
безводные образцы должны храниться при нулевой
влажности или при значениях влажности ниже кри¬
тического предела, в закрытых камерах, содержащих
поглотитель влаги и, предпочтительно, индикатор
эффективности. Если должны проводиться исследо¬
вания кристалличность - влажность, то образец дол¬
жен храниться в закрытой камере, содержащей насы¬
щенный раствор соли для обеспечения определенной
влажности.
07/2016:20264
2.2.64. СПЕКТРОМЕТРИЯ ЯДЕРНОГО
МАГНИТНОГО РЕЗОНАНСА ДЛЯ
ИДЕНТИФИКАЦИИ ПЕПТИДОВ
Данная общая статья при идентификации пепти¬
дов должна использоваться наряду с общей статьей
2.2.33. Спектрометрия ядерного магнитного резо¬
нанса. Описываемый метод является качественным и
основывается на сравнении спектра ядерного магнит¬
ного резонанса (ЯМР) испытуемого образца со спек¬
тром стандартного образца, полученным в идентич¬
ных условиях.
Данная общая статья большей частью приме¬
нима к использованию спектрометрии протонного
магнитного резонанса (ПМР или 1Н ЯМР) для под¬
тверждения подлинности (идентичности) продуктов
небольших пептидов (до около 15 аминокислот). Так¬
же данная статья применима при использовании спек¬
трометрии 13С ЯМР с некоторыми модификациями.
Область применения данной общей статьи ограничи¬
вается одномерной спектрометрией ЯМР.
ОСНОВНЫЕ ПРИНЦИПЫ
Оборудование. Если нет других указаний, ис¬
пользуют прибор с напряженностью поля, обеспечи¬
вающей рабочую частоту для протонного ЯМР не ме¬
нее 300 МГц.
Условия получения спектров и их оптимиза¬
ция. После помещения в магнит образец выдержива¬
ют до температуры уравновешивания, особенно если
испытание проводится при температуре, значительно
отличающейся от комнатной, зачастую значимым ви¬
зуальным средством для контроля данного процесса
является мониторинг блокировки сигнала.
Ширина спектра должна охватывать полный
спектр пептида с пустой спектральной областью с
обеих сторон. Обычно подходящей является ширина
спектра 12 ррт или 16 ррт.
Для улучшения разделения характеристических
пиков могут быть оптимизированы следующие усло¬
вия: главным образом температура и/или величина
рН, а также концентрации буфера и пептида. Контроль
температуры образца является рекомендацией, а не
требованием: если температуру не контролируют, не¬
обходимо валидационными испытаниями установить
влияние небольших температурных изменений на
внешний вид спектра.
Количество записанных точек на спектре должно
быть достаточным для адекватного определения пиков.
Подавление растворителя не рекомендуется, так
как это может повлиять на интенсивности пиков близ¬
ких к резонансу растворителя, что потребует отдель¬
ной валидации при сравнении спектров.
Стандарты химического сдвига. Для образцов
в водных растворах подходящими стандартами явля¬
ются натрия 2,2-диметил-2-силапентан-5-сульфонат
(055), натрия 3-(триметилсилил)пропионат (75Р) или
его дейтерированный аналог (Т5Р-с!4), а химический
сдвиг метильных групп обычно устанавливается на
О ррт. В качестве вторичного стандарта может быть
использован стандартный материал, добавленный в
малых количествах (подходящим является от 10 до
100 ррт) к дейтерированной воде, применяемой для
растворения образца, либо постоянно присутству¬
ющий легко различимый источник внутреннего ре¬
зонанса (такой как ацетат ион). В таком случае для
определения химического сдвига вторичного стандар¬
та используется достоверный спектр, полученный в
тех же условиях.
Количество образца. Обычно используют не¬
сколько миллиграммов образца. Если количество об¬
разца изменяется, то необходимо валидационными
испытаниями установить влияние таких изменений на
внешний вид спектра.
Приготовление образца. Испытуемый образец
и стандартный образец должны быть соизмеримы по
концентрации, величине рН и составу буфера. Обычно
образцы подвергают лиофильной сушке из растворов,
затем высушенные образцы растворяют в дейтериро¬
ванной воде или в буферном растворе в дейтериро¬
ванной воде. Способ однократной или многократной
лиофильной сушки растворов, полученных с дейтери¬
рованной водой («дейтериевый обмен»), может быть
полезным, так как при этом уменьшается интенсив¬
ность сильных сигналов растворителя; летучие приме¬
си, такие как этанол, тоже будут удалены. Использова¬
ние буферных растворов для окончательного приготов¬
ления образца может снизить агрегацию и улучшить
спектральную воспроизводимость путем уменьшения
вариаций значений рН от серии к серии. Некоторые
датчики неустойчивы к высоким концентрациям солей,
при этом нормально переносят ионную силу раствора
до 200 мм натрия хлорида. Высокие концентрации со¬
лей ведут к увеличению длительности 90° импульса.
ПРОВЕРКА ИДЕНТИЧНОСТИ
Определение ключевых спектральных фак¬
торов. Использование качественного подхода не
подразумевает строгого подхода к спектральным па¬
раметрам (например, может быть использована вы¬
сокая частота следования импульсов, так как полная
релаксация не требуется). Использование импуль¬
сов короткой длительности (например, 30° импульс)
и высокой частоты следования импульсов не окажет
значительных негативных эффектов на спектр и по¬
зволит быстрее получить приемлемые соотношения
сигнал/шум. Изменения в длительности импульса и
времени получения спектра (в широких пределах) не
окажут воздействия на возможность сравнения спек¬
тров. Число записанных сканирований должно давать
подходящие соотношения сигнал/шум для резонан¬
сов низкой интенсивности, таким образом, рекомен¬
дуется, чтобы минимальное соотношение сигнал/шум
составляло 50:1.
Идентификация характеристических резонан¬
сов. Допускается использовать для сравнения либо
40
Государственная фармакопея Республики Беларусь
целый спектр, либо его часть. Сравнение спектров ха¬
рактерных образцов позволяет выделить отличитель¬
ные участки спектра; сравнение спектров может быть
ограничено использованием таких участков. Важно
отличать резонансы от примесей, таких как остаточ¬
ные растворители, которые главным образом не свя¬
заны с качеством продукта и которые могут отличать¬
ся по интенсивности от серии к серии.
Сравнение спектров. См. положения общей
статьи 2.2.33.
07/2016:20265
2.2.65. ВОЛЬТАМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
Конечная точка титрования при вольтаметри-
ческом титровании представляет собой величину
изменения напряжения, измеренного между двумя
электродами (либо одного индикаторного электрода
и одного электрода сравнения, либо двумя индика¬
торными электродами), погруженными в испытуемый
раствор при поддержании постоянного тока, как функ¬
цию количества прибавленного титранта.
Прибор. Прибор состоит из регулируемого ис¬
точника тока и вольтметра; система детектирования
обычно включает индикаторный электрод (например,
платиновый электрод, электрод с вращающимися
дисками или угольный электрод) и второй электрод
(например, платиновый электрод, электрод с враща¬
ющимися дисками или угольный электрод).
Методика. Настраивают ток на индикаторном
электроде, как указано в частной статье, и строят гра¬
фик зависимости начального напряжения и значений,
полученных при титровании, от количества прибав¬
ленного титранта. Прибавляют титрант не менее чем
тремя последовательными количествами, равными в
сумме около 80 % от теоретического значения, соот¬
ветствующего предполагаемой точке эквивалентно¬
сти. Эти три значения должны лежать на одной пря¬
мой линии. Продолжают прибавление титранта сверх
предполагаемой точки эквивалентности не менее чем
тремя последовательными порциями. Полученные
значения должны лежать на другой прямой линии.
Точка пересечения этих линий будет соответствовать
конечной точке титрования.
При использовании системы вольтаметрического
титрования с двумя индикаторными электродами за¬
писывается кривая титрования, на которой определя¬
ется конечная точка титрования.
07/2016:20266
2.2.66. ОБНАРУЖЕНИЕ И ИЗМЕРЕНИЕ
РАДИОАКТИВНОСТИ
ВВЕДЕНИЕ
В контексте Фармакопеи понятие «радиоактив¬
ность» применяется для описания явления радиоак¬
тивного распада и выражения физической величины
данного явления. В частных статьях на радиоактив¬
ные фармацевтические препараты обнаружение и из¬
мерение радиоактивности проводятся с различными
целями: контроля свойств, идентификации, опреде¬
ления радионуклидной и радиохимической чистоты, а
также определения радиоактивности вещества (коли¬
чественное определение).
Исходя из вышеуказанного измерение может
быть качественным, количественным или и тем, и
другим в зависимости от того, направлено оно на
идентификацию радионуклида, или определение его
активности (скорости распада) или имеет одновре¬
менно обе цели.
В соответствии с радионуклидным составом ра¬
диоактивные источники могут испускать различные
типы эмиссий, такие как альфа-частицы, электроны,
позитроны, гамма- и рентгеновское излучение.
Каждый радионуклид испускает характерные
эмиссии с определенными значениями энергий и от¬
носительных интенсивностей. Подобные излучения
могут быть обнаружены без дальнейшего определе¬
ния их характеристик в ионизационной камере в ре¬
зультате проявления ионизирующих свойств; при их
обнаружении и анализе с использованием спектро¬
метра получают спектр значений энергии. Детальный
спектральный анализ обычно применяют для иденти¬
фикации радионуклидов, присутствующих в образце.
Спектрометрия может также применяться для количе¬
ственного определения радиоактивности источников,
состоящих из одного радионуклида или смеси радио¬
нуклидов или присутствующих радионуклидов по от¬
дельности.
Измерение радиоактивности обычно осущест¬
вляется счетом числа обнаруженных явлений распада
(эмиссий). Следовательно, геометрические параме¬
тры образца на протяжении измерения радиоактивно¬
сти и время определения оказывают сильное влияние
на результат. В большинстве случаев геометрия изме¬
рения должна соответствовать калиброванной геоме¬
трии, а время определения должно быть достаточно
длительным для получения необходимых статистиче¬
ских данных по счету.
Измерение радиоактивности может быть осу¬
ществлено автономно (например, используя иониза¬
ционную камеру или спектрометр) или в комбинации
с методом разделения (например, радиохроматогра¬
фия) для расчета относительных вкладов различных
видов радиоактивных химических источников, кото¬
рые могут присутствовать в смеси.
ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
Если известна схема распада радионуклида,
может проводиться прямое определение радиоактив¬
ности испытуемого образца в беккерелях (Бк), но на
практике для получения точных результатов требует¬
ся внесение большого количества поправок. В связи с
этим существует возможность проводить измерение с
помощью первичного стандартного источника или ис¬
пользуя измерительные приборы, такие как иониза¬
ционная камера или спектрометр, калиброванные с
использованием подходящих стандартов для конкрет¬
ных радионуклидов.
Спектрометр используется при измерении радио¬
активности радионуклидов в смеси, идентифицируя
каждый радионуклид по его эмиссиям и характерным
для них энергиям.
Все измерения радиоактивности должны коррек¬
тироваться, учитывая потери на мертвое время и вы¬
читая фоновый сигнал в связи с излучением в окружа¬
ющую среду и ложные сигналы, создаваемые самим
оборудованием.
2.2.66. Обнаружение и измерение радиоактивности
41
Радиоактивность лекарственного средства опре¬
деляется при заданных параметрах. Если период
полураспада радионуклида менее 70 дней, также
указывается время. Заключение о радиоактивном со¬
держании должно быть сделано со ссылкой на кон¬
кретный период времени. Радиоактивность при других
значениях времени может быть рассчитана из экспо¬
ненциального уравнения распада или из таблиц.
Обычно для правильного измерения радиоактив¬
ности необходимо уделять внимание некоторым или
всем следующим позициям.
Потери на мертвое время. В связи с ограничен¬
ным временем разрешения (мертвое время) детек¬
тора и соединенного с ним электронного оборудова¬
ния может быть необходимым внесение поправок на
потери при наложении. Время разрешения счетчи¬
ка представляет собой минимальный временной ин¬
тервал, необходимый счетчику для разрешения двух
отдельно регистрируемых импульсов. Эпизодиче¬
ские явления излучения в более короткие интерва¬
лы времени могут не обнаруживаться или обнаружи¬
ваться как отдельное явление с суммарной энергией.
Эти потери иногда называются потерями на мертвое
время. Для подсчитывающей системы с фиксирован¬
ным мертвым временем т после каждого счета досто¬
верную скорость счета в с-1 рассчитывают по следую¬
щей формуле:
Ц
где:
— наблюдаемая скорость счета в секунду;
т— мертвое время, в секундах.
Некоторые приборы вносят данную поправку ав¬
томатически. Внесение поправок на потери, вызван¬
ные наложением, должно быть сделано перед по¬
правками на фоновое излучение.
Поправки на распад, происходящий на протя¬
жении измерения. Если период времени для отдель¬
ного измерения (*т) не является настолько коротким,
что им можно пренебречь, по сравнению с периодом
полураспада радионуклида Тш, то необходимо при¬
нимать во внимание распад, происходящий на про¬
тяжении этого измерения. Например, 5 % суммарных
потерь при счете в связи с распадом в течение счетно¬
го периода представляет собой 15 % от периода полу¬
распада радионуклида.
После корректировки показания прибора (скоро¬
сти счета, ионизационного тока и т.д.) с учетом фоно¬
вых сигналов и при необходимости потерь, вызванных
электронными эффектами, откорректированное пока¬
зание прибора на начало отдельного измерения рас¬
считывается по следующей формуле:
_я(яу_,
1 - (е~Мт)
где:
Я — показание прибора перед внесением по¬
правки на распад, но после поправки на фоновый
сигнал и т. д.;
А — константа распада радионуклида (1п 2/Т1/2);
е — основание натурального логарифма;
1т — продолжительность измерения.
Статистика измерения радиоактивности. Раз¬
личия в результатах определений радиоактивности
проистекают главным образом из хаотичной природы
ядерных превращений. С помощью счета для любого
ограниченного периода времени можно оценить только
достоверную скорость ядерных превращений. Для ком¬
пенсации различий в числе превращений за период
времени должно регистрироваться достаточное коли¬
чество импульсов. В случае измерения радиоактив¬
ности стандартное отклонение зарегистрированных
импульсов является квадратным корнем числа этих
импульсов. Таким образом, не менее 10 000 импульсов
необходимо для получения относительного стандарт¬
ного отклонения, не превышающего 1 %.
Линейность. Линейность измерительного прибо¬
ра представляет собой диапазон значений радиоак¬
тивности для конкретного радионуклида, в пределах
которого его эффективность остается постоянной.
Линейный диапазон значений совокупного измере¬
ния радиоактивности может определяться при повтор¬
ном счете для радиоактивного образца в условиях фик¬
сированной геометрии, так как он распадается начиная
с уровня значений активности, превышающего линей¬
ный диапазон. После внесения поправок на фоновый
сигнал строится график зависимости натурального ло¬
гарифма значений скорости счета от времени, прошед¬
шего после первого измерения (рисунок 2.2.66-1).
1 1 — ■" I 1 1
О 20 40 60 во 100 120
Прошедшее время, ч
Рисунок 2.2.66.-1. Гоафик зависимости измерен-
ной и экстраполированной скорости счета (на¬
туральный логарифм счета в секунду (1п срз))
при использовании источника, содержащего
технеций-99м, от времени начиная с уровня зна¬
чений радиоактивности, превышающих линейный
диапазон измерительного оборудования
Линейный регрессионный анализ центральной
линейной части совокупности данных приводит к на¬
клону кривой, который является константой распада А,
имеющей характерное значение для каждого радио¬
нуклида:
1п срз = -М + с,
где с — натуральный логарифм скорости счета
при ( = 0 в полной мере линейного измерительного
прибора.
Итоговое уравнение регрессии используется для
расчета теоретической скорости счета при каждом
значении времени, при котором были зарегистрирова¬
ны текущие показания прибора. Линейный диапазон
измерительного оборудования является превышен¬
ным в случае, если отклонение измеренной скорости
счета от теоретической неприемлемо высокое.
С другой стороны, может быть произведен ряд
разбавлений раствора радиоактивного вещества с
известным содержанием радиоактивности. Затем от¬
меряют равные объемы каждого разбавленного раст¬
вора с использованием типовых установок геометрии
42
Государственная фармакопея Республики Беларусь
и счетчика. Отношение скорости счета для каждо¬
го образца (после поправки на фоновые сигналы и
распад) к рассчитанной радиоактивности соответ¬
ствующего образца в Бк представляет собой эффек¬
тивность счета. Диапазон, в пределах которого это
отношение имеет постоянное значение, является диа¬
пазоном измерительного оборудования, подходящим
для данного радионуклида.
Предел обнаружения и предел количественного
определения для оборудования и методик, использу¬
емых для измерения радиоактивности, должны уста¬
навливаться перед их повседневным использованием.
Предел обнаружения. Предел обнаружения
(ПО, НтН о!6е1есНоп (/.00)) отдельной методики пред¬
ставляет собой наименьшее содержание радиоактив¬
ности в образце, которое может быть обнаружено,
но не обязательно определено количественно в виде
точного значения. На практике для этого необходимо
определить фоновый сигнал и его стандартное откло¬
нение. ПО обычно принимается равным 3-кратному
стандартному отклонению фонового сигнала.
Предел количественного определения. Предел
количественного определения (ПКО, НтН оI
диапННсаНоп (/.00)) отдельной методики представ¬
ляет собой наименьшее содержание радиоактив¬
ности в образце, которое может быть определено
количественно с соответствующей точностью и пра¬
вильностью. ПКО применяется, в частности, для опре¬
деления примесей и/или продуктов разложения. На
практике ПКО обычно принимается равным 10-крат¬
ному стандартному отклонению фонового сигнала.
ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
С ИСПОЛЬЗОВАНИЕМ ИОНИЗАЦИОННОЙ
КАМЕРЫ
Прибор. В практической радиофармации для из¬
мерения радиоактивности чаще всего применяются
ионизационные камеры (включая калибраторы дозы).
Прибор в большинстве случаев может измерять радио¬
активность от нескольких десятков кБк до сотен ГБк
и обычно содержит плотно закрытую ионизационную
камеру с колодцем и встроенное компьютерное обо¬
рудование для преобразования сигнала детектора в
единицы радиоактивности.
Камера заполняется газом, на который накла¬
дывается электрическое поле. После ионизации газа
под действием излучения, испускаемого источни¬
ком, результирующий ионизационный ток измеряет¬
ся и соотносится с радиоактивностью в ионизацион¬
ной камере. Прикладываемое напряжение, энергия,
интенсивность излучения, природа и давление газа
оказывают влияние на ионизационный ток. Установки
прибора (фактор калибровки) могут быть отрегулиро¬
ваны для создания прямой зависимости между иони¬
зацией в результате излучения конкретного радиону¬
клида и значением радиоактивности, полученным для
каждого условия геометрии измерения.
Ионизационная камера измеряет только резуль¬
тирующий ток, возникающий в итоге общей ионизации
внутри камеры, и не может различать эмиссии раз¬
личных радионуклидов.
Для точного измерения радиоактивности от¬
дельного радионуклида в измерение должны быть
внесены поправки на вклады в ионизационный ток
примесями радионуклидов, присутствующими в ле¬
карственном средстве.
Измеряемые уровни содержания активности за¬
висят от насыщения, диапазона усилителя и дизай¬
на камеры. Диапазон линейности ионизационной
камеры устанавливается, как описано выше в подраз¬
деле «Линейность».
Ионизационная камера должна быть экранирова¬
на для минимизации фоновых сигналов до приемле¬
мого уровня.
Метод. Образец размещают внутри колодца
ионизационной камеры в заданном положении, ис¬
пользуя держатель. После помещения образца в
ионизационную камеру и достижения стабильного
отклика регистрируется значение активности. К полу¬
чению наиболее правильных результатов приводит
измерение радиоактивности образца при условиях
геометрии, абсолютно идентичных калибровочному
источнику. При необходимости доводят измеряемое
лекарственное средство до объема, равного объему
калибровочного источника.
Калибровка. Ионизационная камера калибру¬
ется с учетом формы, размеров, материала контей¬
нера, объема и состава раствора, положения внутри
камеры измеряемого радионуклида. Значения преде¬
лов неопределенности при калибровке можно найти
в государственных или международных нормативах.
Ионизационную камеру калибруют один раз в год
по геометрии, используя радионуклидные источники,
соответствующие государственным или международ¬
ным стандартам, в подходящих контейнерах (виале,
флаконе, шприце). Устанавливают и применяют до¬
полнительные корректирующие факторы для учета
различия в конфигурациях измеряемых радионукли¬
дов. Выполняют контроль линейности отклика прибо¬
ра на протяжении всего диапазона значений энергии
и активности, для измерения которых применяется
данное оборудование.
Для подтверждения калиброванного состояния
ионизационной камеры для каждой установки пара¬
метра и перед каждым применением (как минимум
один раз в день применения) выполняют проверку ее
стабильности, используя стандартные радионуклид¬
ные источники с длительным периодом полураспада.
В день применения ионизационной камеры для под¬
тверждения ее нахождения в калиброванном состо¬
янии должна выполняться проверка на соответствие
источнику сравнения, например цезию-137.
ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
С ИСПОЛЬЗОВАНИЕМ ТВЕРДОТЕЛЬНЫХ
ДЕТЕКТОРОВ
К твердотельным детекторам относятся пласт¬
массовые флуоресцентные и кристаллические сцин¬
тилляторы, а также полупроводники. Кроме исполь¬
зования в спектрометрии, как описано в разделе
«Спектрометрия», твердотельные детекторы могут
применяться для измерения радиоактивности. В
частности, в связи с высокой чувствительностью
пластмассовые и кристаллические сцинтилляцион-
ные детекторы применяются при счете нижних уров¬
ней содержания радиоактивности. Потери на мерт¬
вое время должны быть тщательно изучены для всех
типов детекторов. Полупроводниковые детекторы
применяются при необходимости более высокой диф¬
ференциации энергии, например в случае измерения
радиоактивности смесей радионуклидов или если
предполагаемые примеси радионуклидов испускают
эмиссии с близкими значениями энергии.
Прибор. Прибор состоит из экранированного де¬
тектора, содержащего присоединенный к усилителю и
электронно-вычислительным устройствам пластмас-
2.2.66. Обнаружение и измерение радиоактивности
43
совый или кристаллический сцинтиллятор, соединен¬
ный с фотоумножителем, или полупроводник. Систе¬
ма может иметь регулируемое окошко для значения
энергии, используемое для выбора необходимого для
счета диапазона в спектре энергий радионуклида, ре¬
гулируемого при необходимости оператором.
Измерительные приборы имеют различные пара¬
метры разрешения по энергии и эффективности обна¬
ружения, зависящие от типа детектора, его объемных
и геометрических характеристик. Чем меньше эффек¬
тивность, тем большее время счета необходимо.
Измеряемые образцы должны быть помещены
перед детектором или внутрь колодца детектора с ко¬
лодцем. Камеры для измерения могут быть закрыты
устройством для экранирования детектора, а введе¬
ние образцов по отдельности может осуществляться
с использованием крышки или других систем, позволя¬
ющих размещать образцы в определенном положении
для обеспечения правильной геометрии измерения.
Сцинтилляционный детектор может применять¬
ся для измерения радиоактивности в динамическом
режиме, например в случае когда элюат из жидкост¬
ного хроматографа направляется над детектором или
через него, как описано в подразделе «Обнаружение
и измерение радиоактивности в сочетании с методом
разделения».
Метод. Необходимо удостовериться, что радиоак¬
тивность образца обеспечивает нахождение значения
скорости счета в диапазоне линейности оборудования.
Когда все заслонки находятся на месте или крышка
колодца возвращена в исходное положение и время
счета выбрано таким образом, чтобы получить число
импульсов, достаточное для проведения необходимой
статистической обработки, начинается измерение.
Калибровка. Калибровка детектора должна осу¬
ществляться измерением его эффективности с ис¬
пользованием содержащего анализируемый радиону¬
клид источник, соответствующий государственным или
международным стандартам. При калибровке, исходя
из эффективности, используют такие источники, как
цезий-137, кобальт-60, барий-133, и другие, охватыва¬
ющие необходимый диапазон значений энергии.
ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
С ИСПОЛЬЗОВАНИЕМ ЖИДКОСТНЫХ
СЦИНТИЛЛЯЦИОННЫХ ДЕТЕКТОРОВ
Жидкостной сцинтиллятор обычно используется
для образцов, испускающих бета-частицы, но также
может быть использован и для образцов, испуска¬
ющих альфа-частицы. Принципы обнаружения ра¬
диоактивности с использованием жидкостных сцин-
тилляционных детекторов описаны в подразделе
« Бета-спектрометрия».
Калибровка. Для того чтобы учесть потери эф¬
фективности счета в связи с поглощением, жидкост¬
ной сцинтилляционный счетчик может использовать
внешний источник, обычно барий-133 или европий-152,
который подносится близко к виале с образцом для
высвобождения электронов Комптона. Форма резуль¬
тирующего спектра анализируется автоматически для
вычисления параметра, отвечающего за поглощение.
Затем этот параметр может иметь отношение к источ¬
никам измерения эффективности счета с известной ак¬
тивностью при определенном содержании поглотителя.
Полученная кривая поглощения позволяет определить
активность неизвестного образца при известных значе¬
ниях скорости счета и параметра поглощения.
ОПРЕДЕЛЕНИЕ ПЕРИОДА ПОЛУРАСПАДА
Период полураспада является характеристи¬
кой радионуклида, которая может применяться для
его идентификации. Период полураспада рассчиты¬
вается при измерении изменения радиоактивности
испытуемого образца как функции времени. Изме¬
рения выполняют в диапазоне линейности калибро¬
ванного измерительного прибора.
Прибор. Период полураспада может изме¬
ряться с использованием любого типа детектора
для количественного определения радиоактивно¬
сти при условии, что он используется в пределах
диапазона линейности на протяжении всего диапа¬
зона значений активности, характерного для дан¬
ного измерения, и геометрия не изменяется в тече¬
ние измерения.
Определение приблизительных периодов полу¬
распада для лекарственных средств, содержащих
радионуклид с коротким периодом полураспада, и в
случае указания в частной статье является частью
идентификации.
Метод
Период полураспада. Испытуемое лекарствен¬
ное средство (образец) используется в том виде, в
котором есть, или разводится или высушивается в
капсуле после соответствующего разведения. Радио¬
активный образец подготавливается таким образом,
чтобы избежать потери вещества при работе с ним.
В случае если радиоактивный образец представляет
собой жидкость (раствор), то он содержится в закры¬
той колбе или плотно закрытой пробирке. Если обра¬
зец представляет собой остаток после высушивания
в капсуле, то его помещают в упаковку, состоящую
из слоя клейкой ацетилцеллюлозы или каких-либо
других материалов.
Радиоактивность образца должна быть достаточ¬
но высокой, чтобы обеспечить проведение измере¬
ний на протяжении периода, соответствующего трем
предполагаемым периодам полураспада, но для каж¬
дого измерения она должна быть в пределах диапа¬
зона линейности оборудования. При необходимости
применяется внесение поправок на потери на мерт¬
вое время.
Измерение одного и того же источника проводит¬
ся в идентичных условиях геометрии и с интервалами,
которые обычно составляют не менее половины пред¬
полагаемого периода полураспада. Каждое значение
вносится в таблицу напротив временного интервала,
начиная с первоначального измерения. Во избежание
влияния распада в течение измерения время счета
является одинаковым для всех измерений.
Строят график, откладывая на оси абсцисс зна¬
чения времени, а на оси ординат — логарифм соот¬
ветствующих показаний прибора (например, скорость
счета). Период полураспада рассчитывается начиная
от наклона наиболее линейного участка кривой изме¬
ренных значений относительно значений времени, со¬
ответствующих каждому измерению.
Приблизительный период полураспада. Для его
определения делаются не менее трех измерений на
протяжении не менее ЛА предполагаемого периода по¬
лураспада.
Испытуемый образец и используемый измери¬
тельный прибор должны соответствовать вышеизло¬
женным требованиям. Обработка данных осущест¬
вляется описанным выше способом.
6*. Зак. 1060.
44
Гэсударственная фармакопея Республики Беларусь
СПЕКТРОМЕТРИЯ
Радионуклиды могут быть идентифицирова¬
ны по их спектру эмиссии. Для получения спектра
каждого типа эмиссии (например, альфа-частицы,
бета-частицы и электроны, гамма- и рентгеновское
излучение) требуется определенное оборудование.
Для обеспечения надлежащей работы спектроме¬
тры должны быть откалиброваны; следующие раз¬
делы описывают различное измерительное обору¬
дование и детально излагают основные методики
для надежного измерения.
ГАММА-СПЕКТРОМЕТРИЯ
Основные принципы. В гамма-спектроме¬
трии с использованием сцинтилляционного детек¬
тора абсорбция гамма- и рентгеновского излучения
приводит к образованию света, который преобразу¬
ется с помощью фотоумножителя в электрический
импульс. В гамма-спектрометрии с использованием
полупроводникового детектора абсорбция гамма- и
рентгеновского излучения приводит к незамедли¬
тельному образованию электрического импульса. В
обоих случаях амплитуда импульса пропорциональ¬
на энергии абсорбированного излучения. Наибо¬
лее распространенными детекторами для гамма- и
рентгеновской спектрометрии являются сцинтилля-
ционные счетчики на основе №1, активированного
таллием (Ма1(Т1)), и полупроводниковые детекторы
на основе высокочистого германия (НРСВе).
Спектр гамма-излучения может быть образо¬
ван при получении и анализе достаточного числа
импульсов.
Прибор. Гамма-спектрометр обычно содер¬
жит экранированную измерительную камеру, в кото¬
рую помещается образец, детектор, электрическую
цепь и многоканальный анализатор.
Экранирование камеры может уменьшать фо¬
новый сигнал до уровня, позволяющего осущест¬
влять регистрацию правильного спектра гамма-из¬
лучения.
Измерительная камера имеет подвижную
крышку или выдвижной ящик, позволяющие разме¬
стить образец. Может применяться держатель об¬
разца для обеспечения воспроизводимости геоме¬
трии между измерениями.
Продолжительность измерения зависит от ра¬
диоактивности конкретного радионуклида, и для
достижения необходимой статистики счета может
потребоваться длительный период получения ре¬
зультата. Должны быть тщательно рассмотрены
потери на мертвое время для данного типа детек¬
тора.
Чувствительность детектора на основе Ыа1(Т1)
выше, чем у детектора на основе германия того же
размера. Обычно пики на спектре значений энер¬
гии идентифицируются с неопределенностью в за¬
висимости от значений ширины пика и полови¬
ны его высоты (РИ/У-М, Л/// №1сЛЬ о( Ою реак а1 Из
ЬаК-тах/тит Ье'\дЫ). Разрешение по энергии сцин¬
тилляционного твердотельного детектора намно¬
го хуже, чем для полупроводникового детектора, и,
следовательно, пики, полученные при использова¬
нии полупроводникового детектора, намного уже,
чем в случае применения сцинтилляционного де¬
тектора. На рисунке 2.2.66.-2 показано сравнение
спектров, полученных с использованием одного и
того же источника с двумя типами детекторов.
О 100 200 300 400 500 600 700 800 900 1000
Энергия, кэВ
Рисунок 2.2.66.-2. Сравнительные амплитудные
спектры импульсов, полученные с использова¬
нием сцинтиллятора на основе натрия йодида,
активированного таллием (верхняя кривая), и
полупроводникового детектора на основе высо¬
кочистого германия (нижняя кривая). В качестве
источника использовано гамма- и рентгенов¬
ское излучение, полученное в результате распа¬
да йода-131
Различные характеристики детекторов на основе
№1(Т1) и НРСВе могут послужить ограничением их при¬
менения в некоторых спектрометрических анализах.
Для идентификации радионуклида (радиону¬
клидов) лекарственного средства (образца) и опре¬
деления радионуклидной чистоты оценка риска про¬
цесса получения радионуклида должна включать в
себя оценку возможного присутствия других радио¬
нуклидов со значениями энергии фотона в диапазо¬
не (±10 %), характерном также и для радионуклидов,
присутствующих в лекарственном средстве (образце).
В случае присутствия радионуклидных приме¬
сей, которые испускают гамма- и рентгеновское излу¬
чение с энергией в диапазоне значений, также харак¬
терном и для фотонов, испускаемых радионуклидом
лекарственного средства (образца), для идентифика¬
ции пика достаточной является измеренная энергия
пика, находящаяся в пределах максимального интер¬
вала значений ±2 кэВ или ±2 % (тот, который больше)
по отношению к номинальной энергии пика, как указа¬
но в общей статье 5.7. Таблица физических характе¬
ристик радионуклидов, упоминаемых в Фармакопее.
В случае если не предполагается присутствие
таких примесей, для идентификации пика приемле¬
мым является максимальный интервал ±10кэВ или
±6 % (тот, который больше) по отношению к номи¬
нальной энергии пика.
Метод. Необходимо обеспечить, чтобы скорость
счета для об р азид уменьшалась анутрн диапазо¬
на линейности оборудования. Для жидких образцов
этого можно достичь с помощью соответствующего
разведения; для твердых образцов — увеличением
расстояния между источником и детектором или при¬
менением разжижающего вещества. Испытуемое ле¬
карственное средство (образец) вносят в камеру из¬
мерительного оборудования и снимают спектр после
закрытия заслонки.
Необходимо обеспечить, чтобы контейнер, при¬
меняемый для количественных измерений, был иден¬
тичен по форме, размерам, объему и материалу кон¬
тейнеру с калибровочным стандартом.
Необходимо обеспечить идентичность контейне¬
ра по составу раствора и положению в измерительной
камере контейнеру с калибровочным стандартом для
количественного определения.
2 2 66. Обнаружение и измерение радиоактивности
45
Идентификация радионуклида. Калибруют
спектрометр по энергии. Определение соответ¬
ствия энергии пиков, обнаруженной для образца,
энергиям, указанным в частной статье, является
достоверным испытанием подлинности.
Радионуклидная чистота. Калибруют спек¬
трометр по эффективности и энергии. Опреде¬
ляют ПКО (1-00) и разрешение измерительного
оборудования. Полученные значения должны соот¬
ветствовать предельным содержаниям определяе¬
мых радионуклидов. Снимают спектр лекарствен¬
ного средства (образца).
Идентифицируют радионуклиды, присутствую¬
щие в испытуемом лекарственном средстве (образ¬
це), и определяют их радиоактивность, используя
общую статью 5.7. Таблица физических характе¬
ристик радионуклидов, упоминаемых в Фармако¬
пее. В связи с тем что содержание радионуклидных
примесей в пересчете на процентное содержа¬
ние общей радиоактивности может возрастать или
уменьшаться со временем, измеренная активность
каждой примеси должна пересчитываться на ак¬
тивность в течение срока годности лекарственного
средства. Активности всех радионуклидных приме¬
сей необходимо суммировать (принимая во внима¬
ние предел количественного определения) для по¬
лучения общей радиоактивности лекарственного
средства (образца).
Образец размещается близко к детектору
или внутри детектора с колодцем. Все результа¬
ты в пределах диапазона заданных значений энер¬
гии собираются и выводятся на дисплей измерите¬
ля скорости в виде числа импульсов в секунду или
накапливаются на протяжении заданного периода
времени. В случае достаточного различия значе¬
ний энергии фотонов, испускаемых радионуклидом
(радионуклидами), может использоваться детектор
на основе натрия йодида при установлении его вы¬
сокой чувствительности. Тем не менее, если необ¬
ходимо различить эмиссии с похожими энергиями,
следует применять детектор на основе НРОе или
другой полупроводниковый детектор.
Калибровка. Калибровка по энергии осущест¬
вляется с использованием пиков известных источ¬
ников, соответствующих государственным или меж¬
дународным стандартам, а именно: кобальта-57,
цезия-137, кобальта-60 и других, охватывающих не¬
обходимый диапазон энергий. Параллельно может
быть проведена калибровка по эффективности с
целью получения не только спектра значений энер¬
гии, но в дальнейшем также и значений активности
образца и радионуклидных примесей. Калибровка
по эффективности может выполняться с примене¬
нием радионуклидного источника, соответствующе¬
го государственным или международным стандар¬
там, с пиками энергии, охватывающей необходимый
диапазон, или смешанного радионуклидного стан¬
дарта, соответствующего государственным или
международным стандартам, со значениями энер¬
гии гамма-излучения, охватывающими необходи¬
мый диапазон.
Для получения кривой эффективности отклик
детектора как функции энергии должен измерять¬
ся с использованием геометрии каждого отдельного
образца/детектора. В связи с этим возможно про¬
водить измерение с помощью первичного стандарт¬
ного источника. Первичные стандарты могут быть
неподходящими для радионуклидов с коротким пе¬
риодом полураспада, например некоторых эмит¬
теров позитронов. При измерении образец должен
преимущественно находиться в контейнере и зани¬
мать определенную позицию по отношению к детек¬
тору. В этом случае геометрия образца/детектора
определяется положением образца относительно
детектора и характеристиками контейнера и образ¬
ца, например формой, объемом и плотностью. На
рисунке 2.2.66.-3 показана типичная кривая эффек¬
тивности для детектора на основе НРОе, получен¬
ная для контейнера цилиндрической формы, поме¬
щенного наверху детектора.
Энергия, кэВ
Рисунок 2.2.66.-3. Типичная кривая эффективно¬
сти для детектора на основе НРСе, полученная
при применении специализированного контейне¬
ра, помещенного наверху детектора
БЕТА-СПЕКТРОМЕТРИЯ
В случае бета-эмиттера необходимо, чтобы бета-
спектрометр определял распределение испускаемых
бета-частиц по энергиям. Он аналогичен гамма-спек¬
трометру, но в нем часто используется жидкостной
сцинтиллятор для преобразования энергии бета-ча¬
стиц в свет, который можно обнаружить и затем про¬
анализировать. Бета-спектрометрия заключается пре¬
имущественно в растворении или суспендировании
образца в жидкой сцинтилляционной смеси в прозрач¬
ных или полупрозрачных (стеклянных или пластико¬
вых) контейнерах и последующем счете электрических
импульсов, образованных с помощью фотоумножи¬
теля из излучаемого света. Амплитуда импульса за¬
висит от энергии поглощенного излучения. Спектр
бета-частиц может быть получен при образовании до¬
статочного числа импульсов. Жидкая сцинтилляци-
онная смесь выбирается таким образом, чтобы ми¬
нимизировать ошибки счета вследствие поглощения,
хемилюминесценции, фосфоресценции и т.д. Счет
совпадений при использовании двух или более фо¬
тоумножителей также используется для минимиза¬
ции числа импульсов, к которым приводят фоновое
излучение, электронные эффекты и т.д. Для выявле¬
ния различия между эмиссиями альфа- и бета-частиц
обычно используется селекция импульсов по форме.
Идентификация радионуклида. Определение
соответствия средних и/или максимальных значений
энергии на спектре энергий для образца значениям
энергии, указанным в частной статье, является вали-
дированным испытанием на подлинность.
Калибровка. Общепринятым методом калибров¬
ки по энергии является применение непоглощающего
образца сравнения для определения максимального
значения энергии бета-частиц, испускаемых интере¬
суемым радионуклидом.
' За*. 1060.
46
Гэсударственная фармакопея Республики Беларусь
АЛЬФА-СПЕКТРОМЕТРИЯ
Для идентификации и количественного опре¬
деления альфа-эмиттеров чаще всего применяет¬
ся спектрометрия с использованием жидкостного
сцинтилляционного детектора. Принцип объясняет¬
ся в предыдущем разделе, касающемся бета-спек-
трометрии.
Для идентификации и определения радио¬
нуклидной чистоты альфа-эмиттеров может при¬
меняться спектрометрия с использованием полу¬
проводникового детектора на кремниевых диодах.
При использовании такого детектора поглощение
альфа-частиц приводит к незамедлительному об¬
разованию электрического импульса. Движение
электронно-дырочных пар, образованных при воз¬
действии излучения, индуцирует электрический
заряд, который усиливается и измеряется.
Подготовка образца является исключительно
важной. После химического отделения интересу¬
емого радионуклида образец наносится как элек¬
тролитическое покрытие на диск из нержавеющей
стали в виде очень тонкого слоя вещества для ми¬
нимизации собственной абсорбции. Результатив¬
ность всей процедуры может определяться экспе¬
риментально с добавлением известного количества
индикатора, который должен учитывать эффектив¬
ность разделения, эффективность нанесения элек¬
тролитического покрытия и эффективность счета.
Для обоих типов детекторов амплитуда им¬
пульса зависит от энергии поглощенного излуче¬
ния. Спектр альфа-частиц может быть получен при
образовании достаточного количества импульсов.
Радионуклидная идентификация. Определе¬
ние соответствия энергии пиков, обнаруженных для
образца, энергиям, указанным в частной статье, яв¬
ляется валидированным испытанием на подлин¬
ность.
Калибровка. Альфа-спектрометр должен быть
откалиброван по энергии и эффективности. Кали¬
бровка осуществляется с использованием пиков
известных источников, позволяющих охватить не¬
обходимый диапазон значений энергии, таких как
америций-241 и плутоний-242. Не все альфа-части¬
цы, испускаемые источником, будут образовывать
импульс в системе. Вероятность того, что испускае¬
мые альфа-частицы будут взаимодействовать с ма¬
териалом детектора и образовывать импульс, яв¬
ляется эффективностью детектора, зависящей от
геометрии.
ОБНАРУЖЕНИЕ И ИЗМЕРЕНИЕ
РАДИОАКТИВНОСТИ В СОЧЕТАНИИ
С МЕТОДОМ РАЗДЕЛЕНИЯ
Радиоактивное лекарственное средство (обра¬
зец) может содержать радионуклид в различных хи¬
мических формах, помимо исследуемой. Поэтому
необходимо разделить различные вещества, содер¬
жащие радионуклид, и определить процентное со¬
держание радиоактивности для рассматриваемого
радионуклида, связанного с указанной химической
формой, и вклад рассматриваемого радионуклида,
поступающего из других веществ, в общую радио¬
активность. Для этого измерительные приборы для
обнаружения и измерения радиоактивности исполь¬
зуются в сочетании с физико-химическим методом
разделения. В принципе может быть использован
любой метод разделения.
Частные статьи на радиоактивные фарма¬
цевтические препараты могут включать в себя со¬
вместное применение измерения радиоактивности
и методов бумажной хроматографии (2.2.26), тон¬
кослойной хроматографии (2.2.27), газовой хро¬
матографии (2.2.28), жидкостной хроматографии
(2.2.29), эксклюзионной хроматографии (2.2.30)
или электрофореза (2.2.31).
Во всех случаях радиоактивность каждого ана¬
лизируемого вещества измеряется после разделе¬
ния, проведенного указанным методом.
Измерение радиоактивности может выпол¬
няться с применением детекторов, смонтирован¬
ных последовательно с другими детекторами в ана¬
литических измерительных приборах, например
жидкостных хроматографах с осуществлением по¬
точного (т-Ипе) обнаружения радиоактивности ис¬
пытуемых образцов, или выполняться автономно
(о/7-//ле), то есть после завершения аналитического
разделения измерением радиоактивности фракций
элюата, полученных после разделения равных объ¬
емов с помощью жидкостной хроматографии или в
виде распределения по радиоактивности на под¬
ложках в бумажной хроматографии или тонкослой¬
ной хроматографии.
ПОТОЧНОЕ (М-иМЕ) ОБНАРУЖЕНИЕ
И ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
В СОЧЕТАНИИ С ЖИДКОСТНОЙ
ХРОМАТОГРАФИЕЙ
Прибор. Жидкостная хроматография (2.2.29)
может применяться для отделения основного ради¬
оактивного вещества радиоактивного лекарствен¬
ного средства (образца) от радиохимических при¬
месей или продуктов разложения. Поточное (т-Нпе)
обнаружение обычно осуществляется при исполь¬
зовании сцинтилляционного детектора, соединен¬
ного с измерителем скорости и регистрирующим
устройством. Сцинтилляционный материал детек¬
тора выбирается исходя из обнаруживаемой эмис¬
сии, например пластмассовый сцинтиллятор — для
эмиссий бета-частиц или сцинтилляционные кри¬
сталлы — для гамма- и рентгеновского излучений.
Прибавление жидкой сцинтилляционной смеси
перед достижением элюатом детектора для поточ¬
ного обнаружения радиоактивности может также
использоваться и для радионуклидов, испускаю¬
щих бета-частицы.
Одновременное использование детектора ра¬
диоактивного излучения и других детекторов (уль¬
трафиолетового излучения, показателя преломле¬
ния, кондуктометрического и т. д.), соединенных
последовательно, может применяться для иден¬
тификации вещества, например, по времени удер¬
живания известного стандарта, для определения
количества вещества с использованием подхо¬
дящего стандартного образца и для измерения
радиоактивности данного вещества. При после¬
довательном соединении разных детекторов по¬
следовательно необходимо откорректировать экс¬
периментально полученные значения времени
удерживания с учетом задержки во времени между
детекторами.
В жидкостной хроматографии некоторые ради¬
охимические примеси, например коллоидные при¬
меси, могут удерживаться в колонке. В подобных
случаях необходим отдельный метод для опреде¬
ления содержания удержанных радиохимических
2.2.66. Обнаружение и измерение радиоактивности
47
примесей, а расчетная формула для общей радио¬
химической чистоты должна учитывать относитель¬
ное содержание удержанных радиохимических при¬
месей.
Единственной возможностью для оценки по¬
добного удерживания в процессе валидации метода
является оценка восстановления радиоактивности
после прохождения колонки с помощью измерения
общей радиоактивности, восстановленной после
применения хроматографического оборудования с
колонкой и без нее.
Метод. Образец разводят при необходимости
и затем вносят в колонку в указанном объеме и при
указанных условиях. В данном случае важно под¬
твердить ПО (предел обнаружения, НтН оМе(есНоп
(ИЗО)), ПКО (предел количественного определе¬
ния, НтН риапНЯсаНоп ^00)) и линейность де¬
тектора на протяжении всего диапазона измеряе¬
мых значений активности.
Проточный детектор. Часть трубки, через
которую движется элюат, содержащий радиоак¬
тивные элементы, помещается перед детектором
или внутри него. Эффективность счета можно по¬
высить, используя более длинную часть трубки
(например, осуществляя многократные повороты
перед детектором или внутри него); тем не менее
это будет уменьшать возможность системы к раз¬
делению двух близко расположенных элюирующих
пиков радиоактивности.
При указании в испытании на радиохимическую
чистоту определения общего содержания радио¬
химических примесей или количественного опре¬
деления отдельной примеси важно выбрать соот¬
ветствующую установку предельного содержания и
подходящие условия для интегрирования площа¬
дей пиков. В подобных испытаниях неучитываемый
предел, то есть предел, при котором или ниже ко¬
торого пик не учитывается, зависит от метода и от¬
носится к пределу обнаружения и пределу количе¬
ственного определения. Таким образом, настройка
предельного содержания примеси системы сбора
данных соответствует не менее чем половине не¬
учитываемого предела.
Сигнал детекторов регистрируют как функцию
времени.
Идентификация пиков по радиометрическому
сигналу (радиохроматограмма) осуществляется на
основании времени удерживания анализируемых
веществ. Для этой цели может быть использован
профиль, полученный с использованием других де¬
текторов.
Количественное определение содержания раз¬
личных компонентов профилей хроматограммы и
радиохроматограммы осуществляется исходя из
площадей пиков. Площади пиков обычно получа¬
ются непосредственным интегрированием сигнала
детектора с использованием коммерчески доступ¬
ного программного обеспечения.
АВТОНОМНОЕ (ОЕР-иМЕ) ОБНАРУЖЕНИЕ
И ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
Жидкостная хроматография (2.2.29). При ус¬
ловии, что времена удерживания различных ра¬
диоактивных элементов согласуются по воспро¬
изводимости, применяется альтернативный метод
количественного определения радиоактивности,
заключающийся в сборе элюата в жидкостной хро¬
матографии в виде последовательно выходящих во
времени порций (фракций), для определения со¬
держания радиоактивности автономно (оЯ-Нпе). Ра¬
диоактивность фракций в соответствии с пиками
может быть выражена в виде процентного содержа¬
ния от общей радиоактивности для всех фракций,
учитывая предел количественного определения.
Метод. Образец вносят в колонку в указанном
объеме и при указанных условиях. Фракции соби¬
раются на конечной стадии хроматографирования.
Измеряется объем между детектором, исполь¬
зуемым для идентификации времени удерживания
пиков, и местом сбора фракций, а фактор удер¬
живания рассчитывается на основании скорости
потока элюата и используется для оценки време¬
ни элюирования для каждого пика в месте сбора.
Фракции собираются с учетом фиксированного ин¬
тервала времени или в момент выхода, опреде¬
ленный исходя из времени удерживания таким об¬
разом, чтобы любой из рассматриваемых пиков
относился к одной или более фракциям.
Радиоактивность каждой фракции рассчиты¬
вается с использованием калиброванного средства
измерения, например калибратора доз или сцин-
тилляционного детектора, учитывая предел коли¬
чественного определения и линейность.
Профиль элюирования получают построени¬
ем таблицы, в которой количество импульсов, со¬
ответствующее одной фракции, вносится напротив
времени элюирования или объема. Для определе¬
ния радиохимической чистоты значения активно¬
сти фракций, относящихся к одному и тому же пику,
могут суммироваться, и рассчитывается относитель¬
ное процентное содержание.
Тонкослойная хроматография (2.2.27) и бумаж¬
ная хроматография (2.2.26). При условии что анали¬
тический метод тонкослойной хроматографии или
бумажной хроматографии валидирован для раз¬
деления компонентов радиоактивных препаратов,
число и относительная интенсивность разделен¬
ных пятен могут обнаруживаться и измеряться с ис¬
пользованием детектора радиоактивности, который
может соотносить значение радиоактивности с ха¬
рактерным расположением пятен на хроматографи¬
ческой подложке.
Расположение пятен (пиков) дает возможность
провести химическую идентификацию при сравне¬
нии с растворами этих же химических веществ (не¬
радиоактивных), используя соответствующий метод
обнаружения.
Прибор
Сканирующее устройство. Прибор обычно со¬
держит детектор радиоактивности, например про¬
порциональный счетчик позитронов или коллимиро¬
ванный сцинтилляционный детектор, размещенный
на фиксированном расстоянии от сканирующей
платформы, на которой располагается сканируемая
хроматографическая подложка.
Радиоактивность образца, наносимого на хрома¬
тографическую подложку, должна быть такой, чтобы
значение скорости счета находилось в диапазоне
линейности измерительного оборудования, поэтому
при необходимости образец может быть разведен.
Сканируемая область размещается в соответствую¬
щем положении таким образом, чтобы необходимая
полоса находилась на одной линии с траекторией
сканирования детектора. Необходимо откорректиро¬
вать время сканирования для обеспечения достаточ¬
ного времени счета в течение пробега.
7*. Зак. 1060.
48
Гэсударственная фармакопея Республики Беларусь
Детектор или платформа могут перемещаться
в плоскости вдоль оси х или оси у таким образом,
чтобы вся поверхность полностью была проскани¬
рована в течение одного пробега.
Детектор соединяется с соответствующим счет¬
ным устройством таким образом, чтобы можно было
провести количественное измерение обнаруженной
радиоактивности и соотнести пространственно ско¬
рость счета со сканируемой поверхностью.
Отчет со значением радиоактивности по отно¬
шению к пройденному расстоянию выводится ав¬
томатически, а профиль содержит пики, имеющие
площадь, пропорциональную числу импульсов на
единицу расстояния.
Счетчик радиоактивности. В случае если не¬
обходимо идентифицировать не более трех полно¬
стью разделенных радиохимических компонентов,
то подложка может быть разрезана на равные по¬
лоски, каждая из которых имеет размер, не превы¬
шающий половину длины подложки в соответствии
с различием факторов удерживания двух наибо¬
лее близко расположенных пятен. Каждая отдель¬
ная полоска нумеруется, начиная с изначальной
поверхности, и считается по отдельности. С другой
стороны, для хорошо характеризуемых систем под¬
ложка может быть разрезана на две или более не¬
равные части, размеры которых перед счетом при
необходимости могут быть откорректированы заги¬
банием до ориентировочно равных. Для этой цели
могут быть использованы ионизационная камера
или сцинтилляционный счетчик при условии их при¬
менения в пределах диапазона линейности измери¬
тельного оборудования и свыше его ПКО.
Авторадиография. Авторадиография также
может быть использована для получения изобра¬
жения распределения по радиоактивности на хро¬
матографической подложке. В этом случае необ¬
ходимо продемонстрировать, что отклик системы,
применяемой для получения изображения, напри¬
мер люминофорной системы формирования изо¬
бражения или фотопленки, является линейным по
отношению к радиоактивности, представленной на
хроматограмме. В ином случае систему необходи¬
мо калибровать повторно или параллельно подвер¬
гнуть действию серии радиоактивных источников
сравнения, полученной разведением калибровоч¬
ного стандартного раствора и охватывающей ди¬
апазон ожидаемых значений радиоактивности
компонентов, которые могут присутствовать на
подложке.
Метод. Помещают необходимое количество
образца на оригинал хроматографической подлож¬
ки, высушивая при необходимости во избежание
расплывания пятна. Хроматографирование осу¬
ществляется согласно указанному методу. Если
указано в частной статье, может прибавляться но¬
ситель.
В бумажной и тонкослойной хроматографии
предпочтительно не разводить испытуемое лекар¬
ственное средство (образец), но важно избегать
нанесения вещества с таким содержанием радио¬
активности, которое в течение ее измерения обу¬
словливает потери при счете в связи с наложением
(потери на мертвое время).
После элюирования подложка высушивается и
определяется расположение площадей радиоактив¬
ности измерением радиоактивности на протяжении
всей хроматограммы с использованием соответ¬
ствующего коллимированного счетчика, с помощью
авторадиографии или разрезанием полосок на
части и счетом радиоактивности каждой части.
Радиоактивность может измеряться с помо¬
щью интегрирования с использованием автомати¬
зированного измерительного прибора или счетчика
с цифровой индикацией.
Из отношения площадей пиков получают отно¬
шение значений процентного содержания радиоак¬
тивности для соответствующих радиохимических
веществ.
Если полоски разрезаются на части, из от¬
ношения измеренных значений содержания ра¬
диоактивности определяют отношение значений
процентного содержания радиоактивности для со¬
ответствующих радиохимических веществ.
Калибровка. Подтверждение пределов обна¬
ружения и количественного определения и линей¬
ности детектора на протяжении всего диапазона
измеряемых значений активности и для всех поло¬
жений на подложке хроматографической системы
имеет важное значение. С этой целью применяют
образцы, охватывающие диапазон значений актив¬
ности от 0,1 % до 100 % от ожидаемого диапазо¬
на. Образцы готовят разведением и применяют их
равные объемы, высушивая при необходимости.
После изучения профиля радиоактивности, исполь¬
зуя стандартные установки измерительного обору¬
дования, площади пиков интегрируют для сравне¬
ния с рассчитанным содержанием радиоактивности
для каждого пятна. Контролируют, чтобы отклик де¬
тектора на протяжении всей длины и ширины тра¬
ектории перемещения детектора был одинаковым,
так как он может отличаться в зависимости от поло¬
жения детектора.
На разрешение пиков влияют размер пятна,
общая радиоактивность радионуклида и оснаще¬
ние детектора, что может регулироваться образо¬
ванием пятен при нанесении пробы в объеме 5 мкл,
разделенных расстоянием, возрастающим от 4 мм
до 20 мм с шагом 2 мм. Приблизительное разре¬
шение системы обнаружения может определяться
исходя из профиля радиоактивности как расстоя¬
ние между двумя пятнами, где базовая линия явля¬
ется только слегка четко отделенной.
07/2016: РБ20290
#2.2.90. ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
АНАЛИЗА
ОБЩИЕ ПОЛОЖЕНИЯ
Титриметрические методы анализа широко ис¬
пользуются для определения количественного со¬
держания субстанций для фармацевтического
использования и их компонентов, а также для опре¬
деления некоторых примесей.
Требования к титрованным растворам, их при¬
готовлению, установке титра и хранению описаны в
статье 4.2.2. Титрованные растворы.
В общем случае титрованный раствор (титрант)
добавляют в испытуемый раствор из подходящей
бюретки, выбранной в соответствии с молярной кон¬
центрацией титранта таким образом, чтобы расходу¬
емый на титрование объем находился между 30 %
и 100 % от номинальной вместимости бюретки (ми-
#2.2.90. Титриметрические методы анализа
49
кробюретки). Приближаясь к конечной точке титрова¬
ния, титрант добавляют по каплям для того, чтобы
последняя прибавленная капля позволила устано¬
вить конечную точку титрования и дать возможность
точно зафиксировать использованный объем титран-
та. При необходимости проводят контрольный опыт.
При прямом титровании испытуемый раст¬
вор помещают в подходящую колбу для титрова¬
ния (титруемый раствор) и прибавляют к нему со¬
ответствующий титрант. Количество испытуемого
образца рассчитывают исходя из объема и моляр¬
ной концентрации титранта (с учетом коэффициен¬
та поправки) и значения эквивалента вещества.
При обратном титровании к испытуемому рас¬
твору прибавляют измеренный объем титранта, яв¬
ляющийся избыточным по сравнению с объемом,
необходимым для реакции с испытуемым образ¬
цом. Избыток первого титранта затем оттитровыва-
ют вторым титрантом. Количество испытуемого об¬
разца рассчитывают исходя из объема и молярной
концентрации титранта (с учетом коэффициента
поправки) и значения эквивалента вещества.
Конечная точка титрования. Конечная точка
титрования, определяемая при титровании, нахо¬
дится вблизи точки эквивалентности или совпада¬
ет с ней. Близость нахождения конечной точки ти¬
трования к точке эквивалентности зависит от ряда
факторов, к которым относятся, например, приро¬
да титруемого вещества и концентрация титранта.
Индикаторы. Определение конечной точки ти¬
трования с помощью индикаторов является наибо¬
лее простым и удобным. Индикаторы представляют
собой вещества, обычно имеющие окраску и реаги¬
рующие на изменение условий в растворе перед и
после достижения точки эквивалентности, изменяя
при этом окраску, что может быть визуально при¬
нято за конечную точку титрования, находящуюся
вблизи точки эквивалентности.
Инструментальные методы установления
конечной точки титрования. В связи с тем, что ин¬
дикаторы оказываются непригодными для установ¬
ления конечной точки титрования при титровании
сильно окрашенных или мутных растворов, приме¬
няют другие способы, основанные на наблюдении
свойств раствора, резко меняющихся в точке экви¬
валентности. Большое значение приобрели инстру¬
ментальные методы установления конечной точки
титрования, основанные на измерении при помощи
приборов величин, характеризующих свойство
раствора, которое меняется в процессе титрования
постепенно, а в точке эквивалентности — резко. К
таким методам относятся потенциометрическое ти¬
трование (2.2.20), амперометрическое титрование
(2.2.19), вольтаметрическое титрование (2.2.65),
фотометрическое титрование, кондуктометриче¬
ское титрование и другие.
Контрольный опыт. Для повышения точ¬
ности установления конечной точки титрования
может проводиться контрольный опыт, представля¬
ющий собой титрование, при выполнении которо¬
го все необходимые процедуры повторяются, за ис¬
ключением использования испытуемого образца. В
таких случаях фактический объем титранта, экви¬
валентный количеству испытуемого образца, явля¬
ется разницей между объемами титранта, израсхо¬
дованного на титрование испытуемого образца, и
титранта, израсходованного в контрольном опыте.
ОСНОВНЫЕ ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
АНАЛИЗА
Наиболее распространенными титриметрически-
ми методами анализа являются:
- кислотно-основное титрование;
- окислительно-восстановительное титрование;
- осадительное титрование;
- комплексонометрическое титрование;
- нитритометрическое титрование.
Кислотно-основное титрование. Метод пред¬
назначен для определения веществ, обладающих
кислотными или основными свойствами в водной или
неводной среде. В качестве титрантов применяют ти¬
трованные растворы, обладающие кислотными или
основными свойствами. Рекомендуемые индикаторы
приведены в статье 2.2.4. Зависимость между реак¬
цией раствора, приблизительным значением рН и
цветом индикаторов, при этом точка эквивалентно¬
сти должна находиться в интервале значений рН пе¬
рехода окраски выбранного индикатора.
Титрование в неводных растворителях описано
в статье #2.5.50. Титрование в неводных раствори¬
телях.
Окислительно-восстановительное титрова¬
ние. Окислительно-восстановительное титрование
основано на использовании окислительно-восстано¬
вительных реакций разного типа. В качестве титран¬
тов применяют титрованные растворы, обладающие
окислительными или восстановительными свойства¬
ми. Роль индикатора во многих случаях может играть
сам титрант. В случае когда титрант является также и
индикатором, разница между конечной точкой титро¬
вания и точкой эквивалентности определяется только
способностью аналитика установить изменение окра¬
ски. Обычным примером может служить использова¬
ние перманганат-иона в качестве титранта-окислителя,
небольшой избыток которого может быть легко опре¬
делен по розовому окрашиванию. Примерами других
титрантов, которые могут быть использованы одновре¬
менно и в качестве индикаторов, являются йод, соли
церия (IV), дихромат калия. Однако в большинстве слу¬
чаев более точное определение конечной точки титро¬
вания достигается при использовании соответствую¬
щего окислительно-восстановительного индикатора.
Во многих случаях испытуемый образец необхо¬
димо привести в окисленную или восстановленную
форму перед титрованием с помощью соответствую¬
щего восстановителя или окислителя, избыток которо¬
го должен быть впоследствии удален, например осаж¬
дением. Чаще всего такой способ применяется для
титруемых растворов окислителей, так как большин¬
ство растворов восстановителей постепенно окисля¬
ется кислородом воздуха.
Осадительное титрование. В основе метода
лежит использование реакций осаждения или образо¬
вания малодиссоциированных соединений. Реакции
применимы при условии, что стехиометрическое рав¬
новесие наступает быстро и может быть установлено
с помощью индикаторов или другим способом.
Комплексонометрическое титрование. Метод
описан в статье 2.5.11. Комплексонометрическое ти¬
трование.
Нитритометрическое титрование. Метод
описан в статье 2.5.8. Определение аминного
азота в соединениях, которые содержат первич¬
ную ароматическую аминогруппу.
8. Зак. 1060.
50
Гэсударственная фармакопея Республики Беларусь
2.3. ПОДЛИННОСТЬ
(ИДЕНТИФИКАЦИЯ)
07/2016:20301
2.3.1. РЕАКЦИИ ПОДЛИННОСТИ
(ИДЕНТИФИКАЦИИ) НА ИОНЫ
И ФУНКЦИОНАЛЬНЫЕ ГРУППЫ
АЛКАЛОИДЫ
Несколько миллиграммов или указанное в
частной статье количество испытуемого образца
растворяют в 5 мл воды Р, прибавляют кислоту
хлористоводородную разведенную Р до кислой ре¬
акции раствора (2.2.4), затем 1 мл раствора калия
йодовисмутата Р; тотчас образуется оранжевый
или оранжево-красный осадок.
АЛЮМИНИЙ
Около 15 мг испытуемого образца растворяют
в 2 мл воды Р К полученному раствору или к 2 мл
раствора, указанного в частной статье, прибавля¬
ют 0,5 мл кислоты хлористоводородной разведен¬
ной Р и около 0,5 мл реактива тиоацетамида Р;
осадок не образуется. Затем прибавляют по каплям
раствор натрия гидроксида разведенный Р; об¬
разуется гелеобразный белый осадок, растворяю¬
щийся при последующем прибавлении раствора
натрия гидроксида разведенного Р. К полученному
раствору постепенно прибавляют раствор аммония
хлорида Р; вновь образуется гелеобразный белый
осадок.
АМИНЫ АРОМАТИЧЕСКИЕ ПЕРВИЧНЫЕ
Испытуемый раствор, указанный в частной
статье, подкисляют кислотой хлористоводород¬
ной разведенной Р #(или 0,05 г испытуемого об¬
разца растворяют в кислоте хлористоводородной
разведенной Р)# и прибавляют 0,2 мл раствора
натрия нитрита Р. Через (1—2) мин прибавляют
1 мл раствора р-нафтола Р; появляется интен¬
сивно оранжевое или красное окрашивание, и, как
правило, образуется осадок такого же цвета.
АММОНИЯ СОЛИ
К испытуемому раствору, указанному в частной
статье, прибавляют 0,2 г магния оксида Р. Через
жидкость пропускают воздух и направляют выходя¬
щий газ в смесь из 1 мл 0,1 М раствора кислоты
хлористоводородной и 0,05 мл раствора метило¬
вого красного Р; окраска индикатора переходит в
желтую. Затем прибавляют 1 мл свежеприготовлен¬
ного раствора 100 г/л натрия кобальтинитрита Р;
образуется желтый осадок.
АММОНИЯ СОЛИ И СОЛИ ЛЕТУЧИХ
ОСНОВАНИЙ
Около 20 мг испытуемого образца растворяют
в 2 мл воды Р. К полученному раствору или к 2 мл
раствора, указанного в частной статье, прибавляют
2 мл раствора натрия гидроксида разведенного Р.
При нагревании раствора выделяются пары аммиа¬
ка, которые обнаруживаются по запаху и щелочной
реакции (2.2.4).
АЦЕТАТЫ
a) Испытуемый образец нагревают с равным
количеством кислоты щавелевой Р; выделяет¬
ся кислота уксусная, обнаруживаемая по запаху и
кислой реакции (2.2.4).
b) Около 30 мг испытуемого образца раство¬
ряют в 3 мл воды Р. К полученному раствору или к
3 мл раствора, указанного в частной статье, после¬
довательно прибавляют 0,25 мл раствора ланта¬
на (III) нитрата Р, 0,1 мл 0,05 М раствора йода и
0,05 мл раствора аммиака разведенного Р2. Смесь
осторожно нагревают до кипения; в течение не¬
скольких минут образуется синий осадок или появ¬
ляется синее окрашивание.
#с) К 2 мл нейтрального раствора испытуе¬
мого образца, содержащего (20—60) мг ацетат-
иона (СН3СОО), прибавляют 0,2 мл раствора 30 г/л
железа (III) хлорида Р; появляется красно-коричне¬
вое окрашивание, исчезающее при прибавлении
минеральных разведенных кислот.
#6) 2 мл раствора испытуемого образца, со¬
держащего (20—60) мг ацетат-иона (СН3СОО), на¬
гревают с равным количеством кислоты серной*
концентрированной Р и 0,5 мл 96 % спирта Р; об¬
разуется этилацетат, обнаруживаемый по запаху.
АЦЕТИЛ
Около 15 мг или указанное в частной статье
количество испытуемого образца помещают в про¬
бирку длиной около 180 мм и наружным диаме¬
тром 18 мм и прибавляют 0,15 мл кислоты фос¬
форной Р. Пробирку закрывают пробкой, через
которую пропущена небольшая пробирка длиной
около 100 мм и наружным диаметром 10 мм, со¬
держащая воду Р и выполняющая роль холодиль¬
ника. На внешнюю поверхность меньшей пробирки
помещают 1 каплю раствора лантана (III) нитра¬
та Р. Если субстанция относительно легко гидро¬
лизуется, устройство помещают на 5 мин в водя¬
ную баню, затем вынимают меньшую пробирку.
Для трудно гидролизуемых субстанций смесь мед¬
ленно нагревают на открытом пламени до кипения.
Каплю снимают, смешивают на фарфоровой пла¬
стинке с 0,05 мл 0,01 М раствора йода. На край
капли наносят 0,05 мл раствора аммиака разве¬
денного Р2; через (1—2) мин в месте соединения
двух капель появляется синее окрашивание, кото¬
рое усиливается и сохраняется в течение коротко¬
го промежутка времени.
БАРБИТУРАТЫ (ЗА ИСКЛЮЧЕНИЕМ
Ы-ЗАМЕЩЕННЫХ)
Около 5 мг испытуемого образца растворяют в
3 мл метанола Р, прибавляют 0,1 мл раствора, со¬
держащего 100 г/л кобальта нитрата Р и 100 г/л
кальция хлорида Р, перемешивают и прибавляют при
встряхивании 0,1 мл раствора натрия гидроксида
разведенного Р; появляется фиолетово-синее окра¬
шивание, и образуется осадок.
БЕНЗОАТЫ
а) К 1 мл раствора, указанного в частной
статье, прибавляют 0,5 мл раствора железа (III)
2.3.1. Реакции подлинности (идентификации) на ионы и функциональные группы
51
хлорида Р1\ образуется розовато-желтый осадок,
растворимый в эфире Р.
b) 0,2 г испытуемого образца, при необходимо¬
сти подготовленного как указано в частной статье,
помещают в пробирку, смачивают 0,2 мл или 0,3 мл
кислоты серной Р и осторожно нагревают дно про¬
бирки; на внутренних стенках пробирки появляется
белый налет.
c) 0,5 г испытуемого образца растворяют в
10 мл воды Р. К полученному раствору или к 10 мл
раствора, указанного в частной статье, прибавля¬
ют 0,5 мл кислоты хлористоводородной Р; обра¬
зуется осадок, который после перекристаллиза¬
ции из теплой воды Р и высушивания под вакуумом
(2.2.32) имеет температуру плавления (2.2.14) от
120 °С до 124 °С.
БРОМИДЫ
a) Навеску испытуемого образца, содержа¬
щую около 3 мг бромид-иона (Вг), растворяют в
2 мл воды Р Полученный раствор или 2 мл раст¬
вора, указанного в частной статье, подкисляют кис¬
лотой азотной разведенной Р, прибавляют 0,4 мл
раствора серебра нитрата Р1, перемешивают и
отстаивают; образуется светло-желтый творожи¬
стый осадок. Осадок отделяют центрифугировани¬
ем и промывают тремя порциями воды Р по 1 мл
каждая. Эти операции проводят быстро в защищен¬
ном от яркого света месте, при этом допускается,
чтобы жидкость над осадком не была полностью
прозрачной. Полученный осадок суспендируют в
2 мл воды Р и прибавляют 1,5 мл раствора аммиа¬
ка Р; осадок трудно растворяется.
b) Навеску испытуемого образца, эквивалент¬
ную около 5 мг бромид-иона (Вг), или количество
образца, указанное в частной статье, помещают в
небольшую пробирку, прибавляют 0,25 мл воды Р,
около 75 мг свинца (IV) оксида Р, 0,25 мл кислоты
уксусной Р и осторожно встряхивают. Верхнюю вну¬
треннюю часть пробирки высушивают с помощью
фильтровальной бумаги и выдерживают в течение
5 мин. Полоску фильтровальной бумаги необходимо¬
го размера пропитывают, помещая ее край в каплю
раствора фуксина обесцвеченного Р, и тотчас по¬
мещают пропитанную часть в пробирку. В течение
10 с у нижнего края фильтровальной бумаги появля¬
ется фиолетовое окрашивание, которое четко отли¬
чается от красной окраски фуксина, наблюдаемой в
верхней пропитанной части полоски бумаги.
#с) К 1 мл раствора испытуемого образца, со¬
держащего (2—30) мг бромид-иона (Вг), прибавля¬
ют 1 мл кислоты хлористоводородной разведен¬
ной Р, 0,5 мл свежеприготовленного раствора 50 г/л
хлорамина Р, 1 мл хлороформа Р и встряхивают;
хлороформный слой приобретает желто-коричне¬
вую окраску.
ВИСМУТ
а) 0,5 г испытуемого образца растворяют в
10 мл кислоты хлористоводородной разведен¬
ной Р. Полученный раствор или 10 мл раствора, ука¬
занного в частной статье, кипятят в течение 1 мин,
охлаждают и при необходимости фильтруют. К 1 мл
полученного раствора прибавляют 20 мл воды Р;
образуется белый или светло-желтый осадок, цвет
которого после прибавления от 0,05 мл до 0,1 мл
раствора натрия сульфида Р изменяется на ко¬
ричневый.
Ь) Около 45 мг испытуемого образца раство¬
ряют в 10 мл кислоты азотной разведенной Р. По¬
лученный раствор или 10 мл раствора, указанного
в частной статье, кипятят в течение 1 мин, охлаж¬
дают и при необходимости фильтруют. К 5 мл полу¬
ченного раствора прибавляют 2 мл раствора 100 г/л
тиомочевины Р; появляется желтовато-оранжевое
окрашивание или образуется оранжевый осадок.
Затем прибавляют 4 мл раствора 25 г/л натрия
фторида Р; раствор не обесцвечивается в течение
30 мин.
#с) Навеску испытуемого образца, содержа¬
щую около 50 мг висмут-иона (ВР+), встряхивают с
5 мл кислоты серной разведенной Р и фильтруют.
К фильтрату прибавляют 2 капли раствора калия
йодида Р1\ образуется черный осадок, раствори¬
мый в избытке реактива с образованием раствора
желтовато-оранжевого цвета.
ЖЕЛЕЗО
a) Навеску испытуемого образца, содержащую
около 10 мг железо-иона (Ре2+), растворяют в 1 мл
воды Р К полученному раствору или к 1 мл раст¬
вора, указанного в частной статье, прибавляют
1 мл раствора калия феррицианида Р; образуется
синий осадок, не растворяющийся при прибавлении
5 мл кислоты хлористоводородной разведенной Р.
b) Навеску испытуемого образца, содержащую
около 1 мг железо-иона (Ре3+), растворяют в 30 мл
воды Р К 3 мл полученного раствора или к 3 мл
раствора, указанного в частной статье, прибавляют
1 мл кислоты хлористоводородной разведенной Р
и 1 мл раствора калия тиоцианата Р; появляется
красное окрашивание. Отбирают две порции полу¬
ченного раствора по 1 мл каждая. К одной порции
прибавляют 5 мл спирта изоамилового Р или 5 мл
эфира Р, встряхивают и оставляют до расслоения;
органический слой окрашивается в розовый цвет. К
другой порции прибавляют 2 мл раствора ртути
(II) хлорида Р; красное окрашивание раствора ис¬
чезает.
c) Навеску испытуемого образца, содержа¬
щую не менее 1 мг железо-иона (Ре3+), растворяют
в 1 мл воды Р К полученному раствору или к 1 мл
раствора, указанного в частной статье, прибавляют
1 мл раствора калия ферроцианида Р; образуется
синий осадок, не растворяющийся при прибавлении
5 мл кислоты хлористоводородной разведенной Р
ЙОДИДЫ
a) Навеску испытуемого образца, содержа¬
щую около 4 мг йодид-иона (I ), растворяют в 2 мл
воды Р. Полученный раствор или 2 мл раствора,
указанного в частной статье, подкисляют кислотой
азотной разведенной Р, прибавляют 0,4 мл раст¬
вора серебра нитрата Р1, перемешивают и отста¬
ивают до образования светло-желтого творожисто¬
го осадка. Осадок отделяют центрифугированием
и промывают 3 порциями воды Р по 1 мл каждая.
Эту операцию проводят быстро в защищенном от
яркого света месте; при этом допускается, чтобы
жидкость над осадком не была полностью прозрач¬
ной. Осадок суспендируют в 2 мл воды Р и прибав¬
ляют 1,5 мл раствора аммиака Р; осадок не рас¬
творяется.
b) К 0,2 мл раствора испытуемого образца, со¬
держащего около 5 мг йодид-иона (I ) в 1 мл, или к
0,2 мл раствора, указанного в частной статье, при-
Г За». -060
52
Гэсударственная фармакопея Республики Беларусь
бавляют 0,5 мл кислоты серной разведенной Р,
0,1 мл раствора калия дихромата Р, 2 мл воды Р,
2 мл хлороформа Р, встряхивают в течение не¬
скольких секунд и оставляют до расслоения; хлоро¬
формный слой приобретает фиолетовую или фио¬
летово-красную окраску.
#с) При нагревании 0,1 г испытуемого образца
с 1 мл кислоты серной Р выделяются фиолетовые
пары йода.
КАЛИЙ
a) 0,1 г испытуемого образца растворяют в
2 мл воды Р К полученному раствору или к 2 мл
раствора, указанного в частной статье, прибавляют
1 мл раствора натрия карбоната Р и нагревают;
осадок не образуется. К горячему раствору прибав¬
ляют 0,05 мл раствора натрия сульфида Р; осадок
не образуется. Раствор охлаждают в ледяной воде,
прибавляют 2 мл раствора 150 г/л кислоты винной
Р и отстаивают; образуется белый кристаллический
осадок.
b) Около 40 мг испытуемого образца раство¬
ряют в 1 мл воды Р. К полученному раствору или к
1 мл раствора, указанного в частной статье, прибав¬
ляют 1 мл кислоты уксусной разведенной Р и 1 мл
свежеприготовленного раствора 100 г/л натрия ко-
бальтинитрита Р; тотчас образуется желтый или
оранжево-желтый осадок.
#с) Соль калия, внесенная в бесцветное
пламя, окрашивает его в фиолетовый цвет, или при
рассматривании через синее стекло — в пурпурно¬
красный.
КАЛЬЦИЙ
a) К 0,2 мл нейтрального раствора испытуе¬
мого образца, содержащего около 0,2 мг кальций-
иона (Са2+) в 1 мл, или к 0,2 мл раствора, указан¬
ного в частной статье, прибавляют 0,5 мл раствора
2 г/л глиоксальгидроксианила Р в 96% спирте Р,
0,2 мл раствора натрия гидроксида разведенно¬
го Р и 0,2 мл раствора натрия карбоната Р. Смесь
встряхивают с 1 мл или 2 мл хлороформа Р и при¬
бавляют от 1 мл до 2 мл воды Р; хлороформный
слой приобретает красную окраску.
b) Около 20 мг или указанное в частной статье
количество испытуемого образца растворяют в 5 мл
кислоты уксусной Р. К полученному раствору при¬
бавляют 0,5 мл раствора калия ферроцианида Р;
раствор остается прозрачным. К раствору прибав¬
ляют около 50 мг аммония хлорида Р; образуется
белый кристаллический осадок.
#с) К 1 мл раствора испытуемого образца, со¬
держащего около (2—20) мг кальций-иона (Са2+),
прибавляют 1 мл раствора 40 г/л аммония оксала¬
та Р; образуется белый осадок, не растворимый в
кислоте уксусной разведенной Р, растворе амми¬
ака Р и растворимый в разведенных минеральных
кислотах.
#6) Соль кальция, смоченная кислотой хлори¬
стоводородной Р и внесенная в бесцветное пламя,
окрашивает его в оранжево-красный цвет.
КАРБОНАТЫ И ГИДРОКАРБОНАТЫ
а) 0,1 г испытуемого образца помещают в про¬
бирку и суспендируют в 2 мл воды Р К полученной
суспензии или к 2 мл суспензии, указанной в част¬
ной статье, прибавляют 3 мл кислоты уксусной
разведенной Р Пробирку тотчас закрывают притер¬
той пробкой со стеклянной трубкой, дважды изогну¬
той под прямым углом; наблюдается бурное выде¬
ление пузырьков газа без цвета и запаха. Пробирку
осторожно нагревают и пропускают выделяющийся
газ через 5 мл раствора бария гидроксида Р; обра¬
зуется белый осадок, растворяющийся при прибав¬
лении избытка кислоты хлористоводородной Р1.
#Ь) 0,2 г испытуемого образца растворяют в
2 мл воды Р К полученному раствору прибавляют
0,5 мл насыщенного раствора магния сульфата Р;
образуется белый осадок (в отличие от гидрокар¬
бонатов, растворы которых образуют осадок только
при кипячении смеси).
#с) 0,2 г испытуемого образца растворяют в
2 мл воды Р. К полученному раствору прибавляют
0,05 мл раствора фенолфталеина Р; появляется
красное окрашивание (в отличие от гидрокарбона¬
тов, растворы которых остаются бесцветными).
КСАНТИНЫ
К нескольким миллиграммам или к указанному
в частной статье количеству испытуемого образца
прибавляют 0,1 мл раствора водорода пероксида
концентрированного Р, 0,3 мл кислоты хлористо¬
водородной разведенной Р и выпаривают на водя¬
ной бане до получения сухого желтовато-красного
остатка. К остатку прибавляют 0,1 мл аммиака раз¬
веденного Р2\ цвет остатка изменяется на фиоле¬
тово-красный.
ЛАКТАТЫ
Навеску испытуемого образца, эквивалент¬
ную около 5 мг кислоты молочной, растворяют в
5 мл воды Р. К полученному раствору или к 5 мл
раствора, указанного в частной статье, прибавля¬
ют 1 мл бромной воды Р, 0,5 мл кислоты серной
разведенной Р и нагревают на водяной бане, пе¬
риодически перемешивая стеклянной палочкой, до
обесцвечивания раствора. К раствору прибавля¬
ют 4 г аммония сульфата Р и перемешивают. При¬
бавляют по каплям, не перемешивая, 0,2 мл раст¬
вора 100 г/л натрия нитропруссида Р в кислоте
серной разведенной Р, осторожно прибавляют,
также не перемешивая, 1 мл раствора аммиака
концентрированного Р и выдерживают в течение
30 мин; на границе двух жидкостей образуется тем¬
но-зеленое кольцо.
МАГНИЙ
Около 15 мг испытуемого образца растворяют
в 2 мл воды Р. К полученному раствору или к 2 мл
раствора, указанного в частной статье, прибавля¬
ют 1 мл раствора аммиака разведенного Р1\ об¬
разуется белый осадок, растворяющийся при при¬
бавлении 1 мл раствора аммония хлорида Р. К
полученному раствору прибавляют 1 мл раствора
динатрия гидрофосфата Р; образуется белый кри¬
сталлический осадок.
МЫШЬЯК
а) 5 мл испытуемого раствора нагревают на во¬
дяной бане с равным объемом реактива гипофос¬
фита Р; образуется коричневый осадок.
#Ь) Мышьяк (III) (арсениты). К 0,3 мл раствора
испытуемого образца, содержащего около 30 мг
арсенит-иона (А5033), прибавляют 0,5 мл кисло¬
ты хлористоводородной разведенной Р и 0,1 мл
раствора натрия сульфида Р; образуется желтый
2.3.1. Реакции подлинности (идентификации) на ионы и функциональные группы
53
осадок, не растворимый в кислоте хлористоводо¬
родной Р и растворимый в растворе аммиака Р.
#с) Мышьяк (V) (арсенаты). К 0,3 мл раствора
испытуемого образца, содержащего около 1 мг ар-
сенат-иона (Аз043), прибавляют по 1 мл раствора
100 г/л аммония хлорида Р, раствора аммиака Р
и раствора 100 г/л магния сульфата Р; образует¬
ся белый кристаллический осадок, растворимый в
кислоте хлористоводородной разведенной Р (от¬
личие от арсенитов).
НАТРИЙ
a) 0,1 г испытуемого образца растворяют в
2 мл воды Р. К полученному раствору или к 2 мл
раствора, указанного в частной статье, прибавляют
2 мл раствора 150 г/л калия карбоната Р и нагре¬
вают до кипения; осадок не образуется. К раство¬
ру прибавляют 4 мл раствора калия пироантимо-
ната Р и нагревают до кипения, затем охлаждают в
ледяной воде и при необходимости потирают вну¬
тренние стенки пробирки стеклянной палочкой; об¬
разуется плотный осадок белого цвета.
b) Навеску испытуемого образца, эквивалент¬
ную около 2 мг натрий-иона (14а*), растворяют в
0,5 мл воды Р. К полученному раствору или к 0,5 мл
раствора, указанного в частной статье, прибавля¬
ют 1,5 мл реактива метоксифенилуксусной кис¬
лоты Р, охлаждают в ледяной воде в течение 30
мин; образуется объемный белый кристаллический
осадок. Смесь помещают в воду при температуре
20 °С и перемешивают в течение 5 мин; осадок не
исчезает. К смеси прибавляют 1 мл раствора ам¬
миака разведенного Р1\ осадок полностью раство¬
ряется. К полученному раствору прибавляют 1 мл
раствора аммония карбоната Р; осадок не обра¬
зуется.
#с) Соль натрия, смоченная кислотой хлори¬
стоводородной Р и внесенная в бесцветное пламя,
окрашивает его в желтый цвет.
НИТРАТЫ
а) Навеску испытуемого образца, содержащую
около 1 мг нитрат-иона (МО3'), или количество, ука¬
занное в частной статье, прибавляют к смеси из
0,1 мл нитробензола Р и 0,2 мл кислоты серной Р
и через 5 мин охлаждают в ледяной воде. Продол¬
жая охлаждение, медленно при перемешивании
прибавляют 5 мл воды Р, 5 мл раствора натрия
гидроксида концентрированного Р, 5 мл ацето¬
на Р, взбалтывают и отстаивают; верхний слой при¬
обретает темно-фиолетовую окраску.
#Ь) Раствор испытуемого образца, содержа¬
щий около 2 мг нитрат-иона (1Ч03), не обесцвечи¬
вает раствор 1 г/л калия перманганата Р, подкис¬
ленный кислотой серной разведенной Р (в отличие
от нитритов).
#НИТРИТЫ
a) Несколько кристаллов феназона Р раство¬
ряют в фарфоровой чашке в 0,1 мл кислоты хлори¬
стоводородной разведенной Р, прибавляют 0,1 мл
испытуемого раствора, содержащего около 1 мг ни¬
трит-иона (1\Ю2); появляется зеленое окрашивание
(в отличие от нитратов).
b) К навеске испытуемого образца, эквивалент¬
ной 30 мг нитрит-иона (Ы02), прибавляют 1 мл кис¬
лоты серной разведенной Р; выделяются желто-ко¬
ричневые пары (в отличие от нитратов).
РТУТЬ
a) Около 0,1 мл раствора испытуемого образ¬
ца помещают на тщательно очищенную поверхность
медной фольги; появляется темно-серое пятно, кото¬
рое при натирании становится блестящим. Фольгу вы¬
сушивают и нагревают в пробирке; пятно исчезает.
b) К раствору, указанному в частной статье, при¬
бавляют раствор натрия гидроксида разведенный Р
до сильнощелочной среды (2.2.4); образуется плот¬
ный осадок желтого цвета (ртути (II) соединения).
#с) К 1 мл раствора испытуемого образца, со¬
держащего (10—30) мг ртуть-иона (Нд2+), прибавляют
осторожно по каплям раствор калия йодида Р; обра¬
зуется красный осадок, растворимый в избытке этого
реактива.
САЛИЦИЛАТЫ
a) К 1 мл раствора, указанного в частной
статье, прибавляют 0,5 мл раствора железа (III)
хлорида Р1\ появляется фиолетовое окрашивание,
которое не исчезает после прибавления 0,1 мл кис¬
лоты уксусной Р, #но исчезает при прибавлении
кислоты хлористоводородной разведенной Р; при
этом образуется белый кристаллический осадок са¬
лициловой кислоты#.
b) 0,5 г испытуемого образца растворяют в
10 мл воды Р. К полученному раствору или к 10 мл
раствора, указанного в частной статье, прибавляют
0,5 мл кислоты хлористоводородной Р. Получен¬
ный осадок после перекристаллизации из горячей
воды Р и высушивания под вакуумом (2.2.32) имеет
температуру плавления (2.2.14) от 156 °Сдо 161 °С.
СВИНЕЦ
a) 0,1 г испытуемого образца растворяют в
1 мл кислоты уксусной Р. К полученному раствору
или к 1 мл раствора, указанного в частной статье,
прибавляют 2 мл раствора калия хромата Р; об¬
разуется желтый осадок, растворяющийся при при¬
бавлении 2 мл раствора натрия гидроксида кон¬
центрированного Р.
b) 50 мг испытуемого образца растворяют в
1 мл кислоты уксусной Р. К полученному раствору
или к 1 мл раствора, указанного в частной статье,
прибавляют 10 мл воды Р и 0,2 мл раствора калия
йодида Р, образуется желтый осадок. Смесь кипя¬
тят в течение (1—2) мин; осадок растворяется. Рас¬
твору дают остыть; вновь образуется осадок в виде
блестящих желтых пластинок.
СЕРЕБРО
а) Около 10 мг испытуемого образца раство¬
ряют в 10 мл воды Р. К полученному раствору или
к 10 мл раствора, указанного в частной статье, при¬
бавляют 0,3 мл кислоты хлористоводородной Р1,
образуется белый творожистый осадок, растворя¬
ющийся при прибавлении 3 мл раствора аммиака
разведенного Р1.
#Ь) К 1 мл раствора испытуемого образца, со¬
держащего около 5 мг серебро-иона, прибавляют
раствор аммиака разведенного Р1 до растворения
образующегося вначале осадка, затем прибавля¬
ют 2—3 капли раствора формальдегида Р и нагре¬
вают; на стенках пробирки образуется блестящий
налет металлического серебра.
9 Зак. 1060.
54
Государственная фармакопея Республики Беларусь
СИЛИКАТЫ
а) Количество испытуемого образца, указан¬
ное в частной статье, смешивают в свинцовом или
платиновом тигле с помощью медной проволоки с
примерно 10 мг натрия фторида Р и несколькими
каплями кислоты серной Р до образования суспен¬
зии. Тигель накрывают тонкой прозрачной пласти¬
ковой пластинкой с висящей каплей воды Р и осто¬
рожно нагревают; через короткий промежуток вре¬
мени вокруг капли воды появляется белое кольцо.
#Ь) Около 0,1 г испытуемого образца, содержа¬
щего (50—60) мг силикат-ионов (ЗЮ32), смешивают
с 10 мл раствора аммония хлорида Р, прибавляют
1 мл раствора уксусной кислоты разведенной Р и
0,5 мл раствора метиленового синего Р и встря¬
хивают. Осадок фильтруют и промывают холодной
водой Р. Образуется осадок синего цвета, филь¬
трат — бесцветный.
#с) Навеску испытуемого образца, соответ¬
ствующую 10 мг силикат-ионов (ЗЮ32), суспенди¬
руют в 2 мл воды Р. К полученной смеси прибавля¬
ют 1 мл кислоты азотной Р, 5 мл раствора 50 г/л
кислоты щавелевой Р, 2 мл раствора молибдата
аммония Р, нагревают до начала кипения и затем
охлаждают. К охлажденному раствору прибавляют
2 мл 0,5 % (м/об) раствора бензидина Р в кислоте
уксусной Р. Появляется синее окрашивание.
СУЛЬФАТЫ
a) Около 45 мг испытуемого образца раство¬
ряют в 5 мл воды Р К полученному раствору или
к 5 мл раствора, указанного в частной статье, при¬
бавляют 1 мл кислоты хлористоводородной раз¬
веденной Р и 1 мл раствора бария хлорида Р1\ об¬
разуется белый осадок.
b) К суспензии, полученной в результате реак¬
ции (а), прибавляют 0,1 мл 0,05 М раствора йода\
желтая окраска йода не исчезает (в отличие от суль¬
фитов и дитионитов), но обесцвечивается при при¬
бавлении по каплям раствора олова хлорида Р (в от¬
личие от йодатов). Смесь кипятят; осадок не окра¬
шивается (в отличие от селенатов и вольфраматов).
#СУЛЬФИТЫ
a) К 2 мл раствора испытуемого образца, со¬
держащего (10—30) мг сульфит-иона (З032), при¬
бавляют 2 мл кислоты хлористоводородной раз¬
веденной Р и встряхивают; постепенно выделяется
сернистый газ, обнаруживаемый по характерному
резкому запаху.
b) К указанному в частной статье раствору, со¬
держащему сульфит-ион (З032 ), прибавляют 0,1 мл
0,5 М раствора йода; реактив обесцвечивается.
СУРЬМА
Около 10 мг испытуемого образца растворяют
при нагревании в растворе 0,5 г калия-натрия тар¬
трата Р в 10 мл воды Р и охлаждают. К 2 мл полу¬
ченного раствора или к 2 мл раствора, указанного
в частной статье, прибавляют по каплям раствор
натрия сульфида Р; образуется оранжево-красный
осадок, растворяющийся при прибавлении раст¬
вора натрия гидроксида разведенного Р
ТАРТРАТЫ
а) Около 15 мг испытуемого образца раство¬
ряют в 5 мл воды Р. К полученному раствору или
к 5 мл раствора, указанного в частной статье, при¬
бавляют 0,05 мл раствора 10 г/л железа (II) суль¬
фата Р и 0,05 мл раствора пероксида водорода
разведенного Р; появляется неустойчивое желтое
окрашивание. После обесцвечивания раствора к
нему прибавляют по каплям раствор натрия ги¬
дроксида разведенный Р; появляется интенсивное
синее окрашивание.
Ь) К 0,1 мл раствора испытуемого образца, со¬
держащего около 15 мг/мл кислоты винной, или к
0,1 мл раствора, указанного в частной статье, при¬
бавляют 0,1 мл раствора 100 г/л калия бромида Р,
0,1 мл раствора 20 г/л резорцина Р, 3 мл кислоты
серной Р и нагревают на водяной бане от 5 мин
до 10 мин; появляется темно-синее окрашивание.
Раствор охлаждают и вливают в воду Р; окраска
раствора изменяется на красную.
ФОСФАТЫ (ОРТОФОСФАТЫ)
a) К 5 мл раствора, указанного в частной ста¬
тье, при необходимости нейтрализованного, при¬
бавляют 5 мл раствора серебра нитрата Р1\ об¬
разуется желтый осадок, цвет которого не изме¬
няется при кипячении и который растворяется при
прибавлении раствора аммиака Р
b) 1 мл раствора, указанного в частной статье,
смешивают с 2 мл молибденованадиевого реакти¬
ва Р; появляется желтое окрашивание.
#с) К 1 мл раствора испытуемого образца, со¬
держащего (10—30) мг фосфат-иона (Р043 ) в кис¬
лоте азотной разведенной Р, прибавляют 2 мл
раствора аммония молибдата Р и нагревают; об¬
разуется желтый кристаллический осадок, раство¬
римый в растворе аммиака разведенном Р1.
#6) К 1 мл раствора испытуемого образца, со¬
держащего (10—30) мг фосфат-иона (Р043), при¬
бавляют 1 мл раствора аммония хлорида Р, 1 мл
раствора аммиака разведенного Р1 и 0,5 мл раст¬
вора 100 г/л магния сульфата Р; образуется белый
кристаллический осадок, растворимый в разведен¬
ных минеральных кислотах.
ХЛОРИДЫ
a) Навеску испытуемого образца, содержащую
около 2 мг хлорид-иона (С1), растворяют в 2 мл
воды Р Полученный раствор или 2 мл раствора,
указанного в частной статье, подкисляют кислотой
азотной разведенной Р, прибавляют 0,4 мл раст¬
вора серебра нитрата Р1, перемешивают и от¬
стаивают; образуется белый творожистый осадок,
который центрифугируют и промывают тремя пор¬
циями воды Р по 1 мл каждая. Эту операцию про¬
водят быстро в защищенном от яркого света месте,
при этом допускается, чтобы жидкость над осадком
не была полностью прозрачной. Осадок суспенди¬
руют в 2 мл воды Р и прибавляют 1,5 мл раствора
аммиака Р; осадок быстро растворяется; допуска¬
ется наличие нескольких крупных частиц, раство¬
ряющихся медленно.
b) Навеску испытуемого образца, содержащую
около 15 мг хлорид-иона (С1), или количество, ука¬
занное в частной статье, помещают в пробирку,
прибавляют 0,2 г калия дихромата Р и 1 мл кисло¬
ты серной Р У входного отверстия пробирки поме¬
щают фильтровальную бумагу, пропитанную 0,1 мл
раствора дифенилкарбазида Р (при этом пропи-
111111'
2.4.20. Определение остаточных количеств металлических катализаторов 55
тайная бумага не должна соприкасаться с калия
дихроматом); бумага окрашивается в фиолетово¬
красный цвет.
ЦИНК
а) 0,1 г испытуемого образца растворяют в
5 мл воды Р К полученному раствору или к 5 мл
раствора, указанного в частной статье, прибавля¬
ют 0,2 мл раствора натрия гидроксида концен¬
трированного Р; образуется белый осадок. Затем
прибавляют еще 2 мл раствора натрия гидрокси¬
да концентрированного Р; осадок растворяется.
К полученному раствору прибавляют 10 мл раст¬
вора аммония хлорида Р; раствор остается про¬
зрачным. К раствору прибавляют 0,1 мл раствора
натрия сульфида Р; образуется белый хлопьевид¬
ный осадок.
#Ь) К 2 мл раствора испытуемого образца, со¬
держащего (5—20) мг цинк-иона (2п2+), прибавляют
0,5 мл раствора калия ферроцианида Р; образует¬
ся белый осадок, не растворимый в кислоте хлори¬
стоводородной разведенной Р
ЦИТРАТЫ
а) Навеску испытуемого образца, содержащую
около 50 мг кислоты лимонной, растворяют в 5 мл
воды Р. К полученному раствору или к 5 мл раст¬
вора, указанного в частной статье, прибавляют
0,5 мл кислоты серной Р и 1 мл раствора калия
перманганата Р Раствор нагревают до обесцвечи¬
вания, прибавляют 0,5 мл раствора 100 г/л натрия
нитропруссида Р в кислоте серной разведенной Р,
4 г кислоты сульфаминовой Р К смеси прибавля¬
ют раствор аммиака концентрированный Р до ще¬
лочной реакции среды, прибавляя его по каплям
до полного растворения кислоты сульфаминовой.
Прибавление избытка раствора аммиака концен¬
трированного Р приводит к появлению фиолетово¬
го окрашивания, переходящего в фиолетово-синее.
#Ь) К 1 мл нейтрального раствора испытуемого
образца, содержащего (2—10) мг цитрат-иона, при¬
бавляют 1 мл раствора 200 г/л кальция хлорида Р;
раствор остается прозрачным; при кипячении раст¬
вора образуется белый осадок, растворимый в кис¬
лоте хлористоводородной разведенной Р.
#с) К навеске испытуемого образца, содержа¬
щей (1—2) мг цитрат-иона, прибавляют 0,5 мл ан¬
гидрида уксусного Р и нагревают; через (20—40) с
появляется красное окрашивание.
ЭФИРЫ СЛОЖНЫЕ
К 30 мг или к указанному в частной статье ко¬
личеству испытуемого образца прибавляют 0,5 мл
раствора 70 г/л гидроксиламина гидрохлорида Р в
метаноле Р, 0,5 мл раствора 100 г/л калия гидрок¬
сида Р в 96% спирте Р, нагревают до кипения и
охлаждают. Полученный раствор подкисляют кис¬
лотой хлористоводородной разведенной Р, при¬
бавляют 0,2 мл раствора железа (III) хлорида Р1,
разбавленного в 10 раз; появляется синевато-крас¬
ное или красное окрашивание.
#Другие реакции подлинности (идентификации), в
том числе реакции на другие ионы и функциональные
группы, проводят по методикам, указанным в частных
статьях, с использованием реактивов, эталонных, бу¬
ферных и титрованных растворов, описанных в раз¬
деле 4. Реактивы, если нет других указаний*.
2.4. ИСПЫТАНИЯ
НА ПРЕДЕЛЬНОЕ
СОДЕРЖАНИЕ ПРИМЕСЕЙ
07/2016:20420
2.4.20. ОПРЕДЕЛЕНИЕ ОСТАТОЧНЫХ
КОЛИЧЕСТВ МЕТАЛЛИЧЕСКИХ
КАТАЛИЗАТОРОВ
ИЛИ МЕТАЛЛСОДЕРЖАЩИХ
РЕАКТИВОВ
ВВЕДЕНИЕ
В данной статье описан общий подход определе¬
ния остаточных количеств металлических катализато¬
ров или металлсодержащих реактивов в субстанциях
для фармацевтического использования. Так как хими¬
ческий состав рассматриваемых субстанций и преде¬
лы искомых металлов, указываемые в спецификаци¬
ях, в значительной степени варьируют, описать все
применимые способы пробоподготовки и проведения
измерений не представляется возможным. Вслед¬
ствие этого может быть использован любой метод, от¬
вечающий условиям, приведенным в данной статье.
Результаты анализа являются приемлемыми
только при условии, что пригодность системы была
подтверждена надлежащим способом. Перед ис¬
пользованием методики аналитик должен убедиться
в том, что данная методика подходит для используе¬
мых образцов и приборов. Это достигается проведе¬
нием валидации для методик, не описанных в част¬
ных статьях, или проведением проверки пригодности
системы для методик, описанных в частных статьях.
Схемы принятия решения для выбора способа пробо¬
подготовки и методики проведения измерений пред¬
ставлены на рисунках 2.4.20.-1 и 2.4.20.-2.
МЕТОДИКИ
Так как не существует исходной методики для
каждого из сочетаний металла, матрицы и концентра¬
ции, ответственность за выбор методики на основа¬
нии рисунков 2.4.20.-1 и 2.4.20.-2, включая пробопод-
готовку, способ детектирования и параметры прибора,
лежит на пользователе.
Схему, приведенную на рисунке 2.4.20.-1, ис¬
пользуют для определения способа пробоподготов¬
ки, а схему на рисунке 2.4.20.-2 — для определения
методики проведения измерения. В результате про¬
боподготовки должно получаться достаточное количе¬
ство образца, позволяющее провести количественное
определение каждого металла в специфицированных
пределах, указанных в частной или общей статье.
Для определения остаточных количеств ме¬
таллов могут использоваться все подходящие спо¬
собы пробоподготовки и методики проведения из¬
мерений (например, 2.2.22. Атомно-эмиссионная
спектрометрия (АЭС), 2.2.23. Атомно-абсорбци¬
онная спектрометрия (ААС), 2.2.37. Рентгеноф¬
луоресцентная спектрометрия, 2.2.57. Атомно¬
эмиссионная спектрометрия с использованием
индуктивно связанной плазмы (ИСП-АЭС), 2.2.58.
Масс-спектрометрия с использованием индуктив¬
но связанной плазмы (ИСП-МС), 2.4.2. Мышьяк,
9*. Зак. 1060.
56
Гэсударственная фармакопея Республики Беларусь
Рисунок 2.4.20.-1. Схема принятия решений: пробоподготовка
2.4.8. Тяжелые металлы, 2.4.9. Железо, 2.4.10.
Свинец в сахарах, 2.4.15. Никель в полиолах, 2.4.31.
Никель в гидрогенезированных растительных
маслах) при условии, что перед началом использо¬
вания они были верифицированы путем валидации
или проверки пригодности системы в соответствии
с данной статьей.
Если в частной статье не описана пробопод¬
готовка и/или методика проведения измерения, то
они должны быть разработаны и валидированы (см.
рисунки 2.4.20.-1 и 2.4.20.-2).
ПРОБОПОДГОТОВКА
Для успешного анализа элементов важным мо¬
ментом является пробоподготовка. Многие методи¬
ки, не использующие прямое измерение, в значи¬
тельной степени зависят от переноса образца.
При использовании системы атомизации наи¬
более распространенным способом подачи образ¬
ца является распыление раствора. В таком случае
перед введением в атомизатор твердый образец
должен быть предварительно растворен в любом
подходящем растворителе. Для этих целей на¬
стоятельно рекомендуется использовать водные
растворы или растворы на разведенной азотной
кислоте по причине их минимального влияния по
сравнению с другими растворителями. Для раство¬
рения образцов могут использоваться хлористово¬
дородная кислота, фтористоводородная кислота,
хлорная кислота, серная кислота и пероксид водо¬
рода с различными концентрациями, при этом не¬
обходимо учитывать, что вязкость серной кислоты
выше, чем вязкость других кислот, и это может вли¬
ять на текучесть раствора. Список используемых
растворов включает в себя (но не ограничивается):
разведенные основания, чистые или разведенные
органические растворители, комбинации кислот
или оснований, комбинации органических раство¬
рителей.
Используемые кислоты, основания и пероксид
водорода должны иметь высокую степень чистоты,
2.4.20. Определение остаточных количеств металлических катализаторов
57
Рисунок 2.4.20.-2. Схема принятия решений: измерение
особенно при проведения испытания с помощью
ИСП-МС. Для приготовления водных растворов не¬
обходимо использовать воду деионизированную
дистиллированную Р. При использовании в испы¬
тании растворителей необходимо проверить отсут¬
ствие мешающих воздействий. По причине того, что
не всегда возможно использование органических
растворителей, не содержащих металлы, необходи¬
мо использовать органические растворители с наи¬
большей степенью чистоты относительно содержа¬
ния металлов. Для методик ИСП с введением об¬
разцов в плазму путем распыления раствора важно
рассмотреть потенциальные эффекты матрицы и
мешающие воздействия, вызываемые раствори¬
телем. Если в методиках ИСП-АЭС и ИСП-МС не
достигается необходимая степень правильности и
точности, используют внутренний стандарт и/или
корректируют матрицу стандарта, приближая ее
к матрице испытуемых образцов. В любом случае
при выборе подходящего внутреннего стандарта
необходимо учитывать искомый металл (металлы),
энергию ионизации, длины волн или массы и при¬
роду матрицы испытуемого образца.
Если испытуемый образец не растворяется ни
в одном из приемлемых растворителей, могут быть
использованы различные способы разложения или
сжигания, такие как разложение на горячей плитке,
сжигание и микроволновое разложение в открытых и
закрытых сосудах.
Принятие решения об использовании конкретно¬
го способа разложения зависит от природы образца,
искомого металла и его концентрации. При анализе
летучих металлов не рекомендуется применять раз¬
ложение в открытых сосудах. Пригодность способа
разложения, будь то в открытых или в закрытых со¬
судах, должна быть подтверждена путем открываемо-
сти добавок для проверки того, что при разложении
летучие элементы не теряются сверх допустимых
пределов. Цикл разложения считается пригодным,
если в результате получается прозрачный раствор.
Важным является выбор типа, материала, а также
предварительной обработки и очистки лабораторной
посуды, используемой в элементном анализе. Матери¬
ал должен быть инертным и, в зависимости от предна¬
значения, должен быть устойчивым к действию едких
щелочей, кислот и/или органических растворителей.
При проведении некоторых анализов должны быть
приняты меры предосторожности для предотвращения
адсорбции металлов на поверхности сосуда, особен¬
но это касается анализа ультра-следовых количеств.
Также к неточному результату может приводить загряз¬
нение растворов испытуемого образца металлами и
ионами, присутствующими в составе контейнера.
Использование мерной стеклянной посуды, не
соответствующей требованиям Международного
стандарта Международной организации по стандар¬
тизации (/50) для класса А, допустимо, если при этом
с помощью валидации или проверки пригодности си¬
стемы экспериментально доказано, что метод являет¬
ся подходящим для намеченной цели.
ПРЕДОСТЕРЕЖЕНИЕ: при использовании раз¬
ложения в сосудах под высоким давлением и при ис¬
пользовании микроволнового лабораторного обо¬
рудования необходимо следовать требованиям по
безопасности и инструкциям по использованию про¬
изводителя.
10. Зак. 1060.
58
Гэсударственная фармакопея Республики Беларусь
ИЗМЕРЕНИЕ
Методика. Выбор методики главным образом
зависит от матрицы образца и свойств и специфи¬
цированных пределов искомого металла (метал¬
лов). При настройке программы и длины волны сле¬
дуют инструкциям производителя прибора.
Пригодность системы. Проверка пригодности
системы должна проводиться в день анализа для
подтверждения того, что пробоподготовка и систе¬
ма проведения измерения являются соответствую¬
щими.
Критерий приемлемости для приготовления
раствора испытуемого образца: полученный раст¬
вор является прозрачным.
Критерий приемлемости для системы прове¬
дения измерения: измеренная концентрация стан¬
дартного раствора металла с концентрацией, нахо¬
дящейся в диапазоне используемой калибровочной
кривой, не отличается от истинной концентрации
более чем на 20 %.
Расчеты. При расчете содержания необходи¬
мо учитывать значение, полученное при использо¬
вании контрольного раствора. По окончании про¬
ведения измерений концентрация данного металла
в испытуемом образце рассчитывается с помощью
программного обеспечения прибора исходя из кон¬
центрации металла в испытуемом растворе. Если
таковое программное обеспечение отсутствует или
если в разделе 2.2. Физические и физико-химиче¬
ские методы нет указаний на проведение расче¬
тов, концентрация данного металла в образце мо¬
жет быть рассчитана на основании концентрации
металла в растворе с использованием следующей
формулы:
где:
С — концентрация металла в испытуемом образ¬
це, мкг/г;
А — показываемая прибором концентрация ме¬
талла в растворе испытуемого образца, мкг/мл;
т — масса испытуемого образца, взятая для
приготовления исходного раствора испытуемого об¬
разца, г;
]/1 — объем начального раствора испытуемого
образца, мл;
У2 — общий объем любого проводимого разведе¬
ния, мл;
Уг — объем начального раствора, используемый
для любого проводимого разведения, мл.
ВАЛИДАЦИОННЫЕ ТРЕБОВАНИЯ
Некоторые валидационные требования, приве¬
денные ниже, могут отличаться от требований других
общих статей Фармакопеи (например, 2.2.22 (АЭС),
2.2.23 (ААС), 2.2.57 (ИСП-АЭС), 2.2.58 (ИСП-МС)).
Перед началом использования выбранной
методики аналитик должен убедиться в том, что
пробоподготовка и способ проведения измерения
подходят для определяемого металла (металлов),
матрицы испытуемого образца и используемого
прибора. Это достигается выполнением валидации
перед началом использования методики и провер¬
кой пригодности системы в день проведения испы¬
тания.
При анализе остаточных количеств металла
валидация предельного испытания должна вклю¬
чать в себя специфичность и предел обнаружения.
В нижеприведенном разделе даны определе¬
ния характеристик для количественной методики.
Должно быть представлено экспериментальное
подтверждение того, что методика выдерживает ва¬
лидационные требования и соответствующую про¬
верку пригодности системы с использованием об¬
разцов, содержащих добавки подходящего количе¬
ства стандартного образца. Добавка в испытуемый
образец должна быть осуществлена до начала про-
боподготовки. Например, если испытуемый образец
должен быть предварительно минерализован, до¬
бавление стандартного образца должно проводить¬
ся до начала процедуры разложения.
СПЕЦИФИЧНОСТЬ
Специфичность — это способность подтверж¬
дения того, что аналитические методики для про-
боподготовки и проведения измерений позволяют
достоверно определить металл (металлы) в при¬
сутствии ожидаемых компонентов (например, газ-
носитель, примеси, матрица).
Критерий приемлемости: методика должна
позволять однозначно определять остаточные коли¬
чества каждого металла в присутствии ожидаемых
компонентов, в том числе остаточных количеств
иных металлов, компонентов матрицы, а также и
других источников мешающего воздействия; специ¬
фичность подтверждается выполнением требова¬
ний для правильности для определяемого металла
(металлов).
ДИАПАЗОН
Критерий приемлемости: диапазон подтверж¬
дается выполнением требований для открываемости.
ПРАВИЛЬНОСТЬ
Правильность проверяют с помощью аттесто¬
ванного стандартного материала или путем прове¬
дения проверки открываемости.
Открываемость. Открываемость может быть
определена с использованием испытуемого об¬
разца, к которому добавлено известное количество
стандартного образца металла (три различные
концентрации, лежащие в диапазоне от 50 % до
150 % специфицированного предела, даже если из¬
начальная концентрация стандартного образца на¬
ходится близко к специфицированному пределу) в
трех повторностях.
Критерий приемлемости: открываемость добав¬
ки должна быть от 70 % до 150 % для среднего значе¬
ния трех повторностей для каждой концентрации.
СХОДИМОСТЬ
Испытуемые образцы: либо 6 независимых
испытуемых образцов, содержащих добавки под¬
ходящего стандартного образца с концентрацией
на уровне специфицированного предела, либо три
концентрационных предела, приготовленных в трех
повторностях.
Критерий приемлемости: относительное
стандартное отклонение в обоих случаях не должно
превышать 20 %.
ВНУТРИЛАБОРАТОРНАЯ ТОЧНОСТЬ
Необходимо установить влияние случайных
явлений (внутрилабораторных вариаций) на анали¬
тическую точность метода. Для этого проводят, на¬
пример, анализ сходимости либо в различные дни,
либо с использованием различного оборудования,
либо различными аналитиками. Для подтвержде¬
ния внутрилабораторной точности достаточно про¬
ведения только одного из трех экспериментов.
2.4.27. Тяжелые металлы в лекарственном растительном сырье
59
Критерий приемлемости: относительное стан¬
дартное отклонение не должно превышать 25 %.
ПРЕДЕЛ КОЛИЧЕСТВЕННОГО
ОПРЕДЕЛЕНИЯ
Определяют нижний предел концентрации, отве¬
чающий критерию приемлемости. Используют резуль¬
таты, полученные при изучении правильности.
Критерий приемлемости: предел количествен¬
ного определения должен быть ниже предела специ¬
фикации.
ПРЕДЕЛ ОБНАРУЖЕНИЯ
(ПРИМЕНИМ ТОЛЬКО ДЛЯ ИСПЫТАНИЙ
НА ПРЕДЕЛЬНОЕ СОДЕРЖАНИЕ)
Определяют нижний предел концентрации, да¬
ющей сигнал, который отчетливо отличим от сиг¬
нала, полученного с использованием контрольного
раствора.
Критерий приемлемости: предел обнаруже¬
ния не должен превышать 0,5 концентрации предела
спецификации.
07/2016:20427
2.4.27. ТЯЖЕЛЫЕ МЕТАЛЛЫ
В ЛЕКАРСТВЕННОМ
РАСТИТЕЛЬНОМ
СЫРЬЕ И ПРОДУКТАХ
ИЗ ЛЕКАРСТВЕННОГО
РАСТИТЕЛЬНОГО СЫРЬЯ
ПРЕДУПРЕЖДЕНИЕ: при использовании за¬
крытых реакционных сосудов высокого давления и
микроволнового лабораторного оборудования не¬
обходимо строго придерживаться инструкций по
технике безопасности и эксплуатации оборудова¬
ния, прилагаемых производителем.
ОБОРУДОВАНИЕ
Оборудование обычно состоит из следующих
компонентов:
- закрытых реакционных сосудов, выдержи¬
вающих высокое давление: колб из политетраф¬
торэтилена, перфторёлкоксиполимера, кварца или
стекла вместимостью (20—150) мл, снабженных
герметичными пробками, клапаном, регулирующим
давление внутри емкости, и политетрафторэтиле-
новой трубкой для отвода газа;
- системы для герметичного укупоривания
колб, использующей одинаковую силу закрутки для
каждой из них;
- программируемой микроволновой печи (на¬
пример, с частотой магнетрона 2450 МГц и контро¬
лируемой выходной мощностью от 0 до (1500±70)
Вт с инкрементом 1 %), СВЧ-резонатора, покрытого
политетрафторэтиленом, с вытяжным вентилято¬
ром, работающим в различных режимах, вращаю¬
щейся приводной системы и вытяжки для отвода
испарений;
- атомно-абсорбционного спектрометра
(2.2.23), индуктивно связанной плазмы — атом¬
но-эмиссионного спектрометра (2.2.57) или индук¬
тивно связанной плазмы — масс-спектрометра
(2.2.58).
МЕТОДИКА
Испытание проводят методами атомно-аб¬
сорбционной спектрометрии (ААС) (2.2.23), атом¬
но-эмиссионной спектрометрии с индуктивно-свя¬
занной плазмой (ИСП-АЭС) (2.2.57) или масс-
спектрометрии с индуктивно-связанной плазмой
(ИСП-МС) (2.2.58).
Отступление от условий приготовления об¬
разца и приведенного ниже описания методики
допустимо в случае соблюдения всех валидацион-
ных требований, а также выполнения теста про¬
верки пригодности системы на день проведения
испытания.
Приготовление образца
Перед использованием вся стеклянная посуда
и лабораторное оборудование должны быть обра¬
ботаны раствором 10 г/л кислоты азотной Р.
Испытуемый раствор. Указанное в частной
статье количество испытуемого образца (около
0,50 г измельченного сырья (1400) (2.9.12)) поме¬
щают в колбу для минерализации, прибавляют 6 мл
кислоты азотной, свободной от тяжелых метал¬
лов, Р и 4 мл кислоты хлористоводородной, сво¬
бодной от тяжелых металлов, Р. Герметично уку¬
поривают.
Помещают колбы для минерализации в микро¬
волновую печь и проводят минерализацию в соот¬
ветствии со следующей программой в три стадии,
используя семь колб, содержащих испытуемый
раствор: 80 % мощности в течение 15 мин; 100 %
мощности в течение 5 мин; 80 % мощности в тече¬
ние 20 мин.
В конце цикла колбы охлаждают на воздухе или
в воде. После охлаждения колбы открывают и пере¬
носят полученный прозрачный бесцветный раствор
в мерную колбу вместимостью 50 мл. Каждую колбу
дважды ополаскивают 15 мл кислоты азотной раз¬
веденной, свободной от тяжелых металлов, Р, со¬
бирают промывные воды в мерную колбу и доводят
водой Р до объема 50,0 мл. При необходимости мо¬
гут быть использованы модификаторы (например,
в случае ААС с электротермической атомизацией
1,0 мл раствора 10 г/л магния нитрата Р и 1,0 мл
раствора 100 г/л аммония дигидрофосфата Р) или
стабилизирующие реагенты.
Холостой раствор. 4 мл кислоты хлористо¬
водородной, свободной от тяжелых металлов, Р
и 6 мл кислоты азотной, свободной от тяжелых
металлов, Р помещают в колбу для минерализа¬
ции и обрабатывают аналогично испытуемому рас¬
твору.
ОПРЕДЕЛЕНИЕ МЫШЬЯКА, КАДМИЯ, МЕДИ,
НИКЕЛЯ И СВИНЦА МЕТОДОМ ААС (2.2.23)
С ЭЛЕКТРОТЕРМИЧЕСКОЙ АТОМИЗАЦИЕЙ
Содержание мышьяка, кадмия, меди, никеля
и свинца определяют методом градуировочного
графика (2.2.23, метод I) или методом стандарт¬
ных добавок (2.2.23, метод II), используя растворы
сравнения каждого тяжелого металла при рабочих
параметрах прибора, рекомендованных в таблице
2.4.27.-1.
Величина оптической плотности холостого
раствора вычитается из значения, полученного для
испытуемого раствора.
10*. Зак. 1060.
60
Гэсударственная фармакопея Республики Беларусь
Таблица 2.4.27.-1
Рабочие параметры прибора ААС
с электротермической атомизацией
Аз
Сд
Си
N1
РЬ
Длина волны
нм
193,7
228,8
324,8
232
283,5
Ширина щели
нм
0,5
0,5
0,5
0,2
0,5
Ток лампы
мА
10
6
7
10
5
Температура
сжигания
°С
1400
800
800
800
800
Температура
атомизации
°С
2600
1800
2300
2500
2200
Скорость
потока азота
л/мин
3
3
3
3
3
ОПРЕДЕЛЕНИЕ МЫШЬЯКА И РТУТИ
МЕТОДОМ ААС (2.2.23) С АТОМИЗАЦИЕЙ
ХОЛОДНОГО ПАРА ИЛИ ГИДРИДОВ
Содержание мышьяка и ртути определяют ме¬
тодом градуировочного графика (2.2.23, метод I)
или методом стандартных добавок (2.2.23, метод
II), используя растворы сравнения мышьяка и ртути
с известными концентрациями, применяя автомати¬
ческий генератор гидридного пара этих элементов.
Величина оптической плотности холостого
раствора вычитается из значения, полученного для
испытуемого раствора.
Мышьяк
Раствор образца. К 19,0 мл испытуемого раст¬
вора или холостого раствора, приготовленных как
описано выше, прибавляют 1 мл раствора 200 г/л
калия йодида Р. Выдерживают испытуемый раствор
при комнатной температуре в течение около 50 мин
или при температуре 70 °С в течение около 4 мин.
Кислотный реактив. Кислота хлористоводо¬
родная, свободная от тяжелых металлов, Р.
Восстанавливающий реактив. Раствор 6 г/л
натрия тетрагидробората Р в растворе 5 г/л на¬
трия гидроксида Р.
Могут быть использованы рекомендуемые па¬
раметры прибора, описанные в таблице 2.4.27.-2.
Таблица 2.4.27.-2
Рабочие параметры прибора ААС
с атомизацией холодного пара или гидридов
Аз Нд
Длина волны
нм
193,7
253,7
Ширина щели
нм
0,2
0,5
Сила тока в лампе
мА
10
4
Кислотный реактив
мл/мин
1,0
1,0
Восстанавливаю¬
щий реактив
мл/мин
1,0
1,0
Раствор образца
мл/мин
7,0
7,0
Кювета
Кварцевая
(подогре¬
тая)
Кварцевая
(не подо¬
гретая)
Ток азота
л/мин
0,1
0,1
Ртуть
Раствор образца. К 19,0 мл испытуемого раст¬
вора или холостого раствора, приготовленных как
описано выше, прибавляют 1 мл раствора 200 г/л
калия йодида Р. Выдерживают испытуемый раст¬
вор при комнатной температуре в течение около
50 мин или при температуре 70 °С в течение около
4 мин.
Кислотный реактив. Раствор 515 г/л кислоты
хлористоводородной, свободной от тяжелых ме¬
таллов, Р.
Восстанавливающий реактив. Раствор 10 г/л
олова (II) хлорида Р в кислоте хлористоводород¬
ной разведенной, свободной от тяжелых метал¬
лов, Р.
Могут быть использованы рекомендуемые па¬
раметры прибора, описанные в таблице 2.4.27.-2.
ОПРЕДЕЛЕНИЕ МЫШЬЯКА, КАДМИЯ,
МЕДИ, РТУТИ, НИКЕЛЯ И СВИНЦА
МЕТОДОМ ИСП-АЭС (2.2.57)
Содержание мышьяка, кадмия, меди, ртути,
никеля и свинца определяют методом градуировоч¬
ного графика (2.2.23, метод /), используя растворы
сравнения каждого тяжелого металла при рабочих
параметрах прибора, рекомендованных в таблице
2.4.27.-3.
Величина эмиссии холостого раствора вычи¬
тается из значения, полученного для испытуемого
раствора.
ОПРЕДЕЛЕНИЕ МЫШЬЯКА, КАДМИЯ,
МЕДИ, РТУТИ, НИКЕЛЯ И СВИНЦА
МЕТОДОМ ИСП-МС (2.2.58)
Содержание мышьяка, кадмия, меди, ртути, ни¬
келя и свинца определяют методом градуировочного
графика (2.2.23, метод /), используя растворы срав¬
нения каждого тяжелого металла, а также аналитиче¬
ские изотопы и дополнительные массы, рекомендо¬
ванные в таблице 2.4.27.-4.
Таблица 2.4.27.-4
Рекомендуемые аналитические изотопы
и дополнительные массы для ИСП-МС
Изотоп
Определяемый элемент
75
Мышьяк
106, 108, 111, 114
Кадмий
63, 65
Медь
202
Ртуть
60, 62
Никель
206, 207, 208
Свинец
Величина интенсивности сигнала холостого раст¬
вора вычитается из значения, полученного для испы¬
туемого раствора.
Таблица 2.4.27.-3
Рабочие параметры прибора ИСП-АЭС
Аз
са
Си
нд
N1
РЬ
Длина волны
нм
193,696/
214,438/
324,754/
184,950/
231,604/
220,351/
197,197/
226,502/
327,396/
253,652/
231,997/
283,306/
189,042
228,802
224,700
435,835
352,454
168,215
Аг, контроль
нм
430,010
430,010
430,010
430,010
430,010
430,010
Энергия плазмы
Вт
1200
1200
1200
1200
1200
1200
Алгоритм расчета пиков
с поправкой на фон
Да
да
Да
Да
да
Да
2.5.7. Кислотное число
61
ПРИГОДНОСТЬ СИСТЕМЫ
Проверка пригодности системы должна осу¬
ществляться в день проведения испытания для
подтверждения того, что приготовление образца и
система измерения подходят для заданных целей.
Критерии приемлемости для приготовления
испытуемого раствора: получается прозрачный
раствор.
Критерии приемлемости для системы изме¬
рения: измеренная концентрация раствора сравне¬
ния металла при концентрации, лежащей в преде¬
лах градуировочного графика, не отличается от
действительной концентрации более чем на 20 %.
ВАЛИДАЦИОННЫЕ ТРЕБОВАНИЯ
Аналитическая методика должна быть валиди-
рована в соответствии с использующимися общими
методами ААС (2.2.23), ИСП-АЭС (2.2.57) и ИСП-
МС (2.2.58). Дополнительно должны выполняться
следующие условия.
СПЕЦИФИЧНОСТЬ
Специфичность — возможность удостоверить¬
ся, что аналитические процедуры приготовления
образца для испытаний и измерение позволяют до¬
стоверно определить металл (металлы) в присут¬
ствии других компонентов (например, газ-носитель,
примеси, матрица), которые могут присутствовать
при испытании.
Критерии приемлемости: методика должна
позволять однозначно оценивать каждый опреде¬
ляемый тяжелый металл в присутствии возможных
посторонних компонентов, включая другие тяже¬
лые металлы, компоненты матрицы и другие источ¬
ники интерференции; специфичность подтвержда¬
ется соответствием требований к правильности для
определяемого металла (металлов).
ДИАПАЗОН
Диапазон калибровки для каждого метал¬
ла должен лежать в линейном диапазоне метода;
испытуемые растворы, содержащие примесные
концентрации, выходящие за пределы диапазона
калибровки, могут быть разбавлены до концентра¬
ций, лежащих в пределах диапазона калибровки.
Критерий приемлемости: диапазон подтверж¬
дается соответствием требований к открываемое™.
ПРАВИЛЬНОСТЬ
Правильность проверяют с использованием
аттестованных станадртных веществ (СРМ) или пу¬
тем определения открываемости.
Открываемость. Открываемость может быть
определена на образце исследуемой субстанции,
к которой прибавляют известные количества стан¬
дартного образца металла (три уровня концентра¬
ции в диапазоне от 50 % до 150 % от определяемого
специфицированного предела, даже если исходная
концентрация стандартного образца соответствует
специфицированному значению), в трех повторах.
Критерий приемлемости: открываемость вве¬
денного количества составляет от 70 % до 150 %
для среднего из трех повторов на каждом уровне
концентрации.
ПОВТОРЯЕМОСТЬ
Испытуемые образцы: либо 6 независимых
образцов исследуемой субстанции с прибавлен¬
ным подходящим стандартным образцом на уровне
специфицированной концентрации, либо 3 уровня
концентрации, приготовленные в трех повторах.
Критерий приемлемости: относительное стан¬
дартное отклонение для обоих случаев должно
быть не более, чем указанное в таблице 2.4.27.-5.
Таблица 2.4.27.-5
Диапазон концен¬
траций металла,
мг/кг
Повторяемость
(К8Р), %
Внутрилабо-
раторная точ¬
ность (К8Р), %
0,01 — 1
20
32
> 1
10
16
ВНУТРИЛАБОРАТОРНАЯ ТОЧНОСТЬ
Должно быть установлено воздействие случай¬
ного события (внутрилабораторных отклонений) на
аналитическую точность метода. Допустимыми ис¬
пытаниями при установлении внутрилабораторной
точности является определение повторяемости ре¬
зультатов анализа в различные дни, при использо¬
вании разных приборов или при проведении испы¬
таний разными аналитиками. Для подтверждения
внутрилабораторной точности требуется только
одно из трех перечисленных выше испытаний.
Критерий приемлемости: относительное стан¬
дартное отклонение должно быть не более, чем ука¬
занное в таблице 2.4.27.-5.
ПРЕДЕЛ КОЛИЧЕСТВЕННОГО
ОПРЕДЕЛЕНИЯ
Определяют минимальную концентрацию, от¬
вечающую критерию приемлемости. Используют
результаты из испытания по определению правиль¬
ности.
Критерий приемлемости: предел количе¬
ственного определения должен быть ниже специ¬
фицируемого предела.
ПРЕДЕЛ ОБНАРУЖЕНИЯ (ПРИМЕНИМО
ТОЛЬКО К ПРЕДЕЛЬНЫМ ИСПЫТАНИЯМ)
Определяют минимальную концентрацию, да¬
ющую сигнал, явно отличимый от сигнала, получен¬
ного с контрольным раствором.
Критерий приемлемости: предел обнаруже¬
ния должен быть не выше, чем 0,1-кратная концен¬
трация специфицируемого предела.
2.5. МЕТОДЫ
КОЛИЧЕСТВЕННОГО
ОПРЕДЕЛЕНИЯ
07/2016:20501
2.5.1. КИСЛОТНОЕ ЧИСЛО
Кислотным числом /д называют массу (мг) ка¬
лия гидроксида, необходимую для нейтрализации
свободных кислот, содержащихся в 1 г испытуемого
образца.
10,00 г или указанное в частной статье коли¬
чество испытуемого образца (т, г) растворяют в
50 мл смеси из равных объемов 96 % спирта Р и
петролейного эфира РЗ, предварительно нейтра¬
лизованной 0,1 М раствором калия гидроксида
62
Гэсударственная фармакопея Республики Беларусь
или 0,1 М раствором натрия гидроксида, если в
частной статье нет других указаний, используя в
качестве индикатора 0,5 мл раствора фенолфта¬
леина Р1. При необходимости используют нагрева¬
ние до температуры около 90 °С для растворения
испытуемого образца. После растворения титруют
0,1 М раствором калия гидроксида или 0,1 М рас¬
твором натрия гидроксида до появления розового
окрашивания, не исчезающего в течение 15 с (л мл
титранта). В случае использования нагревания для
достижения растворения при титровании поддер¬
живают температуру около 90 °С.
. 5,611* л
07/2016:20512
2.5.12. ВОДА: ПОЛУМИКРОМЕТОД
Определение воды полумикрометодом основа¬
но на использовании количественной реакции воды
с диоксидом серы и йодом в подходящей безводной
среде в присутствии основания с достаточной бу¬
ферной емкостью.
Прибор
Прибор состоит из сосуда для титрования,
снабженного:
-двумя одинаковыми платиновыми электро¬
дами;
- герметичными входными отверстиями для
ввода растворителя и титранта;
- входным отверстием для доступа воздуха че¬
рез осушитель;
- входным отверстием для испытуемого об¬
разца с плотно прилегающей пробкой или, в случае
жидких образцов, с перегородкой.
Также могут присутствовать системы для по¬
дачи сухого азота или для удаления (отсасывания)
растворителей.
Титрование проводят в соответствии с ин¬
струкцией производителя, прилагаемой к прибору.
При проведении испытания уделяют внимание на
предотвращение воздействия на реактивы и рас¬
творители атмосферной влаги. Конечная точка
титрования определяется с помощью двух одина¬
ковых индикаторных электродов, подсоединенных
к электрическому источнику, поддерживающему
между электродами либо постоянную силу тока
,(2.2.65. Вольтаметрическое титрование), либо
постоянное напряжение (2.2.19. Амперометриче¬
ское титрование). В случае применения прямого
метода титрования (метод А) прибавление титранта
вызывает либо уменьшение напряжения (в случае
если поддерживается постоянная сила тока), либо
увеличение силы тока (в случае если поддержива¬
ется постоянное напряжение) до тех пор, пока не
будет достигнута точка эквивалентности. Проверка
пригодности прибора может проводиться с исполь¬
зованием подходящего сертифицированного стан¬
дартного материала (например, ФСО амоксицилли-
на тригидрата для проверки пригодности).
Установка титра. В сосуд для титрования по¬
мещают высушенный при необходимости мета¬
нол Р или другой растворитель, рекомендованный
производителем титранта. В зависимости от харак¬
теристики прибора удаляют остатки воды из изме¬
рительной ячейки или проводят предварительное
титрование. Прибавляют подходящее количество
воды в подходящем виде (вода Р или сертифици¬
рованный стандартный образец) и титруют при
перемешивании в течение необходимого времени.
Титр используемого титранта должен быть не ме¬
нее 80 % от указанного производителем. Титр уста¬
навливают перед первым использованием и затем
через подходящие интервалы времени.
Если нет других указаний в частной статье, ис¬
пользуют метод А.
Метод А. В сосуд для титрования помещают ме¬
танол Р или растворитель, который указан в частной
статье или рекомендован производителем титранта.
В зависимости от характеристики прибора удаляют
остатки воды из измерительной ячейки или проводят
предварительное титрование. Быстро вносят испы¬
туемый образец и титруют при перемешивании в те¬
чение необходимого для извлечения времени.
Метод В. В сосуд для титрования помещают
метанол Р или растворитель, который указан в
частной статье или рекомендован производителем
титранта. В зависимости от характеристики прибо¬
ра удаляют остатки воды из измерительной ячейки
или проводят предварительное титрование. Быстро
вносят измельченный до необходимой степени ис¬
пытуемый образец. Прибавляют точно отмеренный
объем титранта, взятого в избытке приблизительно
на 1 мл, или объем, указанный в частной статье.
Выдерживают в защищенном от света месте в те¬
чение 1 мин или в течение времени, указанного в
частной статье, при перемешивании. Избыток реак¬
тива титруют метанолом Р или указанным в част¬
ной статье растворителем, содержащим точно из¬
вестное количество воды.
Пригодность. Правильность использования
выбранного титранта для определения содержания
воды должна быть верифицирована для каждой
комбинации испытуемого образца, титранта и рас¬
творителя. Например, в случае образцов, содержа¬
щих воду в количестве от 2,5 мг до 25 мг, примени¬
ма следующая методика.
Проводят определение содержания воды с ис¬
пользованием выбранной системы реактив/раст-
воритель. Соответственно, в тот же самый сосуд
для титрования последовательно прибавляют в
подходящей форме известные количества воды Р
(по крайней мере 5 добавлений), соответствующие
от 50 % до 100 % от ее содержания в испытуемом
веществе, определяя содержание воды после каж¬
дого добавления. Рассчитывают процентное значе¬
ние открываемости (г) после каждого добавления
по формуле:
ш
г = 100—
где:
1/И, — масса прибавленной воды, мг;
У/2 — масса найденной воды, мг.
Рассчитывают среднее процентное значение
открываемости (г). Система реактив/растворитель
считается пригодной, если (г) лежит в интервале от
97,5% до 102,5%.
Строят линию регрессии. На оси х откладыва¬
ют совокупное количество добавленной воды, на
2.5.40. Метил-, этил- и изопропилтолуолсульфонат в субстанциях
63
оси у— сумму начального содержания воды, опре¬
деленной в веществе (М), и совокупного количества
воды, определенного после каждого добавления.
Рассчитывают наклон (Ь), пересечение с осью у (а)
и пересечение экстраполированной калибровочной
кривой с осью х (с/).
Рассчитывают процентные значения ошибок
(е1 и е2) по формулам:
= 100-
а-М
М ’
е2
= 100-
\б\-М
м
где:
а — величина отрезка, отсекаемого калибро¬
вочной прямой на оси ординат, в мг воды;
с1 — величина отрезка, отсекаемого калибро¬
вочной прямой на оси абсцисс, в мг воды;
М — содержание воды в испытуемом образце,
в мг воды.
Используемая система реактив/растворитель
считается пригодной, если:
- |е1| и |е2| не более 2,5 %;
- значение Ь находится в диапазоне от 0,975
до 1,025 (отклонение ±2,5 %).
07/2016:20540
2 5.40. МЕТИЛ-, ЭТИЛ- И ИЗОПРОПИЛ¬
ТОЛУОЛСУЛЬФОНАТ
В ФАРМАЦЕВТИЧЕСКИХ
СУБСТАНЦИЯХ
Следующий метод был валидирован для опре¬
деления метиловых, этиловых и изопропиловых
эфиров толуолсульфоновой кислоты (в диапазоне
концентраций от 0,2 ррт до 5 ррт) в сультамицил-
лина тозилате дигидрате.
Если данный метод предполагается использо¬
вать для определения эфиров толуолсульфоновой
кислоты в других фармацевтических субстанциях,
содержащих отличающиеся концентрации эфиров
толуолсульфоновой кислоты, концентрация испы¬
туемого раствора и раствора сравнения должна
быть соответствующим образом скорректирована и
проведена подходящая валидация.
МЕТОДИКА
Парофазная газовая хроматография {2.2.28),
спаренная с масс-спектрометрией (2.2.43). Испы¬
туемый раствор и раствор сравнения готовят
непосредственно перед использованием.
Смесь растворителей. Вода Р — ацетони¬
трил Р (20:80, об/об). Используемый ацетонитрил
должен иметь соответствующую степень чи¬
стоты.
Раствор А. 30 мг натрия тиосульфата безво¬
дного Р и 60,0 г натрия йодида Р растворяют при
помощи ультразвука в воде Р и доводят до объема
50,0 мл этим же растворителем.
Раствор внутреннего стандарта. Юмкл
ФСО бутилметансульфоната (БМС) доводят сме¬
сью растворителей до объема 10,0 мл. 20 мкл по¬
лученного раствора доводят смесью растворителей
до объема 100,0 мл.
Контрольный раствор. Во флакон для парофаз¬
ного пробоотборника помещают 0,50 мл раствора А и
0,50 мл раствора внутреннего стандарта, немедленно
закрывают флакон силиконовой мембраной с полите-
трафторэтиленовым покрытием и обкатывают алюми¬
ниевым колпачком.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца помещают во флакон для парофазного пробо¬
отборника вместимостью 20 мл, прибавляют 0,50 мл
раствора А и 0,50 мл раствора внутреннего стандар¬
та, немедленно закрывают флакон силиконовой мем¬
браной с политетрафторэтиленовым покрытием и об¬
катывают алюминиевым колпачком. В ходе реакции
дериватизации может образоваться осадок, кото¬
рый не влияет на достоверность количественного
определения.
Раствор сравнения (а). По 25,0 мг метилтолу-
олсульфоната Р (МТС), этилтолуолсульфоната Р
(ЭТО) и изопропилтолуолсульфоната Р (ИТС) раст¬
воряют в толуоле Р и доводят до объема 5,0 мл этим
же растворителем. 50 мкл полученного раствора до¬
водят раствором внутреннего стандарта до объема
25,0 мл.
Раствор сравнения (Ь). 40 мкл раствора срав¬
нения (а) доводят раствором внутреннего стандарта
до объема 20,0 мл. 0,50 мл полученного раствора и
0,50 мл раствора А помещают во флакон для паро¬
фазного пробоотборника вместимостью 20 мл, немед¬
ленно закрывают флакон силиконовой мембраной с
политетрафторэтиленовым покрытием и обкатывают
алюминиевым колпачком.
Раствор сравнения (с). 500 мкл раствора срав¬
нения (а) доводят раствором внутреннего стандарта
до объема 20,0 мл. 0,50 мл полученного раствора и
0,50 мл раствора А помещают во флакон для паро¬
фазного пробоотборника вместимостью 20 мл, немед¬
ленно закрывают флакон силиконовой мембраной с
политетрафторэтиленовым покрытием и обкатывают
алюминиевым колпачком.
Условия хроматографирования:
- колонка: кварцевая капиллярная, длиной 30 м
и внутренним диаметром 0,25 мм, заполненная по¬
лярно деактивированным полиэтиленгликолем Р
(толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р
(использование подводящего лайнера без стеклова¬
ты значительно снижает эффект переноса между
вводами пробы);
- скорость потока: 0,5 мл/мин;
- деление потока: 1:20;
- условия для парофазного пробоотборника:
- температура уравновешивания: 60 °С;
- время уравновешивания: 30 мин;
- температура автоматической линии: 120 °С;
-температура:
Время
(мин)
Температура (°С)
Колонка
0—1
40
1—10
40—>130
Блок ввода проб
220
Детектор: автоматическая
линия
280
источник
250
анализатор
200
В конце анализа поднимают температуру колонки до
240 °С и поддерживают ее в течение 7 мин
64
Гэсударственная фармакопея Республики Беларусь
- детектор: масс-спектрометр, описанный
ниже; корректируют настройку детектора для со¬
ответствия критериям пригодности системы; аль¬
тернативно может быть использован подходящий
детектор с электронной ловушкой;
- квадрупольный масс-спектрометр, снаб¬
женный системой электронной ударной иони¬
зации (70 эВ);
-для режима фрагментометрии (однои-
онный мониторинг (81М)) параметры масс-
спектрометра устанавливают следующим об¬
разом:
Вещество
Ион для количе¬
ственного опре¬
деления
(т/г)
Ион для квали¬
фикации
(т/г)
Бутилйодид (ВиГ)*
184
127
Метилйодид (Ме1)*
142
127
Этилйодид (Е11)*
156
127
Изопропилйодид
(/Рг/)*
170
127
* Получены из БМС, МТС, ЭТС и ИТС в результате реак¬
ции дериватизации
- объем вводимой пробы: по 1 мл равновес¬
ной газовой фазы испытуемого раствора, раствора
сравнения (Ь), раствора сравнения (с) и контроль¬
ного раствора.
Относительное удерживание (по отноше¬
нию к внутреннему стандарту (бутилйодид) (время
удерживания — около 8,5 мин)): метилйодид —
около 0,51; этилйодид — около 0,63; изопропилйо-
дид — около 0,68.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками
этилйодида и изопропилйодида на хроматограмме
раствора сравнения (с);
- отношение сигнал/шум: не менее 10 для пи¬
ков каждого алкилйодида на хроматограмме раст¬
вора сравнения (Ь).
Содержание каждого алкилтолуолсульфоната
(в частях на миллион, ррт) рассчитывают по фор¬
муле:
А21, Щ С 0,05
Д/2И/2 -
где:
/\1 — площадь пика каждого алкилйодида на
хроматограмме раствора сравнения (с);
А2 — площадь пика каждого алкилйодида на
хроматограмме испытуемого раствора;
С — концентрация каждого эфира, в процен¬
тах;
А, — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (с);
/2 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора;
— масса навески каждого эфира, взятой
для приготовления раствора сравнения (а), в мг;
]/У2 — масса навески испытуемого образца,
взятой для приготовления испытуемого раствора,
в мг;
0,05 — коэффициент разведения.
07/2016:20540
2.5.41. МЕТИЛ-, ЭТИЛ- И ИЗОПРОПИЛ¬
БЕНЗОЛ СУЛЬФОНАТ
В ФАРМАЦЕВТИЧЕСКИХ
СУБСТАНЦИЯХ
Следующий метод был валидирован для опре¬
деления метиловых, этиловых и изопропиловых
эфиров бензолсульфоновой кислоты (в диапазоне
концентраций от 2,5 ррт до 40 ррт) в амлодипине
бесилате.
Если данный метод предполагается использо¬
вать для определения эфиров бензолсульфоновой
кислоты в других фармацевтических субстанциях, со¬
держащих отличающиеся концентрации эфиров бен¬
золсульфоновой кислоты, концентрация испытуемого
раствора и раствора сравнения должна быть соот¬
ветствующим образом скорректирована и проведена
подходящая валидация.
Данный метод не подходит для клопидогреля
бесилата, так как было установлено, что в процессе
хроматографического определения образуется ме-
тилбензолсульфонат, как артефакт, появляющийся
вследствие его деструкции.
МЕТОДИКА
Парофазная газовая хроматография (2.2.28), спа¬
ренная с масс-спекгрометрией (2.2.43). Испытуемый
раствор и раствор сравнения готовят непосред¬
ственно перед использованием.
Смесь растворителей. Вода Р — ацетонитрил Р
(20:80, об/об). Используемый ацетонитрил должен
иметь соответствующую степень чистоты.
Раствор А. 30 мг натрия тиосульфата безводно¬
го Р и 60,0 г натрия йодида Р растворяют при помощи
ультразвука в воде Р и доводят до объема 50,0 мл этим
же растворителем.
Раствор внутреннего стандарта. Юмкл ФСО
бутилметансульфоната (БМС) доводят смесью раст¬
ворителей до объема 10,0мл. 20мкл полученного
раствора доводят смесью растворителей до объема
100.0 мл.
Контрольный раствор. Во флакон для парофаз¬
ного пробоотборника помещают 0,50 мл раствора А и
0,50 мл раствора внутреннего стандарта, немедленно
закрывают флакон силиконовой мембраной с полите-
трафторэтиленовым покрытием и обкатывают алюмини¬
евым колпачком.
Испытуемый раствор. 25,0 мг испытуемого образ¬
ца помещают во флакон для парофазного пробоотбор¬
ника вместимостью 20 мл, прибавляют 0,50 мл раствора
А и 0,50 мл раствора внутреннего стандарта, немедлен¬
но закрывают флакон силиконовой мембраной с полите-
трафторэтиленовым покрытием и обкатывают алюмини¬
евым колпачком. В ходе реакции дериватизации может
образоваться осадок, который не влияет на достовер¬
ность количественного определения.
Раствор сравнения (а). По 25,0 мг метилбен-
золсульфоната Р (МБС), этилбензолсульфоната Р
(ЭБС) и изопропилметансульфоната Р (ИМС) раст¬
воряют в толуоле Р и доводят до объема 5,0 мл этим
же растворителем. 50 мкл полученного раствора до¬
водят раствором внутреннего стандарта до объема
25.0 мл.
Раствор сравнения (Ь). 40 мкл раствора срав¬
нения (а) доводят раствором внутреннего стандарта
г
2.6.31. Микробиологические испытания лекарственных средств
65
до объема 20,0 мл. 0,50 мл полученного раствора и
0,50 мл раствора А помещают во флакон для паро¬
фазного пробоотборника вместимостью 20 мл, немед¬
ленно закрывают флакон силиконовой мембраной с
политетрафторэтиленовым покрытием и обкатывают
алюминиевым колпачком.
Раствор сравнения (с). 500 мкл раствора срав¬
нения (а) доводят раствором внутреннего стандарта
до объема 20,0 мл. 0,50 мл полученного раствора и
0,50 мл раствора А помещают во флакон для паро¬
фазного пробоотборника вместимостью 20 мл, немед¬
ленно закрывают флакон силиконовой мембраной с
политетрафторэтиленовым покрытием и обкатывают
алюминиевым колпачком.
Условия хроматографирования:
- колонка: кварцевая капиллярная, длиной 30 м
и внутренним диаметром 0,25 мм, заполненная по¬
лярно деактивированным полиэтиленгликолем Р
(толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р
(использование подводящего лайнера без стеклова¬
ты значительно снижает эффект переноса между
вводами пробы)',
- скорость потока: 0,5 мл/мин;
- деление потока: 1:20;
- условия для парофазного пробоотборника:
- температура уравновешивания: 60 °С;
- время уравновешивания: 30 мин;
- температура автоматической линии: 120 °С;
- температура:
Время (мин)
Температура
(•С)
Колонка
0—1
40
1—10
о
со
7
о
■'Г
Блок ввода проб
220
Детектор: автоматическая
280
линия
источник
250
анализатор
200
В конце анализа поднимают температуру колонки до
240 °С и поддерживают ее в течение 7 мин
- детектор: масс-спектрометр, описанный
ниже; корректируют настройку детектора для соот¬
ветствия критериям пригодности системы; альтерна¬
тивно может быть использован подходящий детектор
с электронной ловушкой;
- квадрупольный масс-спектрометр, снабжен¬
ный системой электронной ударной ионизации
(70 эВ);
-для режима фрагментометрии (одноионный
мониторинг (31М)) параметры масс-спектрометра
устанавливают следующим образом:
Вещество
Ион для количе¬
ственного опре¬
деления
(т/г)
Ион для квали¬
фикации
(т/г)
Бутилйодид (Ви1)*
184
127
Метилйодид (МеГ)*
142
127
Этилйодид (ЕИ)*
156
127
Изопропилйодид
С1РПГ
170
127
* Получены из БМС, МБС, ЭБС и ИМС в результате реак¬
ции дериватизации
- объем вводимой пробы: по 1 мл равновес¬
ной газовой фазы испытуемого раствора, раствора
сравнения (Ь), раствора сравнения (с) и контроль¬
ного раствора.
Относительное удерживание (по отношению к
внутреннему стандарту (бутилйодид) (время удер¬
живания — около 8,5 мин)): метилйодид — около
0,51; этилйодид — около 0,63; изопропилйодид —
около 0,68.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками
этилйодида и изопропилйодида на хроматограмме
раствора сравнения (с);
- отношение сигнал/шум: не менее 10 для пи¬
ков каждого алкилйодида на хроматограмме раст¬
вора сравнения (Ь).
Содержание каждого алкилбензолсульфоната
(в частях на миллион, ррт) рассчитывают по фор¬
муле:
0,05
АЛ Щ 9
где:
А1 — площадь пика каждого алкилйодида на хро¬
матограмме раствора сравнения (с);
А2 — площадь пика каждого алкилйодида на хро¬
матограмме испытуемого раствора;
С — концентрация каждого эфира, в процентах;
!у — площадь пика внутреннего стандарта на хро¬
матограмме раствора сравнения (с);
/2 — площадь пика внутреннего стандарта на хро¬
матограмме испытуемого раствора;
^ — масса навески каждого эфира, взятой для
приготовления раствора сравнения (а), в мг;
IЫ2 — масса навески испытуемого образца, взя¬
той для приготовления испытуемого раствора, в мг;
0,05 — коэффициент разведения.
2.6. БИОЛОГИЧЕСКИЕ
ИСПЫТАНИЯ
07/2016:20631
2.6.31. МИКРОБИОЛОГИЧЕСКИЕ
ИСПЫТАНИЯ ЛЕКАРСТВЕННЫХ
СРЕДСТВ РАСТИТЕЛЬНОГО
ПРОИСХОЖДЕНИЯ
ДЛЯ ВНУТРЕННЕГО
ПРИМЕНЕНИЯ И ЭКСТРАКТОВ,
ИСПОЛЬЗУЮЩИХСЯ для их
ПРИГОТОВЛЕНИЯ
1. ИСПЫТАНИЯ ПО ОПРЕДЕЛЕНИЮ
МИКРОБНОГО ЧИСЛА
Общее количество аэробов (ОКА). Проводят
подсчет, как описано в общей статье 2.6.12.
Общее количество грибов (ОКГ)- Проводят под¬
счет, как описано в общей статье 2.6.12. В отношении про¬
дуктов, описанных в общей статье 5.1.8, с высоким при¬
родным содержанием бактерий может быть использован
декстрозный агар Сабуро, содержащий антибиотики.
66
Гэсударственная фармакопея Республики Беларусь
2. ИСПЫТАНИЕ НА НАЛИЧИЕ
СПЕЦИФИЧЕСКИХ МИКРООРГАНИЗМОВ
2-1. ВВЕДЕНИЕ
Испытания, описанные ниже, позволяют опре¬
делить отсутствие или ограниченное наличие спец¬
ифических микроорганизмов, которые могут быть
обнаружены при описанных условиях. Испытание
в первую очередь предназначено для определения
соответствия продукта, субстанции или лекарствен¬
ного средства (далее — продукта) требованиям ут¬
вержденной спецификации по микробиологической
чистоте. При использовании испытаний для этих
целей следуют приведенным ниже инструкциям,
включая количество образцов для испытания и
оценку результатов.
Могут быть использованы и альтернативные
методы определения микробиологической чистоты,
включая автоматизированные методы, при условии
подтверждения их эквивалентности методам, опи¬
санным в Фармакопее.
2-2. ОБЩИЕ ИСПЫТАНИЯ
Приготовление образца проводят, как описано
в общей статье 2.6.12.
Если испытуемый продукт обладает антими¬
кробным действием, его удаляют или нейтрализу¬
ют, как описано в общей статье 2.6.12.
Если для приготовления образца используют¬
ся поверхностно-активные вещества, отсутствие их
токсичности в отношении микроорганизмов, а также
их совместимость с используемыми инактиватора¬
ми должна быть продемонстрирована, как описано
в общей статье 2.6.12.
2-3. РОСТОВЫЕ И ИНГИБИТОРНЫЕ
СВОЙСТВА СРЕД, ПРИГОДНОСТЬ
ИСПЫТАНИЯ И ОТРИЦАТЕЛЬНЫЕ
КОНТРОЛИ
Способность испытания к определению микро¬
организмов в присутствии испытуемого продукта
должна быть установлена. При внесении изменений
в проведение испытания или в продукт, которые мо¬
гут оказать влияние на результат испытания, долж¬
на быть подтверждена пригодность испытания.
2-3-1. ПРИГОТОВЛЕНИЕ ТЕСТ-ШТАММОВ
Используют стандартизованные стабильные
суспензии тест-штаммов или готовят их, как описа¬
но ниже. Способы сохранения посевного материала
культуры (система посевного материала) использу¬
ют таким образом, чтобы живые микроорганизмы,
используемые для инокуляции, были не более чем
пятым пассажем от главного посевного материала.
2-3-1-1. Аэробные микроорганизмы. Каждый
тест-штамм бактерии выращивают отдельно на со¬
ево-казеиновом бульоне или соево-казеиновым
агаре при температуре от 30 °С до 35 °С в течение
(18—24)ч.
- 81арЬу!ососсиз аигеиз, например АТСС 6538,
1МС1МВ 9518, С1Р 4.83 или ЫВКС 13276;
- Рзеидотопаз аегидтоза, например АТСС 9027,
1ЧС1МВ 8626, С1Р 82.118 или ЫВРС 13275;
- ЕзсНепсЫа со//, например АТСС 8739, ЫС1МВ
8545, С1Р 53.126 или ЫВКС 3972;
- 8а1топе11а еп1епса зиЬзр. еп(епса зегоуаг
ТурЫтипит, например АТСС 14028 или, как альтер¬
нативный, 5. ел/е/7са зиЬзр. ел/елса зегоуаг АЬопу, на¬
пример ИВКС 100797,1ЧСТС 6017 или С1Р 80.39;
- ВасШиз зиЫШз, например АТСС 6633, 1ЧС1МВ
8054, С1Р 52.62 или ИВКС 3134.
Для приготовления тест-суспензий используют
забуференный раствор натрия хлорида — пептона
рН 7,0 или фосфатный буферный раствор рН 7,2.
Суспензии используют в течение 2 ч или, при хра¬
нении при температуре от 2 °С до 8 °С, в течение
24 ч. Вместо приготовления и дальнейшего раз¬
ведения свежеприготовленной суспензии вегета¬
тивных клеток В. зиЫШз может быть приготовлена
суспензия стабильных спор, определенный объем
которой используется для посева. Суспензия ста¬
бильных спор может поддерживаться при темпе¬
ратуре от 2 °С до 8 °С в течение валидированного
срока хранения.
2-3-2. ОТРИЦАТЕЛЬНЫЙ КОНТРОЛЬ
Для верификации условий испытания выпол¬
няется отрицательный контроль с использовани¬
ем выбранного растворителя вместо испытуемого
препарата. Рост организмов наблюдаться не дол¬
жен. Отрицательный контроль проводится также
при испытании продукта, как описано в п. 2-4. При
неудовлетворительном отрицательном контроле
требуется проведение расследования.
2-3-3. РОСТОВЫЕ И ИНГИБИРУЮЩИЕ СВОЙ¬
СТВА СРЕД
Испытанию подлежит каждая серия готовой к
использованию коммерческой питательной среды,
а также каждая серия среды, приготовленной или
из сухой среды, или из описанных компонентов.
Проверку соответствующих свойств сред про¬
водят, как описано в таблице 2.6.31.-1.
Испытание на ростовые свойства, жидкие
питательные среды: в порцию соответствующей
среды вносят небольшое количество (не более 100
КОЕ) соответствующего микроорганизма. Инку¬
бируют при предписанной температуре в течение
наименьшего периода времени, предписанного
испытанием. Соево-казеиновый бульон инкубиру¬
ют при температуре от 30 °С до 35 °С в течение
не более 3 дней. Наблюдается ясно видимый рост
микроорганизмов, сопоставимый с ранее получен¬
ным ростом на ранее испытанной и разрешенной к
использованию серии среды.
Испытание на ростовые свойства, твер¬
дые питательные среды: посев выполняют по¬
верхностным способом, внося на каждую чашку
небольшое количество (не более 100 КОЕ) соот¬
ветствующего микроорганизма. Инкубируют при
предписанной температуре в течение наименьше¬
го периода времени, предписанного испытанием.
Наблюдается ясно видимый рост микроорганиз¬
мов, сопоставимый с ранее полученным ростом на
ранее испытанной и разрешенной к использованию
серии среды.
Испытание на ингибирующие свойства,
жидкие или твердые среды: инокулируют соот¬
ветствующую среду не менее 100 КОЕ соответ¬
ствующего микроорганизма. Инкубируют при пред¬
писанной температуре в течение наибольшего
периода времени, предписанного испытанием. Не
должен наблюдаться рост микроорганизмов.
Испытание на дифференцирующие свой¬
ства: посев выполняют поверхностным способом,
внося на каждую чашку небольшое количество (не
более 100 КОЕ) соответствующего микроорганиз¬
ма. Инкубируют при предписанной температуре
в течение времени, находящегося в диапазоне,
2.6.31. Микробиологические испытания лекарственных средств
67
Таблица 2.6.31.-1
Ростовые, ингибирующие и дифференцирующие свойства сред
Среда
Свойство
Испытуемые штаммы
Соево-казеиновый бульон
Обеспечение роста
3. аигеиз
Р. аегидтоза
В. зиЫШз
Испытание на грамотрица-
тельные бактерии, устойчи¬
вые к желчи
Бульон Мосселя для обогаще-
ния энтеробактерий
Обеспечение роста
Е. соН
Р. аегидтоза
Ингибирование
8. аигеиз
Агаризованная среда с глюко¬
зой, кристаллическим фиоле¬
товым, нейтральным красным
и желчью
Обеспечение роста + диффе¬
ренцирующие свойства
Е. соН
Р. аегидтоза
Соево-казеиновый бульон
Обеспечение роста
Е. соН
Р. аегидтоза
В. зиЫШз
Испытание на ЕзсЬепсЫа соН
Бульон Мак-Конки
Обеспечение роста
Е. соН
Ингибирование
8. аигеиз
Агар Мак-Конки
Обеспечение роста + диффе¬
ренцирующие свойства
Е. соН
Забуференная пептонная
среда
Обеспечение роста
8. еп!епса зиЬзр. ел/елса
зегоуаг ТурЫтипит или 5.
еп!епса зиЬзр. еп(епса зегоуаг
АЬопу
Испытание на 8а1топе11а
Соевый бульон Раппапорта-
Вассилиадиса для обогаще¬
ния 8а!топеЧа
Обеспечение роста
5. ел/елса зиЬзр. ел/елса
зегоуаг ТурЫтипит или 5.
ел/елса зиЬзр. ел/елса зегоуаг
АЬопу
Ингибирование
5. аигеиз
Агар на основе ксилозы,
лизина и дезоксихолата
Обеспечение роста + диффе¬
ренцирующие свойства
8. ел/елса зиЬзр. ел/елса
зегоуаг ТурЫтипит или 5.
ел/елса зиЬзр. еп(епса зегоуаг
АЬопу
предписанном испытанием. Колонии сопоставимы
по внешнему виду и наблюдающимся реакциям с
результатами, полученными на ранее испытанной
и разрешенной к использованию серии среды.
2-3-4. ПРИГОДНОСТЬ МЕТОДИКИ
Для каждого испытуемого продукта выполняют
подготовку образца, как описано в соответствую¬
щем параграфе п. 2-4. Каждый тест-штамм прибав¬
ляют во время смешивания в предписанную пита¬
тельную среду (соево-казеиновый бульон или за-
буференную пептонную среду). Для количествен¬
ного определения грамотрицательных бактерий,
устойчивых к желчи, посев Е. со// и Р. аегидтоза
осуществляют отдельно. Для испытания на Е. соН
и 8а1топе11а посев целевого микроорганизма осу¬
ществляют отдельно.
При наличии у продукта антимикробной актив¬
ности необходимо внесение изменений в методику
испытаний (см. 4-5-3 общей статьи 2.6.12).
Если антимикробная активность продукта в
отношении микроорганизмов, для оценки которых
это испытание предназначено, не может быть ней¬
трализована, принято считать, что ингибирован¬
ный микроорганизм не будет присутствовать в про¬
дукте.
2-3-4-1. Испытание на отсутствие. Использу¬
ют количество микроорганизмов, эквивалентное не
более 100 КОЕ в инокулированном испытуемом об¬
разце. Проводят испытание, как описано в соответ¬
ствующем параграфе п. 2-4, в течение наименьше¬
го периода времени, предписанного испытанием.
Целевые (специфицированные) микроорганизмы
должны обнаруживаться реакциями идентифика¬
ции, описанными в п. 2-4.
2-3-4-2. Количественное определение. По-
луколичественное испытание (метод наиболее ве¬
роятного числа).
Используют количество микроорганизмов, эк¬
вивалентное не более 100 КОЕ в 1 г или 1 мл про¬
дукта. Проводят испытание, как описано в соответ¬
ствующем параграфе п. 2-4, в течение наименьше¬
го периода времени, предписанного испытанием.
Разведения, соответствующие 0,1 г или 0,1 мл,
должны быть положительными.
2-4. ИСПЫТАНИЕ ПРОДУКТА
2-4-1. ГРАМОТРИЦАТЕЛЬНЫЕ БАКТЕРИИ, ТОЛЕ¬
РАНТНЫЕ К ЖЕЛЧИ
2-4-1-1. Метод подсчета. Полуколичественный
метод (метод наиболее вероятного числа).
2-4-1-1-1. Приготовление образцов и предва¬
рительная инкубация. Готовят образец с исполь¬
зованием разведения 1:10 из не менее 1 г испыту¬
емого продукта, как указано в общей статье 2.6.12,
но используя бульон на основе гидролизатов казе¬
ина и соевых бобов в качестве разбавителя, пере¬
мешивают и инкубируют при температуре от 20 °С
до 25 °С в течение времени, достаточного для вос¬
становления бактерий, но недостаточного для зна¬
чительного увеличения числа микроорганизмов (от
2 ч до 3 ч).
2-4-1-1-2. Селекция и пересев. Инокулируют
подходящие количества обогатительного бульона
Мосселя для энтеробактерий приготовленным рас¬
твором, описанным выше, и/или, в зависимости
от предельного содержания, предусмотренного
для испытуемого продукта, тремя из четырех раз-
ведений, содержащих соответственно 0,1 г, 0,01 г,
0,001 г и 0,0001 г (или 0,1 мл, 0,01 мл, 0,001 мл
68
Гэсударственная фармакопея Республики Беларусь
и 0,0001 мл) испытуемого продукта. Инкубиру¬
ют при температуре от 30 °С до 35 °С в течение
(24—48) ч. Пересевают на чашку с агаризованной
средой с глюкозой, кристаллическим фиолетовым,
нейтральным красным и желчью. Инкубируют при
температуре от 30 °С до 35 °С в течение (18—24) ч.
2-4-1-1-3. Оценка результатов. Рост колоний
является положительным результатом. Отмечают
минимальное количество, которое дает положи¬
тельный результат, и максимальное количество,
которое дает отрицательный результат.
Определяют по таблице 2.6.31 .-2 наиболее ве¬
роятное число бактерий.
Таблица 2.6.31 .-2
Оценка результатов
Результаты для соответствующего коли¬
чества продукта
НВЧ в грамме
или миллили-
тре испытуемо¬
го продукта
0,1 г
или
0,1 мл
0,01 г
или
0,01 мл
0,001 г
или
0,001 мл
0,0001 г
или
0,0001 мл
+
+
+
+
> 104
+
+
+
-
< 104 и > 103
+
+
-
-
< 103 и > 102
+
-
-
-
< 102и> 10
-
-
-
-
< 10
2-4-2. Е8СНЕР1СН1А СОИ
2-4-2-1. Испытание на отсутствие
2-4-2-1-1. Приготовление образцов и пред¬
варительная инкубация. Готовят образец с ис¬
пользованием разведения 1:10 из не менее 1 г ис¬
пытуемого продукта, как указано в общей статье
2.6.12, и используют 10 мл или количество, соот¬
ветствующее 1 г или 1 мл, для инокуляции подхо¬
дящего количества (определенного как указано в
подразделе 2-3-4) бульона на основе гидролизатов
казеина и соевых бобов, перемешивают и инкуби¬
руют при температуре от 30 °С до 35 9С в течение
(18—24) ч.
2-4-2-1-2. Селекция и пересев. Контейнер
встряхивают, переносят 1 мл бульона на основе
гидролизотов казеина и соевых бобов в 100 мл бу¬
льона Макконки и инкубируют при температуре от
42 °С до 44 °С в течение (24—48) ч. Пересевают на
чашку с агаризованной средой Макконки и инкуби¬
руют при температуре от 30 °С до 35 °С в течение
(18—72) ч.
2-4-2-1-3. Оценка результатов. Рост колоний
указывает на возможное присутствие Е. соН. Ре¬
зультат подтверждается идентификационными ис¬
пытаниями.
Продукт выдерживает испытание, если подоб¬
ные колонии не обнаруживаются или испытания
идентификации дают отрицательный результат.
2-4-2-2. Метод подсчета. Полуколичествен-
ный метод (метод наиболее вероятного числа).
2-4-2-2-1. Приготовление образцов и предва¬
рительная инкубация. Готовят образец с исполь¬
зованием разведения 1:10 из не менее 1 г испыту¬
емого продукта, как указано в общей статье 2.6.12,
и используют количество, соответствующее 0,1 г,
0,01 г и 0,001 г (или 0,1 мл, 0,01 мл и 0,001 мл),
для инокуляции подходящего количества (опреде¬
ленного как указано в подразделе 2-3-4) бульона
на основе гидролизатов казеина и соевых бобов,
перемешивают и инкубируют при температуре от
30 °С до 35 °С в течение (18—24) ч.
2-4-2-2-2. Селекция и пересев. Контейнер
встряхивают, переносят 1 мл бульона на основе
гидролизатов казеина и соевых бобов в 100 мл бу¬
льона Макконки и инкубируют при температуре от
42 °С 44 °С в течение (24—48) ч. Пересевают на
чашку с агаризованной средой Макконки и инкуби¬
руют при температуре от 30 °С до 35 °С в течение
(18—72)ч.
2-4-2-2-3. Оценка результатов. Рост колоний
указывает на возможное присутствие Е. соН. Ре¬
зультат подтверждается идентификационными ис¬
пытаниями.
Отмечают минимальное количество, которое
дает положительный результат, и максимальное
количество, которое дает отрицательный резуль¬
тат.
Определяют по таблице 2.6.31 .-3 наиболее ве¬
роятное число бактерий.
Таблица 2.6.31.-3
Оценка результатов
Результаты для соответствую¬
щего количества продукта
НВЧ в грамме или
миллилитре испыту-
емого продукта
0,1 г или
0,1 мл
0,01 г или
0,01 мл
0,001 г
или
0,001 мл
+
+
+
> 103
+
+
-
< 103 и > 102
+
-
-
< 102 и > 10
-
-
-
<10
2-4-3. ЗАЬМОЫЕНА
2-4-3-1. Испытание на отсутствие
2-4-3-1-1. Приготовление образцов и предва¬
рительная инкубация. Используют 25 г или 25 мл
испытуемого продукта для инокуляции 225 мл бу¬
феризованной среды на основе пептона и переме¬
шивают (например, гомогенизируют с использова¬
нием блендера), и инкубируют при температуре от
30 °С до 35 °С в течение (18—24) ч.
2-4-3-1-2. Селекция и пересев. Переносят
0,1 мл буферизованной среды на основе пептона в
10 мл обогатительного питательного бульона Рап-
папорта Вассилиадиса для 8а1топе11а и инкубиру¬
ют при температуре от 30 °С до 35 °С в течение
(18—24) ч. Пересевают на чашку с агаризованной
средой на основе ксилозы, лизина, дезоксихолата.
Инкубируют при температуре от 30 °С до 35 °С в
течение (18—48) ч.
2-4-3-1-3. Оценка результатов. На возмож¬
ное присутствие За1топе11а указывает рост хорошо
развитых красных колоний или красных колоний
с черным центром. Присутствие 8а1топе11а под¬
тверждается в идентификационных испытаниях.
Продукт выдерживает испытание, если коло¬
нии описанного типа не обнаруживаются или ис¬
пытания идентификации дают отрицательный ре¬
зультат.
2.8.21. Испытание лекарственного растительного сырья на наличие аристолохиевых кислот 69
Следующий подраздел приводится для ин¬
формации
РЕКОМЕНДУЕМЫЕ РАСТВОРЫ
И ПИТАТЕЛЬНЫЕ СРЕДЫ
Растворы и питательные среды, упомянутые в
этой статье и описанные в общей статье 2.6.13, а так¬
же приведенная ниже забуференная пептонная среда
являются пригодными для целей их использования в
данном разделе. Могут быть использованы и другие
среды при условии демонстрации их пригодности.
Буферизованная среда на основе пептона
Калия дигидрофосфат
1,5 г
Д и натрия гидрофосфат додекагидрат
9,0 г
Натрия хлорид
5,0 г
Пептон
10,0 г
Вода очищенная
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло 7,0±0,2 при
температуре 25 °С. Стерилизуют в автоклаве в со¬
ответствии с валидированной процедурой.
2.8. МЕТОДЫ ФАРМАКОГНОЗИИ
07/2016:20802
2.8.2. ПРИМЕСИ
Допустимыми примесями в лекарственном рас¬
тительном сырье являются примеси, включающие
в себя:
1) несырьевые части растения: части данного
лекарственного растения, не являющиеся лекар¬
ственным растительным сырьем;
2) посторонние примеси: примеси, не относя¬
щиеся к данному лекарственному растению и име¬
ющие растительное ^органические примеси — не¬
ядовитые части других растений, солома, сено и
т.п.#) или минеральное ^минеральные примеси —
земля, песок, камешки#) происхождение.
Лекарственные средства на основе лекар¬
ственного растительного сырья (лекарственное
растительное сырье цельное или измельченное
фасованное, сборы, растительные чаи) не должны
обладать устойчивым посторонним запахом, со¬
держать такие примеси, как стекло, помет грызунов
и птиц, ядовитые растения и т.п.; кроме этого, не
должны обнаруживаться признаки гниения, плесе¬
ни, амбарных вредителей и других примесей живот¬
ного происхождения.#
СУХОЕ ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ
СЫРЬЕ
Отбор проб и подготовка образцов. Исполь¬
зуют требования общей статьи 2.8.20. Лекарствен¬
ное растительное сырье: отбор проб.
Определение содержания примесей. От
100 г до 500 г испытуемого образца или минималь¬
ное количество, указанное в частной статье, рассы¬
пают тонким слоем. Осматривают невооруженным
глазом или с помощью лупы (6*). Каждую группу
примесей отделяют, взвешивают и рассчитывают их
процентное содержание.
СВЕЖЕЕ ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ
СЫРЬЕ
В случае невозможности применения требований
общей статьи 2.8.20. Лекарственное растительное
сырье: отбор проб, используют один из двух соответ¬
ствующих методов: метод А, если испытание может
быть проведено на всей серии сырья; метод В, если
испытание не может быть проведено на всей серии
сырья.
МЕТОДА
Отбор проб и подготовка образцов. Испыта¬
ние проводят на всей серии.
Определение содержания примесей. Рассыпа¬
ют всю серию тонким слоем и проводят определение
примесей путем осмотра невооруженным глазом или
с помощью лупы (6х). Каждую группу посторонних
примесей отделяют, взвешивают и рассчитывают их
процентное содержание.
МЕТОД В
Отбор проб и подготовка образцов. Если не¬
возможно провести испытание на всей серии, посту¬
пают как описано далее.
Объединенная проба. Подготавливают объеди¬
ненную пробу, как указано в общей статье 2.8.20. Ле¬
карственное растительное сырье: отбор проб.
Испытуемый образец. Используют объединен¬
ную пробу или, если масса объединенной пробы
больше 1 кг, уменьшают ее подходящим способом,
позволяющим сохранить репрезентативность объеди¬
ненной пробе.
Определение содержания примесей. Полученный
образец или минимальное количество, указанное в
частной статье, рассыпают тонким слоем. Осматрива¬
ют невооруженным глазом или с помощью лупы (6*).
Каждую группу посторонних примесей отделяют, взве¬
шивают и рассчитывают их процентное содержание.
07/2016:20821
2.8.21. ИСПЫТАНИЕ
ЛЕКАРСТВЕННОГО
РАСТИТЕЛЬНОГО СЫРЬЯ
НА НАЛИЧИЕ
АРИСТОЛОХИЕВЫХ КИСЛОТ
ПРЕДУПРЕЖДЕНИЕ: аристолохиевые кисло¬
ты представляют собой высокотоксичные веще¬
ства, обладающие канцерогенным действием. Во
всех возможных случаях выполнение всех манипуля¬
ций с этими веществами должно проводиться в вы¬
тяжном шкафу. Когда вещество находится в сухой
форме, ввиду особых электростатических свойств
и тенденции проникать через рабочие зоны необ¬
ходимо применение особых предосторожностей,
таких как использование защитного бокса, снабжен¬
ного перчатками-манипуляторами.
На методы А и В даются ссылки в частных ста¬
тьях на лекарственное растительное сырье, которое
в соответствии с современными хемотаксономиче-
скими знаниями не должно содержать аристолохие¬
вых кислот, но может быть фальсифицировано или
заменено на материал растительного происхожде¬
ния, содержащий аристолохиевые кислоты. Методы
А и В предназначены для скрининга лекарственно-
70
Гэсударственная фармакопея Республики Беларусь
го растительного сырья на наличие аристолохиевых
кислот в пределах установленной нормы и обычно
дополняются макроскопическим и/или микроскопиче¬
ским испытаниями для того, чтобы исключить матери¬
ал растительного происхождения, содержащий ари-
столохиевые кислоты.
Метод С не представлен в частных статьях, но
предусмотрен в качестве метода, подтверждающе¬
го присутствие аристолохиевой кислоты I с концен¬
трацией, равной или превышающей 2 ррт. Метод
С может применяться, если результаты хромато¬
графического исследования указывают на присут¬
ствие аристолохиевой кислоты I.
Эти методы не предназначены для включения
в качестве методов количественного определения
в частные статьи на лекарственное растительное
сырье, в котором образуются аристолохиевые кисло¬
ты в качестве вторичных метаболитов; для этой цели
необходимы более чувствительные и валидирован-
ные методы.
МЕТОДА: СКРИНИНГ-ИСПЫТАНИЕ НА
НАЛИЧИЕ АРИСТОЛОХИЕВЫХ КИСЛОТ
Тонкослойная хроматография (2.2.27).
Смесь растворителей. Кислота муравьиная
безводная Р — вода Р — метанол Р (1:9:40, об/об/об).
Испытуемый раствор. К 1,0 г измельченного ле¬
карственного растительного сырья (710) (2.9.12) при¬
бавляют 10,0 мл смеси растворителей, обрабатывают
ультразвуком в течение 10 мин и центрифугируют.
Раствор сравнения (а). Количество ФСО ари-
столохии, соответствующее 0,10 мг аристолохиевой
кислоты I, диспергируют в 20,0 мл смеси раствори¬
телей, обрабатывают ультразвуком в течение 10 мин
и центрифугируют.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят метанолом Р до объема 25,0 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р254 Р ((2—Ю) МКМ):
Подвижная фаза: кислота муравьиная без¬
водная Р — вода Р — этилацетат Р — толуол Р
(3:3:30:60, об/об/об/об); используют верхний слой.
Наносимый объем пробы: по 20 мкл в виде полос
длиной 8 мм.
Фронт подвижной фазы: не менее 6 см от линии
старта.
Высушивание: в потоке холодного воздуха в те¬
чение 5 мин.
Проявление: пластинку опрыскивают раствором
100 г/л олова (II) хлорида Р в кислоте хлористово¬
дородной разведенной Р до тех пор, пока пластинка
не станет слегка мокрой, нагревают при температуре
100 °С в течение 1 мин. Просматривают в ультрафио¬
летовом свете при длине волны 365 нм.
Пригодность хроматографической системы:
- на хроматограмме раствора сравнения (а) об¬
наруживаются две зеленовато-синие зоны, соответ¬
ствующие аристолохиевым кислотам I и II, между
0,35 и /?Р=0,55, которые могут быть не полностью
разделены;
- на хроматограмме раствора сравнения (Ь) об¬
наруживается не менее одной из этих зон (соот¬
ветствует концентрации аристолохиевой кислоты I,
равной 2 ррт).
Результаты: на хроматограмме испытуемого
раствора не обнаруживаются зоны, соответствующие
по расположению и флуоресценции любым зонам
аристолохиевых кислот на хроматограмме раствора
сравнения (а).
Если на хроматограмме испытуемого раствора
обнаруживаются зоны, соответствующие по располо¬
жению и флуоресценции любой из зон аристолохие¬
вых кислот I и II на хроматограмме раствора сравне¬
ния (а), то применяют метод В.
МЕТОД В: ИСПЫТАНИЕ НА ПРЕДЕЛЬНОЕ
СОДЕРЖАНИЕ АРИСТОЛОХИЕВОЙ
КИСЛОТЫ I
Жидкостная хроматография (2.2.29).
Смесь растворителей. Ацетонитрил Р —
вода Р (50:50, об/об).
Испытуемый раствор. 2,0 г измельченного ле¬
карственного растительного сырья (710) (2.9.12)
помещают во флакон из коричневого стекла с за¬
винчивающейся крышкой вместимостью 250 мл и
прибавляют 100,0 мл смеси растворителей. Пере¬
мешивают в течение 30 мин со скоростью около
300 об/мин и фильтруют через мембранный фильтр
(размер пор 0,45 мкм).
Раствор сравнения (а). Содержимое контейнера
с ФСО аристолохиевой кислоты I растворяют в смеси
растворителей так, чтобы получить раствор с концен¬
трацией 0,04 мкг/мл аристолохиевой кислоты I.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО аристолохиевой кислоты для проверки при¬
годности хроматографической системы (содержит
аристолохиевые кислоты I и II) растворяют в смеси
растворителей и доводят до объема 20,0 мл этой же
смесью растворителей.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
2,1 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3,5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: трифторуксусная кисло¬
та Р — вода Р (0,1:99,9, об/об);
- подвижная фаза В: трифторуксусная кисло¬
та Р — ацетонитрил Р (0,1:99,9, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—25
85—>35
15—>65
25—30
35 —> 0
65->100
30—31
0 —> 85
100 — 15
- скорость подвижной фазы: 0,3 мл/мин;
- спектрофотометрический детектор, длина
волны 390 нм;
- объем вводимой пробы: по 25 мкл;
Пригодность хроматографической системы:
- разрешение: не менее 3,0 между пиками ари¬
столохиевых кислот I и II на хроматограмме раствора
сравнения (Ь);
- отношение сигнал/шум: не менее 10 для пика
аристолохиевой кислоты I на хроматограмме раст¬
вора сравнения (а).
Предельное содержание примесей:
- испытуемый образец выдерживает испытание,
если на хроматограмме испытуемого раствора не
обнаруживаются пики, соответствующие по времени
удерживания пику аристолохиевой кислоты I на хро¬
матограмме раствора сравнения (а) (2 ррт).
2.9.10. Содержание этанола
71
МЕТОД С: ПОДТВЕРЖДАЮЩЕЕ ИСПЫТАНИЕ
НА АРИСТОЛОХИЕВУЮ КИСЛОТУ I
Жидкостная хроматография (2.2.29) с использо¬
ванием масс-спектрометрии (2.2.43).
Смесь растворителей. Ацетонитрил Р —
вода Р (50:50, об/об).
Испытуемый раствор. 2,0 г измельченного ле¬
карственного растительного сырья (710) (2.9.12) взве¬
шивают во флакон из коричневого стекла с завинчива¬
ющейся крышкой вместимостью 250 мл и прибавляют
100,0 мл смеси растворителей. Обрабатывают уль¬
тразвуком в течение 30 мин и фильтруют через мем¬
бранный фильтр (номинальный размер пор 0,45 мкм).
Раствор сравнения (а). Содержимое контейнера
с ФСО аристолохиевой кислоты I растворяют в сме¬
си растворителей так, чтобы получить раствор с кон¬
центрацией 0,04 мкг/мл аристолохиевой кислоты I.
Раствор сравнения (Ь). В соответствии с ин¬
струкцией, прилагаемой к ФСО аристолохиевой кис¬
лоты /, готовят раствор, содержащий 0,45 мкг аристо¬
лохиевой кислоты I в 10,0 мл испытуемого раствора.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
2,1 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3,5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: кислота муравьиная без¬
водная Р — раствор 1 г/л аммония ацетата Р
в воде Р (0,1:99,9, об/об);
- подвижная фаза В: кислота муравьиная без¬
водная Р — раствор 1 г/л аммония ацетата Р в
метаноле Р (0,1:99,9, об/об);
Время (мин)
Подвижная фаза А
^об/об\
Подвижная фаза В
(%< об/об)
0—15
о
т
О
о
о
т—
т
о
со
!
15—16
0
100
16—17
о
г-
г
о
о
со
т
о
о
- скорость подвижной фазы: 0,4 мл/мин;
- объем вводимой пробы: по 20 мкл; вводят раст¬
вор сравнения (а) дважды, испытуемый раствор дваж¬
ды, раствор сравнения (а) дважды и затем раствор
сравнения (Ь) дважды;
- масс-детектор: как описано ниже в испыта¬
ниях А или В. Регулируют скорость подвижной фазы,
температуру и настройки детектора таким образом,
чтобы привести в соответствие с критериями пригод¬
ности хроматографической системы.
А. Масс-спектрометр с ионной ловушкой, с ио¬
низацией электрораспылением (Е81) и анализатором
М5п-типа.
Настраивают параметры масс-спектрометра для
М53-типа следующим образом:
Тип
Родительский ион
(т/2)
Ширина
выделения
(т/2)
Относительная
энергия стол¬
кновений (%)
М32
359
[М+ЫН^*
2,0
30
М33
298
2,0
35
-полный диапазон сканирования дочерних ио¬
нов: от т/г 80 до т/г 370;
- контролируемые дочерние ионы: т/г 252, т/г
268 и /77/7 281.
Пригодность хроматографической системы:
-отношение сигнал/шум: не менее 100 для
контролируемых дочерних ионов на хроматограмме
раствора сравнения (а);
-проверка влияния матрицы: среднее значе¬
ние 2 величин отношений для раствора сравнения
(Ь) должно находиться в пределах ±40 % интервала
среднего значения 2 величин отношений для раст¬
вора сравнения (а); в другом случае необходимо из¬
менить настройки детектора.
Результаты: рассчитывают среднее значение
величин отношений (252/268 и 281/268) относитель¬
ной интенсивности для 3 дочерних ионов аристолохи¬
евой кислоты I в испытуемом растворе; рассчитывают
среднее значение 2 величин отношений сигналов при
значении времени удерживания аристолохиевой кис¬
лоты I в растворе сравнения (а); если среднее значе¬
ние 2 величин отношений для испытуемого раствора
находится в пределах ±40 % интервала среднего зна¬
чения 2 величин отношений для раствора сравнения
(a) , то наличие аристолохиевой кислоты I в испытуе¬
мом растворе считается установленным.
В. Масс-спектрометр с тремя квадруполями, с ио¬
низацией электрораспылением (Е31) и анализатором
М5п-типа.
Настраивают параметры масс-спектрометра
для М32-типа следующим образом:
- ион-предшественник: т/г 359 [М+ИН4]+;
- контролируемые дочерние ионы: т/г 265, т/г
281 и т/г 296.
Пригодность хроматографической системы:
-отношение сигнал/шум: не менее 100 для
контролируемых дочерних ионов на хроматограмме
раствора сравнения (а);
-проверка влияния матрицы: среднее значе¬
ние 2 величин отношений для раствора сравнения
(b) должно находиться в пределах ±40 % интервала
среднего значения 2 величин отношений для раст-
случае необходимо из¬
менить настройки детектора.
Результаты: рассчитывают среднее значение
величин отношений (265/281 и 296/281) относитель¬
ной интенсивности для 3 дочерних ионов аристолохи¬
евой кислоты I в испытуемом растворе; рассчитывают
среднее значение 2 величин отношений сигналов при
значении времени удерживания аристолохиевой кис¬
лоты I в растворе сравнения (а); если среднее значе¬
ние 2 величин отношений для испытуемого раствора
находится в пределах ±40 % интервала среднего зна¬
чения 2 величин отношений для раствора сравнения
(а), то наличие аристолохиевой кислоты I в испытуе¬
мом растворе считается установленным.
2.9. ФАРМАЦЕВТИКО¬
ТЕХНОЛОГИЧЕСКИЕ
ИСПЫТАНИЯ
07/2016:20910
2.9.10. СОДЕРЖАНИЕ ЭТАНОЛА
Эти методы предназначены для испытания жид¬
ких лекарственных средств и их ингредиентов, кото¬
рые содержат этанол.
Содержание этанола в жидкости выражают ко¬
личеством объемов этанола, содержащихся в 100
объемах жидкости при температуре (20±0,1)°С. Эта
9
72 Государственная фармакопея Республики Беларусь
характеристика называется «процентное содержание
этанола по объему» (%, об/об). Содержание этанола
можно также выражать в граммах этанола в 100 г жид¬
кости. Эта характеристика называется «процентное
содержание этанола по массе» (%, м/м).
МЕТОДА
В случае присутствия в образцах растворенных
веществ, их отделяют от этанола путем дистилля¬
ции (отгонки). Если при дистилляции кроме этанола
и воды могут отгоняться другие летучие вещества, в
частной статье приводят соответствующие указания.
Соотношение плотности при температуре
(20±0,1)°С, относительной плотности (в вакууме) и
содержания этанола в смеси воды и спирта представ¬
лено в таблицах Международной организации офици¬
альной метрологии (1972), Международная рекомен¬
дация № 22.
Прибор. Прибор (рисунок 2.9.10.-1) представля¬
ет собой колбу с круглым дном (А), имеющую переход¬
ник (В) с улавливателем водяного пара, соединенную
с вертикальным холодильником (С). Нижняя часть хо¬
лодильника соединена с трубкой (О), через которую
дистиллят поступает в нижнюю часть мерной колбы
вместимостью 100 или 250 мл. Во время дистилляции
мерная колба погружена в смесь льда и воды (Е). Для
предотвращения обугливания растворенных веществ
под колбой (А) помещают диск, который имеет кру¬
глое отверстие диаметром 6 см.
(размеры указаны в миллиметрах)
Методика
Пикнометрический метод / метод с использо¬
ванием плотномера с осциллирующим датчиком.
25,0 мл испытуемого образца, измеренных при тем¬
пературе (20±0,1)°С, помещают в дистилляцион-
ную колбу, разводят водой дистиллированной Р до
объема 100 мл или 150 мл и прибавляют несколько
кусочков пемзы. Присоединяют переходник и холо¬
дильник. Отгоняют и собирают в мерную колбу вме¬
стимостью 100 мл не менее 90 мл дистиллята (от¬
гона). Температуру отгона доводят до температуры
(20±0,1)°С и доводят водой дистиллированной Р с
температурой (20±0,1) °С до объема 100 мл. Опреде¬
ляют относительную плотность отгона при температу¬
ре (20±0,1) °С с помощью пикнометра или с помощью
плотномера с осциллирующим датчиком.
По таблице 2.9.10.-1 (колонка 3) находят со¬
держание этанола в отгоне и рассчитывают про¬
центное содержание этанола в лекарственном
средстве (об/об) путем умножения найденного та¬
бличного значения на четыре. Полученный резуль¬
тат округляют до десятичного знака.
Гидрометрический метод. 50,0 мл испытуемого
образца, измеренных при температуре (20±0,1)°С,
помещают в дистилляционную колбу, прибавляют от
200 мл до 300 мл воды дистиллированной Р и выпол¬
няют дистилляцию, как описано выше, собирая в мер¬
ную колбу вместимостью 250 мл не менее 180 мл дис¬
тиллята. Температуру отгона доводят до (20±0,1) °С и
разводят до объема 250,0 мл водой дистиллирован¬
ной Р с температурой (20±0,1) °С.
Помещают отгон в цилиндр, диаметр которого
должен быть на 6 мм шире утолщения ареометра.
Если объем дистиллята недостаточен, удваивают
количество испытуемого лекарственного средства и
дистиллят разводят до объема 500,0 мл дистиллиро¬
ванной водой Р с температурой (20±0,1) °С.
Вносят поправку на разведение путем умноже¬
ния найденного значения на пять. По таблице 2.9.10.-
1 рассчитывают процентное содержание этанола в
лекарственном средстве (об/об) и округляют резуль¬
тат до десятичного знака.
Таблица 2.9.10.-1
Соотношение между плотностью, относительной
плотностью и содержанием этанола
Рго (кг/м3)
Относительная плот¬
ность дистиллята на
воздухе дЦ
Содержание
этанола в про¬
центах (об/об)
при 20 °С
968,0
0,9697
25,09
968,5
0,9702
24,64
969,0
0,9707
24,19
969,5
0,9712
23,74
970,0
0,9717
23,29
970,5
0,9722
22,83
971,0
0,9727
22,37
971,5
0,9733
21,91
972,0
0,9738
21,45
972,5
0,9743
20,98
973,0
0,9748
20,52
973,5
0,9753
20,05
974,0
0,9758
19,59
974,5
0,9763
19,12
975,0
0,9768
18,66
975,5
0,9773
18,19
976,0
0,9778
17,73
Г
2.9.10. Содержание этанола
73
Ри (кг/м3)
Относительная плот¬
ность дистиллята на
воздухе с/22о
Содержание
этанола в про¬
центах (об/об)
при 20 °С
976,5
0,9783
17,25
977,0
0,9788
16,80
977,5
0,9793
16,34
978,0
0,9798
15,88
978,5
0,9803
15,43
979,0
0,9808
14,97
979,5
0,9813
14,52
980,0
0,9818
14,07
980,5
0,9823
13,63
981,0
0,9828
16,18
981,5
0,9833
12,74
982,0
0,9838
12,31
982,5
0,9843
11,87
983,0
0,9848
11,44
983,5
0,9853
11,02
984,0
0,9858
10,60
984,5
0,9863
10,18
985,0
0,9868
9,76
985,5
0,9873
9,35
986,0
0,9878
8,94
986,5
0,9883
8,53
987,0
0,9888
8,13
987,5
0,9893
7,73
988,0
0,9898
7,34
988,5
0,9903
6,95
989,0
0,9908
6,56
989,5
0,9913
6,17
990,0
0,9918
5,79
990,5
0,9923
5,42
991,0
0,9928
5,04
991,5
0,9933
4,67
992,0
0,9938
4,30
992,5
0,9943
3,94
993,0
0,9948
3,58
993,5
0,9953
3,22
994,0
0,9958
2,86
994,5
0,9963
2,51
995,0
0,9968
2,16
995,5
0,9973
1,82
996,0
0,9978
1,47
996,5
0,9983
1,13
997,0
0,9988
0,80
997,5
0,9993
0,46
998,0
0,9998
0,13
#Если испытуемое лекарственное средство со¬
держит летучие вещества, такие как эфир, эфирные
масла, хлороформ, камфору, летучие кислоты или
основания, свободный йод и др., его предварительно
обрабатывают. Испытуемое лекарственное средство,
содержащее эфир, эфирные масла, хлороформ или
камфору, помещают в делительную воронку, прибав¬
ляют равный объем насыщенного раствора натрия
хлорида Р и такой же объем петролейного эфира Р.
Смесь взбалтывают в течение 3 мин. После разде¬
ления слоев водно-спиртовой слой сливают в дру¬
гую делительную воронку и обрабатывают таким же
способом половинным количеством петролейного
эфира Р. Водно-спиртовой слой сливают в дистил-
ляционную колбу. Эфирные извлечения объединяют
и взбалтывают с половинным количеством насыщен¬
ного раствора натрия хлорида Р. После разделения
слоев водно-спиртовой слой присоединяют к жидко¬
сти, находящейся в дистилляционной колбе.
Если жидкость при дистилляции сильно пенится,
прибавляют (2—3) мл кислоты серной Р или кисло¬
ты фосфорной Р, (2—3) г кальция хлорида Р или па¬
рафина Р.
Если испытуемое лекарственное средство со¬
держит менее 30 % спирта, то высаливание проводят
не насыщенным раствором натрия хлорида Р, а 10 г
натрия хлорида Р.
При содержании в испытуемом лекарственном
средстве летучих веществ их нейтрализуют раство¬
ром щелочи, при содержании летучих оснований —
кислотой фосфорной Р или кислотой серной Р.
Испытуемые лекарственные средства, содержа¬
щие свободный йод, перед дистилляцией обрабаты¬
вают порошком цинка Р или рассчитанным количе¬
ством натрия тиосульфата Р до обесцвечивания.
Для связывания летучих сернистых соединений при¬
бавляют несколько капель раствора натрия гидрок¬
сида Р.#
МЕТОД В
Парофазная газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 1,0 мл про¬
панола Р1 доводят водой Р до объема 100,0 мл.
1.0 мл полученного раствора доводят водой Р до объ¬
ема 20,0 мл.
Испытуемый раствор. Количество испытуемого
образца, соответствующее 0,4 г этанола, доводят во¬
дой Р до объема 50,0 мл. 1,0 мл полученного раствора
доводят водой Р до объема 20,0 мл. К 2,0 мл получен¬
ного раствора прибавляют 1,0 мл раствора внутренне¬
го стандарта и доводят водой Р до объема 20,0 мл.
Раствор сравнения (а). 5,0 мл этанола безво¬
дного Р доводят водой Р до объема 100,0 мл. 25,0 мл
полученного раствора доводят водой Р до объема
100.0 мл. 1,0 мл полученного раствора доводят во¬
дой Р до объема 20,0 мл.
Раствор сравнения (Ь). 0,5 мл раствора сравне¬
ния (а) и 1,0 мл раствора внутреннего стандарта дово¬
дят водой Р до объема 20,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (а) и 1,0 мл раствора внутреннего стандарта дово¬
дят водой Р до объема 20,0 мл.
Раствор сравнения (д). 1,5 мл раствора сравне¬
ния (а) и 1,0 мл раствора внутреннего стандарта дово¬
дят водой Р до объема 20,0 мл.
Раствор сравнения (е). 1,0 мл метанола Р2 до¬
водят водой Р до объема 100,0 мл. 1,0 мл полученно¬
го раствора доводят водой Р до объема 20,0 мл.
Раствор сравнения (1). Смешивают 1,0 раствора
внутреннего стандарта, 2,0 мл раствора сравнения
(а), 2,0 мл раствора сравнения (е) и доводят водой Р
до объема 20,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная дли¬
ной 30 м и диаметром 0,53 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 3 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 3 мл/мин;
- деление потока: 1:50;
- параметры парофазного пробоотборника:
- равновесная температура: 85 °С;
- время достижения равновесия: 20 мин;
74
Гэсударственная фармакопея Республики Беларусь
- температура:
Время (мин)
Температура (°С)
Колонка
0—1,6
40
1,6—9,9
40—65
9,9—13,6
65—175
13,6—20,0
175
Блок ввода проб
200
Детектор
200
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1,0 мл газовой фазы
испытуемого раствора и растворов сравнения (Ь), (с),
(б) и (Т) не менее 3 раз каждый.
Порядок выхода пиков: метанол, этанол, пропанол.
Относительное удерживание (по отношению к
этанолу, время удерживания — около 5,3 мин): мета¬
нол — около 0,8; пропанол — около 1,6.
Пригодность хроматографической системы.
раствор сравнения ({):
- разрешение: не менее 5 между пиками метано¬
ла и этанола.
Строят градуировочный график, откладывая по
оси абсцисс концентрации этанола в растворах срав¬
нения (Ь), (с), (д) и (0, а по оси ординат — среднее
значение отношения площадей пиков этанола и вну¬
треннего стандарта на хроматограммах соответству¬
ющих растворов.
Рассчитывают процентное содержание этанола в
испытуемом образце.
МЕТОД С
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 1,0 мл про¬
панола Р1 доводят водой Р до объема 100,0 мл.
Испытуемый раствор. Количество испытуемого
образца, содержащее 1 г этанола, доводят водой Р до
объема 50,0 мл. К 1,0 мл полученного раствора при¬
бавляют 1,0 мл раствора внутреннего стандарта и до¬
водят водой Р до объема 20,0 мл.
Раствор сравнения (а). 1,0 мл этанола безво¬
дного Р доводят водой Р до объема 50,0 мл.
Раствор сравнения (Ь). 1,0 мл метанола Р2 до¬
водят водой Р до объема 100,0 мл. 1,0 мл полученно¬
го раствора доводят водой Р до объема 20,0 мл.
Раствор сравнения (с). Смешивают 1,0 мл раст¬
вора внутреннего стандарта, 1,0 мл раствора сравне¬
ния (а), 2,0 мл раствора сравнения (Ь) и доводят во¬
дой Р до объема 20,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная дли¬
ной 30 м и диаметром 0,53 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 3 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 3 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1,6
40
1,6—9,9
40—65
9,9—13,6
65-175
13,6—20,0
175
Блок ввода проб
200
Детектор
200
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1,0 мкл испытуемо¬
го раствора и раствора сравнения (с) не менее 3 раз
каждый.
Порядок выхода пиков: метанол, этанол, пропанол.
Относительное удерживание (по отношению к
этанолу, время удерживания — около 5,3 мин): мета¬
нол — около 0,8; пропанол — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 5 между пиками метано¬
ла и этанола.
Содержание этанола (в процентах (об/об)) рас¬
считывают по формуле
А,/2Ю0
Аг-М ’
где:
Л1 — площадь пика этанола на хроматограмме
испытуемого раствора;
А2 — площадь пика этанола на хроматограмме
раствора сравнения (с);
^ — площадь пика внутреннего стандарта на хро¬
матограмме испытуемого раствора;
/2 — площадь пика внутреннего стандарта на хро¬
матограмме раствора сравнения (с);
V, — объем испытуемого образца в испытуемом
растворе, в мл.
07/2016:20911
2.9.11. ИСПЫТАНИЕ НА СОДЕРЖАНИЕ
МЕТАНОЛА И 2-ПРОПАНОЛА
МЕТОД А
Парофазная газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 1,0 мл пропа¬
нола Р1 доводят водой Р до объема 100,0 мл воды Р.
1.0 мл полученного раствора доводят водой Р до объ¬
ема 20,0 мл.
Испытуемый раствор. К 4,0 мл испытуемого об¬
разца прибавляют 1,0 мл раствора внутреннего стан¬
дарта и доводят водой Р до объема 20,0 мл.
Раствор сравнения (а). К 1,0 мл метанола Р2
прибавляют 1,0 мл 2-пропанола Р2 и доводят водой Р
до объема 100,0 мл. 1,0 мл полученного раствора до¬
водят водой Р до объема 20,0 мл.
Раствор сравнения (Ь). 5,0 мл этанола безво¬
дного Р доводят водой Р до объема 100,0 мл. 25,0 мл
полученного раствора доводят водой Р до объема
100.0 мл. 1,0 мл полученного раствора доводят во¬
дой Р до объема 20,0 мл.
Раствор сравнения (с). Смешивают 1,0 мл раст¬
вора внутреннего стандарта, 2,0 мл раствора сравне¬
ния (а), 2,0 мл раствора сравнения (Ь) и доводят во¬
дой Р до объема 20,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная дли¬
ной 30 м и диаметром 0,53 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 3 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 3 мл/мин;
- деление потока: 1:50;
- параметры парофазного пробоотборника:
- равновесная температура: 85 °С;
- время достижения равновесия: 20 мин;
2.9.11. Испытание на содержание метанола и 2-пропанола
75
- температура:
Время (мин)
Температура (°С)
Колонка
0—1,6
40
1,6—9,9
40—>65
9,9—13,6
65—>175
13,6—20,0
175
Блок ввода проб
200
Детектор
200
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1,0 мл газовой фазы
испытуемого раствора и раствора сравнения (с) не
менее 3 раз каждый.
Порядок выхода пиков: метанол, этанол, 2-про-
панол, 1-пропанол.
Относительное удерживание (по отношению к
этанолу, время удерживания — около 5,3 мин): мета¬
нол — около 0,8; 2-пропанол — около 1,2; 1-пропа¬
нол — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 5 между пиками метано¬
ла и этанола.
Содержание метанола (в процентах (об/об)) рас¬
считывают по формуле
Д/2
Дг-Л-40 1
где:
А — площадь пика метанола на хроматограмме
испытуемого раствора;
А2 — площадь пика метанола на хроматограмме
раствора сравнения (с);
А, — площадь пика внутреннего стандарта на хро¬
матограмме испытуемого раствора;
/2 — площадь пика внутреннего стандарта на хро¬
матограмме раствора сравнения (с).
Содержание 2-пропанола (в процентах (об/об))
рассчитывают по формуле:
а-/2
А /г 40’
где:
А3 — площадь пика 2-пропанола на хроматограм¬
ме испытуемого раствора;
А4 — площадь пика 2-пропанола на хроматограм¬
ме раствора сравнения (с);
А, — площадь пика внутреннего стандарта на хро¬
матограмме испытуемого раствора;
/2 — площадь пика внутреннего стандарта на хро¬
матограмме раствора сравнения (с).
МЕТОД В
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 1,0 мл про¬
панола Р1 доводят водой Р до объема 100,0 мл.
Испытуемый раствор. К 4,0 мл испытуемого об¬
разца прибавляют 1,0 мл раствора внутреннего стан¬
дарта и доводят водой Р до объема 20,0 мл.
Раствор сравнения (а). К 1,0 мл метанола Р2
прибавляют 1,0 мл 2-пропанола Р2 и доводят водой Р
до объема 100,0 мл. 1,0 мл полученного раствора до¬
водят водой Р до объема 20,0 мл.
Раствор сравнения (Ь). 1,0 мл этанола безво¬
дного Р доводят водой Р до объема 50,0 мл.
Раствор сравнения (с). Смешивают 1,0 мл раст¬
вора внутреннего стандарта, 2,0 мл раствора сравне¬
ния (а), 1,0 мл раствора сравнения (Ь) и доводят во¬
дой Р до объема 20,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная дли¬
ной 30 м и диаметром 0,53 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 3 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 3 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1,6
40
1,6—9,9
40—>65
9,9—13,6
65—>175
13,6—20,0
175
Блок ввода проб
200
Детектор
200
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1,0 мкл испытуемо¬
го раствора и раствора сравнения (с) не менее 3 раз
каждый.
Порядок выхода пиков: метанол, этанол, 2-про-
панол, 1-пропанол.
Относительное удерживание (по отношению к
этанолу, время удерживания — около 5,3 мин): мета¬
нол — около 0,8; 2-пропанол — около 1,2; 1-пропа¬
нол — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 5 между пиками метано¬
ла и этанола.
Содержание метанола (в процентах (об/об)) рас¬
считывают по формуле:
д/2
А /г 40 1
где:
А — площадь пика метанола на хроматограмме
испытуемого раствора;
А, — площадь пика метанола на хроматограмме
раствора сравнения (с);
А, — площадь пика внутреннего стандарта на хро¬
матограмме испытуемого раствора;
/2 — площадь пика внутреннего стандарта на хро¬
матограмме раствора сравнения (с).
Содержание 2-пропанола (в процентах (об/об))
рассчитывают по формуле:
АЛ
А*'г40’
где:
Аз — площадь пика 2-пропанола на хроматограм¬
ме испытуемого раствора;
Д4 — площадь пика 2-пропанола на хроматограм¬
ме раствора сравнения (с);
А, — площадь пика внутреннего стандарта на хро¬
матограмме испытуемого раствора;
/2 — площадь пика внутреннего стандарта на хро¬
матограмме раствора сравнения (с).
76
Государственная фармакопея Республики Беларусь
07/2016:20925
2.9.25. ВЫСВОБОЖДЕНИЕ
ДЕЙСТВУЮЩЕГО ВЕЩЕСТВА
ИЗ ЛЕКАРСТВЕННЫХ
ЖЕВАТЕЛЬНЫХ РЕЗИНОК
ПРИНЦИП
Высвобождение действующего вещества из
лекарственной жевательной резинки определя¬
ют путем механического смешивания пластины
жевательной резинки, помещенной в небольшую
камеру, моделирующую процесс жевания, с опре¬
деленным объемом буферного раствора.
ПРИБОРА
Прибор (рисунок 2.9.25.-1) состоит из следующих
частей:
- 1 камеры, моделирующей процесс жевания;
-1 вертикального поршня;
- 2 горизонтальных поршней с уплотнительными
кольцами и манжетами.
Камера, моделирующая процесс жевания, состо¬
ит из четырех отдельных частей:
-1 центральной камеры;
- 1 воронки (рисунок 2.Э.25.-2);
- 2 направляющих с втулками (рисунок 2.9.25.-3).
Воронка и направляющие смонтированы на
центральной камере. Уплотнительные кольца на¬
ходятся в углублениях на поршнях и окружены
манжетами; манжеты предназначены для дополни¬
тельного предохранения камеры от протекания у
направляющих.
Имитацию жевания резинки обеспечивают го¬
ризонтальные поршни, а вертикальный поршень
предназначен для обеспечения правильного на¬
хождения резинки между горизонтальными порш¬
нями.
Скорость машины контролируют, обеспечивая
постоянство циклов. Один цикл включает в себя
следующие стадии: горизонтальные поршни начи¬
нают свое движение от самых дальних положений
и двигаются до своих самых ближних положений,
затем возвращаются в дальние положения. Один
раз в течение цикла вертикальный поршень дви¬
жется из своего нижнего положения до верхнего
положения и обратно до нижнего.
Ход каждого горизонтального поршня состав¬
ляет 25,0 мм. Максимальное расстояние между
двумя поршнями составляет 50 мм. Минимальное
расстояние между поршнями составляет от 0,1 мм
до 1,0 мм. Ход вертикального поршня составляет
22,0 мм.
Движение горизонтальных поршней происхо¬
дит таким образом, что оба поршня находятся в
своих ближних положениях в один и тот же момент
времени. Вертикальный поршень настраивают
так, чтобы он не мешал работе горизонтальных
поршней.
При необходимости горизонтальные поршни
могут быть настроены так, чтобы они вращались
вокруг своей оси в противоположных друг другу на¬
правлениях в конце каждого движения, имитирую-
A. Горизонтальный поршень. й. Воронка.
B. Направляющая. Е. Вертикальный поршень.
C. Камера, моделирующая процесс жевания.
Рисунок 2.9.25.-1. Прибор А — камера, моделирующая процесс жевания, и поршни
(размеры указаны в миллиметрах)
г
2.9.25. Высвобождение действующего вещества из лекарственных жевательных резинок
77
щего жевание, обеспечивая, таким образом, макси¬
мально эффективное жевание.
Все части прибора, вступающие в контакт с лекар¬
ственным средством или средой растворения, должны
быть химически инертными и не должны адсорбиро¬
вать образец, реагировать и взаимодействовать с ним.
ПРИБОР В
Прибор для моделирования процесса жевания В
(рисунок 2.Э.25.-4) состоит из:
- 1 тест-ячейки (рисунок 2.Э.25.-5 или 2.9.25.-6);
- 1 вертикального штока с верхней поверхностью
моделирования процесса жевания (рисунок 2.9.25.-7
и 2.Э.25.-8);
Рисунок 2.Э.25.-2. Воронка
(размеры указаны в миллиметрах)
Рисунок 2.9.25.-3. Направляющие (секция О-О)
(размеры указаны в миллиметрах)
-1 камеры-основания с нижней поверхностью
моделирования процесса жевания (рисунок 2.9.25.-Э
и 2.9.25.-10);
- 1 устройства для моделирования процесса же¬
вания с движением вверх-вниз;
- 1 вращающегося устройства для вертикально¬
го штока.
Жевательная резинка подвергается «искус¬
ственному жеванию» между двумя поверхностями
моделирования процесса жевания. Скорость при¬
бора контролируется для обеспечения постоян¬
ства рабочего цикла. Расстояние между нижней и
верхней поверхностями моделирования процесса
A. Вращающееся устройство для верхней поверхности
моделирования процесса жевания.
B. Корпус.
C. Тест-ячейка.
Б. Шток.
Е. Верхняя поверхность моделирования процесса жевания.
Р. Нижняя поверхность моделирования процесса жевания.
С. Камера-основание.
Н. Устройство для моделирования процесса жевания с дви¬
жением вверх-вниз.
Рисунок 2.9.25.-4. Прибор В
78
Государственная фармакопея Республики Беларусь
жевания может быть установлено на уровне 5 мм.
Угол поворота вращающегося устройства состав¬
ляет около 20°.
Тест-ячейка может быть также снабжена одной
или двумя стеклянными пробоотборными трубка¬
ми, проходящими через термостатирующие двой¬
ные стенки. Такие трубки также позволяют про¬
вести слив, который может быть необходим для
обеспечения условий проточности в случае уме¬
ренно растворимых веществ.
Для предупреждения распада жевательной ре¬
зинки она обычно помещается между двумя круглы¬
ми пластмассовыми сетками.
Могут быть использованы сетки, изготовлен¬
ные из нейлона (РА6), имеющие размер отверстия
1,4 мм и диаметр волокна 0,405 мм.
Все части прибора, которые могут находить¬
ся в контакте с образцом или средой растворения,
должны быть химически инертными и не абсорби¬
ровать, не реагировать и не взаимодействовать с
образцом.
0 59 ±2
0 59 ±2
Рисунок 2.Э.25.-6. Тест-ячейка (прямая)
(размеры указаны в миллиметрах)
2.9.25. Высвобождение действующего вещества из лекарственных жевательных резинок
79
МЕТОДИКА
Для проведения испытания необходима следу¬
ющая информация:
- тип используемого прибора (А или В);
- состав, объем и температура среды раство¬
рения;
- количество движений, имитирующих жева¬
ние, в минуту;
- время и способ отбора образцов;
- проводят ли анализ остатка жевательной ре¬
зинки или анализируют среду растворения;
Рисунок 2.9.25.-7. Шток
(размеры указаны в миллиметрах)
- описание аналитической методики.
Указанный объем среды растворения, обычно
это 20 мл фосфатного буферного раствора рН
6,0 Р2, помещают в камеру, моделирующую про¬
цесс жевания. Температуру среды поддержива¬
ют при (37±0,5) °С с помощью электрического при¬
способления с внешним контролем (прибор А) или
термостата (прибор В). Выставляют скорость дви¬
жения поршней с указанной частотой движений,
имитирующих жевание, в минуту (обычно 60). В
камеру, моделирующую процесс жевания, помеща-
037.8 т
8.8 (2х)
Н I-, 08.5 с
*! СО)
■■ 1
1 1—1^ 1
со
Л I { л~ттяв\
1 *
—I :!!!: м °
ю
I
Рисунок 2.9.25.-Э. Камера-основание
(размеры указаны в миллиметрах)
Контактирующая
поверхность КА 1,5 - 2,1
Рисунок 2.9.25.-8. Верхняя
поверхность симуляции жевания
(размеры указаны в миллиметрах)
Рисунок 2.9.25.-10. Нижняя поверхность
моделирования процесса жевания
(размеры указаны в миллиметрах)
80
Гэсударственная фармакопея Республики Беларусь
ют точно взвешенную часть жевательной резинки
или целую резинку и запускают прибор.
ОТБОР ПРОБ И ПРОВЕДЕНИЕ АНАЛИЗА
Через указанное время прибор останавливают.
Удаляют остатки жевательной резинки и отбирают
образец среды растворения. Подходящим методом
определяют содержание действующего вещества.
После каждого отбора образца может проводить¬
ся замена среды растворения, при этом при расче¬
тах необходимо учитывать изменение объема или
разведение среды растворения. В качестве альтер¬
нативы может определяться содержание действу¬
ющего вещества в остатке жевательной резинки.
Испытание проводят на 6 лекарственных жеватель¬
ных резинках. Все 6 лекарственных жевательных
резинок должны выдерживать испытание.
Количество действующего вещества, раство¬
рившееся за определенное время, выражают в про¬
центах от указанного на этикетке значения.
07/2016:20939
2.9.39. ВЗАИМОДЕЙСТВИЯ «ВОДА-
ТВЕРДОЕ ВЕЩЕСТВО»:
ПОСТРОЕНИЕ ИЗОТЕРМ
СОРБЦИИ - ДЕСОРБЦИИ
И ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ВОДЫ
ВВЕДЕНИЕ
Твердые субстанции для фармацевтическо¬
го использования в виде сырья или наполнителей
лекарственных форм часто вступают во взаимодей¬
ствие с водой в процессе производства или хране¬
ния. Такие взаимодействия возможны (а) при кри¬
сталлизации, лиофилизации, влажной грануляции
или распылительной сушке; (Ь) в результате воз¬
действия при работе и хранении влаги воздуха или
других веществ лекарственной формы, содержащих
воду и способных к ее передаче другим компонен¬
там. К свойствам, изменяющимся при взаимодей¬
ствии твердых веществ с водой, относятся скорости
химического разложения веществ в твердом состоя¬
нии, роста кристаллов и растворения, дисперсность
и смачиваемость, текучесть порошка, масляни¬
стость, уплотняемость порошка, твердость прессо¬
ванного порошка, микробиологическая чистота.
Принимая во внимание проблемы, к которым
может привести присутствие воды, могут прини¬
маться меры предосторожности, заключающиеся
в удалении всей влаги, уменьшении контакта с
воздухом или контроле относительной влажности
воздуха и, как правило, увеличивающие стоимость
производственного процесса, не гарантируя от¬
сутствие в дальнейшем на протяжении жизненно¬
го цикла продукта проблем, связанных с влагой.
Также важно учитывать, что существует много
ситуаций, когда определенное содержание воды
в твердом веществе необходимо для обеспече¬
ния соответствующих свойств, например плотно¬
сти порошка. С учетом вышеизложенного важно
в наиболее полной мере обладать знанием о воз¬
действии влаги на твердые вещества перед раз¬
работкой стратегий по работе с ними, их хранению
и применению.
К некоторым наиболее важным разделам не¬
обходимой информации, касающейся взаимодей¬
ствий «вода - твердое вещество», относятся:
- общее количество присутствующей воды;
- степень адсорбции и абсорбции;
- наличие гидратной формы;
- площадь удельной поверхности твердого ве¬
щества, а также такие свойства, как степень кри¬
сталличности, степень пористости, стеклование и
температура плавления;
- месторасположение взаимодействия с во¬
дой, степень связывания и степень молекулярной
подвижности;
- воздействие температуры и относительной
влажности;
- преимущественно необратимая гидратация;
- кинетика поглощения влаги;
- различные факторы, вероятно оказывающие
влияние на скорость, с которой водяной пар может
поглощаться твердым веществом;
- для твердых веществ, растворимых в воде и
способных растворяться при сорбции воды, усло¬
вия, при которых будет происходить растворение.
ФИЗИЧЕСКИЕ ФОРМЫ СОРБИРОВАННОЙ
ВОДЫ
Вода может физически взаимодействовать с
твердыми веществами различными способами. Взаи¬
модействие может происходить на поверхности (ад¬
сорбция) или вода может проникать внутрь объемной
структуры твердого вещества (абсорбция). Термин
«сорбция» обычно используется в случае одновре¬
менной адсорбции и абсорбции. В случае большой
площади удельной поверхности адсорбция оказыва¬
ет огромное влияние на свойства твердых веществ.
Большие значения площади удельной поверхности
наблюдаются для твердых веществ с очень малень¬
кими частицами, а также твердых веществ с высокой
степенью пористости внутри частиц. Абсорбция ха¬
рактеризуется связанностью воды в грамме твердого
вещества, намного превышающей связанность, кото¬
рую может образовать мономолекулярный слой на
соответствующей поверхности, и величиной, которая
обычно не зависит от площади удельной поверхности.
Большинство кристаллических твердых ве¬
ществ не будет объемно абсорбировать воду из-за
плотной упаковки и высокой степени упорядочен¬
ности расположения атомов в кристаллической
решетке. При этом было доказано, что степень аб¬
сорбции твердыми веществами, имеющими частич¬
но кристаллическую и частично аморфную струк¬
туру, часто обратно пропорциональна степени кри¬
сталличности. Однако некоторые кристаллические
твердые вещества могут образовывать кристалло¬
гидраты. Эти гидраты могут выражаться стехиоме¬
трическим соотношением исходя из числа связан¬
ных молекул воды на молекулу твердого вещества
или могут быть нестехиометрическими. После деги¬
дратации кристаллогидраты могут сохранять свою
первоначальную структуру, или утрачивать кристал¬
лическую структуру и становиться аморфными или
переходить в новую безводную или менее гидрати¬
рованную форму.
Аморфные или частично аморфные твердые
вещества могут захватывать значительные коли-
2.9.39. Взаимодействия «вода - твердое вещество»
81
чества воды в связи с молекулярной неупорядо¬
ченностью в твердом веществе, достаточной для
поглощения, набухания или растворения. Подоб¬
ным свойством обладает большинство аморфных
полимеров и твердых веществ с небольшими мо¬
лекулярными массами, приведенных в аморфное
состояние в процессе получения, например, ли-
офилизацией или после измельчения. Введение
дефектов в твердые вещества с высокой степенью
кристалличности также будет приводить к проявле¬
нию подобного свойства. Чем больше химическое
сродство воды к твердому веществу, тем большее
общее количество воды может абсорбироваться. В
случае абсорбции воды аморфными твердыми ве¬
ществами их объемные свойства могут значительно
измениться. Установлено, например, что аморфные
твердые вещества в зависимости от температуры
могут существовать по крайней мере в двух состоя¬
ниях: «стеклообразном» или «жидком»; температу¬
ра, при которой одно состояние переходит в другое,
является температурой стеклования 7\
Объемно абсорбируемая твердым веществом
вода, действуя на незаполненный объем твердого
вещества, может играть роль эффективного пла¬
стификатора и уменьшать значение Тд. Поскольку
реологические свойства «жидкого» и «стеклообраз¬
ного» состояний имеют существенные различия,
заключающиеся в том, что вещества в «жидком»
состоянии имеют намного меньшую вязкость, ха¬
рактерную для температуры, превышающей темпе¬
ратуру стеклования, то следует ожидать, что на ряд
важных свойств внутри объема твердого вещества,
зависящих от его реологии, влияет содержание
влаги. Поскольку аморфные твердые вещества яв¬
ляются веществами с небольшими молекулярными
массами, метастабильными по отношению к кри¬
сталлической форме вещества, абсорбированная
влага может способствовать возвращению твердого
вещества в кристаллическое состояние, особенно
если твердое вещество трансформируется сорби¬
рованной водой в «жидкое» состояние. Это лежит в
основе «образования осадка», часто наблюдаемого
в течение процесса лиофилизации. Дополнитель¬
ным свойством, характерным именно для водорас¬
творимых твердых веществ, является их тенденция
расплываться, то есть растворяться в их собствен¬
ной сорбированной воде, при значениях относи¬
тельной влажности ЯН, превышающих ее значение
для насыщенного раствора твердого вещества ЯН0.
Способность расплываться повышается в резуль¬
тате высокой растворимости твердого вещества в
воде и ее значительного влияния на коллигативные
свойства воды. Это динамический процесс, продол¬
жающийся до тех пор, пока ЯН, превышает ЯН0.
В основе понимания влияния, оказываемого
водой на свойства твердых веществ и наоборот, ле¬
жит знание о положении молекулы воды и ее фи¬
зическом состоянии. Вода, связанная с твердым
веществом, может существовать в состоянии, кото¬
рое характеризуется связью с твердым веществом
напрямую, а также в состоянии подвижности, дости¬
гающей подвижности объемной воды. Наблюдение
данного различия в подвижности осуществляется с
помощью измерений, например, теплоты сорбции,
температуры затвердевания, ядерного магнитного
резонанса, диэлектрических свойств и диффузии.
Подобные изменения в подвижности интерпрети¬
руются как возникающие вследствие изменений в
термодинамическом состоянии воды по мере того,
как все большее и большее количество воды сор¬
бируется. Таким образом, вода, напрямую связан¬
ная с твердым веществом, часто рассматривается
в качестве недоступной для оказания влияния на
свойства твердого вещества, тогда как большие
количества сорбированной воды могут в большей
мере объединяться в кластеры и образовывать
воду, в большей степени имеющей подобные, чем
выраженные свойства растворителя. В случае кри¬
сталлогидратов комбинация межмолекулярных сил
(водородные связи) и кристаллической упаковки
может привести к очень сильным взаимодействиям
«вода - твердое вещество». Учитывая, что присут¬
ствие воды в аморфном твердом веществе может
влиять на температуру стеклования и, следова¬
тельно, на физическое состояние твердого веще¬
ства, при низких содержаниях воды большинство
полярных аморфных твердых веществ находится в
«стеклообразном» состоянии с высокой вязкостью
в связи с высокими значениями Тд. Тем не менее
вода является «замороженной» в структуре твер¬
дого вещества и приведенной в состояние непод¬
вижности большим значением вязкости, например
1013 Па с. Так как количество сорбированной воды
возрастает, а значение Тд уменьшается, достигая
температуры окружающей среды, «стеклообраз¬
ное» состояние достигает «жидкого» состояния, и
подвижность воды наряду с собственной подвиж¬
ностью твердого вещества значительно возрастает.
При высоких значениях ЯН степень пластификации
твердого вещества водой может быть достаточно
высокой для того, чтобы вода и твердое вещество
смогли достичь значительной подвижности. Таким
образом, данное описание природы сорбированной
воды помогает в целом объяснить довольно значи¬
тельное влияние, которое может оказать влага на
объемные свойства твердого вещества, такие как
химическая реакционная способность и механиче¬
ская деформация. Это дает возможность предполо¬
жить, что методы для оценки химической и физиче¬
ской стабильности твердых веществ и твердых до¬
зированных форм учитывают влияние сорбирован¬
ной воды на твердое вещество, в частности когда
вода поглощается твердым веществом и действует
в качестве пластификатора.
Скорость поглощения воды. Скорость и сте¬
пень сорбции или десорбции твердыми вещества¬
ми, подвергающимися воздействию окружающей
среды, водяного пара, могут быть критическим фак¬
тором при работе с твердыми веществами. Даже
простое действие взвешивания образцов твердо¬
го вещества на аналитических весах и подвергание
тонкого слоя порошка воздействию атмосферного
воздуха в течение нескольких минут может приве¬
сти к значительной ошибке, например при опреде¬
лении величины потери массы при высушивании.
Установлено, что водорастворимые твердые веще¬
ства, подвергаемые воздействию относительной
влажности, превышающей влажность насыщенного
раствора того же твердого вещества, будут само¬
произвольно растворяться, расплываясь в течение
длительного периода времени. Скорость поглоще¬
ния воды в целом зависит от ряда параметров, ко¬
торые считаются некритическими при измерениях
в условиях равновесия в связи с тем, что скорость
11. Зак. 1060.
82
Гэсударственная фармакопея Республики Беларусь
сорбции главным образом контролируется меха¬
низмами массообмена с сопровождением тепло-
переноса. Таким образом, такие факторы, как ко¬
эффициенты диффузии влаги в воздухе и твердом
веществе, конвективный воздушный поток, пло¬
щадь поверхности, геометрия внутреннего строе¬
ния твердого вещества и окружающая среда, могут
играть важную роль. Более того, метод, применяе¬
мый для проведения измерений, часто может быть
фактором, определяющим скорость, что обусловле¬
но влиянием геометрических параметров и окружа¬
ющей среды.
ПОСТРОЕНИЕ ИЗОТЕРМ
СОРБЦИИ-ДЕСОРБЦИИ
Принцип. Лучшим способом оценки склонно¬
сти к поглощению водяного пара является изме¬
рение сорбции или десорбции в качестве функции
относительной влажности при постоянной темпера¬
туре и условиях, при которых сорбция или десорб¬
ция преимущественно происходит независимо от
времени, то есть в равновесном состоянии. Относи¬
тельная влажность КН определяется по следующей
формуле:
р
— • 100
Ро
где:
Рс — давление водяного пара в системе;
Р0 — давление насыщенного водяного пара при
тех же условиях. р
Отношение ^ называется относительным дав¬
лением. Оценку 0 сорбции, или поглощения, воды
лучше проводить, начиная с высушенных образ¬
цов и подвергая их воздействию известной относи¬
тельной влажности. Десорбцию исследуют начиная
с системы, уже содержащей сорбированную воду и
уменьшая относительную влажность. Из названия
понятно, что изотерма сорбции-десорбции пригод¬
на только для стандартной температуры, следова¬
тельно, для каждой температуры существуют ин¬
дивидуальные изотермы. Обычно при равновесии
содержание влаги при конкретной относительной
влажности должно быть одинаковым независимо от
определения показателей сорбции или десорбции.
Однако обычно наблюдается гистерезис сорбции-
десорбции.
Методы. Образцы могут выдерживаться в ка¬
мерах при различных значениях относительной
влажности (рисунок 2.9.39.-1). Затем определяется
увеличение или уменьшение в массе для каждого
образца. Главное преимущество данного метода —
это удобство, а главные недостатки — низкая ско¬
рость достижения постоянной массы, в частности
при высоких значениях относительной влажности,
и ошибка, возникающая при открывании и закрыва¬
нии камеры в течение взвешивания.
Динамические гравиметрические системы
сорбции воды дают возможность взвешивания об¬
разцов в оперативном режиме в контролируемой
системе для оценки взаимодействия вещества с
влагой при различных программируемых уровнях
относительной влажности при постоянной темпе¬
ратуре. Главное преимущество контролируемой
системы заключается в том, что изотермические
условия могут быть установлены с большей на¬
дежностью и может быть проконтролирована ре¬
акция образца на изменение условий в динамике.
Для построения изотермы сорбции используются
значения величин (например, от 0 % до приблизи¬
тельно 95 % РН, без уменьшения объема), заре¬
гистрированные после установления достаточно
постоянного сигнала, указывающего на то, что об¬
разец достиг равновесия при данном уровне влаж¬
ности. В некоторых случаях (например, расплыва¬
ние) максимальное время может быть ограничено,
несмотря на то что уровень равновесия не достиг¬
нут. Прибор должен в достаточной мере контро¬
лировать температуру для обеспечения хорошей
стабильности исходных данных, а также точного
контроля над установлением относительной влаж¬
ности. Требуемая относительная влажность может
быть достигнута, например, с помощью осторож¬
ного перемешивания осушенного и насыщенного
парами газа с регуляторами потока. Также должно
учитываться электростатическое свойство порош¬
ка. Контроль над температурой и относительной
влажностью (например, с помощью сертифициро¬
ванного гигрометра, растворов сертифицирован¬
ных солей или значений температуры, при которых
происходит расплывание сертифицированных со¬
лей, превышающих соответствующий предел) дол¬
жен осуществляться в соответствии со специфика¬
цией на прибор. Весы должны в достаточной мере
обеспечивать массовое разрешение и длительную
продолжительность стабильности.
Также существует возможность измерить коли¬
чество поглощенной воды, неопределяемой грави¬
метрически, используя методы объемного анали¬
за. В некоторых случаях прямые методы анализа
воды, такие как определение температуры кипения,
определение содержания воды с помощью дистил¬
ляции, потери в массе при высушивании или газо¬
вой хроматографии, могут быть предпочтительны¬
ми. В случае абсорбции для повышения чувстви¬
тельности можно увеличить площадь удельной по¬
верхности образца, уменьшая размер частиц, или
увеличить общую площадь, используя образцы с
большими размерами. Однако важным является то,
что измельчение твердого вещества не изменяет
структуру поверхности твердого вещества, не дела
A. Регулятор влажности.
B. Камера для контроля температуры.
C. Модуль весов.
Э. Модуль, регулирующий влажность.
Е. Стандарт.
Р. Образец.
С. Паровой увлажнитель.
H. Модуль контроля потока.
I. Осушенный газ.
Рисунок 2.9.39.-1. Пример прибора для определения
сорбции воды (возможны другие схемы)
2.9.39. Взаимодействия «вода - твердое вещество»
83
ет его более аморфным или, наоборот, его кристал¬
лическую структуру менее упорядоченной. В слу¬
чае если поглощение воды не зависит от площади
удельной поверхности, только увеличение размера
образца может способствовать абсорбции. Увели¬
чение размера образца, однако, приведет к уве¬
личению времени установления какого-либо типа
равновесия. Для установления точных значений
важно достичь как можно более полной десольва¬
тации образца. Данному процессу способствуют
более высокая температура и более низкое давле¬
ние (вакуум); тем не менее необходимо учитывать
любые возможные негативные процессы, происхо¬
дящие с твердым веществом, например дегидрата¬
цию, химическую дегидратацию или сублимацию.
Кроме того, в связи с возможными ошибками необ¬
ходимо с осторожностью применять более высокие
значения температуры для побуждения десорбции,
так же как и в термогравиметрическом приборе.
Представление и интерпретация получен¬
ных данных. Полученные данные по сорбции
обычно представляются в виде графика зависи¬
мости изменения присоединенной массы в про¬
центах по отношению к массе сухого образца от
относительной влажности или времени. Изотермы
сорбции представляются в табличном и в графиче¬
ском виде. Полученные данные должны содержать
ссылку на метод измерения.
Гистерезис адсорбции-десорбции может ин¬
терпретироваться, например, исходя из пористости
образца, его состояния агломерации (капиллярной
конденсации), образования гидратов, полиморфно¬
го превращения или перехода образца в жидкое со¬
стояние. Определенные типы систем, в частности
системы с микропористыми твердыми веществами
и аморфными твердыми веществами, обладают
способностью сорбировать большие количества
водяных паров. В этом случае количество воды,
связанной с твердым веществом, в то время как от¬
носительная влажность уменьшается, больше ко¬
личества воды, изначально сорбируемой по мере
увеличения относительной влажности. Для микро¬
пористых твердых веществ гистерезис адсорбции-
десорбции водяного пара является равновесным
явлением, связанным с процессом капиллярной
конденсации. Это происходит в связи с сильным
неравномерным искривлением микропор и их «за¬
полненностью» (адсорбция) и «пустотой» (десорб¬
ция) при различных равновесных состояниях. Для
непористых твердых веществ, способных абсорби¬
ровать воду, гистерезис обусловлен изменением
степени взаимодействия «пар - твердое вещество»
в связи с изменением, например, конформации по¬
лимерных цепей в равновесном состоянии твердо¬
го вещества или тем, что для установления струк¬
турного равновесия необходим больший период
времени, чем для десорбции воды. Поэтому при
построении изотерм сорбции-десорбции важно
установить, что состояние, близкое к равновесию,
достигнуто. В частности, для гидрофильных поли¬
меров при высоких значениях относительной влаж¬
ности установление величин сорбции воды или де¬
сорбции независимо от времени является довольно
сложным, так как в данном случае мы имеем дело с
полимером, пластифицированным внутрь его жид¬
кого «состояния», где твердое вещество подверга¬
ется значительному изменению.
В случае образования кристаллогидрата на
кривой поглощения воды в зависимости от дав¬
ления или относительной влажности будет видно
резкое увеличение поглощения при конкретном
давлении, и количество поглощенной воды будет
в большинстве случаев стехиометрическим: мо¬
лярное отношение воды к твердому веществу. Тем
не менее в некоторых случаях кристаллогидраты,
по-видимому, не будут подвергаться фазовому из¬
менению или безводная форма будет выглядеть
аморфной. Следовательно, сорбция или десорбция
воды может выглядеть более похожей на сорбцию,
наблюдаемую при адсорбционных процессах. Кри¬
сталлографический анализ, выполненный методом
рентгенографии, и термический анализ являются в
наибольшей мере применимыми для изучения по¬
добных систем.
Для ситуаций, когда преимущественно проис¬
ходит адсорбция водяных паров, весьма полезным
является измерение площади удельной поверх¬
ности твердого вещества с помощью независимо¬
го метода и выражение адсорбции в виде массы
сорбированной воды на единицу площади поверх¬
ности твердого вещества. Это может применяться
при оценке вероятной значимости влияния сорбции
воды на свойства твердого вещества. Например,
0,5 % (м/м) поглощение воды едва может покрыть
непокрытую поверхность размером 100 м2/г, в то
время как для 1,0 м2/г размер покрываемой поверх¬
ности в 100 раз больше. В случае твердых веществ
для фармацевтического использования, имеющих
площадь удельной поверхности в диапазоне значе¬
ний от 0,01 м2/г до 10 м2/г, кажущееся низкое содер¬
жание воды может представлять собой значитель¬
ное ее количество для данной доступной поверх¬
ности. Так как «площадь сухой поверхности» не
является показателем в абсорбции, сорбция воды
аморфными или частично аморфными твердыми
веществами может выражаться исходя из единицы
массы, откорректированной для кристалличности,
когда кристаллическая форма не сорбирует значи¬
тельное количество воды по сравнению с аморф¬
ными областями.
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ВОДЫ
Принцип. Активность воды представляет
собой отношение давления пара воды в веществе
(Р) к давлению насыщенного водяного пара (Р0) при
одинаковой температуре и численно равно 1/100
относительной влажности (РН), созданной веще¬
ством в замкнутой системе. РН может быть рас¬
считана из прямых измерений парциального дав¬
ления пара, или точки росы или косвенного изме¬
рения датчиками, физические или электрические
характеристики которых изменяются в результате
воздействия РН. Пренебрегая коэффициентами
активности, отношение между А^ и равновесной
относительной влажностью (ЕРЯ) представляется
следующими выражениями:
ЕРЯ(%) = -100.
Метод. Активность воды определяют, помещая
образец в небольшую воздухонепроницаемую ем¬
кость, внутри которой может устанавливаться рав-
•"5а. '06С
84
Гэсударственная фармакопея Республики Беларусь
новесие между водой в твердом веществе и неза¬
полненным пространством. Объем незаполненного
пространства должен быть небольшим относитель¬
но объема образца, чтобы сорбционное состояние
образца в течение испытания оставалось неизмен¬
ным. Установление равновесия, являясь термоди¬
намическим процессом, занимает некоторое время,
но может быть ускорено принудительной циркуля¬
цией внутри ячейки. Полученное значение активно¬
сти воды является действительным только для од¬
новременно определенной температуры. В связи с
этим измерительное оборудование должно содер¬
жать в качестве элемента устройство для точного
измерения температуры. Кроме того, датчик должен
быть термоизолирован для обеспечения постоян¬
ной температуры в течение испытания. Датчик для
измерения влажности воздуха незаполненного про¬
странства над образцом является ключевым эле¬
ментом. Теоретически могут применяться все типы
гигрометров, но для аналитических целей миниатю¬
ризация и надежность являются необходимыми ус¬
ловиями. В качестве конденсирующей поверхности
используют хорошо протертое, охлажденное зерка¬
ло. Охлаждающая система электрически соединена
с фотоэлементом, внутрь которого отражается свет
от конденсирующего зеркала. Поток воздуха, нахо¬
дящегося в состоянии равновесия с испытуемым
образцом, направляется на зеркало, которое его ох¬
лаждает и конденсирует. Температура, при которой
эта конденсация начинается, является точкой росы,
из которой определяется ЕГСН. Коммерчески доступ¬
ные приборы, использующие метод точки росы/ох-
лажденного зеркала или другие методики, при при¬
менении для определения активности воды должны
оцениваться с точки зрения пригодности, поверять¬
ся и калиброваться. Эти приборы обычно кали¬
бруются свыше соответствующего диапазона, на¬
пример с использованием насыщенных растворов
некоторых солей при температуре 25 °С. К таким
растворам относятся насыщенные растворы солей,
пер'ечисленные в таблице 2.9.39.-1.
Таблица 2.9.39.-1
Стандартные насыщенные растворы солей
Насыщенные растворы
солей при температуре 25 °С
ЕВН (%)
к
Калия сульфат (К7504)
97,3
0,973
Бария хлорид (ВаС19)
90,2
0,902
Натрия хлорид (ИаС!)
75,3
0,753
Магния нитрат (Мд(МОд)?)
52,9
0,529
Магния хлорид (МдС1,)
32,8
0,328
Лития хлорид (ЫС1)
11,2
0,112
07/2016:20947
2.9.47. ПОДТВЕРЖДЕНИЕ
ОДНОРОДНОСТИ
ДОЗИРОВАННЫХ ЕДИНИЦ
С ИСПОЛЬЗОВАНИЕМ
БОЛЬШОГО КОЛИЧЕСТВА
ОБРАЗЦОВ
Одной из целей подтверждения однородности
дозированных единиц с использованием большо¬
го количества образцов является оценка лекар¬
ственных средств, производимых с применением
методологии процессно-аналитической техноло¬
гии (ПАТ, ргосезз апа!уИса11ес1зпо1оду— РАТ).
Соответствие требованиям общей статьи
2.9.40. Однородность дозированных единиц может
быть подтверждено описанным далее методом, по¬
зволяющим производить оценку большого количе¬
ства образцов (количество образца п > 100).
Применение данной статьи не является обя¬
зательным требованием. В статье приведено 2
альтернативных испытания (вариант 1 и вариант
2). Выполнение требований любого из двух вари¬
антов является подтверждением соответствия ис¬
пытуемого лекарственного препарата требовани¬
ям общей статьи 2.9.40. Оба варианта считаются
равнозначными для подтверждения соответствия
требованиям общей статьи 2.9.40.
Таблица 2.9.47.-1
Константа приемлемости (к) и допустимое количество дозированных единиц,
выходящих за пределы (1±Ь2 0,01)М (= с2), для количества образцов п
П(>)
к
с2
п(>)
к
с2
/»<>)
к
с2
я(>)
к
с2
п(>)
к
с2
п(>)
к
с2
100
2,15
804
2,26
7
2480
2,29
23
4366
2,30
41
6252
2,31
59
8243
2,31
78
105
2,16
905
2,27
2585
2,29
24
4471
2,30
42
6357
2,31
60
8347
2,31
79
120
2,17
0
908
2,27
8
2690
2,29
25
4576
2,30
43
6462
2,31
61
8452
2,31
80
139
2,18
1013
2,27
9
2794
2,29
26
4680
2,30
44
6566
2,31
62
8557
2,31
81
161
2,19
1118
2,27
10
2899
2,29
27
4785
2,30
45
6671
2,31
63
8662
2,31
82
176
2,19
1223
2,27
11
3004
2,29
28
4890
2,30
46
6776
2,31
64
8767
2,31
83
189
2,20
1
1276
2,28
3109
2,29
29
4995
2,30
47
6881
2,31
65
8871
2,31
84
224
2,21
1328
2,28
12
3171
2,30
5099
2,30
48
6985
2,31
66
8976
2,31
85
270
2,22
1432
2,28
13
3213
2,30
30
5204
2,30
49
7090
2,31
67
9081
2,31
86
280
2,22
1537
2,28
14
3318
2,30
31
5309
2,30
50
7195
2,31
68
9186
2,31
87
328
2,23
2
1642
2,28
15
3423
2,30
32
5414
2,30
51
7300
2,31
69
9290
2,31
88
385
2,23
1747
2,28
16
3528
2,30
33
5519
2,30
52
7404
2,31
70
9395
2,31
89
407
2,24
3
1851
2,28
17
3633
2,30
34
5623
2,30
53
7509
2,31
71
9500
2,31
90
490
2,24
1918
2,29
3737
2,30
35
5728
2,30
54
7614
2,31
72
9605
2,31
91
516
2,25
4
1956
2,29
18
3842
2,30
36
5833
2,30
55
7719
2,31
73
9710
2,31
92
594
2,25
2061
2,29
19
3947
2,30
37
5938
2,30
56
7824
2,31
74
9814
2,31
93
672
2,26
5
2166
2,29
20
4052
2,30
38
6042
2,30
57
7928
2,31
75
9919
2,31
94
699
2,26
6
2270
2,29
21
4156
2,30
39
6136
2,31
8033
2,31
76
2375
2,29
22
4261
2,30
40
6147
2,31
58
8138
2,31
77
%
г
2.9.47. Подтверждение однородности дозированных единиц
85
*
ВАРИАНТ 1 (ПАРАМЕТРИЧЕСКИЙ)
Отбирают не менее 100 единиц в соответствии
с предписанным планом отбора образцов.
Однородность дозированных единиц оцени¬
вается по количественному определению или по
массе, как указано в таблице 2.9.40.-1. Приемле¬
мое значение (АV) рассчитывают по формуле:
|М-Х|±/С5.
Значения величин, указанных в формуле, опре¬
деляются из таблицы 2.9.40.-2, кроме значений вели¬
чины к, зависящих от количества образцов и приве¬
денных в таблице 2.9.47.-1.
КРИТЕРИИ
Используют следующие критерии, если нет
других указаний в частной статье.
Требования к однородности дозированных форм
соблюдаются, если выполняются все следующие условия:
1. Приемлемое значение (АV) должно быть мень¬
шим или равным /_1;
2. При расчете приемлемого значения (АУ) для
метода прямого определения или для расчетно¬
весового метода количество отдельных дозированных
единиц, выходящих за пределы (1 ±/.2*0,01 )М, должно
быть меньшим или равным с2, как указано для коли¬
чества образцов п в таблице 2.9.47.-1.
Таблица 2.Э.47.-2
Допустимое количество отдельных дозированных единиц, выходящих за пределы (1±И0,01)Т (= с1)
и (1±12 0,01)Т (= с2) соответственно, для количества образцов п
/.(>)
с1
с2
п(>)
с1
с2
п<>)
с1
с2
Л(>)
с1
с2
Ий
с1
с2
/»(>)
с1
с2
лС>)
с1
с2
100
3
1432
35
2899
67
4366
98
5833
129
7300
160
8767
191
123
4
0
1476
36
13
2935
68
27
4377
99
41
5835
130
55
7304
161
69
8780
192
83
159
5
1521
37
2981
69
4424
100
5883
131
7351
162
8828
193
176
5
1537
37
3004
69
4471
101
5930
132
7399
163
8871
193
196
6
1
1566
38
14
3027
70
28
4518
102
42
5938
132
7404
163
8875
194
84
234
7
1611
39
3073
71
4565
103
5977
133
56
7447
164
70
8923
195
273
8
1642
39
3109
71
4576
103
6024
134
7494
165
8971
196
280
8
1656
40
15
3120
72
29
4612
104
43
6042
134
7509
165
8976
196
313
9
2
1701
41
3166
73
4658
105
6072
135
57
7542
166
71
9019
197
85
353
10
1746
42
3212
74
4680
105
6119
136
7589
167
9066
198
385
10
1747
42
3213
74
4705
106
44
6147
136
7614
167
9081
198
394
11
3
1791
43
16
3259
75
30
4752
107
6166
137
58
7637
168
72
9114
199
86
434
12
1836
44
3305
76
4785
107
6214
138
7684
169
9162
200
476
13
1851
44
3318
76
4799
108
45
6252
138
7719
169
9186
200
490
13
1882
45
17
3351
77
31
4846
109
6261
139
59
7732
170
73
9210
201
87
517
14
4
1927
46
3398
78
4890
109
6308
140
7779
171
9257
202
559
15
1956
46
3423
78
4893
110
46
6355
141
7824
171
9290
202
594
15
1972
47
18
3444
79
32
4940
111
6357
141
7827
172
74
9305
203
88
601
16
5
2018
48
3491
80
4987
112
6403
142
60
7875
173
9353
204
644
17
2061
48
3528
80
4995
112
6450
143
7922
174
9395
204
686
18
2063
49
19
3537
81
33
5034
113
47
6462
143
7928
174
9401
205
89
699
18
2109
50
3584
82
5081
114
6498
144
61
7970
175
75
9449
206
729
19
6
2154
51
3630
83
5099
114
6545
145
8017
176
9496
207
772
20
2166
51
3633
83
5128
115
48
6566
145
8033
176
9500
207
804
20
2200
52
20
3677
84
34
5175
116
6592
146
62
8056
177
76
9544
208
90
815
21
7
2246
53
3723
85
5204
116
6640
147
8113
178
9592
209
858
22
2270
53
3737
85
5222
117
49
6671
147
8138
178
9605
209
902
23
2291
54
21
3770
86
35
5269
118
6687
148
63
8160
179
77
9640
210
91
908
23
2337
55
3817
87
5309
118
6734
149
8208
180
9688
211
945
24
8
2375
55
3842
87
5317
119
50
6776
149
8243
180
9710
211
989
25
2383
56
22
3863
88
36
5364
120
6782
150
64
8256
181
78
9735
212
92
1013
25
2429
57
3910
89
5411
121
6829
151
8303
182
9783
213
1033
26
9
2475
58
3947
89
5414
121
6877
152
8347
182
9814
213
1077
27
2480
58
3956
90
37
5458
122
51
6881
152
8351
183
79
9831
214
93
1118
27
2520
59
23
4003
91
5505
123
6924
153
65
8399
184
9879
215
1121
28
10
2566
60
4050
92
5519
123
6972
154
8446
185
9919
215
1165
29
2585
60
4052
92
5552
124
52
6985
154
8452
185
9927
216
1209
30
2612
61
24
4097
93
38
5599
125
7019
155
66
8494
186
80
9975
217
1223
30
2658
62
4143
94
5623
125
7067
156
8542
187
10023
218
1253
31
11
2690
62
4156
94
5647
126
53
7090
156
8557
187
1298
32
2704
63
25
4190
95
39
5694
127
7114
157
67
8589
188
81
94
1328
32
2750
64
4237
96
5728
127
7161
158
8637
189
1342
33
2794
64
4261
96
5741
128
7195
158
8662
189
10070
219
12
2796
65
26
4284
97
40
54
7209
159
68
8685
190
1387
34
2843
66
4330
98
5788
129
82
2889
67
7256
160
8732
191
12. Зак. 1060.
86
Гэсударственная фармакопея Республики Беларусь
Если нет других указаний в частных статьях, то
/.1 равно 15,0 и 1.2 равно 25,0.
При использовании таблицы 2.9.47.-1 необходимо
учитывать следующее:
- для количества образцов п = 400 используют
табличные данные для значения п > 385: к = 2,23 и
с2 = 3;
-для количества образцов л = 450 используют
табличные данные для значения п > 407: к = 2,24 и
с2 = 3;
- для количества образца п = 500 используют
табличные данные для значения п > 490: к = 2,24 и
с2 = 4.
ВАРИАНТ 2 (НЕПАРАМЕТРИЧЕСКИЙ)
Отбирают не менее 100 единиц в соответствии с
предписанным планом отбора образцов.
Однородность дозированных единиц оценивает¬
ся по количественному определению или по массе,
как указано в таблице 2.9.40.-1. Проводят количе¬
ственное определение для каждой отдельной едини¬
цы или взвешивают единицы и рассчитывают содер¬
жание, как указано в общей статье 2.9.40. Считают
количество отдельных дозированных единиц, выхо¬
дящих за пределы (1±/.10,01)Г, и количество отдель¬
ных дозированных единиц, выходящих за пределы
(1±/.2-0,01)7". Оценка проводится, если значения на¬
ходятся в пределах, указанных в таблице 2.Э.47.-2.
КРИТЕРИИ
Используют следующие критерии, если нет
других указаний в частной статье.
Требования к однородности дозированных форм
соблюдаются, если выполняются все следующие ус¬
ловия:
1. Количество отдельных дозированных единиц,
выходящих за пределы (1±М-0,01)7, должно быть
меньшим или равным с1 ;
2. Количество отдельных дозированных единиц,
выходящих за пределы (1 ±/.2-0,01 )Т, должно быть
меньшим или равным с2.
Значения с1 и с2 для количества образцов п
приведены в таблице 2.Э.47.-2.
Если нет других указаний в частных статьях, то
71 равно 15,0 и /_2 равно 25,0.
При использовании таблицы 2.9.47.-2 необходимо
учитывать следующее:
- для количества образцов п = 400 используют
табличные данные для значения п >394: с1 = 11 и
с2 = 3;
- для количества образцов п = 450 используют
табличные данные для значения п >434: с1 = 12 и
с2 = 3;
- для количества образцов п = 500 используют
табличные данные для значения п>490: с1 = 13 и
с2 = 4.
4.1.1. Реактивы
87
01/2013:40000
4. РЕАКТИВЫ
Дополнительную информацию о реактивах,
которые могут быть идентифицированы только
по торговым наименованиям или доступность ко¬
торых ограничена, можно найти на сайте ЕйОМ в
разделе Оа^Ьавез: Кпом/едде Оа/аЬазе. Эта инфор¬
мация приводиться только для облегчения поиска
таких реактивов и не несет никаких особых реко¬
мендаций или подтверждения их сертификации Ев¬
ропейской фармакопейной комиссией или Советом
Европы. Допускается использование реактивов из
других источников на основании их соответствия
требованиям Фармакопеи.
07/2016:40101
4.1.1. РЕАКТИВЫ
Алдрин. С12Н8С16. (М.м. 364,9). 1123100. [309-00-2].
Температура кипения: около 145 °С.
Температура плавления: около 104 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
4-Аминофолиевая кислота. С19Н20М8О5. (М.м.
440,4). 1163700. [54-62-6]. (25)-2-[[4-[[(2,4-Диамино-
птеридин-6-ил)метил]амино]бензоиол]амино]пентан-
дионовая кислота. /^-[4-[[(2,4-Диаминоптеридин-6-ил)-
уегил}а1У1ино]бенэоиол}-Ь-глутаминовая кислота. Ами-
носттерин.
Желтоватый порошок.
Температура плавления, около 230 °С.
Р-Амирин. С^Н^О. (М.м. 426,7). 1141800. [559-70-6].
Олеан-12-ен-Зр-ол.
Белый или почти белый порошок.
Температура плавления: от 187 °С до 190 °С.
Аммония сульфида раствор. 1123300.
Насыщают 120 мл раствора аммиака разведен¬
ного Р1 сероводородом Р и прибавляют 80 мл раст¬
вора аммиака разведенного Р1. Готовят непосред¬
ственно перед применением.
Аммония хлорида буферный раствор рН 10,0.
4007300.
5,4 г аммония хлорида Р растворяют в 20 мл
воды Р, прибавляют 35,0 мл раствора аммиака Р и
доводят водой Р до 100,0 мл.
Аммония хлорида буферный раствор рН 9,5.
4007200.
33,5 г аммония хлорида Р растворяют в 150 мл
воды Р, прибавляют 42,0 мл раствора аммиака кон¬
центрированного Р и доводят водой Р до объема
250,0 мл.
Хранят в полиэтиленовом контейнере.
Алилактоза. С12Н2201Г (М.м. 342,3). 1189200.
[20869-27-6]. 4-О-Р-О-Галактопиранозил-О-маннопира-
ноза.
Содержит не менее 98 % апилактозы.
4-Бензилпиридин. С^Н^М. (М.м. 169,2). 1181200.
[2116-65-6].
Содержание: не менее 98,0 %.
Желтая жидкость.
Температура плавления: от 72 °С до 78 °С.
Бромофос. С8Н8ВгС1203РЗ. (М.м. 366,0). 1123700.
[2104-96-3].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
Бромофос-этил. С10Н12ВгС12О3Р5. (М.м. 394,0).
1123800. [4824-78-6].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
Бромтимолового синего раствор Р4. 1012904.
100 мг бромтимолового синего Р растворяют в
смеси из равных объемов 96 % спирта Р и воды Р и
доводят до объема 100 мл этой же смесью раствори¬
телей. При необходимости фильтруют.
Вода дистилированная деионизированная.
1095508
Деионизированную водуР получают дестилля-
цией. Значение удельного сопротивления не менее
0,18 МОм-м.
Гексахлорбензол. С6С18. (М.м. 284,8). 1128200.
[118-74-1].
Температура кипения: около 332 °С.
Температура плавления: около 230 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
а-Гексахлорциклогексан. С6Н6С16. (М.м. 290,8).
1128300. [319-84-6].
Температура кипения: около 288 °С.
Температура плавления: около 158 °С.
Может быть использован подходящий сертифи¬
цированный раствор сравнения (10 нг/мкл в циклогек¬
сане).
В-Гексахлорциклогексан. С„НКС1. (М.м. 290,8).
1128400. [319-85-7].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
б-Гексахлорциклогексан. С6Н6С16. (М.м. 290,8).
1128500. [319-86-8].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Гептафтормасляная кислота. С4НР702. (М.м.
214,0). 162400. [375-22-4]. НЕВА.
Содержание: не менее 99,5 %.
Прозрачная, бесцветная, коррозионно-активная
жидкость.
б2около 1,645.
л^0: около 1,300.
Температура кипения: около 120 °С.
Гептафтор-/У-метил-М-(триметилсилил)бутана-
мид. С8Н12Р7М05|. (М.м. 299,3). 1139500. [53296-64-3].
2,2,3,3,4,4,4-Гептафтор-Л/-метил-Л/-(триметилсилил)бу-
тирамид.
Прозрачная, бесцветная жидкость, легковоспла¬
меняющаяся.
Л^°: около 1,351.
Температура кипения: около 148 °С.
Гептахлор. С10Н5С17. (М.м. 373,3). 1128000. [76-44-8].
Температура кипения: около 135 °С.
Температура плавления: около 95 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Гептахлорэпоксид. С10Н5С17О. (М.м. 389,3).
1128100. [1024-57-3].
Температура кипения: около 200 °С.
Температура плавления: около 160 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
12*. Зак 1060.
88
Государственная фармакопея Республики Беларусь
2-Гидроксибензимидазол. С7Н61Ч20. (М.м. 134,1).
1169600. [615-16-7]. 1Н-Бензимидазол-2-ол.
4-Гидроксибензогидразид. С7Н8Ы202. (М.м. 152,2).
1145900. [5351-23-5]. л-Гидроксибензогидразид.
Глицина ангидрид. С4Н6М202. (М.м. 114,1). 1192200.
[106-57-0]. Пиперазин-2,5-дион (2,5-ОКР).
о,л'-ДДД. С14Н10С14. (М.м. 320,0). 1125200. [53-19-0].
1-(2-Хлорфенил)-1-(4-хлорфенил)-2,2-дихлорэтан.
Может быть использован подходящий сертифи¬
цированный раствор сравнения (10 нг/мкл в циклогек¬
сане).
л,/т'-ДЦД. С14Н10С14. (М.м. 320,0). 1125300. [72-54-8].
1.1 -бис(4-Хл орфенил )-2,2-дихлорэтан.
Температура кипения: около 193 °С.
Температура плавления: около 109 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
о,л'-ДДЕ. С14Н8С14. (М.м. 318,0). 1125400. [3424-82-6].
1 -(2-Хлорфенил)-1 -(4-хлорфенил)-2,2-дихлорэтилен.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
/7,/7,-ДДЕ. С14Н8С14. (М.м. 318,0). 1125500. [72-55-9].
1.1 -бис(4-Хлорфенил)-2,2-дихлорэтилен.
Температура кипения: от 316 °С до 317 °С.
Температура плавления: от 88 °С до 89 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
о,л'-ДДТ. С14Н9С15. (М.м. 354,5). 1125600. [789-02-6].
1- (2-Хлорфенил)-1-(4-хлорфенил)-2,2,2-трихлорэтан.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
/7,/7'-ДДТ. С14Н9С15. (М.м. 354,5). 1125700. [50-29-3].
1.1 -бис(4-Хлорфенил )-2,2,2-трихлорэтан.
Температура кипения: около 260 °С.
Температура плавления: от 108 °С до 109 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
4-Дезоксипиридоксина гидрохлорид. ОдН^О^НС!.
(М.м. 189,6). 1175500. [148-51-6]. б-(Гидроксиметил)-
2,4-диметилпиридин-З-ол.
Дельтаметрин. С^Н^В^МС^. (М.м. 505,2). 1125800.
[52918-63-5].
Температура кипения: около 300 °С.
Температура плавления: около 98 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Диазинон. С12Н21М203Р5. (М.м. 304,3). 1125900.
[333-41-5].
Температура кипения: около 306 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
Диглицин. С4НД03. (М.м. 132,1). 1191700. [556-50-3].
2- [(2-Аминоацетил)амино]уксусная кислота. Глицил-
глицин.
Триглицин. (М.м. 189,2). 1192600. [556-33-2].
2-[[2-[(2-Аминоацетил)амино]ацетил]амино]уксусная
кислота. Глицилглицилглицин.
Дидокозагексаеноин. С47Н6805. (М.м. 713,0).
1142700. [88315-12-2]. Диглицерид докозагексаеноевой
кислоты (С22:6). Глицерина дидокозагексаеноат.
Диэфир пропан-1,2,3-триола и (аИ-7)-докозагексаеновой
кислоты.
М,М'-Диизопропилэтилендиамин. С8Н20М2.
(М.м. 144,3). 1140600. [4013-94-9]. /У,/У'-Бис(1-метилэтил)-
1,2-этандиамин.
Бесцветная или желтоватая, вызывающая корро¬
зию, легковоспламеняющаяся, гигроскопичная жидкость.
с!™: около 0,798.
п1°: около 1,429.
Температура кипения: около 170 °С.
Диметиламинобензальдегид.СдН^МО. (М.м. 149.2).
1029800. [100-10-7]. 4-Диметиламинобензальдегид.
Белые или желтовато-белые кристаллы. Раство¬
рим в 96 % спирте и в разведенных кислотах.
Температура плавления: около 74 °С.
Диметиламинобензальдегида раствор Р9.1029806.
1,0 г диметиламинобензальдегида Р раство¬
ряют в 3,5 мл кислоты хлорной (600 г/л НСЮ4) и мед¬
ленно прибавляют 6,5 мл 2-пропанола Р.
УУ,/У-Д и метил-1_-фенил аланин. С^Н^МО^
(М.м. 193,2). 1164000. [17469-89-5]. (25)-2-(Диметилами-
но)-3-фенилпропановая кислота.
Температура плавления: около 226 °С.
Диталимфос. С12Н141\104Р5. (М.м. 299,3). 1126700.
[5131-24-8]. 0,0-Диэтил(1,3-дигидро-1,3-диоксо-2Н-
изоиндол-2-ил)фосфонотиоат.
Очень мало растворим в воде, в этилацетате и в
этаноле.
Может быть использован подходящий сертифи¬
цированный раствор сравнения.
Дихлофентион. С10Н13С12О3РЗ. (М.м. 315,2).
1126100. [97-17-6].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Диэлдрин. С12Н8С160. (М.м. 380,9). 1126200.
[60-57-1].
Температура кипения: около 385 °С.
Температура плавления: около 176 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
й-Допа. СдИ^О,. (М.м. 197.2). 1164100. [5796-17-8].
(2Я)-2-Амино-3-(3,4-дигидроксифенил)пропановая кис¬
лота. З-Гидрокси-й-тирозин. 3,4-Дигидрокси-О-фенил-
аланин.
[«]о°: От +9,5 до +11,5. Определение проводят,
используя раствор 10 г/л в 1 М растворе кислоты
хлористоводородной.
Температура плавления: около 277 °С.
Железа (ДО) сульфата раствор. 1037901.
50 г железа (III) сульфата Р растворяют в избыт¬
ке воды Р, прибавляют 200 мл кислоты серной Р и
доводят водой Р до объема 1000 мл.
#Железа (III) хлорида и кислоты уксусной реактив.
75 мг железа (III) хлорида Р растворяют в 50 мл
кислоты уксусной безводной Р и при интенсивном
встряхивании и охлаждении прибавляют 50 мл кис¬
лоты серной Р. Используют свежеприготовленный
раствор.
Изодрин. С12Н8С16. (М.м. 364,9). 1128700. [465-73-
6]. 1,2,3,4,10,10-Гексахлор-1,4,4а,5,8,8а-гексагидро-
эндо,эндо- 1,4:5,8-диметанонафталин.
Практически нерастворим в воде, растворим
в распространенных органических растворителях,
таких как ацетон.
Может быть использован подходящий сертифи¬
цированный раствор сравнения.
4.1.1. Реактивы
89
Изолейцин. 380 х 280 х 70 1185000. [73-32-5].
См. статью Изолейцин (0770).
Изоникотинамид. С6НбМ20. (М.м. 122,1). 1193000.
[1453-82-3]. 4-Пиридинкарбоксамид. Пиридин-4-
карбоксамид.
Белый или почти белый кристаллический поро¬
шок. Растворим в воде.
Иминодиуксусная кислота. С4Н71Ч04. (М.м. 133,1).
1192300. [142-73-4]. 2,2-Иминодиуксусная кислота.
Йода раствор Р5. 1045807.
12,7 г йода Р и 20 г калия йодида Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же раст¬
ворителем (0,05 М раствор).
Йодплатината реактив Р1. 1172200.
2,5 мл раствора 50 г/л кислоты хлорплатино-
вой Р смешивают с 22,5 мл раствора 100 г/л калия
йодида Р и 50 мл воды Р.
Хранят в защищенном от света месте при темпе¬
ратуре от 2 °С до 8 °С.
Кадмия нитрат тетрагидрат. Сф1\Ю3)2в4Н20.
1174900. [10022-68-1].
Гигроскопичные орторомбические кристаллы.
Очень легко растворим в воде, растворим в ацетоне
и в 96 % спирте.
Температура плавления: около 59,5 °С.
Карбофенотион. С11Н16СЮ2Р83. (М.м. 342,9).
1016200. [786-19-6]. 0,ОДиэтил-5-[[(4-хлорфенил)тио]
метил]-фосфордитионат.
Желтоватая жидкость. Практически нерастворим
в возе смешивается с органическими растворителями.
й^: около 1,27.
Для частной статьи Ланолин (0134) может быть
использован подходящий сертифицированный раст¬
вор сравнения (10 нг/мкл в изооктане).
Кариофиллена оксид. С15Н240. (М.м. 220,4).
1149000. [1139-30-6]. (-)-Р-Кариофиллена эпоксид.
(1 РАР,6Р, 105)-4,12,12-Триметил-9-метилен-5-оксатри-
цикл о[8.2.0.04 6]додекан.
Бесцветные мелкие кристаллы с комками.
Температура плавления: от 62 °С до 63 °С.
Кариофиллена оксид, используемый в газовой
хроматографии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. Газовая хрома¬
тография (2.2.28), как указано в статье Терпентинное
масло, тип Ртиз р'таз1ег.
Содержание: не менее 99,0 %, рассчитывают ме¬
тодом нормализации.
Карминовая кислота. С22Н20О13. (М.м. 492,4).
1156700. [1260-17-9].
7-а-0-Глюкопиранозил-3,5,6,8-тетрагидрокси-1-
метил-9,10-диоксо-9,10-дигидроантрацен-2-карбоно-
вая кислота.
Темно-красный порошок, очень мало раство¬
рим в воде, растворим в диметилсульфоксиде, очень
мало растворим в 96 % спирте.
Кремнийорганический полимер, аморфный, по¬
лярно-встроенный октадецилсилильный, эндкепи-
рованный. 1150600.
Синтетические сферические гибридные частицы,
состоящие как из неорганических (кремний), так и ор¬
ганических (органосилоксан) компонентов, с поверх¬
ностью, химически модифицированной путем свя¬
зывания полярно-встроенных октадецилсилильных
групп. Для минимизации каких-либо взаимодействий
с соединениями основного характера его тщательно
эндкепируют для закрытия большинства оставшихся
силанольных групп. Размер частиц указывают после
названия реактива в испытании, в котором он исполь¬
зуется.
#Ксиленцианол РР. С25Н27М2Ма0682. (М.м. 538,61).
[2650-17-1]. Ксилен циановый РР. Кислотный синий 147.
Порошок от серо-синего до темно-синего цвета.
Растворим в воде.
Количественное определение. 0,050 г испы¬
туемого образца растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем. 2,0 мл по¬
лученного раствора доводят фосфатным буферным
раствором рН 7,0 Р до объема 50,0 мл.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора в кюветах 1 см при длине волны
максимума поглощения около 614 нм, используя в ка¬
честве компенсационного раствора воду Р. Удельный
показатель поглощения должен быть не менее 55,9,
что соответсвует около 83 % С25Н27Ы2МаОб82.
Потеря в массе при высушивании (2.2.32). Не
более 6,0 %. 1,000 г испытуемого образца сушат при
температуре 110 °С.
Кумафос. С14Н16СЮ5Р8. (М.м. 362,8). 1124800.
[56-72-4].
Температура плавления: от 91 °С до 92 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
Лейцин. 1048500. [61-90-5].
См. статью Лейцин (0771).
Линдан. С6Н6С16. (М.м. 290,8). 1128900. [58-89-9].
у-Гексахлорциклогексан.
Для частной статьи Ланолин (0134) может быть
использован подходящий сертифицированный раст¬
вор сравнения (10 нг/мкл в циклогексане).
Лонгифолен. С15Н24. (М.м. 204, 4). 1150300. [475-
20-7]. (15,ЗаЯ,45,8а5)-4,8,8-Триметил-9-метилендека-
гидро-1,4-метаназулен.
Маслянистая бесцветная жидкость. Практически
нерастворим в воде, смешивается с 96 % спиртом.
0,9319.
/?о°: 1,5050.
[«]“: +42,7.
Температура кипения: от 254 °С до 256 °С.
Лонгифолен, используемый в газовой хромато¬
графии, должен выдерживать следующее дополни¬
тельное испытание.
Количественное определение. Газовая хромато¬
графия (2.2.28), как указано в статье Терпентинное
масло, тип Ртиз ртаз1ег (1627).
Содержание: не менее 98,0 %, рассчитывают ме¬
тодом нормализации.
Лютеолин-7-глюкозид. С21Н20О1Г (М.м. 448,4).
1163400. [5373-11-5]. 2-(3,4-Дигидроксифенил)-7-(Р-0-
глюкопиранозилокси)-5-гидрокси-4/-/-1-бензпиран-4-он.
Желтый порошок.
Оптическая плотность (2.2.25). Раствор в ме¬
таноле Р имеет максимумы поглощения при 255 нм,
267 нм и 350 нм.
Температура плавления: около 247 °С.
Магния силикат для анализа остаточных пе¬
стицидов. 1129100. [1343-88-0].
Магния силикат для хроматографии ((60—100) меш).
13. Зак. 1060.
90
Гэсударстеенная фармакопея Республики Беларусь
Малатион. С10Н19О6Р52. (М.м. 330,3). 1129200.
[121-75-5].
Температура кипения: около 156 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
#Менадион. [58-27-5].
См. статью Менадион (0507).
#2-Метил-1,4-нафтохинон.
См. Менадион Р.
Метилаль. С3Н802. (М.м. 76,1). 1173500. [109-87-5].
Диметоксиметан. Диоксапентан. Формальдегида диме-
тилацеталь. Метилендиметиловый эфир.
Прозрачная, бесцветная, летучая, воспламеняю¬
щаяся жидкость. Образец растворим в воде и смеши¬
вается с 96 % спиртом.
(У22°: 0,860.
Ло°: 1,354.
Температура кипения: около 41 °С.
Метилаль, используемый в газовой хромато¬
графии, должен выдерживать следующее дополни¬
тельное испытание.
Содержание: не менее 99,5 %, при определении
методом газовой хроматографии.
Метилйодид. СН31. (М.м. 141,9). 1166400. [74-88-4].
Йодометан.
3-Метил пентан-2-он. С6Н120. (М.м. 100,2;. 1141100.
[565-61-7].
Бесцветная, легковоспламеняющаяся жидкость.
с/20- около 0,815.
20
П0 : около 1,400.
Температура кипения: около 118 °С.
Л/-Метилпирролидин. СДД (М.м. 85,2). 1164700.
[120-94-5].
Содержание: не менее 97,0 %.
Температура кипения: около 80 °С.
М-Метил-м-толуидин. СДД (М.м. 121,2). 1175200.
[696-44-6]. Д/,3-Диметиланилин. А/,3-Диметилбензамин.
Метил-м-толиамин.
Содержание: не менее 97 %.
/У-Метилтриметилсилил-трифторацетамид.
С6Н12Р^ОЗ|. (М.м. 199,3). 1129600. [24589-78-4].
2,2,2-Трифтор-Л/-метил-Л/-(триметилсилил)ацетамид.
20
п0 : около 1,380.
Температура кипения: от 130 °С до 132 °С.
Метилциклогексан. С7Н14. (М.м. 98,2). 1189900.
[108-87-2].
1_-Метионин сульфоксид. СД^О^. (М.м. 165,2).
1193300. [3226-65-1]. (25)-2-Амино-4-[(Я5)-метилсуль-
финил]бутановая кислота.
6-Метокси-2-нафтойная кислота. С12Н10О3.
(М.м. 202,2). 1184200. [2471-70-7]. 6-Метоксинафтол-2-
карбоновая кислота.
Белый или почти белый кристаллический порошок.
Температура плавления: от 201 °С до 206 °С.
З-Метокси-Ьтирозин. С10Н13МО4Н2О. (М.м. 229,2).
1164400. [200630-46-2].
Порошок белого или почти белого цвета.
Метоксихлор. С16Н15С1303. (М.м. 345,7). 1129300.
[72-43-5]. 1,1-(2,2,2-Трихлорэлилиден)-бис(4-метоксибен-
зол).
Практически нерастворим в воде, легко раство¬
рим в большинстве органических растворителей.
Температура кипения: около 346 °С.
Температура плавления: от 78 °С до 86 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
Монодокозагексаеноин. С25Н3804. (М.м. 402,6).
1143600. [124516-13-8]. Моноглицерид докозагексае-
ноевой кислоты (С22:6). Глицерина монодокозагекса-
еноат. Моноэфир пропан-1,2,3-триола и (а//-2)-докоза-
4,7,10,13,16,19-гексаеновой кислоты.
Натрия пентансульфонат моногидрат.
СД^аОДНА (М.м. 192,2). 1132100. [22767-49-3].
Белое или почти белое кристаллическое твердое
вещество. Растворим в воде.
Никеля нитрат гексагидрат. Ы|(М03)2*6Н20. (М.м.
290,8). 1175300. [13478-00-7].
№-Нитрозодиизопропаноламин. СД4М203. (М.м.
162,2). 1176500. [53609-64-6]. 1,1'-(Нитрозоимино)-
биспропан-2-ол.
Температура кипения: от 122 °С до 124 °С.
Л/-Нитрозодиэтаноламин. СДсЫ203. (М.м. 134,1).
1129800. [1116-54-7]. 2,2'-(Нитрозоимино)диэтанол.
Жидкость желтого цвета. Смешивается с этанолом.
20
П0 : около 1,485.
Температура кипения: около 125 °С.
2,2'-Оксибис(М,М-диметилэтиламин). с8н20м2о.
(М.м. 160,3). 1141200. [3033-62-3]. Бис(2-диметил-
аминоэтиловый) эфир.
Бесцветная, вызывающая коррозию жидкость.
дЦ: около 0,85.
20
п0 : около 1,430.
Пентафторпропановая кислота. С3НР502. (М.м.
164,0). 1151100. [422-64-0].
Прозрачная, бесцветная жидкость.
С^: около 1,561.
П%°: около 1,284.
Температура кипения: около 97 °С.
Перилен. С20Н12. (М.м. 252,3). 1130100. [198-55-0].
Дибенз(с/е,/с/)антрацен.
Порошок оранжевого цвета.
Температура плавления: около 279 °С.
Перметрин. С21Н20С12О3. (М.м. 391,3). 1130000.
[52645-53-1].
Температура плавления: от 34 °С до 35 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Пиримифос-этил. С13Н24М303РЗ. (М.м. 333,4).
1130300. [23505-41-1].
Температура плавления: от 15 °С до 18 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Пирролидин. С4НД (М.м. 71,1). 1165000. [123-75-1].
Содержание: не менее 99 %.
Температура кипения: от 87 °С до 88 °С.
Поли(диметил)(дифенил)силоксан, деактиви¬
рованный по отношению к основаниям. 1176600.
Неподвижная фаза для газовой хроматографии,
деактивированная по отношению к основаниям, спе¬
циально созданная для анализа аминов.
Содержит 95 % метильных групп и 5 % фениль-
ных групп.
Пролин. 1152200. [147-85-3].
См. статью Пролин (0785).
4.1.1. Реактивы
91
Пропанол Р1. 1184400. [71-23-8].
См. статью Пропанол (2036).
2-Пропанол Р2. 1184900. [67-63-0].
См. статью Пропанол (0970).
Пропетамфос. С10Н20МО4Р5. (М.м. 281,3). 1130900.
[31218-83-4].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Силикагель алкил-связанный для использова¬
ния с подвижными фазами с высоким содержани¬
ем воды, эндкепированный, для хроматографии.
1176900.
Очень мелкий силикагель со связанными алкиль¬
ными группами, подходящий для использования с
подвижными фазами с высоким содержанием воды.
Для сведения к минимуму взаимодействия с основны¬
ми соединениями тщательно эндкепируют для устра¬
нения большинства оставшихся силанольных групп.
Размер частиц указывают после названия реактива в
испытании, в котором он используется.
Силикагель для хроматографии, фенилгексил-
силильный, эндкепированный. 1170600.
Силикагель очень тонко измельченный с разме¬
ром частиц 3 мкм и поверхностью, химически моди¬
фицированной фенилгексилсилильными группами.
Для сведения к минимуму взаимодействия с основ¬
ными соединениями тщательно эндкепируют для
устранения большинства силанольных групп. Размер
частиц указывают после названия сорбента в испыта¬
ниях, в которых он используется.
Силикагель АО для хиральных разделений.
1171700.
Силикагель для хроматографии очень тонко из¬
мельченный с размером частиц 5 мкм, покрытый сле¬
дующим производным:
Силикагель для хроматографии, амидогекса-
децилсил ильный. 1170400.
Силикагель очень тонко измельченный с части¬
цами небольшого размера и поверхностью, химиче¬
ски модифицированной амидогексадецилсилильными
группами. Размер частиц указывают после названия
сорбента в испытаниях, в которых он используется.
Силикагель для хроматографии, диизобути-
локтадецилсилильный. 1140000.
Силикагель очень тонко измельченный с поверх¬
ностью, химически модифицированной диизобути-
локтадецилсилильными группами. Размер частиц
указывают после названия реактива в испытаниях, в
которых он используется.
Силикагель для хроматографии, октадецилси-
лильный, поверхностно-пористый, эндкепирован¬
ный. 1193900.
Силикагель со сферическими частицами, состоя¬
щими из непористого ядра диоксида кремния, покры¬
того тонким слоем пористого диоксида кремния, хи¬
мически модифицированного октадецилсилильными
группами. Для сведения к минимуму взаимодействия
с основными соединениями тщательно эндкепируют
для устранения большинства оставшихся силаноль¬
ных групп. Размер частиц указывают после названия
сорбента в испытаниях, в которых он используется.
Силикагель для хроматографии, окгадецилси-
лильный, эндкепированный Р1. 1115401.
Силикагель сверхчистый, очень тонко измель¬
ченный, с размером пор 10 нм и поверхностью, хи¬
мически модифицированной октадецилсилильными
группами (содержит 19 % углерода). Для сведения к
минимуму взаимодействия с основными соединения¬
ми тщательно эндкепируют для устранения большин¬
ства оставшихся силанольных групп. Размер частиц
указывают после названия сорбента в испытаниях, в
которых он используется.
Содержание металлов должно быть менее
20 ррт.
Силикагель для хроматографии, совместимый
с подвижными фазами со 100 % содержанием воды,
октадецилсилильный, эндкепированный. 1188400.
Очень мелкий силикагель со связанными октаде¬
цилсилильными группами, подходящий для использо¬
вания с подвижными фазами с высоким содержанием
воды, включая подвижные фазы со 100 % содержани¬
ем воды. Для сведения к минимуму взаимодействия
с основными соединениями тщательно эндкепируют
для устранения большинства оставшихся силаноль¬
ных групп. Размер частиц указывают после названия
реактива в испытании, в котором он используется.
Силикагель для хроматографии, фенилсилиль-
ный, эндкепированный. 1154900.
Силикагель очень тонко измельченный с разме¬
ром частиц от 5 мкм до 10 мкм и поверхностью, хи¬
мически модифицированной фенильными группами.
Для сведения к минимуму взаимодействия с основны¬
ми соединениями тщательно эндкепируют для устра¬
нения большинства оставшихся силанольных групп.
Размер частиц указывают после названия сорбента в
испытаниях, в которых он используется.
Р-Ситостирол. С29Н50О. (М.м. 414,7). 1140200.
[83-46-5]. Стигмаст-5-ен-Зр-ол. 22,23-Дигидростигма-
стирол.
Белый или почти белый порошок, практически
нерастворим в воде, умеренно растворим в тетраги-
дрофуране.
Содежание: не менее 75,0 % (м/м) (в пересчете
на сухое вещество).
Количественное определение. Газовая хромато¬
графия (2.2.28), как указано в статье Фитостирол.
Испытуемый раствор. 0,100 г испытуемого
образца растворяют в тетрагидрофуране Р и до¬
водят этим же растворителем до объема 10,0 мл.
100 мкл полученного раствора переносят в подхо¬
дящую колбу объемом 3 мл и выпаривают досуха в
потоке азота Р. К полученному остатку прибавляют
100 мкл свежеприготовленной смеси из 50 мкл 1-ме-
тилимидазола Р и 1,0 мл гептафтор-И-метил-
И-(триметилсилил)бутанамида Р. Колбу плотно
закрывают, нагревают при температуре 100 °С в те¬
чение 15 мин и охлаждают.
Объем вводимой пробы: 1 мкл испытуемого раствора.
Станолон. С19Н30О2. (М.м. 290,4). 1154400.
[521-18-6]. 17Р-Гидрокси-5а-андростан-3-он.
Белый или практически белый порошок.
Температура плавления: около 180 °С.
92
Гэсударственная фармакопея Республики Беларусь
Текназол. СбНС14М02. (М.м. 260,9). 1132400.
[117-18-0].
Температура кипения: около 304 °С.
Температура плавления: от 99 °С до 100 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Теобромин. 1138800. [83-67-0].
См. статью Теобромин (0298).
1,2,3,4-Тетра-0-ацетил-(3-0-глюкопираноза.
С14Н20°ю- (Мж 348’3)- 1172600. [13100-46-4].
Белый или почти белый порошок. Растворим в
воде при легком нагревании.
[а]™: + 11, используют раствор 6 г/л в хлорофор¬
ме Р.
Температура плавления: от 126 °С до 128 °С.
1,1,3,3-Тетраметилбутиламин. С8Н19М. (М.м. 129,3).
1141500. [107-45-9]. 2-Амино-2,4,4-триметилпентан.
Прозрачная, бесцветная жидкость.
с!20- около 0,805.
Л™: около 1,424.
Температура кипения: около 140 °С.
Тетрахлорвинфос. С10Н9С14О4Р. (М.м. 366,0).
1132500. [22248-79-9].
Температура плавления: около 95 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
Тридокозагексаеноин. Сб9Н9806. (М.м. 1023,5).
1144900. [124596-98-1]. Триглицерид докозагексаено-
евой кислоты (С22:6). Глицерина тридокозагексаено-
ат. Пропан-1,2,3-триила три-(а//-2)-докоза-4,7,10,13,16,19-
гексаеноат.
Установлено, что реактив производства Ыи-СЬек
Ргер, /появляется подходящим.
Триметилпентан для хроматографии. 1093402.
Должен соответствовать требованиям, как указа¬
но для триметилпентана Р, а также должен выдер¬
живать следующее дополнительное испытание.
Остаток после выпаривания: не более 2 мг/л.
М,0-Бис(триметилсилил)трифторацетамид.
С8Н18Р31Ч0812. (М.м. 257,4). 1133200. [25561-30-2]. БСТФА.
Бесцветная жидкость.
бЦ: около 0,97.
/?о° • около 1,38.
Температура кипения,2ии: около 40 °С.
Триметилсульфония гидроксид. с3н10оз.
(М.м. 94,2). 1145000. [17287-03-5].
с/4°: около 0,81.
Трис(гидроксиметил)аминометана раствор Р1.
1094202.
60,6 мг трис(гидроксиметил)аминометана Р и
0,234 г натрия хлорида Р растворяют в воде Р и до¬
водят этим же растворителем до объема 100 мл.
Хранение: при температуре от 2 °С до 8 °С, ис¬
пользуют в течение 3 дней.
Триэтиламин Р1. С6Н15М. (М.м. 101,2). 1093001.
[121-44-8]. А/,А/-Диэтилэтанамин.
Должен выдерживать требования для триэти-
ламина Р и следующие дополнительные требования.
Содержание: не менее 99,5 % (газовая хромато¬
графия).
Вода. Не более 0,1 %.
Используют свежеперегнанный или непосред¬
ственно после вскрытия контейнера.
Триэтиламин Р2. С6Н15М. (М.м. 101,2). 1093002.
[121-44-8]. А/,А/-Диэтилэтанамин.
Должен выдерживать требования для триэти-
ламина Р и следующие дополнительные требования.
Содержание: не менее 99,5 % (газовая хромато¬
графия).
Вода. Не более 0,2 %.
Подходит для градиентного элюирования в жид¬
костной хроматографии.
Используют свежеперегнанный или непосред¬
ственно после вскрытия контейнера.
ТСХ пластинка со слоем силикагеля для опре¬
деления аминополиэфира. 1172700.
ТСХ пластинку со слоем силикагеля Р погружа¬
ют в реактив йодплатината Р1 на (5—10) с. Сушат
при комнатной температуре в течение 12 ч в защи¬
щенном от света месте.
Хранят в защищенном от света месте в открытом
контейнере. Используют в течение не более 30 дней
после приготовления.
Уксусная кислота разведенная Р1. 1000403.
Содержание: не менее 57,5 г/л и не более 62,5 г/л
(М.м. 60,1).
6 г кислоты уксусной ледяной Р доводят водой Р
до объема 100 мл.
Фенвалерат. С25Н2201МО3. (М.м. 419,9). 1127300.
[51630-58-1].
Температура кипения: около 300 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Фенхлорфос. С8Н8С1303Р5. (М.м. 321,5). 1127200.
[299-84-3].
Температура плавления: около 35 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Фозалон. С12Н15С1М04Р32. (М.м. 367,8). 1130200.
[2310-17-0].
Температура плавления: от 45 °С до 48 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в изооктане).
2-Фтор-2-дезокси-0-глюкоза. С6Н11Р05. (М.м.
182,2). 1113900. [86783-82-6].
Белый или почти белый кристаллический порошок.
Температура плавления: от 174 °С до 176 °С.
2-Фтор-2-дезокси-О-манноза. С^Н^РОд.
(М.м. 182,1). 1172100. [38440-79-8].
Бесцветная мягкая масса.
2-Хлор-2-дезокси-0-глюкоза. СдН^СЮд.
(М.м. 198,6). 1134700. [14685-79-1].
Белый или почти белый кристаллический поро¬
шок. Очень гигроскопичен. Растворим в воде и ди-
метилсульфоксиде, практически нерастворим в 96 %
спирте.
Хлористоводородной кислоты метанольный
раствор. 1043511.
4,0 мл кислоты хлористоводородной Р доводят
метанолом Р2 до объема 1000,0 мл.
Хлорпирифос. С0Н11СГМО,Р5. (М.м. 350,6).
1124400. [2921-88-2].
Температура кипения: около 200 °С.
Температура плавления: от 42 °С до 44 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
4.2.2. Титрованные растворы
93
Хлорпирифос-метил. С7Н7С131\Ю3Р5. (М.м. 322,5).
1124500. [5598-13-0].
Температура плавления: от 45 °С до 47 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Хлорфенвинфос. С12Н14С1304Р. (М.м. 359,6).
1124200. [470-90-6].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Цепгилпиридина хлорид моногидрат. С21Н38С1М Н20.
(М.м. 358,0). 1162800. [6004-24-6]. 1-Гексадецилпири¬
дина хлорид моногидрат.
Белый или практически белый порошок. Легко
растворим в воде и в 96 % спирте.
Температура плавления: от 80 °С до 83 °С.
Циперметрин. С22Н19С12Ш3. (М.м. 416,3). 1125100.
[52315-07-8].
Температура кипения: от 170 °С до 195 °С.
Температура плавления: от 60 °С до 80 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Цихалотрин. С23Н19С1Р3М03. (Мм. 449,9). 1125000.
[91465-08-6].
Температура кипения: от 187 °С до 190 °С.
Температура плавления: около 49 °С.
Может быть использован подходящий сертифи¬
цированный раствор сравнения (10 нг/мкл в циклогек¬
сане).
а-Эндосульфан. 09Н6С1бО33. (М.м. 406,9). 1126800.
[959-98-8].
Температура кипения: около 200 °С.
Температура плавления: около 108 °С.
Может быть использован подходящий сертифи¬
цированный раствор сравнения (10 нг/мкл в циклогек¬
сане).
Р-Эндосульфан. С9Н6С16033. (М.м. 406,9). 1126900.
[33213-65-9].
Температура кипения: около 390 °С.
Температура плавления: около 207 °С.
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Эндрин. С12Н8С160. (М.м. 380,9). 1127000. [72-20-8].
Может быть использован подходящий сертифици¬
рованный раствор сравнения (10 нг/мкл в циклогексане).
Этанол. 1034800. [64-17-5].
См. Этанол безводный Р.
Этанол безводный. 1034800. [64-17-5].
См. статью Этанол безводный (1318).
#Этанол безводный может быть получен из 96 %
спирта Р одним из перечисленных способов.
1. К 1 л 96% спирта Р прибавляют 200 г свеже-
прокаленного кальция оксида Р, встряхивают и кипя¬
тят с обратным холодильником в течение 4 ч.
2. 500 г меди (II) сульфата Р, прокаленного до
изменения цвета на белый с желтоватым оттенком,
помещают в колбу со шлифом и прибавляют 1 л 96 %
спирта Р и укупоривают. Выдерживают в течение
3 суток при периодическом встряхивании. Перед ис¬
пользованием фильтруют.#
Этион. С9Н2204Р254. (М.м. 384,5). 1127100. [563-
12-2].
Температура плавления: от -24 °С до -25 °С.
Может быть использован подходящий сертифи¬
цированный раствор сравнения (10 нг/мкл в циклогек¬
сане).
07/2016:40203
4.1.3. БУФЕРНЫЕ РАСТВОРЫ
0,1 М фосфатный буферный раствор рН 7,0.
4008200.
1,361 г калия дигидрофосфата Р растворяют в
воде Р и доводят до объема 100,0 мл этим же раство¬
рителем. Доводят рН (2.2.3) раствором 135 г/л дина¬
трия гидрофосфата Р.
4.2. РЕАКТИВЫ, ТИТРОВАННЫЕ
РАСТВОРЫ ДЛЯ
ОБЪЕМНОГО АНАЛИЗА
07/2016:40202
4.2.2. ТИТРОВАННЫЕ РАСТВОРЫ
Титрованные растворы должны готовиться в со¬
ответствии с обычными требованиями химического
анализа. Проверяют точность используемого обору¬
дования для того, чтобы удостовериться в его пригод¬
ности для предполагаемого применения.
Концентрацию титрованных растворов выража¬
ют их молярностью. Молярность выражает количе¬
ство вещества, растворенного в 1 л раствора, в виде
количества молей. Раствор, содержащий х молей ве¬
щества в одном литре, обозначают х М раствором.
Концентрация титрованных растворов не должна
отличаться от указанной более чем на 10 %. Моляр¬
ность титрованных растворов определяют с точно¬
стью 0,2 %.
Титрованные растворы стандартизуют описан¬
ными ниже методами. Если титрованный раствор
используют в количественном анализе (#2.2.90), в
котором конечную точку определяют электрохимиче¬
ским методом (например, амперометрии или потен-
циометрии), раствор стандартизуют тем же методом.
Состав среды, в которой стандартизуют титрованный
раствор, должен быть таким же, как и тот, в котором он
будет использован.
Растворы более разведенные, чем описанные
ниже, получают разведением последних водой, сво¬
бодной от углерода диоксида, Р. Поправочные коэф¬
фициенты полученных растворов такие же, как у ис¬
ходных растворов.
коэффициент поправки рекомендуется устанав¬
ливать при температуре 20 °С, при этой же темпера¬
туре применяют титрованные растворы, если нет дру¬
гих указаний.
Если устанавливают коэффициент поправки
и применяют раствор при других температурах, то
вводят температурную поправку в соответствии с та¬
блицами #4.2.2.-1 и #4.2.2.-2. Коэффициент поправки
проверяют не реже одного раза в два месяца, если
раствор устойчив, соблюдены условия хранения (хра¬
нят в помещениях при комнатной температуре в ме¬
стах, защищенных от попадания прямых солнечных
лучей) и нет других указаний.
14. Зак. 1060.
94
Гэсударственная фармакопея Республики Беларусь
Таблица #4.2.2.-1
Приведение объемов растворов при данной
температуре к объемам при 20 °С (для 1000 мл)
Температура (°С)
Вода, растворы кон¬
центрации 0,01 М и
растворы НС1
Растворы концентрации
0,1 М
(кро-
ме
НС1)
0,5 М
НС1
1 М
НС1
2 о
1 М
ЫаОН
1
2
3
4
5
6
7
5
+1,5
+1,7
+1,0
+2,3
+2,35
+3,6
6
+1,5
+1,65
+1,85
+2,2
+2,25
+3,4
7
+1,4
+1,6
+1,8
+2,15
+2,2
+3,2
8
+1,4
+1,55
+1,75
+2,1
+2,15
+3,0
9
+1,4
+1,5
+1,7
+2,0
+2,05
+2,7
10
+1,3
+1,45
+1,6
+1,9
+1,95
+2,5
11
+1,2
+1,35
+1,5
+1,8
+1,8
+2,3
12
+1,1
+1,3
+1,4
+1,6
+1,7
+2,0
13
+1,0
+1,1
+1,2
+1,4
+1,5
+1,8
14
+0,9
+1,0
+1,1
+1,2
+1,3
+1,6
15
+0,8
+0,9
+0,9
+1,0
+1,1
+1,3
16
+0,6
+0,7
+0,8
+0,8
+0,9
+1,1
17
+0,5
+0,6
+0,6
+0,6
+0,7
+0,8
18
+0,3
+0,4
+0,4
+0,4
+0,5
+0,6
19
+0,2
+0,2
+0,2
+0,2
+0,2
+0,3
20
0,0
0,0
0,0
0,0
0,0
0,0
21
-0,2
-0,2
-0,2
-0,2
-0,2
-0,3
22
-0,4
-0,4
-0,4
-0,5
-0,5
-0,5
23
-0,6
-0,6
-0,7
-0,7
-0,8
-0,9
24
-0,8
-0,9
-0,9
-1,0
-1,0
-1,2
25
-1,0
-1,1
-1,1
-1,2
-1,3
-1,5
26
-1,3
-1,4
-1,4
-1,4
-1,5
-1,8
27
-1,5
-1,7
-1,7
-1,7
-1,8
-2,1
28
-1,8
-2,0
-2,0
-2,0
-2,1
-2,4
29
-2,1
-2,3
-2,3
-2,3
-2,4
-2,8
30
-2,3
-2,5
-2,5
-2,6
-2,8
-3,2
Примечание:
1. Числа в графах 2—7 выражают объемы в мил¬
лилитрах, которые следует прибавить (+) к 1000 мл
соответствующей жидкости при 1 °С или вычесть (-)
от 1000 мл, чтобы получить объем титрованного раст¬
вора при 20 °С.
2. Примеры:
2.1. В колбу вместимостью 1000 мл, калиброван¬
ную при 20 °С, необходимо налить при 15 °С 0,1 М
раствор серебра нитрата. По таблице #4.2.2.-1. на¬
ходят, что при 20 °С этот раствор займет объем боль¬
ший на 0,9 мл.
2.2. Из бюретки, калиброванной при 20 °С, при
25 °С израсходовано на титрование 34,75 мл 1 М
раствора натрия гидроксида. При 20 °С расход ти¬
трованного раствора (в мл) составит:
ъд 75.1 5
34,75---- ’= 34,75 - 0,05 = 34,70 мл.
1000
Таблица #4.2.2.-2
Поправки объемов разбавленных растворов при
разных температурах
Тем-
пера-
тура
(°С)
Объем, мл
10
20
25
30
40
50
Поправка
10
+0,01
+0,03
+0,03
+0,04
+0,05
+0,06
12
+0,01
+0,02
+0,03
+0,03
+0,04
+0,06
14
+0,01
+0,02
+0,02
+0,03
+0,04
+0,05
16
+0,01
+0,01
+0,02
+0,02
+0,03
+0,03
18
0,00
+0,01
+0,01
+0,01
+0,01
+0,02
20
0,00
0,00
0,00
0,00
0,00
0,00
22
0,00
-0,01
-0,01
-0,01
-0,02
-0,02
24
-0,01
-0,02
-0,02
-0,02
-0,03
-0,04
26
-0,01
-0,03
-0,03
-0,04
-0,05
-0,06
28
-0,02
-0,03
-0,04
-0,05
-0,07
-0,09
30
-0,02
-0,03
-0,05
-0,07
-0,09
-0,12
Примечание:
Таблицей #4.2.2.-2. пользуются при работе с рас¬
творами 0,1 М концентрации или более разбавленными.
На контейнере с титрованным раствором указы¬
вают: наименование, заданную концентрацию, дату
приготовления, коэффициент поправки, применяе¬
мый индикатор, дату и температуру установления ко¬
эффициента поправки. Допускается вместо заданной
концентрации и коэффициента поправки указывать
значение точной концентрации с четырьмя значащи¬
ми цифрами после запятой. При электрохимическом
титровании вместо индикатора приводят указание о
применении метода (амперометрического, потенцио¬
метрического). Титрованные растворы, в которых при
хранении появились хлопья или осадок, не должны
применяться.#
#0,25 М раствор серной кислоты в спирте.
Медленно, при перемешивании прибавляют
13,9 мл кислоты серной Р к достаточному количеству
этанола безводного Р и доводят до объема 1000,0 мл
этанолом безводным Р. Охлаждают.
Установка титра. 2,500 г трометамина Р} вы¬
сушенного в соответствии с указаниями завода-изгото-
вителя или, при отсутствии таковых, высушенного при
температуре 105 °С в течение 3 ч, растворяют в 50 мл
воды Р и прибавляют 2 капли раствора бромкрезоло-
вого зеленого Р. Титруют приготовленным раствором
кислоты серной до бледно-желтого окрашивания.
1 мл 0,25 М раствора серной кислоты в спирте
соответствует 60,57 мг С^Н^ЫОз.
5.4. Остаточные количества органических растворителей
95
5. ОБЩИЕ ТЕКСТЫ
07/2016:50400
5.4. ОСТАТОЧНЫЕ
КОЛИЧЕСТВА
ОРГАНИЧЕСКИХ
РАСТВОРИТЕЛЕЙ
РЕГЛАМЕНТАЦИЯ СОДЕРЖАНИЯ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ В
СУБСТАНЦИЯХ, ВСПОМОГАТЕЛЬНЫХ
ВЕЩЕСТВАХ И ГОТОВЫХ ЛЕКАРСТВЕННЫХ
СРЕДСТВАХ
На Международной конференции по гармони¬
зации технических требований для регистрации ле¬
карственных средств для использования человеком
(ЮН) было принято Руководство по примесям оста¬
точных растворителей, в котором указаны конкрет¬
ные предельные содержания растворителей, которые
могут оставаться в фармацевтических субстанциях,
вспомогательных веществах и готовых лекарствен¬
ных средствах после обработки. Данное руководство,
текст которого приведен ниже, не распространяется
на продукты, уже присутствующие на рынке. Фарма¬
копея использует те же самые принципы к существую¬
щим фармацевтическим субстанциям, вспомогатель¬
ным веществам и готовым лекарственным средствам
вне зависимости от того, являются они объектом Фар¬
макопеи или нет. Все субстанции или готовые лекар¬
ственные средства должны быть проверены на содер¬
жание возможных остаточных растворителей в них.
Если используемые пределы совпадают с приве¬
денными ниже, испытание на содержание конкретных
остаточных растворителей обычно не указывается в
частной статье, так как используемые растворители
могут отличаться в зависимости от производителя,
а требования данной общей статьи применяются по¬
средством общей статьи Субстанции для фармацев¬
тического использования (2034). Уполномоченный
орган должен быть информирован об используемых в
процессе производства растворителях. Эта информа¬
ция также предоставляется в регистрационном досье
и указывается в сертификате.
Если используются растворители только класса
3, то для контроля их содержания может быть исполь¬
зовано либо испытание «Потеря в массе при высуши¬
вании», либо специфичное испытание на содержание
конкретного растворителя. Если для растворителя
класса 3 обосновано и разрешено предельное содер¬
жание более 0,5 %, то специфичное испытание на со¬
держание растворителя является обязательным.
Для контроля остаточных растворителей класса 1
или 2 (или класса 3 с содержанием более 0,5 %) следу¬
ет использовать, по возможности, методологию, описан¬
ную в общей статье (2.4.24). В противном случае следу¬
ет применять подходящую валидированную методику.
Если проводят количественное определение со¬
держания остаточных растворителей, полученный ре¬
зультат учитывают при расчете количественного содер¬
жания субстанции, за исключением случаев, когда про¬
водят определение потери в массе при высушивании.
1. ВВЕДЕНИЕ
Целью данного руководства является рекомен¬
дация приемлемых для безопасности больного ко¬
личеств остаточных растворителей в лекарственных
средствах. В руководстве рекомендуется использова¬
ние менее токсичных растворителей и описываются
токсикологически обоснованные предельные содер¬
жания некоторых из них.
Остаточные растворители в лекарственных сред¬
ствах определяются согласно данному руководству
как летучие органические вещества, используемые
или образующиеся при производстве субстанций,
вспомогательных веществ или готовых лекарствен¬
ных средств. Эти растворители полностью не удаля¬
ются в применяемом технологическом процессе. Вы¬
бор подходящего растворителя для синтеза субстан¬
ции может увеличить ее выход или обусловить такие
характеристики, как кристаллическая форма, чистота
и растворимость. Таким образом, растворитель ино¬
гда может быть критическим параметром в процессе
синтеза. Данное руководство не распространяется
на растворители, используемые в качестве вспомога¬
тельных веществ или относящиеся по составу к соль¬
ватам. Однако содержание растворителей в таких
препаратах должно обосновываться и определяться.
Поскольку остаточные растворители не имеют
терапевтического действия, адекватного действию
лекарственного вещества, они должны удаляться до
такой степени, чтобы удовлетворять требованиям
спецификации, Надлежащей производственной прак¬
тики (6МР) или другим требованиям к качеству. Ле¬
карственные средства не должны содержать остаточ¬
ные растворители выше нормы, установленной дан¬
ными по безопасности. При производстве субстанций,
вспомогательных веществ и готовых лекарственных
средств следует избегать использования растворите¬
лей, имеющих высокую токсичность (класс 1, табли¬
ца 1), если их применение не может быть достаточно
обоснованным с точки зрения оценки «риск-польза».
Содержание некоторых растворителей, имеющих
меньшую токсичность (класс 2, таблица 2), должно
быть ограничено, чтобы исключить потенциальный
побочный эффект. В идеале на практике должны ис¬
пользоваться менее токсичные растворители (класс
3, таблица 3). Полный список растворителей, вклю¬
ченных в данное руководство, представлен в Прило¬
жении 1.
Данный перечень не является исчерпывающим,
он может дополняться другими используемыми рас¬
творителями. Рекомендуемые предельные содержа¬
ния остаточных растворителей классов 1 и 2, а также
классификация растворителей может изменяться по
мере появления новых данных относительно их без¬
опасности. Обоснование безопасности применения
на рынке нового лекарственного средства, содержа¬
щего новый растворитель, отсутствующий в перечне
растворителей (Приложение 1), может строиться на
концепциях данного руководства.
2. ОБЛАСТЬ ПРИМЕНЕНИЯ
Область применения руководства — остаточные
растворители в фармацевтических субстанциях, вспо¬
могательных веществах и лекарственных средствах.
Определение остаточных растворителей необходимо
проводить, если известно, что процесс производства
или очистки приводит к их появлению. Следует опре¬
делять те растворители, которые используются или
96
Государственная фармакопея Республики Беларусь
производятся в процессе изготовления или очистки
фармацевтических субстанций, вспомогательных ве¬
ществ или лекарственных средств. Для определения
содержания остаточных растворителей возможно как
проведение испытания лекарственного средства, так
и использование совокупного метода расчета исходя
из информации об их содержании во входящих ингре¬
диентах, использованных в процессе производства
лекарственного средства. Если на основании резуль¬
татов вычисления концентрация остаточных раство¬
рителей не превышает предела, рекомендуемого в
настоящей статье, нет необходимости проводить ис¬
пытания готового лекарственного средства на содер¬
жание остаточных растворителей. Однако, если рас¬
четная концентрация выше рекомендуемого предела,
лекарственное средство должно быть проверено, что¬
бы установить, способствует ли процесс изготовления
уменьшению уровня данного растворителя до прием¬
лемого количества. Испытание готового лекарственно¬
го средства также необходимо проводить, если раст¬
воритель используется в процессе его производства.
Данная статья не относится ни к потенциально
новым фармацевтическим субстанциям, вспомога¬
тельным веществам и готовым лекарственным сред¬
ствам, находящимся на стадии клинических испыта¬
ний, ни к зарегистрированным лекарственным сред¬
ствам.
Статья применима ко всем дозированным фор¬
мам и способам применения. В некоторых случаях,
таких как краткосрочное (30 дней или меньше) или
местное применение, могут быть приемлемы более
высокие уровни остаточных растворителей. Оценка
таких уровней должна проводиться в каждом конкрет¬
ном случае.
Дополнительную информацию по остаточным
растворителям см. в Приложении 2.
3. ОБЩИЕ ПОЛОЖЕНИЯ
3.1. КЛАССИФИКАЦИЯ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ
ПО СТЕПЕНИ РИСКА
Международная программа по химической без¬
опасности (1РС8, 1п1егпаИопа1 Ргодгат оп СЬет'юа!
8а?е1у) для описания пределов воздействия токсиче¬
ских химических реагентов использует термин «мак¬
симальнодопустимое ежедневное потребление» (ТО/,
ШегаЫе баНуМаке, или ДЕП), а Всемирная организа¬
ция здравоохранения (ВОЗ) и другие национальные
и международные организации здравоохранения и
институты используют термин «приемлемое еже¬
дневное потребление» (ДО/, ассер1аЫе баНу Шаке,
или ПЕП). Во избежание путаницы с ПЕП некоторых
субстанций в данном руководстве определен новый
термин «допустимая (разрешенная) суточная доза»
(РОЕ, регтМеб баНу ехрозиге, или ДСД) — это макси¬
мально приемлемое суточное потребление остаточ¬
ного растворителя в лекарственном средстве.
Остаточные растворители, которым дает оценку
данное руководство, перечислены в Приложении 1
под общими названиями и структурными формулами.
Они оценивались по степени возможного риска для
здоровья человека и разделены на 3 класса:
Класс 1. Растворители, использования кото¬
рых нужно избегать.
Известные канцерогены для человека; предпо¬
лагаемые канцерогены для человека; растворители,
опасные для окружающей среды.
Класс 2. Растворители, использование кото¬
рых нужно ограничивать.
Негенотоксичные канцерогены для животных или
растворители, которые могут вызвать другие необ¬
ратимые эффекты, такие, как нейротоксичность или
тератогенность.
Растворители с предполагаемой другой суще¬
ственной, но обратимой токсичностью.
Класс 3. Малотоксичные растворители.
Растворители с низким потенциалом токсичности
для человека; для них не требуется устанавливать
предельное содержание, обусловленное информа¬
цией о риске для здоровья человека. Растворители
класса 3 имеют ДСД от 50 мг/сут и выше.
3.3. ПОДХОДЫ УСТАНОВЛЕНИЯ
ПРЕДЕЛЬНОГО
СОДЕРЖАНИЯ РАСТВОРИТЕЛЕЙ КЛАССА 2
Для установления предельного содержания раст¬
ворителей класса 2 могут использоваться два подхода.
Подход 1. Используют предельные концентра¬
ции в ррт из таблицы 2, которые были рассчитаны
с помощью уравнения (1) исходя из предположения,
что суточное потребление лекарственного средства
составляет 10 г.
„ , , 1000 • ДСД т
Концентрация (ррт) = , (Н
доза
где ДСД выражается в мг/сут, а доза — в г/сут.
Эти предельные содержания остаточных раство¬
рителей рассматриваются как приемлемые для всех
субстанций, вспомогательных веществ и готовых
лекарственных средств. Поэтому этот метод может
быть использован, если суточная доза неизвестна
или не установлена. Если содержание остаточных
растворителей во всех вспомогательных веществах
и фармацевтических субстанциях, которые входят в
состав готового лекарственного средства, удовлетво¬
ряет пределам, приведенным в таблице 2, то все эти
компоненты можно использовать в любой пропорции.
Если суточная доза лекарственного средства не пре¬
вышает 10 г, никакие дальнейшие вычисления не тре¬
буются. Определение предельного содержания оста¬
точных растворителей в лекарственных средствах,
которые принимаются в дозах, превышающих 10 г,
должно проводиться с использованием Подхода 2.
Подход 2. Нет необходимости, чтобы содержа¬
ние остаточных растворителей в каждом компоненте
лекарственного средства соответствовало пределам,
регламентированным с использованием Подхода 1.
Определить предельное содержание остаточного
растворителя в лекарственном средстве можно по
формуле (1), используя ДСД (мг/сут), приведенное в
таблице 2, и известное значение максимальной су¬
точной дозы лекарственного средства. Такие пределы
- являются приемлемыми при условии, что показано
снижение содержания остаточного растворителя до
минимума, который достигается практически. Преде¬
лы должны быть реалистичными в отношении анали¬
тической точности, производственной возможности,
разумного изменения производственного процесса
и соответствовать современным производственным
стандартам.
3.2. МЕТОДЫ УСТАНОВЛЕНИЯ
ПРЕДЕЛЬНОГО СОДЕРЖАНИЯ
В Приложении 3 представлен метод для установ¬
ления предельной суточной дозы остаточных раство¬
рителей.
ш
5.4. Остаточные количества органических растворителей
97
Подход 2 предусматривает суммирование коли¬
честв остаточного растворителя, присутствующего
в каждом из компонентов готового лекарственного
средства. Суммарное содержание растворителя в
сутки должно быть меньше, чем ДСД.
Рассмотрим использование Подхода 1 и Подхода
2 для расчета предельного содержания ацетонитрила
в лекарственном средстве. ДСД для ацетонитрила —
4,1 мг/сутки. Таким образом, его предельное содержа¬
ние с использованием Подхода 1 — 410 ррт. Макси¬
мально потребляемая масса лекарственного средства
в сутки — 5,0 г. Лекарственное средство содержит два
вспомогательных вещества. Состав лекарственного
средства и расчетное предельное содержание ацето¬
нитрила приведены в следующей таблице:
Компонент
Количество
в составе, г
Содержание
ацетонитри¬
ла, ррт
Суточное
воздей¬
ствие, мг
Фармацевти¬
ческая суб¬
станция
0,3
800
0,24
Вспомога¬
тельное ве¬
щество 1
0,9
400
0,36
Вспомога¬
тельное ве¬
щество 2
3,8
800
3,04
Готовое ле¬
карственное
средство
5,0
728
3,64
Концентрация ацетонитрила во вспомогательном
веществе 1 удовлетворяет предельному значению,
установленному с использованием Подхода 1, но его
концентрация в фармацевтической субстанции, вспо¬
могательном веществе 2 и готовом лекарственном
средстве не удовлетворяет. Тем не менее содержа¬
ние ацетонитрила в готовом лекарственном средстве,
установленное с использованием Подхода 2, не пре¬
вышает предела 4,1 мг/сутки и, следовательно, соот¬
ветствует рекомендациям данного руководства.
Рассмотрим другой пример для ацетонитрила в
качестве остаточного растворителя. Максимальная
потребляемая масса лекарственного средства в сут¬
ки — 5,0 г, и лекарственное средство содержит два
вспомогательных вещества. Состав лекарственного
средства и расчетное максимальное содержание аце¬
тонитрила приведены в следующей таблице:
Компонент
Количество
в составе, г
Содержание
ацетонитри¬
ла, ррт
Суточное
воздей¬
ствие, мг
Фармацевти¬
ческая суб¬
станция
0,3
800
0,24
Вспомога¬
тельное ве¬
щество 1
0,9
2000
1,80
Вспомога¬
тельное ве¬
щество 2
3,8
800
3,04
Готовое ле¬
карственное
средство
5,0
1016
5,08
В данном случае концентрация ацетонитрила в ле¬
карственном средстве не удовлетворяет предельному
значению ни с использованием Подхода 1, ни Подхода 2.
Производитель может провести испытания лекарствен¬
ного средства, чтобы определить, снижает ли процесс
производства содержание ацетонитрила. Если же содер¬
жание ацетонитрила не уменьшается в процессе произ¬
водства до допустимого предела, производитель должен
предпринять другие шаги для уменьшения концентрации
ацетонитрила в лекарственном средстве. Если все пред¬
принятые меры не приведут к уменьшению содержания
остаточного растворителя, в исключительных случаях
производитель может подготовить резюме о предприня¬
тых усилиях, направленных на уменьшение содержания
растворителя до норм данного руководства, и провести
анализ «риск-польза», чтобы получить разрешение на
использование лекарственного средства, содержащего
более высокий уровень остаточного растворителя.
3.4. АНАЛИТИЧЕСКИЕ МЕТОДИКИ
Остаточные растворители, как правило, опре¬
деляются с использованием хроматографических
методов, в частности газовой хроматографии. Для
определения содержания остаточных растворителей
могут использоваться любые подходящие методики,
описанные в фармакопеях. Иначе говоря, произво¬
дители должны быть свободны в выборе наиболее
подходящей валидированной аналитической методи¬
ки для конкретного применения. Если присутствуют
только растворители класса 3, могут быть использо¬
ваны неспецифические методы контроля, такие как,
например, потеря в массе при высушивании.
Валидация методов контроля остаточных раст¬
ворителей должна соответствовать руководству ЮН
«Валидация аналитических методик» #(см. статью
#5.3.2. Валидация аналитических методик и испы¬
таний (разделы А, В)), а также требованиям соответ¬
ствующих действующих ТНПА#.
3.5. ИНФОРМАЦИЯ О СОДЕРЖАНИИ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ
Для того чтобы соответствовать требованиям
данного руководства, производителям готовых лекар¬
ственных средств необходима точная информация о
содержании остаточных растворителей во вспомога¬
тельных веществах или фармацевтических субстанци¬
ях. Информация о содержании остаточных раствори¬
телей, которая может передаваться производителям
лекарственных средств поставщиками фармацевтиче¬
ских субстанций или вспомогательных веществ, может
быть представлена в следующих вариантах:
- вероятно присутствуют остаточные растворите¬
ли только класса 3. Потеря в массе при высушивании
менее 0,5 %;
- вероятно присутствуют остаточные раство¬
рители только класса 2; содержание каждого из них
не превышает пределов в соответствии с Подходом
1; указывают наименование каждого из остаточных
растворителей;
- вероятно присутствуют остаточные растворите¬
ли классов 2 и 3; содержание каждого из растворите¬
лей класса 2 не превышает пределов в соответствии
с Подходом 1, а содержание растворителей класса
3 — менее 0,5 %.
Если существует вероятность присутствия раст¬
ворителей класса 1, то каждый из них должен быть
идентифицирован и определен количественно.
«Вероятно присутствуют» относится к раствори¬
телям, используемым на заключительном этапе про¬
изводства, и тем, которые используются на ранних
стадиях производства и полностью не удаляются ут¬
вержденным (валидированным) процессом.
Если присутствуют остаточные растворители
класса 2 выше предельной концентрации в соответ¬
ствии с Подходом 1, а содержание остаточных раст-
15 Зак. 1060.
98
Гэсударственная фармакопея Республики Беларусь
ворителей класса 3 превышает 0,5 %, то каждый из
них должен быть идентифицирован и определен ко¬
личественно.
4. ПРЕДЕЛЬНЫЕ СОДЕРЖАНИЯ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ
4.1. РАСТВОРИТЕЛИ, ИСПОЛЬЗОВАНИЯ
КОТОРЫХ НУЖНО ИЗБЕГАТЬ
Растворители класса 1 не должны использоваться
в производстве фармацевтических субстанций, вспо¬
могательных веществ и готовых лекарственных средств
из-за их высокой токсичности и вредного воздействия
на окружающую среду. Однако, если их использование
неизбежно для производства лекарственного средства,
которое имеет сильно выраженный терапевтический
эффект, их количества должны быть ограничены в со¬
ответствии с таблицей 1, если иное не обосновано.
1,1,1-Трихлорэтан включен в таблицу 1, потому что он
опасен для окружающей среды. Установленный предел
в 1500 ррт основан на обзоре данных о безопасности.
Таблица 1
Растворители класса 1
в лекарственных средствах и субстанциях
для фармацевтического использования (раствори-
тели, применения которых нужно избегать).
Растворитель
Концентрационный
предел, ррт
Влияние
Бензол
2
канцероген
Четыреххлористый
углерод
4
токсичен и
опасен для
окружающей
среды
1,2-Дихлорэтан
5
токсичен
1,1-Дихлорэтен
8
токсичен
1,1,1-Трихлорэтан
1500
опасен для
окружающей
среды
4.2. РАСТВОРИТЕЛИ, ИСПОЛЬЗОВАНИЕ
КОТОРЫХ НУЖНО ОГРАНИЧИВАТЬ
Содержание растворителей, приведенных в та¬
блице 2, должно быть ограничено в лекарственных
средствах в связи с их токсичностью. Данные ДСД
даны с точностью до 0,1 мг/сут, а концентрации — с
точностью до 10 ррт. Установленные значения не от¬
ражают необходимую аналитическую точность опре¬
деления. Точность должна быть определена как часть
валидации (проверки правильности) методики.
Таблица 2
Растворители класса 2
в лекарственных средствах и субстанциях
для фармацевтического использования
Растворитель
ДСД, мг/сут
Концентра¬
ционный
предел, ррт
Ацетонитрил
4,1
410
Гексан
2,9
290
Л/, Л/-Д и мети л ацетамид
10,9
1090
Л/,Л/-Диметилформамид
8,8
880
1,2-Диметоксиэтан
1,0
100
1,4-Диоксан
3,8
380
1,2-Дихлорэтен
18,7
1870
Ксилол*
21,7
2170
Кумол
0,7
70
Метанол
30,0
3000
Растворитель
ДСД, мг/сут
Концентра¬
ционный
предел, ррт
Метилбутилкетон
0,5
50
Метиленхлорид
6,0
600
А/-Метил-
пирролидон
5,3
530
Метилциклогексан
11,8
1180
2-Метоксиэтанол
0,5
50
Нитрометан
0,5
50
Пиридин
2,0
200
Сульфолан
1,6
160
Тетра гид рофуран
7,2
720
Тетралин
1,0
100
Толуол
8,9
890
1,1,2-Трихлорэтен
0,8
80
Формамид
2,2
220
Хлорбензол
3,6
360
Хлороформ
0,6
60
Циклогексан
38,8
3880
Этиленгликоль
6,2
620
2-Этоксиэтанол
1,6
160
* Обычно 60 % м-ксилола, 14 % п-ксилола, 9 % о-ксилола
и 17 % этилбензола.
4.3. МАЛО ТОКСИЧНЫЕ РАСТВОРИТЕЛИ
Растворители класса 3 (представлены в таблице 3)
могут быть отнесены к менее токсичным и обладающим
меньшим риском для здоровья человека растворите¬
лям. Класс 3 не включает растворители, известные как
опасные для здоровья человека в концентрациях, кото¬
рые обычно допускаются в лекарственных средствах.
Однако для многих растворителей класса 3 не проводи¬
лось долгосрочное изучение токсичности или канцеро-
генности. Доступные данные указывают на то, что они
менее токсичны в острых или краткосрочных испытани¬
ях и дают отрицательный результат в испытаниях на ге-
нотоксичность (не проявляют генотоксичность). Счита¬
ется, что содержание этих остаточных растворителей,
равное 50 мг/сут или меньше (соответствует 5000 ррт
или 0,5 % по Подходу 1), приемлемо без обоснования.
Более высокие значения также могут быть приемлемы
при условии, что они определяются возможностями
производства, которое отвечает требованиям Надле¬
жащей производственной практики (<ЗМР).
Таблица 3
Растворители класса 3,
которые должны быть ограничены требованиями
ОМР или другими требованиями к качеству
Анизол
2-Метил-1 -пропанол
Ацетон
Мети лэти л кетон
1-Бутанол
Муравьиная кислота
2-Бутанол
Пентан
Бутилацетат
1-Пентанол
тре/л-Бутилметиловый
эфир
1-Пропанол
Гептан
2-Пропанол
Диметилсульфоксид
Пропилацетат
Изобутилацетат
Тетрагидрофуран
Изопропилацетат
Уксусная кислота
Метилацетат
Этанол
3-Метил-1 -бутанол
Этилацетат
Метилизобутилкетон
Этиловый эфир
Этилформиат
5.4. Остаточные количества органических растворителей
99
4.4. РАСТВОРИТЕЛИ, ДЛЯ КОТОРЫХ
ОТСУТСТВУЮТ НЕОБХОДИМЫЕ ДАННЫЕ
О ТОКСИЧНОСТИ
Растворители, представленные в таблице 4, могут
также представлять интерес для производителей вспо¬
могательных веществ, фармацевтических субстанций
или лекарственных средств. Однако для них отсутству¬
ют обоснованные данные о токсичности. Производите¬
ли должны сами обосновывать остаточные содержа¬
ния этих растворителей в лекарственных средствах.
Таблица 4
Растворители, для которых отсутствуют
обоснованные данные о токсичности
1,1 -Диметоксиметан
Метилизопропилкетон
2,2-Диметоксипропан
Метилтетрагидрофуран
1,1-Диэтокси пропан
Петролейный эфир
Изооктан
Трифторуксусная кислота
Изопропиловый эфир
Трихлоруксусная кислота
ТЕРМИНЫ
Генотоксичные канцерогены — канцерогены,
которые вызывают появление злокачественных опу¬
холей, воздействуя на гены или хромосомы.
Коэффициент корреляции (поправочный коэф¬
фициент) (тодНутд /ас^ог) — коэффициент, опреде¬
ленный в соответствии с обоснованным заключением
токсиколога и основанный на экстраполяции на чело¬
века данных по безопасности, полученных при испы¬
таниях на животных.
Минимальный уровень, при котором наблю¬
дается эффект (ШЕЦ 1ошез1-оЬзеп/ес1 еНес1 /еуе/,
МУНЭ), — минимальная доза (вещества) в испытании
или серии испытаний, которая вызывает биологиче¬
ски существенное увеличение в частоте или серьез¬
ности любых эффектов у людей или животных.
Нейротоксичность — способность вещества
оказывать неблагоприятное воздействие на нервную
систему.
Обратимая токсичность — возникновение
вредных эффектов, которые вызваны данным веще¬
ством и исчезают после прекращения действия ве¬
щества.
Предполагаемый канцероген человека — веще¬
ство, для которого нет эпидемиологической очевид¬
ности канцерогенеза, но есть положительные данные
по генотоксичности и очевидность канцерогенеза у
грызунов.
Разрешенная (допустимая) суточная доза (РОЕ,
регтМес! да\\у ехрозиге, ДСД) — максимально прием¬
лемое суточное потребление остаточного растворите¬
ля в лекарственном средстве.
Тератогенность — возникновение структурных
уродств в развивающемся зародыше, когда вещество
принимается в период беременности.
Уровень, при котором не наблюдается эффект
(ИОЕ1-, по-оЬзегуес1-еТГе& ехрозиге, УННЭ), — макси¬
мальная доза вещества, при которой нет биологиче¬
ски существенных увеличений в частоте или серьез¬
ности любых эффектов у людей или животных.
ПРИЛОЖЕНИЕ 1. СПИСОК РАСТВОРИТЕЛЕЙ, ВКЛЮЧЕННЫХ В РУКОВОДСТВО
Растворитель
Второе название
Структура
Класс
Анизол
Метоксибензол
,^/0СНз
и
Класс 3
Ацетон
2-Пропанон, пропан-2-он
СН3СОСН3
Класс 3
Ацетонитрил
сн3см
Класс 2
Бензол
О
Класс 1
1 -Бутанол
н-Бутиловый спирт, бутан-1 -ол
СН3[СН2]3ОН
Класс 3
2-Бутанол
втор-Бутиловый спирт, бутан-2-ол
СН3СН2СН(ОН)СН3
Класс 3
Бутилацетат
Бутиловый эфир уксусной кислоты
СН3СОО[СН2]3СН3
Класс 3
трет-Бутилметиловый эфир
2-Метокси-2-метилпропан
(СН3)3СОСН3
Класс 3
Гексан
н-Гексан
СН3[СН2]4СН3
Класс 2
Гептан
н-Гептан
СН3[СН2]5СН3
Класс 3
Л/,Л/-Диметилацетамид
ДМА
СН3СОМ(СН3)2
Класс 2
Д и мети л сульфоксид
Метилсульфинилметан, метилсульфоксид, ДМСО
(СН3)280
Класс 3
Л/,А/-Диметилформамид
ДМФА
НСОЫ(СН3)2
Класс 2
1,2-Дйметоксиэтан
Диметиловый эфир этиленгликоля, моноглим, диметил-
целлозольв
Н3СОСН2СН2ОСН3
Класс 2
1,4-Диоксан
л-Диоксан, [1,4]диоксан
0
Класс 2
Дихлорметан
Метиленхлорид
СН2С12
Класс 2
1,2-Дихлорэтан
слм-Дихлорэтан, этилендихлорид, этиленхлорид
СН2СЮН2С1
Класс 1
1,1-Дихлорэтен
1,1 -Дихлорэтилен, винилиденхлорид
Н2С=СС12
Класс 1
1,2-Дихлорэтен
1,2-Дихлорэтилен, ацетилендихлорид
С1НС=СНС1
Класс 2
100
Государственная фармакопея Республики Беларусь
Растворитель
Второе название
Структура
Класс
Изобутилацетат
Изобутиловый эфир уксусной кислоты
СН3СООСН2СН(СН3)2
Класс 3
Изопропилацетат
Изопропиловый эфир уксусной кислоты
СН3СООСН(СН3)2
Класс 3
Ксилол*
Диметилбензол
^^СНз
нз°^ 1
Класс 2
Кумол
Изопропилбензол, (1-метилэтил)бензол
сн3
0^3
Класс 2
Метанол
Метиловый спирт
СН3ОН
Класс 2
Метилацетат
Метиловый эфир уксусной кислоты
сн3соосн3
Класс 3
З-Метил-1 -бутанол
Изоамиловый спирт, изопропиловый спирт,
З-метилбутан-1 -ол
(СН3)2СНСН2СН2ОН
Класс 3
Метилбутилкетон
2-Гексанон, гексан-2-он
СН3[СН2]3СОСН3
Класс 2
Метилизобутилкетон
4-Метилпентан-2-он, 4-метил-2-пентанон, МИБК
СН3СОСН2СН(СН3)2
Класс 3
А/-Метилпирролидон
1 -Метилпирролидин-2-он, 1 -метил-2-пирролидинон
СНз
<У°
Класс 2
2-Метил-1 -пропанол
Изобутиловый спирт, 2-метилпропан-1-ол
(СН3)2СНСН2ОН
Класс 3
Метилциклогексан
Циклогексилметан
Класс 2
Метилэтилкетон
2-Бутанон, МЭК, бутан-2-он
сн3сн2сосн3
Класс 3
2-Метоксиэтанол
Метилцеллозольв
СН3ОСН2СН2ОН
Класс 2
Муравьиная кислота
нсоон
Класс 3
Нитрометан
сн3мо2
Класс 2
Пентан
н- Пентан
СН3[СН2]3СН3
Класс 3
1-Пентанол
Амиловый спирт, пентан-1-ол
СН3[СН2]3СН2ОН
Класс 3
Пиридин
.14.
о
Класс 2
1-Пропанол
Пропан-1-ол, пропиловый спирт
сн3сн2сн2он
Класс 3
2-Пропанол
Пропан-2-ол, изопропиловый спирт
(СН3)СНОН
Класс 3
Пропилацетат
Пропиловый эфир уксусной кислоты „
сн3соосн2сн2сн3
Класс 3
Сульфолан
Тетрагидротиофен-1,1 -диоксид
°Ч Р
V
о
Класс 2
Тетрагидрофуран
Тетраметиленоксид, оксациклопентан
О
Класс 2
Тетралин
1,2,3,4-Тетрагидронафталин
со
Класс 2
Толуол
Метилбензол
сг
Класс 2
1,1,1-Трихлорэтан
Метилхлороформ
СН3СС13
Класс 1
1,1,2-Трихлорэтен
Трихлорэтан
НС1С=СС12
Класс 2
Углерод четыреххлористый
Тетрахлорметан
о
о
Класс 1
Уксусная кислота
Этановая кислота
сн3соон
Класс 3
Формамид
Метанамид
нсомн2
Класс 2
5.4. Остаточные количества органических растворителей
101
Растворитель
Второе название
Структура
Класс
Хлоробензол
о
ь
Класс 2
Хлороформ
Трихлорметан
СНС13
Класс 2
Циклогексан
Гексаметилен
О
Класс 2
Этанол
Этиловый спирт
сн3сн2он
Класс 3
Этилацетат
Этиловый эфир уксусной кислоты
сн3соосн2сн3
Класс 3
Этиленгликоль
1,2-Дигидроксиэтан, 1,2-этандиол
носн2сн2он
Класс 2
Этиловый эфир
Диэтиловый эфир, этоксиэтан, 1,1’-оксибисэтан
сн3сн2осн2сн3
Класс 3
Этилформиат
Этиловый эфир муравьиной кислоты
нсоосн2сн3
Класс 3
2-Этоксиэтанол
Целлозольв
сн3сн2осн2сн2он
Класс 2
* Обычно 60 % м-ксилола, 14 % /7-ксилола, 9 % о-ксилола и 17 % этилбензола.
ПРИЛОЖЕНИЕ 2. ДОПОЛНИТЕЛЬНЫЕ
ДАННЫЕ
А2.7. ВЛИЯНИЕ ОРГАНИЧЕСКИХ
ЛЕТУЧИХ РАСТВОРИТЕЛЕЙ НА
ОКРУЖАЮЩУЮ СРЕДУ
Некоторые из остаточных растворителей, часто
используемых в фармацевтическом производстве,
внесены в перечень токсичных химических соедине¬
ний в статьях «Критерии здоровья окружающей сре¬
ды» (ЕНС) и «Объединенная информационная систе¬
ма риска» (/#/5). Цели таких организаций, как Между¬
народная программа по химической безопасности
(1Р8С), Управление по охране окружающей среды
США (И8ЕРА), Управление лекарственных средств и
пищевых продуктов (1/5ЕОА), включают определение
предельного содержания химических веществ. Основ¬
ная их цель — защита человеческого здоровья и окру¬
жающей среды от возможного негативного влияния
химических соединений в результате длительного воз¬
действия. Методы, используемые для оценки макси¬
мальных, безопасных пределов воздействия, обычно
основываются на долгосрочных исследованиях. Когда
данные долгосрочных испытаний недоступны, могут
быть использованы данные краткосрочных испытаний
с модификацией подхода, например использование
более высоких коэффициентов корреляции. Подход,
описанный здесь, относится, прежде всего, к долго¬
срочным воздействиям или воздействиям на продол¬
жительность жизни всего населения окружающей сре¬
ды, в частности воздуха, продовольствия, питьевой
воды и др.
А2.2. ОСТАТОЧНЫЕ РАСТВОРИТЕЛИ В
ЛЕКАРСТВЕННЫХ СРЕДСТВАХ
Пределы воздействия в этом руководстве уста¬
новлены в соответствии с методологией и данными
токсичности, приведенными в монографиях ЕСН и
/Р/8. Однако при установлении пределов воздействия
должны быть приняты некоторые допущения относи¬
тельно остаточных растворителей, которые использу¬
ются в процессе синтеза и изготовления лекарствен¬
ных средств, а именно:
1) пациенты (не все население) используют лекар¬
ственные средства для лечения болезней или для про¬
филактики с целью избежать инфекции или болезни;
2) предположение о воздействии на продолжи¬
тельность жизни пациента необязательно для боль¬
шинства лекарственных средств, но может рассма¬
триваться как рабочая гипотеза, чтобы уменьшить
риск для здоровья человека;
3) остаточные растворители — неизбежные ком¬
поненты фармацевтического производства и зачастую
являются составной частью лекарственных средств;
4) остаточные растворители не должны превы¬
шать рекомендуемые концентрации, кроме исключи¬
тельных обстоятельств;
5) данные о токсикологических испытаниях, ко¬
торые используются для определения приемлемых
концентраций остаточных растворителей, должны
быть зафиксированы с использованием соответству¬
ющих протоколов, описанных, например, в докумен¬
тах ОЕСО и ГОА Ред Воок.
ПРИЛОЖЕНИЕ 3. МЕТОДЫ УСТАНОВЛЕНИЯ
ДОПУСТИМОЙ СУТОЧНОЙ ДОЗЫ
Для оценки риска канцерогенных растворителей
класса 1 используют метод Гейлора-Коделя (6ау1ог,
О Ж апд КодеН, Р.1.: Ипеаг ШегроШюп а1догШип Тог
/оIV дозе аззеззтеп1 оТ 1охю зиЬз^апсе. д. ЕпуКоп.
РаИю1оду, 4, 305, 1980). Для установления ДСД экс¬
траполяцию с использованием математических мо¬
делей следует применять только в тех случаях, когда
есть достоверные данные о канцерогенности. ДСД
для растворителей класса 1 также может определять¬
ся с использованием высокого значения коэффициен¬
та корреляции (например, от 10 000 до 100 000) для
уровня, при котором эффект не наблюдается. Обна¬
ружение и количественное определение этих раство¬
рителей следует проводить валидированными анали¬
тическими методиками.
Предельные содержания для растворителей
класса 2 в этом руководстве были установлены пу¬
тем вычисления значений ДСД согласно методикам
определения предельного содержания растворителей
в лекарственных средствах (РЬагтасоре1а1 Гогит, но¬
ябрь-декабрь 1989 г.) и методам, принятым 1РС8 для
оценки риска химических веществ в отношении здоро¬
вья человека (ЕпПгоптепШ НеаНЬ СгНепа 170, ВОЗ,
1994). Эти методы подобны тем, которые используют
ИВЕРА (/Р/8), И8ГОА (Ред Воок) и др. Метод описан
ниже, чтобы пояснить происхождение значений ДСД.
Чтобы использовать значения ДСД, приведенные в
таблице Раздела 4 этого документа, нет необходимо¬
сти производить эти вычисления.
102
Гэсударственная фармакопея Республики Беларусь
При экспериментах на животных значения ДСД
рассчитывают исходя из уровня, при котором эффект
не наблюдается (УННЭ), или уровня, при котором на¬
блюдается самый низкий эффект (МУНЭ), по формуле:
ппп _ ЭННЭ Масса тела
Д Д~ Р1.Р2 РЗ .Р4 Р5 '
Значение ДСД преимущественно получают на
основании УННЭ. Если значения УННЭ неизвестны,
могут быть использованы значения МУНЭ. Коэффи¬
циенты корреляции, предложенные здесь для экстра¬
поляции на человека данных, полученных на живот¬
ных, — это те же «коэффициенты неопределенности»,
которые использовались в монографии «Критерии
здоровья окружающей среды» (Епмгоптеп1а1 НеаПМ
СгНепа 170, ВОЗ, Женева, 1994) и «коэффициенты
корреляции» или «коэффициенты безопасности» — в
РЬагтасоре1а1 Рогит. Во всех расчетах принимается
предположение о 100 % системном воздействии неза¬
висимо от способа применения лекарств.
Коэффициенты корреляции:
Р1 — коэффициент корреляции для расчета экс¬
траполяции между видами;
Р1 = 5 при экстраполяции на человека дан¬
ных, полученных при исследованиях на кры¬
сах;
Р1 = 12 при экстраполяции на человека дан¬
ных, полученных при исследованиях на мы¬
шах;
Р1 = 2 при экстраполяции на человека дан¬
ных, полученных при исследованиях на со¬
баках;
РI = 2,5 при экстраполяции на человека
данных, полученных при исследованиях на
кроликах;
Р1 = 3 при экстраполяции на человека дан¬
ных, полученных при исследованиях на обе¬
зьянах;
Р1 = 10 при экстраполяции на человека дан¬
ных, полученных при исследованиях на дру¬
гих животных.
р| принимает во внимание отношение площади
поверхности тела к весу тела соответствующих видов
животных и человека. Площадь поверхности рассчи¬
тывается следующим образом:
5 = кт0'67,
где:
т — масса тела;
к— константа, принята равной 10.
Массы тела, используемые в уравнении, пред¬
ставлены в таблице АЗ.-1.
Таблица АЗ.-1
Значения, использованные при расчетах
в данном документе
Масса крысы
425 г
Масса беременной крысы
330 г
Масса мыши
28 г
Масса беременной мыши
30 г
Масса морской свинки
500 г
Масса макаки-резус
2,5 кг
Масса кролика (беременного или нет)
4 кг
Масса гончей собаки (бигль)
11,5 кг
Дыхательный объем крысы
290 л/сут
Дыхательный объем мыши
43 л/сут
Дыхательный объем кролика
1440 л/сут
Дыхательный объем морской свинки
430 л/сут
Дыхательный объем человека
28800 л/сут
Дыхательный объем собаки
9000 л/сут
Дыхательный объем обезьяны
1150 л/сут
Потребление воды мышью
5 мл/сут
Потребление воды крысой
30 мл/сут
Потребление пищи крысой
30 г/сут
Р2 — коэффициент 10, учитывающий индивиду¬
альную изменчивость. Для всех органических раство¬
рителей, приведенных в данном руководстве, коэф¬
фициент обычно принимают равным 10.
РЗ — переменный коэффициент для расчета ис¬
пытаний токсичности кратковременных воздействий.
РЗ = 1 для испытаний, которые длятся, по
меньшей мере, в течение периода, равного
половине продолжительности жизни живот¬
ных (1 год для грызунов и кроликов; 7 лет
для собак, котов и обезьян).
РЗ = 1 для репродуктивных (воспроизводи¬
тельных) испытаний, которые охватывают
весь период органогенеза.
РЗ = 2 для испытаний в течение 6 месяцев
на грызунах или 3,5 лет — не на грызунах.
РЗ = 5 для 3-месячных испытаний на грызу¬
нах или 2-летних — не на грызунах.
РЗ = 10 для испытаний более короткой про¬
должительности.
Для всех промежуточных испытаний необходимо
использовать более высокий коэффициент (напри¬
мер, для 9-месячных испытаний на грызунах исполь¬
зуется коэффициент 2).
Р4 — коэффициент, который может применяться
при высокой токсичности растворителя, например не¬
генотоксичной канцерогенности, нейротоксичности или
тератогенности. В испытаниях репродуктивной токсич¬
ности используются следующие коэффициенты:
Р4 = 1 для эмбриональной токсичности, свя¬
занной с материнской токсичностью (инток¬
сикацией);
Р4 = 5 для эмбриональной токсичности (ин¬
токсикации), не связанной с материнской;
Р4 = 5 для тератогенного эффекта, связан¬
ного с материнской интоксикацией;
Р4 = 10 для тератогенного эффекта, не свя¬
занного с материнской интоксикацией.
Р5 — переменный коэффициент, который может
применяться, если УННЭ (уровень, не вызывающий эф¬
фекта) не был установлен. Когда доступны только дан¬
ные уровня МУНЭ (уровень, вызывающий минималь¬
ный эффект), то в зависимости от уровня токсичности
может использоваться коэффициент вплоть до 10.
Допускается, что масса тела взрослого человека
любого пола равна 50 кг. Этот относительно низкий
вес обеспечивает дополнительный коэффициент без¬
опасности стандартному весу человека 60 кг или 70
кг, который часто используется в таких вычислениях.
Известно, что многие взрослые пациенты весят менее
50 кг, поэтому в этом случае при определении ДСД ис¬
пользуются другие коэффициенты. Если лекарствен¬
ное средство, содержащее растворитель, предназна¬
чено для педиатрии, то необходимо сделать корректи¬
ровку на более низкую массу тела.
Как пример применения этого уравнения рассмо¬
трим испытание токсичности ацетонитрила на мышах,
которое было описано в РЬагтеигора, т. 9, № 1, До¬
полнение, апрель 1997, с. 524. Установлено, что зна-
5.12. Стандартные образцы
103
чение УННЭ — 50,7 мг/кг-сут. ДСД для ацетонитрила
при этом рассчитывали следующим образом:
50,7 мг кг-1 сут-1 • 50 кг
ДСД = -
12-10-5-1-1
= 4,22 мг сут-1.
В этом примере:
Р1 = 12, учитывает экстраполяцию на человека
данных, полученных при исследованиях на мышах;
/=2 = 10, учитывает индивидуальную изменчи¬
вость;
РЗ = 5, так как продолжительность испытаний со¬
ставила только 13 недель;
Р4 = 1, так как с серьезной токсичностью не стал¬
кивались;
Р5 = 1, так как был определен уровень, не вызы¬
вающий эффекта.
Для перерасчета концентраций газов, исполь¬
зуемых в дыхательных (ингаляторных) испытаниях
из ррт в мг/л или мг/м3, использовали уравнение для
идеального газа: РУ = пРГ. Рассмотрим в качестве
примера испытание репродуктивной токсичности кры¬
сы в результате вдыхания четыреххлористого углеро¬
да (М.м. 153,84), описанное в РЬагтеигоре, т. 9, № 1,
Дополнение, апрель 1997, с. 59.
п
V
Р
РТ
300-10-6атм -153840 мг моль 1
0,082 л атм К-1 моль-1 -298 К
46,15 мг
24,45 л
= 1,89 мг/л
Для перевода в мг/м3 используют отношение
1000л = 1 м3.
07/2016:51200
5.12. СТАНДАРТНЫЕ ОБРАЗЦЫ
Статья приводится для информации.
1. ВВЕДЕНИЕ
Термин «стандартный образец» применяется в
данной статье как обобщенное понятие, включающее
стандартные вещества, стандартные препараты и
эталонные спектры.
Стандартные образцы необходимы для прове¬
дения соответствующего контроля качества лекар¬
ственных средств и их компонентов #(субстанций для
фармацевтического использования, лекарственного
растительного сырья, продуктов из лекарственного
растительного сырья и др.)#.
Стандартные образцы утверждаются в установ¬
ленном порядке, а их пригодность для последующего
применения проверяется согласно предписанной про¬
грамме. Необходимость использования стандартного
образца предусматривается в статье Фармакопеи или
нормативном документе производителя по контролю
качества #(например, НД или ФСП)#. Если в частной
или общей статье указан фармакопейный стандарт¬
ный образец, то только он принимается за официаль¬
ный, т.е. единственно надежный в спорных случаях
стандартный образец.
2. ТЕРМИНОЛОГИЯ
Первичный стандартный образец (рптагу
з1апс1агс1). Стандартный образец, который характе¬
ризуется или признается как образец с наилучшими
метрологическими характеристиками, числовые зна¬
чения параметров (свойств) которого установлены
без использования других стандартных образцов с
такими же параметрами (свойствами) или такими же
количественными характеристиками, в рамках кон¬
кретной области применения. Данное определение не
относится к международным стандартным образцам.
Международный стандартный образец
(ЫетаНопа! з1апдагд). Первичный стандартный об¬
разец, обеспечивающий возможность одинакового
выражения результатов биологических и иммуноло¬
гических определений во всем мире. Присвоенное
значение выражается в Международных единицах
(МЕ, 1п1егпаНопа1 ШНз — Ю) или других подходящих
единицах. Единица измерения обычно присваивается
впервые выпускаемому международному стандарт¬
ному образцу Всемирной организацией здравоохра¬
нения (ВОЗ) произвольно на основании международ¬
ного межлабораторного испытания. В применимых
случаях, для замещенных международных стандарт¬
ных образцов, активности в Международных едини¬
цах устанавливают путем сравнения с предыдущими
стандартными образцами.
Вторичный стандартный образец (зесопдагу
з(апс!агсГ). Стандартный образец, числовые значения
свойств которого установлены путем сравнения с пер¬
вичным стандартным образцом, обладающим такими
же параметрами (свойствами) или такими же количе¬
ственными характеристиками.
Стандартный образец Европейской Фарма¬
копеи (Еигореап РЬагтасорое/а ге1егепсе з1апс!агсЗ).
Стандартный образец, утвержденный и одобренный
Европейской фармакопейной комиссией.
Химический стандартный образец Европей¬
ской Фармакопеи (Еигореап Р/пагтасорое/а сНетюа!
геТегепсе 8иЬз1апсе — СР5). Вещество или смесь
веществ, предназначенные для использования, как
указано в частной или общей статье Европейской
Фармакопеи. Данные стандартные образцы являются
первичными стандартными образцами, за исключе¬
нием образцов (особенно антибиотиков), активность
которых установлена в Международных единицах.
Такие стандартные образцы являются вторичными и
прослеживаются до международных стандартных об¬
разцов.
Растительный стандартный образец Евро¬
пейской Фармакопеи (Еигореап РНагтасорое/а ЬегЬаI
ге1егепсе з1апдагд — НР8). Продукт из лекарственно¬
го растительного сырья (обычно экстракт) или лекар¬
ственное растительное сырье, предназначенные для
использования, как указано в частной или общей ста¬
тье Европейской Фармакопеи. Если иное не указано,
данные стандартные образцы являются первичными
для их предполагаемого использования.
Стандартный образец Государственной фар¬
макопеи Республики Беларусь (СО ГФ РБ). Стандарт¬
ный образец, утвержденный и одобренный компетент¬
ным уполномоченным органом.
Фармакопейный стандартный образец (ФСО).
Стандартный образец, предназначенный для исполь¬
зования, как указано в частной или общей статье Фар¬
макопеи.
В качестве фармакопейных стандартных образ¬
цов могут быть использованы стандартные образцы
Европейской Фармакопеи (СР5, НРБ), Государствен¬
ной фармакопеи Республики Беларусь (СО ГФ РБ), а
также других фармакопей при условии их пригодно¬
сти для целей, указанных в частной или общей статье
Фармакопеи/
104
Гэсударственная фармакопея Республики Беларусь
Биологический стандартный препарат Ев¬
ропейской Фармакопеи (Еигореап РЬагтасорое1а
Ыо1од'юа1 ге(егепсе ргерагаНоп — ВЕР). Вещество или
смесь веществ, предназначенные для использования,
как указано в частной или общей статье Европейской
Фармакопеи. Биологические стандартные препара¬
ты — это либо вторичные стандартные образцы, ак¬
тивность которых выражается в Международных еди¬
ницах, либо первичные стандартные образцы, кото¬
рые могут использоваться для определения единицы
действия Европейской Фармакопеи (Р/?. Еиг. II.). Могут
использоваться и другие подходящие единицы измере¬
ния, например вирусный титр или число бактерий.
#Биологический стандартный препарат (БСП).
Биологические стандартные препараты, предназна¬
ченные для использования, как указано в частной или
общей статье Фармакопеи. В качестве биологических
стандартных препаратов могут быть использованы
биологические стандартные образцы Европейской
Фармакопеи (ВНР), а также другие биологические
стандартные образцы при условии их пригодности
для целей, указанных в частной или общей статье
Фармакопеи.#
Специфичность фармакопейных стандартных
образцов была официально признана во введении
150 Си1с1е 34 — Общие требования к компетентно¬
сти изготовителей стандартных образцов. В част¬
ности признано, что в обычном случае измеренная
неопределенность заявленного содержания (припи¬
санного значения) не указывается, так как она незна¬
чительна по отношению к установленным пределам в
метод-специфичных испытаниях, описанных в фар-
макопеях, для которых они предназначены.
Стандартное вещество (геТегепсе та(епа1 —
РМ). Вещество (материал, образец), достаточно од¬
нородное и стабильное в отношении одного или не¬
скольких специфических параметров (свойств), кото¬
рые определены и достаточны для его использования
по назначению в измерительном процессе.
Аттестованное стандартное вещество
{сегИЛед ге(егепсе та(епа1 — СРМ). Стандартное ве¬
щество, значения одного или нескольких параметров
(свойств) которого установлены по метрологически
валидированной методике, сопровождаемое серти¬
фикатом, в котором приводятся значение установлен¬
ного параметра, связанная с ним неопределенность
(погрешность) и заявление о метрологической про¬
слеживаемости.
#ПРИМЕЧАНИЕ: фармакопейные стандартные
образцы необходимо отличать от стандартных ве¬
ществ и аттестованных стандартных веществ. При¬
менение стандартных веществ необходимо или реко¬
мендовано в частных или общих статьях Фармакопеи
в основном для калибровки и контроля удовлетвори¬
тельной эффективности приборов.
3. ИСПОЛЬЗОВАНИЕ ФАРМАКОПЕЙНЫХ
СТАНДАРТНЫХ ОБРАЗЦОВ
Фармакопейные стандартные образцы использу¬
ются для идентификации, в испытаниях на чистоту и
при количественном определении, как указано в част¬
ных и общих статьях Фармакопеи. Фармакопейные
стандартные образцы должны соответствовать непо¬
средственному предназначению; они не обязатель¬
но должны быть пригодны для других целей. Если
фармакопейные стандартные образцы используются
для целей, отличных от тех, для которых они были
созданы, их пригодность должна быть доказана и
полностью описана в документах досье, подаваемого
на регистрацию. Приписанное значение любого по¬
казателя стандартного образца пригодно только для
предписанного использования и не всегда пригодно
для других целей.
Фармакопейные стандартные образцы с установ¬
ленным содержанием/активностью для количествен¬
ного определения субстанции для фармацевтическо¬
го использования могут быть пригодны для количе¬
ственного определения содержания данной субстан¬
ции в лекарственном средстве при соблюдении всех
следующих условий:
- применим хроматографический метод количе¬
ственного определения субстанции для фармацевти¬
ческого использования, описанный в частной статье;
- подтверждена пользователем применимость
данного метода для конкретного лекарственного
средства (отсутствие взаимодействий и других меша¬
ющих влияний);
- любая пробоподготовка образца (например,
экстракция, фильтрование) валидирована для кон¬
кретного лекарственного средства.
Как правило, фармакопейные стандартные об¬
разцы поставляются в соответствующих количествах
для немедленного использования (т. е. необходи¬
мые для проведения испытаний количества указа¬
ны в частных и общих статьях Фармакопеи) после
вскрытия контейнера. За использование стандарт¬
ных образцов в других условиях ответственность
несет аналитик. Если закрытый контейнер хранится
в рекомендуемых условиях, он остается пригодным
в течение срока годности данной серии. Например,
информация о номерах серий стандартных образ¬
цов Европейской Фармакопеи имеется в каталоге
Европейского управления по качеству лекарствен¬
ных средств и здравоохранению при Совете Евро¬
пы (ЕйОМ — Еигореап 01гес1ога1е /ог Ме ОиаШу о/
МесЛстез (СоипсН о! Еигоре)) (Нйр://до.ес1рт.еи/РЗ).
Хранение растворов стандартных образцов не реко¬
мендуется, за исключением случаев, когда пользова¬
телем предварительно подтверждена их пригодность
для использования.
Вторичные стандартные образцы. Вторичные
стандартные образцы обычно разрабатываются для
сокращения использования первичных стандартных
образцов и могут быть использованы для рутинного
контроля качества. Вторичный стандартный образец
должен обладать таким же параметром или пара¬
метрами, как и первичный стандартный образец, до
которого он должен быть прослеживаем. Поэтому
вторичные стандартные образцы должны использо¬
ваться в тех же целях, что и первичные стандартные
образцы.
Международные стандартные образцы обычно
доступны в ограниченных количествах и предназначе¬
ны для использования при определении параметров и
калибровки вторичных стандартных образцов. Такие
вторичные стандарты в свою очередь могут быть ис¬
пользованы в качестве рабочих стандартных образцов.
4. СОЗДАНИЕ СТАНДАРТНЫХ ОБРАЗЦОВ
4-1. ПЕРВИЧНЫЕ СТАНДАРТНЫЕ ОБРАЗЦЫ
Для подтверждения пригодности использования
веществ или препаратов в качестве первичных стан¬
дартных образцов применяются различные аналити¬
ческие методики.
5.12. Стандартные образцы
105
Для стандартных образцов, использующихся для
контроля качества лекарственных средств и их ком¬
понентов #(субстанций для фармацевтического ис¬
пользования, лекарственного растительного сырья,
продуктов из лекарственного растительного сырья и
др.)#, обычно применяются соответствующие части
следующей программы испытаний.
Программа испытаний:
-Описание вещества (структурная характери¬
стика) с помощью соответствующих химических атри¬
бутов, таких как структурная формула, эмпирическая
формула и молекулярная масса. При этом могут быть
использованы различные методы, включая:
- спектрометрию ядерного магнитного резонанса;
- масс-спектрометрию;
- инфракрасную спектрометрию;
- элементный анализ.
- Определение чистоты:
- определение содержания сопутствующих (род¬
ственных) примесей подходящим методом разделения
и/или спектрометрическим методом, где применимо;
- количественное определение воды;
- определение содержания остаточных органи¬
ческих растворителей;
- определение потери в массе при высушива¬
нии, которое может в некоторых случаях заме¬
нить определение воды и остаточных количеств
органических растворителей;
-определение неорганических примесей (на¬
пример, сульфатная зола, атомно-абсорбцион¬
ная спектрометрия, спектрометрия индуктивно
связанной плазмы, спектрометрия рентгенов¬
ской флуоресценции); результаты определения
обычно не используются при количественном
определении устанавливаемого содержания ос¬
новного компонента, за исключением значитель¬
ного влияния на это содержание;
-определение чистоты независимым методом
(например, спектрометрия ядерного магнитного
резонанса (количественный анализ), дифферен¬
циальная сканирующая калориметрия или титро¬
вание, в приемлемых случаях; результаты этих
определений используются для подтверждения ре¬
зультатов, полученных с использованием методов
разделения; они не используются при количествен¬
ном определении приписываемого значения).
Для биологических препаратов рекомендации
по изготовлению, определению параметров и ут¬
верждению международных и других биологических
стандартных образцов приведены в руководстве ВОЗ
(И/НО ТесМт'са! РероН Вепез).
4-2. ХИМИЧЕСКИЕ СТАНДАРТНЫЕ ОБРАЗЦЫ
ЕВРОПЕЙСКОЙ ФАРМАКОПЕИ
Рамки испытаний и число привлеченных лабора¬
торий зависят от применения химического стандарт¬
ного образца (СРВ) и служат для обеспечения пригод¬
ности образца для его использования по назначению.
При проведении межлабораторного испытания
в ходе разработки СРВ каждым участником должен
быть представлен протокол испытания. Только обо¬
снованные результаты, указанные в протоколах, ис¬
пользуются для установления количественного со¬
держания основного компонента (заявленного содер¬
жания, приписанного значения) или для какого-либо
другого подтверждения пригодности.
Обычно используются соответствующие части
приведенной ниже программы.
4-2-1. Подлинность (идентификация). В общем
случае выбранная серия образца должна выдержи¬
вать соответствующие требования частной статьи.
Для первой серии проводится полное структурное
описание.
4-2-2. Испытание на сопутствующие примеси.
Стандартный образец, соответствующий примеси, ха¬
рактеризуется показателями подлинности и чистоты.
В случае использования стандартного образца для
определения содержания заданной примеси пред¬
почтительно, чтобы содержание этой примеси в об¬
разце составляло не менее 95,0 %; если это условие
выполняется, то приписанное значение не указывает¬
ся и содержание принимается за 100,0 %; такое при¬
ближенное значение допустимо, так как оно не будет
оказывать существенного влияния на определение
примесей. Если такое минимальное содержание не
может быть получено, для стандартного образца ука¬
зывается заявленное содержание (приписанное зна¬
чение).
В рамках конкретной частной статьи на субстан¬
цию для определения содержания примеси обычно
используется стандартный образец в той же кислот¬
ной, основной или солевой форме, что и субстанция,
являющаяся объектом этой статьи. В противном слу¬
чае и если нет других указаний в частной статье, ис¬
пользуется соответствующий стехиометрический фак¬
тор (коэффициент) пересчета.
Если примесь недоступна в достаточном для по¬
лучения стандартного образца количестве, использу¬
ются следующие подходы:
- приготовление стандартного образца, который
содержит смесь основного компонента (компонентов)
и примеси или примесей;
- приготовление стандартного образца, содержа¬
щего смесь специфических примесей.
В случае если такая смесь используется и для
определения содержания данной примеси, количе¬
ственное содержание примеси в стандартном образ¬
це устанавливается при помощи стандартных мето¬
дов разделения, а полученное значение приписыва¬
ется стандартному образцу.
4-2-3. Количественное определение
4-2-3-1. Химический метод количественного
определения. Если стандартный образец применяют
для количественного определения субстанции для
фармацевтического использования (стандартный об¬
разец для количественного определения), рамки ис¬
пытания такого стандартного образца шире, чем в
случае другого целевого назначения. В случае когда
субстанция не является высокочистой, совместные
испытания обычно проводят в нескольких лаборато¬
риях. Полученные результаты используют для расче¬
та заявленного содержания. В случае использования
стандартного образца в селективном методе количе¬
ственного определения особо важно определить со¬
держание в нем примесей; при этом для испытаний
выбранной субстанции предпочтительно использо¬
вать дополнительные аналитические научно-обосно¬
ванные методы, включая, где это*возможно, незави¬
симые методы и методы, основанные на различных
принципах.
Если стандартные образцы предназначены для
методов количественного определения, заявленное
содержание обычно рассчитывают исходя из значе¬
ний, полученных при определении примесей (орга¬
нические примеси, неорганические примеси, вода и
106
Гэсударственная фармакопея Республики Беларусь
остаточные органические растворители), используя
принцип материального баланса; могут быть исполь¬
зованы и другие подходящие методы. Если есть воз¬
можность, заявленное содержание подтверждается
путем сравнения с результатами, полученными при
использовании независимого метода.
Если стандартные образцы предназначены для
методов количественного определения, не включа¬
ющих хроматографию (например, колориметрии или
спектрофотометрии в ультрафиолетовой области), то
должна быть проверена относительная реакционная
способность или относительное поглощение приме¬
сей, присутствующих в субстанции, чтобы удостове¬
риться, что эти характеристики значительно не отли¬
чаются от характеристик выбранной субстанции.
Если нет других указаний в частной статье, за¬
явленное содержание для субстанции или препа¬
рата, поставляемого в контейнере, принимается без
дополнительных пересчетов «как есть» («аз /$»), и
содержимое контейнера не требует высушивания
перед использованием. Для стандартных образцов,
использующихся при количественном определении
и приготовленных путем лиофилизации, содержание
чистой субстанции указывается в миллиграммах или
в Международных единицах на контейнер.
4-2-3-2. Микробиологический метод количе¬
ственного определения. Активность выражается в
Международных единицах или единицах действия Ев¬
ропейской Фармакопеи (в случае отсутствия между¬
народного стандартного образца). Заявленная актив¬
ность и доверительные интервалы рассчитываются
исходя из статистически обоснованных результатов
межлабораторного испытания согласно обычным ста¬
тистическим методикам (5.3).
4-2-3-3. Количественное определение компонен¬
тов в лекарственном растительном сырье и про¬
дуктах из лекарственного растительного сырья.
Рамки испытаний стандартных образцов, использу¬
емых в частных статьях на лекарственные средства
на основе лекарственного растительного сырья, ва¬
рьируют в зависимости от типа этих стандартных об¬
разцов.
Активные компоненты или маркеры, используе¬
мые в качестве стандартных образцов, оцениваются
по показателям подлинности и чистоты; значение ко¬
личественного содержания устанавливается незави¬
симо от чистоты.
4-2-4. Отчет о создании стандартного образ¬
ца. Отчет, подготовленный ЕБОМ (содержащий ре¬
зультаты исследований, полученных при разработке
стандартного образца, а также информацию по его
использованию) и согласованный соответствую¬
щей группой экспертов, утверждается Европейской
фармакопейной комиссией. В отчете стандартно¬
го образца, использующегося для количественного
определения, указывают приписанное субстанции
содержание с его логическим обоснованием. Рассчи¬
тывают неопределенность заявленного содержания,
и если эта неопределенность меньше предписанной
величины, которая обоснованно является незначи¬
тельной по отношению к критериям приемлемости
для количественного определения, то результаты ис¬
пытаний принимают. В противном случае могут быть
проведены повторные испытания (целиком или ча¬
стично) либо расширены пределы количественного
содержания фармацевтической субстанции. Неопре¬
деленность полученного заявленного содержания не
приводится как часть информации, предоставляемой
со стандартным образцом, так как точность метода и
неопределенность установленного содержания для
стандартного образца учитываются при установлении
предела (пределов) в частной или общей статье.
4-3. РАСТИТЕЛЬНЫЕ СТАНДАРТНЫЕ
ОБРАЗЦЫ ЕВРОПЕЙСКОЙ ФАРМАКОПЕИ
Растительный стандартный образец использу¬
ется, если соответствующий чистый компонент для
количественного определения отсутствует в доста¬
точном количестве или является неустойчивым. Кро¬
ме использования при количественном определении
растительные стандартные образцы могут создавать¬
ся для определения примесей (в т.ч. фальсификации)
и проверки пригодности системы. Для подтверждения
пригодности использования растительных стандарт¬
ных образцов используются различные аналитиче¬
ские методики; при этом могут применяться соответ¬
ствующие части следующей программы испытаний.
Программа испытаний'.
- внешние признаки;
- микроскопия;
- тонкослойная хроматография;
- газовая хроматография;
- жидкостная хроматография;
- количественное определение воды;
-содержание остаточных органических раство¬
рителей;
- потеря в массе при высушивании;
-допустимые примеси;
- количественное определение компонентов (на¬
пример, компонентов с известным терапевтическим
действием, активных маркеров, аналитических мар¬
керов), необходимых для использования такого стан¬
дартного образца по назначению.
Рамки испытаний и число привлеченных лабо¬
раторий зависят от применения растительного стан¬
дартного образца и его последующего использования
по назначению.
Для растительных стандартных образцов, ис¬
пользуемых для количественных определений, за¬
явленное содержание обычно устанавливается в
межлабораторном испытании по результатам коли¬
чественного определения по методике, описанной
в частной статье и в соответствии с которой данный
растительный стандартный образец используется.
Расчет проводится относительно подходящего чисто¬
го образца компонента или компонентов, для которых
в таком стандартном образце должно быть установле¬
но заявленное содержание.
Отчет о создании стандартного образца. От¬
чет о создании растительного стандартного образца
подготавливается аналогично отчету для химического
стандартного образца (см. подраздел 4-2-4).
4-4. БИОЛОГИЧЕСКИЕ СТАНДАРТНЫЕ
ПРЕПАРАТЫ ЕВРОПЕЙСКОЙ ФАРМАКОПЕИ
И ХИМИЧЕСКИЕ СТАНДАРТНЫЕ
ОБРАЗЦЫ ЕВРОПЕЙСКОЙ ФАРМАКОПЕИ,
ИСПОЛЬЗУЕМЫЕ ДЛЯ БИОЛОГИЧЕСКИХ
ЛЕКРАСТВЕННЫХ СРЕДСТВ И ПРЕПАРАТОВ
Большая часть биологически стандартных пре¬
паратов и химических стандартных образцов, исполь¬
зующихся при проведении испытаний биологических
субстанций и препаратов, разрабатывается по Про¬
грамме биологической стандартизации (Вю1од1са1
ЫапдагсИзаИоп Ргодгатте) под эгидой Совета Евро-
5.12. Стандартные образцы
107
пы и Европейской комиссии. Такие стандартные об¬
разцы обычно представляют собой вторичные стан¬
дартные образцы, калиброванные по соответствую¬
щим международным стандартным образцам ВОЗ,
а в случае отсутствия международного стандартного
образца представляют собой первичные стандартные
образцы с заявленным активностью/содержанием,
выраженной в единицах действия Европейской Фар¬
макопеи или в других подходящих единицах. Такие
первичные стандартные образцы разрабатываются
посредством межлабораторных испытаний, при кото¬
рых лаборатории-участницы испытывают выбранный
материал (вещество) или материалы (вещества) и их
достоверные результаты используются для приписы¬
вания официального значения активности/содержа-
ния. Такие испытания могут проводиться совместно
с другими организациями для создания общего ма¬
териала (вещества) или серии материала (вещества)
в качестве стандартного образца. В этом случае, не¬
смотря на то что составляющий материал (вещество)
стандартного образца Европейской Фармакопеи мо¬
жет быть идентичен международному стандартному
образцу и его использование подтверждено в ана¬
логичных межлабораторных испытаниях, такой стан¬
дартный образец рассматривается как вторичный
стандартный образец, использующийся в качестве
рабочего стандартного вещества.
Отчеты об испытании подписываются участни¬
ками, проводившими испытание, утверждаются со¬
ответствующей группой экспертов Европейской Фар¬
макопеи, где применимо, и Руководящим комитетом
Программы биологической стандартизации. Затем
результаты испытания предоставляются Европейской
фармакопейной комиссии. Стандартные образцы/ма-
териалы/вещества/препараты, утвержденные по Про¬
грамме биологической стандартизации, официально
одобряются Европейской фармакопейной комисси¬
ей. Утвержденные отчеты публикуются в журнале
РЬагтеигора Вю & ЗаепИЛс Л/о/ез (Ийр://рЬагтеигора.
ес!дт.еи/РЬагтеигораВю5М).
4- 5.. ВТОРИЧНЫЕ СТАНДАРТНЫЕ ОБРАЗЦЫ
Вторичный стандартный образец должен прояв¬
лять те же значимые для испытания (испытаний) свой¬
ства, что и первичный стандартный образец. Объем
испытаний вторичного стандартного образца не такой
большой, как требуется при создании первичного стан¬
дартного образца. Вторичный стандартный образец
утверждается путем сравнения с первичным стандарт¬
ным образцом, до которого он должен быть прослежи¬
ваем. В любом возможном случае для утверждения
вторичного стандартного образца используют офици¬
альный первичный стандартный образец.
5. ПРОИЗВОДСТВО, МАРКИРОВКА,
ХРАНЕНИЕ И РАСПРОСТРАНЕНИЕ
СТАНДАРТНЫХ ОБРАЗЦОВ ЕВРОПЕЙСКОЙ
ФАРМАКОПЕИ
5- 1. ПРОИЗВОДСТВО
Все производственные операции осуществляют¬
ся в соответствии с существующими нормами надле¬
жащей практики, для того чтобы гарантировать про¬
слеживаемость и сохранность стандартного образца.
Производственная документация включает информа¬
цию относительно упаковки, маркировки и хранения.
Для обеспечения сохранности стандартных образцов
их помещают в контейнеры при соблюдении соответ¬
ствующих условий наполнения и укупорки. Применяе¬
мые для этих целей контейнеры могут быть одноразо¬
вого или многоразового использования; при этом для
уменьшения риска разложения, контаминации или
попадания воды более предпочтительны одноразо¬
вые контейнеры.
5-2. МАРКИРОВКА
Маркировка включает название стандартного об¬
разца, название и адрес поставщика, номер серии и
количество образца (количество на флакон/ампулу).
Обычно предоставляется сопроводительный листок
(вкладыш), который рассматривается как часть мар¬
кировки.
Если стандартный образец используется для ко¬
личественного определения, дополнительно указы¬
вают:
- заявленное процентное содержание;
- либо содержание химического вещества в мил¬
лиграммах или миллилитрах на контейнер;
-либо заявленную активность (для биологиче¬
ского или микробиологического метода количествен¬
ного определения) в единицах активности на милли¬
грамм или на флакон/ампулу.
Для стандартных образцов Европейской Фарма¬
копеи дату переконтроля или дату истечения срока
годности не приводят, так как в плане переконтроля
(см. раздел 6) предусмотрен контроль непрерывной
пригодности стандартных образцов для использо¬
вания. Указание о пригодности серии (ЬаШ? уаНсМу
з(а(етеп(— ВУЗ) для стандартного образца Европей¬
ской Фармакопеи приводится в базе данных стандарт¬
ных образцов (Нйр://до.ес1цт.еи/ГСЗ).
5-3. ХРАНЕНИЕ И РАСПРОСТРАНЕНИЕ
Стандартные образцы должны храниться и рас¬
пространяться в условиях, гарантирующих наилуч¬
шую стабильность.
Стандартные образцы Европейской Фармакопеи
главным образом хранятся в помещениях с контроли¬
руемым температурным режимом при температуре
(5±3) °С. Однако ряд стандартных образцов, которые
являются относительно нестабильными, хранятся при
температуре -(20±5) °С; или в некоторых случаях (на¬
пример, препараты живых вирусов) — при температу¬
ре -(80±10) °С; клеточные культуры — в жидком азоте
(при температуре от -196 °С до -170 °С).
Для минимизации риска повреждения при транс¬
портировке, возможности хранения стандартного об¬
разца при определенной температуре (при необходи¬
мости) и соответствия действующим руководствам по
транспортировке используют подходящую упаковку.
Стандартные образцы, которые обычно хранят¬
ся при температуре (5±3) °С, отправляются обычной
почтой без дополнительного охлаждения, так как кра¬
тковременное отклонение от температурных условий
долговременного хранения для таких образцов не
опасно. Тем не менее когда повышение температуры
оказывает влияние на стабильность, некоторые стан¬
дартные образцы могут отправляться с температу¬
рой +5 °С в упаковках с охлаждающими элементами.
Стандартные образцы, хранящиеся при температуре
-20 °С, упаковываются в контейнеры с охлаждающи¬
ми элементами или сухим льдом (диоксид углерода в
твердом состоянии) и отправляются экспресс-почтой.
Стандартные образцы, хранящиеся при температуре
-80 °С или в жидком азоте, упаковываются в контейне¬
ры с сухим льдом.
108
Гэсударственная фармакопея Республики Беларусь
Условия доставки, отправки, а также темпера¬
туры хранения стандартных образцов доступны для
ознакомления в Каталоге стандартных образцов
(РеГегепсе 8^апда^дз СаШодие), Условиях поставки
(Тегтз о?8ирр1у) и в базе данных стандартных образ¬
цов Европейской Фармакопеи (Нир://до.ес1дт.еи/РЗ).
6. ПЛАН ПЕРЕКОНТРОЛЯ
Система переконтроля разработана и выполня¬
ется для гарантирования непрерывной пригодности
стандартных образцов для использования. Обычно
в плане переконтроля учитывают известные физи¬
ко-химические свойства и данные по стабильности
стандартного образца. Стандартные образцы пери¬
одически проверяют на стабильность при хранении.
Программа мониторинга призвана обнаруживать лю¬
бые признаки разложения на ранней стадии с исполь¬
зованием соответствующих аналитических методик.
Используемые методы должны быть выбраны из уже
использованных в процессе создания стандартных
образцов.
Периодичность и полнота переконтроля стан¬
дартных образцов зависят от ряда следующих фак¬
торов:
- стабильности;
- контейнера и системы укупоривания;
- условий хранения;
- гигроскопичности;
- физического состояния;
- предполагаемого использования;
- однократного или многократного использования.
Период переконтроля может быть увеличен при
наличии достаточного количества эксперименталь¬
ных обосновывающих данных. Максимально допусти¬
мое отклонение от установленного содержания долж¬
но быть заранее определено; в случае его превыше¬
ния серию необходимо переутвердить или заменить.
07/2016:51500
5.15. ФУНКЦИОНАЛЬНО-
ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
ВСПОМОГАТЕЛЬНЫХ
ВЕЩЕСТВ
Данная общая статья и разделы частных ста¬
тей «Функционально-обусловленные характеристи¬
ки» не являются обязательными и публикуются для
информации и руководства.
ВВЕДЕНИЕ
Вспомогательные вещества, предварительно ис¬
следованные на предмет безопасности, используются
в рецептуре готовых лекарственных средств для при¬
дания им определенных функций. Целью использова¬
ния вспомогательного вещества является гарантиро¬
вание заданных физико-химических и биофармацев-
тических свойств готового лекарственного средства.
Функциональные характеристики вспомогатель¬
ных веществ определяются их физическими и хими¬
ческими свойствами и, в некоторых случаях, содер¬
жанием побочных продуктов или добавок, использо¬
ванных для улучшения желаемых функциональных
характеристик. Кроме того, эти характеристики могут
зависеть от сложных взаимодействий между состав¬
ляющими лекарственного средства и от внешних
воздействий во время производственного процесса.
Поэтому функциональные характеристики вспомога¬
тельных веществ могут быть установлены только в
контексте конкретной рецептуры и процесса произ¬
водства, зачастую с использованием нескольких ана¬
литических методик.
Частные статьи Фармакопеи на вспомогатель¬
ные вещества предназначены для обеспечения их
приемлемого качества для потребителей. Информа¬
ция по внешнему виду и описанию вспомогательных
веществ, требования к идентификации, химической и
микробиологической чистоте и физическим характе¬
ристикам, связанным с химической структурой, таким
как оптическое вращение, приведены в частных ста¬
тьях и в общей статье Субстанции для фармацевти¬
ческого использования (2034).
Однако некоторые свойства вспомогательных
веществ (такие как размер частиц, требующийся для
приготовления твердых лекарственных форм, или мо¬
лекулярная масса полимерного материала, использу¬
емого в качестве добавки, увеличивающей вязкость)
могут относиться к функциональным характеристикам
в более широком понимании. Если на этапе разработ¬
ки была продемонстрирована критическая роль таких
функционально-обусловленных характеристик (ФОХ,
1ипсИопа1Ну-геШес1 сЬагас1епзИсз, РРСз) для про¬
цесса производства и параметров качества готового
лекарственного средства, эти характеристики могут
быть отслежены и для них могут быть установлены
продукт-специфические требования к качеству. Такие
критические ФОХ могут описываться как критические
параметры качества (КПК, сгШсаI диаШу аИпЬи1ез,
СОАз) готового лекарственного средства.
Знание этих характеристик может облегчить при¬
менение процессно-аналитической технологии (ПАТ,
Ргосезз Апа1уИса1 ТесЬпо1оду, РАТ).
ФОХ включены в статьи на вспомогательные ве¬
щества с целью помочь производителям лекарствен¬
ных средств установить требования к качеству, осно¬
ванные на стандартных аналитических методиках. Эти
характеристики позволяют найти взаимопонимание
производителям и потребителям вспомогательных
веществ с целью осуществления поставок вспомога¬
тельных веществ со специфицированными свойства¬
ми. ФОХ могут быть указаны (например, в сертифи¬
кате) производителем вспомогательных веществ со
ссылкой на фармакопейную статью, указывая, таким
образом, метод, использованный для установления
данного свойства. Раздел «Функционально-обуслов¬
ленные характеристики» частных статей описывает
ФОХ, влияющие, как известно, на функциональность
вспомогательных веществ для предполагаемого ис¬
пользования. Из-за широкого применения многих
вспомогательных веществ и разработки новых спосо¬
бов их использования перечисленные ФОХ и способы
их применения не являются исчерпывающими.
РЕГУЛЯТОРНЫЕ РУКОВОДСТВА
В соответствии с действующими регуляторными
руководствами, разработанными в соответствии с?,
например, ЮН 08 «Фармацевтическая разработка»,
в заявке на регистрацию должны быть рассмотрены
выбранные вспомогательные вещества и их концен¬
трация, а также должны быть указаны свойства, кото-
5.15. Функционально-обусловленные характеристики вспомогательных веществ
109
рые могут влиять на качество продукта или на процесс
производства с учетом функций каждого вспомогатель¬
ного вещества. Также должна быть продемонстрирова¬
на способность вспомогательных веществ выполнять
свои функции в течение необходимого срока годности
лекарственного средства. Информация о характери¬
стиках вспомогательных веществ при необходимости
может быть использована для обоснования выбора та¬
ких веществ, а также их показателей качества.
Вспомогательные вещества обычно произво¬
дятся сериями, поэтому у одного и того же произво¬
дителя существует возможность варьирования харак¬
теристик вспомогательных веществ от серии к серии.
Вспомогательные вещества от различных производи¬
телей могут не иметь идентичных свойств, играющих
важную роль при использовании этих вспомогатель¬
ных веществ в конкретной рецептуре. Варьирование
химических и физических свойств вспомогательных
веществ является ключевым моментом, который не¬
обходимо принимать во внимание при разработке ле¬
карственного средства и процесса его производства.
Многие вспомогательные вещества являются веще¬
ствами природного происхождения и представляют
собой сложную смесь химически близких соединений.
Некоторые вспомогательные вещества производятся
на химических заводах, изначально нацеленных на
производство химических веществ не для фармацев¬
тической промышленности. Таким образом, процесс
производства может быть сфокусирован на получении
химических характеристик и некоторых физических
свойств, необходимых для первичного рынка сбыта
производителя. Очень часто производители вспомо¬
гательных веществ не имеют достаточных знаний о
фармацевтическом использовании этих веществ.
Понимание химической и физической природы
фармацевтической субстанции (субстанций) и вспо¬
могательных веществ по отдельности, а также того,
каким образом их свойства влияют на другие ингреди¬
енты и на процесс производства, является ключевым
аспектом создания успешной и надежной рецептуры.
При фармацевтической разработке идентифицируют
свойства, критические для процесса производства и
для характеристик лекарственного средства. После
идентификации критических свойств вспомогатель¬
ных веществ, основываясь предпочтительно на под¬
ходе оценки рисков, в процессе фармацевтической
разработки может быть установлен диапазон при¬
емлемых значений критических параметров веще¬
ства, включая варьирование как физических, так и
химических свойств. Интересующие ФОХ могут не
контролироваться производителем вспомогательно¬
го вещества и, таким образом, быть непостоянными.
Поэтому предпочтительным является планирование
устойчивого процесса производства готового лекар¬
ственного средства, чтобы эффект воздействия на
него нормальной изменчивости вспомогательных ве¬
ществ был ограничен.
Оценка сортов с различными физическими и хи¬
мическими характеристиками и, при необходимости,
установление требований к качеству для критических
характеристик являются частью фармацевтической
разработки независимо от необязательного характера
ФОХ. Разработка должна рассматриваться в свете ре¬
гуляторного руководства по фармацевтической разра¬
ботке и желаемой регуляторной гибкости, основанной
на установлении приемлемого диапазона свойств ве¬
щества внутри пространства проектных параметров.
СОРТА С РАЗЛИЧНЫМИ ФИЗИЧЕСКИМИ
ХАРАКТЕРИСТИКАМИ
Сыпучие твердые вспомогательные вещества
могут быть доступны в виде большого количества
различных сортов с различными физическими харак¬
теристиками (например, распределением частиц по
размерам), которые обычно контролируются постав¬
щиком. Однако ФОХ этих веществ могут затрагивать
другие свойства, обусловленные твердым состояни¬
ем вещества, а также обусловленные сыпучим состо¬
янием твердого вещества, которые могут не контроли¬
роваться поставщиком вспомогательного вещества.
К свойствам сыпучих твердых веществ относятся,
например, распределение частиц по размерам, удель¬
ная площадь поверхности, насыпная плотность, теку¬
честь, смачиваемость и водопоглощение. В зависимо¬
сти от диапазона размеров распределение частиц по
размерам может определяться с помощью ситового
анализа (общая статья 2.9.38. Определение размера
частиц методом аналитического просеивания) или
инструментальных методов, например 2.9.31. Опре¬
деление размера частиц методом дифракции ла¬
зерного излучения. Метод, описанный в общей статье
2. 9.26. Определение удельной площади поверхности
методом газовой адсорбции, основан на методике
Брунауэра-Эмметта-Теллера (БЭТ). Методы оценки
текучести и насыпной плотности порошков описаны в
общих статьях 2.9.36. Текучесть порошков и 2.9.34.
Насыпная плотность и плотность после усадки.
Свойства, обусловленные твердым состоянием веще¬
ства, могут воздействовать на смачиваемость (общая
статья 2.9.45. Смачиваемость пористых твердых
материалов, включая порошки) и взаимодействия
«вода - твердое вещество» (общая статья 2.9.39. Вза¬
имодействия «вода - твердое вещество»: постро¬
ение изотерм сорбции-десорбции и определение ак¬
тивности воды) сыпучих твердых веществ.
Примерами свойств, учитываемых при разработ¬
ке твердой лекарственной формы и обусловленных
твердым состоянием вещества, могут служить поли¬
морфизм, псевдополиморфизм, кристалличность и
плотность. Методики их оценки приведены в общих
статьях 5.9. Полиморфизм, 5.16. Кристалличность и
2.2.42. Плотность твердых тел.
СОРТА С РАЗЛИЧНЫМИ ХИМИЧЕСКИМИ
ХАРАКТЕРИСТИКАМИ
Доступные вспомогательные вещества представ¬
лены в виде сортов с различными химическими харак¬
теристиками и имеют природное, полусинтетическое
или синтетическое происхождение. В частных статьях
обычно контролируется химический состав вспомога¬
тельных веществ, которые представляют собой смесь
близких соединений, например состав жирных кислот
растительных масел или поверхностно-активных ве¬
ществ. Однако в Фармакопее присутствуют статьи,
описывающие класс полимерных материалов, которые
могут варьировать по составу в зависимости от структу¬
ры гомополимеров, блоков полимеров и сополимеров,
степени полимеризации, а, следовательно, по массе
и массовому распределению, степени замещения и, в
некоторых случаях, даже по заместителям на основ¬
ной цепи полимера. Эта вариабельность может иметь
сильное влияние на функциональные характеристики
вспомогательных веществ и должна учитываться при
фармацевтической разработке; желательно установить
диапазон приемлемых значений для каждого параме-
110
Государственная фармакопея Республики Беларусь
тра, критического как для процесса производства, так
и для характеристик готового лекарственного средства.
РАЗДЕЛ «ФУНКЦИОНАЛЬНО-
ОБУСЛОВЛЕННЫЕ ХАРАКТЕРИСТИКИ»
В ЧАСТНЫХ СТАТЬЯХ
Частные статьи на вспомогательные вещества
могут содержать раздел «Функционально-обусловлен¬
ные характеристики», который приводится для инфор¬
мации и не является обязательным. В этом разделе
перечисляются характеристики, которые существенны
для конкретного использования вспомогательного ве¬
щества. При этом указывают вид использования, для
которого данная характеристика важна, а на примене¬
ние в других целях данная характеристика может не
влиять. По этой причине данный раздел не должен рас¬
сматриваться как просто дополнение к частной статье.
Решение о том, как приведенная в разделе «Функцио¬
нально-обусловленные характеристики» информация
будет использована в процессе производства с учетом
применения вспомогательного вещества и данных, по¬
лученных при фармацевтической разработке, прини¬
мает производитель лекарственного средства.
Информация о функционально-обусловленных
характеристиках может быть приведена различными
способами:
- название ФОХ;
- название ФОХ и рекомендованный метод для
ее определения со ссылкой, где это возможно, на об¬
щую статью Фармакопеи;
- название ФОХ с рекомендованным методом
для ее определения и типичными значениями, кото¬
рые могут быть представлены в виде отклонений от
номинального значения.
Данная характеристика может присутствовать
как обязательное требование в частной статье. Если
она является важной для конкретного использования,
то она также указывается в разделе «Функционально-
обусловленные характеристики» как важная харак¬
теристика, которую производитель лекарственного
средства имеет право конкретизировать для точного
указания качества, требуемого для конкретного ле¬
карственного средства.
Раздел, включающий ФОХ, предназначен для от¬
ражения текущих знаний о наиболее важных исполь¬
зованиях вспомогательного вещества. Ввиду широкой
области применения некоторых вспомогательных ве¬
ществ и продолжающейся разработки новых типов их
использования данный раздел может быть неполным.
Кроме этого методы, указанные для определения кон¬
кретных характеристик, приведены в качестве реко¬
мендации и не исключают возможности использова¬
ния других методов.
МЕЖДУНАРОДНАЯ ГАРМОНИЗАЦИЯ
Количество частных фармакопейных статей на
вспомогательные вещества является предметом гар¬
монизации между фармакопеями Европы, Японии и
США. Введение раздела «Функционально-обусловлен¬
ные характеристики» в частные статьи Европейской
Фармакопеи означает, что вид гармонизированных
статей отличается. Испытания физических и химиче¬
ских характеристик, относящихся как к качественным,
так и функционально-обусловленным в Европейкой
Фармакопее, в остальных двух фармакопеях вклю¬
чены только в текст частной статьи. Такой различный
формат не влияет на спецификацию вспомогательно¬
го вещества для производителя лекарственного сред¬
ства. Руководящие документы в настоящее время
рекомендуют идентифицировать и специфицировать
только такие критические свойства, которые влияют на
производственный процесс и качество готового лекар¬
ственного средства. Различная законодательная база
в случае трех фармакопей допускает существование
различных форматов частных фармакопейных статей
без вреда для статуса международной гармонизации.
ГЛОССАРИЙ
Критическая характеристика (сгШса!
сЬагас1епзИс)\ любая физическая или химическая
характеристика вещества, которая в значительной
степени влияет на технологичность и/или качество
лекарственного средства.
Пространство проектных параметров (с/ез/дл
зрасе): многомерная комбинация и взаимодействие
вводимых переменных (например, характеристики ве¬
щества) и параметров процесса, которые обеспечива¬
ют гарантию качества.
Функционально-обусловленная характери¬
стика (IипсИопаШу-геШес! сЬага&епзНс, РРСз)\ кон¬
тролируемая физическая или химическая характери¬
стика вспомогательного вещества, влияющая на его
функциональность.
Процессно-аналитическая технология
(Ргосезз Апа1уИса1 ТесМпо1оду, РАТ): система для
планирования, анализа и контроля производства по¬
средством периодических измерений (т.е. в течение
процесса производства) критических параметров ка¬
чества сырья и веществ, используемых в процессе
производства, и процессов с целью обеспечения ка¬
чества готового продукта.
07/2016:52000
5.20. ОСТАТОЧНЫЕ
КОЛИЧЕСТВА
МЕТАЛЛИЧЕСКИХ
КАТАЛИЗАТОРОВ ИЛИ
МЕТАЛЛСОДЕРЖАЩИХ
РЕАКТИВОВ
Европейское агентство по лекарственным сред¬
ствам (Еигореап МесИстез Адепсу, ЕМА) приняло ру¬
ководство по остаточным количествам металлических
катализаторов или металлсодержащих реактивов,
которые могут содержаться в субстанциях для фар¬
мацевтического использования или в лекарственных
средствах. Данное руководство предписывает преде¬
лы содержания металлов, которые могут оставаться в
субстанциях для фармацевтического использования
и в лекарственных средствах. Представленное руко¬
водство, текст которого приведен ниже (за исключени¬
ем дополнений), применимо как к новым продуктам,
так и к продуктам, уже представленным на рынке.
Согласно Фармакопее, данное руководство при¬
менимо ко всем субстанциям для фармацевтического
использования вне зависимости от того, существуют
ли на эти субстанции частные статьи или нет. Все суб¬
станции должны быть проверены на содержание ме¬
таллов, присутствие которых возможно.
5.20. Остаточные количества металлических катализаторов
111
Если используемые пределы содержания совпа¬
дают с приведенными ниже, испытания на остаточ¬
ные количества металлов обычно не упоминаются в
частных статьях по причине того, что используемые
металлы могут отличаться у разных производителей и
требования данной статьи применяются посредством
общей статьи Субстанции для фармацевтического
использования (2034). Информация о металлах, ис¬
пользованных в виде реактивов или катализаторов в
процессе производства, должна быть представлена
компетентному и уполномоченному органу. Данная
информация должна быть включена в регистрацион¬
ное досье и приведена в сертификате качества*.
По возможности должна использоваться методо¬
логия, описанная в статье 2.4.20. Определение оста¬
точных количеств металлических катализаторов
или металлсодержащих реактивов.
Конец пятилетнего переходного периода, упомя¬
нутого в Разделе 2 «Определение и область приме¬
нения» руководства ЕМ А, приходится на 1 сентября
2013 года.
РУКОВОДСТВО НА СПЕЦИФИКАЦИЮ
ПРЕДЕЛОВ СОДЕРЖАНИЯ
ОСТАТОЧНЫХ КОЛИЧЕСТВ
МЕТАЛЛИЧЕСКИХ КАТАЛИЗАТОРОВ ИЛИ
МЕТАЛЛСОДЕРЖАЩИХ РЕАКТИВОВ
(ЕМЕА/СНМР/8\А/Р/4446/2000)
ОСНОВНЫЕ ПОЛОЖЕНИЯ
1. ВВЕДЕНИЕ
2. ОПРЕДЕЛЕНИЕ И ОБЛАСТЬ ПРИМЕНЕНИЯ
3. ПРАВОВЫЕ ОСНОВЫ
4. ОСНОВНОЙ ТЕКСТ РУКОВОДСТВА
4.1. КЛАССИФИКАЦИЯ
4.2. ПРЕДЕЛ ВОЗДЕЙСТВИЯ
4.3. УСТАНАВЛИВАНИЕ КОНЦЕНТРАЦИОННЫХ
ПРЕДЕЛОВ СОДЕРЖАНИЯ ОСТАТОЧНЫХ КОЛИ¬
ЧЕСТВ МЕТАЛЛОВ
4.3.1. Общие понятия
4.3.2. Лекарственные средства для перорально¬
го, парентерального применения и для ингаляций
4.3.3. Лекарственные средства, применяемые
иным способом
4.3.4. Лекарственные средства, применяемые
кратковременно и для спасения жизни
4.4. АНАЛИТИЧЕСКИЕ МЕТОДИКИ
4.5. РЕЗУЛЬТАТЫ ПРОВЕРКИ СЕРИИ, ЧАСТОТА
ПРОВЕДЕНИЯ ИСПЫТАНИЯ И УДАЛЕНИЕ ИСПЫТА¬
НИЯ ИЗ СПЕЦИФИКАЦИИ
4.6. УЧИТЫВАЕМЫЙ ПРЕДЕЛ
5. ГЛОССАРИЙ
6. ССЫЛКИ (НАУЧНЫЕ И/ИЛИ ЗАКОНОДАТЕЛЬ¬
НЫЕ)
ОСНОВНЫЕ ТРЕБОВАНИЯ
Целью данного руководства является рекомен¬
дация максимально приемлемых концентрационных
пределов содержания остаточных количеств метал¬
лических катализаторов или металлсодержащих
реактивов в субстанциях для фармацевтического
использования или лекарственных средствах. Под
субстанцией для фармацевтического использования
в данном руководстве понимаются как фармацевти¬
ческие субстанции (активные вещества), так и вспо¬
могательные вещества.
Металлы, описанные в данном руководстве,
обычно используются в качестве катализаторов про¬
цессов или в качестве реактивов в синтезе субстанций
для фармацевтического использования. Их использо¬
вание может приводить к тому, что в субстанциях, а
затем и в лекарственных средствах могут обнаружи¬
ваться остатки этих металлов. Такие остатки металлов
не приносят какой-либо терапевтической пользы для
пациента и, следовательно, должны быть оценены, а
их содержание должно быть ограничено на основа¬
нии критериев безопасности и качества. С течением
времени данное руководство может быть дополнено
включением информации и о других металлах.
Согласно приведенному руководству, остаточные
металлы делятся на 3 категории на основании их ин¬
дивидуального уровня безопасности и концентраци¬
онных пределов содержания. Пределы содержания
устанавливаются исходя из максимальной суточной
дозы, продолжительности лечения, пути введения
лекарственного средства, а также допустимой (разре¬
шенной) суточной дозы (регтМед даНу ехрозиге, РОЕ
или ДСД) остаточного металла. В руководстве также
приведены рекомендации по стратегии проведения
испытания, аналитическим методикам и учитывае¬
мым пределам в субстанциях для фармацевтического
использования или лекарственных средствах.
1. ВВЕДЕНИЕ
Появление остаточных металлов в субстанциях
для фармацевтического использования и в лекар¬
ственных средствах может быть обусловлено такими
источниками, как металлические катализаторы и ме¬
таллсодержащие реактивы, используемые в синтезе
фармацевтических субстанций и вспомогательных
веществ, производственное оборудование и трубо¬
проводы, упаковочные материалы для нефасованной
продукции, окружающая среда, чистящие средства и
так далее. Так как остаточные металлы не дают ни¬
какой терапевтической выгоды для пациента, а риск
должен быть сопоставим с оказываемым лекарствен¬
ным средством эффектом, может понадобиться вклю¬
чение пределов содержания остаточных количеств
металлов и валидированный метод их определения
в спецификацию на субстанцию или лекарственное
средство для гарантирования их надлежащего каче¬
ства. Принятие во внимание указанных требований
должно быть основано на критериях безопасности и
качества, а также учитывать требования Надлежа¬
щей производственной практики (СМР), Надлежащей
практики оптовой торговли (СОР) и иных соответству¬
ющих руководств.
Целью данного руководства является рекомен¬
дация максимально приемлемых концентрационных
пределов содержания остаточных количеств метал¬
лов, источником которых являются металлические
катализаторы или металлсодержащие реактивы, ис¬
пользуемые в синтезе фармацевтических субстанций
и вспомогательных веществ. Так как использование
этих металлов ограничивается определенными хими¬
ческими реакциями, обычно достаточно ограничить
их содержания в субстанциях для фармацевтическо¬
го использования. Таким образом, ограничивать со¬
держание остаточных количеств металлов в готовом
лекарственном средстве, как правило, не требуется.
В данном руководстве концентрационные пределы
содержания основываются на критериях безопасно¬
сти и достижения надлежащего качества субстанций
для фармацевтического использования и лекарствен¬
ных средств. Таким образом, не следует ожидать, что
112
Гэсударственная фармакопея Республики Беларусь
фармацевтическая отрасль будет ужесточать концен¬
трационные пределы содержания в регистрационном
досье на основании правил 6МР, возможностей про¬
изводства или любых иных критериев качества.
Так как потенциальный токсический эффект
остаточных металлов не зависит от их источника,
концентрационные пределы содержания, указанные
в данном руководстве, в принципе могут быть при¬
менимы при определении остаточных количеств ме¬
таллов, произошедших не только от металлических
катализаторов или металлсодержащих реактивов, но
и из иных источников. Однако утверждение предель¬
ного содержания и валидированной методики для
остаточных количеств металлов, произошедших из
других источников, является необходимым только в
исключительных случаях, когда известно, что при со¬
блюдении правил ОМР, ОБР и иных соответствующих
руководств не достигается необходимая чистота от
остаточных металлов. Фармацевтические предпри¬
ятия не обязаны проводить интенсивные испытания
на наличие остаточных металлов неизвестного проис¬
хождения для того, чтобы соответствовать требовани¬
ям данного руководства. Они могут полагаться на ин¬
формацию, получаемую от заслуживающих доверия
поставщиков.
Металлы, включенные в настоящее время в дан¬
ное руководство, перечислены в таблице 1. С тече¬
нием времени руководство может быть дополнено и
другими остаточными металлами. Любая заинтересо¬
ванная сторона может подать заявку и предоставить
на рассмотрение соответствующие данные по без¬
опасности. Классификация и концентрационные пре¬
делы содержания металлов, включенных в настоящее
время, также могут изменяться по мере получения но¬
вых данных по безопасности.
При определении концентрационных пределов
были приняты следующие допущения и/или значения
по умолчанию:
- масса тела взрослого человека: 50 кг;
- объем вдыхаемого воздуха для взрослого чело¬
века: 20 м3 в сутки;
-подвергание ингаляционному воздействию на
рабочем месте: 8 ч в сутки;
- пределы воздействия (экспозиции) были уста¬
новлены с использованием фактора неопределенно¬
сти, как указано в приложении к руководству ЮН 03С;
- в практических целях число факторов неопре¬
деленности адаптировано для получения конечных
безопасных и практических ДСД — метод 03С для
фактора неопределенности (I//1) плюс практический
фактор для расчета ДСД;
- допустимый дополнительный риск возникнове¬
ния рака: увеличение риска возникновения рака на 1
на 100 000 был принят СНМР как приемлемый для ге¬
нотоксических примесей в лекарственных средствах.
2. ОПРЕДЕЛЕНИЕ И ОБЛАСТЬ
ПРИМЕНЕНИЯ
Под металлическими катализаторами и металл¬
содержащими реактивами понимают химические ве¬
щества, которые используются для изменения скоро¬
сти химических реакций или которые взаимодейству¬
ют с другими химическими веществами в химических
реакциях. В данном руководстве рассматриваются
металлические катализаторы и металлсодержащие
реактивы, используемые в синтезе фармацевтиче¬
ских субстанций, вспомогательных веществ, а также
любых вспомогательных веществ, используемых при
производстве лекарственного средства, но не вхо¬
дящих в его состав. Остаточные металлы могут при¬
сутствовать либо в исходном виде металла, либо в
форме металлического элемента, видоизмененного
происходящим химическим процессом.
Действие данного руководства распространя¬
ется как на новые, так и на уже присутствующие на
рынке лекарственные средства. Для лекарственных
средств, уже присутствующих на рынке, дается пя¬
тилетний срок на введение данного руководства в
действие, если это невозможно будет сделать ранее.
После этого переходного пятилетнего срока на рынок
будут допускаться только лекарственные средства,
произведенные с использованием субстанций, от¬
вечающих данному руководству. Руководство не ка¬
сается потенциально новых фармацевтических суб¬
станций или вспомогательных веществ, проходящих
стадию клинических испытаний при разработке ново¬
го лекарственного средства. В данный период могут
быть применены более высокие уровни содержания
остаточных металлов.
Также руководство не касается металлов, кото¬
рые вводятся в субстанции специально (например,
противоионы), или металлы, которые входят в состав
лекарственного средства в качестве вспомогательных
веществ (например, краситель оксид железа). Как ука¬
зывалось во введении, руководство, как правило, не
распространяется на чужеродные металлические за¬
грязнения, наличие которых подпадает под действие
иных подходящих документов, таких как ОМР, ОБР
или других руководств, касающихся качества.
Способ применения лекарственного средства
может влиять на фактическое экспонирование орга¬
низма человека воздействию металла. По причине
ограниченной биодоступности металлов при перо¬
ральном приеме в данном руководстве применяются
различные нормы при парентеральном и перораль¬
ном введении. Так как иные способы применения мо¬
гут иметь различные токсикологические последствия,
специальные для некоторых металлов, были установ¬
лены пределы также и для ингаляционного пути вве¬
дения. При кратковременном воздействии (экспони¬
ровании) ДСД может быть адаптирована, как указано
в разделе 4.3.
3. ПРАВОВЫЕ ОСНОВЫ
Данное руководство должно рассматриваться од¬
новременно с Директивой 2001/83/ЕС с изменениями,
а также вместе со всеми релевантными руководящи¬
ми документами СНМР с особым значением следую¬
щих документов:
-Nо^е /ог Ошдапсе оп 1тригШез т А/еи/ Бгид
Рго4ис1з (СРМР/ЮН/2738/99, ЮН 03В (Я));
- Л1о(е Тог ОиМапсе оп 1тригШез ТезИпд: 1тригШез
т /Уем Бгид 8иЬз(апсез (СРМР/ЮН/2737/99, /СНОЗА);
- Ио(е Тог ОиШапсе оп IтригШез: РезМиа! 8оЫеп1з
(СРМР/ЮН/283/95 вместе с СРМР/ЮН/1507/02,
СРМР/ЮНА940ЛЮ согг, СРМР/ОМР/450/03 и СРМР/
ОИ/Р/8567/99);
- ОиМеНпе оп 0ю ЫтИз оТ Оепо1охю 1тригШез
(ЕМЕА/СНМР/0\А/Р/25^ 344/2006 и СРМР/
8)Л/Р/5199/82);
- УаИбаИоп оТ Апа1уИса1 Ргоседигез: Тех1 апс!
Ме(Шо1оду (СНМР/ЮН/381/95, ЮН 02 (Я1)).
5.20. Остаточные количества металлических катализаторов
113
Таблица 1
Классы воздействия (экспозиции) и концентрационные пределы для отдельных металлических
катализаторов и металлсодержащих реактивов
Классификация
Пероральное воздействие
Парентеральное воздействие
Ингаляционное
воздействие*
ДСД (мкг/сут)
Концентрация (ррт)
ДСД (мкг/сут)
Концентрация (ррт)
ДСД (нг/сут)
Металлы с высоким уровнем опас-
ности
Класс 1 А:
Р*, Рс1
100
10
10
1
Р1: 70*
Класс 1В:
1г, ГСП, ГСи, 0$
100**
10**
10**
!**
Класс 1С:
Мо, N1, Сг, V
250
25
25
2,5
100
Сг (VI): 10
Металлы с пониженным уровнем
опасности
Класс 2:
Си, Мп
2500
250
250
25
Металлы с минимальным уровнем
опасности
Класс 3:
Ре, 2п
13 000
1300
1300
130
* См. раздел 4.4.
** Предельные значения для подкласса: общее количество перечисленных металлов не должно превышать указанный предел
4. ОСНОВНОЙ ТЕКСТ РУКОВОДСТВА
4.1. КЛАССИФИКАЦИЯ
Термин «максимально допустимое ежедневное
потребление» (ТО/, /о/егаЬ/е даНу Маке, или ДЕП)
используется Международной программой по хими¬
ческой безопасности (1РС8) для описания пределов
воздействия (экспозиции) токсических химических ве¬
ществ, тогда как термин «приемлемое ежедневное по¬
требление» (АО/, ассер1аЫе даНу Маке, или ПЕП) ис¬
пользуется Всемирной организацией здравоохране¬
ния (ВОЗ) и другими национальными и международ¬
ными организациями здравоохранения и институтами.
В руководстве ЮН 03С на остаточные растворители,
чтобы избежать путаницы в терминах и их определе¬
ниях, был выбран новый термин, который называет¬
ся «допустимая (разрешенная) суточная доза» (РОЕ,
регтШед даНу ехрозиге, или ДСД). В данном руковод¬
стве ДСД обозначает фармацевтически максимально
приемлемое длительное воздействие (экспозицию)
металла, не приносящее вреда здоровью.
Остаточные металлы оцениваются по степени
возможного риска для здоровья человека и разделе¬
ны на 3 класса.
Металлы 1-го класса: металлы с высоким уров¬
нем опасности. Эта группа включает металлы, которые
известны как канцерогены для человека или предполо¬
жительно к ним относятся, а также могут служить источ¬
никами высокой токсичности иного характера.
Металлы 2-го класса: металлы с пониженным
уровнем опасности. Эта группа включает металлы с
пониженной токсичностью для человека. Эти метал¬
лы и их воздействия (экспозиции) обычно допуска¬
ются при применении лекарственных средств. Это
могут быть следы металлов, предназначенных для
пищевых целей или чаще всего входящих в состав пи¬
щевых наполнителей или широко распространенных
пищевых добавок.
Металлы 3-го класса: металлы с минималь¬
ным уровнем опасности. Эта группа включает ме¬
таллы с незначительной токсичностью. Их безопас¬
ность установлена. Они обычно допускаются вплоть
до доз свыше тех, которые обычно вводятся при при¬
менении лекарственных средств. Они повсеместно
встречаются в окружающей среде, в животном и рас¬
тительном мире.
4.2. ПРЕДЕЛ ВОЗДЕЙСТВИЯ (ЭКСПОЗИЦИИ)
Общая оценка безопасности, осуществляемая
исходя из пределов содержания, определяется для
остаточных металлов каждого отдельного класса,
принимая во внимание путь введения.
Таблица 1 представляет информацию по допу¬
стимым предельным значениям ДСД и концентраци¬
ям включенных в данное руководство 14 остаточных
металлов после их перорального, парентерального
и/или ингаляционного воздействий. Металлы, вклю¬
ченные в класс 1, в дальнейшем разделяются на 3
подкласса: 1А, 1В и 1С. Пределы воздействия (экс¬
позиции) для класса 1А (платиноиды) и 1С относят¬
ся к отдельным металлам, тогда как пределы воздей¬
ствия (экспозиции) для класса 1В (также платиноиды)
относятся к общему количеству перечисленных ме¬
таллов. Для металлов платиновой группы класса 1В
был принят традиционный подход, потому что в на¬
стоящее время доступные данные по токсичности до¬
вольно ограничены. Поэтому указанный предел для
класса 1В является пределом для общего количества
металлов платиновой группы, которые предположи¬
тельно присутствуют, принимая во внимание исполь¬
зуемые способы синтеза.
4.3. УСТАНОВЛЕНИЕ КОНЦЕНТРАЦИОННЫХ
ПРЕДЕЛОВ ОСТАТОЧНЫХ МЕТАЛЛОВ
4.3.1. Общие положения
Если известно или предполагается, что процесс
синтеза фармацевтических субстанций приводит к
появлению остаточных металлов вследствие приме¬
нения специфического металлического катализатора
или металлсодержащего реактива, то для всех оста¬
точных металлов должны устанавливаться концентра¬
ционные пределы и валидированное испытание. Все
концентрационные пределы должны быть достовер¬
ными с точки зрения аналитической точности, произ¬
водственной возможности, обоснованных изменений
в производственном процессе. Так как использова¬
ние металлических катализаторов или металлсодер-
114
Гэсударственная фармакопея Республики Беларусь
жащих реактивов в течение синтеза ограничивается
определенными химическими реакциями, то опреде¬
ление предельного содержания остаточных количеств
металлов в фармацевтических субстанциях будет,
как правило, достаточным. Предельное содержание
остаточного количества металла в фармацевтической
субстанции может быть заменено его предельным со¬
держанием в готовом лекарственном средстве, как
описано ниже.
4.3.2. Лекарственные средства для перо¬
рального, парентерального применения и для
ингаляций
При установлении концентрационного предела
для остаточного металла возможны 2 подхода.
Подход 1. Используют концентрационный предел
(частей на миллион (ррт)) из таблицы 1, рассчитан¬
ный по формуле (1), исходя из предположения, что
суточная доза потребления лекарственного средства
составляет 10 г.
ДСД (мкг/сут)
суточная доза (г/сут) ^)
В случае соответствия всех фармацевтических
субстанций в лекарственном средстве по концентраци¬
онному пределу для металлов требованиям Подхода 1
эти субстанции могут быть использованы в любых про¬
порциях в лекарственном средстве в случае, если суточ¬
ная доза лекарственного средства не превышает 10 г в
сутки. Если суточная доза лекарственного средства пре¬
вышает 10 г в сутки, то применяется Подход 2.
Подход 2а. ДСД (мкг/сут), указанные в таблице 1,
могут применяться совместно с фактической суточной
дозой фармацевтической субстанции в лекарственном
средстве для расчета концентрации остаточного метал¬
ла, допускаемой в этой фармацевтической субстанции.
Подход 2Ь. Альтернативный, не является обяза¬
тельным для фармацевтических субстанций, для кото¬
рых концентрационные пределы удовлетворяют тре¬
бованиям Подхода 1 или рассчитаны в соответствии с
Подходом 2.
ДСД (мкг/сут), указанные в таблице 1, могут также
применяться с известными максимальными суточны¬
ми дозами лекарственного средства для определения
концентрации остаточного металла, происходящего из
любой фармацевтической субстанции в данном лекар¬
ственном средстве (не в субстанции). Такое допущение
приемлемо при подтверждении того, что содержание
остаточного количества металла снижено до допусти¬
мого минимума в каждой субстанции. Это допущение
подразумевает, что максимальные уровни содержания
металла в определенной субстанции могут быть выше
пределов, установленных требованиями Подхода 1 или
Подхода 2, но это компенсируется более низкими макси¬
мальными уровнями в других субстанциях.
4.3.3. Лекарственные средства, применяемые
иным способом
Концентрационные пределы устанавливаются с
учетом способа применения лекарственного средства.
Предельные значения ДСД и концентрации для
парентерального применения лекарственного средства
могут использоваться без специфического обоснования
для фармацевтических субстанций, которые применя¬
ются другими путями введения, включая ингаляцион¬
ный. Предельные значения ДСД и концентрации для
перорального приема могут использоваться в том слу¬
чае, если абсорбция при использовании других путей
введения не превышает абсорбцию после перорального
введения. Например, в случае накожного применения
считаются допустимыми предельные значения ДСД и
концентрации, установленные для перорального при¬
менения.
Установлено, что соли платины являются аллер¬
генами, а гексахлорплатиновая кислота — наиболее
сильный из них (Ма1о, Л-/_, 2005). Поэтому был установ¬
лен специфичный предел для этого металла при воз¬
действии путем ингаляции, равный 70 нг/сут (см. при¬
ложение 2 «Монографии на элементы» полной версии
данного руководства). Для хрома (VI) и никеля при воз¬
действии ингаляционным путем проведена ассоциация
с канцерогенами. Поэтому были установлены специфич¬
ные пределы для хрома (VI) и никеля при воздействии
ингаляционным путем, равные 10 нг/сут и 100 нг/сут со¬
ответственно (см. приложение 2 «Монографии на эле¬
менты» полной версии данного руководства).
4.3.4. Лекарственные средства, применяемые
кратковременно и для спасения жизни
В данном руководстве приведены предельные зна¬
чения ДСД и концентрации с учетом длительного воздей¬
ствия (экспозиции), в то время как в случае кратковре¬
менного применения (30 дней или менее) принимаются
более высокие предельные значения ДСД и концентра¬
ции. Это может быть применимо, например, к средствам
для контрастных методов в диагностике, антидотам или
средствам для диагностики, но только если предельные
значения, установленные в Подходе 1 и Подходе 2, не
применимы. В каждом конкретном случае необходимо
проводить оценку с точки зрения «риск-польза», напри¬
мер более высокие предельные значения могут быть
установлены для лекарственных средств, используемых
при показаниях к применению, направленных на спасе¬
ние жизни. В каждом конкретном случае должен быть
индивидуальный и обоснованный подход.
4.4. АНАЛИТИЧЕСКИЕ МЕТОДИКИ
Для определения каждого остаточного металла ис¬
пользуется соответствующая валидированная методика.
Следует обратить внимание на то, что остаточные ме¬
таллы могут находиться в другом виде, чем элемент в
исходных катализаторе или реактиве. Если не доказано
противоположное, проводится специфичное испытание
для каждого элемента. В случае наличия обосновываю¬
щих данных может применяться общая аналитическая
методика для определения одного или более остаточных
металлов с общим концентрационным пределом, если
может быть показано, что предел воздействия (экспози¬
ции) ни для одного из отдельных металлов не превышен.
Для определения предельных концентраций оста¬
точных количеств металлов могут применяться любые
гармонизированные подходящие методы, описываемые
в Фармакопее. Производители свободны в выборе более
подходящей валидированной аналитической методики
для применения в частном случае. Если присутствуют
только остаточные металлы класса 2 и класса 3, может
применяться неспецифичный метод. В частности, это
имеет отношение к платиноидам класса 1 В, где приме¬
няются групповые предельные значения и допускается,
что из-за технических ограничений нижний предел опре¬
деления не может быть ниже 0,5 ррт для отдельных
платиноидов.
Общие полуколичественные методы определения
пределов содержания металлов, основанные на осажде¬
нии при рН 3,5 окрашенных сульфидов металлов, описы¬
ваются в Фармакопее. Подобные испытания не подходят
для количественного определения фактических уровней
отдельных остаточных металлов в фармацевтической
I
5.20. Остаточные количества металлических катализаторов
115
субстанции. В случае наличия подтверждающих данных
(например, полученных с использованием метода стан¬
дартных добавок) и проведения валидации (включая пе¬
рекрестную валидацию с элемент-специфичным испы¬
танием) испытания, в основе которых лежит осаждение
сульфидом, могут быть подходящими для проведения
рутинных испытаний в некоторых случаях.
4.5. РЕЗУЛЬТАТЫ ПРОВЕРКИ СЕРИИ,
ЧАСТОТА ПРОВЕДЕНИЯ ИСПЫТАНИЯ
И УДАЛЕНИЕ ИСПЫТАНИЯ ИЗ
СПЕЦИФИКАЦИИ
Если известно или предполагается, что процесс
синтеза фармацевтических субстанций приводит к по¬
явлению остаточных металлов вследствие применения
специфического металлического катализатора или ме¬
таллсодержащего реактива, то должно быть проведено
элемент-специфичное количественное определение
для измерения фактического количества этих остаточ¬
ных металлов, в частности в течение разработки процес¬
са синтеза.
Если показано, что в результате процессов синтеза
или производства происходит удаление потенциального
остаточного металла, рутинное испытание на этот оста¬
точный металл можно заменить на нерутинное (возмож¬
ность пропускать данное испытание). Остаточный ме¬
талл может считаться в достаточной мере удаленным,
если в 6 последовательных опытно-промышленных (пи¬
лотных) сериях и 3 последовательных промышленных
сериях обнаружено менее 30% от соответствующего
концентрационного предела. Замена рутинного испыта¬
ния на нерутинное не означает, что оно может быть уда¬
лено из спецификации.
Только для металлов класса 3 испытание может
быть удалено из соответствующей спецификации, если
производитель лекарственного средства в достаточной
степени продемонстрировал, что остаточный металл га¬
рантированно удален из фармацевтической субстанции
или лекарственного средства.
4.6. УЧИТЫВАЕМЫЙ ПРЕДЕЛ ОСТАТОЧНЫХ
МЕТАЛЛОВ
Чтобы удовлетворять критериям данного руковод¬
ства, производителям лекарственных средств необхо¬
дима точная информация о содержании остаточных
металлов в фармацевтических субстанциях. Поэтому
необходимо, чтобы производители фармацевтических
субстанций предоставляли отчет производителям ле¬
карственных средств об идентификации и количествен¬
ном содержании всех остаточных металлов, входящих в
состав фармацевтических субстанций. Ниже приведены
примеры подобных отчетов.
-Вероятно присутствуют только металлы класса
3. Содержание каждого из них ниже пределов в соот¬
ветствии с Подходом 1 для <перорального> и <паренте-
рального> способов применения лекарственного сред¬
ства (поставщик приводит способ применения лекар¬
ственного средства: пероральный или парентеральный).
- Вероятно присутствуют только металлы X, У,
класса 2. Содержание каждого из них ниже пределов в
соответствии с Подходом 1 для <перорального> и <па-
рентерального> способов применения лекарственного
средства (поставщик приводит названия металлов клас¬
са 2, представленных как X, У, , и способ примене¬
ния лекарственного средства: пероральный или парен¬
теральный).
- Присутствует металл 2 класса 1. Металл присут¬
ствует в концентрации ш. ррт, которая не превышает
<критерий приемлемости (поставщик идентифицирует
металл, приводит определенную фактическую концен¬
трацию и применяемый критерий приемлемости. Если
установлено, что содержание металла ниже /_00 или
ШО применяемой аналитической методики, то приво¬
дятся /.00 или ШО для данной методики).
Фраза «Вероятно присутствует» относится к метал¬
лам, применяемым на конечной стадии производства, и
к металлам, которые применялись на более ранних ста¬
диях и не удаляются в соответствии с процессом произ¬
водства.
5. ГЛОССАРИЙ
ВОЗ: Всемирная организация здравоохранения
()Л/ог1д НеаНЬ Огдап'иаНоп, И/НО).
ДЕП: максимально допустимое ежедневное по¬
требление (То1егаЫе даНу Маке, Тй1).
ДСД: допустимая (разрешенная) суточная доза
(РегтШед даНу ехрозиге, РОЕ).
Масса тела взрослого человека: масса с по¬
правкой на принятое допущение о том, что масса тела
произвольно выбранного взрослого человека любого
пола составляет 50 кг. Этот относительно низкий вес
предоставляет дополнительный фактор безопасности
к стандартному весу 60 кг или 70 кг, которые часто ши¬
роко используются.
МСД: максимальная суточная доза (Маютит
даНу дозе, МОО).
МУНЭ: минимальный уровень, при котором на¬
блюдается эффект (1-Ошез^оЬзеп/ед еЯес1 /еуе/,
ШЕЕ).
ПЕП: приемлемое ежедневное потребление
(ассер1аЫе даНу Маке, А01).
ПМДСД: предварительная максимально допусти¬
мая суточная доза (Ргомзюпа! тах\тит 1о1егаЫе даНу
т(аке, РМТ01).
СД: суточная доза (максимальная), относящаяся
к массе фармацевтической субстанции или лекар¬
ственного средства.
Субстанция для фармацевтического использо¬
вания: фармацевтическая субстанция или вспомога¬
тельное вещество.
УННЭ: уровень, при котором не наблюдается эф¬
фект (Ио-оЬзегуед еКес1 /еуе/, Л/ОЕ/_).
АСС1Н: Американская ассоциация органов управ¬
ления гигиены в области промышленности (Атепсап
СопТегепсе о? СоVе^птеп^аI 1пдиз1паI Нуд1еп1з(з).
АТ80Р: Организация по токсическим веществам
и регистрации болезней (Адепсу 1ог Тоюс 8иЬз1апсез
апд 0/$еа$е Ред1з(гу).
Е8А001: определенная безопасность и соответ¬
ствующее ежедневное потребление (езИта1ед за/е
апд асУеода/е даНу т1акё).
Р8А: Организация по пищевым стандартам (Роод
81апдагд Адепсу).
СйР: Надлежащая практика оптовой реализации
(для лекарственных средств, применяемых челове¬
ком) (Соод 01з1пЬиИоп РгасИсе).
СМР: Надлежащая производственная практика
(Соод Мапи?ас1иппд РгасИсё).
1РС8: Международная программа по химиче¬
ской безопасности (1МегпаИопа1 Ргодгат оп СЬетюа!
8а(е*у).
/.00: предел обнаружения {ЫтП оШе^есИоп).
ШО: предел количественного определения (ЫтИ
о! ОиапШсаИоп).
ррт: частей на миллион (Радз регтННоп).
116
Гэсударственная фармакопея Республики Беларусь
РЮ: эталонная доза (РеГегепсе Бозе).
ТТС: пороговая токсикологической опасности
('ТЬгезЬоШ оНох1со1од1са1 сопсегп).
1!Р\ фактор неопределенности (11псе|1ат!у /ас/ог).
из ЕРА\ Организация защиты окружающей сре¬
ды Соединенных Штатов Америки (НпНеб ЗШез
ЕпмгоптепШ Рго1есИоп Адепсу).
6. ССЫЛКИ (НАУЧНЫЕ И/ИЛИ
ИНФОРМАЦИОННЫЕ)
-О/лэсйге 2001/83/ЕС (О/гес/л/е 2001/83/ЕС с
внесенными поправками);
- Ои1беИпе оп Изе ИтНз о! <Зепо1ох'ю Iтрип'Иез
(СРМР/1СН/2.737/99, ЮН03А)]
- МаШепапсе Л/о/е Тог Сшбапсе оп 1тригШез:
РезМиа! ЗокегИз (СРМР/1СНД 507/02, ЮНОЗА(М))]
- Ма\о, б-Е ОссираНопа! гЫпШз апс! азМта бие (о
те(а1 заНз. АНегду 60: 138—139, 2005;
-Nо^е Тог Синапсе оп IтригШез т №\н Огид
Ргобис1з (СРМР/1СН/2738/99, 1СНОЗВ (Р));
- Л/о/е /ог <3шнапсе оп /тригШез: РезШиа! Зо1уеп1з
(СРМР/1СН/283/95, /СНОЗС);
- Л/о/е 1ог бшбапсе оп \/аНс1аНоп о? Апа1уИса1
Ме/Мобз: БеЛпНюп апб Тегтто1оду (СРМР/1СНА381/95,
1СН02А)]
- Л/о/е /ог Ошбапсе оп УаИбаНоп о( Апа1уИса1
Ргосебигез: МеИюбо1оду (СРМР//С/-//281/95,1СН02В);
- 1//еге/а/., Соп/ас/ Бегта/Шз, 1995, 32,135—142.
- УаПбаИоп о/ Апа1уИса1 Ргосебигез: 7ех/ апб
Ме0юбо1оду (СНМР/1СН/381/95, /СН02(Я1)).
07/2016:РБ55000
#5.50. КРИТЕРИИ ЧИСТОТЫ ПИЩЕВЫХ КРАСИТЕЛЕЙ
Данная статья публикуется для информации
Целью данной статьи является указание приемлемых критериев чистоты пищевых красителей, используемых
в качестве вспомогательных веществ при производстве лекарственных средств.
Данная статья основана на критериях чистоты пищевых красителей, указанных в Приложении к Директиве Ко¬
митета 95/45/ЕС от 26.07.1995 (с изменениями и дополнениями М1, М2, М3, М4).
А. ОБЩИЕ СПЕЦИФИКАЦИИ ДЛЯ КРАСИТЕЛЕЙ НА ОСНОВЕ АЛЮМИНИЕВЫХ ЛАКОВ
Определение
Получают взаимодействием красителей, соответствующих критериям чисто¬
ты, указанных в данной статье, с алюминия оксидом в водной среде. Алюминия
оксид обычно представляет собой свежеприготовленное невысушенное веще¬
ство, полученное взаимодействием алюминия сульфата или хлорида с карбо¬
натом натрия или кальция или гидрокарбонатом аммония. Образовавшийся лак
фильтруют, промывают водой и сушат. Непрореагировавший алюминия оксид
также может присутствовать в конечном продукте
Вещества, нерастворимые в хлористоводо¬
родной кислоте
Не более 0,5 %
Вещества, экстрагируемые эфиром
Не более 0,2 % (в условиях нейтральной среды)
Применимы специфические критерии чистоты для соответствующих красителей
В. СПЕЦИФИЧЕСКИЕ КРИТЕРИИ ЧИСТОТЫ
Е100
КУРКУМИН (С(У/?С(УМ/АО
Синонимы
С1 Натуральный желтый 3, куркума желтая, дифероилметан
С/ Л/а/ига/ Уе!1о\л/ 3, Тигтепс УеПощ йКегоу! МеХЬапе
Определение
Получают экстракцией растворителями из куркумы, то есть измельченных корне¬
вищ природных видов Сигсита 1опда 1_. Для получения концентрированного по¬
рошка куркумина экстракт очищают кристаллизацией. Состоит преимущественно
из куркуминов, т.е. основного окрашенного компонента (1,7-бис(4-гидрокси-3-
метоксифенил)гепта-1,6-диен-3,5-диона) и его 2 деметокси производных в раз¬
личных соотношениях. Могут присутствовать незначительные количества масел
и смол, встречающихся в природной куркуме.
Для экстракции могут быть использованы только следующие растворители: эти-
лацетат, ацетон, углерода диоксид, метиленхлорид, н-бутанол, метанол, этанол,
гексан
Класс
Дициннамоилметан
Номер индекса цвета
75300
Е/Л/ЕС5<1>
207-280-5
Химическое название
I 1,7-Бис(4-гидрокси-3-метоксифенил)гепта-1,6-диен-3,5-дион;
II 1-(4-гидроксифенил)-7-(4-гидрокси-3-метоксифенил)гепта-1,6-диен-3,5-дион;
III 1,7-бис(4-гидроксифенил)гепта-1,6-диен-3,5-дион
Химическая формула
' С21НюОе;
||с20н18о6;
|"с19н16о4
#5.50. Критерии чистоты пищевых красителей
117
Молекулярная масса
I 368,39;
II 338,39;
III 308,39
Количественное определение
Не менее 90 % от всех окрашенных веществ.
Д1* — ^607 при длине волны около 426 нм в этаноле
Описание
Оранжево-желтый кристаллический порошок
Идентификация
A. Спектрометрия
B. Температура плавления
Максимум при длине волны около 426 нм в этаноле.
От 179 °С до 182 °С
Чистота
Остаточные количества органических раст¬
ворителей
•ч
Этилацетат
Ацетон
н-Бутанол >. не более 50 мг/кг,
Метанол по отдельности или суммарно.
Этанол
Гексан ^
Метиленхлорид: не более 10 мг/кг.
Мышьяк
Не более 3 мг/кг.
Свинец
Не более 10 мг/кг.
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
(1> Регистрационный номер Европейского реестра выпускаемых химических веществ (Еигореап 1теп1огу о{Ех/зИпд СоттепяаI СЛетка!
8иЬз{апсез—Е/Л/ЕС5), который приводится на сайте Европейского бюро по химическим веществам {Еигореап СЬет'юа1з Вигеаи—ЕСВ).
Е 101 (I)
РИБОФЛАВИН* (К/ВОЕМУ/Л/)
Синонимы
Лактофлавин.
1-ас1оЛамп
Класс
Изоаллоксазин
Е/МЕС5
201-507-1
Химическое название
7.8- Диметил-Ю-(0-рибо-2,3,4,5-тетрагидроксипентил)бензо(д)птеридин-
2,4(ЗН,10Н)-дион.
7.8- Диметил-10-(1 '-0-рибитил)изоаллоксазин
Химическая формула
с1Тн„м4о.
Молекулярная масса
376,37
Количественное определение
Не менее 98 % в пересчете на безводное вещество.
— 323 ПРИ Д™46 волны около 444 нм в воде
Описание
Кристаллический порошок от желтого до оранжево-желтого цвета со слабым за¬
пахом
Идентификация
А. Спектрометрия
Отношение оптических плотностей:
А375/А067 — от 0.31 до 0,33
> в воде.
Отношение оптических плотностей:
А444/А267 — от 0,36 до 0,39
Максимум при длине волны около 375 нм в воде
В. Удельное оптическое вращение
Мо°: от -115 до -140 в 0,05 М растворе натрия гидроксида
Чистота
Потеря в массе при высушивании
Сульфатная зола
Первичные ароматические амины
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 1,5 % (105 °С, 4 ч).
Не более 0,1 %.
Не более 100 мг/кг (в пересчете на анилин).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
* Или см. требования статьи Рибофлавин (0292).
118
Гэсударственная фармакопея Республики Беларусь
Е 101 (II)
РИБ ОФЛА ВИН-5 '-ФОСФАТ* (В1В 0Р1-А VIN-5 '-РН08РНА ТЕ)
Синонимы
Рибофлавин-5-фосфат натрия.
МЬоЛамп-5'-р1ю$рШе восИит
Определение
Требования данной статьи применяются к рибофлавин-5'-фосфату, содержаще¬
му незначительное количество рибофлавина в свободном состоянии и рибофла¬
вина дифосфата
Класс
Изоаллоксазин
Е1МЕС8
204-988-6
Химическое название
Мононатрия (2Я,ЗЯ,45)-5-(3')10-дигидро-7',8-диметил-2',4-диоксо-10-бензо[у]-
птеридинил)-2,3,4-тригидроксипентил фосфат.
Мононатриевая соль 5-монофосфорного эфира рибофлавина
Химическая формула
Для дигидратной формы: С^Н^МаОдР-гНзО.
Для безводной формы: С17Н,пЫ4№ОчР
Молекулярная масса
541,36
Количественное определение
Не менее 95 % от всех окрашенных веществ в пересчете на С17Н20Ы4МаО9Р*2Н2О.
А,1* — 250 при длине волны около 375 нм в воде
Описание
Кристаллический порошок от желтого до оранжевого цвета, гигроскопичен, со
слабым запахом и горьким вкусом
Идентификация
А. Спектрометрия
Отношение оптических плотностей:
А375/А067 — от 0,30 до 0,34
гх>/ V в воде.
Отношение оптических плотностей:
А444/А267 — от 0,35 до 0,40
Максимум при длине волны около 375 нм в воде.
В. Удельное оптическое вращение
[а]^° в диапазоне от +38 до +42 в 5 М растворе кислоты хлористоводородной
Чистота
Потеря в массе при высушивании
Сульфатная зола
Неорганические фосфаты
Второстепенные окрашенные вещества
Не более 8 % (100 °С, 5 ч в вакууме над Р2Об) для дигидратной формы.
Не более 25 %.
Не более 1,0 % (в пересчете на Р04 безводного вещества).
Рибофлавин (в свободном состоянии): не более 6 %.
Первичные ароматические амины
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Рибофлавина дифосфат: не более 6 %.
Не более 70 мг/кг (в пересчете на анилин).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
* Или см. требования статьи Рибофлавина натрия фосфат (0786).
Е102
ТАРТРАЗИН (ТАВТРАШЕ)
Синонимы
С! Пищевой желтый 4.
С/ Роод УеИом 4
Определение
Состоит преимущественно из тринатрия 5-гидрокси-1-(4-сульфонатофенил)-4-(4-
сульфонатофенилазо)-Н-пиразол-3-карбоксилата и второстепенных окрашенных
веществ наряду с натрия хлоридом и/или сульфатом в качестве основных не¬
окрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Моноазо
Номер индекса цвета
19140
Е/ЫЕС8
217-699-5
Химическое название
Тринатрия 5-гидрокси-1-(4-сульфонатофенил)-4-(4-сульфонатофенилазо)-Н-
пиразол-3-карбоксилат
Химическая формула
С16Н9^№30952
Молекулярная масса
534,37
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
А,1 * — 530 при длине волны около 426 нм в воде
Описание
Порошок или гранулы светло-оранжевого цвета
Идентификация
A. Спектрометрия
B. Водный раствор желтый
Максимум при длине волны около 426 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
Не более 0,2 %.
Не более 1,0 %.
#5.50. Критерии чистоты пищевых красителей
119
4-гидразинобензолсульфоновая кислота
4- аминобензолсульфоновая кислота
5- оксо-1-(4-сульфофенил)-2-пиразолин-3-
карбоновая кислота
4,4'-диазоаминоди(бензол сульфоновая
кислота)
тетрагидроксиянтарная кислота
Несульфированные первичные
ароматические амины
У- Общее содержание не более 0,5 %.
У
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е104
ХИНОЛИНОВЫЙ ЖЕЛТЫЙ (ОШЫОШЕ УЕ/./.ОИ0
Синонимы
С1 Пищевой желтый 13.
С/ 1=оо(1 Уе//оиг 13
Определение
Получают сульфонированием 2-(2-хинолил)индан-1,3-диона. Состоит преимуще¬
ственно из натриевых солей смеси дисульфонатов (в основном), моносульфона¬
тов и трисульфонатов вышеуказанного вещества и второстепенных окрашенных
веществ наряду с натрия хлоридом и/или сульфатом в качестве основных не¬
окрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Хинофталон
Номер индекса цвета
47005
Е1МЕС5
305-897-5
Химическое название
Динатриевые соли дисульфонатов 2-(2-хинолил)индан-1,3-диона (основной ком¬
понент)
Химическая формула
С18Н9ММа20832 (основной компонент)
Молекулярная масса
477,38 (основной компонент)
Количественное определение
Не менее 70 % от всех окрашенных веществ в пересчете на натриевую соль.
Хинолиновый желтый должен иметь следующий состав окрашенных веществ:
- не менее 80 % динатрия 2-(2-хинолил)индан-1,3-дион-дисульфоната от всех
присутствующих окрашенных веществ;
- не более 15 % натрия 2-(2-хинолил)индан-1,3-дион-моносульфоната от всех
присутствующих окрашенных веществ;
- не более 7,0 % тринатрия 2-(2-хинолил)индан-1,3-дион-трисульфоната от всех
присутствующих окрашенных веществ.
А}* — 865 (основной компонент) при длине волны около 411 нм в растворе кис¬
лоты уксусной
Описание
Порошок или гранулы желтого цвета
Идентификация
A. Спектрометрия
B. Водный раствор желтый
Максимум при длине волны около 411 нм в растворе кислоты уксусной (рН 5)
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашен¬
ных:
Не более 0,2 %.
Не более 4,0 %.
2-метилхинолин
2-метилхинопинсульфоновая кислота
фталевая кислота
2.6- диметилхинолин
2.6- диметилхинолинсульфоновая кислота
У Общее содержание не более 0,5 %.
У
2-(2-хинолил)индан-1,3-дион
Несульфированные первичные ароматиче¬
ские амины
Не более 4 мг/кг.
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
120
Государственная фармакопея Республики Беларусь
Е110
АПЕЛЬСИНОВЫЙ ЖЕЛТЫЙ (ЗиЫЗЕТ УЕЬЬОШ РСР)
Синонимы
С1 Пищевой желтый 3, оранжево-желтый 3, солнечный закат желтый, апельсино¬
вый желтый РСР.
С/ Роод УеИом 3, Огапде УеНом 8
Определение
Состоит преимущественно из динатрия 2-гидрокси-1-(4-сульфонатофенилазо)
нафталин-6-сульфоната и второстепенных окрашенных веществ наряду с натрия
хлоридом и/или сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Моноазо
Номер индекса цвета
15985
Е1МЕС5
220-491-7
Химическое название
Динатрия 2-гидрокси-1-(4-сульфонатофенилазо)нафталин-6-сульфонат
Химическая формула
С,6Н10М2Ма2О732
Молекулярная масса
452,37
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
А« — 555 при длине волны около 485 нм в воде (рН 7)
Описание
Оранжево-красный порошок или гранулы
Идентификация
A. Спектрометрия
B. Водный раствор оранжевый
Максимум при длине волны около 485 нм в воде (рН 7)
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашен¬
ных:
4-аминобензол-1 -сульфоновая кислота
3-гидроксинафталин-2,7-дисульфоновая
кислота
6- гидроксинафталин-2-сульфоновая кис¬
лота
7- гидроксинафталин-1,3-дисульфоновая
кислота
4,4'-диазоаминоди(бензолсульфоновая кис¬
лота)
6,6'-оксиди(нафталин-2-сульфоновая кис¬
лота)
Несульфированные первичные
ароматические амины
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 %.
Не более 5,0 %.
"Л
V- Общее содержание не более 0,5 %.
Не более 0,01 % (в пересчете на анилин).
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е120
КОШЕНИЛЬ, КАРМИНОВАЯ КИСЛОТА, КАРМИНЫ (СОСН1ЫЕАЦ САРМ1Ы1С АСЮ, САРМШЕЗ)
Определение
Кармины и карминовую кислоту получают из водных, водно-спиртовых или спир¬
товых экстрактов кошенили, состоящей из высушенных телец женских особей
насекомого Оас1у1орш$ соссиз Соз1а.
Основной окрашенной составляющей является карминовая кислота.
Могут образовываться алюминиевые лаки карминовой кислоты (кармины), в ко¬
торых алюминий и карминовая кислота присутствуют предположительно в мо¬
лярном соотношении 1:2.
В готовом к использованию красителе основная окрашенная составляющая
представлена в соединении с катионами аммония, кальция, калия или натрия по
отдельности или совместно; кроме того, данные катионы могут присутствовать в
избытке.
Готовые к использованию красители могут также содержать вещества белково¬
го происхождения, извлеченные из источника получения карминов и карминовой
кислоты — насекомых, также может содержать свободный карминат или неболь¬
шие остатки несвязанных катионов алюминия
Класс
Антрахинон
Номер индекса цвета
75470
Е/Л/ЕС5
Кошениль: 215-680-6;
карминовая кислота: 215-023-3;
кармины: 215-724-4
#5.50. Критерии чистоты пищевых красителей
121
Химическое название
7-р -О-глюкопиранозил-3,5, 6,8-тетрагидрокси-1 -метил-9,10-диоксоантра-цин-2-
карбоновая кислота (карминовая кислота); кармин является гидратированным хе¬
латным соединением алюминия с указанной кислотой
Химическая формула
С22Н2о°1з (карминовая кислота)
Молекулярная масса
492,39 (карминовая кислота)
Количественное определение
Не менее 2,0 % карминовой кислоты в содержащих ее экстрактах; не менее 50 %
карминовой кислоты в хелатах
Описание
Хрупкое твердое вещество или порошок от красного до темно-красного цвета.
Экстракт кошенили обычно представляет собой темно-красную жидкость, но
может быть также высушен до состояния порошка
Идентификация
Спектрометрия
Максимум при длине волны около 518 нм в растворе аммиака.
Максимум при длине волны около 494 нм для карминовой кислоты в кислоте
хлористоводородной разведенной
Чистота
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е122
АЗОРУБИН, КАРМУАЗИН (А20РЧВШЕ, САРМ013ШЕ)
Синонимы
С1 Пищевой красный 3.
С/ Роод Кед 3
Определение
Состоит преимущественно из динатрия 4-гидрокси-3-(4-сульфонато-1 -нафтилазо)-
нафталин-1-сульфоната и второстепенных окрашенных веществ наряду с натрия
хлоридом и/или сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Моноазо
Номер индекса цвета
14720
Е1ЫЕС8
222-657-4
Химическое название
Динатрия 4-гидрокси-3-(4-сульфонато-1 -нафтилазо)нафталин-1 -сульфонат
Химическая формула
С,„Н„М,Ма,073,
Молекулярная масса
502,44
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
— 510 при длине волны около 516 нм в воде
Описание
Порошок или гранулы от красного до насыщенного красно-коричневого цвета
Идентификация
A. Спектрометрия
B. Водный раствор красный
Максимум при длине волны около 516 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
Не более 0,2 %.
Не более 2,0 %.
4-аминонафталин-1 -сульфоновая кислота
4-гидроксинафталин-1 -сульфоновая кис¬
лота
Общее содержание не более 0,5 %.
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Е123
АМАРАНТ (АМАРАЫТН)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Синонимы
С1 Пищевой красный 9.
С/ Роод Кед 9
Определение
Состоит преимущественно из тринатрия 2-гидрокси-1-(4-сульфонато-1-нафтила-
зо)нафталин-3,6-дисульфоната и второстепенных окрашенных веществ наряду с
натрия хлоридом и/или сульфатом в качестве основных неокрашенных компонен¬
тов.
Кальциевая и калиевая соли также допускаются
Зак. 1060.
122
Гэсударственная фармакопея Республики Беларусь
Класс
Моноазо
Номер индекса цвета
16185
Е/Л/ЕС5
213-022-2
Химическое название
Тринатрия 2-гидрокси-1 -(4-сульфонато-1 -нафтилазо)нафталин-3,6-дисульфонат
Химическая формула
СаД.и/ицо.д
Молекулярная масса
604,48
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
Кт — 440 при длине волны около 520 нм в воде
Описание
Порошок или гранулы красновато-коричневого цвета
Идентификация
A. Спектрометрия
B. Водный раствор красный
Максимум при длине волны около 520 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашенных:
Не более 0,2 %.
Не более 3,0 %.
4-аминонафталин-1 -сульфоновая кислота
3-гидроксинафталин-2,7-дисульфоновая
кислота
6- гидроксинафталин-2-сульфоновая кис¬
лота
7- гидроксинафталин-1,3-дисульфоновая
кислота
7-гидроксинафталин-1,3,6-трисульфоновая
кислота
V Общее содержание не более 0,5 %.
У
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е124
ПОНС0 4К, КОШЕНИЛОВЫЙ КРАСНЫЙ А (РОИСЕАЧ 4Р, СОСНШЕАЬ ЛЕРА)
Синонимы
С1 Пищевой красный 7, новый кокцин, пунцовый.
С/ Роод Яес1 7, Л/еуу Сосс'те
Определение
Состоит преимущественно из тринатрия 2-гидрокси-1-(4-сульфонато-1-нафтилазо)-
нафталин-6,8-дисульфоната и второстепенных окрашенных веществ наряду с натрия
хлоридом и/или сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Моноазо
Номер индекса цвета
16255
Е/Л/ЕС5
220-036-2
Химическое название
Тринатрия 2-гидрокси-1 -(4-сульфонато-1 -нафтилазо)нафталин-6,8-дисульфонат
Химическая формула
Молекулярная масса
604,48
Количественное определение
Не менее 80 % от всех окрашенных веществ в пересчете на натриевую соль.
Аш — 430 при длине волны около 505 нм в воде
Описание
Порошок или гранулы красноватого цвета
Идентификация
A. Спектрометрия
B. Водный раствор красный
Максимум при длине волны около 505 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашенных:
Не более 0,2 %.
Не более 1,0 %.
4-аминонафтапин-1 -сульфоновая кислота
7-гидроксинафталин-1,3-дисульфоновая
кислота
3-гидроксинафталин-2,7-дисульфоновая
кислота
6- гидроксинафталин-2-сульфоновая кис¬
лота
7- гидроксинафталин-1,3,6-трисульфоновая
кислота
У Общее содержание не более 0,5 %.
У
#5.50. Критерии чистоты пищевых красителей
123
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Е128
КРАСНЫЙ 26 (КЕО 26)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Синонимы
С1 Пищевой красный 10, азогеранин.
С/ Роод Ней 10, Агодегапте
Определение
Состоит преимущественно из динатрия 8-ацетамидо-1-гидрокси-2-фенилазо-
нафталин-3,6-дисульфоната и второстепенных окрашенных веществ наряду с натрия
хлоридом и/или сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Моноазо
Номер индекса цвета
18050
Е/Л/ЕС5
223-098-9
Химическое название
Динатрия 8-ацетамидо-1-гидрокси-2-фенилазонафталин-3,6-дисульфонат
Химическая формула
С18Н13М3Ма20832
Молекулярная масса
509,43
Количественное определение
Не менее 80 % от всех окрашенных веществ в пересчете на натриевую соль.
А* — 620 при длине волны около 532 нм в воде
Описание
Порошок или гранулы красного цвета
Идентификация
A. Спектрометрия
B. Водный раствор красный
Максимум при длине волны около 532 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
Не более 0,2 %.
Не более 2,0 %.
5-ацетамидо-4-гидроксинафталин-2,7-
дисульфоновая кислота
5-амино-4-гидроксинафтапин-2,7-
дисульфоновая кислота
^ Общее содержание не более 0,5 %.
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е129
КРАСНЫЙ ОЧАРОВАТЕЛЬНЫЙ (АЬШЙА ЯЕО АС)
Синонимы
Определение
Класс
Номер индекса цвета
Е/Л/ЕС5
Химическое название
Химическая формула
Молекулярная масса
Количественное определение
С1 Пищевой красный 17, красный очаровательный АС.
С/ Роод Неб 17
Состоит преимущественно из динатрия 2-гидрокси-1-(2-метокси-5-метил-4-суль-
фонатофенилазо)нафталин-6-сульфоната и второстепенных окрашенных ве¬
ществ наряду с натрия хлоридом и/или сульфатом в качестве основных неокра¬
шенных компонентов.
Кальциевая и калиевая соли также допускаются
Моноазо
16035
247-368-0
Динатрия 2-гидрокси-1-(2-метокси-5-метил-4-сульфонатофенилазо)нафталин-6-
сульфонат
С,8Н,ЛМа20832
496,42
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
Аш — 540 при длине волны около 504 нм в водном растворе (рН 7)
16*. Зак. 1060.
124
Гэсударственная фармакопея Республики Беларусь
Описание
Идентификация
Темно-красный порошок или гранулы
A. Спектрометрия
B. Водный раствор красный
Максимум при длине волны около 504 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашенных:
Не более 0,2 %.
Не более 3,0 %.
6-гидрокси-2-нафталинсульфоновой кисло¬
ты натриевая соль
Не более 0,3 %.
4-амино-5-метокси-2-
метилбензолсульфоновая кислота
Не более 0,2 %.
6,6-оксибис (2-нафталинсульфоновой кис¬
лоты) динатриевая соль
Не более 1,0 %.
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (из раствора с рН 7).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е131
ПАТЕНТОВАННЫЙ ГОЛУБОЙ (РАТЕЫТ ВШЕ V)
Синонимы
С! Пищевой синий 5, сульфановый синий, патентованный голубой V.
С/ Еоос/ В1ие 5
Определение
Состоит преимущественно из соединения кальция или натрия с внутренней
солью [4-(а-(4-диэтиламинофенил)-5-гидрокси-2,4-дисульфофенилметилиден)-
2,5-циклогексадиен-1-илиден]диэтиламмония гидроксида и второстепенных окра¬
шенных веществ наряду с натрия хлоридом и/или натрия сульфатом и/или каль¬
ция сульфатом в качестве основных неокрашенных компонентов.
Калиевая соль также допускается
Класс
Триарилметан
Номер индекса цвета
42051
Е/Л/ЕС5
222-573-8
Химическое название
Соединение кальция или натрия с внутренней солью 4-(а-(4-диэтиламинофенил)-
5-гидрокси-2,4-дисульфофенилметилиден)2,5-циклогексадиен-1-илиден]диэти-
ламмония гидроксида
Химическая формула
Соединение кальция: С27Н31М20782Са1/2.
Соединение натрия: С27Н31М20752Ыа
Молекулярная масса
Соединение кальция: 579,72.
Соединение натрия: 582,67
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
К™ — 2000 при длине волны около 638 нм в водном растворе (рН 5)
Описание
Порошок или гранулы темно-синего цвета
Идентификация
A. Спектрометрия
B. Раствор в воде синий
Максимум при длине волны около 638 нм в воде (рН 5)
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашенных:
3-гидроксибензальдегид
Не более 0,2 %.
Не более 2,0 %.
"I
3-гидроксибензойная кислота
З-гидрокси-4-сульфобензойная кислота
А/,Л/-диэтиламинобензолсульфоновая
кислота
V Общее содержание не более 0,5 %.
Бесцветное производное
Несульфированные первичные
ароматические амины
Не более 4,0 %.
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (из раствора, имеющего значение рН 5).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
#5.50. Критерии чистоты пищевых красителей
125
Е132
ИНДИГОКАРМИН, ИНДИГОТИН (ШОЮОТШЕ, Шй/вО САЯМ1ЫЕ)
Синонимы
С1 Пищевой синий 1.
С/ ЕоосУ В1ие 1
Определение
Состоит преимущественно из смеси динатрия 3,3'-диоксо-2,2-бисиндолилиден-
5,5'-д и сульфоната и динатрия 3,3'-диоксо-2,2'-бисиндолилиден-5,7'-дисульфоната
и второстепенных окрашенных веществ наряду с натрия хлоридом и/или сульфа¬
том в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Индигоид
Номер индекса цвета
73015
Е/Л/ЕС5
212-728-8
Химическое название
Динатрия 3,3-диоксо-2,2-бисиндолилиден-5,5-дисульфонат
Химическая формула
С16Н,М2Ма2Ов32
Молекулярная масса
466,36
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
Не более 18 % динатрия 3,3'-диоксо-2,2'-бисиндолилиден-5,7-дисульфоната.
А1ш — 480 при длине волны около 610 нм в воде
Описание
Порошок или гранулы темно-синего цвета
Идентификация
A. Спектрометрия
B. Водный раствор синий
Максимум при длине волны около 610 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме окрашенных:
Не более 0,2 %.
Не более 1,0 %, за исключением динатрия 3,3'-диоксо-2,2-бисиндолилиден-5,7-
дисульфоната.
изатин-5-сульфоновая кислота
5-сульфоантраниловая кислота
антраниловая кислота
Общее содержание не более 0,5 %.
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е133
БРИЛЛИАНТОВЫЙ ГОЛУБОЙ (ВМШАЫТ ВШЕ РСР)
Синонимы
С1 Пищевой синий 2, бриллиантовый голубой РСР.
С/ Роос1 В1ие 2
Определение
Состоит преимущественно из динатрия а-(4-(А/-этил-3-сульфонатобензил-амино)-
фенил)-а-(4-/У-этил-3-сульфонатобензиламино)циклогекса-2,5-диенилиден)-
толуол-2-сульфоната, его изомеров и второстепенных окрашенных веществ
наряду с натрия хлоридом и/или сульфатом в качестве основных неокрашенных
компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Триарил метан
Номер индекса цвета
42090
Е/Л/ЕС5
223-339-8
Химическое название
Динатрия а-(4-(Л/-этил-3-сульфонатобензиламино)фенил)-а-(4-Л/-этил-3-сульфо-
натобензиламино)циклогекса-2,5-диенилиден)толуол-2-сульфонат
Химическая формула
С37НиМ2Ма20933
Молекулярная масса
792,84
Количественное определение
Не менее 85 % от всех окрашенных веществ в пересчете на натриевую соль.
Не более 18 % динатрия 3,3'-диоксо-2,2'-бииндолилиден-5,7-дисульфоната.
— 1630 при длине волны около 630 нм в воде
Описание
Идентификация
Порошок или гранулы красновато-синего цвета
A. Спектрометрия
B. Водный раствор синий
Максимум при длине волны около 630 нм в воде
Чистота
Вещества, нерастворимые в воде
Не более 0,2 %.
17. Зак. 1060.
126
Гэсударственная фармакопея Республики Беларусь
Второстепенные окрашенные вещества
Органические вещества, кроме окрашенных:
сумма 2-, 3- и
4-формилбензолсульфоновых кислот
3-((этил)(4-сульфофенил)амино)метилбен-
золсульфоновая кислота
Бесцветное производное
Несульфированные первичные
ароматические амины
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 6,0 %.
Не более 1,5 %.
Не более 0,3 %.
Не более 5,0 %.
Не более 0,01 % (в пересчете на анилин).
Не более 0,2 % (при рН 7).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е140 (!)
хлорофиллы (Сш.окорнги.з)
Синонимы
С1 Натуральный зеленый 3, магния хлорофилл, магния фаеофитин.
С/ А1а1ига1 бгееп 3, Мадпезшт СЫогорНуН, Мадпез'шт РЬаеорЬуИп
Определение
Получают экстракцией растворителем пригодного в пищу растительного сырья,
природных видов травы, люцерны и крапивы. В течение последующего удале¬
ния растворителя присутствующий в растительном сырье комплексообразова-
тель магний может быть полностью или частично удален из хлорофиллов, давая
соответствующие феофитины. Основными окрашенными веществами являются
феофитины и магния хлорофиллы. Экстракт, из которого удален растворитель,
содержит другие пигменты, такие как каротиноиды, извлеченные из исходного
сырья наряду с маслами, жирами и парафинами. Для экстракции могут быть ис¬
пользованы только следующие растворители: ацетон, метил эти л кетон, метилен-
хлорид, углерода диоксид, метанол, этанол, пропан-2-ол и гексан
Класс
Порфирин
Номер индекса цвета
75810
Е/А/ЕС5
Хлорофиллы: 215-800-7, хлорофилл а: 207-536-6, хлорофилл Ь: 208-272-4
Химическое название
Наиболее важные окрашенные компоненты:
фитил(132Е, 175,185)-3-(8-этил-132-метоксикарбонил-2,7,12,18-тетраметил-13-
оксо-З-винил-131-132-17,18-тетрагидроциклопента[а1]-порфирин-17-ил)пропионат
(феофитин а) или в виде комплексного соединения с магнием (хлорофилл а);
фитил(132/?, 175,185)-3-(8-этил-7-формил-132-метоксикарбонил-2,12,18-
триметил-13'-оксо-3-винил-131-132-17,18-тетрагидроциклопента[а*]-порфирин-
17-ил)пропионат (феофитин Ь) или в виде комплексного соединения с магнием
(хлорофилл Ь)
Химическая формула
Хлорофилл а (комплексное соединение с магнием): 055Н72Мд1Ч4О5.
Хлорофилл а: С55Н74М405.
Хлорофилл Ь (комплексное соединение с магнием): С55Н70МдЫ4О6.
Хлорофилл Ь: С55Н72Ы406
Молекулярная масса
Хлорофилл а (комплексное соединение с магнием): 893,51.
Хлорофилл а: 871,22.
Хлорофилл Ь (комплексное соединение с магнием): 907,49.
Хлорофилл Ь: 885,20
Количественное определение
Общее количество хлорофиллов и их комплексов с магнием — не менее 10 %.
Кш — 700 при длине волны около 409 нм в хлороформе
Описание
Воскообразное твердое вещество от оливково-зеленого до темно-зеленого цвета
в зависимости от содержания комплексов магния
Идентификация
А. Спектрометрия
Максимум при длине волны около 409 нм в хлороформе
Чистота
Остаточные количества органических
растворителей
Ацетон
Метилэтилкетон
Метанол I - __ .
Этанол Г не б°лее 50 мг^кг»
Пропан-2-ол по отдельности или суммарно.
Г ексан
Метиленхлорид: не более 10 мг/кг.
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
#5.50. Критерии чистоты пищевых красителей
127
Е 140 (И)
ХЛОРОФИЛЛИНЫ (СМ.<ЖОРНУШЫВ)
Синонимы
С1 Натуральный зеленый 5, натрия хлорофиллин, калия хлорофиллин.
С/ Д/а/ига/ Огееп 5, ЗосИит СЫогорЬуШп, Ро1аззшт СЫогорЬуШп
Определение
Соли щелочных металлов с хлорофиллинами, полученные омылением экстрак¬
та из пригодного в пищу растительного сырья, природных видов травы, люцер¬
ны и крапивы. При омылении удаляются метиловая и фитоловая группы эфира и
может частично размыкаться кольцо циклопентенила. Кислотные группы нейтра¬
лизуются с образованием солей калия и/или натрия.
Для экстракции могут быть использованы только следующие растворители:
ацетон, метилэтилкетон, метиленхлорид, углерода диоксид, метанол, этанол,
пропан-2-ол и гексан
Класс
Порфирин
Номер индекса цвета
75815
Е/Л/ЕС5
287-483-3
Химическое название
Наиболее важные окрашенные компоненты в кислотной форме:
- 3-(10-карбоксилато-4-этил-1,3,5,8-тетраметил-9-оксо-2-винилфорбин-7-ил)про-
пионат (хлорофиллин а);
- 3-(10-карбоксилато-4-этил-3-формил-1,5,8-триметил-9-оксо-2-винилфорбин-7-
ил)пропионат (хлорофиллин Ь).
В зависимости от степени гидролиза кольцо циклопентенила может разомкнуть¬
ся с образованием третьей карбоксильной группы.
Могут также присутствовать комплексные соединения с магнием
Химическая формула
Хлорофиллин а (кислотная форма): С^Н^^О,..
Хлорофиллин Ь (кислотная форма): Сг4Н„М4Ой
Молекулярная масса
Хлорофиллин а (кислотная форма): 578,68.
Хлорофиллин Ь (кислотная форма): 592,66.
Каждая из них может увеличиваться на 18 дальтон при размыкании кольца ци¬
клопентенила
Количественное определение
Общее содержание хлорофиллинов — не менее 95 % в образце, высушенном
при температуре около 100 °С в течение 1 ч.
А« — 700 при длине волны около 405 нм в водном растворе (рН 9).
Д1 см —140 при длине волны около 653 нм в водном растворе (рН 9)
Описание
Порошок от темно-зеленого до синего/черного цвета
Идентификация
Спектрометрия
Максимумы при длине волны около 405 нм и при длине волны около 653 нм в
водном фосфатном буферном растворе (рН 9)
Чистота
Остаточные количеств^ органических
растворителей
•ч
Ацетон
Метилэтилкетон
Метанол - ,
Этанол Гне б°лее 50 мг/кг,
Пропан-2-ол по отдельности или суммарно.
Гексан
Метиленхлорид: не более 10 мг/кг.
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е 141 (!)
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ МЕДИ С ХЛОРОФИЛЛАМИ (СОРРЕР С0МР1.ЕХЕЗ СЖ.ОКОЯНУ/./.5)
Синонимы
С1 Натуральный зеленый 3, меди хлорофилл, меди фаеофитин.
С/ А1а(ига1 Огееп 3, Соррег СЫогорЬуИ, Соррег РНаеорЬуНп
Определение
Получают добавлением соли меди к веществу, полученному экстракцией раство¬
рителем пригодного в пищу растительного сырья, природных видов травы, лю¬
церны и крапивы. Краситель, из которого удален растворитель, содержит другие
пигменты, такие как каротиноиды, наряду с маслами, жирами и парафинами, из¬
влеченные из исходного сырья. Основными окрашенными компонентами являют¬
ся меди феофитины.
Для экстракции могут быть использованы только следующие растворители:
ацетон, метилэтилкетон, метиленхлорид, углерода диоксид, метанол, этанол,
пропан-2-ол и гексан
Класс
Порфирин
Номер индекса цвета
75815
Е/А/ЕС5
Меди хлорофилл а: 239-830-5.
Меди хлорофилл Ь: 246-020-5
17*. Зак 1060.
128
Гэсударственная фармакопея Республики Беларусь
Химическое название
[Фитил(132Я, 178,185)-3-(8-этил-132-метоксикарбонил-2,7,12,18-тетраметил-13-
оксо-З-винил-134 32-17,18-тетрагидроциклопента[а*]-порфирин-17-ил)пропионат]
меди (II) (меди хлорофилл а).
[Фитил(132Я,175,185)-3-(8-этил-132-метоксикарбонил-2,12,18-триметил-13-оксо-
З-винил-131-132-17,18-тетрагидроциклопента[а1]-порфирин-17-ил)пропионат]
меди (II) (меди хлорофилл Ь)
Химическая формула
Меди хлорофилл а: С55Н72СиМ405.
Меди хлорофилл Ь: С55Н70СиМ4О6
Молекулярная масса
Меди хлорофилл а: 932,75.
Меди хлорофилл Ь: 946,73
Количественное определение
Суммарное содержание меди хлорофиллов — не менее 10 %.
А1^ — 540 при длине волны около 422 нм в хлороформе.
Кш — 300 при длине волны около 652 нм в хлороформе
Описание
Воскообразное твердое вещество от сине-зеленого до темно-зеленого цвета в
зависимости от источника происхождения
Идентификация
Спектрометрия
Максимум при длине волны около 422 нм и при длине волны около 652 нм в хло¬
роформе
Чистота
Остаточные количества органических
растворителей
■ч
Ацетон
Метил эти л кетон
Метанол I - сл ,
Этанол Гне б°лее 50 мг/кг,
Пропан-2-ол по ^Дельности или суммарно.
Г ексан
У
Мышьяк
Свинец
Ртуть
Кадмий
Меди ионы
Общая медь
Метиленхлорид: не более 10 мг/кг.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 200 мг/кг.
Не более 8,0 % от общего содержания меди феофитинов
Е 141 (II)
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ МЕДИ С ХЛОРОФИЛЛИНАМИ (СОРРЕР СОМРЬЕХЕЗ
СН1-01ЮРНУШЫ8\
Синонимы
Натрия меди хлорофиллин, калия меди хлорофиллин, С1 натуральный зеленый 5.
ЗосИит Соррег СЫогорЬуШп, Ро1а88шт Соррег СЫогорЬуШп, С1 Ма(ига1 Сгееп 5
Определение
Соли щелочных металлов меди хлорофиллинов получают прибавлением меди к
веществу, полученному омылением экстракта из пригодного в пищу растительно¬
го сырья, природных видов травы, люцерны и крапивы. При омылении удаляют¬
ся метиловая и фитоловая группы эфира и может частично размыкаться кольцо
циклопентенила. Кислотные группы нейтрализуются с образованием солей
калия и/или натрия.
Для экстракции могут быть использованы только следующие растворители:
ацетон, метилэтилкетон, метиленхлорид, углерода диоксид, метанол, этанол,
пропан-2-ол и гексан
Класс
Порфирин
Номер индекса цвета
75815
Химическое название
Наиболее важные окрашенные компоненты в кислотной форме:
- 3-(10-карбоксилато-4-этил-1,3,5,8-тетраметил-9-оксо-2-винилфорбин-7-ил)про-
пионат, комплексное соединение с медью (меди хлорофиллин а);
- 3-(10-карбоксилато-4-этил-3-формил-1,5,8-триметил-9-оксо-2-винилфорбин-7-
ил)пропионат, комплексное соединение с медью (меди хлорофиллин Ь)
Химическая формула
Меди хлорофиллин а (кислотная форма): С^Н^Си^О^
Меди хлорофиллин Ь (кислотная форма): С^Н^СиМ^
Молекулярная масса
Меди хлорофиллин а: 640,20.
Меди хлорофиллин Ь: 654,18.
Каждая из них может увеличиваться на 18 дальтон при размыкании кольца ци¬
клопентенила
Количественное определение
Общее содержание меди хлорофиллинов — не менее 95 % в образце, высушен¬
ном при 100 °С в течение 1 ч.
А1<^ — 565 при длине волны около 405 нм в фосфатном буфере (рН 7,5).
А1^ —145 при длине волны около 630 нм в фосфатном буфере (рН 7,5)
Описание
Порошок от темно-зеленого до синего/черного цвета
Идентификация
А. Спектрометрия
Максимум при длине волны около 405 нм и при длине волны около 630 нм в
водном фосфатном буфере (рН 7,5)
#5.50. Критерии чистоты пищевых красителей 129
Чистота
Остаточные количества органических
растворителей
Ацетон
Мети л эти л кетон
Метанол с СЛ .
Этанол Уне более 50 мг/кг,
Пропан-2-ол по отДельности или суммарно.
Гексан
Мышьяк
Свинец
Ртуть
Кадмий
Меди ионы
Общая медь
Метиленхлорид: не более 10 мг/кг.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 200 мг/кг.
Не более 8,0 % от общего содержания меди хлорофиллинов
Е142
ЗЕЛЕНЫЙ 3 (вРЕЕЫ 3)
Синонимы
С1 Пищевой зеленый 4, бриллиантовый зеленый ВЗ.
С/ Рооб Огееп 4, ВпШап! (Згееп ВЗ
Определение
Состоит преимущественно из натрия Л/-[4-(диметиламино)фенил]-2-гидрокси-
3,6-дисульфо-1-нафталенил)метилен]-2,5-циклогексадиен-1-илиден]-Л/-
метилметанамина и второстепенных окрашенных веществ наряду с натрия хло¬
ридом и/или сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Триарил метан
Номер индекса цвета
44090
Е/Л/ЕСЗ
221-409-2
Химическое название
Натрия А/-[4-[[4-(диметиламино)фенил](2-гидрокси-3,6-дисульфо-1-нафталенил)-
метилен]-2,5-циклогексадиен-1-илиден]-Л/-метилметанаминий.
Натрия 5-[4-диметиламино-а-(4-диметилиминоциклогекса-2,5-диенилиден)
бензил]-6-гидрокси-7-сульфонато-нафтапин-2-сульфонат (другое химическое
название)
Химическая формула
С27Н25М2№0732
Молекулярная масса
576,63
Количественное определение
Не менее 80 % от всех окрашенных веществ в пересчете на натриевую соль.
А}* — 1720 при длине волны около 632 нм в воде
Описание
Порошок или гранулы темно-синего или темно-зеленого цвета
Идентификация
A. Спектрометрия
B. Водный раствор синий или зеленый
Максимум при длине волны около 632 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
4,4'-бис(диметиламино)-бензгидриловый
спирт
4,4-бис(диметиламино)-бензофенон
3-гид роксинафталин-2,7-дисульфоновая
кислота
Бесцветное производное
Несульфированные первичные
ароматические амины
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 %.
Не более 1,0 %.
Не более 0,1 %.
Не более 0,1 %.
Не более 0,2 %.
Не более 5,0 %.
Не более 0,01 % (в пересчете на анилин).
Не более 0,2 % (в условиях нейтральной среды).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е 150А
КАРАМЕЛЬ (САХАРНЫЙ КОЛЕР I ПРОСТОЙ) (РЬАШ САНАМЕЦ
Определение
Получают контролируемой термической обработкой углеводов (коммерчески до¬
ступных пищевых подсластителей, являющихся мономерами глюкозы и фрук¬
тозы и/или их полимерами, например сиропов глюкозы, сахарозы и/или ин¬
вертированных сиропов, декстрозы). Для ускорения карамелизации возможно
применение кислот, щелочей и солей за исключением соединений аммония и
сульфитов
18. Зак. 1060.
130
Государственная фармакопея Республики Беларусь
Е/Л/ЕС5
232-435-9
Описание
Жидкости или твердые вещества от темно-коричневого до черного цвета
Чистота
Краситель, связывающийся ДЭАЭ-
целлюлозой
Не более 50 %.
Краситель, связывающийся
фосфорилцеллюлозой
Не более 50 %.
Интенсивность цвета*1 >
Общее содержание азота
Общее содержание серы
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
0,01—0,12.
Не более 0,1 %.
Не более 0,2 %.
Не более 1 мг/кг.
Не более 2 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 25 мг/кг
(1) Под интенсивностью цвета подразумевают оптическую плотность 0,1 % (м/об) водного раствора карамели, находящейся в
твердом состоянии, при проведении измерения при длине волны 610 нм с использованием кюветы 1 см.
Е 150В
КАРАМЕЛЬ, ПОЛУЧЕННАЯ ПО ЩЕЛОЧНО-СУЛЬФИТНОЙ ТЕХНОЛОГИИ (САХАРНЫЙ КОЛЕР II,
ПОЛУЧЕННЫЙ ПО ЩЕЛОЧНО-СУЛЬФИТНОЙ ТЕХНОЛОГИИ) (САЦЗТ1С ЗШ.РН1ТЕ САРАМЕЦ
Определение
Получают контролируемой термической обработкой углеводов (коммерчески до¬
ступных пищевых подсластителей, являющихся мономерами глюкозы и фрукто¬
зы и/или их полимерами, например сиропов глюкозы, сахарозы и/или инверти¬
рованных сиропов, декстрозы) с применением или без применения кислот или
щелочей в присутствии сернистой кислоты и ее солей (калия сульфита, калия
бисульфита, натрия сульфита и натрия бисульфита). Соединения аммония не
применяются
Е/Л/ЕС5
232-435-9
Описание
Жидкости или твердые вещества от темно-коричневого до черного цвета
Чистота
Краситель, связывающийся ДЭАЭ-
целлюлозой
Более 50 %.
Интенсивность цвета(1)
Общее содержание азота
Серы диоксид
Общее содержание серы
Сера, связывающаяся ДЭАЭ-целлюлозой
Отношение оптических плотностей краси¬
теля, связывающегося ДЭАЭ-целлюлозой
0,05—0,13.
Не более 0,3 %(2>.
Не более 0,2 %*2>.
(0,3—3,5) %*2>.
Более 40 %.
19—34.
Отношение оптических плотностей (А^А^)
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Более 50.
Не более 1 мг/кг.
Не более 2 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 25 мг/кг
(,) Под интенсивностью цвета подразумевают оптическую плотность 0,1 % (м/об) водного раствора карамели, находящейся в
твердом состоянии, при проведении измерения при длине волны 610 нм с использованием кюветы 1 см.
(2) В пересчете на эквивалентное количество красителя, т.е. количество красителя, имеющего интенсивность, соответствующую
интенсивности цвета 0,1 единиц оптической плотности.
Е150С
КАРАМЕЛЬ, ПОЛУЧЕННАЯ ПО АММИАЧНОЙ ТЕХНОЛОГИИ (САХАРНЫЙ КОЛЕР III, ПОЛУЧЕННЫЙ
ПО АММИАЧНОЙ ТЕХНОЛОГИИ) (АММОША САЯАМЕЦ
Определение
Получают контролируемой термической обработкой углеводов (коммерчески до¬
ступных пищевых подсластителей, являющихся мономерами глюкозы и фрукто¬
зы и/или их полимерами, например сиропов глюкозы, сахарозы и/или инверти¬
рованных сиропов, декстрозы) с применением или без применения кислот или
щелочей в присутствии соединений аммония (аммония гидроксида, аммония
карбоната, аммония гидрокарбоната и аммония фосфата). Производные суль¬
фитов не применяются
Е/Л/ЕС5
232-435-9
Описание
Чистота
Жидкости или твердые вещества от темно-коричневого до черного цвета
Краситель, связываемый ДЭАЭ-
целлюлозой
Не более 50 %.
#5.50. Критерии чистоты пищевых красителей
131
Краситель, связываемый
Более 50 %.
фосфорилцеллюлозой
Интенсивность цвета0}
0,08—0,36.
Содержание азота аммиака и его произво-
Не более 0,3 %<2>.
к дных
4-Метилимидазол
Не более 250 мг/кг<2).
2-Ацетил-4-тетрагидрокси-бутилимидазол
Не более 10 мг/кг<2).
Общее содержание серы
Не более 0,2 %<2>.
Общее содержание азота
(0,7—3,3) %<2>.
Отношение оптических плотностей краси-
13—35.
теля, связываемого фосфорилцеллюлозой
Мышьяк
Не более 1 мг/кг.
Свинец
Не более 2 мг/кг.
Ртуть
Не более 1 мг/кг.
Кадмий
Не более 1 мг/кг.
Тяжелые металлы (в пересчете на РЬ)
Не более 25 мг/кг
(1) Под интенсивностью цвета подразумевают оптическую плотность 0,1 % (м/об) водного раствора карамели, находящейся в
твердом состоянии, при проведении измерения при длине волны 610 нм с использованием кюветы 1 см.
(2) В пересчете на эквивалентное количество красителя, т.е. количество красителя, имеющего интенсивность, соответствующую
интенсивности цвета 0,1 единиц оптической плотности.
Е 1500
КАРАМЕЛЬ, ПОЛУЧЕННАЯ ПО АММИАЧНО-СУЛЬФИТНОЙ ТЕХНОЛОГИИ (САХАРНЫЙ КОЛЕР IV,
ПОЛУЧЕННЫЙ ПО АММИАЧНО-СУЛЬФИТНОЙ ТЕХНОЛОГИИ) (ЗШ-РН1ТЕ АММОЫ1А САПАМ ЕЦ
Определение
Получают контролируемой термической обработкой углеводов (коммерчески до¬
ступных пищевых подсластителей, являющихся мономерами глюкозы и фрук¬
тозы и/или их полимерами, например сиропов глюкозы, сахарозы и/или инвер¬
тированных сиропов, декстрозы) с применением или без применения кислот
или щелочей при одновременном присутствии сульфитов и соединений аммо¬
ния (сернистой кислоты, калия сульфита, калия бисульфита, натрия сульфита,
натрия бисульфита, аммония гидроксида, аммония карбоната, аммония гидро¬
карбоната, аммония фосфата, аммония сульфата, аммония сульфита и аммония
гидросульфита)
Е/Л/ЕС5
232-435-9
Описание
Жидкости или твердые вещества от темно-коричневого до черного цвета
Чистота
Краситель, связываемый ДЭАЭ-
Более 50 %.
целлюлозой
Интенсивность цвета0>
0,10—0,60.
Содержание азота аммиака и его произво-
Не более 0,6 %<2>.
дных
Серы диоксид
Не более 0,2 %(2).
4-Метилимидазол
Не более 250 мг/кг<2).
Общее содержание азота
(0,3—1,7) %<г>.
Общее содержание серы
(0,8—2,5) %<Ч
Отношение содержания азота к содержа-
0,7—2,7.
нию серы в осадке после осаждения спир-
том
Отношение оптических плотностей краси-
8—14.
теля после осаждения спиртом(3)
Отношение оптических плотностей (А^А^)
Не более 50.
Мышьяк
Не более 1 мг/кг.
Свинец
Не более 2 мг/кг.
Ртуть
Не более 1 мг/кг.
Кадмий
Не более 1 мг/кг.
Тяжелые металлы (в пересчете на РЬ)
Не более 25 мг/кг
°> Под интенсивностью цвета подразумевают оптическую плотность 0,1 % (м/об) водного раствора карамели в твердом состоянии
при проведении измерения при длине волны 610 нм с использованием кюветы 1 см.
{2) В пересчете на эквивалентное количество красителя, т.е. количество красителя, имеющего интенсивность, соответствующую
интенсивности цвета 0,1 единиц оптической плотности.
<3> Под отношением оптических плотностей красителя после осаждения спиртом подразумевают оптическую плотность красителя
при длине волны 250 нм, разделенную на оптическую плотность при длине волны 560 нм (кювета 1 см).
Е151
БРИЛЛИАНТОВЫЙ ЧЕРНЫЙ (ВН1ШАЫТ ВЬАСК ВЫ, В1.АСК РЫ)
Синонимы
С1 Пищевой черный 1, бриллиантовый черный РЫ.
С/ Рооб В1аск 1
Ч’ Зак. 1060
132
Государственная фармакопея Республики Беларусь
Определение
Состоит преимущественно из тетранатрия 4-ацетамидо-5-гидрокси-6-
[7-сульфонато-4-(4-сульфонатофенилазо)-1 -нафтилазо] нафталин-1,7-
дисульфоната и второстепенных окрашенных веществ наряду с натрия хлори¬
дом и/или сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Биазо
Номер индекса цвета
28440
Е/Л/ЕС5
219-746-5
Химическое название
Тетранатрия 4-ацетамидо-5-гидрокси-6-[7-сульфонато-4-(4-
сульфонатофенилазо)-1 -нафтилазо]нафталин-1,7-дисульфонат
Химическая формула
С2вН17М5Ма401434
Молекулярная масса
867,69
Количественное определение
Не менее 80 % от всех окрашенных веществ в пересчете на натриевую соль.
Кш — 530 при длине волны около 570 нм в растворе
Описание
Идентификация
Порошок или гранулы черного цвета
A. Спектрометрия
B. Раствор в воде синевато-черный
Максимум при длине волны около 570 нм в воде
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
Не более 0,2 %.
Не более 10 % (в пересчете на окрашенный компонент).
4-ацетамидо-5-гидроксинафталин-1,7-
дисульфоновая кислота
4-амино-5-гидроксинафталин-1,7-
дисульфоновая кислота
8-аминонафталин-2-сульфоновая кислота
4,4'-диазоаминоди-(бензолсульфоновая
кислота)
У- Общее содержание не более 0,8 %.
Несульфированные первичные
ароматические амины
Не более 0,01 % (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % в условиях нейтральной среды.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е153
УГОЛЬ РАСТИТЕЛЬНЫЙ (УЕвЕТАВЬЕ САКВОЫ)
Синонимы
Растительный черный.
\/еде(аЫе Ыаск
Определение
Получают карбонизацией растительного сырья, такого как древесина, отходы
целлюлозы, торф и скорлупа кокосового и других орехов. Сырье карбонизи¬
руют при высоких температурах. Состоит преимущественно из измельченного
угля. Может содержать незначительные количества азота, водорода и кисло¬
рода. Небольшое количество влаги может абсорбироваться на продукте после
производства
Номер индекса цвета
77266
Е1ЫЕС8
215-609-9
Химическое название
Углерод
Химическая формула
С
Молекулярная масса
12,01
Количественное определение
Не менее 95 % углерода в пересчете на безводное и беззольное вещество
Описание
Черный порошок без запаха и вкуса
Идентификация
A. Растворимость
B. Горение
Нерастворим в воде и органических растворителях.
При нагревании докрасна горит медленно без пламени
Чистота
Зола (общая)
Мышьяк
Свинец
Ртуть
Кадмий
Не более 4,0 % (температура прокаливания — 625 °С).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
#5.50. Критерии чистоты пищевых красителей
133
Тяжелые металлы (в пересчете на РЬ)
{Попваромаггмчесхие углеводороды
Не более 40 мг/кг.
Экстракт, полученный экстракцией 1 г образца с помощью 10 г циклогексана в
приборе для непрерывной экстракции, должен быть бесцветным и флуоресцен¬
ция экстракта в ультрафиолетовом свете должна быть не интенсивнее, чем для
раствора 0,100 мг хинина сульфата в 1000 мл 0,01 М раствора серной кислоты.
Потеря в массе при высушивают
Ваищсп1а, ра!’тпп|11—и в щелочах
Не более 12% (120 °С,4ч).
Фильтрат, полученный при нагревании 2 г образца в 20 мл 1 М раствора натрия
гидроксида и фильтровании, должен быть бесцветным
Е154
ЛЕОП1ЧМЕВЫЙ ЯК (ВКОМЫ ЯК)
~т_гг '
С1 Пищевой коричневый 1.
С/ Роос1 Вго\лт 1
Состоит преимущественно из смеси:
I натрия 4-(2,4-диаминофенилазо)бензолсульфоната;
II натрия 4-(4,6-диамино-м-толилазо)бензолсульфоната;
III динатрия 4,4-(4,6-диамино-1,3-фениленбиазо)ди(бензолсульфоната);
IV динатрия 4,4'-(2,4-диамино-1,3-фениленбиазо)ди(бензолсульфоната);
V динатрия 4,4'-(2,4-диамино-5-метил-1,3-фениленбиазо)ди(бензолсульфоната);
VI тринатрия 4,4',4"-(2,4-диаминобензол-1,3,5-триазо)три(бензолсульфоната) и
второстепенных окрашенных веществ наряду с водой, натрия хлоридом и/или
сульфатом в качестве основных неокрашенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Азо (смесь моно-, би- и триазокрасителей)
Химическое название
Смесь из:
I натрия 4-(2,4-диаминофенилазо)бензолсульфоната;
II натрия 4-(4,6-диамино-м-толилазо)бензолсульфоната;
III динатрия 4,4'-(4,6-диамино-1.3-фениленбиазо)ди(бензолсульфоната);
IV динатрия 4,4'-(2,4-диамино-1,3-фениленбиазо)ди(бензолсульфоната);
V динатрия 4,4-(2,4-диамино-5-метил-1,3-фениленбиазо)ди(бензолсульфоната);
VI тринатрия 4,4',4"-(2,4-диаминобензол-1,3,5-триазо)три(бензолсульфоната)
Химическая формула
I С,2Н„М4№033.
II С.3Н,Д№035.
1"С18Н1Д№20652.
1УС)8Н14МвМа20632.
УС19Н,ДМа20832.
VI С2ДД№30953
Молекулярная масса
I 314,30.
II 328,33.
III 520,46.
IV 520,46.
V 534,47.
VI 726,59
Количественное определение
Не менее 70 % от всех окрашенных веществ.
Содержание компонентов от общего содержания не должны превышать:
I 26 %;
II 17%;
II117%;
IV 16 %;
V 20 %;
VI 16%
Описание
Порошок или гранулы красно-коричневого цвета
Идентификация
Раствор от оранжевого до красноватого
цвета
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
4-аминобензол-1-сульфониловая кислота
м-фенилендиамин и 4-метип-м-
фенилендиамин
Несульфированные первичные аромати¬
ческие амины кроме м-фенилецдиамина и
4-метил-м-фенилендиамина
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 %.
Не более 3,5 %.
Не более 0,7 %.
Не более 0,35 %.
Не более 0,007 % (в пересчете на анилин).
Не более 0,2 % из раствора рН 7.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг
Не более 40 мг/кг
19. Зак. 1060.
134
Государственная фармакопея Республики Беларусь
Е155
КОРИЧНЕВЫЙ НТ (ВВОМЫ НТ)
Синонимы
С1 Пищевой коричневый 3.
С/ Рооб Вгомп 3
Определение
Состоит преимущественно из динатрия 4,4'-(2,4-дигидрокси-5-гидроксиметил-1,3-
фениленбиазо)ди(нафталин-1-сульфоната) и второстепенных окрашенных ве¬
ществ наряду с натрия хлоридом и/или сульфатом в качестве основных неокра¬
шенных компонентов.
Кальциевая и калиевая соли также допускаются
Класс
Биазо
Номер индекса цвета
20285
Е/Л/ЕС5
224-924-0
Химическое название
Динатрия 4,4'-(2,4-дигидрокси-5-гидроксиметил-1,3-фениленбиазо)ди(нафталин-
1-сульфонат)
Химическая формула
С27Н18М4№20932
Молекулярная масса
652,57
Количественное определение
Не менее 70 % от всех окрашенных веществ в пересчете на натриевую соль.
Аш — 403 в максимуме при длине волны около 460 нм в водном растворе (рН 7)
Описание
Порошок или гранулы красновато-коричневого цвета
Идентификация
A. Спектрометрия
B. Раствор в воде коричневый
Максимум при длине волны около 460 нм в воде (рН 7)
Чистота
Вещества, нерастворимые в воде
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
4-аминонафталин-1 -сульфоновая кислота
Несульфированные первичные
ароматические амины
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 %.
Не более 10 % (тонкослойная хроматография).
Не более 0,7 %.
Не более 0,01 % (в пересчете на анилин).
Не более 0,2 % в растворе со значением рН 7.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е 160А (I)
СМЕШАННЫЕ КАРОТИНЫ (М1ХЕЭ САВОТЕЫЕЗ)
1. КАРОТИНЫ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ (РЬАИТ САРОТЕИЕЗ)
Синонимы
С1 Пищевой оранжевый 5.
С/ Роод Огапде 5
Определение
Смешанные каротины получают экстракцией растворителем пригодного в пищу
растительного сырья природного происхождения, моркови, растительных масел,
травы, люцерны и крапивы.
Основными окрашенными компонентами являются каротиноиды, представлен¬
ные большей частью р-каротином. Могут присутствовать а-, у-каротин и другие
пигменты. Кроме окрашенных пигментов может содержать масла, жиры и пара¬
фины, являющиеся естественными компонентами исходного сырья.
Для экстракции могут быть использованы только следующие растворители: аце¬
тон, метил эти л кетон, метанол, этанол, пропан-2-ол, гексан(1), метиленхлорид и
углерода диоксид
Класс
Каротиноиды
Номер индекса цвета
75130
Е/А/ЕС5
230-636-6
Химическая формула
Р-Каротин:
Молекулярная масса
Р-Каротин: 536,88
Количественное определение
Содержание каротинов (в пересчете на р-каротин) не менее 5 %. Для веществ,
полученных экстракцией растительных масел: не менее 0,2 % в пригодных в
пищу жирах.
А» — 2500 в максимуме в диапазоне длин волн от 440 нм до 457 нм в циклогек¬
сане
Идентификация
А. Спектрометрия
Максимумы в диапазоне длин волн от 440 нм до 457 нм и от 470 нм до 486 нм в
циклогексане
#5.50. Критерии чистоты пищевых красителей
135
Чистота
Остаточные количества органических
растворителей
Ацетон
Метилэтилкетон
Метанол I - ,
Пропан-2-ол [ не более 50 мг/кг,
Гексан п0 отДельности или суммарно.
Этанол
Свинец
Метиленхлорид: не более 10 мг/кг.
Не более 5 мг/кг
2. КАРОТИНЫ ВОДОРОСЛЕЙ (МвМ САРОТЕИЕ8)
Синонимы
С1 Пищевой оранжевый 5.
С/ Рооб Огапде 5
Определение
Смешанные каротины могут быть также получены из водорослей ОипаНеНа заИпа,
произрастающих в больших соленых озерах, расположенных в городе Уайалле,
Южная Австралия. р-Каротин экстрагируют с использованием эфирного масла.
Готовые красители представляют собой (20—30) % суспензию в масле, пригодном
для пищи. Отношение транс- и цыс-изомеров находится в диапазоне от 50/50 до
71/29.
Основными окрашенными компонентами являются каротиноиды, представлен¬
ные большей частью р-каротином. Могут присутствовать а-каротин, лютеин, зеак-
сантин и р-криптоксантин. Кроме окрашенных пигментов, может содержать масла,
жиры и парафины, являющиеся естественными компонентами исходного сырья
Класс
Каротиноиды
Номер индекса цвета
75130
Химическая формула
Р-Каротин:
Молекулярная масса
Р-Каротин: 536,88
Количественное определение
Содержание каротинов (в пересчете на р-каротин) не менее 20 %.
Аш — 2500 в максимуме в диапазоне длин волн от 440 нм до 457 нм в циклогек¬
сане
А. Спектрометрия
Максимум в диапазоне длин волн от 440 нм до 457 нм и от 474 нм до 486 нм в
циклогексане
Чистота
Нейтральные токоферолы в пищевых
маслах
Свинец
Не более 0,3 %.
Не более 5 мг/кг
{1> Содержание бензола - не более 0,05 %.
Е160А(П)
БЕТА-КАРОТИН ЩЕТА-САКОТЕЫЕ)
1. 8-КАРОТИН (0-САЯОТЕНЕ)
с“—'
С1 Пищевой оранжевый 5.
С/ Роод Огапде 5
Определение
Требования данной статьи применяются преимущественно к аН-шранс-изомерам
р-каротина с незначительным количеством других каротиноидов. Разведенные и
стабилизированные готовые препараты могут иметь разные соотношения цис- и
транс-изомеров
Класс
Каротиноиды
Ноыер индекса цвета
40800
Е1ЫЕСЗ
230-636-6
Химическое название
Р-каротин; р,Р-каротин
Химическая формула
^40^56
Молекулярная масса
536,88
Количественное определение
Не менее 96 % от всех окрашенных веществ (в пересчете на р-каротин).
А1^ — 2500 в максимуме в диапазоне длин волн от 440 нм до 457 нм в
циклогексане
Описание
Кристаллы или кристаллический порошок от красного до коричневато-красного
цвета
Идентификация
А. Спектрометрия
Максимум в диапазоне длин волн от 453 нм до 456 нм в циклогексане
Чистота
Сульфатная зола
Второстепенные окрашенные вещества
С—юц
Не более 0,2 %.
Каротиноиды, кроме р.
Не более 2 мг/кг
•Г За». -ЭВС
136
Государственная фармакопея Республики Беларусь
2. р-КАРОТИН ИЗ В1-АКЕ31.ЕА ТР13Р0РА (р-САРОТЕЫЕ ЕРОМ В1.АКЕ31.ЕА ТР13Р0РА)
Синонимы
С1 Пищевой оранжевый 5.
С/ Роос/ Огапде 5
Определение
Получают с помощью ферментативного процесса с использованием смешанной
культуры скрещивающихся между собой типов (+) и (-) природных штаммов
грибов В1акез1еа 1пзрога. (3-Каротин экстрагируют из биомассы с использованием
этилацетата или изобутилацетата с последующей обработкой изопропиловым
спиртом и кристаллизуют. Кристаллизованный продукт состоит в основном из
транс-р-каротина. По причине природного происхождения продукт содержит
около 3 % смешанных каротиноидов, которые специфичны для конкретного
продукта
Класс
Каротиноиды
Номер индекса цвета
40800
Е1ЫЕС8
230-636-6
Химическое название
р-каротин; р,Р-каротин
Химическая формула
СЛ
Молекулярная масса
536,88
Количественное определение
Не менее 96 % от всех окрашенных веществ (в пересчете на р-каротин).
Кш — 2500 в максимуме в диапазоне длин волн от 440 нм до 457 нм в
циклогексане
Описание
Кристаллы или кристаллический порошок красного, коричневато-красного или
красновато-фиолетового цвета (цвет варьирует в зависимости от использованно¬
го в качестве экстрагента растворителя и от условий кристаллизации)
Идентификация
А. Спектрометрия
Максимум в диапазоне длин волн от 453 нм до 456 нм в циклогексане
Чистота
Остаточные количества органических
растворителей
Этилацетат 1 не более 0,8 %,
Этанол Г по отдельности или суммарно.
Сульфатная зола
Второстепенные окрашенные вещества
Свинец
Микотоксины:
Афлатоксин В1
Трихотецен (Т2)
Охратоксин
Зераленон
Микробиологическая чистота
Плесневые грибы
Дрожжи
8а1топе11а
ЕзсЬепсЫа соН
Изобутилацетат: не более 1,0 %.
Этанол: не более 0,1 %.
Не более 0,2 %.
Каротиноиды, кроме Р-каротина: не более 3,0 % от всех окрашенных веществ.
Не более 2 мг/кг.
Отсутствие.
Отсутствие.
Отсутствие.
Отсутствие.
Не более 100/г.
Не более 100/г.
Отсутствие в 25 г.
Отсутствие в 5 г
Е160В
АННАТТО, БИКСИН, НОРБИКСИН (АЫЫАТТО, В1Х1Ы, ЫОРВ1Х1Ы)
Синонимы
С1 Натуральный оранжевый 4.
С/ А/аЩга/ Огапде 4
Определение
Класс
Каротиноиды
Номер индекса цвета
75120
Е1ЫЕС8
Аннатто: 215-735-4, экстракт семян аннатто: 289-561-2; биксин: 230-248-7
Химическое название
Биксин: б'-метилгидро-Э’-ц^с-б.б-диапокаротин-6,6-диоат.
6'-метилгидро-9-транс-6,6-диапокаротин-6,6-диоат.
Норбиксин: 9-щ/с-6,6-диапокаротин-6,6-дионовая кислота.
9'-транс-6,б'-диапокаротин-б,6-дионовая кислота
Химическая формула
Биксин: С^НэдО^
Норбиксин: С24Н2804
Молекулярная масса
Биксин: 394,51.
Норбиксин: 380,48
Описание
Порошок, суспензия или раствор красновато-коричневого цвета
Идентификация
Спектрометрия
Биксин: максимум при длине волны около 502 нм в хлороформе.
Норбиксин: максимум при длине волны около 482 нм в разведенном растворе
КОН
#5.50. Критерии чистоты пищевых красителей
137
(1) Биксин и норбиксин, полученные экстракцией растворителями
Определение
Биксин получают экстракцией из внешней оболочки семян дерева аннатто (В/ха
огеНапа 1_.) одним или несколькими ниже перечисленными растворителями: аце¬
тоном, метанолом, гексаном или метиленхлоридом, углерода диоксидом с после¬
дующим удалением растворителя.
Норбиксин получают гидролизом щелочным раствором экстрагированного бик-
сина.
Биксин и норбиксин могут содержать и другие вещества, экстрагируемые из
семян аннатто.
Порошок биксина содержит несколько окрашенных компонентов, большую же
часть его составляет биксин, который может присутствовать как в цис-, так и в
транс-форме. Могут также присутствовать продукты термического разложения
биксина.
Порошок норбиксина содержит образованные в результате гидролиза биксина
вещества в виде солей натрия или калия, являющиеся основными окрашенными
компонентами. Могут присутствовать как цис-, так и транс-формы
Количественное определение
Порошок биксина содержит не менее 75 % от всех каротиноидов в пересчете на
биксин.
Порошок норбиксина содержит не менее 25 % от всех каротиноидов в пересчете
на норбиксин.
Биксин: А1^ — 2870 при длине волны около 502 нм в хлороформе.
Норбиксин: А1 См — 2870 при длине волны около 482 нм в растворе КОН
Чистота
Остаточные количества органических
растворителей
Ацетон
Метанол ^ не более 50 мг/кг,
Гексан по отдельности или суммарно.
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Метиленхлорид: не более 10 мг/кг.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
(Н) Аннатто, полученный экстракцией растворами щелочей
Определение
Водорастворимый аннатто получают экстракцией растворами щелочей (натрия
или калия гидроксидом) внешней оболочки семян дерева аннатто (В/ха огеНапа 1.).
Водорастворимый аннатто содержит норбексин, образующийся в результате ги¬
дролиза биксина, в виде солей натрия или калия, являющийся основным окра¬
шенным компонентом. Могут присутствовать как цис-, так и транс-формы
Количественное определение
Не менее 0,1 % от всех каротиноидов в пересчете на норбиксин.
Норбиксин: А1* — 2870 при длине волны около 482 нм в растворе КОН
Чистота
Мышьяк
Не более 3 мг/кг.
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
(Ш) Аннатто, полученный экстракцией маслом
Определение
Экстракт аннатто в масле в виде раствора или суспензии получают экстракцией
внешней оболочки семян дерева аннатто (В/ха огеНапа 1_.) с помощью пригодно¬
го в пищу растительного масла. Экстракт аннатто в масле содержит несколько
окрашенных компонентов, большую часть из которых составляет биксин, кото¬
рый может присутствовать как в цис-, так и в транс-форме.
Могут также присутствовать продукты термического разложения биксина
Количественное определение
Не менее 0,1 % от всех каротиноидов в пересчете на биксин.
Биксин: А« — 2870 при длине волны около 502 нм в хлороформе
Чистота
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
20. Зак. 1060.
138
Государственная фармакопея Республики Беларусь
Е160С
ЭКСТРАКТ ПАПРИКИ, КАПСАНТИН, КАПСОРУБИН (РАРР1КА ЕХТРАСТ, САР5АЫТНШ, САРЗОКиВМ)
Синонимы
Маслосмолы паприки.
Рарпка 01еогев ‘т
Определение
Получают экстракцией растворителем природных видов паприки, состоящей из
стручков земляных плодов (с семенами или без них) Сарзюит аппиит 1_. Содер¬
жит большую часть окрашенных компонентов этой специи. Основными окрашен¬
ными компонентами являются капсантин и капсорубин. Известно, что присутству¬
ет большое разнообразие других окрашенных компонентов.
Для экстракции могут быть использованы только следующие растворители: мета¬
нол, этанол, ацетон, гексан, метиленхпорид, этилацетат и углерода диоксид
Класс
Каротиноиды
Е/Л/ЕС5
Капсантин: 207-364-1; капсорубин: 207-425-2
Химическое название
Капсантин: (ЗЯ?,3'5,57?)-3,3'-дигидрокси-р,/с-каротин-6-он.
Химическая формула
Капсантин: С^Н^Оз-
Капсорубин: С^Н^О,,
Молекулярная масса
Капсантин: 584,85.
Капсорубин: 600,85
Количественное определение
Экстракт паприки: не менее 7,0 % каротиноидов.
Капсантин/капсорубин: не менее 30 % от всех каротиноидов.
Кш — 2100 при длине волны около 462 нм в ацетоне
Описание
Вязкая жидкость темно-красного цвета
Идентификация
A. Спектрометрия
B. Цветная реакция
Максимум при длине волны около 462 нм в ацетоне.
Появляется темно-синее окрашивание при добавлении одной капли серной кис¬
лоты к одной капле испытуемого образца в 2—3 каплях хлороформа
Чистота
Остаточные количества органических
растворителей
Этилацетат^)
Метанол
Этанол ► не более 50 мг/кг,
Ацетон по отдельности или суммарно.
Гексан ^
Капсаицин
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Метиленхпорид: не более 10 мг/кг.
Не более 250 мг/кг.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е160Э
ЛИКОПИН (1.УСОРЕЫЕ)
Синонимы
Натуральный желтый 27.
А/а/ига/ УеЧоу/ 27
Определение
Получают экстракцией растворителем природных видов красных томатов (/_усо-
регвюоп евси1еп1ит 1_.) с последующим удалением растворителя. Могут быть ис¬
пользованы только следующие растворители: метиленхпорид, углерода диоксид,
этилацетат, ацетон, пропан-2-ол, метанол, этанол, гексан. Основным окрашенным
компонентом томатов является ликопин, могут присутствовать незначительные
количества других пигментов каротиноидов. Кроме других пигментов в красителе
могут содержаться масла, жиры, парафины и ароматические компоненты, изна¬
чально присутствующие в томатах
Класс
Каротиноиды
Номер индекса цвета
75125
Химическое название
Ликопин, 'Р, Т-каротин
Химическая формула
С„Н„
Молекулярная масса
536,85
Количественное определение
А™ — 3450 при длине волны около 472 нм в гексане
Описание
Вязкая жидкость темно-красного цвета
Идентификация
Спектрометрия
Максимум в гексане при длине волны около 472 нм
Чистота
Остаточные количества органических
растворителей
Этилацетат 1
Метанол
Этанол ^ не более 50 мг/кг,
Ацетон Г по отдельности или суммарно.
Гексан
Пропан-2н^
Метиленхпорид: не более 10 мг/кг.
#5.50. Критерии чистоты пищевых красителей
139
Сульфатная зола
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,1 %.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е160Е
АЛПО-Г-ЖЛРО ТИН АЛЬ (СЗО) (,9-АРО-8'-САКОТЕИА1. (СЗО))
с-““
С1 Пищевой оранжевый 6.
С/ Еоос/ Огапде 6
Оарсзвелемие
Данные требования применяются преимущественно к аИ-транс-изомерам р
Стасе
Каротиноиды
Номер индекса цвета
40820
ВЫЕСЗ
214-171-6
Химическое название
р-Апо-8'-каротиналь, шранс-р-апо-8-каротинальдегид
Химическая формула
ело
Молекулярная масса
416,65
Количественное определение
Не менее 96 % от всех окрашенных веществ.
А]* — 2640 в диапазоне длин волн от 460 нм до 462 нм в циклогексане
Описание
Темно-фиолетовые кристаллы с металлическим блеском или кристаллический
порошок
Идентификация
Спектрометрия
Максимум в циклогексане в диапазоне длин волн от 460 нм до 462 нм
Чистота
Сульфатная зола
Второстепенные окрашенные вещества
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,1 %.
Каротиноиды, кроме р.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 10 мг/кг
Е160Р
ЭТИЛОВЫЙ ЭФИР/3-АПО-8'-КАРОТИНОЕВОЙ КИСЛОТЫ (СЗО) (ЕТНУ1. ЕЗТЕР ОР/В-АРО-8'-
САР0ТЕЫ01С АСЮ (СЗО))
Синонимы
С1 Пищевой оранжевый 7, Р-апо-8-каротинэфир.
С/ Роод Огапде 7, $-аро-8'-саго1епо'ю е$1ег
Определение
Данные требования применяются преимущественно к аН-шранс-изомерам р-апо-
8'-каротиналя совместно с незначительными количествами других каротиноидов.
Разведенные и стабилизированные формы готовят из р-апо-8'-каротиналя,
удовлетворяющего данным требованиям, и включают в себя растворы или
суспензии р-апо-8'-каротиналя в пригодных в пищу жирах или маслах, эмульсии
и порошки, диспергированные в воде. Данные формы красителя могут иметь
разные соотношения цис- и транс-изомеров
Класс
Каротиноиды
Номер индекса цвета
40825
Е/Л/ЕС5
214-173-7
Химическое название
Этиловый эфир р-апо-8'-каротиноевой кислоты, этил-8'-апо-р-каротин-8'-оат
Химическая формула
ело*
Молекулярная масса
460,70
Количественное определение
Не менее 96 % от всех окрашенных веществ.
А,1 * — 2550 при длине волны около 449 нм в циклогексане
Описание
Кристаллы или кристаллический порошок от красного до фиолетово-красного цвета
Идентификация
Спектрометрия
Максимум при длине волны около 449 нм в циклогексане
Чистота
Сульфатная зола
Второстепенные окрашенные вещества
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,1 %.
Каротиноиды, кроме этилового эфира р
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 10 мг/кг
20*. Зак. 1060
140
Гэсударственная фармакопея Республики Беларусь
Е161В
ЛЮТЕИН (ШТЕ1Ы)
Синонимы
Смешанные каротиноиды, ксантофиллы.
М/хес/ Саго1епо1с1з, ХапИюрЬуНз
Определение
Получают экстракцией растворителем природных видов пригодных в пищу фрук¬
тов и растений, травы, люцерны и (аде(ез егес1а. Основные окрашенные компо¬
ненты состоят из каротиноидов, из которых лютеин и его эфиры жирных кислот
составляют большую часть. Могут также присутствовать различные количества
каротинов. Лютеин может содержать жиры, масла и парафины, являющиеся есте¬
ственными компонентами исходного сырья.
Могут быть использованы только следующие растворители для экстракции: мета¬
нол, этанол, пропан-2-ол, гексан, ацетон, метилэтилкетон, метиленхлорид и угле¬
рода диоксид
Класс
Каротиноиды
Е/Л/ЕС5
204-840-0
Химическое название
3,3'-дигидрокси-0-каротин
Химическая формула
с„нио2
Молекулярная масса
568,88
Количественное определение
Не менее 4 % от всех окрашенных веществ в пересчете на лютеин.
— 2550 при длине волны около 445 нм в смеси хлороформ — этанол (10:90)
или гексан — этанол — ацетон (80:10:10)
Описание
Темная жидкость желтовато-коричневого цвета
Идентификация
Спектрометрия
Максимум при длине волны около 445 нм в смеси хлороформ — этанол (10:90)
Чистота
Остаточные количества органических
растворителей
-\
Ацетон
Метилэтилкетон
Метанол - .
Этанол Хне более 50 мг/кг,
Пропан-2-ол по 0ТДельн0СТИ или суммарно.
Гексан
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Метиленхлорид: не более 10 мг/кг.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е 1610
КАНТАКСАНТИН (САЫТНАХАМТШ)
Синонимы
С1 Пищевой оранжевый 8.
С/ ЕоосУ Огапде 8
Определение
Данные требования применяются преимущественно к аН-транс-изомерам кан-
таксантина совместно с незначительными количествами других каротиноидов.
Разведенные и стабилизированные формы готовят из кантаксантина, удовлетво¬
ряющего данным требованиям, и включают в себя растворы и суспензии кантак¬
сантина в пригодных в пищу жирах или маслах, эмульсии и порошки, дисперги¬
рованные в воде. Данные формы красителя могут иметь разные соотношения
цис- и транс-изомеров
Класс
Каротиноиды
Номер индекса цвета
40850
Е/Л/ЕС5
208-187-2
Химическое название
р-Каротин-4,4'-дион, кантаксантин, 4,4-диоксо-р-каротин
Химическая формула
с40н6А
Молекулярная масса
564,86
Количественное определение
Не менее 96 % от всех окрашенных веществ в пересчете на кантаксантин.
А« — 2200:
- при длине волны около 485 нм в хлороформе;
- в диапазоне длин волн от 468 нм до 472 нм в циклогексане;
- в диапазоне длин волн от 464 нм до 467 нм в петролейном эфире
Описание
Кристаллы или кристаллический порошок темно-фиолетового цвета
Идентификация
Спектрометрия
Максимум при длине волны около 485 нм в хлороформе.
Максимум в диапазоне длин волн от 468 нм до 472 нм в циклогексане.
Максимум в диапазоне длин волн от 464 нм до 467 нм в петролейном эфире
Чистота
Сульфатная зола
Второстепенные окрашенные вещества
Не более 0,1 %.
Каротиноиды, кроме кантаксантина: не более 5,0 % от всех окрашенных веществ.
#5.50. Критерии чистоты пищевых красителей
141
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 3 мг/кг.
Не более 10 мг/кг
Не более 1 мг/кг
Не более 1 мг/кг
Не более 40 мг/кг
Е 162
СВЕКОЛЬНЫЙ КРАСНЫЙ, БЕТАНИН (ВЕЕТЯООТ ЛЕО, ВЕТАЫЩ
Синонимы
Свекольный красный.
Вее^ Кес1
Определение
Получают из корнеплодов природных видов красной свеклы (Ве1а уи!дапз 1_., Vа^.
гиЬга) прессованием измельченной свеклы по мере того как отжимают сок или экс¬
тракцией водой дробленых корнеплодов свеклы с последующим обогащением в
активном составляющем. Цвет образуется различными пигментами, относящими¬
ся к классу беталаина. Основной окрашенный компонент состоит из бетациани-
нов (красный), из которых бетанин составляет (75—95) %. Могут присутствовать
незначительные количества бетаксантина (желтый) и продукты разложения бета-
лаинов (светло-коричневый).
Кроме пигментов красителя сок или экстракт содержит сахара, соли и/или белки,
являющиеся естественными компонентами исходного сырья. Раствор может быть
концентрированным и некоторые из красителей могут очищаться для удаления
большей части сахаров, солей и белков
Класс
Беталаин
Е/Л/ЕС5
231-628-5
Химическое название
($-(/?,Е')-4-(2-(2-карбокси-5(р-0-глюкопиранозилокси)-2,3-дигидро-6-гидрокси-
1 Н-индол-1 -ил)этенил)-2,3-дигидро-2,6-пиридин-дикарбоновая кислота
Химическая формула
1-(2-(2,6-Дикарбокси-1,2,3,4-тетрагидро-4-пиридилиден)этилиден)-5-р-0-
глюкопиранозилокси)-6-гидроксииндолин-2-карбоксилат
Молекулярная масса
550,48
Количественное определение
Содержание красного красителя (в пересчете на бетанин) составляет не менее 0,4 %.
А?м — 1120 при длине волны около 535 нм в водном растворе (рН 5)
Описание
Жидкость, паста, порошок или твердое вещество красного или темно-красного
цвета
Идентификация
Спектрометрия
Максимум при длине волны около 535 нм в воде (рН 5)
Чистота
Нитраты
Не более 2 г нитрат-ионов в 1 г красного красителя (как рассчитано в разделе
«Количественное определение»).
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 3 мг/кг
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е163
АНТОЦИАНИНЫ (АЫТНОСУАШЫ8)
Определение
Получают экстракцией сульфированной водой, подкисленной водой, углерода
диоксидом, метанолом или этанолом из природных видов овощей и пригодных
в пищу фруктов. Содержат обычно компоненты исходного сырья, а именно: ан-
тоцианин, органические кислоты, танины, сахара, минералы и т.д., но не обяза¬
тельно в тех же пропорциях, что и в исходном сырье
Класс
Антоцианин
Е/Л/ЕС5
208-438-6 (цианидин); 205-125-6 (пеонидин); 208-437-0 (дельфинидин); 211-403-8
(малвидин); 205-127-7 (пеларгонидин)
Химическое название
3,3',4',5,7-Пентагидроксифлавилина хлорид (цианидин).
3,4’,5,7-Тетрагидрокси-З-метоксифлавилина хлорид (пеонидин).
3,4',5,7-Тетрагидрокси-3',5-диметоксифлавилина хлорид (малвидин).
3.5.7- Тригидрокси-2-(3,4,5-тригидроксифенил)-1 -бензопирилина хлорид (делфи-
нидин).
3,3',4',5,7-Пентагидрокси-5'-метоксифлавилина хлорид (петунидин).
3.5.7- Тригидрокси-2-(4-гидроксифенил)-1 -бензопирилина хлорид (пеларгонидин)
Химическая формула
Цианидин: С^Н^С^С!.
Пеонидин: С16Н1306С1.
Малвидин: С17Н1507С1.
Делфинидин: С^Н^С^С!.
Петунидин: С16Н1307С1.
Пеларгонидин: С^Н^О^С!.
142
Гэсударственная фармакопея Республики Беларусь
Молекулярная масса
Цианидин: 322,6.
Пеонидин: 336,7.
Малвидин: 366,7.
Делфинидин: 340,6.
Петунидин: 352,7.
Пеларгонидин: 306,7
Количественное определение
К™ — 300 для чистого пигмента в диапазоне длин волн от 515 нм до 535 нм при
рН 3,0
Описание
Багряно-красная жидкость, порошок или паста, имеющие слабый характерный
запах
Идентификация
Спектрометрия
Максимумы поглощения в метаноле с 0,01 % раствором концентрированной НС1:
- цианидин: 535 нм;
- пеонидин: 532 нм;
- малвидин: 542 нм;
- делфинидин: 546 нм;
- петунидин: 543 нм;
- пеларгонидин: 530 нм
Чистота
Остаточные количества органических
растворителей
Метанол не более 60 мг/кг
Этанол по отдельности или суммарно.
Серы диоксид
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 1000 мг/кг в 1 % пигменте.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е170
КАЛЬЦИЯ КАРБОНАТ* (САЬСШМ САЯВОЫАТЕ)
Синонимы
С1 Пигмент белый 18, мел.
С/ Р1дтеп1 ШПе 18, СЬа1к
Определение
Получают из измельченного известняка или осаждением ионов кальция карбо¬
нат-ионами
Класс
Неорганический
Номер индекса цвета
77220
Е1ЫЕС8
Кальция карбонат: 207-439-9.
Известняк: 215-279-6
Химическое название
Кальция карбонат
Химическая формула
СаС03
Молекулярная масса
100,1
Количественное определение
Не менее 98 % в пересчете на безводное вещество
Описание
Белый кристаллический или аморфный порошок без запаха и вкуса
Идентификация
Растворимость
Практически нерастворим в воде и в спирте. Растворяется с бурным выделением
пузырьков газа в кислоте уксусной разведенной, в кислоте хлористоводородной
разведенной и в кислоте азотной разведенной; полученные растворы после ки¬
пячения дают положительные реакции на кальций
Чистота
Потеря в массе при высушивании
Нерастворимые в кислоте вещества
Магний и соли щелочных металлов
Фториды
Сурьма (как ЗР)
Медь (как Си)
Хром (как Сг)
Цинк (как 2п)
Барий (как Ва)
Мышьяк
Свинец
Кадмий
Не более 2,0 % (200 °С, 4 ч).
Не более 0,2 %.
Не более 1,5 %.
Не более 50 мг/кг.
I не более 100 мг/кг,
Г по отдельности или суммарно.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг
* Или см. требования статьи Кальция карбонат (0014).
Е171
ТИТАНА ДИОКСИД* (Т1ТАЫШМ О/ОХ/ОЕ)
Синонимы
С1 Пигмент белый 6.
С/ Р/дтел/ ШПе 6
#5.50. Критерии чистоты пищевых красителей
143
Определение
Состоит преимущественно из анатаза и/или титана диоксида, который может
быть покрыт небольшим количеством алюминия оксида и/или кремния диоксида
для улучшения технологических свойств красителя
Класс
Неорганический
Номер индекса цвета
77891
Е/Л/ЕС5
236-675-5
Химическое название
Титана диоксид
Химическая формула
-по,
Молекулярная масса
79,88
Количественное определение
Не менее 99 % в пересчете на вещество, не содержащее алюминия оксид и
кремния диоксид
Описание
Порошок белого или почти белого цвета
Идентификация
Растворимость
Нерастворим в воде и в органических растворителях. Растворяется медленно в
кислоте фтористоводородной и в горячей кислоте серной концентрированной
Чистота
Потеря в массе при высушивании
Потеря в массе при прокаливании
Не более 0,5% (105 °С, 3 ч).
Не более 1,0 % в пересчете на вещество, не содержащее летучих компонентов
(800 °С).
Алюминия оксид и/или кремния диоксид
Вещества, растворимые в 0,5 М НС1
Общее содержание не более 2,0 %.
Не более 0,5 % в пересчете на вещество, не содержащее алюминия оксид и
кремния диоксид. Дополнительно для веществ, содержащих алюминия оксид и/
или кремния диоксид не более 1,5 % в пересчете на твердое вещество.
Растворимые в воде вещества
Кадмий
Сурьма
Мышьяк
Свинец
Ртуть
Цинк
Не более 0,5 %.
Не более 1 мг/кг.
Не более 50 мг/кг после полного растворения.
Не более 3 мг/кг после полного растворения.
Не более 10 мг/кг после полного растворения.
Не более 1 мг/кг после полного растворения.
Не более 50 мг/кг после полного растворения
* Или см. требования статьи Титана диоксид (0150).
Е172
ЖЕЛЕЗА ОКСИДЫ И ЖЕЛЕЗА ГИДРОКСИДЫ (1Р0Ы 0ХЮЕ8 АЛЮ НУ0Р0ХЮЕ8)
Синонимы
Железа оксид желтый: С1 Пигмент желтый 42 и 43.
Железа оксид красный: С1 Пигмент красный 101 и 102.
Железа оксид черный: С1 Пигмент черный 11.
/гол Ох/'с/е Уе//оиг. С/ Р/дшел/ У<э//оуи 42 апб 43.
1гоп ОхШе Ре&. С1 Р1дтеп1 Кед 101 апб 102.
/гол Ох/бе В1ас1с. С/ Р/дшел/ В1аск 11
Определение
Получают путем синтеза, состоит преимущественно из безводных и/или гидрати¬
рованных железа оксидов. Диапазон оттенков включает желтые, красные, корич¬
невые и черные цвета. Качество железа оксида для пищевых целей отличается
от технического сравнительно низкими уровнями загрязнения другими металла¬
ми. Это достигается путем подбора и контроля сырья железа и/или степенью хи¬
мической очистки в течение процесса производства
Класс
Неорганический
Номер индекса цвета
Железа оксид желтый: 77492.
Железа оксид красный: 77491.
Железа оксид черный: 77499
Е/А/ЕС5
Железа оксид желтый: 257-098-5.
Железа оксид красный: 215-168-2.
Железа оксид черный: 235-442-5
Химическое название
Железа оксид желтый: гидратированный железа (III) оксид.
Железа оксид красный: безводный железа (III) оксид.
Железа оксид черный: железа (II, III) оксид
Химическая формула
Железа оксид желтый: РеО(ОН) ■ хН20.
Железа оксид красный: Ре203.
Железа оксид черный: РеО • Ре203
Молекулярная масса
88,85: РеО(ОН).
159,70: Ре203.
231,55: РеО ■ Ре203
Количественное определение
Желтый: не менее 60 % общего железа в пересчете на железо.
Красный и черный: не менее 68 % общего железа в пересчете на железо
Описание
Порошок оттенков желтого, красного, коричневого или черного цветов
Идентификация
Растворимость
Нерастворим в воде и в органических растворителях. Растворяется в концентри¬
рованных минеральных кислотах
144
Гэсударственная фармакопея Республики Беларусь
Чистота
Растворимые в воде вещества
Не более 1,0 % после полного растворения.
Мышьяк
Не более 5 мг/кг после полного растворения.
Барий
Не более 50 мг/кг после полного растворения.
Кадмий
Не более 5 мг/кг после полного растворения.
Хром
Не более 100 мг/кг после полного растворения.
Медь
Не более 50 мг/кг после полного растворения.
Свинец
Не более 20 мг/кг после полного растворения.
Ртуть
Никель
Цинк
Не более 1 мг/кг после полного растворения.
Не более 200 мг/кг после полного растворения.
Не более 100 мг/кг после полного растворения
Е 173
АЛЮМИНИЙ (АШМ1ЫШМ)
Синонимы
С1 металлический пигмент, А1.
С/ Р1дтеп( Ме(а1, А1
Определение
Порошок алюминия состоит из мелко измельченных частиц алюминия. Измель¬
чение может или не проводиться в присутствии пригодных в пищу растительных
масел и/или жирных кислот. Не содержит примеси других веществ, кроме пригод¬
ных в пищу растительных масел и/или жирных кислот
Номер индекса цвета
77000
Е1ИЕС8
231-072-3
Химическое название
Алюминий
Химическая формула
А1
Атомная масса
26,98
Количественное определение
Не менее 99 % в пересчете на алюминий на свободное от масла вещество
Описание
Серебристо-серый порошок или очень маленькие пластинки
Идентификация
Растворимость
Нерастворим в воде и в органических растворителях. Растворим в кислоте хло¬
ристоводородной разведенной. Конечный раствор дает положительные реакции
на алюминий
Чистота
Потеря в массе при высушивании
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,5 % (105 °С, до постоянной массы).
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
Е174
СЕРЕБРО (5//.УЕК)
Синонимы
Агдеп(ит, Ад
Класс
Неорганический
Номер индекса цвета
77820
Е/Л/ЕС5
231-131-3
Химическое название
Серебро
Химическая формула
Ад
Атомная масса
107,87
Количественное определение
Не менее 99,5 % Ад
Описание
Серебристый порошок или очень маленькие пластинки
Е175
ЗОЛОТО (001.0)
Синонимы
Металлический пигмент 3.
Р1дтеп1 МеШ 3, Аигит, Аи
Класс
Неорганический
Номер индекса цвета
77480
Е/Л/ЕС5
231-165-9
Химическое название
Золото
Химическая формула
Аи
Атомная масса
197,0
Количественное определение
Не менее 90 % Аи
Описание
Золотистый порошок или очень маленькие пластинки
Чистота
Серебро
Не более 7 % 1
Медь
Не более 4 % г после полного растворения
#5.50. Критерии чистоты пищевых красителей
145
Е 180
ЛИТОЛРУБИН (иТНО!-ГШВтЕ ВК)
Синонимы
С1 Пигмент красный 57, рубиновый пигмент, кармин 6В, литолрубин ВК.
С/ Р1дтеп1 Кес157, РиЫпр1дтеп1, Сагт'те 6В
Определение
Состоит преимущественно из кальция 3-гидрокси-4-(4-метил-2-
сульфонатофенилазо)-2-нафталинкарбоксилата и второстепенных окрашенных
веществ наряду с водой, кальция хлоридом и/или сульфатом в качестве основ¬
ных неокрашенных компонентов
Класс
Моноазо
Номер индекса цвета
15850:1
ВЫЕСЗ
226-109-5
Химическое название
Кальция 3-гидрокси-4-(4-метил-2-сульфонатофенилазо)-2-нафталинкарбоксилат
Химическая формула
С18Н12СаГ42Ов5
Молекулярная масса
424,45
Количественное определение
Не менее 90 % от всех окрашенных веществ.
— 200 при длине волны около 442 нм в диметилформамиде
Описание
Красный порошок
Идентификация
Спектрометрия
Максимум при длине волны около 442 нм в диметилформамиде
Чистота
Второстепенные окрашенные вещества
Органические вещества, кроме
окрашенных:
Не более 0,5 %.
2-амино-5-метилбензолсульфоновая кисло¬
та, кальциевая соль
Не более 0,2 %.
З-гидрокси-2-нафталинкарбоновая кислота,
кальциевая соль
Не более 0,4 %.
Несульфированные первичные
ароматические амины
Не более 0,01% (в пересчете на анилин).
Вещества, экстрагируемые эфиром
Мышьяк
Свинец
Ртуть
Кадмий
Тяжелые металлы (в пересчете на РЬ)
Не более 0,2 % из раствора с рН 7.
Не более 3 мг/кг.
Не более 10 мг/кг.
Не более 1 мг/кг.
Не более 1 мг/кг.
Не более 40 мг/кг
146
Гэсударственная фармакопея Республики Беларусь
Радиоактивные фармацевтические препараты
147
ОБЩИЕ СТАТЬИ
07/2016:0125
РАДИОАКТИВНЫЕ
ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
КаФорЬагтасеиИса
ОПРЕДЕЛЕНИЕ
Радиоактивные фармацевтические препараты
или радиофармацевтические препараты представля¬
ют собой лекарственные средства, готовые к меди¬
цинскому применению и содержащие один или более
радионуклидов (радиоактивных изотопов) для меди¬
цинских целей.
В рамках данной общей статьи к радиоактивным
фармацевтическим препаратам также относятся:
- изотопные генераторы — любая система, вклю¬
чающая заданный материнский радионуклид, из кото¬
рого получается дочерний радионуклид, отделяемый
элюированием или любым другим методом и приме¬
няемый в радиоактивном фармацевтическом препа¬
рате;
-комплекты для радиоактивного фармацевти¬
ческого препарата — любое лекарственное средство,
воссоздаваемое или объединяемое с радионукли¬
дами в конечный радиоактивный фармацевтический
препарат, обычно перед его применением;
- радионуклидные прекурсоры — любой радио¬
нуклид, полученный для введения радиоактивной
метки другому веществу перед применением;
-химические прекурсоры — нерадиоактивные
вещества для объединения с радионуклидом.
Радионуклидные прекурсоры могут поставляться
в виде растворов для введения радиоактивной метки.
Нуклид представляет собой разновидность ато¬
ма, характеризующуюся числом протонов и нейтронов
в его ядре (т.е. его атомным номером 2 и массовым
числом А), а также его потенциалом ядерной энергии.
Изотопы элемента — это нуклиды с одинаковым атом¬
ным номером, но различным массовым числом. Ну¬
клиды, содержащие нестабильные комбинации про¬
тонов и нейтронов, могут преобразовываться само¬
произвольно в устойчивые или другие нестабильные
комбинации протонов и нейтронов с постоянной ста¬
тистической вероятностью. Такие нуклиды являются
радиоактивными и называются радионуклидами. На¬
чальный нестабильный нуклид называется материн¬
ским радионуклидом, а конечный нуклид — дочерним
нуклидом.
Радиоактивный распад или преобразование
может повлечь испускание заряженных частиц,
электронный захват (ЭЗ) или изомерный переход
(ИП). Заряженные частицы, исходящие от ядра,
могут быть альфа-частицами (ядрами 4Не) или бе¬
та-частицами (отрицательно заряженными, обычно
называемыми электронами, или положительно за¬
ряженными, обычно называемыми позитронами).
Альфа-распаду обычно подвергаются тяжелые ядра
(2>82). Радионуклиды с недостатком протонов обыч¬
но распадаются, испуская электроны. Радионукли¬
ды с недостатком нейтронов обычно распадаются,
захватывая электроны или испуская позитроны. В
последнем случае радионуклиды называются эмит¬
терами позитронов. Позитроны аннигилируются по¬
сле взаимодействия с электронами в окружающем
пространстве. Аннигиляция приводит к эмиссии двух
гамма-фотонов обычно под углом 180° друг к другу,
каждый из которых имеет энергию 0,511 МэВ (ан¬
нигиляционное излучение). Все способы распада
могут сопровождаться эмиссией гамма-излучения.
Испускание гамма-излучения может быть частич¬
но или полностью замещено выбросом электронов,
которые называются внутренними конверсионными
электронами. Данное явление, подобно процессу за¬
хвата электрона, вызывает вторичную эмиссию гам¬
ма-излучения (вследствие реорганизации электро¬
нов в атоме). Такая вторичная эмиссия может быть
частично замещена выбросом электронов, которые
называются оже-электронами.
Радиоактивность. Обычно термин «радиоактив¬
ность» используется для описания явления радиоак¬
тивного распада и для выражения физической вели¬
чины данного явления.
Радиоактивность лекарственного средства — это
количество ядерных распадов или преобразований в
единицу времени.
В Международной системе единиц (СИ) радиоак¬
тивность выражается в беккорелях (Бк); один бекке-
рель равен одному ядерному превращению в секунду.
Измерения абсолютной радиоактивности могут быть
проведены только в специализированной лаборато¬
рии. Идентификация и количественное измерение
радиоактивности могут быть выполнены соответ¬
ственно путем сравнения измеряемых образцов со
стандартизированными лекарственными средствами
в лабораториях, признанных компетентными уполно¬
моченными органами, или применения калиброванно¬
го измерительного оборудования.
Радиоактивный распад — любой радиоактив¬
ный распад, находящийся в экспоненциальной зави¬
симости от характерной для него константы распада.
Кривая экспоненциального распада (кривая рас¬
пада) описывается следующим уравнением:
где:
Аг— радиоактивность во время ^
А0 — радиоактивность во время (= 0;
А — константа распада, характерная для каждого
радионуклида;
е — основание натурального логарифма.
Период полураспада (ТУ2) — это значение вре¬
мени, при котором заданная радиоактивность (коли¬
чество) радионуклида снижается вдвое от ее перво¬
начального значения.
Период полураспада связан с константой распа¬
да (А) следующим уравнением:
Таким образом, уравнение экспоненциально¬
го распада может быть представлено в следующем
виде, применяемом для быстрой оценки радиоактив¬
ности, оставшейся по истечении времени (:
Проникающая способность каждого излучения
существенно изменяется в зависимости от его приро¬
ды и энергии. Альфа-частицы полностью поглощают¬
ся объектом толщиной от нескольких микрометров до
148
Гэсударственная фармакопея Республики Беларусь
нескольких десятков микрометров. Бета-частицы пол¬
ностью поглощаются объектом толщиной от несколь¬
ких миллиметров до нескольких сантиметров. Гам¬
ма-излучение полностью не поглощается, а только
ослабляется, и, например, для десятикратного умень¬
шения может потребоваться пластина свинца толщи¬
ной несколько сантиметров. В большинстве случаев
с увеличением плотности объекта уменьшается про¬
никающая способность альфа- и бета-частиц и гам¬
ма-излучения.
Каждый радионуклид характеризуется постоян¬
ным периодом полураспада, выраженным в единицах
времени, а также природой и энергией его излучения
или излучений. Энергия выражается в электронволь-
тах (эВ), килоэлектронвольтах (кэВ) или мегаэлек¬
тронвольтах (МэВ).
Радионуклидная чистота — отношение радиоак¬
тивности определенного радионуклида к общей ради¬
оактивности радиоактивного фармацевтического пре¬
парата, выраженное в процентах. Перечень значимых
возможных радионуклидных примесей с их предельны¬
ми содержаниями приводится в частных статьях.
Радиохимическая чистота — отношение ради¬
оактивности определенного радионуклида, который
присутствует в радиоактивном фармацевтическом пре¬
парате в установленной химической форме, к общей
радиоактивности этого же радионуклида, присутствую¬
щего в радиоактивном фармацевтическом препарате,
выраженное в процентах. Перечень значимых возмож¬
ных радионуклидных примесей с их предельными со¬
держаниями приводится в частных статьях.
Химическая чистота — химическая чистота кон¬
тролируется указанием предельных содержаний для
химических примесей в частных статьях на радиоак¬
тивные фармацевтические препараты.
Переносчик изотопа — стабильный изотоп дан¬
ного элемента, присутствующий или добавленный
в радиоактивное лекарственное средство в той же
самой химической форме, что и сам радионуклид.
Лекарственное средство, не содержащее пере¬
носчик изотопа — лекарственное средство, не содер¬
жащее стабильные изотопы того же элемента, что и
данный радионуклид, присутствующего в лекарствен¬
ном средстве в установленной химической форме или
в положении радионуклида в данной молекуле.
Лекарственное средство без добавления пе¬
реносчика изотопа — лекарственное средство, к ко¬
торому намеренно не добавлены стабильные изотопы
того же элемента, что и данный радионуклид, в уста¬
новленной химической форме или в положении ради¬
онуклида в данной молекуле.
Удельная радиоактивность — радиоактив¬
ность радионуклида на единицу массы элемента или
определенной химической формы, например бекке-
рель на грамм или беккерель на моль.
Радиоактивная концентрация — радиоактив¬
ность радионуклида на единицу объема или единицу
массы лекарственного средства. Для растворов ради-
офармацевтических препаратов она выражается в пе¬
ресчете на радиоактивность на единицу объема ле¬
карственного средства.
Общая радиоактивность — радиоактивность
радионуклида в пересчете на единицу измерения
(виалу, флакон, капсулу, ампулу, генератор и т.д.).
Химические прекурсоры для синтеза радио¬
активных веществ — если активная субстанция ра¬
диоактивного фармацевтического препарата не полу¬
чена, то для его синтеза в качестве субстанции для
фармацевтического использования рассматривается
химический прекурсор.
Рекомендуется испытывать каждую партию ве¬
щества химического прекурсора в производственный
период перед его применением в производстве ради¬
оактивных фармацевтических препаратов для гаран¬
тии того, что при заданных условиях производства из
данной субстанции получается радиоактивный фар¬
мацевтический препарат в необходимом количестве и
установленного качества.
Гарантийный срок — время, в течение которого
должны выдерживаться все показатели, указанные в
частной статье.
ПРОИЗВОДСТВО
Радиоактивный фармацевтический препарат со¬
держит радионуклид:
- в качестве элемента в атомной или молекуляр¬
ной форме, например [133Хе], [150]02;
-в качестве иона, например [1311]йодид, [99тТс]
пертехнетат;
- в качестве включенного, поглощенного или
прикрепленного к молекулам с образованием коор¬
динационного комплекса, например [1111п]индийоксин,
или ковалентного соединения, например 2-[18Р]фтор-
2-дезокси-О-глюкоза.
Радионуклиды могут быть получены следующи¬
ми способами:
- при бомбардировке нейтронами (облучение
мишени в ядерных реакторах);
- при бомбардировке заряженными частицами
(облучение мишени с применением акселераторов, в
частности циклотронов);
- их отделением от изотопных генераторов.
Вероятность возникновения ядерной реакции за¬
висит от природы и энергии бомбардирующих частиц
(протонов, нейтронов, дейтронов и т.д.) и природы
ядер, подвергающихся облучению ими. Степень по¬
лучения (выход) данного радионуклида в результа¬
те облучения зависит также от изотопного состава ве¬
щества-мишени и его химической чистоты, в случае
нейтронов — от их истечения, а в случае заряженных
частиц — от тока пучка.
Одновременно с необходимой ядерной реакци¬
ей обычно происходят преобразования. Вероятность
их осуществления задается несколькими факторами,
как упоминалось в предыдущем разделе. В результа¬
те таких одновременных преобразований могут появ¬
ляться радионуклидные примеси.
Ядерная реакция (преобразование) может быть
записана в следующей форме: ядра-мишени (бомбар¬
дирующая частица, излученная частица) произведен¬
ные ядра.
Примеры: 58Ре(п,у)59Ре
180(р,п)18Р.
ОБЛУЧЕНИЕ НЕЙТРОНАМИ
Облучение стабильных радионуклидов в ядер¬
ных реакторах обычно приводит к образованию ядер
с недостатком протонов, т.е. эмиттеров электронов,
в результате (п,у) реакций (также называемых ради¬
ационным захватом). Образующийся радионуклид
является изотопным с ядром-мишенью и, следова¬
тель но, может содержать значительное количество
переносчика.
Ряд нуклидов с большим атомным номером рас¬
щепляется с помощью нейтронов. Ядерное расще-
Радиоактивные фармацевтические препараты
149
пление, называемое (п,Т) реакцией, приводит к об¬
разованию большого числа радионуклидов с различ¬
ными массами и периодами полураспада. Наиболее
часто применяемым является ядерное расщепление
23511. Йод-131, молибден-99 и ксенон-133 могут быть
получены в результате ядерного расщепления 235У в
ядерных реакторах и их отделения от более 200 ради¬
онуклидов, образовавшихся в этом процессе.
ОБЛУЧЕНИЕ ЗАРЯЖЕННЫМИ ЧАСТИЦАМИ
Облучение стабильных радионуклидов заряжен¬
ными частицами обычно приводит к образованию
ядер с недостатком нейтронов, которые распадаются
при электронном захвате или эмиссии позитронов.
Они образуются в частности в (р,хп) реакциях (где х —
число испускаемых электронов). Полученный радио¬
нуклид не является изотопным с ядрами-мишенями
и может иметь удельную радиоактивность, близкую к
радиоактивности лекарственного средства, не содер¬
жащего переносчик изотопа.
ИЗОТОПНЫЕ ГЕНЕРАТОРЫ
Системы изотопных генераторов используют ма¬
теринский радионуклид, который распадается до до¬
чернего радионуклида, обладающего более коротким
периодом полураспада.
Используя химический или физический процесс,
отделяют дочерний радионуклид от материнского, что
позволяет использовать дочерний радионуклид, не¬
смотря на его малый период полураспада, на значи¬
тельном расстоянии от места его получения.
ВЕЩЕСТВА-МИШЕНИ
Относительное процентное содержание основ¬
ного радионуклида и радиоактивных примесей, полу¬
ченных при облучении, зависит от изотопного состава
и чистоты вещества-мишени, а также других факто¬
ров, например природы и энергии бомбардирующих
частиц. Использование искусственно обогащенного
изотопами вещества-мишени может способствовать
увеличению выхода и повысить чистоту необходимого
радионуклида.
Химическая форма, чистота и физическое со¬
стояние вещества-мишени и химических добавок, а
также условия облучения и непосредственная физи¬
ческая и химическая среда определяют химическое
состояние и химическую чистоту полученных радио¬
нуклидов. При производстве радионуклидов и, в част¬
ности, короткоживущих радионуклидов, перед обра¬
боткой и производством радиоактивных фармацевти¬
ческих препаратов может отсутствовать возможность
определить какой-либо из критериев качества. Поэто¬
му оценка качества каждой серии вещества-мишени
проводится перед ее использованием при получении
и производстве радиоактивных фармацевтических
препаратов в установленном порядке.
Бомбардируемое вещество в газообразном, жид¬
ком или твердом виде находится в емкости, обеспе¬
чивающей возможность облучения. Для облучения
нейтронами вещество-мишень обычно помещают в
кварцевые ампулы или контейнеры из алюминия или
титана высокой степени очистки. Необходимо убе¬
диться, что никакого взаимодействия между контей¬
нером и его содержимым в условиях облучения про¬
изойти не может.
Для облучения заряженными частицами емкость
для вещества-мишени конструируют из подходяще¬
го металла, по возможности с входным и выходным
отверстиями, примыкающей системы охлаждения и
обычно окна для мишени иэ тонкой металлической
фольги.
Для оценки всех факторов, воздействующих на
эффективность получения радионуклида с точки зре¬
ния качества и количества, методика получения долж¬
на четко описывать и учитывать вещество-мишень,
конструкцию емкости для вещества-мишени, метод
облучения и отделения необходимого радионуклида.
СВОЙСТВА
Таблица физических характеристик радиону¬
клидов, упоминаемых в Фармакопее (5.7), содержит
наиболее общие физические характеристики радио¬
нуклидов, применяемых в лекарственных средствах,
описанных в частных статьях. Кроме того, таблица
содержит физические характеристики основных воз¬
можных примесей радионуклидов, описанных в част¬
ных статьях.
Под термином «вероятность перехода» подраз¬
умевается вероятность преобразования ядра в дан¬
ном состоянии энергии через определенный переход.
Вместо термина «вероятность» может также исполь¬
зоваться термин «распространенность».
Под термином «вероятность эмиссии» подраз¬
умевают вероятность атома радионуклида вызывать
эмиссию частиц или определенного излучения.
Вне зависимости от того, какое значение подраз¬
умевается, вероятность обычно указывается в про¬
центах.
ИДЕНТИФИКАЦИЯ
Радионуклид обычно идентифицируется по его
периоду полураспада или по природе и энергии его
излучения или излучений, или по обеим характери¬
стикам, как указано в частной статье.
Приблизительный период полураспада —
период полураспада, определяемый на протяжении
относительно небольшого периода времени для раз¬
решения выпуска радиоактивных фармацевтических
препаратов для их применения.
Рассчитанный приблизительный период полурас¬
пада находится в пределах диапазона значений, ука¬
занного в отдельной частной статье.
Определение природы и энергии излучения.
Природа и энергия испускаемого излучения опреде¬
ляются с применением спектрометрии. Природа и
энергия излучения эмиттеров позитронов обычно не
определяются; идентификация в этом случае прово¬
дится посредством определения периода полураспа¬
да и получения спектра гамма-излучения.
ИСПЫТАНИЯ
Если радионуклид лекарственного средства
имеет короткий период полураспада, проведение
некоторых изложенных ниже испытаний перед вы¬
пуском партии для последующего применения в
некоторых случаях представляет сложность. От¬
дельная частная статья устанавливает испыта¬
ния, которые не обязательно должны быть полны¬
ми перед выпуском для последующего применения.
Эти испытания в последующем составляют кон¬
троль качества производства.
Нерадиоактивные вещества и сопутствую¬
щие примеси. Этот раздел описывает определение
нерадиоактивных веществ и сопутствующих приме¬
сей, которые могут присутствовать.
Остаточные количества растворителей.
Предельные содержания остаточных растворителей
150
Гэсударственная фармакопея Республики Беларусь
определяются в соответствии е общей статьей 5.4.
Остаточные количества органических раствори¬
телей с применением методов, указанных в общей
статье 2.4.24. Идентификация и контроль содержа¬
ния остаточных растворителей или других подхо¬
дящих методов.
РАДИОНУКЛИДНАЯ ЧИСТОТА
Радионуклидные примеси могут появляться в
процессе производства и распада радионуклида. Воз¬
можные радионуклидные примеси могут упоминаться
в частных статьях, а их характеристики описываться
в общей статье 5.7. Таблица физических характери¬
стик радионуклидов, упоминаемых в Фармакопее.
В большинстве случаев для определения радио¬
нуклидной чистоты радиоактивного фармацевтиче¬
ского препарата должны быть идентифицированы все
присутствующие радионуклиды и известны значения
их радиоактивности. Обычно наиболее подходящим
методом для проверки радионуклидной чистоты гам¬
ма- и рентгеновских излучений, испускаемых радио¬
нуклидами, является гамма-спектрометрия. Приме¬
нение детекторов на основе натрия иодида может
привести к проблеме: пики для гамма-излучения, ис¬
пускаемого примесями, могут быть скрыты в спектре
основного радионуклида или быть неразрешенными
относительно пиков других радионуклидных приме¬
сей лекарственного средства. Примеси, испускающие
альфа- и бета-частицы и не испускающие гамма- и
рентгеновского излучения, не могут быть определены
с помощью данного метода. Для альфа- и бета-эмит-
теров должны применяться другие методы.
В отдельных частных статьях приводится тре¬
буемая радионуклидная чистота и могут быть уста¬
новлены предельные содержания для отдельных
радионуклидных примесей (например, молибден-99
в технеции-99м). Хотя эти требования необходимы,
они не являются достаточными, чтобы определять
годность лекарственного средства к медицинскому
применению. Производитель должен осуществлять
тщательную проверку лекарственного средства, осо¬
бенно содержащего радионуклиды с коротким перио¬
дом полураспада, на содержание примесей с долгим
периодом полураспада после соответствующего пе¬
риода распада. Таким образом, получают информа¬
цию о пригодности производственных процессов и
методик проведения испытания. В случаях когда два
или более радионуклидов, испускающих позитроны,
необходимо идентифицировать и/или различить, как,
например, 18Р-примеси в 1314-препаратах, определе¬
ние периода полураспада должно быть проведено до¬
полнительно к методу гамма-спектрометрии.
Из-за различий в периодах полураспада различ¬
ных радионуклидов, присутствующих в радиоактив¬
ных фармацевтических препаратах, радионуклидная
чистота изменяется со временем.
РАДИОХИМИЧЕСКАЯ ЧИСТОТА
Радиохимические примеси могут возникать при:
- производстве радионуклида;
- последующих химических процессах;
- неполном подготовительном разделении;
- химических изменениях в период хранения.
Для определения радиохимической чистоты не¬
обходимо провести разделение различных химиче¬
ских веществ, содержащих радионуклид, и определе¬
ние процентного содержания радиоактивности иссле¬
дуемого радионуклида, связанного с указанной хими¬
ческой формой. Раздел отдельной частной статьи по
радиохимической чистоте может содержать значения
предельных содержаний определяемых радиохими¬
ческих примесей, включая изомеры.
В принципе, при определении радиохимической
чистоты может быть использован любой метод анали¬
тического разделения. Например, частные статьи на
радиоактивные фармацевтические препараты могут
содержать методы бумажной хроматографии (2.2.26),
тонкослойной хроматографии (2.2.27), электрофоре¬
за (2.2.31), эксклюзионной хроматографии (2.2.30),
газовой хроматографии (2.2.28) и жидкостной хрома¬
тографии (2.2.29). Конкретное описание данных ана¬
литических методов приводится в частных статьях.
Кроме того, необходимо также принимать во внима¬
ние определенные меры предосторожности при ра¬
боте с радиофармацевтическими препаратами, такие
как защита от радиации, геометрия измерения, детек¬
тор линейности, использование переносчиков, разве¬
дение лекарственного средства.
Удельная радиоактивность
Удельная радиоактивность обычно рассчиты¬
вается, учитывая радиоактивную концентрацию и
концентрацию исследуемого химического вещества
после подтверждения того, что радиоактивность от¬
носится только к определяемым радионуклиду (ради¬
онуклидная чистота) и химическим элементам (радио¬
химическая чистота).
Удельная радиоактивность изменяется со време¬
нем, поэтому в отчете об удельной радиоактивности
должны указываться дата и, при необходимости, вре¬
мя определения.
Физиологическое распределение
По мере возможности необходимо избегать про¬
ведения испытаний, связанных с животными. Если
испытания по определению подлинности и радио¬
химической чистоты не подходят для завершенного
определения и контроля радиохимических элементов
радиоактивного фармацевтического препарата, не¬
обходимо провести испытание по показателю физио¬
логического распределения. Картина распределения
радиоактивности, наблюдаемая в определенных ор¬
ганах, тканях или других частях тела животного под¬
ходящего вида, может быть надежным индикатором
для установления пригодности для предназначенной
цели.
С другой стороны, испытание по показателю фи¬
зиологического распределения может способствовать
установлению биоэквивалентности лекарственного
средства при проведении испытаний аналогичных
лекарственных средств с известной клинической эф¬
фективностью.
В частных статьях приводится подробная инфор¬
мация, касающаяся проведения испытания и требо¬
ваний к физиологическому распределению, которые
должны выполняться.
В общем случае испытание выполняется следу¬
ющим образом.
Исследуемое лекарственное средство вводится
внутривенно каждому из трех животных. В некоторых
случаях может требоваться разведение непосред¬
ственно перед введением.
Незамедлительно после инъекции каждое жи¬
вотное помещается в отдельную клетку, условия со¬
держания в которой позволяют собирать выделения
и предотвращать загрязнение поверхности тела жи¬
вотного. По истечении определенного времени после
Радиоактивные фармацевтические препараты
151
«шъещии животные умерщвляются и расчленяются
соответствующим способом. В отобранных органах
и тканях исследуют радиоактивность. Физиологиче¬
ское распределение рассчитывают и выражают через
процентное содержание введенной радиоактивности,
обнаруженной в каждом из отобранных органов или
тканей, учитывая поправки на радиоактивный распад.
Для некоторых радиоактивных фармацевтических
препаратов требуется определение соотношения ра¬
диоактивности к массе образцов отобранных тканей
(радиоактивность/масса).
Радиоактивный фармацевтический препарат
должен выдерживать требования физиологического
распределения по крайней мере у двух из трех жи¬
вотных.
Не учитывают результаты, полученные для лю¬
бого животного, у которого присутствует кровопод¬
тек от инъекции (наблюдаемый во время инъекции
или выявляемый при последующем количественном
определении радиоактивности ткани). В этом случае
испытание необходимо повторить.
Стерильность
Радиоактивные фармацевтические препараты
должны выдерживать испытание на стерильность. Их
получение должно проводиться при соблюдении мер
предосторожности, предназначенных для исключения
микробиологического загрязнения и обеспечения сте¬
рильности. Испытание на стерильность выполняется,
как описано в общем методе (2.6.1). Особые трудно¬
сти возникают с радиоактивными фармацевтически¬
ми препаратами с коротким периодом полураспада
радионуклидов, малым размером серии и из-за ра¬
диационной опасности. В случае если частная статья
устанавливает, что лекарственное средство может
быть готово к выпуску перед завершением испытания
на стерильность, испытание на стерильность должно
быть начато как можно быстрее в связи с излучением.
При невозможности начать проведение испытания
незамедлительно образцы хранят в условиях, обе¬
спечивающих предотвращение получения ошибочных
отрицательных результатов. Параметрический выпуск
(5.1.1) лекарственного средства, произведенного в ре¬
зультате полностью валидированного процесса, явля¬
ется методом выбора в подобных случаях. Контроль
стерильности должен проводиться при контроле каче¬
ства лекарственного средства в случае асептического
производства.
Если размер выпускаемой серии радиоактивного
фармацевтического препарата ограничен одним или
несколькими образцами, отбор образцов для прове¬
дения испытания на стерильность в соответствие с
рекомендациями общего метода (2.6.1) может не про¬
водиться.
Если период полураспада радионуклида менее 5
мин, прием радиоактивного фармацевтического пре¬
парата пациентом обычно происходит в месте его
производства, осуществляемого в соответствии с ва-
лидированной системой.
Из соображений безопасности (высокий уровень
радиоактивности) невозможно исследование радио¬
активного фармацевтического препарата в количе¬
стве, необходимом для проведения испытания на
стерильность (2.6.1). Метод мембранной фильтрации
является предпочтительным для уменьшения воздей¬
ствия излучения на персонал.
Несмотря на требования по использованию
антимикробных консервантов, указанные в общей
статье Лекарственные средства для парентераль¬
ного применения (0520), их добавление в радиоак¬
тивные фармацевтические препараты, фасованные
в многодозовые контейнеры, не является обязатель¬
ным, если нет других указаний в частной статье.
Бактериальные эндотоксины — пирогены
Радиофармацевтические препараты для парен¬
терального применения должны выдерживать испы¬
тание на бактериальные эндотоксины (2.6.14) или
на пирогенность (2.6.8).
Элюаты генераторов радионуклидов, растворы
для введения радиоактивной метки и комплекты для
радиоактивных фармацевтических препаратов также
должны выдерживать испытание на бактериальные
эндотоксины, если они предназначены для приготов¬
ления радиофармацевтических препаратов для па¬
рентерального применения без дальнейшей очистки.
Радионуклидные прекурсоры и химические
прекурсоры должны выдерживать испытание на
бактериальные эндотоксины, если они предназна¬
чены для производства лекарственных средств для
парентерального применения без дальнейшей под¬
ходящей процедуры удаления бактериальных эндо¬
токсинов.
Испытание на бактериальные эндотоксины про¬
водится, как указано в общем методе (2.6.14), с при¬
менением необходимых мер предосторожности по
защите персонала, проводящего испытание, от облу¬
чения. Значения предельного содержания для бакте¬
риальных эндотоксинов указываются в определенной
частной статье или рассчитываются в соответствии с
общей статьей 5.1.10. Руководство по применению
испытания на бактериальные эндотоксины.
Если природа радиоактивного фармацевтиче¬
ского препарата или прекурсора приводит к интер¬
ференции в испытании на бактериальные эндоток¬
сины путем ингибирования или активации, а уда¬
ление интерферирующего фактора (факторов) не
представляется возможным, может быть отдельно
описано испытание на пирогенность (2.6.8).
ХРАНЕНИЕ
Лекарственные средства, содержащие радиоак¬
тивные вещества, хранят в воздухонепроницаемых
контейнерах, закрытых таким образом, чтобы обеспе¬
чить защиту персонала от облучения первичным или
вторичным излучением, в соответствии с националь¬
ными и международными правилами по хранению
радиоактивных веществ. Во время хранения контей¬
неры из-за облучения могут потемнеть, что не обяза¬
тельно означает ухудшение качества лекарственного
средства.
МАРКИРОВКА
Маркировка радиоактивных фармацевтических
препаратов должна соответствовать национальному
и европейскому законодательству.
Для лекарственных средств, приготовленных в
месте применения, маркировка может быть изменена.
Радиоактивность лекарственного средства уста¬
навливается в указанный день. Если период полурас¬
пада менее 70 дней, то также указывается и время со
ссылкой на временной период. Радиоактивность при
других значениях времени может рассчитываться из
уравнения распада или табличных данных.
В дополнение к вышеприведенному на этикетке
контейнера, упаковке, листке-вкладыше, прилагае¬
мом к упаковке, или в сертификате анализа, прила-
152
Гэсударственная фармакопея Республики Беларусь
гаемом к радиоактивному фармацевтическому препа¬
рату, указывают:
- способ применения;
- где применимо, максимальную рекомендуемую
дозу в миллилитрах;
- наименование и концентрацию любого добав¬
ленного антибактериального консерванта;
-где применимо, любые специальные условия
хранения.
Для химических прекурсоров прилагаемая до¬
кументация рекомендует проведение испытания
субстанции из одной или более партий перед ее при¬
менением для производства радиоактивных фарма¬
цевтических препаратов для гарантии того, что при
заданных условиях производства из данной субстан¬
ции получается радиоактивный фармацевтический
препарат в необходимом количестве и установлен¬
ного качества.
ОБНАРУЖЕНИЕ И ИЗМЕРЕНИЕ
РАДИОАКТИВНОСТИ
Обнаружение и измерение радиоактивности про¬
водится в соответствии с общей статьей 2.2.66. Обна¬
ружение и измерение радиоактивности.
Лекарственные средства для парентерального применения
153
ДОЗИРОВАННЫЕ
ЛЕКАРСТВЕННЫЕ ФОРМЫ
07/2016:0520
ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ
ПАРЕНТЕРАЛЬНОГО ПРИМЕНЕНИЯ
Рагеп1егаИа
Требования данной статьи не обязательно
должны распространяться на лекарственные сред¬
ства, изготовленные из человеческой крови, имму¬
нологические лекарственные средства или радио-
фармацевтические лекарственные средства.
ОПРЕДЕЛЕНИЕ
Лекарственные средства для парентерального
применения — это стерильные лекарственные сред¬
ства, предназначенные для введения путем инъек¬
ций, инфузий или имплантаций в организм человека.
Для приготовления лекарственных средств для па¬
рентерального применения могут использоваться вспо¬
могательные вещества, например, обеспечивающие
изотоничность лекарственных средств относительно
крови, регулирующие рН, улучшающие растворимость
действующих веществ, предотвращающие их разложе¬
ние, обеспечивающие соответствующие антимикробные
свойства. Эти вещества не должны отрицательно влиять
на основное действие лекарственного средства или в
используемых концентрациях вызывать токсичность или
нежелательное местное раздражающее действие.
Контейнеры для лекарственных средств для па¬
рентерального применения должны быть изготовлены
из материалов, которые как можно более прозрачны
и позволяют визуально просмотреть содержимое кон¬
тейнера, за исключением имплантатов и других обо¬
снованных и утвержденных случаев.
Если это применимо, контейнеры для лекар¬
ственных средств для парентерального применения
должны соответствовать требованиям статей разде¬
лов 3.1. Материалы, используемые для производ¬
ства контейнеров и 3.2. Контейнеры.
Лекарственные средства для парентерального
применения, предназначенные для длительного (хро¬
нического) применения или парентерального питания,
должны иметь соответствующие пределы содержания
для определенных компонентов или элементов с уче¬
том их долгосрочной токсичности.
Лекарственные средства для парентерального
применения выпускают в стеклянных (3.2.7) или, на¬
пример, в пластмассовых контейнерах (3.2.2, 3.2.2.7
и 3.2.9) и предварительно наполненных шприцах. Гер¬
метичность контейнера обеспечивают соответствую¬
щими способами. Укупорочные средства должны обе¬
спечивать надежную изоляцию, предотвращать до¬
ступ микроорганизмов и других загрязнений и обычно
позволяют извлекать часть или все содержимое кон¬
тейнера без удаления укупорочного средства. Пласти¬
ковые материалы или эластомеры (3.2.9), из которых
изготавливают укупорочные средства, должны быть
достаточно прочными и эластичными, чтобы при про¬
хождении иглы выделялось наименьшее количество
частиц. Укупорочные средства контейнеров, содержа¬
щих несколько доз лекарственного средства, должны
быть достаточно эластичными, чтобы обеспечивать
герметизацию контейнера при извлечении иглы.
Лекарственные средства для парентерального
применения можно классифицировать как:
- инъекции;
- инфузии;
- концентраты для приготовления инъекций и ин¬
фузий;
- порошки для приготовления инъекций и ин¬
фузий;
- гели для инъекций;
- имплантаты;
*- салфетки лекарственные#.
ПРОИЗВОДСТВО
При разработке лекарственных средств для па¬
рентерального применения, в состав которых входят
антимикробные консерванты, уполномоченному орга¬
ну представляют данные, подтверждающие необхо¬
димость использования и эффективность выбранных
консервантов. Подходящий метод определения и кри¬
терии оценки эффективности консервантов представ¬
лены в статье 5.7.3. Эффективность антимикроб¬
ных консервантов.
Лекарственные средства для парентерального
применения изготавливают с использованием мате¬
риалов и методов, обеспечивающих стерильность
и предотвращающих загрязнение лекарственных
средств и развитие микроорганизмов в соответствии
с требованиями статьи 5.7.7. Методы приготовления
стерильных продуктов.
Вода, используемая при производстве лекар¬
ственных средств для парентерального применения,
должна соответствовать требованиям, указанным в
частной статье Вода для инъекций (0169).
ИСПЫТАНИЯ
Загрязнение механическими включениями:
невидимые частицы (2.9.79). Лекарственные сред¬
ства, предназначенные для введения в организм чело¬
века и представляющие собой растворы для инфузий
и инъекций, должны выдерживать данное испытание.
В случае лекарственных средств, предназначен¬
ных для подкожного или внутримышечного введения,
могут быть применены более широкие допустимые
пределы. Данное испытание не распространяется на
радиоактивные фармацевтические лекарственные
средства. Данное испытание не распространяется
на лекарственное средство, на маркировке которого
указано, что оно используется с фильтром, если пока¬
зано, что фильтр обеспечивает получение раствора,
выдерживающего данное испытание.
Стерильность (2.6.7). Лекарственные средства
для парентерального применения должны выдержи¬
вать испытание на стерильность.
ХРАНЕНИЕ
Хранить в стерильном воздухонепроницаемом
контейнере с контролем первого вскрытия.
МАРКИРОВКА
На этикетке указывают:
- наименование и концентрацию всех добавлен¬
ных антимикробных консервантов;
- если необходимо, использование фильтра для
раствора;
-если необходимо, «не содержит бактериаль¬
ных эндотоксинов» или «апирогенно».
154
Гэсударственная фармакопея Республики Беларусь
Инъекционные лекарственные
средства
ОПРЕДЕЛЕНИЕ
Инъекционные лекарственные средства (инъ¬
екции) — это стерильные растворы, эмульсии или
суспензии. Их готовят путем растворения, эмуль¬
гирования или суспендирования действующего ве¬
щества или веществ и вспомогательных веществ в
воде, в подходящей неводной жидкости, которая,
если обосновано, может быть не стерильна, или в
смеси этих растворителей.
Растворы для инъекций в соответствующих ус¬
ловиях наблюдения должны быть прозрачными и
практически свободными от частиц.
Эмульсии для инъекций не должны обнаружи¬
вать признаков расслоения. В суспензиях для инъ¬
екций может наблюдаться осадок, который должен
быстро диспергироваться при взбалтывании, обра¬
зуя суспензию, достаточно стабильную, чтобы обе¬
спечить необходимую дозу при введении.
Многодозовые лекарственные средства.
Многодозовые водные инъекционные лекарствен¬
ные средства содержат соответствующий анти¬
микробный консервант в необходимой концентра¬
ции, за исключением лекарственных средств, об¬
ладающих соответствующими противомикробными
свойствами. При выпуске лекарственного средства
для парентерального применения в многодозовом
контейнере необходимо указывать меры предосто¬
рожности по его введению и, особенно, по хране¬
нию между отбором доз.
Антимикробные консерванты. Водные ле¬
карственные средства, которые готовят в асепти¬
ческих условиях и которые не могут быть подвер¬
гнуты термической стерилизации, могут содержать
подходящий антимикробный консервант в соответ¬
ствующей концентрации.
Антимикробные консерванты не применяют,
если:
- объем, вводимый в одноразовой дозе, пре¬
вышает 15 мл, если иное не обосновано;
- лекарственное средство предназначено для
введения такими способами, при которых по меди¬
цинским показателям не допускается присутствие
антимикробных консервантов, например для вну-
триполостного, эпидурального, интратекального
введения или другого способа введения, дающего
доступ к спинномозговой жидкости, или интра- или
ретробульбарного введения.
Такие лекарственные средства выпускают в
однодозовых контейнерах.
ПРОИЗВОДСТВО
При производстве инъекционных лекарственных
средств, содержащих дисперсные частицы, принима¬
ют меры, обеспечивающие необходимый размер ча¬
стиц и его контроль.
Однодозовые лекарственные средства. Объем
инъекционного лекарственного средства в однодозо-
вом контейнере должен быть достаточным для отбо¬
ра и введения номинальной дозы при использовании
обычного способа введения (2.9.17).
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые суспензии для инъекций должны
выдерживать испытание на однородность дози¬
рованных единиц (2.9.40) или, если это обосно¬
вано и утверждено, испытание на однородность
содержания, как указано ниже. На лекарственное
растительное сырье и продукты из лекарственно¬
го растительного сырья, представленные в дози¬
рованной форме, требования данного раздела не
распространяются.
Однородность содержания (2.9.6). Одно¬
дозовые суспензии для инъекций с содержанием
действующего вещества менее 2 мг или менее
2 % от общей массы содержимого должны выдер¬
живать испытание на однородность содержания
действующего вещества в единице дозированного
лекарственного средства (тест А), если в частной
статье нет других указаний. Если лекарственное
средство содержит более одного действующе¬
го вещества, это требование распространяется
только на те вещества, содержание которых соот¬
ветствует вышеуказанным условиям.
Бактериальные эндотоксины — пироген-
ность. Инъекционные лекарственные средства
должны выдерживать испытание на бактериаль¬
ные эндотоксины (2.6.14) или, если обосновано и
утверждено, испытание на пирогенность (2.6.8).
Рекомендации по предельно допустимым нормам
содержания бактериальных эндотоксинов приве¬
дены в статье 2.6.14.
Инфузионные лекарственные
средства
ОПРЕДЕЛЕНИЕ
Инфузионные лекарственные средства (ин¬
фузии) — это стерильные водные растворы или
эмульсии с водой в качестве дисперсионной сре¬
ды. Они обычно изотоничны с кровью. Инфузии
преимущественно предназначаются для приме¬
нения в больших объемах. Инфузии не содержат
антимикробных консервантов.
Растворы для инфузий оценивают в соот¬
ветствующих условиях наблюдения, при этом они
должны быть прозрачными и практически свобод¬
ными от частиц.
Эмульсии для инфузий не должны обнаружи¬
вать признаков расслоения.
ПРОИЗВОДСТВО
При производстве инфузий, содержащих дис¬
персные частицы, предпринимают меры, обеспе¬
чивающие необходимый размер частиц и его кон¬
троль.
Объем инфузий в однодозовом контейнере
должен быть достаточен для отбора и введения
номинальной дозы при использовании обычного
способа введения (2.9.17).
ИСПЫТАНИЯ
Бактериальные эндотоксины — пироген¬
ность. Инфузионные лекарственные средства
должны выдерживать испытание на бактериаль¬
ные эндотоксины (2.6.14) или, если обосновано и
утверждено, испытание на пирогенность (2.6.8).
При проведении испытания на пирогенность вво¬
дят 10 мл лекарственного средства на килограмм
веса кролика, если в частной статье нет других ука¬
заний.
Лекарственные средства для парентерального применения
155
Концентраты для приготовления
инъекционных или инфузионных
лекарственных средств
ОПРЕДЕЛЕНИЕ
Концентраты для приготовления инъекционных
лекарственных средств или инфузий представляют
собой стерильные растворы, предназначенные для
инъекций или инфузий после разведения. Концентра¬
ты перед применением разводят до указанного объ¬
ема соответствующей жидкостью. После разведения
полученный раствор должен соответствовать требо¬
ваниям, предъявляемым к инъекционным или инфу¬
зионным лекарственным средствам.
ИСПЫТАНИЯ
Бактериальные эндотоксины — пироген-
ность. Лекарственное средство должно выдерживать
требования, предъявляемые к инъекционным или ин¬
фузионным лекарственным средствам соответствен¬
но, после разведения до определенного объема.
Порошки для приготовления
инъекционных или инфузионных
лекарственных средств
ОПРЕДЕЛЕНИЕ
Порошки для приготовления инъекционных или
инфузионных лекарственных средств =— это твердые
стерильные вещества, помещенные в контейнеры,
которые при встряхивании с указанным объемом
соответствующей стерильной жидкости быстро об¬
разуют или прозрачный, практически свободный от
частиц раствор, или однородную суспензию. После
растворения или суспендирования они должны со¬
ответствовать требованиям, предъявляемым к ле¬
карственным средствам для инъекций или инфузий
соответственно.
Лиофилизированные лекарственные средства
для парентерального применения рассматривают как
порошки для приготовления инъекционных или инфу¬
зионных лекарственных средств.
ПРОИЗВОДСТВО
Однородность содержания и однородность мас¬
сы лиофилизированных лекарственных средств для
парентерального применения обеспечивают в ходе
внутрипроизводственного контроля за количеством
раствора перед лиофилизацией.
ИСПЫТАНИЯ
Однородность дозированных единиц. По¬
рошки для приготовления инъекционных или инфу¬
зионных растворов должны выдерживать испыта¬
ние на однородность дозированных единиц (2.9.40)
или, если это обосновано и утверждено, испытания
на однородность содержания и/или однородность
массы, как указано ниже. На лекарственное рас¬
тительное сырье и продукты из лекрственного рас¬
тительного сырья, представленные в дозированной
форме, требования данного раздела не распростра¬
няются.
Однородность содержания (2.9.6). Порошки
для приготовления инъекционных или инфузионных
растворов с содержанием действующего вещества
менее 2 мг или менее 2 % от общей массы содер¬
жимого или с массой дозированной единицы, равной
40 мг либо менее, должны выдерживать испытание
на однородность содержания действующего веще¬
ства в единице дозированного лекарственного сред¬
ства (тест А), если нет других указаний в частной ста¬
тье. Если лекарственное средство содержит более
одного действующего вещества, это требование рас¬
пространяется только на те вещества, содержание
которых соответствует вышеуказанным условиям.
Однородность массы (2.9.5). Порошки для
приготовления инъекционных или инфузионных рас¬
творов должны выдерживать испытание на одно¬
родность массы для единицы дозированного лекар¬
ственного средства. Испытание на однородность
массы не требуется, если испытание на однород¬
ность содержания предусмотрено для всех действу¬
ющих веществ.
Бактериальные эндотоксины — пироген-
ность. Лекарственное средство должно выдержи¬
вать требования, предъявляемые к инъекционным
или инфузионным лекарственным средствам соот¬
ветственно, после разведения или суспендирования
до определенного объема жидкости.
МАРКИРОВКА
На этикетке указывают способ приготовления ле¬
карственного средства для инъекций и инфузий.
Гели для инъекций
ОПРЕДЕЛЕНИЕ
Гели для инъекций — это стерильные гели с
вязкостью, обеспечивающей модифицированное вы-
свобождение действующего вещества или веществ в
месте инъекции.
Имплантаты
ОПРЕДЕЛЕНИЕ
Имплантаты представляют собой стерильные
твердые лекарственные средства, имеющие подхо¬
дящие для парентеральной имплантации размеры,
форму и высвобождение действующего вещества или
веществ в течение длительного периода времени.
Они упакованы в индивидуальные стерильные кон¬
тейнеры.
ИСПЫТАНИЯ
Проводят подходящие испытания для подтверж¬
дения соответствующего высвобождения действую¬
щего вещества (веществ).
#Салфетки лекарственные
ОПРЕДЕЛЕНИЕ
Салфетки — это стерильная лекарственная фор¬
ма, имеющая фиксированные геометрические разме¬
ры, предназначенная для наружного и имплантацион¬
ного применения.
ПРОИЗВОДСТВО
Салфетки получают путем сорбции лекарствен¬
ных веществ на полотне. Стерилизация салфеток
осуществляется у-лучами, что должно быть указано в
частной статье.
ХРАНЕНИЕ
Хранят в стерильном воздухонепроницаемом,
неповрежденном контейнере, если нет других указа¬
ний в частной статье.
156
Государственная фармакопея Республики Беларусь
Аденозин
157
ЧАСТНЫЕ ФАРМАКОПЕЙНЫЕ
СТАТЬИ НА СУБСТАНЦИИ
ДЛЯ ФАРМАЦЕВТИЧЕСКОГО
ИСПОЛЬЗОВАНИЯ
07/2016:1486
АДЕНОЗИН
Лёвпоекпжп
А0ЕМ081ЫЕ
С„Н13Н504 М.м. 267,2
[58-61-7]
ОПРЕДЕЛЕНИЕ
9-Р"0-Рибофуранозил-9Н-пурин-6-амин.
Содержание: не менее 99,0 % и не более 101,0 %
|в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, растворим в горячей
воде, практически нерастворим в 96 % спирте и мети¬
ленхлориде. Растворяется в разведенных минераль¬
ных кислотах.
Температура плавления: около 234 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО аденозина #или спектр, пред¬
ставленный на рисунке #1486.-1#.
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца суспенди¬
руют в 100 мл воды дистиллированной Р и нагревают
до кипения. Охлаждают, фильтруют под вакуумом и до¬
водят водой дистиллированной Р до объема 100 мл.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность и щелочность. К 10 мл раствора
3 прибавляют 0,1 мл раствора бромкрезолового пур¬
пурового Р и 0,1 мл 0,01 М раствора кислоты хло¬
ристоводородной. Появляется желтое окрашивание.
При прибавлении не более 0,4 мл 0,01 М раствора
натрия гидроксида должно появиться фиолетово-си¬
нее окрашивание.
Удельное оптическое вращение (2.2.7). От -45
до -49 (в пересчете на сухое вещество). 1,25 г испыту¬
емого образца растворяют в 1 М растворе кислоты
хлористоводородной и доводят до объема 50,0 мл
этим же растворителем. Полученный раствор исполь¬
зуют в течение 10 мин после приготовления.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. 6,8 г калия гидросульфа¬
та Р и 3,4 г тетрабутиламмония гидросульфата Р
растворяют в воде Р, доводят до рН 6,5 раствором
60 г/л калия гидроксида Р и доводят до объема
1000 мл этим же растворителем. Используют свеже¬
приготовленную смесь растворителей.
Испытуемый раствор. 20 мг испытуемого образ¬
ца растворяют в подвижной фазе и доводят до объ¬
ема 20 мл этим же растворителем.
Рисунок #1486.-1. Инфракрасный спектр ФСО аденозина
158
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг аденина Р (при¬
месь А) и 5 мг инозина Р (примесь О) растворяют в
подвижной фазе и доводят до объема 50 мл этим же
растворителем. 4 мл полученного раствора доводят
подвижной фазой до объема 100 мл.
Условия хроматографирования:
-колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: вода Р — смесь растворите¬
лей (40:60, об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания аденозина.
Относительное удерживание (по отношению к
аденозину, время удерживания — около 13 мин): при¬
месь А — около 0,3; примесь О — около 0,4.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
месей А и О.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 0,6; для примеси О — 1,4):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-примесь О (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси О, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и О, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
10 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
Раствор 3 должен выдерживать испытание на сульфаты.
Аммония соли (2.4.1, метод В). Не более
0,0010 % (10 ррт). 0,5 г испытуемого образца долж¬
ны выдерживать испытание на соли аммония. Эталон
готовят с использованием 5 мл эталонного раствора
аммония (1 ррт N14) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют, при не¬
обходимости слегка подогревая, в смеси из 20 мл ук¬
сусного ангидрида Р и 30 мл кислоты уксусной без¬
водной Р и титруют 0,1 М раствором кислоты хлор¬
ной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 26,72 мг С10Н13М5О4.
ПРИМЕСИ
Специфицированные примеси: А, 6.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимитиру¬
ется общим критерием приемлемости для других/неспе-
цифицированных примесей и/или общей статьей Суб¬
станции для фармацевтического использования (2034).
Вследствие этого нет необходимости идентифицировать
эти примеси для доказательства соответствия требова¬
ниям. См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования): Р, Н.
Р. 1 -р-0-Рибофуранозилпиримидин-2,4(1 Н,ЗН)-
дион (уридин).
С. 9-Р-0-Рибофуранозил-1,9-дигидро-6Н-пурин-6-
он (инозин).
Н. 2-Амино-9-р-0-рибофуранозил-1,9-дигидро-6Н-
пурин-6-он (гуанозин).
Адипиновая кислота
159
07/2016:1586
АДИПИНОВАЯ КИСЛОТА
ЛсШит асИрюит
А01Р1САСЮ
но2с^-^С02Н
С6Н10О4 М.м. 146,1
[124-04-9]
ОПРЕДЕЛЕНИЕ
Гександиовая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый кристаллический порошок.
Умеренно растворима в воде, растворима в кипя¬
щей воде, лелсо растворима в 96 % спирте и метано¬
ле, растворима в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14). От 151 °С до
154 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО адипиновой кислоты.
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют при нагревании в воде дистиллированной Р и
доводят до объема 50 мл этим же растворителем.
Охлаждают до образования кристаллов и филь¬
труют через стеклянный фильтр (ПОР 40) (2.1.2).
Фильтр промывают водой дистиллированной Р. Со¬
бирают фильтрат и промывные воды до получения
50 мл раствора.
Раствор 51. 1,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 31 должен
быть бесцветным.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,20 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 20 мг кислоты глута-
ровой Р растворяют в 1,0 мл испытуемого раствора и
доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц
5 мкм) с удельной площадью поверхности 350 м2/г и
размером пор 10 нм;
- температура: 30 °С;
- подвижная фаза: ацетонитрил Р — раствор
24,5 г/л кислоты фосфорной разведенной Р (3:97, об/об);
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 209 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания кислоты адипиновой.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 9,0 между пиками кисло¬
ты глутаровой и кислоты адипиновой.
Предельное содержание примесей:
-любая примесь (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площадь любого
пика, кроме основного, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2.5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Нитраты. Не более 0,0030 % (30 ррт). К 1 мл
раствора 3 прибавляют 2 мл раствора аммиака кон¬
центрированного Р, 0,5 мл раствора 10 г/л магния
сульфата Р, 1 мл раствора 10 г/л сульфаниламида Р
и доводят водой Р до объема 20 мл. Прибавляют
0,10 г порошка цинка Р и охлаждают в ледяной бане
в течение 30 мин, периодически встряхивая. Раствор
фильтруют. 10 мл фильтрата охлаждают в ледяной
бане, прибавляют 2,5 мл кислоты хлористоводород¬
ной Р1, 1 мл раствора 10 г/л нафтилэтилендиами-
на дигидрохлорида Р и выдерживают при комнатной
температуре. Через 15 мин окраска смеси должна
быть не интенсивнее окраски раствора сравнения,
приготовленного параллельно с использованием
1.5 мл эталонного раствора нитрата (2 ррт NО^ Р
вместо 1 мл раствора 3. Испытание недействительно,
если окраска контрольного раствора, приготовленно¬
го параллельно с использованием 1 мл воды Р вместо
1 мл раствора 3, интенсивнее окраски раствора 2 мг/л
калия перманганата Р.
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
3 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 5 должны выдерживать испытания на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,2 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Полностью расплавляют 1,0 г испытуемого образца
над пламенем газовой горелки и затем прокаливают
в пламени. После прокаливания, уменьшают пламя
160
Гэсударственная фармакопея Республики Беларусь
или полностью выключают горелку для предотвраще¬
ния кипения расплавленного прокаленного образца и
поддерживают такое пламя до полного обугливания.
Полученный остаток используют для определения со¬
держания сульфатной золы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
60,0 мг испытуемого образца растворяют в 50 мл
воды Р, прибавляют 0,2 мл раствора фенолфталеи¬
на Рп титруют 0,1 М раствором натрия гидроксида.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 7,31 мг С6Н10О4.
ПРИМЕСИ
/К
но2с^
A. К = СН2-С02Н: Кислота пентандиовая (кислота
глутаровая).
B. Р = С02Н: Кислота бутандиовая (кислота ян¬
тарная).
C. Р = [СН2]3-С02Н: Кислота гептандиовая (кисло¬
та пимелиновая).
07/2016:0254
АДРЕНАЛИНА ТАРТРАТ
АйгепаНт 1аг1газ
АОКЕЫАЫЫЕ ТАКТ К АТЕ
[51-42-3]
ОПРЕДЕЛЕНИЕ
(1/?)-1-(3,4-Дигидроксифенил)-2-(метиламино)
этанола гидро-(2Р,ЗР)-2,3-дигидроксибутандиоат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или серовато-белый кристаллический по¬
рошок.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 5 г испытуемого образца растворяют в 50 мл
раствора 5 г/л натрия метабисульфита Р и подще¬
лачивают раствором аммиака Р. Полученную смесь
выдерживают при комнатной температуре не менее
15 мин и фильтруют. Фильтрат используют для иден¬
тификации С. Полученный осадок трижды промывают
метанолом Р порциями по 10 мл и сушат при темпе¬
ратуре 80 °С. Удельное оптическое вращение (2.2.7)
остатка (адреналина основания) от -53,5 до -50,
определенное с использованием раствора 20,0 г/л в
0,5 М растворе кислоты хлористоводородной.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках; используют адренали¬
на основание, полученное в идентификации А.
Сравнение: адреналина основание, получен¬
ное как указано в идентификации А, из 50 мг ФСО
адреналина тартрата, растворенного в 5 мл раст¬
вора 5 г/л натрия метабисульфита Р. Полученную
смесь выдерживают при комнатной температуре не
менее 30 мин. Фильтруют через пористый стеклянный
фильтр (2.1.2).
C. 0,2 мл фильтрата, полученного в идентифика¬
ции А, дают реакцию (Ь) на тартраты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем. Раствор используют немедленно.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)5.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы готовят с защитой от
света.
Смесь растворителей А. 5,0 г калия дигидро¬
фосфата Р растворяют в воде для хроматогра¬
фии Р, в полученном растворе растворяют 2,6 г
натрия октансульфоната Р и доводят водой для
хроматографии Р до объема 1000 мл (для полно¬
го растворения компонентов раствор обычно пере¬
мешивают не менее 30 мин). Доводят до рН 2,8
кислотой фосфорной Р.
Смесь растворителей В. Ацетонитрил Р1 —
смесь растворителей А (130:870, об/об).
Испытуемый раствор. 75 мг испытуемого об¬
разца растворяют в 5 мл 0,1 М раствора кислоты
хлористоводородной и доводят смесью раствори¬
телей В до объема 50 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей В до объ¬
ема 100,0 мл. 1,0 мл полученного раствора дово¬
дят смесью растворителей В до объема 10,0 мл.
Раствор сравнения (Ь). 1,5 мг ФСО норадре¬
налина тартрата (примесь В) и 1,5 мг ФСО адре-
налона гидрохлорида (примесь С) растворяют в
смеси растворителей В, прибавляют 1,0 мл испы¬
туемого раствора и доводят смесью растворителей
В до объема 100,0 мл.
Раствор сравнения (с). Содержимое контей¬
нера с ФСО адреналина смеси примесей (примеси
Р и Е) растворяют в 0,1 мл 0,1 М раствора кисло¬
ты хлористоводородной и 0,9 мл смеси раствори¬
телей В.
Раствор сравнения (д). 7,5 мг ФСО адренали¬
на тартрата с примесью А растворяют в 0,5 мл
0,1 М раствора кислоты хлористоводородной и
доводят смесью растворителей В до объема 5,0 мл.
Холостой раствор. 0,1 М раствор кислоты
хлористоводородной — смесь растворителей В
(1:9, об/об).
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 50 °С;
Адреналина тартрат
161
- подвижная фаза :
- пшвижная фаза А: ацетонитрил Р1 —
смесь растворителей А (5:95, об/об);
- подвижная фаза В: ацетонитрил Р1 —
смесь растворителей А (45:55, об/об);
Вщат (мин)
Подвижная фаза А
Подвижная фаза В
(%, об/об)
(%, об/об)
0—15
92 —* 50
8 — 50
15—20
50 — 92
50 — 8
20—25
92
8
- скорость подвижной фазы: 2,0 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей О и Е, используя хроматограмму
раствора сравнения (с) и хроматограмму, прилага¬
емую к ФСО адреналина смеси примесей; иденти¬
фицируют пик примеси А, используя хроматограмму
раствора сравнения (б) и хроматограмму, прилагае¬
мую к ФСО адреналина тартрата с примесью А.
Относительное удерживание (по отношению к
адреналину, время удерживания — около 4 мин): при¬
месь В — около 0,8; примесь С — около 1,3; примесь
А — около 3,2; примесь О — около 3,3; примесь Е —
около 3,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками при¬
меси В и адреналина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси □ — 0,7; для примеси Е — 0,6):
- примесь А (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примеси В, С (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и С, не должны превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примеси О, Е (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям О и Е, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, О и Е, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,6 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 6-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме в течение 18 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микро6иологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р, при необходимости
слегка подогревая, и титруют 0,1 М раствором кисло¬
ты хлорной до появления синевато-зеленого окраши¬
вания, используя в качестве индикатора 0,1 мл раст¬
вора кристаллического фиолетового Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 33,33 мг С13Н19М09.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере или пред¬
почтительнее в запаянной ампуле под вакуумом или
в среде инертного газа, в защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере или
предпочтительнее в запаянной ампуле под вакуу¬
мом или в среде инертного газа, в защищенном от
света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
А. Структура не установлена.
В. (1 Я)-2-Амино-1 -(3,4-дигидроксифенил)этанол
(норадреналин).
о
С. 1-(3,4-Дигидроксифекил)-2-(метиламино)эта-
нон (адреналон).
й. 4-[(1 Я)-2-(Бензилметиламино)-1-гидроксиэтил]-
бензол-1,2-диол.
Е. 2-(Бензилметиламино)-1 -(3,4-дигидроксифенил)-
этанон.
21. Зак. 1060.
162
Гэсударственная фармакопея Республики Беларусь
07/2016:1649
АЗИТРОМИЦИН
АгНЬготус'тит
АНТНКОМУСШ
С38Н72Ы2012 ■ хН20 М.м. 749 (безводное вещество)
х = 1 или 2
Азитромицин моногидрат: [121470-24-4]
Азитромицин дигидрат: [117772-70-0]
ОПРЕДЕЛЕНИЕ
(2Я,35,4Я,5Я,8Я,10Я,11Я,125,135,14Я)-13-
[(2,6-Дидезокси-3-С-метил-3-0-метил-а-1--рибо-
гексопиранозил)окси]-2-этил-3,4,10-тригидрокси-
3,5,6,8,10,12,14-гептаметил-11 -[[3,4,6-тридезокси-3-
(диметиламино)-Р-0-кс1/ло-гексопиранозил]окси]-1-
окса-6-азациклопентадекан-15-он. Степень гидрата¬
ции — 1 или 2.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в этаноле безводном и метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО азитромицина #или спектр,
представленный на рисунке #1649.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то готовят
растворы 90 г/л испытуемого образца и ФСО азитро¬
мицина в метиленхлориде Р, которые используют
для получения новых спектров.
ИСПЫТАНИЯ
Раствор 3. 0,500 г испытуемого образца раст¬
воряют в этаноле безводном Р и доводят до объема
50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 9,0 до 11,0.0,100 г испытуемого образ¬
ца растворяют в 25,0 мл метанола Р и доводят водой,
свободной от углерода диоксида, Р до объема 50,0 мл.
Удельное оптическое вращение (2.2.7). От -45
до -49 (в пересчете на безводное вещество). Опреде¬
ление проводят с использованием раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Раствор 1,73 г/л аммо¬
ния дигидрофосфата Р доводят до рН 10,0 раство¬
ром аммиака Р. 350 мл полученного раствора пере¬
носят в соответствующий контейнер и прибавляют
300 мл ацетонитрила Р1 и 350 мл метанола Р1 и
тщательно перемешивают.
Рисунок #1649.-1. Инфракрасный спектр ФСО азитромицина
"1111111
Азитромицин
163
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл.
Раствор сравнения (Ь). Содержимое контейне¬
ра с ФСО азитромицина для проверки пригодности
хроматографической системы (содержит примеси Р,
Н и ^ растворяют в 1,0 мл смеси растворителей и об¬
рабатывают ультразвуком в течение 5 мин.
Раствор сравнения (с). 8,0 мг ФСО азитромици¬
на для идентификации пиков (содержит примеси А,
В, С, Е, Р, О, I, 1_, М, N. О и Р) растворяют в 1,0 мл
смеси растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная кремнийорганическим по¬
лимером аморфным октадецилсилильным эндкепи-
рованным для масс-спектрометрии Р с размером
частиц 5 мкм;
- температура: 60 °С;
- подвижная фаза:
- подвижная фаза А: раствор 1,80 г/л динатрия
гидрофосфата безводного Р, доведенный до рН
8,9 кислотой фосфорной разведенной Р или рас¬
твором натрия гидроксида разведенным Р\
- подвижная фаза В: метанол Р1 — ацетони¬
трил Р1 (250:750, об/об)\
Время (мин)
Подвижная фаза А |
(%, об/об)
: Подвижная фаза В
(%.об*>б)
0-25
50 — 45 '
50 — 55
25—30
45 — 40
55 — 60
30—80
40 — 25
60 — 75
80—81
25 — 50
75 — 50
81—93 !
50
50
- опросив» подвижной фазы. 1,0 мл/мин;
- сы&тщххратометрический детектор, длина
волны 2*0 нм:
- объем вводимой пробы: 50 мкл.
Идентификация пиков примесей: идентифициру¬
ют пики примесей А, В, С, Е, Р, С, I, 3, Ц М, Ы, О и Р, ис¬
пользуя хроматограмму, прилагаемую к ФСО азитро¬
мицина для идентификации пиков, и хроматограмму
раствора сравнения (с); идентифицируют пик примеси
Н, используя хроматограмму, прилагаемую к ФСО ази¬
тромицина для проверки пригодности хроматогра¬
фической системы, и хроматограмму раствора срав¬
нения (Ь).
Относительное удерживание (по отношению к
азитромицину, время удерживания — (45—50) мин):
примесь 1_ — около 0,29; примесь М — около 0,37;
примесь Е — около 0,43; примесь Р — около 0,51; при¬
месь й — около 0,54; примесь ^ — около 0,54; при¬
месь I — около 0,61; примесь С — около 0,73; примесь
N — около 0,76; примесь Н — около 0,79; примесь
А— около 0,83; примесь Р — около 0,92; примесь
О— около 1,23; примесь О — около 1,26; примесь
В — около 1,31.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,4
(Нр — высота пика примеси 3 относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Л и
примеси Р).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Р — 0,3; для примеси О — 0,2; для примеси
Н — 0,1; для примеси I. — 2,3; для примеси М — 0,6;
для примеси N — 0,7):
- примесь В (не более 2,0 %): на хроматограм¬
ме испытуемого раствора площадь пика примеси В
не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- примеси А, О, Е, Р, Н, I, М, /V, О, Р (не более
0. 5 %): на хроматограмме испытуемого раствора пло¬
щади пиков, соответствующих примесям А, С, Е, Р, Н,
1, М, 14, О и Р, не должны превышать 0,5 площади ос¬
новного пика на хроматограмме раствора сравнения (а);
- сумма примесей О и 3 (не более 0,5 %): на
хроматограмме испытуемого раствора сумма площа¬
дей пиков примесей й и 3 не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (а);
- примесь О (не более 0,2 %): на хроматограм¬
ме испытуемого раствора площадь пика примеси О
не должна превышать 0,2 площади основного пика на
хроматограмме раствора сравнения (а);
- любая другая примесь (не более 0,2 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С, Р, Е,
Р, С, Н, I, 3, Ц М, N. О и Р, не должна превышать 0,2
площади основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 3,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,1 площади основного пика на хроматограм¬
ме раствора сравнения (а) (0,1 %) и пики, выходящие
перед пиком примеси Ь и после пика примеси В.
Тяжелые металлы (2.4.8, метод В). Не более
0,0025 % (25 ррт). 2,0 г испытуемого образца раство¬
ряют в смеси вода Р — этанол безводный Р (15:85,
об/об) и доводят до объема 20 мл этой же смесью
растворителей. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (2,5 ррт РЬ), полученного путем разведения
эталонного раствора свинца (100 ррт РЬ) Р смесью
вода Р — этанол Р (15:85, об/об).
Вода (2.5.12). Не менее 1,8% и не более 6,5%.
Определение проводят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Раствор А. Смешивают 60 объемов ацетони¬
трила Р1 и 40 объемов раствора 6,7 г/л дикалия
гидрофосфата Р, доведенного до рН 8,0 кислотой
фосфорной Р.
Испытуемый раствор. 53,0 мг испытуемого об¬
разца растворяют в 2 мл ацетонитрила Р1 и доводят
раствором А до объема 100,0 мл.
21*. Зак. 1060.
164
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (а). 53,0 мг ФСО азитроми-
цина растворяют в 2 мл ацетонитрила Р1 и доводят
раствором А до объема 100,0 мл.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 5 мг ФСО азитромицина примеси А растворяют
в 0,5 мл ацетонитрила Р1 и доводят раствором А до
объема 10 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная винилполимером октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 60 объемов аце-
тонитрила Р1 и 40 объемов раствора 6,7 г/л дикалия
гидрофосфата Р, доведенного до рН 11,0 раствором
560 г/л калия гидроксида Р;
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания азитромицина.
Время удерживания: азитромицин — около 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками при¬
меси А и азитромицина.
Содержание С38Н72М2012 рассчитывают в процен¬
тах с учетом содержания азитромицина в ФСО ази¬
тромицина.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят микробиологическим ме¬
тодом (2.7.2, метод А).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, О,
Н, /, Л, Ц М, /V, О, Р
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): К.
А. Р1 = ОН, Р2 = Р6 = Н, РЗ = Р4 = Р5 = СН3: 6-Де-
метилазитромицин.
B. Р1 = Р6 = Н, Р2 = РЗ = Р4 = Р5 = СН3: З-Дезок-
сиазитромицин (азитромицин В).
C. Р1 = ОН, Р2 = РЗ = Р5 = СН3, Р4 = Р6 = Н:
3"-0-Деметилазитромицин (азитромицин С).
й. Р1 = ОН, Р2 = РЗ = Р4 = СН3, Р5 = СН2ОН,
Р6 = Н: 14-Деметил-14-(гидроксиметил)азитромицин
(азитромицин Р).
Р. Р1 = ОН, Р2 = Р4 = Р5 = СН3, РЗ = СНО, Р6 = Н:
3'-Л/-Деметил-3'-/У-формилазитромицин.
I. Р1 = ОН, Р2 = Р4 = Р5 = СН3, РЗ = Р6 = Н:
3'-А/-Деметилазитромицин.
О. Р1 = ОН, Р2 = РЗ = Р4 = Р5 = Р6 = СН3:
2-Дезэтил-2-пропилазитромицин.
он
Е. 3'-(А/,Л/-Дидеметил)азитромицин (аминоазитро-
мицин).
К1 = кладинозил
О. 3'-А/-Деметил-3'-Л/-[(4-метилфенил)сульфонил]
азитромицин.
ГС1 = кладинозил
Н. 3'-А/-[[4-(Ацетиламино)фенил]сульфонил]-3'-А/-
деметилазитромицин.
К1 =Н
ОН
3. 13-О-Декладинозилазитромицин.
Р1 = кладинозил
Р2 = Н,С / .О
он
I.. Азитромицина 3'-Л/-оксид.
к
II
Азота закись
165
К1 = кладинозил
ОН
М. 3'-(А/,А/-Дидеметил)-3,-Л/“формилазитромицин.
Р1 = кладинозил
N. 3'-Де(диметиламино)-3'-оксоазитромицин.
К. С'4,1 "-Эпсжеиазитромицин (азитромицин Е).
Р. Структура не установлена.
07/2016:0416
АЗОТА ЗАКИСЬ
Отйтдет оххкмп
мгтоиз охюе
ЫгО М.м. 44,01
[10024-97-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % (об/об) М20 (азота
(I) оксид) в газовой фазе при температуре 15 °С.
Данная частная статья распространяется на азо¬
та закись для медицинского применения.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветный газ.
Растворимость: при температуре 20 °С и при
давлении 101 кПа 1 объем закиси азота растворяется
примерно в 1,5 объемах воды.
ПРОИЗВОДСТВО
Азота закись получают термическим разложени¬
ем аммония нитрата.
Проводят испытания газовой фазы.
Если проводят испытание баллона, баллон вы¬
держивают при комнатной температуре в течение
не менее 6 ч перед проведением испытаний. Баллон
держат в вертикальном положении таким образом,
чтобы вентиль был расположен сверху.
Углерода диоксид. Газовая хроматография
(2.2.28).
Испытуемый газ. Испытуемый образец.
Гэз сравнения. Смесь, содержащая 300 ррт
(об/об) углерода диоксида Р1 в азота закиси Р.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 3,5 м и
диаметром 2 мм, заполненная сополимером этилви-
нилбензол-дивинилбензола Р\
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 15 мл/мин;
- температура колонки: 40 °С;
- температура детектора: 90 °С;
-- детектор по теплопроводности;
- вводимая проба: через петлевой инжектор.
Условия работы прибора и объем вводимой про¬
бы регулируют таким образом, чтобы высота пика
углерода диоксида на хроматограмме газа сравнения
составляла не менее 35 % от полной шкалы регистри¬
рующего устройства.
Пригодность хроматографической системы:
- пики углерода диоксида и азота закиси на хро¬
матограммах полностью разделяются.
Предельное содержание:
- углерода диоксид (не более 300 ррт (об/об)):
на хроматограмме испытуемого газа площадь пика,
соответствующего углерода диоксиду, не должна пре¬
вышать площадь основного пика на хроматограмме
газа сравнения.
Углерода монооксид. Газовая хроматография
(2.2.28). При проведении испытания газа в баллоне
используют первую порцию отбираемого газа.
Испытуемый газ. Испытуемый образец.
Гзз сравнения. Смесь, содержащая 5 ррт (об/об)
углерода монооксида Р в азота закиси Р.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 2 м и
диаметром 4 мм, заполненная подходящим молеку¬
лярным ситом для хроматографии (0,5 нм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 60 мл/мин;
- температура колонки: 50 °С;
- температура детектора: 130 °С;
- детектор: пламенно-ионизационный детектор
с метанизатором;
- вводимая проба: через петлевой инжектор.
Условия работы прибора и объем вводимой про¬
бы регулируют таким образом, чтобы высота пика
углерода монооксида на хроматограмме газа сравне¬
ния составляла не менее 35 % от полной шкалы реги¬
стрирующего устройства.
Предельное содержание:
- углерода монооксид (не более 5 ррт (об/об)):
на хроматограмме испытуемого газа площадь пика,
соответствующего углерода монооксиду, не должна
превышать площадь основного пика на хроматограм¬
ме газа сравнения.
Азота монооксид и азота диоксид. Не более
2 ррт (об/об) суммарно в газовой и жидкой фазах. Хе¬
милюминесцентный анализ (2.5.26).
Испытуемый газ. Испытуемый образец.
Гэз сравнения (а). Азота закись Р.
Гэз сравнения (Ь). Смесь, содержащая 2 ррт
(об/об) азота монооксида Р в азоте Р1.
Анализаторы калибруют и настраивают чувстви¬
тельность с помощью газов сравнения (а) и (Ь). Изме¬
ряют содержание азота монооксида и азота диоксида
при раздельном исследований газовой и жидкой фаз
испытуемого газа.
Полученный результат умножают на поправоч¬
ный фактор для учета гашения отклика анализато¬
ра, вызываемого матричным эффектом азота заки-
22. Зак. 1060.
166
Гэсударственная фармакопея Республики Беларусь
си. Поправочный фактор определяют на известной
стандартной смеси азота монооксида в азота закиси
и сравнивают фактическое содержание с показания¬
ми анализатора, откалиброванного при помощи стан¬
дартной смеси МО/1М2:
_ „ . Фактическое содержание (азота монооксид)
Поправочный фактор = 5 .
Показания анализатора (азота монооксид)
Вода. Не более 67 ррт (об/об). Определение
проводят на электролитическом гигрометре (2.5.28).
Количественное определение. Инфракрасный
анализатор (2.5.35).
Испытуемый газ. Испытуемый образец фильтру¬
ют, чтобы избежать светорассеяния.
Газ сравнения (а). Азота закись Р.
Газ сравнения (Ь). Смесь, состоящая из 5,0 %
(об/об) азота Р1 и 95,0 % (об/об) азота закиси Р.
Анализатор калибруют и настраивают чувстви¬
тельность при помощи газов сравнения (а) и (Ь). Из¬
меряют содержание азота закиси в испытуемом газе.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
B. В испытуемый газ вносят тлеющую деревян¬
ную лучину. Лучина должна загореться.
C. Испытуемый газ пропускают через раствор
пирогаллола щелочной Р. Не должно появляться ко¬
ричневое окрашивание.
ИСПЫТАНИЯ
Проводят испытания газовой фазы.
Если проводят испытание баллона, баллон вы¬
держивают при комнатной температуре в течение
не менее 6 ч перед проведением испытаний. Баллон
держат в вертикальном положении таким образом,
чтобы вентиль был расположен сверху.
Углерода диоксид (2.1.6). Не более 300 ррт
(об/об). Определение проводят с помощью индика¬
торной трубки для углерода диоксида.
Углерода монооксид (2.1.6). Не более 5 ррт
(об/об). Определение проводят с помощью индика¬
торной трубки для углерода монооксида. При прове¬
дении испытания с использованием баллона исполь¬
зуют первую порцию отбираемого газа.
Азота монооксид и азота диоксид (2.1.6). Не
более 2 ррт (об/об). Определение проводят с помо¬
щью индикаторной трубки для азота монооксида и
азота диоксида.
Пары воды (2.1.6). Не более 67 ррт (об/об).
Определение проводят с помощью индикаторной
трубки для паров воды.
ХРАНЕНИЕ
В виде сжиженного газа под давлением в контей¬
нерах, соответствующих установленным требовани¬
ям. Клапаны и вентили не должны подвергаться об¬
работке маслами и смазочными материалами.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
A. С02: углерода диоксид.
B. СО: углерода монооксид.
C. N0: азота монооксид.
0.1\Ю2: азота диоксид.
Е. Н20: вода.
07/2016:1549
АЗОТНАЯ КИСЛОТА
АсШит пНпсит
МТМСАСЮ
НЫ03 М.м. 63,0
[7697-37-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 68,0 % (м/м) и не более
70,0 % (м/м).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или почти бесцветная
жидкость.
Смешивается с водой.
Относительная плотность: около 1,41.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 мл испытуемого образца доводят водой Р до
объема 100 мл. Полученный раствор имеет сильно¬
кислую реакцию (2.2.4).
B. Раствор, полученный в испытании А, дает ре¬
акцию (а) на нитраты.
ИСПЫТАНИЯ
Раствор 5. 2 мл испытуемого образца доводят
водой Р до объема 10 мл.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)6.
Хлориды (2.4.4). Не более 0,00005 % (0,5 ррт).
К 5 г испытуемого образца прибавляют 10 мл
воды Р, 0,3 мл раствора серебра нитрата Р2 и
выдерживают в защищенном от света месте в те¬
чение 2 мин. Опалесценция полученного раствора
не должна превышать опалесценцию раствора
сравнения, приготовленного аналогично с исполь¬
зованием 13 мл воды Р, 0,5 мл азотной кислоты Р,
0,5 мл эталонного раствора хлорида (5 ррт С1) Р
и 0,3 мл раствора серебра нитрата Р2.
Сульфаты (2.4.13). Не более 0,0010 % (10 ррт).
К 15 г испытуемого образца добавляют 0,2 г
натрия карбоната Р. После испарения углерода
диоксида выпаривают досуха. Остаток растворяют
в 15 мл воды дистиллированной Р. Полученный
раствор должен выдерживать испытание на суль¬
фаты.
Железо (2.4.9). Не более 0,0010 % (10 ррт).
Остаток, полученный при проведении испыта¬
ния «Сульфатная зола», растворяют в 1 мл хлори¬
стоводородной кислоты разведенной Р и доводят
до объема 20 мл водой Р. 1 мл полученного раст¬
вора доводят водой Рдо объема 10 мл. Полученный
раствор должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0004 % (4 ррт).
10,0 г испытуемого образца осторожно выпари¬
вают на водяной бане досуха. Остаток смачивают не¬
сколькими каплями хлористоводородной кислоты
разведенной Р и доводят до объема 20 мл водой Р.
12 мл полученного раствора должны выдерживать
испытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Сульфатная зола. Не более 0,01 %.
Албендазол
167
20,00 г испытуемого образца осторожно выпари¬
вают досуха. Остаток смачивают несколькими капля¬
ми серной кислоты Р и сжигают до красного каления.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,750 г испытуемого образца прибавляют 50 мл
воды Р и титруют 1 М раствором натрия гидроксида
потенциометрически (2.2.20).
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 63,0 мг НМ03.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1386
АЛБЕНДАЗОЛ
А1Ьепс1а2о1ит
А1.ВЕЫ0А201.Е
С12Н15Ыз025 М.м. 265,3
[54965-21-8]
ОПРЕДЕЛЕНИЕ
Метил[5-(пропилсульфанил)-1/-/-бензимидазол-
2-ил]карбамат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок.
Практически нерастворим в воде, легко раство¬
рим в кислоте муравьиной безводной, очень мало
растворим в метиленхлориде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО албендазола.
ИСПЫТАНИЯ
Раствор 5. 0,10 г испытуемого образца раство¬
ряют в смеси из кислоты муравьиной безводной Р и
метиленхлориде Р (1:9, об/об) и доводят до объема
10 мл этой же смесью растворителей.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 8
должна быть не интенсивнее эталона ВУ(КЖ)6.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в 5 мл метанола Р, содержащего
1 % (об/об) кислоты серной Р, и доводят подвижной
фазой до объема 50,0 мл.
Раствор сравнения (а). 10,0 мг испытуемого об¬
разца растворяют в 10 мл метанола Р, содержащего
1 % (об/об) кислоты серной Р, и доводят подвижной
фазой до объема 100,0 мл. 0,5 мл полученного раст¬
вора доводят подвижной фазой до объема 20,0 мл.
Раствор сравнения (Ь). 50,0 мг испытуемого
раствора и 50 мг ФСО оксибендазола растворяют в
5 мл метанола Р, содержащего 1 % (об/об) кисло¬
ты серной Р, и доводят подвижной фазой до объема
100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р (размер частиц 5 мкм) с размером пор
10 нм и содержанием углерода 19 %;
- подвижная фаза: раствор 1,67 г/л аммония ди¬
гидрофосфата Р — метанол Р (300:700, об/об);
- скорость подвижной фазы: 0,7 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания албендазола.
Относительное удерживание (по отношению к
албендазолу): примесь О — около 0,40; примеси В и
С — около 0,43; примесь Е — около 0,47; примесь Р —
около 0,57; примесь А — около 0,80.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 3,0 между пиками ал¬
бендазола и оксибендазола.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р (не более 0,75 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, О, Е, Р, не
должны превышать 1,5-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Во избежание перегревания необходимо тща¬
тельно перемешивать раствор на протяжении
всего титрования и прекратить титрование не¬
замедлительно после достижения конечной точки
титрования.
0,250 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 40 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 26,53 мг С12Н15М3028.
ХРАНЕНИЕ
В защищенном от света месте.
22*. Зак. 1060.
168
Гэсударственная фармакопея Республики Беларусь
ПРИМЕСИ
ОПРЕДЕЛЕНИЕ
Специфицированные примеси: А, В, С, О, Е, Р.
А. В = 3-СН2-СН2-СН3: 5-(Пропилсульфанил)-1Н-
бензимидазол-2-амин.
О. Р = 302-СН2-СН2-СН3: 5-(Пропилсульфонил)-
1 /-/-бензимидазол-2-амин.
B. Р = 30-СН2-СН2-СН3: Метил[5-(пропилсульфи-
нил)-1Н-бензимидазол-2-ил]карбамат.
C. Р = 302-СН2-СН2-СН3: Метил[5-(пропилсульфо-
нил)-1Ж5ензимидазол-2-ил]карбамат.
Е. Р = Н: Метил(1/-/-бензимидазол-2-ил)карбамат.
Р. Р = 3-СН3: Метил[5-(метилсульфанил)-1Н-
бензимидазол-2-ил]карбамат.
07/2016:1288
АЛЛАНТОИН
АНапШпит
АИ.АЫТОШ
и энантиомер
С4нбы403 М.м. 158,1
[97-59-6]
(Р5)-(2,5-Диоксоимидазолидин-4-ил)мочевина.
Содержание: не менее 98,5 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Мало растворим в воде, очень мало растворим в
96 % спирте.
Температура плавления: около 225 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО аллантоина #или спектр, пред¬
ставленный на рисунке #1288.-1#.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси». На хрома¬
тограмме испытуемого раствора (Ь) обнаруживается
основное пятно, соответствующее по расположению,
цвету и размеру основному пятну на хроматограмме
раствора сравнения (а).
C. 20 мг испытуемого образца кипятят в смеси из
1 мл раствора натрия гидроксида разведенного Р и
1 мл воды Р. Охлаждают, прибавляют 1 мл кислоты
хлористоводородной разведенной Р. К 0,1 мл полу¬
ченного раствора прибавляют 0,1 мл раствора 100 г/л
калия бромида Р, 0,1мл раствора 20 г/л резорци¬
на Р и 3 мл кислоты серной Р. Нагревают в течение
(5—10) мин на водяной бане. Появляется темно-си¬
нее окрашивание, переходящее в красное после ох¬
лаждения и прибавления около 10 мл воды Р.
й. Нагревают около 0,5 г испытуемого образца.
Выделяется аммиак, который обнаруживают по сине¬
му окрашиванию красной лакмусовой бумаги Р.
Рисунок #1288.-1. Инфракрасный спектр ФСО аллантоина
Алюминия оксид гидратированный
169
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р,
нагревая при необходимости, и доводят до объема
100 мл этим же растворителем.
Кислотность или щелочность. К 5 мл раствора
3 прибавляют 5 мл воды, свободной от углерода ди¬
оксида, Р, 0,1 мл раствора метилового красного Р и
0,2 мл 0,01 М раствора натрия гидроксида. Должно
появиться желтое окрашивание. При прибавлении не
более 0,4 мл 0,01 М раствора кислоты хлористо¬
водородной окраска раствора должна измениться на
красную.
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. Измеряют угол оптического вращения раст¬
вора 3.
Восстанавливающие вещества. 1,0 г испытуе¬
мого образца встряхивают с 10 мл воды Р в течение
2 мин и фильтруют. К фильтрату прибавляют 1,5 мл
0,02 М раствора калия перманганата. Окраска раст¬
вора должна оставаться фиолетовой в течение не ме¬
нее 10 мин.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого образ¬
ца растворяют в 5,0 мл воды Р при нагревании. Охлажда¬
ют и доводят метанолом Р до объема 10 мл. Раствор
используют немедленно после приготовления.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят смесью из метанола Р и воды Р
(1:1, об/об) до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО аллантои-
на растворяют в смеси из метанола Р и воды Р (1:1,
об/об) и доводят до объема 10 мл этой же смесью
растворителей.
Раствор сравнения (Ь). 10 мг мочевины Р раст¬
воряют в 10 мл воды Р. 1 мл полученного раствора
доводят метанолом Р до объема 10 мл.
Раствор сравнения (с). 1 мл раствора сравнения
(а) смешивают с 1 мл раствора сравнения (Ь).
Пластинка: ТСХ пластинка со слоем подходя¬
щей целлюлозы для хроматографии Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— бутанол Р (15:25:60, об/об/об).
Наносимый объем пробы: Юмкл испытуемого
раствора (а); по 5 мкл испытуемого раствора (Ь) и рас¬
творов сравнения (а), (Ь) и (с).
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром 5 г/л диметиламинобензальдегида Р в смеси из
1 объема кислоты хлористоводородной Р и 3 объемов
метанола Р и сушат в потоке горячего воздуха. Через
30 мин пластинку просматривают при дневном свете.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5%): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,1 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
120,0 мг испытуемого образца растворяют в
40 мл воды Р и титруют 0,1 М раствором натрия ги¬
дроксида потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 15,81 мгС4Нб1Ч403.
ПРИМЕСИ
н..со2н
Т
о
А. Глиоксиловая кислота.
В. Карбамид (мочевина).
07/2016:0311
АЛЮМИНИЯ ОКСИД
ГИДРАТИРОВАННЫЙ
А1иттН ох1с1ит Ьус1псит
АШМ1ЫШМ ОХЮЕ, НУОНАТЕО
ОПРЕДЕЛЕНИЕ
Содержание: не менее 47,0 % и не более 60,0 %
А120 ъ(М.м. 102,0).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый аморфный порошок.
Практически нерастворим в воде. Растворяется
в разведенных минеральных кислотах и растворах ги¬
дроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Раствор 3, приготовленный как указано в разде¬
ле «Испытания», дает реакцию на алюминий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в 15 мл кислоты хлористоводородной Р при
нагревании на водяной бане и доводят водой дистил¬
лированной Р до объема 100 мл.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона <ЗУ(ЗЖ)6.
Щелочные примеси. 1,0 г испытуемого образ¬
ца встряхивают с 20 мл воды, свободной от углеро¬
да диоксида, Р в течение 1 мин и фильтруют. К 10 мл
фильтрата прибавляют 0,1 мл раствора фенолфта¬
леина Р. После прибавления не более 0,3 мл 0,1 М
раствора кислоты хлористоводородной не должно
наблюдаться розовое окрашивание.
Нейтрализующая способность. Испытание
проводят при температуре 37 °С. 0,5 г испытуемо¬
го образца диспергируют в 100 мл воды Р, нагрева¬
ют и прибавляют при непрерывном перемешивании
100,0 мл предварительно подогретого 0,1 Мраствора
23. Зак. 1060.
170
Гэсударственная фармакопея Республики Беларусь
кислоты хлористоводородной. Значения рН (2.2.3)
полученного раствора, измеренные через 10 мин,
15 мин и 20 мин, должны быть не менее 1,8, 2,3 и
3.0 соответственно и не должны превышать 4,5. При¬
бавляют 10,0 мл предварительно подогретого 0,5 М
раствора кислоты хлористоводородной, непре¬
рывно перемешивают в течение 1 ч и титруют 0,1 М
раствором натрия гидроксида до значения рН 3,5.
На титрование должно быть израсходовано не более
35.0 мл 0,1 М раствора натрия гидроксида.
Хлориды (2.4.4). Не более 1 %. 0,1 г испытуемо¬
го образца растворяют при нагревании в 10 мл кис¬
лоты азотной разведенной Р и доводят водой Р до
объема 100 мл. 5 мл полученного раствора доводят
водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 1 %. 4 мл раствора
3 доводят водой дистиллированной Р до объема
100 мл.
Мышьяк (2.4.2, метод А). Не более 0,0004 %
(4 ррт). 10 мл раствора 3 должны выдерживать ис¬
пытания на мышьяк.
Тяжелые металлы (2.4.8, метод А). Не более
0,0060 % (60 ррт). 20 мл раствора 3 нейтрализуют
раствором аммиака концентрированным Р, исполь¬
зуя в качестве внешнего индикатора раствор мета-
нилового желтого Р. При необходимости раствор
фильтруют и доводят водой Р до объема 30 мл. 12 мл
полученного раствора должны выдерживать испыта¬
ния на тяжелые металлы. Эталон готовят с использо¬
ванием 10 мл эталонного раствора свинца (1 ррт
РЬ)Р
Микробиологическая чистота
ОКА: допустимый критерий приемлемости
103 КОЕ/г (2.6.12).
ОКГ: допустимый критерий приемлемости
102 КОЕ/г (2.6.12).
Отсутствие грамотрицательных бактерий, толе¬
рантных к желчи (2.6.13).
Отсутствие ЕзсЬепсЫа соН (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,800 г испытуемого образца растворяют в 10 мл
кислоты хлористоводородной Р1 при нагревании на
водяной бане. Охлаждают и доводят водой Р до объ¬
ема 50,0 мл. К 10,0 мл полученного раствора прибав¬
ляют раствор аммиака разведенного Р1 до появле¬
ния осадка. К полученной смеси прибавляют кислоту
хлористоводородную разведенную Р в минимально
необходимом количестве для растворения осадка,
доводят водой Р до объема 20 мл и проводят ком¬
плексометрическое определение алюминия (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 5,098 мг А12Оэ.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре не выше 30 °С.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик необязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для алюминия оксида гидратированного, исполь¬
зуемого в качестве адсорбента.
Распределение частиц по размерам (2.9.31).
Удельная площадь поверхности (2.9.26).
07/2016:1598
АЛЮМИНИЯ ФОСФАТ
ГИДРАТИРОВАННЫЙ
А1ит1пН р/юзрЬаз Ьубпсиз
АШМШШМ РН08РНАТЕ, НУОРАТЕО
А1Р04 ■ хН20 М.м. 122,0 (безводное вещество)
ОПРЕДЕЛЕНИЕ
Содержание: не менее 94,0 % и не более 102,0 %
А1Р04 (М.м. 122,0) (в пересчете на прокаленное ве¬
щество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде, практически не¬
растворим в 96 % спирте. Растворяется в разведен¬
ных растворах минеральных кислот и гидроксидов
щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакцию (Ь) на фосфаты
(2.3.1).
B. Раствор 3 дает реакцию на алюминий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,00 г испытуемого образца раство¬
ряют в кислоте хлористоводородной разведенной Р
и доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 5,5 до 7,2. 4,0 г испытуемого образ¬
ца встряхивают с водой, свободной от углерода ди¬
оксида, Р и доводят до объема 100 мл этим же раст¬
ворителем.
Хлориды (2.4.4). Не более 1,3 %. 50,0 мг испыту¬
емого образца растворяют в 10 мл кислоты азотной
разведенной Р и доводят водой Р до объема 200 мл.
15 мл полученного раствора должны выдерживать ис¬
пытание на хлориды.
Растворимые фосфаты. Не более 1,0 % в пере¬
счете на РОД
Испытуемый раствор. К 5,0 г испытуемого об¬
разца прибавляют 150 мл воды Р и перемешивают в
течение 2 ч. Фильтруют и промывают фильтр 50 мл
воды Р. Фильтрат и промывные воды объединяют и
Алюминия хлорид гексагидрат
171
доводят водой Рдо объема 250,0 мл. 10,0 мл получен¬
ного раствора доводят водой Р до объема 100,0 мл.
Раствор сравнения (а). 2,86 г калия дигидрофос¬
фата Р растворяют в воде Р и доводят до объема
100 мл этим же растворителем.
Раствор сравнения (Ь). 1 мл раствора сравнения
(а) доводят водой Р до объема 5 мл.
Раствор сравнения (с). 3 мл раствора сравнения
(а) доводят водой Р до объема 5 мл.
Испытуемый раствор и растворы сравнения об¬
рабатывают следующим образом. К 5,0 мл раствора
прибавляют 4 мл кислоты серной разведенной Р,
1 мл раствора аммония молибдата Р, 5 мл воды Р
и 2 мл раствора, содержащего 0,10 г 4-метилами-
нофенола сульфата Р, 0,5 г натрия сульфита без¬
водного Р и 20,0 г натрия метабисульфита Р в
100 мл воды Р, встряхивают и выдерживают в тече¬
ние 15 мин. Полученный раствор доводят водой Р
до объема 25,0 мл и выдерживают в течение 15 мин.
Измеряют оптическую плотность (2.2.25) полученных
растворов при длине волны 730 нм. Содержание рас¬
творимых фосфатов рассчитывают по градуировочно¬
му графику, полученному с использованием обрабо¬
танных растворов сравнения (а), (Ь) и (с).
Сульфаты (2.4.13). Не более 0,6 %. 8 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 100 мл. 15 мл полученного раствора должны вы¬
держивать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0001 %
(1 ррт). 1,0 г испытуемого образца должен выдержи¬
вать испытание на мышьяк.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раство¬
ряют в кислоте хлористоводородной разведенной Р
и доводят до объема 20 мл этим же растворителем.
12 мл полученного раствора должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (1 ррт РЬ) Р
Потеря в массе при прокаливании. Не менее
10,0 % и не более 20,0 %. 1,000 г испытуемого образ¬
ца прокаливают при температуре (800±50) °С.
Нейтрализующая способность. 0,50 г испытуе¬
мого образца прибавляют к 30 мл 0,1 М раствора кис¬
лоты хлористоводородной, предварительно нагре¬
той до температуры 37 °С, и выдерживают при этой
температуре в течение 15 мин при перемешивании.
Значение рН (2.2.3) полученной смеси через 15 мин
при температуре 37 °С должно быть от 2,0 до 2,5.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 10 мл
кислоты хлористоводородной разведенной Р и дово¬
дят водой Р до объема 100,0 мл. К 10,0 мл получен¬
ного раствора прибавляют 10,0 мл 0,1 М раствора
натрия эдетата и 30 мл смеси из равных объемов
раствора аммония ацетата Р и кислоты уксусной
разведенной Р, кипятят в течение 3 мин и охлажда¬
ют. К полученному раствору прибавляют 25 мл 96 %
спирта Р и 1 мл свежеприготовленного раствора
0,25 г/л дитизона Рв96% спирте Р. Избыток натрия
эдетата титруют 0,1 М раствором цинка сульфата до
появления розового окрашивания.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 12,20 мгА1Р04.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0971
АЛЮМИНИЯ ХЛОРИД ГЕКСАГИДРАТ
А/иттН сЫопс1ит ЬехаЬус/псит
АШМШШМ СШ-ОКШЕ НЕХАНУОЕАТЕ
А1С13 ■ 6Н20 М.м. 241,4
[7784-13-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 95,0 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтый кристаллический по¬
рошок или бесцветные кристаллы. Расплывается на
воздухе.
Очень легко растворим в воде, легко растворим в
96 % спирте, растворим в глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,1 мл раствора 32, приготовленного, как ука¬
зано в разделе «Испытания», доводят водой Р до
объема 2 мл. Полученный раствор дает реакцию (а)
на хлориды (2.3.1).
B. 0,3 мл раствора 32 доводят водой Р до объема
2 мл. Полученный раствор дает реакцию на алюминий
(2.3.1).
ИСПЫТАНИЯ
Раствор 31. 10,0 г испытуемого образца раст¬
воряют в воде дистиллированной Р и доводят до
объема 100 мл этим же растворителем.
Раствор 32. 50 мл раствора 31 доводят водой Р
до объема 100 мл.
Прозрачность (2.2.1). Раствор 32 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
32 должна быть не интенсивнее эталона В(К)7.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
Раствор 51 должен выдерживать испытание на суль¬
фаты.
Железо (2.4.9). Не более 0,0010 % (10 ррт). Раст¬
вор 51 должен выдерживать испытание на железо.
Щелочи и щелочноземельные металлы. Не
более 0,5 %. К 20 мл раствора 52 прибавляют 100 мл
воды Р и нагревают до кипения. К горячему раство¬
ру прибавляют 0,2 мл раствора метилового крас¬
ного Р. К полученному раствору прибавляют раст¬
вор аммиака разведенный Р1 до появления желтого
окрашивания, доводят водой Р до объема 150 мл, на¬
гревают до кипения и фильтруют. 75 мл полученно¬
го фильтрата выпаривают на водяной бане досуха и
прокаливают до постоянной массы. Масса полученно¬
го остатка не должна превышать 2,5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 51 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Вода (2.5.12). Не менее 42,0 % и не более 48,0 %.
Определение проводят из 50,0 мг испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
23*. Зак. 1060.
172
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в
25,0 мл воды Р и проводят комплексометрическое
определение алюминия (2.5.11). Титруют 0,1 М рас¬
твором цинка сульфата до перехода окраски раст¬
вора от серовато-зеленой до розовой. Параллельно
проводят контрольный опыт.
1 мп 0,1 М раствора натрия эдетата соответ¬
ствует 24,14 мг А1С13-6Н20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0463
АМАНТАДИНА ГИДРОХЛОРИД
АтаМасИш Ьус1госЫопс1ит
АМАЫТАОШЕ НУОКОСШОКЮЕ
С10Н18С1Ы М.м. 187,7
[665-66-7]
ОПРЕДЕЛЕНИЕ
Трицикло[3.3.1.13 7]декан-1 -амина гидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в 96 % спирте.
При нагревании сублимируется.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амантадина гидрохлорида
#или спектр, представленный на рисунке #0463.-1#.
B. К 0,1 г испытуемого образца прибавляют 1 мл
пиридина Р, перемешивают и прибавляют 0,1 мл ук¬
сусного ангидрида Р. Нагревают до кипения в тече¬
ние 10 с. Выливают горячий раствор в 10 мл кислоты
хлористоводородной разведенной Р, охлаждают до
температуры 5 °С и фильтруют. Осадок промывают
водой Р и сушат при температуре 60 °С в вакууме в
течение 1 ч. Температура плавления (2.2.14) получен¬
ного остатка: от 147 °С до 151 °С.
C. 0,2 г испытуемого образца растворяют в 1 мл
0,1 М раствора кислоты хлористоводородной и при¬
бавляют 1 мл раствора 500 г/л натрия нитрита Р
Образуется белый осадок.
Р. 1 мл раствора 5, приготовленного, как указано
в разделе «Испытания», дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)7.
Кислотность или щелочность. 2 мл раствора 3
доводят водой, свободной от углерода диоксида, Р до
объема 10 мл, прибавляют 0,1 мл раствора метилово¬
го красного Р и 0,2 мл 0,01 М раствора натрия гидрок¬
сида. Должно появиться желтое окрашивание. При при¬
бавлении не более 0,4 мл 0,01 М кислоты хлористово¬
дородной должно появиться красное окрашивание.
Рисунок #0463.-1. Инфракрасный спектр ФСО амантадина гидрохлорида
Амантадина гидрохлорид
173
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Раствор внутреннего стандарта. 0,500 г ада-
мантана Р растворяют в метиленхлориде Р и дово¬
дят до объема 10,0 мл этим же растворителем.
Испытуемый раствор. 0,5 г испытуемого об¬
разца помещают в центрифужную пробирку, при¬
бавляют 9 мл метиленхлорида Р, 10 мл раствора
210 г/л натрия гидроксида Р и встряхивают в те¬
чение 10 мин. Верхний слой отбрасывают. Нижний
слой высушивают с использованием натрия суль¬
фата безводного Р, фильтруют, фильтрат соби¬
рают в мерную колбу, прибавляют 0,1 мл раствора
внутреннего стандарта и доводят метиленхлори-
дом Р до объема 10,0 мл.
Раствор сравнения. 5 мг ФСО амантадина ги¬
дрохлорида помещают в центрифужную пробирку,
прибавляют 9 мл метиленхлорида Р, 10 мл раст¬
вора 210 г/л натрия гидроксида Р и встряхивают в
течение 10 мин. Верхний слой отбрасывают. Ниж¬
ний слой высушивают с использованием натрия
сульфата безводного Р, фильтруют, фильтрат со¬
бирают в мерную колбу, прибавляют 1,0 мл раст¬
вора внутреннего стандарта и доводят метилен-
хлоридом Р до объема 100,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,53 мм, покрытая слоем поли(диметил)
(дифенил)силоксана, деактивированного по отно¬
шению к основаниям, Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 4 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
70
5—23
70 — 250
23-40
250
Блок ввода
проб
220
Детектор
300
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению
к амантадину, время удерживания— около 14 мин):
внутренний стандарт — около 0,8.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 5,0 между пиками вну¬
треннего стандарта и амантадина.
Предельное содержание примесей:
- неспецифицированные примеси (не бо¬
лее 0,10%): используя хроматограмму раствора
сравнения, рассчитывают отношение (Р,) площа¬
ди пика амантадина к площади пика внутреннего
стандарта; используя хроматограмму испытуемого
раствора, рассчитывают отношение площади лю¬
бого пика, кроме основного пика и пика внутрен¬
него стандарта, к площади пика внутреннего стан¬
дарта: полученное значение не должно превышать
значение Р^,
- сумма примесей (не более 0,3 %): используя
хроматограмму раствора сравнения, рассчитыва¬
ют отношение (Р2) 3-кратной площади пика аман¬
тадина к площади пика внутреннего стандарта;
используя хроматограмму испытуемого раствора,
рассчитывают отношение суммы площадей всех
пиков, кроме основного пика и пика внутреннего
стандарта, к площади пика внутреннего стандарта:
полученное значение не должно превышать значе¬
ние Р2;
- неучитываемый предел (0,05%): используя
хроматограмму раствора сравнения, рассчитыва¬
ют отношение (Р3) 0,5 площади пика амантадина
к площади пика внутреннего стандарта; используя
хроматограмму испытуемого раствора рассчиты¬
вают отношение площади любого пика, кроме пи¬
ков амантадина и внутреннего стандарта, к пло¬
щади пика внутреннего стандарта: не учитывают
пики, для которых полученные значения менее
значения Р3.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 2,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в сме¬
си из 5,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 50 мл 96 % спирта Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 18,77 мг С10Н18С1Ы.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, В.
А. 1 -Хл ортрицикло[3.3.1.13-7]декан.
' В. А/-(Трицикло[3.3.1.13’7]дек-1-ил)ацетамид.
24. Зак. 1060.
174
Гэсударственная фармакопея Республики Беларусь
07/2016:1489
АМБРОКСОЛА ГИДРОХЛОРИД
АтЬгохоН ЬубгосЫопбит
дмвроха нуокостокюЕ
С13Н19ВГ2С1Ы20 М.м. 414,6
[23828-92-4]
ОПРЕДЕЛЕНИЕ
транс-4-[(2-Амино-3,5-дибромбензил)амино]ци-
клогексанола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтоватый кристаллический порошок.
Умеренно растворим в воде, растворим в мета¬
ноле, практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 0,05 М растворе кислоты сер¬
ной и доводят до объема 100,0 мл этим же раствори¬
телем. 2,0 мл полученного раствора доводят 0,05 М
раствором кислоты серной до объема 10,0 мл.
Диапазон длин волн: от 200 нм до 350 нм.
Максимумы поглощения: при 245 нм и при 310 нм.
Отношение оптических плотностей:
^Аш — отЗ,2цоЗА.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амброксола гидрохлорида #или
спектр, представленный на рисунке #1489.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения. 50 мг ФСО амброксола ги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
пяРг54Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — пропанол Р — этилацетат Р — гек¬
сан Р (1:10:20:70, об/об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
й. 25 мг испытуемого образца растворяют в
2,5 мл воды Р, смешивают с 1,0 мл раствора аммиа¬
ка разведенного Р1, выдерживают в течение 5 мин и
фильтруют. Фильтрат подкисляют кислотой азотной
разведенной Р. Полученный раствор дает реакцию (а)
на хлориды (2.3.1).
Рисунок #1489.-1. Инфракрасный спектр ФСО амброксола гидрохлорида
Амброксола гидрохлорид
175
ИСПЫТАНИЯ
Раствор 5. 0,75 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 15 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 4,5 до 6,0. 0,2 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 20 мл этим же раство¬
рителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 50 мг испытуемого образ¬
ца растворяют в воде Р и доводят до объема 50,0 мл
этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят водой Р до объема 100,0 мл. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 10,0 мл.
Раствор сравнения (Ь). Для приготовления при¬
меси В /V? зИи 5 мг испытуемого образца растворяют в
0,2 мл метанола Р, прибавляют 0,04 мл смеси раст¬
вор формальдегида Р — вода Р (1:99, об/об), нагре¬
вают при температуре 60 °С в течение 5 мин и выпа¬
ривают досуха в потоке азота. Остаток растворяют в
5 мл воды Р и доводят подвижной фазой до объема
20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецил-
синильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: смесь из равных объемов
ацетонитрила Р и раствора, приготовленного следу¬
ющим образом: 1,32 г аммония фосфата Р раство¬
ряют в 900 мл воды Р, доводят до рН 7,0 кислотой
фосфорной Р и разводят водой Р до объема 1000 мл;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 248 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания амброксола.
Идентификация пиков примесей: идентифици¬
руют пик примеси В, используя хроматограмму раст¬
вора сравнения (Ь).
Относительное удерживание (по отношению к
амброксолу, время удерживания — около 9 мин): при¬
месь В — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками при¬
меси В и амброксола.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 70 мл
96 % спирта Р, прибавляют 5 мл 0,03 футы раст¬
вора кислоты хлористоводородной и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 41,46 мг С13Н19Вг2С11\120.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е.
Вг
А. Аг-СН2ОН: (2-Амино-3,5-дибромфенил)метанол.
В. транс-4-(6,8-Дибром-1,4-дигидрохиназолин-
3(2Н)-ил)циклогексанол.
С. л?ранс-4-[[(Е)-2-Амино-3,5-дибромбензилиден]-
амино]циклогексанол.
24*. Зак. 1060.
176
Гэсударственная фармакопея Республики Беларусь
й. /_/ос-4-[(2-амино-3,5-дибромбензил)амино]ци-
клогеканол.
Е. Аг-СН=0: 2-Амино-3,5-дибромбензальдегид.
07/2016:1289
АМИКАЦИН
Ат/каапит
АМ1КАС1Ы
С22Н4зЫ5013 М.м. 585,6
[37517-28-5]
ОПРЕДЕЛЕНИЕ
б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1-Л/-
[(25)-4-амино-2-гидроксибутаноил]-2-дезокси-0-
стрептамин.
Антимикробное вещество, полученное из кана-
мицина А.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Умеренно растворим в воде, мало растворим в
метаноле, практически нерастворим в ацетоне и в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амикацина.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (а). 25 мг ФСО амикацина
растворяют в воде Р и доводят до объема 10 мл этим
же растворителем.
Раствор сравнения (Ь). 5 мг ФСО канамицина
моносульфата растворяют в 1 мл испытуемого раст¬
вора и доводят водой Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: метиленхлорид Р — раствор
аммиака Р— метанол Р (25:30:40, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р1 и нагревают при температуре 110 °С в
течение 5 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
рН (2.2.3). От 9,5 до 11,5. 0,1 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От +97
до +105 (в пересчете на безводное вещество). 0,50 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
10,0 мл.
Раствор сравнения (с). 5 мг ФСО амикацина для
проверки пригодности хроматографической систе¬
мы (содержит примеси А, В, Р и Н) растворяют в под¬
вижной фазе А и доводят до объема 10 мл этим же
растворителем.
Раствор сравнения (д). 5,0 мг ФСО амикацина
примеси I растворяют в подвижной фазе А и доводят
до объема 20,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят подвижной фазой А до
объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смесь, приготовленная с
использованием воды, свободной от углерода
диоксида, Р, содержащая 1,8 г/л натрия октан-
сульфоната Р, 20 г/л натрия сульфата безво¬
дного Р1, 1,4 % (об/об) тетрагидрофурана Р,
5 % (об/об) 0,2 М раствора калия дигидрофосфа¬
та Р, предварительно доведенного до рН 3,0 кис¬
лотой фосфорной разведенной Р; дегазируют;
- подвижная фаза В: смесь, приготовленная с
использованием воды, свободной от углерода
диоксида, Р, содержащая 1,8 г/л натрия октан-
сульфоната Р, 28 г/л натрия сульфата безво¬
дного Р1, 1,4 % (об/об) тетрагидрофурана Р,
5 % (об/об) 0,2 М раствора калия дигидрофосфа¬
та Р, предварительно доведенного до рН 3,0 кис¬
лотой фосфорной разведенной Р; дегазируют;
Амикацин
177
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
100
10
3—38,0
о
со
т
о
о
т—
о
т
о
38,0—38,1
300 — 0
70-100
38,1—68
0
100
- скорость подвижной фазы: 1,0 мл/мин;
- постколоночный раствор: смесь из раствора
натрия гидроксида, свободного от карбонатов, Р и
предварительно дегазированной воды, свободной от
углерода диоксида, Р (1:24, об/об), прибавляемая не¬
импульсным способом к выходящей из колонки фрак¬
ции с использованием полимерной спирали для сме¬
шивания объемом 375 мкл;
- скорость постколоночного раствора:
0. 3 мл/мин;
- детектирование: импульсный амперометриче¬
ский детектор или аналогичный с золотым индикатор¬
ным электродом, хлорсеребряным электродом срав¬
нения и вспомогательным электродом из нержавею¬
щей стали, являющимся центральной частью ячейки,
в которой устанавливаются соответственно потенциал
детектирования +0,05 В, потенциал окисления +0,75 В
и потенциал восстановления -0,15 В, с длительностью
импульса в соответствии с применяемым прибором;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифициру¬
ют пики примесей А, В, Р и Н, используя хроматограмму
раствора сравнения (с) и хроматограмму, прилагаемую
к ФСО амикацина для проверки пригодности хромато¬
графической системы; идентифицируют пик примеси
1, используя хроматограмму раствора сравнения (б).
Относительное удерживание (по отношению к
амикацину, время удерживания — около 28 мин): при¬
месь I — около 0,13; примесь Р — около 0,92; примесь
В — около 0,95; примесь А— около 1,62; примесь Н —
около 1,95.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 5
(Нр — высота пика примеси В относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси В и пик
амикацина); при необходимости корректируют объем
тетрагидрофурана в подвижной фазе.
Расчет процентных содержаний:
-для примеси I — используют концентрацию
примеси I в растворе сравнения (б);
- для всех примесей, кроме примеси I, — использу¬
ют концентрацию амикацина в растворе сравнения (а).
Предельное содержание примесей:
- примеси А, В, Р, Н и I: не более 0,5 % для каж¬
дой примеси;
- любая другая примесь: не более 0,5 % для каж¬
дой примеси;
- сумма примесей: не более 1,5 %;
- учитываемый предел: 0,1 %.
Вода (2.5.12). Не более 8,5 %. Определение про¬
водят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения. 50,0 мг ФСО амикацина
растворяют в подвижной фазе и доводят этим же
растворителем до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смесь, приготовленная с ис¬
пользованием воды, свободной от углерода диокси¬
да, Р, содержащая 1,8 г/л натрия октансульфона-
та Р, 20 г/л натрия сульфата безводного Р1, 5,8 %
(об/об) ацетонитрила Р1, 5 % (об/об) 0,2 М раст¬
вора калия дигидрофосфата Р, предварительно до¬
веденного до рН 3,0 кислотой фосфорной разведен¬
ной Р\ дегазируют;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 200 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,3-кратное вре¬
мя удерживания амикацина.
Время удерживания: амикацин — около 30 мин.
Пригодность хроматографической системы:
раствор сравнения:
-фактор асимметрии: не более 1,5 для пика
амикацина; при необходимости корректируют коли¬
чество ацетонитрила Р в подвижной фазе; может
наблюдаться разделение пика, когда время удержи¬
вания становится слишком маленьким;
- относительное стандартное отклонение: не
более 1,5 % при 6 повторных вводах пробы.
Содержание С^Н^МдО^ в процентах рассчитыва¬
ют с учетом содержания амикацина в ФСО амикацина.
ПРИМЕСИ
Специфицированные примеси: А, В, Р, Н, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О, Е, 6.
А. 4-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-6-
0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1-Л/-[(25)-4-
амино-2-гидроксибутаноил]-2-дезокси-1-стрептамин.
25. Зак. 1060.
178
Гэсударственная фармакопея Республики Беларусь
В. 4-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-
6-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1,3-Л/-
бис[(25)-4-амино-2-гидроксибутаноил]-2-дезокси-Ь
стрептамин.
С. 4-0-(6-Амино-6-дезокси-а-0-глюкопиранозил)-
6-0-[3-[[(25)-4-амино-2-гидроксибутаноил]амино]-3-
дезокси-а-0-глюкопиранозил]-2-дезокси-0-стрептамин.
О. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-2-дезокси-
О-стрептамин (канамицин).
Е. 4-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-
6-0-[6-[[(25)-4-амино-2-гидроксибутаноил]амино]-6-
дезокси-а-0-глюкопиранозил]-2-дезокси-1_-стрептамин.
Р. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-[6-[(25)-4-амино-2-гидроксибутаноил]амино-6-
дезокси-а-0-глюкопиранозил]-1-Л/-[(25)-4-амино-2-
гидроксибутаноил]-2-дезокси-0-стрептамин.
<3. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1-Л/-[(2Я)-
4-амино-2-гидроксибутаноил]-2-дезокси-0-стрептамин.
Н. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
1-Л/-[(25)-4-амино-2-гидроксибутаноил]-4-0-(2,6-
диамино-2,6-дидезокси-а-0-глюкопиранозил)-2-дезокси-
О-стрептамин.
но2с
N42
I. (25)-4-Амино-2-гидроксибутановая кислота.
07/2016:1290
АМИКАЦИНА СУЛЬФАТ
Ат/касЫ зиКаз
АМ1КАС1Ы ЗШ.РАТЕ
СяН^А, • 2Н2304 М.м, 782
[39831-55-5]
ОПРЕДЕЛЕНИЕ
6-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-4-
0-(6-амино-6-дезокси-а-0-плюкопиранозил)-1-Л/-[(2 5)-
4-амино-2-гидроксибутаноил]-2-дезокси-0-стрептамина
сульфат.
Антимикробное вещество, полученное из кана-
мицина А.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Легко растворим в воде, практически нераство¬
рим в ацетоне и в 96 % спирте.
Амикацина сульфат
179
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амикацина сульфата #или
спектр, представленный на рисунке #1290.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (а). 25 мг ФСО амикацина
сульфата растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО канамицина
моносульфата растворяют в 1 мл испытуемого раст¬
вора и доводят водой Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: метиленхлорид Р — раствор
аммиака Р — метанол Р (25:30:40, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р1 и нагревают при температуре 110 °С в
течение 5 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
C. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 2,0 до 4,0. 0,1 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От +76
до +84 (в пересчете на сухое вещество). 0,50 г испы¬
туемого образца растворяют в воде Р и доводят до
объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 33 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой А до объема 10,0 мл.
Раствор сравнения (с). 5 мг ФСО амикацина для
проверки пригодности хроматографической систе¬
мы (содержит примеси А, В, Р и Н) растворяют в под¬
вижной фазе А и доводят до объема 10 мл этим же
растворителем.
Раствор сравнения (ф. 6,6 мг ФСО амикацина
примеси I растворяют в подвижной фазе А и доводят
до объема 20,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят подвижной фазой А до
объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смесь, приготовленная с
использованием воды, свободной от углерода
диоксида, Р, содержащая 1,8 г/л натрия октан-
сульфоната Р, 20 г/л натрия сульфата безво¬
дного Р1, 1,4% (об/об) тетрагидрофурана Р,
5 % (об/об) 0,2 М раствора калия дигидрофосфа¬
та Р, предварительно доведенного до рН 3,0 кис¬
лотой фосфорной разведенной Р; дегазируют;
Рисунок #1290.-1. Инфракрасный спектр ФСО амикацина сульфата
25* Зак. 1060.
180
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза В: смесь, приготовленная с
использованием воды, свободной от углерода
диоксида, Р, содержащая 1,8 г/л натрия октан-
сульфоната Р, 28 г/л натрия сульфата безво¬
дного Р1, 1,4 % (об/об) тетрагидрофурана Р,
5 % (об/об) 0,2 М раствора калия дигидрофосфа¬
та Р, предварительно доведенного до рН 3,0 кис¬
лотой фосфорной разведенной Р; дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
100
10
3—38,0
100 — 30
0-70
38,0—38,1
о
т
о
со
70 — 100
38,1—68
0
100
- скорость подвижной фазы: 1,0 мл/мин;
- постколоночный раствор: смесь из раствора
-натрия гидроксида, свободного от карбонатов, Р и
предварительно дегазированной воды, свободной от
углерода диоксида, Р (1:24, об/об), прибавляемая не¬
импульсным способом к выходящей из колонки фрак¬
ции с использованием полимерной спирали для сме¬
шивания объемом 375 мкл;
- скорость постколоночного раствора: 0,3 мл/мин;
- детектирование: импульсный амперометри¬
ческий детектор или аналогичный с золотым инди¬
каторным электродом, хлорсеребряным электродом
сравнения и вспомогательным электродом из нержа¬
веющей стали, являющимся центральной частью
ячейки, в которой устанавливаются соответственно
потенциал детектирования +0,05 В, потенциал окис¬
ления +0,75 В и потенциал восстановления -0,15 В,
с длительностью импульса в соответствии с приме¬
няемым прибором;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, Р и Н, используя хромато¬
грамму раствора сравнения (с) и хроматограмму, при¬
лагаемую к ФСО амикацина для проверки пригодно¬
сти хроматографической системы; идентифициру¬
ют пик примеси I, используя хроматограмму раствора
сравнения (с!).
Относительное удерживание (по отношению к
амикацину, время удерживания — около 28 мин): при¬
месь I — около 0,13; примесь Р — около 0,92; примесь
В — около 0,95; примесь А—около 1,62; примесь Н —
около 1,95.
Пригодность хроматографической системы:
раствор сравнения (с):
-коэффициент разделения пиков: не менее 5
(Нр — высота пика примеси В относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси В и пик
амикацина); при необходимости корректируют объем
тетрагидрофурана в подвижной фазе.
Расчет процентных содержаний:
-для примеси I — используют концентрацию
примеси I в растворе сравнения (д);
- для всех примесей, кроме примеси I, — исполь¬
зуют концентрацию амикацина сульфата в растворе
сравнения (а).
Предельное содержание примесей:
- примеси А, В, Р, Н, I: не более 0,5 % для каждой
примеси;
- любая другая примесь: не более 0,5 % для каж¬
дой примеси;
- сумма примесей: не более 1,5 %;
- неучитываемый предел: 0,1 %.
Сульфаты. Не менее 23,3 % и не более 25,8 %
(в пересчете на сухое вещество). 0,250 г испытуемого
образца растворяют в 100 мл воды Р и доводят полу¬
ченный раствор до рН 11, используя раствор аммиа¬
ка концентрированный Р. Прибавляют 10,0 мл 0,1 М
раствора бария хлорида и около 0,5 мг фталеинового
пурпурного Р. Титруют 0,1 М раствором натрия эде-
тата, прибавляют 50 мл 96 % спирта Р, когда окра¬
ска раствора начнет меняться, и продолжают титрова¬
ние до исчезновения фиолетово-голубой окраски.
1 мл 0,1 М раствора бария хлорида соответству¬
ет 9,606 мг сульфатов (304).
Потеря в массе при высушивании (2.2.32). Не
более 13,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С и давлении не более 0,7 кПа в те¬
чение 3 ч.
Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным, если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения без дальнейшей
подходящей процедуры удаления пирогенов. Тест-
доза — 25 мг испытуемого образца в 5 мл воды для
инъекций Р на 1,0 кг массы тела кролика.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения. 50,0 мг ФСО амикацина
сульфата растворяют в подвижной фазе и доводят
до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смесь, приготовленная с ис¬
пользованием воды, свободной от углерода диокси¬
да, Р, содержащая 1,8 г/л натрия октансульфона-
та Р, 20 г/л натрия сульфата безводного Р1, 5,8 %
(об/об) ацетонитрила Р1, 5% (об/об) 0,2 М раст¬
вора калия дигидрофосфата Р, предварительно до¬
веденного до рН 3,0 кислотой фосфорной разведен¬
ной Р; дегазируют;
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 200 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,3-кратное вре¬
мя удерживания амикацина.
Время удерживания: амикацин — около 30 мин.
Пригодность хроматографической системы:
раствор сравнения:
- фактор асимметрии: не более 1,5 для пика
амикацина; при необходимости корректируют коли¬
чество ацетонитрила Р в подвижной фазе; может
наблюдаться разделение пика, когда время удержи¬
вания становится слишком маленьким;
- относительное стандартное отклонение: не
более 1,5 % при 6 повторных вводах пробы.
Содержание С22Н43М5013'2Н23 04 в процентах рас¬
считывают с учетом содержания амикацина сульфата
в ФСО амикацина сульфата.
Амикацина сульфат
181
ХРАНЕНИЕ
Если субстанция стерильная, ее хранят в сте¬
рильном воздухонепроницаемом контейнере с кон¬
тролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, Р, Н, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О, Е, 6.
А. 4-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-
6-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1-Л/-[(25)-
4-амино-2-гидроксибутаноил]-2-дезокси-1_-стрептамин.
В. 4-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-
6-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1,3-А/-
бис[(25)-4-амино-2-гидроксибутаноил]-2-дезокси-1_-
стрептамин.
С. 4-0-(6-Амино-6-дезокси-а-0-глюкопиранозил)-
6-0-[3-[[(25)-4-амино-2-гидроксибутаноил]амино]-3-
дезокси-а-0-глюкопиранозил]-2-дезокси-0-стрептамин.
й. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-2-дезокси-
О-стрептамин (канамицин).
Е. 4-0-(3-Амино-3-дезокси-а-0-глюкопиранозил)-
6-0-[6-[[(25)-4-амино-2-гидроксибутаноил]амино]-6-
дезокси-а-0-глюкопиранозил]-2-дезокси-1_-стрептамин.
Р. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-[6-[(25)-4-амино-2-гидроксибутаноил]амино-6-
дезокси-а-0-глюкопиранозил]-1-А/-[(25)-4-амино-2-
гидроксибутаноил]-2-дезокси-0-стрептамин.
6. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
4-0-(6-амино-6-дезокси-а-0-глюкопиранозил)-1-А/-[(2Е)-
4-амино-2-гидроксибутаноил]-2-дезокси-0-стрептамин.
Н. б-О-(З-Амино-З-дезокси-а-О-глюкопиранозил)-
1-Л/-[(25)-4-амино-2-гидроксибутаноил]-4-0-(2,6-
диамино-2,6-дидезокси-а-0-глюкопиранозил)-2-дезокси-
О-стрептамин.
182
Гэсударственная фармакопея Республики Беларусь
I. (25)-4-Амино-2-гидроксибутановая кислота.
07/2016:РБ0022
#АМИНАЛОН
Ат1па1опит (Аас1ит ат1поЬиИпсит)
АМШОВиТУШСАСЮ
С4Н9Ы02 М.м. 103,1
[56-12-2]
ОПРЕДЕЛЕНИЕ
4-Аминобутановая кислота. у-Аминомасляная кис¬
лота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые кристаллы или кристал¬
лический порошок.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
Температура плавления: от 198 °С до 204 °С с
разложением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: стандартный образец у-аминомасля-
ной кислоты или спектр, представленный на рисунке
РБ0009.-1.
B. 10 мг испытуемого образца растворяют в
10 мл воды Р, прибавляют несколько капель раствора
3 г/л нингидрина Р и нагревают до кипения. Появляет¬
ся фиолетово-синее окрашивание.
С. 50 мг испытуемого образца растворяют в 5 мл
воды Р и нейтрализуют 0,1 М раствором натрия ги¬
дроксида по раствору фенолфталеина Р до розово¬
го окрашивания. К полученному раствору прибавляют
5 мл раствора формальдегида Р, нейтрализованно¬
го 0,1 М раствором натрия гидроксида по раствору
фенолфталеина Р до розового окрашивания. Раст¬
вор обесцвечивается.
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 100,0 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 6,5 до 7,5. Измеряют рН раствора 3.
Хлориды (2.4.4). Не более 0,0040 % (40 ррт).
10.0 г испытуемого образца растворяют в воде Р и
доводят до объема 100,0 мл этим же растворителем.
12,5 мл полученного раствора доводят водой Р до
объема 15 мл. Полученный раствор должен выдержи¬
вать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0050 % (50 ррт).
20.0 г испытуемого образца растворяют в воде Р и
доводят до объема 100,0 мл этим же растворителем.
15 мл полученного раствора должны выдерживать ис¬
пытание на сульфаты.
Кальций (2.4.3). 10,0 г испытуемого образца
растворяют в воде Р и доводят до объема 100,0 мл
этим же растворителем. К 5 мл полученного раствора
прибавляют 5 мл воды Р и 1 мл раствора аммония
оксалата Р. Через 15 мин опалесценция полученного
раствора не должна превышать опалесценцию раст¬
вора, состоящего из 5 мл раствора 3 и 6 мл воды Р.
Рисунок РБ0009.-1. Инфракрасный спектр у-аминомасляной кислоты
Аминокапроноеая кислота
183
Мышьяк {2.4.2, метод А). Не более 0,0001 %
(1 рргп). 1,0 г испытуемого образца должен выдержи¬
вать испытание на мышьяк.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). Эталон готовят с использованием
1 мл эталонного раствора свинца (10 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более 0,07 %. Ис¬
пытание проводят из 1,5 г испытуемого образца.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4. *
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
70,0 мг испытуемого образца растворяют в 10 мл
кислоты уксусной ледяной Р, прибавляют 1 каплю
раствора кристаллического фиолетового Р и титру¬
ют 0,1 М раствором кислоты хлорной до изменения
окраски раствора на зеленую.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 10,31 мгС4Н9М02.
ХРАНЕНИЕ
В защищенном от света и влаги месте.
07/2016:0874
АМИНОКАПРОНОВАЯ КИСЛОТА
АсШит аттосаргоюит
АМ1ЫОСАРРЮ1С АСЮ
С6Н13М02 М.м. 131,2
[60-32-2]
ОПРЕДЕЛЕНИЕ
6-Аминогексановая кислота.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворима в воде, мало растворима в
96 % спирте.
Температура плавления: около 205 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО аминокапроновой кислоты
#или спектр, представленный на рисунке #0874.-1#.
B. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества».
На хроматограмме испытуемого раствора (Ь) обнару¬
живается пятно, соответствующее по расположению,
размеру и цвету основному пятну на хроматограмме
раствора сравнения (а).
C. 0,5 г испытуемого образца растворяют в 4 мл
смеси из равных объемов кислоты хлористоводо¬
родной разведенной Р и воды Р и выпаривают досуха
на водяной бане. Остаток высушивают в эксикаторе,
затем растворяют в около 2 мл кипящего этанола Р,
охлаждают и выдерживают при температуре от 4 °С
до 8 °С в течение 3 ч. Раствор фильтруют при пони¬
женном давлении. Температура плавления (2.2.14)
осадка, промытого около 10 мл ацетона Р и высу¬
шенного при температуре 60 °С в течение 30 мин: от
131 °С до 133 °С.
Рисунок #0874.-1. Инфракрасный спектр ФСО аминокапроновой кислоты
184
Гэсударственная фармакопея Республики Беларусь
0.5 мг испытуемого образца растворяют в 0,5 мл
воды дистиллированной Р, прибавляют 3 мл диме-
тилформамида Р, 2 мл раствора кислоты аскор¬
биновой Р и нагревают на водяной бане. Появляется
оранжевое окрашивание.
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным в течение 24 ч.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
рН (2.2.3). От 7,5 до 8,0. Измеряют рН раствора 3.
Оптическая плотность (2.2.25).
A. Не более 0,10 при длине волны 287 нм и не
более 0,03 при длине волны 450 нм. Измеряют опти¬
ческую плотность раствора 3.
B. Не более 0,15 при длине волны 287 нм и не
более 0,03 при 450 нм. 2,0 г испытуемого образца по¬
мещают ровным слоем в плоскодонную чашку диаме¬
тром 9 см, накрывают и выдерживают при температу¬
ре от 98 °С до 102 °С в течение 72 ч. Остаток раство¬
ряют в воде Р и доводят до объема 10,0 мл этим же
растворителем.
Нингидрин-положительные вещества. Тонкос¬
лойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО аминокапро¬
новой кислоты растворяют в воде Р и доводят до
объема 50 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО аминокапроно¬
вой кислоты и 10 мг ФСО лейцина растворяют в воде Р
и доводят до объема 25 мл этим же растворителем.
Пластинка'. ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: по 5 мкл каждого раст¬
вора. Пластинку высушивают на воздухе.
Фронт подвижной фазы: не менее 15 см от ли-
. нии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 °С
до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1%. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 20 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной до перехода окраски от си¬
невато-фиолетовой до синевато-зеленой, используя в
качестве индикатора 0,1 мл раствора кристалличе¬
ского фиолетового Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 13,12 мг С6Н131Ч02.
07/2016:0803
АМИОДАРОНА ГИДРОХЛОРИД
Атюс1агоп1 ЬудгосЫопс/ит
АМЮОАКОЫЕ НУ0К0СН1.0П10Е
[19774-82-4]
ОПРЕДЕЛЕНИЕ
(2-Бутилбензофуран-3-ил)[4-[2-(диэтиламино)-
этокси]-3,5-дийодфенил]метанона гидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый мелкий кристаллический
порошок.
Очень мало растворим в воде, легко растворим
в метиленхпориде, растворим в метаноле, умеренно
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амиодарона гидрохлорида #или
спектр, представленный на рисунке #0803.-1#.
B. Испытуемый образец дает реакцию (Ь) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона ОУ(ЗЖ)5 или
ВУ(КЖ)5.
рН (2.2.3). От 3,2 до 3,8.1,0 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р, нагревая при температуре 80 °С. Охлаждают
и доводят до объема 20 мл этим же растворителем.
Амиодарона гидрохлорид
185
Примесь Н. Не более 0,02 %. Тонкослойная хро¬
матография (2.2.27). Растворы готовят непосред¬
ственно перед использованием и хранят, защищая
от яркого света.
Испытуемый раствор. 0,500 г испытуемого об¬
разца растворяют в метиленхлориде Р и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг (2-хлорэтил)диэ-
тиламина гидрохлорида Р (примесь Н) растворяют в
метиленхлориде Р и доводят до объема 50,0 мл этим
же растворителем. 2,0 мл полученного раствора дово¬
дят метиленхлоридом Р до объема 20,0 мл.
Раствор сравнения (Ь). Смешивают 2,0 мл ис¬
пытуемого раствора и 2,0 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге-
ля р254 Р.
Подвижная фаза: кислота муравьиная безвод¬
ная Р — метанол Р — метиленхлорид Р (5:10:85,
об/об/об).
Наносимый объем пробы: по 50 мкл испытуемого
раствора и раствора сравнения (а); 100 мкл раствора
сравнения (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку опрыскивают раствором
калия йодовисмутата Р1 и затем раствором водо¬
рода пероксида разведенным Р. Немедленно просма¬
тривают при дневном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживается отчетливо
видимое пятно примеси Н.
Предельное содержание примесей:
- примесь Н (не более 0,02 %): на хроматограм¬
ме испытуемого раствора любое пятно с соответ¬
ствующим примеси Н на хроматограмме раствора
сравнения (Ь), должно быть не интенсивнее пятна на
хроматограмме раствора сравнения (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор рН 4,9. К 800 мл воды Р при¬
бавляют 3,0 мл кислоты уксусной ледяной Р, доводят
до рН 4,9 раствором аммиака разведенным Р1 и до¬
водят водой Р до объема 1000 мл.
Испытуемый раствор. 0,125 г испытуемого об¬
разца растворяют в смеси из равных объемов ацето¬
нитрила Р и воды Р и доводят этой же смесью раст¬
ворителей до объема 25,0 мл.
Раствор сравнения. 5 мг ФСО амиодарона при¬
меси О, 5 мг ФСО амиодарона примеси Е и 5,0 мг ФСО
амиодарона гидрохлорида растворяют в метаноле Р
и доводят до объема 25,0 мл этим же растворителем.
1.0 мл полученного раствора доводят смесью из рав¬
ных объемов ацетонитрила Р и воды Р до объема
20.0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 30 °С;
- подвижная фаза: буферный раствор рН 4,9 —
метанол Р — ацетонитрил Р (30:30:40, об/об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 240 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания амиодарона.
Относительное удерживание (по отношению
к амиодарону, время удерживания — около 24 мин):
примесь А — около 0,26; примесь й — около 0,29;
примесь Е — около 0,37; примесь В — около 0,49;
примесь С — около 0,55; примесь 6 — около 0,62;
примесь Р — около 0,69.
Пригодность хроматографической системы:
раствор сравнения:
3000
1000
2000 1500
Волновое число (см'1)
Рисунок #0803.-1. Инфракрасный спектр ФСО амиодарона гидрохлорида
500
186
Гэсударственная фармакопея Республики Беларусь
- разрешение: не менее 3,5 между пиками при¬
меси О и примеси Е.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р, 6 (не более 0,2 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, О, Е, Р и О,
не должны превышать площадь пика амиодарона на
хроматограмме раствора сравнения;
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, й, Е, Р и О, не должна превышать 0,5
площади пика амиодарона на хроматограмме раст¬
вора сравнения;
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2,5-кратную площадь пика амиодарона на хромато¬
грамме раствора сравнения;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,25 площади пика амиодарона на
хроматограмме раствора сравнения.
Йодиды. Не более 0,0150 % (150 ррт). Испыту¬
емый раствор и раствор сравнения готовят одно¬
временно.
Раствор А. К 40 мл воды Р с температурой 80 °С
прибавляют 1,50 г испытуемого образца и встряхива¬
ют до полного растворения. Охлаждают и доводят во¬
дой Р до объема 50,0 мл.
Испытуемый раствор. К 15,0 мл раствора А при¬
бавляют 1,0 мл 0,1 М раствора кислоты хлористо¬
водородной и 1,0 мл 0,05 М раствора калия йодата
и доводят водой Р до объема 20,0 мл. Раствор выдер¬
живают в защищенном от света месте в течение 4 ч.
Раствор сравнения. К 15,0 мл раствора А при¬
бавляют 1,0 мл 0,1 М раствора кислоты хлористо¬
водородной, 1,0 мл раствора 88,2 мг/л калия йодида Р
и 1,0 мл 0,05 М раствора калия йодата и доводят
водой Р до объема 20,0 мл. Раствор выдерживают в
защищенном от света месте в течение 4 ч.
Компенсационный раствор. К 15,0 мл раствора
А прибавляют 1,0 мл 0,1 М раствора кислоты хлори¬
стоводородной и доводят водой Р до объема 20,0 мл.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора и раствора сравнения при длине
волны 420 нм. Оптическая плотность испытуемого
раствора не должна превышать половину оптической
плотности раствора сравнения.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 50 °С и давлении, не превышающем
0,3 кПа, в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,600 г испытуемого образца растворяют в сме¬
си из 5,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 75 мл 96 % спирта Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 68,18 мг С25Н2912М03-НС1.
ХРАНЕНИЕ
В защищенном от света месте при температуре
не выше 30 °С.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, С, Н.
А. (2-Бутилбензофуран-3-ил)[4-[2-(диэтиламино)-
этокси]фенил]метанон.
В. (2-Бутилбензофуран-3-ил)[4-[2-(этиламино)-
этокси]-3,5-дийодфенил]метанон.
С. (2-Бутилбензофуран-3-ил)[4-[2-(диэтиламино)-
этокси]-3-йодфенил]метанон.
О. (2-Бутилбензофуран-3-ил)(4-гидрокси-3,5-
дийодфенил)метанон.
Р. (2-Бутилбензофуран-3-ил)(4-гидрокси-3-
йодфенил)метанон.
Амитриптилина гидрохлорид
187
О. [4-[2-(Диэтиламино)этокси]-3,5-дийодфенил]-
[2-[(1 Я$)-1 -метоксибутил]бензофуран-3-ил]метанон.
С1
СН3
Н. 2-Хлор-А/,А/-диэтилэтанамин (2-хлортриэтила-
мин, (2-хлорэтил)диэтиламин).
07/2016:0464
АМИТРИПТИЛИНА ГИДРОХЛОРИД
АтИпр1уНп1 Ьус/госЫопдит
АМ1ТШРТУШЕ НУОКОСШОШОЕ
^СНз
N
| • НС1
СНз
С20Н23М ■ НС1 М.м. 313,9
[549-18-8]
ОПРЕДЕЛЕНИЕ
3-(10,11-Дигидро-5Н-дибензо[а,с/][7]аннулен-5-
илиден)-А/,А/-диметилпропан-1 -амина гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Легко растворим в воде, в 96% спирте и метилен-
хлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амитриптилина гидрохлорида
#или спектр, представленный на рисунке #0464.-1#.
B. 20 мг испытуемого образца дают реакцию (а)
на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)7.
Кислотность или щелочность. 0,20 г испытуемо¬
го образца растворяют в воде, свободной от углерода
диоксида, Р и доводят до объема 10 мл этим же раст¬
ворителем. Прибавляют 0,1 мл раствора метилового
красного Р и 0,2 мл 0,01 Мраствора натрия гидрокси¬
да. Появляется желтое окрашивание. При прибавлении
не более 0,4 мл 0,01 М раствора кислоты хлористо¬
водородной должно появиться красное окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Рисунок #0464.-1. Инфракрасный спектр ФСО амитриптилина гидрохлорида
188
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО дибензосуберо-
на (примесь А) и 5,0 мг ФСО циклобензаприна гидрохло¬
рида (примесь В) растворяют в 5,0 мл испытуемого раст¬
вора и доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 50,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная полимером органосили¬
катным аморфным, с введенными полярными груп¬
пами, октадецилсилильным эндкепированным Р с
размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 35 объемов аце¬
тонитрила Р и 65 объемов раствора 5,23 г/л дикалия
гидрофосфата Р, предварительно доведенного до
рН 7,0 кислотой фосфорной Р;
- скорость подвижной фазы: 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания амитриптилина.
Относительное удерживание (по отношению к
амитриптилину, время удерживания — около 14 мин):
примесь В — около 0,9; примесь А— около 2,2.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси В и амитриптилина.
Предельное содержание примесей:
-примесь В (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси В, не должна превышать площадь соответству¬
ющего пика на хроматограмме раствора сравнения (Ь);
- примесь А (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 0,5 площади
соответствующего пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А и В, не должна превышать площадь пика амитрипти¬
лина на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь пика амитриптилина на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади амитриптилина на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 30 мл
96 % спирта Р и титруют 0,1 М раствором натрия
гидроксида потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 31,39 мг С20Н2зЫ НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О, Е, Р, 6.
А. 10,11 -Дигидро-5Н-дибензо[а,с/|[7]аннулен-5-он
(дибензосуберон).
В. 3-(5Н-Дибензо[а,с/][7]аннулен-5-илиден)-Л/,Л/-
диметилпропан-1 -амин (циклобензаприн).
С. 3-(10,11 -Дигидро-5/-/-дибензо[а,с/|[7]аннулен-5-
илиден)-/У-метилпропан-1 -амин (нортриптилин).
О. Р = СН2-СН2-СН2-Ы(СН3)2:5-[3-(Диметиламино)-
пропил]-10,11 -дигидро-5Я-дибензо[а,с/][7]аннулен-5-ол.
6. Р = Н: 10,11-Дигидро-5Н-дибензо[а,с/|[7]аннулен-
5-ол (дибензосуберол).
Амлодипина бесилат
189
Р. (5Е2,10/?5)-5-[3-(Диметиламино)пропилиден]-
10,11 -дигидро-5Н-дибензо[а,с/|[7]аннулен-10-ол.
07/2016:1491
АМЛОДИПИНА БЕСИЛАТ
Ат1осИр1П1 ЬезНаз
АМШ01РШЕ ВЕЗИАТЕ
[111470-99-6]
$03Н
М.м. 567,1
ОПРЕДЕЛЕНИЕ
3-Этил-5-метил-(4Я5)-2-[(2-аминоэтокси)метил]-
4-(2-хлорфенил)-6-метил-1,4-дигидропиридин-3,5-
дикарбоксилата бензолсульфонат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ПРОИЗВОДСТВО
Необходимо учитывать, что алкилбензолсульфо-
наты являются генотоксичными соединениями и могут
присутствовать в качестве примесей в амлодипина
бесилате. Процесс производства должен соблюдать
принципы управления рисками качества, принимая во
внимание требования к качеству исходных материа¬
лов, мощности производственного процесса и валида¬
ции. Производитель может использовать метод, опи¬
санный в статье 2.5.41. Метил- этил- и изпропилбен-
золсульфонат в фармацевтических субстанциях.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Мало растворим в воде, легко растворим в ме¬
таноле, умеренно растворим в этаноле безводном,
мало растворим в 2-пропаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амлодипина бесилата #или
спектр, представленный на рисунке #1491 .-1#.
ИСПЫТАНИЯ
Угол оптического вращения (2.2.7). От 0,10° до
+0,10°. 0,250 г испытуемого образца растворяют в ме-
таноле Р и доводят до объема 25,0 мл этим же раст¬
ворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от света.
Рисунок #1491 .-1. Инфракрасный спектр ФСО амлодипина бесилата
190
Государственная фармакопея Республики Беларусь
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 5,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
100.0 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
10.0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 2,5 мг ФСО амлодипи-
на примеси В и 2,5 мг ФСО амлодипина примеси (3
растворяют в подвижной фазе и доводят до объема
25.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят подвижной фазой до объема
10.0 мл.
Раствор сравнения (с). 2,5 мг ФСО амлодипина
для идентификации пиков (содержит примеси й, Е,
Р) растворяют в 5 мл подвижной фазы.
Раствор сравнения (ф. 5,0 мг ФСО амлодипина
примеси А растворяют в ацетонитриле Р и дово¬
дят до объема 5,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл. 1,0 мл полученного раствора дово¬
дят подвижной фазой до объема 10,0 мл.
Раствор сравнения (е). 50,0 мг ФСО амлоди¬
пина бесилата растворяют в подвижной фазе и до¬
водят до объема 50,0 мл этим же растворителем.
5.0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: раствор 2,3 г/л аммония аце¬
тата Р — метанол Р (30:70, об/об);
- скорость подвижной фазы. 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 237 нм;
- объем вводимой пробы, по 20 мкл испытуе¬
мого раствора (а) и растворов сравнения (а), (Ь),
(с) и (б);
- время хроматографирования: 2-кратное вре¬
мя удерживания амлодипина.
Идентификация пиков примесей: идентифици¬
руют пики примесей О, Е и Р, используя хроматограм¬
му раствора сравнения (с) и хроматограмму, при¬
лагаемую к ФСО амлодипина для идентификации
пиков; идентифицируют пик примеси А, используя
хроматограмму раствора сравнения (б).
Относительное удерживание (по отношению
к амлодипину, время удерживания — около 20 мин):
примесь О — около 0,21; примесь В — около 0,25;
примесь й — около 0,5; примесь Р — около 0,8; при¬
месь Е — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
месей С и В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 1,7; для примеси Р — 0,7):
- примесь О (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси О, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-примесь А (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
1.5- кратную площадь соответствующего пика на хро¬
матограмме раствора сравнения (б);
-примеси Е, Р (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Е и Р, не должны превышать
1.5- кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, й, Е и Р, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей: не более 0,8 %.
- неучитываемый предел: на хроматограмме
испытуемого раствора не учитывают пики с площа¬
дью менее 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а) (0,05 %) и пик бен-
золсульфоната (относительное удерживание — око¬
ло 0,14).
Вода (2.5.12). Не более 0,5%. Определение
проводят из 1,000 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (е).
Содержание С20Н25С1М2О5 С6Н6О33 рассчитывают
в процентах с учетом содержания амлодипина беси¬
лата в ФСО амлодипина бесилата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, О, Е, Е
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацев¬
тического использования (2034). Вследствие этого
нет необходимости идентифицировать эти приме¬
си для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использова¬
ния): В, С, Н.
Аммиака раствор концентрированный
191
А. 3-Этил-5-метил-(4/?5)-4-(2-хлорфенил)-2-[[2-
(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этокси]-
метил]-6-метил-1,4-дигидропиридин-3,5-дикарбоксилат.
В. 3-Этил-5-метил-(4Я5)-4-(2-хлорфенил)-6-метил-
2-[[2-[[2-(метилкарбамоил)бензоил]амино]этокси]метил]-
1,4-дигидропиридин-3,5-дикарбоксилат.
О. 3-Этил-5-метил-2-[(2-аминоэтокси)метил]-4-(2-
хлрорфенил)-6-метилпиридин-3,5-дикарбоксилат.
Е. Диэтил-(4/?5)-2-[(2-аминоэтокси)метил]-4-(2-
хлорфенил)-6-метил-1,4-дигидропиридин-3,5-дикарбо-
ксилат.
Р. Диметил-(4/?5)-2-[(2-аминоэтокси)метил]-4-(2-
хлорфенил)-6-метил-1,4-дигидропиридин-3,5-дикарбо-
ксилат.
О. Диметил-4-(2-хлорфенил)-2,6-диметил-1,4-
дигидропиридин-3,5-дикарбоксилат.
Н. 2-[[2-[[(4/?5)-4-(2-Хлорфенил)-3-(этоксикарбо-
нил)-5-(метоксикарбонил)-6-метил-1,4-дигидро-пиридин-
2-ил]метокси]этил]карбамоил]бензойная кислота.
07/2016:0877
АММИАКА РАСТВОР
КОНЦЕНТРИРОВАННЫЙ
Аттотае зоЫю сопсеп1га1а
АММОША 80ШТЮЫ, СОЫСЕЫТПАТЕО
ЫН3 М.м. 17,03
ОПРЕДЕЛЕНИЕ
Содержание: не менее 25,0 % (м/м) и не более
30,0 % (м/м).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная, очень едкая жидкость.
Смешивается с водой и 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ).
A. Относительная плотность (2.2.5). От 0,892 до
0,910.
B. Испытуемый образец имеет сильнощелочную
реакцию (2.2.4).
C. К 0,5 мл испытуемого образца прибавляют
5 мл воды Р, пропускают воздух и полученную газо¬
вую смесь направляют на поверхность раствора, со¬
держащего 1 мл 0,1 М раствора кислоты хлористо¬
водородной и 0,05 мл раствора метилового крас¬
ного Р. Красное окрашивание раствора переходит
в желтое. К полученному раствору прибавляют 1 мл
раствора натрия кобальтинитрита Р. Образуется
желтый осадок.
ИСПЫТАНИЯ
Раствор 5.220 мл испытуемого образца выпари¬
вают почти досуха на водяной бане, охлаждают, при¬
бавляют 1 мл кислоты уксусной разведенной Р и до¬
водят водой дистиллированной Р до объема 20 мл.
Прозрачность (2.2.1). К 2 мл испытуемого об¬
разца прибавляют 8 мл воды Р. Полученный раствор
должен быть прозрачным.
Цветность (2.2.2, метод II). Раствор, приготов¬
ленный для испытания «Прозрачность», должен быть
бесцветным.
Восстанавливающие вещества. К 100 мл кис¬
лоты серной разведенной Р, при охлаждении, осто¬
рожно прибавляют 8,8 мл испытуемого образца. За¬
тем прибавляют 0,75 мл 0,002 М раствора калия
перманганата. Полученный раствор выдерживают в
течение 5 мин. Раствор должен сохранять слабо-ро¬
зовое окрашивание.
Пиридин и сопутствующие примеси. Не более
0,0002 % (2 ррт) в пересчете на пиридин. Оптическая
плотность (2.2.25) испытуемого образца, измеренная при
192
Гэсударственная фармакопея Республики Беларусь
длине волны 252 нм, должна быть не более 0,06. В каче¬
стве компенсационного раствора используют воду Р.
Карбонаты. Не более 0,0060 % (60 ррт). 10 мл
испытуемого образца помещают в пробирку со сте¬
клянной притертой пробкой, прибавляют 10 мл раст¬
вора кальция гидроксида Р, тотчас закрывают проб¬
кой и перемешивают. Опалесценция полученного
раствора не должна превышать опалесценцию раст¬
вора сравнения, приготовленного параллельно с ис¬
пользованием 10 мл раствора 0,1 г/л натрия карбо¬
ната безводного Р.
Хлориды (2.4.4). Не более 0,0001 % (1 ррт).
5 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0005 % (5 ррт).
3 мл раствора 5 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Железо (2.4.9). Не более 0,000025 % (0,25 ррт).
4 мл раствора 3 доводят водой Р до объема 10 мл.
Полученный раствор должен выдерживать испытание
на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0001 % (1 ррт). 4 мл раствора 3 доводят водой Р до
объема 20 мл. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (2 ррт РЬ) Р.
Остаток после выпаривания. Не более 20 мг/л.
50 мл испытуемого образца выпаривают на водяной
бане и высушивают при температуре от 100 °С до
105 °С в течение 1 ч. Масса сухого остатка не должна
превышать 1 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50,0 мл 1 М раствора кислоты хлористоводо¬
родной помещают в колбу со стеклянной притертой
пробкой, точно взвешивают, прибавляют 2 мл испыту¬
емого образца и повторно взвешивают. Полученный
раствор титруют 1 М раствором натрия гидроксида
до перехода окраски раствора от красной до желтой,
используя в качестве индикатора 0,1 мл раствора ме¬
тилового красного Р.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 17,03 мг 1ЧН3.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре не выше 20 °С.
07/2016:0007
АММОНИЯ ХЛОРИД
АттопН сЫопс1ит
АММОЫШМ СН1-ОРЮЕ
МН4С1 М.м. 53,49
[12125-02-9]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на хлори¬
ды (2.3.1).
B. 10 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», дают реакцию на соли
аммония (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, по¬
лученной из воды дистиллированной Р, и доводят до
объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,05 мл раствора метилового
красного Р. При прибавлении не более 0,5 мл 0,01 М
раствора кислоты хлористоводородной или 0,01 М
раствора натрия гидроксида окраска раствора
должна измениться.
Бромиды и йодиды. К10 мл раствора 3 прибав¬
ляют 0,1 мл кислоты хлористоводородной разведен¬
ной Р и 0,05 мл раствора хлорамина Р. Через 1 мин
к полученному раствору прибавляют 2 мл хлорофор¬
ма Р и интенсивно встряхивают. Слой хлороформа
должен оставаться бесцветным (2.2.2, метод I).
Сульфаты (2.4.13). Не более 0,0150 % (150 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытания на сульфаты.
Кальций (2.4.3). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на кальций.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 5 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0%. 1,00 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют в 20 мл
воды Р, прибавляют смесь из 5 мл раствора фор¬
мальдегида Р, предварительно нейтрализованного
по раствору фенолфталеина Р, и 20 мл воды Р. По¬
лученный раствор через (1—2) мин медленно титруют
1 М раствором натрия гидроксида, используя в каче¬
стве индикатора 0,2 мл раствора фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 53,49 мг МН4С1.
Амоксициллин натрия
193
07/2016:0577
АМОКСИЦИЛЛИН НАТРИЯ
АтохюННпит паМсит
АМОХ1С1ШЫ ЗСЮШМ
С16Н18М3Ма053 М.м. 387,4
[34642-77-8]
ОПРЕДЕЛЕНИЕ
Натрия (25,5Я6Я)-6-[[(2#)-2-амино-2-(4-гидрокси-
фенил)ацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-
азабицикло-[3.2.0]гептан-2-карбоксилат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 89,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Очень гигро¬
скопичен.
Очень легко растворим в воде, умеренно раство¬
рим в этаноле, очень мало растворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 0,250 г испытуемого образца
растворяют в 5 мл воды Р, прибавляют 0,5 мл уксус¬
ной кислоты разведенной Р, взбалтывают и выдер¬
живают в течение 10 мин в ледяной воде. Кристаллы
отфильтровывают и промывают (2—3) мл смеси, со¬
стоящей из воды Р и ацетона Р (1:9, об/об), затем су¬
шат при температуре 60 °С в течение 30 мин.
Сравнение: ФСО амоксициллина тригидрата
#или спектр, представленный на рисунке #0577.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 10 мл раствора натрия гидро-
карбоната Р.
Раствор сравнения (а). 25 мг ФСО амоксицил¬
лина тригидрата растворяют в 10 мл раствора на¬
трия гидрокарбоната Р.
Раствор сравнения (Ь). 25 мг ФСО амоксицилли¬
на тригидрата и 25 мг ФСО ампициллина тригидра¬
та растворяют в 10 мл раствора натрия гидрокар¬
боната Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: 10 объемов ацетона Р смеши¬
вают с 90 объемами раствора 154 г/л аммония ацета¬
та Р, предварительно доведенного кислотой уксус¬
ной ледяной Р до рН 5,0.
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода до появления пятен, затем просматривают в
дневном свете.
Рисунок #0577.-1. Инфракрасный спектр ФСО амоксициллина тригидрата
194
Государственная фармакопея Республики Беларусь
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
С. Около 2 мг испытуемого образца помещают
в пробирку длиной около 150 мм и диаметром около
15 мм, смачивают 0,05 мл воды Р и прибавляют 2 мл
формальдегида раствора в серной кислоте Р. Со¬
держимое пробирки взбалтывают; раствор практиче¬
ски бесцветный. Пробирку помещают в водяную баню
на 1 мин; появляется темное желтое окрашивание.
О. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят этим же растворителем до
объема 10,0 мл. Испытания проводят немедленно по¬
сле растворения.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность. Раствор 3 в течение небольшого
промежутка времени после приготовления имеет ро¬
зовое окрашивание. Через 5 мин его оптическая плот¬
ность (2.2.25) должна быть не более 0,20 при длине
волны 430 нм.
рН (2.2.3). От 8,0 до 10,0.2,0 г испытуемого образца
растворяют в воде, свободной от углерода диоксида, Р
и доводят до объема 20 мл этим же растворителем.
Удельное оптическое вращение (2.2.7). От
+240 до +290 (в пересчете на безводное вещество).
62,5 мг испытуемого образца растворяют в растворе
4 г/л калия гидрофталата Р и доводят этим же раст¬
ворителем до объема 25,0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 30,0 мг испытуемого
образца растворяют в подвижной фазе А и доводят
до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 30,0 мг испытуемого
образца растворяют в подвижной фазе А и доводят
до объема 20,0 мл этим же растворителем. Готовят
непосредственно перед использованием.
Раствор сравнения (а). 30,0 мг ФСО амоксицил-
лина тригидрата растворяют в подвижной фазе А и
доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 4,0 мг ФСО цефадрокси-
ла растворяют в подвижной фазе А и доводят до объ¬
ема 50 мл этим же растворителем. К 5,0 мл получен¬
ного раствора прибавляют 5,0 мл раствора сравнения
(а) и доводят подвижной фазой А до объема 100 мл.
Раствор сравнения (с). 2,0 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
20,0 мл. 5,0 мл полученного раствора доводят под¬
вижной фазой А до объема 20,0 мл.
Раствор сравнения (д). К 0,20 г амоксициллина
тригидрата Р прибавляют 1,0 мл воды Р. Встряхива¬
ют и прибавляют по каплям раствор натрия гидрок¬
сида разведенный Р для получения раствора. Полу¬
ченный раствор должен иметь рН около 8,5. Раствор
хранится при комнатной температуре в течение 4 ч.
0,5 мл полученного раствора доводят подвижной фа¬
зой А до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: смешивают 1 объем аце¬
тонитрила Р и 99 объемов 25 % (об/об) раст¬
вора 0,2 М раствора калия дигидрофосфата Р,
доведенного до рН 5,0 раствором натрия ги¬
дроксида разведенным Р\
- подвижная фаза В: смешивают 20 объемов
ацетонитрила Р и 80 объемов 25 % (об/об)
раствора 0,2 М раствора калия дигидрофосфа¬
та Р, доведенного до рН 5,0 раствором натрия
гидроксида разведенным Р;
Время (мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
92
8
'я-('к + 25)
92 ->0
00
1
о
о
(<я + 25)-(*я + 40)
0
100
(*я + 40)-(1я + 55)
92
8
^ — время удерживания амоксициллина на хроматограмме
раствора сравнения (с).
Если для достижения необходимого разрешения
изменялось соотношение подвижных фаз А и В, то
подвижную фазу полученного состава используют в
начальном времени градиента и для количественного
определения.
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 50 мкп растворов
сравнения (Ь) и (с) хроматографируют изократически,
используя соотношение подвижных фаз А и В началь¬
ного времени градиента; по 50 мкл испытуемого раст¬
вора (Ь) и раствора сравнения (б) хроматографируют
согласно программе градиента; 50 мкл подвижной
фазы А хроматографируют согласно программе гра¬
диента в качестве контрольного опыта.
Идентификация пиков примесей: используя хро¬
матограмму раствора сравнения (с!) идентифициру¬
ют 3 основных пика, элюированных после основного
пика, соответствующих примеси С, димеру амокси¬
циллина (примесь 3; п = 1) и тримеру амоксициллина
(примесь 3] п = 2).
Относительное удерживание (по отношению к
амоксициллину): примесь С — около 3,4; примесь 3
(п = 1) — около 4,1; примесь (п = 2) — около 4,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками амок¬
сициллина и цефадроксила; при необходимости соот¬
ношение подвижных фаз А и В корректируют.
Предельное содержание примесей:
- примесь 3 (п = 1) (не более 3 %): на хромато¬
грамме испытуемого раствора (Ь) площадь пика, со¬
ответствующего примеси (п= *\), не должна превы¬
шать 3-кратную площадь основного пика на хромато¬
грамме раствора сравнения (с);
- любая другая примесь (не более 2 %): на хро¬
матограмме испытуемого раствора (Ь) площадь лю¬
бого пика, кроме основного и пика примеси и (п = 1),
не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (с);
Амоксициллин натрия
195
- сумма примесей (не более 9 %): на хромато¬
грамме испытуемого раствора (Ь) сумма площадей
всех пиков, кроме основного, не должна превышать
9-кратную площадь основного пика на хроматограмме
раствора сравнения (с);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора (Ь) не учитывают пики
с площадью менее 0,1 площади основного пика на
хроматограмме раствора сравнения (с).
№,№Диметиланилин (2.4.26, метод А или В). Не
более 0,0020 % (20 ррт).
2-Этилгексановая кислота (2.4.28). Не более
0,8 % (м/м).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 0,400 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
0,25 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: исходный состав смеси под¬
вижных фаз А и В, при необходимости откорректиро¬
ванный;
- объем вводимой пробы: по 50 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение: не
более 1,0 % при 6 повторных вводах пробы.
Содержание амоксициллина натрия в процен¬
тах рассчитывают, умножая процентное содержание
амоксициллина на 1,060.
ХРАНЕНИЕ
В. (25,5Я,6Я)-6-[[(25)-2-Амино-2-(4-гидроксифе-
нил)ацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-аза-
бицикло[3.2.0]гептан-2-карбоновая кислота (Ьамокси-
циллин).
С. (4$)-2-[5-(4-Гидроксифенил)-3,6-диоксопиперазин-
2-ил]-5,5-диметилтиазолидин-4-карбоновая кислота
(амоксициллина дикетопиперазины).
Э. (45)-2-[[[(2Я)-2-Амино-2-(4-гидроксифенил)аце-
тил]амино]карбоксиметил]-5,5-диметилтиазолидин-4-
карбоновая кислота (пенициллоиновые кислоты амок¬
сициллина).
и эпимер при С*
Е. (2Я5,45)-2-[[[(2Я)-2-Амино-2-(4-гидроксифенил)-
ацетил]амино]метил]-5,5-диметилтиазолидин-4-
карбоновая кислота (пениллоиновые кислоты амокси¬
циллина).
В воздухонепроницаемом контейнере.
Если субстанция стерильная, ее хранят в сте¬
рильном воздухонепроницаемом контейнере с кон¬
тролем первого вскрытия.
ПРИМЕСИ
н н
А. (25,5Я,6Я)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
Р. 3-(4-Гидроксифенил)пиразин-2-ол.
О. (25,5/?,6Я)-6-[[(2Я)-2-[[(2Я)-2-Амино-2-(4-
гидроксифенил)ацетил]амино]-2-(4-гидроксифенил)-
ацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-азабицик-
ло[3.2.0]гептан-2-карбоновая кислота (0-(4-гидроксифе-
нил)глициламоксициллин).
196
Гэсударственная фармакопея Республики Беларусь
Н. (2Я)-2-[(2,2-Диметилпропаноил)амино]-2-(4-
гидроксифенил)уксусная кислота.
I. (2/?)-2-Амино-2-(4-гидроксифенил)уксусная
кислота.
^ Со-олигомеры амоксициллина и пенициллоино-
вых кислот амоксициллина.
К. Олигомеры пенициллоиновых кислот амокси¬
циллина.
07/2016:0260
АМОКСИЦИЛЛИН ТРИГИДРАТ
АтохюННпит МЬус/псит
АМОХ1С1ШЫ тШУОНАТЕ
[61336-70-7]
ОПРЕДЕЛЕНИЕ
(25,5/?,6Я)-6-[[(2Я)-2-Амино-2-(4-гидроксифенил)-
ацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-азабици-
кло[3.2.0]гептан-2-карбоновая кислота тригидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, очень мало растворим в
96 % спирте, практически нерастворим в жирных мас¬
лах. Растворяется в разведенных кислотах и разве¬
денных растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амоксициллина тригидрата
#или спектр, представленный на рисунке #0260.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 10 мл раствора натрия гидро¬
карбоната Р.
Раствор сравнения (а). 25 мг ФСО амоксицил¬
лина тригидрата растворяют в 10 мл раствора на¬
трия гидрокарбоната Р
Раствор сравнения (Ь). 25 мг ФСО амоксицилли¬
на тригидрата и 25 мг ФСО ампициллина тригидра¬
та растворяют в 10 мл раствора натрия гидрокар¬
боната Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированногоР
Подвижная фаза: ацетон Р — раствор 154 г/л
аммония ацетата Р, доведенный до рН 5,0 кисло¬
той уксусной ледяной Р, (10:90, об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода до проявления пятен и просматривают при днев¬
ном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения (а).
C. 2 мг испытуемого образца помещают в про¬
бирку длиной около 150 мм и диаметром около 15 мм.
Увлажняют 0,05 мл воды Р, прибавляют 2 мл раст¬
вора формальдегида в серной кислоте Р и взбал¬
тывают. Раствор практически бесцветный. Пробирку
выдерживают в водяной бане в течение 1 мин. Появ¬
ляется темно-желтое окрашивание.
ИСПЫТАНИЯ
Раствор 5. С помощью ультразвука или при осто¬
рожном нагревании 0,100 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). 1,0 г испытуемого об¬
разца растворяют в 10 мл 0,5 М раствора кислоты
хлористоводородной (раствор А). 1,0 г испытуемого
образца растворяют в 10 мл раствора аммиака раз¬
веденного Р2 (раствор В). Сразу после приготовления
Амоксициллин тригидрат
197
Рисунок #0260.-1. Инфракрасный спектр ФСО амоксициллина тригидрата
растворы А и В по степени мутности не должны пре¬
вышать эталон II.
рН (2.2.3). От 3,5 до 5,5. Измеряют рН раствора 5.
Удельное оптическое вращение (2.2.7). От
+290 до +315 (в пересчете на безводное вещество).
Определение проводят с использованием раствора 5.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор рН 5,0. К 250 мл 0,2 М раст¬
вора калия дигидрофосфата Р прибавляют раствор
натрия гидроксида разведенный Р до рН 5,0 и дово¬
дят водой Р до объема 1000,0 мл.
Испытуемый раствор (а). 30,0 мг испытуемого
образца растворяют в подвижной фазе А и доводят до
объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). Раствор готовят
непосредственно перед использованием. 30,0 мг ис¬
пытуемого образца растворяют в подвижной фазе А и
доводят до объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 30,0 мг ФСО амоксицил¬
лина тригидрата растворяют в подвижной фазе А и
доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 4,0 мг ФСО цефадрокси-
ла растворяют в подвижной фазе А и доводят до объ¬
ема 50 мл этим же растворителем. К 5,0 полученного
раствора прибавляют 5,0 мл раствора сравнения (а) и
доводят подвижной фазой А до объема 100 мл.
Раствор сравнения (с). 2,0 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
20,0 мл. 5,0 мл полученного раствора доводят под¬
вижной фазой А до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — бу¬
ферный раствор рН 5,0 (1:99, об/об);
- подвижная фаза В: ацетонитрил Р — бу¬
ферный раствор рН 5,0 (20:80, об/об);
Время (мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
°-{к
92
8
'„-('« + 25)
92 —► 0
о
о
7
со
(Гя + 25)-(Г„ + 40)
0
100
((к + 40) - (1н + 55)
92
8
— время удерживания амоксициллина на хроматограм¬
ме раствора сравнения (с).
Если для достижения необходимого разрешения из¬
менялось соотношение подвижных фаз А и В, то подвиж¬
ную фазу полученного состава используют в начальном
времени градиента и для количественного определения.
- скорость подвижной фазы. 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм.
- объем вводимой пробы, по 50 мкл растворов
сравнения (Ь) и (с) хроматографируют изократиче-
ски, используя соотношение подвижных фаз А и В
начального времени градиента; 50 мкл испытуемого
раствора (Ь) хроматографируют согласно программе
градиента; 50 мкл подвижной фазы А хроматографи¬
руют согласно программе градиента в качестве кон¬
трольного опыта.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками амок¬
сициллина и цефадроксила; при необходимости изме¬
няют соотношение подвижных фаз А и В.
Предельное содержание примесей:
-любая примесь (не более 1 %): на хромато¬
грамме испытуемого раствора (Ь) площадь любого
пика, кроме основного, не должна превышать пло-
198
Гэсударственная фармакопея Республики Беларусь
щадь основного пика на хроматограмме раствора
сравнения (с).
№,№-Диметиланилин (2.4.26, метод А или В). Не
более 0,0020 % (20 ррт).
Вода (2.5.72). Не менее 11,5 % и не более 14,5 %.
Определение проводят из 0,100 г испытуемого образца.
Сульфатная зола {2.4.14). Не более 1,0%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота {2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: соотношение подвижных фаз
А и В начального времени градиента в соответствии
с параметрами пригодности хроматографической си¬
стемы;
- объем вводимой пробы, по 50 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- сходимость: максимальное относительное
стандартное отклонение не более 1,0 % для площа¬
дей пиков при 6 повторных вводах пробы.
Содержание С16Н19М305$ рассчитывают в про¬
центах с учетом содержания амоксициллина в ФСО
амоксициллина тригидрата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
н н
А. (25,5/?,6Я)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
В. (25,5Я,6#)-6-[[(25)-2-Амино-2-(4-гидроксифе-
нил)ацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-
азабицикло[3.2.0]гептан-2-карбоновая кислота
(Ьамоксициллин).
С. (45)-2-[5-(4-Гидроксифенил)-3,6-диоксопипе-
разин-2-ил]-5,5-диметилтиазолидин-4-карбоновая кис¬
лота (дикетопиперазины амоксициллина).
гидроксифенил)ацетил]амино]карбоксиметил]-5,5-
диметилтиазолидин-4-карбоновая кислота (пеницил-
лоиновые кислоты амоксициллина).
Е. К = Н: (2/?5,45)-2-[[[(2Я?)-2-Амино-2-(4-гидрокси-
фенил)ацетил]амино]метил]-5,5-диметилтиазолидин-
4-карбоновая кислота (пениллоиновые кислоты амок¬
сициллина).
Р. 3-(4-Гидроксифенил)пиразин-2-ол.
С. (25,5Я6/?)-6-[[(2Р?)-2-[[(2/?)-2-Амино-2-(4-гидро-
ксифенил)ацетил]амино]-2-(4-гидроксифенил)ацетил]-
амино]-3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]-
гептан-2-карбоновая кислота (0-(4-гидроксифенил)гли-
циламоксициллин).
Н. (2Я?)-2-[(2,2-Диметилпропаноил)амино]-2-(4-
гидроксифенил)уксусная кислота.
I. (2/?)-2-Амино-2-(4-гидроксифенил)уксусная кис¬
лота.
3. Со-олигомеры амоксициллина и пенициллои-
новых кислот амоксициллина.
Ампициллин натрия
199
К. Олигомеры пенициллоиновых кислот амокси-
циллина.
1_. (2/?,5/?,6^)-6-[[25,5/?,6/?)-6-[[(2/?)-2-Амино-2-(4-
гидроксифенил)ацетил]амино]-3,3-диметил-7-оксо-4-
тиа-1-азабицикло[3.2.0]гептан-2-карбонил]амино]-3,3-
диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-3-
карбоновая кислота (6-АПК амоксициллина амид).
07/2016:0578
АМПИЦИЛЛИН НАТРИЯ
АтрюННпит паНсит
АМР1С1ШЫ 8ООШМ
[69-52-3]
ОПРЕДЕЛЕНИЕ
Натрия (25,5Я6Я)^[[(2#)-2-амино-2-фенилацетил]-
амино]-3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0),
гептан-2-карбоксилат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 91,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде, умеренно растворим в
ацетоне, практически нерастворим в жирных маслах
и вазелиновом масле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 0,250 г испытуемого образца
растворяют в 5 мл воды Р, прибавляют 0,5 мл кисло¬
ты уксусной разведенной Р, взбалтывают, выдержи¬
вают в течение 10 мин в ледяной бане и фильтруют
через стеклянный фильтр (ПОР 40) (2.1.2) под ваку¬
умом. Кристаллы на фильтре промывают (2—3) мл
смеси вода Р — ацетон Р (1:9, об/об) и сушат при
температуре 60 °С в течение 30 мин.
Сравнение: ФСО ампициллина тригидрата #или
спектр, представленный на рисунке #0578.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 10 мл раствора натрия гидро¬
карбоната Р.
Раствор сравнения (а). 25 мг ФСО ампициллина
тригидрата растворяют в 10 мл раствора натрия
гидрокарбоната Р.
Раствор сравнения (Ь). 25 мг ФСО амоксицилли¬
на тригидрата и 25 мг ФСО ампициллина тригидра¬
та растворяют в 10 мл раствора натрия гидрокар¬
боната Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: смесь из 10 объемов ацетона Р
и 90 объемов раствора 154 г/л аммония ацетата Р,
доведенного до рН 5,0 кислотой уксусной ледяной Р.
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода до проявления пятен и просматривают при днев¬
ном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения (а).
C. 2 мг испытуемого образца помещают в про¬
бирку длиной около 150 мм и диаметром около 15 мм.
Увлажняют 0,05 мл воды Р, прибавляют 2 мл раст¬
вора формальдегида в серной кислоте Р и встря¬
хивают. Раствор практически бесцветный. Пробирку
выдерживают в водяной бане в течение 1 мин. Появ¬
ляется темно-желтое окрашивание.
О. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Прозрачность (2.2.1). 1,0 г испытуемого образца
помещают в коническую колбу и медленно прибавля¬
ют при постоянном перемешивании 10 мл 1 М раст¬
вора кислоты хлористоводородной (раствор А). 1,0 г
испытуемого образца растворяют в воде Р и доводят
до объема 10,0 мл этим же растворителем (раст¬
вор В). Сразу после приготовления растворы А и В по
степени мутности не должны превышать эталон II.
Оптическая плотность (2.2.25). Не более 0,15
при 430 нм. Измеряют оптическую плотность раст¬
вора В сразу после приготовления, как указано в ис¬
пытании «Прозрачность».
рН (2.2.3). От 8,0 до 10,0. 2,0 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 20 мл этим же раство¬
рителем. Через 10 мин после растворения измеряют
рН полученного раствора.
200
Гэсударственная фармакопея Республики Беларусь
Рисунок #0578.-1. Инфракрасный спектр ФСО ампициллина тригидрата
Удельное оптическое вращение (2.2.7). От
+258 до +287 (в пересчете на безводное вещество).
62,5 мг испытуемого образца растворяют в растворе
4 г/л калия гидрофталата Р и доводят до объема
25.0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 31,0 мг испытуемого
образца растворяют в подвижной фазе А и доводят
до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). Раствор готовят
непосредственно перед использованием. 31,0 мг ис¬
пытуемого образца растворяют в подвижной фазе А
и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 27,0 мг ФСО ампицил¬
лина безводного растворяют в подвижной фазе А и
доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 2,0 мг ФСО цефрадина
растворяют в подвижной фазе А и доводят до объема
50 мл этим же растворителем. К 5,0 мл полученного
раствора прибавляют 5,0 мл раствора сравнения (а).
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
20.0 мл.
Раствор сравнения (д). К 0,20 г испытуемого
образца прибавляют 1,0 мл воды Р и нагревают при
температуре 60 °С в течение 1 ч. 0,5 мл полученно¬
го раствора доводят подвижной фазой А до объема
50.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: к 0,5 мл кислоты ук¬
сусной разведенной Р прибавляют 50 мл 0,2 М
раствора калия дигидрофосфата Р, 50 мл
ацетонитрила Р и доводят водой Р до объе**а
1000 мл;
- подвижная фаза В: к 0,5 мл кислоты ук¬
сусной разведенной Р прибавляют 50 мл 0,2 М
раствора калия дигидрофосфата Р, 400 мл
ацетонитрила Р и доводят водой Р до объема
1000 мл;
Время (мин)
Подвижная
фаза А
(%,об/об)
Подвижная
фаза В
(%, об/об)
85
15 |
30)
85 —> 0
15->100 !
<4, + 30)-(4, + 45)
0
100
(*„+ 45)-<4,+ 60)
85
15 !
(я — время удерживания ампициллина на хроматограмме
раствора сравнения (с).
Если для достижения необходимого разрешения
изменялось соотношение подвижных фаз А и В, то
подвижную фазу полученного состава используют в
начальном времени градиента и для количественного
определения.
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 50 мкл растворов
сравнения (Ь) и (с) хроматографируют изократически,
используя соотношение подвижных фаз А и В началь¬
ного времени градиента; по 50 мкл испытуемого раст¬
вора (Ь) и раствора сравнения (д) хроматографируют
согласно программе градиента; 50 мкл подвижной
фазы А хроматографируют согласно программе гра¬
диента в качестве контрольного опыта.
Идентификация пиков: идентифицируют пики
ампициллина и димера ампициллина, используя хро¬
матограмму раствора сравнения (д).
Ампициллин натрия
201
Относительное удерживание (по отношению к
ампициллину): димер ампициллина — около 2,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками ампи¬
циллина и цефрадина; при необходимости изменяют
соотношение подвижных фаз А и В;
Предельное содержание примесей:
- димер ампициллина (не более 4,5 %): на хрома¬
тограмме испытуемого раствора (Ь) площадь пика, со¬
ответствующего димеру ампициллина, не должна пре¬
вышать 4,5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (с);
- любая другая примесь (не более 2 %): на хро¬
матограмме испытуемого раствора (Ь) площадь любо¬
го пика, кроме основного и димера ампициллина, не
должна превышать 2-кратную площадь основного пика
на хроматограмме раствора сравнения (с).
№,№Диметиланилин (2.4.26, метод В). Не более
0,0020 % (20 ррт).
2-Этилгексановая кислота (2.4.28). Не более
0,8 % (м/м).
Метиленхлорид. Не более 0,2 % (м/м). Газовая
хроматография (2.2.28).
Раствор внутреннего стандарта. 1,0 мл эти-
ленхлорида Р растворяют в воде Р и доводят до объ¬
ема 500,0 мл этим же растворителем.
Испытуемый раствор (а). 1,0 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 10,0 мл
этим же растворителем.
Испытуемый раствор (Ь). 1,0 г испытуемого об¬
разца растворяют в воде Р, прибавляют 1,0 мл раст¬
вора внутреннего стандарта и доводят водой Р до объ¬
ема 10,0 мл.
Раствор сравнения. 1,0 мл метиленхлорида Р
растворяют в воде Р и доводят до объема 500,0 мл
этим же растворителем. К 1,0 мл полученного раствора
прибавляют 1,0 мл раствора внутреннего стандарта и
доводят водой Р до объема 10,0 мл.
Условия хроматографирования:
- колонка стеклянная длиной 1,5 м и диаметром
4 мм, заполненная диатомитом для газовой хрома¬
тографии Р, импрегнированным 10 % (м/м) макрогола
1000 Р]
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 40 мл/мин;
-температура: колонка — 60 °С, блок ввода
проб — 100 °С,.детектор — 150 °С;
- детектор: пламенно-ионизационный.
Содержание метиленхлорида рассчитывают с
учетом плотности метиленхлорида при 20 °С, равной
1,325 г/мл.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эталон
готовят с использованием 2 мл эталонного раствора
свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 2,0 %. Определение про¬
водят из 0,300 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,15 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
#Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным. Тест-доза — 20 мг испы¬
туемого образца в 1 мл воды для инъекций Р на 1,0 кг
массы тела кролика.
Испытание «Пирогенность» проводят в качестве
альтернативного испытанию «Бактериальные эндо¬
токсины».
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
#Стерильность (2.6.1). Если указано, что суб¬
станция является стерильной, испытуемый образец
должен выдерживать испытание на стерильность.
Ампициллин натрия в условиях испытания обладает
антимикробным действием. Для устранения антими¬
кробного действия посев на питательные среды про¬
водят методом мембранной фильтрации.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: соотношение подвижных фаз А
и В начального времени градиента в соответствии с па¬
раметрами пригодности хроматографической системы;
- объем вводимой пробы: по 50 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение:
не более 1,0 % для площадей пиков при 6 повторных
вводах пробы.
Содержание ампициллина натрия в процентах
рассчитывают, умножая процентное содержание ам¬
пициллина на 1,063.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
н н
А. (25,5Я,6Я)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
В. (25,5#,6/?)-6-[[(25)-2-Амино-2-фенилацетил]-
амино]-3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]-
гептан-2-карбоновая кислота (Ьампициллин).
С. (45)-2-(3,6-Диоксо-5-фенилпиперазин-2-ил)-5,5-
диметилтиазолидин-4-карбоновая кислота (дикетопи-
перазины ампициллина).
202
Государственная фармакопея Республики Беларусь
O. К = С02Н: (45)-2-[[[(2Я?)-2-Амино-2-фенилацетил]-
амино]карбоксиметил]-5,5-диметилтиазолидин-4-карбо-
новая кислота (пенициллоиновые кислоты ампициллина).
P. К = Н: (2Я5,45)-2-[[[(2Я)-2-Амино-2-фенил-
ацетил]-амино]метил]-5,5-диметилтиазолидин-4-карбо-
новая кислота (пениллоиновые кислоты ампициллина).
Е. (2#)-2-[[[(25,5Я,6Я)-6-[[(2Я)-2-Амино-2-
фенилацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-
азабицикло[3.2.0]гепт-2-ил]карбонил]амино]-2-фенил-
уксусная кислота (ампициллинил-О-фенилглицин).
С. (3/?,6Я)-3,6-Дифенилпиперазин-2,5-дион.
Н. З-Фенилпиразин-2-ол.
I. (25,5Я,6Я)-6-[[(2Я)-2-[[(2Я)-2-Амино-2-
фенилацетил]амино]-2-фенилацетил]амино]-3,3-
диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоновая кислота (Р-фенилтицилампициллин).
3. (25,5#,6#)-6-[(2,2-Диметилпропаноил)амино]-
3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоновая кислота.
К. (2#)-2-[(2,2-Диметилпропаноил)амино]-2-
фенилуксусная кислота.
1_. (2Я)-2-Амино-2-фенилуксусная кислота
Р-фенилглицин).
М. Со-олигомеры ампициллина и пенициллоино-
вых кислот ампициллина.
N. Олигомеры пенициллоиновых кислот ампицил¬
лина.
07/2016:0168
АМПИЦИЛЛИН ТРИГИДРАТ
АтрюННпит Шус1псит
АМР1С1ШЫ ТРиНУОРАТЕ
С1бн19ыз045 ■ ЗН20 М.м. 403,5
[7177-48-2]
ОПРЕДЕЛЕНИЕ
(2$,5Я,6Я)-6[[(2Я)-2-Амино-2-фенилацетил]-
амино]-3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]-
гептан-2-карбоновая кислота.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, практически нераство¬
рим в 96 % спирте и в жирных маслах. Растворяется в
разведенных растворах кислот и гидроксидов щелоч¬
ных металлов.
Ампициллин тригидрат
203
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ампициллина тригидрата #или
спектр, представленный на рисунке #0168.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 10 мл раствора натрия гидро¬
карбоната Р.
Раствор сравнения (а). 25 мг ФСО ампициллина
тригидрата растворяют в 10 мл раствора натрия
гидрокарбоната Р.
Раствор сравнения (Ь). 25 мг ФСО амоксицилли-
на тригидрата и 25 мг ФСО ампициллина тригидра¬
та растворяют в 10 мл раствора натрия гидрокар¬
боната Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: смесь из 10 объемов ацето¬
на Р и 90 объемов раствора 154 г/л аммония ацета¬
та Р, доведенного до рН 5,0 кислотой уксусной ле¬
дяной Р.
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода до проявления пятен и просматривают при днев¬
ном саете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты, на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения (а).
С. 2 мг испытуемого образца помещают в про¬
бирку длиной около 150 мм и диаметром около 15 мм,
увлажняют 0,05 мл воды Р, прибавляют 2 мл раст¬
вора формальдегида в серной кислоте Р и встря¬
хивают. Раствор практически бесцветный. Пробирку
выдерживают в водяной бане в течение 1 мин. Появ¬
ляется темно-желтое окрашивание.
О. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Прозрачность (2.2.1). 1,0 г испытуемого об¬
разца растворяют в 10 мл 1 М раствора кислоты
хлористоводородной (раствор А). 1,0 г испытуемого
образца растворяют в 10 мл раствора аммиака раз¬
веденного Р2 (раствор В). Сразу после приготовления
растворы А и В по степени мутности не должны пре¬
вышать эталон II.
рН (2.2.3). От 3,5 до 5,5. 0,1 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 40 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+280 до +305 (в пересчете на безводное вещество).
62,5 мг испытуемого образца растворяют в воде Р и
доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 31,0 мг испытуемого
образца растворяют в подвижной фазе А и доводят
до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). Раствор готовят
непосредственно перед использованием. 31,0 мг ис¬
пытуемого образца растворяют в подвижной фазе А
и доводят до объема 10,0 мл этим же растворителем.
?» -
96 -
96 4
94 -
92 -
90 |
• 88 |
5 I
х $
»> 86 4
о
^ ?
84 4
!
82 I
80 -
78 -
76 т
74 -
/Л
Л
3000
1000
2000 1500
Волновое число (см'1)
Рисунок #0168.-1. Инфракрасный спектр пропускания ФСО ампициллина тригидрата
26’ За* *06С
204
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (а). 27,0 мг ФСО ампицил¬
лина безводного растворяют в подвижной фазе А и
доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 2 мг ФСО цефрадина
растворяют в подвижной фазе А и доводят до объема
50 мл этим же растворителем. К 5 мл полученного
раствора прибавляют 5 мл раствора сравнения (а).
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: к 0,5 мл кислоты уксус¬
ной разведенной Р прибавляют 50 мл 0,2 М раст¬
вора калия дигидрофосфата Р, 50 мл ацетони¬
трила Р и доводят водой Р до объема 1000 мл;
- подвижная фаза В: к 0,5 мл кислоты уксус¬
ной разведенной Р прибавляют 50 мл 0,2 М раст¬
вора калия дигидрофосфата Р, 400 мл ацетони¬
трила Р и доводят водой Р до объема 1000 мл;
Время (мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
85
15
'*-('* + 30)
85 — 0
15 — 100
(1я + 30) - ((к + 45)
0
100
(1я + 45) - (1я + 60)
85
15
— время удерживания ампициллина на хроматограмме
раствора сравнения (с).
Если для достижения необходимого разрешения
изменялось соотношение подвижных фаз А и В, то
подвижную фазу полученного состава используют в
начальном времени градиента и для количественного
определения.
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 50 мкл растворов
сравнения (Ь) и (с) хроматографируют изократиче-
ски, используя соотношение подвижных фаз А и В
начального времени градиента; 50 мкл испытуемого
раствора (Ь) хроматографируют согласно программе
градиента; 50 мкл подвижной фазы А хроматографи¬
руют согласно программе градиента в качестве кон¬
трольного опыта.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками ампи¬
циллина и цефрадина; при необходимости изменяют
соотношение подвижных фаз А и В;
Предельное содержание примесей:
- любая примесь (не более 1,0 %): на хромато¬
грамме испытуемого раствора (Ь) площадь любого
пика, кроме основного, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с).
№,№Диметиланилин (2.4.26, метод В). Не бо¬
лее 0,0020 % (20 ррт).
Вода (2.5.12). Не менее 12,0 % и не более 15,0 %.
Определение проводят из 0,100 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: соотношение подвижных фаз
А и В начального времени градиента в соответствии
с параметрами пригодности хроматографической си¬
стемы;
- объем вводимой пробы: по 50 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение:
не более 1,0 % для площадей пиков при 6 повторных
вводах пробы.
Содержание С16Н19М3045 рассчитывают в про¬
центах с учетом содержания ампициллина в ФСО ам¬
пициллина безводного.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
А. (25,5/?,6Я)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
В. (25,5Я,6Я)-6-[[(25)-2-Амино-2-фенилацетил]-
амино]-3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]-
гептан-2-карбоновая кислота (Ьампициллин).
С. (45)-2-(3,6-Диоксо-5-фенилпиперазин-2-ил)-5,5-
диметилтиазолидин-4-карбоновая кислота (дикетопи-
перазины ампициллина).
й. Р = С02Н: (45)-2-[[[(2Р)-2-Амино-2-фенилацетил]-
амино]карбоксиметил]-5,5-диметилтиазолидин-4-
карбоновая кислота (пенициллоиновые кислоты ампи¬
циллина);
Амфотерицин В
205
Р. К = Н: (2Я$,45)-2-[[[(2Я)-2-Амино-2-фенил-
ацетил]-амино]метил]-5,5-диметилтиазолидин-4-карбо-
новая кислота (пениллоиновые кислоты ампициллина).
Е. (2Я)-2-[[[(25,5Я,6Я)-6-[[(2Я)-2-Амино-2-
фенилацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-
азабицикло[3.2.0]гепт-2-ил]карбонил]амино]-2-фенил-
уксусная кислота (ампициллинил-О-фенилглицин).
М. Со-олигомеры ампициллина и пенициллано-
вых кислот ампициллина.
6. (ЗЯ,6Я)-3,6-Дифенилпиперазин-2,5-дион.
Н. З-Фенилпиразин-2-ол.
I. (25,5Я,6Я)-6-[[(2Я)-2-[[(2Я)-2-Амино-2-
фенилацетил]амино]-2-фенилацетил]амино]-3,3-
диметп-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоновая кислота (О-фенилтицилампициллин).
Л. (25,5Я,6Я)-6-[(2,2-Диметилпропаноил)амино]-
3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоновая кислота.
К. (2Я)-2-[(2,2-Диметилпропаноил)амино]-2-
фенилуксусная кислота.
н ЫИ2
^^^СОгН
Ь. (2Я)-2-Амино-2-фенилуксусная кислота
(О-фенилглицин).
N. (35)-6-[[(2Я)-2-Амино-2-фенилацетил]амино]~
2,2-диметил-7-оксо-2,3,4,7-тетрагидро-1,4-тиазепин-3-
карбоновая кислота.
07/2016:1292
АМФОТЕРИЦИН В
АтрШепстит В
АМРНОТЕШС1Ы В
С47Н73М017 М.м. 924
[1397-89-3]
ОПРЕДЕЛЕНИЕ
Смесь противогрибковых полиенов, обра¬
зуемых посредством роста определенных штам¬
мов 31гер1отусез пос/озиз или полученных дру¬
гими способами. Состоит преимущественно из
амфотерицина В, представляющего собой (1Я,
35,5Я,6Я,9Я, 11 Я, 1 55,16Я, 1 7Я, 185,19Е,21 Е,
23Е,25Е,27Е,29Е,31Е,ЗЗЯ,355,36Я,37$)-33-[(3-
Амино-3,6-дидезокси-р-0-маннопиранозил)окси]-
1,3,5,6,9,11,17,37-октагидрокси-15,16,18-триметил-
13-оксо-14,39-диокса-бицикло[33.3.1]нонатриаконта-
19,21,23,25,27,29,31-гептаен-36-карбоновую кислоту.
Содержание: не менее 750 МЕ/мг (в пересчете на
сухое вещество).
27. Зак. 1060.
206
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Желтый или оранжевый порошок. Гигроскопичен.
Практически нерастворим в воде, растворим в
диметилсульфоксиде и пропиленгликоле, мало рас¬
творим в диметилформамиде, очень мало растворим
в метаноле, практически нерастворим в 96 % спирте.
В разведенных растворах чувствителен к свету.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 25 мг испытуемого образ¬
ца растворяют в 5 мл диметилсульфоксида Р и дово¬
дят метанолом Р до объема 50 мл. 2 мл полученного
раствора доводят метанолом Р до объема 200 мл.
Диапазон длин волн: от 300 нм до 450 нм.
Максимумы поглощения: при 362 нм, 381 нм и
405 нм.
Отношение оптических плотностей:
-^381-°т °.57 до 0,61;
-Л381М405 — от 0,87 Д° 0,93.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО амфотерицина В.
Если спектры отличаются, то сушат испытуемый
образец и ФСО амфотерицина В при температуре
60 °С и давлении не выше 0,7 кПа в течение 1 ч и по¬
лучают новые спектры.
C. К 1 мл раствора 0,5 г/л диметилсульфокси¬
да Р прибавляют, избегая смешивания 2 жидкостей,
5 мл кислоты фосфорной Р для образования нижне¬
го слоя. На границе раздела двух жидкостей незамед¬
лительно появляется синее кольцо. При смешивании
образуется синее окрашивание. Прибавляют 15 мл
воды Р и перемешивают; раствор становится бледно-
желтым.
й. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме
испытуемого раствора при длине волны 383 нм соот¬
ветствует по времени удерживания основному пику на
хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29). Растворы защищают от света и ис¬
пользуют в течение 24 ч после приготовления, за ис¬
ключением раствора сравнения (с), который исполь¬
зуется незамедлительно после его приготовления.
Смесь растворителей. Раствор 10 г/л аммония
ацетата Р — И-метилпирролидон Р — метанол Р
(1:1:2, об/об/об).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 15 мл И-метилпирролидона Р и
в течение 2 ч доводят смесью растворителей до объ¬
ема 50,0 мл. 5,0 мл полученного раствора доводят
смесью растворителей до объема 25,0 мл.
Раствор сравнения (а). 20,0 мг ФСО амфотери¬
цина В растворяют в 15 мл Ы-метилпирролидона Р и
в течение 2 ч доводят смесью растворителей до объ¬
ема 50,0 мл. 5,0 мл полученного раствора доводят
смесью растворителей до объема 25,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят смесью растворителей до объема
100,0 мл.
Раствор сравнения (с). 20,0 мг ФСО нистатина
растворяют в 15 мл Ы-метилпирролидона Р и в те¬
чение 2 ч доводят смесью растворителей до объема
50,0 мл. 5,0 мл полученного раствора доводят рас¬
твором сравнения (а) до объема 25,0 мл. 2,0 мл полу¬
ченного раствора доводят смесью растворителей до
объема 100,0 мл.
Раствор сравнения (д). Для приготовления при¬
месей В и С 10 мг испытуемого образца растворяют
в 5 мл И-метилпирролидона Р ив течение 2 ч при¬
бавляют 35 мл смеси из метанола Р и этанола без¬
водного Р (1:4, об/об). Прибавляют 0,10 мл кислоты
хлористоводородной разведенной Р, перемешивают
и инкубируют при температуре 25 °С в течение 2,5 ч.
Прибавляют 10 мл раствора 10 г/л аммония ацета¬
та Р и перемешивают.
Раствор сравнения (е). 4 мг ФСО амфотерици¬
на В для идентификации пиков (содержит примеси
А и В) растворяют в 5 мл И-метилпирролидона Р ив
течение 2 ч доводят смесью растворителей до объ¬
ема 50 мл.
Холостой раствор. Смесь растворителей.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным, деактивированным по отношению к ос¬
нованиям, эндкепированным для хроматографии Р
(размер частиц 3 мкм);
- температура: 20 °С;
- подвижная фаза: смешивают 35 мл воды Р с
56 мл ацетонитрила Р, выдерживают до установле¬
ния равновесия, доводят объем раствора водой Р до
объема 100 мл и перемешивают;
- подвижная фаза:
- подвижная фаза А: метанол Р — ацетони¬
трил Р — раствор 4,2 г/л кислоты лимонной Р,
предварительно доведенный раствором аммиака
концентрированным до рН 4,7 (1:3:6:, об/об/об);
- подвижная фаза В: метанол Р — раствор
4,2 г/л кислоты лимонной Р, предварительно
доведенный раствором аммиака концентриро¬
ванным до рН 3,9 — ацетонитрил Р (12:20:68,
об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
100
0
3—23
о
N.
Т
О
о
0 — 30
23—33
70 — 0
о
о
т
со
33—40
0
100
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор:
- длина волны 303 нм: определение тетраенов;
- длина волны 383 нм: определение гептаенов;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с), (д) и (е).
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (е) и хроматограмму, прилага¬
емую к ФСО амфотерицина В для идентификации
пиков.
Относительное удерживание (по отношению
к амфотерицину В, время удерживания — около
16 мин): примесь В — около 0,75; примесь А— около 0,8;
нистатин — около 0,85.
Пригодность хроматографической системы
при длине 383 нм: раствор сравнения (д):
Амфотерицин В
207
-разрешение: не менее 1,5 между 2 пиками,
имеющими относительное удерживание около 0,7.
Предельное содержание примесей:
- примесь А при длине волны 303 нм: на хрома¬
тограмме испытуемого раствора площадь пика, со¬
ответствующего примеси А при длине волны 303 нм,
не должна превышать 2,5-кратную площадь основ¬
ного пика на хроматограмме раствора сравнения
(с) (5,0 %) и не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с)
(2,0 %), если субстанция предназначена для произ¬
водства лекарственных средств для парентерально¬
го применения;
- любая другая примесь при длине волны 303 нм
(не более 1,0 %): на хроматограмме испытуемо¬
го раствора площадь любого пика при длине волны
303 нм, кроме основного и пика примеси А при дли¬
не волны 303 нм, не должна превышать 0,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примесь В при длине волны 383 нм (не более
4.0 %): на хроматограмме испытуемого раствора пло¬
щадь пика, соответствующего примеси В при длине
волны 383 нм, не должна превышать 4-кратную пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- любая другая примесь при длине волны 383 нм
(не более 2,0 %): на хроматограмме испытуемо¬
го раствора площадь любого пика при длине волны
383 нм, кроме основного и пика примеси В при дли¬
не волны 383 нм, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей при длинах волн 303 и 383 нм:
не более 15,0 %;
- неучитываемый предел при длине волны
303 нм (0,1 %): на хроматограмме испытуемого раст¬
вора не учитывают пики с площадью менее 0,05 пло¬
щади основного пика на хроматограмме раствора
сравнения (с);
- неучитываемый предел при длине волны
383 нм (0,1 %): на хроматограмме испытуемого раст¬
вора не учитывают пики с площадью менее 0,1 пло¬
щади основного пика на хроматограмме раствора
сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 60 °С и давлении не выше 0,7 кПа.
Сульфатная зола (2.4.14). Не более 3,0 % и не
более 0,5 %, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения. Определение проводят из 1,0 г
испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
1.0 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Растворы защищают от света на протяжении
всего количественного определения. 25,0 мг испыту¬
емого образца растворяют в диметилсульфоксиде Р
и, встряхивая, доводят до объема 25,0 мл этим же
растворителем. Полученный исходный раствор при
постоянном перемешивании разводят диметилсуль-
фоксидом Р для получения растворов с соответствую¬
щими концентрациями (подходящими концентрациями
являются следующие: 44,4, 66,7 и 100 МЕ/мл). Конеч¬
ные растворы готовят разведением 1:20 с помощью
0,2 М фосфатного буферного раствора рН 10,5 таким
образом, чтобы они содержали 5 % (об/об) диметил-
сульфоксида. Раствор сравнения и испытуемый раст¬
вор готовят одновременно. Проводят количественное
определение антибиотиков микробиологическим ме¬
тодом (2.7.2).
ХРАНЕНИЕ
В защищенном от света месте при температуре
от 2 °С до 8 °С в воздухонепроницаемом контейнере.
Если субстанция стерильная, ее хранят в стерильном
контейнере с контролем первого вскрытия.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. Амфотерицин А (28,29-дигидро-амфотерицин В).
27*. Зак. 1060.
208
Гэсударственная фармакопея Республики Беларусь
07/2016:2406
АНАСТРОЗОЛ
Апа81гою1илп
ОПРЕДЕЛЕНИЕ
2,2'-[5-(1 Н-1,2,4-Триазол-1 -илметил)бензол-1,3-
диил]бис(2-метилпропаннитрил).
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде, легко растворим
в этаноле, практически нерастворим в циклогексане.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО анастрозола #или спектр, пред¬
ставленный на рисунке #2406.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, различаются, то испыту¬
емый образец и ФСО анастрозола растворяют по от¬
дельности в ацетоне Р и выпаривают до сухих остат¬
ков, которые используют для получения новых спектров.
АЫА8ТК0201-Е
сы
С17н19м5 М.м. 293,4
[120511-73-1]
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р1 —
вода для хроматографии Р (50:50, об/об).
Испытуемый раствор (а). 25 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 25,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 200,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
30 1
25 |
20 :
3000 2000 1500 1000 500
Волновое число (см'1)
Рисунок #2406.-1. Инфракрасный спектр ФСО анастрозола
Анастрозол
209
Раствор сравнения (Ь). 2,5 мг ФСО анастрозола
примеси Е растворяют в 20,0 мл смеси растворите¬
лей. 1,0 мл полученного раствора доводят испытуе¬
мым раствором (а) до объема 50,0 мл.
Раствор сравнения (с). 25,0 мг ФСО анастрозо¬
ла растворяют в смеси растворителей и доводят до
объема 200,0 мл этой же смесью растворителей.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная полимером органосили¬
катным аморфным, с введенными полярными груп¬
пами, октадецилсилильным зндкепированным Р с
размером частиц 3,5 мкм;
- подвижная фаза:
- подвижная фаза А: кислота фосфорная Р —
вода для хроматографии Р (0,1:100, об/об);
- подвижная фаза В: кислота фосфорная Р —
ацетонитрил Р1 (0,1:100, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
95
5
2—54
95 — 35
5 — 65
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (Ь).
Идентификация пиков примесей: идентифици¬
руют пик примеси Е, используя хроматограмму раст¬
вора сравнения (Ь).
Относительное удерживание (по отношению
к анастрозолу, время удерживания — около 29 мин):
примесь Е — около 1,05.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,5 между пиками ана¬
строзола и примеси Е.
Расчет процентного содержания:
- рассчитывают процентное содержание каждой
примеси с учетом концентрации анастрозола в рас¬
творе сравнения (а).
Предельное содержание примесей:
- неспецифицированные примеси: не более 0,10 %;
- сумма примесей: не более 0,2 %;
- учитываемый предел: 0,05 %.
Вода (2.5.32). Не более 0,3 %. Определение про¬
водят из 50,0 мг испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С17Н191М5 в процентах рассчитывают с
учетом содержания анастрозола в ФСО анастрозола.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): А, В, С, О, Е, Р, 6, Н, I.
си
А. 2-[3-[(1 /?5)-1 -Цианоэтил]-5-(1 Н-1,2,4-триазол-1 -
илметил)фенил]-2-метилпропаннитрил.
В. (2/?5>)-2,3-Бис[3-(1-циано-1-метилэтил)-5-(1 Н-
1,2,4-триазол-1 -илметил)фенил]-2-метилпропаннитрил.
си
С. 2,2’-[5-(Бромметил)бензол-1,3-диил]бис(2-
метилпропаннитрил).
Вг сы
О. 2,2’-[5-(Дибромметил)бензол-1,3-диил]бис(2-
метилпропаннитрил).
си
Е. 2,2’-[5-(Гидроксиметил)бензол-1,3-диил]бис(2-
метилпропаннитрил).
Р. 4-Метилбензолсульфоновая кислота.
28. Зак. 1060.
210
Гэсударственная фармакопея Республики Беларусь
О. 2,2’-[5-(4Н-1,2,4-Триазол-4-илметил)бензол-1,3-
диил]бис(2-метилпропаннитрил).
сы
Н. 2,2’-(5-Метилбензол-1,3-диил)бис(2-метилпро-
паннитрил).
см
I. 2,2’-[5-(Хлорметил)бензол-1,3-диил]бис(2-
метилпропаннитрил).
07/2016:0804
АНИСОВОЕ МАСЛО
Фронт подвижной фазы: не менее 15 см от
линии старта (для обычных ТСХ пластинок) или не
менее б см от линии старта (для ТСХ пластинок с
маленьким размером частиц).
Высушивание: на воздухе.
Проявление А: пластинку просматривают в
ультрафиолетовом свете при длине волны 254 нм.
Результаты А: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения
и испытуемого раствора. На хроматограмме испы¬
туемого раствора могут обнаруживаться и другие
зоны.
Верх хроматограс
эической пластинки
Анетол: зона поглощения
Анисовый альдегид: зона
поглощения
Зона очень сильного
поглощения (анетол)
Зона поглощения
Зона поглощения (анисовый
альдегид)
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку опрыскивают реак¬
тивом метил-4-ацетилбензоата Р и нагревают
при температуре от 100 °С до 105 °С в течение
10 мин; просматривают еще горячую хроматограм¬
му при дневном свете в течение 5 мин.
Результаты В: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения
и испытуемого раствора. На хроматограмме испы¬
туемого раствора могут обнаруживаться и другие
зоны.
Лл/5/ аеМего1еит
АЫ13Е О//.
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное методом перегон¬
ки с водяным паром из высушенных зрелых плодов
Р/шр/ле//а амзит 1_.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная или бледно-желтая жид¬
кость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 г испытуемого образца
растворяют в толуоле Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения. 10 мкл линалола Р, 30 мкл
анисового альдегида Р и 200 мкл анетола Р раство¬
ряют в толуоле Р и доводят до объема 15 мл этим же
растворителем. 1 мл полученного раствора доводят
толуолом Р до объема 5 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: этилацетат Р — толуол Р
(7:93, об/об).
Наносимый объем пробы: 5 мкл в виде полос
длиной 10 мм (для обычных ТСХ пластинок) или 2 мкл
в виде полос длиной 10 мм (для ТСХ пластинок с ма¬
леньким размером частиц).
Верх хроматографической пластинки
Анетол: коричневая зона
Фиолетово-коричневая зона
(монотерпеновые углеводо¬
роды)
(фронт растворителя)
Очень интенсивная
коричневая зона (анетол),
отчетливо отделенная
Анисовый альдегид: желтая
зона
Серая зона
Желтая зона (анисовый
альдегид)
Линалол: серая зона
Серая зона (линалол)
Серая зона
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, получен¬
ные в испытании «Хроматографический профиль».
Результаты: характерные пики на хромато¬
грамме испытуемого раствора по времени удержи¬
вания соответствуют характерным пикам на хрома¬
тограмме раствора сравнения.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,980 до
0,990.
Показатель преломления (2.2.6). От 1,552 до
1,561.
Температура затвердевания (2.2.18). От 15 °С
до 19 °С.
Анисовое масло
211
Фенхон. Газовая хроматография (2.2.28), как ука¬
зано в испытании «Хроматографический профиль»,
со следующими изменениями.
Испытуемый раствор. 400 мкп испытуемого об¬
разца растворяют в 2,0 мл гексана Р.
Раствор сравнения (а). 10 мкл фенхона Р дово¬
дят гексаном Р до 1,2 г.
Раствор сравнения (Ь). 100 мкл раствора срав¬
нения (а) доводят гексаном Р до объема 100 мл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- отношение сигнал/шум: не менее 10 для ос¬
новного пика.
Предельное содержание примесей:
- фенхон: не более 0,01 %.
Феникулин. Газовая хроматография (2.2.28), как
указано в испытании «Сопутствующие примеси», со
следующими изменениями.
Испытуемый раствор. Испытуемый образец.
Раствор сравнения (а). 10 мг испытуемого раст¬
вора доводят гексаном Р до 1,000 г. 0,5 мл получен¬
ного раствора доводят гексаном Р до объема 100 мл.
Раствор сравнения (Ь). ФСО феникулина для
идентификации пиков.
Пригодность хроматографической системы:
- хроматограмма раствора сравнения (Ь) должна
соответствовать хроматограмме, прилагаемой к ФСО
феникулина для идентификации пиков',
- отношение сигнал/шум: не менее 10 для основ¬
ного пика на хроматограмме раствора сравнения (а).
Предельное содержание примесей: обнаружи¬
вают пик, принадлежащий феникулину, с помощью
хроматограммы, прилагаемой к ФСО феникулина для
идентификации пиков:
- феникулин: не более 0,01 %.
Жирные и минеральные масла (2.8.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 200 мкл испытуемого об¬
разца растворяют в 1,0 мл гексана Р.
Раствор сравнения. К 1,0 мл гексана Р прибав¬
ляют 20 мкл линалола Р, 20 мкл эстрагола Р, 20 мкл
а-терпинеола Р, 60 мкл анетола Р и 30 мкл анисово¬
го альдегида Р.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и диаметром 0,25 мм, покрытая слоем макрогола
20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
60
5—80
60 — 210
80—95
210
Блок ввода
проб
200
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,2 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения; отмечают времена
удерживания всех веществ.
1. Линалол. 3. а-Терпинеол. 5. Транс-анетол. 7. Псевдоизоэвгенил-2-метилбутират.
2. Эстрагол. 4. Цис-анетол. 6. Анисальдегид.
Рисунок #0804.-1 .Хроматограмма для испытания «Хроматографический профиль анисового масла»
28*. Зак. 1060.
212
Гэсударственная фармакопея Республики Беларусь
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,5 между пиками эстра-
гола и а-терпинеола.
По временам удерживания компонентов раствора
сравнения определяют состав испытуемого раствора;
цис-анетол и псевдоизоэвгенил-2-метилбутират иден¬
тифицируют, используя хроматограмму, приведенную
на рисунке 0804.-1 (не учитывают пик гексана).
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- линалол: не более 1,5 %;
- эстрагол: от 0,5 % до 5,0 %;
- а-терпинеол: не более 1,2 %;
- цис-анетол: от 0,1 % до 0,4 %;
- транс-анетол: от 87 % до 94 %;
- анисовый альдегид: от 0,1 % до 1,4 %;
- псевдоизоэвгенил-2-метилбутират: от 0,3 %
до 2,0 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:1811
АПЕЛЬСИНОВОЕ МАСЛО
АигапШ с1и1с18 ае01его1еит
51УЕЕ7" ОРАЫСЕ 011.
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное механическим ме¬
тодом без нагревания из свежей цедры плода СИгиз
81пеп818 (1_.) ОзЬеск (СИгиз аигапИит 1_. уаг. ди1аз 1_.).
Может содержать подходящий антиоксидант.
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная, бледно-желтого или оранжевого цвета
подвижная жидкость с характерным запахом свежей
цедры апельсина. Может мутнеть при охлаждении.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Просматривают хроматограммы, полученные в
испытании «Бергаптен».
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора.
Верх хроматографической пластинки
Бергаптен: зеленовато-жел¬
тая флуоресцирующая зона
Многочисленные голубые
флуоресцирующие зоны
Раствор сравнения
Испытуемый раствор
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Линалилацетат:
коричневато-оранжевая
флуоресцирующая зона
Коричневая
флуоресцирующая зона
Тусклая коричневато¬
оранжевая
флуоресцирующая зона
(линалилацетат)
Линалол:коричневато¬
оранжевая
флуоресцирующая зона
Бергаптен: тусклая
зеленовато-желтая
флуоресцирующая зона
Многочисленные оранжевые
флуоресцирующие зоны
Коричневато-оранжевая
флуоресцирующая зона
(линалол)
Многочисленные
коричневато-оранжевые
флуоресцирующие зоны
Многочисленные голубые
флуоресцирующие зоны
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Результаты: характерные пики на хроматограм¬
ме испытуемого раствора по времени удерживания
соответствуют характерным пикам на хроматограмме
раствора сравнения.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,842до 0,850.
Показатель преломления (2.2.6). От 1,470 до 1,476.
Угол оптического вращения (2.2.7). От +94° до +99°.
Перекисное (пероксидное) число (2.5.5, ме¬
тод В). Не более 20.
Жирные и минеральные масла (2.8.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Бергаптен. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,2 мл испытуемого об¬
разца растворяют в 1 мл 96 % спирта Р.
Раствор сравнения. 2 мг бергаптена Р, 10 мкл
линалола Р и 20 мкл линалилацетата Р растворяют
в 10 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — толуол Р
(15:85, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 365 нм.
Результаты А: на хроматограмме испытуемого
раствора не должна обнаруживаться зона с зеленова¬
то-желтой флуоресценцией, соответствующая зоне с
зеленовато-желтой флуоресценцией на хроматограм¬
ме раствора сравнения.
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р и нагревают при темпера¬
туре от 100 °С до 105 °С в течение 10 мин; просматри¬
вают хроматограмму в ультрафиолетовом свете при
длине волны 365 нм.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 300 мкл испытуемого об¬
разца растворяют в 1 мл ацетона Р.
Аргинин
213
Раствор сравнения (а). 10 мкл а-пинена Р, 10 мкл
Р-линена Р, 10 мкл сабинена Р, 20 мкл 0-мирцена Р,
800 мкл лимонена Р, 10 мкл октаналя Р, 10 мкл де-
каналя Р, 10 мкл линалола Р, 10 мкл цитраля Р (со¬
ставляющие нераль и гераниапь) и 10 мкл валенце-
на Р растворяют в 1 мл ацетона Р.
Раствор сравнения (Ь). 5 мкл р-пинена Р раство¬
ряют в 10 мл ацетона Р. 0,5 мл полученного раствора
доводят ацетоном Р до объема 10 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренне* диаметром 0,53 мм, покрытая слоем ма-
ярогола 20 000 Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—6
50
6—31
50 — 150
31—41
150-180
41—55
180
I Блок ввода
проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,5 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения (а); отмечают време¬
на удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,9 для пиков (3-пинена
и сабинена; не менее 1,5 для пиков валенцена и ге-
раниапя.
По временам удерживания компонентов раствора
сравнегмя (а) определяют состав испытуемого раствора.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- а-пинен: от 0,4 % до 0,6 %;
- Р-пинен: от 0,02 % до 0,3 %;
- сабинен: от 0,2 % до 1,1 %;
- р-мирцен: от 1,7 % до 2,5 %;
- лимонен: от 92,0 % до 97,0 %;
- октаналь: от 0,1 % до 0,4 %;
- деканаль: от 0,1 % до 0,4 %;
- линалол: от 0,2 % до 0,7 %;
- нераль: от 0,02 % до 0,10 %;
- валенцен: от 0,02 % до 0,5 %;
- гераниаль: от 0,03 % до 0,20 %.
Неучитываемый предел (0,01 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади пика на хроматограмме
раствора сравнения (Ь).
Остаток после выпаривания. От 1,0 % до 5,0 %.
5,0 г испытуемого образца выпаривают на водяной
бане и сушат при температуре от 100 °С до 105 °С в
течение 4 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
#В заполненном доверху воздухонепроницаемом
контейнере, в защищенном от света месте# при тем¬
пературе не выше 25 °С.
#МАРКИРОВКА
Указывают наименование и концентрацию до¬
бавленного антиоксиданта.
07/2016:0806
АРГИНИН
Агдттит
АКвМШЕ
ин
С,Н14Ы402 М.м. 174,2
[74-79-3]
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-5-гуанидинпентановая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Гигроскопичен.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», имеет сильнощелочную реакцию
(2.2.4).
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО аргинина.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО аргинина высушивают при
температуре 105 °С и записывают новые спектры.
Р. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в растворе 10,3 г/л кислоты хлори¬
стоводородной Р и доводят до объема 50 мл этим же
растворителем.
Раствор сравнения. 10 мг ФСО аргинина раство¬
ряют в растворе 10,3 г/л кислоты хлористоводород¬
ной Р и доводят до объема 50 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: аммиака раствор концентри¬
рованный Р — 2-пропанол Р (30:70, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 105 °С до полно¬
го удаления аммиака.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 15 мин.
29. Зак. 1060.
214
Гэсударственная фармакопея Республики Беларусь
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения.
Е. 25 мг испытуемого образца растворяют в
2 мл воды Р, прибавляют 1 мл раствора а-нафто-
лаР и 2 мл смеси из равных объемов раствора
натрия гипохлорита концентрированного Р и
воды Р. Появляется красное окрашивание.
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность {2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От
+25,5 до +28,5 (в пересчете на сухое вещество). 2,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 25,0 мл этим
же растворителем.
Нингидрин-положительные вещества. Анализ
аминокислот (2.2.56). Для анализа используют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Вода Р или буферный раствор для
приготовления образца, подходящий для применяе¬
мого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт NН^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (д). 30 мг изолейцина Р и 30 мг
лейцина Р растворяют в растворе А и доводят до объ¬
ема 50,0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят раствором А до объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
вводят в аминокислотный анализатор. Запускают под¬
ходящую программу для определения физиологиче¬
ских аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм, — ис¬
пользуют концентрацию аргинина в растворе сравне¬
ния (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм, — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
- любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт). К
5 мл раствора 3 прибавляют 0,5 мл кислоты азотной
разведенной Р и доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 %(300 ррт).
К 10 мл раствора 3 прибавляют 1,7 мл кислоты хло¬
ристоводородной разведенной Р и доводят водой
дистиллированной Р до 15 мл. Полученный раствор
должен выдерживать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Аргинина аспартат
215
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
воды Р и титруют 0,1 М раствором кислоты хлори¬
стоводородной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 17,42 мг С6НиЫ402.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи-
. щенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
друпа/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при-
мео» для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): А, В, С.
Н МН2
Н21М^ ^ ^ "со2н
А. (гЗ^г.б-Диаминогексановая кислота (лизин).
Н МН2
Н2М М ^.
хо2н
B. (25)-2-Амино-5-(карбамоиламино)пентановая
кислота (цитруллин).
N42
^со2н
C. (25)-2,5-Диаминопентановая кислота (орнитин).
н2мх
07/2016:2096
АРГИНИНА АСПАРТАТ
Агдти?/ азраг1аз
АРвШМЕ АЗРАРТАТЕ
н ын2
Н2М. .Й. • но2с.
со2н
с*н«н<о2сан7но<
[7675-83-4]
Н ЫН2
со2н
М.м. 307,3
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-5-гуанидинпентановой кислоты
(25)-2-аминобутандиоат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые гранулы или порошок.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО аргинина аспартата #или
спектр, представленный на рисунке #2096.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживаются два пятна, соответству-
Рисунок #2096.-1. Инфракрасный спектр ФСО аргинина аспартата
г?' ъ=* ■ :е:
216
Гэсударственная фармакопея Республики Беларусь
ющие по расположению, цвету и размеру двум основ¬
ным пятнам на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)7.
рН (2.2.3). От 6,0 до 7,0. Измеряют рН раствора 5.
Удельное оптическое вращение (2.2.7). От +25
до +27 (в пересчете на сухое вещество). 2,50 г испы¬
туемого образца растворяют в кислоте хлористово¬
дородной разведенной Р и доводят до объема 25,0 мл
этим же растворителем.
Нингидрин-положительные вещества. Тонкос¬
лойная хроматография (2.2.27).
Испытуемый раствор (а). 0,20 г испытуемого
образца растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 10 мл.
Раствор сравнения (а). 25 мг аргинина Р и 25 мг
кислоты аспарагиновой Р растворяют в воде Р и до¬
водят до объема 25 мл этим же растворителем.
Раствор сравнения (Ь). 2 мл раствора сравнения
(а) доводят водой Р до объема 50 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р.
Подвижная фаза: раствор аммиака Р — пропа¬
нол Р (36:64, об/об).
Наносимый объем пробы: 5 мкп.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 105 °С в течение
10 мин.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Предельное содержание примесей:
-любая примесь (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
двух основных пятен, должно быть не интенсивнее
каждого из двух основных пятен на хроматограмме
раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2,5 мл раствора 5 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
К 0,5 г испытуемого образца прибавляют 2,5 мл кис¬
лоты хлористоводородной разведенной Р и доводят
водой дистиллированной Р до объема 15 мл. Испы¬
тание проводят через 30 мин после приготовления
раствора.
Аммония соли (2.4.1). Не более 0,0100%
(100 ррт). Определение проводят из 100 мг испыту¬
емого образца.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 60 °С в течение 24 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
80,0 мг испытуемого образца растворяют в 2 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 10,24 мг СбН14М402 С4Н7Ы04.
07/2016:0805
АРГИНИНА ГИДРОХЛОРИД
Агдтт1 ЬуйгосЫогШит
АКв/МЫЕ НУОКОСШОПЮЕ
Н N4,
Н
Т
N4
Сбн14ы402 ■ НС1 М.м. 210,7
[1119-34-2]
ОПРЕДЕЛЕНИЕ
(5)-2-Амино-5-гуанидинпентановой кислоты ги¬
дрохлорид.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: А, С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО аргинина гидрохлорида #или
спектр, представленный на рисунке #0805.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
О. 25 мг испытуемого образца растворяют в 2 мл
воды Р, прибавляют 1 мл раствора а-нафтола Р и
Аргинина гидрохлорид
217
3000
1000
2000 1500
Волновое число (см'1)
Рисунок #0805.-1. Инфракрасный спектр ФСО аргинина гидрохлорида
2 мл смеси из равных объемов раствора натрия ги¬
похлорита концентрированного Р и воды Р. Появля¬
ется красное окрашивание.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)е.
Удельное оптическое вращение (2.2.7). От
+21,0 до +23,5 (в пересчете на сухое вещество). 2,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 25,0 мл этим
же растворителем.
Нингидрин-положительные вещества. Испы¬
тание проводят одним из приведенных ниже методов/
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Вода Р или буферный раствор для
приготовления образца, подходящий для применяе¬
мого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт ЫН^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (с1). 30 мг изолейцина Р и
30 мг лейцина Р растворяют в растворе А и доводят
до объема 50,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
вводят в аминокислотный анализатор. Запускают под¬
ходящую программу для определения физиологиче¬
ских аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм, — ис¬
пользуют концентрацию аргинина в растворе сравне¬
ния (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм, — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
30. Зак. 1060.
218
Государственная фармакопея Республики Беларусь
Предельное содержание примесей:
- любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО аргинина ги¬
дрохлорида растворяют в воде Р и доводят до объема
50 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО аргинина ги¬
дрохлорида и 10 мг ФСО лизина гидрохлорида раст¬
воряют в воде Р и доводят до объема 25 мл этим же
растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: аммиака раствор концентри¬
рованный Р— 2-пропанол Р (30:70, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре 105 °С до полно¬
го удаления аммиака.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
-любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до 15 мл. Полученный раствор должен выдерживать
испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хрома¬
тографии, то испытание «Аммония соли» (2.4.1, ме¬
тод В) проводят следующим образом. 50 мг испытуе¬
мого образца должны выдерживать испытание на ам¬
мония соли. Эталон готовят с использованием 0,1 мл
эталонного раствора аммония (100 ррт NН^ Р.#
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,180 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 21,07 мг СбН14М402 НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства'-соответствия требованиям.
См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования):
А, В, С.
н2ы
А. (25)-2,6-Диаминогексановая кислота (лизин).
Н ЫНз
С02Н
B. (25)-2-Амино-5-(карбамоиламино)пентановая
кислота (цитруллин).
н мн2
н2*к ЗС
со2н
C. (25)-2,5-Диаминопентановая кислота (орнитин).
Артикаина гидрохлорид
219
07/2016:1688
АРТИКАИНА ГИДРОХЛОРИД
АгИсат/ ЬудгосЫопдит
АКТ1СА1ЫЕ НУОКОСШ-ОШОЕ
С13Н20М2О35 ■ НС1 М.м. 320,8
[23964-57-0]
ОПРЕДЕЛЕНИЕ
Метил-4-метил-3-[[(2Р5)-2-(пропиламино)пропа-
ноил]амино]тиофен-2-карбоксилата гидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в растворе 1 г/л кислоты хлори¬
стоводородной Р и доводят до объема 100,0 мл этим
же растворителем. 5,0 мл полученного раствора дово¬
дят раствором 1 г/л кислоты хлористоводородной Р
до объема 100,0 мл.
Диапазон длин волн: от 200 нм до 350 нм.
Максимум поглощения: при 272 нм.
Удельный показатель поглощения в максимуме:
от 290 до 320.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 20 мкп испытуемого раствора
по каплям наносят на диск массой 300 мг.
0,1 г испытуемого образца растворяют в 5 мл
воды Р, прибавляют 3 мл насыщенного раствора на¬
трия гидрокарбоната Р и дважды встряхивают с
метиленхлоридом Р порциями по 2 мл. Метилен-
хлоридные слои объединяют, доводят метиленхло¬
ридом Р до объема 5,0 мл и высушивают с помощью
натрия сульфата безводного Р.
Сравнение: ФСО артикаина гидрохлорида.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 5 мл 96 % спирта Р.
Раствор сравнения. 20 мг ФСО артикаина ги¬
дрохлорида растворяют в 5 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р.
Подвижная фаза: триэтиламин Р — этилаце-
татР — гептан Р (10:35:65, об/об/об).
Наносимый объем пробы: 5 мкп.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
й. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,50 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод /). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 4,2 до 5,2. 0,20 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 20,0 мл этим же раст¬
ворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 10,0 мг ФСО артика¬
ина примеси А и 5,0 мг ФСО артикаина примеси Е
растворяют в подвижной фазе и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (с). К 50,0 мг ФСО артикаи¬
на гидрохлорида прибавляют 1,0 мл раствора сравне¬
ния (Ь) и доводят подвижной фазой до объема 50 мл.
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (Ь) доводят подвижной фазой до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная сферическим силикагелем окта-
децилсилильным эндкепированным для хроматогра¬
фии Р (размер частиц 5 мкм) с удельной площадью
поверхности 335 м2/г и содержанием углерода 19 %;
- температура: 45 °С;
-подвижная фаза: смешивают 25 объемов аце¬
тонитрила Р и 75 объемов раствора, приготовленного
следующим образом: 2,02 г натрия гептансульфона-
таР и 4,08 г калия дигидрофосфата Р растворяют в
воде Р и доводят до объема 1000 мл этим же раство¬
рителем; доводят до рН 2,0 кислотой фосфорной Р;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 276 нм;
- объем вводимой пробы: по 10 мкп испытуемого
раствора и растворов сравнения (а), (с) и (с!);
- время хроматографирования: 5-кратное вре¬
мя удерживания артикаина.
Относительное удерживание (по отношению к
артикаину, время удерживания — около 9 мин): при¬
месь А — около 0,8; примесь Е — около 0,86.
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 1,2 между пиками при¬
меси А и примеси Е.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству-
30*. Зак. 1060.
220
Гэсударственная фармакопея Республики Беларусь
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (й);
- неспецифицированные примеси (не более
0,10 %) на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (а);
- сумма неспецифицированных примесей (не более
0,5 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
А, не должна превышать 5-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 4,0 г испытуемого образца раство¬
ряют в 20,0 мл воды Р. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
В. 4-Метил-3-[[(2Р?5)-2-(пропиламино)пропаноил]-
амино]тиофен-2-карбоновая кислота (артикаиновая
кислота).
С. 1 -Метилэтил-4-метил-3-[[(2#5)-2-(пропиламино)-
пропаноил]амино]тиофен-2-карбоксилат (артикаина
изопропиловый эфир).
О. Метил-3-[[(2Я5)-2-(этиламино)пропаноил]амино]-
4-метилтиофен-2-карбоксилат (этилартикаин).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в сме¬
си из 5,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 50 мл 96 % спирта Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 32,08 мг С13Н201\12О35НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): В, С, О, Е, Р, 6, Н, I, 3.
Е. Метил-4-метил-3-[[(2Я5)-2-[(1-метилэтил)ами-
но]пропаноил]амино]тиофен-2-карбоксилат(изопропи-
лартикаин).
Р. 4-Метил-Л/-пропил-3-[[(2Я5)-2-(пропиламино)-
пропаноил]амино]тиофен-2-карбоксамид (артикаино-
вой кислоты пропионамид).
6. Метил-3-[[(2Я5)-2-(бутиламино)пропаноил]-
амино]-4-метилтиофен-2-карбоксилат(бутилартикаин).
и энантиомер
Н. Метил-3-[[(2Я5)-2-(дипропиламино)пропаноил]-
амино}4-метилтиофен-2-карбоксилат(дипропилартикаин).
.о
А. Метил-3-[[2-(пропиламино)ацетил]амино]-4-
метилтиофен-2-карбоксилат (ацетамидоартикаин).
I. Метил-3-амино-4-метилтиофен-2-карбоксилат
(3-аминоартикаин).
Аскорбиновая кислота
221
3. Метил-3-[[(2/?5)-2-бромпропаноил]амино]-4-
метитиофен-2-карбоксилат (бром-производное).
07/2016:0253
АСКОРБИНОВАЯ КИСЛОТА
Ас1с1ит азсогЫсит
А8СОКВ1С АСЮ
С6Н806 М.м. 176,1
[50-81-7]
ОПРЕДЕЛЕНИЕ
(5Я)-5-[(15)-1,2-Дигидроксиэтил]-3,4-дигидрокси-
фуран-2(5А/)-он.
Содержание: не менее 99,0 % и не более 100,5 %.
Температура плавления: около 190 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в водеР и немедленно доводят
до объема 100,0 мл этим же растворителем. К 10 мл
0,1 М раствора кислоты хлористоводородной при¬
бавляют 1,0 мл полученного раствора и доводят во¬
дой Р до объема 100,0 мл.
Максимум поглощения: при 243 нм; определение
проводят немедленно после приготовления испытуе¬
мого раствора.
Удельный показатель поглощения в максимуме:
от 545 до 585.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО аскорбиновой кислоты #или
спектр, представленный на рисунке #0253.-1#.
C. рН (2.2.3): от 2,1 до 2,6. Измеряют рН раст¬
вора 3, приготовленного как указано в разделе «Ис¬
пытания».
О. К1 мл раствора 3 прибавляют 0,2 мл кислоты
азотной разведенной Р и 0,2 мл раствора серебра
нитрата Р2. Образуется серый осадок.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. При воздействии воз¬
духа и влаги становится бесцветным.
Легко растворим в воде, умеренно растворим в
96 % спирте.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Рисунок #0253.-1. Инфракрасный спектр ФСО аскорбиновой кислоты
222
Гэсударственная фармакопея Республики Беларусь
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)7.
Удельное оптическое вращение (2.2.7). От
+20,5 до +21,5. 2,50 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем.
Примесь Е. Не более 0,2 %.
Испытуемый раствор. 0,25 г испытуемого об¬
разца растворяют в 5 мл воды Р и нейтрализуют рас¬
твором натрия гидроксида разведенным Р, прибав¬
ляют 1 мл кислоты уксусной разведенной Р и 0,5 мл
раствора кальция хлорида Р.
Раствор сравнения. 70 мг кислоты щавелевой
Р растворяют в воде Р и доводят до объема 500 мл
этим же растворителем. К 5 мл полученного раствора
прибавляют 1 мл кислоты уксусной разведенной Р и
0,5 мл раствора кальция хлорида Р.
Растворы выдерживают в течение 1 ч. Опалес¬
ценция испытуемого раствора должна быть не более
интенсивной, чем опалесценция раствора сравнения.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Фосфатный буферный раствор. 6,8 г калия ди¬
гидрофосфата Р растворяют в воде Р и доводят до
объема около 175 мл этим же растворителем, филь¬
труют (размер пор 0,45 мкм) и доводят водой Р до
объема 1000 мл.
Испытуемый раствор. 0,500 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО аскорбино¬
вой кислоты примеси С растворяют в подвижной фазе
и доводят до объема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО аскорбино¬
вой кислоты примеси О и 5,0 мг ФСО аскорбиновой
кислоты растворяют в подвижной фазе, прибавляют
2.5 мл раствора сравнения (а) и доводят подвижной
фазой до объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл. К
1,0 мл полученного раствора прибавляют 1,0 мл раст¬
вора сравнения (а).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4.6 мм, заполненная силикагелем аминопропилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 45 °С;
- подвижная фаза: фосфатный буферный раст¬
вор — ацетонитрил Р1 (25:75, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 2,5-кратное
время удерживания аскорбиновой кислоты.
Идентификация пиков примесей: идентифици¬
руют пики примесей С и О, используя хроматограмму
раствора сравнения (Ь).
Относительное удерживание (по отношению к
аскорбиновой кислоте, время удерживания — около 11
мин): примесь О — около 0,4; примесь С — около 1,7.
Пригодность хроматографической системы:
- разрешение: не менее 3,0 между пиками аскор¬
биновой кислоты и примеси С на хроматограмме
раствора сравнения (с);
- отношение сигнал/шум: не менее 20 для пика
примеси С на хроматограмме раствора сравнения (Ь);
Предельное содержание примесей:
- примеси С и О (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков приме¬
сей С и О не должны превышать 1,5-кратные площади
соответствующих пиков на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей С и О, не должна превышать площадь пика
аскорбиновой кислоты на хроматограмме раствора
сравнения (Ь);
- сумма примесей, кроме примесей С и О (не бо¬
лее 0,2 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пиков
примесей С и О, не должна превышать 2-кратную пло¬
щадь пика аскорбиновой кислоты на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади пика аскорбиновой
кислоты на хроматограмме раствора сравнения (Ь).
Медь. Не более 0,0005 % (5 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод /).
Испытуемый раствор. 2,0 г испытуемого образ¬
ца растворяют в 0,1 М растворе кислоты азотной и
доводят до объема 25,0 мл этим же растворителем.
Растворы сравнения. Готовят растворы сравне¬
ния, содержащие 0,2 ррт, 0,4 ррт и 0,6 ррт Си, раз¬
ведением эталонного раствора меди (10 ррт Си) Р
0,1 М раствором кислоты азотной.
Источник излучения: лампа с полым катодом
для определения меди.
Длина волны: 324,8 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
В качестве холостого раствора используют 0,1 М
раствор кислоты азотной.
Железо. Не более 0,0002 % (2 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод /).
Испытуемый раствор. 5,0 г испытуемого образ¬
ца растворяют в 0,1 М растворе кислоты азотной и
доводят до объема 25,0 мл этим же растворителем.
Растворы сравнения. Готовят растворы сравне¬
ния, содержащие 0,2 ррт, 0,4 ррт и 0,6 ррт Ре, раз¬
ведением эталонного раствора железа (20 ррт Ре) Р
0,1 М раствором кислоты азотной.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
В качестве холостого раствора используют 0,1 М
раствор кислоты азотной.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Аспарагиновая кислота
223
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в сме¬
си из 10 мл кислоты серной разведенной Р и 80 мл
воды, свободной от углерода диоксида, Р, прибавля¬
ют 1 мл раствора крахмала Р и титруют 0,05 М рас¬
твором йода до получения устойчивого фиолетово¬
синего окрашивания.
1 мл 0,05 Мраствора йода соответствует 8,81 мг
сен8о6.
ХРАНЕНИЕ
В неметаллическом контейнере в защищенном
от света месте.
ПРИМЕСИ
Специфицированные примеси: С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, Р, С, Н.
А. 2-Фуральдегид.
он он о
он
6. Метил-(2Я)-2-[(2Я)-3,4-дигидрокси-5-оксо-2,5-
дигидрофуран-2-ил]-2-гидроксиацетат.
07/2016:0797
АСПАРАГИНОВАЯ КИСЛОТА
Аас1ит азрагИсит
АЗРАКТ1С АСЮ
но2с.
ЫНг
"С02Н
С4Н7Ы04 М.м. 133,1
[56-84-8]
ОПРЕДЕЛЕНИЕ
(25)-2-Аминобутандиовая кислота.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Мало растворима в воде, практически нераство¬
рима в 96 % спирте. Растворяется в разведенных рас¬
творах минеральных кислот и разведенных растворах
гидроксидов щелочных металлов.
он он о
С. О-Ксило-гекс-2-улосоновая кислота (О-сорбо-
соновая кислота).
он он о
О. Метил-й-ксило-гекс-2-улосонат (метил-й-
сорбосонат).
о
Е. Щавелевая кислота.
он
но' он
Р. (5/?)-5-[(1 Р)-1,2-Дигидроксиэтил]-3,4-дигидро-
ксифуран-2(5Н)-он.
О. (2Я)-2-[(2#)-3,4-Дигидрокси-5-оксо-2,5-ди-
гидрофуран-2-ил]-2-гидроксиуксусная кислота.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Суспензия 1 г испытуемого образца в 10 мл
воды Р имеет сильнокислую реакцию (2.2.4).
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО кислоты аспарагиновой.
й. Просматривают хроматограмму, полученную
в испытании «Нингидрин-положительные вещества».
Результаты: на хроматограмме испытуемого
раствора (Ь) основное пятно по расположению, цвету
и размеру соответствует основному пятну на хромато¬
грамме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют в 1 М растворе кислоты хлористоводородной
и доводят до объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От
+24,0 до +26,0 (в пересчете на сухое вещество).
2,000 г испытуемого образца растворяют в кислоте
хлористоводородной Р1 и доводят до объема 25,0 мл
этим же растворителем.
224
Гэсударственная фармакопея Республики Беларусь
Нингидрин-положительные вещества. Тонко¬
слойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в 2 мл раствора аммиака Р и до¬
водят водой Р до объема 10 мл.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО кислоты
аспарагиновой растворяют в 2 мл раствора аммиака
разведенного Р1 и доводят водой Р до объема 50 мл.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО кислоты
аспарагиновой и 10 мг ФСО кислоты глутаминовой
растворяют в 2 мл раствора аммиака разведенно¬
го Р1 и доводят водой Р до объема 25 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром нингидрина Р и высушивают при температуре от
100 °С до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в 3 мл кис¬
лоты азотной разведенной Р и доводят водой Р до
объема 15 мл. Полученный раствор должен выдержи¬
вать испытание на хлориды. Вместо кислоты азот¬
ной разведенной Р прибавляют 1 мл воды Р.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
0,5 г испытуемого образца растворяют в 4 мл кис¬
лоты хлористоводородной Р и доводят до объема
15 мл водой дистиллированной Р. Полученный раст¬
вор должен выдерживать испытание на сульфаты.
Опалесценцию испытуемого раствора сравнивают с
опалесценцией эталона через 30 мин.
Соли аммония (2.4.1, метод В). Не более
0,0200 % (200 ррт). 50 мг испытуемого образца долж¬
ны выдерживать испытание на соли аммония. Эталон
готовят с использованием 0,1 мл эталонного раст¬
вора аммония (100 ррт NН^ Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре от 100 °С до 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 50 мл
воды, свободной от углерода диоксида, Р, слегка на¬
гревая при необходимости, охлаждают и прибавляют
0,1 мл раствора бромтимолового синего Р1. Титру¬
ют 0,1 М раствором натрия гидроксида до перехода
окраски раствора от желтой до синей.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 13,31 мгС4Н7М04.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0973
АСПАРТАМ
АзраЛатит
А8РАКТАМЕ
С14н18м205 М.м. 294,3
[22839-47-0]
ОПРЕДЕЛЕНИЕ
(35)-3-Амино-4-[[(25)-1 -метокси-1 -оксо-3-фенил-
пропан-2-ил]амино]-4-оксобутановая кислота (метил-
а-Ь-аспартил-Ь-фенилаланинат).
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Слегка гигроскопичен.
Умеренно растворим или мало растворим в воде
и в 96 % спирте, практически нерастворим в гексане и
в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,1 г испытуемого образ¬
ца растворяют в 96% спирте Р и доводят до объема
100,0 мл этим же растворителем.
Диапазон длин волн: от 230 нм до 300 нм.
Аспартам
225
Максимумы поглощения: при 247 нм, при 252 нм,
при 258 нм и при 264 нм.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО аспартама.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 15 мг испытуемого об¬
разца растворяют в 2,5 мл воды Р и доводят кисло¬
той уксусной Р до объема 10 мл.
Раствор сравнения. 15 мг ФСО аспартама раст¬
воряют в 2,5 мл воды Р и доводят кислотой уксус¬
ной Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: вода Р — кислота муравьи¬
ная безводная Р — метанол Р — метиленхлорид
(2:4:30:64, об/об/об/об).
Наносимый объем пробы: 20 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 °С
до 105 °С в течение 15 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
й. 20 мг испытуемого образца растворяют в 5 мл
метанола Р, прибавляют 1 мл раствора гидроксила-
мина щелочного Р1 и нагревают на водяной бане в
течение 15 мин. Раствор охлаждают, доводят до рН 2
кислотой хлористоводородной разведенной Р и при¬
бавляют 0,1 мл раствора железа (III) хлорида Р1. По¬
является коричневато-красное окрашивание.
ИСПЫТАНИЯ
Раствор 3. 0,8 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят объем раствора этим же растворителем до
100 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона СУ(ЗЖ)6.
Удельная электропроводность (2.2.38). Не бо¬
лее 30 мкСм см-1. 0,8 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100,0 мл этим же растворителем.
Измеряют удельную электропроводность испыту¬
емого раствора (С,) и воды, из которой был приготов¬
лен испытуемый раствор (С2). Стабильность измерен¬
ных значений должны быть в пределах 1 % в течение
30 сек.
Рассчитывают удельную электропроводность по
формуле:
С1 - 0,992С2.
Удельное оптическое вращение (2.2.7). От
+14,5 до +16,5 (в пересчете на сухое вещество). 2,00 г
испытуемого образца растворяют в растворе 690 г/л
кислоты муравьиной безводной Р и доводят до объ¬
ема 50,0 мл этим же растворителем. Измерение про¬
водят не позднее 30 мин после приготовления испы¬
туемого раствора.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,60 г испытуемого об¬
разца растворяют в смеси из 1,5 объемов кислоты
уксусной ледяной Р и 98,5 объемов воды Р и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (а). 4,5 мг ФСО примеси А
аспартама растворяют в смеси из 1,5 объемов кис¬
лоты уксусной ледяной Р и 98,5 объемов воды Р и
доводят до объема 50,0 мл этой же смесью раство¬
рителей.
Раствор сравнения (Ь). 30,0 мг фенилаланина Р
(примесь С) растворяют в смеси из 15 объемов кисло¬
ты уксусной ледяной Р и 85 объемов воды Р и дово¬
дят до объема 100,0 мл этой же смесью растворите¬
лей. 1,0 мл полученного раствора доводят водой Р до
объема 10,0 мл.
Раствор сравнения (с). 5,0 мл испытуемого
раствора доводят водой Р до объема 10,0 мл. 3,0 мл
полученного раствора доводят водой Р до объема
100.0 мл.
Раствор сравнения (д). 30,0 мг 1--аспартил-1--
фенилаланина Р (примесь В) растворяют в смеси из
15 объемов кислоты уксусной ледяной Р и 85 объ¬
емов воды Р и доводят до объема 100,0 мл этой же
смесью растворителей. 1,0 мл полученного раствора
доводят водой Р до объема 10,0мл. Смешивают
1.0 мл полученного раствора с 1,0 мл раствора срав¬
нения (Ь).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
(5—10) мкм;
- подвижная фаза: ацетонитрил Р — раствор
6,8 г/л калия дигидрофосфата Р, доведенного до рН
3,7 кислотой фосфорной Р (10:90, об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания аспартама.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не мене 3,5 между пиками приме¬
си В и примеси С.
Предельное содержание примесей:
- примесь А (не более 1,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси С, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения
(Ь);
- сумма примесей, кроме примесей А и С (не бо¬
лее 1,5 %): на хроматограмме испытуемого раствора
площадь всех пиков, кроме основного и пиков приме¬
сей А и С, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с);
-неучитываемый предел: не учитывают пики
растворителя.
226
Гэсударственная фармакопея Республики Беларусь
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 4,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 1,5 мл
кислоты муравьиной безводной Р, прибавляют 60 мл
кислоты уксусной безводной Р и незамедлительно
титруют 0,1 М раствором кислоты хлорной потенцио-
метрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 29,43 мг С14Н181Ч205.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В.
А. 2-[(25,55)-5-Бензил-3,6-диоксопиперазин-2-ил]-
уксусная кислота.
но2с
B. (35)-3-Амино-4-[[(15)-1-карбокси-2-фенилэтил]-
амино]-4-оксобутановая кислота (а-1_-аспартил-1_-
фенилаланин).
(I I Н\/МНг
C. (25)-2-Амино-3-фенилпропановая кислота
(1_.-фенилаланин).
07/2016:0703
АТЕНОЛОЛ
А1епо!о!ит
АГЕЛ/0/.0/1
н он
\/ н
Х " СНз
и энантиомер
I 12П1
С14Н22М203 М.М. 266’3
[29122-68-7]
ОПРЕДЕЛЕНИЕ
2-[4-[(2РЗ)-2-Гидрокси-3-[(1 -метилэтил)амино]про-
покси]фенил]ацетамид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Умеренно растворим в воде, растворим в этано¬
ле безводном, мало растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 152 °С до
155 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
100 мл этим же растворителем. 10,0 мл полученного
раствора доводят метанолом Р до объема 100 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 275 нм и при
282 нм.
Отношение оптических плотностей:
^275^282 — от 1,15 до 1,20.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО атенолола #или спектр, пред¬
ставленный на рисунке #0703.-1#.
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 1 мл метанола Р.
Раствор сравнения. 10 мг ФСО атенолола раст¬
воряют в 1 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р254 Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р1 — метанол Р (1:99, об/об).
Наносимый объем пробы: 10 мкл.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Раствор 5. 0,10 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Атенолол
227
Рисунок #0703.-1. Инфракрасный спектр ФСО атенолола
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее шестого эталона шкалы
наиболее подходящего цвета.
Угол оптического вращения (2.2.7). От
+0,10° до -0,10°. Измеряют оптическое вращение
раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 20 мл подвижной фазы и доводят
до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО атенолола
для проверки пригодности хроматографической си¬
стемы (содержит примеси В, Р, О, I и ^ растворяют в
1.0 мл подвижной фазы.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,125м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: 1,0 г натрия октансульфона-
таР\л 0,4 г тетрабутиламмония гидросульфата Р
растворяют в 1 л смеси тетрагидрофуран Р — ме¬
танол Р2 — раствор 3,4 г/л калия дигидрофосфа¬
та Р (20:180:800, об/об/об) и доводят до рН 3,0 кис¬
лотой фосфорной Р\
- скорость подвижной фазы: 0,6 мл/мин;
-спектрофотометрический детектор, длина
волны 226 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 5-кратное вре¬
мя удерживания атенолола.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, Р, С, I и 3, используя хрома¬
тограмму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО атенолола для проверки пригод¬
ности хроматографической системы.
Относительное удерживание (по отношению к
атенололу, время удерживания — около 8 мин): при¬
месь В — около 0,3; примесь и — около 0,7; примесь
I — около 0,8; примесь Р — около 2,0 (сдвоенный пик);
примесь О — около 3,5.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,4 между пиками при¬
меси >) (неидентифицируемая примесь) и I.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси I — 1,5):
- примесь В (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
-примеси Р, О, I (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Р, 6 и I, не должны превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
В, Р, 6 и I, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
228
Гэсударственная фармакопея Республики Беларусь
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,1 %. 50 мг испыту¬
емого образца растворяют в смеси из 1 мл кислоты
азотной разведенной Р и 15 мл воды Р. Получен¬
ный раствор без дальнейшего прибавления кислоты
азотной разведенной Р должен выдерживать испыта¬
ния на хлориды.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 80 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 26,63 мг С14Н221Ч203.
но2с.
и энантиомер
О. 2-[4-[(2Я5)-2-Гидрокси-3-[(1-метилэтил)амино]-
пропокси]фенил]уксусная кислота.
N0.
^СН3
СН3
и энантиомер
Н. 2-[4-[(2Я5)-2-Гидрокси-3-[(1-метилэтил)амино]-
пропокси]фенил]ацетонитрил.
и энантиомер
I. 2-[4-[(2Я5)-3-(Этиламино)-2-гидроксипропокси]-
фенил]ацетамид.
07/2016:2191
АТОРВАСТАТИН КАЛЬЦИЯ
ТРИГИДРАТ
ПРИМЕСИ
Специфицированные примеси: В, Р, С, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, О, Е, Н.
н2м
А. Р-Н: 2-(4-Гидроксифенил)ацетамид.
н он
и энантиомер
В. 2-[4-[(2Я5)-2,3-Дигидроксипропокси]фенил]аце-
тамид.
н он
и энантиомер
О. 2-[4-[(2/?5)-3-Хлор-2-гидроксипропокси]фенил]-
ацетамид.
он
Е. 2,2'-[(2-Гидроксипропан-1,3-диил)бис(окси-4,1-
фенилен)]диацетамид.
н3с.
он
Р. 2,2-[[(1 -Метилэтил)имино]бис[(2-гидроксипропан-
3,1 -диил )окси-4,1 -фенилен]]диацетамид.
А1оп/а$Ш'тит са1асит ММус1псит
АТОКУАЗТАТШ СА1.СШМ ТШНУОПАТЕ
[344423-98-9]
ОПРЕДЕЛЕНИЕ
Кальция (ЗР,5Р)-7-[2-(4-фторфенил)-5-(1 -метил-
этил )-3-фенил-4-(фенилкарбамоил)-1 Н-пиррол-1 -ил]-
3,5-дигидроксигептаноат тригидрат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде, мало растворим в
96 % спирте, практически нерастворим в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кальция аторвастатина три-
гидрата #или спектр, представленный на рисунке
#2191.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, различаются, то испыту¬
емый образец и ФСО кальция аторвастатина три-
гидрата растворяют по отдельности в метаноле Р и
выпаривают до сухих остатков, которые используют
для получения новых спектров.
Аторвастатин кальция тригидрат
229
Рисунок #1291.-1. Инфракрасный спектр ФСО кальция аторвастатина тригидрата
B. Испытуемый образец выдерживает испытание
«Энантиомерная чистота», как указано в разделе «Ис¬
пытания».
C. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
О. Сжигают. Остаток дает реакцию (Ь) на кальций
(2.3.1). Если остаток не растворяется полностью, то
раствор фильтруют.
ИСПЫТАНИЯ
Энантиомерная чистота. Жидкостная хромато¬
графия (2.2.29).
Смесь растворителей. Этанол Р — метанол Р
(50:50, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 4 мл смеси растворителей и до¬
водят гексаном Р до объема 10,0 мл.
Раствор сравнения (а). 2 мг ФСО аторвастати¬
на примеси Е растворяют в метаноле Р и доводят до
объема 20,0 мл этим же растворителем (раствор А).
10 мг испытуемого образца растворяют в 1,25 мл
метанола Р, прибавляют 0,75 мл раствора А и 2 мл
этанола Р и доводят гексаном Р до объема 10,0 мл.
Раствор сравнения (Ь). К 2,0 мл испытуемого
раствора прибавляют 40,0 мл смеси растворителей
и доводят гексаном Р до объема 100,0 мл. К 3,0 мл
полученного раствора прибавляют 5 мл смеси раст¬
ворителей и доводят гексаном Р до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем для хромато¬
графии, модифицированным амилозой Р с размером
частиц 10 мкм;
- подвижная фаза: кислота трифторуксус-
ная Р — этанол Р — гексан Р (0,1:6:94, об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 244 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,2-кратное вре¬
мя удерживания аторвастатина.
Относительное удерживание (по отношению к
аторвастатину, время удерживания — около 44 мин):
примесь Е — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси Е и аторвастатина.
Предельное содержание примесей:
- примесь Е (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси Е, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 40,0 мг испытуемого
образца растворяют в диметилформамиде Р и дово¬
дят до объема 100,0 мл этим же растворителем.
Испытуемый раствор (Ь). 50 мг испытуемого об¬
разца растворяют в диметилформамиде Р и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 40,0 мг ФСО кальция
аторвастатина тригидрата растворяют в диме¬
тилформамиде Р и доводят до объема 100,0 мл этим
же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (Ь) доводят диметилформамидом Р до объема
100,0 мл. 1,0 мл полученного раствора доводят диме¬
тилформамидом Р до объема 10,0 мл.
Раствор сравнения (с). 2,5 мг ФСО аторваста¬
тина примеси А, 2,5 мг ФСО аторвастатина приме¬
си В, 2,5 мг ФСО аторвастатина примеси С, 2,5 мг
ФСО аторвастатина примеси О и 2,5 мг испытуе¬
мого образца растворяют в диметилформамиде Р и
доводят до объема 50,0 мл этим же растворителем.
230
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм);
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: тетрагидрофуран Р —
ацетонитрил Р — раствор 3,9 г/л аммония аце¬
тата Р, доведенный кислотой уксусной ледя¬
ной Р до рН 5,0 (12:21:67, об/об/об);
- подвижная фаза В: тетрагидрофуран Р —
раствор 3,9 г/л аммония ацетата Р, доведенный
кислотой уксусной ледяной Р до рН 5,0 — аце¬
тонитрил Р (12:27:61, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—40
100
0
40—70
100 — 20
о
00
т
о
70—85
о
г
о
см
00
0
1
О
о
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 244 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и растворов сравнения (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С и й, используя хромато¬
грамму раствора сравнения (с).
Относительное удерживание (по отношению к
аторвастатину, время удерживания — около 33 мин):
примесь А — около 0,8; примесь В — около 0,9; при¬
месь С — около 1,2; примесь й — около 2,1.
При необходимости корректируют состав подвиж¬
ной фазы, увеличивая или уменьшая процентное содер¬
жание ацетонитрила или рН раствора аммония ацетата
для достижения времени удерживания около 33 мин
для аторвастатина. Например, повышение рН вызывает
уменьшение времени удерживания аторвастатина.
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 1,5 между пиками при¬
меси В и аторвастатина.
Предельное содержание примесей:
- примеси А, В (не более 0,3 %): на хроматограм¬
ме испытуемого раствора (Ь) площади пиков, соот¬
ветствующих примесям А и В, не должны превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (Ь);
-примеси С, О (не более 0,15%): на хромато¬
грамме испытуемого раствора (Ь) площади пиков,
соответствующих примесям С и Ь, не должны превы¬
шать 1,5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (Ь)
площадь любого пика, кроме основного и пиков приме¬
сей А, В, С и О, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 1,5 %): на хромато¬
грамме испытуемого раствора (Ь) сумма площадей
всех пиков, кроме основного, не должна превышать
15-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (Ь) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Натрий. Не более 0,4 % (в пересчете на безво¬
дное вещество).
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Смесь растворителей. Хлористоводородная
кислота Р— вода Р — метанол Р (2:25:75, об/об/об).
Испытуемый раствор. 5,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Растворы сравнения. Готовят разведениями
эталонного раствора натрия (50 ррт А1а) Р смесью
растворителей.
Источник излучения: лампа с полым катодом
для определения натрия.
Длина волны: 589,0 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Тяжелые металлы (2.4.8, метод Н). Не более
0,0020 % (20 ррт).
Смесь растворителей. Вода Р — метанол Р
(10:90, об/об).
Испытание проводят со следующими изменениями.
Испытуемый раствор. 0,250 г испытуемого об¬
разца растворяют в 30 мл смеси растворителей.
Раствор сравнения. 0,5 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р доводят смесью раствори¬
телей до объема 30 мл.
Контрольный раствор. 30 мл смеси раствори¬
телей.
Вода (2.5.12). Не менее 3,5 % и не более 5,5 %.
Определение проводят из 0,130 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и раствора сравнения (а).
Содержание С66Н68СаР21Ч4О10 в процентах рас¬
считывают с учетом содержания кальция аторваста¬
тина тригидрата в ФСО кальция аторвастатина
тригидрата.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Р, С, Н.
А. (3/?,5/?)-3,5-Дигидрокси-7-[5-(1 -метилэтил)-2,3-
дифенил-4-(фенилкарбамоил )-1 Н-пиррол-1 -ил]гепта-
новая кислота (дезфтораторвастатин).
Атропина сульфат
231
В. (ЗЯ5,53Я)-7-[2-(4-Фторфенил)-5-(1 -метилэтил)-
3-фенил-4-(фенилкарбамоил )-1 Н-пиррол-1 -ил]-3,5-
дигидроксигептановая кислота.
С. (ЗЯ,5Я)-7-[2,3-Бис(4-фторфенил)-5-(1 -метил-
этил )-4-(фенилкарбамоил)-1 Н-пиррол-1 -ил]-3,5-дигид ро-
ксигептановая кислота (фтораторвастатин).
й. 3-[(4-Фторфенил)карбонил]-2-(2-метилпро-
паноил)-А/,3-дифенилоксиран-2-карбоксамид.
Е. (35,53)-7-[2-(4-Фторфенил)-5-(1-метилэтил)-3-
фенил-4-(фенилкарбамоил)-1 Н-пиррол-1 -ил]-3,5-диги-
дроксигептановая кислота (энантиомер аторвастатина).
НО н н он
но2с^
Р. (ЗЯ,5Я)-7-[[(ЗЯ,5Я)-7-[2-(4-Фторфенил)-5-(1 -
метилэтил)-3-фенил-4-(фенилкарбамоил)-1Н-пиррол-
1-ил]-3,5-дигидроксигептаноил]амино]-3,5-дигидро-
ксигелтановая кислота.
С. (ЗЯ,5Я)-7-[2-(4-Фторфенил)-5-(1 -метилэтил)-3-
фенил-4-(фенилкарбамоил)-1 Н-пиррол-1 -ил]-5-гидрокси-
3-метоксигептановая кислота (З-О-метилаторвастатин).
Н. (4/?,6Н)-6-[2-[2-(4-Фторфенил)-5-(1 -метилэтил)-
3-фенил-4-(фенилкарбамоил)-1 Н-пиррол-1 -ил]этил]-4-
гидрокситетрагидро-2Н-пиран-2-он.
07/2016:0068
АТРОПИНА СУЛЬФАТ
А&орт1 зиНаз
АТРОРШЕ 8Ш.РАТЕ
[5906-99-6]
ОПРЕДЕЛЕНИЕ
Бис[(1Я,Зг,55)-8-метил-8-азабицикло[3.2.1]окт-
3-ил-(2/?5)-3-гидрокси-2-фенилпропаноата] сульфат
моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: С, О, Е, Р.
A. Испытуемый образец выдерживает испытание
«Угол оптического вращения», как указано в разделе
«Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО атропина сульфата.
232
Гэсударственная фармакопея Республики Беларусь
С. 50 мг испытуемого образца растворяют в 5 мл
воды Р и прибавляют 5 мл раствора пикриновой
кислоты Р. Осадок промывают водой Р и сушат при
температуре от 100 °С до 105 °С в течение 2 ч. Тем¬
пература плавления (2.2.14) полученного остатка: от
174 °С до 179 °С.
O. К 1 мг испытуемого образца прибавляют
0,2 мл кислоты азотной дымящейся Р и выпарива¬
ют досуха в водяной бане. Остаток растворяют в 2 мл
ацетона Р и прибавляют 0,1 мл раствора 30 г/л калия
гидроксида Р в метаноле Р. Появляется фиолетовое
окрашивание.
Е. Испытуемый образец дает реакции на сульфа¬
ты (2.3.7).
P. Испытуемый образец дает реакцию на алкало¬
иды (2.3.7).
ИСПЫТАНИЯ
рН (2.2.3). От 4,5 до 6,2. 0,6 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 30 мл этим же раство¬
рителем.
Угол оптического вращения (2.2.7). От -0,50°
до +0,05°. 2,50 г испытуемого образца растворяют в
воде Р и доводят до объема 25,0 мл этим же раст¬
ворителем. Определение проводят в трубке длиной
2 дм.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 24 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО атропина при¬
меси В растворяют в испытуемом растворе и доводят
до объема 20 мл этим же растворителем. 5 мл полу¬
ченного раствора доводят подвижной фазой А до объ¬
ема 25 мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО атропина для идентификации примесей (со¬
держит примеси А, й, Е, Р, 6 и Н) растворяют в 1 мл
подвижной фазы А.
Раствор сравнения (ф. 5 мг кислоты тропо-
вой Р (примесь С) растворяют в подвижной фазе А
и доводят до объема 10 мл этим же растворителем.
1 мл полученного раствора доводят подвижной фазой
А до объема 100 мл. 1 мл полученного раствора дово¬
дят подвижной фазой А до объема 10 мл.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: 3,5 г натрия додецил-
сульфата Р растворяют в 606 мл раствора
7,0 г/л калия дигидрофосфата Р, предваритель¬
но доведенного до рН 3,3 0,05 М раствором кис¬
лоты фосфорной, и смешивают с 320 мл ацето¬
нитрила Р1\
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
95
5
2—20
95 -> 70
о
со
т
Ю
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, О, Е, Р, С и Н, используя хро¬
матограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО атропина для идентификации
примесей; идентифицируют пик примеси В, используя
хроматограмму раствора сравнения (Ь), и пик примеси
С, используя хроматограмму раствора сравнения (б).
Относительное удерживание (по отношению к
атропину, время удерживания — около 11 мин): при¬
месь С — около 0,2; примесь Е — около 0,67; примесь
О — около 0,73; примесь Р — около 0,8; примесь В —
около 0,89; примесь Н — около 0,93; примесь <3 —
около 1,1; примесь А—около 1,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками при¬
меси В и атропина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 0,6; для примеси С — 0,6):
- примеси Е, Н (не более 0,3 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Е и Н, не должны превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примеси А, В, С, О, Р, О (не более 0,2 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, й, Р, О, не
должны превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е, Р, 6 и Н, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а).
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Вода (2.5.72). От 2,0 % до 4,0 %. Определение
проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в 30 мл
кислоты уксусной безводной Р, при необходимо¬
сти подогревая. Раствор охлаждают и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 67,68 мг С34Н46М206 Н2504.
Ацетилсалициловая кислота
233
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Е, С, Н.
А. (1 Д,Зг,55)-8-Метил-8-азабицикло[3.2.1 ]окт-3-ил-
2-фенилпропеноат (апоатропин).
и энантиомер
В. (1Я,Зг,55)-8-Азабицикло[3.2.1]окт-3-ил-(2#5)-3-
гидрокси-2-фенилпропаноат (норатропин).
С. (2#5)-3-Гидрокси-2-фенилпропановая кислота
(троповая кислота).
и эпимер при *С
О. (1К,35,5Я6Я5)-6-Гидрокси-8-метил-8-азаби-
цикло[3.2.1]окт-3-ил-(25)-3-гидрокси-2-фенилпропаноат
(6-гидроксигиосциамин).
цикло[3.2.1]окт-3-ил-(25)-3-гидрокси-2-фенилпропаноат
(7-гидроксигиосциамин).
кло[3.3.1.024]нон-7-ил-(23)-3-гидрокси-2-фенил пропаноат
(гиосцин).
6. (1 Я,Зг,55)-8-Метил-8-азабицикло[3.2.1]окт-3-ил-
(2#3)-2-гидрокси-3-фенилпропаноат (литторин).
Н. Структура не установлена.
07/2016:0309
АЦЕТИЛСАЛИЦИЛОВАЯ КИСЛОТА
Ас1с1ит асе1у!$аИсуНсит
АСЕТПЗАиСУиС АСЮ
С9НйО. М.м. 180,2
[50-78-2]
ОПРЕДЕЛЕНИЕ
2-(Ацетилокси)бензойная кислота.
Содержание: не менее 99,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Мало растворим в воде, легко растворим в 96 %
спирте.
Температура плавления: около 143 °С (метод
мгновенного плавления).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ацетилсалициловой кислоты
#или спектр, представленный на рисунке #0309.-1#.
B. К 0,2 г испытуемого образца прибавляют 4 мл
раствора натрия гидроксида разведенного Р и кипя¬
тят в течение 3 мин. Охлаждают и прибавляют 5 мл
кислоты серной разведенной Р. Образуется кристал¬
лический осадок. Осадок отфильтровывают, промы¬
вают и высушивают при температуре от 100 °С до
105 °С. Температура плавления (2.2.14) полученных
кристаллов: от 156 °С до 161 °С.
C. В пробирке смешивают 0,1 г испытуемого об¬
разца и 0,5 г кальция гидроксида Р. Смесь нагревают
и в выделяющиеся пары помещают фильтровальную
бумагу, импрегнированную 0,05 мл раствора нитро-
бензальдегида Р. На бумаге появляется зеленовато¬
синее или зеленовато-желтое окрашивание. Бумагу
смачивают кислотой хлористоводородной разведен¬
ной Р. Появляется синее окрашивание.
й. 20 мг осадка, полученного в идентификации В,
растворяют при нагревании в 10 мл воды Р и охлаж¬
дают. Полученный раствор дает реакцию (а) на сали-
цилаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 9 мл 96 % спирта Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
234
Гэсударственная фармакопея Республики Беларусь
Рисунок #0309.-1. Инфракрасный спектр ФСО ацетилсалициловой кислоты
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в ацетонитриле для хроматогра¬
фии Р и доводят до объема 10,0 мл этим же раство¬
рителем.
Раствор сравнения (а). 50,0 мг салициловой кис¬
лоты Р (примесь С) растворяют в подвижной фазе и
доводят до объема 50,0 мл этим же растворителем.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (Ь). 10 мг салициловой кис¬
лоты Р (примесь С) растворяют в подвижной фазе и
доводят до объема 10,0 мл этим же растворителем.
К 1,0 мл полученного раствора прибавляют 0,2 мл ис¬
пытуемого раствора и доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). Содержимое контейнера
ФСО ацетилсалициловой кислоты для идентифика¬
ции пиков (содержит примеси А, В, О, Е и Р) раство¬
ряют с использованием ультразвука в 1,0 мл ацето¬
нитрила Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
-подвижная фаза: кислота фосфорная Р —
ацетонитрил для хроматографии Р — вода Р
(2:400:600, об/об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 237 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 7-кратное вре¬
мя удерживания ацетилсалициловой кислоты.
Идентификация пиков примесей: идентифици¬
руют пик примеси С, используя хроматограмму раст¬
вора сравнения (а); идентифицируют пики примесей
А, В, О, Е и Р, используя хроматограмму раствора
сравнения (с) и хроматограмму, прилагаемую к ФСО
ацетилсалициловой кислоты для идентификации
пиков.
Относительное удерживание (по отношению к
ацетилсалициловой кислоте, время удерживания —
около 5 мин): примесь А — около 0,7; примесь В —
около 0,8; примесь С — около 1,3; примесь О — около
2,3; примесь Е — около 3,2; примесь Р — около 6,0.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 6,0 между пиками аце¬
тилсалициловой кислоты и примеси С.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р (не более 0,15 %): на
хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного, не должна превышать
1.5- кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, Э, Е, и Р, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 0,25 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2.5- кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раство¬
ряют в 12 мл ацетона Р и доводят водой Р до объема
20 мл. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон го-
Ацетилцистеин
235
товят с использованием эталонного раствора свинца
(1 ррт РЬ), приготовленного путем разведения эта¬
лонного раствора свинца (100 ррт РЬ) Р смесью из
6 объемов воды Р и 9 объемов ацетона Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца помещают в колбу
со стеклянной притертой пробкой, растворяют в 10 мл
96 % спирта Р, прибавляют 50,0 мл 0,5 М раствора
натрия гидроксида, закрывают колбу пробкой и вы¬
держивают в течение 1 ч. Титруют 0,5 М раствором
кислоты хлористоводородной, используя в качестве
индикатора 0,2 мл раствора фенолфталеина Р.
Параллельно проводят контрольный опыт.
1 мл 0,5 М раствора натрия гидроксида соот¬
ветствует 45,04 мг С9Н804.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
В. 4-Гидроксибензол-1,3-дикарбоновая кислота
(4-гидроксиизофталевая кислота).
С. 2-Гидроксибензойная кислота (салициловая кис¬
лота).
О. 2-[[2-(Ацетилокси)бензоил]окси]бензойная кис¬
лота (ацетилсалицилсалициловая кислота).
Е. 2-[(2-Гидроксибензоил)окси]бензойная кислота
(салсалат, салицилсалициловая кислота).
Р. 2-(Ацетилокси)бензойный ангидрид (ацетилса¬
лициловый ангидрид).
07/2016:0967
АЦЕТИЛЦИСТЕИН
Асе1у1су$1е'тит
АСЕТПСУ8ТЕШЕ
С$Н,1Ч035 М.м. 163,2
[616-91-1]
ОПРЕДЕЛЕНИЕ
(2/?)-2-(Ацетиламино)-3-сульфанилпропановая
кислота.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде и в 96 % спирте, практи¬
чески нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Температура плавления (2.2.14): от 104 °С до
110 °С.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках с калия бромидом Р.
Сравнение: ФСО ацетилцистеина #или спектр,
представленный на рисунке #0967.-1#.
й. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме
испытуемого раствора (Ь) по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (Ь).
Е, К 0,5 мл раствора 5, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 0,05 мл
раствора 50 г/л натрия нитропруссида Р и 0,05 мл
раствора аммиака концентрированного Р. Появля¬
ется темно-фиолетовое окрашивание.
236
Гэсударственная фармакопея Республики Беларусь
Рисунок #0967.-1. Инфракрасный спектр ФСО ацетилцистеина
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 2,0 до 2,8. К 2 мл раствора 3 при¬
бавляют 8 мл воды, свободной от углерода диокси¬
да, Р и перемешивают.
Удельное оптическое вращение (2.2.7). От
+21,0 до +27,0 (в пересчете на сухое вещество). К
1,25 г испытуемого образца прибавляют 1 мл раст¬
вора 10 г/л натрия эдетата Р, перемешивают, при¬
бавляют 7,5 мл раствора 40 г/л натрия гидроксида Р,
перемешивают до растворения и доводят фосфат¬
ным буферным раствором рН 7,0 Р2 до объема
25.0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Если не указано иное, растворы
готовят непосредственно перед использованием.
Испытуемый раствор (а). 0,80 г испытуемого
образца суспендируют в 1 мл 1 М раствора кислоты
хлористоводородной и доводят водой Р до объема
100.0 мл.
Испытуемый раствор (Ь). 5,0 мл испытуемого
раствора (а) доводят водой Р до объема 100,0 мл.
5.0 мл полученного раствора доводят водой Р до объ¬
ема 50,0 мл.
Испытуемый раствор (с). Используют испытуе¬
мый раствор (а) после хранения не менее 1 ч.
Раствор сравнения (а). 4,0 мг ФСО ацетил¬
цистеина, 4,0 мг Ь-цистина Р (примесь А), 4,0 мг
[--цистеина Р (примесь В), 4,0 мг ФСО ацетилцисте¬
ина примеси С и 4,0 мг ФСО ацетилцистеина при¬
меси О суспендируют в 1 мл 1 М раствора кислоты
хлористоводородной и доводят водой Р до объема
100.0 мл.
Раствор сравнения (Ь). 4,0 мг ФСО ацетилци¬
стеина суспендируют в 1 мл 1 М раствора кислоты
хлористоводородной и доводят водой Р до объема
100.0 мл.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,25 м
и внутренним диаметром 4 мм, заполненная силика¬
гелем октадецилсилильным для хроматографии Р с
размером частиц 5 мкм;
-подвижная фаза: смесь ацетонитрил Р —
вода Р (3:97, об/об), доведенная кислотой фосфор¬
ной Рцо рН 3,0;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 20 мкл каждого
раствора и 0,01 М раствора кислоты хлористоводо¬
родной;
- время хроматографирования: 5-кратное вре¬
мя удерживания ацетилцистеина.
Времена удерживания: примесь А — около
2,2 мин; примесь В — около 2,4 мин; 2-метил-2-
тиазолин-4-карбоновая кислота, образующаяся в ис¬
пытуемом растворе (с), — около 3,3 мин; ацетилци-
стеин — около 6,4 мин; примесь С — около 12 мин;
примесь О — около 14 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками при¬
меси А и примеси В; не менее 2,0 между пиками при¬
меси С и примеси О.
Содержание известных (Т.,) и неизвестных при¬
месей (Т2) в процентах рассчитывают по формулам:
Д ш2 -100
1 А2тл '
Ацетон
237
А3 т3 -100
А4т,
где:
Л1 — площадь пика известной примеси (приме¬
сей А, В, С и й) на хроматограмме испытуемого раст¬
вора (а);
А2 — площадь соответствующего пика известной
примеси (примесей А, В, С и О) на хроматограмме
раствора сравнения (а);
А3 — площадь пика неизвестной примеси на хро¬
матограмме испытуемого раствора (а);
Ал — площадь пика ацетилцистеина на хромато¬
грамме раствора сравнения (Ь);
тл — масса навески испытуемого образца, взя¬
тая для приготовления испытуемого раствора (а), мг;
т2 — масса навески индивидуальной известной
примеси, взятая для приготовления раствора сравне¬
ния (а), мг;
т3 — масса навески ацетилцистеина, взятая для
приготовления раствора сравнения (Ь), мг.
Предельное содержание примесей:
- примеси А, В, С, О: не более 0,5 %;
- любая другая примесь: не более 0,5 %;
- сумма примесей: не более 0,5 %;
-неучитываемый предел: на хроматограмме
испытуемого раствора (а) не учитывают пик со вре¬
менем удерживания около 3,3 мин, соответствующий
2-метил-2-тиазолин-4-карбоновой кислоте, и пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь) (0,05 %).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Цинк. Не более 0,0010% (10 ррт). Атомно-аб¬
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в 0,001 М растворе кислоты хло¬
ристоводородной и доводят до объема 50,0 мл этим
же растворителем.
Растворы сравнения. Растворы сравнения гото¬
вят с использованием эталонного раствора цинка
(5 мг/мл 1п) Р, разведенного 0,001 М раствором кис¬
лоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,8 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Проводят коррекцию неселективного поглощения.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 70 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,140 г испытуемого образца растворяют в 60 мл
воды Р, прибавляют 10 мл кислоты хлористоводо¬
родной разведенной Р, охлаждают в ледяной воде,
прибавляют 10 мл раствора калия йодида Р и титру¬
ют 0,05 М раствором йода, используя в качестве ин¬
дикатора 1 мл раствора крахмала Р.
1 мл 0,05 М раствора йода соответствует
16,32 мг С5Н9М038.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
н нн2
но2с^
"со2н
Н ИНг
A. 3,3-Дисульфандиилбис[(2Р)-2-аминопропановая
кислота] (Ьцистин).
н ын2
НЗ. ЗС
хо2н
B. (2Р)-2-Амино-3-сульфанилпропановая кислота
(Ьцистеин).
СН3
о=Ч
н N4
Н02С.
С02Н
Н ЫН
0=<
СН3
С. (2Р,2Р7-3,3-Дисульфандиилбис[2-(ацетиламино)-
пропановая кислота] (Л/,Л/'-диацетил-1_-цистин).
рн3
Н 1ЧН
НзС.^
т
со2н
О. (2Р)-2-(Ацетиламино)-3-(ацетилсульфанил)про-
пановая кислота (Л/,5-диацетил-1_-цистеин).
07/2016:0872
АЦЕТОН
Асе1опит
АСЕТОЫЕ
О
Н3СГ сн3
С3Н,0 М.м. 58,08
[67-64-1]
ОПРЕДЕЛЕНИЕ
Пропанон.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная, летучая жидкость.
Смешивается с водой, 96 % спиртом.
Пары огнеопасны.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
238
Гэсударственная фармакопея Республики Беларусь
B. К 1 мл испытуемого образца прибавляют
3 мл раствора натрия гидроксида разведенного Р и
0,3 мл раствора 25 г/л натрия нитропруссида Р. По¬
является интенсивное красное окрашивание, которое
переходит в фиолетовое при прибавлении 3,5 мл кис¬
лоты уксусной Р.
C. К 10 мл раствора 0,1 % (об/об) испытуемого
образца в спирте (50 %, об/об) Р прибавляют 1 мл
раствора 10 г/л нитробензальдегида Р в спирте
(50 %, об/об) Р и 0,5 мл раствора натрия гидроксида
концентрированного Р. Полученный раствор выдер¬
живают в течение 2 мин и подкисляют кислотой уксус¬
ной Р. Появляется зеленовато-синее окрашивание.
ИСПЫТАНИЯ
Раствор 5. К 10 мл испытуемого образца при¬
бавляют 10 мл воды Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 5 мл испыту¬
емого образца прибавляют 5 мл воды, свободной от
углерода диоксида, Р, 0,15 мл раствора фенолфта¬
леина Р и 0,5 мл 0,01 М раствора натрия гидрокси¬
да. Раствор должен быть розовым. При добавлении
0,7 мл 0,01 М раствора кислоты хлористоводород¬
ной и 0,05 мл раствора метилового красного Р долж¬
но появиться красное или оранжевое окрашивание.
Относительная плотность (2.2.5). От 0,790 до
0,793.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. Испытуемый образец.
Раствор сравнения (а). К 0,5 мл метанола Р
прибавляют 0,5 мл 2-пропанола Р и доводят испы¬
туемым раствором до объема 100,0 мл. 1,0 мл полу¬
ченного раствора доводят испытуемым раствором до
объема 10,0 мл.
Раствор сравнения (Ь). 100 мкл бензола Р до¬
водят испытуемым раствором до объема 100,0 мл.
0,20 мл полученного раствора доводят испытуемым
раствором до объема 100,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 50 м и
внутренним диаметром 0,3 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость газа-носителя: 21 см/с;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—11
45-100
11—20
100
Блок ввода проб
150
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Время удерживания: примесь С — около 7,5 мин.
Пригодность хроматографической системы:
- разрешение: не менее 5,0 между пиком приме¬
си А (второй пик) и пиком примеси В (третий пик) на
хроматограмме раствора сравнения (а);
- отношение сигнал-шум: не менее 5 для пика
примеси С на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
- примеси А, В (не более 0,05 %, об/об): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А и В, не должны превы¬
шать разницу площадей соответствующих пиков на
хроматограмме раствора сравнения (а) и на хромато¬
грамме испытуемого раствора;
- примесь С (не более 0,0002 % (2 ррт), об/об):
на хроматограмме испытуемого раствора площадь
пика, соответствующего примеси С, не должна пре¬
вышать разницу площадей соответствующего пика на
хроматограмме раствора сравнения (Ь) и на хромато¬
грамме испытуемого раствора;
- любая другая примесь (не более 0,05 %, об/об):
на хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пиков примесей А, В
и С, не должна превышать разницу между площадью
пика примеси А на хроматограмме раствора сравне¬
ния (а) и площадью соответствующего пика на хрома¬
тограмме испытуемого раствора.
Вещества, нерастворимые в воде. 1,0 мл ис¬
пытуемого образца доводят водой Р до объема 20 мл.
Полученный раствор должен быть прозрачным (2.2.1).
Остаток после выпаривания. Не более
0,0050 % (50 ррт). 20,0 г испытуемого образца выпа¬
ривают досуха на водяной бане и сушат при темпе¬
ратуре от 100 °С до 105 °С. Масса сухого остатка не
должна превышать 1 мг.
Вода (2.5.12). Не более 3 г/л. Определение про¬
водят из 10,0 мл испытуемого образца. В качестве рас¬
творителя используют 20 мл пиридина безводного Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
A. СН3-ОН: метанол.
B. СН3-СНОН-СН3: пропан-2-ол (изопропанол).
C. С6Н6: бензол.
07/2016:0968
АЦИКЛОВИР
Аас!ошит
АСIС^ОVт
СДДО, М.м. 225,2
[59277-89-3]
ОПРЕДЕЛЕНИЕ
2-Амино-9-[(2-гидроксиэтокси)метил]-1,9-дигидро-
6Н-пурин-6-он.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на безводное вещество).
Ацикловир
239
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, очень мало растворим
в 96 % спирте, практически нерастворим в гептане.
Растворяется в разведенных растворах минеральных
кислот и гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ацикловира #или спектр, пред¬
ставленный на рисунке #0968.-1#.
ИСПЫТАНИЯ
Раствор 5. 0,25 г испытуемого образца раство¬
ряют в растворе 4 г/л натрия гидроксида Р и доводят
до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)7.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед применением.
Смесь растворителей: диметилсульфок-
сид Р — вода Р (20:80, об/об).
Фосфатный буферный раствор рН 2,5. 3,48 г
дикалия гидрофосфата Р растворяют в 1000 мл
воды Р и доводят до рН 2,5 кислотой фосфорной Р.
Фосфатный буферный раствор рН 3,1. 3,48 г
дикалия гидрофосфата Р растворяют в 1000 мл
воды Р и доводят до рН 3,1 кислотой фосфорной Р.
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 5,0 мл диметилсульфоксида Р и
доводят водой Р до объема 25,0 мл.
Раствор сравнения (а). 5 мг ФСО ацикловира
для пригодности хроматографической системы
(содержит примеси А, В, б, К, N. О и Р) растворяют
в 1 мл диметилсульфоксида Р и доводят водой Р до
объема 5,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). Содержимое контей¬
нера ФСО ацикловира для идентификации пиков 1
(содержит примеси С и I) растворяют в 200 мкл ди¬
метилсульфоксида Р и доводят водой Р до объема
1.0 мл.
Раствор сравнения (д). Содержимое контейнера
ФСО ацикловира для идентификации пиков 2 (со¬
держит примеси Р и С) растворяют в 1,0 мл раствора
сравнения (а).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — фос¬
фатный буферный раствор рН 3,1 (1:99, об/об);
- подвижная фаза В: ацетонитрил Р — фос¬
фатный буферный раствор рН 2,5 (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
100
0
5—27
о
00
г
о
о
т—
т
о
27-40
80
20
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (б).
Рисунок #0968.-1. Инфракрасный спектр ФСО ацикловира
240
Гэсударственная фармакопея Республики Беларусь
Идентификация пиков примесей: идентифици¬
руют пики примесей С и I, используя хроматограмму
раствора сравнения (с), и хроматограмму, прилагае¬
мую к ФСО ацикловира для идентификации пиков 1\
идентифицируют пики примесей А, В, Р, С, ^ К, N. О и
Р, используя хроматограмму раствора сравнения (с!) и
хроматограмму, прилагаемую к ФСО ацикловира для
идентификации пиков 2.
Относительное удерживание (по отношению к
ацикловиру, время удерживания — около 13 мин): при¬
месь В — около 0,4; примесь Р — около 0,7; примесь
С — около 0,9; примесь N — около 1,37; примеси О и
О — около 1,42; примесь I — около 1,57; примесь 3 —
около 1,62; примесь Р — около 1,7; примесь А— около
1,8; примеси К и Р — около 2,5; примесь 6 — около 2,6.
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками при¬
меси С и ацикловира на хроматограмме раствора
сравнения (с); не менее 1,5 между пиками примесей
Р и А и не менее 1,5 между пиками примесей К и С на
хроматограмме раствора сравнения (с1).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси! —1,5): .
- примесь В (не более 0,7 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 7-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей О и О (не более 0,3 %): на хро¬
матограмме испытуемого раствора сумма площадей
пиков примесей О и О не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- сумма примесей К и Я (не более 0,2 %): на хро¬
матограмме испытуемого раствора сумма площадей
пиков примесей К и К не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- примеси А, О, Л/, Р (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, С, 3, N и Р, не долж¬
ны превышать 2-кратную площадь основного пика на
хроматограмме раствора сравнения (Ь);
- примеси С, Р, I (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С, Р, I, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, Р, О, I, К, N1, О, Р, О, Я, не должна
превышать 0,5 площади основного пика на хромато¬
грамме раствора сравнения (Ь);
- сумма примесей (не более 1,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
15-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Вода (2.5.12). Не более 6,0 %. Определение про¬
водят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14, ме¬
тод О). Менее 0,50 МЕ/мг, если субстанция предна¬
значена для производства лекарственных средств
для парентерального применения без дальнейшей
подходящей процедуры удаления бактериальных эн¬
дотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 22,52 мг СдН^И.Рз.
ПРИМЕСИ
Специфицированные примеси: А, В, С, Р, в, I, 3,
К, Ы, О, Р, О, Я.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Ь, М.
А. 2-[(2-Амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-
метокси]этилацетат.
В. 2-Амино-1,7-дигидро-6Н-пурин-6-он (гуанин).
С. 2-Амино-7-[(2-гидроксиэтокси)метил]-1,7
дигидро-6Н-пурин-6-он.
Р. Л/-[9-[(2-Гидроксиэтокси)метил]-6-оксо-6,9-
дигидро-1 Н-пурин-2-ил]ацетамид.
Бария сульфат
241
Н2М
<3. 2-[[2-(Ацетиламино)-6-оксо-1,6-дигидро-9Н-
пурин-9-ил]метокси]этилацетат.
I. 2-Амино-7-[[2-[(2-амино-6-оксо-1,6-дигидро-9Н-
пурин-9-ил)метокси]этокси]метил]-1,7-дигидро-6Н-пурин-
6-он.
9,9'-[Этиленбис(оксиметилен)]бис(2-амино-1,9-
дигидро-6Н-пурин-6-он).
О. Смесь 2-амино-9-[[2-(гидроксиметокси)этокси]-
метил]-1,9-дигидро-6Н-пурин-6-он и 2-амино-9-[[2-
(гидроксиэтокси)метокси]метил]-1,9-дигидро-6Н-пурин-
6-он.
Я. 9,9'-[Метилен-бис(оксиэтиленокси-метилен)]-
бис(2-амино-1,9-дигидро-6Н-пурин-6-он).
07/2016:0010
БАРИЯ СУЛЬФАТ
Ваги зиКаз
ВАВШМ 5Ш.РАГЕ
К. 2,2-(Метилендиимино)бис[9-[(2-гидроксиэтокси)-
метил]-1,9-дигидро-бН-пурин-б-он].
о
Ва$04 М.м. 233,4
[7727-43-7]
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый мелкий порошок без зер¬
нистости.
Практически нерастворим в воде и в органиче¬
ских растворителях. Очень мало растворим в кисло¬
тах и в растворах гидроксидов щелочных металлов.
Ь. А/-(9-Ацетил-6-оксо-6,9-дигидро-1 Н-пурин-2-ил)-
ацетамид (А/^Э-диацетилгуанин).
M. 2-[[2-(Ацетиламино)-6-оксо-1,6-дигидро-7Н-
пурин-7-ил]метокси]этилацетат.
N. Структура не установлена.
O. Структура не установлена.
Р. 2-Амино-9-(2-гидроксиэтил)1,9-дигидро-6Н-
пурин-6-он.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Кипятят суспензию 0,2 г испытуемого образца
в 5 мл раствора 500 г/л натрия карбоната Р в тече¬
ние 5 мин, прибавляют 10 мл воды Р, фильтруют и
подкисляют часть фильтрата кислотой хлористово¬
дородной разведенной Р. Полученный раствор дает
реакцию на сульфаты (2.3.1).
B. Остаток, собранный на фильтре в предыду¬
щем испытании, промывают трижды последовательно
небольшими порциями воды Р. К остатку прибавляют
5 мл кислоты хлористоводородной разведенной Р,
фильтруют и прибавляют к фильтрату 0,3 мл кислоты
серной разведенной Р. Образуется белый осадок, не¬
растворимый в растворе натрия гидроксида разве¬
денном Р.
ИСПЫТАНИЯ
Раствор 5. К 20,0 г испытуемого образца прибав¬
ляют 40 мл воды дистиллированной Р и 60 мл кисло¬
ты уксусной разведенной Р. Кипятят в течение 5 мин,
фильтруют и доводят охлажденный фильтрат водой
дистиллированной Р до объема 100 мл.
г
242
Государственная фармакопея Республики Беларусь
Кислотность или щелочность. К 5,0 г испытуе¬
мого образца прибавляют 20 мл воды, свободной от
углерода диоксида, Р, нагревают на водяной бане в
течение 5 мин и фильтруют. К 10 мл фильтрата при¬
бавляют 0,05 мл раствора бромтимолового сине¬
го Р1. При прибавлении не более 0,5 мл 0,01 М раст¬
вора кислоты хлористоводородной или 0,01 М раст¬
вора натрия гидроксида окраска должна измениться.
Вещества, растворимые в кислоте. Не более
0,3 %. 25 мл раствора 3 выпаривают досуха на водя¬
ной бане и сушат до постоянной массы при темпера¬
туре от 100 °С до 105 °С. Масса остатка должна быть
не более 15 мг.
Окисляемые серосодержащие вещества.
Встряхивают 1,0 г испытуемого образца с 5 мл воды Р
в течение 30 с и фильтруют. К фильтрату прибавля¬
ют 0,1 мл раствора крахмала Р, в полученной смеси
растворяют 0,1 г калия йодида Р, прибавляют 1,0 мл
свежеприготовленного раствора 3,6 мг/л калия йода¬
та Р и 1 мл 1 М раствора кислоты хлористоводо¬
родной и интенсивно встряхивают. Окраска получен¬
ного раствора должна быть интенсивнее эталона,
приготовленного параллельно без использования ка¬
лия йодата.
Растворимые соли бария. Не более 0,0010%
(10 ррт). К 2,5 мл раствора 0,2 мг/л бария нитрата Р
в смеси из 95 % спирта Р и воды Р (30:70, об/об)
прибавляют 10 мл кислоты серной разведенной Р.
Встряхивают и выдерживают в течение 5 мин. К 1 мл
полученного раствора прибавляют 10 мл раствора 3.
Эталон готовят с использованием 10 мл эталонного
раствора бария (2 ррт Ва) Р вместо раствора 3. Че¬
рез 10 мин испытуемый раствор по степени мутности
не должен превышать эталон.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 10 мл раствора 3 доводят водой Р
до объема 20 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Потери при прокаливании. Не более 2,0 %.
Определение проводят из 1,0 г испытуемого образца
при температуре (600±50) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:0654
БЕКЛОМЕТАЗОНА ДИПРОПИОНАТ
БЕЗВОДНЫЙ
Вес1оте(азоп1 сИргорюпаз апЬус/псиз
ВЕС1.0МЕТА50ЫЕ 01РВОРЮЫАТЕ,
АЫНУОРОиЗ
С28Н37СЮ7 М.м. 521,0
[5534-09-8]
Волновое число (см'1)
Рисунок #0654.-1. Инфракрасный спектр ФСО беклометазона дипропионата безводного
Беклометазона дипропионат безводный
243
ОПРЕДЕЛЕНИЕ
9-Хлор-11 р-гид рокси-16р-метил-3,20-диоксопрегна-
1,4-диен-17,21-диилдипропаноат.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, умеренно растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО беклометазона дипропионата
безводного #или спектр, представленный на рисунке
#0654.-1#.
B. 25 мг испытуемого образца сжигают в колбе с
кислородом (2.5.10). Для поглощения продуктов горе¬
ния используют смесь из 1 мл 1 М раствора натрия
гидроксида и 20 мл воды Р. Полученный раствор дает
реакцию (а) на хлориды (2.3.1).
C. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+108 до +115 (в пересчете на сухое вещество). 0,100 г
испытуемого образца растворяют в 96% спирте Р и
доводят до объема 10,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Подвижная фаза А —
подвижная фаза В (45:55, об/об).
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в 28 мл подвижной фазы В и до¬
водят подвижной фазой А до объема 50,0 мл.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 50,0 мл.
Раствор сравнения (а). 5,0 мл испытуемого раст¬
вора (Ь) доводят смесью растворителей до объема
100,0 мл.
Раствор сравнения (Ь). 5 мг ФСО бекломета¬
зона дипропионата для проверки пригодности хро¬
матографической системы (содержит примесь О)
растворяют в 3 мл подвижной фазы В и доводят под¬
вижной фазой А до объема 5 мл.
Раствор сравнения (с). 5 мг ФСО бекломета¬
зона дипропионата для идентификации пиков (со¬
держит примеси А, В, С, Ь и М) растворяют в 3 мл
подвижной фазы В и доводят подвижной фазой А до
объема 5 мл. 1 мл полученного раствора используют
для растворения содержимого контейнера с ФСО бе¬
клометазона дипропионата примесей Р и N.
Раствор сравнения (ф. 50,0 мг ФСО бекломета¬
зона дипропионата безводного растворяют в 28 мл
подвижной фазы В и доводят подвижной фазой А до
объема 50,0 мл. 1,0 мл полученного раствора доводят
смесью растворителей до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим дифункцио¬
нально связанным силикагелем октадецилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 50 °С;
- подвижная фаза:
- подвижная фаза А: раствор 2,72 г/л калия ди¬
гидрофосфата Р, доведенный до рН 2,35 кисло¬
той фосфорной Р;
- подвижная фаза В: тетрагидрофуран Р —
ацетонитрил Р — метанол Р (5:23:25, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—4
40
60
4—12
40 — 45
60 — 55
12—59
45
55
- скорость подвижной фазы: 1,4 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, Р, Ц М и N. используя хро¬
матограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО беклометазона дипропионата
для идентификации пиков; идентифицируют пик при¬
меси Р, используя хроматограмму раствора сравне¬
ния (Ь) и хроматограмму, прилагаемую к ФСО бекло¬
метазона дипропионата для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
беклометазона дипропионату, время удерживания —
около 25 мин): примесь А — около 0,3; примесь В —
около 0,6; примесь й — около 1,1; примесь М — около
1,2; примесь 1_ — около 1,3; примесь С — около 1,8;
примесь N — около 2,0; примесь Р — около 2,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси Э относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси Р и пик
беклометазона дипропионата).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Р — 1,3; для примеси М — 2,0):
- примесь /. (не более 0,6 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси Ц не должна превышать 6-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примеси В, Р, М (не более 0,5 %): на хрома¬
тограмме испытуемого раствора (а) площади пиков,
соответствующих примесям В, Р, М, не должны пре¬
вышать 5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- примеси А, О, N (не более 0,2 %): на хрома¬
тограмме испытуемого раствора (а) площади пиков,
соответствующих примесям А, О, N. не должны пре¬
вышать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- примесь С (не более 0,15 %): на хроматограм¬
ме испытуемого раствора (а) площадь пика, соответ¬
ствующего примеси С, не должна превышать 1,5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков при-
31*. Зак. 1060.
244
Гэсударственная фармакопея Республики Беларусь
месей А, В, С, й, Р, 1_, М, N. не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 1,5 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
15-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (б).
Содержание С28Н37СЮ7 в процентах рассчитыва¬
ют с учетом содержания беклометазона дипропиона¬
та в ФСО беклометазона дипропионата безводного.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Р, Ц М, N.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): Е, Н, I, 3, О, О, Р, 8, II, V.
Н К1
A. К1 = КЗ = Н, К2 = С1, К4 = СО-С2Н5: 9-Хлор-
11 р, 17-дигидрокси-16р-метил-3,20-диоксопрегна-1,4-
диен-21-илпропаноат (беклометазона 21-пропионат).
B. К1 = Н, К2 = С1, КЗ = СО-С2Н5, К4 = СО-СН3:
21 -(Ацетилокси)-9-хлор-11 р-гидрокси-16р-метил-3,20-
диоксопрегна-1,4-диен-17-илпропаноат (беклометазо¬
на 21-ацетат 17-пропионат).
C. К1 = Н, К2 = С1, КЗ = СО-С2Н5> К4 = СО-СН2-
СН2-СН3:9-Хлор-11 р-гидрокси-1 бр-метил-3,20-диоксо-
17-(пропаноилокси)-прегна-1,4-диен-21-илбутаноат (бе¬
клометазона 21-бутират 17-пропионат).
й. К1 = Н, К2 = Вг, КЗ = К4 = СО-С2Н5:9-Бром-11р-
гидрокси-1 бр-метил-3,20-диоксопрегна-1,4-диен-17,21 -
диилдипропаноат.
Р. К1 = Вг, К2 = С!, КЗ = К4 = СО-С2Н5:6а-Бром-9-
хлор-11 р-гидрокси-1 бр-метил-3,20-диоксопрегна-1,4-
диен-17,21-диилдипропаноат.
Е. К1 = С1, К2 = СО-С2Н5:6а,9-Дихлор-11 р-гидрокси-
1бр-метил-3,20-диоксопрегна-1,4-диен-17,21 -диилди¬
пропаноат.
Н. К1 = К2 =Н: 9-Хлор-11р,21-дигидрокси-16р-
метил-3,20-диоксопрегна-1,4-диен-17-илпропаноат (бе¬
клометазона 17-пропионат).
1.1 бр-Метил-3,20-диоксопрегна-1,4,9(11 }-триен-
17,21 -диилдипропаноат.
3. К1 = К2 = СО-С2Н5: 9,11р-Эпокси-1бр-метил-
3,20-диоксо-9р-прегна-1,4-диен-17,21 -диилдипропаноат.
К. К1 = К2 =Н: 9,11р-Эпокси-17,21-дигидрокси-1бр-
метил-9р-прегна-1,4-диен-3,20-дион.
II. К1 = СО-С2Н5, К2 = Н: 9,11р-Эпокси-21-гидрокси-
1 бр-метил-3,20-диоксо-9р-прегна-1,4-диен-17-илпро-
паноат.
V. К1 = Н, К2 = СО-С2Н5: 9,11р-Эпокси-17-гидрокси-
16р-мешп-3,20-диоксо-9р-прегна-1,4-диен-21 -илпропаноат.
1_. 9-Хлор-11 р-гидрокси-1 бр-метил-3,20-диоксо-
прегн-4-ен-17,21-диилдипропаноат.
М. 9-Хлор-11 р-гидрокси-1 бр-метил-3,20-диок-
сопрегна-4,6-диен-17,21 -диилдипропаноат.
Беклометазона дипропионат моногидрат
245
N. 2-Бром-9-хлор-11 р-гидрокси-1 бр-метил-3,20-
диоксопрета-1,4-диен-17,21 -диилдипропаноат.
О. = П2 = С1: 9,11р-Дихлор-1бр-метил-3,20-
диоксопрегна-1,4-диен-17,21-диилдипропаноат.
О. Н1 = П2 = Н: 16р-Метил-3,20-диоксопрегна-1,4-
диен-17,21 -диилдипропаноат.
5. П1 = 0-С0-С2Н5, Н2 = С1: 9-Хлор-1бр-метил-
3,20-диоксопрегна-1,4-диен-11р, 17,21 -триилтрипропа-
ноат (беклометазона трипропионат).
07/2016:1709
БЕКЛОМЕТАЗОНА ДИПРОПИОНАТ
МОНОГИДРАТ
Вес1оте1азот сИргорюпаз топоЬудпсиз
ВЕС1-ОМЕТАЗОЫЕ 01РКОРЮЫАТЕ
МОЫОНУОРАТЕ
о
С28Н37СЮ7 • н20 М.м. 539,1
ОПРЕДЕЛЕНИЕ
9-Хлор-11 Р-гидрокси-1 бр-метил-3,20-диоксопрегна-
1,4-диен-17,21-диилдипропаноат моногидрат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, умеренно растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО беклометазона дипропионата
моногидрата.
B. 25 мг испытуемого образца сжигают в колбе с
кислородом (2.5.10). Для поглощения продуктов горе¬
ния используют смесь из 1 мл 1 М раствора натрия
гидроксида и 20 мл воды Р. Полученный раствор дает
реакцию (а) на хлориды (2.3.1).
C. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+108 до +115 (в пересчете на сухое вещество). 0,100 г
испытуемого образца растворяют в 96 % спирте Р и
доводят до объема 10,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Подвижная фаза А —
подвижная фаза В (45:55, об/об).
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в 28 мл подвижной фазы В и до¬
водят подвижной фазой А до объема 50,0 мл.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 50,0 мл.
Раствор сравнения (а). 5,0 мл испытуемого раст¬
вора (Ь) доводят смесью растворителей до объема
100,0 мл.
Раствор сравнения (Ь). 5 мг ФСО бекломета¬
зона дипропионата для проверки пригодности хро¬
матографической системы (содержит примесь О)
растворяют в 3 мл подвижной фазы В и доводят под¬
вижной фазой А до объема 5 мл.
Раствор сравнения (с). 5 мг ФСО беклометазо¬
на дипропионата для идентификации пиков (содер¬
жит примеси В, С и 1_) растворяют в 3 мл подвижной
фазы В и доводят подвижной фазой А до объема 5 мл.
1 мл полученного раствора используют для растворе¬
ния содержимого контейнера с ФСО беклометазона
дипропионата примесей Р и N.
Раствор сравнения (д). 50,0 мг ФСО бекломета¬
зона дипропионата безводного растворяют в 28 мл
подвижной фазы В и доводят подвижной фазой А до
объема 50,0 мл. 1,0 мл полученного раствора доводят
смесью растворителей до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим дифункцио¬
нально связанным силикагелем октадецилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 50 °С;
- подвижная фаза:
- подвижная фаза А: раствор 2,72 г/л калия ди¬
гидрофосфата Р, доведенный до рН 2,35 кисло¬
той фосфорной Р\
- подвижная фаза В: тетрагидрофуран Р —
ацетонитрил Р—метанол Р (5:23:25, об/об/об)]
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-^
40
60
4—12
40 —»45
60 —* 55
12—59
45
55
- скорость подвижной фазы: 1,4 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
32. Зак. 1060.
246
Гэсударственная фармакопея Республики Беларусь
- объем ееодимой пробы, по 20 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с).
Идентификация пиков примесей: идентифи¬
цируют пики примесей В, С, Р и 1_, используя хрома¬
тограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО беклометазона дипропионата
для идентификации пиков; идентифицируют пик при¬
меси О, используя хроматограмму раствора сравне¬
ния (Ь) и хроматограмму, прилагаемую к ФСО бекло¬
метазона дипропионата для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
беклометазона дипропионату, время удерживания —
около 25 мин): примесь В — около 0,6; примесь О —
около 1,1; примесь Ь — около 1,3; примесь С — око¬
ло 1,8; примесь Р — около 2,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси О относительно базовой
линии; — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси О и пик
беклометазона дипропионата).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Р — 1,3):
- примесь В (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси В, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-примеси С, Р, Ь (не более 0,15 %): на хрома¬
тограмме испытуемого раствора (а) площади пиков,
соответствующих примесям С, Р, Ц не должны превы¬
шать 1,5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков приме¬
сей В, С, Р и I», не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
менее 2,8 % и не более 3,8 %. 1,000 г испытуемого об¬
разца сушат при температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с!).
Содержание С28На7СЮ7 в процентах рассчитыва¬
ют с учетом содержания беклометазона дипропиона¬
та в ФСО беклометазона дипропионата безводного.
ПРИМЕСИ
Специфицированные примеси: В, С, Р, I.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимити¬
руется общим критерием приемлемости для других/не-
специфицированных примесей и/или общей статьей
Субстанции для фармацевтического использования
(2034). Вследствие этого нет необходимости иденти¬
фицировать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Контроль
примесей в субстанциях для фармацевтического ис¬
пользования): А, О, Е, Н, /, и, М, Л/, О, О, Я Я Ц V.
Н К1
А. Р1 = РЗ = Н, Р2 = С1, Р4 = СО-С2Н5: 9-Хлор-
11Р, 17-дигидрокси-1 бр-метил-3,20-диоксопрегна-1,4-
д иен-21-илпропаноат (беклометазона 21-пропионат).
й. Р1 = Н, Р2 = Вг, РЗ = Р4 = СО-С2Н5:9-Бром-11Э-
гидрокси-1 бр-метил-3,20-диоксопрегна-1,4-диен-17,21 -
диилдипропаноат.
Е. Р1 = Р2 =С1, РЗ = Р4 =СО-С2Н5: 6а,9-Дихлор-
11 р-гидрокси-1 бр-метил-3,20-диоксопрегна-1,4-диен-
17,21 -диилдипропаноат.
Н. Р1 = Р4 = Н, Р2 = С1, РЗ = СО-С2Н5: 9-Хлор-
11 р,21 -дигидрокси-1 бр-метил-3,20-диоксопрегна-1,4-
диен-17-илпропаноат (беклометазона 17-пропионат).
B. Р1 = Н, Р2 = СО-СН3: 21 -(Ацетилокси)-9-хлор-
11 р-гидрокси-16р-метил-3,20-диоксопрегна-1,4-диен-17-
илпропаноат (беклометазона 21-ацетат 17-пропионат).
C. Р1 = Н, Р2 = СО-СН2-СН2-СН3: 9-Хлор-11р-
гидрокси-1 бр-метил-3,20-диоксо-17-(пропаноилокси)-
прегна-1,4-диен-21-илбутаноат (беклометазона 21-бу¬
тират 17-пропионат).
Р. Р1 = Вг, Р2 = СО-С2Н5: 6а-Бром-9-хлор-11р-
гидрокси-16р-метил-3,20-диоксопрегна-1,4-диен-17,21 -
диилдипропаноат.
I. 1 бр-Метил-3,20-диоксопрегна-1,4,9(11 )-триен-
17,21 -диилдипропаноат.
#Бендазола гидрохлорид
247
3. Я1 = Я2 = СО-С2Н5: 9,11р-Эпокси-1бр-метил-
3,20-диоксо-9р-лрегна-1,4-диен-17,21-диилдипропаноат.
Я. Я1 = Я2 =Н: 9,11р-Эпокси-17,21-дигидрокси-1бр-
меткп-9р-прегна-1,4-диен-3,20-дион.
И Н1 = СО-С2Н5, Н2 = Н: 9,11р-Эпокси-21-гидрокси-
16р-метил-3,20-диоксо-9р-прегна-1,4-диен-17-илпро-
паноат.
V. Р1 = Н, Н2 = СО-С2Н5:9,11р-Эпокси-17-гидрокси-
1бр-метил-3,20-диоксо-9р-прегна-1,4-диен-21-илпро-
паноат.
I. 9-Хлор-11р-гидрокси-1бр-метил-3,20-диок-
сопрегн-4-ен-17,21 -диилдипропаноат.
о
М. 9-Хлор-11 р-гидрокси-1 бр-метил-3,20-диок-
сопрегна-4,6-диен-17,21-диилдипропаноат.
N. Н1 = Вг, Я2 = ОН, ЯЗ = С1: 2-Бром-9-хлор-11р-
гидрокси-1 бр-метил-3,20-диоксопрегна-1,4-диен-17,21-
диилдипропаноат.
O. Я1 = Н, Я2 = ЯЗ =С1: 9,11р-Дихлор-1бр-метил-
3,20-диоксопрегна-1,4-диен-17,21 -диилдипропаноат.
О. Я1 = Я2 = ЯЗ = Н: 16р-Метил-3,20-диоксопрегна-
1,4-диен-17,21-диилдипропаноат.
5. Я1 = Н, Я2 = 0-С0-С2Н5, ЯЗ = С1: 9-Хлор-16р-
метил-3,20-диоксопрегна-1,4-диен-11р, 17,21-триилтри-
пропаноат (беклометазона трипропионат).
07/2016:РБ0017
#БЕНДАЗОЛА ГИДРОХЛОРИД
ВепйагоН ЬуйгосЫопдит
ВЕЫОАЮ1.Е НУОКОСНШКЮЕ
• НС1
С14Н12М2НС1
[621-72-7]
М.м. 244,7
ОПРЕДЕЛЕНИЕ
2-(Фенилметил)-1 Н-бензимидазола гидрохлорид.
Дибазол.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или белый со слегка сероватым или жел¬
товатым оттенком кристаллический порошок. Гигро¬
скопичен.
Умеренно растворим в воде, легко растворим в
96 % спирте, практически нерастворим в эфире.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 100,0 мл этим же растворителем. К 5 мл получен¬
ного раствора прибавляют 30 мл 96 % спирта Р, 1 мл
0,1 М раствора натрия гидроксида и доводят 96 %
спиртом Р до объема 50,0 мл.
Диапазон длин волн: от 225 нм до 300 нм.
Максимумы поглощения: при 244 нм, 275 нм,
281 нм.
Минимумы поглощения: при 230 нм, 259 нм,
279 нм.
B. 20 мг испытуемого образца растворяют в 5 мл
воды Р, прибавляют 0,15 мл кислоты хлористоводо¬
родной разведенной Р, 0,15 мл 0,05 М раствора йода
и встряхивают. Образуется красновато-серебристый
осадок.
C. 20 мг испытуемого образца растворяют в 5 мл
воды Р, прибавляют 1 мл раствора аммиака разве¬
денного Р1 и фильтруют. Полученный фильтрат дает
реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют в 100 мл воды Р, нагревают на водяной бане при
температуре 60 °С в течение 10 мин и охлаждают.
Прозрачность (2.2.7). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
рН (2.2.3). От 3,0 до 4,0. Измеряют рН раствора 3.
Температура плавления (2.2.14). От 182 °С до
186 °С.
1,2-Фенилендиамин. Не более 0,05 %. 0,5 г ис¬
пытуемого образца растворяют в 10 мл воды Р при на¬
гревании до температуры 90 °С и подкисляют 0,5 мл
0,1 М раствора кислоты хлористоводородной. При
32*. Зак. 1060.
248
Гэсударственная фармакопея Республики Беларусь
прибавлении 0,05 мл раствора 10 г/л железа (III) хло¬
рида Р не должно появляться желтое окрашивание.
Органические примеси (2.2.2, метод II). 0,3 г
испытуемого образца растворяют в 5 мл кислоты
серной концентрированной Р. Окраска полученного
раствора должна быть не интенсивнее эталона или
У(Ж)6, или В(К)б, или ВУ(КЖ)6.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). Определение проводят из 1,0 г ис¬
пытуемого образца. Эталон готовят с использованием
1 мл эталонного раствора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 1,5 %. 0,500 г испытуемого образца сушат при
температуре от 70 °С до 80 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
2 мл кислоты муравьиной безводной Р, прибавля¬
ют 40 мл уксусного ангидрида Р, 0,3 мл раствора
кристаллического фиолетового Р и титруют 0,1 М
раствором кислоты хлорной до появления зеленого
окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 24,47 мг С14Н12М2 НС1.
ХРАНЕНИЕ
В защищенном от влаги месте при температуре
от 15 °С до 25 °С.
07/2016:0372
БЕНЗАЛКОНИЯ ХЛОРИД
Вепга1копН сЫопдит
ВЕЫ1А1.КОЫШМ СНШВЮЕ
НзС^ уЗНз
'%СНЗ
СП
[8001-54-5]
ОПРЕДЕЛЕНИЕ
Смесь хлоридов алкилбензилдиметиламмония,
алкильная группа которых имеет преимущественно
цепочки С12, С14 и С16.
Содержание: не менее 95,0 % и не более 104,0 %
хлоридов алкилбензилдиметиламмония (в пересчете
на безводное вещество), рассчитанных с использова¬
нием средней относительной молекулярной массы,
определенной как указано в разделе «Испытания».
ОПИСАНИЕ(СВОЙСТВА)
Белый или желтовато-белый порошок или же¬
латинообразные желтовато-белые фрагменты. Ги¬
гроскопичен. При нагревании образуется прозрачная
расплавленная масса.
Очень легко растворим в воде и в 96 % спирте.
Водный раствор при встряхивании образует обиль¬
ную пену.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 80 мг испытуемого образ¬
ца растворяют в воде Р и доводят до объема 100 мл
этим же растворителем.
Диапазон длин волн: от 220 нм до 350 нм.
Максимумы поглощения: при 257 нм, при 263 нм
и при 269 нм.
Плечо: при длине волны около 250 нм.
B. Просматривают хроматограммы, полученные
в испытании «Средняя относительная молекулярная
масса и отношение алкильных компонентов».
Результаты: основные пики на хроматограмме
испытуемого раствора по временам удерживания со¬
ответствуют основным пикам на хроматограмме раст¬
вора сравнения.
C. К 2 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,1 мл кисло¬
ты уксусной ледяной Р и по каплям 1 мл раствора
натрия тетрафенилбората Р. Образуется белый
осадок. Полученную смесь фильтруют, осадок раст¬
воряют в смеси из 1 мл ацетона Р и 5 мл 96 % спир¬
та Р при нагревании до температуры не выше 70 °С.
В горячий раствор по каплям прибавляют воду Р до
появления легкой опалесценции. Раствор слегка по¬
догревают до получения прозрачного раствора и ох¬
лаждают. Образуется белый кристаллический оса¬
док. Раствор фильтруют, осадок трижды промывают
водой Р порциями по 10 мл и высушивают в вакууме
над фосфора (V) оксидом Р или силикагелем безвод¬
ным Р при температуре не выше 50 °С. Температура
плавления (2.2.14) полученных кристаллов от 127 °С
до 133 °С.
й. К 5 мл раствора гидроксида натрия разве¬
денного Р прибавляют 0,1 мл раствора бромфеноло-
вого синего Р1, 5 мл метиленхлорида Р и встряхива¬
ют. Слой метиленхлорида — бесцветный. Прибавля¬
ют 0,1 мл раствора 3 и встряхивают. Слой метилен¬
хлорида окрашивается в синий цвет.
Е. К 2 мл раствора 3 прибавляют 1 мл кислоты
азотной разведенной Р. Образуется белый осадок,
который растворяют в 5 мл 96 % спирта Р. Получен¬
ный раствор дает реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)б.
Кислотность или щелочность. К 50 мл раст¬
вора 5 прибавляют 0,1 мл раствора бромкрезолово-
го пурпурного Р При прибавлении не более 0,1 мл
0,1 М раствора кислоты хлористоводородной или
0,1 М раствора натрия гидроксида окраска раствора
должна измениться.
Средняя относительная молекулярная масса
и отношение алкильных компонентов. Жидкостная
хроматография (2.2.29).
Испытуемый раствор. 0,400 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем.
Бензалкония хлорид
249
Раствор сравнения. Содержимое контейнера
ФСО бензалкония хлорида для проверки пригодно¬
сти хроматографической системы растворяют в
5,0 мл воды Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем нитрильным
эндкепированным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза: смесь из 45 объемов ацето¬
нитрила Р и 55 объемов раствора 13,6 г/л натрия
ацетата Р, предварительно доведенного до рН 5,0
кислотой уксусной ледяной Р;
- скорость подвижной фазы. 2,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Идентификация гомологов: идентифицируют
пики гомологов С12> С14 и С16, используя хроматограм¬
му раствора сравнения и хроматограмму, прилагае¬
мую к ФСО бензалкония хлорида для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению к
гомологу С12, время удерживания — около 6 мин): го¬
молог С14 — около 1,3; гомолог С1б — около 1,7.
Пригодность хроматографической системы.
раствор сравнения:
- разрешение: не менее 1,5 между пиками гомо¬
логов С12 и С14.
Рассчитывают среднюю относительную молеку¬
лярную массу испытуемого образца, суммируя про¬
изведения для каждого из гомологов, полученные по
формуле:
где:
А — площадь пика, соответствующего данному
гомологу, на хроматограмме испытуемого раствора;
В — сумма площадей пиков, соответствующих
всем гомологам, на хроматограмме испытуемого
раствора;
\Л/ — относительная молекулярная масса данного
гомолога: 340,368 и 396 для С12, С14 и С16 соответственно.
Рассчитывают содержание каждого гомолога в
процентах по формуле:
где:
С — произведение относительной молекулярной
массы данного гомолога на площадь соответствующе¬
го пика на хроматограмме испытуемого раствора;
О — сумма значений С для всех гомологов, коли¬
чественно определенных.
Предельное содержание:
- гомолог С12: не менее 40 %;
- гомолог С ■ не менее 20 %;
- сумма гомологов С12 и С14: не менее 70 %.
Примеси А, В и С. Жидкостная хроматография
(2.2.29). Растворы готовят непосредственно перед
использованием.
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в метаноле Р1 и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (а). 25,0 мг ФСО бензилово-
го спирта (примесь А) растворяют в метаноле Р1 и
доводят до объема 100,0 мл этим же растворителем.
Раствор сравнения (Ь). 75,0 мг ФСО бензаль-
дегида (примесь В) растворяют в метаноле Р1 и до¬
водят до объема 100,0 мл этим же растворителем.
1,0 мл полученного раствора доводят метанолом Р1
до объема 10,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (а) доводят метанолом Р1 до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: 1,09 г натрия гексан-
сульфоната Р и 6,9 г натрия дигидрофосфата
моногидрата Р растворяют в воде Р, доводят
до рН 3,5 кислотой фосфорной Р и доводят до
объема 1000,0 мл этим же растворителем;
- подвижная фаза В: метанол Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
80
20
10—14
00
0
1
сл
о
20 —* 50
14—35
50
50
35—36
50 —► 20
сл
0
1
00
о
36—55
20
80
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм для примесей А и С и 257 нм для при¬
меси В;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению к
примеси А, время удерживания — около 10 мин): при¬
месь В — около 1,3; примесь С — около 2,4.
Пригодность хроматографической системы:
при длине волны 210 нм:
- отношение сигнал/шум: не менее 10 для основ¬
ного пика на хроматограмме раствора сравнения (с);
-фактор асимметрии: не менее 0,6 для пика
примеси А на хроматограмме раствора сравнения (а).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С—1,3):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (а);
-примесь В (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (Ь);
- примесь С (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 0,1 площади
основного пика на хроматограмме раствора сравне¬
ния (а).
Амины и соли аминов. 5,0 г испытуемого образ¬
ца растворяют при нагревании в 20 мл смеси из 3 объ¬
емов 1 М раствора кислоты хлористоводородной и
97 объемов метанола Р и прибавляют 100 мл 2-про¬
панола Р. Через раствор медленно пропускают поток
азота Р. Титруют 0,1 М раствором тетрабутилам-
33. Зак. 1060.
250
Гэсударственная фармакопея Республики Беларусь
мония гидроксида, прибавляя его вплоть до 12,0 мл,
и регистрируют кривую потенциометрического титро¬
вания (2.2.20). Если на кривой титрования определя¬
ются две точки перегиба, то объем титранта, который
был добавлен между этими точками, должен быть не
более 5,0 мл. Если на кривой титрования нет ни одной
точки перегиба, то испытуемый образец не выдержи¬
вает требования данного испытания. Если на кривой
титрования определяется одна точка перегиба, ис¬
пытание повторяют, предварительно добавив перед
титрованием 3,0 мл раствора 25,0 г/л диметилдеци-
ламина Р в 2-пропаноле Р. Если на кривой титрования
после прибавления 12,0 мл титранта определяется
только одна точка перегиба, то испытуемый образец
не соответствует требованиям данного испытания.
Вода (2.5.12). Не более 10 %. Определение про¬
водят из 0,300 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,00 г испытуемого образца растворяют в воде Р
и доводят до объема 100,0 мл этим же растворителем.
25,0 мл полученного раствора помещают в делитель¬
ную воронку, прибавляют 25 мл метиленхлорида Р,
10 мл 0,1 М раствора натрия гидроксида и 10,0 мл
свежеприготовленного раствора 50 г/л калия йодида Р.
Полученную смесь энергично встряхивают и дают от¬
стояться до полного разделения слоев. Слой метилен¬
хлорида удаляют. Водный слой трижды встряхивают
с метиленхлоридом Р порциями по 10 мл; слои ме¬
тиленхлорида удаляют. К водному слою прибавляют
40 мл кислоты хлористоводородной Р, охлаждают
и титруют 0,05 М раствором калия йодата до почти
полного исчезновения темно-коричневого окрашива¬
ния. Прибавляют 5 мл метиленхлорида Р и продолжа¬
ют титрование, энергично встряхивая, до прекращения
изменения окраски слоя метиленхлорида.
Проводят контрольный опыт с использованием
смеси из 10,0 мл свежеприготовленного раствора
50 г/л калия йодида Р, 20 мл воды Р и 40 мл кислоты
хлористоводородной Р.
^1 мл 0,05 М раствора калия йодата соответству¬
ет ^ мг бензалкония хлорида, где х — средняя отно¬
сительная молекулярная масса испытуемого образца.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. Бензиловый спирт.
В. Бензальдегид.
07/2016:0705
БЕНЗИЛБЕНЗОАТ
ВепгуНз Ьепгоаз
ВЕЫ2П. ВЕЫЮАТЕ
[120-51-4]
ОПРЕДЕЛЕНИЕ
Фенилметилбензоат.
Содержание: не менее 99,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветные или почти бесцветные кристаллы
или бесцветная или почти бесцветная маслянистая
жидкость.
Практически нерастворим в воде, смешивается
с 96 % спиртом, с метиленхлоридом и с жирными и
эфирными маслами.
Температура кипения: около 320 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр бензилбензоата
по Европейской Фармакопее.
B. К 2 г испытуемого образца прибавляют 25 мл
раствора калия гидроксида спиртового Р и кипятят с
обратным холодильником в течение 2 ч. Выпаривают
этанол на водяной бане, прибавляют 50 мл воды Р и
перегоняют, собирая около 25 мл дистиллята, кото¬
рый используют для идентификации С. Оставшуюся
в перегонной колбе жидкость подкисляют кислотой
хлористоводородной разведенной Р. Образуется
осадок белого цвета, который промывают водой Р
и высушивают в вакууме. Температура плавления
(2.2.14) полученного остатка: от 121 °С до 124 °С.
C. К дистилляту, полученному в идентифика¬
ции В, прибавляют 2,5 г калия перманганата Р, 5 мл
раствора натрия гидроксида разведенного Р, ки¬
пятят с обратным холодильником в течение 15 мин,
охлаждают и фильтруют. Фильтрат подкисляют кисло¬
той хлористоводородной разведенной Р. Образует¬
ся белый осадок, который промывают водой Р и вы¬
сушивают в вакууме. Температура плавления (2.2.14)
полученного остатка: от 121 °С до 124 °С.
ИСПЫТАНИЯ
Кислотность. 2,0 г испытуемого образца раство¬
ряют в 96% спирте Р, доводят до объема 10 мл этим
же растворителем и прибавляют раствор фенолфта¬
леина Р в качестве индикатора. При прибавлении не
более 0,2 мл 0,1 М раствора натрия гидроксида
окраска раствора должна измениться на розовую.
Относительная плотность (2.2.5). От 1,118 до
1,122.
Показатель преломления (2.2.6). От 1,568 до
1,570.
С. (Хлорметил)бензол.
Бензиновый спирт
251
Температура затвердевания (2.2.18). Не менее
17.0 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 2,000 г испытуемого образца прибавляют
50.0 мл 0,5 М раствора калия гидроксида спиртово¬
го и осторожно кипятят с обратным холодильником в
течение 1 ч. Горячий раствор титруют 0,5 М раство¬
ром кислоты хлористоводородной, используя в ка¬
честве индикатора 1 мл раствора фенолфталеина Р.
Параллельно проводят контрольный опыт.
1 мл 0,5 М раствора калия гидроксида спирто¬
вого соответствует 106,1 мг С14Н1202.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом
контейнере в защищенном от света месте.
07/2016:0256
БЕНЗИНОВЫЙ СПИРТ
А1со1ю1 ЬепгуНсиз
ВЕЫ2У1. А1.СОН01.
С7Н,0 М.м. 108,1
[ЮО-51-6]
ОПРЕДЕЛЕНИЕ
Фенилметанол.
Содержание: не менее 98,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная, маслянистая жидкость.
Растворим в воде, смешивается с 96 % спиртом,
с жирными и эфирными маслами.
Относительная плотность: от 1,043 до 1,049.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО спирта бензинового #или
спектр, представленный на рисунке #0256.-1#.
ИСПЫТАНИЯ
Раствор 5. 2,0 мл испытуемого образца встряхи¬
вают с 60 мл воды Р. Испытуемый образец полностью
растворяется.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 8 должен
быть бесцветным.
Кислотность. К 10 мл испытуемого образца
прибавляют 10 мл 96% спирта Р и 1 мл раствора
фенолфталеина Р. При прибавлении не более 1 мл
0,1 М раствора натрия гидроксида должно появить¬
ся розовое окрашивание.
Показатель преломления (2.2.6). От 1,538 до
1,541.
Перекисное (пероксидное) число (2.5.5). Не бо¬
лее 5.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. Испытуемый образец.
Стандартный раствор (а). 0,100 г этилбензо¬
ла Р растворяют в испытуемом растворе и доводят до
Рисунок #0256.-1. Инфракрасный спектр ФСО спирта бензинового
33*. Зак. 1060.
252
Гэсударственная фармакопея Республики Беларусь
объема 10,0 мл этим же растворителем. 2,0 мл полу¬
ченного раствора доводят испытуемым раствором до
объема 20,0 мл.
Стандартный раствор (Ь). 2,000 г дициклогек¬
сила Р растворяют в испытуемом растворе и доводят
до объема 10,0 мл этим же растворителем. 2,0 мл по¬
лученного раствора доводят испытуемым раствором
до объема 20,0 мл.
Раствор сравнения (а). 0,750 г бензальдегида Р
и 0,500 г циклогексилметанола Р растворяют в испы¬
туемом растворе и доводят до объема 25,0 мл этим
же растворителем. 1,0 мл полученного раствора при¬
бавляют к смеси из 2,0 мл стандартного раствора (а)
и 3,0 мл стандартного раствора (Ь) и доводят испыту¬
емым раствором до объема 20,0 мл.
Раствор сравнения (Ь). 0,250 г бензальдегида Р
и 0,500 г циклогексилметанола Р растворяют в испы¬
туемом растворе и доводят до объема 25,0 мл этим
же растворителем. 1,0 мл полученного раствора при¬
бавляют к смеси из 2,0 мл стандартного раствора (а)
и 2,0 мл стандартного раствора (Ь) и доводят испыту¬
емым раствором до объема 20,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 25 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—34
50 — 220
34—69
220
Блок ввода проб
200
Детектор
310
- детектор: пламенно-ионизационный.
Бензиновый спирт, не предназначенный для па¬
рентерального применения.
Объем вводимой пробы: по 0,1 мкл испытуемого
раствора и раствора сравнения (а).
Относительное удерживание (по отношению
к спирту бензиловому, время удерживания — около
26 мин): этилбензол — около 0,28; дициклогексил — око¬
ло 0,59; примесь А— около 0,68, примесь В—около 0,71.
Пригодность хроматографической системы:
раствор сравнения (а):
- - разрешение: не менее 3,0 между пиками при¬
меси А и примеси В.
Если на хроматограмме испытуемого раствора
обнаруживается пик со временем удерживания, со¬
впадающим со временем удерживания этилбензола
или дициклогексила, площадь такого пика вычитают
из площади пика с таким же временем удерживания
на хроматограмме раствора сравнения (а) или (Ь)
(корректируют площади пиков этилбензола дицикло¬
гексила). Такие пики на хроматограмме испытуемого
раствора должны быть учтены при определении сум¬
мы других примесей.
Предельное содержание примесей:
- примесь А (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать разницу
площадей соответствующего пика на хроматограмме
раствора сравнения (а) и на хроматограмме испытуе¬
мого раствора;
- примесь В (не более 0,10 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси В, не должна превышать разницу площадей со¬
ответствующего пика на хроматограмме раствора срав¬
нения (а) и на хроматограмме испытуемого раствора;
- сумма других примесей с относительным вре¬
менем удерживания меньшим, чем время удержива¬
ния пика бензинового спирта (не более 0,04 %): на
хроматограмме испытуемого раствора сумма площа¬
дей всех пиков с относительным временем удержива¬
ния меньшим, чем время удерживания пика бензино¬
вого спирта, кроме пиков примесей А и В, не должна
превышать 4-кратную площадь пика этилбензола на
хроматограмме раствора сравнения (а), при необхо¬
димости скорректированную как указано выше;
- сумма примесей с относительным временем
удерживания большим, чем время удерживания пика
бензинового спирта (не более 0,3 %): сумма площа¬
дей пиков с относительным временем удерживания
большим, чем время удерживания пика бензилового
спирта, не должна превышать площадь пика дицикло¬
гексила на хроматограмме раствора сравнения (а), при
необходимости скорректированную как указано выше;
- неучитываемый предел (0,0001 %): на хрома¬
тограмме испытуемого раствора не учитывают пики с
площадью менее 0,01 площади пика этилбензола на
хроматограмме раствора сравнения (а), при необхо¬
димости скорректированной как указано выше.
Бензиловый спирт, предназначенный для парен¬
терального применения.
Объем вводимой пробы: по 0,1 мкл испытуемого
раствора и раствора сравнения (Ь).
Относительное удерживание (по отношению
к спирту бензиловому, время удерживания — около
26 мин): этилбензол — около 0,28; дициклогексил — око¬
ло 0,59; примесь А— около 0,68; примесь В—около 0,71.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,0 между пиками при¬
меси А и примеси В.
Если на хроматограмме испытуемого раствора
обнаруживается пик со временем удерживания, со¬
впадающим со временем удерживания этилбензола
или дициклогексила, площадь такого пика вычитают
из площади пика с таким же временем удерживания
на хроматограмме раствора сравнения (а) или (Ь)
(корректируют площади пиков этилбензола дицикло¬
гексила). Такие пики на хроматограмме испытуемого
раствора должны быть учтены при определении сум¬
мы других примесей.
Предельное содержание примесей:
- примесь А (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать разницу
площадей соответствующего пика на хроматограмме
раствора сравнения (Ь) и на хроматограмме испытуе¬
мого раствора;
-примесь В (не более 0,10 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси В, не должна превышать разницу
площадей соответствующего пика на хроматограмме
раствора сравнения (Ь) и на хроматограмме испытуе¬
мого раствора;
- сумма других примесей с относительным вре¬
менем удерживания меньшим, чем время удержива¬
ния пика бензилового спирта (не более 0,02 %): на
хроматограмме испытуемого раствора сумма площа¬
дей всех пиков с относительным временем удержива¬
ния меньшим, чем время удерживания пика бензило-
Бензилпенициллин натрия
253
вого спирта, кроме пиков примесей А и В, не должна
превышать 2-кратную площадь пика этилбензола на
хроматограмме раствора сравнения (Ь), при необхо¬
димости скорректированную как указано выше;
- сумма примесей с относительным временем
удерживания большим, чем время удерживания пика
бензинового спирта (не более 0,2 %): сумма площа¬
дей пиков с относительным временем удерживания
большим, чем время удерживания пика бензилового
спирта, не должна превышать площадь пика дицикло¬
гексила на хроматограмме раствора сравнения (Ь), при
необходимости скорректированную как указано выше;
- неучитываемый предел (0,0001 %): на хрома¬
тограмме испытуемого раствора не учитывают пики с
площадью менее 0,01 площади пика этилбензола на
хроматограмме раствора сравнения (Ь), при необхо¬
димости скорректированной как указано выше.
Остаток после выпаривания. Не более 0,05 %.
Если испытуемый образец выдерживает испытание
«Перекисное (пероксидное) число», 10,0 г испытуе¬
мого образца выпаривают досуха в предварительно
взвешенном кварцевом, фарфоровом или плати¬
новом тигле на горячей плитке при температуре не
выше 200 °С, не допуская кипения. Сушат на плитке в
течение 1 ч и охлаждают в эксикаторе. Масса остатка
не должна превышать 5 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,900 г (т, г) испытуемого образца прибавляют
15,0 мл свежеприготовленной смеси ангидрид уксус¬
ный Р — пиридин безводный Р (1:7, об/об) и кипятят
с обратным холодильником на водяной бане в тече¬
ние 30 мин. Охлаждают и прибавляют 25 мл воды Р.
Титруют 1 М раствором натрия гидроксида (пг мл),
используя в качестве индикатора 0,25 мл раствора
фенолфталеина Р.
Параллельно проводят контрольный опыт (л2, мл).
Рассчитывают содержание С7Н80 в процентах по
формуле:
10,81 (п2-п,)
т
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в атмос¬
фере азота, в защищенном от света месте при темпе¬
ратуре от 2 °С до 8 °С.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. Бензальдегид.
В. Циклогексилметанол.
07/2016:0114
БЕНЗИЛПЕНИЦИЛЛИН НАТРИЯ
Веп2у1ретсИНпит паМсит
ВЕЛ/2У/.РЕЛ//С/И./Л/ 8СЮШМ
0
С16Н17Ы2Ыа043 М.м. 356,4
[69-57-8]
ОПРЕДЕЛЕНИЕ
Натрия (25,5Я,6#)-3,3-диметил-7-оксо-6-[(фенил-
ацетил)амино]-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоксилат.
Получают с использованием штаммов Рет’сИНит
по1а1ит или родственных организмов или любым дру¬
гим способом.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень легко растворим в воде, практически не¬
растворим в жирных маслах и в вазелиновом масле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая подлинность (идентификация): А, О.
Вторая подлинность (идентификация): В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бензилпенициллина натрия
#или спектр, представленный на рисунке #0114.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 5 мл воды Р.
Раствор сравнения (а). 25 мг ФСО бензилпени¬
циллина натрия растворяют в 5 мл воды Р.
Раствор сравнения (Ь). 25 мг ФСО бензилпени¬
циллина натрия и 25 мг ФСО феноксиметилпени-
циллина калия растворяют в 5 мл воды Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: смешивают 30 объемов ацето¬
на Р с 70 объемами раствора 154 г/л аммония ацета¬
та Р, предварительно доведенного до рН 5,0 кисло¬
той уксусной ледяной Р.
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода до появления пятен и просматривают при днев¬
ном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
34. Зак. 1060.
254
Государственная фармакопея Республики Беларусь
3000 2000 1500 1000
Волновое число (см1)
Рисунок #0114.-1. Инфракрасный спектр ФСО бензилпенициллина натрия
С. 2 мг испытуемого образца помещают в про¬
бирку длиной около 150 мм и диаметром около
15 мм, увлажняют 0,05 мл воды Р, прибавляют 2 мл
раствора формальдегида в серной кислоте Р и
перемешивают. Раствор практически бесцветный.
Пробирку выдерживают на водяной бане в течение
1 мин. Появляется красновато-коричневое окраши¬
вание.
О. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 5,5 до 7,5. 2,0 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 20 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+285 до +310 (в пересчете на сухое вещество). 0,500 г
испытуемого образца растворяют в воде, свободной
от углерода диоксида, Р и доводят до объема 25,0 мл
этим же растворителем.
Оптическая плотность (2.2.25). 90,0 мг испы¬
туемого образца растворяют в воде Р и доводят до
объема 50,0 мл этим же растворителем. Измеряют
оптическую плотность раствора при 325 нм, 280 нм
и в максимуме поглощения при 264 нм; при необхо¬
димости раствор разводят для измерения оптической
плотности при 264 нм. Оптическая плотность при
325 нм и 280 нм не должна превышать 0,10; оптиче¬
ская плотность в максимуме при 264 нм — от 0,80 до
0,88 в пересчете на неразведенный раствор (1,80 г/л).
Определяют разрешающую способность спектрофо¬
тометра (2.2.25): отношение оптических плотностей
должно быть не менее 1,7.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем.
Испытуемый раствор (Ь). 80,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
20.0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг ФСО бензилпе¬
нициллина натрия растворяют в воде Р и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО бензилпени¬
циллина натрия и 10 мг фенилуксусной кислоты Р
(примесь В) растворяют в воде Р и доводят до объема
50 мл этим же растворителем.
Раствор сравнения (с). 4,0 мл раствора сравне¬
ния (а) доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
-подвижная фаза А: смешивают 10 объемов
раствора 68 г/л калия дигирофосфата Р, дове¬
денного до рН 3,5 раствором 500 г/л кислоты
фосфорной разведенной Р, 30 объемов метано¬
ла Р и 60 объемов воды Р;
-подвижная фаза В: смешивают 10 объемов
раствора 68 г/л калия дигирофосфата Р, дове¬
денного до рН 3,5 раствором 500 г/л кислоты
фосфорной разведенной Р, 40 объемов воды Р и
50 объемов метанола Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
70
30
о
см
+
1
70 — 0
30 — 100
(Гк + 20)-((„ + 35)
0
100
(*я + 35)-(4, + 50)
70
30
1Я — время удерживания бензилпенициллина на хромато¬
грамме раствора сравнения (с).
Бензилпенициллин натрия
255
М
Если для достижения необходимого разрешения
изменялось соотношение подвижных фаз А и В, то
подвижную фазу полученного состава используют в
начальном времени градиента и для количественного
определения.
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: по 20 мкл растворов
сравнения (Ь) и (с) хроматографируют изократически,
используя соотношение подвижных фаз А и В началь¬
ного времени градиента; 20 мкл испытуемого раст¬
вора (Ь) хроматографируют согласно программе гра¬
диента; 20 мкл воды Р хроматографируют согласно
программе градиента в качестве контрольного опыта.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 6,0 между пиками при¬
меси В и бензилпенициллина; при необходимости из¬
меняют соотношение подвижных фаз А и В.
Предельное содержание примесей:
-любая примесь (не более 1 %): на хромато¬
грамме испытуемого раствора (Ь) площадь любого
пика, кроме основного, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с).
2-Этилгексановая кислота (2.4.28). Не более
0,5 % (м/м).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Бактериальные эндотоксины (2.6.14, метод ЕЕ).
Менее 0,16 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей процеду¬
ры удаления бактериальных эндотоксинов.
#Пирогенность (2.6.8). Испытание «Пироген-
ность» проводят в качестве альтернативного ис¬
пытанию «Бактериальные эндотоксины». Испытуе¬
мый образец должен быть апирогенным. Тест-доза —
20 000 ЕД в 1 мл воды для инъекций Р на 1,0 кг массы
тела кролика, внутривенно.
#Стерильность (2.6.1). Если указано, что ис¬
пытуемый образец должен быть стерильным.
Бензилпенициллин натрия в условиях испытания
обладает антимикробным действием. При посеве на
питательные среды используют метод мембранной
фильтрации.
#Аномальная токсичность (2.6.9). Испытуе¬
мый образец должен быть нетоксичным. Тест-доза —
5000 ЕД на мышь в 0,5 мл воды для инъекций Р, вну¬
тривенно. Срок наблюдения — 24 ч.
#Антимикробная активность (2.7.2, метод А).
Не менее 1600 ЕД/мг в пересчете на сухое вещество,
где применимо.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: соотношение подвижных фаз
А и В начального времени градиента в соответствии
с параметрами пригодности хроматографической си¬
стемы;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и раствора сравнения (а).
Содержание С16Н17М2Ма043 в процентах рассчи¬
тывают с учетом содержания бензилпенициллина на¬
трия в ФСО бензилпенициллина натрия.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
\
н2м--
н н
Н С02Ма
м--Дхн3
^8/^СНз
А. (25,5Я6Я)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
ГГ'
со2н
В. Фенилуксусная кислота.
С. (2$,5Я,6#)-6-[[(4-Гидроксифенил)ацетил]амино]-
3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоновая кислота.
О. (35,77?,7а/?)-5-Бензил-2,2-диметил-2,3,7,7а-
тетрагидроимидазо[5,1 -Ь]тиазол-3,7-дикарбоновая кис¬
лота (пениллиновая кислота бензилпенициллина).
Е. (45)-2-[Карбокси[(фенилацетил)амино]метил]-
5,5-диметилтиазолидин-4-карбоновая кислота (пени-
циллоиновые кислоты бензилпенициллина).
и эпимер у С*
Р. (2/?5,45)-2-[[(Фенилацетил)амино]метил]-5,5-
диметилтиазолидин-4-карбоновая кислота (пениллои-
новые кислоты бензилпенициллина).
34*. Зак. 1060.
256
Гэсударственная фармакопея Республики Беларусь
07/2016:0066
БЕНЗОЙНАЯ КИСЛОТА
АсШит Ьепго 'юит
ВЕИ201С АСЮ
С7Н602 М.м. 122,1
[65-85-0]
ОПРЕДЕЛЕНИЕ
Бензолкарбоновая кислота.
Содержание: не менее 99,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Мало растворима в воде, растворима в кипящей
воде, легко растворима в 96 % спирте и в жирных маслах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ).
A. Температура плавления (2.2.14): от 121 °С до
124 °С.
B. Раствор 5, приготовленный как указано в разде¬
ле «Испытания», дает реакцию (а) на бензоаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в 96 % спирте Р и доводят до объема 100 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Обугливающиеся вещества. 0,5 г испытуемого
образца растворяют при встряхивании в 5 мл кис¬
лоты серной Р. Через 5 мин окраска полученного
раствора должна быть не интенсивнее эталона У(Ж)5
(2.2.2, метод I).
Окисляемые вещества. 0,2 г испытуемого об¬
разца растворяют в 10 мл кипящей воды Р, охлажда¬
ют, встряхивают и фильтруют. К фильтрату прибавляют
1 мл кислоты серной разведенной Р и 0,2 мл 0,02 М
раствора калия перманганата. Розовая окраска раст¬
вора должна сохраняться в течение не менее 5 мин.
4 Галогенопроизводные и галогениды. Не бо¬
лее 0,0300 % (300 ррт).
Вся используемая стеклянная посуда не должна
содержать хлориды. Для этой цели рекомендуется
выдерживать стеклянную посуду в течение ночи в
растворе 500 г/л кислоты азотной Р, промывать во¬
дой Р и хранить полностью погруженной в воду Р. Ре¬
комендуется использовать отдельную стеклянную
посуду исключительно для данного испытания.
Раствор (а). 6,7 г испытуемого образца раст¬
воряют в смеси из 40 мл 1 М раствора натрия ги¬
дроксида и 50 мл 96 % спирта Р и доводят водой Р
до объема 100,0 мл. К 10,0 мл полученного раствора
прибавляют 7,5 мл раствора натрия гидроксида раз¬
веденного Р и 0,125 г никель-алюминиевого спла¬
ва Р и нагревают на водяной бане в течение 10 мин.
Охлаждают до комнатной температуры, фильтруют
в мерную колбу вместимостью 25 мл и промывают
тремя порциями по 2 мл 96% спирта Р. Фильтрат и
промывные воды доводят водой Р до объема 25,0 мл.
Полученный раствор используют для приготовления
раствора А.
Раствор (Ь). Готовят аналогично раствору (а), но
без испытуемого образца. Полученный раствор ис¬
пользуют для приготовления раствора В.
В четыре мерные колбы вместимостью 25 мл по¬
мещают по отдельности 10 мл раствора (а), 10 мл
раствора (Ь), 10 мл эталонного раствора хлорида
(8 ррт С!) Р (используют для приготовления раст¬
вора С) и 10 мл воды Р. В каждую колбу прибавляют по
5 мл раствора железа (III) аммония сульфата Р5, пере¬
мешивают, прибавляют по каплям при взбалтывании по
2 мл кислоты азотной Р и по 5 мл раствора ртути (II)
тиоцианата Р и встряхивают. Доводят содержимое
каждой мерной колбы водой Р до объема 25,0 мл. Полу¬
ченные растворы выдерживают в водяной бане при тем¬
пературе 20 °С в течение 15 мин. Измеряют оптическую
плотность (2.2.25) раствора А, используя раствор В в ка¬
честве компенсационного раствора, и оптическую плот¬
ность раствора С, используя раствор, полученный из
10 мл водыР, в качестве компенсационного раствора.
Оптическая плотность раствора А не должна превы¬
шать оптическую плотность раствора С.
Тяжелые металлы (2.4.8, метод В). Не более
0,0010% (10 ррт). 12 мл раствора 3 должны вы¬
держивать испытание на тяжелые металлы. Эталон
готовят с использованием смеси из 5 мл эталонного
раствора свинца (1 ррт РЬ) Р и 5 мл 96 % спирта Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 20 мл
96 % спирта Р и титруют 0,1 М раствором натрия
гидроксида до изменения окраски раствора от желтой
до фиолетово-красной, используя в качестве индика¬
тора 0,1 мл раствора фенолового красного Р.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 12,21 мгС7Н602.
07/2016:0011
БЕНЗОКАИН
Ветосатит
ВЕЫТ.ОСАШЕ
[94-09-7]
ОПРЕДЕЛЕНИЕ
Этил-4-аминобензоат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
г
Бензокаин
257
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень мало растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 89 °С до
92 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бензокаина #или спектр, пред¬
ставленный на рисунке #0011.-1#.
C. 50 мг испытуемого образца помещают в
пробирку и прибавляют 0,2 мл раствора 500 г/л
хрома (VI) оксида Р. Пробирку накрывают филь¬
тровальной бумагой, смоченной свежеприготов¬
ленной смесью из равных объемов раствора 50 г/л
натрия нитропруссида Р и раствора 200 г/л пипе¬
разина гидрата Р, и осторожно кипятят в течение
не менее 30 с. На фильтровальной бумаге появля¬
ется синее окрашивание.
й. 50 мг испытуемого образца растворяют в
96 % спирте Р и доводят до объема 100 мл этим
же растворителем. 2 мл полученного раствора
дают реакцию на первичные ароматические амины
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 96% спирте Р и доводят до объема 20 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность или щелочность. 0,5 г испытуе¬
мого образца растворяют в 10 мл 96 % спирта Р,
предварительно нейтрализованного (с использо¬
ванием 0,05 мл раствора фенолфталеина Р), и
прибавляют 10 мл воды, свободной от углерода
диоксида, Р. Раствор должен быть бесцветным.
При прибавл+ении не более 0,5 мл 0,01 М раст¬
вора натрия гидроксида окраска раствора должна
измениться.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,00 г испытуемого образца сушат
в вакууме.
Сульфатная зола (2.4.14). Не более 0,1 %. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ.
0,400 г испытуемого образца растворяют в
смеси из 25 мл кислоты хлористоводородной Р
и 50 мл воды Р и проводят определение аминного
азота в соединениях, которые содержат первичную
ароматическую аминогруппу (2.5.8).
1 мл 0,1 М раствора натрия нитрита соот¬
ветствует 16,52 мг СдН^О^
ХРАНЕНИЕ
В защищенном от света месте.
Рисунок #0011 .-1. Инфракрасный спектр ФСО бензокаина
35. Зак. 1060.
258
Гэсударственная фармакопея Республики Беларусь
07/2016:0974
БЕНЗЭТОНИЯ ХЛОРИД
ВепгеПюпН сЫопс1ит
ВЕЫ1ЕТНОЫШМ СШ-ОКЮЕ
С27Н42С1Ы02 М.м. 448,1
[121-54-0]
ОПРЕДЕЛЕНИЕ
А/-Бензил-Л/,Л/-диметил-2-[2-[4-(1,1,3,3-тетраме-
тилбутил)фенокси]этокси]этанаминия хлорид.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок.
Очень легко растворим в воде и в 96 % спирте,
легко растворим в метиленхлориде.
Водный раствор при встряхивании дает обиль¬
ное пенообразование.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14). От 158 °С до
164 °С, определяют после высушивания при темпера¬
туре 105 °С в течение 4 ч.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения. 25 мг ФСО бензэтония хло¬
рида растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
пяР254 Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— метанол Р (5:5:100, об/об/об).
Наносимый объем пробы: 20 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основно¬
му пятну на хроматограмме раствора сравнения.
C. К 5 мл раствора натрия гидроксида раз¬
веденного Р прибавляют 0,1 мл раствора бром-
фенолового синего Р1, 5 мл метиленхлорида Р и
встряхивают. Нижний слой должен быть бесцвет¬
ным. Прибавляют 0,1 мл раствора 3, приготовлен¬
ного как указано в разделе «Испытания», и встря¬
хивают. В нижнем слое появляется синее окраши¬
вание.
О, К 2 мл раствора 3 прибавляют 1 мл кис¬
лоты азотной разведенной Р. Образуется белый
осадок, который растворяется при прибавлении
5 мл 96% спирта Р. Полученный раствор дает ре¬
акцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Кислотность или щелочность. К 25 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфтале¬
ина Р. Раствор должен быть бесцветным. Прибав¬
ляют 0,3 мл 0,01 М раствора натрия гидроксида.
Должно появиться розовое окрашивание. Прибав¬
ляют 0,1 мл раствора метилового красного Р и
0,5 мл 0,01 М раствора кислоты хлористоводо¬
родной. Должно появиться оранжево-красное окра¬
шивание.
Летучие основания и соли летучих основа¬
ний (2.4.1, метод В). Не более 0,0050 % (50 ррт).
0,20 г испытуемого образца должны выдерживать
испытание на аммония соли. Эталон готовят с ис¬
пользованием 0,1 мл эталонного раствора аммо¬
ния (100 ррт Д/Н^ Р. Магния оксид тяжелый заме¬
няют на 2,0 мл раствора натрия гидроксида кон¬
центрированного Р.
Потеря в массе при высушивании (2.2.32).
Не более 5,0 %. 1,000 г испытуемого образца су¬
шат при температуре от 100 °С до 105 °С в течение
4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца растворяют в
воде Р и доводят до объема 100,0 мл этим же раст¬
ворителем. 25,0 мл полученного раствора пере¬
носят в делительную воронку, прибавляют 10 мл
раствора 4 г/л натрия гидроксида Р, 10,0 мл све¬
жеприготовленного раствора 50 г/л калия йодида Р
и 25 мл метиленхлорида Р. Интенсивно встряхи¬
вают, оставляют до разделения слоев и отбрасы¬
вают нижний слой. Верхний слой трижды встряхи¬
вают с метиленхлоридом Р порциями по 10 мл,
каждый раз отбрасывая нижний слой. К верхнему
слою прибавляют 40 мл кислоты хлористоводо¬
родной Р, охлаждают и титруют 0,05 М раствором
калия йодата до практически полного исчезнове¬
ния темно-коричневой окраски. Затем прибавляют
4 мл метиленхлорида Р и продолжают титрование
при интенсивном встряхивании до исчезновения
коричневой окраски нижнего слоя.
Параллельно проводят контрольный опыт с ис¬
пользованием смеси из 10,0 мл свежеприготовлен¬
ного раствора 50 г/л калия йодида Р, 20 мл воды Р и
40 мл кислоты хлористоводородной Р.
1 мл 0,05 М раствора калия йодата соответ¬
ствует 44,81 мг С27Н42СИЧ02.
ХРАНЕНИЕ
В защищенном от света месте.
Бентонит
259
07/2016:0467
БЕНТОНИТ
Веп1опПит
ВЕЫТОШТЕ
ОПРЕДЕЛЕНИЕ
Натуральная глина, состоящая большей частью
из монтмориллонита, природного гидратированного
алюминия силиката, в котором некоторые атомы алю¬
миния и кремния могут быть замещены другими ато¬
мами, например, магния и железа.
ОПИСАНИЕ (СВОЙСТВА)
Очень мелкий гомогенный серовато-белый поро¬
шок с большим или меньшим желтоватым или розова¬
тым оттенком.
Практически нерастворим в воде и водных рас¬
творах.
Набухает в небольшом количестве воды, образуя
вязкую массу.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,5 г испытуемого образца помещают в
металлический тигель и прибавляют 1 г калия ни¬
трата Р и 3 г натрия карбоната Р и нагревают
до плавления смеси. Охлаждают. К полученному
остатку прибавляют 20 мл кипящей воды Я, пере¬
мешивают и фильтруют. Нерастворимый остаток
промывают, используя 50 мл воды Р. К получен¬
ному остатку прибавляют 1 мл кислоты хлори¬
стоводородной Р и 5 мл воды Р и фильтруют. К
полученному фильтрату прибавляют 1 мл раст¬
вора натрия гидроксида концентрированного Р и
фильтруют. К полученному фильтрату прибавляют
3 мл раствора аммония хлорида Р. Образуется
студенистый белый осадок.
B. 2,0 г испытуемого образца прибавляют
двадцатью порциями к 100 мл раствора 10 г/л на¬
трия лаурилсульфата Р, помещенного в мерный
цилиндр, градуированный до 100 мл и диаметром
около 30 мм. После прибавления каждой порции
содержимому цилиндра дают отстояться 2 мин.
Выдерживают в течение 2 ч. Кажущийся объем
осадка составляет не менее 22 мл.
C. 0,25 г испытуемого образца дают реакцию (а)
на силикаты (2.3.7).
ИСПЫТАНИЯ
Щелочность. К 2 г испытуемого образца при¬
бавляют 100 мл воды, свободной от углерода ди¬
оксида, Р и встряхивают в течение 5 мин. К 5 мл
полученной суспензии прибавляют 0,1 мл раст¬
вора тимолфталеина Р. Жидкость должна при¬
обрести голубоватую окраску. При прибавлении
0,1 мл 0,1 М раствора кислоты хлористоводо¬
родной жидкость обесцвечивается в течение 5 мин.
Крупнозернистые частицы. Не более 0,5 %.
К 20 г прибавляют 1000 мл воды Р и переме¬
шивают в течение 15 мин, используя высокоско¬
ростную мешалку, обеспечивающую при работе не
менее 5000 об/мин. Переносят суспензию на ув¬
лажненное сито (75), предварительно взвешенное
после высушивания при температуре от 100 °С до
105 °С. Трижды промывают водой Р порциями по
500 мл, обеспечивая диспергирование всех агло¬
мератов. Высушивают сито при температуре от
100 °С до 105 °С и взвешивают. Масса частиц на
сите не должна превышать 0,1 г.
Тяжелые металлы (2.4.8, метод А). Не более
0,0050 % (50 ррт). К 5,0 г испытуемого образца
прибавляют 7,5 мл кислоты хлористоводород¬
ной разведенной Р и 27,5 мл воды Р и кипятят в
течение 5 мин. Центрифугируют и фильтруют на-
досадочную жидкость. Промывают осадок, образо¬
ванный в результате центрифугирования, водой Р
и фильтруют. Объединенные фильтраты доводят
водой Р до объема 50,0 мл. К 5 мл полученного
раствора прибавляют 5 мл воды Р, 10 мл кислоты
хлористоводородной Р и 25 мл метилизобутил-
кетона Р и встряхивают в течение 2 мин. Разде¬
ляют слои. Выпаривают водный слой досуха на во¬
дяной бане. Остаток растворяют в 1 мл кислоты
уксусной Р, доводят водой Р до объема 25 мл и
фильтруют. 12 мл фильтрата должны выдерживать
испытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 15 %. 1,000 г испытуемого образца сушат
при температуре 105 °С.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным и для
подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для бентонита, используемого в качестве
вещества, повышающего вязкость, и суспендиру¬
ющего вещества.
Объем седиментации. К 6,0 г испытуемого
образца прибавляют 200 мл воды Р и перемешива¬
ют в течение 20 мин, используя высокоскоростную
мешалку, обеспечивающую при работе не менее
10 000 об/мин. 100 мл полученной суспензии пере¬
носят в мерный цилиндр и выдерживают в течение
24 ч. Объем прозрачной надосадочной жидкости
не должен превышать 2 мл.
Степень набухания в воде (см. подлинность В).
35*. Зак/1060.
260
Гэсударственная фармакопея Республики Беларусь
07/2016:1665
БЕТАГИСТИНА ДИГИДРОХЛОРИД
Ве1аЫвИп1 сШ1ус1госМопс1ит
ВЕТАН18Т1ЫЕ 0/НУОЯОСЯ/.ОЯ/ОЕ
2НС1
С8Н12Ы2 • 2НС1 М.м. 209,1
[5579-84-0]
ОПРЕДЕЛЕНИЕ
Л/-Метил-2-(пиридин-2-ил)этанамина дигидрохло-
рид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтый порошок. Очень гигро¬
скопичен.
Очень легко растворим в воде, растворим в 96 %
спирте, практически нерастворим в 2-пропаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 150 °С до
154 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бетагистина дигидрохлорида
#или спектр, представленный на рисунке #1665.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 2 мл 96% спирта Р.
Раствор сравнения. 10 мг ФСО бетагистина
дигидрохлорида растворяют в 2 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ вР254 Р'
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетатР — метанол Р
(0,75:15:30, об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 110 °С в течение
10 мин.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)8.
рН (2.2.3). От 2,0 до 3,0. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО бетагистина
дигидрохлорида и 10 мг 2-винилпиридина Р раство¬
ряют в подвижной фазе и доводят до объема 50,0 мл
*
8.
б
Волновое число (см'1)
Рисунок #1665.-1. Инфракрасный спектр ФСО бетагистина дигидрохлорида
I
ш
*
А
Бетагистина мезилат
261
этим же растворителем. 2,0 мл полученного раствора
доводят подвижной фазой до объема 50,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 2,0 мл раствора сравне¬
ния (Ь) доводят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 3,0 мм, заполненная силикагелем октадецил-
сипильным деактивированным по отношению к ос¬
нованиям эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: 2,0 г натрия додецилсуль-
фатаР растворяют в смеси из 15 мл 10% (об/об)
раствора кислоты серной Р, 35 мл раствора 17 г/л
тетрабутиламмония гидросульфата Р и 650 мл
воды Р; доводят до рН 3,3 раствором натрия гидрок¬
сида разведенным Р и перемешивают с 300 мл аце¬
тонитрила Р;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания бетагистина.
Относительное удерживание (по отношению к
бетагистину, время удерживания — около 7 мин): при¬
месь В — около 0,2; примесь А — около 0,3; примесь
С — около 3.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,5 между пиками 2-ви-
нилпиридина и бетагистина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,4):
- примеси А, В, С (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, В, С, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (с);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
80,0 мг испытуемого образца растворяют в 50 мл
96 % спирта Р и титруют 0,1 М раствором натрия
гидроксида потенциометрически (2.2.20). Отмечают
объем титранта во второй точке перегиба на кривой
титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 10,46 мг С8Н12М2-2НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
^|^СН2
А. 2-Этенилпиридин (2-винилпиридин).
.IV .он
В. 2-(Пиридин-2-ил)этанол.
СН3
с"
.14.
N.
С. А/-Метил-2-(пиридин-2-ил)-Л/-[2-(пиридин-2-ил)-
этил]этанамин.
07/2016:1071
БЕТАГИСТИНА МЕЗИЛАТ
Ве1аЫзИп'1 тезНаз
ВЕТАН18Т1ЫЕ МЕЗ ПАТЕ
Н
I СН3 2 Н3С—З03н
С8н12м2 ■ 2СН4035 М.м. 328,4
[54856-23-4]
ОПРЕДЕЛЕНИЕ
Л/-Метил-2-(пиридин-2-ил)этанамина бис(метан-
сульфонат).
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ПРОИЗВОДСТВО
Необходимо учитывать, что алкилметансульфо-
наты являются генотоксичными соединениями и могут
присутствовать в качестве примесей в бетагистина
мезилате. При разработке процесса производства не¬
обходимо соблюдать принципы управления рисками
качества, принимая во внимание качество исходных
материалов, мощность производственного процесса
и валидацию. Подходящими для применения произ¬
водителями являются методы 2.5.37 Метил-, этил- и
изопропилметансульфонат в метансульфоновой кис¬
лоте, 2.5.38 Метил-, этил- и изопропилметансульфо¬
нат в фармацевтических субстанциях и 2.5.39 Ме-
тансульфонилхлорид в метансульфоновой кислоте.
262
Государственная фармакопея Республики Беларусь
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Очень гигроскопичен.
Очень легко растворим в воде, легко растворим
в 96 % спирте, очень мало растворим в 2-пропаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 108 °С до
112 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бетагистина мезилата.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 96 % спирте Р и доводят до объ¬
ема 2 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО бетагистина
мезилата растворяют в 96 % спирте Р и доводят до
объема 2 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ляРг5АР.
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетат Р — метанол Р
(0,75:15:30, об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: при температуре 110 °С в течение
10 мин.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
О. К 0,1 г испытуемого образца прибавляют
5 мл кислоты хлористоводородной разведенной Р
и встряхивают в течение около 5 мин. Прибавляют
1 мл раствора бария хлорида Р1. Раствор остается
прозрачным. К другой навеске испытуемого образца
массой 0,1 г прибавляют 0,5 г натрия карбоната без¬
водного Р, перемешивают и прокаливают до получе¬
ния белого остатка. Охлаждают и растворяют остаток
в 7 мл воды Р. Полученный раствор дает реакцию (а)
на сульфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 2,0 до 3,0. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО бетагисти¬
на мезилата и 10 мг 2-винилпиридина Р (примесь А)
растворяют в подвижной фазе и доводят до объема
50,0 мл этим же растворителем. 2,0 мл полученно¬
го раствора доводят подвижной фазой до объема
50,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 2,0 мл раствора сравне¬
ния (Ь) доводят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- подвижная фаза: 2,0 г натрия додецилсульфа-
та Р растворяют в смеси 10 % (об/об) раствор кисло¬
ты серной Р — раствор 17 г/л тетрабутиламмония
гидросульфата Р — вода Р (15:35:650, об/об/об); до¬
водят до рН 3,3 раствором натрия гидроксида раз¬
веденным Р и смешивают с 300 мл ацетонитрила Р\
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания бетагистина мезилата.
Время удерживания: бетагистина мезилат — око¬
ло 8 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,5 между пиками при¬
меси А и бетагистина мезилата.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси А, не должна превышать 0,1 площади основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (Ь).
2-Пропанол (2.4.24). Не более 0,5 %.
Хлориды (2.4.4). Не более 0,0035 % (35 ррт).
К 14 мл раствора 3 прибавляют 1 мл воды Р. Полу¬
ченный раствор должен выдерживать испытания на
хлориды.
Сульфаты (2.4.13). Не более 0,0250 % (250 ррт).
6 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Вода (2.5.12). Не более 2,0 %. Определение про¬
водят из 0,50 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бетаксолола гидрохлорид
263
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,140 г испытуемого образца растворяют в 50 мл
смеси из кислоты уксусной безводной Р и уксусного
ангидрида Р (1:7, об/об). Титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,42 мг С8Н12Ы2-2СН4033.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Спетфицированные примеси: А.
А. 2-Этенилпиридин (2-винилпиридин).
07/2016:1072
БЕТАКСОЛОЛА ГИДРОХЛОРИД
Ве1ахо1оН ЬудюсЫогШит
ВЕТАХОЮ1. НУОВОСН1.СЖЮЕ
н он
и энантиомер
с18н29мо3
НС1
[63659-19-8]
НС!
М.м. 343,9
ОПРЕДЕЛЕНИЕ
(2/?5)-1-[4-[2-(Циклопропилметокси)этил]фенокси]-
3-[(1-метилэтил)амино]пропан-2-ола гидрохлорид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Очень гигроскопичен.
Очень легко растворим в воде, легко растворим в
96 % спирте, растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 113 °С до
117 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бетаксолола гидрохлорида
#или спектр, представленный на рисунке #1072.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 1 мл метанола Р.
Раствор сравнения (а). 20 мг ФСО бетаксолола
гидрохлорида растворяют в 2 мл метанола Р.
Раствор сравнения (Ь). 10 мг ФСО окспренолола
гидрохлорида растворяют в 1 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: кислота хлорная Р — мета¬
нол Р — вода Р (0,5:50:50, об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Рисунок #1072.-1. Инфракрасный спектр ФСО бетаксолола гидрохлорида
264
Гэсударственная фармакопея Республики Беларусь
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ-
ствущее по расположению и размеру основному пят¬
ну на хроматограмме раствора сравнения (а).
Проявление В: пластинку опрыскивают раство¬
ром 50 г/л ванилина Р в смеси из кислоты серной Р,
кислоты уксусной ледяной Р и метанола Р (5:10:85,
об/об/об), нагревают при температуре от 100 °С до
105 °С до достижения максимально интенсивной
окраски пятен ((10—15) мин) и просматривают при
дневном свете.
Результаты В: основное пятно на хроматограм¬
ме испытуемого раствора соответствует по располо¬
жению, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность и щелочность. 0,20 г испытуемого
образца растворяют в воде, свободной от углерода
диоксида, Р и доводят до объема 20 мл этим же раст¬
ворителем. При прибавлении 0,2 мл раствора мети-
лового красного Р и 0,2 мл 0,01 М раствора кисло¬
ты хлористоводородной должно появиться красное
окрашивание. При прибавлении к полученному рас¬
твору 0,4 мл 0,01 М раствора натрия гидроксида
должно появиться желтое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы сравнения (с) и (д) го¬
товят непосредственно перед использованием.
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (а). 8 мг испытуемого образ¬
ца и 4 мг ФСО бетаксолола примеси А растворяют в
20,0 мл подвижной фазы.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 2 мг ФСО бетаксолола
примеси С растворяют в 50 мл подвижной фазы. 5 мл
полученного раствора доводят подвижной фазой до
объема 20 мл.
Раствор сравнения (д). 10 мг ФСО бетаксолола
для идентификации пиков (содержит примеси В, О и
Е) растворяют в 5 мл раствора сравнения (с).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
-подвижная фаза: смешивают 175мл ацето¬
нитрила Р и 175 мл метанола Р и доводят до объ¬
ема 1 л раствором 3,4 г/л калия дигидрофосфата Р,
предварительно доведенным до рН 3,0 кислотой
фосфорной Р;
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 273 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а), (Ь) и (б);
- время хроматографирования: 4,5-кратное вре¬
мя удерживания бетаксолола.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (а); идентифицируют пики примесей
В, С, О и Е, используя хроматограмму раствора срав¬
нения (б) и хроматограмму, прилагаемую к ФСО бе¬
таксолола для идентификации пиков.
Относительное удерживание (по отношению
к бетаксололу, время удерживания — около 8 мин):
примесь В — около 0,3; примесь А — около 0,8; при¬
месь О — около 1,5; примесь Е — около 2,2; примесь
С — около 4,1.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси А и бетаксолола.
Предельное содержание примесей:
- примеси А, В, С, О, Е (не более 0,3 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, В, С, О и Е, не должны
превышать 0,3 площади основного пика на хромато¬
грамме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О и Е, не должна превышать 0,1 пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
-сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (Юррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл воды Р. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием 10 мл эталон¬
ного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в сме¬
си из 10,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 50 мл 96% спирта Р. Титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 34,39 мг С18Н30С1МО3.
ХРАНЕНИЕ
В защищенном от света месте.
V
Бетаметазона валериат
265
ПРИМЕСИ
07/2016:0811
Специфицированные примеси: А, В, С, О, Е.
н3с
СНз
и энантиомер
СН3
А. (2Я5)-1-(4-Этилфенокси)-3-[(1-метилэтил)ами-
но]пропан-2-ол.
НО'
В. (2Я5)-1 -[4-(2-Гидроксиэтил)фенокси)-3-[(1 -
метилэтил)амино]пропан-2-ол.
БЕТАМЕТАЗОНА ВАЛЕРИАТ
Ве1атеИпа$оп'1 VаIе^аз
ВЕТАМЕТНА80ЫЕ УА1.ЕКАТЕ
ОН
С. (2Я5)-2-[[4-[2-(Циклопропилметокси)этил]фе-
нокси]метил]оксиран.
й. 4-[2-(Циклопропилметокси)этил]фенол.
Е. (2Я5)-1-[4-(2-Бутоксиэтил)фенокси]-3-[(1-ме-
тилэтил)амино]пропан-2-ол.
С27Н37РОв М.м. 476,6
[2152-44-5]
ОПРЕДЕЛЕНИЕ
9-Фтор-11 р,21 -дигидрокси-1 бр-метил-3,20-диоксо-
прегна-1,4-диен-17-илпентаноат.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне и метилехлориде, растворим в 96 %
спирте.
Температура плавления: около 192 °С с разложением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бетаметазона 17-валериата
#или спектр, представленный на рисунке #0811.-1#.
Рисунок #0811.-1. Инфракрасный спектр ФСО бетаметазона 17-валериата
266
Гэсударственная фармакопея Республики Беларусь
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО бетаметазона 17-валериата
растворяют по отдельности в минимальном количе¬
стве метиленхлорида Р и выпаривают на водяной
бане до сухих остатков, которые используют для по¬
лучения новых спектров.
В. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (Ь).
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От +77
до +83 (в пересчете на сухое вещество). 0,250 г испы¬
туемого образца растворяют в этаноле Р и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от света. Растворы готовят непосредственно пе¬
ред использованием.
Смесь растворителей: кислота уксусная ледя¬
ная Р— подвижная фаза (1:1000, об/об)
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 12,5 мг ФСО бетаме¬
тазона валериата для проверки пригодности хро¬
матографической системы (содержит примеси й и
О) растворяют в 5,0 мл смеси растворителей. 1,0 мл
полученного раствора используют для растворения
содержимого контейнера ФСО бетаметазона вале¬
риата смеси примесей (содержит примеси С, Н и I).
Раствор сравнения (с). 6 мг ФСО бетаметазона
(примесь А) и 3 мг ФСО бетаметазона 21-валериата
(примесь Е) растворяют в 30,0 мл смеси растворите¬
лей. 1,0 мл полученного раствора доводят смесью
растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 20 °С;
-подвижная фаза: ацетонитрилР — водаР
(50:50, об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 239 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания бетаметазона валериата.
Идентификация пиков примесей: идентифици¬
руют пики примесей С, О, 6, Н и I, используя хрома¬
тограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО бетаметазона валериата для
проверки пригодности хроматографической систе¬
мы, идентифицируют пики примесей А и Е, используя
хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению к
бетаметазона валериату, время удерживания — око¬
ло 20 мин): примесь А— около 0,3; примесь I — около
0,6; примесь С — около 0,8; примесь Н — около 1,3;
примесь й — около 1,4; примесь Е — около 1,6; при¬
месь 6 — около 2,0.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,7 между пиками при¬
месей Н и О.
Предельное содержание примесей:
- примесь А (не более 0,7 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 7-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примеси Е, в (не более 0,3 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Е и С, не должны превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примеси С, Н, I (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С, Н и I, не должны превы¬
шать 1,5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, С, Е, 6, Н и I, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 1,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
15-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50,0 мг испытуемого образца растворяют в 96 %
спирте Р и доводят до объема 100,0 мл этим же
растворителем. 2,0 мл полученного раствора дово¬
дят 96 % спиртом Р до объема 50,0 мл. Измеряют
оптическую плотность (2.2.25) полученного раствора
в максимуме при длине волны 240 нм.
Содержание С27Н37Р06 рассчитывают с учетом
удельного показателя поглощения, равного 325.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, С, Е, 6, Н, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до-
Бетаметазона дипропионат
267
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, О, Р.
А. К1 = КЗ = К5 = Кб = Н, Я2 = Р, К4 = СН3:9-Фтор-
11 (3,17,21 -тригидрокси-1 бр-метилпрегна-1,4-диен-3,20-
дион (бетаметазон).
С. Р1 = Р4 = Р6 = Н, В2 = Р, КЗ = СН3, К5 = СО-
[СН2]3-СН3: 9-Фтор-11 р,21 -дигидрокси-1 ба-метил-3,20-
диоксопрегна-1,4-диен-17-илпентаноат (дексаметазо-
на 17-валериат).
Е. К1 = КЗ = К5 = Н, К2 = Р, К4 = СН3, Кб = СО-
[СН2]3-СН3: 9-Фтор-11 р,17-дигидрокси-1 бр-метил-3,20-
диоксопрегна-1,4-диен-21-илпентаноат (бетаметазона
21-валериат).
106. К1 = Вг, К2 = Р, КЗ = Кб = Н, К4 = СН3, К5 = СО-
[СН2]3-СН3: 6а-Бром-9-фтор-11Р,21-дигидрокси-1 бр-
метил-3,20-диоксопрегна-1,4-диен-17-илпентаноат
(ба-бромбетаметазона валериат).
Н. К1 = КЗ = Кб = Н, К2 = С1, К4 = СН3, К5 = СО-
[СН2]3-СН3: 9-Хлор-11 р,21 -дигидрокси-1 бр-метил-3,20-
диоксопрегна-1,4-диен-17-илпентаноат (беклометазо-
на 17-валериат).
|. К1 = КЗ = К4 = Кб = Н, К2 = Р, К5 = СО-[СН2]3-
СН3:9-Фтор-11 р,21 -дигидрокси-3,20-диоксопрегна-1,4-
диен-17-илпентаноат (9-фторпреднизолона 17-вале-
риат).
яз
В. К1 = Р, К2 = КЗ = Н: 9-Фтор-11р,17-дигидрокси-
1 бр-метилпрегна-1,4-диен-3,20-дион (21-дезоксибета-
метазон).
О. К1 = Вг, К2 = СО-[СН2]3-СН3, КЗ = ОН: 9-Бром-
11Р,21 -дигидрокси-1 бр-метил-3,20-диоксопрегна-1,4-
диен-17-илпентаноат (9-бромбетаметазона валериат).
он
Р. 21-Гидрокси-1бр-метил-3,20-диоксопрегна-
1,4,9(11)-триен-17-илпентаноат (бетаметазона валери¬
ат 5-9(11)).
07/2016:0809
БЕТАМЕТАЗОНА ДИПРОПИОНАТ
Ве1ате111а80п1 сИргорюпаз
ВЕТАМЕТНА80ЫЕ 01РК0РЮЫАТЕ
С28Н37Р07 М-М’ 504’6
[5593-20-4]
ОПРЕДЕЛЕНИЕ
9-Фтор-11 р-гид рокси-1 бр-метил-3,20-диоксопрегна-
1,4-диен-17,21 -диилдипропаноат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне и в метиленхлориде, умеренно раство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О, Е.
A. 10,0 мг испытуемого образца растворяют в
этаноле безводном Р и доводят до объема 100,0 мл
этим же растворителем. 2,0 мл полученного раст¬
вора помещают в пробирку с притертой стеклянной
пробкой, прибавляют 10,0 мл раствора фенилгидра-
зина в кислоте серной Р, перемешивают, нагрева¬
ют в водяной бане при температуре 60 °С в течение
20 мин и немедленно охлаждают. Оптическая плот¬
ность (2.2.25) полученного раствора при длине волны
419 нм не более 0,10.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бетаметазона дипропионата
#или спектр, представленный на рисунке #0809.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 25 мг испытуемого об¬
разца растворяют в метаноле Р при слабом нагрева¬
нии и доводят до объема 5 мл этим же растворителем
(раствор А). 2 мл раствора А доводят метиленхлори-
дом Р до объема 10 мл.
Испытуемый раствор (Ь). 2 мл раствора А по¬
мещают в стеклянную пробирку вместимостью 15 мл
с притертой стеклянной пробкой или пробкой из по¬
литетрафторэтилена, прибавляют 10 мл раствора ка¬
лия гидрокарбоната метанольного насыщенного Р
и немедленно пропускают струю азота Р через раст¬
вор в течение 5 мин. Пробирку закрывают пробкой,
нагревают в водяной бане при температуре 45 °С, за¬
щищая раствор от света, в течение 2 ч и охлаждают.
268
Гэсударственная фармакопея Республики Беларусь
Рисунок #0809.-1. Инфракрасный спектр ФСО бетаметазона дипропионата
Раствор сравнения (а). 25 мг ФСО бетаметазо¬
на дипропионата растворяют в метаноле Р при сла¬
бом нагревании и доводят до объема 5 мл этим же
растворителем (раствор В). 2 мл раствора В доводят
метиленхлоридом Р до объема 10 мл.
Раствор сравнения (Ь). 2 мл раствора В помеща¬
ют в стеклянную пробирку вместимостью 15 мл с при¬
тертой стеклянной пробкой или пробкой из полите¬
трафторэтилена, прибавляют 10 мл раствора калия
гидрокарбоната метанольного насыщенного Р и
немедленно быстро пропускают струю азота Р через
раствор в течение 5 мин. Пробирку закрывают проб¬
кой, нагревают в водяной бане при температуре 45 °С,
защищая раствор от света, в течение 2 ч и охлаждают.
Пластинка: ТСХ пластинка со слоем силикаге-
ляРгмР.
Подвижная фаза: к смеси из 15 объемов эфира Р
и 77 объемов метиленхлорида Р прибавляют смесь
из 1,2 объема воды Р и 8 объемов метанола Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: основные пятна на хроматограм¬
мах испытуемых растворов соответствуют по распо¬
ложению и размеру основным пятнам на хромато¬
граммах соответствующих растворов сравнения.
Проявление В: пластинку опрыскивают раствором
кислоты серной спиртовым Р, нагревают при темпе¬
ратуре 120 °С в течение 10 мин или до проявления пя¬
тен и охлаздают. Просматривают при дневном свете и в
ультрафиолетовом свете при длине волны 365 нм.
Результаты В: основные пятна на хроматограм¬
мах испытуемых растворов соответствуют по распо¬
ложению и цвету при просматривании при дневном
свете и флуоресценции при просматривании в ультра¬
фиолетовом свете при длине волны 365 нм и размеру
основным пятнам на хроматограммах соответствую¬
щих растворов сравнения. Значения /?, основных пя¬
тен на хроматограммах испытуемого раствора (Ь) и
раствора сравнения (Ь) заметно ниже значений ос¬
новных пятен на хроматограммах испытуемого раст¬
вора (а) и раствора сравнения (а).
й. 2 мг испытуемого образца прибавляют к 2 мл
кислоты серной Р и встряхивают до полного раст¬
ворения. В течение 5 мин появляется насыщенное
красновато-коричневое окрашивание. Полученный
раствор прибавляют к 10 мл воды Р и перемешивают.
Окраска исчезает, а раствор остается прозрачным.
Е. 5 мг испытуемого образца смешивают с 45 мг
магния оксида тяжелого Р и сжигают в тигле до по¬
лучения почти белого остатка (обычно менее 5 мин).
Охлаждают, прибавляют 1 мл воды Р, 0,05 мл раст¬
вора фенолфталеина Р1 и около 1 мл кислоты хло¬
ристоводородной разведенной Р до обесцвечивания
раствора. Фильтруют. 1,0 мл полученного фильтрата
прибавляют к свежеприготовленной смеси из 0,1 мл
раствора ализарина 3 Р и 0,1 мл раствора цирко-
нила нитрата Р, перемешивают и выдерживают в
течение 5 мин. Окраску раствора сравнивают с кон¬
трольным раствором, приготовленным аналогичным
способом. Окраска испытуемого раствора желтая, а
контрольного раствора — красная.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От +84
до +88 (в пересчете на сухое вещество). 0,250 г испы¬
туемого образца растворяют в этаноле безводном Р
и доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 60,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Бетаметазона дипропионат
269
Испытуемый раствор (Ь). 1,0 мл испытуемого раст¬
вора (а) доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (а). 5 мг ФСО бетаметазо¬
на дипропионата для проверки пригодности хрома¬
тографической системы (содержит примеси В, С, Э,
Е и О) растворяют в подвижной фазе и доводят до
объема 2,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
100.0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 10,0 мл.
Раствор сравнения (с). 60,0 мг ФСО бетамета¬
зона дипропионата растворяют в подвижной фазе и
доводят до объема 25,0 мл этим же растворителем.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (д). 5 мг ФСО бетаметазо¬
на дипропионата для идентификации пиков (содер¬
жит примесь Н) растворяют в подвижной фазе и дово¬
дят до объема 2,0 мл этим же растворителем.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,10 м
и внутренним диаметром 2,0 мм, заполненная силика¬
гелем октадецилсилильным для хроматографии Р с
размером частиц 2,5 мкм;
- температура: (20±2) °С;
- подвижная фаза: смешивают 35 мл воды Р и
56 мл ацетонитрила Р, выдерживают до установле¬
ния равновесия, доводят водой Р до объема 100 мл и
перемешивают;
- скорость подвижной фазы: 0,2 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 5 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с!);
- время хроматографирования: 3-кратное вре¬
мя удерживания бетаметазона дипропионата.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, О, Е и С, используя хромато¬
грамму раствора сравнения (а) и хроматограмму, при¬
лагаемую к ФСО бетаметазона дипропионата для
проверки пригодности хроматографической систе¬
мы, идентифицируют пик примеси Н, используя хро¬
матограмму раствора сравнения (д) и хроматограмму,
прилагаемую к ФСО бетаметазона дипропионата
для идентификации пиков.
Относительное удерживание (по отношению к
бетаметазона дипропионату, время удерживания —
около 10 мин): примесь В — около 0,4; примесь С —
около 0,5; примесь О — около 0,7; примесь Е — около
1,2; примесь Н — около 1,7; примесь (В — около 2,1.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 4,0
(Нр — высота пика примеси Е относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси Е и пик
бетаметазона дипропионата).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 1,3; для примеси Н — 1,4):
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси С, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В и Н (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям В и Н, не должны превы¬
шать 3-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- примеси О, Е и О (не более 0,2 %): на хрома¬
тограмме испытуемого раствора (а) площади пиков,
соответствующих примесям О, Е и О, не должны пре¬
вышать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков приме¬
сей В, С, О, Е, О и Н, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С28Н37Р07 в процентах рассчитыва¬
ют с учетом содержания бетаметазона дипропионата
в ФСО бетаметазона дипропионата.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С, О, Е, 6, Н.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет необ¬
ходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, Р.
А. 9-Фтор-11 р, 17,21 -тригидрокси-16р-метилпрегна-
1,4-диен-3,20-дион (бетаметазон).
270
Гэсударственная фармакопея Республики Беларусь
он
В. 9-Фтор-11р,21-дигидрокси-16р-метил-3,20-
диоксопрегна-1,4-диен-17-илпропаноат (бетаметазона
17-пропионат).
С. 9-Фтор-11 р, 17-дигидрокси-1 бр-метил-3,20-диок-
сопрегна-1,4-диен-21-илпропаноат (бетаметазона
21-пропионат).
О. 21 -(Ацетокси)-9-фтор-11 р-гидрокси-1 бр-метил-
3,20-диоксопрегна-1,4-диен-17-илпропаноат (бетаме¬
тазона 21-ацетат 17-пропионат).
Е. 9-Хлор-11 Р-гидрокси-16р-метил-3,20-диоксо-
прегна-1,4-диен-17,21-диилдипропаноат (бекломета-
зона дипропионат).
Р. 9,11 р-Эпокси-16р-метил-3,20-диоксо-9Р-прегна-
1,4-диен-17,21-диилдипропаноат (9р,11 р-эпоксибета-
метазона дипропионат).
6. 9-Фтор-1 бр-метил-3,20-диоксопрета-1,4-диен-
11р,17,21-триилтрипропаноат (бетаметазона трипро-
пионат).
Н. 6а-Бром-9-фтор-11 р-гидрокси-1 бр-метил-3,20-
диоксопрегна-1,4-диен-17,21-диилдипропаноат
(ба-бромбетаметазона дипропионат).
07/2016:0595
БИСАКОДИЛ
В1засос1у1ит
В18АСООП
СИН19Ы04 М.м. 361,4
1603-50-91
ОПРЕДЕЛЕНИЕ
4,4'-(Пиридин-2-илметилен)дифенилдиацетат.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, растворим в
ацетоне, умеренно растворим в 96 % спирте. Раство¬
ряется в разведенных минеральных кислотах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 131 °С до
135 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Бисакодил
271
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в растворе 6 г/л калия гидрокси¬
да Р в метаноле Р и доводят до объема 100,0 мл
этим же растворителем. 10,0 мл полученного раст¬
вора доводят раствором 6 г/л калия гидроксида Р в
метаноле Р до объема 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 248 нм.
Плечо: при 290 нм.
Удельный показатель поглощения в максимуме:
от 632 до 672.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бисакодила #или спектр, пред¬
ставленный на рисунке #0595.-1#.
Если полученные спектры отличаются, то испытуе¬
мый образец и ФСО бисакодила растворяют по отдель¬
ности в хлороформе Р и выпаривают до сухих остатков,
которые используют для получения новых спектров.
Р. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО бисакодила раст¬
воряют в ацетоне Р и доводят до объема 10 мл этим
же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ вР254 Р-
Подвижная фаза: метилэтилкетон Р — кси¬
лол Р (50:50, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе, при необходимости
нагревают при температуре от 100 °С до 105 °С.
Проявление: пластинку опрыскивают смесью из
равных объемов 0,05 М раствора йода и кислоты
серной разведенной Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Кислотность или щелочность. К 1,0 г испыту¬
емого образца прибавляют 20 мл воды, свободной
от углерода диоксида, Р, встряхивают, нагревают
до кипения, охлаждают и фильтруют. Прибавляют
0,2 мл 0,01 М раствора натрия гидроксида и 0,1 мл
раствора метилового красного Р. Появляется жел¬
тое окрашивание. При прибавлении не более 0,4 мл
0,01 М раствора кислоты хлористоводородной
должно появиться красное окрашивание.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Смесь растворителей: кислота уксусная ледя¬
ная Р— ацетонитрил Р— вода Р (4:30:66, об/об/об).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 25 мл ацетонитрила Р и доводят
смесью растворителей до объема 50,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 2,0 мг ФСО бисакодила
для проверки пригодности хроматографической си¬
стемы (содержит примеси А, В, С, О и Е) растворяют
в 1,0 мл ацетонитрила Р и доводят смесью раство¬
рителей до объема 2,0 мл.
Раствор сравнения (с). 5,0 мг ФСО бисакоди¬
ла для идентификации пиков (содержит примесь Р)
растворяют в 2,5 мл ацетонитрила Р и доводят сме¬
сью растворителей до объема 5,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
Рисунок #0595.-1. Инфракрасный спектр ФСО бисакодила
272
Гэсударственная фармакопея Республики Беларусь
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: смесь из 45 объемов ацето¬
нитрила Р и 55 объемов раствора 1,58 г/л аммония
формиата Р, предварительно доведенного до рН 5,0
кислотой муравьиной безводной Р;
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 265 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания бисакодила.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, О и Е, используя хрома¬
тограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО бисакодила для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению
к бисакодилу, время удерживания— около 13 мин):
примесь А — около 0,2; примесь В — около 0,4; при¬
месь С — около 0,45; примесь О — около 0,8; примесь
Е — около 0,9; примесь Р — около 2,6.
Пригодность хроматографической системы.
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси Е относительно базовой
линии; Н}/ — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Е и
бисакодила).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 0,7):
- примеси А, В (не более 0,1 %): на хромато¬
грамме улсгтыт^емоге раствора
ветствующих примесям А и В, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- примеси С, Е (не более 0,5 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С и Е, не должны превышать
5-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примесь О (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь Е (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Р, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е и Р, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 0,500 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 36,14 мг С22Н19М04.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
A. Р1 = РЗ = ОН, Р2 = Н: 4,4'-(Пиридин-2-илметилен)-
дифенол.
B. Р1 = Н, Р2 = РЗ = ОН: 2-[(Р$)-(4-Гидроксифенил)-
(пиридин-2-ил)метил]фенол.
C. Р1 = ОН, Р2 = Н, РЗ = 0-СО-СН3:4-[(Р5)-(4-Гидро-
ксифенил)(пиридин-2-ил)метил]фенилацетат.
Е. Р1 = Н, Р2 = РЗ = 0-СО-СН3:2-[(Я5)-[4-(Ацетил-
окси)фенил](пиридин-2-ил)метил]фенилацетат.
O. Структура не установлена.
P. Структура не установлена.
07/2016:1710
БИСОПРОЛОЛА ФУМАРАТ
В18орго1оН Титагаз
В180РК01-01- РУ МАРАТЕ
(с18н31мо4)2 с4н4о4
[104344-23-2]
но2с
/
со2н
М.м. 767
ОПРЕДЕЛЕНИЕ
(2Р5)-1 -[4-[[2-(1 -Метилэтокси)этокси]метил]фенок-
си]-3-[(1 -метилэтил)амино]пропан-2-ола фумарат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
Бисопролола фумарат
273
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Слегка гигро¬
скопичен.
Очень легко растворим в воде, легко растворим
в метаноле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бисопролола фумарата #или
спектр, представленный на рисунке #1710.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО бисопролола фумарата раст¬
воряют по отдельности в метаноле Р, выпаривают до
сухих остатков, которые сушат при температуре 60 °С
и давлении, не превышающем 0,7 кПа, и используют
для получения новых спектров.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р1 —
вода для хроматографии Р (20:80, об/об).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 2,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО бисопролола для идентификации пиков (содер¬
жит примеси А и Е) растворяют в 1,0 мл смеси раст¬
ворителей.
Раствор сравнения (с). Содержимое контейнера
ФСО бисопролола для проверки пригодности хрома¬
тографической системы (содержит примесь 6) раст¬
воряют в 1,0 мл смеси растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- температура: (20±2) °С;
- подвижная фаза:
-подвижная фаза А: раствор 10 г/л кислоты
фосфорной Р\
-подвижная фаза В: раствор 10 г/л кислоты
фосфорной Р в ацетонитриле Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-^
95
5
4—8
95 — 80
5 — 20
8—15
80
20
15—34
80 — 20
20 — 80
34—36
20
80
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и Е, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО бисопролола для идентификации пиков;
идентифицируют пик примеси (В, используя хромато¬
грамму раствора сравнения (с) и хроматограмму, при¬
лагаемую к ФСО бисопролола для проверки пригод¬
ности хроматографической системы.
Относительное удерживание (по отношению к
бисопрололу, время удерживания — около 18 мин):
примесь А — около 0,5; примесь С — около 1,1; при¬
месь Е — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (с):
Волновое число (см1)
Рисунок #1710.-1. Инфракрасный спектр ФСО бисопролола фумарата
274
Государственная фармакопея Республики Беларусь
- коэффициент разделения пиков: не менее 2,5
(Нр — высота пика примеси 6 относительно базовой
линии; — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси <3 и
бисопролола).
Предельное содержание примесей:
- примесь 6 (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси 6, не должна превышать 2,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь А (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь Е (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси Е, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, Е и 6, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а); не учитывают
пик фумаровой кислоты.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,000 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 38,35 мг (С18Н31М04)2 С4Н404.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, Е, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Р, К, Ь,
/V, О, Я 5, Т, И.
и энантиомер
А. (2Я5)-1 -(4-Гидроксиметилфенокси)-3-изопро-
пиламинопропан-2-ол.
В. (2/?5)-1-Изопропиламино-3-[4-(2-пропокси-
этоксиметил)фенокси]пропан-2-ол.
С. 1-[4-[4-(2-Гидрокси-3-изопропиламинопропокси)-
бензил]фенокси]-3-изопропиламинопропан-2-ол.
н он
0.1 -[4-[4-(2-Гидрокси-3-изопропиламинопропокси)-
бензилоксилметил]фенокси]-3-изопропиламинопропан-
2-ол.
Е. (Е2)-[3-[4-(2-Изопропоксиэтоксиметил)фенокси]-
аллил]изопропиламин.
Р. (2/?5)-2-[4-(2-Изопропоксиэтоксиметил)фенокси]-
З-изопропиламинопропан-2-ол.
О. (2Я5)-1 -[4-[[(2-Изопропоксиэтокси)метокси]ме-
тил]фенокси]-3-изопропиламинопропан-2-ол.
Бифоназол
275
БИФОНАЗОЛ
ВПопаго/ит
В1Р0ЫА201.Е
07/2016:1395
К. 2-Изопропоксиэтил-4-[[(2#$)-2-Гидрокси-3-(изо-
пропиламино)пропил]окси]бензоат.
Ь. 4-[[(2Я5)-2-Гидрокси-3-(изопропиламино)про-
пил]окси]бензальдегид.
N. (2Я5)-1 -[4-[(2-Этоксиэтокси)метил]фенокси]-3-
изопропиламинопропан-2-ол.
О. (2/?5)-1 -(Изопропиламино)-3-[4-(2-метокси-
этокси)метил]феноксипропан-2-ол.
С22Н18Ы2 М.м. 310,4
[60628-96-8]
ОПРЕДЕЛЕНИЕ
1 -[(Я5)-(Бифенил-4-ил)фенилметил]-1 Н-имидазол.
Содержание: не менее 98,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, умеренно рас¬
творим в этаноле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бифоназола #или спектр, пред¬
ставленный на рисунке #1395.-1
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО бифоназола растворяют по от¬
дельности в минимальном количестве 2-пропанола Р
и выпаривают до сухих остатков, которые используют
для получения новых спектров.
ИСПЫТАНИЯ
Я. (2Я5)-1 -(Изопропиламино)-3-(4-метилфенокси)-
пропан-2-ол.
3. 4-Гидроксибензальдегид.
Т. 4-[(3-Изопропил-2-оксо-1,3-оксазолидин-5-ил)-
метокси]бензальдегид.
II. 5-[[4-(Гидроксиметил)фенокси]метил]-3-изо-
пропил-1,3-оксазолидин-2-он.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор рН 3,2. 2,0 мл кислоты фос¬
форной Р смешивают с 980 мл воды Р, доводят до рН
3,2 (2.2.3) триэтиламином Р и разводят водой Р до
объема 1000,0 мл.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 25 мл ацетонитрила Р и доводят
буферным раствором рН 3,2 до объема 50,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят буферным раствором рН 3,2 до объема
100.0 мл. 1,0 мл полученного раствора доводят бу¬
ферным раствором рН 3,2 до объема 10,0 мл.
Раствор сравнения (Ь). 2 мг ФСО бифоназола
для проверки пригодности хроматографической си¬
стемы (содержит примеси А, В, С, О и Е) растворяют
в 2 мл ацетонитрила Р и доводят буферным раство¬
ром рН 3,2 до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаметром
4.0 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 — бу¬
ферный раствор рН 3,2 (20:80, об/об);
276
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза В: буферный раствор
рН 3,2 — ацетонитрил Р1 (20:80, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—8
60
40
8—12
О)
0
1
о
0
1
СО
о
12—30
10
90
- скорость подвижной фазы. 1 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 50 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, О и Е, используя хрома¬
тограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО бифоназола для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению к
бифоназолу, время удерживания — около 4 мин): при¬
месь С — около 0,2; примесь А — около 3,2; примесь
О — около 3,6; примесь Е — около 5,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками при¬
меси В и бифоназола.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 2):
- примеси В, О (не более 0,5 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и О, не должны превышать
5-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примеси А, С (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, С, не должны превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примесь Е (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси Е, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, О и Е, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 80 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 31,04 мг С22Н18М2.
Бромгексина гидрохлорид
277
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
и энантиомер
А. (#5)-(Бифенил)-4“ил)Фенилметанол-
и энантиомер
В. 4-[(Я5)-(Бифенил-4-ил)фенилметил]-1 /-/-ими¬
дазол.
О. 1,3-Бис[(бифенил-4-ил)фенилметил]-1 Н-ими-
дазолия.
Е. 1,4-Бис[(бифенил-4-ил)фенилметил]-1 Н- ими¬
дазол.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,1 г испытуемого образца растворяют при
осторожном нагревании в 5 мл метанола Р, прибав¬
ляют 0,1 мл кислоты серной Р и поджигают получен¬
ный раствор. Пламя имеет зеленую кайму.
B. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», имеет кислую реакцию (2.2.4).
ИСПЫТАНИЯ
Раствор 5. 3,3 г испытуемого образца раство¬
ряют в 80 мл кипящей воды дистиллированной Р,
охлаждают и доводят водой, свободной от углерода
диоксида, Р, приготовленной из воды дистиллирован¬
ной Р, до объема 100 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 3,8 до 4,8. Измеряют рН раствора 3.
Растворимость в 96 % спирте. 1,0 г испыту¬
емого образца растворяют в 10 мл кипящего 96%
спирта Р. Полученный раствор по степени мутности
не должен превышать эталон II (2.2.1) и должен быть
бесцветным (2.2.2, метод II).
Органические вещества. Испытуемый образец
не должен темнеть при постепенном нагревании до
тусклого красного каления.
Сульфаты (2.4.13). Не более 0,0450 % (450 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0015 % (15 ррт). 12 мл раствора 3 должны выдержи¬
вать испытание на тяжелые металлы. Эталонный раст¬
вор готовят с использованием смеси из 2,5 мл эталон¬
ного раствора свинца (2 ррт РЬ) Р и 7,5 мл воды Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют при на¬
гревании в 100 мл водыР, содержащей 15 г манни¬
та Р, и титруют 1 М раствором натрия гидроксида до
появления розового окрашивания, используя в каче¬
стве индикатора 0,5 мл раствора фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 61,8 мг Н3В03.
07/2016:0001
07/2016:0706
БОРНАЯ КИСЛОТА
АсШит Ьопсит
вошсасю
Н3ВО3 М.м. 61,8
[10043-35-3]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок, бесцветные блестящие жирные на ощупь пла¬
стинки или белые или почти белые кристаллы.
Растворима в воде и в 96 % спирте, легко раство¬
рима в кипящей воде и в 85 % глицерине.
БРОМГЕКСИНА ГИДРОХЛОРИД
ВготЬехт/ Нус1госЫопдит
ВКОМНЕХ1ЫЕ НУОКОСШ-ОКЮЕ
С14Н20Вг2Ы2 • НС1 М.м. 412,6
[611-75-6]
278
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
А/-(2-Амино-3,5-дибромбензил)-Л/-метилцикло-
гексанамина гидрохлорид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень мало растворим в воде, мало растворим в
96 % спирте и в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО бромгексина гидрохлорида
#или спектр, представленный на рисунке #0706.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО бромгексина гидрохлорида
растворяют по отдельности в метаноле Р и выпари¬
вают до сухих остатков, которые используют для по¬
лучения новых спектров.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО бромгексина ги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯР254 Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (17:17:66, об/об/об).
Наносимый объем пробы: 20 мкп.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
С. 25 мг испытуемого образца растворяют в сме¬
си из 1 мл кислоты серной разведенной Р и 50 мл
воды Р, прибавляют 2 мл метиленхлорида Р и 5 мл
раствора хлорамина Р и встряхивают. Нижний слой
окрашивается в коричневато-желтый цвет.
0. 1 мг испытуемого образца растворяют в 3 мл
0,1 М раствора хлористоводородной кислоты. По¬
лученный раствор дает реакцию на первичные арома¬
тические амины (2.3.1).
Е. 20 мг испытуемого образца растворяют в 1 мл
метанола Р и прибавляют 1 мл воды Р. Полученный
раствор дает реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,6 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор. 1,26 г аммония формиата Р
растворяют в 850 мл воды Р, доводят муравьиной
кислотой безводной Р до рН 4,4 и разводят водой Р
до объема 1000,0 мл.
Смесь растворителей. Ацетонитрил Р —
вода Р (50:50, об/об).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). 10 мг ФСО бромгексина
для проверки пригодности хроматографической си-
Рисунок #0706.-1. Инфракрасный спектр ФСО бромгексина гидрохлорида
Бутилгидроксианизол
279
стемы (содержит примеси С и О) растворяют в сме¬
си растворителей и доводят до объема 2,0 мл этой же
смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 2,1 мм, заполненная силикагелем октадецилси-
лильным поверхностно-пористым эндкепированным
для хроматографии Р с размером частиц 2,6 мкм;
- температура: 30 °С;
- подвижная фаза: ацетонитрил Р — буферный
раствор (40:60, об/об);
- скорость подвижной фазы: 0,2 мл/мин;
-спектрофотометрический детектор, длина
волны 248 нм;
- объем вводимой пробы. 3,0 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания бромгексина.
Идентификация пиков примесей: идентифици¬
руют пики примесей С и О, используя хроматограмму
раствора сравнения (а) и хроматограмму, прилагае¬
мую к ФСО бромгексина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
бромгексину, время удерживания— около 10 мин):
примесь С — около 0,2; примесь О — около 0,3.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси С и Ь.
Расчет процентных содержаний:
-поправочный коэффициент: умножают пло¬
щадь пика примеси С на 1,6;
- для каждой примеси — используют концентра¬
цию бромгексина в растворе сравнения (Ь).
Предельное содержание примесей:
- примесь С: не более 0,15 %;
- неспецифицированные примеси: для каждой
примеси не более 0,10 %;
- сумма примесей: не более 0,2 %;
- учитываемый предел: 0,05 %.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 70 мл
96 % спирта Р, прибавляют 1 мл 0,1 М раствора кис¬
лоты хлористоводородной и титруют 0,1 М раство¬
ром натрия гидроксида потенциометрически (2.2.20).
Отмечают объем титранта между двумя точками пе¬
региба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 41,26 мг С^Н^Вг^-НС!.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимити¬
руется общим критерием при емлемости для других/
неспецифицированных примесей и/или общей статьей
Субстанции для фармацевтического использования
(2034). Вследствие этого нет необходимости иденти¬
фицировать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Контроль
примесей в субстанциях для фармацевтического ис¬
пользования): А, В, Д Е.
А. 2-Амино-3,5-дибромфенил)метанол.
сно
С. 2-[[Циклогексил(метил)амино]метил]анилин.
0.4-Бром-2-[[циклогексил(метил)амино]метил]ани-
лин.
и энантиомер
Е. (ЗР5)-6,8-Дибром-3-циклогексил-3-метил-1,2,3,4-
тетрагидрохиназолин-3-ий.
07/2016:0880
БУТИЛГИДРОКСИАНИЗОЛ
ВШу111удгохуап18о1ит
В11ТУ1-НУ01ЮХУАЫ1801-Е
С„Н1вОг М.м. 180,3
[25013-16-5]
280
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Бутилгидроксианизол представляет собой 2-(1,1 -
диметилэтил)-4-метоксифенол, содержащий не более
10 % 3-(1,1-диметилэтил)-4-метоксифенола.
ОПИСАНИЕ (СВОЙСТВА)
Белый, желтоватый или слегка розоватый кри¬
сталлический порошок.
Практически нерастворим в воде, очень легко
растворим в метиленхлориде, легко растворим в 96 %
спирте и жирных маслах. Растворяется в разведенных
растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, со¬
ответствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а).
B. К 0,5 мл раствора 8, приготовленного как
указано в разделе «Испытания», прибавляют 10 мл
раствора аминопиразолона Р и 1 мл раствора ка¬
лия феррицианида Р. Перемешивают и прибавляют
10 мл метиленхлорида Р. Энергично встряхивают.
После разделения органический слой окрашивает¬
ся в красный цвет.
C. 10 мг испытуемого образца растворяют в
2 мл 96 % спирта Р. Прибавляют 1 мл раствора
1 г/л тестостерона пропионата Р в 96 % спирте
Р и 2 мл раствора натрия гидроксида разведенно¬
го Р. Нагревают в водяной бане при температуре
80 °С в течение 10 мин и охлаждают. Появляется
красное окрашивание.
ИСПЫТАНИЯ
Раствор 8. 2,5 г испытуемого образца раство¬
ряют в 96% спирте Р и доводят до объема 25 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 8
должна быть не интенсивнее, чем пятый по интенсив¬
ности эталон наиболее подходящего цвета.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,25 г испытуемого
образца растворяют в метиленхлориде Р и доводят
до объема 10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого раст¬
вора (а) доводят метиленхлоридом Р до объема 10 мл.
Раствор сравнения (а). 25 мг ФСО бутилгидрок-
сианизола растворяют в метиленхлориде Р и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 1 мл раствора сравнения
(а) доводят метиленхлоридом Р до объема 20 мл.
Раствор сравнения (с). 50 мг гидрохинона Р раст¬
воряют в 5 мл 96 % спирта Р и доводят до объема
100 мл метиленхлоридом Р. 1 мл полученного раст¬
вора доводят метиленхлоридом Р до объемаЮ мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: метиленхлорид Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают свеже¬
приготовленной смесью из 10 объемов раствора
калия феррицианида Р, 20 объемов раствора же¬
леза (III) хлорида Р1 и 70 объемов воды Р и про¬
сматривают при дневном свете.
Предельное содержание примесей:
- 3-(1,1-диметилэтил)-4-метоксифенол (не
более 10 %): на хроматограмме испытуемого раст¬
вора (а) пятно фиолетово-синего цвета со значе¬
нием Рг около 0,35 должно быть не интенсивнее
основного пятна на хроматограмме раствора срав¬
нения (а);
- гидрохинон (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) пятно, соответ¬
ствующее гидрохинону, должно быть не интенсив¬
нее пятна основного пятна на хроматограмме раст¬
вора сравнения (с);
- любая другая примесь (не более 0,5 %): на
хроматограмме испытуемого раствора любое пят¬
но, кроме основного и пятен 3-(1,1-диметилэтил)-
4-метоксифенола и гидрохинона, должно быть не
интенсивнее основного пятна на хроматограмме
раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод С). Не болем
0,0010 % (10 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 1 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12,
2.6.13). Испытуемый образец должен выдерживать
требования статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. 1,4-Дигидроксибензол (гидрохинон).
07/2016:РБ0023
#ВАЗЕЛИН
УазеИпит
РАРАРНЫ, 80РТ
ОПРЕДЕЛЕНИЕ
Вазелин представляет собой очищенную смесь
твердых и жидких углеводородов, получаемых в ре¬
зультате переработки нефти.
ОПИСАНИЕ (СВОЙСТВА)
Белая или желтая тянущаяся нитями мазеобраз¬
ная масса. Расплавленная субстанция при дневном
свете слегка флуоресцирует.
Практически нерастворим в воде и в 96 % спир¬
те, мало растворим в эфире, умеренно растворим в
хлороформе.
Валсартан
281
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура каплепадения (2.2.17): от 37 °С
до 50 °С.
B. 2 г испытуемого образца расплавляют и после
получения гомогенной фазы прибавляют 2 мл воды Р
и 0,2 мл 0,05 М раствора йода. Встряхивают и охлаж¬
дают. В твердом верхнем слое появляется фиолето¬
во-розовое или коричневое окрашивание.
ИСПЫТАНИЯ
Кислотность или щелочность. К 5 г расплав¬
ленного испытуемого образца прибавляют 20 мл ки¬
пящей воды Р и интенсивно встряхивают в течение
1 мин. Выдерживают до охлаждения и разделения
фаз. К 10 мл водного слоя прибавляют 0,1 мл раст¬
вора фенолфталеина Р. Раствор должен быть бес¬
цветным. При прибавлении не более 0,05 мл 0,1 М
раствора натрия гидроксида должно появиться
красное окрашивание.
Вязкость (2.2.9). Не менее 1,6-10-5 м2/с при тем¬
пературе 60 °С.
Кислотное число (2.5.1). Не более 0,1.5,00 г ис¬
пытуемого образца растворяют, при необходимости
подогревая с обратным холодильником, в смеси из
равных объемов 96 % спирта Р и эфира Р, предва¬
рительно нейтрализованной 0,1 М раствором калия
гидроксида, используя в качестве индикатора 0,5 мл
раствора фенолфталеина Р.
Органические примеси. К 3 г испытуемого об¬
разца прибавляют 6 мл кислоты серной Р, тщатель¬
но перемешивают в фарфоровой ступке и выдержи¬
вают в течение 30 мин. Не должно появляться черное
окрашивание. Допускается появление коричневого
окрашивания.
Жиры и смолы. К 3 г испытуемого образца при¬
бавляют 10 мл раствора 100 г/л натрия гидроксида Р
и кипятят в течение 2 мин при встряхивании. Выдер¬
живают до охлаждения и разделения слоев. Водный
слой подкисляют кислотой хлористоводородной Р.
Не должен образовываться осадок и не должна появ¬
ляться муть.
Восстанавливающие вещества. Испытуемый
образец должен выдерживать испытание на восста¬
навливающие вещества, если субстанцию используют
для приготовления глазных мазей. 1,0 г испытуемого
образца смешивают с 5 мл воды очищенной Р, 2 мл
кислоты хлористоводородной разведенной Р, 0,1 мл
0,02 М раствора калия перманганата и нагревают на
водяной бане в течение 5 мин. Должна сохраняться
розовая окраска водного слоя.
Сульфиды. Смесь из 3 г испытуемого образца,
2 капель раствора свинца (II) ацетата основного Р и
96 % спирта Р нагревают в водяной бане при темпе¬
ратуре 70 °С в течение 10 мин. Не должно появляться
потемнение.
Сульфатная зола (2.4.14, метод А). Не более
0,05 %. Определение проводят из 2,0 г испытуемого
образца
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре от 8 °С до
15 °С.
07/2016:2423
ВАЛСАРТАН
Уа1заг1апит
М4*.$АЯГАА/
СмН»М,Оз М.м. 435,5
[137862-53-4]
ОПРЕДЕЛЕНИЕ
(25)-3-Метил-2-[пентаноил[[2'-(1Н-тетразол-5-ил)
бифенил-4-ил]метил]амино]бутановая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Практически нерастворим в воде, легко растворим
в этаноле, умеренно растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Проводят определение подлинности А, В или А, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО валсартана #или спектр, пред¬
ставленный на рисунке #2423.-1#.
B. Испытуемый образец выдерживает испытание
«Энантиомерная чистота», как указано в разделе «Ис¬
пытания».
C. Удельное оптическое вращение (2.2.7). От -64,0
до -69,0 (в пересчете на безводное вещество). 0,200 г
испытуемого образца растворяют в метаноле Р и до¬
водят до объема 20,0 мл этим же растворителем.
ИСПЫТАНИЯ
Энантиомерная чистота. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО валсарта¬
на для идентификации пиков (содержит примесь А)
растворяют в подвижной фазе и доводят до объема
5,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем ОО для хи¬
ральных разделений Р\
- подвижная фаза: кислота трифторуксус-
ная Р — 2-пропанол Р — гексан Р(0,1:15:85, об/об/об);
- скорость подвижной фазы: 0,8 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
ш
282 Гэсударственная фармакопея Республики Беларусь
3000 2000 1500 1000 500
Волновое число (см'1)
Рисунок #2423.-1. Инфракрасный спектр ФСО валсартана
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания валсартана.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (а) и хроматограмму, прилагаемую к
ФСО валсартана для идентификации пиков.
Относительное удерживание (по отношению
к валсартану, время удерживания— около 13 мин):
примесь А — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси А и валсартана.
Предельное содержание примесей:
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО валсартана для проверки пригодности хро¬
матографической системы (содержит примесь С)
растворяют в 1,0 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 3,0 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: кислота уксусная ледяная Р —
ацетонитрил Р1 — вода Р (1:500:500, об/об/об);
- скорость подвижной фазы: 0,4 мл/мин;
- спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 6-кратное вре¬
мя удерживания валсартана.
Идентификация пиков примесей: идентифици¬
руют пик примеси С используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО валсартана для проверки пригодности хрома¬
тографической системы.
Относительное удерживание (по отношению к
валсартану, время удерживания — около 5 мин): при¬
месь С — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками при¬
меси С и валсартана.
Предельное содержание примесей:
- примесь С (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси С, не должна превышать 2-кратную пло¬
щадь основного пика на хроматограмме раствора срав¬
нения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си С, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Валин
283
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт).
1,0 г испытуемого образца растворяют в смеси из
воды Р и ацетона Р (15:85, об/об) и доводят до объ¬
ема 20 мл этой же смесью растворителей. 12 мл полу¬
ченного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
10 мл эталонного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 2.0 %. Определение про¬
водят из 0,500 г испытуемого образца.
Гу »епия зама (2.4.14). Не более 0,1%.
гъ^чмципмчя проводят из 1,0 г испытуемого образца.
1Чмрнби11И11111 ни IIII чистота (2.6.12, 2.6.13).
«Чсгъггуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,170 г испытуемого образца растворяют в 70 мл
2-пропанопа Р и титруют 0,1М раствором тетрабути-
лашюния гидроксида в 2-пропаноле потенциометриче-
ао« {2.2.20). Все действия проводят в атмосфере азота.
1 мл 0,1 М раствора тетрабутиламмония
гидроксида в 2-пропаноле соответствует 21,78 мг
с24н29м5о3.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В.
А. К1 = Н, В2 = С02Н, КЗ = СН3: (2/?)-3-Метил-2-
[пентаноил[[2,-(1Н-тетразол-5-ил)бифенил-4-ил]метил]-
амино]бутановая кислота.
С. В1 = С02Н, В2 = КЗ = Н: (25)-2-[Бутаноил[[2’-
(1Н-тетразол-5-ил)бифенил-4-ил]метил]амино]-3-
метилбутановая кислота.
В. Бензил-(25)-3-метил-2-[пентаноил[[2'-(1 Н-
тетразол-5-ил)бифенил-4-ил]метил]амино]бутаноат.
07/2016:0796
ВАЛИН
ЧаНпит
ЧАИНЕ
СвН11Ы02 М.м. 117,1
[72-18-4]
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-3-метилбутановая кислота.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Растворим в воде, очень мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО валина #или спектр, представ¬
ленный на рисунке #0796.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 100 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)в.
Удельное оптическое вращение (2.2.7). От
+26,5 до +29,0 (в пересчете на сухое вещество). 2,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 25,0 мл этим
же растворителем.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов.#
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен-
36*. Зак. 1060.
284
Гэсударственная фармакопея Республики Беларусь
Рисунок #0796.-1. Инфракрасный спектр ФСО валина
Пригодность хроматографической системы.
раствор сравнения (й):
- разрешение: не менее 1,5 между пиками при¬
месей В и С.
Расчет процентных содержаний:
-для примеси В — используют концентрацию
примеси В в растворе сравнения (е);
-для любого нингидрин-положительного веще¬
ства, детектируемого при 570 нм — используют кон¬
центрацию валина в растворе сравнения (а);
-для любого нингидрин-положительного веще¬
ства, детектируемого при 440 нм — используют кон¬
центрацию пролина в растворе сравнения (Ь); если
пик выше учитываемого предела при обеих длинах
волн, для расчетов используют результат, полученный
при 570 нм.
Предельное содержание примесей:
- примесь В при 570 нм: не более 0,4 % для каж¬
дой примеси;
- любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 1,0 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в кислоте хлористоводородной
разведенной Р и доводят до объема 10 мл этим же
растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО валина раство¬
ряют в 0,1 М растворе кислоты хлористоводородной
и доводят до объема 50 мл этим же растворителем.
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная разве¬
денная Р1 или буферный раствор для приготовления об¬
разцов, подходящий для используемого оборудования.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят раствором А до объ¬
ема 10,0 мл.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (ф. 30 мг изолейцина Р (при¬
месь В) и 30 мг лейцина Р (примесь С) растворяют
в растворе А и доводят до объема 50,0 мл этим же
растворителем. 1,0 мл полученного раствора доводят
раствором А до объема 200,0 мл.
Раствор сравнения (е). 30,0 мг изолейцина Р
(примесь В) растворяют в растворе А и доводят до
объема 100,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
250.0 мл.
Холостой раствор. Раствор А.
В аминокислотный анализатор вводят подходя¬
щие равные объемы испытуемого раствора, холосто¬
го раствора и растворов сравнения. Запускают подхо¬
дящую программу для определения физиологических
аминокислот.
Ванилин
285
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО валина и 10 мг
ФСО фенилаланина растворяют в 0,1 М растворе
ямиюты хлористоводородной и доводят до объема
25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкп.
Фронт подвижной фазы, не менее 15 см от ли-
тт. стяс^э.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром ттгидрина Р и высушивают при температуре от
100 вС до 105 X в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
10 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
0,5 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 15 мл этим же
растворителем. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
- вводимая проба: испытуемый раствор, раствор
сравнения (с), холостой раствор.
Предельное содержание:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато¬
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 50 мг испытуемого
образца должны выдерживать испытание на аммония
соли. Эталон готовят с использованием 0,1 мл эта¬
лонного раствора аммония (100 ррт N14^ Р*
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010% (10 ррт).
Растворитель: вода Р.
0,25 г испытуемого образца должны выдержи¬
вать испытание на тяжелые металлы. Эталон гото¬
вят с использованием 0,25 мл эталонного раствора
свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 X.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в Змл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 11,71 мгС^Н^О^
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): А, С.
н 1ЧН2
НзС^^СОгН
А. (2Э)-2-Аминопропановая кислота (аланин).
Н СН3
В. (25,35)-2-Амино-3-метилпентановая кислота
(изолейцин).
сн-
’з Н
,МН2
н3с со2н
С. (25)-2-Амино-3-метилпентановая кислота (лей¬
цин).
07/2016:0747
ВАНИЛИН
УапННпит
УАШШЫ
С8н803 М.м. 152,1
[121-33-5]
37 Зак. 1060.
286
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
4-Гидрокси-З-метоксибензальдегид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический порошок или игольчатые кри¬
сталлы белого или белого с желтоватым оттенком
цвета.
Мало растворим в воде, легко растворим в 96 %
спирте и в метаноле. Растворяется в разведенных
растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14). От 81 °С до
84 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО ванилина.
C. Хроматограммы, полученные в испытании
«Сопутствующие примеси», после опрыскивания про¬
сматривают при дневном свете.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
й. К 5 мл насыщенного раствора испытуемого об¬
разца прибавляют 0,2 мл раствора железа (III) хлори¬
да Р1. Появляется синее окрашивание. Полученный
раствор нагревают до температуры 80 °С. Появляется
коричневое окрашивание. При охлаждении образует¬
ся белый осадок.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 96 % спирте Р и доводят до объема 20 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона В(К)6.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,1 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят метанолом Р до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО ванилина
растворяют в метаноле Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения (Ь). 0,5 мл испытуемого раст¬
вора (а) доводят метанолом Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ вР254 Р■
Подвижная фаза: кислота уксусная ледяная Р—
метанол Р—метиленхлорид Р (0,5:1:98,5, об/об/об);
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку сначала просматривают
в ультрафиолетовом свете при длине волны 254 нм,
после чего опрыскивают раствором динитрофенил¬
гидразина уксусно-хлористоводородного Р и просма¬
тривают при дневном свете.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) при просматривании
в ультрафиолетовом свете и при дневном свете любое
пятно, кроме основного, должно быть не интенсивнее
пятна на хроматограмме раствора сравнения (Ь).
Реакция с кислотой серной. 50 мг испытуемого
образца растворяют в 5 мл кислоты серной Р. Через
5 мин окраска полученного раствора должна быть не
интенсивнее окраски смеси из 4,9 мл желтого основ¬
ного раствора и 0,1 мл красного основного раствора
или смеси из 4,9 мл желтого основного раствора и
0,1 мл синего основного раствора (2.2.2, метод I).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в
эксикаторе в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,05%.
Определение проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в 20 мл
96 % спирта Р, прибавляют 60 мл воды, свободной
от углерода диоксида, Р и титруют 0,1 М раствором
натрия гидроксида потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 15,21 мгС8Н803.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1058
ВАНКОМИЦИНА ГИДРОХЛОРИД
Уапсотуст! ЬудгосЫопдит
УАЫСОМУСШ НУОКОСНШКЮЕ
СмН75С12М9024 • НС1 М.м. 1486
Ванкомицина гидрохлорид
287
ОПРЕДЕЛЕНИЕ
Смесь гидрохлоридов родственных гликопепти¬
дов, состоящая в основном из моногидрохлорида (35,
6/?, 7Я,22Я,235,265,30а5а, 36/?, 38аЯ)-3-(2-амино-2-
оксоэтилН4-[[2-0-(3-амино-2,3,6-тридезокси-3-С-метил-
а-1-л(У/ссо-гексопиранозил)-Р-0-глюкопиранозил]окси]-
10,19-дихлор-7,22,28,30,32-пентагидрокси-6-{[(2#)-4-
метил-2-(метиламино)пентаноил]амино]-2,5,24,38,39-
пентаоксо-2,3,4,5,6,7,23,24,25,26,36,37,38,38а-тетра-
декагидро-22Н-8,11:18,21 -диэтен-23,36-(иминометан)-
13,16:31,35-диметен-1 Н,13Н-[1,6,9]оксадиаза-
циклогексадецин[4,5-ш][10,2,16]-бензоксадиазацик-
лотетракозин-26-карбоновой кислоты (ванкомицин В).
Получают с использованием штаммов
Атусо1а(орз18 опеп1аНз или любым другим способом.
Активность: не менее 1050 МЕ/мг (в пересчете
на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в испытании «Ванкомицин В».
Результаты, основной пик на хроматограмме
испытуемого раствора (а) по времени удерживания
соответствует основному пику на хроматограмме
раствора сравнения.
B. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,10
при длине волны 450 нм. Измеряют оптическую плот¬
ность раствора 3.
рН (2.2.3). От 2,5 до 4,5. 0,50 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Ванкомицин В. Не менее 93,0 %. Жидкостная
хроматография (2.2.29). Растворы используют в те¬
чение 4 ч после приготовления.
Испытуемый раствор (а). 10,0 мг испытуемого
образца растворяют в подвижной фазе А и доводят
до объема 5,0 мл этим же растворителем.
Испытуемый раствор (Ь). 2,0 мл испытуемого
раствора (а) доводят подвижной фазой А до объема
50.0 мл.
Испытуемый раствор (с). 0,5 мл испытуемого
раствора (Ь) доводят подвижной фазой А до объема
20.0 мл.
Раствор сравнения. Содержимое контейнера
ФСО ванкомицина гидрохлорида растворяют в воде Р
и разводят этим же растворителем до получения кон¬
центрации 0,5 мг/мл. Нагревают при температуре
65 °С в течение 24 ч. Охлаждают.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: к 4 мл триэтиламина Р
прибавляют 1996 мл воды Р и доводят до рН 3,2
кислотой фосфорной Р; к 920 мл полученного
раствора прибавляют 10 мл тетрагидрофура-
на Р и 70 мл ацетонитрила Р;
- подвижная фаза В: к 4 мл триэтиламина Р
прибавляют 1996 мл воды Р и доводят до рН 3,2
кислотой фосфорной Р; к 700 мл полученного
раствора прибавляют 10 мл тетрагидрофура-
наР и 290 мл ацетонитрила Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—13
100
0
13—22
о
0
1
о
0 — 100
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл.
Пригодность хроматографической системы:
- разрешение: не менее 5,0 между двумя основ¬
ными пиками на хроматограмме раствора сравнения;
- отношение сигнал/шум: не менее 5 для основ¬
ного пика на хроматограмме испытуемого раствора (с);
-фактор асимметрии: не более 1,6 для пика
ванкомицина на хроматограмме испытуемого раст¬
вора (Ь).
Содержание ванкомицина В гидрохлорида в про¬
центах рассчитывают по формуле:
А6 •100
где:
Аь — площадь пика ванкомицина В на хромато¬
грамме испытуемого раствора (Ь);
А(— сумма площадей пиков примесей на хрома¬
тограмме испытуемого раствора (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29), как указано в испытании «Ванкоми¬
цин В», со следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемых
растворов (а), (Ь) и (с).
Содержание каждой примеси в процентах рас¬
считывают по формуле:
где:
Д — площадь пика примеси на хроматограмме
испытуемого раствора (а);
Аь — площадь пика ванкомицина В на хромато¬
грамме испытуемого раствора (Ь);
Д— сумма площадей пиков примесей на хрома¬
тограмме испытуемого раствора (а).
Предельное содержание примесей:
- любая примесь: не более 4,0 %;
- сумма примесей: не более 7,0 %;
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики
с площадью менее площади основного пика на хро¬
матограмме испытуемого раствора (с).
37*. За*. 1060
288
Гэсударственная фармакопея Республики Беларусь
Тяжелые металлы (2.4.8, метод С). Не более
0,0030 % (30 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 3,0 мл эталон-
ного раствора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 5,0%. Определение
проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 1,0%.
Определение проводят из 1,00 г испытуемого об¬
разца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 0,25 МЕ/мг, если субстанция предназначена
для производства лекарственных средств для па¬
рентерального применения без последующей про¬
цедуры удаления бактериальных эндотоксинов.
#Стерильность (2.6.1). Если субстанция
предназначена для производства лекарственных
средств для парентерального применения без по¬
следующей процедуры стерилизации, испытуемый
образец должен выдерживать испытание на сте¬
рильность методом мембранной фильтрации.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят количественное определение анти¬
биотиков микробиологическим методом (2.7.2). В
качестве стандартного образца используют хими¬
ческий стандартный образец ФСО ванкомицина
гидрохлорида.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте. Если субстанция стериль¬
ная — в стерильном воздухонепроницаемом кон¬
тейнере с контролем первого вскрытия.
ПРИМЕСИ
А. Р2 = МН2, РЗ = Н:Л/-Деметилванкомицин В.
С. Р1 = Н, Р2 = 1ЧН2, РЗ = СНЗ: Аглюкованкоми-
цин В.
О. Р2 = ЫН2, РЗ = СНЗ: Дезванкозаминилванко-
мицин В.
В. (45,7Я,8Я,23/?,245,275,31 а$а,37Я,39а/?)-45-[[2-
0-(3-Амино-2,3,6-тридезокси-3-С-метил-а-Ь-ликсо-
гексопиранозил)-р-0-глюкопиранозил]окси]-11,20-
дихлор-8,23,29,31,33-пентагидрокси-7-[[(2Р)-4-метил-
2-(метиламин)пентаноил]амино]-2,6,25,39,40-пентаоксо-
1,2,3,4,5,6,7,8,24,25,26,27,37,-38,39,39а-гексадекагидро-
23Н-9,12:19,22-диэтен-24,37-(иминометан)-14,17:32,36-
диметен-14Н-[1,6,10]оксадиазациклогептадецин[4,5-ш]
[10,2,16]бензоксадиазациклотетракозин-4,27-дикарбоно-
вая кислота ([Р Азр3]ванкомицин В).
07/2016:0698
ВАРФАРИН НАТРИЯ
)Л/агГаппит па1псит
И/АКРАМИ 800ШМ
и энантиомер
С19Н15Ма04 М.м. 330,3
[129-06-6]
ОПРЕДЕЛЕНИЕ
Натрия 2-оксо-3-[(1 Я5)-3-оксо-1 -фенилбутил]-2Н-
1 -бензопиран-4-олат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ш
Варфарин натрия
289
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый аморфный порошок. Ги¬
гроскопичен.
Очень легко растворим в воде и в 96 % спирте,
растворим в ацетоне, очень мало растворим в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО варфарина натрия *или спектр,
грерставпенный на рисунке #0698.-1#.
B. Испытуемый образец выдерживает испытание
«2-Пропанол», как указано в разделе «Испытания».
C. Испытуемый образец дает реакцию (Ь) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор Б. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 7,6 до 8,6.1,0 г испытуемого образца
растворяют в воде, свободной от углерода диоксида, Р
и доводят до объема 100 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Метанол Р — вода Р
(25:75, об/об).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (а). 2 мг 4-гидроксикумари-
на Р (примесь В) и 2 мг бензалацетона Р (примесь С)
растворяют в 25 мл метанола Р и доводят водой Р до
объема 100 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем цианосилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30°С;
- подвижная фаза: кислота уксусная ледя¬
ная Р — ацетонитрил Р—вода Р (1:25:75, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания варфарина.
Идентификация пиков примесей: идентифици¬
руют пики примесей В и С, используя хроматограмму
раствора сравнения (а).
Относительное удерживание (по отношению к
варфарину, время удерживания — около 9 мин): при¬
месь В — около 0,4; примесь С — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
месей В и С.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,5; для примеси С — 0,4):
-примеси В, С (не более 0,15%): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и С, не должны превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
Рисунок #0698.-1. Инфракрасный спектр ФСО варфарина натрия в дисках с калия брони*
38. Зак. 1060.
290
Гэсударственная фармакопея Республики Беларусь
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Фенольные кетоны. Абсорбционная спектро¬
фотометрия в ультрафиолетовой и видимой областях
(2.2.25) . 1,25 г испытуемого образца растворяют в
растворе 20 г/л натрия гидроксида Р и доводят до
объема 10,0 мл этим же растворителем. Оптическая
плотность полученного раствора при длине волны
385 нм в течение 15 мин после приготовления раст¬
вора не должна превышать 0,20.
2-Пропанол (2.4.24, система А). Не более 0,5 %.
Вода (2.5.12). Не более 4,0 %. Определение про¬
водят из 0,750 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 0,01 М
растворе натрия гидроксида и доводят до объема
100,0 мл этим же растворителем. 10,0 мл полученного
раствора доводят 0,01 М раствором натрия гидрок¬
сида до объема 100,0 мл. 10,0 мл полученного раст¬
вора доводят 0,01 М раствором натрия гидроксида
до объема 100,0 мл. Измеряют оптическую плотность
(2.2.25) в максимуме при длине волны 308 нм.
Рассчитывают содержание С19Н15№04, исполь¬
зуя удельный показатель поглощения, равный 431.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
он
В. 4-Гидрокси-2Н-1-бензопиран-2-он (4-гидрокси-
кумарин).
С. (ЗЕ)-4-Фенилбут-3-ен-2-он (бензалацетон).
07/2016:0699
ВАРФАРИНА НАТРИЯ КЛАТРАТ
И/агГаппит паМсит с1а01га(ит
МАВРАМИ ЗОйШМ аАТНВАТЕ
ЯН3
СН3
ОПРЕДЕЛЕНИЕ
Представляет собой смесь в форме клатрата
варфарина натрия (натрия 2-оксо-3-[(1/?5)-3-оксо-1-
фенилбутил]-2Н-1-бензопиран-4-олат) и 2-пропанола
в молекулярном соотношении 2:1 (соответствует со¬
держанию варфарина натрия около 92 %).
Содержание:
- варфарин натрия: не менее 98,0 % и не более
102,0% (в пересчете на безводное и свободное от
2-пропанола вещество);
- 2-пропанол: не менее 8,0 % и не более 8,5 %.
!
ПРИМЕСИ
Специфицированные примеси: В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А.
А. (5Я5)-3-(2-Гидроксифенил)-5-фенилциклогекс-
2-енон.
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Очень легко растворим в воде, легко растворим
в 96 % спирте, растворим в ацетоне, очень мало рас¬
творим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО варфарина натрия клатрата
#или спектр, представленный на рисунке #0699.-1#.
B. Испытуемый образец выдерживает испыта¬
ние «2-Пропанол», как указано в разделе «Испы¬
тания».
C. Испытуемый образец дает реакцию (Ь) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Варфарина натрия клатрат
291
Рисунок #0699.-1. Инфракрасный спектр ФСО варфарина натрия клатрата в дисках с калия бромидом Р
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 7,6 до 8,6.1,0 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 100 мл этим же раство¬
рителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Метанол Р — вода Р
(25:75, об/об).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (а). 2 мг 4-гидроксикумари-
на Р (примесь В) и 2 мг бензалацетона Р (примесь С)
растворяют в 25 мл метанола Р и доводят водой Р до
объема 100 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем цианосилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: кислота уксусная ледя¬
ная Р — ацетонитрил Р— вода Р (1:25:75, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы. 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания варфарина.
Идентификация пиков примесей: идентифици¬
руют пики примесей В и С, используя хроматограмму
раствора сравнения (а).
Относительное удерживание (по отношению к
варфарину, время удерживания — около 9 мин): при¬
месь В — около 0,4; примесь С — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
месей В и С.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,5; для примеси С — 0,4):
-примеси В, С (не более 0,15%): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и С, не должны превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Фенольные кетоны. Абсорбционная спектро¬
фотометрия в ультрафиолетовой и видимой областях
(2.2.25). 1,25 г испытуемого образца растворяют в
растворе 20 г/л натрия гидроксида Р и доводят до
объема 10,0 мл этим же растворителем. Оптическая
плотность полученного раствора при длине волны
385 нм в течение 15 мин после приготовления раст¬
вора не должна превышать 0,20.
38*. Зак. 1060.
292
Гэсударственная фармакопея Республики Беларусь
2-Пропанол (2.4.24, система А). Не менее 8,0 %
и не более 8,5 %.
Вода (2.5.12). Не более 0,3 %. Определение про¬
водят из 2,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 0,01 М
растворе натрия гидроксида и доводят до объема
100,0 мл этим же растворителем. 10,0 мл полученного
раствора доводят 0,01 М раствором натрия гидрок¬
сида до объема 100,0 мл. 10,0 мл полученного раст¬
вора доводят 0,01 М раствором натрия гидроксида
до объема 100,0 мл. Измеряют оптическую плотность
(2.2.25) в максимуме при длине волны 308 нм.
Содержание варфарина натрия (С19Н15Ма04) рас¬
считывают с учетом удельного показателя поглоще¬
ния, равного 431.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А.
А. (5Р8)-3-(2-Гидроксифенил)-5-фенилциклогекс-
2-енон.
/О. .о
он
В. 4-Гидрокси-2Н-1-бензопиран-2-он (4-гидрокси-
кумарин).
С. (ЗЕ)-4-Фенилбут-3-ен-2-он (бензалацетон).
07/2016:0573
ВЕРАПАМИЛА ГИДРОХЛОРИД
УегаратШ ЬудгосЫопдит
УЕКАРАМИ- НУОТЮСт.ОПЮЕ
С27Н38М204 ■ НС1 М.м. 491,1
[152-11-4]
ОПРЕДЕЛЕНИЕ
(2/?5)-2-(3,4-Диметоксифенил)-5-[[2-(3,4-димето-
ксифенил)этил](метил)амино]-2-(1-метилэтил)пентан-
нитрила гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Растворим в воде, легко растворим в метаноле,
умеренно растворим в 96 % спирте.
Температура плавления: около 144 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем. 5,0 мл полученного раствора дово¬
дят 0,01 М раствором кислоты хлористоводородной
до объема 50,0 мл.
Диапазон длин волн: от 210 нм до 340 нм.
Максимумы поглощения: при 229 нм и при 278 нм.
Плечо: при 282 нм.
Отношение оптических плотностей:
А^/А^ — от °’35 Д° °»39-
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО верапамила гидрохлорида #или
спектр, представленный на рисунке #0573.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метиленхлориде Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО верапамила
гидрохлорида растворяют в метиленхлориде Р и до¬
водят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО папаверина
гидрохлорида растворяют в растворе сравнения (а) и
доводят до объема 5 мл этим же раствором.
Пластинка: ТСХ пластинка со слоем силикаге-
пяР254 Р.
Подвижная фаза: диэтиламин Р — циклогек¬
сан Р (15:85, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Верапамила гидрохлорид
293
Рисунок #0573.-1. Инфракрасный спектр ФСО верапамила гидрохлорида
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
О. Испытуемый образец дает реакцию (Ь) хлори¬
ды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р при
осторожном нагревании и доводят до объема 20,0 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должна
быть бесцветным.
рН (2.2.3). От 4,5 до 6,0. Измеряют рН раствора 5.
Угол оптического вращения (2.2.7). От -0,10° до
+0,10°. Измеряют угол оптического вращения раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Подвижная фаза А —
подвижная фаза В (37:63, об/об).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). 5 мг ФСО верапами¬
ла гидрохлорида, 5 мг ФСО верапамила примеси I и
5 мг ФСО верапамила примеси М растворяют в сме¬
си растворителей и доводят до объема 20 мл этой же
смесью растворителей. 1 мл полученного раствора
доводят смесью растворителей до объема 10 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная кремнийорганическим аморфным
полярно-встроенным октадецилсилильным полиме¬
ром эндкепированным Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 6,97 г/л дикалия
гидрофосфата Р, доведенный до рН 7,20 кисло¬
той фосфорной Р;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—22
63
37
22—27
ю
со
т
со
со
ю
со
т
1^
со
27—35
35
65
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 278 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к верапамилу, время удерживания— около 15 мин):
примесь I — около 1,3; примесь М — около 2,4.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 5,0 между пиками вера¬
памила и примеси I;
- примесь М должна элюироваться из колонки.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
39. Зак. 1060.
294
Гэсударственная фармакопея Республики Беларусь
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 50 мл
этанола безводного Р, прибавляют 5,0 мл 0,01 М
раствора кислоты хлористоводородной и титруют
0,1 М раствором натрия гидроксида потенциометри-
чески (2.2.20). Отмечают объем титранта между дву¬
мя точками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 49,11 мгС27Н38М204 НС1.
СНз
С. 2-(3,4-Диметоксифенил)-А/,А/-диметилэтанамин.
Э. 3-Хлор-Л/-[2-(3,4-Диметоксифенил)этил]-Л/-ме-
тилпропан-1 -амин.
Е. (3,4-Диметоксифенил)метанол.
Р. (2Я5)-2-(3,4-Диметоксифенил)-5-(метиламино)-
2-(1 -метилэтил)пентаннитрил.
О. 3,4-Диметоксибензальдегид.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимити¬
руется общим критерием приемлемости для других/
неспецифицированных примесей и/или общей статьей
Субстанции для фармацевтического использования
(2034). Вследствие этого нет необходимости иденти¬
фицировать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Контроль
примесей в субстанциях для фармацевтического ис¬
пользования): А, В, С, О, Е, Р, 6, Н, I, 3, К, /_, М, /V, О, Р.
А. А/,Л/'-Бис[2-(3,4-диметоксифенил)этил]-Л/,Л/'-
диметилпропан-1,3-диамин.
НзСО.
Н. (2Я5)-2-(3,4-Диметоксифенил)-5-[[2-(3,4-диме-
токсифенил)этил](метил)амино]-2-этилпентаннитрил.
I. (2Я5)-2-(3,4-Диметоксифенил)-2-[2-[[2-(3,4-диме-
токсифенил)этил](метил)амино]этил]-3-метил-бутан-
нитрил.
Н3СО'
сн3
В. 2-(3,4-Диметоксифенил)-А/-метилэтанамин.
3. (2Я5)-2-(3,4-Диметоксифенил)-5-[[2-(3,4-диме-
токсифенил)этил]амино]-2-(1-метилэтил)пентаннитрил
(А/-норверапамил).
Винная кислота
295
ОСНз
ОСНз
СМ и энантиомер
07/2016:0460
ВИННАЯ КИСЛОТА
АсШит ^а^^а^^сит
ТАКТАШСАСЮ
К. (2Я5)-2-(3,4-Диметоксифенил)-3-метилбутан-
нитрил.
1-(3,4-Диметоксифенил)-2-метилпропан1-он.
М. 5,5'-[[2-(3,4-Диметоксифенил)этил]ими-
но]бис[2-(3,4-диметоксифенил)-2-(1-метилэтил)пен-
таннитрил].
N. 5,5'-(Метилимино)бис[2-(3,4-диме-токсифе-
нил)-2-(1-метилэтил)пентаннитрил].
О. (2Я$)-2-(3,4-Диметоксифенил)-5-[[2-(3,4-ди-
метоксифенил)этил](метил)амино]-2-пропилпен-
таннитрил.
Р. 2,6-бис(3,4-Диметоксифенил)-2,6-бис(1 -метил-
этил )-гептан-1 ,7-динитрил.
С4Н6Об М.м. 150,1
[87-69-4]
ОПРЕДЕЛЕНИЕ
(2/?,3/?)-2,3-Дигидроксибутандиовая кислота.
Содержание: не менее 99,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень легко растворима в воде, легко раствори¬
ма в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 5, приготовленный как указано в разде¬
ле «Испытания», имеет сильнокислую реакцию (2.2.4).
B. Испытуемый образец дает реакции на тартра¬
ты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Удельное оптическое вращение (2.2.7). От
+12,0 до +12,8 (в пересчете на сухое вещество). 5,00 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Щавелевая кислота. Не более 0,0350 %
(350 ррт) (в пересчете на безводную щавелевую кис¬
лоту). 0,80 г испытуемого образца растворяют в 4 мл
воды Р. Прибавляют 3 мл кислоты хлористоводо¬
родной Р и 1 г цинка Р в гранулах. Кипятят в течение
1 мин. Выдерживают в течение 2 мин. Переносят надо-
садочную жидкость в пробирку, содержащую 0,25 мл
раствора 10 г/л фенилгидразина гидрохлорида Р и на¬
гревают до кипения. Быстро охлаждают, переносят в
мерный цилиндр и прибавляют такой же объем кисло¬
ты хлористоводородной Р и 0,25 мл раствора 50 г/л
калия феррицианида Р. Встряхивают и выдерживают
в течение 30 мин. Розовое окрашивание раствора
должно быть не интенсивнее, чем окраска раствора
сравнения, приготовленного параллельно с использо¬
ванием 4 мл раствора 0,1 г/л кислоты щавелевой Р.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0150 % (150 ррт).
10 мл раствора 5 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
39*. Зак. 1060.
296
Гэсударственная фармакопея Республики Беларусь
Кальций (2.4.3). Не более 0,0200 % (200 ррт). К
5 мл раствора 3 прибавляют 10 мл раствора 50 г/л на¬
трия ацетата Р в воде дистиллированной Р. Полу¬
ченный раствор должен выдерживать испытание на
кальций.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,2 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,650 г испытуемого образца растворяют в
25 мл воды Р и титруют 1 М раствором натрия
гидроксида до появления розового окрашивания,
используя в качестве индикатора 0,5 мл раствора
фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 75,05 мг С4Н606.
07/2016:2107
ВИНОРЕЛБИНА ТАРТРАТ
Утоге/Ыт 1аг1газ
УШСЖЕ1.ВШЕ ТАРТРАТЕ
С^Н^О, ■ 204Н6О6 М.м. 1079
[125317-39-7]
ОПРЕДЕЛЕНИЕ
Метил-(ЗаР,4Р,55,5аР, 1ОЬР, 1ЗаР)-4-(ацетилокси)-
За-этил-9-[(6/?,85)-4-этил-8-(метоксикарбонил)-
1,3,6,7,8,9-гексагидро-2,6-метано-2Н-азаци-
клодецино[4,3-Ь]индол-8-ил]-5-гидрокси-8-метокси-6-
метил-3а,4,5,5а,6,11,12,1 За-октагидро-1 Н-индоли-зи-
но[8,1 -сфарбазол-5-карбоксилата дигидро-бис[(2Р,ЗР)-
2,3-дигидроксибутандиоат].
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде и в метаноле, практиче¬
ски нерастворим в гексане.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 10 мг испытуемого образца
растворяют в 5 мл воды Р, прибавляют 0,5 мл раст¬
вора натрия гидроксида Р и экстрагируют, используя
5 мл метиленхлорида Р. Органический слой сушат
над натрия сульфатом безводным Р, фильтруют,
упаривают до объема 0,5 мл и наносят на диск из ка¬
лия бромида Р. Выпаривают и снимают спектр.
Сравнение: ФСО винорелбина тартрата, обра¬
ботанный как описано выше, #или спектр, представ¬
ленный на рисунке #2107.-1#.
В. Испытуемый образец дает реакцию (Ь) на тар¬
траты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. Количество испытуемого образца, эк¬
вивалентное 0,140 г безводного вещества, растворяют
в воде Р и доводят до объема 10,0 мл этим же раство¬
рителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Оптическая плотность (2.2.25). Оптическая
плотность раствора 3 при длине волны 420 нм не
должна превышать 0,030.
рН (2.2.3). От 3,3 до 3,8. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят методом нор¬
мализации.
Испытуемый раствор. 35,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25 мл этим же растворителем.
Раствор сравнения (а). 7 мг ФСО винорелбина
примеси В растворяют в воде Р и доводят до объема
50 мл этим же растворителем. К 1 мл полученного
раствора прибавляют 14 мг ФСО винорелбина тар¬
трата, растворяют в воде Р и доводят до объема
10 мл этим же растворителем. Полученный раствор
подвергают воздействию ксеноновой лампы при дли¬
не волны (310—880) нм в течение часа, обеспечивая
дозу 1600 кДж/м2 с плотностью потока 500 Вт/м2 для
образования примеси А.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят водой Р до объема 20,0 мл. 1,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 3,9 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р (размер частиц 5 мкм) с удельной пло¬
щадью поверхности 125 м2/г, размером пор 30 нм и
содержанием углерода 7 %;
- температура: (35±5) °С;
- подвижная фаза: 1,22 г натрия декансульфо-
ната Р растворяют в 620 мл метанола Р и прибавля¬
ют 380 мл раствора 7,80 г/л натрия дигидрофосфа¬
та Р, предварительно доведенного до рН 4,2 кисло¬
той фосфорной разведенной Р;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 267 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания винорелбина.
Относительное удерживание (по отношению к
винорелбину, время удерживания — около 14 мин):
примесь А — около 0,8; примесь В — около 1,2.
Пригодность хроматографической системы:
-коэффициент разделения пиков: не менее 4
(Нр — высота пика примеси В относительно базовой ли¬
нии; Ну — расстояние между базовой линией и нижней
Винорелбина тартрат
297
Рисунок #2107.-1. Инфракрасный спектр ФСО винорелбина тартрата
точкой кривой, разделяющей пик примеси В и пик вино¬
релбина на хроматограмме раствора сравнения (а));
- отношение сигнал/шум: не менее 10 для основ¬
ного пика на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
- примесь А: не более 0,3 %;
-любая другая примесь: не более 0,2 %;
- сумма примесей, кроме примеси А: не более 0,7 %;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади основного пика на хроматограмме
раствора сравнения (Ь).
Бор. Не более 0,0050 % (50 ррт).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в 2 мл воды Р. Медленно прибав¬
ляют 10,0 мл кислоты серной Р при охлаждении в
ледяной бане. Полученный раствор перемешивают и
выдерживают до достижения комнатной температу¬
ры, затем прибавляют 10,0 мл раствора 0,5 г/л кисло¬
ты карминовой Р в кислоте серной Р.
Раствор сравнения. 2,5 мл раствора 0,572 г/л
кислоты борной Р доводят водой Р до объема
100,0 мл. К 2,0 мл полученного раствора медленно
прибавляют 10,0 мл кислоты серной Р при охлажде¬
нии в ледяной бане. Полученный раствор перемеши¬
вают и выдерживают до достижения комнатной тем¬
пературы, затем прибавляют 10,0 мл раствора 0,5 г/л
кислоты карминовой Р в кислоте серной Р.
Контрольный раствор. К 2,0 мл воды Р медлен¬
но прибавляют 10,0 мл кислоты серной Р при охлаж¬
дении в ледяной бане. Полученный раствор переме¬
шивают и выдерживают до достижения комнатной
температуры, затем прибавляют 10,0 мл раствора
0,5 г/л кислоты карминовой Р в кислоте серной Р.
Через 45 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора и раствора сравнения
в диапазоне от 560 нм до 650 нм, используя контроль¬
ный раствор в качестве компенсационного раствора.
Максимальное значение оптической плотности для
испытуемого раствора должно быть не более чем
максимальное значение оптической плотности для
раствора сравнения.
Фториды. Не более 0,0050 % (50 ррт).
Потенциометрическое определение (2.2.36, ме¬
тод /) с использованием фторселекгивного индикатор¬
ного электрода и хлорсеребряного электрода сравнения.
Испытуемый раствор. 0,19 г испытуемого об¬
разца растворяют в 20 мл воды Р, прибавляют 5,0 мл
буфера для регулирования ионной силы Р и доводят
водой Р до объема 50 мл.
Растворы сравнения. К 0,6 мл, 0,8 мл, 1,0 мл, 1,2 мл
и 1,4 мл эталонного раствора фторида (10 ррт Р) Р
прибавляют по 5,0 мл буфера для регулирования ион¬
ной силы Р и доводят водой Р до объема 50 мл.
Погружают электроды в растворы сравнения и
выдерживают в течение 5 мин. Определяют разность
потенциалов между электродами через 1 мин стаби¬
лизации. Используя полулогарифмическую бумагу,
графически отображают разность потенциалов для
каждого раствора сравнения как функцию концентра¬
ции фторид-иона. В точно таких же условиях проводят
определение разности потенциалов для испытуемого
раствора и рассчитывают содержание фторид-иона.
Серебро. Не более 0,0005 % (5 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Испытуемый раствор. 0,500 г испытуемого об¬
разца растворяют в 10,0 мл воды Р.
Растворы сравнения. Готовят разведениями эта¬
лонного раствора серебра (5 ррт Ад) Р 6,5 % (об/об)
раствором кислоты азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения серебра.
Длина волны: 328,1 нм.
Атомизатор: воздушно-ацетиленовое пламя.
40. Зак. 1060.
298
Гэсударственная фармакопея Республики Беларусь
Вода (2.5.12). Не более 4,0 %. Определение про¬
водят из 0,250 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
2 МЕ/мг (в пересчете на основание винорелбина),
если субстанция предназначена для производства
лекарственных средств для парентерального приме¬
нения без дальнейшей подходящей процедуры удале¬
ния бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 40 мл
кислоты уксусной ледяной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ-
ствует 53,96 мг С^Н^О.-гСДО..
ХРАНЕНИЕ
В среде инертного газа в защищенном от света
месте при температуре не выше -15 °С.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): В, С, О, Е, Е, 6, Н, I, 3.
А. Метил-(ЗаЗ,4Я,5аЯ, 1ОЬЯ, 1ЗаЯ)-4-(ацетилокси)-
3,5-эпокси-За-этил-9-[(6Я,85)-4-этил-8-(мето-
ксикарбонил)-1,3,б,7,8,9-гексагидро-2,6-метано-2Н-
азациклодецино[4,3-Ь]индол-8-ил]-8-метокси-6-метил-
За,4,5,5а,6,11,12,1За-октагидро-ЗН-индолизино[8,1 -ссЦ-
карбазол-5-карбоксилат.
В. Р1 = РЗ = Н, Р2 = СН3: Метил-(За/?,4Я,55,5аЯ,-
1ОЬЯ, 1ЗаЯ)-За-этил-9-[(6Я,85)-4-этил-8-(метокси-
карбонил)-1,3,6,7,8,9-гексагидро-2,6-метано-2Н-аза-
циклодецино[4,3-Ь]индол-8-ил]-4,5-дигидрокси-8-мето-
кси-6-метил-3а,4,5,5а,6,11, 12,1 За-октагидро-1 Н-
индол изино[8,1 -сс/|карбазол-5-карбоксилат.
H. Р1 = Р2 = Н, РЗ = СО-СН3: (ЗаЯ,4Я,55,5аЯ,-
1ОЬЯ, 1ЗаЯ)-4-(Ацетилокси)-За-этил-9-[(6Я,85)-4-этил-
8-(метоксикарбонил)-1,3,6,7,8,9-гексагидро-2,6-метано-
2Я-азациклодецино[4,3-Ь]индол-8-ил]-5-гидрокси-8-
метокси-6-метил-За,4,5,5а,6,11,12,1 За-октагидро-1 Н-
индолизино[8,1-сс/]карбазол-5-карбоновая кислота.
I. Р1 = Вг, Р2 = СН3, РЗ = СО-СН3: Метил-(ЗаЯ,4Я,-
55,5аЯ, 1ОЬЯ, 1ЗаЯ)-4-(ацетилокси)-7-бром-За-этил-9-
[(6Я,85)-4-этил-8-(метоксикарбонил)-1,3,6,7,8,9-
гексагидро-2,6-метано-2Н-азациклодецино[4,3-Ь]индол-
8-ил]-5-гидрокси-8-метокси-6-метил-За,4,5,5а,6,11
12,1 За-октагидро-1 Н-индолизино[8,1-сс/]карбазол-5-
карбоксилат.
С. Метил-(ЗаЯ,4Я,55,5аЯ, 1ОЬЯ, 1ЗаЯ)-4-(ацетило-
кси)-За-этил-9-[(6Я,8Я)-4-этил-8-(метоксикарбонил)-
1,3,6,7,8,9-гексагидро-2,6-метано-2Н-азацикло-
децино[4,3-Ь]индол-8-ил]-5-гидрокси-8-метокси-6-метил-
За,4,5,5а,6,11,12,1 За-октагидро-1 Н-индолизино[8,1 -сд\-
карбазол-5-карбоксилат.
й. Метил-(ЗаЯ,4Я,55,5аЯ,10ЬЯ,13аЯ)-4-(ацетил-
окси)-За-этил-9-[(2Я5,6Я,85)-4-этил-8-(метоксикар-
бонил)-2-оксидо-1,3,6,7,8,9-гексагидро-2,6-метано-2Н-
азациклодецино[4,3-Ь]индол-8-ил]-5-гидрокси-8-метокси-
6-метил-За,4,5,5а,6,11,12,1 За-октагидро-1 Н-индоли-
зино[8,1-сс/]карбазол-5-карбоксилат.
Е. X = СН2-СН2: Метил-( 1 а5,115,133,1ЗаЯ)-11 -
[(ЗаЯ,4Я,55,5аЯ, 1ОЬЯ, 1ЗаЯ)-4-(ацетилокси)-За-этил-5-
гидрокси-8-метокси-5-(метоксикарбонил)-6-метил-
За,4,5,5а,6,11,12,1 За-октагидро-1 Н-индолизино[8,1 -сс/]-
карбазол-9-ил]-1 а-этил-1 а,4,5,10,11,12,13,1 За-октагидро-
2Н-3,13-метанооксирено[9,10]азациклоундецино[5,4-Ь]-
индол-11-карбоксилат (леурозин).
О. X = СН2: Метил-(1а5,105,125,12аЯ)-10-
[(ЗаЯ,4Я,55,5аЯ, 1ОЬЯ, 1ЗаЯ)-4-(ацетилокси)-За-этил-5-
гидрокси-8-метокси-5-(метоксикарбонил)-6-метил-
Винпоцетин
299
За,4,5,5а,6,11,12,13а-октагидро-1Н-индолизино[8,1-сс/]-
карбазол-9-ил]-1 а-этил-1 а,2,4,9,10,11,12,12а-октагидро-
3,12-метано-ЗН-оксирено[8,9]азациклодецино[4,3-Ь]-
индол-1О-карбоксилат.
Р. (2Я5,6Я,85)-8-[(ЗаЯ,4Я,55,5аЯ, 1ОЬЯ,1ЗаЯ)-4-
(Ацетилокси)-За-этил-5-гидрокси-8-метокси-5-
(метоксикарбонил )-6-метил-За,4,5,5а,6,11,12,1 За-
октагидро-1 Н-индолизино[8,1-ссУ]карбазол-9-ил]-4-этил-
8-(метоксикарбонил)-2-метил-1,3,6,7,8,9-гексагидро-
2,6-метано-2Н-азациклодецино[4,3-Ь]индолий.
^. Метил-(За/?,4/?,55,5аЯ, 10ЬЯ, 1ЗаЯ)-4-(аце-
тилокси)-За-этил-9-[(7Я,95)-5-этил-9-(метоксикарбонил)-
1,4,7,8,9,10-гексагидро-2/-/-3,7-метаноазацикло-
ундецино[5,4-Ь]индол-9-ил]-5-гидрокси-8-метокси-6-
метил-За,4,5,5а,6,11,12,13а-октагидро-1Н-индо-
лизино[8,1-с</|карбазол-5-карбоксилат.
07/2016:2139
ВИНПОЦЕТИН
УтросеИпит
УШРОСЕТШЕ
С22Н26М202 М.М. 350»5
[42971-09-5]
ОПРЕДЕЛЕНИЕ
Этил-(13а5,13Ь5)-13а-этил-2,3,5,6,13а,13Ь-
гексагидро-1Н-индол[3,2,1-с/е]пиридо[3,2,1-(/][1,5]-
нафтиридин-12-карбоксилат.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтый кристаллический по¬
рошок.
Практически нерастворим в воде, растворим в ме-
тиленхлориде, мало растворим в этаноле безводном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
90 -I
85 I
80 ]
I
75 1
|
70 {
« |
60 -I
55 1
50 \
I
45 -I
40 -}
35 -!
3000
2000
Волновое число (см'1)
1500
1000
Рисунок #2139.-1. Инфракрасный спектр ФСО еинпоцетина
40*. Зак. 1060.
300
Гэсударственная фармакопея Республики Беларусь
В. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО винпоцетина #или спектр,
представленный на рисунке #2139.-1#.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+127 до +134 (в пересчете на сухое вещество). 0,25 г
испытуемого образца растворяют в диметилформа-
миде Р и доводят до объема 25,0 мл этим же раство¬
рителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО винпоцети¬
на примеси В, 6,0 мг ФСО винпоцетина примеси А,
5,0 мг ФСО винпоцетина примеси С и 5,0 мг ФСО
винпоцетина примеси О растворяют в подвижной
фазе и доводят до объема 50,0 мл этим же раство¬
рителем.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (а) и 1,0 мл раствора сравнения (Ь) доводят под¬
вижной фазой до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: раствор 15,4 г/л аммония аце¬
тата Р — ацетонитрил Р (45:55, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 15 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания винпоцетина.
Относительное удерживание (по отношению к
винпоцетину, время удерживания — около 16 мин):
примесь А — около 0,4; примесь й — около 0,68; при¬
месь В — около 0,75; примесь С — около 0,83.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 2,0 между пиками при¬
месей О и В.
Предельное содержание примесей:
- примесь А (не более 0,6 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
-примеси В, О (не более 0,5%): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и О, не должны превышать
площади соответствующих пиков на хроматограмме
раствора сравнения (с);
- примесь С (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси С, не должна превышать 0,6 площади
соответствующего пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С и О, не должна превышать площадь
пика винпоцетина на хроматограмме раствора срав¬
нения (с);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь пика винпоцетина на хромато¬
грамме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади пика винпоцетина на
хроматограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 100 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
смеси из равных объемов уксусного ангидрида Р
и кислоты уксусной безводной Р и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 35,05 мг С22Н261Ч202.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. Этил(125,13а5,13Ь5)-1 За-этил-12-гидрокси-
2,3,5,6,12,13,1 За,13Ь-октагидро-1 Н-индол[3,2,1 -с/е]-
пиридо[3,2,1-/)][1,5]нафтиридин-12-карбоксилат (этил-
винкаминат).
В. Метил(1 За5,13Ь5)-13а-этил-2,3,5,6,13а, 13Ь-
гексагидро-1Н-индол [3,2,1 -</е]пиридо[3,2,1 -/у][1,5]наф-
тиридин-12-карбоксилат (аповинкамин).
Н3СО
С. Этил(13а5,1 ЗЬ5)-1 За-этил-10-метокси-
2,3,5,6,13а,13Ь-гексагидро-1Н-индол[3,2,1-с/е]пи-
ридо[3,2,1 -/у][1,5]нафтиридин-12-карбоксилат (метокси-
винпоцетин).
Висмута нитрат основной, тяжелый
301
О. Этил(12Я5.1 ЗаЯ5,1 ЗЬ/?5)-13а-этил-2,3,5,-
6,12,13,1 За,1 ЗЬ-октагидро-1 Н-индол[3,2,1 -с/е]пири-
до[3,2,1-/у][1,5]нафтиридин-12-карбоксилат (дигидро-
винпоцетин).
07/2016:1494
ВИСМУТА НИТРАТ ОСНОВНОЙ,
ТЯЖЕЛЫЙ
В1зти011 зиЬпПгаз ропс1егозиз
В/5ЛШ7Н 811ВЫ1ТКАТЕ, НЕАУУ
4[В|Ы03(ОН)2], ВЮ(ОН) М.м. 1462
[1304-85-4]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 71,0 % и не более 74,0 %
В| (А.м. 209,0) (в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде и в 96 % спирте.
Растворяется в минеральных кислотах с разложением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 мл раствора 51, приготовленного как указано
в разделе «Испытания», доводят водой Р до объема
5 мл и прибавляют 0,3 мл раствора калия йодида Р.
Образуется черный осадок, который растворяется при
прибавлении 2 мл раствора калия йодида Р с образо¬
ванием оранжевого раствора.
B. Испытуемый образец дает реакцию (Ь) на вис¬
мут (2.3.1).
C. Испытуемый образец дает реакцию (а) на ни¬
траты (2.3.1).
й. рН (2.2.3): не более 2,0. Измеряют рН раст¬
вора 52, приготовленного как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Раствор 51. 5,0 г испытуемого образца встря¬
хивают с 10 мл воды Р при осторожном нагревании,
прибавляют 20 мл кислоты азотной Р, нагревают до
растворения, охлаждают и доводят водой Р до объ¬
ема 100 мл.
Раствор 52.1,00 г испытуемого образца помеща¬
ют в мерную колбу вместимостью 20 мл и прибавляют
2,0 мл кислоты азотной, свободной от свинца, Р.
Выдерживают без нагревания, слегка подогревая при
необходимости в конце реакции для полного растворе¬
ния испытуемого образца. Прибавляют 10 мл воды Р,
встряхивают и прибавляют небольшими порциями
4,5 мл раствора аммиака, свободного от свинца, Р,
встряхивают и охлаждают. Полученный раствор дово¬
дят водой Р до объема 20,0 мл, снова встряхивают и
выдерживают до оседания твердых частиц. Использу¬
ют прозрачную надосадочную жидкость.
Кислотность. 1,0 г испытуемого образца су¬
спендируют в 15 мл воды Р и встряхивают несколько
раз. Выдерживают в течение 5 мин и фильтруют. К
10 мл фильтрата прибавляют 0,5 мл раствора фе¬
нолфталеина Р1. При прибавлении не более 0,5 мл
0,1 М раствора натрия гидроксида окраска раствора
должна измениться на розовую.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт). К
5,0 мл раствора 51 прибавляют 3 мл кислоты азот¬
ной Р и доводят водой Р до объема 15 мл.
Медь. Не более 0,0050 % (50 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод /).
Испытуемый раствор. Раствор 52.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора меди
(10 ррт Си) Р с помощью 37 % (об/об) раствора кис¬
лоты азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения меди.
Длина волны: 324,7 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Свинец. Не более 0,0020 % (20 ррт). Атомно-аб¬
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Раствор 52.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора свинца
(10 ррт РЬ) Р с помощью 37 % (об/об) раствора кис¬
лоты азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 283,3 нм (или 217,0 нм, в зависи¬
мости от используемого оборудования).
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Серебро. Не более 0,0025 % (25 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. Раствор 52.
Раствор сравнения. Готовят соответствующими
разведениями эталонного раствора серебра (5 ррт
Ад) Р с помощью 37 % (об/об) раствора кислоты
азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения серебра.
Длина волны: 328,1 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Вещества, не осаждаемые раствором аммиа¬
ка. Не более 1,0 %. К 20 мл раствора 51 прибавляют
раствор аммиака концентрированный Р до получе¬
ния щелочной реакции и фильтруют. Осадок промы¬
вают водой Р. Фильтрат и промывные воды объеди¬
няют и выпаривают досуха на водяной бане. К сухому
остатку прибавляют 0,3 мл кислоты серной разведен¬
ной Р и прокаливают. Масса полученного остатка не
должна превышать 10 мг.
Потеря в массе при высушивании (2.2.32). Не
более 3,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют при
нагревании в 10 мл смеси из 2 объемов кислоты
хлорной Р и 5 объемов воды Р. К горячему раство¬
ру прибавляют 200 мл воды Р и 50 мг индикаторной
смеси ксиленолового оранжевого Р и титруют 0,1 М
302
Гэсударственная фармакопея Республики Беларусь
раствором натрия эдетата до появления желтого
окрашивания.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 20,90 мг В|.
07/2016:1493
ВИСМУТА СУБГАЛЛАТ
ЕИзти1Ы зиЬдаИаз
В13М1ЛН БиВСАНАТЕ
[99-26-3]
ОПРЕДЕЛЕНИЕ
Комплексное соединение висмута и галловой
кислоты.
Содержание: не менее 48,0 % и не более 51,0 %
В| (А.м. 209,0) (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый порошок.
Практически нерастворим в воде и в 96 % спирте.
Растворяется в минеральных кислотах с разложени¬
ем и в растворах гидроксидов щелочных металлов с
образованием красновато-коричневой жидкости.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,1 г испытуемого образца смешивают с 5 мл
водыР и 0,1 мл кислоты фосфорной Р. Нагревают
до кипения и кипятят в течение 2 мин. Охлаждают
и фильтруют. При прибавлении к фильтрату 1,5 мл
раствора железа (III) хлорида Р1 появляется чернова¬
то-синее окрашивание.
B. Испытуемый образец дает реакцию (Ь) на вис¬
мут (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца помеща¬
ют в фарфоровую или кварцевую чашку и сжигают,
постепенно повышая температуру. Нагревают в му¬
фельной печи при температуре (600±50) °С в течение
2 ч. Охлаждают, полученный остаток растворяют при
подогревании в 4 мл смеси из равных объемов кис¬
лоты азотной, свободной от свинца, Р и воды Р и
доводят водой Р до объема 20 мл.
Кислотность. 1,0 г испытуемого образца встря¬
хивают в течение 1 мин с 20 мл воды Р и фильтруют. К
фильтрату прибавляют 0,1 мл раствора метилового
красного Р. При прибавлении не более 0,15 мл 0,1 М
раствора натрия гидроксида должно появиться жел¬
тое окрашивание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
К 0,5 г испытуемого образца прибавляют 10 мл кис¬
лоты азотной разведенной Р. Нагревают на водяной
бане в течение 5 мин и фильтруют. 5 мл фильтрата
доводят водой Р до объема 15 мл. Полученный раст¬
вор должен выдерживать испытание на хлориды.
Нитраты. Не более 0,2%. К 1,0 г испытуемого
образца прибавляют 25 мл воды Р, затем 25 мл смеси
из кислоты серной Р и воды Р (2:9, об/об). Нагревают
при температуре около 50 °С в течение 1 мин при пе¬
ремешивании и фильтруют. К 10 мл фильтрата осто¬
рожно прибавляют 30 мл кислоты серной Р. Корич¬
невато-желтое окрашивание полученного раствора
должно быть не интенсивнее окрашивания эталона,
приготовленного параллельно следующим образом:
к 0,4 г кислоты галловой Р прибавляют 20 мл эта¬
лонного раствора нитрата (100 ррт NО^ Р и 30 мл
смеси из кислоты серной Р и воды Р (2:9, об/об) и
фильтруют. К 10 мл полученного фильтрата осторож¬
но прибавляют 30 мл кислоты серной Р.
Медь. Не более 0,0050 % (50 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Готовят путем разведения
эталонного раствора меди (10 ррт Си) Р 6,5 % (об/об)
раствором кислоты азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения меди.
Длина волны: 324,7 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Свинец. Не более 0,0020 % (20 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. Раствор 5.
Растворы сравнения. Готовят путем разведения
эталонного раствора свинца (10 ррт РЬ) Р 6,5%
(об/об) раствором кислоты азотной, свободной от
свинца, Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 283,3 нм (в зависимости от прибо¬
ра может быть использована линия поглощения при
длине волны 217,0 нм).
Атомизатор: воздушно-ацетиленовое пламя.
Серебро. Не более 0,0025 % (25 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Готовят путем разведения
эталонного раствора серебра (5 ррт Ад) Р 6,5 %
(об/об) раствором кислоты азотной, свободной от
свинца, Р.
Источник излучения: лампа с полым катодом
для определения серебра.
Длина волны: 328,1 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Вещества, не осаждаемые раствором аммиа¬
ка. Не более 1,0 %. 2,0 г испытуемого образца поме¬
щают в фарфоровую или кварцевую чашку, сжигают,
постепенно повышая температуру до (600±50) °С,
и охлаждают. Полученный остаток смачивают 2 мл
кислоты азотной Р, выпаривают досуха на водя¬
ной бане, затем осторожно нагревают и сжигают при
температуре (600±50) °С. После охлаждения остаток
растворяют в 5 мл кислоты азотной Р и доводят во¬
дой Р до объема 20 мл. К10 мл полученного раствора
прибавляют раствор аммиака концентрированный Р
до щелочной реакции и фильтруют. Промывают оста¬
ток водой Р и выпаривают объединенные фильтрат
и промывные воды досуха на водяной бане. К полу¬
ченному остатку прибавляют 0,3 мл кислоты серной
разведенной Р и прокаливают. Масса остатка после
прокаливания не должна превышать 10 мг.
Вода высокоочищенная
303
Потеря в массе при высушивании (2.2.32). Не
менее 7,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,300 г испытуемого образца прибавляют
10 мл смеси из равных объемов кислоты азотной Р
и воды Р, нагревают до кипения и кипятят в течение
2 мин. Прибавляют 0,1 г калия хлората Р, нагревают
до кипения и кипятят в течение 1 мин. Прибавляют
10 мл воды Р и нагревают до обесцвечивания раст¬
вора. К горячему раствору прибавляют 200 мл воды Р
и 50 мг ксиленолового оранжевого индикаторной
смеси Р. Титруют 0,1 М раствором натрия эдетата
до желтого окрашивания.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 20,90 мг В|.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1927
ВОДА ВЫСОКООЧИЩЕННАЯ
Адиа уа/с/е ригШса1а
И(АТЕК, Н/6Н/.У Р1ЛУР1ЕО
НгО М.м. 18,02
ОПРЕДЕЛЕНИЕ
Вода высокоочищенная предназначена для при¬
готовления лекарственных средств, если необходима
вода повышенного биологического качества, кроме
тех случаев, в которых необходимо использование
только Воды для инъекций.
ПРОИЗВОДСТВО
Воду высокоочищенную получают из воды питье¬
вой, отвечающей всем требованиям, установленным
соответствующими регуляторными органами.
В настоящее время для получения воды высоко-
очищенной применяют такие методы, как, например,
двойной обратный осмос, используемый с другими
подходящими методами, такими как ультрафильтрация
и деионизация. Необходимо надлежащее содержание
и техническое обслуживание системы очищения воды.
С целью обеспечения необходимого качества
воды применяются валидированные методики, вну¬
трипроцессный контроль электрической проводимо¬
сти и регулярный мониторинг микробиологической
чистоты.
Воду высокоочищенную /л Ьи1к хранят и исполь¬
зуют в условиях, позволяющих избежать роста микро¬
организмов и проникновения каких-либо других за¬
грязнений.
Мониторинг микробиологической чистоты.
Во время производства и дальнейшего хранения со¬
ответствующим образом контролируют и отслежива¬
ют суммарное количество жизнеспособных аэробов.
Чтобы предупредить неблагоприятные тенденции,
устанавливают соответствующие предупреждающие
и нормативные пределы. Обычно соответствующий
нормативный предел равен 10 КОЕ в 100 мл при
определении методом мембранной фильтрации через
фильтр с порами не более 0,45 мкм, используя не ме¬
нее 200 мл воды высокоочищенной и агаризованную
среду Р2А. Инкубирование проводят при температуре
от 30 °С до 35 °С в течение не менее 5 дней.
Агаризованная среда Р2А
Экстракт дрожжевой
0,5 г
Протеозный пептон
0,5 г
Г идролизат казеина
0,5 г
Глюкоза
0,5 г
Крахмал
0,5 г
Дикалия гидрофосфат
0,3 г
Магния сульфат безводный
0,024 г
Натрия пируват
0,3 г
Агар
15,0 г
Вода очищенная
до 1000 мл
Доводят рН таким образом, чтобы после стери¬
лизации его значение было 7,2±0,2. Стерилизуют ав¬
токлавированием при температуре 121 °С в течение
15 мин.
Активация роста на агаризованной среде Р2А
- Приготовление тест-штаммов. Исполь¬
зуют стабильные стандартизованные суспензии
тест-штаммов или готовят их, как указано в таблице
1927.-1. Используется техника сохранения эталонного
банка культур микроорганизмов (системы эталонного
банка) таким образом, чтобы живые микроорганизмы,
использованные для инокуляции, были удалены не
более чем на 5 пересевов от оригинального контроль¬
ного банка культур микроорганизмов. Выращивают
каждый из бактериальных штаммов по отдельности,
как указано в таблице 1927.-1. Для приготовления ис¬
пытуемой суспензии используют забуференный раст¬
вор натрия хлорида и пептона рН 7,0 или фосфатный
буферный раствор рН 7,2. Приготовленные суспензии
Таблица 1927.-1
Активация роста на агаризованной среде Р2А
Микроорганизм
Приготовление испытуемого штамма
Активация роста
Рзеис/отопаз аегидтоза, такие как
Агаризованная среда на основе
Агаризованная среда Р2А
АТСС 9027
гидролизата казеина и соевых бобов
< 100 КОЕ
А1С1МВ 8626
или бульон на основе гидролизата
(30—35) °С
С/Р 82.118
казеина и соевых бобов
й 3 дней
/7ВРС13275
(30—35) °С
(18—24)ч
ВасШиз зиЫШз
Агаризованная среда на основе
Агаризованная среда Р2А
АТСС 6633
гидролизата казеина и соевых бобов
< 100 КОЕ
ЫС1МВ 8054
или бульон на основе гидролизата
(30—35) °С
С/Р 52.62
казеина и соевых бобов
< 3 дней
ЫВРС 3134
(30—35) °С
(18—24)ч
304
Гэсударственная фармакопея Республики Беларусь
используют в течение 2 ч или в течение 24 ч при хра¬
нении при температуре от 2 °С до 8 °С. В качестве
альтернативы приготовлению и последующему разве¬
дению свежей суспензии вегетативных клеток ВасШиз
зиЫШз для инокуляции может быть использован необ¬
ходимый объем стабильной суспензии спор. Стабиль¬
ная суспензия спор хранится при температуре от 2 °С
до 8 °С в течение валидированного периода времени.
-Активация роста. Проводят испытание каж¬
дой серии готовой среды и каждой серии среды, при¬
готовленной из сухой среды или из указанных ингре¬
диентов. Пластинки с агаризованной средой Р2А ино-
кулируют по отдельности небольшим количеством (не
более 100 КОЕ) микроорганизмов, указанных в табли¬
це 1927.-1, и инкубируют в условиях, указанных в та¬
блице. Обнаруживаемый рост не должен отличаться
от значения, найденного для стандартизованного ино¬
кулята, более чем в 2 раза. Рост микроорганизмов для
свежеприготовленного инокулята должен быть срав¬
ним с ростом, который был получен с использованием
ранее испытанной и утвержденной серией среды.
Содержание общего органического углерода
(2.2.44). Не более 0,5 мг/л.
Электропроводность. Проводят в режиме о/7-
Нпе или т-Нпе в следующих условиях.
ОБОРУДОВАНИЕ
Измерительная камера:
-электроды должны быть из соответствующего
материала, например из нержавеющей стали;
- постоянная камеры: постоянная камеры обыч¬
но сертифицируется производителем и затем про¬
веряется через установленные интервалы времени
с использованием сертифицированного стандартно¬
го раствора, имеющего электропроводность менее
1500 мкСм-см'1, или в сравнении с камерой с серти¬
фицированным значением постоянной камеры; по¬
стоянная камеры считается подтвержденной, если
найденное значение находится в пределах 2 % от
сертифицированного значения; при несоответствии
необходимо проводить повторную градуировку.
Кондуктометр: точность 0,1 мкСм-см1 или луч¬
ше в самом низком диапазоне.
Система градуировки (измерительная камера и
кондуктометр):
- относительно одного или более соответствую¬
щих сертифицированных стандартных растворов;
- точность: в пределах 3 % от измеренной элек¬
тропроводности плюс 0,1 мкСм-см-1.
Градуировка кондуктометра: проводят градуи¬
ровку для каждого используемого диапазона измере¬
ний после отсоединения камеры и с использованием
сертифицированных резисторов точности или анало¬
гичных приспособлений с неопределенностью не бо¬
лее 0,1 % от сертифицированного значения.
Если измерительная камера т-Нпе не может
быть демонтирована, система градуировки может
быть проведена относительно калиброванного изме¬
рительного прибора с измерительной камерой, рас¬
положенной близко к калибруемой камере в водном
потоке.
Измерение температуры: отклонение ±2 °С.
МЕТОДИКА
Первая стадия
1. Измеряют электропроводность без темпе¬
ратурной компенсации, одновременно регистрируя
температуру. Измерения с температурной компен¬
сацией могут проводиться после соответствующей
валидации.
2. Используя таблицу 1927.-2, находят самое
близкое табличное значение температуры к изме¬
ренной, но не выше измеренной температуры. Соот¬
ветствующее значение электропроводности является
пределом для данной температуры.
Таблица 1927.-2
Первая стадия
Требуемые значения электропроводности при опре¬
деленной температуре (измерение электропровод¬
ности без температурной компенсации)
Температура (°С)
Электропроводность
(мкСм*см~1)
0
0,6
5
0,8
10
0,9
15
1,0
20
1,1
25
1,3
30
1,4
35
1,5
40
1,7
45
1,8
50
1,9
55
2,1
60
2,2
65
2,4
70
2,5
75
2,7
80
2,7
85
2,7
90
2,7
95
2,9
100
3,1
3. Если измеренная электропроводность не пре¬
вышает значение, указанное в таблице 1927.-2, то
испытуемая вода удовлетворяет требованиям к элек¬
тропроводности. Если значения электропроводности
выше табличного, переходят ко второй стадии.
Вторая стадия
4. Переносят определенное количество испытуе¬
мой воды (100 мл или более) в подходящий контейнер
и перемешивают испытуемый образец. Корректируют
температуру, если необходимо, и, поддерживая ее на
уровне (25±1)°С, начинают энергично взбалтывать
испытуемый образец, периодически измеряя электро¬
проводность. Записывают значение электропровод¬
ности, когда изменение электропроводности в тече¬
ние 5 мин (из-за захвата углерода диоксида атмосфе¬
ры) составит менее 0,1 мкСм-см1.
5. Если электропроводность не превышает
2,1 мкСм-см1, то испытуемая вода удовлетворяет требо¬
ваниям к электропроводности. Если электропроводность
превышает 2,1 мкСм-см1, переходят к третьей стадии.
Третья стадия
6. Проводят данное испытание в течение при¬
близительно 5 мин с момента определения электро¬
проводности, как указано в пункте 5 стадии 2, поддер¬
живая температуру испытуемого образца (25±1) °С. К
испытуемому образцу прибавляют свежеприготовлен¬
ный насыщенный раствор калия хлорида Р (0,3 мл на
100 мл испытуемого образца) и определяют рН (2.2.3)
с точностью до 0,1.
Вода для инъекций
305
7. Используя таблицу 1927.-3, определяют пре¬
дельное значение электропроводности при измерен¬
ном, как указано в пункте 6, значении рН. Если из¬
меренная электропроводность, как указано в пункте
4 стадии 2, не превышает требуемое значение элек¬
тропроводности для определенного рН, то испыту¬
емая вода выдерживает испытание на электропро¬
водность. Если же измеренная электропроводность
превышает это значение или значение рН выходит за
пределы 5,0—7,0, то испытуемая вода не выдержива¬
ет испытание на электропроводность.
Таблица 1927.-3
Третья стадия
Требуемые значения электропроводности при опреде¬
ленном значении рН для образцов в уравновешенном
состоянии (температура и атмосферное давление)
рн
Электропроводность
(МКСМ’СМ*1)
5,0
4,7
5,1
4,1
5,2
3,6
5,3
3,3
5,4
3,0
5,5
2,8
5,6
2,6
5,7
2,5
5,8
2,4
5,9
2,4
6,0
2,4
6,1
2,4
6,2
2,5
6,3
2,4
6,4
2,3
6,5
2,2
6,6
2,1
6,7
2,6
6,8
3,1
6,9
3,8
7,0
4,6
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная жидкость.
ИСПЫТАНИЯ
Нитраты. Не более 0,00002 % (0,2 ррт). 5 мл
испытуемого образца помещают в пробирку, погру¬
женную в ледяную баню, прибавляют 0,4 мл раствора
100 г/л калия хлорида Р, 0,1 мл раствора дифенила¬
мина Р и по каплям при перемешивании 5 мл кислоты
серной, свободной от азота, Р. Пробирку переносят в
водяную баню, нагретую до температуры 50 °С. Через
15 мин голубое окрашивание испытуемого раствора
должно быть не интенсивнее окрашивания эталона,
приготовленного параллельно с использованием сме¬
си из 4,5 мл воды, свободной от нитратов, Р и 0,5 мл
эталонного раствора нитрата (2 ррт NО^ Р.
Алюминий (2.4.17). 0,000001 % (10 ррЬ), если
вода высокоочищенная предназначена для производ¬
ства растворов для диализа.
Испытуемый раствор. К 400 мл испытуемой воды
прибавляют 10 мл ацетатного буферного раствора
рН 6,0 Р и 100 мл воды дистиллированной Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферного
раствора рН 6,0 Р и 98 мл воды дистиллированной Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды дистиллированной Р.
Бактериальные эндотоксины (2.6.14). Менее
0,25 МЕ/мл.
#Пирогенность. На испытуемой воде высоко-
очищенной готовят раствор 9 г/л натрия хлорида Р.
Тест-доза 10 мл на 1 кг массы животного.
Испытание «Пирогенность» проводится в каче¬
стве альтернативного испытанию «Бактериальные
эндотоксины».
МАРКИРОВКА
При необходимости указывают:
-вода высокоочищенная пригодна для произ¬
водства растворов для диализа.
07/2016:0169
ВОДА ДЛЯ ИНЪЕКЦИЙ
Лдиа ас/ /л/ес/аЬ//е
И(АТЕК РОК ШЕСТЮЫ8
н20 М.м. 18,02
ОПРЕДЕЛЕНИЕ
Вода для инъекций предназначена для изготов¬
ления лекарственных средств для парентерального
применения, когда вода для инъекций используется
в качестве носителя (вода для инъекций /л Ьи1к), или
для растворения или разведения субстанций или ле¬
карственных средств для парентерального примене¬
ния (вода для инъекций стерильная).
Вода для инъекций т Ьи1к
ПРОИЗВОДСТВО
Воду для инъекций т Ьи1к получают из воды пи¬
тьевой, отвечающей всем требованиям, установлен¬
ным соответствующими регуляторными органами, или
из воды очищенной методом дистилляции на оборудо¬
вании, контактирующая с водой поверхность которого
изготовлена из нейтрального стекла, кварца или под¬
ходящего металла. Оборудование должно быть обе¬
спечено эффективным приспособлением для предот¬
вращения захвата капель. Необходимы надлежащее
содержание и техническое обслуживание оборудова¬
ния. Первую порцию воды, полученную в начале рабо¬
ты, отбрасывают, затем дистиллят собирают.
С целью обеспечения необходимого качества
воды применяются валидированные методики, внутри-
процессный контроль электрической проводимости и
регулярный мониторинг микробиологической чистоты.
Воду для инъекций /л Ьи1к хранят и используют в
условиях, позволяющих избежать роста микроорганиз¬
мов и проникновения каких-либо других загрязнений.
Мониторинг микробиологической чистоты. В
течение производства и дальнейшего хранения со¬
ответствующим образом контролируют и отслежива¬
ют суммарное количество жизнеспособных аэробов.
Чтобы предупредить неблагоприятные тенденции,
306
Гэсударственная фармакопея Республики Беларусь
устанавливают соответствующие предупреждающие
и нормативные пределы. Обычно соответствующий
нормативный предел равен 10 КОЕ в 100 мл при
определении методом мембранной фильтрации че¬
рез фильтр с порами не более 0,45 мкм, используя
не менее 200 мл воды для инъекций /л Ьи1к и агари-
зованную среду Р2А. Инкубирование проводят при
температуре от 30 °С до 35 °С в течение не менее 5
дней. При производстве воды для инъекций т Ьи1к,
используемой в дальнейшем для асептического из¬
готовления лекарственных средств, может возникнуть
необходимость установить более жесткие предупреж¬
дающие пределы.
Агаризованная среда Р2А
Экстракт дрожжевой
0,5 г
Протеозный пептон
0,5 г
Гидролизат казеина
0,5 г
Глюкоза
0,5 г
Крахмал
0,5 г
Дикалия гидрофосфат
0,3 г
Магния сульфат безводный
0,024 г
Натрия пируват
0,3 г
Агар
15,0 г
Вода очищенная
до 1000 мл
Доводят рН таким образом, чтобы после стери¬
лизации его значение было 7,2±0,2. Стерилизуют ав¬
токлавированием при температуре 121 °С в течение
15 мин.
Активация роста на агаризованной среде Р2А
- Приготовление тест-штаммов. Исполь¬
зуют стабильные стандартизованные суспензии
тест-штаммов или готовят их, как указано в таблице
0169.-1. Используется техника сохранения эталонного
банка культур микроорганизмов (системы эталонного
банка) таким образом, чтобы живые микроорганизмы,
использованные для инокуляции, были удалены не
более чем на 5 пересевов от оригинального контроль¬
ного банка культур микроорганизмов. Выращивают
каждый из бактериальных штаммов по отдельности,
как указано в таблице 0169-1. Для приготовления ис¬
пытуемой суспензии используют забуференный раст¬
вор натрия хлорида и пептона рН 7,0 или фосфатный
буферный раствор рН 7,2. Приготовленные суспензии
используют в течение 2 ч или в течение 24 ч при хра¬
нении при температуре от 2 °С до 8 °С. В качестве
альтернативы приготовлению и последующему разве¬
дению свежей суспензии вегетативных клеток ВасШиз
зиЫШз для инокуляции может быть использован необ¬
ходимый объем стабильной суспензии спор. Стабиль¬
ная суспензия спор хранится при температуре от 2 °С
до 8 °С в течение валидированного периода времени.
- Активация роста. Проводят испытание каж¬
дой серии готовой среды и каждой серии среды, при¬
готовленной из сухой среды или из указанных ингре¬
диентов. Пластинки с агаризованной средой Р2А ино-
кулируют по отдельности небольшим количеством (не
более 100 КОЕ) микроорганизмов, указанных в табли¬
це 0169.-1, и инкубируют в условиях, указанных в та¬
блице. Обнаруживаемый рост не должен отличаться
от значения, найденного для стандартизованного ино¬
кулята, более чем в 2 раза. Рост микроорганизмов для
свежеприготовленного инокулята должен быть срав¬
ним с ростом, который был получен с использованием
ранее испытанной и утвержденной серией среды.
Содержание общего органического углерода
(2.2.44). Не более 0,5 мг/л.
Электропроводность. Проводят в режиме о/7-
Гте или т-Нпе в следующих условиях.
ОБОРУДОВАНИЕ
Измерительная камера:
- электроды должны быть из соответствующего
материала, например из нержавеющей стали;
- постоянная камеры: постоянная камеры обыч¬
но сертифицируется производителем и затем про¬
веряется через установленные интервалы времени
с использованием сертифицированного стандартно¬
го раствора, имеющего электропроводность менее
1500 мкСм см1, или в сравнении с камерой с серти¬
фицированным значением постоянной камеры; по¬
стоянная камеры считается подтвержденной, если
найденное значение находится в пределах 2 % от
сертифицированного значения; при несоответствии
необходимо проводить повторную градуировку.
Кондуктометр: точность 0,1 мкСмсм-1 или луч¬
ше в самом низком диапазоне.
Система градуировки (измерительная камера и
кондуктометр) :
- относительно одного или более соответствую¬
щих стандартных растворов;
- точность: в пределах 3 % от измеренной элек¬
тропроводности плюс 0,1 мкСмсм-1.
Градуировка кондуктометра: проводят градуи¬
ровку для каждого используемого диапазона измере¬
ний после отсоединения камеры и с использованием
сертифицированных резисторов точности или анало¬
гичных приспособлений с неопределенностью не бо¬
лее 0,1 % от сертифицированного значения.
Если измерительная камера т-Нле не может быть
демонтирована, система градуировки может быть про-
Таблица 0169.-1
Активация роста на агаризованной среде Н2А
Микроорганизм
Приготовление испытуемого штамма
Активация роста
Рзеидотопаз аегидтоза, такие как
Агаризованная среда на основе
Агаризованная среда Р2А
АТСС 9027
гидролизата казеина и соевых бобов
<100 КОЕ
Л/СМВ 8626
или бульон на основе гидролизата
(30—35) °С
С/Р 82.118
казеина и соевых бобов
< 3 дней
Л/ВРС 13275
(30—35) °С
(18—24)ч
ВасШиз зиЫШз
Агаризованная среда на основе
Агаризованная среда Р2А
АТСС 6633
гидролизата казеина и соевых бобов
< 100 КОЕ
ЫС1МВ 8054
или бульон на основе гидролизата
(30—35) °С
С/Р 52.62
казеина и соевых бобов
< 3 дней
Л/ВРС 3134
(30—35) °С
(18—24) ч
Вода для инъекций
307
ведена относительно калиброванного измерительного
прибора с измерительной камерой, расположенной
близко к калибруемой камере в водном потоке.
Измерение температуры: отклонение ±2 °С.
МЕТОДИКА
Первая стадия
1. Измеряют электропроводность без температур¬
ной компенсации, одновременно регистрируя темпера¬
туру. Измерения с температурной компенсацией могут
проводиться после соответствующей валидации.
2. Используя таблицу 0169.-2, находят самое
близкое табличное значение температуры к изме¬
ренной, но не выше измеренной температуры. Соот¬
ветствующее значение электропроводности является
пределом для данной температуры.
Таблица 0169.-2
Первая стадия
Требуемые значения электропроводности при опре¬
деленной температуре (измерение электропровод¬
ности без температурной компенсации)
Температура (°С)
Электропроводность
(мкСм-см'1)
0
0,6
5
0,8
10
0,9
15
1,0
20
1,1
25
1,3
30
1,4
35
1,5
40
1,7
45
1,8
50
1,9
55
2,1
60
2,2
65
2,4
70
2,5
75
2,7
80
2,7
85
2,7
90
2,7
95
2,9
100
3,1
3. Если измеренная электропроводность не
превышает значение, указанное в таблице 0169.-2,
то испытуемая вода удовлетворяет требованиям к
электропроводности. Если значения электропровод¬
ности выше табличного, переходят ко второй стадии.
Вторая стадия
4. Переносят определенное количество ис¬
пытуемой воды (100 мл или более) в подходящий
контейнер и перемешивают испытуемый образец.
Корректируют температуру, если необходимо, и, под¬
держивая ее на уровне (25±1) °С, начинают энергич¬
но взбалтывать испытуемый образец, периодически
измеряя электропроводность. Записывают значение
электропроводности, когда изменение электропро¬
водности в течение 5 мин (из-за повышения уровня
углерода диоксида в атмосфере) составит менее
0,1 мкСмсм*1.
5. Если электропроводность не превышает
2,1 мкСм-см1, то испытуемая вода удовлетворяет
требованиям к электропроводности. Если электро¬
проводность превышает 2,1 мкСм см1, переходят к
третьей стадии.
Третья стадия
6. Проводят данное испытание в течение при¬
близительно 5 мин с момента определения электро¬
проводности, как указано в пункте 5 стадии 2, поддер¬
живая температуру испытуемого образца (25±1) °С. К
испытуемому образцу прибавляют свежеприготовлен¬
ный насыщенный раствор калия хлорида Р (0,3 мл на
100 мл испытуемого образца) и определяют рН (2.2.3)
с точностью до 0,1.
7. С использованием таблицы 0169.-3 определя¬
ют предельное значение электропроводности при из¬
меренном, как указано в пункте 6, значении рН. Если
измеренная электропроводность, как указано в пункте
4 стадии 2, не превышает требуемое значение элек¬
тропроводности для определенного рН, то испыту¬
емая вода выдерживает испытание на электропро¬
водность. Если же измеренная электропроводность
превышает это значение или значение рН выходит за
пределы 5,0-7,0, то испытуемая вода не выдерживает
испытание на электропроводность.
Таблица 0169.-3
Третья стадия
Требуемые значения электропроводности при опреде¬
ленном значении рН для образцов в уравновешенном
состоянии (температура и атмосферное давление)
рН
Электропроводность
(мкСм-см*1)
5,0
4,7
5,1
4,1
5,2
3,6
5,3
3,3
5,4
3,0
5,5
2,8
5,6
2,6
5,7
2,5
5,8
2,4
5,9
2,4
6,0
2,4
6,1
2,4
6,2
2,5
6,3
2,4
6,4
2,3
6,5
2,2
6,6
2,1
6,7
2,6
6,8
3,1
6,9
3,8
7,0
4,6
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная, бесцветная жидкость.
ИСПЫТАНИЯ
Нитраты. Не более 0,00002 % (0,2 ррпп). 5 мл
испытуемого образца помещают в пробирку, погру¬
женную в ледяную баню, прибавляют 0,4 мл раст¬
вора 100 г/л калия хлорида Р, 0,1 мл раствора дифе¬
ниламина Р и по каплям, при перемешивании, 5 мл
кислоты серной, свободной от азота, Р. Пробирку
переносят в водяную баню, нагретую до температу¬
ры 50 °С. Через 15 мин голубое окрашивание испы¬
туемого раствора должно быть не интенсивнее окра-
308
Гэсударственная фармакопея Республики Беларусь
шивания эталона, приготовленного параллельно с
использованием смеси из 4,5 мл воды, свободной от
нитратов, Р и 0,5 мл эталонного раствора нитра¬
та (2 ррт NО^ Р.
Алюминий (2.4.17). 0,000001 % (10 ррЬ), если
вода для инъекций предназначена для производства
растворов для диализа.
Испытуемый раствор. К 400 мл испытуемого об¬
разца прибавляют 10 мл ацетатного буферного раст¬
вора рН 6,0 Р и 100 мл воды дистиллированной Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферного
раствора рН 6,0 Р и 98 мл воды дистиллированной Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды дистиллированной Р.
Бактериальные эндотоксины (2.6.14). Менее
0,25 МЕ/мл.
#Пирогенность. На испытуемой воде для инъ¬
екций готовят раствор 9 г/л натрия хлорида Р. Тест-
доза 10 мл на 1 кг массы животного.
Тест «Пирогенность» проводится в качестве аль¬
тернативного тесту «Бактериальные эндотоксины».
Вода для инъекций стерильная
ОПРЕДЕЛЕНИЕ
Вода для инъекций стерильная — это вода для
инъекций т Ьи1к, расфасованная в подходящие кон¬
тейнеры, укупоренная и стерилизованная нагревани¬
ем в условиях, которые гарантируют, что полученный
продукт выдерживает испытание на бактериальные
эндотоксины. Вода для инъекций стерильная не
должна содержать никаких дополнительных веществ.
Вода для инъекций стерильная должна быть про¬
зрачной и бесцветной при просматривании в подходя¬
щих условиях видимости.
Каждый контейнер должен вмещать достаточное
количество воды для инъекций, чтобы обеспечить
возможность извлечения ее номинального объема.
ИСПЫТАНИЯ
Кислотность или щелочность. К 20 мл испыту¬
емого образца прибавляют 0,05 мл раствора фено¬
лового красного Р. Если раствор окрашивается в жел¬
тый цвет, то при прибавлении не более 0,1 мл 0,01 М
раствора натрия гидроксида должно появиться
красное окрашивание. Если раствор окрашивается в
красный цвет, то при прибавлении не более 0,15 мл
0,01 М раствора кислоты хлористоводородной
должно появиться желтое окрашивание.
Электропроводность. Не более 25 мкСм см1
для контейнеров с номинальным объемом 10 мл или
менее; не более 5 мкСм-см1 для контейнеров с номи¬
нальным объемом более 10 мл.
Используют оборудование и методику градуиров¬
ки, как указано в разделе «Вода для инъекций т Ьи1к».
Определение проводят при температуре испытуемого
образца (25±1) °С.
Восстанавливающие вещества. Для контейне¬
ров с номинальным объемом менее 50 мл: к 100 мл
испытуемого образца прибавляют 10 мл кислоты
серной разведенной Р, доводят до кипения, прибав¬
ляют 0,4 мл 0,02 М раствора калия перманганата и
кипятят в течение 5 мин. Раствор должен сохранять
слабо-розовое окрашивание.
Для контейнеров с номинальным объемом
50 мл или более: к 100 мл испытуемого образца
прибавляют 10 мл кислоты серной разведенной Р,
доводят до кипения, прибавляют 0,2 мл 0,02 М
раствора калия перманганата и кипятят в течение
5 мин. Раствор должен сохранять слабо-розовое
окрашивание.
Хлориды (2.4.4). Не более 0,00005 % (0,5 ррт)
для воды для инъекций в контейнерах с номиналь¬
ным объемом 100 мл или менее. 15 мл испытуемого
образца должны выдерживать испытание на хлори¬
ды. Эталон готовят с использованием смеси из 1,5 мл
эталонного раствора хлорида (5 ррт С!) Р и 13,5 мл
воды Р. Опалесценцию полученных растворов срав¬
нивают по вертикальной оси пробирок.
Для контейнеров с номинальным объемом более
100 мл используют следующее испытание: к 10 мл ис¬
пытуемого образца прибавляют 1 мл кислоты азот¬
ной разведенной Р и 0,2 мл раствора серебра нитра¬
та Р2. В течение 15 мин не должно быть видимых из¬
менений раствора.
Нитраты. Не более 0,00002 % (0,2 ррт). 5 мл
испытуемого образца помещают в пробирку, погру¬
женную в ледяную баню, прибавляют 0,4 мл раст¬
вора 100 г/л калия хлорида Р, 0,1 мл раствора дифе¬
ниламина Р и по каплям, при перемешивании, 5 мл
кислоты серной, свободной от азота, Р. Пробирку
помещают в водяную баню, нагретую до температу¬
ры 50 °С. Через 15 мин голубое окрашивание испы¬
туемого раствора должно быть не интенсивнее окра¬
шивания эталона, приготовленного параллельно с
использованием смеси из 4,5 мл воды, свободной от
нитратов, Р и 0,5 мл эталонного раствора нитра¬
та (2 ррт NО3) Р.
Сульфаты. К 10 мл испытуемого образца при¬
бавляют 0,1 мл кислоты хлористоводородной раз¬
веденной Р и 0,1 мл раствора бария хлорида Р1.
В течение 1 ч не должно быть видимых изменений
раствора.
Алюминий (2.4.17). 0,000001 % (10 мкг/л), если
вода для инъекций предназначена для производства
растворов для диализа.
Испытуемый раствор. К 400 мл испытуемого
образца прибавляют 10 мл ацетатного буферного
раствора рН 6,0 Р и 100мл воды дистиллирован¬
ной Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды дистиллирован¬
ной Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды дистиллированной Р.
Соли аммония. Для воды для инъекций в кон¬
тейнерах с номинальным объемом менее 50 мл: не
более 0,00006 % (0,6 ррт); для воды для инъекций в
контейнерах с номинальным объемом 50 мл и более:
не более 0,00002 % (0,2 ррт).
Для воды для инъекций в контейнерах с номи¬
нальным объемом менее 50 мл: к 20 мл испытуемого
образца прибавляют 1 мл раствора калия тетра-
йодомеркурата щелочного Р. Через 5 мин исследуют
раствор вдоль вертикальной оси пробирки. Окраска
полученного раствора должна быть не интенсивнее
окраски эталона, приготовленного параллельно, при¬
бавляя 1 мл раствора калия тетрайодомеркурата
щелочного Р к смеси из 4 мл эталонного раствора
аммония (3 ррт N4^ Р и 16 мл воды, свободной от
аммиака, Р.
Вода очищенная
309
Для воды для инъекций в контейнерах с номи¬
нальным объемом 50 мл и более: к 20 мл испытуемо¬
го образца прибавляют 1 мл раствора калия тетра-
йодомеркурата щелочного Р. Через 5 мин исследуют
раствор вдоль вертикальной оси пробирки. Окраска
полученного раствора должна быть не интенсивнее
окраски эталона, приготовленного параллельно, при¬
бавляя 1 мл раствора калия тетрайодомеркурата
щелочного Р к смеси из 4 мл эталонного раствора
аммония (1 ррт N4^ Р и 16 мл воды, свободной от
аммиака, Р.
Кальций и магний. К 100 мл испытуемого об¬
разца прибавляют 2 мл аммиачного буферного раст¬
вора рН 10,0 Р, 50 мг протравного черного 11 инди¬
каторной смеси Р и 0,5 мл 0,01 М раствора натрия
эдетата. Появляется синее окрашивание.
Остаток после выпаривания. Для воды для
инъекций в контейнерах с номинальным объемом
10 мл или менее: не более 4 мг (0,004 %); для воды
для инъекций в контейнерах с номинальным объемом
более 10 мл: не более 3 мг (0,003 %). 100 мл испыту¬
емого образца выпаривают досуха на водяной бане и
сушат при температуре от 100 °С до 105 °С.
Механические включения: невидимые части¬
цы (2.9.19). Вода для инъекций должна выдерживать
требования испытаний А или В, где применимо.
Стерильность (2.6.1). Вода для инъекций долж¬
на выдерживать требования испытания на стериль¬
ность.
Бактериальные эндотоксины (2.6.14). Менее
0,25 МЕ/мл.
#Пирогенность. На испытуемой воде для инъ¬
екций готовят раствор 9 г/л натрия хлорида Р. Тест-
доза 10 мл на 1 кг массы животного.
Испытание «Пирогенность» проводится в каче¬
стве альтернативного испытанию «Бактериальные
эндотоксины».
07/2016:0008
ВОДА ОЧИЩЕННАЯ
Адиа ригШса1а
И(АТЕК, РУ/?//=/ЕО
Н20 М.м. 18,02
ОПРЕДЕЛЕНИЕ
Вода, предназначенная для приготовления ле¬
карственных средств, кроме тех, которые должны
быть стерильными и апирогенными, если нет других
указаний и разрешений компетентного уполномочен¬
ного органа.
Вода очищенная т Ьи\к
ПРОИЗВОДСТВО
Воду очищенную получают из воды питьевой,
отвечающей всем требованиям, установленным со¬
ответствующими регуляторными органами, методами
дистилляции, ионного обмена, обратного осмоса или
каким-нибудь другим подходящим способом.
Воду очищенную т Ьи1к хранят и используют в ус¬
ловиях, позволяющих избежать роста микроорганиз¬
мов и проникновения каких-либо других загрязнений.
Мониторинг микробиологической чистоты.
В течение производства и дальнейшего хранения
соответствующим образом контролируют и отслежи¬
вают суммарное количество жизнеспособных аэро¬
бов. Чтобы предупредить неблагоприятные тенден¬
ции, устанавливают соответствующие предупреж¬
дающие и нормативные пределы. Обычно соответ¬
ствующий нормативный предел равен 100 КОЕ/мл
при определении методом мембранной фильтрации
через фильтр с порами не более 0,45 мкм, исполь¬
зуя агаризованную среду Р2А. Инкубирование про¬
водят при температуре от 30 °С до 35 °С в течение
не менее 5 дней. Размер испытуемого образца вы¬
бирают исходя из ожидаемого результата.
Агаризованная среда Р2А
Экстракт дрожжевой
0,5 г
Протеозный пептон
0,5 г
Гидролизат казеина
0,5 г
Глюкоза
0,5 г
Крахмал
0,5 г
Дикалия гидрофосфат
0,3 г
Магния сульфат безводный
0,024 г
Натрия пируват
0,3 г
Агар
15,0 г
Вода очищенная
до 1000 мл
Доводят рН таким образом, чтобы после стери¬
лизации его значение было 7,2±0,2. Стерилизуют ав¬
токлавированием при температуре 121 °С в течение
15 мин.
Активация роста на агаризованной среде Р2А
- Приготовление тест-штаммов. Исполь¬
зуют стабильные стандартизованные суспензии
тест-штаммов или готовят их, как указано в таблице
0008.-1. Используется техника сохранения эталонного
банка культур микроорганизмов (системы эталонного
банка) таким образом, чтобы живые микроорганизмы,
Активация роста на агаризованной среде Р2А
Таблица 0008.-1
Микроорганизм
Приготовление испытуемого штамма
Активация роста
Рзеидотопаз аегидтоза, такие как
Агаризованная среда на основе
Агаризованная среда Р2А
АТСС 9027 .
гидролизата казеина и соевых бобов или
< 100 КОЕ
А1С1МВ 8626
бульон на основе гидролизата казеина и
(30—35) °С
С/Р 82.118
соевых бобов
^ 3 дней
Л/ВРС 13275
(30—35) °С
(18—24)ч
ВасШиз зиЫШз
Агаризованная среда на основе гидроли¬
Агаризованная среда Р2А
АТСС 6633
зата казеина и соевых бобов или бульон
* 100 КОЕ
Л/С/МВ 8054
на основе гидролизата казеина и соевых
(30—35) °С
С/Р 52.62
бобов
^ 3 дней
Л/БРС 3134
(30—35) °С
(18—24)ч
310
Гэсударственная фармакопея Республики Беларусь
использованные для инокуляции, были удалены не
более чем на 5 пересевов от оригинального контроль¬
ного банка культур микроорганизмов. Выращивают
каждый из бактериальных штаммов по отдельности,
как указано в таблице 0008.-1. Для приготовления ис¬
пытуемой суспензии используют забуференный раст¬
вор натрия хлорида и пептона рН 7,0 или фосфатный
буферный раствор рН 7,2. Приготовленные суспензии
используют в течение 2 ч или в течение 24 ч при хра¬
нении при температуре от 2 °С до 8 °С. В качестве
альтернативы приготовлению и последующему разве¬
дению свежей суспензии вегетативных клеток ВасШиз
зиЫШз для инокуляции может быть использован необ¬
ходимый объем стабильной суспензии спор. Стабиль¬
ная суспензия спор хранится при температуре от 2 °С
до 8 °С в течение валидированного периода времени.
- Активация роста. Проводят испытание каж¬
дой серии готовой среды и каждой серии среды, при¬
готовленной из сухой среды или из указанных ингре¬
диентов. Пластинки с агаризованной средой Р2А ино-
кулируют по отдельности небольшим количеством (не
более 100 КОЕ) микроорганизмов, указанных в табли¬
це 0008.-1, и инкубируют в условиях, указанных в та¬
блице. Обнаруживаемый рост не должен отличаться
от значения, найденного для стандартизованного ино¬
кулята, более чем в 2 раза. Рост микроорганизмов для
свежеприготовленного инокулята должен быть срав¬
ним с ростом, который был получен с использованием
ранее испытанной и утвержденной серией среды.
Содержание общего органического углерода
или восстанавливающие вещества. Определяют
содержание общего органического углерода (2.2.44) с
предельным содержанием 0,5 мг/л или проводят ис¬
пытание на восстанавливающие вещества: к 100 мл
испытуемого образца прибавляют 10 мл кислоты
серной разведенной Р, 0,1 мл 0,02 М раствора калия
перманганата и кипятят в течение 5 мин. Раствор
должен оставаться слабо-розовым.
Электропроводность. Проводят в режиме о/Т-
Нпе или т-Нпе в следующих условиях.
ОБОРУДОВАНИЕ
Измерительная камера:
- электроды должны быть из соответствующего
материала, например из нержавеющей стали;
- постоянная камеры: постоянная камеры обыч¬
но сертифицируется производителем и затем прове¬
ряется через установленные интервалы времени с
использованием сертифицированного стандартного
раствора, имеющего электропроводность менее 1500
мкСм см'1, или в сравнении с камерой с сертифици¬
рованным значением постоянной камеры; постоянная
камеры считается подтвержденной, если найденное
значение находится в пределах 2 % от сертифици¬
рованного значения; при несоответствии необходимо
проводить повторную градуировку.
Кондуктометр: точность 0,1 мкСм см'1 или луч¬
ше в самом низком диапазоне.
Система градуировки (измерительная камера и
кондуктометр):
- относительно одного или более соответствую¬
щих сертифицированных стандартных растворов;
- точность: в пределах 3 % от измеренной элек¬
тропроводности плюс 0,1 мкСм-см-1.
Градуировка кондуктометра: проводят градуи¬
ровку для каждого используемого диапазона измере¬
ний после отсоединения камеры и с использованием
сертифицированных резисторов точности или анало¬
гичных приспособлений с неопределенностью не бо¬
лее 0,1 % от сертифицированного значения.
Если измерительная камера ю-Нпе не может
быть демонтирована, система градуировки может
быть проведена относительно калиброванного изме¬
рительного прибора с измерительной камерой, рас¬
положенной близко к калибруемой камере в водном
потоке.
Измерение температуры: отклонение ±2 °С.
МЕТОДИКА
Измеряют электропроводность без температур¬
ной компенсации, одновременно регистрируя темпе¬
ратуру. Измерения с температурной компенсацией мо¬
гут проводиться после соответствующей валидации.
Испытуемая вода удовлетворяет требованиям,
если измеренная электропроводность при измерен¬
ной температуре не превышает значение, указанное
в таблице 0008.-2.
Таблица 0008.-2
Требуемые значения электропроводности
при определенной температуре
Температура (°С)
Электропроводность
(мкСм-см'1)
0
2,4
10
3,6
20
4,3
25
5,1
30
5,4
40
6,5
50
7,1
60
8,1
70
9,1
75
9,7
80
9,7
90
9,7
100
10,2
Если значение температуры не указано в табли¬
це 0008.-2, то рассчитывают максимально возможную
электропроводность методом интерполяции между
следующим наименьшим и следующим наибольшим
табличными значениями.
Тяжелые металлы. Если вода очищенная т Ьи1к
выдерживает испытание «Электропроводность», как
указано в статье Вода для инъекций (0169) (т Ьи1к), то
нет необходимости проводить испытание «Тяжелые
металлы», как указано ниже.
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная, бесцветная жидкость.
ИСПЫТАНИЯ
Нитраты. Не более 0,00002 % (0,2 ррт). 5 мл
испытуемого образца помещают в пробирку, погру¬
женную в ледяную баню, прибавляют 0,4 мл раствора
100 г/л калия хлорида Р, 0,1 мл раствора дифенила¬
мина Р и по каплям, при перемешивании, 5 мл кисло¬
ты серной, свободной от азота, Р Пробирку помеща¬
ют в водяную баню, нагретую до температуры 50 °С.
Через 15 мин голубое окрашивание испытуемого раст¬
вора должно быть не интенсивнее окрашивания эта¬
лона, приготовленного параллельно с использовани¬
ем смеси из 4,5 мл воды, свободной от нитратов, Р и
0,5 мл эталонного раствора нитрата (2 ррт NО^ Р.
Водорода пероксида 3 % раствор
311
Алюминий (2.4.17). 0,000001 % (10 ррЬ), если
вода очищенная предназначена для производства
растворов для диализа.
Испытуемый раствор. К 400 мл испытуемой
воды прибавляют 10 мл ацетатного буферного
раствора рН 6,0 Р и 100мл воды дистиллирован¬
ной Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 рртА1) Р, 10 мл ацетатного буферного
раствора рН 6,0 Р и 98 мл воды дистиллированной Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды дистиллированной Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,00001 % (0,1 ррт). К 200 мл испытуемого образца
прибавляют 0,15 мл 0,1 М раствора кислоты азот¬
ной и нагревают в стеклянной выпарительной чашке
на водяной бане до объема 20 мл. 12 мл полученного
концентрированного раствора должны выдерживать
испытание на тяжелые металлы. Эталон готовят с ис¬
пользованием 10 мл эталонного раствора свинца
(1 ррт РЬ) Р, прибавляя к нему 0,075 мл 0,1 М раст¬
вора кислоты азотной. Контрольный раствор гото¬
вят, добавляя к нему 0,075 мл 0,1 М раствора кисло¬
ты азотной.
Бактериальные эндотоксины (2.6.14). Менее
0,25 МЕ/мл, если вода очищенная предназначена для
производства растворов для диализа без дальнейшей
процедуры удаления бактериальных эндотоксинов.
МАРКИРОВКА
При необходимости указывают:
-вода очищенная пригодна для производства
растворов для диализа.
Вода очищенная в контейнерах
ОПРЕДЕЛЕНИЕ
Вода очищенная в контейнерах — это вода очи¬
щенная т Ьи1к, расфасованная в подходящие контей¬
неры, которые хранятся в условиях, обеспечивающих
микробиологическую чистоту. Вода очищенная в кон¬
тейнерах не должна содержать никаких дополнитель¬
ных веществ.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная жидкость.
ИСПЫТАНИЯ
Вода очищенная в контейнерах должна выдер¬
живать испытания, как указано в разделе «Вода очи¬
щенная т Ьи1к», и следующие дополнительные испы¬
тания.
Кислотность или щелочность. К 10 мл испыту¬
емого образца, свежепрокипяченного в колбе из бо¬
росиликатного стекла и охлажденного, прибавляют
0,05 мл раствора метилового красного Р. Раствор
не должен окрашиваться в красный цвет.
К 10 мл испытуемого образца прибавляют 0,1 мл
раствора бромтимолового синего Р1. Раствор не
должен окрашиваться в синий цвет.
Восстанавливающие вещества. К 100 мл испы¬
туемого образца прибавляют 10 мл кислоты серной
разведенной Р, 0,1 мл 0,02 М раствора калия пер¬
манганата и кипятят в течение 5 мин. Раствор дол¬
жен сохранять слабо-розовое окрашивание.
Хлориды. К 10 мл испытуемого образца прибав¬
ляют 1 мл кислоты азотной разведенной Р и 0,2 мл
раствора серебра нитрата Р2. В течение 15 мин не
должно быть видимых изменений раствора.
Сульфаты. К 10 мл испытуемого образца при¬
бавляют 0,1 мл кислоты хлористоводородной раз¬
веденной Р и 0,1 мл раствора бария хлорида Р1. В
течение 1 ч не должно быть видимых изменений раст¬
вора.
Соли аммония. Не более 0,00002 % (0,2 ррт). К
20 мл испытуемого образца прибавляют 1 мл раст¬
вора калия тетрайодомеркурата щелочного Р. Че¬
рез 5 мин исследуют раствор вдоль вертикальной
оси пробирки. Окраска полученного раствора должна
быть не интенсивнее окраски эталона, приготовлен¬
ного параллельно, прибавляя 1 мл раствора калия
тетрайодомеркурата щелочного Р к смеси из 4 мл
эталонного раствора аммония (1 ррт NН^ Р и 16 мл
воды, свободной от аммиака, Р.
Кальций и магний. К 100 мл испытуемого об¬
разца прибавляют 2 мл аммиачного буферного раст¬
вора рН 10,0 Р, 50 мг протравного черного 11 инди¬
каторной смеси Р и 0,5 мл 0,01 М раствора натрия
эдетата. Появляется синее окрашивание.
Остаток после выпаривания. Не более 0,001 %.
100 мл испытуемого образца выпаривают досуха на
водяной бане и сушат при температуре от 100 °С до
105 °С. Масса остатка не должна превышать 1 мг.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/мл
(2.6.12). Определение проводят с использованием
агаризованной среды на основе гидролизата казеина
и соевых бобов.
МАРКИРОВКА
При необходимости указывают:
-вода очищенная пригодна для производства
растворов для диализа.
07/2016:0395
ВОДОРОДА ПЕРОКСИДА
3 % РАСТВОР
Нус1годепН регох1с1ит 3 рег сеп1ит
НУОР06ЕМ РЕКОХЮЕ ЗОШТЮЫ
(3 РЕП СЕЫТ)
ОПРЕДЕЛЕНИЕ
Содержание: не менее 2,5 % (м/м) и не более
3,5 % (м/м) Н202 (М.м. 34,01).
1 объем 3 % раствора водорода пероксида соот¬
ветствует около 10 объемам кислорода.
Может содержать подходящий стабилизатор.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 2 мл испытуемого образца прибавляют
0,2 мл кислоты серной разведенной Р и 0,2 мл 0,02 М
раствора калия перманганата. Через 2 мин раствор
становится бесцветным либо слегка розовым.
B. К 1 мл испытуемого образца прибавляют
0,1 мл кислоты хлористоводородной разведенной Р
и 0,1 мл раствора калия йодида Р. Появляется корич¬
невое окрашивание. Могут образовываться черные
частицы.
312
Государственная фармакопея Республики Беларусь
С. Испытуемый образец выдерживает требова¬
ния по количественному содержанию Н202.
ИСПЫТАНИЯ
Кислотность. К 10 мл испытуемого образца при¬
бавляют 20 мл воды Р и 0,25 мл раствора метилово¬
го красного Р. При прибавлении не менее 0,05 мл и
не более 1,0 мл 0,1 М раствора натрия гидроксида
окраска раствора должна измениться.
Органические стабилизаторы. Не более
0,0250 % (250 ррт). 20 мл испытуемого образца
встряхивают с 10 мл хлороформа Р, затем еще дваж¬
ды с хлороформом Р порциями по 5 мл. Хлороформ¬
ные слои объединяют, выпаривают при пониженном
давлении и при температуре, не превышающей 25 °С,
и высушивают в эксикаторе. Масса полученного
остатка не должна превышать 5 мг.
Нелетучий остаток. Не более 2 г/л. 10 мл испы¬
туемого образца выдерживают в платиновом тигле до
прекращения выделения пузырьков газа, выпаривают
на водяной бане досуха и высушивают при темпера¬
туре от 100 °С до 105 °С. Масса полученного остатка
не должна превышать 20 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,0 г испытуемого образца доводят водой Р до
объема 100,0 мл. К 10,0 мл полученного раствора
прибавляют 20 мл кислоты серной разведенной Р и
титруют 0,02 М раствором калия перманганата до
появления розового окрашивания.
1 мл 0,02 М раствора калия перманганата соот¬
ветствует 1,701 мг Н202 или 0,56 мл кислорода.
ХРАНЕНИЕ
В защищенном от света месте и, если раствор не
содержит стабилизатор, при температуре ниже 15 °С.
МАРКИРОВКА
Если раствор содержит стабилизатор, то указы¬
вают, что содержимое стабилизировано. По требова¬
нию компетентного уполномоченного органа указыва¬
ют наименование стабилизатора.
ПРЕДОСТЕРЕЖЕНИЕ
Разлагается при контакте с окисляющимися ор¬
ганическими веществами, некоторыми металлами и в
щелочной среде.
07/2016:0396
ВОДОРОДА ПЕРОКСИДА
30 % РАСТВОР
НуйгодепН регохШит 30 рег сеп1ит
НУОКООЕЫ РЕКОХЮЕ ЗОШТЮЫ
(30 РЕК СЕЫТ)
[7722-84-1]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 29,0 % (м/м) и не более
31,0 % (м/м) Н202 (М.м. 34,01).
1 объем 30 % раствора водорода пероксида со¬
ответствует около 110 объемам кислорода.
Может содержать подходящий стабилизатор.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 1 мл испытуемого образца прибавляют
0,2 мл кислоты серной разведенной Р и 0,25 мл
0,02 М раствора калия перманганата. Раствор
обесцвечивается с выделением пузырьков газа.
B. К 1 мл испытуемого образца прибавляют
0,1 мл кислоты хлористоводородной разведенной Р
и 0,1 мл раствора калия йодида Р Появляется корич¬
невое окрашивание. Могут образовываться черные
частицы.
C. Испытуемый образец выдерживает требова¬
ния по количественному содержанию Н202.
ИСПЫТАНИЯ
Кислотность. К 10 мл испытуемого образца при¬
бавляют 100 мл воды Р и 0,25 мл раствора метило¬
вого красного Р. При прибавлении не менее 0,05 мл и
не более 0,5 мл 0,1 М раствора натрия гидроксида
окраска раствора должна измениться.
Органические стабилизаторы. Не более
0,0500 % (500 ррт). 20 мл испытуемого образца
встряхивают с 10 мл хлороформа Р, затем еще дваж¬
ды с хлороформом Р порциями по 5 мл. Хлороформ¬
ные слои объединяют, выпаривают при пониженном
давлении и при температуре, не превышающей 25 °С,
и высушивают в эксикаторе. Масса полученного
остатка не должна превышать 10 мг.
Нелетучий остаток. Не более 2 г/л. 10 мл испы¬
туемого образца выдерживают в платиновом тигле до
прекращения выделения пузырьков газа, охлаждая
при необходимости. Выпаривают на водяной бане
досуха и высушивают при температуре от 100 °С до
105 °С. Масса полученного остатка не должна превы¬
шать 20 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,00 г испытуемого образца доводят водой Р до
объема 100,0 мл. К 10,0 мл полученного раствора
прибавляют 20 мл кислоты серной разведенной Р и
титруют 0,02 М раствором калия перманганата до
появления розового окрашивания.
1 мл 0,02 М раствора калия перманганата соот¬
ветствует 1,701 мг Н202 или 0,56 мл кислорода.
ХРАНЕНИЕ
В защищенном от света месте и, если раствор не
содержит стабилизатор, при температуре ниже 15 °С.
МАРКИРОВКА
Если раствор содержит стабилизатор, то указы¬
вают, что содержимое стабилизировано. По требова¬
нию компетентного уполномоченного органа указыва¬
ют наименование стабилизатора.
ПРЕДОСТЕРЕЖЕНИЕ
Энергично разлагается при контакте с окисляю¬
щимися органическими веществами, некоторыми ме¬
таллами и в щелочной среде.
Воск пчелиный желтый
313
07/2016:0069
ВОСК ПЧЕЛИНЫЙ БЕЛЫЙ
Сега а!Ьа
ВЕЕ8ШХ, Ш1ТЕ
ОПРЕДЕЛЕНИЕ
Воск пчелиный белый получают отбеливанием
воска пчелиного желтого.
ОПИСАНИЕ(СВОЙСТВА)
Белые или желтовато-белые кусочки или пла¬
стинки, полупрозрачные в тонком слое, разлом мел¬
козернистый, матовый, не кристаллический. При на¬
гревании в руке становятся мягкими и податливыми.
Имеет запах, аналогичный воску пчелиному жел¬
тому, но более слабый, без примеси прогорклости. Не
обладает вкусом и не прилипает к зубам.
Практически нерастворим в воде, частично рас¬
творим в горячем спирте (90 %, об/об) и полностью
растворим в жирных и эфирных маслах.
Относительная плотность: около 0,960.
ИСПЫТАНИЯ
Температура каплепадения (2.2.17). От 61 °С
до 66 °С. Воск плавят, нагревая на водяной бане, вы¬
ливают на стеклянную пластину и выдерживают до
застывания в полутвердую массу. Наполняют метал¬
лическую чашечку, вдавливая широкую часть в воск, и
повторяют процедуру до тех пор, пока воск не начнет
выталкиваться из узкого отверстия. Избыток удаляют
шпателем и сразу же вставляют термометр. Вытес¬
ненный воск удаляют. Выдерживают при комнатной
температуре не менее 12 ч и затем определяют тем¬
пературу каплепадения.
Кислотное число. От 17,0 до 24,0. 2,00 г (ш, г)
испытуемого образца помещают в коническую колбу
вместимостью 250 мл с обратным холодильником,
прибавляют 40 мл ксилола Р и несколько стеклянных
центров кипения. Нагревают до растворения испыту¬
емого образца, прибавляют 20 мл 96 % спирта Р и
0,5 мл раствора фенолфталеина Р1. Горячий раст¬
вор титруют 0,5 М спиртовым раствором калия ги¬
дроксида до появления красной окраски, устойчивой
не менее 10 с (л1? мл). Параллельно проводят кон¬
трольный опыт (п2, мл).
„ 28,05 (п1 ~п2)
Кислотное число = — —•
т
Эфирное число (2.5.2). От 70 до 80.
Число омыления. От 87 до 104. 2,00 г (т, г) ис¬
пытуемого образца помещают в коническую колбу
вместимостью 250 мл с обратным холодильником,
прибавляют 30 мл смеси из равных объемов 96 %
спирта Р и ксилола Р и несколько стеклянных цен¬
тров кипения. Нагревают до растворения испытуемо¬
го образца. Прибавляют 25,0 мл 0,5 М спиртового
раствора калия гидроксида и нагревают с обратным
холодильником в течение 3 ч. Горячий раствор не¬
медленно титруют 0,5 М раствором кислоты хлори¬
стоводородной (п^ мл), используя в качестве инди¬
катора 1 мл раствора фенолфталеина Р1. Во время
титрования раствор несколько раз повторно доводят
до кипения.
Параллельно проводят контрольный опыт (п2, мл).
Число омыления рассчитывают по формуле:
,, 28,05* (л2 - л,)
Число омыления = — —-
т
Церезин, парафин и некоторые другие воски.
3,0 г испытуемого образца помещают в круглодон¬
ную колбу вместимостью 100 мл, прибавляют 30 мл
раствора 40 г/л калия гидроксида Р в 96% спирте,
свободном от альдегидов, Р и осторожно кипятят с
обратным холодильником в течение 2 ч. Холодильник
отсоединяют и сразу же вставляют термометр. Колбу
помещают в водяную баню с температурой 80 °С и
выдерживают до охлаждения при постоянном пере¬
мешивании. До температуры 65 °С не должно обра¬
зовываться осадка, хотя раствор может слегка опа¬
лесцировать. Начиная с температуры 65 °С раствор
может помутнеть и может образоваться осадок. При
температуре 59 °С раствор должен быть мутным.
Глицерин и другие многоатомные спирты. Не
более 0,5 % (м/м) в пересчете на глицерин. К 0,20 г
испытуемого образца прибавляют 10 мл раствора ка¬
лия гидроксида спиртового Р и нагревают на водяной
бане с обратным холодильником в течение 30 мин.
Прибавляют 50 мл кислоты серной разведенной Р,
охлаждают и фильтруют. Колбу и фильтр промывают
кислотой серной разведенной Р. Фильтрат и промыв¬
ную жидкость объединяют и доводят кислотой сер¬
ной разведенной Р до объема 100,0 мл. 1,0 мл полу¬
ченного раствора помещают в пробирку, прибавляют
0,5 мл раствора 10,7 г/л натрия перйодата Р, пере¬
мешивают и выдерживают в течение 5 мин. Прибав¬
ляют 1,0 мл раствора фуксина обесцвеченного Р и
перемешивают. Любой осадок должен исчезнуть. Про¬
бирку помещают в стакан с водой, нагретой до тем¬
пературы 40 °С. Производят наблюдение в процессе
охлаждения в течение (10—15) мин. Фиолетово-синяя
окраска раствора не должна быть интенсивнее такой
же окраски эталона, приготовленного параллельно с
использованием 1,0 мл раствора 10 мг/л глицерина Р
в кислоте серной разведенной Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:0070
ВОСК ПЧЕЛИНЫЙ ЖЕЛТЫЙ
Сега //ауа
ВЕЕ8ШАХ, УЕИ.ОУУ
ОПРЕДЕЛЕНИЕ
Воск, полученный при расплавлении с горячей
водой стенок медовых сот, произведенных медонос¬
ной пчелой Ар/'з теНКега и очищенный от посто¬
ронних веществ.
ОПИСАНИЕ (СВОЙСТВА)
Желтые или светло-коричневые кусочки или пла¬
стинки, разлом мелкозернистый, матовый, не кристал¬
лический. При нагревании в руке становятся мягкими
и податливыми.
Имеет слабый медовый запах. Не обладает вку¬
сом и не прилипает к зубам.
Практически нерастворим в воде, частично рас¬
творим в горячем спирте (90 %, об/об) и полностью
растворим в жирных и эфирных маслах.
Относительная плотность: около 0,960.
314
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Температура каплепадения (2.2.17). От 61 °С
до 66 °С. Воск плавят, нагревая на водяной бане, вы¬
ливают на стеклянную пластину и выдерживают до
застывания в полутвердую массу. Наполняют метал¬
лическую чашечку, вдавливая широкую часть в воск, и
повторяют процедуру до тех пор, пока воск не начнет
выталкиваться из узкого отверстия. Избыток удаляют
шпателем и сразу же вставляют термометр. Вытес¬
ненный воск удаляют. Выдерживают при комнатной
температуре не менее 12 ч и затем определяют тем¬
пературу каплепадения.
Кислотное число. От 17,0 до 22,0. 2,00 г (т, г)
испытуемого образца помещают в коническую колбу
вместимостью 250 мл с обратным холодильником,
прибавляют 40 мл ксилола Р и несколько стеклянных
центров кипения. Нагревают до растворения испыту¬
емого образца, прибавляют 20 мл 96 % спирта Р и
0,5 мл раствора фенолфталеина Р1. Горячий раст¬
вор титруют 0,5 М спиртовым раствором калия ги¬
дроксида до появления красной окраски, устойчивой
не менее 10 с (л^ мл).
Параллельно проводят контрольный опыт (л2, мл).
Кислотное число рассчитывают по формуле:
Кислотное число =
28,05-(л, -л2)
т
Эфирное число (2.5.2). От 70 до 80.
Число омыления. От 87 до 102. 2,00 г (т, г) ис¬
пытуемого образца помещают в коническую колбу
вместимостью 250 мл с обратным холодильником,
прибавляют 30 мл смеси из равных объемов 96 %
спирта Р и ксилола Р и несколько стеклянных цен¬
тров кипения. Нагревают до растворения испытуе¬
мого образца. Прибавляют 25,0 мл 0,5 М спиртового
раствора калия гидроксида и нагревают с обратным
холодильником в течение 3 ч. Горячий раствор немед¬
ленно титруют 0,5 М раствором кислоты хлористо¬
водородной (л1Т мл), используя в качестве индикатора
1 мл раствора фенолфталеина Р1. Во время титро¬
вания раствор несколько раз повторно доводят до ки¬
пения.
Параллельно проводят контрольный опыт (л2, мл).
Число омыления рассчитывают по формуле:
Число омыление =
28,05 (п2 -п1)
т
Церезин, парафин и некоторые другие воски.
3,0 г испытуемого образца помещают в круглодон¬
ную колбу вместимостью 100 мл, прибавляют 30 мл
раствора 40 г/л калия гидроксида Р в 96% спирте,
свободном от альдегидов, Р и осторожно кипятят с
обратным холодильником в течение 2 ч. Холодильник
отсоединяют и сразу же вставляют термометр. Колбу
помещают в водяную баню с температурой 80 °С и
выдерживают до охлаждения при постоянном пере¬
мешивании. До температуры 65 °С не должно обра¬
зовываться осадка, хотя раствор может слегка опа¬
лесцировать. Начиная с температуры 65 °С раствор
может помутнеть и может образоваться осадок. При
температуре 59 °С раствор должен быть мутным.
Глицерин и другие многоатомные спир¬
ты. Не более 0,5 % (м/м) в пересчете на глицерин.
К 0,20 г испытуемого образца прибавляют 10 мл
раствора калия гидроксида спиртового Р и нагре¬
вают на водяной бане с обратным холодильником
в течение 30 мин. Прибавляют 50 мл кислоты сер¬
ной разведенной Р, охлаждают и фильтруют. Колбу и
фильтр промывают кислотой серной разведенной Р.
Фильтрат и промывную жидкость объединяют и до¬
водят кислотой серной разведенной Р до объема
100.0 мл. 1,0 мл полученного раствора помещают в
пробирку, прибавляют 0,5 мл раствора 10,7 г/л на¬
трия перйодата Р, перемешивают и выдерживают
в течение 5 мин. Прибавляют 1,0 мл раствора фук¬
сина обесцвеченного Р и перемешивают. Любой оса¬
док должен исчезнуть. Пробирку помещают в стакан
с водой, нагретой до температуры 40 °С. Произво¬
дят наблюдение в процессе охлаждения в течение
(10—15) мин. Фиолетово-синяя окраска раствора не
должна быть интенсивнее такой же окраски этало¬
на, приготовленного параллельно с использованием
1.0 мл раствора 10 мг/л глицерина Р в кислоте сер¬
ной разведенной Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:1215
ГАЛАКТОЗА
Оа1ас1озит
ОА1.АСТ08Е
СвН12Ов М.м. 180,2
[59-23-4]
ОПРЕДЕЛЕНИЕ
О-Галактопираноза.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
мелко гранулированный порошок.
Очень легко растворима в воде, очень мало рас¬
творима в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО галактозы.
B. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(2:3, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20 мл этой же смесью растворителей.
Раствор сравнения (а). 10 мг ФСО галактозы
растворяют в смеси растворителей и доводят до объ¬
ема 20 мл этой же смесью растворителей.
Галоперидол
315
Раствор сравнения (Ь). 10 мг галактозы Р, 10 мг
глюкозы Р и 10 мг лактозы Р растворяют в смеси
растворителей и доводят до объема 20 мл этой же
смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: вода Р — пропанол Р (15:85,
об/об).
Наносимый объем пробы: 2 мкл; тщательно вы¬
сушивают нанесенные пробы.
Фронт подвижной фазы: камеру не насыщают
парами подвижной фазы, не менее 3/4 высоты пла¬
стинки.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р. Нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются три полно¬
стью разделенных пятна.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
С. 0,1 г испытуемого образца растворяют в 10 мл
воды Р. Прибавляют 3 мл раствора медно-тартрат-
ного Р и нагревают. Образуется осадок оранжевого
или красного цвета.
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца при на¬
гревании в водяной бане растворяют в воде, сво¬
бодной от углерода диоксида, Р, приготовленной
из воды дистиллированной Р, и доводят до объема
50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)6.
Кислотность или щелочность. К 30 мл раст¬
вора 3 прибавляют 0,3 мл раствора фенолфталеи¬
на Р. Раствор бесцветный. При прибавлении не более
1,5 мл 0,01 М раствора натрия гидроксида должно
появиться розовое окрашивание.
Удельное оптическое вращение (2.2.7). От
+78,0 до +81,5 (в пересчете на безводное вещество).
10,00 г испытуемого образца растворяют в 80 мл
воды Р, прибавляют 0,2 мл раствора аммиака разве¬
денного Р1, выдерживают в течение 30 мин и доводят
водой Р до объема 100,0 мл.
Барий. 5 мл раствора 3 разводят водой дистил¬
лированной Р до объема 10 мл и прибавляют 1 мл
кислоты серной разведенной Р. Опалесценция полу¬
ченного раствора сразу после приготовления и через
1 ч должна быть не интенсивнее опалесценции раст¬
вора, состоящего из 5 мл раствора 3 и 6 мл воды дис¬
тиллированной Р.
Свинец (2.4.10). Не более 0,00005 % (0,5 ррт).
Вода (2.5.12). Не более 1,0 %. Определение про¬
водят из 1,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
К 5 мл раствора 3 прибавляют 2 мл кислоты
серной Р, выпаривают досуха на водяной бане и про¬
каливают до постоянной массы. Масса остатка не
должна превышать 1 мг.
Микробиологическая чистота
ОКА: допустимый критерий приемлемости
102 КОЕ/г (2.6.12).
07/2016:0616
ГАЛОПЕРИДОЛ
На1орепс1о1ит
НА1.0РЕКЮ01.
С21Н23С1РЫ02 М.м. 375,9
[52-86-8]
ОПРЕДЕЛЕНИЕ
4-[4-(4-Хлорфенил)-4-гидроксипиперидин-1-ил]-
1 -(4-фторфенил)бутан-1 -он.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, мало раство¬
рим в 96 % спирте, в метаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: 3, Е.
Вторая идентификация: А, С, О, Е.
A. Температура плавления (2.2.14). От 150 °С до
153 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО галоперидола.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО галоперидола
растворяют в метаноле Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО галоперидола
и 10 мг ФСО бромперидола растворяют в метаноле Р
и доводят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р.
Подвижная фаза: тетрагидрофуран Р — мета¬
нол Р — раствор 58 г/л натрия хлорида Р (10:45:45,
об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки; камеру предварительно не насыщают па¬
рами подвижной фазы.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два пятна,
которые могут быть разделены не полностью.
316
Гэсударственная фармакопея Республики Беларусь
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
Р. Около 10 мг испытуемого образца раство¬
ряют в 5 мл этанола Р и прибавляют 0,5 мл раст¬
вора динитробензола Р и 0,5 мл 2 М раствора
калия гидроксида спиртового Р. Появляется фио¬
летовое окрашивание, которое через 20 мин стано¬
вится коричневато-красным.
Е. 0,1 г испытуемого образца помещают в пла¬
тиновый тигель и прибавляют 0,5 г натрия карбо¬
ната безводного Р. Нагревают над открытым пла¬
менем в течение 10 мин, охлаждают, прибавляют
к остатку 5 мл кислоты азотной разведенной Р,
перемешивают и фильтруют. К 1 мл фильтрата при¬
бавляют 1 мл воды Р. Полученный раствор дает ре¬
акцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,2 г испытуемого образца раство¬
ряют в 20 мл 1 % (об/об) раствора кислоты молоч¬
ной Р.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона У(Ж)7.
Сопутствующие примеси. Жидкостная хро-
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием и с защитой
от света.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО галопери-
дола для проверки пригодности хроматографиче¬
ской системы (содержит примеси В и О) растворяют
в 1,0 мл метанола Р.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
1,0 мл полученного раствора доводят метанолом Р
до объема 10,0 мл.
Раствор сравнения (с). 10 мг ФСО галоперидо-
ла для идентификации пиков (содержит примеси О
и Н) растворяют в 1,0 мл метанола Р.
Условия хроматографирования:
-колонка длиной 0,1 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным деактивированным по отношению к ос¬
нованиям эндкепированным для хроматографии Р
с размером частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 17 г/л тетрабу-
тиламмония гидросульфата Р1\
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
90
10
2—17
90 —* 50
10 — 50
17—22
50
50
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифи¬
цируют пики примесей В и О, используя хромато¬
грамму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО галоперидола для провер¬
ки пригодности хроматографической системы,
идентифицируют пики примесей 6 и Н, используя
хроматограмму раствора сравнения (с) и хрома¬
тограмму, прилагаемую к ФСО галоперидола для
идентификации пиков.
Относительное удерживание (по отношению к
галоперидолу, время удерживания — около 8 мин):
примесь В — около 0,9; примесь О — около 1,6; при¬
месь О — около 1,8; примесь Н— около 2,0.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 3,0 между пиками при¬
меси В и галоперидола.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,7):
- примесь О (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси О и Н (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям О и Н, не должны превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В, О, (3 и Н, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 1,0%.
Определение проводят из 1,0 г испытуемого образца
в платиновом тигле.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
смеси из 1 объема кислоты уксусной безводной Р и
7 объемов метилэтилкетона Р и титруют 0,1 М рас¬
твором кислоты хлорной, используя в качестве ин¬
дикатора 0,2 мл раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 37,59 мг С21Н2зС1РЫ02.
Г
Гз айфенезин
317
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, О, 6, Н.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, С, Е, Р.
А. 1 -(4-Фторфенил)-4-(4-гидрокси-4-фенил-
пиперидин-1-ил)бутан-1-он.
В. 4-[4-(4-Хлорфенил)-4-гидроксипиперидин-1-ил]-
1-(2-фторфенил)бутан-1-он.
Р. 4-[4-(3’-Хлорбифенил-4-ил)-4-гидрокси-
пиперидин-1 -ил]-1 -(4-фторфенил)бутан-1 -он.
6. Структура не установлена.
Н. Структура не установлена.
07/2016:0615
ГВАЙФЕНЕЗИН
Оиайепезтит
С11А1ЕЕЫЕ81Ы
С1вн1404 М.м. 198,2
[93-14-1]
ОПРЕДЕЛЕНИЕ
(2Я5)-3-(2-Метоксифенокси)пропан-1,2-диол.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Умеренно растворим в воде, растворим в 96 %
спирте.
С. 4-[4-(4-Хлорфенил)-4-гидроксипиперидин-1 -ил]-
1-(3-этил-4-фторфенил)бутан-1-он.
й. 4-[4-(4-Хлорфенил)-4-гидроксипиперидин-1 -ил]-
1 -[4-[4-(4-хлорфенил)-4-гидроксипиперидин-1 -ил]фе-
нил]бутан-1-он.
Е. 4-[4-(4’-Хлорбифенил-4-ил)-4-гидроксипи-
перидин-1 -ил]-1 -(4-фторфенил)бутан-1 -он.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Температура плавления (2.2.14): от 79 °С до 83 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гвайфенезина #или спектр,
представленный на рисунке #0615.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 30 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 30 мг ФСО гвайфенезина
растворяют в метаноле Р и доводят до объема 10 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: метиленхлорид Р — пропанол
Р (20:80, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают смесью из
равных объемов раствора 10 г/л калия феррициани-
да Р и раствора 200 г/л железа (III) хлорида Р и 96%
спирта Р.
318
Гэсударственная фармакопея Республики Беларусь
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, размеру и цвету основному пятну на
хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при
необходимости слегка нагревая, и доводят до объема
50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,05 мл раствора фенолфталеи¬
на Р1. При прибавлении не более 0,1 мл 0,01 Мраст¬
вора натрия гидроксида окраска раствора должна
измениться. К 10 мл раствора 3 прибавляют 0,15 мл
раствора метилового красного Р. При прибавлении
не более 0,1 мл 0,01 М раствора кислоты хлористо¬
водородной окраска раствора должна измениться на
красную.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в ацетонитриле Р и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят ацетонитрилом Р до объема 20,0 мл.
1.0 мл полученного раствора доводят ацетонитри¬
лом Р до объема 10,0 мл.
Раствор сравнения (Ь). 10,0 мг гвайякола Р
растворяют в ацетонитриле Р и доводят до объема
50.0 мл этим же растворителем. 0,5 мл полученно¬
го раствора доводят ацетонитрилом Р до объема
50.0 мл.
Раствор сравнения (с). 50,0 мг гвайякола Р
растворяют в ацетонитриле Р и доводят до объема
50.0 мл этим же растворителем. 5,0 мл полученного
раствора доводят испытуемым раствором до объема
10.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц 5
мкм;
- подвижная фаза:
- подвижная фаза А: кислота уксусная ледя¬
ная Р — вода Р (10:990, об/об);
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—32
о
ю
т
о
со
20 —► 50
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 276 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
гвайфенезину, время удерживания — около 8 мин):
примесь В — около 0,9; примесь А — около 1,4; при¬
месь С —- около 3,1; примесь О — около 3,7.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 3,0 между пиками гвай-
фенезина и примеси А.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь);
- примесь В (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству-
Гзоздичное масло
319
ющего примеси В, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- любая другая примесь (не более 0,5 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А и В, не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (а);
- сумма примесей (кроме примеси В) (не более
1.0 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
В, не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Хлориды и монохлоргидрины. Не более
0,0250 % (250 ррт). К 10 мл раствора 5 прибавляют
2 мл раствора натрия гидроксида разведенного Р и
нагревают на водяной бане в течение 5 мин. Охлаж¬
дают и прибавляют 3 мл раствора кислоты азотной
разведенной Р. Полученный раствор должен выдер¬
живать испытание на хлориды (2.4.4).
Тяжелые металлы (2.4.8, метод В). Не более
0,0025 % (25 ррт). 2,0 г испытуемого образца раство¬
ряют в смеси вода Р—96% спирт Р (1:9, об/об) и до¬
водят до объема 25 мл этим же растворителем. 12 мл
полученного раствора должны выдерживать испыта¬
ние на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (2 ррт РЬ),
полученного путем разведения эталонного раст¬
вора свинца (100 ррт РЬ) Р смесью вода Р — 96%
спирт Р (1:9, об/об).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г (т, г) испытуемого образца растворяют в
10.0 мл свежеприготовленной смеси ангидрид уксус¬
ный Р — пиридин Р (1:7, об/об) и кипятят с обратным
холодильником в течение 45 мин. Охлаждают, прибав¬
ляют 25 мл воды Р и титруют 1 М раствором натрия
гидроксида (п^ мл), используя в качестве индикатора
0,25 мл раствора фенолфталеина Р.
Параллельно проводят контрольный опыт (л2, мл).
Содержание С10Н14О4 в процентах рассчитывают
по формуле:
19,82 (п2-п,)
2т
ПРИМЕСИ
A. Р = Н: 2-Метоксифенол (гвайякол).
B. Р = СН(СН2ОН)2:2-(2-Метоксифенокси)пропан-
1,3-диол (В-изомер).
он он
Э. 1,3-Бис(2-метоксифенокси)пропан-2-ол.
07/2016:1091
ГВОЗДИЧНОЕ МАСЛО
СагуорЬуШ Лопз ае№его1еит
С ШУЕ ОН
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное методом пере¬
гонки с водяным паром из сухих цветочных буто¬
нов Зугудшт аготаИсит (1_.) Мегг. е* Ь.М.Реггу (син.
Еидета сагуорЬуНиз (Зргепд.) ВиНоск е! З.С.Нагпзоп).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, желтая жидкость, которая становит¬
ся коричневой на воздухе.
Смешивается с метиленхлоридом, толуолом и
жирными маслами.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мкл испытуемого об¬
разца растворяют в 2,0 мл толуола Р.
Раствор сравнения. 15 мкл эвгенола Р и 15 мкл
ацетилэвгенола Р растворяют в 2,0 мл толуола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР294Р.
Подвижная фаза: толуол Р, камеру не насыща¬
ют парами подвижной фазы.
Наносимый объем пробы: 20 мкл испытуемого
раствора и 15 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: хроматографируют
дважды не менее 10 см от линии старта; между двумя
последовательными хроматографированиями пла¬
стинку выдерживают в течение 5 мин.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора в средней части обнаруживается зона по¬
глощения (эвгенол), соответствующая по располо¬
жению зоне поглощения на хроматограмме раствора
сравнения; непосредственно ниже зоны эвгенола об¬
наруживается зона слабого поглощения (ацетилэвге-
нол), соответствующая по расположению зоне ацети¬
лэвгенола на хроматограмме раствора сравнения.
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р, нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин и про¬
сматривают при дневном свете.
320
Гэсударственная фармакопея Республики Беларусь
Результаты В: на хроматограммах испытуемо¬
го раствора и раствора сравнения обнаруживаются
зоны эвгенола интенсивного коричневато-фиолето¬
вого цвета; на хроматограмме испытуемого раствора
обнаруживается зона ацетилэвгенола бледного фио¬
летово-синего цвета; на хроматограмме испытуемо¬
го раствора обнаруживаются и другие окрашенные
зоны, в частности бледная красная зона в нижней
части хроматограммы и красновато-фиолетовая зона
(р-кариофиллен) в верхней части.
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Результаты: на хроматограмме испытуемо¬
го раствора обнаруживаются три основных пика,
соответствующие по времени удерживания трем
основным пикам на хроматограмме раствора срав¬
нения.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 1,030 до
1,063.
Показатель преломления (2.2.6). От 1,528 до
1,537.
Угол оптического вращения (2.2.7). От -2°до 0°.
Жирные и минеральные масла (2.8.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Растворимость в спирте (2.8.10). 1,0 мл испы¬
туемого образца должен растворяться не менее чем в
2,0 мл спирта (70 %, об/об) Р.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 0,2 г испытуемого образ¬
ца растворяют в 10 г гексана Р.
Раствор сравнения. 7 мг р-кариофиллена Р,
80 мг эвгенола Р и 4 мг ацетилэвгенола Р раство¬
ряют в 10 г гексана Р.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м и
внутренним диаметром 0,25 мм, покрытая слоем ма-
крогола 20 000 Р;
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—8
60
8—48
60—► 180
48—53
180
Блок ввода проб
270
Детектор
270
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1,0 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения; отмечают времена
удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,5 между пиками эвге¬
нола и ацетилэвгенола;
- число теоретических тарелок, не менее
30 000, рассчитанных для пика р-кариофиллена при
температуре 110 °С.
По временам удерживания компонентов раст¬
вора сравнения определяют состав испытуемого
раствора.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- р-кариофиллен: от 5,0 % до 14,0 %;
- эвгенол: от 75,0 % до 88,0 %;
- ацетилэвгенол: от 4,0 % до 15,0 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
#В заполненном доверху воздухонепроницаемом
контейнере, в защищенном от света месте при темпе¬
ратуре не выше 25 °С.# Защищать от нагревания.
07/2016:1545
ГЕКСАМЕТИЛЕНТЕТРАМИН
МеОюпаттит (#иго1гортит)
МЕТНЕЫАМШЕ
С6Н12Ы4 М.м. 140.2
[100-97-0]
ОПРЕДЕЛЕНИЕ
1,3,5,7-Тетраазотрицикло[3.3.1.137]декан.
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, растворим в 96 % спирте
и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гексаметилентетрамина.
B. К 1 мл раствора 5, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 1 мл кис¬
лоты серной Р и немедленно нагревают до кипения.
Охлаждают. К 1 мл раствора прибавляют 4 мл воды Р
и 5 мл реактива ацетилацетона Р1 и нагревают на
водяной бане в течение 5 мин. Появляется интенсив¬
ное желтое окрашивание.
C. К 1 мл раствора 3 прибавляют 1 мл кислоты
серной разведенной Р и немедленно нагревают до ки¬
пения. Раствор дает реакцию на аммония соли и соли
летучих оснований (2.3.1).
й. 10 мг испытуемого образца растворяют в 5 мл
воды Р, подкисляют кислотой хлористоводородной
разведенной Р и прибавляют 1 мл раствора калия
йодовисмутата Р. Сразу образуется осадок оранже¬
вого цвета.
Гэнтамицина сульфат
321
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 5 мл раствора
3 прибавляют 0,1 мл раствора фенолфталеина Р.
При прибавлении не более 0,2 мл 0,1 М раствора
кислоты хлористоводородной или 0,1 М раствора
натрия гидроксида окрашивание раствора должно
измениться.
Свободный формальдегид. Не более
0,0050 % (50 ррт). 0,8 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 8 мл этим
же растворителем. Прибавляют 2 мл аммиачного
раствора серебра нитрата Р. Серая окраска, по¬
явившаяся в растворе через 5 мин, должна быть не
интенсивнее окраски эталона, приготовленного па¬
раллельно с использованием смеси из 8 мл свеже¬
приготовленного эталонного раствора формаль¬
дегида (5 ррт СН20) Р и 2 мл аммиачного раст¬
вора серебра нитрата Р.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
15 мл раствора 3 должны выдерживать испытание на
сульфаты.
Аммония соли (2.4.1). Не более 0,0050%
(50 ррт). 2 мл свежеприготовленного раствора 3 до¬
водят водой Р до объема 13 мл. Прибавляют 2 мл
раствора натрия гидроксида разведенного Р. Полу¬
ченный раствор должен выдерживать испытание на
соли аммония.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 2,0%. 1,000 г испытуемого образца сушат в
эксикаторе.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 30 мл
метанола Р. Титруют 0,1 М раствором кислоты
хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 14,02 мг С6Н12Ы4.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0331
ГЕНТАМИЦИНА СУЛЬФАТ
Оеп(ат1ст1 зиКаз
ОЕЫТАМЮШ 81Л.РАТЕ
Гентамицин
Молекулярная
формула
Р1
К2
КЗ
С1
с21н„м5о7
сн3
со
X
о
н
С1а
с,9н39м5о7
н
н
н
С2
СзоН.ЛО,
н
сн3
н
С2а
с20н<ло7
н
н
со
X
О
С2Ь
СЛ.МА
СН3
н
н
[1405-41-0]
ОПРЕДЕЛЕНИЕ
Смесь сульфатов антимикробиологических ве¬
ществ, продуцируемых Мюготопозрога ригригеа, ос¬
новными компонентами которых являются гентамици¬
ны С1, С1а, С2, С2а и С2Ь.
Содержание: не менее 590 МЕ/мг (в пересчете на
безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде, практически нераство¬
рим в ацетоне и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, С.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения. Содержимое контейнера с
ФСО гентамицина сульфата растворяют в воде Р и
доводят до объема 5 мл этим же растворителем.
Пластинка: ТСХпластинка со слоем силикагеля Р.
Подвижная фаза: нижний слой смеси из равных
объемов раствора аммиака концентрированного Р,
метанола Р и метиленхлорида Р.
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р1 и нагревают при температуре 110 °С в
течение 5 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются три основных пятна, соответ¬
ствующие по расположению, цвету и размеру трем ос¬
новным пятнам на хроматограмме раствора сравнения.
41. Зак. 1060.
322
Гэсударственная фармакопея Республики Беларусь
B. Просматривают хроматограммы, полученные
в испытании «Состав».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживаются пять основных пиков,
соответствующих по времени удерживания пяти основ¬
ным пикам на хроматограмме раствора сравнения (а).
C. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.7).
ИСПЫТАНИЯ
Раствор 5. 0,8 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.7). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее шестого эталона шкалы
наиболее подходящего цвета.
рН (2.2.3). От 3,5 до 5,5. Измеряют рН раствора 5.
Удельное оптическое вращение (2.2.7). От
+107 до +121 (в пересчете на безводное вещество).
2.5 г испытуемого образца растворяют в воде Р и до¬
водят до объема 25,0 мл этим же растворителем.
Состав. Жидкостная хроматография (2.2.29). Ис¬
пользуют метод нормализации, учитывая только пики,
соответствующие гентамицинам С1, С1а, С2, С2а и
С2Ь.
Испытуемый раствор (а). 25,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Испытуемый раствор (Ь). 5,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
25,0 мл.
Раствор сравнения (а). 5 мг ФСО гентамици¬
на для идентификации пиков (содержит примесь В)
растворяют в подвижной фазе и доводят до объема
25 мл этим же растворителем.
Раствор сравнения (Ь). 20,0 мг ФСО сисомицина
сульфата (примесь А) растворяют в подвижной фазе
и доводят до объема 20,0 мл этим же растворителем.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (д). К 1 мл раствора сравне¬
ния (Ь) прибавляют 5 мл испытуемого раствора (а) и
доводят подвижной фазой до объема 50 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4.6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 35 °С;
- подвижная фаза: к 900 мл воды, свободной от
углерода диоксида, Р прибавляют 7,0 мл кислоты
трифторуксусной Р, 250,0 мкл кислоты пентаф-
торпропановой Р и 4,0 мл раствора натрия гидрок¬
сида, свободного от карбонатов, Р, выдерживают до
установления равновесия и доводят до рН 2,6 рас¬
твором натрия гидроксида, свободным от карбона¬
тов, Р, разведенным 1 к 25. Прибавляют 15 мл аце¬
тонитрила Р и доводят водой, свободной от углеро¬
да диоксида, Рдо объема 1000,0 мл.
- скорость подвижной фазы: 1,0 мл/мин;
- постколоночный раствор: раствор натрия
гидроксида, свободный от карбонатов, Р, разведен¬
ный 1 к 25 и предварительно дегазированный, при¬
бавляемый неимпульсным способом к выходящей из
колонки фракции с использованием полимерной спи¬
рали для смешивания объемом 375 мкл;
- скорость постколоночного раствора:
0,3 мл/мин;
- детектирование: импульсный амперометриче¬
ский детектор или аналогичный с золотым индикатор¬
ным электродом, хлорсеребряным электродом срав¬
нения и вспомогательным электродом из нержавею¬
щей стали, являющимся центральной частью ячейки,
в которой устанавливаются соответственно потенциал
детектирования +0,05 В, потенциал окисления +0,75 В
и потенциал восстановления -0,15 В, с длительностью
импульса в соответствии с применяемым прибором;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и растворов сравнения (а), (с) и (с1);
- время хроматографирования: 1,2-кратное вре¬
мя удерживания гентамицина С1.
Идентификация пиков: идентифицируют пики
гентамицинов С1, С1а, С2, С2а и С2Ь, используя хро¬
матограмму, прилагаемую к ФСО гентамицина для
идентификации пиков.
Относительное удерживание (по отношению к
примеси А, время удерживания — около 23 мин): ген¬
тамицин С1а — около 1,1; гентамицин С2 — около 1,8;
гентамицин С2Ь — около 2,0; гентамицин С2а — око¬
ло 2,3; гентамицин С1 — около 3,0.
Пригодность хроматографической системы:
-разрешение: не менее 1,2 между пиками при¬
меси А и гентамицина С1а и не менее 1,5 между пи¬
ками гентамицина С2 и гентамицина С2Ь на хрома¬
тограмме раствора сравнения (6); при необходимости
корректируют объем ацетонитрила Р в подвижной
фазе, общий объем прибавленного ацетонитрила Р
может быть вплоть до 50 мл на 1 л подвижной фазы;
- отношение сигнал/шум: не менее 20 для основ¬
ного пика на хроматограмме раствора сравнения (с).
Предельное содержание:
-гентамицин С1: не менее 25,0% и не более
45.0 %;
-гентамицин С1а: не менее 10,0 % и не более
30.0 %;
- сумма гентамицинов С2, С2а и С2Ь: не менее
35.0 % и не более 55,0 %.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29), как указано в испытании «Состав»,
со следующими изменениями; для расчета содержа¬
ния примесей в процентах используют раствор срав¬
нения (с).
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (с).
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (с); идентифицируют пик примеси В,
используя хроматограмму раствора сравнения (а) и
хроматограмму, прилагаемую к ФСО гентамицина
для идентификации пиков.
Предельное содержание примесей:
- примеси А, В (не более 3,0 %): на хроматограм¬
ме испытуемого раствора (а) площади пиков, соот¬
ветствующих примесям А и В, не должны превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (с);
-любая другая примесь (не более 3,0 %): не
должна превышать 3-кратную площадь основного
пика на хроматограмме раствора сравнения (с);
-сумма примесей (не более 10%): не должна
превышать 10-кратную площадь основного пика на
хроматограмме раствора сравнения (с);
Гэпарин натрия
323
- неучитываемый предел (0,5 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (с).
Метанол (2.4.24, система В). Не более 1,0 %.
Сульфаты. Не менее 32,0 % и не более 35,0 %
(в пересчете на безводное вещество). 0,250 г испыту¬
емого образца растворяют в 100 мл воды дистилли¬
рованной Р и доводят до рН 11 раствором аммиака
концентрированным Р. Прибавляют 10,0 мл 0,1 М
раствора бария хлорида Р и около 0,5 мг фталеино-
вого пурпурного Р. Титруют 0,1 М раствором натрия
эдетата, после начала изменения окраски раствора
прибавляют 50 мл 96 % спирта Р и продолжают ти¬
тровать до исчезновения фиолетово-голубой окраски.
1 мл 0,1 М раствора бария хлорида Р соответ¬
ствует 9,606 мг 804.
Вода (2.5.12). Не более 15,0%. Определение
проводят из 0,300 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 1,0 %. Опре¬
деление проводят из 0,50 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,71 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят количественное определение антибио¬
тиков микробиологическим методом (2.7.2).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере. Стериль¬
ную субстанцию хранят в стерильном воздухонепро¬
ницаемом контейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О, Е.
А. 2-Дезокси-4-0-[3-дезокси-4-С-метил-3-(метил-
амино)-р-Ьарабинопиранозил]-6-0-(2,6-диамино-2,3,4,6-
тетрадезокси-а-0-глицеро-гекс-4-енопиранозил)-1_-
стрептамин (сисомицин).
В. 2-Дезокси-4-0-[3-дезокси-4-С-метил-3-(метил-
амино)-р-1_-арабинопиранозил]-1.-стрептамин (гарамин).
С. 4-0-(6-Амино-6,7-дидезокси-0-глицеро-а-0-
глюко-гептопиранозил)-2-дезокси-6-0-[3-дезокси-4-С-
метил-3-(метиламино)-р-1_-арабинопиранозил]-0-
стрептамин (гентамицин В.,).
О. 2-Дезокси-4-0-[3-дезокси-4-С-метил-3-(метил-
амино)-р-1--арабинопиранозил]-6-0-(2,6-диамино-2,6-
дидезокси-а-0-г/7кжо-гексопиранозил>М_-стрептамин.
Е. 2-Дезоксистрептамин.
07/2016:0333
ГЕПАРИН НАТРИЯ
Нераппит паМсит
НЕРАК1Ы ЗОйШМ
ОПРЕДЕЛЕНИЕ
Гепарин натрия содержит натриевую соль суль-
фатированных гликозаминогликанов, присутствую¬
щих в тканях млекопитающих. Получают из легочной
ткани крупного рогатого скота или слизистой кишеч¬
ника свиней, крупного рогатого скота и овец. При
полном гидролизе высвобождаются О-глюкозамин,
О-глюкуроновая кислота, Ь-идуроновая кислота, ук¬
сусная кислота и серная кислота. Обладает специфи¬
ческим свойством удлинять время свертывания све¬
жей крови.
Активность: не менее 180 МЕ/мг (в пересчете
на сухое вещество).
41*. Зак. 1060.
324
Гэсударственная фармакопея Республики Беларусь
ПРОИЗВОДСТВО
Животные, из которых получают гепарин, должны
выдерживать требования к животным для потребле¬
ния человеком. Все стадии производственного про¬
цесса и получения сырья должны быть выполнены в
соответствии с соответствующими системами менед¬
жмента качества. С помощью подходящего испытания
во время производства проверяют тождественность
вида животного, являющегося источником сырья, и
отсутствие сырья от других видов.
При производстве используют методы, позволя¬
ющие уменьшить либо удалить вещества, снижаю¬
щие кровяное давление.
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец замедляет свертывание
обогащенной кальцием и цитратом овечьей плазмы,
как указано в разделе «Количественное определение».
B. Спектрометрия ядерного магнитного резонан¬
са (2.2.33).
Раствор А. Раствор в дейтерия оксиде Р, содер¬
жащий 20 мкг/мл дейтерированного натрия триме-
тилсилилпропионата Р и, если сигнал при 5,22 ррт
менее 80 % сигнала при 5,44 ррт, 12 мкг/мл натрия
эдетата Р.
Приготовление: 20 мг испытуемого образца
растворяют в растворе А.
Сравнение: 20 мг ФСО гепарина натрия для
идентификации с помощью ЯМР растворяют в 0,7 мл
раствора А.
При хранении растворы натрия эдетата и дей¬
терированного натрия триметилсилилпропионата
должны находиться в бутылках из полиэтилена вы¬
сокой плотности.
Прибор: спектрометр, работающий минимум при
300 МГц.
Получение спектра Н1-ЯМР:
-количество сканирований: не менее 16;
устанавливают таким образом, чтобы отношение
сигнал/шум было не менее 1000:1 для сигнала метила
гепарина при 2,04 ррт;
- температура: около 25 °С; спектры испытуе¬
мого образца и стандартного образца должны быть
получены при одной и той же температуре;
- время получения спектра: не менее 2 с;
-время повторения (снятие спектра плюс за¬
держка): не менее 4 с;
-спектральная ширина: (10—12) ррт, центри¬
рование вокруг 4,5 ррт;
-ширина пульса: чтобы получался переверну¬
тый угол между 30° и 90°.
Обработка:
-экспоненциальная функция уширения линий:
0,3 Гц;
- фурье-преобразование;
- установление значения 0,00 ррт с помощью
триметилсилилпропионата.
Результаты:
-должны наблюдаться высокие сигналы гепа¬
рина натрия: 2,04 ррт, 3,27 ррт (дублет), 4,34 ррт,
5,22 ррт и 5,42 ррт, для всех ±0,03 ррт;
-после нормализации, выполненной таким об¬
разом, чтобы спектры имели близкие интенсивности,
Н1-ЯМР спектры испытуемого образца и ФСО гепарина
натрия для идентификации с помощью ЯМР сравнивают
количественно; может обнаруживаться дерматансуль¬
фат с сигналом метила при (2,08±0,02) ррт; не должны
наблюдаться неидентифицированные сигналы более
4 % от высоты сигнала гепарина при 5,42 в диапазонах
(0,10—2,00) ррт, (2,10—3,10) ррт и (5,70—8,00) ррт;
могут обнаруживаться идентифицированные сигналы
растворителя и веществ, связанных с процессом; может
происходить варьирование интенсивности сигналов в
некоторых областях спектра гепарина: области варьи¬
рования интенсивности расположены между 3,35 ррт и
4,55 ррт, в которых картина сигналов примерно сохра¬
няется, однако интенсивности варьируют.
С. Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со сле¬
дующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и раствора сравнения (с).
Относительное удерживание (по отношению к
гепарину, время удерживания — около 26 мин): дер¬
матансульфат и хондроитинсульфат — около 0,9;
сверхсульфатированный хондроитинсульфат — око¬
ло 1,3.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 1,3
(Нр — высота пика дерматансульфат + хондроитин¬
сульфат относительно базовой линии; Ну — расстоя¬
ние между базовой линией и нижней точкой кривой,
разделяющей пик дерматансульфат + хондроитин¬
сульфат и пик гепарина).
Результаты: основной пик на хроматограмме
испытуемого раствора (а) по времени удерживания
и форме соответствует основному пику на хромато¬
грамме раствора сравнения (с).
О. Испытуемый образец выдерживает испытание
«Натрий», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. Количество испытуемого образца,
эквивалентное 50 000 МЕ, растворяют в воде Р и
доводят до 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее пятого эталона шка¬
лы наиболее подходящего цвета.
рН (2.2.3). От 5,5 до 8,0. 0,1 г испытуемого об¬
разца растворяют в воде, свободной от углерода
диоксида, Р и доводят до объема 10 мл этим же
растворителем.
Белковые и нуклеотидные примеси. 40 мг
испытуемого образца растворяют в 10 мл воды Р.
Оптическая плотность (2.2.25) полученного раст¬
вора, измеренная при длине волны 260 нм, должна
быть не более 0,15.
Белок. Не более 0,5 % (в пересчете на сухое
вещество).
Раствор А. 2 объема раствора 10 г/л натрия ги¬
дроксида Р смешивают с 2 объемами раствора 50 г/л
натрия карбоната Р и доводят водой Р до 5 объемов.
Раствор В. 2 объема раствора 12,5 г/л меди
сульфата Р смешивают с 2 объемами раствора
29,8 г/л натрия тартрата Р и доводят водой Р до
5 объемов.
Гэпарин натрия
325
Раствор С. 1 объем раствора В смешивают с 50
объемами раствора А.
Раствор О. Фосфорномодибденово-вольфра-
мовый реагент разводят водой Р в 2—4 раза. Разво¬
дят таким образом, чтобы после прибавления рас¬
творов С и О к испытуемому раствору и растворам
сравнения полученные растворы имели значение
рН 10,25±0,25.
Испытуемый раствор. Испытуемый образец
растворяют в воде Р, чтобы получить концентрацию
5 мг/мл.
Растворы сравнения. Бычий альбумин Р раство¬
ряют в воде Р, чтобы получить концентрацию 100 мг/
мл. Готовят разведения водой Р, как указано в разде¬
ле 2.5.33, метод 2.
Контрольный раствор. Вода Р.
Методика. К 1 мл каждого раствора сравнения,
испытуемого раствора и контрольного раствора при¬
бавляют по 5 мл раствора С и выдерживают в тече¬
ние 10 мин. Прибавляют 0,5 мл раствора О, переме¬
шивают и выдерживают при комнатной температуре
в течение 30 мин. Определяют оптическое поглоще¬
ние (2.2.25) полученных растворов при 750 нм, ис¬
пользуя в качестве компенсационной жидкости раст¬
вор, полученный с использованием контрольного
раствора.
Расчеты. Рассчитывают содержание белка, как
указано в разделе 2.5.33, метод 2.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы сравнения стабильны
при комнатной температуре в течение 24 ч.
Испытуемый раствор (а). Точно взвешенную на¬
веску испытуемого образца массой около 50 мг раст¬
воряют в 5,0 мл воды для хроматографии Р. Пере¬
мешивают с использованием вихревой мешалки до
полного растворения.
Испытуемый раствор (Ь). Точно взвешенную на¬
веску испытуемого образца массой около 0,1 г раст¬
воряют в 1,0 мл воды для хроматографии Р. Пере¬
мешивают с использованием вихревой мешалки до
полного растворения. 500 мкл полученного раствора
смешивают с 250 мкл 1 М раствора кислоты хлори-
имяадародмю, прибавляют 50 мкл раствора 250 г/л
натрия нитрита Р, перемешивают и выдерживают
при комнатной температуре в течение 40 мин, после
чего прибавляют 200 мкл 1 М раствора натрия ги¬
дроксида для остановки реакции.
Раствор сравнения (а). 250 мг ФСО гепари¬
на для физико-химического анализа растворяют в
2,0 мл воды для хроматографии Р. Перемешивают с
использованием вихревой мешалки до полного раст¬
ворения.
Раствор сравнения (Ь). 1200 мкл раствора срав¬
нения (а) прибавляют к 300 мкл ФСО дерматансуль¬
фата и сверхсульфатированного хондроитинсуль-
фата. Перемешивают с использованием вихревой
мешалки до однородного состояния.
Раствор сравнения (с). 100 мкл раствора срав¬
нения (Ь) прибавляют к 900 мкл воды для хромато¬
графии Р. Перемешивают с использованием вихре¬
вой мешалки до однородного состояния.
Раствор сравнения (<Л). 400 мкл раствора срав¬
нения (а) прибавляют к 100 мкл воды для хромато¬
графии Р и перемешивают с использованием вих¬
ревой мешалки. Полученный раствор смешивают с
250 мкл 1 М раствора кислоты хлористоводород¬
ной, прибавляют 50 мкл раствора 250 г/л натрия ни¬
трита Р, перемешивают и выдерживают при комнат¬
ной температуре в течение 40 мин, после чего прибав¬
ляют 200 мкл 1 М раствора натрия гидроксида для
остановки реакции.
Раствор сравнения (е). К 500 мкл раствора срав¬
нения (Ь) прибавляют 250 мкл 1 М раствора кислоты
хлористоводородной, затем 50 мкл раствора 250 г/л
натрия нитрита Р, перемешивают и выдерживают
при комнатной температуре в течение 40 мин, после
чего прибавляют 200 мкл 1 М раствора натрия ги¬
дроксида для остановки реакции.
Условия хроматографирования:
- предколонка длиной 0,05 м и внутренним диа¬
метром 2 мм, заполненная анионообменной смолой Р
с размером частиц 13 мкм;
- колонка длиной 0,25 м и внутренним диаме¬
тром 2 мм, заполненная анионообменной смолой Р с
размером частиц 9 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: 0,40 г натрия дигидро¬
фосфата Р растворяют в 1 л воды для хромато¬
графии,Р и доводят до рН 3,0 кислотой фосфор¬
ной разведенной Р;
- подвижная фаза В: 0,40 г натрия дигидро¬
фосфата Р растворяют в 1 л воды для хромато¬
графии Р, прибавляют 140 г натрия перхлора¬
та Р и доводят до рН 3,0 кислотой фосфорной
разведенной Р; фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
75
25
10—35
75—0
25—100
35—40
0
100
- скорость подвижной фазы: 0,22 мл/мин;
- спектрофотометрический детектор, длина
волны 202 нм;
-уравновешивание: не менее 15 мин;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и растворов сравнения (с!) и (е).
Относительное удерживание (по отношению к
гепарину, время удерживания — около 26 мин): дерма¬
тансульфат и хондроитинсульфат — около 0,9; сверх-
сульфатированный хондроитинсульфат — около 1,3.
Пригодность хроматографической системы:
- на хроматограмме раствора сравнения (с!) не
должен обнаруживаться пик, соответствующий по
времени удерживания пику гепарина;
- разрешение: не менее 3,0 между пиком дерма¬
тансульфат + хондроитинсульфат и пиком сверхсуль¬
фатированного хондроитинсульфата на хроматограм¬
ме раствора сравнения (е).
Предельное содержание примесей:
- сумма дерматансульфата и хондроитин¬
сульфата (не более 2,0 %): на хроматограмме ис¬
пытуемого раствора (Ь) площадь пика дерматан¬
сульфат + хондроитинсульфат не должна превышать
площадь соответствующего пика на хроматограмме
раствора сравнения (е);
- любая другая примесь: на хроматограмме ис¬
пытуемого раствора (Ь) не должны обнаруживаться
пики, кроме основного и пика дерматансульфат + хон¬
дроитинсульфат.
Азот (2.5.9) Не менее 1,5 % и не более 2,5 % (в
пересчете на сухое вещество). Определение прово¬
дят из 0,100 г испытуемого образца.
42. Зак. 1060.
326
Гэсударственная фармакопея Республики Беларусь
Натрий. Не менее 9,5 % и не более 12,5 % (в пе¬
ресчете на сухое вещество). Атомно-абсорбционная
спектрометрия (2.2.23, метод I).
Испытуемый образец. 50 мг испытуемого образ¬
ца растворяют в растворе 1,27 мг/мл цезия хлорида Р
в 0,1 М растворе кислоты хлористоводородной и
доводят до объема 100,0 мл этим же растворителем.
Растворы сравнения. Готовят растворы сравне¬
ния, содержащие 25 ррт, 50 ррт и 75 ррт №, раз¬
ведением эталонного раствора натрия (200 ррт
Ыа) Р раствором 1,27 мг/мл цезия хлорида Р в 0,1 М
растворе кислоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения натрия.
Длина волны: 330,3 нм.
Гэнератор атомного пара: пламя подходящего
состава (например, воздух — 11 л/мин, ацетилен —
2 л/мин).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0030 % (30 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 3,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 8,0 %. 1,000 г испытуемого образца сушат при
температуре 60 °С над фосфора (V) оксидом Р и дав¬
лении, не превышающем 670 Па, в течение 3 ч.
Бактериальные эндотоксины (2.6.14). Менее
0,01 МЕ в Международной единице гепарина, если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения
без последующей процедуры удаления бактериаль¬
ных эндотоксинов.
#Пирогенность (2.6.8). Субстанция должна быть
апирогенной. Тест-доза — 2000 МЕ в 1 мл раствора
9 г/л натрия хлорида Р на 1 кг массы тела кролика.
Раствор вводят внутривенно.
Тест «Пирогенность» проводится в качестве аль¬
тернативного тесту «Бактериальные эндотоксины».
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят количественное определение гепарина
(2.7.5). Полученная активность должна быть не менее
90 % и не более 111 % от заявленной активности. Ре¬
зультаты испытаний считаются достоверными, если
границы доверительного интервала определения
(Р = 0,95) составляют от 80 % до 125 % от заявленно¬
го значения активности.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
МАРКИРОВКА
Указывают:
-количество Международных единиц в милли¬
грамме;
- вид животного, из которого получен гепарин.
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:0335
ГИДРОКОРТИЗОН
НудгосогНзопит
НУОКОСОКТ180ЫЕ
С21нзо05 М.м. 362,5
[50-23-7]
ОПРЕДЕЛЕНИЕ
11Р, 17,21 -Тригидроксипрегн-4-ен-3,20-дион.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, умеренно рас¬
творим в ацетоне и в 96 % спирте, мало растворим в
метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гидрокортизона #или спектр,
представленный на рисунке #0335.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО гидрокортизона растворяют
по отдельности в минимальном объеме ацетона Р и
выпаривают на водяной бане до сухих остатков, кото¬
рые используют для получения новых спектров.
B. Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со сле¬
дующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (с).
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (с).
C. Тонкослойная хроматография (2.2.27).
Раствор А. 25 мг испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 5 мл этим
же растворителем.
Раствор В. 25 мг ФСО гидрокортизона раство¬
ряют в метаноле Р и доводят до объема 5 мл этим
же растворителем.
Испытуемый раствор (а). 2 мл раствора А дово¬
дят метиленхлоридом Р до объема 10 мл.
Испытуемый раствор (Ь). 0,4 мл раствора А по¬
мещают в стеклянную пробирку длиной 100 мм и диа¬
метром 20 мм с подходящей стеклянной притертой
Гидрокортизон
327
Волновое число (см'1)
Рисунок #0335.-1. Инфракрасный спектр ФСО гидрокортизона
пробкой или пробкой из политетрафторэтилена и вы¬
паривают растворитель при слабом нагревании в по¬
токе азота Р. Прибавляют 2 мл раствора 15 % (об/об)
кислоты уксусной ледяной Р и 50 мг натрия вишу-
тата Р, пробирку закрывают и встряхивают получен¬
ную суспензию в течение 1 ч на механическом встря-
хивателе, защищая раствор от света. Прибавляют
2 мл раствора 15 % (об/об) кислоты уксусной ледя¬
ной Р и фильтруют в делительную воронку вместимо¬
стью 50 мл, промывая фильтр двумя порциями воды Р
по 5 мл каждая. Прозрачный фильтрат встряхивают с
10 мл метиленхлорида Р. Органический слой промы¬
вают 5 мл 1 М раствора натрия гидроксида и двумя
порциями воды Р по 5 мл каждая и высушивают с ис¬
пользованием натрия сульфата безводного Р.
Раствор сравнения (а). 2 мл раствора В доводят
метиленхлоридом Р до объема 10 мл.
Раствор сравнения (Ь). 0,4 мл раствора В поме¬
щают в стеклянную пробирку длиной 100 мм и диаме¬
тром 20 мм с подходящей стеклянной притертой проб¬
кой или пробкой из политетрафторэтилена и выпари¬
вают растворитель при слабом нагревании в потоке
азота Р. Прибавляют 2 мл раствора 15 % (об/об) кис¬
лоты уксусной ледяной Р и 50 мг натрия висмута-
та Р, пробирку закрывают и встряхивают полученную
суспензию в течение 1 ч на механическом встряхи-
вателе, защищая раствор от света. Прибавляют 2 мл
раствора 15 % (об/об) кислоты уксусной ледяной Р и
фильтруют в делительную воронку вместимостью 50
мл, промывая фильтр двумя порциями воды Р по 5 мл
каждая. Прозрачный фильтрат встряхивают с 10 мл
метиленхлорида Р. Органический слой промывают
5 мл 1 М раствора натрия гидроксида и двумя пор¬
циями воды Р по 5 мл каждая и высушивают с исполь¬
зованием натрия сульфата безводного Р
Пластинка: ТСХ пластинка со слоем силикаге-
Подвижная фаза А: смесь из 1,2 объема воды Р
и 8 объемов метанола Р прибавляют к смеси из 15
объемов эфира Р\л77 объемов метиленхлорида Р.
Подвижная фаза В: бутанол Р, насыщенный во¬
дой Р — толуол Р — эфир Р (5:15:80, об/об/об).
Наносимый объем пробы: по 5 мкл испытуемого
раствора (а) и раствора сравнения (а) и по 25 мкл ис¬
пытуемого раствора (Ь) и раствора сравнения (Ь), по¬
следние два раствора наносят малыми количествами
для получения пятен небольшого размера.
Фронт подвижной фазы, не менее 15 см от ли¬
нии старта в подвижной фазе А и не менее 15 см в
подвижной фазе В.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: основные пятна на хроматограм¬
мах испытуемых растворов (а) и (Ь) соответствуют по
расположению и размеру основным пятнам на хрома¬
тограммах соответствующих растворов сравнения.
Проявление В: пластинку опрыскивают раство¬
ром кислоты серной спиртовым Р и нагревают при
температуре 120 °С в течение 10 мин или до проявле¬
ния пятен и охлаждают. Просматривают при дневном
свете и в ультрафиолетовом свете при длине волны
365 нм.
Результаты В: основные пятна на хроматограм¬
мах испытуемых растворов (а) и (Ь) соответствуют по
расположению и цвету при просматривании при днев¬
ном свете и флуоресценции при просмотре в ультра¬
фиолетовом свете при длине волны 365 нм и размеру
основным пятнам на хроматограммах соответствую¬
щих растворов сравнения. Значения основных пя¬
тен на хроматограммах испытуемого раствора (Ь) и
раствора сравнения (Ь) заметно выше значений Рг ос¬
новных пятен на хроматограммах испытуемого раст¬
вора (а) и раствора сравнения (а).
42*7 Зак. 1060.
328
Гэсударственная фармакопея Республики Беларусь
О. 2 мг испытуемого образца прибавляют к 2 мл
кислоты серной Р и встряхивают до растворения. В
течение 5 мин появляется интенсивное коричнева¬
то-красное окрашивание и зеленая флуоресценция,
очень интенсивная при просматривании в ультрафи¬
олетовом свете при длине волны 365 нм. Полученный
раствор прибавляют к 10 мл воды Р и перемешивают.
Окраска исчезает, а раствор остается прозрачным.
Флуоресценция при просматривании в ультрафиоле¬
товом свете при длине волны 365 нм не исчезает.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+162 до +168 (в пересчете на сухое вещество). 0,200 г
испытуемого образца растворяют в метаноле Р, до¬
водят до объема 25,0 мл этим же растворителем и об¬
рабатывают ультразвуком в течение 10 мин.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р —
вода Р (40:60, об/об).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в смеси растворителей, доводят до
объема 10,0 мл этой же смесью растворителей и об¬
рабатывают ультразвуком в течение 10 мин.
Раствор сравнения (а). 4 мг ФСО преднизолона
(примесь А), 2 мг ФСО кортизона (примесь В), 8 мг
ФСО гидрокортизона ацетата (примесь С) и 6 мг
вещества Рейхштейна 8 Р (примесь Р) растворяют в
40 мл ацетонитрила Р и доводят водой Р до объема
100.0 мл. 0,5 мл полученного раствора доводят испы¬
туемым раствором до объема 5,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 2 мг ФСО гидрокортизо¬
на растворяют в 1,0 мл смеси растворителей и обра¬
батывают ультразвуком в течение 10 мин.
Раствор сравнения (д). 2 мг ФСО гидрокортизо¬
на для идентификации пиков (содержит примеси й,
Е, С, Н, I и N1) растворяют в 1,0 мл смеси растворите¬
лей и обрабатывают ультразвуком в течение 10 мин.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: вода Р\
- подвижная фаза В: ацетонитрил Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—18
74
26
18—32
74 —► 55
26 —* 45
32-48
55 —* 30
45 —* 70
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора, растворов сравнения (а), (Ь) и (б).
Идентификация пиков примесей: идентифици¬
руют пики примесей О, Е, О, Н, I и N1, используя хро¬
матограмму раствора сравнения (с!) и хроматограмму,
прилагаемую к ФСО гидрокортизона для идентифи¬
кации пиков] идентифицируют пики примесей А, В, С и
Р, используя хроматограмму раствора сравнения (а).
Относительное удерживание (по отношению к
гидрокортизону, время удерживания — около 24 мин):
примесь О — около 0,2; примесь Н — около 0,3; при¬
месь I — около 0,5; примесь 6 — около 0,8; примесь
Е — около 0,86; примесь А—около 0,96; примесь В —
около 1,1; примесь Р — около 1,4; примесь С — около
1,5; примесь N — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 3,0
(Нр — высота пика примеси А относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси А и пик
гидрокортизона).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 1,8; для примеси Е — 2,7):
- примеси С, О, Е, I (не более 0,5 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С, О, Е и I, не должны превы¬
шать 5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- примесь О (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси 6, не должна превышать 4-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь Р (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Р, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, В (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А и В, не должны превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (Ь);
-примеси Н, N (не более 0,15%): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Н и N. не должны превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е, Р, 6, Н, I и N. не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 2,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
20-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Гидрокортизон
329
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 96 %
спирте Р и доводят до объема 100,0 мл этим же
растворителем. 2,0 мл полученного раствора доводят
96 % спиртом Р до объема 100,0 мл. Измеряют оп¬
тическую плотность (2.2.25) полученного раствора в
максимуме при длине волны 241,5 нм.
Содержание С21Н30О5 рассчитывают с учетом
удельного показателя поглощения, равного 440.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, С,
Н, I, N.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): 3, К, Ц М, О.
А. 11 Р,17,21-Тригидроксипрегна-1,4-диен-3,20-дион
(преднизолон).
В. 17,21-Дигидроксипрегн-4-ен-3,11,20-трион (кор¬
тизон).
С. 11Р, 17-Дигидрокси-3,20-диоксопрегн-4-бн-21 -
илацетат (гидрокортизона ацетат).
О. Р1 = РЗ = ОН, Р2 = Р4 = Н, Р5 = СН2ОН: бр,11р,-
17,21 -Тетрагидроксипрегн-4-ен-3,20-дион (бр-гидрокси-
гидрокортизон).
Р. Р1 = Р2 = РЗ = Р4 = Н, Р5 = СН2ОН: 17,21-Дигид-
роксипрегн-4-ен-3,20-дион (вещество Рейхштейна 3).
0. Р1 = Р2 = Р4 = Н, РЗ = ОН, Р5 =СНО: 11р,17-
Дигидрокси-3,20-диоксопрегн-4-ен-21-аль (гидрокор¬
тизон-21-альдегид).
Н. Р1 = Р4 = Н, Р2 = РЗ = ОН, Р5 = СН2ОН: 7а,11р,-
17.21 -Тетрагидроксипрегн-4-ен-3,20-дион (7а-гидрокси-
гидрокортизон).
1. Р1 = Р2 = Н, РЗ = Р4 = ОН, Р5 = СН2ОН: 11р,14,-
17.21 -Тетрагидроксипрегн-4-ен-3,20-дион (14а-гидрокси-
гидрокортизон).
К. Р1 = Р2 = РЗ = Р4 = Н, Р5 = СН2-0-С0-СН3:
17-Гидрокси-3,20-диоксопрегн-4-ен-21 -илацетат (веще¬
ство Рейштейна 3-21-ацетат).
Е. 11 р, 17,21 -Тригидроксипрегна-4,6-диен-3,20-дион
(Аб-гидрокортизон).
3. Р1 = Н, Р2 = СО-СН3; РЗ = ОН: 11р,21-Дигид-
рокси-3,20-диоксопрегн-4-ен-17-илацетат (гидрокор¬
тизон-17-ацетат).
1~ Р1 = Р2 = РЗ = Н: 11р,17-Дигидроксипрегн-4-ен-
3,20-дион (оксенол).
О. Р1 = РЗ = ОН, Р2 = Н: 11р, 17,19,21-Тетрагидро-
ксипрегн-4-ен-3,20-дион (19-гидроксигидрокортизон).
М. 11а,17,21-Тригидроксипрегн-4-ен-3,20-дион (эпи¬
гидрокортизон).
N. 11 р, 17,21 -Тригидрокси-21 -(11 р, 17,21 -тригидро-
кси-3,20-диоксопрегн-4-ен-21-ил)прегн-4-ен-3,20-дион
(димер гидрокортизона).
43. Зак. 1060.
330
Гэсударственная фармакопея Республики Беларусь
07/2016:0334
ГИДРОКОРТИЗОНА АЦЕТАТ
НубгосогИзоп/ асе^а8
ЯУ0Я0С0ЯГ/50Л/Е А СЕТАТЕ
С23Н3206 М.м. 404,5
[50-03-3]
ОПРЕДЕЛЕНИЕ
110,17-Дигидрокси-3,2О-диоксопрегн-4-ен-21-
илацетат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, мало раство¬
рим в этаноле безводном и в метиленхпориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: С, О, Е.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гидрокортизона ацетата #или
спектр, представленный на рисунке #0334.-1#.
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора (Ь) по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (с1).
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 25 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем (раствор А). 2 мл полу¬
ченного раствора доводят метиленхлоридом Р до
объема 10 мл.
Испытуемый раствор (Ь). 2 мл раствора А по¬
мещают в стеклянную пробирку вместимостью 15 мл
со стеклянной притертой пробкой или подходящей
пробкой из политетрафторэтилена, прибавляют 10 мл
раствора калия гидрокарбоната метанольного на¬
сыщенного Р и немедленно пропускают струю азо¬
та Р через раствор в течение 5 мин. Пробирку закры¬
вают пробкой, нагревают в водяной бане при темпера¬
туре 45 °С, защищая раствор от света, в течение 2 ч
30 мин и охлаждают.
Раствор сравнения (а). 25 мг ФСО гидрокорти¬
зона ацетата растворяют в метаноле Р и доводят
до объема 5 мл этим же растворителем (раствор В).
2 мл полученного раствора доводят метиленхлори¬
дом Р до объема 10 мл.
Раствор сравнения (Ь). 2 мл раствора В поме¬
щают в стеклянную пробирку вместимостью 15 мл
со стеклянной притертой пробкой или подходящей
пробкой из политетрафторэтилена, прибавляют 10 мл
раствора калия гидрокарбоната метанольного на¬
сыщенного Р и немедленно пропускают струю азо¬
та Р через раствор в течение 5 мин. Пробирку закры¬
вают пробкой, нагревают в водяной бане при темпера¬
туре 45 °С, защищая раствор от света, в течение 2 ч
30 мин и охлаждают.
зооо
2000 1500
Волновое число (см1)
1000
Рисунок #0334.-1. Инфракрасный спектр ФСО гидрокортизона ацетата
Гидрокортизона ацетат
331
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Г 2!4Р.
Подвижная фаза: смесь из 1,2 объема воды Р и 8
объемов метанола Р прибавляют к смеси из 15 объ¬
емов эфира Р и 77 объемов метиленхлорида Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе?
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: основные пятна на хроматограм¬
мах испытуемых растворов соответствуют по распо¬
ложению и размеру основным пятнам на хромато¬
граммах соответствующих растворов сравнения.
Проявление В: пластинку опрыскивают раство¬
ром кислоты серной спиртовым Р и нагревают при
температуре 120 °С в течение 10 мин или до проявле¬
ния пятен и охлаждают. Просматривают при дневном
свете и в ультрафиолетовом свете при длине волны
365 нм.
Результаты В: основные пятна на хроматограм¬
мах испытуемых растворов соответствуют по рас¬
положению, цвету при просматривании при дневном
свете и флуоресценции при просматривании в ультра¬
фиолетовом свете при длине волны 365 нм и размеру
основным пятнам на хроматограммах соответствую¬
щих растворов сравнения. Значения К, основных пя¬
тен на хроматограммах испытуемого раствора (Ь) и
раствора сравнения (Ь) заметно ниже значений /?, ос¬
новных пятен на хроматограммах испытуемого раст¬
вора (а) и раствора сравнения (а).
й. 2 мг испытуемого образца прибавляют к 2 мл
кислоты серной Р и встряхивают до растворения. В
течение 5 мин появляется интенсивное коричневато-
красное окрашивание с зеленой флуоресценцией,
особенно интенсивной при просматривании в ультра¬
фиолетовом свете при длине волны 365 нм. Полу¬
ченный раствор прибавляют к 10 мл воды Р и пере¬
мешивают. Окрашивание исчезает, а флуоресценция
в ультрафиолетовом свете не исчезает.
Е. Около 10 мг испытуемого образца дают реак¬
цию на ацетил (2.3.1).
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+158 до +167 (в пересчете на сухое вещество). 0,250 г
испытуемого образца растворяют в диоксане Р и до¬
водят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Уксусная кислота Р —
вода Р — метанол Р (1:10:90, об/об/об).
Испытуемый раствор (а). 25,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 10,0 мл.
Раствор сравнения (а). 2 мг ФСО гидрокортизо¬
на ацетата и 2 мг ФСО преднизолона ацетата (при¬
месь С) растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 5 мг ФСО гидрокорти¬
зона ацетата для идентификации пиков (содержит
примеси А, В, О, Е и (3) растворяют в 2,0 мл смеси
растворителей.
Раствор сравнения (д). 25,0 мг ФСО гидрокор¬
тизона ацетата растворяют в смеси растворителей
и доводят до объема 25,0 мл этой же смесью раст¬
ворителей. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: 400 мл ацетонитрила Р
смешивают с 550 мл воды Р, выдерживают до уста¬
новления равновесия, доводят водой Р до объема
1000,0 мл и перемешивают;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с);
- время хроматографирования: 4-кратное вре¬
мя удерживания гидрокортизона ацетата.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, О, Е и 6, используя хрома¬
тограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО гидрокортизона ацетата для
идентификации пиков; идентифицируют пик примеси
С, используя хроматограмму раствора сравнения (а).
Относительное удерживание (по отношению к
гидрокортизона ацетату, время удерживания — около
10 мин): примесь А — около 0,4; примесь В — около
0,7; примесь С — около 0,9; примесь О — около 1,2;
примесь С — около 1,8; примесь Е — около 2,3.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками при¬
меси С и гидрокортизона ацетата.
Предельное содержание примесей:
- примесь С (не более 0,6 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси С, не должна превышать 6-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси А, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В, О и Е (не более 0,3 %): на хрома¬
тограмме испытуемого раствора (а) площади пиков,
соответствующих примесям В, О и Е, не должны пре¬
вышать 3-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
-примесь О (не более 0,15 %): на хроматограм¬
ме испытуемого раствора (а) площадь пика, соответ¬
ствующего примеси <3, не должна превышать 1,5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е и (3, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
43*. Зак. 1060.
332
Гэсударственная фармакопея Республики Беларусь
- сумма примесей (не более 1,5 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
15-кратную ллощадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (б);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания гидрокортизона ацетата.
Время удерживания: гидрокортизона ацетат —
около 10 мин.
Содержание С23Н3206 в процентах рассчитывают
с учетом содержания гидрокортизона ацетата в ФСО
гидрокортизона ацетата.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е
А. 11р,17,21-Тригидроксипрегн-4-ен-3,20-дион (ги¬
дрокортизон).
В. 11 р, 17-Дигидроксипрегн-4-ен-3,20-дион (оксенол).
С. 11 р, 17-Дигидрокси-3,20-диоксопрегна-1,4-диен-
21-илацетат (преднизолона ацетат).
О. 17-Гидрокси-3,11,20-триоксопрегн-4-ен-21 -ил-
ацетат (кортизона ацетат).
Е. 17-Гидрокси-3,20-диоксопрегна-4,9(11 )-диен-21 -
илацетат.
Р. 11 а, 17-Дигидрокси-3,20-диоксопрегн-4-ен-21 -
илацетат (эпи-гидрокортизона ацетат).
О. 17-Гидрокси-3,20-диоксопрегн-4-ен-11р,21-
диилдиацетат.
07/2016:1616
ГИДРОКСИКАРБАМИД
НубгохусагЬатШит
НУйКОХУСАНВАМЮЕ
О
СН4М202 М.м. 76,1
[127-07-1]
ОПРЕДЕЛЕНИЕ
А/-Гидроксимочевина.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
Гидроксикарбамид
333
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гидроксикарбамида #или
спектр, представленный на рисунке #1616.-1#.
Если спектры, полученные с использованием образ¬
цов в твердом состоянии, отличаются, то испытуемый об¬
разец и ФСО гидроксикарбамида растворяют по отдель¬
ности в 96 % спирте Р, выпаривают до сухих остатков,
которые используют для получения новых спектров.
B. Просматривают хроматограммы, полученные
в испытании «Мочевина». На хроматограмме испы¬
туемого раствора обнаруживается основное пятно,
соответствующее по расположению и размеру ос¬
новному пятну на хроматограмме раствора сравне¬
ния (с).
ИСПЫТАНИЯ
Мочевина. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого образ¬
ца растворяют в воде Р и доводят до объема 1,0 мл
этим же растворителем.
Раствор сравнения (а). 12,5 мг мочевины Р раст¬
воряет в воде Р и доводят до объема 50 мл этим же
растворителем.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 5 мг мочевины Р растворяют в воде Р и доводят
до объема 20 мл этим же растворителем.
Раствор сравнения (с). 50 мг ФСО гидроксикар¬
бамида растворяют в воде Р и доводят до объема
1 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: пиридин Р — вода Р — этила-
цетат Р (2:2:10, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
10 г/л диметиламинобензальдегида Р в 1 М раство¬
ре кислоты хлористоводородной.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме должны обнаруживаться два
полностью разделенных пятна.
Предельное содержание примесей:
- мочевина (не более 0,5 %): на хроматограмме
испытуемого раствора пятно, соответствующее моче¬
вине, должно быть не интенсивнее соответствующего
пятна на хроматограмме раствора сравнения (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Испытуемый раствор (Ь). 5,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
50.0 мл.
Раствор сравнения (а). Раствор готовят непо¬
средственно перед использованием. 0,100 г гидрок-
силамина гидрохлорида Р и 5 мг испытуемого образ¬
ца растворяют в подвижной фазе и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 0,1 мл испытуемого
раствора (а) доводят подвижной фазой до объема
100.0 мл.
Раствор сравнения (с). 0,100 г ФСО гидрокси¬
карбамида растворяют в подвижной фазе и доводят
Рисунок #1616.-1. Инфракрасный спектр ФСО гидроксикарбамида
44. Зак. 1060.
334
Гэсударственная фармакопея Республики Беларусь
до объема 10,0 мл этим же растворителем. 5,0 мл
полученного раствора доводят подвижной фазой до
объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: метанол Р — вода Р (5:95,
об/об);
- скорость подвижной фазы: 0,5 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (Ь);
- время хроматографирования: 3-кратное вре¬
мя удерживания гидроксикарбамида (которое состав¬
ляет около 5 мин).
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,0 между пиками при¬
меси А и гидроксикарбамида.
Предельное содержание примесей:
-любая примесь (не более 0,1 %): на хромато¬
грамме испытуемого раствора (а) площадь любого
пика, кроме основного, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,02 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики
с площадью менее 0,2 площади основного пика раст¬
вора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0050 % (50 ррт).
1,0 г испытуемого образца растворяют в воде Р и до¬
водят до объема 15 мл этим же растворителем. Полу¬
ченный раствор должен выдерживать испытание на
хлориды.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 2,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
А. Н2М-ОН: Гидроксиламин.
07/2016:0337
ГИДРОКСИПРОПИЛЦЕЛЛЮЛОЗА
Нус1гохургору1се11и1озит
НУОРОХУРРОРУ1-СЕ1.ИЛ.ОЗЕ
[9004-64-2]
ОПРЕДЕЛЕНИЕ
Частично 0-(2-гидроксипропилированная) цел¬
люлоза.
Содержание: не менее 53,4 % и не более 80,5 %
гидроксипропоксигрупп (в пересчете на сухое веще¬
ство).
Может содержать противослеживающие компо¬
ненты, например кремния диоксид (5Ю2).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок или грану¬
лы. Слегка гигроскопична.
Растворима в холодной воде, 96 % спирте, про-
пиленгликоле с образованием коллоидных растворов,
практически нерастворима в горячей воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца растворяют в воде Р
и доводят до объема 100 мл этим же растворителем.
1 мл полученного раствора помещают на предметное
стекло. После выпаривания воды образуется тонкая
пленка.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гидроксипропилцеллюлозы.
Не учитывают полосы с волновым числом около
1719 см1.
C. 1,0 г испытуемого образца равномерно рас¬
пределяют в 100 мл кипящей воды Р и смесь переме¬
шивают с помощью магнитной мешалки с магнитным
стержнем длиной около 25 мм. 50 мл полученного
раствора помещают в лабораторный стакан с 50 мл
воды Р. В полученный раствор помещают термометр,
перемешивают с помощью магнитной мешалки на го¬
рячей плите и нагревают со скоростью (2—5) °С/мин.
При температуре свыше 40 °С раствор становится
мутным или образуется хлопьевидный осадок. Раст¬
вор снова становится прозрачным при охлаждении.
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 8,5.1,0 г испытуемого образ¬
ца равномерно распределяют в 100 мл кипящей воды,
свободной от углерода диоксида, Р, смесь переме¬
шивают с помощью магнитной мешалки
Вязкость (2.2.10). Не менее 75% и не более
140 % от значения, указанного на этикетке. 6,00 г ис¬
пытуемого образца (в пересчете на сухое вещество)
растворяют при постоянном перемешивании в 150 г
воды Р, нагретой до температуры 90 °С. Переме¬
шивают при помощи лопастной мешалки в течение
10 мин. Продолжая перемешивание, колбу помещают
в ледяную баню и оставляют на 40 мин до полного
растворения. Доводят массу полученного раствора
до 300 г и раствор центрифугируют для вытеснения
пузырьков воздуха. Доводят раствор до температуры
Гидроксипропилцеллюлоза
335
(20±0,1)°С. Определяют вязкость при помощи рота¬
ционного вискозиметра при температуре 20 °С и со
скоростью сдвига 10с1.
В случае низкой вязкости, раствор для испыта¬
ния готовят в концентрации, указанной на этикетке.
Кремния диоксид. Не более 0,6 %. Если на эти¬
кетке есть информация о присутствии кремния диок¬
сида и определенное при испытании «Сульфатная
зола» значение превышает 0,2 %, то остаток, получен¬
ный при испытании «Сульфатная зола», смачивают
водой Р и прибавляют к нему небольшими порциями
5 мл кислоты фтористоводородной Р Выпаривают
при температуре от 95 °С до 105 °С досуха, избегая
разбрызгивания. Охлаждают и омывают стенки плати¬
нового тигля 5 мл кислоты фтористоводородной Р.
Прибавляют 0,5 мл кислоты серной Р и выпаривают
досуха. Постепенно повышают температуру до тех
пор, пока все кислоты улетучатся, и сжигают остаток
при температуре (1000±25) °С. Охлаждают в эксика¬
торе и взвешивают. Разница между массой остатка,
полученного при испытании «Сульфатная зола», и
массой полученного остатка представляет собой со¬
держание кремния диоксида в испытуемом образце.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,8%.
Определение проводят из 1,0 г испытуемого образца,
используя платиновый тигель.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. К 10 мл
о-ксилсла Р прибавляют 1,0 мл метилциклогекса-
на Р и доводят до объема 50,0 мл о-ксилолом Р.
Испытуемый раствор. В реакционный сосуд
вместимостью 5,0 мл помещают 30,0 мг испытуемо¬
го образца (в пересчете на сухое вещество) и при¬
бавляют 60 мг кислоты адипиновой Р. Прибавляют
2,00 мл раствора внутреннего стандарта и 1,0 мл
кислоты йодистоводородной Р Закрывают септой и
точно взвешивают реакционный сосуд (общая масса
перед нагреванием). Сосуд помещают в сушильный
шкаф или нагревают в подходящем термостате, обе¬
спечивающем поддержание внутренней температуры
(115±2)°С в течение 70 мин, при постоянном пере¬
мешивании. Реакционный сосуд охлаждают и точно
взвешивают (общая масса после нагревания). Если
разница значений общей массы до нагревания и по¬
сле нагревания составляет более 10 мг, то готовят
новый раствор. После фазового разделения прокалы¬
вают септу сосуда с помощью охлажденного шприца
и отделяют объем верхней фазы, достаточный для ис¬
пользования в качестве испытуемого раствора.
Раствор сравнения. 60 мг кислоты адипино¬
вой Р и 2,00 мл раствора внутреннего стандарта по¬
мещают в реакционный сосуд вместимостью 5,0 мл,
прибавляют 1,0 мл кислоты йодистоводородной Р,
закрывают септой и точно взвешивают. 25 мкл изопро-
пилйодида Р вводят через септу и опять точно взве¬
шивают. Тщательно перемешивают. После фазового
разделения прокалывают септу сосуда с помощью ох¬
лажденного шприца и отделяют объем верхней фазы,
достаточный для использования в качестве раствора
сравнения.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,53 мм, покрытая слоем поли(диметил)-
силоксана Р (толщина слоя 3 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 7 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—3
40
3—9
40 — 100
9—12
100 — 250
12—15
250
Блок ввода проб
180
Детектор
280
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 2 мкл.
Относительное удерживание (по отношению
к метилциклогексану, время удерживания — около
8 мин): изопропилйодид — около 0,8.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 2,0 между пиками изо-
пропилйодида и метилциклогексана на хроматограм¬
ме раствора сравнения;
- сходимость: максимальное относительное
стандартное отклонение фактора отклика для основ¬
ного пика не более 2,0 % при 6 вводах пробы.
Фактор отклика Р рассчитывают по формуле:
А-ЩС
А2-Ш ’
где:
/\1 — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения;
А2 — площадь пика изопропилйодида на хрома¬
тограмме раствора сравнения;
Щ — масса изопропилйодида Р в растворе срав¬
нения, мг;
С — процентное содержание изопропилйодида Р.
Содержание гидроксипропильных групп в про¬
центах (м/м) рассчитывают по формуле:
1,15 -А^П-М^Ш
А3ЩМ2 9
где:
А3 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора;
А4 — площадь пика изопропилйодида на хрома¬
тограмме испытуемого раствора;
Р — фактор отклика;
— молекулярная масса гидроксипропокси-
группы (75,1);
М2 — молекулярная масса изопропилиодида
(170,0);
IУ2 — масса образца (в пересчете на сухое веще¬
ство) в испытуемом растворе, мг;
1,15 — коэффициент пересчета.
44*. Зак. 1060.
336
Гэсударственная фармакопея Республики Беларусь
МАРКИРОВКА
Указывают:
- вязкость 2 % (м/м) раствора, мПа-с;
- в случае низкой вязкости — используемую кон¬
центрацию раствора и вязкость, мПа-с;
- при необходимости указывают, что субстанция
содержит оксид кремния.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомогатель¬
ного вещества (см. статью 5.15). Некоторые харак¬
теристики, описанные в данном разделе, могут быть
также представлены в обязательном разделе част¬
ной статьи, так как они представляют собой обяза¬
тельные показатели качества. В этом случае данный
раздел содержит ссылку на испытания, описанные в
обязательной части. Контроль данных характери¬
стик может вносить вклад в качество лекарствен¬
ного средства путем достижения постоянства
производственного процесса и улучшения свойств
лекарственного средства при его использовании.
Предлагаемые методы испытаний являются подхо¬
дящими для указанных целей, однако другие методы
также могут быть использованы. В случае указания
результатов конкретных характеристик требуется
указывать также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для гидроксипропилцеллюлозы, используемой в
качестве связующего, повышающего вязкость ком¬
понента или пленкообразователя.
Вязкость. См. раздел «Испытания».
Степень замещения. См. раздел «Количествен¬
ное определение».
Следующие характеристики могут быть важ¬
ны для гидроксипропилцеллюлозы, используемой для
получения матрицы для таблеток с пролонгирован¬
ным высвобождением.
Вязкость. См. раздел «Испытания».
Степень замещения. См. раздел «Количествен¬
ное определение».
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошков (2.9.36).
07/2016:0394
ГИДРОХЛОРТИАЗИД
Нус1госЫого1Ыа21с1ит
НУОКОСН1.(ЖОТН1А110Е
н
С7Н8С1Ыз0452 М.м. 297,7
[58-93-5]
ОПРЕДЕЛЕНИЕ
6-Хлор-3,4-дигидро-2Н-1,2,4-бензотиадиазин-7-
сульфонамид-1,1 -диоксид.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
зооо
2000
1500
1000
Волновое число (см1)
Рисунок #0394.-1. Инфракрасный спектр ФСО гидрохлортиазида
Гидрохлортиазид
337
Очень мало растворим в воде, растворим в аце¬
тоне, умеренно растворим в 96 % спирте. Растворя¬
ется в разведенных растворах гидроксидов щелочных
металлов.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 10 мл 0,1 М раствора натрия
гидроксида и доводят водой Р до объема 100,0 мл.
2,0 мл полученного раствора доводят 0,01 М раство¬
ром натрия гидроксида до объема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм.
Максимумы поглощения: при 273 нм и при
323 нм.
Отношение оптических плотностей:
—от5-4 Д°5'7-
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гидрохлортиазида #или спектр,
представленный на рисунке #0394.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то испыту¬
емый образец и ФСО гидрохлортиазида растворяют
по отдельности в минимальном количестве этано¬
ла Р1 и выпаривают до сухих остатков, которые ис¬
пользуют для получения новых спектров.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 50 мг ФСО гидрохлорти¬
азида растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 25 мг хлортиазида Р
растворяют в растворе сравнения (а) и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: этилацетат Р.
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 1/2 высоты
пластинки.
Высушивание: в потоке воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения (а).
О. К около 1 мг испытуемого образца прибавляют
2 мл свежеприготовленного раствора 0,5 г/л натрие¬
вой соли хромотроповой кислоты Р в охлажденной
смеси вода Р — кислота серная Р (35:65, об/об) и
осторожно нагревают. Появляется фиолетовое окра¬
шивание.
ИСПЫТАНИЯ
Кислотность или щелочность. 0,5 г измель¬
ченного испытуемого образца встряхивают с 25 мл
воды Р в течение 2 мин и фильтруют. К 10 мл получен¬
ного фильтрата прибавляют 0,2 мл 0,01 М раствора
натрия гидроксида и 0,15 мл раствора метилового
красного Р. Должно появиться желтое окрашивание.
При прибавлении не более 0,4 мл 0,01 М раствора
кислоты хлористоводородной должно появиться
красное окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. 50,0 мл смеси из равных
объемов ацетонитрила Р1 и метанола Р2 доводят
фосфатным буферным раствором рН 3,2 Р1 до объ¬
ема 200,0 мл.
Испытуемый раствор (а). 30,0 мг испытуемого
образца растворяют в 5 мл смеси из равных объемов
ацетонитрила Р1 и метанола Р2, при необходимо¬
сти используя ультразвук, и доводят фосфатным бу¬
ферным раствором рН 3,2 Р1 до объема 20,0 мл.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят фосфатным буферным рас¬
твором рН 3,2 Р1 до объема 20,0 мл.
Раствор сравнения (а). 3 мг ФСО хлортиази¬
да (примесь А) и 3 мг ФСО гидрохлортиазида раст¬
воряют в 5 мл смеси из равных объемов ацетони¬
трила Р1 и метанола Р2, используя ультразвук при
необходимости, и доводят фосфатным буферным
раствором рН 3,2 Р1 до объема 20,0 мл. 5,0 мл полу¬
ченного раствора доводят смесью растворителей до
объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 30,0 мг ФСО гидрохлор¬
тиазида растворяют в 5 мл смеси равных объемов
ацетонитрила Р1 и метанола Р2, используя уль¬
тразвук при необходимости, и доводят фосфатным
буферным раствором рН 3,2 Р1 до объема 20,0 мл.
1.0 мл полученного раствора доводят фосфатным
буферным раствором рН 3,2 Р1 до объема 20,0 мл.
Раствор сравнения (ф. 3 мг ФСО гидрохлорти¬
азида для идентификации пиков (содержит примеси
В и С) растворяют в 0,5 мл смеси из равных объемов
ацетонитрила Р1 и метанола Р2, используя ультра¬
звук при необходимости, и доводят фосфатным бу¬
ферным раствором рН 3,2 Р1 до объема 2,0 мл.
Условия хроматографирования:
- колонка длиной 0,1 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: к 940 мл фосфатно¬
го буферного раствора рН 3,2 Р1 прибавляют
60,0 мл метанола Р2, 10,0 мл тетрагидрофу-
рана Р и перемешивают;
- подвижная фаза В: к смеси из 500 мл мета¬
нола Р2 и 500 мл фосфатного буферного раст¬
вора рН 3,2 Р1 прибавляют 50,0 мл тетрагидро-
фурана Р и перемешивают;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—17
17—30
100 — 55
55
0 — 45
45
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 224 нм;
45. Зак. 1060.
338
Гэсударственная фармакопея Республики Беларусь
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с!).
Идентификация пиков примесей. Идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (а); идентифицируют пики примесей
В и С, используя хроматограмму раствора сравнения
(с!) и хроматограмму, прилагаемую к ФСО гидрохлор-
тиазида для идентификации пиков.
Относительное удерживание (по отношению
к гидрохлортиазиду, время удерживания — около
8 мин): примесь В — около 0,7, примесь А — около
0,9, примесь С — около 2,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,5 между пиками при¬
меси А и гидрохлортиазида.
Предельное содержание примесей:
- примеси А, В, С (не более 0,5 %): на хрома¬
тограмме испытуемого раствора (а) площади пиков,
соответствующих примесям А, В и С, не должны пре¬
вышать 5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Хлориды {2.4.4). Не более 0,0100% (100 ррт).
1,0 г испытуемого образца растворяют в 25 мл ацето¬
на Р и доводят водой Р до объема 30 мл. Эталон го¬
товят с использованием 10 мл эталонного раствора
хлорида (5 ррт С1) Р и 5 мл ацетона Р, содержащего
15 % (об/об) воды Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза:
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—4
80
20
4—10
00
0
1
м
о
о
00
т
8
- скорость подвижной фазы: 1,6 мл/мин;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (с).
Относительное удерживание (по отношению
к гидрохлортиазиду, время удерживания — около
2,2 мин): примесь А — около 0,9.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси А и гидрохлортиазида.
Содержание С7Н8С1М30432 в процентах рассчиты¬
вают с учетом содержания гидрохлортиазида в ФСО
гидрохлортиазида.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. 6-Хлор-2Н-1,2,4-бензотиадиазин-7-сульфонамид-
1,1-диоксид (хлортиазид).
В. 4-Амино-6-хлорбензол-1,3-дисульфонамид (са-
ламид).
С. 6-Хлор-Л/-[(6-хлор-7-сульфамоил-2,3-дигидро-
4Н-1,2,4-бензотиадиазин-4-ил-1,1-диоксид)метил]-3,4-
дигидро-2Н-1,2,4-бензотиадиазин-7-сульфонамид-1,1-
диоксид.
07/2016:0501
ГИОСЦИАМИНА СУЛЬФАТ
Нуозсуатт! зи1Таз
НУ08СУАМ1ЫЕ ЗШ-РАТЕ
СздНдвМА • н2304 - 2НгО М.м. 713
[620-61-1]
ОПРЕДЕЛЕНИЕ
Бис[(1/?,Зг,55)-8-метил-8-азабицикло[3.2.1]окт-3-
ил-(25)-3-гидрокси-2-фенилпропаноата] сульфат диги-
драт.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные игольчатые кристаллы.
Гиосциамина сульфат
339
Очень легко растворим в воде, умеренно раство¬
рим или растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гиосциамина сульфата.
C. К 0,5 мл раствора 5, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 2 мл кис-
лоты уксусной разведенной Р и нагревают. К горяче¬
му раствору прибавляют 4 мл раствора пикриновой
кислоты Р и охлаждают при периодическом встряхи¬
вании. Собирают кристаллы, дважды промывают ле¬
дяной водой Р порциями по 3 мл и сушат при темпе¬
ратуре от 100 °С до 105 °С. Температура плавления
(2.2.14) полученных кристаллов: от 164 °С до 168 °С.
О. К 1 мг испытуемого образца прибавляют
0,2 мл кислоты азотной дымящейся Р и выпаривают
на водяной бане досуха. Остаток растворяют в 2 мл
ацетона Р и прибавляют 0,2 мл раствора 30 г/л калия
гидроксида Р в метаноле Р. Появляется фиолетовое
окрашивание.
Е. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50,0 мл этим же
растворителем.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 4,5 до 6,2. 0,5 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 25 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От -24
до -29 (в пересчете на безводное вещество). Измеря¬
ют угол оптического вращения раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 60,0 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 50,0 мл этим же растворителем. 10,0 мл по¬
лученного раствора доводят подвижной фазой А до
объема 50,0 мл.
Раствор сравнения (а). 5,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
5.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 50,0 мл.
Раствор сравнения (Ь). 5,0 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
25.0 мл.
Раствор сравнения (с). 5,0 мг ФСО гиосциами¬
на примеси Е растворяют в испытуемом растворе и
доводят до объема 20,0 мл этим же растворителем.
5.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 25,0 мл.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
3 мкм;
- температура: (25±1) °С;
- подвижная фаза:
- подвижная фаза А: 3,5 г натрия додецил-
сульфата Р растворяют в 606 мл раствора
7,0 г/л калия дигидрофосфата Р, предваритель¬
но доведенного до рН 3,3 0,05 М раствором кис¬
лоты фосфорной, и смешивают с 320 мл ацето¬
нитрила Р;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2,0
95
5
2,0—20,0
95 — 70
5—► 30
20,0—20,1
70 —► 95
30 — 5
20,1—25,0
95
5
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (пр отношению к
гиосциамину, время удерживания — около 10,5 мин):
примесь А— около 0,2; примесь В — около 0,67; при¬
месь С — около 0,72; примесь О — около 0,8; примесь
Е — около 0,9; примесь Р — около 1,1; примесь 6 —
около 1,8.
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 2,5 между пиками при¬
меси Е и гиосциамина;
- фактор асимметрии: не более 2,5 для пика
гиосциамина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А— 0,3; для примеси 6 — 0,6):
- примесь Е (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Е, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, В, С, О, Р, С (не более 0,2 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, О, Р, О, не
должны превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, Р, Е, Р, 6, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Вода (2.5.12). Не менее 2,0 % и не более 5,5 %.
Определение проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
45*. Зак. 1060.
340
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
07/2016:0106
0,500 г испытуемого образца растворяют в 25 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 67,7 мг С34Н4вМ20е-Н2304.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, /=• 6.
А. (2Я5)-3-Гидрокси-2-фенилпропановая кислота
рЬтроповая кислота).
B. К = ОН, Р’ = Н: (1Я,35,5Я,6Я5)-6-Гидрокси-8-
метил-8-азабицикло[3.2.1]-окт-3-ил-(25)-3-гидрокси-2-
фенилпропаноат (7-гидроксигиосциамин).
C. Р = Н, Р’ = ОН: (15,ЗЯ,55,6Я5)-6-Гидрокси-8-
метил-8-азабицикло[3.2.1]окт-3-ил-(25)-3-гидрокси-2-
фенилпропаноат (6-гидроксигиосциамин).
цикло[3.3.1.02>4]нон-7-ил-(25)-3-гидрокси-2-фенил-
пропаноат (гиосцин).
Е. Р1 = СН2ОН, Р2 = РЗ = Н: (1Я,Зг,55)-8-Азабици-
кло[3.2.1 ]окт-3-ил-(25)-3-гидрокси-2-фенил пропаноат
(норгиосциамин).
О. Р1 + Р2 = СН2, РЗ = СН3: (1Я,Зг,55)-8-Метил-8-
азабицикло[3.2.1 ]окт-3-ил-2-фенилпроп-2-еноат (апо-
атропин).
Р. (1 Р,Зг,55)-8-Метил-8-азабицикпо[3.2.1]окт-3-ил-
(2Р)-2-гидрокси-2-фенилпропаноат (литорин).
ГИОСЦИНА ГИДРОБРОМИД
Нуозс1П1 ЬудгоЬготШит
(#8соро1атт1 ЬудгоЬготШит)
НУ08СШЕ НУОКОВКОМЮЕ
НВг зн2о
С17Н21Ы04 • НВг • ЗН20 М.м. 438,3
[6533-68-2]
ОПРЕДЕЛЕНИЕ
(1Р,2Р,45,55,7$)-9-Метил-3-окса-9-азатрицикло-
[3.3.1.02>4]нон-7-ил-(25)-3-гидрокси-2-фенил пропаноата
гидробромид тригидрат. #Скополамина гидробромид#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Выветривается на
воздухе.
Легко растворим в воде, растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гиосцина гидробромида.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то 3 мг ис¬
пытуемого образца растворяют в 1 мл 96 % спирта Р
и выпаривают досуха на водяной бане; остаток раст¬
воряют в 0,5 мл метиленхлорида Р, прибавляют 0,2 г
калия бромида Р и 15 мл эфира Р и выдерживают в
течение 5 мин при частом встряхивании; осадок отде¬
ляют и сушат на водяной бане до удаления раство¬
рителей; используя полученный остаток готовят диск,
который сушат при температуре от 100 °С до 105 °С в
течение 3 ч. Аналогично поступают с ФСО гиосцина
гидробромида и получают новые спектры.
C. 50 мг испытуемого образца растворяют в 5 мл
воды Р и прибавляют по каплям 5 мл раствора пи¬
криновой кислоты Р при постоянном встряхивании.
Полученный осадок промывают водой Р и сушат при
температуре от 100 °С до 105 °С в течение 2 ч. Тем¬
пература плавления (2.2.14) полученного остатка от
188 °С до 193 °С.
й. К 1 мг испытуемого образца прибавляют
0,2 мл кислоты азотной дымящейся Р и выпаривают
на водяной бане досуха. Остаток растворяют в 2 мл
ацетона Р и прибавляют 0,1 мл раствора 30 г/л калия
гидроксида Р в метаноле Р. Появляется фиолетовое
окрашивание.
Е. Испытуемый образец дает реакцию (а) на бро¬
миды (2.3.1).
Гиосцина гидробромид
341
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50,0 мл этим же растворителем.
рН (2.2.3). От 4,0 до 5,5. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От -24
до -27 (в пересчете на безводное вещество). Измеря¬
ют угол оптического вращения раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 70,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 2,0 мл испытуемого раст¬
вора доводят до объема 100,0 мл подвижной фазой.
5,0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 25,0 мл.
Раствор сравнения (с). 5,0 мг ФСО гиосцина
гидробромида примеси В растворяют в подвижной
фазе, прибавляют 5,0 мл испытуемого раствора и до¬
водят подвижной фазой до объема 50,0 мл. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- температура: (25±1) °С;
- подвижная фаза: смешивают 330 мл ацетони¬
трила Р с 670 мл раствора 2,5 г/л натрия додецил-
сульфата Р, предварительно доведенного до рН 2,5
3 М раствором кислоты фосфорной;
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 5 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания гиосцина.
Относительное удерживание (по отношению к
гиосцину, время удерживания — около 5,0 мин): при¬
месь О — около 0,2; примесь В — около 0,9; примесь
А — около 1,3; примесь С — около 2,4.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 1,5 между пиками при¬
меси В и гиосцина;
- фактор асимметрии: не более 2,5 для пика
гиосцина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 0,3; для примеси С — 0,6):
- примесь В (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси В, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- примеси А, С, О (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, С, й, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С, О, не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (Ь);
- сумма примесей (не более 0,7 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
1,4-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а); не учитывают пик, соот¬
ветствующий бромид-иону, который обнаруживается
около пика растворителя;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Вода (2.5.12). Не менее 10,0% и не более
13,0 %. Определение проводят из 0,20 г испытуемого
образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в сме¬
си, состоящей из 5,0 мл 0,01 М раствора кислоты
хлористоводородной и 50 мл 96 % спирта Р, и титру¬
ют 0,1 М раствором натрия гидроксида, свободного
от карбонатов, потенциометрически (2.2.20). Отмеча¬
ют объем титранта между двумя точками перегиба на
кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 38,43 мг С17Н211\Ю4-НВг.
ХРАНЕНИЕ
В заполненных доверху воздухонепроницаемых
контейнерах малой емкости в защищенном от света
месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. (1 Я?,Зг,55)-8-Метил-8-азабицикло[3.2.1]окт-3-ил-
(25)-3-гидрокси-2-фенилпропаноат (гиосциамин).
B. Р1 = СН2ОН, Р2 = КЗ = Н: (1Я,2Я,45,55,75)-3-
Окса-9-азатрицикло[3.3.1.02’4]нон-7-ил-(25)-3-гидрокси-
2-фенилпропаноат (норгиосцин).
C. Р1 + Р2 = СН2, РЗ = СН3: (1 Я.2Я,48,55,7з)-9-
Метил-3-окса-9-азатрицикло[3.3.1.02-4]нон-7-ил-2-
фенилпроп-2-еноат (апогиосцин).
О. (2Я5)-3-Гидрокси-2-фенилпропановая кислота
рЬтроповая кислота).
342
Гэсударственная фармакопея Республики Беларусь
07/2016:0348
ГИПРОМЕЛЛОЗА
НурготеНозит
НУРКОМЕИОЗЕ
[9004-65-3]
ОПРЕДЕЛЕНИЕ
Гидроксипропилметил целлюлоза. 2-Гидроксипро-
пилметиловый эфир целлюлозы.
Частично О-метилированная и 0-(2-гидроксипро-
пилированная) целлюлоза.
Содержание: в таблице указаны содержания
метокси- (-ОСН3; М.м. 31,03) и гидроксипропокси-
(-ОСН3Н6ОН; М.м. 75,09) групп (в пересчете на сухое
вещество), подтверждающие тип гипромеллозы.
Тип
замещения
Метокси- (%)
Гидроксипропокси- (%)
1828
от 16,5 до 20,0
от 23,0 до 32,0
2208
от 19,0 до 24,0
от 4,0 до 12,0
2906
от 27,0 до 30,0
от 4,0 до 7,5
2910
от 28,0 до 30,0
от 7,0 до 12,0
ОПИСАНИЕ(СВОЙСТВА)
Белый, желтовато-белый или серовато-белый по¬
рошок или гранулы. Гигроскопичен после высушивания.
Практически нерастворим в горячей воде, в аце¬
тоне, в этаноле и в толуоле. Растворяется в холодной
воде с получением коллоидного раствора.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1,0 г испытуемого образца равномерно рас¬
пределяют по поверхности 100 мл воды Р в лабора¬
торном стакане, при необходимости осторожно посту¬
кивая по его верхней части для получения однород¬
ного слоя на поверхности, и выдерживают в течение
(1—2) мин: на поверхности воды образуются агрегаты
из порошкообразного вещества.
B. 1,0 г испытуемого образца равномерно вносят в
100 мл кипящей воды Р и перемешивают смесь, исполь¬
зуя магнитную мешалку с магнитом длиной 25 мм: об¬
разуется суспензия и частицы не растворяются. Суспен¬
зию охлаждают до температуры 10 °С и перемешивают,
используя магнитную мешалку: образуется прозрачный
или слегка мутный раствор, консистенция которого зави¬
сит от вязкости испытуемого вещества.
C. К 0,1 мл раствора, приготовленного для под¬
линности В, прибавляют 9 мл 90 % (об/об) раствора
кислоты серной Р, встряхивают, нагревают на водя¬
ной бане в течение ровно 3 мин, немедленно охлаж¬
дают в ледяной бане, осторожно прибавляют 0,6 мл
раствора 20 г/л нингидрина Р, встряхивают и выдер¬
живают при температуре 25 °С: сначала появляется
красное окрашивание, переходящее в фиолетовое в
течение 100 мин.
й. (2—3) мл раствора, полученного в подлинно¬
сти В, наносят тонким слоем на предметное стекло и
позволяют воде испариться: на поверхности стекла
должна образоваться единая прозрачная пленка.
Е. 50,0 мл раствора, приготовленного для подлин¬
ности В, прибавляют к 50,0 мл воды Р в лабораторном
стакане. В раствор помещают термометр. Раствор
перемешивают с помощью магнитной мешалки на го¬
рячей плитке и начинают нагревать, повышая темпе¬
ратуру со скоростью от 2 до 5 °С в минуту. Определяют
температуру, при которой раствор начинает мутнеть,
обозначаемую как температура флоккуляции: темпе¬
ратура флоккуляции должна быть выше 50 °С.
ИСПЫТАНИЯ
Раствор 5. Испытуемый образец в количестве,
эквивалентом 1,0 г сухого вещества, при перемеши¬
вании вносят в 50 г воды, свободной от углерода ди¬
оксида, Р, нагретой до температуры 90 °С. Охлажда¬
ют, доводят массу раствора до 100 г водой, свободной
от углерода диоксида, Р и перемешивают до полного
растворения.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон III.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 5,0 до 8,0. Используют раствор,
приготовленный как указано в испытании «Вязкость».
Результаты записывают через (5±0,5) мин после по¬
гружения пробы.
Вязкость. Не менее 80 % и не более 120 % от
номинального значения для гипромеллозы с вязко¬
стью менее 600 мПа-с (метод 1); не менее 75 % и не
более 140 % от номинального значения для образцов
с вязкостью 600 мПа-с или выше (метод 2).
Метод 1, для образцов с вязкостью менее
600 мПа-с. Точную навеску испытуемого образца в
количестве, соответствующем 4,000 г сухого веще¬
ства, помещают в колбу с широким горлом и доводят
общую массу испытуемого образца и воды до 200,0 г
горячей водой Р (90—99) °С. Колбу закрывают и пере¬
мешивают механически при (400±50) об/мин в течение
(10—20) мин до полного диспергирования и смачива¬
ния частиц. При необходимости по внутренней стенке
колбы проводят шпателем, чтобы убедиться в отсут¬
ствии нерастворенного вещества на стенках колбы, и
продолжают перемешивание в охлажденной водяной
бане при температуре ниже 10 °С в течение последу¬
ющих (20—40) мин. При необходимости доводят массу
раствора до 200,0 г холодной водой Р. При необходи¬
мости раствор центрифугируют для удаления пузырь¬
ков воздуха. При образовании пены ее удаляют шпа¬
телем. Определяют вязкость полученного раствора,
используя метод капиллярной вискозиметрии (2.2.9)
для определения кинематической вязкости (у). Отдель¬
но определяют плотность (р) (2.2.5) раствора и рассчи¬
тывают динамическую вязкость (г/) по формуле г]=ру.
Метод 2, для образцов с вязкостью 600 мПа-с
и более. Точную навеску испытуемого образца в ко¬
личестве, эквивалентном 10,00 г сухого вещества, пе¬
реносят в колбу с широким горлом и доводят общую
массу испытуемого образца и воды до 500,0 г горя¬
чей водой Р (90—99) °С. Колбу закрывают и переме¬
шивают механически при (400±50) об/мин в течение
(10—20) мин до полного диспергирования и смачива¬
ния частиц. При необходимости по внутренней стенке
колбы проводят шпателем, чтобы убедиться в отсут¬
ствии нерастворенного вещества на стенках колбы, и
продолжают перемешивание в охлажденной водяной
бане при температуре ниже 10 °С в течение после¬
дующих (20—40) мин. При необходимости доводят
массу раствора до 500,0 г холодной водой Р. При не¬
обходимости раствор центрифугируют для удаления
пузырьков воздуха. При образовании пены ее удаля¬
ют шпателем. Определяют вязкость (2.2.10) получен¬
ного раствора при температуре (20±0,1)°С с помо¬
щью ротационного вискозиметра.
Гипромеллоза
343
Таблица 0348.-1.
Номинальная вязкость* (мПа-с)
Номер ротора
Число оборотов (об/мин)
Расчетный множитель
От 600 до менее 1400
3
60
20
От 1400 до менее 3500
3
12
100
От 3500 до менее 9500
4
60
100
От 9500 до менее 99 500
4
6
1000
99 500 и более
4
3
2000
* Значение номинальной вязкости основывается на спецификации производителя.
Прибор: одноцилиндровый шпиндельный виско¬
зиметр.
Номер ротора, число оборотов и расчетный
множитель: определение проводят при условиях,
приведенных в таблице 0348.-1.
Шпиндель вращают в течение 2 мин перед из¬
мерением. Период покоя между последовательными
измерениями составляет 2 мин. Измерения проводят
трижды и определяют среднее значение результатов
по 3 измерениям.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Сульфатная зола (2.4.14). Не более 1,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Прибор:
- реакционный флакон: герметичный флакон
вместимостью 5 мл, высотой 50 мм, внешним диа¬
метром 20 мм и внутренним диаметром 13 мм в гор¬
ловине, снабженный герметичной бутилкаучуковой
мембранной пробкой, покрытой политетрафторэтиле¬
ном, и алюминиевой закручивающейся крышкой или
другим плотно закрывающим приспособлением, обе¬
спечивающим достаточную воздухонепроницаемость;
- термоблок: нагревательный модуль с ква¬
дратной алюминиевой подставкой с отверстиями
диаметром 20 мм и глубиной 32 мм, пригодными для
установки реакционных флаконов; перемешивание
содержимого флакона осуществляют или с помощью
магнитной мешалки, встроенной в термоблок, или при
помощи возвратно-поступательного встряхивателя,
совершающего приблизительно 100 циклов/мин.
Раствор внутреннего стандарта: раствор
30 г/л октана Р в ксилоле Р.
Испытуемый раствор. 65,0 мг испытуемого об¬
разца помещают в реакционный флакон, прибавляют
(0,06—0,10) г кислоты адипиновой Р, 2,0 мл раствора
внутреннего стандарта и 2,0 мл кислоты йодистово¬
дородной Р, незамедлительно плотно укупоривают
флакон и точно взвешивают. Содержимое флакона
непрерывно перемешивают в течение 60 мин при на¬
гревании в термоблоке, обеспечивая температуру со¬
держимого на уровне (130±2) °С. При невозможности
использования возвратно-поступательного встряхи¬
вателя или магнитной мешалки флакон встряхивают
вручную с 5-минутными интервалами после первых
30 мин нагревания. Затем флакон охлаждают и снова
точно взвешивают. Если потеря в массе составляет
менее 0,50 % от массы содержимого и не наблюда¬
ется нарушения герметичности, верхний слой смеси
используют в качестве испытуемого раствора.
Раствор сравнения. (0,06-0,10) г кислоты ади¬
пиновой Р, 2,0 мл раствора внутреннего стандарта и
2,0 мл кислоты йодистоводородной Р помещают в
другой реакционный флакон, плотно закрывают и точ¬
но взвешивают. Прибавляют (15—22) мкл изопропи-
лйодида Р проколом пробки с помощью шприца, точно
взвешивают, прибавляют 45 мкл метилйодида Р ана¬
логичным образом и снова точно взвешивают. Тща¬
тельно встряхивают реакционный флакон и использу¬
ют верхний слой в качестве раствора сравнения.
Условия хроматографирования:
- колонка длиной (1,8—3) м и внутренним диаме¬
тром (3—4) мм, заполненная диатомитом для газо¬
вой хроматографии Р ((125—150) мкм), импрегниро-
ванным (10—20) % поли(диметил)силоксана Р;
- температура: 100 °С;
- газ-носитель: гелий для хроматографии Р
(детектор по теплопроводности); гелий для хромато¬
графии Р или азот для хроматографии Р (пламен¬
но-ионизационный детектор);
- скорость газа-носителя: подбирают таким об¬
разом, чтобы время удерживания внутреннего стан¬
дарта составляло около 10 мин;
-детектор: пламенно-ионизационный или де¬
тектор по теплопроводности;
- объем вводимой пробы: (1—2) мкл.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: пики метилйодида (1-й пик), изо-
пропилйодида (2-й пик) и внутреннего стандарта
(3-й пик) должны быть хорошо разделены.
Расчеты:
- на хроматограмме испытуемого раствора рас¬
считывают отношения (01 и 02) площадей пиков мети¬
лйодида и изопропилйодида к площади пика внутрен¬
него стандарта; по хроматограмме раствора сравне¬
ния рассчитывают отношения (03 и 04) площадей пи¬
ков метилйодида и изопропилйодида к площади пика
внутреннего стандарта.
Процентное содержание метокси-групп рассчи¬
тывают по следующей формуле:
02 т
процентное содержание гидроксипропокси-групп
рассчитывают по следующей формуле:
”>-44,17.
04 т
где:
/771 — масса метилйодида в растворе срав¬
нения, мг;
т2 — масса изопропилйодида в растворе срав¬
нения, мг;
т — масса испытуемого образца (в пересчете на
сухое вещество), мг.
344
Гэсударственная фармакопея Республики Беларусь
МАРКИРОВКА
На этикетке указывают:
- номинальную вязкость (мПа-с);
- тип замещения.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Некоторые
из характеристик, описанных в разделе «Функцио¬
нально-обусловленные характеристики», могут
также присутствовать в части фармакопейной
статьи, обязательной для выполнения, так как од¬
новременно они являются и показателями качества.
В таком случае в разделе «Функционально-обуслов¬
ленные характеристики» дается перекрестная
ссылка на это испытание в части фармакопейной
статьи, обязательной для выполнения. Контроль
данных характеристик может вносить вклад в каче¬
ство лекарственного средства путем достижения
постоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, однако
другие методы также могут быть использованы. В
случае указания результатов конкретных характе¬
ристик требуется указывать также и методы про¬
ведения испытаний.
Следующие характеристики могут быть важ¬
ны для гипромеллозы, используемой в качестве свя¬
зывающего вещества, вещества, повышающего
вязкость, или пленкообразующего вещества.
Вязкость: см. раздел «Испытания».
Степень замещения: см. раздел «Количествен¬
ное определение».
Следующие характеристики могут быть важ¬
ны для гипромеллозы, используемой в качестве ма¬
трицы в лекарственных средствах пролонгирован¬
ного действия.
Вязкость: см. раздел «Испытания».
Степень замещения: см. раздел «Количествен¬
ное определение».
Молекулярно-массовое разделение (2.2.30).
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошка (2.9.36).
07/2016:0143
ГИСТАМИНА ДИГИДРОХЛОРИД
Н15{ат'п1 сННус1госЫопс1ит
Н15ТАМ1ЫЕ 01НУ0РЮСНЦЭР1ЮЕ
NN I 2НС1
Хч-/^"ын2
С5Н,М3 ■ 2НС1 М.м. 184,1
[56-92-8]
ОПРЕДЕЛЕНИЕ
2-(1 Н-Имидазол-4-ил)этанамина дигидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Гигроскопичен.
Очень легко растворим в воде, растворим в спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, Б.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках; используют 1 мг испы¬
туемого образца.
Сравнение: ФСО гистамина дигидрохлорида.
B. Просматривают хроматограммы, полученные
в испытании «Гистидин».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
C. 0,1 г испытуемого образца растворяют в 7 мл
воды Р и прибавляют 3 мл раствора 200 г/л натрия
гидроксида Р. 50 мг сульфаниловой кислоты Р раство¬
ряют в смеси из 0,1 мл кислоты хлористоводородной Р
и 10 мл воды Р и прибавляют 0,1 мл раствора натрия
нитрита Р. Прибавляют второй раствор к первому и
перемешивают. Появляется красное окрашивание.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 2,85 до 3,60. Измеряют рН раст¬
вора 3.
Гистидин. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,5 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Испытуемый раствор (Ь). 2 мл испытуемого
раствора (а) доводят водой Р до объема 10.
Раствор сравнения (а). 0,1 г ФСО гистамина ди¬
гидрохлорида растворяют в воде Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (Ь). 50 мг ФСО гистидина
моногидрохлорида растворяют в воде Р и доводят до
объема 100 мл этим же растворителем.
Раствор сравнения (с). 1 мл испытуемого раст¬
вора (а) смешивают с 1 мл раствора сравнения (Ь).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: аммиака раствор концентри¬
рованный Р — вода Р — ацетонитрил Р (5:20:75,
об/об/об).
Наносимый объем пробы: по 1 мкл испытуемых
растворов (а) и (Ь), растворов сравнения (а) и (Ь);
2 мкл раствора сравнения (с).
Гистидин
345
Фронт подвижной фазы: дважды; не менее
15 см от линии старта.
Высушивание: в потоке воздуха после каждого
хроматографирования.
Проявление: пластинку опрыскивают раствором
нингидрина Р1 и нагревают при температуре 110 °С в
течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание:
- гистидин (не более 1 %): на хроматограмме
испытуемого раствора (а) пятно, соответствующее ги¬
стидину, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (Ь).
Сульфаты (2.4.13). Не более 0,1 %. 3 мл раст¬
вора 3 доводят до объема 15 мл водой дистиллиро¬
ванной Р. Полученный раствор должен выдерживать
испытания на сульфаты.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 0,20 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 0,5 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,080 г испытуемого образца растворяют в смеси
из 5,0 мл 0,01 М раствором кислоты хлористоводо¬
родной и 50 мл спирта Р и титруют 0,1 М раствором
натрия гидроксида потенциометрически (2.2.20). От¬
мечают объем титранта между двумя точками пере¬
гиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 9,203 мг С5НдМ3-2НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0911
гистидин
ШзИсИпит
Н13ТЮ1ЫЕ
/=Н
н мн2
\ /
тю2н
С6Н9Ы302 М.м. 155,2
[71-00-1]
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-3-(1Я-имидазол-4-ил)пропановая
кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Растворим в воде, очень мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гистидина.
Если полученные спектры отличаются, то ис¬
пытуемый образец и ФСО гистидина растворяют
по отдельности в минимальном количестве воды Р
и выпаривают до сухих остатков при температуре
60 °С, которые используют для получения новых
спектров.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 50 мл
этим же растворителем.
Раствор сравнения. 10 мг ФСО гистидина раст¬
воряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и высушивают при температуре 105 °С
в течение 15 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. Около 0,1 г испытуемого образца растворяют
в 7 мл воды Р, прибавляют 3 мл раствора 200 г/л на¬
трия гидроксида Р. 50 мг кислоты сульфаниловой Р
растворяют в смеси из 0,1 мл кислоты хлористово¬
дородной Ри 10 мл воды Р и прибавляют 0,1 мл раст¬
вора натрия нитрита Р. Прибавляют второй раствор
к первому и перемешивают. Появляется оранжево¬
красное окрашивание.
ИСПЫТАНИЯ
Раствор 3. 2,5 испытуемого образца растворяют
в воде дистиллированной Р, нагревая в водяной бане,
и доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
Удельное оптическое вращение (2.2.7). От
+11,4 до +12,4 (в пересчете на сухое вещество). 2,75 г
испытуемого образца растворяют в 12,0 мл кислоты
хлористоводородной Р1 и доводят водой Р до объ¬
ема 25,0 мл.
Нингидрин-положительные вещества. Ана¬
лиз аминокислот (2.2.56). Для анализа используют
Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон-
346
Гэсударственная фармакопея Республики Беларусь
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Вода Р или буферный раствор для
приготовления образца, подходящий для применяе¬
мого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят раствором А до объ¬
ема 10,0 мл.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт Л/Н^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (д). 30 мг изолейцина Р и
30 мг лейцина Р растворяют в растворе А и доводят
до объема 50,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
200.0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
вводят в аминокислотный анализатор. Запускают под¬
ходящую программу для определения физиологиче¬
ских аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с!):
-разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию гистидина в растворе срав¬
нения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
10 мл раствора 5 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
- вводимая проба: испытуемый раствор, раствор
сравнения (с) и холостой раствор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в смеси из 3 мл кислоты хлористоводородной
разведенной Р и 15 мл воды Р, слегка нагревая при
необходимости, и доводят водой Р до объема 20 мл.
12 мл полученного раствора должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с ис¬
пользованием эталонного раствора свинца (1 ррт
РЬ)Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,130 г испытуемого образца растворяют в 50 мл
воды Р и титруют 0,1 М раствором кислоты хлори¬
стоводородной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 15,52 мг 06Н9М3О2.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемлемо¬
сти для других/неспецифицированных примесей.
Вследствие этого нет необходимости идентифици¬
ровать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Кон¬
троль примесей в субстанциях для фармацевти¬
ческого использования): А.
| ^ н мн2
^^^^^СОгН
А. (25)-2-Амино-3-(4-гидроксифенил)пропановая
кислота (тирозин).
Гистидина гидрохлорид моногидрат
347
07/2016:0910
ГИСТИДИНА ГИДРОХЛОРИД
МОНОГИДРАТ
НИзИсИп! ЬудгосЫопдит топоЬудпсит
Н15ТЮ1ЫЕ НУОКОСШОКЮЕ
МОЫОНУОПАТЕ
н
мн2
• НС1 • н2о
^со2н
С6Н9М302 ■ НС1 ■ н20 М.м. 209,6
[5934-29-2]
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-3-(1Я-имидазол-4-ил)пропановой
кислоты гидрохлорид моногидрат.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, С, Р.
Вторая идентификация: А, В, О, Е, Р.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО гистидина гидрохлорида моно¬
гидрата.
й. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 50 мл
этим же растворителем.
Раствор сравнения. 10 мг ФСО гистидина ги¬
дрохлорида моногидрата растворяют в воде Р и до¬
водят до объема 50 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и высушивают при температуре 105 °С
в течение 15 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
Е. 0,1 г испытуемого образца растворяют в 7 мл
воды Р, прибавляют 3 мл раствора 200 г/л натрия
гидроксида Р. 50 мг кислоты сульфаниловой Р раст¬
воряют в смеси из 0,1 мл кислоты хлористоводород¬
ной Р и 10 мл воды Р и прибавляют 0,1 мл раствора
натрия нитрита Р Прибавляют второй раствор к
первому и перемешивают. Появляется оранжево¬
красное окрашивание.
Р. Около 20 мг испытуемого образца дают реак¬
цию на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 испытуемого образца растворяют
в воде, свободной от углерода диоксида, Р, приготов¬
ленной из воды дистиллированной Р, и доводят до
объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод И). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)б.
рН (2.2.3). От 3,0 до 5,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От +9,2
до +10,6 (в пересчете на сухое вещество). 2,75 г ис¬
пытуемого образца растворяют в 12,0 мл кислоты
хлористоводородной Р1 и доводят водой Р до объ¬
ема 25,0 мл.
Нингидрин-положительные вещества. Анализ
аминокислот (2.2.56). Для анализа используют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Вода Р или буферный раствор для
приготовления образца, подходящий для применяе¬
мого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят раствором А до объ¬
ема 10,0 мл.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт Л/Н^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (ф. 30 мг изолейцина Р и 30 мг
лейцина Р растворяют в растворе А и доводят до объ¬
ема 50,0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят раствором А до объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
вводят в аминокислотный анализатор. Запускают под¬
ходящую программу для определения физиологиче¬
ских аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с!):
-разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис-
348
Гэсударственная фармакопея Республики Беларусь
пользуют концентрацию гистидина в растворе срав¬
нения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
10 мл раствора 5 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
- вводимая проба: испытуемый раствор, раствор
сравнения (с) и холостой раствор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в смеси из 3 мл кислоты хлористоводородной
разведенной Р и 15 мл воды Р, слегка нагревая при
необходимости, и доводят водой Р до объема 20 мл.
12 мл полученного раствора должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с ис¬
пользованием эталонного раствора свинца (1 ррт
РЬ)Р.
Потеря в массе при высушивании (2.2.32). Не
менее 7,0 % и не более 10,0 %. 1,000 г испытуемого
образца сушат при температуре (145—150) °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,160 г испытуемого образца растворяют в 50 мл
воды, свободной от углерода диоксида, Р и титруют
0,1 М раствором натрия хлорида потенциометриче-
ски (2.2.20).
1 мл 0,1 М раствора натрия хлорида соответ¬
ствует 19,16 мг С6Н9Ы302*НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): А.
|| ^ Н МН2
^^^^^^СОгН
А. (25)-2-Амино-3-(4-гидроксифенил)пропановая
кислота (тирозин).
07/2016:0718
ГЛИБЕНКЛАМИД
ОНЬепс1ат1с1ит
СиВЕЫО-АМЮЕ
С23Н28С1М3055 М.м. 494,0
[10238-21-8]
ОПРЕДЕЛЕНИЕ
1-[[4-[2-[(5-Хпор-2-метоксибензоил)амино]этил]-
фенил]сульфонил]-3-циклогексилмочевина.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, умеренно рас¬
творим в метиленхлориде, мало растворим в 96 %
спирте и метаноле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О, Е.
A. Температура плавления (2.2.14): от 169 °С до
174 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в метаноле Р, при необходимости
используют ультразвук, и доводят до объема 50,0 мл
этим же растворителем. К 10,0 мл полученного раст¬
вора прибавляют 1,0 мл раствора 103 г/л кислоты
хлористоводородной Р и доводят метанолом Р до
объема 100,0 мл.
Гпибенкламид
349
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 300 нм и менее вы¬
раженный при 275 нм.
Удельный показатель поглощения в максимуме:
при 300 нм — от 61 до 65; при 275 нм — от 27 до 32.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО глибенкламида #или спектр,
представленный на рисунке #0718.-1*.
Если полученные спектры отличаются, то испыту¬
емый образец и ФСО глибенкламида увлажняют мета¬
нолом Р, растирают, сушат при температуре от 100 °С
до 105 °С и используют для получения новых спектров.
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси из равных объемов ме¬
танола Р и метиленхлорида Р и доводят до объема
10 мл этой же смесью растворителей.
Раствор сравнения. 10 мг ФСО глибенкламида
растворяют в смеси из равных объемов метанола Р
и метиленхлорида Р и доводят до объема 10 мл этой
же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ 6Р254 Р-
Подвижная фаза: 96 % спирт Р — кислота ук¬
сусная ледяная Р — циклогексан Р — метиленхло-
рид Р (5:5:45:45, об/об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
Е. 20 мг испытуемого образца растворяют в 2 мл
кислоты серной Р. Раствор прозрачный и обладает
синей флуоресценцией в ультрафиолетовом свете
при 365 нм. В полученном растворе растворяют 0,1 г
хлоралгидрата Р. В течение 5 мин цвет раствора из¬
меняется на ярко-желтый, а через 20 мин появляется
коричневатый оттенок.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед применением или хранят при темпе¬
ратуре 5 °С не более 40 часов.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в метаноле Р, при необходимости
применяя ультразвук, и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (а). 3,0 мг ФСО глибенкла¬
мида примеси А и 3 мг ФСО глибенкламида приме¬
си В растворяют в метаноле Р и доводят до объема
100.0 мл этим же растворителем. 5,0 мл полученного
раствора доводят метанолом Р до объема 20,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
1.0 мл полученного раствора доводят метанолом Р
до объема 10,0 мл.
Раствор сравнения (с). 12,5 мг ФСО глибенкла¬
мида для идентификации пиков (содержит примесь
С) растворяют в метаноле Р, при необходимости при¬
меняя ультразвук, и доводят до объема 5,0 мл этим
же растворителем.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным, деактивированным по отно¬
шению к основаниям, эндкепированным для хрома¬
тографии Р с размером частиц 3 мкм;
Рисунок #0718.-1. Инфракрасный спектр ФСО глибенкламида
350
Гэсударственная фармакопея Республики Беларусь
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 20 мл раст¬
вора 100,0 г/л триэтиламина Р2, предвари¬
тельно доведенного до рН 3,0 кислотой фос¬
форной Р, и 50 мл ацетонитрила Р; доводят
водой Р до объема 1000 мл;
- подвижная фаза В: подвижная фаза А —
вода Р — ацетонитрил Р (20:6,5:91,5, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
45
55
15—30
45 — 5
55 — 95
30-40
5
95
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (а) и хроматограмму, прилагае¬
мую к ФСС глибенкламида для идентификации пи¬
ков’, идентифицируют пик примеси С, используя хро¬
матограмму раствора сравнения (с).
Относительное удерживание (по отношению к
глибенкламиду, время удерживания — около 5 мин):
примесь А — около 0,5; примесь В — около 0,6; при¬
месь С — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 2,0 между пиками при¬
месей А и В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 1,8):
- примесь А (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси А, не должна превышать площадь соответству¬
ющего пика на хроматограмме раствора сравнения (а);
- примесь С (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,8 %);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод 6). Не более
0,0020 % (20 ррт). 0,250 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 0,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют при
нагревании в 100 мл 96 % спирта Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 49,40 мг С23Н28С11Ч3055.
ПРИМЕСИ
Специфицированные примеси: А, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, О, Е.
А. 5-Хлор-2-метокси-Л/-[2-(4-сульфамоилфенил)-
этил]бензамид.
В. Метил-[[4-[2-[(5-хлор-2-метоксибензоил)амино]-
этил]фенил]сульфонил]карбамат.
С. 1 -Циклогексил-3-[[4-[2-[(циклогексилкарбамоил)
амино]этил]фенил]сульфонил]мочевина.
0.1 -Бутил-3-[[4-[2-[(5-хлор-2-метоксибензоил)ами-
но]этил]фенил]сульфонил]мочевина.
Е. 1 -Циклогексил-3-[[4-[2-[(3,5-дихлор-2-метокси-
бензоил)амино]этил]фенил]сульфонил]мочевина.
Гпиклазид
351
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Смесь растворителей. Ацетонитрил Р —
вода Р (45:55, об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 23 мл ацетонитрила Р и доводят
водой Р до объема 50,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 10,0 мл полученного раствора доводят сме¬
сью растворителей до объема 100,0 мл.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 15 мг ФСО гликлазида примеси Р растворяют в
23 мл ацетонитрила Р и доводят водой Р до объема
50 мл. 1 мл полученного раствора доводят смесью
растворителей до объема 20 мл.
Раствор сравнения (с). 10,0 мг ФСО гликлази¬
да примеси Р растворяют в 45 мл ацетонитрила Р
и доводят водой Р до объема 100,0 мл. 1,0 мл полу¬
ченного раствора доводят смесью растворителей до
объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: триэтиламин Р — кислота
трифторуксусная Р — ацетонитрил Р — вода Р
(0,1:0,1:45:55, об/об/об/об);
- скорость подвижной фазы: 0,9 мл/мин;
- спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания гликлазида.
Относительное удерживание (по отношению к
гликлазиду, время удерживания — около 16 мин): при¬
месь Р — около 0,9.
Рисунок #1524.-1. Инфракрасный спектр ФСО гликлазида
07/2016:1524
ГЛИКЛАЗИД
ОНс/агМит
СиС1_А2/0Е
С15Н21М3035 М.м. 323,4
[21187-98-4]
ОПРЕДЕЛЕНИЕ
1-(Гексагидроциклопента[с]пиррол-2(1Н)-ил)-3-
[(4-метилфенил)сульфонил]мочевина.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, умеренно растворим в ацето¬
не, мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО гликлазида #или спектр, пред¬
ставленный на рисунке #1524.-1#.
352
Гэсударственная фармакопея Республики Беларусь
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,8 между пиками при¬
меси Р и гликпазида.
Предельное содержание примесей:
-примесь Р (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси Р, не должна превышать площадь соответству¬
ющего пика на хроматограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си Р, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
-сумма примесей, кроме примеси Р (не более
O, 2 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
P, не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,02 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (а).
Примесь В. Не более 0,0002 % (2 ррт). Жид¬
костная хроматография (2.2.29), как указано в испы¬
тании «Сопутствующие примеси», со следующими
изменениями.
Испытуемый раствор. 0,400 г испытуемого об¬
разца растворяют в 2,5 мл диметилсульфоксида Р и
доводят водой Р до объема 10,0 мл. Перемешивают в
течение 10 мин, выдерживают при температуре 4 °С в
течение 30 мин и фильтруют.
Раствор сравнения. 20,0 мг ФСО гликлазида
примеси В растворяют в диметилсульфоксиде Р и
доводят до объема 100,0 мл этим же растворителем.
К 1,0 мл полученного раствора прибавляют 12 мл ди¬
метилсульфоксида Р и доводят водой Р до объема
50,0 мл. К 1,0 мл полученного раствора прибавляют
12 мл диметилсульфоксида Р и доводят водой Р до
объема 50,0 мл.
Условия хроматографирования:
- объем вводимой пробы: 50 мкл.
Время удерживания: примесь В — около 8 мин.
Предельное содержание примесей:
- примесь В: на хроматограмме испытуемого
раствора площадь пика примеси В не должна превы¬
шать площадь соответствующего пика на хромато¬
грамме раствора сравнения.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0010 % (10 ррт). 1,5 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 1,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,25 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
*\ мп 0,1 М раствора кислоты хлорной соответ¬
ствует 32,34 мг С15Н21М3035.
ПРИМЕСИ
Специфицированные примеси: В, Р
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, С, О, Е, С.
А. Р-Н: 4-Метилбензолсульфонамид.
B. 2-Нитрозо-октагидроциклопента[с]пиррол.
C. Р-С0-0-С2Н5: Этил[(4-метилфенил)сульфонил]-
карбамат.
О. А/-[(4-Метилфенил)сульфонил]гексагидро-
циклопента[с]пиррол-2(1Я)-карбоксамид.
Е. 1 -[(4-Метилфенил)сульфонил]-3-(3,За,4,6а-
тетрагидроциклопента[с]пиррол-2(1Н)-ил)мочевина.
Р. 1-(Гексагидроциклопента[с]пиррол-2(1Н)-ил)-3-
[(2-метилфенил)сульфонил]мочевина.
С. А/-[(4-Метилфенил)сульфонил]-1,4а,5,6,7,7а-
гексагидро-2Н-циклопента[с/|пиридазин-2-карбоксамид.
Гпимепирид
353
07/2016:2223
ГЛИМЕПИРИД
ОНтертдит
симЕРтюЕ
СмНмЫ40.8 М.м. 490,6
[93479-97-1]
ОПРЕДЕЛЕНИЕ
1 -[[4-[2-(3-Этил-4-метил-2-оксо-3-пирролин-1 -
карбоксамидо)-этил]фенил]сульфонил]-3-транс-(4-
метилциклогексил)мочевина.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, растворим в
диметилформамиде, мало растворим в метиленхло-
риде, очень мало растворим в метаноле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО глимепирида #или спектр, пред¬
ставленный на рисунке #2223.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, различаются, то испы¬
туемый образец и ФСО глимепирида растворяют по
отдельности в диметилформамиде Р и выпаривают
до сухих остатков, которые используют для получения
новых спектров.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы хранят при темпера¬
туре не выше 12 °С в течение не более 15 ч.
Смесь растворителей. Вода для хроматографии Р—
ацетонитрил для хроматографии Р (1:4, об/об).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (а). Содержимое контейнера
с ФСО глимепирида для проверки пригодности хро¬
матографической системы (содержит примеси В, С
и О) растворяют в 2,0 мл испытуемого раствора.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 20,0 мг ФСО глимепири¬
да растворяют в смеси растворителей и доводят до
объема 100,0 мл этой же смесью растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 4 мкм;
- подвижная фаза: 0,5 г натрия дигидрофосфа¬
та Р растворяют в 500 мл воды для хроматографии Р
и доводят до рН 2,5 кислотой фосфорной Р. Прибав¬
ляют 500 мл ацетонитрила для хроматографии Р\
- скорость подвижной фазы: 1,0 мл/мин;
Рисунок #2223.-1. Инфракрасный спектр ФСО глимепирида
354
Гэсударственная фармакопея Республики Беларусь
-спектрофотометрический детектор, длина
волны 228 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а) и (Ь);
- время хроматографирования: 2,5-кратное
время удерживания глимепирида.
Относительное удерживание (по отношению к
глимепириду, время удерживания — около 17 мин):
примесь В — около 0,2; примесь С — около 0,3; при¬
месь О— около 1,1.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 4,0 между пиками при¬
месей В и С.
Предельное содержание примесей:
- примесь В (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 4-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь О (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В и О, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
-сумма примесей, кроме примеси В (не более
0,5 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
В, не должна превышать 5-кратную площадь основного
пика на хроматограмме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Примесь А. Жидкостная хроматография (2.2.29).
Растворы готовят непосредственно перед исполь¬
зованием.
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в 5 мл метиленхлорида Р и дово¬
дят подвижной фазой до объема 20,0 мл.
Раствор сравнения (а). 0,8 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 2,0 мг ФСО глимепири¬
да для идентификации примеси А растворяют в 1 мл
метиленхлорида Р и доводят подвижной фазой до
объема 4,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 3 мм, заполненная силикагелем диольным для
хроматографии Р с размером частиц 5 мкм;
-подвижная фаза: кислота уксусная безвод¬
ная Р—2-пропанол Р—гептан Р (1:100:899, об/об/об);
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 228 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания глимепирида.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО глимепирида для идентификации примеси А.
Относительное удерживание (по отношению к
глимепириду, время удерживания — около 14 мин):
примесь А — около 0,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика примеси А относительно базовой
линии; Н,— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси А и пик
глимепирида).
Предельное содержание примесей:
- примесь А (не более 0,8 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а).
Вода (2.5.32). Не более 0,5 %. 0,250 г испытуе¬
мого образца растворяют в диметилформамиде Р и
доводят этим же растворителем до объема 5,0 мл.
Испытание проводят из 1,0 мл полученного раствора.
Проводят контрольный опыт.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (с).
Содержание С^Н^Ы^З в процентах рассчиты¬
вают с учетом площадей пиков и содержания глиме¬
пирида в ФСО глимепирида.
ПРИМЕСИ
Специфицированные примеси: А, В, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): С, Е, Р, С, Н, I, 3.
А. 1 -[[4-[2[[(3-Этил-4-метил-2-оксо-2,3-дигидро-1 Н-
пиррол-1-ил)карбонил]амино]этил]фенил]сульфонил]-
3-(щ/с-4-метилциклогексил)мочевина.
Глицерин
355
В. 3-Этил-4-метил-2-оксо-Л/-[2-(4-сульфамоил-
фенил)этил]-2,3-дигидро-1 Н-пиррол-1-карбоксамид.
С. Метил-[[4-[2-[[(3-этил-4-метил-2-оксо-2,3-
дигидро-1 Н-пиррол-1 -ил)карбонил]амино]этил]фенил]-
сульфонил]карбамат.
О. 1 -[[3-[2-[[(3-Этил-4-метил-2-оксо-2,3-дигидро-
1 Н-пиррол-1 -ил)карбонил]амино]этил]фенил]-суль-
фонил]-3-(шранс-4-метилциклогексил)мочевина.
Е. 3-Этил-4-метил-2-оксо-Л/-[2-(3-сульфамоил-
фенип)этил]-2,3-дигидро-1 Н-пиррол-1 -карбоксамид.
Р. Метил-[[2-[2-Ц(3-этил-4-метил-2-оксо-2,3-дигидро-
1 Н-пиррол-1 -ил)карбонил]амино]этил]фенил]сул ьфо-
нил]карбамат.
<3. Метил-[[4-[2-[[(3-этил-4-метил-2-оксо-2,3-ди-
гидро-1 Н-пиррол-1 -ил)карбонил]амино]этил]фенил]-
сульфонил]метилкарбамат.
Н. 1 -[[4-[2-[[(3-Этил-4-метил-2-оксо-2,3-дигидро-
1 Н-пиррол-1 -ил)карбонил]амино]этил]фенил]суль-
фонил]-3-(4-метилфенил)мочевина.
1.1 -[[2-[2-[[(3-Этил-4-метил-2-оксо-2,3-дигидро-1 Н-
пиррол-1-ил)карбонил]амино]этил]фенил]сульфонил]-
3-(транс-4-метилциклогексил)мочевина.
3.1 -[[4-(2-Аминоэтил)фенил]сульфонил]-3-(транс-
4-метилциклогексил)мочевина.
07/2016:0496
ГЛИЦЕРИН
01усего1ит
е^уСЕ/?о^
Сзн,0, М.м. 92,1
[56-81-5]
ОПРЕДЕЛЕНИЕ
Пропан-1,2,3-триол.
Содержание: не менее 98,0 % (м/м) и не более
101,0 % (м/м) (в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Сиропообразная маслянистая на ощупь бесц¬
ветная или почти бесцветная прозрачная жидкость.
Очень гигроскопичен.
Мало растворим в ацетоне, практически нераст¬
ворим в жирных и эфирных маслах. Смешивается с
водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
356
Гэсударственная фармакопея Республики Беларусь
A. Испытуемый образец выдерживает испытание
«Показатель преломления», как указано в разделе
«Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: к 5 мл испытуемого образца
прибавляют 1 мл воды Р и тщательно перемешивают.
Сравнение: эталонный спектр глицерина 85 %
по Европейской Фармакопее #или спектр, представ¬
ленный на рисунке #0496.-1#.
C. 1 мл испытуемого образца смешивают с
0,5 мл кислоты азотной Р. На полученный раствор
наслаивают 0,5 мл раствора калия дихромата Р. На
границе раздела жидкостей появляется синее кольцо.
В течение 10 мин синее окрашивание не переходит в
нижний слой.
й. В выпарительной чашке нагревают 1 мл ис¬
пытуемого образца с 2 г калия гидросульфата Р. Вы¬
деляющиеся пары (акролеин) окрашивают фильтро¬
вальную бумагу, импрегнированную щелочным рас¬
твором калия тетрайодмеркурата Р, в черный цвет.
ИСПЫТАНИЯ
Раствор 5.100,0 г испытуемого образца доводят
водой, свободной от углерода диоксида, Р до объема
200,0 мл.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). 10 мл раствора 3
доводят водой Р до объема 25 мл. Полученный раст¬
вор должен быть бесцветным.
Кислотность или щелочность. К 50 мл раст¬
вора 3 прибавляют 0,5 мл раствора фенолфтале¬
ина Р. Раствор должен быть бесцветным. При при¬
бавлении не более 0,2 мл 0,1 М раствора натрия
гидроксида должно появиться розовое окрашивание.
#Раствор сохраняют для испытания «Эфиры»/
Показатель преломления (2.2.6). От 1,470 до
1,475.
Альдегиды. Не более 0,0010% (Юррт). 7,5 мл
раствора 3 помещают в колбу со стеклянной притертой
пробкой, прибавляют 7,5 мл воды Р и 1,0 мл обесцве¬
ченного раствора парарозанилина Р. Колбу укупорива¬
ют и выдерживают при температуре (25±1) °С в течение
1 ч. Оптическая плотность (2.2.25) полученного раст¬
вора, измеренная при 552 нм, не должна превышать
оптическую плотность раствора сравнения, приготов¬
ленного параллельно с использованием 7,5 мл эталон¬
ного раствора формальдегида (5 ррт СН20) Р и 7,5 мл
водыР. Испытание считают недействительным, если
раствор сравнения не имеет розового окрашивания.
Эфиры. К конечному раствору, полученному в
испытании «Кислотность или щелочность», прибав¬
ляют 10,0 мл 0,1 М раствора натрия гидроксида,
кипятят с обратным холодильником в течение 5 мин,
охлаждают и прибавляют 0,5 мл раствора фенол¬
фталеина Р. На титрование полученного раствора
должно пойти не менее 8,0 мл 0,1 М раствора кисло¬
ты хлористоводородной.
Примесь А и сопутствующие примеси. Газовая
хроматография (2.2.28).
Испытуемый раствор. 10,0 мл раствора 3 дово¬
дят водой Р до объема 100,0 мл.
Раствор сравнения (а). 10,0 г глицерина Р1 до¬
водят водой Р до объема 20,0 мл. 10,0 мл полученно¬
го раствора доводят водой Р до объема 100,0 мл.
Раствор сравнения (Ь). 1,000 г диэтиленгли¬
коля Р растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят раствором сравнения (а) до объема
10.0 мл. 1,0 мл полученного раствора доводят раство¬
ром сравнения (а) до объема 20,0 мл.
Раствор сравнения (ф. 1,0 мл испытуемого раст¬
вора смешивают с 5,0 мл раствора сравнения (Ь) и до¬
водят водой Р до объема 100,0 мл. 1,0 мл полученно¬
го раствора доводят водой Р до объема 10,0 мл.
Рисунок #0496.-1. Инфракрасный спектр глицерина 85 %
Глицерин
357
Раствор сравнения (е). 5,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 30 м и диаметром 0,53 мм, по¬
крытая слоем полимера с 6 % полицианопропилфе-
нилсилоксана и 94 % полидиметилсилоксана;
- газ-носитель: гелий для хроматографии Р;
- деление потока: 1:10;
- линейная скорость: 38 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0
100
0—16
100 -> 220
16—20
220
Блок ввода
проб
220
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,5 мкл.
Порядок выхода пиков: примесь А, глицерин.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 7,0 между пиками при¬
меси А и глицерина.
Предельное содержание примесей:
-примесь А (не более 0,1 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь соответствующего пика на хроматограмме
раствора сравнения (с);
- любая другая примесь со временем удержи¬
вания меньшим, чем время удерживания глицерина
(не более 0,1 %): на хроматограмме испытуемого
раствора площадь любого пика со временем удер¬
живания меньшим, чем время удерживания глицери¬
на, не должна превышать площадь пика примеси А
на хроматограмме раствора сравнения (с);
- сумма примесей со временами удерживания
большими, чем время удерживания глицерина (не
более 0,5 %): на хроматограмме испытуемого раст¬
вора сумма площадей пиков с временами удержива¬
ния большими, чем время удерживания глицерина,
не должна превышать 5-кратную площадь пика при¬
меси А на хроматограмме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,05 площади пика примеси А на
хроматограмме раствора сравнения (е).
Галогенпроизводные. Не более 0,0035%
(35 ррт). К 10 мл раствора 5 прибавляют 1мл
раствора натрия гидроксида разведенного Р, 5 мл
воды Р и 50 мг никель-алюминиевого сплава, сво¬
бодного от галогенов, Р. Смесь нагревают на водя¬
ной бане в течение 10 мин, охлаждают и фильтруют.
Колбу и фильтр промывают водой Р до получения
25 мл фильтрата. К 5 мл фильтрата прибавляют
4 мл 96% спирта Р, 2,5 мл воды Р, 0,5 мл кисло¬
ты азотной Р и 0,05 мл раствора серебра нитра¬
та Р2 и перемешивают. Через 2 мин полученный
раствор по степени мутности не должен превышать
эталон, приготовленный в то же самое время путем
смешивания 7,0 мл эталонного раствора хлорида
(5 ррт С1) Р, 4 мл 96% спирта Р, 0,5 мл воды Р,
0,5 мл кислоты азотной Р и 0,05 мл раствора се¬
ребра нитрата Р2.
Сахара. К 10 мл раствора 5 прибавляют 1 мл
кислоты серной разведенной Р и нагревают на во¬
дяной бане в течение 5 мин. Прибавляют 3 мл раст¬
вора натрия гидроксида Р, свободного от карбона¬
тов (приготовленного как указано для 1 М раствора
натрия гидроксида, свободного от карбонатов),
перемешивают и прибавляют по каплям 1 мл све¬
жеприготовленного раствора меди сульфата Р.
Раствор должен быть прозрачным и иметь синее
окрашивание. Продолжают нагревание на водяной
бане в течение 5 мин. Не должно образовываться
осадка, должно сохраняться синее окрашивание.
Хлориды (2.4.4). Не более 0,0010 % (10 ррт).
1 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испыта¬
ние на хлориды. Эталон готовят с использованием
1 мл эталонного раствора хлорида (5 ррт С1) Р,
доведенного водой Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 8 мл раствора 3 доводят водой Р
до объема 20 мл. 12 мл полученного раствора
должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием эталон¬
ного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 2,0%. Определение
проводят из 1,000 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,01 %.
Определение проводят из 5,0 г испытуемого образ¬
ца после нагревания до кипения и прокаливания.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,075 г испытуемого образца тщательно пере¬
мешивают с 45 мл воды Р, прибавляют 25,0 мл
смеси из 1 объема 0,1 М раствора кислоты сер¬
ной и 20 объемов 0,1 М раствора натрия перйо¬
дата. Выдерживают в защищенном от света месте
в течение 15 мин. Прибавляют 5,0 мл раствора
500 г/л этиленгликоля Р и выдерживают в защи¬
щенном от света месте в течение 20 мин. Титруют
0,1 М раствором натрия гидроксида, используя в
качестве индикатора 0,5 мл раствора фенолфта¬
леина Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 9,21 мг С3Н803.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
A. 2,2'-Оксидиэтанол (диэтиленгликоль).
/ОН
но
B. Этан-1,2-диол (этиленгликоль).
н он
/ОН и энантиомер
Н3С^
C. (Р5)-Пропан-1,2-диол (пропиленгликоль).
358
Гэсударственная фармакопея Республики Беларусь
07/2016:0497
ГЛИЦЕРИН 85%
01усего1ит (85 рег сеп1ит)
в1-УСЕ1Ю1- (85 РЕК СЕЫТ)
ОПРЕДЕЛЕНИЕ
Водный раствор пропан-1,2,3-триола.
Содержание: не менее 83,5 % (м/м) и не более
88,5 % (м/м) пропан-1,2,3-триола (С3Н803; М.м. 92,1).
ОПИСАНИЕ (СВОЙСТВА)
Сиропообразная маслянистая на ощупь бесц¬
ветная или почти бесцветная прозрачная жидкость.
Очень гигроскопичен.
Мало растворим в ацетоне, практически нераст¬
ворим в жирных и эфирных маслах. Смешивается с
водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Показатель преломления», как указано в разделе
«Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр глицерина 85 %
по Европейской Фармакопее #или спектр, представ¬
ленный на рисунке #0497.-1*.
C. 1 мл испытуемого образца смешивают с
0,5 мл кислоты азотной Р. На полученный раствор
наслаивают 0,5 мл раствора калия дихромата Р. На
границе раздела жидкостей появляется синее кольцо.
В течение 10 мин синее окрашивание не переходит в
нижний слой.
О. В выпарительной чашке нагревают 1 мл ис¬
пытуемого образца с 2 г калия гидросульфата Р. Вы¬
деляющиеся пары (акролеин) окрашивают фильтро¬
вальную бумагу, импрегнированную щелочным рас¬
твором калия тетрайодмеркурата Р, в черный цвет.
ИСПЫТАНИЯ
Раствор 5.117,6 г испытуемого образца доводят
водой, свободной от углерода диоксида, Р до объема
200,0 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). 10 мл раствора 3
доводят водой Р до объема 25 мл. Полученный раст¬
вор должен быть бесцветным.
Кислотность или щелочность. К 50 мл раст¬
вора 3 прибавляют 0,5 мл раствора фенолфтале¬
ина Р. Раствор должен быть бесцветным. При при¬
бавлении не более 0,2 мл 0,1 М раствора натрия
гидроксида должно появиться розовое окрашивание.
Фаствор сохраняют для испытания «Эфиры».#
Показатель преломления (2.2.6). От 1,449 до
1,455.
Альдегиды. Не более 0,0010 % (10 ррт). 7,5 мл
раствора 3 помещают в колбу со стеклянной при¬
тертой пробкой, прибавляют 7,5 мл воды Р и 1,0 мл
обесцвеченного раствора парарозанилина Р. Кол¬
бу укупоривают и выдерживают при температуре
(25±1) °С в течение 1 ч. Оптическая плотность (2.2.25)
полученного раствора, измеренная при 552 нм, не
должна превышать оптическую плотность раствора
сравнения, приготовленного параллельно с использо¬
ванием 7,5 мл эталонного раствора формальдегида
(5 ррт СН20) Р и 7,5 мл воды Р. Испытание считают
недействительным, если раствор сравнения не имеет
розового окрашивания.
Эфиры. К конечному раствору, полученному в
испытании «Кислотность или щелочность», прибав¬
ляют 10,0 мл 0,1 М раствора натрия гидроксида,
кипятят с обратным холодильником в течение 5 мин,
Рисунок #0497.-1. Инфракрасный спектр глицерина 85 %
Гпицерин 85 %
359
охлаждают и прибавляют 0,5 мл раствора фенол¬
фталеина Р. На титрование полученного раствора
должно пойти не менее 8,0 мл 0,1 М раствора кисло¬
ты хлористоводородной.
Примесь А и сопутствующие примеси. Газовая
хроматография (2.2.28).
Испытуемый раствор. 10,0 мл раствора 3 дово¬
дят водой Р до объема 100,0 мл.
Раствор сравнения (а). 11,8 г глицерина 85 % Р1
доводят водой Р до объема 20,0 мл. 10,0 мл получен¬
ного раствора доводят водой Р до объема 100,0 мл.
Раствор сравнения (Ь). 1,000 г диэтиленгли¬
коля Р растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят раствором сравнения (а) до объема
10.0 мл. 1,0 мл полученного раствора доводят раство¬
ром сравнения (а) до объема 20,0 мл.
Раствор сравнения (д). 1,0 мл испытуемого раст¬
вора смешивают с 5,0 мл раствора сравнения (Ь) и до¬
водят водой Р до объема 100,0 мл. 1,0 мл полученно¬
го раствора доводят водой Р до объема 10,0 мл.
Раствор сравнения (е). 5,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 30 м и диаметром 0,53 мм, по¬
крытая слоем полимера с б % полицианопропилфе-
нилсилоксана и 94 % полидиметилсилоксана;
- газ-носитель: гелий для хроматографии Р;
- деление потока: 1:10;
- линейная скорость: 38 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0
100
0—16
100 —► 220
16—20
220
Блок ввода
проб
220
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,5 мкл.
Порядок выхода пиков: примесь А, глицерин.
Пригодность хроматографической системы:
раствор сравнения (с!):
-разрешение: не менее 7,0 между пиками при¬
меси А и глицерина.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
- любая другая примесь со временем удержива¬
ния меньшим, чем время удерживания глицерина (не
более 0,1 %): на хроматограмме испытуемого раст¬
вора площадь любого пика со временем удержива¬
ния меньшим, чем время удерживания глицерина, не
должна превышать площадь пика примеси А на хро¬
матограмме раствора сравнения (с);
-сумма примесей со временами удерживания
большими, чем время удерживания глицерина (не
более 0,5 %): на хроматограмме испытуемого раст¬
вора сумма площадей пиков с временами удержива¬
ния большими, чем время удерживания глицерина, не
должна превышать 5-кратную площадь пика примеси
А на хроматограмме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,05 площади пика примеси А на
хроматограмме раствора сравнения (е).
Галогенпроизводные Не более 0,0030%
(30 ррт). К 10 мл раствора 3 прибавляют 1 мл раст¬
вора натрия гидроксида разведенного Р, 5 мл
воды Р и 50 мг никель-алюминиевого сплава, свобод¬
ного от галогенов, Р. Смесь нагревают на водяной
бане в течение 10 мин, охлаждают и фильтруют. Кол¬
бу и фильтр промывают водой Р до получения 25 мл
фильтрата. К 5 мл фильтрата прибавляют 4 мл 96%
спирта Р, 2,5 мл воды Р, 0,5 мл кислоты азотной Р
и 0,05 мл раствора серебра нитрата Р2 и переме¬
шивают. Через 2 мин полученный раствор по степени
мутности не должен превышать эталон, приготовлен¬
ный параллельно путем смешивания 7,0 мл эталон¬
ного раствора хлорида (5 ррт С1) Р, 4 мл 96% спир¬
та Р, 0,5 мл воды Р, 0,5 мл кислоты азотной Р и
0,05 мл раствора серебра нитрата Р2.
Сахара. К10 мл раствора 3 прибавляют 1 мл кисло¬
ты серной разведенной Р и нагревают на водяной бане
в течение 5 мин. Прибавляют 3 мл раствора натрия ги¬
дроксида Р, свободного от карбонатов (приготовленного
как указано для 1М раствора натрия гидроксида, сво¬
бодного от карбонатов), перемешивают и прибавляют
по каплям 1 мл свежеприготовленного раствора меди
сульфата Р. Раствор должен быть прозрачным и иметь
синее окрашивание. Продолжают нагревание на водя¬
ной бане в течение 5 мин. Не должно образовываться
осадка, должно сохраняться синее окрашивание.
Хлориды (2.4.4). Не более 0,0010 % (10 ррт).
1 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды. Эталон готовят с использованием 1 мл
эталонного раствора хлорида (5 ррт С1) Р, доведен¬
ного водой Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 8 мл раствора 3 доводят водой Р до
объема 20 мл. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не менее 12,0% и не более
16,0 %. Определение проводят из 0,200 г испытуемо¬
го образца.
Сульфатная зола (2.4.14). Не более 0,01 %.
Определение проводят из 5,0 г испытуемого образца
после нагревания до кипения и прокаливания.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,075 г испытуемого образца тщательно пере¬
мешивают с 45 мл воды Р, прибавляют 25,0 мл сме¬
си из 1 объема 0,1 М раствора кислоты серной и 20
объемов 0,1 М раствора натрия перйодата. Выдер¬
живают в защищенном от света месте в течение 15
мин. Прибавляют 5,0 мл раствора 500 г/л этиленгли¬
коля Р и выдерживают в защищенном от света месте
в течение 20 мин. Титруют 0,1 М раствором натрия
гидроксида, используя в качестве индикатора 0,5 мл
раствора фенолфталеина Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 9,21 мг С3Н803.
360
Государственная фармакопея Республики Беларусь
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
A. 2,2-Оксидиэтанол (диэтиленгликоль).
/ОН
нет
B. Этан-1,2-диол (этиленгликоль).
С. (Я5)-Пропан-1,2-диол (пропиленгликоль).
07/2016:0495
ГЛИЦЕРИНА МОНОСТЕАРАТ 40-55
(31усегоН топоз1еагаз 40-55
вОГСЕЯОк- МОЫ08ТЕАКАТЕ 40-55
ОПРЕДЕЛЕНИЕ
Смесь моноацилгпицеринов, в основном моно-
стеароилглицерина, с переменными количествами
ди- и триацилглицеринов. Получают частичным гли-
церолизом растительных масел, преимущественно
содержащих триацилглицерины, образованные паль¬
митиновой (гексадекановой) или стеариновой (окта¬
декановой) кислотой, либо этерификацией глицерина
стеариновой кислотой. Происхождение жирных кис¬
лот может быть растительным или животным.
Содержание:
- моноацилглицерины: от 40,0 % до 55,0 %;
- диацилглицерины: от 30,0 % до 45,0 %;
- триацилглицерины: от 5,0 % до 15,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Твердая воскообразная масса, или маслянистый
порошок или хлопья белого или почти белого цвета.
Практически нерастворим в воде, растворим в
96 % спирте при температуре 60 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С, О.
Вторая идентификация: А, В.
A. Температура плавления (2.2.15). От 54 °С до
66 °С. Расплавленный испытуемый образец вводят в
капиллярные трубки и выдерживают в течение 24 ч в
плотно укупоренном контейнере.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,5 г испытуемого образ¬
ца растворяют в метиленхлориде Р при слабом на¬
гревании и доводят до объема 10 мл этим же раство¬
рителем.
Раствор сравнения. 0,5 г ФСО глицерина моно-
стеарата 40-55 растворяют в метиленхлориде Р
при слабом нагревании и доводят до объема 10 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: гексан Р—эфир Р (30:70, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Проявление: пластинку опрыскивают раство¬
ром 0,1 г/л родамина В Р в 96 % спирте Р и про¬
сматривают в ультрафиолетовом свете при длине
волны 365 нм.
Пригодность хроматографической системы:
раствор сравнения:
- на хроматограмме обнаруживаются 4 полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются пятна, соответствующие
по расположению пятнам на хроматограмме раствора
сравнения.
С. Испытуемый образец выдерживает испыта¬
ние «Состав жирных кислот», как указано в разделе
«Испытания», в соответствии с типом, указанным на
этикетке.
й. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение» по со¬
держанию моноацилгпицеринов.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 3,0. Опре¬
деление проводят из 1,0 г испытуемого образца. В
качестве растворителя используют смесь равных
объемов 96 % спирта Р и толуола Р. Испытание
проводят с использованием слабого нагревания.
Йодное число (2.5.4, метод А). Не более 3,0.
Число омыления (2.5.6). От 158 до 177. Опреде¬
ление проводят из 2,0 г испытуемого образца. Титро¬
вание проводят при нагревании.
Свободный глицерин. Не более 6,0 %. Испыта¬
ние проводят, как указано в разделе «Количественное
определение».
Состав жирных кислот (2.4.22, метод С). Ис¬
пользуют смесь веществ для градуировки согласно
таблице 2.4.22.-1.
Фракция испытуемого образца, состоящая из
жирных кислот, должна иметь следующий состав:
Глицерина
моностеа¬
рат 40-55
Состав жирных кислот
Тип I
Кислота стеариновая: от 40,0 % до 60,0 %.
Суммарное содержание пальмитиновой и
стеариновой кислот: не менее 90,0 %
Тип II
Кислота стеариновая: от 60,0 % до 80,0 %.
Суммарное содержание пальмитиновой и
стеариновой кислот: не менее 90,0 %
Тип III
Кислота стеариновая: от 80,0 % до 99,0 %.
Суммарное содержание пальмитиновой и
стеариновой кислот: не менее 96,0 %
Никель (2.4.31). Не более 0,0001 % (1 ррт).
Вода (2.5.12). Не более 1,0 %. Определение про¬
водят из 1,00 г испытуемого образца при слабом на¬
гревании. В качестве растворителя используют пири¬
дин Р.
Общая зола (2.4.16). Не более 0,1 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эксклюзионная хроматография (2.2.30).
Испытуемый раствор. 0,200 г испытуемого об¬
разца (ш) помещают в предварительно взвешенную
колбу вместимостью 15 мл, прибавляют 5,0 мл те-
трагидрофурана Р и встряхивают до растворения.
Гпицин
361
Снова взвешивают колбу и вычисляют общую массу
растворителя и испытуемого образца (М).
Растворы сравнения. В четыре предварительно
взвешенные колбы вместимостью 15 мл помещают
2,5 мг, 5,0 мг, 10,0 мг и 20,0 мг глицерина Р. В каждую
из четырех колб прибавляют по 5,0 мл тетрагидро-
фурана Р. Колбы снова взвешивают и вычисляют кон¬
центрацию глицерина в мг/г для каждого из растворов
сравнения.
Условия хроматографирования:
- колонка длиной 0,6 м и внутренним диаметром
7 мм, заполненная сополимером стирол-дивинилбен-
зола Р (размер частиц 5 мкм) с размером пор 10 нм;
- подвижная фаза: тетрагидрофуран Р\
- скоростью подвижной фазы: 1 мл/мин;
-детектор: дифференциальный рефрактоме¬
трический;
- объем вводимой пробы: 40 мкл.
Относительное удерживание (по отношению к
глицерину, время удерживания — около 15 мин): три-
ацилглицерины — около 0,75; диацилглицерины —
около 0,80; моноацилглицерины — около 0,85.
Расчеты:
- свободный глицерин: с помощью градуировоч¬
ного графика находят концентрацию глицерина (С, мг/г)
в испытуемом растворе и рассчитывают его содержа¬
ние в процентах в испытуемом образце по формуле:
СМ .
ал -10 *
- моно-, ди- и триацилглицерины: рассчитывают
процентное содержание в испытуемом образце мето¬
дом нормализации.
МАРКИРОВКА
Указывают тип глицерина моностеарата 40-55.
07/2016:0614
глицин
О/устит
енгсшЕ
НгМ^^СОгН
С2Н5КЮ2 М.м. 75,1
[56-40-6]
ОПРЕДЕЛЕНИЕ
2-Аминоуксусная кислота.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A, Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО глицина #или спектр, представ¬
ленный на рисунке #0614.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО глицина растворяют по от¬
дельности в минимальном количестве спирта (60%,
об/об) Р и выпаривают до сухих остатков, которые ис¬
пользуют для получения новых спектров.
B. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Рисунок #0614.-1. Инфракрасный спектр ФСО глицина
46. Зак. 1060.
362
Гэсударственная фармакопея Республики Беларусь
Результаты, на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
С. 50 мг испытуемого образца растворяют в 5 мл
воды Р, прибавляют 1 мл раствора натрия гипо¬
хлорита концентрированного Р и кипятят в течение
2 мин. Прибавляют 1 мл кислоты хлористоводород¬
ной Р и кипятят в течение (4—5) мин. Прибавляют
2 мл кислоты хлористоводородной Р и 1 мл раст¬
вора 20 г/л резорцина Р, кипятят в течение 1 мин и
охлаждают. Прибавляют 10 мл воды Р и перемеши¬
вают. К 5 мл полученного раствора прибавляют б мл
раствора натрия гидроксида разведенного Р. Полу¬
ченный раствор имеет фиолетовую окраску и зелено¬
вато-желтую флуоресценцию. Через несколько минут
окраска изменяется на оранжевую, а затем на жел¬
тую. Интенсивная флуоресценция сохраняется.
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)7.
рН (2.2.3). От 5,9 до 6,4. 10 мл раствора 3 до¬
водят водой, свободной от углерода диоксида, Р до
объема 20 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. К 500 мл подвижной
фазы прибавляют 1,5 мл кислоты фосфорной Р.
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора смесью раст¬
ворителей до объема 10,0 мл.
Раствор сравнения (Ь). 80 мг иминодиуксусной
кислоты Р (примесь А) и 80 мг испытуемого образца
растворяют в смеси растворителей и доводят до объ¬
ема 50,0 мл этой же смесью растворителей.
Раствор сравнения (с). 50,0 мг глицина ангидри¬
да Р (примесь В), 50,0 мг диглицина Р (примесь Н) и
50.0 мг триглицина Р (примесь I) растворяют в смеси
растворителей и доводят до объема 50,0 мл этой же
смесью растворителей. 1,0 мл полученного раствора
доводят смесью растворителей до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 25 °С;
- подвижная фаза: 1,4 г натрия пентансульфо-
ната Р растворяют в 900 мл воды Р, доводят до рН
2,2 кислотой фосфорной Р и доводят водой Р до объ¬
ема 1 л;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания глицина.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, Н и I, используя хроматограм¬
му раствора сравнения (с).
Относительное удерживание (по отношению к
глицину, время удерживания — около 5,5 мин): при¬
месь А— около 0,7; примесь В — около 0,75; примесь
Н — около 1,7; примесь I — около 2,0.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5,0 между пиками при¬
меси А и глицина.
Расчет процентных содержаний:
- для каждой примеси — используют концентра¬
цию глицина гидрохлорида в растворе сравнения (а);
-для примесей В, Н и I — используют концентра¬
цию соответствующей примеси в растворе сравнения (с).
Предельное содержание примесей:
-примеси В, Н, I: не более 0,10 % для каждой
примеси;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,2 %;
- учитываемый предел: 0,05 %.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов/
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Вода Р или буферный раствор для
приготовления образца, подходящий для применяе¬
мого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт АIН^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (ф. 30 мг изолейцина Р и
30 мг лейцина Р (примесь А) растворяют в растворе А
и доводят до объема 50,0 мл этим же растворителем.
1.0 мл полученного раствора доводят раствором А до
объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь), (б) вводят в аминокислотный анализатор. За¬
пускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с!):
Глицин
363
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию глицина в растворе сравне¬
ния (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,10 % для каждой примеси;
- сумма примесей: не более 1,0 %;
- учитываемый предел: 0,05 %.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в воде Р и доводят до объема
10,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят водой Р до объема 10,0 мл.
Раствор сравнения (а). 10 мг ФСО глицина раст¬
воряют в воде Р и доводят до объема 10,0 мл этим же
растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (а) доводят водой Р до объема 200 мл.
Раствор сравнения (с). 10 мг ФСО глицина и
10 мг ФСО аланина растворяют в воде Р и доводят до
объема 25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем целлюлозы
для хроматографии Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 80 °С в течение
30 мин.
Проявление: пластинку опрыскивают раство¬
ром нингидрина Р и высушивают при температуре от
100 °С до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0075 % (75 ррт).
0,67 г испытуемого образца растворяют в воде Р и до¬
водят до объема 15 мл этим же растворителем.
#Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
#Сульфаты (2.4.13). Не более 0,0065%
(65 ррт). 2,31 г испытуемого образца растворяют в
воде Р и доводят до объема 15 мл этим же раство¬
рителем. Полученный раствор должен выдерживать
испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато¬
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 50 мг испытуемого
образца должны выдерживать испытание на аммония
соли. Эталон готовят с использованием 0,1 мл эта¬
лонного раствора аммония (100 ррт N14^ Я#
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
70,0 мг испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
кислоты уксусной безводной Р и немедленно по¬
сле растворения титруют 0,1 М раствором кислоты
хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 7,51 мг С2Н5М02.
ПРИМЕСИ
Специфицированные примеси: В, Н, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования):
А, С, О, Е, Г, С.
н
НОгС^^^ N ^^С02Н
А. 2,2'-Иминодиуксусная кислота.
B. Пиперазин-2,5-дион (глицина ангидрид).
С1^^С02Н
C. 2-Хлоруксусная кислота.
46*. Зак. 1060.
364
Гэсударственная фармакопея Республики Беларусь
го
О. 1,3,5,7-Тетраазатрицикло[3.3.1.137]декан.
хо2н
Н2М
Е. З-Аминопропановая кислота (р-аланин).
н3с^
^N1. Х02Н
Р. 2-(Метиламино)уксусная кислота (саркозин).
н мн2
со2н
(3. (25)-2-Амино-3-гидроксипропановая кислота
(серин).
Н21Ч
N. Х02Н
О
Н. 2-[(2-Аминоацетил)амино]уксусная кислота (ди¬
глицин).
I. 2-[[2-[(2-аминоацетил)амино]ацетил]амино]-
уксусная кислота (триглицин).
07/2016:0750
ГЛУТАМИНОВАЯ КИСЛОТА
Аас1ит дЫатюит
6ШТАМ1САСЮ
но2с
С5Н9Ы04 М.м. 147,1
[56-86-0]
ОПРЕДЕЛЕНИЕ
(25)-2-Аминопентандиовая кислота.
Содержание: не менее 98,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в кипящей воде, мало раство¬
рим в холодной воде, практически нерастворим в ук¬
сусной кислоте, в ацетоне и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО глутаминовой кислоты #или
спектр, представленный на рисунке #0750.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО глутаминовой кислоты раст¬
воряют по отдельности в минимальном количестве
воды Р, выпаривают при температуре 60 °С до сухих
остатков, которые используют для получения новых
спектров.
С. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества».
На хроматограмме испытуемого раствора (Ь) обнару¬
живается пятно, соответствующее по расположению,
цвету и размеру основному пятну на хроматограмме
раствора сравнения (а).
й. К 2,0 мл раствора 3, приготовленного как
указано в разделе «Испытания», прибавляют 0,1 мл
раствора фенолфталеина Р и от 3,0 мл до 3,5 мл
1 М раствора натрия гидроксида до появления крас¬
ного окрашивания. К полученному раствору прибавля¬
ют смесь из 3 мл раствора формальдегида Р, 3 мл
воды, свободной от углерода диоксида, Р и 0,1 мл
раствора фенолфталеина Р, к которой прибавлен
1 М раствор натрия гидроксида до появления розо¬
вого окрашивания. Раствор обесцвечивается. Прибав¬
ляют 1 М раствор натрия гидроксида до появления
красного окрашивания. Общий объем израсходован¬
ного 1 М раствора натрия гидроксида составляет от
4.0 мл до 4,7 мл.
ИСПЫТАНИЯ
Раствор 3. 5,00 г испытуемого образца раство¬
ряют в 1 М растворе кислоты хлористоводородной
при осторожном нагревании и доводят до объема
50.0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
+30,5 до +32,5 (в пересчете на сухое вещество). Опре¬
деляют удельное оптическое вращение раствора 3.
Нингидрин-положительные вещества. Тонко¬
слойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в 5 мл раствора аммиака разве¬
денного Р2 и доводят водой Р до объема 10 мл.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО глутамино¬
вой кислоты растворяют в воде Р и доводят до объ¬
ема 50 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО глутамино¬
вой кислоты и 10 мг ФСО аспарагиновой кислоты
растворяют в воде Р и доводят до объема 25 мл этим
же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл; пластинку су¬
шат в потоке воздуха в течение 15 мин.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром нингидрина Р и высушивают при температуре от
100 °С до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Гпюкоза безводная
365
Рисунок #0750.-1. Инфракрасный спектр ФСО глутаминовой кислоты
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в 3 мл кис¬
лоты азотной разведенной Р и доводят водой Р до
объема 15 мл. Раствор, к которому прибавляют 1 мл
воды Р вместо кислоты азотной разведенной Р, дол¬
жен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли (2.4.1, метод В). Не более
0,0200 % (200 ррт). 50 мг испытуемого образца долж¬
ны выдерживать испытание на аммония соли. Эталон
готовят с использованием 0,1 мл эталонного раст¬
вора аммония (100 ррт Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,130 г испытуемого образца растворяют в 50 мл
воды, свободной от углерода диоксида, Р при осторож¬
ном нагревании и охлаждают. Титруют 0,1 Мраствором
натрия гидроксида до изменения окраски раствора от
желтой до синей, используя 0,1 мл раствора бромти-
молового синего Р1 в качестве индикатора.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 14,71 мг С5Н91Ч04.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0177
ГЛЮКОЗА БЕЗВОДНАЯ
Сз1исозит апНудпсит
ОШС08Е, АЫНУОКОиЗ
С'Н12Ов М.м. 180,2
[50-99-7]
47. Зак. 1060.
366
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
0-Глкжопираноза.
Получают из крахмала.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворима в воде, очень мало раствори¬
ма в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: С, О.
A. Удельное оптическое вращение (2.2.7): от
+52,5 до +53,3 (в пересчете на безводное вещество).
10,0 г испытуемого образца растворяют в 80 мл
воды Р, прибавляют 0,2 мл раствора аммиака разве¬
денного Р1, выдерживают в течение 30 мин и доводят
водой Р до объема 100,0 мл.
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
C. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(2:3, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20 мл этой же смесью растворителей.
Раствор сравнения (а). 10 мг ФСО глюкозы
моногидрата растворяют в смеси растворителей и
доводят до объема 20 мл этой же смесью раствори¬
телей.
Раствор сравнения (Ь). 10 мг фруктозы Р, 10 мг
глюкозы Р, 10 мг лактозы Р и 10 мг сахарозы Р раст¬
воряют в смеси растворителей и доводят до объема
20 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: вода Р — метанол Р — кис¬
лота уксусная безводная Р — этиленхлорид Р
(10:15:25:50, об/об/об/об); объемы веществ отмеряют
точно, так как небольшой избыток воды вызывает по¬
мутнение.
Наносимый объем пробы: 2 мкл; тщательно вы¬
сушивают нанесенные пробы.
Фронт подвижной фазы А: не менее 3/4 высоты
пластинки.
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 3/4 высоты пластинки, после замены подвижной
фазы на новую.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку обрабатывают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р и нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-на хроматограмме обнаруживаются 4 полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
О. 0,1 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют 3 мл раствора медно-тартрат-
ного Р и нагревают. Образуется красный осадок.
Е. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в 15 мл воды Р, подогревая на водяной бане.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)7.
Электропроводность (2.2.38). Не более
20 мкСм см1.
20,0 г испытуемого образца растворяют в воде,
свободной от углерода диоксида, Р, приготовленной
из воды дистиллированной Р, и доводят до объема
100.0 мл этим же растворителем. Измеряют электро¬
проводность полученного раствора при осторожном
перемешивании на магнитной мешалке.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,300 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 0,330 г ФСО глюкозы мо¬
ногидрата растворяют в воде Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят водой Р до объема 250,0 мл.
Раствор сравнения (с). 25,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 200,0 мл.
Раствор сравнения (д). 5 мг фруктозы Р (при¬
месь О), 5 мг мальтозы моногидрата Р (примесь А)
и 5 мг мальтотриозы Р (примесь С) растворяют в
воде Р и доводят до объема 50 мл этим же раство¬
рителем.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
7,8 мм, заполненная катионообменной смолой силь¬
ной (кальциевая форма) Р с размером частиц 9 мкм;
- температура: (85±1) °С;
- подвижная фаза: дегазированная вода Р\
- скорость подвижной фазы: 0,3 мл/мин;
- рефрактометрический детектор, при посто¬
янной температуре (например, 40 °С);
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (б);
- время хроматографирования: 1,5-кратное
время удерживания глюкозы.
Относительное удерживание (по отношению к
глюкозе, время удерживания — около 21 мин): при¬
месь С — около 0,7; примеси А и В — около 0,8; при¬
месь й — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (д):
-разрешение: не менее 1,3 между пиками при¬
месей С и А.
Предельное содержание примесей:
- сумма примесей А и В (не более 0,4 %): не бо¬
лее площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
- примесь С (не более 0,2 %): не более 0,5 пло¬
щади основного пика на хроматограмме раствора
сравнения (Ь);
Гпюкоза моногидрат
367
-примесь О (не более 0,15 %): не более 3-крат-
ной площади основного пика на хроматограмме раст¬
вора сравнения (с);
- неспецифицированные примеси (не более
0,10 %): для каждой примеси не более 2-кратной пло¬
щади основного пика на хроматограмме раствора
сравнения (с);
- сумма примесей (не более 0,5 %): не более
1,25-кратной площади основного пика на хромато¬
грамме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): не учитыва¬
ют пики с площадью менее площади основного пика
на хроматограмме раствора сравнения (с).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Декстрин. К 1 г тонкоизмельченного испытуемо¬
го образца прибавляют 20 мл 96 % спирта Р и нагре¬
вают с обратным холодильником. Испытуемый обра¬
зец должен растворяться полностью.
Растворенный крахмал, сульфиты. Не более
0,0015% (15 ррт).
6,7 г испытуемого образца растворяют в 15,0 мл
воды Р при нагревании на водяной бане. Охлаждают
и прибавляют 25 мкл раствора йода Р5. Раствор дол¬
жен быть желтым.
Вода (2.5.12). Не более 1,0 %. Определение про¬
водят из 0,50 г испытуемого образца.
Пирогенность (2.6.8). Если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения больших объемов
без дальнейшей подходящей процедуры удаления
бактериальных эндотоксинов, компетентный уполно¬
моченный орган может потребовать, чтобы она вы¬
держивала испытание на пирогенность. Вводят на 1
кг массы кролика 10 мл раствора, содержащего 50 мг
испытуемого образца в 1 мл воды для инъекций Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С6Н1206 в процентах рассчитывают
с учетом содержания глюкозы в ФСО глюкозы моно¬
гидрата.
#МАРКИРОВКА
При необходимости указывают:
- субстанция апирогенна.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
он он
А. 4-О-а-О-Глюкопиранозил-О-глюкопираноза
(мальтоза).
В. 6-Оа-О-Глюкопиранозил-О-глюкопираноза (изо¬
мальтоза).
он он он
С. а-0-Глюкопиранозил-(1-^4)-а-0-глюкопиранозил-
(1 —>4)-0-ткжопираноза (мальтотриоза).
о
он
I ОН
ОН
ОН
он
Р. О-арабино-Гекс-2-улопираноза (фруктоза).
07/2016:0178
ГЛЮКОЗА МОНОГИДРАТ
С1исозит топо/1ус1псит
СШС08Е МОЫОНУОКАТЕ
НО.
°\
^он
>
^он • Н20
ОН '
С
С Н О ■ Н О
Ч#6П12Ч/6 2^
[77938-63-7]
М.м. 198,2
ОПРЕДЕЛЕНИЕ
й-Гпюкопираноза моногидрат.
Получают из крахмала.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворима в воде, очень мало раствори¬
ма в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: С, О.
А. Удельное оптическое вращение (2.2.7): от
+52,5 до +53,3 (в пересчете на безводное вещество).
10,0 г испытуемого образца растворяют в 80 мл
воды Р, прибавляют 0,2 мл раствора аммиака разве-
47’. Зак. 1060.
368
Гэсударственная фармакопея Республики Беларусь
денного Р1, выдерживают в течение 30 мин и доводят
водой Р до объема 100,0 мл.
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
C. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(2:3, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20 мл этой же смесью растворителей.
Раствор сравнения (а). 10 мг ФСО глюкозы
моногидрата растворяют в смеси растворителей и
доводят до объема 20 мл этой же смесью раствори¬
телей.
Раствор сравнения (Ь). 10 мг фруктозы Р, 10 мг
глюкозы Р 10 мг лактозы Р и 10 мг сахарозы Р раст¬
воряют в смеси растворителей и доводят до объема
20 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: вода Р — метанол Р — кис¬
лота уксусная безводная Р — этиленхлорид Р
(10:15:25:50, об/об/об/об)\ объемы веществ отмеряют
точно, так как небольшой избыток воды вызывает по¬
мутнение.
Наносимый объем пробы: 2 мкл; тщательно вы¬
сушивают нанесенные пробы.
Фронт подвижной фазы А: не менее 3/4 высоты
пластинки.
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 3/4 высоты пластинки, после замены подвижной
фазы на новую.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку обрабатывают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р и нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются 4 полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
Э. 0,1 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют 3 мл раствора медно-тартрат-
ного Р и нагревают. Образуется красный осадок.
Е. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в 15 мл воды Р, подогревая на водяной бане.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
Электропроводность (2.2.38). Не более
20 мкСм см-1.
20,0 г испытуемого образца растворяют в воде,
свободной от углерода диоксида, Р, приготовлен¬
ной из воды дистиллированной Р, и доводят до
объема 100,0 мл этим же растворителем. Измеря¬
ют электропроводность полученного раствора при
осторожном перемешивании на магнитной мешалке.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 0,330 г испытуемого
образца растворяют в воде Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 0,330 г ФСО глюкозы
моногидрата растворяют в воде Р и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят водой Р до объема 250,0 мл.
Раствор сравнения (с). 25,0 мл раствора срав¬
нения (Ь) доводят водой Р до объема 200,0 мл.
Раствор сравнения (д). 5 мг фруктозы Р (при¬
месь О), 5 мг мальтозы моногидрата Р (примесь
А) и 5 мг мальтотриозы Р (примесь С) растворяют
в воде Р и доводят до объема 50 мл этим же раст¬
ворителем.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаме¬
тром 7,8 мм, заполненная катионообменной смо¬
лой сильной (кальциевая форма) Р с размером ча¬
стиц 9 мкм;
- температура: (85±1) °С;
- подвижная фаза: дегазированная вода Р;
- скорость подвижной фазы: 0,3 мл/мин;
- рефрактометрический детектор, при по¬
стоянной температуре (например, 40 °С);
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и растворов сравнения (Ь), (с) и (с!);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания глюкозы.
Относительное удерживание (по отношению
к глюкозе, время удерживания — около 21 мин):
примесь С — около 0,7; примеси А и В — около
0,8; примесь й — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 1,3 между пиками
примесей С и А.
Предельное содержание примесей:
- сумма примесей А и В (не более 0,4 %): не
более площади основного пика на хроматограмме
раствора сравнения (Ь);
- примесь С (не более 0,2 %): не более 0,5
площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
-примесь О (не более 0,15%): не более
3-кратной площади основного пика на хромато¬
грамме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10 %): для каждой примеси не более 2-кратной
площади основного пика на хроматограмме раст¬
вора сравнения (с);
- сумма примесей (не более 0,5 %): не более
1,25-кратной площади основного пика на хромато¬
грамме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): не учиты¬
вают пики с площадью менее площади основного
пика на хроматограмме раствора сравнения (с).
Пределы, указанные в разделе «Сопутству¬
ющие примеси» (таблица 2034.-1) статьи Суб¬
станции для фармацевтического использования
(2034), не применяют.
Гпюкозамина гидрохлорид
369
Декстрин. К 1 г тонкоизмельченного испытуе¬
мого образца прибавляют 20 мл 96 % спирта Р и
нагревают с обратным холодильником. Испытуе¬
мый образец должен растворяться полностью.
Растворенный крахмал, сульфиты. Не бо¬
лее 0,0015 % (15 ррт).
7,4 г испытуемого образца растворяют в
15,0 мл воды Р при нагревании на водяной бане.
Охлаждают и прибавляют 25 мкл раствора
йода Р5. Раствор должен быть желтым.
Вода (2.5.12). Не менее 7,5% и не более
9,5 %. Определение проводят из 0,25 г испытуемо¬
го образца.
Пирогенность (2.6.8). Если субстанция пред¬
назначена для производства лекарственных
средств для парентерального применения боль¬
ших объемов без дальнейшей подходящей про¬
цедуры удаления бактериальных эндотоксинов,
компетентный уполномоченный орган может по¬
требовать, чтобы она выдерживала испытание на
пирогенность. Вводят на 1 кг массы кролика 10 мл
раствора, содержащего 50 мг испытуемого образ¬
ца в 1 мл воды для инъекций Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С6Н1206 в процентах рассчитывают
с учетом содержания глюкозы в ФСО глюкозы моно¬
гидрата.
#МАРКИРОВКА
При необходимости указывают:
- субстанция апирогенна.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
он он
А. 4-О-а-О-Глюкопиранозил-О-глюкопираноза
(мальтоза).
В. 6-О-а-О-Глюкопиранозил-О-глюкопираноза (изо¬
мальтоза).
С. а-й-Гпюкопиранозил-(1-*4)-а-Р-глюкопиранозил-
(1 -+4)-0-глюкопираноза (мальтотриоза).
он
О. О-арабшо-Гекс-2-улопираноза (фруктоза).
07/2016:2446
ГЛЮКОЗАМИНА ГИДРОХЛОРИД
01исо8атт1 ЬудгосЫопдит
6ШС08АМШЕ НУОКОСНМЖЮЕ
С6Н1}Ы05 • НС1 М.м. 215,6
[66-84-2]
ОПРЕДЕЛЕНИЕ
2-Амино-2-дезокси-0-глюкопиранозы гидрохло¬
рид.
Выделяют из природных источников или получа¬
ют с использованием ферментации.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ПРОИЗВОДСТВО
Животные, из которых получают глюкозамина ги¬
дрохлорид, должны выдерживать требования к здоро¬
вью животных, используемых для потребления чело¬
веком.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворим в воде, мало растворим в мета¬
ноле, практически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО глюкозамина гидрохлорида
#или спектр, представленный на рисунке #2446.-1#.
B. 1 мл раствора 5, приготовленного как указано
в разделе «Испытания», дает реакцию (а) на хлориды
(2.3.1).
C. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
48. Зак. 1060.
370
Гэсударственная фармакопея Республики Беларусь
Рисунок #2446.-1. Инфракрасный спектр ФСО глюкозамина гидрохлорида
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
рН (2.2.3). От 3,0 до 5,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
+70,0 до +74,0 (в пересчете на сухое вещество). Из¬
меряют удельное оптическое вращение раствора 3
через 3 ч после приготовления.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. К 0,300 г испытуемого об¬
разца прибавляют 80 мл подвижной фазы и обраба¬
тывают ультразвуком в течение 10 мин, охлаждают до
комнатной температуры и доводят подвижной фазой
до объема 100,0 мл.
Раствор сравнения (а). 25,0 мг ФСО 2-метил-
пиразина растворяют в подвижной фазе и доводят
до объема 10,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 10,0 мл.1,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 15 мг ФСО глюкозами¬
на для проверки пригодности хроматографической
системы (содержит примеси В и С) растворяют в
подвижной фазе и доводят до объема 5,0 мл этим же
растворителем.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 30 °С;
- подвижная фаза: 0,5 г натрия гептансульфо-
ната Р растворяют в воде для хроматографии Р,
прибавляют 0,5 мл фосфорной кислоты Р и 4 мл
раствора 56 г/л калия гидроксида Р и доводят водой
для хроматографии Р до объема 1000 мл; к 1000 мл
полученного раствора прибавляют 50 мл ацетони¬
трила Р1\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 195 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания 2-метилпиразина.
Время удерживания: 2-метилпиразин — около
9 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,5 между пиками при¬
месей В и С.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010% (10 ррт).
Растворитель: вода Р.
Глюкозамина сульфат натрия хлорид
371
1,0 г испытуемого образца должен выдерживать
испытание на тяжелые металлы. Эталон готовят с
использованием 1 мл эталонного раствора свинца
(10 рртРЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
Отсутствие ЕзсНепсЫа со11 (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в
50 мл воды Р, прибавляют 1,0 мл 0,1 М раствора
хлористоводородной кислоты и титруют 0,1 М
раствором натрия гидроксида потенциометриче-
ски (2.2.20). Отмечают объем титранта между дву¬
мя точками перегиба на кривой титрования.
0,1 мл 0,1 М раствора натрия гидроксида со¬
ответствует 21,56 мг С6Н13М05НС1.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, В, С, Е.
ОНО. /О,
'ОН
Е. 5-(Гидроксиметил)фуран-2-карбальдегид
(5-гидроксиметилфурфураль).
07/2016:2447
ГЛЮКОЗАМИНА СУЛЬФАТ
НАТРИЯ ХЛОРИД
С1исозатт1 зи1/аз па1гН сЫопдит
СШС08АММЕ 8Ш.РАТЕ 800ШМ
СШОПЮЕ
[С§Н„Ы0^2 • Н250, • 2ЫаС1 М.м. 573,3
ОПРЕДЕЛЕНИЕ
Бис(2-амино-2-дезокси-0-глюкопиранозы) суль¬
фат бис(натрия хлорид).
Субстанция, состоящая из глюкозамина гидрох¬
лорида, выделенного из природного сырья или полу¬
ченного ферментацией, и натрия сульфата.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ПРОИЗВОДСТВО
Животные, являющиеся источником получения
глюкозамина сульфата натрия хлорида, должны соот¬
ветствовать требованиям к здоровью животных, под¬
ходящих для использования человеком.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, умеренно растворим в
метаноле, практически нерастворим в ацетоне.
А. 2-(Ацетиламино)-2-дезокси-0-глюкопираноза
(А/-ацетилглюкозамин).
В. (1 Я,ТЯ,25,2'5,ЗЯ,3'Я)-1 ,1 '-Пиразин-2,5-диил-
бис(бутан-1,2,3,4-тетрол) (фруктозазин).
С. (1 /?,25,ЗР)-1 -[5-[(25,3Р)-2,3,4-Тригидроксибу-
тил]-пиразин-2-ил]бутан-1,2,3,4-тетрол (дезоксифрук-
тозазин).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО глюкозамина сульфата
натрия хлорида #или спектр, представленный на ри¬
сунке #2447.-1#.
B. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
C. 1 мл раствора 5, приготовленного как указано в
разделе «Испытания», дает реакцию (а) на натрий (2.3.1).
О. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
Е. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
48*. Зак. 1060.
372
Гэсударственная фармакопея Республики Беларусь
Рисунок #2447.-1. Инфракрасный спектр ФСО глюкозамина сульфата натрия хлорида
Раствор 51. 5,0 мл раствора 5 доводят водой Р
до объема 25,0 мл.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
+50,0 до +55,0 (в пересчете на сухое вещество). Из¬
меряют оптическое вращение раствора 5. Испытание
проводят через 3 ч после приготовления раствора 5.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. К 0,400 г испытуемого об¬
разца прибавляют 80 мл подвижной фазы и обраба¬
тывают ультразвуком в течение 10 мин. Охлаждают до
комнатной температуры и доводят подвижной фазой
до объема 100,0 мл.
Раствор сравнения (а). 25,0 мг ФСО 2-метилпи-
разина растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем. 1,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 10,0 мл. 1,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 15 мг ФСО глюкозами¬
на для проверки пригодности хроматографической
системы (содержит примеси В и С) растворяют в
подвижной фазе и доводят до объема 5,0 мл этим же
растворителем.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 30 °С;
- подвижная фаза: 0,5 г натрия гептансульфо-
ната Р растворяют в воде для хроматографии Р,
прибавляют 0,5 мл кислоты фосфорной Р и 4 мл
раствора 56 г/л калия гидроксида Р и доводят водой
для хроматографии Р до объема 1000 мл; к 1000 мл
полученного раствора прибавляют 50 мл ацетони¬
трила Р1\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 195 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания 2-метилпиразина.
Время удерживания: 2-метилпиразин — около 9 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
месей В и С.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010% (10 ррт).
Растворитель: вода Р.
1,0 г испытуемого образца должен выдерживать
испытание на тяжелые металлы. Эталон готовят с
использованием 1 мл эталонного раствора свинца
(10 ррт РЬ) Р.
#Деготь березовый
373
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не менее 23,5 % и не
более 26,0 %. Определение проводят из 1,0 г испыту¬
емого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсНепсЫа соН: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
воды Р и прибавляют 1,0 мл 0,1 М раствора кислоты
хлористоводородной, затем титруют 0,1 М раство¬
ром натрия гидроксида потенциометрически (2.2.20).
Отмечают объем титранта между двумя точками пе¬
региба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 28,67 мг [С6Н13М05]2 Н2504-2МаС1.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, Е.
А. 2-(Ацетиламино)-2-дезокси-0-глюкопираноза)
(Л/-ацетил-глюкозамин).
В. (т,ГР,28,2'8,ЗР,3'Р)-'1,'\'-Пираз]лн-2,5-
диилбис(бутан-1,2,3,4-тетрол) (фруктозазин).
С. (1 /?,25,ЗЯ)-1 -[5-[(25,3/?)-2,3,4-Тригидроксибутил]-
пиразин-2-ил]бутан-1,2,3,4-тетрол (дезоксифруктоза-
зин).
Е. 5-(Гидроксиметил)фуран-2-карбальдегид (5-ги-
дроксиметилфурфурол).
07/2016:РБ0016
#ДЕГОТЬ БЕРЕЗОВЫЙ
Р/х //ди/сУа ВеЫае (01еит Низа)
В1КСН ТАК
ОПРЕДЕЛЕНИЕ
Деготь березовый представляет собой продукт
сухой перегонки наружной части коры (отборной бе¬
ресты) березы.
ОПИСАНИЕ (СВОЙСТВА)
Густая маслянистая неклейкая на ощупь жид¬
кость черного цвета, в отраженном свете имеет голу¬
бовато-зеленый или зеленовато-синий отлив. Запах
специфический.
Смешивается с эфиром, хлороформом, раство¬
ряется в этаноле и в растворах гидроксидов щелоч¬
ных металлов.
Относительная плотность: от 0,925 до 0,950.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца встряхивают с 10 мл
воды Р и прибавляют 2 капли раствора 30 г/л железа
(III) хлорида Р. Появляется зеленоватое окрашивание.
B. 1 г испытуемого образца встряхивают с 10 мл
воды Р и прибавляют 10 мл раствора кальция ги¬
дроксида Р. Появляется темно-коричневое или корич¬
невато-красное окрашивание.
ИСПЫТАНИЯ
Чистота. Каплю испытуемого образца помеща¬
ют на фильтровальную бумагу. Должно образоваться
светлое маслянистое пятно без узора или налета.
Наличие осадка. 5 г испытуемого образца по¬
мещают в пробирку и выдерживают при температу¬
ре 20 °С в течение 24 ч. Не должен образовываться
осадок.
Смолы и осиновый деготь. К 5 г испытуемого
образца прибавляют 50 мл воды Р, взбалтывают и
выдерживают до полного разделения слоев. Окраска
водного слоя не должна изменяться.
Нефть и минеральные масла. К 1,0 г испытуе¬
мого образца прибавляют 50 мл ацетона Р и встря¬
хивают. Испытуемый образец должен полностью рас¬
твориться.
Нерастворимые в бензине вещества. Не более
6,0 %. 2,000 г испытуемого образца помещают в пред¬
варительно высушенную при температуре 105 °С и
взвешенную пробирку, прибавляют 16 мл бензина Р,
тщательно перемешивают, выдерживают до оседа¬
ния частиц и сливают надосадочную жидкость. Оса¬
док высушивают сначала на водяной бане, а затем в
сушильном шкафу при температуре 105 °С в течение
3 ч. Масса полученного остатка не должна превышать
120 мг.
Кислотное число (2.5.1). От 15 до 25.
Число омыления (2.5.6). От 36 до 60.
Эфирное число (2.5.2). Не более 45.
Вода (2.2.13). Не более 0,5 %. Определение про¬
водят из 200 г испытуемого образца. Навеску испыту¬
емого образца помещают в сухую колбу (при опреде¬
лении не используют толуол Р и воду Р для первого
отгона). Содержание воды в процентах рассчитывают
по формуле:
49. Зак. 1060.
374
Гэсударственная фармакопея Республики Беларусь
100 V
т
где:
V — объем воды, определенный при отгоне, мл;
т — масса навески испытуемого образца, г.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0549
ДЕКСАМЕТАЗОНА НАТРИЯ
ФОСФАТ
ОехатеМазоп/ па(гИ рЬозрЬаз
ОЕХАМЕТНА80ЫЕ ЗСЮШМ
РН08РНАТЕ
С22Н20РМаАР М “- 516>4
[2392-39-4]
ОПРЕДЕЛЕНИЕ
9-Фтор-11 (3,17-дигидрокси-16а-метил-3,20-диоксо-
прегна-1,4-диен-21 -илфосфат динатрия.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Очень гигро¬
скопичен.
Легко растворим в воде, мало растворим в 96 %
спирте, практически нерастворим в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, 6.
Вторая идентификация: А, С, О, Е, Р.
A. 10,0 мг испытуемого образца растворяют
в 5 мл воды Р и доводят этанолом Р до объема
100,0 мл. 2,0 мл полученного раствора помещают
в пробирку со шлифом, прибавляют 10,0 мл раст¬
вора фенилгидразина в серной кислоте Р, переме¬
шивают, нагревают в водяной бане при температуре
60 °С в течение 20 мин и немедленно охлаждают.
Оптическая плотность (2.2.25) полученного раст¬
вора, измеренная в максимуме при 419 нм, — не
менее 0,20.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО дексаметазона натрия фосфа¬
та* или спектр, представленный на рисунке#0549.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то испытуе¬
мый образец и ФСО дексаметазона натрия фосфа¬
та растворяют по отдельности в минимальном коли¬
честве 96 % спирта Р, выпаривают на водяной бане
до сухих остатков, которые используют для получения
новых спектров.
Рисунок #0549.-1. Инфракрасный спектр ФСО дексаметазона натрия фосфата
Дексаметазона натрия фосфат
375
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО дексамета¬
зона натрия фосфата растворяют в метаноле Р и
доводят до объема 20 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО преднизолона
натрия фосфата растворяют в растворе сравнения
(а) и доводят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля ^254 Р-
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
Проявление В: пластинку опрыскивают спирто¬
вым раствором кислоты серной Р, нагревают при тем¬
пературе 120 °С в течение 10 мин или до проявления
пятен и охлаждают. Просматривают при дневном свете
и в ультрафиолетовом свете при длине волны 365 нм.
Результаты В: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, размеру и цвету при просматривании
при дневном свете и флуоресценции при просматри¬
вании в ультрафиолетовом свете при длине волны
365 нм основному пятну на хроматограмме раствора
сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два пятна,
которые могут быть не полностью разделены.
й. К 2 мл кислоты серной Р прибавляют 2 мг ис¬
пытуемого образца и встряхивают до растворения. В
течение 5 мин появляется бледное желтовато-корич¬
невое окрашивание. Полученный раствор прибавля¬
ют к 10 мл воды Р и перемешивают. Окраска тускнеет,
а раствор остается прозрачным.
Е. 5 мг испытуемого образца смешивают с 45 мг
магния оксида тяжелого Р и прокаливают в тигле
до получения почти белого остатка (обычно менее 5
мин). Охлаждают, прибавляют 1 мл воды Р, 0,05 мл
раствора фенолфталеина Р1, около 1 мл кислоты
хлористоводородной разведенной Р до получения
бесцветного раствора и фильтруют. К свежеприготов¬
ленной смеси из 0,1 мл раствора ализарина 8Р и
0,1 мл раствора цирконила нитрата Р прибавляют
1,0 мл полученного фильтрата, смешивают, выдержи¬
вают в течение 5 мин и сравнивают окраску получен¬
ного раствора с окраской контрольного опыта, приго¬
товленного таким же образом. Окраска испытуемого
раствора желтая, а контрольного опыта — красная.
Р. К 40 мг испытуемого образца прибавляют 2 мл
кислоты серной Р и осторожно нагревают до выде¬
ления белых паров, прибавляют по каплям кислоту
азотную Р, продолжают нагревание до получения
почти бесцветного раствора и охлаждают. Прибав¬
ляют 2 мл воды Р, нагревают до тех пор, пока белые
пары начнут снова выделяться, охлаждают, прибавля¬
ют 10 мл воды Р и нейтрализуют раствором аммиака
разведенного Р1 по красной лакмусовой бумаге Р.
Полученный раствор дает реакцию (а) на натрий
(2.3.1) и реакцию (Ь) на фосфаты (2.3.1).
С. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (Ь).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона В(К)7.
рН (2.2.3). От 7,5 до 9,5.1 мл раствора 5 доводят во¬
дой, свободной от углерода диоксида, Р до объема 5 мл.
Удельное оптическое вращение (2.2.7). От +75
до +83 (в пересчете на безводное вещество). 0,250 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Раствор А. 7,0 г аммония ацетата Р раство¬
ряют в 1000 мл воды Р.
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО дексаметазо¬
на натрия фосфата и 2 мг ФСО бетаметазона на¬
трия фосфата (примесь В) растворяют в подвижной
фазе А и доводят до объема 100,0 мл этим же раст¬
ворителем.
Раствор сравнения (Ь). 2 мг ФСО дексамета¬
зона натрия фосфата для идентификации пиков
(содержит примеси А, С, Е, Р и О) растворяют в под¬
вижной фазе А и доводят до объема 2,0 этим же раст¬
ворителем.
Раствор сравнения (с). 1,0 мл испытуемо¬
го образца доводят подвижной фазой А до объема
100,0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой А до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 300 мл раст¬
вора А и 350 мл воды Р, доводят до рН 3,8 кис¬
лотой уксусной Р и прибавляют 350 мл метано¬
ла Р\
- подвижная фаза В: 300 мл раствора А дово¬
дят до рН 4,0 кислотой уксусной Р и прибавляют
700 мл метанола Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3,5
90
10
3,5—23,5
90 — 60
10 — 40
23,5—34,5
60 — 5
40 — 95
34,5—50
5
95
49*. Зак. 1060.
376
Гэсударственная фармакопея Республики Беларусь
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифициру¬
ют пики примесей А, С, О, Е, Р и 6, используя хромато¬
грамму раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО дексаметазона натрия фосфата для
идентификации пиков; идентифицируют пик примеси
В, используя хроматограмму раствора сравнения (а).
Относительное удерживание (по отношению
к дексаметазона натрия фосфату, время удержива¬
ния — около 22 мин): примесь С — около 0,5; примесь
О — около 0,6; примесь Е — около 0,8; примесь Р —
около 0,92; примесь В — около 0,95; примесь А— око¬
ло 1,37; примесь О — около 1,41.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси В и дексаметазона натрия фосфата.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—0,75):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющая примеси А, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примесь 6 (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющая примеси О, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примеси В, С, О, Е, Г (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, С, О, Е и Р, не долж¬
ны превышать 2-кратную площадь основного пика на
хроматограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е, Р и О, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Неорганические фосфаты. Не более 1 %. 50 мг
испытуемого образца растворяют в воде Р и доводят
до объема 100 мл этим же растворителем. К 10 мл
полученного раствора прибавляют 5 мл реактива мо¬
либденовованадиевого Р, перемешивают и выдержи¬
вают в течение 5 мин. Желтое окрашивание получен¬
ного раствора должно быть не интенсивнее эталона,
приготовленного в то же самое время и таким же об¬
разом с использованием 10 мл эталонного раствора
фосфата (5 ррт РО) Р.
Этанол (2.4.24, система А). Не более 1,5 %.
Вода (2.5.12). Не более 10,0%. Определение
проводят из 0,200 г испытуемого образца.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем. 5,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 50,0 мл.
Раствор сравнения (а). 2 мг ФСО дексаметазо¬
на (примесь А) и 2 мг ФСО дексаметазона натрия
фосфата растворяют в 2 мл тетрагидрофурана и до¬
водят подвижной фазой до объема 100,0 мл. 5,0 мл
полученного раствора доводят подвижной фазой до
объема 50,0 мл.
Раствор сравнения (Ь). 30,0 мг ФСО дексамета¬
зона натрия фосфата растворяют в подвижной фазе
и доводят до объема 50,0 этим же растворителем.
5,0 мл полученного раствора доводят подвижной фа¬
зой до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: смешивают 520 мл воды Р и
2 мл кислоты фосфорной Р, доводят до температу¬
ры 20 °С, затем доводят до рН 2,6 натрия гидрокси¬
дом Р. Полученный раствор смешивают с 36 мл те¬
трагидрофурана Р и 364 мл метанола Р;
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания дексаметазона натрия фосфата.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (а).
Относительное удерживание (по отношению
к дексаметазона натрия фосфату, время удержива¬
ния — около 8 мин): примесь А— около 2,0.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 6,0 между пиками декса¬
метазона натрия фосфата и примеси А.
Содержание С22Н28РМа208Р в процентах рассчи¬
тывают, используя хроматограмму раствора сравне¬
ния (Ь) и с учетом содержания дексаметазона натрия
фосфата в ФСО дексаметазона натрия фосфата.
ХРАНЕНИЕ
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не-
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Декспантенол
377
г
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Н.
А. 9-Фтор-11Р, 17,21 -тригидрокси-1 ба-метилпрегна-
1,4-диен-3,20-дион (дексаметазон).
В. 9-Фтор-11 р, 17-дигидрокси-16р-метил-3,20-
диоксопрегна-1,4-диен-21-илдигидрофосфат (бетаме-
тазона фосфат).
С, О, Е, Р. Каждая примесь представляет собой
диастереоизомер (диастереоизомеры) (9-фтор-11р,17а-
дигидрокси-16-метил-3,17-диоксо-0-гомо-андроста-1,4-
диен-17а-ил)метилдигидрофосфата (стереохимия у
атомов С-16 и С-17а неизвестна) или (9-фтор-11р,17-
дигидрокси-16а-метил-3,17а-диоксо-0-гомо-андроста-
1,4-диен-17-ил)метилдигидрофосфата (стереохимия у
атома С-17 неизвестна).
С. 9-Фтор-11 р, 17-дигидрокси-16а-метил-3-оксо-
андроста-1,4-диен-17Р-карбоновая кислота.
Н. 9-Фтор-11р,17-дигидрокси-16а-метил-3,20-
диоксопрегн-4-ен-21 -илдигидрофосфат.
07/2016:0761
ДЕКСПАНТЕНОЛ
ОехрапМепо/ит
ОЕХЯАЛ/77УЕЛЮ/-
С9Н19М0д М.м. 205,3
[81-13-0]
ОПРЕДЕЛЕНИЕ
(2Я)-2,4-Дигидрокси-Л/-(3-гидроксипропил)-3,3-
диметилбутанамид.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная или слегка желтоватая вязкая гигро¬
скопичная жидкость или кристаллический порошок
белого или почти белого цвета.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испыта¬
ние «Удельное оптическое вращение», как указано
в разделе «Испытания».
B. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Приготовление: испытуемый образец и раст¬
вор сравнения по отдельности растворяют в 1,0 мл
этанола безводного Р для получения раствора с
концентрацией 5 мг/мл. 0,5 мл полученного раст¬
вора по каплям наносят на поверхность диска из
калия бромида Р. Диск сушат при температуре от
100 °С до 105 °С в течение 15 мин.
Сравнение: ФСО декспантенола #или спектр,
представленный на рисунке #0761 .-1#.
C. Просматривают хроматограммы, получен¬
ные в испытании «З-Аминопропанол». На хромато¬
грамме испытуемого раствора (Ь) обнаруживается
основное пятно, соответствующее по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
О. К 1 мл раствора 5, приготовленного как
указано в разделе «Испытания», прибавляют 1 мл
раствора натрия гидроксида разведенного Р и
0,1 мл раствора меди сульфата Р. Появляется
синее окрашивание.
ИСПЫТАНИЯ
Раствор 5. 2,500 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р
и доводят до объема 50,0 мл этим же растворите¬
лем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
50. Зак. 1060.
378
Гэсударственная фармакопея Республики Беларусь
Рисунок #0761 .-1. Инфракрасный спектр ФСО декспантенола
Цветность (2.2.2, метод II). Окраска раствора
5 должна быть не интенсивнее эталона В(К)6.
рН (2.2.3). Не более 10,5. Измеряют рН раст¬
вора 3.
Удельное оптическое вращения (2.2.7). От
+29,0 до +32,0 (в пересчете на безводное веще¬
ство). Определение проводят с использованием
раствора 5.
З-Аминопропанол. Не более 0,5 %. Тонко¬
слойная хроматография (2.2.27).
Испытуемый раствор (а). 0,25 г испытуемого
образца растворяют в этаноле безводном Р и до¬
водят до объема 5 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят этанолом безводным Р до
объема 10 мл.
Раствор сравнения (а). Содержимое контей¬
нера с ФСО декспантенола растворяют в 1,0 мл
этанола безводного Р для получения раствора с
концентрацией 5 мг/мл.
Раствор сравнения (Ь). 25 мг 3-аминопропа-
нола Р растворяют в этаноле безводном Р и до¬
водят до объема 100 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля О Р.
Подвижная фаза: раствор аммиака кон¬
центрированный Р — метанол Р — бутанол Р
(20:25:55, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от
линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром 100 г/л кислоты трихлоруксусной Р в ме¬
таноле Р и нагревают при температуре 150 °С в
течение 10 мин, опрыскивают раствором 1 г/л нин-
гидрина Р в метаноле Р и нагревают при темпера¬
туре 120 °С до появления окрашивания.
Результаты: на хроматограмме испытуемого
раствора (а) пятно, соответствующее 3-аминопро-
панолу, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны вы¬
держивать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Вода (2.5.72). Не более 1,0%. Определение
проводят из 1,000 г испытуемого образца.
Сульфатная зола (2.4.74). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.7.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,400 г испытуемого образца прибавляют
50,0 мл 0,1 М раствора кислоты хлорной, кипятят
с обратным холодильником в течение 5 ч, предо¬
храняя от попадания влаги, охлаждают и прибав¬
ляют 50 мл диоксана Р, промывая холодильник
и предохраняя от попадания влаги. Прибавляют
0,2 мл раствора нафтолбензеина Р и титруют
0,1 М раствором калия гидрофталата до перехо¬
да окраски от зеленой до желтой.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соот¬
ветствует 20,53 мг С9Н19!Ч04.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Декстран 40 для инъекций
379
07/2016:0999
ДЕКСТРАН 40 ДЛЯ ИНЪЕКЦИЙ
Оех(гапит 40 ад т1ес1аЫ1е
ОЕХТКАЫ 40 РОК 1ШЕСТЮЫ
ОПРЕДЕЛЕНИЕ
Декстран 40 для инъекций представляет со¬
бой смесь полисахаридов преимущественно а-1,6-
глюканового типа.
Средняя относительная молекулярная масса:
около 40 000.
ПРОИЗВОДСТВО
Получают путем гидролиза и фракциони¬
рования декстранов, полученных ферментаци¬
ей сахарозы штаммом иеисопоз!ос тезеп1его1с1ез
ЫРРЬ В-512 = С1Р 78,59 или его подштаммами (на¬
пример тезеп1его1с1ез В-512Р = N010 10817).
Производят в условиях минимального риска ми¬
кробного загрязнения.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень легко растворим в воде, очень мало рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Удельное оптическое вращение (2.2.7): от
+195 до +201 (в пересчете на сухое вещество). 1,0 г
испытуемого образца растворяют в воде Р при нагре¬
вании на водяной бане и доводят до объема 50,0 мл
этим же растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО декстрана #или спектр, пред¬
ставленный на рисунке #0999.-1#.
С. Испытуемый образец выдерживает испыта¬
ние «Молекулярно-массовое распределение декстра¬
нов», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р при нагревании
на водяной бане и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфтале¬
ина Р. Раствор должен оставаться бесцветным. При
прибавлении 0,2 мл 0,01 М раствора натрия гидрок¬
сида должно появиться красное окрашивание. При
прибавлении к полученному раствору 0,4 мл 0,01 М
раствора кислоты хлористоводородной раствор
должен обесцветиться. При прибавлении 0,1 мл
раствора метилового красного Р должно появиться
красное или оранжевое окрашивание.
Азотсодержащие вещества. Не более 0,0110 %
N (110 ррт Ы). Проводят определение азота после
минерализации серной кислотой (2.5.9), используя
0,200 г испытуемого образца и нагревание в течение
2 ч. Дистиллят собирают в смесь из 0,5 мл раствора
бромкрезолового зеленого Р, 0,5 мл раствора ме¬
тилового красного Р и 20 мл воды Р. На титрование
должно пойти не более 0,15 мл 0,01 М раствора кис¬
лоты хлористоводородной.
Остаточные количества органических раство¬
рителей. Газовая хроматография (2.2.28).
Внутренний стандарт: пропанол Р.
Испытуемый раствор. 5 г испытуемого образ¬
ца растворяют в 100 мл воды Р и перегоняют. Со-
Рисунок #0999.-1. Инфракрасный спектр ФСО декстрана
50*. Зак. 1060.
380
Гэсударственная фармакопея Республики Беларусь
бирают первые 45 мл дистиллята. К полученному
дистилляту прибавляют 1 мл раствора 25 г/л про¬
панола Р и доводят водой Р до объема 50 мл.
Раствор сравнения. К 0,5 мл раствора 25 г/л
этанола безводного Р прибавляют 0,5 мл раст¬
вора 25 г/л пропанола Р и 0,5 мл раствора 2,5 г/л
метанола Р и доводят водой Р до объема 25,0 мл.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 1,8 м
и внутренним диаметром 2 мм, заполненная со¬
полимером этилвинилбензол-дивинилбензола Р
((125—150) мкм);
- газ-носитель: азот для хроматографии Р\
- скорость газа носителя: 25 мл/мин;
- температура: колонка — 190 °С; блок вво¬
да проб — 240 °С; детектор — 210 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: вводят равные объ¬
емы испытуемого раствора и раствора сравнения.
Предельное содержание примесей:
- этанол (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площадь пика этанола
не должна превышать площадь соответствующего
пика на хроматограмме раствора сравнения;
- метанол (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика метанола
не должна превышать площадь соответствующего
пика на хроматограмме раствора сравнения;
- сумма растворителей, кроме этанола, ме¬
танола и пропанола (не более 0,5 % в пересче¬
те на пропанол): на хроматограмме испытуемого
раствора сумма площадей пиков всех раствори¬
телей, кроме метанола, этанола и пропанола, не
должна превышать площадь пика пропанола на
хроматограмме раствора сравнения.
Молекулярно-массовое распределение
декстранов (2.2.39). Средняя молекулярная мас¬
са (Мш) должна быть от 35 000 до 45 000. Средняя
молекулярная масса 10% высокомолекулярной
фракции не должна превышать 110 000. Средняя
молекулярная масса 10 % низкомолекулярной
фракции должна быть не менее 7000.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны вы¬
держивать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 7,0 %. 0,200 г испытуемого образца су¬
шат при температуре (105±2) °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,3%.
Определение проводят из 0,50 г испытуемого об¬
разца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 10 МЕ/г.
#Пирогенность (2.6.8). Субстанция должна
быть апирогенной. Тест-доза — 1,0 г (в пересчете
на сухое вещество) в 10 мл раствора 9 г/л натрия
хлорида Р на 1 кг массы тела кролика. Раствор вво¬
дят внутривенно в течение 2 мин.
Тест «Пирогенность» проводится в качестве
альтернативного тесту «Бактериальные эндоток¬
сины».
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
07/2016:1000
ДЕКСТРАН 60 ДЛЯ ИНЪЕКЦИЙ
Оех1гапит 60 ас! 1тес1аЫ1е
ОЕХТКАЫ 60 РОК 1ШЕСТЮЫ
ОПРЕДЕЛЕНИЕ
Декстран 60 для инъекций представляет со¬
бой смесь полисахаридов преимущественно а-1,6-
глюканового типа.
Средняя относительная молекулярная масса:
около 60 000.
ПРОИЗВОДСТВО
Получают путем гидролиза и фракциони¬
рования декстранов, полученных ферментаци¬
ей сахарозы штаммом 1~еисопоз1ос тезе^его1дез
ЫКР1_ В-512 = С1Р 78,59 или его подштаммами (на¬
пример тезеМегоМез В-512Р = 1ЧСТС 10817).
Производят в условиях минимального риска ми¬
кробного загрязнения.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень легко растворим в воде, очень мало рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Удельное оптическое вращение (2.2.7): от
+195 до +201 (в пересчете на сухое вещество). 1,0 г
испытуемого образца растворяют в воде Р при нагре¬
вании на водяной бане и доводят до объема 50,0 мл
этим же растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО декстрана #или спектр, пред¬
ставленный на рисунке #1000.-1#.
C. Испытуемый образец выдерживает испыта¬
ние «Молекулярно-массовое распределение декстра¬
нов», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р при нагревании
на водяной бане и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод //). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфта¬
леина Р. Раствор должен оставаться бесцветным.
При прибавлении 0,2 мл 0,01 М раствора натрия
гидроксида должно появиться красное окраши¬
вание. При прибавлении к полученному раствору
0,4 мл 0,01 М раствора кислоты хлористоводо¬
родной раствор должен обесцветиться. При при¬
бавлении 0,1 мл раствора метилового красно¬
го Р должно появиться красное или оранжевое
окрашивание.
Азотсодержащие вещества. Не более 0,0110 %
N (110 ррт N1). Проводят определение азота после
минерализации серной кислотой (2.5.9), используя
0,200 г испытуемого образца и нагревание в течение
2 ч. Дистиллят собирают в смесь из 0,5 мл раствора
бромкрезолового зеленого Р, 0,5 мл раствора ме-
Декстран 60 для инъекций
381
Рисунок #1000.-1. Инфракрасный спектр ФСО декстрана
типового красного Р и 20 мл воды Р. На титрование
должно пойти не более 0,15 мл 0,01 М раствора кис-
лоты хлористоводородной.
Остаточные количества органических раство¬
рителей. Газовая хроматография (2.2.28).
Внутренний стандарт: пропанол Р.
Испытуемый раствор. 5 г испытуемого образца
растворяют в 100 мл воды Р и перегоняют. Собирают
первые 45 мл дистиллята. К полученному дистилляту
прибавляют 1 мл раствора 25 г/л пропанола Р и дово¬
дят водой Р до объема 50 мл.
Раствор сравнения. К 0,5 мл раствора 25 г/л
этанола безводного Р прибавляют 0,5 мл раствора
25 г/л пропанола Р и 0,5 мл раствора 2,5 г/л метано¬
ла Р и доводят водой Р до объема 25,0 мл.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 1,8 м
и внутренним диаметром 2 мм, заполненная со¬
полимером этилвинилбензол-дивинилбензола Р
((125—150) мкм);
- газ-носитель: азот для хроматографии Р\
- скорость газа носителя: 25 мл/мин;
- температура: колонка — 190 °С; блок ввода
проб — 240 °С; детектор — 210 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: вводят равные объемы
испытуемого раствора и раствора сравнения.
Предельное содержание примесей:
- этанол (не более 0,5 %): на хроматограмме ис¬
пытуемого раствора площадь пика этанола не должна
превышать площадь соответствующего пика на хро¬
матограмме раствора сравнения;
- метанол (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика метанола не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения;
- сумма растворителей, кроме этанола, мета¬
нола и пропанола (не более 0,5 % в пересчете на про¬
панол): на хроматограмме испытуемого раствора сум¬
ма площадей пиков всех растворителей, кроме ме¬
танола, этанола и пропанола, не должна превышать
площадь пика пропанола на хроматограмме раствора
сравнения.
Молекулярно-массовое распределение дек-
странов (2.2.39). Средняя молекулярная масса (Мш)
должна быть от 54 000 до 66 000. Средняя молеку¬
лярная масса 10% высокомолекулярной фракции
не должна превышать 180 000. Средняя молекуляр¬
ная масса 10 % низкомолекулярной фракции должна
быть не менее 14 000.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 0,200 г испытуемого образца сушат при
температуре (105±2) °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,3 %. Опре¬
деление проводят из 0,50 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
16 МЕ/г.
#Пирогенность (2.6.8). Субстанция должна быть
апирогенной. Тест-доза — 1,0 г (в пересчете на сухое
вещество) в 10 мл раствора 9 г/л натрия хлорида Р
на 1 кг массы тела кролика. Раствор вводят внутри¬
венно в течение 2 мин.
Тест «Пирогенность» проводится в качестве аль¬
тернативного тесту «Бактериальные эндотоксины».
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
382
Гэсударственная фармакопея Республики Беларусь
07/2016:1001
ДЕКСТРАН 70 ДЛЯ ИНЪЕКЦИЙ
Оех&апит 70 аб т1ес1аЫ1е
ОЕХТКАЫ 70 РОК 1ШЕСТЮЫ
ОПРЕДЕЛЕНИЕ
Декстран 70 для инъекций представляет со¬
бой смесь полисахаридов преимущественно а-1,6-
глюканового типа.
Средняя относительная молекулярная масса:
около 70 000.
ПРОИЗВОДСТВО
Получают путем гидролиза и фракциони¬
рования декстранов, полученных ферментаци¬
ей сахарозы штаммом /.еисолоз/ос тезеп1егоШез
ЫРРЬ В-512 = С1Р 78,59 или его подштаммами (на¬
пример I. тезеп1его1дез В-512Р = ИСТО 10817).
Производят в условиях минимального риска ми¬
кробного загрязнения.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень легко растворим в воде, очень мало рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Удельное оптическое вращение (2.2.7): от
+195 до +201 (в пересчете на сухое вещество). 1,0 г
испытуемого образца растворяют в воде Р при нагре¬
вании на водяной бане и доводят до объема 50,0 мл
этим же растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО декстрана #или спектр, пред¬
ставленный на рисунке #1001.-1#.
С. Испытуемый образец выдерживает испыта¬
ние «Молекулярно-массовое распределение декстра¬
нов», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р при нагревании
на водяной бане и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфтале¬
ина Р. Раствор должен оставаться бесцветным. При
прибавлении 0,2 мл 0,01 М раствора натрия гидрок¬
сида должно появиться красное окрашивание. При
прибавлении к полученному раствору 0,4 мл 0,01 М
раствора кислоты хлористоводородной раствор
должен обесцветиться. При прибавлении 0,1 мл
раствора метилового красного Р должно появиться
красное или оранжевое окрашивание.
Азотсодержащие вещества. Не более 0,0110 %
N (110 ррт Ы). Проводят определение азота после
минерализации серной кислотой (2.5.9), используя
0,200 г испытуемого образца и нагревание в течение
2 ч. Дистиллят собирают в смесь из 0,5 мл раствора
бромкрезолового зеленого Р, 0,5 мл раствора ме¬
тилового красного Р и 20 мл воды Р. На титрование
должно пойти не более 0,15 мл 0,01 М раствора кис¬
лоты хлористоводородной.
Остаточные количества органических раство¬
рителей. Газовая хроматография (2.2.28).
Внутренний стандарт: пропанол Р.
Испытуемый раствор. 5 г испытуемого образца
растворяют в 100 мл воды Р и перегоняют. Собирают
Рисунок #1001 .-1. Инфракрасный спектр ФСО декстрана
Декстрин
383
первые 45 мл дистиллята. К полученному дистилляту
прибавляют 1 мл раствора 25 г/л пропанола Р и дово¬
дят водой Р до объема 50 мл.
Раствор сравнения. К 0,5 мл раствора 25 г/л
этанола безводного Р прибавляют 0,5 мл раствора
25 г/л пропанола Р и 0,5 мл раствора 2,5 г/л метано¬
ла Р и доводят водой Р до объема 25,0 мл.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 1,8 м и
диаметром 2 мм, заполненная сополимером этилви-
нилбензол-дивинилбензола Р ((125—150) мкм);
- газ-носитель: азот для хроматографии Р;
- скорость газа носителя: 25 мл/мин;
- температура: колонка — 190 °С; блок ввода
проб — 240 °С; детектор — 210 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: вводят равные объемы
испытуемого раствора и раствора сравнения.
Предельное содержание примесей:
- этанол (не более 0,5 %): на хроматограмме ис¬
пытуемого раствора площадь пика этанола не должна
превышать площадь соответствующего пика на хро¬
матограмме раствора сравнения;
- метанол (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика метанола не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения;
- сумма растворителей, кроме этанола, мета¬
нола и пропанола (не более 0,5 % в пересчете на про¬
панол): на хроматограмме испытуемого раствора сум¬
ма площадей пиков всех растворителей, кроме ме¬
танола, этанола и пропанола, не должна превышать
площадь пика пропанола на хроматограмме раствора
сравнения.
Молекулярно-массовое распределение дек-
странов (2.2.39). Средняя молекулярная масса (Мш)
должна быть от 64 000 до 76 000. Средняя молеку¬
лярная масса 10% высокомолекулярной фракции
не должна превышать 185 000. Средняя молекуляр¬
ная масса 10 % низкомолекулярной фракции должна
быть не менее 15 000.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 0,200 г испытуемого образца сушат при
температуре (105±2) °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,3 %. Опре¬
деление проводят из 0,50 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
16 МЕ/г.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
07/2016:1507
ДЕКСТРИН
ОехМпит
ОЕХТКШ
ОПРЕДЕЛЕНИЕ
Декстрин представляет собой кукурузный, кар¬
тофельный или маниоковый крахмал, частично ги¬
дролизованный и модифицированный нагреванием
с кислотами, щелочами, рН-контролирующими веще¬
ствами или без них.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый, легко сыпучий порошок.
Очень легко растворим в кипящей воде с образо¬
ванием вязкого раствора, медленно растворим в хо¬
лодной воде, практически нерастворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца суспендируют в 50 мл
воды Р, кипятят в течение 1 мин и охлаждают. К 1 мл
полученного раствора прибавляют 0,05 мл раствора
йода Р1. Появляется темно-синяя или красновато-ко¬
ричневая окраска, исчезающая при нагревании.
B. 5 мл вязкого раствора, полученного в подлин¬
ности А, центрифугируют. К верхнему слою прибав¬
ляют 2 мл раствора натрия гидроксида разведенно¬
го Р и, по каплям, при встряхивании, 0,5 мл раствора
меди сульфата Р и кипятят. Образуется красный оса¬
док.
C. Испытуемый образец очень легко растворим
в кипящей воде Р с образованием вязкого раствора.
ИСПЫТАНИЯ
рН (2.2.3). От 2,0 до 8,0. 5,0 г испытуемого образ¬
ца растворяют в 100 мл воды, свободной от углерода
диоксида, Р.
Хлориды. Не более 0,2 %. 2,5 г испытуемого об¬
разца растворяют в 50 мл кипящей воды Р, доводят
водой Р до объема 100 мл и фильтруют. 1 мл фильтра¬
та доводят до объема 15 мл, прибавляют 1 мл кисло¬
ты азотной разведенной Р, прибавляют полученную
смесь, в один прием, к 1 мл раствора серебра нитра¬
та Р2 и выдерживают в течение 5 мин в защищенном
от света месте. При поперечном просмотре на черном
фоне опалесценция полученного раствора не должна
превышать по интенсивности опалесценцию раствора
сравнения, приготовленного параллельно с использо¬
ванием смеси из 10 мл эталонного раствора хлори¬
да (5 ррт С1) Р и 5 мл воды Р.
Восстанавливающие сахара. Не более 10%
в пересчете на глюкозу СбН12Об. К 2,0 г испытуемого
образца (в пересчете на сухое вещество) прибавля¬
ют 100 мл воды Р, встряхивают в течение 30 мин,
доводят водой Р до объема 200,0 мл и фильтруют. К
10,0 мл раствора медно-тартратного щелочного Р
прибавляют 20,0 мл фильтрата, перемешивают и на¬
гревают на плитке, отрегулированной таким образом,
чтобы раствор был доведен до кипения в течение
3 мин. Кипятят 2 мин и немедленно охлаждают. При¬
бавляют 5 мл раствора 300 г/л калия йодида Р и 10 мл
1 М раствора кислоты серной, перемешивают и сра¬
зу же титруют 0,1 М раствором натрия тиосульфа¬
та, используя в качестве индикатора раствор крах¬
мала Р, добавляемый в конце титрования. Процедуру
повторяют, начиная со слов «К 10,0 мл...», используя
вместо фильтрата 20,0 мл точно приготовленного
раствора 1,0 г/л глюкозы Р. Параллельно проводят
контрольный опыт. Значение (Ук - Уд) не должно пре¬
вышать значение (V - V), где Уд, У и Ук — объемы
(мл) 0,1 М раствора натрия тиосульфата, израсхо¬
дованные на титрование декстрина, глюкозы и в кон¬
трольном опыте соответственно.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца вы¬
держивает испытание на тяжелые металлы. Эта-
384
Гэсударственная фармакопея Республики Беларусь
лон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32).
Не более 13,0 %. 1,000 г испытуемого образца су¬
шат при температуре от 130 °С до 135 °С в течение
90 мин.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для декстрина, используемого в качестве
наполнителя и связующего вещества при изго¬
товлении таблеток и капсул.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошков (2.9.36).
Следующая характеристика может быть
важна для декстрина, используемого в качестве
вещества, повышающего вязкость.
Кажущаяся вязкость (2.2.10). Обычно от
100 мПа-с до 350 мПа-с (в пересчете на сухое ве¬
щество), в зависимости от сорта декстрина.
В химическом стакане готовят (10—50) % ка¬
шицу таким образом, чтобы значение вязкости
было в диапазоне от 100 мПа-с до 350 мПа-с. Об¬
щая масса образца вместе с водой должна состав¬
лять 600 г. Полученную кашицу перемешивают с
помощью пластмассовой палочки до достижения
гомогенности, после чего химический стакан поме¬
щают в водяную баню с температурой (100±1) °С,
вводят в него лопасть мешалки и закрывают стакан
крышкой. Как можно скорее начинают перемеши¬
вание со скоростью 250 об/мин и перемешивают
точно в течение 30 мин. Полученную пасту немед¬
ленно переносят во второй химический стакан,
в котором будет проводиться измерение и кото¬
рый помещен в водяную баню при температуре
(40±1) °С. Пасту перемешивают до тех пор, пока ее
температура не будет составлять (40±1) °С, после
чего проводят определение кажущейся вязкости со
скоростью вращения 100 об\мин и с использовани¬
ем шпинделя № 2.
07/2016:0020
ДЕКСТРОМЕТОРФАНА
ГИДРОБРОМИД
ОехХготеХЬогрЬап! ЬудгоЪгот'\6ит
ОЕХТПОМЕТНОЕРНАЫ
НУОКОВКОМЮЕ
/СНз
С18Н25МО ■ НВг ■ Н20 М.м. 370,3
[6700-34-1]
ОПРЕДЕЛЕНИЕ
энш-З-Метокси-17-метилморфинана гидробромид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Почти белый кристаллический порошок.
Умеренно растворим в воде, легко растворим в
96 % спирте.
Температура плавления: около 125°С с разложе¬
нием.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО декстрометорфана гидробро¬
мида #или спектр, представленный на рисунке #0020.-1 *
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 25 мг ФСО декстрометор¬
фана гидробромида растворяют в метаноле Р и до¬
водят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: раствор аммиака концентриро¬
ванный Р — метиленхлорид Р — метанол Р — эти-
лацетат Р — толуол Р(2:10:13:20:55, об/об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
калия йодовисмутата Р2.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
Декстрометорфана гидробромид
385
Рисунок #0020.-1. Инфракрасный спектр ФСО декстрометорфана гидробромида
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
Э. Испытуемый образец дает реакцию (а) на бро¬
миды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в 96% спирте Р и доводят до объема до 20 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. 0,4 г испытуемо¬
го образца растворяют в воде, свободной от углерода
диоксида, Р при слабом нагревании, охлаждают и до¬
водят до объема 20 мл этим же растворителем. При¬
бавляют 0,1 мл раствора метилового красного Р и
0,2 мл 0,01 М раствора натрия гидроксида. Должно
появиться желтое окрашивание. При прибавлении не
более 0,4 мл 0,01 М раствора кислоты хлористово¬
дородной должно появиться красное окрашивание.
Удельное оптическое вращения (2.2.7). От +28
до +30 (в пересчете на безводное вещество). 0,200 г
испытуемого образца растворяют в 0,1 М растворе
кислоты хлористоводородной и доводят до объема
10,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО декстрометор¬
фана примеси А растворяют в 2 мл испытуемого раст¬
вора и доводят подвижной фазой до объема 25,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц 5
мкм;
-подвижная фаза: 3,11 г докузата натрия Р
растворяют в смеси из 400 мл воды Р и 600 мл ацето¬
нитрила Р, прибавляют 0,56 г аммония нитрата Р и
доводят до рН 2,0 кислотой уксусной ледяной Р\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания декстрометорфана.
Относительное удерживание (по отношению
к декстрометорфану, время удерживания — около
22 мин): примесь В — около 0,4; примесь С — около
0,8; примесь О — около 0,9; примесь А — около 1,1.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиком приме¬
си А и пиком декстрометорфана.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 0,2):
- примеси А, В, С, О: на хроматограмме испы¬
туемого раствора площади пиков примесей А, В, С и
О, не должны превышать площадь основного пика на
хроматограмме раствора сравнения (Ь) (0,5 %); пло¬
щадь не более одного из таких пиков может превы¬
шать 0,5 площади основного пика на хроматограмме
раствора сравнения (Ь) (0,25 %);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
386
Гэсударственная фармакопея Республики Беларусь
примесей А, В, С и й, не должна превышать 0,2
площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,1 площади основного
пика на хроматограмме раствора сравнения (Ь).
Л/,М-Диметиланилин. Не более 0,0010%
(Юррт). 0,5 г испытуемого образца растворяют
при нагревании в 20 мл воды Р, охлаждают, при¬
бавляют 2 мл кислоты уксусной разведенной Р и
1 мл раствора 10 г/л натрия нитрита Р и дово¬
дят водой Р до объема 25 мл. Окраска полученно¬
го раствора должна быть не интенсивнее окраски
раствора сравнения, приготовленного аналогично
и в то же самое время с использованием 20 мл
раствора 0,25 мг/л диметиланилина Р.
Вода (2.5.12). Не менее 4,0% и не более
5,5 %. Определение проводят из 0,200 г испытуе¬
мого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в
смеси из 5,0 мл 0,01 М раствора кислоты хлори¬
стоводородной и 20 мл 96 % спирта Р и титруют
0,1 М раствором натрия гидроксида потенциоме-
трически (2.2.20). Отмечают объем титранта между
двумя точками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 35,23 мг С18Н251ЧО-НВг.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. энш-З-Метоксиморфинан.
В. энш-17-Метилморфинан-З-ол.
С. эн/77-3-Метокси-17-метилморфинан-10-он.
О. энл?-(145)-3-Метокси-17-метилморфинан.
07/2016:0022
ДИАЗЕПАМ
01агератит
Р1А2ЕРАМ
С1вН13СШ20 М.м. 284,7
[439-14-5]
ОПРЕДЕЛЕНИЕ
7-Хлор-1 -метил-5-фенил-1,3-дигидро-2Н-1,4-
бензодиазепин-2-он.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок.
Очень мало растворим в воде, растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО диазепама #или спектр, пред¬
ставленный на рисунке #0022.-1#.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы готовят с защитой от
яркого света.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в 0,5 мл ацетонитрила Р и дово¬
дят подвижной фазой до объема 50,0 мл.
Диазепам
387
Рисунок #0022.-1. Инфракрасный спектр ФСО диазепама
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО диазепама для проверки пригодности хромато¬
графической системы (содержит примеси А, В и Е)
растворяют в 1,0 мл подвижной фазы.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- температура: 30 °С;
-подвижная фаза: смешивают 22 объема аце¬
тонитрила Р, 34 объема метанола Р и 44 объема
раствора 3,4 г/л калия дигидрофосфата Р, доведен¬
ного до рН 5,0 раствором натрия гидроксида разве¬
денным Р;
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания диазепама.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и Е, используя хроматограм¬
му раствора сравнения (Ь) и хроматограмму, прила¬
гаемую к ФСО диазепама для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
диазепаму, время удерживания — около 9 мин): при¬
месь Е — около 0,7; примесь А — около 0,8; примесь
В — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 2,5 между пиками при¬
меси Е и примеси А; не менее 6,0 между пиками при¬
меси А и диазепама.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 1,3; для примеси Е — 1,3):
-примеси А, В, Е (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В, и Е, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, и Е, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 4 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
388
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 50 мл
уксусного ангидрида Р и титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 28,47 мг С16Н13С1М20.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О, Р.
А. 7-Хлор-5-фенил-1,3-дигидро-2Н-1,4-бензодиа-
зепин-2-он (нордиазепам).
В. Р = СО-СН2-С1: 2-Хлор-Л/-(4-хлор-2-бензоил-
фенил)-А/-метилацетамид.
О. П = Н: [5-Хлор-2-(метиламино)фенил]фенилме-
танон.
С. 3-Амино-6-хлор-1 -метил-4-фенилхинол ин-2-
(1Н)-он.
Е. 6-Хлор-1 -метил-4-фенилхиназолин-2-(1 Н)-он.
Р. 7-Хлор-2-метокси-5-фенил-ЗН-1,4-бензодиа-
зепин.
07/2016:0079
дигоксин
0\дох\пит
о/еох/л/
С41нм014 М.м. 781
[20830-75-5]
ОПРЕДЕЛЕНИЕ
ЗР-[(2,6-Дидезокси-р-0-рмбо-гексопиранозил-(1—>4)-
2,6-дидезокси-р-0-рибо-гексопиранозил-(1->4)-2,6-
дидезокси-р-О-рибо-гексопиранозил )окси]-12р, 14-
дигидрокси-5р-кард-20(22)-енолид.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Практически нерастворим в воде, легко раство¬
рим в смеси из равных объемов метанола и метилен-
хлорида, мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО дигоксина.
ИСПЫТАНИЯ
Раствор 5. 50 мг испытуемого образца раство¬
ряют в смеси из равных объемов метанола Р и ме-
тиленхлорида Р и доводят до объема 10 мл этой же
смесью растворителей.
Дигоксин
389
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод /). Раствор 3 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
+13,9 до +15,9 (в пересчете на сухое вещество). 0,50 г
испытуемого образца растворяют в смеси из равных
объемов метанола Р и метиленхлорида Р и доводят
до объема 25,0 мл этой же смесью растворителей.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 100,0 мл метанола Р.
Раствор сравнения (а). 10,0 мг ФСО дигокси-
на растворяют в метаноле Р и доводят до объема
20.0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят метанолом Р до объема 100,0 мл.
Раствор сравнения (с). 2,5 мг ФСО дигоксигени-
на (примесь С) растворяют в метаноле Р и доводят
до объема 5,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят метанолом Р до объема
50.0 мл. 1,0 мл полученного раствора доводят мета¬
нолом Р до объема 10,0 мл.
Раствор сравнения (д). 50,0 мг ланатозида С Р
(примесь Н) растворяют в метаноле Р и доводят до
объема 100,0 мл этим же растворителем. К 1,0 мл по¬
лученного раствора прибавляют 1,0 мл испытуемого
раствора и доводят метанолом Р до объема 20,0 мл.
Раствор сравнения (е). 5,0 мг ФСО дигоксина
для идентификация пиков растворяют в метаноле Р
и доводят до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
-колонка длиной 0,15 м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц 5
мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р— вода Р
(10:90, об/об);
- подвижная фаза В: вода Р— ацетонитрил Р
(10:90, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
78
22
5—15
о
со
т
со
г-
22 —> 70
- скорость подвижной фазы: 1,5 мл/миин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь), (с), (б) и (е).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, Е, Р, С и К, используя хро¬
матограмму раствора сравнения (е) и хроматограмму,
прилагаемую к ФСО дигоксина для идентификация
пиков.
Относительное удерживание (по отношению к
дигоксину, время удерживания — около 4,3 мин): при¬
месь С — около 0,3; примесь Е — около 0,5; примесь
Р — около 0,6; примесь 6 — около 0,8; примесь 1_ —
около 1,4; примесь К — около 1,6; примесь В — око¬
ло 2,2; примесь А — около 2,6.
Пригодность хроматографической системы:
раствор сравнения (д):
-разрешение: не менее 1,5 между пиками при¬
меси Н и дигоксина.
Предельное содержание примесей:
- примесь Р (не более 2,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси Р, не должна превышать 2,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь С (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 5-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (с);
- примеси Е, К (не более 1,0 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Е и К, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь 6 (не более 0,8 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси О, не должна превышать 0,8 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- примеси А, В (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площади пиков, соответ¬
ствующих примесям А и В, не должны превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (Ь);
- примесь /_ (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси 1_, не должна превышать 0,3 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- любая другая примесь (не более 0,2 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С, Е,
Р, О, К и Ц не должна превышать 0,2 площади основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей, кроме примесей А, В, С, Е, Р,
О, К и I- (не более 0,7 %): на хроматограмме испы¬
туемого раствора сумма площадей всех пиков, кроме
основного и пиков примесей А, В, С, Е, Р, О, К и 1_,
не должна превышать 0,7 площади основного пика на
хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 3,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
3,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (Ь).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 0,500 г испытуемого образца сушат в ва¬
кууме.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из остатка, полученного в ис¬
пытании «Потеря в массе при высушивании».
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
390
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
— объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С41Н64014 рассчитывают с учетом со¬
держания дигоксина в ФСО дигоксина.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, Е, Р, О, К,
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О, Н, I, 3.
2,6-дидезокси-
Э-О-глюкопиранозил
р-О-дигитоксозил
С1и = “(ОН
НО
ОН
р-0-глюкопиранозил
2.6- дидезокси-р-0-р*убо-гексопиранозил-(1-*4)-2,6-
дидезокси-р-О-рибо-гексопиранозил )окси]-12р, 14-
дигидрокси-5р-кард-20(22)-енолид (а-ацетилдигоксин).
3. Р1 = ОН, Р2 = КЗ = Н, Р4 = СО-СН3: Зр-[(4-0-
Ацетил-2,6-дидезокси-р-0-рибо-гексопиранозил-(1—>4)-
2.6- дидезокси-р-0-рибо-гексопиранозил-(1-»4)-2,6-
дидезокси-р-0-р(убо-гексопиранозил)окси]-12р,14-
дигидрокси-5р-кард-20(22)-енолид (Р-ацетилдигоксин).
К. Р1 = ОН, Р2 = РЗ = Н, Р4 = 0\д: зр-[(2,6-
Дидезокси-р-0-р1/бо-гексопиранозил-(1-»4)-2,6-
дидезокси-р-0-рабо-гексопиранозил-(1-»4)-2,6-
дидезокси-р-0-рибо-гексопиранозил-(1-»4)-2,6-
дидезокси-Р-0-р(/бо-гексопиранозил)окси]-12р,14-
дигидрокси-5р-кард-20(22)-енолид (дигоксигенинтетра-
кисдигитоксозид).
С. Р = Н: Зр,12р,14-Тригидрокси-5Р-кард-20(22)-
енолид (дигоксигенин).
O. Р = 0\д: ЗР-(2,6-Дидезокси-р-0-р(/бо-
гексопиранозилокси)-12(3, 14-дигидрокси-5Р-кард-20(22)-
енолид (дигоксигенинмонодигитоксозид).
P. Р = 0|д-(1 —►4)-Э-01д: зр-[(2,6-Дидезокси-р-0-
р*убо-гексопиранозил-(1—5>4)-2,6-цпцезоксм-$-0-рибо-
гексопиранозил)окси]-12р, 14-дигидрокси-5р-кард-20(22)-
енолид (дигоксигенинбисдигитоксозид).
О. Р = Обб-(1->4)-01д-(1-*4)-0|д: Зр-[(2,6-Д-
идезокси-р-0-араб(/но-гексопиранозил-(1—>4)-2,6-
дидезокси-р-0-рибо-гексопиранозил-(1-»4)-2,6-
дидезокси-р-0-рабо-гексопиранозил)окси]-12р,14-
дигидрокси-5р-кард-20(22)-енолид (неодигоксин).
Ь. Структура не установлена.
07/2016:1003
A. Р1 = Р2 = РЗ = Р4 = Н: Зр-[(2,6-Дидезокси-р-0-
рибо-гексопиранозил-(1->4)-2,6-дидезокси-Р-0-рибо-
гексопиранозил-(1—»4)-2,6-дидезокси-р-0-рбУбо-гексо-
пиранозил)окси]-14-гидрокси-5р-кард-20(22)-енолид
(дигитоксин).
B. Р1 = РЗ = Р4 = Н, Р2 = ОН: Зр-[(2,6-Дидезокси-
Р-0-р1/бо-гексопиранозил-(1—>4)-2,6-дидезокси-р-0-
р1/бо-гексопиранозил-(1->4)-2,6-дидезокси-Р-0-р(убо-
гексопиранозил)окси]-14,1бр-дигидрокси-5р-кард-20(22)-
енолид (гитоксин).
Е. Р1 = Р2 = ОН, РЗ = Р4 = Н: Зр-[(2,6-Дидезокси-
р-0-р(/бо-гексопиранозил-(1—>4)-2,6-дидезокси-р-0-
рыбо-гексопиранозил-(1->4)-2,6-дидезокси-р-0-р1/бо-
гексопиранозил )окси]-12Р,14,1 бр-тригидрокси-5р-кард-
20(22)-енолид (дигинатин).
H. Р1 = ОН, Р2 = Н, РЗ = СО-СН3, Р4 = 01и: Зр-[р-
0-Глюкопиранозил-(1—^-З-О-ацетил-г.б-дидезокси-р-
О-рибо-гексопиранозил-О—>4)-2,6-дидезокси-р-0-рибо-
гексопиранозил-(1 -^4)-2,6-дидезокси-р-0-р1/бо-
гексопиранозил)окси]-12р,14-дигидрокси-5р-кард-20(22)-
енолид (ланатозид С).
I. Р1 = ОН, Р2 = Р4 = Н, РЗ = СО-СН3: Зр-[(3-0-
Ацетил-2,6-дидезокси-р-0-р1Убо-гексопиранозил-(1—>4)-
ДИКАЛИЯ ФОСФАТ
0//са/// рНозрЬаз
01РОТА88ШМ РНОЗРНАТЕ
^НРО,, М.м. 174,2
[7758-11-4]
ОПРЕДЕЛЕНИЕ
#Калия гидрофосфат*.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы. Очень гигроскопичен.
Очень легко растворим в воде, очень мало рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в разде¬
ле «Испытания», имеет слабощелочную реакцию (2.2.4).
B. Раствор 3 дает реакцию (Ь) на фосфаты (2.3.1).
C. Раствор 3 дает реакцию (а) на калий (2.3.1).
Диклофенак натрия
391
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Восстанавливающие вещества. Смесь 5 мл
раствора 3, 5 мл кислоты серной разведенной Р и
0,25 мл 0,02 М раствора калия перманганата нагре¬
вают на водяной бане в течение 5 мин. Раствор дол¬
жен сохранить бледно-розовое окрашивание.
Калия дигидрофосфат. Не более 2,5 %. Исходя
из объемов 1 М раствора кислоты хлористоводород¬
ной (10,0 мл) и 1 М раствора натрия гидроксида (пг
мл, и п2, мл), израсходованных при количественном
определении, рассчитывают соотношение по формуле:
л2 -10
10-^ ‘
Полученное соотношение не должно превышать
0,025.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт). К
2,5 мл раствора 3 прибавляют 10 мл кислоты азот¬
ной разведенной Р и доводят водой Р до объема
15 мл. Полученный раствор должен выдерживать ис¬
пытание на хлориды.
Сульфаты (2.4.13). Не более 0,1%. К 1,5 мл
раствора 3 прибавляют 2 мл кислоты хлористоводо¬
родной разведенной Р и доводят водой дистиллиро¬
ванной Р до объема 15 мл. Полученный раствор дол¬
жен выдерживать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 5 мл раствора 3 должны выдерживать испы¬
тание на мышьяк.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 3 должны выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в 8 мл воды Р, подкисляют с использованием
около б мл кислоты хлористоводородной разведен¬
ной Р до значения рН от 3 до 4 и доводят водой Р до
объема 20 мл. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Натрий. Не более 0,1 %, если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения. Атомно-эмиссион¬
ная спектрометрия (2.2.22, метод I).
Испытуемый раствор: 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят этим же раство¬
рителем до 100,0 мл.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора натрия
(200 ррт А1а) Р водой Р.
Длина волны: 589 нм.
Потеря в массе при высушивании (2.2.32). Не
более 2,0 %. 1,000 г испытуемого образца сушат при
температуре от 125 °С до 130 °С.
Бактериальные эндотоксины (2.6.14). Менее
1,1 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дополнительной процедуры удале¬
ния бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,800 г испытуемого образца (т) растворяют в
40 мл воды, свободной от углерода диоксида, Р, при¬
бавляют 10,0 мл 1 М раствора кислоты хлористово¬
дородной и титруют 1 М раствором натрия гидрокси¬
да потенциометрически (2.2.20) до первого скачка по¬
тенциала на кривой титрования (пг мл). Продолжают
титрование до второго скачка потенциала на кривой
титрования (п2, мл — общий объем 1 М раствора на¬
трия гидроксида, израсходованного при титровании).
Содержание ^НРС^ в процентах рассчитывают
по формуле:
1742 (10-п,)
ш (ЮО-сУ) ’
где:
д— потеря в массе при высушивании, %.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:1002
ДИКЛОФЕНАК НАТРИЯ
Ою1о?епасит паМсит
ОЮЮРЕЫАС ЗСЮШМ
С14Н10С12Ы№Ог М.м. 318,1
[15307-79-6]
ОПРЕДЕЛЕНИЕ
Натрия 2-[(2,6-дихлорфенил)амино]фенил]ацетат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или белый с желтоватым оттенком кри¬
сталлический порошок. Слегка гигроскопичен.
Умеренно растворим в воде, легко растворим в
метаноле, растворим в 96 % спирте, мало растворим
в ацетоне.
Температура плавления: около 280 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
392
Гэсударственная фармакопея Республики Беларусь
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО диклофенака натрия #или
спектр, представленный на рисунке #1002.-1*
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения (а). 25 мг ФСО диклофенака
натрия растворяют в метаноле Р и доводят до объ¬
ема 5 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг индометацина Р
растворяют в растворе сравнения (а) и доводят до
объема 2 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
лявР254 Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р — метанол Р — этилацетат Р
(10:10:80, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 1/2 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
C. 10 мг испытуемого образца растворяют в 10 мл
96 % спирта Р. К 1 мл полученного раствора прибав¬
ляют 0,2 мл приготовленной непосредственно перед
использованием смеси из равных объемов раствора
6 г/л калия феррицианида Р и раствора 9 г/л железа
(III) хлорида Р и выдерживают в течение 5 мин в за¬
щищенном от света месте. Прибавляют 3 мл раствора
10 г/л кислоты хлористоводородной Р и выдерживают
в течение 15 мин в защищенном от света месте. По¬
является синее окрашивание и образуется осадок.
Р. 60 мг испытуемого образца растворяют в
0,5 мл метанола Р, прибавляют 0,5 мл воды Р. Полу¬
ченный раствор дает реакцию (Ь) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,25 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 25,0 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,05
при 440 нм. Измеряют оптическую плотность раст¬
вора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО диклофенака для проверки пригодности хрома¬
тографической системы (содержит примеси А и Р)
растворяют в 1,0 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
Рисунок #1002.-1. Инфракрасный спектр ФСО диклофенака натрия
Диклофенак натрия
393
- подвижная фаза: 34 объема раствора, содер¬
жащего 0,5 г/л кислоты фосфорной Р и 0,8 г/л на¬
трия дигидрофосфата Р, предварительно доведен¬
ного до рН 2,5 кислотой фосфорной Р, смешивают с
66 объемами метанола Р;
- скорость подвижной фазы: 1,0 мл/мин:
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,6-кратное вре¬
мя удерживания диклофенака.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и Р, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО диклофенака для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
диклофенаку, время удерживания — около 25 мин):
примесь А — около 0,4; примесь Р — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиком приме¬
си Р и пиком диклофенака.
Расчет процентных содержаний:
- для примеси А — умножают площадь пика на
поправочный коэффициент 0,7;
- для примеси Р — умножают площадь пика на
поправочный коэффициент 0,3;
- для каждой примеси — используют концентра¬
цию диклофенака в растворе сравнения (а).
Предельное содержание примесей:
- примесь А: не более 0,2 %;
- примесь Р: не более 0,15 %;
- неспецифицированные примеси: не более
0,10 % (для каждой примеси);
- сумма примесей: не более 0,4 %;
- учитываемый предел: 0,05 %.
Тяжелые металлы (2.4.8, метод И). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в 20 мл метанола Р. Полученный раствор дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р, полученный разведением
эталонного раствора свинца (100 ррт РЬ) Р мета¬
нолом Р
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 31,81 мг С14Н10С12ММаО2.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Е.
А. 1-(2,6-Дихлорфенил)-1,3-дигидро-2Н-индол-
2-он.
О. 2-[2-[(2-Бром-6-хлорфенил)амино]фенил]уксус-
ная кислота.
Е. 1,3-Дигидро-2Н-индол-2-он.
Р. Л/-(4-Хлорфенил)-2-(2,6-дихлорфенил)ацета-
мид.
394
Государственная фармакопея Республики Беларусь
07/2016:0763
ДИМЕТИЛСУЛЬФОКСИД
0/ше//?у//5 зиНохШит
01МЕТНП 8Ш-РОХЮЕ
Н3С^ ^СН3
С2Н605 М.м. 78,1
[67-68-5]
ОПРЕДЕЛЕНИЕ
Сульфинилбисметан.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная жидкость или бесцветные кристал¬
лы. Гигроскопичен.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Показатель преломления», как указано в разделе
«Испытания».
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО диметилсульфоксида #или
спектр, представленный на рисунке #0763.-1
й. 50 мг никеля хлорида Р растворяют в 5 мл ис¬
пытуемого образца. Появляется зеленовато-желтое
окрашивание. Полученный раствор нагревают в водя¬
ной бане при температуре 50 °С. Появляется синева¬
то-зеленое или зеленое окрашивание, которое при ох¬
лаждении раствора переходит в зеленовато-желтое.
ИСПЫТАНИЯ
Кислотность. 50,0 г испытуемого образца раст¬
воряют в 100 мл воды, свободной от углерода диок¬
сида, Р, прибавляют 0,1 мл раствора фенолфталеи¬
на Р1. При прибавлении не более 5,0 мл 0,01 М раст¬
вора натрия гидроксида должно появиться розовое
окрашивание.
Относительная плотность (2.2.5). От 1,100 до
1,104.
Показатель преломления (2.2.6). От 1,478 до
1,479.
Температура затвердевания (2.2.18). Не более
18,3 °С.
Оптическая плотность (2.2.25). Через испытуе¬
мый образец пропускают азот Р в течение 15 мин. Из¬
меряют оптическую плотность, используя в качестве
компенсационного раствора воду. Оптическая плот¬
ность при длине волны 275 нм не должна превышать
0,30; Оптическая плотность при длинах волн 285 нм
и 295 нм не должна превышать 0,20. В интервале от
270 до 350 нм спектр поглощения испытуемого образ¬
ца не должен иметь максимумов поглощения.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Раствор внутреннего стандарта. 0,125 г ди¬
бензила Р растворяют в ацетоне Р и доводят объем
раствора этим же растворителем до 50 мл.
Испытуемый раствор (а). 5,0 г испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10,0 мл этим же растворителем.
Испытуемый раствор (Ь). 5,0 г испытуемого об¬
разца растворяют в ацетоне Р, прибавляют 1,0 мл
раствора внутреннего стандарта и доводят ацето¬
ном Р до объема 10,0 мл.
Рисунок #0763.-1. Инфракрасный спектр ФСО диметилсульфоксида
Динатрия фосфат дигидрат
395
Раствор сравнения. 50,0 мг испытуемого образ¬
ца и 50 мг диметилсульфона Р растворяют в ацето¬
не Р, прибавляют 10,0 мл раствора внутреннего стан¬
дарта и доводят ацетоном Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 1,5 м и внутренним диаметром
4 мм, заполненная диатомитом для газовой хрома¬
тографии Р (размер частиц (125—180) мкм), импрег-
нированным 10 % (м/м) полиэтиленгликольадипина-
том Р;
- газ-носитель: азот для хроматографии Р\
- скорость газа-носителя: 30 мл/мин;
- температура колонки: 165 °С;
- температура блока ввода пробы и детекто¬
ра: 190 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания диметилсульфоксида.
Порядок выхода пиков: диметилсульфоксид, ди-
метилсульфон, дибензил.
Время удерживания: диметилсульфоксид — око¬
ло 5 мин.
Пригодность хроматографической системы:
- разрешение: не менее 3 между пиками диме¬
тилсульфоксида и диметилсульфона на хроматограм¬
ме раствора сравнения;
-на хроматограмме испытуемого раствора (а)
должен отсутствовать пик со временем удерживания
таким же, как у внутреннего стандарта.
Предельное содержание примесей:
- сумма примесей (не более 0,1 %): на хромато¬
грамме раствора сравнения рассчитывают отношение
К площади пика диметилсульфоксида к площади пика
дибензила; на хроматограмме испытуемого раствора
(Ь) рассчитывают отношение суммы площадей всех
пиков, кроме пиков диметилсульфоксида, дибензила
и ацетона, к площади пика дибензила: это отношение
не должно превышать К.
Вода (2.5.12). Не более 0,2 %. Определение про¬
водят из 10,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом стеклянном контейне¬
ре, в защищенном от света месте.
07/2016:0602
ДИНАТРИЯ ФОСФАТ ДИГИДРАТ
О/па/г/7 рЬозрНаз сИМус1псиз
01800ШМ РН08РНАТЕ 01НУОРАТЕ
Ма2НР04-2Н20 М.м. 178,0
[10028-24-7]
ОПРЕДЕЛЕНИЕ
#Натрия гидрофосфат дигидрат*.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцвет¬
ные кристаллы.
Растворим в воде, практически нерастворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», имеет слабощелочную реакцию
(2.2.4).
B. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
C. Раствор 3 дает реакцию (Ь) на фосфаты (2.3.1).
Р. Раствор 3 дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Восстанавливающие вещества. К 5 мл раст¬
вора 3 прибавляют 5 мл кислоты серной разведен¬
ной Р, 0,25 мл 0,02 М раствора калия перманганата
и нагревают на водяной бане в течение 5 мин. Окра¬
ска перманганата не должна полностью исчезать.
Натрия дигидрофосфат. Не более 2,5 %. Исхо¬
дя из объемов 1 М раствора кислоты хлористово¬
дородной (25 мл) и 1 М раствора натрия гидроксида
(пл, мл, и п2, мл), израсходованных при количествен¬
ном определении, рассчитывают соотношение по
формуле:
п2 -25
25 - п, ’
Полученное соотношение не должно превышать
0,025.
Хлориды (2.4.4). Не более 0,0400 % (400 ррт). К
2,5 мл раствора 3 прибавляют 10 мл кислоты азот¬
ной разведенной Р и доводят водой Р до объема
15 мл. Полученный раствор должен выдерживать ис¬
пытание на хлориды.
Сульфаты (2.4.13). Не более 0,1 %. К 3 мл раст¬
вора 3 прибавляют 2 мл кислоты хлористоводород¬
ной разведенной Р и доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0004 %
(4 ррт). 5 мл раствора 3 должны выдерживать испы¬
тание на мышьяк.
Железо (2.4.9). Не более 0,0040 % (40 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 19,5 % и не более 21,0 %. 1,000 г испытуемого
образца сушат при температуре 130 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
396
Государственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца (т) растворяют в
50 мл воды Р, прибавляют 25,0 мл 1 М раствора кис¬
лоты хлористоводородной и титруют 1 М раство¬
ром натрия гидроксида потенциометрически (2.2.20)
до первого скачка потенциала на кривой титрования
(л^ мл). Продолжают титрование до второго скачка
потенциала на кривой титрования (л2, мл — общий
объем 1 М раствора натрия гидроксида, израсходо¬
ванного на титрование).
Содержание №2НР04 в процентах рассчитывают
по формуле:
1420 (25-л,)
я?-(100-с/) ’
где:
д — потеря в массе при высушивании, %.
07/2016:0118
ДИНАТРИЯ ФОСФАТ
ДОДЕКАГИДРАТ
О/ла/г/7 р/юзрНаз йодесаЬудпсиз
0/500/Ш РНОЗРНАТЕ
ОООЕСАНУОРАТЕ
№2НР04 - 12Н20 М.м. 358,1
[10039-32-4]
ОПРЕДЕЛЕНИЕ
^Натрия гидрофосфат додекагидрат*.
Содержание: не менее 98,5 % и не более 102,5 %.
ОПИСАНИЕ(СВОЙСТВА)
Бесцветные прозрачные кристаллы. Очень легко
выветриваются на воздухе.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», имеет слабощелочную реакцию
(2.2.4).
B. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
C. Раствор 3 дает реакцию (Ь) на фосфаты (2.3.1).
О. Раствор 3 дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод И). Раствор 5 должен
быть бесцветным.
Восстанавливающие вещества. К 5 мл раст¬
вора 3 прибавляют 5 мл кислоты серной разведен¬
ной Р и 0,25 мл 0,02 М раствора калия перманга¬
ната и нагревают на водяной бане в течение 5 мин.
Цвет перманганата не должен полностью исчезать.
Мононатрия фосфат. Не более 2,5 %. Ис¬
пользуя объемы 1 М кислоты хлористоводородной
(25 мл) и гидроксида натрия (пЛ, мл, и л2, мл) из коли¬
чественного определения, рассчитывают следующее
соотношение:
л2- 25
25-л,'
Соотношение должно быть не более 0,025.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт). К
2,5 мл раствора 3 прибавляют 10 мл кислоты азот¬
ной разведенной и доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
К 3 мл раствора 3 прибавляют 2 мл кислоты хлори¬
стоводородной разведенной Р и доводят водой дис¬
тиллированной Р до объема 15 мл.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 5 мл раствора 3 должны выдерживать испы¬
тание на мышьяк.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят до объема 10 мл водой Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Вода (2.5.12). Не менее 57,0 % и не более 61,0 %.
Определение проводят из 50,0 мг испытуемого образ¬
ца. В качестве растворителя используют смесь, со¬
стоящую из 10 объемов метанола безводного Р и 40
объемов формамида Р1.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
4,00 г (т) испытуемого образца растворяют в
25 мл воды Р и прибавляют 25,0 мл 1 М раствора
кислоты хлористоводородной. Титруют 1 М раство¬
ром натрия гидроксида потенциометрически (2.2.20).
Отмечают объем титранта в первой точке перегиба
(л,, мл). Продолжают титровать до второй точки пере¬
гиба на кривой титрования (общий затраченный объ¬
ем 1 М раствора натрия гидроксида, п2, мл).
Процентное содержание Ыа2НР04-12Н20 рассчи¬
тывают по формуле:
3581(25-л,)
т-Ш
07/2016:0232
ДИНАТРИЯ ЭДЕТАТ
йтаи7/ ес/е/аз
0/500/СШ ЕОЕТАТЕ
,со2н
.Ы. ^С02Ыа
Ыа02С N ’ 2НгО
Н02С
С^Н^Ы^О, ■ 2НгО М.м. 372,2
ОПРЕДЕЛЕНИЕ
Динатрия дигидро(этилендинитрило)тетраацетат
дигидрат. "Динатриевая соль этилендиаминтетраук-
сусной кислоты, трилон Б#.
Динатрия эдетат
397
Содержание: не менее 98,5 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде, практически нерастворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификациям, В, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО динатрия эдетата.
B. 2 г испытуемого образца растворяют в 25 мл
воды Р, прибавляют 6 мл раствора свинца нитра¬
та Р, перемешивают и прибавляют 3 мл раствора
калия йодида Р. Не должен образовываться желтый
осадок. Полученный раствор подщелачивают раство¬
ром аммиака разведенным Р2 по красной лакмусовой
бумаге Р и прибавляют 3 мл раствора аммония окса¬
лата Р. Осадок не образуется.
C. 0,5 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют 0,5 мл раствора кальция хлори¬
да Р, подщелачивают раствором аммиака разведен¬
ным Р2 по красной лакмусовой бумаге Р и прибавля¬
ют 3 мл раствора аммония оксалата Р. Осадок не
образуется.
й. Испытуемый образец дает реакции (а) и (Ь) на
натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,0 до 5,5. Измеряют рН раствора 3.
Примесь А. Жидкостная хроматография (2.2.29).
Испытание проводят с защитой от света.
Смесь растворителей. 10,0 г железа (III) суль¬
фата пентагидрата Р растворяют в 20 мл 0,5 М
раствора кислоты серной и прибавляют 780 мл
воды Р. Доводят до рН 2,0 1 М раствором натрия ги¬
дроксида и разводят водой Р до объема 1000 мл.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в растворяющей смеси и доводят
до 25,0 мл этим же растворителем.
Раствор сравнения. 40,0 мг нитрилотриуксус-
ной кислоты Р растворяют в растворяющей смеси и
доводят до объема 100,0 мл этим же растворителем.
К 1,0 мл полученного раствора прибавляют 0,1 мл ис¬
пытуемого раствора и доводят до объема 100,0 мл
растворяющей смесью.
Условия хроматографирования;
- колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим углеродом
графитированным для хроматографии Р1 (размер
частиц 5 мкм) с удельной площадью поверхности
120 м2/г и размером пор 25 нм;
- подвижная фаза: 50,0 мг железа сульфата
пентагидрата Р растворяют в 50 мл 0,5 М раствора
кислоты серной и прибавляют 750 мл воды Р. Дово¬
дят рН раствора до значения 1,5 при помощи 0,5 М
раствора кислоты серной или 1 М раствора натрия
гидроксида, прибавляют 20 мл этиленгликоля Р и
доводят водой Р до объема 1000 мл;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 273 нм;
- объем вводимой пробы: 20 мкл; растворы
фильтруют и вводят немедленно;
- время хроматографирования: 4-кратное вре¬
мя удерживания железного комплекса примеси А.
Время удерживания: железный комплекс приме¬
си А— около 5 мин; комплекс железа с кислотой эти-
лендиаминтетрауксусной — около 10 мин.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 7 между пиками желез¬
ного комплекса примеси А и железного комплекса кис¬
лоты эдетатовой;
- отношение сигнал/шум: не менее 50 для пика
примеси А.
Предельное содержание примесей:
- примесь А (не более 0,1 %): площадь пика при¬
меси А на хроматограмме испытуемого раствора не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения.
Железо (2.4.9). Не более 0,0080 % (80 ррт). 2,5 мл
раствора 3 доводят водой Р до объема 10 мл. Получен¬
ный раствор должен выдерживать испытание на желе¬
зо. К каждому раствору перед прибавлением кислоты
тиогликолевой Р добавляют 0,25 г кальция хлорида Р.
#Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
К 10 мл раствора 3 прибавляют 3 мл кислоты азот¬
ной разведенной Р и фильтруют. Объем фильтрата
доводят водой Р до 15 мл. Полученный раствор дол¬
жен выдерживать испытание на хлориды.
#Сульфаты (2.4.13). Не более 0,0600%
(600 ррт). 5 мл раствора 3 доводят водой дистил¬
лированной Р до объема 15 мл. Полученный раствор
должен выдерживать испытание на сульфаты.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в воде Р
и доводят до объема 300 мл этим же растворителем. К
полученному раствору прибавляют 2 г гексаметилен¬
тетрамина Р, 2 мл кислоты хлористоводородной
разведенной Р и титруют 0,1 М раствором свинца ни¬
трата, используя в качестве индикатора около 50 мг
индикаторной смеси ксиленолового оранжевого Р.
1 мл 0,1 М раствора свинца нитрата соответ¬
ствует 37,22 мг С10Н14Ы2Ма2О8-2Н2О.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
!М V
но2с—/ со2н
А. Нитрилотриуксусная кислота.
398
Государственная фармакопея Республики Беларусь
07/2016:0023
ДИФЕНГИДРАМИНА ГИДРОХЛОРИД
О/рЬелЛус/гат/л/ ЬудгосЫопбит
01РНЕИНУ0ЯАМ1ЫЕ НУОРОСН1-ОРЮЕ
ч/
С17Н21ЫО НС1 М.м. 291,8
[147-24-0]
ОПРЕДЕЛЕНИЕ
2-(Дифенилметокси)-Л/,Л/-диметипэтанамина ги¬
дрохлорид. #Димедрол#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С, О.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 168 °С до
172 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 96 % спирте Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 253 нм, при 258 нм
и при 264 нм.
Отношение оптических плотностей:
“ ^58^53-~от 1’1 Д° 1’3«
— А258^А2в4 ОТ 1,2 ДО 1,4.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО дифенгидрамина гидрохлори¬
да #или спектр, представленный на рисунке #0023.-1#.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 и пятикратно
разведенный раствор 8 должны быть прозрачными.
Цветность (2.2.2, метод II). Окраска раствора 8
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность или щелочность. К 10 мл раст¬
вора 8 прибавляют 0,15 мл раствора метилового
красного Р и 0,25 мл 0,01 М раствора кислоты хло¬
ристоводородной. Должно появиться розовое окра¬
шивание. При прибавлении не более 0,5 мл 0,01 М
раствора натрия гидроксида должно появиться жел¬
тое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 70 мг испытуемого образ¬
ца растворяют в подвижной фазе и доводят до объема
20,0 мл этим же растворителем. 2,0 мл полученного
раствора доводят подвижной фазой до объема 10,0 мл.
Рисунок #0023.-1. Инфракрасный спектр ФСО дифенгидрамина гидрохлорида
Щиэтаноламин
399
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 10,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (Ь). 5 мг ФСО дифенгидра-
мина примеси А и 5 мг дифенилметанола Р раство¬
ряют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем. К 2,0 мл полученного раст¬
вора прибавляют 1,5 мл испытуемого раствора и до¬
водят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — раствор
5,4 г/л калия дигидрофосфата Р, доведенный до рН
3.0 кислотой фосфорной Р (35:65, об/об);
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 7-кратное вре¬
мя удерживания дифенгидрамина.
Относительное удерживание (по отношению
к дифенгидрамину, время удерживания — около
6 мин): примесь А — около 0,9; примесь В — около
1,5; примесь С — около 1,8; примесь О — около 2,6;
примесь Е — около 5,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 2,0 между пиками ди¬
фенгидрамина и примеси А.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 0,7):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- любая другая примесь (не более 0,3 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пика примеси А, не должна
превышать 0,6 площади оствновного пика на хрома¬
тограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
96 % спирта Р, прибавляют 5 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 29,18 мг С17Н21МО-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
A. Р = Р' = Н: 2-(Дифенилметокси)-Л/-метилэтан-
амин.
B. Р = Р* = СН3:2-[(Р5)-(4-Метилфенил)фенилме-
токси]-Л/,Л/-диметилэтанамин.
C. Р = Вг, Р' = СН3. 2-[(Р5)-(4-Бромфенил)фенил-
метокси]-Л/, Л/-д и метилэтам ин.
й. Р = ОН, Р' = Н: Дифенилметанол (бензгидрол).
Е. Р + Р' = О: Дйфенилметанон (бензофенон).
07/2016.РБ0018
«ДИЭТАНОЛАМИН
0\е1Ъапо1ат'\пит
01ЕТНА N01. А МШЕ
САН"Ы02 М.м. 105,1
[111-42-2]
ОПРЕДЕЛЕНИЕ
Смесь этаноламинов, содержащая в основном
диэтаноламин (2,2-иминобисэтанол).
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на безводный С4Н11Ы02).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная или слегка желтоватая
вязкая жидкость или кристаллы, расплывающиеся на
воздухе.
Смешивается с водой, с ацетоном и с 96 % спир¬
том.
Относительная плотность: около 1,09.
Температура плавления кристаллов: около
28 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО диэтаноламина.
400
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
рН (2.2.3). От 10,0 до 11,5. Измеряют рН 5 % (м/об)
раствора испытуемого образца в воде, свободной от
углерода диоксида, Р.
Показатель преломления (2.2.6). От 1,473 до
1,476 при температуре 30 °С.
Триэтаноламин. Не более 1,0 % (м/м).
Раствор индикатора. Раствор, содержащий
1,5 г/л метилового оранжевого Р и 0,8 г/л ксиленци-
анола РРР в воде Р.
В коническую колбу с притертой стеклянной
пробкой вместимостью 500 мл помещают 100 мл ме¬
танола Р, прибавляют 6—8 капель раствора инди¬
катора и нейтрализуют 0,1 М спиртовым раствором
серной кислоты, полученным при разведении 2,5 М
раствора серной кислоты в-спирте, или 0,1 М спир¬
товым раствором калия гидроксида. Нейтральный
раствор при просматривании в проходящем свете
имеет желтоватое окрашивание, а в отраженном све¬
те — красно-коричневое окрашивание. В полученный
нейтрализованный раствор добавляют 20 г испытуе¬
мого образца, осторожно прибавляют 75 мл уксусного
ангидрида Р и перемешивают вращением до полного
растворения образца. При необходимости охлаждают
до комнатной температуры и выдерживают при этой
температуре в течение 30 мин. Полученный раствор
титруют 0,25 М раствором серной кислоты в спирте.
Параллельно проводят контрольный опыт.
1 мл 0,25 М раствора серной кислоты в спирте
соответствует 74,6 мг триэтаноламина.
Вода (2.5.12). Не более 0,15%. Определение
проводят из 20,00 г испытуемого образца. В качестве
растворителя используют смесь из 25 мл кислоты ук¬
сусной ледяной Р и 40 мл метанола Р.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
3,000 г испытуемого образца растворяют в 50 мл
воды Р и титруют 1 М раствором кислоты хлори¬
стоводородной до розового окрашивания, используя
в качестве индикатора 0,1 мл раствора метилового
оранжевого Р.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 105,1 мг С4Н11Ы02.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
07/2016:1589
ДОКСИЛАМИНА ГИДРОСУКЦИНАТ
Ооху1атт1 Ьубгодепозисс'таз
ООХУ1-АМ1ЫЕ НУйРЮвЕЫ ЗиССШАТЕ
но2с
,/-\^С°2Н
С21Н28М205 М.м. 388,5
[562-10-7]
ОПРЕДЕЛЕНИЕ
А/,Л/-Диметил-2-[(1 Р5)-1 -фенил-1 -(пиридин-2-ил)-
этошфтанамингидробутандиоат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО доксиламина гидросукцината.
Если спектры отличаются, то испытуемый обра¬
зец и ФСО доксиламина гидросукцината растворяют
по отдельности в метаноле Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Раствор 5. 0,4 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. 0,650 г испытуемого об¬
разца растворяют в 20 мл раствора 10,3 г/л кислоты
хлористоводородной Р, прибавляют 3 мл раствора
100 г/л натрия гидроксида Р и экстрагируют тремя
порциями по 25 мл метиленхлорида Р. Объединяют
метиленхлоридные экстракты и фильтруют с исполь¬
зованием гидрофобной для разделения фаз фильтро¬
вальной бумаги. Промывают фильтр 10 мл метилен¬
хлорида Р и объединяют промывные воды и метилен¬
хлоридные экстракты. Выпаривают растворитель при
пониженном давлении и температуре не выше 40 °С.
Остаток растворяют в 20,0 мл этанола безводного Р.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят этанолом безводным Р до объема
100,0 мл. 1,0 мл полученного раствора доводят эта¬
нолом безводным Р до объема 10,0 мл.
Раствор сравнения (Ь). 50 мг ФСО доксилами¬
на для проверки пригодности хроматографической
системы (содержит примесь С) растворяют 10 мл
раствора 10,3 г/л кислоты хлористоводородной Р,
прибавляют 1,5 мл раствора 100 г/л натрия гидрок¬
сида Р и экстрагируют тремя порциями по 25 мл ме-
тиленхлорида Р. Объединяют метиленхлоридные
экстракты и фильтруют с использованием гидрофоб¬
ной для разделения фаз фильтровальной бумаги.
Промывают фильтр 10 мл метиленхлорида Р и объ¬
единяют промывные воды и метиленхлоридные экс¬
тракты. Выпаривают растворитель при пониженном
давлении и температуре не выше 40 °С. Остаток раст¬
воряют в 5,0 мл этанола безводного Р.
Условия хроматографирования:
- колонка длиной 30 м и внутренним диаметром
0,53 мм, заполненная поли(диметил)(дифенил)си-
локсаном Р (толщина пленки 1,5 мкм);
- газ-носитель : гелий для хроматографии Р;
"НИ
Доксициклина гиклат
401
- скорость газа-носителя: 7 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—12
160 —> 220
12—27
220
Блок ввода
проб
250
Детектор
250
- детектор: пламенноионизационный;
- объем вводимой пробы: 1 мкл.
Идентификация пиков примесей: идентифици¬
руют пик примеси С, используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО доксиламина для проверки пригодности хрома¬
тографической системы.
Относительное удерживание (по отношению к
доксиламину, время удерживания — около 12 мин):
примесь С — около 0,96.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси С и доксиламина.
Предельное содержание примесей:
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си С, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,7 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 7-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 2,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 19,43 мг С^Н^О...
ПРИМЕСИ
Специфицированные примеси: С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, О.
и энантиомер
А. Л/,Л/-Диметил-2-[(1 Д5)-1 -фенил-1 -(пиридин-4-
ил)этокси]этанамин.
и энантиомер
В. (17?5)-1 -Фенил-1 -(пиридин-2-ил)этанол.
С. Л/,А/-Диметил-2-[(1Я5)-1 -фенил-1 -(пиридин-2-
ил)метокси]этанамин.
О. Фенил(пиридин-2-ил)метанон (2-бензоилпи-
ридин).
07/2016:0272
ДОКСИЦИКЛИНА ГИКЛАТ
ОохусусИп! Ьус!аз
ООХУСУШЫЕ НУС1-АТЕ
(С22Н24М2°8 ■ нс0 - V* С2Н6° " % Н20 М.м. 512,9
[24390-14-5]
ОПРЕДЕЛЕНИЕ
(45,4а#,55,5а/?,6Я,12а5)-4-(Диметиламино)-
3,5,10,12,12а-пентагидрокси-6-метил-1,11 -диоксо-
1,4,4а,5,5а,6,11,12а-октагидротетрацен-2-карбоксамида
гидрохлорид гемиэтанол гемигидрат.
Получают из окситетрациклина или метациклина
или любым другим способом.
51. Зак. 1060
402
Гэсударственная фармакопея Республики Беларусь
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное и свободное от этанола ве¬
щество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый кристаллический порошок. Гигроскопичен.
Легко растворим в воде и метаноле, умеренно
растворим в 96 % спирте. Растворяется в растворах
гидроксидов и карбонатов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
B. К около 2 мг испытуемого образца прибавляют
5 мл кислоты серной Р. Появляется желтое окраши¬
вание.
C. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 2,0 до 3,0. 0,1 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Оптическая плотность (2.2.25). Не более 0,07
при длине волны 490 нм (в пересчете на безводное и
свободное от этанола вещество).
0,10 г испытуемого образца растворяют в смеси
из 1 объема раствора 103 г/л кислоты хлористово¬
дородной Р и 99 объемов метанола Р и доводят до
объема 10,0 мл этой же смесью растворителей. Изме¬
ряют оптическую плотность в течение 1 ч после при¬
готовления раствора.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Раствор А. 111,6 г натрия эдетата Р суспенди¬
руют в 900 мл воды Р, доводят до рН 7,0 концентри¬
рованным раствором аммиака Р для достижения пол¬
ного растворения и доводят водой Р до объема 1 л.
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в растворе 1 г/л кислоты хлори¬
стоводородной Р и доводят до объема 25,0 мл этим
же растворителем.
Раствор сравнения (а). 20,0 мг ФСО доксици-
клина гиклата растворяют в растворе 1 г/л кислоты
хлористоводородной Р и доводят до объема 25,0 мл
этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО доксициклина
для проверки пригодности хроматографической си¬
стемы (содержит примеси А, В, С и Р) растворяют в
растворе 1 г/л кислоты хлористоводородной Р и до¬
водят до объема 10 мл этим же растворителем.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят до объема 100,0 мл раствором 1 г/л кис¬
лоты хлористоводородной Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная полимером органосили¬
катным аморфным, с введенными полярными груп¬
пами, октадецилсилильным эндкепированным Р с
размером частиц 5 мкм;
- температура: 35 °С;
- подвижная фаза: смесь ацетонитрил Р —
вода Р — раствор 67,9 г/л тетрабутиламмония ги¬
дросульфата Р, предварительно доведенный рас¬
твором аммиака концентрированным Р до рН 7,0
— раствор А (13:17:35:35, об/об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 2-кратное вре¬
мя удерживания доксициклина.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С и Р, используя хромато¬
грамму раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО доксициклина для проверки пригод¬
ности хроматографической системы.
Относительное удерживание (по отношению к
доксициклину, время удерживания — около 21 мин):
примесь С — около 0,4; примесь А — около 0,7; при¬
месь В — около 0,8; примесь Р — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
месей А и В; не менее 2,0 между пиками примеси В и
доксициклина.
Расчет процентных содержаний:
- для каждой примеси — используют концентра¬
цию доксициклина в растворе сравнения (с).
Предельное содержание примесей:
- примесь А: не более 2,0 %;
- примесь Р: не более 1,2 %;
- примесь В: не более 0,5 %;
- примесь С: не более 0,2 %;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 3,0 %;
- учитываемый предел: 0,05 %.
Этанол. Не менее 4,3 % и не более 6,0 %. Газо¬
вая хроматография (2.2.28).
Раствор внутреннего стандарта. 0,50 мл про¬
панола Р доводят водой Р до объема 1,0 л.
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в растворе внутреннего стандарта
и доводят до объема 10,0 мл этим же раствором.
Раствор сравнения. 0,5 мл этанола безводно¬
го Р доводят раствором внутреннего стандарта до
объема 100,0 мл. 1,0 мл полученного раствора до¬
водят раствором внутреннего стандарта до объема
10,0 мл.
Условия хроматографирования:
- колонка капиллярная длиной 50 м и внутренним
диаметром 0,32 мм, покрытая слоем поли(диметил)
силоксана Р (толщина слоя 5 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 37 см/с;
- деление потока: 1:10;
- температура:
Время (мин)
Температура (°С)
Колонка
0—7
70
7—13
о
(О
7
о
г-
13—15
160
Блок ввода
проб
200
Детектор
250
Доксициклина гиклат
403
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению
к пропанолу, время удерживания — около 7 мин):
этанол — около 0,6.
Рассчитывают содержание этанола, принимая
плотность (2.2.25) равной 0,790 г/мл при темпера¬
туре 20 °С.
Тяжелые металлы (2.4.8, метод С). Не бо¬
лее 0,0050 % (50 ррт). 0,5 г испытуемого образца
должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием 2,5 мл
эталонного раствора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 1,4% и не более
2,8 %. Определение проводят из 1,20 г испытуемо¬
го образца.
Сульфатная зола (2.4.14). Не более 0,4%.
Определение проводят из 1,0 г испытуемого об¬
разца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 1,14 МЕ/мг, если субстанция предназначена
для производства лекарственных средств для па¬
рентерального применения без последующей про¬
цедуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и раствора сравнения (а).
Содержание С22Н241М208-НС1 в процентах рас¬
считывают с учетом содержания доксициклина ги-
клата в ФСО доксициклина гиклата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Стерильная субстанция — в стерильном воз¬
духонепроницаемом контейнере с контролем пер¬
вого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, С, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): О, Е.
А. (45,4а/?,55,5а/:?,65,12а5)-4-(Диметиламино)-
3,5,10,12,12а-пентагидрокси-6-метил-1,11 -диоксо-
1,4,4а,5,5а,6,11,12а-октагидротетрацен-2-карбоксамид
(6-эпидоксициклин).
В. (4$,4аЯ,55,5аЯ,12а5)-4-(Диметиламино)-
3,5,10,12,12а-пентагидрокси-6-метилен-1,11 -диоксо-
1,4,4а,5,5а,6,11,12а-октагидротетрацен-2-карбоксамид
(метациклин).
С. (4#,4а/?,55,5аЯ,6Я,12а5)-4-(Диметиламино)-
3,5,10,12,12а-пентагидрокси-6-метил-1,11 -диоксо-
1,4,4а,5,5а,6,11,12а-октагидротетрацен-2-карбоксамид
(4-эпидоксициклин).
О. (4/?,4а/?,55,5а/?,65,12аЗ)-4-(Диметиламино)-
3,5,10,12,12а-пентагидрокси-6-метил-1,11 -диоксо-
1,4,4а,5,5а,6,11,12а-октагидротетрацен-2-карбоксамид
(4-эпи-б-эпидоксициклин).
Е. (45,4аЯ55,5а#,65,12а5)-4-(Диметиламино)-
3,5,6,10,12,12а-гексагидрокси-6-метил-1,11 -диоксо-
1,4,4а,5,5а,6,11,12а-октагидротетрацен-2-карбоксамид
(окситетрациклин).
Р. (45,4аЯ,53,5аЯ,6Я,12а5)-2-Ацетил-4-
(диметиламино)-3,5,10,12,12а-пентагидрокси-6-метил-
4а,5а,6,12а-тетрагидротетрацен-1,11(4Н,5Н)-дион
(2-ацетил-2-декарбамоилдоксициклин).
51*. Зак. 1060.
404
Гэсударственная фармакопея Республики Беларусь
07/2016:0714 ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
ДОКСОРУБИЦИНА ГИДРОХЛОРИД
ОохогиЫст/ Ьус1госЫопс1ит
ООХОР11В1С1Ы НУОГЮСШ-ОШОЕ
С27Н29Ы011 ■ НС1 М.м. 580,0
[25316-40-9]
ОПРЕДЕЛЕНИЕ
(85,105)-10-[(3-Амино-2,3,6-тридезокси-а-Ьл1//ссо-
гексопиранозил)окси]-6,8,11 -тригидрокси-8-(гидро-
ксиацетил )-1 -метокси-7,8,9,10-тетрагидротетрацен-5,12-
диона гидрохлорид.
Получают с использованием отдельных штам¬
мов 8&ер№тусе$ соеги1еогиЫдиз или 81гер1отусез
реисеИиз или любым другим способом.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Оранжево-красный кристаллический порошок.
Гигроскопичен.
Растворим в воде, мало растворим в метаноле.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО доксорубицина гидрохлорида
#или спектр, представленный на рисунке #0714.-1#.
B. 10 мг испытуемого образца растворяют в
0,5 мл кислоты азотной Р, прибавляют 0,5 мл
воды Р и нагревают над открытым пламенем в
течение 2 мин. Охлаждают и прибавляют 0,5 мл
раствора серебра нитрата Р1. Образуется белый
осадок.
ИСПЫТАНИЯ
рН (2.2.3). От 4,0 до 5,5. 50 мг испытуемого об¬
разца растворяют в воде, свободной от углерода
диоксида, Р и доводят до объема 10 мл этим же
растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием.
Испытуемый раствор (а). 50,0 мг испытуемо¬
го образца растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 10,0 мл испытуемо¬
го раствора (а) доводят подвижной фазой до объ¬
ема 100,0 мл.
Раствор сравнения (а). 10,0 мг ФСО доксо¬
рубицина гидрохлорида и 10 мг ФСО эпирубицина
гидрохлорида растворяют в подвижной фазе и до¬
водят до объема 50,0 мл этим же растворителем.
10.0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Раствор сравнения (Ь). 5,0 мл раствора срав¬
нения (а) доводят подвижной фазой до объема
20.0 мл.
Рисунок #0714.-1. Инфракрасный спектр ФСО доксорубицина гидрохлорида
Докузат натрия
405
Раствор сравнения (с). 50,0 мг ФСО доксо-
рубицина гидрохлорида растворяют в подвижной
фазе и доводят до объема 50,0 мл этим же раст¬
ворителем. 10,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,0 мм, заполненная силикагелем октаде-
цилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- подвижная фаза: смешивают равные объ¬
емы ацетонитрила Р и раствора, содержащего
2,88 г/л натрия лаурилсульфата Р и 2,25 г/л кис¬
лоты фосфорной Р;
- скорость подвижной фазы. 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 254 нм;
- объем вводимой пробы: по 5 мкл испытуемо¬
го раствора (а) и растворов сравнения (а) и (Ь);
- время хроматографирования: 3,5-кратное вре¬
мя удерживания доксорубицина.
Время удерживания: доксорубицин — около
8 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками
доксорубицина и эпирубицина.
Предельное содержание примесей:
-любая примесь (не более 0,5 %): на хрома¬
тограмме испытуемого раствора (а) площадь лю¬
бого пика, кроме основного, не должна превышать
площадь пика доксорубицина на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора (а) не учитывают
пики с площадью менее 0,1 площади пика доксору¬
бицина на хроматограмме раствора сравнения (Ь).
Этанол (2.4.24, система В). Не более 1,0 %.
Вода (2.5.12). Не более 4,0%. Определение
проводят из 0,100 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 2,2 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без последующей процедуры
удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемо¬
го раствора (Ь) и раствора сравнения (с).
Содержание С27Н29М01(|'НС1 рассчитывают в
процентах.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
А. (85,105)-8-Ацетил-10-[(3-амино-2,3,6-тридезокси-
а-1_-Л1ЖСО-гексопиранозил)окси]-6,8,11 -тригидрокси-1 -
метокси-7,8,9,10-тетрагидротетрацен-5,12-дион (дау-
норубицин).
B. Р = ОСН3: (85,105)-10-[(3-Амино-2,3,6-тридезо-
кси-а-1-л*жсогексопиранозил)окси]-8-(2-бром-1,1 -диме-
токсиэтил)-6,8,11 -тригидрокси-1 -метокси-7,8,9,10-тетра-
гидротетрацен-5,12-дион.
C. Р + Р = О: (85,105)-10-[(3-Амино-2,3,6-триде-
зокси-а-1_-лб/кео-гексопиранозил)окси]-8-(бромацетил)-
6,8,11 -тригидрокси-1 -метокси-7,8,9,10-тетрагидро-
тетрацен-5,12-дион.
О. (85,105)-6,8,10,11 -Тетрагидрокси-8-(гидрокси-
ацетил)-1 -метокси-7,8,9,10-тетрагидротетрацен-5,12-
дион (агликон доксорубицина, доксорубицинон).
07/2016:1418
ДОКУЗАТ НАТРИЯ
А/а/г/7 с1осизаз
ООСиЗАТЕ ЗООШМ
С20Н37ЫаО73 М.м. 444,6
[577-11-7]
ОПРЕДЕЛЕНИЕ
Натрия 1,4-бис[(2-этилгексил)окси]-1,4-диоксобутан-
2-сульфонат.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
52. Зак. 1060.
406
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ(СВОЙСТВА)
Белая или почти белая парафиноподобная масса
или хлопья. Гигроскопичен.
Умеренно растворим в воде, легко растворим в
96 % спирте и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: около 3 мг испытуемого об¬
разца помещают на пластину из натрия хлорида,
прибавляют 0,05 мл ацетона Р и немедленно на¬
крывают другой пластиной из натрия хлорида.
Пластины потирают друг относительно друга для
растворения испытуемого образца, затем разъеди¬
няют и выдерживают до испарения ацетона.
Сравнение: ФСО натрия докузата.
B. 0,75 г испытуемого образца помещают в
тигель и сжигают в присутствии кислоты серной
разведенной Р до получения почти белого остатка.
Охлаждают, к остатку прибавляют 5 мл воды Р и
фильтруют. 2 мл полученного фильтрата дают ре¬
акцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Щелочность. 1,0 г испытуемого образца раст¬
воряют в 100 мл смеси из равных объемов метано¬
ла Р и воды Р, предварительно нейтрализованной
по раствору метилового красного Р. Прибавляют
0,1 мл раствора метилового красного Р. При при¬
бавлении не более 0,2 мл 0,1 М раствора кисло¬
ты хлористоводородной цвет индикатора должен
измениться на красный.
Сопутствующие неионизированные веще¬
ства. Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 10 мг ме-
тилбегената Р растворяют в гексане Р и доводят
до объема 50 мл этим же растворителем.
Испытуемый раствор (а). 0,10 г испытуе¬
мого образца растворяют в 2,0 мл раствора вну¬
треннего стандарта и доводят до объема 5,0 мл
гексаном Р. Раствор пропускают со скоростью
1,5 мл/мин через колонку внутренним диаметром
10 мм, заполненную 5 г алюминия оксида основно¬
го Р и предварительно промытую 25 мл гексана Р.
Элюируют 5 мл гексана Р и отбрасывают элюат.
Элюируют 20 мл смеси, состоящей из равных объе¬
мов эфира Р и гексана Р. Элюат выпаривают досуха
и сухой остаток растворяют в 2,0 мл гексана Р.
Испытуемый раствор (Ь). Готовят аналогично
испытуемому раствору (а), но растворяя 0,10 г ис¬
пытуемого образца в гексане Р и доводя до объ¬
ема 5,0 мл этим же растворителем; используют
новую колонку.
Раствор сравнения. 2,0 мл раствора внутрен¬
него стандарта доводят до объема 5,0 мл гекса¬
ном Р.
Условия хроматографирования:
- колонка стеклянная длиной 2 м и диаметром
2 мм, заполненная диатомитом силанизирован-
ным для газовой хроматографии Р, импрегниро-
ванным 3 % (м/м) полиметилфенилсилоксаном Р;
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 30 мл/мин;
- температура колонки: 230 °С;
- температура блока ввода проб и детекто¬
ра: 280 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания внутреннего стандарта.
Пригодность хроматографической системы:
- на хроматограмме испытуемого раствора (Ь)
должен отсутствовать пик со временем удержива¬
ния, равным времени удерживания внутреннего
стандарта.
Предельное содержание примесей:
- любая примесь (не более 0,4 %): на хрома¬
тограмме испытуемого раствора (а) площадь лю¬
бого пика, кроме основного, не должна превышать
площадь пика внутреннего стандарта.
Хлориды. Не более 0,0350 % (350 ррт). 5,0 г
испытуемого образца растворяют в 50 мл спирта
(50 %, об/об) Р и титруют 0,01 М раствором сере¬
бра нитрата потенциометрически (2.2.20).
1 мл 0,01 М раствора серебра нитрата соот¬
ветствует 0,3545 мг С1.
Натрия сульфат. Не более 2 %. 0,25 г испы¬
туемого образца растворяют в 40 мл смеси из 20
объемов воды Р и 80 объемов 2-пропанола Р и до¬
водят раствором хлорной кислоты Р до рН 2,5—
4,0. Прибавляют 0,4 мл раствора нафтарзона Р и
0,1 мл раствора 0,125 г/л метиленового синего Р.
При прибавлении не более 1,5 мл 0,025 М раст¬
вора бария перхлората цвет индикатора должен
измениться с желтовато-зеленого на желтовато¬
розовый.
Тяжелые металлы (2.4.8, метод В). Не бо¬
лее 0,0010 % (10 ррт). 4,0 г испытуемого образца
растворяют в спирте (80 %, об/об) Р и доводят
до объема 20 мл этим же растворителем. 12 мл
полученного раствора должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца
(2 ррт РЬ), полученного путем разведения эталон¬
ного раствора свинца (100 ррт РЬ)Рспиртом (80 %,
об/об) Р.
Вода (2.5.12). Не более 3,0%. Определение
проводят из 0,250 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца помещают в
коническую колбу вместимостью 250 мл, прибав¬
ляют 25,0 мл 0,5 М спиртового раствора калия
гидроксида и нагревают на водяной бане с об¬
ратным холодильником в течение 45 мин. Охлаж¬
дают, прибавляют 0,25 мл раствора фенолфта¬
леина Р1 и титруют 0,5 М раствором кислоты
хлористоводородной до исчезновения красного
окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,5 М спиртового раствора калия гидрок¬
сида соответствует 0,1112 г С20Н37МаО75.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Допамина гидрохлорид
407
07/2016:0664
ДОПАМИНА ГИДРОХЛОРИД
Оорат1т Ьус1гос111опс1ит
ООРАМШЕ НУОПОСН1-(Ж10Е
СД^О, ■ НС1 М.м. 189,6
[62-31-7]
ОПРЕДЕЛЕНИЕ
4-(2-Аминоэтил)бензол-1,2-диола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворим в воде, растворим в 96 % спирте,
умеренно растворим в ацетоне и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в 0,1 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем. 10,0 мл полученного раствора до¬
водят 0,1 М раствором кислоты хлористоводород¬
ной до объема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 280 нм.
Удельный показатель поглощения в максимуме:
от 136 до 150.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО допамина гидрохлорида.
C. 5 мг испытуемого образца растворяют в смеси
из 5 мл 1 М раствора кислоты хлористоводородной
и 5 мл воды Р. При прибавлении к полученному рас¬
твору 0,1 мл раствора натрия нитрита Р, содер¬
жащего 100 г/л аммония молибдата Р, появляется
желтое окрашивание, которое изменяется на красное
при прибавлении раствора натрия гидроксида кон¬
центрированного Р.
О. 2 мг испытуемого образца растворяет в 2 мл
воды Р и прибавляют 0,2 мл раствора железа (III)
хлорида Р2. Появляется зеленое окрашивание, кото¬
рое изменяется на голубовато-фиолетовое при при¬
бавлении 0,1 г гексаметилентетрамина Р.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,4 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)6 или У(Ж)6.
Кислотность или щелочность. 0,5 г испыту¬
емого образца растворяют в воде, свободной от
углерода диоксида, Р и доводят до объема 10 мл
этим же растворителем. К полученному раствору
прибавляют 0,1 мл раствора метилового красно¬
го Р и 0,75 мл 0,01 М раствора натрия гидрок¬
сида. Появляется желтое окрашивание. При при¬
бавлении к полученному раствору 1,5 мл 0,01 М
раствора кислоты хлористоводородной должно
появиться красное окрашивание.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы защищают от
света.
Буферный раствор. 21 г кислоты лимонной Р
растворяют в 200 мл 1 М раствора натрия ги¬
дроксида и доводят водой Р до объема 1000 мл. К
600 мл полученного раствора прибавляют 400 мл
0,1 М раствора кислоты хлористоводородной.
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят
до объема 25 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят подвижной фазой А до объема
100,0 мл. 1,0 мл полученного раствора доводят
подвижной фазой А до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг З-О-метилдопа-
мина гидрохлорида Р (примесь В) и 10 мг 4-О-ме-
тилдопамина гидрохлорида Р (примесь А) раст¬
воряют в подвижной фазе А и доводят до объема
100 мл этим же растворителем. 6 мл полученного
раствора доводят подвижной фазой А до объема
25 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 3,9 мм, заполненная сферическим силикаге¬
лем октадецилсилильным эндкепированным для
хроматографии Р с размером частиц 4 мкм;
- подвижная фаза:
-подвижная фаза А: 1,08 г натрия октан-
сульфоната Р растворяют в 880 мл буферно¬
го раствора и прибавляют 50 мл метанола Р и
70 мл ацетонитрила Р;
-подвижная фаза В: 1,08 г натрия октан-
сульфоната Р растворяют в 700 мл буферно¬
го раствора и прибавляют 100 мл метанола Р
и 200 мл ацетонитрила Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
90
10
5—20
со
0
1
-Еь
О
10 — 60
20—25
40
60
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 10 мкл.
Время удерживания: допамин — около 5 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5,0 между пиками при¬
месей В и А.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
52*. Зак. 1060.
408
Государственная фармакопея Республики Беларусь
- сумма примесей (не более 0,2 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г должен выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с ис¬
пользованием 2 мл эталонного раствора свинца
(10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца су¬
шат при температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Во избежание перегрева реакционной среды
в течение титрования осуществляют интенсив¬
ное перемешивание и прекращают титрование
немедленно после достижения конечной точки
титрования.
0,150 г испытуемого образца растворяют в
10 мл кислоты муравьиной безводной Р. К полу¬
ченному раствору прибавляют 50 мл уксусного
ангидрида Р и титруют 0,1 М раствором кислоты
хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соот¬
ветствует 18,96 мг С8Н11М02 НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в атмос¬
фере азота в защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, В, С.
A. Р = СН3, Р' = Н: 5-(2-Аминоэтил)-2-метоксифе-
нол (4-О-метилдопамин).
B. Р = Н, Р' = СН3: 4-(2-Аминоэтил)-2-метоксифе-
нол (З-О-метилдопамин).
C. Р = Р' = СН3:2-(3,4-Диметоксифенил)этанамин.
07/2016:2593
ДОЦЕТАКСЕЛ БЕЗВОДНЫЙ
Оосе1ахе1ит апЬудпсит
РОСЕТАХЕЦ аынуоксшз
С„НиЫО.. М.м. 808
[114977-28-5]
ОПРЕДЕЛЕНИЕ
5р,20-Эпокси-1,7р, 10р-тригидрокси-9-оксотакс-11 -
ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2Р,35)-3-
[[(1,1-диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат.
Содержание: не менее 97,5 % и не более 102,0 **
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Практически нерастворим в воде, легко раствск
рим в этаноле, растворим в метиленхлориде.
ПОДЛИННОСТЬ(ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО доцетаксела безводного.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в этаноле Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора Б
должна быть не интенсивнее эталона В(К)5.
Удельное оптическое вращение (2.2.7). О
-41,5 до -38,5 (в пересчете на безводное веществе.
0,250 г испытуемого образца растворяют в метано¬
ле Р и доводят до объема 25,0 мл этим же раствори¬
телем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Кислота уксусная Р —
ацетонитрил Р1 — вода Р (0,05:50:50, об/об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл этанола Р и доводят сме¬
сью растворителей до объема 50,0 мл.
Раствор сравнения (а). 50,0 мг ФСО доцетаюь-
ла тригидрата растворяют в 2,5 мл этанола Р и от¬
водят смесью растворителей до объема 50,0 мл.
Доцетаксел безводный
409
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 5 мг ФСО доцетаксела
для проверки пригодности хроматографической
системы (содержит примеси А, В и С) растворяют в
0,25 мл этанола Р и доводят смесью растворителей
до объема 5,0 мл.
Раствор сравнения (д), 5 мг ФСО доцетаксела
примеси Е растворяют в 2,5 мл этанола Р и доводят
смесью растворителей до объема 50,0 мл. 1,0 мл по¬
лученного раствора доводят смесью растворителей
до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3,5 мкм;
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: вода Р;
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—9
72
28
9—39
72 —* 28
28 —► 72
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 232 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (б).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и С, используя хроматограм¬
му раствора сравнения (с) и хроматограмму, прилага¬
емую к ФСО доцетаксела для проверки пригодности
хроматографической системы; идентифицируют
пик примеси Е, используя хроматограмму раствора
сравнения (с1).
Относительное удерживание (по отношению
к доцетакселу, время удерживания — около 27 мин):
примесь Е — около 0,2; примесь А— около 0,97; при¬
месь В — около 1,08; примесь С — около 1,13.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 3,0 между пиками при¬
меси А и доцетаксела.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—1,6):
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
-примесь С (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
-примесь Е (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси Е, не должна превышать
1,5-кратную площадь соответствующего пика на
хроматограмме раствора сравнения (с1);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А, В, С и Е, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей: не более 0,8 %;
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт).
Смесь растворителей. Вода Р — диметил-
формамид Р (15:85, об/об).
1,0 г испытуемого образца растворяют с ис¬
пользованием ультразвука в смеси растворителей
и доводят до объема 20 мл этой же смесью раст¬
ворителей. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ), полученного разведением
эталонного раствора свинца (100 ррт РЬ) Р сме¬
сью растворителей.
Вода (2.5.32). Не более 1,5 %. Вводят 800 мкл
раствора 25 мг/мл испытуемого образца в мета¬
ноле Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 0,3 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без дальнейшей подходящей
процедуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуе¬
мого раствора и раствора сравнения (а).
Содержание С43Н53Ы014 в процентах рассчиты¬
вают с учетом содержания доцетаксела в ФСО до¬
цетаксела тригидрата.
ХРАНЕНИЕ
В защищенном от света месте в воздухонепрони¬
цаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма-
53. Зак. 1060.
410
Гэсударственная фармакопея Республики Беларусь
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): О, Рг С.
А. 5р,20-Эпокси-1,7р,10р-тригидрокси-9-оксотакс-
11 -ен-2а,4,1За-триил-4-ацетата 13-[(2Я,35)-3-[[( 1,1-
диметилэтокси)карбонил]амино]-2-гидрокси-3-фенил-
пропаноат] 2-[(2Е)-2-метилбут-2-еноат] (2-О-тиглилдо-
цетаксел).
В. 5р,20-Эпокси-1,7р-дигидрокси-9,10-диоксотакс-
11-ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2/?,3 5)-
3-[[(1,1-диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] (10-дезокси-10-оксодоцетаксел).
С. 5р,20-Эпокси-1,7а, 10Р-тригидрокси-9-оксотакс-
11-ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2Я,35)-
3-[[( 1,1 -диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] (7-эла-доцетаксел).
О. 5р,20-Эпокси-1,7а-дигидрокси-9,10-диоксотакс-
11-ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2/?,3 5)-
3-[[(1,1 -диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] (10-дезокси-10-оксо-7-эга/-доцетаксел).
Е. 5р,20-Эпокси-4-(ацетилокси)-1,7р, 10р,1 Зс~
тетрагидрокси-9-оксотакс-11 -ен-2а-илбензоат (1 0-ле-
зацетил-баккатин III).
Р. 5р,20-Эпокси-1,7р-дигидрокси-9-оксотакс-11
2а,4,1 ор, 13а-тетраил-4,10-диацетата 2-бензоа~
13-[(2Я35)-3-(бензоиламино)-2-гидрокси-3-фенилпрс~
паноат] (паклитаксел).
О. 5р,20-Эпокси-1,7р-дигидрокси-9-оксотакс-1 ~
ен-2а,4,10р, 13а-тетраил-4,10-диацетата 2-бензоаг
13-[(2/?,35)-3-[[(1,1 -диметилэтокси)карбонил]амино]-2-
гидрокси-3-фенилпропаноат] (10-ацетилдоцетаксел|»
07/2016:2441
ДОЦЕТАКСЕЛ ТРИГИДРАТ
ОосеШе1ит Шуйпсит
ООСЕТАХЕ1. ТШНУОКАТЕ
Доцетаксел тригидрат
411
ОПРЕДЕЛЕНИЕ
5р,20-Эпокси-1,7р, 10р-тригидрокси-9-оксотакс-11 -
ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2Я,35)-3-
[[(1,1-диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] тригидрат.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в этаноле, растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО доцетаксела тригидрата #или
спектр, представленный на рисунке #2449.-1#.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в этаноле Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)5.
Удельное оптическое вращение (2.2.7). От
-41,5 до “38,5 (в пересчете на безводное вещество).
0,250 г испытуемого образца растворяют в метано¬
ле Р и доводят до объема 25,0 мл этим же раствори¬
телем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Кислота уксусная Р —
ацетонитрил Р1 — вода Р (0,05:50:50, об/об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл этанола Р и доводят сме¬
сью растворителей до объема 50,0 мл.
Раствор сравнения (а). 50,0 мг ФСО доцетаксе¬
ла тригидрата растворяют в 2,5 мл этанола Р и до¬
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 5 мг ФСО доцетаксела
для проверки пригодности хроматографической
системы (содержит примеси А, В и С) растворяют в
0,25 мл этанола Р и доводят смесью растворителей
до объема 5,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3,5 мкм;
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: вода Р\
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—9
72
28
9—39
72 —► 28
28—>72
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 232 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и С, используя хроматограм-
Рисунок #2449.-1. Инфракрасный спектр ФСО доцетаксела тригидрата
53*. Зак. 1060.
412
Гэсударственная фармакопея Республики Беларусь
му раствора сравнения (с) и хроматограмму, прилага¬
емую к ФСО доцетаксела для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению
к доцетакселу, время удерживания — около 27 мин):
примесь А—около 0,97; примесь В — около 1,08; при¬
месь С — около 1,13.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 3,0 между пиками при¬
меси А и доцетаксела.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—1,6):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В, С (не более 0,3 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и С, не должны превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт).
Смесь растворителей. Вода Р — диметилфор-
мамид Р (15:85, об/об).
1,0 г испытуемого образца растворяют с исполь¬
зованием ультразвука в смеси растворителей и дово¬
дят до объема 20 мл этой же смесью растворителей.
12 мл полученного раствора должны выдерживать
испытание на тяжелые металлы. Эталон готовят с
использованием эталонного раствора свинца (1 ррт
РЬ), полученного разведением эталонного раствора
свинца (100 ррт РЬ) Р смесью растворителей.
Вода (2.5.32). Не менее 5,0 % и не более 7,0 %.
Вводят 200 мкл раствора 100 мг/мл испытуемого об¬
разца в диметилформамиде Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,3 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С43Н53М014 в процентах рассчиты¬
вают с учетом содержания доцетаксела тригидрата в
ФСО доцетаксела тригидрата.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимитиру¬
ется общим критерием приемлемости для других/неспец-
ифицированных примесей и/или общей статьей Суб¬
станции для фармацевтического использования (2034).
Вследствие этого нет необходимости идентифицировать
эти примеси для доказательства соответствия требова¬
ниям. См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования): О.
А. 5(3,20-Эпокси-1,7р,10р-тригидрокси-9-оксотакс-
11 -ен-2а,4,13а-триил-4-ацетата 13-[(2Я,35)-3-[[(1,1 -
диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] 2-[(2Е)-2-метилбут-2-еноат]-
(2-0-дезбензоил-2-0-тиглилдоцетаксел).
В. 5р,20-Эпокси-1,7р-дигидрокси-9,10-диоксотакс-
11-ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2Я,35)-
3-[[(1,1 -диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] (10-дезокси-10-оксодоцетаксел).
С. 5р,20-Эпокси-1,7а,10Р-тригидрокси-9-оксотакс-
11-ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2Я,35)-
3-[[(1,1 -диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] (7-эпи-цоцетаксел).
Дроперидол
413
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в диметилформамиде и в метиленхлориде, уме¬
ренно растворим в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО дроперидола #или спектр, пред¬
ставленный на рисунке #1010.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО дроперидола растворяют по
отдельности в минимальном количестве ацетона Р,
выпаривают на водяной бане до сухих остатков, кото¬
рые используют для получения новых спектров.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 30 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения (а). 30 мг ФСО дроперидола
растворяют в подвижной фазе и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 30 мг ФСО дроперидола
и 30 мг ФСО бенперидола растворяют в подвижной
фазе и доводят до объема 10 мл этим же раствори¬
телем.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ 6/Г2« Р-
Подвижная фаза: ацетон Р — метанол Р
(10:90, об/об).
Рисунок #1010.-1. Инфракрасный спектр ФСО дроперидола
О. 5р,20-Эпокси-1,7а-дигидрокси-9,10-диоксотакс-
11-ен-2а,4,13а-триил-4-ацетата 2-бензоат 13-[(2Р,35)-
3-[[(1,1 -диметилэтокси)карбонил]амино]-2-гидрокси-3-
фенилпропаноат] (10-дезокси-10-оксо-7-эш-доцетаксел).
07/2016:1010
ДРОПЕРИДОЛ
Огорепс1о1ит
ОКОРЕР1Ю01-
С^РМзО,
[548-73-2]
ОПРЕДЕЛЕНИЕ
1 -[1 -[4-(4-Фторфенил)-4-оксобутил]-1,2,3,6-тетра-
гидропиридин-4-ил]-1,3-дигидро-2Н-бензимидазол-2-он.
54. Зак. 1060.
414
Гэсударственная фармакопея Республики Беларусь
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два пол¬
ностью разделенных пятна.
Результаты: на хроматограмме испытуемо¬
го раствора обнаруживается основное пятно, со¬
ответствующее по расположению и размеру ос¬
новному пятну на хроматограмме раствора срав¬
нения (а).
С. 10 мг испытуемого образца растворяют в
5 мл этанола Р, прибавляют 0,5 мл раствора ди-
нитробензола Р и 0,5 мл 2 М спиртового раст¬
вора калия гидроксида Р. Появляется фиолетовое
окрашивание, переходящее через 20 мин в корич¬
невато-красное.
О. 5 мг испытуемого образца смешивают с
45 мг магния оксида тяжелого Р и прокаливают в
тигле до получения почти белого остатка (обычно
менее 5 мин). Охлаждают, прибавляют 1 мл воды Р,
0,05 мл раствора фенолфталеина Р1, около 1 мл
кислоты хлористоводородной разведенной Р до
получения бесцветного раствора и фильтруют. К
свежеприготовленной смеси из 0,1 мл раствора
ализарина 8Р и 0,1 мл раствора циркон ила ни¬
трата Р прибавляют 1,0 мл полученного филь¬
трата, смешивают, выдерживают в течение 5 мин и
сравнивают окраску полученного раствора с окра¬
ской контрольного опыта, приготовленного таким
же образом. Окраска испытуемого раствора жел¬
тая, а контрольного опыта — красная.
ИСПЫТАНИЯ
Раствор 5. 0,20 г испытуемого образца раст¬
воряют в метиленхлориде Р и доводят до объема
20,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона ВУ(КЖ)5.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием.
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в диметилформамиде Р и дово¬
дят до объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 2,5 мг ФСО дропери-
дола и 2,5 мг ФСО бенперидола растворяют в ди¬
метилформамиде Р и доводят до объема 100,0 мл
этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят диметилформамидом Р до объ¬
ема 100,0 мл. 5,0 мл полученного раствора дово¬
дят диметилформамидом Р до объема 20,0 мл.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем окта-
децилсилильным, деактивированным по отноше¬
нию к основаниям, для хроматографии Р с разме¬
ром частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р\
- подвижная фаза В: раствор 10 г/л тетрабу-
тиламмония гидросульфата Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
0
1
О
о
0
1
О)
о
15—20
40
60
20—25
40 -+ 0
о>
о
О
о
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 275 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
дроперидолу, время удерживания — около 7 мин):
примесь А — около 0,2; примесь В — около 0,85;
бенперидол — около 0,9; примесь С — около 0,95;
примесь О — около 1,2; примесь Е — около 1,5.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками бен¬
перидола и дроперидола.
Предельное содержание примесей:
- примеси А, В, С, О, Е (не более 0,25 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, О и Е, не
должны превышать площадь основного пика на хро¬
матограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О и Е, не должна превышать 0,4 пло¬
щади основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,2 площади основного пика на
хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца сушат
в вакууме при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
смеси из 1 объема кислоты уксусной безводной Р и
7 объемов метилэтилкетона Р и титруют 0,1 М рас¬
твором кислоты хлорной, используя 0,2 мл раст¬
вора нафтолбензола Р в качестве индикатора, до
перехода окраски раствора от оранжево-желтой до
зеленой.
#Дротаверина гидрохлорид
415
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 37,94 мг С^Н^РЫзО^
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
А. 1-(1,2,3,б-Тетрагидропиридин-4-ил)-1,3-дигидро-
2Н-бензимидазол-2-он.
В. 1 -[1 -[4-(2-Фторфенил Н-оксобутил]-1,2,3,6-тетра¬
гид ропиридин-4-ил]-1,3-дигццро-2Н-бензимидазол-2-он.
С. 1 -[4-(4-Фторфенил)-4-оксобутил]-4-(2-оксо-2,3-
дигидро-1 Н-бензимидазол-1 -ил)пиридина хлорид.
0.(1 ЯЗ)-1 -[4-(4-Фторфенил)-4-оксобутил]-4-(2-
оксо-2,3-дигидро-1 /-/-бензимидазол-1 -ил)-1,2,3,6-тетра¬
гидропиридина 1-оксид.
Е. 1-[1-[4-[4-[4-(2-Оксо-2,3-дигидро-1Н-бензими-
дазол-1 -ил )-3,6-ди гидропиридин-1 (2Н)-ил]-1 -оксобутил]-
фенил]-1,2,3,6-тетрагидропиридин-4-ил]-1,3-дигидро-
2Н-бензимидазол-2-он.
07/2016:РБ0027
#ДРОТАВЕРИНА ГИДРОХЛОРИД
ОгоШегЫ ЬудгосЫопдит
ОКОТАУЕКШЕ НУОКОСНШКЮЕ
СН3
СмН31Ы04 ■ НС1 М.м. 434,0
ОПРЕДЕЛЕНИЕ
1-[(3,4-Диэтоксифенил)метилен]-6,7-диэтокси-
3,4-дигидро-2/-/-изохинолина гидрохлорид.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное и свободное от этанола ве¬
щество).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический порошок от светло-желтого до
зеленовато-желтого цвета.
Умеренно растворим в воде, растворим в 96 %
спирте, легко растворим в хлороформе.
Температура плавления: от 208 °С до 211 °С (без
предварительного подсушивания) с разложением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО дротаверина гидрохлорида
или спектр, представленный на рисунке РБ0027.-1.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25). 1,5 мг ис¬
пытуемого образца растворяют в 0,01 М растворе
кислоты хлористоводородной и доводят до объема
100,0 мл этим же растворителем. Спектр поглоще¬
ния полученного раствора в диапазоне длин волн
от 220 нм до 400 нм имеет максимумы при 241 нм,
302 нм и 353 нм.
C. 10 мг испытуемого образца растворяют в
5 мл кислоты серной Р, прибавляют 0,5 мл раствора
30 г/л железа (III) хлорида Р и нагревают при темпе¬
ратуре 100 °С в течение 3 мин. Появляется зеленое
окрашивание. После охлаждения прибавляют 0,5 мл
кислоты азотной разведенной Р. Появляется корич¬
невато-красное окрашивание.
й. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в 50,0 мл воды, свободной от углерода диокси¬
да, Р.
54*. Зак. 1060.
416
Гэсударственная фармакопея Республики Беларусь
Волновое число (см'1)
Рисунок РБ0027.-1. Инфракрасный спектр пропускания ФСО дротаверина гидрохлорида
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона <ЗУ(ЗЖ)2.
рН (2.2.3). От 3,5 до 5,5. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор. 10,88 г натрия ацетата Р
растворяют в 500 мл воды Р, прибавляют 30 мл кис¬
лоты уксусной ледяной Р и доводят водой Р до объ¬
ема 1000,0 мл.
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 5 мл подвижной фазы.
Раствор сравнения (а). 2,5 мл испытуемого раст¬
вора доводят подвижной фазой до объема 25,0 мл.
0,5 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 20 мг испытуемого об¬
разца растворяют в 19 мл воды Р при температуре
от 40 °С до 50 °С, прибавляют 5 мг растертого калия
перманганата Р, перемешивают в течение 2 мин,
прибавляют 1 мл кислоты фосфорной Р и переме¬
шивают до получения прозрачного раствора.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным деактивированным по отношению к ос¬
нованиям эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: метанол Р — ацетонитрил
для хроматографии Р — буферный раствор (2:12:11,
об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
-объем вводимой пробы: по Юмкл раствора
сравнения (а) и испытуемого раствора и по 5 мкл
раствора сравнения (Ь);
- время хроматографирования: 3-кратное вре¬
мя удерживания дротаверина.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-после пика дротаверина элюируются 3 пика
примесей (продукты окисления) на хроматограмме
раствора сравнения (Ь);
-разрешение: не менее 3,0 между соседними
пиками на хроматограмме раствора сравнения (Ь);
- фактор асимметрии: не более 1,3 для пика дро¬
таверина на хроматограмме раствора сравнения (Ь);
- число теоретических тарелок, не менее 3000
для пика дротаверина на хроматограмме раствора
сравнения (Ь).
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт) свинца или железа. 1,0 г испыту¬
емого образца должен выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
2 мл эталонного раствора свинца (10 ррт РЬ) Р
Этанол (2.4.24, система А). Не более 5,0 %.
Вода (2.5.12). Не более 3,0 %. 0,500 г испытуемо¬
го образца растворяют в 10 мл смеси хлороформ Р —
метанол Р (9:1, об/об).
Сульфатная зола (2.4.14, метод А). Не более
0,1 %. Определение проводят из 1,0 г испытуемого
образца.
Бактериальные эндотоксины (2.6.14). Менее
4,3 МЕ/г, если субстанция предназначена для произ-
Дутастерид
417
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 20 мл
кислоты уксусной ледяной Р, прибавляют 3 мл раст¬
вора 50 г/л ртути (II) ацетата Р в кислоте уксусной
ледяной Р и титруют 0,1 М раствором кислоты хлор¬
ной до появления зеленого окрашивания, используя
0,1 мл раствора кристаллического фиолетового Р в
качестве индикатора.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствором кислоты хлорной соответ¬
ствует 43,40 мг С24Н31М04 НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищен¬
ном от света месте при температуре не выше 25 °С.
07/2016:2641
ДУТАСТЕРИД
0и1аз1епс1ит
ОиТАЗТЕКЮЕ
С27Н30Р6М2О2 М.М. 528,5
[164656-23-9]
ОПРЕДЕЛЕНИЕ
Л/-[2,5-Бис(трифторметил)фенил]-3-оксо-4-аза-5а-
андрост-1 -ен-17р-карбоксамид.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или бледно-желтый порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, растворим или умеренно рас¬
творим в этаноле безводном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО дутастерида.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+33,0 до +39,0 (в пересчете на безводное вещество).
0,100 г испытуемого образца растворяют в этаноле
безводном Р и доводят до объема 20,0 мл этим же
растворителем.
Сопутствующие примеси
А. Жидкостная хроматография (2.2.29).
Смесь растворителей. Вода для хроматогра¬
фии Р — ацетонитрил Р1 (40:60, об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО дутастерида
для проверки пригодности хроматографической си¬
стемы (содержит примеси А, В, С, Е, Р, 6, Н и I) раст¬
воряют в смеси растворителей и доводят до объема
10 мл этой же смесью растворителей.
Раствор сравнения (с). 50,0 мг ФСО дутастери¬
да растворяют в смеси растворителей и доводят до
объема 100,0 мл этой же смесью растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным эндкепированным для хроматографии Р
(размер частиц 5 мкм);
- температура: 35 °С;
- подвижная фаза: смесь трифторуксусная кис¬
лота Р — вода для хроматографии Р — ацетони¬
трил Р1 (0,25:480:520, об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а) и (Ь);
- время хроматографирования: 1,6-кратное вре¬
мя удерживания дутастерида.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, Е, Р и 6, используя хро¬
матограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО дутастерида для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению
к дутастериду, время удерживания — около 36 мин):
примесь А—около 0,10; примесь В — около 0,11; при¬
месь С — около 0,4; примесь Е — около 0,9; примесь
Р — около 1,1; примесь 6 — около 1,2.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками при¬
меси Е и дутастерида и не менее 1,5 между пиками
примесей А и В на хроматограмме раствора сравне¬
ния (Ь);
- отношение сигнал/шум: не менее 30 для пика
дутастерида на хроматограмме раствора сравнения (а).
Расчет предельного содержания примесей: для
каждой примеси используют концентрацию дутасте¬
рида в растворе сравнения (а).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,7; для примеси Р — 3,0):
- примесь Р: не более 0,4 %;
- примеси Е и О: для каждой примеси не более
0,3 %;
- примеси А и С: для каждой примеси не более
0,2 %;
55. Зак. 1060.
418
Гэсударственная фармакопея Республики Беларусь
- примесь В: не более 0,15 %;
- неспецифицированные примеси: для каждой
примеси не более 0,10 %;
- учитываемый предел: 0,05 %.
В. Жидкостная хроматография (2.2.29), как ука¬
зано в испытании А «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилсилиль-
ным для хроматографии Р (размер частиц 5 мкм);
- подвижная фаза: вода для хроматогра¬
фии Р— ацетонитрил Р1 (20:80, об/об);
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а) и (Ь);
- время хроматографирования: 5-кратное вре¬
мя удерживания дутастерида.
Идентификация пиков примесей: идентифици¬
руют пики примесей Н и I, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО дутастерида для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
дутастериду, время удерживания — около 4 мин): при¬
месь Н — около 3,4; примесь I — около 3,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
месей Н и I.
Расчет предельного содержания примесей: для
каждой примеси используют концентрацию дутасте¬
рида в растворе сравнения (а).
Предельное содержание примесей:
- примесь I: не более 0,5 %;
- примеси Н: не более 0,3 %;
- неспецифицированные примеси, элюируемые по¬
сле дутастерида: для каждой примеси не более 0,10 %;
- учитываемый предел: 0,05 %.
Предельное содержание примесей:
- общее содержание примесей для испытаний А
и В: не более 1,5 %.
Вода (2.5.32). Не более 0,2 %. Определение про¬
водят из 0,100 г испытуемого образца с применением
метода выпаривания:
- температура: 180 °С ;
- время нагревания: 4 мин.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца
в платиновом тигле.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании А «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (с).
Содержание С27Н30Рв1Ч2О2 в процентах рассчиты¬
вают с учетом содержания дутастерида в ФСО дута¬
стерида.
ПРИМЕСИ
Специфицированные примеси: А, В, С, Е, Р, О, Н, I.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимитиру¬
ется общим критерием приемлемости для других/неспец-
ифицированных примесей и/или общей статьей Суб¬
станции для фармацевтического использования (2034).
Вследствие этого нет необходимости идентифицировать
эти примеси для доказательства соответствия требова¬
ниям. См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования): О.
А. 3-Оксо-4-аза-5а-андрост-1 -ен-17р-карбоновая
кислота.
В. А/,А/-Диметил-3-оксо-4-аза-5а-андрост-1 -ен-17Р-
карбоксамид.
С. Этил-3-оксо-4-аза-5а-андрост-1 -ен-17р-карбо-
ксилат.
О. Л/-[2,5-Бис(трифторметил)фенил]-3-оксо-4-
азаандрост-1,5-диен-17а-карбоксамид.
Е. А/-[2,5-Бис(трифторметил)фенил]-3-оксо-4-аза-
5а-андрост-1 -ен-17а-карбоксамид.
Р. А/-[2,5-Бис(трифторметил)фенил]-1а-хлор-3-
оксо-4-аза-5а-андростан-17р-карбоксамид.
Желатин
419
О. Л/-[2,5-Бис(трифторметил)фенил]-3-оксо-4-
азаандрост-1,5-диен-17р-карбоксамид.
Н. Л/-[2,5-Бис(трифторметил)фенил]-3-оксо-4-[3-
оксо-4-аза-5а-андрост-1 -ен-17а-карбонил]-4-аза-5а-
андрост-1-ен-17р-карбоксамид (димер 1 дутастерида).
I. Л/-[2,5-Бис(трифторметил )фенил]-3-оксо-4-[3-
оксо-4-аза-5а-андрост-1 -ен-17р-карбонил]-4-аза-5а-
андрост-1-ен-17р-карбоксамид (димер 2 дутастерида).
07/2016:0330
ЖЕЛАТИН
6е1аИпа
вЕ!-АТт
ОПРЕДЕЛЕНИЕ
Очищенный белок, полученный неполным ще¬
лочным и/или кислотным гидролизом и/или фермент¬
ным гидролизом или термическим гидролизом живот¬
ного коллагена.
Производитель должен гарантировать соответ¬
ствие желатина требованиям статьи 5.2.8. Снижение
риска передачи возбудителей губчатой энцефалопа¬
тии животных при применении медицинских лекар¬
ственных средств*
В результате гидролиза получают гелеобразую¬
щий или не образующий геля виды желатина. Требо¬
вания данной статьи распространяются на оба вида.
ОПИСАНИЕ (СВОЙСТВА)
- Гэлеобразующий вид: твердое вещество свет¬
ло-желтого или светлого желтовато-коричневого цве¬
та, обычно в виде полупрозрачных листков, кусков,
гранул или порошка. Практически нерастворим в
обычных органических растворителях; набухает в хо¬
лодной воде и при нагревании образует коллоидный
раствор, который дает при охлаждении более или ме¬
нее твердый гель;
-не образующий геля вид: светло-желтые или
белые гранулы или порошок. Растворим в холодной
или теплой воде, практически нерастворим в обычных
органических растворителях.
Различные формы водных растворов желатина
могут различаться по прозрачности и окраске. Для
конкретного применения обычно используют подходя¬
щую спецификацию по испытаниям «Прозрачность» и
«Цветность».
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Гэлеобразующий вид: А, В.
Не образующий геля вид: А, В, С.
A. К 2 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,05 мл раст¬
вора меди сульфата Р. Перемешивают и прибавляют
0,5 мл раствора натрия гидроксида разведенного Р.
Появляется фиолетовое окрашивание.
B. 0,5 г испытуемого образца помещают в про¬
бирку с внутренним диаметром около 15 мм и прибав¬
ляют 10 мл воды Р. Выдерживают в течение 10 мин,
затем нагревают при температуре 60 °С в течение
15 мин и выдерживают вертикально в течение 6 ч при
температуре 0 °С. Переворачивают пробирку. Для не
образующего геля вида — содержимое вытекает сра¬
зу, а для гелеобразующего — не сразу.
C. 0,5 г испытуемого образца помещают во фла¬
кон вместимостью 250 мл и прибавляют 10 мл воды Р и
5 мл кислоты серной Р. Флакон частично, но не полно¬
стью закрытый (например, с использованием часового
стекла) помещают в печь при температуре 105 °С на
4 ч. Охлаждают, прибавляют 200 мл воды Р и доводят
до рН 6,0—8,0 раствором 200 г/л натрия гидроксида Р.
2 мл полученного раствора помещают в пробирку и при¬
бавляют 2 мл приготовленного непосредственно перед
применением раствора 14 г/л хлорамина Р в фосфат¬
ном буферном растворе рН 6,8 Р. Перемешивают и
выдерживают в течение 20 мин, прибавляют 2 мл раст¬
вора диметиламинобензальдегида Р9. Перемешивают
и помещают в водяную баню при температуре 60 °С на
15 мин. Появляется красное окрашивание.
ИСПЫТАНИЯ
Раствор 3. 1,00 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р при
температуре около 55 °С, доводят до объема 100 мл
этим же растворителем и при этой температуре про¬
водят испытания.
рН (2.2.3). От 3,8 до 7,6. Измеряют рН раствора 5
при температуре 55 °С.
Удельная электропроводность (2.2.38). Не
более 1 мС см1 в 1,0 % растворе при температуре
(30±1,0) °С (без применения температурного компен¬
сатора).
Серы диоксид (2.5.29). Не более 0,0050 %
(50 ррт).
Пероксиды. Не более 0,0010 % (10 ррт). Опре¬
деление проводят с использованием пероксидных
тест-полосок Р.
55’. Зак. 1060.
420
Гэсударственная фармакопея Республики Беларусь
Органический окислительно-восстановитель¬
ный индикатор тест-полоски при окислении при¬
обретает синее окрашивание. Интенсивность
окраски пропорциональна количеству пероксида и
для определения его концентрации может сравни¬
ваться с цветовой шкалой, прилагаемой к тест-
полоскам.
Пригодность испытания. Погружают тест-полоску
на 1 с в эталонный раствор водорода пероксида
(2 ррт Н20^ Р таким образом, чтобы реакционная зона
была как следует смочена. Вынимают тест-полоску,
стряхивают избыток жидкости и через 15 с сравнивают
с цветовой шкалой. Цвет тест-полоски должен соответ¬
ствовать концентрации 2 ррт по шкале.
Испытание. (20,0±0,1)г испытуемого образ¬
ца помещают в лабораторный стакан и прибавляют
(80,0±0,2) мл воды Р. Перемешивают до полного ув¬
лажнения желатина и выдерживают при комнатной
температуре в течение (1—3) ч. Накрывают стакан
часовым стеклом. Если растворение неполное, то
помещают стакан в водяную баню с температурой
(65±2) °С на (20±5) мин для растворения образца.
Перемешивают содержимое стакана стеклянной па¬
лочкой до получения однородного раствора. Погружа¬
ют тест-полоску на 1 с в испытуемый раствор таким
образом, чтобы реакционная зона была должным об¬
разом смочена. Вынимают тест-полоску, стряхивают
избыток жидкости и через 15с сравнивают с цветовой
шкалой. Концентрацию, определенную по цветовой
шкале, умножают на 5 для расчета содержания перок¬
сида в испытуемом образце в ррт.
Сила (прочность) геля (число Блюм). От 80 %
до 120 % от указанного номинального значения для
гелеобразующего вида.
Сила (прочность) геля выражается в массе в
граммах, необходимой для создания такой силы, ко¬
торая при приложении к плунжеру диаметром 12,7 мм
дает погружение 4 мм в студень с концентрацией
6,67 % (м/м) и выдержанный при температуре 10 °С.
Оборудование. Текстурный анализатор или же-
лометр состоит из следующих частей:
- цилиндрического плунжера диаметром
(12,7±0,1)мм с ровной напорной поверхностью с
острыми краями;
-бутыли с внутренним диаметром (59±1)мм и
высотой 85 мм.
Настраивают прибор согласно инструкции про¬
изводителя. Параметры: расстояние 4 мм, скорость
0,5 мм/с.
Методика. 7,5 г испытуемого образца помещают
во флакон, прибавляют 105 мл воды Р. Бутыль закры¬
вают и выдерживают в течение (1—4) ч. Нагревают
в водяной бане в течение 15 мин при температуре
(65±2) °С, помешивая стеклянной палочкой. Убежда¬
ются, что раствор однородный и что весь конденсат на
внутренних стенках бутыли инкорпорирован. Охлаж¬
дают при комнатной температуре в течение 15 мин
и помещают бутыль в термостат с контролируемой
температурой (10,0±0,1) °С и приспособлением, удер¬
живающим платформу, на которой стоит бутыль, в
строго горизонтальном положении. Закрывают бутыль
резиновой пробкой и выдерживают в течение (17±1)
ч. Вынимают бутыль, быстро протирают ее внешнюю
поверхность насухо. Бутыль помещают в центр плат¬
формы прибора таким образом, чтобы плунжер со¬
прикасался с серединой образца настолько близко,
насколько это возможно. Проводят измерение.
Железо. Не более 0,0030 % (30 ррт). Атомно-аб¬
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 5,00 г испытуемого образ¬
ца помещают в коническую колбу и прибавляют 10 мл
кислоты хлористоводородной Р. Закрывают колбу и
помещают в водяную баню при температуре от 75 °С
до 80 °С на 2 ч (при необходимости для надлежащей
солюбилизации допускается набухание желатина по¬
сле прибавления кислоты и перед нагреванием, может
продлеваться время нагревания и использоваться бо¬
лее высокая температура). Охлаждают и доводят со¬
держимое колбы водой Р до массы 100,0 г.
Растворы сравнения. Готовят разведением эта¬
лонного раствора железа (8 ррт Ре) Р с использова¬
нием воды Р.
Длина волны: 248,3 нм.
Хром. Не более 0,0010% (10 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Готовят, как описано в ис¬
пытании на железо.
Растворы сравнения. Готовят разведением эта¬
лонного раствора хрома (100 ррт Сг) Р с использо¬
ванием воды Р.
Длина волны: 357,9 нм.
Цинк. Не более 0,0030 % (30 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Готовят, как описано в ис¬
пытании на железо.
Растворы сравнения. Готовят разведением эта¬
лонного раствора цинка (10 ррт 1п) Р с использова¬
нием воды Р.
Длина волны: 213,9 нм.
Потеря в массе при высушивании (2.2.32). Не
более 15,0 %. 5,000 г испытуемого образца сушат при
температуре 105 °С в течение 16 ч.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа соН: отсутствие (2.6.13).
8а1топе11а: отсутствие (2.6.13).
ХРАНЕНИЕ
В защищенном от тепла и влаги месте.
МАРКИРОВКА
Указывают силу (прочность) геля (число Блюм)
или то, что это не образующий геля вид.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Некоторые характеристики, описанные в раз¬
деле «Функционально-обусловленные характе¬
ристики», могут быть также представлены в
обязательной части частной статьи, так как
они представляют собой обязательные крите¬
рии качества. В подобных случаях перекрестная
ссылка на испытания, описанные в обязательной
части, включена в раздел «Функционально-обу¬
словленные характеристики». Контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
Желатин
421
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, од¬
нако другие методы также могут быть использо¬
ваны. В случае указания результатов конкретных
характеристик требуется указывать также и ме¬
тоды проведения испытаний.
Следующие характеристики могут быть важ¬
ны для гелеобразующего вида желатина, использу¬
емого в качестве повышающего вязкость агента,
связующего агента или для микрокапсулирования.
Сила (прочность) геля (число Блюм). Как ука¬
зано в разделе «Испытания».
Следующие характеристики могут быть важ¬
ны для желатина, используемого для производства
капсул.
#рН 6,67% (м/м) раствора желатина (2.2.3).
Определяют рН 6,67 % (м/м) раствора желатина при
температуре 60 °С.
рН 6,67 % (м/м) раствора желатина типов А и В с
разным числом Блюм (В1) должен быть следующим:
-желатин типа А (из свиных шкур) 270 В1 — от
5.0 до 5,7;
- желатин типа В (из говяжьих шкур и говяжьих
костей) 250 В1 — от 5,0 до 6,0;
-желатин типа А (из свиных шкур) 175 В1 — от
5.0 до 5,7;
- желатин типа В (из говяжьих шкур и говяжьих
костей) 150 В1 — от 5,5 до 6,0.
#Размер частиц (2.9.38). 100,0 г испытуемого
образца просеивают в течение 3 мин со скоростью
1 удар в секунду.
Не менее 90 % порошка должно пройти через
сито (5000) и остаться на сите (500).
#Прозрачность (2.2.1). Раствор 3, полученный
из желатина типов А и В, по степени мутности не дол¬
жен превышать эталон IV.
#Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона У(Ж)4.
Продолжительность растворения. Не более
25 мин. 10,0 г испытуемого образца, взвешенного с
точностью 0,01 г, помещают в лабораторный стакан,
прибавляют 100 мл воды Р с температурой от 15 °С
до 18 °С и оставляют набухать при этой же темпера¬
туре в течение 30 мин. Затем стакан с содержимым
помещают в термостат с температурой (40±1) °С и
выдерживают при помешивании до полного раство¬
рения желатина.
Время, прошедшее с момента доведения темпе¬
ратуры в стакане до 40 °С до полного растворения
желатина, принимают за продолжительность раство¬
рения.
#Динамическая вязкость. Динамическая вяз¬
кость 6,67 % (м/м) раствора желатина при 60 °С в
пересчете на желатин с содержанием влаги 11,5%
должна быть следующая:
-желатин типа А (из свиных шкур) 270 В1 — от
4,2 до 4,8 мПа с;
- желатин типа В (из говяжьих шкур и говяжьих
костей) 250 В1 — от 4,2 до 4,8 мПа с;
-желатин типа А (из свиных шкур) 175 В1 — от
2,7 до 3,2 мПа с;
- желатин типа В (из говяжьих шкур и говяжьих
костей) 150 В! — от 3,5 до 4,0 мПа с.
Вязкость 6,67 % (м/м) раствора желатина
определяется при температуре 60 °С измерением
времени истечения 100 мл раствора через
стандартную пипетку.
Вязкость желатина определяется как динамиче¬
ская вязкость 6,67 % (м/м) раствора желатина в воде
при температуре 60 °С, выраженная в мПа с.
Для калибровки при температуре 60 °С
используют два стандартных калибровочных масла
с вязкостью в пределах (2—10) сСт (1 сСт = 0,01 Ст).
Вязкость масел должна различаться как минимум в
два раза.
Оборудование:
1. Пипетка вместимостью 100 мл с определен¬
ным диаметром входного и выходного отверстия и от¬
меткой нижнего уровня на стекле (рисунок #0330.-1).
Рисунок #0330.-1. Пипетка
для определения вязкости
Могут использоваться другие аналогичные типы
вискозиметра (например, 11-образный вискозиметр,
рисунок #0330.-2).
Рисунок #0330.-2. С1-образный вискозиметр
422
Гэсударственная фармакопея Республики Беларусь
2. Термостатическая баня для пипетки с возмож¬
ностью для термостатического перемешивания и тем¬
пературой (60±0,1) °С.
3. Точный термометр (ртутный или электронный)
с ценой деления 0,1 °С, с длинным тонким корпусом
для измерения температуры внутри пипетки.
4. Секундомер с точностью до 0,1 с.
5. Термостатируемая водяная баня для испытуе¬
мого образца, поддерживающая температуру на уров¬
не (65±2) °С.
6. Весы с порогом чувствительности 0,01 г.
Испытание. (7,50±0,01)г испытуемого образца
помещают в коническую колбу вместимостью 150 мл,
прибавляют (105±0,2) мл воды Р, перемешивают до
полного смачивания желатина, закрывают колбу ре¬
зиновой пробкой или часовым стеклом и выдержива¬
ют при комнатной температуре в течение (1,5—2) ч.
Колбу с набухшим желатином помещают в водяную
баню с температурой 65 °С на время, не превыша¬
ющее 20 мин, до растворения образца. Для полного
растворения желатина и получения гомогенного раст¬
вора колбу периодически осторожно, не поднимая
пену, встряхивают (при закрытии резиновой пробкой)
или помешивают стеклянной палочкой (при закры¬
тии часовым стеклом). Когда температура раствора
достигнет (61—62) °С и образец полностью раство¬
рится, термометр достают и раствор быстро, исклю¬
чая возможность захватывания воздуха, наливают в
пипетку для определения вязкости до уровня на 1 см
выше верхней отметки, удерживая раствор на вы¬
ходном отверстии пальцем. Колбу с оставшимся рас¬
твором помещают непосредственно под выходное
отверстие пипетки. Проверяют температуру раствора
желатина в пипетке термометром. Когда температура
достигнет уровня 60 °С, производят замер времени
истечения раствора от верхней до нижней отметки.
Регистрируют время, необходимое для истечения че¬
рез пипетку 100 мл раствора желатина.
Перед каждым из трех измерений, а также перед
использованием пипетку необходимо промыть 25 мл
воды Р, нагретой до температуры 62 °С, и полностью
высушить.
Динамическую вязкость раствора р в мПа с рас¬
считывают по формуле:
А1-
где:
^ — время истечения раствора, с;
р — плотность 6,67 % раствора желатина, 60 °С,
равная 1,001 г/см3;
А, В — константы пипетки.
Калибровка. Калибровка пипетки производится
с использованием двух стандартных масел с раз¬
личной вязкостью (для определения констант А и
В). Перед использованием пипетка должна быть
тщательно вымыта и высушена с использованием
ацетона Р.
Каждое масло нагревают при перемешивании
в бане с постоянной температурой (63—64) °С,
переносят в пипетку, удерживая выходное отверстие
пальцем до тех пор, пока температура масла не
станет равной 60 °С. Измеряют секундомером время
истечения масла от верхней до нижней отметки.
Повторяют измерение не менее трех раз для
каждого масла. При смене масла пипетку каждый
раз промывают полностью соответствующим
органическим растворителем для удаления масла,
затем промывают ацетоном Р и высушивают.
Константы А и В рассчитывают по формулам:
В
и*2-{П2*,-П:*2)
г2 -I2
12 Ч
где:
р1 — кинематическая вязкость калибровочного
масла с низкой вязкостью, сСт;
р2 — кинематическая вязкость калибровочного
масла с высокой вязкостью, сСт;
^ — среднее время истечения калибровочного
масла с низкой вязкостью, с;
\2 — среднее время истечения калибровочного
масла с высокой вязкостью, с.
Коррекция результатов с учетом влаги. Вяз¬
кость 6,67 % (м/м) раствора желатина при 60 °С в
пересчете на желатин с содержанием влаги 11,5%
рассчитывают по формуле:
р1 = р + р0,02(М, -11,5),
где:
П — вязкость раствора при влажности М7;
р1 — вязкость раствора при влажности 11,5 %.
Примечания
1. Допускается производить определение динами¬
ческой вязкости раствора желатина с использованием
капиллярного вискозиметра с внутренним диаметром
трубки от 0,99 до 1,47 мм (см. рисунок 2.2.9.-1.).
2. Допускается динамическую вязкость раствора
желатина р в мПа с рассчитывать по формуле:
П = 1рк,
где:
I — время истечения раствора, с;
р — плотность раствора при 60 °С, г/см3;
к— постоянная вискозиметра, мм2/с2.
#Падение динамической вязкости 12,5 % (м/м)
раствора желатина за 17 часов. Не более 24 %.
12,50 г испытуемого образца взвешивают в ко¬
ническую колбу вместимостью 250 мл, прибавля¬
ют необходимое количество воды Р для получения
12,5% (м/м) раствора желатина, закрывают колбу
пробкой и выдерживают раствор при комнатной
температуре в течение (1,5—2) ч для набухания же¬
латина. Колбу с набухшим желатином помещают в
водяную баню с температурой 65 °С до растворения
образца, контролируя, чтобы температура испытуе¬
мого раствора не превышала 60 °С, периодически
встряхивая или помешивая раствор стеклянной па¬
лочкой (не поднимая пену) для достижения полной
равномерности концентрации. Время, необходимое
для полного растворения желатина, не должно пре¬
вышать 20 мин. Когда температура раствора дости¬
гает (61—62) °С и образец полностью растворится,
термометр достают и раствор быстро, исключая
возможность захватывания воздуха, перемещают
в пипетку для определения вязкости до уровня на
1 см выше верхней отметки, удерживая раствор на
выходном отверстии пальцем. Колбу с оставшим¬
ся раствором помещают непосредственно под вы¬
ходное отверстие пипетки. Проверяют температуру
раствора желатина в пипетке термометром. Когда
температура достигнет уровня 60 °С, измеряют вре-
Желатин
423
мя истечения раствора от верхней до нижней отмет¬
ки. Регистрируют время, необходимое для истече¬
ния через пипетку 100 мл раствора желатина.
Перед каждым измерением, а также перед ис¬
пользованием пипетку необходимо промыть 25 мл
воды Р, нагретой до 62 °С, и полностью высушить.
Динамическую вязкость раствора (/7, мПас) рас¬
считывают по формуле:
а*-вА
П = —,
Р
где:
(— время истечения раствора, с;
р — плотность 6,67 % (м/м) раствора желатина,
60 °С, равная 1,001 г/см3;
А, В — константы пипетки.
Калибровка. Калибровка пипетки производится с
использованием двух стандартных масел с различной
вязкостью (для определения констант А и В). Перед
использованием пипетка должна быть тщательно вы¬
мыта и высушена с использованием ацетона Р.
Каждое масло нагревают при перемешивании
на бане с постоянной температурой (63—64) °С,
переносят в пипетку, удерживая выходное отверстие
пальцем до тех пор, пока температура масла не
станет равной 60 °С. Измеряют секундомером
время истечения масла от верхней до нижней
отметки. Повторяют измерение не менее трех
раз для каждого масла. При смене масла пипетку
каждый раз промывают полностью соответствующим
органическим растворителем для удаления масла,
затем промывают ацетоном Р и высушивают.
Константы Айв рассчитывают по формулам:
^ * ^2 * (0г ‘ ~ Па • ^2 )
±2 +2 »
где:
— кинематическая вязкость калибровочного
масла с низкой вязкостью, сСт;
г)2 — кинематическая вязкость калибровочного
масла с высокой вязкостью, сСт;
/1 — среднее время истечения калибровочного
масла с низкой вязкостью, с;
12 — среднее время истечения калибровочного
масла с высокой вязкостью, с.
Коррекция результатов с учетом влаги. Вяз¬
кость 12,5% (м/м) раствора желатина при 60 °С в
пересчете на желатин с содержанием влаги 11,5%
рассчитывают по формуле:
Па =П + П‘0,02• (А/, -11,5),
где:
р — вязкость раствора при влажности Ц;
— вязкость раствора при влажности 11,5 %.
Помещают испытуемый раствор в термостат с
температурой 60 °С на 17 ч. Затем еще раз опреде¬
ляют динамическую вязкость раствора, как указано
выше.
Падение динамической вязкости (X) в процентах
вычисляют по формуле:
х_(п,-п2у™ о
где:
гу1 — вязкость раствора желатина 12,5 % до вы¬
держки в термостате, мПа с;
г\2 — вязкость раствора желатина 12,5 % после
выдержки в термостате, мПа с.
Примечания
1. Допускается производить определение
динамической вязкости раствора желатина с ис¬
пользованием капиллярного вискозиметра с вну¬
тренним диаметром трубки от 0,99 до 1,47 мм (см.
рисунок 2.2.9.-1).
2. Допускается динамическую вязкость раст¬
вора желатина р в мПа с рассчитывать по формуле:
П = 1рк,
где:
I — время истечения раствора, с;
р — плотность раствора при 60 °С, г/см3;
к— постоянная вискозиметра, мм2/с*.
#Общая зола (2.4.16). Не более 2 %.
#Мышьяк (2.4.2, метод А). Не более 0,0001 %
(1 ррт). К 1,0 г испытуемого образца прибавляют
смесь 10 мл воды Р, 2,5 мл кислоты серной Р,
2,5 мл кислоты азотной Р и небольшой избыток
бромной воды Р. Выдерживают в течение 30 мин,
затем кипятят с обратным холодильником в тече¬
ние 1 ч. Полученный раствор должен выдерживать
испытание на мышьяк.
Вместо ртутно-бромидной бумаги Р допуска¬
ется использование бумаги, пропитанной раство¬
ром ртути (II) хлорида Р (беззольную фильтро¬
вальную бумагу смачивают насыщенным спирто¬
вым раствором ртути (II) хлорида Р, дают спирту
испариться, повторяют это 4—5 раз, после чего
бумагу высушивают при комнатной температуре;
хранят в плотно укупоренных контейнерах).
#Желатиноразжижающие бактерии. Нали¬
чие желатиноразжижающих бактерий в 1 г желати¬
на: не допускается.
20,0 г желатина помещают в коническую кол¬
бу вместимостью 250 мл, прибавляют 180 мл сте¬
рильного фосфатного буферного раствора рН
7,0 Р и оставляют набухать при температуре от
5 °С до 10 °С в течение (1,0—1,5) ч. Колбу с набух¬
шим желатином помещают в водяную баню, нагре¬
тую до температуры (40±2) °С, выдерживают в ней
до полного растворения желатина. Затем в колбу
помещают стерильный магнит и ставят на магнит¬
ную мешалку, нагретую до (38±2) °С, и перемеши¬
вают в течение (5—10) мин. Магнит стерилизуют
кипячением в колбе в течение (15—20) мин.
По 10,0 мл полученного раствора желатина
вносят в две чашки Петри. Желатин распределяют
по дну чашек легкими вращательными движениями
и выдерживают при температуре (24±1) °С. Через
48 ч чашки с желатином просматривают. Колонии
желатиноразжижающих бактерий имеют вид ма¬
леньких прозрачных пузырьков за счет разжижения
желатина. При выдерживании чашек с желатином
более 4 сут колония увеличивается. При наклоне
чашки на месте колонии желатин сползает.
Через 96 ч колонии желатиноразжижающих
бактерий подсчитывают в каждой чашке Петри.
За окончательный результат анализа принимают
среднее арифметическое значение двух результа¬
тов подсчета.
424
Гэсударственная фармакопея Республики Беларусь
07/2016:2340
ЖЕЛЕЗА (II) СУЛЬФАТ
ВЫСУШЕННЫЙ
Ре/гоз/ зиКаз сУез/сса/из
РЕЯЯО(/5 81ЛРАТЕ, ОЯ/ЕО
Ре304 М.м. 151,9
ОПРЕДЕЛЕНИЕ
Гидратированный железа (II) сульфат, часть ги-
дратной воды которого удалена при высушивании.
Содержание: не менее 86,0 % и не более 90,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Серовато-белый порошок.
Медленно, но легко растворим в воде, очень лег¬
ко растворим в кипящей воде, практически нераство¬
рим в 96 % спирте.
При окислении на влажном воздухе становится
коричневым.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
B. Испытуемый образец дает реакцию (а) на же¬
лезо (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 2,00 г испытуемого образца раство¬
ряют в 5 % (об/об) растворе кислоты азотной, сво¬
бодной от свинца, Р и доводят до объема 100,0 мл
этим же растворителем.
рН (2.2.3). От 3,0 до 4,0.1,0 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 20 мл этим же раство¬
рителем.
Хлориды (2.4.4). Не более 0,0300 % (300 ррт).
2,5 г испытуемого образца растворяют в воде Р, при¬
бавляют 0,5 мл кислоты серной разведенной Р и до¬
водят водой Р до объема 50 мл. 3,3 мл полученного
раствора доводят водой Р до объема 10 мл и прибав¬
ляют 5 мл кислоты азотной разведенной Р. Эталон
готовят с использованием смеси из 10 мл эталонного
раствора хлорида (5 ррт С1) Р и 5 мл кислоты азот¬
ной разведенной Р. В испытании используют 0,15 мл
раствора серебра нитрата Р2.
Хром. Не более 0,0100 % (100 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Готовят путем разведения
эталонного раствора хрома (100 ррт Сг) Р 5 % (об/об)
раствором кислоты азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения хрома с использованием полосы
пропускания предпочтительно 1 нм.
Длина волны: 357,9 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Медь. Не более 0,0050 % (50 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Готовят путем разведе¬
ния эталонного раствора меди (0,1 % Си) Р 5 %
(об/об) раствором кислоты азотной, свободной от
свинца, Р.
Источник излучения: лампа с полым катодом
для определения меди с использованием полосы про¬
пускания предпочтительно 1 нм.
Длина волны: 324,7 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Ионы железа (III). Не более 0,5 %.
В колбе с притертой пробкой растворяют 5,00 г
испытуемого образца в смеси из 10 мл кислоты
хлористоводородной Р и 100 мл воды, свободной
от углерода диоксида, Р. К полученному раство¬
ру прибавляют 3 г калия йодида Р, закрывают колбу
и выдерживают в темноте в течение 5 мин. Титруют
выделившийся йод 0,1 М раствором натрия тио¬
сульфата, используя в качестве индикатора 0,5 мл
раствора крахмала Р, прибавляемого в конце титро¬
вания. Параллельно проводят контрольный опыт. На
титрование должно израсходоваться не более 4,5 мл
0,1 М раствора натрия тиосульфата.
Марганец. Не более 0,1 %.
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. 1,0 мл раствора 3 дово¬
дят 5 % (об/об) раствором кислоты азотной, свобод¬
ной от свинца, Р до объема 20,0 мл.
Растворы сравнения. Готовят путем разведения
эталонного раствора марганца (1000 ррт Мп) Р 5 %
(об/об) раствором кислоты азотной, свободной от
свинца, Р.
Источник излучения: лампа с полым катодом
для определения марганца с использованием полосы
пропускания предпочтительно 1 нм предпочтительно
1 нм.
Длина волны: 279,5 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Никель. Не более 0,0100 % (100 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Готовят путем разведе¬
ния эталонного раствора никеля (10 ррт М) Р 5 %
(об/об) раствором кислоты азотной, свободной от
свинца, Р.
Источник излучения: лампа с полым катодом
для определения никеля с использованием полосы
пропускания предпочтительно 1 нм.
Длина волны: 232,0 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Цинк. Не более 0,0100 % (100 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Готовят путем разведе¬
ния эталонного раствора цинка (100 ррт 1п) Р 5 %
(об/об) раствором кислоты азотной, свободной от
свинца, Р.
Источник излучения: лампа с полым катодом
для определения цинка с использованием полосы
пропускания предпочтительно 1 нм.
Длина волны: 213,9 нм.
Атомизатор: воздушно-ацетиленовое пламя.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Железа сульфат гептагидрат
425
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,5 г натрия гидрокарбоната Р растворяют в
смеси из 150 мл воды Р и 10 мл кислоты серной Р.
После прекращения выделения пузырьков газа к по¬
лученному раствору прибавляют 0,140 г испытуемого
образца и растворяют при осторожном встряхивании.
Прибавляют 0,1 мл ферроина Р и титруют 0,1 М рас-
твором аммония церия нитрата до исчезновения
красного окрашивания.
1 мл 0,1 М раствора аммония церия нитрата
соответствует 15,19мг Ре304.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0083
ЖЕЛЕЗА СУЛЬФАТ ГЕПТАГИДРАТ
Геггоз/ зи№аз Ьер1аЬус1пси8
РЕ К КО и 8 8Ш.РАТЕ НЕРТАНУОКАТЕ
РеЗО„ • 7Н20 М.м. 278,0
[7782-63-0]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 105,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический порошок светло-зеленого цвета
или голубовато-зеленые кристаллы. Выветривается
на воздухе.
Легко растворим в воде, очень легко растворим в
кипящей воде, практически нерастворим в 96 % спирте.
Железа сульфат гептагидрат окисляется во влаж¬
ном воздухе, окрашиваясь в коричневый цвет.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
B. Испытуемый образец дает реакцию (а) на же¬
лезо (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 4,0 г испытуемого образца раство¬
ряют в 5 % (об/об) растворе кислоты азотной, сво¬
бодной от свинца, Р и доводят до объема 100,0 мл
этим же растворителем.
рН (2.2.3). От 3,0 до 4,0.1,0 г испытуемого образца
растворяют в воде, свободной от углерода диоксида, Р
и доводят до объема 20 мл этим же растворителем.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 5 доводят водой Р до объема 10 мл и
прибавляют 5 мл кислоты азотной разведенной Р.
Полученный раствор должен выдерживать испытание
на хлориды. Эталон готовят с использованием сме¬
си из 2 мл воды Р, 5 мл кислоты азотной разведен¬
ной Р и 8 мл эталонного раствора хлорида (5 ррт
С1) Р. При испытании используют 0,15 мл раствора
серебра нитрата Р2.
Хром. Не более 0,0050 % (50 ррт). Атомно-аб¬
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Растворы сравнения го¬
товят разведением эталонного раствора хрома
(100 ррт Сг) Р 5 % (об/об) раствором кислоты азот¬
ной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения хрома, предпочтительно используя
ширину щели 1 нм.
Длина волны: 357,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Медь. Не более 0,0050 % (50 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Растворы сравнения го¬
товят разведением эталонного раствора меди
(0,1 % Си) Р 5 % (об/об) раствором кислоты азотной,
свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения меди, предпочтительно используя
ширину щели 1 нм.
Длина волны: 324,7 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Железа (III) ионы. Не более 0,3 %. 5,00 г испы¬
туемого образца помещают в колбу с притертой сте¬
клянной пробкой и растворяют в смеси из 10 мл кис¬
лоты хлористоводородной Р и 100 мл воды, свобод¬
ной от углерода диоксида, Р, прибавляют 3 г калия
йодида Р, колбу закрывают и выдерживают в темном
месте в течение 5 мин. Выделившийся йод титруют
0,1 М раствором натрия тиосульфата, используя
в качестве индикатора 0,5 мл раствора крахмала Р,
который прибавляют в конце титрования.
Параллельно проводят контрольный опыт.
На титрование испытуемого раствора должно
быть использовано не более 2,7 мл 0,1 М раствора
натрия тиосульфата с учетом количества титранта,
использованного в контрольном опыте.
Марганец. Не более 0,1 %. Атомно-абсорбцион¬
ная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 1,0 мл раствора 3 дово¬
дят 5 % (об/об) раствором кислоты азотной, свобод¬
ной от свинца, Р до объема 20,0 мл.
Растворы сравнения. Растворы сравнения го¬
товят разведением эталонного раствора марган¬
ца (1000 ррт Мп) Р 5 % (об/об) раствором кислоты
азотной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения марганца, предпочтительно исполь¬
зуя ширину щели 1 нм.
Длина волны: 279,5 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Никель. Не более 0,0050 % (50 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Раствор 3.
Растворы сравнения. Растворы сравнения го¬
товят разведением эталонного раствора никеля
(10 ррт Щ Р 5 % (об/об) раствором кислоты азот¬
ной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения никеля, предпочтительно используя
ширину щели 1 нм.
Длина волны: 232,0 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Цинк. Не более 0,0050 % (50 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Раствор 3.
426
Гэсударственная фармакопея Республики Беларусь
Растворы сравнения. Растворы сравнения го¬
товят разведением эталонного раствора цинка
(100 ррт 2.п) Р 5 % (об/об) раствором кислоты азот¬
ной, свободной от свинца, Р.
Источник излучения: лампа с полым катодом
для определения цинка, предпочтительно используя
ширину щели 1 нм.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
#Мышьяк (2.4.2, метод А). Не более 0,0003 %
(3 ррт). 1,0 г испытуемого образца должен выдер¬
живать испытание на мышьяк. Эталон готовят с ис¬
пользованием 3 мл эталонного раствора мышьяка
(1 ррт Аз) Р и 22 мл воды Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,5 г натрия гидрокарбоната Р растворяют в
смеси из 150 мл воды Р и 10 мл кислоты серной Р.
По окончании бурного выделения пузырьков к раство¬
ру прибавляют 0,500 г испытуемого образца и раство¬
ряют, осторожно перемешивая. К полученному рас¬
твору прибавляют 0,1 мл ферроина Р и титруют 0,1 М
раствором аммония церия нитрата до исчезнове¬
ния красного окрашивания.
1 мл 0,1 М раствора аммония церия нитрата
соответствует 27,80 мг Ре304 • 7Н20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:1515
ЖЕЛЕЗА ХЛОРИД ГЕКСАГИДРАТ
Регп сЫопдит ЬехаЬуйпсит
РЕктс снижюЕ нехануокате
РеС13 • 6Н20 М.м. 270,3
[10025-77-1]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 102,0 %
РеС13-6Н20.
ОПИСАНИЕ (СВОЙСТВА)
Кристаллическая масса или оранжево-желтые
или коричневато-желтые кристаллы. Очень гигроско¬
пичен.
Очень легко растворим в воде и 96 % спирте, лег¬
ко растворим в глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
B. Испытуемый образец дает реакцию (с) на же¬
лезо (2.3.1).
ИСПЫТАНИЯ
Раствор 3. Юг испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 100 мл этим же растворителем.
Кислотность. 3,0 г калия фторида Р помеща¬
ют в подходящий полиэтиленовый контейнер, раст¬
воряют в 15 мл воды Р и титруют 0,1 М раствором
натрия гидроксида, используя 0,1 мл раствора
фенолфталеина Р в качестве индикатора, до по¬
явления розового окрашивания. К полученному
раствору прибавляют 10 мл раствора 3, выдер¬
живают в течение 3 ч и фильтруют. Используют
12,5 мл полученного фильтрата. При прибавлении
не более 0,30 мл 0,1 М раствора натрия гидрок¬
сида должно появиться розовое окрашивание.
Свободный хлор. Нагревают 5 мл раствора 3.
Выделяющиеся пары не должны окрашивать йодкрах-
мальную бумагу Р в синий цвет.
Сульфаты (2.4.13). Не более 0,0100%
(100 ррт). 15 мл раствора 3 нагревают на водяной
бане и прибавляют 5 мл раствора натрия гидрок¬
сида концентрированного Р. Охлаждают и филь¬
труют. Полученный фильтрат нейтрализуют кисло¬
той хлористоводородной Р1 по синей лакмусовой
бумаге Р и выпаривают до объема 15 мл.
Железа (II) ионы. Не более 0,0050 % (50 ррт).
К 10 мл раствора 3 прибавляют 1мл воды Р,
0,05 мл раствора калия феррицианида Р и 4 мл
кислоты фосфорной Р. Через 10 мин синяя окра¬
ска полученного раствора должна быть не интен¬
сивнее окраски раствора, приготовленного парал¬
лельно с использованием 10 мл воды Р и 1 мл све¬
жеприготовленного раствора 0,250 г/л железа (II)
сульфата Р.
Тяжелые металлы (2.4.8, метод А). Не бо¬
лее 0,0050 % (50 ррт). 1,0 г испытуемого образца
растворяют в 10 мл кислоты хлористоводород¬
ной Р1, прибавляют 2 мл раствора водорода пе¬
роксида концентрированного Р и выпаривают до
объема 5 мл. Охлаждают, доводят кислотой хло¬
ристоводородной Р1 до объема 20 мл и перено¬
сят полученный раствор в делительную воронку.
Встряхивают трижды, каждый раз в течение 3 мин,
с метилизобутилкетоном Р1 порциями по 20 мл.
Нижний слой отделяют, выпаривают до половины
объема и доводят водой Р до объема 25 мл. 10 мл
полученного раствора нейтрализуют раствором
аммиака разведенным Р1 по красной лакмусо¬
вой бумаге Р и доводят водой Р до объема 20 мл.
12 мл полученного раствора должны выдерживать
испытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца помещают в ко¬
ническую колбу с притертой стеклянной пробкой,
растворяют в 20 мл воды Р, прибавляют 10 мл кис¬
лоты хлористоводородной разведенной Р, 2 г ка¬
лия йодида Р, закрывают пробкой и выдерживают в
защищенном от света месте в течение 1 ч. Титруют
0,1 М раствором натрия тиосульфата, исполь¬
зуя 5 мл раствора крахмала Р в качестве индика¬
тора, который прибавляют в конце титрования.
1 мл 0,1 М раствора натрия тиосульфата со¬
ответствует 27,03 мг РеС13-6Н20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Зопиклон
427
07/2016:0462
ЖИР ТВЕРДЫЙ
Ас/ерз зоНс1из
НАНО РАТ
ОПРЕДЕЛЕНИЕ
Смесь триглицеридов, диглицеридов и моногли¬
церидов, которые могут быть получены этерификаци¬
ей жирных кислот природного происхождения глице¬
рином или переэтерификацией природных жиров.
Каждый тип твердого жира характеризуется сво¬
ими номинальными значениями температуры плавле¬
ния, гидроксильного числа, числа омыления.
Твердый жир не содержит добавок.
ОПИСАНИЕ (СВОЙСТВА)
Хрупкая воскообразная масса белого или почти
белого цвета.
Практически нерастворим в воде, мало раство¬
рим в этаноле безводном.
При нагревании до температуры 50 °С субстан¬
ция плавится с образованием бесцветной или слегка
желтоватой жидкости.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в этиленхлориде Р и доводят до объ¬
ема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: эфир Р — этиленхлорид Р
(10:90, об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают парами
йода до проявления пятен и просматривают при днев¬
ном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно с Рг около 0,6, со¬
ответствующее триглицеридам (#8, 1); могут обнару¬
живаться пятна, соответствующие 1,3-диглицеридам
(Рз{ 0,5), 1,2-диглицеридам 0,3) и 1-моноглицери-
дам (Яз, 0,05). Если пятна моно- и диглицеридов не
обнаруживаются, для подтверждения подлинности
дополнительно проводят испытания «Температура
плавления» и «Гидроксильное число», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Щелочные примеси. 2,00 г испытуемого об¬
разца растворяют в смеси из 1,5 мл 96 % спирта Р и
3,0 мл эфира Р, прибавляют 0,05 мл раствора бром-
фенолового синего Р. При прибавлении не более
0,15 мл 0,01 М раствора кислоты хлористоводород¬
ной должно появиться желтое окрашивание раствора.
Температура плавления (2.2.15). От 30 °С до
45 °С. Температура плавления не должна отличаться
более чем на 2 °С от номинального значения. Испыту¬
емый образец расплавляют, заполняют капилляр и вы¬
держивают при температуре ниже 10 °С в течение 24 ч.
Кислотное число (2.5.1). Не более 0,5. 5,0 г ис¬
пытуемого образца растворяют в 50 мл предписанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). Не более
50. Значение гидроксильного числа не должно отли¬
чаться от номинального значения более чем на 5 еди¬
ниц. Если номинальное значение гидроксильного числа
меньше 5, педроксильное число должно быть не более 5.
Йодное число (2.5.4, метод А). Не более 3.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 3.
Число омыления (2.5.6). От 210 до 260. Опреде¬
ление проводят из 2,0 г испытуемого образца. Число
омыления не должно отличаться более чем на 5 % от
номинального значения.
Неомыляемые вещества (2.5.7). Не более 0,6 %.
Определение проводят из 5,0 г испытуемого образца.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Общая зола (2.4.16). Не более 0,05 %. Опреде¬
ление проводят из 2,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте #при температуре
от 2 °С до 8 °С#.
МАРКИРОВКА
Указывают:
- номинальное значение температуры плавления;
- номинальное значение гидроксильного числа;
- номинальное значение числа омыления.
07/2016:1060
ЗОПИКЛОН
2ор'1с1опит
ЮР1С1-ОМЕ
С17Н17С1Ы603 М.м. 388,8
[43200-80-2]
ОПРЕДЕЛЕНИЕ
(5Я5)-6-(5-Хлорпиридин-2-ил)-7-оксо-6,7-дигидро-
5/-/-пирроло[3,4-Ь]пиразин-5-ил-4-метилпиперазин-1-
карбоксилат.
Содержание: не менее 98,5 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, умеренно растворим в ацето¬
не, практически нерастворим в 96 % спирте. Раство¬
ряется в разведенных минеральных кислотах.
Температура плавления: около 177 °С с разло¬
жением.
428
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в растворе 3,5 г/л кислоты хлори¬
стоводородной Р и доводят до объема 100,0 мл этим
же растворителем. 2,0 мл полученного раствора до¬
водят раствором 3,5 г/л кислоты хлористоводород¬
ной Р до объема 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 303 нм.
Удельный показатель поглощения в максимуме:
от 340 до 380.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО зопиклона #или спектр, пред¬
ставленный на рисунке #1060.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метиленхлориде Р и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО зопиклона раст¬
воряют в метиленхлориде Р и доводят этим же раст¬
ворителем до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ вР254 Р■
Подвижная фаза: триэтиламин Р — аце¬
тон Р — этилацетат Р (2:50:50, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в диметилформамиде Р и доводят до объема
20.0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее пятого эталона шкалы
наиболее подходящего цвета.
Угол оптического вращения (2.2.7). От -0,05°
до +0,05°. 10,0 мл раствора 5 доводят диметилфор-
мамидом Р до объема 50,0 мл.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 3,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (с). 4,0 мг ФСО зопиклона
оксида (примесь А) растворяют в подвижной фазе и
доводят этим же растворителем до объема 10,0 мл.
К 10,0 мл полученного раствора прибавляют 1,0 мл
испытуемого раствора и доводят подвижной фазой
до объема 100,0 мл.
Рисунок #1060.-1. Инфракрасный спектр ФСО зопиклона
Ибупрофен
429
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 30 °С;
- подвижная фаза: смешивают 38 объемов аце¬
тонитрила Р и 62 объема раствора, содержащего
8,1 г/л натрия лаурилсульфата Р и 1,6 г/л натрия ди¬
гидрофосфата Р, доведенного до рН 3,5 10 % (об/об)
раствором кислоты фосфорной Р\
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 303 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания зопиклона.
Время удерживания: зопиклон — от 27 мин до
31 мин; при необходимости корректируют содержание
ацетонитрила в подвижной фазе (при увеличении кон¬
центрации уменьшается время удерживания).
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 3,0 между пиками при¬
меси А и зопиклона; при необходимости доводят под¬
вижную фазу до рН 4,0 10 % (об/об) раствором кисло¬
ты фосфорной Р
Предельное содержание примесей:
- примеси А, В, С: на хроматограмме испытуемо¬
го раствора площади пиков, соответствующих приме¬
сям А, В и С, не должны превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а)
(0,3 %) и площади не более двух из таких пиков могут
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь) (0,1 %).
2-Пропанол. Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 5 мл этано¬
ла Р1 доводят этиленхлоридом Р до объема 100 мл.
1 мл полученного раствора доводят этиленхлори¬
дом Р до объема 10 мл.
Испытуемый раствор. 0,25 г испытуемого об¬
разца растворяют в этиленхлориде Р, прибавляют
0,5 мл раствора внутреннего стандарта и доводят
этиленхлоридом Р до объема 5,0 мл.
Раствор сравнения. 4,5 мл 2-пропанола Р дово¬
дят этиленхлоридом Р до объема 100,0 мл. К 1,0 мл
полученного раствора прибавляют 10,0 мл раствора
внутреннего стандарта и доводят этиленхлоридом Р
до объема 100,0 мл.
Условия хроматографирования:
-колонка кварцевая капиллярная длиной 10 м и
внутренним диаметром около 0,53 мм, покрытая сло¬
ем сополимера стирол-дивинилбензола Р (толщина
слоя 20 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 4 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
50
5—10
сл
0
1
о
10—14
70
14—20,5
70 -> 200
20,5—27,5
200
Блок ввода
проб
150
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Содержание 2-пропанола в процентах (м/м) рас¬
считывают с учетом его плотности, равной 0,785 г/мл
при температуре 20 °С.
Предельное содержание примесей:
- 2-пропанол: не более 0,7 % (м/м).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в смеси
из 10 мл кислоты уксусной безводной Р и 40 мл ук¬
сусного ангидрида Р и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 38,88 мг С17Н17С11\1603.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
A. (5Р5)-6-(5-Хлорпиридин-2-ил)-7-оксо-6,7-диги-
дро-5/-/-пирроло[3,4-Ь]пиразин-5-ил-4-метилпиперазин-
1-карбоксилата 4-оксид (зопиклона оксид).
B. Р-ОН и энантиомер: (7Р5)-6-(5-Хлорпиридин-2-ил)-
7-гид рокси-6,7-дигидро-5Н-пирроло[3,4-Ь]пиразин-5-он.
C. Р-Н: 6-(5-Хлорпиридин-2-ил)-6,7-дигидро-5Н-
пирроло[3,4-Ь]пиразин-5-он.
07/2016:0721
ИБУПРОФЕН
1Ьирго(епит
1ВЧРКОРЕЫ
^цН1В02
[15687-27-1]
и энантиомер
М.м. 206,3
430
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
(2Я5)-2-[4-(2-Метилпропил)фенил]пропановая
кислота.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, в метаноле и в метиленхлориде. Рас¬
творяется в разведенных растворах гидроксидов и
карбонатов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 75 °С до
78 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в растворе 4 г/л натрия гидрокси¬
да Р и доводят до объема 100,0 мл этим же раство¬
рителем.
Диапазон длин волн: от 240 нм до 300 нм. Ис¬
пользуют спектрофотометр с шириной щели 1,0 нм и
скоростью сканирования не более 50 нм/мин.
Максимумы поглощения: при 264 нм и 272 нм.
Плечо: при 258 нм.
Отношение оптических плотностей:
- — от 1 «20 Д° 1.30;
- А^А^д — от 1,00 до 1,10.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ибупрофена #или спектр, пред¬
ставленный на рисунке #0721.-1#.
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в метиленхлориде Р и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения. 50 мг ФСО ибупрофена
растворяют в метиленхлориде Р и доводят до объ¬
ема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная безводная Р—
этилацетат Р— гексан Р (5:24:71, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре 120 °С в течение
30 мин.
Проявление: пластинку слегка опрыскивают
раствором 10 г/л калия перманганата Р в кислоте
серной разведенной Р, нагревают при температуре
120 °С в течение 20 мин и просматривают в ультра¬
фиолетовом свете при длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Угол оптического вращения (2.2.7). От -0,05°
до +0,05°. 0,50 г испытуемого образца растворяют в
метаноле Р и доводят до объема 20,0 мл этим же
растворителем.
Волновое число (см'1)
Рисунок #0721 .-1. Инфракрасный спектр ФСО ибупрофена
Ибупрофен
431
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 2 мл ацетонитрила Р и доводят
подвижной фазой А до объема 10,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). 1,0 мл ФСО ибупрофе-
на примеси В доводят ацетонитрилом Р1 до объема
10.0 мл (раствор А). 20 мг ФСО ибупрофена растворяют
в 2 мл ацетонитрила Р1, прибавляют 1,0 мл раствора
А и доводят подвижной фазой А до объема 10,0 мл.
Раствор сравнения (с). Содержимое контейнера
ФСО ибупрофена для идентификации пиков (содер¬
жит примеси А, и N1) растворяют в 1 мл ацетонитри¬
ла Р1 и доводят подвижной фазой А до объема 5 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза:
- подвижная фаза А: смешивают 0,5 мл кис¬
лоты фосфорной Р, 340 мл ацетонитрила Р1
и 600 мл водыР, выдерживают до установле¬
ния равновесия и доводят водой Р до объема
1000 мл;
- лодвюоия фаза В: ацетонитрил Р1\
Время (мм)
(%, огыоб)
аА
Подвижная фаза В
(%,об/об)
0—25
100
! о
| 25—55 |
100—15
! 0 — 85
| 55—70 |
15
|
| 85
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, 3 и N1, используя хроматограм¬
му раствора сравнения (с) и хроматограмму, прилага¬
емую к ФСО ибупрофена для идентификации пиков.
Относительное удерживание (по отношению
к ибупрофену, время удерживания — около 21 мин):
примесь и — около 0,2; примесь N — около 0,3; при¬
месь А — около 0,9; примесь В — около 1,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси В относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси В и
ибупрофена); при необходимости корректируют кон¬
центрацию ацетонитрила в подвижной фазе А.
Предельное содержание примесей:
- примеси А, 3, N (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, ^ и N. не должны превы¬
шать 1,5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, ^ и N. не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Примесь Р. Не более 0,1 %. Газовая хромато¬
графия (2.2.28): определение проводят методом вну¬
тренней нормализации.
Метилирующий раствор. 1 мл Ы^-диметил-
формамида диметилацеталя Р и 1 мл пиридина Р
доводят этилацетатом Р до объема 10 мл.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца помещают в герметично укупориваемый кон¬
тейнер, растворяют в 1,0 мл этилацетата Р и при¬
бавляют 1 мл метилирующего раствора. Контейнер
герметично укупоривают и нагревают в термостате
при температуре 100 °С в течение 20 мин. После ох¬
лаждения раствор выпаривают при комнатной тем¬
пературе в токе азота досуха. Остаток растворяют в
5 мл этилацетата Р.
Раствор сравнения (а). 0,5 мг ФСО ибупрофена
примеси Р растворяют в этилацетате Р и доводят
до объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 50,0 мг ФСО ибупрофена
помещают в герметично укупориваемый контейнер,
растворяют в 1 мл раствора сравнения (а) и прибав¬
ляют 1 мл метилирующего раствора. Контейнер гер¬
метично укупоривают и нагревают в термостате при
температуре 100 °С в течение 20 мин. После охлаж¬
дения раствор выпаривают досуха при комнатной
температуре в токе азота. Остаток растворяют в 5 мл
этилацетата Р.
Условия хроматографирования:
- колонка капиллярная кварцевая длиной 25 м и
внутренним диаметром 0,53 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 2 мкм);
-температура: колонка — 150 °С, блок ввода
проб — 200 °С, детектор — 250 °С;
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 5,0 мл/мин;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора и раствора сравнения (Ь);
- время хроматографирования: 2-кратное вре¬
мя удерживания ибупрофена.
Пригодность хроматографической системы:
-относительное удерживание (по отношению
к ибупрофену, время удерживания — около 17 мин):
примесь Р — около 1,5.
Тяжелые металлы (2.4.8, метод В). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон го¬
товят с использованием эталонного раствора свинца
(1 ррт РЬ), полученного путем разведения эталонно¬
го раствора свинца (100 ррт РЬ) Р метанолом Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
432
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,450 г испытуемого образца растворяют в 50 мл
метанола Р, прибавляют 0,4 мл раствора фенол¬
фталеина Р1 и титруют 0,1 М раствором натрия ги¬
дроксида до появления красного окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 20,63 мг С13Н1802.
ПРИМЕСИ
Специфицированные примеси: А, Р, 3, N.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Е, С, Н,
I, К, Ь, М, О, Р, О, Р-
A. Р1 = ОН, Р2 = СН2-СН(СН3)2, РЗ = Н: (2Я5)-2-
[3-(2-Метилпропил)фенил]пропановая кислота.
B. Р1 = ОН, Р2 = Н, РЗ = [СН2]3-СН3: (2Я5)-2-(4-Бу-
тилфенил)пропановая кислота.
C. Р1 = !\1Н2, Р2 = Н, РЗ = СН2-СН(СН3)2: (2Р8)-2-
[4-(2-Метилпропил)фенил]пропанамид.
О. Р1 = ОН, Р2 = Н, РЗ = СН3: (2Я5)-2-(4-
Метилфенил)пропановая кислота.
о
Е. 1 -[4-(2-Метилпропил)фенил]этанон.
н3с ^ ^
Р. 3-[4-(2-Метилпропил)фенил]пропановая кис¬
лота.
н3с
О. (1Я5,4Я5)-7-(2-Метилпропил)-1-[4-(2-метил-
пропил)фенил]-1,2,3,4-тетрагидронафтален-1,4-дикар-
боновая кислота.
и энантиомер
H. X = О: (ЗЯ5)-1,3-бис[4-(2-метилпропил)фенил]-
бутан-1-он.
I. X = Н2:1 -(2-Метилпропил)-4-[(ЗЯ5)-3-[4-(2-метил-
пропил)фенил]бутил]бензол.
1 Р = СО-СН(СН3)2: (2Я5)-2-[4-(2-Метилпропаноил)-
фенил]пропановая кислота.
К. Р = СНО: (2Я5)-2-(4-Формилфенил)пропановая
кислота.
1_. Р = СНОН-СН(СН3)2: 2-[4-(1 -Гидрокси-2-
метилпропил)фенил]пропановая кислота.
N. Р = С2Н5: (2Я5)-2-(4-Этилфенил)пропановая
кислота.
O. Р = СН(СН3)-С2Н5:2-[4-(1-Метилпропил)фенил]-
пропановая кислота.
М. Р1= ОН, Р2 = СН3, РЗ = С02Н: (2Я$)-2-Гидрокси-
2-[4-(2-метилпропил)фенил]пропановая кислота.
Р. Р1= Н, Р2 = СН3, РЗ = СН2ОН: (2Я5)-2-[4-(2-
Метилпропил)фенил]пропан-1-ол.
О. Р1= Р2 = Н, РЗ = СН2ОН: 2-[4-(2-Метилпропил)-
фенил]этанол.
Р. 1,1 '-(Зтан-1,1 -диил)-4,4'-(2-метил пропил )дибен-
зол.
07/2016:0669
ИДОКСУРИДИН
МохипсИпит
ЮОХС1КЮ1ЫЕ
С9Н111М205 М.м. 354,1
[54-42-2]
Идоксуридин
433
ОПРЕДЕЛЕНИЕ
5-Йод-1-(2-дезокси-Р-0-эрг/тро-пентафуранозил)
пиримидин-2,4(1 Н,ЗАУ)-дион.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Мало растворим в воде и в 96 % спирте. Раство¬
рим в разведенных растворах гидроксидов щелочных
металлов.
Температура плавления: около 180 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках; 1 мг испытуемого об¬
разца или стандартного образца прибавляют к 0,3 мг
калия бромида Р.
Сравнение: ФСО идоксуридина.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению и размеру основному пятну на хро¬
матограмме раствора сравнения (с).
C. Около 5 мг испытуемого образца помещают в
пробирку и нагревают на открытом пламени. Выделя¬
ется фиолетовый газ.
й. 2 мг испытуемого образца диспергируют в
1 мл воды Р и прибавляют 2 мл раствора дифенила¬
мина Р2. Нагревают в водяной бане в течение 10 мин.
Появляется устойчивое светло-голубое окрашивание.
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раство¬
ряют в 1 М растворе натрия гидроксида и доводят
до объема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 5,5 до 6,5. 0,10 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 100 мл этим же раст¬
ворителем.
Удельное оптическое вращение (2.2.7). От +28
до +32 (в пересчете на сухое вещество). Измеряют
угол оптического вращения раствора 3.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,20 г испытуемого
образца растворяют в смеси из раствора аммиака
концентрированного Р и метанола Р (1:5, об/об) и
доводят до объема 5 мл этой же смесью раствори¬
телей.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят смесью из раствора аммиака
концентрированного Р и метанола Р (1:5, об/об) до
объема 10 мл.
Раствор сравнения (а). 20 мг 5-йодураци-
ла Р, 20 мг 2-дезоксиуридина Р и 20 мг 5-бром-2-
дезоксиуридина Р растворяют в смеси из раствора
аммиака концентрированного Р и метанола Р (1:5,
об/об) и доводят до объема 100 мл этой же смесью
растворителей.
Раствор сравнения (Ь). 0,20 г испытуемого об¬
разца растворяют в 5 мл раствора сравнения (а).
Раствор сравнения (с). 20 мг ФСО идоксуриди¬
на растворяют в смеси из раствора аммиака концен¬
трированного Р и метанола Р (1:5, об/об) и доводят
до объема 5 мл этой же смесью растворителей.
Раствор сравнения (д). 1 мл испытуемого раст¬
вора (Ь) доводят до объема 20 мл смесью, состоящей
из аммиака концентрированного Р и метанола Р
(1:5, об/об).
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля с флуоресцентным индикатором, имею¬
щим максимальную интенсивность при 254 нм.
Подвижная фаза: раствор аммиака концен¬
трированный Р — хлороформ Р — 2-пропанол Р
(10:40:50, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: дважды; не менее 15 см
от линии старта.
Высушивание: в потоке холодного воздуха после
каждого хроматографирования.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме раствора сравнения (Ь) об¬
наруживаются четыре полностью разделенных пятна.
Предельное содержание примесей:
- сопутствующие примеси (не более 0,5 %): на
хроматограмме испытуемого раствора (а) пятна, со¬
ответствующие 5-йодурацилу, 2-дезоксиуридину и
5-бром-2'-дезоксиуридину, должны быть не интенсив¬
нее соответствующих пятен на хроматограмме раст¬
вора сравнения (а);
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного и пятен 5-йодурацила, 2'-дезоксиуридина и
5-бром-2'-дезоксиуридина, должно быть не интенсив¬
нее пятна на хроматограмме раствора сравнения (с1).
Йодиды. Не более 0,1 %. 0,25 г испытуемого об¬
разца растворяют в 25 мл 0,1 М раствора натрия
гидроксида Р, прибавляют 5 мл кислоты хлористово¬
дородной разведенной Р, доводят водой Р до объема
50 мл, выдерживают в течение 10 мин и фильтруют. К
25 мл фильтрата прибавляют 5 мл раствора водоро¬
да пероксида разведенного Р и 10 мл хлороформа Р
и встряхивают. Розовое окрашивание органического
слоя должно быть не интенсивнее эталона, приготов¬
ленного параллельно с использованием 1 мл раствора
0,33 г/л калия йодида Р вместо испытуемого образца.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,3000 г испытуемого образца растворяют в
20 мл диметилформамида Р и титруют 0,1 М раство¬
ром тетрабутиламмония гидроксида потенциоме-
трически (2.2.20).
434
Гэсударственная фармакопея Республики Беларусь
1 мл 0,1 М раствора тетрабутиламмония ги¬
дроксида соответствует 35,41 мг С9Н111М205.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0770
ИЗОЛЕЙЦИН
1зо1еистит
/50/-Е1Ю/МЕ
н мн2
Н3С X" со2н
н сн3
С6Н13Ы02 М.м. 131,2
[73-32-5]
(25,35)-2-Амино-3-метилпентановая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или чешуйки.
Умеренно растворим в воде, мало растворим в
96 % спирте. Растворяется в разведенных минераль¬
ных кислотах и в разведенных растворах гидроксидов
щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО изолейцина #или спектр, пред¬
ставленный на рисунке #0770.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
На хроматограмме испытуемого раствора (Ь) об¬
наруживается основное пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в растворе 103 г/л кислоты хлористоводород¬
ной Р и доводят до объема 10 мл этим же раствори¬
телем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От
+40,0 до +43,0 (в пересчете на сухое вещество). 1,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 25,0 мл этим
же растворителем.
Нингидрин-положительные вещества
Испытание проводят одним из приведенных
ниже методов/
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемых растворов и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон-
Рисунок #0770.-1. Инфракрасный спектр ФСО изолейцина
Изолейцин
435
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образцов, подходящий для используемого обору¬
дования.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят раствором А до объ¬
ема 10,0 мл.
Раствор сравнения (Ь). 30,0 мг валина Р (при¬
месь А) растворяют в растворе А и доводят до объема
100.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (д). 30,0 мг лейцина Р (при¬
месь С) растворяют в растворе А и доводят до объема
100.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят раствором А до объема 250,0 мл.
Раствор сравнения (е). 6,0 мл эталонного раст¬
вора аммония (100 ррт ЫН) Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (1). 30 мг изолейцина Р и
30 мг лейцина Р (примесь С) растворяют в растворе
А и доводят до объема 50,0 мл. 1,0 мл полученного
раствора доводят раствором А до объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь), (с), (с!) и (1) вводят в аминокислотный анали¬
затор. Запускают подходящую программу для опреде¬
ления физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (1):
-разрешение: не менее 1,5 между пиками изо¬
лейцина и примеси С.
Расчет процентных содержаний:
-для примеси А — используют концентрацию
примеси А в растворе сравнения (Ь);
-для примеси С — используют концентрацию
примеси С в растворе сравнения (д);
-для любого нингидрин-положительного веще¬
ства, детектируемого при 570 нм — используют кон¬
центрацию изолейцина в растворе сравнения (а);
-для любого нингидрин-положительного веще¬
ства, детектируемого при 440 нм — используют кон¬
центрацию пролина в растворе сравнения (с); если
пик выше учитываемого предела при обеих длинах
волн, для расчетов используют результат, полученный
при 570 нм.
Предельное содержание примесей:
- примеси А и С при 570 нм: не более 0,3 % для
каждой примеси;
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 1,0 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в растворе 10,3 г/л кислоты хло¬
ристоводородной Р и доводят до объема 10 мл этим
же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО изолейцина
растворяют в растворе 10,3 г/л кислоты хлористово¬
дородной Р и доводят до объема 50 мл этим же раст¬
ворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО изолейцина и
10 мг ФСО валина растворяют в растворе 10,3 г/л кис¬
лоты хлористоводородной Р и доводят до объема
25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и высушивают при температуре 105 °С
в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в воде Р и до¬
водят до объема 15 мл этим же растворителем. Полу¬
ченный раствор должен выдерживать испытание на
хлориды.
Сульфаты (2.4.13). Не более 0,0300%
(300 ррт). 0,5 г испытуемого образца растворяют в
3 мл кислоты хлористоводородной разведенной Р и
доводят водой дистиллированной Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(е) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хрома-
436
Гэсударственная фармакопея Республики Беларусь
тографии, то испытание «Аммония соли» (2.4.1, ме¬
тод В) проводят следующим образом. 50 мг испытуе¬
мого образца должны выдерживать испытание на ам¬
мония соли. Эталон готовят с использованием 0,1 мл
эталонного раствора аммония (100 ррт NН^ Я#
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод И). Не более
0,0010% (10 ррт).
Растворитель. Вода Р.
0,25 г испытуемого образца должны выдержи¬
вать испытание на тяжелые металлы. Эталон гото¬
вят с использованием 0,25 мл эталонного раствора
свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20), #или используя 0,1 мл раствора нафтолбен-
зеина Р в качестве индикатора, до перехода окраски
раствора от коричневато-желтой до зеленой#.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 13,12 мг С6Н13М02.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемлемо¬
сти для других/неспецифицированных примесей.
Вследствие этого нет необходимости идентифици¬
ровать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Кон¬
троль примесей в субстанциях для фармацевти¬
ческого использования): В, О.
н гчн2
Н3(
С02Н
цин).
СН3 н N42
ИъС^^^^С02И
С. (25)-2-Амино-4-метилпентановая кислота (лей-
ню.
0. (25)-2-Амино-4-(метилсульфанил)бутановая
кислота (метионин).
07/2016:0146
ИЗОНИАЗИД
1воп1агМит
180Ы1А2Ю
СвН7Ы,0 М.м. 137,1
[54-85-3]
ОПРЕДЕЛЕНИЕ
Пиридин-4-карбогидразид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, умеренно растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В
Вторая идентификация: А, С.
A. Температура плавления (2.2.14): от 170 °С до
174 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО изониазида #или спектр, пред¬
ставленный на рисунке #0146.-1#.
C. 0,1 г испытуемого образца растворяют в 2 мл
воды Р, прибавляют 10 мл теплого раствора 10 г/л
ванилина Р, раствор отстаивают и, при необходимо¬
сти, потирают внутренние стенки пробирки стеклян¬
ной палочкой. Образуется желтый осадок, который
после перекристаллизации из 5 мл спирта (70 %,
об/об) Р и высушивания при температуре от 100 °С
до 105 °С имеет температуру плавления (2.2.14) от
226 °С до 231 °С.
ИСПЫТАНИЯ
сн3
A. (25)-2-Амино-3-метилбутановая кислота (валин).
Н МН2
ИЗчХ
со2н
B. (2Я?)-2-Амино-3-сульфанилпропановая кислота
(цистеин).
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
рН (2.2.3). От 6,0 до 8,0. Измеряют рН раствора 3.
Изопропилмиристат
437
Рисунок #0146.-1. Инфракрасный спектр ФСО изониазида
Гидразин и сопутствующие примеси. Гидра¬
зин: не более 0,05 %; любая другая примесь: не более
0,2 %. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1,0 г испытуемого образца
растворяют в смеси из ацетона Р и воды Р( 1:1, об/об)
и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения. 50,0 мг гидразин сульфа¬
та Р растворяют в 50 мл воды Р и доводят ацето¬
ном Р до объема 100,0 мл. К 10,0 мл полученного
раствора прибавляют 0,2 мл испытуемого раствора и
доводят смесью из ацетона Р и воды Р( 1:1, об/об) до
объема 100,0 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: вода Р — ацетон Р — мета¬
нол Р— этилацетат Р (10:20:20:50, об/об/об/об).
Наносимый объем пробы. 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора любое пятно, кроме основного, должно быть
не интенсивнее пятна на хроматограмме раствора
сравнения.
Проявление В: пластинку опрыскивают раство¬
ром диметиламинобензальдегида Р1 и просматрива¬
ют при дневном свете.
Результаты В: на хроматограмме раствора
сравнения проявляется дополнительное пятно (ги¬
дразин). На хроматограмме испытуемого раствора
пятно, соответствующее гидразину, должно быть не
интенсивнее пятна гидразина на хроматограмме раст¬
вора сравнения.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют
в воде Р и доводят до объема 100,0 мл этим же
растворителем. К 20,0 мл полученного раствора
прибавляют 100 мл воды Р, 20 мл кислоты хлори¬
стоводородной Р, 0,2 г калия бромида Р, 0,05 мл
раствора метилового красного Р и титруют по
каплям 0,0167 М раствором калия бромата при
постоянном перемешивании до исчезновения крас¬
ной окраски.
1 мл 0,0167 М раствора калия бромата соответ¬
ствует 3,429 мг С6Н7М30.
07/2016:0725
ИЗОПРОПИЛМИРИСТАТ
/зоргоруНз тупз1аз
130РРОРУ1. МУР13ТАТЕ
С17нм02 М.м. 270,5
#[110-27-0]*
438
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
1-Метилэтилтетрадеканоат с разным количе¬
ством изопропиловых эфиров других жирных кислот.
Содержание: не менее 90,0 % С^Н^О^
ОПИСАНИЕ(СВОЙСТВА)
Бесцветная, прозрачная маслянистая жидкость.
Не смешивается с водой, смешивается с 96 %
спиртом, метиленхлоридом, жирными кислотами и
маслом вазелиновым.
Относительная плотность: около 0,853.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испытания».
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения.
C. 2 мл раствора 1 г/л испытуемого образца в
96 % спирте Р наслаивают на свежеприготовленный
раствор 20 мг диметиламинобензальдегида Р в 2 мл
кислоты серной Р. Через 2 мин желтовато-красное
окрашивание на границе двух жидкостей постепенно
переходит в красное.
ИСПЫТАНИЯ
Раствор 8. 2,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 8
должна быть не интенсивнее эталона У(Ж)7
Показатель преломления (2.2.6). От 1,434 до
1,437.
Вязкость (2.2.9). От 5 мПа-с до 6 мПа-с.
Кислотное число (2.5.1). Не более 1,0.
Йодное число (2.5.4). Не более 1,0.
Число омыления (2.5.6). От 202 до 212.
Вода (2.5.12). Не более 0,1 %. Определение про¬
водят из 5,0 г испытуемого образца.
Общая зола (2.4.16). Не более 0,1 %. Определе¬
ние проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 50,0 мг три-
козана Р растворяют в гептане Р и доводят до объ¬
ема 250,0 мл этим же растворителем.
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в растворе внутреннего стандарта и
доводят до объема 100,0 мл этим же растворителем.
Раствор сравнения. 20,0 мг ФСО изопропилте-
традеканоата растворяют в растворе внутренне¬
го стандарта и доводят до объема 100,0 мл этим же
растворителем.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 50 м
и внутренним диаметром 0,2 мм, покрытая сло¬
ем поли(цианопропил)силоксана Р (толщина слоя
0,2 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:40;
- температура:
Время (мин)
Температура (°С)
Колонка
0—6
125-185
6—16
185
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 2 мкл.
Рассчитывают содержание С17Н3402 в испытуе¬
мом образце в процентах.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0970
ИЗОПРОПИЛОВЫЙ СПИРТ
Л/со/ю/ 1зоргоруНсиз
180РРОРУ1. А1.СОН01.
он
С3НвО М.м. 60,1
[67-63-0]
ОПРЕДЕЛЕНИЕ
Пропан-2-ол.
ОПРЕДЕЛЕНИЕ(СВОЙСТВА)
Бесцветная, прозрачная жидкость.
Смешивается с водой и 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, В, О.
A. Относительная плотность (2.2.5). От 0,785 до
0,789.
B. Показатель преломления (2.2.6). От 1,376 до
1,379.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО изопропилового спирта #или
спектр, представленный на рисунке #0970.-1#.
6. К 1 мл испытуемого образца прибавляют 4 мл
воды Р и перемешивают. Осторожно прибавляют 2 мл
раствора 10 г/л диметиламинобензальдегида Р в
кислоте серной Р, избегая перемешивания жидко¬
стей; на границе раздела двух жидкостей незамед¬
лительно образуется яркое красновато-фиолетовое
кольцо. Через (2—5) мин весь слой серной кислоты
становится фиолетовым.
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным. 1 мл испытуемого образца до¬
водят водой Р до объема 20 мл. Полученный раствор
через 5 мин должен быть прозрачным.
Изопропиловый спирт
439
Рисунок #0970.-1. Инфракрасный спектр ФСО изопропилового спирта
Цветность (2.2.2, метод II). Испытуемый обра¬
зец должен быть бесцветным.
Кислотность или щелочность. 25 мл испыту¬
емого образца осторожно кипятят в течение 5 мин,
прибавляют 25 мл воды, свободной от углерода ди¬
оксида, Р, выдерживают до охлаждения, защищая от
диоксида углерода воздуха. К полученному раство¬
ру прибавляют 0,1 мл раствора фенолфталеина Р.
Раствор бесцветный. При прибавлении не более
0,6 мл 0,01 М раствора натрия гидроксида должно
появиться бледно-розовое окрашивание.
Оптическая плотность (2.2.25). Не более 0,30
при длине волны 230 нм, не более 0,10 при длине
волны 250 нм, не более 0,03 при длине волны 270 нм,
не более 0,02 при длине волны 290 нм и не более 0,01
при длине волны 310 нм.
Оптическую плотность измеряют в области от
230 нм до 310 нм, используя в качестве компенсаци¬
онного раствора водуР Спектр представляет собой
падающую кривую без видимых пиков и плеч.
Бензол и сопутствующие примеси. Газовая
хроматография (2.2.28).
Испытуемый раствор (а). Испытуемый образец.
Испытуемый раствор (Ь). 1,0 мл 2-бутано-
ла Р1 доводят испытуемым раствором (а) до объема
50.0 мл. 5,0 мл полученного раствора доводят испы¬
туемым раствором (а) до объема 100,0 мл.
Раствор сравнения (а). 0,5 мл 2-бутанола Р1
и 0,5 мл пропанола Р доводят испытуемым раство¬
ром (а) до объема 50,0 мл. 5,0 мл полученного раст¬
вора доводят испытуемым раствором (а) до объема
50.0 мл.
Раствор сравнения (Ь). 100 мкл бензола Р дово¬
дят испытуемым раствором (а) до объема 100,0 мл.
0,20 мл полученного раствора доводят испытуемым
раствором (а) до объема 100,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и внутренним диаметром 0,32 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 1,8 мкм);
- газ-носитель: гелий для хроматографии Р;
- продувка детектора: гелий для хроматогра¬
фии Р или азот для хроматографии Р;
- линейная скорость: 35 см/с;
- деление потока: 1:5;
- температура:
Время (мин)
Температура (°С)
Колонка
0—12
40
12—32
40 -> 240
32—42
240
Блок ввода проб
280
Детектор
280
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Время удерживания: бензол — около 10 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 10 между первым (про¬
панол) и вторым (2-бутанол) пиками.
Предельное содержание примесей:
- бензол (не более 0,0002 % (2 ррт)): на хро¬
матограмме испытуемого раствора (а) площадь
пика бензола не должна превышать половины пло¬
щади соответствующего пика на хроматограмме
раствора сравнения (Ь) (чувствительность систе¬
мы регулируют таким образом, чтобы высота пика
бензола на хроматограмме раствора сравнения
(Ь), составляла не менее 10 % шкалы регистриру¬
ющего устройства);
- сумма примесей, кроме 2-бутанола (не более
0,3 %): на хроматограмме испытуемого раствора (Ь)
сумма площадей всех пиков, кроме пика 2-бутанола,
440
Гэсударственная фармакопея Республики Беларусь
не должна превышать 3-кратную площадь пика 2-бу¬
танола на хроматограмме испытуемого раствора (Ь)
(чувствительность системы регулируют таким обра¬
зом, чтобы высота двух пиков, следующих за основ¬
ным пиком, на хроматограмме раствора сравнения (а)
составляла не менее 50 % шкалы регистрирующего
устройства).
Пероксиды. 8 мл раствора крахмала с калия
йодидом Р помещают в пробирку с притертой сте¬
клянной пробкой вместимостью 12 мл и диаметром
около 15 мм, заполняют полностью испытуемым об¬
разцом и интенсивно перемешивают. Выдерживают в
защищенном от света месте в течение 30 мин. Раст¬
вор не должен окрашиваться.
Нелетучие вещества. Не более 0,0020 %
(20 ррт). 100 г испытуемого образца, если он вы¬
держал испытание «Пероксиды», выпаривают на во¬
дяной бане досуха и остаток сушат при температуре
от 100 °С до 105 °С. Масса сухого остатка не должна
превышать 2 мг.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 5,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. Пропанон (ацетон).
В. Бензол.
к я
С. К = СН3: 2-(1-Метилэтокси)пропан (диизопро-
пиловый эфир).
O. Р = Н: Этоксиэтан (диэтиловый эфир).
Е. СН3-ОН: Метанол.
юн
н3с^
P. Пропан-1-ол (н-пропанол).
07/2016:1117
ИЗОСОРБИДА ДИНИТРАТ
РАЗВЕДЕННЫЙ
/зозогЫсИ сИпПгаз сШиЫз
18030КВЮЕ ОШПЙАТЕ, 01ШТЕО
СвН8Ы2Ов М.м. 236,1
ОПРЕДЕЛЕНИЕ
Сухая смесь изосорбида динитрата и Лактозы
моногидрата (0187) или Маннита (0559).
Содержание: не менее 95,0 % (м/м) и не более
105,0% (м/м) 1,4:3,б-диангидро-0-глюцитола 2,5-ди-
нитрата от заявленного содержания.
МЕРЫ ПРЕДОСТОРОЖНОСТИ. Изосорбида
динитрат неразведенный под воздействием уда¬
ра или при нагревании может взрываться. Должны
быть приняты необходимые меры предосторожно¬
сти. Работать следует только с очень небольши¬
ми количествами неразведенного изосорбида дини¬
трата.
ОПИСАНИЕ (СВОЙСТВА)
Неразведенный изосорбида динитрат — мелкий
кристаллический порошок белого или почти белого
цвета.
Неразведенный изосорбида динитрат очень
мало растворим в воде, очень легко растворим в аце¬
тоне, умеренно растворим в 96 % спирте.
Растворимость изосорбида динитрата разведен¬
ного зависит от разбавителя и его концентрации.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках; используют остаток,
полученный в идентификации Э.
Сравнение: ФСО изосорбида динитрата #или
спектр, представленный на рисунке #1117.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 10 мг изосорбида динитрата,
встряхивают с 10 мл 96 % спирта Р в течение 5 мин
и фильтруют.
Раствор сравнения. Навеску ФСО изосорбида
динитрата, соответствующую 10 мг изосорбида ди¬
нитрата, встряхивают с 10 мл 96 % спирта Р в тече¬
ние 15 мин и фильтруют.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: метанол Р — метиленхло-
рид Р (5:95, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке воздуха.
Проявление: пластинку опрыскивают свежеприго¬
товленным раствором калия йодида с крахмалом Р,
выдерживают в ультрафиолетовом свете при длине
волны 254 нм в течение 15 мин и просматривают при
дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 0,10 г лактозы или маннита,
встряхивают с 10 мл воды Р и при необходимости
фильтруют.
Раствор сравнения (а). 0,10 г лактозы Р раст¬
воряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Аххарбида динитрат разведенный 441
3000 2000 1500 1000 500
Волновое число (см'1)
Рисунок #1117.-1. Инфракрасный спектр ФСО изосорбида динитрата
Раствор сравнения (Ь). 0,10 г маннита Р раст¬
воряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Раствор сравнения (с). Смешивают равные объ¬
емы растворов сравнения (а) и (Ь).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: вода Р — метанол Р — кис¬
лота уксусная безводная Р — этиленхлорид Р
(10:15:25:50, об/об/об/об). Объемы компонентов под¬
вижной фазы отмеряют точно, так как небольшой из¬
быток воды приводит к помутнению.
Наносимый объем пробы: 1 мкл (место нанесе¬
ния пробы тщательно высушивают).
Фронт подвижной фазы А: не менее 15 см от ли¬
нии старта.
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 15 см от линии старта, после обновления подвиж¬
ной фазы.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
кислоты 4-аминобензойной Р и сушат в потоке хо¬
лодного воздуха до исчезновения запаха ацетона, за¬
тем пластинку выдерживают при температуре 100 °С
в течение 15 мин, охлаждают, опрыскивают раствором
2 г/л натрия перйодата Р, сушат в потоке холодного
воздуха и выдерживают при температуре 100 °С в те¬
чение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, окраске и размеру основному пятну
на хроматограмме раствора сравнения (а), если раз¬
бавителем является лактоза, или пятну на хромато¬
грамме раствора сравнения (Ь), если разбавителем
является маннит.
Навеску испытуемого образца, соответствую¬
щую 25 мг изосорбида динитрата, встряхивают с 10 мл
ацетона Р в течение 5 мин, фильтруют и упаривают
досуха при температуре не выше 40 °С. Полученный
остаток сушат над фосфора (V) оксидом Р при давле¬
нии, не превышающем 0,7 кПа, в течение 16 ч. Темпе¬
ратура плавления (2.2.14) остатка: от 69 °С до 72 °С.
ИСПЫТАНИЯ
Примесь А. Не более 0,5 % в пересчете на калия
нитрат. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 0,10 г изосорбида динитра¬
та, встряхивают с 5 мл 96 % спирта Р и фильтруют.
Раствор сравнения. 10 мг калия нитрата Р
растворяют в 1 мл воды Р и доводят 96 % спиртом Р
до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р —
ацетон Р — толуол Р (15:30:60, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке воздуха до полного ис¬
чезновения запаха уксусной кислоты.
Проявление: пластинку обильно опрыскивают
свежеприготовленным раствором калия йодида с
крахмалом Р, выдерживают в ультрафиолетовом све¬
те при длине волны 254 нм в течение 15 мин и про¬
сматривают при дневном свете.
Предельное содержание примесей:
-примесь А: на хроматограмме испытуемого
раствора пятно, соответствующее примеси А, должно
56. Зак. 1060.
442
Гэсударственная фармакопея Республики Беларусь
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения.
Примеси В и С. Жидкостная хроматография (2.2.29).
Испытуемый раствор (а). К навеске испытуемого
образца, соответствующей 25,0 мг изосорбида дини¬
трата, прибавляют 20 мл подвижной фазы, обрабаты¬
вают ультразвуком в течение 15 мин и доводят подвиж¬
ной фазой до объема 25,0 мл. Полученный раствор
фильтруют через подходящий мембранный фильтр.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
10,0 мл.
Раствор сравнения (а). К навеске ФСО изосорби¬
да динитрата, соответствующей 25,0 мг изосорбида
динитрата, прибавляют 20 мл подвижной фазы, обраба¬
тывают ультразвуком в течение 15 мин и доводят под¬
вижной фазой до объема 25,0 мл. Полученный раствор
фильтруют через подходящий мембранный фильтр.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (с). 10,0 мг ФСО изосорби¬
да 2-нитрата (примесь В) растворяют в подвижной
фазе и доводят до объема 10,0 мл этим же раствори¬
телем. 0,1 мл полученного раствора доводят подвиж¬
ной фазой до объема 20,0 мл.
Раствор сравнения (ф. 10,0 мг ФСО изосорбида
мононитрата (примесь С) растворяют в подвижной
фазе и доводят до объема 10,0 мл этим же раствори¬
телем. 0,1 мл полученного раствора доводят подвиж¬
ной фазой до объема 20,0 мл.
Раствор сравнения (е). 5 мг ФСО изосорби¬
да 2-нитрата (примесь В) растворяют в подвижной
фазе и доводят до объема 10 мл этим же растворите¬
лем. К 1 мл полученного раствора прибавляют 0,5 мл
раствора сравнения (а) и доводят подвижной фазой
до объема 10 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем аминопропил-
метилсилильным для хроматографии Р с размером
частиц 10 мкм;
- подвижная фаза: этанол Р — триметилпен-
тан Р (15:85, об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны (210—215) нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а), растворов сравнения (с), (б) и (е).
Времена удерживания: изосорбида динитрат —
около 5 мин; примесь В — около 8 мин; примесь С —
около 11 мин.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 6,0 между пиками изо¬
сорбида динитрата и примеси В.
Предельное содержание примесей:
- примесь В (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответствую¬
щего примеси В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с);
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответствую¬
щего примеси С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (б).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Примеси В и С», со следующими изме¬
нениями.
Условия хроматографирования:
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (Ь).
Если площади пиков на двух последовательных
хроматограммах раствора сравнения (Ь) отличаются
более чем на 1,0 %, то повторяют хроматографирова¬
ние еще 4 раза и рассчитывают относительное стан¬
дартное отклонение для 6 хроматограмм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- относительное стандартное отклонение:
не более 2,0 % для площадей пиков при 6 повторных
вводах пробы.
Содержание изосорбида динитрата рассчитыва¬
ют в процентах от заявленного содержания.
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
Указывают содержание изосорбида динитрата в
процентах.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. Неорганические нитраты.
В. 1,4:3,6-Диангидро-0-глюцитола 2-нитрат (изо¬
сорбида 2-нитрат).
С. 1,4:3,6-Диангидро-0-глюцитола 5-нитрат (изо¬
сорбида 5-нитрат, изосорбида мононитрат).
07/2016:1118
ИЗОСОРБИДА МОНОНИТРАТ
РАЗВЕДЕННЫЙ
1зозогЫсЛ топопИгаз сШи1из
18030КВЮЕ МОЫОШТПАТЕ, 01ШТЕО
С.Н.ИО,
М.м. 191,1
Изосорбида мононитрат разведенный
443
ОПРЕДЕЛЕНИЕ
Сухая смесь изосорбида мононитрата и Лакто¬
зы моногидрата (0187) или Маннита (0559).
Содержание: не менее 95,0 % (м/м) и не более
105,0 % (м/м) 1,4:3,6-диангидро-0-глюцитола 5-нитра¬
та от заявленного содержания.
ОПИСАНИЕ (СВОЙСТВА)
Неразведенный изосорбида мононитрат — кри¬
сталлический порошок белого или почти белого цвета.
Неразведенный изосорбида мононитрат легко
растворим в воде, в ацетоне, в 96 % спирте и в мети-
ленхлориде.
Растворимость изосорбида мононитрата разве¬
денного зависит от разбавителя и его концентрации.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках; используют остаток,
полученный в идентификации О.
Сравнение: ФСО изосорбида мононитрата #или
спектр, представленный на рисунке #1118.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 10 мг изосорбида монони¬
трата, встряхивают с 10 мл 96 % спирта Р в течение
5 мин и фильтруют.
Раствор сравнения. Навеску ФСО изосорбида
мононитрата, соответствующую 10 мг изосорбида
мононитрата, встряхивают с 10 мл 96% спирта Р в
течение 15 мин и фильтруют.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: метанол Р — метиленхло-
рид Р (5:95, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке воздуха.
Проявление: пластинку опрыскивают свежеприго¬
товленным раствором калия йодида с крахмалом Р,
выдерживают в ультрафиолетовом свете при длине
волны 254 нм в течение 15 мин и просматривают при
дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 0,10 г лактозы или маннита,
встряхивают с 10 мл воды Р и при необходимости
фильтруют.
Раствор сравнения (а). 0,10 г лактозы Р раст¬
воряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Раствор сравнения (Ь). 0,10 г маннита Р раст¬
воряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Раствор сравнения (с). Смешивают равные объ¬
емы растворов сравнения (а) и (Ь).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: вода Р — метанол Р — кис¬
лота уксусная безводная Р — этиленхлорид Р
(10:15:25:50, об/об/об/об). Объемы компонентов под¬
вижной фазы отмеривают точно, так как небольшой
избыток воды приводит к помутнению.
Наносимый объем пробы: 1 мкл (место нанесе¬
ния пробы тщательно высушивают).
Фронт подвижной фазы А: не менее 15 см от ли¬
нии старта.
Рисунок #1118.-1. Инфракрасный спектр ФСО изосорбида мононитрата
56*. Зак. 1060.
444
Гэсударственная фармакопея Республики Беларусь
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 15 см от линии старта, после обновления подвиж¬
ной фазы.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раство¬
ром кислоты 4-аминобензойной Р и сушат в потоке
холодного воздуха до исчезновения запаха ацетона.
Пластинку выдерживают при температуре 100 °С в
течение 15 мин, охлаждают, опрыскивают раствором
2 г/л натрия перйодата Р, сушат в потоке холодного
воздуха и выдерживают при температуре 100 °С в те¬
чение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, окраске и размеру основному пятну
на хроматограмме раствора сравнения (а), если раз¬
бавителем является лактоза, или пятну на хромато¬
грамме раствора сравнения (Ь), если разбавителем
является маннит.
О. Навеску испытуемого образца, соответству¬
ющую 25 мг изосорбида мононитрата, встряхивают с
10 мл ацетона Р в течение 5 мин, фильтруют и упари¬
вают досуха при температуре не выше 40 °С. Получен¬
ный остаток сушат над фосфора (V) оксидом Р при дав¬
лении, не превышающем 0,7 кПа, в течение 16 ч. Тем¬
пература плавления (2.2.14) остатка: от 89 °С до 91 °С.
ИСПЫТАНИЯ
Примесь А. Не более 0,5 % в пересчете на калия
нитрат. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 0,10 г изосорбида монони¬
трата, встряхивают с 5 мл 96 % спирта Р и фильтруют.
Раствор сравнения. 10 мг калия нитрата Р
растворяют в 1 мл воды Р и доводят 96 % спиртом Р
до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р —
ацетон Р— толуол Р (15:30:60, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке воздуха до полного ис¬
чезновения запаха уксусной кислоты.
Проявление: пластинку обильно опрыскивают
свежеприготовленным раствором калия йодида с
крахмалом Р, выдерживают в ультрафиолетовом све¬
те при длине волны 254 нм в течение 15 мин и про¬
сматривают при дневном свете.
Предельное содержание примесей:
-примесь А: на хроматограмме испытуемого
раствора пятно, соответствующее примеси А, должно
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения.
Примеси В и С. Жидкостная хроматография (2.2.29).
Испытуемый раствор (а). К навеске испытуе¬
мого образца, соответствующей 25,0 мг изосорбида
мононитрата, прибавляют 20 мл подвижной фазы,
обрабатывают ультразвуком в течение 15 мин и дово¬
дят подвижной фазой до объема 25,0 мл. Полученный
раствор фильтруют через подходящий мембранный
фильтр.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
10,0 мл.
Раствор сравнения (а). 25,0 мг ФСО изосорбида
мононитрата растворяют в подвижной фазе и дово¬
дят до объема 25,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 10,0 мл.
Раствор сравнения (Ь). 10,0 мг ФСО изосорби¬
да 2-нитрата (примесь С) растворяют в подвижной
фазе и доводят до объема 10,0 мл этим же раствори¬
телем. 0,1 мл полученного раствора доводят подвиж¬
ной фазой до объема 20,0 мл.
Раствор сравнения (с). К навеске ФСО изосорби¬
да динитрата (примесь В), соответствующей 10,0 мг
изосорбида динитрата, прибавляют 15 мл подвижной
фазы, обрабатывают ультразвуком в течение 15 мин
и доводят подвижной фазой до объема 20,0 мл. По¬
лученный раствор фильтруют через подходящий мем¬
бранный фильтр. 0,1 мл полученного раствора дово¬
дят подвижной фазой до объема 10,0 мл.
Раствор сравнения (б). 5 мг ФСО изосорбида
мононитрата и 5 мг ФСО изосорбида 2-нитрата
(примесь С) растворяют в подвижной фазе и доводят
до объема 10 мл этим же растворителем. 1 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 10 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем аминопропил-
метилсилильным для хроматографии Р с размером
частиц 10 мкм;
- подвижная фаза: этанол Р — триметилпен-
тан Р (15:85, об/об);
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны (210—215) нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) растворов сравнения (Ь), (с) и (б).
Времена удерживания: примесь В — около 5 мин;
примесь С — около 8 мин; изосорбида 5-нитрат —
около 11 мин.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 4,0 между пиками при¬
меси С и изосорбида 5-нитрата.
Предельное содержание примесей:
- примесь В (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответствую¬
щего примеси В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с);
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответствую¬
щего примеси С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Примеси В и С», со следующими изме¬
нениями.
Условия хроматографирования:
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (а).
Имипенем моногидрат
445
Если площади пиков на двух последовательных
хроматограммах раствора сравнения (а) отличаются
более чем на 1,0 %, то повторяют хроматографирова¬
ние еще 4 раза и рассчитывают относительное стан¬
дартное отклонение для 6 .хроматограмм.
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение:
не более 2,0 % для площадей пиков при 6 повторных
вводах пробы.
Содержание изосорбида мононитрата рассчиты¬
вают в процентах от заявленного содержания.
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
Указывают содержание изосорбида мононитрата
в процентах.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. Неорганические нитраты.
В. 1,4:3,6-Диангидро-0-глюцитола 2,5-динитрат
(изосорбида динитрат).
С. 1,4:3,6-Диангидро-0-глюцитола 2-динитрат (изо¬
сорбида 2-нитрат).
07/2016:1226
ИМИПЕНЕМ МОНОГИДРАТ
1т1репетит топоЬудпсит
1М1РЕЫЕМ МОЫОНУОКАТЕ
С12Н17М3045 • Н20 М.м. 317,4
[74431-23-5]
ОПРЕДЕЛЕНИЕ
(5Я65)-6-[(#)-1 -Гидроксиэтил]-3-[[2-(иминометил)-
амино]этил]сульфанил]-7-оксо-1-азабицикло[3.2.0]гепт-
2-ен-2-карбоновая кислота моногидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации или любыми другими способами.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый или бледно-желтый по¬
рошок. Слегка гигроскопичен.
Мало растворим в воде и в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО имипенема.
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раство¬
ряют в фосфатном буферном растворе рН 7,0 РЗ и
доводят этим же растворителем до объема 50 мл.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее шестого эталона шкалы
наиболее подходящего цвета.
рН (2.2.3). От 4,5 до 7,5. 0,500 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 100,0 мл этим же раст¬
ворителем.
Удельное оптическое вращение (2.2.7). От +90
до +95 (в пересчете на безводное вещество) при тем¬
пературе 25 °С. Растворы готовят непосредствен¬
но перед использованием.
0,125 г растворяют в фосфатном буферном рас¬
творе рН 7,0 РЗ и доводят до объема 25,0 мл этим же
растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Буферный раствор А. 0,32 г натрия дигидро-
фосфата безводного Р и 1,04 г динатрия гидрофос¬
фата безводного Р растворяют в 900 мл воды Р и до¬
водят до рН 7,3 кислотой фосфорной разведенной Р.
Полученный раствор доводят водой Р до объема
1000 мл.
Буферный раствор В. 0,11 г динатрия гидро¬
фосфата безводного Р растворяют в 900 мл воды Р
и доводят до рН 6,8 кислотой фосфорной разведен¬
ной Р. Полученный раствор доводят водой Р до объ¬
ема 1000 мл.
Смесь растворителей. Ацетонитрил Р — бу¬
ферный раствор В (0,7:99,3, об/об).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Раствор сравнения (а). 25,0 мг ФСО имипенема
растворяют в смеси растворителей и доводят до объ¬
ема 25,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл.
Раствор сравнения (с). 5 мг испытуемого об¬
разца растворяют в 8 мл смеси из кислоты серной
разведенной Р и воды Р (1:200, об/об). Через 5 мин
прибавляют 10 мг натрия карбоната Р и доводят во¬
дой Р до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 30 °С;
- подвижная фаза:
57. Зак. 1060.
446
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза А: ацетонитрил Р1 — бу¬
ферный раствор А (0,7:99,3, об/об);
- подвижная фаза В: ацетонитрил Р1 — бу¬
ферный раствор А (25:75, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—9
100
0
9—24
100 —* 68
см
со
т
о
24—24,5
68 —* 50
32 —> 50
24,5—29
50
50
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики эпимеров примеси В, используя хромато¬
грамму раствора сравнения (с).
Относительное удерживание (по отношению к
имипенему, время удерживания — около 8 мин): эпи¬
мер I примеси В — около 0,33; эпимер II примеси В —
около 0,35; примесь А— около 0,8.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика эпимера I примеси В относительно
базовой линии; Ну — расстояние между базовой лини¬
ей и нижней точкой кривой, разделяющей пик эпимера
I примеси В и пик эпимера II примеси В).
Расчет процентного содержания:
- для расчета содержания примеси А умножают
площадь соответствующего пика на поправочный ко¬
эффициент — 2,4;
- рассчитывают процентное содержание каждой
примеси с учетом концентрации имипенема в раство¬
ре сравнения (Ь).
Предельное содержание примесей:
- примесь А: не более 1,0 %;
- примесь В (каждого эпимера): не более 0,3 %;
- неспецифицированные примеси: не более 0,10 %;
- сумма примесей: не более 1,5 %;
- учитываемый предел: 0,05 %.
Вода (2.5.12). Не менее 5,0 % и не более 8,0 %.
Определение проводят из 0,100 г испытуемого образ¬
ца. Используют йодсернистый реактив, содержащий
имидазол вместо пиридина, а также чистый контейнер
для каждого определения.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,17 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С12Н17М3045 в процентах рассчиты¬
вают с учетом содержания имипенема в ФСО имипе¬
нема.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре от 2 °С до 8 °С. Если субстанция стерильная,
ее хранят в стерильном воздухонепроницаемом кон¬
тейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. (5Я,6$)-3-[(2-Аминоэтил)сульфанил]-6-[(#)-1-
гидроксиэтил]-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-
карбоновая кислота (тиенамицин).
В. (2Р,4РЗ)-2-[(15,2Я)-1 -Карбокси-2-гидро-
ксипропил]-4-[[2-[(иминометил)амино]этил]сульфанил]-
3,4-дигидро-2Н-пиррол-5-карбоновая кислота (имипе-
немовая кислота).
07/2016:1108
ИНДАПАМИД
1пдаратШит
ШОАРАМЮЕ
С16Н16СШ3035 М.м. 365,8
[26807-65-8]
ОПРЕДЕЛЕНИЕ
4-Хлор-Л/-[(2/?5)-2-метил-2,3-дигидро-1 Н-индол-1 -
ил]-3-сульфамоилбензамид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого образ¬
ца растворяют в 96% спирте Р и доводят до объема
100,0 мл этим же растворителем. 2,0 мл полученного
раствора доводят 96 % спиртом Р до объема 100,0 мл.
Индапамид
447
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 242 нм.
Плечи: при 279 нм и при 287 нм.
Удельный показатель поглощения в максимуме:
от 590 до 630.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках с калия бромидом Р.
Сравнение: ФСО индапамида #или спектр, пред¬
ставленный на рисунке #1108.-1
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО индапамида
растворяют в 96% спирте Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО индометаци-
на растворяют в 5 мл раствора сравнения (а) и дово¬
дят 96 % спиртом Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ля в Р254 Р.
Подвижная фаза : кислота уксусная ледяная Р —
ацетон Р — толуол Р (1:20:79, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения (а).
ИСПЫТАНИЯ
Угол оптического вращения (2.2.7). От -0,02°
до +0,02°. 0,250 г испытуемого образца растворяют в
этаноле Р и доводят до объема 25,0 мл этим же раст¬
ворителем.
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29). Испытание проводят с защитой от
попадания света. Растворы хранят при температуре
4 °С или используют свежеприготовленные растворы.
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 7 мл смеси из равных объемов
ацетонитрила Р и метанола Р и доводят раствором
0,2 г/л натрия эдетата Р до объема 20,0 мл.
Раствор сравнения (а). 3,0 мг ФСО индапамида
примеси В растворяют в 3,5 мл смеси из равных объ¬
емов ацетонитрила Р и метанола Р и доводят рас¬
твором 0,2 г/л натрия эдетата Р до объема 10,0 мл.
К 1,0 мл полученного раствора прибавляют 35 мл
смеси из равных объемов ацетонитрила Р и мета¬
нола Р и доводят раствором 0,2 г/л натрия эдета¬
та Р до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью ацетонитрил Р — ме¬
танол Р — раствор 0,2 г/л натрия эдетата Р
(17,5:17,5:65, об/об/об) до объема 50,0 мл. 1,0 мл
полученного раствора доводят смесью ацетони¬
трил Р — метанол Р — раствор 0,2 г/л натрия эде¬
тата Р (17,5:17,5:65, об/об/об) до объема 20,0 мл.
Раствор сравнения (с). 20,0 мг ФСО индапамида
растворяют в 7 мл смеси из равных объемов ацето¬
нитрила Р и метанола Р и доводят раствором 0,2 г/л
натрия эдетата Р до объема 20,0 мл.
Раствор сравнения (ф. 25,0 мг ФСО индапамида
и 45,0 мг ФСО метилнитрозоиндолина (примесь А)
растворяют 17,5 мл смеси из равных объемов ацето¬
нитрила Р и метанола Р и доводят раствором 0,2 г/л
натрия эдетата Р до объема 50,0 мл.
Рисунок #1108.-1. Инфракрасный спектр ФСО индапамида
57*. Зак. 1060.
448
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
- колонка длиной 0,20 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: кислота уксусная ледяная —
ацетонитрил Р — метанол Р — раствор 0,2 г/л на¬
трия эдетата Р (0,1:17,5:17,5:65, об/об/об/об);
- скорость подвижной фазы: 2 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания индапамида.
Время удерживания: индапамид — около 11 мин.
Пригодность хроматографической системы:
- разрешение: не менее 4,0 между пиками ин¬
дапамида и примеси А на хроматограмме раствора
сравнения (с!);
- отношение сигнал/шум: не менее 6 для основ¬
ного пика на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика примеси В не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си В, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Примесь А. Не более 0,0005 % (5 ррт). Жид¬
костная хроматография (2.2.29). Испытание прово¬
дят с защитой от света.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в 1 мл ацетонитрила Р и доводят
водой Р до объема 10,0 мл. Встряхивают в течение
15 мин, выдерживают при температуре 4 °С в течение
1 ч и фильтруют.
Раствор сравнения. 25,0 мг испытуемого образ¬
ца растворяют в 1,0 мл раствора 0,125 мг/л ФСО ме-
тилнитрозоиндолина (примесь А) в ацетонитриле Р
и доводят водой Р до объема 10,0 мл. Встряхивают в
течение 15 мин, выдерживают при температуре 4 °С в
течение 1 ч и фильтруют.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: смесь из 7 объемов ацетони¬
трила Р, 20 объемов тетрагидрофурана Р и 73 объ¬
ема раствора 1,5 г/л триэтиламина Р, доведенного
кислотой фосфорной Р до рН 2,8;
- скорость подвижной фазы: 1,4 мл/мин;
-спектрофотометрический детектор, длина
волны 305 нм;
- объем вводимой пробы: 0,1 мл.
Пригодность хроматографической системы:
раствор сравнения:
- отношение сигнал/шум: не менее 3 для пика
примеси А, выходящего непосредственно перед ос¬
новным пиком;
- коэффициент разделения пиков: не менее 6,7
(Нр — высота пика примеси А относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси А и ос¬
новной пик).
Предельное содержание примесей:
-примесь А: на хроматограмме испытуемого
раствора площадь пика примеси А не должна превы¬
шать разницу между площадями пиков примеси А на
хроматограммах раствора сравнения и испытуемого
раствора.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца выдер¬
живают испытание на тяжелые металлы. Эталон го¬
товят с использованием 2 мл эталонного раствора
свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение
проводят из 0,10 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (с).
Пригодность хроматографической системы:
раствор сравнения (с):
- относительное стандартное отклонение:
не более 1,0 % для площадей пиков при 6 повторных
вводах пробы.
Содержание С16Н16С1М3033 в процентах рассчи¬
тывают с учетом содержания индапамида в ФСО ин¬
дапамида.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
н
ом
А. (2К5)-2-Метил-1 -нитрозо-2,3-дигидро-1 Н-индол.
В. 4-Хлор-Л/-(2-метил-1 Н-индол-1 -ил]-3-сульфамо-
илбензамид.
Индометацин
449
07/2016:0092
ИНДОМЕТАЦИН
1пс1оте1астит
ШООМЕТАС1Ы
С19Н16С1М04 М.м. 357,8
[53-86-1]
ОПРЕДЕЛЕНИЕ
[1-(4-Хлорбензоил)-5-метокси-2-метилиндол-3-ил]-
уксусная кислота.
Содержание: не менее 98,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический порошок от белого до желто¬
го цвета.
Практически нерастворим в воде, умеренно рас¬
творим в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О, Е.
A. Температура плавления (2.2.14): от 158 °С
до 162 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 25 мг испытуемого образ¬
ца растворяют в смеси 7 М раствор кислоты хлори¬
стоводородной — метанол Р (1:9, об/об) и доводят
до объема 100,0 мл этим же растворителем. 10,0 мл
полученного раствора доводят смесью 7 М раствор кис¬
лоты хлористоводородной — метанол Р( 1:9, об/об) до
объема 100,0 мл.
Диапазон длин волн: от 300 нм до 350 нм.
Максимумы поглощения: при 318 нм.
Удельный показатель поглощения в максимуме:
от 170 до 190.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: определяют подлинность об¬
разца в твердом состоянии, без перекристаллизации.
Сравнение: ФСО индометацина #или спектр,
представленный на рисунке #0092.-1#.
О. 0,1 г испытуемого образца растворяют в 10 мл
96 % спирта Р, при необходимости слегка подогрева¬
ют. К 0,1 мл полученного раствора прибавляют 2 мл
свежеприготовленной смеси из 1 объема раствора
250 г/л гидроксиламина гидрохлорида Р и 3 объемов
раствора натрия гидроксида разведенного Р. При¬
бавляют 2 мл кислоты хлористоводородной разве¬
денной Р, 1 мл раствора железа хлорида Р2 и переме¬
шивают. Появляется фиолетово-розовое окрашивание.
Е. К 0,5 мл спиртового раствора, приготовленного
в идентификации О, прибавляют 0,5 мл раствора ди-
метиламинобензальдегида Р2. Образуется осадок,
который растворяется при встряхивании. Нагревают
на водяной бане. Появляется голубовато-зеленое
окрашивание. Продолжают нагревать в течение 5 мин
и охлаждают в ледяной воде в течение 2 мин. Обра¬
зуется осадок, и окраска изменяется на светло-серо¬
зеленую. Прибавляют 3 мл 96% спирта Р. Раствор
становится прозрачным, и появляется фиолетово-ро¬
зовое окрашивание.
Рисунок #0092.-1. Инфракрасный спектр ФСО индометацина
58. Зак. 1060.
450
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р — вода
для хроматографии Р (50:50, об/об).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое виалы с
ФСО индометацина смеси примесей (примеси I и <))
растворяют в 1,0 мл смеси растворителей.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилгексил-
силильным эндкепированным для хроматографии Р
с размером частиц 3 мкм;
- температура: 40 °С;
- подвижная фаза:
-подвижная фаза А: раствор 10 г/л кислоты
уксусной Р;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
70
30
2—11
0
1
сл
о
30-50
11—12
50
50
12
сл
о
^1
о
50-30
12—21
70 — 30
30 — 70
21—27
30
70
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей I и 3, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО индометацина смеси примесей.
Относительное удерживание (по отношению к
индометацину, время удерживания — около 18 мин):
примесь I — около 1,3; примесь 3 — около 1,4.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
месей I и 3.
Расчет процентных содержаний:
- для каждой примеси — используют концентра¬
цию индометацина в растворе сравнения (а).
Предельное содержание примесей:
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,3 %;
- учитываемый предел: 0,05 %.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 4 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 75 мл
ацетона Р, через который предварительно пропуска¬
ют азот Р, свободный от углерода диоксида, в тече¬
ние 15 мин. Поддерживают постоянный поток азота
через полученный раствор. Прибавляют 0,1 мл раст¬
вора фенолфталеина Р и титруют 0,1 М раствором
натрия гидроксида.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 35,78 мг С19Н16С1М04.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): А, В, С, О, Е, Р, <3, Н, I, 3.
А. 4-Хлорбензойная кислота.
В. (5-Метокси-2-метил-1/7-индол-3-ил)уксусная кис¬
лота.
С. 4-Хлор-Л/-(4-метоксифенил)бензамид.
й. [1 -(2-Хлорбензоил)-5-метокси-2-метил-1 /7-индол-
3-ил]уксусная кислота.
Инсулин аспарт
451
Е. [1 -(3-Хлорбензоил)-5-метокси-2-метил-1 Н-индол-
3-ил]уксусная кислота.
Р. 4-Хлор-Л/’-(4-хлорбензоил)-М-(4-метоксифенил)
бензогидразид.
6. [1 -(3,4-Дихлорбензоил)-5-метокси-2-метил-1 Н-
индол-3-ил]уксусная кислота.
Н. Метил [1-(4-хлорбензоил )-5-метокси-2-метил-
1 Н-индол-3-ил]ацетат.
I. Этил[1 -(4-хлорбензоил)-5-метокси-2-метил-1 Н-
индол-3-ил]ацетат.
^ 4-Хлор-Л/'-[2-[1-(4-хлорбензоил)-5-метокси-2-
метил-1Н-индол-3-ил]ацетил]-Л/-(4-метоксифенил)бен-
зогидразид.
07/2016:2084
ИНСУЛИН АСПАРТ
1пзиНпит азраПит
Ш8Ш-1Ы А8РАКТ
I 1
Н-С1у-11е-Уа1-С1и-С1п-Суз-Суз-Т11Г-5ег-11е-Суз-5ег-
| 10
Ьеи-Туг-С1п-Ьеи-С1и-Азп-Туг-Суз-Азп-0Н
20|
Н-РЬе-Уа1-Азп-С1п-Н1з-Ьеи-Суз--С1у-Зег-Н1з-Ъеи-
10
Уа1-61и-А1а-Ъеи-Туг-Ъеи-Уа1-Суз-С1у-С1и-Агд-
20
СХу-РЬе-РЬе-Туг-ТЪг-Азр-Ъуз-ТЪг-ОН
30
С2«Н381М6507936 М.м.5826
ОПРЕДЕЛЕНИЕ
28в-/_-Аспартат инсулина (человеческий).
2-цепочечный пептид, содержащий 51 амино¬
кислоту. А-цепочка состоит из 21 аминокислоты,
а В-цепочка — из 30 аминокислот. Его первичная
структура идентична человеческому инсулину, за ис¬
ключением того, что вместо пролина в положении 28
В-цепочки находится аспарагиновая кислота. Так же
как и человеческий инсулин, инсулина аспарт содержит
2 дисульфидные связи, соединяющие пептидные цепи,
и одну дисульфидную связь, находящуюся в цепи.
Содержание: не менее 90,0 % и не более 104,0 %
суммы инсулина аспарта С256Н381М6507936, А21Азр ин¬
сулина аспарта, ВЗАзр инсулина аспарта, ВЗ|'зоАзр
инсулина аспарта и В28|ЗоАзр инсулина аспарта
(в пересчете на сухое вещество).
В целях маркировки лекарственных средств, со¬
держащих инсулин аспарт, принято, что 0,0350 мг ин¬
сулина аспарта эквивалентны 1 МЕ инсулина.
ПРОИЗВОДСТВО
Инсулин аспарт получают методом, основанным
на технологии рекомбинантной ДНК (рДНК), в услови¬
ях, направленных на минимизацию степени микроб¬
ной контаминации.
Предварительно перед реализацией проводят¬
ся следующие испытания на каждой партии готового
нефасованного продукта в случае отсутствия осво¬
бождения от их проведения, предоставленного компе¬
тентным уполномоченным органом.
Белки кпеток-хозяев. Предел утверждается
компетентным уполномоченным органом.
Одноцепочечный прекурсор. Предел утверж¬
дается компетентным уполномоченным органом. Ис¬
пользуется метод с подходящей чувствительностью.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в 96 % спирте, в мета¬
ноле и в водных растворах, имеющих рН около 5,1. В
водных растворах, имеющих рН ниже 3,5 или выше
6,5, растворимость больше или равна 25 мг/мл.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со-
58*. Зак. 1060.
452
Гэсударственная фармакопея Республики Беларусь
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
В. Пептидное картирование (2.2.55).
СЕЛЕКТИВНОЕ РАСЩЕПЛЕНИЕ ПЕПТИДНЫХ
СВЯЗЕЙ
Испытуемый раствор. Готовят раствор
2,0 мг/мл испытуемого образца в 0,01 М растворе
кислоты хлористоводородной. 25 мкл полученного
раствора помещают в чистую пробирку, прибавля¬
ют 100 мкл буферного (НЕРЕ8) раствора рН 7,5 Р
и 20 мкл раствора 1 мг/мл протеазы 81ар11у1ососсиз
аигеиз штамм \/8, тип Х\/11-В, Р. Закрывают пробир¬
ку и инкубируют при температуре 25 °С в течение 6 ч.
Останавливают реакцию прибавлением 145 мкл суль¬
фатного буферного раствора рН 2,0 Р.
Раствор сравнения. Готовят параллельно с ис¬
пытуемым раствором, используя ФСО инсулина
аспарта вместо испытуемого образца.
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ. Жид¬
костная хроматография (2.2.29).
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц
3 мкм) с размером пор 8 нм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 100 мл аце¬
тонитрила для хроматографии Р, 200 мл суль¬
фатного буферного раствора рН 2,0 Р и 700 мл
воды Р, фильтруют и дегазируют;
- подвижная фаза В: смешивают 200 мл суль¬
фатного буферного раствора рН 2,0 Р, 400 мл
ацетонитрила для хроматографии Р и 400 мл
воды Р, фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
90 —* 30
10 — 70
60—65
30 — 0
^1
о
о
о
65—70
0
100
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 214 нм;
-уравновешивание колонки: в начальных ус¬
ловиях не менее 15 мин. Проводят холостой прогон,
используя вышеуказанную программу градиентного
элюирования;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы
(на хроматограмме раствора сравнения идентифи¬
цируют пики, соответствующие расщепленным фраг¬
ментам I, II и III):
-хроматограмма испытуемого раствора и раст¬
вора сравнения должны качественно соответствовать
хроматограмме расщепленного инсулина аспарта,
прилагаемой к ФСО инсулина аспарта;
- фактор асимметрии: не более 1,5 для пиков
фрагментов II и III;
- разрешение: не менее 8,0 между пиками фраг¬
ментов II и III.
Результаты: профиль хроматограммы испытуе¬
мого раствора соответствует профилю хроматограм¬
мы раствора сравнения.
ПРИМЕЧАНИЕ: время удерживания фрагмен¬
тов I, II и IV такое же, как и для инсулина человече¬
ского. Время удерживания фрагмента III отличает¬
ся от времени удерживания для человеческого инсу¬
лина в связи с замещением пролина аспарагиновой
кислотой.
ИСПЫТАНИЯ
Примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина аспарта. Экс-
клюзионная хроматография (2.2.30): определение
проводят методом внутренней нормализации.
Испытуемый раствор. Готовят раствор 4 мг/мл
испытуемого образца в 0,01 М растворе кислоты
хлористоводородной. Раствор хранят при температу¬
ре от 2 °С до 8 °С и используют в течение 48 ч.
Раствор сравнения. Используют раствор около
4 мг/мл инсулина, содержащего более 0,4 % белков с
высокой молекулярной массой. Может использовать¬
ся инъекционный препарат инсулина, как раствор, так
и суспензия, растворенная в достаточным количестве
6 М растворе кислоты хлористоводородной, содер¬
жащий обнаруживаемое количество белков с высокой
молекулярной массой, или раствор, приготовленный
из субстанции инсулина, растворенной в 0,01 М рас¬
творе кислоты хлористоводородной. Инсулин, со¬
держащий обнаруживаемое количество белков с вы¬
сокой молекулярной массой, может быть приготовлен,
если выдержать порошок инсулина при комнатной
температуре в течение 10 дней. Раствор хранят при
температуре от 2 °С до 8 °С и используют в течение
7 дней.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
не менее 7,8 мм, заполненная силикагелем гидро¬
фильным для хроматографии Р (размер частиц от
5 мкм до 10 мкм, размер пор от 12 нм до 12,5 нм), при¬
годным для разделения мономера инсулина от диме¬
ра и полимеров;
- подвижная фаза: смешивают кислоту уксус¬
ную ледяную Р, ацетонитрил для хроматографии Р,
раствор 1,0 г/л аргинина Р (15:20:65, об/об/об); филь¬
труют и дегазируют;
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 276 нм;
-уравновешивание: не менее 3 введений раст¬
вора сравнения; колонку считают уравновешенной,
если после двух последовательных введений получа¬
ют воспроизводимые результаты;
- объем вводимой пробы: 100 мкл;
- время хроматографирования: около 35 мин.
Времена удерживания: полимеры инсулина
аспарта — от 13 мин до 17 мин; димер инсулина
аспарта — около 17,5 мин; мономер инсулина аспар¬
та — около 20 мин; соли — около 22 мин.
Пригодность хроматографической системы:
раствор сравнения:
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика димера относительно базовой ли¬
нии; Ну — расстояние между базовой линией и нижней
точкой кривой, разделяющей пики димера и мономера).
Предельное содержание примесей:
-примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина аспарта (не бо¬
лее 0,5 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков со временем удержи¬
вания меньшим, чем время удерживания основного
пика, не должна превышать 0,5 % от суммы площадей
всех пиков;
Инсулин бычий
453
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики со временем
удерживания большим, чем время удерживания пика
мономера инсулина аспарта.
Родственные белки. Жидкостная хроматогра¬
фия (2.2.29.), как указано в разделе «Количественное
определение». Применяют метод нормализации.
Предельное содержание примесей:
- В281зоАзр инсулин аспарт: не более 1,0%;
- сумма А21Азр инсулина аспарта, ВЗАзр инсу¬
лина аспарта и ВЗ/зоАзр инсулина аспарта: не бо¬
лее 2,0 %;
- сумма других примесей: не более 1,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 0,200 г испытуемого образца сушат при
температуре 105 °С в течение 24 ч.
Сульфатная зола (2.4.14). Не более 6,0 % (в пе¬
ресчете на сухое вещество). Определение проводят
из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
10 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без последующей процедуры удаления
бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. Испытуемый образец
растворяют в 0,01 М растворе кислоты хлористово¬
дородной до получения концентрации 4,0 мг/мл. Раст¬
вор хранят при температуре от 2 °С до 8 °С и исполь¬
зуют в течение 24 ч.
Раствор сравнения (а). Содержимое контейнера
с ФСО инсулина аспарта растворяют в 0,01 М рас¬
творе кислоты хлористоводородной до получения
концентрации 4,0 мг/мл. Раствор хранят при темпера¬
туре от 2 °С до 8 °С и используют в течение 48 ч.
Раствор сравнения (Ь). Используют соответству¬
ющий раствор с содержанием ВЗАзр инсулина аспар¬
та и А21Азр инсулина аспарта не менее 1 %. Такой
раствор может быть получен при хранении раствора
сравнения (а) при комнатной температуре в течение
периода ориентировочно от 1 до 3 дней. Раствор хра¬
нят при температуре от 2 °С до 8 °С и используют в
течение 72 ч.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц
5 мкм);
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: 142,0 г натрия сульфа¬
та безводного Р растворяют в воде Р, прибав¬
ляют 13,5 мл кислоты фосфорной Р и доводят
водой Р до объема 5000 мл, при необходимости
доводят раствором натрия гидроксида концен¬
трированным Р до рН 3,6; фильтруют, дегазиру¬
ют. Смешивают 9 объемов полученного раствора
с 1 объемом ацетонитрила для хроматогра¬
фии Р\ фильтруют, дегазируют;
- подвижная фаза В: смешивают равные
объемы воды Р и ацетонитрила для хромато¬
графии Р; фильтруют, дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—35
58
42
35—40
58 —* 20
42 — 80
40—45
20
80
45—46
20-58
80 — 42
46—60
58
42
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
инсулина аспарту, время удерживания — от 20 мин
до 24 мин): В2813оАзр инсулин аспарт — около 0,9;
сумма ВЗАзр инсулина аспарта и А21Азр инсулина
аспарта (в основном совместно элюируются) — около
1,3; В313оАзр инсулин аспарт — около 1,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками инсу¬
лина аспарта, А21Азр инсулина аспарта и ВЗАзр ин¬
сулина аспарта.
Суммарное содержание инсулина аспарта
С256Нз8^65^7956» В28|ЗоАзр инсулина аспарта, А21Азр
инсулина аспарта, ВЗАзр инсулина аспарта и ВЗ|'зоАзр
инсулина аспарта рассчитывают из площадей соот¬
ветствующих пиков на хроматограммах испытуемого
раствора и раствора сравнения (а) и известного сум¬
марного содержания инсулина аспарта, В28|ЗоАзр ин¬
сулина аспарта, А21 Азр инсулина аспарта, ВЗАзр ин¬
сулина аспарта и В313оАзр инсулина аспарта в ФСО
инсулина аспарта.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре, равной или
ниже -18 °С, до момента реализации производите¬
лем. После оттаивания инсулин аспарт может хра¬
ниться при температуре (5±3) °С и использоваться
для приготовления лекарственных средств в течение
короткого промежутка времени. Для предотвращения
абсорбции влаги из воздуха в процессе взвешивания
инсулин аспарт должен быть комнатной температуры.
07/2016:1637
ИНСУЛИН БЫЧИЙ
1п8иНпит Ьомпит
1Ы8Ш.1Ы, ВОУШЕ
Н-01у—Не—Уа1—Ои-О1п-Суз—Су$—А1а—8ег-Уа1-
I ' 10
Су& - Зег - Ьеи - Туг - С!л—Ьеи—61и - Азл—Туг - Суз-
201
Азп—ОН
Н-РГ1в-Уа1-А$п-61п—Н*з—Ьеи—Суз-С1у—$ег-Ню-
1_еи-Уа1 - 31и - А1а—1-еи—Туг—1_еи—\7а1—Суз -<31у -
20
С1и- Агд - 01у - РИе -РНе -Туг—ТЬг—Рго—Ьуз—А1а—ОН!
С254Нз7Л50755б М.м. 5734
ОПРЕДЕЛЕНИЕ
Природное антидиабетическое вещество, полу¬
ченное из бычьей печени и очищенное.
59. Зак. 1060.
454
Гэсударственная фармакопея Республики Беларусь
Содержание: не менее 93,0 % и не более 105,0 %
суммы инсулина бычьего С254 Н377Ы6507536 и А21-дез-
амидоинсулина бычьего (в пересчете на сухое веще¬
ство).
В целях маркировки лекарственных средств, со¬
держащих инсулин, принято, что 0,0342 мг инсулина
бычьего эквивалентны 1 МЕ инсулина.
ПРОИЗВОДСТВО
Животные, используемые для получения инсули¬
на бычьего, должны выдерживать требования, предъ¬
являемые к здоровью животных, используемых чело¬
веком в пищу.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде и спирте. Рас¬
творяется в разведенных минеральных кислотах и с
разложением в разведенных растворах гидроксидов
щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (с).
B. Пептидное картирование.
Испытуемый раствор. Готовят раствор
2,0 мг/мл испытуемого образца в 0,01 М растворе
кислоты хлористоводородной. 500 мкл полученно¬
го раствора помещают в чистую пробирку, прибавля¬
ют 2,0 мл буферного (НЕРЕ8) раствора рН 7,5 Р и
400 мкл раствора 1 мг/мл протеазы 81арЬу1ососсиз
аигеиз штамм \/8, тип XVI1-В, Р. Закрывают пробирку
и инкубируют при температуре 25 °С в течение 6 ч.
Останавливают реакцию прибавлением 2,9 мл суль¬
фатного буферного раствора рН 2,0 Р.
Раствор сравнения. Готовят параллельно с ис¬
пытуемым раствором, используя ФСО инсулина бы¬
чьего вместо испытуемого образца.
Продукты расщепления исследуют с использова¬
нием жидкостной хроматографии (2.2.29).
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р (размер частиц 3 мкм);
- температура: 40 °С;
- подвижная фаза:
-подвижная фаза А: смешивают 100 мл аце¬
тонитрила для хроматографии Р, 700 мл воды
Р и 200 мл сульфатного буферного раствора
рН 2,0 Р, фильтруют и дегазируют;
- подвижная фаза В: смешивают 400 мл аце¬
тонитрила для хроматографии Р, 400 мл воды
Р и 200 мл сульфатного буферного раствора
рН 2,0 Р, фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
90 -> 30
о
т
о
т-
60—65
о
т
о
со
70->100
65—70
0
100
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
-уравновешивание колонки: в начальных ус¬
ловиях не менее 15 мин. Проводят холостой прогон,
используя вышеуказанную программу градиентного
элюирования;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы
(на хроматограмме раствора сравнения идентифи¬
цируют пики, соответствующие расщепленным фраг¬
ментам I, II и III):
- хроматограмма испытуемого раствора и раст¬
вора сравнения должны качественно соответствовать
хроматограмме расщепленного инсулина бычьего,
прилагаемой к ФСО инсулина бычьего;
- фактор асимметрии: не более 1,5 для пиков
фрагментов II и III;
- разрешение: не менее 1,9 между пиками фраг¬
ментов II и III.
Результаты: профиль хроматограммы испытуе¬
мого раствора соответствует профилю хроматограм¬
мы раствора сравнения.
ПРИМЕЧАНИЕ: время удерживания фрагмента
I одинаково для инсулина свиного и человеческого.
Время удерживания фрагментов II и IV одинаково
для всех инсулинов. Время удерживания фрагмента
III одинаково для свиного и бычьего инсулинов.
ИСПЫТАНИЯ
Примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина. Эксклюзион-
ная хроматография (2.2.30): определение проводят
методом внутренней нормализации. Растворы хра¬
нят при температуре от 2 °С до 10 °С и использу¬
ют в течение 7 дней. При использовании автомати¬
ческого инжектора поддерживают его температу¬
ру от 2 °С до 10 °С.
Испытуемый раствор. 4 мг испытуемого образ¬
ца растворяют в 1,0 мл 0,01 М раствора кислоты
хлористоводородной.
Раствор сравнения. Используют раствор около
4 мг/мл инсулина, содержащего более 0,4 % белков с
высокой молекулярной массой. Может использовать¬
ся инъекционный препарат инсулина, как раствор, так
и суспензия, растворенная в достаточным количестве
6 М кислоты хлористоводородной, содержащий об¬
наруживаемое количество белков с высокой молеку¬
лярной массой, или раствор, приготовленный из суб¬
станции инсулина, растворенной в 0,01 М растворе
кислоты хлористоводородной. Инсулин, содержа¬
щий обнаруживаемое количество белков с высокой
молекулярной массой, может быть приготовлен, если
выдержать порошок инсулина при комнатной темпе¬
ратуре в течение 10 дней.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
не менее 7,5 мм, заполненная силикагелем гидро¬
фильным для хроматографии Р (размер частиц от
5 мкм до 10 мкм), пригодным для разделения моно¬
мера инсулина от димера и полимеров;
- подвижная фаза: смешивают кислоту уксусную
ледяную Р, ацетонитрил Р и раствор 1,0 г/л аргини¬
на Р (15:20:65, об/об/об); фильтруют и дегазируют;
- скорость подвижной фазы: 0,5 мл/мин;
- спектрофотометрический детектор, длина
волны 276 нм;
- уравновешивание: перед использованием но¬
вой колонки для хроматографического анализа урав¬
новешивают ее путем повторных введений раствора
Инсулин бычий
455
инсулина, содержащего белки высокой молекулярной
массы (не менее трех повторных введений раствора
сравнения). Колонку считают уравновешенной, если
после двух последовательных введений получают
воспроизводимые результаты;
- объем вводимой пробы: 100 мкл;
- время хроматографирования: около 35 мин.
Времена удерживания: полимерные инсулино¬
вые комплексы — от 13 мин до 17 мин; ковалентный
димер инсулина — около 17,5 мин; мономер инсулина
— около 20 мин; соли — около 22 мин.
Пригодность хроматографической системы:
раствор сравнения:
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика димера относительно базовой ли¬
нии; Ну — расстояние между базовой линией и нижней
точкой кривой, разделяющей пики димера и мономера).
Предельное содержание примесей:
-примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина (не более 1,0 %):
на хроматограмме испытуемого раствора сумма пло¬
щадей всех пиков со временем удерживания меньшим,
чем время удерживания основного пика, не должна
превышать 1,0 % от суммы площадей всех пиков;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики со временем
удерживания большим, чем время удерживания пика
инсулина.
Родственные белки. Жидкостная хроматогра¬
фия (2.2.29.), как указано в разделе «Количественное
определение», со следующей программой градиент¬
ного элюирования:
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
42
58
30—44
42 —> 11
58 —► 89
44—50
11
89
Растворы хранят при температуре от 2 °С до
10 °С и используют в течение 24 ч.
Пригодность хроматографической системы
указана в разделе «Количественное определение».
При необходимости изменяют относительные соот¬
ношения подвижных фаз для обеспечения полного
элюирования А21-дезамидоинсулина бычьего перед
началом градиента. Профиль градиента также может
регулироваться для обеспечения полного элюирова¬
ния всех родственных инсулину примесей.
Объем вводимой пробы: по 20 мкл раствора
сравнения (с) и испытуемого раствора. При необхо¬
димости изменяют объем вводимой пробы от 10 мкл
до 20 мкл в соответствии с результатами определения
линейности для проверки пригодности хроматографи¬
ческой системы, как указано в разделе «Количествен¬
ное определение».
Время хроматографирования: около 50 мин.
Относительное удерживание (по отношению к
основному пику): А21-дезамидоинсулин бычий (не¬
большой пик, элюирующийся после основного пика на
хроматограмме раствора сравнения (с)) — около 1,3.
Предельное содержание примесей:
- А21-дезамидоинсулин бычий (не более 3,0 %):
на хроматограмме испытуемого раствора площадь
пика А21-дезамидоинсулина бычьего не должна пре¬
вышать 3,0 % от суммы площадей всех пиков;
- сумма примесей (не более 3,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного и пика А21-дезамидоинсулина
бычьего, не должна превышать 3,0 % от суммы пло¬
щадей всех пиков.
Проинсулин-подобные иммунореактивные
примеси (РЦ). Не более 0,0010 % (10 ррт) (в пере¬
счете на сухое вещество). Определение проводят
подходящим по чувствительности иммунохимическим
методом (2.7.1), например радиоиммунологическим
анализом, с использованием Международного стан¬
дартного реагента проинсулина бычьего для кали¬
бровки метода.
Цинк. Не более 1,0 % (в пересчете на сухое
вещество). Атомно-абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хло¬
ристоводородной и доводят до объема 25,0 мл этим
же растворителем. При необходимости разводят
0,01 М раствором кислоты хлористоводородной
до получения подходящей концентрации (например,
от 0,4 мкг/мл до 1,6 мкг/мл 2п).
Растворы сравнения. Используют свежепри¬
готовленные растворы, содержащие 0,40 мкг/мл,
0,80 мкг/мл, 1,00 мкг/мл, 1,20 мкг/мл и 1,60 мкг/мл
2п. Растворы готовят путем разведения эталонного
раствора цинка (5 мг/мл 7.п) Р 0,01 М раствором кис¬
лоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя подходящего состава (например, воз¬
дух — 11 л/мин, ацетилен — 2 л/мин).
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 0,200 г испытуемого образца сушат при
температуре 105 °С в течение 24 ч.
Сульфатная зола (2.4.14). Не более 2,5 % (в пе¬
ресчете на сухое вещество). Определение проводят
из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
10 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без последующей процедуры удаления
бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. Необходимое количество
испытуемого образца растворяют в 0,01 М растворе
кислоты хлористоводородной до получения концен¬
трации 4,0 мг/мл.
Раствор сравнения (а). Содержимое контейнера
с ФСО инсулина человеческого растворяют в 0,01 М
растворе кислоты хлористоводородной до получе¬
ния концентрации 4,0 мг/мл.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО инсулина свиного для проверки пригодности
системы растворяют в 0,01 М растворе кислоты
хлористоводородной до получения концентрации
4 мг/мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО инсулина бычьего растворяют в 0,01 М рас¬
творе кислоты хлористоводородной до получения
концентрации 4,0 мг/мл.
59*. Зак. 1060.
456
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (с) доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 10,0 мл.
Раствор сравнения (е). Смешивают 1,0 мл раст¬
вора сравнения (а) и 1,0 мл раствора сравнения (Ь).
Растворы хранят при температуре от 2 °С до 10 °С и
используют в течение 48 ч. При использовании авто¬
матического инжектора поддерживают его температу¬
ру от 2 °С до 10 °С.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р (размер частиц 5 мкм);
- температура: 40 °С;
- подвижная фаза: смешивают подвижную фазу
А и подвижную фазу В (42:58, об/об), при необходимо¬
сти состав корректируют;
следующие растворы готовят и хранят при
температуре не ниже 20 °С:
- подвижная фаза А: 28,4 г натрия сульфа¬
та безводного Р растворяют в воде Р и дово¬
дят этим же растворителем до объема 1000 мл;
прибавляют 2,7 мл кислоты фосфорной Р; при
необходимости доводят этаноламином Р до рН
2,3; фильтруют и дегазируют;
- подвижная фаза В: 550 мл подвижной фазы
А смешивают с 450 мл ацетонитрила Р. Полу¬
ченный раствор подогревают до температуры
не ниже 20 °С для предотвращения выпадения
осадка (смешивание подвижной фазы А с аце¬
тонитрилом — эндотермический процесс), филь¬
труют и дегазируют;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора, растворов сравнения (Ь), (с), (б) и (е).
Пригодность хроматографической системы:
- разрешение (на хроматограмме раствора срав¬
нения (е) пик, соответствующий основному пику на
хроматограмме раствора сравнения (Ь), должен быть
четким. На хроматограмме раствора сравнения (е)
идентифицируют пики, соответствующие свиному и
человеческому инсулину): не менее 1,2 между пиками
человеческого и свиного инсулина на хроматограмме
раствора сравнения (е). При необходимости изменя¬
ют концентрацию ацетонитрила в подвижной фазе;
- линейность: площадь основного пика на хрома¬
тограмме раствора сравнения (с) должна превышать
площадь основного пика на хроматограмме раствора
сравнения (б) в (10±0,5) раз. При необходимости из¬
меняют объем вводимой пробы до (10—20) мкл, в за¬
висимости от уровня линейности детектора.
Суммарное содержание инсулина бычьего
(С254Нз77|^б5°753б) и А21-дезамидоинсулина бычьего
рассчитывают из площади основного пика и пика, со¬
ответствующего А21-дезамидоинсулину бычьему на
хроматограммах испытуемого раствора и раствора
сравнения (с), и известного суммарного содержания
инсулина бычьего и А21-дезамидоинсулина бычьего в
ФСО инсулина бычьего.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, защищен¬
ном от света, при температуре -20 °С до момента ре¬
ализации производителем. После оттаивания инсулин
может храниться при температуре (5±3) °С и исполь¬
зоваться для приготовления лекарственных средств в
течение короткого промежутка времени. Для предот¬
вращения поглощения влаги из воздуха в процессе взве¬
шивания инсулин должен быть комнатной температуры.
07/2016:0831
ИНСУЛИН ДВУХФАЗОВЫЙ
ДЛЯ ИНЪЕКЦИЙ
1п8иНпит ЫрЬазюит 1мес(аЫ1е
1Ы8Ш.1Ы 1ШЕСТ10Ы, В1РНА81С
Инсулин двухфазовый для инъекций должен со¬
ответствовать требованиям статьи Инъекцион¬
ные лекарственные средства инсулина (0854) с из¬
менениями, приведенными ниже.
ОПРЕДЕЛЕНИЕ
Стерильная суспензия кристаллов, содержащая
инсулин бычий в растворе инсулина свиного.
ОПИСАНИЕ(СВОЙСТВА)
Белая или почти белая суспензия. Изучение с по¬
мощью микроскопа показывает, что большая часть ча¬
стиц является ромбоэдрическими кристаллами, име¬
ющими при измерении от одного угла кристалла до
другого максимальный размер более 10 мкм, а иногда
более 40 мкм.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные в
разделе «Количественное определение». Пики обоих
инсулинов на хроматограмме испытуемого раствора
по положению соответствуют основным пикам на хро¬
матограмме соответствующего раствора сравнения.
ИСПЫТАНИЯ
рН (2.2.3). От 6,6 до 7,2. Измеряют рН испытуе¬
мой суспензии.
Содержание инсулина в надосадочном слое.
От 22,0 % до 28,0 % от содержания инсулина в рас¬
творе. Определяют с помощью метода, описанного в
статье Инъекционные лекарственные средства ин¬
сулина (0854).
Общий цинк. От 26,0 мкг до 37,5 мкг на 100 МЕ
инсулина, определенного как описано статье Инъек¬
ционные лекарственные средства инсулина (0854).
07/2016:0832
ИНСУЛИН ИЗОФАН ДВУХФАЗОВЫЙ
ДЛЯ ИНЪЕКЦИЙ
IпзиНпит 1’зор11апит ЫрМаз1сит 1т'ес(аЫ1е
М81Л-1Ы ШЕСТЮЫ, В1РНА31С
180РНАЫЕ
Инсулин изофан двухфазовый для инъекций
должен соответствовать требованиям статьи
Инъекционные лекарственные средства инсулина
(0854), за исключением испытания «Содержание ин¬
сулина в надосадочном слое» и учитывая изменения,
приведенные ниже.
ОПРЕДЕЛЕНИЕ
Стерильная забуференная суспензия комплекса
инсулина свиного или инсулина человеческого с про-
Инсулин лизпро
457
тамином сульфатом или другим подходящим прота-
мином в растворе того же инсулина.
ПРОИЗВОДСТВО
Получают проведением действий, описанных в
статье Инъекционные лекарственные средства ин¬
сулина (0854).
Получают перемешиванием растворимого инсу¬
лина для инъекций и инсулина изофана для инъек¬
ций в заданных пропорциях. Соответствие заданных
пропорций указанным на этикетке должно быть под¬
тверждено испытанием, утвержденным компетент¬
ным уполномоченным органом.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая суспензия, при отстаи¬
вании которой образуется осадок белого или почти
белого цвета и бесцветная или почти бесцветная на-
досадочная жидкость; осадок легко ресуспендирует-
ся при осторожном встряхивании. Изучение с помо¬
щью микроскопа показывает, что частицы являются
стержнеобразными кристаллами, большая часть ко¬
торых имеет максимальный размер более 1 мкм, а
иногда — более 60 мкм, не образующими большие
агрегаты.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные в
разделе «Количественное определение». Пик инсули¬
на на хроматограмме испытуемого раствора по поло¬
жению соответствует основному пику на хроматограм¬
ме соответствующего раствора сравнения.
ИСПЫТАНИЯ
Общий цинк. Не более 40,0 мкг на 100 МЕ инсу¬
лина, определенного как описано в статье Инъекцион¬
ные лекарственные средства инсулина (0854).
МАРКИРОВКА
На этикетке дополнительно к положениям, изло¬
женным в статье Инъекционные лекарственные сред¬
ства инсулина (0854), указывают соотношение раство¬
римого инсулина для инъекций и инсулина изофана
для инъекций, используемое в процессе производства
инсулина изофана двухфазового для инъекций.
07/2016:0833
ИНСУЛИН ИЗОФАН ДЛЯ ИНЪЕКЦИЙ
1п8иНпит 13ор11апит 1тес1аЫ1е
1Ы81Л.1Ы ШЕСТЮЫ, 130РНАЫЕ
Инсулин изофан для инъекций должен соот¬
ветствовать требованиям статьи Инъекционные
лекарственные средства инсулина (0854) с измене¬
ниями, приведенными ниже.
ОПРЕДЕЛЕНИЕ
Стерильная суспензия комплекса инсулина бы¬
чьего, инсулина свиного или инсулина человеческо¬
го с протамином сульфатом или другим подходящим
протамином.
ПРОИЗВОДСТВО
Получают проведением действий, описанных в
статье Инъекционные лекарственные средства ин¬
сулина (0854).
Количество протамина основывается на извест¬
ной пропорции изофана и эквивалентно количеству,
составляющему не менее 0,3 мг и не более 0,6 мг про¬
тамина сульфата на каждые 100 МЕ инсулина в инсу-
лин-протаминовом комплексе.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая суспензия, при отстаи¬
вании которой образуется осадок белого или почти
белого цвета и бесцветная или почти бесцветная
надосадочная жидкость; осадок легко ресуспенди-
руется при осторожном встряхивании. Изучение с
помощью микроскопа показывает, что частицы яв¬
ляются стержнеобразными кристаллами, большая
часть которых имеет максимальный размер более
1 мкм, а иногда— более 60 мкм, не образующими
большие агрегаты.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные
в разделе «Количественное определение». Пик ин¬
сулина на хроматограмме испытуемого раствора
по положению соответствует основному пику на
хроматограмме соответствующего раствора срав¬
нения.
ИСПЫТАНИЯ
Общий цинк. Не более 40,0 мкг на 100 МЕ инсу¬
лина, определенного как описано в статье Инъекцион¬
ные лекарственные средства инсулина (0854).
07/2016:2085
ИНСУЛИН ЛИЗПРО
1пзиНпит Изргит
1Ы811Ш Ы8РРО
I 1
Н-01у-11е-Уа1-61и-С1п-Суз-Суз-ТЬг-Зег-11е-Суз-Зег-
I 10
Ъеи-Туг-С1п-Ьеи-С1и-Азп-Туг-Суз-Азп-0Н
20)
Н-Р]зе-Уа1-Азп-С1п-Н1з-Ьеи-Су5-01у-5ег-Н1з-Ьеи-
10
I
Уа1-С1и-А1а-Ьеи-Туг-Ьеи-Уа1-Суз-С1у-С1и-Агд-
20
СХу-РЬе-РЪе-Туг-ТЪг-Ъуз-Рго-ТЪг-ОН
30
С257Н38зМ6507756 М.м.5808
ОПРЕДЕЛЕНИЕ
28в-/--Лизин-29в-/.-пролин инсулина (человеческий).
2-цепочечный пептид, содержащий 51 амино¬
кислоту. А-цепочка состоит из 21 аминокислоты,
а В-цепочка — из 30 аминокислот. Его первичная
структура идентична человеческому инсулину, от¬
личие состоит только в последовательности ами¬
нокислот в положениях 28 и 29 В-цепочки. Если в
человеческом инсулине эта последовательность
Рго(В28), 1_уз(В29), то в инсулине лизпро — Ьуз(В28),
Рго(В29). Как и человеческий инсулин, инсулин лиз¬
про содержит 2 дисульфидные связи, соединяющие
пептидные цепи, и одну дисульфидную связь, нахо¬
дящуюся в цепи.
Содержание: не менее 94,0 % и не более 104,0 %
(в пересчете на сухое вещество).
60. Зак. 1060.
458
Гэсударственная фармакопея Республики Беларусь
В целях маркировки лекарственных средств, со¬
держащих инсулин лизпро, принято, что 0,0347 мг ин¬
сулина лизпро эквивалентны 1 МЕ.
ПРОИЗВОДСТВО
Инсулин лизпро получают методом, основан¬
ным на технологии рекомбинантной ДНК (рДНК), в
условиях, направленных на минимизацию степени
микробной контаминации.
Предварительно перед реализацией прово¬
дятся следующие испытания на каждой партии
готового нефасованного продукта в случае отсут¬
ствия освобождения от их проведения, предостав¬
ленного компетентным уполномоченным органом.
Белки клеток-хозяев. Предел утверждается
компетентным уполномоченным органом.
Одноцепочечный прекурсор. Предел ут¬
верждается компетентным уполномоченным орга¬
ном. Используется метод с подходящей чувстви¬
тельностью.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде и в 96 %
спирте. Растворяется в разведенных минеральных
кислотах и с разложением в разведенных раство¬
рах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, получен¬
ные в разделе «Количественное определение».
Результаты, основной пик на хроматограмме
испытуемого раствора по времени удерживания
соответствует основному пику на хроматограмме
раствора сравнения.
B. Пептидное картирование (2.2.55).
СЕЛЕКТИВНОЕ РАСЩЕПЛЕНИЕ ПЕПТИДНЫХ
СВЯЗЕЙ
Испытуемый раствор. Готовят раствор
2,0 мг/мл испытуемого образца в 0,01 М растворе
кислоты хлористоводородной. 500 мкл полученно¬
го раствора помещают в чистую пробирку, прибавля¬
ют 2,0 мл буферного (НЕРЕ8) раствора рН 7,5 Р и
400 мкл раствора 1 мг/мл протеазы 3!арЬу1ососсиз
аигеиз штамм \/8, тип Х\/11-В, Р. Закрывают пробир¬
ку и инкубируют при температуре 25 °С в течение 6 ч.
Останавливают реакцию прибавлением 2,9 мл суль¬
фатного буферного раствора рН 2,0 Р.
Раствор сравнения. Готовят параллельно с
испытуемым раствором, используя ФСО инсулина
лизпро вместо испытуемого образца.
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ.
Жидкостная хроматография (2.2.29).
Условия хроматографирования:
-колонка длиной 0,10 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем окта-
децилсилильным для хроматографии Р (размер
частиц 3 мкм) с размером пор 8 нм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 100 мл аце¬
тонитрила для хроматографии Р, 200 мл
сульфатного буферного раствора рН 2,0 Р и
700 мл воды Р, фильтруют и дегазируют;
-подвижная фаза В: смешивают 200 мл
сульфатного буферного раствора рН 2,0 Р,
400 мл ацетонитрила для хроматографии Р
и 400 мл воды Р, фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
90 — 30
о
^1
о
60—65
30->0
70—100
65—70
0
100
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 214 нм;
- уравновешивание колонки: в начальных ус¬
ловиях не менее 15 мин. Проводят холостой про¬
гон, используя вышеуказанную программу гради¬
ентного элюирования;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы
(на хроматограмме раствора сравнения иденти¬
фицируют пики, соответствующие расщепленным
фрагментам I, II и III):
- хроматограмма испытуемого раствора и
раствора сравнения должны качественно соответ¬
ствовать хроматограмме расщепленного инсулина
лизпро, прилагаемой к ФСО инсулина лизпро;
- фактор асимметрии: не более 1,5 для пи¬
ков фрагментов II и III;
- разрешение: не менее 8,0 между пиками
фрагментов II и III.
Результаты: профиль хроматограммы испы¬
туемого раствора соответствует профилю хромато¬
граммы раствора сравнения.
ПРИМЕЧАНИЕ: время удерживания фраг¬
ментов I, II и IV такое же, как и для инсулина че¬
ловеческого. Время удерживания фрагмента III
отличается от времени удерживания для чело¬
веческого инсулина в связи с различием в после¬
довательности аминокислот в положениях 28 и
29 В-цепочки.
ИСПЫТАНИЯ
Примеси с молекулярной массой, превыша¬
ющей молекулярную массу инсулина лизпро.
Эксклюзионная хроматография (2.2.30): определе¬
ние проводят методом внутренней нормализации.
Испытуемый раствор. Готовят раствор
4 мг/мл испытуемого образца в 0,01 М растворе
кислоты хлористоводородной. Раствор хранят
при температуре от 2 °С до 8 °С и используют в те¬
чение 48 ч.
Раствор сравнения. Используют раствор око¬
ло 4 мг/мл инсулина, содержащего более 0,4 %
белков с высокой молекулярной массой. Может
использоваться инъекционный препарат инсули¬
на, как раствор, так и суспензия, растворенная в
достаточным количестве 6 М раствора кислоты
хлористоводородной, содержащий обнаружива¬
емое количество белков с высокой молекулярной
массой, или раствор, приготовленный из субстан¬
ции инсулина, растворенной в 0,01 М растворе
кислоты хлористоводородной. Инсулин, содержа¬
щий обнаруживаемое количество белков с высокой
молекулярной массой, может быть приготовлен,
если выдержать порошок инсулина при комнатной
температуре в течение 10 дней. Раствор хранят
при температуре от 2 °С до 8 °С и используют в те¬
чение 8 дней.
Условия хроматографирования:
- колонка длиной 0,30 м и внутренним диа¬
метром 7,8 мм, заполненная силикагелем гидро¬
фильным для хроматографии Р (размер частиц от
Инсулин лизпро
459
5 мкм до 10 мкм, размер пор от 12 нм до 12,5 нм),
пригодным для разделения мономера инсулина от
димера и полимеров;
- подвижная фаза: смешивают кислоту уксус¬
ную ледяную Р, ацетонитрил для хроматографии Р,
раствор 1,0 г/л аргинина Р (15:20:65, об/об/об); филь¬
труют и дегазируют;
- скорость подвижной фазы. 0,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 276 нм;
- уравновешивание: не менее 3 введений
раствора сравнения; колонку считают уравнове¬
шенной, если после двух последовательных вве¬
дений получают воспроизводимые результаты;
- объем вводимой пробы: 100 мкл;
- время хроматографирования: около 35 мин.
Времена удерживания: полимеры инсулина
лизпро — от 13 мин до 17 мин; димер инсулина
лизпро — около 17,5 мин; мономер инсулина лиз¬
про — около 20 мин; соли — около 22 мин.
Пригодность хроматографической системы:
раствор сравнения:
- коэффициент разделения пиков: не менее
2,0 (Н — высота пика димера относительно базо¬
вой линии; Ну— расстояние между базовой линией
и нижней точкой кривой, разделяющей пики диме¬
ра и мономера);
- фактор асимметрии: не более 2,0 для пика
инсулина лизпро.
Предельное содержание примесей:
- примеси с молекулярной массой, превыша¬
ющей молекулярную массу инсулина лизпро (не
более 0,25 %): на хроматограмме испытуемого
раствора сумма площадей всех пиков со временем
удерживания меньшим, чем время удерживания
основного пика, не должна превышать 0,25 % от
суммы площадей всех пиков;
- неучитываемый предел: на хроматограмме
испытуемого раствора не учитывают пики со вре¬
менем удерживания большим, чем время удержи¬
вания пика мономера инсулина лизпро.
Родственные белки. Жидкостная хромато¬
графия (2.2.29.): применяют метод нормализации.
Испытуемый раствор. 3,5 мг испытуемого об¬
разца растворяют в 1,0 мл 0,01 М раствора кисло¬
ты хлористоводородной. Раствор хранят при тем¬
пературе от 2 °С до 8 °С и используют в течение
56 ч.
Раствор сравнения. 3,5 мг испытуемого об¬
разца растворяют в 1,0 мл 0,01 М раствора кисло¬
ты хлористоводородной, выдерживают при ком¬
натной температуре до получения раствора, содер¬
жащего от 0,8% до 11 % А21-дезамидоинсулина
лизпро.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем окта-
децилсилильным для хроматографии Р (размер
частиц 5 мкм) с размером пор 30 нм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 82 объема
раствора 28,4 г/л натрия сульфата безводно¬
го Р, доведенного кислотой фосфорной Р до
рН 2,3, и 18 объемов ацетонитрила для хрома¬
тографии Р\ фильтруют и дегазируют;
-подвижная фаза В: смешивают равные
объемы раствора 28,4 г/л натрия сульфата
безводного Р, доведенного кислотой фос¬
форной Р до рН 2,3, и ацетонитрила для хро¬
матографии Р; фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
81
19
60—83
81 — 51
19 — 49
83—84
51—81
49—19
84-94
81
19
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 214 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: около 35 мин.
Время удерживания: инсулин лизпро — кор¬
ректируют состав подвижной фазы для получе¬
ния времени удерживания около 41 мин; А21-дез-
амидоинсулин лизпро элюируется близко к началу
градиента элюирования.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: не менее 1,5 между первым
пиком (инсулин лизпро) и вторым пиком (А21-дез-
амидоинсулин лизпро);
- фактор асимметрии: не более 2,0 для пика
инсулина лизпро.
Предельное содержание примесей:
- А21-дезамидоинсулин лизпро: не более
1,0 %;
-любая другая примесь: не более 0,50 %;
- сумма примесей, кроме примеси А21: не бо¬
лее 2,0 %.
Цинк. Не более 1,0% (в пересчете на сухое
вещество). Атомно-абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. Не менее 50 мг испы¬
туемого образца растворяют в 0,01 М растворе
кислоты хлористоводородной и доводят этим же
растворителем до объема 25 мл. При необходимо¬
сти разводят 0,01 М раствором кислоты хлори¬
стоводородной до получения подходящей концен¬
трации (например, от 0,4 мкг/мл до 0,6 мкг/мл 2п).
Растворы сравнения. Используют свежепри¬
готовленные растворы с концентрацией в преде¬
лах ожидаемой концентрации цинка в образцах,
например от 0,2 мкг/мл до 0,8 мкг/мл 2п. Растворы
готовят путем разведения эталонного раствора
цинка (5 мг/мл 1п) Р 0,01 М раствором кислоты
хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацети¬
леновое пламя подходящего состава (например,
воздух — 11 л/мин, ацетилен — 2 л/мин).
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 0,200 г испытуемого образца су¬
шат при температуре 105 °С в течение 16 ч.
Сульфатная зола (2.4.14). Не более 2,5 % (в
пересчете на сухое вещество). Определение про¬
водят из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14, ме¬
тод О). Менее 10 МЕ/мг, если субстанция предна¬
значена для производства лекарственных средств
60*. Зак. 1060.
460
Гэсударственная фармакопея Республики Беларусь
для парентерального применения без последую¬
щей процедуры удаления бактериальных эндоток¬
синов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. Испытуемый образец
растворяют в 0,01 М растворе кислоты хлористово¬
дородной до получения концентрации 0,8 мг/мл. Раст¬
вор хранят при температуре от 2 °С до 8 °С и исполь¬
зуют в течение 48 ч.
Раствор сравнения (а). Содержимое контейнера
с ФСО инсулина лизпро растворяют в 0,01 М раство¬
ре кислоты хлористоводородной до получения кон¬
центрации 0,8 мг/мл. Раствор хранят при температуре
от 2 °С до 8 °С и используют в течение 48 ч.
Раствор сравнения (Ь). Около 10 мг испытуе¬
мого образца растворяют в 10 мл 0,01 М раствора
кислоты хлористоводородной. Выдерживают при
комнатной температуре до получения раствора, со¬
держащего от 0,8 % до 11 % А21-дезамидоинсулина
лизпро. Раствор хранят при температуре от 2 °С до
8 °С и используют в течение 14 дней.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц
3 мкм) с размером пор 8 нм;
- температура: 40 °С;
-подвижная фаза: смешивают 745 объемов
раствора 28,4 г/л натрия сульфата безводного Р,
доведенного кислотой фосфорной Р до рН 2,3, и
255 объемов ацетонитрила для хроматографии Р;
фильтруют и дегазируют;
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: 20 мкл.
Время удерживания: инсулин лизпро — около
24 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,8 между первым
пиком (инсулин лизпро) и вторым пиком (А21-дез-
амидоинсулин лизпро);
- относительное стандартное отклонение: не
более 1,1 % при 3 вводах раствора сравнения (а).
Содержание инсулина лизпро С257Н383М650778б
рассчитывают с учетом хроматограмм испытуемого
раствора и раствора сравнения (а) и содержания
инсулина лизпро С257Н383М6507786 в ФСО инсулина
лизпро.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре, равной
или ниже -18 °С. После оттаивания инсулин лиз¬
про хранится и взвешивается в условиях, опреде¬
ленных производителем для сохранения качества
продукта, и используется для приготовления ле¬
карственных средств в течение короткого проме¬
жутка времени. Для предотвращения абсорбции
влаги из воздуха в процессе взвешивания инсулин
лизпро должен быть комнатной температуры.
07/2016:0834
ИНСУЛИН РАСТВОРИМЫЙ
ДЛЯ ИНЪЕКЦИЙ
1п8иНпит зо1иЬНе т1ес1аЬНе
/ызииы шестюи, зошвбе
Инсулин растворимый для инъекций должен со¬
ответствовать требованиям статьи Инъекцион¬
ные лекарственные средства инсулина (0854) с из¬
менениями, приведенными ниже.
ОПРЕДЕЛЕНИЕ
Нейтральный стерильный раствор инсулина бы¬
чьего, инсулина свиного или инсулина человеческого.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость, не содержа¬
щая посторонние вещества. В процессе хранения мо¬
жет образоваться незначительное количество осадка.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные
в разделе «Количественное определение». Пик ин¬
сулина на хроматограмме испытуемого раствора
по положению соответствует основному пику на
хроматограмме соответствующего раствора срав¬
нения.
ИСПЫТАНИЯ
Общий цинк. Не более 40,0 мкг на 100 МЕ инсу¬
лина, определенного как описано в статье Инъекцион¬
ные лекарственные средства инсулина (0854).
Используют следующий испытуемый раствор.
Испытуемый раствор. Количество испыту¬
емого образца, содержащего 200 МЕ, осторожно
встряхивают и доводят водой Р до объема 25,0 мл.
При необходимости разводят водой Р до подхо¬
дящей концентрации (например, от 0,4 мкг/мл до
1,6 мкг/мл 2п).
07/2016:1638
ИНСУЛИН свиной
IпзиНпит рогстит
1Ы811Ш, РОНСШЕ
Н-е1у-Ие-Уа1-в1и-ат-Суз—Сув-Ш—Зег-Ие-
I ' 10
Суз - Зег-1_еи - Туг—С1п—Ьеи—С1и - Авп—Туг-Су$ -
Азп—ОН
Н-Р119-Уа1-Мп-01п—Н13—1еи—Суз-С1у—8ег-Н5-
1.еи-\/а1-С1и-А1а-ии-Туг—Сеи-Уа1—Суз-01у-
20
С1и - Агд - <31у - Р&е - РЬе -Туг—ТЪг-Рго—Ьуз - А|а—ОН
С25вНзвЛ507б5е М.м.5778
ОПРЕДЕЛЕНИЕ
Инсулин свиной — натуральный антидиабетиче¬
ский продукт, полученный из свиной поджелудочной
железы и подвергнутый очистке.
Содержание:
Инсулин свиной
461
- сумма инсулина свиного (С256Н3811\1б50765б) и
А21-дезамидоинсулина свиного: не менее 95,0 % и не
более 105,0 % (в пересчете на сухое вещество).
В целях маркировки лекарственных средств, со¬
держащих инсулин, принято, что 0,0345 мг инсулина
свиного эквивалентны 1 МЕ инсулина.
ПРОИЗВОДСТВО
Животные, используемые для получения инсули¬
на свиного, должны выдерживать требования, предъ¬
являемые к здоровью животных, используемых чело¬
веком в пищу.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде и этаноле. Рас¬
творяется в разведенных минеральных кислотах и, с
разрушением структуры, в разведенных растворах ги¬
дроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (Ь).
B. Пептидное картирование.
Испытуемый раствор. Готовят раствор
2,0 мг/мл испытуемого образца в 0,01 М растворе
кислоты хлористоводородной. 500 мкл полученного
раствора помещают в чистую пробирку, прибавля¬
ют 2,0 мл буферного (НЕРЕ8) раствора рН 7,5 Р и
400 мкл раствора 1 мг/мл протеазы 81арЬу1ососсиз
аигеиз штамма \/8, типХУН-В, Р. Закрывают пробир¬
ку и инкубируют при температуре 25 °С в течение 6 ч.
Останавливают реакцию прибавлением 2,9 мл суль¬
фатного буферного раствора рН 2,0 Р.
Раствор сравнения. Готовят параллельно с ис¬
пытуемым раствором, используя ФСО инсулина сви¬
ного для проверки пригодности системы вместо ис¬
пытуемого образца.
Продукты расщепления исследуют с использова¬
нием жидкостной хроматографии (2.2.29).
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- температура: 40 °С;
- подвижная фаза:
-подвижная фаза А: смешивают 100мл аце¬
тонитрила для хроматографии Р, 700 мл
воды Р и 200 мл сульфатного буферного раст¬
вора рН 2,0 Р, фильтруют и дегазируют;
- подвижная фаза В: смешивают 400 мл аце¬
тонитрила для хроматографии Р, 400 мл
воды Р и 200 мл сульфатного буферного раст¬
вора рН 2,0 Р, фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
90 —* 30
10 —* 70
60—65
о
т
о
со
70—100
65—70
0
100
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
-уравновешивание колонки: в начальных ус¬
ловиях не менее 15 мин. Проводят холостой прогон,
используя вышеуказанную программу градиентного
элюирования;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы
(на хроматограмме раствора сравнения идентифи¬
цируют пики, соответствующие расщепленным фраг¬
ментам I, N и III);
- хроматограмма испытуемого раствора и раст¬
вора сравнения должны качественно соответствовать
хроматограмме расщепленного инсулина свиного,
прилагаемой к ФСО инсулина свиного для проверки
пригодности системы.
- фактор асимметрии: не более 1,5 для пиков
фрагментов II и III;
- разрешение: не менее 1,9 между пиками фраг¬
ментов II и III.
Результаты: профиль хроматограммы испытуе¬
мого раствора соответствует профилю хроматограм¬
мы раствора сравнения.
ПРИМЕЧАНИЕ: время удерживания фрагмента
I одинаково для инсулина свиного и человеческого.
Время удерживания фрагмента II и IV одинаково для
всех инсулинов. Время удерживания фрагмента III
одинаково для свиного и бычьего инсулинов.
ИСПЫТАНИЯ
Примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина. Эксклюзион-
ная хроматография (2.2.30): определение проводят
методом внутренней нормализации. Растворы хра¬
нят при температуре от 2 °С до 10 °С и использу¬
ют в течение 7 дней. При использовании автомати¬
ческого инжектора, поддерживают его температу¬
ру от 2 °С до 10 °С.
Испытуемый раствор. 4 мг испытуемого образ¬
ца растворяют в 1,0 мл 0,01 М раствора кислоты
хлористоводородной.
Раствор сравнения. Используют раствор около
4 мг/мл инсулина, содержащего более 0,4 % белков с
высокой молекулярной массой. Может использовать¬
ся инъекционный препарат инсулина, как раствор, так
и суспензия, растворенная в достаточным количестве
6 М кислоты хлористоводородной, содержащий об¬
наруживаемое количество белков с высокой молеку¬
лярной массой, или раствор, приготовленный из суб¬
станции инсулина, растворенной в 0,01 М растворе
кислоты хлористоводородной. Инсулин, содержа¬
щий обнаруживаемое количество белков с высокой
молекулярной массой, может быть приготовлен, если
выдержать порошок инсулина при комнатной темпе¬
ратуре в течение около 10 дней.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
не менее 7,5 мм, заполненная силикагелем гидро¬
фильным для хроматографии Р (размер частиц от
5 мкм до 10 мкм), пригодным для разделения моно¬
мера инсулина от димера и полимеров;
-подвижная фаза: смешивают 15 объемов кис¬
лоты уксусной ледяной Р, 20 объемов ацетонитри¬
ла Р, 65 объемов раствора 1,0 г/л аргинина Р; филь¬
труют и дегазируют;
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 276 нм;
-уравновешивание: перед использованием но¬
вой колонки для хроматографического анализа урав-
462
Гэсударственная фармакопея Республики Беларусь
новешивают ее путем повторных введений раствора
инсулина, содержащего белки высокой молекулярной
массы (например, не менее трех повторных введений
раствора сравнения). Колонку считают уравновешен¬
ной, если после двух последовательных введений по¬
лучают воспроизводимые результаты;
- объем вводимой пробы. 100 мкл;
- время хроматографирования: около 35 мин.
Времена удерживания: полимерные инсулино¬
вые комплексы — от 13 мин до 17 мин; ковалентный
димер инсулина — около 17,5 мин; мономер инсулина
— около 20 мин; соли — около 22 мин.
Пригодность хроматографической системы.
раствор сравнения:
- коэффициент разделения пиков: не менее 2,0
(Н — высота пика димера относительно базовой ли¬
нии; Ну — расстояние между базовой линией и нижней
точкой кривой, разделяющей пики димера и мономера).
Предельное содержание примесей:
-примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина (не более 1,0 %):
на хроматограмме испытуемого раствора сумма пло¬
щадей всех пиков со временем удерживания мень¬
шим, чем время удерживания основного пика, не
должна превышать 1,0% от суммы площадей всех
пиков;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики со временем
удерживания большим, чем время удерживания пика
инсулина.
Родственные белки. Жидкостная хроматогра¬
фия (2.2.29), как указано в разделе «Количественное
определение», со следующей программой градиент¬
ного элюирования:
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
42
58
30-44
42 —► 11
О)
00
т
00
ю
44—50
11
89
Растворы хранят при температуре от 2 °С до
10 °С и используют в течение 24 ч.
Пригодность хроматографической системы
указана в разделе «Количественное определение».
При необходимости изменяют относительные пропор¬
ции подвижных фаз А и В для обеспечения полного
элюирования А21-дезамидоинсулина свиного перед
началом градиента. Профиль градиента также может
регулироваться для обеспечения полного элюирова¬
ния всех родственных инсулину примесей.
Объем вводимой пробы: по 20 мкл раствора
сравнения (Ь) и испытуемого раствора. При необхо¬
димости изменяют объем вводимой пробы от 10 мкл
до 20 мкл в соответствии с результатами определения
линейности для проверки пригодности хроматографи¬
ческой системы, как указано в разделе «Количествен¬
ное определение».
Время хроматографирования: около 50 мин.
Относительное удерживание (по отношению к
основному пику): А21-дезамидоинсулин свиной (не¬
большой пик, элюирующийся после основного пика на
хроматограмме раствора сравнения (Ь)) — около 1,3.
Предельное содержание примесей:
- А21-дезамидоинсулин свиной (не более 2,0 %):
на хроматограмме испытуемого раствора площадь
пика А21-дезамидоинсулина свиного не должна пре¬
вышать 2,0 % от суммы площадей всех пиков;
- сумма примесей (не более 2,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного и пика А21-дезамидоинсулина
свиного, не должна превышать 2,0 % от суммы пло¬
щадей всех пиков.
Проинсулин-подобные иммунореактивные
примеси (Ри). Не более 10 ррт (в пересчете на су¬
хое вещество). Определение проводят подходящим
по чувствительности иммунохимическим методом
(2.7.1), например радиоиммунологическим анализом,
с использованием Международного стандартного ре¬
агента проинсулина свиного для калибровки метода.
Цинк. Не более 1,0% (в пересчете на сухое
вещество). Атомно-абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в 0,01 М растворе кислоты
хлористоводородной и доводят до объема 25,0 мл
этим же растворителем. При необходимости доводят
0,01 М раствором кислоты хлористоводородной до
получения подходящей концентрации (например, от
0,4 мкг/мл до 1,6 мкг/мл 2п).
Растворы сравнения. Используют свежепри¬
готовленные растворы, содержащие 0,40 мкг/мл,
0,80 мкг/мл, 1,00 мкг/мл, 1,20 мкг/мл и 1,60 мкг/мл
2п. Растворы готовят путем разведения эталонного
раствора цинка (5 мг/мл 2.п) Р 0,01 М раствором кис¬
лоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя подходящего состава (например, воз¬
дух — 11 л/мин, ацетилен — 2 л/мин).
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 0,200 г испытуемого образца сушат при
температуре 105 °С в течение 24 ч.
Сульфатная зола (2.4.14). Не более 2,5 % (в пе¬
ресчете на сухое вещество). Определение проводят
из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
10 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без последующей процедуры удаления
бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Растворы
хранят при температуре от 2 °С до 10 °С и исполь¬
зуют в течение 48 ч. При использовании автомати¬
ческого инжектора поддерживают его температу¬
ру от 2 °С до 10 °С.
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 10,0 мл этим же
растворителем.
Раствор сравнения (а). Содержимое контейнера
ФСО инсулина человеческого растворяют в 0,01 М
растворе кислоты хлористоводородной до получе¬
ния концентрации 4,0 мг/мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО инсулина свиного растворяют в 0,01 М раство¬
ре кислоты хлористоводородной до получения кон¬
центрации 4,0 мг/мл.
Инсулин человеческий
463
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 10,0 мл.
Раствор сравнения (д). К 1,0 мл раствора срав¬
нения (а) прибавляют 1,0 мл раствора сравнения (Ь).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: подвижная фаза А — подвиж¬
ная фаза В (42:58, об/об), при необходимости состав
корректируют;
следующие растворы готовят и хранят при
температуре не ниже 20 °С:
- подвижная фаза А: 28,4 г натрия сульфата
безводного Р растворяют в воде Р и доводят до
объема 1000 мл этим же растворителем; прибав¬
ляют 2,7 мл кислоты фосфорной Р; при необ¬
ходимости доводят до рН 2,3 этаноламином Р;
фильтруют и дегазируют;
- подвижная фаза В: 550 мл подвижной фазы
А смешивают с 450 мл ацетонитрила Р. По¬
лученный раствор нагревают до температуры
не ниже 20 °С для предотвращения выпадения
осадка (смешивание подвижной фазы А с аце¬
тонитрилом — эндотермический процесс), филь¬
труют и дегазируют;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: по 20 мкл растворов
сравнения (Ь), (с), (б) и испытуемого раствора.
Пригодность хроматографической системы:
- разрешение (на хроматограмме раствора срав¬
нения (б) пик, соответствующий основному пику на
хроматограмме раствора сравнения (Ь), должен быть
четким. На хроматограмме раствора сравнения (б)
идентифицируют пики, соответствующие свиному и
человеческому инсулину): не менее 1,2 между пиками
человеческого и свиного инсулина на хроматограмме
раствора сравнения (б). При необходимости изменя¬
ют концентрацию ацетонитрила в подвижной фазе;
-линейность: площадь основного пика на хро¬
матограмме раствора сравнения (Ь) должна превы¬
шать площадь основного пика на хроматограмме раст¬
вора сравнения (с) в (10±0,5) раз. При необходимости
изменяют объем вводимой пробы ((10—20) мкл) в за¬
висимости от уровня линейности детектора.
Суммарное содержание инсулина свиного
(С25бНз8^б5°7б5б) и А21-дезамидоинсулина свиного
рассчитывают из площадей соответствующих пиков
на хроматограммах испытуемого раствора и раствора
сравнения (Ь) и известного суммарного содержания
инсулина свиного и А21-дезамидоинсулина свиного в
ФСО инсулина свиного.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, защи¬
щенном от света, при температуре -20 °С до момен¬
та реализации производителем. После оттаивания
инсулин может храниться при температуре (5±3) °С
и использоваться для приготовления лекарственных
средств в течение короткого промежутка времени.
Для предотвращения абсорбции влаги из воздуха в
процессе взвешивания инсулин должен быть комнат¬
ной температуры.
07/2016:0838
ИНСУЛИН ЧЕЛОВЕЧЕСКИЙ
IпзиНпит Ьитапит
шзиш, ш/млы
Н-С1у-11еЛ/а1-С1и-С1п-Суз-Суз-Т11г-Зег-11е-
10
Су5-5ег-1еи-Туг-С1п-1_еи-С1и-Азп-Туг-Су5-
20"—|
Азп-ОН
Н-РЬеЛ/а1-Азп-С1п-Н1з-1-еи-Суз-<31у-5ег-Н18-
10
1_еи-\/а1-С1и-А1а-1-еи-Туг-1_еи-\/а1-Су5-01у-
20
С1и-Агд-С1у-РЬе-РНе-Туг-ТЬг-Рго-1_уз-ТНг-ОН
30
М.м.5808
ОПРЕДЕЛЕНИЕ
Инсулин человеческий — двухцепочечный пеп¬
тид, имеющий структуру антидиабетического гормона,
продуцируемого поджелудочной железой человека.
Содержание:
- сумма инсулина человеческого (С257Н383М6507736)
и А21-дезамидоинсулина человеческого: не менее
95,0 % и не более 105,0 % (в пересчете на сухое ве¬
щество).
В целях маркировки лекарственных средств, со¬
держащих инсулин, принято, что 0,0347 мг инсулина
человеческого эквивалентны 1 МЕ инсулина.
ПРОИЗВОДСТВО
Инсулин человеческий производят либо с помо¬
щью ферментативной модификации и подходящей
очистки инсулина, полученного из поджелудочной же¬
лезы свиней, либо методом, основанным на техноло¬
гии рекомбинантной ДНК (рДНК).
Где это применимо, животные, используемые
для получения инсулина человеческого, должны вы¬
держивать требования, предъявляемые к здоровью
животных, используемых человеком в пищу.
При производстве инсулина человеческого ис¬
пользуют методы, позволяющие уменьшить степень
микробиологической контаминации.
Для инсулина человеческого, полученного пу¬
тем ферментативной модификации и подходящей
очистки инсулина из поджелудочной железы свиней,
производственный процесс валидируется, чтобы
подтвердить удаление любой остаточной протео¬
литической активности. Компетентный уполномо¬
ченный орган может предъявлять дополнительные
требования.
Для инсулина человеческого, полученного ме¬
тодом, основанным на технологии рекомбинантной
ДНК, перед реализацией на каждой партии готового
нефасованного продукта проводятся следующие ис¬
пытания, если иное не утверждено компетентным
уполномоченным органом.
Белки клетки-хозяина. Предельное содержа¬
ние утверждается компетентным уполномоченным
органом.
Одноцепочечный прекурсор. Предельное со¬
держание утверждается компетентным уполномочен¬
ным органом. Используют метод с подходящей чув¬
ствительностью.
464
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде и в 96 % спирте.
Растворяется в разведенных минеральных кислотах
и, с разрушением структуры, в разведенных раство¬
рах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
B. Пептидное картирование (2.2.55).
СЕЛЕКТИВНОЕ РАСЩЕПЛЕНИЕ ПЕПТИДНЫХ
СВЯЗЕЙ
Испытуемый раствор. Готовят раствор 2,0 мг/мл
испытуемого образца в 0,01 М растворе кислоты
хлористоводородной. 500 мкл полученного раствора
помещают в чистую пробирку, прибавляют 2,0 мл бу¬
ферного (НЕРЕЗ) раствора рН 7,5 Р и 400 мкл раст¬
вора 1 мг/мл протеазы 31ар11у1ососсиз аигеиз штамм
\/8, тип Х\/11-В, Р. Закрывают пробирку и инкубируют
при температуре 25 °С в течение 6 ч. Останавливают
реакцию прибавлением 2,9 мл сульфатного буфер¬
ного раствора рН 2,0 Р.
Раствор сравнения. Готовят параллельно с ис¬
пытуемым раствором, используя ФСО инсулина чело¬
веческого вместо испытуемого образца.
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ.
Жидкостная хроматография (2.2.29).
Условия хроматографирования:
-колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц
3 мкм) с размером пор 8 нм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 100 мл аце¬
тонитрила для хроматографии Р, 700 мл воды
Р и 200 мл сульфатного буферного раствора
рН 2,0 Р, фильтруют и дегазируют;
- подвижная фаза В: смешивают 400 мл аце¬
тонитрила для хроматографии Р, 400 мл воды
Р и 200 мл сульфатного буферного раствора
рН 2,0 Р, фильтруют и дегазируют;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
90 — 30
10 — 70
60—65
о
т
8
70 — 100
65—70
0
100
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
-уравновешивание колонки: в начальных ус¬
ловиях не менее 15 мин. Проводят холостой прогон,
используя вышеуказанную программу градиентного
элюирования;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы
(на хроматограмме раствора сравнения идентифи¬
цируют пики, соответствующие расщепленным фраг¬
ментам I, I! и III):
-хроматограмма испытуемого раствора и раст¬
вора сравнения должны качественно соответствовать
хроматограмме расщепленного инсулина человече¬
ского, прилагаемой к ФСО инсулина человеческого;
- фактор асимметрии: не более 1,5 для пиков
фрагментов II и III;
- разрешение: не менее 3,4 между пиками фраг¬
ментов II и III.
Результаты: профиль хроматограммы испытуе¬
мого раствора соответствует профилю хроматограм¬
мы раствора сравнения.
ПРИМЕЧАНИЕ: время удерживания фрагмента
I одинаково для инсулина свиного и человеческого.
Время удерживания фрагментов II и IV одинаково
для всех инсулинов. Время удерживания фрагмента
III одинаково для свиного и бычьего инсулинов.
ИСПЫТАНИЯ
Примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина. Эксклюзион-
ная хроматография (2.2.30): определение проводят
методом внутренней нормализации. Растворы хра¬
нят при температуре от 2 °С до 8 °С и используют
в течение 7 дней. При использовании автоматиче¬
ского инжектора поддерживают его температуру
от 2 °С до 8 °С.
Испытуемый раствор. 4 мг испытуемого образ¬
ца растворяют в 1,0 мл 0,01 М раствора кислоты
хлористоводородной.
Раствор сравнения. Используют раствор около
4 мг/мл инсулина, содержащего более 0,4 % белков с
высокой молекулярной массой. Может использовать¬
ся инъекционный препарат инсулина, как раствор, так
и суспензия, растворенная в достаточным количестве
6 М кислоты хлористоводородной, содержащий об¬
наруживаемое количество белков с высокой молеку¬
лярной массой, или раствор, приготовленный из суб¬
станции инсулина, растворенной в 0,01 М растворе
кислоты хлористоводородной. Инсулин, содержа¬
щий обнаруживаемое количество белков с высокой
молекулярной массой, может быть приготовлен, если
выдержать порошок инсулина при комнатной темпе¬
ратуре в течение 10 дней.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
не менее 7,5 мм, заполненная силикагелем гидро¬
фильным для хроматографии Р (размер частиц от
5 мкм до 10 мкм, размер пор от 12 нм до 12,5 нм), при¬
годным для разделения мономера инсулина от диме¬
ра и полимеров;
-подвижная фаза: смешивают 15 объемов кис¬
лоты уксусной ледяной Р, 20 объемов ацетонитри¬
ла Р, 65 объемов раствора 1,0 г/л аргинина Р\ филь¬
труют и дегазируют;
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 276 нм;
-уравновешивание: перед использованием но¬
вой колонки для хроматографического анализа урав¬
новешивают ее путем повторных введений раствора
инсулина, содержащего белки высокой молекулярной
массы (например, не менее трех повторных введений
раствора сравнения). Колонку считают уравновешен¬
ной, если после двух последовательных введений по¬
лучают воспроизводимые результаты;
- объем вводимой пробы: 100 мкл;
- время хроматографирования: около 35 мин.
Времена удерживания: полимерные инсулино¬
вые комплексы — от 13 мин до 17 мин; ковалентный
Инсулин человеческий
465
димер инсулина — около 17,5 мин; мономер инсули¬
на — около 20 мин; соли — около 22 мин.
Пригодность хроматографической системы:
раствор сравнения:
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика димера относительно базовой ли¬
нии; Ну — расстояние между базовой линией и нижней
точкой кривой, разделяющей пики димера и мономера).
Предельное содержание примесей:
-примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина (не более 1,0 %):
на хроматограмме испытуемого раствора сумма пло¬
щадей всех пиков со временем удерживания мень¬
шим, чем время удерживания основного пика, не
должна превышать 1,0 % от суммы площадей всех
пиков;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики со временем
удерживания большим, чем время удерживания пика
инсулина.
Родственные белки. Жидкостная хроматогра¬
фия (2.2.29), как указано в разделе «Количественное
определение», со следующей программой градиент¬
ного элюирования:
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
42
58
30—44
42 —> 11
58—>89
44—50
11
89
Растворы хранят при температуре от 2 °С до
8 °С и используют в течение 24 ч.
Пригодность хроматографической системы
указана в разделе «Количественное определение».
При необходимости изменяют относительные пропор¬
ции подвижных фаз А и В для обеспечения полного
элюирования А21-дезамидоинсулина человеческого
перед началом градиента. Профиль градиента также
может регулироваться для обеспечения полного элю¬
ирования всех родственных инсулину примесей.
Объем вводимой пробы: по 20 мкл растворов
сравнения (а), (Ь), (с) и испытуемого раствора. При
необходимости изменяют объем вводимой пробы
от 10 мкл до 20 мкл в соответствии с результатами
определения линейности для проверки пригодности
хроматографической системы, как указано в разделе
«Количественное определение».
Время хроматографирования: около 50 мин.
Относительное удерживание (по отношению к
основному пику): А21-дезамидоинсулин (небольшой
пик, элюирующийся после основного пика на хрома¬
тограмме раствора сравнения (а)) — около 1,3.
Предельное содержание примесей:
- А21-дезамидоинсулин человеческий (не более
2,0 %): на хроматограмме испытуемого раствора пло¬
щадь пика А21-дезамидоинсулина человеческого не
должна превышать 2,0 % от суммы площадей всех
пиков;
- сумма примесей (не более 2,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного и пика А21-дезамидоинсулина
человеческого, не должна превышать 2,0 % от суммы
площадей всех пиков;
только для полусинтетического инсулина чело¬
веческого:
- инсулин свиной (не более 1,0 %): на хрома¬
тограмме испытуемого раствора площадь пика, со¬
ответствующего основному пику на хроматограмме
раствора сравнения (Ь), не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с).
Следующее испытание проводят только для
инсулина человеческого, полученного путем фер¬
ментативной модификации инсулина свиного.
Проинсулин-подобные иммунореактивные
примеси (Р/./). Не более 10 ррпл (в пересчете на су¬
хое вещество). Определение проводят подходящим
по чувствительности иммунохимическим методом
(2.7.1), например радиоиммунологическим анализом,
с использованием Международного стандартного ре¬
агента проинсулина свиного для калибровки метода.
Цинк. Не более 1,0 % в пересчете на сухое веще¬
ство. Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хло¬
ристоводородной и доводят до объема 25,0 мл этим
же растворителем. При необходимости разводят
0,01 М раствором кислоты хлористоводородной до
получения подходящей концентрации (например, от
0,4 мкг/мл до 1,6 мкг/мл 2п).
Растворы сравнения. Используют свежепри¬
готовленные растворы, содержащие 0,40 мкг/мл,
0,80 мкг/мл, 1,00 мкг/мл, 1,20 мкг/мл и 1,60 мкг/мл
2.п. Растворы готовят путем разведения эталонного
раствора цинка (5 мг/мл 1п) Р 0,01 М раствором кис¬
лоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя подходящего состава (например, воз¬
дух — 11 л/мин, ацетилен — 2 л/мин).
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 0,200 г испытуемого образца сушат при
температуре 105 °С в течение 24 ч.
Сульфатная зола (2.4.14). Не более 2,5 % (в пе¬
ресчете на сухое вещество). Определение проводят
из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
10 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без последующей процедуры удаления
бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Растворы
хранят при температуре от 2 °С до 8 °С и использу¬
ют в течение 48 ч. При использовании автоматиче¬
ского инжектора поддерживают его температуру
от 2 °С до 8 °С.
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 10,0 мл этим же
растворителем.
Раствор сравнения (а). Содержимое контейнера
ФСО инсулина человеческого растворяют в 0,01 М
растворе кислоты хлористоводородной до получе¬
ния концентрации 4,0 мг/мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО инсулина свиного для проверки пригодности
466
Гэсударственная фармакопея Республики Беларусь
системы растворяют в 0,01 М растворе кислоты
хлористоводородной до получения концентрации
4 мг/мл.
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (Ь) доводят 0,01 М раствором кислоты хло¬
ристоводородной до объема 50,0 мл. К 1,0 мл полу¬
ченного раствора прибавляют 1,0 мл раствора срав¬
нения (а).
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (а) доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 10,0 мл.
Раствор сравнения (е). К 1,0 мл раствора срав¬
нения (а) прибавляют 1,0 мл раствора сравнения (Ь).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: подвижная фаза А — подвиж¬
ная фаза В (42:58, об/об), при необходимости состав
корректируют;
следующие растворы готовят и хранят при
температуре не ниже 20 °С:
- подвижная фаза А: 28,4 г натрия сульфата
безводного Р растворяют в воде Р и доводят до
объема 1000 мл этим же растворителем; прибав¬
ляют 2,7 мл фосфорной кислоты Р\ при необ¬
ходимости доводят до рН 2,3 этаноламином Р;
фильтруют и дегазируют;
- подвижная фаза В: 550 мл подвижной фазы
А смешивают с 450 мл ацетонитрила Р. Полу¬
ченный раствор подогревают до температуры
не ниже 20 °С для предотвращения выпадения
осадка (смешивание подвижной фазы А с аце¬
тонитрилом — эндотермический процесс), филь¬
труют и дегазируют;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 214 нм;
- объем вводимой пробы: по 20 мкп растворов
сравнения (а), (Ь), (б), (е) и испытуемого раствора.
Пригодность хроматографической системы:
- разрешение (на хроматограмме раствора срав¬
нения (е) пик, соответствующий основному пику на
хроматограмме раствора сравнения (Ь), должен быть
четким. На хроматограмме раствора сравнения (е)
идентифицируют пики, соответствующие свиному и
человеческому инсулину): не менее 1,2 между пиками
человеческого и свиного инсулина на хроматограмме
раствора сравнения (е). При необходимости изменя¬
ют концентрацию ацетонитрила в подвижной фазе;
- линейность: площадь основного пика на хрома¬
тограмме раствора сравнения (а) должна превышать
площадь основного пика на хроматограмме раствора
сравнения (б) в (10±0,5) раз. При необходимости из¬
меняют объем вводимой пробы ((10—20) мкл) в зави¬
симости от уровня линейности детектора.
Суммарное содержание инсулина человече¬
ского (С257Н383М6507786) и А21-дезамидоинсулина
человеческого рассчитывают из площадей соответ¬
ствующих пиков на хроматограммах испытуемого
раствора и раствора сравнения (а) и известного сум¬
марного содержания инсулина человеческого и А21-
дезамидоинсулина человеческого в ФСО инсулина
человеческого.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, защи¬
щенном от света, при температуре не выше -18 °С
до момента реализации производителем. После от¬
таивания инсулин хранится при температуре (5±3) °С
и используется для приготовления лекарственных
средств в течение короткого промежутка времени.
Для предотвращения поглощения влаги из воздуха в
процессе взвешивания инсулин должен быть комнат¬
ной температуры.
МАРКИРОВКА
Указывают метод получения инсулина человече¬
ского: метод ферментативной модификации инсулина
свиного или метод, основанный на технологии реком¬
бинантной ДНК.
07/2016:0854
ИНСУЛИНА ИНЪЕКЦИОННЫЕ
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
РгаерагаНопез тзиНп/ 'т1ес1аЫ1ез
1ызииы РРЕРАРАТЮЫ8,1ШЕСТАВ1Е
Инъекционные лекарственные средства инсу¬
лина должны соответствовать требованиям к инъ¬
екционным лекарственным средствам, описанным в
статье Лекарственные средства для парентераль¬
ного применения (0520).
ОПРЕДЕЛЕНИЕ
Стерильные лекарственные средства Инсулина
человеческого (0838), Инсулина бычьего (1637), Ин¬
сулина свиного (1638).
Содержание: не менее 90,0 % и не более 110,0 %
от количества инсулина, указанного на этикетке.
Представляют собой растворы или суспензии, или
их получают объединением растворов и суспензий.
ПРОИЗВОДСТВО
Методы получения должны обеспечивать прида¬
ние соответствующих свойств, обеспечивающих на¬
чало и длительность терапевтического воздействия.
В зависимости от метода получения в подходящей
последовательности проводятся следующие действия:
-добавление соответствующих антимикробных
консервантов;
-добавление соответствующего вещества или
веществ для изотонирования;
-добавление соответствующего вещества или
веществ для изменения рН до подходящего значения;
- определение активности инсулинсодержащего
компонента или компонентов с последующей, при не¬
обходимости, корректировкой для получения необхо¬
димого количества Международных единиц в 1 мл в
готовом лекарственном средстве;
- стерилизация посредством фильтрования ин¬
сулинсодержащего компонента или компонентов; по¬
сле ее проведения все последующие процедуры про¬
водятся в асептических условиях, с использованием
материалов, стерилизованных с помощью подходя¬
щего метода.
Кроме этого, в соответствующих случаях добав¬
ляются подходящие вспомогательные вещества и
осуществляется подходящий порядок действий для
придания соответствующей формы инсулинсодержа-
Инсулина инъекционные лекарственные средства
467
щему компоненту или компонентам. Готовое лекар¬
ственное средство помещается в асептических усло¬
виях в стерильные контейнеры, которые закрываются
таким образом, чтобы исключить микробиологиче¬
скую контаминацию.
ИСПЫТАНИЯ
рН (2.2.3). От 6,9 до 7,8. Измеряют рН раствора или
суспензии, если не указывается иное в частной статье.
Содержание инсулина в надосадочном слое.
Не более 2,5 % от общего содержания инсулина — для
лекарственного средства инсулина в виде суспензии,
если не указывается иное. 10 мл суспензии центрифу¬
гируют с ускорением 1500 д в течение 10 мин и осто¬
рожно разделяют надосадочную жидкость и осадок.
Определяют содержание инсулина в надосадочной
жидкости (5) с помощью соответствующего метода, на¬
пример хроматографического, как описано в разделе
«Количественное определение». Содержание инсули¬
на в растворе в процентах рассчитывают по формуле:
1005
Т ’
где:
Т — общее содержание инсулина, определенное,
как описано в разделе «Количественное определение».
Примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина. Эксклюзион-
ная хроматография (2.2.30).
Испытуемый раствор. 4 мкп 6 М раствора
кислоты хлористоводородной прибавляют к 1 мл
испытуемого образца в виде суспензии или раст¬
вора для получения прозрачного раствора инсу¬
лина в кислоте. При использовании испытуемого
образца в виде суспензии перед применением его
встряхивают для обеспечения однородности. Если
суспензия не становится прозрачной в течение
5 мин после первоначального прибавления кислоты
хлористоводородной, то прибавляют дополнитель¬
но небольшие количества кислоты (менее 4 мкл/мл)
до получения раствора. Лекарственные средства с
концентрацией большей, чем 100 МЕ/мл, необходи¬
мо разбавлять 0,01 М раствором кислоты хлори¬
стоводородной во избежание переполнения колон¬
ки мономером инсулина.
Раствор сравнения. Используют раствор около
4 мг/мл инсулина, содержащего более 0,4 % белков с
высокой молекулярной массой. Может использовать¬
ся инъекционное лекарственное средство инсулина,
как раствор, так и суспензия, растворенная в доста¬
точным количестве 6М раствора кислоты хлори¬
стоводородной, содержащее обнаруживаемое коли¬
чество белков с высокой молекулярной массой, или
раствор, приготовленный из субстанции инсулина,
растворенной в 0,01 М растворе кислоты хлористо¬
водородной. Инсулин, содержащий обнаруживаемое
количество белков с высокой молекулярной массой,
может быть приготовлен, если выдержать порошок
инсулина при комнатной температуре в течение около
10 дней. Растворы хранят при температуре от 2 °С до
10 °С и используют в течение 30 ч (растворимый инсу¬
лин для инъекций) или 7 дней (другие лекарственные
средства инсулина). При использовании автоматиче¬
ского инжектора поддерживают температуру от 2 °С
до 10 °С.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
не менее 7,5 мм, заполненная силикагелем гидро¬
фильным для хроматографии Р (размер частиц от
5 мкм до 10 мкм), пригодным для разделения моно¬
мера инсулина от димеров и полимеров;
-подвижная фаза: смешивают кислоту уксус¬
ную ледяную Р, ацетонитрил Р, раствор 1,0 г/л арги¬
нина Р (15:20:65, об/об/об); фильтруют и дегазируют;
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 276 нм;
-уравновешивание: не менее 3 введений раст¬
вора сравнения; колонку считают уравновешенной,
если после двух последовательных введений полу¬
чают воспроизводимые результаты; для протаминсо-
держащих испытуемых образцов уравновешивание
колонки проводится с использованием раствора, со¬
держащего протамин;
- объем вводимой пробы: по 100 мкл испытуемо¬
го раствора и раствора сравнения;
- время хроматографирования: около 35 мин.
Времена удерживания: полимерные инсулино¬
вые комплексы или ковалентный инсулин-протами-
новый комплекс — от около 13 мин до 17 мин; кова¬
лентный димер инсулина — около 17,5 мин; мономер
инсулина — около 20 мин; соли — около 22 мин; если
раствор испытуемого образца содержит консерванты,
например метилпарабен, м-крезол или фенол, то они
элюируются позже.
Пригодность хроматографической системы:
раствор сравнения:
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика димера относительно базовой ли¬
нии; Н]/ — расстояние между базовой линией и нижней
точкой кривой, разделяющей пики димера и мономера).
Предельное содержание примесей:
-примеси с молекулярной массой, превышаю¬
щей молекулярную массу инсулина: на хроматограм¬
ме испытуемого раствора сумма площадей всех пиков
со временем удерживания меньшим, чем время удер¬
живания пика инсулина, не должна превышать 3,0 %
(протаминсодержащие лекарственные средства) или
2,0 % (не содержащие протамин лекарственные сред¬
ства) от суммы площадей всех пиков;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики со временем
удерживания большим, чем время удерживания пика
инсулина.
Родственные белки. Жидкостная хроматогра¬
фия (2.2.29), как указано в разделе «Количественное
определение», со следующей программой градиент¬
ного элюирования:
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
42
58
30—44
42—11
58 — 89
44—50
11
89
Растворы хранят при температуре от 2 °С до
10 °С и используют в течение 24 ч.
Пригодность хроматографической системы
(разрешение, линейность) указана в разделе «Ко¬
личественное определение». При необходимости
изменяют относительные соотношения подвижных
фаз для обеспечения полного элюирования А21-дез-
амидоинсулина свиного перед началом градиента.
Профиль градиента также может регулироваться для
обеспечения полного элюирования всех родственных
инсулину примесей.
468
Гэсударственная фармакопея Республики Беларусь
Объем вводимой пробы: по 20 мкп испытуемого
раствора и раствора сравнения (а) для лекарствен¬
ных средств инсулина, содержащих 100 МЕ/мл, или
раствора сравнения (Ь) для лекарственных средств
инсулина, содержащих 40 МЕ/мл. При необходимости
изменяют объем вводимой пробы до объема от 10 мкл
до 20 мкл в соответствии с результатами определе¬
ния линейности для проверки пригодности хромато¬
графической системы, как указано в разделе «Коли¬
чественное определение». При необходимости про¬
водят дальнейшую корректировку подвижной фазы
для обеспечения полного отделения антимикробных
консервантов, присутствующих в испытуемом раство¬
ре, от инсулина и меньшего времени их удерживания.
Небольшое уменьшение концентрации ацетонитрила
вызывает увеличение времени удерживания пиков
инсулина в большей степени, чем времени удержива¬
ния пиков консервантов.
Время хроматографирования: около 50 мин.
Относительное удерживание (по отношению к
основному пику, соответствующему инсулину): А21-дез-
амидоинсулин (небольшой пик, элюирующийся после
основного пика на хроматограмме раствора сравне¬
ния (а) или (Ь)) — около 1,3.
Предельное содержание примесей:
- А21-дезамидоинсулин (не более 5,0%); на
хроматограмме испытуемого раствора площадь пика
А21-дезамидоинсулина не должна превышать 5,0%
от суммы площадей всех пиков;
-сумма примесей, кроме примеси А21-дезами-
доинсулина (не более 6,0 %): на хроматограмме испыту¬
емого раствора сумма площадей всех пиков, кроме пиков
инсулина и А21-дезамидоинсулина, не должна превы¬
шать 6,0 % от суммы площадей всех пиков;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики консервантов
и протамина, которые элюируются ранее.
Общий цинк. Не более содержания, указанного
в частной статье и определенного с помощью атомно¬
абсорбционной спектрометрии (2.2.23, метод /).
Используют следующий метод, если не указы¬
вается иное в частной статье.
Испытуемый раствор. Лекарственное средство
осторожно встряхивают и объем, содержащий 200 МЕ
инсулина, доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 25,0 мл. При необходимо¬
сти разводят 0,01 М раствором кислоты хлористо¬
водородной до получения подходящей концентрации
цинка (например, от 0,4 мкг/мл до 1,6 мкг/мл 2п).
Растворы сравнения. Используют свежепри¬
готовленные растворы, содержащие 0,40 мкг/мл,
0,80 мкг/мл, 1,00 мкг/мл, 1,20 мкг/мл и 1,60 мкг/мл
2п. Растворы готовят путем разведения эталонного
раствора цинка (5 мг/мл 2п) Р 0,01 М раствором кис¬
лоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя подходящего состава (например, воз¬
дух — 11 л/мин, ацетилен — 2 л/мин).
Содержание цинка в растворе. Где применимо,
не более содержания, указанного в частной статье
и определенного с помощью атомно-абсорбционной
спектрометрии (2.2.23, метод I).
Испытуемый раствор. Испытуемый образец
центрифугируют и 1 мл прозрачной надосадоч-
ной жидкости доводят водой Р до объема 25,0 мл.
При необходимости разводят водой Р рр получе¬
ния подходящей концентрации цинка (например, от
0,4 мкг/мл до 1,6 мкг/мл 2п).
Растворы сравнения. Используют свежепри¬
готовленные растворы, содержащие 0,40 мкг/мл,
0,80 мкг/мл, 1,00 мкг/мл, 1,20 мкг/мл и 1,60 мкг/мл
2п. Растворы готовят путем разведения эталонного
раствора цинка (5 мг/мл 2п) Р 0,01 М раствором кис¬
лоты хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя подходящего состава (например, воз¬
дух — 11 л/мин, ацетилен — 2 л/мин).
Бактериальные эндотоксины (2.6.14). Менее
80 ЕЭ на 100 МЕ инсулина.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 4 мкл 6 М раствора
кислоты хлористоводородной прибавляют к 1 мл
испытуемого образца в виде суспензии или раст¬
вора для получения прозрачного раствора. При ис¬
пользовании испытуемого образца в виде суспензии
перед применением его встряхивают для обеспече¬
ния однородности. Если суспензия не становится
прозрачной в течение 5 мин после первоначального
прибавления кислоты, то дополнительно прибавляют
небольшие количества кислоты (менее 4 мкл/мл) до
получения раствора. Лекарственные средства с кон¬
центрацией большей, чем 100 МЕ/мл, необходимо
дополнительно разбавлять 0,01 М раствором кисло¬
ты хлористоводородной во избежание переполне¬
ния колонки.
Раствор сравнения (а). Для лекарственного
средства, содержащего один вид инсулина, содер¬
жимое контейнера соответственно с ФСО инсулина
человеческого, ФСО инсулина свиного или ФСО ин¬
сулина бычьего растворяют в 0,01 М растворе кисло¬
ты хлористоводородной до получения концентрации
4,0 мг/мл. Для лекарственного средства, содержащего
инсулин свиной и инсулин бычий, смешивают 1,0 мл
раствора, содержащего 4,0 мг ФСО инсулина бычьего
в 1 мл 0,01 М раствора кислоты хлористоводород¬
ной, и 1,0 мл раствора, содержащего 4,0 мг ФСО инсу¬
лина свиного в 1 мл 0,01 М раствора кислоты хлори¬
стоводородной. Раствор сравнения (а) применяется
для количественного определения лекарственных
средств инсулина, содержащих 100 МЕ/мл.
Раствор сравнения (Ь). 4,0 мл раствор сравнения
(а) доводят 0,01 Мраствором кислоты хлористоводо¬
родной до объема 10,0 мл. Раствор сравнения (Ь) при¬
меняется для количественного определения лекар¬
ственных средств инсулина, содержащих 40 МЕ/мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО инсулина человеческого растворяют в 0,01 М
растворе кислоты хлористоводородной до получе¬
ния концентрации 4,0 мг/мл.
Раствор сравнения (ф. Содержимое контейнера
с ФСО инсулина свиного для проверки пригодности
системы растворяют в 0,01 М растворе кислоты хло¬
ристоводородной до получения концентрации 4 мг/мл.
Раствор сравнения (е). 1,0 мл раствора сравне¬
ния (а) доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 10,0 мл.
Инсулина цинка суспензия (аморфная) для инъекций
469
Раствор сравнения (I). 1,0 мл раствора сравне¬
ния (Ь) доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 10,0 мл.
Раствор сравнения (д). Смешивают 1,0 мл раст¬
вора сравнения (с) и 1,0 мл раствора сравнения (б).
Растворы хранят при температуре от 2 °С до
10 °С и используют в течение 48 ч. При использо¬
вании автоматического инжектора поддерживают
температуру от 2 °С до 10 °С.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц —
5 мкм);
- скорость подвижной фазы: 1 мл/мин;
- подвижная фаза: подвижная фаза А— подвиж¬
ная фаза В (42:58, об/об), при необходимости состав
корректируют;
следующие растворы готовят и хранят при
температуре не ниже 20 °С:
- подвижная фаза А: 28,4 г натрия сульфа¬
та безводного Р растворяют в воде Р и дово¬
дят этим же растворителем до объема 1000 мл,
прибавляют 2,7 мл кислоты фосфорной Р и при
необходимости доводят этаноламином Р до
рН 2,3; фильтруют и дегазируют;
- подвижная фаза В: смешивают 550 мл под¬
вижной фазы А и 450 мл ацетонитрила Р. На¬
гревают полученный раствор до температуры
не ниже 20 °С во избежание образования осадка
(смешивание подвижной фазы и ацетонитрила
является эндотермическим процессом); фильтру¬
ют и дегазируют;
- спектрофотометрический детектор, длина
волны 214 нм;
- температура: 40 °С;
- объем вводимой пробы, по 20 мкл испытуемого
раствора, растворов сравнения (б) и (д) и растворов
сравнения (а) и (е) (для лекарственных средств ин¬
сулина, содержащих 100 МЕ/мл) или растворов срав¬
нения (Ь) и (Т) (для лекарственных средств инсулина,
содержащих 40 МЕ/мл). При необходимости проводят
дальнейшую корректировку подвижной фазы для обе¬
спечения полного отделения антимикробных консер¬
вантов, присутствующих в испытуемом растворе, от
инсулина и меньшего времени их удерживания. Не¬
большое уменьшение концентрации ацетонитрила
вызывает увеличение времени удерживания инсули¬
на в большей степени, чем времени удерживания кон¬
сервантов. При необходимости после хроматографи¬
рования промывают колонку смесью из равных объ¬
емов ацетонитрила Р и воды Р в течение времени,
достаточного для обеспечения элюирования любых
интерферирующих веществ перед введением следу¬
ющего раствора.
Идентификация пиков примесей: идентифици¬
руют пики инсулина свиного и инсулина человеческо¬
го, используя хроматограмму раствора сравнения (д).
Пригодность хроматографической системы:
раствор сравнения (д):
-должен наблюдаться четкий пик, соответству¬
ющий основному пику на хроматограмме раствора
сравнения (б);
- разрешение: не менее 1,2 между пиками инсу¬
лина человеческого и инсулина свиного. При необхо¬
димости корректируют концентрацию ацетонитрила в
подвижной фазе;
- линейность: площадь основного пика на хро¬
матограмме раствора сравнения (а) или (Ь) должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (е) или (1) в (10±0,5) раз. При
необходимости изменяют объем вводимой пробы от
10 мкл до 20 мкл в зависимости от уровня линейности
детектора.
Суммарное содержание инсулина и А21-дез-
амидоинсулина рассчитывают из площадей пиков
инсулинов бычьего, свиного или человеческого и
А21-дезамидоинсулина и известного суммарного со¬
держания инсулина и А21-дезамидоинсулина в ФСО
инсулина бычьего, ФСО инсулина свиного или ФСО
инсулина человеческого соответственно. Для ле¬
карственных средств, содержащих инсулин бычий и
инсулин свиной, используют сумму площадей пиков
инсулина бычьего и инсулина свиного и пиков произ¬
водных А21-дезамидоинсулина (100 МЕ соответству¬
ют 3,47 мг инсулина человеческого, 3,45 мг инсулина
свиного и 3,42 мг инсулина бычьего).
ХРАНЕНИЕ
В стерильном воздухонепроницаемом контейне¬
ре с контролем первого вскрытия, в защищенном от
света месте при температуре от 2 °С до 8 °С, если не
указывается иное. Лекарственные средства инсулина
не должны замораживаться.
МАРКИРОВКА
На этикетке указывают:
- активность в Международных единицах на мл;
-концентрацию, исходя из количества инсули¬
на в мг/мл (для лекарственных средств, содержащих
инсулин бычий и инсулин свиной, концентрация уста¬
навливается как суммарное количество обоих инсули¬
нов);
-где применимо, что вещество получено фер¬
ментативной модификацией инсулина свиного;
- где применимо, что вещество получено с помо¬
щью технологии рекомбинантной ДНК;
-где применимо, животные виды, являющиеся
источником происхождения;
- лекарственное средство не должно быть замо¬
рожено;
- где применимо, что лекарственное средство дол¬
жно быть ресуспендировано перед использованием.
07/2016:0835
ИНСУЛИНА ЦИНКА СУСПЕНЗИЯ
(АМОРФНАЯ) ДЛЯ ИНЪЕКЦИЙ
1пзиНт яла атогрЫ зизрепзю т1ес(аЬШз
Ш8Ш-Ш ШС ШЕСТАВ1.Е
ЗиЗРЕЫЗЮЫ (АМОРРНСШ5)
Суспензия (аморфная) цинка инсулина для инъ¬
екций должна соответствовать требованиям ста¬
тьи Инъекционные лекарственные средства инсу¬
лина (0854) с изменениями, приведенными ниже.
ОПРЕДЕЛЕНИЕ
Стерильная нейтральная суспензия комплекса
инсулина бычьего, инсулина свиного или инсулина
человеческого с соответствующей солью цинка. Инсу¬
лин находится в форме, которая практически нерас¬
творима в воде.
470
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая суспензия, при отстаи¬
вании которой образуются осадок белого или почти
белого цвета и бесцветная или почти бесцветная на-
досадочная жидкость; осадок легко ресуспендируется
при осторожном встряхивании. Изучение с помощью
микроскопа показывает, что частицы имеют неодно¬
родную форму и их максимальный размер иногда пре¬
вышает 2 мкм.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные в
разделе «Количественное определение». Пик инсули¬
на на хроматограмме испытуемого раствора по поло¬
жению соответствует основному пику на хроматограм¬
ме соответствующего раствора сравнения.
ИСПЫТАНИЯ
Общий цинк. От 0,12 мг до 0,25 мг на 100 МЕ
инсулина, определенного как описано в статье Инъек¬
ционные лекарственные средства инсулина (0854).
Содержание цинка в растворе. От 20 % до 65 %
общего цинка, находящегося в растворе. Определяют
с помощью метода, описанного в статье Инъекцион¬
ные лекарственные средства инсулина (0854).
07/2016:0836
ИНСУЛИНА ЦИНКА СУСПЕНЗИЯ
(КРИСТАЛЛИЧЕСКАЯ)
ДЛЯ ИНЪЕКЦИЙ
1п$иИп1 г\пс\ спз(аШп1 зизрепзю т1ес1аЫИз
ШС ШЕСТАВ1.Е 5(У5РЕА/5/ОЛ/
(СКУ8ТАШЫЕ)
Суспензия (кристаллическая) цинка инсулина
для инъекций должна соответствовать требовани¬
ям статьи Инъекционные лекарственные средства
инсулина (0854) с изменениями, описанными ниже.
ОПРЕДЕЛЕНИЕ
Стерильная нейтральная суспензия комплекса
инсулина бычьего, инсулина свиного или инсулина
человеческого с соответствующей солью цинка. Инсу¬
лин находится в форме, которая практически нерас¬
творима в воде.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая суспензия, при отстаи¬
вании которой образуются осадок белого или почти
белого цвета и бесцветная или почти бесцветная на-
досадочная жидкость; осадок легко ресуспендируется
при осторожном встряхивании. Изучение с помощью
микроскопа показывает, что частицы являются ром¬
боэдрическими кристаллами, большая часть которых
имеет при измерении от одного угла кристалла до
другого максимальный размер более 10 мкм, а ино¬
гда — более 40 мкм.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные в
разделе «Количественное определение». Пик инсули¬
на на хроматограмме испытуемого раствора по поло¬
жению соответствует основному пику на хроматограм¬
ме соответствующего раствора сравнения.
ИСПЫТАНИЯ
Инсулин, не экстрагирующийся забуферен-
ным ацетоновым раствором. Не более 90 % от
общего содержания инсулина. Центрифугируют ис¬
пытуемый образец, содержащий 200 МЕ инсулина,
и отделяют надосадочную жидкость. Суспендируют
осадок в 1,65 мл воды Р, прибавляют 3,3 мл забуфе-
ренного ацетонового раствора Р, перемешивают в
течение 3 мин, опять центрифугируют, отделяют надо¬
садочную жидкость и повторяют все описанные выше
действия, используя осадок. Осадок растворяют с по¬
мощью соответствующей методики, например раство¬
ряют в 0,1 М растворе кислоты хлористоводородной
до получения конечного объема 2,0 мл. Определяют
содержание инсулина в осадке (Р) и общее содержа¬
ние инсулина (7) в таком же объеме суспензии под¬
ходящим методом. Содержание инсулина, не экстра¬
гирующегося забуференным ацетоновым раствором,
в процентах рассчитывают по формуле:
100Я
Т '
Общий цинк. От 0,12 мг до 0,25 мг на 100 МЕ
инсулина, определенного как описано в статье Инъек¬
ционные лекарственные средства инсулина (0854).
Содержание цинка в растворе. От 20 % до 65 %
общего цинка, находящегося в растворе. Определяют
с помощью метода, описанного в статье Инъекцион¬
ные лекарственные средства инсулина (0854).
07/2016:1110
ИНТЕРФЕРОНА АЛЬФА-2
КОНЦЕНТРИРОВАННЫЙ РАСТВОР
1МегГегоп1 аИа-2 $о1иНо сопсеп1га1а
ШТЕНРЕПОЫ А1РА-2 СОЫСЕЫТПАТЕО
ЗОШТЮЫ
СОЪРОТНЗЪС
ЕЕГС№ЕЩА
ЬОКЕУТЕЬУО
ЮГЕ<2К1ТЬУЪ
ЬКЗКЕ
ЗККТЬМЬЬАО
ЕТ1РУЬНЕМ1
ОЬЫОЬЕАСУ1
I
КЕККУЗРСАЮ
МЮС113ЬР5СЪ
001Г1ЯЪЕ5ТК
ОСУ^ТЕТРЬ
ЕУУКАЕШКЗ
КОК1ШЕСЕР0
ЛЗЗААШЭЕТЬ
МКЕ051ЬАУК
ГЗЬЗТЫЬОЕЗ
ОПРЕДЕЛЕНИЕ
Раствор белка, полученного в соответствии с ин¬
формацией, закодированной альфа-2 подвидом гена
интерферона альфа, и вызывающего неспецифичную
противовирусную активность по крайней мере в гомо¬
логичных клетках посредством клеточных метаболи¬
ческих процессов, включающих синтез как рибонукле¬
иновой кислоты, так и белка. Вызывает антипролифе-
ративную активность. Различные типы интерферона
альфа-2 различаются аминокислотным остатком в
положении 23 и обозначаются строчной буквой.
Обозначение
Остаток в позиции 23 (X,)
альфа-2а
Лизин (1_уз)
альфа-2Ь
Аргинин (Агд)
Данная статья применяется по отношению к кон¬
центрированным растворам интерферона альфа-2а и
альфа-2Ь.
Активность концентрированного раствора интер¬
ферона альфа-2 составляет не менее 1,4-108 МЕ в
Интерферона альфа-2 концентрированный раствор
471
1 мг белка. Концентрированный раствор интерферо¬
на альфа-2 содержит не менее 2-108 МЕ интерферона
альфа-2 в 1 мл.
ПРОИЗВОДСТВО
Концентрированный раствор интерферона аль-
фа-2 получают методом, основанным на технологии
рекомбинантной ДНК (рДНК), используя бактерии в
качестве клеток-хозяев. Его получают в условиях, на¬
правленных на минимизацию микробной контамина¬
ции продукта.
Концентрированный раствор интерферона аль-
фа-2 должен выдерживать следующие дополнитель¬
ные требования.
Белки клеток-хозяев. Предельное содержание
должно быть согласовано с компетентным уполномо¬
ченным органом.
ДНК клеток-хозяев или векторов. Предельное
содержание должно быть согласовано с компетент¬
ным уполномоченным органом.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слегка желтоватая
жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец обладает предполагае¬
мой биологической активностью, определяемой как
указано в разделе «Количественное определение».
B. Изоэлектрическое фокусирование.
Испытуемый раствор. Испытуемый образец до¬
водят водой Р до концентрации белка 1 мг/мл.
Раствор сравнения. Готовят раствор с концен¬
трацией 1 мг/мл подходящего ФСО альфа-2 интер¬
ферона в воде Р.
Гоадуировочный раствор для определения изо-
электрической точки с диапазоном р1 от 3,0 до 10,0.
Готовят и используют в соответствии с инструкциями
производителя.
Используют подходящий прибор вместе с цирку¬
ляционной водяной баней с контролем температуры,
установленной на значение 10 °С, и гели для изоэлек-
трического фокусирования с градиентом рН от 3,5 до
9,5. Работу с прибором осуществляют в соответствии
с инструкциями производителя. В качестве анодного
раствора используют кислоту фосфорную Р (98 г/л
Н3Р04), а в качестве катодного раствора — 1 М раст¬
вор натрия гидроксида. Образцы наносят на гель с по¬
мощью фильтровальной бумаги. Импрегнированные
фильтры наносят на поверхность геля около катода.
Наносят 15мкл испытуемого раствора и 15мкл
раствора сравнения. Начинают изоэлектрическое фо¬
кусирование при 1500 В и 50 мА. Через 30 мин вы¬
ключают источник тока, удаляют импрегнированные
фильтры и снова пропускают ток в течение 1 ч. В те¬
чение процесса фокусирования мощность удержива¬
ют постоянной. После фокусирования погружают гель
в подходящий объем раствора, содержащего 115 г/л
кислоты трихлоруксусной Р и 34,5 г/л кислоты суль-
фосалициловой Р в воде Р, и осторожно взбалтывают
контейнер в течение 60 мин. Переносят гель в смесь
кислота уксусная ледяная Р — этанол Р — вода Р
(32:100:268, об/об/об) и вымачивают в течение 5 мин.
Погружают гель на 10 мин в окрашивающий раст¬
вор, предварительно нагретый до температуры 60 °С
и представляющий собой вышеуказанную смесь из
кислоты уксусной ледяной, этанола и воды, к которой
прибавлен 1,2 г/л кислотный синий 83 Р. Промывают
гель в нескольких контейнерах с вышеуказанной сме¬
сью из кислоты уксусной ледяной, этанола и воды и
выдерживают в ней гель до тех пор, пока фон не ста¬
нет прозрачным (от 12 ч до 24 ч). После достаточного
обесцвечивания гель выдерживают в течение 1 ч в
10 % (об/об) растворе глицерина Р в вышеуказанной
смеси из кислоты уксусной ледяной, этанола и воды.
На электрофореграмме испытуемого раствора
обнаруживаются основные полосы, соответствующие
по расположению основным полосам на электрофо¬
реграмме раствора сравнения. Строят график зави¬
симости расстояний миграции маркеров изоэлектри-
ческих точек от значений их изоэлектрических точек и
определяют изоэлектрические точки основных компо¬
нентов испытуемого раствора и раствора сравнения.
Они не должны отличаться более чем на 0,2 р1. Испы¬
тание считают пригодным, если маркеры изоэлектри¬
ческих точек равномерно распределены по всей дли¬
не геля, а значения изоэлектрических точек основных
полос на электрофореграмме раствора сравнения на¬
ходятся между значениями 5,8 и 6,3.
С. Просматривают электрофореграммы, полу¬
ченные как указано в испытании «Примеси с молеку¬
лярными массами, отличающимися от молекулярной
массы интерферона альфа-2» в восстанавливающих
условиях.
Результаты: на электрофореграмме испытуемого
раствора (а) обнаруживается основная полоса, соот¬
ветствующая по расположению основной полосе на
электрофореграмме раствора сравнения (а).
О. Пептидное картирование.
Испытуемый раствор. Испытуемый образец
доводят водой Р до концентрации белка 1,5 мг/мл.
25 мкл полученного раствора переносят в полипро¬
пиленовую или стеклянную пробирку вместимостью
1,5 мл, прибавляют 1,6 мкл 1 М фосфатного буфер¬
ного раствора рН 8,0 Р, 2,8 мкл свежеприготовленно¬
го раствора 1,0 мг/мл трипсина для пептидного кар¬
тирования Р в воде Р и 3,6 мкл воды Р и интенсивно
перемешивают. Закрывают пробирку и выдерживают
ее в водяной бане при температуре 37 °С в течение
18 ч, затем прибавляют 100 мкл раствора 573 г/л гу¬
анидина гидрохлорида Р и тщательно перемешивают.
К полученному раствору прибавляют 7 мкл раствора
154,2 г/л дитиотреитола Р и тщательно перемеши¬
вают. Закрытую пробирку выдерживают в кипящей
воде в течение 1 мин и охлаждают до комнатной тем¬
пературы.
Раствор сравнения. Готовят параллельно с испы¬
туемым раствором, но используют раствор 1,5 мг/мл
подходящего ФСО интерферона альфа-2 в воде Р.
Испытание проводят с помощью жидкостной хро¬
матографии (2.2.29).
Условия хроматографирования:
-колонка из нержавеющей стали длиной 0,10 м
и внутренним диаметром 4,6 мм, заполненная силика¬
гелем октадецилсилильным для хроматографии Р
(размер частиц 5 мкм) с размером пор 30 нм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: 1 мл кислоты трифто-
руксусной Р доводят водой Р до объема 1000 мл;
- подвижная фаза В: к 100 мл воды Р прибав¬
ляют 1 мл кислоты трифторуксусной Р и дово¬
дят ацетонитрилом для хроматографии Р до
объема 1000 мл;
472
Государственная фармакопея Республики Беларусь
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—8
100
0
8—68
о
т
о
о
8
Т
О
68—72
40
60
72—75
40 —* 100
60-*0
75—80
100
0
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 214 нм;
- уравновешивание: не менее 15 мин с помощью
подвижной фазы А;
- объем вводимой пробы: 100 мкл.
Пригодность хроматографической системы:
- хроматограммы испытуемого раствора и раст¬
вора сравнения должны соответствовать по каче¬
ственным характеристикам хроматограмме расще¬
пленного интерферона альфа-2, прилагаемой к ФСО
интерферона альфа-2.
Результаты: хроматографический профиль ис¬
пытуемого раствора должен соответствовать хрома¬
тографическому профилю хроматограммы раствора
сравнения.
ИСПЫТАНИЯ
Примеси с молекулярными массами, отлича¬
ющимися от молекулярной массы интерферона
альфа-2. Электрофорез с натрием додецилсульфа-
том в полиакриламидном геле (ДСН-ПАГ, 808-РАСЕ)
(2.2.31). Испытание проводят в восстанавливающих и
невосстанавливающих условиях, используя раздели¬
тельные гели с 14 % акриламида и окрашивание се¬
ребром для обнаружения белка.
Буфер образца (невосстанавливающие усло¬
вия). Смешивают равные объемы воды Р и буфер¬
ного образцового раствора (концентрированного)
для электрофореза в системе натрия додецилсуль-
фат — полиакриламидный гель (808-РАСЕ) Р.
Буфер образца (восстанавливающие условия).
Смешивают равные объемы воды Р и буферного об¬
разцового раствора (концентрированного) для элек¬
трофореза в системе натрия додецилсульфат —
полиакриламидный гель (808-РАСЕ) для восстано¬
вительных условий Р, содержащего 2-меркаптоэта-
нол в качестве восстанавливающего агента.
Испытуемый раствор (а). Испытуемый образец
доводят буфером образца до концентрации белка
0,5 мг/мл.
Испытуемый раствор (Ь). 0,20 мл испытуемого
раствора (а) доводят буфером образца до объема
1 мл.
Раствор сравнения (а). Готовят раствор
0,625 мг/мл подходящего ФСО интерферона альфа-2
в буфере образца.
Раствор сравнения (Ь). 0,20 мл раствора срав¬
нения (а) доводят буфером образца до объема 1 мл.
Раствор сравнения (с). 0,20 мл раствора срав¬
нения (Ь) доводят буфером образца до объема 1 мл.
Раствор сравнения (ф. 0,20 мл раствора срав¬
нения (с) доводят буфером образца до объема 1 мл.
Раствор сравнения (е). 0,20 мл раствора срав¬
нения (с1) доводят буфером образца до объема 1 мл.
Раствор сравнения ((). Используют раствор
стандартов молекулярных масс, подходящий для гра¬
дуирования ДСН-ПАГ гелей в диапазоне от 15 кДа до
67 кДа.
Закрытые пробирки с испытуемым раствором и
растворами сравнения выдерживают на водяной бане
в течение 2 мин.
10 мкл раствора сравнения (V) и по 50 мкл осталь¬
ных растворов наносят в лунки концентрирующего
геля. Электрофорез проводят в условиях, рекомендо¬
ванных производителем оборудования. Белки обнару¬
живают в геле с помощью окрашивания серебром.
Пригодность системы:
- должны соблюдаться критерии валидации
(2.2.31)\
- на электрофореграмме раствора сравнения (е)
должна обнаруживаться полоса;
- наблюдается градация интенсивности окраши¬
вания на электрофореграммах испытуемых раство¬
ров (а) и (Ь) и растворов сравнения от (а) до (е) соот¬
ветственно.
На электрофореграмме испытуемого раствора
(а) , полученной в восстанавливающих условиях, кро¬
ме основной полосы могут наблюдаться менее интен¬
сивные полосы с более низкими по сравнению с ос¬
новной полосой молекулярными массами. Из них ни
одна полоса не должна быть интенсивнее основной
полосы на электрофореграмме раствора сравнения
(б) (1,0 %) и не более 3 таких полос могут быть ин¬
тенсивнее основной полосы на электрофореграмме
раствора сравнения (е) (0,2 %).
На электрофореграмме испытуемого раствора
(а), полученной в невосстанавливающих условиях,
кроме основной полосы могут наблюдаться менее
интенсивные полосы с более высокими по сравнению
с основной полосой молекулярными массами. Из них
ни одна полоса не должна быть интенсивнее основ¬
ной полосы на электрофореграмме раствора сравне¬
ния (с!) (1,0 %) и не более 3 таких полос могут быть
интенсивнее основной полосы на электрофореграм¬
ме раствора сравнения (е) (0,2 %).
Сопутствующие белки. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. Испытуемый образец до¬
водят водой Р до концентрации белка 1 мг/мл.
0,25 % (м/м) раствор водорода пероксида. Раст¬
вор водорода пероксида разведенный Р доводят во¬
дой Р до концентрации 0,25 % (м/м).
Раствор сравнения. К испытуемому раствору
прибавляют 0,25 % (м/м) раствор водорода перок¬
сида в таком объеме, чтобы получить конечную кон¬
центрацию водорода пероксида 0,005 % (м/м), и вы¬
держивают при комнатной температуре в течение 1 ч
или такого периода времени, в течение которого об¬
разуется около 5 % окисленного интерферона. При¬
бавляют 12,5 мг (.-метионина Р на 1 мл раствора.
Выдерживают при комнатной температуре в течение
1 ч. Растворы хранят не более 24 ч при температуре
от 2 °С до 8 °С.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,25 м
и внутренним диаметром 4,6 мм, заполненная силика¬
гелем октадецилсилильным для хроматографии Р
(размер частиц 5 мкм) с размером пор 30 нм;
- подвижная фаза:
- подвижная фаза А: к 700 мл воды Р при¬
бавляют 2 мл кислоты трифторуксусной Р и
300 мл ацетонитрила для хроматографии Р;
- подвижная фаза В: к 200 мл воды Р при¬
бавляют 2 мл кислоты трифторуксусной Р и
800 мл ацетонитрила для хроматографии Р;
Интерферона альфа-2 концентрированный раствор
473
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—1
72
28
1—5
72—>67
28 ->33
5—20
со
со
т
со
со
т
со
со
20—30
63 —> 57
со
I
со
30—40
57—>40
43 —> 60
40-42
40
60
42—50
40->72
60 —> 28
50—60
72
28
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
-уравновешивание: не менее 15 мин;
- объем вводимой пробы: 50 мкл.
Время удерживания: интерферон альфа-2 —
около 20 мин. На хроматограмме раствора сравне¬
ния обнаруживается пик окисленного интерферона
со временем удерживания около 0,9 по отношению к
основному пику.
Пригодность хроматографической системы:
- разрешение: не менее 1,0 между пиками окис¬
ленного интерферона и интерферона.
Предельное содержание примесей (учитывают
пики, времена удерживания которых составляют от
0,7 до 1,4 по отношению к времени удерживания ос¬
новного пика):
- любая примесь: на хроматограмме испытуемого
раствора площадь любого пика, кроме основного, не
должна превышать 3 % от общей площади всех пиков;
- сумма примесей: сумма площадей пиков, кро¬
ме основного, не должна превышать 5 % от общей
площади всех пиков.
Бактериальные эндотоксины (2.6.14). Менее
100 МЕ в объеме, содержащем 1,0 мг белка.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Белки
Испытуемый раствор. Испытуемый образец
доводят водой Р до концентрации около 0,5 мг/мл
интерферона альфа-2.
Растворы сравнения. Готовят основной раст¬
вор 0,5 мг/мл альбумина бычьего Р. Из основного
раствора путем разведения готовят 8 растворов
сравнения, содержащих от 3 мг/мл до 30 мг/мл аль¬
бумина бычьего Р.
Проводят 30-кратное и 50-кратное разведе¬
ния испытуемого раствора. Прибавляют по 1,25 мл
смеси, приготовленной в тот же день смешивани¬
ем 2,0 мл раствора 20 г/л меди (II) сульфата Р в
воде Р, 2,0 мл раствора 40 г/л натрия тартрата Р
в воде Р и 96,0 мл раствора 40 г/л натрия карбона¬
та Р в 0,2 М растворе натрия гидроксида, в про¬
бирки, содержащие 1,5 мл воды Р (контрольный
раствор), 1,5 мл растворов разных разведений ис¬
пытуемого раствора или 1,5 мл растворов сравне¬
ния. Перемешивают после каждого прибавления.
Приблизительно через 10 мин прибавляют в каж¬
дую пробирку по 0,25 мл смеси из равных объемов
воды Р и фосфорномолибденово-вольфрамового
реактива Р. Перемешивают после каждого при¬
бавления. Приблизительно через 30 мин измеряют
оптическую плотность (2.2.25) каждого раствора
при длине волны 750 нм, используя контрольный
раствор в качестве компенсационного раствора.
Строят градуировочный график зависимости опти¬
ческих плотностей 8 растворов сравнения от соот¬
ветствующих концентраций белка и находят содер¬
жание белка в испытуемом растворе.
Активность
Активность интерферона альфа-2 оценивают
сопоставлением его эффективности в защите кле¬
ток от вирусного цитопатического воздействия с
эффективностью соответствующего Международ¬
ного стандартного образца человеческого реком¬
бинантного интерферона альфа-2 или стандарт¬
ного образца, калиброванного в Международных
единицах.
За Международную единицу принимают актив¬
ность в установленном количестве соответству¬
ющего Международного стандартного образца.
Эквивалентность Международного стандартного
образца в Международных единицах устанавлива¬
ется Всемирной организацией здравоохранения.
Проводят количественное определение с по¬
мощью подходящей методики, основанной на сле¬
дующих положениях.
В условиях стандартизированных культур ис¬
пользуют установленную клеточную линию, чув¬
ствительную к цитопатическому воздействию под¬
ходящего вируса (подходит человеческая дипло¬
идная фибробластная клеточная линия, свободная
от микробной контаминации, реагирующая на ин-
теферон и чувствительная к вирусу энцефаломи¬
окардита).
Подходящими могут быть следующие кле¬
точные культуры и вирусы: МйВК клетки (АТСС
№ СС/_22) или Моизе Т клетки (Л/СТС клон 929;
АТСС № СС/_ 1) в качестве клеточной культуры
и везикулярного вируса стоматита УЗУ, /псИапа
штамм (АТСС № УР-158) в качестве инфекцион¬
ного агента; или А-549 клетки (АТСС № СС7-185),
реагирующие на интерферон как клеточная куль¬
тура, и энцефаломиокардитический вирус (АТСС
№ УР-129В) как инфекционный агент.
Как минимум в 4 сериях инкубируют клетки с
3 или более различными концентрациями испыту¬
емого образца и стандартного образца на микро-
титрической пластине и включают в каждую серию
подходящие контроли необработанных клеток.
Выбирают концентрации испытуемого образца
таким образом, чтобы наименьшая концентрация
обеспечивала небольшую защиту, а наибольшая
концентрация обеспечивала защиту от вирусного
цитопатического эффекта меньше максимальной.
В подходящее время прибавляют цитопатический
вирус во все лунки, за исключением достаточного
количества лунок в каждой серии, которые остают¬
ся с неинфицированными контрольными клетками.
Проводят количественное определение цитопати¬
ческого воздействия вируса с помощью подходя¬
щего метода. Определяют активность испытуемого
образца с помощью обычных статистических мето¬
дов для количественного определения параллель¬
ных линий.
Определенная активность должна быть не
менее 80 % и не более 125 % от заявленной ак¬
тивности. Доверительный интервал определенной
активности (Р = 0,95) должен быть не менее 64 % и
не более 156 % от заявленной активности.
474
Гэсударственная фармакопея Республики Беларусь
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре не более
~20 °С.
МАРКИРОВКА
На этикетке указывают:
- тип интерферона (альфа-2а или альфа-2Ь);
- тип производства.
07/2016:1335
ИТРАКОНАЗОЛ
Игасопаго1ит
1ТКА СОЫА201.Е
С35НмС12М,04 М.М. 706
[84625-61-6]
ОПРЕДЕЛЕНИЕ
4-[4-[4-[4-[[цис-2-(2,4-Дихлорфенил)-2-(1 Н-1,2,4-
триазол-1 -илметил)-1,3-диоксолан-4-ил]метокси]фе-
нил]пиперазин-1 -ил]фенил]-2-[(1 /?5)-1 -метилпропил]-
2,4-дигидро-ЗР/-1,2,4-триазол-3-он.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, очень мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО интраконазола.
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раст¬
воряют в метиленхлориде Р и доводят до объема
20.0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
8 должна быть не интенсивнее эталона Р(Кр)6
или В(К)б.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием.
Испытуемый раствор. 0,100 г испытуемо¬
го образца растворяют в метанольном раство¬
ре кислоты хлористоводородной Р и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят метанольным раствором кис¬
лоты хлористоводородной Р до объема 100,0 мл.
1.0 мл полученного раствора доводят метаноль¬
ным раствором кислоты хлористоводородной Р
до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг ФСО итракона-
зола для проверки пригодности хроматографиче¬
ской системы (содержит примеси В, С, й, Е, Р и
О) растворяют в 1,0 мл метанольного раствора
кислоты хлористоводородной Р.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем окта-
децилсилильным, деактивированным по отноше¬
нию к основаниям, эндкепированным для хрома¬
тографии Р с размером частиц 3 мкм или 3,5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: раствор 27,2 г/л те-
трабутиламмония гидросульфата Р1\
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
80
20
2—22
80 —* 50
20 — 50
22—27
50
50
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, й, Е, Р и С, используя хро¬
матограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО итраконазола для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению к
итраконазолу, время удерживания — около 14 мин):
примесь В — около 0,7; примеси СиР — около 0,8;
примесь Е — около 0,9; примесь Р — около 1,05; при¬
месь С — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси Р относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси Р и пик
итраконазола).
Предельное содержание примесей:
Итраконазол
475
- примеси В, О (не более 0,3 %): на хроматограм¬
ме испытуемого раствора площади пиков, соответ¬
ствующих примесям В и 6, не должны превышать
3-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- примесь Е (не более 0,2 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси Е, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей С и О (не более 0,3 %): на
хроматограмме испытуемого раствора сумма пло¬
щадей пиков, соответствующих примесям С и Э, не
должна превышать 3-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей В, С, О, Е и 6, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 0,8 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
8-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца сушат
при температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в
70 мл смеси из 1 объема кислоты уксусной без¬
водной Р и 7 объемов метилэтилкетона Р при
интенсивном перемешивании в течение не менее
10 мин. Титруют 0,1 М раствором кислоты хлор¬
ной Р потенциометрически (2.2.20). Отмечают объ¬
ем титранта во второй точке перегиба на кривой
титрования.
1 мл 0,1 М раствора кислоты хлорной соот¬
ветствует 35,3 мг Сз5Н38С121\1804.
ПРИМЕСИ
Специфицированные примеси: В, С, О, Е, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, Р.
N—N
о»
К1
н РН3 СН3
и энантиомер Н2 ~ СН3
А. 4-[4-[4-(4-Метоксифенил)пиперазин-1 -ил]фенил]-2-
[(1 Я5)-1-метил пропил]-2,4-дигидро-ЗН-1,2,4-триазол-З-он.
К1
/
н рНз рн3
и энантиомер
В. 4-[4-[4-[4-[[цг/с-2-(2,4-Дихлорфенил)-2-(4Н-1,2,4-
триазол-4-илметил)-1,3-диоксолан-4-ил]метокси]фе-
нил]пиперазин-1 -ил]фенил]-2-[(1 Я5)-1 -метилпропил]-
2,4-дигидро-ЗН-1,2,4-триазол-З-он.
С. 4-[4-[4-[4-[[цб/с-2-(2,4-Дихлорфенил)-2-(1/-/-1,2,4-
триазол-1 -илметил )-1,3-диоксолан-4-ил]метокси]фе-
нил]пиперазин-1-ил]фенил]-2-пропил-2,4-дигидро-ЗН-
1,2,4-триазол-З-он.
0.4-[4-[4-[4-[[щ/с-2-(2,4-Дихлорфенил)-2-(1 Н-1,2,4-
триазол-1 -илметил )-1,3-диоксолан-4-ил]метокси]фе-
нил]пиперазин-1 -ил]фенил]-2-( 1 -метилэтил )-2,4-дигидро-
ЗН-1,2,4-триазол-З-он.
Е. 4-[4-[4-[4-[[шранс-2-(2,4-Дихлорфенил)-2-(1 Н-
1.2.4- триазол-1-илметил )-1,3-диоксолан-4-ил]метокси]-
фенил]пиперазин-1 -ил]фенил]-2-[(1 /?5)-1 -метилпропил]-
2.4- дигидро-ЗН-1,2,4-триазол-З-он.
476
Гэсударственная фармакопея Республики Беларуси
Р. 2-Бутил-4-[4-[4-[4-[[цыс-2-(2,4-дихлорфенил)-2-
(1/-/-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]ме-
токси]фенил]пиперазин-1-ил]фенил]-2,4-дигидро-ЗН-
1,2,4-триазол-З-он.
С. 4-[4-[4-[4-[[цос-2-(2,4-Дихлорфенил)-2-(1 Н-1,2,4-
триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фе-
нил]пиперазин-1-ил]фенил]-2-[[щ/с-2-(2,4-дихлорфенил)-
2-(1 /-/-1,2,4-триазол-1 -ил метил )-1,3-диоксолан-4-ил]-
метил]-2,4-дигидро-ЗН-1,2,4-триазол-З-он.
07/2016:0917
ИХТИОЛ
1сШатто1ит (#1сЫ11уо1ит)
1СНТНАММ01-
ОПРЕДЕЛЕНИЕ
Ихтиол получают дистилляцией из некоторых ви¬
дов битумных сланцев с последующим сульфирова¬
нием дистиллята и нейтрализацией полученного про¬
дукта раствором аммиака.
Содержание:
- сухое вещество: от 50,0 % (м/м) до 56,0 % (м/м)]
-общее количество аммиака (МН3, М.м. 17,03):
от 4,5 % (м/м) до 7,0 % (м/м) (в пересчете на сухое ве¬
щество);
-органически связанная сера: не менее 10,5 %
(м/м) (в пересчете на сухое вещество);
- сера в форме сульфата: не более 20,0 % (м/м)
от общей серы.
ОПИСАНИЕ (СВОЙСТВА)
Густая черновато-коричневая жидкость.
Смешивается с водой и глицерином, мало рас¬
творим в 96 % спирте, жирных маслах и вазелиновом
масле. Образует однородные смеси с ланолином и
вазелином.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1,5 г испытуемого образца растворяют в 15 мл
воды Р (раствор А). К 2 мл раствора А прибавляют
2 мл кислоты хлористоводородной Р. Образуется
смолистый осадок. Сливают надосадочный слой жид¬
кости. Осадок частично растворяется в эфире Р.
B. 2 мл раствора А, полученного в идентифика¬
ции А, дают реакцию на аммония соли и соли летучих
оснований (2.3.1).
C. Смесь раствора А и раствора натрия гидрок¬
сида разведенного Р, полученную в идентификации
В, выпаривают досуха и прокаливают. К полученному
остатку прибавляют 5 мл кислоты хлористоводород¬
ной разведенной Р. Выделяется газ, окрашивающий
свинцово-ацетатную бумагу Р в коричневый или
черный цвет. Полученный раствор фильтруют. Филь¬
трат дает реакцию (а) на сульфаты (2.3.1).
ИСПЫТАНИЯ
Кислотность или щелочность. К 10,0 мл про¬
зрачного фильтрата, полученного как указано в раз¬
деле «Количественное определение. Общее количе¬
ство аммиака», прибавляют 0,05 мл раствора мети¬
лового красного Р. При прибавлении не более 0,2 мл
0,02 М раствора кислоты хлористоводородной или
0,02 М раствора натрия гидроксида окраска раст¬
вора должна измениться.
Относительная плотность (2.2.5). От 1,040 дс
1,085. Определяют относительную плотность смеси
из равных объемов испытуемого образца и воды Р.
Сульфатная зола (2.4.14). Не более 0,3 %. Опре¬
деление проводят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Сухое вещество. 1,000 г испытуемого образа
помещают в предварительно взвешенную колбу, со¬
держащую небольшую стеклянную палочку и 2 г пе¬
ска Р, предварительно высушенного до постоянней
массы. Нагревают на водяной бане в течение 2 ч пси
частом перемешивании и высушивают при температу¬
ре от 100 °С до 105 °С до достижения разницы межд;
двумя последовательными взвешиваниями не более
2,0 мг; второе взвешивание проводят после 1 ч пс-
вторного высушивания.
Общее количество аммиака. 2,50 г испытуеме-
го образца растворяют в 25 мл теплой воды Р. Полу¬
ченный раствор количественно переносят в мери.-:
колбу вместимостью 250 мл, прибавляют 200 м.-
раствора натрия хлорида Р, доводят водой Р д:
объема 250,0 мл и фильтруют, отбрасывая первое
20 мл фильтрата. К 100,0 мл прозрачного фильтрз-в
прибавляют 25 мл раствора формальдегида Р, ней
трализованного по раствору фенолфталеина Р1 -
титруют 0,1 М раствором натрия гидроксида до
явления розового окрашивания.
1 мл 0,1 М раствора натрия гидроксида сос-
ветствует 1,703 мг 1МН3.
Органически связанная сера. В фарфоровое
тигле вместимостью около 50 мл смешивают 0,50С *
испытуемого образца, 4 г натрия карбоната бе:
водного Р и 3 мл метиленхлорида Р, нагревакг *■
перемешивают до полного испарения метиленхг:
рида. Прибавляют 10 г грубоизмельченного мег.
(II) нитрата Р, тщательно перемешивают и оче- =
осторожно нагревают на слабом огне. После п:а-
кращения начальной реакции температуру медпе-
но повышают до почернения большей части сме>
Охлаждают, помещают тигель в большой ста*;з-
прибавляют 20 мл кислоты хлористоводородно1 -
и, после прекращения реакции, 100 мл воды Р. •>-
пятят до полного растворения оксида меди. Пег »
ченный раствор фильтруют, прибавляют 400 мг
воды Р, нагревают до кипения, прибавляют 20 м.-
раствора бария хлорида Р1, выдерживают в те-~-
ние 2 ч и фильтруют. Остаток на фильтре промыва¬
ют водой Р, сушат и прокаливают при температура
около (600±50) °С до достижения разницы ме>^»
Макао масло
477
двумя последовательными взвешиваниями не бо¬
лее 0,2 % от массы остатка.
1 г остатка соответствует 0,1374 г общего количе¬
ства серы.
Рассчитывают общее процентное содержание
серы и вычитают процентное содержание серы в фор¬
ме сульфата.
Сера в форме сульфата. 2,000 г испытуемого
образца растворяют в 100 мл воды Р, прибавляют 2 г
меди (II) хлорида Р, растворенного в 80 мл воды Р,
доводят водой Р до объема 200,0 мл, встряхивают
и фильтруют. 100,0 мл фильтрата нагревают почти
до кипения, прибавляют 1 мл кислоты хлористово¬
дородной Р и по каплям 5 мл раствора бария хло¬
рида Р1 и нагревают на водяной бане. Фильтруют,
остаток на фильтре промывают водой Р, сушат и про¬
каливают при температуре около (600±50) °С до до¬
стижения разницы между двумя последовательными
взвешиваниями не более 0,2 % от массы остатка.
1 г остатка соответствует 0,1374 г серы в форме
сульфата.
Содержание серы в форме сульфата рассчиты¬
вают в процентах.
07/2016:0031
йод
!одит
ЮОШЕ
12 М.м. 253,8
[7553-56-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,5 % и не более 100,5 % I.
ОПИСАНИЕ(СВОЙСТВА)
Серовато-фиолетовые хрупкие пластинки или
мелкие кристаллы с металлическим блеском.
Очень мало растворим в воде, очень легко рас¬
творим в концентрированных растворах йодидов, рас¬
творим в 96 % спирте, мало растворим в глицерине.
Медленно улетучивается при комнатной темпе¬
ратуре.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Несколько кусочков испытуемого образца на¬
гревают в пробирке. Выделяются пары фиолетового
цвета, и образуется кристаллический сублимат сине¬
вато-черного цвета.
B. К насыщенному раствору испытуемого образ¬
ца прибавляют раствор крахмала Р. Появляется си¬
нее окрашивание. Нагревают до обесцвечивания. При
охлаждении окрашивание появляется снова.
ИСПЫТАНИЯ
Раствор 5. 3,0 г испытуемого образца растирают
с 20 мл воды Р, фильтруют, фильтр промывают во¬
дой Р и фильтрат доводят до объема 30 мл этим же
растворителем. К полученному раствору прибавляют
1 г порошка цинка Р. После обесцвечивания раствор
фильтруют, фильтр промывают водой Р и фильтрат
доводят до объема 40 мл этим же растворителем.
Бромиды и хлориды. Не более 0,0250 %
(250 ррт). К 10 мл раствора 3 прибавляют Змл
раствора аммиака Р и 6 мл раствора серебра ни¬
трата Р2. Фильтруют, фильтр промывают водой Р и
фильтрат доводят до объема 20 мл этим же раство¬
рителем. К 10 мл полученного раствора прибавляют
1,5 мл кислоты азотной Р. Через 1 мин полученный
раствор по степени мутности не должен превышать
раствор, приготовленный одновременно путем сме¬
шивания 10,75 мл воды Р, 0,25 мл 0,01 М раствора
кислоты хлористоводородной, 0,2 мл кислоты
азотной разведенной Р и 0,3 мл раствора серебра
нитрата Р2.
Нелетучие вещества. Не более 0,1 %. 1,00 г ис¬
пытуемого образца нагревают в фарфоровой чашке
на водяной бане до удаления йода. Остаток сушат при
температуре от 100 °С до 105 °С. Масса полученного
остатка не должна превышать 1 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца помещают в колбу,
содержащую 1 г калия йодида Р и 2 мл воды Р, и при¬
бавляют 1 мл кислоты уксусной разведенной Р. По¬
сле полного растворения прибавляют 50 мл воды Р и
титруют 0,1 М раствором натрия тиосульфата, ис¬
пользуя в качестве индикатора раствор крахмала Р.
1 мл 0,1 М раствора натрия тиосульфата со¬
ответствует 12,69 мг I.
#ХРАНЕНИЕ
В воздухонепроницаемых стеклянных контейне¬
рах в защищенном от света месте при температуре от
8 °С до 15 °С.
07/2016: РБ0020
#КАКАО МАСЛО
Сасао о1еит
СОСОА ВиТТЕЙ
ОПРЕДЕЛЕНИЕ
Жирное масло, получаемое прессованием
поджаренных и освобожденных от кожуры семян
ТЬеоЬгота сасао 1_.
ОПИСАНИЕ (СВОЙСТВА)
Плотная однородная масса желтоватого цвета,
со слабым ароматным запахом какао, хрупкая при
комнатной температуре.
Легко растворим в эфире и мало растворим в
96 % спирте.
ИСПЫТАНИЯ
Температура плавления (2.2.15). От 31 °С до
35 °С. 50 г испытуемого образца расплавляют при
температуре от 50 °С до 60 °С и охлаждают при тем¬
пературе 25 °С, непрерывно перемешивая до об¬
разования пастообразной консистенции, избегая
образования воздушных пузырей. Расплавленную и
охлажденную массу нагревают на водяной бане при
температуре от 32 °С до 33 °С, продолжая переме¬
шивать до приобретения ею температуры водяной
бани и превращения в жидкую кремообразную массу
(нагревание около 30 минут). Расплавленную массу
переливают в выпарительную чашку и оставляют за¬
твердевать при комнатной температуре на 2 ч.
478
Гэсударственная фармакопея Республики Беларусь
Кислотное число (2.5.1). Не более 2,8.
Показатель преломления (2.2.6). От 1,454 до
1,459. Определение проводят при температуре 40 °С.
Йодное число (2.5.4). От 33 до 42.
Число омыления (2.5.6). От 188 до198.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте при температуре от 8 °С до
15 °С.
07/2016:1139
КАЛИЯ АЦЕТАТ
КаШ асе1аз
РОТАЗЗШМ АСЕТАТЕ
С2Н,К02 М.м. 98,1
[127-08-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок или бесцветные кристаллы. Расплывается на
воздухе.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на аце¬
таты (2.3.1).
B. Испытуемый образец дает реакцию (а) на ка¬
лий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 7,5 до 9,0. 1,0 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 20 мл этим же раство¬
рителем.
Восстанавливающие вещества. 10 мл раст¬
вора 3 доводят водой Р до объема 100 мл. К полу¬
ченному раствору прибавляют 5 мл кислоты серной
разведенной Р и 0,5 мл раствора 0,32 г/л калия пер¬
манганата Р. Раствор перемешивают и осторожно
кипятят в течение 5 мин. Раствор должен сохранять
розовое окрашивание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2.5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
7.5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на сульфаты.
Алюминий (2.4.17). Не более 0,0001 % (1 ррт),
если субстанция предназначена для приготовления
растворов для перитонеального диализа, гемодиали¬
за или гемофильтрации.
Предписанный раствор. 2,0 г испытуемого об¬
разца растворяют в 50 мл воды Р и прибавляют 5 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смесь из 1 мл эталонного раствора
алюминия (2 ррт А!) Р, 5 мл ацетатного буферного
раствора рН 6,0 Р и 49 мл воды Р.
Контрольный раствор. Смесь из 5 мл ацетат¬
ного раствора рН 6,0 Р и 50 мл воды Р.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Получен¬
ный раствор должен выдерживать испытание на железо.
Натрий. Не более 0,5 %. Атомно-эмиссионная
спектрометрия (2.2.22, метод II).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора натрия
(200 ррт Ыа) Р водой Р.
Длина волны: 589 нм.
#Кальций (2.4.3). Не более 0,0300 % (300 ррт).
2 мл раствора 3 должны выдерживать испытание на
кальций. Эталон готовят с использованием 6 мл эта¬
лонного раствора кальция (10 ррт Са) Р и 9 мл воды
дистиллированной Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0004 % (4 ррт). 5,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 3,0 %. 1,000 г испытуемого образца сушат при
температуре от 100 °С до 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
80,0 мг испытуемого образца растворяют в 20 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной, используя в качестве ин¬
дикатора 0,2 мл раствора нафтолбензеина Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 9,81 мг С2Н3К02.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0184
КАЛИЯ БРОМИД
КаШ ЬготМит
РОТАЗЗШМ ВРОМЮЕ
КВг М.м. 119,0
[7758-02-3]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
Калия бромид
479
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде и в глицерине, мало рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на бро¬
миды (2.3.1).
B. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», дает реакции на калий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора бромтимоло-
вого синего Р1. При прибавлении не более 0,5 мл
0,01 М раствора хлористоводородной кислоты или
0,01 М раствора натрия гидроксида окраска раст¬
вора должна измениться.
Броматы. К 10 мл раствора 3 прибавляют 1 мл
раствора крахмала Р, 0,1 мл раствора 100 г/л калия
йодида Р, 0,25 мл 0,5 М раствора серной кислоты и
выдерживают в течение 5 мин в защищенном от света
месте. Не должно образовываться синее или фиоле¬
товое окрашивание раствора.
Хлориды и сульфаты. Жидкостная хроматогра¬
фия (2.2.29).
Испытуемый раствор (а). 0,400 г испытуемого об¬
разца растворяют в 50 мл воды для хроматографии Р
и доводят до объема 100,0 мл этим же растворителем.
Испытуемый раствор (Ь). 25,0 мл испытуемого
раствора (а) доводят водой для хроматографии Р до
объема 50,0 мл.
Раствор сравнения (а). К 25,0 мл испытуемого
раствора (а) прибавляют 1,0 мл эталонного раст¬
вора сульфата (10 ррт 30) Р и 12,0 мл эталонного
раствора хлорида (50 ррт С!) Р и доводят водой для
хроматографии Р до объема 50,0 мл.
Раствор сравнения (Ь). 10,0 мл испытуемого
раствора (а) доводят водой для хроматографии Р
до объема 100,0 мл. К 2,0 мл полученного раствора
прибавляют 8,0 мл эталонного раствора хлорида
(50 ррт С1) Р и доводят водой для хроматографии Р
до объема 20,0 мл.
Холостой раствор. Вода для хроматографии Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
2 мм, заполненная анионообменной смолой сильноос¬
новной для хроматографии Р с размером частиц 13 мкм;
- подвижная фаза: 0,600 г калия гидроксида Р
растворяют в воде для хроматографии Р и доводят
до объема 1000,0 мл этим же растворителем;
- скорость подвижной фазы: 0,4 мл/мин;
- кондуктометрический детектор, оборудо¬
ванный подходящим подавителем ионов;
- объем вводимой пробы: по 50 мкл испытуемого
раствора (Ь), растворов сравнения (а) и (Ь) и холосто¬
го раствора;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания бромидов.
Время удерживания: хлориды — около 5 мин;
бромиды — около 8 мин; сульфаты — около 16 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 8,0 между пиками хло¬
ридов и бромидов.
Расчет процентного содержания примесей:
- хлориды: используют концентрацию хлоридов
в растворе сравнения (а); корректируют площадь
пика, соответствующего хлоридам, на хроматограмме
раствора сравнения (а) вычитанием площади пика,
соответствующего хлоридам, на хроматограмме ис¬
пытуемого раствора (Ь);
- сульфаты: используют концентрацию сульфа¬
тов в растворе сравнения (а); корректируют площадь
пика, соответствующего сульфатам, на хроматограм¬
ме раствора сравнения (а) вычитанием площади
пика, соответствующего сульфатам, на хроматограм¬
ме испытуемого раствора (Ь).
Предельное содержание примесей:
- хлориды (не более 0,6 %);
- сульфаты (не более 0,01 %).
Йодиды. К 5 мл раствора 3 прибавляют 0,15 мл
раствора железа (III) хлорида Р1, 2 мл метиленхло-
рида Р и встряхивают. После разделения слоев ниж¬
ний слой должен быть бесцветным (2.2.2, метод /).
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Получен¬
ный раствор должен выдерживать испытание на железо.
Магний и щелочноземельные металлы (2.4.7).
Не более 0,0200 % (200 ррт) в пересчете на кальций.
10,0 г испытуемого образца должны выдерживать ис¬
пытание на магний и щелочноземельные металлы.
Объем использованного 0,01 М раствора натрия
эдетата не должен превышать 5,0 мл.
#Барий. К 5 мл раствора 3 прибавляют 5 мл
воды дистиллированной Р и 1 мл кислоты серной
разведенной Р. Через 15 мин опалесценция в полу¬
ченном растворе должна быть не интенсивнее смеси
из 5 мл раствора 3 и 6 мл воды дистиллированной Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
100,0 мг испытуемого образца растворяют в
воде Р, прибавляют 5 мл раствора кислоты азотной
разведенной Р и доводят водой Р до объема 50 мл.
Титруют 0,1 М раствором серебра нитрата потенци-
ометрически (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 11,90 мг КВг.
Содержание КВг в процентах рассчитывают по
формуле:
а- 3,357Ь,
где:
а — содержание КВг и КС1, полученное при ко¬
личественном определении, рассчитанное как содер¬
жание КВг;
Ь — содержание хлора, полученное в испытании
на хлориды.
480
Гэ сударственная фармакопея Республики Беларусь
07/2016:2076
КАЛИЯ ГИДРОАСПАРТАТ
ГЕМИГИДРАТ
КаШ ЬудгодепоазраЛаз ЬетФуйнсиз
РОТА38ШМ НУОПОвЕЫ АЗРАЯТАТЕ
НЕМ1НУОЯАТЕ
н ИНг
но2с. V • 1/2н2о
хо2к
С4Н6КЫОд ■ 1лн20 М.м. 180,2
[1115-63-5]
ОПРЕДЕЛЕНИЕ
Калия гидро-(25)-2-аминобутандиоат гемигидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или кристалли¬
ческий порошок или бесцветные кристаллы.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испыта¬
ние «Удельное оптическое вращение», как указано в
разделе «Испытания».
B. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, со¬
ответствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а).
C. Испытуемый образец дает реакцию (Ь) на ка¬
лий (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 8 должен
быть бесцветным.
рН (2.2.3). От 6,0 до 7,5. Измеряют рН раствора 8.
Удельное оптическое вращение (2.2.7). От
+18,0 до +20,5 (в пересчете на безводное вещество).
0,50 г испытуемого образца растворяют в смеси из
равных объемов кислоты хлористоводородной Р и
воды Р и доводят до объема 25,0 мл этой же смесью
растворителей.
Нингидрин-положительные вещества. Тонкос¬
лойная хроматография (2.2.27).
Испытуемый раствор (а). Раствор 8.
Испытуемый раствор (Ь). 1,0 мл раствора 8 до¬
водят водой Р до объема 10,0 мл.
Раствор сравнения (а). 25 мг ФСО калия гидроа-
спартата гемигидрата растворяют в воде Р и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20,0 мл.
Раствор сравнения (с). 10 мг ФСО глутамино¬
вой кислоты и 10 мг испытуемого образца раство¬
ряют в воде Р и доводят до объема 25 мл этим же
растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 °С
до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт). К
10 мл раствора 8 прибавляют 5 мл воды Р.
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
К 12 мл раствора 8 прибавляют 3 мл воды дистилли¬
рованной Р.
Аммония соли (2.4.1, метод В). Не долее
0,0200 % (200 ррт). 50 мг испытуемого образца долж¬
ны выдерживать испытание на аммония соли. Эталон
готовят с использованием 0,1 мл эталонного раст¬
вора аммония (100 ррт Р.
Железо (2.4.9). Не более 0,0030 % (30 ррт).
0,33 г испытуемого образца помещают в делительную
воронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл воды Р. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не менее 4,0 % и не более 6,0 %.
Определение проводят из 0,200 г испытуемого образ¬
ца. Испытуемый образец растворяют в 10 мл форма-
мида Р1 и прибавляют 10 мл метанола безводного Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
70,0 мг испытуемого образца растворят в 5 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 8,56 мг С4Н6К1Ч04.
Калия дигидрофосфат
481
07/2016:0840
КАЛИЯ ГИДРОКСИД
КаШ ЬудгохШит
РОТА83ШМ НУйРОХЮЕ
КОН М.м. 56,11
[1310-58-3]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 85,0 % и не более 100,5 %
общего содержания гидроксидов щелочных металлов
в пересчете на КОН.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая кристаллическая твердая
масса, поставляемая в виде брусков, гранул или ку¬
сков неправильной формы. Расплывается на воздухе.
Гигроскопична. Поглощает диоксид углерода.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. рН (2.2.3): не менее 10,5.0,1 г испытуемого об¬
разца растворяют в 10 мл воды Р (раствор А, исполь¬
зуемый в идентификации В). 1 мл полученного раст¬
вора доводят водой Р до объема 100 мл.
B. 1 мл раствора А, приготовленный как указано
в идентификации А, дает реакцию (Ь) на калий (2.3.1).
ИСПЫТАНИЯ
Раствор 51. 2,5 г испытуемого образца раство¬
ряют в 10 мл воды Р. Осторожно прибавляют 2 мл
кислоты азотной Р при охлаждении и доводят кис¬
лотой азотной разведенной Р до объема 25 мл.
Раствор 52. Юг испытуемого образца раство¬
ряют в 15 мл воды дистиллированной Р, осторожно
прибавляют 12 мл кислоты хлористоводородной Р
при охлаждении и доводят кислотой хлористоводо¬
родной разведенной Р до объема 50 мл.
Раствор 53. 5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 53 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 53 должен
быть бесцветным.
Карбонаты. Не более 2,0 % в пересчете на
^СОд. Определяют, как указано в разделе «Количе¬
ственное определение».
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2,5 мл раствора 51 доводят водой Р до объема 15 мл.
Фосфаты (2.4.11). Не более 0,0100 % (100 ррт).
1 мл раствора 51 доводят водой Р до объема 100 мл.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
5 мл раствора 52 доводят водой дистиллированной Р
до объема 20 мл.
Алюминий (2.4.17). Не боле 0,00002 % (0,2 ррт),
если субстанция предназначена для производства
растворов для гемодиализа.
Предписанный раствор. 20 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды дистиллированной Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 5 мл
раствора 52 доводят водой Р до объема 10 мл.
Натрий. Не более 1,0 %.
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. 1,00 г испытуемого раст¬
вора растворяют в 50 мл воды Р, прибавляют 5 мл
кислоты серной Р и доводят водой Р до объема
100,0 мл. 1,0 мл полученного раствора доводят во¬
дой Р до объема 10,0 мл.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора натрия
(200 ррт А1а) Р водой Р.
Источник излучения: лампа с полым катодом
для определения натрия.
Длина волны: 589 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 10 мл раствора 52 доводят во¬
дой Р до объема 20 мл. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца растворяют в 25 мл
воды, свободной от углерода диоксида, Р, прибав¬
ляют 25 мл свежеприготовленного раствора бария
хлорида Р1, 0,3 мл раствора фенолфталеина Р и
медленно, при перемешивании, 25,0 мл 1 М раст¬
вора кислоты хлористоводородной. Продолжают
титрование 1 М раствором кислоты хлористоводо¬
родной до обесцвечивания розового раствора. При¬
бавляют 0,3 мл раствора бромфенолового синего Р
и продолжают титрование 1 М раствором кислоты
хлористоводородной до перехода окраски раствора
от фиолетово-голубой до желтой.
1 мл 1 М раствора кислоты хлористоводород¬
ной, используемой во второй части титрования, соот¬
ветствует 69,11 мг К2С03.
1 мл 1 М раствора кислоты хлористоводород¬
ной, используемой в объединенном титровании, со¬
ответствует 56,11 мг общего содержания гидроксидов
щелочных металлов в пересчете на КОН.
ХРАНЕНИЕ
В воздухонепроницаемом неметаллическом кон¬
тейнере.
МАРКИРОВКА
При необходимости на этикетке указывают, что
субстанция пригодна для использования в производ¬
стве растворов для гемодиализа.
07/2016:0920
КАЛИЯ ДИГИДРОФОСФАТ
КаШ сИНу^годепор/юзрМаз
РОТА83ШМ О/НУОКОСЕЛ/ РНОЗРНАТЕ
КН2Р04 М.м. 136,1
[7778-77-0]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 100,5 %
(в пересчете на сухое вещество).
61. Зак. 1060.
482
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 5, приготовленный как указано в
разделе «Испытания», имеет слабокислую реакцию
(2.2.4).
B. Раствор 5 дает реакцию (Ь) на фосфаты (2.3.1).
C. 0,5 мл раствора 3 дают реакцию (Ь) на калий
(2.3.1).
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,2 до 4,5. К 5 мл раствора 3 прибав¬
ляют 5 мл воды, свободной от углерода диоксида, Р.
Восстанавливающие вещества. К 5 мл раст¬
вора 3 прибавляют 5 мл кислоты серной разведен¬
ной Р, 0,25 мл 0,02 М раствора калия перманганата
и нагревают на водяной бане в течение 5 мин. Раст¬
вор не должен полностью обесцвечиваться.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2,5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
К 5 мл раствора 3 прибавляют 0,5 мл кислоты хлори¬
стоводородной Р и доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Железо (2.4.9). Не более 0,0010% (10 ррт).
Раствор 3 должен выдерживать испытание на железо.
Натрий. Не более 0,1 %, если калия дигидро¬
фосфат предназначен для производства лекарствен¬
ных средств для парентерального применения. Атом¬
но-эмиссионная спектрометрия (2.2.22, метод /).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в водеР и доводят до объема
100,0 мл этим же растворителем.
Растворы сравнения. Готовят соответствующи¬
ми разведениями с помощью воды Р раствора, при¬
готовленного следующим образом: 0,5084 г натрия
хлорида Р, предварительно высушенного при темпе¬
ратуре от 100 °С до 105 °С в течение 3 ч, растворяют
в воде Р и доводят до объема 1000,0 мл этим же раст¬
ворителем (200 мкг/мл №).
Длина волны: 589 нм.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 2,0 %. 1,000 г испытуемого образца сушат при
температуре от 125 °С до 130 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют в 50 мл
воды, свободной от углерода диоксида, Р и титруют
1 М раствором натрия гидроксида, свободным от
карбонатов, потенциометрически (2.2.20).
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 0,1361 гКН2Р04.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:0186
КАЛИЯ ЙОДИД
КаШ ЫМит
РОТАЗЗШМ /ОО/ОЕ
К1 М.м. 166,0
[7681-11-0]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Очень легко растворим в воде, легко растворим в
глицерине, растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции (а) и (Ь) на йодиды
(2.3.1) .
B. Раствор 3 дает реакции (а) и (Ь) на калий
(2.3.1) .
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Щелочность. К 12,5 мл раствора 3 прибавляют
0,1 мл раствора бромтимолового синего Р1. При
прибавлении не более 0,5 мл 0,01 М раствора кис¬
лоты хлористоводородной окраска раствора должна
измениться.
Йодаты. К 10 мл раствора 3 прибавляют 0,25 мл
раствора крахмала, свободного от йодидов, Р,
0,2 мл кислоты серной разведенной Р и выдержива¬
ют в защищенном от света месте в течение 2 мин. Не
должно появляться синее окрашивание.
Сульфаты (2.4.13). Не более 0,0150 % (150 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
Тиосульфаты. К 10 мл раствора 3 прибавляют
0,1 мл раствора крахмала Р и 0,1 мл 0,005 М раст¬
вора йода. Должно появиться синее окрашивание.
*
Р'
Калия клавуланат
483
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 5 доводят водой Р до объема 10 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0%. 1,00 г предварительно измельченного
испытуемого образца сушат при температуре 105 °С
в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,500 г испытуемого образца растворяют в
воде Р и доводят до объема 100,0 мл этим же раство¬
рителем. К 20,0 мл полученного раствора прибавля¬
ют 40 мл кислоты хлористоводородной Р и титруют
0,05 М раствором калия йодата до изменения окра¬
шивания от красного до желтого. Прибавляют 5 мл
хлороформа Р и продолжают титрование, интенсивно
встряхивая, до обесцвечивания хлороформного слоя.
1 мл 0,05 М раствора калия йодата соответ¬
ствует 16,60 мг К1.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1140
КАЛИЯ КЛАВУЛАНАТ
КаШ с1ауи1апаз
РОТА83ШМ а А ИАТЕ
СД.КЫО, М.м. 237,3
[61177-45-5]
ОПРЕДЕЛЕНИЕ
Калия (2Р,32,5Р)-3-(2-гидроксиэтилиден)-7-оксо-
4-окса-1-азабицикло[3,2,0]гептан-2-карбоксилат. Ка¬
лиевая соль субстанции, продуцируемой некоторыми
видами 31гер1отусез с^иНдегиз или получаемой лю¬
быми другими способами.
Содержание: не менее 96,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, мало растворим в 96 %
спирте, очень мало растворим в ацетоне.
ПРОИЗВОДСТВО
Методы получения, экстракции и очистки должны
обеспечивать отсутствие клавам-2-карбоксилата или
содержание его не выше 0,01 %.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр калия клавулана-
та по Европейской Фармакопее.
B. Испытуемый образец дает реакцию (Ь) на ка¬
лий (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 0,400 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20,0 мл этим же растворителем.
рН (2.2.3). От 5,5 до 8,0. 5 мл раствора 8 доводят
водой, свободной от углерода диоксида, Р до объема
10 мл.
Удельное оптическое вращение (2.2.7). От +53
до +63 (в пересчете на безводное вещество). Опреде¬
ляют удельное оптическое вращение раствора 8.
Полимерные и другие примеси, поглощаю¬
щие при длине волны 278 нм. 50,0 мг испытуемого
образца растворяют в 0,1 М фосфатном буферном
растворе рН 7,0 Р и доводят до объема 50,0 мл этим
же растворителем. Незамедлительно измеряют опти¬
ческую плотность. Оптическая плотность (2.2.25) по¬
лученного раствора, определенная при длине волны
278 нм, не должна превышать 0,40.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 0,250 г испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (Ь). 10 мг ФСО лития кла-
вуланата и 10 мг ФСО амоксициллина тригидрата
растворяют в подвижной фазе А и доводят до объема
100 мл этим же растворителем.
Раствор сравнения (с). 2 мг ФСО калия клавулана-
та примеси 6 растворяют в 20 мл подвижной фазы А.
Условия хроматографирования:
-колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: раствор 7,8 г/л натрия
дигидрофосфата Р, доведенный до рН 4,0 кис¬
лотой фосфорной Р;
- подвижная фаза В: смесь из равных объемов
метанола Р и подвижной фазы А;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—4
100
0
4—15
о
ю
т
о
о
0—>50
15—18
50
50
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению к
кпавуланату, время удерживания — около 3 мин): при¬
месь Е — около 2,3; примесь О — около 3,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
61*. Зак. 1060.
484
Гэсударственная фармакопея Республики Беларусь
- разрешение: не менее 13 между пиками клаву-
ланата (1-й пик) и амоксициллина (2-й пик).
Предельное содержание примесей:
-примеси Е и О (не более 1,0 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Е и О, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- любая другая примесь (не более 0,2 %): на
хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного и пиков примесей Е и О,
не должна превышать 0,2 площади основного пика на
хроматограмме раствора сравнения (а);
- сумма примесей (не более 2,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (а).
Алифатические амины. Газовая хроматогра¬
фия (2.2.28).
Метод, описанный ниже, может быть исполь¬
зован для определения следующих алифатиче¬
ских аминов: 1,1-диметилэтиламина, тетрамети-
лэтилендиамина, 1,1,3,3-тетраметилбутиламина,
Л/,Л/-диизопропилэтилендиамина, 2,2-оксибис(А/,А/-
диметилэтиламина).
Раствор внутреннего стандарта. 50 мкп
З-метилпентан-2-она Р растворяют в воде Р и дово¬
дят до объема 100,0 мл этим же растворителем.
Испытуемый раствор. 1,00 г испытуемого об¬
разца помещают в центрифужную пробирку и при¬
бавляют 5,0 мл раствора внутреннего стандарта,
5.0 мл раствора натрия гидроксида разведенного Р,
10.0 мл воды Р, 5,0 мл 2-метилпропанола Р и 5 г на-
трия хлорида Р. Интенсивно встряхивают в течение
1 мин, затем центрифугируют для разделения слоев.
Раствор сравнения. По 80,0 мг каждого из сле¬
дующих аминов: 1,1-диметилэтиламина Р, тетра-
метилэтилендиамина Р, 1,1,3,3-тетраметилбу¬
тиламина Р, Ы^'-диизопропилэтилендиамина Р,
2,2'-оксибис(М,М-диметилэтиламина) Р растворяют
в кислоте хлористоводородной разведенной Р и до¬
водят этой же кислотой до объема 200,0 мл. 5,0 мл
полученного раствора помещают в центрифужную
пробирку и прибавляют 5,0 мл раствора внутренне¬
го стандарта, 10,0 мл раствора натрия гидроксида
разведенного Р, 5,0 мл 2-метилпропанола Р и 5 г на¬
трия хлорида Р. Интенсивно встряхивают в течение
1 мин, затем центрифугируют для разделения слоев.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 50 м
и внутренним диаметром 0,53 мм, покрытая слоем
поли(диметил)(дифенил)силоксана Р (толщина слоя
5 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 8 мл/мин;
- деление потока: 1:10;
- температура:
Время
Температура
(мин)
(°С)
Колонка
0—7
35
7—10,8
35-150
10,8—25,8
150
Блок ввода проб
200
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл верхних сло¬
ев, полученных после центрифугирования испытуе¬
мого раствора и раствора сравнения.
Относительное удерживание (по отношению
к З-метилпентан-2-ону, время удерживания — около
11,4 мин): примесь Н — около 0,55; примесь и — око¬
ло 1,07; примесь К — около 1,13; примесь I- — около
1,33; примесь М — около 1,57.
Предельное содержание примесей:
- сумма алифатических аминов: не более 0,2 %.
2-Этилкапроевая кислота (2.4.28). Не более
0,8 %.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
0,03 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Растворы
готовят непосредственно перед использованием.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в растворе 4,1 г/л натрия ацета¬
та Р, предварительно доведенном кислотой уксус¬
ной ледяной Р до рН 6,0, и разводят этим же раство¬
ром до объема 50,0 мл.
Раствор сравнения (а). 50,0 мг ФСО лития
клавуланата растворяют в растворе 4,1 г/л натрия
ацетата Р, предварительно доведенном кислотой
уксусной ледяной Р до рН 6,0, и разводят до объема
50,0 мл этим же раствором.
Раствор сравнения (Ь). 10 мг ФСО амоксицилли¬
на тригидрата растворяют в 10 мл раствора сравне¬
ния (а).
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц
10 мкм;
- подвижная фаза: смесь метанола Р1 и раст¬
вора 15 г/л натрия дигидрофосфата Р, предвари¬
тельно доведенного кислотой фосфорной разведен¬
ной Р до рН 4,0, (5:95, об/об).
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл.
Пригодность хроматографической системы
раствор сравнения (Ь):
- разрешение: не менее 3,5 между пиками клаву¬
ланата (1-й пик) и амоксициллина (2-й пик).
1 мг С8Н91Ч05 соответствует 1,191 мгС8Н8КМ05.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре от 2 °С до 8 °С. Если субстанция стерильная
ее хранят в стерильном воздухонепроницаемом кон¬
тейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: Е, С, Н, 3, К, М
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным исгь-
Калия клавуланат разведенный
485
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): А, В, С, О, Р.
С помощью жидкостной хроматографии: А, В,
С, О, Е, Р, С.
С помощью жидкостной хроматографии: Н, 3,
К, Ц М.
^ /ОН
НО'
A. К = Н: 2,2'-(Пиразин-2,5-диил)диэтанол.
B. К = СН2-СН2-С02Н: 3-[3,6-Бис(2-гидроксиэтил)
пиразин-2-ил]пропановая кислота.
C. К = СН2-СН3: 2,2'-(3-Этилпиразин-2,5-диил)ди-
этанол.
ни
О. 4-(2-Гидроксиэтил)-1Н-пиррол-3-карбоновая
кислота.
Е. (2Я,4/?,52)-2-(Карбоксиметил)-5-(2-гидрокси-
этилиден)-3-[[(2Я32,5Я)-3-(2-гидроксиэтилиден)-7-оксо-
4-окса-1-азабицикло[3.2.0]гепт-2-ил]карбонил]-
оксазолидин-4-карбоновая кислота.
9 со2н
ни
г>
он
Р. 4-[[[[4-(2-Гидроксиэтил)-1 Н-пиррол-3-ил]карбо-
нил]окси]метил]-1 Н-пиррол-З-карбоновая кислота.
^он
но2д н
ны
Н02С'
6. 4-[[(15)-1-Карбокси-2-(4-гидроксифенил)этил]-
амино]-4-оксобутановая кислота (Л/-(гидросукцинил Ти¬
розин).
н3с.
СНз
X
Н3С ЫНг
Н. 2-Метилпропан-2-амин (2-амино-2-метилпропан,
шреш-бутиламин, этилдиметиламин).
СН3
сн3
3. А/,Л/,/У',А/-Тетраметилэтан-1,2-диамин (1,2-бис-
(диметиламино)этан, Л/,А/,Л/',Л/-тетраметилэтилен-
диамин).
Н3С Г^СНз
СН3 СНз
К. 2,4,4-Триметилпентан-2-амин (2-амино-2,4,4-
триметилпентан, 1,1,3,3-тетраметил бутиламин).
1_. А/,Л/-Диизопропилэтан-1,2-диамин (А/,Л/'-бис(1-
метилэтил)этан-1,2-диамин).
Н3сх
СН3 СНз
М. 2,2,-0ксибис(/\/,Л/-диметилэтанамин) (бис[2-(ди-
метиламино)этиловый] эфир, Л/,Л/,Л/',Л/'-тетраме-
тил(оксидиэтилен)диамин).
07/2016:1653
КАЛИЯ КЛАВУЛАНАТ
РАЗВЕДЕННЫЙ
КаШ с1ауи1апаз сИМиз
РОТА88ШМ С1-А\/Ш-АЫАТЕ, 01ШТЕ0
с,н,кмо5
ОПРЕДЕЛЕНИЕ
Сухая смесь Калия клавуланата (1140) и Цел¬
люлозы микрокристаллической (0316), или Крем¬
ния диоксида коллоидного безводного (0434), или
Кремния диоксида коллоидного гидратированного
(0738).
Содержание: не менее 91,2% и не более
107,1 % от содержания калия клавуланата, указан¬
ного на этикетке.
ОПИСАНИЕ (СВОЙСТВА)
Внешний вид калия клавуланата разведенного:
белый или почти белый порошок; гигроскопичен.
Растворимость калия клавуланата: легко рас¬
творим в воде, мало растворим в 96 % спирте, очень
мало растворим в ацетоне.
Растворимость калия клавуланата разведенного
зависит от разбавителя и его концентрации.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: на хроматограмме испытуемо¬
го раствора основной пик соответствует по времени
удерживания основному пику на хроматограмме раст¬
вора сравнения (а).
B. Испытуемый образец дает реакцию (Ь) на ка¬
лий (2.3.1).
C. В зависимости от применяемого разбавителя
проводят соответствующие испытания (а) или (Ь).
62. Зак. 1060.
486
Гэсударственная фармакопея Республики Беларусь
(a) Испытуемый образец в количестве, соответ¬
ствующем 20 мг целлюлозы, при помещении на часо¬
вое стекло и диспергировании в 4 мл раствора цинка
хлорида йодированного Р окрашивается в фиолето¬
во-синий цвет.
(b) Испытуемый образец дает реакцию (а) на си¬
ликаты (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 4,8 до 8,0. Испытуемый образец
в количестве, соответствующем 0,200 г калия кла-
вуланата, суспендируют в 20 мл воды, свободной
от углерода диоксида, Р.
Полимерные и другие примеси, поглощаю¬
щие при длине волны 278 нм. Испытуемый обра¬
зец в количестве, соответствующем 50,0 мг калия
клавуланата, диспергируют в ТО мл 0,1 М фосфат¬
ного буферного раствора рН 7,0 Р, доводят до
объема 50,0 мл этим же растворителем и фильтру¬
ют. Незамедлительно измеряют оптическую плот¬
ность. Оптическая плотность (2.2.25) раствора,
определенная при длине волны 278 нм, не должна
превышать 0,40.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием.
Испытуемый раствор. Испытуемый образец
в количестве, соответствующем 0,250 г калия кла¬
вуланата, диспергируют в 5 мл подвижной фазы А,
доводят до объема 25,0 мл этим же растворителем
и фильтруют.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят подвижной фазой А до объема
100,0 мл.
Раствор сравнения (Ь). 10 мг ФСО амоксицил-
лина тригидрата растворяют в 1 мл испытуемого
раствора и доводят подвижной фазой А до объема
100 мл.
Раствор сравнения (с). 2 мг ФСО калия клаву¬
ланата примеси О растворяют в 20 мл подвижной
фазы А.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: раствор 7,8 г/л натрия
дигидрофосфата Р, доведенный до рН 4,0
кислотой фосфорной Р;
- подвижная фаза В: смесь из равных объ¬
емов подвижной фазы А и метанола Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-4
100
0
4—15
100 — 50
о
ю
т
о
15-18
50
50
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению к
клавуланату, время удерживания — около 3 мин): при¬
месь Е — около 2,3; примесь О — около 3,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 13 между пиками кла¬
вуланата (1-й пик) и амоксициллина (2-й пик).
Предельное содержание примесей:
- примеси Е и 6 (не более 1,0 %): на хромато¬
грамме испытуемого раствора площади пиков, со¬
ответствующих примесям Е и О, не должны превы¬
шать площадь основного пика на хроматограмме
раствора сравнения (а);
-любая другая примесь (не более 0,2 %): на
хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пиков примесей Е
и 6, не должна превышать 0,2 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 2,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,05 площади основного
пика на хроматограмме раствора сравнения (а).
Вода (2.5.12). Не более 2,5%. Определение
проводят из 1,000 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Раство¬
ры готовят непосредственно перед использова¬
нием.
Испытуемый раствор. Испытуемый образец
в количестве, соответствующем 50,0 мг калия кла¬
вуланата, диспергируют в растворе 4,1 г/л натрия
ацетата Р, предварительно доведенном кисло¬
той уксусной ледяной Р до рН 6,0, и разводят этим
же раствором до объема 50,0 мл, затем фильтруют.
Раствор сравнения (а). 50,0 мг ФСО лития
клавуланата растворяют в растворе 4,1 г/л на¬
трия ацетата Р, предварительно доведенном
кислотой уксусной ледяной Р до рН 6,0, и разво¬
дят этим же раствором до объема 50,0 мл.
Раствор сравнения (Ь). 10 мг ФСО амоксицил¬
лина тригидрата растворяют в 10 мл раствора
сравнения (а).
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 10 мкм;
- подвижная фаза: смесь метанола Р1 и раст¬
вора 15 г/л натрия дигидрофосфата Р, предвари¬
тельно доведенного кислотой фосфорной разве¬
денной Р до рН 4,0, (5:95, об/об).
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 230 нм;
- объем вводимой пробы: 10 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 3,5 между пиками
клавуланата (1-й пик) и амоксициллина (2-й пик).
1 мгС8Н9М05 соответствует 1,191 мг С8Н8К1Ч05
»
Калия метабисульфит
487
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
Указывают:
- процентное содержание калия клавуланата и
разбавитель, применяемый для приготовления смеси.
ПРИМЕСИ
Специфицированные примеси: Е, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или об¬
щей статьей Субстанции для фармацевтического
использования. Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацев¬
тического использования): А, В, С, О, Е.
A. Р = Н: 2,2'-(Пиразин-2,5-диил)диэтанол.
B. Р = СН2-СН2-С02Н: 3-[3,6-Бис(2-гидроксиэтил)
пиразин-2-ил]пропановая кислота.
C. Р = СН2-СН3: 2,2'-(3-Этилпиразин-2,5-диил)ди-
этанол.
О. 4-(2-Гидроксиэтил)-1Н-пиррол-3-карбоновая
кислота.
Е. (2Я,4/?,52)-2-(Карбоксиметил)-5-(2-гидро-
ксиэтилиден)-3-[[(2Р,32,5Р)-3-(2-гидроксиэтилиден)-7-
оксо-4-окса-1-азабицикло[3.2.0]гепт-2-ил]карбонил]-
оксазолидин-4-карбоновая кислота.
Р. 4-[[[[4-(2-Гидроксиэтил)-1Н-пиррол-3-ил]карбо-
нил]окси]метил]-1 Н-пиррол-З-карбоновая кислота.
О. 4-[[(1$)-1-Карбокси-2-(4-гидроксифенил)этил]-
амино]-4-оксобутановая кислота (Л/-(гидросукцинил Ти¬
розин).
07/2016:2075
КАЛИЯ МЕТАБИСУЛЬФИТ
КаШ те1аЫзиШз
РОТА88ШМ МЕТАВ18Ш-Р1ТЕ
КЛВЛОш М.м. 222,3
[16731-55-8]
ОПРЕДЕЛЕНИЕ
Калия метабисульфит (калия дисульфит).
Содержание: не менее 95,0 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
B. К 5 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,5 мл 0,05 М
раствора йода. Полученная смесь бесцветная и дает
реакцию (а) на сульфаты (2.3.1).
C. Раствор 3 дает реакцию (а) на калий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод /). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 3,0 до 4,5. Измеряют рН раствора 3.
Тиосульфаты. К 2,00 г испытуемого образца
прибавляют 25 мл раствора 42,5 г/л натрия гидрок¬
сида Р и 75 мл воды Р. Встряхивают до растворения,
прибавляют 10 мл формальдегида Р и 10 мл кис¬
лоты уксусной Р. Через 5 мин титруют 0,05 М рас¬
твором йода, используя в качестве индикатора 1 мл
раствора крахмала Р. Параллельно проводят кон¬
трольный опыт. Разность между объемом, израсходо¬
ванным на титрование испытуемого образца, и объе¬
мом, израсходованным на титрование в контрольном
опыте, должна быть не более 0,15 мл.
Железо. Не более 0,0010 % (10 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод /).
Испытуемый раствор. 20 мл раствора 3 дово¬
дят водой Р до объема 50 мл.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора железа
(20 ррт Ре) Р водой Р.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Селен. Не более 0,0010 % (10 ррт). К 3,0 г ис¬
пытуемого образца прибавляют 10 мл формальдеги¬
да Р \л осторожно, малыми порциями, 2 мл кислоты
хлористоводородной Р. Нагревают на водяной бане
в течение 20 мин. Розовая окраска полученного раст¬
вора должна быть не интенсивнее окраски раствора,
приготовленного параллельно с использованием 1,0 г
испытуемого образца и 0,2 мл эталонного раствора
селена (100 ррт 8е) Р.
62*. Зак. 1060.
488
Гэсударственная фармакопея Республики Беларусь
Цинк. Не более 0,0025 % (25 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 20 мл раствора 3 дово¬
дят водой Р до объема 50 мл.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора цинка
(100 ррт 1п) Р водой Р.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Тяжелые металлы (2.4.8, метод Е). Не более
0,0010% (10 ррт). 40 мл раствора 3 помещают в
кварцевый тигель, прибавляют 10 мл кислоты хло¬
ристоводородной Р и выпаривают досуха. Получен¬
ный остаток растворяют в 19 мл воды Р и прибавляют
1 мл раствора 40 г/л натрия фторида Р. Полученный
раствор должен выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием 20 мл
эталонного раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
В коническую колбу вместимостью 500 мл по¬
мещают 50,0 мл 0,05 М раствора йода, прибавляют
0,150 г испытуемого образца и 5 мл кислоты хлори¬
стоводородной Р. Избыток йода титруют 0,1 М рас¬
твором натрия тиосульфата, используя в качестве
индикатора 0,1мл раствора крахмала Р, который
прибавляют перед концом титрования.
1мл 0,05 М раствора йода соответствует
5,558 мг К^О,..
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0121
КАЛИЯ ПЕРМАНГАНАТ
КаШ регтапдапаз
РОТАЗЗШМ РЕКМАЫСАЫАТЕ
КМп04 М.м. 158,0
[7722-64-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
ОПИСАНИЕ(СВОЙСТВА)
Темно-пурпурный или коричневато-черный грану¬
лированный порошок или темно-пурпурные или почти
черные кристаллы обычно с металлическим блеском.
Растворим в холодной воде, легко растворим в
кипящей воде.
При контакте с некоторыми органическими веще¬
ствами разлагается.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Около 50 мг испытуемого образца растворяют
в 5 мл воды Р и прибавляют 1 мл 96 % спирта Р и
0,3 мл раствора натрия гидроксида разведенного Р.
Появляется зеленое окрашивание. Нагревают до ки¬
пения. Образуется темно-коричневый осадок.
В. Фильтруют смесь, полученную в идентифика¬
ции А. Фильтрат дает реакцию (Ь) на калий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,75 г испытуемого образца раство¬
ряют в 25 мл воды дистиллированной Р, прибавляют
3 мл 96% спирта Р и кипятят в течение (2—3) мин.
Полученный раствор охлаждают, доводят водой дис¬
тиллированной Р до объема 30 мл и фильтруют.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Нерастворимые в воде вещества. Не более
1,0 %. 0,5 г испытуемого образца растворяют в 50 мл
воды Р и нагревают до кипения. Фильтруют через
предварительно взвешенный стеклянный фильтр
(ПОР 16) (2.1.2), промывают водой Р до получения
бесцветного фильтрата и высушивают фильтр с остат¬
ком при температуре от 100 °С до 105 °С. Масса полу¬
ченного остатка не должна превышать 5 мг.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
12 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в
воде Р и доводят до объема 100,0 мл этим же раство¬
рителем. К 20,0 мл полученного раствора прибавляют
20 мл воды Р, 1 г калия йодида Р и 10 мл кислоты
хлористоводородной разведенной Р. Выделившийся
йод титруют 0,1 М раствором натрия тиосульфата,
используя в качестве индикатора 1 мл раствора крах¬
мала Р.
1 мл 0,1 М раствора натрия тиосульфата со¬
ответствует 3,160 мг КМп04.
07/2016:0618
КАЛИЯ СОРБАТ
КаШ зогЬаз
РОТАЗЗШМ ЗОРВАТЕ
С6Н7К02 М.м. 150,2
[590-00-1]
ОПРЕДЕЛЕНИЕ
Калия (Е,Е)-гекса-2,4-диеноат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или гранулы.
Очень легко растворим в воде, мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
250,0 мл этим же растворителем. 2,0 мл полученного
Калия хлорид
489
раствора доводят 0,1 М раствором кислоты хлори¬
стоводородной до объема 200,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 264 нм.
Удельный показатель поглощения в максимуме:
от 1650 до 1900.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО калия сорбата.
C. 1,0 г испытуемого образца растворяют в 50 мл
воды Р, прибавляют 10 мл кислоты хлористоводо¬
родной разведенной Р и встряхивают. Отфильтровы¬
вают кристаллический осадок, промывают водой Р и
сушат в вакууме над кислотой серной Р в течение 4 ч.
Температура плавления (2.2.14) полученного остатка:
от 132 °С до 136 °С.
й. 0,2 г испытуемого образца растворяют в 2 мл
воды Р и прибавляют 2 мл кислоты уксусной разве¬
денной Р и фильтруют. Полученный раствор дает ре¬
акцию (Ь) на калий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)5.
Кислотность или щелочность. К 20 мл раствора
3 прибавляют 0,1 мл раствора фенолфталеина Р.
При прибавлении не более 0,25 мл 0,1 Мраствора на¬
трия гидроксида или 0,1 М раствора кислоты хлори¬
стоводородной окраска раствора должна измениться.
Альдегиды. Не более 0,15% в пересчете на
ацетальдегид. 1,0 г испытуемого образца растворяют
в смеси из 30 мл воды Р и 50 мл 2-пропанола Р, дово¬
дят 1 М раствором кислоты хлористоводородной до
рН 4 и разводят раствор водой Р до объема 100 мл.
К 10 мл приготовленного раствора прибавляют 1 мл
обесцвеченного раствора фуксина Р и выдерживают
в течение 30 мин. Окраска раствора должна быть не
интенсивнее окраски эталона, параллельно приготов¬
ленного прибавлением 1 мл обесцвеченного раст¬
вора фуксина Р к смеси из 1,5 мл эталонного раст¬
вора ацетальдегида (100 ррт С2Н40) Р, 4 мл 2-про-
панола Р и 4,5 мл воды Р.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре от 100 °С до 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в 20 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной до перехода окраски от
фиолетовой до голубовато-зеленой, используя 0,1 мл
раствора кристаллического фиолетового Р в каче¬
стве индикатора.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 15,02 мг С6Н7К02.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0185
КАЛИЯ ХЛОРИД
КаШ сЫогМит
РОТА38ШМ СШ-ОКЮЕ
КС1 М.м. 74,6
[7447-40-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %
КС1 (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, практически нераство¬
рим в этаноле безводном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на хлори¬
ды (2.3.1).
B. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции (а) и (Ь) на калий
(2.3.1).
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 50 мл раст¬
вора 3 прибавляют 0,1 мл раствора бромтимоло-
вого синего Р1. При прибавлении не более 0,5 мл
0,01 М раствора кислоты хлористоводородной или
0,01 М раствора натрия гидроксида окраска раст¬
вора должна измениться.
Бромиды. Не более 0,1 %. 1,0 мл раствора 3
доводят водой Р до объема 50 мл. К 5,0 мл получен¬
ного раствора прибавляют 2,0 мл раствора феноло¬
вого красного Р2, 1,0 мл раствора хлорамина Р1 и
сразу перемешивают. Точно через 2 мин прибавляют
0,15 мл 0,1 М раствора натрия тиосульфата, пере¬
мешивают и доводят водой Р до объема 10,0 мл. Оп¬
тическая плотность (2.2.25) полученного раствора,
измеренная при длине волны 590 нм, не должна пре¬
вышать оптическую плотность эталона, приготовлен¬
ного параллельно с использованием 5 мл раствора
3,0 мг/л калия бромида Р. В качестве раствора срав¬
нения используют воду Р.
Йодиды. 5 г испытуемого образца увлажняют,
прибавляя по каплям свежеприготовленную смесь
из 0,15 мл раствора натрия нитрита Р, 2 мл 0,5 М
раствора кислоты серной, 25 мл раствора крахма¬
ла, свободного от йодидов, Р и 25 мл воды Р и через
5 мин просматривают при дневном свете. Не должно
появиться синего окрашивания.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
63. Зак. 1060.
490
Гэсударственная фармакопея Республики Беларусь
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Алюминий (2.4.17). Не более 0,00010%
(1,0 ррт), если субстанция предназначена для приго¬
товления растворов для растворов для гемодиализа.
Предписанный раствор. 4 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смесь из 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смесь из 10 мл ацетат¬
ного раствора рН 6,0 Р и 100 мл воды Р.
Барий. К 5 мл раствора 3 прибавляют 5 мл воды
дистиллированной Р и 1 мл кислоты серной разве¬
денной Р. Через 15 мин опалесценция полученного
раствора не должна превышать опалесценцию смеси
из 5 мл раствора 3 и 6 мл воды дистиллированной Р.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Магний и щелочноземельные металлы (2.4.7).
Не более 0,0200 % (200 ррт) в пересчете на Са.
10.0 г испытуемого образца должны выдерживать
испытание на магний и щелочноземельные металлы
(используют 0,15 г индикаторной смеси протравно¬
го черного 11 Р). Объем пошедшего на титрование
0,01 М раствора натрия эдетата не должен превы¬
шать 5,0 мл.
Натрий. Не более 0,1 %, если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения или растворов для
гемодиализа. Атомно-эмиссионная спектрометрия
(2.2.22, метод /).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Растворы сравнения. Готовят соответствующими
разведениями раствора, содержащего 200 мкг/мл № и
приготовленного следующим образом: 0,5084 г натрия
хлорида Р, предварительно высушенного при темпера¬
туре 105 °С в течение 3 ч, растворяют в воде Р и дово¬
дят до объема 1000,0 мл этим же растворителем.
Длина волны: 589 нм.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 выдерживают ис¬
пытание на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (1 ррт РЬ) Р.
#Мышьяк (2.4.2, метод А). Не более 0,0001 %
(1 ррт). 10 мл раствора 3 должны выдерживать ис¬
пытание на мышьяк.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
60,0 мг испытуемого образца растворяют в
воде Р, прибавляют 5 мл кислоты азотной разведен¬
ной Р и доводят до объема 50 мл водой Р. Титруют
0,1 М раствором серебра нитрата потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 7,46 мг КС1.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют в воде Р
и доводят до объема 50,0 мл этим же растворителем.
К 5,0 мл полученного раствора прибавляют 40 мл
воды Р и титруют 0,1 М раствором серебра нитрата
до оранжево-желтого окрашивания, используя в каче¬
стве индикатора 0,5 мл раствора калия хромата Р.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 7,46 мг КС1.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения;
- субстанция пригодна для производства раство¬
ров для гемодиализа.
07/2016:0400
КАЛИЯ ЦИТРАТ
КаШ сЛгаз
РОТА88ШМ С1ТКАТЕ
он со2к
КОгО-^Зч^^СОгК Нг0
С6Н5К307 ■ НгО М.м. 324,4
[6100-05-6]
ОПРЕДЕЛЕНИЕ
Трикалия 2-гидроксипропан-1,2,3-трикарбоксилат
моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый гранулированный поро¬
шок или прозрачные кристаллы. Гигроскопичен.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 4 мл воды Р.
Полученный раствор дает реакцию (а) на цитраты
(2.3.1) .
B. 0,5 мл раствора 3 дает реакцию (Ь) на калий
(2.3.1) .
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раствора
3 прибавляют 0,1 мл раствора фенолфталеина Р. При
прибавлении не более 0,2 мл 0,1 М раствора кислоты
хлористоводородной или 0,1 М раствора натрия ги¬
дроксида должна измениться окраска индикатора.
Легкообугливающиеся вещества. К 0,20 г
измельченного испытуемого образца прибавляют
Кальция глицерофосфат
491
10 мл кислоты серной Р и нагревают в водяной
бане при температуре (90±1) °С в течение 60 мин.
Быстро охлаждают. Окраска полученного раствора
(2.2.2, метод II) должна быть не интенсивнее эта¬
лона У(Ж)2 или ОУ(ЗЖ)2.
Хлориды (2.4.4). Не более 0,0050 % (50 рргп).
10 мл раствора 5 доводят водой Р до объема
15 мл.
Оксалаты. Не более 0,0300 % (300 ррт). 0,50 г
испытуемого образца растворяют в 4 мл воды Р,
прибавляют 3 мл кислоты хлористоводородной Р
и 1 г гранулированного цинка Р и нагревают на во¬
дяной бане в течение 1 мин. Оставляют на 2 мин.
Жидкость декантируют в пробирку, содержащую
0,25 мл раствора 10 г/л фенилгидразина гидрох¬
лорида Р, и нагревают до кипения. Быстро охлаж¬
дают, переносят в мерный цилиндр и прибавляют
такой же объем кислоты хлористоводородной Р
и 0,25 мл раствора калия феррицианида Р. Пере¬
мешивают и выдерживают в течение 30 мин. Ро¬
зовое окрашивание раствора должно быть не ин¬
тенсивнее эталона, приготовленного параллельно
с использованием 4 мл раствора 0,05 г/л кислоты
щавелевой Р и такого же количества реактивов.
Сульфаты (2.4.13). Не более 0,0150 %(150 ррт).
К 10 мл раствора 5 прибавляют 2 мл раствора кис¬
лоты хлористоводородной Р1 и доводят водой дис¬
тиллированной Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Натрий. Не более 0,3 %.
Атомно-эмиссионная спектрометрия (2.2.22, ме¬
тод II).
Испытуемый раствор. К 10 мл раствора 3 при¬
бавляют 1 мл кислоты хлористоводородной разве¬
денной Р и доводят водой дистиллированной Р до
объема 100 мл.
Растворы сравнения. Готовят соответствующи¬
ми разведениями раствора натрия хлорида Р, содер¬
жащего 1 мг/мл Ыа, водой дистиллированной Р.
Длина волны: 589 нм.
Вода (2.5.12). Не менее 4,0% и не более
7,0 %. Определение проводят из 0,250 г испытуе¬
мого образца. В качестве растворителя используют
смесь, состоящую из формамида Р и метанола Р
(1:2, об/об). После внесения испытуемого образца
перед началом титрования раствор перемешивают
в течение 15 мин.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 20 мл
кислоты уксусной безводной Р при нагревании до
температуры 50 °С и охлаждают. Титруют 0,1 М рас¬
твором кислоты хлорной до появления зелено¬
го окрашивания, используя в качестве индикатора
0,25 мл раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 10,21 мг С6Н5К307.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0980
КАЛЬЦИЯ ГЛИЦЕРОФОСФАТ
Са1сН д!усегор1юзрЬа8
СА1.СШМ О-УСЕ1ЮРН08РНАТЕ
С,Н7СаОвР М.м. 210,1
[27214-00-2]
ОПРЕДЕЛЕНИЕ
Смесь (/?5)-2,3-дигидроксипропилфосфата каль¬
ция и 2-гидрокси-1-(гидроксиметил)этилфосфата каль¬
ция в различных соотношениях, которая может быть
гидратирована.
Содержание: не менее 18,6 % и не более 19,4 %
Са (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Умеренно растворим в воде, практически нераст¬
ворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца смешивают с 1 г калия
гидросульфата Р в пробирке, снабженной стеклянной
трубкой. Сильно нагревают и направляют белые пары
на кусочек фильтровальной бумаги, импрегнирован-
ной свежеприготовленным раствором 10 г/л натрия
нитропруссида Р. Фильтровальная бумага приобрета¬
ет синее окрашивание при контакте с пиперидином Р.
B. 0,1 г испытуемого образца прокаливают в ти¬
гле. К остатку прибавляют 5 мл кислоты азотной Р,
перемешивают, нагревают на водяной бане в течение
1 мин и фильтруют. Полученный фильтрат дает реак¬
цию (Ь) на фосфаты (2.3.1).
C. Испытуемый образец дает реакцию (Ь) на
кальций (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,5 г испытуемого образца раство¬
ряют при комнатной температуре в воде, свободной
от углерода диоксида, Р, приготовленной из воды
дистиллированной Р, и доводят до объема 150 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон III.
Кислотность или щелочность. К 100 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфталеи¬
на Р. При прибавлении не более 1,5 мл 0,1 М раст¬
вора кислоты хлористоводородной или 0,5 мл 0,1 М
раствора натрия гидроксида окраска раствора
должна измениться.
Лимонная кислота. 5,0 г испытуемого образца
встряхивают с 20 мл воды, свободной от углерода
диоксида, Р и фильтруют. К полученному фильтрату
прибавляют 0,15 мл кислоты серной Р и снова филь¬
труют. К полученному фильтрату прибавляют 5 мл
раствора ртути сульфата Р, нагревают до кипения,
прибавляют 0,5 мл раствора 3,2 г/л калия перманга¬
ната Р и снова нагревают до кипения. Не должен об¬
разовываться осадок.
Глицерин и вещества, растворимые в 96 %
спирте. Не более 0,5 %. 1,000 г испытуемого образца
встряхивают с 25 мл 96 % спирта Р в течение 1 мин
и фильтруют. Полученный фильтрат выпаривают на
водяной бане и остаток высушивают при температу¬
ре 70 °С в течение 1 ч. Масса полученного остатка не
должна превышать 5 мг.
63’. Зак. 1060.
492
Гэсударственная фармакопея Республики Беларусь
Хлориды (2.4.4). Не более 0,0500 % (500 ррт).
0,1 г испытуемого образца растворяют в смеси из
2 мл кислоты уксусной Р и 8 мл воды Р и доводят во¬
дой Р до объема 15 мл.
Фосфаты (2.4.11). Не более 0,0400 % (400 ррт).
2,5 мл раствора 5 доводят водой Р до объема 100 мл.
Сульфаты (2.4.13). Не более 0,1 %. Определе¬
ние проводят с использованием раствора 3.
Мышьяк (2.4.2, метод А). Не более 0,0003 %
(3 ррт). 0,33 г испытуемого образца растворяют в
воде Р и доводят до объема 25 мл этим же раство¬
рителем.
Железо (2.4.9). Не более 0,0050 % (50 ррт).
Определение проводят из 0,20 г испытуемого образца.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 2,0 г испытуемого образца раст¬
воряют в буферном растворе рН 3,5 Р и доводят во¬
дой Р до объема 20 мл. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 12,0 %. 1,000 г испытуемого образца сушат при
температуре 150 °С в течение 4 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в воде Р
и проводят комплексометрическое определение каль¬
ция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 4,008 мг Са.
07/2016:0172
КАЛЬЦИЯ ГЛЮКОНАТ
Са/с/7 д1исопаз
САТСШМ вШСОЫАТЕ
[18016-24-5]
ОПРЕДЕЛЕНИЕ
Кальция бис[(2Р,35,4/?,5Р)-2,3,4,5,6-пентагидро-
ксигексаноат] моногидрат (кальция ди(О-глюконат) мо¬
ногидрат).
Содержание: не менее 98,5 % и не более 102,0 %
С12Н22Са014Н20.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
гранулированный порошок.
Умеренно растворим в воде, легко растворим в
кипящей воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 1 мл воды Р, при необходимости
нагревая до температуры 60 °С.
Раствор сравнения. 20 мг ФСО кальция глюко¬
ната растворяют в 1 мл воды Р, при необходимости
нагревая до температуры 60 °С.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетат Р — вода Р — 96%
спирт Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 100 °С в течение
20 мин и охлаждают.
Проявление: пластинку опрыскивают раствором,
содержащим 10 г/л церия сульфата Р и 25 г/л аммо¬
ния молибдата Р в кислоте серной разведенной Р и
нагревают при температуре 105 °С в течение 10 мин.
Результаты: через 5 мин на хроматограмме ис¬
пытуемого раствора обнаруживается пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
B. Раствор 3, приготовленный как указано в разделе
«Испытания», дает реакции (а) и (Ь) на кальций (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р, нагретой до температуры 60 °С, и до¬
водят до объема 50 мл этим же растворителем.
Цветность (2.2.2, метод II). Окраска раствора 3
при температуре 60 °С должна быть не интенсивнее
эталона У(Ж)6.
Прозрачность (2.2.1). Раствор 3 после охлаждения
по степени мутности не должен превышать эталон II.
Органические примеси и борная кислота. 0,5 г
испытуемого образца помещают в фарфоровую чаш¬
ку, которую предварительно ополоснули кислотой
серной Р и поместили в ледяную баню. Прибавляют
2 мл охлажденной кислоты серной Р и перемеши¬
вают. Не должно появляться желтое или коричневое
окрашивание. Прибавляют 1 мл раствора хромотро¬
па IIВ Р. Появляется фиолетовое окрашивание, кото¬
рое не должно переходить в темно-синее. Окрашива¬
ние полученного раствора должно быть не интенсив¬
нее окраски смеси из 1 мл раствора хромотропа и
В Р и 2 мл охлажденной кислоты серной Р.
Сахароза и восстанавливающие сахара. 0,5 '
испытуемого образца растворяют в смеси из 2 мг
кислоты хлористоводродной Р1 и 10 мл воды Р. Ки¬
пятят в течение 5 мин, охлаждают, прибавляют 10 мг
раствора натрия карбоната Р и выдерживают. По¬
лученный раствор доводят водой Р до объема 25 мг
и фильтруют. К 5 мл фильтрата прибавляют 2 м.-
раствора медно-тартратного Р и кипятят в течение
1 мин. Выдерживают в течение 2 мин. Не должен об¬
разовываться осадок красного цвета.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт
12,5 мл раствора 3 доводят водой Р до объема 15 мг-
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт
10,0 г испытуемого образца растворяют при нагрева¬
нии в смеси из 10 мл кислоты уксусной Р и 90 м.-
водь/ дистиллированной Р.
Кальция глюконат безводный
493
Магний и щелочные металлы. Не более 0,4 %.
1,00 г испытуемого образца растворяют в 100 мл ки¬
пящей воды Р, прибавляют 10 мл раствора аммония
хлорида Р, 1 мл раствора аммиака Р и, по каплям,
50 мл горячего раствора аммония оксалата Р. По¬
лученный раствор выдерживают в течение 4 ч, дово¬
дят водой Р до объема 200 мл и фильтруют. 100 мл
полученного фильтрата выпаривают досуха и прока¬
ливают. Масса полученного остатка не должна пре¬
вышать 2 мг.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Испы¬
туемый образец постепенно и осторожно нагревают
до тех пор, пока он почти полностью не превратится
в белую массу, и затем прокаливают. Эталон готовят
с использованием 2 мл эталонного раствора свинца
(10 ррт РЬ) Р.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,8000 г испытуемого образца растворяют в
20 мл горячей воды Р, охлаждают и доводят водой Р
до объема 300 мл и проводят комплексометрическое
определение кальция (2.5.11).
1мл 0,1 М раствора натрия эдетата соответ¬
ствует 44,84 мг С^Н^СаО^ НзО.
07/2016:2364
КАЛЬЦИЯ ГЛЮКОНАТ БЕЗВОДНЫЙ
Са/с/7 д!исопа$ апЬуёпсиз
СА1.СШМ вШСОЫАТЕ, АЫНУОКСШЗ
С12НиСа014 М.м. 430,4
[299-28-5]
ОПРЕДЕЛЕНИЕ
Кальция О-глюконат безводный.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
гранулированный порошок.
Умеренно растворим в воде, легко растворим в
кипящей воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 1 мл воды Р, при необходимости
нагревая в водяной бане при температуре 60 °С.
Раствор сравнения. 20 мг ФСО кальция глюко¬
ната растворяют в 1 мл воды Р, при необходимости
нагревая в водяной бане при температуре 60 °С.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: аммиака раствор концен¬
трированный Р — этилацетат Р — вода Р — 96%
спирт Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 100 °С в течение
20 мин и охлаждают.
Проявление: пластинку опрыскивают раствором,
содержащим 25 г/л аммония молибдата Р и 10 г/л
церия сульфата Р в кислоте серной разведенной Р,
и нагревают при температуре от 100 °С до 105 °С в
течение 10 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
B. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции (а) и (Ь) на кальций
(2.3.1).
C. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде Р, нагретой до температуры 60 °С, и до¬
водят до объема 50 мл этим же растворителем.
Цветность (2.2.2, метод II). Окраска раствора 3
при температуре 60 °С должна быть не интенсивнее
эталона У(Ж)6.
Прозрачность (2.2.1). Раствор 3 после охлаждения
по степени мутности не должен превышать эталон II.
Органические примеси и борная кислота. 0,5 г
испытуемого образца помещают в фарфоровую чаш¬
ку, которую предварительно ополоснули кислотой
серной Р и поместили в ледяную баню. Прибавляют
2 мл охлажденной кислоты серной Р и перемеши¬
вают. Не должно появляться желтое или коричневое
окрашивание. Прибавляют 1 мл раствора хромотро¬
па IIВ Р. Появляется фиолетовое окрашивание, кото¬
рое не должно переходить в темно-синее. Сравнива¬
ют окраску полученного раствора с окраской смеси из
1 мл раствора хромотропа II В Р и 2 мл охлажден¬
ной кислоты серной Р.
Сахароза и восстанавливающие сахара. 0,5 г
испытуемого образца растворяют в смеси из 2 мл
кислоты хлористоводродной Р1 и 10 мл воды Р. Ки¬
пятят в течение 5 мин, охлаждают, прибавляют 10 мл
раствора натрия карбоната Р и выдерживают в те¬
чение 10 мин. Полученный раствор доводят водой Р
до объема 25 мл и фильтруют. К 5 мл фильтрата при¬
бавляют 2 мл раствора медно-тартратного Р и ки¬
пятят в течение 1 мин. Выдерживают в течение 2 мин.
Не должен образовываться осадок красного цвета.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
12,5 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
10,0 г испытуемого образца растворяют при нагрева¬
нии в смеси из 10 мл кислоты уксусной Р и 90 мл
воды дистиллированной Р.
Магний и щелочные металлы. Не более 0,4 %
(в пересчете на МдО). 1,00 г испытуемого образца
растворяют в 100 мл кипящей воды Р, прибавляют
64. Зак. 1060.
494
Гэсударственная фармакопея Республики Беларусь
10 мл раствора аммония хлорида Р, 1 мл раствора
аммиака Р и, по каплям, 50 мл горячего раствора ам¬
мония оксалата Р. Полученный раствор выдержива¬
ют в течение 4 ч, доводят водой Р до объема 200 мл и
фильтруют. 100 мл полученного фильтрата выпарива¬
ют досуха и прокаливают. Масса полученного остатка
не должна превышать 2 мг. ,
Тяжелые металлы {2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Испы¬
туемый образец постепенно и осторожно нагревают
до тех пор, пока он почти полностью не превратится
в белую массу, и затем прокаливают. Эталон готовят
с использованием 2 мл эталонного раствора свинца
(ЮрртРЬ)Р.
Потеря в массе при высушивании (2.2.32). Не
более 2,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 16 ч.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г {2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г {2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 20 мл
горячей воды Р, охлаждают, доводят водой Р до объ¬
ема 300 мл и проводят комплексометрическое опре¬
деление кальция {2.5.11).
1мл 0,1 М раствора натрия эдетата соответ¬
ствует 43,04 мг С12Н22Са014.
07/2016:0979
КАЛЬЦИЯ ГЛЮКОНАТ
ДЛЯ ИНЪЕКЦИЙ
Са/с/7 д/исопаз аХ 1тесХаЫ1е
САТСШМ вШСОИАТЕ РОК ШЕСТЮЫ
[18016-24-5]
ОПРЕДЕЛЕНИЕ
Кальция бис[(2Р,35,4Р,5Р)-2,3,4,5,6-пентагидро-
ксигексаноат] моногидрат (кальция ди(О-глюконат) мо¬
ногидрат).
Содержание: не менее 99,0 % и не более 101,0 %
С12Н22Са014Н20.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
гранулированный порошок.
Умеренно растворим в воде, легко растворим в
кипящей воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 1 мл воды Р, при необходимости
нагревая до температуры 60 °С.
Раствор сравнения. 20 мг ФСО кальция глюко¬
ната растворяют в 1 мл воды Р, при необходимости
нагревая до температуры 60 °С.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р {{2—10) мкм)].
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетат Р — вода Р — 96%
спирт Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 100 °С в течение
20 мин и охлаждают.
Проявление: пластинку опрыскивают раствором,
содержащим 10 г/л церия сульфата Р и 25 г/л аммо¬
ния молибдата Р в кислоте серной разведенной Р,
и нагревают при температуре 105 °С в течение около
10 мин.
Результаты: через 5 мин на хроматограмме ис¬
пытуемого раствора обнаруживается пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
В. Около 20 мг испытуемого образца дают реак¬
цию (Ь) на кальций {2.3.1).
ИСПЫТАНИЯ
Раствор 8. К 10,0 г испытуемого образца при¬
бавляют 90 мл кипящей воды дистиллированной Р
и кипятят при перемешивании в течение не более
10 с до полного растворения, затем доводят до объ¬
ема 100,0 мл этим же растворителем.
Цветность {2.2.2, метод II). Окраска раствора 5
при температуре 60 °С должна быть не интенсивнее
эталона В(К)7.
Прозрачность {2.2.1). Раствор 5 после охлаж¬
дения до температуры 20 °С по степени мутности не
должен превышать эталон II.
рН (2.2.3). От 6,4 до 8,3. 1,0 г испытуемого об¬
разца растворяют при нагревании на водяной бане в
20 мл воды, свободной от углерода диоксида, Р.
Органические примеси и борная кислота. 0,5 г
испытуемого образца помещают в фарфоровую чаш¬
ку, предварительно сполоснутую кислотой серной Р
и помещенную в ледяную баню. Прибавляют 2 мл
охлажденной кислоты серной Р и перемешивают.
Не должно появляться желтое или коричневое окра¬
шивание. Прибавляют 1 мл раствора хромотропа II
ВР. Появляется фиолетовое окрашивание, которое
не должно переходить в темно-синее. Окраска полу¬
ченного раствора должна быть не интенсивнее окра¬
ски смеси из 1 мл раствора хромотропа IIВ Р и 2 мл
охлажденной кислоты серной Р.
Оксалаты. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде для хроматографии Р и до¬
водят до объема 100,0 мл этим же растворителем.
Раствор сравнения. 1,00 г испытуемого образца
растворяют в воде для хроматографии Р, прибавля¬
ют 0,5 мл раствора 0,152 г/л натрия оксалата Р в
воде для хроматографии Р и доводят водой для хро¬
матографии Р до объема 100,0 мл.
Условия хроматографирования:
- предколонка длиной 30 мм и внутренним диа¬
метром 4 мм, заполненная подходящей смолой ани¬
онообменной сильноосновной с размером частиц
(30—50) мкм;
Кальция карбонат
495
- колонки 1 и 2 длиной 0,25 м и внутренним диа¬
метром 4 мм, заполненные подходящей смолой ани¬
онообменной сильноосновной с размером частиц
(30—50) мкм;
- анион-подавляющая колонка: расположена
между предколонкой и аналитическими колонками и
снабжена микромембраной, которая отделяет под¬
вижную фазу от регенерирующего раствора, двигаю¬
щегося противотоком подвижной фазе;
-подвижная фаза: 0,212 г натрия карбоната
безводного Р и 63 мг натрия гидрокарбоната Р
растворяют в воде для хроматографии Р и доводят
до объема 1000,0 мл этим же растворителем;
- скорость подвижной фазы: 2 мл/мин;
- регенерирующий раствор: раствор 1,23 г/л
кислоты серной Р в воде для хроматографии Р;
- скорость регенерирующего раствора: 4 мл/мин;
- кондуктометрический детектор;
- объем вводимой пробы: по 50 мкл каждого
раствора трижды.
Пригодность хроматографической системы:
раствор сравнения:
- относительное стандартное отклонение: не
более 2,0 % для пика оксалата при 5 повторных вво¬
дах пробы.
Содержание оксалатов в частях на миллион
(ррт) рассчитывают по формуле:
5Т -50
5*-8т’
где:
5Т — площадь пика оксалата на хроматограмме
испытуемого раствора;
5^ — площадь пика оксалата на хроматограмме
раствора сравнения.
Предельное содержание примесей:
- оксалаты: не более 0,0100 % (100 ррт).
Сахароза и восстанавливающие сахара. 0,5 г
испытуемого образца растворяют в смеси из 2 мл
кислоты хлористоводродной Р1 и 10 мл воды Р. Ки¬
пятят в течение 5 мин, охлаждают, прибавляют 10 мл
раствора натрия карбоната Р и выдерживают в те¬
чение 10 мин. Полученный раствор доводят водой Р
до объема 25 мл и фильтруют. К 5 мл фильтрата при¬
бавляют 2 мл раствора медно-тартратного Р и ки¬
пятят в течение 1 мин. Выдерживают в течение 2 мин.
Не должен образовываться осадок красного цвета,
л Хлориды (2.4.4). Не более 0,0050 % (50 ррт). К
10 мл предварительно профильтрованного раствора
3 прибавляют 5 мл воды Р.
Фосфаты (2.4.11). Не более 0,0100 % (100 ррт).
1 мл раствора 5 доводят водой Р до объема 100 мл.
Сульфаты (2.4.13). Не более 0,0050 % (50 ррт).
Предварительно профильтрованный раствор 3 дол¬
жен выдерживать испытание на сульфаты. Эталон
готовят с использованием смеси из 7,5 мл эталонно¬
го раствора сульфата (10 ррт 80^ Р и 7,5 мл воды
дистиллированной Р.
Железо. Не более 0,0005 % (5 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 2,0 г испытуемого об¬
разца помещают в политетрафторэтиленовый лабо¬
раторный стакан вместимостью 100 мл, прибавляют
5 мл кислоты азотной Р и кипятят почти досуха. К
остатку прибавляют 1 мл раствора водорода перокси¬
да концентрированного Р и выпаривают почти досу¬
ха. Обработку водорода пероксидом проводят до по¬
лучения прозрачного раствора. Полученный раствор
переносят в мерную колбу вместимостью 25,0 мл с
помощью 2 мл кислоты азотной Р и доводят кисло¬
той хлористоводородной разведенной Р до объема
25.0 мл.
Компенсационный раствор. Готовят аналогично
испытуемому раствору с использованием 0,65 г каль¬
ция хлорида Р1 вместо испытуемого образца.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора железа
(20 ррт Ре) Р кислотой хлористоводородной разве¬
денной Р.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Коррекция фона: дейтериевая лампа.
Магний и щелочные металлы. Не более 0,4 %.
К 0,50 г испытуемого образца прибавляют смесь из
1.0 мл кислоты уксусной разведенной Р и 10,0 мл
воды Р и быстро кипятят, встряхивая, до полно¬
го растворения. К кипящему раствору прибавляют
5.0 мл раствора аммония оксалата Р и выдержива¬
ют в течение не менее 6 ч. Фильтруют через стеклян¬
ный фильтр (ПОР 1,6) (2.1.2) в фарфоровый тигель,
осторожно выпаривают досуха и прокаливают. Масса
полученного остатка не должна превышать 2 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Бактериальные эндотоксины (2.6.14). Менее
167МЕ/Г.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 20 мл
горячей воды Р, охлаждают и доводят водой Р до объ¬
ема 300 мл и проводят комплексометрическое опре¬
деление кальция (2.5.11). Используют 50 мг индика¬
торной смеси кальконкарбоновой кислоты Р.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 44,84 мг С12Н22Са014 Н20.
07/2016:0014
КАЛЬЦИЯ КАРБОНАТ
Са/с/7 сагЬопаз
СА1СШМ САКВОЫАТЕ
СаС03 М.м. 100,1
[471-34-1]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакцию (а) на кар¬
бонаты (2.3.1).
64* Зак. 1060.
496
Государственная фармакопея Республики Беларусь
В. 0,2 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», дают реакции (а) и (Ь) на
кальций (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в 80 мл кислоты уксусной разведенной Р. По¬
сле прекращения выделения пузырьков газа раствор
кипятят в течение 2 мин, охлаждают, доводят объем
кислотой уксусной разведенной Р до объема 100 мл
и, если необходимо, фильтруют через стеклянный
фильтр (2.1.2).
Вещества, нерастворимые в кислоте уксус¬
ной. Не более 0,2 %. Осадок, полученный при приго¬
товлении раствора 3, промывают четырьмя порциями
по 5 мл горячей воды Р и сушат при температуре от
100 °С до 105 °С в течение 1 ч. Масса сухого остатка
не должна превышать 10 мг.
Хлориды (2.4.4). Не более 0,0330 % (330 ррт).
3 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,25%. 1,2 мл
раствора 3 доводят водой дистиллированной Р до
объема 15 мл. Полученный раствор должен выдержи¬
вать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0004 %
(4 ррт). 5 мл раствора 3 должны выдерживать испы¬
тание на мышьяк.
Барий. К 10 мл раствора 3 прибавляют 10 мл
раствора кальция сульфата Р. Не менее чем через
15 мин опалесценция полученного раствора не долж¬
на превышать опалесценцию смеси из 10 мл раст¬
вора 3 и 10 мл воды дистиллированной Р.
Железо (2.4.9). Не более 0,0200 % (200 ррт).
50 мг испытуемого образца растворяют в 5 мл кисло¬
ты хлористоводородной разведенной Р и доводят
водой Р до объема 10 мл. Полученный раствор дол¬
жен выдерживать испытание на железо.
Магний и щелочные металлы. Не более 1,5 %.
1,0 г испытуемого образца растворяют в 12 мл кисло¬
ты хлористоводородной разведенной Р, кипятят в
течение 2 мин и прибавляют 20 мл воды Р, 1 г аммо¬
ния хлорида Р и 0,1 мл раствора метилового крас¬
ного Р. К полученному раствору прибавляют раствор
аммиака разведенный Р1 до перехода окраски инди¬
катора и еще избыток в количестве 2 мл, нагревают
до кипения и прибавляют 50 мл горячего раствора
аммония оксалата Р. Выдерживают в течение 4 ч, до¬
водят водой Р до объема 100 мл и фильтруют через
подходящий фильтр. К 50 мл фильтрата прибавляют
0,25 мл кислоты серной Р, выпаривают досуха на во¬
дяной бане и сжигают до постоянной массы при тем¬
пературе (600±50) °С. Масса сухого остатка не долж¬
на превышать 7,5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 2,0 %. 1,000 г испытуемого образца сушат при
температуре (200±10) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в смеси
из 3 мл кислоты хлористоводородной разведенной Р
и 20 мл воды Р, кипятят в течение 2 мин, охлаждают и
доводят водой Р до объема 50 мл. Проводят комплек¬
сометрическое определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 10,01 мгСаС03.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для кальция карбоната, используемого в каче¬
стве наполнителя при изготовлении таблеток ь
капсул.
Распределение частиц по размерам (2.9.3"
или 2.9.38).
Текучесть порошков (2.9.36).
07/2016:2118
КАЛЬЦИЯ ЛАКТАТ БЕЗВОДНЫЙ
Са/с// 1ас1аз апЬудпсиз
СА1.СШМ 1.АСТАТЕ, АЫНУйКСШЗ
Са2+
Н3С
Н ОН
и энантиомер
2
С6Н10СаОб М.м. 218 2
[814-80-2]
ОПРЕДЕЛЕНИЕ
Кальция бис(2-гидроксипропаноат) или сме:;
кальция (2Р)-, (25)- и (2Р5)-2-гидроксипропаноатоБ
Содержание: не менее 98,0 % и не более 102 С :
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический
гранулированный порошок.
Растворим в воде, легко растворим в кипя_~
воде, очень мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец выдерживает испьга-
«Потеря в массе при высушивании», как указа-: *
разделе «Испытания».
Кальция лактат моногидрат
497
B. Испытуемый образец дает реакцию на лакта¬
ты (2.3.7).
C. Испытуемый образец дает реакцию (Ь) на
кальций (2.3.7).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют при нагревании в воде, свободной от углерода
диоксида, Р, приготовленной из воды дистиллирован¬
ной Р, охлаждают и доводят до объема 100 мл этим
же растворителем.
Прозрачность (2.2.7). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфтале¬
ина Р и 0,5 мл 0,01 М раствора кислоты хлористо¬
водородной. Раствор должен быть бесцветным. При
прибавлении не более 2,0 мл 0,01 М раствора на¬
трия гидроксида должно появиться розовое окраши¬
вание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.73). Не более 0,0400 % (400 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Барий. К 10 мл раствора 3 прибавляют 1мл
раствора кальция сульфата Р и выдерживают в те¬
чение 15 мин. Опалесценция полученного раствора
не должна превышать опалесценцию смеси из 1 мл
воды дистиллированной Р и 10 мл раствора 3.
Железо (2.4.9). Не более 0,0050 % (50 ррт). 4 мл
раствора 5 доводят водой Р до объема 10 мл.
Соли магния и щелочных металлов. Не более
1 %. К 20 мл раствора 3 прибавляют 20 мл воды Р, 2 г
аммония хлорида Р и 2 мл раствора аммиака разве¬
денного Р1. Нагревают до кипения и быстро прибав¬
ляют 40 мл горячего раствора аммония оксалата Р.
Выдерживают в течение 4 ч, доводят водой Р до объ¬
ема 100,0 мл и фильтруют. К 50,0 мл полученного
фильтрата прибавляют 0,5 мл кислоты серной Р, вы¬
паривают досуха и сжигают остаток при температуре
(600±50) °С до постоянной массы. Масса полученного
остатка не должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 3,0 %. 0,500 г испытуемого образца сушат при
температуре 125 °С.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в
воде Р, доводят до объема 300 мл этим же раствори¬
телем и проводят комплексометрическое определе¬
ние кальция (2.5.11).
1 мп 0,1 М раствора натрия эдетата соответ¬
ствует 21,82 мг С6Н10СаО6.
07/2016:2117
КАЛЬЦИЯ ЛАКТАТ МОНОГИДРАТ
Са/с/7 /ас?а$ топоЬуёпсиз
СА1.СШМ 1.АСТАТЕ МОЫОНУОКАТЕ
Са2+
Н3С
Н ОН
и энантиомер
• Н20
С6Н10СаОе ■ Н20 М.м. 236,0
[41372-22-9]
ОПРЕДЕЛЕНИЕ
Кальция бис(2-гидроксипропаноат) моно¬
гидрат или смесь кальция (2Р)-, (28)- и (2Р8)-2-
гидроксипропаноатов моногидратов.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
гранулированный порошок.
Растворим в воде, легко растворим в кипящей
воде, очень мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
B. Испытуемый образец дает реакцию на лакта¬
ты (2.3.7).
C. Испытуемый образец дает реакцию (Ь) на
кальций (2.3.7).
ИСПЫТАНИЯ
Раствор 5. 5,4 г испытуемого образца (соответ¬
ствует 5,0 г сухого вещества) растворяют при нагре¬
вании в воде, свободной от углерода диоксида, Р,
приготовленной из воды дистиллированной Р, охлаж¬
дают и доводят до объема 100 мл этим же раствори¬
телем.
Прозрачность (2.2.7). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность или щелочность. К10 мл раствора
3 прибавляют 0,1 мл раствора фенолфталеина Р и
0,5 мл 0,01 М раствора кислоты хлористоводород¬
ной. Раствор должен быть бесцветным. При прибавле¬
нии не более 2,0 мл 0,01 М раствора натрия гидрок¬
сида должно появиться розовое окрашивание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.73). Не более 0,0400 % (400 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Барий. К 10 мл раствора 3 прибавляют 1мл
раствора кальция сульфата Р и выдерживают в те¬
чение 15 мин. Опалесценция полученного раствора
не должна превышать опалесценцию смеси из 1 мл
воды дистиллированной Р и 10 мл раствора 3.
Железо (2.4.9). Не более 0,0050 % (50 ррт). 4 мл
раствора 3 доводят водой Р до объема 10 мл.
Соли магния и щелочных металлов. Не более
1 %. К 20 мл раствора 5 прибавляют 20 мл воды Р, 2 г
65. Зак. 1060.
498
Гэсударственная фармакопея Республики Беларусь
аммония хлорида Р и 2 мл раствора аммиака разве¬
денного Р1. Нагревают до кипения и быстро прибав¬
ляют 40 мл горячего раствора аммония оксалата Р.
Выдерживают в течение 4 ч, доводят водой Р до объ¬
ема 100,0 мл и фильтруют. К 50,0 мл полученного
фильтрата прибавляют-0,5 мл кислоты серной Р, вы¬
паривают досуха и сжигают остаток при температуре
(600±50) °С до постоянной массы. Масса полученного
остатка не должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). Навеску испытуемого образца, со¬
ответствующую 2,0 г сухого вещества, растворяют в
воде Р и доводят до объема 20 мл этим же раство¬
рителем. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 5,0 % и не более 8,0 %. 0,500 г испытуемого об¬
разца сушат при температуре 125 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Навеску испытуемого образца, соответствующую
0,200 г сухого вещества, растворяют в воде Р, доводят
до объема 300 мл этим же растворителем и проводят
комплексометрическое определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 21,82 мг С6Н10СаО6.
07/2016:0468
КАЛЬЦИЯ ЛАКТАТ ПЕНТАГИДРАТ
Са1сН 1ас(аз реМаЬус1пси8
СА1.СШМ 1-АСТАТЕ РЕЫТАНУОКАТЕ
Са'
,2+
НзС^^^СОг
А
н он
и энантиомер • 5Н20
С6Н10СаО6 ■ 5Н20 М.м. 308,3
[5743-47-5]
ОПРЕДЕЛЕНИЕ
Кальция бис(2-гидроксипропаноат) пентагидрат
или смесь кальция (2/?)-, (25)- и (2#5)-2-гидрок-
сипропаноатов пентагидратов.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический или
гранулированный порошок. Слегка выветривается на
воздухе.
Растворим в воде, легко растворим в кипящей
воде, очень мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Потеря в масде при высушивании», как указано в
разделе «Испытания».
B. Испытуемый образец дает реакцию на лакта¬
ты (2.3.1).
С. Испытуемый образец дает реакцию (Ь) на
кальций (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 771 г испытуемого образца (соответ¬
ствует 5,0 г сухого вещества) растворяют при нагре¬
вании в воде; своббдной от углерода диоксида, Р.
приготовленной из воды дистиллированной Р, охлаж¬
дают и доводят до объема 100 мл этим же раствори¬
телем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)б.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфтале¬
ина Р и 0,5 мл 0,01 М раствора кислоты хлористо¬
водородной. Раствор должен быть бесцветным. При
прибавлении не более 2,0 мл 0,01 М раствора на¬
трия гидроксида должно появиться розовое окраши¬
вание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт)
5 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0400 % (400 ррт)
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Барий. К 10 мл раствора 3 прибавляют 1 мг
раствора кальция сульфата Р и выдерживают в те¬
чение 15 мин. Опалесценция полученного раствора
не должна превышать опалесценцию смеси из 1 мг
воды дистиллированной Р и 10 мл раствора 3.
Железо (2.4.9). Не более 0,0050 % (50 ррт). 4 мг
раствора 3 доводят водой Р до объема 10 мл.
Соли магния и щелочных металлов. Не более
1 %. К 20 мл раствора 3 прибавляют 20 мл воды Р, 2 -
аммония хлорида Р и 2 мл раствора аммиака разве¬
денного Р1. Нагревают до кипения и быстро прибав¬
ляют 40 мл горячего раствора аммония оксалата Р
Выдерживают в течение 4 ч, доводят водой Р до объ¬
ема 100,0 мл и фильтруют. К 50,0 мл полученнсг:
фильтрата прибавляют 0,5 мл кислоты серной Р, вь-
паривают досуха и сжигают остаток при температуре
(600±50) °С до постоянной массы. Масса получение:
остатка не должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). Навеску испытуемого образца, со¬
ответствующую 2,0 г сухого вещества, растворяют =
воде Р и доводят до объема 20 мл этим же раство¬
рителем. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора евин~ г
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 22,0 % и не более 27,0 %. 0,500 г испытуемо-:
образца сушат при температуре 125 °С.
#Микробиологическая чистота (2.6.12, 2.6.1:
Испытуемый образец должен выдерживать требе не¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Навеску испытуемого образца, соответствую^.. *:
0,200 г сухого вещества, растворяют в воде Р, дово¬
дят до объема 300 мл этим же растворителем и про¬
водят комплексометрическое определение каль«о=
(2.5.11).
1 мл 0,1 М раствора натрия эдетата соотее--
ствует 21,82 мг С6Н10СаО6.
Кальция стеарат
499
07/2016:0469
КАЛЬЦИЯ ЛАКТАТ ТРИГИДРАТ
Са/с// /ас/аа Шудпсиз
СА1.СШМ 1-АСТАТЕ ТШНУОКАТЕ
Са2+
Н3С
Н ОН
и энантиомер
• ЗН20
С6Н10СаОв ■ ЗН20 М.м. 272,3
[41372-22-9]
ОПРЕДЕЛЕНИЕ
Кальция бис(2-гидроксипропаноат) тригидрат
или смесь кальция (2/?)-, (25)- и (2Я5)-2-гидрок-
сипропаноатов тригидратов.
Содержание: не менее 98,0 % и не более 102,0 %
в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
гранулированный порошок.
Растворим в воде, легко растворим в кипящей
воде, очень мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
B. Испытуемый образец дает реакцию на лакта¬
ты (2.3.1).
C. Испытуемый образец дает реакцию (Ь) на
кальций (2.3.1).
аммония хлорида Р и 2 мл раствора аммиака разве¬
денного Р1. Нагревают до кипения и быстро прибав¬
ляют 40 мл горячего раствора аммония оксалата Р.
Выдерживают в течение 4 ч, доводят водой Р до объ¬
ема 100,0 мл и фильтруют. К 50,0 мл полученного
фильтрата прибавляют 0,5 мл кислоты серной Р, вы¬
паривают досуха и сжигают остаток при температуре
(600±50) °С до постоянной массы. Масса полученного
остатка не должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). Навеску испытуемого образца, со¬
ответствующую 2,0 г сухого вещества, растворяют в
воде Р и доводят до объема 20 мл этим же раство¬
рителем. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 15,0 % и не более 20,0 %. 0,500 г испытуемого
образца сушат при температуре 125 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Навеску испытуемого образца, соответствующую
0,200 г сухого вещества, растворяют в воде Р, дово¬
дят до объема 300 мл этим же растворителем и про¬
водят комплексометрическое определение кальция
(2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 21,82 мг С6Н10СаО6.
07/2016:0882
ИСПЫТАНИЯ
Раствор 5. 6,2 г испытуемого образца (соответ¬
ствует 5,0 г сухого вещества) растворяют при нагрева¬
нии в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, охлаждают
и доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)б.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфтале¬
ина Р и 0,5 мл 0,01 М раствора кислоты хлористо¬
водородной. Раствор должен быть бесцветным. При
прибавлении не более 2,0 мл 0,01 М раствора на¬
трия гидроксида должно появиться розовое окраши¬
вание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0400 % (400 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Барий. К 10 мл раствора 3 прибавляют 1мл
раствора кальция сульфата Р и выдерживают в те¬
чение 15 мин. Опалесценция полученного раствора
не должна превышать опалесценцию смеси из 1 мл
воды дистиллированной Р и 10 мл раствора 3.
Железо (2.4.9). Не более 0,0050 % (50 ррт). 4 мл
раствора 3 доводят водой Р до объема 10 мл.
Соли магния и щелочных металлов. Не более
1 %. К 20 мл раствора 3 прибавляют 20 мл воды Р, 2 г
КАЛЬЦИЯ СТЕАРАТ
Са/сн 5/еагаз
САШвМ ЗТЕАЯАТЕ
[1592-23-0]
ОПРЕДЕЛЕНИЕ
Смесь солей кальция различных жирных кислот,
содержащих главным образом стеариновую (октаде¬
кановую) [(С17Н35СОО)2Са; М.м. 607] и пальмитиновую
(гексадекановую) кислоты [(С15Н31СОО)2Са; М.м. 550,9]
с минимальным содержанием других жирных кислот.
Содержание:
- кальций: от 6,4 % до 7,4 % (А.м. 40,08) (в пере¬
счете на сухое вещество);
- стеариновая кислота во фракции жирных кис¬
лот: не менее 40,0 %;
- сумма стеариновой и пальмитиновой кислот:
не менее 90,0 %.
ОПИСАНИЕ(СВОЙСТВА)
Мелкий, белый или почти белый кристалличе¬
ский порошок.
Практически нерастворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С, О.
Вторая идентификация: А, В, О.
А. Остаток, полученный при приготовлении раст¬
вора 3, как указано в разделе «Испытания», имеет
температуру затвердевания (2.2.18) не ниже 53 °С.
65*. Зак. 1060.
500
Гэсударственная фармакопея Республики Беларусь
B. Кислотное число (2.5.1). От 195 до 210. 0,200 г
остатка, полученного при приготовлении раствора 5,
растворяют в 25 мл предписанной смеси растворите¬
лей.
C. Просматривают хроматограммы, полученные
при определении состава жирных кислот.
Результаты, основные пики на хроматограмме
испытуемого раствора по времени удерживания соот¬
ветствуют основным пикам на хроматограмме раст¬
вора сравнения.
й. 5 мл раствора 5 нейтрализуют раствором на¬
трия гидроксида концентрированным Р по красной
лакмусовой бумаге Р. Полученный раствор дает ре¬
акцию (Ь) на кальций (2.3.1).
ИСПЫТАНИЯ
Раствор 3. К 5,0 г испытуемого образца при¬
бавляют 50 мл эфира, свободного от перокси¬
дов, Р, 20 мл кислоты азотной разведенной Р и
20 мл воды дистиллированной Р и кипятят с об¬
ратным холодильником до полного растворения
испытуемого образца. Раствор охлаждают, с помо¬
щью делительной воронки отделяют водный слой.
Эфирный слой встряхивают дважды с водой дис¬
тиллированной Р порциями по 5 мл. Объединяют
водные слои, промывают 15 мл эфира, свободного
от пероксидов, Р и доводят водой дистиллирован¬
ной Р до объема 50 мл (раствор 5). Эфирный слой
выпаривают досуха и сушат остаток при темпера¬
туре от 100 °С до 105 °С. Остаток используют для
проведения подлинности А и В.
Кислотность или щелочность. К 1,0 г испы¬
туемого образца прибавляют 20 мл воды, свобод¬
ной от углерода диоксида, Р, кипятят в течение
1 мин при постоянном перемешивании, охлажда¬
ют и фильтруют. К 10 мл фильтрата прибавляют
0,05 мл раствора бромтимолового синего Р1.
При прибавлении не более 0,5 мл 0,01 Мраствора
кислоты хлористоводородной или 0,01 М раст¬
вора натрия гидроксида окраска раствора должна
измениться.
Хлориды (2.4.4). Не более 0,1 %. 0,5 мл раст¬
вора 5 доводят водой Р до объема 15 мл. Получен¬
ный раствор должен выдерживать испытание на
хлориды.
Сульфаты (2.4.13). Не более 0,3%. 0,5 мл
раствора 3 доводят водой дистиллированной Р до
объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Кадмий. Не более 0,0003 % (3 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 50,0 мг испытуемого
образца помещают в политетрафторэтиленовый
сосуд для минерализации, прибавляют 0,5 мл сме¬
си, состоящей из 1 объема кислоты хлористово¬
дородной Р и 5 объемов кислоты азотной, свобод¬
ной от свинца и кадмия, Р, выдерживают в течение
5 ч при температуре 170 °С и охлаждают. Получен¬
ный остаток растворяют в воде Р и доводят до объ¬
ема 5,0 мл этим же растворителем.
Растворы сравнения. Готовят разведением эта¬
лонного раствора кадмия (10 ррт Сд) Р с использо¬
ванием 1 % (об/об) раствора кислоты хлористоводо¬
родной Р.
Источник излучения: лампа с полым катодом
для определения кадмия.
Длина волны: 228,8 нм.
Гэнератор атомного пара: графитовая печь.
Свинец. Не более 0,0010 % (10 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Используют раствор,
приготовленный в испытании на кадмий.
Растворы сравнения. Готовят разведением эта¬
лонного раствора свинца (10 ррт РЬ) Р с использо¬
ванием воды Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 283,3 нм; в зависимости от прибо¬
ра может использоваться длина волны 217,0 нм.
Гэнератор атомного пара: графитовая печь.
Никель. Не более 0,0005 % (5 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Используют раствор,
приготовленный в испытании на кадмий.
Растворы сравнения. Готовят разведением эта¬
лонного раствора никеля (10 ррт Щ Р с использова¬
нием воды Р.
Источник излучения: лампа с полым катодом
для определения никеля.
Длина волны: 232,0 нм.
Гэнератор атомного пара: графитовая печь.
Потеря в массе при высушивании (2.2.32). Не
более 6,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа соН: отсутствие (2.6.13).
8а!топеИа: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Кальций. 0,500 г испытуемого образца поме¬
щают в коническую колбу вместимостью 250 мл,
прибавляют 50 мл смеси, состоящей из равных
объемов этанола безводного Р и бутанола Р,
5 мл раствора аммиака концентрированного Р,
Змл аммиачного буферного раствора рН 10,0 Р,
30.0 мл 0,1 М раствора натрия эдетата и 15 мг
индикаторной смеси протравного черного 11 Р
(*эриохрома черного Р*), и нагревают при темпе¬
ратуре от 45 °С до 50 °С до получения прозрачно¬
го раствора. Охлаждают. Титруют 0,1 М раствором
цинка сульфата до изменения окраски раствора от
синей до фиолетовой.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 4,008 мг Са.
Состав жирных кислот. Газовая хроматогра¬
фия (2.2.28). Определение проводят методом норма¬
лизации.
Испытуемый раствор. 0,10 г испытуемого об¬
разца помещают в колбу со шлифом и растворяю-
в 5 мл раствора бора фторида в метаноле Р.
Колбу присоединяют к обратному холодильнику
и кипятят в течение 10 мин. Затем в колбу через
холодильник прибавляют 4 мл гептана Р, кипя¬
тят с обратным холодильником в течение 10 ми-
и охлаждают. В колбу прибавляют 20 мл раст¬
вора натрия хлорида насыщенного Р, встряхива¬
ют и оставляют до разделения слоев. Около 2 мг
органического слоя отделяют и сушат с помощь-:
0,2 г натрия сульфата безводного Р. 1,0 мл поле¬
ченного раствора доводят гептаном Р до объема
10.0 мл.
Кальция фолинат
501
Раствор сравнения. Готовят точно так же, как и
испытуемый раствор, используя вместо испытуемо¬
го образца 50,0 мг ФСО кислоты пальмитиновой и
50,0 мг ФСО кислоты стеариновой.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 2,4 мл/мин;
- температура:
Время
Температура
(мин)
<°С)
Колонка
0—2
70
2—36
70 — 240
36-^И
240
Блок ввода проб
220
Детектор
260
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению к
метилстеарату): метилпальмитат — около 0,9.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: не менее 5,0 между пиками ме-
тилпальмитата и метилстеарата.
Рассчитывают содержание пальмитиновой и сте¬
ариновой кислот. Пики растворителей во внимание не
принимают.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для кальция стеарата, используемого в ка¬
честве смазывающего вещества при изготовле¬
нии таблеток и капсул.
Распределение частиц по размерам (2.9.31).
Удельная площадь поверхности (2.9.26, ме¬
тод /). Определяют удельную площадь поверхности
в диапазоне Р/Р0 от 0,05 до 0,15.
Дегазация образца: в течение 2 ч при темпера¬
туре 40 °С.
07/2016:0978
КАЛЬЦИЯ ФОЛИНАТ
Са/с// ТоНпаз
САЬСШМ РОиЫАТЕ
С20Н21Са1Ч7О7 • хН20 М.м. 511,5 (безводное
вещество)
ОПРЕДЕЛЕНИЕ
Кальция (25)-2-[[4-[[[(6Р8)-2-амино-5-формил-4-
оксо-1,4,5,6,7,8-гексагидроптеридин-6-ил]метил]ами-
но]бензоил]амино]пентандиоат.
Содержание:
-кальция фолинат (С20Н21СаМ7О7): не менее
97,0 % и не более 102,0 % (в пересчете на безводное
вещество);
-кальций (Са, А.м. 40,08):ьне менее 7,54 % и не
более 8,14 % (в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или светло-желтый аморфный или кри¬
сталлический порошок. Гигроскопичен.
Умеренно растворим в воде, практически нераст¬
ворим в ацетоне и в 96 % спирте.
Аморфная форма может давать пересыщенные
растворы в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Б, О.
Вторая идентификация: А, С, Б.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО кальция фолината #или спектр,
представленный на рисунке #0978.-1#.
Если полученные спектры отличаются, то ис¬
пытуемый образец и ФСО кальция фолината раст¬
воряют по отдельности в минимальном количестве
воды Р, прибавляют по каплям достаточное коли¬
чество ацетона Р для получения осадков и вы¬
держивают в течение 15 мин. Осадки собирают с
помощью центрифугирования, дважды промывают
малым количеством ацетона Р, высушивают и ис¬
пользуют для получения новых спектров.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 15 мг испытуемого об¬
разца растворяют в 3 % (об/об) растворе аммиа¬
ка Р и доводят до объема 5 мл этим же раствори¬
телем.
Раствор сравнения. 15 мг ФСО кальция фоли¬
ната растворяют в 3 % (об/об) растворе аммиака Р и
доводят до объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем целлюлозы
для хроматографии Р254 Р.
Подвижная фаза: используют нижний слой сме¬
си из 1 объема изоамилового спирта Р и 10 объемов
502
Гэсударственная фармакопея Республики Беларусь
Рисунок #0978.-1. Инфракрасный спектр ФСО кальция фолината
раствора 50 г/л лимонной кислоты Р, предваритель¬
но доведенного до рН 8 раствором аммиака Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
О. Испытуемый образец дает реакцию (Ь) на
кальций (2.3.1).
Испытания и количественное определение про¬
водят как можно быстрее и с защитой от света, спо¬
собного вызвать фотохимические превращения.
ИСПЫТАНИЯ
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при
необходимости нагревая до 40 °С, и доводят до объ¬
ема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,60
при 420 нм. Измеряют оптическую плотность раст¬
вора 3, используя в качестве компенсационного раст¬
вора воду Р.
рН (2.2.3). От 6,8 до 8,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От +14,4
до +18,0 (в пересчете на безводное вещество). Опре¬
деляют удельное оптическое вращение раствора 3.
Ацетон, этанол и метанол. Газовая хроматогра¬
фия (2.2.28) с использованием парофазного пробоот¬
борника. Используют метод стандартных добавок.
Испытуемый раствор. 0,25 г испытуемого об¬
разца растворяют в водеР и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения. 0,125 г ацетона Р, 0,750 г
этанола Р и 0,125 г метанола Р растворяют в воде Р и
доводят до объема 1000,0 мл этим же растворителем.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 10 м и
диаметром 0,32 мм, покрытая слоем сополимера сти-
рола-дивинилбензола Р;
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 4 мл/мин;
- параметры парофазного пробоотборника:
- равновесная температура: 80 °С,
- время достижения равновесия: 20 мин,
- время приложения избыточного давления: 30 с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—6
125 — 185
6—15
185
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: проводят не менее 3
вводов.
Предельное содержание:
- ацетон: не более 0,5 %;
- этанол: не более 3,0 %;
- метанол: не более 0,5 %.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО кальция
фолината растворяют в воде Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят водой Р до объема 100,0 мл.
Кальция фолинат
503
Раствор сравнения (с). 10,0 мг ФСО формил-
фолиевой кислоты (примесь й) растворяют в под¬
вижной фазе и доводят до объемэ-100,0 мл этим же
растворителем. 1,0 мл полученного раствора доводят
водой Р до объема 10,0 мл.
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 10,0 мл.
Раствор сравнения (е). 5*€ч\/ит раствора сравне¬
ния (с) доводят раствором сравнения (Ь) до объема
10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
пильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 220 мл метано¬
ла Р и 780 мл раствора, содержащего 2,0 мл раст¬
вора тетрабутиламмония гидроксида (400 г/л) Р и
2,2 г дикалия гидрофосфата Р, предварительно до¬
веденного до рН 7,8 кислотой фосфорной Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь), (с), (б) и (е);
- время хроматографирования: 2,5-кратное вре¬
мя удерживания фолината.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 2,2 между пиками фоли¬
ната и примеси О.
Предельное содержание примесей:
- примесь О (не более 1 %): на хроматограмме ис¬
пытуемого раствора площадь пика, соответствующего
примеси й, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с);
- примеси А, В, С, Е, Р, <3 (не более 1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пика примеси О, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- сумма примесей кроме примеси О (не более
2,5 %): на хроматограмме испытуемого раствора сум¬
ма площадей всех пиков, кроме основного и пика при¬
меси О, не должна превышать 2,5-кратную площадь
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (б).
Хлориды. Не более 0,5 %. 0,300 г испытуемо¬
го образца растворяют в 50 мл воды Р, при необхо¬
димости нагревая до 40 °С. К полученному раствору
прибавляют 10 мл 2 М раствора кислоты азотной и
титруют 0,005 М раствором серебра нитрата потен-
циометрически (2.2.20).
1 мл 0,005 М раствора серебра нитрата соот¬
ветствует 0,177 мг С1.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0050 % (50 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 5 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Платина. Не более 0,0020 % (20 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в^водеР и доводят до объема
100,0 мл этим же растворителем.%
Растворы сравнения. Готовят растворы сравне¬
ния с использованием эталонного раствора платины
(30 ррт РI) Р (при необходимости разводят смесью из
1 объема кислоты азотной Р и 99 объемов воды Р).
Источник излучения: лампа с полым катодом
для определения платины.
Длина волны: 265,9 нм.
Вода (2.5.12). Не более 17,0 %. 0,100 г испытуе¬
мого образца растворяют в смеси из 50 мл раствори¬
теля и 15 мл формамида Р. Перед титрованием смесь
перемешивают в течение 6 мин. Используют подходя¬
щий титрант, не содержащий пиридин.
Бактериальные эндотоксины (2.6.14). Менее
0,5 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без последующей процедуры удаления
бактериальных эндотоксинов.
#Стерильность (2.6.1). Если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения без дополнитель¬
ной процедуры стерилизации, то она должна выдер¬
живать испытание на стерильность.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Кальций. 0,400 г испытуемого образца раст¬
воряют в 150 мл воды Р, доводят до объема 300 мл
этим же растворителем и проводят комплексометри¬
ческое определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 4,008 мг Са.
Кальция фолинат. Жидкостная хроматография
(2.2.29), как указано в испытании «Сопутствующие
примеси», со следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемого
раствора и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение: не
более 2,0 % для площади основного пика при 6 по¬
вторных вводах.
Содержание С20Н21СаЫ7О7 рассчитывают с уче¬
том содержания кальция фолината в ФСО кальция
фолината.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, О.
А. (25)-2-[(4-Аминобензоил)амино]пентандиовая
кислота.
504
Гэсударственная фармакопея Республики Беларусь
В. (25)-2-[[4-[[[(6Я5)-2-Амино-5-формил-4-оксо-
1,4,5,6,7,8-гексагидроптеридин-6-ил]метил]формила-
мино]бензоил]амино]пентандиовая кислота (5,10-ди-
формилтетрагидрофолиевая кислота).
С. (25)-2-[[4-[[(2-Амино-4-оксо-1,4-дигидропте-
ридин-6-ил)метил]амино]бензоил]амино]пентандиовая
кислота (фолиевая кислота).
й. (25)-2-[[4-[[(2-Амино-4-оксо-1,4-дигидро-
птеридин-6-ил)метил]формиламино]бензоил]амино]-
пентандиовая кислота (10-формилфолиевая кислота).
Е. 4-[[[(6/?5)-2-Амино-5-формил-4-оксо-1,4,5,6,7,8-
гексагидроптеридин-6-ил]метил]амино]бензойная кис¬
лота (5-формилтетрагидроптероевая кислота).
Р. К = ОНО: (2$)-2-[[4-[[(2-Амино-4-оксо-1,4,7,8-
тетрагидроптеридин-6-ил)метил]формиламино]бензо-
ил]амино]пентандиовая кислота (10-формилдигидро-
фолиевая кислота).
О. К = Н: (25)-2-[[4-[[(2-Амино-4-оксо-1,4,7,8-
тетрагидроптеридин-6-ил)метил]амино]бензоил]ами-
но]пентандиовая кислота (дигидрофолиевая кислота).
07/2016:0707
КАЛЬЦИЯ ХЛОРИД ГЕКСАГИДРАТ
Са/с/У сЫопйит ЬехаЪудпсит
СА1-СШМ СШ.ОПЮЕ НЕХА Н У ОНА ТЕ
СаС12 • 6Н20 М.м. 219,1
[7774-34-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 97,0 % и не более 103,0 %
СаС12-6Н20.
ОПИСАНИЕ(СВОЙСТВА)
Белая или почти белая кристаллическая масса
или бесцветные кристаллы.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
Затвердевает при температуре около 29 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в
разделе «Испытания», дает реакцию (а) на хлориды
(2.3.1).
B. Испытуемый образец дает реакции (а) и (Ь) на
кальций (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 15,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Кислотность или щелочность. К 10 мл свеже¬
приготовленного раствора 5 прибавляют 0,1 мл раст¬
вора фенолфталеина Р. При прибавлении не более
0,2 мл 0,01 М раствора кислоты хлористоводород¬
ной окраска раствора должна измениться с красной
на бесцветную или при прибавлении не более 0,2 мл
0,01 М раствора натрия гидроксида окраска раст¬
вора должна измениться с бесцветной на красную.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Алюминий. К 10 мл раствора 3 прибавляют 2 мл
раствора аммония хлорида Р, 1 мл раствора аммиа¬
ка разведенного Р1 и нагревают раствор до кипения.
Не должно быть помутнения или выпадения осадка.
Если субстанция используется для приготовле¬
ния растворов для диализа, вместо описанного выше
испытания она должна выдерживать испытание на
алюминий (2.4.17): не более 0,0001 % (1 ррт).
Предписанный раствор. 6 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смесь из 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смесь из 10 мл ацетат¬
ного буферного раствора рН 6,0 Р и 100 мл воды Р.
Барий. К 10 мл раствора 3 прибавляют 1мл
раствора кальция сульфата Р. Не менее чем через
15 мин опалесценция раствора должна быть не ин¬
тенсивнее опалесценции смеси из 10 мл раствора 3
и 1 мл воды дистиллированной Р.
Железо (2.4.9). Не более 0,0007 % (7 ррт). 10 мг
раствора 3 должны выдерживать испытание на железо.
Магний и щелочные металлы. Не более 0,3 %. К
смеси из 20 мл раствора 3 и 80 мл воды Р прибавляю-
2 г аммония хлорида Р и 2 мл раствора аммиака раз-
Кальция хлорид дигидрат
505
веденного Р1, нагревают до кипения и вливают в кипя¬
щий раствор горячий раствор 5 г аммония оксалата Р в
75 мл воды Р. Выдерживают раствор в течение 4 ч, до¬
водят водой Р до объема 200 мл и фильтруют через под¬
ходящий фильтр. К100 мл фильтрата прибавляют 0,5 мл
кислоты серной Р. Полученный раствор выпаривают
досуха на водяной бане и прокаливают до постоянной
массы при температуре (600±50) °С. Масса остатка не
должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0015 % (15 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в
100 мл воды Р. Проводят комплексометрическое
определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 21,91 мг СаС12-6Н20.
МАРКИРОВКА
При необходимости указывают:
-субстанция пригодна для приготовления рас¬
творов для диализа.
07/2016:0015
КАЛЬЦИЯ ХЛОРИД ДИГИДРАТ
Са1сН сЫопдит сИЬус1псит
СА/.С/1/М СШ.ОКЮЕ ОIНУАТЕ
СаС12 • 2Н20 М.м. 147,0
[10035-04-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 97,0 % и не более 103,0 %
СаС12-2Н20.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в
разделе «Испытания», дает реакцию (а) на хлориды
(2.3.1).
B. Испытуемый образец дает реакции (а) и (Ь) на
кальций (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Кислотность или щелочность. К 10 мл свеже¬
приготовленного раствора 3 прибавляют 0,1 мл раст¬
вора фенолфталеина Р. При прибавлении не более
0,2 мл 0,01 М раствора кислоты хлористоводород¬
ной окраска раствора должна измениться с красной
на бесцветную или при прибавлении не более 0,2 мл
0,01 М раствора натрия гидроксида окраска раст¬
вора должна измениться с бесцветной на красную.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Алюминий. К 10 мл раствора 3 прибавляют 2 мл
раствора аммония хлорида Р, 1 мл раствора аммиа¬
ка разведенного Р1 и нагревают раствор до кипения.
Не должно быть помутнения или выпадения осадка.
Если субстанция используется для приготовле¬
ния растворов для диализа, вместо описанного выше
испытания она должна выдерживать испытание на
алюминий (2.4.17): не более 0,0001 % (1 ррт).
Предписанный раствор. 4 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смесь из 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смесь из 10 мл ацетат¬
ного буферного раствора рН 6,0 Р и 100 мл воды Р.
Барий. К 10 мл раствора 3 прибавляют 1 мл
раствора кальция сульфата Р. Через 15 мин опа¬
лесценция раствора должна быть не интенсивнее
опалесценции смеси из 10 мл раствора 3 и 1 мл воды
дистиллированной Р.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 3 должны выдерживать испытание на
железо.
Магний и щелочные металлы. Не более 0,5 %.
К смеси из 20 мл раствора 3 и 80 мл воды Р прибав¬
ляют 2 г аммония хлорида Р и 2 мл раствора аммиа¬
ка разведенного Р1, нагревают до кипения и вливают
в кипящий раствор горячий раствор 5 г аммония окса¬
лата Р в 75 мл воды Р. Выдерживают раствор в тече¬
ние 4 ч, доводят водой Р до объема 200 мл и фильтру¬
ют. К 100 мл фильтрата прибавляют 0,5 мл кислоты
серной Р. Полученный раствор выпаривают досуха
на водяной бане и прокаливают до постоянной массы
при температуре (600±50) °С. Масса сухого остатка не
должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,280 г испытуемого образца растворяют в
100 мл воды Р. Проводят комплексометрическое
определение кальция (2.5.11).
1 мп 0,1 М раствора натрия эдетата соответ¬
ствует 14,70 мг СаС12-2Н20.
МАРКИРОВКА
При необходимости указывают:
-субстанция пригодна для приготовления рас¬
творов для диализа.
ХРАНЕНИЕ
В плотно закрытом контейнере.
506
Гэсударственная фармакопея Республики Беларусь
07/2016:1400
й-КАМФОРА
О-СатрЬога
й-САМРНОЯ
СН3
Н
С10Н16О М.м. 152,2
[464-49-3]
ОПРЕДЕЛЕНИЕ
(1Я,4Я)-1,7,7-Триметилбицикло[2.2.1]гептан-2-он.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или хрупкая кристаллическая масса.
Очень летуча даже при комнатной температуре.
Мало растворима в воде, очень легко раствори¬
ма в 96 % спирте и в петролейном эфире, легко рас¬
творима в жирных маслах, очень мало растворима в
глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Температура плавления (2.2.14). От 175 °С до
179 °С.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО камфоры рацемической.
0.1,0 г испытуемого образца растворяют в 30 мл
метанола Р. Прибавляют 1,0 г гидроксиламина ги¬
дрохлорида Р и 1,0 г натрия ацетата безводного Р.
Кипятят с обратным холодильником в течение 2 ч,
охлаждают, прибавляют 100 мл воды Р и фильтруют.
Полученный осадок промывают 10 мл воды Р и пере-
кристаллизовывают из 10 мл смеси 96 % спирт Р —
вода Р (4:6, об/об). Температура плавления (2.2.14)
полученных кристаллов, высушенных в вакууме: от
118 °С до 121 °С.
ИСПЫТАНИЯ
Взвешивание и растворение производят бы¬
стро.
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в 10 мл 96% спирта Р и доводят до объема
25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод //). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К10 мл раствора
3 прибавляют 0,1 мл раствора фенолфталеина Р1.
Полученный раствор должен быть бесцветным. При
прибавлении не более 0,2 мл 0,1 М раствора натрия
гидроксида окраска раствора должна измениться.
Удельное оптическое вращение (2.2.7). От
+41,0 до +44,0. Используют раствор 3.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. 2,50 г испытуемого об¬
разца растворяют в гептане Р и доводят до объема
25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят гептаном Р до объема 100,0 мл.
Раствор сравнения (Ь). 10,0 мл раствора срав¬
нения (а) доводят гептаном Р до объема 20,0 мл.
Раствор сравнения (с). 0,50 г борнеола Р раст¬
воряют в гептане Р и доводят до объема 25,0 мл
этим же растворителем. 5,0 мл полученного раствора
доводят гептаном Р до объема 50,0 мл.
Раствор сравнения (д). 50 мг линалола Р и 50 мг
борнилацетата Р растворяют в гептане Р и доводят
до объема 100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 30 м и внутренним диаметром
0,25 мм, покрытая слоем макрогола 20 000 Р (толщи¬
на слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- деление потока: 1:70;
- скорость потока: 45 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
50
10—35
50->100
35—45
100 ->200
45—55
200
Блок ввода проб
220
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 3,0 между пиками бор¬
нилацетата и линалола.
Предельное содержание примесей:
- борнеол (не более 2,0 %): на хроматограмме ис¬
пытуемого раствора площадь пика, соответствующего
борнеолу, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с);
- любая другая примесь (не более 0,5 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пика борнеола, не должна
превышать 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 4,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 4-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Галогены. Не более 0,0100 % (100 ррт). 1,0 г
испытуемого образца помещают в колбу для пере¬
гонки и растворяют в 10 мл 2-пропанола Р. Прибав¬
ляют 1,5 мл раствора натрия гидроксида разве-
ш
Камфора рацемическая
507
денного Р и 50 мг никель-алюминиевого сплава Р.
Нагревают на водяной бане до испарения 2-пропа¬
нола Р. Охлаждают и прибавляют 5 мл воды Р. Пе¬
ремешивают и фильтруют через влажный фильтр,
предварительно промытый водой Р до удаления
хлоридов. Фильтрат доводят водой Р до 10,0 мл. К
5,0 мл полученного раствора прибавляют по каплям
кислоту азотную Р до повторного растворения вы¬
павшего осадка, затем разводят водой Р до 15 мл.
Полученный раствор должен выдерживать испыта¬
ние на хлориды (2.4.4).
Остаток после выпаривания (2.8.9). Не более
0,05 %. 2,0 г испытуемого образца выпаривают на
водяной бане и сушат при температуре от 100 °С до
105 °С в течение 1 ч. Масса остатка не должна пре¬
вышать 1 мг.
Вода. 1 г испытуемого образца растворяют в
10 мл петролейного эфира Р. Полученный раствор
должен быть прозрачным (2.2.1).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ПРИМЕСИ
сн3
н.
н3с
н3с
и энантиомер
А. 2,6,6-Триметилбицикло[3.1.1]гепт-2-ен (а-пинен).
гГ
,сн3
-СН3
и энантиомер
СН2
О. Р1 = Н, К2 = ОН, КЗ = СН3: экзо-2,3,3-Триметил-
бицикло[2.2.1]гептан-2-ол (камфена гидрат).
Н. Р1 = Н, Р2 = СН3, КЗ = ОН: эндо-2,3,3-Триметил-
бицикло[2.2.1]гептан-2-ол (метилкамфенилол).
9нз
к
I. Р = ОН, Р' = Н: экзо-1,7,7-Триметилбицикло[2.2.1]-
гептан-2-ол (экзо-борнеол).
1 Р = Н, Р' = ОН: эндо-1,7,7-Триметилбицикло[2.2.1]-
гептан-2-ол (эндо-борнеол).
07/2016:0655
КАМФОРА РАЦЕМИЧЕСКАЯ
СатрЬога гасетюа
САМРНОК, КАСЕМ1С
СН3
С10Н1вО М.м. 152,2
[76-22-2]
ОПРЕДЕЛЕНИЕ
(1 Р5,4Р5)-1,7,7-Триметилбицикло[2.2.1]гептан-2-он.
н
В. 2,2-Диметил-3-метиленбицикло[2.2.1]гептан
(камфен).
и энантиомер
Н3с ;
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или хрупкая кристаллическая масса.
Очень летуча даже при комнатной температуре.
Мало растворима в воде, очень легко раствори¬
ма в 96 % спирте и в петролейном эфире, легко рас¬
творима в жирных маслах, очень мало растворима в
глицерине.
С. 6,6-Диметил-2-метиленбицикло[3.1.1]гептан
(р-пинен).
сн3
сн3
сн3
й. 1,3,3-Триметил-2-оксабицикло[2.2.2]октан (ци-
неол).
$1
Н
Р*2
и энантиомер
СН3
Е. Н1 = СН3, К2 + НЗ = О: 1,3,3-Триметилбицик-
ло[2.2.1]гептан-2-он (фенхон).
Р. Р1 = СН3, Р2 = ОН, НЗ = Н: экзо-1,3,3-Триметил-
бицикло[2.2.1]гептан-2-ол (фенхол).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О.
A. Испытуемый образец выдерживает испытание
«Оптическое вращение», как указано в разделе «Ис¬
пытания».
B. Температура плавления (2.2.14). От 172 °С до
180 °С.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в суспензиях в вазелиновом
масле Р.
Сравнение: ФСО камфоры рацемической.
й. 1,0 г испытуемого образца растворяют в 30 мл
метанола Р. Прибавляют 1,0 г гидроксиламина ги¬
дрохлорида Р и 1,0 г натрия ацетата безводного Р.
Кипятят с обратным холодильником в течение 2 ч,
охлаждают и прибавляют 100 мл воды Р. Образовав¬
шийся осадок отфильтровывают, промывают 10 мл
воды Р и перекристаллизовывают из 10 мл смеси
96% спирт Р — вода Р (4:6, об/об). Температура
ч
508
Государственная фармакопея Республики Беларусь
плавления (2.2.14) полученных кристаллов, высушен¬
ных в вакууме: от 118 °С до 121 °С.
ИСПЫТАНИЯ
Взвешивание производят быстро.
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в 10 мл 96% спирта Р и доводят до объема
25.0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфталеи¬
на Р1. Полученный раствор должен быть бесцветным.
При прибавлении не более 0,2 мл 0,1 М раствора на¬
трия гидроксида окрашивание раствора должно из¬
мениться.
Оптическое вращение (2.2.7). От -0,15° до
+0,15°. Используют раствор 3.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в гексане Р и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 50 мг испытуемого образ¬
ца и 50 мг борнилацетата Р растворяют в гексане Р
и доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят гексаном Р до объема 200,0 мл.
Условия хроматографирования:
- колонка длиной 2 м и внутренним диаметром
2 мм, заполненная диатомитом для газовой хрома¬
тографии Р, импрегнированным 10 % (м/м) макрого-
ла 20 000 Р;
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 30 мл/мин;
- температура колонки: 130 °С;
- температура блока ввода проб и детектора:
200 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания камфоры.
Чувствительность системы: регулируют таким
образом, чтобы высота основного пика на хромато¬
грамме испытуемого раствора составляла не менее
80 % шкалы регистрирующего устройства.
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками кам¬
форы и борнилацетата на хроматограмме раствора
сравнения (а);
- отношение сигнал-шум: не менее 5 для основ¬
ного пика на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
-любая примесь: на хроматограмме испытуе¬
мого раствора площадь любого пика, кроме основно¬
го, не должна превышать 2 % от площади основного
пика;
- сумма примесей: на хроматограмме испыту¬
емого раствора сумма площадей всех пиков, кроме
основного, не должна превышать 4 % от площади ос¬
новного пика;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади основного пика на хроматограмме
раствора сравнения (Ь).
Галогены. Не более 0,0100% (100 ррт). 1,0 г
испытуемого образца помещают в колбу для перегон¬
ки и растворяют в 10 мл 2-пропанола Р. Прибавляют
1,5 мл раствора натрия гидроксида разведенного Р
и 50 мг никель-алюминиевого сплава Р. Нагревают
на водяной бане до испарения 2-пропанола Р. Ох¬
лаждают и прибавляют 5 мл воды Р. Перемешивают
и фильтруют через влажный фильтр, предварительно
промытый водой Р до удаления хлоридов. Фильтрат
разводят водой Р до 10,0мл. К 5,0 мл полученного
раствора прибавляют по каплям кислоту азотную Р
до повторного растворения выпавшего осадка, затем
доводят водой Р до 15 мл. Полученный раствор дол¬
жен выдерживать испытание на хлориды (2.4.4).
Вода. 1 г испытуемого образца растворяют в
10 мл петролейного эфира Р. Полученный раствор
должен быть прозрачным (2.2.1).
Остаток после выпаривания. Не более 0,05 %.
2,0 г испытуемого образца выпаривают на водяной
бане и высушивают при температуре от 100 °С до
105 °С в течение 1 ч. Масса остатка не должна пре¬
вышать 1 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:0503
КАОЛИН ТЯЖЕЛЫЙ
КаоНпит ропёегозит (#Во1из а1Ьа)
КАО 1.1 И, НЕАУУ
ОПРЕДЕЛЕНИЕ
"Глина белая*.
Очищенный природный гидратированный сили¬
кат алюминия различного состава.
ОПИСАНИЕ (СВОЙСТВА)
Мелкий белый или серовато-белый маслянистый
на ощупь порошок.
Практически нерастворим в воде и в органиче¬
ских растворителях.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,5 г испытуемого образца помещают в метал¬
лический тигель, прибавляют 1 г калия нитрата Р, 3 г
натрия карбоната Р и нагревают до расплавления
смеси. Охлаждают, к остатку прибавляют 20 мл кипящей
воды Р, перемешивают, фильтруют и промывают оста¬
ток 50 мл воды Р. К остатку прибавляют 1 мл кислоты
хлористоводородной Р, 5 мл воды Р и фильтруют. К
фильтрату прибавляют 1 мл раствора натрия гидрок¬
сида концентрированного Р и фильтруют. К полученно¬
му фильтрату прибавляют 3 мл раствора аммония хло¬
рида Р. Образуется белый гелеобразный осадок.
B. В мерный цилиндр вместимостью 100 мл и
диаметром около 30 мм помещают 100 мл раствора
10 г/л натрия лаурилсульфата Р и прибавляют 2,0 г
испытуемого образца двадцатью порциями. После
прибавления каждой порции испытуемого образца
раствор выдерживают в течение 2 мин для оседания
частиц. Выдерживают в течение 2 ч. Кажущийся объ¬
ем осадка должен быть не более 5 мл.
C. 0,25 г испытуемого образца дают реакцию (а)
на силикаты (2.3.1).
Шапсулы твердые желатиновые
509
ИСПЫТАНИЯ
Раствор 5. К 4 г испытуемого образца прибавля¬
ют смесь из б мл кислоты уксусной Р и 34 мл воды
дистиллированной Р, встряхивают в течение 1 мин и
фильтруют.
Кислотность или щелочность. К 1,0 г испытуе¬
мого образца прибавляют 20 мл воды, свободной от
углерода диоксида, Р, встряхивают в течение 2 мин
и фильтруют. К 10 мл полученного фильтрата прибав¬
ляют 0,1 мл раствора фенолфталеина Р. Раствор
должен быть бесцветным. При прибавлении не более
0,25 мл 0,01 М раствора натрия гидроксида должно
появиться розовое окрашивание.
Органические примеси. 0,3 г испытуемого об¬
разца нагревают до красного каления. Полученный
остаток может быть лишь незначительно более окра¬
шен, чем исходный испытуемый образец.
Адсорбционная способность. 1,0 г испытуе¬
мого образца помещают в пробирку со шлифом, при¬
бавляют 10,0 мл раствора 3,7 г/л метиленового си¬
него Р, встряхивают в течение 2 мин и выдерживают
до оседания частиц. Центрифугируют и разводят во¬
дой Р в 100 раз. Окраска полученного раствора долж¬
на быть не интенсивнее окраски раствора 0,03 г/л ме¬
тиленового синего Р.
Способность к набуханию. 2 г испытуемого об¬
разца растирают с 2 мл воды Р. Полученная смесь не
должна быть текучей.
Вещества, растворимые в минеральных кис¬
лотах. Не более 1 %. К 5,0 г испытуемого образца
прибавляют 7,5 мл кислоты хлористоводородной
разведенной Р, 27,5 мл воды Р и кипятят в течение
5 мин. Фильтруют, остаток на фильтре промывают во¬
дой Р. Фильтрат и промывные воды доводят водой Р
до объема 50,0 мл. К 10,0 мл полученного раствора
прибавляют 1,5 мл кислоты серной разведенной Р,
выпаривают досуха на водяной бане и сжигают. Мас¬
са полученного остатка не должна превышать 10 мг.
Хлориды (2.4.4). Не более 0,0250 % (250 ррт).
2 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,1 %. 1,5 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл.
Кальций (2.4.3). Не более 0,0250 % (250 ррт).
4 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
Экстрагируемые тяжелые металлы (2.4.8, ме¬
тод А). Не более 0,0050 % (50 ррт). К 5 мл раствора,
приготовленного как указано в испытании «Вещества,
растворимые в минеральных кислотах», прибавляют
5 мл воды Р, 10 мл кислоты хлористоводородной Р,
25 мл метилизобутилкетона Р и встряхивают в те¬
чение 2 мин. Слои разделяют; водный слой выпари¬
вают на водяной бане досуха. Полученный остаток
растворяют в 1 мл кислоты уксусной Р, доводят во¬
дой Р до объема 25 мл и фильтруют. 12 мл получен¬
ного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (1 ррт РЬ) Р.
Если субстанция предназначена для внутреннего
применения, определение тяжелых металлов прово¬
дят следующим методом (2.4.8, метод А). Не более
0,0025 % (25 ррт). К10 мл раствора, приготовленного
как указано в испытании «Вещества, растворимые в
минеральных кислотах», прибавляют 10 мл воды Р,
20 мл кислоты хлористоводородной Р, 25 мл мети¬
лизобутилкетона Р и встряхивают в течение 2 мин.
Слои разделяют; водный слой выпаривают на водя¬
ной бане досуха. Полученный остаток растворяют в
1 мл кислоты уксусной Р, доводят водой Р до объ¬
ема 25 мл и фильтруют. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для внутреннего приме¬
нения.
07/2016:РБ0021
#КАПСУЛЫ ТВЕРДЫЕ
ЖЕЛАТИНОВЫЕ
Сарзи1ае бигае де!аИпозае
САР8Ш-Е8, НАНО, вЕ1.АТ1МОи8
ОПРЕДЕЛЕНИЕ
Капсулы твердые желатиновые представляют
собой желатиновые оболочки для дозированного на¬
полнения порошкообразными или гранулированными
лекарственными веществами.
ПРОИЗВОДСТВО
Основные требования к составу капсул твердых
желатиновых (оболочек капсул) приведены в статье
Капсулы (0016).
Основные типоразмеры твердых желатиновых
капсул указаны в таблице РБ0021.-1.
Таблица РБ0021.-1
Основные типоразмеры твердых
желатиновых капсул
Характе-
ристика
Номера капс^
т
000
00
0
1
2
3
4
5
Длина за¬
крытой на
«замок»
капсулы,
мм
26,0
±0,3
23,3
±0,3
21,8
±0,8
19,2
±0,8
17,7
±0,5
15,5
±0,3
13,9
±0,3
11,1
±0,4
Средняя
вмести¬
мость, мл*
1,37
0,95
0,68
0,50
0,37
0,30
0,21
0,13
— приводится для информации.
ОПИСАНИЕ(СВОЙСТВА)
Капсулы твердые желатиновые состоят из двух
частей цилиндрической формы, один конец которых
закруглен и закрыт, а другой конец открыт. Обе части
свободно входят одна в другую, не образуя зазоров.
Они могут иметь специальные канавки и выступы
для обеспечения «замка», могут быть прозрачными и
окрашенными различными красителями.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,2 г капсул полностью растворяют в 20 мл
воды Р при нагревании до температуры от 60 °С до
70 °С после предварительного набухания и фильтру¬
ют (раствор А). К 2 мл раствора А прибавляют 0,5 мл
510
Гэсударственная фармакопея Республики Беларусь
раствора 50 г/л таниновой кислоты Р. Выпадает
хлопьевидный осадок.
B. К 5 мл раствора А, полученного в идентифи¬
кации А, прибавляют (5—7) капель раствора натрия
гидроксида Р и (2—3) капли раствора меди (II) суль¬
фата Р. Появляется фиолетовое окрашивание.
C. Для идентификации органических красителей
используют подходящий валидированный метод. В
случае присутствия в капсуле органических краси¬
телей, указанных в таблице РБ0021.-2, допустимо
использовать следующую методику определения:
(10—20) капсул (в зависимости от интенсивности
окраски капсул) помещают в коническую колбу вме¬
стимостью 250 мл, прибавляют 100 мл воды Р, 2 мл
раствора аммония ацетата Р и встряхивают в те¬
чение 10 мин. Раствор фильтруют через фильтр «бе¬
лая лента» и снимают спектр поглощения (2.2.25)
полученного раствора в области от 350 нм до 650 нм
в кювете с толщиной слоя 10 мм, используя в каче¬
стве раствора сравнения смесь из воды Р и раствора
аммония ацетата Р (1:1, об/об). Спектр поглощения
полученного раствора имеет максимумы поглощения
при длинах волн, указанных в таблице РБ0021.-2. До¬
пустимое отклонение максимумов поглощения для
каждого красителя — ±3 нм.
Таблица РБ0021.-2
Наименование красителя
Максимум
поглощения
при длине
волны, нм
Удельный
показатель
поглоще¬
ния
Хинолиновый желтый, Е-104
414
865
Апельсиновый желтый,
Е-110
482
555
Кармуазин, Е-122
514
510
Понсо4Р, Е-124
506
430
Красный очаровательный,
Е-129
500
540
Индигокармин, Е-132
610
480
Бриллиантовый голубой,
Е-133
630
1630
Зеленый 5, Е-142
634
1720
Если цвет крышки и корпуса различается, то го¬
товят отдельно раствор из крышек капсулы и раствор
из корпусов.
При комбинированном использовании красите¬
лей, указанных в таблице РБ0021.-2, их концентрация
может существенно отличаться. В этом случае сле¬
дует произвести коррекцию концентрации раствора,
чтобы оптическая плотность искомого красителя ле¬
жала в пределах 0,2—0,8.
О. В случае присутствия в капсуле железа оксида
(Е-172) и титана диоксида (Е-171) определение их на¬
личия проводят качественными химическими реакци¬
ями с зольным остатком, полученным при проведении
испытания «Общая зола».
Зольный остаток делят на равные части.
Половину остатка, полученного после прокали¬
вания, переносят в коническую колбу вместимостью
50 мл, растворяют в 10 мл воды Р и фильтруют через
бумажный фильтр. К фильтрату добавляют на кончи¬
ке шпателя небольшое количество аммония персуль¬
фата Р, а затем аммония роданида Р и взбалтывают.
Постепенно появляется темно-красное окрашивание
(железа оксид).
Оставшуюся часть остатка, полученного после
прокаливания, сплавляют в тигле с калия сульфа¬
том Р. Затем в тигель прибавляют 6 мл 2 Мраствора
кислоты серной и несколько капель раствора водо¬
рода пероксида Р. Постепенно появляется желтое
окрашивание (титана диоксид).
ИСПЫТАНИЯ
Внешний вид. Оценка производится визуально.
Осмотру подлежит 100 % капсул при сортировке и 300
капсул из каждой серии готового продукта. Капсулы
должны иметь гладкую поверхность и не содержать
видимых воздушных или механических включений и
повреждений (вмятин, отверстий, трещин) и других де¬
фектов («звездочек», морщин, неровной обрезки края).
При наличии более 3 % капсул, не соответствую¬
щих требованиям, проводят повторное испытание на
удвоенном количестве капсул.
Капсулы выдерживают испытание, если не менее
97 % капсул соответствуют требованиям.
Масса 100 капсул. Взвешивают 100 капсул с точ¬
ностью до второго знака после запятой. Отклонение в
массе 100 капсул не должно превышать установлен¬
ного производителем предела.
Длина закрытой на «замок» капсулы. Опреде¬
ление проводят на 60 капсулах подходящим толщи-
нометром, штангенциркулем или микрометром. При
наличии более 2 капсул, не соответствующих требо¬
ваниям, указанным в таблице РБ0021.-1, испытание
проводят на следующих 240 капсулах.
Капсулы выдерживают испытание, если не менее
97 % капсул соответствуют требованиям.
Распадаемость (2.9.1). Капсулы должны распа¬
даться в течение не более 20 мин. Допускается нали¬
чие фрагментов оболочки на сетке.
Потеря в массе при высушивании (2.2.32). От
12 % до 17 %. 1,000 г мелко нарезанных капсул сушат
при температуре (107±2) °С.
Общая зола (2.4.16). Не более 2,0% (прозрач¬
ные капсулы), не более 7,0 % (непрозрачные капсу¬
лы), не более 5,0 % (прозрачные/непрозрачные кап¬
сулы).
Тяжелые металлы. Не более 0,005 % (50 ррт).
Испытуемый раствор. 1,0 г измельченных кап¬
сул помещают в фарфоровый тигель, смачивают
(0,5—1) мл кислоты серной концентрированной Р
и осторожно нагревают на сетке или песчаной бане
до удаления паров кислоты серной. Затем прокали¬
вают при температуре около 500 °С, избегая сплав¬
ления золы и спекания ее со стенками тигля. Тигель
охлаждают и зольный остаток обрабатывают при на¬
гревании на сетке 2 мл насыщенного раствора аммо¬
ния ацетата Р (растворяют достаточное количество
аммония ацетата Р в воде Р до получения раствора,
содержащего не менее 61,5 % (м/об) аммония ацета¬
та Р), нейтрализованного раствором 300 г/л натрия
гидроксида Р (насыщенный раствор ацетата аммо¬
ния Р нейтрализуют следующим образом: сначала
прибавляют раствор 300 г/л натрия гидроксида Р до
розового окрашивания по фенолфталеину, а затем из¬
быток натрия гидроксида нейтрализуют насыщенным
раствором ацетата аммония до слабо-розового окра¬
шивания), прибавляют 3 мл воды Р и фильтруют в
пробирку через беззольный фильтр небольшого диа¬
метра, предварительно промытый 1 % раствором кис¬
лоты уксусной Р (2 мл кислоты уксусной разведен¬
ной Р разбавляют водой Р до 24 мл), а затем горячей
водой Р. Тигель и фильтр промывают 5 мл воды Р.
пропуская ее через тот же фильтр в ту же пробирку.
Каптоприл
511
Раствор сравнения. В тигель помещают серную
кислоту концентрированную Р в количестве, взятом
для сжигания капсул, и далее поступают как описано
в приготовлении испытуемого раствора, но промыва¬
ние фильтра и тигля производят 4,5 мл воды Р, по¬
сле чего к фильтрату прибавляют 0,5 мл эталонного
раствора свинца (100 ррт РЬ) Р, перемешивают.
К полученным 10 мл испытуемого раствора и
раствора сравнения прибавляют по 1 мл раствора
300 г/л кислоты уксусной Р, по 2 капли раствора
20 г/л натрия сульфида Р, перемешивают и через
1 мин сравнивают окраску растворов по оси пробирки
диаметром около 1,5 см на белой поверхности. Окра¬
ска испытуемого раствора не должна превышать по
интенсивности окраску раствора сравнения.
Метилпарагидроксибензоат и пропилпараги-
дроксибензоат. Если на маркировке не указано на¬
личие консервантов, проведение испытания также
обязательно. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,000 г измельченных
капсул растворяют при температуре от 30 °С до 40 °С
в (20—30) мл воды Р. Охлаждают до комнатной тем¬
пературы, переносят в мерную колбу вместимостью
50 мл и доводят объем раствора водой Р до метки.
10.0 мл полученного раствора помещают в делитель¬
ную воронку вместимостью 50 мл, прибавляют 15 мл
эфира Р и встряхивают в течение 3 мин. Экстракцию
проводят трижды. Эфирные вытяжки объединяют
в мерную колбу вместимостью 50 мл, доводят объ¬
ем раствора эфиром Р до метки и фильтруют через
фильтр с натрия сульфатом безводным Р, отбра¬
сывая первые (15—20) мл фильтрата. 15,0 мл полу¬
ченного раствора выпаривают досуха. Остаток с по¬
мощью 96 % спирта Р количественно переносят в
мерную колбу вместимостью 25 мл и доводят этим же
растворителем до метки.
Раствор сравнения. 100 мг метилпарагидрокси-
бензоата Р и 25 мг пропилпарагидроксибензоата Р
растворяют в 96% спирте Р и доводят до объема
100.0 мл этим же растворителем. 1,0 мл получен¬
ного раствора доводят 96 % спиртом Р до объема
100.0 мл. Раствор используют свежеприготовленным.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,25 м
и внутренним диаметром 3,0 мм, заполненная силика¬
гелем октадецилсилильным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — вода для
хроматографии Р (40:60, об/об);
- скорость подвижной фазы: 0,6 мл/мин;
- спектрофотометрический детектор, длина
волны 260 нм;
- температура колонки: 20 °С;
- объем вводимой пробы: 5 мкл.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: не менее 10 между пиками ме-
тилпарагидроксибензоата и пропилпарагидрокси¬
бензоата.
Предельное содержание:
- метилпарагидроксибензоат: не более 0,4 %;
- пропилпарагидроксибензоат: не более 0,1 %.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от влаги и света месте при темпе¬
ратуре от 15 °С до 25 °С.
07/2016:1079
КАПТОПРИЛ
Сар1орп!ит
САРТОРКП.
[62571-86-2]
ОПРЕДЕЛЕНИЕ
(25)-1-[(25)-2-Метил-3-сульфанилпропаноил]-
пирролидин-2-карбоновая кислота.
Содержание: не менее 98,0 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде, легко растворим в метилен-
хлориде и в метаноле. Растворяется в разведенных
растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО каптоприла #или спектр, пред¬
ставленный на рисунке #1079.-1#.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 2,0 до 2,6. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
-127 до -132 (в пересчете на сухое вещество). 0,250 г
испытуемого образца растворяют в этаноле Р и дово¬
дят до объема 25,0 мл этим же растворителем.
Примесь Р. Газовая хроматография (2.2.28).
Раствор реактива. 2,8 мл ацетилхлорида Р по
каплям прибавляют к 17,2 мл метанола безводного Р
при температуре 0 °С и перемешивают. Перед исполь¬
зованием выдерживают при комнатной температуре в
течение 20 мин.
Испытуемый раствор. 20,0 мг испытуемого об¬
разца помещают в виалу, прибавляют 1,0 мл раствора
реактива, перемешивают, нагревают при температуре
60 °С в течение 30 мин и выпаривают досуха в токе
азота Р. Полученный остаток растворяют в 0,5 мл
этилацетата Р, прибавляют 0,5 мл пентафторпро-
пионового ангидрида Р, перемешивают, нагревают
при температуре 60 °С в течение 30 мин и выпарива-
512
Государственная фармакопея Республики Беларусь
Волновое число (см'1)
Рисунок #1079.-1. Инфракрасный спектр ФСО каптоприла
ют досуха в токе азота Р. Полученный остаток раст¬
воряют в 1,0 мл бутилацетата Р.
Раствор сравнения (а). Содержимое контейнера
с ФСО каптоприла для проверки хроматографиче¬
ской системы (содержит примесь Р) растворяют в
1,0 мл раствора реактива и обрабатывают получен¬
ный раствор аналогично испытуемому раствору.
Раствор сравнения (Ь). Смешивают 0,25 мл
раствора сравнения (а) и 0,75 мл бутилацетата Р.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 25 м и
диаметром 0,32 мм, покрытая слоем поли(диметил)-
(дифенил)силоксана Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1,2 мл/мин;
- деление потока: 1:20;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
200
10—14
200 240
14—34
240
Блок ввода проб
270
Детектор
300
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению к
каптоприлу, время удерживания — около 6 мин): при¬
месь Р — около 0,96.
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками при¬
меси Р и каптоприла на хроматограмме раствора
сравнения (а);
- отношение сигнал/шум: не менее 10 для пика
примеси Р на хроматограмме раствора сравнения (Ь).
Процентное содержание примеси Р рассчитыва¬
ют по формуле:
где:
А — площадь пика примеси Р на хроматограмме
испытуемого раствора;
В — площадь пика каптоприла на хроматограм¬
ме испытуемого раствора.
Предельное содержание примесей:
- примесь Р: не более 0,2 %.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Фосфорная кислота Р —
ацетонитрил Р1 — вода Р (0,08:10:90, об/об/об).
Испытуемый раствор. 0,125 г испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Раствор сравнения (а). 4,0 мг ФСО каптоприла
примеси 5,0 мг ФСО каптоприла примеси В, 5,0 мг
ФСО каптоприла примеси С и 5,0 мг ФСО каптопри¬
ла примеси О растворяют в смеси растворителей и
доводят до объема 50,0 мл этой же смесью раствори¬
телей. 1,0 мл полученного раствора доводят смесью
растворителей до объема 20,0 мл. Раствор готовят
непосредственно перед использованием.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 5 мг ФСО капторила примеси Е растворяют в
ацетонитриле Р и доводят до объема 25,0 мл этим
же растворителем. 4 мл полученного раствора дово¬
дят смесью растворителей до объема 50,0 мл.
Раствор сравнения (с). Для приготовления при¬
меси А /л зйи 1,0 мл испытуемого раствора помеща¬
ют в мерную колбу вместимостью 50 мл и прибавля¬
ют 230 мкл 0,05 М раствора йода. Если раствор не
обесцветился, прибавляют 0,1 Мраствор натрия ти¬
осульфата до обесцвечивания. Полученный раствор
доводят смесью растворителей до объема 50,0 мл.
5,0 мл полученного раствора доводят смесью раство¬
рителей до объема 20,0 мл.
Каптоприл
513
Раствор сравнения (ф. 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
3,9 мм, заполненная силикагелем октадецилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 10 мкм;
- температура: 50 °С;
- подвижная фаза:
- подвижная фаза А: фосфорная кислота Р —
вода Р (0,08:100, об/об);
- подвижная фаза В: фосфорная кислота Р —
ацетонитрил Р1 — вода Р (0,08:50:50, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
90
10
5—20
со
0
1
сл
о
10 —► 50
20—45
50
50
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 25 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, О и б, используя хромато¬
грамму раствора сравнения (а); идентифицируют пик
примеси Е, используя хроматограмму раствора срав¬
нения (Ь); идентифицируют пик примеси А, используя
хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению
к каптоприлу, время удерживания — около 15 мин):
примесь С — около 0,6; примесь й — около 0,8; при¬
месь Е--около 0,9; примесь В — около 1,17; примесь
б — около 1,22; примесь А — около 1,7.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками при¬
меси В и примеси б на хроматограмме раствора срав¬
нения (а);
- разрешение: не менее 2,0 между пиками при¬
меси Е и капторила на хроматограмме раствора срав¬
нения (Ь).
Предельное содержание примесей:
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 10-кратную
площадь основного пика на хроматограмме раствора
сравнения (б);
- примесь ^ (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси б, не должна превышать 2,5-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (а);
- примеси В, С, О (не более 0,15 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В, С и О, не должны превы¬
шать 1,5-кратные площади соответствующих пиков на
хроматограмме раствора сравнения (а);
-примесь Е (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси Е, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (б);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, О, Е и б, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (б);
- сумма примесей: не более 1,2 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (б).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0020 % (20 ррт).
Растворитель: вода Р.
0,5 г испытуемого образца должны выдерживать
испытания на тяжелые металлы. Эталон готовят с
использованием 1 мл эталонного раствора свинца
(ЮрртРЬ)Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 30 мл
воды Р и титруют 0,05 М раствором йода потенцио-
метрически (2.2.20), используя комбинированный пла¬
тиновый электрод.
1 мл 0,05 М раствора йода соответствует
21,73 мг С9Н15М038.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, 3.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): О, Н, I, /., М, Л/, О.
А. 1,1 -|Дисульфандиилбис[(25)-2-метил-1 -оксопро-
пан-3,1 -диил]]-бис[(25)-пиррол идин-2-карбоновая] кис¬
лота (каптоприл-дисульфид).
В. (25)-1-[(25)-3-Бром-2-метилпропаноил]пирроли-
дин-2-карбоновая кислота.
Н5‘
С02Н
и энантиомер
Н СН3
С. (2/?5)-2-Метил-3-сульфанилпропановая кислота.
Вг
со2н
и энантиомер
Н ЪН3
О. (2#5)-3-Бром-2-метилпропановая кислота.
514
Гэсударственная фармакопея Республики Беларусь
Е. (25)-1 -(2-Метилпропаноил)пирролидин-2-карбо-
новая кислота.
О. 1 ,1'-[Пропан-2,2-диилбис[сульфандиил[(25)-2-
метил-1 -оксопропан-3,1 -диил]]]-бис[(25)-пирролидин-
2-карбоновая] кислота.
07/2016:1081
Р. (25)-1 -[(2Я)-2-Метил-3-сульфанилпропаноил]-
пирролидин-2-карбоновая кислота (эга/-каптоприл).
о
со2н
и энантиомер
н сн3
6. (2Я5)-3-(Ацетилсульфанил)-2-метилпропановая
кислота.
Н. (2 5)-1 -[(2 5)-3-[[(2Я)-3-( Ацетил сул ьфа н и л )-2-
метилпропаноил]сульфанил]-2-метилпропаноил]пир-
ролидин-2-карбоновая кислота.
I. (25)-1-[(25)-3-[[[(25)-1-[(25)-2-Метил-3-
сульфанилпропаноил]пирролидин-2-ил]карбонил]суль-
фанил]-2-метилпропаноил]пирролидин-2-карбоновая
кислота.
Л. (25)-1-[(25)-3-(Ацетилсульфанил)-2-метил-
пропаноил]пирролидин-2-карбоновая кислота (ацетил-
каптоприл).
Ь. 1,1 '-[Метиленбис[сульфандиил[(25)-2-метил-1 -
оксопропан-3,1 -диил]]]-бис[(25)-пирролидин-2-карбо-
новая] кислота.
М. (25)-1-[(25)-3-[[(25)-2-Карбоксипропил]дисуль-
фанил]-2-метилпропаноил]пирролидин-2-карбоновая
кислота.
НзД Н
Н02С'
•С02Н
Н СНз
N. 3,3'-Дисульфандиилбис[(25)-2-метилпропано-
вая] кислота.
КАРБОПЛАТИН
СагЬор1аНпит
САКВОР1.АТ1Ы
С6Н12М204Р1 М.м. 371,3
[41575-94-4]
ОПРЕДЕЛЕНИЕ
(5Р-4-2)-Диамин[циклобутан-1,1-дикарбоксила-
то(2-)-0,0]платина.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Бесцветный кристаллический порошок.
Умеренно растворим в воде, очень мало раство¬
рим в ацетоне и в 96 % спирте.
Температура плавления: около 200 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр карбоплатина
по Европейской Фармакопее #или спектр, представ¬
ленный на рисунке #1081.-1*.
ИСПЫТАНИЯ
Раствор §. 0,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II)- Раствор 3 должен
быть бесцветным.
Примесь В и кислотность. Не более 0,5 % (в
пересчете на примесь В).
К 10 мл раствора 3 прибавляют 0,1 мл раст¬
вора фенолфталеина Р1, полученный раствор дол¬
жен быть бесцветным. При прибавлении к раствору
не более 0,7 мл 0,01 М раствора натрия гидроксида
должно появиться розовое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в смеси из равных объемов аце¬
тонитрила Р и воды Р и доводят до объема 20,0 мл
этой же смесью растворителей.
Карбоплатин
515
Рисунок #1081 .-1. Инфракрасный спектр карбоплатина
Раствор сравнения. 0,5 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем аминопропилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: вода Р — ацетонитрил Р
(13:87, об/об);
- скорость подвижной фазы: 2 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное
время удерживания карбоплатина.
Относительное удерживание (по отношению к
карбоплатину, время удерживания — около 7 мин):
примесь А — около 0,3.
Пригодность хроматографической системы:
испытуемый раствор:
- число теоретических тарелок: не менее 5000;
при необходимости корректируют концентрацию аце¬
тонитрила в подвижной фазе;
- коэффициент распределения масс: не менее
4,0; при необходимости корректируют концентрацию
ацетонитрила в подвижной фазе;
- фактор асимметрии: не более 2,0; при необ¬
ходимости корректируют концентрацию ацетонитрила
в подвижной фазе.
Предельное содержание примесей:
- примесь А (не более 0,25 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения;
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
0,5 г испытуемого образца растворяют в воде Р, слег¬
ка нагревая при необходимости, доводят этим же
растворителем до объема 20 мл и фильтруют при
необходимости. 10 мл полученного раствора дово¬
дят водой Р до объема 15 мл. Эталон готовят с ис¬
пользованием 5 мл эталонного раствора хлорида
(5 ррт С1) Р.
Аммоний (2.4.1, метод В). Не более 0,0100%
(100 ррт). Определение проводят из 0,20 г ис¬
пытуемого образца. Эталон готовят с использо¬
ванием 0,2 мл эталонного раствора аммония
(100 ррт N14^ Р.
Серебро. Не более 0,0010% (10 ррт). Атомно¬
эмиссионная спектрометрия с использованием индук¬
тивно-связанной плазмы (2.2.57).
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в 1 % (об/об) растворе кислоты
азотной Р и доводят этим же раствором до объема
50,0 мл.
Растворы сравнения. Готовят с использованием
эталонного раствора серебра (5 ррт Ад) Р путем
его разведения 1 % (об/об) раствором кислоты азот¬
ной Р.
Длина волны: 328,1 нм.
Растворимый барий. Не более 0,0010%
(10 ррт). Атомно-эмиссионная спектрометрия с ис¬
пользованием индуктивно-связанной плазмы (2.2.57).
Испытуемый раствор. Используют раствор, по¬
лученный как указано в испытании «Серебро».
Растворы сравнения. Готовят с использованием
эталонного раствора бария (50 ррт Ва) Р путем его
разведения 1 % (об/об) раствором кислоты азотной Р.
Длина волны: 455,4 нм.
516
Гэсударственная фармакопея Республики Беларусь
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
ЪДЪЪ т остатка, полученного в испытании «"Поте¬
ря в массе при высушивании», прокаливают до посто¬
янной массы при температуре (800±50) °С.
1 мг остатка соответствует 1,903 мг С6Н12М204Р1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
С1
С1
з
з
А. цас-Диаминдихлорплатина (И) (цисплатин).
. /0О2Н
со2н
В. Циклобутан-1,1-дикарбоновая кислота.
07/2016:1745
КАРВЕДИЛОЛ
Сап/ес1Но1ит
САКУЕОИСП.
Сг4Н26М204 М.м. 406,5
[72956-09-3]
ОПРЕДЕЛЕНИЕ
(2#5)-1-(9Н-Карбазол-4-илокси)-3-[[2-(2-метокси-
фенокси)этил]амино]пропан-2-ол.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, умеренно рас¬
творим в метиленхлориде, мало растворим в 96 %
спирте. Практически не растворяется в разведенных
кислотах.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО карведилола #или спектр, пред¬
ставленный на рисунке #1745.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО карведилола растворяют в
2-пропаноле Р и выпаривают до сухих остатков, кото¬
рые используют для получения новых спектров.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО карведило¬
ла примеси С растворяют в 5,0 мл подвижной фазы и
доводят до объема 100,0 мл этим же растворителем.
4.0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл. 1,0 мл полученного раствора
доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (с). 5 мг ФСО карведилола
для проверки пригодности хроматографической си¬
стемы (содержит примеси А и О) растворяют в под¬
вижной фазе и доводят до объема 50,0 мл этим же
растворителем.
Условия хроматографирования:
-колонка длиной 0,150 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 55 °С;
- подвижная фаза: 1,77 г калия дигидрофосфа¬
та Р растворяют в воде Р, разводят водой Р до объе¬
ма 650 мл, доводят до рН 2,0 кислотой фосфорной Р
и прибавляют 350 мл ацетонитрила Р;
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 240 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 6-кратное вре¬
мя удерживания карведилола.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и О, используя хроматограмму
раствора сравнения (с) и хроматограмму, прилагае¬
мую к ФСО карведилола для проверки пригодности
хроматографической системы, идентифицируют
пик примеси С, используя хроматограмму раствора
сравнения (Ь).
Относительное удерживание (по отношению
к карведилолу, время удерживания — около 4 мин):
примесь А — около 0,5; примесь С — около 2,9; при¬
месь В — около 3,8.
Пригодность хроматографической системы:
-разрешение: не менее 3,5 между пиками при¬
меси А и карведилола на хроматограмме раствора
сравнения (с);
- отношение сигнал/шум: не менее 10 для пика
примеси С на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 2,0):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
Карведилол 517
3000 2000 1500 1000
Волновое число (см1)
Рисунок #1745.-1. Инфракрасный спектр ФСО карведилола
-примесь О (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси Ь, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь С (не более 0,02 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, С и й, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
-сумма примесей кроме примеси С (не более
0,5 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
С, не должна превышать 5-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010 % (10 ррт).
Растворитель: диметилсульфоксид Р.
2,0 г испытуемого образца должны выдерживать
испытание на тяжелые металлы. Эталон готовят с
использованием 2 мл эталонного раствора свинца
(ЮрртРЬ)Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в
60 мл кислоты уксусной безводной Р и титруют
0,1 М раствором кислоты хлорной потенциоме-
трически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соот¬
ветствует 40,65 мг С24Н2бМ204.
ПРИМЕСИ
Специфицированные примеси: А, С, й.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В.
А. 1 -[[9-[2-Гидрокси-3-[[2-(2-метоксифенокси)эти л]-
амино]пропил]-9Н-карбазол-4-ил]окси]-3-[[2-(2-метокси-
фенокси)этил]амино]пропан-2-ол.
518
Гэсударственная фармакопея Республики Беларусь
В. 1,1 *-[[2-(2-Метоксифенокси)этил]нитрило]бис[3-
(9Н-карбазол-4-илокси)пропан-2-ол].
С. (2Р5)-1 -[Бензил[2-(2-метоксифенокси)этил]-
амино]-3-(9Н-карбазол-4-илокси)пропан-2-ол.
он
0.1 -(9Н-Карбазол-4-илокси)-3-[4-[2-гидрокси-3-[[2-
(2-метоксифенокси)этил]амино]пропокси]-9Н-карбазол-
9-ил]пропан-2-ол.
07/2016:0355
КАРТОФЕЛЬНЫЙ КРАХМАЛ
Зо1ат‘ ату1ит
РОТАТО ЗТАРСН
формы, обычно размером от 30 мкм до 100 мкм,
иногда больше 100 мкм; или округлой формы
размером от 10 мкм до 35 мкм. Могут встречать¬
ся сложные зерна, имеющие от 2 до 4 зернышек
(рисунок 0355.-1). Овальные и грушевидные зерна
имеют эксцентрический центр наслоения, окру¬
глые зерна — нецентрированный или слегка экс¬
центрический. У всех зерен наблюдается четкая
концентрическая слоистость. При рассмотрении
между скрещенными призмами Николя, на зернах
присутствует отчетливый черный крестик на цен¬
тре наслоения.
ОПРЕДЕЛЕНИЕ
Картофельный крахмал получают из клубня кар¬
тофеля 8о1апит (иЬегозит 1_.
ОПИСАНИЕ (СВОЙСТВА)
Очень мелкий, белый или почти белый порошок,
при сжатии между пальцами скрипит.
Практически нерастворим в холодной воде и в
96 % спирте.
Картофельный крахмал не должен содержать
зерен крахмала другого происхождения, может содер¬
жать в минимальном количестве фрагменты тканей
клубня.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Проводят микроскопический анализ (2.8.23)
с использованием 50 % (об/об) раствора глице¬
рина Р. Испытуемый образец представляет собой
зерна неправильной, овальной или грушевидной
Рисунок 0344.-1. Иллюстрация к испытанию А
раздела «Подлинность (идентификация)»
для картофельного крахмала
B. 1 г испытуемого образца суспендируют в 50 мл
воды Р, кипятят в течение 1 мин и охлаждают. Образу¬
ется густой опалесцирующий клейстер.
C. К 1 мл клейстера, полученного в подлинности
В, прибавляют 0,05 мл раствора йода Р1. Появляет¬
ся окрашивание от оранжево-красного до темно-сине¬
го, исчезающее при нагревании.
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 8,0. К 5,0 г испытуемого об¬
разца прибавляют 25,0 мл воды, свободной от угле¬
рода диоксида, Р, взбалтывают в течение 60 с и вы¬
держивают в течение 15 мин.
Касторовое масло нерафинированное
519
Посторонние вещества. При изучении под ми¬
кроскопом с использованием 50 % (об/об) раствора
глицерина Р должны обнаруживаться не более чем
следовые количества веществ, отличных от зерен
крахмала. Присутствие зерен крахмала любого друго¬
го происхождения не допускается.
Окисляющие вещества (2.5.30). Не более
0,0020 % (20 ррт), в пересчете на Н202.
Серы диоксид (2.5.29). Не более 0,0050 %
(50 ррт).
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,5 г
испытуемого образца встряхивают с 15 мл кислоты
хлористоводородной разведенной Р и фильтруют.
Фильтрат должен выдерживать испытание на железо.
Потеря в массе при высушивании (2.2.32). Не
более 20,0 %. 1,000 г испытуемого образца сушат при
температуре 130 °С течение 90 мин.
Сульфатная зола (2.4.14). Не более 0,6%.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсРепсЫа соН: отсутствие (2.6.13).
8а1топе11а: отсутствие (2.6.13).
07/2016:0051
КАСТОРОВОЕ МАСЛО
НЕРАФИНИРОВАННОЕ
Я/с/п/ о1еит V^^д^паIе
сазтоп ощ утвт
ОПРЕДЕЛЕНИЕ
Жирное масло, полученное методом холодного
отжима из семян Р1стиз соттит’з 1_. Может содер¬
жать подходящий антиоксидант.
ПРОИЗВОДСТВО
На стадии отжима температура масла не должна
превышать 50 °С.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная (при температуре 40 °С), светло-
желтая, вязкая, гигроскопичная жидкость.
Мало растворимо в петролейном эфире, смеши¬
вается с 96 % спиртом и с ледяной уксусной кислотой.
Относительная плотность: около 0,958.
Показатель преломления: около 1,479.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, В.
A. 2 мл испытуемого образца смешивают с 8 мл
96 % спирта Р. Полученный раствор прозрачный (2.2.1).
B. Испытуемый образец выдерживает испытание
«Удельный показатель поглощения», как указано в
разделе «Испытания».
C. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Угол оптического вращения (2.2.7). От +3,5° до
+6,0°.
Удельный показатель поглощения (2.2.25). Не
более 0,7, определенный в максимуме при длине вол¬
ны 270 нм. 1,00 г испытуемого образца растворяют в
96 % спирте Р и доводят до объема 100,0 мл этим же
растворителем.
Кислотное число (2.5.1). Не более 1,5. 5,00 г ис¬
пытуемого образца растворяют в 25 мл предписанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). Не ме¬
нее 160.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 10,0.
Неомыляемые вещества (2.5.7). Не более
0,8 %. Определение проводят из 5,0 г испытуемого
образца.
Состав жирных кислот. Газовая хроматография
(2.4.22) со следующими изменениями.
Применяют смесь веществ для калибровки в со¬
ответствии с таблицей 2.4.22.-3.
Испытуемый раствор. 75 мг испытуемого об¬
разца помещают в центрифужную пробирку вме¬
стимостью 10 мл с навинчивающейся крышкой и
растворяют в 2 мл 1,1-диметилэтилметилового
эфира Р1 при встряхивании и осторожном нагре¬
вании ((50—60) °С). В теплый раствор прибавляют
1 мл раствора 12 г/л натрия Р в метаноле безво¬
дном Р, приготовленного с необходимыми мерами
предосторожности, и тщательно перемешивают не
менее 5 мин. Затем прибавляют 5 мл воды дистил¬
лированной Р и тщательно перемешивают в тече¬
ние 30 с. Центрифугируют с ускорением 1500 д в
течение 15 мин. Используют верхний слой.
Раствор сравнения. 50 мг ФСО метилрицино-
леата и 50 мг ФСО метилстеарата растворяют в
10,0 мл 1,1-диметилэтилметилового эфира Р1.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,25 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 0,9 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—55
215
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Рассчитывают содержание каждой жирной
кислоты в процентах методом внутренней норма¬
лизации.
Корректируют площадь пика, соответствующего
метилрицинолеату, умножением на поправочный ко¬
эффициент (Р), рассчитанный по формуле:
Д/п2’
где:
ш1 — масса метилрицинолеата в растворе срав¬
нения;
т2 — масса метилстеарата в растворе сравне¬
ния;
А1 — площадь пика метилрицинолеата на хрома¬
тограмме раствора сравнения;
А2 — площадь пика метилстеарата на хромато¬
грамме раствора сравнения. ^
520
Гэсударственная фармакопея Республики Беларусь
Состав фракции жирных кислот масла:
- кислота пальмитиновая: не более 2,0 %;
- кислота стеариновая: не более 2,5 %;
- кислота олеиновая: от 2,5 % до 6,0 %;
- кислота линолевая: от 2,5 % до 7,0 %;
- кислота линоленовая: не более 1,0%;
- кислота эйкозеновая: не более 1,0 %;
- кислота рицинолевая: от 85,0 % до 92,0 %;
- любая другая жирная кислота: не более 1,0 %.
Вода (2.5.32). Не более 0,3 %. Определение про¬
водят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом
контейнере, в защищенном от света месте.
#МАРКИРОВКА
Указывают наименование и концентрацию до¬
бавленного антиоксиданта.
07/2016:2367
КАСТОРОВОЕ МАСЛО
РАФИНИРОВАННОЕ
Я/с/л/ о1еит гаШпа1ит
САЗТОК ОЩ ЯЕР/А/ЕО
ОПРЕДЕЛЕНИЕ
Жирное масло, полученное методом холодного
отжима из семян Рютиз соттит'з Ь и очищенное.
Может содержать подходящий антиоксидант.
ПРОИЗВОДСТВО
На стадии отжима температура масла не должна
превышать 50 °С.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, почти бесцветная или слегка желто¬
ватая, вязкая, гигроскопичная жидкость.
Мало растворимо в петролейном эфире, смеши¬
вается с 96 % спиртом и с ледяной уксусной кислотой.
Относительная плотность: около 0,958.
Показатель преломления: около 1,479.
Вязкость: около 1000 мПа-с.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, В.
A. 2 мл испытуемого образца смешивают с 8 мл
96 % спирта Р. Полученный раствор прозрачный (2.2.1).
B. Испытуемый образец выдерживает испытание
«Удельный показатель поглощения», как указано в
разделе «Испытания».
C. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Окраска испытуе¬
мого образца должна быть не интенсивнее окраски
20 мл смеси из 0,25 мл исходного синего раствора,
0,25 мл исходного красного раствора, 0,8 мл исход¬
ного желтого раствора и 18,7 мл раствора, приготов¬
ленного путем разведения 4,0 мл кислоты хлори¬
стоводородной Р1 до 100,0 мл водой Р.
Угол оптического вращения (2.2.7). От +3,5° до
+6,0°.
Удельный показатель поглощения (2.2.25). Не
более 0,7, определенный в максимуме при длине вол¬
ны 270 нм. 1,00 г испытуемого образца растворяют в
96 % спирте Р и доводят до объема 100,0 мл этим же
растворителем.
Кислотное число (2.5.1). Не более 0,8. 5,00 г ис¬
пытуемого образца растворяют в 25 мл предписанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). Не ме¬
нее 160.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 5,0.
Неомыляемые вещества (2.5.7). Не более
0,8 %. Определение проводят из 5,0 г испытуемого
образца.
Масло, полученное экстракцией и подме¬
шиванием. В пробирке длиной около 125 мм и
внутренним диаметром около 18 мм с притертой
пробкой тщательно смешивают 3 мл испытуемого
образца с 3 мл углерода дисульфида Р. Встряхи¬
вают с 1 мл кислоты серной Р в течение 3 мин.
Окраска полученной смеси должна быть не ин¬
тенсивнее окраски свежеприготовленной смеси из
3,2 мл раствора железа хлорида Р1, 2,3 мл воды Р
и 0,5 мл раствора аммиака разведенного Р.
Состав жирных кислот. Газовая хроматогра¬
фия (2.4.22) со следующими изменениями.
Применяют смесь веществ для калибровки в
соответствии с таблицей 2.4.22.-3.
Испытуемый раствор. 75 мг испытуемого об¬
разца помещают в центрифужную пробирку вме¬
стимостью 10 мл с навинчивающейся крышкой и
растворяют в 2 мл 1,1-диметилэтилметилового
эфира Р1 при встряхивании и осторожном нагре¬
вании ((50—60) °С). В теплый раствор прибавляют
1 мл раствора 12 г/л натрия Р в метаноле безво¬
дном Р, приготовленного с необходимыми мера¬
ми предосторожности, и тщательно перемешива¬
ют не менее 5 мин. Затем прибавляют 5 мл воды
дистиллированной Р и тщательно перемешива¬
ют в течение 30 с. Центрифугируют с ускорением
1500 д в течение 15 мин. Используют верхний слой.
Раствор сравнения. 50 мг ФСО метилрицино-
леата и 50 мг ФСО метилстеарата растворяют в
10,0 мл 1,1-диметилэтилметилового эфира Р1.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,25 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 0,9 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—55
215
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
Кетоконазол
521
- объем вводимой пробы: 1 мкл.
Рассчитывают содержание каждой жирной кис¬
лоты в процентах методом внутренней нормализации.
Корректируют площадь пика, соответствующего
метилрицинолеату, умножением на поправочный ко¬
эффициент (Я), рассчитанный по формуле:
МАРКИРОВКА
Где применимо, указывают, что субстанция при¬
годна для производства лекарственных средств для
парентерального применения.
Дт2’
где:
тл — масса метилрицинолеата в растворе срав¬
нения;
т2 — масса метилстеарата в растворе сравнения;
А1 — площадь пика метилрицинолеата на хрома¬
тограмме раствора сравнения;
А2 — площадь пика метилстеарата на хромато¬
грамме раствора сравнения.
Состав фракции жирных кислот масла:
- кислота пальмитиновая: не более 2,0 %;
- кислота стеариновая: не более 2,5 %;
- кислота олеиновая: от 2,5 % до 6,0 %;
- кислота линолевая: от 2,5 % до 7,0 %;
- кислота линоленовая: не более 1,0 %;
- кислота эйкозеновая: не более 1,0 %;
- кислота рицинолевая: от 85,0 % до 92,0 %;
- любая другая жирная кислота: не более 1,0 %.
Вода (2.5.32). Не более 0,3 % или не более
0,2 %, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения. Определение проводят из 1,00 г испыту¬
емого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом
контейнере, в защищенном от света месте.
07/2016:0921
КЕТОКОНАЗОЛ
Ке1осопаго\ит
КЕТОСОЫАЮ1-Е
С26Н28С12М404 М.м. 531,4
[65277-42-1]
ОПРЕДЕЛЕНИЕ
1-Ацетил-4-[4-[[(2Я5,45Я)-2-(2,4-дихлорфенил)-2-
(1 Н-имидазол-1 -илметил)-1,3-диоксолан-4-ил]метокси]-
фенил]пиперазин.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, растворим в метаноле, уме¬
ренно растворим в 96 % спирте.
Рисунок #0921 .-1. Инфракрасный спектр ФСО кетоконазола
66. За* 1060
>я Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификациям, С, О.
A. Температура плавления (2.2.14): от 148 °С
до 152 °С.
B. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО кетоконазола #или спектр,
представленный на рисунке #0921.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 30 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (а). 30 мг ФСО кетокона¬
зола растворяют в подвижной фазе и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 30 мг ФСО кетокона¬
зола и 30 мг ФСО эконазола нитрата растворяют
в подвижной фазе и доводят до объема 5 мл этим
же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля октадецилсилильного Р.
Подвижная фаза: раствор аммония аце¬
тата Р — диоксан Р — метанол Р (20:40:40,
об/об/об).
Наносимой объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от
линии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку выдерживают в парах
йода до появления пятен и просматривают при
дневном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два пол¬
ностью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пят¬
ну на хроматограмме раствора сравнения (а).
Л. 30 мг испытуемого образца помещают в
фарфоровый тигель, прибавляют 0,3 г натрия
карбоната безводного Р и нагревают на открытом
пламени в течение 10 мин. Охлаждают, получен¬
ный остаток растворяют в 5 мл кислоты азотной
разведенной Р и фильтруют. К 1 мл фильтрата при¬
бавляют 1 мл воды Р. Полученный раствор дает
реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в метиленхлориде Р и доводят до объема 10 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)4.
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. Измеряют угол оптического вращения раст¬
вора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 2,5 мг ФСО кетокона¬
зола и 2,5 мг ФСО лоперамида гидрохлорида раст¬
воряют в метаноле Р и доводят до объема 50,0 мл
этим же растворителем.
Раствор сравнения (Ь). 5,0 мл испытуемо¬
го раствора доводят метанолом Р до объема
100,0 мл. 1,0 мл полученного раствора доводят
метанолом Р до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 —
раствор 3,4 г/л тетрабутиламмония гидросуль¬
фата Р (5:95, об/об)]
- подвижная фаза В: ацетонитрил Р1 —
раствор 3,4 г/л тетрабутиламмония гидросуль¬
фата Р (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100 —>0
0 — 100
10—15
0
100
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
-уравновешивание колонки: ацетонитрил Р в
течение около 30 мин, затем подвижная фаза А в те¬
чение не менее 5 мин;
- объем вводимой пробы: по 10 мкл испытуемого
раствора, растворов сравнения (а) и (Ь) и метанола Р
в качестве контрольного опыта.
Времена удерживания: кетоконазол — около 6
мин; лоперамид — около 8 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 15 между пиками кетоко¬
назола и лоперамида; при необходимости изменяют
концентрацию ацетонитрила в подвижной фазе или
время линейного градиента.
Предельное содержание примесей:
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Кетопрофен
523
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворят в 70 мл
смеси кислота уксусная безводная Р — метилэтил-
кетон Р (1:7, об/об) и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 26,57 мг С26Н28С12М404.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. 1 -Ацетил-4-[4-[[(2/?5,45/?)-2-(2,4-дихлорфенил)-
2-(1 Н-имидазол-1 -илметил)-1,3-диоксолан-4-ил]меток-
си]фенил]-1,2,3,4-тетрагидропиразин.
В. 1 -Ацетил-4-[4-[[(2/?5,45/:?)-2-(2,4-дихлорфенил)-
2- (1 Н-имидазол-1 -илметил )-1,3-диоксолан-4-ил]метокси]-
3- [4-(4-ацетилпиперазин-1-ил)фенокси]фенил]пипера-
зин.
С. 1 -Ацетил-4-[4-[[(2Я5,4Я5)-2-(2,4-дихлорфенил)-
2-(1 Н-имидазол-1 -илметил)-1,3-диоксолан-4-ил]меток-
си]фенил]пиперазин.
й. 1-[4-[[(2Я5,45Я)-2-(2,4-Дихлорфенил)-2-(1Н-
имидазол-1 -илметил)-1,3-диоксолан-4-ил]метокси]фе-
нил]пиперазин.
Е. [(2Н5,45Н)-2-(2,4-Дихлорфенил)-2-(1 Н-ими-
дазол-1 -илметил)-1,3-диоксолан-4-ил]метил 4-метил-
бензолсульфонат.
07/2016:0922
КЕТОПРОФЕН
Ке1орго1епит
КЕТОР КОРЕИ
С1вн1403 М.м. 254,3
[22071-15-4]
ОПРЕДЕЛЕНИЕ
(2Я5)-2-(3-Бензоилфенил)пропановая кислота.
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, в 96 % спирте и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 94 °С до
97 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 100,0 мл этим же растворителем. 1,0 мл получен¬
ного раствора доводят 96 % спиртом Р до объема
50,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 255 нм.
Удельный показатель поглощения в максимуме:
от 615 до 680.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кетопрофена #или спектр,
представленный на рисунке #0922.-1#.
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО кетопрофена
растворяют в ацетоне Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО индомета-
цина растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем. К 1 мл полученного
раствора прибавляют 1 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге-
ля вР254 Р.
Подвижная фаза: кислота уксусная ледяная Р —
метиленхлорид Р — ацетон Р (1:49:50, об/об/об).
66*. Зак. 1060.
524
Государственная фармакопея Республики Беларусь
Волновое число (см'1)
Рисунок #0922.-1. Инфракрасный спектр ФСО кетопрофена
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в ацетоне Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО кетопрофе¬
на примеси А растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 50,0 мл.
Раствор сравнения (с). 5,0 мг ФСО кетопрофе¬
на примеси С растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 50,0 мл.
Раствор сравнения (д). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл. К
1,0 мл полученного раствора прибавляют 1,0 мл раст¬
вора сравнения (Ь).
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р (размер частиц
5 мкм) с удельной площадью поверхности 350 м2/г и
размером пор 10 нм;
-подвижная фаза: свежеприготовленный фос¬
фатный буферный раствор рН 3,5 Р — ацетони¬
трил Р — вода Р (2:43:55, об/об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 233 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 7-кратное вре¬
мя удерживания кетопрофена.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (Ь); идентифицируют пик примеси С,
используя хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению
к кетопрофену, время удерживания — около 7 мин);
примесь С — около 0,3; примесь Е — около 0,69; при¬
месь В — около 0,73; примесь О — около 1,35; при¬
месь А— около 1,5; примесь Р — около 2,0.
Пригодность хроматографической системы:
раствор сравнения (д):
- разрешение: не менее 7,0 между пиками кето¬
профена и примеси А.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе-
Кетопрофен
525
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь);
- примесь С (не более 0,2 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси С, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- примеси В, О, Е, Р (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, О, Е и Р, не должны
превышать площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А, В, С, й, Е, и Р, не должна превышать
0,5 площади основного пика на хроматограмме
раствора сравнения (а);
- сумма примесей кроме примесей А и С (не бо¬
лее 0,4 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пи¬
ков примесей А и С, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 60 °С и давлении, не превышающем
0,67 кПа.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 25 мл
96 % спирта Р, прибавляют 25 мл воды Р и титруют
0,1 М раствором натрия гидроксида потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 25,43 мг С16Н1403.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): 6, Н, I, 3, К, Е
А. 1-(3-Бензоилфенил)этанон.
В. (З-Бензоилфенил)уксусная кислота.
С. 3-[(1Р5)-1-Карбоксиэтил]бензойная кислота.
О. (2Р5)-2-[3-(4-Метилбензоил)фенил]пропановая
кислота.
Е. (2Р5)-2-(3-Бензоилфенил)пропанамид.
и энантиомер
Р. (2Р5)-2-(3-Бензоилфенил)пропанонитрил.
С. 3-[(1Р5)-1-Цианоэтил]бензойная кислота.
'СИ
но ^о
Н. 3-(Цианометил)бензойная кислота.
67. Зак. 1060.
526
Гэсударственная фармакопея Республики Беларусь
I. (З-Бензоилфенил)этанонитрил.
и. (2Я5)-2-[3-(2,4-Диметилбензоил)фенил]пропа-
новая кислота.
К. Смесь из (2#5)-2-[3-(2,3,4-триметилбензоил)-
фенил]пропановой кислоты и (2Я8)-2-[3-(3|4,5“Триметил-
бензоил)фенил]пропановой кислоты.
1_. (2Я$)-2-[3-(2,4,5-Триметилбензоил)фенил]про-
пановая кислота.
07/2016:1755
КЕТОРОЛАК ТРОМЕТАМИН
Ке1ого!асит 1готе1ато1ит
КЕТ0К01-АС ТК0МЕТАМ01-
С15Н13Ы03 • М.м. 376,4
[74103-07-4]
ОПРЕДЕЛЕНИЕ
2-Амино-2-(гидроксиметил)пропан-1,3-диол-
(1Я$)-5-бензоил-2,3-дигидро-1Н-пирролизин-1-
карбоксилат.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в метаноле, мало рас¬
творим в 96 % спирте, практически нерастворим в ме-
тиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кеторолака трометамина
#или спектр, представленный на рисунке #1755.-1#.
ИСПЫТАНИЯ
Раствор 3. 0,75 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
рН (2.2.3). От 5,7 до 6,7. 5 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объема
15 мл.
Оптическая плотность (2.2.25). Не более 0,10.
Измеряют оптическую плотность раствора 3 при дли¬
не волны 430 нм.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы защищают от яркого
света.
Смесь растворителей. Тетрагидрофуран Р —
вода Р (30:70, об/об).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
10,0 мл. 1,0 мл полученного раствора доводят смесью
растворителей до объема 100,0 мл.
Раствор сравнения (Ь). 2 мг ФСО кеторолака
трометамина для идентификации пиков (содержит
примеси А, В, С и Э) растворяют в 5 мл смеси раст¬
ворителей.
Условия хроматографии:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм:
- температура: 40 °С;
- подвижная фаза: смешивают 30 объемов те-
трагидрофурана Р и 70 объемов раствора, приготов¬
ленного следующим образом: 5,75 г аммония диги-
дрофосфата Р растворяют в 900 мл воды Р, доводя'
кислотой фосфорной Р до рН 3,0 и доводят водой Р
до объема 1000 мл;
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 313 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания кеторолака.
Идентификация примесей: идентифицируют при¬
меси А, В, С и О, используя хроматограмму растворе
сравнения (Ь) и хроматограмму, прилагаемую к ФСС
кеторолака трометамина для идентификации пиков
Относительные времена удерживания (по отно¬
шению к кеторолаку, время удерживания — около * I
мин): примесь С — около 0,5; примесь А— около 0 €
примесь О — около 0,7; примесь В — около 0,9.
Кеторолак трометамин
527
Рисунок #1755.-1. Инфракрасный спектр ФСО кеторолака трометамина
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси В и кеторолака.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 0,67; для примеси В — 0,52; для при¬
меси С — 2,2):
- примеси А, В, С, О (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В, С и О, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С и О, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррш). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 37,64 мг ^ДзЫОз-СД^Оз.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е, Р, 6, Н, I, Л.
A. Р1 = Н, Р2 = ОН: (1Я5)-5-Бензоил-2,3-дигцдро-
1 Н-пирролизин-1 -ол.
B. Р1 + Р2 = О: 5-Бензоил-2,3-дигидро-1Н-пирро-
лизин-1-он.
67*. Зак. 1060.
528
Государственная фармакопея Республики Беларусь
0. ГС1 = С02Н, Р2 = ОСН3: (1Я?5)-5-Бензоил-1-ме-
токси-2,3-дигидро-1 Н-пирролизин-1 -карбоновая кислота.
Е. К1 = Н, Р*2 = СО-МН-С(СН2ОН)3: (1Я5)-5-Бензоил-
Л/-[2-гидрокси-1,1 -бис(гидроксиметил)этил]-2,3-дигидро-
1 Н-пирролизин-1 -карбоксамид.
<3. К1 = С02СН3, Н2 = ОН: Метил-(1Я$)-5-бензоил-
1 -гидрокси-2,3-дигидро-1 Н-пирролизин-1 -карбоксилат.
Н. К1 = Н, К2 = С02СН3: Метил-(1Я$)-5-бензоил-
2.3- дигидро-1 Н-пирролизин-1 -карбоксилат.
1. К1 = Н2 = Н: Фенил(2,3-дигидро-1Н-пирролизин-
5-ил)метанон.
3. К1 = Н, К2 = С02С2Н3: Этил-(1Я5)-5-бензоил-
2.3- дигидро-1 Н-пирролизин-1 -карбоксилат.
^ К7
-со2н
и энантиомер
С. Кб = СО-СбН5, К7 = Н: (1Я$)-6-Бензоил-2,3-
дигидро-1 Н-пирролизин-1-карбоновая кислота.
Р. Кб = Н, К7 = СО-С6Н5: (1Я5)-7-Бензоил-2,3-
дигидро-1 Н-пирролизин-1-карбоновая кислота.
07/2016:1592
КЕТОТИФЕНА ГИДРОФУМАРАТ
КеШЯет ЬусИгодепоТитагаз
КЕТОТ1РЕЫ НУйНОвБЫ ПМАЙАТЕ
С18Н19ЫОЗ ■ С4Н404 М.м. 425,5
[34580-14-8]
ОПРЕДЕЛЕНИЕ
4-(1-Метил пиперидин-4-ил иден)-4,9-дигидро-
10Н-бензо[4,5]циклогепта[1,2-Ь]тиофен-10-она гидро-
(Е)-бутендиоат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Мелкий кристаллический порошок от белого до
коричневато-желтого цвета.
Умеренно растворим в воде, мало растворим в
метаноле, практически нерастворим в гептане.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кетотифена гидрофуМарата.
ИСПЫТАНИЯ
Раствор 5. 0,2 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 10 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раст¬
вора 5 должна быть не интенсивнее эталона У(Ж)4,
ВУ(КЖ)4 или В(К)4.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Испытание проводят с за¬
щитой от света.
Смесь растворителей. Метанол Р — вода Р
(50:50, об/об).
Испытуемый раствор. 30,0 мг испытуемого
образца растворяют в смеси растворителей и до¬
водят до объема 100,0 мл этой же смесью раство¬
рителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объ¬
ема 100,0 мл. 1,0 мл полученного раствора дово¬
дят смесью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контей¬
нера с ФСО кетотифена примеси 6 растворяют в
1.0 мл раствора, приготовленного следующим об¬
разом: смешивают 1,0 мл испытуемого раствора с
9.0 мл смеси растворителей. Обрабатывают уль¬
тразвуком до полного растворения примеси С.
Раствор сравнения (с). 5 мг ФСО кетотифе¬
на для идентификации пиков (содержит примесь
А) растворяют в смеси растворителей и доводят до
объема 5,0 мл этой же смесью растворителей.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диа¬
метром 4,0 мм, заполненная силикагелем октаде-
цилсилильным деактивированным по отношению
к основаниям эндкепированным для хроматогра¬
фии Р с размером частиц 3 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 175 мкл три-
этиламина Р и 500 мл воды Р\
- подвижная фаза В: смешивают 175 мкл три-
этиламина Р и 500 мл метанола Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—12
40
60
12—20
о
7
о
о
05
т
о
со
20—25
10
90
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 297 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифи¬
цируют пик примеси А, используя хроматограмм>
раствора сравнения (с) и хроматограмму, прила¬
гаемую к ФСО кетотифена для идентификации
пиков; идентифицируют пик примеси 6, используя
хроматограмму раствора сравнения (Ь).
Относительное удерживание (по отношение
к кетотифену, время удерживания — около 11 мин
фумаровая кислота — около 0,1; примесь С — окс-
ло 0,8; примесь А — около 1,9.
Пригодность хроматографической систем&
раствор сравнения (Ь):
-разрешение: не менее 1,5 между пиками ке¬
тотифена и примеси (3.
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиксг
Кетотифена гидрофумарат
529
на соответствующие поправочные коэффициенты:
для примеси О — 1,4):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь С (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а); не учитывают
пик фумаровой кислоты.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
В. (4Я5)-10-Метокси-4-(1 -метилпиперидин-4-ил)-
4Н-бензо[4,5]циклогепта[1,2-Ь]тиофен-4-ол.
С. (4/?5)-4-Гидрокси-4-(1-метилпиперидин-4-ил)-
4,9-дигидро-10Н-бензо[4,5]циклогепта[1,2-Ь]тиофен-
10-он.
й. 4-[(а#а5)-1 -Метилпиперидин-4-илиден]-4,9-
дигидро-10Н-бензо[4,5]циклогепта[1,2-Ь]тиофен-10-он
А/-ОКСИД (кетотифен Л/-оксид).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в смеси
из 30 мл кислоты уксусной безводной Р и 30 мл ук¬
сусного ангидрида Р и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 42,55 мг С19Н19ЫОЗ С4Н404.
ПРИМЕСИ
Специфицированные примеси: А, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Е, Р.
Е. 10-(1 -Метилпиперидин-4-илиден)-5,10-дигидро-
4Р/-бензо[5,6]циклогепта[1,2-Ь]тиофен-4-он.
Р. 4-(1-Метилпиперидин-4-илиден)-4,10-дигидро-
9Н-бензо[4,5]циклогепта[1,2-Ь]тиофен-9-он.
А. 4-(4/7-Бензо[4,5]циклогепта[1,2-Ь]тиофен-4-
илиден)-1 -метилпиперидин.
С. 4-(1 -Метилпиперидин-4-илиден)-4Н-бензо[4,5]-
циклогепта[1,2-Ь]тиофен-9,10-дион.
68. Зак. 1060.
530
Гэсударственная фармакопея Республики Беларусь
07/2016:РБ0001
«КИСЛОРОД ГАЗООБРАЗНЫЙ
Охудетит дазеозиз
ОХУОЕЫ, ОА1ЕОЗС18
02 М.м. 32,00
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,5 % (об/об) 02.
Требования данной статьи распространяются на
кислород, использующийся для медицинских целей.
Если нет других указаний в частной статье, про¬
бу кислорода из баллона отбирают при давлении
(14,7±0,5) МПа в прибор для анализа с помощью ре¬
дуктора или вентиля тонкой регулировки и соедини¬
тельной трубки от баллона до прибора для анализа.
Соединительную трубку продувают не менее чем де¬
сятикратным объемом испытуемого газа.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветный газ, без запаха.
1 объем кислорода растворяется в около 32 объ¬
емах воды при температуре 20 °С и давлении 101 кПа.
ПРОИЗВОДСТВО
Кислород получают из атмосферного воздуха
способом низкотемпературной ректификации.
Запах. Должен отсутствовать. Запах кислорода,
выпускаемого через слегка открытый вентиль кисло¬
рода, определяют органолептически.
Углерода диоксид. Не более 0,03 % (300 ррт)
(об/об) при определении с использованием инфра¬
красного анализатора (2.5.24) или не более 0,01 %
(100 ррт) (об/об) при определении с использованием
контрольного раствора натрия гидрокарбоната.
Определение углерода диоксида с использова¬
нием инфракрасного анализатора (2.5.24).
Испытуемый газ. Испытуемый образец фильтру¬
ют для исключения явления побочного свечения.
Гэз сравнения (а). Кислород Р.
Гэз сравнения (Ь). Смесь, содержащая 300 ррт
(об/об) углерода диоксида Р1 в азоте Р1.
Градуируют прибор и устанавливают его чувстви¬
тельность с использованием газов сравнения (а) и (Ь).
Определяют содержание углерода диоксида в испы¬
туемом газе.
Определение углерода диоксида с использовани¬
ем контрольного раствора натрия гидрокарбоната.
Поглотительный раствор. 5 г бария гидрокси¬
да Р растворяют в 100 мл воды, свободной от угле¬
рода диоксида, Р. Раствор быстро фильтруют через
плотный бумажный фильтр и хранят в колбе, закры¬
той пробкой. В пробку вставлена стеклянная трубка,
соединенная с промывной склянкой с 20 % (м/м) рас¬
твором натрия гидроксида Р или 20 % (м/м) раство¬
ром калия гидроксида Р.
Испытуемый раствор. Подходящую склянку для
промывания газов продувают в течение (1—2) мин ис¬
пытуемым газом, который отбирают из баллона с по¬
мощью редуктора через резиновую трубку. В склянку
для промывания газов вливают 100,0 мл поглотитель¬
ного раствора. Через раствор пропускают 1000 мл
испытуемого газа в течение (15—20) мин. Объем ис¬
пытуемого газа, пропущенный через поглотительный
раствор, измеряют с помощью склянки с тубусом или
прибора для отбора проб газа, присоединенного к ко¬
роткой трубке склянки на выходе газа.
Контрольный раствор. Готовят одновременно с
проведением испытания. К 100,0 мл поглотительного
раствора прибавляют 1,0 мл раствора 0,40 г/л натрия
гидрокарбоната Р (соответствует 0,01 % (об/об) угле¬
рода диоксида).
Опалесценция испытуемого раствора не должна
быть интенсивнее опалесценции контрольного раст¬
вора при просматривании в проходящем свете.
Углерода монооксид. Не более 0,0005 %
(5 ррт) (об/об) при определении с использованием
инфракрасного анализатора (2.5.25) или индикатор¬
ной трубки для монооксида углерода (2.1.6) или ис¬
пытуемый образец должен выдерживать испытание
при пропускании его через насыщенный аммиачный
раствор серебра нитрата.
Определение углерода монооксида с использо¬
ванием инфракрасного анализатора (2.5.25).
Испытуемый газ. Испытуемый образец фильтру¬
ют для исключения явления побочного свечения.
Гэз сравнения (а). Кислород Р.
Гзз сравнения (Ь). Смесь, содержащая 5 ррт
(об/об) углерода монооксида Р в азоте Р1.
Градуируют прибор и устанавливают его чувстви¬
тельность с использованием газов сравнения (а) и (Ь).
Определяют содержание углерода монооксида в ис¬
пытуемом газе.
Определение углерода монооксида путем про¬
пускания через насыщенный аммиачный раствор се¬
ребра нитрата.
Насыщенный аммиачный раствор серебра
нитрата. 5,0 г серебра нитрата Р растворяют в
100 мл воды Р и прибавляют по каплям при постоян¬
ном помешивании аммиака раствор разведенный Р1
пока осадок не будет почти (но не полностью) раство¬
рен. Раствор фильтруют и хранят в плотно закрытом
флаконе из темного стекла в защищенном от света
месте (поглотительный раствор).
Подходящую склянку для промывания газов проду¬
вают в течение (1—2) мин испытуемым газом, который
отбирают из баллона с помощью редуктора через рези¬
новую трубку. В склянку для промывания газов помеща¬
ют 100,0 мл слабо нагретого насыщенного аммиачного
раствора серебра нитрата. Через раствор пропускаю*
2000 мл испытуемого газа в течение (30—35) мин. Объ¬
ем испытуемого газа, пропущенный через поглотитель¬
ный раствор, измеряют с помощью склянки с тубусом
или прибора для отбора проб газа, присоединенного *
короткой трубке склянки на выходе газа.
Раствор должен оставаться бесцветным и про¬
зрачным.
Вода. Не более 0,0067 % (67 ррт) (об/об) пр/
определении с использованием электролитического
гигрометра (2.5.28) или не более 0,007 % (70 рргг
(об/об) при определении с использованием подходя¬
щего кулонометрического влагомера (относительная
погрешность не более ±5 % в диапазоне измеряемы *
концентраций водяных паров).
Газообразные кислоты и основания. Испыту¬
емый газ должен выдерживать испытание, описан¬
ное ниже.
Три склянки для промывания газов продуваю"
(1—2) мин испытуемым газом, который отбирают
баллона с помощью редуктора через резиновую труб¬
ку; склянки нумеруют. В три пронумерованные склян¬
ки для промывания газов наливают по 100 мл водь
свободной от углерода диоксида, Р и прибавляют =
каждую из них по (3—4) капли 0,2 % (м/м) раствооа
Шислород газообразный
531
метилового красного Р в спирте (60 %, м/м) Р. За¬
тем в склянку № 2 с помощью пипетки прибавляют
0,2 мл 0,01 М раствора хлористоводородной кисло¬
ты, в склянку № 3 — 0,4 мл 0,01 М раствора хлори¬
стоводородной кислоты. Через раствор в склянке
№ 2 пропускают 2000 мл испытуемого газа в течение
(30—35) мин. Объем испытуемого газа, пропущенный
через раствор, измеряют с помощью склянки с тубу¬
сом или прибора для отбора проб газа, присоединен¬
ного к короткой трубке склянки на выходе газа.
Сравнивают окраску раствора в склянке № 2 с
окраской растворов в склянках № 1 и № 3.
Окраска раствора в склянке № 2 должна сохра¬
нять розовый цвет в отличие от раствора в склянке
№ 1, окрашенного в желтый цвет (выдерживает ис¬
пытание по содержанию оснований); розовая окраска
раствора в склянке № 2 должна быть менее интенсив¬
ной, чем в склянке № 3 (выдерживает испытание по
содержанию кислот).
Пороговая чувствительность метода 0,001 г/моль
газообразных кислоты или основания в 1 м3 кислорода.
Озон и другие газы-окислители. Испытуемый
газ должен выдерживать испытание, описанное ниже.
Смешанный раствор крахмала. 0,5 г калия йо¬
дида Р растворяют при нагревании в 95 мл воды Р;
0,5 г крахмала растворимого Р размешивают в 5 мл
холодной воды. Раствор крахмала медленно вливают
при помешивании в кипящий раствор калия йодида и
кипятят в течение (2—3) мин.
Подходящую склянку для промывания газов
продувают в течение (1—2) мин испытуемым газом,
который отбирают из баллона с помощью редуктора
через резиновую трубку. В склянку для промывания
газов помещают 100 мл свежеприготовленного сме¬
шанного раствора крахмала и прибавляют 1 каплю
кислоты уксусной ледяной Р. Через наполненную
склянку для промывания газов пропускают 2000 мл
испытуемого газа в течение (30—35) мин. Объем ис¬
пытуемого газа, пропущенный через раствор, изме¬
ряют с помощью склянки с тубусом или прибора для
отбора проб газа, присоединенного к короткой трубке
склянки на выходе газа.
Раствор должен оставаться бесцветным (отсут¬
ствие озона и других газов-окислителей).
Количественное определение. Определяют
концентрацию кислорода с использованием пара¬
магнитного анализатора (2.5.27) или на подходящем
измерительном приборе (см. рисунок РБ0001.-1), как
описано ниже.
Аммиачный раствор аммония хлорида. 750 г ам¬
мония хлорида Р растворяют в 1000 мл воды Р и при¬
бавляют 1000 мл раствора аммиака Р (поглотитель¬
ный раствор). Поглотительный раствор в пипетке при¬
бора заменяют после проведения 20—30 испытаний.
Цилиндрическую часть пипетки заполняют мед¬
ной круглой электротехнической проволокой диа¬
метром (0,8—1,0) мм в виде спиралей длиной около
10 мм и диаметром витка около 5 мм и закрывают
пробкой. В пипетку и уравнительную склянку залива¬
ют поглотительный раствор.
Кран бюретки смазывают и соединяют отдельные
части прибора резиновыми трубками, проверяют при¬
бор на герметичность по постоянству уровня жидко¬
сти в бюретке при закрытом кране и нижнем положе¬
нии уравнительной склянки.
Перед проведением анализа заполняют поглоти¬
тельным раствором цилиндрическую часть пипетки с
1. Бюретка.
2. Двухходовой кран.
3. 4. Отростки крана.
5, 6. Капиллярные
стеклянные трубки.
7. Поглотительная пипетка
с капиллярной трубкой.
8. Штатив.
9. Уравнительная склянка.
10. 11. Резиновые трубки.
Рисунок РБ0001 .-1. Измерительный прибор для
определения объемной доли кислорода
(размеры указаны в миллиметрах)
капиллярной трубкой, капиллярную трубку 5, бюретку,
проходы и капиллярные отростки крана.
Жидкость в пипетке и бюретке прибора пере¬
мещается подъемом или опусканием уравнительной
склянки с поглотительным раствором. При этом пово¬
ротом крана соединяют внутренний объем бюретки с
поглотительной пипеткой или атмосферой.
Отбирают в бюретку прибора через отросток 3
крана чуть более 100 мл испытуемого газа.
Для приведения объема газа в бюретке к атмос¬
ферному давлению устанавливают уровень поглоти¬
тельного раствора в уравнительной склянке против
нулевого давления бюретки. Пережимают резиновую
трубку 10 и быстрым поворотом крана выпускают из
бюретки избыток газа в атмосферу. Затем поворотом
крана соединяют бюретку с пипеткой и, поднимая урав¬
нительную склянку, вытесняют весь кислород из бю¬
ретки в цилиндрическую часть пипетки. После запол¬
нения раствором капиллярной трубки кран закрывают.
Для лучшего поглощения кислорода измеритель¬
ный прибор осторожно встряхивают. Через (2—3) мин
поглощение кислорода обычно заканчивается. Пово¬
ротом крана соединяют бюретку с пипеткой и, мед¬
ленно опуская уравнительную склянку, переводят в
бюретку непоглощенный остаток пробы. Как только
поглотительный раствор начинает поступать в бюрет¬
ку, кран закрывают. Газ в бюретке приводят к атмос¬
ферному давлению, устанавливая на одной высоте
уровни жидкости в бюретке и уравнительной склянке.
Объем остаточных газов в бюретке измеряют через
(1—2) мин, выжидая, пока жидкость стечет со стенок
бюретки.
68*. Зак. 1060.
532
Гэсударственная фармакопея Республики Беларусь
Деление, соответствующее уровню жидкости
в бюретке, показывает объемную долю кислорода в
процентах (об/об) в испытуемом образце кислорода.
Поглощение кислорода повторяют Испытание за¬
канчивают, если после повторного поглощения измене¬
ние объема остаточных газов не превышает 0,05 мл.
За результат испытания принимают среднее
арифметическое результатов двух параллельных
определений, абсолютное расхождение между ко¬
торыми не должно превышать 0,05 % (допускаемое
расхождение). Допускаемая абсолютная суммарная
погрешность результата испытания ±0,05 % при дове¬
рительной вероятности Р = 0,95.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Испытуемый образец выдерживает требования
раздела «Количественное определение».
ИСПЫТАНИЯ
Углерода диоксид. Не более 0,03 % (300 ррт)
(об/об) при определении с использованием индика¬
торной трубки для диоксида углерода (2.1.6) или не
более 0,01 % (100 ррт) (об/об) при определении с ис¬
пользованием контрольного раствора натрия гидро-
карбоната как указано в разделе «Производство».
Углерода монооксид. Не более 0,0005 % (5 ррт)
(об/об) при определении с использованием индика¬
торной трубки для монооксида углерода (2.1.6) или
испытуемый образец должен выдерживать испытание
при пропускании его через насыщенный аммиачный
раствор серебра нитрата, как указано в разделе «Про¬
изводство».
Вода. Не более 0,0067 % (67 ррт) (об/об). Опре¬
деление проводят с использованием индикаторной
трубки для паров воды (2.1.6) или не более 0,007 %
(70 ррт) (об/об) при определении с использованием
подходящего кулонометрического влагомера, как ука¬
зано в разделе «Производство».
ХРАНЕНИЕ
Хранят в сосудах, работающих под давлением, в
предназначенных для данных целей помещениях. Тре¬
бования к сосудам и помещениям хранения должны от¬
вечать требованиям действующего законодательства.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
A. С02: Углерода диоксид.
B. СО: Углерода монооксид.
C. Н20: Вода.
07/2016:2174
КЛАДРИБИН
С1абпЫпит
адоривмЕ
С10Н12С1М5О3 М.м. 285,7
[4291-63-8]
ОПРЕДЕЛЕНИЕ
2-Хлор-9-(2-дезокси-р-0-эритро-пентофуранозил)-
9Н-пурин-6-амин.
Рисунок #2174.-1. Инфракрасный спектр ФСО кладрибина
Кладрибин
533
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, растворим в диметил-
сульфоксиде, мало растворим в метаноле, практиче¬
ски нерастворим в ацетонитриле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кладрибина #или спектр, пред¬
ставленный на рисунке #2174.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то испыту¬
емый образец и ФСО кладрибина растворяют по от¬
дельности в минимальном количестве метанола Р,
выпаривают до сухих остатков, которые сушат при
температуре 100 °С в течение 2 ч и используют для
получения новых спектров.
ИСПЫТАНИЯ
Раствор 5. 0,15 г испытуемого образца диспер¬
гируют в воде Р, доводят до объема 50 мл этим же
растворителем и обрабатывают ультразвуком до пол¬
ного растворения.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
-21,0 до -27,0 (в пересчете на безводное вещество).
0,25 г испытуемого образца растворяют в диметил-
сульфоксиде Р и доводят до объема 25,0 мл этим же
растворителем.
Примесь Е. Не более 0,3 %. Тонкослойная хро¬
матография (2.2.27).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в диметилформамиде Р и доводят
до объема 2,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг 2-дезокси-О-
рибозы Р (примесь Е) растворяют в диметилформа¬
миде Р и доводят до объема 25,0 мл этим же раство¬
рителем. 3,0 мл полученного раствора доводят диме-
тилформамидом Р до объема 10,0 мл.
Раствор сравнения (Ь). 10,0 мг 2-дезокси-й-
рибозы Р (примесь Е) растворяют в диметилформа¬
миде Р и доводят до объема 5,0 мл этим же раство¬
рителем. К 9 объемам полученного раствора прибав¬
ляют 1 объем испытуемого раствора.
Пластинка: ТСХ пластинка со слоем силикаге-
ляРг54Р.
Подвижная фаза: аммиака раствор концен¬
трированный Р — 96% спирт Р — этилацетат Р
(20:40:40, об/об/об).
Наносимый объем пробы: по 5 мкл в виде полос
длиной 10 мм. Тщательно высушивают в потоке те¬
плого воздуха.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе, затем при температу¬
ре 45 °С в течение 10 мин.
Проявление: пластинку опрыскивают раствором,
содержащим 0,5 г тимола Р в смеси из 5 мл кислоты
серной Р и 95 мл 96 % спирта Р, и нагревают при темпе¬
ратуре 110 °С в течение 20 мин или до появления пятен.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
-примесь Е: на хроматограмме испытуемого
раствора пятно, соответствующее примеси Е, должно
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р —
вода Р (10:90, об/об).
Испытуемый раствор (а). 25,0 мг испытуемого
образца растворяют в смеси растворителей и дово¬
дят до объема 5,0 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 20,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (а). 20,0 мг ФСО кладрибина
растворяют в смеси растворителей и доводят до объ¬
ема 100,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100.0 мл.
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (Ь) доводят смесью растворителей до объема
10.0 мл.
Раствор сравнения (ф. 1,0 мг ФСО кладрибина
примеси С растворяют в растворе сравнения (Ь) и до¬
водят до объема 25,0 мл этим же растворителем.
Раствор сравнения (е). 5,0 мл раствора срав¬
нения (с) доводят смеськграстворителей до объема
10.0 мл.
Раствор сравнения (I). 3 мг ФСО кладрибина для
идентификации пиков (содержит примеси А, В, С и О)
растворяют в 2 мл смеси растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: вода для хроматогра¬
фии Р;
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р\
- подвижная фаза С: раствор 50 г/л кислоты
фосфорной Р в воде для хроматографии Р;
Время
(мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
Подвижная
фаза С
(%, об/об)
0—10
80 — 70
10 — 20
10
10—25
70 — 20
20 — 70
10
25—30
20
70
10
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 252 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (с), (б), (е) и (I).
Идентификация пиков примесей: идентифициру¬
ют пики примесей А, В, С и О, используя хроматограмму
раствора сравнения (Г) и хроматограмму, прилагаемую к
ФСО кладрибина для идентификации пиков.
69. Зак. 1060.
534
Гэсударственная фармакопея Республики Беларуси
Относительное удерживание (по отношению
к кладрибину, время удерживания — около 10 мин):
примесь А — около 0,33; примесь В — около 0,44;
примесь С — около 0,73; примесь О — около 0,92.
Пригодность хроматографической системы:
раствор сравнения (с1):
- разрешение: не менее 4,5 между пиками при¬
меси С и кладрибина.
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси В — 1,7, для примеси С — 0,8):
- примеси А, С (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям А и С, не должны пре¬
вышать 3-кратную площадь основного пика на хро¬
матограмме раствора сравнения (с);
- примеси В, О (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям В и О, не должны пре¬
вышать 2-кратную площадь основного пика на хро¬
матограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пиков
примесей А, В, С и О, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора (а) сумма площа¬
дей всех пиков, кроме основного, не должна превы¬
шать 10-кратную площадь основного пика на хро¬
матограмме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора (а) не учитывают
пики с площадью менее площади основного пика
на хроматограмме раствора сравнения (е).
Вода (2.5.32). Не более 0,5 %. Определение
проводят из 0,100 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 3 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без последующей процедуры
удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства ле¬
карственных средств для парентерального приме¬
нения.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): Е, О.
A. Р = ЫН2: 9-(2-Дезокси-Р-0-эрш7?ро-пенто-
фуранозил)-9Н-пурин-2,6-диамин.
B. Р = ОСН3: 9-(2-Дезокси-Р-0-эрашро-пенто-
фуранозил)-2-метокси-9Н-пурин-6-амин.
С. 2-Хлор-7Н-пурин-6-амин (2-хлораденин).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29) как указа¬
но в испытании «Сопутствующие примеси».
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора (Ь) и раствора сравнения (а).
Содержание С10Н12С11Ч5О3 в процентах рассчи¬
тывают с учетом содержания кладрибина в ФСО
кладрибина.
ХРАНЕНИЕ
В защищенном от света месте при температу¬
ре от 2 °С до 8 °С.
Стерильная субстанция — в стерильном воз¬
духонепроницаемом контейнере с контролем пер¬
вого вскрытия.
й. 2-Хлор-9-(2-дезокси-а-0-эрш?7ро-пентофу-
ранозил)-9Н-пурин-6-амин.
но.
.а
он
он
Е. 2-Дезокси-0-эрш?ро-пентофураноза (2-дезокси-
О-рибоза).
о
н3<г
р
Р. Р = [ЧН2: 4-Метилбензамид.
С. Р = ОСН3: Метил-4-метилбензоат.
Кларитромицин
535
07/2016:1651
КЛАРИТРОМИЦИН
С1агШ1готус'тит
сшттнкомусш
Сз8Нв9М013
[81103-11-9]
ОПРЕДЕЛЕНИЕ
(ЗЯ,45,55,6Я,7Я,9Я, 11 Я, 12Я, 135,14Я>4-[(2,6-Диде-
зокси-З-С-метил-З-О-метил-а-Ьрабо-гексапиранозил)
окси]-14-этил-12,13-дигидрокси-7-метокси-3,5,7,9,11,13-
гексаметил-6-[[3,4,б-тридезокси-3-(диметиламино)-(3-
0-/ссш7о-гексапиранозил]окси]оксациклотетрадекан-
2,10-дион (6-О-метилэритромицин А).
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, растворим в аце¬
тоне и в метиленхлориде, мало растворим в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кларитромицина #или спектр,
представленный на рисунке #1651.-1#.
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раст¬
воряют в метиленхлориде Р и доводят до объема
50.0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным или по степени мутности не должен пре¬
вышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)7.
Удельное оптическое вращение (2.2.7). От-94
до -102 (в пересчете на безводное вещество). Опре¬
деляют удельное оптическое вращение раствора 5.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 75,0 мг испытуемого об¬
разца растворяют в 25 мл ацетонитрила Р1 и дово¬
дят водой Р до объема 50,0 мл.
Раствор сравнения (а). 75,0 мг ФСО кларитро¬
мицина растворяют в 25 мл ацетонитрила Р1 и до¬
водят водой Р до объема 50,0 мл.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят смесью из равных объемов ацетони¬
трила Р1 и воды Р до объема 100,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят смесью из равных объемов ацетони¬
трила Р1 и воды Р до объема 10,0 мл.
Раствор сравнения (ф. 15,0 мг ФСО кларитро¬
мицина для идентификации пиков растворяют в
5.0 мл ацетонитрила Р1 и доводят водой Р до объ¬
ема 10,0 мл.
Холостой раствор. 25,0 мл ацетонитрила Р1
доводят водой Р до объема 50,0 мл и перемешивают.
Волновое число (см'1)
Рисунок #1651 .-1. Инфракрасный спектр ФСО кларитромицина
69*. Зак. 1060.
536
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3,5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 4,76 г/л калия ди¬
гидрофосфата Р, доведенный кислотой фос¬
форной разведенной Р или раствором 45 г/л
калия гидроксида Р до рН 4,4 и профильтрован¬
ный через фильтрационный комплект С18;
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—32
75 —► 40
25 —► 60
32—34
40
60
- скорость подвижной фазы: 1,1 мл/мин;
- температура: 40 °С;
-спектрофотометрический детектор, длина
волны 205 нм;
-объем вводимой пробы: по Юмкп холостого
раствора, испытуемого раствора, растворов сравне¬
ния (Ь), (с) и (с!).
Относительное удерживание (по отношению
к кпаритромицину, время удерживания — около
11 мин): примесь I — около 0,38; примесь А — около
0,42; примесь ^ — около 0,63; примесь 1_ — около 0,74;
примесь В — около 0,79; примесь М — около 0,81;
примесь С — около 0,89; примесь О — около 0,96;
примесь N — около 1,15; примесь Е — около 1,27;
примесь Р — около 1,33; примесь Р — около 1,35; при¬
месь О— около 1,41; примесь К — около 1,59; при¬
месь С — около 1,72; примесь Н — около 1,82.
Пригодность хроматографической системы:
-фактор асимметрии: не более 1,7 для пика
кларитромицина на хроматограмме раствора сравне¬
ния (Ь);
-коэфициент разделения пиков: не менее 3,0
(Нр — высота пика примеси Э относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси О и
кларитромицина на хроматограмме раствора сравне¬
ния (б)).
Идентификация пиков примесей: используют
хроматограмму, прилагаемую к ФСО кларитромици¬
на для идентификации пиков.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 0,27; для примеси Н — 0,15):
- любая примесь: на хроматограмме испытуемо¬
го раствора площадь любого пика, соответствующего
примеси, не должна превышать 2-кратную площадь
основного пика на хроматограмме раствора сравне¬
ния (с) (не более 1,0 %); площади не более четырех из
таких пиков могут превышать 0,8 площади основного
пика на хроматограмме раствора сравнения (с) (0,4 %);
- сумма примесей (не более 3,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 7-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,2 площади основного пика на хроматограмме
раствора сравнения (с) (0,1 %) и пики, выходящие до
пика примеси I и после пика примеси Н.
Тяжелые металлы (2.4.8, метод В). Не более
0,0020% (20 ррт). 1,0 г испытуемого образца раст¬
воряют в смеси вода Р — диоксан Р (15:85, об/об) и
доводят до объема 20 мл этой же смесью раствори¬
телей. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон го¬
товят с использованием эталонного раствора свинца
(1 ррт РЬ), полученного путем разведения эталонно¬
го раствора свинца (100 ррт РЬ) Р смесью вода Р —
диоксан Р (15:85, об/об).
Вода (2.5.12). Не более 2,0 %. Определение про¬
водят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 0,5 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкп испытуемого
раствора и раствора сравнения (а).
Содержание С^Н^АЮ^ рассчитывают в процентах.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, О,
Н, I, 3, К, /_, М, А/, О, Р.
СН3
A. Р1 = СН3, Р2 = ОН, РЗ = Н: 2-Деметил-2-
(гидроксиметил)-б-О-метилэритромицин А (кларитро-
мицин Р).
B. Р1 = Р2 = РЗ = Н: 6-0-Метил-15-норэритро-
мицин А.
Р. Р1 = РЗ = СН3, Р2 = Н: 4',6-Ди-О-метилэритро-
мицин А.
С. Р1 = Р2 = СН3, РЗ = Н: 6-О-Метилэритромицин
А (Е)-9-оксим.
О. Р1 = Р2= РЗ = СН3: 6-О-Метилэритромицин А
(Е)-Э-(О-метилоксим).
3. Р1 = СН3, Р2 = РЗ = Н: Эритромицин А (Е)-9-оксим.
Кленбутерола гидрохлорид
537
й. К1 = К2 = КЗ = Н: 3"-Л/-Деметил-6-0-метил-
эритромицин А.
Е. К1 = К2 = СН3, КЗ = Н: 6,11-Ди-О-метил-
эритромицин А.
Р. К1 = КЗ = СН3, К2 = Н: 6,12-Ди-О-метил-
эритромицин А.
Н. К1 = ОНО, К2 = КЗ =Н: 3"-Л/-Деметил-3'-/У-фор-
мил-6-О-метилэритромицин А.
I. З-О-Декладинозил-б-О-метилэритромицин А.
К. (13,2Р,5П,63,75,8Н,9П, 112)-2-Этил-6-гидрокси-
9-метокси-1,5,7,9,11,13-гексаметил-8-[[3,4,6-тридезокси-
3-(диметиламино)-р-0-кш/7О-гексапиранозил]окси]-3,15-
диоксабицикло[10.2.1 ]пентадека-11,13-диен-4-он
(З-О-декладинозил-8,9:10,11 -диангидро-6-О-метил-
эритромицин-А-9,12-гемикетал.
СН3
1_. К = Н: 6-О-Метилэритромицин А(2)-9-оксим.
О. К = СН3: 6-О-Метилэритромицин А (2)-9-(0-
метилоксим).
07/2016:1409
КЛЕНБУТЕРОЛА ГИДРОХЛОРИД
С1епЬи(егоН Ьус1госЫопс1ит
С1.ЕМВ11ТЕК01- НУОРЮСН1.0ЯЮЕ
С.Д.С1ДО • НС1 М.м. 313,7
[21462-39-5]
ОПРЕДЕЛЕНИЕ
(1Я8)-1-(4-Амино-3,5-дихлорфенил)-2-[(1,1-ди-
метилэтил)амино]этанола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Растворим в воде и в 96 % спирте, мало раство¬
рим в ацетоне.
Температура плавления: около 173 °С с разложе¬
нием.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кленбутерола гидрохлорида #или
спектр, представленный на рисунке #1409.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого образца
растворяют в 10 мл метанола Р.
Раствор сравнения. 10 мг ФСО кленбутерола ги¬
дрохлорида растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р254 Р.
Подвижная фаза: раствор аммиака Р — эта¬
нол Р— толуол Р (0,15:10:15, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 10 см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
10 г/л натрия нитрита Р в 1М растворе кислоты хло¬
ристоводородной. Через 10 мин пластинку погружают
в раствор 4 г/л нафтилэтилендиамина дигидрохлори¬
да Р в метаноле Р и высушивают на воздухе.
70. Зак. 1060.
538
Гэсударственная фармакопея Республики Беларусь
Рисунок #1409.-1. Инфракрасный спектр ФСО кленбутерола гидрохлорида
Результаты: на хроматограмме испытуемого раст¬
вора обнаруживается пятно, соответствующее по распо¬
ложению, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения.
С. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5.0,5 г испытуемого образца растворяют в
10 мл воды, свободной от углерода диоксида, Р.
Прозрачность (2.2.1). Раствор 5 по степени мутно¬
сти не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее окраски эталона У(Ж)6.
рН (2.2.3). От 5,0 до 7,0. Измеряют рН раствора 3.
Оптическое вращение (2.2.7). От-0,10° до +0,10°.
0,30 г испытуемого образца растворяют в воде Р, дово¬
дят до объема 10,0 мл этим же растворителем и, при не¬
обходимости, фильтруют.
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. 100,0 мг испытуемого об¬
разца диспергируют в подвижной фазе и доводят до объ¬
ема 50,0 мл этим же растворителем.
Раствор сравнения (а). 0,1 мл испытуемого раст¬
вора доводят водой Р до объема 100,0 мл.
Раствор сравнения (Ь). 5 мг ФСО кленбутерола
примеси В растворяют в 10 мл подвижной фазы, прибав¬
ляют 2,5 мл испытуемого раствора и доводят подвижной
фазой до объема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилсилильным
эндкепированным для хроматографии Р с размером
частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 200 объемов аце¬
тонитрила Р, 200 объемов метанола Р и 600 объемов
раствора, приготовленного следующим образом: 3,0 г
натрия декансульфоната Р и 5,0 г калия дигидрофос¬
фата Р растворяют в 900 мл воды Р, доводят кисло¬
той фосфорной разведенной Р до рН 3,0 и разводят
водой Р до объема 1000 мл;
- скорость подвижной фазы: 0,5 мл/мин;
- спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: 5 мкл;
- время хроматографирования: 1,5-кратное время
удерживания кленбутерола.
Время удерживания: кленбутерол — около 29 мин.
Пригодность хроматографической системы.
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками примеси
В и кленбутерола.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р (не более 0,1 %): на хро¬
матограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В, С, й, Е и Р, не должны
превышать площадь основного пика на хроматограмме
раствора сравнения (а);
- любая другая примесь (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площадь любого пика,
кроме основного и пиков примесей А, В, С, О, Е и Р, не
должна превышать площадь основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех пи¬
ков, кроме основного, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-неучитываемый предел (0,05%): на хромато¬
грамме испытуемого раствора не учитывают пики с пло¬
щадью менее 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а).
Вода (2.5.12). Не более 1,0 %. Определение прово¬
дят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13)
Испытуемый образец должен выдерживать требования
статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мг
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
Клиндамицина фосфат
539
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 31,37 мг С12Н18С12М2О НС1.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
A. Р1 = Н, Р2 = С1:4-Амино-3,5-дихлорбензальдегцд.
B. Р1 = СН -1\1Н-С(СН3)3, К2 = С1: 1-(4-Амино-3,5-
дихлорфенил)-2-[(1,1-диметилэтил)амино]этанон.
C. Р1 = СН3, Р2 = С1:1-(4-Амино-3,5-дихлорфенил)-
этанон.
й. Р1 = СИ3, Н2 = Н: 1-(4-Аминофенил)этанон.
Е. Р1 = СН2Вг, Р2 = С1: 1-(4-Амино-3,5-дихлор-
фенил)-2-бромэтанон.
Р. (1 Я5)-1-(4-Амино-3-бром-5-хлорфенил)-2-[(1,1-
диметилэтил)амино]этанол.
07/2016:0996
КЛИНДАМИЦИНА ФОСФАТ
СНпйатуат' рЬозрЬаз
СиЫОАМУСШ РН08РНАТЕ
[24729-96-2]
ОПРЕДЕЛЕНИЕ
Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4/?)-1-
метил-4-пропилпирролидин-2-ил]карбонил]ами-но]-1-
тио-1_-/лрео-а-0-галак/тю-октопиранозид-2-дигидро-
фосфат.
Полусинтетический продукт, полученный из продук¬
та ферментации.
Содержание: не менее 96,0 % и не более 102,0 % (в
пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Слегка гигро¬
скопичен.
Легко растворим в воде, очень мало растворим
в 96 % спирте, практически нерастворим в метилен-
хлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО клиндамицина фосфата.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то в одну про¬
бирку помещают 50 мг испытуемого образца, в другую—
50 мг ФСО клиндамицина фосфата. В каждую пробирку
прибавляют 0,2 мл воды Р и нагревают до полного раст¬
ворения. Выпаривают при пониженном давлении досуха
и сушат при температуре от 100 °С до 105 °С в течение
2 ч. Используют полученные сухие остатки для получе¬
ния новых спектров.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого образца
растворяют в метаноле Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (а). 20 мг ФСО клиндамицина
фосфата растворяют в метаноле Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО линкомицина ги¬
дрохлорида растворяют в 5 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикагеля Н Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты пла¬
стинки.
Высушивание: при температуре от 100 °С до 105 °С
в течение 30 мин.
Проявление: пластинку опрыскивают раствором
1 г/л калия перманганата Р
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два основных
пятна.
Результаты: на хроматограмме испытуемого раст¬
вора обнаруживается пятно, соответствующее по распо¬
ложению, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
C. 10 мг испытуемого образца растворяют в 2 мл
кислоты хлористоводородной разведенной Р, нагрева¬
ют на водяной бане в течение 3 мин, прибавляют 4 мл
раствора натрия карбоната Р и 1 мл раствора 20 г/л
натрия нитропруссида Р. Аналогично готовят раствор
сравнения, используя ФСО клиндамицина фосфата
вместо испытуемого образца. Окраска испытуемого
раствора соответствует окраске раствора сравнения.
О. К 0,1 г испытуемого образца прибавляют смесь
из 5 мл раствора натрия гидроксида концентрирован¬
ного Р и 5 мл воды Р и кипятят с обратным холодиль¬
ником в течение 90 мин. Охлаждают, прибавляют 5 мл
кислоты азотной Р и экстрагируют тремя порциями,
по 15 мл каждая, метиленхлорида Р. Нижний слой от¬
брасывают, верхний слой фильтруют через бумажный
фильтр. Фильтрат дает реакцию (Ь) на фосфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,00 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р (при
необходимости при слабом нагревании). Охлаждают
и доводят водой, свободной от углерода диоксида, Р
до объема 25,0 мл.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен быть
бесцветным.
70*. Зак. 1060.
540
Государственная фармакопея Республики Беларусь
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. 30,0 мг испытуемого образ¬
ца растворяют в подвижной фазе А и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 30,0 мг ФСО клиндамицина
фосфата растворяют в подвижной фазе А и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фазой
А до объема 10,0 мл.
Раствор сравнения (с). 3,0 мг ФСО клиндамицина
фосфата для проверки пригодности хроматографи¬
ческой системы (содержит примеси В, Е, Р, СВ, I, <3, К и
1_) растворяют в подвижной фазе А и доводят до объема
1.0 мл этим же растворителем.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилильным
эндкепированным для хроматографии Р с размером ча¬
стиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 — раст¬
вор 13,6 г/л калия дигидрофосфата Р, пред¬
варительно доведенный до рН 6,0 раствором
450 г/л калия гидроксида Р (21:79, об/об);
- подвижная фаза В: раствор 13,6 г/л калия ди¬
гидрофосфата Р, предварительно доведенный
до рН 6,0 раствором 450 г/л калия гидроксида Р
- ацетонитрил Р1 (40:60, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—13
100
0
13—18
100 —> 50
0
1
сл
о
18—39
50
50
- скорость подвижной фазы: 1,1 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Идентификация пиков примесей: идентифициру¬
ют пики примесей В, Е, Р, О, I, 3, К и 1_, используя хро¬
матограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО клиндамицина фосфата для про¬
верки пригодности хроматографической системы.
Относительное удерживание (по отношению к
клиндамицину фосфату, время удерживания — около
12 мин): примесь Р — около 0,15; примесь О — около
0,19; примесь I — около 0,34; примесь В — около 0,45;
примесь — около 0,64; примесь 3 — около 1,20; при¬
месь Е — около 1,73; примесь К — около 1,90.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 2,0 между пиками при¬
месей Р и О.
Расчет процентных содержаний:
- для каждой примеси — используют концентра¬
цию клиндамицина фосфата в растворе сравнения (Ь).
Предельное содержание примесей:
- примеси В, Ь: не более 1,0 % для каждой при¬
меси;
- примеси Е, Р: не более 0,5 % для каждой при¬
меси;
- примеси О, /, 3, К: не более 0,2 % для каждой
примеси;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 2,0 %;
- учитываемый предел: 0,05 %.
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,6 ЕД/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дополнительной процедуры удале¬
ния бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в разделе «Сопутствующие примеси», со следующи¬
ми изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- фактор асимметрии: не более 3,0 для пика
клиндамицина фосфата.
Содержание С18Н34С1М2ОвРЗ в процентах рассчи¬
тывают с учетом содержания клиндамицина фосфата
в ФСО клиндамицина фосфата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Стерильная субстанция — в стерильном контей¬
нере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: В, Е, Р, С, 1, 3, К, Е
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, С, О, Н.
А. Метил-6,8-дидезокси-6-[[[(25,4Я)-1 -метил-4-
пропилпирролидин-2-ил]карбонил]амино]-1-тио-0-
эритро-а-О-галакто-октотраногт (линкомицин).
В. Метил-7-хлор-6,7,8-тридезокси-6-[[[(2В,4Я)-4-
этил-1-метилпирролидин-2~ил]карбонил]амино]-2-0-
фосфоноИ-тио-Ь/лрео-а-О-галзшю-октопиранозид
(клиндамицина В 2-фосфат).
И
Клиндамицина фосфат
541
С. Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4Я)-1-
метил-4-пропилпирролидин-2-ил]карбонил]амино]-3-
0-фосфоно-1-тио-1_-трео-а-0-галакл7о-октопиранозид
(клиндамицина 3-фосфат).
О. Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4#)-1-
метил-4-пропилпирролидин-2-ил]карбонил]амино]-4-
0-фосфоно-1-тио-1_-л7рео-а-0-галаклго-октопиранозид
(клиндамицина 4-фосфат).
Е. Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4Я)-1-
метил-4-пропилпирролидин-2-ил]карбонил]амино]-1-
тио-1_-л7рео-а-0-галакл7о-октопиранозид (клиндами-
цин).
Р. Метил-6,8-дидезокси-6-[[[(25,4Я)-1-метил-4-
пропилпирролидин-2-ил]карбонил]амино]-2-0-фосфоно-
1 -тио-О-эритро-а-О-галакто-октопиранозид (линко-
мицина 2-фосфат).
(3. Метил-6,8-дидезокси-2,4-0(гидроксифосфорил)-
6-[[[(25,4/?)-1-метил-4-пропилпирролидин-2-ил]карбо-
нил]амино]-1-тио-0-эрш7ро-а-0-гала/сл?о-окгопиранозид
(2,4-фосфатидиллинкомицин).
Н. Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4Я)-1-
метил-4-пропилпирролидин-2-ил]карбонил]амино]-2,3-
ди-0-фосфоно-1-тио-1_-л?рео-а-0-галакл7о-октопира-
нозид (клиндамицина 2,3-бифосфат).
I. Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4Я)-1-
метил-4-пропилпирролидин-2-ил]карбонил]амино]-2,4-
ди-О-фосфоно-1-тио-Ьл7рео-а-0-галахл7О-октопира-
нозид (клиндамицина 2,4-бифосфат).
3. Метил-7-хлор-6,7,8-тридезокси-6-[[[(2$)-1 -метил-
4-пропилиденпирролидин-2-ил]карбонил]амино]-2-0-
фосфоно-1-тио-1_-л7рео-а-0-галаклю-октопиранозид
(пропилиденовый аналог клиндамицина 2-фосфата).
К. 2,2'-оксибис(гидроксифосфорил)бис[метил-7-
хлор-6,7,8-тридезокси-6-[[[(25,4/?)-1-метил~4-пропил-пир-
ролидин-2-ил]карбонил]амино]-1-тио-1_.-/7)рео-а-0-галак-
/ло-октопиранозид (диклиндамицина пирофосфат).
Ь. Метил-7-хлор-6,7,8-тридезокси-6-[[[(25,4Я)-1-
метил-4-пропилпирролидин-2-ил]карбонил]амино]-2-
О-фосфоно-1 -тмо-О-эритро-а-О-галакто-окгопмранозмц
(7-эпиклиндамицина 2-фосфат).
542
Гэсударственная фармакопея Республики Беларусь
07/2016:1191
ИСПЫТАНИЯ
КЛОЗАПИН
С1о1артит
С1-02АРШЕ
С18Н19С1Ы4 М.м. 326,8
[5786-21-0]
ОПРЕДЕЛЕНИЕ
8-Хлор-11 -(4-метилпиперазин-1 -ил)-5Н-дибензо[Ь,е]
[1,4]диазепин.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, растворим в 96 % спирте, рас¬
творим в разведенной кислоте уксусной.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14): от 182 °С до
186 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО клозапина #или спектр, пред¬
ставленный на рисунке #1191.-1#.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Вода Р — метанол Р2
(20:80, об/об).
Раствор А. 2,04 г калия дигидрофосфата Р
растворяют в 1000 мл воды Р и доводят до рН 2,4±0.05
кислотой фосфорной разведенной Р.
Испытуемый раствор. 75 мг испытуемого об¬
разца растворяют в 80 мл метанола Р2 и доводят во¬
дой Рцо объема 100 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
10,0 мл. 1,0 мл полученного раствора доводят смесью
растворителей до объема 100,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО клозапина для идентификации пиков (содер¬
жит примеси А, В, С и О) растворяют в 1,0 мл смеси
растворителей.
Условия хроматографирования:
-колонка длиной 0,125 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил для хрома¬
тографии Р — метанол Р2 — раствор А (1:1:8
об/об/об);
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р — метанол Р2 — раствор А (4:4:2
об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-4
100
0
4—24
100-0
о
о
т
о
24—29
0
100
Рисунок #1191.-1. Инфракрасный спектр ФСО клозапина
Клонидина гидрохлорид
543
- скорость подвижной фазы: 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 257 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифициру¬
ют пики примесей А, В, С и й, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО клозапина для идентификации пиков.
Относительное удерживание (по отношению к
клозапину, время удерживания — около 11 мин): при¬
месь С — около 0,9; примесь О — около 1,1; примесь А
— около 1,6, примесь В — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиком примеси
С и пиком клозапина;
-хроматограмма раствора сравнения (Ь) должна
соответствовать хроматограмме, прилагаемой к ФСО
клозапина для идентификации пиков.
Предельное содержание примесей (для расчета со¬
держания примесей умножают площади пиков на соот¬
ветствующие поправочные коэффициенты: для примеси
О — 2,7):
-примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- примеси В и О (не более 0,2 %): на хроматограм¬
ме испытуемого раствора площади пиков, соответству¬
ющих примесям В и О, не должны превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь С (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси С, не должна превышать 3-кратную площадь ос¬
новного пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей А,
В, С и О, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
-сумма примесей (не более 0,6%): на хромато¬
грамме испытуемого раствора сумма площадей всех пи¬
ков, кроме основного, не должна превышать 6-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-неучитываемый предел (0,05%): на хромато¬
грамме испытуемого раствора не учитывают пики с пло¬
щадью менее 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а).
Тяжелые металлы {2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эталон
готовят с использованием 2 мл эталонного раствора
свинца (10 ррт РЬ) Я
Потеря в массе при высушивании (2.2.32). Не бо¬
лее 0,5 %. 1,000 г испытуемого образца сушат при тем¬
пературе 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требования
статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,34 мг С18Н19С11Ч4.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. 8-Хлор-5,10-дигидро-11Н-дибензо[Ь,е][1,4]-
диазепин-11-он.
В. 11,11 '-(Пиперазин-1,4-диил)бис(8-хлор-5Н-
дибензо[Ь,е][1,4]диазепин).
С. 8-Хлор-11-(пиперазин-1-ил)-5Н-дибензо[Ь,е]-
[1,4]диазепин.
О. 1 -[2-[(2-Амино-4-хлорфенил)амино]бензоил]-
4-метилпиперазин.
07/2016:0477
КЛОНИДИНА ГИДРОХЛОРИД
С1оп1сИт ЬудгосЫопс1ит
С1.0ЫЮШЕ НУОНОСШОЕЮЕ
С9Н9С12Ы3 • НС1 М.м. 266,6
[4205-91-8]
ОПРЕДЕЛЕНИЕ
2,6-Дихлор-Л/-(имидазолидин-2-илиден)анилина
гидрохлорид. #Клофелин#.
544
Гэсударственная фармакопея Республики Беларусь
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Растворим в воде и в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем.
Диапазон длин волн: от 245 нм до 350 нм.
Максимумы поглощения: при 272 нм и при 279 нм.
Точка перегиба: при 265 нм.
Удельный показатель поглощения в максимуме:
- при 272 нм — около 18;
- при 279 нм — около 16.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО клонидина гидрохлорида #или
спектр, представленный на рисунке #0477.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения. 5 мг ФСО клонидина ги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: кислота уксусная ледяная Р —
бутанол Р — вода Р (10:40:50, об/об/об); выдержива¬
ют до расслоения, верхний слой фильтруют и полу¬
ченный фильтрат используют в качестве подвижной
фазы.
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
калия йодовисмутата Р2, высушивают на воздухе
в течение 1 ч, снова опрыскивают раствором калия
йодовисмутата Р2 и немедленно опрыскивают рас¬
твором 50 г/л натрия нитрита Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3. “/).
ИСПЫТАНИЯ
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)7.
рН (2.2.3). От 4,0 до 5,0. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 50 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Рисунок #0477.-1. Инфракрасный спектр ФСО клонидина гидрохлорида
Клопидогреля гидросульфат
545
Раствор сравнения (Ь). 5 мг ФСО клонидина при¬
меси В растворяют в 2 мл ацетонитрила Р и доводят
подвижной фазой А до объема 5 мл. К 1 мл получен¬
ного раствора прибавляют 1 мл испытуемого раст¬
вора и доводят подвижной фазой А до объема 10 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 3,0 мм, заполненная силикагелем пропилси-
лильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: 4 г калия дигидрофосфа¬
та Р растворяют в 1000 мл воды Р и доводят до
рН 4,0 кислотой фосфорной Р\
- подвижная фаза В: подвижная фаза А — аце¬
тонитрил Р1 (25:75, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0
90
10
0—15
90 — 30
о
т
о
15—15,1
30 — 90
70 — 10
сл
I
о
90
10
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 5 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5 между пиками клони¬
дина и примеси В.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 70 мл
96 % спирта Р и титруют 0,1 М раствором натрия ги¬
дроксида этанольным потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида эта-
нольного Р соответствует 26,66 мг С9Н9Су\13НС1.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С.
А. 1-Ацетилимидазолидин-2-он.
В. 1 -Ацетил-2-[(2,6-дихлорфенил)амино]-4,5-
дигидро-1 Я-имидазол.
С1
С. 2,6-Дихлоранилин.
07/2016:2531
КЛОПИДОГРЕЛЯ ГИДРОСУЛЬФАТ
С1ор1с1одгеН ЬубгодепозиКаз
С1-ОРЮООКЕ1. НУОКОвЕЫ 8Ш-РАТЕ
С1вН16С1М023 ■ Н,50. М.м. 419,9
[120202-66-6]
ОПРЕДЕЛЕНИЕ
Метил(25)-(2-хлорфенил)[6,7-дигидротиено[3,2-с]-
пиридин-5(4/7)-ил]ацетата сульфат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Легко растворим в воде и в метаноле, практиче¬
ски нерастворим в циклогексане.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Проводят определение подлинности (идентифи¬
кацию) испытаниями А, В, й или В, С, О.
А. Удельное оптическое вращение (2.2.7). От
+54,0 до +58,0 (в пересчете на безводное вещество).
546
Гэсударственная фармакопея Республики Беларусь
0,250 г испытуемого образца растворяют в метано¬
ле Р и доводят этим же растворителем до объема
25,0 мл.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО клопидогреля гидросульфата
#или спектр, представленный на рисунке #2531 .-1#.
Если спектры отличаются, то испытуемый обра¬
зец и ФСО клопидогреля гидросульфата растворяют
по отдельности в этаноле Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров (испытуемый образец может прилипнуть к
поверхности используемой посуды для выпаривания).
C. Испытуемый образец выдерживает испытание
«Энантиомерная чистота», как указано в разделе «Ис¬
пытания».
О. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят этим же растворителем
до объема 20,0 мл.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод I). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Энантиомерная чистота. Жидкостная хромато¬
графия (2.2.29). Используют метод нормализации.
Испытуемый раствор. 0,1 г испытуемого образ¬
ца растворяют в 25,0 мл этанола Р и доводят гепта¬
ном Р до объема 50,0 мл.
Раствор сравнения. 10 мг ФСО клопидогреля
для проверки пригодности хроматографической си¬
стемы (содержит примеси В и С) растворяют в 2,5 мл
этанола Р и доводят гептаном Р до объема 5,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем 03 для хи¬
ральных разделений Р с размером частиц 10 мкм;
- подвижная фаза: этанол Р — гептан Р
(15:85, об/об);
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,25-кратное вре¬
мя удерживания клопидогреля.
Идентификация пиков примесей: идентифици¬
руют пики примесей В и С, используя хроматограмму
раствора сравнения и хроматограмму, прилагаемую к
ФСО клопидогреля для проверки пригодности хро¬
матографической системы.
Относительное удерживание (по отношению к
клопидогрелю, время удерживания — около 18 мин):
примесь С — около 0,6; примесь В — около 0,7.
Пригодность хроматографической системы.
раствор сравнения:
- разрешение: не менее 2,0 между пиками при¬
месей С и В;
- отношение сигнал/шум: не менее 20 для пика
примеси С.
Предельное содержание примесей:
- примесь С: не более 0,5 %.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Подвижная фаза А —
ацетонитрил Р1 (40:60, об/об).
Испытуемый раствор. 65 мг испытуемого об¬
разца растворяют в смеси растворителей и доводя'
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). 5 мг ФСО клопидогреля
примеси А растворяют в смеси растворителей и дово¬
дят этой же смесью растворителей до объема 25,0 мг
Волновое число (см'1)
Рисунок #2531 .-1. Инфракрасный спектр ФСО клопидогреля гидросульфата
Клопидогреля гидросульфат
547
Раствор сравнения (Ь). 32 мг ФСО клопидогреля
для проверки пригодности хроматографической си¬
стемы (содержит примеси В и С) растворяют в смеси
растворителей, прибавляют 0,5 мл раствора сравне¬
ния (а) и доводят этой же смесью растворителей до
объема 5,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: смесь из метанола Р2 и
раствора 0,96 г/л натрия пентансульфоната моно¬
гидрата Р, доведенного кислотой фосфорной Р до
рН 2,5 (5:95, об/об);
- подвижная фаза В: метанол Р2 — ацетони¬
трил Р1 (5:95, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
3-48
48—68
89.5
89,5 — 31,5
31.5
10.5
10,5 — 68,5
68.5
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО клопидогреля для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
кпопидогрелю, время удерживания — около 25 мин):
примесь А— около 0,4, примесь В — около 1,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-коэффициент разделения пиков: не менее 10
(Нр — высота пика примеси В относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик клопидогреля и
пик примеси В).
Предельное содержание примесей:
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,00 г испытуемого образца. Растворитель
заменяют после каждого титрования.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,160 г испытуемого образца растворяют в сме¬
си из 10 мл ацетона Р, 10 мл метанола Р и 30 мл
воды Р и титруют 0,1 М раствором натрия гидрокси¬
да потенциометрически (2.2.20). В течение титрова¬
ния может образоваться осадок.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 20,99 мг С16Н16С1М025-Н2504. .
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О.
А. (25)-(2-Хлорфенил)[6,7-дигидротиено[3,2-с]-
пиридин-5(4Н)-ил]уксусная кислота.
В. Метил(25)-(2-хлорфенил)[4,7-дигидротиено-
[2,3-с]пиридин-6(5Н)-ил]ацетат.
548
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
1-[(2-Хпорфенил)дифенилметил]-1/-/-имидазол.
Содержание: не менее 98,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
С. Метил(2/?)-(2-хлорфенил)[6,7-дигидротиено-
[3,2-с]пиридин-5(4/-/)-ил]ацетат.
О. Метил(2Я)-(2-хлорфенил)[(2$)-(2-хлорфенил)-
[6,7-дигидротиено[3,2-с]пиридин-5(4Н)-ил]ацетилокси]-
ацетат.
07/2016:0757
КЛОТРИМАЗОЛ
С1о1птаю1ит
аОТШМА201-Е
Белый или бледно-желтый кристаллический по¬
рошок.
Практически нерастворим в воде, растворим в
96 % спирте и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Температура плавления (2.2.14): от 141 °С до
145 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО клотримазола #или спектр,
представленный на рисунке #0757.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 5 мл этим же растворителем.
Раствор сравнения. 50 мг ФСО клотримазола
растворяют в 96% спирте Р и доводят до объема
5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР2ЫР
Подвижная фаза: раствор аммиака концентри¬
рованный Р1 — пропанол Р — толуол Р (0,5:10:90.
об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Волновое число (см1)
Рисунок #0757.-1. Инфракрасный спектр ФСО клотримазола
Клотримазол
549
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в ацетонитриле Р1 и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят ацетонитрилом Р1 до объема
100.0 мл. 1,0 мл полученного раствора доводят аце¬
тонитрилом Р1 до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО клотримазола для идентификации пиков (со¬
держит примеси А, В и Р) растворяют в 1,0 мл ацето¬
нитрила Р1.
Раствор сравнения (с). 5,0 мг ФСО имидазола
(примесь й) и 5,0 мг ФСО клотримазола примеси Е
растворяют в ацетонитриле Р1 и доводят до объема
100.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят ацетонитрилом Р1 до объема
25.0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: 1,0 г калия дигидрофос¬
фата Р и 0,5 г тетрабутиламмония гидро¬
сульфата Р1 растворяют в воде Р и доводят до
объема 1000 мл этим же растворителем;
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
75
25
3—25
75 —* 20
25 —► 80
25—30
20
80
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
клотримазолу, время удерживания — около 12 мин):
примесь й — около 0,1; примесь Р — около 0,9; при¬
месь В — около 1,1; примесь Е — около 1,5; примесь
А—около 1,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси Р и клотримазола;
-полученная хроматограмма аналогична хро¬
матограмме, прилагаемой к ФСО клотримазола для
идентификации пиков.
Предельное содержание примесей:
- примеси А и В (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А и В, не должны превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примеси О и Е (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям О и Е, не должны превышать
площади соответствующих пиков на хроматограмме
раствора сравнения (с);
-примесь Р (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси Р, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, Р, Е и Р, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 80 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной до перехода окраски от ко¬
ричневато-желтой до зеленой, используя в качестве
индикатора 0,3 мл раствора нафтолбензеина Р
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 34,48 мг С22Н17С1М2.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, О, Е, Е
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. Р = ОН, Р' = С6Н5: (2-Хлорфенил)дифенилме¬
танол.
С. Р = С1, Р' = С6Н5:1-Хлор-2-(хлордифенилметил)-
бензол.
Е. Р + Р' = О: (2-Хлорфенил)фенилметанон (2-хлор-
бензофенол).
550
Гэсударственная фармакопея Республики Беларусь
В. Р = С1: 1-[(4-Хлорфенил)дифенилметил]-1Н-
имидазол.
Р. К = Н: 1-(Трифенилметил)-1Н-имидазол (дез-
хлорклотримазол).
о
О. Имидазол.
КОДЕИН
Собетит
СООЕ/А/Е
07/2016:0076
Н20
М.м. 317,4
[6059-47-8]
ОПРЕДЕЛЕНИЕ
7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба-ол моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Растворим в кипящей воде, легко растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О, Е.
A. Температура плавления (2.2.14): от 155 °С до
159 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. К 2,0 мл раствора 5, при¬
готовленного как указано в разделе «Испытания», при¬
бавляют 50 мл воды Р, 10 мл 1 М раствора натрия
гидроксида и доводят водой Р до объема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм.
Максимум поглощения: при 284 нм.
Удельный показатель поглощения в максимуме:
около 50 (в пересчете на сухое вещество).
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: высушенная субстанция в дис¬
ках с калия бромидом Р.
Сравнение: ФСО кодеина #или спектр, представ¬
ленный на рисунке #0076.-1#.
й. К 10 мг испытуемого образца прибавляют
1 мл кислоты серной Р, 0,05 мл раствора железа
(III) хлорида Р2 и нагревают на водяной бане. По¬
является синее окрашивание. Прибавляют 0,05 мл
Волновое число (см*1)
Рисунок #0076.-1. Инфракрасный спектр ФСО кодеина
Кодеин
551
кислоты азотной Р. Окрашивание раствора изме¬
няется на красное.
Е. Испытуемый образец дает реакцию на алка¬
лоиды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 50 мг испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 10,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
-142 до -146 (в пересчете на сухое вещество). 0,50 г
испытуемого образца растворяют в 96% спирте Р и
доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца и 0,100 г натрия октансульфоната Р раство¬
ряют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО кодеина при¬
меси А растворяют в подвижной фазе и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 20,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
5,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (д). К 0,25 мл испытуемого
раствора прибавляют 2,5 мл раствора сравнения (а).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- подвижная фаза: 1,08 г натрия октансульфо¬
ната Р растворяют в смеси из 20 мл кислоты уксус¬
ной ледяной Р и 250 мл ацетонитрила Р и доводят
водой Р до объема 1000 мл;
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 245 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 10-кратное вре¬
мя удерживания кодеина.
Относительное удерживание (по отношению
к кодеину, время удерживания — около 6 мин): при¬
месь В — около 0,6; примесь Е — около 0,7; при¬
месь А— около 2,0; примесь С — около 2,3; при¬
месь О — около 3,6.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 3 между пиками кодеина
и примеси А.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 0,25):
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В, С, О, Е (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям В, С, Э и Е, не должны пре¬
вышать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О и Е, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (с);
- сумма примесей, кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пика
примеси А, не должна превышать 10-кратную пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
менее 4,0 % и не более 6,0 %. 1,000 г испытуемого об¬
разца сушат при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в кис¬
лоте уксусной безводной Р, прибавляют 20 мл диок-
сана Р и титруют 0,1 М раствором кислоты хлорной,
используя в качестве индикатора 0,05 мл раствора
кристаллического фиолетового Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 29,94 мг С18Н211\Ю3.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Р, С.
А. 7,8-Дидегидро-4,5а-эпокси-3,6а-диметокси-17-
метилморфинан (метилкодеин).
/
552
Гэсударственная фармакопея Республики Беларусь
В. 7,8-Дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а-диол (морфин).
С. 7,7',8,8'-Тетрадегидро-4,5а:4',5'а-диэпокси-3,3'-
диметокси-17,17'-диметил-2,2'-биморфинанил-6а,6'а-
диол (димер кодеина).
й. 7,8-Дидегидро-2-[(7,8-дидегидро-4,5а-эпокси-
6а-гидрокси-17-метилморфинан-3-ил)окси]-4,5а-эпокси-
З-метокси-17-метилморфинан-6а-ол (3-0-(кодеин-2-
ил)морфин).
Е. 7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба, 10-диол.
Р. 7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба, 14-диол.
(В. 6,7,8,14-Тетрадегидро-4,5а-эпокси-3,6-диме-
токси-17-метилморфинан (тебаин).
07/2016:0074
КОДЕИНА ФОСФАТ ГЕМИГИДРАТ
Сос/е/л/ рЬозрЬаз Ьет/Нус/псиз
СООЕШЕ РНОЗРНАТЕ НЕМ1НУОРАТЕ
с18н21мо3 • Н3Р04 • %н2о
[41444-62-6]
Н3Р04 • 1/2 Н20
М.м. 406,4
ОПРЕДЕЛЕНИЕ
7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-6а-ола фосфат гемигидрат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или мелкие бесцветные кристаллы.
Легко растворим в воде, мало растворим или
очень мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е, Р.
Вторая идентификация: А, С, О, Е, Р, С.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 1,0 мл раствора 3, при¬
готовленного как указано в разделе «Испытания», до¬
водят водой Р до объема 100,0 мл. К 25,0 мл получен¬
ного раствора прибавляют 25 мл воды Р, 10 мл 1 М
раствора натрия гидроксида и доводят водой Р до
объема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм.
Максимум поглощения: при 284 нм.
Удельный показатель поглощения в максимуме:
около 38 (в пересчете на сухое вещество).
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 0,20 г испытуемого образца
растворяют в 4 мл воды Р, прибавляют 1 мл смеси
из равных объемов раствора натрия гидроксида
концентрированного Р и воды Р, инициализируют
кристаллизацию, потирая внутренние стенки про¬
бирки стеклянной палочкой, и охлаждают в ледяной
бане. Полученный осадок промывают водой Р и вы¬
сушивают при температуре от 100 °С до 105 °С. Сухой
остаток используют для приготовления дисков с калия
бромидом Р.
Сравнение: эталонный спектр кодеина по Евро¬
пейской Фармакопее #или спектр, представленный на
рисунке #0074.-1#.
C. 0,20 г испытуемого образца растворяют в 4 мл
воды Р, прибавляют 1 мл смеси из равных объемов
раствора натрия гидроксида концентрированного Р
и воды Р, инициализируют кристаллизацию, потирая
внутренние стенки пробирки стеклянной палочкой, и
охлаждают в ледяной бане. Полученный осадок про-
4
Кодеина фосфат гемигидрат
553
Рисунок #0074.-1. Инфракрасный спектр кодеина
мывают водой Р и высушивают при температуре от
100 °С до 105 °С. Температура плавления (2.2.14) по¬
лученного остатка: от 155 °С до 159 °С.
O. К 10 мг испытуемого образца прибавляют 1 мл
кислоты серной Р, 0,05 мл раствора железа (III) хло¬
рида Р2 и нагревают на водяной бане. Появляется си¬
нее окрашивание. Прибавляют 0,05 мл кислоты азот¬
ной Р. Окрашивание раствора изменяется на красное.
Е. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
P. Раствор 3 дает реакцию (а) на фосфаты (2.3.1).
О. Испытуемый образец дает реакцию на алка¬
лоиды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,00 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 25,0 мл этим же растворителем.
рН (2.2.3). От 4,0 до 5,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От -98
до -102 (в пересчете на сухое вещество). 5,0 мл раст¬
вора 3 доводят водой Р до объема 10,0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца и 0,100 г натрия октансульфоната Р раство¬
ряют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО кодеина при¬
меси А растворяют в подвижной фазе и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 20,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
5,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (д). К 0,25 мл испытуемого
раствора прибавляют 2,5 мл раствора сравнения (а).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
-подвижная фаза: 1,08 г натрия октансульфо¬
ната Р растворяют в смеси из 20 мл кислоты уксус¬
ной ледяной Р и 250 мл ацетонитрила Р и доводят
водой Р до объема 1000 мл;
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 245 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 10-кратное вре¬
мя удерживания кодеина.
Относительное удерживание (по отношению к
кодеину, время удерживания — около 6 мин): примеси
В и Е — около 0,7; примесь А — около 2,0; примесь
С — около 2,3; примесь О — около 3,6.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 3 между пиками кодеина
и примеси А.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 0,25):
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
554
Гэсударственная фармакопея Республики Беларусь
- сумма примесей В и Е (не более 0,4 %): на
хроматограмме испытуемого раствора сумма пло¬
щадей пиков, соответствующих примесям В и Е, не
должна превышать 4-кратную площадь основного
пика на хроматограмме раствора сравнения (с);
- примеси С, О (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, со¬
ответствующих примесям С и О, не должны превы¬
шать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А, В, С, О и Е, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- сумма примесей, кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и
пика примеси А, не должна превышать 10-кратную
площадь основного пика на хроматограмме раст¬
вора сравнения (с);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (с).
Сульфаты (2.4.13). Не более 0,1%. 5 мл
раствора 5 доводят водой дистиллированной Р до
объема 20 мл. 15 мл полученного раствора долж¬
ны выдерживать испытание на сульфаты.
Потеря в массе при высушивании (2.2.32).
Не менее 1,5 % и не более 3,0 %. 1,000 г испытуе¬
мого образца сушат при температуре 105 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в смеси
из 10 мл кислоты уксусной безводной Р и 20 мл диок-
сана Р и титруют 0,1 М раствором кислоты хлорной,
используя в качестве индикатора 0,05 мл раствора
кристаллического фиолетового Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 39,74 мг С18Н21М03 Н3Р04.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): Р, С.
А. 7,8-Дидегидро-4,5а-эпокси-3,6а-диметокси-17-
метилморфинан (метилкодеин).
В. 7,8-Дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а-диол (морфин).
С. 7,7',8,8'-Тетрадегидро-4,5а:4',5'а-диэпокси-3,3 -
диметокси-17,17'-диметил-2,2'-биморфинанил-6а,6'а-
диол (димер кодеина).
О. 7,8-Дидегидро-2-[(7,8-дидегидро-4,5а-эпокс**-
6а-гидрокси-17-метилморфинан-3-ил)оксиН,5а-эпокс^
З-метокси-17-метилморфинан-6а-ол (3-0-(кодеин-2-
ил)морфин).
Е. 7,8-Дидегидро-4,5а-эпокси-3-метокси-1 ~-
метилморфинан-6а,10-диол.
Р. 7,8-Дидегидро-4,5а-эпокси-3-метокси-'~-
метилморфинан-6а,14-диол.
Кодеина фосфат сесквигидрат
555
С. 6,7,8,14-Тетрадегидро-4,5а-эпокси-3,6-димето-
кси-17-метилморфинан (тебаин).
07/2016:0075
КОДЕИНА ФОСФАТ СЕСКВИГИДРАТ
Содет/ рЬозрЬаз зездиИ1ус1псиз
СОИЕШЕ РНОЗРНАТЕ
ЗЕ80ШНУ0ЯАТЕ
С18Н21Ы03 • Н3Р04 ■ 1 'ЛН20 М.м. 424,4
[5913-76-8]
ОПРЕДЕЛЕНИЕ
7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-6а-ола фосфат сесквигидрат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или мелкие бесцветные кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е, Е
Вторая идентификация: А, С, О, Е, Р, С.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 1,0 мл раствора 5, при¬
готовленного как указано в разделе «Испытания», до¬
водят водой Р до объема 100,0 мл. К 25,0 мл получен¬
ного раствора прибавляют 25 мл воды Р, 10 мл 1 М
раствора натрия гидроксида и доводят водой Р до
объема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм.
Максимум поглощения: при 284 нм.
Удельный показатель поглощения в максимуме:
около 38 (в пересчете на сухое вещество).
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 0,20 г испытуемого образца раст¬
воряют в 4 мл воды Р, прибавляют 1 мл смеси из рав¬
ных объемов раствора натрия гидроксида концентри¬
рованного Р и воды Р, инициализируют кристаллиза¬
цию, потирая внутренние стенки пробирки стеклянной
палочкой, и охлаждают в ледяной бане. Полученный
осадок промывают водой Р и высушивают при темпера¬
туре от 100 °С до 105 °С. Сухой остаток используют для
приготовления дисков с калия бромидом Р.
Сравнение: эталонный спектр кодеина по Евро¬
пейской Фармакопее #или спектр, представленный на
рисунке #0075.-1#.
C. 0,20 г испытуемого образца растворяют в 4 мл
воды Р, прибавляют 1 мл смеси из равных объемов
Рисунок #0075.-1. Инфракрасный спектр кодеина
556
Гэсударственная фармакопея Республики Беларусь
раствора натрия гидроксида концентрированного Р
и воды Р, инициализируют кристаллизацию, потирая
внутренние стенки пробирки стеклянной палочкой, и
охлаждают в ледяной бане. Полученный осадок про¬
мывают водой Р и высушивают при температуре от
100 °С до 105 °С. Температура плавления (2.2.14) по¬
лученного остатка: от 155 °С до 159 °С.
O. К 10 мг испытуемого образца прибавляют 1 мл
кислоты серной Р, 0,05 мл раствора железа (III) хло¬
рида Р2 и нагревают на водяной бане. Появляется
синее окрашивание. Прибавляют 0,05 мл кислоты
азотной Р. Окрашивание раствора изменяется на
красное.
Е. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
P. Раствор 5 дает реакцию (а) на фосфаты (2.3.7).
С. Испытуемый образец дает реакцию на алка¬
лоиды (2.3.7).
ИСПЫТАНИЯ
Раствор 3. 1,00 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 25,0 мл этим же растворителем.
рН (2.2.3). От 4,0 до 5,0. Измеряют рН раствора 5.
Удельное оптическое вращение (2.2.7). От-98
до -102 (в пересчете на сухое вещество). 5,0 мл раст¬
вора 3 доводят водой Р до объема 10,0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца и 0,100 г натрия октансульфоната Р раство¬
ряют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО кодеина при¬
меси А растворяют в подвижной фазе и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 20,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
5,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (д). К 0,25 мл испытуемого
раствора прибавляют 2,5 мл раствора сравнения (а).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- подвижная фаза: 1,08 г натрия октансульфо¬
ната Р растворяют в смеси из 20 мл кислоты уксус¬
ной ледяной Р и 250 мл ацетонитрила Р и доводят
водой Р до объема 1000 мл;
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 245 нм;
- объем вводимой пробы: 10 мкп;
- время хроматографирования: 10-кратное вре¬
мя удерживания кодеина.
Относительное удерживание (по отношению к ко¬
деину, время удерживания — около 6 мин): примесь В —
около 0,6; примесь Е — около 0,7; примесь А — около
2,0; примесь С — около 2,3; примесь О — около 3,6.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 3 между пиками кодеина
и примеси А.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 0,25):
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В, С, О, Е (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям В, С, О и Е, не должны пре¬
вышать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, О и Е, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с);
- сумма примесей, кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора сум¬
ма площадей всех пиков, кроме основного и пика при¬
меси А, не должна превышать 10-кратную площадь
основного пика на хроматограмме раствора сравне¬
ния (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Сульфаты (2.4.13). Не более 0,1 %. 5 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 20 мл. 15 мл полученного раствора должны вы¬
держивать испытание на сульфаты.
Потеря в массе при высушивании (2.2.32). Не
менее 5,0 % и не более 7,5 %. 0,500 г испытуемого об¬
разца сушат при температуре 105 °С.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в смеси
из 10 мл кислоты уксусной безводной Р и 20 мл диок-
сана Р и титруют 0,1 М раствором кислоты хлорной.
используя в качестве индикатора 0,05 мл раствора
кристаллического фиолетового Р.
1 мг\ 0,1 М раствора кислоты хлорной соответ¬
ствует 39,74 мг С18Н21М03-Н3Р04.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Р, С.
Кокосовое масло рафинированное
557
А. 7,8-Дидегидро-4,5а-эпокси-3,6а-диметокси-17-
метилморфинан (метилкодеин).
В. 7,8-Дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а-диол (морфин).
С. 7,7',8,8'-Тетрадегидро-4,5а:4',5'а-диэпокси-3,3-
диметокси-17,17'-диметил-2,2'-биморфинанил-6а,6'а-
диол (димер кодеина).
О. 7,8-Дидегидро-2-[(7,8-дидегидро-4,5а-эпокси-6а-
гидрокси-17-метилморфинан-3-ил)окси]-4,5а-эпокси-3-
метокси-17-метилморфинан-6а-ол (3-0-(кодеин-2-ил)-
морфин).
Е. 7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба, 10-диол.
Р. 7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба, 14-диол.
О. 6,7,8,14-Тетрадегидро-4,5а-эпокси-3,6-ди-
метокси-17-метилморфинан (тебаин).
07/2016:1410
КОКОСОВОЕ МАСЛО
РАФИНИРОВАННОЕ
Сосо/5 о1еит гаШпа1ит
сосоыит ОН, КЕРШЕй
[8001-31-8]
ОПРЕДЕЛЕНИЕ
Жирное масло, полученное из высушенной твер¬
дой части эндосперма Сосоз писйега Ь. с последую¬
щим рафинированием.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая маслянистая масса.
Практически нерастворимо в воде, легко раство¬
римо в метиленхлориде и петролейном эфире (тем¬
пература кипения: от 65 °С до 70 °С), очень мало рас¬
творимо в 96 % спирте.
Показатель преломления: около 1,449. Опреде¬
ление проводят при температуре 40 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ).
A. Испытуемый образец выдерживает испыта¬
ние «Температура плавления», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Температура плавления (2.2.14). От 23 °С до
26 °С.
Кислотное число (2.5.1). Не более 0,5. Опреде¬
ление проводят из 20,0 г испытуемого образца.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 5,0.
Неомыляемые вещества (2.5.7). Не более
1,0%. Определение проводят из 5,0 г испытуемого
образца.
Щелочные примеси в жирных маслах (2.4.19).
Испытуемый образец должен выдерживать испыта¬
ние на щелочные примеси в жирных маслах.
Состав жирных кислот (2.4.22, метод В). Перед
испытанием кокосовое масло, осторожно нагревая,
расплавляют до однородной жидкости.
Раствор сравнения. 15,0 мг ФСО трикапрои-
на, 80,0 мг ФСО тристеарина, 0,150 г ФСО три-
каприна, 0,200 г ФСО трикаприлина, 0,450 г ФСО
тримиристина и 1,25 г ФСО трилаурина раство¬
ряют в смеси метиленхлорид Р — гептан Р (2:8,
об/об) и доводят до объема 50 мл этой же смесью
растворителей при нагревании при температуре от
45 °С до 50 °С. 2 мл полученного раствора пере¬
носят в пробирку для центрифугирования вмести¬
мостью 10 мл с закручивающейся крышкой и вы¬
паривают растворитель в потоке азота Р. Остаток
растворяют в смеси из 1 мл гептана Р и 1 мл ди-
метилкарбоната Р и интенсивно перемешивают
при осторожном нагревании (при температуре от
50 °С до 60 °С). К еще теплому раствору прибав¬
ляют 1 мл раствора 12 г/л натрия Р в метаноле
безводном Р, приготовленного с необходимой осто¬
рожностью, и энергично перемешивают в течение
приблизительно 5 мин. Прибавляют 3 мл воды дис¬
тиллированной Р и энергично перемешивают в
558
Гэсударственная фармакопея Республики Беларусь
течение 30 с. Центрифугируют в течение 15 мин с
ускорением 1500 д. Хроматографируют 1 мкл орга¬
нического слоя.
Рассчитывают содержание жирных кислот в про¬
центах по формуле:
Ау,5,С
Х,8,С
•100 %(м/м),
где:
Ахзс — скорректированная площадь пиков каж¬
дой жирной кислоты в испытуемом растворе:
^Х,5,С = ^Х,5 * «с.
Рс — корригирующий коэффициент для пиков,
соответствующих метиловым эфирам капроновой,
каприловой, каприновой, лауриновой и миристиновой
кислот:
*с =
К т1.г'
где:
тхг — масса трикапроина, трикапролина, три-
каприна, трилаурина или тримиристина в растворе
сравнения, мг;
т1г — масса тристеарина в растворе сравне¬
ния, мг;
Ахг — площадь пиков, соответствующих метило¬
вым эфирам капроновой, каприловой, каприновой,
лауриновой и миристиногвой кислот в растворе срав¬
нения;
Аи — площадь пика метилового эфира стеарино¬
вой кислоты в растворе сравнения;
АХ8 —• площадь пиков метиловых эфиров любых
специфицированных или неспецифицированных жир¬
ных кислот;
Рс — 1 для пиков, соответствующих каждому
другому специфическому метиловому эфиру жирных
кислот или какому-либо неспецифическому метило¬
вому эфиру жирной кислоты.
Состав фракции жирных кислот масла:
- капроновая кислота (Р^ 0,11): не более 1,5 %;
- каприловая кислота (Р^ 0,23): от 5,0 % до 11,0 %;
- каприновая кислота (Р^ 0,56): от 4,0 % до 9,0 %;
-лауриновая кислота (Р^ 0,75): от 40,0% до
50.0 %;
- миристиновая кислота (Р^ 0,85): от 15,0% до
20.0 %;
- пальмитиновая кислота (Р^ 0,93): от 7,0% до
12,0%;
- стеариновая кислота (Р^ 1,00): от 1,5 % до 5,0 %;
- олеиновая кислота (Р^ 1,01): от 4,0 % до 10,0 %;
- линолевая кислота (Р^ 1,03): от 1,0 % до 3,0 %;
- линоленовая кислота (Р^ 1,06): не более 0,2 %;
- арахидоновая кислота (Р^ 1,10): не более 0,2 %;
- эйкозеновая кислота (Р^ 1,11): не более 0,2 %.
Вода (2.5.32). Не более 0,1 %. Определение про¬
водят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В заполненном доверху контейнере, в защищен¬
ном от света месте.
07/2016:0891
коповидон
СороуШопит
СОРОУЮОЫЕ
(С6Н9ЫО)„ (С4нб02)т М.м. (111,1), + (86,1)ш
[25086-89-9]
ОПРЕДЕЛЕНИЕ
Сополимер 1-этенилпирролидин-2-она и этени-
лацетата в массовой пропорции 3:2.
Содержание:
- азот (N1; А.м. 14,01): не менее 7,0 % и не более
8,0 % (в пересчете на сухое вещество);
- этенилацетат (С4Н602; М.м. 86,10): не менее
35,3 % и не более 42,0 % (в пересчете на сухое веще¬
ство).
К-значение: не менее 90,0 % и не более 110,0 %
от значения, указанного на маркировке.
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок или хло¬
пья. Гигроскопичен.
Легко растворим в воде, в 96 % спирте и в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр коповидона пс
Европейской Фармакопее.
B. К 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 5 мл воды Р
и 0,2 мл 0,05 М раствора йода. Появляется красное
окрашивание.
C. 0,7 г еидроксиламина гидрохлорида Р раст¬
воряют в 10 мл метанола Р, прибавляют 20 м.-
раствора 40 г/л натрия гидроксида Р и, при необхс-
димости, фильтруют. К 5 мл полученного раствора
прибавляют 0,1 г испытуемого образца и кипятят =
течение 2 мин. 50 мкл полученного раствора поме¬
щают на фильтровальную бумагу, прибавляют 0,1 м.-
смеси равных объемов раствора железа (III) хлори¬
да Р1 и кислоты хлористоводородной Р. Появляе--
ся фиолетовое окрашивание.
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 100 мл этим же
растворителем. Испытуемый образец прибавляют в
воду малыми порциями при постоянном перемеши¬
вании.
Коповидон
559
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон III.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)5, Р(Кр)5 или
ВУ(КЖ)5.
Вязкость, обозначенная как К-значение. 5,0 мл
раствора 3 доводят водой Р до объема 50,0 мл. Вы¬
держивают в течение 1 ч и определяют вязкость (2.2.9)
раствора при температуре 25 °С, используя вискозо-
метр № 1 с минимальным временем вытекания 100 с.
Рассчитывают К-значение по формуле:
1,5 • 1од/7 — 1 V300 с-1од^ + (с + 1,5с1одл])2
0,15+ 0,003 с + 0,15 с+ 0,003 с2
где:
с — концентрация испытуемого образца в пере¬
счете на сухое вещество, г/100 мл;
г]—вязкость раствора относительно вязкости воды.
Альдегиды. Не более 0,0500 % (500 ррт), в пе¬
ресчете на ацетальдегид.
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в фосфатном буферном растворе рН
9.0 Р и доводят до объема 100,0 мл этим же раство¬
рителем. Закрывают колбу пробкой, нагревают при
температуре 60 °С в течение 1 ч и охлаждают.
Раствор сравнения. 0,140 г ацетальдегидамми-
ака тримера тригидрата Р растворяют в воде Р и
доводят до объема 200,0 мл этим же растворителем.
1.0 мл полученного раствора доводят фосфатным
буферным раствором рН 9,0 Р до объема 100,0 мл.
В 3 одинаковые отдельные спектрофотометри¬
ческие кюветы с длиной оптического пути 1 см поме¬
щают 0,5 мл испытуемого раствора, 0,5 мл раствора
сравнения и 0,5 мл воды Р (контрольный раствор). В
каждую кювету прибавляют 2,5 мл фосфатного бу¬
ферного раствора с рН 9,0 Р и 0,2 мл раствора ни-
котинамид-аденин динуклеотида Р. Перемешивают
и плотно закрывают пробкой. Выдерживают при тем¬
пературе (22±2) °С в течение (2—3) мин и измеряют
оптическую плотность (2.2.25) каждого раствора при
340 нм, используя воду Р в качестве компенсационно¬
го раствора. В каждую кювету прибавляют по 0,05 мл
раствора альдегиддегидрогеназы Р, смешивают,
плотно закрывают пробкой и выдерживают при темпе¬
ратуре (22±2) °С в течение 5 мин. Измеряют оптиче¬
скую плотность каждого раствора при 340 нм, исполь¬
зуя воду Р в качестве компенсационного раствора.
Рассчитывают содержание альдегидов по фор¬
муле:
(А2-Л„)-(Л62-ЛЬ1) 100000 с
(А,2-Ава)-(АЬ2-АЬ1) т
где:
— оптическая плотность испытуемого раст¬
вора до прибавления альдегиддегидрогеназы;
Аа — оптическая плотность испытуемого раст¬
вора после прибавления альдегиддегидрогеназы;
А51 — оптическая плотность раствора сравнения
до прибавления альдегиддегидрогеназы;
Аз2 — оптическая плотность раствора сравнения
после прибавления альдегиддегидрогеназы;
ЛЬ1 — оптическая плотность контрольного раст¬
вора до прибавления альдегиддегидрогеназы;
АЬ2 — оптическая плотность контрольного раст¬
вора после прибавления альдегиддегидрогеназы;
т — масса испытуемого образца, в пересчете на
сухое вещество, г;
С — концентрация ацетальдегида в растворе
сравнения, рассчитанная из массы ацетальдегидам-
миака тримера тригидрата с учетом коэффициента
0,72, мг/мл.
Пероксиды. Не более 0,0400 % (400 ррт) в пе¬
ресчете на Н202. 10 мл раствора 3 доводят водой Р
до объема 25 мл, прибавляют 2 мл реактива тита¬
на хлорида (III) и кислоты серной Р и выдерживают
в течение 30 мин. Измеряют оптическую плотность
(2.2.25) полученного раствора при 405 нм, исполь¬
зуя в качестве компенсационного раствора смесь из
25 мл раствора 40 г/л испытуемого образца и 2 мл
13 % (об/об) раствора кислоты серной Р. Оптическая
плотность не должна превышать 0,35.
Гидразин. Не более 0,0001 % (1 ррт). Тонкос¬
лойная хроматография (2.2.27). Используют свеже¬
приготовленные растворы.
Испытуемый раствор. К 25 мл раствора 3 при¬
бавляют 0,5 мл раствора 50 г/л салицилового альде¬
гида Р в метаноле Р, смешивают и нагревают на во¬
дяной бане при температуре 60 °С в течение 15 мин.
Охлаждают, прибавляют 2,0 мл ксилола Р, перемеши¬
вают в течение 2 мин и центрифугируют. Используют
верхний прозрачный слой жидкости.
Раствор сравнения. 9 мг салицилового альдеги¬
да азина Р растворяют в ксилоле Р и доводят до объ¬
ема 100 мл этим же растворителем. 1 мл полученного
раствора доводят ксилолом Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: вода Р — метанол Р (20:80,
об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 365 нм.
Предельное содержание примесей:
- гидразин (не более 1 ррт): на хроматограмме
испытуемого раствора пятно, соответствующее пятну
салицилового альдегида азина, должно быть не интен¬
сивнее пятна на хроматограмме раствора сравнения.
Мономеры. Не более 0,1 %. 10,0 г испытуемого
образца растворяют в 30 мл метанола Р и медлен¬
но прибавляют 20,0 мл раствора йода бромида Р.
Выдерживают в защищенном от света месте в тече¬
ние 30 мин, периодически перемешивая. Прибавля¬
ют 10 мл раствора 100 г/л калия йодида Р и титруют
0,1 М раствором натрия тиосульфата до появле¬
ния желтого окрашивания. Продолжают титрование,
прибавляя 0,1 М раствор натрия тиосульфата по
каплям, до обесцвечивания раствора.
Параллельно проводят контрольный опыт.
На титрование должно пойти не более 1,8 мл
0,1 М раствора натрия тиосульфата.
Примесь А. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения. 0,100 г 2-пирролидона Р
(примесь А) растворяют в воде Р и доводят до объема
100 мл этим же растворителем. 1,0 мл полученного
раствора доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- предколонка длиной 0,025 м и внутренним диа¬
метром 4 мм, заполненная силикагелем октадецил-
560
Государственная фармакопея Республики Беларусь
синильным эндкепированным для хроматографии Р
с размером частиц 5 мкм;
- колонка из нержавеющей стали длиной 0,25 м
и внутренним диаметром 4,0 мм, заполненная сфери¬
ческим силикагелем аминогексадецилсилильным для
хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: вода Р, доведенная до рН 2,4
кислотой фосфорной Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 205 нм; детектор устанавливают между пред-
колонкой и аналитической колонкой; второй детектор
устанавливают после аналитической колонки;
- объем вводимой пробы: 10 мкл; после выхода
примеси А из предколонки (примерно 1,2 мин) пере¬
ключают поток напрямую от насоса к аналитической
колонке; до начала следующей хроматограммы про¬
мывают предколонку противотоком.
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этенилацетат. Определяют число омыления
(2.5.6) из 2,00 г испытуемого образца. Рассчитывают
содержание этенилацетата в процентах, умножая по¬
лученный результат на 0,1534.
Азот. Проводят определение азота (2.5.9), ис¬
пользуя 30,0 мг испытуемого образца и 1 г смеси из 3
частей меди сульфата Р и 997 частей калия сульфа¬
та Р. Нагревают до образования прозрачного светло-
зеленого раствора и продолжают нагревать в течение
45 мин.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
Указывают К-значение.
ПРИМЕСИ
ХУ
А. Пирролидин-2-он (2-пирролидон).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной стать ~
проверка указанных характеристик не обязательна
Однако контроль данных характеристик можег
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важнь
для коповидона, используемого в качестве связываю¬
щего вещества при производстве таблеток и гранул
Вязкость (2.2.9). Определяют динамическую
вязкость с использованием капиллярного вискозиме¬
тра и 10 % раствора (в пересчете на сухое вещество
или 20 % раствора (в пересчете на сухое вещество
при температуре 25 °С. Обычно вязкость составляет
около 8 мПа-с или около 23 мПа с соответственно.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Насыпная плотность и плотность после усад¬
ки (2.9.34).
Следующие характеристики могут быть важ¬
ны для коповидона, используемого в качестве плен¬
кообразующего вещества при производстве дозиро¬
ванных форм, покрытых оболочкой, и аэрозолей.
Вязкость (2.2.9): см. выше.
07/2016:0267
КОФЕИН
СоЯетит
САРРЕШЕ
С8Н10М4О2 М.м. 194,2
[58-08-2]
ОПРЕДЕЛЕНИЕ
1,3,7-Триметил-3,7-дигидро-1Н-пурин-2,6-дион.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок либо белые или почти белые шелковистые кри¬
сталлы.
Умеренно растворим в воде, легко растворим в
кипящей воде, мало растворим в 96 % спирте. Раство¬
ряется в концентрированных растворах бензоатов и
салицилатов щелочных металлов.
Легко сублимируется.
Кофеин
561
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: А, С, О, Е, Е
A. Температура плавления (2.2.14): от 234 °С до
239 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кофеина #или спектр, представ¬
ленный на рисунке #0267.-1#.
C. К 2 мл насыщенного раствора испытуемого
образца прибавляют 0,05 мл раствора калия йоди¬
да йодированного Р. Раствор остается прозрачным.
К полученному раствору прибавляют 0,1 мл кислоты
хлористоводородной разведенной Р. Образуется ко¬
ричневый осадок. Нейтрализуют раствором натрия
гидроксида разведенным Р. Осадок растворяется.
O. В пробирку с притертой пробкой помещают
10 мг испытуемого образца, растворяют в 0,25 мл
смеси из 0,5 мл ацетилацетона Р и 5 мл раствора
натрия гидроксида разведенного Р, нагревают в во¬
дяной бане при температуре 80 °С в течение 7 мин,
охлаждают и прибавляют 0,5 мл раствора димети-
ламинобензальдегида Р2. Нагревают в водяной бане
при температуре 80 °С в течение 7 мин, охлаждают
и прибавляют 10 мл воды Р. Появляется интенсивное
синее окрашивание.
Е. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
P. Испытуемый образец дает реакцию на ксанти¬
ны (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют при нагревании в воде, свободной от углерода
диоксида, Р, приготовленной из воды дистиллирован¬
ной Р, охлаждают и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность. К 10 мл раствора 3 прибавляют
0,05 мл раствора бромтимолового синего Р1. Появля¬
ется зеленое или желтое окрашивание. При прибавле¬
нии не более 0,2 мл 0,01 М раствора натрия гидрокси¬
да окрашивание раствора должно измениться на синее.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до объ¬
ема 50,0 мл этим же растворителем. 1,0 мл полученного
раствора доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (а). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО кофеина для
проверки пригодности хроматографической си¬
стемы (содержит примеси А, С, О и Р) растворяют в
подвижной фазе и доводят до объема 5 мл этим же
растворителем. 2 мл полученного раствора доводят
подвижной фазой до объема 10 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: смешивают 20 объемов те-
трагидрофурана Р, 25 объемов ацетонитрила Р и
955 объемов раствора 0,82 г/л натрия ацетата без¬
водного Р, предварительно доведенного до рН 4,5
кислотой уксусной ледяной Р\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 275 нм;
Волновое число (см'1)
Рисунок #0267.-1. Инфракрасный спектр ФСО кофеина
71. Зак. 1060.
562
Гэсударственная фармакопея Республики Беларусь
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания кофеина.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, С, О и Р, используя хромато¬
грамму раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО кофеина для проверки пригодности
хроматографической системы.
Время удерживания: кофеин — около 8 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками при¬
меси С и примеси й; не менее 2,5 между пиками при¬
меси Р и примеси А.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 0,1 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05%): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
15 мл раствора 3 должны выдерживать испытание на
сульфаты. Эталон готовят с использованием смеси
из 7,5 мл эталонного раствора сульфата (10 ррт
80) Р и 7,5 мл воды дистиллированной Р.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,170 г испытуемого образца растворяют при на¬
гревании в 5 мл кислоты уксусной безводной Р, ох¬
лаждают, прибавляют 10 мл уксусного ангидрида Р,
20 мл толуола Р и титруют 0,1 М раствором кисло¬
ты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 19,42 мг С8Н10М4О2.
А. 1,3-Диметил-3,7-дигидро-1 /-/-пурин-2,6-дион (те-
офиллин).
В. А/-(6-Амино-1,3-диметил-2,4-диоксо-1,2,3,4-тетра-
гидропиримидин-5-ил)формамид.
С. 1,3,9-Триметил-3,9-дигидро-1Н-пурин-2,6-дион
(изокофеин).
й. 3,7-Диметил-3,7-дигидро-1Н-пурин-2,6-дион (тео¬
бромин).
Е. А/, 1 -Диметил-4-(метиламино)-1 Н-имидазол-5-
карбоксамид (кофеидин).
Р. 1,7-Диметил-3,7-дигидро-1Н-пурин-2,6-дион.
07/2016:0434
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е, Р.
КРЕМНИЯ ДИОКСИД коллоидный
БЕЗВОДНЫЙ
5///са соНоШаНз апЬус/пса
81ЫСА, СОЦ-ОЮА1.АЫНУ01Юи8
ЗЮ2 М.м. 60,1
[7631-86-9]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
ЗЮ2 (в пересчете на прокаленное вещество).
Кремния диоксид коллоидный гидратированный
563
ОПИСАНИЕ (СВОЙСТВА)
Легкий мелкий аморфный порошок белого или
почти белого цвета с размером частиц около 15 нм.
Практически нерастворим в воде и минеральных
кислотах, кроме фтористоводородной кислоты. Рас¬
творяется в горячих растворах гидроксидов щелочных
металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
20 мг испытуемого образца дают реакцию (а) на
силикаты (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 3,5 до 5,5.1,0 г испытуемого образ¬
ца встряхивают с 30 мл воды, свободной от углерода
диоксида, Р.
Хлориды (2.4.4). Не более 0,0250 % (250 ррт). К
1.0 г испытуемого образца прибавляют смесь из 20 мл
азотной кислоты разведенной Р и 30 мл воды Р, на¬
гревают на водяной бане в течение 15 мин, часто
встряхивая. При необходимости доводят водой Р до
объема 50 мл, фильтруют и охлаждают. 10 мл филь¬
трата доводят водой Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0025 % (25 ррт). 2,5 г испытуемого образца суспен¬
дируют в достаточном количестве воды Р для получе¬
ния полужидкой суспензии. Высушивают при темпера¬
туре 140 °С. Когда сухое вещество становится белым,
массу измельчают стеклянной палочкой. Прибавляют
25 мл 1 М раствора кислоты хлористоводородной
и осторожно кипятят в течение 5 мин, часто пере¬
мешивая суспензию стеклянной палочкой. Центри¬
фугируют в течение 20 мин и фильтруют надосадоч-
ную жидкость через мембранный фильтр. К остатку в
пробирке для центрифугирования прибавляют 3 мл
кислоты хлористоводородной разведенной Р, 9 мл
воды Р и кипятят. Центрифугируют в течение 20 мин и
фильтруют надосадочную жидкость через тот же мем¬
бранный фильтр. Остаток промывают небольшими
порциями воды Р, объединяют фильтраты и промыв¬
ные воды и доводят водой Р до объема 50 мл. К 20 мл
раствора прибавляют 50 мг аскорбиновой кислоты Р
и 1 мл раствора аммиака концентрированного Р.
Нейтрализуют раствором аммиака разведенным Р2.
Доводят водой Р до объема 25 мл. 12 мл полученного
раствора должны выдерживать испытание на тяже¬
лые металлы. Эталон готовят с использованием эта¬
лонного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при прокаливании. Не более
5.0 %. 0,200 г испытуемого образца прокаливают в
платиновом тигле при температуре (900±50) °С в те¬
чение 2 ч. Перед взвешиванием охлаждают в эксика¬
торе.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К остатку, полученному в испытании «Потеря в
массе при прокаливании», прибавляют 0,2 мл кис¬
лоты серной Р и 96% спирт Р в количестве, доста¬
точном для полного смачивания остатка. Прибавляют
6 мл кислоты фтористоводородной Р и выпари¬
вают досуха на плитке при температуре от 95 °С до
105 °С, не допуская разбрызгивания. Стенки чашки
промывают 6 мл кислоты фтористоводородной Р
и выпаривают досуха. Прокаливают при температуре
(900±50) °С, охлаждают в эксикаторе и взвешивают.
Количество 5Ю2 в навеске испытуемого образца рас¬
считывают как разность между массой остатка, полу¬
ченного при проведении испытания «Потеря в массе
при прокаливании», и массой окончательного остатка.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для кремния диоксида коллоидного безводного,
используемого в качестве скользящего вещества
при производстве таблеток и капсул.
Удельная площадь поверхности (2.9.26, ме¬
тод I). Определяют удельную площадь поверхности
в диапазоне Р/Р0 от 0,05 до 0,30.
Дегазация образца: в течение 20 мин при темпе¬
ратуре 160 °С.
07/2016:0738
КРЕМНИЯ диоксид коллоидный
ГИДРАТИРОВАННЫЙ
8Шса соИоМаНз Ьудпса
81ЫСА, СОи.ОЮА1- НУй Я АТЕЭ
[63231-67-4]
ОПРЕДЕЛЕНИЕ
Получают осаждением или гелеобразованием.
Содержание: не менее 98,0 % и не более 100,5 %
ЗЮ2 (М.м. 60,1) (в пересчете на прокаленное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Легкий мелкий аморфный порошок белого или
почти белого цвета.
Практически нерастворим в воде и в минераль¬
ных кислотах, кроме фтористоводородной кислоты.
Растворяется в горячих растворах гидроксидов ще¬
лочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
20 мг испытуемого образца дают реакцию (а) на
силикаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. К 2,5 г испытуемого образца при¬
бавляют 50 мл кислоты хлористоводородной Р и
перемешивают. Нагревают на водяной бане в тече-
71*. Зак. 1060.
564
Гэсударственная фармакопея Республики Беларусь
ние 30 мин, периодически перемешивая. Доводят до
первоначального объема кислотой хлористоводо¬
родной разведенной Р. Выпаривают досуха. К остат¬
ку прибавляют смесь из 8 мл кислоты хлористово¬
дородной разведенной Р и 24 мл воды Р. Нагревают
до кипения и фильтруют при пониженном давлении
через стеклянный фильтр (ПОР 16) (2.1.2). Остаток на
фильтре промывают горячей смесью из 3 мл кислоты
хлористоводородной разведенной Р и 9 мл воды Р.
Промывают небольшими порциями воды Р, объеди¬
няют фильтрат и промывные воды и доводят водой Р
до объема 50 мл.
рН (2.2.3). От 4,0 до 7,0.1,0 г испытуемого образца
суспендируют в 30 мл раствора 75 г/л калия хлорида Р.
Водопоглощающая способность. 5 г испытуе¬
мого образца растирают в ступке, прибавляя по ка¬
плям 5 мл воды Р. Смесь должна оставаться порош¬
кообразной.
Вещества, растворимые в хлористоводород¬
ной кислоте. Не более 2,0 %. 10,0 мл раствора 3 по¬
мещают в платиновую чашку, выпаривают досуха и
высушивают при температуре от 100 °С до 105 °С до
постоянной массы. Масса остатка не должна превы¬
шать 10 мг.
Хлориды (2.4.4). Не более 0,1 %. 0,5 г испытуе¬
мого образца смешивают с 50 мл воды Р и нагревают
на водяной бане в течение 15 мин. Доводят водой Р
до объема 100 мл и центрифугируют с ускорением
1500 д в течение 5 мин. 10 мл надосадочной жидкости
доводят водой Р до объема 15 мл. Полученный раст¬
вор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 1 %. 2 мл раствора
3 доводят водой дистиллированной Р до объема
100 мл. 15 мл полученного раствора должны выдер¬
живать испытание на сульфаты.
Железо (2.4.9). Не более 0,0300 % (300 ррт). К
2 мл раствора 3 прибавляют 28 мл воды Р. 10 мл по¬
лученного раствора должны выдерживать испытание
на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0025 % (25 ррт). К 20 мл раствора 3 прибавляют
50 мг гидроксиламина гидрохлорида Р и 1 мл раст¬
вора аммиака концентрированного Р. Доводят до
рН 3,5 (контролируют потенциометрически) раство¬
ром аммиака разведенным Р2. Доводят водой Р до
объема 25 мл. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при прокаливании. Не более
20,0 %. 0,200 г испытуемого образца нагревают в пла¬
тиновом тигле при температуре от 100 °С до 105 °С
в течение 1 ч и затем прокаливают при температуре
(900±50) °С в течение 2 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К остатку, полученному в испытании «Потеря в
массе при прокаливании», прибавляют 0,2 мл кис¬
лоты серной Р и 96% спирт Р в количестве, доста¬
точном для полного смачивания остатка. Прибавляют
6 мл кислоты фтористоводородной Р и выпари¬
вают досуха на плитке при температуре от 95 °С до
105 °С, не допуская разбрызгивания. Стенки чашки
промывают 6 мл кислоты фтористоводородной Р
и выпаривают досуха. Прокаливают при температуге
(900±50) °С, охлаждают в эксикаторе и взвешивают.
Количество ЗЮ2 в навеске испытуемого образца рас¬
считывают как разность между массой остатка, полу¬
ченного при проведении испытания «Потеря в массе
при прокаливании», и массой окончательного остатка.
07/2016:0985
КРОСКАРМЕЛЛОЗА НАТРИЯ
СагтеНозит паМсит сопехит
СК08САКМЕИ.ОЗЕ ЗООШМ
ОПРЕДЕЛЕНИЕ
Перекрестно-сшитая натрия карбоксиметилцел-
люлоза.
Натриевая соль перекрестно-сшитой частично
О-карбоксиметилированной целлюлозы.
ОПИСАНИЕ (СВОЙСТВА)
Белый или серовато-белый порошок.
Практически нерастворима в ацетоне, этаноле
безводном и толуоле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца перемешивают со
100 мл раствора 0,004 г/л (4 ррт) метиленового си¬
него Р, взбалтывают смесь и дают отстояться. Испы¬
туемый образец абсорбирует метиленовый синий и
оседает в виде синей волокнистой массы.
B. 1 г испытуемого образца перемешивают с
50 мл воды Р. 1 мл полученной смеси переносят в
пробирку, прибавляют 1 мл воды Р и 0,05 мл свеже¬
приготовленного раствора 40 г/л а-нафтола Р в ме¬
таноле Р. Наклоняют пробирку и осторожно прибав¬
ляют 2 мл кислоты серной Р вниз по стенке так, что¬
бы она образовала нижний слой. На границе раздела
появляется красновато-фиолетовое окрашивание.
C. Раствор, приготовленный из сульфатной золы,
полученной в испытании «Тяжелые металлы», дает
реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 7,0.1 г испытуемого образца
встряхивают с 100 мл воды, свободной от углерода
диоксида, Р в течение 5 мин.
Натрия хлорид и натрия гликолят. Не более
0,5 % (в пересчете на сухое вещество) суммарного
процентного содержания натрия хлорида и натрия
гликолята.
Натрия хлорид. 5,00 г испытуемого образца по¬
мещают в коническую колбу вместимостью 250 мл.
прибавляют 50 мл воды Р и 5 мл раствора водоро¬
да пероксида концентрированного Р и нагревают на
водяной бане в течение 20 мин, периодически пере¬
мешивая для достижения полной гидратации. Охлаж¬
дают, прибавляют 100 мл воды Р и 10 мл кислоты
азотной Р. Титруют при постоянном перемешивании
0,05 М раствором серебра нитрата потенциометри¬
чески (2.2.20). Используют серебряный индикаторный
электрод и двойной электрод сравнения, содержащий
раствор 100 г/л калия нитрата Р во внешней ячейке
и стандартный раствор во внутренней ячейке.
1 мл 0,05 М раствора серебра нитрата соот¬
ветствует 2,922 мг №01.
Натрия гликолят. 0,500 г испытуемого образца
(в пересчете на сухое вещество) помещают в лабо-
Кроскармеллоза натрия
565
раторный стакан вместимостью 100 мл, прибавляют
5 мл кислоты уксусной ледяной Р и 5 мл воды Р и
перемешивают до полной гидратации (около 15 мин).
Прибавляют 50 мл ацетона Р и 1 г натрия хлорида Р.
Перемешивают в течение нескольких минут до полно¬
го осаждения карбоксиметилцеллюлозы. Фильтруют
через быстрофильтрующую фильтровальную бумагу,
пропитанную ацетоном Р, в мерную колбу. Ополаски¬
вают стакан и фильтр 30 мл ацетона Р и фильтрат
доводят до объема 100,0 мл этим же растворителем.
Выдерживают в течение 24 ч без перемешивания. Ис¬
пользуют прозрачную надосадочную жидкость в каче¬
стве испытуемого раствора.
Стандартные растворы готовят следующим об¬
разом: 0,100 г кислоты гликолевой Р, предваритель¬
но высушенной под вакуумом над фосфора (V) ок¬
сидом Р при комнатной температуре в течение ночи,
помещают в мерную колбу вместимостью 100 мл,
растворяют в воде Р и доводят до объема 100,0 мл
этим же растворителем. Используют раствор в тече¬
ние 30 дней. 1,0 мл, 2,0 мл, 3,0 мл и 4,0 мл получен¬
ного раствора переносят в отдельные мерные кол¬
бы; доводят содержимое каждой колбы водой Р до
объема 5,0 мл, прибавляют 5 мл кислоты уксусной
ледяной Р, доводят ацетоном Р до объема 100,0 мл
и перемешивают.
Переносят 2,0 мл испытуемого раствора, по
2,0 мл каждого стандартного раствора и для приго¬
товления контрольного раствора 2,0 мл раствора, со¬
держащего 5 % (об/об) кислоты уксусной ледяной Р и
5 % (об/об) воды Р в ацетоне Р, в отдельные мерные
колбы вместимостью 25 мл. Нагревают незакрытые
колбы в течение 20 мин на водяной бане до удаления
ацетона. Охлаждают и прибавляют по 5,0 мл раст¬
вора 2,7-дигидроксинафталинаР в каждую колбу.
Перемешивают, прибавляют по 15,0 мл раствора
2,7-дигидроксинафталина Р и снова перемешивают.
Закрывают колбы алюминиевой фольгой и нагревают
на водяной бане в течение 20 мин. Охлаждают и дово¬
дят кислотой серной Р до объема 25,0 мл.
Измеряют оптическую плотность (2.2.25) каждо¬
го раствора при длине волны 540 нм. Строят граду¬
ировочный график, используя оптические плотности
стандартных растворов. По градуировочному графику
и оптической плотности испытуемого раствора опре¬
деляют массу (а, мг) кислоты гликолевой в испытуе¬
мом образце и рассчитывают содержание натрия гли-
колята по формуле:
101,29 а
(100-Ь) т ’
где:
1,29 — коэффициент пересчета кислоты гликоле¬
вой в натрия гликолят;
Ь — потеря в массе при высушивании, %;
т — масса испытуемого образца, г.
Растворимые в воде вещества. Не более
10,0%. 10,00 г испытуемого образца диспергируют
в 800,0 мл воды Р и перемешивают по 1 мин через
каждые 10 мин в течение первых 30 мин. Выдержи¬
вают в течение 1 ч и при необходимости центрифу¬
гируют. Декантируют 200 мл надосадочной жидкости
на быстрофильтрующую фильтровальную бумагу в
вакуумную фильтрационную воронку, подают вакуум
и собирают 150,0 мл фильтрата. Выпаривают досуха
и сушат осадок при температуре от 100 °С до 105 °С
в течение 4 ч.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). К осадку, полученному при опре¬
делении сульфатной золы, прибавляют 1 мл кислоты
хлористоводородной Р и выпаривают на водяной
бане. Прибавляют к осадку 20 мл воды Р. 12 мл полу¬
ченного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 6 ч.
Сульфатная зола (2.4.14). От 14,0 % до 28,0 %
(в пересчете на сухое вещество). Определение прово¬
дят из 1,0 г испытуемого образца; используют смесь,
состоящую из равных объемов кислоты серной Р и
воды Р.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа со//: отсутствие (2.6.13).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для кроскармеллозы натрия, используемой в ка¬
честве разрыхлителя.
Объем осадка. От 10,0 мл до 30,0 мл. 75 мл
воды Р помещают в мерный цилиндр вместимостью
100 мл и прибавляют 1,5 г испытуемого образца пор¬
циями по 0,5 г, энергично встряхивая после каждого
прибавления. Доводят водой Р до объема 100,0 мл и
снова встряхивают до равномерного распределения
образца. Выдерживают в течение 4 ч. Измеряют объ¬
ем осадка.
Степень замещения. От 0,60 до 0,85 (в пересче¬
те на сухое вещество). 1,000 г испытуемого образца
помещают в коническую колбу вместимостью 500 мл,
прибавляют 300 мл раствора 100 г/л натрия хлори¬
да Р, 25,0 мл 0,1 М раствора натрия гидроксида,
закрывают колбу и выдерживают в течение 5 мин, пе¬
риодически перемешивая. Прибавляют 0,05 мл раст¬
вора м-крезолового пурпурового Р и около 15 мл
0,1 М раствора кислоты хлористоводородной из
бюретки. Закрывают пробкой и перемешивают. Если
раствор имеет фиолетовую окраску, прибавляют 0,1 М
раствор кислоты хлористоводородной порциями по
1 мл до тех пор, пока окраска не станет желтой, пере¬
мешивая после каждого прибавления. Титруют 0,1 М
раствором натрия гидроксида до изменения окраски
на фиолетовую.
72. Зак. 1060.
566
Гэсударственная фармакопея Республики Беларусь
Рассчитывают число миллиэквивалентов (М) ос¬
нования, необходимых для нейтрализации 1 г сухого
вещества (испытуемого образца).
Рассчитывают степень замещения карбоксиме-
тиловой кислотой (А) по формуле:
115 ОМ
~ (7102-412М-80С)’
где:
С — сульфатная зола, %.
Рассчитывают степень замещения натриевой со¬
лью карбоксиметиловой кислоты (5) по формуле:
5 (162 + 58А) С
(7102-80С) *
Степень замещения представляет собой сумму
А и 5.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошков (2.9.36).
07/2016:0892
КРОСПОВИДОН
СгозроуШопит
СКОЗРОУЮОЫЕ
(С6Н9ЫО)„ М.м. (111,1)л
[9003-39-8]
ОПРЕДЕЛЕНИЕ
Поперечно-сшитый гомополимер 1-этенилпирро-
лидин-2-она.
Содержание: не менее 11,0 % и не более 12,8 %
азота (Ы; А.м. 14,01) (в пересчете на сухое вещество).
В зависимости от размера частиц различают два
типа: тип А и тип В.
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок или хло¬
пья. Гигроскопичен.
Практически нерастворим в воде, в 96 % спирте
и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кросповидона.
B. 1 г испытуемого образца суспендируют в 10 мл
воды Р, прибавляют 0,1 мл 0,05 М раствора йода и
встряхивают в течение 30 с. Прибавляют 1 мл раст¬
вора крахмала Р и снова встряхивают. Синее окраши¬
вание не появляется в течение 30 с.
C. К 10 мл воды Р прибавляют 0,1 г испытуемого
образца и встряхивают. Образуется суспензия, кото¬
рая остается мутной в течение 15 мин.
О. Аналитические сита должны быть чистыми
и сухими. Для этого сита промывают в горячей воде
и сушат в течение ночи при температуре 105 °С.
20 г (в пересчете на сухое вещество) испытуемо¬
го образца помещают в коническую колбу вместимо¬
стью 1000 мл, прибавляют 500 мл воды Р и встряхи¬
вают полученную суспензию в течение 30 мин. Полу¬
ченную суспензию пропускают через предварительно
взвешенное аналитическое сито с размером ячейки
63 мкм и промывают водой Р до получения прозрач¬
ного фильтрата. Сито с остатком сушат без циркуля¬
ции воздуха при температуре 105 °С в течение 5 ч. Ох¬
лаждают в эксикаторе в течение 30 мин и взвешивают.
Рассчитывают количество вещества, оставшего¬
ся на сите в процентах (фракция частиц испытуемого
образца диаметром более 63 мкм) по формуле:
т2
где:
ш1 — масса сита и остатка на сите после высуши¬
вания в течение 5 ч, г;
т2 — начальная масса навески испытуемого об¬
разца, г;
тг — масса сита, г.
Если остаток на сите составляет более 15 %, суб¬
станция классифицируется как тип А; если остаток на
сите составляет 15 % или менее, субстанция класси¬
фицируется как тип В.
ИСПЫТАНИЯ
Пероксиды. Тип А: не более 0,0400 % (400 ррт)
в пересчете на Н202. Тип В: не более 0,1000 %
(1000 ррт) в пересчете на Н202.
2,0 г испытуемого образца суспендируют в 50 мл
воды Р. К 25 мл полученной суспензии (тип А) или к
10 мл полученной суспензии, доведенным водой Р до
объема 25 мл (тип В), прибавляют 2 мл реактива ти¬
тана (III) хлорида и кислоты серной Р. Выдерживают
в течение 30 мин и фильтруют. Оптическая плотность
(2.2.25) фильтрата при 405 нм не должна превышать
0,35. В качестве компенсационного раствора исполь¬
зуют смесь из 25 мл профильтрованной суспензии
испытуемого образца с концентрацией 40 г/л и 2 мл
13 % (об/об) раствора кислоты серной Р.
Растворимые в воде вещества. Не более
1,5 %. 25,0 г испытуемого образца помещают в лабо¬
раторный стакан, вместимостью 400 мл, прибавля¬
ют 200 мл воды Р и перемешивают в течение 1 ч на
магнитной мешалке. Переносят суспензию в мерную
колбу вместимостью 250,0 мл, ополаскивая стакан во¬
дой Р, и доводят объем до метки этим же растворите¬
лем. Дают частицам вещества осесть. Отфильтровы¬
вают около 100 мл наиболее прозрачной надосадоч-
ной жидкости через мембранный фильтр с диаметром
пор 0,45 мкм, защищенный мембранным фильтром с
диаметром пор 3 мкм. В процессе фильтрации пере¬
мешивают жидкость над фильтром вручную или с
помощью механической мешалки, соблюдая осто¬
рожность, чтобы не повредить фильтр. 50,0 мл про¬
зрачного фильтрата переносят в предварительно вы¬
сушенный и взвешенный лабораторный стакан вме¬
стимостью 100 мл, выпаривают досуха и сушат при
температуре от 105 °С до 110 °С в течение 3 ч. Масса
остатка должна быть не более 75 мг.
Примесь А. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,250 г испытуемого об¬
разца суспендируют в 50,0 мл метанола Р и встря¬
хивают в течение 60 мин. Дают частицам вещества
осесть и фильтруют через мембранный фильтр с раз¬
мером пор 0,2 мкм.
Кросповидон
567
Раствор сравнения (а). 50 мг 1-винилпирролидин-
2-она Р (примесь А) растворяют в метаноле Р и до¬
водят до объема 100,0 мл этим же растворителем.
1,0 мл полученного раствора доводят метанолом Р
до объема 100,0 мл. 5,0 мл полученного раствора до¬
водят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 10 мг 1-винилпирролидин-
2-она Р (примесь А) и 0,50 г винилацетата Р раство¬
ряют в метаноле Р и доводят до объема 100 мл этим
же растворителем. 1,0 мл полученного раствора дово¬
дят подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- предколонка длиной 0,025 м и внутренним диа¬
метром 4 мм, заполненная силикагелем октадецил-
синильным для хроматографии Р с размером частиц
5 мкм;
- колонка длиной 0,25 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: ацетонитрил Р — вода Р
(10:90, об/об);
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: 50 мкл. После каждого
введения испытуемого раствора промывают предко-
лонку, пропуская подвижную фазу противотоком с та¬
кой же скоростью, как в испытании, в течение 30 мин.
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками при¬
меси А и винилацетата на хроматограмме раствора
сравнения (Ь);
- относительное стандартное отклонение:
не более 2,0 % для площадей пиков при 6 повторных
вводах раствора сравнения (а).
Расчет процентного содержания:
- процентное содержание примеси А рассчиты¬
вают с учетом концентрации примеси А в растворе
сравнения (а).
Предельное содержание примесей:
- примесь А: не более 0,0010 % (10 ррт).
Тяжелые металлы {2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца выдер¬
живают испытание на тяжелые металлы. Эталон го¬
товят с использованием 2 мл эталонного раствора
свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца (т, мг) помещают в
колбу для сжигания, прибавляют 5 г смеси из 1 г меди
сульфата Р, 1 г титана диоксида Р и 33 г калия
сульфата Р и 3 стеклянных центра кипения. Смы¬
вают прилипшие частицы с горла колбы небольшим
количеством воды Р. Прибавляют 7 мл кислоты сер¬
ной Р по стенкам колбы. Постепенно нагревают колбу
до получения прозрачного желтовато-зеленого раст¬
вора и до отсутствия на стенках колбы обуглившихся
частиц, после чего нагревают ее в течение 45 мин и
охлаждают. Осторожно прибавляют 20 мл воды Р и
присоединяют колбу к перегонному аппарату, который
был предварительно очищен пропусканием через него
пара. В поглотительную колбу помещают 30 мл раст¬
вора 40 г/л кислоты борной Р, 0,15 мл смешанного
раствора бромкрезолового зеленого и метилового
красного Р и воду Р в количестве, необходимом для
погружения в раствор нижнего конца алонжа. В пере¬
гонную колбу прибавляют 30 мл натрия гидроксида
концентрированного Р с помощью воронки, осторож¬
но промывают воронку 10 мл воды Р и немедленно
закрывают зажим, присоединенный к резиновой труб¬
ке, после чего начинают перегонку с водяным паром
до получения объема дистиллята (80—100) мл. По¬
глотительную колбу убирают от нижнего конца алон¬
жа, ополаскивают нижний конец алонжа небольшим
количеством воды Р и титруют полученный дистил¬
лят 0,025 М раствором кислоты серной до перехода
окраски от зеленой через бледную серовато-голубую
до бледной сероватой красно-фиолетовой.
Проводят контрольный опыт.
1 мл 0,025 М раствора кислоты серной соответ¬
ствует 0,700 мг N.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
Указывают тип кросповидона (тип А или тип В).
ПРИМЕСИ
А. 1-Этенилпирролидин-2-он (1-винилпирролидин-
2-он).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для кросповидона, используемого в качестве раз¬
рыхлителя.
Влагопоглощающая способность. Обычно от 3
до 9. 2,0 г испытуемого образца помещают в центри¬
фужную пробирку вместимостью 100 мл, прибавляют
40 мл воды Р, интенсивно встряхивают до получения
суспензии, затем повторно встряхивают через 5 мин и
72*. Зак. 1060.
568
Гэсударственная фармакопея Республики Беларусь
10 мин, после чего центрифугируют в течение 15 мин
при ускорении 750 д. Надосадочную жидкость слива¬
ют и взвешивают полученный остаток. Влагопоглоща¬
ющую способность рассчитывают как отношение мас¬
сы полученного остатка к массе навески испытуемого
образца.
Распределение частиц по размерам (2.9.31).
Текучесть порошков (2.9.36).
Следующие характеристики могут быть важ¬
ны для кросповидона, используемого в качестве ста¬
билизатора суспензии.
Объем осадка. Обычно более 60 мл. 10 мл ис¬
пытуемого образца помещают в градуированный
цилиндр вместимостью 100 мл, прибавляют 90 мл
воды Р и интенсивно встряхивают. Доводят объем до
100 мл водой Р, смывая частицы порошка со стенок
цилиндра. Выдерживают в течение 24 ч. Измеряют
объем осадка.
07/2016:1277
КСАНТАНОВАЯ КАМЕДЬ
ХапМам дитт/
ХАЫТНАЫ С(/М
ОПРЕДЕЛЕНИЕ
Ксантановая камедь — высокомолекулярный
анионный полисахарид, полученный ферментаци¬
ей углеводов с помощью культуры ХапИютопаз
сатрезМз. Состоит из основной цепи остатков
0-глюкозы, соединенных по положению р(1—>4), с
трисахаридными боковыми цепями, чередуемыми
ангидроглюкозными звеньями, состоящими из од¬
ного звена глюкуроновой кислоты, заключенного
между двумя маннозными звеньями. Большинство
конечных звеньев содержат остатки пировиноград-
ной кислоты, а маннозное звено, присоединенное к
основной цепи, может быть ацетилировано в поло¬
жении С-6.
Относительная молекулярная масса ксантано-
вой камеди составляет примерно 1-106. Ксантановую
камедь получают в виде солей натрия, калия или
кальция.
Содержание: не менее 1,5 % остатков пировино-
градной кислоты (С3Н302; М.м. 71,1) (в пересчете на
сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или желтовато-белый сыпучий порошок.
Растворима в воде с образованием сильно вяз¬
кого раствора, практически нерастворима в органиче¬
ских растворителях.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца помещают в колбу
и суспендируют в 15 мл 0,1 М раствора кислоты
хлористоводородной. Закрывают колбу пробкой,
оснащенной ферментационным сосудом, содержа¬
щим раствор бария гидроксида Р, и осторожно на¬
гревают в течение 5 мин. В растворе бария гидрок¬
сида появляется мутность белого цвета.
B. 300 мл воды Р, предварительно нагретой
до 80 °С, помещают в лабораторный стакан вме¬
стимостью 400 мл, прибавляют при энергичном
механическом перемешивании в точку максималь¬
ного перемешивания жидкости высушенную смесь
из 1,5 г испытуемого образца и 1,5 г камеди бобов
рожкового дерева Р. Перемешивают до получения
раствора и затем продолжают перемешивание еще
в течение не менее 30 мин. В процессе перемеши¬
вания температуру смеси поддерживают на уров¬
не не ниже 60 °С. Прекращают перемешивание и
оставляют смесь минимум на 2 ч. При температуре
ниже 40 °С образуется прочный каучукообразный
гель. В контрольном 1 % растворе, приготовлен¬
ном параллельно, но без добавления камеди бо¬
бов рожкового дерева Р, такой гель не образуется.
ИСПЫТАНИЯ
рН (2.2.3). От 6,0 до 8,0. Измеряют рН раствора
испытуемого образца с концентрацией 10,0 г/л.
2-Пропанол. Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 0,50 г
2-метил-2-пропанола Р растворяют в 500 мл воды Р.
Испытуемый раствор. 200 мл воды Р по¬
мещают в круглодонную колбу вместимостью
1000 мл, прибавляют 5,0 г испытуемого образца и
1 мл эмульсии 10 г/л диметикона Р в масле вазе¬
линовом Р. Колбу закрывают и встряхивают в тече¬
ние 1 ч. Колбу соединяют с холодильником, отгоня¬
ют около 90,0 мл дистиллята, прибавляют 4,0 мл
раствора внутреннего стандарта и доводят водой Р
до объема 100,0 мл.
Раствор сравнения. Необходимое точно взве¬
шенное количество 2-пропанола Р растворяют в
воде Р до получения раствора с концентрацией
2-пропанола около 1 мг/мл. К 4,0 мл полученного
раствора прибавляют 4,0 мл раствора внутреннего
стандарта и доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 1,8 м и внутренним диаметром
4,0 мм, заполненная сополимером стирол-дивинил-
бензола Р с размером частиц (149—177) мкм;
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 30 мл/мин;
- температура колонки: 165 °С;
- температура детектора и блока ввода проб:
200 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 5 мкл.
Относительное удерживание (по отношению к
2-пропанолу): 2-метил-2-пропанол — около 1,5.
Предельное содержание примесей:
- 2-пропанол: не более 0,0750 % (750 ррт).
Другие полисахариды. Тонкослойная хромато¬
графия (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца помещают в толстостенную центрифужную
пробирку, прибавляют 2 мл раствора 230 г/л кис¬
лоты трифторуксусной Р, энергично встряхивают
до растворения образовавшегося геля, пробирку
закрывают и нагревают смесь при температуре
120 °С в течение 1 ч. Гидролизат центрифугируют,
осторожно переносят прозрачный надосадочный
слой в колбу вместимостью 50 мл, прибавляют
10 мл воды Р и выпаривают досуха при понижен¬
ном давлении. Полученный таким образом остаток
растворяют в 10 мл воды Р, выпаривают досуха при
пониженном давлении. Промывают трижды мета¬
нолом Р порциями по 20 мл и выпаривают досуха
при пониженном давлении. К полученной прозрач¬
ной пленке, не имеющей запаха кислоты уксусной,
Ксантановая камедь
569
прибавляют 0,1 мл воды Р и 1 мл метанола Р. Цен¬
трифугируют до отделения аморфного осадка. На-
досадочную жидкость, при необходимости, доводят
метанолом Р до объема 1 мл.
Раствор сравнения. 10 мг глюкозы Р и 10 мг
маннозы Р растворяют в 2 мл воды Р и доводят ме¬
танолом Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: раствор 16 г/л натрия гидро¬
фосфата Р — бутанол Р — ацетон Р (10:40:50,
об/об/об).
Наносимый объем пробы. 5 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Проявление: пластинку опрыскивают раствором
0,5 г дифениламина Р в 25 мл метанола Р, к которо¬
му было прибавлено 0,5 мл анилина Р и 2,5 мл кис¬
лоты фосфорной Р. Пластинку нагревают в течение
5 мин при температуре 120 °С и просматривают при
дневном свете.
Пригодность хроматографической системы:
раствор сравнения:
- в средней трети хроматограммы обнаружи¬
ваются две полностью разделенных зоны серова¬
то-коричневого цвета, соответствующие глюкозе и
маннозе.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются две зоны, соответствую¬
щие зонам глюкозы и маннозы на хроматограмме
раствора сравнения. Кроме того, на хроматограмме
испытуемого раствора могут обнаруживаться одна
красноватая зона и две тусклые голубовато-серые
зоны непосредственно над линией старта и одна
или две зоны голубовато-серого цвета в верхней
четверти хроматограммы. Не допускается наличие
дополнительных зон.
Потеря в массе при высушивании (2.2.32). Не
более 15,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2,5 ч.
Общая зола (2.4.16). От 6,5 % до 16,0 %.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор. 120,0 мг испытуемого об¬
разца, в пересчете на сухое вещество, растворяют в
воде Р и доводят до объема 20,0 мл этим же раство¬
рителем.
Раствор сравнения. 45,0 мг кислоты пировино-
градной Р растворяют в воде Р и доводят до объема
500.0 мл этим же растворителем.
10,0 мл испытуемого раствора помещают в кру¬
глодонную колбу вместимостью 50 мл, прибавляют
20.0 мл 0,1 М раствора кислоты хлористоводо¬
родной и взвешивают. Кипятят на водяной бане с об¬
ратным холодильником в течение 3 ч. Взвешивают и
доводят до первоначальной массы водой Р. 2,0 мл
полученного раствора помещают в делительную во¬
ронку, содержащую 1,0 мл раствора динитрофенил-
гидразина хлористоводородного Р. Выдерживают в
течение 5 мин и прибавляют 5,0 мл этилацетата Р.
Встряхивают и выдерживают до оседания твердых ча¬
стиц. Отделяют верхний слой и встряхивают трижды с
раствором натрия карбоната Р порциями по 5,0 мл.
Объединенные водные слои доводят раствором на¬
трия карбоната Р до объема 50,0 мл. Перемешива¬
ют. Параллельно аналогично обрабатывают 10,0 мл
раствора сравнения.
Незамедлительно измеряют оптическую плот¬
ность (2.2.25) полученных растворов при 375 нм,
используя раствор натрия карбоната Р в каче¬
стве компенсационного раствора.
Оптическая плотность испытуемого раствора
должна быть не меньше оптической плотности
раствора сравнения, что соответствует содержа¬
нию остатков пировиноградной кислоты не менее
1,5 %.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для ксантановой камеди, используемой в
качестве вещества, повышающего вязкость.
Кажущаяся вязкость (2.2.10). Обычно не ме¬
нее 600 мПа-с при температуре (24±1) °С. 250 мл
раствора 12 г/л калия хлорида Р помещают в лабо¬
раторный стакан вместимостью 500 мл и прибав¬
ляют при перемешивании лопастным миксером со
скоростью 800 об/мин 3,0 г испытуемого образца в
течение (45—90) с. При прибавлении испытуемо¬
го образца следят за тем, чтобы агломераты были
разрушены. Прибавляют дополнительно 44 мл
воды Р, ополаскивая стенки стакана. Перемешива¬
ют раствор со скоростью 800 об/мин в течение 2 ч,
поддерживая температуру (24±1)°С. Определяют
вязкость раствора в течение 15 мин при температу¬
ре (24±1) °С и 60 об/мин с помощью ротационного
вискозиметра, оснащенного вращающимся шпин¬
делем с диаметром 12,7 мм и высотой 1,6 мм, со¬
единенным с осью диаметром 3,2 мм. Расстояние
от верхней части цилиндра до нижней точки оси со¬
ставляет 25,4 мм, глубина погружения — 50,0 мм.
Следующие характеристики могут быть
важны для ксантановой камеди, используемой в
качестве матрицеобразующего вещества при
производстве таблеток с пролонгированным вы¬
свобождением.
Кажущаяся вязкость: см. выше.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошков (2.9.36).
73. Зак. 1060.
570
Государственная фармакопея Республики Беларусь
07/2016:1162
КСИЛОМЕТАЗОЛИНА
ГИДРОХЛОРИД
Ху1оте1а2оНп‘1 ЬудгосЫопдит
ХП0МЕТА20ЫЫЕ НУОПОСШОКЮЕ
С1вН24М2 ■ НС1 М.М. 280,8
[1218-35-5]
ОПРЕДЕЛЕНИЕ
2-[4-( 1,1 -Диметилэтил)-2,6-диметилбензил]-4,5-
дигидро-1 Н-имидазола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок.
Легко растворим в воде, 96 % спирте и метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ксилометазолина гидрохлори¬
да #или спектр, представленный на рисунке #1162.-1*
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО ксилометазоли¬
на гидрохлорида растворяют в метаноле Р и доводят
до объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — метанол Р (5:100, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Обработка хлором: на дно хроматографической
камеры помещают химический стакан, содержащий
смесь кислота хлористоводородная Р1 — водаР—
раствор 15 г/л калия перманганата Р (1:1:2, об/об/об).
Камеру закрывают и выдерживают в течение 15 мин.
Высушенную пластинку выдерживают в закрытой ка¬
мере в парах хлора в течение 5 мин. Затем пластинку
обрабатывают струей холодного воздуха до удаления
избытка хлора (пластинка ниже точек нанесения рас¬
творов не должна окрашиваться в синий цвет от при¬
бавления капли раствора калия йодида и крахмала Р).
Проявление: пластинку опрыскивают раствором
калия йодида и крахмала Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
C. 0,5 мг испытуемого образца растворяют в 1 мл
метанола Р, прибавляют 0,5 мл свежеприготовлен¬
ного раствора 50 г/л натрия нитропруссида Р, 0,5 мл
раствора 20 г/л натрия гидроксида Р и выдерживают
в течение 10 мин. Прибавляют 1 мл раствора 80 г/л
натрия гидрокарбоната Р. Появляется фиолетовое
окрашивание.
число (см'1)
Рисунок #1162.-1. Инфракрасный спектр ФСО ксилометазолина гидрохлорида
Ксилометазолина гидрохлорид
571
О. 0,2 г испытуемого образца растворяют в 1 мл
воды Р, прибавляют 2,5 мл 96 % спирта Р, 2 мл 1 М
раствора натрия гидроксида и тщательно переме¬
шивают. При просмотре в ультрафиолетовом свете
при длине волны 365 нм полученный раствор не об¬
ладает флуоресценцией, либо его флуоресценция не
интенсивнее флуоресценции контрольного раствора,
приготовленного аналогично, но без испытуемого об¬
разца. Результаты испытаний считаются достоверны¬
ми, если раствор, приготовленный аналогично, но с
использованием вместо испытуемого образца ФСО
нафазолина гидрохлорида, обладает отчетливой си¬
неватой флуоресценцией.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.7).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50,0 мл этим же
растворителем.
Прозрачность (2.2.7). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Кислотность или щелочность. 0,25 г испытуе¬
мого образца растворяют в воде, свободной от угле-
рода диоксида, Р и доводят до объема 25 мл этим
же растворителем. Прибавляют 0,1 мл раствора
метилового красного Р и 0,1 мл 0,01 М раствора
кислоты хлористоводородной. Должно появиться
красное окрашивание. При прибавлении не более
0,2 мл 0,01 М раствора натрия гидроксида должно
появиться желтое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем. Перед хроматогра¬
фированием раствор выдерживают в течение 1 ч.
Раствор сравнения (а). 5,0 мл испытуемого
раствора доводят водой Р до объема 100,0 мл. 2,0 мл
полученного раствора доводят водой Р до объема
100.0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО ксиломета¬
золина примеси А и 5 мг испытуемого образца раст¬
воряют в воде Р и доводят до объема 50,0 мл этим же
растворителем. 10,0 мл полученного раствора дово¬
дят водой Р до объема 50,0 мл.
Раствор сравнения (с). 5,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, с введенными полярными группами,
эндкепированным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 1,36 г/л калия ди¬
гидрофосфата Р, доведенный кислотой фос¬
форной Р до рН 3,0;
- подвижная фаза В: ацетонитрил Р1;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
70
30
5—20
70 —► 15
30-> 85
20—35
15
85
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к ксилометазолину, время удерживания — около
7,2 мин): примесь А—около 0,79.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками при¬
меси А и ксилометазолина.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворят в 25 мл
кислоты уксусной безводной Р, прибавляют 10 мл ан¬
гидрида уксусного Р и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 28,08 мг С16Н24Ы2-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацев¬
тического использования. Вследствие этого нет
необходимости идентифицировать эти примеси
для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использова¬
ния): В, С, О, Е, Р.
73*. Зак. 1060.
572
Гэсударственная фармакопея Республики Беларусь
A. В = СН2-СО-МН-СН2-СН2-1\1Н2: Л/-(2-Аминоэтил)-
2-[4-(1,1-диметилэтил)-2,6-диметилфенил]ацетамид.
B. К = СН2-С1:2-(Хлорметил)-5-(1,1-диметилэтил)-
1,3-диметилбензол.
C. К = СН2-СМ: [4-(1,1-Диметилэтил)-2,6-диметил-
фенил]ацетонитри л.
O. К = Н: 1-(1,1-Диметилэтил)-3,5-диметилбензол.
P. К = СН2-С02Н: [4-(1,1-Диметилэтил)-2,6-диметил-
фенил]уксусная кислота.
Е. Этан-1,2-диамина моно(4-метилбензол-суль-
фонат).
07/2016:1342
КУКУРУЗНОЕ МАСЛО
РАФИНИРОВАННОЕ
МаусИз о1еит га№па1ит
МАНЕ ОН, КЕРШЕО
ОПРЕДЕЛЕНИЕ
Рафинированное кукурузное масло представ¬
ляет собой жирное масло, полученное из семян 2еа
тауз Ь. методом отжима или экстракцией с последую¬
щим рафинированием.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачное светло-желтое или желтое масло.
Практически нерастворимо в воде и 96 % спирте,
смешивается с петролейным эфиром (температура
кипения от 40 °С до 60 °С) и метиленхлоридом.
Относительная плотность: около 0,920.
Показатель преломления: около 1,474.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Тонкослойная хроматография (2.3.2).
Результаты: хроматограмма испытуемого раст¬
вора соответствует хроматограмме раствора сравнения.
B. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 0,5 или не
более 0,3, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения. Определение проводят из 10,0 г испыту¬
емого образца.
Перекисное (пероксидное) число (2.5.5, метод
А). Не более 10,0 или не более 5,0, если субстанция
предназначена для производства лекарственных
средств для парентерального применения.
Неомыляемые вещества (2.5.7). Не более
2,8 %. Определение проводят из 5,0 г испытуемого
образца.
Щелочные примеси в жирных маслах (2.4.19).
Испытуемый образец должен выдерживать испыта¬
ние на щелочные примеси в жирных маслах.
Состав жирных кислот (2.4.22, метод А). При¬
меняют смесь веществ для калибровки в соответ¬
ствии с таблицей 2.4.22.-3.
Состав фракции жирных кислот масла:
- насыщенные жирные кислоты с длиной цепи
менее С16: не более 0,6 %;
- пальмитиновая кислота: от 8,6 % до 16,5 %;
- стеариновая кислота: не более 3,3 %;
- олеиновая кислота: от 20,0 % до 42,2 %;
- линолевая кислота: от 39,4 % до 65,6 %;
- линоленовая кислота: от 0,5 % до 1,5 %;
- арахидоновая кислота: не более 0,8 %;
- эйкозеновая кислота: не более 0,5 %;
- бегеновая кислота: не более 0,5 %;
- другие жирные кислоты: не более 0,5 %.
Стерины (2.4.23). Стериновая фракция масла
должна содержать не более 0,3 % брассикастерола.
Вода (2.5.32). Не более 0,1 %. Определение про¬
водят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте, при температуре
не выше 25 °С.
МАРКИРОВКА
Указывают:
- метод получения масла (механический отжим
или экстракция).
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:0344
КУКУРУЗНЫЙ КРАХМАЛ
МаусИз ату1ит
МАНЕ ЗТАКСН
ОПРЕДЕЛЕНИЕ
Крахмал кукурузный получают из зерен 1еа тауз II.
ОПИСАНИЕ (СВОЙСТВА)
Очень мелкий матовый порошок от белого до
слегка желтоватого цвета, при сжатии между пальца¬
ми скрипит.
Практически нерастворим в холодной воде и в
96 % спирте.
Наличие зерен крахмала с обломанными или не¬
ровными краями возможно в исключительных случаях.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Проводят микроскопический анализ (2.8.23) с
использованием 50 % (об/об) раствора глицерина Р.
Испытуемый образец представляет собой либо за¬
остренные многогранные зерна различных размеров
с диаметром приблизительно от 2 мкм до 23 мкм,
либо округлые или сферические зерна различных
размеров с диаметром приблизительно от 25 мкм до
35 мкм (рисунок 0344.-1). Центральное ядро состо¬
ит из отдельной полости или лучевидной трещины,
Лавандовое масло
573
состоящей из (2—5) лучей; не должно наблюдаться
концентрической бороздчатости. При рассмотрении
между перпендикулярно ориентированными поляри¬
зующими пластинками или призмами на зернах при¬
сутствует отчетливый черный крестик с пересечением
в области ядра.
07/2016:1338
ЛАВАНДОВОЕ МАСЛО
1.ачапди1ае аеНюго/еит
1.АУЕЫОЕК ОН
Рисунок 0344.-1. Рисунок кукурузного крахмала
(как указано в испытании В на подлинность)
B. 1 г испытуемого образца суспендируют в 50 мл
воды Р, кипятят в течение 1 мин и охлаждают. Образу¬
ется мутный водянистый клейстер.
C. К 1 мл клейстера, полученного в подлинности
В, прибавляют 0,05 мл раствора йода Р1. Появляет¬
ся окраска от оранжево-красной до темно-синей, ис¬
чезающая при нагревании.
ИСПЫТАНИЯ
рН (2.2.3). От 4,0 до 7,0. 5,0 г испытуемого образ¬
ца встряхивают с 25,0 мл воды, свободной от углеро¬
да диоксида, Р в течение 60 с при небольшой скоро¬
сти. Выдерживают в течение 15 мин.
Посторонние вещества. При изучении под ми¬
кроскопом с использованием 50 % (об/об) раствора
глицерина Р должны обнаруживаться не более чем
следовые количества частиц, отличных от зерен крах¬
мала. Присутствие зерен крахмала любого другого
происхождения не допускается.
Окисляющие вещества (2.5.30). Не более
0,0020 % (20 ррт), в пересчете на Н202.
Серы диоксид (2.5.29). Не более 0,0050 %
(50 ррт).
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,5 г
испытуемого образца встряхивают с 15 мл кислоты
хлористоводородной разведенной Р и фильтруют.
Фильтрат должен выдерживать испытание на железо.
Потеря в массе при высушивании (2.2.32). Не
более 15,0 %. 1,000 г испытуемого образца сушат при
температуре 130 °С в течение 90 мин.
Сульфатная зола (2.4.14). Не более 0,6%.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа соИ: отсутствие (2.6.13).
8а1топе11а: отсутствие (2.6.13).
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное перегонкой с во¬
дяным паром из цветущих верхушек 1.ауапс1и1а
апдизШоНа МШ. (1.ауапби1а оШтаНз СНа1х).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная или бледно-желтая жид¬
кость со сложным запахом, напоминающим запах ли-
налилацетата.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мкл испытуемого об¬
разца растворяют в 1 мл толуола Р.
Раствор сравнения. 10 мкл линалола Р, 10 мкл
цинеола Р и 10 мкл линалилацетата Р растворяют в
1 мл толуола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 10 мкл [или 2 мкл] в
виде полос длиной 10 мм [или 6 мм].
Фронт подвижной фазы: не менее 10 см [или
8 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин; просматри¬
вают незамедлительно при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны фио¬
летово-красного или зеленовато-коричневого цвета,
расположенные между зоной линолилацетата и фрон¬
том подвижной фазы.
Верх хроматографической пластинки
Зона фиолетово-красного
или зеленовато-коричнево¬
го цвета
Линалилацетат: зона фио¬
летового или коричневого
цвета
Зона фиолетового или ко¬
ричневого цвета (линалила-
цетат)
Зона фиолетово-красного
цвета
1,8-Цинеол: зона фиолето¬
во-коричневого цвета
Линалол: зона фиолетового
или коричневого цвета
Возможно наличие неинтен¬
сивной зоны фиолетово-ко¬
ричневого цвета (1,8-цинеол)
Зона фиолетового или ко¬
ричневого цвета (линалол)
Неинтенсивная зона желто¬
вато-коричневого цвета
Несколько неразделенных
зон
Раствор сравнения
Испытуемый раствор
74. Зак. 1060.
574
Гэсударственная фармакопея Республики Беларусь
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Результаты: характерные пики на хроматограм¬
ме испытуемого раствора соответствуют по времени
удерживания характерным пикам на хроматограмме
раствора сравнения (а).
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,878 до
0,892.
Показатель преломления (2.2.5). От 1,455 до
1,466.
Угол оптического вращения (2.2.7). От -12,5°
до -6,0°.
Кислотное число (2.5.1). Не более 1,0. Опреде¬
ление проводят из 5,0 г испытуемого образца, раство¬
ренного в 50 мл предписанной смеси растворителей.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 200 мкл испытуемого об¬
разца растворяют в гептане Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 5 мкл лимонена Р, 5 мкл
цинеола Р, 5 мкл 3-октанона Р, 5 мг камфоры Р,
40 мкл линалола Р, 50 мкл линалилацетата Р, 10 мкл
терпинен-4-ола Р, 5 мкл лавандулилацетата Р,
5 мкл лавандулола Р и 5 мг а-терпинеола Р раство¬
ряют в гептане Р и доводят до объема 10 мл этим же
растворителем.
Раствор сравнения (Ь). 5 мкл лимонена Р раст¬
воряют в гептане Р и доводят до объема 50,0 мл
этим же растворителем. 0,5 мл полученного раствора
доводят гептаном Р до объема 5,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м и
внутренним диаметром 0,25 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—15
70
15—70
о
со
■с—
т
о
I
I
Блок ввода проб
220
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения (а); отмечают време¬
на удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,4 между пиками тер-
пинен-4-ола и лавандулилацетата.
По временам удерживания компонентов раст¬
вора сравнения (а) определяют состав испытуемого
раствора.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- лимонен: не более 1,0 %;
- 1,8-цинеол: не более 2,5 %;
- 3-октанон: от 0,1 % до 5,0 %;
- камфора: не более 1,2 %;
- линапол: от 20,0 % до 45,0 %;
- линалилацетат: от 25,0 % до 47,0 %;
- терпинен-4-ол: от 0,1 % до 8,0 %;
- лавандулилацетат: не менее 0,2 %;
- лавандулол: не менее 0,1 %;
- а-терпинеол: не более 2,0 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (Ь).
Хиральная чистота. Газовая хроматография
(2.2.28).
Испытуемый раствор. 0,02 г испытуемого об¬
разца растворяют в пентане Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 10 мкл линалола Р (смесь
из (Я)-линалола и (З)-линалола), 10 мкл линали¬
лацетата Р (смесь (Я)-линалилацетата и ($)-
линалилацетата) и 5 мг борнеола Р растворяют в
пентане Р и доводят до объема 10 мл этим же раст¬
ворителем.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 25 м и
внутренним диаметром 0,25 мм, покрытая слоем мо¬
дифицированного р-циклодекстрина для хиральной
хроматографии Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1,3 мл/мин;
- деление потока: 1:30;
- температура:
Время (мин)
Температура (°С)
Колонка
0—65
50 — 180
Блок ввода проб
230
Детектор
230
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: (Я)-линалол, (З)-линалол,
борнеол, (/?)-линалилацетат и (З)-линалилацетат; в
зависимости от условий проведения испытания и со¬
стояния колонки борнеол может элюироваться до или
после (З)-линалола.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 5,5 между пиками (Я)-
линалола и (З)-линалола; не менее 2,9 между пиками
(З)-линалола и борнеола; не менее 2,0 между пиками
(Я)-линалилацетата и (З)-линалилацетата.
Содержание специфицированных (^-энантиоме¬
ров в процентах рассчитывают по формуле:
где:
А3 — площадь пика соответствующего (5)-
энантиомера;
Ап — площадь пика соответствующего (Р)-
энантиомера.
Предельное содержание примесей:
- (З)-линалол: не более 12 %;
- (З)-линалилацетат: не более 1 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Лактоза безводная
575
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:1061
ЛАКТОЗА БЕЗВОДНАЯ
1-ас1озит апЬус/псит
1-АСТ08Е, АЫНУОК0118
он он
а-лактоза р-лактоза
М.м. 342.3
[63-42-3]
ОПРЕДЕЛЕНИЕ
0-р-0-галактопиранозил-(1-*4)-р-0-глюкопираноза
или смесь из 0-р-0-галактопиранозил-(1-*4)-а-0-
глюкопиранозы и 0-р-0-галактопиранозил-(1->4)-р-0-
глюкопиранозы.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворима в воде, практически нераство¬
рима в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лактозы безводной.
B. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(40:60, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО лактозы без¬
водной растворяют в смеси растворителей и дово¬
дят до объема 20 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг фруктозы Р, 10 мг
глюкозы Р, 10 мг лактозы Р и 10 мг сахарозы Р раст¬
воряют в смеси растворителей и доводят до объема
20 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Поавижная <$та\ аойаР — «атт? —
кислота уксусная ледяная Р — этиленхлорид Р
(10:15:25:50, об/об/об/об); отмеряют объемы точно,
так как небольшой избыток воды вызывает помут-
нение.
Наносимый объем пробы: 2 мкл; тщательно вы¬
сушивают нанесенные пробы.
Фронт подвижной фазы А: не менее 3/4 высоты
пластинки.
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 3/4 высоты пластинки, после замены подвижной
фазы на новую.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р и нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются четыре
полностью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
С. 0,25 г испытуемого образца растворяют в 5 мл
воды Р, прибавляют 5 мл раствора аммиака Р и на¬
гревают на водяной бане при температуре 80 °С в те¬
чение 10 мин. Появляется красное окрашивание.
й. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в кипящей воде Р, охлаждают и доводят до объ¬
ема 10,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
Кислотность или щелочность. 6,0 г испытуемо¬
го образца растворяют при нагревании в 25 мл воды,
свободной от углерода диоксида, Р, охлаждают и
прибавляют 0,3 мл раствора фенолфталеина Р1.
Раствор бесцветный. При прибавлении не более
0,4 мл 0,1 М раствора натрия гидроксида должно
появиться розовое или красное окрашивание.
Удельное оптическое вращение (2.2.7). От
+54,4° до +55,9° (в пересчете на безводное вещество).
10,0 г испытуемого образца растворяют в 80 мл воды Р,
нагревают до 50 °С и охлаждают. Прибавляют 0,2 мл
раствора аммиака разведенного Р1, выдерживают в
течение 30 мин и доводят до объема 100,0 мл водой Р.
Оптическая плотность (2.2.25).
Испытуемый раствор (а). Раствор 3.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят до объема 10,0 мл водой Р.
Диапазон длин волн: 400 нм для испытуемого раст¬
вора (а) и (210—300) нм для испытуемого раствора (Ь).
Результаты:
- при 400 нм: не более 0,04 для испытуемого
раствора (а);
- в диапазоне длин волн от 210 нм до 220 нм: не
более 0,25 для испытуемого раствора (Ь);
- в диапазоне длин волн от 270 нм до 300 нм: не
более 0,07 для испытуемого раствора (Ь).
ттм
0,0005 % (5 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 1,0%. Определение
проводят из 1,00 г испытуемого образца, используя в
качестве растворителя смесь формамид Р — мета¬
нол Р (1:2, об/об).
74*. Зак. 1060.
576
Государственная фармакопея Республики Беларусь
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕвсЬепсЫа соН: отсутствие (2.6.13).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важны
для лактозы безводной, используемой в качестве на-
полнителя/разбавителя при производстве твердых
дозированных форм (прессованных и в порошках).
Распределение частиц по размерам (2.9.31
или 2.9.38).
Насыпная плотность и плотность после усад¬
ки (2.9.34). Определяют насыпную плотность и плот¬
ность после усадки. Отношение Хауснера рассчиты¬
вают по формуле:
Ун
V,'
где:
У0 — насыпной объем до усадки;
V)— конечный объем после усадки.
а-Лактоза и р-лактоза. Газовая хроматография
(2.2.28).
Силилирующий реактив. Диметилсульфоксид
Р — А1-триметилсилилимидазол Р — пиридин Р
(19,5:22:58,5, об/об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца помещают в контейнер с завинчивающейся
крышкой, прибавляют 4 мл силилирующего реакти¬
ва и обрабатывают ультразвуком в течение 20 мин
при комнатной температуре. Охлаждают, переносят
400 мкл полученного раствора в контейнер для ввода
проб, прибавляют 1 мл пиридина Р, закрывают кон¬
тейнер и тщательно перемешивают.
Раствор сравнения. Готовят смесь а-лактозы
моногидрата Р и Р-лактозы Р, имеющих аномер-
ное соотношение около 1:1 исходя из номинального
аномерного содержания а-лактозы моногидрата и
Р-лактозы. 10 мг приготовленной смеси помещают в
контейнер с завинчивающейся крышкой, прибавля¬
ют 4 мл силилирующего реактива и обрабатывают
ультразвуком в течение 20 мин при комнатной темпе¬
ратуре. Охлаждают, переносят 400 мкл полученного
раствора во флакон для автосамплера, прибавляют
1 мл пиридина Р, закрывают контейнер и тщательно
перемешивают.
Условия хроматографирования:
- предколонка деактивированная кварцевая
среднеполярная длиной 2 м и внутренним диаметром
0,53 мм;
- колонка кварцевая капиллярная длиной 15м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)(дифенил)силоксана Р (толщина плен¬
ки 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 2,8 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1
80
1—3
80 — 150
3—15,5
о
о
со
т
ю
15,5—17,5
300
Блок ввода проб
275 или используют
холодный ввод проб
Детектор
325
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,5 мкл, без деления
потока или холодным вводом пробы.
Относительное удерживание (по отношению
к р-лактозе, время удерживания — около 12 мин):
а-лактоза — около 0,9.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 3,0 между пиками
а-лактозы и р-лактозы.
Содержание а-лактозы рассчитывают в процен¬
тах по формуле:
Ю0-$а
Содержание р-лактозы рассчитывают в процен¬
тах по формуле:
100-5Ь
5.+5/
где:
5а — площадь пика а-лактозы;
5Ь — площадь пика р-лактозы.
Потеря в массе при высушивании (2.2.32).
1,000 г испытуемого образца сушат при температуре
80 °С в течение 2 ч.
ЛАКТОЗА МОНОГИДРАТ
1.ас1озит топоЬудпсит
1.АСТОЗЕ МОЫОНУОКАТЕ
ОНО но
*'1212
07/2016:0187
Н20
М.м. 360,3
Лактоза моногидрат
577
ОПРЕДЕЛЕНИЕ
0-Р-0-Галакгопиранозил-(1—м^-а-О-глюкопираноза
моногидрат.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворима в воде, практически нераство¬
рима в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лактозы моногидрата.
B. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(2:3, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО лактозы мо¬
ногидрата растворяют в смеси растворителей и до¬
водят до объема 20 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг фруктозы Р, 10 мг
глюкозы Р, 10 мг лактозы Р и 10 мг сахарозы Р раст¬
воряют в смеси растворителей и доводят до объема
20 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: вода Р—метанол Р—кислота
уксусная ледяная Р — этиленхлорид Р (10:15:25:50,
об/об/об/об); отмеряют объемы точно, так как неболь¬
шой избыток воды вызывает помутнение.
Наносимый объем пробы: 2 мкл; тщательно вы¬
сушивают нанесенные пробы.
Фронт подвижной фазы А: не менее 3/4 высоты
пластинки.
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 3/4 высоты пластинки, после замены подвижной
фазы на новую.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р и нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются четыре
полностью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
C. 0,25 г испытуемого образца растворяют в 5 мл
воды Р, прибавляют 5 мл раствора аммиака Р и на¬
гревают на водяной бане при температуре 80 °С в те¬
чение 10 мин. Появляется красное окрашивание.
О. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в кипящей воде Р, доводят до объема 10,0 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть не интенсивнее эталона ВУ(КЖ)7.
Кислотность или щелочность. 6,0 г испыту¬
емого образца растворяют при нагревании в 25 мл
воды, свободной от углерода диоксида, Р, охлажда¬
ют, прибавляют 0,3 мл раствора фенолфталеина Р1.
Раствор должен быть бесцветным. При прибавлении
не более 0,4 мл 0,1 М раствора натрия гидроксида
должно появиться розовое или красное окрашивание.
Удельное оптическое вращение (2.2.7). От
+54,4 до +55,9 (в пересчете на безводное вещество).
10,0 г испытуемого образца растворяют в 80 мл
воды Р, нагревают до 50 °С и охлаждают. Прибавляют
0,2 мл раствора аммиака разведенного Р1, выдер¬
живают в течение 30 мин и доводят водой Р до объ¬
ема 100,0 мл.
Оптическая плотность (2.2.25).
Испытуемый раствор (а). Раствор 5.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят водой Р до объема 10,0 мл.
Диапазон длин волн: 400 нм для испытуемого
раствора (а) и (210—300) нм для испытуемого раст¬
вора (Ь).
Результаты:
- при 400 нм: не более 0,04 для испытуемого
раствора (а);
- в диапазоне длин волн от 210 нм до 220 нм: не
более 0,25 для испытуемого раствора (Ь);
- в диапазоне длин волн от 270 нм до 300 нм: не
более 0,07 для испытуемого раствора (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 4,0 г испытуемого образца раство¬
ряют в воде Р при нагревании, прибавляют 1 мл 0,1 М
раствора кислоты хлористоводородной и доводят
водой Р до объема 20 мл. 12 мл полученного раст¬
вора должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Вода (2.5.72). Не менее 4,5 % и не более 5,5 %.
Определение проводят из 0,50 г испытуемого образ¬
ца, используя в качестве растворителя смесь форма-
мид Р — метанол Р (1:2, об/об).
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсНепсЫа соН: отсутствие (2.6.13).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными параме¬
трами, связанными с одной или более функцией суб¬
станции, используемой в качестве вспомогательного
вещества (см. статью 5.15). Данный раздел не явля¬
ется обязательным, и для подтверждения соответ¬
ствия требованиям частной статьи проверка указан¬
ных характеристик не обязательна. Однако контроль
данных характеристик может вносить вклад в каче¬
ство лекарственного средства путем достижения по¬
стоянства производственного процесса и улучшения
свойств лекарственного средства при его использова¬
нии. Предлагаемые методы испытаний являются под¬
ходящими для указанных целей, однако другие методы
также могут быть использованы. В случае указания
результатов конкретных характеристик требуется
указывать также и методы проведения испытаний.
75. Зак. 1060.
578
Гэсударственная фармакопея Республики Беларусь
Следующие характеристики могут быть важ¬
ны для лактозы моногдирата, используемой в каче¬
стве наполнителя/разбавителя при производстве
твердых дозированных форм (прессованных или в
порошках).
Распределение частиц по размерам (2.9.31
или 2.9.38).
Насыпная плотность и плотность после усад¬
ки (2.9.34). Определяют насыпную плотность и плот¬
ность после усадки. Отношение Хауснера рассчиты¬
вают по формуле:
ч
где:
\/0 — насыпной объем до усадки;
Vг— конечный объем после усадки.
07/2016:1230
ЛАКТУЛОЗА
ЕасЮ/озит
1-АСТШ-08Е
СпН220„ М.м. 342,3
[4618-18-2]
ОПРЕДЕЛЕНИЕ
4-0-@-0-Галактопиранозил-0-арабшо-гекс-2-уло-
фураноза.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворим в воде, умеренно растворим в
метаноле, практически нерастворим в толуоле.
Температура плавления: около 168 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С, О, Е.
Вторая идентификация: А, С, О, Е.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения. 50,0 мг ФСО лактулозы
растворяют в водеР и доводят до объема 10,0 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: кислота уксусная ледяная Р —
раствор 50 г/л кислоты борной Р — метанол Р —
этилацетат Р (10:15:20:55, об/об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: при температуре от 100 °С до
105 °С в течение 5 мин и охлаждают.
Проявление: пластинку опрыскивают раствором
1.0 г/л 1,3-дигидроксинафталина Р в смеси из кисло¬
ты серной Р и метанола Р (10:90, об/об) и нагревают
при температуре 110 °С в течение 5 мин.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения.
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (Ь).
C. 50 мг испытуемого образца растворяют в 10 мл
воды Р, прибавляют 3 мл раствора медно-тартрат-
ного Р и нагревают. Образуется красный осадок.
0.0,125 г испытуемого образца растворяют в
5 мл воды Р, прибавляют 5 мл раствора аммиака Р
и нагревают на водяной бане при температуре 80 °С
в течение 10 мин. Появляется красное окрашивание.
Е. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
ИСПЫТАНИЯ
Раствор 3. 3,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)5.
рН (2.2.3). От 3,0 до 7,0. К 10 мл раствора 3 при¬
бавляют 0,1 мл насыщенного раствора калия хлори¬
да Р.
Удельное оптическое вращение (2.2.7). От
-46,0 до -50,0 (в пересчете на безводное вещество).
1,25 г испытуемого образца растворяют в воде Р, при¬
бавляют 0,2 мл раствора аммиака концентрирован¬
ного Р и доводят водой Р до объема 25,0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в 10 мл воды Р. К полученному рас¬
твору прибавляют 12,5 мл ацетонитрила Р, слегка
нагревая, и доводят водой Р до объема 25,0 мл.
Раствор сравнения (а). К 3,0 мл испытуемого
раствора прибавляют 47,5 мл ацетонитрила Р, слег¬
ка нагревая, и доводят водой Р до объема 100,0 мл.
Раствор сравнения (Ь). 1,00 г ФСО лактулозы
растворяют в 10 мл воды Р. К полученному раствору
прибавляют 12,5 мл ацетонитрила Р, слегка нагре¬
вая, и доводят водой Р до объема 25,0 мл.
Раствор сравнения (с). 10 мг лактулозы Р, 10 мг
апилактозы Р (примесь А) и 10 мг лактозы Р (при¬
месь С) растворяют в воде Р и доводят до объема
5.0 мл этим же растворителем.
Раствор сравнения (д). К 5,0 мл испытуемого
Лактулоза
579
раствора прибавляют 47,5 мл ацетонитрила Р, слег¬
ка нагревая, и доводят водой Р до объема 100,0 мл.
5.0 мл полученного раствора доводят до объема
100.0 мл смесью из равных объемов ацетонитри¬
ла Р и воды Р.
Условия хроматографирования:
- колонка 1 длиной 0,05 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем аминопропил-
силильным для хроматографии Р с размером частиц
3 мкм;
- температура: (38±1) °С;
- колонка 2 длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем аминопропил-
синильным для хроматографии Р с размером частиц
3 мкм;
- температура: (38±1) °С; колонки 1 и 2 распо¬
лагаются последовательно;
- подвижная фаза: 0,253 г натрия дигидрофос¬
фата Р растворяют в 200 мл воды Р и доводят аце¬
тонитрилом Р до объема 1000 мл;
- скорость подвижной фазы. 1,0 мл/мин;
- рефрактометрический детектор, при посто¬
янной температуре;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а), (с) и (б);
- время хроматографирования: 2-кратное вре¬
мя удерживания лактулозы.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и С, используя хроматограмму
раствора сравнения (с).
Относительное удерживание (по отношению к
лактулозе, время удерживания — около 18 мин): при¬
месь А — около 0,9; примесь С — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 5,0
(Нр — высота пика примеси А относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси А и пик
лактулозы).
Предельное содержание примесей:
- примесь С (не более 3,0 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси С, не должна превышать пло¬
щадь пика лактулозы на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
С, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (б);
- сумма примесей (не более 3,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь пика лактулозы на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,25 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади пика лактулозы на хрома¬
тограмме раствора сравнения (б).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Метанол. Парофазная газовая хроматография
(2.2.28).
Раствор внутреннего стандарта. 0,5 мл пропа¬
нола Р смешивают с 100,0 мл воды Р. 1,0 мл получен¬
ного раствора доводят водой Р до объема 100,0 мл.
5.0 мл полученного раствора доводят водой Р до объ¬
ема 50,0 мл.
Испытуемый раствор. 79 мг испытуемого об¬
разца помещают в контейнер вместимостью 20 мл,
прибавляют 1,0 мл раствора внутреннего стандарта и
5 мкл 0,1 % (об/об) раствора метанола Р.
Раствор сравнения. 1,0 мл раствора внутренне¬
го стандарта помещают в контейнер вместимостью
20 мл и прибавляют 5 мкл 0,1 % (об/об) раствора ме¬
танола Р.
Условия хроматографирования:
- колонка длиной 2 м и диаметром 2 мм, запол¬
ненная сополимером этилвинилбензол-дивинилбен-
зола Р с размером гранул 180 мкм;
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 30 мл/мин;
- параметры парофазного пробоотборника:
- равновесная температура: 60 °С;
- время достижения равновесия: 1 ч;
- время приложения избыточного давления. 1 мин;
- температура:
- колонка: 140 °С;
- блок ввода проб: 200 °С;
- детектор: 220 °С.
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мл газовой фазы.
Рассчитывают содержание метанола, принимая
его плотность (2.2.5) равной 0,79 г/мл при 20 °С.
Предельное содержание примесей:
- метанол (не более 0,0050 % (50 ррт)): исполь¬
зуя хроматограмму раствора сравнения рассчитыва¬
ют отношение (Р) площади пика метанола к площади
пика внутреннего стандарта; используя хроматограм¬
му испытуемого раствора рассчитывают отношение
площади пика метанола к площади пика внутреннего
стандарта: полученное значение не должно превы¬
шать значение 2Р.
Бор. Не более 0,0009 % (9 ррт). По возможно¬
сти необходимо избегать использования стеклян¬
ной посуды.
Раствор сравнения. 50,0 мг кислоты борной Р
растворяют в воде Р и доводят до объема 100,0 мл
этим же растворителем. 5,0 мл полученного раствора
доводят водой Р до объема 100,0 мл. Полученный
раствор необходимо хранить в плотно закрытом
полиэтиленовом контейнере.
В 4 полиэтиленовые колбы вместимостью 25 мл
помещают по отдельности:
- 0,50 г испытуемого образца, растворенного в
2.0 мл воды Р (раствор А);
- 0,50 г испытуемого образца, растворенного в
1.0 мл раствора сравнения и 1,0 мл воды Р (раствор В);
- 1,0 мл раствора сравнения и 1,0 мл воды Р
(раствор С);
- 2,0 мл воды Р (раствор И).
К содержимому каждой колбы прибавляют по
4.0 мл ацетатно-эдетатного буферного раствора
рН 5,5 Р и перемешивают. К полученным растворам
прибавляют по 4,0 мл свежеприготовленного раст¬
вора азометина Н Р, перемешивают и выдерживают в
течение 1 ч. Измеряют оптическую плотность (2.2.25)
растворов А, В и С при длине волны 420 нм, используя
раствор й в качестве компенсационного раствора. Ис¬
пытание считается недействительным, если оптиче¬
ская плотность раствора С составляет менее 0,25. Оп-
75*. Зак. 1060.
580
Гэсударственная фармакопея Республики Беларусь
тическая плотность раствора В должна быть не менее
2-кратной оптической плотности раствора А.
Свинец (2.4.10). Не более 0,5 ррт.
Вода (2.5.12). Не более 2,5 %. Определение про¬
водят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсНепсЫа со//: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы, по 20 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С^Н^О^ в процентах рассчитывают
с учетом содержания лактулозы в ФСО лактулозы.
ПРИМЕСИ
Специфицированные примеси: С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, О, Е.
А. 4-О-р-О-Галактопиранозил-О-маннопираноза
(эпилактоза).
В. 0-Галактопираноза (галактоза).
С. 4-О-р-О-Галактопиранозил-О-глюкопираноза
(лактоза).
о ^—он
ой/
он>—* он
он
й. О-арабино-Гекс-2-улопираноза (фруктоза).
он
Е. О-ликсо-Гекс-2-улопираноза (тагатоза).
07/2016:1756
ЛАМОТРИДЖИН
1-атоМдтит
ШМОТМв1ЫЕ
С9Н7С12Ы5 М.м. 256,1
[84057-84-1]
ОПРЕДЕЛЕНИЕ
6-(2,3-Дихлорфенил)-1,2,4-триазин-3,5-диамин.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде, мало растворим
в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ламотриджина #или спектр,
представленный на рисунке #1756.-1#.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 5 мл метанола Р и доводят рас¬
твором 10,3 г/л кислоты хлористоводородной Р до
объема 100,0 мл.
Раствор сравнения (а). 5 мг ФСО ламотриджи¬
на для проверки пригодности хроматографической
системы (содержит примесь С) растворяют в 2,5 мл
метанола Р и доводят раствором 10,3 г/л кислоты
хлористоводородной Р до объема 25,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят раствором 10,3 г/л кислоты хлористо¬
водородной Р до объема 100,0 мл. 2,0 мл полученно¬
го раствора доводят раствором 10,3 г/л кислоты хло¬
ристоводородной Р до объема 10,0 мл.
Раствор сравнения (с). 5,0 мг ФСО ламотриджи¬
на примеси Е растворяют в смеси из 0,25 мл кислоты
хлористоводородной Р и 45 мл метанола Р и дово-
Ламотриджин
581
Волновое число (см'1)
Рисунок #1756.-1. Инфракрасный спектр ФСО ламотриджина
дят метанолом Р до объема 50,0 мл. 5,0 мл получен¬
ного раствора доводят раствором 10,3 г/л кислоты
хлористоводородной Р до объема 100,0 мл. К 4,0 мл
полученного раствора прибавляют 5 мл метанола Р и
доводят раствором 10,3 г/л кислоты хлористоводо¬
родной Р до объема 100,0 мл.
Раствор сравнения (д). 10 мг ФСО ламотрид¬
жина для идентификации пиков (содержит примеси
А, Е, Р) растворяют в 2,5 мл метанола Р и доводят
раствором 10,3 г/л кислоты хлористоводородной Р
до объема 50,0 мл.
Холостой раствор. Смесь метанола Р и раствора
10,3 г/л кислоты хлористоводородной Р (5:95, об/об).
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: смешивают триэтила-
мин Р и раствор 2,7 г/л калия дигидрофосфа¬
та Р (1:150, об/об); доводят до рН 2,0 кислотой
фосфорной Р;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—4
85
15
4—14
85—>20
15 — 80
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 270 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора, растворов сравнения (а), (Ь) и (б) и холосто¬
го раствора.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, Е и Р, используя хроматограм¬
му раствора сравнения (б) и хроматограмму, прила¬
гаемую к ФСО ламотриджина для идентификации
пиков; идентифицируют пик примеси <3, используя
хроматограмму раствора сравнения (а) и хромато¬
грамму, прилагаемую к ФСО ламотриджина для про¬
верки пригодности хроматографической системы.
Относительное удерживание (по отношению к
ламотриджину, время удерживания — около 7 мин):
примесь О — около 1,1; примесь А— около 1,3; при¬
месь Е — около 1,7; примесь Р — около 1,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 1,2
(Нр — высота пика примеси О относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик ламотриджина и
пик примеси 6).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Р —1,3):
- примесь Р (не более 0,2 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси Р, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
-примеси А и О (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А и О, не должны превышать
0,5 площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, С и Р, не должна превышать 0,5 площади
582
Гэсударственная фармакопея Республики Беларусь
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 0,2 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь); не учитывают
пики холостого раствора и примеси Е.
Примесь Е. Жидкостная хроматография (2.2.29),
как указано в испытании «Сопутствующие примеси»,
со следующими изменениями.
Условия хроматографирования:
- подвижная фаза: ацетонитрил для хромато¬
графии Р — подвижная фаза А (36:65, об/об);
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (б) и (с);
- время хроматографирования: 10 мин.
Время удерживания: примесь Е — около 5,5 мин;
примесь Р — около 8,5 мин.
Пригодность хроматографической системы:
раствор сравнения (б):
- хроматограмма раствора сравнения (б) должна
соответствовать хроматограмме, прилагаемой к ФСО
ламотриджина для идентификации пиков.
Предельное содержание примесей:
- примесь Е (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Е, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с).
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). К остатку, полученному как указа¬
но в испытании «Сульфатная зола», прибавляют 2 мл
кислоты хлористоводородной Р и медленно выпа¬
ривают на водяной бане досуха. Смачивают остаток
0,05 мл кислоты хлористоводородной Р, прибавляют
10 мл кипящей воды Р и нагревают смесь в течение
10 мин на водяной бане. Охлаждают до комнатной
температуры, фильтруют при необходимости, филь¬
трат и промывные воды доводят водой Р до объема
20 мл. 12 мл полученного раствора должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием 10 мл эталонного раствора свин¬
ца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 2,000 г испытуемого образца сушат при
температуре 105 °С и давлении не более 0,7 кПа в те¬
чение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20). Проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 25,61 мг С9Н7С12М5.
ПРИМЕСИ
Специфицированные примеси: А, Е, Р, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
А. 3-Амино-6-(2,3-дихлорфенил)-1,2,4-триазин-
5(4Н)-он.
н2м^^мн2
Т
В. (2Е)-[2-(Диаминометилиден)диазанилиден]-
(2,3-дихлорфенил)ацетонитрил.
С. (27)-[2-(Диаминометилиден)диазанилиден]-
(2,3-дихлорфенил)ацетонитрил.
О. 6-(2,3-Дихлорфенил)-1,2,4-триазин-3,5(2Я,4Я)-
дион.
С1
Е. 2,3-Дихлорбензойная кислота.
Р. А/-[5-Амино-6-(2,3-дихлорфенил)-1,2,4-триазин-
3-ил]-2,3-дихлорбензамид.
Ланолин
583
(3. 6-(2,4-Дихлорфенил)-1,2,4-триазин-3,5-диамин.
07/2016:0134
ЛАНОЛИН
Ас/ерз 1апае
УУ001. РАТ
ОПРЕДЕЛЕНИЕ
Очищенное, безводное, воскообразное веще¬
ство, получаемое из шерсти овцы (Омз апез). Может
содержать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
#Коричнево-желтая густая вязкая масса с харак¬
терным запахом#. При плавлении превращается в
прозрачную или почти прозрачную желтую жидкость.
Раствор в петролейном эфире опалесцирует.
Практически нерастворим в воде, мало раство¬
рим в кипящем этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,5 г испытуемого образца помещают в про¬
бирку, растворяют в 5 мл метиленхлорида Р и при¬
бавляют 1 мл уксусного ангидрида Р и 0,1 мл кисло¬
ты серной Р. Появляется зеленое окрашивание.
B. 50 мг испытуемого образца растворяют в 5 мл
метиленхлорида Р, прибавляют 5 мл кислоты сер¬
ной Р и встряхивают. Появляется красное окрашива¬
ние, в нижнем слое — интенсивная зеленая флуорес¬
ценция при освещении источником дневного света,
расположенным позади наблюдателя.
ИСПЫТАНИЯ
Растворимые в воде кислоты или щелочи.
5,0 г испытуемого образца расплавляют на водяной
бане и энергично встряхивают в течение 2 мин с
75 мл воды Р, предварительно нагретой до темпе¬
ратуры (90—95) °С. Охлаждают и фильтруют через
бумажный фильтр, предварительно промытый во¬
дой Р. К 60 мл фильтрата (который может не быть
прозрачным) прибавляют 0,25 мл раствора бром-
тимолового синего Р1. При прибавлении не более
0,2 мл 0,02 М раствора кислоты хлористоводород¬
ной или не более 0,15 мл 0,02 М раствора натрия
гидроксида окраска раствора должна измениться.
Водопоглощающая способность. Юг рас¬
плавленного испытуемого образца помещают
в ступку и охлаждают до комнатной температу¬
ры. Взвешивают ступку с испытуемым образцом
и прибавляют порциями воду Р из бюретки по
(0,2—0,5) мл. После каждого прибавления воды Р
энергично растирают массу цилиндрическим стерж¬
нем из полипропилена высокой плотности (напри¬
мер, длиной 120 мм и диаметром 10 мм). Концом
испытания является появление видимых капель
воды Р, которые не могут быть поглощенными.
Взвешивают ступку и по разности определяют коли¬
чество абсорбированной воды. Должно абсорбиро¬
ваться не менее 20 г воды Р.
Кислотное число (2.5.1). Не более 1,0. 5,0 г ис¬
пытуемого образца растворяют в 25 мл предписанной
смеси растворителей.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 20. Перед прибавлением 0,5 мл
насыщенного раствора калия йодида Р полученный
раствор охлаждают до комнатной температуры.
Число омыления (2.5.6). От 90 до 105. Опреде¬
ляют из 2,00 г испытуемого образца. Нагревают с об¬
ратным холодильником в течение 4 ч.
Растворимые в воде восстанавливающие ве¬
щества. К 10 мл фильтрата, полученного в испытании
на растворимые в воде кислоты или щелочи, прибав¬
ляют 1 мл кислоты серной разведенной Р и 0,1 мл
0,02 М раствора калия перманганата. Через 10 мин
раствор должен сохранять розовое окрашивание.
Парафины. Не более 1,0 %. Используемые кран
и ватные пробки не должны содержать смазку. Подго¬
тавливают колонку длиной 0,23 м и диаметром 20 мм,
содержащую алюминия оксид безводный, прибавляя
суспензию алюминия оксида безводного Р в петролей¬
ном эфире Р1 в стеклянную трубку с краником, содер¬
жащую петролейный эфир Р1 (перед использованием
алюминия оксид безводный дегидратируют, выдержи¬
вая его в печи при температуре 600 °С в течение 3 ч).
Дают суспензии отстояться и уменьшают слой раство¬
рителя над колонкой до 40 мм. 3,0 г испытуемого об¬
разца растворяют в 50 мл теплого петролейного эфи¬
ра Р1, охлаждают, пропускают раствор через колонку
со скоростью 3 мл/мин и промывают 250 мл петролей¬
ного эфира Р1. Собранные элюаты и промывные рас¬
творы концентрируют до минимального объема путем
дистилляции, выпаривают досуха на водяной бане и
нагревают остаток при температуре 105 °С периода¬
ми по 10 мин до тех пор, пока разница между двумя
последовательными взвешиваниями не станет менее
1 мг. Масса остатка не должна превышать 30 мг.
Остаточные количества пестицидов. Не более
0,05 ррт каждого хлорорганического пестицида, не
более 0,5 ррт любого другого пестицида и не более
1 ррт суммы всех пестицидов.
Вся используемая стеклянная посуда должна
быть тщательно промыта от пестицидов при по¬
мощи моющих средств (детергентов), не содержа¬
щих фосфор. Посуду замачивают в растворе моюще¬
го средства (5% в деионизированной воде) в течение
24 ч. Моющее средство отмывают большим количе¬
ством ацетона или гексана для анализа пестицидов.
Стеклянную посуду для анализа пестицидов хранят
отдельно. Стеклянная посуда не должна содержать
хлорсодержащих растворителей, пластиковых и
резиновых материалов, в особенности фталатных
пластификаторов, окисленных соединений и азотсо¬
держащих растворителей, например ацетонитрила.
Используемые гексан, толуол, ацетон должны быть
специального качества для анализа пестицидов;
этилацетат, циклогексан и вода должны иметь ка¬
чество для ВЭЖХ анализа.
При испытании проводят выделение остатков
пестицидов методом эксклюзионной хроматографии
(2.2.30) с последующей твердофазной экстракцией и
идентификацией методом газовой хроматографии с
использованием детектора электронного захвата или
термоионного детектора.
ВЫДЕЛЕНИЕ ОСТАТОЧНЫХ ПЕСТИЦИДОВ. В
качестве детектора для калибровки колонки для гель-
проникающей хроматографии используют УФ/Вид-
спектрофотометр.
584
Государственная фармакопея Республики Беларусь
Калибровка для гель-проникающей хрома¬
тографии очень важна для проверки постоянства
давления, скорости подвижной фазы, соотношения
растворителей, температуры и сосотояния колон¬
ки. Колонка для гель-проникающей хроматографии
должна регулярно калиброваться с использовани¬
ем смеси стандартов, приготовленной следующим
образом: в мерную колбу вместимостью 1000 мл
помещают 50,00 г масла кукурузного Р, 0,20 г ме-
токсихлора Р и 50,0 мг перилена Р и доводят сме¬
сью из равных объемов циклогексана Р и этила-
цетата Р до объема 1000,0 мл
Для калибровки колонки подают подвижную
фазу, состоящую из равных объемов циклогекса¬
на Р и этилацетата Р, со скоростью 5 мл/мин.
Вводят 5 мкл смеси стандартов и регистрируют
хроматограмму. Времена удерживания аналитов
между калибровками не должны отличаться более
чем на ±5 %. Если время удерживания сдвинулось
более чем на ±5 %, предпринимают корректирую¬
щие действия. Чрезмерный сдвиг времени удержи¬
вания может быть вызван:
- ненадлежащим лабораторным температур¬
ным контролем;
- содержанием воздуха в насосе; это может
быть проверено путем измерения скорости под¬
вижной фазы: собирают 25 мл колоночного элюата
в мерную колбу и отмечают время ((300±5) с);
- протечками в системе.
Изменение давления, скорости подвижной
фазы или температуры колонки, а также загрязне¬
ние колонки могут влиять на времена удержива¬
ния пестицидов и поэтому должны отслеживаться.
Если скорость подвижной фазы или давление ко¬
лонки находятся за пределами заданного диапазо¬
на, то предколонка или колонка подлежат замене.
Испытуемый раствор. 1 г точно взвешенного
испытуемого образца помещают в мерную колбу,
растворяют в смеси из 1 объема этилацетата Р
и 7 объемов циклогексана Р, прибавляют 1 мл вну¬
треннего стандарта (2 ррт, либо изодрин Р, либо
диталимфос Р) и доводят до объема 20 мл этой
же смесью растворителей.
Растворы внутренних стандартов исполь¬
зуются для подтверждения того, что открыва-
емость пестицидов на стадиях очистки с помо¬
щью гель-проникающей хроматографии, выпа¬
ривания и твердофазной экстракции находится
на приемлемом уровне. Уровни открываемости
растворов внутренних стандартов из ланолина
устанавливают путем сравнения площадей пи¬
ков экстрактов ланолина с площадями пиков рас¬
творов внутренних стандартов.
Условия хроматографирования:
- предколонка длиной 0,075 м и внутренним
диаметром 21,2 мм, заполненная сополимером
стирол-дивинилбензола Р с размером гранул
5 мкм;
- колонка для гель-проникающей хромато¬
графии длиной 0,3 м и внутренним диаметром
21,2 мм, заполненная сополимером стирол-диви¬
нилбензола Р с размером гранул 5 мкм;
- подвижная фаза: этилацетат Р — циклогек¬
сан Р (10:70, об/об);
- скорость подвижной фазы: 5 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм.
Вводят 5 мл испытуемого раствора. Первые
95 мл (19 мин) элюата, содержащего испытуемый
образец, отбрасывают, собирая последующие
155 мл элюата (31 мин), где содержатся остаточ¬
ные количества пестицидов, в сосуд для выпари¬
вания.
155 мл элюата, собранного в результате гель-
проникающей хроматографии, выпаривают при
температуре 45 °С и давлении азота 55 кПа до по¬
лучения 0,5 мл остатка.
Для приготовления картриджей для твердо¬
фазной экстракции магния силикат для анализа
остаточных пестицидов Р нагревают в муфель¬
ной печи при температуре 700 °С в течение 4 ч для
удаления влаги и полихлорированных бифенилов.
Далее охлаждают магния силикат в течение 2 ч,
переносят в сушильный шкаф с температурой от
100 °С до 105 °С и выдерживают в течение 30 мин.
Магния силикат переносят в стеклянный сосуд с
пробкой и выдерживают для уравновешивания в
течение 48 ч. Полученный материал может быть
использован в течение 2 недель. После этого срока
магния силикат должен быть реактивирован нагре¬
ванием в муфельной печи при температуре 600 °С
в течение 2 ч. Магния силикат достают из муфель¬
ной печи и хранят в закрытом стеклянном сосуде.
Магния силикат деактивируют путем прибавления
1 % воды Р и последующего периодического встря¬
хивания не менее чем за 15 мин перед использова¬
нием. Деактивированный магния силикат пригоден
для использования в течение 1 недели. Для испы¬
тания используют только деактивированный маг¬
ния силикат.
1 г деактивированного магния силиката по¬
мещают в пустой картридж для твердофазной экс¬
тракции вместимостью 6 мл.
Дальнейшая очистка фракции, полученной по¬
сле гель-проникающей хроматографии, необходи¬
ма, так как на этой стадии она все еще содержит
около 10% испытуемого образца. Проводятся от¬
дельные процедуры выделения для а) хлороргани-
ческих и синтетических пиретроидных пестицидов
и Ь) фосфорорганических пестицидов. Прекондици-
онированный картридж для твердофазной экстрак¬
ции, содержащий 1 г деактивированного магния
силиката для анализа остаточных пестицидов Р,
помещают на вакуумный коллектор.
Кондиционируют картридж, прибавляя 10 мл
толуола Р и позволяя растворителю элюироваться
через картридж. На прекондиционированный кар¬
тридж наносят 0,5 мл фракции, полученной после
выпаривания растворителей. Фракции пестицидов
элюируют с картриджей с помощью 20 мл одной из
двух систем растворителей, описанных ниже:
a) для определения хлорорганических и син¬
тетических птиретроидных пестицидов: толуол Р;
при этом элюируется очень небольшое количество
испытуемого образца;
b) для определения фосфорорганических пе¬
стицидов: смесь из 2 объемов ацетона Р и 98 объ¬
емов толуола Р; данная система растворителей
используется для элюирования всех пестицидов,
включая более полярные фосфорорганические пе¬
стициды; к сожалению, при этом элюируются неко¬
торые компоненты испытуемого образца, которые
могут обнаруживаться с помощью детектора элек¬
тронного захвата.
Ланолин
585
С картриджей для экстракции собирают элюат
в виалы вместимостью 25 мл. Затем элюат количе¬
ственно переносят в сосуд для выпаривания, триж¬
ды промывая виалы гексаном Р порциями по 10 мл.
Содержимое сосуда для выпаривания упари¬
вают до объема 0,5 мл при температуре 45 °С и
давлении азота 55 кПа.
Полученные остатки исследуют с помощью
газовой хроматографии (2.2.28) с использованием
детектора электронного захвата и термоионного
детектора, как описано ниже.
Открываемость. Рассчитывают поправочный
коэффициент открываемости (РсГ) внутренних стан¬
дартов (диталимфос Р или изодрин Р), добавлен¬
ных к испытуемому раствору, по формуле:
— •100,
А
где:
/\1 — площадь пика внутреннего стандарта 1 ррт
в растворе;
А2 — площадь внутреннего стандарта, экстраги¬
рованного из испытуемого раствора.
5 мл из 20 мл испытуемого раствора, содержа¬
щего 1 мл 2 ррт раствора внутреннего стандарта,
сконцентрированного до 0,5 мл, соответствует 1 ррт
внутреннего стандарта в растворе.
Открываемость внутреннего стандарта должна
находиться в диапазоне от 70 % до 110 %.
Растворы сравнения. Готовят растворы сравне¬
ния пестицидов, используя стандарты пестицидов с
концентрацией 0,5 ррт (см. составы растворов срав¬
нения А—О в таблице 0134.-1). Могут быть использо¬
ваны коммерчески доступные стандарты. Отдельные
стандарты имеют концентрацию 10 ррт.
В то же самое время готовят растворы пести¬
цидов, эквивалентные пределу обнаружения метода
(см. рекомендованные составы в таблице 0134.-1).
Эти растворы сравнения используются для оптимиза¬
ции детектора электронного захвата и термоионного
детектора для достижения предела обнаружения ме¬
тода (растворы сравнения Е и Р).
Для приготовления растворов сравнения различ¬
ных концентраций используют калиброванные пипет¬
ки и мерные колбы. Для приготовления растворов вну¬
тренних стандартов 6 и Н используют аналитические
весы до четвертого знака, пипетки и мерные колбы.
Таблица 0134.-1.
Состав растворов сравнения
Раствор сравнения А
(0,5 ррт или 0,5 мг/л)
(хлорорганические и син¬
тетические пиретроидные
пестициды)
Раствор сравнения В
(0,5 ррт или 0,5 мг/л)
(хлорорганические и син¬
тетические пиретроидные
пестициды)
Гзксахлорбензол Р
Гэптахпор Р
Гептахлорэпоксид Р
о, п'-ДДЕ Р
п, п’-ДДЕ Р
п,п'-ДДТ Р
Дельтаметрин Р
ЛинданР
Текназол Р
Циперметрин Р
Цихалотрин Р
ЭндринР
АлдринР
а-Гэксахлорциклогексан Р
Р-Гексахлорциклогексан Р
5-Гэксахлорциклогексан Р
ол'-ДДДР
п,п’-ДДДР
о.п’-ДДТ Р
Диэлдрин Р
Метоксихлор Р
Перметрин Р
Фенвалерат Р
а-Эндосульфан Р
Р-Эндосульфан Р
Раствор сравнения С
(0,5 ррт или 0,5 мг/л)
(фосфорорганические пе¬
стициды)
Раствор сравнения Р
(0,5 ррт или 0,5 мг/л)
(фосфорорганические пе¬
стициды)
Бромофос-этил Р
Диазинон Р
Дихлофентион Р
Карбофенотион Р
Малатион Р
Пропетамфос Р
Фенхлорфос Р
Хлорфенвинфос Р
Этион Р
Бромофос Р
Кумафос Р
Пиримифос-этил Р
Тетрахлорвинфос Р
Фозалон Р
Хлорпирифос Р
Хлорпирифос-метил Р
Раствор сравнения Е
(калибровочная смесь для
детектора электронного
захвата)
Раствор сравнения Р
(калибровочная смесь для
термоионного детектора)
Алдрин Р (0,01 мг/л)
р-Гэксахлорциклогексан Р
(0,01 мг/л)
о,п'-ДДД Р (0,01 мг/л)
Дельтаметрин Р (0,1 мг/л)
Циперметрин Р (0,1 мг/л)
Эндрин Р (0,01 мг/л)
Диазинон Р (0,05 мг/л)
Пропетамфос Р (0,05 мг/л)
Фенхлорфос Р (0,05 мг/л)
Хлорфенвинфос Р
(0,05 мг/л)
Этион Р (0,05 мг/л)
Раствор сравнения С
(раствор внутреннего
стандарта для фосфорор-
ганических пестицидов)
Раствор сравнения Н
(раствор внутреннего
стандарта для хлорорга-
нических пестицидов)
Диталимфос Р
(2 ррт или 2,0 мг/л)
Диталимфос Р
(1 ррт или 1,0 мг/л)
ИзоодринР
(2 ррт или 2,0 мг/л)
Изоодрин Р
(1 ррт или 1,0 мг/л)
ИДЕНТИФИКАЦИЯ И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ОСТАТОЧНЫХ КОЛИЧЕСТВ
ПЕСТИЦИДОВ
Для идентификации остаточных пестицидов по¬
лученные хроматограммы сравнивают с хроматограм¬
мами растворов сравнения А—О.
Подлинность пестицидов может быть подтвержде¬
на вводом стандартного образца в испытуемый раст¬
вор или путем наложения хроматограмм с помощью
программного обеспечения. Интерпретация результа¬
тов при анализе следовых количеств пестицидов явля¬
ется чрезвычайно сложной. На детекторы, в частности
детектор электронного захвата, мешающее действие
могут оказывать как непосредственно испытуемый об¬
разец, так и растворители, реактивы и инструменты,
используемые для экстракции. Эти пики могут быть
легко неправильно интерпретированы или могут быть
расценены как ложноположительные. Подтверждение
подлинности пестицидов может быть достигнуто с по¬
мощью проведения анализа испытуемых образцов и
стандартных образцов на различных капиллярных ко¬
лонках (см. хроматографические системы А и В, опи¬
санные ниже). Пики могут быть идентифицированы с
помощью информации, приведенной в таблице 0134.-2.
Таблица 0134.-2.
Порядок выхода пиков пестицидов
в хроматографических системах А и В
Хроматографическая
Хроматографическая
система А
система В
Текназол
Текназол
а-Гэксахлорциклогексан
Гексахлорбензол
Гексахл орбензол
а-Гексахлорциклогексан
Р-Гексахлорциклогексан
Диазинон
Линдан
Линдан
Пропетамфос
Пропетамфос
5-Гэксахлорциклогексан
Гептахлор
586
Гэсударственная фармакопея Республики Беларусь
Хроматографическая
Хроматографическая
система А
система В
Диазинон
Дихлофентион
Дихлофентион
Алдрин
Хлорпирифос-метил
Хлорпирифос-метил
Гептахлор
Фенхлорфос
Фенхлорфос
Р-Гексахлорциклогексан
Алдрин
б-Гексахл орцикл огексан
Малатион
Пиримифос-этил
Хлорпирифос
Хлорпирифос
Бромофос
Бромофос
Пиримифос-этил
Малатион
Гептахл орэпоксид
Гептахлорэпоксид
Хлорфенвинфос (Е)
о,л-ДДЕ
Хлорфенвинфос (2)
Хлорфенвинфос (Е)
Бромофос-этил
а-Эндосульфан
о.п’-ДДЕ
Хлорфенвинфос (2)
а-Эндосульфан
Бромофос-этил
Тетрахлорвинфос
ал-ддЕ
Диэлдрин
Диэлдрин
л,л’-ДДЕ
Тетрахлорвинфос
о,л'-ДДТ
о,л’-ДДТ
Эндрин
Эндрин
р-Эндосульфан
о.л-ДДД
о,л'-ДДД
л,л'-ДДД
л,л-ДДД
Р-Эндосульфан
Этион
Этион
Карбофенотион
л,л-ДДТ
л,л'-ДДТ
Карбофенотион
Метоксихлор
Метоксихлор
Фозалон
Цихалотрин
Цихалотрин (2 изомера)
щ/с-Перметрин
щ/с-Перметрин
Фозалон
транс- Перметрин
транс- Перметрин
Кумафос
Циперметрин (4 изомера)
Циперметрин (4 изомера)
Кумафос
Фенвалерат (2 изомера)
Фенвалерат (2 изомера)
Дельтаметрин
Дельтаметрин
Знание различий откликов пестицидов на двух
детекторах полезно при идентификации неизвестных
пиков.
После того как пестициды были идентифициро¬
ваны, проводят количественный расчет содержания
каждого пестицида по формуле:
с _РР Р Се 100
Р6 ' К*9
где:
Ср — концентрация идентифицированного пести¬
цида (ррт);
Рр — площадь пика отдельного пестицида, най¬
денная при анализе испытуемого образца;
Се — концентрация отдельного пестицида во
внешнем стандарте (ррт);
Ре — площадь пика отдельного пестицида во
внешнем стандарте;
О — фактор разведения;
Ро(— поправочный коэффициент открываемости.
Фактор разведения находят по следующей фор¬
муле:
где:
V, — объем образца, полученный после второй
стадии выпаривания;
/л — масса испытуемого образца;
У2 — объем вводимой пробы для гель-
проникающей хроматографии;
Уъ — объем мерной колбы для образца.
Хроматографическая система А:
- предколонка кварцевая (деактивированная)
длиной 4,5 м и внутренним диаметром 0,53 мм;
- колонка кварцевая капиллярная длиной 60 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)(дифенил)силоксана Р (толщина слоя
0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 25 см/с;
- давление: 180 кПа;
- температура:
Время (мин)
Температура (°С)
0—1
75
1—5
75 — 175
Колонка
5—30
175 — 275
30-^0
275 — 285
40—55
285
Блок ввода проб
300
Детектор
350
- детектор: электронного захвата или специфи¬
ческий термоионный;
- объем вводимой пробы: 2 мкл.
Хроматографическая система В (может быть
использована для подтверждения результатов ана¬
лиза):
- предколонка кварцевая (деактивированная)
длиной 4,5 м и внутренним диаметром 0,53 мм;
- колонка кварцевая капиллярная длиной 60 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(цианопропил) (7) (фенил) (7) (метил) (86) силокса-
на Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 25 см/с;
- давление: 180 кПа;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1
75
1—5
75 — 175
5—30
175 — 275
30—40
275 — 285
40—55
285
Блок ввода проб
300
Детектор
350
- детектор: электронного захвата или специфи¬
ческий термоионный;
- объем вводимой пробы: 2 мкл.
Хлориды. Не более 0,0150 % (150 ррт). 1,0 г
испытуемого образца помещают в круглодон¬
ную колбу со шлифом, прибавляют 20 мл спирта
(90 %, об/об) Р, присоединяют обратный холодиль¬
ник и кипятят в течение 5 мин. Охлаждают, прибав¬
ляют 40 мл воды Р, 0,5 мл кислоты азотной Р и
фильтруют. К фильтрату прибавляют 0,15 мл раст¬
вора 10 г/л серебра нитрата Р в спирте (90 %,
об/об) Р и выдерживают в течение 5 мин в защи¬
щенном от света месте. Опалесценция полученно¬
го раствора должна быть не интенсивнее эталона,
приготовленного параллельно путем прибавле¬
ния 0,15 мл раствора 10 г/л серебра нитрата Р
в спирте (90 %, об/об) Р к смеси из 0,2 мл 0,02 М
раствора кислоты хлористоводородной, 20 мл
спирта (90 %, об/об) Р, 40 мл воды Р и 0,5 мл кис¬
лоты азотной Р.
Ланолин водный
587
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Сульфатная зола (2.4.14). Не более 0,15 %. Сжи¬
гают 5,0 г испытуемого образца. Полученный остаток
используют для определения сульфатной золы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Ланолин в условиях испытания не обладает анти¬
микробным действием. Испытуемый образец должен
выдерживать требования статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Некоторые
из характеристик, описанных в разделе «Функци¬
онально-обусловленные характеристики», могут
также присутствовать в части фармакопейной
статьи, обязательной для выполнения, так как од¬
новременно они являются и показателями качества.
В таком случае в разделе «Функционально-обуслов¬
ленные характеристики» дается перекрестная
ссылка на это испытание в части фармакопейной
статьи, обязательной для выполнения. Контроль
данных характеристик может вносить вклад в каче¬
ство лекарственного средства путем достижения
постоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, однако
другие методы также могут быть использованы. В
случае указания результатов конкретных характе¬
ристик требуется указывать также и методы про¬
ведения испытаний.
Следующие характеристики могут быть важ¬
ны для ланолина, используемого в производстве во¬
доэмульсионных мазей и лиофильных кремов.
Водопоглощающая способность (см. раздел
«Испытания»).
Температура каплепадения (2.2.17, метод А).
Для заполнения металлической чашечки испытуемый
образец расплавляют на водяной бане, охлаждают до
температуры около 50 °С, заполняют чашечку и вы¬
держивают при температуре от 15 °С до 25 °С в тече¬
ние 24 ч. Температура каплепадения обычно состав¬
ляет от 38 °С до 44 °С.
07/2016:0135
ЛАНОЛИН ВОДНЫЙ
Айерз 1апае сит адиа
ИГОО!. РАТ, ИУОЯ01/5
ОПРЕДЕЛЕНИЕ
Смесь из 75 % (м/м) ланолина и 25 % (м/м) воды.
Его получают путем постепенного прибавления воды
к расплавленному ланолину при постоянном растира¬
нии. Может содержать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Бледно-желтая густая вязкая масса.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,5 г испытуемого образца помещают в про¬
бирку, растворяют в 5 мл метиленхлорида Р и при¬
бавляют 1 мл уксусного ангидрида Р и 0,1 мл кисло¬
ты серной Р. Появляется зеленое окрашивание.
B. 50 мг испытуемого образца растворяют в 5 мл
метиленхлорида Р, прибавляют 5 мл кислоты сер¬
ной Р и встряхивают. Появляется красное окрашива¬
ние; при освещении источником дневного света, рас¬
положенным позади наблюдателя, в нижнем слое на¬
блюдается интенсивная зеленая флуоресценция.
ИСПЫТАНИЯ
Растворимые в воде кислоты и щелочи. На
водяной бане расплавляют 6,7 г испытуемого образ¬
ца и энергично встряхивают в течение 2 мин с 75 мл
воды Р, предварительно нагретой до температуры
(90—95) °С. Охлаждают и фильтруют через бумажный
фильтр, предварительно промытый водой Р. К 60 мл
фильтрата (который может быть непрозрачным) при¬
бавляют 0,25 мл раствора бромтимолового сине¬
го Р1. При прибавлении не более 0,2 мл 0,02 М раст¬
вора кислоты хлористоводородной или не более
0,15 мл 0,02 М раствора натрия гидроксида окраска
раствора должна измениться.
Температура каплепадения (2.2.17). Не менее
38 °С и не более 44 °С. Определяют из остатка, по¬
лученного в испытании «Содержание ланолина». Для
заполнения металлической чашечки остаток расплав¬
ляют на водяной бане, охлаждают до температуры
около 50 °С. Заполняют чашечку и выдерживают при
температуре (15—20) °С в течение 24 ч.
Водопоглощающая способность. В ступку по¬
мещают 10 г остатка, полученного в испытании «Со¬
держание ланолина», прибавляют из бюретки воду Р
порциями по (0,2—0,5) мл, энергично растирая, после
каждого прибавления воды Р. Появление видимых
капель воды Р, которые не поглощаются ланолином,
является концом испытания. Должно абсорбировать¬
ся не менее 20 мл воды Р.
Кислотное число (2.5.1). Не более 0,8. 5,0 г ис¬
пытуемого образца растворяют в 25 мл предписанной
смеси растворителей.
Перекисное (пероксидное) число (2.5.5, метод
А). Не более 15.
Число омыления (2.5.6). Не менее 67 и не бо¬
лее 79. Определение проводят из 2,00 г испытуемого
образца. Нагревают с обратным холодильником в те¬
чение 4 ч.
Растворимые в воде восстанавливающие ве¬
щества. К 10 мл фильтрата, полученного в испытании
«Растворимые в воде кислоты и щелочи», прибав¬
ляют 1 мл кислоты серной разведенной Р и 0,1 мл
0,02 М раствора калия перманганата. Через 10 мин
розовая окраска раствора должна сохраниться.
Парафины. Не более 1,0%. Применяемые
кран и ватные пробки не должны содержать
смазку. Подготавливают колонку длиной 230 мм и
диаметром 20 мм с алюминия оксидом безводным,
прибавляя суспензию алюминия оксида безводно¬
го Р в петролейном эфире Р1 в стеклянную труб¬
ку с краном, содержащую петролейный эфир Р1.
Дают суспензии отстояться и уменьшают высоту
слоя растворителя над колонкой до около 40 мм.
588
Гэсударственная фармакопея Республики Беларусь
3,0 г остатка, полученного в испытании «Содержа¬
ние ланолина», растворяют в 50 мл теплого петро-
лейного эфира Р1, охлаждают, пропускают раствор
через колонку со скоростью 3 мл/мин и промывают
250 мл петролейного эфира Р1. Объединенный
элюат и промывные воды концентрируют до ми¬
нимального объема путем перегонки, выпаривают
досуха на водяной бане и нагревают остаток при
температуре 105 °С в течение 10 мин до тех пор,
пока разница между двумя последовательными
взвешиваниями будет не более 1 мг. Масса остатка
не должна превышать 30 мг.
Хлориды. Не более 0,0115 % (115 ррт). К 1,3 г
испытуемого образца прибавляют 20 мл спирта
(90 %, об/об) Р и кипятят с обратным холодильни¬
ком в течение 5 мин. Охлаждают, прибавляют 40 мл
воды Р, 0,5 мл кислоты азотной Р и фильтруют.
К фильтрату прибавляют 0,15 мл раствора 10 г/л
серебра нитрата Р в спирте (90 %, об/об) Р. Вы¬
держивают в течение 5 мин в защищенном от света
месте. Опалесценция раствора должна быть не ин¬
тенсивнее эталона, приготовленного параллельно
путем прибавления 0,15 мл раствора 10 г/л сере¬
бра нитрата Р в спирте (90 %, об/об) Р к смеси
из 0,2 мл 0,02 М раствора кислоты хлористо¬
водородной, 20 мл спирта (90 %, об/об) Р, 40 мл
воды Р и 0,5 мл кислоты азотной Р.
Сульфатная зола (2.4.14). Не более 0,1 %. Сжи¬
гают 5,0 г испытуемого образца. Полученный остаток
используют для определения сульфатной золы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
Содержание ланолина. Не менее 72,5 % и не
более 77,5 %. 30,0 г испытуемого образца нагревают
на водяной бане в предварительно взвешенной ем¬
кости со стеклянной палочкой при постоянном поме¬
шивании до постоянной массы. Взвешивают остаток.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:0969
ЛАНОЛИН
ГИДРОГЕНИЗИРОВАННЫЙ
Ас/ерз 1апае Ьудгодепа1из
У/001 ЕАТ, НУОЕОвЕЫАТЕО
ОПРЕДЕЛЕНИЕ
Смесь из высших алифатических спиртов и сти-
ролов, полученная методом прямой гидрогенизации
Ланолина (0134) при высоком давлении и высокой
температуре, при которых присутствующие эфиры
и кислоты восстанавливаются до соответствующих
спиртов. Может содержать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Белая или бледно-желтая густая вязкая масса.
Практически нерастворим в воде, растворим в
кипящем этаноле безводном и петролейном эфире.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Испытуемый образец выдерживает испыта¬
ние «Температура плавления», как указано в разделе
«Испытания».
B. Просматривают хроматограммы, полученные в
испытании «Содержание жирных спиртов и стеролов».
Результаты: основные пики на хроматограмме
испытуемого раствора по времени удерживания и ве¬
личине соответствуют основным пикам на хромато¬
грамме раствора сравнения (а).
C. 50 мг испытуемого образца растворяют в 5 мл
метиленхлорида Р, прибавляют 1 мл уксусного анги¬
дрида Р и 0,1 мл кислоты серной Р. Появляется зеле¬
ное окрашивание.
ИСПЫТАНИЯ
Температура плавления (2.2.15). От 45 °С до
55 °С. Испытуемый образец выдерживают при темпе¬
ратуре 20 °С в течение 16 ч.
Кислотное число (2.5.1). Не более 1,0. Опреде¬
ление проводят из 5,0 г испытуемого образца.
Гидроксильное число (2.5.3, метод А). От 140
до 180.
Число омыления (2.5.6). Не более 8,0. Испыту¬
емый образец нагревают с обратным холодильником
в течение 4 ч.
Содержание жирных спиртов и стеролов. Га¬
зовая хроматография (2.2.28).
Испытуемый раствор. 0,25 г испытуемого об¬
разца растворяют в 60 мл этанола безводного Р и
доводят до объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 0,25 г ФСО ланолина
гидрогенизированного растворяют в 60 мл этанола
безводного Р и доводят до объема 100,0 мл этим же
растворителем.
Раствор сравнения (Ь). 50 мг ФСО цетилового
спирта и 50 мг ФСО стеарилового спирта растворяют
в 60 мл этанола безводного Р и доводят до объема
100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)силоксана Р или другой неполярной
фазой (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
100
5—45
100 -> 300
45—60
300
Блок ввода проб
325
Детектор
350
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Результаты: хроматограмма испытуемого раст¬
вора не должна значительно отличаться от хромато¬
граммы раствора сравнения (а) (см. рисунок 0969.-1)
и не должна иметь увеличенных пиков со значениями
времен удерживания, соответствующими цетиловому
спирту и стеариловому спирту, присутствующих на
хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Лансопразол
589
Рисунок 0969.-1. Хроматограмма раствора сравнения (а) для испытания «Содержание жирных спиртов
и стеролов» в ланолине гидрогенизированном
ОПРЕДЕЛЕНИЕ
2-[(Я5)-[[3-Метил-4-(2,2,2-трифторэтокси)пиридин-
2-ил]метил]сульфинил]-1Н-бензимидазол.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или коричневатый порошок.
Практически нерастворим в воде, растворим в
этаноле, очень мало растворим в ацетонитриле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лансопразола.
Если спектры, полученные с использованием
образцов в твердом состоянии различаются, то ис¬
пытуемый образец и ФСО лансопразола растворяют
по отдельности в этаноле Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в диметилформамиде Р и доводят до объема
20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона В(К)2 или
ВУ(КЖ)2.
Потеря в массе при высушивании (2.2.32). Не
более 3,0 %. 2,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Общая зола (2.4.16). Не более 0,1 %. Определе¬
ние проводят из 5,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В заполненном доверху контейнере, в защищен¬
ном от света месте.
07/2016:2219
ЛАНСОПРАЗОЛ
[.апзоргазо/ит
1.АЫ80РКА201.Е
С16Н14Р3М3025 М.м. 369,4
[103577-45-3]
590
Гэсударственная фармакопея Республики Беларусь
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед применением и обеспечивают их за¬
щиту от света.
Смесь растворителей. Смесь триэтила-
мин Р — вода Р (1:60, об/об) доводят кислотой фос¬
форной Р до рН 10,5. Полученный раствор смешива¬
ют с 40 объемами ацетонитрила Р1.
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
этой же смесью растворителей до объема 10 мл.
Раствор сравнения (а). Содержимое контейне¬
ра с ФСО лансопразола для идентификации пиков
(содержит примеси А и В) растворяют в 1,0 мл смеси
растворителей.
Раствор сравнения (Ь). 2,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 5 мг 2-гидроксибензими-
дазола Р (примесь Р) и 5 мг 2-меркаптобензимидазо-
ла Р (примесь Е) растворяют в смеси растворителей
и доводят до объема 100 мл этой же смесью раство¬
рителей. 1 мл полученного раствора доводят смесью
растворителей до объема 10 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем амидогекса-
децилсилильным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза: смесь триэтиламин Р —
вода Р (1:60, об/об) доводят кислотой фосфорной Р
до рН 6,2; полученный раствор смешивают с 40 объ¬
емами ацетонитрила Р1\
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 285 нм;
- объем вводимой пробы: по 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания лансопразола.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (а) и хроматограмму, прилагае¬
мую к ФСО лансопразола для идентификации пиков;
идентифицируют пики примесей О и Е, используя
хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению к
лансопразолу, время удерживания — около 7 мин):
примесь О — около 0,4; примесь А — около 0,5; при¬
месь Е — около 0,6; примесь В — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,0 между пиками лан¬
сопразола и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Е — 0,4):
- примесь В (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, О и Е (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, О и Е, не должны превы¬
шать 0,5 площади основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, О, Е, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 0,6 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь).
Вода (2.5.32). Не более 0,1 %. Определение про¬
водят из (0,150—0,200) г испытуемого образца, ис¬
пользуя методику испарения:
- температура: от 50 °С до 70 °С;
- время нагревания: 15 мин;
- скорость потока: 150 мл/мин.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 40 мл
96 % спирта Р, доводят водой Р до объема 50 мл и
титруют 0,1 М раствором натрия гидроксида потен-
циометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 36,94 мг С1бН14Р3М3023.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, Р.
А. 2-[(Р$)-[[3-Метил-1 -оксидо-4-(2,2,2-трифторэто-
кси)пнрщт-2-ип\метт\сульфтил1-1 ^/-бензимидазол.
Л№:
|Ц||й:"
Леводопа
591
B. X = 502: 2-[[[3-Метил-4-(2,2,2-трифторэтокси)-
пиридин-2-ил]метил]сульфонил]-1Н-бензимидазол.
C. X = 5: 2-[[[3-Метил-4-(2,2,2-трифторэтокси)-
пиридин-2-ил]метил]сульфанил]-1Н-бензимидазол.
й. Р = ОН: 1Н-Бензимидазол-2-ол.
Е. К =5Н: 1Н-Бензимидазол-2-тиол.
Р. 2-[(#5)-[(4-Хлор-3-метилпиридин-2-ил)метил]-
сульфинил]-1 Н-бензимидазол.
07/2016:0038
ЛЕВОДОПА
1_еуос1орит
1-Е\/ОООРА
С,Н11Н04 М.м. 197,2
[59-92-7]
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-3-(3,4-дигидроксифенил)пропано-
вая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Мало растворим в воде, практически нераство¬
рим в 96 % спирте. Легко растворим в 1 М растворе
кислоте хлористоводородной и умеренно растворим
в 0,1 М растворе кислоты хлористоводородной.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотомерия в инфракрас¬
ной области (2.2.24).
Сравнение: ФСО леводопы.
ИСПЫТАНИЯ
Цветность (2.2.2, метод //). Окраска раствора
должна быть не интенсивнее эталона ВУ(КЖ)6. 1,0 г
испытуемого образца растворяют в растворе 103 г/л
кислоты хлористоводородной Р и доводят до объ¬
ема 25 мл этим же растворителем.
рН (2.2.3). От 4,5 до 7,0. 0,10 г испытуемого об¬
разца встряхивают с 10 мл воды, свободной от угле¬
рода диоксида, Р в течение 15 мин.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Используют свежеприготовлен¬
ные растворы.
Раствор А. Раствор 10,3 г/л кислоты хлористо¬
водородной Р.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в растворе А и доводят до объема
25 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 50,0 мл. 5,0 мл
полученного раствора доводят А до объема 100,0 мл.
Раствор сравнения (Ь). 8 мг тирозина Р (при¬
месь В) и 4 мг 3-метокси-1--тирозина Р (Ьизомер
примеси С) растворяют в 2 мл испытуемого раствора
и доводят раствором А до объема 50 мл. 5 мл полу¬
ченного раствора доводят раствором А до объема
100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
ди-изобутилоктадецилсилильным для хроматогра¬
фии Р (размер частиц 5 мкм) с размером пор 8 нм.
- подвижная фаза:
- подвижная фаза А: 0,1 М фосфатный бу¬
ферный раствор рН 3,0 Р;
- подвижная фаза В: метанол Р — 0,1 М фос¬
фатный буферный раствор рН 3,0 Р (18:85, об/о6)\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—18
90
10
18—22
90 — 0
0
1
о
о
22—35
0
100
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В и С, используя хроматограмму
раствора сравнения (Ь).
Относительное удерживание (по отношению к
леводопе, время удерживания — около 6 мин): при¬
месь А — около 0,7; примесь В — около 2; примесь
С — около 3,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 10 между пиками лево¬
допы и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 2,2):
- примесь В (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь С (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
592
Гэсударственная фармакопея Республики Беларусь
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,05 %): для каждой примеси площадь пика, кроме ос¬
новного и пиков примесей А, В и С, не должна превы¬
шать 0,5 площади основного пика на хроматограмме
раствора сравнения (а);
- сумма примесей (не более 1,0%): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Энантиометрическая чистота. Жидкостная хро¬
матография (2.2.29). Используют свежеприготов¬
ленные растворы.
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 50,0 мл.
Раствор сравнения (Ь). 10 мг О-допа Р (примесь
О) растворяют в 10 мл испытуемого раствора. 1 мл
полученного раствора доводят подвижной фазой до
объема 100 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 3,9 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р с размером частиц 5 мкм;
- подвижная фаза: 200 мг меди (II) ацетата Р и
387 мг М,М-диметил-1--фенилаланина Р растворяют
по отдельности в 250 мл воды Р\ смешивают 2 раст¬
вора и немедленно доводят до рН 4,0 кислотой ук¬
сусной Р\ прибавляют 50 мл метанола Р и доводят
водой Р до объема 1000 мл; смешивают и фильтруют;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания леводопы.
Относительное удерживание (по отношению к
леводопе, время удерживания — около 7 мин): при¬
месь й — около 0,4.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5 между пиками приме¬
си О и леводопы.
Предельное содержание примесей:
- примесь О (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси О, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, при необходимо¬
сти нагревая. Прибавляют 50 мл кислоты уксусной
безводной Р. Титруют 0,1 М раствором кислоты
хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 19,72 мг С9Н11М04.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
он
А. (25)-2-Амино-3-(2,4,5-тригидроксифенил)про-
пановая кислота.
В. (25)-2-Амино-3-(4-гидроксифенил)пропановая
кислота (тирозин).
со2н
и энантиомер
ОСН3
С. (2/?5)-2-Амино-3-(4-гидрокси-3-метоксифенил)-
пропановая кислота (3-метокси-0!_-тирозин).
со2н
он
О. (2/?)-2-Амино-3-(3,4-дигидроксифенил)пропа-
новая кислота (О-допа).
07/2016:0619
ЛЕВОМЕНТОЛ
1.еуотепМо1ит
1-ЕУОМЕМТН01-
С10НмО М.м. 156,3
[2216-51-5]
Левоментол
593
ОПРЕДЕЛЕНИЕ
(1 /?,25,5/?)-5-Метил-2-(1 -метилэтил)циклогексанол.
ОПИСАНИЕ (СВОЙСТВА)
Призматические или игольчатые бесцветные
блестящие кристаллы.
Практически нерастворим в воде, очень легко
растворим в 96 % спирте и в петролейном эфире, лег¬
ко растворим в жирных маслах и в вазелиновом мас¬
ле, очень мало растворим в глицерине.
Температура плавления: около 43 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения. 25 мг ФСО ментола раство¬
ряют в метаноле Р и доводят до объема 5 мл этим же
растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе до удаления раство¬
рителей.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин. Просма¬
тривают при дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме
испытуемого раствора (Ь) по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (с).
О. 0,20 г испытуемого образца растворяют в
0,5 мл пиридина безводного Р и прибавляют 3 мл
раствора 150 г/л динитробензоилхлорида Р в пири¬
дине безводном Р. Нагревают на водяной бане в те¬
чение 10 мин, прибавляют 7,0 мл воды Р небольши¬
ми порциями при перемешивании и выдерживают в
ледяной бане в течение 30 мин. Образуется осадок.
Раствор отстаивают и сливают надосадочную жид¬
кость. Осадок дважды промывают ледяной водой Р
порциями по 5 мл, перекристаллизовывают из 10 мл
ацетона Р, промывают ледяным ацетоном Р и сушат
при температуре 75 °С и давлении, не превышающем
2,7 кПа, в течение 30 мин. Температура плавления
(2.2.14) полученных кристаллов: от 154 °С до 157 °С.
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в 10 мл 96% спирта Р и доводят до объема
25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. 1,0 г испытуемо¬
го образца растворяют в 96 % спирте Р и доводят до
объема 10 мл этим же растворителем. Прибавляют
0,1 мл раствора фенолфталеина Р. Раствор оста¬
ется бесцветным. При прибавлении не более 0,5 мл
0,01 М раствора натрия гидроксида должно по¬
явиться розовое окрашивание.
Удельное оптическое вращение (2.2.7). От -48
до -51. Определяют удельное оптическое вращение
раствора 3.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор (а). 0,20 г испытуемого
образца растворяют в метиленхлориде Р и доводят
до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят метиленхлоридом Р до объема
10.0 мл.
Раствор сравнения (а). 40,0 мг испытуемого об¬
разца и 40,0 мг изоментола Р растворяют в мети¬
ленхлориде Р и доводят до объема 100,0 мл этим же
растворителем.
Раствор сравнения (Ь). 0,10 мл испытуемого
раствора (а) доводят метиленхлоридом Р до объ-
ема 100,0 мл.
Раствор сравнения (с). 40,0 мг ФСО ментола
растворяют в метиленхлориде Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка стеклянная длиной 2,0 м и внутренним
диаметром 2 мм, заполненная диатомитом для газо¬
вой хроматографии Р, импрегнированным 15 % (м/м)
макрогола 1500 Р;
- газ-носитель: азот для хроматографии Р\
- скорость газа-носителя: 30 мл/мин;
-температура: колонка — 120 °С, блок ввода
проб — 150 °С, детектор — 200 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания ментола.
Пригодность хроматографической системы:
- разрешение: не менее 1,4 между пиками мен¬
тола и изоментола на хроматограмме раствора срав¬
нения (а);
- отношение сигнал/шум: не менее 5 для основ¬
ного пика на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей: испытуемый
раствор (а):
- сумма примесей: не более 1 % площади основ¬
ного пика;
-неучитываемый предел: не учитывают пики,
площадь которых менее 0,05 % площади основного
пика на хроматограмме испытуемого раствора (а).
Остаток после выпаривания. Не более 0,05 %.
2.00 г испытуемого образца выпаривают на водяной
бане и сушат при температуре от 100 °С до 105 °С
в течение 1 ч. Масса остатка не должна превышать
1.0 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре от 8 °С до 15 °С.
594
Гэсударственная фармакопея Республики Беларусь
07/2016:0401
ЛЕВОТИРОКСИН НАТРИЯ
иеуоМугох'тит паМсит
1-ЕУОТНУ1ЮХШЕ 8СЮШМ
С15Н1014ЫМаО4 хН20; х*5 М.м. 799
(безводное вещество)
[25416-65-3]
ОПРЕДЕЛЕНИЕ
Натрия (25)-2-амино-3-[4-(4-гидрокси-3,5-дийодо-
фенокси)-3,5-дийодофенил]пропионат. ^Тироксин натрия#.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
Содержит переменное количество воды.
ОПИСАНИЕ(СВОЙСТВА)
Почти белый или слегка коричневато-желтый мел¬
кий кристаллический порошок. Слегка гигроскопичен.
Очень мало растворим в воде, мало растворим в
96 % спирте. Растворяется в разведенных растворах
гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО левотироксина натрия #или
спектр, представленный на рисунке #0401.-1*.
В. К 200 мг испытуемого образца прибавляют
2 мл кислоты серной разведенной Р, нагревают на
водяной бане, а затем осторожно нагревают над от¬
крытым пламенем, постепенно увеличивая темпе¬
ратуру до (600±50) °С. Продолжают сжигание до ис¬
чезновения большинства черных частиц. Остаток
растворяют в 2 мл воды Р. Полученный раствор дает
реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раст¬
воряют в 23 мл слабо кипящей смеси 1 М раствор
кислоты хлористоводородной — 96% спирт Р (1:4,
об/об). Охлаждают и доводят смесью 1 М раствор
кислоты хлористоводородной — 96% спирт Р (1:4,
об/об) до объема 25,0 мл.
Цветность (2.2.2, метод II). Окраска свежепри¬
готовленного раствора 5 должна быть не интенсивнее
эталона ВУ(КЖ)3.
Удельное оптическое вращение (2.2.7). От +16
до +20 (в пересчете на безводное вещество). Опреде¬
ляют удельное оптическое вращение свежеприготов¬
ленного раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы защищают от света в
течение всего испытания.
Смесь растворителей. Подвижная фаза А —
96 % спирт Р (1:2, об/об).
Испытуемый раствор. 25,0 мг испытуемого
образца растворяют в смеси растворителей и дово¬
дят до объема 50,0 мл этой же смесью растворите¬
лей. 10,0 мл полученного раствора доводят до объема
25,0 мл смесью растворителей.
Рисунок #0401 .-1. Инфракрасный спектр ФСО левотироксина натрия
Левотироксин натрия
595
Раствор сравнения (а). 2,5 мг ФСО левоти-
роксина натрия и 2,5 мг ФСО лиотиронина натрия
(примесь А) растворяют в смеси растворителей и
доводят до объема 25,0 мл этой же смесью раство¬
рителей. 1,0 мл полученного раствора доводят до
объема 50,0 мл смесью растворителей.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят до объема 10,0 мл смесью раст¬
ворителей.
Раствор сравнения (с). 25,0 мг ФСО левоти-
роксина натрия растворяют в смеси растворителей
и доводят до объема 50,0 мл этой же смесью раст¬
ворителей. 10,0 мл полученного раствора доводят
до объема 25,0 мл смесью растворителей.
Раствор сравнения (д). 2,0 мг ФСО левотирок-
сина для идентификации пиков (содержит примеси
Р и 6) растворяют в 10,0 мл смеси растворителей и
обрабатывают ультразвуком в течение 10 мин.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диа¬
метром 4,0 мм, заполненная силикагелем октаде-
цилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 3 мкм;
- подвижная фаза:
-подвижная фаза А: 1,97 г кислоты фос¬
форной Р растворяют в воде Р и доводят до
объема 2 л этим же растворителем;
-подвижная фаза В: 1,97 г кислоты фос¬
форной Р растворяют в ацетонитриле Р1 и
доводят до объема 2 л этим же растворителем;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
70
30
10—40
70 — 20
30 — 80
40—50
20
80
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: по 25 мкл испытуемого
раствора и растворов сравнения (а), (Ь) и (д).
Идентификация пиков примесей: идентифици¬
руют пики примесей Р и 6, используя хроматограм¬
му раствора сравнения (б) и хроматограмму, прила¬
гаемую к ФСО левотироксина для идентификации
пиков.
Относительное удерживание (по отноше¬
нию к левотироксину, время удерживания — около
11 мин): примесь А — около 0,5; примесь Р — около
2,0; примесь С — около 2,4.
Пригодность хроматографической системы.
раствор сравнения (а):
- разрешение: не менее 5,0 между пиками при¬
меси А и левотироксина.
Предельное содержание примесей:
-примесь А (не более 1,0%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
площадь соответствующего пика на хроматограмме
раствора сравнения (а);
- примесь Р (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси Р, не должна превышать 5-крат¬
ную площадь пика, соответствующего левотирокси¬
ну на хроматограмме раствора сравнения (Ь);
- примесь С (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси 6, не должна превышать 3-кратную
площадь пика, соответствующего левотироксину на
хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,2 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А, Р и Н, не должна превышать 2-крат¬
ную площадь пика, соответствующего левотирок¬
сину на хроматограмме раствора сравнения (Ь);
- сумма примесей: не более 2,0 %;
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади пика, соот¬
ветствующего левотироксину на хроматограмме
раствора сравнения (Ь).
Пределы, указанные в разделе «Сопутству¬
ющие примеси» (таблица 2034.-1) статьи Суб¬
станции для фармацевтического использования
(2034), не применяют.
Вода (2.5.32). Не менее 6,0 % и не более
12,0 %. Определение проводят из 0,100 г испыту¬
емого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 25 мкл испытуемого
раствора и раствора сравнения (с).
Содержание С15Н1014ЫМаО4 в процентах рассчи¬
тывают с учетом содержания левотироксина натрия в
ФСО левотироксина натрия.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре от 2 °С до
8 °С.
ПРИМЕСИ
Специфицированные примеси: А, Р, 6.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацев¬
тического использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования):
В, С, О, Е, Н, 3, К.
А. (25)-2-Амино-3-[4-(4-гидрокси-3-йодфенокси)-
3,5-дийодфенил]пропановая кислота (лиотиронин).
596
Гэсударственная фармакопея Республики Беларусь
В. (25)-2-Амино-3-[4-(3-хлор-4-гидрокси-5-йодфе-
нокси)-3,5-дийодфенил]пропановая кислота.
С. [4-(4-Гидрокси-3-йодфенокси)-3,5-дийодфенил]-
уксусная кислота (трийодтироуксусная кислота).
О. [4-(4-Гид рокси-3,5-д и йодфенокси )-3,5-д и йодфе-
нил]уксусная кислота (тетрайодтироуксусная кислота).
Е. (25)-2-Амино-3-[4-(4-гидроксифенокси)-3,5-
дийодфенил]пропановая кислота (дийодтиронин).
Р. (25)-2-Амино-3-[4-[4-(4-гидрокси-3,5-дийодфе-
нокси)-3,5-дийодфенокси]-3,5-дийодфенил]пропановая
кислота.
СВ. Структура не установлена.
Н. 4-(4-Гидрокси-3,5-дийодфенокси)-3,5-дийодбен-
зойная кислота.
I
3. (25)-2-Амино-3-[4-(4-гидрокси-3-йодфенокси)-3-
йодфенил]пропановая кислота.
К. (25)-2-Амино-3-[4-(4-гидрокси-3,5-дийодфе-
нокси)-3-йодфенил]пропановая кислота.
07/2016:0771
ЛЕЙЦИН
1-еиапит
/.ЕС/С/Л/Е
С6Н13Ы02 М.м 131,2
[61-90-5]
ОПРЕДЕЛЕНИЕ
(5)-2-Амино-4-метилпентановая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или блестящие чешуйки.
Умеренно растворим в воде, практически нераст¬
ворим в 96 % спирте. Растворяется в разведенных
минеральных кислотах и в разведенных растворах
гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ) .
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО лейцина #или спектр, представ¬
ленный на рисунке #0771.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5.0,5 г испытуемого образца растворяют
в растворе 103 г/л кислоты хлористоводородной Р и
доводят до объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От
+14,5 до +16,5 (в пересчете на сухое вещество). 1,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 25,0 мл этим
же растворителем.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов.#
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемых растворов и раство¬
ров сравнения могут быть изменены в зависимости от
Лейцин
597
Рисунок #0771 .-1. Инфракрасный спектр ФСО лейцина
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образцов, подходящий для используемого обору¬
дования.
Испытуемый раствор (а). 30,0 мг испытуемого
образца растворяют в растворе А и доводят до объ¬
ема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят раствором А до объема 25,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора (а) доводят раствором А до объема 100,0 мл.
2.0 мл полученного раствора доводят раствором А до
объема 10,0 мл.
Раствор сравнения (Ь). 30,0 мг изолейцина Р
(примесь А) растворяют в растворе А и доводят до
объема 100,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
250.0 мл. 1,0 мл полученного раствора доводят рас¬
твором А до объема 10,0 мл.
Раствор сравнения (с). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (д). 6,0 мл эталонного раст¬
вора аммония (100 ррт ЫН} Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (е). 30 мг изолейцина Р (при¬
месь А) и 30 мг лейцина Р растворяют в растворе А и
доводят до объема 50,0 мл. 1,0 мл полученного раст¬
вора доводят раствором А до объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора (а), холостого раствора и растворов сравне¬
ния (а), (Ь), (с) и (е) вводят в аминокислотный анали¬
затор. Запускают подходящую программу для опреде¬
ления физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 1,5 между пиками при¬
меси А и лейцина.
Расчет процентных содержаний:
-для примеси А в испытуемом растворе (Ь) —
используют концентрацию примеси А в растворе
сравнения (Ь);
-для любого нингидрин-положительного веще¬
ства, детектируемого при 570 нм в испытуемом рас¬
творе (а) — используют концентрацию лейцина в рас¬
творе сравнения (а);
-для любого нингидрин-положительного веще¬
ства, детектируемого при 440 нм в испытуемом рас¬
творе (а) — используют концентрацию пролина в
растворе сравнения (с); если пик выше учитываемого
предела при обеих длинах волн, для расчетов исполь¬
зуют результат, полученный при 570 нм.
Предельное содержание примесей:
- примесь А при 570 нм: не более 0,8 %;
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 1,0 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в растворе 10,3 г/л кислоты хло-
598
Гэсударственная фармакопея Республики Беларусь
ристоводородной Р и доводят до объема 10 мл этим
же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО лейцина раст¬
воряют в растворе 10,3 г/л кислоты хлористоводо¬
родной Р и доводят до объема 50 мл этим же раст¬
ворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО лейцина и
10 мг ФСО валина растворяют в растворе 10,3 г/л кис¬
лоты хлористоводородной Р и доводят до объема
25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и высушивают при температуре 105 °С
в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в воде Р и до¬
водят до объема 15 мл этим же растворителем. Полу¬
ченный раствор должен выдерживать испытание на
хлориды.
Сульфаты (2.4.13). Не ^олее 0,0300%
(300 ррт). 0,5 г испытуемого образца растворяют в
3 мл кислоте хлористоводородной разведенной Р и
доводят водой дистиллированной Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора (а), холостого раствора и раствора сравне¬
ния (6) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато-
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 50 мг испытуемого
образца должны выдерживать испытание на аммония
соли. Эталон готовят с использованием 0,1 мл эта¬
лонного раствора аммония (100 ррт NН^ Я#
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010% (10 ррт).
Растворитель. Вода Р.
0,25 г испытуемого образца должны выдержи¬
вать испытание на тяжелые металлы. Эталон гото¬
вят с использованием 0,25 мл эталонного раствора
свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20) #или используя в качестве индикатора 0,1 мл
раствора нафтолбензеина Р до изменения окраски
раствора от коричневато-желтой до зеленой.#
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 13,12 мг С6Н131Ч02.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования):
В, С, О, Е.
Н СН3
А. (25,35)-2-Амино-3-метилпентановая кислота
(изолейцин).
В. 128)-2-Амино-4-(метилсульфанил)бутановая
кислота (метионин).
С. (25)-2-Амино-3-фенилпропановая кислота (фе¬
нилаланин).
Лидокаина гидрохлорид
599
СН3
Н3С
со2н
Н N42
й. (25)-2-Амино-5-метилгексановая кислота (5-ме-
тил-норлейцин).
н ын2
НзС\.Х
СОгН
СНз
Е. (25}-2-Амино-3-метилбутановая кислота (валин).
07/2016:0227
ЛИДОКАИНА ГИДРОХЛОРИД
Шоса/п/ Ьус1госЫопс1ит
иоосмыЕ нуокосш-отоЕ
• н2о
С14Н22Ы2° * НС1 - н20 М.м. 288,8
[6108-05-0]
ОПРЕДЕЛЕНИЕ
2-(Диэтиламино)-Л/-(2,6-диметилфенил)ацетами-
да гидрохлорид моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Температура плавления {2.2.14): от 74 °С до
79 °С. Определение проводят без предварительного
высушивания испытуемого образца.
B. Абсорбционная спектрофотометрия в инфра¬
красной области {2.2.24).
Сравнение: ФСО лидокаина гидрохлорида #или
спектр, представленный на рисунке #0227.-1#.
C. К 5 мг испытуемого образца прибавляют 0,5 мл
азотной кислоты дымящейся Р, выпаривают на во¬
дяной бане досуха, охлаждают и растворяют остаток
в 5 мл ацетона Р. Прибавляют 0,2 мл спиртового
раствора калия гидроксида Р. Появляется зеленое
окрашивание.
О. Испытуемый образец дает реакцию (а) на хло¬
риды {2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность {2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,0 до 5,5.1 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объема
10 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Рисунок #0227.-1. Инфракрасный спектр ФСО лидокаина гидрохлорида
600
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг 2,6-диметилани-
лина Р (примесь А) растворяют в подвижной фазе и
доводят до объема 100,0 мл этим же растворителем.
10.0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Раствор сравнения (Ь). 5 мг 2-хлор-М-(2,6-
диметилфенил)ацетамида Р (примесь Н) раство¬
ряют в подвижной фазе и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (ф. К 1,0 мл раствора срав¬
нения (а) прибавляют 1,0 мл раствора сравнения (Ь),
1.0 мл раствора сравнения (с) и доводят подвижной
фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 3,9 мм, заполненная полимером органосили¬
катным аморфным, с введенными полярными груп¬
пами, октадецилсилильным эндкепированным Р с
размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: ацетонитрил для хромато¬
графии Р — раствор 4,85 г/л калия дигидрофосфа¬
та Р, доведенный до рН 8,0 раствором натрия ги¬
дроксида концентрированным Р, (30:70, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания лидокаина.
Относительное удерживание (по отношению к
лидокаину, время удерживания — около 17 мин): при¬
месь Н — около 0,37; примесь А — около 0,40.
Пригодность хроматографической системы:
раствор сравнения (с():
- разрешение: не менее 1,5 между пиками при¬
меси Н и примеси А.
Предельное содержание примесей:
- примесь А (не более 0,01 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (б);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь пика лидокаина
на хроматограмме раствора сравнения (б);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь пика лидокаина на хроматограмме раст¬
вора сравнения (б);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади пика лидокаина на
хроматограмме раствора сравнения (б).
Тяжелые металлы (2.4.8, метод Е). Не более
0,0005 % (5 ррт). 1,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 25 мл этим же
растворителем. Проводят предфильтрацию раствора.
10 мл предфильтрата должны выдерживать испытание
на тяжелые металлы. Эталон готовят с использовани¬
ем 2 мл эталонного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не менее 5,5 % и не более 7,0 %.
Определение проводят из 0,25 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,220 г испытуемого образца растворяют в 50 мл
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 27,08 мг С14Н221М20-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): В, С, О, Е, Р, 6, Н, I, */, К.
А. Р = Н: 2,6-Диметиланилин.
С. Р = СО-СН3: А/-(2,6-Диметилфенил)ацетамид.
О. Р = СО-СН2-1ЧН-С2Н5: А/-(2,6-Диметилфенил)-2-
(этиламино)ацетамид.
С. Р = СО-СН2-МН-СН(СН3)2: АА-(2,6-Димегилфенил)-
2-[(1 -метилэтил )амино]ацетамид.
Н. Р = СО-СН2-С1: 2-Хлор-А/-(2,6-диметилфенил)-
ацетамид.
К. Р = СО-СН2-1Ч(СН3)С2Н5: А/-(2,6-Диметилфенил)-
2-(метилэтиламино)ацетамид.
В. 2-(Диэтилазиноил)-А/-(2,б-диметилфенил)аце-
тамид (лидокаин А^-оксид).
Е. 2-2'-(Азандиил)бис[А/-(2,6-диметилфенил)аце-
тамид].
Лизина гидрохлорид
601
К1
Р. Р1 = СН3, К2 = КЗ = Н: 2-(Диэтиламино)-Л/-(2,3-
диметил фенил )ацетамид.
I. К1 = КЗ = Н, К2 = СН3: 2-(Диэтиламино)-Л/-(2,4-
диметилфенил)ацетамид.
3. К1 =К2 =Н, КЗ = СН3: 2-(Диэтиламино)-Л/-(2,5-
диметилфенил)ацетамид.
07/2016:0930
ЛИЗИНА ГИДРОХЛОРИД
/.уз/л/ ЬудгосЫопдит
1.У81ЫЕ НУ0К0СН1.0КЮЕ
н мн2
Зч. НС1
н2н хо2н
С6Н,4Ы2Ог • НС1 М.м. 182,7
[657-27-2]
ОПРЕДЕЛЕНИЕ
(5)-2,6-Диаминогексановой кислоты гидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или'бесцветные кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: А, С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО лизина гидрохлорида #или
спектр, представленный на рисунке #0930.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО лизина гидрохлорида раст¬
воряют по отдельности в минимальном количестве
воды Р, выпаривают при температуре 60 °С до сухих
остатков, которые используют для получения новых
спектров.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
й. К 0,1 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 2 мл воды Р
и 1 мл раствора 50 г/ л фосфорномолибденовой кис¬
лоты Р. Образуется желтовато-белый осадок.
Е. К 0,1 мл раствора 3 прибавляют 2 мл воды Р.
Полученный раствор дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Рисунок #0930.-1. Инфракрасный спектр ФСО лизина гидрохлорида
76. Зак. 1060.
602
Гэсударственная фармакопея Республики Беларусь
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона В(К)7 или
ОУ(ЗЖ)7.
Удельное оптическое вращение (2.2.7). От
+21,0 до +22,5 (в пересчете на сухое вещество). 2,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 25,0 мл этим
же растворителем.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов*
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Вода Р или буферный раствор для
приготовления образца, подходящий для применяе¬
мого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт ИН) Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (д). 30 мг изолейцина Р и
30 мг лейцина Р (примесь А) растворяют в растворе А
и доводят до объема 50,0 мл этим же растворителем.
1.0 мл полученного раствора доводят раствором А до
объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь) и (с!) вводят в аминокислотный анализатор.
Запускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и примеси А.
Расчет процентных содержаний:
— для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию лизина гидрохлорида в рас¬
творе сравнения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 1,0 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для фар¬
мацевтического использования (2034), не применяют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО лизина ги¬
дрохлорида растворяют в воде Р и доводят до объема
50 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО лизина гидрох¬
лорида и 10 мг ФСО аргинина растворяют в воде Р и
доводят до объема 25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: аммиака раствор концентри¬
рованный Р— 2-пропанол Р (30:70, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре 105 °С до полно¬
го удаления аммиака.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два четко
разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
5 мл раствора 5 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато¬
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 50 мг испытуемого
образца должны выдерживать испытание на аммония
соли. Эталон готовят с использованием 0,1 мл эта¬
лонного раствора аммония (100 ррт NН^ Я#
Лизиноприл дигидрат
603
Железо (2.4.9). Не более 0,0030 % (30 ррт).
0,33 г испытуемого образца помещают в делительную
воронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
#Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 18,27 мг С6Н14М202-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования):
В, С, О, Е.
сн3 н т2
н3с
со2н
цин).
А. (25)-2-Амино-4-метилпентановая кислота (лей-
B. (25)-2-Амино-3-фенилпропановая кислота (фе¬
нилаланин).
н ,мн2
Н3с.
уГ со2н
н он
C. (25,3#)-2-Амино-3-гидроксибутановая кислота
(треонин).
07/2016:1120
ЛИЗИНОПРИЛ ДИГИДРАТ
изторгНит сШ1ус1псит
иЗШОРГШ. О/НУОКАТЕ
С21Н31Ы305 2Н20 М.м. 441,5
[83915-83-7]
ОПРЕДЕЛЕНИЕ
(25)-1 -[(25)-6-Амино-2-[[(1 $)-1 -карбокси-3-фенил-
пропил]амино]гексаноил]пирролидин-2-карбоновая кис¬
лота.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде, практически нерастворим в
этаноле безводном и гептане.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Удельное оптическое вращение (2.2.27): от
-47 до -43 (в пересчете на безводное вещество). 0,5 г
испытуемого образца растворяют в растворе цин¬
ка ацетата Р и доводят до объема 50,0 мл этим же
растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лизиноприла дигидрата #или
спектр, представленный на рисунке #1120.-1#.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО лизиноприла
для проверки пригодности хроматографической си¬
стемы (содержит примеси А и Е) растворяют в под¬
вижной фазе А и доводят до объема 1,0 мл этим же
растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО лизиноприла примеси Р растворяют в 1,0 мл
подвижной фазы А.
Раствор сравнения (д). 5 мг ФСО лизинопри¬
ла для идентификации пиков (содержит примесь 6)
растворяют в подвижной фазе А и доводят до объема
1.0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
76*. Зак. 1060.
604
Гэсударственная фармакопея Республики Беларусь
синильным, деактивированным по отношению к ос¬
нованиям, эндкепированным для хроматографии Р
(размер частиц 5 мкм);
- температура: 50 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 — раст¬
вор 3,12 г/л натрия дигидрофосфата Р, дове¬
денный кислотой фосфорной разведенной Р до
рН 3,8, (3:97, об/об);
- подвижная фаза В: ацетонитрил Р1 — раст¬
вор 3,12 г/л натрия дигидрофосфата Р, дове¬
денный кислотой фосфорной разведенной Р до
рН 3,5, (20,5:79,5, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
100
0
2—37
100 — 0
о
о
т—
Т
О
37—62
0
100
- скорость подвижной фазы: 1,6 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 50 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и Е, используя хроматограмму
раствора сравнения (а) и хроматограмму, прилагае¬
мую к ФСО лизиноприла для проверки пригодности
хроматографической системы, идентифицируют
пик примеси Р, используя хроматограмму раствора
сравнения (с); идентифицируют пик примеси С, ис¬
пользуя хроматограмму раствора сравнения (б) и
хроматограмму, прилагаемую к ФСО лизиноприла для
идентификации пиков.
Относительное удерживание (по отношению к
лизиноприлу, время удерживания — около 14 мин):
примесь А — около 0,7; примесь Е — около 1,2; при¬
месь Р — около 1,9; примесь О — около 2,9.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками ли¬
зиноприла и примеси Е на хроматограмме раствора
сравнения (а);
- отношение сигнал/шум: не менее 45 для ос¬
новного пика на хроматограмме раствора сравне¬
ния (Ь).
Расчет процентных содержаний:
-поправочный коэффициент: умножают пло¬
щадь пика примеси Р на 2,1;
- для каждой примеси — используют концентра¬
цию лизиноприла в растворе сравнения (Ь).
Предельное содержание примесей:
- примеси А, Е, Р: не более 0,2 % для каждой
примеси;
- примесь С: не более 0,15 %;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %; не учитывают
пики со временем удерживания менее 3 мин.
Вода (2.5.12). Не менее 8,0 % и не более 9,5 %.
Определение проводят из 0,200 г испытуемого об¬
разца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 50 мл
воды Р и титруют 0,1 М раствором натрия гидрокси¬
да потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 40,55 мг С21Н311Ч305.
Рисунок #1120.-1. Инфракрасный спектр ФСО лизиноприла дигидрата
Лимонная кислота безводная
605
ПРИМЕСИ
Специфицированные примеси: А, Е, Р, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Н, I.
А. (2Я5)-2-Амино-4-фенилбутановая кислота.
В. 4-Метилбензолсульфоновая кислота.
С. (25)-2-[(35,8а5)-3-(4-Аминобутил)-1,4-диоксоге-
ксагидропирроло[1,2-а]пиразин-2(1 Н)-ил]-4-фенилбута-
новая кислота (5,5,5-дикетопиперазин).
О. (25)-2-[(35,8аЯ)-3-(4-Аминобутил)-1,4-диоксогек-
сагидропирроло[1,2-а]пиразин-2(1 Н)-ил]-4-фенилбу-
тановая кислота (Я,5,5-дикетопиперазин),
Е. (25)-1 -[(25)-6-Амино-2-[[(1 Я)-1 -карбокси-3-фе-
нилпропил]амино]гексаноил]пирролидин-2-карбоновая
кислота (лизиноприла Я,5,5-изомер).
Р. (25)-1-[(25)-6-Амино-2-[[(15)-1-карбокси-3-
циклогексилпропил]амино]гексаноил]пирролидин-2-
карбоновая кислота (циклогексиловый аналог).
С. (25)-1 -[(25)-6-Амино-2-[[(25)-1 -[[[(55)-5-[[(15)-1 -
карбокси-3-фенилпропил]амино]-6-[(25)-2-карбокси-
пирролидин-1-ил]-6-оксогексил]амино]карбонил]-3-
фенилпропил]амино]гексаноил]пирролидин-2-карбоно-
вая кислота (димер лизиноприла).
Н. (25)-6-Амино-2-[[(15)-1 -карбокси-3-фенилпропил]-
амино]гексановая кислота.
I. (25)-1-[(25)-2,6-Бис[[(15)-1-карбокси-3-фенил-
пропил]амино]гексаноил]пирролидин-2-карбоновая кис¬
лота.
07/2016:0455
ЛИМОННАЯ КИСЛОТА БЕЗВОДНАЯ
АсШит сИпсит апЬудпсит
С1ТШСАСЮ, АМУУОЯО(У5
С6Н807 М.м. 192,1
[77-92-9]
ОПРЕДЕЛЕНИЕ
2-Гидроксипропан-1,2,3-трикарбоновая кислота.
Содержание: не менее 99,5 % и не более 100,5 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок, бесцветные кристаллы или гранулы.
Очень легко растворима в воде, легко раствори¬
ма в 96 % спирте.
Температура плавления: около 153 °С с раз¬
ложением.
77. Зак. 1060.
606
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. 1 г испытуемого образца растворяют в 10 мл
воды Р. Полученный раствор имеет сильнокислую ре¬
акцию (2.2.4).
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец и стан¬
дартный образец сушат при температуре от (105±2) °С
в течение 2 ч.
Сравнение: ФСО лимонной кислоты безводной.
C. Около 5 мг испытуемого образца прибавляют
к смеси из 1 мл уксусного ангидрида Р и 3 мл пириди¬
на Р. Появляется красное окрашивание.
Б. 0,5 г испытуемого образца растворяют в 5 мл
воды Р, нейтрализуют 1 М раствором натрия ги¬
дроксида (около 7 мл), прибавляют 10 мл раствора
кальция хлорида Р и нагревают до кипения. Образу¬
ется осадок белого цвета.
Е. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.7). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным или окраска раствора 3 должна
быть не интенсивнее эталона У(Ж)7, ВУ(КЖ)7 или
6У(ЗЖ)7.
Легкообугливающиеся вещества (2.2.2, метод
I). 1,0 г испытуемого образца помещают в чистую про¬
бирку, прибавляют 10 мл кислоты серной Р и смесь
немедленно нагревают в водяной бане при темпера¬
туре (90±1)°С в течение 60 мин. Незамедлительно
после этого быстро охлаждают. Окраска полученного
раствора должна быть не интенсивнее, чем смесь из
1 мл исходного красного раствора и 9 мл исходного
желтого раствора.
Щавелевая кислота. Не более 0,0360 % (360 ррт),
в пересчете на безводную щавелевую кислоту.
0,80 г испытуемого образца растворяют в 4 мл
воды Р. Прибавляют 3 мл кислоты хлористоводо¬
родной Р и 1 г цинка Р в гранулах. Кипятят в тече¬
ние 1 мин. Выдерживают в течение 2 мин. Переносят
надосадочную жидкость в пробирку, содержащую
0,25 мл раствора 10 г/л фенилгидразина гидрохло¬
рида Р, и нагревают до кипения. Немедленно охлаж¬
дают, переносят в мерный цилиндр и прибавляют
такой же объем кислоты хлористоводородной Р
и 0,25 мл раствора 50 г/л калия феррицианида Р.
Встряхивают и выдерживают в течение 30 мин. Ро¬
зовое окрашивание раствора должно быть не ин¬
тенсивнее, чем окраска стандарта, приготовленного
параллельно с использованием 4 мл раствора 0,1 г/л
кислоты щавелевой Р.
Сульфаты(2.4.73). Неболее0,0150 %(150 ррт).
2,0 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 30 мл этим же
растворителем. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Алюминий (2.4.17). Не более 0,00002 % (0,2 ррт),
если субстанция предназначена для производства
растворов для диализа.
Предписанный раствор. 20 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А1) Р, 10 мл ацетатного буферного
раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 5,0 г испытуемого образца раство¬
ряют в 39 мл (добавляют порциями) натрия гидрок¬
сида разведенного Р и доводят водой дистиллиро¬
ванной Р до объема 50 мл. 12 мл полученного раст¬
вора должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Вода (2.5.72). Не более 1,0 %. Определение про¬
водят из 2,000 г испытуемого образца.
Сульфатная зола (2.4.74). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,5 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.7.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,550 г испытуемого образца растворяют в 50 мл
воды Р и титруют 7 М раствором натрия гидрокси¬
да, используя в качестве индикатора 0,5 мл раствора
фенолфталеина Р.
1 мл 7 М раствора натрия гидроксида соответ¬
ствует 64,03 мг С6Н807.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства раство¬
ров для диализа.
07/2016:0456
ЛИМОННАЯ КИСЛОТА
МОНОГИДРАТ
АсШит сНпсит топоЬудпсит
С1ТШСАСЮ МОЫОНУОКАТЕ
но со2н
Н02С.З<{.С02Н ‘ Нг°
С,Н,07 ■ Н-0 М.м. 210,1
[5949-29-1]
ОПРЕДЕЛЕНИЕ
2-Гидроксипропан-1,2,3-трикарбоновая кислота
моногидрат.
Содержание: не менее 99,5 % и не более 100,5 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок, бесцветные кристаллы или гранулы. На воздухе
выветривается.
Лимонное масло
607
Очень легко растворима в воде, легко раствори¬
ма в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. 1 г испытуемого образца растворяют в 10 мл
воды Р. Полученный раствор имеет сильнокислую ре¬
акцию (2.2.4).
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец и стан¬
дартный образец сушат при температуре (105±2) °С
в течение 2 ч.
Сравнение: ФСО лимонной кислоты моноги¬
драта.
C. Около 5 мг испытуемого образца прибавляют
к смеси из 1 мл уксусного ангидрида Р и 3 мл пириди¬
на Р. Появляется красное окрашивание.
0. 0,5 г испытуемого образца растворяют в 5 мл
воды Р, нейтрализуют 1 М раствором натрия ги¬
дроксида (около 7 мл), прибавляют 10 мл раствора
кальция хлорида Р и нагревают до кипения. Образу¬
ется осадок белого цвета.
Е. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)7, ВУ(КЖ)7
или ОУ(ЗЖ)7.
Легкообугливающиеся вещества (2.2.2, ме¬
тод I). 1,0 г испытуемого образца помещают в
чистую пробирку, прибавляют 10 мл кислоты сер¬
ной Р и смесь немедленно нагревают в водяной
бане при температуре (90±1) °С в течение 60 мин.
Быстро охлаждают. Окраска полученного раствора
должна быть не интенсивнее, чем смесь из 1 мл ис¬
ходного красного раствора и 9 мл исходного желто¬
го раствора.
Щавелевая кислота. Не более 0,0360 %
(360 ррт), в пересчете на безводную щавелевую
кислоту.
0,80 г испытуемого образца растворяют в 4 мл
воды Р. Прибавляют 3 мл кислоты хлористоводо¬
родной Р и 1 г цинка Р в гранулах. Кипятят в тече¬
ние 1 мин. Выдерживают в течение 2 мин. Пере¬
носят надосадочную жидкость в пробирку, содер¬
жащую 0,25 мл раствора 10 г/л фенилгидразина
гидрохлорида Р, и нагревают до кипения. Немед¬
ленно охлаждают, переносят в мерный цилиндр и
прибавляют такой же объем кислоты хлористово¬
дородной Р и 0,25 мл раствора 50 г/л калия ферри-
цианида Р. Встряхивают и выдерживают в течение
30 мин. Розовое окрашивание раствора должно
быть не интенсивнее, чем окраска стандарта, при¬
готовленного параллельно с использованием 4 мл
раствора 0,1 г/л кислоты щавелевой Р
Сульфаты (2.4.13). Не более 0,0150 % (150 ррт).
2,0 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 30 мл этим же
растворителем. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Алюминий (2.4.17). Не более 0,00002%
(0,2 ррт), если субстанция предназначена для произ¬
водства растворов для диализа.
Предписанный раствор. 20 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р.
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А1) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Рм 98 мл воды Р
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН6,0Р\л 100 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 5,0 г испытуемого образца (до¬
бавляют порциями) растворяют в 39 мл раствора
натрия гидроксида разведенного Р и доводят водой
дистиллированной Р до объема 50 мл. 12 мл полу¬
ченного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не менее 7,5 % и не более 9,0 %.
Определение проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,5 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,550 г испытуемого образца растворяют в 50 мл
воды Р и титруют 1 М раствором натрия гидрокси¬
да, используя в качестве индикатора 0,5 мл раствора
фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 64,03 мг С6Н807.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства раство¬
ров для диализа.
07/2016:0620
ЛИМОННОЕ МАСЛО
Итотв аеМего/еит
ЕЕМОЫ О/Е
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное соответствующим
механическим методом без помощи нагревания из
свежей цедры СНгиз Нтоп (1_.) Вигтап Я1.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, подвижная, светло-желтая или зе¬
леновато-желтая жидкость с характерным запахом.
Может мутнеть при охлаждении.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
77*. Зак. 1060.
608
Гэсударственная фармакопея Республики Беларусь
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 мл испытуемого образ¬
ца смешивают с 1 мл толуола Р.
Раствор сравнения. 10 мг цитроптена Р и
50 мкл цитраля Р растворяют в толуоле Р и дово¬
дят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика-
геля 6Р254 Р.
Подвижная фаза: этилацетат Р —- толуол Р
(15:85, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от
линии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в
ультрафиолетовом свете при длине волны 254 нм.
Результаты А: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения
и испытуемого раствора.
Верх хроматографической пластинки
Цитраль: зона поглощения
Цитроптен: светло-голубая
флуоресцирующая зона
Зона поглощения (берга-
мотин)
Зона поглощения (цитраль)
Зона темно-синего
цвета (5-геранилокси-7-
метоксикумарин)
Светло-голубая флуоресци¬
рующая зона (цитроптен)
Зона поглощения (произво¬
дное псоралена)
Зона поглощения (биакан-
гелицин)
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 365 нм.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Цитраль: зона поглощения
Цитроптен: яркая синяя
флуоресцирующая зона
Желтая флуоресцирующая
зона (бергамотин)
Зона поглощения (цитраль)
Яркая синяя флуоресцирую¬
щая зона (5-геранилокси-7-
метоксикумарин)
Яркая фиолетово-синяя
флуоресцирующая зона (ци¬
троптен)
Желтая флуоресцирующая
зона (производное псора¬
лена)
Зона оранжевого цвета (би-
акангелицин)
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Результаты: характерные пики на хроматограм¬
ме испытуемого раствора по времени удерживания
соответствуют характерным пикам на хроматограмме
раствора сравнения.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,850 до
0,858.
Показатель преломления (2.2.6). От 1,473 до
1,476.
Угол оптического вращения (2.2.7). От +57° до
+70°.
Оптическая плотность (2.2.25). 0,250 г испы¬
туемого образца растворяют в 96 % спирте Р, пе¬
ремешивают и доводят до объема 100,0 мл этим же
растворителем. Оптическую плотность определяют
в интервале от 260 нм до 400 нм. Если измерения
проводят на приборе, не позволяющем автомати¬
чески записать спектр, то поступают следующим
образом: от 260 нм до достижения длины волны,
которая приблизительно на 12 нм меньше длины
предполагаемого максимума, измерения проводят
с интервалом 5 нм. Затем выполняют три измере¬
ния с интервалом 3 нм. Затем интервал составляет
1 нм до тех пор, пока не достигают длины волны,
на 5 нм превышающей длину волны максимума.
Дальше измерения выполняют с интервалом 10 нм
до длины волны 400 нм. Строят график, на котором
по оси ординат откладывают значения оптической
плотности, а по оси абсцисс — значения длины
волны. Через точки А и Б проводят касательную
линию (см. рисунок 0620.-1). Максимальная опти¬
ческая плотность в точке С отмечается при длине
волны (315±3) нм. Из точки С опускают перпендику¬
ляр на ось абсцисс, который пересекает линию АВ
в точке О. Из значения оптической плотности, соот¬
ветствующего точке С, вычитают значение оптиче¬
ской плотности, соответствующее точке О. Разница
С - О должна быть от 0,20 до 0,96. Для лимонного
масла итальянского типа это значение должно быть
не менее 0,45.
Длина волны, нм
Рисунок 0620.-1. Типичный спектр поглощения
масла лимонного для испытания
«Оптическая плотность»
Жирные и минеральные масла (2.6.7). Ис¬
пытуемый образец должен выдерживать испыта¬
ние на жирные и минеральные масла.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят мето¬
дом внутренней нормализации.
Испытуемый раствор. Испытуемый образец.
Линкомицина гидрохлорид
609
Раствор сравнения. 20 мкл р-пинена Р, Юмкл
сабинена Р, 100 мкл лимонена Р, 10 мкл у-терпинена Р,
5 мкл р-кариофиллена Р, 20 мкл цитраля Р, 5 мкл
а-терпинеола Р, 5 мкл нерилацетата Р и 5 мкл
геранилацетата Р растворяют в 1 мл ацетона Р
Условия хроматографирования:
- колонка капиллярная кварцевая длиной от 30 м
(толщина слоя 1 мкм) до 60 м (толщина слоя 0,2 мкм)
и внутренним диаметром (0,25—0,53) мм, покрытая
слоем макрогола 20 000 Рш,
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,0 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—6
45
6—21
45 —* 90
21—39
90 — 180
39—55
180
Блок ввода проб
220
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,5 мкл раствора срав¬
нения и 0,2 мкл испытуемого раствора.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения; отмечают времена
удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: не менее 1,5 между пиками
Р-пинена и сабинена; не менее 1,5 между пиками ге-
раниаля и геранилацетата.
По временам удерживания компонентов раст¬
вора сравнения определяют состав испытуемого
раствора.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- Р-пинен: от 7,0 % до 17,0 %;
- сабинен: от 1,0 % до 3,0 %;
- лимонен: от 56,0 % до 78,0 %;
- у-терпинен: от 6,0 % до 12,0 %;
- р-кариофиллен: не более 0,5 %;
- нераль: от 0,3 % до 1,5 %;
- а-терпинеол: не более 0,6 %;
- нерилацетат: от 0,2 % до 0,9 %;
- гераниаль: от 0,5 % до 2,3 %;
- геранилацетат: от 0,1 % до 0,8 %.
Остаток после выпаривания (2.8.9). Не менее
1,8 % и не более 3,6 %. Нагревают на водяной бане в
течение 4 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
МАРКИРОВКА
При необходимости указывают:
- масло лимонное итальянского типа.
07/2016:0583
ЛИНКОМИЦИНА ГИДРОХЛОРИД
Ипсотуст/ ЬудгосЫогШит
иысомуст ж/ояосш.оя/ое
Линкомицин В Н С17Н32М2065 НС1Н20 447,0
Линкомицина гидрохлорид моногидрат [7179-49-9]
ОПРЕДЕЛЕНИЕ
Смесь антибиотиков, продуцируемых 81гер1отусез
Нпсо1пепз1з уаг. Нпсо1пепз1з или полученных дру¬
гим способом, состоящая в основном из метил-6,8-
дидезокси-6-[[[(25,4#)-1-метил-4-про-пилпирролидин-
2-ил]карбонил]-амино]-1-тио-0-эр1/шро-а-0-гала/сшо-
октопиранозида (линкомицина) гидрохлорида моно¬
гидрата.
Содержание:
-суммарное количество линкомицина гидрохло¬
рида и линкомицина В гидрохлорида: не менее 96,0 % и
не более 102,0 % (в пересчете на безводное вещество);
-линкомицина В гидрохлорид: не более 5,0% (в
пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень легко растворим в воде, мало рартворим в
96 % спирте, очень мало растворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО линкомицина гидрохлорида
#или спектр, представленный на рисунке #0583.-1#.
B. 0,1 г испытуемого образца растворяют в
воде Р и доводят до объема 10 мл этим же раство¬
рителем. Полученный раствор дает реакцию (а) на
хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца растворяют
в воде, свободной от углерода диоксида, Р и доводят
до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть про¬
зрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)б.
Удельное оптическое вращение (2.2.7). От +135
до +150 (в пересчете на безводное вещество). 1,000 г
испытуемого образца растворяют в воде Р и доводят до
объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
78. Зак. 1060.
610
Гэсударственная фармакопея Республики Беларусь
Рисунок #0583.-1. Инфракрасный спектр ФСО линкомицина гидрохлорида
Раствор сравнения (а). 25,0 мг ФСО линкоми¬
цина гидрохлорида растворяют в подвижной фазе и
доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО линкомицина
гидрохлорида для проверки пригодности хромато¬
графической системы (содержит примеси А, В и С)
растворяют в 2 мл подвижной фазы.
Раствор сравнения (с). 2,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (с) доводят подвижной фазой до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октилсилильным,
деактивированным по отношению к основаниям,
эндкепированным для хроматографии Р с размером
частиц 5 мкм;
- температура: 50 °С;
-буферный раствор рН 6,1: 34 г фосфорной
кислоты Р растворяют в 900 мл воды для хромато¬
графии Р, доводят до рН 6,1 раствором аммиака
концентрированным Р и разводят водой для хрома¬
тографии Р до объема 1000 мл;
-подвижная фаза: метанолР — ацетони¬
трил Р1 —буферный раствор рН 6,1 (8:17:75, об/об/об);
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (б);
- время хроматографирования: 5,5-кратное
время удерживания линкомицина.
Относительное удерживание (по отношению к
линкомицину, время удерживания — около 10 мин):
примесь С — около 0,4; линкомицин В — около 0,5;
примесь А — около 0,7; примесь В — около 1,2 и 1,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,8 между пиком линко¬
мицина и первым пиком примеси В.
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограмме ис¬
пытуемого раствора площадь пика, соответствующего
примеси А, не должна превышать 5-кратную площадь ос¬
новного пика на хроматограмме раствора сравнения (б);
- сумма площадей пиков, соответствующих
примеси В (не более 0,5 %): на хроматограмме ис¬
пытуемого раствора сумма площадей пиков, соответ¬
ствующих примеси В, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (б);
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (с!);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (б);
- сумма примесей (не более 2,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с!).
Тяжелые металлы (2.4.8, метод С). Не более
0,0005 % (5 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Ловастатин
611
Вода (2.5.12). Не менее 3,1 % и не более 4,6 %.
Определение проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а) и (с).
Содержание С^Н^^ДЗ-НС! (линкомицин) и
С17Н32М206ЗНС1 (линкомицин В) в процентах рассчи¬
тывают с учетом содержания ДдН^НДДНС! в ФСО
линкомицина гидрохлорида. Содержание линкоми-
цина определяют путем сравнения с площадью пика
линкомицина на хроматограмме раствора сравнения
(а). Содержание линкомицина В определяют путем
сравнения с площадью пика линкомицина на хрома¬
тограмме раствора сравнения (с).
ХРАНЕНИЕ
При температуре не выше 30 °С. Стерильная
субстанция — в стерильном воздухонепроницаемом
контейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О, Е, Р.
А. Метил-6,8-дидезокси-6-[[[(2Я,4Я)-1 -метил-4-
пропилпирролидин-2-ил]карбонил]амино]-1-тио-0-
эритро-сх-О-галакто-октопмранозмд (а-амид эпимер).
В. Метил-6,8-дидезокси-6-[[[(25,4Е2)-1 -метилы-
пропил иденпирролидин-2-ил]карбонил]амино]-1-тио-
О-эрг/глро-а-О-гала/сшо-октопиранозид (пропилидено-
вые аналоги).
С. Метил-6,8-дидезокси-6-[[[(25,4Я)-4-пропил-
пирролидин-2-ил]карбонил]амино]-1-тио-0-эр1/тро-а-
0-галаклю-октопиранозид (Л/-деметил линкомицин).
й. Метил-6,8-дидезокси-6-[[[(25,4Я)-1 -метил-4-
пропилпирролидин-2-ил]карбонил]амино]-1-тио-1_-/лрео-
а-О-гала/тшо-октопиранозид (7-эла-линкомицин).
Е. (25,4/?)-1 -Метил-4-пропилпирролидин-2-карбо-
новая кислота.
Р. Метил-6-амино-6,8-дидезокси-1 -тт-О-эритро-
а-О-галактсюктопиранозид (метил-1 -тиолинкозаминид).
07/2016:1538
ЛОВАСТАТИН
1.оуа8(аЫпит
1.0УА8ТАТШ
78*. Зак. 1060.
612
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
(15,3/?,75,85,8аА?)-8-[2-[(2Я4Я?Н-Гидрокси-6-оксо-
тетрагидро-2/-/-пиран-2-ил]этил]-3,7-диметил-1,2,3,-
7,8,8а-гексагидронафтален-1-ил-(25)-2-метилбутаноат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок.
Практически нерастворим в воде, растворим в
ацетоне, умеренно растворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ловастатина #или спектр,
представленный на рисунке #1538.-1#.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+325 до +340 (в пересчете на сухое вещество). 0,125 г
испытуемого образца растворяют в ацетонитриле Р
и доводят до объема 25,0 мл этим же растворителем.
Примесь Е. Не более 0,5 %. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в ацетонитриле Р1 и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого
раствора доводят ацетонитрилом Р1 до объема
100,0 мл. 5,0 мл полученного раствора доводят аце¬
тонитрилом Р1 до объема 50,0 мл.
Раствор сравнения (Ь). 4 мг ФСО ловастатина
для идентификации пиков (содержит примеси А, В, С,
О, Е и Р) растворяют в ацетонитриле Р1 и доводят
до объема 10,0 л этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 7 объемов раст¬
вора 1,1 г/л фосфорной кислоты Р и 13 объемов аце¬
тонитрила Р1;
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 200 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания ловастатина.
Идентификация пиков примесей: идентифици¬
руют пик примеси Е, используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО ловастатина для идентификации пиков.
Относительное удерживание (по отношению к
ловастатину, время удерживания — около 5 мин): при¬
месь Е — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5,0 между пиками лова¬
статина и примеси Е.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Е —1,6):
-примесь Е: на хроматограмме испытуемого
раствора площадь пика, соответствующего приме¬
си Е, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Рисунок #1538.-1. Инфракрасный спектр ФСО ловастатина
Ловастатин
613
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в ацетонитриле Р и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 20,0 мг ФСО ловаста-
тина растворяют в ацетонитриле Р и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 0,5 мл испытуемого раст¬
вора доводят ацетонитрилом Р до объема 100,0 мл.
5.0 мл полученного раствора доводят до объема
50.0 мл.
Раствор сравнения (с). 4 мг ФСО ловастатина
для идентификации пиков(содержит примеси А, В, С,
О, Е и Р) растворяют в ацетонитриле Р и доводят до
объема 10,0 л этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: 0,1 % (об/об) раствор кис¬
лоты фосфорной Р\
- подвижная фаза В: ацетонитрил Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—7
40
60
7—9
40 —> 35
ю
со
т
о
со
9—15
35 —> 10
65 — 90
15—20
10
90
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 238 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Идентификация пиков примесей: идентифициру¬
ют пики примесей А, В, С, О и Р, используя хроматограм¬
му раствора сравнения (с) и хроматограмму, прилагае¬
мую к ФСО ловастатина для идентификации пиков.
Относительное удерживание (по отношению
к ловастатину, время удерживания — около 7 мин):
примесь В — около 0,6; примесь А — около 0,8; при¬
месь Р — около 0,9; примесь С — около 1,6; примесь
й — около 2,3.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 3,0
(Нр — высота пика примеси Р относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси Р и пик
ловастатина).
Предельное содержание примесей:
- примеси А, В, С, О (не более 0,3 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, В, С и О, не должны
превышать 0,6 площади основного пика на хромато¬
грамме раствора сравнения (Ь);
- примесь Р (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси Р, не должна превышать 0,3 пло¬
щади основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О и Р, не должна превышать 0,2 площа¬
ди основного пика на хроматограмме раствора срав¬
нения (Ь);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С24Н3605 рассчитывают с учетом со¬
держания ловастатина в ФСО ловастатина.
ХРАНЕНИЕ
При температуре от 2 °С до 8 °С в среде азота.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
А. (15,7С,85,8аЯ)-8-[2-[(2Я,4Я)-4-Гидрокси-6-оксо-
тетрагидро-2Я-пиран-2-ил]этил]-7-метил-1,2,3,7,8,8а-
гексагидронафтален-1-ил-(25)-2-метилбутаноат (мева-
статин).
В. (ЗЯ,5Я)-7-[(15,25,6Я,85,8аЯ)-2,6-Диметил-8-
[[(25)-2-метилбутаноил]окси]-1,2,6,7,8,8а-гекса-гид-
ронафтален-1 -ил]-3,5-дигидроксигептановая кислота
(гидроксикислота ловастатина).
79. Зак. 1060.
614
Гэсударственная фармакопея Республики Беларусь
С. (15,3/?,75,85,8а«>-3,7-Диметил-8-[2-[(2«)-6-оксо-
3,6-дигидро-2Н-пиран-2-ил]этил]-1,2,3,7,8,8а-гекса-
гидронафтален-1 -ил-(23)-2-метилбутаноат (дегидро-
ловастатин).
О. (2Я,4Я)-2-[2-[(15,23,6К,85,8аК)-2,6-Диметил-8-
[[(25)-2-метилбутаноил]окси]-1,2,6,7,8,8а-гекса-
гидронафтален-1-ил]этил]-6-оксотетрагидро-2Н-пиран-
4-ил-(3«,5К)-7-[(13,23,6/?,85,8а/?)-2,6-диметил-8-[[(25)-
2-метилбуганоил]окси]-1,2,6,7,8,8а-гексагидронафтапен-
1-ил]-3,5-дигидроксигептаноат (димер ловастатина).
Е. (15,35,4а/?,75,85,8а5)-8-[2-[(2/?,4/?)-4-гидрокси-
6-оксотетрагидро-2/-/-пиран-2-ил]этил]-3,7-диметил-
1,2,3,4,4а,7,8,8а-октагидронафтален-1-ил-(25)-2-
метилбутаноат (4,4-дигидроловастатин).
Р. (15,ЗЯ,78,85,8а/?)-8-[2-[(2Я,4/?)-4-гидрокси-6-
оксотетрагидро-2Н-пиран-2-ил]этил]-3,7-диметил-
1,2,3,7,8,8а-гексагидронафтапен-1 -ил-(22)-2-метилбут-
2-еноат.
07/2016:0929
ЛОПЕРАМИДА ГИДРОХЛОРИД
1-орегат1сИ Ьус1гос111опс1ит
ЕОРЕРАМЮЕ НУОПОСШОПЮЕ
С-Н-СМ-О. - НС1 М.м. 513,5
[34552-83-5]
ОПРЕДЕЛЕНИЕ
4-[4-(4-Хлорфенил)-4-гидроксипиперидин-1-ил]-
А/,А/-диметил-2,2-дифенилбутанамида гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Мало растворим в воде, легко растворим в 96 %
спирте и в метаноле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лоперамида гидрохлорида #или
спектр, представленный на рисунке #0929.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО лоперамида гидрохлорида
растворяют по отдельности в минимальном объеме
метиленхлорида Р и выпаривают до сухих остатков,
которые используют для получения новых спектров.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО лоперами¬
да гидрохлорида для проверки пригодности хрома¬
тографической системы растворяют в метаноле Р
и доводят до объема 1,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят метанолом Р до объема 20,0 мл. 1,0 мл
полученного раствора доводят метанолом Р до объ¬
ема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
Лоперамида гидрохлорид
615
Рисунок #0929.-1. Инфракрасный спектр ФСО лоперамида гидрохлорида
синильным, деактивированным по отношению к
основаниям, для хроматографии Р с размером ча¬
сти ц 3 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: раствор 17,0 г/л тетра-
бутиламмония гидросульфата Р1\
- подвижная фаза В: ацетонитрил Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
о
со
т
о
05
о
г*-
т
о
15—17
30
70
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси С относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси О и
примеси Н);
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси Е относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Е и
примеси А);
-хроматограмма должна соответствовать хро¬
матограмме, прилагаемой к ФСО лоперамида гидрох¬
лорида для проверки пригодности хроматографиче¬
ской системы.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 1,3; для примеси О — 1,7):
- примеси А, В, С, О, Е, Р, <3, Н (не более 0,2 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, С, й, Е, Р, 6 и
H, не должны превышать площадь основного пика на
хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е, Р, С и Н, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,3 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
I, 5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 50 мл
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 51,35 мг С29Н33С1М202 НС1.
79*. Зак. 1060.
616
ХРАНЕНИЕ
В защищенном от света месте.
Гэсударственная фармакопея Республики Беларусь
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Е,
О, Н.
А. 4-[4-(4'-Хлорбифенил-4-ил)-4-гидрокси-
пиперидин-1-ил]-Л/,Л/-диметил-2,2-дифенилбутанамид.
В. 4-(4-Хлорфенил)-1,1-бис[4-(диметиламино)-4-
оксо-3,3-дифенилбутил]-4-гидроксипиперидиний.
Е. 4-(4-Хлорфенил)-1-[4-[4-(4-хлорфенил)-4-гид-
роксипиперидин-1-ил]-2,2-дифенилбутаноил]пиперидин-
4-о л.
Р. 4-[л?ранс-4-(4-Хлорфенил)-4-гидрокси-1-окси-
допиперидин-1-ил]-Л/>А/-диметил-2,2-дифенилбутанамид
(лоперамида оксид).
О. 4-[дбУС-4-(4-Хлорфенил)-4-гидрокси-1-окси-
допиперидин-1-ил]-Л/,А/-диметил-2,2-дифенилбутанамид.
С!
О. 4-(4-Гидрокси-4-фенилпиперидин-1-ил)-Л/,Л/-
диметил-2,2-дифенилбутанамид.
Н. 4-[4-(4-Хлорфенил)-3,6-дигидропиридин-1 (2Н)-
ил]-Л/,А/-диметил-2,2-дифенилбутанамид.
Лоперамида оксид моногидрат
617
07/2016:1729
ЛОПЕРАМИДА ОКСИД
МОНОГИДРАТ
1-орегат1сИ омдит топоЬудпсит
ШРЕКАМЮЕ ОХЮЕ МОЫОНУОЕАТЕ
С29Н33С1М203 • н20 М.м. 511,1
[106900-12-3]
ОПРЕДЕЛЕНИЕ
4-[шранс-4-(4-Хлорфенил)-4-гидрокси-1-оксидо-
пиперидин-1-ил]-Л/,Л/-диметил-2,2-дифенилбутанамид
моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Слегка гигроско¬
пичен.
Практически нерастворим в воде, легко раство¬
рим в 96 % спирте и в метиленхлориде.
Температура плавления: около 152 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лоперамида оксида моногидра¬
та #или спектр, представленный на рисунке #1729.-1#.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО лоперамида
гидрохлорида растворяют в метаноле Р, прибавляют
0,5 мл испытуемого раствора и доводят метанолом Р
до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят метанолом Р до объема 20,0 мл. 1,0 мл
полученного раствора доводят метанолом Р до объ¬
ема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 3 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: раствор 17,0 г/л тетра-
бутиламмония гидросульфата Р1\
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
15—17
90 — 30
30
10 — 70
70
- скорость подвижной фазы. 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл.
Рисунок #1729.-1. Инфракрасный спектр ФСО лоперамида оксида моногидрата
80. Зак. 1060.
618
Гэсударственная фармакопея Республики Беларусь
Относительное удерживание (по отношению
к лоперамида оксиду, время удерживания — около
7 мин): примесь А — около 0,9; примесь В — около
1,11; примесь С — около 1,13.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,8 между пиками лопе¬
рамида оксида и примеси А.
Предельное содержание примесей:
- примеси А, В, С (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, В и С, не должны превы¬
шать площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 0,3 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь).
Вода (2.5.12). Не менее 3,4 % и не более 4,2 %.
Определение проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 50 мл
смеси кислота уксусная безводная Р — метилэтил-
кетон Р (1:7, об/об) и титруют 0,1 М раствором кис-
лоты хлорной с использованием 0,2 мл раствора на-
фтолбензеина Р в качестве индикатора.
1 мп 0,1 М раствора кислоты хлорной соответ¬
ствует 49,30 мг С29Н33С1М203.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. 4-[4-(4-Хлорфенил )-4-гидроксипиперидин-1 -ил]-
Л/,А/-диметил-2,2-дифенилбутанамид (лоперамид).
В. 4-[цш>4-(4-Хлорфенил)-4-гидрокси-1 -окси-
допиперидин-1-ил]-Л/,Л/-диметил-2,2-дифенилбутанамид.
С1
С. 4-[4-(4-Хпорфенил)-3,6-дигидропиридин-1 (2Н)-
ил]-Л/,Л/-диметил-2,2-дифенилбутанамид.
07/2016:2124
ЛОРАТАДИН
1-Ога1асИпит
1.0КАТАОШЕ
С22Н23С1Ы202 М.м. 382,9
[79794-75-5]
ОПРЕДЕЛЕНИЕ
Этил-4-(8-хлор-5,6-дигидро-11Н- бензо[5,6]-
циклогепта-[1,2-Ь]пиридин-11 -илиден)пиперидин-1 -
карбоксилат.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне и в метаноле.
Обладает полиморфизмом (5.9).
Лоратадин
619
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО лоратадина #или спектр, пред¬
ставленный на рисунке #2124.-1#.
Если спектры, полученные с использованием
образцов в твердом состоянии, отличаются, то испы¬
туемый образец и ФСО лоратадина растворяют по
отдельности в минимальном объеме ацетона Р и вы¬
паривают до сухих остатков, которые используют для
получения новых спектров.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20,0 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)5.
Примесь Н. Не более 0,1 %. Газовая хроматогра¬
фия (2.2.28).
Раствор внутреннего стандарта. 25 мг изо-
амилбензоата Р растворяют в метиленхлориде Р и
доводят до объема 100 мл этим же растворителем.
5.0 мл полученного раствора доводят метиленхлори-
дом Р до объема 50 мл.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в метиленхлориде Р, прибавляют
1.0 мл раствора сравнения (а), 1,0 мл раствора вну¬
треннего стандарта и доводят метиленхлоридом Р
до объема 5,0 мл.
Раствор сравнения (а). 25,0 мг ФСО лората¬
дина примеси Н растворяют в метиленхлориде Р и
доводят до объема 100,0 мл этим же растворителем.
5.0 мл полученного раствора доводят метиленхлори¬
дом Р до объема 50,0 мл.
Раствор сравнения (Ь). К 1,0 мл раствора срав¬
нения (а) прибавляют 1,0 мл раствора внутреннего
стандарта и доводят метиленхлоридом Р до объема
5,0 мл.
Условия хроматографирвоания:
- колонка капиллярная кварцевая длиной 25 м и
диаметром 0,32 мм, покрытая слоем поли(диметил)
силоксана Р (толщина слоя 0,52 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,0 мл/мин;
- деление потока: 1:30;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1
80
1—23
80 -► 300
23—33
300
Блок ввода проб
260
Детектор
300
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора и раствора сравнения (Ь).
Относительное удерживание (по отношению
к лоратадину, время удерживания — около 32 мин):
примесь Н — около 0,33; изоамилбензоат —
около 0,37.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси Н и изоамилбензоата;
- отношение сигнал/шум: не менее 10 для пика
примеси Н.
Предельное содержание примесей:
- примесь Н: на хроматограмме раствора срав¬
нения (Ь) рассчитывают отношение (/?) площади пика
примеси Н к площади пика изоамилбензоата; на хро-
Рисунок #2124.-1. Инфракрасный спектр ФСО лоратадина
80*. Зак. 1060.
620
Гэсударственная фармакопея Республики Беларусь
матограмме испытуемого раствора рассчитывают от¬
ношение площади пика примеси Н к площади пика
изоамилбензоата: полученное отношение не должно
превышать 2-кратное значение Р.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО лоратадина
примеси Р растворяют в подвижной фазе и доводят
до объема 25 мл этим же растворителем. 1 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 10 мл.
Раствор сравнения (Ь). 5 мг ФСО лоратадина
для проверки пригодности хроматографической си¬
стемы (содержит примеси А и Е) растворяют в под¬
вижной фазе, прибавляют 0,5 мл раствора сравнения
(а) и доводят подвижной фазой до объема 5 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р с размером частиц 5 мкм с очень низкой
силанольной активностью;
- температура: 40 °С;
- подвижная фаза: метанол Р — раствор
6,8 г/л калия дигидрофосфата Р (доведенный до
рН (2,80±0,05) кислотой фосфорной Р) — ацетони¬
трил Р (30:35:40, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 5-кратное вре¬
мя удерживания лоратадина.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и Е, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО лоратадина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению
к лоратадину, время удерживания — около 12 мин):
примесь й — около 0,2; примесь В — около 0,4; при¬
месь Р — около 0,9; примесь Е — около 1,1; при¬
месь А— около 2,4; примесь С — около 2,7.
Пригодность хроматографической системы:
раствора сравнения (Ь):
- коэффициент разделения пиков: не менее 2,5
(Нр — высота пика примеси Е относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Е и
лоратадина).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 1,7; для примеси Е — 3,4; для приме¬
си Р — 1,6):
- примесь Р (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Р, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примеси А, В, С, Ц Е (не более 0,1 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, В, С, О и Е, не должны
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, О, Е и Р, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Сульфаты (2.4.13). Не более 0,0150%
(150 ррт). 1,33 г испытуемого образца прокаливают
при температуре (800±25) °С. Полученный остаток
растворяют в 20 мл воды дистиллированной Р и, при
необходимости, фильтруют через бумажный фильтр,
свободный от сульфатов. Повторяют фильтрование с
новыми бумажными фильтрами до получения немут¬
ного фильтрата.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
кислоты уксусной ледяной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 38,29 мг С22Н2зС11Ч202
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, Н.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. П = ОН: Этил-4-[(11Я5)-8-хлор-11-гидрокси-6,11-
дигидро-5Н-бензо[5,6]циклогепта[1,2-Ь]пиридин-11 -ил]-
пиперидин-1 -карбоксилат.
Магния аспартат дигидрат
621
Р. К = Р: Этил-4-[( 11 Я5)-8-хлор-11 -фтор-6,11 -дигид-
ро-5Н-бензо[5,6]циклогепта[1,2-Ь]пиридин-11 -ил]-
пиперидин-1 -карбоксилат.
В. 8-Хлор-5,6-дигидро-11 Н-бензо[5,6]циклогеп-
та[1,2-Ь]пиридин-11 -он.
С. Я = С1, К' = СО-ОС2Н5: Этил-4-(4,8-дихпор-5,6-
дигидро-11 Н-бензо[5,6]циклогепта[1,2-Ь]пиридин-11 -
илиден)пиперидин-1 -карбоксилат.
й. Я = Я' = Н: 8-Хлор-11-(пиперидин-4-илиден)-
6,11-дигидро-5/-/-бензо[5,6]цикпогепта[1,2-Ь]пиридин.
6. В = Н, К' = СН3:8->Оюр-11-(1-метилпиперидин-
4-ил иден)-6,11 -дигидро-5Н-бензо[5,6]циклогепта[1,2-Ь]-
пиридин.
Е. Этил^4-[(11 Я5)-8-хлор-6,11 -дигидро-5Н-бензо[5,6]-
циклогепта[1,2-Ь]пиридин-11-ил]-3,6-дигидропиридин-
1 (2Я)-карбоксилат.
о
з
И. Этил-4-оксопиперидин-1 -карбоксилат.
07/2016:1445
МАГНИЯ АСПАРТАТ Д И ГИДРАТ
МадпезН азраЛаз сШтудпсиз
МАСЫЕ8ШМ А8 РАНТ АТЕ Р1НУОНАТЕ
Мд2+
о2с
Н
1ЧН2
со2н
• 2 Н20
С,Н1гМд^Ов • 2НгО М.м. 324,5
#[2068-80-6]*
ОПРЕДЕЛЕНИЕ
Магния ди[(5)-2-аминогидробутан-1,4-диоат] ди¬
гидрат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества».
Результат: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
C. 15 мг испытуемого образца прокаливают до
образования белого остатка. Полученный остаток
растворяют в 1 мл кислоты хлористоводородной
разведенной Р, нейтрализуют раствором натрия
гидроксида разведенным Р по красной лакмусовой
бумаге Р и, при необходимости, фильтруют. Получен¬
ный раствор дает реакцию на магний (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 6,0 до 8,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
+22,0 до +24,0 (в пересчете на безводное вещество).
0,50 г испытуемого образца растворяют в растворе
515 г/л кислоты хлористоводородной Р и доводят до
объема 25,0 мл этим же растворителем.
Нингидрин-положительные вещества. Тонкос¬
лойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО магния аспар-
тата дигидрата растворяют в воде Р и доводят до
объема 50 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО магния аспар-
тата дигидрата и 10 мг ФСО глутаминовой кисло¬
ты растворяют в 2 мл воды Р и доводят до объема
25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкп.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 *С
до 105 °С в течение 15 мин.
622
Гэсударственная фармакопея Республики Беларусь
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Предельное содержание примесей:
-любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт). К
10 мл раствора 8 прибавляют 5 мл воды Р.
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
К 12 мл раствора 8 прибавляют 3 мл воды дистилли¬
рованной Р и выдерживают в течение 30 мин.
Аммония соли (2.4.1, метод В). Не более
0,0200 % (200 ррт). 50 мг испытуемого образца долж¬
ны выдерживать испытание на аммония соли. Эталон
готовят с использованием 0,1 мл эталонного раст¬
вора аммония (100 ррт Л/Н^ Р.
Железо (2.4.9). Не более 0,0050 % (50 ррт).
0,20 г испытуемого образца помещают в делительную
воронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в 20 мл воды Р при слабом нагревании. 12 мл
полученного раствора должны выдерживать испыта¬
ние на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (1 ррт РЬ) Р
Вода (2.5.12). Не менее 10,0% и не более
14,0 %. Определение проводят из 0,100 г испытуемо¬
го образца. Испытуемый образец растворяют в 10 мл
формамида Р1 при температуре 50 °С предохраняя
от попадания влаги, прибавляют 10 мл метанола без¬
водного Р и охлаждают. Параллельно проводят кон¬
трольный опыт.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,260 г испытуемого образца растворят в 10 мл
воды Р и проводят комплексометрическое определе¬
ние магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 28,85 мг С8Н12МдЫ208.
ПРИМЕСИ
н т2
Н°2С\.Х
со2н
А. (28)-2-Аминобутандиовая кислота (аспараги¬
новая кислота).
07/2016:2035
МАГНИЯ АЦЕТАТ ТЕТРАГИДРАТ
МадпезН асе1аз ШгаЬубпсиз
МАСЫЕЗШМ АСЕТАТЕ ТЕТКАНУОКАТЕ
Мд(СН3СОО)2 - 4Н20 М.м. 214,5
[16674-78-5]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Бесцветные кристаллы или белый или почти бе¬
лый кристаллический порошок.
Легко растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Около 100 мг испытуемого образца раство¬
ряют в 2 мл воды Р. К полученному раствору при¬
бавляют 1 мл раствора аммиака разведенного Р1 и
нагревают. Образуется белый осадок, который мед¬
ленно растворяется при прибавлении 5 мл раствора
аммония хлорида Р. При прибавлении 1 мл раствора
динатрия гидрофосфата Р образуется белый кри¬
сталлический осадок.
B. Испытуемый образец дает реакцию (Ь) на аце¬
таты (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 7,5 до 8,5. 2,5 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 50 мл этим же раство¬
рителем.
Хлориды (2.4.4). Не более 0,0330 % (330 ррт).
1.0 г испытуемого образца растворяют в воде Р и до¬
водят до объема 100 мл этим же растворителем.
Нитраты. Не более 0,0003% (3 ррт). 1,0 г ис¬
пытуемого образца растворяют в воде дистиллиро¬
ванной Р и доводят до объема 10 мл этим же раство¬
рителем, прибавляют 5 мг натрия хлорида Р, 0,05 мл
раствора индигокармина Р и, при перемешивании,
10 мл кислоты серной, свободной от азота, Р. По¬
является голубое окрашивание, которое сохраняется
в течение не менее 10 мин.
Сульфаты (2.4.13). Не более 0,0600 % (600 ррт).
0,25 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 15 мл этим же
растворителем.
Алюминий (2.4.17). Не более 0,0001 % (1 ррт).
Предписанный раствор. 4,0 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100 мл этим же растворителем. К полученному рас¬
твору прибавляют 10 мл ацетатного буферного
раствора рН 6,0 Р.
Раствор сравнения. Смешивают 2 мл эталон¬
ного раствора алюминия (2 ррт А!) Р, 10 мл аце¬
татного буферного раствора рН 6,0 Р и 98 мл
воды Р.
Холостой раствор. Смешивают 10 мл ацетат¬
ного буферного раствора рН 6,0 Р и 100 мл воды Р.
Кальций (2.4.3). Не более 0,0100% (100 ррт).
1.0 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 15 мл этим же
растворителем.
Калий. Не более 0,1 %. Атомно-эмиссионная
спектрометрия (2.2.22, метод II).
Испытуемый раствор. 0,5 г испытуемого образ¬
ца растворяют в воде Р и доводят этим же раствори¬
телем до объема 100 мл.
Растворы сравнения. Готовят путем разведения
эталонного раствора калия (600 ррт К) Р водой Р.
Длина волны: 766,5 нм.
Натрий. Не более 0,5 %. Атомно-эмиссионная
спектрометрия (2.2.22, метод II).
Магния гидроксид
623
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в воде Р и доводят этим же раствори¬
телем до объема 100 мл.
Растворы сравнения. Готовят путем разведения
эталонного раствора натрия (200 ррт Ыа) Р во¬
дой Р.
Длина волны: 589,0 нм.
Тяжелые металлы (2.4.8, метод А). Не более
0,0040 % (40 ррт). 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят этим же растворителем до
объема 20 мл. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (2 ррт РЬ) Р.
Легкоокисляемые вещества. 2,0 г испытуемого
образца растворяют в 100 мл кипящей воды Р, при¬
бавляют 6 мл раствора 150 г/л кислоты серной Р и
0,3 мл 0,02 М раствора калия перманганата. Пере¬
мешивают и кипятят осторожно в течение 5 мин. Ро¬
зовое окрашивание не должно полностью исчезнуть.
Вода (2.5.12). Не менее 33,0% и не более
35,0 %. Определение проводят из 0,100 г испытуемо¬
го образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют 300 мл
воды Р и проводят комлексометрическое определе¬
ние магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 14,24 мг С4Н6Мд04.
07/2016:0039
МАГНИЯ ГИДРОКСИД
МадпезИ НудгохМит
МАОЫЕ8ШМ НУОКОХЮЕ
Мд(ОН)2 М.м. 58,32
[1309-42-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 95,0 % и не более 100,5 %
Мд(ОН)2.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый мелкий аморфный поро¬
шок.
Практически нерастворим в воде. Растворяется в
разведенных кислотах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 15 мг испытуемого образца растворяют в 2 мл
кислоты азотной разведенной Р и нейтрализуют
раствором натрия гидроксида разведенным Р. По¬
лученный раствор дает реакцию на магний (2.3.1).
B. Испытуемый образец выдерживает испытание
«Потеря в массе при прокаливании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в смеси из 50 мл кислоты уксусной Р и 50 мл
воды дистиллированной Р, при этом допускается не¬
значительное выделение пузырьков газа. Кипятят в
течение 2 мин, охлаждают и доводят кислотой уксус¬
ной разведенной Р до объема 100 мл. При необходи¬
мости фильтруют через предварительно прокаленный
и взвешенный фарфоровый или кварцевый фильтр с
подходящим размером пор для получения прозрач¬
ного фильтрата. #Фильтр с остатком используют для
испытания «Вещества, нерастворимые в кислоте ук¬
сусной»^
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)3.
Растворимые вещества. Не более 2,0 %. 2,00 г
испытуемого образца смешивают с 100 мл воды Р и
кипятят в течение 5 мин. Горячую смесь фильтруют
через пористый стеклянный фильтр (ПОР 40) (2.1.2).
Фильтрат охлаждают и доводят водой Р до объема
100 мл. 50 мл полученного раствора выпаривают до¬
суха и сушат остаток при температуре от 100 °С до
105 °С. Масса полученного остатка не должна превы¬
шать 20 мг.
Вещества, нерастворимые в кислоте уксус¬
ной. Не более 0,1 %. Остаток, полученный при филь¬
тровании раствора 3, промывают, высушивают и про¬
каливают при температуре (600±50) °С. Масса полу¬
ченного остатка не должна превышать 5 мг.
Хлориды (2.4.4). Не более 0,1 %. 1 мл раствора
3 доводят водой Р до объема 15мл. Полученный
раствор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 1,0 %. 0,3 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл. Полученный раствор должен выдерживать
испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0004 %
(4 ррт). 5 мл раствора 5 должны выдерживать испы¬
тание на мышьяк.
Кальций (2.4.3). Не более 1,5 %. 1,3 мл раствора
3 доводят водой дистиллированной Р до объема
150 мл. 15 мл полученного раствора должны выдер¬
живать испытание на кальций.
Железо (2.4.9). Не более 0,07 %. 0,15 г испыту¬
емого образца растворяют в 5 мл кислоты хлори¬
стоводородной разведенной Р и доводят водой Р до
объема 10 мл. 1 мл полученного раствора доводят во¬
дой Р до объема 10 мл. Полученный раствор должен
выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0030 % (30 ррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл кислоты хлористоводородной Р1 и
встряхивают с 25 мл метилизобутилкетона Р в те¬
чение 2 мин. Выдерживают до разделения слоев. Вы¬
паривают водный слой досуха, полученный остаток
растворяют в 30 мл воды Р. 12 мл полученного раст¬
вора должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием эталонного
раствора свинца (2 ррт РЬ) Р.
Потеря в массе при прокаливании. Не менее
29,0 % и не более 32,5 %. 0,5 г испытуемого образца
постепенно прокаливают при температуре (900±50) °С
до постоянной массы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в смеси
из 20 мл воды Р и 2 мл кислоты хлористоводород¬
ной разведенной Р и проводят комплексометрическое
определение магния (2.5.11).
624
Гэсударственная фармакопея Республики Беларусь
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 5,832 мг Мд(ОН)2.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0042
МАГНИЯ КАРБОНАТ ОСНОВНОЙ,
ЛЕГКИЙ
Мадпези зиЬсагЬопаз /еу/з
МАОИЕЗШМ САКВОЙ АТЕ, ЫвНТ
ОПРЕДЕЛЕНИЕ
Гидратированный магния карбонат основной.
Содержание: не менее 40,0 % и не более 45,0 % в
пересчете на МдО (М.м. 40,30).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде. Растворим в
разведенных растворах кислот, процесс сопровожда¬
ется бурным выделением пузырьков.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Насыпная плотность (2.9.34): не более 0,15 г/мл.
B. Испытуемый образец дает реакцию (а) на кар¬
бонаты (2.3.1).
C. 15 мг испытуемого образца растворяют в 2 мл
кислоты азотной разведенной Р и нейтрализуют
раствором натрия гидроксида разведенного Р. По¬
лученный раствор дает реакцию на магний (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в 100 мл кислоты уксусной разведенной Р. По¬
сле прекращения выделения пузырьков газа кипятят
в течение 2 мин, охлаждают и доводят до объема
100 мл кислотой уксусной разведенной Р. Фильтру¬
ют, если необходимо, через предварительно прока¬
ленный и взвешенный фарфоровый или кварцевый
фильтр с подходящим размером пор до получения
прозрачного фильтрата.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона В(К)4.
Растворимые вещества. Не более 1,0 %. 2,00 г
испытуемого образца смешивают с 100 мл воды Р и
кипятят в течение 5 мин. Фильтруют горячий раствор
через стеклянный фильтр (ПОР 40) (2.1.2), фильтрат
охлаждают и доводят водой Р до объема 100 мл. 50 мл
полученного раствора выпаривают и остаток сушат
при температуре от 100 °С до 105 °С. Масса остатка
после высушивания не должна превышать 10 мг.
Вещества, нерастворимые в кислоте уксус¬
ной. Не более 0,05 %. Масса осадка, полученно¬
го при приготовлении раствора 3, после промыва¬
ния, высушивания и прокаливания при температуре
(600±50) °С, не должна превышать 2,5 мг.
Хлориды (2.4.4). Не более 0,0700 % (700 ррт).
1,5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,3 %. 1 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл. Полученный раствор должен выдерживать
испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 10 мл раствора 3 должны выдерживать ис¬
пытания на мышьяк.
Кальций (2.4.3). Не более 0,75 %. 2,6 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 150 мл. 15 мл полученного раствора должны вы¬
держивать испытание на кальций.
Железо (2.4.9). Не более 0,0400 % (400 ррт).
0,1 г испытуемого образца растворяют в 3 мл кисло¬
ты хлористоводородной разведенной Р, доводят во¬
дой Р до объема 10 мл. 2,5 мл полученного раствора
доводят водой Р до объема 10мл. Раствор должен
выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). К 20 мл раствора 3 прибавля¬
ют 15 мл кислоты хлористоводородной Р1, 25 мл
метилизобутилкетона Р и встряхивают в течение
2 мин. Выдерживают до разделения слоев, затем от¬
деляют водный слой и выпаривают досуха. Остаток
растворяют в 1 мл кислоты уксусной Р и доводят во¬
дой Р до объема 20 мл. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в смеси
из 20 мл воды Р и 2 мл кислоты хлористоводород¬
ной разведенной Р. Проводят комплексометрическое
определение магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 4,030 мг МдО.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Некоторые
из характеристик, описанных в разделе «Функци¬
онально-обусловленные характеристики», могут
также присутствовать в части фармакопейной
статьи, обязательной для выполнения, так как од¬
новременно они являются и показателями качества.
В таком случае в разделе «Функционально-обуслов¬
ленные характеристики» дается перекрестная
ссылка на это испытание в части фармакопейной
статьи, обязательной для выполнения. Контроль
данных характеристик может вносить вклад в каче¬
ство лекарственного средства путем достижения
постоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, однако
другие методы также могут быть использованы. В
случае указания результатов конкретных характе¬
ристик требуется указывать также и методы про¬
ведения испытаний.
Следующие характеристики могут быть важ¬
ны для магния карбоната основного, легкого, ис¬
пользуемого в качестве наполнителя при производ¬
стве твердых дозированных лекарственных форм
для внутреннего применения.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Магния карбонат основной, тяжелый
625
Насыпная плотность и плотность после усад¬
ки (2.9.34).
07/2016:0043
МАГНИЯ КАРБОНАТ ОСНОВНОЙ,
ТЯЖЕЛЫЙ
МадпезН зиЬсагЬопаз ропс1егозиз
МАСЫЕ8ШМ САКВОИАТЕ, НЕАУУ
ОПРЕДЕЛЕНИЕ
Гидратированный магния карбонат основной.
Содержание: не менее 40,0 % и не более 45,0 % в
пересчете на МдО (М.м. 40,30).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде. Растворим в
разведенных кислотах, процесс сопровождается бур¬
ным выделением пузырьков.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Насыпная плотность (2.9.34): не менее 0,25 г/мл.
B. Испытуемый образец дает реакцию (а) на кар¬
бонаты (2.3.1).
C. Около 15 мг испытуемого образца раство¬
ряют в 2 мл кислоты азотной разведенной Р и
нейтрализуют раствором натрия гидроксида раз¬
веденным Р. Полученный раствор дает реакцию на
магний (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в 100 мл кислоты уксусной разведенной Р.
После прекращения выделения пузырьков кипятят
в течение 2 мин, охлаждают и доводят кислотой
уксусной разведенной Р до объема 100 мл. Филь¬
труют, если необходимо, через предварительно
прокаленный и взвешенный фарфоровый или квар¬
цевый фильтр с подходящим размером пор до по¬
лучения прозрачного фильтрата.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)4.
Растворимые вещества. Не более 1,0%.
2,00 г испытуемого образца смешивают с 100 мл
воды Р и кипятят в течение 5 мин. Фильтруют го¬
рячий раствор через стеклянный фильтр (ПОР 40)
(2.1.2), фильтрат охлаждают и доводят водой Р до
объема 100 мл. 50 мл полученного раствора выпа¬
ривают и остаток сушат при температуре от 100 °С
до 105 °С. Масса остатка после высушивания не
должна превышать 10 мг.
Вещества, нерастворимые в уксусной кисло¬
те. Не более 0,05 %. Масса осадка, полученного при
приготовлении раствора 3, после промывания, высу¬
шивания и прокаливания при температуре (600±50) °С
не должна превышать 2,5 мг.
Хлориды (2.4.4). Не более 0,0700 % (700 ррт).
1,5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,6 %. 0,5 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл. Полученный раствор должен выдерживать
испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 10 мл раствора 3 должны выдерживать
испытания на мышьяк.
Кальций (2.4.3). Не более 0,75 %. 2,6 мл раст¬
вора 3 доводят водой дистиллированной Р до
объема 150 мл. 15 мл полученного раствора долж¬
ны выдерживать испытание на кальций.
Железо (2.4.9). Не более 0,0400 % (400 ррт).
0,1 г испытуемого образца растворяют в 3 мл кис¬
лоты хлористоводородной разведенной Р, дово¬
дят водой Р до объема 10 мл. 2,5 мл полученного
раствора доводят водой Р до объема 10 мл. Раст¬
вор должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). К 20 мл раствора 3 прибавля¬
ют 15 мл кислоты хлористоводородной Р1, 25 мл
метилизобутилкетона Р и встряхивают в течение
2 мин. Выдерживают до разделения слоев, затем
отделяют водный слой и выпаривают досуха. Оста¬
ток растворяют в 1 мл кислоты уксусной Р и дово¬
дят водой Р до объема 20 мл. 12 мл полученного
раствора должны выдерживать испытание на тя¬
желые металлы. Эталон готовят с использованием
эталонного раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в смеси
из 20 мл воды Р и 2 мл кислоты хлористоводород¬
ной разведенной Р. Проводят комплексометрическое
определение магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 4,030 мг МдО.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для магния карбоната основного, тяже¬
лого, используемого в качестве наполнителя при
производстве таблеток.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Насыпная плотность и плотность после
усадки (2.9.34).
626
Государственная фармакопея Республики Беларусь
07/2016:0040
МАГНИЯ ОКСИД ЛЕГКИЙ
МадпезИ охМит /еуе
МАвЫЕЗШМ охюе, ивнт
МдО М.м. 40,30
[1309-48-4]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 100,5 %
МдО (в пересчете на прокаленное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Мелкий, белый или почти белый аморфный по¬
рошок.
Практически нерастворим в воде. Растворяется
в разведенных кислотах, в большинстве случаев со
слабым выделением пузырьков.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Насыпная плотность (2.9.34): не более 0,15 г/мл.
B. Около 15 мг испытуемого образца раство¬
ряют в 2 мл кислоты азотной разведенной Р и
нейтрализуют раствором натрия гидроксида раз¬
веденным Р. Полученный раствор дает реакцию на
магний (2.3.7).
C. Испытуемый образец выдерживает испыта¬
ние «Потеря в массе при прокаливании», как ука¬
зано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в смеси из 30 мл воды дистиллированной Р
и 70 мл кислоты уксусной Р. Кипятят в течение
2 мин, охлаждают и доводят кислотой уксусной
разведенной Р до объема 100 мл. При необходи¬
мости фильтруют через предварительно прокален¬
ный и взвешенный фарфоровый или кварцевый
фильтр с подходящим размером пор до получения
прозрачного фильтрата.
Цветность (2.2.2, метод //). Окраска раствора
5 должна быть не интенсивнее эталона В(К)2.
Растворимые вещества. Не более 2,0 %.
2,00 г испытуемого образца смешивают с 100 мл
воды Р и кипятят в течение 5 минут. Горячую смесь
фильтруют через стеклянный фильтр (ПОР 40)
(2.1.2), охлаждают и доводят водой Р до объема
100 мл. 50 мл фильтрата выпаривают досуха и су¬
шат при температуре от 100 °С до 105 °С. Масса
остатка не должна превышать 20 мг.
Вещества, нерастворимые в уксусной кис¬
лоте. Не более 0,1 %. Остаток, полученный при
приготовлении раствора 5, промывают, высуши¬
вают и прокаливают при температуре (600±50) °С.
Масса полученного остатка не должна превышать
5 мг.
Хлориды (2.4.4). Не более 0,15%. 0,7 мл
раствора 5 доводят водой Р до объема 15 мл. По¬
лученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 1,0%. 0,3 мл
раствора 5 доводят водой дистиллированной Р до
объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0004 %
(4 ррт). 5 мл раствора 3 должны выдерживать ис¬
пытание на мышьяк.
Кальций (2.4.3). Не более 1,5 %. 1,3 мл раст¬
вора 3 доводят водой дистиллированной Р до
объема 150 мл. 15 мл полученного раствора долж¬
ны выдерживать испытание на кальций.
Железо (2.4.9). Не более 0,1 %. 50 мг испытуемо¬
го образца растворяют в 5 мл кислоты хлористово¬
дородной разведенной Р и доводят водой Р до объе¬
ма 10 мл. 2 мл полученного раствора доводят водой Р
до объема 10 мл. Полученный раствор должен выдер¬
живать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0030 % (30 ррт). К 20 мл раствора 3 прибавляют
15 мл кислоты хлористоводородной Р1 и встряхива¬
ют с 25 мл метилизобутилкетона Р в течение 2 мин.
Выдерживают до разделения слоев, затем отделяют
водный слой и выпаривают досуха. Остаток раство¬
ряют в 1,5 мл кислоты уксусной Р и доводят водой Р
до объема 30 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Потеря в массе при прокаливании. Не более
8,0%. 1,00 г испытуемого образца прокаливают при
температуре (900±25) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,320 г испытуемого образца растворяют в 20 мл
кислоты хлористоводородной разведенной Р и дово¬
дят водой Р до объема 100,0 мл. Используя 20,0 мл
полученного раствора, проводят комплексометриче¬
ское определение магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 4,030 мг МдО.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, од¬
нако другие методы также могут быть использо¬
ваны. В случае указания результатов конкретных
характеристик требуется указывать также и ме¬
тоды проведения испытаний.
Следующие характеристики могут быть важ¬
ны для магния оксида легкого, используемого в ка¬
честве наполнителя при производстве твердых до¬
зированных лекарственных форм для внутреннего
применения.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Насыпная плотность и плотность после усад¬
ки (2.9.34).
Магния оксид тяжелый
627
07/2016:0041
МАГНИЯ ОКСИД ТЯЖЕЛЫЙ
МадпезН охМит ропбегозит
МА6ЫЕ8ШМ ОХЮЕ, НЕАУУ
МдО М.м. 40,30
[1309-48-4]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 100,5 %
МдО (в пересчете на прокаленное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Мелкий, белый или почти белый порошок.
Практически нерастворим в воде. Растворяется
в разбавленных кислотах, в большинстве случаев со
слабым выделением пузырьков.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Насыпная плотность (2.9.34): не менее 0,25 г/мл.
B. 15 мг испытуемого образца растворяют в 2 мл
кислоты азотной разведенной Р и нейтрализуют
раствором натрия гидроксида разведенным Р. По¬
лученный раствор дает реакцию на магний (2.3.1).
C. Испытуемый образец выдерживает испыта¬
ние «Потеря в массе при прокаливании», как ука¬
зано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в смеси из 30 мл воды дистиллированной Р
и 70 мл кислоты уксусной Р. Кипятят в течение
2 мин, охлаждают и доводят кислотой уксусной
разведенной Р до объема 100 мл. При необходи¬
мости фильтруют через предварительно прокален¬
ный и взвешенный фарфоровый или кварцевый
фильтр с подходящим размером пор до получения
прозрачного фильтрата.
Цветность (2.2.2, метод //). Окраска раствора
3 должна быть не интенсивнее эталона В(К)3.
Растворимые вещества. Не более 2,0 %.
2,00 г испытуемого образца смешивают с 100 мл
воды Р и кипятят в течение 5 минут. Горячую смесь
фильтруют через стеклянный фильтр (ПОР 40)
(2.12), охлаждают и доводят водой Р до объема
100 мл. 50 мл фильтрата выпаривают досуха и су¬
шат при температуре от 100 °С до 105 °С. Масса
остатка не должна превышать 20 мг.
Вещества, нерастворимые в уксусной кис¬
лоте. Не более 0,1 %. Остаток, полученный при
приготовлении раствора 3, промывают, высуши¬
вают и прокаливают при температуре (600±50) °С.
Масса полученного остатка не должна превышать
5 мг.
Хлориды (2.4.4). Не более 0,1 %. 1 мл раст¬
вора 3 доводят водой Р до объема 15 мл. Полу¬
ченный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 1,0%. 0,3 мл
раствора 3 доводят водой дистиллированной Р до
объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0004 %
(4 ррт). 5 мл раствора 3 должны выдерживать ис¬
пытание на мышьяк.
Кальций (2.4.3). Не более 1,5 %. 1,3 мл раст¬
вора 3 доводят водой дистиллированной Р до
объема 150 мл. 15 мл полученного раствора долж¬
ны выдерживать испытание на кальций.
Железо (2.4.9). Не более 0,07%. 0,15 г ис¬
пытуемого образца растворяют в 5 мл кислоты
хлористоводородной разведенной Р и доводят
водой Р до объема 10 мл. 1 мл полученного раст¬
вора доводят водой Р до объема 10 мл. Получен¬
ный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0030 % (30 ррт). К 20 мл раствора 3 прибавляют
15 мл кислоты хлористоводородной Р1 и встря¬
хивают с 25 мл метилизобутилкетона Р в тече¬
ние 2 мин. Выдерживают до разделения слоев, за¬
тем отделяют водный слой и выпаривают досуха.
Остаток растворяют в 1 мл кислоты уксусной Р и
доводят водой Р до объема 30 мл. 12 мл получен¬
ного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использовани¬
ем эталонного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при прокаливании. Не более
8,0%. 1,00 г испытуемого образца прокаливают
при температуре (900±25) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,320 г испытуемого образца растворяют в 20 мл
кислоты хлористоводородной разведенной Р и
доводят водой Р до объема 100,0 мл. Используя
20,0 мл полученного раствора, проводят комплек¬
сометрическое определение магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соот¬
ветствует 4,030 мг МдО.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, од¬
нако другие методы также могут быть использо¬
ваны. В случае указания результатов конкретных
характеристик требуется указывать также и ме¬
тоды проведения испытаний.
Следующие характеристики могут быть важ¬
ны для магния оксида тяжелого, используемого в
качестве наполнителя при производстве твердых
дозированных лекарственных форм для внутренне¬
го применения.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Насыпная плотность и плотность после усад¬
ки (2.9.34).
628
Гэсударственная фармакопея Республики Беларусь
07/2016:0229
МАГНИЯ СТЕАРАТ
МадпезН з!еагаз
МАвИЕЗШМ 8ТЕАКАТЕ
ОПРЕДЕЛЕНИЕ
Соединение магния со смесью твердых органи¬
ческих кислот, состоящее главным образом из магния
стеарата и магния пальмитата в различных пропор¬
циях, полученных из источников растительного или
животного происхождения.
Содержание:
- магний (Мд; А.м. 24,305): не менее 4,0 % и не
более 5,0% (в пересчете на сухое вещество);
- стеариновая кислота во фракции жирных кис¬
лот: не менее 40,0 %;
- сумма стеариновой кислоты и пальмитино¬
вой кислоты во фракции жирных кислот: не менее
90,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Очень мелкий, легкий порошок белого или почти
белого цвета, жирный на ощупь.
Практически нерастворим в воде и этаноле без¬
водном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С, О.
Вторая идентификация: А, В, О.
A. Температура затвердевания (2.2.18): не ме¬
нее 53 °С. Определение проводят с использовани¬
ем остатка, полученного при приготовлении раст¬
вора 5, как указано в разделе «Испытания».
B. Кислотное число жирных кислот (2.5.1):
от 195 до 210. Определение проводят из 0,200 г
остатка, полученного при приготовлении раствора
5, растворяя его в 25 мл предписанной смеси раст¬
ворителей.
C. Просматривают хроматограммы, получен¬
ные при количественном определении стеарино¬
вой кислоты и пальмитиновой кислоты.
Результаты: два основных пика на хромато¬
грамме испытуемого раствора по времени удержи¬
вания соответствуют двум основным пикам на хро¬
матограмме раствора сравнения.
О. К 1 мл раствора 3 прибавляют 1 мл раст¬
вора аммиака разведенного Р1. Образуется белый
осадок, который растворяется при прибавлении
1 мл раствора аммония хлорида Р. К полученному
раствору прибавляют 1 мл раствора 120 г/л дина¬
трия гидрофосфата Р. Образуется белый кри¬
сталлический осадок.
ИСПЫТАНИЯ
Раствор 5. К 5,0 г испытуемого образца при¬
бавляют 50 мл эфира, свободного от перокси¬
дов, Р, 20 мл кислоты азотной разведенной Р,
20 мл воды Р и нагревают с обратным холодильни¬
ком до полного растворения испытуемого образца.
Раствор охлаждают, помещают в делительную во¬
ронку и удаляют водный слой. Эфирный слой дваж¬
ды встряхивают с водой Р порциями по 4 мл. Объ¬
единяют водные слои, промывают 15 мл эфира,
свободного от пероксидов, Р и доводят водой Р до
объема 50,0 мл (раствор 3). Органический слой вы¬
паривают досуха и сушат остаток при температуре
от 100 °С до 105 °С. Остаток используют для про¬
ведения испытаний А и В на подлинность.
Кислотность или щелочность. К 1,0 г испытуе¬
мого образца прибавляют 20 мл воды, свободной от
углерода диоксида, Р и кипятят в течение 1 мин при
постоянном встряхивании, охлаждают и фильтруют. К
10 мл фильтрата прибавляют 0,05 мл раствора бром-
тимолового синего Р4. При прибавлении не более
0,05 мл 0,1 М раствора кислоты хлористоводород¬
ной или 0,1 М раствора натрия гидроксида окраска
раствора должна измениться.
Хлориды. Не более 0,1 %. 10,0 мл раствора 3
доводят водой Р до объема 40 мл и при необходи¬
мости нейтрализуют кислотой азотной Р, используя
лакмус Р в качестве индикатора. Прибавляют 1 мл
кислоты азотной Р и 1 мл 0,1 М раствора сере¬
бра нитрата и доводят водой Р до объема 50 мл.
Перемешивают и выдерживают в течение 5 мин в
защищенном от света месте. Полученный раствор
по степени мутности не должен превышать раствор,
содержащий 1,4 мл 0,02 М раствора кислоты хлори¬
стоводородной.
Сульфаты. Не более 1,0 %. 6,0 мл раствора 5
доводят водой Р до объема 40 мл и при необходимо¬
сти нейтрализуют кислотой хлористоводородной Р,
используя лакмус Р в качестве индикатора. Прибавля¬
ют 1 мл 3 М раствора кислоты хлористоводородной
и 3 мл раствора 120 г/л бария хлорида Р и доводят во¬
дой Р до объема 50 мл. Перемешивают и выдержива¬
ют в течение 10 мин. Полученный раствор по степени
мутности не должен превышать раствор, содержащий
3,0 мл 0,02 М раствора кислоты серной.
Кадмий. Не более 0,0003 % (3 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Для ополаскивания стеклянной посуды перед
использованием и для приготовления всех водных
растворов применяют воду, предварительно про¬
пущенную через сильнокислотную, сильноосновную
смешанную ионообменную смолу. Используют реак¬
тивы с максимально низким содержанием кадмия,
свинца и никеля и хранят все растворы реактивов в
контейнерах из боросиликатного стекла. Стеклян¬
ную посуду перед использованием очищают путем
замачивания в теплом 8 М растворе кислоты азот¬
ной в течение 30 мин и последующим ополаскивани¬
ем деионизированной водой.
Контрольный раствор. 25 мл кислоты азотной,
свободной от свинца и кадмия, Р доводят водой Р до
объема 100,0 мл.
Раствор модификатора. 20 г аммония дигидро¬
фосфата Р и 1 г магния нитрата Р растворяют в
воде Р и доводят до объема 100 мл этим же раство¬
рителем. Альтернативно может использоваться под¬
ходящий матричный модификатор, рекомендованный
производителем графитовой печи атомно-абсорбци¬
онного спектрометра.
Испытуемый раствор. 0,100 г испытуемого об¬
разца помещают в политетрафторэтиленовый сосуд
для минерализации, прибавляют 2,5 мг\у кислоты
азотной, свободной от свинца и кадмия, Р и плотно
укупоривают в соответствии с инструкциями произ¬
водителя (перед использованием сосуда для мине¬
рализации необходимо внимательно ознакомиться
с инструкциями по технике безопасности и по при¬
менению. Строго выполняют инструкции произво¬
дителя по эксплуатации и обращению с этими кол¬
бами. Нельзя использовать колбы с металлической
Магния стеарат
629
оплеткой или лайнеры, использованные при работе
с соляной кислотой, так как это может привести
к загрязнению образца вследствие коррозии ме¬
талла). Сосуд нагревают при температуре 170 °С в
течение 3 ч и медленно охлаждают на воздухе до ком¬
натной температуры в соответствии с инструкциями
производителя. Так как могут выделяться едкие газы,
сосуд осторожно открывают в вытяжном шкафу. По¬
лученный остаток растворяют в воде Р и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения. Готовят раствор с концен¬
трацией 0,0030 мкг/мл кадмия соответствующими раз¬
ведениями раствора 0,00825 мкг/мл кадмия нитрата
тетрагидрата Р в контрольном растворе.
1,0 мл испытуемого раствора доводят кон¬
трольным раствором до объема 10,0 мл. Готовят
смеси из полученного раствора, раствора сравне¬
ния и контрольного раствора в следующих соотно¬
шениях: (1,0:0:1,0, об/об/об), (1,0:0,5:0,5, об/об/об),
(1,0:1,0:0, об/об/об). К каждой смеси прибавляют
50 мкл раствора модификатора и перемешивают.
Полученные растворы содержат соответственно
0 мкг/мл, 0,00075 мкг/мл и 0,0015 мкг/мл кадмия из
раствора сравнения (оставшийся испытуемый раст¬
вор сохраняют для проведения испытаний «Свинец»
и «Никель»).
Источник излучения: лампа с полым катодом
для определения кадмия.
Длина волны: 228,8 нм.
Атомизатор: графитовая печь.
Платформа: с пиропокрытием с интегрирован¬
ной кюветой.
Условия проведения испытания: используют
температурную программу для кадмия, рекомендуе¬
мую производителем атомно-абсорбционного спек¬
трометра. Ниже приведен пример температурных ус¬
ловий для определения кадмия методом атомно-аб¬
сорбционной спектрометрии в графитовой печи.
Стадия
Конечная
температура
(°С)
Время под¬
нятия тем¬
пературы (с)
Время удер¬
живания
температу-
ры (С)
Высушива¬
ние
110
10
20
Озоление
600
10
30
Атомизация
1800
0
5
Свинец. Не более 0,0010 % (10 ррт). Атомно-аб¬
сорбционная спектрометрия (2.2.23, метод II).
Для ополаскивания стеклянной посуды перед
использованием и для приготовления всех водных
растворов применяют воду, предварительно про¬
пущенную через сильнокислотную, сильноосновную
смешанную ионообменную смолу. Используют реак¬
тивы с максимально низким содержанием кадмия,
свинца и никеля и хранят все растворы реактивов в
контейнерах из боросиликатного стекла. Стеклян¬
ную посуду перед использованием очищают путем
замачивания ее в теплом 8М растворе кислоты
азотной в течение 30 мин и последующим ополаски¬
ванием деионизированной водой.
Контрольный раствор. Используют раствор,
приготовленный как указано в испытании «Кадмий».
Раствор модификатора. Используют раствор,
приготовленный как указано в испытании «Кадмий».
Испытуемый раствор. Используют раствор,
приготовленный как указано в испытании «Кадмий».
Раствор сравнения. Готовят раствор с кон¬
центрацией 0,100 мкг/мл свинца соответствующи¬
ми разбавлениями эталонного раствора свинца
(100 ррт РЬ) Р контрольным раствором.
Готовят смеси из испытуемого раствора, раст¬
вора сравнения и контрольного раствора в следую¬
щих соотношениях: (1,0:0:1,0, об/об/об), (1,0:0,5:0,5,
об/об/об), (1,0:1,0:0, об/об/об). К каждой смеси при¬
бавляют 50 мкл раствора модификатора и перемеши¬
вают. Полученные растворы содержат соответственно
0 мкг/мл, 0,025 мкг/мл и 0,05 мкг/мл свинца из раст¬
вора сравнения.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 283,3 нм.
Атомизатор: графитовая печь.
Платформа: с пиропокрытием с интегрирован¬
ной кюветой.
Условия проведения испытания: используют
температурную программу для свинца, рекомендуе¬
мую производителем атомно-абсорбционного спек¬
трометра. Ниже приведен пример температурных
условий для определения свинца методом атомно-аб¬
сорбционной спектрометрии в графитовой печи.
Стадия
Конечная
температура
ГС)
Время под¬
нятия тем¬
пературы (с)
Время удер¬
живания
температу-
ры (С)
Высушива¬
ние
110
10
20
Озоление
450
10
30
Атомизация
2000
0
5
Никель. Не более 0,0005 % (5 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Для ополаскивания стеклянной посуды перед
использованием и для приготовления всех водных
растворов применяют воду, предварительно про¬
пущенную через сильнокислотную, сильноосновную
смешанную ионообменную смолу. Используют реак¬
тивы с максимально низким содержанием кадмия,
свинца и никеля и хранят все растворы реактивов в
контейнерах из боросиликатного стекла. Стеклян¬
ную посуду перед использованием очищают путем
замачивания ее в теплом 8М растворе кислоты
азотной в течение 30 мин и последующим ополаски¬
ванием деионизированной водой.
Контрольный раствор. Используют раствор,
приготовленный как указано в испытании «Кадмий».
Раствор модификатора. 20 г аммония дигидро¬
фосфата Р растворяют в воде Р и доводят до объема
100 мл этим же растворителем. Альтернативно может
использоваться матричный модификатор, рекомендо¬
ванный производителем графитовой печи атомно-аб¬
сорбционного спектрометра.
Испытуемый раствор. Используют раствор,
приготовленный как указано в испытании «Кадмий».
Раствор сравнения. Готовят раствор с концен¬
трацией 0,050 мкг/мл никеля соответствующими раз¬
ведениями раствора 0,2477 мкг/мл никеля нитрата
гексагидрата Р в контрольном растворе.
Готовят смеси из испытуемого раствора, раст¬
вора сравнения и контрольного раствора в следую¬
щих соотношениях: (1,0:0:1,0, об/об/об), (1,0:0,5:0,5,
об/об/об), (1,0:1,0:0, об/об/об). К каждой смеси при¬
бавляют 50 мкл раствора матричного модификатора
и перемешивают. Полученные растворы содержат со-
630
Гэсударственная фармакопея Республики Беларусь
ответственно 0 мкг/мл, 0,0125 мкг/мл, и 0,025 мкг/мл
никеля из раствора сравнения.
Источник излучения: лампа с полым катодом
для определения никеля.
Длина волны: 232,0 нм.
Атомизатор: графитовая печь.
Платформа: с пиропокрытием с интегрирован¬
ной кюветой.
Условия проведения испытания: используют
температурную программу для никеля, рекомендуе¬
мую производителем атомно-абсорбционного спек¬
трометра. Ниже приведен пример температурных
условий для определения никеля методом атомно-аб¬
сорбционной спектрометрии в графитовой печи.
Стадия
Конечная
температура
(°С)
Время под¬
нятия тем¬
пературы (с)
Время под¬
держивания
температу¬
ры (с)
Высушива¬
ние
110
10
20
Озоление
1000
20
30
Атомизация
2300
0
5
Потеря в массе при высушивании (2.2.32). Не
более 6,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа соН: отсутствие (2.6.13).
8а1топеИа: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Магний. 0,500 г испытуемого образца помещают
в коническую колбу вместимостью 250 мл, прибавляют
50 мл смеси из равных объемов бутанола Р и этано¬
ла безводного Р, 5 мл раствора аммиака концентри¬
рованного Р, 3 мл аммиачного буферного раствора
рН 10,0 Р, 30,0 мл 0,1 М раствора натрия эдетата и
15 мг индикаторной смеси протравного черного 11 Р
и нагревают при температуре (45—50) °С до получе¬
ния прозрачного раствора. Титруют 0,1 М раствором
цинка сульфата до изменения окраски раствора от
синей до фиолетовой.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 2,431 мг Мд.
Стеариновая кислота и пальмитиновая кисло¬
та. Газовая хроматография (2.2.28): испытание прово¬
дят методом внутренней нормализации.
Испытуемый раствор. 0,10 г испытуемого об¬
разца помещают в коническую колбу и растворяют в
5 мл раствора бора фторида в метаноле Р. Колбу
присоединяют к обратному холодильнику и кипятят в
течение 10 мин. Затем в колбу через холодильник при¬
бавляют 4 мл гептана Р, кипятят в течение 10 мин и
охлаждают. В колбу прибавляют 20 мл раствора на¬
трия хлорида насыщенного Р, встряхивают и вы¬
держивают до разделения слоев. Органический слой
сушат с использованием 0,1 г натрия сульфата без¬
водного Р (предварительно промытого гептаном Р).
1.0 мл полученного раствора доводят гептаном Р до
объема 10,0 мл.
Раствор сравнения. Готовят точно так же, как и
испытуемый раствор, используя вместо испытуемо¬
го образца 50,0 мг ФСО пальмитиновой кислоты и
50.0 мг ФСО стеариновой кислоты.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 2,4 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—2
70
2—36
70 -»240
36—41
240
Блок ввода проб
220
Детектор
260
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению к
метилстеарату): метилпальмитат — около 0,9.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 5,0 между пиками ме-
тилпальмитата и метилстеарата;
- относительное стандартное отклонение: не
более 3,0 % для площадей пиков метилпальмитата
и метилстеарата при 6 повторных вводах пробы; не
более 1,0 % для отношения площадей пиков метил¬
пальмитата к площадям пиков метилстеарата при 6
повторных вводах пробы.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Некоторые
из характеристик, описанных в разделе «Функци¬
онально-обусловленные характеристики», могут
также присутствовать в части фармакопейной
статьи, обязательной для выполнения, так как од¬
новременно они являются и показателями качества.
В таком случае в разделе «Функционально-обуслов¬
ленные характеристики» дается перекрестная
ссылка на это испытание в части фармакопейной
статьи, обязательной для выполнения. Контроль
данных характеристик может вносить вклад в каче¬
ство лекарственного средства путем достижения
постоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, однако
другие методы также могут быть использованы. В
случае указания результатов конкретных характе¬
ристик требуется указывать также и методы про¬
ведения испытаний.
Следующие характеристики могут быть важ¬
ны для магния стеарата, используемого в качестве
смазывающего вещества при производстве табле¬
ток и капсул.
Распределение частиц по размерам (2.9.31).
Удельная площадь поверхности (2.9.26, ме¬
тод /). Определяют удельную площадь поверхности
в диапазоне Р1Р0 от 0,05 до 0,15.
Образец дегазации: в течение 20 мин при темпе¬
ратуре 40 °С.
Термогравиметрия (2.2.34).
Магния хлорид 4,5-гидрат
631
07/2016:0044
МАГНИЯ СУЛЬФАТ ГЕПТАГИДРАТ
Мадпези зиКаз Ьер(а11ус1псиз
МАОЫЕ8ШМ ЗШ-РАТЕ НЕРТАНУОКАТЕ
Мд304 ■ 7Н20 М.м. 246,5
[10034-99-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или блестящие бесцветные кристаллы.
Легко растворим в воде, очень легко растворим в
кипящей воде, практически нерастворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
B. Испытуемый образец дает реакцию на магний
(2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,05 мл раствора фенолового
красного Р. При прибавлении не более 0,2 мл 0,01 М
раствора кислоты хлористоводородной или 0,01 М
раствора натрия гидроксида окраска раствора
должна измениться.
Хлориды (2.4.4). Не более 0,0300 % (300 ррт).
1,7 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 48,0 % и не более 52,0 %. 0,500 г испытуемого
образца сушат при температуре от 110 °С до 120 °С в
течение 1 часа, затем при температуре 400 °С до по¬
стоянной массы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,450 г испытуемого образца растворяют в
100 мл воды Р и проводят комплексометрическое
определение магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 12,04 мг Мд304.
07/2016:1341
МАГНИЯ ХЛОРИД 4,5-ГИДРАТ
Мадпези сЫопс1ит 4,5-Ьус1псит
МАвЫЕЗШМ СШОРЮЕ 4,5-НУйКАТЕ
МдС12 - хН20 (х * 4,5) М.м. 95,21
(безводное вещество)
ОПРЕДЕЛЕНИЕ
Содержание', не менее 52,5 % и не более 55,5 %
(рассчитанное как есть, без учета содержания воды).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый гранулированный поро¬
шок. Гигроскопичен.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
B. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
C. Испытуемый образец дает реакцию на магний
(2.3.1).
ИСПЫТАНИЯ
Раствор §. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 5 мл раст¬
вора 3 прибавляют 0,05 мл раствора фенолово¬
го красного Р. При прибавлении не более 0,3 мл
0,01 М раствора кислоты хлористоводородной
или 0,01 М раствора натрия гидроксида окраска
раствора должна измениться.
Бромиды. Не более 0,0500 % (500 ррт).
2.0 мл раствора 3 доводят водой Р до объема
10,0мл. К 1,0 мл полученного раствора прибав¬
ляют 4,0 мл воды Р, 2,0 мл раствора фенолового
красного РЗ, 1,0 мл раствора хлорамина Р2 и сра¬
зу же перемешивают. Точно через 2 мин прибавля¬
ют 0,30 мл 0,1 М раствора натрия тиосульфа¬
та, перемешивают и доводят водой Р до объема
10.0 мл.
Оптическая плотность (2.2.25) полученно¬
го испытуемого раствора, измеренная при длине
волны 590 нм, не должна превышать оптическую
плотность раствора, приготовленного параллель¬
но с использованием 5,0 мл раствора 3 мг/л калия
бромида Р. В качестве компенсационной жидкости
используют воду Р.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
15 мл раствора 3 должны выдерживать испытание на
сульфаты.
Алюминий (2.4.17). 0,0001 % (1 ррт), если суб¬
станция предназначена для производства растворов
632
Гэсударственная фармакопея Республики Беларусь
для перитонеального диализа, гемодиализа или ге¬
мофильтрации.
Предписанный раствор. 4 г испытуемого образ¬
ца растворяют в 100 мл воды Р и прибавляют 10 мл
ацетатного буферного раствора рН 6,0 Р
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды Р.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Кальций (2.4.3). Не более 0,1 %. 1 мл раствора
3 доводят водой дистиллированной Р до объема
15 мл. Полученный раствор должен выдерживать ис¬
пытание на кальций.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 3 должны выдерживать испытание на
железо.
Калий. Не более 0,0500 % (500 ррт), если суб¬
станция предназначена для производства лекар¬
ственных средств для парентерального применения.
Атомно-эмиссионная спектрометрия (2.2.22, ме¬
тод I).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Растворы сравнения: готовят соответствующи¬
ми разбавлениями водой Р раствора, приготовлен¬
ного следующим образом: 1,144 г калия хлорида Р,
предварительно высушенного при температуре от
100 °С до 105 °С в течение 3 ч, растворяют в воде Р и
доводят до объема 1000,0 мл этим же растворителем
(600 мкг/мл К).
Длина волны: 766,5 нм.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Вода (2.5.12). Не менее 44,0% и не более
48.0 %. Определение проводят из 50,0 мг испытуемо¬
го образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
воды Р. Проводят комплексометрическое определе¬
ние магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 9,521 мг МдС12.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства рас¬
творов для перитонеального диализа, гемодиализа
или гемофильтрации;
- субстанция пригодна для производства ле¬
карственных средств для парентерального приме¬
нения.
07/2016:0402
МАГНИЯ ХЛОРИД ГЕКСАГИДРАТ
МадпезН сЫопс1ит ЬехаЬубпсит
МАСЫЕЗШМ СШ.ОМОЕ НЕХАНУОПАТЕ
МдС12 ■ 6Н20 М.м. 203,3
[7791-18-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 101,0 %
МдС12-6Н20.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветные кристаллы. Гигроскопичен.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
B. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
C. Испытуемый образец дает реакцию на магний
(2.3.1).
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раст¬
воряют в воде, свободной от диоксида углеро¬
да, Р, приготовленной из воды дистиллирован¬
ной Р, и доводят до объема 100,0 мл этим же раст¬
ворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 5 мл раст¬
вора 3 прибавляют 0,05 мл раствора фенолово¬
го красного Р. При прибавлении не более 0,3 мл
0,01 М раствора кислоты хлористоводородной
или 0,01 М раствора натрия гидроксида окраска
раствора должна измениться.
Бромиды. Не более 0,0500 % (500 ррт).
2.0 мл раствора 3 доводят водой Р до объема
10,0мл. К 1,0 мл полученного раствора прибав¬
ляют 4,0 мл воды Р, 2,0 мл раствора фенолового
красного РЗ, 1,0 мл раствора хлорамина Р2 и сра¬
зу же перемешивают. Точно через 2 мин прибавля¬
ют 0,30 мл 0,1 М раствора натрия тиосульфа¬
та, перемешивают и доводят водой Р до объема
10.0 мл.
Оптическая плотность (2.2.25) полученно¬
го испытуемого раствора, измеренная при длине
волны 590 нм, не должна превышать оптическую
плотность раствора, приготовленного параллель¬
но с использованием 5,0 мл раствора 3 мг/л калия
бромида Р. В качестве компенсационной жидкости
используют воду Р.
Сульфаты (2.4.13). Не более 0,0100%
(100 ррт). 15 мл раствора 3 должны выдерживать
испытание на сульфаты.
Алюминий (2.4.17). 0,0001 % (1 ррт), если
субстанция предназначена для производства рас¬
творов для перитонеального диализа, гемодиали¬
за или гемофильтрации.
Магния цитрат безводный
633
Предписанный раствор. 4 г испытуемого об¬
разца растворяют в 100 мл воды Р и прибавляют
10 мл ацетатного буферного раствора рН 6,0 Р.
Эталон. Смешивают 2 мл эталонного раст¬
вора алюминия (2 ррт А1) Р, 10 мл ацетатного
буферного раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100 мл
воды Р.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдер¬
живать испытание на мышьяк.
Кальций (2.4.3). Не более 0,1 %. 1 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл. Полученный раствор должен выдержи¬
вать испытание на кальций.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 3 должны выдерживать испытание
на железо.
Калий. Не более 0,0500 % (500 ррт), если
субстанция предназначена для производства ле¬
карственных средств для парентерального приме¬
нения.
Атомно-эмиссионная спектрометрия (2.2.22,
метод I).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Растворы сравнения: готовят соответствующи¬
ми разбавлениями водой Р раствора, приготовлен¬
ного следующим образом: 1,144 г калия хлорида Р,
предварительно высушенного при температуре от
100 °С до 105 °С в течение 3 ч, растворяют в воде Р
и доводят до объема 1000,0 мл этим же раствори¬
телем (600 мкг/мл К).
Длина волны: 766,5 нм.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 12 мл раствора 3 должны вы¬
держивать испытание на тяжелые металлы. Эталон
готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не менее 51,0% и не более
55.0 %. Определение проводят из 50,0 мг испыту¬
емого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
воды Р. Проводят комплексометрическое определе¬
ние магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 20,33 мг МдС12-6Н20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства рас¬
творов для перитонеального диализа, гемодиализа
или гемофильтрации;
- субстанция пригодна для производства ле¬
карственных средств для парентерального приме¬
нения.
07/2016:2339
МАГНИЯ ЦИТРАТ БЕЗВОДНЫЙ
МадпезНсНгаз апЬубпсиз
МАОЫЕ8ШМ С1ТКАТЕ, АЫНУОК0118
Мд,(С,Н507)2 М.м. 451,1
[3344-18-1]
ОПРЕДЕЛЕНИЕ
Тримагния бис(2-гидроксипропан-1,2,3-трикар-
боксилат).
Содержание: не менее 15,0 % и не более 16,5 %
Мд (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый мелкий порошок. Слегка
гигроскопичен.
Растворим в воде, практически нерастворим в
96 % спирте. Растворяется в разведенной хлористо¬
водородной кислоте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на ци¬
траты (2.3.1).
B. Испытуемый образец дает реакцию на магний
(2.3.1).
C. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
О. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, на¬
гревая при температуре 60 °С, охлаждают и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон III.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона У(Ж)7 или
ВУ(КЖ)6.
рН (2.2.3). От 6,0 до 8,5. Измеряют рН раствора 3.
Оксалаты. Не более 0,0280 % (280 ррт). 0,50 г
испытуемого образца растворяют в 4 мл воды Р, при¬
бавляют 3 мл кислоты хлористоводородной Р, 1 г
цинка активированного Р и выдерживают в течение
5 мин. Жидкость переносят в пробирку, содержащую
0,25 мл раствора 10 г/л фенилгидразина гидрохло¬
рида Р, нагревают до кипения, быстро охлаждают,
переносят в мерный цилиндр, прибавляют равный
объем кислоты хлористоводородной Р, 0,25 мл
раствора калия феррицианида Р, встряхивают и
выдерживают в течение 30 мин. Розовая окраска
полученного раствора должна быть не интенсивнее
эталона, приготовленного аналогично и в то же са¬
мое время с использованием 4 мл раствора 50 мг/л
кислоты щавелевой Р.
Сульфаты (2.4.13). Не более 0,2 %. 1,5 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл.
634
Гэсударственная фармакопея Республики Беларусь
Кальций (2.4.3). Не более 0,2%. 1,0 мл раст¬
вора 3 доводят водой дистиллированной Р до объ¬
ема 15 мл.
Железо (2.4.9). Не более 0,0100% (100 ррт).
2,0 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 10 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 5,0 г испытуемого образца раст¬
воряют в 15 мл кислоты хлористоводородной разве¬
денной Р при нагревании, доводят раствором аммиа¬
ка Р до рН 3,5 и разводят водой дистиллированной Р
до объема 50 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 3,5 %. 1,000 г испытуемого образца сушат при
температуре (180±10) °С в течение 5 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворят в 50 мл
воды Р и проводят комплексометрическое определе¬
ние магния (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 2,431 мг Мд.
ХРАНЕНИЕ
В неметаллическом воздухонепроницаемом кон¬
тейнере.
07/2016:1123
МАКРОГОЛА ЦЕТОСТЕАРИЛОВЫЙ
ЭФИР
МасгодоН аеМег се1оз1еагуИсиз
МАСК0001. СЕТ08ТЕАКУ1. ЕТНЕК
ОПРЕДЕЛЕНИЕ
Смесь эфиров разных макроголов и линейных
жирных спиртов, в основном спирта цетостеарилово-
го. Может содержать несколько свободных макрого¬
лов и разное количество свободного цетостеарилово-
го спирта. Число молей прореагировавшего этиленок-
сида на моль цетостеарилового спирта составляет от
2 до 33 (номинальное значение).
ОПИСАНИЕ(СВОЙСТВА)
Белая или желтовато-белая воскообразная мас¬
лянистая масса, гранулы, мелкие шарики или хлопья.
Макрогола цетостеариловый эфир с низким зна¬
чением числа молей прореагировавшего этиленок-
сида на моль вещества: практически нерастворим в
воде, растворим в 96 % спирте и метиленхпориде.
Макрогола цетостеариловый эфир с высоким зна¬
чением числа молей прореагировавшего этиленокси-
да на моль вещества: диспергируется или растворим
в воде, растворим в 96 % спирте и метиленхпориде.
Затвердевает при температуре от 32 °С до 52 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец выдерживает испытание
«Гидроксильное число», как указано в разделе «Ис¬
пытания».
B. Испытуемый образец выдерживает испытание
«Йодное число», как указано в разделе «Испытания».
C. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испытания».
й. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Количество испытуемо¬
го образца, указанное в таблице, приведенной ниже,
растворяют в смеси из 1 объема воды Р и 9 объемов
метанола Р и доводят до объема 75 мл этой же сме¬
сью растворителей.
Число молей прореаги¬
ровавшего этиленоксида
на моль вещества (номи¬
нальное значение)
Количество испытуемого
образца, г
2—6
5,0
10—22
10,0
25—33
15,0
Прибавляют 60 мл гексана Р и встряхивают в
течение 3 мин. Образующуюся пену гасят прибав¬
лением нескольких капель 96 % спирта Р. Верхний
слой фильтруют через фильтр, на который помещают
натрия сульфат безводный Р. Фильтр промывают
трижды гексаном Р порциями по 10мл. Объединен¬
ные фильтраты выпаривают досуха. 0,05 г остатка
растворяют в 10 мл метанола Р (допускается опалес¬
ценция раствора).
Раствор сравнения. 25 мг ФСО стеарилового
спирта растворяют в метаноле Р и доводят до объ¬
ема 25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: этилацетат Р.
Наносимый объем пробы: 20 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилин — кислота серная, приготовленным следу¬
ющим образом: 0,5 г ванилина Р растворяют в 50 мл
96 % спирта Р и доводят до объема 100 мл кислотой
серной Р\ пластинку сушат на воздухе, нагревают при
температуре около 130 °С в течение 15 мин и охлаж¬
дают на воздухе.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются несколько пятен, одно из
которых по расположению соответствует основному
пятну на хроматограмме раствора сравнения.
Е. 0,1 г испытуемого образца растворяют или
диспергируют в 5 мл 96% спирта Р, прибавляют
2 мл воды Р, 10 мл кислоты хлористоводородной
разведенной Р, 10 мл раствора бария хлорида Р1 и
10 мл раствора 100 г/л кислоты фосфорномолибде¬
новой Р. Образуется осадок.
ИСПЫТАНИЯ
Цветность (2.2.2, метод II). 5,0 г испытуемого
образца растворяют в 96% спирте Р и доводят до
объема 50 мл этим же растворителем. Окраска полу¬
ченного раствора должна быть не интенсивнее этало¬
на ВУ(КЖ)5.
Щелочность. 2,0 г испытуемого образца раство¬
ряют в горячей смеси из 10 мл воды Р и 10 мл 96 %
спирта Р, прибавляют 0,1 мл раствора бромтимо-
нового синего Р1. При прибавлении не более 0,5 мл
0,1 М раствора кислоты хлористоводородной долж¬
но появиться желтое окрашивание.
Макроголглицерина гидроксистеарат
635
Кислотное число (2.5.1). Не более 1,0. Опреде¬
ление проводят из 5,0 г испытуемого образца.
Гидроксильное число (2.5.3, метод А).
Число молей прореаги¬
ровавшего этиленоксида
на моль вещества (номи¬
нальное значение)
Гидроксильное число
2
150—180
3
135—155
5—6
100—134
10
75—90
12
67—77
15
58—67
20—22
40—55
25
36—46
30—33
32—40
Йодное число (2.5.4, метод А). Не более 2,0.
Число омыления (2.5.6). Не более 3,0. Опреде¬
ление проводят из 10,0 г испытуемого образца.
Этиленоксид и диоксан (2.4.25). Не более
0,0001 % (1 ррт) этиленоксида и не более 0,0010 %
(10 ррт) диоксана.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 2,00 г испытуемого образца.
Общая зола (2.4.16). Не более 0,2 %. Определе¬
ние проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
Указывают:
- число молей прореагировавшего этиленок¬
сида на моль цетостеарилового спирта (номиналь¬
ное значение).
07/2016:1083
МАКРОГОЛГЛИЦЕРИНА
ГИДРОКСИСТЕАРАТ
Масгодо1д1усегоН Ьудгохув1еагаз
МАСКОв01-в1-УСЕ1Ю1.
НУОПОХУ5ТЕАПАТЕ
ОПРЕДЕЛЕНИЕ
#Г идрированное полиоксильное касторовое
масло*.
Состоит в основном из трис(12-гидрокси-
стеарил)глицерина, этоксилированного 7—60 мо¬
лекулами этиленоксида (номинальное значение),
с небольшими количествами гидростеарата макро-
гола и соответствующими свободными гликолями.
Получают в результате взаимодействия гидриро¬
ванного касторового масла и этиленоксида.
ОПИСАНИЕ (СВОЙСТВА)
Макроголглицерина гидроксистеарат с числом
единиц этиленоксида в молекуле менее 10: желто¬
ватая, мутная, вязкая жидкость.
Практически нерастворим в воде, растворим в
ацетоне и диспергируется в 96 % спирте.
Макроголглицерина гидроксистеарат с числом
единиц этиленоксида в молекуле более 20: белая
или желтоватая полужидкая или пастообразная
масса.
Легко растворим в воде, ацетоне и 96 % спирте,
практически нерастворим в петролейном эфире.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испыта¬
ние «Йодное число», как указано в разделе «Ис¬
пытания».
B. Испытуемый образец вьщерживает испытание
«Число омыления», как указано в разделе «Испытания».
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1 г испытуемого об¬
разца прибавляют 100 мл раствора 100 г/л калия
гидроксида Р и кипятят с обратным холодильником
в течение 30 мин. Раствор охлаждают и подкисля¬
ют 20 мл кислоты хлористоводородной Р. Встря¬
хивают смесь с 50 мл эфира Р и оставляют для раз¬
деления слоев. Прозрачный верхний слой перено¬
сят в пробирку, прибавляют 5 г натрия сульфата
безводного Р, закрывают пробирку и выдерживают
в течение 30 мин. Раствор фильтруют и фильтрат
выпаривают досуха на водяной бане. 50 мг полу¬
ченного остатка растворяют в 25 мл эфира Р.
Раствор сравнения. 50 мг кислоты 12-гидрок-
систеариновой Р растворяют в метиленхлориде Р и
доводят до объема 25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р.
Подвижная фаза: метиленхлорид Р — кислота
уксусная ледяная Р — ацетон Р (10:40:50, об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 8 см от линии
старта.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку опрыскивают раствором
80 г/л кислоты фосфорномолибденовой Р в 2-пропа-
ноле Р и нагревают при температуре 120 °С в течение
(1—2) мин.
Результаты: основное пятна на хроматограмме
испытуемого раствора соответствует по расположе¬
нию и цвету пятну на хроматограмме раствора срав¬
нения.
О. 2 г испытуемого образца помещают в пробир¬
ку, прибавляют 0,2 мл кислоты серной Р и закрывают
пробирку пробкой, через которую проходит стеклян¬
ная трубка, изогнутая дважды под прямым углом. На¬
гревают пробирку до появления паров белого цвета.
Пары при прохождении через 1 мл раствора ртути
(II) хлорида Р образуют осадок белого цвета, а филь¬
тровальная бумага, пропитанная щелочным раство¬
ром калия тетрайодмеркурата Р, окрашивается в
черный цвет.
ИСПЫТАНИЯ
Раствор 8. 5,0 г испытуемого образца с числом
единиц этиленоксида в молекуле менее 40 раство¬
ряют в смеси из 50 объемов ацетона Р и 50 объемов
этанола Р и доводят до объема 50 мл этой же сме¬
сью растворителей.
5,0 г испытуемого образца с числом единиц эти¬
леноксида в молекуле 40 и более растворяют в воде,
свободной от углерода диоксида, Р и доводят до
объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 по степени мут¬
ности не должен превышать эталон III.
636
Гэсударственная фармакопея Республики Беларусь
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6
Щелочность. К 2 мл раствора 3 прибавляют
0,5 мл раствора бромтимолового синего Р1. Не
должно появляться синее окрашивание.
Кислотное число (2.5.1). Не более 2,0. Опреде¬
ление проводят из 5,0 г испытуемого образца.
Гидроксильное число (2.5.3, метод А). См. та¬
блицу 1083.-1.
Йодное число (2.5.4). Не более 5,0.
Число омыления (2.5.6). См. таблицу 1083.-1.
Таблица 1083.-1.
Число единиц
этиленоксида в
молекуле (номи¬
нальное значе¬
ние)
Гидроксильное
число
Число омыления
7
115—135
125—140
25
•'ч!
О
о
70—90
40
57—80
45—69
60
45—67
40—51
Остаточные количества этиленоксида и диок-
сана (2.4.25). Не более 0,0001 % (1 ррт) остаточного
этиленоксида и не более 0,0010 % (10 ррт) остаточ¬
ного диоксана.
Тяжелые металлы (2.4.8).
Субстанции, растворимые в ацетоне/этано-
ле: не более 0,0010 % (10 ррт). 12 мл раствора 3
должны выдерживать испытание на тяжелые ме¬
таллы (метод В). Эталон готовят с использованием
эталонного раствора свинца (1 ррт РЬ), получен¬
ного разведением эталонного раствора свинца
(100 ррт РЬ) Р смесью из равных объемов ацето-
на Р и этанола Р.
Субстанции, растворимые в воде: не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы (метод А).
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 2,000 г испытуемого образца.
Общая зола (2.4.16). Не более 0,3 %. Определе¬
ние проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
МАРКИРОВКА
Указывают число единиц этиленоксида в молеку¬
ле (номинальное значение).
07/2016:1444
МАКРОГОЛЫ
Масгодо1а
МАСК0601.3
ОПРЕДЕЛЕНИЕ
Смесь полимеров с общей формулой Н-[ОСН2-
СН2]л-ОН, где п — среднее число оксиэтиленовых
групп. Тип макрогола обозначают числом, указыва¬
ющим среднюю относительную молекулярную мас¬
су. Может содержать подходящий стабилизатор.
ОПИСАНИЕ(СВОЙСТВА)
Тип ма¬
крогола
Внешний вид
Растворимость
300
400
600
Прозрачная,
вязкая, бесцвет¬
ная или почти
бесцветная гигро¬
скопичная жид¬
кость
Смешивается с водой, очень
легко растворим в ацетоне,
96 % спирте и метиленхпо-
риде, практически нераство¬
рим в жирных и минераль¬
ных маслах
1000
Белое или почти
белое гигроско¬
пичное твердое
воскообразное
или парафино¬
образное веще¬
ство
Очень легко растворим в
воде, легко растворим в
96 % спирте и метиленхло-
риде, практически нераство¬
рим в жирных и минераль¬
ных маслах
1500
Белое или почти
белое твердое
воскообразное
или парафино¬
образное веще¬
ство
Очень легко растворим в
воде и метиленхлориде,
легко растворимо в 96 %
спирте, практически нераст¬
ворим в жирных и минераль¬
ных маслах
3000
3350
Белое или почти
белое твердое
воскообразное
или парафино¬
образное веще¬
ство
Очень легко растворим в
воде и метиленхлориде,
очень мало растворим в
96 % спирте, практически
нерастворим в жирных и ми¬
неральных маслах
4000
6000
8000
Белое или почти
белое твердое
воскообразное
или парафино¬
образное веще¬
ство
Очень легко растворим в
воде и метиленхлориде,
практически нерастворим в
96 % спирте, жирных и мине¬
ральных маслах
20 000
35 000
Белое или почти
белое твердое
воскообразное
или парафино¬
образное веще¬
ство
Очень легко растворим в
воде, растворим в метилен¬
хлориде, практически не¬
растворим в 96 % спирте,
жирных и минеральных
маслах
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Вязкость», как указано в разделе «Испытания».
B. 1 г испытуемого образца помещают в пробир¬
ку, прибавляют 0,5 мл кислоты серной Р, закрывают
пробирку пробкой, оснащенной отводной трубкой,
и нагревают до появления белых паров. Собирают
пары через отводную трубку в 1 мл раствора ртути
(II) хлорида Р. Образуется обильный белый кристал¬
лический осадок.
C. К 0,1 г испытуемого образца прибавляют 0,1 г
калия тиоцианата Р и 0,1 г кобальта нитрата Р,
тщательно перемешивают стеклянной палочкой.
Прибавляют 5 мл метиленхлорида Р и встряхивают.
Слой жидкости окрашивается в синий цвет.
ИСПЫТАНИЯ
Раствор 5. 12,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность или щелочность. 5,0 г испытуе¬
мого образца растворяют в 50 мл воды, свободной
от углерода диоксида, Р и прибавляют 0,15 мл
Макроголы
637
раствора бромтимолового синего Р1. Получен¬
ный раствор имеет желтую или зеленую окраску.
При прибавлении не более 0,1 мл 0,1 М раствора
натрия гидроксида должно появиться синее окра¬
шивание.
Вязкость (2.2.9). Вязкость рассчитывают по
плотности, приведенной в таблице 1444.-1.
Таблица 1444.-1.
Тип
макро-
гопа
Кинематиче¬
ская вязкость
(мм2-с-1)
Динамическая
вязкость
(мПа-с)
Плотность*
(г/мл)
300
71—94
80—105
1,120
400
94—116
105—130
1,120
600
13,9—18,5
15—20
1,080
1000
20,4 —27,7
22—30
1,080
1500
31—46
34—50
1,080
3000
69—93
75—100
1,080
3350
76—110
83—120
1,080
4000
00
ю
Ч—
I
см
о
110—170
1,080
6000
185—250
200—270
1,080
8000
240—472
260—510
1,080
20 000
2500—3200
2700—3500
1,080
35 000
10 000—13 000
11 000—14 000
1,080
*Для макроголов 300 и 400 указана плотность вещества.
Для других макроголов — плотность 50 % (м/м) растворов.
Для макроголов с относительной молекулярной
массой большей, чем 400, определяют вязкость 50 %
(м/м) раствора испытуемого образца.
Температура затвердевания (2.2.18). См. табли¬
цу 1444.-2.
Таблица 1444.-2.
Тип макрогола
Температура затвердевания (°С)
600
15—25
1000
35-40
1500
42—48
3000
50—56
3350
53—57
4000
53—59
6000
55—61
8000
55—62
20 000
Не менее 57
35 000
Не менее 57
Гидроксильное число. Количество испытуе¬
мого образца (ш, г), указанное в таблице 1444.-3,
помещают в сухую коническую колбу, оснащенную
обратным холодильником. Прибавляют 25,0 мл
раствора фталевого ангидрида Р, перемешивают
вращательными движениями до растворения и ки¬
пятят с обратным холодильником в течение 60 мин
на горячей плитке. Охлаждают, ополаскивают хо¬
лодильник сначала 25 мл пиридина Р, затем 25 мл
воды Р, прибавляют 1,5 мл раствора фенолфта¬
леина Р и титруют 1 М раствором натрия гидрок¬
сида до появления бледно-розового окрашивания
(пл, мл).
Параллельно проводят контрольный опыт (л2, мл).
Гидроксильное число рассчитывают по формуле:
56,1 • (п2 - п,)
т
Таблица 1444.-3.
Тип макрогола
Гидроксильное число
ш(г)
300
340—394
1,5
400
264—300
1.9
600
178—197
3,5
1000
107—118
5,0
1500
70—80
7,0
3000
34-42
12,0
3350
30—38
12,0
4000
25—32
14,0
6000
16—22
18,0
8000
12—16
24,0
20 000
—
—
35 000
—
—
Для макроголов с относительной молекулярной
массой более 1000 и с содержанием воды более
0,5 % сушат соответствующую массу испытуемого об¬
разца при температуре от 100 °С до 105 °С в течение
2 ч и определяют гидроксильное число, используя по¬
лученный сухой образец.
Восстанавливающие вещества (2.2.2, метод I).
1 г испытуемого образца растворяют в 1 мл раствора
10 г/л резорцина Р, при необходимости осторожно на¬
гревая. Прибавляют 2 мл кислоты хлористоводород¬
ной Р. Через 5 мин окраска раствора должна быть не
интенсивнее эталона К(Кр)3.
Формальдегид. Не более 0,0030 % (30 ррт).
Испытуемый раствор. К 1,00 г испытуемого об¬
разца прибавляют 0,25 мл раствора натриевой соли
кислоты хромотроповой Р, охлаждают в ледяной
бане и прибавляют 5,0 мл кислоты серной Р. Выдер¬
живают в течение 15 мин и медленно доводят водой Р
до объема 10 мл.
Раствор сравнения. 0,860 г раствора фор¬
мальдегида Р доводят водой Р до объема 100 мл.
1,0 мл полученного раствора доводят водой Р до объ¬
ема 100 мл. В колбе вместимостью 10 мл смешива¬
ют 1,00 мл полученного раствора и 0,25 мл раствора
натриевой соли кислоты хромотроповой Р, охлаж¬
дают в ледяной бане и прибавляют 5,0 мл кислоты
серной Р. Выдерживают в течение 15 мин и медленно
доводят водой Р до объема 10 мл.
Контрольный раствор. В колбе вместимостью
10 мл смешивают 1,00 мл воды Р и 0,25 мл раствора
натриевой соли кислоты хромотроповой Р, охлаж¬
дают в ледяной бане и прибавляют 5,0 мл кислоты
серной Р. Медленно доводят водой Р до объема
10 мл.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора и раствора сравнения при длине
волны 567 нм относительно контрольного раствора.
Оптическая плотность испытуемого раствора не
должна превышать оптическую плотность раствора
сравнения.
Если применение макроголов с высоким содер¬
жанием формальдегида может оказать вредное воз¬
действие, компетентный уполномоченный орган мо¬
жет установить предел не более 0,0015 % (15 ррт).
Этиленгликоль и диэтиленгликоль. Испыта¬
ние проводят только для макроголов, имеющих от¬
носительную молекулярную массу менее 1000.
638
Гэсударственная фармакопея Республики Беларусь
Газовая хроматография (2.2.28).
Испытуемый раствор. 5,00 г испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения. 0,10 г этиленгликоля Р и
0,50 г диэтиленгликоля Р растворяют в ацетоне Р и
доводят до объема 100,0 мл этим же растворителем.
1.0 мл полученного раствора доводят ацетоном Р до
объема 10,0 мл.
Условия хроматографирования:
- колонка стеклянная длиной 1,8 м и внутренним
диаметром 2 мм, заполненная диатомитом силани-
зированным для газовой хроматографии Р, импрег-
нированным 5 % (м/м) раствором макрогола 20 000 Р;
- газ-носитель: азот для хроматографии Р;
- скорость потока: 30 мл/мин;
-температура колонки: при необходимости,
предварительно нагревают колонку при темпера¬
туре 200 °С в течение 15 часов; устанавливают на¬
чальную температуру колонки таким образом, чтобы
время удерживания диэтиленгликоля составляло
(14—16) мин; повышают температуру колонки при¬
близительно на 30 °С (но не выше 170 °С) со скоро¬
стью 2 °С/мин;
- температура блока ввода проб и детектора:
250 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 2 мкл.
Проводят 5 повторных введений для проверки
воспроизводимости системы.
Предельное содержание примесей:
-сумма содержания этиленгликоля и диэти¬
ленгликоля: не более 0,4 %.
Этиленоксид и диоксан (2.4.25). Не более
0,0001 % (1 ррт) этиленоксида и не более 0,0010 %
(10 ррт) диоксана.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (2 ррт РЬ) Р.
Вода (2.5.12). Не более 2,0 % для макроголов с
относительной молекулярной массой не более 1000;
не более 1,0 % для макроголов с относительной моле¬
кулярной массой более 1000. Определение проводят
из 2,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
Указывают:
- тип макрогола;
- содержание формальдегида.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Некоторые
из характеристик, описанных в разделе «Функци¬
онально-обусловленные характеристики», могут
также присутствовать в части фармакопейной
статьи, обязательной для выполнения, так как од¬
новременно они являются и показателями качества.
В таком случае в разделе «Функционально-обуслов¬
ленные характеристики» дается перекрестная
ссылка на это испытание в части фармакопейной
статьи, обязательной для выполнения. Контроль
данных характеристик может вносить вклад в каче¬
ство лекарственного средства путем достижения
постоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, однако
другие методы также могут быть использованы. В
случае указания результатов конкретных характе¬
ристик требуется указывать также и методы про¬
ведения испытаний.
Следующая характеристика может быть важ¬
на для макроголов, используемых в качестве раст¬
ворителей.
Вязкость (см. раздел «Испытания»).
Следующая характеристика может быть важ¬
на для макроголов, используемых в качестве стаби¬
лизаторов суспензий и загустителей.
Вязкость (см. раздел «Испытания»).
Следующая характеристика может быть важ¬
на для макроголов, используемых в качестве скольз¬
ящих веществ при изготовлении таблеток.
Распределение частиц по размерам (2.9.31).
Следующие характеристики могут быть важ¬
ны для макроголов, используемых в качестве основы
суппозиториев, и для макроголов, используемых при
изготовлении гидрофильных мазей.
Вязкость (см. раздел «Испытания»).
Температура плавления (2.2.15).
07/2016:0559
МАННИТ(МАННИТОЛ)
МаппНо1ит
МАЫштои
С6Н14Ов М.м. 182,2
[69-65-8]
ОПРЕДЕЛЕНИЕ
О-Маннитол.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белые или почти белые кристаллы или порошок.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
Обладает полиморфизмом (5.9).
Маннит (маннитол)
639
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Удельное оптическое вращение (2.2.7): от +23
до +25 (в пересчете на сухое вещество). 2,00 г испы¬
туемого вещества и 2,6 г динатрия тетрабората Р
растворяют в 20 мл воды Р при температуре 30 °С и
встряхивают в течение (15—30) мин без дальнейшего
нагревания. Полученный прозрачный раствор дово¬
дят водой Р до объема 25,0 мл.
B. Испытуемый образец выдерживает испыта¬
ние «Температура плавления», как указано в разделе
«Испытания».
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО маннита #или спектр, пред¬
ставленный на рисунке #0559.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то 25 мг ис¬
пытуемого образца и 25 мг ФСО маннита помещают
по отдельности в стеклянные сосуды и растворяют в
0,25 мл воды дистиллированной Р без нагревания.
Полученные растворы должны быть прозрачными.
Выпаривают досуха в микроволновой печи с мощно¬
стью (600—700) Вт в течение 20 мин или в сушиль¬
ном шкафу при температуре 100 °С в течение 1 ч с
последующим постепенным приложением вакуума до
получения сухого остатка. Образуются нелипкие по¬
рошки белого или слегка желтоватого цвета, которые
используют для получения новых спектров.
й. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (а). 25 мг ФСО маннита
растворяют в воде Р и доводят до объема 10 мл этим
же растворителем.
Раствор сравнения (Ь). 25 мг маннита Р и 25 мг
сорбита Р растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: вода Р — этилацетат Р —
пропанол Р (10:20:70, об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром 4-аминобензойной кислоты Р, высушивают в
потоке холодного воздуха до удаления запаха аце¬
тона, нагревают при температуре 100 °С в течение
15 мин, охлаждают, опрыскивают раствором 2 г/л на¬
трия перйодата Р, высушивают в потоке холодного
воздуха и нагревают при температуре 100 °С в тече¬
ние 15 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Рисунок #0559.-1. Инфракрасный спектр пропускания ФСО маннита в дисках с калия бромидом Р
640
Государственная фармакопея Республики Беларусь
N(2.2.38). Не бо¬
лее 20 мкСм см'1. 20,0 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р,
приготовленной из воды дистиллированной Р, при
нагревании от 40 °С до 50 °С и доводят до объема
100.0 мл этим же растворителем. Охлаждают и из¬
меряют удельную электропроводность при слабом
перемешивании на магнитной мешалке.
Температура плавления (2.2.14). От 165 °С до
170 °С.
Восстанавливающие сахара. Не более 0,1 %
(в пересчете на глюкозу). К 7,0 г испытуемого образца
прибавляют 13 мл воды Р, 40 мл медно-цитратного
раствора Р, осторожно кипятят в течение 3 мин и за¬
тем выдерживают в течение 2 мин. Образуется оса¬
док. Фильтруют через стеклянный пористый фильтр
(16) (2.1.2), покрытый диатомитом Р, или стеклянный
пористый фильтр (10) (2.1.2). Осадок промывают го¬
рячей водой Р ((50—60) °С) до отсутствия щелочи в
промывных водах, которые фильтруют через тот же
стеклянный пористый фильтр. Фильтрат отбрасывают.
Остаток незамедлительно растворяют в 20 мл раст¬
вора железа (III) сульфата Р, фильтруют через тот же
стеклянный пористый фильтр и промывают фильтр
(15—20) мл воды Р. Объединяют фильтрат и промыв¬
ные воды, нагревают до температуры 80 °С и титруют
0,02 М раствором калия перманганата. При прибав¬
лении не более 3,2 мл 0,02 М раствора калия перман¬
ганата окраска раствора должна измениться с зеле¬
ной на розовую и сохраняться в течение не менее 10 с.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в 2,5 мл воды Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (а). 0,50 г ФСО маннита
растворяют в 2,5 мл воды Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (Ь). 2,0 мл испытуемого раст¬
вора доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). 0,5 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 20,0 мл.
Раствор сравнения (д). 0,25 г маннита Р и 0,25 г
сорбита Р (примесь А) растворяют в 5 мл воды Р и
доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (е). 0,5 г мальтита Р (при¬
месь В) и 0,5 г изомальта Р (примесь С) растворяют
в 5 мл воды Р и доводят до объема 100 мл этим же
растворителем. 2 мл полученного раствора доводят
водой Р до объема 10 мл.
Условия хроматографирования:
- колонка длиной 0,3 м и диаметром 7,8 мм, за¬
полненная катионообменной смолой сильной (каль¬
циевой формой) Р с размером частиц 9 мкм;
- температура: (85±2) °С;
- подвижная фаза: дегазированная вода Р\
- скорость подвижной фазы: 0,5 мл/мин;
- рефрактометрический детектор, уравновешен¬
ный до постоянной температуры (например, до 40 °С);
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с), (с1) и (е);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания маннита.
Идентификация пиков примесей: идентифициру¬
ют пик примеси А, используя хроматограмму раствора
сравнения (с!); идентифицируют пики примесей В и С,
используя хроматограмму раствора сравнения (е).
Относительное удерживание (по отношению к
манниту, время удерживания — около 20 мин): при¬
месь С (первый пик) — около 0,6; примесь В — около
0,7; примесь С (второй пик) — около 0,73; примесь
А— около 1,2. Примесь С элюируется двумя пиками,
при этом может наблюдаться совместное элюирова¬
ние примеси В и второго пика примеси С.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 2,0 между пиками ман¬
нита и примеси А.
Предельное содержание примесей:
- примесь А (не более 2,0 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь);
- сумма примесей ВиС( не более 2,0 %): на хро¬
матограмме испытуемого раствора сумма площадей
пиков, соответствующих примесям В и С, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать 2-кратную пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- сумма примесей (не более 2,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
Никель (2.4.15). Не более 0,0001 % (1 ррт).
10,0 г испытуемого образца суспендируют в 30,0 мл
кислоты уксусной разведенной Р и доводят водой Р
до объема 100,0 мл. Используют насыщенный водой
метилизобутилкетон Р.
Тяжелые металлы. Не более 0,0005 % (5 ррт).
Испытуемый раствор. 5,0 г испытуемого образ¬
ца помещают в пробирку, подходящую для опреде¬
ления окрашивания жидкостей, вместимостью 50 мл
и растворяют в 40 мл воды Р. Прибавляют 2 мл кис¬
лоты уксусной разведенной Р1 и доводят водой Р до
объема 50 мл.
Раствор сравнения. 2,5 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р помещают в пробирку, под¬
ходящую для определения окрашивания жидкостей,
вместимостью 50 мл, прибавляют 2 мл кислоты ук¬
сусной разведенной Р1 и доводят водой Р до объема
50 мл.
В испытуемый раствор и раствор сравнения при¬
бавляют по 50 мкл раствора натрия сульфида Р1,
тщательно перемешивают и выдерживают в течение
5 мин. Просматривают пробирки вдоль вертикально
оси или горизонтально (перпендикулярно оси) на бе¬
лом фоне. Окраска испытуемого раствора не должна
быть интенсивнее окраски раствора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Бактериальные эндотоксины (2.6.14).
- Менее 4 МЕ/г, если субстанция предназначена
для производства лекарственных средств для парен¬
терального применения с концентрацией маннита не
Марганца глюконат
641
более 100 г/л без последующей подходящей процеду¬
ры удаления бактериальных эндотоксинов.
— Менее 2,5 МЕ/г, если субстанция предназначе¬
на для производства лекарственных средств для па¬
рентерального применения с концентрацией маннита
более 100 г/л без последующей подходящей процеду¬
ры удаления бактериальных эндотоксинов.
Микробиологическая чистота
Ест субсгамры предназначена для производ¬
ств имарсшанд средств для парентерального
10РКОВН2&12).
для проиэ-
среоств для парентерального
- ОКА. критерий приемлемости 103 КОЕ/г(2.6.72);
- ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12);
- ЕзсЬепсЫа соН: отсутствие (2.6.13);
- 8а1топе11а: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями:
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
г Содержание СбН1406 в процентах рассчитывают с
учетом содержания маннита в ФСО маннита.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомогатель¬
ного вещества (см. статью 5.15). Данный раздел не
является обязательным, и для подтверждения соот¬
ветствия требованиям частной статьи проверка
указанных характеристик не обязательна. Однако
контроль данных характеристик может вносить
вклад в качество лекарственного средства путем
достижения постоянства производственного про¬
цесса и улучшения свойств лекарственного сред¬
ства при его использовании. Предлагаемые методы
испытаний являются подходящими для указанных
цепей, однако другие методы также могут быть ис¬
пользованы. В случае указания результатов конкрет¬
ных характеристик требуется указывать также и
методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для маннита, используемого в качестве наполни¬
теля таблеток и капсул.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошков (2.9.36).
07/2016:2162
МАРКИРОВКА
При необходимости указывают:
- максимальное содержание бактериальных эн¬
дотоксинов;
- субстанция предназначена для производства ле¬
карственных средств для парентерального применения.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
В. 4-О-о-О-Глюкопиранозил-О-глюцитол (О-мальтит).
С. Смесь б-О-а-Р-глюкопиранозил-О-глюцитола и
1-О-а-О-гпюкопиранозил-О-маннита (изомальт).
МАРГАНЦА ГЛЮКОНАТ
Мандат д1исопаз
МАЫвАИЕЗЕ вШСОЫАТЕ
С^Н^МпО^ ■ хН20 М.м. 445,2
(безводное вещество)
ОПРЕДЕЛЕНИЕ
Марганца (II) О-глюконат безводный или гидрати¬
рованный.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или бледно-розовый кристаллический по¬
рошок. Слегка гигроскопичен.
Растворим в воде, практически нерастворим в
этаноле, нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 1 мл воды Р.
Раствор сравнения. 20 мг ФСО кальция глюко¬
ната растворяют в 1 мл воды Р, при необходимости
нагревая в водяной бане при температуре 60 °С.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
*- за*. -:ес
642
Гэсударственная фармакопея Республики Беларусь
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетат Р —вода Р — 96%
спирт Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: при температуре 105 °С в течение
20 мин и охлаждают до комнатной температуры.
Проявление: пластинку опрыскивают раствором,
содержащим 25 г/л аммония молибдата Р и 10 г/л це¬
рия (IV) сульфата Р в кислоте серной разведенной Р
и нагревают при температуре 105 °С в течение около
10 мин.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения.
В. 50 мг испытуемого образца растворяют в 5 мл
воды Р и прибавляют 0,5 мл раствора аммония суль¬
фида Р. Образуется бледно-розовый осадок, который
растворяется при прибавлении 1 мл кислоты уксус¬
ной ледяной Р.
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее шестого эталона шкалы
наиболее подходящего цвета.
Сахароза и восстанавливающие сахара. 0,5 г
испытуемого образца растворяют в смеси из 2 мл кис¬
лоты хлористоводородной Р1 и 10 мл воды Р. Кипя¬
тят в течение 5 мин, охлаждают, прибавляют 10 мл
раствора натрия карбоната Р и выдерживают в те¬
чение 10 мин. Полученный раствор доводят водой Р
до объема 25 мл и фильтруют. К 5 мл фильтрата при¬
бавляют 2 мл раствора медно-тартратного Р, кипя¬
тят в течение 1 мин и выдерживают в течение 2 мин.
Не должен образовываться красный осадок.
Хлориды (2.4.4). Не более 0,0500 % (500 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0500%
(500 ррт). 2,0 г испытуемого образца растворяют в
смеси из 10 мл кислоты уксусной Р и 90 мл воды дис¬
тиллированной Р.
Цинк. Не более 0,0050 % (50 ррт). К 10 мл раст¬
вора 3 прибавляют 1 мл кислоты серной Р и 0,1 мл
раствора калия ферроцианида Р. Через 30 с полу¬
ченный раствор по степени мутности не должен пре¬
вышать смесь из 1,0 мл эталонного раствора цинка
(10 ррт 1п) Р, 9 мл воды Р, 1 мл кислоты серной Р и
0,1 мл раствора калия ферроцианида Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл воды Р, нагревая в водяной бане
при температуре 60 °С. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Вода (2.5.32). Не более 9,0 %. Определение про¬
водят из 80 мг испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют 50 мл
воды Р, прибавляют 10 мг кислоты аскорбиновой Р,
20 мл аммония хлорида буферного раствора рН 10,0
Р и 0,2 мл раствора 2 г/л протравного черного 11 Р
в триэтаноламине Р. Титруют 0,1 М раствором на¬
трия эдетата Р до перехода окраски раствора от
фиолетовой до чисто-голубой.
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 44,52 мг 012Н22МпО14.
ХРАНЕНИЕ
В неметаллическом воздухонепроницаемом кон¬
тейнере.
07/2016:0845
МЕБЕНДАЗОЛ
МеЬепс1а1о1ит
МЕВЕЫ0А201-Е
[31431-39-7]
ОПРЕДЕЛЕНИЕ
Метил(5-бензоил-1Н-бензимидазол-2-ил)карба-
мат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, в 96 % спирте
и в метиленхлориде.
Обладает полиморфизмом (5.9). Приемлемая
кристаллическая форма соответствует ФСО мебен-
дазола.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО мебендазола.
Приготовление: используют субстанции без
предварительной обработки.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в диметилформамиде Р и доводят
этой же смесью растворителей до объема 25,0 мл.
Раствор сравнения (а). 5,0 мг ФСО мебендазо¬
ла для проверки пригодности хроматографической
системы (содержит примеси А, В, С, О, Е, Р и С) раст¬
воряют в диметилформамиде Р и доводят до объема
5,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят диметилформамидом Р до объема
Мебендаэал
643
100,0 ми. 5,0 мл полученного раствора доводят диме-
ямяяфармамидом Р до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаме¬
тром 4.6 мм, заполненная силикагелем октадецил-
сшшпьным деактивированным по отношению к
основаниям для хроматографии Р (размер частиц
-твмпержурас 40 *С;
— шуршмая фаза А: раствор 7,5 г/л аммония
■1ГПТ1-Т ~
- -сданная фаза В: ацетонитрил Р;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
- . . Подвижная фаза А
^ И**» | (% об/об)
Подвижная фаза В
(%, об/об)
I 0—15 | 80 — 70
20 — 30
| 15—20 70-10
30 — 90
1 20—25 | 10
90
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 250 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: иденти¬
фицируют пики примесей А, В, С, О, Е, Р и С, ис¬
пользуя хроматограмму раствора сравнения (а) и
хроматограмму, прилагаемую к ФСО мебендазола
йш протерши пригодности хроматографической
€пшвосяшштмот удершятание (по апюше-
мшт ж ■ебацазпя^ время удерживания — около
0.5: примесь С — около 0.7; примесь О — около
1.1: примесь Е — около 1,3; примесь Р — около 1,4;
примесь С — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения ликов: не менее 4
(Н^ — высота пика примеси О относительно базо¬
вом линии; Ну— расстояние между базовой линией
и нижней точкой кривой, разделяющей пик примеси
О и пик мебендазола).
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси (3 — 1,4):
- примесь 6 (не более 0,5 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси 6, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- примеси А, В, С, О, Е, Е (не более 0,25 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, С, О, Е, Р,
не должны превышать площадь основного пика на
хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А, В, С, О, Е, Р и О, не должна превы¬
шать 0,4 площади основного пика на хроматограм¬
ме раствора сравнения (Ь);
- сумма примесей (не более А ,0 %у. на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
4-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
смеси из кислоты уксусной безводной Р и метил-
этилкетона Р( 1:7, об/об) и титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 29,53 мг С16Н131Ч303.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Е, О.
о
А. (2-Амино-1 Н-бензимидазол-5-ил)фенилмета-
нон.
о
В. (2-Гидрокси-1Н-бензимидазол-5-ил)фенилме-
танон.
С. (2-Амино-1 -метил-1 Н-бензимидазол-5-ил)фе-
нилметанон.
О. Метил (5-бензоил-1 -метил-1 Н-бе нзимидазол-2-
ил)карбамат.
Е. Этил(5-бензоил-1 /-/-бензимидазол-2-ил)карба-
мат.
81*. Зак. 1060.
644
Гэсударственная фармакопея Республики Беларусь
Р. Метил[5-(4-метилбензоил)-1 Н-бензимидазол-2-
ил]карбамат.
О. Л/,Л/-Бис(5-бензоил-1 Я-бензимидазол-2-илМо¬
чевина.
07/2016:0893
МЕДИ СУЛЬФАТ БЕЗВОДНЫЙ
Сирп зиКаз апЬубпсиз
СОРРЕК 8Ш.РНАТЕ, АЛ/НУОЯО(75
Си804 М.м. 159,6
[7758-98-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Зеленовато-серый порошок. Очень гигроскопичен.
Легко растворим в воде, мало растворим в мета¬
ноле, практически нерастворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 1 мл раствора 5, приготовленного как ука¬
зано в разделе «Испытания», прибавляют несколько
капель раствора аммиака разведенного Р2. Образу¬
ется синий осадок, который растворяется при даль¬
нейшем прибавлении раствора аммиака разведенно¬
го Р2 и появляется темно-синее окрашивание.
B. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
C. 1 мл раствора 3 доводят водой Р до объема
5 мл. Полученный раствор дает реакцию (а) на суль¬
фаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,6 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Хлориды (2.4.4). Не более 0,0150% (150 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Железо. Не более 0,0150 % (150 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 0,32 г испытуемого об¬
разца растворяют в 10 мл воды Р, прибавляют 2,5 мл
кислоты азотной, свободной от свинца, Р и доводят
водой Р до объема 25,0 мл.
Растворы сравнения. Для приготовления рас¬
творов сравнения используют эталонный раствор
железа (20 ррт Ре) Р и 2,5 мл кислоты азотной,
свободной от свинца, Р. Доводят объем полученного
раствора до 25,0 мл водой Р.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Медь при взаимодействии с ацетиленом может
образовывать взрывоопасные ацетилиниды. Во из¬
бежание этого тщательно очищайте горелку, пре¬
дотвращая высыхание загрязнений.
Свинец. Не более 0,0080 % (80 ррт). Атомно-аб¬
сорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 1,6 г испытуемого об¬
разца растворяют в 10 мл воды Р, прибавляют 2,5 мл
кислоты азотной, свободной от свинца, Р и доводят
водой Р до объема 25,0 мл.
Раствор сравнения. Для приготовления рас¬
творов сравнения используют эталонный раствор
свинца (100 ррт РЬ) Р и 2,5 мл кислоты азотной,
свободной от свинца, Р. Доводят объем полученного
раствора до 25,0 мл водой Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 217,0 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Медь при взаимодействии с ацетиленом может
образовывать взрывоопасные ацетилиниды. Во из¬
бежание этого тщательно очищайте горелку, пре¬
дотвращая высыхание загрязнений.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 0,500 г испытуемого образца сушат при
температуре (250±10) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,125 г испытуемого образца растворяют в 50 мл
воды Р, прибавляют 2 мл кислоты серной Р, 3 г ка¬
лия йодида Р и титруют 0,1 М раствором натрия
тиосульфата, используя в качестве индикатора 1 мл
раствора крахмала Р, который прибавляют в конце
титрования.
1 мл 0,1 М раствора натрия тиосульфата со¬
ответствует 15,96 мг Си304.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0894
МЕДИ СУЛЬФАТ ПЕНТАГИДРАТ
Сирп зиНаз реМаЬуйпсиз
СОРРЕР 8Ш.РНАТЕ, РЕЫТАНУОРАТЕ
Си304 • 5Н,0 М.м. 249,7
[7758-99-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %.
Меклозина дигидрохлорид
645
ОПИСАНИЕ (СВОЙСТВА)
Синий кристаллический порошок или прозрач¬
ные синие кристаллы.
Легко растворим в воде, растворим в метаноле,
практически нерастворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 1 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», прибавляют несколько
капель раствора аммиака разведенного Р2. Образу¬
ется синий осадок, который растворяется при даль¬
нейшем прибавлении раствора аммиака разведенно¬
го Р2Ти появляется темно-синее окрашивание.
B. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
C. 1 мл раствора 3 доводят водой Р до объема
5 мл. Полученный раствор дает реакцию (а) на суль¬
фаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5 г испытуемого образца растворяют
в воде Р и доводят до объема 100 мл этим же раство¬
рителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Железо. Не более 0,0100 % (100 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 0,5 г испытуемого об¬
разца растворяют в 10 мл воды Р, прибавляют 2,5 мл
кислоты азотной, свободной от свинца, Р и доводят
водой Р до объема 25,0 мл.
Растворы сравнения. Для приготовления рас¬
творов сравнения используют эталонный раствор
железа (20 ррт Ре) Р и 2,5 мл кислоты азотной,
свободной от свинца, Р. Доводят объем полученного
раствора до 25,0 мл водой Р.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Медь при взаимодействии с ацетиленом может
образовывать взрывоопасные ацетилиниды. Во из¬
бежание этого тщательно очищайте горелку, пре¬
дотвращая высыхание загрязнений.
Свинец. Не более 0,0050 % (50 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 2,5 г испытуемого об¬
разца растворяют в 10 мл воды Р, прибавляют 2,5 мл
кислоты азотной, свободной от свинца, Р и доводят
водой Р до объема 25,0 мл.
Раствор сравнения. Для приготовления рас¬
творов сравнения используют эталонный раствор
свинца (100 ррт РЬ) Р и 2,5 мл кислоты азотной,
свободной от свинца, Р. Доводят объем полученного
раствора до 25,0 мл водой Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 217,0 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Медь при взаимодействии с ацетиленом может
образовывать взрывоопасные ацетилиниды. Во из¬
бежание этого тщательно очищайте горелку, пре¬
дотвращая высыхание загрязнений.
Потеря в массе при высушивании (2.2.32). Не
менее 35,0 % и не более 36,5 %. 0,500 г испытуемого
образца сушат при температуре (250±10) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 50 мл
воды Р, прибавляют 2 мл кислоты серной Р, 3 г ка¬
лия йодида Р и титруют 0,1 М раствором натрия
тиосульфата, используя в качестве индикатора 1 мл
раствора крахмала Р, который прибавляют в конце
титрования.
1 мл 0,1 М раствора натрия тиосульфата со¬
ответствует 24,97 мг Си304-5Н20.
07/2016:0622
МЕКЛОЗИНА ДИГИДРОХЛОРИД
Мес1о2т'1 сНЬудгосЫопдит
МЕСШ2ШЕ 01НУОКОСШОВЮЕ
и энантиомер
2НС1
С25Н27С1Ы2 - 2НС1 М.м. 463,9
[1104-22-9]
ОПРЕДЕЛЕНИЕ
1-[(/?5)-(4-Хлорфенил)фенилметил]-4-[(3-метил-
фенил)метил]пиперазина дигидрохлорид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый кристаллический
порошок. Слегка гигроскопичен.
Мало растворим в воде, растворим в 96 % спирте
и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 15,0 мг испытуемого об¬
разца растворяют в 0,1 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем. 10,0 мл полученного раствора до¬
водят 0,1 М раствором кислоты хлористоводород¬
ной до объема 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 232 нм.
Удельный показатель поглощения в максимуме:
от 345 до 380 (в пересчете на безводное вещество).
Раствор также имеет слабую абсорбцию без опре¬
деленного максимума в диапазоне от 260 нм до 300 нм.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
82. Зак. 1060.
646
Гэсударственная фармакопея Республики Беларусь
Сравнение: ФСО меклозина дигидрохлорида.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 50 мг ФСО меклозина диги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля <ЗР254 Р.
Подвижная фаза: диэтиламин Р — толуол Р —
циклогексан Р (10:15:75, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке теплого воздуха в тече¬
ние 5 мин.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
О. 15 мг испытуемого образца растворяют в 2 мл
96 % спирта Р. Раствор дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Кислотность или щелочность. Значение А долж¬
но быть не менее -0,3 мл и не более 0,3 мл. Определе¬
ние проводят из 0,350 г испытуемого образца.
Кислотность или щелочность рассчитывают по
формуле, используя объемы титранта, полученные в
разделе «Количественное определение»:
А = \/2 - 2Ц,
где:
V, — объем 0,1 М раствора натрия гидроксида
в первой точке перегиба на кривой титрования;
У2 — объем 0,1 М раствора натрия гидроксида
во второй точке перегиба на кривой титрования.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р —
вода Р (50:50, об/об).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 7,5 мг ФСО меклозина
примеси В и 7,5 мг ФСО меклозина примеси Н раст¬
воряют в смеси растворителей и доводят до объема
100.0 мл этой же смесью растворителей. 1,0 мл полу¬
ченного раствора доводят смесью растворителей до
объема 100,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная аморфным кремнийоргани-
ческим полимером октадецилсилильным эндкепиро-
ванным Р с размером частиц 3,5 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: 0,1 % (об/об) раствор ам¬
миака концентрированного Р\
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
60
40
3—13
60 — 15
40 — 85
13—23
15 — 5
85— 95
23—33
5
95
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В и Н, используя хроматограмму
раствора сравнения (Ь).
Относительное удерживание (по отношению к
меклозину, время удерживания — около 18 мин): при¬
месь В — около 0,45; примесь Н — около 0,49.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,5 между пиками при¬
меси В и примеси Н.
Предельное содержание примесей:
- примесь В (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 1,5-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си В, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (0,3 %): на хроматограмме
испытуемого раствора сумма площадей всех пиков,
кроме основного, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 50 мл
96 % спирта Р и титруют 0,1 М раствором натрия
гидроксида потенциометрически (2.2.20). Отмечают
объем титранта между двумя точками перегиба на
кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 46,39 мг С25Н27С1!\12-2НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированная примесь: В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемлемо-
Мелоксикам
647
сти для других/неспецифицированных примесей и/
или общей статьей Субстанции для фармацевти¬
ческого использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): С,
Д Е, Рг Н.
В. (Я5)-(4-Хлорфенил)фенилметанол.
С. (4-Хлорфенил)фенилметанон (4-хлорбензофе-
нон).
й. 1,1 '-(1,3-Фениленбисметилен)бис[4-[(4-хпорфе-
нил)фенилметил]пиперазин].
Е. 1 -(Дифенилметил)-4-[(3-метилфенил)метил]пи-
перазин.
Р. 1,4-Бис[(4-хлорфенил)фенилметил]пиперазин.
Н. 1 -[(/?5)-(4-Хлорофенил)фенилметил]пиперазин.
07/2016:2373
МЕЛОКСИКАМ
Ме1охюатит
МЕШХЮАМ
С14Н13М30432 М.м. 351,4
[71125-38-7]
ОПРЕДЕЛЕНИЕ
4-Гидрокси-2-метил-А/-(5-метилтиазол-2-ил)-2Н-
1,2-бензотиазин-З-карбоксамида 1,1-диоксид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Бледно-желтый порошок.
Практически нерастворим в воде, растворим в
диметилформамиде, очень мало растворим в 96 %
спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО мелоксикама #или спектр, пред¬
ставленный на рисунке #2373.-1#.
Если спектры, полученные с использованием
образцов в твердом состоянии, различаются, то ис¬
пытуемый образец и ФСО мелоксикама растворяют
по отдельности в ацетоне Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 40 мг испытуемого образ¬
ца растворяют в смеси из 5 мл метанола Р и 0,3 мл
1 М раствора натрия гидроксида и доводят метано¬
лом Р до объема 20,0 мл.
Раствор сравнения (а). 2,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
5,0 мл полученного раствора доводят метанолом Р
до объема 100,0 мл.
Раствор сравнения (Ь). 2 мг испытуемого образ¬
ца, 2 мг ФСО мелоксикама примеси А, 2 мг ФСО мелок¬
сикама примеси В, 2 мг ФСО мелоксикама примеси С и
2 мг ФСО мелоксикама примеси О растворяют в смеси
из 5 мл метанола Р и 0,3 мл 7 М раствора натрия ги¬
дроксида и доводят метанолом Р до объема 25 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
82*. Зак. 1060.
648
Гэсударственная фармакопея Республики Белару<
Рисунок #2373.-1. Инфракрасный спектр ФСО мелоксикама
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: раствор 1 г/л калия ди¬
гидрофосфата Р, доведенного 1 М раствором
натрия гидроксида до рН 6,0;
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
60
40
2—10
о
со
т
о
со
40 —> 70
10—15
30
70
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длины
волн 260 нм и 350 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к мелоксикаму, время удерживания — около 7 мин):
примесь В — около 0,5; примесь А — около 1,4; при¬
месь С — около 1,7; примесь О — около 1,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками ме¬
локсикама и примеси А при длине волны 350 нм; не
менее 3,0 между пиками примеси В и мелоксикама
при длине волны 260 нм.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—2,0):
- примесь А при длине волны 350 нм (не более
0,1 %): на хроматограмме испытуемого раствора пло¬
щадь пика, соответствующего примеси А, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а) при длине волны 350 нм;
-примесь В при длине волны 260 нм (не бо¬
лее 0,1 %): на хроматограмме испытуемого раст¬
вора площадь пика, соответствующего примеси В,
не должна превышать площадь основного пика на
хроматограмме раствора сравнения (а) при длине
волны 350 нм;
- примеси С, О при длине волны 350 нм (не бо¬
лее 0,05 %): на хроматограмме испытуемого раствора
площади пиков, соответствующих примесям С, О, не
должны превышать 0,5 площади основного пика на
хроматограмме раствора сравнения (а) при длине
волны 350 нм;
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С и О, при длине волны, устанавливаю¬
щей наибольший размер пика примеси, не должна
превышать площадь основного пика на хромато¬
грамме раствора сравнения (а) при той же длине
волны;
- сумма примесей: не более 0,3 %;
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а) при той же длине
волны.
Тяжелые металлы {2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Мельдоний дигидрат
649
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Во избежание перегревания в течение титро¬
вания необходимо тщательно перемешивать на
протяжении всего периода титрования и прекра¬
тить титрование сразу же после достижения ко¬
нечной точки.
0,250 г испытуемого образца растворяют в смеси
из 5 мл кислоты муравьиной безводной Р и 50 мл кис¬
лоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 35,14 мг С14Н131Ч30432.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е, Р.
Р. Я = СН(СН3)2: Изопропил-4-гидрокси-2-метил-
2Н-1,2-бензотиазин-З-карбоксилата 1,1-диоксид.
07/2016:2624
МЕЛЬДОНИЙ ДИГИДРАТ
Ме1с1општ сИМус/псит
МЕ1-ООМШМ 01НУОКАТЕ
Н3СЧ 7СНз
02С. 2Н20
N ХН3
Н 3
Сбниы202 ■ 2НгО М.м. 182,2
[86426-17-7]
ОПРЕДЕЛЕНИЕ
3-(2,2,2-Триметилгидразин-2-ий-1-ил) пропаноата
дигидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые кристаллы или кристал¬
лический порошок. Расплывается на воздухе.
Очень легко растворим в воде, легко растворим в
метаноле, практически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО мельдония дигидрата.
ИСПЫТАНИЯ
А. Этил 4-гидрокси-2-метил-2Н-1,2-бензотиазин-
3-карбоксилата 1,1 -диоксид.
В. 5-Метилтиазол-2-амин.
С. Я = СН3: Л/-[(22)-3,5-Диметилтиазол-2(ЗЯ)-
илиден]-4-гидрокси-2-метил-2Н-1,2-бензотиазин-3-
карбоксамида 1,1-диоксид.
О. Р = С2Н5: А/-[(22)-3-Этил-5-метилтиазол-2(ЗН)-
илиден]-4-гидрокси-2-метил-2Н-1,2-бензотиазин-3-
карбоксамида 1,1 -диоксид.
Е. Р = СН3: Метил-4-гидрокси-2-метил-2Н-1,2-
бензотиазин-3-карбоксилата 1,1-диоксид.
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в 50 мл воды дистиллированной Р.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон I.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)9.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29) с использованием масс-
спектрометрии (2.2.43).
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО мельдония
примеси А, 10,0 ж ФСО мельдония примеси В, 10,0 мг
ФСО мельдония примеси С, 10,0 мг ФСО мельдо¬
ния примеси О, 10,0 мг ФСО мельдония примеси Е и
10.0 мг ФСО мельдония примеси Р растворяют в под¬
вижной фазе А и доводят до объема 100,0 мл этим же
растворителем.
Раствор сравнения (Ь). 1,5 мл раствора срав¬
нения (а) доводят подвижной фазой А до объема
100.0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 3,0 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3,5 мкм;
83. Зак. 1060.
650
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза:
- подвижная фаза А: 0,1 % (об/об) раствор геп-
тафтормасляной кислоты Р в воде для хрома¬
тографии Р;
- подвижная фаза В: 0,1 % (об/об) раствор геп-
тафтормасляной кислоты Р в метаноле Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—7
90 — 65
10-35
7—12
65 — 40
35 — 60
12—18
40 — 25
60-75
- скорость подвижной фазы: 0,2 мл/мин;
- масс-детектор (тройной квадруполь): приво¬
димые параметры указываются в качестве примера
подходящих рабочих настроек; если детектор имеет
другие рабочие параметры, их настраивают таким об¬
разом, чтобы выполнить условия пригодности хрома¬
тографической системы:
- ионизация: электрораспылительная иониза¬
ция в режиме положительных ионов;
- обнаружение:
- режим многократного мониторинга реак¬
ции (МРМ)\
- регистрируемое отношение т/г: 50—300;
- температура источника ионов: 110 °С;
- температура десольватации: 220 °С;
- напряжение фрагментатора: 15 В;
- напряжение капилляра (Усар): 3 кВ;
- СЮ газ: аргон;
- давление СЮ газа: 2,7*1 О*3 мБар.
Параметры режима МРМ:
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,3 %;
- учитываемый предел: 0,05 %.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
5 мл раствора 5 доводят водой дистиллирован¬
ной Р до объема 30 мл. Полученный раствор дол¬
жен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0100%
(100 ррт). 10 мл раствора 3 доводят водой дис¬
тиллированной Р до объема 20 мл. Полученный
раствор должен выдерживать испытание на суль¬
фаты.
Вода (2.5.12). Не менее 19,7% и не более
21,0%. Определение проводят из 0,120 г испыту¬
емого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в
40 мл кислоты уксусной ледяной Р и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 14,62 мг С6Н141Ч202.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Вещество
Каналы МЯМ
Энергия коллизии
(еУ) для интервала
(0—25) мин
Мельдоний
147,00 — 59,00
18,00
Примесь А
59,97 — 44,98
18,00
Примесь В
74,99 - 59,04
15,00
Примесь Р
115,19 — 71,92
19,00
Примесь С
161,19 — 59,00
23,0
Примесь О
175,19 — 58,07
23,0
Примесь Е
189,26 — 58,01
22,0
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
Относительное удерживание (по отношению к
мельдонию, время удерживания — около 9 мин): при¬
месь Р — около 0,7; примесь В — около 0,8; примесь
А— около 0,9; примесь С — около 1,5; примесь О —
около 1,8; примесь Е — около 2,0.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- отношение сигнал/шум: не менее 10 для при¬
меси А; не менее 50 для примеси В; не менее 200 для
примеси С; не менее 1000 для примеси О] не менее
1000 для примеси Е; не менее 50 для примеси Р;
- сходимость: максимальное относительное
стандартное отклонение не более 10 % после 6 вво¬
дов пробы.
Расчет процентных содержаний:
- для примесей А, В, С, О, Е и Р (режим МРМ) —
используют концентрацию соответствующей примеси
в растворе сравнения (Ь);
-для примесей, кроме примесей А, В, С, О, Е и Р
(режим полного спектра) — используют концентрацию
мельдония в растворе сравнения (с).
Предельное содержание примесей:
- примеси А, В, С, О, Е и Р: не более 0,15 % для
каждой примеси;
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
СН3
.т*
Н3С ''СНз
А. Л/,Л/-Диметилметанаминий.
н3сч уЗНз
Н2Ы ХН3
В. 1,1,1-Триметилгидразин-1-ий.
Н3СО'
Н3С^ ^сн3
N ХН3
н 3
С. 2-(3-Метокси-3-оксопропил)-1,1,1-триметил-
гидразин-1-ий.
о
н3с^
Н3СХ ^Нз
N -СНз
й. 2-(3-Этокси-3-оксопропил)-1,1,1 -триметил-
гидразин-1-ий.
сн3 о
н3с
НзС^ уЗНз
.X
N СН3
н 3
Е. 1,1,1 -Триметил-2-[3-(1 -метилэтокси)-3-оксопро-
пил]гидразин-1 -ий.
/
Г Ж
СНз
СНз
Р. 1,1-Диметил-4,5-дигидро-1Н-пиразол-1-ий-3-
олат.
#Менадиона натрия бисульфит
651
07/2016:0507
МЕНАДИОН
МепасИопит
МЕЫАОЮЫЕ
С„Н602 М.м. 172,2
[58-27-5]
ОПРЕДЕЛЕНИЕ
2-Метилнафталин-1,4-дион.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Бледно-желтый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в толуоле, умеренно растворим в 96 % спирте и
в метаноле.
Нестабилен на свету.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 105 °С до
108 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО менадиона.
C. 1 мг испытуемого образца растворяют в 5 мл
96 % спирта Р, прибавляют 2 мл раствора амми¬
ака Р и 0,2 мл этилцианоацетата Р. Появляется
интенсивное голубовато-фиолетовое окрашивание.
Прибавляют 2 мл кислоты хлористоводородной Р.
Раствор обесцвечивается.
Э. 10 мг испытуемого образца растворяют в 1 мл
96 % спирта Р, прибавляют 1 мл кислоты хлористо¬
водородной Р и нагревают в водяной бане. Появляет¬
ся красное окрашивание.
ИСПЫТАНИЯ
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27). Испытание проводят с защитой
от яркого света.
Испытуемый раствор. 0,2 г испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 0,5 мл испытуемого раст¬
вора доводят ацетоном Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: нитрометан Р — ацетон Р —
этиленхлорид Р — циклогексан Р (1:2:5:90, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: трижды; не менее 15 см
от линии старта.
Высушивание: в потоке горячего воздуха после
каждого хроматографирования.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат над
фосфора (V) оксидом Р при давлении от 2 кПа до
3 кПа в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца помещают в колбу с
пробкой, снабженной клапаном, и растворяют в 15 мл
кислоты уксусной ледяной Р. Прибавляют 15 мл
кислоты хлористоводородной разведенной Р и 1 г
цинка порошка Р, колбу закрывают и выдерживают
в течение 60 мин в защищенном от света месте при
периодическом взбалтывании. Раствор фильтруют че¬
рез ватный тампон и трижды промывают водой, сво¬
бодной от углерода диоксида, Р порциями по 10 мл.
К объединенному фильтрату и промывным водам при¬
бавляют 0,1 мл ферроина Р и немедленно титруют
0,1 М раствором аммония церия нитратом.
1 мл 0,1 М раствора аммония церия нитрат со¬
ответствует 8,61 мг С„Н802.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:РБ0019
#МЕНАДИОНА НАТРИЯ
БИСУЛЬФИТ
МепасИоп! па\п\ ЫзиШз
МЕЫАОЮЫЕ ЗОИШМ В18Ш-Р1ТЕ
С^НдМаОдЗ • хН20 М.м. 276,2 (безводный)
ОПРЕДЕЛЕНИЕ
Натрия 1,2,3,4-тетрагидро-2-метил-1,4-диоксо-2-
нафталинсульфонат гидрат. Викасол.
Содержание: не менее 95,0 % (в пересчете на
безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или белый с желтоватым оттенком кри¬
сталлический порошок.
Легко растворим в воде, мало растворим в 96 %
спирте.
83*. Зак. 1060.
652
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: спектр, представленный на рисунке
РБ0019.-1.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор (а). 2 мг испытуемого об¬
разца растворяют в 100 мл воды Р.
Испытуемый раствор (Ь). 2,5 мл испытуемого
раствора (а) доводят водой Р до объема 10 мл.
Спектр поглощения испытуемого раствора (а)
в области от 280 нм до 340 нм имеет максимум при
305 нм. Спектр поглощения испытуемого раствора (Ь)
в области от 220 нм до 280 нм имеет максимум при
230 нм и 265 нм и минимум при 248 нм.
C. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,2 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор $ должен
быть бесцветным.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27). Испытание проводят с защитой
от яркого света.
Испытуемый раствор. 350 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема
5,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг менадиона Р раст¬
воряют в ацетоне Р и доводят до объема 25,0 мл
этим же растворителем.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят ацетоном Р до объема 10,0 мл.
Раствор сравнения (с). 5,0 мл раствора сравне¬
ния (Ь) доводят ацетоном Р до объема 10,0 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ 6Р254 Р'
Подвижная фаза: хлороформ Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживается пятно.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (а);
- сумма примесей: не более 2 % (для опреде¬
ления содержания единичных примесей используют
пятна, полученные на хроматограммах растворов
сравнения (а), (Ь) и (с)).
Натрия 2-метил-1,4-дигидрокси-З-нафтал инсул ь-
фонат. К 5 мл раствора 3 прибавляют 2 капли ферро-
ина Р. Полученный раствор должен быть прозрачным.
Натрия гидросульфит. Не более 2,0 %. 1 г ис¬
пытуемого образца растворяют в 30 мл воды Р, при¬
бавляют 20 мл 0,1 М раствора кислоты серной и
30 мл 0,1 М раствора йода. Избыток йода титруют
0,1 М раствором натрия тиосульфата, используя в
качестве индикатора 1 мл раствора крахмала Р.
1 мл 0,1 М раствора йода соответствует 5,203 мг
№Н303.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта-
Рисунок РБ0019.-1. Инфракрасный спектр менадиона натрия бисульфита
Ментол рацемический
653
пон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 12,0 % и не более 16,5 %.
Определение проводят из 0,100 г испытуемого образца.
Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным, если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения. Тест-доза — 3 мг
испытуемого образца в 0,5 мл раствора 9 г/л натрия
хлорида Р в воде для инъекций Р на 1 кг массы тела
кролика.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 20 мл
воды Р, переносят в делительную воронку, быстро
прибавляют 17 мл 0,1 М раствора натрия гидрокси¬
да и немедленно экстрагируют хлороформом Р триж¬
ды порциями по 20 мл. Хлороформные извлечения
объединяют, промывают 10 мл воды Р, фильтруют че¬
рез бумажный фильтр, смоченный хлороформом Р, и
промывают фильтр 5 мл хлороформа Р. Фильтрат вы¬
паривают досуха в вакууме при комнатной темпера¬
туре. Остаток растворяют в 15 мл кислоты уксусной
ледяной Р, прибавляют 15 мл кислоты хлористово¬
дородной разведенной Р, 3 г порошка цинка Р, вы¬
держивают в течение 30 мин в защищенном от света
месте при периодическом перемешивании и быстро
фильтруют. Осадок в колбе и на фильтре немедленно
промывают водой Р трижды порциями по 10 мл. К по¬
лученному фильтрату прибавляют 2—3 капли ферро-
ина Р и титруют 0,1 М раствором церия сульфата до
появления устойчивого зеленого окрашивания..
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора церия сульфата соответ¬
ствует 13,81 мг С^Н^аС^З.
ХРАНЕНИЕ
В защищенном от света и влаги месте при темпе¬
ратуре не выше 25 °С.
07/2016:0623
МЕНТОЛ РАЦЕМИЧЕСКИЙ
Меп0ю1ит гасетюит
МЕИТНОЦ КАСЕМ1С
ОПРЕДЕЛЕНИЕ
Смесь равных частей (1/?5,25Я,5Я5)-5-метил-2-
(1-метилэтил)циклогексанола.
ОПИСАНИЕ (СВОЙСТВА)
Текучий или агломерированный кристаллический
порошок либо призматические или игольчатые бес¬
цветные блестящие кристаллы.
Практически нерастворим в воде, очень легко
растворим в 96 % спирте и в петролейном эфире,
легко растворим в жирных маслах и в вазелиновом
масле, очень мало растворим в глицерине.
Температура плавления: около 34 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, О.
A. Испытуемый образец выдерживает испы¬
тание «Угол оптического вращения», как указано в
разделе «Испытания».
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объ¬
ема 5 мл этим же растворителем.
Раствор сравнения. 25 мг ФСО ментола раст¬
воряют в метаноле Р и доводят до объема 5 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля С Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе до удаления раство¬
рителей.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р и нагревают при тем¬
пературе от 100 °С до 105 °С в течение (5—10) мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения.
C. Просматривают хроматограммы, получен¬
ные в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме
испытуемого раствора (Ь) соответствует по распо¬
ложению и величине основному пику на хромато¬
грамме раствора сравнения (с).
0. 0,20 г испытуемого образца растворяют в
0,5 мл пиридина безводного Р и прибавляют 3 мл
раствора 150 г/л динитробензоилхлорида Р в пи¬
ридине безводном Р. Нагревают на водяной бане в
течение 10 мин, прибавляют 7,0 мл воды Р неболь¬
шими порциями при перемешивании и выдержива¬
ют в ледяной бане в течение 30 мин. Образуется
осадок. Раствор отстаивают и сливают надосадоч-
ную жидкость. Осадок дважды промывают ледяной
водой Р порциями по 5 мл, перекристаллизовывают
из 10 мл ацетона Р, промывают ледяным ацето¬
ном Р и сушат при температуре 75 °С и давлении,
не превышающем 2,7 кПа, в течение 30 мин. Темпе¬
ратура плавления (2.2.14) полученных кристаллов:
от 130 °С до 131 °С.
ИСПЫТАНИЯ
Раствор 3. 2,50 г испытуемого образца раство¬
ряют в 10 мл 96% спирта Р и доводят до объема
25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
84. Зак. 1060.
654
Гэсударственная фармакопея Республики Беларусь
Кислотность или щелочность. 1,0 г испыту¬
емого образца растворяют в 96 % спирте Р и до¬
водят до объема 10 мл этим же растворителем.
Прибавляют 0,1 мл раствора фенолфталеина Р.
Раствор остается бесцветным. При прибавлении
не более 0,5 мл 0,01 М раствора натрия гидрок¬
сида должно появиться розовое окрашивание.
Угол оптического вращения (2.2.7). От -0,2°
до +0,2°. Измеряют угол оптического вращения
раствора 5.
Сопутствующие примеси. Газовая хромато¬
графия (2.2.28).
Испытуемый раствор (а). 0,20 г испытуемого
образца растворяют в метиленхлориде Р и дово¬
дят до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят метиленхлоридом Р до объ¬
ема 10,0 мл.
Раствор сравнения (а). 40,0 мг испытуемо¬
го образца и 40,0 мг изоментола Р растворяют в
метиленхлориде Р и доводят до объема 100,0 мл
этим же растворителем.
Раствор сравнения (Ь). 0,10 мл испытуемого
раствора (а) доводят метиленхлоридом Р до объ¬
ема 100,0 мл.
Раствор сравнения (с). 40,0 мг ФСО ментола
растворяют в метиленхлориде Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка стеклянная длиной 2,0 м и внутрен¬
ним диаметром 2 мм, заполненная диатомитом
для газовой хроматографии Р, импрегнированным
15 % [м/м) макрогола 1500 Р;
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 30 мл/мин;
- температура: колонка — 120 °С, блок ввода
проб -—150 °С, детектор — 200 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания ментола.
Пригодность хроматографической системы:
-разрешение: не менее 1,4 между пиками
ментола и изоментола на хроматограмме раствора
сравнения (а);
- отношение сигнал/шум: не менее 5 для ос¬
новного пика на хроматограмме раствора сравне¬
ния (Ь).
Предельное содержание примесей: испытуе¬
мый раствор (а):
- сумма примесей: не более 1 %;
- неучитываемый предел: не учитывают пики,
площадь которых менее 0,05 % площади основного
пика на хроматограмме испытуемого раствора (а).
Остаток после выпаривания. Не более
0,05 %. 2,00 г испытуемого образца выпаривают на
водяной бане и сушат при температуре от 100 °С
до 105 °С в течение 1 ч. Масса остатка не должна
превышать 1,0 мг.
#Микробиологическая чистота [2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре от 8 °С до 15 °С.
07/2016:2234
МЕРОПЕНЕМ ТРИГИДРАТ
Мегорепетит НМус/псит
МЕКОРЕЫЕМ ТЕ1НУОЕАТЕ
С17Н25Ы3055 • ЗН20 М.м. 437,5
[119478-56-7]
ОПРЕДЕЛЕНИЕ
(4/?,55,65)-3-[[(35,5$)-5-[(Диметиламино)карбо-
нил]пирролидин-3-ил]сульфанил]-6-[(1 Я)-1 -гидрокси-
этил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-
карбоновая кислота тригидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации, или синтетический продукт.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или светло-желтый кристаллический по¬
рошок.
Умеренно растворим в воде, практически нераст¬
ворим в 96 % спирте и метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО меропенема.
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в 20 мл раствора 50 г/л натрия гидрокарбона¬
та Р.
Прозрачность [2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)5.
рН (2.2.3). От 4,0 до 6,0. 0,20 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят этим же растворителем до объ¬
ема 20 мл.
Удельное оптическое вращение (2.2.7). От -17
до -21 (в пересчете на безводное вещество). 0,125 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография [2.2.29). Испытуемые растворы (а) и (Ь)
и раствор сравнения (с) готовят непосредственно
перед использованием. Раствор сравнения (а) гото¬
вят и хранят при температуре 4 °С и используют в
течение 6 ч.
Смесь растворителей. К 1,0 мл триэтилами-
на Р прибавляют 900 мл воды для хроматографии Р.
Полученный раствор доводят кислотой фосфорной
разведенной Р до рН 5,0 и затем разводят водой для
хроматографии Р до объема 1000,0 мл.
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Меропенем тригидрат
655
Испытуемый раствор (Ь). 50,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Для приготовления при¬
месей А и В т-зИи 10 мл испытуемого раствора (а) на¬
гревают до температуры 60 °С в течение около 20 мин
или в качестве альтернативы выдерживают 10 мл ис¬
пытуемого раствора (а) при температуре окружающей
среды в течение 8 ч.
Раствор сравнения (с). 50,0 мг ФСО меропенема
тригидрата растворяют в подвижной фазе и доводят
до объема 100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 40 °С;
-подвижная фаза: ацетонитрил Р1 — смесь
растворителей (7:100, об/об);
- скорость подвижной фазы: 1,6 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (а) и (Ь);
- время хроматографирования: 4-кратное вре¬
мя удерживания меропенема.
Относительное удерживание (по отношению к
меропенему, время удерживания — около 7 мин): при¬
месь А— около 0,5, примесь В — около 2,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5,0 между пиками при¬
меси А и меропенема.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А— 1,6):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси А, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси В, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пиков
примесей А и В, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей, кроме примесей А и В (не бо¬
лее 0,3 %): на хроматограмме испытуемого раствора
(а) сумма площадей всех пиков, кроме основного и пи¬
ков примесей А и В, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Для меропенема тригидрата, полученного пу¬
тем полного синтеза:
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пиков
примесей А и В, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 0,50 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 11,4% и не более
13,4 %. Определение проводят из 0,100 г испытуемо¬
го образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
0,125 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С17Н25Ы3055 в процентах рассчиты¬
вают с учетом содержания меропенема тригидрата в
ФСО меропенема тригидрата.
ХРАНЕНИЕ
Если субстанция стерильная, ее хранят в сте¬
рильном воздухонепроницаемом контейнере с кон¬
тролем первого вскрытия.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. (4Я,55)-5-[(15,2Я)-1-Карбокси-2-гидроксипро-
пил]-3-[[(3$,5$)-5-[(диметиламино)карбонил]пирролидин-
3-ил]сульфанил]-4-метил-4,5-дигидро-1Н-пиррол-2-
карбоновая кислота.
84*. Зак. 1060.
656
Государственная фармакопея Республики Беларусь
В. (4/?,55,65)-3-[[(35,55)-1-[(25,ЗК)-2-[(25,ЗК)-5-
Карбокси-4-[[(35,55)-5-[(диметиламино)карбонил]-
пирролидин-3-ил]сульфанил]-3-метил-2,3-дигидро-1Н-
пиррол-2-ил]-3-гидроксибутаноил]-5-[(диметиламино)-
карбонил]пирролидин-3-ил]сульфанил]-6-[(1 К)-1 -
гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-
2-ен-2-карбоновая кислота.
07/2016:0096
МЕРКАПТОПУРИН
Мегсар1ориппит
МЕЯСАРторитыЕ
5Н
[6112-76-1]
/
N
Н20
М.м. 170,2
ОПРЕДЕЛЕНИЕ
7Н-Пурин-6-тиол.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Желтый кристаллический порошок.
Практически нерастворим в воде, мало раство¬
рим в 96 % спирте. Растворяется в растворах гидрок¬
сидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20 мг испытуемого образ¬
ца растворяют в 5 мл диметилсульфоксида Р и дово¬
дят 0,1 М раствором кислоты хлористоводородной
до объема 100 мл. 5 мл полученного раствора дово¬
дят 0,1 М раствором кислоты хлористоводородной
до объема 200 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения, только один максимум
поглощения при 325 нм.
B. 20 мг испытуемого образца растворяют в 20 мл
96 % спирта Р, нагретого до температуры 60 °С, и
прибавляют 1 мл насыщенного раствора ртути аце¬
тата Рв96% спирте Р. Образуется белый осадок.
C. 20 г испытуемого образца растворяют в 20 мл
96 % спирта Р, нагретого до температуры 60 °С, и
прибавляют 1 мл раствора 10 г/л свинца ацетата Р в
96 % спирте Р. Образуется желтый осадок.
ИСПЫТАНИЯ
Примесь А. Не более 2,0 %. Тонкослойная хро¬
матография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 1 мл диметилсульфоксида Р и
доводят метанолом Р до объема 10 мл.
Раствор сравнения. 10 мг гипоксантина Р раст¬
воряют в 10 мл диметилсульфоксида Р и доводят ме¬
танолом Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
6р254 р.
Подвижная фаза: аммиака раствор концентри¬
рованный Р — вода Р — ацетон Р (3:7:90, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- примесь А: на хроматограмме испытуемо¬
го раствора пятно, соответствующее гипоксантину,
должно быть не интенсивнее пятна на хроматограмме
раствора сравнения.
Вода (2.5.12). Не менее 10,0% и не более
12,0 %. Определение проводят из 0,250 г испытуемо¬
го образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 50 мл
диметилформамида Р и титруют 0,1 М раствором
тетрабутиламмония гидроксида потенциометриче-
ски (2.2.20).
1 мл 0,1 М раствора тетрабутиламмония ги¬
дроксида соответствует 15,22 мг С5Н4М43.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
о
/>
А. 1,7-Дигидро-6Н-пурин-6-он (гипоксантин).
07/2016:2077
МЕТАКРЕЗОЛ
Ме(асгезо1ит
МЕТАСКЕ801.
С7Н„0 М.м. 108,1
[108-39-4]
Метакрезол
657
ОПРЕДЕЛЕНИЕ
З-Метилфенол.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная или желтоватая жидкость.
Умеренно растворим в воде, смешивается с 96 %
спиртом и с метиленхлоридом.
Относительная плотность: около 1,03.
Температура плавления: около 11 °С.
Температура кипения: около 202 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр метакрезола по
Европейской Фармакопее #или спектр, представлен¬
ный на рисунке #2077.-1#.
ИСПЫТАНИЯ
Раствор 3. 1,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Свежеприготовленный
раствор 5 по степени мутности не должен превышать
эталон III.
Цветность (2.2.2, метод II). Окраска свежепри¬
готовленного раствора 8 должна быть не интенсивнее
эталона ВУ(КЖ)7.
Кислотность. К 25 мл раствора 8 прибавляют
0,15 мл раствора метилового красного Р. Раствор
должен быть красным. При прибавлении не более
0,5 мл 0,01 М раствора натрия гидроксида должно
появиться желтое окрашивание.
Сопутствующие примеси. Газовая хромато¬
графия (2.2.28): определение проводят методом вну¬
тренней нормализации.
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения (а). 0,10 г крезола Р, 0,10 г
п-крезола Р и 0,10 г испытуемого образца растворяют
в метаноле Р и доводят до объема 20,0 мл этим же
растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
1.0 мл полученного раствора доводят метанолом Р
до объема 20,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 25 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли[(цианопропил) (метил)][(фен ил) (метил)]силок-
сана Р (толщина слоя 0,2 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,8 мл/мин;
- деление потока: 1:30;
- температура:
Время (мин)
Температура (°С)
Колонка
0—35
100
35—40
100 —И 50
40—50
150
Блок ввода проб
200
Детектор
200
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1,0 мкл.
Относительное удерживание (по отношению к
метакрезолу, время удерживания — около 28 мин):
примесь В — около 0,75; примесь С — около 0,98.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,4 между пиками при¬
меси С и метакрезола.
85. Зак. 1060.
658
Государственная фармакопея Республики Беларусь
Предельное содержание примесей:
- примеси В и С: не более 0,5 %;
- любая другая примесь: не более 0,1 %;
- сумма примесей: не более 1,0 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади пика на хроматограмме
раствора сравнения (Ь).
Остаток после выпаривания. Не более 0,1 %.
2,0 г испытуемого образца выпаривают на водяной
бане в вытяжном шкафу и сушат при температуре
от 100 °С до 105 °С в течение 1 ч. Масса остатка не
должна превышать 2 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, О, Е, Р, С, Н,
/, */, К, Ц М.
A. Р2 = КЗ =К4 = К5 = Кб = Н: фенол.
B. К2 = СН3, КЗ = К4 = К5 = Кб = Н: 2-Метилфе-
нол (о-крезол, крезол).
C. К2 = КЗ = К5 = Кб = Н, К4 = СН3: 4-Метилфе-
нол (л-крезол).
й. К2 = Кб = СН3, КЗ = К4 = К5 = Н: 2,6-Диметил-
фенол (2,6-ксиленол).
Е. К2 = С2Н5, КЗ = К4 = К5 = Кб = Н: 2-Этилфенол
(о-этилфенол).
Р. К2 = К4 = СН3, КЗ = К5 = Кб = Н: 2,4-Диметил-
фенол (2,4-ксиленол).
<3. К2 = К5 = СН3, КЗ = К4 = Кб = Н: 2,5-Диметил-
фенол (2,5-ксиленол).
H. К2 = СН(СН3)2, КЗ = К4 = К5 = Кб = Н: 2-(1-Ме¬
ти лэти л )фенол.
I. К2 = КЗ = СН3, К4 = К5 = Кб = Н: 2,3-Диметил-
фенол (2,3-ксиленол).
3. К2 = К4 = Кб = Н, КЗ = К5 = СН3: 3,5-Диметил-
фенол (3,5-ксиленол).
К. К2 = КЗ = К5 = Кб = Н, К4 = С2Н5:4-Этилфенол
(л-этилфенол).
I. К2 = К5 = Кб = Н, КЗ = К4 = СН3: 3,4-Диметил-
фенол (3,4-ксиленол).
М. К2 = КЗ = К5 = СН3, К4 = Кб = Н: 2,3,5-Триме-
тилфенол.
07/2016:1128
МЕТАКРИЛОВОЙ КИСЛОТЫ И
ЭТИЛАКРИЛАТА СОПОЛИМЕР (1:1)
А с/с// теМасгуНа е^ е(НуНз асгуШИз
ро1утепза1ит 1:1
МЕТНА СЯУЫС А СЮ — ЕТНУ1.
АСКПАТЕ СОР01УМЕК (1:1)
ОПРЕДЕЛЕНИЕ
Сополимер метакриловой кислоты и этилакрила-
та со средней относительной молекулярной массой
около 250 000. Отношение карбоксильных и сложно¬
эфирных групп составляет приблизительно 1:1. По¬
лучают в виде кислотной формы (тип А) и кислотной
формы, частично нейтрализованной натрия гидрок¬
сидом (тип В). Может содержать подходящие поверх¬
ностно-активные вещества, такие как натрия доде-
цилсульфат и полисорбат 80.
Содержание:
- тип А: не менее 46,0 % и не более 50,6 % еди¬
ниц метакриловой кислоты (в пересчете на сухое ве¬
щество);
- тип В: не менее 43,0 % и не более 48,0 % еди¬
ниц метакриловой кислоты (в пересчете на сухое ве¬
щество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый легко сыпучий порошок.
Практически нерастворим в воде (тип А) или дис¬
пергируется в воде (тип В), легко растворим в этано¬
ле, практически нерастворим в этилацетате. Легко
растворяется в растворе 40 г/л натрия гидроксида.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 0,1 г испытуемого образца раст¬
воряют в 1 мл спирта (90 %, об/об) Р. 2 капли полу¬
ченного раствора помещают на пластинку натрия хло¬
рида, высушивают до получения пленки и накрывают
другой пластинкой натрия хлорида.
Сравнение: ФСО сополимера (1:1) метакрило¬
вой кислоты и этилакрилата (тип А или тип В).
B. Испытуемый образец выдерживает требова¬
ния, указанные в разделе «Количественное опреде¬
ление».
C. Испытуемый образец выдерживает испыта¬
ние «Сульфатная зола», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Вязкость (2.2.10).
- Тип А: от 100 мПа сдо 200 мПа с.
37,5 г испытуемого образца, в пересчете на су¬
хое вещество, растворяют в смеси из 7,9 г воды Р и
254,6 г 2-пропанола Р. Определяют вязкость, исполь¬
зуя ротационный вискозиметр при температуре 20 °С
и скорости сдвига 10с1.
- Тип В: не более 100 мПа-с.
80,0 г испытуемого образца, в пересчете на су¬
хое вещество, диспергируют в воде Р и доводят массу
раствора до 320 г этим же растворителем. Перемеши¬
вают в течение 3 ч и определяют вязкость, используя
ротационный вискозиметр при температуре 23 °С и
скорости вращения шпинделя 100 об/мин.
Метакриловой кислоты и этилакрилата сополимер (1:1), 30% дисперсия
659
Размеры шпинделя: диаметр — 47,0 мм; высо¬
та — 27,0 мм, диаметр стержня — 3,18 мм.
Внешний вид пленки. 1 мл раствора (тип А) или
дисперсии (тип В) испытуемого образца, приготовлен¬
ного как указано в испытании «Вязкость», наносят на
стеклянную пластину и дают высохнуть. Образуется
прозрачная хрупкая пленка.
Этилакрилат и метакриловая кислота. Жид¬
костная хроматография (2.2.29).
Контрольный раствор. К 50,0 мл метанола Р
прибавляют 25,0 мл подвижной фазы.
Испытуемый раствор. 40 мг испытуемого об¬
разца растворяют в 50,0 мл метанола Р и прибавля¬
ют 25,0 мл подвижной фазы.
Раствор сравнения. По 10 мг этилакрилата Р и
метакриловой кислоты Р растворяют в метаноле Р
и доводят до объема 50,0 мл этим же растворителем.
0,1 мл полученного раствора доводят метанолом Р до
объема 50,0 мл и прибавляют 25,0 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилсилильным
для хроматографии Р с размером частиц 5 мкм;
-подвижная фаза: метанол Р — фосфатный
буферный раствор рН 2,0 Р (30:70, об/об);
- скорость подвижной фазы: 2,5 мл/мин;
-спектрофотометрический детектор, длина
волны 202 нм;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками эти¬
лакрилата и метакриловой кислоты на хроматограмме
раствора сравнения;
-на хроматограмме контрольного раствора не
должны обнаруживаться пики с временами удержива¬
ния такими же, как у пиков этилакрилата и метакрило¬
вой кислоты.
Предельное содержание примесей:
-суммарное содержание этилакрилата и ме¬
такриловой кислоты: не более 0,1 %.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 6 ч.
Сульфатная зола (2.4.14). Не более 0,4 % (тип
А); не менее 0,5 % и не более 3,0 % (тип В). Опреде¬
ление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют в сме¬
си из 40 мл воды Р и 60 мл 2-пропанола Р. Медленно
титруют при постоянном перемешивании 0,5 М рас¬
твором натрия гидроксида. В качестве индикатора
используют раствор фенолфталеина Р.
1 мл 0,5 М раствора натрия гидроксида соот¬
ветствует 43,05 мг С4Н602 (единиц метакриловой кис¬
лоты).
МАРКИРОВКА
Указывают:
- тип сополимера (тип А или тип В).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомогатель¬
ного вещества (см. статью 5.15). Некоторые из ха¬
рактеристик, описанных в разделе «Функционально-
обусловленные характеристики», могут также при¬
сутствовать в части фармакопейной статьи, обяза¬
тельной для выполнения, так как одновременно они
являются и показателями качества. В таком случае
в разделе «Функционально-обусловленные характери¬
стики» дается перекрестная ссылка на это испыта¬
ние в части фармакопейной статьи, обязательной
для выполнения. Контроль данных характеристик
может вносить вклад в качество лекарственного
средства путем достижения постоянства произ¬
водственного процесса и улучшения свойств лекар¬
ственного средства при его использовании. Предла¬
гаемые методы испытаний являются подходящими
для указанных целей, однако другие методы также
могут быть использованы. В случае указания резуль¬
татов конкретных характеристик требуется указы¬
вать также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для сополимера (1:1) метакриловой кислоты и
этилакрилата, используемого в качестве качестве
компонента оболочки, обусловливающего устойчи¬
вость к воздействию желудочного сока.
Вязкость (см. раздел «Испытания»).
Внешний вид пленки (см. раздел «Испытания»).
Растворимость пленки. Кусочек пленки, полу¬
ченной как указано в испытании «Внешний вид плен¬
ки», помещают в колбу, содержащую 0,1 М раствор
кислоты хлористоводородной, и перемешивают.
Пленка не должна растворяться в течение 2 ч. Другой
кусочек этой же пленки помещают в колбу с фосфат¬
ным буферным раствором рН 6,0 Р и перемешивают.
В течение 1 ч пленка должна раствориться.
07/2016:1129
МЕТАКРИЛОВОЙ КИСЛОТЫ И
ЭТИЛАКРИЛАТА СОПОЛИМЕР (1:1),
30% ДИСПЕРСИЯ
АасИ теМасгуНа е1 еИпуНз асгу/аИз
ро1утепзаИ 1:1 сИзрегзю 30 рег сеп1ит
МЕТНАСРУЫС АСЮ — ЕТНУ1.
АСРУ1-АТЕ С0Р01.УМЕК (1:1)
018РЕР8ЮЫ 30 РЕК СЕЫТ
ОПРЕДЕЛЕНИЕ
Дисперсия в воде сополимера метакриловой
кислоты и этилакрилата со средней относительной
молекулярной массой около 250 000. Соотношение
карбоксильных и сложноэфирных групп составляет
приблизительно 1:1.
Содержание: не менее 46,0 % (м/м) и не более
50,6 % (м/м) единиц метакриловой кислоты (в пере¬
счете на остаток после выпаривания).
Может содержать подходящие поверхностно-ак¬
тивные вещества, такие как натрия додецилсульфат
и полисорбат 80.
ОПИСАНИЕ (СВОЙСТВА)
Непрозрачная белая или почти белая слегка вяз¬
кая жидкость.
85*. Зак. 1060.
660
Гэсударственная фармакопея Республики Беларусь
Смешивается с водой. При прибавлении таких
растворителей, как ацетон, этанол или 2-пропанол,
образуется осадок, который растворяется при прибав¬
лении избыточного количества растворителя. Смеши¬
вается с раствором 40 г/л натрия гидроксида.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр 30 % дисперсии
сополимера (1:1) метакриловой кислоты и этила-
крилата по Европейской Фармакопее.
B. Испытуемый образец выдерживает требова¬
ния, указанные в разделе «Количественное опреде¬
ление».
ИСПЫТАНИЯ
Вязкость (2.2.10). Не более 15 мПас. Опреде¬
ление проводят, используя ротационный вискозиметр
при температуре 20 °С и скорости сдвига 50 с1.
Внешний вид пленки. 1 мл испытуемого образ¬
ца наносят на стеклянную пластину и дают высохнуть.
Должна образоваться прозрачная хрупкая пленка.
Макрочастицы. 100,0 г испытуемого образца
фильтруют через предварительно взвешенное сито
(90) из нержавеющей стали. Отмывают водой Р до
получения прозрачного фильтрата и сушат при темпе¬
ратуре от 100 °С до 105 °С. Масса остатка не должна
превышать 1,00 г.
Этилакрилат и метакриловая кислота. Жид¬
костная хроматография (2.2.29).
Контрольный раствор. К 50,0 мл метанола Р
прибавляют 25,0 мл подвижной фазы.
Испытуемый раствор. 40 мг испытуемого об¬
разца растворяют в 50,0 мл метанола Р и прибавля¬
ют 25,0 мл подвижной фазы.
Раствор сравнения. По 10 мг этилакрилата Р и
метакриловой кислоты Р растворяют в метаноле Р
и доводят до объема 50,0 мл этим же растворителем.
0,1 мл полученного раствора доводят метанолом Р
до объема 50,0 мл и прибавляют 25,0 мл подвижной
фазы.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: метанол Р — фосфатный
буферный раствор рН 2,0 Р (30:70, об/об);
- скорость подвижной фазы: 2,5 мл/мин;
-спектрофотометрический детектор, длина
волны 202 нм;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы:
- разрешение: не менее 2 между пиками этила¬
крилата и метакриловой кислоты на хроматограмме
раствора сравнения;
-на хроматограмме контрольного раствора не
должны обнаруживаться пики со временами удержи¬
вания пиков этилакрилата и метакриловой кислоты.
Предельное содержание примесей:
-суммарное содержание этилакрилата и ме¬
такриловой кислоты: не более 0,1 %.
Остаток после выпаривания. Не менее 28,5 %
и не более 31,5 %. 1,000 г испытуемого образца сушат
при температуре 110 °С в течение 5 ч. Масса сухого
остатка должна составлять не менее 0,285 г и не бо¬
лее 0,315 г.
Сульфатная зола (2.4.14). Не более 0,2°:
Определение проводят из 1,0 г испытуемого образца
Микробиологическая чистота
ОКА: критерий приемлемости: 103 КОЕ/г (2.6.12)
ОКГ: критерий приемлемости: 102 КОЕ/г (2.6.12)
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,500 г испытуемого образца растворяют в сме¬
си из 40 мл воды Р и 60 мл 2-пропанола Р. Медленно
титруют при постоянном перемешивании 0,5 М рас¬
твором натрия гидроксида. В качестве индикатора
используют раствор фенолфталеина Р.
1 мл 0,5 М раствора натрия гидроксида соот¬
ветствует 43,05 мг 04НбО2 (единиц метакриловой кис¬
лоты).
ХРАНЕНИЕ
При температуре от 5 °С до 25 °С. Не заморажи¬
вать.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация с
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Некоторые из характеристик, описанных в раз¬
деле «Функционально-обусловленные характери¬
стики», могут также присутствовать в части
фармакопейной статьи, обязательной для выпол¬
нения, так как одновременно они являются и по¬
казателями качества. В таком случае в разделе
«Функционально-обусловленные характеристики»
дается перекрестная ссылка на это испытание
в части фармакопейной статьи, обязательной
для выполнения. Контроль данных характеристик
может вносить вклад в качество лекарственного
средства путем достижения постоянства про¬
изводственного процесса и улучшения свойств
лекарственного средства при его использовании.
Предлагаемые методы испытаний являются под¬
ходящими для указанных целей, однако другие ме¬
тоды также могут быть использованы. В случае
указания результатов конкретных характери¬
стик требуется указывать также и методы про¬
ведения испытаний.
Следующие характеристики могут быть
важны для 30 % дисперсии сополимера (1:1) мета¬
криловой кислоты и этилакрилата, используемой
в качестве компонента оболочки, обусловливаю¬
щего устойчивость к воздействию желудочного
сока.
Вязкость (см. раздел «Испытания»).
Внешний вид пленки (см. раздел «Испыта¬
ния»).
Растворимость пленки. Кусочек пленки, по¬
лученной как указано в испытании «Внешний вид
пленки», помещают при перемешивании в колбу
содержащую раствор 10,3 г/л кислоты хлористо¬
водородной Р. Пленка не должна растворяться
в течение 2 ч. Другой кусочек этой же пленки по¬
мещают при перемешивании в колбу, содержащую
фосфатный буферный раствор рН 6,0 Р. В тече¬
ние 1 ч пленка должна раствориться.
Метамизол натрия моногидрат
661
07/2016:1346
МЕТАМИЗОЛ НАТРИЯ МОНОГИДРАТ
Ме^ат^IоIит паНсит топоЬус/псит
МЕТАМ1201-Е ЗООШМ МОИОНУОКАТЕ
С13Н16М3Ыа045 ■ Н20 М.м. 351,4
[5907-38-0]
ОПРЕДЕЛЕНИЕ
Натрия [(1,5-диметил-3-оксо-2-фенил-2,3-дигидро-
1 Н-пиразол-4-ил)-Л/-метиламино]метансульфонат мо¬
ногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Очень легко растворим в воде, растворим в 96 %
спирте, практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификациям, О.
Вторая идентификация: В, С, О.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО метамизола натрия #или
спектр, представленный на рисунке #1346.-1#.
B. 50 мг испытуемого образца растворяют в 1 мл
раствора водорода пероксида концентрированно¬
го Р. Появляется синее окрашивание, которое быстро
тускнеет и в течение нескольких минут переходит в
интенсивное красное окрашивание.
C. 0,10 г испытуемого образца помещают в
пробирку, прибавляют несколько стеклянных бусин
и растворяют в 1,5 мл воды Р Прибавляют 1,5 мл
кислоты хлористоводородной разведенной Р и
помещают на открытом конце пробирки фильтро¬
вальную бумагу, смоченную раствором 20 мг калия
йодата Р в 2 мл раствора крахмала Р, и осторож¬
но нагревают. Выделяющиеся пары серы диоксида
окрашивают фильтровальную бумагу в синий цвет.
После осторожного нагревания в течение 1 мин в от¬
верстие пробирки помещают стеклянную палочку с
каплей раствора 10 г/л натриевой соли хромотро-
повой кислоты Р в кислоте серной Р. В течение
10 мин в капле реактива появляется сине-фиолето¬
вое окрашивание.
О. 0,5 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», дают реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 40 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод /). Окраска свежеприго¬
товленного раствора 3 должна быть не интенсивнее
эталона ВУ(КЖ)6.
Кислотность или щелочность. К 5 мл раствора
3 прибавляют 0,1 мл раствора фенолфталеина Р1.
Раствор должен быть бесцветным. При прибавлении
не более 0,1 мл 0,02 М раствора натрия гидроксида
должно появиться розовое окрашивание.
Рисунок #1346.-1. Инфракрасный спектр ФСО метамизола натрия
662
Гэсударственная фармакопея Республики Беларусь
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29). Растворы готовят непосредственно
перед использованием.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО метамизола
примеси А растворяют в метаноле Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравнения
(а) доводят подвижной фазой до объема 50,0 мл. 1,0 мл
полученного раствора используют для растворения со¬
держимого флакона с ФСО метамизола примеси Е.
Раствор сравнения (с). Для получения примеси С
/л зКи 40 мг испытуемого образца растворяют в метано¬
ле Р, доводят до объема 20 мл этим же растворителем
и кипятят с обратным холодильником в течение 10 мин.
Охлаждают до комнатной температуры и доводят мета¬
нолом Рцо объема 20 мл.
Раствор сравнения (ф. 1,0 мл раствора сравнения
(а) доводят метанолом Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,05 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии эндкепированным Р с разме¬
ром частиц 1,8 мкм;
- подвижная фаза: смесь из 28 объемов метано¬
ла Р и 78 объемов буферного раствора, приготовленного
следующим образом: смешивают 1000 объемов раст¬
вора 6,0 г/л натрия дигидрофосфата Р и 1 объем три-
этиламина Р и доводят до рН 7,0 раствором натрия
гидроксида концентрированным Р;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
-объем вводимой пробы: по Юмкл испытуемого
раствора и растворов сравнения (Ь), (с) Ь (б);
- время хроматографирования: 4,5-кратное время
удерживания метамизола.
Идентификация пиков примесей: идентифицируют
пики примесей А и Е, используя хроматограмму раствора
сравнения (Ь); идентифицируют пик примеси С, исполь¬
зуя хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению к ме-
тамизолу, время удерживания — около 2 мин): примесь
А—около 0,7; примесь Е — около 0,8; примесь С — око¬
ло 2,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 3,0 (Нр
— высота пика примеси А относительно базовой линии;
Ну—расстояние между базовой линией и нижней точкой
кривой, разделяющей пик примеси А и пик примеси Е).
Предельное содержание примесей (для расчета со¬
держания примесей умножают площади пиков на соот¬
ветствующие поправочные коэффициенты: для примеси
Е —1,5):
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси С, не должна превышать 5-кратную площадь ос¬
новного пика на хроматограмме раствора сравнения (б);
-примесь Е (не более 0,15%): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси Е, не должна превышать 1,5-кратную площадь
основного пика на хроматограмме раствора сравнения (с!);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей С
и Е, не должна превышать 0,5 площади основного пика
на хроматограмме раствора сравнения (б);
-сумма примесей (не более 0,5%): на хромато¬
грамме испытуемого раствора сумма площадей всех пи¬
ков, кроме основного, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения (б);
-неучитываемый предел (0,03%): на хромато¬
грамме испытуемого раствора не учитывают пики с пло¬
щадью менее 0,3 площади основного пика на хромато¬
грамме раствора сравнения (б).
Сульфаты (2.4.13). Не более 0,1 %. 0,150 г испыту¬
емого образца растворяют в воде дистиллированной Р
и доводят до объема 15 мл этим же растворителем.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020% (20ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл свежеприготовленного раствора
должны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раствора
свинца (2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не ме¬
нее 4,9 % и не более 5,3 %. 1,000 г испытуемого образца
сушат при температуре 105 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требования
статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 10 мл
0,01 М раствора кислоты хлористоводородной,
предварительно охлажденной в ледяной бане, и не¬
медленно титруют 0,05 М раствором йода по каплям,
растворяя образующийся осадок перемешиванием
раствора после каждого прибавления титранта. В
конце титрования прибавляют 2 мл раствора крахма¬
ла Р и титруют до тех пор, пока синяя окраска раст¬
вора не будет сохраняться в течение не менее 2 мин.
Температура раствора во время титрования не долж¬
на превышать 10 °С.
1мл 0,05 М раствора йода соответствует
16,67 мг С13Н161Ч3№045.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацевти¬
ческого использования (2034). Вследствие этого нет
необходимости идентифицировать эти примеси для
доказательства соответствия требованиям. См. так¬
же статью 5.10. Контроль примесей в субстанциях
для фармацевтического использования): А, В, О.
А. 4-(Формиламино)-1,5-диметил-2-фенил-2,3-
дигидро-1 Н- пиразол-3-он.
Метилпарагидроксибензоат
663
07/2016:0409
МЕТИЛПАРАГИДРОКСИБЕНЗОАТ
МеМуНз рагаЬудгохуЬепгоаз
МЕТНП. РАКАНУОКОХУВЕИЮАТЕ
В. 4-Амино-1,5-диметил-2-фенил-2,3-дигидро-1Н-
пиразол-3-он.
С8Н803
[99-76-3]
М.м. 152,1
С. 1,5-Диметил-4-(метиламино)-2-фенил-2,3-
дигидро-1 Н-пиразол-З-он.
О. 1,5-Диметил-4-(диметиламино)-2-фенил-2,3-
дигидро-1 Н-пиразол-З-он.
Е. [(1,5-Диметил-3-оксо-2-фенил-2,3-дигидро-1 Н-
пиразол-4-ил)амино]метансульфоновая кислота
(4-Л/-деметилметамизол).
ОПРЕДЕЛЕНИЕ
Метил-4-гидроксибензоат. #Метилпарабен, нипа-
гин#.
Содержание: не менее 98,0 % и не более 102,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень мало растворим в воде, легко растворим в
96 % спирте и метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Температура плавления {2.2.14): от 125 °С до
128 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО метилпарагидроксибензоата
#или спектр, представленный на рисунке #0409.-1
C. Тонкослойная хроматография (2.2.27).
Рисунок #0409.-1. Инфракрасный спектр ФСО метилпарагидроксибензоата
664
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в ацетоне Р и доводят до объ¬
ема 10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО метилпара-
гидроксибензоата растворяют в ацетоне Р и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО этилпара-
гидроксибензоата растворяют в 1 мл испытуемого
раствора (а) и доводят ацетоном Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р — метанол Р (1:30:70, об/об/об).
Наносимый объем пробы: 2 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 96% спирте Р и доводят до объема 10 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность. К 2 мл раствора 3 прибавляют
3 мл 96 % спирта Р, 5 мл воды, свободной от угле¬
рода диоксида, Р и 0,1 мл раствора бромкрезолового
зеленого Р. При прибавлении не более 0,1 мл 0,1 М
раствора натрия гидроксида Р должно появиться
синее окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл метанола Р и доводят
подвижной фазой до объема 50,0 мл. 10,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Раствор сравнения (а). 5 мг кислоты 4-гидрок-
сибензойной Р (примесь А) и 5 мг испытуемого образ¬
ца растворяют в подвижной фазе и доводят до объ¬
ема 100,0 мл этим же растворителем. 1,0 мл получен¬
ного раствора доводят подвижной фазой до объема
10.0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО метилпара-
гидроксибензоата растворяют в 2,5 мл метанола Р и
доводят подвижной фазой до объема 50,0 мл. 10,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: раствор 6,8 г/л калия диги¬
дрофосфата Р — метанол Р (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а) и (с);
- время хроматографирования: 5-кратное вре¬
мя удерживания метилпарагидроксибензоата.
Относительное удерживание (по отношению к
метилпарагидроксибензоату, время удерживания —
около 2,3 мин): примесь А— около 0,6.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 2,0 между пиками при¬
меси А и метилпарагидроксибензоата.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А на 1,4):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения
(с);
- неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (с);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (с).
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С8Н803 в процентах рассчитывают
с учетом содержания метилпарагидроксибензоата в
ФСО метилпарагидроксибензоата.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
Метилтиониния хлорид
665
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
А. 4-Гидроксибензойная кислота.
С. Пропил-4-гидроксибензоат (пропилпарагидрок-
сибензоат).
О. Бутил-4-гидроксибензоат (бутилпарагидрокси-
бензоат).
07/2016:1132
МЕТИЛТИОНИНИЯ ХЛОРИД
МеМуНЫоптН сЫопс1ит
(#Ме01у1епит соеги1еит)
МЕТНПТНЮШЫШМ СШОШОЕ
СН3 СН3
С16Н18С1Ы3§ - хН20 М.м. 319,9 (безводный)
ОПРЕДЕЛЕНИЕ
3,7-Бис(диметиламино)фенотиазин-5-илия хло¬
рид (метиленовый синий).
Содержание: не менее 93,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Темно-синий или темно-зеленый кристалличе¬
ский порошок с металлическим блеском. Гигроскопи¬
чен.
Мало растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в кислоте хлористоводородной
разведенной Р и доводят до объема 100 мл этим же
растворителем. 5 мл полученного раствора доводят
кислотой хлористоводородной разведенной Р до
объема 100 мл.
Диапазон длин волн: от 240 нм до 800 нм.
Максимумы поглощения: при (255—260) нм,
(285—290) нм, (670—680) нм и (740—750) нм.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем. 1 мл полученного раст¬
вора доводят метанолом Р до объема 10 мл.
Раствор сравнения. 10 мг ФСО метилтиониния
хлорида растворяют в метаноле Р и доводят до объ¬
ема 10 мл этим же растворителем. 1 мл полученного
раствора доводят до объема 10 мл метанолом Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — пропанол Р (20:80, об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 1/2 высоты
пластинки.
Высушивание: на воздухе в защищенном от све¬
та месте.
Проявление: пластинку просматривают при днев¬
ном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения. На обе¬
их хроматограммах допускается наличие вторичного
пятна выше основного пятна.
C. 50 мг испытуемого образца прокаливают с
0,5 г натрия карбоната безводного Р. После охлаж¬
дения остаток растворяют в 10 мл азотной кислоты
разведенной Р и фильтруют. Фильтрат, без дальней¬
шего прибавления кислоты азотной разведенной Р,
дает реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р — под¬
вижная фаза А (30:70, об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этим же растворителем. Обраба¬
тывают ультразвуком в течение 5 мин.
Раствор сравнения (а). 50,0 мг ФСО метилти¬
ониния хлорида растворяют в смеси растворителей и
доводят до объема 50,0 мл этим же растворителем.
Обрабатывают ультразвуком в течение 5 мин.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл.
Раствор сравнения (с). 10 мг ФСО метилтиони¬
ния для проверки пригодности хроматографической
системы (содержит примесь А) растворяют в смеси
растворителей и доводят до объема 10,0 мл этим же
растворителем.
Раствор сравнения (д). 1,0 мл раствора срав¬
нения (Ь) доводят смесью растворителей до объема
10.0 мл.
Условия хроматографирования:
666
Государственная фармакопея Республики Беларусь
-колонка длиной 0,10м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилси-
лильным для хроматографии Р с размером частиц
3,5 мкм;
- подвижная фаза:
- подвижная фаза А: 0,1 % (об/об) раствор кис¬
лоты трифторуксусной Р;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
80
20
5—25
80 — 30
о
г-
т
о
см
25—32
30
70
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 246 нм;
- объем вводимой пробы: по 5 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (с!).
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму, при¬
лагаемую к ФСО метилтиониния для проверки при¬
годности хроматографической системы, и хрома¬
тограмму раствора сравнения (с).
Относительное удерживание (по отношению к
метилтионинию, время удерживания — около 11 мин):
примесь А — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 3,5 между пиком приме¬
си В и метилтионинием.
Относительное удерживание (по отношению к
метилтионинию, время удерживания — около 11 мин):
примесь А — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 1,5 между пиком при¬
меси А и пиком метилтиониния; при необходимости
изменяют концентрацию ацетонитрила в подвижной
фазе.
Расчет процентных содержаний:
-для примеси А — используют концентрацию
метилтиониния в растворе сравнения (Ь);
- для каждой примеси, кроме примеси А — ис¬
пользуют концентрацию метилтиониния в растворе
сравнения (с1).
Предельное содержание примесей:
- примесь А: не более 5,0 %;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей, кроме примеси А: не более
0,5 %;
- учитываемый предел: 0,05 %.
Металлы. Атомно-эмиссионная спектроме¬
трия (2.2.22) в плазме аргона, в качестве детектора
используют стандартную оптическую систему или
масс-спектрометр. В случае использования масс-
спектрометра в качестве внутреннего стандарта ис¬
пользуют индий.
Испытуемый раствор. В мерной колбе вмести¬
мостью 10 мл растворяют при перемешивании 100 мг
испытуемого образца в 9 мл воды Р, прибавляют
100,0 мкл раствора 10 мкг/мл индия, приготовленного
из индия исходного эталонного раствора для атом¬
ной спектрометрии (1,000 г/л) Р в азотной кисло¬
те Р, разведенного в пятьдесят раз водой Р. Полу¬
ченный раствор доводят водой Р до объема 10,0 мл.
Раствор сравнения. В мерную колбу вместимо¬
стью 100 мл вносят 10,0 мл эталонного раствора, при¬
готовленного из исходных эталонных растворов для
атомной спектрометрии (1,000 г/л) Р соответству¬
ющих элементов и разведенного водой Р, содержа¬
щего 1,00 мкг/мл каждого определяемого элемента. К
полученному раствору прибавляют 1,00 мл раствора
10 мкг/мл индия, приготовленного из индия исходного
эталонного раствора для атомной спектрометрии
(1,000 г/л) Р в азотной кислоте Р, разведенного в
пятьдесят раз водой Р. Полученный раствор доводят
водой Р до объема 100,0 мл.
Холостой раствор. Раствор 10 мкг/мл индия, ис¬
пользуемый для приготовления испытуемого раствора
и раствора сравнения, разбавляют в сто раз водой Р.
Оптический детектор
Масс-детектор
Элемент
Сигнал
(нм)
Фон 1
(нм)
Фон 2
(нм)
Изотоп
Алюминий
396,15
396,05
396,25
27
Железо
238,20
238,27
238,14
*
Кадмий
214,44
214,37
214,51
114
Марганец
260,57
260,50
260,64
55
Медь
327,40
327,31
327,48
65
Молибден
202,03
202,02
202,04
95
Никель
231,60
231,54
231,66
60
Олово
190,00**
189,90
190,10
118
Ртуть
253,70***
253,60
253,80
200
Свинец
217,00**
216,90
217,10
208
Хром
283,56
283,49
283,64
*
Цинк
213,86
213,80
213,91
66
Индий
115
* Трудноопределяемый или неопределяемый элемент при
использовании масс-спектрометра в качестве детектора.
** Предельная чувствительность при оптическом детекти¬
ровании.
*** Ртуть зачастую невозможно определить, используя
стандартный оптический детектор, в таком случае исполь¬
зуют устройство для определения гидридов.
Элемент
Предельно-допустимое содержание
(ррт)
Алюминий
100
Железо
200
Кадмий
1
Марганец
10
Медь
300
Молибден
10
Никель
10
Олово
10
Ртуть
1
Свинец
10
Хром
100
Цинк
100
Потеря в массе при высушивании (2.2.32). Не
менее 8,0 % и не более 24,0 %. 1,000 г испытуемого
образца сушат при температуре 105 °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,25%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Метилцеллюлоза
667
Ч
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемого
раствора и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- фактор асимметрии: не более 3,0 для основ¬
ного пика.
Содержание С18Н18С1М33 в процентах рассчиты¬
вают с учетом содержания метилтиониния хлорида в
ФСО метилтиониния хлорида.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре не выше
30 °С.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С.
А. 3-(Диметиламино)-7-(метиламино)фенотиазиН'
5-илий (азур В).
сн3
В. 3-Амино-7-(диметиламино)фенотиазин-5-илий
(азур А).
С. 3-Амино-7-(метиламино)фенотиазин-5-илий
(азур С).
07/2016:0345
МЕТИЛЦЕЛЛЮЛОЗА
Ме1Ьу1се11и1озит
МЕТНП.СЕШЛ.ОЗЕ
[9004-67-5]
ОПРЕДЕЛЕНИЕ
Частично О-метилированная целлюлоза. Мети¬
ловый эфир целлюлозы.
Содержание: не менее 26,0 % и не более 33,0 %
метокси-групп (-ОСН3; М.м. 31,03) (в пересчете на су¬
хое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый, желтовато-белый или серовато-белый
порошок или гранулы. Гигроскопична после высуши¬
вания.
Практически нерастворима в горячей воде, аце¬
тоне, этаноле безводном и толуоле. Растворяется в
холодной воде с образованием коллоидного раствора.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. В лабораторный стакан помещают 100 мл
воды Р, равномерно насыпают на поверхность воды
1,0 г испытуемого образца и выдерживают в течение
(1—2) мин. Порошок агрегирует на поверхности.
B. В лабораторный стакан помещают 100 мл ки¬
пящей воды Р, равномерно насыпают на поверхность
воды 1,0 г испытуемого образца и перемешивают на
магнитной мешалке с перемешивающим элементом
длиной 25 мм. Образуется взвесь, частицы не рас¬
творяются. Полученную взвесь охлаждают до темпе¬
ратуры 5 °С и перемешивают на магнитной мешалке.
Образуется прозрачный или слегка мутный (в зависи¬
мости от вязкости) раствор.
C. К 0,1 мл раствора, полученного в подлинности
В, прибавляют 9 мл 90 % (об/об) кислоты серной Р,
встряхивают, нагревают на водяной бане в течение
ровно 3 мин, быстро охлаждают в ледяной бане, осто¬
рожно прибавляют 0,6 мл раствора 20 г/л нингидри-
на Р, встряхивают и выдерживают при температуре
25 °С. Появляется красное окрашивание, не перехо¬
дящее в красно-фиолетовое в течение 100 мин.
О. (2—3) мл раствора, полученного в подлинно¬
сти В, помещают тонким слоем на стеклянную пла¬
стинку и выдерживают до испарения воды. На поверх¬
ности стеклянной пластинки образуется прозрачная
пленка.
Е. В лабораторный стакан помещают 50,0 мл
воды Р и прибавляют 50,0 мл раствора, полученно¬
го в подлинности В. В полученный раствор помеща¬
ют термометр. Раствор перемешивают на магнитной
мешалке с подогревом и нагревают со скоростью
(2—5) °С/мин. Отмечают температуру, при которой на¬
чинает увеличиваться мутность (температура флоку¬
ляции). Температура флокуляции выше 50 °С.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца (в пере¬
счете на сухое вещество) помещают при перемешива¬
нии в 50 г воды, свободной от углерода диоксида, Р,
нагретой до температуры 90 °С. Охлаждают, доводят
водой, свободной от углерода диоксида, Р до мас¬
сы 100 г и перемешивают до полного растворения.
Перед проведением испытаний «Прозрачность» и
«Цветность» раствор выдерживают при температуре
от 2 °С до 8 °С в течение 1 ч.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон III.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 5,0 до 8,0. Измеряют рН раствора,
приготовленного как указано в испытании «Кажу¬
щаяся вязкость». Значение рН регистрируют через
(5±0,5) мин после погружения электродов в раствор.
Вязкость. Не менее 80 % и не более 120 % от
номинального значения для образцов с вязкостью ме¬
нее 600 мПа-с (метод 1); не менее 75 % и не более
140 % от номинального значения для образцов с вяз¬
костью 600 мПа*с или более (метод 2).
668
Гэсударственная фармакопея Республики Беларусь
Метод 1, применяется для образцов с вязко¬
стью менее бООмПа с. Взвешивают количество ис¬
пытуемого образца, эквивалентное 4,000 г сухого ве¬
щества, переносят в широкогорлую колбу и доводят
массу содержимого горячей водой Р (90—91)°С до
200.0 г. Колбу закрывают, перемешивают содержимое
с помощью механического приспособления со скоро¬
стью (400±50) об/мин в течение (10—20) мин до тех
пор, пока частицы не будут полностью диспергиро¬
ваны и смочены. Убеждаются, что на стенках колбы
не осталось нерастворенных частиц, при необходи¬
мости потирая внутренние стенки колбы шпателем,
и продолжают перемешивание в течение следующих
(20—40) мин при охлаждении в водяной бане с темпе¬
ратурой ниже 5 °С. Полученный раствор при необхо¬
димости доводят холодной водой Р до массы 200,0 г.
При необходимости полученный раствор центрифуги¬
руют для удаления воздушных пузырьков. Появившу¬
юся пену удаляют с помощью шпателя. С помощью
метода капиллярной визкозиметрии (2.2.9) находят
кинематическую вязкость (V) полученного раствора.
Отдельно определяют плотность этого раствора (р)
(2.2.5) и рассчитывают динамическую вязкость по
формуле г} = р-у.
Метод 2, применяется для образцов с вязко¬
стью бООмПас или более. Взвешивают количество
испытуемого образца, эквивалентное 10,00 г сухого
вещества, помещают в широкогорлую колбу и дово¬
дят массу содержимого горячей водой Р (90—91) °С до
500.0 г. Колбу закрывают, перемешивают содержимое
с помощью механического приспособления со скоро¬
стью (400±50) об/мин в течение (10—20) мин до тех
пор, пока частицы не будут полностью диспергированы
и смочены. Убеждаются, что на стенках колбы не оста¬
лось нерастворенных частиц, при необходимости поти¬
рая внутренние стенки колбы шпателем, и продолжают
перемешивание в течение следующих (20—40) мин
при охлаждении в водяной бане с температурой ниже
5 °С. Полученный раствор при необходимости доводят
холодной водой Р до массы 500,0 г. При необходимо¬
сти полученный раствор центрифугируют для удале¬
ния воздушных пузырьков. Появившуюся пену удаля¬
ют с помощью шпателя. Определяют вязкость (2.2.10)
полученного раствора при температуре (20±0,1)°С с
использованием ротационного вискозиметра.
Прибор: шпиндельный вискозиметр одноцилин¬
дрового типа.
Роторное число, скорость вращения и расчет¬
ный коэффициент: используют условия, указанные в
таблице 0345.-1.
Таблица 0345.-1
Номинальная вяз¬
кость*
(мПа-с)
Ротор¬
ное
число
Ско¬
рость
враще¬
ния
(об/мин)
Расчет¬
ный ко¬
эффици¬
ент
От 600 до менее 1400
3
60
20
От 1400 до менее 3500
3
12
100
От 3500 до менее 9500
4
60
100
От 9500 до менее 99 500
4
6
1000
99 500 или выше
4
3
2000
* Номинальная вязкость указывается в спецификациях
производителя.
Перед снятием показания вращают шпиндель в
течение 2 мин. Между двумя последовательными из¬
мерениями делают перерыв 2 мин. Измерение повто¬
ряют еще два раза и определяют среднее значение по
трем полученным показаниям.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Сульфатная зола (2.4.14). Не более 1,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Прибор:
- реакционный сосуд: герметично закрываю¬
щийся сосуд вместимостью 5 мл, высотой 50 мм, с
внешним диаметром 20 мм и внутренним диаметром
горлышка 13 мм с герметично закрывающейся мем¬
бранной крышкой из бутилового каучука, покрытой по¬
литетрафторэтиленом и надежно закрепленной с по¬
мощью обжимного алюминиевого колпачка или иного
приспособления, обеспечивающего достаточную воз¬
духонепроницаемость;
- нагреватель: нагревающий модуль с квадрат¬
ным алюминиевым блоком, имеющим отверстия диа¬
метром 20 мм и глубиной 32 мм, подходящими для
реакционных сосудов; перемешивание содержимого
реакционного сосуда в нагревающем модуле осу¬
ществляется с помощью магнитной мешалки или с
помощью возвратно-поступательного встряхивателя,
производящего около 100 циклов в минуту.
Раствор внутреннего стандарта. Раствор
30 г/л октана Р в о-ксилоле Р.
Испытуемый раствор. 65,0 мг испытуемого об¬
разца помещают в реакционный сосуд, прибавляют
(0,06—0,10) г кислоты адипиновой Р, 2,0 мл раствора
внутреннего стандарта и 2,0 мл кислоты йодистово¬
дородной Р, немедленно герметично укупоривают и
точно взвешивают. Содержимое контейнера переме¬
шивают в течение 60 мин при нагревании блока та¬
ким образом, чтобы температура содержимого была
(130±2)°С. При отсутствии магнитной мешалки или
возвратно-поступательного встряхивателя реакцион¬
ные. сосуды интенсивно встряхивают вручную с пя¬
тиминутными интервалами в течение первых 30 мин
нагревания. Сосуды охлаждают и снова точно взве¬
шивают. Если потеря массы составляет менее 0,50 %
от массы содержимого и если отсутствуют признаки
протекания, верхний слой смеси используют в каче¬
стве испытуемого раствора.
Раствор сравнения. (0,06—0,10) г кислоты ади¬
пиновой Р, 2,0 мл раствора внутреннего стандарта и
2,0 л кислоты йодистоводородной Р помещают в
другой реакционный сосуд, герметично укупоривают и
точно взвешивают. Прокалывая мембрану с помощью
шприца, прибавляют 45 мкл метилйодида Р и точно
взвешивают. Реакционный сосуд встряхивают и ис¬
пользуют верхний слой в качестве раствора сравнения.
Условия хроматографирования:
- колонка длиной (1,8—3) м и внутренним диа¬
метром (3—4) мм, заполненная диатомитом для
О^Метионин
669
газовой хроматографии Р, импрегнированным
(10—20) % поли(диметил)силоксаном Р (толщина
слоя (125—150) мкм);
- температура: 100 °С;
- газ-носитель: гелий для хроматографии Р
или азот для хроматографии Р (пламенно-иониза¬
ционный детектор); гелий для хроматографии Р (де¬
тектор по теплопроводности);
-поток газа-носителя: устанавливают таким
образом, чтобы время удерживания внутреннего
стандарта составляло около 10 мин;
-детектор: пламенно-ионизационный или по
теплопроводности;
- объем вводимой пробы: (1—2) мкл.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: хорошо разделенные пики мети-
лйодида (первый пик) и внутреннего стандарта (вто¬
рой пик).
Рассчитывают отношение (О) площади пика ме-
тилйодида к площади пика внутреннего стандарта на
хроматограмме испытуемого раствора и отношение
(С^) площади пика метил йодида к площади пика вну¬
треннего стандарта на хроматограмме раствора срав¬
нения.
Рассчитывают процентное содержание метокси-
групп по формуле:
0^.21*64.
О, т
где:
ш1 — масса навески метилйодида, взятой для
приготовления раствора сравнения, мг;
т — масса навески испытуемого образца (в пе¬
ресчете на сухое вещество), мг.
МАРКИРОВКА
Указывают вязкость в мПа-с.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испытаний
являются подходящими для указанных целей, од¬
нако другие методы также могут быть использо¬
ваны. В случае указания результатов конкретных
характеристик требуется указывать также и ме¬
тоды проведения испытаний.
Следующие характеристики могут быть важ¬
ны для метилцеллюлозы, используемой в качестве
связующего вещества, увеличивающего вязкость
агента или пленкообразующего вещества.
Вязкость. См. раздел «Испытания».
Степень замещения. См. раздел «Количествен¬
ное определение».
07/2016:0624
ОЫМЕТИОНИН
01--Ме1Ыоп'тит
01.-МЕТН10Ы1ЫЕ
Н3с.
н мн2
со2н
и энантиомер
СД^О.З М.м. 149,2
[59-51-8]
ОПРЕДЕЛЕНИЕ
(2Я5)-2-Амино-4-(метилсульфанил)бутановая
кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Почти белый кристаллический порошок или мел¬
кие хлопья.
Умеренно растворим в воде, очень мало раство¬
рим в 96 % спирте. Растворяется в разведенных кис¬
лотах и разведенных растворах гидроксидов щелоч¬
ных металлов.
Температура плавления: около 270 °С (метод
мгновенного плавления).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец и стандарт¬
ный образец высушивают при температуре 105 °С.
Сравнение: ФСО а-метионина.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
C. 2,50 г испытуемого образца растворяют в 1 М
растворе кислоты хлористоводородной и доводят
до объема 50,0 мл этим же растворителем. Угол опти¬
ческого вращения (2.2.7): от -0,05° до +0,05°.
й. К 0,1 г испытуемого образца прибавляют 0,1 г
глицина Р и растворяют в 4,5 мл раствора натрия
гидроксида разведенного Р. Прибавляют 1 мл раст¬
вора 25 г/л натрия нитропруссида Р и нагревают при
температуре 40 °С в течение 10 мин. Охлаждают и
прибавляют 2 мл смеси из кислоты фосфорной Р и
кислоты хлористоводородной Р (1:9, об/об). Появля¬
ется насыщенное красное окрашивание.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 5,4 до 6,1. Измеряют рН раствора 3.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
670
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор (а). 0,2 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 20 мг ФСО 01,-метионина
растворяют в воде Р и доводят до объема 50 мл этим
же растворителем.
Раствор сравнения (Ь). 1 мл раствора сравнения
(а) доводят водой Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
СР.
Подвижная фаза : кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 °С
до 105 °С в течение 15 мин.
Предельное содержание примесей:
- любая примесь (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды. Не более 0,0200 % (200 ррт). 0,25 г
испытуемого образца растворяют в 35 мл воды Р,
прибавляют 5 мл кислоты азотной разведенной Р,
10 мл раствора серебра нитрата Р2 и выдерживают
в течение 5 мин в защищенном от света месте. Опа¬
лесценция полученного раствора не должна превы¬
шать опалесценцию эталона, приготовленного таким
же образом и в то же время с использованием 10 мл
эталонного раствора хлорида (5 ррт С1) Р и 25 мл
воды Р. Пробирки просматривают на черном фоне го¬
ризонтально (перпендикулярно оси пробирок).
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
1,0 г испытуемого образца растворяют в 20 мл воды
дистиллированной Р при нагревании до температуры
60 °С. Охлаждают до температуры 10 °С и фильтруют.
15 мл полученного раствора должны выдерживать ис¬
пытание на сульфаты.
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,140 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р и прибавляют
30 мл кислоты уксусной безводной Р. После раство¬
рения немедленно титруют 0,1 М раствором кисло¬
ты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 14,92 мг С5Н11М023.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1027
МЕТИОНИН
МеОи'оптит
МЕТНЮШЫЕ
СД^З М.м. 149,2
[63-68-3]
ОПРЕДЕЛЕНИЕ
(25)-2-Амино-4-(метилсульфанил)бутановая кис¬
лота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Растворим в воде, очень мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО метионина #или спектр, пред¬
ставленный на рисунке #1027.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в кислоте хлористоводородной
разведенной Р и доводят до объема 50 мл этим же
растворителем.
Раствор сравнения. 10 мг ФСО метионина
растворяют в кислоте хлористоводородной разве¬
денной Р и доводят до объема 50 мл этим же раст¬
ворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 15 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
О. 0,1 г испытуемого образца и 0,1 г глицина Р
растворяют в 4,5 мл раствора натрия гидроксида
разведенного Р, прибавляют 1 мл раствора 25 г/л
натрия нитропруссида Р, нагревают до температу¬
ры 40 °С в течение 10 мин, охлаждают и прибавляют
2 мл смеси из 1 объема кислоты фосфорной Р и 9
объемов кислоты хлористоводородной Р. Появляет¬
ся темно-красное окрашивание.
Метионин
671
Рисунок #1027.-1. Инфракрасный спектр ФСО метионина
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 5,5 до 6.5. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
+22,5 до +24,0 (в пересчете на сухое вещество). 1,00 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной Р1 и доводят до объема 50,0 мл этим
же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,30 г испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 10 мг метионина сульфоксида Р (примесь А)
растворяют в подвижной фазе А и доводят до объема
10.0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 — раст¬
вор 23 г/л кислоты фосфорной Р — вода для
хроматографии Р (0,5:12,5:87, об/об/об);
- подвижная фаза В: раствор 23 г/л кислоты
фосфорной Р — вода для хроматографии Р —
ацетонитрил Р1 (12,5:40:47,5, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—6
100
0
6—50
100 — 0
0 — 100
50—60
0
100
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 205 нм;
- объем вводимой пробы: 50 мкл;
Относительное удерживание (по отношению к
метионину, время удерживания — около 6 мин): при¬
месь А — около 0,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5,0 между пиками при¬
меси А и метионина.
Расчет процентных содержаний:
- для каждой примеси — используют концентра¬
цию метионина в растворе сравнения (а).
Предельное содержание примесей:
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,3 %;
- учитываемый предел: 0,05 %.
Хлориды. Не более 0,0200 % (200 ррт). 0,5 г ис¬
пытуемого образца растворяют в 5 мл кислоты азот¬
ной разведенной Р и доводят до объема 10 мл этим
же растворителем. Прибавляют 10 мл концентриро¬
ванного раствора водорода пероксида Р и нагрева¬
ют на водяной бане в течение 30 мин. Охлаждают,
доводят водой Р до объема 50 мл, прибавляют 1 мл
раствора серебра нитрата Р2, перемешивают и вы¬
держивают в защищенном от света месте в течение
672
Гэсударственная фармакопея Республики Беларусь
5 мин. Опалесценция испытуемого раствора должна
быть не интенсивнее эталона, приготовленного па¬
раллельно с использованием 2 мл эталонного раст¬
вора хлоридов (50 ррт С1) Р. Пробирки просматрива¬
ют перпендикулярно оси пробирки на черном фоне.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
0,5 г испытуемого образца растворяют в 3 мл кислоты
хлористоводородной разведенной Р и доводят водой
дистиллированной Р до объема 15мл. Полученный
раствор должен выдерживать испытание на сульфаты.
Соли аммония (2.4.1, метод В). Не более
0,0200% (200 ррт). 0,10 г испытуемого образца
должны выдерживать испытание на соли аммония.
Эталон готовят с использованием 0,2 мл эталонного
раствора аммония (100 ррт NН^ Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,125 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 14,92 мг СдН^ИОзЗ.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е.
Н ^Н2
Н’С\д/\^Х. и эпимер у 3
II
О
А. (25)-2-Амино-4-[(Р5)-метилсульфинил]бутано-
вая кислота (Ь-метионин сульфоксид).
Н3С^
А
о о
1\1Н2
С02Н
В. (25)-2-Амино-4-(метилсульфонил)бутановая
кислота.
Н3С-
НзСк
NN и энантиомер
^со2н
С. (2Р5)-2-(Ацетиламино)-4-(метилсульфанил)бу-
тановая кислота.
и эпимер у С*
й. (2Р)-2-[[(2Р5)-2-(Ацетиламино)-4-(метилсульфа-
нил)бутаноил]амино]-4-(метилсульфанил)бутановая
кислота.
Е. (25)-2-[[(2Р$)-2-(Ацетиламино)-4-(метилсульфа-
нил)бутаноил]амино]-4-(метилсульфанил)бутановая
кислота.
07/2016:0674
МЕТОКЛОПРАМИДА ГИДРОХЛОРИД
Ме(ос1оргат1сИ Ьус1госМопс1ит
МЕТОС1.0РКАМЮЕ НУОПОСШ-ОКШЕ
С14Н22С1М302 ■ НС1 ■ НгО М.м. 354,3
[54143-57-6]
ОПРЕДЕЛЕНИЕ
4-Амино-5-хлор-А/-[2-(диэтиламино)этил]-2-метокси-
бензамида гидрохлорид моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или кристаллы.
Очень легко растворим в воде, легко растворим в
96 % спирте, умеренно растворим в метиленхлориде.
Температура плавления: около 183 °С с разло¬
жением.
Метоклопрамида гидрохлорид
673
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: А, С, О, Е.
A. рН (2.2.3): от 4,5 до 6,0. Измеряют рН раст¬
вора 3, приготовленного как указано в разделе «Ис¬
пытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках с калия хлоридом Р.
Сравнение: ФСО метоклопрамида гидрохлори¬
да #или спектр, представленный на рисунке #0674.-1#.
* С. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси», в ультра¬
фиолетовом свете перед опрыскиванием раствором
диметиламинобензальдегида Р1.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
й. 1 мл раствора 3 доводят водой Р до объема
2 мл. Полученный раствор дает реакцию (а) на хло¬
риды (2.3.7).
Е. 2 мг испытуемого образца растворяют в 2 мл
воды Р. Полученный раствор дает реакцию на пер¬
вичные ароматические амины (2.3.7).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.7). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,40 г испытуемого
образца растворяют в метаноле Р и доводят до объ¬
ема 10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят метанолом Р до объема 10 мл.
Раствор сравнения (а). 20 мг ФСО метоклопра¬
мида гидрохлорида растворяют в метаноле Р и дово¬
дят до объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (а) доводят метанолом Р до объема 100 мл.
1 мл полученного раствора доводят метанолом Р до
объема 10 мл.
Раствор сравнения (с). 10 мг Л/,Л/-
диэтилэтилендиамина Р растворяют в метаноле Р
и доводят до объема 50 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля
НР!54Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — диоксан Р — метанол Р — метилен-
хлорид Р (2:10:14:90, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора (а) любое пятно, кроме основного, должно
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения (Ь) (0,5 %).
Проявление В: пластинку опрыскивают раство¬
ром диметиламинобензальдегида Р1 и сушат на воз¬
духе.
Результаты В: на хроматограмме испытуемо¬
го раствора (а) любое пятно, кроме пятен, обнару¬
женных при просматривании в ультрафиолетовом
свете при длине волны 254 нм, должно быть не ин-
Рисунок #0674.-1. Инфракрасный спектр ФСО метоклопрамида гидрохлорида
674
Гэсударственная фармакопея Республики Беларусь
тенсивнее пятна на хроматограмме раствора срав¬
нения (с) (0,5 %).
Тяжелые металлы {2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2рртРЬ)Р.
Вода {2.5.12). Не менее 4,5 % и не более 5,5 %.
Определение проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,2500 г испытуемого образца растворяют в
смеси из 5,0 мл 0,01 М раствора хлористоводород¬
ной кислоты и 50 мл 96 % спирта Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 33,63 мг С14Н22С1М302*НС1.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1028
МЕТОПРОЛОЛА ТАРТРАТ
Ме1орго!оИ 1аг1газ
МЕТ0РК01-01- ТАРТРАТЕ
(С15Н25М03)2 ■ С4Н606 М.м. 685
[56392-17-7]
ОПРЕДЕЛЕНИЕ
Бис[(2Я5)-1 -[4-(2-метоксиэтил )фенокси]-3-[(1 -ме-
тилэтил)амино]пропан-2-ол] (2Я,ЗЯ)-2,3-дигидрокси-
бутандиоат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
В. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО метопролола тартрата #или
спектр, представленный на рисунке #1028.-1#.
Если спектры, полученные с использовани¬
ем образцов в твердом состоянии, различаются,
то отдельно готовят растворы 100 г/л испытуемого
образца и ФСО метопролола тартрата в мети-
ленхлориде Р. По 25 мкл полученных растворов на¬
носят на поверхность диска калия бромида Р, вы¬
паривают растворитель и немедленно записывают
новые спектры.
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р
и доводят до объема 25,0 мл этим же растворите¬
лем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)8.
рН (2.2.3). От 6,0 до 7,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От +7,0
до +10,0 (в пересчете на сухое вещество). Определя¬
ют удельное оптическое вращение раствора 3.
Примеси М, Ы, О. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 1 мл испытуемого раст¬
вора доводят метанолом Р до объема 20 мл. 5 мл по¬
лученного раствора доводят метанолом Р до объема
50 мл.
Раствор сравнения (Ь). 4 мл раствора сравнения
(а) доводят метанолом Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: метанол Р — этилацетат Р
(20:80, об/об). В хроматографическую камеру с под¬
вижной фазой помещают два лабораторных стакана,
каждый из которых содержит по 30 объемов раствора
аммиака концентрированного Р. Насыщают камеру в
течение не менее 1 ч.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе в течение не менее 3 ч.
Проявление: пластинку обрабатывают парами
йода в течение не менее 15 ч.
Предельное содержание примесей: _
- любая примесь: на хроматограмме испытуемо¬
го раствора любое пятно, кроме основного, должно
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения (а) (0,5 %) и не более одного из таких
пятен может быть интенсивнее пятна на хроматограм¬
ме раствора сравнения (Ь) (0,2 %).
На хроматограмме испытуемого раствора не учи¬
тывают пятна на линии старта.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 1,5 мг ФСО метопроло¬
ла примеси А и 2,5 мг ФСО метопролола тартрата
Метопролола тартрат
675
Рисунок #1028.-1. Инфракрасный спектр ФСО метопролола тартрата
растворяют в подвижной фазе и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 50,0 мл.
Условия хроматографирования:
-колонка длиной 0,15 м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецил-
силильным для хроматографии эндкепированным Р
(размер частиц 5 мкм);
- подвижная фаза: 3,9 г аммония ацетата Р
растворяют в 810 мл воды Р, прибавляют 2,0 мл три-
этиламина Р, 10,0 мл кислоты уксусной ледяной Р,
3.0 мл кислоты фосфорной Р, 146 мл ацетонитри¬
ла Р и перемешивают;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания метопролола.
Относительное удерживание (по отношению
к метопрололу, время удерживания — около 7 мин):
примесь Н — около 0,3; примесь С — около 0,4; при¬
месь О — около 0,45; примесь Р — около 0,7; при¬
месь А— около 0,8; примесь ^ — около 1,4; примесь
О — около 1,6; примесь Е — около 1,8; примесь В —
около 2.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 6,0 между пиками при¬
меси А и метопролола.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 0,1):
- примеси А, В, С, О, Е, Р, С, Н, 3 (не более
0,3 %): на хроматограмме испытуемого раствора
площади пиков, соответствующих примесям А, В, С,
О, Е, Р, О, Н и ^ не должны превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, й, Е, Р, (3, Н и ^, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь).
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (Ь), а также пик
винной кислоты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (Юррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл воды Р. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5%. 1,000 г испытуемого образца сушат в
вакууме над кальция хлоридом безводным Р в тече¬
ние 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
676
Государственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворят в 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 34,24 мг (С15Н25М03)2-С4Н606.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р,
О, Н, 3.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования); М, А/, О.
А. (2/?5)-1 -(Этиламино)-3-[4-(2-метоксиэтил)фе-
нокси]пропан-2-ол.
он
н3с.
О'
В. 4-(2-Метоксиэтил)фенол.
и энантиомер СН3
ОН<
С. 4-[(2#5)-2-Гидрокси-3-[( 1 -метилэтил)амино]про-
покси]бензальдегид.
н рн
У /ОН
н3с^
и энантиомер
О. (2/?5)-3-[4-(2-Метоксиэтил)фенокси]пропан-1,2-
диол.
н он
сн3 сн3
и энантиомер
Е. (2Я5)-1-[2-(2-Метоксиэтил)фенокси]-3-[(1-
метилэтил)амино]пропан-2-ол.
н рн
сн3
сн3
и энантиомер
Р. (2Я5)-1 -[(1-Метилэтил)амино]-3-феноксипропан-
2-ол.
он
НО'
С. 2-(4-Гидроксифенил)этанол.
И. (2Я5)-1 -[4-(2-Гидроксиэтил)фенокси]-3-[(1 -
метилэтил)амино]пропан-2-ол.
н он
н он
Н3СО'
„СН3
и энантиомер СН3
и его эпимер в положении С*
и их энантиомеры
3. Смесь 4 стереоизомеров 1-[2-гидрокси-3-[(1-
метилэтил)амино]пропокси]-3-[4-(2-метоксиэтил)фе-
нокси]пропан-2-ола.
сн3 сн3
М. 1,3-Бис[(1-метилэтил)амино]пропан-2-ол.
сн3
и энантиомер
N. (2Я?5)-3-[(1-Метилэтил)амино]пропан-1,2-диол.
О. Смесь 3 стереоизомеров 1,1-[(1-метилэтил)-
имино]бис[3-[4-(2-метоксиэтил)фенокси]пропан-2-ола].
07/2016:0560
МЕТОТРЕКСАТ
Ме1Шгеха1ит
МЕТНОТПЕХАТЕ
С20Н22М8О5 М.М. 454>4
[59-05-2]
ОПРЕДЕЛЕНИЕ
(25)-2-[[4-[[(2,4-Диаминоптеридин-6-ил)метил]-
метиламино]бензоил]амино]пентандионовая кислота.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый или оранжевый кристаллический поро¬
шок. Гигроскопичен.
Практически нерастворим в воде, в 96 %
спирте и в метиленхлориде. Растворяется в раз-
Метотрексат
677
веденных минеральных кислотах и разведенных
растворах гидроксидов щелочных металлов и кар¬
бонатов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО метотрексата.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 40,0 мг испытуе¬
мого образца растворяют в смеси из 0,5 мл раст¬
вора аммиака разведенного Р1 и 5 мл подвижной
фазы А и доводят подвижной фазой А до объема
100.0 мл.
Испытуемый раствор (Ь). 25,0 мг испытуемо¬
го образца растворяют в смеси из 0,5 мл раствора
аммиака разведенного Р1 и 5 мл подвижной фазы
А и доводят подвижной фазой А до объема 50,0 мл.
5.0 мл полученного раствора доводят подвижной
фазой А до объема 50,0 мл.
Раствор сравнения (а). 25,0 мг ФСО мето¬
трексата растворяют в смеси из 0,5 мл раствора
аммиака разведенного Р1 и 5 мл подвижной фазы
А и доводят подвижной фазой А до объема 50,0 мл.
5.0 мл полученного раствора доводят подвижной
фазой А до объема 50,0 мл.
Раствор сравнения (Ь). 5,0 мл испытуемого
раствора (а) доводят подвижной фазой А до объ¬
ема 100,0 мл. 5,0 мл полученного раствора доводят
подвижной фазой А до объема 50,0 мл.
Раствор сравнения (с). 5,0 мл раствора срав¬
нения (Ь) доводят подвижной фазой А до объема
25.0 мл.
Раствор сравнения (д). 5 мг испытуемого об¬
разца, 5 мг 4-аминофолиевой кислоты Р (примесь
В), 5 мг ФСО метотрексата примеси С, 5 мг ФСО
метотрексата примеси О и 5 мг ФСО метотрек¬
сата примеси Е растворяют в смеси из 0,5 мл
раствора аммиака разведенного Р1 и 5 мл подвиж¬
ной фазы А и доводят подвижной фазой А до объ¬
ема 100 мл.
Раствор сравнения (е). 8 мг ФСО метотрек¬
сата для идентификации пиков (содержит приме¬
си Н и I) растворяют в смеси из 0,1 мл раствора
аммиака разведенного Р1 и 1 мл подвижной фазы
А и доводят подвижной фазой А до объема 20 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,0 мм, заполненная силикагелем октаде-
цилсилильным эндкепированным для хроматогра¬
фии Р (размер частиц 5 мкм);
- подвижная фаза:
- подвижная фаза А: смешивают 5 объемов
ацетонитрила для хроматографии Р и 95
объемов раствора 3,4 г/л натрия дигидрофос¬
фата безводного Р, предварительно доведен¬
ного раствором 42 г/л натрия гидроксида Р до
рН 6,0;
- подвижная фаза В: смешивают 50 объемов
ацетонитрила для хроматографии Р и 50
объемов раствора 3,4 г/л натрия дигидрофос¬
фата безводного Р, предварительно доведен¬
ного раствором 42 г/л натрия гидроксида Р до
рН 6,0;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100
0
10—20
о
0
1
СО
сл
0—>5
20—28
95—>50
5—>50
28—37
50
50
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (Ь), (с), (с!) и (е).
Идентификация пиков примесей: идентифици¬
руют пики примесей Н и I, используя хроматограмму,
прилагаемую к ФСО метотрексата для идентифи¬
кации пиков, и хроматограмму раствора сравнения
(е); идентифицируют пики примесей В, С и Е, исполь¬
зуя хроматограмму раствора сравнения (д).
Относительное удерживание (по отношению к
метотрексату, время удерживания — около 18 мин):
примесь В — около 0,3; примесь С — около 0,4; при¬
месь О — около 0,9; примесь Е — около 1,4; примесь
I — около 1,5; примесь Н — около 1,6.
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками при¬
месей В и С и не менее 1,5 между пиками примеси Р
и метотрексата на хроматограмме раствора сравне¬
ния (б); не менее 1,5 между пиками примесей I и Н на
хроматограмме раствора сравнения (е); если разре¬
шение между пиками примеси О и метотрексата не со¬
ответствует, увеличивают скорость подвижной фазы.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Е — 0,8, для примеси I — 1,4):
- примесь С (не более 0,5 %): на хроматограм¬
ме испытуемого раствора (а) площадь пика, соот¬
ветствующего примеси С, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В и Е (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям В и Е, не должны превы¬
шать 0,6 площади основного пика на хроматограмме
раствора сравнения (Ь);
- примеси Н и I (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) площади пиков, со¬
ответствующих примесям Н и I, не должны превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (с);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков при¬
месей В, Е, С, Н и I, не должна превышать 0,5 площа¬
ди основного пика на хроматограмме раствора срав¬
нения (с);
- сумма примесей, кроме примесей В, С и Е (не
более 0,5 %): на хроматограмме испытуемого раст¬
вора (а) сумма площадей всех пиков, кроме основно¬
го и пиков примесей В, С и Е, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (с).
Энантиомерная чистота. Жидкостная хромато¬
графия (2.2.29).
678
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 4,0 мг ФСО метотрек¬
сата для проверки пригодности хроматографиче¬
ской системы (содержит примесь Р) (Р5) растворяют
в подвижной фазе и доводят до объема 20,0 мл этим
же растворителем.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем для хромато¬
графии Р со связанным альбумином бычьим Р (раз¬
мер частиц 7 мкм) с размером пор 30 нм;
-подвижная фаза: 500мл раствора 7,1 г/л ди¬
натрия гидрофосфата безводного Р прибавляют
к 600 мл раствора 6,9 г/л натрия дигидрофосфата
моногидрата Р, перемешивают и доводят раствором
натрия гидроксида разведенного Р до рН 6,9; к 920 мл
полученной смеси прибавляют 80 мл пропанола Р]
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 302 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пик примеси Р, используя хроматограмму раст¬
вора сравнения (Ь).
Относительное удерживание (по отношению
к метотрексату, время удерживания — около 4 мин):
примесь Р — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками мето¬
трексата и примеси Р.
Предельное содержание примесей:
- примесь Р (не более 3,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Р, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0050 % (50 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 5 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 13,0%. Определение
проводят из 0,10 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (а).
Содержание С20Н22М8О5 в процентах рассчитыва¬
ют с учетом содержания метотрексата в ФСО мето¬
трексата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С, Е, Р, Н, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемлемо¬
сти для других/неспецифицированных примесей и/
или общей статьей Субстанции для фармацевти¬
ческого использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): А,
О, 6, 3, К, Ь.
А. (2,4-Диаминоптеридин-6-ил)метанол.
В. Р1 = Ж2, К2 = РЗ = Р4 = Н: (25)-2-[[4-[[(2,4-Диа-
миноптеридин-6-ил)метил]амино]бензоил]амино]пен-
тандионовая кислота (4-аминофолиевая кислота, ами-
ноптерин).
H. Р1 = 1ЧН2, Р2 = К4 = СН3, КЗ = Н: (25)-2-[[4-[[(2,4-
Диаминоптеридин-6-ил)метил]метиламино]бензоил]-
амино]-5-метокси-5-оксопентановая кислота (5-мети-
ловый эфир метотрексата).
I. К1 = К1Н,, Р2 = КЗ = СН3, Р4 = Н: (45)-4-[[4-[[(2,4-
Диаминоптеридин-6-ил)метил]метиламино]бензоил]-
амино]-5-метокси-5-оксопентановая кислота (1-мети¬
ловый эфир метотрексата).
3. ГС1 = 1ЧН2, Р2 = КЗ = П4 = СН3: Диметил-(25)-2-
[[4-[[(2,4-диаминоптеридин-6-ил)метил]метиламино]-
бензоил]амино]пентандиоат (диметиловый эфир мето¬
трексата).
С. (25)-2-[[4-[[(2-Амино-4-оксо-1,4-дигидропте-
ридин-6-ил)метил]метиламино]бензоил]амино]пентан-
дионовая кислота (Л/-метилфолиевая кислота, метоп-
терин).
О. 4-[[(2-Амино-4-оксо-1,4-дигидроптеридин-6-ил)-
метил]метиламино]бензойная кислота (Л/™-метилпте-
роевая кислота).
Метронидазол
679
Е. 4-[[(2,4-Диаминоптеридин-6-ил)метил]метила-
мино]бензойная кислота (4-амино-Л/*°-метилптероевая
кислота, АРА).
Р. (2Я)-2-[[4-[[(2,4-Диаминоптеридин-6-ил)метил]-
метиламино]бензоил]амино]пентандионовая кислота
((Я)-метотрексат).
6. (25)-2-[[4-[[4-[[(2,4-Диаминоптеридин-6-ил)ме-
тил]метиламино]бензоил]метиламино]бензоил]амино]-
пентандионовая кислота.
К. К = Н: (25)-2-[(4-Аминобензоил)амино]пентан-
дионовая кислота.
1~ Я = СН3: (25)-2-[[4-(Метиламино)бензоил]ами-
но]пентандионовая кислота.
07/2016:0675
МЕТРОНИДАЗОЛ
Ме&отс1а1о1ит
МЕТРЮЫЮА2.01Е
С6Н9Ы30з М.м. 171,2
[443-48-1]
ОПРЕДЕЛЕНИЕ
2-(2-Метил-5-нитро-1 Н-имидазол-1 -ил)этанол.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтоватый кристаллический порошок.
Мало растворим в воде, в ацетоне, в 96 % спирте
и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 159 °С до
163 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в 0,1 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем. 5,0 мл полученного раствора дово¬
дят 0,1 М раствором кислоты хлористоводородной
до объема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 277 нм.
Минимум поглощения: при 240 мн.
Удельный показатель поглощения в максимуме:
от 365 до 395.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО метронидазола #или спектр,
представленный на рисунке #0675.-1#.
й. К 10 мг испытуемого образца прибавляют
10 мг порошка цинка Р, 1 мл воды Р, 0,25 мл кисло¬
ты хлористоводородной разведенной Р, нагревают
на водяной бане в течение 5 мин и охлаждают. Полу¬
ченный раствор дает реакцию на первичные аромати¬
ческие амины (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в 1 М растворе кислоты хлористоводородной
и доводят до объема 20 мл этим же растворителем.
Прозрачность. (2.2.1). Раствор 5 по степени
мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона СУ(ЗЖ)6.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы готовят с защитой от
света.
Испытуемый раствор. 0,05 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО метронида¬
зола примеси А растворяют в подвижной фазе, при¬
бавляют 10,0 мл испытуемого раствора и доводят
подвижной фазой до объема 100,0 мл. 1,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: метанол Р— раствор 1,36 г/л
калия дигидрофосфата Р (30:70, об/об);
- скорость подвижной фазы: 1 мл/мин;
680
Гэсударственная фармакопея Республики Беларусь
Рисунок #0675.-1. Инфракрасный спектр ФСО метронидазола
-спектрофотометрический детектор, длина
волны 315 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания метронидазола.
Относительное удерживание (по отношению к
метронидазолу, время удерживания — около 7 мин):
примесь А — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками ме¬
тронидазола и примеси А.
Предельное содержание примесей:
-любая примесь (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площадь любого
пика, кроме основного, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,01 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 17,12 мг С6Н9Ы303.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Я4
A. Р1 = Р4 = Н, Р2 = СН3, РЗ = И02: 2-Метил-4-
нитроимидазол.
B. Р1 = Р2 = Р4 = Н, КЗ = 1\Ю2:4-Нитроимидазол.
C. Р1 = СН2-СН2-ОН, Р2 = Р4 = Н, РЗ = N0,,:
2-(4- Н итро-1Н- и м ид азол-1 -и л )этанол.
O. Р1 = СН2-СН2-ОН, Р2 = РЗ = Н, Р4 = 1Ч02:
2-(5-Нитро-1 Я-имидазол-1 -ил) этанол.
Е. Р1 = СН2-СН2-ОН, Р2 = СН3, РЗ = Ы02, Р4 = Н:
2-(2-Метил-4-нитро-1 Я-имидазол-1 -ил)этанол.
P. Р1 = СН2-СН2-0-СН2-СН2-0Н, Р2 = СН3, РЗ = Н,
Р4 = 1\Ю2:2-[2-(2-Метил-5-нитро-1 Я-имидазол-1 -ил Это¬
кси] этанол.
О. Р1= СН2-С02Н, Р2 = СН3, РЗ = Н, Р4 = N0,,:
2-(2-Метил-5-нитро-1 Я-имидазол-1 -ил)уксусная кис¬
лота.
Метронидазола бензоат
681
07/2016:0934
МЕТРОНИДАЗОЛА БЕНЗОАТ
Ме&от'с1а2оН Ьепгоаз
МЕТКОЫЮА201.Е ВЕЫ20АТЕ
С13Н13М304 М.м. 275,3
[13182-89-3]
ОПРЕДЕЛЕНИЕ
2-(2-Метил-5-нитро-1 Н-имидазол-1 -ил)этилбен-
зоат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый кристаллический
порошок или хлопья.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, растворим в ацетоне, мало
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 99 °С до
102 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в растворе 103 г/л кислоты хлори¬
стоводородной Р и доводят до объема 100,0 мл этим
же растворителем. 1,0 мл полученного раствора до¬
водят раствором 103 г/л кислоты хлористоводород¬
ной Р до объема 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимумы поглощения: при 232 нм и при
275 нм.
Удельный показатель поглощения в максимуме
при длине волны 232 нм: от 525 до 575.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр метронидазола
бензоата по Европейской Фармакопее #или спектр,
представленный на рисунке #0934.-1#.
Р. К 10 мг испытуемого образца прибавляют око¬
ло 10 мг цинка порошка Р, 1 мл воды Р и 0,3 мл кис¬
лоты хлористоводородной Р, нагревают на водяной
бане в течение 5 мин и охлаждают. Полученный раст¬
вор дает реакцию на первичные ароматические ами¬
ны (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в диметилформамиде Р и доводят этим же
растворителем до объема 10 мл.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ОУ(ЗЖ)3.
Кислотность. 2,0 г испытуемого образца раст¬
воряют в смеси из 20 мл диметилформамида Р и
20 мл воды Р, предварительно нейтрализованной
0,02 М раствором кислоты хлористоводородной
или 0,02 М раствором натрия гидроксида с исполь¬
зованием 0,2 мл раствора метилового красного Р.
Рисунок #0934.-1. Инфракрасный спектр метронидазола бензоата
682
Гэсударственная фармакопея Республики Беларусь
При прибавлении не более 0,25 мл 0,02 М раствора
натрия гидроксида должна измениться окраска инди¬
катора.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Подвижная фаза В —
подвижная фаза А (45:55, об/об).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл
Раствор сравнения (Ь). 5,0 мг ФСО метрони-
дазола (примесь А), 5,0 мг ФСО метронидазола при¬
меси А (примесь В) и 5,0 мг ФСО кислоты бензойной
(примесь С) растворяют в смеси растворителей и до¬
водят до объема 50,0 мл этой же смесью раствори¬
телей. 1,0 мл полученного раствора доводят смесью
растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
диизобутилоктадецилсилильным для хроматогра¬
фии Р (размер частиц 5 мкм) с удельной площадью
поверхности 180 м2/г, размером пор 8 нм и содержа¬
нием углерода 10 %;
- подвижная фаза:
- подвижная фаза А: раствор 1,5 г/л калия ди¬
гидрофосфата Р, доведенный до рН 3,2 кисло¬
той фосфорной Р)
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
80
20
5—15
80 —* 55
20 —► 45
15-40
55
45
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
метронидазола бензоату, время удерживания — око¬
ло 20 мин): примесь В — около 0,17; примесь А— око¬
ло 0,20; примесь С — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
месей А и В.
Предельное содержание примесей:
- примеси А, В и С (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, В и С, не должны превы¬
шать площади соответствующих пиков на хромато¬
грамме раствора сравнения (Ь);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В и С, не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,01 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020% (20 ррт). 1,0 г испытуемого раствора дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 80 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 27,53 мг С13Н13Н304.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
о?ы
он
А. 2-(2-Метил-5-нитро-1 Я-имидазол-1-ил)этанол
(метронидазол).
02М
В. 2-Метил-4-нитроимидазол.
со2н
С. Бензол карбоновая кислота (бензойная кислота).
07/2016:0931
МЕТФОРМИНА ГИДРОХЛОРИД
МеНогт'т'1 ЬудгосЫопдит
МЕТРОКМШ НУОКОСН1.0МОЕ
СЛЛ-НС! М.м. 165,6
[1115-70-4]
Метформина гидрохлорид
683
ОПРЕДЕЛЕНИЕ
1,1 -Диметилбигуанида гидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте, практически нерастворим в ацетоне и в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. Температура плавления (2.2.14): от 222 °С до
226 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО метформина гидрохлорида
#или спектр, представленный на рисунке #0931.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения. 20 мг ФСО метформина ги¬
дрохлорида растворяют в воде Р и доводят до объема
5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р.
Подвижная фаза: верхний слой смеси кислота
уксусная ледяная Р — бутанол Р — вода Р (10:40:50,
об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: при температуре от 100 °С до
105 °С в течение 15 мин.
Проявление: пластинку опрыскивают смесью из
равных объемов раствора 100 г/л натрия нитропрус-
сидаР, раствора 100 г/л калия феррицианида Р и
раствора 100 г/л натрия гидроксида Р (приготовлен¬
ный раствор выдерживают в течение 20 мин).
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. 5 мг испытуемого образца растворяют в
воде Р и доводят до объема 100 мл этим же раство¬
рителем. К 2 мл полученного раствора прибавляют
0,25 мл раствора натрия гидроксида концентри¬
рованного Р, 0,10 мл раствора а-нафтола Р, пере¬
мешивают и выдерживают в ледяной воде в течение
15 мин. Прибавляют 0,5 мл раствора натрия гипо-
бромита Р и перемешивают. Появляется розовое
окрашивание.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 подогревают до
50 °С и охлаждают до комнатной температуры. Полу¬
ченный раствор должен быть прозрачным.
Цветность (2.2.2, метод II). Раствор 3 подогре¬
вают до 50 °С и охлаждают до комнатной температу¬
ры. Полученный раствор должен быть бесцветным.
Примесь Р. Не более 0,05 %. Жидкостная хрома¬
тография (2.2.29).
Раствор для дериватизации. Раствор готовят
непосредственно перед использованием. 1 мл фтор-
динитробензола Р растворяют в 100,0 мл ацетони¬
трила для хроматографии Р.
Рисунок #0931 .-1. Инфракрасный спектр ФСО метформина гидрохлорида
86*. Зак. 1060.
684
Гэсударственная фармакопея Республики Беларусь
Холостой раствор. К 5,0 мл ацетонитрила для
хроматографии Р прибавляют 100 мкл триэтилами-
наР1 и 1,0 мл раствора для дериватизации. Хорошо
встряхивают и греют при температуре 60 °С в течение
30 мин. После охлаждения доводят ацетонитрилом
для хроматографии Р до объема 10,0 мл.
Испытуемый раствор. Раствор готовят непо¬
средственно перед использованием. 10,0 мг испыту¬
емого образца суспендируют в 5,0 мл ацетонитрила
для хроматографии Р, выдерживают в ультразвуко¬
вой ванне в течение 5 мин, прибавляют 100 мкл три-
этиламина Р1 и 1,0 мл раствора для дериватизации.
Хорошо встряхивают и греют при температуре 60 °С
в течение 30 мин. После охлаждения доводят ацето¬
нитрилом для хроматографии Р до объема 10,0 мл.
Перед использованием фильтруют или центрифугиру¬
ют при 800 д в течение 5 мин.
Раствор сравнения. 1,0 мл ФСО метформина
примеси Р растворяют в 100,0 мл ацетонитрила
для хроматографии Р. 2,5 мл полученного раствора
доводят ацетонитрилом для хроматографии Р до
объема 100,0 мл. К 1,0 мл полученного раствора по¬
следовательно прибавляют 5,0 мл ацетонитрила
для хроматографии Р, 100 мкл триэтиламина Р1 и
1,0 мл раствора для дериватизации. Хорошо встряхи¬
вают и греют при температуре 60 °С в течение 30 мин.
После охлаждения доводят ацетонитрилом для хро¬
матографии Р до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 3 мм, заполненная сферическим силикагелем
октадецилсилильным для хроматографии эндкепи-
рованным Р1 (размер частиц 5 мкм);
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: кислота фосфорная Р —
вода Р (0,1:99,9, об/об);
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
60 -* 45
40 —> 55
10—11
45 —> 25
55 —> 75
11—15
25
75
- скорость подвижной фазы: 0,7 мл/мин;
- спектрофотометрический детектор, длина
волны 380 нм;
- объем вводимой пробы: 5 мкл.
Идентификация пиков примесей: идентифици¬
руют пик производного примеси Р, используя хрома¬
тограммы холостого раствора и раствора сравнения.
Время удерживания: производное примеси Р —
около 4 мин.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 3,0 между пиком произ¬
водного примеси Р и ближайшими пиками деривати-
зационного реактива.
Предельное содержание примеси:
- примесь Р: на хроматограмме испытуемого раст¬
вора площадь пика, соответствующего производному
примеси Р, не должна превышать площадь соответству¬
ющего пика на хроматограмме раствора сравнения.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 20,0 мг ФСО метформи¬
на примеси А растворяют в воде Р и доводят до объ¬
ема 100,0 мл этим же растворителем. 1,0 мл получен¬
ного раствора доводят подвижной фазой до объема
200.0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (с). 10 мг меламина Р (при¬
месь Э) растворяют в 90 мл воды Р, прибавляют 5 мл
испытуемого раствора и доводят водой Р до объема
100 мл. 1 мл полученного раствора доводят подвиж¬
ной фазой до объема 50 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная катионо-обменным силь¬
нокислотным силикагелем для хроматографии Р
(размер частиц 10 мкм);
- подвижная фаза: раствор 17 г/л аммония диги¬
дрофосфата Р, доведенный кислотой фосфорной Р
до рН 3,0;
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 218 нм;
- объем вводимой пробы. 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания метформина.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (а).
Относительное удерживание (по отношению к
метформину, время удерживания — около 10 мин):
примесь А — около 0,2; примесь й — около 0,3.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 10 между пиками приме¬
си О и метформина.
Предельное содержание примесей:
- примесь А (не более 0,02 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси А, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,2 %);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (Ь); учитывают пик
примеси А.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Миконазола нитрат
685
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворят в 4 мл
кислоты муравьиной безводной Р, прибавляют 80 мл
ацетонитрила Р и немедленно титруют 0,1 Мраство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,56 мг С4Н„Ы5 НС\.
ПРИМЕСИ
Специфицированные примеси: А, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Е.
А. Цианогуанидин.
B. В = 1\1Н-С(=МН)-МН2: (4,6-Диамино-1,3,5-триазин-
2-ил)гуанидин.
C. Р = М(СН3)2: А/,Л/-Диметил-1,3,5-триазин-2,4,6-
триамин (А/,А/-диметилмеламин).
й. Р = Ш2:1 Д5-Триазин-2,4,6-триамин (меламин).
Е. 1-Метилбигуанид.
Р. СН3-1ЧН-СН3: АРМетилметанамин (диметиламин).
07/2016:0513
МИКОНАЗОЛА НИТРАТ
МюопаюН пПгаз
М1С0ЫА201-Е ШТКАТЕ
С18Н14С14Ы20 ■ НМ03 М.м. 479,1
[22832-87-7]
ОПРЕДЕЛЕНИЕ
1-[(2#5)-2-[(2,4-Дихлорбензил)окси]-2-(2,4-ди-
хлорфенил)этил]-1 Н-имидазола нитрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде, умеренно раство¬
рим в метаноле, мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 178 °С до
184 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО миконазола нитрата #или
спектр, представленный на рисунке #0513.-1*.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 30 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (а). 30 мг ФСО миконазола
нитрата растворяют в подвижной фазе и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 30 мг ФСО миконазола
нитрата и 30 мг ФСО эконазола нитрата (примесь
В) растворяют в подвижной фазе и доводят до объема
5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р.
Подвижная фаза: раствор аммония ацета¬
та Р — диоксан Р — метанол Р (20:40:40, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: в потоке теплого воздуха в тече¬
ние 15 мин.
Проявление: пластинку обрабатывают парами
йода до проявления пятен. Просматривают при днев¬
ном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения (а).
О. Испытуемый образец дает реакцию на нитра¬
ты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,1 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 10 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)7.
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. Определяют угол оптического вращения
раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
87. Зак. 1060.
686
Гэсударственная фармакопея Республики Беларусь
Рисунок #0513.-1. Инфракрасный спектр ФСО миконазола нитрата
Раствор сравнения (а). 2,5 мг ФСО миконазола
нитрата и 2,5 мг ФСО эконазола нитрата (примесь
В) растворяют в подвижной фазе и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
5.0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- подвижная фаза: 6,0 г аммония ацетата Р
растворяют в смеси из 300 мл ацетонитрила Р,
320 мл метанола Р и 380 мл воды Р;
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,2-кратное вре¬
мя удерживания миконазола.
Относительное удерживание (по отношению к ми-
коназолу, время удерживания — около 20 мин): примесь
А— около 0,1; примесь Е — около 0,3; примесь С — око¬
ло 0,4; примесь В — около 0,6; примесь й — около 0,75;
примесь Р — около 0,85; примесь <3 — около 0,9.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 10 между пиками приме¬
си В и миконазола.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р и О (не более 0,25 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, С, О, Е, Р и
6, не должны превышать площадь основного пика на
хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей А,
В, С, й, Е, Р и 6, не должна превышать 0,4 площади ос¬
новного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
-неучитываемый предел (0,05%): на хромато¬
грамме испытуемого раствора не учитывают пик, соот¬
ветствующий нитрат-иону, и пики с площадью менее 0,2
площади основного пика на хроматограмме раствора
сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола {2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 75 мл
кислоты уксусной ледяной Р, при необходимости
слегка нагревая, и титруют 0,1 М раствором хлорной
кислоты потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора хлорной кислоты соответ¬
ствует 47,91 мг С^Н^С^О^НМОз.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Е, О.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе-
Моксифлоксацина гидрохлорид
687
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимитиру¬
ется общим критерием приемлемости для других/неспец-
ифицированных примесей и/или общей статьей Суб¬
станции для фармацевтического использования (2034).
Вследствие этого нет необходимости идентифицировать
эти примеси для доказательства соответствия требова¬
ниям. См. также статью 5.10. Контроль примесей в суб¬
станциях для фармацевтического использования) : Н, I.
с*
/
НО'
и энантиомер
А. (1 Я5)-1 -(2,4-Дихлорфенил)-2-(1 Н-имидазол-1 -
ил)этанол.
и энантиомер
В. 1-{(2/?5)-2-{(4-Хлор6ензил)окси]-2-(2,4-дихлор-
фенил )этил}-1 Н-имидазол.
С. (2/?5)-2-{(2,4-ДихлорбензилХна>1]-2-{2,4-дихлоро-
фенил)этанамин.
и энантиомер
й. 1 -[(2Я5)-2-[(2,6-Дихлорбензил)окси]-2-(2,4-ди-
хлорфенил)этил]-1 Н-имидазол.
Е. 2-[1 -[(2Я5)-2-[(2,4-Дихлорбензил)окси]-2-(2,4-
дихлорфенил)этил]-1Н-имидазол-3-ио]-2-метилпропи-
онат.
и энантиомер
Р. 1-[(2Я5)-2-[(3,4-Дихлорбензил)окси]-2-(2,4-
дихлорфенил)этил]-1 Н-имидазол.
и энантиомер
С. 1 -[(2Я5)-2-[(2,5-Дихлорбензил)окси]-2-(2,4-
дихлорфенил)этил]-1 Н-имидазол.
С1
и энантиомер
Н. 1-[(2Я5)-2-Бензилокси-2-(2,4-дихлорфенил)-
этил]-1 Н-имидазол.
I. 1 -[(2Я5)-2-[(2-Хлорбензил)окси]-2-(2,4-дихлор-
фенил)этил]-1 Н-имидазол.
07/2016:2254
МОКСИФЛОКСАЦИНА
ГИДРОХЛОРИД
МохШохасЫ ЬудгосЫопдит
М0X1Р1. ОХА СШ ЯУОЯО СШ ОКЮЕ
87*. Зак. 1060.
688
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
1-Циклопропил-6-фтор-8-метокси-7-[(4а5,7а5)-
октагидро-6Н-пирроло[3,4-Ь]пиридин-6-ил]-4-оксо-1,4-
дигидрохинолин-3-карбоновая кислота гидрохлорид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ПРОИЗВОДСТВО
Способ получения должен обеспечивать удовлет¬
ворительную энантиомерную чистоту конечного про¬
дукта, что должно быть подтверждено валидацией.
ОПИСАНИЕ(СВОЙСТВА)
Светло-желтый или желтый порошок или кри¬
сталлы. Слегка гигроскопичен.
Умеренно растворим в воде, мало растворим в
96 % спирте, практически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО моксифлоксацина гидрохлори¬
да #или спектр, представленный на рисунке #2254.-1#.
C. 50 мг испытуемого образца растворяют в 5 мл
воды Р, прибавляют 1 мл кислоты азотной разведен¬
ной Р, перемешивают, выдерживают в течение 5 мин
и фильтруют. Фильтрат дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 20 мл раствора натрия гидроксида разведен¬
ного Р.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II. В случае если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения,
раствор 3 должен быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ОУ2(ЗЖ)2.
рН (2.2.3). От 3,9 до 4,6. 0,10 г испытуемого об¬
разца растворяют в 50 мл воды, свободной от угле¬
рода диоксида, Р.
Удельное оптическое вращение (2.2.7). От
-125 до -138 (в пересчете на безводное вещество).
0,200 г испытуемого образца растворяют в 20,0 мл
смеси из равных объемов ацетонитрила Р и воды Р.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от света.
Раствор А. 0,50 г тетрабутиламмония гидро¬
сульфата Р и 1,0 г калия дигидрофосфата Р раство¬
ряют в около 500 мл воды Р. К полученному раствору
прибавляют 2 мл кислоты фосфорной Р и 0,050 г на¬
трия сульфита безводного Р, затем доводят водой Р
до объема 1000,0 мл.
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в растворе А и доводят до объ¬
ема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 2,0 мл испытуемого
раствора (а) доводят раствором А до объема 20,0 мл.
Раствор сравнения (а). 50,0 мг ФСО моксиф¬
локсацина гидрохлорида растворяют в растворе А и
доводят до объема 50,0 мл этим же растворителем.
2,0 мл полученного раствора доводят раствором А до
объема 20,0 мл.
Раствор сравнения (Ь). 5 мг ФСО моксифлокса¬
цина для идентификации пиков (содержит примеси
А, В, С, О и Е) растворяют в растворе А и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора (а) доводят раствором А до объема 100,0 мл.
Рисунок #2254.-1. Инфракрасный спектр ФСО моксифлоксацина гидрохлорида
Моксонидин
689
1,0 мл полученного раствора доводят раствором А до
объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 45 °С;
- подвижная фаза: смешивают 28 объемов мета¬
нола Р и 72 объема раствора, содержащего 0,5 г/л те-
трабутиламиония гидросульфата Р, 1,0 г/л калия
дигидрофосфата Р и 3,4 г/л кислоты фосфорной Р,
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 293 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (Ь) и (с);
- время хроматографирования: 2,5-кратное вре¬
мя удерживания моксифлоксацина.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, О и Е, используя хрома¬
тограмму, прилагаемую к ФСО моксифлоксацина для
идентификации пиков, и хроматограмму раствора
сравнения (Ь).
Относительное удерживание (по отношению
к моксифлоксацину, время удерживания — около 14
мин): примесь А— около 1,1; примесь В — около 1,3;
примесь С — около 1,4; примесь О — около 1,6; при¬
месь Е — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками мок¬
сифлоксацина и примеси А;
-хроматограмма должна соответствовать хро¬
матограмме, прилагаемой к ФСО моксифлоксацина
для идентификации пиков.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 1,4; для примеси Е — 3,5):
- примеси А, В, С, О, Е (не более 0,1 %): на хро¬
матограмме испытуемого раствора (а) площади пи¬
ков, соответствующих примесям А, В, С, О и Е, не
должны превышать площадь основного пика на хро¬
матограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков приме¬
сей А, В, С, О и Е, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения (с);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Вода (2.5.12). Не более 4,5 %. Определение про¬
водят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца
в платиновом тигле.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора (Ь) и раствора сравнения (а).
Содержание С21Н24РЫ304 НС1 в процентах рас¬
считывают с учетом содержания моксифлоксацина ги¬
дрохлорида в ФСО моксифлоксацина гидрохлорида.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
A. Р = Р' = Р: 1 -Циклопропил-6,8-дифтор-7-
[(4а5,7а$)-октагидро-6Н-пирроло[3,4-Ь]пиридин-6-ил]-
4-оксо-1,4-дигидрохинолин-З-карбоновая кислота.
B. Р = Р' = ОСН3: 1 -Циклопропил-6,8-диметокси-
7-[(4а5,7а5)-октагидро-6Н-пирроло[3,4-Ь]пиридин-6-
ил]-4-оксо-1,4-дигидрохинолин-З-карбоновая кислота.
C. Р = Р, Р' = ОС2Н5: 1-Циклопропил-8-этокси-6-
фтор-7-[(4а$,7а5)-октагидро-6Н-пирроло[3,4-Ь]пиридин-
6-ил]-4-оксо-1,4-дигидрохинолин-З-карбоновая кислота.
О. Р = ОСН3, Р' = Р: 1-Циклопропил-8-фтор-6-
метокси-7-[(4а5,7а$)-октагидро-6Н-пирроло[3,4-б]пи-
ридин-6-ил]-4-оксо-1,4-дигидрохинолин-З-карбоновая
кислота.
Е. Р = Р, Р' = ОН: 1-Циклопропил-6-фтор-8-
гидрокси-7-[(4а5,7а5)-октагидро-6А/-пирроло[3,4-Ь]пи-
ридин-6-ил]-4-оксо-1,4-дигидрохинолин-З-карбоновая
кислота.
07/2016:1758
моксонидин
МохотсНпит
МОХОЫЮ1ЫЕ
С-Н.-С^.О М.м. 241,7
[75438-57-2]
ОПРЕДЕЛЕНИЕ
4-Хлор-А/-(имидазолидин-2-илиден)-6-метокси-2-
метилпиримидин-5-амин.
88. Зак. 1060.
690
Гэсударственная фармакопея Республики Беларусь
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде, умеренно раство¬
рим в метаноле, мало растворим в метиленхлориде,
очень мало растворим в ацетонитриле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО моксонидина.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в смеси из равных объемов мета¬
нола Р и воды Р и доводят до объема 100,0 мл этой
же смесью растворителей.
Раствор сравнения (а). 10,0 мг ФСО моксониди¬
на растворяют в смеси из равных объемов метано¬
ла Р и воды Р и доводят до объема 10,0 мл этой же
смесью растворителей.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят смесью из равных объемов метано¬
ла Р и воды Рдо объема 100,0 мл. 2,0 мл полученного
раствора доводят смесью из равных объемов мета¬
нола Р и воды Р до объема 20,0 мл.
Раствор сравнения (с). 5,0 мг ФСО моксониди¬
на примеси А растворяют в смеси из равных объемов
метанола Р и воды Р и доводят до объема 100,0 мл
этой же смесью растворителей.
Раствор сравнения (д). 6,0 мл раствора сравне¬
ния (с) доводят смесью из равных объемов метано¬
ла Р и воды Р до объема 100,0 мл.
Раствор сравнения (е). 2,5 мл раствора сравнения
(а) доводят раствором сравнения (с) до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
-подвижная фаза: смешивают 136 объемов
ацетонитрила Р и 1000 объемов раствора 3,48 г/л
натрия пентансульфоната Р, предварительно до¬
веденного до рН 3,5 кислотой серной разведенной Р;
- скорость подвижной фазы: 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: по 20 мкл холостого
раствора, испытуемого раствора и растворов сравне¬
ния (Ь), (д) и (е);
- время хроматографирования: 2-кратное вре¬
мя удерживания моксонидина.
Относительное удерживание (по отношению к
моксонидину, время удерживания — около 11,6 мин):
примесь А— около 0,9; примесь В — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 2 между пиками приме¬
си А и моксонидина.
Предельное содержание примесей:
- примесь А (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с1);
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05%): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (Ь); не учитыва¬
ют пики, соответствующие пикам на хроматограмме
холостого раствора.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С9Н12С1Ы50 в процентах рассчиты¬
вают с учетом площадей пиков и содержания моксо¬
нидина в ФСО моксонидина.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): С, О.
А. Р4 = Р6 = С1: 4,6-Дихлор-А/-(имидазолидин-2-
илиден)-2-метилпиримидин-5-амин (6-хлормоксони-
дин).
(З)-Молочная кислота
691
B. К4 = Кб = ОСН3: Л/-(Имидазолидин-2-илиден)-
4,6-диметокси-2-метилпиримидин-5-амин (4-метокси-
моксонидин).
C. В4 = ОН, Кб = ОСН3: 5-[(Имидазолидин-2-или-
ден)амино]-6-метокси-2-метилпиримидин-4-ол (4-ги-
дроксимоксонидин).
Э. К4 = ОН, Кб = С1: 6-Хлор-5-[(имидазолидин-2-
илиден)амино]-2-метилпиримидин-4-ол (6-дезметил-
моксонидин).
07/2016:0458
МОЛОЧНАЯ КИСЛОТА
АсШит 1асИсит
1-АСТ1С АСЮ
н3с^.со2н
у\ и энантиомер
н ОН
С3Н603 М.м. 90,1
ОПРЕДЕЛЕНИЕ
Смесь 2-гидроксипропановой кислоты, продуктов
ее конденсации, таких как лактоилмолочная кислота
и полимолочные кислоты, и воды. Равновесие между
молочной кислотой и полимолочными кислотами за¬
висит от концентрации и температуры. Обычно раце¬
мат ((ЯЗ)-молочная кислота).
Содержание: не менее 88,0 % (м/м) и не более
92,0 % (м/м) С3Нб03.
ОПИСАНИЕ(СВОЙСТВА)
Бесцветная или слегка желтоватая сиропообраз¬
ная жидкость.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца растворяют в 10 мл
воды Р Раствор имеет сильнокислую реакцию (2.2.4).
B. Относительная плотность (2.2.5): от 1,20 до
1,21.
C. Дает реакцию на лактаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в 42 мл 1 М раствора натрия гидроксида и до¬
водят водой дистиллированной Р до объема 50,0 мл.
Цветность(2.2.2, метод II). Окраска испытуе¬
мого образца должна быть не интенсивнее эталона
у(Ж)6.
Вещества, нерастворимые в эфире. 1,0 г ис¬
пытуемого образца растворяют в 25 мл эфира Р. Опа¬
лесценция полученного раствора должна быть не ин¬
тенсивнее опалесценции растворителя.
Сахара и другие восстанавливающие веще¬
ства. К 1 мл раствора 3 прибавляют 1 мл 1 М раст¬
вора кислоты хлористоводородной, нагревают до
кипения и охлаждают. К полученному раствору при¬
бавляют 1,5 мл 1 М раствора натрия гидроксида и
2 мл медно-тартратного раствора Р и нагревают
до кипения. Не должен образовываться красный или
зеленоватый осадок.
Метанол (2.4.24). Не более 0,0050 % (50 ррт),
если субстанция предназначена для производства
парентеральных лекарственных средств.
Лимонная, щавелевая и фосфорная кисло¬
ты. К 5 мл раствора 3 прибавляют раствор аммиака
разведенный Р1 до слабощелочной среды (2.2.4). За¬
тем прибавляют 1 мл раствора кальция хлорида Р.
Нагревают на водяной бане 5 мин. Как до, так и по¬
сле нагревания опалесценция полученного раствора
должна быть не интенсивнее опалесценции смеси,
состоящей из 1 мл воды Р и 5 мл раствора 3.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Кальций (2.4.3). Не более 0,0200 % (200 ррт).
5 мл раствора 5 доводят водой дистиллированной Р
до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
5 МЕ/г, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов. Перед ис¬
пользованием испытуемый раствор доводят до рН
7,0—7,5 раствором натрия гидроксида концентри¬
рованным Р и энергично встряхивают.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца помещают в колбу
с притертой пробкой и прибавляют 10 мл воды Р и
20,0 мл 1 М раствора натрия гидроксида. Колбу за¬
крывают пробкой и выдерживают в течение 30 мин.
Титруют 1 М раствором кислоты хлористоводород¬
ной до исчезновения розового окрашивания, исполь¬
зуя в качестве индикатора 0,5 мл раствора фенол¬
фталеина Р.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 90,1 мгС3Н603.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:1771
(3)-МОЛОЧНАЯ КИСЛОТА
АсМит (3)-1асИсит
(8)-1.АСТ1С АСЮ
н3с
^ч^со2н
X
н он
С3Н603 М.м. 90,1
ОПРЕДЕЛЕНИЕ
Смесь (5)-2-гидроксипропановой кислоты, про¬
дуктов ее конденсации, таких как лактоилмолочная
кислота и полимолочные кислоты, и воды. Равнове-
88*. Зак. 1060.
692
Гэсударственная фармакопея Республики Беларусь
сие между молочной кислотой и полимолочными кис¬
лотами зависит от концентрации и температуры.
Содержание: не менее 88,0 % (м/м) и не более
92,0 % (м/м) С3Н603, из которых не менее 95,0 % со¬
ставляет (З)-энантиомер.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная или слегка желтоватая, сиропообраз¬
ная жидкость.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца растворяют в 10 мл
воды Р. Раствор имеет сильнокислую реакцию (2.2.4).
B. Относительная плотность (2.2.5): от 1,20 до 1,21.
C. Испытуемый образец дает реакцию на лакта¬
ты (2.3.1).
й. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в 42 мл 1 М раствора натрия гидроксида и до¬
водят водой Р до объема 50 мл.
Цветность (2.2.2, метод II). Окраска испытуе¬
мого образца должна быть не интенсивнее эталона
У(Ж)6.
Вещества, нерастворимые в эфире. 1,0 г ис¬
пытуемого образца растворяют в 25 мл эфира Р. Опа¬
лесценция полученного раствора не должна превы¬
шать опалесценцию растворителя.
Сахара и другие восстанавливающие веще¬
ства. К 1 мл раствора 3 прибавляют 1 мл 1 М раст¬
вора кислоты хлористоводородной, нагревают до
кипения и охлаждают. К полученному раствору при¬
бавляют 1,5 мл 1 М раствора натрия гидроксида и
2 мл медно-тартратного раствора Р и нагревают
до кипения. Не должен образовываться красный или
зеленоватый осадок.
Метанол (2.4.24). Не более 0,0050 % (50 ррт),
если субстанция предназначена для производства
парентеральных лекарственных средств.
Лимонная, щавелевая и фосфорная кислоты.
К 5 мл раствора 3 прибавляют аммиака раствор раз¬
веденный Р1 до слабощелочной среды (2.2.4). При¬
бавляют 1 мл раствора кальция хлорида Р и нагрева¬
ют на водяной бане в течение 5 мин. Как до, так и по¬
сле нагревания опалесценция полученного раствора
должна быть не интенсивнее опалесценции смеси,
состоящей из 1 мл воды Р и 5 мл раствора 3.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Кальций (2.4.3). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
5 МЕ/г, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов. Перед ис¬
пользованием испытуемый раствор доводят до рН
7,0—7,5 раствором натрия гидроксида концентри¬
рованным Р и энергично встряхивают.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца помещают в колбу
с притертой пробкой и прибавляют 10 мл воды Р и
20,0 мл 1 М раствора натрия гидроксида. Колбу за¬
крывают пробкой и выдерживают в течение 30 мин.
Титруют 1 М раствора кислоты хлористоводород¬
ной Р до исчезновения розового окрашивания, ис¬
пользуя в качестве индикатора 0,5 мл раствора фе¬
нолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 90,1 мг С3Н603.
(З)-Энантиомер
Количество испытуемого образца, эквивалент¬
ное 2,00 г молочной кислоты, помещают в круглодон¬
ную колбу, прибавляют 25 мл 1 М раствора натрия
гидроксида и осторожно нагревают в течение 15 мин.
Охлаждают и доводят до рН 7,0 1 М раствором кис¬
лоты хлористоводородной. Прибавляют 5,0 г аммо¬
ния молибдата Р, растворяют и доводят водой Р до
объема 50,0 мл. Раствор фильтруют и измеряют угол
оптического вращения (2.2.7).
Содержание (З)-энантиомера в процентах рас¬
считывают по формуле:
50+ 24,18 а-
2,222 90^
т с)
где:
а — угол оптического вращения (абсолютная ве¬
личина);
т — масса навески испытуемого образца, г;
с — содержание С3Нб03 в испытуемом образ¬
це, %.
Комплекс (З)-молочной кислоты, образовавший¬
ся в условиях данного испытания, является левовра¬
щающим.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:0097
МОРФИНА ГИДРОХЛОРИД
МогрЫп/ Ьус1госЫопс1ит
МОКРН1ЫЕ НУОПОСШОКЮЕ
С17Н19ГЮ, • НС1 ■ ЗН20 М.м. 375,8
[6055-06-7]
Морфина гидрохлорид
693
ОПРЕДЕЛЕНИЕ
7,8-Дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а-диола гидрохлорид тригидрат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок или бесцветная шелковистая игольчатая или
кубическая масса. Выветривается при воздействии
сухого воздуха.
Растворим в воде, мало растворим в 96 % спир¬
те, практически нерастворим в толуоле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО морфина гидрохлорида триги-
драта.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Раствор А. 25,0 мг испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем.
Испытуемый раствор (а). 10,0 мл раствора А
доводят водой Р до объема 100,0 мл.
Испытуемый раствор (Ь). 10,0 мл раствора А
доводят 0,1 М раствором натрия гидроксида до объ¬
ема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм для ис¬
пытуемых растворов (а) и (Ь).
Максимум поглощения: при 285 нм для испытуе¬
мого раствора (а); при 298 нм для испытуемого раст¬
вора (Ь).
Удельный показатель поглощения в максимуме:
от 37 до 43 для испытуемого раствора (а); от 64 до 72
для испытуемого раствора (Ь).
C. 1 мг измельченного испытуемого образца в
фарфоровой ступке прибавляют 0,5 мл раствора
формальдегида в серной кислоте Р. Появляется
красно-фиолетовое окрашивание, переходящее в фи¬
олетовое.
О. Испытуемый образец дает реакцию на алка¬
лоиды (2.3.1).
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона У(Ж)6 или
ВУ(КЖ)6.
Кислотность или щелочность. К 10 мл раст¬
вора 5 прибавляют 0,05 мл раствора метилового
красного Р. При прибавлении не более 0,2 мл 0,02 М
раствора натрия гидроксида или 0,02 М раствора
кислоты хлористоводородной окраска раствора
должна измениться.
Удельное оптическое вращение (2.2.7). От -110
до -115 в пересчете на безводное вещество. Опреде¬
ляют удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,Т25 г испытуемого образ¬
ца растворяют в 1 % (об/об) растворе кислоты уксус¬
ной Р и доводят до объема 50 мл этим же раствором.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят 1 % (об/об) раствором кислоты уксус¬
ной Р до объема 100,0 мл. 2,0 мл полученного раст¬
вора доводят 1 % (об/об) раствором кислоты уксус¬
ной Р до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО морфина для
проверки пригодности хроматографической систе¬
мы (содержит примеси В, С, Е, Р) растворяют в 1 %
(об/об) растворе кислоты уксусной Р и доводят до
объема 2 мл этим же раствором.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 35 °С;
- подвижная фаза:
-подвижная фаза А: раствор 1,01 г/л натрия
гептансульфоната Р, доведенный до рН 2,6
50 % (об/об) раствором кислоты фосфорной Р;
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
85
15
2—35
85 — 50
15 — 50
35—40
50
50
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, Е и Р, используя хромато¬
грамму раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО морфина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
морфину, время удерживания — около 12,5 мин): при¬
месь Р — около 0,95; примесь Е — около 1,1; примесь
С — около 1,6; примесь В — около 1,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-коэффициент разделения пиков: не менее 2
(Нр — высота пика примеси Р относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Р и
морфина).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,25; для примеси С — 0,4; для примеси
Е — 0,5):
- примесь В (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примеси С, Е (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С и Е, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
89. Зак. 1060.
694
Гэсударственная фармакопея Республики Беларусь
- любая другая примесь (не более 0,2 %): на
хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного и пиков примесей В, С
и Е, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Пределы, указанные в разделе «Сопутствую¬
щие примеси» (таблица 2034.-1) статьи Субстан¬
ции для фармацевтического использования (2034),
не применяют.
Вода (2.5.12). Не менее 12,5% и не более
15,5%. Определение проводят из 0,10 г испытуе¬
мого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в
смеси из 5 мл 0,01 М раствора кислоты хлори¬
стоводородной и 30 мл 96 % спирта Р и титруют
0,1 М раствором натрия гидроксида потенциоме-
трически (2.2.20). Отмечают объем титранта между
двумя точками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 32,18 мг С17Н19М03 НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, О, Р.
В. 7,7\8,8-Тетрадегидро-4,5а:4',5'а-диэпокси-17,17 -
диметил-2,2'-биморфинанил-3,3',6а,6'а-тетрол (2,2-би-
морфин).
С. 6,7,8,14-Тетрадегидро-4,5а-эпокси-6-метокси-
17-метилморфинан-З-ол (орипавин).
О. 7,8-Дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а, 10а-триол (105-гидроксиморфин).
Е. 7,8-Дидегидро-4,5а-эпокси-3-гидрокси-17-метил-
морфинан-6-он (морфинон).
Р. (175)-7,8-Дидегидро-4,5а-эпокси-17-метилмор-
финан-3,6а-диол 17-оксид (морфин А/-оксид).
07/2016:1244
МОРФИНА СУЛЬФАТ
МогрЫп/ зиКаз
МОВРН1ЫЕ 5111-РАТЕ
А. 7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба-ол (кодеин).
С Н N О - Н 30 • 5Н О
34П38 2 6 ‘ 2^^4
[6211-15-0]
М.м. 759
Морфина сульфат
695
ОПРЕДЕЛЕНИЕ
Ди(7,8-дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а-диола) сульфат пентагидрат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде, очень мало растворим в 96 %
спирте, практически нерастворим в толуоле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 20 мг испытуемого образца
растворяют в 1 мл воды Р, прибавляют 0,05 мл 1 М
раствора натрия гидроксида и встряхивают. Полу¬
ченный осадок отфильтровывают, промывают двумя
порциями по 0,5 мл воды Р и высушивают при темпе¬
ратуре 145 °С в течение 1 ч. Сухой остаток использу¬
ют для приготовления дисков.
Сравнение: выполняют описанное выше приго¬
товление, используя 20 мг ФСО морфина сульфата.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Раствор А. 25,0 мг испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем.
Испытуемый раствор (а). 10,0 мл раствора А
доводят водой Р до объема 100,0 мл.
Испытуемый раствор (Ь). 10,0 мл раствора А
доводят 0,1 М раствором натрия гидроксида до объ¬
ема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм для ис¬
пытуемых растворов (а) и (Ь).
Максимум поглощения: при 285 нм для испытуе¬
мого раствора (а); при 298 нм для испытуемого раст¬
вора (Ь).
Удельный показатель поглощения в максимуме:
от 37 до 43 для испытуемого раствора (а); от 64 до 72
для испытуемого раствора (Ь).
C. К 1 мг измельченного испытуемого образца
в фарфоровой ступке прибавляют 0,5 мл раствора
формальдегида в серной кислоте Р. Появляется
красно-фиолетовое окрашивание, переходящее в фи¬
олетовое.
Р. Испытуемый образец дает реакцию на алка¬
лоиды (2.3.1).
Е. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
5 должна быть не интенсивнее эталона У(Ж)6 или
ВУ(КЖ)6.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,05 мл раствора метилового
красного Р. При прибавлении не более 0,2 мл 0,02 М
раствора натрия гидроксида или 0,02 М раствора
кислоты хлористоводородной окраска раствора
должна измениться.
Удельное оптическое вращение (2.2.7). От -107
до “110 в пересчете на безводное вещество. Опреде¬
ляют удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,125 г испытуемого образ¬
ца растворяют в 1 % (об/об) растворе кислоты уксус¬
ной Р и доводят до объема 50 мл этим же раствором.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят 1 % (об/об) раствором кислоты уксус¬
ной Р до объема 100,0 мл. 2,0 мл полученного раст¬
вора доводят 1 % (об/об) раствором кислоты уксус¬
ной Р до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО морфина для
проверки пригодности хроматографической систе¬
мы (содержит примеси В, С, Е, Р) растворяют в 1 %
(об/об) растворе кислоты уксусной Р и доводят до
объема 2 мл этим же раствором.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 35 °С;
- подвижная фаза:
-подвижная фаза А:.раствор 1,01 г/л натрия
гептансульфоната Р, доведенный до рН 2,6
50 % (об/об) раствором кислоты фосфорной Р;
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
85
15
2—35
85 — 50
15 — 50
35—40
50
50
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, Е и Р, используя хромато¬
грамму раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО морфина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
морфину, время удерживания — около 12,5 мин): при¬
месь Р — около 0,95; примесь Е — около 1,1; примесь
С — около 1,6; примесь В — около 1,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-коэффициент разделения пиков: не менее 2
(Нр — высота пика примеси Р относительно базовой
линии; Н]/ — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Р и
морфина).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,25; для примеси С — 0,4; для примеси
Е —0,5):
- примесь В (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
89*, Зак. 1060.
696
Гэсударственная фармакопея Республики Беларусь
- примеси С, Е (не более 0,2 %): на хроматограмме
испытуемого раствора площади пиков, соответствующих
примесям С и Е, не должны превышать площадь основ¬
ною пика на хроматограмме раствора сравнения (а);
- любая другая примесь (не более 0,2 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей В, С и Е, не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для фар¬
мацевтического использования (2034), не применяют.
Железо (2.4.9). Не более 0,0005 % (5 ррт). Оста¬
ток, полученный в испытании «Сульфатная зола»,
растворяют в водеР и доводят до объема 10,0 мл
этим же растворителем.
Вода (2.5.12). Не менее 10,4 % и не более 13,4 %.
Определение проводят из 0,10 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в
120мл кислоты уксусной безводной Р и титруют
0,1 М раствором кислоты хлорной потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 66,88 мг С34Н38М206-Н2504.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, О, Р.
А. 7,8-Дидегидро-4,5а-эпокси-3-метокси-17-метил-
морфинан-ба-ол (кодеин).
В. 7,7',8,8-Тетрадегидро-4,5а:4',5'а-диэпокси-17,17-
диметил-2,2'-биморфинанил-3,3',6а,6'а-тетрол (2,2-би¬
морфин).
С. 6,7,8,14-Тетрадегидро-4,5а-эпокси-6-метокси-
17-метилморфинан-З-ол (орипавин).
н
н
Ап3
N
но-%
н—
□Л
Уч
\ !*Н
но7
О'
Н ОН
й. 7,8-Дидегидро-4,5а-эпокси-17-метилморфинан-
3,6а, 10а-триол (105-гидроксиморфин).
Е. 7,8-Дидегидро-4,5а-эпокси-3-гидрокси-17-метил-
морфинан-6-он (морфинон).
Р. (175)-7,8-Дидегидро-4,5а-эпокси-17-метилмор-
финан-3,6а-диол 17-оксид (морфин Л/-оксид).
МОЧЕВИНА
игеит
иНЕА
СН4Ы20
[57-13-6]
МН2
07/2016:0743
М.м. 60,1
ОПРЕДЕЛЕНИЕ
Карбамид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
Мочевина
697
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок
или прозрачные кристаллы. Слегка гигроскопична.
Очень легко растворима в воде, растворима в
96 % спирте, практически нерастворима в метилен-
хлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 132 °С до
135 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО мочевины #или спектр, пред¬
ставленный на рисунке #0743.-1#.
C. 0,1 г испытуемого образца растворяют в 1 мл
воды Р, прибавляют 1 мл кислоты азотной Р. Обра¬
зуется белый кристаллический осадок.
О. 0,5 г испытуемого образца нагревают в про¬
бирке до расплавления и помутнения жидкости, ох¬
лаждают и растворяют в смеси из 10 мл воды Р и
1 мл раствора натрия гидроксида разведенного Р. К
полученному раствору прибавляют 0,05 мл раствора
меди (II) сульфата Р. Появляется красно-фиолетовое
окрашивание.
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Раствор 31. К 2,5 мл раствора 3 прибавляют
7,5 мл воды Р.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 31 должен
быть бесцветным.
Щелочность. К 2,5 мл раствора 3 прибавляют
7,5 мл воды Р, 0,1 мл раствора метилового красно¬
го Рп 0,4 мл 0,01 М раствора кислоты хлористово¬
дородной. Появляется окрашивание раствора от крас¬
ного до оранжевого.
Биурет. Не более 0,1 %. К 10 мл раствора 3 при¬
бавляют 5 мл воды Р, 0,5 мл раствора 5 г/л меди (II)
сульфата Р, 0,5 мл раствора натрия гидроксида
концентрированного Р и выдерживают в течение
5 мин. Красновато-фиолетовое окрашивание раст¬
вора должно быть не интенсивнее эталона, приготов¬
ленного параллельно с использованием 10 мл раст¬
вора 0,2 г/л биурета Р.
Аммония соли (2.4.1). Не более 0,0500%
(500 ррт). 0,1 мл раствора 3 должен выдерживать ис¬
пытание на соли аммония.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 10 мл раствора 3 доводят водой Р
до объема 20 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,2000 г испытуемого образца растворяют в
воде Р и доводят до объема 50,0 мл этим же раство¬
рителем. 1,0 мл полученного раствора помещают в
колбу для сжигания, прибавляют 4 г смеси порошка
из 100 г калия сульфата Р, 5 г меди (II) сульфата Р
и 2,5 г селена Р и 3 стеклянных шарика. Со стенок
колбы смывают прилипшие частицы, прибавляя 5 мл
Рисунок #0743.-1. Инфракрасный спектр ФСО мочевины
90. Зак. 1060.
698
Гэсударственная фармакопея Республики Беларусь
кислоты серной Р по стенкам колбы, и перемешива¬
ют содержимое колбы вращательными движениями.
Неплотно закрывают колбу, например стеклянной гру¬
шеподобной пробкой с коротким концом, во избежание
избыточных потерь серной кислоты. Колбу постепенно
нагревают, а затем повышают температуру до бурного
кипения и появления на горле колбы конденсата кисло¬
ты серной. Необходимо следить, чтобы верхняя часть
колбы не перегревалась. Нагревают в течение 30 мин.
Колбу охлаждают, растворяют твердый остаток осто¬
рожным прибавлением 25 мл воды Р. Снова охлазда-
ют и присоединяют колбу к аппарату для перегонки с
водяным паром. Прибавляют 30 мл раствора натрия
гидроксида концентрированного Р и сразу перего¬
няют, пропуская пар через смесь. Собирают дистил¬
лят в колбу-приемник, содержащую 15 мл раствора
40 г/л кислоты борной Р, 0,2 мл метилового красного
смешанного раствора Р и воду Р в количестве, до¬
статочном для того, чтобы нижний конец трубки холо¬
дильника был погружен в раствор. После завершения
перегонки опускают колбу-приемник так, чтобы конец
трубки холодильника находился над поверхностью
раствора кислоты. Соблюдают осторожность, чтобы
вода с внешней поверхности трубки холодильника не
попадала в содержимое колбы-приемника. Дистиллят
титруют 0,01 М раствором кислоты серной.
1 мл 0,01 М раствора кислоты серной соответ¬
ствует 0,6006 мг СН4М20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016.РБ0026
«МУРАВЬИНАЯ КИСЛОТА
АсШит 1огт1сит
Г О КМ IС АСЮ
О
СН202 М.м. 46,03
ОПРЕДЕЛЕНИЕ
Содержание: не менее 85,0 % (м/м) СН202
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная, прозрачная, едкая жидкость.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 2 мл испытуемого образца прибавляют
10 мл воды Р, 0,5 г ртути (II) оксида Р и перемеши¬
вают. Собирают надосадочную жидкость и нагревают
до кипения. Выделяются пузырьки газа, и образуется
осадок серого цвета.
B. К 1 мл испытуемого образца прибавляют 3 мл
воды Р и 3 мл раствора свинца (II) ацетата Р. Обра¬
зуется кристаллический осадок белого цвета.
ИСПЫТАНИЯ
Плотность (2.2.5). От 1,192 до 1,220 г/см3.
Оксалаты. К 1,0 мл испытуемого образца прибав¬
ляют 10 мл воды Р, предварительно нейтрализованной
раствором аммиака концентрированным Р (по крас¬
ной лакмусовой бумаге Р), 0,25 мл раствора 100 г/л
кислоты хлористоводородной Р. После охлаждения
раствора прибавляют 2 мл раствора 2 г/л кальция хло¬
рида Р. Раствор должен оставаться прозрачным.
Ацетаты. К 0,5 мл испытуемого образца при¬
бавляют 10 мл воды Р, 3,0 г ртути (II) оксида Р и на¬
гревают с обратным холодильником на водяной бане
до окончания выделения пузырьков газа. Прибавляют
2.0 г ртути (II) оксида Р и нагревают на водяной бане
в течение 20 мин. Охлаждают, фильтруют. Осадок
на фильтре промывают водой Р до получения 40 мл
фильтрата. К 1,0 мл 0,1 М раствора натрия гидрок¬
сида прибавляют 0,1 мл раствора фенолфталеи¬
на Р. К полученному раствору прибавляют фильтрат.
Раствор должен иметь розовую окраску.
Сульфиты. К 0,5 мл испытуемого образца при¬
бавляют 3,5 мл раствора 120 г/л натрия гидрокси¬
да Р и доводят водой Р до объема 10 мл. Прибавляют
0,25 мл 0,01 М раствора йода. Раствор должен иметь
желтую окраску, которая не исчезает в течение 30 с.
Сульфаты (2.4.13). Не более 0,0050 % (50 ррт).
3.0 г испытуемого образца выпаривают на водяной
бане досуха. Остаток растворяют в воде дистиллиро¬
ванной Р и доводят до объема 15 мл этим же раство¬
рителем. Полученный раствор должен выдерживать
испытание на сульфаты. Эталон готовят с исполь¬
зованием 15 мл эталонного раствора сульфата
(10 ррт 80} Р.
Хлориды (2.4.4). Не более 0,0010% (10 ррт).
1.0 г испытуемого образца доводят водой Р до объ¬
ема 15 мл. Полученный раствор должен выдерживать
испытание на хлориды. Эталон готовят с использова¬
нием 2 мл эталонного раствора хлорида (5 ррт С!) Р
и 13 мл воды Р.
Железо (2.4.9). Не более 0,0005 % (5 ррт). 1,0 г
испытуемого образца прибавляют к 5 мл воды Р, ней¬
трализуют раствором аммиака Р и доводят водой Р
до объема 10мл. Полученный раствор должен вы¬
держивать испытание на железо. Эталон готовят с
использованием 5 мл эталонного раствора железа
(1 ррт Ре) Р и 5 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 8,0 г испытуемого образца выпари¬
вают на водяной бане досуха. Остаток растворяют в
воде Р и доводят до объема 20 мл этим же растворите¬
лем. Полученный раствор должен выдерживать испы¬
тание на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (2 ррт РЬ) Р.
Нелетучий остаток. Не более 0,02 % (м/об).
10.0 мл испытуемого образца выпаривают на водяной
бане досуха. Остаток высушивают при температуре от
100 °С до 105 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10 мл воды Р помещают в коническую колбу,
точно взвешивают, быстро прибавляют около 1 мл
испытуемого образца и снова взвешивают. Прибав¬
ляют 50 мл воды Р и титруют 1 М раствором натрия
гидроксида, используя в качестве индикатора 0,5 мл
раствора фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 46,03 мг СН202.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
#Мыло зеленое (мыло калийное)
699
07/2016: РБ0025
«МУРАВЬИНАЯ КИСЛОТА
БЕЗВОДНАЯ
Ас1с1ит Iогтюит апНус/псиз
ГО КМ IС АСЮ АМНУОКО(75
О
сн202 М.м. 46,03
[64-18-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % (м/м) СН202
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная, прозрачная, едкая жидкость.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 2 мл испытуемого образца прибавляют
10 мл воды Р, 0,5 г ртути (II) оксида Р и перемеши¬
вают. Собирают надосадочную жидкость и нагревают
до кипения. Выделяются пузырьки газа и образуется
осадок серого цвета.
B. К 1 мл испытуемого образца прибавляют 3 мл
воды Р и 3 мл раствора свинца (II) ацетата Р. Обра¬
зуется кристаллический осадок белого цвета.
ИСПЫТАНИЯ
Плотность (2.2.5). От 1,215 до 1,221 г/см3.
Оксалаты. К1,0 мл испытуемого образца прибав¬
ляют 10 мл воды Р, предварительно нейтрализованной
раствором аммиака концентрированным Р (по крас¬
ной лакмусовой бумаге Р), 0,25 мл раствора 100 г/л
кислоты хлористоводородной Р. После охлаждения
раствора прибавляют 2 мл раствора 2 г/л кальция хло¬
рида Р. Раствор должен оставаться прозрачным.
Ацетаты. К 0,5 мл испытуемого образца при¬
бавляют 10 мл воды Р, 3,0 г ртути (II) оксида Р и на¬
превают с обратным холодильником на водяной бане
до окончания выделения пузырьков газа. Прибавля¬
ют 2,0 г ртути (II) оксида Р и нагревают на водяной
бане в течение 20 мин. Охлаждают, фильтруют. Оса¬
док на фильтре промывают водой Р до получения
40 мл фильтрата. К 1,0 мл 0,1 М раствора натрия
гидроксида прибавляют 0,1 мл раствора фенол¬
фталеина Р. К полученному раствору прибавляют
фильтрат. Раствор должен иметь розовую окраску.
Сульфиты. К 0,5 мл испытуемого образца при¬
бавляют 4 мл раствора 120 г/л натрия гидроксида Р
и доводят водой Р до объема 10мл. Прибавляют
0,25 мл 0,01 М раствора йода. Раствор должен иметь
желтую окраску, которая не исчезает в течение 30 с.
Сульфаты (2.4.13). Не более 0,0050 % (50 ррт).
3,0 г испытуемого образца выпаривают на водяной
бане досуха. Остаток растворяют в воде дистиллиро¬
ванной Р и доводят до объема 15 мл этим же раство¬
рителем. Полученный раствор должен выдерживать
испытание на сульфаты. Эталон готовят с исполь¬
зованием 15 мл эталонного раствора сульфата
(10 ррт ЗО^Р
Хлориды (2.4.4). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды. Эталон готовят с использованием 2 мл эта¬
лонного раствора хлорида (5 ррт С!) и 13 мл воды Р.
Железо (2.4.9). Не более 0,0005% (5 ррт). 1,0 г
испытуемого образца прибавляют к 5 мл воды Р, ней¬
трализуют раствором аммиака Р и доводят водой Р до
объема 10мл. Полученный раствор должен выдержи¬
вать испытание на железо. Эталон готовят с использо¬
ванием 5 мл эталонного раствора железа (1 ррт Ре) Р
и 5 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 8,0 г испытуемого образца выпари¬
вают на водяной бане досуха. Остаток растворяют в
воде Р и доводят до объема 20 мл этим же растворите¬
лем. Полученный раствор должен выдерживать испы¬
тание на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (2 ррт РЬ) Р.
Нелетучий остаток. Не более 0,02 % (м/об).
10,0 мл испытуемого образца выпаривают на водяной
бане досуха. Остаток высушивают при температуре от
100 °С до 105 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10 мл воды Р помещают в коническую колбу,
точно взвешивают, быстро прибавляют около 1 мл
испытуемого образца и снова взвешивают. Прибав¬
ляют 50 мл воды Р и титруют 1 М раствором натрия
гидроксида, используя в качестве индикатора 0,5 мл
раствора фенолфталеина Р
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 46,03 мг СН202.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:РБ0028
#МЫЛО ЗЕЛЕНОЕ
(МЫЛО КАЛИЙНОЕ)
8аро мпсИз
вКЕЕЫ 80АР
ОПРЕДЕЛЕНИЕ
Мыло зеленое получают омылением раститель¬
ных масел раствором калия гидроксида.
Содержание: не менее 40,0 % жирных кислот.
ОПИСАНИЕ (СВОЙСТВА)
Мягкая, пастообразная, прозрачная масса корич¬
невато-желтоватого или зеленоватого цвета со свое¬
образным запахом.
Легко растворимо в воде и в 96 % спирте с обра¬
зованием прозрачных и сильнопенящихся при взбал¬
тывании растворов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Испытуемый образец выдерживает требования
раздела «Количественное определение».
ИСПЫТАНИЯ
Минеральные примеси. Не более 2,0 %. 6,000 г
испытуемого образца растворяют в 25 мл горяче¬
го 96 % спирта Р и фильтруют через высушенный и
взвешенный фильтр. Фильтр с осадком промывают
90*. Зак. 1060.
700
Гэсударственная фармакопея Республики Беларусь
горячим 96 % спиртом Р. Собранный фильтрат остав¬
ляют для проведения испытания «Органические при¬
меси», а фильтр с осадком высушивают до постоян¬
ной массы и определяют массу сухого остатка.
Органические примеси. К спиртовому филь¬
трату, полученному в испытании «Минеральные при¬
меси», прибавляют 2,5 мл 0,1 М раствора кислоты
хлористоводородной и взбалтывают. Жидкость не
должна мутнеть.
Щелочность. 5,0 г испытуемого образца раство¬
ряют в 25 мл горячей воды Р, прибавляют 20 мл ней¬
трализованного по фенолфталеину раствора 50 г/л ба¬
рия хлорида Р и смесь кипятят несколько минут, пока
бариевое мыло отделится и жидкость не станет про¬
зрачной. Осадок отфильтровывают через складчатый
фильтр и промывают 2—3 раза водой Р. Полученный
фильтрат охлаждают и прибавляют 3—4 капли раст¬
вора фенолфталеина Р. При прибавлении не более
0,2 мл 0,1 М раствора кислоты серной Р слабо-розо¬
вое окрашивание жидкости должно исчезать.
Потеря в массе при высушивании (2.2.32). Не
более 45,0 %. В выпарительную чашку, предваритель¬
но высушенную до постоянной массы, отвешивают
2,000 г испытуемого образца, прибавляют 10 мл 96 %
спирта Р и тщательно перемешивают. Полученный
раствор выпаривают на водяной бане, остаток сушат
при температуре 105 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,000 г испытуемого образца помещают в кони¬
ческую колбу вместимостью 200 мл и растворяют в
100 мл воды Р, прибавляют 10 мл раствора 160 г/л кис¬
лоты серной Р и нагревают на водяной бане до выде¬
ления на поверхности жидкости прозрачного слоя жир¬
ных кислот. Смесь переносят в делительную воронку
и жирные кислоты экстрагируют эфиром Р, прибавляя
сначала 30 мл эфира Р, а затем 3 раза по 20 мл. Эфир¬
ные извлечения фильтруют через фильтр с натрия
сульфатом безводным Р во взвешенную коническую
колбу вместимостью 200 мл. Эфир отгоняют, остаток
высушивают при температуре 75 °С до постоянной
массы и охлаждают в эксикаторе, а затем взвешивают.
ХРАНЕНИЕ
В плотно укупоренном контейнере при темпера¬
туре от 8 °С до 15 °С.
07/2016: РБ0029
#МЫЛО ХОЗЯЙСТВЕННОЕ
ТВЕРДОЕ
М(7Л/0/?У 80АР, НАРЮ
ОПРЕДЕЛЕНИЕ
Содержание: не менее 64,0 % жирных кислот.
ОПИСАНИЕ (СВОЙСТВА)
Твердые на ощупь куски прямоугольной формы
без деформации, трещин и твердых инородных вклю¬
чений; от светло-желтого до коричневого цвета. За¬
пах специфичный мыльный, не должно быть запаха
продуктов разложения органических веществ, прогор¬
клых жиров и других неприятных запахов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Пенообразующая способность», как указано в разде¬
ле «Испытания».
B. Испытуемый образец выдерживает испытание
«Температура затвердевания», как указано в разделе
«Испытания».
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Свободные щелочи. Не более 0,2 %. 5,000 г
испытуемого образца помещают в коническую кол¬
бу вместимостью 250 мл, прибавляют 100 мл 96%
спирта Р, предварительно нейтрализованного по
фенолфталеину. Колбу присоединяют к обратному хо¬
лодильнику и нагревают на водяной бане до раство¬
рения мыла. В горячий раствор прибавляют раствор
100 г/л бария хлорида Р и титруют при интенсивном
взбалтывании 0,1 М раствором кислоты хлори¬
стоводородной. В качестве индикатора используют
0,1 мл раствора фенолфталеина Р1.
Содержание свободных щелочей (X,) в процен¬
тах рассчитывают по формуле:
^ V К-0,004• 100
где:
V — объем 0,1 М раствора кислоты хлористо¬
водородной, израсходованный на титрование испыту¬
емого раствора, мл;
К — поправочный коэффициент к 0,1 М раство¬
ру кислоты хлористоводородной;
0,004 — количество натрия гидроксида, соответ¬
ствующее 1 мл 0,1 М раствора кислоты хлористо¬
водородной, г;
т — масса навески испытуемого образца, г.
Свободный натрия карбонат. Не более 1,0 %.
5,000 г испытуемого образца помещают в коническую
колбу вместимостью 250 мл, прибавляют 75 мл 96 %
спирта Р, предварительно нейтрализованного по фе¬
нолфталеину. Колбу соединяют с обратным холодиль¬
ником и содержимое нагревают на водяной бане до
растворения мыла. После охлаждения раствор титру¬
ют 0,1 М раствором кислоты хлористоводородной.
В качестве индикатора используют 0,1 мл раствора
фенолфталеина Р1.
Содержание свободного натрия карбоната (Х2) в
процентах рассчитывают по формуле:
X,
ооцоо. )
х т '
где:
V — объем 0,1 М раствора кислоты хлористо¬
водородной, израсходованный на титрование испыту¬
емого раствора, мл;
К — поправочный коэффициент к 0,1 М раство¬
ру кислоты хлористоводородной;
0,004 — количество натрия гидроксида, соответ¬
ствующее 1 мл 0,1 М раствора кислоты хлористо¬
водородной, г;
т — масса навески испытуемого образца, г;
X, — содержание свободных щелочей, %;
2,65 — коэффициент пересчета свободных ще¬
лочей на натрия карбонат.
Неомыляемые органические вещества и не¬
омыляемый жир. Не более 3,5% от содержания
жирных кислот. 10,000 г испытуемого образца помеща¬
ют в коническую колбу и растворяют в 75 мл спирта
(60 %, об/об) Р при нагревании на водяной бане. Раст-
#Мыло хозяйственное твердое
701
вор переносят в делительную воронку № 1 вместимо¬
стью 250 мл. Колбу ополаскивают 25 мл спирта (60 %,
об/об) Р. Промывную жидкость помещают в делитель¬
ную воронку № 1. К содержимому воронки прибавляют
50 мл петролейного эфира Р1, сильно встряхивают
и дают отстояться. Нижний слой сливают в делитель¬
ную воронку № 2 вместимостью 250 мл и обрабаты¬
вают 50 мл петролейного эфира Р1 (если образова¬
лась стойкая эмульсия, ее разрушают прибавлением
(1—3)мл 96% спирта Р). Нижний слой удаляют, а
эфирное извлечение помещают в делительную во¬
ронку № 1. Эфирное извлечение промывают спиртом
(60 %, об/об) Р при легком встряхивании до полного
удаления остатков мыла (промывная жидкость, разбав¬
ленная водой и подогретая, не должна окрашиваться
в присутствии фенолфталеина). Эфирное извлечение
фильтруют в предварительно взвешенную коническую
колбу через бумажный фильтр, на который помещают
5,0 г натрия сульфата безводного Р. Фильтр с натрия
сульфатом промывают петролейным эфиром Р1 и
эфир выпаривают на водяной бане при температуре от
35 °С до 45 °С досуха. Полученный остаток сушат при
температуре от 73 °С до 75 °С до постоянной массы.
Содержание суммы неомыляемых органических
веществ и неомыляемого жира (Х3) в процентах от со¬
держания жирных кислот рассчитывают по формуле:
^ тл 100-100
Л О .
т-Х
где:
/771 — масса полученного остатка, г;
т — масса навески испытуемого образца, г;
X — содержание жирных кислот, %.
Пенообразующая способность. Начальная вы¬
сота столба пены должна быть не менее 300 мм. Пе¬
нообразующую способность определяют на приборе
(см. рисунок РБ0029.-1) при температуре 50 °С.
1. Маховик.
2. Шатун.
3. Зажимное кольцо.
4. Гнездо для делительной
воронки.
5. Делительная воронка
(1000 мл).
6. Направляющие планки.
7. Электродвигатель.
8. Редуктор.
Рисунок РБ0029.-1. Прибор для определения
пенообразующей способности мыла
Испытуемый раствор. Навеску испытуемого об¬
разца, соответствующую 1,5 г жирных кислот и взве¬
шенную с точностью 0,001 г, растворяют в нагретой до
50 °С жесткой воде, охлаждают и доводят до объема
300 мл этим же растворителем. Раствор готовят непо¬
средственно перед испытанием.
Приготовление жесткой воды. 0,194 г кальция
хлорида Р и 0,219 г магния сульфата Р растворяют
в воде дистиллированной Р, доводят объем раствора
этим же растворителем до объема 1000 мл и переме¬
шивают.
100.0 мл испытуемого раствора помещают в де¬
лительную воронку прибора, закрывают ее пробкой
и встряхивают в течение 1 мин (около 180 встряхи¬
ваний). Затем быстро вынимают пробку и сразу из¬
меряют объем пены в делительной воронке и ее ко¬
нусной части.
Температура затвердевания (2.2.18). От 35 °С
до 42 °С. 40,0 г измельченного испытуемого образца
растворяют в 300 мл горячей воды Р, прибавляют
0,2 мл раствора метилового оранжевого Р и прибав¬
ляют раствор 300 г/л кислоты серной Р до появления
розового окрашивания водного слоя. Полученную
смесь нагревают до тех пор, пока жирные кислоты не
всплывут наверх в виде прозрачного слоя. Водно-кис¬
лотный слой отбрасывают, а жирные кислоты промы¬
вают горячей водой Р до нейтральной реакции среды
промывных вод по метиловому оранжевому.
После того как жирные кислоты промыты, их ох¬
лаждают и с образовавшейся лепешки удаляют влагу
фильтровальной бумагой. Жирные кислоты помеща¬
ют в сушильный шкаф с температурой выше предпо¬
лагаемой температуры затвердевания на (10—15) °С.
После расплавления жирные кислоты фильтруют че¬
рез двойной складчатый фильтр и определяют темпе¬
ратуру затвердевания.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5.000 г испытуемого образца помещают в кони¬
ческую колбу и растворяют в 60 мл нагретой до ки¬
пения воды дистиллированной Р, раствор охлаждают
до температуры (35—40) °С и переносят в делитель¬
ную воронку № 1 вместимостью 250 мл. К раствору
прибавляют 0,2 мл раствора метилового оранжево¬
го Р и по каплям прибавляют раствор 200 г/л кисло¬
ты хлористоводородной Р до появления неисчеза¬
ющего розового оттенка водного слоя. Содержимое
делительной воронки № 1 перемешивают круговым
вращением и после охлаждения и выделения жирных
кислот прибавляют 50 мл эфира Р. Колбу ополаски¬
вают поочередно водой дистиллированной Р двумя
порциями по 25 мл, 5 мл кислоты хлористоводород¬
ной Р и затем 25 мл эфира Р. Промывные жидкости
помещают в делительную воронку № 1. Содержимое
воронки перемешивают круговым вращением и дают
отстояться. Водно-кислотный слой помещают в дели¬
тельную воронку № 2 вместимостью 250 мл, обраба¬
тывают 30 мл эфира Р и дают отстояться. Водно-кис¬
лотный слой помещают в делительную воронку № 3,
а эфирное извлечение помещают в делительную
воронку № 1. Кислотно-водный слой в делительной
воронке № 3 экстрагируют 25 мл эфира Р. Отстояв¬
шийся водно-кислотный слой удаляют, а эфирное
извлечение помещают в делительную воронку № 1.
702
Гэсударственная фармакопея Республики Беларусь
Делительную воронку №2 ополаскивают эфиром Р
и промывную жидкость помещают в делительную во¬
ронку № 1. Эфирное извлечение в делительной во¬
ронке № 1 промывают раствором 100 г/л натрия хло¬
рида Р порциями по 30 мл до нейтральной реакции
промывной воды по метиловому оранжевому. Эфир¬
ное извлечение фильтруют в предварительно взве¬
шенную коническую колбу через бумажный фильтр,
на который помещают 5,0 г натрия сульфата без¬
водного Р. Делительную воронку и фильтр с натрия
сульфатом промывают эфиром Р и эфир выпаривают
на водяной бане при температуре от 35 °С до 45 °С
досуха. Полученный остаток растворяют в 40 мл 96 %
спирта Р, предварительно нейтрализованного по фе¬
нолфталеину, и титруют 0,5 М раствором натрия ги¬
дроксида этанольным (см. примечание). В качестве
индикатора используют 0,1 мл раствора фенолфта¬
леина Р1. Спирт отгоняют на кипящей водяной бане и
колбу сушат при температуре от 117 °С до 123 °С до
постоянной массы.
Содержание жирных кислот (X) в процентах рас¬
считывают по формуле:
V, _ (т, -\ЛК • 0,011)-100
У\ — ,
т
где:
т — масса навески испытуемого образца, г;
V — объем 0,5 М раствора натрия гидроксида
этанольного, израсходованного на титрование испы¬
туемого раствора, мл;
К — поправочный коэффициент к 0,5 М раствору
натрия гидроксида этанольному;
0,011 — количество жирных кислот, соответству¬
ющее 1 мл 0,5 М раствора натрия гидроксида эта¬
нольного, г;
т1 — масса полученного остатка, г.
Примечание
Приготовление 0,5 М раствора натрия гидрок¬
сида этанольного. К 250 мл этанола Р прибавляют
16,5 г раствора натрия гидроксида концентриро¬
ванного Р.
Установка титра. 40,0 мг кислоты бензойной Р
растворяют в смеси из 2 мл воды Р и 10 мл 96 % спир¬
та Р. Титруют приготовленным 0,5 М раствором на¬
трия гидроксида этанольным. В качестве индикатора
используют 0,2 мл раствора фенолфталеина Р1.
Титр устанавливают непосредственно перед исполь¬
зованием.
1 мл 0,5 М раствора натрия гидроксида этаноль¬
ного соответствует 61,05 мг С7Н602.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от влаги месте.
07/2016:0405
МЯТЫ ПЕРЕЧНОЙ МАСЛО
Меп1Ьае р1регИае ае01его1еит
РЕРРЕРМШТ он.
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное методом перегонки с
водяным паром из свежих надземных частей, собран¬
ных в период цветения, растения МепХЬа * р/регПа Ь.
ОПИСАНИЕ(СВОЙСТВА)
Бесцветная, бледно-желтого или бледного зеле¬
новато-желтого цвета жидкость. Имеет характерный
запах и вкус, оставляющий ощущение холода.
Смешивается с 96 % спиртом и метиленхлори-
дом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А, Просматривают хроматограммы, полученные
в испытании «Мятное масло. А».
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Тимол: зона поглощения
Могут обнаруживаться зоны
поглощения (карвон, пулегон)
Раствор сравнения
Испытуемый раствор
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие менее ин¬
тенсивно окрашенные зоны.
Верх хроматографической пластинки
Зона интенсивного фиолето¬
во-красного цвета (располо¬
жена близко к фронту раст¬
ворителей) (углеводороды)
Зона коричневато-желтого
цвета (ментофуран).
Ментилацетат: зона фиоле¬
тово-синего цвета
Тимол: розовая зона
1,8-Цинеол: зона фиолето¬
во-синего или коричневого
цвета
Зона фиолетово-синего
цвета (ментилацетат)
Зона зеленовато-синего
цвета (ментон)
Могут обнаруживаться зоны
светло-розового, серовато-
синего или серовато-зеле¬
ного цвета (карвон, пулегон,
изоментон)
Слабая зона фиолетово-си¬
него или коричневого цвета
(1,8-цинеол)
Ментол : интенсивная зона
синего или фиолетового
цвета
Интенсивная зона синего
или фиолетового цвета
(ментол)
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Результаты: характерные пики, соответствую¬
щие лимонену, 1,8-цинеолу, ментону, ментофурану,
изоментону, ментилацетату и ментолу, на хромато¬
грамме испытуемого раствора по времени удержи¬
вания соответствуют характерным пикам на хрома¬
тограмме раствора сравнения (а). На хроматограмме
испытуемого раствора могут присутствовать изопуле-
гол, пулегон и карвон.
Напроксен
703
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От0,900 до 0,916.
Показатель преломления (2.2.6). От 1,457 до 1,467.
Угол оптического вращения (2.2.7). От -30° до -10°.
Кислотное число (2.5.1). Не более 1,4. Опреде¬
ление проводят из 5,0 г испытуемого образца, раство¬
ренного в 50 мл предписанной смеси растворителей.
Жирные и минеральные масла (2.6.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Мятное масло
A. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,1 г испытуемого об¬
разца смешивают с толуолом Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 50 мг метанола Р, 20 мкл
цинеола Р 10 мг тимола Р и 10 мкл ментилацета-
та Р растворяют в толуоле Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р254 Р ((2—10) мкм)].
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Объем наносимой пробы: 10 мкл [или 1 мкл]
раствора сравнения и 20 мкл [или 2 мкл] испытуемого
раствора в виде полос длиной 10 мм [или 8 мм].
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Проявление В: пластинку обрабатывают раство¬
ром анисового альдегида Р, нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин и сразу
же просматривают при дневном свете.
Результаты В: на хроматограмме испытуемого
раствора не должна обнаруживаться зона голубого
цвета между зонами, соответствующими 1,8-цинеолу
и ментолу.
B. Просматривают хроматограммы, полученные как
указано в испытании «Хроматографический профиль».
Результаты: на хроматограмме испытуемого
раствора не должен обнаруживаться пик со временем
удерживания изопулегола и площадью более 0,2 % от
общей площади всех пиков.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 0,20 мл испытуемого об¬
разца смешивают с гептаном Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 10 мкл лимонена Р,
20 мкл цинеола Р, 40 мкл ментона Р, 10 мкл менто-
фуранаР, 10 мкл изоментона Р, 40 мкл ментила-
цетата Р, 20 мкл изопулегола Р, 60 мг ментола Р,
20 мкл пулегонаР, 10 мкл пиперитона Р и 10 мкл
карвона Р растворяют в гептане Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 5 мкл изопулегола Р
растворяют в гептане Р и доводят до объема 10,0 мл
этим же растворителем. 0,1 мл полученного раствора
доводят гептаном Р до объема 5,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м и
внутренним диаметром 0,25 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
60
10—70
о>
0
1
00
о
70—75
180
Блок ввода проб
200
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения (а); отмечают време¬
на удерживания всех веществ.
Идентификация пиков: по временам удержива¬
ния компонентов раствора сравнения (а) идентифи¬
цируют компоненты испытуемого раствора.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками лимо¬
нена и 1,8-цинеола; не менее 1,5 между пиками пипе¬
ритона и карвона.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- лимонен: от 1,0 % до 3,5 %;
- 1,8-цинеол: от 3,5 % до 8,0 %;
- ментон: от 14,0 % до 32,0 %;
- ментофуран: от 1,0 % до 8,0 %;
- изоментон: от 1,5 % до 10,0 %;
- ментилацетат: от 2,8 % до 10,0 %;
- изопулегол: не более 0,2 %;
- ментол: от 30,0 % до 55,0 %;
- пулегон: не более 3,0 %;
- карвон: не более 1,0 %;
- неучитываемый предел (0,05 %): не учитыва¬
ют пики с площадью менее площади основного пика
на хроматограмме раствора сравнения (Ь).
Отношение содержания 1,8-цинеола к содержа¬
нию лимонена должно быть не менее 2.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:0731
НАПРОКСЕН
Маргохепит
704
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
ИСПЫТАНИЯ
(25)-2-(6-Метоксинафтален-2-ил)пропановая
кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, растворим в
96 % спирте и в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: А, В, С.
A. Удельное оптическое вращение (2.2.7): от +59
до +62 (в пересчете на сухое вещество). 0,50 г испы¬
туемого образца растворяют в 96 % спирте Р и дово¬
дят до объема 25,0 мл этим же растворителем.
B. Температура плавления (2.2.14): от 154 °С до
158 °С.
C. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объ¬
ема 100,0 мл этим же растворителем. 10,0 мл полу¬
ченного раствора и доводят метанолом Р до объема
100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 262 нм; при 271 нм;
при 316 нм; при 331 нм.
Удельный показатель поглощения в максимуме:
при 262 нм — от 216 до 238; при 271 нм — от 219 до
241; при 316 нм — от 61 до 69; при 331 нм — от 79 до 87.
О. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО напроксена #или спектр, пред¬
ставленный на рисунке #0731 .-1#.
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 25 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
Энантиомерная чистота. Жидкостная хрома¬
тография (2.2.29). Растворы защищают от воздей¬
ствия света.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в тетрагидрофуране Р и доводят
до объема 50,0 мл этим же растворителем. 2,0 мл
полученного раствора доводят подвижной фазой до
объема 20,0 мл.
Раствор сравнения (а). 2,5 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 5 мг ФСО напроксена
рацемического растворяют в 10 мл тетрагидро-
фурана Р и доводят подвижной фазой до объема
100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем тт-акцептором/тт-
донором для хиральных разделений Р (8,8) с разме¬
ром частиц 5 мкм;
- температура: 25 °С;
- подвижная фаза: кислота уксусная ледя¬
ная Р — ацетонитрил Р — 2-пропанол Р — гексан Р
(0,5:5:10:84,5; об/об/об/об);
- скорость подвижной фазы: 2 мл/мин;
- спектрофотометрический детектор, длина
волны 263 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания напроксена.
Рисунок #0731 .-1. Инфракрасный спектр ФСО напроксена
Напроксен
705
Время удерживания: напроксен — около 5 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3 между пиками приме¬
си О и напроксена.
Предельное содержание примеси:
- примесь 6 (не более 2,5 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси С, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы защищают от воздей¬
ствия света.
Испытуемый раствор. 12 мг испытуемого образ¬
ца растворяют в подвижной фазе и доводят до объ¬
ема 20 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (Ь). 6 мг бромометоксинаф-
тола Р (примесь И), 6,0 мг ФСО напроксена приме¬
си 6 мг 6-метокси-2-нафтойной кислоты Р (при¬
месь О), 6 мг (1Р8)-1-(6-метоксинафтален-2-ил)
этанола Р (примесь К) растворяют в ацетонитри¬
ле Р и доводят до объема 10,0 мл этим же раство¬
рителем. К 1,0 мл полученного раствора прибавляют
1.0 мл испытуемого раствора и доводят подвижной
фазой до объема 50,0 мл. 1,0 мл полученного раст¬
вора доводят подвижной фазой до объема 20,0 мл.
Условия хроматографирования:
-колонка длиной 0,10 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 50 °С;
-подвижная фаза: смешивают 42 объема аце¬
тонитрила Р и 58 объемов раствора 1,36 г/л калия
дигидрофосфата Р, предварительно доведенного
кислотой фосфорной Р до рН 2,0;
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания примеси N.
Идентификация пиков примесей: идентифици¬
руют пики примесей К, 1_, N и О, используя хромато¬
грамму раствора сравнения (Ь).
Относительное удерживание (по отношению
к напроксену, время удерживания — около 2,5 мин):
примесь О — около 0,8; примесь К — около 0,9; при¬
месь Ь — около 1,4; примесь N — около 5,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,2 между пиками при¬
меси К и напроксена.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 2,0):
-примесь О (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-примесь I- (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси 1_, не должна превышать
1,5-кратную площадь соответствующего пика на
хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси Ц не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 3-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррпл). 1,0 г испытуемого образца
должен выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием 2 мл
эталонного раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца су¬
шат при температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в смеси
из 25 мл воды Р и 75 мл метанола Р и титруют 0,1 М
раствором натрия гидроксида, используя в качестве
индикатора 1 мл раствора фенолфталеина Р.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 23,03 мг С14Н1403.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, /_, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е, Р,
Н, I, 3, К, М, N.
А. (25)-2-(6-Гидроксинафтален-2-ил)пропановая
кислота.
706
Гэсударственная фармакопея Республики Беларусь
НзССГ
В. (25)-2-(5-Хлор-6-метоксинафтален-2-ил)пропа-
новая кислота.
К рн3
Н3СО'
со2н
С. (25)-2-(5-Бром-6-метоксинафтален-2-ил)пропа-
новая кислота.
Н3СО'
О. (25)-2-(5-Йод-6-метоксинафтален-2-ил)пропа-
новая кислота.
Е. Метил-(25)-2-(6-метоксинафтален-2-ил)пропа-
ноат.
Р. Этил-(25)-2-(6-метоксинафтален-2-ил)пропаноат.
С. (2Я)-2-(6-Метоксинафтален-2-ил)пропановая
кислота.
Н*СО
H. б-Метоксинафтален-2-ол.
I. (6-Метоксинафтален-2-ил)уксусная кислота.
2-Этил-6-метоксинафтол.
о
М. 2-Метоксинафтол (неролин).
N. 2-Бром-6-метоксинафтол.
со2н
н3са
О. 6-Метоксинафтол-2-карбоновая кислота
(6-метокси-2-нафтойная кислота).
07/2016:1993
НАТРИЯ АМИНОСАЛИЦИЛАТ
ДИГИДРАТ
Л/а/г/7 аттозаНсу1аз сШ1ус1псиз
ЗОйШМ АМШОЗАиСПАТЕ
О/НУОЯДГЕ
^/С02Ма
2 Н20
С7Н6ЫЫа03 • 2Н20 М.м. 211,2
ОПРЕДЕЛЕНИЕ
Натрия 4-амино-2-гидроксибензоат дигидрат.
Содержание не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или кристаллы. Слегка гигроскопичен.
Легко растворим в воде, умеренно растворим в
96 % спирте, практически нерастворим в метиленхло-
риде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО натрия аминосалицилата ди¬
гидрата #или спектр, представленный на рисунке
#1993.-1#.
B. 0,3 г испытуемого образца помещают в фар¬
форовый тигель и осторожно нагревают на слабом
огне до выделения паров. Тигель накрывают часовым
стеклом, на котором образуется сублимат. Темпера¬
тура плавления (2.2.14) полученного сублимата: от
120 °С до 124 °С.
К. (1 #5)-1-(6-Метоксинафтален-2-ил)этанол.
Натрия аминосалицилат дигидрат
707
Рисунок #1993.-1. Инфракрасный спектр натрия аминосалицилата дигидрата
С. К 0,1 мл раствора 3, приготовленного как
указано в разделе «Испытания», прибавляют 5 мл
воды Р и 0,1 мл раствора хлорида железа Р1. По¬
является красновато-коричневое окрашивание.
й. 2 мл раствора 3 дают реакцию на первич¬
ные ароматические амины {2.3.1).
Е. 0,5 мл раствора 3 дают реакцию (а) на на¬
трий {2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,50 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р
и доводят до объема 25 мл этим же растворителем.
Раствор 51. 2,5 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 25 мл этим
же растворителем.
Прозрачность {2.2.1). Свежеприготовленный
раствор 31 должен быть прозрачным.
Цветность {2.2.2, метод II). Окраска свеже¬
приготовленного раствора 31 должна быть не ин¬
тенсивнее эталона В(К)5.
рН (2.2.3). От 6,5 до 8,5. Измеряют рН раст¬
вора 3.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Используют свежеприготов¬
ленные растворы и подвижную фазу.
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг 3-аминофено-
ла Р (примесь А) растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО месалази-
на (примесь В) растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем. К 10,0 мл
полученного раствора прибавляют 1,0 мл раст¬
вора сравнения (а) и доводят водой Р до объема
50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикаге¬
лем октилсилильным, деактивированным по от¬
ношению к основаниям, для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: 2,2 г кислоты хлор¬
ной Р и 1,0 г кислоты фосфорной Р растворят
в воде Р и доводят до объема 1000,0 мл этим
же растворителем;
-подвижная фаза В: 1,7 г кислоты хлор¬
ной Р и 1,0 г кислоты фосфорной Р раство¬
рят в ацетонитриле Р и доводят до объема
1000,0 мл этим же растворителем;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
15—30
100
100 — 40
0
0 — 60
- скорость подвижной фазы: 1,25 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 220 нм;
- объем вводимой пробы: по 10 мкл испытуе¬
мого раствора и раствора сравнения (Ь).
Относительное удерживание (по отношению
к 4-аминосалицилату, время удерживания — около
12 мин): примесь А — около 0,30; примесь В — око¬
ло 0,37.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками при¬
меси А и примеси В.
708
Гэсударственная фармакопея Республики Беларусь
Предельное содержание примесей:
-примесь А (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
1,5-кратную площадь соответствующего пика на
хроматограмме раствора сравнения (Ь);
- примесь В (не более 1,0 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси В, не должна превышать пло¬
щадь соответствующего пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А и В, не должна превышать 0,1-кратную
площадь пика примеси В, на хроматограмме раст¬
вора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать площадь пика примеси В на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,05 площади пика при¬
меси В на хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод С). Не бо¬
лее 0,0010% (10 ррт) 2,0 г испытуемого образ¬
ца должны выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием 2 мл
эталонного раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не менее 16,0 % и не более 17,5 %. 1,000 г испыту¬
емого образца сушат при температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
20 мл воды Р, прибавляют 10 мл раствора 500 г/л
натрия бромида Р и 25 мл кислоты уксусной ле¬
дяной Р. Прибавляют 5 мл 0,1 М раствора натрия
нитрита и титруют этим же титрантом потенцио-
метрически (2.2.20).
1 мл 0,1 М раствора натрия нитрита соот¬
ветствует 17,52 мг 07Н6ММаО3.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Стерильная субстанция — в воздухонепроницае¬
мом контейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В.
A. К1 = РЗ = Н, Р2 = 1ЧН2: З-Аминофенол.
B. П1 = С02Н, Р2 = Н, РЗ = ИН2: 5-Амино-2-гид-
роксибензойная кислота (месалазин).
07/2016:0411
НАТРИЯ АЦЕТАТ ТРИГИДРАТ
АШт асе(аз Шудпсиз
ЗОйШМ АСЕТАТЕ ТШНУОКАТЕ
О
С2Н,№02 ■ ЗН20 М.м. 136,1
[6131-90-4]
ОПРЕДЕЛЕНИЕ
Натрия этаноат тригидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень легко растворим в воде, растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», дает реакции (Ь) #и (с)# на
ацетаты (2.3.1).
B. 1 мл раствора 3 дает реакцию (а) на натрий (2.3.1).
C. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 7,5 до 9,0. 5 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объема
10 мл. Измеряют рН полученного раствора.
Восстанавливающие вещества. 5,0 г испытуе¬
мого образца растворяют в 50 мл воды Р, прибавляют
5 мл кислоты серной разведенной Р и 0,5 мл 0,002 М
раствора калия перманганата. Раствор должен со¬
хранять розовое окрашивание в течение не менее 1 ч.
Проводят контрольный опыт с аналогично приготов¬
ленным раствором, но без испытуемого образца.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2.5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
7.5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на сульфаты.
Алюминий (2.4.17). Не более 0,00002%
(0,2 ррт), если субстанция предназначена для произ¬
водства растворов для диализа.
Предписанный раствор. 20 г испытуемого образца
растворяют в 100 мл воды Р и доводят до рН 6,0 1Мрас¬
твором кислоты хлористоводородной (около 10 мл).
Эталон. Смешивают 2 мл эталонного раствора
алюминия (2 ррт А!) Р, 10 мл ацетатного буферно¬
го раствора рН 6,0 Р и 98 мл воды Р.
Натрия бензоат
709
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН6,0Р\л 100 мл воды Р.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Кальций и магний. Не более 0,0050 % (50 ррт)
в пересчете на Са. К 200 мл воды Р прибавляют 10 мл
аммиачного буферного раствора рН 10,0 Р, 0,1 г ин¬
дикаторной смеси протравного черного 11 Р, 2,0 мл
0,05 М раствора цинка хлорида и по каплям 0,02 М
раствор натрия эдетата до изменения окрашива¬
ния от фиолетового до голубого. К полученному рас¬
твору прибавляют 10,0 г испытуемого образца, встря¬
хивают до растворения и титруют 0,02 М раствором
натрия эдетата до восстановления голубого окра¬
шивания. Объем израсходованного 0,02 М раствора
натрия эдетата не должен превышать 0,65 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 10 мл
раствора 3 должны выдерживать испытание на железо.
Потеря в массе при высушивании (2.2.32). Не
менее 39,0 % и не более 40,5 %. 1,000 г испытуемого
образца сушат при температуре 130 °С. Испытуемый
образец помещают в сушильный шкаф перед нагре¬
ванием.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р, прибавляют 5 мл ук¬
сусного ангидрида Р, перемешивают и выдерживают
в течение 30 мин. Полученный раствор титруют 0,1 М
раствором кислоты хлорной до зеленого окрашива¬
ния, используя в качестве индикатора 0,3 мл раст¬
вора нафтолбензеина Р #(или 0,05 мл раствора кри¬
сталлического фиолетового Р)*.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 8,20 мг С2Н3Ыа02.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства раство¬
ров для диализа.
07/2016:0123
НАТРИЯ БЕНЗОАТ
Ыа1п1 Ьепгоаз
800ШМ ВЕЫ20АТЕ
^^^С02Ыа
С7Н5Ыа02 М.м. 144,1
[532-32-1]
ОПРЕДЕЛЕНИЕ
Натрия бензилкарбоксилат.
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический или гранулированный порошок
либо хлопья белого или почти белого цвета. Слегка
гигроскопичен.
Легко растворим в воде, умеренно растворим в
90 % (об/об) спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции (Ь) и (с) на
бензоаты (2.3.1).
B. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
Кислотность или щелочность. К10 мл раствора
3 прибавляют 10 мл воды, свободной от углерода ди¬
оксида, Р и 0,2 мл раствора фенолфталеина Р. При
прибавлении не более 0,2 мл 0,1 М раствора натрия
гидроксида или 0,1 М раствора кислоты хлористо¬
водородной окраска раствора должна измениться.
Галогенопроизводные
Вся используемая стеклянная посуда должна
быть свободна от хлоридов. Для этого посуду вы¬
держивают в течение ночи в растворе 500 г/л кис¬
лоты азотной Р, промывают водой Р и хранят по¬
груженной в воду Р. Рекомендуется использовать
отдельную стеклянную посуду исключительно для
данного испытания.
Испытуемый раствор. К 20,0 мл раствора 3
прибавляют 5 мл воды Р и доводят 96 % спиртом Р
до объема 50,0 мл.
Определение хлорид-иона. Не более 0,0200 %
(200 ррт).
В трех мерных колбах вместимостью 25 мл гото¬
вят следующие растворы.
Раствор (а). К 4,0 мл испытуемого раствора при¬
бавляют 3 мл раствора натрия гидроксида разве¬
денного Р и 3 мл 96 % спирта Р. Полученный раствор
используют для приготовления раствора А.
Раствор (Ь). К 3 мл раствора натрия гидрок¬
сида разведенного Р прибавляют 2 мл воды Р и 5 мл
96 % спирта Р. Полученный раствор используют для
приготовления раствора В.
Раствор (с). К 4,0 мл эталонного раствора хлори¬
да (8 ррт С1) Р прибавляют 6,0 мл воды Р. Полученный
раствор используют для приготовления раствора С.
В четвертую мерную колбу вместимостью 25 мл
помещают 10 мл воды Р.
В каждую колбу прибавляют по 5 мл раствора
железа (!!!) аммония сульфата Р5 и перемешивают.
К полученным растворам по каплям при перемеши¬
вании вращательными движениями прибавляют по
2 мл кислоты азотной Р и по 5 мл раствора ртути
(II) тиоцианата Р. Колбы встряхивают и содержимое
каждой колбы доводят водой Р до объема 25,0 мл.
710
Гэсударственная фармакопея Республики Беларусь
Полученные растворы выдерживают в водяной бане
при температуре 20 °С в течение 15 мин. Измеряют
оптическую плотность (2.2.25) раствора А при дли¬
не волны 460 нм в кювете с толщиной слоя 2 см, ис¬
пользуя раствор В в качестве компенсационного раст¬
вора, и оптическую плотность раствора С, используя
в качестве компенсационного раствора раствор, полу¬
ченный с использованием 10 мл воды Р. Оптическая
плотность раствора А не должна превышать оптиче¬
скую плотность раствора С.
Определение общего хлора. Не более 0,0300 %
(300 ррт).
Раствор (а). К 10,0 мл испытуемого раствора
прибавляют 7,5 мл раствора натрия гидроксида
разведенного Р, 0,125 г никель-алюминиевого спла¬
ва Р и нагревают на водяной бане в течение 10 мин.
Охлаждают до комнатной температуры, фильтруют в
мерную колбу вместимостью 25 мл и фильтр промы¬
вают трижды 96 % спиртом Р порциями по 2 мл (воз¬
можно образование едва заметного осадка, который
растворяется при подкислении). Фильтрат и промыв¬
ные воды объединяют и доводят водой Р до объема
25,0 мл. Полученный раствор используют для приго¬
товления раствора А.
Раствор (Ь). Готовят аналогично, используя вме¬
сто испытуемого раствора смесь из 5 мл 96 % спир¬
та Р и 5 мл воды Р Полученный раствор используют
для приготовления раствора В.
Раствор (с). К 6,0 мл эталонного раствора хлори¬
да (8 ррт С!) Р прибавляют 4,0 мл воды Р Полученный
раствор используют для приготовления раствора С.
В четыре мерных колбы вместимостью 25 мл
помещают по отдельности по 10 мл раствора (а),
раствора (Ь), раствора (с) и воды Р. В каждую колбу
прибавляют по 5 мл раствора железа (III) аммония
сульфата Р5 и перемешивают. К полученным раство¬
рам по каплям, при перемешивании вращательными
движениями, прибавляют по 2 мл кислоты азотной Р
и по 5 мл раствора ртути (II) тиоцианата Р Колбы
встряхивают и содержимое каждой доводят водой Р
до объема 25,0 мл. Полученные растворы выдержи¬
вают в водяной бане при температуре 20 °С в течение
15 мин. Измеряют оптическую плотность (2.2.25) раст¬
вора А при длине волны 460 нм в кювете с толщиной
слоя 2 см, используя раствор В в качестве компенса¬
ционного раствора, и оптическую плотность раствора
С, используя в качестве компенсационного раствора
раствор, полученный с использованием 10 мл воды Р.
Оптическая плотность раствора А не должна превы¬
шать оптическую плотность раствора С.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 2,0%. 1,00 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 20 мл
кислоты уксусной безводной Р, при необходимости
нагревая до температуры 50 °С, охлаждают и титруют
0,1 М раствором кислоты хлорной до зеленого окра¬
шивания, используя в качестве индикатора 0,05 мл
раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 14,41 мг С7Н5Ма02.
07/2016:0190
НАТРИЯ БРОМИД
А/а^г/7 Ьгот1с1ит
300ШМ ВКОМЮЕ
ЫаВг М.м. 102,9
[7647-15-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый гранулированный поро¬
шок или мелкие бесцветные прозрачные или непро¬
зрачные кристаллы. Слегка гигроскопичен.
Легко растворим в воде, растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на бро¬
миды (2.3.1).
B. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», дает реакции на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 8 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 8 прибавляют 0,1 мл раствора бромтимоло-
вого синего Р1. При прибавлении не более 0,5 мл
0,01 М раствора хлористоводородной кислоты или
0,01 М раствора натрия гидроксида окраска раст¬
вора должна измениться.
Броматы. К 10 мл раствора 8 прибавляют 1 мл
раствора крахмала Р, 0,1 мл раствора 100 г/л калия
йодида Р, 0,25 мл 0,5 М раствора серной кислоты и
выдерживают в течение 5 мин в защищенном от света
месте. Не должно появляться синее или фиолетовое
окрашивание раствора.
Хлориды и сульфаты. Жидкостная хроматогра¬
фия (2.2.29).
Испытуемый раствор (а). 0,400 г испытуемого
образца растворяют в 50 мл воды для хроматогра¬
фии Р и доводят до объема 100,0 мл этим же раство¬
рителем.
Испытуемый раствор (Ь). 25,0 мл испытуемого
раствора (а) доводят водой для хроматографии Р до
объема 50,0 мл.
Раствор сравнения (а). К 25,0 мл испытуемого
раствора (а) прибавляют 1,0 мл эталонного раст¬
вора сульфата (10 ррт 80^ Р и 12,0 мл эталонного
раствора хлорида (50 ррт С1) Р и доводят водой для
хроматографии Р до объема 50,0 мл.
Раствор сравнения (Ь). 10,0 мл испытуемого
раствора (а) доводят водой для хроматографии Р
до объема 100,0 мл. К 2,0 мл полученного раствора
Натрия гиалуронат
711
прибавляют 8,0 мл эталонного раствора хлорида
(50 ррт С1) Р и доводят водой для хроматографии Р
до объема 20,0 мл.
Холостой раствор. Вода для хроматографии Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 2 мм, заполненная анионообменной смолой
сильноосновной для хроматографии Р с размером
частиц 13 мкм;
- подвижная фаза: 0,600 г калия гидроксида Р
растворяют в воде для хроматографии Р и доводят
до объема 1000,0 мл этим же растворителем;
- скорость подвижной фазы: 0,4 мл/мин;
- кондуктометрический детектор, оборудо¬
ванный подходящим подавителем ионов;
- объем вводимой пробы: по 50 мкл испытуемого
раствора (Ь), растворов сравнения (а) и (Ь) и холосто¬
го раствора;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания бромидов.
Время удерживания: хлориды — около 5 мин;
бромиды — около 8 мин; сульфаты — около 16 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 8,0 между пиками хло¬
ридов и бромидов.
Расчет процентных содержаний:
-для хлоридов — используют концентрацию
хлоридов в растворе сравнения (а); корректируют
площадь пика, соответствующего хлоридам на хрома¬
тограмме раствора сравнения (а), вычитая площадь
пика, соответствующего хлоридам на хроматограмме
испытуемого раствора (Ь);
-для сульфатов — используют концентрацию
сульфатов в растворе сравнения (а); корректируют
площадь пика, соответствующего сульфатам на хро¬
матограмме раствора сравнения (а), вычитая пло¬
щадь пика, соответствующего сульфатам на хромато¬
грамме испытуемого раствора (Ь).
Предельное содержание примесей:
- хлориды: не более 0,6 %;
- сульфаты: не более 0,01 %.
Йодиды. К 5 мл раствора 3 прибавляют 0,15 мл
раствора железа (III) хлорида Р1, 2 мл метиленхло-
рида Р и встряхивают. После разделения слоев ниж¬
ний слой должен быть бесцветным (2.2.2, метод /).
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Магний и щелочноземельные металлы (2.4.7).
Не более 0,0200 % (200 ррт) в пересчете на кальций.
10,0 г испытуемого образца должны выдерживать ис¬
пытание на магний и щелочноземельные металлы.
Объем использованного 0,01 М раствора натрия
эдетата не должен превышать 5,0 мл.
#Барий. К 5 мл раствора 3 прибавляют 5 мл
воды дистиллированной Р и 1 мл кислоты серной
разведенной Р. Через 15 мин опалесценция в полу¬
ченном растворе должна быть не интенсивнее сме¬
си из 5 мл раствора 3 и 6 мл воды дистиллирован¬
ной Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 3,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
85,0 мг испытуемого образца растворяют в
воде Р, прибавляют 5 мл кислоты азотной разведен¬
ной Р, доводят водой Р до объема 50 мл и титруют
0,1 М раствором серебра нитрата потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 10,29 мг ИаВг.
Содержание ЫаВг в процентах рассчитывают по
формуле:
а-2,902Ь,
где:
а — содержание №Вг и №С1, полученное при
количественном определении, рассчитанное как со¬
держание №Вг;
Ь — содержание хлора, полученное в испытании
на хлориды.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:1472
НАТРИЯ ГИАЛУРОНАТ
Л/аУг/У Ьуа/игопаз
500/1Ш НУАШКОЫАТЕ
(С^Н^МЫаО,^
[9067-32-7]
ОПИСАНИЕ (СВОЙСТВА)
Натриевая соль гиалуроновой кислоты, глико-
заминогликан, состоящий из дисахаридных звеньев
й-глюкуроновой кислоты и Л/-ацетил-0-глюкозамина.
Экстрагируют из петушиных гребней или получают
ферментацией из 81гер1ососс1 групп А и С по класси¬
фикации Лэнсфилд.
Содержание: не менее 95,0 % и не более 105,0 %
(в пересчете на сухое вещество).
Характеристическая вязкость: не менее 90 % и
не более 120 % от указанной на этикетке.
ПРОИЗВОДСТВО
Если применимо, животные, используемые для
получения натрия гиалуроната, должны выдерживать
712
Гэсударственная фармакопея Республики Беларусь
требования к животным, пригодным для потребления
человеком.
При получении с помощью ферментации грампо-
ложительных бактерий должно быть показано, что в
ходе процесса происходит уменьшение или удаление
пирогенных или воспалительных компонентов клеточ¬
ной оболочки.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или волокни¬
стые скопления. Очень гигроскопичен.
Умеренно растворим или растворим в воде, прак¬
тически нерастворим в ацетоне и этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр натрия гиалуро-
ната по Европейской Фармакопее.
B. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. К количеству испытуемого образца,
эквивалентному 0,10 г сухого вещества, прибавляют
30.0 мл раствора 9 г/л натрия хлорида Р. Осторожно
перемешивают на шейкере до растворения (около 12 ч).
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,01.
Измеряют оптическую плотность раствора 3 при дли¬
не волны 600 нм.
рН (2.2.3). От 5,0 до 8,5. Готовят раствор испыту¬
емого образца с концентрацией 5 мг/мл (в пересчете
на сухое вещество) в воде, свободной от углерода
диоксида, Р.
Характеристическая вязкость. Натрия гиалу-
ронат очень гигроскопичен и должен быть защищен
от влаги в течение взвешивания.
Буферный раствор (0,15 М раствор натрия
хлорида в 0,01 М фосфатном буферном растворе
рН 7,0). 0,78 г натрия дигидрофосфата Р и 4,50 г
натрия хлорида Р растворяют в воде Р и доводят до
объема 500,0 мл этим же растворителем (раствор А).
1,79 г динатрия гидрофосфата Р и 4,50 г натрия
хлорида Р растворяют в воде Р и доводят этим же
растворителем до объема 500,0 мл (раствор В). Сме¬
шивают растворы А и В до установления рН 7,0, затем
фильтруют через стеклянный фильтр (4) (2.1.2).
Испытуемый раствор (а). 0,200 г (т0р) испыту¬
емого образца взвешивают (примечание: приведен¬
ное значение является ориентировочным и должно
быть откорректировано после первоначального
измерения вязкости испытуемого раствора (а)) и
разводят с помощью 50,0 г (т0а) буферного раствора
при температуре 4 °С. Раствор встряхивают при тем¬
пературе 4 °С в течение 24 ч. Взвешивают 5,00 г (т1р)
полученного раствора и разводят с помощью 100,0 г
(ти) буферного раствора при температуре 25 °С.
Полученный раствор встряхивают в течение 20 мин.
Раствор фильтруют через стеклянный фильтр (100)
(2.1.2), отбрасывая первые 10 мл.
Испытуемый раствор (Ь). 30,0 г (т2р) испытуе¬
мого раствора (а) взвешивают и разводят с помощью
10.0 г (л725) буферного раствора при температуре
25 °С. Полученный раствор встряхивают в течение
20 мин и затем фильтруют через стеклянный фильтр
(100) (2.1.2), отбрасывая первые 10 мл.
Испытуемый раствор (с). 20,0 г (т3р) испытуе¬
мого раствора (а) взвешивают и разводят с помощью
20.0 г (т3з) буферного раствора при температуре
25 °С. Полученный раствор встряхивают в течение
20 мин, затем фильтруют через стеклянный фильтр
(100) (2.1.2), отбрасывая первые 10 мл.
Испытуемый раствор (д). 10,0 г (т4р) испытуе¬
мого раствора (а) взвешивают и разводят с помощью
30.0 г (тА$) буферного раствора при температуре
25 °С. Полученный раствор встряхивают в течение
20 мин, затем фильтруют через стеклянный фильтр
(100) (2.1.2), отбрасывая первые 10 мл.
Определяют значения времени вытекания (2.2.9)
для буферного раствора (/0) и для 4 испытуемых рас¬
творов (^, Х2, *3 и /4) при температуре (25,00±0,03) °С.
Используют соответствующий вискозиметр с вися¬
чим уровнем (технические условия: постоянная ви¬
скозиметра — около 0,005 мм^с2, кинематическая
вязкость— (1—5)мм2/с, внутренний диаметр труб¬
ки/? — 0,53 мм, объем расширения С — 5,6 мл, вну¬
тренний диаметр трубки N — (2,8—3,2) мм) с ворон¬
кообразным нижним капиллярным концом. Для всех
измерений используют один и тот же вискозиметр;
все значения времени вытекания жидкости опреде¬
ляют трижды. Испытание считается пригодным, если
результаты отличаются не более чем на 0,35 % от
среднего значения и значение времени вытекания
^ составляет не менее 1,6 и не более 1,8 значения
времени удерживания /0. В ином случае корректируют
значение л?0ри повторяют испытание.
Расчет относительных вязкостей.
Так как плотности растворов натрия гиалуроната
и растворителя почти равны, относительные вязкости
рн (а именно рл, ра, рл и р^) могут рассчитываться из
отношения значений времени вытекания для соответ¬
ствующих растворов (а именно (2, /3 и /4) к времени
вытекания растворителя /0, но принимая во внима¬
ние корректирующий фактор кинетической энергии
для капилляра (В = 30 800 с2), используя следующую
формулу:
Расчет концентраций.
Концентрацию сг кг/м3, натрия гиалуроната в ис¬
пытуемом растворе (а) рассчитывают по формуле:
т0р ■ х • (100 - Л) ■ т1р ■ д25
Ю0 -100 • (т0р + /Т705 ) • (/л1р + ти) ’
где:
х — содержание натрия гиалуроната, определен¬
ного как указано в разделе «Количественное опреде¬
ление», %;
/? — потеря в массе при высушивании, %;
р25 — 1005 кг/м3 (плотность испытуемого раст¬
вора при температуре 25 °С).
Концентрацию с2, кг/м3, натрия гиалуроната в ис¬
пытуемом растворе (Ь) рассчитывают по формуле:
с тзр
1 ™3*+т3р'
Концентрацию с3, кг/м3, натрия гиалуроната в ис¬
пытуемом растворе (с) рассчитывают по формуле:
Натрия гиалуронат
713
с, ^—.
™3* + т3р
Концентрацию с4, кг/м3, натрия гиалуроната в ис¬
пытуемом растворе (с!) рассчитывают по формуле:
с т'р
1 т*з + тАр '
Расчет характеристической вязкости.
Характеристическую вязкость [р] рассчитывают
методом наименьших квадратов линейного регресси¬
онного анализа, используя уравнение Мартина:
!од^^ = 1од[//]+/ф;]с.
Десятичный антилогарифм значения в точке
пересечения с осью ординат является характери¬
стической вязкостью, кг/м3.
Сульфатированные гликозаминогликаны.
Не более 1 %, если продукт экстрагируется из пе¬
тушиных гребней.
Необходимо соблюдать меры предосторож¬
ности при работе с хлорной кислотой при повы¬
шенной температуре.
Испытуемый раствор. Испытуемый образец
в количестве, эквивалентном 50,0 мг сухого веще¬
ства, помещают в пробирку длиной 150 мм и вну¬
тренним диаметром 16 мм и растворяют в 1,0 мл
кислоты хлорной Р.
Раствор сравнения. 0,149 г натрия сульфата
безводного Р растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем. 10,0 мл
полученного раствора доводят водой Р до объема
100.0 мл. 1,0 мл полученного раствора выпарива¬
ют в пробирке длиной 150 мм и внутренним диа¬
метром 16 мм в нагревательном блоке при темпе¬
ратуре от 90 °С до 95 °С и остаток растворяют в
1.0 мл кислоты хлорной Р.
Закупоривают каждую пробирку кусочком сте¬
кловолокна. Пробирки помещают в нагреватель¬
ный блок или силиконовую масляную баню, под¬
держивающую температуру 180 °С, и нагревают до
образования прозрачных, бесцветных растворов
(около 12 ч). Вынимают пробирки, охлаждают до
комнатной температуры и в каждую пробирку при¬
бавляют 3,0 мл раствора 33,3 г/л бария хлорида Р,
закрывают и интенсивно встряхивают. Пробирки
выдерживают в течение 30 мин, затем встряхивают
повторно каждую пробирку и определяют оптиче¬
скую плотность (2.2.25) при длине волны 660 нм,
используя воду Р в качестве компенсационного
раствора.
Оптическая плотность испытуемого раствора
должна быть не больше оптической плотности
раствора сравнения.
Нуклеиновые кислоты. Оптическая плот¬
ность (2.2.25) раствора 5 при длине волны 260 нм
должна быть не более 0,5.
Белок. Не более 0,3 %. Не более 0,1 %, если
субстанция предназначена для производства ле¬
карственных средств для парентерального приме¬
нения.
Испытуемый раствор (а). Готовят раствор
испытуемого образца с концентрацией 10 мг/мл (в
пересчете на сухое вещество) в воде Р.
Испытуемый раствор (Ь). Смешивают равные
объемы испытуемого раствора (а) и воды Р.
Растворы сравнения. Готовят исходный
раствор альбумина бычьего Р с концентрацией
0,5 мг/мл в воде Р. ,Путем разведений исходно¬
го раствора готовят 5 растворов с концентрацией
альбумина бычьего Р между 5 мкг/мл и 50 мкг/мл.
По 2,5 мл свежеприготовленного медно-тар-
тратного раствора РЗ помещают в пробирки, со¬
держащие 2,5 мл воды Р (компенсационный раст¬
вор), по 2,5 мл испытуемых растворов (а) или (Ь)
или по 2,5 мл растворов сравнения. Перемеши¬
вают после каждого прибавления. Приблизитель¬
но через 10 мин прибавляют в каждую пробирку
0,50 мл смеси из равных объемов фосфорномо-
либденово-вольфрамового реактива Р и воды Р,
приготовленной непосредственно перед приме¬
нением. Перемешивают после каждого прибавле¬
ния. Через 30 мин измеряют оптическую плотность
(2.2.25) каждого раствора по отношению к компен¬
сационному раствору при длине волны 750 нм. По
градуировочному графику, полученному с исполь¬
зованием 5 растворов сравнения, определяют со¬
держание белка в испытуемых растворах.
Хлориды (2.4.4). Не более 0,5 %. 67 мг испы¬
туемого образца растворяют в 100 мл воды Р.
Железо. Не более 0,0080 % (80 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. Испытуемый образец
в количестве, эквивалентном 0,25 г в пересчете на
сухое вещество, растворяют в 1 мл кислоты азот¬
ной Р, нагревая на водяной бане. Охлаждают и до¬
водят водой Р до объема 10,0 мл.
Растворы сравнения. Готовят 2 раствора
сравнения таким же образом, как и испытуемый
раствор, прибавляя соответственно 1,0 мл и 2,0 мл
эталонного раствора железа (10 ррт Ре) Р к рас¬
творенному испытуемому образцу.
Источник излучения: лампа с полым катодом
для определения железа с использованием шири¬
ны щели 0,2 нм.
Длина волны: 248,3 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020% (20 ррт); не более 0,0010% (10 ррт),
если субстанция предназначена для производства
лекарственных средств для парентерального при¬
менения. 1,0 г испытуемого образца должен вы¬
держивать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 20,0 %. 0,500 г испытуемого образца су¬
шат при температуре от 100 °С до 110 °С над фос¬
фора (V) оксидом Р в течение 6 ч.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
Определение проводят из 1 г испытуемого об¬
разца.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 0,5 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без дальнейшей подходящей
процедуры удаления бактериальных эндотоксинов;
менее 0,05 МЕ/мг, если субстанция предназначена
для производства внутриглазных лекарственных
средств или внутриглазных лекарственных средств
без дальнейшей подходящей процедуры удаления
бактериальных эндотоксинов.
714
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Содержание кислоты глюкуроновой определяют
с помощью реакции взаимодействия с карбазолом,
как описано ниже.
Реактив А. 0,95 г динатрия тетрабората Р
растворяют в 100,0 мл кислоты серной Р.
Реактив В. 0,125 г карбазола Р растворяют в
100.0 мл этанола Р.
Испытуемый раствор. Данный раствор гото¬
вят в трех повторностях. 0,170 г испытуемого об¬
разца растворяют в воде Р и доводят до массы
100.0 г этим же растворителем. 10,0 г полученного
раствора доводят водой Р до массы 200,0 г.
Исходный раствор сравнения. 0,100 г кисло¬
ты О-глюкуроновой Р, предварительно высушен¬
ной до постоянной массы в вакууме над фосфора
(V) оксидом Р (2.2.32), растворяют в воде Р и дово¬
дят до массы 100,0 г этим же растворителем.
Растворы сравнения. Готовят 5 раство¬
ров сравнения с концентрациями кислоты
О-глюкуроновой Р в диапазоне от 6,5 мг/г до 65 мг/г
путем разведения исходного раствора сравнения.
25 пробирок, пронумерованных от 1 до 25, по¬
мещают в ледяную баню. В пробирки от 1 до 15 при¬
бавляют по 1,0 мл 5 растворов сравнения в трех
повторностях (пробирки с раствором сравнения), в
пробирки от 16 до 24 прибавляют по 1,0 мл 3 испыту¬
емых раствора трижды (пробирки с испытуемым об¬
разцом) и в пробирку 25 прибавляют 1,0 мл воды Р
(компенсационный раствор). В каждую пробирку
прибавляют по 5,0 мл свежеприготовленного реакти¬
ва А, предварительно охлажденного в ледяной бане.
Пробирки плотно закрывают пластиковыми пробка¬
ми, встряхивают и помещают на водяную баню точно
на 15 мин. Охлаждают в ледяной бане и прибавляют
в каждую пробирку по 0,20 мл реактива В. Повторно
закрывают пробирки, встряхивают и помещают на
водяную баню точно на 15 мин. Охлаждают до ком¬
натной температуры и измеряют оптическую плот¬
ность (2.2.25) растворов по отношению к компенса¬
ционному раствору при длине волны 530 нм.
По градуировочному графику, построенному
по средним значениям оптической плотности для
каждого раствора сравнения, определяют средние
значения концентрации кислоты О-глюкуроновой в
испытуемых растворах.
Содержание натрия гиалуроната в процентах
рассчитывают по формуле:
сд 7 100 401,3
с$ 100-/) 194,1 ’
где:
сд — средние значения концентрации кислоты
О-глюкуроновой в испытуемых растворах, мг/г;
с5 — средние значения концентрации испытуемо¬
го образца в испытуемых растворах, мг/г;
2 — определенное содержание СбН10О7 в кисло¬
те О-глюкуроновой Р, %;
/7 — потеря в массе при высушивании, %;
401,3 — относительная молекулярная масса дис-
ахаридного фрагмента;
194,1 —относительная молекулярная масса кис¬
лоты глюкуроновой.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света и влаги месте. Стерильную субстан¬
цию хранят в стерильном воздухонепроницаемом
контейнере с контролем первого вскрытия.
МАРКИРОВКА
Указывают:
- характеристическую вязкость;
- источник получения субстанции;
- для каких целей предназначена субстанция.
При необходимости указывают:
- субстанция пригодна для парентерального при¬
менения, кроме внутрисуставного введения;
- субстанция пригодна для парентерального при¬
менения, включая внутрисуставное введение;
- субстанция пригодна для внутриглазного вве¬
дения.
07/2016:0195
НАТРИЯ ГИДРОКАРБОНАТ
Л/а?л/ ЬудгодепосагЬопаз
800ШМ НУОНОвЕЫ САКВОЫАТЕ
ЫаНСО, М.м. 84,0
[144-55-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде, практически нерастворим в
96 % спирте.
При нагревании в сухом виде или в растворе по¬
степенно превращается в натрия карбонат.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 5 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,1 мл раст¬
вора фенолфталеина Р. Появляется бледно-розовое
окрашивание. Нагревают. Выделяются пузырьки газа,
и появляется красное окрашивание.
B. Испытуемый образец дает реакцию (а) на кар¬
бонаты и гидрокарбонаты (2.3.1).
C. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в 90 мл воды, свободной от углерода диокси¬
да, Р и доводят до объема 100,0 мл этим же раство¬
рителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Карбонаты. Значение рН свежеприготовленного
раствора 3 не должно превышать 8,6.
Хлориды (2.4.4). Не более 0,0150% (150 ррт).
К 7 мл раствора 3 прибавляют 2 мл кислоты азот¬
ной Р и доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0150 % (150 ррт).
К суспензии из 1,0 г испытуемого образца и 10 мл
воды дистиллированной Р прибавляют кислоту
хлористоводородную Р до нейтральной реакции
среды и еще 1 мл кислоты хлористоводородной Р.
Доводят водой дистиллированной Р до объема 15 мл.
Натрия дигидрофосфат дигидрат
715
Аммония соли (2.4.7). Не более 0,0020%
(20ррт). 10 мл раствора 5 доводят водой Р до
объема 15мл. Эталон готовят с использованием
смеси из 5 мл воды Р и 10 мл эталонного раствора
аммония (1 ррт N4^ Р.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). Определение проводят из 0,5 г испытуемого
образца.
Кальций (2.4.3). Не более 0,0100% (100 ррт).
К суспензии из 1,0 г испытуемого образца и 10 мл
воды дистиллированной Р прибавляют кислоту
хлористоводородную Рдо нейтральной реакции среды
и доводят водой дистиллированной Рдо объема 15 мл.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 0,5 г
испытуемого образца растворяют в 5 мл кислоты
хлористоводородной разведенной Р и доводят
водой Рдо объема 10 мл.
Тяжелые металлы (2.4.8, метод А). Не
более 0,0010% (10 ррт). 2,0 г испытуемого
образца растворяют в смеси из 2 мл кислоты
хлористоводородной Р и 18 мл воды Р. 12 мл
полученного раствора должны выдерживать испытание
на тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.7.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,500 г испытуемого образца растворяют в 50 мл
воды, свободной от углерода диоксида, Р и титруют
7 М раствором кислоты хлористоводородной,
используя в качестве индикатора 0,2 мл раствора
метилового оранжевого Р.
1 мл 7 Мраствора кислоты хлористоводородной
соответствует 84,0 мг №НС03.
07/2016:0677
НАТРИЯ ГИДРОКСИД
Л/а/г/У Ьус1гох1с1ит
800ШМ НУОНОХЮЕ
№ОН М.м. 40,00
[1310-73-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 97,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая кристаллическая масса в
виде гранул, палочек или пластинок. Расплывается на
воздухе, легко поглощая диоксид углерода.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. рН (2.2.3): не менее 11,0. 0,1 г испытуемого об¬
разца растворяют в 10 мл воды Р. 1 мл полученного
раствора доводят водой Р до объема 100 мл.
B. 2 мл раствора 3, приготовленного как указано в
разделе «Испытания», дают реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. Раствор готовят с осторожно¬
стью. 5,0 г испытуемого образца растворяют в 12 мл
воды Р и прибавляют 17 мл кислоты хлористоводо¬
родной Р1. Доводят до рН 7 раствором 103 г/л кис¬
лоты хлористоводородной Р и разводят водой Р до
объема 50 мл.
Раствор 51. 1,0 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.7). Раствор 51 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 51 должен
быть бесцветным.
Карбонаты. Не более 2,0 % в пересчете на
Ма2С03. Испытания проводят, как указано в разделе
«Количественное определение».
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в 5 мл воды Р,
подкисляют раствор 4 мл кислоты азотной Р и дово¬
дят водой Рдо объема 15 мл. Полученный раствор без
дальнейшего прибавления кислоты азотной разве¬
денной Р должен выдерживать испытание на хлориды.
Сульфаты (2.4.73). Не более 0,0200 % (200 ррт).
0,75 г испытуемого образца растворяют в 6 мл воды
дистиллированной Р. Доводят до рН 7 кислотой хло¬
ристоводородной Р (около 7,5 мл) и доводят водой
дистиллированной Р до объема 15мл. Полученный
раствор должен выдерживать испытание на сульфаты.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 5 должны выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца растворяют в 80 мл
воды, свободной от углерода диоксида, Р, прибавля¬
ют 0,3 мл раствора фенолфталеина Р и титруют 7 М
раствором кислоты хлористоводородной #до обесц¬
вечивания раствора#. Затем прибавляют 0,3 мл раст¬
вора метилового оранжевого Р и продолжают титро¬
вать 7 М раствором кислоты хлористоводородной
#до перехода окраски раствора от желтой до красной#.
1 мл 7 М раствора кислоты хлористоводород¬
ной, израсходованной на вторую часть титрования,
соответствует 0,1060 г Ма2С03.
1 мл 7 /14 раствора кислоты хлористоводород¬
ной, израсходованной на все титрование, соответ¬
ствует 40,00 мг общего содержания щелочи в пере¬
счете на №ОН.
ХРАНЕНИЕ
В воздухонепроницаемом неметаллическом кон¬
тейнере.
07/2016:0194
НАТРИЯ ДИГИДРОФОСФАТ
Д И ГИДРАТ
Л/а^г/7 сПЬуйгодепорЬозрЬаз сПЬуйпсиз
3(ЮШМ 01НУ0К06ЕЫ РНОЗРНАТЕ
01НУОРАТЕ
№Н2Р04 • 2Н20 М.м. 156,0
[13472-35-0]
716
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Очень легко растворим в воде, очень мало рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в
разделе «Испытания», имеет слабокислую реакцию
(2.2.4).
B. Раствор 3 дает реакции (а) и (Ь) на фосфаты
(2.3.1).
C. Раствор 3, предварительно нейтрализован¬
ный раствором 100 г/л калия гидроксида Р, дает реак¬
цию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р,
полученной из воды дистиллированной Р, и доводят
этим же растворителем до объема 100 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,2 до 4,5. К 5 мл раствора 3 при¬
бавляют 5 мл воды, свободной от углерода диокси¬
да, Р. Измеряют рН полученного раствора.
Восстанавливающие вещества. К 5 мл раст¬
вора 3 прибавляют 0,25 мл 0,02 М раствора калия
перманганата и 5 мл кислоты серной разведенной Р
и нагревают в водяной бане в течение 5 мин. Окраска
перманганата не должна полностью исчезать.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
2.5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
К 5 мл раствора 3 прибавляют 0,5 мл кислоты хлори¬
стоводородной Р и доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 3 должны выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). От
21.5 % до 24,0 %. 0,50 г испытуемого образца сушат
при температуре 130 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,500 г испытуемого образца растворяют в 40 мл
воды Р и титруют 1 М раствором натрия гидрокси¬
да, свободным от карбонатов, потенциометрически
(2.2.20).
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 0,120 г МаН2Р04.
07/2016:0196
НАТРИЯ ЙОДИД
Л/а&7/ юёШит
8001СМ КЮЮЕ
N31 М.м. 149,9
[7681-82-5]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Гигроскопичен.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции (а) и (Ь) на йодиды
(2.3.1) .
B. Раствор 3 дает реакции (а) и (Ь) на натрий
(2.3.1) .
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Щелочность. К 12,5 мл раствора 3 прибавляют
0,1мл раствора бромтимолового синего Р1. При
прибавлении не более 0,7 мл 0,01 М раствора кис¬
лоты хлористоводородной цвет индикатора должен
измениться.
Йодаты. К 10 мл раствора 3 прибавляют 0,25 мл
раствора крахмала, свободного от йодидов, Р и
0,2 мл кислоты серной разведенной Р и выдержива¬
ют в течение 2 мин в защищенном от света месте. Не
должно появляться синее окрашивание.
Сульфаты (2.4.13). Не более 0,0150 %(150 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл.
Тиосульфаты. К 10 мл раствора 3 прибавляют
0,1 мл раствора крахмала Р и 0,1 мл 0,005 М раст¬
вора йода. Должно появиться синее окрашивание.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 3,0 %. 1,000 г испытуемого образца сушат при
105 °С в течение 3 ч.
Натрия каприлат
717
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,300 г испытуемого образца растворяют в
воде Р и доводят до объема 100,0 мл этим же раство¬
рителем. К 20,0 мл полученного раствора прибавля¬
ют 40 мл кислоты хлористоводородной Р и титруют
0,05 М раствором калия йодата до перехода окраски
от красной до желтой. Прибавляют 5 мл хлорофор¬
ма Р и продолжают титрование при энергичном встря¬
хивании до обесцвечивания хлороформного слоя.
1 мл 0,05 М раствора калия йодата соответ¬
ствует 14,99 мг №1.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1471
НАТРИЯ КАПРИЛАТ
Л/а&У/ саргу1аз
8СЮШМ С АР гт. АТЕ
н,сг
Х02Ыа
С8Н15Ыа02 М.м. 166,2
[1984-06-1]
ОПРЕДЕЛЕНИЕ
Натрия октаноат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень легко растворим или легко растворим в
воде, легко растворим в уксусной кислоте, умеренно
растворим в 96 % спирте, практически нерастворим в
ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора обнаруживается основной пик, соответству¬
ющий по времени удерживания и величине основному
пику на хроматограмме раствора сравнения (а).
B. К 0,2 мл раствора 3, приготовленного как
указано в разделе «Испытания», прибавляют 0,3 мл
воды Р. Полученный раствор дает реакцию (Ь) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г растворяют в воде, свободной
от углерода диоксида, Р и доводят до объема 25 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод //). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 8,0 до 10,5. Измеряют рН раствора 3.
Сопутствующие примеси. Газовая хромато¬
графия (2.2.28): определение проводят методом вну¬
тренней нормализации.
Испытуемый раствор. 0,116 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем. К полученному раствору при¬
бавляют 1 мл 2,8 % (об/об) раствора кислоты сер¬
ной Р и встряхивают с 10 мл этилацетата Р. Орга¬
нический слой отделяют и высушивают при помощи
натрия сульфата безводного Р.
Раствор сравнения (а). 0,10 г ФСО каприловой
кислоты растворяют в этилацетате Р и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 1 мл испытуемого раст¬
вора доводят этилацетатом Р до объема 100 мл.
5 мл полученного раствора доводят этилацета¬
том Р до объема 50 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,25 мм, покрытая слоем макрогола 20 000
2-нитротерефталата Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1
100
1—25
100 — 220
25—35
220
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- отношение сигнал/шум: не менее 5 для основ¬
ного пика.
Предельное содержание примесей:
-любая примесь: не более 0,3 %;
- сумма примесей: не более 0,5 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме испытуемого раствора (Ь).
Тяжелые металлы (2.4.8, метод В). Не более
0,0010% (Юррт). 2,0 г испытуемого образца раст¬
воряют в уксусной кислоте ледяной Р, доводят до
объема 10 мл этим же растворителем и прибавляют
10 мл 96% спирта Р. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием 1 мл эталон¬
ного раствора свинца (10 ррт РЬ) Р и 9 мл смеси из
равных объемов кислоты уксусной ледяной Р и 96 %
спирта Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 1,000 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
кислоты уксусной ледяной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,62 мг С8Н15Ма02.
718
Гэсударственная фармакопея Республики Беларусь
ПРИМЕСИ
НзС^^СОгН
A. п = 4: Гексановая кислота.
B. л = 5: Гептановая кислота.
C. п = 7: Нонановая кислота.
О. п = 8: Декановая кислота.
Нз°\ хо2н
н3с.
Е. 2-Пропилпентановая кислота (вальпроевая кис¬
лота).
о
Р. Н = ОСН3, п = 6: Метилоктаноат.
С. Р = ОС2Н5, л = 6: Этилоктаноат.
H. Р = ОСН3, л = 8: Метилдеканоат.
I. Р = СН3, л = 8: Ундекан-2-он.
Нз° Н'
и энантиомер
ч). (Я5)-5-Бутилтетрагидрофуран-2-он (лактон
у-гидроксиоктановой кислоты).
07/2016:0773
НАТРИЯ КАРБОНАТ БЕЗВОДНЫЙ
А/а/г/7 сагЬопаз апНус/псиз
ЗОйШМ САКВОЫАТЕ, АЛ/НУ0Я01У5
Ыа2С03 М.м. 106,0
[497-19-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый, слегка гранулированный
порошок. Гигроскопичен.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца растворяют в воде Р
и доводят до объема 10 мл этим же растворителем.
Полученный раствор имеет сильнощелочную реак¬
цию (2.2.4).
B. Раствор, приготовленный для испытания А,
дает реакцию (а) на карбонаты (2.3.1).
C. Раствор, приготовленный для испытания А,
дает реакцию (а) на натрий (2.3.1).
О. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца порциями
растворяют в смеси из 5 мл кислоты хлористоводо¬
родной Р и 25 мл воды дистиллированной Р. Приго¬
товленный раствор нагревают до кипения и охлаж¬
дают. К полученному раствору прибавляют раствор
натрия гидроксида разведенный Р до нейтральной
реакции и доводят водой дистиллированной Р до
объема 50 мл.
Раствор 51. 2,0 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.1). Раствор 51 должен быть
прозрачным.
Цветность (2.2.2, метод I). Окраска раствора 51
должна быть не интенсивнее эталона У(Ж)6.
Гидроксиды и гидрокарбонаты щелочных
металлов. 0,4 г испытуемого образца растворяют
в 20 мл воды Р, прибавляют 20 мл раствора бария
хлорида Р1 и фильтруют. К 10 мл фильтрата прибав¬
ляют 0,1 мл раствора фенолфталеина Р. Не должно
появляться красное окрашивание. Оставшуюся часть
фильтрата кипятят в течение 2 мин. Раствор должен
оставаться прозрачным (2.2.1).
Хлориды (2.4.4). Не более 0,0125% (125 ррт).
0,4 г испытуемого образца растворяют в воде Р, при¬
бавляют 4 мл кислоты азотной разведенной Р и до¬
водят водой Р до объема 15 мл. Полученный раствор
должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0250 % (250 ррт).
15 мл раствора 5 должны выдерживать испытание на
сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0005 %
(5 ррт). 5 мл раствора 5 должны выдерживать испы¬
тание на мышьяк.
Железо (2.4.9). Не более 0,0050 % (50 ррт). 5 мл
раствора 5 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0050 % (50 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре (300±15) °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют в 25 мл
воды Р, прибавляют 0,2 мл раствора метилового
оранжевого Р и титруют 1 М раствором кислоты
хлористоводородной до перехода окраски от желтой
к красной.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 52,99 мг №2С03.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0191
НАТРИЯ КАРБОНАТ ДЕКАГИДРАТ
ЫаМ1 сагЬопаз бесаЬубпсиз
8СЮШМ САКВОЫАТЕ ОЕСАНУОЙАТЕ
Ыа2С03 ■ ЮН20 М.м. 286,1
[6132-02-1]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 36,7 % и не более 40,0 %
№2С03.
Натрия карбонат моногидрат
719
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные прозрачные кристаллы. Выве¬
тривается на воздухе.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца растворяют в воде Р
и доводят до объема 10 мл этим же растворителем.
Полученный раствор имеет сильнощелочную реак¬
цию (2.2.4).
B. Раствор, приготовленный для испытания А,
дает реакцию (а) на карбонаты (2.3.1).
C. Раствор, приготовленный для испытания А,
дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор §. 5,0 г испытуемого образца порциями
растворяют в смеси из 5 мл кислоты хлористоводо¬
родной Р и 25 мл воды дистиллированной Р, нагре¬
вают до кипения и охлаждают. К полученному рас¬
твору прибавляют раствор натрия гидроксида раз¬
веденный Р до нейтральной реакции и доводят водой
дистиллированной Р до объема 50 мл.
Раствор 51. 4,0 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.1). Раствор 51 должен быть
прозрачным.
Цветность (2.2.2, метод Г). Окраска раствора 51
должна быть не интенсивнее эталона У(Ж)6.
Гидроксиды и гидрокарбонаты щелочных
металлов. 1,0 г испытуемого образца растворяют
в 20 мл воды Р, прибавляют 20 мл раствора бария
хлорида Р1 и фильтруют. К 10 мл фильтрата прибав¬
ляют 0,1 мл раствора фенолфталеина Р. Не должно
появляться красное окрашивание. Оставшуюся часть
фильтрата нагревают до кипения в течение 2 мин.
Раствор должен оставаться прозрачным (2.2.1).
Хлориды (2.4.4). Не более 0,0050 % (50 ррт).
1,0 г испытуемого образца растворяют в воде Р, при¬
бавляют 4 мл кислоты азотной разведенной Р и до¬
водят водой Р до объема 15 мл. Полученный раствор
должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
15 мл раствора 5 должны выдерживать испытание на
сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 5 мл раствора 5 должны выдерживать испы¬
тание на мышьяк.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 5 мл
раствора 5 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 5 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца растворяют в 25 мл
воды Р и титруют 1 М раствором кислоты хлористо¬
водородной #до перехода окраски от желтой до крас¬
ной* используя в качестве индикатора 0,2 мл раст¬
вора метилового оранжевого Р.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 52,99 мг Ма2С03.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0192
НАТРИЯ КАРБОНАТ МОНОГИДРАТ
Л/а&И сагЬопаз топоЬубпсиз
ЗООЮМ САПВОЫАТЕ МОЫОНУОКАТЕ
№2СО, ■ Н20 М.м. 124,0
[5968-11-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 83,0 % и не более 87,5 %
Ма2С03.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 г испытуемого образца растворяют в воде Р
и доводят до объема 10 мл этим же растворителем.
Полученный раствор имеет сильнощелочную реак¬
цию (2.2.4).
B. Раствор, приготовленный для испытания А,
дает реакцию (а) на карбонаты (2.3.1).
C. Раствор, приготовленный для испытания А,
дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца порциями
растворяют в смеси из 5 мл кислоты хлористоводо¬
родной Р и 25 мл воды дистиллированной Р. Приго¬
товленный раствор нагревают до кипения и охлаждают.
К полученному раствору прибавляют раствор натрия
гидроксида разведенный Р до нейтральной реакции и
доводят водой дистиллированной Р до объема 50 мл.
Раствор 51. 2,0 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.1). Раствор 51 должен быть
прозрачным.
Цветность (2.2.2, метод I). Окраска раствора 51
должна быть не интенсивнее эталона У(Ж)б.
Гидроксиды и гидрокарбонаты щелочных
металлов. 0,4 г испытуемого образца растворяют
в 20 мл воды Р, прибавляют 20 мл раствора бария
хлорида Р1 и фильтруют. К 10 мл фильтрата прибав¬
ляют 0,1 мл раствора фенолфталеина Р. Не должно
появляться красное окрашивание. Оставшуюся часть
фильтрата нагревают до кипения в течение 2 мин.
Раствор должен оставаться прозрачным (2.2.1).
Хлориды (2.4.4). Не более 0,0125% (125 ррт).
0,4 г испытуемого образца растворяют в воде Р, при¬
бавляют 4 мл кислоты азотной разведенной Р и до¬
водят водой Р до объема 15 мл. Полученный раствор
должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0250 % (250 ррт).
15 мл раствора 5 должны выдерживать испытание на
сульфаты.
720
Гэсударственная фармакопея Республики Беларусь
Мышьяк (2.4.2, метод А). Не более 0,0005 %
(5 ррт). 5 мл раствора 5 должны выдерживать испы¬
тание на мышьяк.
Железо (2.4.9). Не более 0,0050 % (50 ррт). 5 мл
раствора 3 доводят водой Р до объема 10 мл. Полу¬
ченный раствор должен выдерживать испытание на
железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0050 % (50 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца растворяют в 25 мл
воды Р и титруют 1 М раствором кислоты хлористо¬
водородной до перехода окраски от желтой до крас¬
ной, используя в качестве индикатора 0,2 мл раст¬
вора метилового оранжевого Р.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 52,99 мг №2С03.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0983
НАТРИЯ КРАХМАЛГЛИКОЛЯТ (ТИП А)
СагЬохуте1Ьу1ату1ит паМсит А
ЗСЮШМ 8ТАКСН ОЦУС01.АТЕ (ТУРЕ А)
ОПРЕДЕЛЕНИЕ
Натриевая соль поперечно-сшитого частично
О-карбоксиметилированного крахмала.
Содержание: не менее 2,8 % и не более 4,2 % на¬
трия (Ыа; А.м. 22,99), в пересчете на промытое 80 %
(об/об) спиртом и высушенное вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый, мелкий, легкосыпучий
порошок. Очень гигроскопичен.
Практически нерастворим в метиленхлориде.
Образует полупрозрачную суспензию в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
B. Готовят, встряхивая и не нагревая смесь из
4,0 г испытуемого образца и 20 мл воды, свободной
от углерода диоксида, Р. Полученная смесь имеет
вид геля. Прибавляют 100 мл воды, свободной от
углерода диоксида, Р и встряхивают. Образуется су¬
спензия, которая оседает после отстаивания.
C. К подкисленному раствору прибавляют раст¬
вор калия йодида йодированный Р1. Раствор окраши¬
вается в синий или фиолетовый цвет.
О. Раствор 32, приготовленный как указано в раз¬
деле «Испытания», дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 51. Центрифугируют суспензию, полу¬
ченную в подлинности В, с ускорением 2500 д в те¬
чение 10 мин. Тщательно собирают надосадочную
жидкость.
Раствор 52. 2,5 г испытуемого образца помеща¬
ют в кварцевый или платиновый тигель и прибавляют
2 мл раствора 500 г/л кислоты серной Р. Нагревают
на водяной бане, затем осторожно над открытым пла¬
менем, постепенно увеличивая температуру, и сжига¬
ют в муфельной печи при температуре (600±25) °С.
Продолжают нагревать до исчезновения черных ча¬
стиц. Охлаждают, прибавляют несколько капель кис¬
лоты серной разведенной Р, нагревают и сжигают,
как указано выше. Затем охлаждают, прибавляют не¬
сколько капель раствора аммония карбоната Р, вы¬
паривают досуха и осторожно сжигают. Охлаждают и
растворяют полученный остаток в 50 мл воды Р.
Прозрачность раствора 51 (2.2.1). Раствор 31
должен быть прозрачным.
Цветность раствора 51 (2.2.2, метод II). Раст¬
вор 31 должен быть бесцветным.
рН (2.2.3). От 5,5 до 7,5.1,0 г испытуемого образ¬
ца диспергируют в 30 мл воды, свободной от углеро¬
да диоксида, Р.
Натрия гликолят. Не более 2,0 %. Испытание
проводят в защищенном от света месте.
Испытуемый раствор. 0,20 г испытуемого об¬
разца помещают в лабораторный стакан, прибавляют
5 мл кислоты уксусной Р и 5 мл воды Р. Перемешива¬
ют до растворения (около 10 мин). Прибавляют 50 мл
ацетона Р, 1 г натрия хлорида Р и фильтруют через
быстрофильтрующий бумажный фильтр, смоченный
ацетоном Р. Промывают стакан и фильтр ацето¬
ном Р. Фильтрат и промывную жидкость объединяют
и доводят ацетоном Р до объема 100,0 мл. Получен¬
ный раствор выдерживают в течение 24 ч не встряхи¬
вая. Используют прозрачную надосадочную жидкость.
Раствор сравнения. 0,310 г кислоты гликоле¬
вой Р, предварительно высушенной в вакууме над
фосфора (V) оксидом Р при комнатной температуре
в течение ночи, растворяют в воде Р и доводят до
объема 500,0 мл этим же растворителем. К 5,0 мл
полученного раствора прибавляют 5 мл кислоты ук¬
сусной Р и выдерживают в течение 30 мин. Прибав¬
ляют 50 мл ацетона Р, 1 г натрия хлорида Р и филь¬
труют через быстрофильтрующий бумажный фильтр,
смоченный ацетоном Р. Промывают стакан и фильтр
ацетоном Р. Фильтрат и промывную жидкость объ¬
единяют и доводят ацетоном Р до объема 100,0 мл.
Полученный раствор выдерживают в течение 24 ч, не
встряхивая. Используют прозрачную надосадочную
жидкость.
2,0 мл испытуемого раствора нагревают на водя¬
ной бане в течение 20 мин. Охлаждают до комнатной
температуры, прибавляют 20,0 мл раствора 2,7-диги-
дроксинафталина Р, встряхивают и нагревают в во¬
дяной бане в течение 20 мин. Охлаждают под струей
воды, помещают в мерную колбу и доводят кислотой
серной Р до объема 25,0 мл, удерживая колбу под
струей воды. В течение 10 мин измеряют оптическую
плотность полученного раствора (2.2.25) при длине
волны 540 нм, используя в качестве компенсацион¬
ного раствора водуР. Оптическая плотность испыту¬
емого раствора должна быть не больше оптической
плотности раствора, приготовленного параллельно с
использованием 2,0 мл раствора сравнения.
Натрия хлорид. Не более 7,0 %. 0,500 г испы¬
туемого образца помещают в лабораторный стакан и
суспендируют в 100 мл воды Р. Прибавляют 1 мл кис¬
лоты азотной Р и титруют 0,1 М раствором серебра
нитрата потенциометрически (2.2.20), используя се-
Натрия крахмалгликолят (тип В)
721
ребряный индикаторный электрод и электрод сравне¬
ния, содержащий раствор 100 г/л калия нитрата Р во
внешнем корпусе и эталонный раствор во внутреннем
корпусе.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,844 мг №С1.
Железо (2.4.9). Не более 0,0020 % (20 ррт).
10 мл раствора 32 должны выдерживать испытание
на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 1,000 г испытуемого образца сушат при
температуре 130 °С в течение 1,5 ч.
Микробиологическая чистота
Испытуемый образец должен выдерживать испы¬
тания на ЕзсЬепсЫа соН и 8а1топе11а (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца встряхивают с
20 мл спирта (80 %, об/об) Р, перемешивают в те¬
чение 10 мин и фильтруют. Повторяют операцию до
отрицательной реакции на хлориды в фильтрате с ис¬
пользованием раствора серебра нитрата Р2. Оста¬
ток сушат при температуре 105 °С до постоянной мас¬
сы. К 0,700 г высушенного остатка прибавляют 80 мл
кислоты уксусной ледяной Р и нагревают с обратным
холодильником в течение 2 ч. Раствор охлаждают до
комнатной температуры и титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 2,299 мг №.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света.
07/2016:0984
НАТРИЯ КРАХМАЛГЛИКОЛЯТ (ТИП В)
СагЪохуте1Ьу1ату1ит паМсит В
ЗСЮШМ 5ТАЯСН 01.УС01.АТЕ (ТУРЕ В)
ОПРЕДЕЛЕНИЕ
Натриевая соль поперечно-сшитого частично
О-карбоксиметилированного крахмала.
Содержание: не менее 2,0 % и не более 3,4 % на¬
трия (№; А.м. 22,99), в пересчете на промытое 80 %
(об/об) спиртом и высушенное вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый, мелкий, легкосыпучий
порошок. Очень гигроскопичен.
Практически нерастворим в метиленхлориде.
Образует полупрозрачную суспензию в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
B. Готовят, встряхивая и не нагревая смесь из
4,0 г испытуемого образца и 20 мл воды, свободной
от углерода диоксида, Р. Полученная смесь имеет
вид геля. Прибавляют 100 мл воды, свободной от
углерода диоксида, Р и встряхивают. Образуется су¬
спензия, которая оседает после отстаивания.
С. К подкисленному раствору прибавляют раст¬
вор калия йодида йодированный Р1. Раствор окраши¬
вается в синий или фиолетовый цвет.
Р. Раствор 32, приготовленный как указано в раз¬
деле «Испытания», дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 51. Центрифугируют суспензию, полу¬
ченную в подлинности В, с ускорением 2500 д в те¬
чение 10 мин. Тщательно собирают надосадочную
жидкость.
Раствор 32. 2,5 г испытуемого образца помеща¬
ют в кварцевый или платиновый тигель, прибавляют
2 мл раствора 500 г/л кислоты серной Р. Нагревают
сначала на водяной бане, затем осторожно над от¬
крытым пламенем, постепенно увеличивая темпера¬
туру, и сжигают в муфельной печи при температуре
(600±25) °С до исчезновения черных частиц. Охлаж¬
дают, прибавляют несколько капель кислоты серной
разведенной Р, нагревают и сжигают, как указано
выше. Затем охлаждают, прибавляют несколько ка¬
пель раствора аммония карбоната Р, выпаривают
досуха и осторожно сжигают. Охлаждают и раство¬
ряют полученный остаток в 50 мл воды Р.
Прозрачность раствора 51 (2.2.1). Раствор 31
должен быть прозрачным.
Цветность раствора 51 (2.2.2, метод II). Раст¬
вор 31 должен быть бесцветным.
рН (2.2.3). От 3,0 до 5,0.1,0 г испытуемого образ¬
ца диспергируют в 30 мл воды, свободной от углеро¬
да диоксида, Р.
Натрия гликолят. Не более 2,0 %. Испытание
проводят в защищенном от света месте.
Испытуемый раствор. 0,20 г испытуемого об¬
разца помещают в лабораторный стакан, прибавляют
5 мл кислоты уксусной Р и 5 мл воды Р. Перемешива¬
ют до растворения (около 10 мин). Прибавляют 50 мл
ацетона Р, 1 г натрия хлорида Р и фильтруют через
быстрофильтрующий бумажный фильтр, смоченный
ацетоном Р. Промывают стакан и фильтр ацето¬
ном Р. Фильтрат и промывную жидкость объединяют
и доводят ацетоном Р до объема 100,0 мл. Получен¬
ный раствор выдерживают в течение 24 ч не стряхи¬
вая. Используют прозрачную надосадочную жидкость.
Раствор сравнения. 0,310 г кислоты гликоле¬
вой Р, предварительно высушенной в вакууме над
фосфора (V) оксидом Р при комнатной температуре в
течение ночи, растворяют в воде Р, доводят этим же
растворителем до объема 500,0 мл. К 5,0 мл получен¬
ного раствора прибавляют 5 мл кислоты уксусной Р
и выдерживают в течение 30 мин. Прибавляют 50 мл
ацетона Р, 1 г натрия хлорида Р и фильтруют через
быстрофильтрующий бумажный фильтр, смоченный
ацетоном Р. Промывают стакан и фильтр ацето¬
ном Р. Фильтрат и промывную жидкость объединяют
и доводят ацетоном Р до объема 100,0 мл. Получен¬
ный раствор выдерживают в течение 24 ч не встряхи¬
вая. Используют прозрачную жидкость.
2,0 мл испытуемого раствора нагревают на водя¬
ной бане в течение 20 мин. Охлаждают до комнатной
температуры, прибавляют 20,0 мл раствора 2,7-ди-
гидроксинафталина Р. Встряхивают и нагревают в
водяной бане в течение 20 мин. Охлаждают под стру¬
ей воды. Раствор переносят количественно в мер-
91. Зак. 1060
722
Гэсударственная фармакопея Республики Беларусь
ную колбу и доводят кислотой серной Р до объема
25,0 мл, удерживая колбу под струей воды. В течение
10 мин измеряют оптическую плотность полученного
раствора (2.2.25) при длине волны 540 нм, используя
в качестве компенсационного раствора воду Р.
Оптическая плотность испытуемого раствора
должна быть не больше оптической плотности раст¬
вора, приготовленного параллельно с использовани¬
ем 2,0 мл раствора сравнения.
Натрия хлорид. Не более 7,0 %. 0,500 г испы¬
туемого образца помещают в лабораторный стакан и
суспендируют в 100 мл воды Р. Прибавляют 1 мл кис¬
лоты азотной Р и титруют 0,1 М раствором серебра
нитрата потенциометрически (2.2.20), используя се¬
ребряный индикаторный электрод и электрод сравне¬
ния, содержащий раствор 100 г/л калия нитрата Р во
внешнем корпусе и эталонный раствор во внутреннем
корпусе.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,844 мг №01.
Железо (2.4.9). Не более 0,0020 % (20 ррт).
10 мл раствора 52 должны выдерживать испытание
на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 1,000 г испытуемого образца сушат при
температуре 130 °С в течение 1,5 ч.
Микробиологическая чистота. Испытуемый об¬
разец должен выдерживать испытания на ЕзсЬепсЫа
со//'и 8а1топе11а (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца встряхивают с
20 мл спирта (80 %, об/об) Р, перемешивают в те¬
чение 10 мин и фильтруют. Повторяют операцию до
отрицательной реакции на хлориды в фильтрате с ис¬
пользованием раствора серебра нитрата Р2. Оста¬
ток сушат при температуре 105 °С до постоянной мас¬
сы. К 0,700 г высушенного остатка прибавляют 80 мл
кислоты уксусной ледяной Р и нагревают с обратным
холодильником в течение 2 ч. Раствор охлаждают до
комнатной температуры и титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 2,299 мг Иа.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
07/2016:1566
НАТРИЯ КРАХМАЛГЛИКОЛЯТ (ТИП С)
СагЬохуте111у1ату1ит паНсит С
3ООШМ 8ТАКСН 01УС01-АТЕ (ТУРЕ С)
ОПРЕДЕЛЕНИЕ
Натриевая соль поперечно-сшитого физической
дегидратацией частично О-карбоксиметилированного
крахмала.
Содержание: не менее 2,8 % и не более 5,0 % на¬
трия (Ыа; А.м. 22,99), в пересчете на промытое 80 %
(об/об) спиртом и высушенное вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый, мелкий, легкосыпучий
порошок. Очень гигроскопичен.
Микроскопия: зерна неправильной, яйцевидной
или грушевидной формы, размером от 30 мкм до
100 мкм, или округлые, размером от 10 мкм до 35 мкм;
соединения зерен, состоящие из 2—4 зернышек,
встречается редко; зерна имеют эксцентрический
центр наслоения и четко видимую концентрическую
бороздчатость. При рассмотрении между скрещенны¬
ми призмами Николя, на зернах присутствует отчетли¬
вый черный крестик, пересекающий центр наслоения.
На поверхности зерен видны маленькие кристаллы.
При контакте с водой зерна значительно набухают.
Растворим в воде, практически нерастворим в ме-
тиленхлориде. В воде образует полупрозрачный гель.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
B. Готовят, встряхивая и не нагревая, смесь из
4,0 г испытуемого образца и 20 мл воды, свободной
от углерода диоксида, Р. Полученная смесь имеет
вид геля. Прибавляют 100 мл воды, свободной от
углерода диоксида, Р и встряхивают: гель стабилен в
отличие от типов А и В. Полученный гель оставляют
для испытаний «рН» и «Цветность».
C. К 5 мл геля, полученного в подлинности В, при¬
бавляют 0,05 мл раствора йода Р1. Раствор окраши¬
вается в темно-синий цвет.
О. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца помеща¬
ют в кварцевый или платиновый тигель, прибавляют
2 мл раствора 500 г/л кислоты серной Р. Нагревают
на водяной бане, затем осторожно над открытым пла¬
менем, постепенно увеличивая температуру, и сжига¬
ют в муфельной печи при температуре (600±25) °С до
исчезновения черных частиц. Охлаждают, прибавля¬
ют несколько капель кислоты серной Р, нагревают
и сжигают, как указано выше. Затем охлаждают, при¬
бавляют несколько капель раствора аммония кар¬
боната Р, выпаривают досуха и осторожно сжигают.
Охлаждают и растворяют полученный остаток в 50 мл
воды Р.
Цветность (2.2.2, метод II). Гель, полученный в
подлинности В, должен быть бесцветным.
рН (2.2.3). От 5,5 до 7,5. Измеряют рН геля, полу¬
ченного в подлинности В.
Натрия гликолят. Не более 2,0 %. Испытание
проводят в защищенном от света месте.
Испытуемый раствор. 0,20 г испытуемого об¬
разца помещают в лабораторный стакан, прибавляют
5 мл кислоты уксусной Р и 5 мл воды Р. Перемешива¬
ют до растворения (около 10 мин). Прибавляют 50 мл
ацетона Р, 1 г натрия хлорида Р и фильтруют через
быстрофильтрующий бумажный фильтр, смоченный
ацетоном Р. Промывают стакан и фильтр ацето¬
ном Р. Фильтрат и промывную жидкость объединяют
и доводят ацетоном Р до объема 100,0 мл. Получен¬
ный раствор выдерживают в течение 24 ч не встряхи¬
вая. Используют прозрачную надосадочную жидкость.
Натрия (З)-лактата раствор
723
Раствор сравнения. 0,310 г кислоты гликоле¬
вой Р, предварительно высушенной в вакууме над
фосфора (V) оксидом, растворяют в воде Р, доводят
этим же растворителем до объема 500,0 мл. К 5,0 мл
полученного раствора прибавляют 5 мл кислоты ук¬
сусной Р и выдерживают в течение 30 мин. Прибавля¬
ют 50 мл ацетона Р, 1 г натрия хлорида Р и доводят
этим же растворителем до объема 100,0 мл.
2,0 мл испытуемого раствора нагревают на водя¬
ной бане в течение 20 мин. Охлаждают до комнатной
температуры, прибавляет 20,0 ип раствора 2,7-диги-
фопмвфтапсма Р, встряхивает и нагревают в во-
дяюи бане в течение 20 мин. Охлаждают под струей
воды Раствор помещают в мерную колбу и доводят
кислотой серной Р до объема 25,0 мл, держа колбу
под струей воды. В течение 10 мин измеряют опти¬
ческую плотность полученного раствора (2.2.25) при
длине волны 540 нм, используя в качестве компен¬
сационного раствора водуР. Оптическая плотность
испытуемого раствора должна быть не больше опти¬
ческой плотности раствора, приготовленного парал¬
лельно с использованием 2,0 мл раствора сравнения.
Натрия хлорид. Не более 1,0 %. 1,00 г испыту¬
емого образца встряхивают с 20 мл спирта (80 %,
об/об) Р в течение 10 мин и фильтруют. Повторяют
процедуру 4 раза. Высушивают остаток до постоянной
массы при температуре 100 °С и оставляют для ко¬
личественного определения. Фильтраты объединяют,
выпаривают досуха, полученный остаток растворяют
в воде Р и доводят этим же растворителем до объема
25,0 мл. К 10,0 мл полученного раствора прибавляют
30 мл воды Р и 5 мл кислоты азотной разведенной Р.
Титруют 0,1 М раствором серебра нитрата потенци-
ометрически (2.2.20), используя серебряный индика¬
торный электрод и электрод сравнения, содержащий
раствор 100 г/л калия нитрата Р во внешнем корпу¬
се и эталонный раствор во внутреннем корпусе.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,844 мг ИаС!.
Железо (2.4.9). Не более 0,0020 % (20 ррт). 10 мл
раствора 3 должны выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0.0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Микробиологическая чистота. Испытуемый об¬
разец должен выдерживать испытания на ЕзсЬепсЫа
соН и 8а1топе11а (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,500 г высушенного и измельченного остатка,
полученного в испытании «Натрия хлорид», как ука¬
зано в разделе «Испытания», прибавляют 80 мл кис¬
лоты уксусной безводной Р и нагревают с обратным
холодильником в течение 2 ч. Раствор охлаждают до
комнатной температуры и титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20). Па¬
раллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 2,299 мг На.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
07/2016:2033
НАТРИЯ (З)-ЛАКТАТА РАСТВОР
Л/а/г/7 (8)-1асШ1$ зоМю
ЗСЮШМ (ЗПАСТАТЕ ЗОШТЮМ
ОПРЕДЕЛЕНИЕ
Содержание: не менее 50,0 % (м/м) натрия (5)-2-
гедроксипропаноата (С3Н5№03; М.м. 112,1); не менее
96.0 % и не более 104,0 % натрия лактата от содер¬
жания, указанного на этикетке, из которых не менее
95.0 % составляет (5)-энантиомер.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная, слегка сиропообразная
жидкость.
Смешивается с водой и 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 0,1 мл испытуемого образца прибавляют
10 мл воды Р. 5 мл полученного раствора дают реак¬
цию на лактаты (2.3.1).
B. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. Количество испытуемого образца,
эквивалентное 40,0 г натрия лактата, доводят водой
дистиллированной Р до объема 200 мл.
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Окраска испытуе¬
мого образца должна быть не интенсивнее эталона
ВУ(КЖ)7.
рН (2.2.3). От 6,5 до 9,0. Измеряют рН испытуе¬
мого образца.
Восстанавливающие сахара и сахароза. К 5 мл
испытуемого образца прибавляют 2 мл раствора на¬
трия гидроксида разведенного Р и 0,2 мл раствора
меди (II) сульфата Р. Раствор должен быть прозрач¬
ным и иметь голубую окраску. Окраска раствора долж¬
на сохраняться при кипячении. К горячему раствору
прибавляют 4 мл кислоты хлористоводородной Р
и кипятят в течение 1 мин. Затем прибавляют 6 мл
раствора натрия гидроксида концентрированного Р
и снова нагревают до кипения. Раствор должен быть
прозрачным и иметь голубую окраску.
Метанол. Газовая хроматография (2.4.24).
Предельное содержание примесей:
- метанол: не более 0,0050 % (50 ррт) по отно¬
шению к натрия лактату, если субстанция предназна¬
чена для производства лекарственных средств для
парентерального применения, диализа, гемодиализа
или гемофильтрации.
Хлориды (2.4.4). Не более 0,0050 % (50 ррт) по
отношению к натрия лактату. 5 мл раствора 5 доводят
водой Р до объема 15 мл. Раствор должен выдержи¬
вать испытание на хлориды.
Оксалаты и фосфаты. К 1 мл испытуемого об¬
разца прибавляют 15 мл 96 % спирта Р и 2 мл раст¬
вора кальция хлорида Р. Нагревают при температуре
75 °С в течение 5 мин. Степень мутности полученного
раствора должна быть не интенсивнее степени мут¬
ности стандартного раствора, приготовленного парал¬
лельно с использованием смеси 1 мл испытуемого об¬
разца, 15 мл 96% спирта Р и 2 мл воды Р.
9-Г Зак 1060.
724
Гэсударственная фармакопея Республики Беларусь
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт)
по отношению к натрия лактату. К 7,5 мл раствора 3
прибавляют 1,9 мл кислоты хлористоводородной Р1
и доводят водой дистиллированной Р до объема
15мл. Раствор должен выдерживать испытание на
сульфаты без добавления 0,5 мл кислоты уксус¬
ной Р. Для подкисления эталонного раствора исполь¬
зуют 0,05 мл кислоты хлористоводородной Р1 вме¬
сто 0,5 мл кислоты уксусной Р.
Алюминий. Не более 0,00001 % (0,1 ррт), если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения,
диализа, гемодиализа или гемофильтрации.
Атомно-абсорбционная спектрометрия (2.2.23,
метод I). При приготовлении растворов используют
посуду, не содержащую алюминия, или посуду, ко¬
торая не выделяет алюминий при ее использовании
(стеклянную, полиэтиленовую и т.д.).
Раствор модификатора. 100,0 г аммония нитра¬
та Р растворяют в смеси из 50 мл воды Р и 4 мл кис¬
лоты азотной Р и доводят водой Р до объема 200 мл.
Контрольный раствор. 2,0 мл раствора моди¬
фикатора доводят водой Р до объема 25,0 мл.
Испытуемый раствор. К 1,25 г испытуемого об¬
разца прибавляют 2,0 мл раствора модификатора и
доводят водой Р до объема 25,0 мл.
Растворы сравнения. От 0,010 ррт до 0,050 ррт
алюминия. Готовят непосредственно перед использо¬
ванием, используя эталонный раствор алюминия
(200 ррт А!) Р#путем его разведения, добавляя такие
же реактивы, как в испытуемом растворе#.
Источник излучения: лампа с полым катодом
для определения алюминия.
Длина волны: 309,3 нм.
Атомизатор: графитовая печь.
Газ-носитель: аргон Р.
Условия: прибор оборудован системой коррекции
неспецифического поглощения. Нагревают печь до
120 °С в течение такого количества секунд, которое
соответствует количеству микролитров раствора, вве¬
денного в прибор, затем нагревают при температуре
1000 °С в течение 30 с и окончательно при температу¬
ре 2700 °С в течение 6 с.
Барий. К 10 мл раствора 3 прибавляют 10 мл
раствора кальция сульфата Р и выдерживают в те¬
чение 30 мин. Степень мутности (2.2.1) полученного
раствора должна быть не интенсивнее степени мут¬
ности раствора, приготовленного параллельно с ис¬
пользованием смеси 10 мл раствора 3 и 10 мл воды
дистиллированной Р.
Железо (2.4.9). Не более 0,0010% (10 ррт) по
отношению к натрия лактату. 5 мл раствора 3 доводят
водой Р до объема 10 мл. Раствор должен выдержи¬
вать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт) по отношению к натрия лактату.
12 мл раствора 3 должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (2 ррт РЬ) Р.
Бактериальные эндотоксины (2.6.14). Не бо¬
лее 5 МЕ/г, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Количество испытуемого образца, эквивалент¬
ное 75,0 мг натрия лактата, растворяют в смеси 10 мл
кислоты уксусной ледяной Р, 20 мл ангидрида уксус¬
ного Р и выдерживают в течение 15 мин. Прибавляют
1 мл раствора нафтолбензеина Р и титруют 0,1 М
раствором кислоты хлорной.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 11,21 мгС3Н5Ма03.
(З)-Энантиомер. Количество испытуемого об¬
разца, эквивалентное 2,50 г натрия лактата, помеща¬
ют в мерную колбу вместимостью 50 мл, прибавля¬
ют около 30 мл воды Р, 5,0 г аммония молибдата Р,
растворяют и доводят водой Р до объема 50,0 мл. Из¬
меряют угол оптического вращения (2.2.7).
Содержание (З)-энантиомера в процентах рас¬
считывают по формуле:
50+ 24,04 а*
5,0 50
т
где:
а — угол оптического вращения (абсолютное зна¬
чение);
/77 — масса навески испытуемого образца, г;
с — содержание С3Н5МаОэ в испытуемом образ¬
це, %.
Образующийся в описанных выше условиях ком¬
плекс натрия (З)-лактата является левовращающим.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства раство¬
ров для диализа, гемодиализа или гемофильтрации;
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения;
- содержание натрия лактата.
07/2016:1151
НАТРИЯ ЛАКТАТА РАСТВОР
Л/а?п/ 1ас(аИз зо1иИо
ЗООШМ 1.АСТАТЕ ЗОШТЮИ
ОПРЕДЕЛЕНИЕ
Раствор смеси энантиомеров натрия 2-гидрокси-
пропаноата в приблизительно равных пропорциях.
Содержание: заявленное содержание не менее
50 % (м/м) натрия 2-гидроксипропаноата (С3Н5Ма03;
М.м. 112,1); не менее 96,0 % и не более 104,0 % на¬
трия лактата от содержания, указанного на этикетке.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная, слегка сиропообразная
жидкость.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 0,1 мл испытуемого образца прибавляют
10 мл воды Р. 5 мл полученного раствора дают харак¬
терную реакцию на лактаты (2.3.1).
B. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. Количество испытуемого образца,
эквивалентное 40,0 г натрия лактата, доводят водой
дистиллированной Р до объема 200 мл.
Натрия лаурилсульфат
725
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Окраска испытуе¬
мого образца должна быть не интенсивнее эталона
ВУ(КЖ)7.
рН (2.2.3). От 6,5 до 9,0. Измеряют рН испытуе¬
мого образца.
Восстанавливающие сахара и сахароза. К
5 мл испытуемого образца прибавляют 0,2 мл раст¬
вора меди (II) сульфата Р и 2 мл раствора натрия
гидроксида разведенного Р. Раствор должен быть
прозрачным и иметь голубую окраску. Окраска раст¬
вора должна сохраняться при кипячении. К горячему
раствору прибавляют 4 мл кислоты хлористоводо¬
родной Р и кипятят в течение 1 мин. Прибавляют 6 мл
раствора натрия гидроксида концентрированного Р
и снова нагревают до кипения. Раствор должен быть
прозрачным и иметь голубую окраску.
Метанол. Газовая хроматография (2.4.24).
Предельное содержание примесей:
- метанол: не более 0,0050 % (50 ррт) по отно¬
шению к натрия лактату, если субстанция предназна¬
чена для производства лекарственных средств для
парентерального применения, диализа, гемодиализа
или гемофильтрации.
Хлориды (2.4.4). Не более 0,0050 % (50 ррт) по
отношению к натрия лактату. 5 мл раствора 5 доводят
водой Р до объема 15 мл. Полученный раствор дол¬
жен выдерживать испытание на хлориды.
Оксалаты и фосфаты. К 1 мл испытуемого об¬
разца прибавляют 15 мл 96 % спирта Р и 2 мл раст¬
вора кальция хлорида Р. Нагревают при температуре
75 °С в течение 5 мин. Мутность раствора должна быть
не интенсивнее мутности раствора, приготовленного
параллельно с использованием смеси из 1 мл испы¬
туемого образца, 15 мл 96 % спирта Р и 2 мл воды Р.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт)
по отношению к натрия лактату. К 7,5 мл раствора
5 прибавляют 1,9 мл кислоты хлористоводород¬
ной Р1 и доводят водой дистиллированной Р до объ¬
ема 15мл. Раствор должен выдерживать испытание
на сульфаты без добавления 0,5 мл кислоты уксус¬
ной Р. Для подкисления эталонного раствора исполь¬
зуют 0,05 мл кислоты хлористоводородной Р1 вме¬
сто 0,5 мл кислоты уксусной Р.
Алюминий. Не более 0,00001 % (0,1 ррт), если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения,
диализа, гемодиализа или гемофильтрации.
Атомно-абсорбционная спектрометрия (2.2.23,
метод I). При приготовлении растворов используют
посуду, не содержащую алюминия, или посуду, ко¬
торая не выделяет алюминий при ее использовании
(стеклянную, полиэтиленовую и т.д.).
Раствор модификатора. 100,0 г аммония нитра¬
та Р растворяют в смеси из 50 мл воды Р и 4 мл кис¬
лоты азотной Р и доводят водой Р до объема 200 мл.
Контрольный раствор. 2,0 мл раствора моди¬
фикатора доводят водой Р до объема 25,0 мл.
Испытуемый раствор. К 5,0 г испытуемого об¬
разца прибавляют 2,0 мл раствора модификатора и
доводят водой Р до объема 25,0 мл.
Растворы сравнения. От 0,010 ррт до 0,050 ррт
алюминия. Готовят непосредственно перед использо¬
ванием, используя эталонный раствор алюминия
(200 ррт А!) Р#путем его разведения, добавляя такие
же реактивы, как в испытуемом растворе#.
Источник излучения: лампа с полым катодом
для определения алюминия.
Длина волны: 309,3 нм.
Атомизатор: графитовая печь.
Газ-носитель: аргон Р.
Условия: прибор оборудован системой коррекции
неспецифического поглощения. Нагревают печь до
120 °С в течение такого количества секунд, которое
соответствует количеству микролитров раствора, вве¬
денного в прибор, затем нагревают при температуре
1000 °С в течение 30 с и окончательно при температу¬
ре 2700 °С в течение 6 с.
Барий. К 10 мл раствора 3 прибавляют 10 мл
раствора кальция сульфата Р. Выдерживают в те¬
чение 30 мин. Степень мутности (2.2.1) полученного
раствора должна быть не интенсивнее степени мут¬
ности раствора, приготовленного параллельно с ис¬
пользованием смеси 10 мл раствора 3 и 10 мл воды
дистиллированной Р.
Железо (2.4.9). Не более 0,0010% (10 ррт) по
отношению к натрия лактату. 5 мл раствора 3 доводят
водой Р до объема 10 мл. Раствор должен выдержи¬
вать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт) по отношению к натрия лактату.
12 мл раствора 3 должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (2 ррт РЬ) Р.
Бактериальные эндотоксины (2.6.14). Менее
5 МЕ/г, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый образец в количестве, эквивалент¬
ном 75,0 мг натрия лактата, растворяют в смеси из
10 мл кислоты уксусной ледяной Р и 20 мл ангидри¬
да уксусного Р и выдерживают в течение 15 мин. При¬
бавляют 1 мл раствора нафтолбензеина Р и титруют
0,1 М раствором кислоты хлорной.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 11,21 мгС3Н5Ма03.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства раство¬
ров для диализа, гемодиализа или гемофильтрации;
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения;
- заявленное содержание натрия лактата.
07/2016:0098
НАТРИЯ ЛАУРИЛСУЛЬФАТ
ЛУа^г/7/аигНзиКаз
ЗОйШМ 1-А1тИ-81П.1=АТЕ
Натрия додециясуяьфат: [151-21-3].
ОПРЕДЕЛЕНИЕ
Смесь натрия аякилсульфатов, состоящая в
основном из натрия додецилсульфата С^Н^ЫаО^З
(М.м. 288,4).
726
Гэсударственная фармакопея Республики Беларусь
Содержание:
- натрия алкилсульфаты: не менее 85,0 % в
пересчете на С12Н25Ыа043.
ОПИСАНИЕ(СВОЙСТВА)
Белый или бледно-желтый порошок или кристаллы.
Легко растворим в воде с образованием опа¬
лесцирующего раствора, частично растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,1 г испытуемого образца растворяют в 10 мл
воды Р и встряхивают. Образуется обильная пена.
B. К 0,1 мл раствора, приготовленного для иден¬
тификации А, прибавляют 0,1 мл раствора 1 г/л мети¬
ленового синего Р, 2 мл раствора кислоты серной
разведенной Р, 2 мл метиленхлорида Р и встряхива¬
ют. В слое метиленхлорида появляется интенсивное
синее окрашивание.
C. Около 10 мг испытуемого образца смешивают
с 10 мл этанола безводного Р. Нагревают до кипе¬
ния на водяной бане, часто встряхивая. Немедлен¬
но фильтруют и выпаривают этанол. Полученный
остаток растворяют в 8 мл воды Р, прибавляют 3 мл
раствора кислоты хлористоводородной разведен¬
ной Р, упаривают раствор до половины объема и ох¬
лаждают. Застывшие жирные спирты отделяют филь¬
трованием. К фильтрату прибавляют 1 мл раствора
бария хлорида Р1. Образуется белый кристалличе¬
ский осадок.
й. Прокаливают 0,5 г испытуемого образца #(при
температуре 800 °С)#. Остаток дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Щелочность. 1,0 г испытуемого образца раст¬
воряют в 100 мл воды, свободной от углерода диок¬
сида, Р и прибавляют 0,1 мл раствора фенолового
красного Р. При прибавлении не более 0,5 мл 0,1 М
кислоты хлористоводородной окраска раствора
должна измениться.
Неэтерифицированные спирты. Не более
4 %. 10 г испытуемого образца растворяют в 100 мл
воды Р, прибавляют 100 мл 96% спирта Р и триж¬
ды встряхивают с пентаном Р порциями по 50 мл
каждая, прибавляя для ускорения разделения слоев
натрия хлорид Р. Объединенные органические слои
трижды промывают водой Р порциями по 50 мл каж¬
дая #(при необходимости для разделения двух сло¬
ев прибавляют натрия хлорид Р)#, фильтруют через
фильтр с натрия сульфатом безводным Р и выпа¬
ривают на водяной бане до испарения растворителя.
Нагревают остаток при температуре 105 °С в течение
15 мин и охлаждают. Масса остатка не должна превы¬
шать 0,4 г.
Натрия хлорид и натрия сульфат. Не более
8,0 % суммы ЫаС1 и Ма2304.
Натрия хлорид. 5,00 г испытуемого образца
растворяют в 50 мл воды Р, прибавляют по каплям
раствор кислоты азотной разведенной Р до ней¬
трализации раствора по синей лакмусовой бумаге Р,
прибавляют 2 мл раствора калия хромата Р и титру¬
ют 0,1 М раствором серебра нитрата.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,844 мг №01.
Натрия сульфат. 0,500 г испытуемого образца
растворяют в 20 мл воды Р, при необходимости слег¬
ка подогревая, и прибавляют 1 мл раствора 0,5 г/л
дитизонаР1 в ацетоне Р. Если раствор красного
цвета, прибавляют по каплям 1 М раствор кислоты
азотной до голубовато-зеленой окраски раствора.
Прибавляют 2,0 мл раствора кислоты дихлоруксус-
ной Р и 80 мл ацетона Р. Титруют 0,01 М раствором
свинца нитрата до появления стойкого фиолетово¬
красного или оранжево-красного окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,01 М раствора свинца нитрата соответ¬
ствует 1,420 мг №23 04.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,15 г испытуемого образца растворяют в воде Р,
при необходимости подогревая, и доводят до объема
1000,0 мл этим же растворителем. К 20,0 мл полу¬
ченного раствора прибавляют 15 мл хлороформа Р
и 10 мл смешанного раствора димидия бромида и
сульфанового синего Р. Титруют 0,004 М раствором
бензэтония хлорида, энергично встряхивая и остав¬
ляя до разделения слоев перед каждым прибавлени¬
ем титранта, до полного исчезновения розового окра¬
шивания хлороформного слоя и появления серовато-
голубого окрашивания.
1 мл 0,004 М раствора бензэтония хлорида со¬
ответствует 1,154 мг натрия алкилсульфатов в пере¬
счете на С12Н25№045.
07/2016:0849
НАТРИЯ МЕТАБИСУЛЬФИТ
А/а/г/7 те1аЫзи1Лз
ЗООШМ МЕТАВ181Л-Р1ТЕ
№2$205 М.м. 190,1
[7681-57-4]
ОПРЕДЕЛЕНИЕ
#Натрия дисульфит#
Содержание: не менее 95,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
B. К 0,4 мл раствора калия йодида йодирован¬
ного Р прибавляют 8 мл воды дистиллированной Р и
1 мл раствора 3, приготовленного как указано в раз¬
деле «Испытания», разведенного водой дистиллиро¬
ванной Р в 10 раз. Полученный раствор бесцветный и
дает реакцию (а) на сульфаты (2.3.1).
C. Раствор 3 дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Г"
Натрия метилпарагидроксибензоат
727
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 3,5 до 5,0. Измеряют рН раствора 3.
Тиосульфаты. К 5 мл раствора 3 прибавляют
5 мл кислоты хлористоводородной разведенной Р.
Раствор должен быть прозрачным (2.2.1) в течение не
менее 15 мин.
Мышьяк (2.4.2, метод А). Не более 0,0005%
(5 ррт). 0,20 г испытуемого образца смешивают в чаш¬
ке с 2 мл еодыР. Прибавляют по каплям 1,5 мл кис-
лоты азотной Рш выпаривают смесь на водяной бане
досуха и нагревают на огне до окончания выделения
паров. Полученный остаток смывают 25 мл воды Р.
Железо (2.4.9). Не более 0,0020% (20 ррт).
Раствор 3 должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 40 мл раствора 3 помещают в
кварцевый тигель, прибавляют 10 мл кислоты хло¬
ристоводородной Р и выпаривают досуха. Получен¬
ный остаток растворяют в 19 мл воды Р и прибавляют
1 мл раствора 40 г/л натрия фторида Р. 12 мл полу¬
ченного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
эталонного раствора свинца (2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в
50.0 мл 0,05 М раствора йода, прибавляют 5 мл кис¬
лоты хлористоводородной разведенной Р и титруют
избыток йода 0,1 М раствором натрия тиосульфа¬
та, используя в качестве индикатора 1 мл раствора
крахмала Р, который прибавляют в конце титрования.
1 мл 0,05 М раствора йода соответствует
4,753 мг N823205.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1262
НАТРИЯ
МЕТИЛПАРАГИДРОКСИБЕНЗОАТ
МеИзуИз рагаЬудгохуЬепгоаз паМсиз
500/1УМ МЕТНП
РАРАНУ0Р0ХУВЕЫ20АТЕ
[5026-62-0]
ОПРЕДЕЛЕНИЕ
Натрия метил-4-гидроксибензоат.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, умеренно растворим в 96 %
спирте, практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификациям, С, О.
Рисунок #1262.-1. Инфракрасный спектр ФСО метилпарагидроксибензоата
92*. Зак. 1060.
728
Гэсударственная фармакопея Республики Беларусь
A. 0,5 г испытуемого образца растворяют в 50 мл
воды Р и немедленно прибавляют 5 мл кислоты хло¬
ристоводородной Р1. Фильтруют, промывают осадок
водой Р. Сушат в вакууме при температуре 80 °С в те¬
чение 2 ч. Температура плавления (2.2.14): от 125 °С
до 128 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: используют осадок, полученный
в идентификации А.
Сравнение: ФСО метилпарагидроксибензоата
#или спектр, представленный на рисунке #1262.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемо¬
го образца растворяют в 10 мл воды Р, немедленно
прибавляют 2 мл кислоты хлористоводородной Р и
встряхивают с 50 мл 1,1-диметилэтилметилового
эфира Р. Выпаривают верхний слой досуха, остаток
растворяют в 10 мл ацетона Р.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО метилпара¬
гидроксибензоата растворяют в ацетоне Р и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО этилпара-
гидроксибензоата растворяют в 1 мл испытуемого
раствора (а) и доводят ацетоном Рцо объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — метанол Р (1:30:70, об/об/об).
Наносимый объем пробы: 5 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
О. К 1 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», прибавляют 1 мл воды Р.
Полученный раствор дает реакцию (а) на натрий
(2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5, приготовлен¬
ный непосредственно перед испытанием, должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3, приготовленного непосредственно перед испыта¬
нием, должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 9,5 до 10,5. 1 мл раствора 3 до¬
водят водой, свободной от углерода диоксида, Р до
объема 100 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл метанола Р и доводят
подвижной фазой до объема 50,0 мл. 10,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Раствор сравнения (а). 5 мг кислоты 4-гидрок-
сибензойной Р (примесь А) и 5 мг испытуемого образ¬
ца растворяют в подвижной фазе и доводят до объ¬
ема 100,0 мл этим же растворителем. 1,0 мл получен¬
ного раствора доводят подвижной фазой до объема
10.0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО метилпара¬
гидроксибензоата растворяют в 2,5 мл метанола Р и
доводят подвижной фазой до объема 50,0 мл. 10,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: раствор 6,8 г/л калия диги¬
дрофосфата Р— метанол Р (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а) и (с);
- время хроматографирования: 5-кратное вре¬
мя удерживания метилпарагидроксибензоата.
Относительное удерживание (по отношению к
метилпарагидроксибензоату, время удерживания —
около 2,3 мин): примесь А— около 0,6.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси А и метилпарагидроксибензоата.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А на 1,4):
- примесь А (не более 3,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 6-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (с);
- сумма примесей, кроме примеси А (не более
1.0 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пика
примеси А, не должна превышать 2-кратную площадь
основного пика на хроматограмме раствора сравне¬
ния (с);
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (с).
Хлориды (2.4.4). Не более 0,0350 % (350 ррт).
К 10 мл раствора 3 прибавляют 30 мл воды Р, 1 мл
Натрия пикосульфат
729
кислоты азотной Р и доводят водой Р до объема
50 мл. Встряхивают и фильтруют. 10 мл фильтрата
доводят водой Р до объема 15 мл. Эталон готовят с
использованием 14 мл эталонного раствора хло¬
ридов (5 ррт С1) Р и 1 мл воды Р.
Сульфаты (2.4.13). Не более 0,0300 %
(300 ррт). К 25 мл раствора 5 прибавляют 5 мл
воды дистиллированной Р, 10 мл кислоты хлори¬
стоводородной Р и доводят водой дистиллирован¬
ной Р до объема 15 мл. Встряхивают и фильтруют.
10 мл фильтрата доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор дол¬
жен выдерживать испытание на сульфаты.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы, по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С8Н7Ма03 в процентах рассчитыва¬
ют с учетом содержания метилпарагидроксибензоа-
та в ФСО метилпарагидроксибензоата и умножают
полученный результат на поправочный коэффициент
1,145.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Пш^^тГТ^гш,ш ш* /|УМРПГ А
Другие обнаруживаемые примеси (следующие
аемества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В, С, О.
А. 4-Гидроксибензойная кислота.
В. Этил-4-гидроксибензоат (этилпарагидроксибен-
зоат).
С. Пропил-4-гидроксибензоат (пропилпарагидрок-
сибензоат).
О. Бутил-4-гидроксибензоат (бутилпарагидрокси-
бензоат).
07/2016:1031
НАТРИЯ ПИКОСУЛЬФАТ
А/а/г/7 рюозиНаз
ЗОйШМ Р1С08Ш-РАТЕ
С18Н13МЫа20852 ■ Н20 М.м. 499,4
ОПРЕДЕЛЕНИЕ
4,4-(Пиридин-2-илметилен)дифенил-бис(натрия
сульфат) моногидрат.
Содержание: не менее 98,5 % и не более 100,5 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО натрия пикосульфата #или
спектр, представленный на рисунке #1031.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до 5 мл
этим же растворителем.
Раствор сравнения. 20 мг ФСО натрия пико-
сульфата растворяют в метаноле Р и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
пя6Р254 Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метанол Р — этилацетат Р
(2,5:12,5:25:60, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 1/2 высоты
пластинки.
93 За*. '060
730
Гэсударственная фармакопея Республики Беларусь
Рисунок #1031 .-1. Инфракрасный спектр ФСО натрия пикосульфата
Высушивание: в потоке горячего воздуха в тече¬
ние 15 мин.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
С. К 5 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 1 мл кислоты
хлористоводородной разведенной Р, нагревают до
кипения и прибавляют 1 мл раствора бария хлори¬
да Р1. Образуется белый осадок.
Р. Раствор 3 дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона <ЗУ(ЗЖ)7.
Кислотность и щелочность. К 10 мл раствора
3 прибавляют 0,05 мл раствора фенолфталеина Р.
Раствор должен быть бесцветным. При прибавлении
не более 0,25 мл 0,01 М раствора натрия гидрокси¬
да должно появиться розовое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
10,0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Раствор сравнения (Ь). 1 Содержимое контей¬
нера ФСО пикосульфата для проверки пригодности
хроматографической системы (содержит примеси А
и В) растворяют в 0,1 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р1 с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: 2,3 г динатрия гидрофосфата
дигидрата Р растворяют в 800 мл воды для хромато¬
графии Р, прибавляют 0,2 мл цетилтриметиламмо¬
ния бромида Р, доводят до рН 7,5 кислотой фосфор¬
ной Р и разводят водой для хроматографии Р до объе¬
ма 1000,0 мл; смешивают 550 мл полученного раствора
и 450 мл ацетонитрила Р (при необходимости, чтобы
выполнить требования по разрешению, изменяют соот¬
ношение вода/ацетонитрил с шагом 10 мл);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 263 нм;
- объем вводимой пробы: 40 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания пикосульфата.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО пикосульфата для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
пикосульфату, время удерживания — около 7,4 мин):
примесь В — около 0,5; примесь А— около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками при¬
месей В и А.
Натрия пропилпарагидроксибензоат
731
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А— 0,7; для примеси В — 0,5):
- примеси А и В (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А и В, не должны превышать
2-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0400 % (400 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на сульфаты.
Вода (2.5.12). Не менее 3,0 % и не более 5,0 %.
Определение проводят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворят в 80 мл
метанола Р и титруют 0,1 М раствором кислоты
хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 48,14 мг С18Н13Ы№20832.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. Р = 303Ма: 4-[(Р$)-(4-Гидроксифенил)(пиридин-
2-ил)метил]фенилнатрия сульфат.
В. Р = Н: 4,4'-[(Пиридин-2-ил)метилен]дифенол.
С. 2[(Р5)-(Пиридин-2-ил)[4-(сульфанатоокси)фе-
нил]метил]фенилдинатрия сульфат.
07/2016:1263
НАТРИЯ
ПРОПИЛПАРАГИДРОКСИБЕНЗОАТ
РгоруНз рагаЬубгохуЬепгоаз паМсиз
ЗООШМ РКОРУ1-
РАРАНУ0Р0ХУВЕЫ20АТЕ
С^Н^аОз М.м. 202,2
[35285-69-9]
ОПРЕДЕЛЕНИЕ
Натрия 4-(пропоксикарбонил)феноат.
Содержание: не менее 94,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, умеренно растворим в
96 % спирте, практически нерастворим в метиленхло-
риде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. 0,5 г испытуемого образца растворяют в 50 мл
воды Р и немедленно прибавляют 5 мл кислоты хло¬
ристоводородной Р1. Фильтруют, промывают осадок
водой Р. Сушат в вакууме при температуре 80 °С в
течение 2 ч. Температура плавления (2.2.14): от 96 °С
до 99 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: используют осадок, полученный
в идентификации А.
Сравнение: ФСО пропилпарагидроксибензоата
#или спектр, представленный на рисунке #1263.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемо¬
го образца растворяют в 10 мл воды Р, немедленно
прибавляют 2 мл кислоты хлористоводородной Р и
встряхивают с 50 мл 1,1-диметилэтилметилового
эфира Р. Выпаривают верхний слой досуха, остаток
растворяют в 10 мл ацетона Р.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
93*. Зак. 1060.
732
Гэсударственная фармакопея Республики Беларусь
Рисунок #1263.-1. Инфракрасный спектр ФСО пропилпарагидроксибензоата
Раствор сравнения (а). 10 мг ФСО пропилпара¬
гидроксибензоата растворяют в ацетоне Р и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО этилпара-
гидроксибензоата растворяют в 1 мл испытуемого
раствора (а) и доводят ацетоном Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р — метанол Р (1:30:70, об/об/об).
Наносимый объем пробы: 5 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
О. К1 мл раствора 5, приготовленного как указано
в разделе «Испытания», прибавляют 1 мл воды Р. По¬
лученный раствор дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8, приготовлен¬
ный непосредственно перед испытанием, должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
5, приготовленного непосредственно перед испыта¬
нием, должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 9,5 до 10,5. 1 мл раствора 8 до¬
водят водой, свободной от углерода диоксида, Р до
объема 100 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл метанола Р и доводят
подвижной фазой до объема 50,0 мл. 10,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Раствор сравнения (а). 5 мг ФСО этилпара-
гидроксибензоата (примесь С), 5 мг кислоты 4-ги-
дроксибензойной Р (примесь А) и 5 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем. 1,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 10,0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО пропилпа¬
рагидроксибензоата растворяют в 2,5 мл метано¬
ла Р и доводят подвижной фазой до объема 50,0 мл.
10.0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
-подвижная фаза: раствор 6,8г/л калия диги¬
дрофосфата Р — метанол Р (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
Натрия салицилат
733
-спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а) и (с);
- время хроматографирования: 2,5-кратное вре¬
мя удерживания пропилпарагидроксибензоата.
Относительное удерживание (по отношению к
пропилпарагидроксибензоату, время удерживания —
около 4,5 мин): примесь А — около 0,3; примесь С —
около 0,7.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 5,0 между пиками при¬
меси С и пропилпарагидроксибензоата.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—1,4):
- примесь А (не более 4,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 8-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (с);
- сумма примесей, кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
А, не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (с);
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (с).
Хлориды (2.4.4). Не более 0,0350 % (350 ррт). К
10 мл раствора 5 прибавляют 30 мл воды Р, 1 мл кис¬
лоты азотной Р и доводят водой Р до объема 50 мл.
Встряхивают и фильтруют. 10 мл фильтрата доводят
водой Р до объема 15 мл. Эталон готовят с использо¬
ванием 14 мл эталонного раствора хлоридов (5 ррт
С1) Р и 1 мл воды Р.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
К 25 мл раствора 5 прибавляют 5 мл воды дистилли¬
рованной Р, 10 мл кислоты хлористоводородной Р и
доводят водой дистиллированной Р до объема 15 мл.
Встряхивают и фильтруют. 10 мл фильтрата доводят во¬
дой дистиллированной Рдо объема 15 мл. Полученный
раствор должен выдерживать испытание на сульфаты.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Содержание С^Н^МаОд в процентах рассчиты¬
вают с учетом содержания пропилпарагидроксибен¬
зоата в ФСО пропилпарагидроксибензоата и умно¬
жают полученный результат на поправочный коэф¬
фициент 1,122.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В, С, О.
А. 4-Гидроксибензойная кислота.
С. Этил-4-гидроксибензоат (этилпарагидроксибен-
зоат).
О. Бутил-4-гидроксибензоат (бутилпарагидрокси-
бензоат).
07/2016:0413
НАТРИЯ САЛИЦИЛАТ
Ыа(гИ заНсу1аз
8СЮШМ 8АЫСУ1-АТЕ
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
С.Н №0
[54-21-7]
^^^С02На
М.м. 160,1
94. Зак. 1060.
734
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Натрия 2-гидроксибензолкарбоксилат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или мелкие бесцветные кристаллы или блестя¬
щие чешуйки.
Легко растворим в воде, умеренно растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО натрия салицилата #или
спектр, представленный на рисунке #0413.-1#.
B. Раствор 5, приготовленный как указано в
разделе «Испытания», дает реакции на сапицилаты
(2.3.1).
C. Испытуемый образец дает реакцию (Ь) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность. К 20 мл раствора 3 прибавляют
0,1 мл раствора фенолового красного Р. Должно по¬
явиться желтое окрашивание. При прибавлении не
более 2,0 мл 0,01 М раствора натрия гидроксида
должно появиться фиолетово-красное окрашивание.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
К 5 мл раствора 3 прибавляют 5 мл воды Р, 10 мл
кислоты азотной разведенной Р и фильтруют. 10 мл
полученного фильтрата доводят водой Р до объема
15 мл.
Сульфаты (2.4.13). Не более 0,0600 % (600 ррт).
2,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 1,6 г испытуемого образца раство¬
ряют в 16 мл смеси из 5 объемов воды Р и 10 объемов
96 % спирта Р. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (2 ррт РЬ), приготовленного путем разведения
эталонного раствора свинца (100 ррт РЬ) Р смесью
из 5 объемов воды Р и 10 объемов 96 % спирта Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5%. 1,00 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,130 г испытуемого образца растворяют в 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,01 мг С7Н5Ыа03.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Рисунок #0413.-1. Инфракрасный спектр ФСО натрия салицилата
Натрия сульфат декагидрат
735
07/2016:0099
НАТРИЯ СУЛЬФАТ БЕЗВОДНЫЙ
А/а/г/7 зиКаз апЬус/псиз
800ШМ 8Ш.РАТЕ, АЛ/ЯУОКОиЗ
Ыа2304 М.м. 142,0
[7757-82-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
B. Испытуемый образец дает реакции (а) и (Ь) на
натрий (2.3.1).
C. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 2,2 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора бромтимолового
синего Р1. При прибавлении не более 0,5 мл 0,01 М
раствора кислоты хлористоводородной или 0,01 М
раствора натрия гидроксида окраска раствора
должна измениться.
Хлориды (2.4.4). Не более 0,0450 % (450 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Кальций (2.4.3). Не более 0,0450 % (450 ррт),
если субстанция предназначена для производства
лекарственных средств для парентерального приме¬
нения.
10 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на кальций.
Железо (2.4.9). Не более 0,0090 % (90 ррт), если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения.
5 мл раствора 3 доводят водой Р до объема
10 мл. Полученный раствор должен выдерживать ис¬
пытание на железо.
Магний. Не более 0,0200 % (200 ррт), если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения.
К 10 мл раствора 3 прибавляют 1 мл глицери¬
на (85 %) Р, 0,15 мл раствора титанового желто¬
го Р, 0,25 мл раствора аммония оксалата Р и 5 мл
раствора натрия гидроксида разведенного Р и
встряхивают. Розовая окраска полученного раствора
должна быть не интенсивнее окраски эталона, при¬
готовленного параллельно с использованием смеси
из 5 мл эталонного раствора магния (10 ррт Мд) Р
и 5 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0045 % (45 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 130 °С.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 40 мл
воды Р, прибавляют 0,2 мл 0,1 М раствора кисло¬
ты хлористоводородной и 80 мл метанола Р. По¬
лученный раствор титруют 0,1 М раствором свинца
нитрата потенциометрически (2.2.20), используя в
качестве индикаторного электрода свинец-селектив-
ный электрод, а в качестве электрода сравнения —
хлорсеребряный.
1 мл 0,1 М раствора свинца нитрата соответ¬
ствует 14,20 мг Ма2304.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для приготовления лекар¬
ственных средств для парентерального применения.
07/2016:0100
НАТРИЯ СУЛЬФАТ ДЕКАГИДРАТ
Ыа1гН зийаз десаЬудпсиз
8СЮШМ 8Ш.РАТЕ ИЕСАНУОРАТЕ
№2304 • 10Н2О М.м. 322,2
[7727-73-3]
ОПРЕДЕЛЕНИЕ
#Глауберова соль#.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные прозрачные кристаллы.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте. Частично растворяется в кристал¬
лизационной воде при температуре около 33 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
B. Испытуемый образец дает реакции (а) и (Ь) на
натрий (2.3.1).
C. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при-
94*. Зак. 1060.
736
Гэсударственная фармакопея Республики Беларусь
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора бромтимолового
синего Р1. При прибавлении не более 0,5 мл 0,01 М
раствора кислоты хлористоводородной или 0,01 М
раствора натрия гидроксида окраска раствора
должна измениться.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Кальций (2.4.3). Не более 0,0200 % (200 ррт),
если субстанция предназначена для производства ле¬
карственных средств для парентерального примене¬
ния. 10 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 15 мл. Полученный раствор должен
выдерживать испытание на кальций.
Железо (2.4.9). Не более 0,0040 % (40 ррт),
если субстанция предназначена для производства
лекарственных средств для парентерального приме¬
нения. 5 мл раствора 3 доводят водой Р до объема
10 мл. Полученный раствор должен выдерживать ис¬
пытание на железо.
Магний. Не более 0,0100 % (100 ррт), если суб¬
станция предназначена для производства лекарствен¬
ных средств для парентерального применения. К10 мл
раствора 3 прибавляют 1 мл глицерина (85 %) Р,
0,15 мл раствора титанового желтого Р, 0,25 мл
раствора аммония оксалата Р и 5 мл раствора на¬
трия гидроксида разведенного Р и встряхивают. Ро¬
зовая окраска полученного раствора должна быть не
интенсивнее окраски эталона, приготовленного парал¬
лельно с использованием смеси из 5 мл эталонного
раствора магния (10 ррт Мд) Р и 5 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 52,0 % и не более 57,0 %. 1,000 г испытуемого
образца сушат при температуре 30 °С в течение 1 ч,
затем при температуре 130 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 40 мл
воды Р, прибавляют смесь из 0,2 мл 0,1 М раствора
кислоты хлористоводородной и 80 мл метанола Р.
Полученный раствор титруют 0,1 М раствором свин¬
ца нитрата потенциометрически (2.2.20), используя
в качестве индикаторного электрода свинец-селек-
тивный электрод, а в качестве электрода сравне¬
ния — хлорсеребряный.
1 мл 0,1 М раствора свинца нитрата соответ¬
ствует 14,20 мг Ма2304.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для изготовления лекар¬
ственных средств для парентерального применения.
07/2016:0775
НАТРИЯ СУЛЬФИТ БЕЗВОДНЫЙ
АШгН зиШз апЬудпсиз
ЗСЮШМ 31Л.Р1ТЕ, АЛ/НУОЯО(75
№2503 М.м. 126,0
[7757-83-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 95,0 % и не более 100,5 %
Ма2303.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», имеет слабощелочную реакцию
(2.2.4).
B. К 5 мл раствора 3 прибавляют 0,5 мл 0,05 М
раствора йода. Раствор бесцветный, дает реакцию
(а) на сульфаты (2.3.1).
C. Раствор 3 дает реакцию (а) на натрий (2.3.1).
О. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
#Е. Раствор 3 дает реакцию (а) на сульфиты
(2.3.1).
ИСПЫТАНИЕ
Раствор 5. 5 г испытуемого образца растворяют
в воде Р и доводят до объема 100 мл этим же раство¬
рителем.
Раствор 51. К 10,0 г испытуемого образца при¬
бавляют 25 мл воды Р. Встряхивают до растворения
большей части испытуемого образца и осторожно и
постепенно прибавляют 15 мл кислоты хлористово¬
дородной Р. Нагревают до кипения. Охлаждают и до¬
водят водой Р до объема 100,0 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод /). Раствор 3 должен
быть бесцветным.
Тиосульфаты. Не более 0,1 %. К 2,00 г испытуе¬
мого образца прибавляют 100 мл воды Р. Встряхива¬
ют и прибавляют 10 мл раствора формальдегида Р и
10 мл кислоты уксусной Р. Выдерживают в течение
5 минут. Прибавляют 0,5 мл раствора крахмала Р и
титруют 0,05 М раствором йода.
Параллельно проводят контрольный опыт.
Разность между объемами титранта не должна
превышать 0,15 мл.
Железо (2.4.9). Не более 0,0010% (10 ррт).
10 мл раствора 31 должны выдерживать испытание
на железо.
Селен. Не более 0,0010 % (10 ррт). К 3,0 г ис¬
пытуемого образца прибавляют 10 мл раствора фор¬
мальдегида Р и осторожно и постепенно прибавляют
2 мл кислоты хлористоводородной Р. Нагревают
на водяной бане в течение 20 мин. Розовая окраска
раствора не должна превышать окраску эталона, при¬
готовленного параллельно с использованием 1,0 г ис¬
пытуемого образца и 0,2 мл эталонного раствора
селена (100 ррт Зе) Р.
Цинк. Не более 0,0025 % (25 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Натрия сульфит гептагидрат
737
Испытуемый раствор. 2,0 мл раствора 31 дово¬
дят водой Р до объема 10,0 мл.
Растворы сравнения. Растворы сравнения го¬
товят разведением эталонного раствора цинка
(100 ррт 2.п) Р с использованием воды Р.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Тяжелые металлы (2.4.8, метод А). Не более
0.0010 % (10 ррт). 20 мл раствора 31 выпаривают
почти досуха. Прибавляют 10 мл воды Р, нейтрали¬
зуют раствором аммиака концентрированным Р и
доводят водой Р до объема 20 мл. 12 мл полученного
раствора должны выдерживать испытание на тяже¬
лые металлы. Эталон готовят с использованием эта¬
лонного раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца помещают в кони¬
ческую колбу вместимостью 500 мл, содержащую
50,0 мл 0,05 М раствора йода. Встряхивают до пол¬
ного растворения. Прибавляют 1 мл раствора крах¬
мала Р и титруют избыток йода 0,1 М раствором на¬
трия тиосульфата.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует 6,30 мг
Ма2303.
07/2016:0776
НАТРИЯ СУЛЬФИТ ГЕПТАГИДРАТ
Л/а^г/7 зиШз Ьер&Ьудпсиз
ЗООШМ 51Л-ИТЕ, НЕРТАНУОНАТЕ
№2803 ■ 7Н20 М.м. 252,2
[10102-15-5]
Содержание: не менее 48,0 % и не более 52,5 %
Ма2303.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветные кристаллы.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», имеет слабощелочную реакцию
(2.2.4).
B. К 5 мл раствора 3 прибавляют 0,5 мл 0,05 М
раствора йода. Раствор бесцветный, дает реакцию
(а) на сульфаты (2.3.1).
C. Раствор 3 дает реакцию (а) на натрий (2.3.1).
й. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
#Е. Раствор 3 дает реакцию (а) на сульфиты (2.3.1).
ИСПЫТАНИЕ
Раствор 5. Юг испытуемого образца раство¬
ряют в воде Р и доводят до объема 100 мл этим же
растворителем.
Раствор 31. К 20,0 г испытуемого образца при¬
бавляют 25 мл воды Р. Встряхивают до растворения
большей части испытуемого образца и осторожно и
постепенно прибавляют 15 мл кислоты хлористово¬
дородной Р. Нагревают до кипения. Охлаждают и до¬
водят водой Р до объема 100,0 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод /). Раствор 3 должен
быть бесцветным.
Тиосульфаты. Не более 0,05 %. К 4,00 г ис¬
пытуемого образца прибавляют 100 мл воды Р.
Встряхивают до растворения и прибавляют 10 мл
раствора формальдегида Р и 10 мл кислоты уксус¬
ной Р. Выдерживают в течение 5 минут. Прибавляют
0,5 мл раствора крахмала Р и титруют 0,05 М рас¬
твором йода.
Параллельно проводят контрольный опыт.
Разность между объемами титранта не должна
превышать 0,15 мл.
Железо (2.4.9). Не более 0,0005 % (5 ррт). 10 мл
раствора 31 должны выдерживать испытание на же¬
лезо.
Селен. Не более 0,0005 % (5 ррт). К 6,0 г ис¬
пытуемого образца прибавляют 10 мл раствора
формальдегида Р и осторожно и постепенно при¬
бавляют 2 мл кислоты хлористоводородной Р. На¬
гревают на водяной бане в течение 20 мин. Розовая
окраска раствора не должна превышать окраску
эталона, приготовленного параллельно с исполь¬
зованием 2,0 г испытуемого образца, к которому
прибавлено 0,2 мл эталонного раствора селена
(100 ррт Зе) Р.
Цинк. Не более 0,0012 % (12 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. 2,0 мл раствора 31 дово¬
дят водой Р до объема 10,0 мл.
Растворы сравнения. Растворы сравнения го¬
товят соответствующими разведениями эталонного
раствора цинка (100 ррт 2п) Р водой Р.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 213,9 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). 20 мл раствора 31 выпаривают
почти досуха. Прибавляют 10 мл воды Р, нейтрали¬
зуют раствором аммиака концентрированным Р и
доводят водой Р до объема 20 мл. 12 мл получен¬
ного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использовани¬
ем эталонного раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца помещают в кони¬
ческую колбу вместимостью 500 мл, содержащую
50,0 мл 0,05 М раствора йода. Встряхивают до пол¬
ного растворения. Прибавляют 1 мл раствора крах¬
мала Р и титруют избыток йода 0,1 М раствором на¬
трия тиосульфата.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует 6,30 мг
Ма25 03.
95. Зак. 1060.
738
Гэсударственная фармакопея Республики Беларусь
07/2016:0013
НАТРИЯ ТЕТРАБОРАТ
Богах
ВОКАХ
№2В407 ■ ЮН20 М.м. 381,4
[1303-96-4]
ОПРЕДЕЛЕНИЕ
Динатрия тетраборат декагидрат.
Содержание: не менее 99,0 % и не более 103,0 %
№2В407ЮН20.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок, бесцветные кристаллы или кристаллическая
масса. Выветривается на воздухе.
Растворим в воде, очень легко растворим в кипя¬
щей воде, легко растворим в глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,1 мл кисло¬
ты серной Р, 5 мл метанола Р и поджигают. Пламя
имеет зеленую кайму.
B. К 5 мл раствора 3 прибавляют 0,1 мл раст¬
вора фенолфталеина Р. Появляется красное окра¬
шивание. При прибавлении 5 мл глицерина (85 %) Р
окрашивание исчезает.
C. Раствор 3 дает реакции (а) и (Ь) на натрий (2.3.7).
ИСПЫТАНИЯ
Раствор 3. 4,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, по¬
лученной из воды дистиллированной Р, и доводят до
объема 100 мл этим же растворителем.
Прозрачность (2.2.7). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 9,0 до 9,6. Измеряют рН раствора 3.
Сульфаты (2.4.73). Не более 0,0050 % (50 ррт).
Определение проводят из 15 мл раствора 3. Исполь¬
зуют 1,0 мл кислоты уксусной Р. Эталон готовят с ис¬
пользованием 3 мл эталонного раствора сульфата
(10 ррт 80^) Р и 12 мл воды дистиллированной Р.
Аммония соли (2.4.7). Не более 0,0010%
(10 ррт). 6 мл раствора 3 доводят водой Р до объ¬
ема 14 мл. Полученный раствор должен выдерживать
испытание на соли аммония. Эталон готовят с ис¬
пользованием 2,5 мл эталонного раствора аммония
(1 ррт N14) Р и 7,5 мл воды Р.
Мышьяк (2.4.2, метод А). Не более 0,0005 %
(5 ррт). 5 мл раствора 3 должны выдерживать испы¬
тание на мышьяк.
Кальций (2.4.3). Не более 0,0100% (100 ррт).
15 мл раствора 3 должны выдерживать испытание на
кальций. Эталон готовят с использованием смеси из
6 мл эталонного раствора кальция (10 ррт Са) Р и
9 мл воды дистиллированной Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0025 % (25 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.7.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
20 г маннита Р растворяют в 100 мл воды Р,
нагревая при необходимости, охлаждают, прибав¬
ляют 0,5 мл раствора фенолфталеина Р и ней¬
трализуют 0,1 М раствором натрия гидроксида до
появления розового окрашивания. К полученному
раствору прибавляют 3,00 г испытуемого образца,
нагревают до полного растворения, охлаждают и
титруют 7 М раствором натрия гидроксида до по¬
явления розового окрашивания.
1 мл 7 М раствора натрия гидроксида соот¬
ветствует 0,1907 г Ыа2В407 ЮН20.
07/2016:0414
НАТРИЯ ТИОСУЛЬФАТ
ЫаЬ7/ МозиКаз
8СЮШМ ТНЮ31Н.РАТЕ
Ыа25203 • 5Н20 М.м. 248,2
[10102-17-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %
Ма23203-5Н20.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачные бесцветные кристаллы. Выветрива¬
ется в сухом воздухе.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте. Растворяется в кристалли¬
зационной воде при нагревании до температуры око¬
ло 49 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец обесцвечивает калия
йодида йодированный раствор Р
B. К 0,5 мл раствора 3, приготовленного как
указано в разделе «Испытания», прибавляют 0,5 мл
воды Р и 2 мл раствора серебра нитрата Р2. Об¬
разуется белый осадок, окраска которого быстро
переходит в желтоватую, а затем в черную.
C. К 2,5 мл раствора 3 прибавляют 2,5 мл
воды Р и 1 мл кислоты хлористоводородной Р. Вы¬
падает осадок серы и выделяется газ, окрашиваю¬
щий в синий цвет йодаткрахмальную бумагу Р.
О. 1 мл раствора 3 дает характерную реакцию
(а) на натрий (2.3.7).
ИСПЫТАНИЯ
Раствор 3. 10,0 г испытуемого образца раст¬
воряют в воде, свободной от углерода диокси¬
да, Р, приготовленной из воды дистиллирован¬
ной Р, и доводят до объема 100 мл этим же раст¬
ворителем.
Раствор 31. 10,0 г испытуемого образца раст¬
воряют в 50 мл воды дистиллированой Р, прибав¬
ляют 1 мл 0,1 М раствора натрия гидроксида и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.7). Свежеприготовленный
раствор 31 должен быть прозрачным.
Цветность (2.2.2, метод II). Свежеприготов¬
ленный раствор 31 должен быть бесцветным.
рН (2.2.3). От 6,0 до 8,4. Измеряют рН свеже¬
приготовленного раствора 5.
Натрия фторид
739
#Хлориды(2.4.4).Неболее0,0200 % (200 ррт).
К 5 мл раствора 3 прибавляют 15 мл кислоты
азотной разведенной Р, осторожно кипятят (3—-4)
мин, охлаждают и фильтруют. Фильтрат доводят
водой Р до объема 25 мл. 12,5 мл полученного
раствора доводят водой Р до объема 15 мл. Раст¬
вор должен выдерживать испытание на хлориды.
Сульфаты и сульфиты (2.4.13). Не более 0,2 %.
2,5 мл свежеприготовленного раствора 3 доводят
водой дистиллированной Р до объема 10мл. К
3 мл полученного раствора сначала прибавляют
2 мл йодированного раствора калия йодида Р, за¬
тем продолжают прибавлять его по каплям до по¬
лучения очень слабого устойчивого желтого окра¬
шивания. Полученный раствор доводят водой дис¬
тиллированной Р до объема 15 мл. Раствор дол¬
жен выдерживать испытание на сульфаты.
Сульфиды. К 10 мл раствора 3 прибавляют
0,05 мл свежеприготовленного раствора 50 г/л на¬
трия нитропруссида Р. Не должно появляться фи¬
олетовое окрашивание.
Тяжелые металлы. Не более 0,0010 % (10 ррт).
К 10 мл раствора 3 прибавляют 0,05 мл раствора
натрия сульфида Р. Через 2 мин коричневое окра¬
шивание раствора не должно быть интенсивнее
окрашивания эталона, приготовленного парал¬
лельно с использованием 10 мл эталонного раст¬
вора свинца (1 ррт РЬ) Р
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в
20 мл воды Р и титруют 0,05 М раствором йода,
используя в качестве индикатора 1 мл раствора
крахмала Р, прибавляемого в конце титрования.
1мл 0,05 М раствора йода соответствует
24,82 мг Ма23203-5Н20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0514
НАТРИЯ ФТОРИД
Ыа1гИ Лиопс/ит
8СЮШМ РШОПЮЕ
№Р М.м. 41,99
[7681-49-4]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Растворим в воде, практически нерастворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 0,5 мл
раствора кальция хлорида Р. Образуется гелео¬
бразный белый осадок, который растворяется при
добавлении 5 мл раствора железа (III) хлорида Р1.
B. К 4 мг испытуемого образца прибавляют
смесь из 0,1 мл раствора ализарина 3 Р и 0,1 мл
раствора цирконила нитрата Р и перемешивают.
Окраска раствора изменяется от красной до жел¬
той.
C. Раствор 3 дает реакцию (а) на натрий
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р
без нагревания и доводят до объема 100 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. 2,5 г калия
нитрата Р растворяют в 40 мл раствора 3 и до¬
водят водой, свободной от углерода диоксида, Р
до объема 50 мл. Охлаждают до температуры 0 °С
и прибавляют 0,2 мл раствора фенолфталеи¬
на Р. Если раствор бесцветный, при прибавлении
не более 1,0 мл 0,1 М раствора натрия гидрок¬
сида должно появиться красное окрашивание, не
исчезающее в течение 15 с. Если раствор красный,
при прибавлении не более 0,25 мл 0,1 М раствора
кислоты хлористоводородной цвет индикатора
должен измениться.
Хлориды (2.4.4). Не более 0,0200 %
(200 ррт). 10 мл раствора 5 доводят водой Р до
объема 15 мл.
Фторсиликаты. Нейтрализованный раствор,
полученный в испытании «Кислотность или щелоч¬
ность», нагревают до кипения и титруют в горячем
виде. При прибавлении не более 0,75 мл 0,1 М
раствора натрия гидроксида цвет индикатора
должен измениться на красный.
Сульфаты (2.4.13). Не более 0,0200%
(200 ррт). 0,25 г испытуемого образца растворяют
в 10 мл раствора 223 г/л алюмния нитрата Р. При¬
бавляют 5 мл воды дистиллированной Р и 0,6 мл
кислоты хлористоводородной Р1. Эталонный
раствор готовят с использованием смеси из 0,6 мл
кислоты хлористоводородной Р1, 5 мл эталон¬
ного раствора сульфата (10 ррт 30) Р и 10 мл
раствора 223 г/л алюмния нитрата Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца су¬
шат при температуре 130 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в
воде Р и доводят до объема 60 мл этим же раст¬
ворителем. Титруют 0,1 М раствором лантана ни¬
трата потенциометрически (2.2.20) с использова¬
нием фтор-селективного индикаторного электрода
и хлорсеребряного электрода сравнения.
1 мл 0,1 М раствора лантана нитрата соот¬
ветствует 12,60 мг ЫаР.
95*. Зак. 1060.
740
Гэсударственная фармакопея Республики Беларусь
07/2016:0193
НАТРИЯ ХЛОРИД
А/а/г/7 сЫогМит
ЗСЮШМ СШ.ОКЮЕ
№С1 М.м. 58,44
[7647-14-5]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы или белые или почти
белые крупинки.
Легко растворим в воде, практически нераство¬
рим в этаноле безводном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
B. Испытуемый образец дает реакции (а) и (Ь) на
натрий (2.3.1).
ИСПЫТАНИЯ
Если испытуемый образец представляет собой
крупинки, перед испытанием его размалывают.
Раствор 5. 20,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 20 мл раст¬
вора 3 прибавляют 0,1 мл раствора бромтимоло-
вого синего Р1. При прибавлении не более 0,5 мл
0,01 М раствора кислоты хлористоводородной или
0,01 М раствора натрия гидроксида окраска раст¬
вора должна измениться.
Бромиды. Не более 0,0100 % (100 ррт). К 0,5 мл
раствора 3 прибавляют 4,0 мл воды Р, 2,0 мл раст¬
вора фенолового красного Р2,1,0 мл раствора 0,1 г/л
хлорамина Р и немедленно перемешивают. Точно че¬
рез 2 мин прибавляют 0,15 мл 0,1 Мраствора натрия
тиосульфата, перемешивают и доводят водой Р до
объема 10,0мл. Оптическая плотность (2.2.25) по¬
лученного раствора, измеренная при длине волны
590 нм, не должна превышать оптическую плотность
эталона, приготовленного параллельно с испытуе¬
мым раствором с использованием 5 мл раствора 3,0
мг/л калия бромида Р. В качестве компенсационного
раствора используют воду Р.
Ферроцианиды. 2,0 г испытуемого образца
растворяют в 6 мл воды Р, прибавляют 0,5 мл смеси
из 5 мл раствора 10 г/л железа (III) аммония сульфа¬
та Р в растворе 2,5 г/л кислоты серной Р и 95 мл
раствора 10 г/л железа (II) сульфата Р. В течение
10 мин не должно появиться синее окрашивание.
Йодиды. 5 г испытуемого образца увлажняют,
прибавляя каплями свежеприготовленную смесь из
0,15 мл раствора натрия нитрита Р, 2 мл 0,5 М
раствора кислоты серной, 25 мл раствора крах¬
мала, свободного от йодидов, Р и 25 мл воды Р.
Полученный раствор через 5 мин просматривают
при дневном освещении. Не должно появиться си¬
нее окрашивание.
Нитриты. К 10 мл раствора 3 прибавляют 10 мл
воды Р. Оптическая плотность (2.2.25) полученного
раствора, измеренная при длине волны 354 нм, не
должна превышать 0,01.
Фосфаты (2.4.11). Не более 0,0025% (25 ррт).
2 мл раствора 3 доводят водой Р до объема 100 мл.
Полученный раствор должен выдерживать испытание
на фосфаты.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
7,5 мл раствора 3 доводят водой дистиллирован¬
ной Р до объема 30 мл. 15 мл полученного раствора
должны выдерживать испытание на сульфаты.
Алюминий (2.4.17). Не более 0,00002%
(0,2 ррт), если субстанция предназначена для про¬
изводства растворов для перитонеального диализа,
гемодиализа или гемофильтрации.
Раствор испытуемого образца. 20,0 г испыту¬
емого образца растворяют в 100 мл воды Р и при¬
бавляют 10 мл ацетатного буферного раствора
рН 6,0 Р.
Раствор сравнения. Смешивают 2 мл эталонно¬
го раствора алюминия (2 ррт А!) Р, 10 мл ацетат¬
ного буферного раствора рН 6,0 Р и 98 мл воды Р.
Контрольный раствор. Смешивают 10 мл аце¬
татного буферного раствора рН 6,0 Р и 100мл
воды Р.
Мышьяк (2.4.2, метод А). Не более 0,0001 %
(1 ррт). 5 мл раствора 3 должны выдерживать испы¬
тание на мышьяк.
Барий. К 5 мл раствора 3 прибавляют 5 мл воды
дистиллированной Р и 2 мл кислоты серной разве¬
денной Р. Через 2 ч опалесценция полученного раст¬
вора не должна превышать опалесценцию смеси из
5 мл раствора 3 и 7 мл воды дистиллированной Р.
Железо (2.4.9). Не более 0,0002 % (2 ррт). 10 мл
раствора 3 должны выдерживать испытание на желе¬
зо. Эталон готовят с использованием смеси из 4 мл
эталонного раствора железа (1 ррт Ре) Р и 6 мл
воды Р.
Магний и щелочноземельный металлы (2.4.7).
Не более 0,0100% (100 ррт) в пересчете на Са.
10.0 г испытуемого образца должны выдерживать ис¬
пытание на магний и щелочноземельные металлы.
Используют 0,150 г индикаторной смеси протрав¬
ного черного 11 Р. Объем пошедшего на титрование
0,01 М раствора натрия эдетата не должен превы¬
шать 2,5 мл.
Калий. Не более 0,0500 % (500 ррт), если суб¬
станция предназначена для производства лекар¬
ственных средств для парентерального применения
или растворов для гемодиализа, перитонеального
диализа и гемофильтрации.
Атомно-эмиссионная спектрометрия (2.2.22, ме¬
тод I).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения. Готовят соответствующими
разведениями раствора, приготовленного следующим
образом: 1,144 г калия хлорида Р, предварительно
высушенного при температуре от 100 °С до 105 °С в
течение 3 ч, растворяют в воде Р и доводят до объема
1000.0 мл этим же растворителем (600 мкг/мл К).
Интенсивность эмиссии измеряют при длине вол¬
ны 766,5 нм.
Натрия цетостеарилсульфат
741
Тяжелые металлы (2.4.8, метод А). Не более
0,0005% (5ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Бактериальные эндотоксины (2.6.14). Менее
5 МЕ/г, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без последующей процедуры удаления
бактериальных эндотоксинов.
#Микробиологмческая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50,0 мг испытуемого образца растворяют в
воде Р и доводят до объема 50 мл этим же раствори¬
телем и титруют 0,1 М раствором серебра нитрата
потенциометрически (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,844 мг ЫаС1.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения;
- субстанция пригодна для производства раство¬
ров для перитонеального диализа, гемодиализа и ге¬
мофильтрации.
07/2016:0847
НАТРИЯ ЦЕТОСТЕАРИЛСУЛЬФАТ
Л/а/г/7 се/у/о- е1 з1еагу1ози1Таз
500/(УМ СЕТОЗТЕАКП. ЗШ-РАТЕ
ОПРЕДЕЛЕНИЕ
Смесь натрия цетилсульфата (С16Н33Ма043; М.м.
344.5) и натрия стеарилсульфата (С18Н37Ма043; М.м.
372.5) . Может быть добавлен подходящий буфер.
Содержание:
- натрия цетостеарилсульфат: не менее
90,0 % (в пересчете на безводное вещество);
- натрия цетилсульфат: не менее 40,0 % (в
пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или бледно-желтый аморфный или кри¬
сталлический порошок.
Растворим в горячей воде с образованием опа¬
лесцирующего раствора, практически нерастворим в
холодной воде, частично растворяется в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О, Р.
Вторая идентификация: А, С, О, Е, Р.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 10 мл спирта (70 %, об/об) Р при
нагревании на водяной бане.
Раствор сравнения. 50 м г ФСО натрия цето-
стеарилсульфата растворяют в 10 мл спирта (70 %,
об/об) Р при нагревании на водяной бане.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: вода Р — ацетон Р — мета¬
нол Р (20:40:40, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
50 г/л кислоты фосфорномолибденовой Р в 96%
спирте Р и нагревают при температуре 120 °С до по¬
явления пятен (около 5 мин).
Результаты: на хроматограмме испытуемого
раствора обнаруживаются основные пятна, соответ¬
ствующие по расположению и цвету основным пятнам
на хроматограмме раствора сравнения.
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определе¬
ние».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживаются 2 основных пика, соот¬
ветствующих по времени удерживания 2 основным
пикам на хроматограммах растворов сравнения (а) и
(Ь)-
C. 0,1 г испытуемого образца растворяют в 10 мл
воды Р и встряхивают. Образуется пена.
Б. Испытуемый образец окрашивает бесцветное
пламя в желтый цвет.
Е. К 0,1 мл раствора, приготовленного как указа¬
но в идентификации С, прибавляют 0,1 мл раствора
1 г/л метиленового синего Р и 2 мл кислоты серной
разведенной Р. Прибавляют 2 мл метиленхлорида Р
и встряхивают. Слой метиленхлорида имеет интен¬
сивный синий цвет.
Р. 10 мг испытуемого образца смешивают с 10 мл
этанола Р и нагревают до кипения на водяной бане
при частом встряхивании. Немедленно фильтруют
и выпаривают досуха. Остаток растворяют в 7 мл
воды Р, прибавляют 3 мл кислоты хлористоводо¬
родной разведенной Р, выпаривают раствор до полу¬
чения половины объема, охлаждают и фильтруют. К
фильтрату прибавляют 1 мл раствора бария хлори¬
да Р1. Образуется белый кристаллический осадок.
ИСПЫТАНИЯ
Кислотность или щелочность. 0,5 г испытуе¬
мого образца растворяют при нагревании в смеси из
10 мл воды Р и 15 мл спирта (90 %, об/об) Р и при¬
бавляют 0,1 мл раствора фенолфталеина Р1. Раст¬
вор бесцветный. Прибавляют 0,1 мл 0,1 М раствора
натрия гидроксида. Появляется красное окрашива¬
ние.
Натрия хлорид и натрия сульфат. Суммарное
процентное содержание: не более 8,0 %.
Натрия хлорид. 5,00 г испытуемого образца
растворяют в 50 мл воды Р, прибавляют по каплям
кислоту азотную разведенную Р до нейтральной по
лакмусовой бумаге синей Р реакции раствора. Титру¬
ют 0,1 М раствором серебра нитрата потенциоме¬
трически (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,844 мг №С1.
Натрия сульфат. 0,500 г испытуемого образца
растворяют в 20 мл воды Р, при необходимости слег¬
ка подогревая, и прибавляют 1 мл раствора 0,5 г/л ди-
тизона Р в ацетоне Р. Если цвет раствора красный,
прибавляют по каплям 1 М раствор кислоты азот-
742
Гэсударственная фармакопея Республики Беларусь
ной до появления голубовато-зеленого окрашивания.
Прибавляют 2,0 мл раствора кислоты дихлоруксус-
ной Р и 80 мл ацетона Р. Титруют 0,01 М раствором
нитрата свинца до появления неисчезающего оран¬
жево-красного окрашивания.
1 мл 0,01 М раствора нитрата свинца соответ¬
ствует 1,420 мг Ма2504.
Свободный цетостеариловый спирт. Не более
4,0 %.
Используя хроматограмму испытуемого раствора
(а), полученную как указано в разделе «Количествен¬
ное определение», рассчитывают процентное содер¬
жание свободного цетостеарилового спирта в испы¬
туемом образце, принимая во внимание заявленное
содержание в ФСО:
Аг /771 100
где:
А1 — сумма площадей пиков цетилового и сте-
арилового спиртов на хроматограмме испытуемого
раствора (а);
А2 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора (а);
дп1 — масса навески испытуемого образца, взя¬
того для приготовления испытуемого раствора (а), мг;
т2 — масса навески внутреннего стандарта, взя¬
того для приготовления раствора внутреннего стан¬
дарта, мг.
Вода (2.5.12). Не более 1,5 %. Определение про¬
водят из 5,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ления слоев. Объединенные верхние слои промывают
двумя порциями по 30 мл воды Р, сушат с использова¬
нием натрия сульфата безводного Р и фильтруют.
Раствор сравнения (Ь). 0,100 г ФСО стеарино¬
вого спирта растворяют в 25,0 мл раствора внутрен¬
него стандарта. Прибавляют 25 мл воды Р и встряхи¬
вают с четырьмя порциями по 25 мл пентана Р, при¬
бавляя при необходимости натрия хлорид Р для об¬
легчения разделения слоев. Объединенные верхние
слои промывают двумя порциями по 30 мл воды Р,
сушат с использованием натрия сульфата безводно¬
го Р и фильтруют.
Условия хроматографирования:
- колонка кварцевая стеклянная длиной 25 м и
диаметром 0,25 мм, покрытая слоем поли(диметил)
силоксана Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—20
150->250
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок элюирования: цетиловый спирт, стеари-
ловый спирт, 1-нонадеканол.
Рассчитывают процентное содержание натрия
цетилсульфата в испытуемом образце по формуле,
принимая во внимание заявленное содержание на¬
трия цетилсульфата в ФСО цетилового спирта:
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 0,200 г ФСО
1-нонадеканола растворяют в этаноле Р и доводят
до объема 100,0 мл этим же растворителем.
Испытуемый раствор (а). 0,300 г испытуемого
образца растворяют в 50,0 мл этанола Р и прибавля¬
ют 2,0 мл раствора внутреннего стандарта и 48,0 мл
воды Р. Встряхивают с четырьмя порциями по 25 мл
пентана Р, при необходимости прибавляя натрия
хлорид Р для облегчения разделения слоев. Верхние
слои объединяют. Нижний слой оставляют для при¬
готовления испытуемого раствора (Ь). Объединенные
верхние слои дважды промывают двумя порциями по
30 мл воды Р, сушат с использованием натрия суль¬
фата безводного Р и фильтруют.
Испытуемый раствор (Ь). 25,0 мл нижнего слоя,
полученного при приготовлении испытуемого раст¬
вора (а), переносят в колбу вместимостью 200 мл,
которую можно присоединить к обратному холодиль¬
нику. Прибавляют 10,0 мл раствора внутреннего стан¬
дарта и 20 мл кислоты хлористоводородной Р. Кипя¬
тят с обратным холодильником в течение 2 ч. Охлаж¬
дают и встряхивают с четырьмя порциями по 20 мл
пентана Р. Объединяют верхние слои, промывают
двумя порциями по 20 мл воды Р, сушат с использо¬
ванием натрия сульфата безводного Р и фильтруют.
Раствор сравнения (а). 0,100 г ФСО цетилово¬
го спирта растворяют в 25,0 мл раствора внутреннего
стандарта. Прибавляют 25 мл воды Р и встряхивают с
четырьмя порциями по 25 мл пентана Р, прибавляя при
необходимости натрия хлорид Р для облегчения разде-
^..^.1.-1004 — Р,
А А,У
т
2,5
где:
Ах — площадь пика цетилового спирта на хрома-
тограммме испытуемого раствора (Ь);
Аху— площадь пика ФСО цетилового спирта на
хроматограмме раствора сравнения (а);
— площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора (Ь);
А2 — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (а);
Рх — фактор пересчета между цетиловым спир¬
том и натрия цетилсульфатом (1,421);
т — масса навески испытуемого образца, взято¬
го для приготовления испытуемого раствора (а), мг;
тху — масса навески ФСО цетилового спирта,
взятого для приготовления раствора сравнения (а), мг.
Рассчитывают процентное содержание натрия
стеарилсульфата в испытуемом образце по фор¬
муле, принимая во внимание заявленное содержа¬
ние натрия стеарилсульфата в ФСО стеаринового
спирта:
А тг,
А А.у т
—-100 4—Р,
2,5
где:
Аг — площадь пика стеарилового спирта на хро-
матограммме испытуемого раствора (Ь);
Агу — площадь пика ФСО стеарилового спирта
на хроматограмме раствора сравнения (Ь);
Л1 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора (Ь);
Натрия цитрат
743
А3 — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (Ь);
Рг — фактор пересчета между стеариновым
спиртом и натрия стеарилсульфатом (1,377);
т — масса навески испытуемого образца, взято¬
го для приготовления испытуемого раствора (а), мг;
тгу — масса навески ФСО стеарилового спир¬
та, взятого для приготовления раствора сравнения
(Ь), мг.
Процентное содержание натрия цетостеарил-
сульфата соответствует сумме процентных содержа¬
ний натрия цетил сульфата и натрия стеарилсул ьфата.
МАРКИРОВКА
При необходимости указывают:
- название и концентрацию добавленного буфера.
07/2016:0412
НАТРИЯ ЦИТРАТ
А/а^г/7 сНгаз
ЗООШМ С1ТКАТЕ
НО С02Ыа
\/ * 2 Н20
Ма02С^>\^С02Ма
С6Н5Ыа307 ■ 2Н20 М.м. 294,1
[6132-04-3]
ОПРЕДЕЛЕНИЕ
Тринатрия 2-гидроксипропан-1,2,3-трикарбоксилат
дигидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или белые или почти белые зернистые кристал¬
лы. Слегка расплывается во влажном воздухе.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 1 мл раствора 5, полученному как указано
в разделе «Испытания», прибавляют 4 мл воды Р.
Полученный раствор дает реакцию (а) на цитраты
(2.3.1) .
B. 1 мл раствора 5 дает реакцию (а) на натрий
(2.3.1) .
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р,
приготовленной из воды дистиллированной Р, и до¬
водят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 5 прибавляют 0,1 мл раствора фенолфта¬
леина Р. При прибавлении не более 0,2 мл 0,1 М
кислоты хлористоводородной или 0,1 М раствора
натрия гидроксида окраска раствора должна изме¬
ниться.
Легкообугливающиеся вещества. К 0,20 г
измельченного испытуемого образца прибавляют
10 мл кислоты серной Р, нагревают на водяной
бане при температуре (90±1) °С в течение 60 мин и
быстро охлаждают. Окраска полученного раствора
должна быть не интенсивнее эталона У(Ж)2 или
<ЗУ(ЗЖ)2 (2.2.2, метод II).
Хлориды (2.4.4). Не более 0,0050 % (50 ррт).
10 мл раствора 5 доводят водой Р до объема 15 мл.
Оксалаты. Не более 0,0300 % (300 ррт).
0,50 г испытуемого образца растворяют в 4 мл
воды Р, прибавляют 3 мл кислоты хлористоводо¬
родной Р, 1 г гранулированного цинка Р и нагрева¬
ют на водяной бане в течение 1 мин. Выдерживают
в течение 2 мин, сливают надосадочную жидкость
в пробирку, содержащую 0,25 мл раствора 10 г/л
фенилгидразина гидрохлорида Р, нагревают до
кипения, быстро охлаждают, переносят в мерный
цилиндр, прибавляют равный объем кислоты
хлористоводородной Р, 0,25 мл раствора калия
феррицианида Р, встряхивают и выдерживают
в течение 30 мин. Розовая окраска полученного
раствора должна быть не интенсивнее эталона,
приготовленного аналогично и в то же самое время
с использованием 4 мл раствора 50 мг/л кислоты
щавелевой Р.
Сульфаты (2.4.13). Не более 0,0150%
(150 ррт). К 10 мл раствора 5 прибавляют 2 мл
кислоты хлористоводородной Р1 доводят водой
дистиллированной Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны вы¬
держивать испытание на тяжелые металлы. Эталон
готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не менее 11,0% и не более
13,0 %. Определение проводят из 0,300 г испыту¬
емого образца. Используют смесь растворителей
из 20 мл метанола Р, 30 мл формамида Р и 5 г
салициловой кислоты Р. После прибавления испы¬
туемого образца раствор перемешивают в течение
15 мин и титруют.
Пирогенность (2.6.8). Если субстанция пред¬
назначена для производства лекарственных
средств для парентерального применения боль¬
ших объемов, испытуемый образец должен выдер¬
живать испытание на пирогенность. Тест-доза —
10 мл свежеприготовленного раствора в воде для
инъекций Р, содержащего 10,0мг/мл испытуемого
образца и 7,5 мг/мл кальция хлорида Р, свободного
от пирогенов, на 1,0 кг массы тела кролика.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворят в 20 мл
кислоты уксусной безводной Р, нагревая до темпера¬
туры около 50 °С, охлаждают и тируют 0,1 М раство¬
ром кислоты хлорной до появления зеленого окра¬
шивания, используя в качестве индикатора 0,25 мл
раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 8,602 мг С6Н5Ма307.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
744
Гэсударственная фармакопея Республики Беларусь
07/2016:2134
НАТРИЯ
ЭТИЛПАРАГИДРОКСИБЕНЗОАТ
ЕОзуНз рагаЬудгохуЬетоаз паМсиз
800ШМ ЕТНУ1-
РАПАНУОПОХУВЕМгОАТЕ
С9Н9Ыа03 М.м. 188,2
[35285-68-8]
ОПРЕДЕЛЕНИЕ
Натрия 4-(этоксикарбонил)феноат.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, растворим в безводном
этаноле, практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификациям, С, О.
A. 0,5 г испытуемого образца растворяют в 50 мл
воды Р и немедленно прибавляют 5 мл кислоты хло¬
ристоводородной Р1. Фильтруют, промывают осадок
водой Р. Сушат в вакууме при температуре 80 °С в те¬
чение 2 ч. Температура плавления (2.2.14): от 115 °С
до 118 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: используют осадок, полученный
в идентификации А.
Сравнение: ФСО этилпарагидроксибензоата.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемо¬
го образца растворяют в 10 мл воды Р, немедленно
прибавляют 2 мл кислоты хлористоводородной Р и
встряхивают с 50 мл 1,1 -диметилэтилметилового
эфира Р. Выпаривают верхний слой досуха, остаток
растворяют в 10 мл ацетона Р.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
Раствор сравнения (а). 5 мг ФСО этилпараги¬
дроксибензоата растворяют в ацетоне Р и доводят
до объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО метилпара-
гидроксибензоата (примесь В) растворяют в 0,5 мл
испытуемого раствора (а) и доводят ацетоном Р до
объема 5 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза : кислота уксусная ледяная Р—
вода Р — метанол Р (1:30:70, об/об/об).
Наносимый объем пробы: 5 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
О. К1 мл раствора 3, приготовленного как указано
в разделе «Испытания», прибавляют 1 мл воды Р. По¬
лученный раствор дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3, приготовлен¬
ный непосредственно перед испытанием, должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3, приготовленного непосредственно перед испыта¬
нием, должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 9,5 до 10,5. 1 мл раствора 3 до¬
водят водой, свободной от углерода диоксида, Р до
объема 100 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл метанола Р и доводят
подвижной фазой до объема 50,0 мл. 10,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Раствор сравнения (а). 5 мг кислоты 4-гидрок-
сибензойной Р (примесь А), 5 мг метилпарагидрокси-
бензоата Р (примесь В) и 5 мг испытуемого образца
растворяют в подвижной фазе и доводят до объема
100.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО этилпара¬
гидроксибензоата растворяют в 2,5 мл метанола Р и
доводят подвижной фазой до объема 50,0 мл. 10,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: раствор 6,8 г/л калия диги¬
дрофосфата Р — метанол Р (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
-спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а) и (с);
- время хроматографирования: 4-кратное вре¬
мя удерживания этилпарагидроксибензоата.
Относительное удерживание (по отношению
к этилпарагидроксибензоата время удерживания —
Нафазолина гидрохлорид
745
около 3 мин): примесь А — около 0,5; примесь В —
около 0,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси В и этилпарагидроксибензоата.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А на 1,4):
- примесь А (не более 3,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 6-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (с);
- сумма примесей, кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора сумма
площадей всех пиков, кроме основного и пика примеси
А, не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (с);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (с).
Хлориды (2.4.4). Не более 0,0350 % (350 ррт). К
10 мл раствора 3 прибавляют 30 мл воды Р, 1 мл кис¬
лоты азотной Р и доводят водой Р до объема 50 мл.
Встряхивают и фильтруют. 10 мл фильтрата доводят
водой Р до объема 15 мл. Эталон готовят с использо¬
ванием 14 мл эталонного раствора хлоридов (5 ррт
С1) Р и 1 мл воды Р.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
К 25 мл раствора 3 прибавляют 5 мл воды дистилли¬
рованной Р, 10 мл кислоты хлористоводородной Р и
доводят водой дистиллированной Р до объема 15 мл.
Встряхивают и фильтруют. 10 мл фильтрата доводят во¬
дой дистиллированной Рдо объема 15 мл. Полученный
раствор должен выдерживать испытание на сульфаты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20,0 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
После прибавления буферного раствора рН
3,5 Р субстанция выпадает в осадок. Доводят этано¬
лом безводным Р до объема 40 мл. Субстанция пол¬
ностью растворяется.
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С9Н9№03 в процентах рассчитыва¬
ют с учетом содержания этилпарагидроксибензоата в
ФСО этилпарагидроксибензоата и умножают полу¬
ченный результат на поправочный коэффициент 1,132.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
А. 4-Гидроксибензойная кислота.
В. Метил-4-гидроксибензоат (метилпарагидрокси-
бензоат).
С. Пропил-4-гидроксибензоат (этилпарагидрокси-
бензоат).
О. Бутил-4-гидроксибензоат (бутилпарагидрокси-
бензоат).
07/2016:0730
НАФАЗОЛИНА ГИДРОХЛОРИД
ЫарЬагоНт ЬудгосМогШит
ЫАРНА20ШЕ НУОРЮСШ-ОНЮЕ
С14н14ы2 • НС1 М.м. 246,7
[550-99-2]
746
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
2-(Нафтален-1-илметил>415-дигидро-1 Н- имидазола
гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворим в воде, растворим в 96 % спирте.
Температура плавления: около 259 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 250,0 мл этим
же растворителем. 25,0 мл полученного раствора до¬
водят 0,01 М раствором кислоты хлористоводород¬
ной до объема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 270 нм, при 280 нм,
при 287 нм и при 291 нм.
Отношение оптических плотностей:
- ^(/Лио — от °'82 Д° °.86;
- — от 0,67 до 0,70;
- А291/А280 — от 0,65 до 0,69.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО нафазолина гидрохлорида #или
спектр, представленный на рисунке #0730.-1#.
C. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диокида, Р и до¬
водят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 20 мл раст¬
вора 3 прибавляют 0,2 мл 0,01 М раствора натрия
гидроксида и 0,1 мл раствора метилового красно¬
го Р. Раствор должен быть желтым. При прибавлении
не более 0,6 мл 0,01 М раствора кислоты хлористо¬
водородной должно появиться красное окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг 1-нафтилуксусной
кислоты Р растворяют в подвижной фазе, прибавля¬
ют 5 мл испытуемого раствора и доводят подвижной
фазой до объема 100 мл.
Раствор сравнения (Ь). 5,0 мг ФСО нафазолина
примеси А растворяют в подвижной фазе и доводят
до объема 100,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 10,0мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, эндкепированным для хроматографии Р (размер
частиц 4 мкм) с размером пор 6 нм;
Нафазолина нитрат
747
- подвижная фаза: 1,1 г натрия октансульфона-
та Р растворяют в смеси из 5 мл кислоты уксусной
ледяной Р, 300 мл ацетонитрила Р и 700 мл воды Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания нафазолина.
Время удерживания: нафазолин — около 14 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 5,0 между пиками нафа¬
золина и примеси В.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (с);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
B. Р = С02Н: (Нафтален-1-ил)уксусная кислота
(1-нафтилуксусная кислота).
C. Р = СЫ: (Нафтален-1-ил)ацетонитрил (1-нафти-
лацетонитрил).
О. 2-(Нафтален-2-илметил)-4,5-дигидро-1 Н-
имидазол (Р-нафазолин).
07/2016:0147
НАФАЗОЛИНА НИТРАТ
МарЬагоНт пИгаз
ЫАРНА20ШЕ ШТКАТЕ
С14н14ы2 ■ НЫОэ М.м. 273,3
[5144-52-5}
ОПРЕДЕЛЕНИЕ
2-(Нафтален-1 -илметил >4,5-дигидро-1 Н-имидазола
нитрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Умеренно растворим в воде, растворим в 96 %
спирте.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в сме¬
си из 5,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 50 мл 96 % спирта Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 24,67 мг С14Н14Ы2 НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси: В, С, О.
А. Р = СО-МН-[СН2]2-1ЧН2: Л/-(2-Аминоэтил)-2-(наф-
тален-1-ил)ацетамид (нафтилацетилэтилендиамин).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 167 °С до
170 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 250,0 мл этим
же растворителем. 25,0 мл полученного раствора до¬
водят 0,01 М раствором кислоты хлористоводород¬
ной до объема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 270 нм, при 280 нм,
при 287 нм и при 291 нм.
Отношение оптических плотностей:
- Агус/Агво “ от °>82 Д° °’8б>
- А^/Ааи — от 0,65 до 0,69.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО нафазолина нитрата #или
спектр,
748
Гэсударственная фармакопея Республики Беларусь
Рисунок #0147.-1. Инфракрасный спектр ФСО нафазолина нитрата
0.45 мг испытуемого образца растворяют в 2 мл
воды Р, прибавляют 1 мл кислоты серной Р, осто¬
рожно встряхивают и выдерживают до охлаждения.
Прибавляют каплями по стенке пробирки 1 мл раст¬
вора железа (II) сульфата Р2. На границе раздела
двух жидкостей появляется коричневое окрашивание.
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диокида, Р и до¬
водят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 5,0 до 6,5. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг 1-нафтилуксусной
кислоты Р растворяют в подвижной фазе, прибавля¬
ют 5 мл испытуемого раствора и доводят подвижной
фазой до объема 100 мл.
Раствор сравнения (Ь). 5,0 мг ФСО нафазолина
примеси А растворяют в подвижной фазе и доводят
до объема 100,0 мл этим же растворителем. 5,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 10,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, эндкепированным для хроматографии Р (размер
частиц 4 мкм) с размером пор 6 нм;
- подвижная фаза: 1,1 г натрия октансульфона-
та Р растворяют в смеси из 5 мл кислоты уксусной
ледяной Р, 300 мл ацетонитрила Р и 700 мл воды Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания нафазолина.
Относительное удерживание (по отношению
к нафазолину, время удерживания — около 14 мин):
примесь А— около 0,76; примесь О — около 1,24; при¬
месь В — около 1,27; примесь С — около 2,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 5,0 между пиками нафа¬
золина и примеси В.
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,1 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси А,
не должна превышать 0,5 площади основного пика на
хроматограмме раствора сравнения (с);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
Никотинамид
749
менее 0,25 площади основного пика на хроматограм¬
ме раствора сравнения (с) (0,05 %) и пик нитрат-иона.
Хлориды (2.4.4). Не более 0,0330 % (330 ррт).
Раствор 3 должен выдерживать испытания на хлориды.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 27,33 мг С14Н14М2-НМ03.
A. К = СО-МН-[СН2]2-МН2: Л/-(2-Аминоэтил)-2-(наф-
тален-1 -ил)ацетамид (нафтилацетилэтилендиамин).
B. Р = С02Н: (Нафтален-1-ил)уксусная кислота
(1-нафтилуксусная кислота).
C. Р = СЫ: (Нафтален-1-ил)ацетонитрил (1-нафти-
лацетонитрил).
й. 2-(Нафтален-2-илметил)-4,5-дигидро-1 Н-
имидазол (р-нафазолин).
ХРАНЕНИЕ
В защищенном от света месте. 07/2016:0047
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
НИКОТИНАМИД
№соИпатШит
ШСОТШАМЮЕ
СДЛ-О М.м. 122,1
[98-92-0]
Рисунок #0047.-1. Инфракрасный спектр ФСО никотинамида
750
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Пиридин-З-карбоксамид. #Витамин В3, Витамин РР#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде и в этаноле безводном,
мало растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
A. Температура плавления (2.2.14): от 128 °С до
131 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО никотинамида #или спектр,
представленный на рисунке #0047.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образ¬
ца растворяют в смеси из равных объемов 96 % спир¬
та Р и воды Р и доводят до 5,0 мл этой же смесью
растворителей.
Раствор сравнения. 5 мг ФСО никотинамида
растворяют в смеси из равных объемов 96 % спир¬
та Р и воды Р и доводят до 5,0 мл этой же смесью
растворителей.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: вода Р — 96% спирт Р — ме-
тиленхлорид Р (4:45:48, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию и размеру основному пятну на хроматограмме
раствора сравнения.
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
рН (2.2.3). От 6,0 до 7,5. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,150 г испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 150,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг изоникотинамида Р
(примесь й) растворяют в испытуемом растворе и до¬
водят до объема 100,0 мл испытуемым раствором.
1.0 мл полученного раствора доводят испытуемым
раствором до объема 25,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным для хроматографии, деактивированным
по отношению к основаниям, эндкепированным Р с
размером частиц 3 мкм;
- подвижная фаза:
-подвижная фаза А: 5,0мл кислоты уксус¬
ной разведенной Р смешивают с 900 мл воды
для хроматографии Р, прибавляют 30 мл раст¬
вора аммиака разведенного РЗ, 15 мл ацетони¬
трила Р и доводят до 1 л водой для хромато¬
графии Р;
- подвижная фаза В: ацетонитрил Р1 — под¬
вижная фаза А (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
98
2
20—25
о
т
00
о>
2 — 100
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 264 нм;
- объем вводимой пробы: 20 мкл;
Идентификация пиков примесей: идентифици¬
руют пик примеси О, используя хроматограмму раст¬
вора сравнения (Ь).
Относительное удерживание (по отношению к
никотинамиду, время удерживания — около 7 мин):
примесь О — около 0,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси О и никотинамида.
Расчет процентных содержаний:
- для каждой примеси — используют концентра¬
цию никотинамида в растворе сравнения (а).
Предельное содержание примесей:
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,2 %;
- учитываемый предел: 0,05 %.
Тяжелые металлы (2.4.8, метод А). Не более
0,0030 % (30 ррт). 12 мл раствора 3 доводят водой Р
до объема 18 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме в течение 18 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 12,21 мг С6Н6Н20.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
Никотиновая кислота
751
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е.
ю2н
А. Пиридин-З-карбоновая кислота (никотиновая
кислота).
.си
В. Пиридин-З-карбонитрил.
о
ИНг
С. Пиридин-2-карбоксамид (пиколинамид).
Е. Пиридин-З-карбоксамид 1-оксид (никотинами-
да Л/-оксид).
07/2016:0459
НИКОТИНОВАЯ КИСЛОТА
АсШит тсоИтсит
Ы1СОТ1Ы1С АСЮ
N
С02Н
СвН.М02 Мм. 123,1
[59-67-6]
ОПРЕДЕЛЕНИЕ
Пиридин-З-карбноновая кислота. #Витамин В3,
Витамин РР#.
Содержание: не менее 99,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворима в кипящей воде и в кипящем 96 %
спирте, умеренно растворима в воде. Растворяется
в разведенных растворах гидроксидов и карбонатов
щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Температура плавления (2.2.14): от 234 °С до
240 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО никотиновой кислоты #или
спектр, представленный на рисунке #0459.-1 #.
Рисунок #0459.-1. Инфракрасный спектр ФСО никотиновой кислоты
752
Гэсударственная фармакопея Республики Беларусь
С. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Смесь растворителей. 6,8 г калия дигидрофос¬
фата Р растворяют в 900 мл воды Р, доводят до рН
7.0 раствором натрия гидроксида разведенным Р и
разводят водой Р до объема 1000 мл.
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
1.0 мл полученного раствора доводят смесью раство¬
рителей до объема 25,0 мл.
Диапазон длин волн: от 237 нм до 262 нм.
Максимумы поглощения: при 262 нм.
Минимум поглощения: при 237 нм.
Отношение оптических плотностей:
А2Ъ7/А262 — от 0,46 до 0,50.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,120 г испытуемого об¬
разца растворяют в 200 мкл раствора аммиака раз¬
веденного Р1 и доводят подвижной фазой А до объ¬
ема 10,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО никотиновой кислоты смеси примесей (приме¬
си А и В) растворяют в 1,0 мл подвижной фазы А.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем алкил-свя-
занным для использования с подвижными фазами с
высоким содержанием воды, эндкепированным, для
хроматографии Р с размером частиц 4 мкм;
- температура: 15 °С;
- подвижная фаза:
- подвижная фаза А: 2 мл кислоты уксусной Р
разодят в 950 мл воды Р, доводят до рН 5,6 рас¬
твором аммиака разведенным Р1 и разводят
водой Р до объема 1000 мл;
- подвижная фаза В: ацетонитрил Р — мета¬
нол Р (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100
0
10—30
о
см
т
о
о
0
1
00
о
30—35
20
80
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 250 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО никотиновой кислоты смеси примесей.
Относительное удерживание (по отношению
к никотиновой кислоте, время удерживания — около
6 мин): примесь А — около 2,7; примесь В — около
2,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
месей А и В.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать 0,5 площади основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 0,05 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в воде Р при
нагревании на водяной бане и доводят до объема
15 мл этим же растворителем. Полученный раствор
должен выдерживать испытание на хлориды.
Тяжелые металлы (2.4.8, метод С). Не более
O, 0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 1 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют 50 мл
воды Р и титруют 0,1 М раствором натрия гидрокси¬
да до появления розового окрашивания, используя в
качестве индикатора 0,25 мл раствора фенолфтале¬
ина Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 12,31 мгС6Н51Ч02.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет необ¬
ходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е,
P, в, Н, I.
А. 6-Метилпиридин-З-карбоновая кислота (6-ме-
тилникотиновая кислота).
Пропускание
Нимесулид
753
Н02С'
со2н
В. 2,2'-Бипиридин-5,5'-дикарбоновая кислота
(6,6-диникотиновая кислота).
N СН3
Н3С'
С. 5-Этил-2-метилпиридин.
н3с
й. Пиридин-2,5-дикабоновая кислота.
со2н
Е. Пиридидн-4-карбоновая кислота (изоникотино-
вая кислота).
,со2н
N02
Р. 5-Нитропиридин-З-карбоновая кислота (5-нитро-
никотиновая кислота).
О. Пиридин.
.N02
Н. 3-Нитропиридин.
.N02
N02
I. 3,5-Динитропиридин.
07/2016:1548
НИМЕСУЛИД
Ы'1тези1Шит
Ы1МЕЗШ.ЮЕ
С.М.М.О.5 М.м. 308,3
[51803-78-2]
ОПРЕДЕЛЕНИЕ
А/-(4-Нитро-2-феноксифенил)метансульфонамид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
Рисунок #1548.-1. Инфракрасный спектр ФСО нимесулида
754
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ(СВОЙСТВА)
Желтоватый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, мало растворим в этаноле.
Температура плавления: около 149 °С.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО нимесулида #или спектр,
представленный на рисунке #1548.-1#.
Если полученные спектры отличаются, то ис¬
пытуемый образец и ФСО нимесулида растворяют
по отдельности в ацетоне Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Оптическая плотность (2.2.25). Не более 0,50
при 450 нм. 1,0 г испытуемого образца растворяют
в ацетоне Р и доводят до объема 10,0 мл этим же
растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 8 мл ацетонитрила Р и дово¬
дят водой Р до объема 20,0 мл.
Раствор сравнения (а). 5 мг 2-феноксианили-
на Р (примесь С) растворяют в 10 мл ацетонитри¬
ла Р и доводят водой Р до объема 25,0 мл. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 50,0 мл. 1,0 мл полученного раствора сме¬
шивают с содержимым контейнера с ФСО нимесу¬
лида примеси О, предварительно растворенного в
1.0 мл ацетонитрила Р.
Раствор сравнения (Ь). 1,0 мл испытуемо¬
го раствора доводят подвижной фазой до объема
10.0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 100,0 мл.
Раствор сравнения (с). 4 мг ФСО нимесулида
для идентификации пиков (содержит примеси А, В,
Е и Р) растворяют в 4,0 мл ацетонитрила Р и дово¬
дят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диа¬
метром 4,0 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — раствор
1,15 г/л аммония дигидрофосфата Р, доведенный
раствором аммиака Р до рН 7,0, (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 230 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 7-кратное вре¬
мя удерживания нимесулида.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, Е и Р, используя хрома¬
тограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО нимесулида для идентифика¬
ции пиков; идентифицируют пики примесей С и О,
используя хроматограмму раствора сравнения (а).
Относительное удерживание (по отношению
к нимесулиду, время удерживания — около 5 мин):
примесь А — около 0,3; примесь В — около 2,4; при¬
месь С — около 3,2; примесь й — около 3,7; при¬
месь Е — около 4,2; примесь Р — около 6,1.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
месей С и О.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р (не более 0,15 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, О, Е и Р, не
должны превышать 1,5-кратную площадь основного
пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е и Р, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей (не более 0,5 %); на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца сушат
при температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12,2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,240 г испытуемого образца растворяют в
30 мл предварительно нейтрализованного ацето¬
на Р, прибавляют 20 мл воды Р и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 30,83 мг С13Н121Ч2055.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): С.
Нистатин
755
К4 К1
A. К1 = 502-СН3, К2 = Н, КЗ = К4 = Ы02: Л/-(2,4-Ди-
нитро-6-феноксифенил)метансульфонамид.
B. К1 = 502-СН3> К2 = КЗ = К4 = Н: АК2-Фенокси-
фенил)метансульфонамид.
C. К1 = Н2 = КЗ = Я4 = Н: 2-Феноксианилин.
й. Я1 = К2 = Я4 = Н, КЗ = 1Ч02:4-Нитро-З-фенокси-
анилин.
Е. К1 = Р2 = 302-СН3, КЗ = К4 = Н: /ЧЛ/-Бис(метил-
сульфонил)-2-феноксианилик.
Р. К1 = К2 = 302-СН3> КЗ = Ы02, К4 = Н: А/,Л/-Бис(ме-
тилсульфонил)-4-нитро-2-феноксианилин.
О. 4-Нитро-2-феноксифенол.
07/2016:0517
НИСТАТИН
МузШ'тит
ЫУЗТАШ
С47Н75М017 М.м. 926
ОПРЕДЕЛЕНИЕ
Противогрибковая субстанция, полученная путем
ферментации с использованием определенных штам¬
мов 8&ер№тусе$ поигзе/ в качестве продуцирующих
микроорганизмов. Содержит в основном тетраены, глав¬
ной составляющей которых является (15,ЗК,4К,7К,
9Я, 11 К, 155,16Я, 17Я,185,19Е,21 Е,25Е,27Е,29Е,
31Е,ЗЗК,355,36К,375)-33-[(3-амино-3,6-дидезокси-Р-
0-маннопиранозил)окси]-1,3,4,7,9,11,17,37-октагидрокси-
15,1 б, 18-триметил-13-оксо-14,39-диоксабицикло[33.3.1 ]-
нонатриаконта-19,21,25,27,29,31 -гексаен-36-карбоновая
кислота (нистатин А1).
Содержание: не менее 4400 МЕ/мг (в пересчете
на сухое вещество); если предназначен для орально¬
го применения — не менее 5000 МЕ/мг (в пересчете
на сухое вещество).
ПРОИЗВОДСТВО
Если нистатин не предназначен для наружного
применения, метод производства должен быть вали-
дирован, чтобы при испытании субстанция отвечала
следующему требованию.
Аномальная токсичность (2.6.9). Испытуемый
образец должен быть нетоксичным. Тест-доза не ме¬
нее 600 МЕ в 0,5 мл раствора 5 г/л гуммиарабика Р на
мышь, внутрибрюшинно.
ОПИСАНИЕ (СВОЙСТВА)
Желтый или слегка коричневатый порошок. Ги¬
гроскопичен.
Практически нерастворим в воде, легко раство¬
рим в диметилформамиде и в диметилсульфоксиде,
мало растворим в метаноле, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в уль¬
трафиолетовой и видимой областях (2.2.25). Спектр
поглощения раствора, приготовленного как указа¬
но в испытании «Оптическая плотность», в области
длин волн от 220 нм до 350 нм имеет максимумы при
230 нм, 291 нм, 305 нм и 319 нм и плечо при 280 нм.
Отношение оптической плотности в максимуме при
291 нм к оптической плотности в максимуме при
305 нм: от 0,61 до 0,73. Отношение оптической плот¬
ности в максимуме при 319 нм к оптической плотности
в максимуме при 305 нм: от 0,83 до 0,96. Отношение
оптической плотности в максимуме при 230 нм к опти¬
ческой плотности в плече при 280 нм: от 0,83 до 1,25.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО нистатина #или спектр, пред¬
ставленный на рисунке #0517.-1#.
C. К 2 мг испытуемого образца прибавляют
0,1 мл кислоты хлористоводородной Р. Появляется
коричневое окрашивание.
О. К 2 мг испытуемого образца прибавляют 0,1 мл
кислоты серной Р. Появляется коричневое окраши¬
вание, которое постепенно переходит в фиолетовое.
Е. Просматривают хроматограммы, полученные
в испытании «Состав».
Результаты: время удерживания основного
пика на хроматограмме испытуемого раствора соот¬
ветствует времени удерживания основного пика на
хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Оптическая плотность (2.2.25). Не более 0,60
при 305 нм. 0,10 г испытуемого образца растворяют
в смеси из 5,0 мл кислоты уксусной ледяной Р и
50 мл метанола Р и доводят метанолом Р до объ¬
ема 100,0 мл. 1,0 мл полученного раствора доводят
метанолом Р до объема 100,0 мл. Измеряют опти¬
ческую плотность в максимуме при 305 нм в течение
30 мин после приготовления.
Состав. Жидкостная хроматография (2.2.29):
определение проводят методом внутренней нормали¬
зации. Испытание проводят с защитой от света.
756
Гэсударственная фармакопея Республики Беларусь
Рисунок #0517.-1. Инфракрасный спектр ФСО нистатина
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в диметилсульфоксиде Р и до¬
водят до объема 50 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО нистати¬
на растворяют в диметилсульфоксиде Р и доводят
до объема 50 мл этим же растворителем.
Раствор сравнения (Ь). 20 мг испытуемого об¬
разца растворяют в 25 мл метанола Р и доводят
водой Р до объема 50 мл. К 10,0 мл полученного
раствора прибавляют 2,0 мл кислоты хлористо¬
водородной разведенной Р и выдерживают при
комнатной температуре в течение 1 ч.
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (а) доводят диметилсульфоксидом Р до
объема 100,0 мл. 1,0 мл полученного раствора до¬
водят диметилсульфоксидом Р до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем окта-
децилсилильным, деактивированным по отноше¬
нию к основаниям, эндкепированным для хрома¬
тографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р —
раствор 3,85 г/л аммония ацетата Р (29:71,
об/об);
- подвижная фаза В: раствор 3,85 г/л аммония
ацетата Р — ацетонитрил Р (40:60, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—25
100
0
25—35
100 — 0
0—100
35-45
0
100
45—50
о
о
т—
т
о
о
г
о
о
50—55
100
0
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 305 нм;
- объем вводимой пробы: 20 мкл.
Время удерживания пиков: нистатин А1 — около
14 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,5 между двумя ос¬
новными пиками (время удерживания около 13 мин
и 19 мин).
Состав:
- нистатин А1: не менее 85,0 %;
- любое другое соединение: не более 4,0 %.
На хроматограмме испытуемого раствора не учи¬
тывают пики со временем удерживания менее 2 мин и
пики с площадью менее площади основного пика на
хроматограмме раствора сравнения (с).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 60 °С над фосфора (V) оксидом Р при
давлении, не превышающем 0,1 кПа, в течение 3 ч.
Сульфатная зола (2.4.14). Не более 3,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Количественное определение антибиотиков ми¬
кробиологическим методом (2.7.2). Испытание про¬
водят с защитой от света.
Нитрофурал
757
Испытуемый образец и ФСО нистатина раство¬
ряют по отдельности в диметилформамиде Р и раз¬
водят смесью из диметилформамида Р и буферного
раствора рН 6,0 (5:95, об/об).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
МАРКИРОВКА
При необходимости указывают:
- только для наружного применения.
07/2016:1135
НИТРОФУРАЛ
МПгоТига1ит
МТКОРиКА!.
02М. уО,
н
N
гмн2
С6Н6Ы404 М.м. 198,1
[59-87-0]
ОПРЕДЕЛЕНИЕ
2-[(5-Нитрофуран-2-ил)метилен]диазанкарбокса-
мид. #Фурацилин#.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый или коричневато-желтый кристалличе¬
ский порошок.
Очень мало растворим в воде, мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25). Испытание
проводят с защитой от яркого света.
Испытуемый раствор. Используют испытуемый
раствор, приготовленный для количественного опре¬
деления.
Диапазон длин волн: от 220 нм до 400 нм.
Максимумы поглощения: при 260 нм и при 375 нм.
Отношение оптических плотностей:
Л^Л-во — ОТ 1,15 до 1,30.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО нитрофурала #или спектр,
представленный на рисунке #1135.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО нитрофурала
растворяют в метаноле Р и доводят до объема 10 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р
Подвижная фаза: метанол Р — нитрометан Р
(10:90, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
фенилгидразина гидрохлорида Р.
Рисунок #1135.-1. Инфракрасный спектр нитрофурала
758
Гэсударственная фармакопея Республики Беларусь
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
О. 1 мг испытуемого образца растворяют в 1 мл
диметилформамида Р и прибавляют 0,1мл раст¬
вора калия гидроксида спиртового Р. Появляется
фиолетово-красное окрашивание.
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 7,0. К 1,0 г испытуемого об¬
разца прибавляют 100 мл воды, свободной от угле¬
рода диоксида, Р, встряхивают #(в течение не менее
15 мин)# и фильтруют.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО нитрофу-
рала примеси В растворяют в подвижной фазе и дово¬
дят до объема 20,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (Ь). 10 мг испытуемого об¬
разца и 10 мг нитрофурантоина Р растворяют в под¬
вижной фазе и доводят до объема 100,0 мл этим же
растворителем. 5,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО нитрофурала для идентификации примесей
(содержит примеси А и В) растворяют с использовани¬
ем ультразвука в 1,0 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
-подвижная фаза: ацетонитрил Р — вода Р
(40:60, об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 310 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 10-кратное вре¬
мя удерживания нитрофурала.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (с).
Относительное удерживание (по отношению
к нитрофуралу, время удерживания — около 4 мин):
нитрофурантоин — около 1,2; примесь В — около 4,0;
примесь А — около 7,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками нитро¬
фурала и нитрофурантоина.
Предельное содержание примесей:
- примесь А, В (не более 0,5 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А и В, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и В, не должна превышать 0,2 площади основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от яркого света.
60,0 мг испытуемого образца растворяют в
20 мл диметилфорамида Р и доводят водой Р до
объема 500,0 мл. 5,0 мл полученного раствора дово¬
дят водой Р до объема 100,0 мл. Аналогично готовят
раствор сравнения с использованием 60,0 мг ФСО
нитрофурала. Измеряют оптическую плотность
(2.2.25) полученных растворов в максимуме при дли¬
не волны 375 нм.
Содержание С6НбМ404 рассчитывают, исходя из
значений оптических плотностей и концентраций рас¬
творов.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. Бис[(5-нитрофуран-2-ил)метилен]диазан.
В. (5-Нитрофуран-2-ил)метилендиацетат.
07/2016:0101
НИТРОФУРАНТОИН
ШгоТигаМотит
ытюрикАЫТош
СДД.О, М.м. 238,2
[67-20-9]
Нифедипин
759
ОПРЕДЕЛЕНИЕ
1-[[(5-Нитрофуран-2-ил)метилен]амино]имидозо-
лидин-2,4-дион. #Фурадонин#.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый кристаллический порошок или желтые
кристаллы.
Очень мало растворим в воде и в 96 % спирте,
растворим в диметилформамиде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытание проводят с защитой от яркого
света. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. Используют раствор,
приготовленный как указано в разделе «Количествен¬
ное определение».
Диапазон длин волн: от 220 нм до 400 нм.
Максимумы поглощения: при 266 нм и при 367 нм.
Отношение оптических плотностей:
^Зб/Д-бв — ОТ 1 -36 Д° 1 >42-
B. 10 мг испытуемого образца растворяют в
10 мл диметилформамида Р. К 1 мл раствора при¬
бавляют 0,1 мл 0,5 М спиртового раствора калия ги¬
дроксида. Появляется коричневое окрашивание.
ИСПЫТАНИЯ
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 0,25 г испытуемого об¬
разца растворяют в минимальном количестве диме¬
тилформамида Р и доводят ацетоном Р до объема
10 мл.
Раствор сравнения. 1 мл испытуемого раствора
доводят ацетоном Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
НРШР.
Подвижная фаза: метанол Р — нитрометан Р
(10:90, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе, затем при температу¬
ре от 100 °С до 105 °С в течение 5 мин.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм, опрыски¬
вают раствором фенилгидразина гидрохлорида Р и
нагревают при температуре от 100 °С до 105 °С в те¬
чение 10 мин.
Предельное содержание примесей:
-любая примесь (не более 1,0 %): при просма¬
тривании в ультрафиолетовом свете и после опры¬
скивания на хроматограмме испытуемого раствора
любое пятно, кроме основного, должно быть не ин¬
тенсивнее пятна на хроматограмме раствора срав¬
нения.
Потеря в массе при высушивании (2.2.32). Не
более 1,0%. 1,00 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %. Опре¬
деление проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от яркого све¬
та. 0,120 г испытуемого образца растворяют в 50 мл
диметилформамида Р и доводят водой Р до объ¬
ема 1000,0 мл. 5,0 мл полученного раствора доводят
раствором, содержащим 18 г/л натрия ацетата Р и
0,14 % (об/об) кислоты уксусной ледяной Р, до объе¬
ма 100,0 мл. Измеряют оптическую плотность (2.2.25)
полученного раствора в максимуме при длине волны
367 нм, используя раствор натрия ацетата, описанный
выше, в качестве компенсационного раствора.
Содержание С8Н6Ы405 рассчитывают с учетом
удельного показателя поглощения, равного 765.
ХРАНЕНИЕ
В защищенном от света месте при температуре
не выше 25 °С.
07/2016:0627
НИФЕДИПИН
ШесИр'тит
ШРЕ01РШЕ
С17н18ы206 М.м. 346,3
[21829-25-4]
ОПРЕДЕЛЕНИЕ
Диметил-2,6-диметил-4-(2-нитрофенил)-1,4-ди-
гидропиридин-3,5-дикарбоксилат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, умеренно растворим в этаноле.
При воздействии дневного и искусственного све¬
та определенных длин волн легко превращается в ни-
трозофенилпиридиновое производное. Воздействие
ультрафиолетового излучения приводит к образова¬
нию нитрофенилпиридинового производного.
Растворы защищают от света и готовят не¬
посредственно перед использованием в темном ме¬
сте или при длинноволновом свете (более 420 нм).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 171 °С до
175 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
760
Гэсударственная фармакопея Республики Беларусь
Сравнение: ФСО нифедипина #или спектр, пред¬
ставленный на рисунке #0627.-1#.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО нифедипина
растворяют в метаноле Р и доводят до объема 10 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ ^254
Подвижная фаза: этилацетат Р — циклогек¬
сан Р (40:60, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, интенсивности поглоще¬
ния при 254 нм и размеру основному пятну на хрома¬
тограмме раствора сравнения.
О. 25 мг испытуемого образца помещают в про¬
бирку и растворяют при слабом нагревании в 10 мл
смеси кислота хлористоводородная Р — вода Р —
96% спирт Р (1,5:3,5:5, об/об/об). Прибавляют 0,5 г
цинка Р в гранулах, выдерживают в течение 5 мин
при периодическом перемешивании и фильтруют во
вторую пробирку. К фильтрату прибавляют 5 мл раст¬
вора 10 г/л натрия нитрита Р и выдерживают в тече¬
ние 2 мин. Прибавляют 2 мл раствора 50 г/л аммония
сульфамата Р, интенсивно и осторожно встряхивают
и прибавляют 2 мл раствора 5 г/л нафтилэтиленди-
амина дигидрохлорида Р. Появляется интенсивное
красное окрашивание, не исчезающее в течение 5 мин.
ИСПЫТАНИЯ
Примесь О и другие примеси основного харак¬
тера. Не более 0,14 %. 4 г испытуемого образца по¬
мещают в коническую колбу вместимостью 250 мл и
растворяют в 160 мл кислоты уксусной ледяной Р, ис¬
пользуя ультразвуковую баню. Титруют 0,1 М раство¬
ром кислоты хлорной, используя в качестве индикато¬
ра 0,25 мл раствора нафтолбензеина Р, до перехода
окраски раствора от коричневато-желтого до зеленого.
На титрование должно быть израсходовано не более
0,48 мл 0,1 М раствора кислоты хлорной.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в 20 мл метанола Р и доводят под¬
вижной фазой до объема 50,0 мл.
Раствор сравнения (а). 10 мг ФСО нифедипина
примеси А растворяют в метаноле Р и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО нифедипина
примеси В растворяют в метаноле Р и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (с). К 1,0 мл раствора срав¬
нения (а) прибавляют 1,0 мл раствора сравнения (Ь),
0,1 мл испытуемого раствора и доводят подвижной
фазой до объема 20,0 мл. 2,0 мл полученного раст¬
вора доводят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — метанол
Р — вода Р (9:36:55, об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (с);
Рисунок #0627.-1. Инфракрасный спектр ФСО нифедипина
Нифуроксазид
761
- время хроматографирования: 2-кратное вре¬
мя удерживания нифедипина.
Порядок выхода пиков: примесь А, примесь В,
нифедипин.
Время удерживания: нифедипин—около 15,5 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 1,5 между пиками при¬
меси А и примеси В; не менее 1,5 между пиками при¬
меси В и нифедипина.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
- примесь В (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
-любая другая примесь (не более 0,1 %): на
хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного и пиков примесей А и В,
не должна превышать площадь пика нифедипина на
хроматограмме раствора сравнения (с);
- сумма примесей: не более 0,3 %;
- неучитываемый предел (0,01 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,1300 г испытуемого образца растворяют в сме¬
си из 25 мл 2-метил-2-пропанола Р и 25 мл раствора
кислоты хлорной Р и титруют 0,1 М раствором церия
сульфата, используя в качестве индикатора 0,1 мл
ферроина Р, до исчезновения розового окрашивания
(титруют медленно при приближении к конечной точке
титрования).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора церия сульфата соответ¬
ствует 17,32 мг С17Н181\12Об.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. Р = 1\Ю2: Диметил-2,6-диметил-4-(2-нитрофенил)-
пиридин-3,5-дикарбоксилат (аналог нитрофенилпири-
дина).
В. Р = N0: Диметил-2,6-диметил-4-(2-нитрозо-фе-
нил)пиридин-3,5-дикарбоксилат (аналог нитрозофе-
нилпиридина).
N02
С. Метил-2-(2-нитробензилиден)-3-оксобутаноат.
о
й. Метил-З-аминобут-2-еноат.
07/2016:1999
НИФУРОКСАЗИД
М1Тигоха21'с1ит
МРШЮХА2ЮЕ
02М
с12н9м3о5
[965-52-6]
ОПРЕДЕЛЕНИЕ
(Е)-4-Гидрокси-А/'-[(5-нитрофуран-2-ил)метили-
ден]бензогидразид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Ярко-желтый кристаллический порошок.
Практически нерастворим в воде, мало раство¬
рим в 96 % спирте, практически нерастворим в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО нифуроксазида #или спектр,
представленный на рисунке #1999.-1#.
ИСПЫТАНИЯ
Удельный показатель поглощения (2.2.25). Не
менее 940 и не более 1000 в максимуме поглощения
при длине волны 367 нм. Испытание проводят в усло¬
виях защиты от света.
10,0 мг испытуемого образца растворяют 10 мл
этиленгликоля монометилового эфира Р и доводят
метанолом Р до объема 100,0 мл. 5,0 мл полученного
раствора доводят метанолом Р до объема 100,0 мл.
Примесь А. Не более 0,05 %.
Испытуемый раствор (а). 1,0 г испытуемого об¬
разца растворяют в диметилсульфоксиде Р и дово¬
дят до объема 10,0 мл этим же растворителем.
96. Зак. 1060.
762
Гэсударственная фармакопея Республики Беларусь
Рисунок #1999.-1. Инфракрасный спектр ФСО нифуроксазида
Испытуемый раствор (Ь). 50,0 мл воды Р
прибавляют к 5,5 мл испытуемого раствора (а) при
перемешивании, выдерживают в течение 15 мин и
фильтруют.
Раствор сравнения. 5,0 мл раствора 50 мг/л
4-гидроксибензогидразида Р (примесь А) в диме-
тилсульфоксиде Р прибавляют к 0,5 мл испыту¬
емого раствора (а). К полученному раствору при¬
бавляют 50,0 мл воды Р при перемешивании, вы¬
держивают в течение 15 мин и фильтруют.
0,5 мл фосфорномолибденово-вольфрамово-
го реактива Р и 10,0 мл раствора натрия карбо¬
ната Р прибавляют по отдельности к 10,0 мл ис¬
пытуемого раствора (Ь) и 10,0 мл раствора срав¬
нения, выдерживают в течение 1 ч. Оптическая
плотность (2.2.25) при длине волны 750 нм для
испытуемого раствора (Ь) не должна превышать
оптическую плотность раствора сравнения.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Если не указывается иное,
используют мерные колбы из темного стекла.
Смесь растворителей. Ацетонитрил Р —
вода Р (40:60, об/б).
Испытуемый раствор. 10,0 мг испытуемого
образца растворяют в смеси растворителей, ис¬
пользуя обработку ультразвуком в течение не бо¬
лее 5 мин, и доводят до объема 100,0 мл этой же
смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объ¬
ема 100,0 мл. 1,0 мл полученного раствора дово¬
дят смесью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Для приготовления
примеси Е /л зНи 5 мг испытуемого образца раство¬
ряют в смеси растворителей в бесцветной мерной
колбе, используя обработку ультразвуком в тече¬
ние 5 мин, доводят смесью растворителей до объ¬
ема 50 мл, и выдерживают при естественном свете
в течение 1 ч.
Раствор сравнения (с). 5,0 мг ФСО метил-
парагидроксибензоата (примесь В) растворяют
в смеси растворителей и доводят до объема
100,0 мл этой же смесью растворителей. 1,0 мл
полученного раствора доводят смесью раствори¬
телей до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикаге¬
лем октадецилсилильным для хроматографии Р
с размером частиц 5 мкм;
- температура: 10 °С;
- подвижная фаза:
- подвижная фаза А: тетрагидрофуран Р —
вода Р (5:95, об/об);
- подвижная фаза В: ацетонитрил Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
67
33
10—30
67 — 43
ю
т
со
со
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 280 нм;
- объем вводимой пробы: 50 мкл.
Относительное удерживание (по отношению к
нифуроксазиду, время удерживания — около 8 мин):
примесь А (кетоенольные таутомеры) — около 0,36
и 0,39; примесь Е — около 0,9; примесь В — около
1,2; примесь С — около 2,6; примесь О — около 3,4.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси Е и нифуроксазида.
Носкапина гидрохлорид
763
Предельное содержание примесей:
- примесь Е (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Е, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примеси В, С и О: на хроматограмме испытуе¬
мого раствора площади пиков, соответствующих при¬
месям В, С и О, не должны превышать 0,6 площади
основного пика на хроматограмме раствора сравне¬
ния (с) (не более 0,3 %) и не более одного из таких
пиков мажет иметь площадь более 0,2 площади ос¬
новного пика на хроматограмме раствора сравнения
(с) (0,1 %);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В, С, О и Е, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей, кроме примеси Е (не более
0,5 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пика
примеси Е, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,1 площади основного пика на
хроматограмме раствора сравнения (с); не учитывают
пики примеси А.
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
тнкробнапот^скал чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют, на¬
гревая при необходимости, в 30 мл диметилформа-
мида Р, прибавляют 20 мл воды Р и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 27,52 мг С12Н91Ч305.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
^ /ОН
к.
о
A. Я = МН-[МН2:4-Гидроксибензогидразид (л-гидро-
ксибензогидразид).
B. Я = ОСН3: Метил-4-гидроксибензоат (метилпа-
рагидроксибензоат).
о
С. (5-Нитрофуран-2-ил)метилидендиацетат.
О. (Е,Е)-Л/,Л/-Бис[(5-нитрофуран-2-ил)метилиден]-
гидразин (5-нитрофурфуральазин).
Е. (2)-4-Гидрокси-А/-[(5-нитрофуран-2-ил)метили-
ден]бензогидразид.
07/2016:0515
НОСКАПИНА ГИДРОХЛОРИД
Д/о5сар/'п/ ЬудгосЫопдит
ЫОЗСАРШЕ НУОКОСШ-ОРЮЕ
!з
О
•НС1Н20
• НС1 • н20 М.м. 467,9
ОПРЕДЕЛЕНИЕ
(35)-6,7-Диметокси-3-[(5Р)-4-метокси-6-метил-
5,6,7,8-тетрагидро-1,3-диоксоло[4,5-д]изохинолин-5-ил]-
изобензофуран-1(ЗН)-она гидрохлорид моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Гигроксопичен.
Легко растворим в воде и в 96 % спирте. Водные
растворы имеют слегка кислую реакцию; при стоянии
основание может выпадать в осадок.
Температура плавления: около 200 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С, Е.
Вторая идентификация: А, В, О, Е.
А. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
96*. Зак. 1060.
764
Гэсударственная фармакопея Республики Беларусь
B. Температура плавления (2.2.14) осадка, полу¬
ченного в идентификации Е: от 174 °С до 177 °С.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: используют осадок, полученный
в идентификации Е.
Сравнение: ФСО носкапина.
й. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 100 мл этим же растворителем.
Раствор сравнения. 22 мг ФСО носкапина раст¬
воряют в ацетоне Р и доводят до объема 100 мл этим
же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — 96% спирт Р — ацетон Р — толу¬
ол Р (1:3:20:20, об/об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
калия йодовисмутата разведенным Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
Е. 40 мг испытуемого образца растворяют в сме¬
си, состоящей из 2 мл воды Р и 3 мл 96 % спирта Р,
и прибавляют 1 мл раствора аммиака разведенно¬
го Р2. Нагревают до полного растворения. Охлажда¬
ют, потирая стенки пробирки стеклянной палочкой.
Фильтруют. Полученный фильтрат дает реакцию (а)
на хлориды (2.3.7). Промывают осадок водой Р, сушат
при температуре от 100 °С до 105 °С и используют
для идентификации В и С.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде Р, прибавляют 0,3 мл 0,1 М раствора
кислоты хлористоводородной и доводят до объема
25 мл водой Р.
Прозрачность (2.2.7). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
5 должна быть не интенсивнее эталона У(Ж)6 или
ВУ(КЖ)6.
рН (2.2.3). Не менее 3,0.0,2 г испытуемого образ¬
ца растворяют в 10 мл воды, свободной от углерода
диоксида, Р.
Удельное оптическое вращение (2.2.7). От
+38,5 до +44,0 (в пересчете на сухое вещество).
0,500 г испытуемого образца растворяют в 0,01 М
растворе кислоты хлористоводородной и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 8 мл метанола Р при помощи
ультразвука и доводят до объема 10,0 мл этим же
растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят метанолом Р до объема 20,0 мл. 1,0 мл
полученного раствора доводят до объема 10,0 мл ме¬
танолом Р.
Раствор сравнения (Ь). 5 мг папаверина гидрох¬
лорида Р растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем. 1,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 10,0 мл.
Раствор сравнения (с). 1,5 г папаверина гидро¬
хлорида Р растворяют в 10 мл испытуемого раствора
и доводят подвижной фазой до объема 25 мл.
Условия хроматографирования:
-колонка длиной 0,125м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем нитрильным
для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: метанол Р — фосфатный
буферный раствор с рН 6,0 Р1 (350:650, об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 240 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания носкапина.
Относительное удерживание (по отношению к
носкапину, время удерживания — около 10 мин): при¬
месь А — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 2 между пиками носка¬
пина и примеси А.
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- любая другая примесь (не более 0,2 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пика примеси А, не должна
превышать 0,4 площади основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей, кроме примеси А (не более
0,5 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пика
примеси А, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
менее 2,5 % и не более 6,5 %. 0,200 г испытуемого об¬
разца сушат при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в сме¬
си из 5,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 50 мл 96% спирта Р. Титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
0,1 мл 0,7 М раствора натрия гидроксида соот¬
ветствует 44,99 мг С22Н23Н07 НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Оксалиплатин
765
А. 1 -(3,4-Диметоксибензил)-6,7-диметоксиизо-
хинопин (папаверин).
07/2016:2017
ОКСАЛИПЛАТИН
ОхаНр1аНпит
охАириьтт
V-0'
\ /
Р1
/ \
С#Н14Ы204Р1 М.м. 397,3
[61825-94-3]
ОПРЕДЕЛЕНИЕ
(ЗР-4-2Н(1 Я2/?)-Циклогексан-1,2-диаминчсЛ/,кЛГ)-
[этандиоато(2-)-кОт,к02]платина.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, очень мало растворим в
метаноле, практически нерастворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО оксалиплатина #или спектр,
представленный на рисунке #2017.-1#.
B. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
ИСПЫТАНИЯ
Раствор 5. 0,10 г испытуемого образца раство¬
ряют в воде Р и доводят этим же растворителем до
объема 50 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность. 0,10 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р,
доводят до объема 50 мл этим же растворителем и
прибавляют 0,5 мл раствора фенолфталеина Р1.
Раствор должен быть бесцветным. При прибавлении
не более 0,60 мл 0,01 М раствора натрия гидрокси¬
да должно появиться розовое окрашивание.
Удельное оптическое вращение (2.2.7). От
+74,5 до +78,0 (в пересчете на сухое вещество).
0,250 г испытуемого образца растворяют в воде Р и
доводят этим же растворителем до объема 50,0 мл.
Примесь О. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 30 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО оксалиплати¬
на примеси О растворяют в метаноле Р и доводят до
объема 100,0 мл этим же растворителем.
Рисунок #2017.-1. Инфракрасный спектр ФСО оксалиплатина
97. Зак. 1060.
766
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (Ь). 15,0 мл раствора срав¬
нения (а) доводят метанолом Р до объема 50,0 мл.
Раствор сравнения (с). 75 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения (ф. 5,0 мл раствора сравне¬
ния (с) доводят метанолом Р до объема 100,0 мл.
Раствор сравнения (е). К 40 мл раствора срав¬
нения (с) прибавляют 1,0 мл раствора сравнения (Ь) и
доводят метанолом Р до объема 50,0 мл.
Раствор сравнения ((). К 4,0 мл раствора срав¬
нения (а) прибавляют 5,0 мл раствора сравнения (с!) и
доводят метанолом Р до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем ОС для хи¬
ральных разделений Р,
- температура: 40 °С;
- подвижная фаза: этанол Р — метанол Р
(30:70, об/об);
- скорость подвижной фазы: 0,3 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (е) и (7);
- время хроматографирования: 2-кратное вре¬
мя удерживания оксалиплатина.
Время удерживания: оксалиплатин — около
14 мин; примесь й — около 16 мин.
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками окса¬
липлатина и примеси О на хроматограмме раствора
сравнения (0;
- отношение сигнал/шум: не менее 10 для пика
примеси О на хроматограмме раствора сравнения (е).
Предельное содержание примесей:
- примесь О (не более 0,15 %): не более 3-крат¬
ной разницы между высотами пиков примеси й на
хроматограмме раствора сравнения (е) и испытуемо¬
го раствора.
Сопутствующие примеси
А. Примесь А. Жидкостная хроматография
(2.2.29). Для растворения испытуемого образца ис¬
пользуют интенсивное встряхивание и очень кра¬
тковременную обработку ультразвуком. Испыту¬
емый раствор вводят в течение 20 мин после при¬
готовления.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 14,0 мг кислоты щаве¬
левой Р (примесь А) растворяют в воде Р и доводят
до объема 250,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят водой Р до объема 200,0 мл.
Раствор сравнения (с). 12,5 мг натрия ни¬
трата Р растворяют в воде Р и доводят до объема
250.0 мл этим же растворителем. Смесь из 2,0 мл по¬
лученного раствора и 25,0 мл раствора сравнения (а)
доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 20 объемов аце¬
тонитрила Р и 80 объемов раствора, приготовленного
следующим образом: к 10 мл раствора 320 г/л тетра-
бутиламмония гидроксида Р прибавляют 1,36 г ка¬
лия дигидрофосфата Р, доводят водой Р до объема
1000 мл и доводят кислотой фосфорной Р до рН 6,0;
- скорость подвижной фазы: 2 мл/мин;
- спектрофотометрический детектор, длина
волны 205 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 2-кратное вре¬
мя удерживания примеси А.
Время удерживания: нитрат — около 2,7 мин;
примесь А — около 4,7 мин.
Пригодность хроматографической системы:
- разрешение: не менее 9 между пиками нитрата
и примеси А на хроматограмме раствора сравнения (с);
- отношение сигнал/шум: не менее 10 для пика
примеси А на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
- примесь А (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь).
В. Примесь В. Жидкостная хроматография
(2.2.29). Для растворения испытуемого образца
используют интенсивное встряхивание и очень
кратковременную обработку ультразвуком. Испы¬
туемый раствор вводят в течение 20 мин после
приготовления. Для приготовления и введения всех
растворов используют подходящие полипропилено¬
вые контейнеры. Для разбавления растворов могут
быть использованы стеклянные пипетки.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в водеР и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО оксалипла¬
тина примеси В прибавляют к 25 мл метанола Р,
доводят водой Р до объема 100,0 мл и обрабатывают
ультразвуком в течение около 1,5 ч до растворения
(раствор А). 3,0 мл раствора А доводят водой Р до
объема 200,0 мл.
Раствор сравнения (Ь). Для приготовления при¬
меси Е /7? $Ни 50,0 мл раствора А доводят раствором
0,2 г/л натрия гидроксида Р до рН 6,0, нагревают при
температуре 70 °С в течение 4 ч и охлаждают.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 20 объемов аце¬
тонитрила Р и 80 объемов раствора, приготовленно¬
го следующим образом: 1,36 г калия дигидрофосфа¬
та Р и 1 г натрия гептансульфоната Р растворяют
в 1000 мл воды Р и доводят кислотой фосфорной Р
до рН (3,0±0,05);
- скорость подвижной фазы: 2,0 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания примеси В.
Оксалиплатин
767
Время удерживания: примесь В — около 4,3 мин;
примесь Е — около 6,4 мин.
Пригодность хроматографической системы:
- разрешение: не менее 7 между пиками приме¬
сей В и Е на хроматограмме раствора сравнения (Ь);
- отношение сигнал/шум: не менее 10 для пика
примеси В на хроматограмме раствора сравнения (а).
Предельное содержание примесей:
- примесь В (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 4-кратную
площадь основного пика на хроматограмме раствора
сравнения (а).
С. Примесь С и другие сопутствующие примеси.
Жидкостная хроматография (2.2.29). Для растворе¬
ния испытуемого образца используют интенсивное
встряхивание и очень кратковременную обработку
ультразвуком. Испытуемый раствор вводят в те¬
чение 20 мин после приготовления.
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем.
Испытуемый раствор (Ь). 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
500.0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мг ФСО оксалипла-
тина и 5,0 мг ФСО оксалиплатина примеси С раст¬
воряют в воде Р и доводят до объема 50,0 мл этим же
растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). 25,0 мг ФСО оксали¬
платина растворяют в воде Р и доводят до объема
250.0 мл этим же растворителем.
Раствор сравнения (ф. 5,0 мг ФСО дихлорди-
аминоциклогексанплатины растворяют в растворе
сравнения (с) и доводят до объема 50,0 мл этим же
растворителем.
Раствор сравнения (е). 5 мл раствора сравнения
(д) доводят водой Р до объема 50,0 мл.
Раствор сравнения (I). К 0,100 г испытуемого об¬
разца прибавляют 1,5 мл раствора сравнения (а) и до¬
водят водой Р до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 1 объем ацето¬
нитрила Р и 99 объемов раствора, приготовленного
следующим образом: 0,6 мл кислоты фосфорной
разведенной Р разводят в 1000 мл воды Р и доводят
раствором натрия гидроксида Р или кислотой фос¬
форной Р до рН 3,0;
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (Ь), (е) и (I);
- время хроматографирования: 3-кратное вре¬
мя удерживания оксалиплатина.
Время удерживания: примесь С — около 4,4 мин;
дихлордиаминоциклогексанплатина — около 6,9 мин;
оксалиплатин — около 8,0 мин.
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками дих-
лордиаминоциклогексанплатины и оксалиплатина на
хроматограмме раствора (е);
- отношение сигнал/шум: не менее 50 для
пика примеси С и не менее 10 для пика оксалипла¬
тина на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
-примесь С (не более 0,15%): на хромато¬
грамме испытуемого раствора (а) площадь пика,
соответствующего примеси С, не должна превы¬
шать 0,5 площади соответствующего пика на хро¬
матограмме раствора сравнения (!);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пика
примеси С, не должна превышать 2-кратную пло¬
щадь пика оксалиплатина на хроматограмме раст¬
вора сравнения (Ь);
- сумма неспецифицированных примесей (не
более 0,15%): на хроматограмме испытуемого
раствора (а) сумма площадей всех пиков, кроме
основного и пика примеси С, не должна превышать
3-кратную площадь пика оксалиплатина на хрома¬
тограмме раствора сравнения (Ь);
- неучитываемый предел: на хроматограм¬
ме испытуемого раствора (а) не учитывают пики с
площадью менее площади пика оксалиплатина на
хроматограмме раствора сравнения (Ь) (0,05 %);
не учитывают пики со временем удерживания ме¬
нее 2 мин.
й. Сумма примесей, кроме примеси О: не бо¬
лее 0,30 %.
Серебро. Не более 0,0005 % (5 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, Метод II).
Испытуемый раствор. 0,1000 г испытуемого
образца растворяют в воде Р и доводят до объема
50,0 мл этим же растворителем. 20 мкл получен¬
ного раствора доводят 0,5 М раствором кислоты
азотной до 40 мкл.
Раствор сравнения (а). Раствор серебра ни¬
трата Р, содержащий 1000 ррт серебра в 0,5 М
растворе кислоты азотной, разводят 0,5 М рас¬
твором кислоты азотной до получения раствора,
содержащего 10 ррЬ серебра.
Раствор сравнения (Ь). Смешивают 20 мкл
испытуемого раствора и 8 мкл раствора сравнения
(а) и доводят 0,5 М раствором кислоты азотной
до объема 40 мкл.
Раствор сравнения (с). Смешивают 20 мкл ис¬
пытуемого раствора и 16 мкл раствора сравнения
(а) и доводят 0,5 М раствором кислоты азотной
до объема 40 мкл.
Источник излучения: лампа с полым катодом
для определения серебра.
Длина волны: 328,1 нм.
Атомизатор: графитовая печь.
Измеряют оптическую плотность испытуемого
раствора и растворов сравнения (Ь) и (с).
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца су¬
шат при температуре 105 °С в течение 2 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 1,0 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без дальнейшей подходящей
процедуры удаления бактериальных эндотоксинов.
97*. Зак. 1060.
768
Государственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указа¬
но в испытании «Примесь С и другие сопутствующие
примеси», со следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и растворов сравнения (с) и (6).
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками дих-
лордиаминоциклогексанплатины и оксалиплатина на
хроматограмме раствора сравнения (с1);
- относительное стандартное отклонение:
раствор сравнения (с).
Содержание оксалиплатина в процентах рассчиты¬
вают, используя хроматограмму раствора сравнения (с).
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е.
.он
о^ он
А. Этандиовая кислота (щавелевая кислота).
В. (ЗР-4-2)-Диаква[(1Я2Я)-циклогексан-1,2-диамин-
кЛ/,кА/]платина (диакводиаминоциклогексанплатина).
С. (ОС-6-33)-[(1 Я,2Я)-Циклогексан-1,2-диамин-
кЛ/,кЛ/][этандиоато(2-)-к07, кО^ди гид рокси плати на.
й. (5Р-4-2)-[(15,25)-Циклогексан-1,2-диамин-
кЛ/,кЛДэтандиоато(2-)-кО*, кО^платина (5,5-энантиомер
оксалиплатина).
н
н2 н2
-\,'° /"
-УЧА.
н, Н-
Е. (5Р-4-2)-Ди-гт)-оксобис[(1 Р,2Р)-циклогексан-1,2-
диамин-кА/.кА/Тдиплатина (диакводиаминоциклогексан-
платины димер).
07/2016:2260
ОКСАЦИЛЛИН НАТРИЯ
МОНОГИДРАТ
ОхасННпит паМсит топоЬубпсит
ОХАС1ШЫ ЗОйШМ МОЫОНУОКАТЕ
С19Н18М3Ма055 ■ Н20 М.м. 441,4
[7240-38-2]
ОПРЕДЕЛЕНИЕ
Натрия (25,5Р,6Р)-3,3-диметил-6-[[(5-метил-3-
фенилизоксазол-4-ил)карбонил]амино]-7-оксо-4-тиа-1-
азабицикло[3.2.0]гептан-2-карбоксилат моногидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый ли почти белый порошок.
Легко растворим в воде, растворим в метаноле,
практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО оксациллина натрия моно¬
гидрата #или спектр, представленный на рисунке
#2260.-1*.
B. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,50 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,10
при 430 нм. Измеряют оптическую плотность раст¬
вора 5.
рН (2.2.3). От 4,5 до 7,5. 0,30 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+196 до +212 (в пересчете на безводное вещество).
0,250 г испытуемого образца растворяют в воде Р и
доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Оксациллин натрия моногидрат
769
Рисунок #2260.-1. Инфракрасный спектр ФСО оксациллина натрия моногидрата
Испытуемый раствор (Ь). 5,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
50.0 мл.
Раствор сравнения (а). 50,0 мг ФСО оксацил¬
лина натрия моногидрата растворяют в подвижной
фазе и доводят до объема 50,0 мл этим же раствори¬
телем. 5,0 мл полученного раствора доводят подвиж¬
ной фазой до объема 50,0 мл.
Раствор сравнения (Ь). 5,0 мл испытуемого раст¬
вора (Ь) доводят подвижной фазой до объема 50,0 мл.
Раствор сравнения (с). 5 мг ФСО клоксациллина
натрия (примесь Е) и 5 мг ФСО оксациллина натрия
моногидрата растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем.
Раствор сравнения (д). 25 мг испытуемого об¬
разца растворяют в 1 мл 0,05 М раствора натрия
гидроксида. Через 3 мин доводят подвижной фазой
до объема 100 мл (получение примесей В и От зПи).
Вводят немедленно.
Раствор сравнения (е). 5 мг ФСО оксациллина
для идентификации пиков (содержит примеси Е, Р, С,
I и б) растворяют в 5 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4.0 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — раствор
2,7 г/л калия дигидрофосфата Р, доведенный рас¬
твором натрия гидроксида разведенным Р до рН
5,0, (25:75, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а), растворов сравнения (Ь), (с), (б) и (е);
- время хроматографирования: 7-кратное вре¬
мя удерживания оксациллина.
Идентификация пиков примесей:
-два основных пика на хроматограмме раствора
сравнения (б), элюирующиеся перед главным пиком,
соответствуют примесям Вий;
- идентифицируют пики примесей Е, Р, С, I и б,
используя хроматограмму раствора сравнения (е) и
хроматограмму, прилагаемую к ФСО оксациллина для
идентификации пиков.
Относительное удерживание (по отношению
к оксациллину, время удерживания — около 5 мин):
примесь А— около 0,3; примесь В (изомер 1) — око¬
ло 0,4; примесь В (изомер 2) — около 0,5; примесь
С — около 0,65; примесь О (2 изомера) — около 0,9;
примесь Е — около 1,5; примесь Р — около 1,9; при¬
месь О — около 2,1; примесь I — около 3,8; примесь
б — около 5,8.
Пригодность хроматографической системы:
-разрешение: не менее 2,5 между пиками ок¬
сациллина и примеси Е на хроматограмме раствора
сравнения (с);
- хроматограмма раствора сравнения (е) должна
соответствовать хроматограмме, прилагаемой к ФСО
оксациллина для идентификации пиков.
Предельное содержание примесей:
- примесь В (не более 1,5 %): на хроматограмме
испытуемого раствора (а) сумма площадей пиков, со¬
ответствующих двум изомерам примеси В, не должна
превышать 1,5-кратную площадь основного пика на
хроматограмме раствора сравнения (Ь);
- примесь Е (не более 1,0 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответствую¬
щего примеси Е, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- примеси О (сумма двух изомеров), Р, С, I, ^ (не
более 0,5 %): на хроматограмме испытуемого раст¬
вора (а) площади пиков, соответствующих примесям
Р (сумма двух изомеров), Р, О, I и б, не должны превы-
770
Гэсударственная фармакопея Республики Беларусь
шать 0,5 площади основного пика на хроматограмме
раствора сравнения (Ь);
-любая другая примесь (не более 0,5 %): на
хроматограмме испытуемого раствора (а) площадь
любого пика, кроме основного и пиков примесей В,
О, Е, Р, О, I и и, не должна превышать 0,5 площади
основного пика на хроматограмме раствора срав¬
нения (Ь);
- сумма примесей (не более 3,0 %): на хрома¬
тограмме испытуемого раствора (а) сумма площа¬
дей всех пиков, кроме основного, не должна превы¬
шать 3-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора (а) не учитывают
пики с площадью менее 0,05 площади основного
пика на хроматограмме раствора сравнения (Ь).
Этилацетат и бутилацетат. Парофазная газо¬
вая хроматография (2.2.28).
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в 6,0 мл воды Р.
Раствор сравнения. 83 мг этилацетата Р и
83 мг бутилацетата Р растворяют в воде Р и дово¬
дят до объема 250,0 мл этим же растворителем. Ис¬
пользуют 6,0 мл полученного раствора.
Флаконы немедленно плотно закрывают ре¬
зиновыми пробками, покрытыми политетраф¬
торэтиленом, и обжимают алюминиевыми кол¬
пачками. Встряхивают до получения однородного
раствора.
Условия хроматографирования:
- колонка капиллярная кварцевая длиной 50 м
и диаметром 0,32 мм, слоем поли(диметил)силокса-
на Р (толщина слоя 5 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость подвижной фазы: 2 мл/мин;
- параметры парофазного пробоотборника:
- равновесная температура: 80 °С;
- время достижения равновесия: 60 мин;
- температура передающей линии: 140 °С;
- время приложения избыточного давления: 30 с;
- температура:
Время (мин)
Температура (вС)
Колонка
0—6
70
6—16
70 — 220
16—18
220
Блок ввода проб
140
Детектор
250
- детектор: пламенно-ионизационный.
Времена удерживания: этилацетат — около
10 мин; бутилацетат — около 15,5 мин.
Предельное содержание примесей:
- этилацетат: не более 1,0 %;
- бутилацетат: не более 1,0 %.
М,М-Диметиланилин (2.4.26, метод В). Не бо¬
лее 0,0020 % (20 ррт).
2-Этилгексановая кислота (2.4.28). Не более 0,8 %.
Вода (2.5.12). Не менее 3,5 % и не более 5,0 %.
Определение проводят из 0,300 г испытуемого об¬
разца.
Бактериальные эндотоксины (2.6.14). Менее
0,20 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без последующей процедуры
удаления бактериальных эндотоксинов.
#Аномальная токсичность (2.6.9). Испытуемый
образец должен быть нетоксичным. Тест-доза —10 мг
испытуемого образца в 0,5 мл воды для инъекций Р
на мышь, внутривенно. Срок наблюдения — 24 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (а).
Содержание С19Н18М3Ма055 в процентах рассчи¬
тывают с учетом содержания оксациллина натрия в
ФСО оксациллина натрия моногидрата.
ПРИМЕСИ
Специфицированные примеси: В, О, Е, Е, 6, /, й.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, С.
н н
А. (25,5Р,6Р)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
В. Р = С02Н: (45)-2-[Карбокси[[(5-метил-3-фенил-
изоксазол-4-ил)карбонил]амино]метил]-5,5-диметил-
тиазолидин-4-карбоновая кислота (пенициллоиновая
кислота оксациллина).
О. Р = Н: (2Р5,45)-5,5-Диметил-2-[[[(5-метил-3-
фенилизоксазол-4-ил)карбонил]амино]метил]тиазоли-
дин-4-карбоновая кислота (пениллоиновая кислота ок¬
сациллина).
С. 5-Метил-3-фенилизоксазол-4-карбоновая кис¬
лота.
Оксиметазолина гидрохлорид
771
Е. (25,5Я,6/?)-6-[[[3-(2-Хлорфенил)-5-метилизо-
ксазол-4-ил]карбонил]амино]-3,3-диметил-7-ош>4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (клок-
сациллин).
Р. Я1 = ЗН, Я2 = Н: (гЯ.бЯб/^-З.З-Диметил-б-Мб-
метил-3-фенилизоксазол-4-ил)карбонил]амино]-7-оксо-
4- тиа-1 -азабицикло[3.2.0]гептан-2-тиокарбоновая кис¬
лота (тиооксациллин).
С. Я1 = ОН, Я2 = С1: (25,5Я,6Я)-6-[[[3-(Хлорфенил)-
5- метилизоксазол-4-ил]карбонил]амино]-3,3-диметил-
7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-карбоновая
кислота (изомер клоксациллина).
I. (23,5Я6Я)-6-[[(25,5Я,6/?)-3,3-Диметил-6-[[(5-ме-
тил-3-фенилизоксазол-4-ил)карбонил]амино]-7-оксо-4-
тиа-1-азабицикло[3.2.0]гептан-2-карбонил]амино]-3,3-
диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-2-
карбоновая кислота (6-АПК оксациллина амид).
3. (25,5Я,6/?)-6-[[(2Я)-[(2Я?,45И-Карбокси-5,5-
диметилтиазолидин-2-ил][[(5-метил-3-фенипизоксазол-
4-ил)карбонил]амино]ацетил]амино]-3,3-диметил-7-
оксо-4-тиа-1 -азабицикло[3.2.0]гептан-2-карбоновая кис¬
лота (озоламид 6-АПК димера).
07/2016:0943
ОКСИМЕТАЗОЛИНА ГИДРОХЛОРИД
Охуте1агоИп'1 Ьус1госЫопс1ит
ОХУМЕТАЮиЫЕ НУОКОСШ-ОКЮЕ
С16Н2«М2° • НС1 М.м. 296,8
[2315-02-8]
Рисунок #0943.-1. Инфракрасный спектр ФСО оксиметазолина гидрохлорида
98*. Зак. 1060.
772
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
3-[(4,5-Дигидро-1Я-имидазол-2-ил)метил]-6-(1,1-
диметилэтил)-2,4-диметилфенола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО оксиметазолина гидрохлорида
#или спектр, представленный на рисунке #0943.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в смеси из равных объемов эти-
лацетата Р и метанола Р и доводят до объема 5 мл
этой же смесью растворителей.
Раствор сравнения. 20 мг ФСО оксиметазолина
гидрохлорида растворяют в смеси из равных объемов
этилацетата Р и метанола Р и доводят до объема
5 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: диэтиламин Р — циклогек¬
сан Р — этанол Р (6:15:79, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке теплого воздуха в тече¬
ние 5 мин и выдерживают до охлаждения.
Проявление: пластинку опрыскивают свежепри¬
готовленным раствором 5,0 г/л калия феррицианица
Р в растворе железа (III) хлорида Р2 и просматрива¬
ют при дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
C. 2 мг испытуемого образца растворяют в 1 мл
воды Р, прибавляют 0,2 мл раствора 50 г/л натрия
нитропруссида Р, 0,2 мл раствора натрия гидрок¬
сида разведенного Р, выдерживают в течение 10 мин
и прибавляют 2 мл раствора натрия гидрокарбона¬
та Р. Появляется фиолетовое окрашивание.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)7.
Кислотность или щелочность. 0,25 г испытуе¬
мого образца растворяют в воде, свободной от угле¬
рода диоксида, Р и доводят до объема 25 мл этим же
растворителем. Прибавляют 0,1 мл раствора мети¬
лового красного Р и 0,2 мл 0,01 М раствора кислоты
хлористоводородной. Появляется красное окрашива¬
ние. При прибавлении не более 0,4 мл 0,01 М раст¬
вора натрия гидроксида должно появиться желтое
окрашивание.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого
раствора доводят водой Р до объема 100,0 мл. 2,0 мл
полученного раствора доводят водой Р до объема
100.0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО оксиметазо¬
лина примеси А и 5 мг испытуемого образца раство¬
ряют в воде Р и доводят до объема 50,0 мл этим же
растворителем. 10,0 мл полученного раствора дово¬
дят водой Р до объема 50,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, с введенными полярными группами,
эндкепированным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 1,36 г/л калия ди¬
гидрофосфата Р, доведенный до рН 3,0 кисло¬
той фосфорной Р\
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
70
30
5—20
70 — 15
30 — 85
20—35
15
85
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к оксиметазолину, время удерживания — около 5,0
мин): примесь А — около 0,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками при¬
меси А и оксиметазолина.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Вода (2.5.32). Не более 0,3 %. Испытание прово¬
дят из 1,00 г испытуемого образца.
Окситоцин
773
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворят в смеси
из 20 мл кислоты уксусной безводной Р и 20 мл анги¬
дрида уксусного Р и титруют 0,1 М раствором кисло¬
ты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 29,68 мг С16Н24М2О НС1.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): В, С, О, Е.
А. А/-(2-Аминоэтил)-2-[4-(1,1-диметилэтил)-3-гидро-
кси-2,6-диметилфенил]ацетамид.
В. 2-[4-(1,1-Диметилэтил)-2,6-диметилбензил]-4,5-
дигидро-1 Н-имидазол (ксилометазолин).
С. К = СО-МН2:2-[4-(1,1-Диметилэтил)-3-гидрокси-
2,6-диметилфенил]ацетамид.
О. Я = С02Н: [4-(1,1-Диметилэтил)-3-гидрокси-2,6-
диметилфенил]уксусная кислота.
Е. К = С№ [4-(1,1-Диметилэтил)-3-гидрокси-2,6-ди-
метилфенил]ацетонитрил.
07/2016:0780
ОКСИТОЦИН
Оху1остит
ОХУТОСШ
Н-С^уз-Туг-Ие-СХп-Азп-Суз-Рго-Ьеи-СХу-Ы^
М.м. 1007
[50-56-6]
ОПРЕДЕЛЕНИЕ
ЬЦистеинил-Ь-тирозил-Е-изолейцил-Ьглутамил-
1--аспарагинил-1_-цистеинил-1_-пролил-1--лейцил-
глицинамид циклический (1—>6)-дисульфид.
Синтетический циклический нонапептид, име¬
ющий структуру гормона, произведенного задней
долей гипофиза, который стимулирует сокращение
матки и выделение молока при кормлении. Досту¬
пен в лиофилизированной форме в виде ацетата.
Содержание: не менее 93,0 % и не более
102,0% (в пересчете на безводное и не содержа¬
щее уксусной кислоты вещество).
В целях маркировки лекарственных средств,
содержащих окситоцин, принято, что 1 мг пептида
(^43^66^12^12^2) эквивалентен 600 МЕ биологиче¬
ской активности.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Очень легко растворим в воде. Растворяется
в разведенных растворах кислоты уксусной и 96 %
спирта.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, получен¬
ные в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания
соответствует основному пику на хроматограмме
раствора сравнения.
B. Анализ аминокислот (2.2.56). Для гидроли¬
за используют метод 1, для анализа используют
метод 1.
Содержание каждой аминокислоты выражают
в молях. Относительные отношения аминокислот
рассчитывают, принимая, что 1/6 суммы числа мо¬
лей аспарагиновой кислоты, глутаминовой кислоты,
пролина, глицина, изолейцина и лейцина равна 1.
Полученные результаты должны находиться в
следующих пределах: аспарагиновая кислота: от
0,90 до 1,10; глутаминовая кислота: от 0,90 до 1,10;
пролин: от 0,90 до 1,10; глицин: от 0,90 до 1,10;
лейцин: от 0,90 до 1,10; изолейцин: от 0,90 до 1,10;
тирозин: от 0,7 до 1,05; полуцистин: от 1,4 до 2,1.
Другие аминокислоты должны присутствовать не
более чем в следовых количествах.
ИСПЫТАНИЯ
рН (2.2.3). От 3,0 до 6,0. 0,200 г испытуемого
образца растворяют в воде, свободной от углеро¬
да диоксида, Р и доводят до объема 10,0 мл этим
же растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29.): используют метод нормали¬
зации.
Испытуемый раствор. Готовят раствор
0,25 мг/мл испытуемого образца в растворе
15,6 г/л натрия дигидрофосфата Р.
Раствор для определения разрешения. Содер¬
жимое контейнера с ФСО смеси окситоцина/дес-
мопрессина для валидации растворяют в 1 мл
раствора 15,6 г/л натрия дигидрофосфата Р.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
99. Зак. 1060.
774
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза:
-подвижная фаза А: раствор 15,6 г/л натрия
дигидрофосфата Р;
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р— вода Р (50:50, об/об);
Время (мин)
Подвижная фаза А
(%г об/об)
Подвижная фаза В
(%, об/об)
0—30
70 — 40
30-60
30—30,1
40 — 70
60 — 30
30,1-^5
70
30
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 220 нм;
- объем вводимой пробы: 50 мкл.
Время удерживания: окситоцин — около
7,5 мин; десмопрессин — около 10 мин.
Пригодность хроматографической системы:
раствор для определения разрешения:
-разрешение: не менее 5,0 между пиками
десмопрессина и окситоцина.
Предельное содержание примесей:
- любая примесь: не более 1,5 %;
- сумма примесей: не более 5 %;
- неучитываемый предел: 0,1 %.
Уксусная кислота (2.5.34). От 6,0 % до 10,0 %.
Испытуемый раствор. 15,0 мг испытуемого
образца растворяют в смеси из 5 объемов под¬
вижной фазы В и 95 объемов подвижной фазы А
и доводят до объема 10,0 мл этой же смесью под¬
вижных фаз.
Вода (2.5.12). Не более 5,0%. Определение
проводят из не менее 50 мг испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Ме¬
нее 300 МЕ/мг, если субстанция предназначена
для производства лекарственных средств для па¬
рентерального применения без последующей про¬
цедуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Раствор сравнения. Содержимое контейнера с
ФСО окситоцина растворяют в растворе 15,6 г/л на¬
трия дигидрофосфата Р с получением концентра¬
ции 0,25 мг/мл.
Условия хроматографирования:
- объем вводимой пробы: 25 мкл.
Содержание окситоцина (С43Н661\11201252) рассчи¬
тывают с учетом содержания окситоцина в ФСО ок¬
ситоцина.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре от 2 °С до
8 °С. Если субстанция стерильная, ее хранят в сте¬
рильном воздухонепроницаемом контейнере с кон¬
тролем первого вскрытия.
МАРКИРОВКА
Указывают:
- содержание пептида окситоцина (С^Н^И^О^З^.
07/2016:0779
ОКСИТОЦИНА РАСТВОР
КОНЦЕНТРИРОВАННЫЙ
Оху1ос1т зо!иИо сопсеп1га1а
ОХУТОСШ СОМСЕЫТКАТЕО 30ШТ10Ы
Н-(?у5-Туг-11е-С1п-А5п-Су5-Рго-Ъеи-С1у-Ш2
С43Н6вМ12°12$2 М.м. 1007
ОПРЕДЕЛЕНИЕ
ЬЦистеинил-Ь-тирозил-Ь-изолейцил-Ь-глутамил-
Ь-аспарагинил-Ьцистеинил-Ьпролил-Ьлейцилгли-
цинамид циклический (1—6)-дисульфид.
Раствор окситоцина, синтетического цикличе¬
ского нонапептида, имеющего структуру гормона,
произведенного задней долей гипофиза, который
стимулирует сокращение матки и выделение мо¬
лока при кормлении. Доступен в виде раствора
с указанной концентрацией окситоцина не менее
0,25 мг/мл в растворителе, который может содер¬
жать подходящий антибактериальный консервант.
Содержание: не менее 95,0 % и не более
105,0 % от указанного количества пептида в 1 мл.
В целях маркировки лекарственных средств,
содержащих окситоцин, принято, что 1 мг пептида
окситоцина (С43Н66Ы1201232) эквивалентен 600 МЕ
биологической активности.
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, получен¬
ные в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания
соответствует основному пику на хроматограмме
раствора сравнения.
B. Анализ аминокислот (2.2.56). Для гидроли¬
за используют метод 1, для анализа используют
метод 1.
Содержание каждой аминокислоты выражают
в молях. Относительные отношения аминокислот
рассчитывают, принимая, что 1/6 суммы числа
молей аспарагиновой кислоты, глутаминовой кис¬
лоты, пролина, глицина, изолейцина и лейцина
равна 1.
Полученные результаты должны находиться
в следующих пределах: аспарагиновая кислота:
от 0,90 до 1,10; глутаминовая кислота: от 0,90 до
1,10; пролин: от 0,90 до 1,10; глицин: от 0,90 до
1,10; лейцин: от 0,90 до 1,10; изолейцин: от 0,90 до
1,10; тирозин: от 0,7 до 1,05; полуцистин: от 1,4 до
2,1. Другие аминокислоты должны присутствовать
не более чем в следовых количествах.
ИСПЫТАНИЯ
рН (2.2.3). От 3,0 до 5,0.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29.): используют метод нормализации.
Испытуемый раствор. Испытуемый образец.
Раствор для определения разрешения. Содер¬
жимое контейнера с ФСО смеси окситоцина/десмо-
Октилдодеканал
775
прессина для валидации растворяют в 1 мл раствора
15,6 г/л натрия дигидрофосфата Р.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 15,6 г/л натрия
дигидрофосфата Р\
- подвижная фаза В: ацетонитрил для хрома-
тографии Р — вода Р (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
70 — 40
30 — 60
30—30,1
40 — 70
60 — 30
30,1—45
70
30
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 220 нм;
- объем вводимой пробы: 50 мкл.
Время удерживания: окситоцин — около
7,5 мин; десмопрессин — около 10 мин.
Пригодность хроматографической системы:
раствор для определения разрешения:
-разрешение: не менее 5,0 между пиками
десмопрессина и окситоцина.
Предельное содержание примесей:
- любая примесь: не более 1,5 %;
- сумма примесей: не более 5 %;
- неучитываемый предел: 0,1 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
Бактериальные эндотоксины {2.6.14). Менее
300 МЕ/мг в объеме, содержащем 1 мг окситоцина,
если субстанция предназначена для производства
лекарственных средств для парентерального при¬
менения без последующей процедуры удаления
бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Раствор сравнения. Содержимое контейнера
с ФСО окситоцина растворяют в растворе 15,6 г/л
натрия дигидрофосфата Р с получением концен¬
трации 0,25 мг/мл.
Условия хроматографирования:
- объем вводимой пробы: 25 мкл.
Содержание окситоцина (С43Н661Ч1201232) рас¬
считывают с учетом содержания окситоцина в ФСО
окситоцина.
ХРАНЕНИЕ
При температуре от 2 °С до 8 °С в защищенном
от света месте. Если субстанция стерильная, ее хра¬
нят в стерильном воздухонепроницаемом контейнере
с контролем первого вскрытия.
МАРКИРОВКА
Указывают:
- содержание пептида окситоцина (С^Н^^О^З,,)
в мг/мл.
99*. Зак. 1060.
07/2016:1136
ОКТИЛДОДЕКАНОЛ
Ос1уШоёесапо1ит
0СТУ1.000ЕСАЛЮ/.
он
и энантиомер
[5333-42-6]
ОПРЕДЕЛЕНИЕ
Конденсированный продукт насыщенных жидких
жирных спиртов.
Содержание: не менее 90 % (2Я?5)-2-октилдодекан-
1-ола (С20Н42О; М.м. 298,6), остальное преимуществен¬
но составляют родственные спирты.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная или желтоватая масля¬
нистая жидкость.
Практически нерастворим в воде, смешивается с
96 % спиртом.
Относительная плотность: около 0,840.
Показатель преломления: около 1,455.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Гидроксильное число», как указано в разделе «Ис¬
пытания».
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,20 г испытуемого об¬
разца растворяют в толуоле Р и доводят до объема
20 мл этим же растворителем.
Раствор сравнения. 0,20 г ФСО октилдодека-
нола растворяют в толуоле Р и доводят до объема
20 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают 7 мл смеси
из 1 объема раствора 25 г/л ванилина Рв 96% спир¬
те Р\л А объемов кислоты серной Р и нагревают при
температуре 130 °С в течение (5—10) мин.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию, размеру и цвету основному пятну на хромато¬
грамме раствора сравнения.
ИСПЫТАНИЯ
Кислотность или щелочность. 5,0 г испытуемого
образца тщательно перемешивают в течение 1 мин со
смесью из 0,1 мл раствора бромтимолового синего Р1,
2 мл гептана Р и 10 мл воды Р. Если водный слой окра¬
шен в синий цвет, то при прибавлении не более 0,15 мл
0,01 М раствора кислоты хлористоводородной долж¬
но появиться желтое окрашивание. Если водный слой
окрашен в желтый цвет, прибавляют 0,45 мл 0,01 М
раствора натрия гидроксида и энергично встряхивают.
После отстаивания до полного разделения слоев во¬
дный слой должен быть окрашен в синий цвет.
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. 2,50 г испытуемого образца растворяют в
776
Государственная фармакопея Республики Беларусь
96 % спирте Р и доводят до объема 25 мл этим же
растворителем.
Гидроксильное число (2.5.3, метод А). От 175
до 190.
Йодное число (2.5.4, метод А). Не более 8,0.
Перекисное (пероксидное) число (2.5.5, метод
А). Не более 5,0.
Число омыления (2.5.6). Не более 5,0.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 2,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 0,4 г тетра¬
декана Р растворяют в гексане Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в растворе внутреннего стандарта
и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения. 0,100 г ФСО октилдодека-
нола растворяют в растворе внутреннего стандарта и
доводят до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 60 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)(дифенил)(дивинил)силоксана Р (тол¬
щина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость потока: 0,68 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—2
180
2—22
180 —► 280
22—52
280
Блок ввода проб
290
Детектор
300
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Рассчитывают содержание С20Н42О в испытуе¬
мом образце.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:2063
ОМЕГА-З-КИСЛОТЫ ЭТИЛОВЫЕ
ЭФИРЫ 60
Отеда-3 асМогит ез^е^^ еМуНс! 60
ОМЕвА-З-АСЮ ЕТНУ1- ЕЗТЕК8 60
ОПРЕДЕЛЕНИЕ
Этиловые эфиры альфа-линоленовой кислоты
(С18:3 п-3), мороктовой кислоты (С18:4 п-3), эйко-
затетраеновой кислоты (С20:4 п-3), тимнодоновой
(эйкозапентаеновой) кислоты (С20:5 п-3; ЭПК), генэй-
козапентаеновой кислоты (021:5 п-3), клупанодоно-
вой кислоты (С22:5 п-3) и цервоновой (докозагекса-
еновой) кислоты (С22:6 п-3; ДГК). Этиловые эфиры
60 омега-3-кислоты получают с помощью транс¬
этерификации масла, полученного из рыб семейств
ЕпдгаиНдае, СагапдШае, СШреШае, Озтепдае,
8а!топШае и ЗсотЬпдае или животных класса
СерЬа!орода, и последующих процессов физико-хи¬
мической очистки, включая молекулярную дистил¬
ляцию. Минимальное общее содержание этиловых
эфиров омега-3-кислоты и минимальное содержание
этиловых эфиров омега-3-кислот ЭПК и ДГК указаны
в таблице 2063.-1.
Таблица 2063.-1
Сумма эти¬
ловых
эфиров
омега-3-
кислоты
Этиловые
эфиры ЭПК
и ДГК
Этиловые
эфиры ЭПК
Этиловые
эфиры ДГК
Минимальное содержание (%)
65
50
25
20
60
50
-
40
55
50
40
-
Может быть добавлен подходящий антиоксидант.
ПРОИЗВОДСТВО
Содержание диоксинов и диоксинподобных ПХД
(полихлориновых дифенилов) контролируют, исполь¬
зуя методы и предельные содержания в соответствии
с требованиями, Предъявляемыми национальным
законодательством или другими применимыми доку¬
ментам^.
ОПИСАНИЕ (СВОЙСТВА)
Светло-желтая жидкость со слабым рыбным за¬
пахом.
Практически нерастворимы в воде, очень легко
растворимы в ацетоне, в 96 % спирте, в гептане и в
метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение
(этиловые эфиры ЭПК и ДГК)».
Результаты: на хроматограмме испытуемого
раствора (Ь) пики этилового эфира эйкозапентаено¬
вой кислоты и этилового эфира докозагексаеновой
кислоты по времени удерживания соответствуют со¬
ответствующим пикам на хроматограммах растворов
сравнения (а.,) и (а2).
B. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение (сумма
этиловых эфиров омега-3-кислоты)».
ИСПЫТАНИЯ
Оптическая плотность (2.2.25). Не более 0,60
при длине волны 233 нм. 0,300 г испытуемого образца
доводят триметилпентаном Р до объема 50,0 мл.
2,0 мл полученного раствора доводят триметилпен¬
таном Р до объема 50,0 мл.
Кислотное число (2.5.1). Не более 2,0. Опреде¬
ление проводят из 10 г в 50 мл предписанной смеси
растворителей.
Анизидиновое число (2.5.36). Не более 20,0.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 10,0.
Омега-З-кислоты этиловые эфиры 60
777
Олигомеры и моно- и диглицериды. Эксклюзи-
онная хроматография (2.2.30).
Испытуемый раствор. 50,0 мг испытуемого образ¬
ца доводят тетрагидрофураном Р до объема 10,0 мл.
Раствор сравнения. 50 мг монодокозагексаенои-
на Р, 30 мг дидокозагексаеноина Р и 20 мг тридокоза-
гексаеноина Р растворяют в тетрагидрофуране Р и
доводят до объема 100,0 мл этим же растворителем.
Условия хроматографирования:
- соединенные в определенной последователь¬
ности 3 колонки длиной 0,3 м и внутренним диаме¬
тром 7,8 мм, заполненные сополимером стирол-ди-
винилбензола Р с размером частиц 5 мкм и со следу¬
ющим размером пор:
- колонка 1: 50 нм;
- колонка 2: 10 нм;
- колонка 3: 5 нм;
- последовательность соединения: инжектор —
колонка 1 — колонка 2 — колонка 3 — детектор;
- подвижная фаза: тетрагидрофуран Р;
- скорость подвижной фазы: 0,8 мл/мин;
- детектор: дифференциальный рефрактометр;
- объем вводимой пробы: 40 мкл.
Пригодность хроматографической системы:
раствор сравнения:
-порядок выхода пиков: тридокозагексаеноин,
дидокозагексаеноин, монодокозагексаеноин;
-разрешение: не менее 2,0 между пиками дидо¬
козагексаеноина и монодокозагексаеноина; не менее
1,0 между пиками тридокозагексаеноина и дидокоза¬
гексаеноина.
Содержание суммы олигомеров и моно- и дигли¬
церидов в процентах рассчитывают по формуле:
— •100,
А
где:
А — сумма площадей всех пиков на хромато¬
грамме;
В — сумма площадей пиков с временем удержи¬
вания меньшим, чем время удерживания пиков этило¬
вых эфиров.
Пики этиловых эфиров, которые могут быть
представлены в форме неразделенного двойного
пика, являются основными пиками на хроматограмме
(рисунок 2063.-1).
А
1. Олигомеры. 2. Моноглицериды. 3. Этиловые эфиры жирной кислоты.
Рисунок 2063.-1. Хроматограмма для испытания по определению содержания олигомеров
и моно- и диглицеридов в этиловых эфирах 60 омега-3-кислоты
1. С16:0
2. С18:0
3. С18:1 п-9
18. С22:5 п-3
19. ДГК
6. С18.3 п-3
9. С20:1 п-9
11. С20:4 п-6
14.022:1 п-11 17. С22:5 п-6
Рисунок 2063.-2. Хроматограмма для количественного определения этиловых эфиров 60 омега-3-кислоты
100. Зак. 1060.
778
Гэсударственная фармакопея Республики Беларусь
Предельное содержание примесей:
- сумма олигомеров и моно- и диглицеридов: не
более 7,0 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.14.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этиловые эфиры ЭПК и ДГК (2.4.29). Для иден¬
тификации пиков используют рисунок 2063.-2.
Сумма этиловых эфиров омега-3-кислоты
(2.4.29). Для идентификации пиков используют рису¬
нок 2063.-2.
ХРАНЕНИЕ
В среде инертного газа в воздухонепроницаемом
контейнере в защищенном от света месте.
МАРКИРОВКА
На этикетке указывают:
- общее содержание этиловых эфиров омега-3-
кислоты;
- содержание этилового эфира ЭПК и этилового
эфира ДГК.
07/2016:1250
ОМЕГА-З-КИСЛОТЫ ЭТИЛОВЫЕ
ЭФИРЫ 90
Отеда-3 асШогит ез1еп еМуЧа 90
ОМЕвА-З-АСЮ ЕТНП Е8ТЕКЗ 90
ОПРЕДЕЛЕНИЕ
Этиловые эфиры альфа-линоленовой кислоты
(С18:3 п-3), мороктовой кислоты (С18:4 п-3), эйко-
затетраеновой кислоты (С20:4 п-3), тимнодоновой
(эйкозапентаеновой) кислоты (С20:5 п-3; ЭПК), ге-
нэйкозапентаеновой кислоты (С21:5 п-3), клупано-
доновой кислоты (С22:5 п-3) и цервоновой (докоза-
гексаеновой) кислоты (С22:6 п-3; ДГК). Этиловые
эфиры омега-3-кислоты получают с помощью транс¬
этерификации масла, полученного из рыбы таких
семейств, как ЕпдгаиНдае, СагапдШае, СШреШае,
Озтепдае, За!топШае и ЗсотЬпдае, или животных
класса СерШороба, и последующих процессов фи¬
зико-химической очистки, включая фракциониро¬
вание с использованием мочевины с последующей
молекулярной дистилляцией.
Содержание:
- этиловые эфиры ЭПК и ДГК: не менее 80 %,
при этом не менее 40 % этиловых эфиров ЭПК и не
менее 34 % этиловых эфиров ДГК;
- сумма этиловых эфиров омега-3-кислоты:
не менее 90 %.
Может быть добавлен подходящий антиокси¬
дант.
ПРОИЗВОДСТВО
Содержание диоксинов и диоксинподобных
ПХД (полихлориновых дифенилов) контролируется
с использованием методов и предельных содержа¬
ний в соответствии с требованиями, Предъявляе¬
мыми национальным законодательством или дру¬
гими применимыми документами!
ОПИСАНИЕ (СВОЙСТВА)
Светло-желтая жидкость.
Практически нерастворима в воде, очень легко
растворима в ацетоне, в 96 % спирте, в гептане и в
метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, получен¬
ные как указано в разделе «Количественное опре¬
деление (этиловые эфиры ЭПК и ДГК)».
Результаты: на хроматограмме испытуемого
раствора (Ь) пики этилового эфира эйкозапентае¬
новой кислоты и этилового эфира докозагексаено-
вой кислоты по времени удерживания соответству¬
ют соответствующим пикам на хроматограммах
растворов сравнения (а^ и (а2).
B. Испытуемый образец выдерживает тре¬
бования раздела «Количественное определение
(сумма этиловых эфиров омега-3-кислоты)».
ИСПЫТАНИЯ
Оптическая плотность (2.2.25). Не более
0,55 при длине волны 233 нм. 0,300 г испытуемого
образца доводят триметилпентаном Р до объ¬
ема 50,0 мл. 2,0 мл полученного раствора доводят
триметилпентаном Р до объема 50,0 мл.
Кислотное число (2.5.1). Не более 2,0. Опре¬
деление проводят из 10 г в 50 мл указанной смеси
растворителей.
Анизидиновое число (2.5.36). Не более 20,0.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 10,0.
Олигомеры. Эксклюзионная хроматография
(2.2.30).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 10,0 мл тетрагидрофурана Р.
Раствор сравнения. 50 мг монодокозагекса-
еноина Р, 30 мг дидокозагексаеноина Р и 20 мг
тридокозагексаеноина Р растворяют в тетраги-
дрофуране Р и доводят до объема 100,0 мл этим
же растворителем.
Условия хроматографирования (3 колонки,
которые соединяются в определенной последова¬
тельности):
Условия хроматографирования:
- соединенные в определенной последова¬
тельности 3 колонки длиной 0,3 м и внутренним ди¬
аметром 7,8 мм, заполненные сополимером сти-
рол-дивинилбензола Р с размером частиц 5 мкм и
со следующим размером пор:
- колонка 1: 50 нм;
-колонка 2: 10 нм;
- колонка 3: 5 нм;
- последовательность соединения: инжек¬
тор — колонка 1 — колонка 2 — колонка 3 — де¬
тектор;
- подвижная фаза: тетрагидрофуран Р\
- скорость подвижной фазы: 0,8 мл/мин;
- детектор: дифференциальный рефракто¬
метр;
- объем вводимой пробы: 40 мкп.
Пригодность хроматографической системы:
раствор сравнения:
- порядок выхода пиков: тридокозагексаено-
ин, дидокозагексаеноин, монодокозагексаеноин;
Омега-З-кислоты этиловые эфиры 90
779
- разрешение: не менее 2,0 между пикам-и ди-
докозагексаеноина и монодокозагексаеноина; не
менее 1,0 между пиками тридокозагексаеноина и
дидокозагексаеноина.
Содержание олигомеров в процентах рассчиты¬
вают по формуле:
вт,
А
где:
А — сумма площадей всех пиков на хромато¬
грамме;
В —- сумма площадей пиков с временем удержи¬
вания меньшим, чем время удерживания пиков этило¬
вых эфиров.
Пики этиловых эфиров, которые могут быть
представлены в форме неразделенного двойного
пика, являются основными пиками на хроматограмме
(рисунок 1250.-1).
Если полученный результат превышает предель¬
ное содержание вследствие присутствия моноглице¬
ридов, испытание проводят в соответствии со следу¬
ющей методикой.
Испытуемый раствор. Взвешивают 50,0 мг
испытуемого образца и помещают в кварцевую
пробирку. Прибавляют 1,5 мл раствора 20 г/л на¬
трия гидроксида Р в метаноле Р, наполняют
азотом Р, герметично укупоривают крышкой с по-
литетрафторэтиленовой прокладкой, перемешива¬
ют и нагревают на водяной бане в течение 7 мин.
Охлаждают, прибавляют 2 мл раствора бора хло¬
рида в метаноле Р, наполняют азотом Р, герме¬
тично укупоривают, перемешивают и нагревают
на водяной бане в течение 30 мин. Охлаждают до
температуры (40—50) °С, прибавляют 1 мл триме-
тилпентана Р, укупоривают и интенсивно встря¬
хивают в течение не менее 30 с. Затем без про¬
медления прибавляют 5 мл насыщенного раст-
1. Олигомеры. 2. Этиловые эфиры.
Рисунок 1250.-1. Хроматограмма
для испытания по определению олигомеров
в этиловых эфирах 90 омега-3-кислоты:
испытуемый образец, содержащий олигомеры
1 .Фитановая
кислота.
2.016:3 п-4.
3.016:4 п-1.
4. С18:3 п-6.
мин
21.022:5 п-6.
22. Фурановая
кислота 11.
23. С22:5 п-3.
24. ДГК.
*
5. 018:3 п-4.
6. 018:3 п-3.
7.018:4 п-3.
8.018:4 п-1.
9. Фурановая
кислота 5.
10. С19:5.
11. 020:3 п-6.
12. С20:4 п-6.
13. Фурановая
кислота 7.
14. 020:4 п-3.
15. Фурановая
кислота 8.
16. ЭПК.
17. Фурановая
кислота 9.
18. С21:5 п-3.
19. 022:4.
20. Фурановая
кислота 10.
Рисунок 1250.-2. Хроматограмма для количественных определений этиловых эфиров 90 омега-3-кислоты
100*. Зак. 1060.
780
Гэсударственная фармакопея Республики Беларусь
вора натрия хлорида Р, наполняют азотом Р, уку¬
поривают и интенсивно встряхивают в течение не
менее 15 с. Переносят верхний слой в отдельную
пробирку, еще раз встряхивают слой метанола с
1 мл триметилпентана Р. Объединенные триме-
тилпентановые экстракты дважды промывают во¬
дой Р порциями по 1 мл. Растворитель осторожно
выпаривают в токе азота Р, к полученному остатку
прибавляют 10,0 мл тетрагидрофурана Р. При¬
бавляют небольшое количество натрия сульфата
безводного Р и фильтруют.
Содержание олигомеров в процентах рассчиты¬
вают по формуле:
В-100,
А
где:
А — сумма площадей всех пиков на хромато¬
грамме;
В’— сумма площадей пиков с временем удержи¬
вания меньшим, чем время удерживания пиков мети¬
ловых эфиров.
Предельное содержание примесей:
- олигомеры: не более 1,0 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этиловые эфиры ЭПК и ДГК (2.4.29). Для иден¬
тификации пиков используют рисунок 1250.-2.
Сумма этиловых эфиров омега-3-кислоты
(2.4.29). Для идентификации пиков используют рису¬
нок 1250.-2.
ХРАНЕНИЕ
В среде инертного газа в воздухонепроницаемом
контейнере в защищенном от света месте.
07/2016:2016
ОНДАНСЕТРОНА ГИДРОХЛОРИД
ДИГИДРАТ
Опдапзе1гот Ьус1госЫопс1ит сПЬус/псит
ОЫОАЫЗЕТКОМ НУОКОСЖ.ОЯ/ОЕ
01НУ0КАТЕ
С18Н19М30 ■ НС! ■ 2Н,0 М.м. 365,9
[103639-04-9]
ОПРЕДЕЛЕНИЕ
(ЗЯ5)-9-Метил-3-[(2-метил-1 Я-имидазол-1 -ил)ме-
тил]-1,2,3,9-тетрагидро-4Н-карбазол-4-она гидрохло¬
рид дигидрат.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Умеренно растворим в воде и в 96 % спирте, рас¬
творим в метаноле, мало растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ондансетрона гидрохлорида
дигидрата #или спектр, представленный на рисун¬
ке #2016.-1#.
B. Испытуемый образец дает реакцию (а) на
хлориды (2.3.1).
ИСПЫТАНИЯ
Примесь В. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,125 г испытуемо¬
го образца растворяют в смеси раствор аммиака
концентрированный Р — 96 % спирт Р — мета¬
нол Р (0,5:100:100, об/об/об) и доводят до объема
10.0 мл этой же смесью растворителей.
Раствор сравнения (а). 12,5 мг ФСО ондан¬
сетрона для проверки пригодности системы
ТСХ (содержит примеси А и В) растворяют в смеси
растворителей и доводят до объема 1,0 мл этой же
смесью растворителей.
Раствор сравнения (Ь). 1 мл испытуемого
раствора доводят смесью растворителей до объ¬
ема 100 мл. 4,0 мл полученного раствора доводят
смесью растворителей до объема 10,0 мл.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р254 Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р—метанол Р—этилацетат Р —
метиленхлорид Р (2:40:50:90, об/об/об/об).
Наносимый объем пробы: 20 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Коэффициент удерживания: примесь А — око¬
ло 0,3; примесь В — около 0,4; ондансетрон — око¬
ло 0,6.
Пригодность хроматографической системы:
раствор сравнения (а):
- на хроматограмме обнаруживаются три пол¬
ностью разделенных пятна.
Предельное содержание примесей:
- примесь В (не более 0,4 %): на хроматограм¬
ме испытуемого раствора пятно, соответствующее
примеси В, не должно быть интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор (а). 50,0 мг испытуемо¬
го образца растворяют в подвижной фазе и дово¬
дят до объема 100,0 мл этим же растворителем.
Испытуемый раствор (Ь). 90,0 мг испытуемо¬
го образца растворяют в подвижной фазе и дово¬
дят до объема 100,0 мл этим же растворителем.
10.0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Раствор сравнения (а). 2,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
100.0 мл. 10,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Ондансетрона гидрохлорид дигидрат
781
Раствор сравнения (Ь). 5,0 мг ФСО ондансе¬
трона примеси Е и 5 мг ФСО ондансетрона при¬
меси А растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (с). 5 мг ФСО ондансе¬
трона для проверки пригодности хроматографи¬
ческой системы (содержит примеси С и й) раст¬
воряют в подвижной фазе и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (д). 5,0 мг ФСО ондансе¬
трона примеси О растворяют в подвижной фазе и
доводят до объема 100,0 мл этим же растворите¬
лем. 1,0 мл полученного раствора доводят подвиж¬
ной фазой до 100,0 мл.
Раствор сравнения (е). 90,0 мг ФСО ондансе¬
трона гидрохлорида дигидрата растворяют в под¬
вижной фазе и доводят до объема 100,0 мл этим
же растворителем. 10,0 мл полученного раствора
доводят подвижной фазой до 100,0 мл.
Раствор сравнения (Г). 5,0 мг ФСО ондансе¬
трона примеси Р и 5 мг ФСО ондансетрона при¬
меси О растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (д). К 1,0 мл раствора
сравнения (Ь) прибавляют 1,0 мл раствора срав¬
нения (!) и доводят подвижной фазой до объема
100.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная сферическим силика¬
гелем нитрильным для хроматографии Р с раз¬
мером частиц 5 мкм (удельная площадь 220 м2/г и
размер пор 8 нм);
- подвижная фаза: 20 объемов ацетонитри¬
ла Р1 и 80 объемов раствора 2,8 г/л натрия ди¬
гидрофосфата моногидрата Р, предварительно
доведенного до рН 5,4 раствором 40 г/л натрия
гидроксида Р\
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 216 нм;
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора (а) и растворов сравнения (а), (Ь),
(с), (с1), (!) и (д);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания ондансетрона.
Идентификация пиков примесей: идентифи¬
цируют пики примесей С и О, используя хромато¬
грамму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО ондансетрона для проверки
пригодности хроматографической системы,
идентифицируют пики примесей А и Е, используя
хроматограмму раствора сравнения (Ь); идентифи¬
цируют пики примесей Р и <3, используя хромато¬
грамму раствора сравнения (!).
Относительное удерживание (по отношению
к ондансетрону; время удерживания около 18 мин):
примесь Е — около 0,17; примесь Р — около 0,20
(примеси Е и Р могут элюироваться совместно);
примесь С — около 0,35; примесь О — около 0,45;
примесь А — около 0,80; примесь О — около 0,89
(примеси А и 6 могут элюироваться совместно или
порядок их выхода может быть инвертирован).
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 2,5 между пиками
примесей С и О.
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси С — 0,6):
- примесь С (не более 0,2 %): на хроматограм¬
ме испытуемого раствора (а) площадь пика, соот¬
ветствующего примеси С, не должна превышать
Волновое число (см‘1)
Рисунок #2016.-1. Инфракрасный спектр ФСО ондансетрона гидрохлорида дигидрата
782
Гэсударственная фармакопея Республики Беларусь
площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- примесь О (не более 0,15%): на хромато¬
грамме испытуемого раствора (а) площадь пика,
соответствующего примеси О, не должна превы¬
шать 1,5-кратную площадь основного пика на хро¬
матограмме раствора сравнения (с1);
- сумма примесей А и 6 (не более 0,2 %): на
хроматограмме испытуемого раствора (а) сумма
площадей пиков, соответствующих примесям А и
O, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей Е и Р (не более 0,2 %): на
хроматограмме испытуемого раствора (а) сумма
площадей пиков, соответствующих примесям Е и
P, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (д);
- неспецифицироеанные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пиков
примесей А, С, О, Е, Р и С, не должна превышать
0,5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей: не более 0,4 %;
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,25 площади основного
пика на хроматограмме раствора сравнения (а).
Вода (2.5.12). Не менее 9,0% и не более
10,5 %. Определение проводят из 0,200 г испыту¬
емого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12,2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
ваниям. См. также статью 5.10. Контроль приме-
сей в субстанциях для фармацевтического ис¬
пользования): Н.
А. (3#5)-3-[(Диметиламино)метил]-9-метил-1,2,3,9-
тетрагидро-4Н-карбазол-4-он.
В. 6,6-Метиленбис[(ЗЯ5)-9-метил-3-[(2-метил-1 Н-
имидазол-1 -ил)метил]-1,2,3,9-тетрагидро-4Н-карбазол-
4-он].
С. 9-Метил-1,2,3,9-тетрагидро-4Н-карбазол-4-он.
О. 9-Метил-3-метилен-1,2,3,9-тетрагидро-4Я-
карбазол-4-он.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора (Ь) и раствора сравнения (е).
Содержание С18Н19Ы3О НС1 в процентах рас¬
считывают с учетом содержания ондансетрона
гидрохлорида дигидрата в ФСО ондансетрона ги¬
дрохлорида дигидрата.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
Е. 1Н-Имидазол.
Н3С
ны
N
С. (ЗЯ5)-3-[(1Н-Имидазол-1-ил)метил]-9-метил-
1,2,3,9-тетрагидро-4Н-карбазол-4-он (С-деметил-
ондансетрон).
и энантиомер
Н. (ЗЯ5)-3-[(2-Метил-1Н-имидазол-1-ил)метил]-
1,2,3,9-тетрагидро-4Н-карбазол-4-он (А/-деметил-
ондансетрон).
Осельтамивира фосфат
783
07/2016:2422
ОСЕЛЬТАМИВИРА ФОСФАТ
ОзеПатмп рНозрЬаз
08Е1.ТАММР1 РН08РНАТЕ
С16Н28М204-Н3Р04 М.м. 410,4
[204255-11-8]
ОПРЕДЕЛЕНИЕ
Этил-(3/?,4Я,55)-4-ацетамидо-5-амино-3-(1-
этилпропокси)-циклогекс-1 -ен-1 -карбоксилата фосфат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Легко растворим в воде и в метаноле, практиче¬
ски нерастворим в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО осельтамивира фосфата (без
примеси В).
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО осельтамивира фосфата
(без примеси В) растворяют по отдельности в мета¬
ноле Р и выпаривают до сухих остатков, которые ис¬
пользуют для получения новых спектров.
C. 200 мг испытуемого образца растворяют в
10 мл воды Р. Испытуемый образец дает реакцию (Ь)
на фосфаты (2.3.1).
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
-30,7 до -32,6 (в пересчете на безводное вещество),
измерение проводят при температуре 25 °С. 0,50 г ис¬
пытуемого образца растворяют в воде Р и доводят до
объема 50,0 мл этим же растворителем.
Примесь В. Жидкостная хроматография (2.2.29)
с использованием масс-спектрометрии (2.2.43).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в воде для хроматографии Р и до¬
водят до объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 2,5 мг ФСО осельта¬
мивира примеси В растворяют в 5,0 мл этанола Р
и доводят водой для хроматографии Р до объема
50,0 мл. 2,0 мл полученного раствора доводят водой
для хроматографии Р до объема 100,0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО осельтами¬
вира фосфата (без примеси В) растворяют в раство¬
ре сравнения (а) и доводят до объема 5,0 мл этим же
растворителем.
Условия хроматографирования:
- колонка длиной 0,05 м и внутренним диаме¬
тром 3,0 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: смешивают 10 объемов раст¬
вора 1,54 г/л аммония ацетата Р в воде для хрома¬
тографии Р, 30 объемов ацетонитрила Р1 и 60 объ¬
емов воды для хроматографии Р;
- скорость подвижной фазы: 1,5 мл/мин;
- постколоночное деление потока: используют
деление потока, подходящее для масс-детектора (на¬
пример, 1:3);
- масс-детектор: приводимые параметры ука¬
зываются в качестве примера подходящих рабочих
настроек; если детектор имеет другие рабочие па¬
раметры, их настраивают таким образом, чтобы вы¬
полнить условия пригодности хроматографической
системы:
- ионизация: электрораспылительная иониза¬
ция в режиме положительных ионов;
- регистрируемое отношение т1г. 356,2;
- время пребывания на массе: 580 мс;
- усиление электронного умножителя: 1;
- напряжение фрагментатора: 120 В;
- температура газового потока: 350 °С;
- скорость газа-осушителя: 13 л/мин;
- давление распылителя: 345 кПа;
- напряжение капилляра: 3 кВ;
- объем вводимой пробы: по 1 мкл испытуемого
раствора и раствора сравнения (Ь);
- время хроматографирования: 3 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- относительное стандартное отклонение: не
более 15 % при 6 повторных вводах пробы.
Предельное содержание примесей:
- примесь В (не более 100 ррт): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (Ь).
Примесь Н. Газовая хроматография (2.2.28).
Силилирующий реактив. 1,0 мл хлортриметил-
силана Р, 2,0 мл гексаметилдисилазана Р и 10,0 мл
пиридина безводного Р.
Испытуемый раствор. 15,0 мг испытуемого об¬
разца помещают в контейнер объемом 2 мл и при¬
бавляют 1,0 мл силилирующего реактива. Контейнер
закрывают, встряхивают, нагревают при температуре
60 °С в течение 20 мин и центрифугируют. Осадок от¬
брасывают.
Раствор сравнения. 15,0 мг ФСО осельтамиви¬
ра примеси Н помещают в контейнер объемом 2 мл
и прибавляют 1,0 мл пиридина безводного Р. Контей¬
нер закрывают и встряхивают (раствор А). (Примеча¬
ние: примесь Н является гигроскопичной). 15,0 мг ис¬
пытуемого образца помещают в контейнер объемом
2 мл и прибавляют 1,0 мл силилирующего реактива.
Контейнер закрывают, встряхивают, нагревают при
температуре 60 °С в течение 20 мин, центрифугируют
и отделяют осадок (раствор В). 10,0 мкл раствора А
и 10,0 мкл раствора В помещают в мерную колбу и
доводят пиридином безводным Р до объема 10,0 мл.
784
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и внутренним диаметром 0,32 мм, покрытая слоем
поли(диметил)(дифенил)силоксана Р (толщина слоя
0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1,2 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—2
180
2—11
180 — 250
11—21
250
Блок ввода проб
260
Детектор
260
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению к
осельтамивира фосфату, время удерживания — око¬
ло 10 мин): примесь Н — около 0,5.
Пригодность хроматографической системы:
раствор сравнения:
- относительное стандартное отклонение: не
более 5 % для площадей пиков примеси Н при 6 по¬
вторных вводах пробы.
Предельное содержание примесей:
- примесь Н (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси Н, не должна превышать 1,5-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р1 — ме¬
танол Р2—вода для хроматографии Р (135:245:620,
об/об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО осельтамиви¬
ра примеси А и 5,0 мг ФСО осельтамивира примеси С
растворяют в смеси растворителей и доводят до объ¬
ема 50,0 мл этой же смесью растворителей. 1,0 мл
полученного раствора доводят смесью растворителей
до объема 10,0 мл.
Раствор сравнения (с). 50,0 мг ФСО осельтами¬
вира фосфата Р (без примеси В) растворяют в смеси
растворителей и доводят до объема 50,0 мл этой же
смесью растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р (раз¬
мер частиц 5 мкм);
- температура: 50 °С;
-подвижная фаза: смешивают 135 объемов
ацетонитрила Р1, 245 объемов метанола Р2 и 620
объемов раствора 6,8 г/л калия дигидрофосфата Р
в воде для хроматографии Р, доведенного 1 М рас¬
твором калия гидроксида до рН 6,0;
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 207 нм;
- объем вводимой пробы: по 15 мкл испытуемого
раствора и растворов сравнения (а) и (Ь);
- время хроматографирования: 2-кратное вре¬
мя удерживания осельтамивира фосфата.
Относительное удерживание (по отношению к
осельтамивира фосфату, время удерживания — око¬
ло 17 мин): примесь А—около 0,16; примесь С — око¬
ло 0,17.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,5 между пиками при¬
месей А и С.
Предельное содержание примесей:
- примесь С (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси С, не должна превышать 0,3 площади
соответствующего пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си С, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,7 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 7-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 15 мкл испытуемого
раствора и раствора сравнения (с).
Содержание С16Н28М204-Н3Р04 в процентах рас¬
считывают с учетом содержания осельтамивира
фосфата в ФСО осельтамивира фосфата (без при¬
меси В).
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С, Н.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацев¬
тического использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): А,
О, Е, Р, 6.
Офлоксацин
785
А. (ЗЯ,4Я,55)-5-Ацетамидо-4-амино-3-(1 -этил про-
покси)циклогекс-1 -ен-1 -карбоновая кислота.
В. Этил-(1 Я,2/?,35,4/?,55)-4-ацетамидо-5-амино-
2-азидо-3-(1-этилпропокси)циклогексанкарбоксилат.
С. (3#,4Я,55)-4-Ацетамидо-5-амино-3-(1 -этилпро-
покси)циклогекс-1 -ен-1 -карбоновая кислота.
й. Этил-4-ацетамидо-З-гидроксибензоат.
Е. Метил-(ЗЯ?,4/?,55)-4-ацетамидо-5-амино-3-(1-
этилпропокси)циклогекс-1 -ен-1 -карбоксилат.
Р. Этил-(ЗЯ,4Я,5$)-4-ацетамидо-5-амино-3-(1 -
метилпропокси)циклогекс-1 -ен-1 -карбоксилат.
<3. Этил-(ЗЯ,4Я,55)-5-ацетамидо-4-амино-3-(1-
этилпропокси)циклогекс-1 -ен-1 -карбоксилат.
Н. Трибутилфосфана оксид.
07/2016:1455
ОФЛОКСАЦИН
ОЛохастит
ОР1-ОХАС1Ы
н
и энантиомер
С18Н20™3°4 М.М. 361,4
[82419-36-1]
ОПРЕДЕЛЕНИЕ
(ЗЯ5)-9-Фтор-3-метил-10-(4-метилпиперазин-1 -
ил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-с/е]-1,4-бензо-
ксазин-6-карбоновая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический порошок от бледно-желтого до
ярко-желтого цвета.
Мало растворим в воде, растворим в ледяной
уксусной кислоте, мало растворим или растворим в
метиленхлориде, мало растворим в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО офлоксацина #или спектр, пред¬
ставленный на рисунке #1455.-1#.
ИСПЫТАНИЯ
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. 0,300 г испытуемого образца растворяют в
смеси из 10 объемов метанола Р и 40 объемов ме-
тиленхлорида Р и доводят до объема 10,0 мл этой же
смесью растворителей.
Оптическая плотность (2.2.25). Не более 0,25
при 440 нм. 0,5 г испытуемого образца растворяют в
0,1 М растворе кислоты хлористоводородной и до¬
водят до объема 100,0 мл этим же растворителем.
Примесь А. Не более 0,2 %. Тонкослойная хро¬
матография (2.2.27).
Смесь растворителей. Метанол Р — метилен-
хлорид Р (10:40, об/об).
Испытуемый раствор. 0,250 г испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 5,0 мл этим же растворителем.
Раствор сравнения. 10,0 мг ФСО офлоксацина
примеси А растворяют в смеси растворителей и дово¬
дят до объема 100,0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля СР254 Р ((2—10) мкм).
786
Гэсударственная фармакопея Республики Беларусь
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — этилацетат Р (10:10:20, об/об/об).
Объем наносимой пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- примесь А: на хроматограмме испытуемого
раствора пятно примеси А должно быть не интенсив¬
нее соответствующего пятна на хроматограмме раст¬
вора сравнения.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Смесь растворителей. Ацетонитрил Р —
вода Р (10:60, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
50,0 мл. 1,0 мл полученного раствора доводят смесью
растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг ФСО офлоксацина
примеси Е растворяют в смеси растворителей и дово¬
дят до объема 100,0 мл этой же смесью растворите¬
лей. К 10 мл полученного раствора прибавляют 5 мл
испытуемого раствора и доводят смесью растворите¬
лей до объема 50,0 мл. 1,0 мл полученного раствора
доводят смесью растворителей до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным для хроматографии Р с размером частиц
5 мкм;
- температура: 45 °С;
- подвижная фаза: 4,0 г аммония ацетата Р
и 7,0 г натрия перхлората Р растворяют в 1300 мл
воды Р, доводят до рН 2,2 кислотой фосфорной Р и
прибавляют 240 мл ацетонитрила Р\
- скорость подвижной фазы: подбирают таким
образом, чтобы время удерживания офлоксацина со¬
ставляло около 20 мин;
- спектрофотометрический детектор, длина
волны 294 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания офлоксацина.
Идентификация пиков примесей: идентифици¬
руют пик примеси Е, используя хроматограмму раст¬
вора сравнения (Ь).
Относительное удерживание (по отношению к
офлоксацину, время удерживания — около 20 мин):
примесь В — около 0,3; примесь С — около 0,5; при¬
месь О — около 0,7; примесь Е — около 0,9; примесь
Р — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси Е и офлоксацина.
Предельное содержание примесей:
- примеси В, С, О, Е, Р (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, С, О, Е и Р, не должны
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В, С, О, Е, и Р, не должна превышать 0,5 площа¬
ди основного пика на хроматограмме раствора срав¬
нения (а);
Паклитаксел
787
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
-неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,2 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в
100 мл кислоты уксусной безводной Р и титруют
0,1 М раствором кислоты хлорной потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 36,14 мг С18Н20РЫ3О4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
А. (ЗЯ5)-9,10-Дифтор-3-метил-7-оксо-2,3-дигидро-
7Н-пиридо[1,2,3-с/е]-1,4-бензоксазин-6-карбоновая кис¬
лота (ФПК).
В. (3/?5)-9-Фтор-3-метил-10-(4-метилпиперазин-1 -
ил)-2,3-дигидро-7Н-пиридо[1,2,3-(Уе]-1,4-бензоксазин-
7-он.
и энантиомер
со2н
С. (З/^-З-МетилИ 0-(4-метилпиперазин-1 -ил)-7-
оксо-2,3-дигидро-7Н-пиридо[1,2,3-с/е]-1,4-бензоксазин-
6-карбоновая кислота.
й. (3#5)-10-Фтор-3-метил-9-(4-метилпиперазин-1-
ил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-</е]-1,4-бензо-
ксазин-6-карбоновая кислота.
Е. (3/?5)-9-Фтор-3-метил-7-оксо-10-(пиперазин-1-
ил)-2,3-дигидро-7/-/-пиридо[1,2,3-с/е]-1,4-бензоксазин-
6-карбоновая кислота.
Р. 4-[(ЗЯ5)-6-Карбокси-9-фтор-3-метил-7-оксо-2,3-
дигидро-7Н-пиридо[1,2,3-сУе]-1,4-бензоксазин-10-ил]-1-
метилпиперазина 1-оксид.
07/2016:1794
ПАКЛИТАКСЕЛ
Рас1Нахе1ит
РАСЫТАХЕ1_
ОПРЕДЕЛЕНИЕ
5р,20-Эпокси-1,7р-дигидрокси-9-оксотакс-11 -ен-
2а,4,10р, 13а-тетраил-4,10-диацетат-2-бензоат-13-
[(2Я?,35)-3-(бензоиламино)-2-гидрокси-3-фенилпро-
паноат].
Паклитаксел выделяют из природных источников
или получают методом ферментации или полусинте-
тическим способом.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
788
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, растворим в
метаноле, легко растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО паклитаксела.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то по 10 мг
испытуемого образца и ФСО паклитаксела раство¬
ряют по отдельности в 0,4 мл метиленхлорида Р и
выпаривают досуха. Остатки используют для получе¬
ния новых спектров.
ИСПЫТАНИЯ
Раствор 5. 0,1 г испытуемого образца раство¬
ряют в 10 мл метанола Р.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
-49,0 до -55,0 (в пересчете на безводное вещество).
0,250 г испытуемого образца растворяют в метано¬
ле Р и доводят до объема 25,0 мл этим же раствори¬
телем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
А. Паклитаксел, выделенный из природных ис¬
точников или полученный методом ферментации.
Испытуемый раствор (а). 20,0 мг испытуемого
образца растворяют в ацетонитриле Р1 и доводят
до объема 10,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят ацетонитрилом Р1 до объема
20.0 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора (а) доводят ацетонитрилом Р1 до объема
10.0 мл. 1,0 мл полученного раствора доводят ацето¬
нитрилом Р1 до объема 100,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО паклитаксе¬
ла растворяют в ацетонитриле Р1 и доводят до объ¬
ема 5,0 мл этим же растворителем. 2,0 мл полученно¬
го раствора доводят ацетонитрилом Р1 до объема
20.0 мл.
Раствор сравнения (с). 2,0 мг ФСО паклитаксе¬
ла примеси С растворяют в ацетонитриле Р1 и дово¬
дят до объема 20,0 мл этим же растворителем.
Раствор сравнения (ф. 1,0 мл раствора срав¬
нения (с) доводят ацетонитрилом Р1 до объема
50.0 мл.
Раствор сравнения (е). К 1 мл раствора сравне¬
ния (Ь) прибавляют 1 мл раствора сравнения (с).
Раствор сравнения (I). 5 мг ФСО природного
паклитаксела для идентификации пиков (содержит
примеси А, В, С, й, Е, Р, Н, О, Р, О, Р) растворяют в
ацетонитриле Р1 и доводят до объема 5 мл этим же
растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем диизопропил-
цианопропилсилильным для хроматографии Р (раз¬
мер частиц 5 мкм) с удельной площадью поверхноз~*
180 м2/г и диаметром пор 8 нм;
- температура: (20±1) °С;
- подвижная фаза:
- подвижная фаза А: метанол Р — вода ~
(200:800, об/об);
- подвижная фаза В: метанол Р — ацетони¬
трил для хроматографии Р (200:800, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—60
85 —* 56
15—>44
60—61
56 —* 85
44 —> 15
61—75
85
15
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 227 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (а), (б), (е) и (Т).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, О, Е, Р, Н, О, Р, О и Р
используя хроматограмму раствора сравнения (1) и
хроматограмму, прилагаемую к ФСО природного па¬
клитаксела для идентификации пиков.
Относительное удерживание (по отношению к
паклитакселу, время удерживания — около 50 мин):
примеси А и В — около 0,90; примесь Р — около 0,93;
примесь И — около 0,96; примеси О и Р — около 1,02:
примесь С — около 1,05; примесь О — около 1,07;
примеси О и Е — около 1,15; примесь Р — около 1,20.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 3,5 между пиками пакли¬
таксела и примеси С.
Предельное содержание примесей:
- сумма примесей Е и О (не более 0,5 %): на
хроматограмме испытуемого раствора (а) сумма пло¬
щадей пиков, соответствующих примесям Е и О, не
должна превышать 5-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- примесь Р (не более 0,5 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси К, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей А и В (не более 0,4 %): на хро¬
матограмме испытуемого раствора (а) сумма площа¬
дей пиков, соответствующих примесям А и В, не долж¬
на превышать 4-кратную площадь основного пика на
хроматограмме раствора сравнения (а);
- примесь С (не более 0,3 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси С, не должна превышать 3-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (б);
- примесь О (не более 0,2 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси О, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей Р и О (не более 0,2 %): на
хроматограмме испытуемого раствора (а) сумма пло¬
щадей пиков, соответствующих примесям Р и О, не
должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- примесь Р (не более 0,1 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответствую-
Паклитаксел
789
щего примеси Р, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с1);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е, Р, О, Р, О и Р, не должна пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- сумма примесей (не более 1,5 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
15-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
В. Паклитаксел, полученный полусинтетическим
способом.
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в ацетонитриле Р1 и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят ацетонитрилом Р1 до объема 10,0 мл.
1,0 мл полученного раствора доводят ацетонитри¬
лом Р1 до объема 100,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО паклитаксе-
ла растворяют в ацетонитриле Р1 и доводят до объ¬
ема 5,0 мл этим же растворителем.
Раствор сравнения (с). 5 мг ФСО полусинте-
тического паклитаксела для идентификации пиков
(содержит примеси А, О, I и I) растворяют в ацето¬
нитриле Р1 и доводят до объема 5 мл этим же раст¬
ворителем.
Раствор сравнения (д). Содержимое контейнера
ФСО полусинтетического паклитаксела для про¬
верки пригодности хроматографической системы
(содержит примеси Е, Н и N1) растворяют в 1 мл аце¬
тонитрила Р1.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным эндкепированным для хроматографии Р
(размер частиц 3 мкм) с удельной площадью поверх¬
ности 300 м2/г и диаметром пор 12 нм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил для хрома¬
тографии Р— вода Р (400:600, об/об);
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—20
100
0
20—60
100 —> 10
0 90
60—62
о
о
о
90 —► 0
62—70
100
0
- скорость подвижной фазы: 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 227 нм;
- объем вводимой пробы: по 15 мкл испытуемого
раствора и растворов сравнения (а), (с) и (б).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, (3, I и Ц используя хромато¬
грамму раствора сравнения (с) и хроматограмму, при¬
лагаемую к ФСО полусинтетического паклитаксела
для идентификации пиков; идентифицируют пики
примесей Е, Н и N. используя хроматограмму раст¬
вора сравнения (б) и хроматограмму, прилагаемую
к ФСО полусинтетического паклитаксела для про¬
верки пригодности хроматографической системы.
Относительное удерживание (по отношению к
паклитакселу, время удерживания — около 23 мин):
примесь N — около 0,2; примесь О — около 0,5; при¬
месь А— около 0,8; примеси М, б, Н — около 0,9; при¬
месь Е — около 1,3; примесь I — около 1,4; примесь
1- — около 1,5; примесь К — около 2,2.
Пригодность хроматографической системы.
раствор сравнения (б):
- разрешение: не менее 1,5 между пиками при¬
меси Н и паклитаксела.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси N — 1,29):
- примесь А (не более 0,7 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 7-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь /_ (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси 1_, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
-- примеси Е, I (не более 0,4 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Е и I, не должны превышать
4-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- сумма примесей Н, б и М (не более 0,4 %): на
хроматограмме испытуемого раствора сумма площа¬
дей пиков, соответствующих примесям Н, б и М, не
должна превышать 4-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
-примеси О, К, N (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С, К, N. не должны превышать
2- кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, Е, О, Н, I, б, К, 1_, М и N. не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 1,2 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
12-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл испытуемого
раствора и 10 мл эталонного раствора свинца (1 ррт
РЬ), полученного разведением эталонного раствора
790
Гэсударственная фармакопея Республики Беларусь
свинца (100 ррт РЬ) Р метанолом Р. К12 мл каждого
раствора прибавляют 2 мл буферного раствора рН
3,5 Р, перемешивают и прибавляют 1,2 мл тиоацета-
мидного реактива Р. Субстанция выпадает в осадок.
Доводят метанолом Р до объема 40 мл — субстан¬
ция полностью растворяется. Полученный раствор
фильтруют через мембранный фильтр с размером
пор 0,45 мкм. Сравнивают окраски мембранных филь¬
тров, полученные в опытах с разными растворами.
Коричневато-черная окраска мембранного фильтра,
полученная в опыте с испытуемым раствором, долж¬
на быть не интенсивнее окраски мембранного филь¬
тра, полученной в опыте с раствором сравнения.
Вода (2.5.32). Не более 3,0 %. Определение про¬
водят из 0,050 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
O, 4 МЕ/мг.
Микробиологическая чистота
ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
A. Паклитаксел, выделенный из природных ис¬
точников или полученный методом ферментации.
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси. А», со следу¬
ющими изменениями.
Объем вводимой пробы: по 10 мкл испытуемого
раствора (Ь) и раствора сравнения (Ь).
Содержание С47Н511Ч014 в процентах рассчитыва¬
ют с учетом содержания паклитаксела в ФСО пакли-
таксела.
B. Паклитаксел, полученный полусинтетическим
способом.
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси. В», со следу¬
ющими изменениями.
Объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С47Н51М014 в процентах рассчитыва¬
ют с учетом содержания паклитаксела в ФСО пакли¬
таксела.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
МАРКИРОВКА
Указывают способ получения:
- выделен из природных источников;
- получен методом ферментации;
- получен полусинтетическим способом.
ПРИМЕСИ
Для испытания «Сопутствующие примеси. А»
Специфицированные примеси: А, В, С, О, Е, Р, О,
P, О, я.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Н.
Для испытания «Сопутствующие примеси. В»
Специфицированные примеси: А, Е, 6, Н, I, 3, К,
Ц М, N.
A. К1 = Тд, К2 = Ас, КЗ = Вг, К4 = Кб = Н, К5 = ОН:
2-0-Дебензоил-2-0-тиглоилпаклитаксел.
B. К1 = Вг, К2 = Ас, КЗ = Тд, К4 = Кб = Н, К5 = ОН:
А/-Дебензоил-Л/-тиглоилпаклитаксел (цефаломаннин).
C. К1 = Вг, К2 = Ас, КЗ = Нх, К4 = Кб = Н, К5 = ОН:
/У-Дебензоил-Л/-гексаноилпаклитаксел (паклитаксел С).
Э. К1 = Вг, К2 = Ас, КЗ = Тд, К4 = К5 = Н, Кб = ОН:
Л/-Дебензоил-А/-тиглоил-7-эл1/-паклитаксел (7-эпи-це-
фаломаннин).
Е. К1 = КЗ = Вг, К2 = Ас, К4 = К5 = Н, Кб = ОН:
7-эпи- Паклитаксел.
Р. К1 = Вг, К2 = Ас, КЗ = Нх, К4 = СН3, К5 = ОН,
Кб = Н: Л/-Дебензоил-Л/-гексаноил-Л/-метил паклитаксел
(А/-метилпаклитаксел С).
0. К1 = КЗ = Вг, К2 = К4 = Кб = Н, К5 = ОН:
10-О-Дезацетилпаклитаксел.
H. К1 = КЗ = Вг, К2 = К4 = К5 = Н, Кб = ОН:
10-О-Дезацетил-7-эл(;-паклитаксел.
1. К1 = КЗ = Вг, К2 = Ра, К4 = Кб = Н, К5 = ОН:
Ю-0-[(2К,35)-3-(Бензоиламино)-2-гидрокси-3-
фенилпропаноил]-Ю-0-дезацетилпаклитаксел.
и. К1 = КЗ = Вг, К2 = Аа, К4 = Кб = Н, К5 = ОН:
10-О-Дезацетил-10-О-(3-оксобутаноил)паклитаксел.
К. К1 = КЗ = Вг, К2 = Ас, К4 = Кб = Н,
К5 = 0-8|(С2Н5)3: 7-0-(Триэтилсиланил)паклитаксел.
I. К1 = КЗ = Вг, К2 = Ас, К4 = Кб = Н, К5 = О-СО-
СН3: 7-О-Ацетилпаклитаксел.
O. К1 = Вг, К2 = Ас, КЗ = Сп, К4 = Кб = Н, К5 = ОН:
Л/-Циннамоил-Л/-дебензоилпаклитаксел.
P. К1 = Вг, К2 = Ас, КЗ = Ва, К4 = Кб = Н, К5 = ОН:
Л/-Дебензоил-Л/-(фенилацетил)паклитаксел.
О. К1 = Вг, К2 = Ас, КЗ = Не, К4 = Кб = Н, К5 = ОН:
Л/-Дебензоил-Л/-[(ЗЕ)-гекс-3-еноил]паклитаксел.
К. К1 = Вг, К2 = Ас, КЗ = МЬ, К4 = Кб = Н, К5 = ОН:
А/-Дебензоил-А/-[(25)-2-метилбутаноил]паклитаксел.
Панкреатина порошок
791
М. 1,2а,4,7р-Дигидрокси-9-оксотакс-11-ен-
5р, 10р, 1За,20-тетраил-5,10-диацетат-20-бензоат-13-
[(2Я,35)-3-(бензоиламино)-2-гидрокси-3-фенилпро-
паноат].
N. 13-0-Де[(2/?,35)-3-(бензоиламино)-2-гидрокси-
3-фенилпропаноил]паклитаксел (баккатин III).
07/2016:0350
ПАНКРЕАТИНА ПОРОШОК
Рапсгеаз риМз
РАЫСРЕА5 РОМО ЕР
ОПРЕДЕЛЕНИЕ
Панкреатина порошок получают из свежих или
замороженных поджелудочных желез млекопитаю¬
щих. Порошок содержит различные ферменты, обла¬
дающие протеолитической, липолитической и амило¬
литической активностями.
1 мг панкреатина порошка содержит не менее
1,0 РН.Еиг.11. общей протеолитической активности, не
менее 15 РН.ЕиШ. липолитической активности и не
менее 12 РЬ.ЕиШ. амилолитической активности.
ПРОИЗВОДСТВО
Животные, из поджелудочных желез которых вы¬
деляют панкреатина порошок, должны выдерживать
требования к здоровью животных, пригодных для по¬
требления человеком.
ОПИСАНИЕ (СВОЙСТВА)
Слегка коричневый аморфный порошок.
Частично растворим в воде, практически нераст¬
ворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,5 г испытуемого образца растирают с 10 мл
воды Р и доводят до рН 8 с помощью 0,1 М раст¬
вора натрия гидроксида, используя в качестве ин¬
дикатора 0,1 мл раствора крезолового красного Р.
Суспензию делят на 2 равные части (суспензию
(а) и суспензию (Ь)). Суспензию (а) кипятят. К каж¬
дой суспензии прибавляют по 10 мг фибрина конго
красного Р, нагревают до температуры (38—40) °С
и выдерживают при этой температуре в течение 1 ч.
Суспензия (а) бесцветная или слегка розовая, су¬
спензия (Ь) имеет заметно более красное окраши¬
вание.
В. 0,25 г испытуемого образца растирают с
10 мл воды Р и доводят до рН 8 с помощью 0,1 М
раствора натрия гидроксида, используя в качестве
индикатора 0,1 мл раствора крезолового красно¬
го Р. Суспензию делят на 2 равные части (суспен¬
зию (а) и суспензию (Ь)). Суспензию (а) кипятят. 0,1 г
крахмала растворимого Р растворяют в 100мл
кипящей воды Р, кипятят в течение 2 минут, охлаж¬
дают и доводят до объема 150 мл водой Р. К 75 мл
раствора крахмала прибавляют суспензию (а) и к
оставшимся 75 мл прибавляют суспензию (Ь). Каж¬
дую смесь нагревают до температуры (38—40) °С
и выдерживают при этой температуре в течение
5 мин.
К 1 мл каждой смеси прибавляют по 10 мл
раствора йода Р2. Смесь, полученная из суспензии
(а), окрашивается в интенсивный сине-фиолетовый
цвет; смесь, полученная из суспензии (Ь), имеет
окраску раствора йода.
ИСПЫТАНИЯ
Содержание жира. Не более 5,0 %. 1,0 г испы¬
туемого образца обрабатывают петролейным эфи¬
ром Р1 в течение 3 ч в экстракционном аппарате.
Растворитель выпаривают и остаток сушат при темпе¬
ратуре от 100 °С до 105 °С в течение 2 ч. Масса остат¬
ка не должна превышать 50 мг.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 0,50 г испытуемого образца сушат при
температуре 60 °С в вакууме при давлении не более
670 Па в течение 4 ч.
Микробиологическая чистота
ОКА: критерий приемлемости 104 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсНепсЫа соН: отсутствие (2.6.13).
За1топе11а: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Общая протеолитическая активность. Об¬
щую протеолитическую активность порошка пан¬
креатина определяют путем сравнения количества
пептидов, неосаждаемых раствором 50 г/л кисло¬
ты трихлоруксуной Р, высвобождаемых в течение
1 минуты из субстрата казеинового раствора, с ко¬
личеством таких же пептидов, высвобождаемых
БСП панкреатина порошка (протеазы) из точно
такого же субстрата в точно таких же условиях.
Раствор казеина. Количество БСП казеина,
эквивалентное 1,25 г сухого вещества, суспенди¬
руют в 5 мл воды Р, прибавляют 10 мл 0,1 М раст¬
вора натрия гидроксида и перемешивают в тече¬
ние 1 мин. (До испытания определяют содержание
воды в БСП казеина, нагревая его при температуре
60 °С в вакууме в течение 4 ч). Прибавляют 60 мл
воды Р и перемешивают на магнитной мешалке до
получения практически прозрачного раствора. Раст¬
вор доводят до рН 8,0 с помощью 0,1 М раствора
натрия гидроксида или 0,1 М раствора кислоты
хлористоводородной. Раствор доводят водой Р до
объема 100,0 мл. Раствор используют в день при¬
готовления.
Раствор энтерокиназы. 50 мг БСП энтероки¬
назы растворяют в 0,02 М растворе кальция хлори¬
да Р и доводят до объема 50,0 мл этим же раство¬
рителем. Раствор используют в день приготовления.
792
Гэсударственная фармакопея Республики Беларусь
Таблица 0350.-1
Пробирки
««
«1
«2ь
5,ь
Т
В
Буферный раствор
2
2
1
1
1
1
3
Суспензия сравнения
1
1
2
2
3
3
Испытуемая суспензия
2
2
Раствор кислоты трихлоруксусной
5
5
5
5
5
Перемешивание
+
+
+
+
+
Водяная баня с температурой 35 °С
+
+
+
+
+
+
+
+
+
Раствор казеина
2
2
2
2
2
Перемешивание
+
+
+
+
+
Раствор казеина
2
2
2
2
Перемешивание
+
+
+
+
Водяная баня с температурой 35 °С, 30 мин
+
+
+
+
+
+
+
+
+
Раствор кислоты трихлоруксусной
5
5
5
5
Перемешивание
+
+
+
+
Комнатная температура, 20 мин
+
+
+
+
+
+
+
+
+
Фильтрование
+
+
+
+
+
+
+
+
+
Во избежание абсорбции воды, образуемой при
конденсации, перед вскрытием контейнеров испы¬
туемый образец и образец сравнения выдерживают
до достижения комнатной температуры.
Приготовление и разведение испытуемой су¬
спензии и суспензии сравнения проводят при темпе¬
ратуре от 0 °С до 4 °С.
Испытуемая суспензия. 0,100 г испытуемого об¬
разца растирают в течение 5 мин, постепенно при¬
бавляя 25 мл 0,02 М раствора кальция хлорида Р.
Количественно переносят в мерную колбу и доводят
0,02 М раствором кальция хлорида Р до объема
100.0 мл. К 10,0 мл полученной суспензии прибавля¬
ют 10,0 мл раствора энтерокиназы и нагревают на
водяной бане при температуре (35±0,5) °С в течение
15 мин. Охлаждают и разводят боратным буферным
раствором рН 7,5 Р при температуре (5±3) °С до кон¬
центрации около 0,065 РН.Еиг.11. общей протеолити¬
ческой активности в 1 мл, рассчитанной на основании
заявленной активности.
Суспензия сравнения. Готовят суспензию БСП
панкреатина порошка (протеазы), как описано
для испытуемой суспензии, но без прибавления эн¬
терокиназы, чтобы получить концентрацию около
0,065 РН.Еиг.11. в 1 мл, рассчитанную на основании за¬
явленной активности.
Маркируют пробирки (по две каждой): Т, Ть, 5,,
®1Ь» ^2» ^2Ь' ^3> ^3Ь’ маркируют пробирку В.
В пробирки прибавляют боратный буферный
раствор рН 7,5 Р, как указано ниже:
В: 3,0 мл;
и 51Ь: по 2,0 мл;
52, 32Ь, Т и Ть: по 1,0 мл.
В пробирки прибавляют раствор сравнения, как
указано ниже:
и $1Й: по 1,0 мл;
52 и 52Ь: по 2,0 мл;
53 и 5ЗЬ: по 3,0 мл.
В пробирки Т и Ть прибавляют по 2,0 мл испыту¬
емой суспензии.
В пробирки В, 51Ь, 52Ь, 5зь и Ть прибавляют по
5.0 мл раствора 50 г/л кислоты трихлоруксусной Р и
встряхивают.
Пробирки и колбу с раствором казеина поме¬
щают в водяную баню с температурой (35±0,5) °С. В
каждую пробирку помещают стеклянную палочку. По¬
сле уравновешивания температуры в пробирки В, 51Ь,
52Ь, 5зй и Ть прибавляют по 2,0 мл раствора казеина.
Перемешивают. Начиная с момента времени «ноль»
в пробирки 51? 52> $3и Т последовательно с интерва¬
лом в 30 с прибавляют по 2,0 мл раствора казеина.
После прибавления раствора содержимое пробирки
сразу перемешивают. Ровно через 30 мин после при¬
бавления раствора казеина, с учетом временных про¬
межутков между внесением субстрата, в пробирки
52, $3и 7 прибавляют по 5,0 мл раствора 50 г/л кис¬
лоты трихлоруксусной Р и перемешивают. Пробирки
извлекают из водяной бани и выдерживают при ком¬
натной температуре в течение 20 мин.
Содержимое каждой пробирки фильтруют дваж¬
ды через одинаковую фильтровальную бумагу, пред¬
варительно промытую раствором 50 г/л кислоты
трихлоруксусной Р, затем водой Р и высушенную.
Используемая фильтровальная бумага должна
выдерживать следующее испытание: через круг белой
фильтровальной бумаги диаметром 7 см фильтруют
5 мл раствора 50 г/л кислоты трихлоруксусной Р\
оптическая плотность (2.2.25) полученного фильтра¬
та, измеренная при длине волны 275 нм с использова¬
нием нефильтрованного раствора кислоты трихло¬
руксусной Р в качестве компенсационного раствора,
должна быть менее 0,04.
Схематическое представление выше описанных
операций представлено в таблице 0350.-1.
Измеряют оптическую плотность (2.2.25) филь¬
тратов при длине волны 275 нм, используя в качестве
компенсационного раствора фильтрат, полученный из
пробирки В.
Корректируют средние значения оптической
плотности фильтратов, полученных из пробирок 5^ 52
и 53, путем вычитания средних значений оптической
плотности фильтратов из соответствующих пробирок
$1Й, 52Ь и 5зь. Строят калибровочный график зависимо¬
сти откорректированных значений оптической плотно¬
сти от используемого объема суспензии сравнения.
Определяют активность испытуемого образца,
используя откорректированную оптическую плотность
испытуемой суспензии (Г-ГЛ) и калибровочный гра¬
фик, учитывая коэффициенты разведения.
Результаты испытания являются недостоверны¬
ми, если откорректированные значения оптической
плотности находятся вне диапазона от 0,15 до 0,60.
Липолитическая активность. Липолитическую
активность определяют сравнением скорости, с ко-
<11
Панкреатина порошок
793
торой суспензия порошка панкреатина гидролизует
субстрат эмульсии оливкового масла, со скоростью, с
которой суспензия БСП панкреатина порошка (липа¬
зы) гидролизует этот же субстрат в этих же условиях.
Испытание проводят в атмосфере азота.
Исходная эмульсия оливкового масла. В лабо¬
раторный стакан вместимостью 800 мл, диаметром
9 см помещают 40 мл оливкового масла Р, 330 мл
раствора гуммиарабика Р и 30 мл воды Р. На дно
лабораторного стакана помещают перемешиваю¬
щий элемент. Стакан помещают в сосуд, содержа¬
щий 96 % спирт Р и достаточное количество льда
в качестве охлаждающей смеси. Эмульгируют, ис¬
пользуя мешалку со средней скоростью вращения
(1000—2000) об/мин. Охлаждают до температуры
(5—10) °С. Увеличивают скорость вращения мешал¬
ки до 8000 об/мин. Перемешивают в течение 30 мин,
поддерживая температуру ниже 25 °С, постоян¬
но прибавляя в охлаждающую смесь колотый лед.
(Также подходит смесь кальция хлорида и колотого
льда). Исходную эмульсию хранят в холодильнике и
используют в течение 14 суток. Эмульсия не должна
расслаиваться на 2 отдельных слоя. Под микроско¬
пом контролируют диаметр частиц эмульсии. Не ме¬
нее 90 % частиц должны иметь диаметр менее 3 мкм,
а частицы с диаметром более 10 мкм должны отсут¬
ствовать. Перед приготовлением субстрата эмульсии
эмульсию тщательно встряхивают.
Эмульсия оливкового масла. Для 10 опреде¬
лений смешивают в указанном порядке следующие
растворы: 100 мл исходной эмульсии, 80 мл раст¬
вора трис(гидроксиметил)аминометана Р1, 20 мл
свежеприготовленного раствора 80 г/л БСП натрия
таурохолата и 95 мл воды Р. Используют в день при¬
готовления.
Прибор. Используют реакционный сосуд вмести¬
мостью около 50 мл, снабженный:
- устройством, поддерживающим температуру
при (37±0,5) °С;
- магнитной мешалкой;
- крышкой с отверстиями для введения электро¬
дов, кончика бюретки, трубок для доступа азота и вве¬
дения реактивов.
Можно использовать прибор для автоматическо¬
го или ручного титрования. В последнем случае бю¬
ретка должны иметь цену деления, равную 0,005 мл,
а рН-метр должен иметь широкий диапазон шкалы и
быть снабженным стеклянным-каломельным или сте-
клянным-хлорсеребряным электродом. После каж¬
дого испытания реакционный сосуд освобождают от
содержимого отсосом и промывают несколько раз во¬
дой Р, каждый раз удаляя промывные воды отсосом.
Во избежание абсорбции воды, образуемой при
конденсации, перед вскрытием контейнеров испы¬
туемый образец и образец сравнения выдерживают
до достижения комнатной температуры.
Приготовление и разведение испытуемой су¬
спензии и суспензии сравнения проводят при темпе¬
ратуре от 0 °С до 4 °С.
Испытуемая суспензия. В небольшой ступке, ох¬
лажденной до температуры (0—4) °С, тщательно рас¬
тирают количество испытуемого образца, эквивалент¬
ное около 2500 РЬ.Еиг.11. липолитической активности,
с 1 мл охлажденного малеатного буферного раст¬
вора рН 7,0 Р (растворитель липазы) до получения
очень мелкой суспензии. Суспензию разводят мале-
атным буферным раствором рН 7,0 Р, количествен¬
но переносят в мерную колбу и доводят буферным
раствором до объема 100,0 мл. Во время титрования
колбу, содержащую испытуемую суспензию, выдер¬
живают в ледяной бане.
Суспензия сравнения. Готовят суспензию БСП
панкреатина порошка (липазы), как описано для ис¬
пытуемой суспензии, используя количество, эквива¬
лентное около 2500 РИ.Еиг.1).
Сразу после приготовления испытуемой суспен¬
зии и суспензии сравнения проводят титрование.
29,5 мл эмульсии оливкового масла помещают в ре¬
акционный сосуд, уравновешенный при температуре
(37±0,5) °С. К сосуду присоединяют электроды, ме¬
шалку и бюретку (кончик бюретки погружают в эмуль¬
сию оливкового масла). Крышку опускают на место
и включают установку. Осторожно при перемешива¬
нии прибавляют 0,1 М раствор натрия гидроксида
и доводят до рН 9,2. Используя градуированную пи¬
петку, переносят около 0,5 мл предварительно гомо¬
генизированной суспензии сравнения, включают се¬
кундомер и непрерывно прибавляют 0,1 М раствор
натрия гидроксида для поддержания рН 9,0. Ровно
через 1 мин отмечают объем израсходованного 0,1 М
раствора натрия гидроксида. Измерение повторяют
еще 4 раза. Данные за первую минуту отбрасывают и
определяют среднее значение 4 последующих опре¬
делений (5^. Проводят еще 2 определения (52 и 53).
Рассчитывают среднее значение величин 5^ 52 и 53.
Средний объем израсходованного 0,1 М раствора на¬
трия гидроксида должен составлять около 0,12 мл/
мин в диапазоне от 0,08 мл до 0,16 мл.
Аналогично проводят 3 определения для испыту¬
емой суспензии (ТГТ2 и Т3). Если количество израсхо¬
дованного на титрование 0,1 М раствора натрия ги¬
дроксида выходит за пределы (0,08—0,16) мл/мин, то
количественное определение повторяют с более под¬
ходящим объемом испытуемой суспензии, который
находится в пределах от 0,4 мл до 0,6 мл. В против¬
ном случае подбирают количество испытуемого об¬
разца, обеспечивающее соблюдение условий испыта¬
ния. Рассчитывают среднее значение по полученным
данным 7^ Т2 и Т3. Рассчитывают активность в едини¬
цах Европейской Фармакопеи в 1 мг (РЬ.Еиг.11./мг) по
следующей формуле:
Пт'.А,
пл-т
где:
п — средний объем 0,1 М раствора натрия ги¬
дроксида, израсходованного за 1 мин в течение титро¬
вания испытуемой суспензии, мл;
л1 — средний объем 0,1 М раствора натрия ги¬
дроксида, израсходованного за 1 мин во время титро¬
вания суспензии сравнения, мл;
т — масса навески испытуемого образца, мг;
ш1 — масса навески стандартного препарата, мг;
А — активность БСП панкреатина порошка (ли¬
пазы), Рй.Еиг.Шмг.
Амилолитическая активность. Амилолитиче¬
скую активность определяют сравнением скорости, с
которой суспензия порошка панкреатина гидролизует
субстрат раствора крахмала, со скоростью, с которой
суспензия БСП панкреатина порошка (амилазы) ги¬
дролизует тот же субстрат в тех же условиях.
Раствор крахмала. К количеству БСП крахмала,
эквивалентному 2,0 г сухого вещества, прибавляют
10 мл воды Р и перемешивают. (До испытания опре-
794
Гэсударственная фармакопея Республики Беларусь
деляют содержание воды в БСП крахмала, нагревая
его при температуре 120 °С в течение 4 ч). Полу¬
ченную суспензию при постоянном перемешивании
прибавляют к 160 мл кипящей воды Р. Контейнер
несколько раз подряд промывают по 10 мл водой Р
и прибавляют промывные воды к горячему раствору
крахмала. Нагревают до кипения, постоянно переме¬
шивая. Охлаждают до комнатной температуры и до¬
водят водой Р до объема 200 мл. Раствор используют
в день приготовления.
Во избежание абсорбции воды, образуемой при
конденсации, перед вскрытием контейнеров испы¬
туемый образец и образец сравнения выдерживают
до достижения комнатной температуры.
Приготовление и разведения испытуемой су¬
спензии и суспензии сравнения проводят при темпе¬
ратуре от 0 °С до 4 °С.
Испытуемая суспензия. Количество испытуе¬
мого образца, эквивалентное около 1500 РН.ЕиШ.
амилолитической активности, растирают с 60 мл фос¬
фатного буферного раствора рН 6,8 Р1 в течение
15 мин. Количественно переносят в мерную колбу
и доводят фосфатным буферным раствором рН
6.8 Р1 до объема 100,0 мл.
Суспензия сравнения. Готовят суспензию БСП
панкреатина порошка (амилазы), как описано для ис¬
пытуемой суспензии, используя количество, эквива¬
лентное около 1500 РИ.Еиг.11.
В пробирку с притертой пробкой длиной 200 мм
и диаметром 22 мм помещают 25,0 мл раствора крах¬
мала, 10,0 мл фосфатного буферного раствора рН
6.8 Р1 и 1,0 мл раствора 11,7 г/л натрия хлорида Р.
Пробирку закрывают, встряхивают, помещают в водя¬
ную баню при температуре (25,0±0,1) °С. Когда темпе¬
ратура уравновесится, прибавляют 1,0 мл испытуемой
суспензии и включают секундомер. Перемешивают
содержимое пробирки и помещают пробирку в водя¬
ную баню. Ровно через 10 мин прибавляют 2 мл 1 М
раствора кислоты хлористоводородной Р. Смесь ко¬
личественно переносят в коническую колбу с притер¬
той пробкой вместимостью 300 мл. При непрерывном
встряхивании прибавляют 10,0 мл 0,05 М раствора
йода и сразу после этого 45 мл 0,1 М раствора натрия
гидроксида. Выдерживают в темноте при температуре
от 15 °С до 25 °С в течение 15 мин. Прибавляют 4 мл
смеси серной кислоты Р и воды Р (1:4, об/об). Избыток
йода титруют 0,1 М раствором натрия тиосульфата,
используя микробюретку. Проводят контрольный опыт,
прибавляя 2 мл 1 М раствора кислоты хлористово¬
дородной перед введением испытуемой суспензии.
Аналогично проводят титрование суспензии
сравнения.
Испытание является пригодным, если значения
(п'-п) и (п^'-п^) находятся между значениями 1,9 мл
и 3,6 мл.
Рассчитывают амилолитическую активность в
РЬ.ЕиШ./мг по следующей формуле:
{п'-п}т, А
{п'-п,)т
где:
п — объем 0,1 М раствора натрия тиосульфа¬
та, израсходованного на титрование испытуемой су¬
спензии, мл;
л1 — объем 0,1 М раствора натрия тиосульфа¬
та, израсходованного на титрование суспензии срав¬
нения, мл;
л' — объем 0,1 М раствора натрия тиосульфа¬
та, израсходованного на контрольное титрование ис¬
пытуемой суспензии, мл;
п\ — объем 0,1 М раствора натрия тиосуль¬
фата, израсходованного на контрольное титрование
суспензии сравнения, мл;
т — масса навески испытуемого образца, мг;
л?1 — масса навески стандартного препарата, мг;
А — активность БСП панкреатина порошка
(амилазы), РИ.Еиг.У./мг.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0102
ПАПАВЕРИНА ГИДРОХЛОРИД
Рарауепп/ Нус1госЫопс1ит
РАРАУЕШЫЕ НУОРОСШ-ОРЮЕ
С20Н21МО4 ■ НС| М.м. 375,9
[61-25-6]
ОПРЕДЕЛЕНИЕ
1-(3,4-Диметоксибензил)-6,7-диметоксиизохинолина
гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или белые или почти белые кристаллы.
Умеренно растворим в воде, мало растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО папаверина гидрохлорида #или
спектр, представленный на рисунке #0102.-1*.’
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 5 мг ФСО папаверина ги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6Р254 Р.
Подвижная фаза: диэтиламин Р — этилаце-
тат Р — толуол Р (10:20:70, об/об/об).
Наносимый объем пробы: 10 мкп.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Папаверина гидрохлорид
795
Рисунок #0102.-1. Инфракрасный спектр ФСО папаверина гидрохлорида
Высушивание: при температуре от 100 °С до
105 °С в течение 2 ч.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и величине основному
пятну на хроматограмме раствора сравнения.
С. К10 мл раствора 3, приготовленного как указан¬
но в разделе «Испытания», прибавляют по каплям 5 мл
раствора аммиака Р и выдерживают в течение 10 мин.
Осадок после промывания и высушивания имеет тем¬
пературу плавления (2.2.14) от 146 °С до 149 °С.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,4 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при
необходимости слегка нагревая, и доводят до объема
20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 3,0 до 4,0. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р — под¬
вижная фаза А (20:80, об/об).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 12 мг ФСО носкапина
растворяют в 1,0 мл испытуемого раствора и доводят
смесью растворителей до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 3,4 г/л калия ди¬
гидрофосфата Р, доведенный кислотой фос¬
форной разведенной Р до рН 3,0;
- подвижная фаза В: ацетонитрил Р;
- подвижная фаза С: метанол Р\
Время (мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
Подвижная
фаза С
(%, об/об)
0—5
85
5
10
5—12
00
СП
1
О)
о
5
ю
со
т
о
12—20
60
5
35
20—24
60 — 40
5 — 20
35 — 40
24—27
40
20
40
27—32
ю
00
т
о
20 — 5
40-10
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 238 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к папаверину, время удерживания — около 24 мин):
примесь Е — около 0,7; примесь С — около 0,75; при¬
месь В — около 0,8; примесь А — около 0,9; примесь
Р — около 1,1; примесь О — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси А и папаверина.
796
Гэсударственная фармакопея Республики Беларусь
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси А — 6,2; для примеси С — 2,7; для
примеси й — 0,5):
-любая примесь (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площадь любого
пика, кроме пика папаверина, не должна превы¬
шать площадь основного пика на хроматограмме
раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, за исключением пика папаверина, не
должна превышать 5-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца су¬
шат при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из остатка, полученного в
испытании «Потеря в массе при высушивании».
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в
смеси из 5,0 мл 0,01 М раствора хлористоводо¬
родной кислоты и 50 мл 96 % спирта Р и титруют
0,1 М раствором натрия гидроксида потенциоме-
трически (2.2.20). Отмечают объем титранта между
двумя точками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 37,59 мг С20Н21МО4-НС1.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
А. (35)-6,7-Диметокси-3-[(5Я)-4-метокси-6-метил-
5,6,7,8-тетрагидро-1,3-диоксоло[4,5-д]изохинолин-5-ил]-
изобензофуран-1(ЗА/)-он (носкапин).
В. (Я5)-(3,4-Диметоксифенил)(6,7-диметокси-
изохинолин-1-ил)метанол (папаверинол).
С. 1-(3,4-Диметоксибензил)-6,7-диметокси-3,4-
дигидроизохинолин (дигидропапаверин).
О. (3,4-Диметоксифенил)(6,7-диметоксиизохинолин-
1-ил)метанон (папавералдин).
Е. (1 Я5)-1 -(3,4-Диметоксибензил)-6,7-диметокси-
1,2,3,4-тетрагидроизохинолин (тетрагидропапаверин).
Р. 2-(3,4-Диметоксифенил)-А/-[2-(3,4-диметокси-
фенил)этил]ацетамид.
07/2016:0239
ПАРАФИН ЖИДКИЙ
РагаШпит НриМит (#01еит уазеИт)
рараррш, исшю
ОПРЕДЕЛЕНИЕ
*Вазелиновое масло#.
Очищенная смесь жидких насыщенных углеводо¬
родов, полученных из нефти.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная, прозрачная, маслянистая жидкость,
не флуоресцирует при дневном свете.
Практически нерастворим в воде, мало раство¬
рим в 96 % спирте, смешивается с углеводородами.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая подлинность: А, С.
Вторая подлинность: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр парафина жид¬
кого по Европейской Фармакопее #или спектр, пред¬
ставленный на рисунке #0239.-1#.
B. 1 мл испытуемого образца помещают в про¬
бирку, прибавляют 1 мл 0,1 М раствора натрия
1арафин жидкий
797
Рисунок #0239.-1. Инфракрасный спектр парафина жидкого
гидроксида и осторожно кипятят в течение 30 с при
непрерывном встряхивании. После охлаждения до
комнатной температуры отделяют две фазы. К водной
фазе прибавляют 0,1 мл раствора фенолфталеи¬
на Р. Окраска раствора изменяется на красную.
С. Испытуемый образец выдерживает испытание
«Вязкость», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Кислотность или щелочность. К 10 мл испы¬
туемого образца прибавляют 20 мл кипящей воды Р
и энергично встряхивают в течение 1 мин. Отделяют
водный слой и фильтруют. К 10 мл фильтрата прибав¬
ляют 0,1 мл раствора фенолфталеина Р. Раствор
должен оставаться бесцветным. При прибавлении
не более 0,1 мл 0,1 М раствора натрия гидроксида
должно появиться розовое окрашивание.
Относительная плотность (2.2.5). От 0,827 до
0,890.
Вязкость (2.2.9). От 110 мПа с до 230 мПа-с.
Полициклические ароматические угле¬
водороды. Используют реактивы для УФ-
спектрофотометрии.
25,0 мл испытуемого образца помещают в де¬
лительную воронку вместимостью 125 мл с несма¬
занными пришлифованными частями (пробка, кран).
Прибавляют 25 мл гексана Р, который предваритель¬
но встряхивают дважды с одной пятой от его объема
диметилсульфоксидом Р. Смешивают и прибавляют
5,0 мл диметилсульфоксида Р. Энергично встряхи¬
вают в течение 1 мин и выдерживают до полного раз¬
деления и получения прозрачных слоев. Переносят
нижний слой во вторую делительную воронку, прибав¬
ляют 2 мл гексана Р и смесь энергично встряхивают.
Выдерживают до полного разделения и получения
прозрачных слоев. Отделяют нижний слой и измеряют
оптическую плотность (2.2.25) в интервале от 260 нм
до 420 нм, используя в качестве компенсационного
раствора нижний слой, полученный при энергичном
встряхивании 5,0 мл диметилсульфоксида Р с 25 мл
гексана Р в течение 1 мин. Готовят раствор сравнения
7,0 мг/л нафталина Р в триметилпентане Р и из¬
меряют оптическую плотность раствора при 275 нм,
используя в качестве компенсационного раствора
триметилпентан Р. Ни при одной длине волны в ин¬
тервале от 260 нм до 420 нм оптическая плотность ис¬
пытуемого раствора не должна превышать одну треть
оптической плотности раствора сравнения при 275 нм.
Легкообугливающиеся вещества. Используют
стеклянную пробирку с пришлифованной пробкой
длиной 125 мм и внутренним диаметром 18 мм, гра¬
дуированную на 5 мл и 10 мл; промывают горячей
(не ниже 60 °С) водой Р, ацетоном Р, гептаном Р
и еще раз ацетоном Р, высушивают при темпера¬
туре от 100 °С до 110 °С и охлаждают в эксикаторе.
В пробирку помещают 5 мл испытуемого образца и
прибавляют 5 мл кислоты серной, свободной от азо¬
та, Р1. Закрывают пробкой и встряхивают настолько
энергично, насколько это возможно, в продольном на¬
правлении пробирки в течение 5 с. Ослабляют пробку,
немедленно помещают пробирку на водяную баню,
избегая контакта пробирки с дном или боковыми ча¬
стями бани, и нагревают в течение 10 мин. Через
2 мин, 4 мин, 6 мин и 8 мин вынимают пробирку из
бани и энергично встряхивают, насколько это возмож¬
но, в продольном направлении пробирки в течение
5 с. После 10 мин нагревания вынимают пробирку из
водяной бани и выдерживают в течение 10 мин. Цен¬
трифугируют с ускорением 2000 д в течение 5 мин.
Окраска нижнего слоя должна быть не интенсивнее
(2.2.2, метод /), чем окраска смеси из 0,5 мл синего
исходного раствора, 1,5 мл красного исходного раст¬
вора, 3,0 мл желтого исходного раствора и 2 мл раст¬
вора 10 г/л кислоты хлористоводородной Р.
798
Гэсударственная фармакопея Республики Беларусь
Твердые парафины. Сушат подходящее коли¬
чество испытуемого образца при температуре 100 °С
в течение 2 ч и охлаздают в эксикаторе над кислотой
серной Р. Помещают в стеклянную пробирку с внутрен¬
ним диаметром 25 мм, закрывают пробирку и погружа¬
ют в ледяную баню. Через 4 ч жидкость должна быть
достаточно прозрачной для того, чтобы при просмотре
вдоль вертикальной оси пробирки на белом фоне была
хорошо видна черная полоса шириной 0,5 мм.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1799
ПАРАФИН МЯГКИЙ БЕЛЫЙ
УазеНпит а\Ьит
РАКАРРШ, МН1ТЕ ЗОРТ
ОПРЕДЕЛЕНИЕ
Парафин мягкий белый (#вазелин белый#) пред¬
ставляет собой очищенную и обесцвеченную или почти
обесцвеченную смесь полутвердых углеводородов, по¬
лучаемых из нефти. Может содержать подходящий ан¬
тиоксидант. Парафин мягкий белый описанный в данной
статье, не предназначен для внутреннего применения.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая полупрозрачная мягкая
мазеобразная масса. Расплавленная субстанция при
дневном свете слегка флуоресцирует.
Практически нерастворим в воде, мало раство¬
рим в метиленхлориде, практически нерастворим в
96 % спирте и в глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: А, С, О.
A. Температура каплепадения (2.2.17): от 35 °С
до 70 °С и не отличается более чем на 5 °С от номи¬
нального значения, указанного на этикетке. Использу¬
ют следующую методику заполнения чашечки. Испы¬
туемый образец нагревают до температуры не выше
80 °С при постоянном перемешивании до получения
однородной массы. Металлическую чашечку нагре¬
вают в сушильном шкафу при температуре не выше
80 °С, затем помещают на чистую тарелку или кера¬
мическую плитку и заполняют расплавленным испы¬
туемым образцом до краев. Заполненную чашечку
охлаждают на тарелке или керамической плитке в
течение 30 мин и выдерживают в водяной бане при
температуре от 24 °С до 26 °С в течение (30—40) мин.
Выравнивают поверхность образца, избегая его сдав¬
ливания, одним движением лезвия ножа или бритвы.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: около 2 мг испытуемого образ¬
ца и ФСО парафина мягкого, белого помещают по от¬
дельности на пластинки из натрия хлорида Р, равно¬
мерно распределяют при помощи других пластинок из
натрия хлорида Р и удаляют одну из пластинок.
Сравнение: ФСО парафина мягкого, белого.
С. 2 г испытуемого образца расплавляют и после
получения гомогенной фазы прибавляют 2 мл воды Р
и 0,2 мл 0,05 М раствора йода. Встряхивают и охлаж¬
дают. В твердом верхнем слое появляется фиолето¬
во-розовое или коричневое окрашивание.
О. Испытуемый образец выдерживает испытание
«Цветность», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Цветность (2.2.2, метод II). Испытуемый обра¬
зец белый. 12 г испытуемого образца расплавляют на
водяной бане. Окраска расплавленной массы должна
быть не интенсивнее смеси из 1 объема исходного
желтого раствора и 9 объемов раствора 10 г/л кисло¬
ты хлористоводородной Р.
Кислотность или щелочность. К Юг испыту¬
емого образца прибавляют 20 мл кипящей воды Р и
интенсивно встряхивают в течение 1 мин. Выдержи¬
вают до охлаждения и разделения фаз. К 10 мл во¬
дного слоя прибавляют 0,1 мл раствора фенолфта¬
леина Р. Раствор должен быть бесцветным. При при¬
бавлении не более 0,5 мл 0,01 М раствора натрия
гидроксида должно появиться красное окрашивание.
Консистенция (2.9.9). От 60 до 300.
Полициклические ароматические углеводо¬
роды. Используют реактивы, подходящие для спек¬
трофотометрии в ультрафиолетовой области.
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в 50 мл гексана Р, который предвари¬
тельно встряхивали дважды с 10 мл диметилсуль-
фоксида Р. Полученный раствор переносят в дели¬
тельную воронку вместимостью 125 мл со шлифами
(краник и пробка) без смазки, прибавляют 20 мл ди-
метилсульфоксида Р, интенсивно встряхивают в те¬
чение 1 мин и выдерживают до образования двух про¬
зрачных слоев. Нижний слой переносят во вторую де¬
лительную воронку. Повторяют экстракцию верхнего
слоя с 20 мл диметилсульфоксида Р. Объединенные
нижние слои интенсивно встряхивают с 20 мл гекса¬
на Р в течение 1 мин и выдерживают до образования
двух прозрачных слоев. Нижний слой отделяют и до¬
водят диметилсульфоксидом Р до объема 50,0 мл.
Раствор сравнения. Раствор, содержащий
6,0 мг/л нафталина Р в диметилсульфоксиде Р.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора в диапазоне от 260 нм до 420 нм в
кювете 4 см, используя в качестве компенсационного
раствора прозрачный нижний слой, полученный по¬
сле интенсивного встряхивания 10 мл диметилсуль¬
фоксида Р с 25 мл гексана Р в течение 1 мин.
Измеряют оптическую плотность (2.2.25) раст¬
вора сравнения в максимуме при 278 нм в кювете
4 см, используя в качестве компенсационного раст¬
вора диметилсульфоксид Р.
Оптическая плотность испытуемого раствора в
диапазоне от 260 нм до 420 нм не должна превышать
оптическую плотность раствора сравнения при 278 нм.
Сульфатная зола (2.4.14). Не более 0,05%.
Определение проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
Указывают номинальное значение температуры
каплепадения.
Парафин твердый
799
07/2016:1554
ПАРАФИН МЯГКИЙ ЖЕЛТЫЙ
УазеНпит #ауит
РАПАРРШ, УЕЦ-0]/У 80РТ
ОПРЕДЕЛЕНИЕ
Парафин мягкий желтый (#вазелин желтый#)
представляет собой очищенную смесь полутвердых
углеводородов, получаемых из нефти. Может содер¬
жать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Желтая полупрозрачная мягкая мазеобразная
масса. Расплавленная субстанция при дневном свете
слегка флуоресцирует.
Практически нерастворим в воде, мало раство¬
рим в метиленхлориде, практически нерастворим в
96 % спирте и в глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: А, С, О.
A. Температура каплепадения (2.2.17): от 40 °С
до 60 °С и не отличается более чем на 5 °С от но¬
минального значения. Используют следующую ме¬
тодику заполнения чашечки. Испытуемый образец
нагревают при температуре от 118 °С до 122 °С при
постоянном перемешивании до получения однород¬
ной массы и охлаждают до температуры от 100 °С
до 107 °С. Металлическую чашечку нагревают в
сушильном шкафу при температуре от 103 °С до
107 °С, затем помещают на чистую тарелку или
керамическую плитку и заполняют расплавленным
испытуемым образцом до краев. Заполненную ча¬
шечку охлаждают на тарелке или керамической
плитке в течение 30 мин и выдерживают в водяной
бане при температуре от 24 °С до 26 °С в течение
(30—40) мин. Выравнивают поверхность образца,
избегая его сдавливания, одним движением лезвия
ножа или бритвы.
B. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Приготовление: около 2 мг испытуемого об¬
разца и ФСО парафина мягкого, желтого помеща¬
ют по отдельности на пластинки из натрия хлори¬
да Р, равномерно распределяют при помощи других
пластинок из натрия хлорида Р и удаляют одну из
пластинок.
Сравнение: ФСО парафина мягкого, желтого.
C. 2 г испытуемого образца расплавляют и по¬
сле получения гомогенной фазы прибавляют 2 мл
воды Р и 0,2 мл 0,05 М раствора йода. Встряхива¬
ют и охлаждают. В твердом верхнем слое появля¬
ется фиолетово-розовое или коричневое окраши¬
вание.
й. Испытуемый образец выдерживает испыта¬
ние «Цветность», как указано в разделе «Испыта¬
ния».
ИСПЫТАНИЯ
Цветность (2.2.2, метод II). Испытуемый об¬
разец желтый. 12 г испытуемого образца расплавля¬
ют на водяной бане. Окраска расплавленной массы
должна быть не интенсивнее смеси из 7,6 объемов
исходного желтого раствора и 2,4 объемов исходного
красного раствора.
Кислотность или щелочность. К Юг испыту¬
емого образца прибавляют 20 мл кипящей воды Р и
интенсивно встряхивают в течение 1 мин. Выдержи¬
вают до охлаждения и разделения фаз. К 10 мл во¬
дного слоя прибавляют 0,1 мл раствора фенолфта¬
леина Р. Раствор должен быть бесцветным. При при¬
бавлении не более 0,5 мл 0,01 М раствора натрия
гидроксида должно появиться красное окрашивание.
Консистенция (2.9.9). От 100 до 300.
Полициклические ароматические углеводо¬
роды. Используют реактивы, подходящие для спек¬
трофотометрии в ультрафиолетовой области.
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в 50 мл гексана Р, который предвари¬
тельно встряхивали дважды с 10 мл диметилсуль-
фоксида Р. Полученный раствор переносят в дели¬
тельную воронку вместимостью 125 мл со шлифами
(краник и пробка) без смазки, прибавляют 20 мл ди-
метилсульфоксида Р, интенсивно встряхивают в те¬
чение 1 мин и выдерживают до образования двух про¬
зрачных слоев. Нижний слой переносят во вторую де¬
лительную воронку. Повторяют экстракцию верхнего
слоя с 20 мл диметилсульфоксида Р. Объединенные
нижние слои интенсивно встряхивают с 20 мл гекса¬
на Р в течение 1 мин и выдерживают до образования
двух прозрачных слоев. Нижний слой отделяют и до¬
водят диметилсульфоксидом Р до объема 50,0 мл.
Раствор сравнения. Раствор, содержащий
9,0 мг/л нафталина Р в диметилсульфоксиде Р.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора в диапазоне от 260 нм до 420 нм в
кювете 4 см, используя в качестве компенсационного
раствора прозрачный нижний слой, полученный по¬
сле интенсивного встряхивания 10 мл диметилсуль¬
фоксида Р с 25 мл гексана Р в течение 1 мин.
Измеряют оптическую плотность (2.2.25) раст¬
вора сравнения в максимуме при 278 нм в кювете
4 см, используя в качестве компенсационного раст¬
вора диметилсульфоксид Р.
Оптическая плотность испытуемого раствора в
диапазоне от 260 нм до 420 нм не должна превышать
оптическую плотность раствора сравнения при 278 нм.
Сульфатная зола (2.4.14). Не более 0,05%.
Определение проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
Указывают номинальное значение температуры
каплепадения.
07/2016:1034
ПАРАФИН ТВЕРДЫЙ
РагаШпит зоНёит
РАРАРНЫ, НАРЮ
ОПРЕДЕЛЕНИЕ
Парафин представляет собой смесь твердых
углеводородов предельного ряда, получаемых в ос¬
новном в результате переработки нефти. Может со¬
держать подходящий антиоксидант.
800
Государственная фармакопея Республики Беларуси
ОПИСАНИЕ(СВОЙСТВА)
Бесцветная или белая или почти белая масса.
Расплавленная субстанция при дневном свете не об¬
ладает флуоресценцией.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, практически нерастворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: около 2 мг испытуемого образ¬
ца и ФСО парафина твердого помещают по отдель¬
ности на пластинки из натрия хлорида Р, нагревают
при температуре 100 °С в течение 10 мин, равномер¬
но распределяют при помощи других пластинок из на¬
трия хлорида Р и удаляют одну из пластинок.
Сравнение: ФСО парафина твердого.
B. Испытуемый образец выдерживает испытание
«Кислотность или щелочность», как указано в разде¬
ле «Испытания».
C. Температура плавления (2.2.16): от 50 °С до 61 °С.
ИСПЫТАНИЯ
Кислотность или щелочность. К 15 г испыту¬
емого образца прибавляют 30 мл кипящей воды Р и
интенсивно встряхивают в течение 1 мин. Выдержи¬
вают до охлаждения и разделения фаз. К 10 мл во¬
дного слоя прибавляют 0,1 мл раствора фенолфта¬
леина Р. Раствор должен быть бесцветным. При при¬
бавлении не более 1,0 мл 0,01 М раствора натрия
гидроксида должно появиться красное окрашивание.
К другим 10 мл водного слоя прибавляют 0,1 мл раст¬
вора метилового красного Р. Должно появиться жел¬
тое окрашивание. При прибавлении не более 0,5 мл
0,01 М раствора кислоты хлористоводородной
должно появиться красное окрашивание.
Полициклические ароматические углеводо¬
роды. Используют реактивы, подходящие для спек¬
трофотометрии в ультрафиолетовой области.
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в 25 мл гептана Р. Полученный
раствор переносят в делительную воронку вмести¬
мостью 125 мл со шлифами (краник и пробка) без
смазки, прибавляют 5,0 мл диметилсульфоксида Р,
интенсивно встряхивают в течение 1 мин и выдержи¬
вают до образования двух прозрачных слоев. Нижний
слой переносят во вторую делительную воронку, при¬
бавляют 2 мл гептана Р, интенсивно встряхивают и
выдерживают до образования двух прозрачных сло¬
ев. Используют нижний слой.
Раствор сравнения. Раствор, содержащий
7,0 мг/л нафталина Р в диметилсульфоксиде Р.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора в диапазоне от 265 нм до 420 нм,
используя в качестве компенсационного раствора
прозрачный нижний слой, полученный после встряхи¬
вания 5,0 мл диметилсульфоксида Р с 25 мл гепта¬
на Р в течение 1 мин.
Измеряют оптическую плотность (2.2.25) раствора
сравнения в максимуме при 278 нм, используя в качестве
компенсационного раствора диметилсульфоксид Р.
Оптическая плотность испытуемого раствора в ди¬
апазоне от 265 нм до 420 нм не должна превышать 1/3
оптической плотности раствора сравнения при 278 нм.
Сульфаты (2.4.13). Не более 0,0150 % (150 рргг
2,0 г расплавленного испытуемого образца помещаю*
в делительную воронку вместимостью 50 мл со шли¬
фом, прибавляют 30 мл кипящей воды дистиллиро¬
ванной Р, интенсивно встряхивают в течение 1 мин и
фильтруют.
#Микробиологическая чистота (2.6.12, 2.6.13)
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0049
ПАРАЦЕТАМОЛ
Рагасе1ато!ит
РАКАСЕТАМ01.
С.Н'ЫО, М.м. 151,2
[103-90-2]
ОПРЕДЕЛЕНИЕ
Л/-(4-Гидроксифенил)ацетамид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Умеренно растворим в воде, легко растворим в
96 % спирте, очень мало растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О, Е.
A. Температура плавления (2.2.14): от 168 °С до
172 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25). 0,1 г испы¬
туемого образца растворяют в метаноле Р и доводят
до объема 100,0 мл этим же растворителем. К 1,0 мл
полученного раствора прибавляют 0,5 мл раствора
10,3 г/л кислоты хлористоводородной Р и доводят
метанолом Р до объема 100,0 мл. Полученный раст¬
вор защищают от яркого света и немедленно изме¬
ряют оптическую плотность в максимуме при 249 нм.
Удельный показатель поглощения в максимуме: от
860 до 980.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО парацетамола #или спектр,
представленный на рисунке #0049.-1#.
О. К 0,1 г испытуемого образца прибавляют 1 мл
кислоты хлористоводородной Р, нагревают до кипе¬
ния в течение 3 мин, прибавляют 1 мл воды Р и ох¬
лаждают в ледяной бане. Осадок не образуется. При¬
бавляют 0,05 мл раствора 4,9 г/л калия дихромата Р.
Появляется фиолетовое окрашивание, которое не
переходит в красное.
Парацетамол
801
Рисунок #0049.-1. Инфракрасный спектр ФСО парацетамола
Е. Испытуемый образец дает реакцию на ацетил
(2.3.1). Нагревают на открытом пламени.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в 2,5 мл метанола Р, содержа¬
щего 4,6 г/л раствора 400 г/л тетрабутиламмония
гидроксида Р, и доводят смесью из равных объемов
раствора 17,9 г/л динатрия гидрофосфата Р и раст¬
вора 7,8 г/л натрия дигидрофосфата Р до объема
10.0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
5.0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (с). 5,0 мг 4-аминофенола Р,
5 мг ФСО парацетамола и 5,0 мг хлорацетанилида Р
растворяют в метаноле Р и доводят до объема 20,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят подвижной фазой до объема 250,0 мл.
Раствор сравнения (ф. 20,0 мг 4-нитрофено-
ла Р растворяют в метаноле Р и доводят до объема
50.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят подвижной фазой до объема
20.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 35 °С;
-подвижная фаза: раствор 17,9 г/л динатрия
гидрофосфата Р — раствор 7,8 г/л натрия диги¬
дрофосфата Р — метанол Р, содержащий 4,6 г/л
раствора 400 г/л тетрабутиламмония гидроксида Р,
(375:375:250, об/об/об)\
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 245 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 12-кратное вре¬
мя удерживания парацетамола.
Относительное удерживание (по отношению к
парацетамолу, время удерживания — около 4 мин):
примесь К — около 0,8; примесь Р — около 3; при¬
месь Л — около 7.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 4,0 между пиками при¬
меси К и парацетамола;
- отношение сигнал/шум: не менее 50 для пика
примеси 3.
Предельное содержание примесей:
- примесь Л (не более 0,0010 % (10 ррт)): на хро¬
матограмме испытуемого раствора площадь пика, со¬
ответствующего примеси не должна превышать 0,2
площади соответствующего пика на хроматограмме
раствора сравнения (с);
-примесь К (не более 0,0050% (50ррт)): на
хроматограмме испытуемого раствора площадь пика,
соответствующего примеси К, не должна превышать
площадь соответствующего пика на хроматограмме
раствора сравнения (с);
- примесь Р (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси Р, не должна превышать 0,5 площади
соответствующего пика на хроматограмме раствора
сравнения (д);
- любая другая примесь (не более 0,05%): на
хроматограмме испытуемого раствора площадь лю-
101. Зак. 1060.
802
Гэсударственная фармакопея Республики Беларусь
бого пика, кроме основного и пиков примесей Р, ^ и К,
не должна превышать 0,5 площади основного пика на
хроматограмме раствора сравнения (а);
- сумма других примесей (не более 0,1 %): на
хроматограмме испытуемого раствора сумма площа¬
дей всех пиков, кроме основного и пиков примесей Р,
^ и К, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,01 %): при расчете
суммы других примесей на хроматограмме испытуе¬
мого раствора не учитывают пики с площадью менее
площади основного пика на хроматограмме раствора
сравнения (Ь).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раст¬
воряют в смеси вода Р — ацетон Р (15:85, об/об) и
доводят до объема 20 мл этой же смесью раствори¬
телей. 12 мл полученного раствора должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца (1 ррт
РЬ), полученного разведением эталонного раствора
свинца (100 ррт РЬ) Р смесью водаР — ацетон Р
(15:85, об/об).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в сме¬
си из 10 мл воды Р и 30 мл кислоты серной разве-
денной Р, кипятят с обратным холодильником в те¬
чение 1 ч, охлаждают и доводят водой Р до объема
100,0 мл. К 20,0 мл полученного раствора прибавляют
40 мл воды Р, 40 г льда, 15 мл кислоты хлористово¬
дородной разведенной Р, 0,1 мл ферроина Р и титру¬
ют 0,1 М раствором церия сульфата до появления
зеленовато-желтого окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора церия сульфата соответ¬
ствует 7,56 мг С8Н9М02.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
КЗ
Н2
К4
Я1<
A. Р1 = КЗ = К4 = Н, Р2 = ОН: А/-(2-Гидроксифенил)-
ацетамид.
B. Р1 = СН3, Р2 = РЗ = Н, В4 = ОН: А/-(4-
Гидроксифенил)пропанамид.
C. Р1 = Р2 = Н, КЗ = С1, Р4 = ОН: Л/-(3-Хлор-4-
гидроксифенил)ацетамид.
О. Р1 = Р2 = РЗ = Р4 = Н: Л/-Фенилацетамид.
Н. Р1 = Р2 = РЗ = Н, Р4 = 0-СО-СН3: 4-(Ацетила-
мино)фенилацетат.
^ Р1 = Р2 = РЗ = Н, Р4 = С1: А/-(4-Хлорфенил)аце-
тамид (хлорацетанилид).
К2.
,Р4
X
Е. X = О, Р2 = Н, Р4 = ОН: 1-(4-Гидроксифенил)эта-
нон.
0. X = Ы-ОН, Р2 - Н, Р4 = ОН: 1-(4-Гидроксифе-
нил)этанона оксим.
1. X = О, Р2 = ОН, Р4 = Н: 1-(2-Гидроксифенил)эта-
нон.
/ОН
к
Р. Р = Ы02: 4-Нитрофенол.
К. Р = 1ЧН2: 4-Аминофенол.
07/2016:0851
ПЕНТОКСИФИЛЛИН
Реп1охНуШпит
РЕИТОЮРУШИЕ
С13Н18Ы403 М.м. 278,3
[6493-05-6]
ОПРЕДЕЛЕНИЕ
3,7-Диметил-1 -(5-оксогексил)-3,7-дигидро-1 Н-пурин-
2,6-дион.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА) >
Белый или почти белый кристаллический порошок. \
Растворим в воде, легко растворим в метилен-
хлориде, умеренно растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 103 °С до
107 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пентоксифиллина #или спектр,
представленный на рисунке #0851 .-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО пентоксифил¬
лина растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р.
Подвижная фаза: метанол Р — этилацетат Р
(15:85, об/об).
Наносимый объем пробы: 5 мкл.
Пентоксифиллин
803
Рисунок #0861 .-1. Инфракрасный спектр ФСО пентоксифиллина
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
□. Испытуемый образец дает реакцию на ксан¬
тины (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
этим же растворителем до объема 50 мл.
Прозрачность (2.2.1). 40% (об/об) раствор 5
должен быть прозрачным.
Цветность (2.2.2, метод II). Окраска 40 % (об/об)
раствора 3 должна быть не интенсивнее эталона У(Ж)7.
Кислотность. К 8 мл раствора 3 прибавляют
12 мл воды, свободной от углерода диоксида, Р и
0,05 мл раствора бромтимолового синего Р1. Долж¬
но появиться зеленое или желтое окрашивание. При
прибавлении не более 0,2 мл 0,01 Мраствора натрия
гидроксида должно появиться синее окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Смесь из равных объ¬
емов раствора 5,44 г/л калия дигидрофосфата Р и
метанола Р.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Раствор сравнения (а). 2,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 5,0 мл полученного раствора доводят сме¬
сью растворителей до объема 100,0 мл.
Раствор сравнения (Ь). 10,0 мл раствора сравнения
(а) доводят смесью растворителей до объема 50,0 мл.
Раствор сравнения (с). 2 мг кофеина Р (примесь
Р) и 2 мг теофиллина Р (примесь С) растворяют в сме¬
си растворителей, прибавляют 1 мл испытуемого раст¬
вора и доводят смесью растворителей до объема 10 мл.
Раствор сравнения (д). 5,0 мг кофеина Р (при¬
месь Р), 5,0 мг теобромина Р (примесь А) и 5,0 мг
теофиллина Р (примесь С) растворяют в смеси раст¬
ворителей и доводят этой же смесью растворителей
до объема 100,0 мл. 1,0 мл полученного раствора до¬
водят смесью растворителей до объема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 30 объе¬
мов метанола Р и 70 объемов раствора 5,44 г/л
калия дигидрофосфата Р;
- подвижная фаза В: смешивают 30 объемов
раствора 5,44 г/л калия дигидрофосфата Р и 70
объемов метанола Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—6
85
15
6—13
85- 10
15 — 90
13—30
10
90
30—35
ю
со
т
о
90—15
35-45
85
15
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 272 нм;
ЮГ. Зак. 1060.
/
804
Гэсударственная фармакопея Республики Беларусь
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к пентоксифиллину, время удерживания — около
12 мин): примесь А— около 0,3; примесь С — около
0,4; примесь Р — около 0,5; примесь 3 — около 1,6.
Пригодность хроматографической системы:
раствор сравнения (с):
- время удерживания: примесь Р — от 4 мин до
7 мин; пентоксифиллин — от 9 мин до 13 мин; при не¬
обходимости подбирают необходимую скорость сме¬
шивания подвижных фаз;
- разрешение: не менее 4 между пиками приме¬
си С и примеси Р.
Предельное содержание примесей:
- примеси А, С и Г (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, С и Р, не должны превы¬
шать площади соответствующих пиков на хромато¬
грамме раствора сравнения (б);
- примесь ^ (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси 3, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, С, Р и ^ не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,02 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0100% (100 ррт).
20 мл раствора 5 помещают в делительную воронку и
встряхивают дважды с 2-метилпропанолом Р порци¬
ями по 20 мл. 10 мл водного слоя доводят водой Р до
объема 15 мл.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
Определение проводят из 15 мл раствора 5.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат над
фосфора (V) оксидом Р при температуре 60 °С и дав¬
лении не более 700 Па.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 5 мл
кислоты уксусной безводной Р, прибавляют 20 мл ук¬
сусного ангидрида Р и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 27,83 мг С13Н18М403.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, С, Р, Л.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): В, О, Е, С, Н, I, К.
СН3
A. Р = Н, Р' = СН3: Теобромин.
B. Р = Р' = Н: 3-Метил-3,7-дигидро-1Н-пурин-2,6-
дион.
C. Р = СН3, Р' = Н: Теофиллин.
0. Р = СН2-СН2-СН2-ОН, Р' = СН3: 1-(3-Гидрокси-
пропил)-3,7-диметил-3,7-дигидро-1Н-пурин-2,6-дион.
Р. Р = Р' = СН3: Кофеин.
Н. Р = Р' = СН2-[СН2]3-СО-СН3:3-Метил-1,7-бис(5-
оксогексил)-3,7-дигидро-1Я-пурин-2,6-дион.
1. Р = СН2-С6Н5, Р' = СН3: 1-Бензил-3,7-диметил-
3,7-дигидро-1 Я-пурин-2,6-дион.
Е. X = СН2:1,1-Метиленбис(3,7-диметил-3,7-дигид-
ро-1 Н-пурин-2,6-дион.
К. X = СН2-СН2-СН2:1,1'-(Пропан-1,3-диил)бис(3,7-
диметил-3,7-дигидро-1Н-пурин-2,6-дион).
о
С. 3,7-Диметил-6-(5-оксогексилокси)-3,7-дигидро-
2Н-пурин-2-он.
*}. 1-[(5Е)-11-(3,7-Диметил-2,6-диоксо-2,3,6,7-
тетрагидро-1 Н-пурин-1 -ил)-5-метил-7-оксоундек-5-енил]-
3,7-диметил-3,7-дигидро-1Я-пурин-2,6-дион.
Пепсина порошок
805
07/2016:0682
ПЕПСИНА ПОРОШОК
Рерз/л/ риМз
РЕР81Ы РОУУОЕР
[9001-75-6]
ОПРЕДЕЛЕНИЕ
Получают из слизистой оболочки желудка сви¬
ней, крупного рогатого скота или овец. Содержит
желудочные протеиназы, активные в кислой среде
(рН от 1 до 5).
Активность: не менее 0,5 РЬ.Еиг.11./мг (в пе¬
ресчете на сухое вещество).
ПРОИЗВОДСТВО
Животные, из тканей которых получают пеп¬
сина порошок, должны выдерживать требования
к здоровью животных, пригодных для потребления
человеком.
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый кристаллический
или аморфный порошок. Гигроскопичен.
Растворим в воде, практически нерастворим в
96 % спирте. Раствор в воде может быть слегка мут¬
ным и давать слабокислую реакцию.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
30 мг фибрина синего Р измельчают в ступке и
суспендируют в 20 мл кислоты хлористоводород¬
ной разведенной Р2. Суспензию фильтруют через
фильтровальную бумагу и промывают осадок на
фильтре кислотой хлористоводородной разведен¬
ной Р2 до получения бесцветного фильтрата. Про¬
дырявливают фильтровальную бумагу и смывают
через отверстие фибрин синий Р в коническую кол¬
бу, используя 20 мл кислоты хлористоводородной
разведенной Р2. Перед использованием встряхи¬
вают. Испытуемый образец в количестве, эквива¬
лентном не менее 20 РН.Еиг.1!., растворяют в 2 мл
кислоты хлористоводородной разведенной Р2,
раствор доводят до рН 1,6±0,1. 1 мл полученного
раствора помещают в пробирку, содержащую 4 мл
суспензии фибрина синего, перемешивают и поме¬
щают в водяную баню с температурой 25 °С, осто¬
рожно встряхивая. Параллельно готовят контроль¬
ный раствор, используя 1 мл воды Р. Через 15 мин
выдержки в водяной бане контрольный раствор
остается бесцветным, испытуемый раствор приоб¬
ретает синее окрашивание.
ИСПЫТАНИЯ
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 0,500 г испытуемого образца сушат при
температуре 60 °С над фосфора (V) оксидом Р при
давлении не более 670 Па в течение 4 ч.
Микробиологическая чистота
ОКА: критерий приемлемости 104 КОЕ/г {2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г {2.6.12).
ЕзсМепсЫа соН: отсутствие {2.6.13).
За1топе11а: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Активность порошка пепсина определяют пу¬
тем сравнения количества пептидов, неосаждае-
мых раствором кислоты трихлоруксуной Р, вы¬
свобождаемых в течение 1 минуты из субстрата
раствора гемоглобина Р, количественное опре¬
деление которых проводят с использованием фос-
форномолибденово-фольфрамового реактива Р,
с количеством таких же пептидов, высвобождае¬
мых БСП пепсина порошка из такого же субстрата
в таких же условиях.
Приготовление и разведение испытуемого и
раствора сравнения проводят при температуре
от 0 °С до 4 °С.
При приготовлении испытуемого и раствора
сравнения необходимо избегать встряхивания и
пенообразования.
Испытуемый раствор. Непосредственно пе¬
ред использованием готовят раствор с ожидаемой
концентрацией 0,5 РИ.Еиг.1)./мл испытуемого об¬
разца в кислоте хлористоводородной разведен¬
ной Р2\ при необходимости перед разведением до
заданного объема раствор доводят до рН 1,6±0,1
1 М раствором кислоты хлористоводородной.
Раствор сравнения. Менее чем за 15 мин пе¬
ред использованием готовят раствор с концентра¬
цией 0,5 РЬ.Еиг.11./мл БСП пепсина порошка в рас¬
творе кислоты хлористоводородной разведен¬
ной Р2\ при необходимости перед разведением до
заданного объема раствор доводят до рН 1,6±0,1
1 М раствором кислоты хлористоводородной.
Маркируют пробирки (по две каждой): Г, Ть, 5,,
$1Ь. 82’ 82Ь’ 53’ 5ЗЬ> маркируют пробирку В.
В пробирки прибавляют кислоту хлористоводо¬
родную разведенную Р2, как указано ниже:
В: 1,0 мл;
31 и 31Ь: по 0,5 мл;
32, 32Ь, Т и Ть: по 0,25 мл.
В пробирки прибавляют раствор сравнения, как
указано ниже:
3, и 31Ь: по 0,5 мл;
32 и 52Ь: по 0,75 мл;
$3 и Ззь: по 1,0 мл.
В пробирки Т\лТь прибавляют по 0,75 мл испыту¬
емого раствора.
В пробирки 31Ь, 32Ь, 5зь, Ть и В прибавляют по
10,0 мл раствора кислоты трихлоруксусной Р и
встряхивают.
Пробирки и колбу с раствором гемоглоби¬
на Р помещают в водяную баню с температурой
(25±0,1) °С. После уравновешивания температуры
в пробирки В, 31Ь, 32Ь, 5ЗЬ и Ть прибавляют по 5,0 мл
раствора гемоглобина Р и перемешивают.
Начиная с момента времени «ноль» в пробирки
5.,, 32, 53 и Т последовательно с интервалом в 30 с
прибавляют по 5,0 мл раствора гемоглобина Р.
После прибавления раствора содержимое
пробирки немедленно перемешивают.
Точно через 10 мин после прибавления раст¬
вора гемоглобина Р, с интервалом в 30 с, реакцию
останавливают прибавлением по 10,0 мл раствора
кислоты трихлоруксусной Р в пробирки 5^ 32, 53 и
Т (рекомендуется использовать пипетку с быстрым
вытеканием жидкости или пипетку на слив) и пере¬
мешивают. Содержимое каждой пробирки (образ¬
цы и контрольные растворы) фильтруют дважды
через одинаковую подходящую фильтровальную
бумагу, предварительно промытую 50 г/л раство¬
ром кислоты трихлоруксусной Р, затем водой Р и
высушенную. Первые 5 мл фильтрата отбрасыва-
102. Зак. 1060.
/
806
Гэсударственная фармакопея Республики Беларусь
Таблица 0682.-1
Пробирки
5,
«1»
«2
«2
**
Т
Т,
В
Кислота хлористоводородная разведенная
Р2, мл
0,5
0,5
0,25
0,25
0,25
0,25
1,0
Раствор сравнения, мл
0,5
0,5
0,75
0,75
1,0
1,0
Испытуемый раствор, мл
0,75
0,75
Раствор кислоты трихлоруксусной Р, мл
10,0
10,0
10,0
10,0
10,0
Перемешивание
+
+
+
+
+
Водяная баня с температурой 25 °С
+
+
+
+
+
+
+
+
+
Раствор гемоглобина Р, мл
5,0
5,0
5,0
5,0
5,0
Перемешивание
+
+
+
+
+
Раствор гемоглобина Р, мл
5,0
5,0
5,0
5,0
Перемешивание
+
+
+
+
Водяная баня с температурой 25 °С, 10 мин
+
+
+
+
+
+
+
+
+
Раствор кислоты трихлоруксусной Р, мл
10,0
10,0
10,0
10,0
Перемешивание
+
+
+
+
Фильтрование
+
+
+
+
+
+
+
+
+
ют, затем по 3,0 мл каждого из фильтратов по от¬
дельности помещают в пробирки, содержащие по
20 мл воды Р и перемешивают.
Подходящая фильтровальная бумага должна
выдерживать следующее испытание: через круг
белой фильтровальной бумаги диаметром 7 см
фильтруют 5 мл раствора 50 г/л кислоты трихлор-
уксусной Р\ оптическая плотность (2.2.25) полу¬
ченного фильтрата, измеренная при длине волны
275 нм с использованием нефильтрованного раст¬
вора кислоты трихлоруксусной Р в качестве ком¬
пенсационного раствора, должна быть менее 0,04.
В каждую пробирку прибавляют по 1,0 мл
раствора натрия гидроксида Р и по 1,0 мл фос-
форномолибденово-вольфрамового реактива Р,
начиная с контрольных растворов, затем в пробир¬
ки с образцами каждой серии в установленной по¬
следовательности.
Схематическое отображение описанных выше
действий представлено в Таблице 0682.-1.
Через 15 мин измеряют оптическую плотность
(2.2.25) растворов 5,, 32, 53, 31Ь, 32Ь, 53;Ь и Т при
длине волны 540 нм, используя в качестве компен¬
сационного раствора фильтрат из пробирки В. Кор¬
ректируют средние значения оптической плотности
для фильтратов из пробирок 5^ 52 и 53 путем вычи¬
тания средних значений оптической плотности для
фильтратов из соответствующих пробирок 51Ь, 32Ь
и 5ЗЬ. Строят калибровочный график зависимости
откорректированной оптической плотности от ис¬
пользованного объема раствора сравнения. Опре¬
деляют активность испытуемого образца, исполь¬
зуя откорректированную оптическую плотность ис¬
пытуемого раствора (7"-Гь) и калибровочный гра¬
фик, принимая во внимание факторы разведения.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в за¬
щищенном от света месте при температуре от 2 °С
до 8 °С.
МАРКИРОВКА
Указывают:
- активность, выраженную в Единицах действия
Европейской Фармакопеи в 1 мг вещества.
07/2016:2019
ПЕРИНДОПРИЛ ТРЕТ-БУТИЛАМИН
РеппдоргШ ^е^^-Ьи^уIат^пит
РЕКШООРРШ. ТЕКТ-ВиТПАМШЕ
С19Нз2Ы205 ■ СДЫ М.м. 441,6
[107133-36-8]
ОПРЕДЕЛЕНИЕ
2-Метилпропан-2-амина (25,За5,7а5)-1 -[(25)-
2-[[(15)-1 -(этоксикарбонил )бутил]амино]пропаноил]
октагидро-1 Н-индол-2-карбоксилат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Слегка гигроскопичен.
Легко растворим в воде и 96 % спирте, раство¬
рим или умеренно растворим в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Удельное оптическое вращение (2.2.7): от -66
до -69 (в пересчете на безводное вещество). 0,250 г
испытуемого образца растворяют в 96 % спирте Р и
доводят до объема 25,0 мл этим же растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО периндоприла трет-
бутиламина #или спектр, представленный на рисунке
#2019.-1#.
Если полученные спектры отличаются, то ис¬
пытуемый образец и ФСО периндоприла трет-
бутиламина растворяют по отдельности в метилен¬
хлориде Р и выпаривают до сухих остатков, которые
используют для получения новых спектров.
C. Просматривают хроматограммы, полученные
в испытании «Примесь А».
Периндоприл трет-бутиламин
807
Рисунок #2019.-1. Инфракрасный спектр ФСО периндоприла трет-бутиламина
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее
по значению Р1 пятну с максимальным значением Р,
на хроматограмме раствора сравнения (с) (трет-
бутиламин).
ИСПЫТАНИЯ
Примесь А. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,20 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО периндоприла
примеси А растворяют в метаноле Р и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят метанолом Р до объема 20,0 мл.
Раствор сравнения (с). К 5 мл раствора сравне¬
ния (а) прибавляют 5 мл раствора 20 г/л 1,1-диметиэ-
тиламина Р в метаноле Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
толуол Р — метанол Р (1:40:60, об/об/об).
Наносимый объем пробы, по 10 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку обрабатывают парами
йода в течение не менее 20 ч.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- примесь А (не более 0,25 %): на хроматограм¬
ме испытуемого раствора пятно, соответствующее
примеси А, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Стереохимическая чистота. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 96 % спирте Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят 96 % спиртом Р до объема 100,0 мл.
1,0 мл полученного раствора доводят 96 % спир¬
том Р до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг ФСО периндопри¬
ла для определения стереохимической чистоты (со¬
держит примесь I) растворяют в 96 % спирте Р и до¬
водят до объема 5,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным для хроматографии Р (размер
частиц 5 мкм);
- температура: 50 °С для колонки и не менее
30 см трубки, предшествующей колонке;
- подвижная фаза: смешивают в следующем по¬
рядке 21,7 объема ацетонитрила Р, 0,3 объема пен-
танола Р и 78 объемов раствора 1,50 г/л натрия геп-
тансульфоната Р, доведенного до рН 2,0 смесью из
равных объемов кислоты хлорной Р и воды Р;
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 215 нм;
- время установления равновесия: не менее 4 ч;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания периндоприла.
Идентификация пиков примесей: идентифици¬
руют пик примеси I, используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
102*. Зак. 1060.
808
Гэсударственная фармакопея Республики Беларусь
ФСО периндоприла для определения стереохимиче-
ской чистоты.
Относительное удерживание (по отношению к
периндоприлу, время удерживания — около 100 мин):
примесь I — около 0,9.
Пригодность хроматографической системы:
- хроматограмма раствора сравнения (Ь) должна
соответствовать хроматограмме ФСО периндоприла
для определения стереохимической чистоты;
- отношение сигнал/шум: не менее 3 для основ¬
ного пика на хроматограмме раствора сравнения (а);
- коэффициент разделения пиков: не менее 3
(Нр — высота пика примеси I относительно базовой ли¬
нии; Ну — расстояние между базовой линией и нижней
точкой кривой, разделяющей пики примеси I и перин¬
доприла на хроматограмме раствора сравнения (Ь)).
Предельное содержание примесей:
-примесь I (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси I, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си I, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики, относитель¬
ное удерживания которых (относительно пика перин¬
доприла) менее 0,6 и более 1,4.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием или хранят при тем¬
пературе не выше 10 °С.
Испытуемый раствор. 60 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 3 мг ФСО периндоприла
для идентификации пиков (содержит примеси В, Е, Р,
Н и К) растворяют в 1 мл подвижной фазы А.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 200,0 мл.
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (Ь) доводят подвижной фазой А до объема
10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4 мм, заполненная сферическим силикагелем
октилсилильным эндкепированным для хромато¬
графии Р с размером частиц 5 мкм и размером пор
15 нм;
- температура: 60 °С для колонки и трубки,
предшествующей колонке;
- подвижная фаза:
- подвижная фаза А: вода Р, доведенная до рН
2,5 смесью равных объемов кислоты хлорной Р
и воды Р;
- подвижная фаза В: раствор 0,03 % (об/об)
кислоты хлорной Р в ацетонитриле Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
О-(5-0
95
5
(5~0—(60-()
95 — 40
5 — 60
(60-0—(65-0
40-►95
60 — 5
Изократическая стадия описана для хромато¬
графической системы со свободным объемом (О)
2 мл. Если О отличается от 2 мл, времена гради¬
ента корректируют с помощью следующего выра¬
жения:
0-2 .
скорость подвижной фазы ’
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, Е, Р, Н и К, используя хрома¬
тограмму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО периндоприла для идентифи¬
кации пиков.
Относительное удерживание (по отношению
к периндоприлу, время удерживания около 25 мин):
примесь В — около 0,68; примесь К — около 0,72;
примесь Е — около 1,2; примесь Р — около 1,6; при¬
месь Н — около 1,8 (примесь Н может элюироваться
одним или двумя пиками).
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 3
(Нр — высота пика примеси В относительно базовой
линии; Ну — расстояние между базовой линией и
нижней точкой кривой, разделяющей пики примеси В
и примеси К).
Предельное содержание примесей:
- примесь Е (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси Е, не должна превышать 0,8 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси В, не должна превышать 0,6 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- примеси Е и Н (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям Р и Н, не должны превышать
0,4 площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%); на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В, Е, Р и Н, не должна превышать 0,2 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
-сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
Вода (2.5.12). Не более 1,0%. Определение
проводят из 0,50 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Периндоприл трет-бутиламин
809
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,160 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 22,08 мг С^Нз^О^Н^.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, Е, Р, Н, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О, С, 3, К, Ц
М, А/, О, Р, О, Р, 5, Т, и, V, Щ У, 2, АА, ВВ, СС.
А. (25,За5,7а5)-Октагидро-1 Н-индол-2-карбоновая
кислота.
н3с.
ро2н
В. К = Н: (25,За5,7а5)-1 -[(25)-2-[[(15)-1 -Карбокси-
бутил]амино]пропаноил]октагидро-1Й-индол-2-карбо-
новая кислота (периндоприлат).
Е. Р = СН(СН3)2: (25,За5,7а5Ж(25^2-[[(1$Ж(1-
Метилэтокси)карбонил]бутил]амино]пропаноил]-
октагидро-1 А/-индол-2-карбоновая кислота.
С. (25)-2-[(35,5а5,9а5,10а5)-3-Метил-1,4-диоксо-
декагидропиразино[1,2-а]индол-2(1 Н)-ил]пентановая
кислота.
О. (25)-2-[(35,5аЗ,9а5,10аР)-3-Метил-1,4-диоксо-
декагидропиразино[1,2-а]индол-2(1 Н)-ил]пентановая
кислота.
Р. Этил-(25)-2-[(33,5а5,9а5,10а5)-3-метил-1,4-ди-
оксодекагидропиразино[1,2-а] индол-2(1 Н)-ил]пента-
ноат.
С. (25,За5,7а5)-1-[(25)-2-[(5Р5)-3-Циклогексил-
2,4-диоксо-5-пропилимидазолидин-1-ил]пропаноил]-
октагидро-1 Н-индол-2-карбоновая кислота.
Н. (25,За5,7а5)-1 -[(25)-2-[(5Р5)-3-Циклогексил-2-
(циклогексилимино)-4-оксо-5-пропилимидазолидин-1-
ил]пропаноил]октагидро-1 Н-индол-2-карбоновая кис¬
лота.
I. (2Р5,ЗаР5,7аР8)-1-[(2Р8)-2-[[(18Р)-1-(Этокси-
карбонил )бутил]амйно]пропаноил]октагидро-1Н-индол-
2-карбоновая кислота ((±)-1'-эли-периндоприл).
± Р = ИН,,; Р' = СН3: (28,За5,7а5)-1-[(28)-2-амино-
пропаноил]октагидро-1 Н-индол-2-карбоновая кислота.
1_. Р = Р' = Н: (25,За8,7а8)-1-Ацетилоктагидро-1Н-
индол-2-карбоновая кислота.
К. (35,5а5,9а5,10а5)-3-Метилдекагидро-пирази-
но[1,2-а]индол-1,4-дион.
103. Зак. 1060.
/
810
Гэсударственная фармакопея Республики Беларусь
М. (25,За5,7а5)-1-[(25)-2-[[(15)-1-(Метоксикар-
бонил)бутил]амино]пропаноил]октагидро-1Н-индол-2-
карбоновая кислота.
N. (25)-3-Циклогексил-2-[[(25)-2-[[(15)-1-(этоксикар-
бонил)бутил]амино]пропаноил]амино]пропановая кис¬
лота.
О. (25,ЗаЗ,7аЗ)-1 -[[(25,За5,7а$)-1-[(25)-2-[[(15)-1-
(Этоксикарбонил)бутил]амино]пропаноил]октагидро-
1 /-/-индол-2-ил]карбонил]октагидро-1 Н- индол-2-
карбоновая кислота.
1 -[2-[[1 -(Этоксикарбонил )бутил]амино]пропаноил]-
октагидро-1 Н-индол-2-карбоновая кислота.
Р. (2Я5,ЗаЯ5,7аЯ5)-, (2'5Я)-, (1"ЯЗ)-: (±)-2’-Эпи-
периндоприл.
О. (2Я$,ЗаЯ$,7а5Я)-, (2X8)-, (1"Я5)-: (±)-7а-Эпи-
периндоприл.
Я. (2Я5,За5Я,7аЯ5)-, (2'Я5)-, (1 "Я5)-: (±)-За-Эли-
периндоприл.
5. (25Я,ЗаЯ5,7аЯ5)-, (2'Я5)-, (1"Я5)-: (±)-2-Эпи-
периндоприл.
Т. (2Я5,ЗаЯ5,7аЯ5)-, (2'$Я)-, (1"5Я)-: (±)-1",2'-Ди-
эга-периндоприл.
I). (2Я5,ЗаЯ5,7а5Я)-, (2'Я5)-, (1"5Я)-: (±)-1",7а-Ди-
элы-периндоприл.
V. (25Я,За5Я,7аЯ5)-, (2'Я5)-, (1"Я5)-: (±)-2,За-Ди-
элл-периндоприл.
\Л/. (25Я,ЗаЯ5,7аЯ5)-, (2'Я5)-, (1"5Я)-: (±)-1",2-Ди-
элл-периндоприл.
X. (25Я,ЗаЯ5,7а$Я)-, (2’ЯЗ)-, (ГЯ5)-: (±)-2,7а-Ди-
элл-периндоприл.
У. (2$Я,ЗаЯ5,7аЯ5)-, (2'5Я)-, (1"Я5)-: (±)-2,2'-Ди-
элл-периндоприл.
2. (2Я5,За5Я,7аЯ5)-, (2'РЗу, (1"5Я)-: (±)-1",За-Ди-
элл-периндоприл.
АА. (2Я5,За5Я,7а5Я)-, (2'Я5)-, (ГЯ5)-: (±)-За,7а-
Ди-элл-периндоприл.
ВВ. (2Я5,За$Я,7аЯ5)-, (2'5Я)-, (1"Я5)-: (±)-2',За-Ди-
элл-периндоприл.
СО. (2Я5,ЗаЯ5,7а$Я)-, (2'5Я)-, (1"Я5)-: (±)-2',7а-Ди-
элл-периндоприл.
07/2016: 0633
ПИЛОКАРПИНА ГИДРОХЛОРИД
РНосагрт/ Ьус1госЫопс1ит
Р1ТОСАЯРШЕ НУОТЮСШОНЮЕ
■ НС1 М.м. 244,7
[54-71-7]
ОПРЕДЕЛЕНИЕ
(35,4Я)-3-Этил-4-[(1 -метил-1 Н-имидазол-5-ил)-
метил]дигидрофуран-2(ЗН)-она гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Гигроскопичен.
Очень легко растворим в воде и легко растворим
в 96 % спирте.
Температура плавления: около 203 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пилокарпина гидрохлорида.
Если испытуемый и стандартный образцы иссле¬
дуются в виде дисков, для их приготовления исполь¬
зуют калия хлорид Р.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
2 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО пилокарпина ги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 2 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р — метанол Р — метиленхлорид Р
(1:14:85, об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: нагревают при температуре от
100 °С до 105 °С в течение 10 мин и охлаждают.
Проявление: пластинку опрыскивают раствором
калия йодовисмутата разведенным Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
й. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
Пилокарпина гидрохлорид
811
ИСПЫТАНИЯ
Раствор 3. 2,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)7.
рН (2.2.3). От 3,5 до 4,5. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От +89
до +93 (в пересчете на сухое вещество). Определяют
удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого
раствора доводят водой Р до объема 100,0 мл. 2,0 мл
полученного раствора доводят водой Р до объема
20.0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО пилокарпина
нитрата для проверки пригодности хроматографи¬
ческой системы (содержит примесь А) растворяют в
воде Р и доводят до объема 50,0 мл этим же раство¬
рителем.
Раствор сравнения (с). К 5 мл испытуемого раст¬
вора прибавляют 0,1 мл раствора аммиака Р, нагре¬
вают на водяной бане в течение 30 мин, охлаждают и
доводят водой Р до объема 25 мл. 3 мл полученного
раствора доводят водой Р до объема 25 мл. При этом
образуется в основном пилокарпиновая кислота (при¬
месь В).
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р1 (размер частиц 5 мкм) с
размером пор 10 нм и содержанием углерода 19 %;
- подвижная фаза: смешивают 55 объемов ме¬
танола Р, 60 объемов ацетонитрила Р и 885 объ¬
емов раствора 0,679 г/л тетрабутиламмония диги¬
дрофосфата Р, предварительно доведенного рас¬
твором аммиака разведенным Р2 до рН 7,7;
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания пилокарпина.
Порядок выхода пиков: примесь В, примесь С,
примесь А, пилокарпин.
Время удерживания: пилокарпин — около 20 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,6 между пиками при¬
меси А и пилокарпина.
Предельное содержание примесей:
- примесь А (не более 1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей А и В (не более 1,5 %): на хро¬
матограмме испытуемого раствора сумма площадей
пиков, соответствующих примесям А и В, не должна
превышать 3-кратную площадь основного пика на
хроматограмме раствора сравнения (а);
- сумма примесей кроме примесей А и В (не бо¬
лее 0,5 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пиков
примесей А и В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- неучитываемый предел (0,2 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,4 площади основного пика на хро¬
матограмме раствора сравнения (а).
Железо (2.4.9). Не более 0,0010% (Юррт).
Раствор 3 должен выдерживать испытание на железо.
Эталон готовят с использованием 5 мл эталонного
раствора железа (1 ррт Ре) Р и 5 мл воды Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 50 мл
96 % спирта Р, прибавляют 5 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 24,47 мг С11Н16Ы202 НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. (ЗР,4Р)-3-Этил-4-[(1 -метил-1 Н-имидазол-5-ил)-
метил]дигидрофуран-2(ЗН)-он (изопилокарпин).
он
B. Р = С2Н5, Р' = Н: (25,ЗЯ)-2-Этил-3-(гидрокси-
метил)-4-(1 -метил-1 Н-имидазол-5-ил)бутановая кисло¬
та (пилокарпиновая кислота).
C. Р = Н, Р' = С2Н5: (2Р,ЗР)-2-Этил-3-(гидрокси-ме-
тил)-4-(1 -метил-1 Н-имидазол-5-ил)бутановая кислота
(изопилокарпиновая кислота).
103*. Зак. 1060.
/
812
Гэсударственная фармакопея Республики Беларусь
07/2016:0423
ПИПЕРАЗИНА АДИПИНАТ
Р/регаг/п/ асИраз
Р1РЕКА21ЫЕ А01РАТЕ
С4Н10М2 - С6Н10О4 М.м. 232,3
[142-88-1]
ОПРЕДЕЛЕНИЕ
Пиперазина гександиоат.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Растворим в воде, практически нерастворим в
96 % спирте.
Температура плавления: около 250 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО пиперазина адипината #или
спектр, представленный на рисунке #0423.-1*.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси», после обра¬
ботки раствором нингидрина.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
С. К 10 мл раствора 3, приготовленному как ука¬
занно в разделе «Испытания», прибавляют 5 мл кис-
лоты хлористоводородной Р и трижды встряхивают
с эфиром Р порциями по 10 мл. Эфирные слои объ¬
единяют и выпаривают досуха. Остаток промывают
5 мл воды Р и сушат при температуре от 100 °С до
105 °С. Температура плавления (2.2.14) полученного
остатка: от 150 °С до 154 °С.
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)8.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 1,0 г испытуемого образ¬
ца растворяют в 6 мл раствора аммиака концентриро¬
ванного Р и доводят этанолом Р до объема 10 мл.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят до объема 10 мл смесью из эта¬
нола Р и раствора аммиака концентрированного Р
(2:3, об/об).
Раствор сравнения (а). 0,1 г ФСО пиперазина
адипината растворяют в смеси из этанола Р и раст¬
вора аммиака концентрированного Р (2:3, об/об) и
доводят до объема 10 мл этой же смесью раствори¬
телей.
Раствор сравнения (Ь). 25 мг этилендиами-
на Р растворяют в смеси из этанола Р и раствора
Рисунок #0423.-1. Инфракрасный спектр ФСО пиперазина адипината
Пиперазин гидрат
813
аммиака концентрированного Р (2:3, об/об) и до¬
водят до объема 100 мл этой же смесью раство¬
рителей.
Раствор сравнения (с). 25 мг триэтилендиа-
мина Р растворяют в смеси из этанола Р и раст¬
вора аммиака концентрированного Р (2:3, об/об)
и доводят до объема 100 мл этой же смесью раст¬
ворителей.
Раствор сравнения (ф. 12,5 мг триэтилен-
диамина Р растворяют в 5,0 мл испытуемого раст¬
вора (а) и доводят до объема 50 мл смесью из
этанола Р и раствора аммиака концентрирован¬
ного Р (2:3, об/об).
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля.
Подвижная фаза: свежеприготовленная смесь из
раствора аммиака концентрированного Р и ацето¬
на Р (20:80, об/об).
Объем наносимой пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре 105 °С в течение
10 мин.
Проявление А: пластинку последовательно опры¬
скивают раствором 3 г/л нингидрина Р в смеси из кисло¬
ты уксусной безводной Р и бутанола Р (3:100, об/об) и
раствором 1,5 г/л нингидрина Р в этаноле Р. Нагре¬
вают при температуре 105 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (б):
- на хроматограмме обнаруживаются два четко
разделенных пятна.
Результаты А: на хроматограмме испытуемого
раствора (а) любое пятно, кроме основного, должно
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения (Ь) (0,25 %).
Проявление В: пластинку опрыскивают 0,05 М
раствором йода и выдерживают в течение 10 мин.
Результаты В: на хроматограмме испытуемого
раствора (а) пятно, соответствующее триэтилендиа-
мину, должно быть не интенсивнее пятна на хромато¬
грамме раствора сравнения (с) (0,25 %).
Не учитывают пятна, расположенные на линии
старта.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Вода (2.5.72). Не более 0,5 %. Определение про¬
водят из 1,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 10 мл
кислоты уксусной безводной Р при легком нагрева¬
нии и доводят до объема 70 мл этим же раствори¬
телем. Титруют 0,1 М раствором хлорной кислоты,
используя в качестве индикатора 0,25 мл раствора
нафтолбензеина Р, до перехода окраски раствора от
коричневато-желтой до зеленой.
1 мл 0,1 М раствора хлорной кислоты соответ¬
ствует 11,61 мг С4Н10М2-СбН10О4.
07/2016:0425
ПИПЕРАЗИН ГИДРАТ
Р/регагтит Ьуйпсит
Р1РЕКАШЕ НУ О РАТЕ
С4Н10М2-6Н2О М.м. 194,2
[142-63-2]
ОПРЕДЕЛЕНИЕ
Пиперазин гексагидрат.
Содержание: не менее 98,0 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветные расплывающиеся на воздухе кри¬
сталлы.
Легко растворим в воде и в 96 % спирте.
Температура плавления: около 43 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец и стан¬
дартный образец сушат в вакууме над фосфора (V)
оксидом Р в течение 48 ч, образцы растирают в по¬
рошок, избегая попадания воды, готовят в дисках и
незамедлительно снимают спектры.
Сравнение: ФСО пиперазина гидрата.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси», после обра¬
ботки раствором нингидрина.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
C. 0,5 г испытуемого образца растворяют 5 мл
раствора натрия гидроксида разведенного Р, при¬
бавляют 0,2 мл бензоилхлорида Р и перемешивают.
Продолжают прибавлять бензоилхлорид Р порциями
по 0,2 мл до прекращения образования осадка. Оса¬
док фильтруют и промывают 10 мл воды Р, прибав¬
ляемой небольшими порциями. Осадок растворяют в
2 мл горячего 96 % спирта Р и полученный раствор
вливают в 5 мл воды Р. Выдерживают в течение 4 ч,
фильтруют, кристаллы промывают водой Р и сушат
при температуре от 100 °С до 105 °С. Температура
плавления (2.2.14) полученных кристаллов: от 191 °С
до 196 °С.
ИСПЫТАНИЯ
Раствор 5. 1,0 испытуемого образца растворяют
в воде, свободной от углерода диоксида, Р и доводят
до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)8.
рН (2.2.3). От 10,5 до 12,0. Измеряют рН раст¬
вора 3.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
104. Зак. 1060.
/
814
Гэсударственная фармакопея Республики Беларусь
Смесь растворителей. Этанол Р — раствор
аммиака концентрированный Р (40:60, об/об).
Испытуемый раствор (а). 1,0 г испытуемого об¬
разца растворяют в 6 мл раствора аммиака концен¬
трированного Р и доводят этанолом Р до объема
10 мл.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 10 мл.
Раствор сравнения (а). 0,1 г ФСО пиперазина
гидрата растворяют в смеси растворителей и дово¬
дят до объема 10 мл этой же смесью растворителей.
Раствор сравнения (Ь). 25 мг этилендиамина Р
растворяют в смеси растворителей и доводят до объ¬
ема 100 мл этой же смесью растворителей.
Раствор сравнения (с). 25 мг триэтилендиами-
на Р растворяют в смеси растворителей и доводят до
объема 100 мл этой же смесью растворителей.
Раствор сравнения (ф. 12,5 мг триэтилендиа-
мина Р растворяют в 5,0 мл испытуемого раствора (а)
и доводят смесью растворителей до объема 50 мл.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля.
Подвижная фаза: свежеприготовленная смесь
раствор аммиака концентрированного Р — аце¬
тон Р (20:80, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: 105 °С.
Проявление А: пластинку последовательно опры¬
скивают раствором 3 г/л нингидрина Р в смеси кисло¬
та уксусная безводная Р — бутанол Р (3:100, об/об)
и раствором 1,5 г/л нингидрина Р в этаноле Р. Пла¬
стинку сушат при 105 °С в течение 10 мин.
Предельное содержание примесей А:
- любая примесь (не более 0,25 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Проявление В: пластинку опрыскивают 0,05 М
раствором йода Р и выдерживают в течение около
10 мин.
Предельное содержание примесей В:
- триэтилендиамин (не более 0,25 %): на хрома¬
тограмме испытуемого раствора (а) пятно, соответству¬
ющее триэтилендиамину, должно быть не интенсивнее
пятна на хроматограмме раствора сравнения (с).
Пригодность хроматографической системы:
раствор сравнения (с1):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Сульфатная зола (2.4.14}. Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
80,0 мг испытуемого образца растворяют в 10 мл
кислоты уксусной безводной Р, осторожно нагревая,
и доводят этим же растворителем до объема 70 мл.
Титруют 0,1 М раствором кислоты хлорной до пере¬
хода окраски раствора от коричневато-желтого до
зеленого, используя в качестве индикатора 0,25 мл
раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 9,705 мг С4Н10М2-6Н2О.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:1680
ПИРАНТЕЛА ЭМБОНАТ
РугаМеН етЬопаз
РУКАЫТЕ1- ЕМВОЫАТЕ
он он
[22204-24-6]
ОПРЕДЕЛЕНИЕ
1 -Метил-2-[(Е)-2-(тиофен-2-ил)этенил]-1,4,5,6-
тетрагидропиримидина гидро-4,4'-метиленбис(3-
гидроксинафтален-2-карбоксилат). #Пирантела памоат*
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Порошок от бледно-желтого до желтого цвета.
Практически нерастворим в воде, растворим в
диметилсульфоксиде, практически нерастворим в ме¬
таноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пирантела эмбоната #или
спектр, представленный на рисунке #1680.-1#.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием и на всех стадиях
испытания защищают от попадания света.
Смесь растворителей. Смешивают при охлаж¬
дении 5 объемов кислоты уксусной ледяной Р с 5
объемами воды Р и 2 объемами диэтиламина Р.
Испытуемый раствор. 80 мг испытуемого об¬
разца растворяют в 7 мл смеси растворителей и до¬
водят ацетонитрилом Р до объема 100,0 мл.
Раствор сравнения (а). 10,0 мг ФСО пирантела
примеси А растворяют в смеси растворителей, при¬
бавляют 2,5 мл испытуемого раствора и доводят сме¬
сью растворителей до объема 50,0 мл. 2,0 мл полу¬
ченного раствора доводят смесью растворителей до
объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл.
Условия хроматографирования:
Пирантела эмбонат
815
Рисунок #1680.-1. Инфракрасный спектр ФСО пирантела эмбоната
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем для хромато¬
графии Р с размером частиц 5 мкм;
- подвижная фаза: смесь растворителей — аце¬
тонитрил для хроматографии Р (72:928, об/об);
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 288 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания пирантела.
Относительное удерживание (по отношению к
пирантелу, время удерживания — около 11 мин): кис¬
лота эмбоновая — около 0,5; примесь А — около 1,3;
примесь В — около 1,8 (примесь А также приводит к
увеличению пика эмбоната).
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 4,0 между пиками пи¬
рантела и примеси А.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,4):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (а);
- примесь В (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси В, не должна превышать 0,4 площади основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и В, не должна превышать 0,2 площади основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей, кроме примесей А и В (не
более 0,3 %): на хроматограмме испытуемого раст¬
вора сумма площадей пиков, кроме основного и пи¬
ков примесей А и В, не должна превышать 0,6 пло¬
щади основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0360 % (360 ррт).
К 0,46 г испытуемого образца прибавляют 10 мл кис¬
лоты азотной разведенной Р и 30 мл воды Р, нагре¬
вают на водяной бане в течение 5 мин, охлаждают,
доводят водой Р до объема 50 мл, тщательно пере¬
мешивают и фильтруют. 15 мл полученного раствора
должны выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,1%. К 0,50 г
испытуемого образца прибавляют 2,5 мл кислоты
азотной разведенной Р, доводят водой дистилли¬
рованной Р до объема 50 мл, нагревают на водяной
бане в течение 5 мин, перемешивают в течение 2 мин,
охлаждают и фильтруют. 15 мл полученного раствора
должны выдерживать испытание на сульфаты.
Железо (2.4.9). Не более 0,0075 % (75 ррт).
0,66 г испытуемого образца прокаливают при темпе¬
ратуре (800±50) °С в течение 2 ч. Полученный остаток
растворяют в 2,5 мл кислоты хлористоводородной
разведенной Р при слабом нагревании в течение 10
мин, охлаждают и доводят водой Р до объема 50 мл.
10 мл полученного раствора должны выдерживать ис¬
пытание на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0020% (20 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
104*. Зак. 1060.
816
Гэсударственная фармакопея Республики Беларусь
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,450 г испытуемого образца прибавляют 10 мл
ангидрида уксусного Р и 50 мл кислоты уксусной ле¬
дяной Р, нагревают при температуре 50 °С, перемеши¬
вают в течение 10 мин и охлаждают (раствор непро¬
зрачный). Полученный раствор титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 59,47 мг С11Н14М28 С23Н1606.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. 1-Метил-2-[(2)-2-(тиофен-2-ил)этенил]-1,4,5,6-
тетрагидропиримидин.
В. (Е)-Л/-[3-(Метиламино)пропил]-3-(тиофен-2-ил)-
проп-2-енамид.
07/2016:1733
ПИРАЦЕТАМ
Р1гасе\атит
Р1КАСЕТАМ
С6Н10М2°2 М.М. 142’2
[7491-74-9]
ОПРЕДЕЛЕНИЕ
2-(2-Оксопирролидин-1-ил)ацетамид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Легко растворим в воде, растворим в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пирацетама #или спектр, пред¬
ставленный на рисунке #1733.-1#.
Если спектры, полученные с использованием
образцов в твердом состоянии, отличаются, то испы¬
туемый образец и ФСО пирацетама растворяют по
отдельности в 96% спирте Р и выпаривают на водя¬
ной бане до сухих остатков, которые используют для
получения новых спектров.
Рисунок #1733.-1. Инфракрасный спектр ФСО пирацетама
Пиридоксина гидрохлорид
817
X
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Полученный раствор дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 50,0 мг испытуемо¬
го образца растворяют в смеси ацетонитрил Р1 —
вода Р (10:90, об/об) и доводят до объема 100,0 мл
этой же смесью растворителей.
Испытуемый раствор (Ь). 10,0 мл испытуемо¬
го раствора (а) доводят смесью ацетонитрил Р1 —
вода Р (10:90, об/об) до объема 50,0 мл.
Раствор сравнения (а). 5 мг испытуемого об¬
разца и 10 мкп 2-пирролидона Р растворяют в смеси
ацетонитрил Р1 — вода Р (10:90, об/об) и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора (а) доводят смесью ацетонитрил Р1 - вода Р
(10:90, об/об) до объема 100,0 мл. 5,0 мл полученного
раствора доводят смесью ацетонитрил Р1 — вода Р
(10:90, об/об) до объема 50,0 мл.
Раствор сравнения (с). 50,0 мг ФСО пирацета¬
ма растворяют в смеси ацетонитрил Р1 — вода Р
(10:90, об/об) и доводят до объемаЮО.О мл этой же
смесью растворителей. 10,0 мл полученного раствора
доводят смесью ацетонитрил Р1 — вода Р (10:90,
об/об) до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
-подвижная фаза: ацетонитрилР1 — раствор
1,0 г/л дикалия гидрофосфата Р (10:90, об/об); рН сме¬
си доводят до 6,0 кислотой фосфорной разведенной Р,
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 205 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (Ь);
- время хроматографирования: 8-кратное вре¬
мя удерживания пирацетама.
Относительное удерживание (по отношению к
пирацетаму, время удерживания — около 4 мин): при¬
месь й— около 0,8; примесь А—около 1,15; примесь
В — около 2,8; примесь С — около 6,3.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,0 между пиками пира¬
цетама и примеси А;
- фактор асимметрии: не более 2,0 для пика
пирацетама.
Предельное содержание примесей:
-примеси А, В, С, О (не более 0,1 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В, С и О, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более 0,1 %):
на хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного пика и пиков примесей А, В,
С и О, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного пика, не должна превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (Юррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл воды Р. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С6Н10Ы2О2 рассчитывают в процентах.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
о
A. Р = Н: Пирролидин-2-он (2-пирролидон).
B. Р = СН2-С0-0-СН3: Метил-(2-оксопирролидин-
1-ил)ацетат.
C. Р = СН2-С0-0-С2Н5: Этил-(2-оксопирролидин-
1-ил)ацетат.
Э. Р = СН2-С02Н: (2-Оксопирролидин-1-ил)уксус-
ная кислота.
07/2016:0245
ПИРИДОКСИНА ГИДРОХЛОРИД
Рупбохт'1 ЬудгосЫопдит
РУРЮОХМЕ НУОРОСШОРЮЕ
НС1
С.Н..ЫО, • НС1
[58-56-0]
М.м. 205,6
105. Зак. 1060.
818
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
(5-Гидрокси-6-метилпиридин-3,4-диил)диметано-
ла гидрохлорид. #Витамин В6#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, мало растворим в 96 %
спирте.
Температура плавления: около 205 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Раствор А. 1,0 мл раствора 3, приготовленного как
указано в разделе «Испытания», доводят 0,1 Мраство¬
ром кислоты хлористоводородной до объема 50,0 мл.
Раствор В. 1,0 мл раствора А доводят 0,1 М рас¬
твором кислоты хлористоводородной до объема
100,0 мл.
Раствор С. 1,0 мл раствора А доводят раствором
0,025 М калия дигидрофосфата + 0,025 М динатрия
гидрофосфата, описанным в разделе 2.2.3, до объ¬
ема 100,0 мл.
Диапазон длин волн: от 250 нм до 350 нм для
раствора В; от 220 нм до 350 нм для раствора С.
Максимумы поглощения: при (288—296) нм для
раствора В; при (248—256) нм и (320—327) нм для
раствора С.
Удельный показатель поглощения в максимумах'.
- от 425 до 445 для раствора В при (288—296) нм;
- от 175 до 195 для раствора С при (248—256) нм;
- от 345 до 365 для раствора С при (320—327) нм.
B. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Сравнение: ФСО пиридоксина гидрохлорида
#или спектр, представленный на рисунке #0245.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1,0 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
10 мл этим же растворителем. 1 мл полученного
раствора доводят водой Р до объема 10 мл.
Раствор сравнения. 0,10 г ФСО пиридоксина
гидрохлорида растворяют в воде Р и доводят до
объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля О Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р — метиленхлорид Р — тетраги-
дрофуран Р — ацетон Р (9:13:13:65, об/об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: в ненасыщенной
хроматографической камере не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают рас¬
твором 50 г/л натрия карбоната Р в смеси 96 %
спирт Р— вода Р (30:70, об/об), высушивают в по¬
токе воздуха, опрыскивают раствором 1 г/л дихлор-
хинонхлоримида Р в 96% спирте Р и немедленно
просматривают.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения.
й. Раствор 3, приготовленный как указано в
разделе «Испытания», дает реакцию (а) на хлори¬
ды (2.3.1).
Рисунок #0245.-1. Инфракрасный спектр ФСО пиридоксина гидрохлорида
Пиридоксина гидрохлорид
819
ИСПЫТАНИЯ
Раствор 3. 2,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, и
доводят до объема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона У(Ж)7.
рН (2.2.3). От 2,4 до 3,0. Измеряют рН раст¬
вора 3.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема
10.0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемо¬
го раствора доводят водой Р до объема 100,0 мл.
1.0 мл полученного раствора доводят водой Р до
объема 10,0 мл.
Раствор сравнения (Ь). 2,5 мг ФСО пиридок¬
сина примеси А и 2,5 мг 4-дезоксипиридоксина
гидрохлорида Р (примесь В) растворяют в воде Р
и доводят до объема 10,0 мл этим же растворите¬
лем. 2,0 мл полученного раствора доводят водой Р
до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным, деактивированным по отношению
к основаниям, эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- подвижная фаза: 2,72 г калия дигидрофос¬
фата Р растворяют в 900 мл воды Р, доводят до
рН 3,0 кислотой фосфорной разведенной Р и раз¬
водят водой Р до объема 1000 мл;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 210 нм;
- объем вводимой пробы: 5 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания пиридоксина.
Идентификация пиков примесей: идентифи¬
цируют пики примесей А и В, используя хромато¬
грамму раствора сравнения (Ь).
Относительное удерживание (по отношению к
пиридоксину, время удерживания — около 12 мин):
примесь А — около 1,7; примесь В — около 1,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
месей А и В.
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси В — 1,5):
- примесь В (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси В, не должна превышать
1,5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси В, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт), 12 мл раствора 3 должны вы¬
держивать испытание на тяжелые металлы. Эталон
готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца сушат
при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Чтобы избежать перегрева реакционной сре¬
ды, раствор тщательно перемешивают в про¬
цессе титрования и прекращают титрование
немедленно после достижения конечной точки
титрования.
0,150 г испытуемого образца растворяют в
5 мл кислоты муравьиной безводной Р, прибавля¬
ют 50 мл уксусного ангидрида Р и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора хлорной кислоты соответ¬
ствует 20,56 мг СД^Оз-НС!.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А.
А. 6-Метил-1,3-дигидрофуро[3,4-с]пиридин-7-ол.
сн3
В. 5-(Гидроксиметил)-2,4-диметилпиридин-3-ол.
105*. Зак. 1060.
820
Гэсударственная фармакопея Республики Беларусь
07/2016:0853
ПЛАЗМА ЧЕЛОВЕЧЕСКАЯ
ДЛЯ ФРАКЦИОНИРОВАНИЯ
Р1а8та Ьитапит ад зерагаНопет
НЦМАЫ Р1-АЗМА РОК РРАСТЮИАТЮМ
ОПРЕДЕЛЕНИЕ
Плазма человеческая для фракционирования
представляет собой жидкую часть крови человека,
полученную посредством извлечения клеточных эле¬
ментов из крови, содержащей антикоагулянт, либо
отделения посредством фильтрации или центрифуги¬
рования цельной крови при аферезе. Предназначена
для изготовления лекарственных средств на основе
плазмы.
ПОЛУЧЕНИЕ
ДОНОРЫ
Для изготовления лекарственных средств на
основе плазмы может быть использована только
плазма здоровых доноров, отобранных в результа¬
те медицинского осмотра, изучения медицинского
анамнеза, лабораторного обследования крови и
отсутствия определяемых агентов инфекционных
заболеваний, переносимых с кровью. Соответству¬
ющие рекомендации определены Советом Европы
[РесоттепдаНоп N0. Р (95) 15 оп (Не ргерагаНоп,
изе апб циаШу аззигапсе оТ Ыоод Сотропеп(з, и по¬
следующие дополнения] и Европейским союзом:
Сотт'/ззюп 0/гес//Уе 2004/33/ЕС о! 22 тагсН 2004
1тр1етеп(тд й1гес(Ые 2002/98/ЕС (Не Еигореап
РагНатеп( апс! СоипсН ап гедагбз сег(ат (есНпюа1
гедшгетеп(з 1ог Ыоос! апб Ыоос! сотропеп(з, #или
действующими нормативными документами Мини¬
стерства здравоохранения Республики Беларусь*.
Иммунизация доноров. Иммунизация доноров
для получения иммуноглобулинов специфической
активности производится, когда иммунная характе¬
ристика плазмы обычных доноров не удовлетворяет
требованиям производителя. Рекомендации по им¬
мунизации сформулированы Всемирной организаци¬
ей здравоохранения (Редшгетеп(з 1ог (Не соИесИоп,
ргосеззтд апс! диаН(у соп(го1 о?Ыоос!, Ыоос! сотропеп(з
апб р!азта бегмаСыез, IШО ТесНпюа1 Рерог(, N0. 840,
1994, и последующие дополнения) #или действующи¬
ми нормативными документами Министерства здра¬
воохранения Республики Беларусь*.
Документирование. Сведения о донорах и до-
нациях должны быть зафиксированы таким образом,
чтобы, сохраняя требуемую степень конфиденци¬
альности, они позволяли идентифицировать донора,
определять происхождение каждой порции плазмы,
включенной в пул, и проследить результаты всех про¬
изведенных процедур и лабораторных тестов.
Лабораторные тесты. Лабораторные тесты
должны быть выполнены для каждой донации с опре¬
делением следующих маркеров:
1. Антитела/антиген вируса иммунодефицита че¬
ловека 1 (апй-Н1\/-1);
2. Антитела/антиген вируса иммунодефицита че¬
ловека 2 (апй-Н1\/-2, #р 24#);
3. Поверхностный антиген гепатита В (НВзАд);
4. Антитела/антиген вируса гепатита С (апй-НСУ);
#5. Антитела к Тгеропета раШбит;
#6. Содержание аланинаминотрансферазы (АН).
Применяемые методы тестирования должны
быть достаточно чувствительны, специфичны и ут¬
верждены уполномоченными органами. Если какой-
либо из данных тестов повторно показывает положи¬
тельные результаты, то такая плазма не может быть
использована.
ИНДИВИДУАЛЬНАЯ ПОРЦИЯ ПЛАЗМЫ
Плазма должна быть получена методом, позво¬
ляющим наиболее полно удалить клетки и их фраг¬
менты. Плазма, полученная из цельной крови или
методом плазмафереза, должна быть отделена от
клеточных элементов методом, обеспечивающим
стерильность продукта. К плазме не добавляют ан¬
тибактериальные или противогрибковые средства.
Контейнер должен соответствовать требованиям к
стеклянным контейнерам (3.2.1) или пластмассовым
контейнерам для крови и ее компонентов (3.2.3). Кон¬
тейнер должен быть герметичен для исключения кон¬
таминации микроорганизмами.
Если перед замораживанием объединяются две
или более дозы, то процесс их объединения должен
проводиться с применением стерильных соединяю¬
щих приспособлений либо в асептических условиях
с использованием контейнеров, которые не были в
употреблении.
Плазма, полученная из цельной крови (после
отделения от клеточных элементов) или методом
плазмафереза и предназначенная для выделения
лабильных белков, не позже чем через 24 ч после
донации должна быть заморожена быстрым охлаж¬
дением #(при температуре -30 °С или ниже в течение
60 мин)#. Процесс замораживания должен быть вали-
дирован таким образом, чтобы температура плазмы
при замораживании в течение 12 ч составляла -25 °С
или ниже.
Плазма, полученная методом плазмафереза и
предназначенная только для выделения нелабиль¬
ных белков, не позже чем через 24 ч после донации
должна быть заморожена быстрым охлаждением при
температуре -20 °С или ниже.
Плазма, полученная из цельной крови и пред¬
назначенная только для выделения нелабильных в
отделенной от клеточных элементов плазме белков,
не позже чем через 72 ч после донации должна быть
заморожена быстрым охлаждением при температуре
-20 °С или ниже.
Приведенное ниже определение общего белка и
фактора свертывания крови VIII не означает, что
оно должно выполняться для каждой порции плазмы.
Эти тесты приводятся как ориентиры для Надле¬
жащей производственной практики (ОМР). Испыта¬
ние по определению фактора свертывания VIII важ¬
но для плазмы, предназначенной для изготовления
концентратов лабильных белков.
Содержание общего белка в порции плазмы за¬
висит от содержания белка в сыворотке донора и
степени разведения в ходе донорской процедуры.
Если плазма получена от подходящего донора и
использована предписанная пропорция раствора
антикоагулянта, содержание общего белка состав¬
ляет не менее 50 г/л. Если объем крови или плазмы
меньше регламентированного, то есть происходит
большее разведение раствором антикоагулянта,
то такую порцию нежелательно присоединять в
пул для фракционирования. Целью Надлежащей про¬
изводственной практики (ОМР) является соблюде¬
ние предписанных правил для всех донаций.
Повидон
821
Сохранность фактора свертывания VIII при
донации зависит от процесса забора плазмы и по¬
следующего обращения с кровью или плазмой. При
надлежащей практике выполнения процедуры ак¬
тивность составляет 0,7 МЕ/мл, но и при меньшей
активности в отдельной порции плазмы она может
быть использована для получения концентрата
фактора свертывания крови. Целью всех стадий
процесса производства является получение плазмы
надлежащего качества и максимальное сохранение
лабильных белков.
Общий белок. Не менее 50 г/л. Испытание прово¬
дят с использованием пула, содержащего не менее 10
порций плазмы. Подходящий объем полученного пре¬
парата разводят раствором 9 г/л натрия хлорида Р
для получения раствора, содержащего 15 мг белка в
2 мл. 2,0 мл полученного раствора помещают в кру¬
глодонную центрифужную пробирку, прибавляют 2 мл
раствора 75 г/л натрия молибдата Р и 2 мл смеси из
1 объема кислоты серной, свободной от азота, Р и
30 объемов воды Р. Перемешивают, центрифугируют
в течение 5 мин, декантируют надосадочную жид¬
кость и извлекают содержимое пробирки на фильтро¬
вальную бумагу для высушивания. Определяют азот в
полученном остатке методом минерализации серной
кислотой (2.5.9). При расчете содержания белка ум¬
ножают полученное содержание азота на 6,25. Ис¬
пытание может быть проведено биуретовым методом
(2.5.33, метод 5)#.
Фактор свертывания VIII (2.7.4). Активность не
менее 0,7 МЕ/мл. Испытание проводят с использова¬
нием пула, содержащего не менее 10 порций плазмы.
При необходимости испытуемый образец оттаивают
при температуре 37 °С. Проводят количественное
определение фактора свертывания VIII с использова¬
нием стандартной плазмы, калиброванной по Между¬
народному стандарту фактора свертывания VIII чело¬
века в плазме.
внутренний контрольный образец, который готовят
путем прибавления подходящего маркера к образцу
пула плазмы. Испытание считается недействитель¬
ным, если положительный контрольный образец
нереактивен или если результат, полученный с вну¬
тренним контрольным образцом, выявит присутствие
ингибиторов. Испытуемый образец пула плазмы вы¬
держивает испытание, если в нем не выявляется ре¬
активности вирусной РНК гепатита С.
В качестве положительного контрольного образ¬
ца может быть использован БСП вирусной РНК ге¬
патита С для метода амплификации нуклеиновых
кислот.
ОПИСАНИЕ
До замораживания — прозрачная или слегка опа¬
лесцирующая жидкость без видимых следов гемоли¬
за, от светло-желтого до зеленого цвета.
МАРКИРОВКА
Каждая индивидуальная порция плазмы долж¬
на содержать сведения, позволяющие проследить ее
происхождение (до конкретного донора).
07/2016:0685
ПОВИДОН
Роу/с/олшл
РОУЮОЫЕ
ХРАНЕНИЕ И ТРАНСПОРТИРОВКА
Замороженная плазма должна храниться и
транспортироваться в условиях, предусматривающих
поддерживание температуры -20 °С и ниже; из-за
временных/случайных причин температура хране¬
ния может подниматься один или более раз во время
хранения и транспортировки; при этом плазма может
быть использована для фракционирования, если вы¬
полнены все нижеследующие условия:
- общий период времени, в течение которого тем¬
пература поднималась выше-20 °С, не превышал 72 ч;
- температура поднималась выше -15 °С не бо¬
лее одного раза;
- температура ни разу не поднималась выше
-5 °С.
ПЛАЗМА, ОБЪЕДИНЕННАЯ В ПУЛ
При производстве лекарственных средств на
основе плазмы первый гомогенный пул плазмы (на¬
пример, после удаления криопреципитата) должен
давать отрицательный результат на НЬз-антиген и
ВИЧ-антитела при использовании подходящих по чув¬
ствительности и специфичности методов.
Кроме этого, пул плазмы должен быть испытан
на вирусную РНК гепатита С с использованием вали-
дированного метода амплификации нуклеиновых кис¬
лот (2.6.21). В испытании используют положительный
контрольный образец, который содержит 100 МЕ РНК
вируса гепатита С, и, для обнаружения ингибиторов,
СвЛ*АА
[9003-39-8]
ОПРЕДЕЛЕНИЕ
а-Гидро-ы-гидрополи[1 -(2-оксопирролидин-1 -ил)-
этилен]. #Поливинилпирролидон#. Состоит из линей¬
ных полимеров 1-этенилпирролидин-2-она.
Содержание: не менее 11,5 % и не более 12,8 %
азота (14; А.м. 14,01) (в пересчете на безводное
вещество).
Различные типы повидона отличаются вязкостью
приготовленных из него растворов, обозначаемой как
К-значение.
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок или
хлопья. Гигроскопичен.
Легко растворим в воде, в 96 % спирте и
метаноле, очень мало растворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец предвари¬
тельно сушат при температуре 105 °С в течение 6 ч; за¬
писывают спектр, используя 4 мг испытуемого образца.
Сравнение: ФСО повидона.
822
Гэсударственная фармакопея Республики Беларусь
B. К 0,4 мл раствора 51, приготовленного как
указано в разделе «Испытания», прибавляют 10 мл
воды Р, 5 мл кислоты хлористоводородной разве¬
денной Р и 2 мл раствора калия дихромата Р. Обра¬
зуется оранжево-желтый осадок.
C. К 1 мл раствора 51 прибавляют 0,2 мл
раствора диметиламинобензальдегида Р1 и 0,1 мл
кислоты серной Р. Появляется розовое окрашивание.
й. К 0,1 мл раствора 51 прибавляют 5 мл воды Р
и 0,2 мл 0,05 М раствора йода. Появляется красное
окрашивание.
Е. К 0,5 г испытуемого образца прибавляют
10 мл водыР и встряхивают. Испытуемый образец
растворяется.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20,0 мл этим же растворителем.
Испытуемый образец прибавляют в воду малыми пор¬
циями, при перемешивании на магнитной мешалке.
Раствор 31. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем. Ис¬
пытуемый образец прибавляют в воду малыми порци¬
ями, при перемешивании на магнитной мешалке.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона В(К)б, ВУ(КЖ)6
или Р(Кр)6.
рН (2.2.3). От 3,0 до 5,0 для раствора 5 повидона
с заявленным К-значением не более 30; от 4,0 до 7,0
для раствора 5 повидона с заявленным К-значением
более 30.
Вязкость, выраженная через К-значение
К-значение повидона с заявленным К-значением
15 или менее должно составлять от 85,0 % до 115,0 %
от заявленного значения.
К-значение повидона с заявленным К-значением
или со средним значением заявленного диапазона
К-значений более 15 должно составлять от 90,0%
до 108,0 % от заявленного значения или от среднего
значения заявленного диапазона.
Из испытуемого образца с заявленным
К-значением 18 или менее готовят раствор с
концентрацией 50 г/л. Из испытуемого образца с
заявленным К-значением более 18 готовят раствор
с концентрацией 10 г/л. Из испытуемого образца
повидона с заявленным К-значением более 95 готовят
раствор с концентрацией 1,0 г/л. Выдерживают в
течение 1 ч и определяют вязкость (2.2.9) раствора
при температуре 25 °С, используя вискозометр № 1 с
минимальным временем вытекания 100 с.
К-значение рассчитывают по формуле:
1,5 • 1од/7 — 1 т/зОО • с • !од?у + (с +1,5 • с • !од^)2
0,15 + 0,003 • с + 0,15 с+ 0,003 с2
где:
с — концентрация испытуемого образца в
пересчете на безводное вещество, г/100 мл;
П — кинематическая вязкость раствора
относительно вязкости воды Р.
Альдегиды. Не более 0,0500 % (500 ррт) в
пересчете на ацетальдегид.
Испытуемый раствор. 1,0 г испытуемого
образца растворяют в фосфатном буферном
растворе рН 9,0 Р и доводят до объема 100,0 мл этим
же растворителем. Плотно закрывают колбу пробкой
и нагревают при температуре 60 °С в течение 1 ч.
Охлаждают до комнатной температуры.
Раствор сравнения. 0,140 г ацетальдегидам-
миака тримера тригидрата Р растворяют в воде Р и
доводят до объема 200,0 мл этим же растворителем.
1.0 мл полученного раствора доводят фосфатным
буферным раствором рН 9,0 Р до объема 100,0 мл.
В 3 одинаковые спектрофотометрические кюветы
с толщиной слоя 1 см помещают 0,5 мл испытуемого
раствора, 0,5 мл раствора сравнения и 0,5 мл
воды Р (контрольный раствор). В каждую кювету
прибавляют 2,5 мл фосфатного буферного раствора
с рН 9,0 Р и 0,2 мл раствора никотинамида-аденина
динуклеотида Р. Перемешивают и плотно закрывают
пробкой. Выдерживают при температуре (22±2)°С в
течение (2—3) мин и измеряют оптическую плотность
(2.2.25) каждого раствора при длине волны 340 нм,
используя воду Рв качестве компенсационной жидкости.
В каждую кювету прибавляют по 0,05 мл раствора
альдегиддегидрогеназы Р, перемешивают и плотно
закрывают пробкой, выдерживают при температуре
(22±2)°С в течение 5 мин. Измеряют оптическую
плотность каждого раствора при 340 нм, используя
воду Р в качестве компенсационной жидкости.
Содержание альдегидов рассчитывают по
формуле:
(Д2 - А1) - (А* - А>1) юоооо-с
(Аг ” А1)— (А2 ~ А1) т
где:
А" — оптическая плотность испытуемого
раствора до прибавления альдегиддегидрогеназы;
Аа — оптическая плотность испытуемого
раствора после прибавления альдегиддегидрогеназы;
Аз1 — оптическая плотность раствора сравнения
до прибавления альдегиддегидрогеназы;
Аз2 — оптическая плотность раствора сравнения
после прибавления альдегиддегидрогеназы;
А1 — оптическая плотность контрольного
раствора до прибавления альдегиддегидрогеназы;
АЬ2 — оптическая плотность контрольного
раствора после прибавления альдегиддегидрогеназы;
т — масса навески повидона в пересчете на
безводное вещество, г;
С — концентрация ацетальдегида в
растворе сравнения, рассчитанная из массы
ацетальдегидаммиака тримера тригидрата с учетом
коэффициента 0,72, мг/мл.
Пероксиды. Не более 0,0400 % (400 ррт) в
пересчете на Н202.
Количество испытуемого образца, эквивалентное
4.0 г безводного вещества, растворяют в воде Р и
доводят до объема 100,0 мл этим же растворителем
(исходный раствор). К 25,0 мл исходного растворе
прибавляют 2,0 мл реактива титана хлорида (III) и
кислоты серной Р и выдерживают в течение 30 мин
Измеряют оптическую плотность (2.2.25) полученного
раствора при длине волны 405 нм, используя е
качестве компенсационного раствора смесь из
25.0 мл исходного раствора и 2,0 мл 13% (об/об
раствора кислоты серной Р. Оптическая плотность
не должна превышать 0,35.
Муравьиная кислота. Не более 0,5 %. Жидкостная
хроматография (2.2.29).
Повидон
823
Испытуемый раствор. Количество испытуемого
образца, эквивалентное 2,0 г безводного вещества,
растворяют в воде Р и доводят до объема 100,0 мл
этим же растворителем (исходный испытуемый раст¬
вор). Суспензию сильнокислотной ионообменной
смолы Р для колоночной хроматографии в воде Р
помещают в колонку с внутренним диаметром около
0,8 см для получения слоя сорбента высотой около
20 мм и следят за тем, чтобы сильнокислотная ионо¬
обменная смола была постоянно погружена в воду Р.
Прибавляют 5 мл воды Р и настраивают скорость по¬
тока таким образом, чтобы она составляла около 20
капель воды в минуту. Когда уровень воды опустится
почти до верхнего края сорбента, в колонку помеща¬
ют исходный испытуемый раствор. После истечения
2 мл раствора собирают 1,5 мл элюата и используют
его в качестве испытуемого раствора.
Раствор сравнения. 0,100 г кислоты муравьиной
безводной Р растворяют в воде Р и доводят до объ¬
ема 100,0 мл этим же растворителем. 1,0 мл получен¬
ного раствора доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной (0,25—0,30) м и внутренним
диаметром (4—8) мм, заполненная ионообменной
смолой сильнокислотной Р для колоночной хромато¬
графии с размером частиц (5—10) мкм;
- температура: 30 °С;
- подвижная фаза: 5 мл кислоты хлорной Р до¬
водят водой Р до объема 1000 мл;
- скорость подвижной фазы: устанавливают та¬
ким образом, чтобы время удерживания муравьиной
кислоты было около 11 мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 50 мкл.
Пригодность хроматографической системы:
раствор сравнения:
- относительное стандартное отклонение: не
более 2,0 % при 6 повторных вводах пробы.
Предельное содержание примесей:
- муравьиная кислота: на хроматограмме испы¬
туемого раствора площадь пика, соответствующего
муравьиной кислоте, не должна превышать 10-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения.
Гидразин. Не более 0,0001 % (1 ррт). Тонкос¬
лойная хроматография (2.2.27). Используют свеже¬
приготовленные растворы.
Испытуемый раствор. Количество испытуемо¬
го образца, эквивалентное 2,5 г безводного веще¬
ства, растворяют в 25 мл воды Р, прибавляют 0,5 мл
раствора 50 г/л салицилового альдегида Р в мета¬
ноле Р, перемешивают и нагревают в водяной бане
при температуре 60 °С в течение 15 мин. Охлажда¬
ют, прибавляют 2,0 мл толуола Р, встряхивают в те¬
чение 2 мин и центрифугируют. Используют верхний
слой жидкости.
Раствор сравнения. 90 мг салицилового альде¬
гида азина Р растворяют в толуоле Р и доводят до
объема 100 мл этим же растворителем. 1 мл получен¬
ного раствора доводят толуолом Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р254 Р.
Подвижная фаза: вода Р—метанол Р (1:2, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 365 нм.
Фактор подвижности: азин салицилового аль¬
дегида — около 0,3.
Предельное содержание примесей:
- гидразин: на хроматограмме испытуемого
раствора пятно, соответствующее азину салицилово¬
го альдегида, должно быть не интенсивнее пятна на
хроматограмме раствора сравнения.
Примесь А. Не более 0,0010 % (10 ррт). Жид¬
костная хроматография (2.2.29).
Испытуемый раствор. Количество испытуемого
образца, эквивалентное 0,250 г безводного вещества,
растворяют в подвижной фазе и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг 1-винилпирро-
лидин-2-она Р (примесь А) растворяют в метаноле Р и
доводят до объема 100,0 мл этим же растворителем.
1.0 мл полученного раствора доводят метанолом Р до
объема 100,0 мл. 5,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 10 мг 1-винилипирро-
лидин-2-она Р и 0,5 г винилацетата Р растворяют в
метаноле Р и доводят до объема 100,0 мл этим же
растворителем. 1,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- предкопонка длиной 0,025 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- колонка длиной 0,25 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
-подвижная фаза: ацетонитрил Р — вода Р
(10:90, об/об);
- скорость подвижной фазы: регулируют таким
образом, чтобы время удерживания примеси А со¬
ставляло около 10 мин;
-спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: 50 мкл; через 2 мин
после каждого введения испытуемого раствора про¬
мывают предколонку, пропуская подвижную фазу про¬
тивотоком с такой же скоростью, как в испытании, в
течение 30 мин.
Пригодность хроматографической системы:
-разрешение не менее 2,0 между пиками при¬
меси А и винилацетата на хроматограмме раствора
сравнения (Ь);
- относительное стандартное отклонение:
не более 2,0 % при 6 повторных вводах раствора
сравнения (а).
Предельное содержание примесей:
-примесь А: на хроматограмме испытуемого
раствора площадь пика, соответствующего примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (а).
Примесь В. Не более 3,0 %. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. Количество испытуемого
образца, эквивалентное 0,100 г безводного вещества,
растворяют в воде Р и доводят до объема 50,0 мл
этим же растворителем.
824
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения. 0,100 г 2-пирролидона Р
(примесь В) растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем. 3,0 мл полученного
раствора доводят водой Р до объема 50,0 мл.
Условия хроматографирования:
- предколонка длиной 0,025 м и внутренним
диаметром 3 мм, заполненная силикагелем
октадецилсилильным эндкепированным для
хроматографии Р с размером частиц 5 мкм;
- колонка длиной 0,25 м и внутренним диаметром
3 мм, заполненная силикагелем октадецилсилильным
эндкепированным для хроматографии Р с размером
частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: вода Р, доведенная до рН 2,4
кислотой фосфорной Р;
- скорость подвижной фазы: регулируют таким
образом, чтобы время удерживания примеси В
составляло около 11 мин;
- спектрофотометрический детектор, длина
волны 205 нм;
- объем вводимой пробы: 50 мкл; после каждого
введения испытуемого раствора промывают предко-
лонку от полимерных материалов повидона, пропу¬
ская подвижную фазу противотоком с такой же скоро¬
стью, как в испытании, в течение 30 мин.
Пригодность хроматографической системы:
раствор сравнения:
- относительное стандартное отклонение: не
более 2,0 % при 6 повторных вводах пробы.
Предельное содержание примесей:
-примесь В: на хроматограмме испытуемого
раствора площадь пика, соответствующего примеси
В, не должна превышать площадь основного пика на
хроматограмме раствора сравнения.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 5,0%. Определение
проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать
требования статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца (т, мг) помещают в
колбу для сжигания, прибавляют 5 г смеси из 1 г меди
сульфата Р, 1 г титана диоксида Р и 33 г калия суль¬
фата Р и 3 стеклянных шарика. Смывают прилипшие
частицы с горла колбы внутрь колбы небольшим ко¬
личеством воды Р. Прибавляют 7 мл кислоты сер¬
ной Р по стенкам колбы. Постепенно нагревают колбу
до получения прозрачного желтовато-зеленого раст¬
вора и исчезновения обуглившихся частиц с внутрен¬
них стенок колбы, и затем продолжают нагревание
еще в течение 45 мин. Охлаждают, затем осторожно
прибавляют 20 мл воды Р и помещают в аппарат для
перегонки, предварительно очищенный пропусканием
через него водяного пара. В колбу-приемник прибав¬
ляют 30 мл раствора 40 г/л борной кислоты Р, 3 кап¬
ли раствора бромкрезолового зеленого и метилово¬
го красного Р и воду в количестве, достаточном для
того, чтобы нижний конец трубки холодильника был
погружен в раствор. Прибавляют 30 мл раствора на¬
трия гидроксида концентрированного Р через во¬
ронку, воронку промывают осторожно 10 мл воды Р,
немедленно закрывают зажим на резиновой трубке,
затем перегоняют с водяным паром до получения от
80 мл до 100 мл дистиллята. Перемещают колбу-при¬
емник от нижнего конца трубки холодильника, опола¬
скивая конец трубки небольшим количеством воды Р.
Дистиллят титруют 0,025 М раствором кислоты сер¬
ной до перехода окраски раствора от зеленой через
бледную серовато-голубую до бледной серовато-
красновато-пурпурной.
Параллельно проводят контрольный опыт.
1 мл 0,025 М раствора кислоты серной соответ¬
ствует 0,7004 мг N.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
МАРКИРОВКА
Указывают К-значение.
ПРИМЕСИ
А. 1-Этенилпирролидин-2-он (1-винил пиррол идин-
2-он).
В. Пирролидин-2-он (2-пирролидон).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомогатель¬
ного вещества (см. статью 5.15). Некоторые из ха¬
рактеристик, описанных в разделе «Функционально-
обусловленные характеристики», могут также при¬
сутствовать в части фармакопейной статьи, обяза¬
тельной для выполнения, так как одновременно они
являются и показателями качества. В таком случае
в разделе «Функционально-обусловленные характери¬
стики» дается перекрестная ссылка на это испыта¬
ние в части фармакопейной статьи, обязательной
для выполнения. Контроль данных характеристик
может вносить вклад в качество лекарственного
средства путем достижения постоянства произ¬
водственного процесса и улучшения свойств лекар¬
ственного средства при его использовании. Предла¬
гаемые методы испытаний являются подходящими
для указанных целей, однако другие методы также
могут быть использованы. В случае указания резуль¬
татов конкретных характеристик требуется указы¬
вать также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для повидона, используемого в качестве солюби¬
лизатора и стабилизатора при производстве жид¬
ких дозированных лекарственных форм.
Вязкость (2.2.9). Определяют динамическую
вязкость 10 % раствора (в пересчете на сухое веще¬
ство) при температуре 25 °С с использованием капил-
Повидон-йод
825
лярного вискозиметра. Типичные значения указаны в
таблице 0685.-1.
Молекулярная масса (см. испытание «Вязкость,
выраженная через К-значение»). Типичные значения
указаны в таблице 0685.-1.
Следующая характеристика может быть важна
для повидона, используемого в качестве связующего
вещества при производстве таблеток и гранул.
Молекулярная масса (см. испытание «Вязкость,
выраженная через К-значение»). Типичные значения
указаны в таблице 0685.-1.
ПРОИЗВОДСТВО
В производстве используют повидон, который
выдерживает требования статьи Повидон, с содержа¬
нием не более 2,0 % кислоты муравьиной и не более
8,0 % воды.
ОПИСАНИЕ (СВОЙСТВА)
Аморфный порошок от желтовато-коричневого
до красновато-коричневого цвета.
Растворим в воде и в 96 % спирте, практически
нерастворим в ацетоне.
Таблица 0685.-1.
Типичные диапазоны вязкости и диапазоны
вязкости, выраженной через К-значение
Диапазон вязкости
(мПа-с)
Молекулярная
масса: вязкость,
выраженная через
К-значение
Повидон К 12
1,3—2,3
11—14
Повидон К 17
1,5—3,5
16—18
Повидон К 25
3,5—5,5
24—27
Повидон К 30
5,5—8,5
28—32
Повидон К 90
300—700
85—95
07/2016:1142
повидон-йод
РоуМопит ю<Япа1ит
РО)/ЮОЫЕ, ЮйШАТЕО
ОПРЕДЕЛЕНИЕ
Комплекс йода и повидона.
Содержание: от 9,0 % до 12,0 % доступного йода
(в пересчете на сухое вещество).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО повидон-йода #или спектр,
представленный на рисунке #1142.-1#.
B. 10 мг испытуемого образца растворяют в
10 мл воды Р, прибавляют 1 мл раствора крахма¬
ла Р. Появляется интенсивное синее окрашивание.
ИСПЫТАНИЯ
рН (2.2.3). От 1,5 до 5,0.1,0 г испытуемого образ¬
ца растворяют в 10 мл воды, свободной от углерода
диоксида, Р.
Йодиды. Не более 6,0 % (в пересчете на сухое
вещество). 0,500 г испытуемого образца растворяют
в 100 мл воды Р, прибавляют натрия метабисуль¬
фит Р до исчезновения окраски йода. Прибавляют
25,0 мл 0,1 М раствора серебра нитрата, 10 мл кис¬
лоты азотной Р и 5 мл раствора железа (III) аммо¬
ния сульфата Р2 и титруют 0,1 М раствором аммо¬
ния тиоцианата.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора аммония тиоцианата соот¬
ветствует 12,69 мг общего содержания йода. Для рас¬
чета содержания йодидов из полученного значения, в
Рисунок #1142.-1. Инфракрасный спектр ФСО повидон-йода
826
Гэсударственная фармакопея Республики Беларусь
пересчете на сухое вещество, вычитают процентное
содержание доступного йода, полученное в разделе
«Количественное определение».
Потеря в массе при высушивании (2.2.32). Не
более 8,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца помещают в сте¬
клянную колбу с притертой пробкой, содержащую
150 мл воды Р, и перемешивают в течение 1 ч. При¬
бавляют 0,1 мл кислоты уксусной разведенной Р и
титруют 0,1 М раствором натрия тиосульфата, ис¬
пользуя в качестве индикатора раствор крахмала Р.
1 мл 0,1 М раствора натрия тиосульфата со¬
ответствует 12,69 мг доступного йода.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1371
ПОДСОЛНЕЧНОЕ МАСЛО
РАФИНИРОВАННОЕ
НеНапМ)/ аппш о1еит гаШпа1ит
5С/А/Е/.01УЕК ОН, РЕЕШЕй
ОПРЕДЕЛЕНИЕ
Жирное масло, полученное из семян НеНапМиз
аппииз 1_. методом механического отжима или экс¬
тракцией, с последующим рафинированием. Масло
может содержать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная светло-желтая жидкость.
Практически нерастворимо в воде и 96 % спирте,
смешивается с петролейным эфиром (температура
кипения от 40 °С до 60 °С).
Относительная плотность: около 0,921.
Показатель преломления: около 1,474.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Идентификация жирных масел методом тонкос¬
лойной хроматографии (2.3.2).
Результаты: хроматограмма испытуемого об¬
разца аналогична соответствующей хроматограмме,
представленной на рисунке 2.3.2.-1.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 0,5. Опреде¬
ление проводят из 10,0 г испытуемого образца.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 10,0.
Неомыляемые вещества (2.5.7). Не более 1,5 %.
Определение проводят из 5,0 г испытуемого образца.
Щелочные примеси в жирных маслах (2.4.19).
Испытуемый образец должен выдерживать испыта¬
ние на щелочные примеси в жирных маслах.
Состав жирных кислот (2.4.22, метод А). Ис¬
пользуют градуировочную смесь, указанную в табли¬
це 2.4.22.-3.
Состав фракции жирных кислот масла:
- пальмитиновая кислота: от 4,0 % до 9,0 %;
- стеариновая кислота: от 1,0 % до 7,0 %;
- олеиновая кислота: от 14,0 % до 40,0 %;
- линолевая кислота: от 48,0 % до 74,0 %.
Вода (2.5.32). Не более 0,1 %. Определение про¬
водят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом
контейнере в защищенном от света месте.
МАРКИРОВКА
Указывают:
-метод получения масла (механический отжим
или экстракция).
07/2016:1961
ПОЛИВИНИЛОВЫЙ СПИРТ
Ро1у(а1соНо1 VIтуНсив)
Р01У(УШУ1. АМОНОЦ
ОПРЕДЕЛЕНИЕ
Поливиниловый спирт получают полимериза¬
цией винилацетата с последующим частичным или
почти полным гидролизом поливинилацетата в при¬
сутствии каталитического количества щелочей или
минеральных кислот.
Полимеры поливинилового спирта соответствуют
индексу:
0 < — < 0,35
т
Средняя относительная молекулярная масса:
от 20 000 до 150 000.
Вязкость: от 3 мПа-с до 70 мПа-с.
Эфирное число, характеризующее степень ги¬
дролиза: не более 280.
ОПИСАНИЕ(СВОЙСТВА)
Желтовато-белый порошок или полупрозрачные
гранулы.
Растворим в воде, мало растворим в этаноле,
практически нерастворим в ацетоне.
Существуют различные виды поливинилового
спирта, которые отличаются степенью полимериза¬
ции и степенью гидролиза, что обуславливает раз¬
личие их физических свойств. Они характеризуются
вязкостью и эфирным числом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Полисорбат 20
827
Сравнение: ФСО поливинилового спирта.
Интенсивности полос поглощения при около
1720 см-1 и 1260 см"1 обратно пропорциональны сте¬
пени гидролиза.
В. Испытуемый образец выдерживает испытание
«Вязкость», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 250 мл воды Р помещают в кругло¬
донную колбу из боросиликатного стекла с обратным
холодильником и мешалкой и нагревают на водяной
бане. Прибавляют 10,0 г испытуемого образца (с уче¬
том потери в массе при высушивании) и продолжают
нагревать в течение 30 мин, непрерывно перемеши¬
вая. Снимают колбу с водяной бани и продолжают
перемешивать до охлаждения раствора до комнатной
температуры.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)7.
рН (2.2.3). От 4,5 до 6,5. Измеряют рН раствора 5.
Вязкость (2.2.49). Не менее 85 % и не более
115% от указанного номинального значения. Изме¬
ряют вязкость раствора 3 сразу после приготовления
при температуре (20±0Г1) °С с помощью вискозиметра
с падающим шариком.
Кислотное число. Не более 3,0. К 50 мл раст¬
вора 3 прибавляют 1 мл раствора фенолфталеи¬
на Р и титруют 0,05 М раствором калия гидроксида
до появления розового окрашивания, не исчезающего
в течение 15 с.
Кислотное число рассчитывают по формуле:
2,805 V
2 ’
где:
V — объем 0,05 М раствора калия гидроксида,
пошедший на титрование, мл.
Эфирное число (2.5.2). Не менее 90 % и не
более 110% от указанного номинального значения.
Омыляют 1,00 г испытуемого образца в смеси из
25,0 мл 0,5 М раствора калия гидроксида спиртово¬
го и 25,0 мл воды Р (2.5.6).
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 1,0%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
МАРКИРОВКА
Указывают:
- вязкость раствора с концентрацией 40 г/л;
- эфирное число.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Некоторые из характеристик, описанных в раз¬
деле «Функционально-обусловленные характери¬
стики», могут также присутствовать в части
фармакопейной статьи, обязательной для выпол¬
нения, так как одновременно они являются и по¬
казателями качества. В таком случае в разделе
«Функционально-обусловленные характеристики»
дается перекрестная ссылка на это испытание
в части фармакопейной статьи, обязательной
для выполнения. Контроль данных характеристик
может вносить вклад в качество лекарственного
средства путем достижения постоянства про¬
изводственного процесса и улучшения свойств
лекарственного средства при его использовании.
Предлагаемые методы испытаний являются под¬
ходящими для указанных целей, однако другие ме¬
тоды также могут быть использованы. В случае
указания результатов конкретных характеристик
требуется указывать также и методы проведе¬
ния испытаний.
Следующие характеристики могут быть важ¬
ны для поливинилового спирта, используемого в ка¬
честве добавки, повышающей вязкость, связующего
вещества или пленкообразователя.
Вязкость (см. Испытания).
Эфирное число (см. Испытания).
Абсорбционная спектрофотометрия в инфра¬
красной области (см. Идентификация А).
07/2016:0426
ПОЛИСОРБАТ 20
Ро!у80гЬа1ит 20
Р01-УЗОКВАТЕ 20
ОПРЕДЕЛЕНИЕ
Смесь неполных (частично этерифицированных)
эфиров жирных кислот, в основном лауриновой (до-
декановой) кислоты, с сорбитолом и его ангидрида¬
ми, этоксилированных таким образом, что на каждый
моль сорбитола и ангидридов сорбитола приходится
около 20 моль этиленоксида.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная или слегка опалесцирующая, мас¬
лянистая жидкость желтого или коричневато-желтого
цвета.
Растворим в воде, в этаноле, в этилацетате и в
метаноле, практически нерастворим в жирных маслах
и в вазелиновом масле.
Относительная плотность: около 1,10.
Вязкость: около 400 мПа-с при 25 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр натрия полисор-
бата 20 по Европейской Фармакопее.
B. Испытуемый образец выдерживает испытание
«Гидроксильное число», как указано в разделе «Ис¬
пытания».
828
Государственная фармакопея Республики Беларусь
С. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испытания».
О. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
Е. 0,1 г испытуемого образца растворяют в 5 мл
метиленхлорида Р. Прибавляют 0,1 г калия тиоциа¬
ната Р и 0,1 г кобальта нитрата Р. Перемешивают
стеклянной палочкой. Появляется синее окрашивание
раствора.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 2,0. 5,0 г ис¬
пытуемого образца растворяют в 50 мл предписанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). От 96
до 108.
Перекисное (пероксидное) число. Не более
10,0. 10,0 г испытуемого образца помещают в хими¬
ческий стакан вместимостью 100 мл и растворяют в
20 мл кислоты уксусной ледяной Р. Прибавляют 1 мл
раствора калия йодида насыщенного Р, перемеши¬
вают и выдерживают в течение 1 мин. Прибавляют
50 мл воды, свободной от углерода диоксида, Р и пе¬
ремешивают на магнитной мешалке. Титруют 0,01 М
раствором натрия тиосульфата потенциометриче-
ски (2.2.20).
Параллельно проводят контрольный опыт.
Рассчитывают перекисное число по формуле:
(л1 -п2)МЮ00
т
где:
л1 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование испытуемого образ¬
ца, мл;
п2 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование в контрольном опы¬
те, мл;
М — молярная концентрация титрованного раст¬
вора натрия тиосульфата, моль/л;
т — масса навески испытуемого образца, г.
Число омыления (2.5.6). От 40 до 50. Определе¬
ние проводят из 4,0 г испытуемого образца. Использу¬
ют 15,0 мл 0,5 М спиртового раствора калия гидрок¬
сида и перед проведением титрования доводят 96 %
спиртом Р до объема 50 мл. Нагревают с обратным
холодильником в течение 60 мин.
Состав жирных кислот (2.4.22, метод С). Готовят
раствор сравнения (а), как указано в таблице 2.4.22.-2.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 50 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—14
80 - 220
14—54
220
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Состав фракции жирных кислот испытуемого
образца:
- капроновая кислота: не более 1,0 %;
- каприловая кислота: не более 10,0 %;
- каприновая кислота: не более 10,0 %;
- лауриновая кислота: от 40,0 % до 60,0 %;
- миристиновая кислота: от 14,0 % до 25,0 %;
- пальмитиновая кислота: от 7,0 % до 15,0 %;
- стеариновая кислота: не более 7,0 %;
- олеиновая кислота: не более 11,0 %;
- линолевая кислота: не более 3,0 %.
Этиленоксид и диоксан (2.4.25, метод А). Не
более 0,0001 % (1 ррт) этиленоксида и не более
0,0010 % (10 ррт) диоксана.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 1,00 г испытуемого образца.
Общая зола (2.4.16). Не более 0,25 %. Опреде¬
ление проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:1914
ПОЛИСОРБАТ 40
Ро1у$огЬа1ит 40
Р01Х80РВАТЕ 40
ОПРЕДЕЛЕНИЕ
Смесь неполных (частично этерифицированных)
эфиров жирных кислот, в основном пальмитиновой
кислоты (см. Пальмитиновая кислота (1904)), с сор¬
битолом и его ангидридами, этоксилированных таким
образом, что на каждый моль сорбитола и ангидридов
сорбитола приходится около 20 моль этиленоксида.
ОПИСАНИЕ (СВОЙСТВА)
Вязкая маслянистая жидкость желтоватого или
коричневато-желтого цвета.
Смешивается с водой, этанолом, этилацетатом и
метанолом, практически нерастворим в жирных мас¬
лах и вазелиновом масле.
Относительная плотность: около 1,10.
Вязкость: около 400 мПа-с при 30 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр натрия полисор-
бата 40 по Европейской Фармакопее.
B. Испытуемый образец выдерживает испытание
«Гидроксильное число», как указано в разделе «Ис¬
пытания».
C. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испыта¬
ния».
Полисорбат 60
829
Р. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
Е. 0,1 г испытуемого образца растворяют в 5 мл
метиленхлорида Р. Прибавляют 0,1 г калия тиоциа¬
ната Р и 0,1 г кобальта нитрата Р. Перемешивают
стеклянной палочкой. Появляется синее окрашивание
раствора.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 2,0. 5,0 г ис¬
пытуемого образца растворяют в 50 мл указанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). От 89
до 105.
Перекисное (пероксидное) число. Не более
10,0. 10,0 г испытуемого образца помещают в хими¬
ческий стакан вместимостью 100 мл и растворяют в
20 мл кислоты уксусной ледяной Р. Прибавляют 1 мл
раствора калия йодида насыщенного Р, перемеши¬
вают и выдерживают в течение 1 мин. Прибавляют
50 мл воды, свободной от углерода диоксида, Р и пе¬
ремешивают на магнитной мешалке. Титруют 0,01 М
раствором натрия тиосульфата потенциометриче-
ски (2.2.20).
Параллельно проводят контрольный опыт.
Рассчитывают перекисное число по формуле:
(л1 - л2)'М'Ю00
т
где:
л1 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование испытуемого образ¬
ца, мл;
п2 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование в контрольном опы¬
те, мл;
М — молярная концентрация титрованного раст¬
вора натрия тиосульфата, моль/л;
т — масса навески испытуемого образца, г.
Число омыления (2.5.6). От 41 до 52. Определе¬
ние проводят из 4,0 г испытуемого образца. Использу¬
ют 15,0 мл 0,5 М спиртового раствора калия гидрок¬
сида и перед проведением титрования доводят 96 %
спиртом Р до объема 50 мл. Нагревают с обратным
холодильником в течение 60 мин.
Состав жирных кислот (2.4.22, метод С). Го¬
товят раствор сравнения (а), как указано в таблице
2.4.22.-1.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 50 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—14
80 220
14—54
220
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Состав фракции жирных кислот испытуемого
образца:
- пальмитиновая кислота: не менее 92,0 %.
Этиленоксид и диоксан (2.4.25, метод А). Не
более 0,0001 % (1 ррт) этиленоксида и 0,0010%
(10 ррт) диоксана.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 1,00 г испытуемого образца.
Общая зола (2.4.16). Не более 0,25 %. Опреде¬
ление проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0427
ПОЛИСОРБАТ 60
Ро1узогЬа1ит 60
Р01-УЗОКВАТЕ 60
ОПРЕДЕЛЕНИЕ
Смесь неполных (частично этерифицированных)
эфиров жирных кислот, в основном стеариновой кис¬
лоты 50 (см. статью Стеариновая кислота 50 (1474)),
с сорбитолом и его ангидридами, этоксилированных
таким образом, что на каждый моль сорбитола и ан¬
гидридов сорбитола приходится около 20 моль этиле¬
ноксида.
ОПИСАНИЕ (СВОЙСТВА)
Желтовато-коричневая желатинообразная мас¬
са, при температуре выше 25 °С — прозрачная жид¬
кость.
Растворим в воде, в этаноле, в этилацетате и в
метаноле, практически нерастворим в жирных маслах
и в вазелиновом масле.
Относительная плотность: около 1,10.
Вязкость: около 400 мПа-с при 30 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр натрия полисор-
бата 60 по Европейской Фармакопее.
B. Испытуемый образец выдерживает испытание
«Гидроксильное число», как указано в разделе «Ис¬
пытания».
C. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испытания».
О. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
Е. 0,1 г испытуемого образца растворяют в 5 мл
метиленхлорида Р. Прибавляют 0,1 г калия тиоциа¬
ната Р и 0,1 г кобальта нитрата Р. Перемешивают
стеклянной палочкой. Появляется синее окрашивание
раствора.
830
Государственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 2,0. 5,0 г ис¬
пытуемого образца растворяют в 50 мл предписанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). От 81
до 96.
Перекисное (пероксидное) число. Не более 10,0.
10,0 г испытуемого образца помещают в химический
стакан вместимостью 100 мл и растворяют в 20 мл кис¬
лоты уксусной ледяной Р. Прибавляют 1 мл раствора
калия йодида насыщенного Р, перемешивают и вы¬
держивают в течение 1 мин. Прибавляют 50 мл воды,
свободной от углерода диоксида, Р и перемешивают
на магнитной мешалке. Титруют 0,01 М раствором на¬
трия тиосульфата потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
Рассчитывают перекисное число по формуле:
(п,-п2)М-тО
т
где:
пЛ — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование испытуемого образ¬
ца, мл;
п2 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование в контрольном опы¬
те, мл;
М — молярная концентрация титрованного раст¬
вора натрия тиосульфата, моль/л;
т — масса навески испытуемого образца, г.
Число омыления (2.5.6). От 45 до 55. Определе¬
ние проводят из 4,0 г испытуемого образца. Использу¬
ют 15,0 мл 0,5 М спиртового раствора калия гидрок¬
сида и перед проведением титрования доводят 96 %
спиртом Р до объема 50 мл. Нагревают с обратным
холодильником в течение 60 мин.
Состав жирных кислот (2.4.22, метод С). Го¬
товят раствор сравнения (а), как указано в таблице
2.4.22.-1.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р\
- линейная скорость: 50 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—14
80 — 220
14—54
220
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Состав фракции жирных кислот испытуемого
образца:
- стеариновая кислота: от 40,0 % до 60,0 %;
- суммарное содержание пальмитиновой и сте¬
ариновой кислот: не менее 90,0 %.
Этиленоксид и диоксан (2.4.25, метод А). Не
более 0,0001 % (1 ррт) этиленоксида и не более
0,0010 % (10 ррт) диоксана.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 1,00 г испытуемого образца.
Общая зола (2.4.16). Не более 0,25 %. Опреде¬
ление проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0428
ПОЛИСОРБАТ 80
Ро1у$огЬа1ит 80
Р01Х80ПВАТЕ 80
ОПРЕДЕЛЕНИЕ
Смесь неполных (частично этерифицированных)
эфиров жирных кислот, в основном олеиновой кисло¬
ты (см. статью Олеиновая кислота (0799)) с сорби¬
толом и его ангидридами, этоксилированных таким
образом, что на каждый моль сорбитола и ангидридов
сорбитола приходится около 20 моль этиленоксида.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная или слегка опалесцирующая, масля¬
нистая жидкость от бесцветной до коричневато-жел¬
того цвета.
Диспергируется в воде, в этаноле, в этилацета-
те и в метаноле, практически нерастворим в жирных
маслах и вазелиновом масле.
Относительная плотность: около 1,10.
Вязкость: около 400 мПа-с при 25 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О, Е.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр натрия полисор-
бата 80 по Европейской Фармакопее.
B. Испытуемый образец выдерживает испытание
«Гидроксильное число», как указано в разделе «Ис¬
пытания».
C. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испытания».
О. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
Е. 0,1 г испытуемого образца растворяют в 5 мл
метиленхлорида Р. Прибавляют 0,1 г калия тиоциа¬
ната Р и 0,1 г кобальта нитрата Р. Перемешивают
стеклянной палочкой. Появляется синее окрашивание
раствора.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 2,0. 5,0 г ис¬
пытуемого образца растворяют в 50 мл предписанной
смеси растворителей.
Гидроксильное число (2.5.3, метод А). От 65
до 80.
Перекисное (пероксидное) число. Не более 10,0.
10,0 г испытуемого образца помещают в химический
стакан вместимостью 100 мл и растворяют в 20 мл кис-
Полисорбат 80
831
лоты уксусной ледяной Р. Прибавляют 1 мл раствора
калия йодида насыщенного Р, перемешивают и вы¬
держивают в течение 1 мин. Прибавляют 50 мл воды,
свободной от углерода диоксида, Р и перемешивают
на магнитной мешалке. Титруют 0,01 М раствором на¬
трия тиосульфата потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
Рассчитывают перекисное число по формуле:
(/г, -л2)МЮ 00
т
где:
л1 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование испытуемого образ¬
ца, мл;
п2 — объем 0,01 М раствора натрия тиосуль¬
фата, пошедший на титрование в контрольном опы¬
те, мл;
М — молярная концентрация титрованного раст¬
вора натрия тиосульфата, моль/л;
т — масса навески испытуемого образца, г.
Число омыления (2.5.6). От 45 до 55. Опреде¬
ление проводят из 4,0 г испытуемого образца. Ис¬
пользуют 30,0 мл 0,5 М спиртового раствора калия
гидроксида, нагревают с обратным холодильником в
течение 60 мин и перед проведением титрования при¬
бавляют 50 мл этанола Р.
Состав жирных кислот (2.4.22, метод С). Ис¬
пользуют смесь веществ для градуировки, как указано
в таблице 2.4.22.-3.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 50 см/с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—14
80 -► 220
14—54
220
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Состав фракции жирных кислот испытуемого
образца:
- миристиновая кислота: не более 5,0 %;
- пальмитиновая кислота: не более 16,0 %;
- пальмитолеиновая кислота: не более 8,0 %;
- стеариновая кислота: не более 6,0 %;
- олеиновая кислота: не менее 58,0 %;
-линолевая кислота: не более 18,0 %;
- линоленовая кислота: не более 4,0 %.
Этиленоксид и диоксан. Не более 0,0001 %
(1 ррт) этиленоксида и не более 0,0010 % (10 ррт)
диоксана.
Парофазная газовая хроматография (2.2.28).
Исходный раствор этиленоксида. 0,5 мл ком¬
мерчески доступного раствора этиленоксида в мети-
ленхлориде (50 мг/мл) доводят водой Р до объема
50,0 мл. (ПРИМЕЧАНИЕ: раствор стабилен в течение
3 месяцев при хранении в виалах, закрытых крышкой
с силиконовой мембраной, покрытой политетрафторэ¬
тиленом, при температуре -20 °С). Выдерживают до
достижения комнатной температуры. 1,0 мл получен¬
ного раствора доводят водой Р до объема 250,0 мл.
Исходный раствор диоксана. 1,0 мл диоксана Р
доводят водой Р до объема 200,0 мл. 1,0 мл получен¬
ного раствора доводят водой Р до объема 100,0 мл.
Исходный раствор ацетальдегида. Около
0,100 г ацетальдегида Р помещают в мерную колбу
вместимостью 100 мл и доводят водой Р до объема
100.0 мл.
Эталонный раствор. К 6,0 мл исходного раст¬
вора этиленоксида прибавляют 2,5 мл исходного раст¬
вора диоксана и доводят водой Р до объема 25,0 мл.
Испытуемый раствор (а). 1,00 г испытуемого об¬
разца помещают в виалу для парофазной газовой хро¬
матографии вместимостью 10 мл, прибавляют 2,0 мл
воды Р и немедленно закрывают виалу силиконовой
мембраной, покрытой политетрафторэтиленом, и алю¬
миниевым колпачком. Тщательно перемешивают.
Испытуемый раствор (Ь). 1,00 г испытуемого
образца помещают в виалу для парофазной газовой
хроматографии вместимостью 10 мл, прибавляют
2.0 мл эталонного раствора и немедленно закрыва¬
ют виалу силиконовой мембраной, покрытой полите¬
трафторэтиленом, и алюминиевым колпачком. Тща¬
тельно перемешивают.
Раствор сравнения. 2,0 мл исходного раствора
ацетальдегида и 2,0 мл исходного раствора этиле¬
ноксида помещают в виалу для парофазной газовой
хроматографии вместимостью 10 мл и немедленно
закрывают виалу силиконовой мембраной, покрытой
политетрафторэтиленом, и алюминиевым колпачком.
Тщательно перемешивают.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 50 м и
диаметром 0,53 мм, покрытая слоем поли(диметил)-
(дифенил)силоксана Р (толщина слоя 5 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 4,0 мл/мин;
- деление потока: 1:3,5;
- параметры парофазного пробоотборника:
- равновесная температура: 80 °С;
- время достижения равновесия: 60 мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—18
70 -► 250
18—23
250
Блок ввода проб
85
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1,0 мкл испытуемых
растворов (а) и (Ь) и раствора сравнения.
Относительное удерживание (по отношению к
этиленоксиду, время удерживания — около 6,5 мин):
ацетальдегид — около 0,9; диоксан — около 1,9.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: не менее 2,0 между пиками аце¬
тальдегида и этиленоксида.
Содержание этиленоксида рассчитывают по
формуле:
2 • Сео • Аа
Аь~Аа 9
где:
СЕО — концентрация этиленоксида, добавленно¬
го в испытуемый раствор (Ь), мкг/мл;
Аа — площадь пика этиленоксида на хромато¬
грамме испытуемого раствора (а);
832
Гэсударственная фармакопея Республики Беларусь
Аь — площадь пика этиленоксида на хромато¬
грамме испытуемого раствора (Ь).
Содержание диоксана рассчитывают по формуле:
2 • 1,03 • С0 • Аа,
Аь.-А,
где:
С0 — концентрация диоксана, добавленного в ис¬
пытуемый раствор (Ь), мкл/мл;
1,03 — плотность диоксана, г/мл;
Аа, — площадь пика диоксана на хроматограмме
испытуемого раствора (а);
Аь, — площадь пика диоксана на хроматограмме
испытуемого раствора (Ь).
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 1,00 г испытуемого образца.
Общая зола (2.4.16). Не более 0,25 %. Опреде¬
ление проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:2416
ПРАМИПЕКСОЛА ДИГИДРОХЛОРИД
МОНОГИДРАТ
Ргат/рехоН сШ1ус1госЫопс1ит топоЬус/псит
РКАМ1РЕХ01-Е О/НУОРОСШОРЮЕ
МОЫОНУОРАТЕ
С10Н19С12М35 ■ н20 М.м. 302,3
[191217-81-9]
ОПРЕДЕЛЕНИЕ
(65)-6-Л/-Пропил-4,5,6,7-тетрагидро-1,3-бензотиа-
зол-2,6-диамина дигидрохпорид моногидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, растворим в метано¬
ле, умеренно растворим или мало растворим в 96 %
спирте, практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Проводят определение подлинности (идентифи¬
кацию) испытаниями В, С, О или А, В, й.
А. Удельное оптическое вращение (2.2.7): от -69,5
до -67,0 (в пересчете на безводное вещество). 0,250 г
испытуемого образца растворяют в метаноле Р и до¬
водят до объема 25,0 мл этим же растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО прамипексола дигидрохлорида
моногидрата.
C. Испытуемый образец выдерживает испытание
«Энантиомерная чистота», как указано в разделе «Ис¬
пытания».
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,1 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 8
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 2,8 до 3,4. 0,4 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 20 мл этим же раство¬
рителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор. 5 г натрия октансульфо-
ната моногидрата Р и 9,1 г калия дигидрофосфа¬
та Р растворяют в 900 мл воды Р. Доводят кислотой
фосфорной Р до рН 3,0 и доводят водой Р до объема
1000 мл.
Смесь растворителей. Ацетонитрил Р — бу¬
ферный раствор (20:80, об/об).
Испытуемый раствор. 75 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 7,5 мг ФСО прамипексо¬
ла для проверки пригодности хроматографической
системы (содержит примеси А, В и С) растворяют в
5.0 мл смеси растворителей.
Условия хроматографирования:
-колонка длиной 0,125 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным эндкепированным для хроматографии Р
(размер частиц 5 мкм);
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: буферный раствор;
- подвижная фаза В: ацетонитрил Р — бу¬
ферный раствор (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
60 —►20
о
00
т
о
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 264 нм;
- объем вводимой пробы: 5 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и С, используя хроматограм¬
му раствора сравнения (Ь) и хроматограмму, прила¬
гаемую к ФСО прамипексола для проверки пригодно¬
сти хроматографической системы.
Прамипексола дигидрохлорид моногидрат
833
Относительное удерживание (по отношению к
прамипексолу, время удерживания — около 6 мин):
примесь А— около 0,7; примесь В — около 1,5; при¬
месь С — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 6,0 меаду пиками при¬
меси А и прамипексола.
Предельное содержание примесей:
- примеси А, В и С (не более 0,15 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В и С, не должны превы¬
шать 1,5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Энантиомерная чистота. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. 6 мг испытуемого образ¬
ца растворяют в 5 мл этанола безводного Р и дово¬
дят подвижной фазой до объема 20,0 мл.
Раствор сравнения (а). 2 мг ФСО прамипексола
примеси О растворяют в подвижной фазе и доводят
подвижной фазой до объема 10 мл. К 1 мл полученно¬
го раствора прибавляют 1 мл испытуемого раствора и
доводят подвижной фазой до объема 20 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем АО для хи¬
ральных разделений Р;
- подвижная фаза: диэтиламин Р—этанол без¬
водный Р — гексан Р (0,1:15:85, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 75 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания прамипексола.
Относительное удерживание (по отношению к
прамипексолу, время удерживания — около 11 мин):
примесь О — около 0,5.
Пригодность хроматографической системы:
-разрешение: не менее 5 между пиками при¬
меси О и прамипексола на хроматограмме раствора
сравнения (а);
- фактор асимметрии: не более 2,4 для пика пра¬
мипексола на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
- примесь О (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси О, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010% (10 ррт).
Растворитель: вода Р.
0,500 г испытуемого образца должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием 0,5 мл эталонного раствора свин¬
ца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 5,0 % и не более 7,0 %.
Определение проводят из 0,500 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в
150 мл воды Р, прибавляют 10 мл 3 М раствора кис¬
лоты азотной и титруют 0,1 М раствором серебра
нитрата потенциометрически (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 14,213 мг С10Н19С12М35.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е.
А. (65)-4,5,6,7-Тетрагидро-1,3-бензотиазол-2,6-
диамин.
н3с
С. Смесь диастереоизомеров (65)-6-А/-[3-[[(65)-2-
амино-4,5,6,7-тетрагидро-1,3-бензотиазол-6-ил]амино]-
1 -этил-2-метилпропил]-4,5,6,7-тетрагидро-1,3-бензотиа-
зол-2,6-диамина.
н
Н3С
О. (6/?)-6-Л/-Пропил-4,5,6,7-тетрагидро-1,3-бензо-
тиазол-2,6-диамин.
834
Гэсударственная фармакопея Республики Беларусь
Н3С
ын2
о
Е. Л/-[(65)-2-Амино^,5,6,7-тетрагидро-1,3-бензотиа-
зол-б-ил]пропанамид.
07/2016:0353
ПРЕДНИЗОЛОН
Ргес1п1зо1опит
РКЕОШЗОШЫЕ
С21н2805 М.м. 360,4
[50-24-8]
ОПРЕДЕЛЕНИЕ
11 р, 17,21 -Тригидроксипрегна-1,4-диен-3,20-дион.
Содержание: не менее 96,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Очень мало растворим в воде, растворим в 96 %
спирте и в метаноле, умеренно растворим в ацетоне,
мало растворим в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО преднизолона #или спектр,
представленный на рисунке #0353.-1#.
Если спектры, полученные с использованием
образцов в твердом состоянии, отличаются, то испы¬
туемый образец и ФСО преднизолона растворяют по
отдельности в минимальном объеме ацетона Р и вы¬
паривают на водяной бане до сухих остатков, которые
используют для получения новых спектров.
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора (Ь) по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (б).
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+113 до +119 (в пересчете на сухое вещество). 0,250 г
испытуемого образца растворяют в 96 % спирте Р и
доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29). Испытание проводят с защитой от
света.
Смесь растворителей. Ацетонитрил Р —
вода Р (40:60, об/об).
Испытуемый раствор (а). 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 25,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
1,0 мл полученного раствора доводят смесью раство¬
рителей до объема 10,0 мл.
Раствор сравнения (а). 5 мг ФСО преднизоло¬
на для проверки пригодности хроматографической
Рисунок #0353.-1. Инфракрасный спектр ФСО преднизолона
Преднизолон
835
системы (содержит примеси А, В и С) растворяют в
смеси растворителей и доводят до объема 10,0 мл
этой же смесью растворителей.
Раствор сравнения (Ь). 5 мг ФСО преднизолона
для идентификации пиков (содержит примеси Р и 3)
растворяют в смеси растворителей и доводят до объ¬
ема 10,0 мл этой же смесью растворителей.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (д). 25,0 мг ФСО преднизо¬
лона растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
1.0 мл полученного раствора доводят смесью раство¬
рителей до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: вода Р;
- подвижная фаза В: ацетонитрил Р — мета¬
нол Р (50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—14
60
40
14—20
60 — 20
40 — 80
20—25
20
80
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и С, используя хроматограм¬
му раствора сравнения (а) и хроматограмму, прилага¬
емую к ФСО преднизолона для проверки пригодности
хроматографической системы, идентифицируют
пики примесей Р и и, используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО преднизолона для идентификации пиков.
Относительное удерживание (по отношению к
преднизолону, время удерживания — около 12 мин):
примесь Р — около 0,7; примесь В — около 0,9; при¬
месь А — около 1,05; примесь ^ — около 1,5; примесь
С — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 3
(Нр — высота пика примеси А относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси А и пик
преднизолона).
Предельное содержание примесей:
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси А, не должна превышать 10-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примесь Р (не более 0,5 %): на хроматограмме ис¬
пытуемого раствора (а) площадь пика, соответствующего
примеси Р, не должна превышать 5-кратную площадь ос¬
новного пика на хроматограмме раствора сравнения (с);
- примеси В, С, 3 (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям В, С и 3, не должны
превышать 3-кратную площадь основного пика на
хроматограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пиков
примесей А, В, С, Р и 3, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
- сумма примесей (не более 1,5 %): на хрома¬
тограмме испытуемого раствора (а) сумма площа¬
дей всех пиков, кроме основного, не должна превы¬
шать 15-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора (а) не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 0,500 г испытуемого образца сушат при
температуре 105 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора (Ь) и раствора сравнения (б).
Содержание С21Н2805 в процентах рассчитывают
с учетом содержания преднизолона в ФСО предни¬
золона.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, Р, 3.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемлемо¬
сти для других/неспецифицированных примесей и/
или общей статьей Субстанции для фармацевти¬
ческого использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): О,
Е, С, Н, I.
А. 11(3,17,21-Тригидроксипрегн-4-ен-3,20-дион (ги¬
дрокортизон).
836
Гэсударственная фармакопея Республики Беларусь
В. 17,21 -Дигидроксипрегна-1,4-диен-3,11,20-трион
(преднизон).
С. 11Р, 17-Дигидрокси-3,20-диоксопрегна-1,4-диен-
21-илацетат (преднизолона ацетат).
I. 11р,21-Дигидроксипрегна-1,4-диен-3,20-дион
(17-дезоксипреднизолон).
3. 17,21 -Дигидроксипрегна-1,4-диен-3,20-дион
(11 -дезоксипреднизолон).
07/2016:0050
ПРОКАИНА ГИДРОХЛОРИД
Ргосат/ Ьус1госЫопс1ит
РКОСАШЕ НУОКОСН1-ОКЮЕ
О. 6р, 11 р, 17,21 -Тетрагидроксипрегна-1,4-диен-
3,20-дион (бр-гидроксипреднизолон).
Е. 11 р, 14а, 17,21 -Тетрагидроксипрегна-1,4-диен-
3,20-дион (14а-гидроксипреднизолон).
Р. 11а,17,21-Тригидроксипрегна-1,4-диен-3,20-дион
(11 -эга-преднизолон).
С. 11Р, 17,20р, 21 -Тетрагидроксипрегна-1,4-диен-
3-он (20р-гидроксипреднизолон).
Н. 11 р, 17,21 -Тригидроксипрегна-1,4,6-триен-3,20-
дион (А6-преднизолон).
С13Н20М2О2 ■ НС1 М.М. 272,8
[51-05-8]
ОПРЕДЕЛЕНИЕ
2-(Диэтиламино)этил-4-аминобензоата гидрохло¬
рид. #Новокаин#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень легко растворим в воде, растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: А, С, О, Е, Р.
A. Температура плавления (2.2.14): от 154 °С до
158 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО прокаина гидрохлорида #или
спектр, представленный на рисунке #0050.-1#.
C. К 5 мг испытуемого образца прибавляют 0,5 мл
кислоты азотной дымящей Р, выпаривают на во¬
дяной бане и охлаждают. Остаток растворяют в 5 мл
ацетона Р. К полученному раствору прибавляют 1 мл
0,1 М раствора калия гидроксида спиртового. Появ¬
ляется только коричневато-красное окрашивание.
Р. К 0,2 мл раствора 5, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 2 мл воды Р
Прокаина гидрохлорид
837
Рисунок #0050.-1. Инфракрасный спектр ФСО прокаина гидрохлорида
и 0,5 мл кислоты серной разведенной Р и встряхи¬
вают. К полученному раствору прибавляют 1 мл раст¬
вора 1 г/л калия перманганата Р. Окрашивание сразу
же исчезает.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
Р. 1 мл раствора 5 доводят водой Р до объема
100 мл. 2 мл полученного раствора дают реакцию на
первичные ароматические амины (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2; метод II). Раствор 5 должен
быть бесцветным.
рН (2.2.3). От 5,0 до 6,5. 4 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объема
10 мл.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения. 50 мг кислоты 4-аминобен-
зойной Р растворяют в воде Р и доводят до объема
100 мл этим же растворителем. 1мл полученного
раствора доводят водой Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: кислота уксусная ледяная Р —
гексан Р — дибутиловый эфир Р (4:16:80, об/об/об).
Наносимый объем пробы: 5 мкп.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 10 мин.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- любая примесь (не более 0,05 %): на хрома¬
тограмме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения.
На хроматограмме испытуемого раствора основ¬
ное пятно остается на линии старта.
Тяжелые металлы (2.4.8, метод Е). Не более
0,0005 % (5 ррт). 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем. Проводят предфильтрацию. 10 мл
предфильтрата должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
5 мл эталонного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,00 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 50 мл
кислоты хлористоводородной разведенной Р и про¬
водят определение аминного азота в соединениях,
которые содержат первичную ароматическую ами¬
ногруппу (2.5.8) #(в качестве индикатора используют
раствор нейтрального красного Р (0,1 мл в начале
и 0,1 мл в конце титрования) — титруют до перехода
окраски раствора от красно-фиолетовой до синей, или
раствор тропеолина 00 Р в смеси с метиленовым
синим Р (0,2 мл раствора тропеолина 00 Р и 0,1 мл
838
Гэсударственная фармакопея Республики Беларусь
раствора метиленового синего Р) — титруют до
перехода окраски от красно-фиолетовой до голубой)#
1 мл 0,1 М раствора натрия нитрита соответ¬
ствует 27,28 мг С13Н20М2О2 НС1.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0567
ПРОКАИНАМИДА ГИДРОХЛОРИД
Ргосатат1сИ Ьус1госЫогИит
РКОСА1ЫАМЮЕ НУОЯОСНЮРЮЕ
С13Н31М30 • НС1 М.м. 271,8
[614-39-1]
ОПРЕДЕЛЕНИЕ
4-Амино-Л/-[2-(диэтиламино)этил]бензамида ги¬
дрохлорид. ^овокаинамид*
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый кристаллический
порошок. Гигроскопичен.
Очень легко растворим в воде, легко растворим в
96 % спирте, мало растворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С, О.
Вторая идентификация: А, В, О, Е.
A. Температура плавления (2.2.14): от 166 °С до
170 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в 0,1 М растворе натрия гидрок¬
сида и доводят до объема 100,0 мл этим же раство¬
рителем. 10,0 мл полученного раствора доводят 0,1 М
раствором натрия гидроксида до объема 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 273 нм.
Удельный показатель поглощения в максимуме:
от 580 до 610.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО прокаинамида гидрохлорида
#или спектр, представленный на рисунке #0567.-1#.
й. 1 мл раствора 5, приготовленного как указано
в разделе «Испытания», доводят водой Р до объема
5 мл. Полученный раствор дает реакцию (а) на хло¬
риды (2.3.1).
Е. 1 мл раствора 5 доводят водой Р до объема
2 мл. 1 мл полученного раствора дает реакцию на
первичные ароматические амины (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2; метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)б.
рН (2.2.3). От 5,6 до 6,3. Измеряют рН раствора 5.
Рисунок #0567.-1. Инфракрасный спектр ФСО прокаинамида гидрохлорида
Пролин
839
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения. 1 мл испытуемого раствора
доводят 96 % спиртом Р до объема 200 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (15:30:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Бактериальные эндотоксины (2.6.14). Менее
0,35 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без последующей процедуры удале¬
ния бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ.
0,2500 г испытуемого образца растворяют в
50 мл кислоты хлористоводородной разведенной Р
и проводят определение аминного азота в соедине¬
ниях, которые содержат первичную ароматическую
аминогруппу (2.5.8).
1 мл 0,1 М раствора натрия нитрита соответ¬
ствует 27,18 мг С13Н21М3О НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0785
ПРОЛИН
РгоИпит
РПОШЕ
С5Н9М02 М.м. 115,1
[147-85-3]
ОПРЕДЕЛЕНИЕ
(5)-Пирролидин-2-карбоновая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пролина.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
-84,0 до -86,0 (в пересчете на сухое вещество). 1,00 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов.#
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образца, подходящий для применяемого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 30,0 мг аланина Р (при¬
месь А) растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят до объема 100,0 мл этим же раст¬
ворителем. 1,0 мл полученного раствора доводят до
объема 10,0 мл этим же растворителем.
840
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят раствором А до объ¬
ема 10,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт Л/Н^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (ф. 30 мг изолейцина Р и
30 мг лейцина Р растворяют в растворе А и доводят
до объема 50,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь) и (с1) вводят в аминокислотный анализатор.
Запускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с1):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию примеси А в растворе срав¬
нения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в 0,1 М растворе хлористоводо¬
родной кислоты и доводят до объема 10 мл этим же
растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО пролина раст¬
воряют в 0,1 М растворе хлористоводородной кисло¬
ты и доводят до объема 50 мл этим же растворите¬
лем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 ФСО пролина и 10 мг
ФСО треонина растворяют в 0,1 М растворе хлори¬
стоводородной кислоты и доводят до объема 25 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
-любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хро¬
матограмме испытуемого раствора площадь пика,
соответствующего аммонию, не должна превышать
площадь соответствующего пика на хроматограмме
раствора сравнения (с), учитывая пик, соответству¬
ющий аммонию на хроматограмме холостого раст¬
вора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хрома¬
тографии, то испытание «Аммония соли» (2.4.1, ме¬
тод В) проводят следующим образом. 50 мг испы¬
туемого образца должны выдерживать испытание
на аммония соли. Эталон готовят с использовани¬
ем 0,1 мл эталонного раствора аммония (100 ррт
А/Н^ Р.*
Железо (2.4.9). Не более 0,0010% (10 ррт).
1,0 г испытуемого образца помещают в делитель¬
ную воронку и прибавляют 10 мл кислоты хлори¬
стоводородной разведенной Р. Полученный раст¬
вор трижды встряхивают, каждый раз в течение
3 мин, с метилизобутилкетоном Р1 порциями по
10мл. К объединенным органическим слоям при¬
бавляют 10 мл воды Р и встряхивают в течение
3 мин. Водный слой должен выдерживать испыта¬
ние на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раст¬
воряют в воде Р и доводят до объема 20,0 мл этим
же растворителем. 12 мл полученного раствора
должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием эталон¬
ного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
Прометазина гидрохлорид
841
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:0524
ПРОМЕТАЗИНА ГИДРОХЛОРИД
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором хлорной кислоты потенциометрически
(2.2.20).
1 мл 0,1 М раствора хлорной кислоты соответ¬
ствует 11,51 мг С5Н9М02.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемлемо¬
сти для других/неспецифицированных примесей.
Вследствие этого нет необходимости идентифици¬
ровать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Кон¬
троль примесей в субстанциях для фармацевти¬
ческого использования): А, В.
Н МН2
НзС'^^СОгН
А. (25)-2-аминопропановая кислота (аланин).
нА мн2
Н3Ск
^со2н
СНз
В. (25)-2-амино-3-метилбутановая кислота (валин).
РготеМаят ЬудгосЫопдит
РКОМЕТНАШЕ НУОКОСШОКЮЕ
и энантиомер - НС1
М.м. 320,9
(2/?5)-Л/,Л/-Диметил-1-(10Н-фенотиазин-10-ил)
пропан-2-амина гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый кристаллический
порошок.
Очень легко растворим в воде, легко растворим в
96 % спирте и в метиленхлориде.
Температура плавления: около 222 °С с разло¬
жением.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: В, С, О.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО прометазина гидрохлорида
#или спектр, представленный на рисунке #0524.-1#.
С17Н20М25 - НС1
[58-33-3]
ОПРЕДЕЛЕНИЕ
106. Зак. 1060.
842
Гэсударственная фармакопея Республики Беларусь
B. Идентификация фенотиазинов методом тон¬
кослойной хроматографии (2.3.3). Для приготовления
раствора сравнения используют ФСО прометазина
гидрохлорида.
C. 0,1 г испытуемого образца растворяют в 3 мл
воды Р и прибавляют по каплям кислоту азотную Р.
Образуется осадок, который быстро растворяется, и
получается раствор красного цвета, который переходит
в оранжевый и затем в желтый. Полученный раствор
нагревают до кипения. Появляется оранжевое окраши¬
вание, и образуется осадок оранжево-красного цвета.
О. Испытуемый образец дает реакцию (Ь) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 4,0 до 5,0.1,0 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диокси¬
да, Р и доводят до объема 10 мл этим же растворите¬
лем. Измеряют рН немедленно после приготовления
раствора.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытания проводят с защитой
от света. Растворы готовят непосредственно пе¬
ред использованием.
Смесь растворителей. Триэтиламин Р — ме¬
танол Р (1:1000, об/об).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (а). 2,5 мг ФСО прометази¬
на для идентификации пиков (содержит примеси А, В
и С) растворяют в смеси растворителей и доводят до
объема 5 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 5,0 мг ФСО прометази¬
на примеси О растворяют в смеси растворителей и
доводят до объема 100,0 мл этой же смесью раство¬
рителей. 1 мл полученного раствора доводят смесью
растворителей до объема 100 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
4,6 мм, заполненная силикагелем октилсилильным, с
введенными полярными группами, эндкепированным
для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: смешивают 20 объемов ме¬
танола Р, 30 объемов ацетонитрила Р и 50 объемов
раствора 3,4 г/л калия дигидрофосфата Р, предвари¬
тельно доведенного до рН 7,0 калия гидроксидом Р;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания прометазина.
Идентификация пиков примесей: идентифи¬
цируют пики примесей А, В и С, используя хромато¬
грамму раствора сравнения (а) и хроматограмму, при¬
лагаемую к ФСО прометазина для идентификации
пиков; идентифицируют пик примеси О, используя
хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению к
прометазину, время удерживания — около 18 мин):
примесь й — около 0,2; примесь С — около 0,5; при¬
месь В — около 1,4; примесь А — около 1,8.
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками при¬
меси В и примеси А на хроматограмме раствора срав¬
нения (а);
- хроматограмма раствора сравнения (а) должна
соответствовать хроматограмме, прилагаемой к ФСО
прометазина для идентификации пиков.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—0,5):
- примесь В (не более 0,8 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 8-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь С (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь А (не более 0,1 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь О (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С и Р, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 1,2 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
12-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод Е). Не более
0,0010% (Юррт). 1,0 г испытуемого образца раст¬
воряют в 5 мл воды Р, прибавляют 5 мл ацетона Р
5 мл буферного раствора рН 3,5 Р и проводят пред-
фильтрацию раствора. Полученный фильтрат должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 5 мл эталонного раст¬
вора свинца (2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат пр*'
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1
Определение проводят из 1,0 г испытуемого образца
#Микробиологическая чистота (2.6.12, 2.6.12
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в сме¬
си из 5,0 мл 0,01 М раствора кислоты хлористо¬
водородной и 50 мл 96 % спирта Р и титруют 0 ' у
Пропиленгликоль
843
раствором натрия гидроксида потенциометриче-
ски (2.2.20). Отмечают объем титранта между двумя
точками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 32,09 мг С^Н^^З НС!.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
мн
А. Фенотиазин.
В. (2/?5)-Л/,Л/-Диметил-2-(10Н-фенотиазин-10-ил)-
пропан-1-амин (изопрометазин).
С. Р = Н, X = 5: (2Я5)-Л/-Метил-1-(10Н-фенотиазин-
10-ил)пропан-2-амин.
О. Р = СН3> X = 30: (2Я5)-А/,Л/-Диметил-1-(10Н-
фенотиазин-10-ил)пропан-2-амина 5-оксид.
07/2016:0430
ПРОПИЛЕНГЛИКОЛЬ
Ргору1епд1усо1ит
РКОРП-ЕЫЕ С/.УССИ1
Н ОН
н3с
.он
и энантиомер
с3н8о2
[57-55-6]
М.м. 76,1
ОПРЕДЕЛЕНИЕ
(Р5)-Пропан-1,2-диол.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная вязкая жидкость. Гигро¬
скопичен.
Смешивается с водой и с 96 % спиртом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Показатель преломления», как указано в разделе
«Испытания».
С. Температура кипения (2.2.12): от 184 °С до
189 °С.
О. К 0,5 мл испытуемого образца прибавляют
5 мл пиридина Р и 2 г тонко измельченного нитробен-
зоилхлорида Р. Кипятят в течение 1 мин, затем нали¬
вают, встряхивая, в 15 мл холодной воды Р. Получен¬
ную смесь фильтруют, осадок на фильтре промывают
20 мл насыщенного раствора натрия гидрокарбона¬
та Р, затем водой Р и высушивают. Полученный оста¬
ток растворяют в кипящем спирте (80 %, об/об) Р и
горячий раствор фильтруют. После охлаждения обра¬
зовавшиеся кристаллы высушивают при температуре
от 100 °С до 105 °С. Температура плавления (2.2.14)
полученных кристаллов: от 121 °С до 128 °С.
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Испытуемый обра¬
зец должен быть бесцветным.
Относительная плотность (2.2.5). От 1,035 до
1,040.
Показатель преломления (2.2.6). От 1,431 до
1,433.
Кислотность. К 10 мл испытуемого образца при¬
бавляют 40 мл воды Р и 0,1 мл раствора бромти-
молового синего Р1. Появляется зеленовато-желтое
окрашивание. При прибавлении не более 0,05 мл
0,1 М раствора натрия гидроксида окрашивание
раствора должно измениться на синее.
Окисляющие вещества. К 10 мл испытуемого
образца прибавляют 5 мл воды Р, 2 мл раствора
калия йодида Р и 2 мл кислоты серной разведен¬
ной Р и выдерживают в колбе с притертой стеклян¬
ной пробкой в защищенном от света месте в течение
15 мин. Титруют 0,05 Мраствором натрия тиосуль¬
фата, используя в качестве индикатора 1 мл раст¬
вора крахмала Р. На титрование раствора должно
пойти не более 0,2 мл 0,05 М раствора натрия ти¬
осульфата.
Восстанавливающие вещества. К 1 мл испыту¬
емого образца прибавляют 1 мл раствора аммиака
разведенного Р1 и нагревают в водяной бане при тем¬
пературе 60 °С в течение 5 мин. Не должно появлять¬
ся желтое окрашивание. К полученному раствору при¬
бавляют 0,15 мл 0,1 М раствора серебра нитрата и
выдерживают в течение 5 мин. Внешний вид раствора
не должен изменяться.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (м/об) (5 ррт (м/об)). 4 мл испытуемого об¬
разца смешивают с 16 мл воды Р. 12 мл полученного
раствора должны выдерживать испытание на тяже¬
лые металлы. Эталон готовят с использованием эта¬
лонного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 0,2 %. Определение про¬
водят из 5,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,01 %. 50 г
испытуемого образца сжигают и прокаливают. После
охлаждения остаток смачивают кислотой серной Р и
прокаливают; процедуру повторяют. Масса остатка не
должна превышать 5 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
106*. Зак. 1060.
844
Гэсударственная фармакопея Республики Беларусь
07/2016:0431
ПРОПИЛПАРАГИДРОКСИБЕНЗОАТ
РгоруНз рагаЬус/гохуЬепгоаз
РКОРП. РАРАНУОРОХУВЕИЮАТЕ
ОПРЕДЕЛЕНИЕ
Пропил-4-гидроксибензоат. #Пропилпарабен, ни-
пазол*.
Содержание: не менее 98,0 % и не более 102,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень мало растворим в воде, легко растворим в
96 % спирте и метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Температура плавления (2.2.14). От 96 °С до 99 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пропилпарагидроксибензоата
#или спектр, представленный на рисунке #0431.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в ацетоне Р и доводят до объ¬
ема 10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО пропилпара¬
гидроксибензоата растворяют в ацетоне Р и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО этилпара-
гидроксибензоата растворяют в 1 мл испытуемого
раствора (а) и доводят ацетоном Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — метанол Р (1:30:70, об/об/об).
Наносимый объем пробы: по 2 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
- на хроматограмме раствора сравнения (Ь) об¬
наруживаются два полностью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 96% спирте Р и доводят до объема 10 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность. К 2 мл раствора 3 прибавля¬
ют 3 мл 96 % спирта Р, 5 мл воды, свободной от
Рисунок #0431 .-1. Инфракрасный спектр ФСО пропилпарагидроксибензоата
Пропилпарагидроксибензоат
845
углерода диоксида, Р и 0,1 мл раствора бромкре-
эолового зеленого Р. При прибавлении не более
0,1 мл 0,1 М раствора натрия гидроксида Р окра¬
шивание раствора должно измениться на синее.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в 2,5 мл метанола Р и дово¬
дят подвижной фазой до объема 50,0 мл.10,0 мл
полученного раствора доводят подвижной фазой
до объема 100,0 мл.
Раствор сравнения (а). 5 мг 4-гидроксибен-
зойной кислоты Р (примесь А), 5 мг этилпараги-
дроксибензоата Р (примесь С) и 5 мг испытуемого
образца растворяют в подвижной фазе и доводят
до объема 100,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой
до объема 10,0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО пропил-
парагидроксибензоата растворяют в 2,5 мл ме¬
танола Р и доводят подвижной фазой до объема
50.0 мл. 10,0 мл полученного раствора доводят
подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 1,0 мл испытуемо¬
го раствора доводят подвижной фазой до объема
20.0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- подвижная фаза: раствор 6,8 г/л калия диги¬
дрофосфата Р — метанол Р (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 272 нм;
-объем вводимой пробы: по 10 мкл испытуе¬
мого раствора и растворов сравнения (а) и (с);
- время хроматографирования: 2,5-кратное вре¬
мя удерживания пропилпарагидроксибензоата.
Относительное удерживание (по отношению
к пропилпарагидроксибензоату, время удержива¬
ния — около 4,5 мин): примесь А — около 0,3; при¬
месь С — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,0 между пиками при¬
меси С и пропилпарагидроксибензоата.
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси А— 1,4):
- примесь А (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
^ - неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси А, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (с);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики
с площадью менее 0,2 площади основного пика на
хроматограмме раствора сравнения (с).
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С10Н12О3 в процентах рассчитывают
с учетом содержания пропилпарагидроксибензоата в
ФСО пропилпарагидроксибензоата.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В, С, О.
А. 4-Гидроксибензойная кислота.
В. Метил-4-гидроксибензоат (метилпарагидрокси-
бензоат).
С. Этил-4-гидроксибензоат (этилпарагидроксибен-
зоат).
О. Бутил-4-гидроксибензоат (бутилпарагидрокси-
бензоат).
107. Зак. 1060.
846
Гэсударственная фармакопея Республики Беларусь
07/2016: РБ0009
#ПРОПОЛИС
РгороНз
ркороиз
ОПРЕДЕЛЕНИЕ
Прополис — продукт жизнедеятельности пчел.
ОПИСАНИЕ (СВОЙСТВА)
Темно-серая масса с зеленоватым или коричне¬
ватым оттенком, неоднородная в изломе, с характер¬
ным смолистым запахом.
Практически нерастворим в воде, в эфире, в хло¬
роформе, в 96 % спирте и в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 2 мл раствора 8, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,2 мл раст¬
вора свинца (II) ацетата основного Р. Образуется
осадок желтого цвета.
B. К 2 мл раствора 8 прибавляют 0,05 г порошка
магния Р и, по каплям, 0,5 мл кислоты хлористово¬
дородной концентрированной Р. Появляется красное
окрашивание.
ИСПЫТАНИЯ
Порошок прополиса. 10 г испытуемого образца
выдерживают в холодильнике при температуре -6 °С
в течение (10—15) мин и измельчают подходящим
способом.
Раствор 8. К 1,000 г порошка прополиса прибав¬
ляют 20,0 мл 96 % спирта Р, перемешивают на маг¬
нитной мешалке в течение 30 мин и фильтруют через
бумажный фильтр. Используют фильтрат.
Механические примеси. Не более 15%. К
1,000 г порошка прополиса (т) прибавляют 50 мл
96 % спирта Р и нагревают до кипения на водяной
бане при перемешивании. Горячую смесь фильтру¬
ют через бумажный фильтр, предварительно вы¬
сушенный до постоянной массы при температуре
50 °С (т0). Осадок на фильтре промывают горячим
96 % спиртом Р до тех пор, пока в капле фильтрата,
помещенной на предметное стекло, при охлаждении
не перестанет появляться белый осадок. Фильтрат
используют для испытания «Воск». Фильтр с осад¬
ком сушат до постоянной массы при температуре
50 °С (т,).
Содержание механических примесей в процен¬
тах рассчитывают по формуле:
(тЛ — /770 ) -100
т
Воск. Не более 20 %. Фильтрат, полученный в
испытании «Механические примеси», охлаждают при
температуре -5 °С. Выделившийся осадок (воск) от¬
фильтровывают через бумажный фильтр, предвари¬
тельно высушенный до постоянной массы при темпе¬
ратуре 50 °С (т0). Осадок на фильтре промывают хо¬
лодным 96 % спиртом Р до тех пор, пока промывной
спирт не станет бесцветным. Фильтр с осадком сушат
до постоянной массы при температуре 50 °С (т,).
Содержание воска в процентах рассчитывают по
формуле:
(ш1 -то)-100
/77
где:
т — масса навески порошка прополиса, исполь¬
зованная в испытании «Механические примеси».
Фенольные соединения. Не менее 25 % суммы
фенольных соединений. К 0,050 г порошка прополиса
прибавляют 10 мл 96% спирта Р и перемешивают
на магнитной мешалке в течение 10 мин. Полученную
смесь фильтруют через бумажный фильтр, фильтр
промывают 96 % спиртом Р и доводят до объема
50,0 мл этим же растворителем. 1,0 мл полученного
раствора доводят 96 % спиртом Рдо объема 25,0 мл.
Измеряют оптическую плотность (2.2.25) полученного
раствора при 290 нм, используя 96 % спирт Р в каче¬
стве компенсационного раствора.
Содержание суммы фенольных соединений в
процентах рассчитывают по формуле:
А-1250
т-510 ’
где:
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески порошка прополиса, г;
510 — коэффициент пропорциональности опти¬
ческой плотности и концентрации суммы фенольных
соединений прополиса при длине волны 290 нм.
Антимикробная активность. Концентрация
испытуемого образца, подавляющая рост тест-
микроорганизма: не более 0,08 %.
Антимикробную активность определяют методом
последовательных разведений в среде № 1 (2.6.13).
Тест-культуру ВасШиз сегеиз ЫСТС 8035 со¬
храняют на среде N8 1 (2.7.2) от 15 до 30 дней при
температуре от 4 °С до 10 °С, после чего перено¬
сят на свежую питательную среду. Культуру можно
сохранять и в лиофилизированном состоянии. При
применении лиофилизированных культур их вы¬
севают в пробирки на жидкую среду № 8 (2.6.13) и
инкубируют при температуре (37±1)°С в течение
(18—20) ч (рост на среде №8 — равномерное по¬
мутнение с осадком на дне). Со среды № 8 культуру
пересевают на среду № 1 (2.7.2) так, чтобы получить
изолированные колонии, и инкубируют при темпе¬
ратуре (37±1) °С в течение (18—20) ч. По окончании
инкубации выросшие колонии просматривают и от¬
бирают типичные: гладкие, круглые, приподнятые, с
зубчатым краем. В мазке агаровой культуры, окра¬
шенной по Граму, должны наблюдаться палочки с
закругленными концами, располагающиеся по оди¬
ночке и длинными цепочками, с хорошо выражен¬
ной грамположительной окраской.
В первую стерильную коническую колбу вмести¬
мостью 100 мл помещают 59 мл расплавленной сре¬
ды № 1 (2.6.13) при температуре от 80 °С до 90 °С.
Во вторую и третью колбы вместимостью по 100 мл
помещают по 30 мл среды № 1 (2.6.13). В первую
колбу вносят 1,0 мл раствора 8 и быстро перемеши¬
вают (концентрация 0,08%) (разведение 1). 30 мл
полученной смеси помещают во вторую колбу и пе¬
ремешивают (концентрация 0,04 %) (разведение 2).
В третью колбу помещают 30 мл смеси из второй
колбы и перемешивают (концентрация 0,02 %) (раз-
ведение 3). Содержимое каждой колбы разливают
по 15 мл в две стерильные чашки Петри диаметром
100 мм. В то же самое время в две стерильные чаш¬
ки вносят по 15 мл среды N8 1 (2.6.13), содержащей
2% 96% спирта Р (контрольный опыт).
Пропранолола гидрохлорид
847
Тест-микроорганизм (суточная бульонная куль¬
тура, выросшая на жидкой среде № 1 (2.6.13)) с по¬
мощью петли или штампа репликатора наносят на
поверхность застывшей и подсушенной среды № 1
(2.6.13), содержащей испытуемый образец и кон¬
трольный опыт. Чашки инкубируют при температуре
(37±1) °С в течение (18—24) ч. Отмечают последнее
разведение с полной видимой задержкой роста тест-
микроорганизма при хорошем росте в контроле.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 20 °С.
07/2016:0568
ПРОПРАНОЛОЛА ГИДРОХЛОРИД
Ргоргапо1оН Нус1гос111опс1ит
РК0РКАЫ01-01- НУОКОСШ-ОКЮЕ
н ОН
V н
Т и энантиомер • НС1
СН3
С16Н21Ы02 ■ НС1
[318-98-9]
М.м. 295,8
ОПРЕДЕЛЕНИЕ
(2/?5)-1 -[(1 -Метилэтил )амино]-3-(нафтален-1 -
илокси)пропан-2-ола гидрохлорид. #Анаприлин#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок.
Растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 163 °С до
166 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО пропранолола гидрохлорида
#или спектр, представленный на рисунке #0568.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 1 мл метанола Р.
Раствор сравнения. 10 мг ФСО пропранолола
гидрохлорида растворяют в 1 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р1 — метанол Р (1:99, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С, пока интенсивность пятен не ста¬
нет максимальной (в течение (10—15) мин).
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
Рисунок #0568.-1. Инфракрасный спектр ФСО пропранолола гидрохлорида
107*. Зак. 1060.
848
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона (6) по шкале
наиболее близкого цвета.
Кислотность или щелочность. 0,20 г испы¬
туемого образца растворяют в воде, свободной от
углерода диоксида, Р, доводят до объема 20 мл этим
же растворителем и прибавляют 0,2 мл раствора
метилового красного Р. При прибавлении не более
0,2 мл 0,01 М раствора хлористоводородной кисло¬
ты должно появится красное окрашивание. При при¬
бавлении не более 0,4 мл 0,01 М раствора натрия
гидроксида должно появиться желтое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО пропрано-
пола для проверки пригодности хроматографиче¬
ской системы растворяют в подвижной фазе и дово¬
дят до объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: 1,6 г натрия лаурилсульфа-
таР и 0,31 г тетрабутиламмония дигидрофосфа¬
та Р растворяют в смеси из 1 мл кислоты серной Р,
450 мл воды Р и 550 мл ацетонитрила Р и доводят до
рН 3,3 раствором натрия гидроксида разведенным Р\
- скорость подвижной фазы: 1,8 мл/мин;
- спектрофотометрический детектор, длина
волны 292 нм;
- время установления равновесия: не менее 30 мин;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 7-кратное вре¬
мя удерживания пропранолола.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму, при¬
лагаемую к ФСО пропранолола для проверки пригод¬
ности хроматографической системы.
Пригодность хроматографической системы:
раствор сравнения (а):
- разделение между пиками примеси А и пропра¬
нолола до базовой линии;
Предельное содержание примесей:
-любая примесь (не более 0,1 %): на хромато¬
грамме испытуемого раствора площадь любого пика,
кроме основного, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 0,4 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раство¬
ряют в смеси из воды Р и метанола Р (15:85, об/об) и
доводят до объема 20 мл этой же смесью раствори¬
телей. 12 мл полученного раствора должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца (1 рргг
РЬ), приготовленного разведением эталонного раст¬
вора свинца (100 ррт РЬ) Р смесью из воды Р и ме¬
танола Р (15:85, об/об).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 25 мл
96 % спирта Р и титруют 0,1 М раствором натрия
гидроксида потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 29,58 мг С1бН21М02 НС1.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
А. (2Я5)-3-(Нафтален-1-илокси)пропан-1,2-диог
(диоловое производное).
В. 1,1 '-[(1 -Метилэтил)имино]бис[3-(нафтален-1 -
илокси)пропан-2-ол] (производное третичного амина)
С. 1,3-бис(Нафтален-1-илокси)пропан-2-ол (про¬
изводное бис-эфира).
07/2016:056$
ПРОТАМИНА СУЛЬФАТ
Рго1ат1п1 зи1Газ
РРОТАМШЕ 8Ш.РАТЕ
[9009-65-8]
ОПРЕДЕЛЕНИЕ
Протамина сульфат состоит из сульфатов основ¬
ных пептидов, извлеченных из семенной жидкое-*
Протамина сульфат
849
или икры рыб, обычно видов 8а1тот'с1ае и С1иреШае.
В растворе связывается с гепарином, ингибируя его
антикоагуляционную активность; в условиях количе¬
ственного определения данная связь обуславливает
образование осадка. 1 мг протамина сульфата осаж¬
дает не менее 100 МЕ гепарина (в пересчете на сухое
вещество).
ПРОИЗВОДСТВО
Животные, являющиеся источником для полу¬
чения протамина сульфата, должны удовлетворять
требованиям, предъявляемым к здоровью животных,
пригодных для применения человеком.
Процесс производства должен быть валидиро-
ван для подтверждения того, что субстанция будет
выдерживать требования следующего испытания в
случае его проведения.
Аномальная токсичность (2.6.9). 0,5 мг испыту¬
емого образца, растворенного в 0,5 мл воды для инъ¬
екций Р, вводят каждой мыши.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Умеренно растворим в воде, практически нераст¬
ворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Удельное оптическое вращение (2.2.7): от -85
до -65 (в пересчете на сухое вещество). 1,000 г ис¬
пытуемого образца растворяют в 0,1 М растворе
кислоты хлористоводородной и доводят до объема
100,0 мл этим же растворителем.
B. В условиях испытания «Количественное опре¬
деление» протамин сульфат способствует образова¬
нию осадка.
C. К 0,5 мл раствора 3, приготовленного как
указано в разделе «Испытания», прибавляют 4,5 мл
воды Р, 1,0 мл раствора 100 г/л натрия гидроксида Р
и 1,0 мл раствора 0,2 г/л а-нафтола Р и перемеши¬
вают. Смесь охлаждают до температуры 5 °С и при¬
бавляют 0,5 мл раствора натрия гипобромита Р.
Появляется интенсивное красное окрашивание.
й. 2 мл раствора 3 нагревают в водяной бане
при температуре 60 °С, прибавляют 0,1 мл раствора
ртути (II) сульфата Р и перемешивают. Осадок не
образуется. Смесь охлаждают в ледяной воде. Обра¬
зуется осадок.
Е. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,20 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10,0 мл этим же
растворителем.
Раствор 31. К 2,5 мл раствора 3 прибавляют
7,5 мл воды Р.
Прозрачность (2.2.1). Раствор 31 по степени
мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора
31 должна быть не интенсивнее эталона ВУ(КЖ)б или
У(Ж)6.
Оптическая плотность (2.2.25). Не более 0,1.
Испытуемый раствор. 2,5 мл раствора 3 дово¬
дят водой Р до объема 5,0 мл.
Диапазон длин волн: от 260 нм до 280 нм.
Сульфаты. От 16 % до 24 % (в пересчете на су¬
хое вещество). 0,150 г испытуемого образца помеща¬
ют в лабораторный стакан и растворяют в 15 мл воды
дистиллированной Р. Прибавляют 5 мл кислоты
хлористоводородной разведенной Р, нагревают до
кипения и медленно прибавляют к кипящему раствору
10 мл раствора 100 г/л бария хлорида Р. Накрывают
стакан и нагревают на водяной бане в течение 1 ч и
фильтруют. Осадок несколько раз промывают неболь¬
шим количеством горячей воды Р, сушат и прокали¬
вают остаток при температуре (600±50) °С до посто¬
янной массы. 1,0 г остатка соответствует 0,4117 г 304.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца растворяют при нагревании в
воде Р и доводят до объема 10 мл этим же раство¬
рителем. Полученный раствор должен выдерживать
испытание на железо.
Ртуть. Не более 0,0010 % (10 ррт).
2,0 г испытуемого образца помещают в кони¬
ческую колбу с притертой пробкой вместимостью
250 мл и прибавляют 20 мл смеси равных объемов
кислоты азотной Р и кислоты серной Р. Кипятят с
обратным холодильником в течение 1 ч, охлаждают
и осторожно разводят водой Р. Кипятят до видимо¬
го прекращения выделения азотистых паров. Раст¬
вор охлаждают, осторожно доводят водой Р до объ¬
ема 200,0 мл, перемешивают и фильтруют. 50,0 мл
полученного фильтрата переносят в делительную
воронку. Последовательно встряхивают с малыми
количествами хлороформа Р до получения бесцвет¬
ного хлороформного слоя. Хлороформные слои от¬
брасывают. К водному слою прибавляют 25 мл кис¬
лоты серной разведенной Р, 115 мл воды Р и 10 мл
раствора 200 г/л гидроксиламина гидрохлорида Р.
Титруют раствором дитизона Р2; после каждого
прибавления смесь встряхивают 20 раз; перед кон¬
цом титрования хлороформный слой отделяют и от¬
брасывают. Титруют до появления голубовато-зеле¬
ного окрашивания. Рассчитывают содержание ртути,
используя эквивалент в микрограммах ртути на 1 мл
титранта, определенного при стандартизации раст¬
вора дитизона Р2.
Азот. От 21,0 % до 26,0 % (в пересчете на су¬
хое вещество). Проводят определение азота после
минерализации серной кислотой (2.5.9). Используют
10.0 мг испытуемого образца и нагревают в течение
(3—4)ч.
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
7.0 МЕ/мг, если субстанция предназначена для произ¬
водства лекарственных средств для парентерального
применения без дальнейшей подходящей процедуры
удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор (а). 15,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Испытуемый раствор (Ь). 2,0 мл испытуемого
раствора (а) доводят водой Р до объема 3,0 мл.
108. Зак. 1060.
850
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор (с). 1,0 мл испытуемого
раствора (а) доводят водой Р до объема 3,0 мл.
В качестве титранта используют 6-кратно раз¬
веденный БСП натрия гепарина в воде Р (напри¬
мер, 1,7 мл доводят водой Р до объема 10,0 мл).
Титруют каждый испытуемый раствор дважды, как
указано далее: точно отмеренный объем титруемо¬
го раствора, например 1,5 мл, помещают в кювету
подходящего колориметра, устанавливают на при¬
боре подходящую длину волны (не является крити¬
ческой) в видимой области спектра. Небольшими
объемами прибавляют титрант до резкого увели¬
чения оптической плотности и регистрируют объем
прибавленного титранта.
Выполняют 3 независимых количественных
определения. Для каждого отдельного титрования
рассчитывают количество Международных единиц
гепарина в объеме прибавленного титранта в ко¬
нечной точке титрования на 1 мг испытуемого об¬
разца. Рассчитывают активность субстанции как
среднее значение результатов 18 определений.
Оценивают линейность отклика обычными стати¬
стическими методами (например, 5.3). Рассчитыва¬
ют 3 стандартных отклонения для результатов каж¬
дого из 3 испытуемых растворов. Рассчитывают 3
стандартных отклонения для результатов каждого
из 3 независимых количественных определений.
Результаты испытания являются достоверны¬
ми, если каждое из 6 стандартных отклонений яв¬
ляется менее 5 % от среднего значения.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере с кон¬
тролем первого вскрытия. Стерильную субстанцию
хранят в стерильном воздухонепроницаемом кон¬
тейнере с контролем первого вскрытия.
07/2016:1367
ПСЕВДОЭФЕДРИНА ГИДРОХЛОРИД
Р8еис1оерЬес1пп1 Ьус/госЫопбит
Р5ЕШЮЕРНЕОЯ/Л/Е НУОРОСШ-ОРШЕ
н он
С10Н15ЫО -НС1 М.м. 201,7
[345-78-8]
ОПРЕДЕЛЕНИЕ
(15,25)-2-(Метиламино)-1-фенилпропан-1-ола
гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде и в 96 % спирте, умерен¬
но растворим в метиленхлориде.
Температура плавления: около 184 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО псевдоэфедрина гидрохлорида
#или спектр, представленный на рисунке #1367.-1#.
Рисунок #1367.-1. Инфракрасный спектр ФСО псевдоэфедрина гидрохлорида
Псевдоэфедрина гидрохлорид
851
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО псевдоэфе¬
дрина гидрохлорида растворяют в метаноле Р и до¬
водят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО эфедрина
гидрохлорида растворяют в растворе сравнения (а)
и доводят объем раствора тем же растворителем до
5 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: метиленхлорид Р — раст¬
вор аммиака концентрированный Р — 2-пропанол Р
(5:15:80, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 110 °С в
течение 5 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а).
О. Раствор 5, приготовленный как указано в
разделе «Испытания», дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность или щелочность. 2 мл раствора 3
доводят водой, свободной от углерода диоксида, Р
до объема 10 мл, прибавляют 0,1 мл раствора мети¬
лового красного Р и 0,1 мл 0,01 М раствора натрия
гидроксида. Должно появиться желтое окрашивание.
При прибавлении не более 0,2 мл 0,01 М раствора
кислоты хлористоводородной окраска раствора
должно появиться красное окрашивание.
Удельное оптическое вращения (2.2.7). От
+61,0 до +62,5 (в пересчете на сухое вещество). Опре¬
деляют удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 20,0 мг ФСО эфедрина
гидрохлорида (примесь А) растворяют в подвижной
фазе и доводят до объема 20,0 мл этим же раство¬
рителем. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 50,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл.
Раствор сравнения (с). 10 мг ФСО эфедрина ги¬
дрохлорида (примесь А) растворяют в 5 мл испытуе¬
мого раствора и доводят подвижной фазой до объема
100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: метанол Р — раствор 11,6 г/л
аммония ацетата Р, предварительно доведенный до
рН 4,0 кислотой уксусной ледяной Р, (6:94, об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 257 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания псевдоэфедрина.
Относительное удерживание (по отношению
к псевдоэфедрину, время удерживания — около
18 мин): примесь А— около 0,9.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 2,0 между пиками при¬
меси А и псевдоэфедрина; при необходимости умень¬
шают концентрацию метанола в подвижной фазе.
Предельное содержание примесей:
- примесь А (не более 1,0 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (а);
- любая другая примесь (не более 0,5 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пика примеси А, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- сума примесей кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пика
примеси А, не должна превышать 2-кратную пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,170 г испытуемого образца растворяют в 30 мл
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 20,17 мг С10Н151\Ю*НС1.
ХРАНЕНИЕ
В защищенном от света месте.
108*. Зак. 1060.
852
Гэсударственная фармакопея Республики Беларусь
ПРИМЕСИ
Специфицированные примеси: А.
А. (1Я,25)-2-(Метиламино)-1-фенилпропан-1-ол
(эфедрин).
07/2016:0359
ПШЕНИЧНЫЙ КРАХМАЛ
ТгШа ату1ит
ШНЕАТ 8ТАКСН
ОПРЕДЕЛЕНИЕ
Крахмал пшеничный получают из зерен ТгШсит
ае§1Ыит 1_. (I VиIда^е VIII.).
ОПИСАНИЕ (СВОЙСТВА)
Очень мелкий белый или почти белый порошок,
при сжатии между пальцами скрипит.
Практически нерастворим в холодной воде и в
96 % спирте.
Пшеничный крахмал не должен содержать зерна
крахмала другого происхождения, может содержать в
минимальном количестве фрагменты тканей расте¬
ния, из которого он получен.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Проводят микроскопический анализ (2.8.23) с
использованием 50 % (об/об) раствора глицерина Р. Ис¬
пытуемый образец представляет собой большие и ма¬
ленькие зерна и, очень редко, зерна средних размеров
(рисунок 0359.-1). Большие зерна диаметром от 10 мкм
до 60 мкм имеют дисковидную или иногда почковидную
форму при рассмотрении с поверхности. Центральное
ядро и бороэдчатость незаметны или едва заметны,
зерна иногда имеют трещины на концах. При рассмо¬
трении сбоку зерна эллиптические и веретеновидные,
ядро представляет собой продольный разрез вдоль
Рисунок 0359.-1. Рисунок пшеничного крахмала
(как указано в испытании А на подлинность)
главной оси. Небольшие зерна диаметром от 2 мкм до
10 мкм имеют округлую или многогранную форму. При
просматривании между скрещенными поляризующими
пластинками или призмами на зернах присутствует от¬
четливый черный крестик в центре.
B. 1 г испытуемого образца суспендируют в 50 мл
воды Р, кипятят в течение 1 мин и охлаждают. Образу¬
ется мутный водянистый клейстер.
C. К 1 мл клейстера, полученного в подлинности
В, прибавляют 0,05 мл раствора йода Р1. Появляет¬
ся темно-синяя окраска, исчезающая при нагревании.
ИСПЫТАНИЯ
рН (2.2.3). От 4,5 до 7,0. 5,0 г испытуемого образца
встряхивают с 25,0 мл воды, свободной от углерода ди¬
оксида, Р в течение 60 с. Выдерживают в течение 15 мин.
Посторонние вещества. При изучении под ми¬
кроскопом с использованием 50 % (об/об) раствора
глицерина Р должны обнаруживаться не более чем
следовые количества веществ, отличных от зерен
крахмала. Присутствие зерен крахмала любого друго¬
го происхождения не допускается.
Общий белок. Не более 0,3 % (соответствует
0,048 % 1Ч2, фактор пересчета 6,25). Определяют из
6,0 г испытуемого образца методом определения азота
после минерализации серной кислотой (2.5.9) со сле¬
дующими изменениями: смывают прилипшие к горлу
колбы частицы 25 мл кислоты серной Р; нагревают до
получения прозрачного раствора; прибавляют 45 мл
раствора натрия гидроксида концентрированного Р.
Окисляющие вещества (2.5.30). Не более
0,0020 % (20 ррт), в пересчете на Н202.
Серы диоксид (2.5.29). Не более 0,0050 %
(50 ррт).
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,5 г
испытуемого образца встряхивают с 15 мл кислоты
хлористоводородной разведенной Р и фильтруют.
Фильтрат должен выдерживать испытание на железо.
Потеря в массе при высушивании (2.2.32). Не
более 15,0 %. 1,000 г испытуемого образца сушат при
температуре 130 °С в течение 90 мин.
Сульфатная зола (2.4.14). Не более 0,6%.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа со//: отсутствие (2.6.13).
8а1топе11а: отсутствие (2.6.13).
07/2016:1368
РАМИПРИЛ
Кат1рп1ит
НАМ1РРШ.
С23Н32М205 М.М. 416 5
[87333-19-5]
Рамиприл
853
ОПРЕДЕЛЕНИЕ
(25,За5,6а5)-1 -[(25)-2-[[(15)-1 -(Этоксикарбонил )-
3-фенилпропил]амино]пропаноил]октагидроцикло-
пента[Ь]пиррол-2-карбоновая кислота.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Умеренно растворим в воде, легко растворим в
метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО рамиприла.
ИСПЫТАНИЯ
Раствор 3. 0,1 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 10 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От
+32,0 до +38,0 (в пересчете на сухое вещество).
0,250 г испытуемого образца растворяют в смеси кис¬
лота хлористоводородная Р1 — метанол Р (14:86,
об/об) и доводят до объема 25,0 мл этой же смесью
растворителей.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО рамиприла при¬
меси А, 2 мг ФСО рамиприла примеси В, 2 мг ФСО ра¬
миприла примеси С и 2 мг ФСО рамиприла примеси О
растворяют в подвижной фазе А и доводят до объема
25.0 мл этим же растворителем. К 1,0 мл полученного
раствора прибавляют 5,0 мл испытуемого раствора и
доводят подвижной фазой В до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мл испытуемого раст¬
вора доводят подвижной фазой В до объема 100,0 мл.
5.0 мл полученного раствора доводят подвижной фа¬
зой В до объема 50,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (Ь) доводят подвижной фазой В до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4.0 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р (размер частиц 3 мкм);
- температура: 65 °С;
- подвижная фаза:
- подвижная фаза А: 2,0 г натрия перхлора¬
та Р растворяют в смеси из 0,5 мл триэти-
ламина Р и 800 мл воды Р; доводят кислотой
фосфорной Р до рН 3,6 и прибавляют 200 мл
ацетонитрила Р1\
- подвижная фаза В: 2,0 г натрия перхлора¬
та Р растворяют в смеси из 0,5 мл триэти-
ламина Р и 300 мл воды Р; доводят кислотой
фосфорной Р до рН 2,6 и прибавляют 700 мл
ацетонитрила Р1.
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—6
90
10
6—7
90—>75
10 — 25
7—20
75 — 65
25 — 35
20—30
65-25
35-75
30—50
25
75
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
-уравновешивание колонки: в начальных ус¬
ловиях не менее 35 мин. Если не удается добиться
устойчивой базовой линии, необходимо использовать
триэтиламин другой степени чистоты;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С и О, используя хромато¬
грамму раствора сравнения (а).
Относительное удерживание (по отношению к
рамиприлу, время удерживания — около 18 мин): при¬
месь А — около 0,8; примесь В — около 1,3; примесь
С — около 1,5; примесь й — около 1,7.
Пригодность хроматографической системы:
- разрешение: не менее 3,0 между пиками при¬
меси А и рамиприла на хроматограмме раствора
сравнения (а);
- отношение сигнал/шум: не менее 3 для основ¬
ного пика на хроматограмме раствора сравнения (с);
- фактор асимметрии: от 0,8 до 2,0 для пика
рамиприла на хроматограмме испытуемого раствора.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 2,4):
- примеси А, В, С, О (не более 0,5 %): на хромато¬
грамме испытуемого раствора площадь любого пика,
соответствующего примесям А, В, С и Э, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С и О, не должна превышать 0,2 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
Палладий. Не более 0,0020 % (20 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. 0,200 г испытуемого
образца растворяют в смеси кислота азотная Р —
вода Р (0,3:99,7, об/об) и доводят до объема 100,0 мл
этой же смесью растворителей.
Растворы сравнения. Используют свежеприго¬
товленные растворы, содержащие 0,02 мкг, 0,03 мкг
и 0,05 мкг палладия в миллилитре, путем разведения
эталонного раствора палладия (0,5 ррт Рд) Р сме¬
сью кислота азотная Р— вода Р (0,3:99,7, об/об).
109. Зак. 1060.
854
Гэсударственная фармакопея Республики Беларусь
Раствор модификатора. 0,150 г магния ни¬
трата Р растворяют в смеси кислота азотная Р —
вода Р (0,3:99,7, об/об) и доводят до объема 100,0 мл
этой же сметью растворителей.
Объем вводимой пробы: 20 мкл испытуемого
раствора и 10 мкл раствора модификатора.
Источник излучения: лампа с полым катодом для
определения палладия с использованием полосы про¬
пускания преимущественно в 1 нм и графитовой кюветы.
Длина волны: 247,6 нм.
Потеря в массе при высушивании (2.2.32). Не
более 0,2 %. 1,000 г испытуемого образца сушат в глу¬
боком вакууме при температуре 60 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 25 мл
метанола Р, прибавляют 25 мл воды Р и титруют
0,1 М раствором натрия гидроксида потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 41,65 мг С23Н32М205.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): Е, Р, С, Н, I, */, К, Ц М, Ы, О.
A. К = СН3: (2$,За$,6а$)-1 -[(25)-2-[[(15)-1 -(Метокси-
карбонил)-3-фенилпропил]амино]пропаноил]октагидро-
циклопента[Ь]пиррол-2-карбоновая кислота (метило¬
вый эфир рамиприла).
B. К = СН(СН3)2: (25,За5,6а5)-1-[(25)-2-[[(15)-1-[(1-
Метилэтокси)карбонил]-3-фенилпропил]амино]пропа-
ноил]октагидроциклопента[Ь]пиррол-2-карбоновая кис¬
лота (изопропиловый эфир рамиприла).
С. (23,За5,6а5)-1-[(25)-2-[[(15)-3-Циклогексил-1-
(этоксикарбонил)-пропил]амино]пропаноил]октагидро-
циклопента[Ь]пиррол-2-карбоновая кислота (гексаги-
дрорамиприл).
О. Этил(25)-2-[(35,5а5,8а5,9а5)-3-метил-1,4-
диоксодекагидро-2Я-циклопента[4,5]пирроло[1,2-а]пи-
разин-2-ил]-4-фенилбутаноат (рамиприл дикетопипе-
разин).
Е. (25,За5,6аЗ)-1 -[(25)-2-[[(15)-1 -Карбокси-З-фе-
нилпропил]амино]пропаноил]октагидроциклопента[Ь]-
пиррол-2-карбоновая кислота (рамиприл дикислота).
Р. (25)-2-[[(15)-1 -(Этоксикарбонил )-3-фенилпропил]-
амино]пропановая кислота.
(3. Метилбензол (толуол).
Н. (25,За5,6а5)-1 -[(25)-2-[[(1 Я)-1 -(Этоксикарбонил)-
3-фенилпропил]амино]пропаноил]октагидроцикло-
пента[Ь]пиррол-2-карбоновая кислота ((Р,5,3,3,8)-
эпимер рамиприла).
I. (25,За$,6а5)-1-[(2Я)-2-[[(15)-1-(Этоксикарбонил)-
3-фенилпропил]амино]пропаноил]октагидроцикло-
пента[Ь]пиррол-2-карбоновая кислота ((5,Я$,5,5)-
эпимер рамиприла).
3. (2Я,За#,6а/?)-1-[(2/?)-2-[[(1К)-1-(Этоксикарбонил)-
3-фенилпропил]амино]пропаноил]октагидроцикло-
пента[Ь]пиррол-2-карбоновая кислота (энантиомер ра¬
миприла).
9 н
,со2н
Ранитидина гидрохлорид
855
К. (25)-2-[(35,5аЗ,8а5,9а5)-3-Метил-1,4-ди-
оксодекагидро-2Н-циклопента[4,5]пирроло[1,2-а]-
пиразин-2-ил]-4-фенилбутановая кислота (рамиприл
дикетопиперазин кислота).
I. Этил(25)-2-[(3$,5аЗ,8а5,9а5)-9а-гидрокси-3-
метил-1,4-диоксодекагидро-2Н-циклопента[4,5]-
пирроло[1,2-а]пиразин-2-ил]-4-фенилбутаноат (рами¬
прил гидроксидикетопиперазин).
М. (2Р,ЗР)-2,3-Бис(бензоилокси)бутандиовая кис¬
лота (дибензоилвинная кислота).
N. (2Р,ЗаР,6аР)-1-[(25)-2-[[(1 $)-1 -(Этоксикарбонил)-
3-фенилпропил]амино]пропаноил]октагидроцикло-
пента[Ь]пиррол-2-карбоновая кислота ((5,3,Р,/?,/?)-
изомер рамиприла).
О. Диэтил-2,2-(2,5-диметил-3,6-диоксопиперазин-
1,4-диил)бис(4-фенилбутаноат).
07/2016:0946
РАНИТИДИНА ГИДРОХЛОРИД
КапШсИт Ьус1госЫопс1ит
ПАШТЮШЕ НУОЕОСНШНЮЕ
н3сх
С.,Н, N.0,5 • НС1 М.м. 350,9
[66357-59-3]
ОПРЕДЕЛЕНИЕ
А/-[2-[[[5-[(Диметиламино)метил]фуран-2-ил]-
метил]сул ьфанил]этил]-ЛГ-метил-2-нитроэтен-1,1 -
диамина гидрохлорид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или бледно-желтый кристаллический по¬
рошок.
Легко растворим в воде, умеренно растворим
или мало растворим в этаноле, очень мало раство¬
рим в метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ранитидина гидрохлорида #или
спектр, представленный на рисунке #0946.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то 10 мг ис¬
пытуемого образца и 10 мг ФСО ранитидина гидрох¬
лорида растворяют по отдельности в 0,5 мл метано¬
ла Р в агатовой ступке, выпаривают досуха в потоке
азота Р и сушат в вакууме в течение 30 мин. К по¬
лученному остатку прибавляют 3 капли вазелинового
масла Р, растирают до тех пор, пока смеси не станут
молочно-белыми. Полученные смеси помещают меж¬
ду двумя пластинками, прозрачными для инфракрас¬
ного излучения, и снимают новые спектры.
B. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют воде, свободной от углерода диоксида, Р и до¬
водят до объема 100,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)5.
рН (2.2.3). От 4,5 до 6,0. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор. 6,8 г калия дигидрофос¬
фата Р растворяют в 950 мл воды Р, доводят до рН
7,1 раствором натрия гидроксида концентрирован¬
ным Р. Полученный раствор разводят водой Р до объ¬
ема 1000 мл.
Испытуемый раствор. 13 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 6,5 мг ФСО ранитидина
для идентификации примеси А растворяют в подвиж¬
ной фазе А и доводят до объема 50,0 мл этим же раст¬
ворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (с). Содержимое флакона с
ФСО ранитидина примеси 3 растворяют в 1,0 мл ис¬
пытуемого раствора.
Раствор сравнения (д). Для получения примеси
О и Н /п зНи 6,5 мг испытуемого образца растворяют в
2,5 мл раствора 40 г/л натрия гидроксида Р, нагрева¬
ют при температуре 60 °С в течение 5 мин и доводят
подвижной фазой А до объема 50,0 мл.
109*. Зак. 1060.
856
Гэсударственная фармакопея Республики Беларусь
Рисунок #0946.-1. Инфракрасный спектр ФСО ранитидина гидрохлорида
Условия хроматографирования:
- колонка длиной 0,1 м и внутренним диаметром
4,6 мм, заполненная аморфным органосиликатным
полимером аморфным октадецилсилильным Р с раз¬
мером частиц 3,5 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — бу¬
ферный раствор (2:98, об/об);
- подвижная фаза В: ацетонитрил Р — бу¬
ферный раствор (22:78, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100 0
о
о
Т
о
10—15
0
100
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а), (Ь), (с), (б) и под¬
вижной фазы А (контрольный опыт).
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму, при¬
лагаемую к ФСО ранитидина для идентификации
примеси А] идентифицируют пик примеси 3, используя
хроматограмму раствора сравнения (с); идентифици¬
руют пики примесей ОиН, используя хроматограмму
раствора сравнения (б).
Относительное удерживание (по отношению к
ранитидину, время удерживания — около 7 мин): при¬
месь Н — около 0,1; примесь (3 — около 0,2; примесь
Р — около 0,4; примесь В — около 0,5; примесь С —
около 0,6; примесь Е — около 0,7; примесь й — около
0,8; примесь 3 — около 0,9; примесь I — около 1,3;
примесь А — около 1,7.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками при¬
меси 3 и ранитидина на хроматограмме раствора
сравнения (с);
- на хроматограмме контрольного опыта не дол¬
жен обнаруживаться пик с относительным временем
удерживания, соответствующим времени удержива¬
ния пика примеси А на хроматограмме раствора срав¬
нения (а).
Предельное содержание примесей (для расчета
содержания примеси 3 умножают площадь соответ¬
ствующего пика на 2):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- примеси В, С, О, Е, Р, О, Н, I, Л (не более 0,2 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям В, С, О, Е, Р, О, Н,
I и 3, не должны превышать 0,2 площади основного
пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е, Р, С, Н, I и 3, не должна превышать
0,1 площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
- сумма примесей кроме примеси А (не более
0,5 %): на хроматограмме испытуемого раствора сум¬
ма площадей всех пиков, кроме основного и пика при¬
меси А, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики, соответству¬
ющие пикам на хроматограмме контрольного опыта и
пики с площадью менее 0,05 площади основного пика
на хроматограмме раствора сравнения (Ь) (0,05 %).
Резорцин
857
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,75%. 1,000 г испытуемого образца сушат в
глубоком вакууме при температуре 60 °С.
Сульфатная зола (2.4.14). Не более 0,1%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,280 г испытуемого образца растворяют в 35 мл
воды Р и титруют 0,1 М раствором натрия гидрокси¬
да потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 35,09 мг С^Нд^ОзЗ-НС!.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, С, Н, 1,1
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): К.
А. Л/,Л/-Бис[2-[[[5-[(диметиламино)метил]фуран-2-
ил]метил]сульфанил]этил]-2-нитроэтилен-1,1 -диамин.
В. Р = 3-СН2-СН2-МН2: 2-[[[5-[(Диметиламино)ме-
тил]фуран-2-ил]метил]сульфанил]этанамин.
й. Р = 3-СН2-СН2-МН-С0-СН2-Ы02: М-[2-[[[5-[(Диме-
тиламино)метил]фуран-2-ил]метил]сульфанил]этил]-
2-нитроацетамид.
Р. Р = ОН: [5-[(Диметиламино)метил]фуран-2-ил]-
метанол.
С. А/-[2-[[[5-[(Диметиламино)метил]фуран-2-ил]ме-
тил]сульфанил]этил]-ЛГ-метил-2-нитроэтен-1,1 -диамин.
Е. А/-[2-[[[5-[(Диметилоксидоамино)метил]фуран-
2-ил]метил]сул ьфанил]этил]-Л/'-метил-2-нитроэтен-1,1 -
диамин.
О. 3-(Метиламино)-5,6-дигидро-2Н-1,4-тиазин-2-
он-оксим.
N02
о'
/СНз
Н. А/-Метил-2-нитроацетамид.
I. 2,2'-Метиленбис[1Ч-[21-[[[5-[(диметиламино)ме-
тил]фуран-2-ил]метил]сульфанил]этил]-Л/'-метил-2-
нитроэтилен-1,1-диамин].
N02 М02
н3с^
СН3
3.1,1'-А/-[Метиленбис(сульфандиилэтилен)]бис(ЛГ-
метил-2-нитроэтилен-1,1 -диамин).
ио2
Н3С. ХН3
5 N
н
К. А/-Метил-1 -метилтио-2-нитроэтиленамин.
07/2016:0290
РЕЗОРЦИН
Кезогсюо1ит
КЕЗОКС1ЫСН.
С,н402 М.м. 110,1
[108-46-3]
ОПРЕДЕЛЕНИЕ
Бензол-1,3-диол.
Содержание: не менее 98,5 % и не более 101.0 %
(в пересчете на сухое вещество).
110. Зак. 1060.
858
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ(СВОЙСТВА)
Бесцветный или светло-розовато-серый кристал¬
лический порошок или кристаллы. При воздействии
света и воздуха краснеет.
Очень легко растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14): от 109 °С до
112 °С.
B. 0,1 г испытуемого образца растворяют в 1 мл
воды Р, прибавляют 1 мл раствора натрия гидрок¬
сида концентрированного Р, 0,1 мл хлороформа Р,
нагревают и охлаждают. Появляется интенсивное
темно-красное окрашивание. Прибавляют небольшой
избыток хлористоводородной кислоты Р. Появляет¬
ся бледно-желтое окрашивание.
C. 10 мг мелкоизмельченного испытуемого об¬
разца тщательно перемешивают с 10 мг мелкоиз¬
мельченного калия гидрофталата Р и нагревают над
открытым пламенем до появления оранжево-желтого
окрашивания. Охлаждают, прибавляют 1 мл раствора
натрия гидроксида разведенного Р, 10 мл воды Р и
встряхивают до растворения. Полученный раствор
имеет интенсивную зеленую флуоресценцию.
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)5 или П(Кр)5
и должна оставаться таковой после нагревания в во¬
дяной бане в течение 5 мин.
Кислотность или щелочность. К 10 мл раст¬
вора 5 прибавляют 0,05 мл раствора бромфеноло-
вого синего Р2. При прибавлении не более 0,05 мл
0,1 М раствора кислоты хлористоводородной или
0,1 М раствора натрия гидроксида окраска раствора
должна измениться.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 0,5 г испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 0,1 мл испытуемого раст¬
вора доводят метанолом Р до объема 20 мл.
Пластинка: ТСХ пластинка со слоем силикагеля С Р.
Подвижная фаза: этилацетат Р — гексан Р
(40:60, об/об).
Наносимый объем пробы: по 2 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе в течение 15 мин.
Проявление: пластинку выдерживают в парах йода.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения.
Пирокатехин. К 2 мл раствора 3 прибавляют
1 мл раствора аммония молибдата Р2 и перемеши¬
вают. Желтая окраска полученного раствора должна
быть не интенсивнее окраски раствора, приготовлен¬
ного таким же образом и в то же самое время с ис¬
пользованием 2 мл раствора 0,1 г/л пирокатехина Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0%. 1,00 г измельченного испытуемого об¬
разца сушат в эксикаторе в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в
воде Р и доводят до объема 250,0 мл этим же раство¬
рителем. 25,0 мл полученного раствора помещают в
колбу со шлифом, прибавляют 1,0 г калия бромида Р,
50,0 мл 0,0167 М раствора калия бромата, 15 мл
хлороформа Р, 15,0 мл кислоты хлористоводород¬
ной Р1, закрывают пробкой, встряхивают и выдержи¬
вают в защищенном от света месте в течение 15 мин
при периодическом встряхивании. Прибавляют 10 мл
раствора 100 г/л калия йодида Р, тщательно встряхи¬
вают, выдерживают в течение 5 мин и титруют 0,1 М
раствором натрия тиосульфата, используя в каче¬
стве индикатора 1 мл раствора крахмала Р.
1 мл 0,0167 М раствора калия бромата соответ¬
ствует 1,835 мг С6Н602.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:2109
РИБАВИРИН
рЦЪачтпит
твАУтш
С8н12ы405 М.м. 244,2
[36791-04-5]
ОПРЕДЕЛЕНИЕ
1-Р-0-Рибофуранозил-1 Н-1,2,4-триазол-З-карбо-
ксамид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, мало растворим в 96 %
спирте, мало растворим или очень мало растворим в
метиленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО рибавирина #или спектр, пред¬
ставленный на рисунке #2109.-1#.
Если спектры, полученные с использованием
образцов в твердом состоянии, отличаются, то ис¬
пытуемый образец и ФСО рибавирина растворяют по
Рибавирин
859
Рисунок #2109.-1. Инфракрасный спектр ФСО рибавирина
отдельности в метиленхлориде Р и выпаривают до
сухих остатков, которые используют для получения
новых спектров.
ИСПЫТАНИЯ
рН (2.2.3). От 4,0 до 6,5. 0,200 г испытуемого
образца растворяют в воде, свободной от углерода
диоксида, Р и доводят этим же растворителем до объ¬
ема 10,0 мл.
Удельное оптическое вращение (2.2.7). От -33
до -37 (в пересчете на сухое вещество). 0,250 г испы¬
туемого образца растворяют в воде Р и доводят этим
же растворителем до объема 25,0 мл. Удельное опти¬
ческое вращение определяют в течение 10 мин после
приготовления раствора.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в воде для хроматографии Р и до¬
водят до объема 100,0 мл этим же растворителем.
Раствор сравнения (а). Для получения приме¬
си А т зНи смешивают 5,0 мл испытуемого раствора
и 5,0 мл раствора 42 г/л натрия гидроксида Р и вы¬
держивают в течение 90 мин. Полученный раствор
нейтрализуют 5,0 мл раствора 103 г/л кислоты хло¬
ристоводородной Р и тщательно перемешивают.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят водой для хроматографии Р до объема
100,0 мл. 1,0 мл полученного раствора доводят водой
для хроматографии Р до объема 10,0 мл.
Раствор сравнения (с). 50,0 мг ФСО рибавирина
растворяют в воде для хроматографии Р и доводят
до объема 100,0 мл этим же растворителем.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р (размер частиц 3 мкм), подходящим для
применения с подвижными фазами с высоким содер¬
жанием воды;
- температура: 25 °С;
- подвижная фаза:
- подвижная фаза А: 1,0 г натрия сульфата
безводного Р растворяют в 950 мл воды для хро¬
матографии Р, прибавляют 2,0 мл 5 % (об/об)
раствора кислоты фосфорной Р, доводят 5 %
(об/об) раствором кислоты фосфорной Р до рН
2,8 и доводят водой для хроматографии Р до
объема 1000 мл;
- подвижная фаза В: ацетонитрил Р1 — под¬
вижная фаза А (5:95, об/об).
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
100
0
15—25
100 — 0
0—100
25—35
0
100
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 5 мкп испытуемого
раствора и растворов сравнения (а) и (Ь).
Относительное удерживание (по отношению к
рибавирину, время удерживания — около 6 мин): при¬
месь А — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 4,0 между пиками при¬
меси А и рибавирина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А— 2,3):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
110*. Зак. 1060.
860
Государственная фармакопея Республики Беларусь
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (Юррт). 4,0 г испытуемого образца раст¬
воряют в 20 мл воды Р, нагревая при необходимости.
12 мл полученного раствора должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с ис¬
пользованием 10 мл эталонного раствора свинца
(2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемого
раствора и раствора сравнения (с).
Содержание С8Н12Ы405 в процентах рассчитыва¬
ют с учетом содержания рибавирина в ФСО рибави-
рина.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Р, О.
А. 1-р-0-Рибофуранозил-1/7-1,2,4-триазол-З-
карбоновая кислота.
В. 1-а-0-Рибофуранозил-1/7-1,2,4-триазол-З-кар-
боксамид (аномер).
но2с
ГД
С. 1/-/-1,2,4-Триазол-3-карбоновая кислота,
о
О. 1/7-1,2,4-Триазол-З-карбоксамид.
Р. 1-(5-0-Ацетил-р-0-рибофуранозил)-1 Н-1,2,4-
триазол-3-карбоксамид (5-О-ацетилрибавирин).
6. 1-р-0-Рибофуранозил-1/7-1,2,4-триазол-5-
карбоксамид (А/-изомер).
07/2016:0292
РИБОФЛАВИН
РИЬоЛампит
тВОР^АVIN
С17Н20М4Ов М.м. 376,4
[83-88-5]
Рибофлавин
861
ОПРЕДЕЛЕНИЕ
7,8-Диметил-10-[(25,35,4/?)-2,3,4,5-тетрагидро-
ксипентил]бензо[д]птеридин-2,4-(ЗН, 10Н)-дион. вита¬
мин В2#.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый или оранжево-желтый кристаллический
порошок.
Очень мало растворим в воде, практически не¬
растворим в 96 % спирте.
Растворы неустойчивы при хранении на свету,
особенно в присутствии щелочи.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца суспендируют в 10 мл воды Р, встряхивают в
течение 5 мин и фильтруют для удаления не раство¬
рившихся частиц.
Раствор сравнения. 25 мг ФСО рибофлавина
суспендируют в 10 мл воды Р, встряхивают в течение
5 мин и фильтруют для удаления не растворившихся
частиц.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((2—10) мкм).
Подвижная фаза: вода Р.
Наносимый объем пробы, наносят пробы в ука¬
занном порядке и сушат в потоке холодного воздуха
после каждого нанесения:
- нанесение 1: 2 мкл метиленхлорида Р, затем
2 мкл испытуемого раствора;
- нанесение 2: 2 мкл метиленхлорида Р, затем
2 мкл раствора сравнения.
Фронт подвижной фазы: не менее 6 см от линии
старта.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
C. 1 мг испытуемого образца растворяют в
100 мл воды Р. Раствор в проходящем свете имеет
бледное зеленовато-желтое окрашивание, а в от¬
раженном свете — интенсивную желтовато-зеленую
флуоресценцию, которая исчезает при прибавлении
минеральных кислот и щелочей.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
-115 до -135 (в пересчете на сухое вещество). 50,0 мг
испытуемого образца растворяют в 0,05 М растворе
натрия гидроксида, свободном от карбонатов, и до¬
водят до объема 10,0 мл этим же растворителем. Оп¬
тическое вращение измеряют в течение 30 мин после
приготовления раствора.
Оптическая плотность (2.2.25).
Испытуемый раствор. Конечный раствор, при¬
готовленный как указано в разделе «Количественное
определение», разводят равным объемом воды Р.
Максимумы поглощения: при 223 нм, при 267 нм,
при 373 нм и при 444 нм.
Отношение оптических плотностей:
~ ^373^267 “ 0Т 0’31 Д° 0>33;
- Аж/Л267 — от 0,36 ДО 0,39.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием и защищают от
света.
Раствор А. Раствор 13,6 г/л натрия ацетата Р.
Испытуемый раствор. 0,120 г испытуемого об¬
разца растворяют при помощи ультразвука в 10 мл
воды Р и доводят раствором А до объема 100 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 10,0 мл. 1,0 мл
полученного раствора доводят раствором А до объ¬
ема 100,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО рибофлавина для идентификации пиков (со¬
держит примеси С и й) растворяют при помощи уль¬
тразвука в 1,0 мл смеси из 1 объема подвижной фазы
В и 9 объемов подвижной фазы А.
Раствор сравнения (с). Для приготовления при¬
месей А и В /л зНи 10 мг испытуемого образца раст¬
воряют в 1 мл 0,5 М раствора натрия гидроксида и
подвергают воздействию света в течение 1,5 ч. При¬
бавляют 0,5 мл кислоты уксусной Р и доводят во¬
дой Р до объема 100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: кислота фосфорная Р —
вода Р (1:1000, об/об);
- подвижная фаза В: ацетонитрил Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
90
10
5—20
(О
0
1
00
о
10 —► 20
20—25
80
20
25—35
80 —► 50
20 —► 50
35—45
50
50
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 267 нм;
- объем вводимой пробы: 15 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей С и О, используя хроматограм¬
му раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО рибофлавина для идентификации
пиков.
Относительное удерживание (по отношению к
рибофлавину, время удерживания — около 16 мин):
примесь С — около 0,2; примесь О — около 0,5; при¬
месь А — около 1,4; примесь В — около 1,9.
Пригодность хроматографической системы:
- разрешение: не менее 5 между пиками приме¬
си А и примеси В на хроматограмме раствора сравне¬
ния (с);
- хроматограмма раствора сравнения (Ь) должна
соответствовать хроматограмме, прилагаемой к ФСО
рибофлавина для идентификации пиков.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
862
Гэсударственная фармакопея Республики Беларусь
соответствующие поправочные коэффициенты: для
примеси А— 0,7; для примеси В — 1,4; для примеси
С — 2,3; для примеси О — 1,4):
- примесь А (не более 0,025 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
0,25 площади основного пика на хроматограмме
раствора сравнения (а);
- примеси В, С и О (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, С и О, не должны
превышать 2-кратную площадь основного пика на
хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С и О, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (а);
- неучитываемый предел для пиков, кроме
пика примеси А (0,05 %): на хроматограмме испыту¬
емого раствора не учитывают пики с площадью ме¬
нее 0,5 площади основного пика на хроматограмме
раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 1,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из остатка, полученного в ис¬
пытании «Потеря в массе при высушивании».
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от света.
Испытуемый раствор. 65,0 мг испытуемо¬
го образца помещают в мерную колбу из темного
стекла вместимостью 500 мл, суспендируют в 5 мл
воды Р до полного увлажнения пробы и растворяют
в 5 мл раствора натрия гидроксида разведенно¬
го Р. Сразу после растворения прибавляют 100 мл
воды Р, 2,5 мл кислоты уксусной ледяной Р и до¬
водят водой Р до объема 500,0 мл. 20,0 мл полу¬
ченного раствора помещают в мерную колбу из
темного стекла вместимостью 200 мл, прибавляют
3,5 мл раствора 14 г/л натрия ацетата Р и дово¬
дят водой Р до объема 200,0 мл.
Измеряют оптическую плотность (2.2.25) ис¬
пытуемого раствора в максимуме при 444 нм.
Содержание С17Н20М4О6 рассчитывают с ис¬
пользованием удельного показателя поглощения,
равного 328.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
о
А. 7,8,10-Триметилбензо[д]птеридин-2,4(ЗН, 10Н)-
дион (люмифлавин).
о
В. 7,8-Диметилбензо[д]пртеридин-2,4(1 Н,ЗН)-дион.
С. 6,7-Диметил-8-[(25,35,4#)-2,3,4,5-тетрагидро-
ксипентил]птеридин-2,4(ЗН,8Н)-дион.
О. 8-(Гидроксиметил)-7-метил-10-[(25,35,4Я)-
2,3,4,5-тетрагидроксипентил]бензо[д]птеридин-
2,4(ЗН,10Н)-дион.
07/2016:0786
РИБОФЛАВИНА НАТРИЯ ФОСФАТ
Р^^Ьо^1аV^п^ па\гИ рНозрЬаз
ШВОПАУШ зоошм рнозрнате
С17Н20Ы4№О9Р М.м. 478,3
[130-40-5]
ОПРЕДЕЛЕНИЕ
Смесь, содержащая в качестве основного ком¬
понента 5'-(натрия гидрофосфат), а также другие ри¬
бофлавина натрия монофосфаты.
Рибофлавина натрия фосфат
863
Содержание: не менее 73,0 % и не более 79,0 %
рибофлавина (С17Н20М4О6; М.м. 376,4) (в пересчете на
сухое вещество).
Содержит непостоянное количество воды.
ОПИСАНИЕ (СВОЙСТВА)
Желтый или оранжево-желтый кристаллический
порошок. Гигроскопичен.
Растворим в воде, очень мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотомерия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор, 50,0 мг испытуемого об¬
разца растворяют в фосфатном буферном раство¬
ре рН 7,0 Р и доводят до объема 100,0 мл этим же
растворителем. 2,0 мл полученного раствора доводят
фосфатным буферным раствором рН 7,0 Р до объ¬
ема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимумы поглощения: при 266 нм. ,
Удельный показатель поглощения в максимуме:
от 580 до 640.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора основной пик по расположению и величи¬
не соответствует основному пику на хроматограмме
раствора сравнения (Ь).
C. 10 мг испытуемого образца растворяют в рас¬
творе натрия гидроксида разведенного Р и доводят
до объема 100 мл этим же растворителем. 1 мл по¬
лученного раствора выдерживают в ультрафиолето¬
вом свете при длине волны 254 нм в течение 5 мин,
прибавляют кислоту уксусную Р до кислой реакции
среды по бумаге лакмусовой синей Р и встряхивают с
2 мл метиленхлорида Р. Нижний слой имеет желтую
флуоресценцию.
О. К 0,5 г испытуемого образца прибавляют 10 мл
кислоты азотной Р и выпаривают смесь на водяной
бане досуха. Полученный остаток сжигают до белого
цвета, растворяют в 5 мл воды Р и фильтруют. Филь¬
трат дает реакцию (а) на натрий и реакцию (Ь) на фос¬
фаты (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 6,5. 0,5 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 50 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+38,0 до +43,0 (в пересчете на сухое вещество).
0,300 г испытуемого образца растворяют в 18,2 мл
кислоты хлористоводородной Р1 и доводят водой Р
до объема до 25,0 мл.
Примесь Е. К 35 мг испытуемого образца при¬
бавляют 10 мл метиленхлорида Р, перемешивают
в течение 5 мин и фильтруют. Окраска полученно¬
го фильтрата должна быть не интенсивнее эталона
ВУ(КЖ)б (2.2.2, метод II).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от актиничного света.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в 50 мл воды Р и доводят подвижной
фазой до объема 100,0 мл. 8,0 мл полученного раст¬
вора доводят подвижной фазой до объема 50,0 мл.
Раствор сравнения (а). 60 мг ФСО рибофлавина
(примесь О) растворяют в 1 мл кислоты хлористо¬
водородной Р и доводят водой Р до объема 250,0 мл.
4,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (Ь). 0,100 г ФСО рибофла¬
вина натрия фосфата растворяют в 50 мл воды Р и
доводят объем подвижной фазой до 100,0 мл. 8,0 мл
полученного раствора доводят подвижной фазой до
объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: метанол Р—раствор 7,35 г/л
калия дигидрофосфата Р (150:850, об/об);
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 266 нм;
- объем вводимой пробы: 100 мкл;
- время хроматографирования: до полного вы¬
хода пика рибофлавина.
Относительное удерживание (по отношению
к рибофлавин-5'-монофосфату, время удержива¬
ния — около 20 мин): примесь А— около 0,2; примесь
В — около 0,3; примесь С — около 0,5; рибофлавин-
З'-монофосфат — около 0,7; рибофлавин-4-мо-
нофосфат — около 0,9; примесь О — около 2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиком рибо-
флавин-4'-монофосфата и пиком рибофлавин-5-мо-
нофосфата.
Содержание свободного рибофлавина (примесь
О) и рибофлавина в форме дифосфатов (примеси А,
В, С) рассчитывают в процентах исходя из площадей
пиков на хроматограмме испытуемого раствора и ко¬
личества свободного рибофлавина в растворе срав¬
нения (а).
Предельное содержание примесей:
- примесь О: не более 6,0 % (в пересчете на су¬
хое вещество);
- сумма примесей А, В, С: не более 6,0 % (в пере¬
счете на сухое вещество).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для фар¬
мацевтического использования (2034), не применяют.
Неорганические фосфаты. Не более 1,5 %.
Испытуемый раствор. 0,10 г испытуемого образ¬
ца растворяют в воде Р и доводят до объема 100 мл
этим же растворителем. К 5 мл полученного раствора
прибавляют 10 мл воды Р, 5 мл забуференого раст¬
вора меди сульфата рН 4,0 Р, 2 мл раствора 30 г/л
аммония молибдата Р, 1 мл свежеприготовленного
раствора, содержащего 20 г/л 4-метиламинофено-
ла сульфата Р и 50 г/л натрия метабисульфита Р,
прибавляют 1 мл 3 % (об/об) раствора кислоты хлор¬
ной Р и доводят водой Р до объема 25,0 мл.
Раствор сравнения. К 15 мл эталонного раст¬
вора фосфата (5 ррт РО) Р прибавляют 5 мл забу¬
ференого раствора меди сульфата рН 4,0 Р, 2 мл
раствора 30 г/л аммония молибдата Р, 1 мл свежепри¬
готовленного раствора, содержащего 20 г/л 4-метила-
минофенола сульфата Р и 50 г/л натрия метабисуль¬
фита Р, прибавляют 1 мл 3 % (об/об) раствора кисло¬
ты хлорной Р и доводят водой Р до объема 25,0 мл.
864
Гэсударственная фармакопея Республики Беларусь
Компенсационный раствор. Готовят аналогично
испытуемому раствору, но без прибавления испытуе¬
мого образца.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора и раствора сравнения при длине вол¬
ны 800 нм не позднее, чем через 15 мин после при¬
готовления. Оптическая плотность испытуемого раст¬
вора не должна превышать оптическую плотность
раствора сравнения.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (Юррт). 2,0 г испытуемого образца поме¬
щают в кварцевый тигель, прибавляют 2 мл кислоты
азотной Р, по каплям 0,25 мл кислоты серной Р,
осторожно нагревают до появления белых паров и
прокаливают. Охлажденный остаток дважды экстра¬
гируют кислотой хлористоводородной Р порциями
по 2 мл и полученное извлечение упаривают на во¬
дяной бане до сухого остатка. Полученный остаток
растворяют в 2 мл кислоты уксусной разведенной Р
и доводят водой Р до объема 20 мл. 12 мл получен¬
ного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
10 мл эталонного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 8,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С и давлении не более 0,7 кПа в те¬
чение 5 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от света.
0,100 г испытуемого образца растворяют в 150 мл
воды Р, прибавляют 2 мл кислоты уксусной ледяной Р
и доводят водой Р до объема 1000,0 мл. К 10,0 мл по¬
лученного раствора прибавляют 3,5 мл раствора 14 г/л
натрия ацетата Р и доводят водой Р объема 50,0 мл.
Измеряют оптическую плотность (2.2.25) полученного
раствора в максимуме при длине волны 444 нм.
Содержание С17Н20М4О6 рассчитывают с учетом
удельного показателя поглощения, равного 328.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
A. КЗ = Р4 = Р03Н2, К5 = Н: Рибофлавин-3',4'-
дифосфат.
B. КЗ = К5 = Р03Н2, К4 = Н: Рибофлавин-3',5'-
дифосфат.
C. КЗ = Н, К4 = К5 = Р03Н2: Рибофлавин-4',5'-
дифосфат.
й. КЗ = К4 = К5 = Н: Рибофлавин.
Е. 7,8,10-Триметилбензо[д]птеридин-2,4(ЗН,10Н)-
дион (люмифлавин).
07/2016:0349
РИСОВЫЙ КРАХМАЛ
Огугае ату1ит
ШСЕ ЗТАКСН
ОПРЕДЕЛЕНИЕ
Крахмал рисовый получают из зерен Огуга зайУа 1_.
ОПИСАНИЕ (СВОЙСТВА)
Очень мелкий белый или почти белый порошок,
при сжатии между пальцами скрипит.
Практически нерастворим в холодной воде и
96 % спирте.
Рисовый крахмал не должен содержать зерен
крахмала другого происхождения, может содержать в
минимальном количестве фрагменты ткани эндоспер¬
ма зерен.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. При изучении под микроскопом (2.8.23) с ис¬
пользованием 50 % (об/об) раствора глицерина Р
испытуемый образец представляет собой много¬
гранные простые зерна размером от 1 мкм до 10 мкм
(преимущественно от 4 мкм до 6 мкм) (рисунок 0349.-
1). Эти простые зерна часто объединяются в эллип¬
соидальные сложные зерна диаметром от 50 мкм до
100 мкм. Зерна имеют плохо различимое централь¬
ное ядро; не наблюдается концентрическая борозд-
чатость. При просматривании между перпендикуляр¬
но расположенными поляризующими пластинками
или призмами на зернах присутствует отчетливый
черный крестик в ядре.
Рисунок 0349.-1. Иллюстрация к испытанию А
раздела «Подлинность (идентификация)»
для рисового крахмала
Рисперидон
865
B. 1 г испытуемого образца суспендируют в 50 мл
воды Р, кипятят в течение 1 мин и охлаждают. Образу¬
ется мутный водянистый клейстер.
C. К 1 мл клейстера, полученного в подлинности
В, прибавляют 0,05 мл раствора йода Р1. Появляет¬
ся окраска от оранжево-красной до темно-синей, ис¬
чезающая при нагревании.
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 8,0. 5,0 г испытуемого об¬
разца встряхивают с 25,0 мл воды, свободной от
углерода диоксида, Р в течение 60 с. Выдерживают в
течение 15 мин.
Железо (2.4.9). Не более 0,0010% (Юррт) в
фильтрате. 1,5 г испытуемого образца встряхивают с
15 мл кислоты хлористоводородной разведенной Р
и фильтруют.
Посторонние вещества. При изучении под ми¬
кроскопом с использованием 50 % (об/об) раствора
глицерина Р должны обнаруживаться не более чем
следовые количества веществ, отличных от зерен
крахмала. Присутствие зерен крахмала любого друго¬
го происхождения не допускается.
Потеря в массе при высушивании (2.2.32). Не
более 15,0 %. 1,00 г испытуемого образца сушат при
температуре 130 °С в течение 90 мин.
Сульфатная зола (2.4.14). Не более 0,6%.
Определение проводят из 1,0 г испытуемого образца.
Окисляющие вещества (2.5.30). Не более
0,002 % в пересчете на Н202.
Серы диоксид (2.5.29). Не более 0,0050 %
(50 ррт).
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсНепсЫа соН: отсутствие (2.6.13).
8а1топе11а: отсутствие (2.6.13).
07/2016:1559
РИСПЕРИДОН
РИзрепс/опит
Я/5РЕЯЮОЛ/Е
[106266-06-2]
ОПРЕДЕЛЕНИЕ
3-[2-[4-(6-Фтор-1,2-бензизоксазол-3-ил)пиперидин-
1 -ил]этил]-2-метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]
пиримидин-4-он.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, умеренно растворим в 96 %
спирте, растворяется в разведенных растворах мине¬
ральных кислот.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО рисперидона.
Если полученные спектры отличаются, то ис¬
пытуемый образец и ФСО рисперидона растворяют
по отдельности в ацетоне Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Раствор 3. 0,1 г испытуемого образца раство¬
ряют в растворе 7,5 г/л кислоты винной Р и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО рисперидона
для проверки пригодности хроматографической си¬
стемы (содержит примеси А, В, С, О и Е) растворяют
в 1,0 мл метанола Р.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
5.0 мл полученного раствора доводят метанолом Р
до объема 25,0 мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО рисперидона примеси К растворяют в 1,0 мл
метанола Р.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, для хроматографии Р с размером частиц
3 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 5 г/л аммония
ацетата Р;
- подвижная фаза В: метанол Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
70
30
2—17
о
со
т
о
I4-
о
Г"-
т
о
со
17—22
30
70
- скорость подвижной фазы. 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, О и Е, используя хрома¬
тограмму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО рисперидона для проверки при¬
годности хроматографической системы, иденти¬
фицируют пик примеси К, используя хроматограмму
раствора сравнения (с).
866
Государственная фармакопея Республики Беларусь
Относительное удерживание (по отноше¬
нию к рисперидону, время удерживания — около
12 мин): примесь А— около 0,7; примесь В — око¬
ло 0,75; примесь С — около 0,8; примесь К — око¬
ло 0,9; примесь О — около 0,94; примесь Е — око¬
ло 1,1.
Пригодность хроматографической системы:
раствор сравнения (а):
- хроматограмма раствора сравнения (а)
должна соответствовать хроматограмме, прилага¬
емой к ФСО рисперидона для проверки пригодно¬
сти хроматографической системы,
- коэффициент разделения пиков: не менее
1,5 (Нр — высота пика примеси О относительно ба¬
зовой линии; Ну— расстояние между базовой лини¬
ей и нижней точкой кривой, разделяющей пик при¬
меси й и пик рисперидона).
Предельное содержание примесей:
- примеси А, В, С, О и Е (не более 0,2 %): на
хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, С, О и Е,
не должны превышать площадь основного пика на
хроматограмме раствора сравнения (Ь);
-примесь К (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси К, не должна превышать
0,75 площади основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать 0,5 площади основного пика на хрома¬
тограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
1,5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца сушат
при температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образ¬
ца в платиновом тигле.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,160 г испытуемого образца растворяют в
70 мл смеси из кислоты уксусной безводной Р и
метилэтилкетона Р (1:7, об/об) и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 20,53 мг С23Н27РМ402.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацев¬
тического использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): Р, Н,
I, 3, Ц М.
А. 3-[2-[4-[(Е^-(2,4-Дифторфенил)(гидроксиимино)-
метил]пиперидин-1-ил]этил]-2-метил-6,7,8,9-тетрагидро-
4Н-пиридо[1,2-а]пиримидин-4-он.
В. 3-[2-[4-[(2)-(2,4-Дифторфенил)(гидроксиимино)-
метил]пиперидин-1-ил]этил]-2-метил-6,7,8,9-тетрагидро-
4Н-пиридо[1,2-а]пиримидин-4-он.
С. (9/?5)-3-[2-[4-(6-Фтор-1,2-бензисоксазол-З-ил)-
пиперидин-1-ил]этил]-9-гидрокси-2-метил-6,7,8,9-
тетрагидро-4Н-пиридо[1,2-а]пиримидин-4-он.
О. 3-[2-[4-(5-Фтор-1,2-бензисоксазол-3-ил)пипе-
ридин-1-ил]этил]-2-метил-6,7,8,9-тетрагидро-4Н-
пиридо[1,2-а]пиримидин-4-он.
Специфицированные примеси: А, В, С, О, Е, К.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
Е. (6Я5)-3-[2-[4-(6-Фтор-1,2-бензисоксазол-З-ил)-
пиперидин-1-ил]этил]-2,6-диметил-2-метил-6,7,8,9-
тетрагидро-4/-/-пиридо[1,2-а]пиримидин-4-он.
Рифабутин
867
Р. 2-[2-Метил-4-оксо-6,7,8,9-тетрагидро-4Н-пири-
до[1,2-а]пиримидин-3-ил]этил-4-(6-фтор-1,2-бензи-
соксазол-3-ил)пиперидин-1-карбоксилат.
Н. 3-[2-[4-(2,4-Дифторбензоил)пиперидин-1-ил]-
этил]-2-метил-6,7,8,9-тетрагидро-4Я-пиридо[1,2-а]-
пиримидин-4-он.
I. 3-[2-[4-[4-Фтор-2-[4-(6-фтор-1,2-бензисоксазол-
3- ил)пиперидин-1 -ил]бензоил]пиперидин-1 -ил]этил]-2-
метил-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-
4- он.
Л. 3-[2-[4-[(2)-[4-Фтор-2-[4-(6-фтор-1,2-бензи-
соксазол-3-ил)пиперидин-1-ил]фенил](гидроксиимино)-
метил]пиперидин-1-ил]этил]-2-метил-6,7,8,9-тетрагидро-
4Н-пиридо[1,2-а]пиримидин-4-он.
К. 3-[2-[4-(1,2-Бензисоксазол-3-ил)пиперидин-1-
ил]этил]-2-метил-6,7>8,9-тетрагидро-4/-/-пиридо[1,2-а]-
пиримидин-4-он (дезфторрисперидон).
С1
о
1_. 3-(2-Хлорэтил)-2-метил-6,7,8,9-тетрагидро-4Н-
пиридо[1,2-а]пиримидин-4-он (пиперидопиримидинон
интермедиат).
М. 6-Фтор-3-(пиперидин-4-ил)-1,2-бензисоксазол.
07/2016:1657
РИФАБУТИН
РМаЬиИпит
мрлвитш
М.м. 847
[72559-06-9]
ОПРЕДЕЛЕНИЕ
(95,12 Е, 145,15/?, 165,17 Я, 18Я, 19Я,205,215,22Е,
242)-6,18,20-Тригидрокси-14-метокси-7,9,15,17,19,21,25-
гептаметил-1 -(2-метилпропил)-5,10,26-триоксо-3,5,9,10-
тетрагидроспиро[9,4-(эпоксипентадека[1,11,13]триени-
мино)-2Н-фуро[2',3':7,8]нафто[1,2-а(]имидазол-2,4'-
пиперидин]-16-илацетат.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Аморфный порошок красновато-фиолетового
цвета.
Мало растворим в воде, растворим в метаноле,
мало растворим в 96 % спирте.
868
Государственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО рифабутина #или спектр, пред¬
ставленный на рисунке #1657.-1#.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме ис¬
пытуемого раствора соответствует по времени удер¬
живания и величине основному пику на хроматограм¬
ме раствора сравнения (а).
ИСПЫТАНИЯ
Примесь А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,100 г испытуемого
образца растворяют в смеси из равных объемов
метанола Р и метиленхлорида Р и доводят до
объема 10 мл этой же смесью растворителей.
Раствор сравнения. 10 мг ФСО рифабутина
примеси А растворяют в смеси из равных объемов
метанола Р и метиленхлорида Р и доводят до
объема 10 мл этой же смесью растворителей. 3 мл
полученного раствора доводят смесью из равных
объемов метанола Р и метиленхлорида Р до объ¬
ема 100 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР2в4Р
Подвижная фаза: ацетон Р — петролейный
эфир Р (23:77, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода в течение 5 мин, опрыскивают раствором калия
йодида и крахмала Р и выдерживают в течение 5 мин.
Предельное содержание примесей:
- примесь А (не более 0,3 %): на хроматограм¬
ме испытуемого раствора пятно, соответствующее
примеси А, должно быть не интенсивнее пятна на
хроматограмме раствора сравнения.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в подвижной фазе и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг ФСО рифабу¬
тина растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой до объема
100,0 мл.
Раствор сравнения (с). 10 мг ФСО рифабу¬
тина растворяют в 2 мл метанола Р, прибавляют
1 мл раствора натрия гидроксида разведенно¬
го Р и выдерживают в течение около 4 мин. При¬
бавляют 1 мл кислоты хлористоводородной раз¬
веденной Р и доводят подвижной фазой до объема
50 мл.
Условия хроматографирвоания:
- колонка длиной 0,110 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- подвижная фаза: смесь из равных объемов
ацетонитрила Р и раствора 13,6 г/л калия диги¬
дрофосфата Р, доведенного до рН 6,5 раствором
натрия гидроксида разведенным Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 254 нм;
- объем вводимой пробы: 20 мкл;
-время хроматографирования: 2,5-кратное
время удерживания рифабутина.
Рисунок #1657.-1. Инфракрасный спектр ФСО рифабутина
Рифампицин
869
Относительное удерживание (по отношению к
рифабутину, время удерживания — около 9 мин): при¬
месь Е — около 0,5; примесь В — около 0,6; примесь
О — около 0,9; примесь С — около 1,3.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 2,0 между вторым пиком
из трех пиков продуктов распада и пиком рифабутина.
Предельное содержание примесей:
-любая примесь: на хроматограмме испыту¬
емого раствора площадь любого пика, кроме пика
рифабутина, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь)
(1,0 %) и только один из таких пиков может иметь пло¬
щадь более 0,5 площади основного пика на хромато¬
грамме раствора сравнения (Ь) (0,5 %);
- сумма примесей (не более 3,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме пика рифабутина, не должна превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (Ь).
Вода (2.5.12). Не более 2,5 %. Определение про¬
водят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,3%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание рифабутина рассчитывают в про¬
центах.
ПРИМЕСИ
А. 1-(2-Метилпропил)пиперидин-4-он.
В. X = О: З-Аминорифамицин 5.
Р. X = ЫН: З-Амино-4-имидорифамицин 3.
С. К1 = СО-СН3, К2 + КЗ = СН2:21,31-Дидегидро-
рифабутин.
Е. К1 = КЗ = Н, К2 = СН3: 16-Дезацетилрифабу-
тин.
07/2016:0052
РИФАМПИЦИН
Шатр1с'тит
Я1РАМР1С1Ы
С43Н58М4012 М.м. 823
[13292-46-1]
ОПРЕДЕЛЕНИЕ
(25,122,14Е, 165,175,18/?,19К,20/?,215,22/?,
233,24Е)-5,6>9,17,19-Пентагидрокси-23-метокси-
2,4,12,16,18,20,22-гептаметил-8-[[(4-метилпиперазин-
1-ил)имино]метил]-1,11-диоксо-1,2-дигидро-2,7-
(эпоксипентадека[1,11,13]триенимино)нафто[2,1 -Ь]-
фуран-21 -илацетат.
Полусинтетический антибиотик, полученный из
рифамицина 5\/.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Красновато-коричневый или коричневато-крас¬
ный кристаллический порошок.
Мало растворим в воде, растворим в метаноле,
мало растворим в ацетоне и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в 50 мл метанола Р. 1 мл полу-
870
Гэсударственная фармакопея Республики Беларусь
ченного раствора доводят фосфатным буферным
раствором рН 7,4 Р до объема 50 мл.
Диапазон длин волн: от 220 нм до 500 нм.
Максимумы поглощения: при 237 нм; при
254 нм; при 334 нм; при 475 нм.
Отношение оптических плотностей:
АиАи — около 1'75'
B. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Приготовление: суспензия в вазелиновом
масле Р.
Сравнение: ФСОрифампицина.
C. 25 мг испытуемого образца суспендиру¬
ют в 25 мл воды Р, встряхивают в течение 5 мин
и фильтруют. К 5 мл фильтрата прибавляют 1 мл
раствора 100 г/л аммония персульфата Р в фос¬
фатном буферном растворе рН 7,4 Р и встряхи¬
вают в течение нескольких минут. Окраска изме¬
няется с оранжево-желтой на фиолетово-красную,
осадок при этом не образуется.
ИСПЫТАНИЯ
рН (2.2.3). От 4,5 до 6,5. Измеряют рН суспен¬
зии 10 г/л испытуемого образца в воде, свободной
от углерода диоксида, Р.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Испытуемый раствор и
раствор сравнения готовят непосредственно
перед использованием.
Смесь растворителей. К 10 объемам раст¬
вора 210,1 г/л кислоты лимонной Р прибавляют
23 объема раствора 136,1 г/л калия дигидрофос¬
фата Р, 77 объемов раствора 174,2 г/л дикалия
гидрофосфата Р, 250 объемов ацетонитрила Р
и 640 объемов воды Р.
Испытуемый раствор. 20,0 мг испытуемого
образца растворяют в ацетонитриле Р и доводят
до объема 10,0 мл этим же растворителем. 5,0 мл
полученного раствора доводят смесью растворите¬
лей до объема 50,0 мл.
Раствор сравнения. 20,0 мг ФСО рифампици¬
на хинона (примесь А) растворяют в ацетонитри¬
ле Р и доводят до объема 100,0 мл этим же раство¬
рителем. К 1,0 мл полученного раствора прибавля¬
ют 1,0 мл испытуемого раствора и доводят смесью
растворителей до объема 100,0 мл.
Условия хроматографирования:
-колонка длиной 0,12м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- подвижная фаза: смешивают 35 объемов
ацетонитрила Р и 65 объемов раствора, содержа¬
щего 0,1 % (об/об) кислоты фосфорной Р, 1,9 г/л
натрия перхлората Р, 5,9 г/л кислоты лимон¬
ной Р и 20,9 г/л калия дигидрофосфата Р;
- скорость подвижной фазы. 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания рифампицина.
Пригодность хроматографической системы.
раствор сравнения:
-разрешение: не менее 4,0 между пиками
рифампицина и примеси А; при необходимости из¬
меняют концентрацию ацетонитрила в подвижной
фазе.
Предельное содержание примесей:
-примесь А (не более 1,5 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
1,5-кратную площадь соответствующего пика на
хроматограмме раствора сравнения;
-любая другая примесь (не более 1,0 %): на
хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пика примеси А, не
должна превышать площадь пика рифампицина на
хроматограмме раствора сравнения;
- сумма примесей, кроме примеси А (не бопее
3,5 %)*. на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и
пика примеси А, не должна превышать 3,5-кратную
площадь пика рифампицина на хроматограмме
раствора сравнения;
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,05 площади пика рифам¬
пицина на хроматограмме раствора сравнения.
Тяжелые металлы (2.4.8, метод С). Не бо¬
лее 0,0020 % (20 ррт). 1,0 г испытуемого образ¬
ца должны выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием 2 мл
эталонного раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 1,0 %. 1,000 г испытуемого образца су¬
шат при температуре 80 °С и давлении не выше
0,67 кПа в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 2,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в ме¬
таноле Р и доводят до объема 100,0 мл этим же
растворителем. 2,0 мл полученного раствора до¬
водят фосфатным буферным раствором рН 7,4 Р
до объема 100,0 мл. Измеряют оптическую плот¬
ность (2.2.25) полученного раствора в максиму¬
ме при 475 нм, используя фосфатный буферный
раствор рН 7,4 Р в качестве компенсационного
раствора.
Содержание С43Н58Ы4012 рассчитывают в про¬
центах с учетом удельного показателя поглощения,
равного 187.
ХРАНЕНИЕ
В атмосфере азота в воздухонепроницаемом
контейнере в защищенном от света месте при темпе¬
ратуре не выше 25 °С.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следую¬
щие вещества, если они присутствуют в значи¬
тельных количествах, следует определять тем или
Рокситромицин
871
иным испытанием, описанным в частной статье.
Их содержание лимитируется общим критерием
приемлемости для других/неспецифицирован-
ных примесей и/или общей статьей Субстанции
для фармацевтического использования (2034).
Вследствие этого нет необходимости идентифици¬
ровать эти примеси для доказательства соответ¬
ствия требованиям. См. также статью 5.10. Кон¬
троль примесей в субстанциях для фармацевти¬
ческого использования): В.
А. Рифампицин хинон.
В. Рифампицин Л/-оксид.
07/2016:1146
РОКСИТРОМИЦИН
КохПЬготус'тит
ГЮХ1ТНКОМУСМ
С41н7вн2015 М.м. 837
[80214-83-1]
ОПРЕДЕЛЕНИЕ
(ЗК,45,55,6К,7К,9К, 115,1 2Я, 135,14Я)-4-[(2,6-
Дидезокси-3-С-метил-3-0-метил-а-1_-р(убо-гексо-
пиранозил)окси]-14-этил-7,12,13-тригид рокси-10-[(Е)-
[(2-метоксиэтокси)метокси]имино]-3,5,7,9,11,13-
гексаметил-6-[[3,4,6-тридезокси-3-(диметиламино)-Р-0-
/сшло-гексопиранозил]окси]оксациклотетрадекан-2-он
(эритромицин-9-(Е)-[0-[(2-метоксиэтокси)метил]оксим]).
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
72 I
70 !
3000 2000 1500 1000
Волновое число (см'1)
Рисунок #1146.-1. Инфракрасный спектр ФСО рокситромицина
872
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень мало растворим в воде, легко растворим в
ацетоне, в 96 % спирте и в метиленхлориде, мало рас¬
творим в разведенной хлористоводородной кислоте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО рокситромицина #или спектр,
представленный на рисунке #1146.-1#.
Если полученные спектры отличаются, то полу¬
чают новые спектры, используя растворы 90 г/л в ме¬
тиленхлориде Р.
B. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 0,2 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Удельное оптическое вращение (2.2.7). От -93
до -96 (в пересчете на безводное вещество). 0,500 г
испытуемого образца растворяют в ацетоне Р и до¬
водят до объема 50,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Раствор А. 30 объемов ацетонитрила Р сме¬
шивают с 70 объемами раствора 48,6 г/л аммония
дигидрофосфата Р, доведенного раствором натрия
гидроксида разведенным Р до рН до 5,3.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
25.0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг ФСО рокситро¬
мицина растворяют в растворе А и доводят до объема
25.0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят раствором А до объема 100,0 мл.
Раствор сравнения (с). 2,0 мг ФСО рокситроми¬
цина для проверки пригодности хроматографиче¬
ской системы растворяют в растворе А и доводят до
объема 1,0 мл этим же растворителем.
Раствор сравнения (д). 1,0 мл толуола Р дово¬
дят ацетонитрилом Рдо объема 100,0 мл. 0,2 мл по¬
лученного раствора доводят раствором А до объема
200.0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р (размер частиц 5 мкм) с размером пор
10 нм, содержащим около 19 % углерода;
- температура: колонка — 15 °С, блок ввода
проб — 8 °С;
- подвижная фаза:
- подвижная фаза А: 26 объемов ацетонитри¬
ла Р смешивают с 74 объемами раствора 59,7 г/л
аммония дигидрофосфата Р, доведенного рас¬
твором натрия гидроксида разведенным Р до
рН 4,3;
- подвижная фаза В: вода Р — ацетонитрил Р
(30:70, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—50
100
0
50—51
100 —*> 90
0 — 10
51—80
90
10
80—81
90 — 100
10-0
81—100
100
0
- скорость подвижной фазы: 1,1 мл/мин;
- спектрофотометрический детектор, длина
волны 205 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (с1).
Относительное удерживание (по отношению
к рокситромицину, время удерживания — около
22 мин): примесь А — около 0,28; примесь В — око¬
ло 0,31; примесь С — около 0,33; примесь О — око¬
ло 0,62; примесь Е — около 0,67; примесь Р — около
0,83; примесь С — около 1,15; примесь К — около 1,7;
примесь И — около 1,85; примесь 3 — около 2,65; при¬
месь I — около 3,1.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика примеси О относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси (3 и
рокситромицина).
Предельное содержание примесей:
- примесь 6 (не более 1,0 %); на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси О, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, В, С, О, Е, Р, Н, I, 3 (не более 0,5 %);
на хроматограмме испытуемого раствора площадь
пика, соответствующего примеси, не должна превы¬
шать 0,5 площади основного пика на хроматограмме
раствора сравнения (Ь);
- сумма примесей (не более 3,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме пика рокситромицина, не должна превы¬
шать 3-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пик, соответствую¬
щий пику толуола на хроматограмме раствора срав¬
нения (с!), и пики с площадью менее 0,05 площади ос¬
новного пика на хроматограмме раствора сравнения
(Ь) (0,05 %).
Тяжелые металлы (2.4.8, метод В). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в смеси из воды Р и ацетона Р (15:85, об/об) и
доводят до объема 20 мл этой же смесью раствори¬
телей. 12 мл полученного раствора должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием эталонного раствора свинца (1 ррт
РЬ), полученного разведением эталонного раствора
свинца (100 ррт РЬ) Р смесью из воды Р и ацетона Р
(15:85, об/об).
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 0,200 г испытуемого образца.
Рокситромицин
873
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- колонка длиной 0,25 м;
- подвижная фаза: 307 объемов ацетонитри¬
ла Р смешивают с 693 объемами раствора 49,1 г/л
аммония дигидрофосфата Р, доведенного до рН 5,3
раствором натрия гидроксида разведенного Р\
- скорость подвижной фазы: 1,5 мл/мин;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а) и (с).
Время удерживания: рокситромицин — около
12 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси С относительно базовой
линии; Н}/ — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси С и
рокситромицина).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р,
в, Н, I, 3.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): К.
А. (ЗЯ,45,55,6Я,7Я,9Я, 11 Я, 12Я, 135,14Я)-4-[(2,6-
Дидезокси-З-С-метил-З-О-метил-а-Ь-рибо-гексопи-
ранозил)окси]-14-этил-7,12,13-тригидрокси-3,5,7,9,11,13-
гексаметил-6-[[3,4,6-тридезокси-3-(диметиламино)-р-
0-ксш70-гексопиранозил]окси]оксациклотетрадекан-
2,10-дион (эритромицин А).
В. 3-0-Де(2,6-дидезокси-3-С-метил-3-0-метил-а-
1-рыбо-гексопиранозил)-эритромицин-9-(Е)-[0-[(2-
метоксиэтокси)метил]оксим].
С. В = Н: Эритромицин-9-(Е)-оксим.
О. В = СН2-0-СН2-0-СН2-СН2-0СН3: Эритромицин-
9-(Е)-[0-[[(2-метоксиэтокси)метокси]метил]оксим].
3. В = СН2-0-СН2-СН2С1: Эритромицин-9-(Е)-[0-[(2-
хлорэтокси)метил]оксим].
К. В = СН2-0-СН2-СН2-0-СН20Н: Эритромицин-9-
(Е)-[0-[[(2-гидроксиметокси)этокси]метил]оксим].
Е. В = Н, В' = СН3: 3"-0-Деметилэритромицин-9-
(Е)-[0-[(2-метоксиэтокси)-метил]оксим].
Р. В = СН3, В' = Н: 3-Л/-Деметилэротромицин-9-(Е)-
[0-[(2-метоксиэтокси)метил]оксим].
СН3
H. В = В' = Н: 12-Дезоксиэритромицин-9-(Е)-[0-[(2-
метоксиэтокси)метил]оксим].
I. В = ОН, В' = СН2-0-СН2-СН2-0СН3: 2'-0-[(2-Ме-
токсиэтокси)метил]эритромицин-9-(Е)-[0-[(2-метокси-
этокси)метил]оксим].
874
Гэсударственная фармакопея Республики Беларусь
07/2016:0120
РТУТИ ХЛОРИД
Нубгагдуп сИсЫопс1ит
АТЕЯС1УК/С СШ-ОКЮЕ
НдС12 М.м. 271,5
[7487-94-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,5 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок или бесцветные, или белые, или почти белые
кристаллы или тяжелая кристаллическая масса.
Растворим в воде и в глицерине, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакции на хлори¬
ды (2.3.1).
B. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», дает реакции (а) и (Ь) на ртуть
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора метилового
красного Р. Должно появиться красное окрашивание.
К полученному раствору прибавляют 0,5 г натрия
хлорида Р. Должно появиться желтое окрашивание.
При прибавлении не более 0,5 мл 0,01 М раствора
кислоты хлористоводородной должно появиться
красное окрашивание.
Ртути (I) хлорид. 1,0 г испытуемого образца
растворяют в 30 мл эфира Р. Раствор не должен быть
мутным.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 2,00 г испытуемого образца сушат в ва¬
кууме в течение 24 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в 100 мл
воды Р. К полученному раствору прибавляют 20,0 мл
0,1 М раствора натрия эдетата и 5 мл буферного
раствора рН 10,9 Р. Выдерживают в течение 15 мин.
Прибавляют 0,1 г индикаторной смеси протравного
черного 11 Р и титруют 0,1 М раствором цинка суль¬
фата до появления фиолетового окрашивания. При¬
бавляют 3 г калия йодида Р, выдерживают в течение
2 мин, прибавляют еще 0,1 г индикаторной смеси
протравного черного 11 Р и титруют 0,1 М раство¬
ром цинка сульфата.
1 мл 0,1 М раствора цинка сульфата, использо¬
ванного во втором титровании, соответствует 27,15 мг
НдС12.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1795
РУТОЗИД ТРИГИДРАТ
Ки&озШит Шус/псит
РШТОЗЮЕ ТШНУОКАТЕ
С27Н30О16 ■ ЗН20 М.м. 665
[250249-75-3]
ОПРЕДЕЛЕНИЕ
3-[[6-0-(6-Дезокси-а-1_-маннопиранозил)-р-0-
глюкопиранозил]окси]-2-(3,4-дигидрофенил)-5,7-
дигидрокси-4Н-1 -бензопиран-4-он. #Рутин#.
Содержание: не менее 95,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый или зеленовато-желтый кристалличе¬
ский порошок.
Практически нерастворим в воде, растворим в
метаноле, умеренно растворим в 96% спирте, практи¬
чески нерастворим в метиленхлориде. Растворяется
в растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
250,0 мл этим же растворителем и, при необходимо¬
сти, фильтруют. 5,0 мл полученного раствора доводят
метанолом Р до объема 50,0 мл.
Диапазон длин волн: от 210 нм до 450 нм.
Максимумы поглощения: при 257 нм и при 358 нм.
Удельный показатель поглощения в максимуме
при 358 нм: от 305 до 330 (в пересчете на безводное
вещество).
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО рутозида тригидрата #или
спектр, представленный на рисунке #1795.-1#.
C. Тонкослойная хроматография (2.2.27).
Рутозид тригидрат
875
Рисунок #1795.-1. Инфракрасный спектр ФСО рутозида тригидрата
Испытуемый раствор: 25 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения: 25 мг ФСО рутозида три-
гидрата растворяют в метаноле Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля 6 Р
Подвижная фаза: бутанол Р — кислота уксус¬
ная безводная Р — вода Р — метилэтилкетон Р —
этилацетат Р (5:10:10:30:50, об/об/об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают смесью из
7,5 мл раствора 10 г/л калия феррицианида Р и 2,5 мл
раствора железа (III) хлорида Р1 и просматривают в
течение 10 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
0.10 мг испытуемого образца растворяют в 5 мл
96 % спирта Р, прибавляют 1 г порошка цинка Р и
2 мл кислоты хлористоводородной Р1. Появляется
красное окрашивание.
ИСПЫТАНИЯ
Светопоглощающие примеси (2.2.25). Погло¬
щение должно быть не более 0,10 в диапазоне длин
волн от 450 нм до 800 нм. 0,200 г испытуемого образ¬
ца растворяют в 40 мл 2-пропанола Р, перемешивают
в течение 15 мин, доводят до объема 50,0 мл этим же
растворителем и фильтруют.
Примеси, нерастворимые в метаноле. Не бо¬
лее 3 %. К 2,5 г испытуемого образца прибавляют
50 мл метанола Р и встряхивают в течение 15 мин при
температуре от 20 °С до 25 °С. Фильтруют при пони¬
женном давлении через предварительно высушенный
при температуре от 100 °С до 105 °С в течение 15 мин
и взвешенный стеклянный фильтр (ПОР 1,6) (2.12).
Фильтр трижды промывают метанолом Р порциями
по 20 мл и высушивают при температуре от 100 °С до
105 °С в течение 30 мин. Охлаждают и взвешивают.
Масса остатка на фильтре не должна превышать 75 мг.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в 20 мл метанола Р и доводят под¬
вижной фазой В до объема 100,0 мл.
Раствор сравнения (а). 10,0 мг ФСО рутозида
тригидрата растворяют в 2,0 мл метанола Р и дово¬
дят подвижной фазой В до объема 10,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой В до объема
50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С.
- подвижная фаза:
- подвижная фаза А: смесь из 5 частей те-
трагидрофурана Р и 95 частей раствора 15,6 г/л
натрия дигидрофосфата Р, доведенного до рН
3.0 кислотой фосфорной Р;
- подвижная фаза В: смесь из 40 частей те-
трагидрофурана Р и 60 частей раствора 15,6 г/л
натрия дигидрофосфата Р, доведенного до рН
3.0 кислотой фосфорной Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
сл
0
1
о
сл
0
1
О
о
10—15
0
100
15—16
о
*
сл
о
100 —► 50
16—20
50
50
876
Гэсударственная фармакопея Республики Беларусь
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 280 нм;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению
к рутозиду, время удерживания — около 7 мин):
примесь В — около 1,1; примесь А — около 1,2;
примесь С — около 2,5.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,5 между пиками ру-
тозида и примеси В.
Предельное содержание примесей (пики ло¬
кализируют путем сравнения с хроматограммой,
прилагаемой к ФСО рутозида тригидрата) (для
расчета содержания примесей умножают площа¬
ди пиков на соответствующие поправочные ко¬
эффициенты: для примеси А — 0,8; для приме¬
си С — 0,5):
- примеси А, В, С (не более 2,0 %): на хрома¬
тограмме испытуемого раствора площади пиков,
соответствующих примесям А, В и С, не должны
превышать площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- сумма примесей (не более 4,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика
на хроматограмме раствора сравнения (Ь) (0,1%).
Вода (2.5.12). Не менее 7,5% и не более
9,5 %. Определение проводят из 0,100 г испытуе¬
мого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 20 мл дс/-
метилформамида Р и титруют 0,1М раствором тетра-
бутиаммония гидроксида потенциометрически (2.2.20).
1 мл 0,1 М раствора тетрабутиаммония ги¬
дроксида соответствует 30,53 мг С^Н^О^.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. 2-(3,4-Дигидроксифенил)-3-(Р-0-гпюкопиранозил-
окси)-5,7-дигидрокси-4Н-1 -бензопиран-4-он (изоквер-
цитрозид).
В. 3-[[6-0-(6-Дезокси-а-1_-маннопиранозил)-Р-0-
глюкопиранозил]окси]-5,7-дигидроокси-2-(4-гидрокси-
фенил >4Н-1 -бензопиран-4-он (кемпферол-3-рутинозид).
С. 2-(3,4-Дигидроксифенил)-3,5,7-тригидрокси-4Н-
1-бензопиран-4-он (кверцетин).
07/2016:2579
САБАЛЯ МЕЛКОПИЛЬЧАТОГО
ЭКСТРАКТ
ЗаЬаНз зеггиШае ех1гасШт
ЗАМРА1.МЕТТО ЕХТКАСТ
ОПРЕДЕЛЕНИЕ
Экстракт, полученный из плодов сабаля мелко¬
пильчатого (Зегепоа герепз (\Л/. Вагёгат) 5та11 (зуп.
ЗаЬа! зеггиШа (МюЬаиз)Т. ИиКа! ех ЗсЬиГСез&ЗсНиКез).
Содержание:
- общее содержание жирных кислот: не менее
80,0 % (в пересчете на безводшй экстракт);
- лауриновая кислота (С^Н^О^ М.м. 200,3): не
менее 23,0 % (в пересчете на безводный экстракт);
- общее содержание стеринов в пересчете на
р-ситостерин (С^Н^О; М.м. 414,7): не менее 0,20 %
(в пересчете на безводный экстракт);
-Р-ситостерин (С^Н^О; М.м. 414,7): не менее
0,10 % (в пересчете на безводный экстракт).
ПРОИЗВОДСТВО
Получают из лекарственного растительного сы¬
рья с помощью подходящей методики, используя
спирт этиловый (не менее 90 % (об/об)), или сверх¬
критический углерода диоксид, или смесь преимуще¬
ственно из н-гексана и метилпентанов (температура
кипения: (65—70) °С).
ОПИСАНИЕ(СВОЙСТВА)
Спиртовой экстракт представляет собой тем¬
ную зеленовато-коричневую маслянистую жидкость;
экстракт, полученный с помощью сверхкритического
углерода диоксида, представляет собой желтовато-
коричневую или оранжево-коричневую маслянистую
жидкость; гексановый экстракт представляет собой
Сабаля мелкопильчатого экстракт
877
маслянистую жидкость от желтовато-зеленого до
оранжево-желтого цвета.
Имеет сильный, но не прогорклый запах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,25 г испытуемого экс¬
тракта растворяют в 20 мл 96 % спирта Р.
Раствор сравнения. 4 мг р-амирина Р и 10 мг
Р-ситостерина Р растворяют в 10 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((2—10) мкм).
Подвижная фаза: кислота уксусная безводная
Р — этилацетат Р — толуол Р (1:30:70, об/об/об).
Наносимый объем пробы: 2 мкл, в виде полос
длиной 8 мм.
Фронт подвижной фазы: не менее 6 см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раство¬
ром анисового альдегида Р, нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин и про¬
сматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора, главным образом в нижней трети, могут об¬
наруживаться и другие неинтенсивные зоны.
Верх хроматографической пластинки
Зона синего цвета
Зона синего цвета
р-Амирин: синяя зона
р-Ситостерин: синяя зона
Интенсивная синевато-фио¬
летовая зона
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
при количественном определении общего содержа¬
ния жирных кислот.
Результаты: пики метилкапроата, метилкапри-
лата, метилкапрата, метиллаурата, метилмиристата,
метилпалмитолеата, метилпалмитата, метиллиноле-
ата, метиллинолената, метилолеата и метилстеара-
та на хроматограмме испытуемого раствора по вре¬
мени удерживания соответствуют соответствующим
пикам на хроматограмме раствора сравнения (Ь);
пики метиллаурата и метилолеата являются основ¬
ными пиками.
ИСПЫТАНИЯ
Вода (2.5.12, метод А). Не более 3,0 %. Опреде¬
ление проводят из 0,5 г испытуемого образца.
Относительная плотность (2.2.5). От 0,850 до 0,950.
Показатель преломления (2.2.6). От 1,40 до 1,50.
Кислотное число (2.5.1). От 150,0 до 220,0.
Йодное число (2.5.4, метод А). От 30,0 до 60,0.
Перекисное (пероксидное) число (2.5.5). Не бо¬
лее 5,0.
Число омыления (2.5.6). От 220,0 до 250,0.
Растворители. Остаточные количества органи¬
ческих растворителей контролируются, как указано в
разделе 5.4, если не указано иное.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Общее содержание жирных кислот. Газовая
хроматография (2.2.28).
Раствор внутреннего стандарта. 0,47 г ме-
тилмаргарата Р растворяют в 20 мл диметилфор-
мамидаР и доводят до объема 100,0 мл этим же
растворителем.
Испытуемый раствор. 0,25 г испытуемого об¬
разца диспергируют в 10 мл диметилформамида Р,
прибавляют 4,0 мл раствора внутреннего стандарта и
доводят диметилформамидом Р до объема 25,0 мл.
0,4 мл полученного раствора смешивают с 0,6 мл
раствора 18,84 г/л триметилсульфония гидрокси¬
да Р в метаноле Р.
Раствор сравнения (а). 0,699 г ФСО кислоты
лауриновой и 0,870 г ФСО кислоты олеиновой раст¬
воряют в диметилформамиде Р и доводят до объема
10,0 мл этим же растворителем. К 1,0 мл полученно¬
го раствора прибавляют 4,0 мл раствора внутреннего
стандарта и доводят диметилформамидом Р до объ¬
ема 25,0 мл. 0,4 мл полученного раствора смешива¬
ют с 0,6 мл раствора 18,84 г/л триметилсульфония
гидроксида Р в метаноле Р.
Раствор сравнения (Ь). 0,25 г ФСО сабаля мел¬
копильчатого экстракта диспергируют в 10 мл ди¬
метилформамида Р, прибавляют 4,0 мл раствора
внутреннего стандарта и доводят диметилформами¬
дом Р до объема 25,0 мл. 0,4 мл полученного раст¬
вора смешивают с 0,6 мл раствора 18,84 г/л триме¬
тилсульфония гидроксида Р в метаноле Р.
Условия хроматографирования:
- колонка капиллярная кварцевая длиной 25 м
и внутренним диаметром 0,20 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 0,33 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 0,5 мл/мин;
- деление потока: 1:40;
- температура:
Время (мин)
Температура (°С)
Колонка
0—2
150
2—7
150 —> 190
7—12
190
12—22
190 — 220
22—32
220
Блок ввода проб
300
Детектор
300
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Идентификация пиков: идентифицируют пики
метилкапроата, метилкаприлата, метилкапрата, ме¬
тиллаурата, метилмиристата, метилпальмитолеата,
метилпапьмитата, метиллинолеата, метиллинолена¬
та, метилолеата, метилстеарата и метилмаргарата,
используя хроматограмму раствора сравнения (Ь) и
хроматограмму, прилагаемую к ФСО сабаля мелко¬
пильчатого экстракта.
Пригодность хроматографической системы:
раствор сравнения (Ь):
878
Гэсударственная фармакопея Республики Беларусь
- коэффициент разделения пиков: не менее 1,2
(Нр — высота пика метиллинолената относительно ба¬
зовой линии; Ну — расстояние между базовой линией
и нижней точкой кривой, разделяющей пик примеси
метиллинолената и пик метиллинолеата).
Рассчитывают общее содержание жирных кислот
в процентах, учитывая, что капроновая, каприловая,
каприновая, лауриновая, миристиновая, пальмитоле-
иновая, пальмитиновая и стеариновая кислоты пере¬
считываются на кислоту лауриновую (С12Н2402; М.м.
200,3), а линолевая, линоленовая и олеиновая кисло¬
ты пересчитываются на кислоту олеиновую (С18Н3402;
М.м. 282,5), по формуле:
А, АЛ т2 р, 0,Ч + А5 А, т3р2 0,1
А2 А3 ■т1 А6А3тл
где:
Л1 — сумма площадей пиков метилкапроата, ме-
тилкаприлата, метилкапрата, метиллаурата, метилми-
ристата, метилпальмитолеата, метильпалмитата и ме¬
ти л стеарата на хроматограмме испытуемою раствора;
А2 — площадь пика метиллаурата на хромато¬
грамме раствора сравнения (а);
Аг — площадь пика метилмаргарата на хромато¬
грамме испытуемого раствора;
Аа — площадь пика метилмаргарата на хромато¬
грамме раствора сравнения (а);
А6 — сумма площадей пиков метиллинолеата,
метиллинолената и метилолеата на хроматограмме
испытуемого раствора;
А6 — площадь пика метилолеата на хромато¬
грамме раствора сравнения (а);
ш1 — масса навески испытуемого образца, ис¬
пользованной для приготовления испытуемого раст¬
вора, г;
т2 — масса навески ФСО кислоты лауриновой,
использованной для приготовления раствора сравне¬
ния (а), г;
т3 — масса навески ФСО кислоты олеиновой,
использованной для приготовления раствора сравне¬
ния (а), г;
р1 — содержание кислоты лауриновой в ФСО
кислоты лауриновой, %;
р2 — содержание кислоты олеиновой в ФСО кис¬
лоты олеиновой, %.
Рассчитывают содержание кислоты лауриновой
в процентах по формуле:
А, • Ал т2 - р - 0,1
А2А3т1
где:
Ал — площадь пика метиллаурата на хромато¬
грамме испытуемого раствора;
А2 — площадь пика метиллаурата на хромато¬
грамме раствора сравнения (а);
Аъ — площадь пика метилмаргарата на хромато¬
грамме испытуемого раствора;
Аа — площадь пика метилмаргарата на хромато¬
грамме раствора сравнения (а);
/771 — масса навески испытуемого экстракта, ис¬
пользованной для приготовления испытуемого раст¬
ворам;
т2 — масса навески ФСО кислоты лауриновой,
использованной для приготовления раствора сравне¬
ния (а), г;
р — содержание кислоты лауриновой в ФСО кис¬
лоты лауриновой, %.
Общее содержание стеринов. Газовая хромато¬
графия (2.2.28).
Раствор для дериватизации (а). Хлортриметил-
силанР — И, 0-бис(тримет илсилил) ацетамид Р —
Ы-триметилсилилимидазол Р (2:3:3, об/об/об).
Раствор для дериватизации (Ь). Раствор для
дериватизации (а) — М,0-бис(триметилсилил)три-
фторацетамид Р — пиридин Р( 1:1:1, об/об/об).
Раствор внутреннего стандарта. 0,25 г холе¬
стерина Р растворяют в 25,0 мл метиленхлорида Р.
Испытуемый раствор. 1,0 мл раствора внутрен¬
него стандарта помещают в круглодонную колбу объе¬
мом 50 мл и выпаривают досуха. В круглодонную кол¬
бу прибавляют 3,35 г точно взвешенного испытуемого
образца и 20 мл раствора, приготовленного следую¬
щим образом: 130 г калия гидроксида Р растворяют
в 200 мл воды Р и доводят метанолом Р до объема
1000 мл. Нагревают с обратным холодильником в те¬
чение 2 ч, переносят количественно в мерную колбу и
доводят водой Р до объема 25,0 мл. 3,0 мл получен¬
ного раствора переносят в картридж, содержащий ди¬
атомит Р, способный удерживать 3 мл водной фазы.
Засасывают раствор в колонку с помощью вакуума.
Вакуум поддерживают в течение не менее 20 мин до
возвращения колонки к комнатной температуре, что
означает полное испарение метанола. Промывают
колонку 90 мл метиленхлорида Р и элюат выпарива¬
ют досуха. Полученный остаток растворяют в 1,0 мл
раствора для дериватизации (Ь).
Раствор сравнения (а). К 9,0 мг ФСО Р-си-
тостерина прибавляют 1,0 мл раствора внутреннего
стандарта и доводят метиленхлоридом Р до объема
5.0 мл. 0,6 мл полученного раствора выпаривают до¬
суха в потоке азота Р. Остаток растворяют в 1,0 мл
раствора для дериватизации (Ь).
Раствор сравнения (Ь). 1,0 мл раствора внутрен¬
него стандарта помещают в круглодонную колбу объ¬
емом 50 мл и выпаривают досуха. В круглодонную
колбу прибавляют 3,35 г точно взвешенного ФСО са¬
баля мелкопильнатого экстракта и 20 мл раствора,
приготовленного следующим образом: 130 г калия
гидроксида Р растворяют в 200 мл воды Р и доводят
метанолом Р до объема 1000 мл. Нагревают с обрат¬
ным холодильником в течение 2 ч, переносят количе¬
ственно в мерную колбу и доводят водой Р до объема
25.0 мл. 3,0 мл полученного раствора переносят в кар¬
тридж, содержащий диатомит Р, способный удержи¬
вать 3 мл водной фазы. Засасывают раствор в колонку
с помощью вакуума. Вакуум поддерживают в течение
не менее 20 мин до возвращения колонки к комнатной
температуре, что означает полное испарение метано¬
ла. Промывают колонку 90 мл метиленхлорида Р и
элюат выпаривают досуха. Полученный остаток раст¬
воряют в 1,0 мл раствора для дериватизации (Ь).
Условия хроматографирования:
- колонка капиллярная кварцевая длиной 25 м
и внутренним диаметром 0,20 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 0,33 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 0,5 мл/мин;
- деление потока: 1:40;
Время (мин)
Температура (°С)
Колонка
0—3
200
3—13
200 - 300
13—35
300
Блок ввода проб
325
Детектор
325
Салициловая кислота
879
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Идентификация пиков: идентифицируют пики
триметилсилильных производных холестерина, кам-
пестерина, стигмастерина, р-ситостерина и стигма-
станола, используя хроматограмму раствора сравне¬
ния (Ь) и хроматограмму, прилагаемую к ФСО сабаля
мелкопильчатого экстракта.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,6 между пиками три¬
метилсилильных производных Р-ситостерина и стиг-
мастанола.
Рассчитывают общее содержание стеринов (кам-
пестерина, стигмастерина, р-ситостерина и стигма-
станола) в процентах в пересчете на Р-ситостерин по
формуле:
А,- А±т2р
А2 • А3 • т^
где:
А1 — сумма площадей пиков триметилсилиль¬
ных производных кампестерина, стигмастерина,
Р-ситостерина и стигмастанола на хроматограмме ис¬
пытуемого раствора;
А2 — площадь пика триметилсилильного произ¬
водного Р-ситостерина на хроматограмме раствора
сравнения (а);
А3 — площадь пика триметилсилильного произ¬
водного холестерина на хроматограмме испытуемого
раствора;
А4 — площадь пика триметилсилильного про¬
изводного холестерина на хроматограмме раствора
сравнения (а);
ш1 — масса навески испытуемого экстракта, ис¬
пользованной для приготовления испытуемого раст¬
вора, г;
т2 — масса навески ФСО р-ситостерина, исполь¬
зованной для приготовления раствора сравнения (а), г;
р — содержание Р-ситостерина в ФСО Р-ситос-
терина, %.
Рассчитывают содержание Р-ситостерина в про¬
центах по формуле:
АЛАЛтгр
АгАг-тл
где:
А1 — площадь пика триметилсилильного произ¬
водного Р-ситостерина на хроматограмме испытуемо¬
го раствора;
А2 — площадь пика триметилсилильного произ¬
водного р-ситостерина на хроматограмме раствора
сравнения (а);
А3 — площадь пика триметилсилильного произ¬
водного холестерина на хроматограмме испытуемого
раствора;
А4 — площадь пика триметилсилильного про¬
изводного холестерина на хроматограмме раствора
сравнения (а);
т1 — масса навески испытуемого экстракта, ис¬
пользованной для приготовления испытуемого раст¬
вора, г;
т2 — масса навески ФСО Р-ситостерина, исполь¬
зованной для приготовления раствора сравнения (а), г;
р — содержание р-ситостерина в ФСО р-ситос-
терина, %.
07/2016:0366
САЛИЦИЛОВАЯ КИСЛОТА
АсШит заНсуНсит
ЗАЫСУиСАСЮ
ОДО, М.м. 138,1
[69-72-7]
ОПРЕДЕЛЕНИЕ
2-Гидроксибензолкарбоновая кислота.
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или белые или бесцветные игольчатые кристаллы.
Мало растворима в воде, легко растворима в
96 % спирте, умеренно растворима в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Температура плавления (2.2.14): от 158 °С до
161 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО салициловой кислоты #или
спектр, представленный на рисунке #0366.-1#.
C. 30 мг испытуемого образца растворяют в 5 мл
0,05 М раствора натрия гидроксида, при необходи¬
мости нейтрализуют и доводят водой Р до объема
20 мл. 1 мл полученного раствора дает реакцию (а) на
салицилаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в 50 мл кипящей воды дистиллированной Р, ох¬
лаждают и фильтруют.
Раствор 31. 1 г испытуемого образца раство¬
ряют в 10 мл 96 % спирта Р.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 31 должен
быть бесцветным.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 10 мг фенола Р (примесь
С) растворяют в подвижной фазе и доводят до объ¬
ема 100,0 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО салициловой
кислоты примеси В растворяют в подвижной фазе и
доводят до объема 20,0 мл этим же растворителем.
Раствор сравнения (с). 50 мг 4-гидроксибензой-
ной кислоты Р (примесь А) растворяют в подвижной
фазе и доводят до объема 100,0 мл этим же раство¬
рителем.
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 10,0 мл.
880 Гэсударственная фармакопея Республики Беларусь
— * ~1й<кГ * 1500 ^ Т 1000 ~ 500
Волновое число (см1)
Рисунок #0366.-1. Инфракрасный спектр ФСО салициловой кислоты
Раствор сравнения (е). По 1,0 мл растворов
сравнения (а), (Ь) и (с) смешивают и доводят подвиж¬
ной фазой до объема 10,0 мл.
Раствор сравнения ({). По 0,1 мл растворов
сравнения (а), (Ь) и (с) смешивают и доводят подвиж¬
ной фазой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная недеактивированным си¬
ликагелем октадецилсилильным для хроматогра¬
фии Р с размером частиц 5 мкм;
- подвижная фаза: кислота уксусная ледя¬
ная Р — метанол Р — вода Р (1:40:60, об/об/об);
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 270 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения ((1), (е) и (I).
Относительное удерживание (по отношению к
примеси С): примесь А — около 0,70; примесь В —
около 0,90.
Пригодность хроматографической системы:
раствор сравнения (е):
- третий пик на хроматограмме должен соответ¬
ствовать пику фенола на хроматограмме раствора
сравнения (б);
- разрешение: не менее 1,0 между пиками при¬
меси В и примеси С; при необходимости изменяют
концентрацию кислоты уксусной в подвижной фазе.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (0;
- примесь В (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (0;
- примесь С (не более 0,02 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (Т);
- любая другая примесь (не более 0,05 %): на
хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного и пиков примесей А, В и
С, не должна превышать площадь пика примеси В на
хроматограмме раствора сравнения (Т);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь пика примеси А на хроматограмме раст¬
вора сравнения (Т);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,01 площади основного пика на хроматограм¬
ме раствора сравнения (Г).
Хлориды (2.4.4). не более 0,0100% (100 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Сульфаты. Не более 0,0200 % (200 ррт). 1,0 г
испытуемого образца растворяют в 5 мл диметил-
формамида Р, прибавляют 4 мл воды Р и тщательно
перемешивают. Прибавляют 0,2 мл кислоты хлори¬
стоводородной разведенной Р и 0,5 мл 25 % (м/м)
раствора бария хлорида Р. Полученный раствор че¬
рез 15 мин по степени мутности не должен превы¬
шать эталон, приготовленный следующим образом
к 2 мл эталонного раствора сульфата (ЮОрргг
80^ Р прибавляют 0,2 мл кислоты хлористоводо¬
родной разведенной Р и 0,5 мл 25 % (м/м) раствора
бария хлорида Р, 3 мл воды Р и 5 мл диметилфсс-
мамида Р.
Сальбутамола сульфат
881
Тяжелые металлы {2.4.8, метод В). Не более
0,0020 % (20 ррт). 2,0 г испытуемого образца раст¬
воряют в 15 мл 96% спирта Р и прибавляют 5 мл
воды Р. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон го¬
товят с использованием эталонного раствора свинца
(2 ррт РЬ), приготовленного путем разведения эта¬
лонного раствора свинца (100 ррт РЬ) Р смесью из 5
объемов воды Р и 15 объемов 96 % спирта Р.
Потеря в массе при высушивании {2.2.32). Не
более 0,5%. 1,000 г испытуемого образца сушат в
эксикаторе.
Сульфатная зола {2.4.14). Не более 0,1 %.
Определение проводят из 2,0 г испытуемого образца.
#Микробиологическая чистота {2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в 96 %
спирте Р, прибавляют 20 мл воды Р и титруют 0,1 М
раствором натрия гидроксида, используя в качестве
индикатора 0,1 мл раствора фенолового красного Р.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 13,81 мгС7Н603.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
A. Р = Н: 4-Гидроксибензойная кислота.
B. Р = С02Н: 4-Гидроксиизофталевая кислота.
С. Фенол.
07/2016:0687
САЛЬБУТАМОЛА СУЛЬФАТ
8а1Ьи1атоН зиКаз
8А1-ВиТАМ011 5 Ш.РАГЕ
С26Н42М20б * Н23 04 М.М. 576»7
[51022-70-9]
ОПРЕДЕЛЕНИЕ
Бис[(1 Р5)-2-[(1,1 -диметилэтил )амино]-1 -[4-гидро-
кси-3-(гидроксиметил)фенил]этанола] сульфат.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде, практически нераство¬
рим или очень мало растворим в 96 % спирте и в ме-
тиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимых областях (2.2.25).
Испытуемый раствор. 80,0 мг испытуемого об¬
разца растворяют в растворе 10 г/л кислоты хлори¬
стоводородной Р и доводят до объема 100,0 мл этим
же растворителем. 10,0 мл полученного раствора до¬
водят раствором 10 г/л кислоты хлористоводород¬
ной Р до объема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 276 нм.
Удельный показатель поглощения в максимуме:
от 55 до 64.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сальбутамола сульфата #или
спектр, представленный на рисунке #0687.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 12 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения. 12 мг ФСО сальбутамола
сульфата растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — вода Р — этилацетат Р — 2-про¬
панол Р — метилизобутилкетон Р (3:18:35:45:50,
об/об/об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром 1 г/л метилбензотиазолонгидразона гидрохло¬
рида Р в 90 % (об/об) растворе метанола Р, затем
раствором 20 г/л калия феррицианида Р в смеси из 1
объема раствора аммиака концентрированного Р1
и 3 объемов воды Р и затем раствором 1 г/л метил¬
бензотиазолонгидразона гидрохлорида Р в 90 %
(об/об) растворе метанола Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, цвету и размеру основному пятну на
хроматограмме раствора сравнения.
О. 10 мг испытуемого образца растворяют в
50 мл раствора 20 г/л динатрия тетрабората Р,
прибавляют 1 мл раствора 30 г/л аминопиразолона Р,
10 мл метиленхлорида Р и 10 мл раствора 20 г/л
калия феррицианида Р, перемешивают и оставляют
до разделения слоев. Появляется оранжево-красное
окрашивание в слое метиленхлорида.
Е. Испытуемый образец дает реакцию (а) на
сульфаты {2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,250 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
111. Зак. 1060.
882
Гэсударственная фармакопея Республики Беларусь
Рисунок #0687.-1. Инфракрасный спектр ФСО сальбутамола сульфата
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. Измеряют угол оптического вращения раст¬
вора 3.
Кислотность или щелочность. К10 мл раствора
3 прибавляют 0,15 мл раствора метилового красно¬
го Р и 0,2 мл 0,01 М раствора натрия гидроксида.
Появляется желтое окрашивание. При прибавлении
не более 0,4 мл 0,01 М раствора кислоты хлористо¬
водородной должно появиться красное окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 3,0 мг ФСО сальбутамола
примеси О и 3,0 мг ФСО сальбутамола примеси Р раст¬
воряют в подвижной фазе А и доводят до объема 50,0 мл
этим же растворителем. 2,0 мл полученного раствора
доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО сальбутамола примеси и растворяют с исполь¬
зованием ультразвука в 1,0 мл испытуемого раствора.
Раствор сравнения (д). 1 мг ФСО сальбутамола
примеси О растворяют в подвижной фазе А и доводят
до объема 100,0 мл этим же растворителем.
Раствор сравнения (е). 4 мг ФСО сальбутамола
сульфата для проверки пригодности хроматогра¬
фической системы (содержит примеси С, Р, N и О)
растворяют в подвижной фазе, прибавляют 0,4 мл
раствора сравнения (с!) и доводят подвижной фазой А
до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 3 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: 3,45 г натрия дигидро¬
фосфата моногидрата Р растворяют в 900 мл
0,05 % (об/об) раствора триэтиламина Р, дово¬
дят до рН 3,0 кислотой фосфорной разведен¬
ной Р и доводят раствором 0,05 % (об/об) триэ¬
тиламина Рдо объема 1000 мл;
- подвижная фаза В: метанол Р — ацетони¬
трил Р (35:65, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
95
5
5—30
95—» 10
сл
4
СО
о
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 273 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а), (Ь), (с) и (е).
Относительное удерживание (по отношению к
сальбутамолу, время удерживания — около 7 мин):
примесь Л — около 0,9; примесь С — около 1,6; при¬
месь N — около 1,67; примесь О — около 1,68; при¬
месь Р — около 1,77; примесь О — около 1,82.
Идентификация пиков примесей: идентифици¬
руют пики примесей С, й, Р, N и О, используя хрома¬
тограмму раствора сравнения (е) и хроматограмму,
прилагаемую к ФСО сальбутамола сульфата для
проверки пригодности хроматографической систе-
Сальбутамола сульфат
883
мы; идентифицируют пик примеси 3, используя хро¬
матограмму раствора сравнения (с).
Пригодность хроматографической системы:
- коэффициент разделения пиков: не менее 1,2
(/-/р — высота пика примеси N относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси N и пик
примеси О на хроматограмме раствора сравнения
(е)); не менее 2,0 (Н — высота пика примеси 3 отно¬
сительно базовой линии; Ну — расстояние между ба¬
зовой линией и нижней точкой кривой, разделяющей
пик примеси ^ и пик сальбутамола на хроматограмме
раствора сравнения (с)).
Предельное содержание примесей:
- примеси О, Р (не более 0,3 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям О и Р, не должны превышать
площади соответствующих пиков на хроматограмме
раствора сравнения (а);
- примеси С, Л/, О (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям С, N и О, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
С, й, Р, N и О, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей: не более 0,9 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Бор. Не более 0,0050 % (50 ррт).
Испытуемый раствор. К 50 мг испытуемого об¬
разца прибавляют 5 мл раствора, содержащего 13 г/л
натрия карбоната безводного Р и 17 г/л калия кар¬
боната Р, и выпаривают на водяной бане при темпе¬
ратуре 120 °С до сухого остатка. Полученный остаток
быстро сжигают до тех пор, пока органическое веще¬
ство не разрушится, охлаждают и прибавляют 0,5 мл
воды Р и 3,0 мл свежеприготовленного раствора
1,25 г/л куркумина Р в кислоте уксусной ледяной Р.
Осторожно нагревают до получения раствора, охлаж¬
дают, прибавляют 3,0 мл смеси, приготовленной мед¬
ленным прибавлением при перемешивании 5 мл кис¬
лоты серной Р к 5 мл кислоты уксусной ледяной Р,
перемешивают и выдерживают в течение 30 мин. По¬
лученный раствор доводят 96 % спиртом Р до объ¬
ема 100,0 мл, фильтруют и используют фильтрат.
Раствор сравнения. 0,572 г кислоты борной Р
растворяют в 1000,0 мл воды Р. 1,0 мл полученно¬
го раствора доводят водой Р до объема 100,0 мл. К
2,5 мл полученного раствора прибавляют 5 мл раст¬
вора, содержащего 13 г/л натрия карбоната безво¬
дного Р и 17 г/л калия карбоната Р, и далее поступа¬
ют аналогично испытуемому раствору.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора и раствора сравнения в максимуме
при длине волны около 555 нм. Оптическая плотность
испытуемого раствора не должна превышать оптиче¬
скую плотность раствора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, прибавляют 35 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 57,67 мг С26Н42М206 Н2304.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, О, Р, Л/, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): А, В, Е, С, /, */, К, Ь, М.
А. [5-[(1/?5)-2-[(1,1-Диметилэтил)амино]-1-метокси-
этил]-2-гидроксифенил]метанол.
В. (1 /?5)-2-[(1,1 -Диметилэтил)амино]-1 -(4-гидрокси-
фенил)этанол.
С. (1Р8)-2-[(1,1 -Диметилэтил)амино]-1 -(4-гидрокси-
3-метилфенил)этанол.
О. 5-[(1 #5)-2-[(1,1 -Диметилэтил)амино]-1 -гидрокси-
этил]-2-гидроксибензальдегид.
111*. Зак. 1060.
884
Гэсударственная фармакопея Республики Беларусь
М. (1 Я5)-2-[(1,1 -Диметилэтил)амино]-1 -[4-гидрокси-
3-(метоксиметил)фенил]этанол.
Е. (1 /?5)-2-[Бензил(1,1 -д и метил эти л )а ми но]-1 -[4-
гидрокси-3-(гидроксиметил)фенил]этанол.
он
он
Р. 1,1 -[Оксибис[метилен(4-гидрокси-1,3-фенилен)]]-
бис[2-[(1,1-диметилэтил )амино]этанол].
С. 2-[Бензил(1,1 -диметилэтилэтил )амино]-1 -[4-
гидрокси-3-(гидроксиметил)фенил]этанон.
N. 2-[(1,1-Диметилэтил)амино]-1-[3-[[5-[2-[(1,1-
диметилэтил)амино]-1-гидроксиэтил]-2-гидроксифенил]-
метил]-4-гидрокси-5-(гидроксиметил)фенил]этанол.
O. Структура не установлена.
07/2016:0787
САХАРИН НАТРИЙ
ЗассЬаппит паНсит
8АССНАШЫ 800ШМ
I. (1 Я5)-2-[(1,1-Диметилэтил)амино]-1-[4-(бензил-
окси)-3-(гидроксиметил)фенил]этанол.
3. 2-[(1,1-Диметилэтил)амино]-1-[4-гидрокси-З-
(гидроксиметил)фенил]этанон (сальбутамон).
о
К. 2-[(1,1-Диметилэтил)амино]-1-[3-хлор-4-гидрокси-
5-(гидроксиметил)фенил]этанон.
Ь. (1/?5)-2-[(1,1-Диметилэтил)амино]-1-[З-хлор-4-
гидрокси-5-(гидроксиметил)фенил]этанол.
С7Н4ЫЫа038 М.м. 205,2 (безводное вещество)
Сахарин натрий безводный: [128-44-9]
ОПРЕДЕЛЕНИЕ
2-Натрий-1,2-бензизотиазол-3(2Н)-она 1,1-диоксид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
Может быть безводным или содержать перемен¬
ное количество воды.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Выветривается на
сухом воздухе.
Легко растворим в воде, умеренно растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
А. Температура плавления {2.2.14): от 226 °С до
230 °С.
К 5 мл раствора 8, приготовленного как указано
в разделе «Испытания», прибавляют 3 мл кислоты
хлористоводородной разведенной Р. Образуется бе¬
лый осадок. Полученную смесь фильтруют, осадок
промывают водой Р и сушат при температуре 105 °С
до постоянной массы.
Сахарин натрий
885
Рисунок #0787.-1. Инфракрасный спектр ФСО сахарина натрия
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках; испытуемый образец
перед использованием сушат при температуре от
100 °С до 105 °С.
Сравнение: ФСО сахарина натрия #или спектр,
представленный на рисунке #0787.-1#.
C. 10 мг испытуемого образца смешивают с око¬
ло 10 мг резорцина Р, прибавляют 0,25 мл кислоты
серной Р. Полученную смесь осторожно нагревают
над открытым пламенем до появления темно-зеле¬
ного окрашивания. Охлаждают, прибавляют 10 мл
воды Р и раствор натрия гидроксида разведенный Р
до щелочной реакции раствора. Появляется интен¬
сивная зеленая флуоресценция.
О. К 0,2 г испытуемого образца прибавляют
1,5 мл раствора натрия гидроксида разведенного Р и
выпаривают досуха. Остаток осторожно нагревают до
расплавления, не допуская обугливания. Охлаждают,
растворяют полученную массу в около 5 мл воды Р,
прибавляют кислоту хлористоводородную разведен¬
ную Р до слабо кислой реакции и, при необходимости,
фильтруют. К фильтрату прибавляют 0,2 мл раствора
железа (III) хлорида Р2. Появляется фиолетовое окра¬
шивание.
Е. Раствор 5 дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50,0 мл этим же растворителем.
Раствор 31. 2,0 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.1). Раствор 51 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 51 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 5 прибавляют около 0,05 мл раствора 10 г/л фе¬
нолфталеина Р в 96% спирте Р. Не должно появ¬
ляться розовое или красное окрашивание раствора.
При прибавлении 0,1 мл 0,1 М раствора натрия
гидроксида должно появиться розовое или красное
окрашивание.
о- и /7-Толуолсульфонамид. Газовая хромато¬
графия (2.2.28).
Раствор внутреннего стандарта. 25 мг кофе¬
ина Р растворяют в метиленхлориде Р и доводят до
объема 100 мл этим же растворителем.
Испытуемый раствор. 10,0 г испытуемого об¬
разца растворяют в 50 мл воды Р, при необходимости
доводят до рН 7—8 1 М раствором натрия гидрокси¬
да или 1 М раствором кислоты хлористоводород¬
ной. Полученный раствор встряхивают с 4 порциями,
по 50 мл каждая, метиленхлорида Р. Объединяют
нижние слои, высушивают над натрия сульфатом
безводным Р и фильтруют. Фильтр и натрия сульфат
промывают 10 мл метиленхлорида Р. Объединяют
раствор и промывные воды и выпаривают в водяной
бане при температуре не выше 40 °С почти досуха.
Небольшим количеством метиленхлорида Р остаток
количественно переносят в пробирку вместимостью
10 мл, выпаривают досуха в потоке азота Р и при¬
бавляют 1,0 мл раствора внутреннего стандарта.
Контрольный раствор. 200 мл метиленхлори¬
да Р выпаривают в водяной бане при температуре не
выше 40 °С досуха. Остаток растворяют в 1 мл мети¬
ленхлорида Р.
Раствор сравнения. 20,0 мг о-толуолсульфона-
мида Р и 20,0 мг толуолсульфонамида Р растворяют
в метиленхлориде Р и доводят до объема 100,0 мл
этим же растворителем. 5,0 мл полученного раствора
доводят метиленхлоридом Р до объема 50,0 мл.
112. Зак. 1060.
886
Гэсударственная фармакопея Республики Беларусь
5,0 мл полученного раствора выпаривают досуха в
потоке азота Р. Остаток растворяют в 1,0 мл раст¬
вора внутреннего стандарта.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 10 м и
внутренним диаметром 0,53 мм, покрытая слоем по-
лиметилфенилсилоксана Р (толщина слоя 2 мкм);
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 10 мл/мин;
- деление потока: 1:2;
- температура колонки: 180 °С;
- температура блока ввода проб и детектора:
250 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: о-толуолсульфонамид,
п-толуолсульфонамид, кофеин.
Пригодность хроматографической системы:
-разрешение: не менее 1,5 между пиками
о-толуолсульфонамида и л-толуолсульфонамида на
хроматограмме раствора сравнения;
- на хроматограмме контрольного раствора не
должны обнаруживаться пики со временами удержи¬
вания такими же, как у пиков внутреннего стандарта,
о-толуолсульфонамида и /7-толуолсульфонамида.
Предельное содержание примесей:
-о-толуолсульфонамид (не более 0,0010%
(Юррт)): отношение площадей пиков о-толуолсуль-
фонамида и внутреннего стандарта на хроматограмме
испытуемого раствора должно быть не больше, чем на
хроматограмме раствора сравнения;
- п-толуолсульфонамид (не более
0,0010% (Юррт)): отношение площадей пиков
п-толуолсульфонамида и внутреннего стандарта на
хроматограмме испытуемого раствора должно быть
не больше, чем на хроматограмме раствора сравне¬
ния.
Легкообугливающиеся вещества. 0,200 г ис¬
пытуемого образца растворяют в 5 мл кислоты сер¬
ной Р и выдерживают при температуре (48—50) °С
в течение 10 мин. При рассмотрении на белом фоне
окраска полученного раствора должна быть не интен¬
сивнее окраски смеси из 0,1 мл исходного красного
раствора, 0,1 мл исходного синего раствора, 0,4 мл
исходного желтого раствора (2.2.2) и 4,4 мл воды Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Вода (2.5.12). Не более 15,0%. Определение
проводят из 0,200 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р, при необходимости
слегка подогревая, и титруют 0,1 Мраствором кисло¬
ты хлорной потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 20,52 мг С7Н4НЫа033.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0204
САХАРОЗА
ЗассЬагит
51/СЯ05Е
М.м. 342,3
[57-50-1]
ОПРЕДЕЛЕНИЕ
Р-Э-фруктофуранозил-а-О-глюкопиранозил.
Не содержит добавок.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или блестящие, бесцветные или белые или почти
белые кристаллы.
Очень легко растворима в воде, мало раствори¬
ма в 96 % спирте, практически нерастворима в этано¬
ле безводном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сахарозы #или спектр, пред¬
ставленный на рисунке #0204.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси вода Р — метанол Р (2:3,
об/об) и доводят до объема 20 мл этой же смесью
растворителей.
Раствор сравнения (а). 10 мг ФСО сахарозы
растворяют в смеси вода Р — метанол Р (2:3, об/об)
и доводят до объема 20 мл этой же смесью раство¬
рителей.
Раствор сравнения (Ь). 10 мг фруктозы Р, 10 мг
глюкозы Р, 10 мг лактозы Р и 10 мг сахарозы Р раст¬
воряют в смеси вода Р — метанол Р (2:3, об/об) и
доводят до объема 20 мл этой же смесью раствори¬
телей.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
Подвижная фаза: раствор кислоты борной,
насыщенный, охлажденный Р — 60 % (об/об) раст¬
вор кислоты уксусной ледяной Р — этанол Р —
ацетон Р — этилацетат Р (10:15:20:60:60.
об/об/об/об/об) (камеру не насыщают парами под¬
вижной фазы).
Сахароза
887
Рисунок #0204.-1. Инфракрасный спектр ФСО сахарозы
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р и нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-на хроматограмме обнаруживаются 4 полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а).
С. 1 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», доводят водой Р до объ¬
ема 100 мл. К 5 мл полученного раствора прибавля¬
ют 0,15 мл свежеприготовленного раствора меди (II)
сульфата Р и 2 мл свежеприготовленного раствора
натрия гидроксида разведенного Р. Полученный
раствор прозрачный и имеет синее окрашивание.
Раствор доводят до кипения — прозрачность и си¬
нее окрашивание сохраняются. К горячему раство¬
ру прибавляют 4 мл кислоты хлористоводородной
разведенной Р и кипятят в течение 1 мин, прибав¬
ляют 4 мл раствора натрия гидроксида разведен¬
ного Р. Немедленно образуется осадок оранжевого
цвета.
ИСПЫТАНИЯ
Раствор 5. 50,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 100 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Удельная электропроводность (2.2.38). Не
более 35 мкСм см1 при температуре 20 °С. 31,3 г ис¬
пытуемого образца растворяют в воде, свободной от
углерода диоксида, Р, приготовленной из воды дис¬
тиллированной Р, и доводят до объема 100 мл этим
же растворителем. Измеряют электропроводность по¬
лученного раствора (СЛ) при слабом перемешивании
на магнитной мешалке и электропроводность воды,
использованной для приготовления раствора (С2).
Показания прибора должны быть устойчивы (±1 %) в
течение 30 с. Удельную электропроводность раствора
испытуемого образца рассчитывают по формуле:
С1 - 0,35 • С2.
Удельное оптическое вращение (2.2.7). От
+66,3 до +67,0. 26,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 100,0 мл этим же
растворителем.
Цветовое число. Не более 45. 50,0 г испытуемо¬
го образца растворяют в 50,0 мл воды Р, перемеши¬
вают, фильтруют (диаметр пор 0,45 мкм) и дегазируют.
Измеряют оптическую плотность (2.2.25) полученного
раствора при 420 нм в кювете с длиной оптического
пути не менее 4 см (предпочтительно 10 см и более).
Цветовое число рассчитывают по формуле:
А-1000
Ьс
где:
А — оптическая плотность при 420 нм;
Ь — длина оптического пути, см;
с — концентрация раствора, рассчитанная по по¬
казателю преломления раствора, г/мл (2.2.6); исполь¬
зуют таблицу 0204.-1 и интерполируют значения при
необходимости.
112*. Зак. 1060.
888
Гэсударственная фармакопея Республики Беларусь
Таблица 0204.-1
л20
"о
с, г/мл
1,4138
0,570
1,4159
0,585
1,4179
0,600
1,4200
0,615
1,4221
0,630
1,4243
0,645
1,4264
0,661
Пригодность системы:
- сходимость: абсолютная разница между двумя
полученными значениями не должна превышать 3.
Декстрины. Если субстанция предназначена
для производства лекарственных средств для па¬
рентерального применения больших объемов, ис¬
пытуемый образец должен выдерживать испытание
на декстрины. К 2 мл раствора 5 прибавляют 8 мл
воды Р, 0,05 мл кислоты хлористоводородной раз¬
веденной Р и 0,05 мл 0,05 М раствора йода. Раствор
должен сохранять желтое окрашивание.
Восстанавливающие сахара. 5 мл раствора 5
помещают в пробирку длиной 150 мм и диаметром
16 мм, прибавляют 5 мл воды Р, 1,0 мл 1 М раст¬
вора натрия гидроксида и 1,0 мл раствора 1 г/л ме¬
тиленового синего Р. Полученный раствор переме¬
шивают, выдерживают в водяной бане ровно 2 мин
и немедленно просматривают. Синее окрашивание
не должно исчезать полностью. Не учитывают синее
окрашивание на границе воздух/раствор.
Сульфиты. Не более 0,0010 % (10 ррпл) в пере¬
счете на 302. Определение содержания сульфитов
проводят подходящим ферментативным методом,
основанным на следующих реакциях. Сульфиты окис¬
ляются сульфитоксидазой до сульфатов и пероксида
водорода, который, в свою очередь, восстанавлива¬
ется никотинамидадениндинуклеотидпероксидазой в
присутствии восстановленного никотинамидаденин-
динуклеотида (Л/АОН). Количество окисленного Л/АОН
пропорционально количеству сульфитов.
Испытуемый раствор. 4,0 г испытуемого образ¬
ца растворяют в свежеприготовленной воде дистил¬
лированной Р и доводят до объема 10,0 мл этим же
растворителем.
Раствор сравнения. 4,0 г испытуемого образца
растворяют в свежеприготовленной воде дистилли¬
рованной Р, прибавляют 0,5 мл эталонного раствора
сульфита (80 ррт 80^ Р и доводят свежеприготов¬
ленной водой дистиллированной Р до объема 10,0.
Контрольный раствор. Свежеприготовленная
вода дистиллированная Р.
По 2,0 мл испытуемого раствора, раствора срав¬
нения и контрольного раствора помещают по отдель¬
ности в кюветы с длиной оптического пути 10 мм и
прибавляют реактивы согласно инструкции, прилагае¬
мой к набору для определения сульфитов. Измеряют
оптическую плотность (2.2.25) растворов в максимуме
при 340 нм до и после реакции и вычитают значение,
полученное с контрольным раствором.
Разница оптических плотностей испытуемого
раствора до и после реакции не должна превышать
0,5 разницы оптических плотностей раствора срав¬
нения.
Потеря в массе при высушивании (2.2.32). Не
более 0,1 %. 2,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Бактериальные эндотоксины {2.6.14). Менее
0,25 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения больших объемов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
МАРКИРОВКА
При необходимости указывают:
-субстанция предназначена для производства
лекарственных средств для парентерального приме¬
нения больших объемов.
07/2016:2269
СЕВОФЛУРАН
8еVоПи^апит
8Е\/ОРШКАМЕ
С4Н3Р70 М.м. 200,1
[28523-86-6]
ОПРЕДЕЛЕНИЕ
1,1,1,3,3,3-Гексафтор-2-(фторметокси)пропан.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, бесцветная, летучая жидкость.
Мало растворим в воде, смешивается с 96 %
спиртом.
Относительная плотность: около 1,52.
Температура кипения: около 59 °С.
Не воспламеняется.
Разлагается в присутствии кислот Льюиса. Разло¬
жение ингибируется достаточным количеством воды.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец исследу¬
ют в газообразном или жидком состоянии.
Сравнение: ФСО севофлурана.
ИСПЫТАНИЯ
Кислотность или щелочность. 20,0 мл испыту¬
емого образца и 20 мл воды, свободной от углерода
диоксида, Р помещают в делительную воронку, встря¬
хивают в течение 3 мин и выдерживают. Собирают
верхний водный слой и добавляют 0,2 мл раствора
бромкрезолового пурпурового Р. При прибавлении
не более 0,10 мл 0,01 М раствора натрия гидрокси¬
да или не более 0,60 мл 0,01 М раствора кислоты
хлористоводородной должна измениться окраска
индикатора.
Показатель преломления (2.2.6). От 1,2745 до
1,2760.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Внутренний стандарт. Метилаль Р.
Испытуемый раствор. 20,0 мл испытуемого
образца помещают в виалу, накрывают мембраной
и плотно укупоривают колпачком. Используя микро-
Севофлуран
889
шприц, прибавляют 5 мкл внутреннего стандарта и
тщательно перемешивают.
Раствор сравнения (а). 2,0 мл этиленхлорида Р
помещают в виалу с завинчивающейся крышкой, не¬
замедлительно накрывают мебраной и завинчивают
крышкой. Используя микрошприц, прибавляют око¬
ло 20 мкл испытуемого образца и записывают при¬
бавленное количество (М2), в миллиграммах. Затем,
используя микрошприц, прибавляют около 20 мкл
внутреннего стандарта и записывают прибавленное
количество (Ц), в миллиграммах.
Раствор сравнения (Ь). ФСО севофлурана (со¬
держит примеси А и В).
Раствор сравнения (с). 20,0 мл этиленхлори¬
да Р помещают в виалу, накрывают мембраной и
плотно укупоривают колпачком. Используя микро¬
шприц, прибавляют 20 мкл испытуемого образца и
тщательно перемешивают. 0,5 мл полученного раст¬
вора доводят этиленхлоридом Р до объема 100,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и диаметром 0,32 мм, покрытая слоем поли[(циано-
пропил)(фенил)][диметил]силоксана Р (толщина слоя
3 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,0 мл/мин;
- деление потока: 1:20;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
40
10—26
40 — 200
26—40
200
Блок ввода проб
200
Детектор
225
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 2 мкл.
Перед введением растворов сравнения необхо¬
димо промыть шприц раствором, содержащим эти-
ленхлорид Р. Перед введением испытуемого раствора
необходимо промыть шприц испытуемым раствором.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилагае¬
мую к ФСО севофлурана.
Относительное удерживание (по отношению к
севофлурану, время удерживания — около 6,6 мин):
примесь А— около 0,78; примесь В — около 0,83; вну¬
тренний стандарт — около 1,35.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
месей А и В.
Рассчитывают относительный коэффициент от¬
клика (Р^ для раствора сравнения (а) по следующей
формуле:
Ц-К
М2 ’
где:
— масса внутреннего стандарта в растворе
сравнения (а), мг;
М2 — масса испытуемого образца в растворе
сравнения (а), мг;
Р — отношение площади пика севофлурана к
площади пика внутреннего стандарта на хромато¬
грамме раствора сравнения (а).
Рассчитывают количество каждой примеси в ис¬
пытуемом образце в ррт по следующей формуле:
0,859 • Р, • 250
1,52 • Я,
где:
0,859 — относительная плотность внутреннего
стандарта;
1,52 — относительная плотность севофлурана;
Рл — отношение площади пика примеси к пло¬
щади пика внутреннего стандарта на хроматограмме
испытуемого раствора;
Ру — относительный коэффициент отклика для
раствора сравнения (а).
Предельное содержание примесей:
- примесь А: не более 0,0025 % (25 ррт);
- примесь В: не более 0,0100 % (100 ррт);
- неспецифицированные примеси: не более
0,0100 % (100 ррт) для каждой примеси;
- сумма примесей: не более 0,0300 % (300 ррт);
- неучитываемый предел (0,0005 % (5 ррт)): на
хроматограмме испытуемого раствора не учитывают
пики с площадью менее площади пика севофлурана
на хроматограмме раствора сравнения (с).
Фториды. Не более 2 мкг/мл. Проводят потен¬
циометрическое определение концентрации ионов с
использованием ионоселективных электродов (2.2.36,
метод I). При проведении испытания используют
пластиковую лабораторную посуду.
Буферный раствор. 0,5 г натрия цитрата Р и
55 г натрия хлорида Р растворяют в 350 мл воды Р.
Прибавляют осторожно 75 г натрия гидроксида Р и
встряхивают до достижения растворения. Охлаждают
до комнатной температуры и осторожно прибавляют
при перемешивании 225 мл кислоты уксусной ледя¬
ной Р. Охлаждают, прибавляют 300 мл изопропилово¬
го спирта Р и доводят водой Р до объема 1000,0 мл.
Измеренное значение рН полученного раствора нахо¬
дится в пределах от 5,0 до 5,5.
Испытуемый раствор. 50,0 мл испытуемого
образца и 50,0 мл воды Р помещают в делительную
воронку, интенсивно встряхивают в течение 3 мин и
выдерживают до полного разделения слоев. 25,0 мл
верхнего водного слоя доводят буферным раствором
до объема 50,0 мл.
Фторида эталонный раствор (1000 ррт Р).
221.0 мг натрия фторида Р, предварительно высу¬
шенного при температуре 150 °С в течение 4 ч, раст¬
воряют в воде Р, прибавляют 1,0 мл 0,01 М раствора
натрия гидроксида и доводят водой Р до объема
100.0 мл.
Исходные растворы сравнения. Готовят соот¬
ветствующим разведением фторида эталонного раст¬
вора (1000 ррт Р) водой Р до получения растворов с
известными концентрациями фторида около 5 мкг/мл,
2 мкг/мл, 0,5 мкг/мл и 0,2 мкг/мл.
Индикаторный электрод. Фторид-селективный.
Электрод сравнения. Хлор-серебряный.
Прибор. Вольтметр с минимальной воспроизво¬
димостью ±0,2 мВ.
Измерения проводят с использованием раство¬
ров сравнения и испытуемого раствора. Для проведе¬
ния измерения в стакан емкостью 100 мл, в котором
находится магнитный стержень с политетрафторэти-
леновым покрытием, помещают раствор и погружают
электроды. Раствор перемешивают с помощью маг¬
нитной мешалки до достижения равновесия (около
113. Зак. 1060.
890
Гэсударственная фармакопея Республики Беларусь
(2—3) мин) и записывают значение потенциала. Осто¬
рожно, во избежание повреждения ионселективного
электрода, ополаскивают электроды буферным рас¬
твором и высушивают.
Рассчитывают концентрацию фторидов с исполь¬
зованием калибровочного графика.
Нелетучий остаток. Не более 100 мг/л.
10,0 мл испытуемого образца помещают в пред¬
варительно взвешенную выпарительную чашку, выпа¬
ривают досуха на водяной бане и сушат остаток при
температуре 105 °С в течение 2 ч. Масса остатка не
должна превышать 1,0 мг.
Вода (2.5.12). Не более 0,050 % (м/м). Определе¬
ние проводят из 10,0 мл испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере из нержа¬
веющей стали, в защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. 1,1,3,3,3-Пентафтор-2-(фторметокси)проп-1-ен.
В. 1,1,1,3,3,3-Гексафтор-2-метоксипропан.
С. 1,1,1,3,3,3-Гексафторпропан-2-ол.
07/2016:0953
СЕРА ДЛЯ НАРУЖНОГО
ПРИМЕНЕНИЯ
ЗиНиг ас! изит ех1егпит
51Н.Я1/Я РОЯ ЕХТЕКЫА1. иЗЕ
В А.м. 32,07
[7704-34-9]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0%.
ОПИСАНИЕ (СВОЙСТВА)
Желтый порошок.
Практически не растворима в веде, растворима в
сероуглероде, мало растворима в растительных маслах.
Температура плавления: около 120 °С.
Размер большинства частиц не более 20 мкм, а
почти всех — не более 40 мкм.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Нагреваемый в присутствии воздуха испыту¬
емый образец горит синим пламенем, выделяя серы
диоксид, который изменяет цвет влажной синей лак¬
мусовой бумаги Р на красный.
B. К 0,1 г испытуемого образца прибавляют
0,5 мл бромной воды Р и нагревают до обесцвечива¬
ния. Прибавляют 5 мл воды Р и фильтруют. Получен¬
ный раствор дает реакцию (а) на сульфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. К 5 г испытуемого образца прибавля¬
ют 50 мл воды, свободной от углерода диоксида, Р,
приготовленной из воды дистиллированной Р. Вы¬
держивают в течение 30 мин при частом встряхива¬
нии и фильтруют.
Цветность (2.2.2, метод И). Раствор 8 должен
быть бесцветным.
Определение запаха (2.3.4). Испытуемый обра¬
зец не должен иметь ощутимого запаха сероводорода.
Кислотность или щелочность. К 5 мл раствора
8 прибавляют 0,1 мл раствора фенолфталеина Р1.
Раствор должен быть бесцветным. Прибавляют 0,2 мл
0,01 М раствора натрия гидроксида. Раствор дол¬
жен быть красным. Прибавляют 0,3 мл 0,01 М кисло¬
ты хлористоводородной. Раствор должен быть бес¬
цветным. Прибавляют 0,15 мл раствора метилового
красного Р. Раствор должен быть оранжево-красным.
Хлориды (2.4.4). Не более 0,0100% (100 ррт).
5 мл раствора 8 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
15 мл раствора 8 должны выдерживать испытание на
сульфаты.
Сульфиды. К 10 мл раствора 8 прибавляют
2 мл буферного раствора рН 3,5 Р и 1 мл свежепри¬
готовленного раствора 1,6 г/л свинца (II) нитрата Р
в воде, свободной от углерода диоксида, Р. Встряхи¬
вают. Через 1 мин окраска раствора должно быть не
более интенсивной, чем окраска раствора сравнения,
приготовленного в то же самое время с использова¬
нием 1 мл эталонного раствора свинца (10 ррт РЬ),
9 мл воды, свободной от углерода диоксида Р, 2 мл
буферного раствора рН 3,5 Р и 1,2 мл реактива ти-
оацетамида Р.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят методом сжигания в кол¬
бе с кислородом (2.5.10), используя 60,0 мг испыту¬
емого образца и огнеупорную колбу вместимостью
1000 мл. В качестве поглощающей жидкости исполь¬
зуют смесь из 5 мл раствора водорода пероксида
разведенного Р и 10 мл воды Р Нагревают до кипе¬
ния, осторожно кипятят в течение 2 мин и охлажда¬
ют. Титруют 0,1 М раствором натрия гидроксида до
перехода окраски раствора от бесцветной до красной,
#Серебра протеинат
891
используя в качестве индикатора 0,2 мл раствора
фенолфталеина Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 1,603 мг 5.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0009
СЕРЕБРА НИТРАТ
АгдепИ пНгаэ
8ЧУЕК ШТКАТЕ
АдЫ03 М.м. 169,9
[7761-88-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или прозрачные бесцветные кристаллы.
Очень легко растворим в воде, растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 10 мг испытуемого образца дают реакцию (а)
на нитраты (2.3.1).
B. 10 мг испытуемого образца дают реакцию (а)
на серебро (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность или щелочность. К 2 мл раствора
3 прибавляют 0,1 мл раствора бромкрезолового зе¬
леного Р. Появляется голубое окрашивание. К 2 мл
раствора 3 прибавляют 0,1 мл раствора фенолового
красного Р. Появляется желтое окрашивание.
Посторонние соли. Не более 0,3 %. К 30 мл
раствора 3 прибавляют 7,5 мл кислоты хлористово¬
дородной разведенной Р, энергично встряхивают, на¬
гревают на водяной бане в течение 5 мин и фильтру¬
ют. 20 мл полученного фильтрата выпаривают досуха
на водяной бане и сушат при температуре от 100 °С
до 105 °С. Масса остатка должна быть не более 2 мг.
Алюминий, свинец, медь и висмут. 1,0 г испы¬
туемого образца растворяют в смеси из 4 мл раст¬
вора аммиака концентрированного Р и 6 мл воды Р.
Раствор должен быть прозрачным (2.2.1) и бесцвет¬
ным (2.2.2, метод II).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 50 мл
воды Р, прибавляют 2 мл кислоты азотной разведен¬
ной Р и 2 мл раствора железа (!!!) аммония сульфа¬
та Р2. Титруют 0,1 М раствором аммония тиоциана¬
та до появления красновато-желтого окрашивания.
1 мл 0,1 М раствора аммония тиоцианата соот¬
ветствует 16,99 мгАд1\Ю3.
ХРАНЕНИЕ
В неметаллическом контейнере в защищенном
от света месте.
07/2016:РБ0010
#СЕРЕБРА ПРОТЕИНАТ
АгдеМит рго1ет1сит (Рго(агдо1ит)
8И-УЕК РПОТЕ1Ы
ОПРЕДЕЛЕНИЕ
Протаргол.
Содержание: не менее 7,5 % и не более 8,5 % Ад
(в пересчете на высушенное до постоянной массы ве¬
щество).
ОПИСАНИЕ (СВОЙСТВА)
Мелкий порошок от темно-желтого до коричнево¬
го цвета. Слегка гигроскопичен.
Легко растворим (медленно) в воде, практически
нерастворим в 96 % спирте и в эфире.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 5 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 2 мл смеси из
1 объема кислоты хлористоводородной Р1 и 2 объ¬
емов воды Р, выдерживают в течение 5 мин и филь¬
труют. К прозрачному фильтрату прибавляют 5 мл
раствора 100 г/л натрия гидроксида Р и перемеши¬
вают, затем прибавляют 0,2 мл раствора 100 г/л меди
(II) сульфата Р и снова перемешивают. Появляется
фиолетовое окрашивание.
B. К 5 мл раствора 3 прибавляют 5 мл воды Р и
0,5 мл раствора ртути (II) хлорида Р. Образуется
белый осадок, надосадочная жидкость становится
бесцветной или почти бесцветной.
C. Испытуемый образец обугливают и прокалива¬
ют. При обугливании распространяется запах жжено¬
го рога. К остатку, полученному после прокаливания,
прибавляют 1 мл кислоты азотной Р, нагревают, до¬
водят водой Р до объема 10 мл и фильтруют. Полу¬
ченный раствор дает реакцию (а) на серебро (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25 мл этим же
растворителем.
Внешний вид раствора. Раствор 3 выдержива¬
ют в течение 30 мин в защищенном от света месте.
Должен отсутствовать осадок.
Щелочность. 1-2 капли раствора 3 наносят на
фильтровальную бумагу, предварительно смоченную
раствором фенолфталеина Р1 и высушенную. Не
должно появляться розовое окрашивание.
Ионы серебра. К 1,0 г испытуемого образца при¬
бавляют 10 мл 96 % спирта Р, встряхивают в течение
1 мин и фильтруют. К полученному фильтрату при¬
бавляют 2 мл кислоты хлористоводородной Р1. Не
должна появляться опалесценция.
Вещества, осаждаемые натрия хлоридом.
К 5 мл раствора 3 прибавляют 5 мл насыщенного
раствора натрия хлорида Р. Не должны появляться
опалесценция или образовываться осадок.
113*. Зак. 1060.
892
Гэсударственная фармакопея Республики Беларусь
Продукты разложения белка. К 4 мл раствора
3 прибавляют 10 мл воды Р и 0,5 мл раствора 100 г/л
натрия гидроксида Р и нагревают до кипения. Выде¬
ляющиеся пары не должны вызывать посинение крас¬
ной лакмусовой бумаги Р.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого образца помещают в колбу
Кьельдаля вместимостью 100 мл, прибавляют 10 мл
кислоты серной Р, неплотно закрывают пробкой и
кипятят в течение 5 мин. Осторожно по каплям при¬
бавляют 3 мл кислоты азотной Р и нагревают в тече¬
ние 30 мин, не доводя до кипения. Прибавляют 1 мл
кислоты азотной Р и кипятят, пока раствор не станет
светло-желтым, а после охлаждения — бесцветным.
Охлажденный раствор количественно переносят в
коническую колбу вместимостью 250 мл, прибавляют
100 мл воды Р и титруют 0,1 М раствором аммония
тиоцианата до появления желтовато-розового окра¬
шивания, используя в качестве индикатора 0,2 мл
раствора железа (III) аммония сульфата Р.
1 мл 0,1 М раствора аммония тиоцианата соот¬
ветствует 10,79 мгАд.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:2281
СЕРЕБРО КОЛЛОИДНОЕ
ДЛЯ НАРУЖНОГО ПРИМЕНЕНИЯ
АгдеМит соНоМа/е ад изит ех1етит
(#Со11агдо1ит)
5//.УЕЯ, СОиОЮАЦ РОЕ ЕХТЕКЫА1.
1У5Е
ОПРЕДЕЛЕНИЕ
Коллоидное металлическое серебро, содержа¬
щее белок. #Колларгол#.
Содержание: не менее 70,0 % и не более 80,0 %
Ад (в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Зеленые или синевато-черные пластинки с ме¬
таллическим блеском или порошок. Гигроскопично.
Легко растворимо или растворимо в воде, прак¬
тически нерастворимо в 96 % спирте и в метиленхло-
риде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 5 мл фильтрата, полученного в испытании
«Щелочность», прибавляют 0,05 мл раствора меди
(II) сульфата Р, 1 мл раствора натрия гидроксида
разведенного Р и встряхивают. В течение 15 мин по¬
является фиолетовое окрашивание.
B. К 1 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 2 мл раст¬
вора натрия хлорида Р. Образуется осадок, который
растворяется в избытке воды.
C. 0,05 г испытуемого образца прокаливают, по¬
лученный остаток растворяют в 10 мл кислоты азот¬
ной Р и фильтруют. Полученный раствор дает реак¬
цию (а) на серебро (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50,0 мл этим же растворителем.
Полученный раствор выдерживают в течение 5 мин,
затем интенсивно встряхивают, выдерживают в те¬
чение 30 мин и фильтруют через предварительно
взвешенный стеклянный фильтр (ПОР 16) (2.1.2).
Щелочность. К 40,0 мл раствора 3 прибавляют
10.0 мл 0,05 М раствора кислоты серной, 2,0 г на¬
трия сульфата безводного Р, встряхивают и филь¬
труют при необходимости несколько раз. К 25,0 мл
полученного прозрачного и бесцветного раствора
прибавляют 0,1мл раствора фенолфталеина Р.
При прибавлении не более 1,5 мл 0,1 М раствора
натрия гидроксида должно появиться розовое
окрашивание.
Ионы серебра. К 0,50 г испытуемого образца
прибавляют 5 мл этанола Р, встряхивают в течение
1 мин и фильтруют. К полученному фильтрату при¬
бавляют 2 мл кислоты хлористоводородной Р. Не
должен образовываться осадок.
Чувствительность к электролитам. 0,1 г ис¬
пытуемого образца растворяют в 100 мл воды Р.
Часть полученного раствора переносят в пробирку.
При просматривании перпендикулярно оси пробир¬
ки раствор прозрачный и имеет красновато-корич¬
невую окраску. При просматривании вдоль оси про¬
бирки раствор мутный и имеет зеленовато-коричне¬
вую флуоресценцию. К 5 мл раствора прибавляют
5 мл раствора 0,50 г/л натрия хлорида Р и встря¬
хивают в течение 1 мин. При просматривании пер¬
пендикулярно оси пробирки раствор должен оста¬
ваться прозрачным и иметь красновато-коричневую
окраску.
Нерастворимые в воде вещества. Не более
1.0 %. Осадок на фильтре, полученный при приго¬
товлении раствора 3, промывают 5 раз порциями по
10 мл воды Р. Фильтр высушивают до постоянной
массы при температуре от 100 °С до 105 °С. Масса
полученного остатка не должна превышать 12,5 мг.
Потеря в массе при высушивании (2.2.32).
Не более 8,0 %. 0,500 г испытуемого образца сушат
при температуре 80 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца прокаливают при
температуре (650±50) °С до получения белого остат¬
ка. Охлаждают, прибавляют 10 мл смеси из равных
объемов кислоты азотной Р и воды Р и кипятят в те¬
чение 1 мин. Содержимое тигля количественно пере¬
носят в колбу и титруют 0,1 М раствором аммония
тиоцианата до появления красновато-коричневого
окрашивания, используя в качестве индикатора 50 мг
железа (III) сульфата Р.
1 мл 0,1 М раствора аммония тиоцианата соот¬
ветствует 10,79 мгАд.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере #в защи¬
щенном от света месте#.
Серин
893
07/2016:0788
СЕРИН
Зеппит
ВЕРНЫЕ
СгИ7НОг
[56-45-1]
ОПРЕДЕЛЕНИЕ
(5)-2-Амино-3-гидроксипропановая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, практически нераство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО серина #или спектр, представ¬
ленный на рисунке #0788.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Н ЫИ2
НО
со2н
М.м. 105,1
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
О. 1мл раствора 10 г/л испытуемого образца
помещают в пробирку и прибавляют 5 мл раствора
20 г/л натрия перйодата Р. Нагревают на водяной
бане и собирают пар в стекловату, увлажненную во-
дой Р и вставленную в горлышко пробирки. После на¬
гревания в течен'ие 5 мин стекловату переносят в про¬
бирку, содержащую 1 мл раствора 15 г/л натриевой
соли кислоты хромотроповой Р и 3 мл кислоты сер¬
ной Р, и нагревают на водяной бане в течение 10 мин.
Появляется фиолетово-красное окрашивание.
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От
+14,0 до +16,0 (в пересчете на сухое вещество). 2,50 г
испытуемого образца растворяют в кислоте хлори¬
стоводородной разведенной Р и доводят до объема
25,0 мл этим же растворителем.
Нингидрин-положительные вещества
Испытание проводят одним из приведенных
ниже методов/
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
Рисунок #0788.-1. Инфракрасный спектр ФСО серина
114. Зак. 1060.
894
Гэсударственная фармакопея Республики Беларусь
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образца, подходящий для применяемого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт Л/Н^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (ф. 30 мг изолейцина Р и
30 мг лейцина Р растворяют в растворе А и доводят
до объема 50,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
200.0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь) и (д) вводят в аминокислотный анализатор.
Запускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентного содержания:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — исполь¬
зуют концентрацию серина в растворе сравнения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в 0,1 М растворе кислоты хло¬
ристоводородной и доводят до объема 10 мл этим же
растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО серина раст¬
воряют в 0,1 М растворе кислоты хлористоводо¬
родной и доводят до объема 50 мл этим же раство¬
рителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО метионина
и 10 мг ФСО серина растворяют в 0,1 М растворе
кислоты хлористоводородной и доводят до объема
25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза : кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 105 °С в
течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
5 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хрома¬
тограмме испытуемого раствора площадь пика, со¬
ответствующего аммонию, не должна превышать
площадь соответствующего пика на хроматограмме
раствора сравнения (с), учитывая пик, соответству¬
ющий аммонию на хроматограмме холостого раст¬
вора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато¬
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 50 мг испытуемо¬
го образца должны выдерживать испытание на соли
аммония. Эталон готовят с использованием 0,1 мл
эталонного раствора аммония (100 ррт А/Н^ Р.#
Железо (2.4.9). Не более 0,0010% (10 ррт).
1,0 г испытуемого образца помещают в делительную
воронку, растворяют в 10 мл кислоты хлористо¬
водородной разведенной Р, встряхивают трижды,
каждый раз в течение 3 мин, с метилизобутилкето-
ном Р1 порциями по 10мл. Объединенные органи¬
ческие слои встряхивают с 10 мл воды Р в течение
3 мин. Водный слой должен выдерживать испытание
на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раст-
Серная кислота
895
воряют в воде Р и доводят до объема 20 мл этим же
растворителем. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная маслянистая жидкость. Очень гигро¬
скопична. При смешивании с водой и 96 % спиртом
сильно разогревается.
Относительная плотность: около 1,84.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 мл испытуемого образца осторожно при¬
бавляют к 100 мл воды Р. Полученный раствор имеет
сильнокислую реакцию (2.2.4).
B. Раствор, полученный в испытании А, дает ре¬
акцию (а) на сульфаты (2.3.1).
ИСПЫТАНИЯ
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р. Прибавляют
30 мл кислоты уксусной безводной Р. Титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 10,51 мг С3Н7МОэ.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей. Вслед¬
ствие этого нет необходимости идентифицировать
эти примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль примесей
в субстанциях для фармацевтического использова¬
ния): А, В, С.
А. (25)-2-Аминопропановая кислота (аланин).
н2м .со2н
В. Аминоуксусная кислота (глицин).
=М н нн2
нм
со2н
С. (25)-2-Амино-3-(имидазол-4-ил)пропановая кис¬
лота (гистидин).
07/2016:1572
СЕРНАЯ КИСЛОТА
Ас\6ит зиКипсит
вш-римс асю
Н2304 М.м. 98,1
[7664-93-9]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 95,0 % (м/м) и не более
100,5 % (м/м).
Раствор 3. Осторожно при охлаждении вливают
5 мл испытуемого образца в 30 мл воды Р и доводят
до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Хлориды (2.4.4). Не более 0,0050 % (50 ррт).
3,3 г испытуемого образца осторожно смешивают
при охлаждении с 30 мл воды Р. Нейтрализуют рас¬
твором аммиака Р и доводят водой Р до объема
50 мл. 15 мл полученного раствора должны выдер¬
живать испытание на хлориды.
Нитраты. 5 мл испытуемого образца прибавля¬
ют к 5 мл воды Р. Охлаждают до комнатной темпе¬
ратуры и прибавляют 0,5 мл раствора индигокар-
мина Р. Синее окрашивание раствора должно сохра¬
няться не менее 1 мин.
Мышьяк (2.4.2, метод А). Не более 0,0001 %
(1 ррт). 1 г испытуемого образца смешивают при ох¬
лаждении с 20 мл воды Р и доводят до объема 25 мл
этим же растворителем. Раствор должен выдержи¬
вать испытание на мышьяк.
Железо (2.4.9). Не более 0,0025 % (25 ррт).
10,0 г испытуемого образца осторожно выпаривают,
затем прокаливают до тусклого красного каления.
Остаток после прокаливания растворяют в 1 мл кис¬
лоты хлористоводородной разведенной Р при сла¬
бом нагревании и доводят водой Р до объема 25 мл.
1 мл полученного раствора доводят водой Р до объ¬
ема 10 мл. Раствор должен выдерживать испытание
на железо.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0005 % (5 ррт). 4,0 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Точно взвешивают колбу с притертой стеклян¬
ной пробкой, содержащую 30 мл воды Р. Прибавляют
0,2 мл испытуемого образца, охлаждают и взвешива¬
ют. Титруют 1 М раствором натрия гидроксида по¬
тенциометрически (2.2.20).
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 49,04 мг Н2304.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
114*. Зак. 1060.
896
Государственная фармакопея Республики Беларусь
07/2016:1148
СЕРТАКОНАЗОЛА НИТРАТ
8ег1асопагоН пНгаз
8ЕКТАС0ЫА201.Е Ы1ТКАТЕ
и энантиомер
С20Н15С13М2ОЗ • НЛОз М.м. 500,8
[99592-39-9]
ОПРЕДЕЛЕНИЕ
(Я$)-1 -[2-[(7-Хлор-1 -бензотиофен-3-ил)метокси]-
2-(2,4-дихлорфенил )этил]-1 Н-имидазола нитрат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, растворим в
метаноле, умеренно растворим в 96 % спирте и в ме-
тиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О, Е.
A. Температура плавления (2.2.14): от 156 °С до
161 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,1 г испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
100 мл этим же растворителем. 10 мл полученного
раствора доводят метанолом Р до объема 100 мл.
Диапазон длин волн: от 240 нм до 320 нм.
Максимумы поглощения: при 260 нм, 293 нм и
302 нм.
Отношение оптических плотностей:
Лю2/Л>9з“-°т1’16Д° 1’28'
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: испытуемый образец высуши¬
вают при температуре от 100 °С до 105 °С в течение
2 ч и проводят испытание с использованием дисков из
калия бромида Р.
Сравнение: ФСО сертаконазола нитрата.
О. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Раствор аммиака кон¬
центрированный Р— метанол Р (10:90, об/об).
Испытуемый раствор. 40 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 10 мл этой же смесью растворителей.
Раствор сравнения (а). 40 мг ФСО сертаконазо¬
ла нитрата растворяют в смеси растворителей и до¬
водят до объема 10 мл этой же смесью растворителей.
Раствор сравнения (Ь). 20 мг ФСО миконазола
нитрата растворяют в растворе сравнения (а) и до¬
водят до объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля <3 Р.
Подвижная фаза: раствор аммиака концентриро¬
ванный Р — толуол Р — диоксан Р (1:40;60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке воздуха в течение 15 мин.
Проявление: пластинку обрабатывают парами
йода в течение 30 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствуют по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
Е. Около 1 мг испытуемого образца дает реакцию
(а) на нитраты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,1 г испытуемого образца раство¬
ряют в 96 % спирте Р и доводят до объема 10 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)5.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО сертакона¬
зола нитрата и 5,0 мг ФСО миконазола нитрата
растворяют в подвижной фазе и доводят до объема
20.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят подвижной фазой до объема
50.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем нитрильным
для хроматографии Р1 с размером частиц 10 мкм;
- подвижная фаза: ацетонитрил Р1 — раствор
1,5 г/л натрия дигидрофосфата Р (37:63, об/об);
- скорость подвижной фазы: 1,6 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,3-кратное вре¬
мя удерживания сертаконазола.
Время удерживания: нитрат-ион — около 1 мин;
миконазол — около 17 мин; сертаконазол — около
19 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками мико¬
назола и сертаконазола.
Предельное содержание примесей:
- примеси А, В и С (не более 0,25 %): на хро¬
матограмме испытуемого раствора площади пиков,
Силденафила цитрат
897
соответствующих примесям А, В и С, не должны
превышать площадь основного пика на хромато¬
грамме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 2-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,2 площади основного
пика на хроматограмме раствора сравнения (а); не
учитывают пик, соответствующий нитрат-иону.
Вода (2.5.72). Не более 1,0%. Определение
проводят из 0,50 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 50 мл
смеси из равных объемов кислоты уксусной без¬
водной Р и метилэтилкетона Р и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 50,08 мг С20Н15С13М2О8Н1\Ю3.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
а
С1
НО'
и энантиомер
А. (1 Р5)-1 -(2,4-Дихлорфенил)-2-(1 Н-имидазол-1 -
ил)этанол.
B. Р = Вг: 3-(Бромметил)-7-хлор-1-бензотиофен.
C. Р = ОН: (7-Хлор-1-бензотиофен-3-ил)метанол.
07/2016:2270
СИЛДЕНАФИЛА ЦИТРАТ
8Нс1епаЛН сНгаз
811.0ЕЫАН1. С1ТЯАТЕ
С^НзЛО.З ■ С,Н,07 М.м. 667
[171599-83-0]
Рисунок #2270.-1. Инфракрасный спектр ФСО силденафила цитрата
115. Зак. 1060.
898
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
5-[2-Этокси-5-[(4-метилпиперазин-1-ил)сульфо-
нил]фенил]-1 -метил-З-пропил-1,6-дигидро-7Н-пиразо-
ло[4,3-с/]пиримидин-7-она дигидро-2-гидроксипропан-
1,2,3-трикарбоксилат.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Слегка гигроскопичен.
Мало растворим в воде и метаноле, практически
нерастворим в гексане.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО силденафила цитрата #или
спектр, представленный на рисунке #2270.-1#.
ИСПЫТАНИЯ
Примесь Е. Не более 0,1 %. Тонкослойная хро¬
матография (2.2.27).
Смесь растворителей. Аммиака раствор кон-
центрированный Р — вода Р — метанол Р (5:25:75,
об/об/об).
Испытуемый раствор. 35,0 мг испытуемого об¬
разца растворяют, при необходимости используя уль¬
тразвук, в 2,0 мл смеси растворителей.
Раствор сравнения (а). 7,0 мг ФСО имидазола
(примесь Е) растворяют в смеси растворителей и до¬
водят до объема 20,0 мл этой же смесью раствори¬
телей. 1,0 мл полученного раствора доводят смесью
растворителей до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мл раствора срав¬
нения (а) доводят смесью растворителей до объема
10,0 мл.
Раствор сравнения (с). 1 мл испытуемого раст¬
вора смешивают с 1 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((2—10) мкм).
Подвижная фаза: раствор аммиака концентри¬
рованный Р— 96% спирт Р—этилацетат Р—ме-
тиленхлорид Р (1:20:30:50, об/об/об/об).
Наносимый объем пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь) и (с) в виде полос
6 мм.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре 100 °С в течение
15 мин.
Проявление: пластинку обрабатывают парами
йода до тех пор, пока пластинка не станет светло-ко¬
ричневой, и затем просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Факторы подвижности: цитрат — около 0; при¬
месь Е — около 0,25; силденафил — около 0,4.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- примесь Е (не более 0,1 %): на хроматограмме
испытуемого раствора зона, соответствующая приме¬
си Е, должна быть не интенсивнее основной зоны на
хроматограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 35,0 мг испытуемого
образца растворяют, при необходимости используя
ультразвук, в подвижной фазе и доводят до объема
50.0 мл этим же растворителем.
Испытуемый раствор (Ь). 2,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
50.0 мл.
Раствор сравнения (а). 35,0 мг ФСО силдена¬
фила цитрата растворяют, при необходимости ис¬
пользуя ультразвук, в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем. 2,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 50,0 мл.
Раствор сравнения (Ь). 5,0 мл испытуемого
раствора (Ь) доводят подвижной фазой до объема
100.0 мл.
Раствор сравнения (с). Для приготовления при¬
меси В /л зПи 70 мг испытуемого образца растворяют
в 1 мл смеси из 1 объема кислоты муравьиной без¬
водной Р и 2 объемов стабилизированного раствора
водорода пероксида концентрированного Р, выдер¬
живают не менее 10 мин и доводят подвижной фазой
до объема 250 мл.
Раствор сравнения (ф. 3 мг ФСО силденафила
примеси А растворяют в подвижной фазе и доводят
до объема 20,0 мл этим же растворителем. 1 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 20,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 30 °С;
-подвижная фаза: ацетонитрил Р — мета¬
нол Р — 0,7 % (об/об) раствор триэтиламина Р, пред¬
варительно доведенный кислотой фосфорной Р до
рН (3,0±0,1) (17:25:58, об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 290 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (Ь), (с) и (б);
- время хроматографирования: 3-кратное вре¬
мя удерживания силденофила.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (д).
Относительное удерживание (по отношению к
силденафилу, время удерживания — около 7 мин):
примесь В — около 1,2; примесь А— около 1,7.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 2,5 между пиками сил¬
денафила и примеси В.
Расчет процентного содержания примесей:
-для расчета содержания каждой примеси ис¬
пользуют концентрацию силденафила в растворе
сравнения (Ь).
Предельное содержание примесей:
- примесь А: не более 0,3 %;
- неспецифицированные примеси: не более
0,10 %;
- сумма неспецифицированных примесей: не бо¬
лее 0,3 %;
Симвастатин
899
- сумма примесей: не более 0,5 %;
- неучитываемый предел: 0,05 %.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 2,5 %. Определение про¬
водят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (а).
Содержание С22Н30М6О45*С6Н8О7 в процентах
рассчитывают с учетом содержания силденафила ци¬
трата в ФСО силденафила цитрата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В, С, О.
А. 5-[2-Этокси-5-[(4-метилпиперазин-1 -ил)сульфо-
нил]фенил]-1 -метил-3-(2-метилпропил)-1,6-дигидро-
7Н-пиразоло[4,3-сУ]пиримидин-7-он.
В. 5-[2-Этокси-5-[(4-метил-4-оксидопиперазин-1-
ил )сульфонил]фенил]-1 -метил-З-пропил-1,6-дигидро-
7Н-пиразоло[4,3-с/]пиримидин-7-он.
С. 5-[2-Гидрокси-5-[(4-метилпиперазин-1 -ил)суль-
фонил]фенил]-1 -метил-З-пропил-1,6-дигидро-7Н-
пиразоло[4,3-сУ]пиримидин-7-он.
й. 4-Этокси-3-(1-метил-7-оксо-3-пропил-6,7-дигид-
ро-1/-/-пиразоло[4,3-с(]пиримидин-5-ил)бензолсульфо-
новая кислота.
Е. 1Н-Имидазол.
07/2016:1563
СИМВАСТАТИН
81туазШтит
81М\/А8ТАТ1Ы
О
ОПРЕДЕЛЕНИЕ
(15,ЗК,75,85,8аЯ)-8-[2-[(2К,4К)-4-Гидрокси-6-
оксотетрагидро-2А7-пиран-2-ил]этил]-3,7-диметил-
1,2,3,7,8,8а-гексагидронафталин-1 -ил-2,2-диметил-
бутаноат.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
Может содержать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, очень легко
растворим в метиленхлориде, легко растворим в 96 %
спирте.
115*. Зак. 1060.
900
Государственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО симвастатина #или спектр,
представленный на рисунке #1563.-1#.
ИСПЫТАНИЯ
Раствор 5. 0,20 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)7.
Удельное оптическое вращение (2.2.7). От
+285 до +300 (в пересчете на сухое вещество). 0,125 г
испытуемого образца растворяют в ацетонитриле Р
и доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Смесь растворителей. 40 объемов раствора
1,4 г/л калия дигидрофосфата Р, доведенного до рН
4.0 кислотой фосфорной Р, смешивают с 60 объема¬
ми ацетонитрила Р и фильтруют.
Испытуемый раствор. 75,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (а). 1,0 мг ФСО симваста¬
тина и 1,0 мг ФСО ловастатина (примесь Е) раст¬
воряют в смеси растворителей и доводят до объема
50.0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 0,5 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл.
Раствор сравнения (с). 75,0 мг ФСО симваста¬
тина растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (д). 5 мг ФСО симвастатина
для идентификации пиков (содержит примеси А, В, С,
О, Е, Р и О) растворяют в 5,0 мл смеси растворителей.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,033 м
и внутренним диаметром 4,6 мм, заполненная сили¬
кагелем октадецилсилильным эндкепированным для
хроматографии Р с размером частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — 0,1 %
(об/об) раствор кислоты фосфорной Р (50:50,
об/об);
- подвижная фаза В: 0,1 % (об/об) раствор кис¬
лоты фосфорной Р в ацетонитриле Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—4,5
100
0
4,5—4,6
100 — 95
0 — 5
4,6—8,0
95 — 25
5 — 75
8,0—11,5
25
75
- скорость подвижной фазы: 3,0 мл/мин;
- спектрофотометрический детектор, длина
волны 238 нм;
- объем вводимой пробы: по 5 мкл испытуемого
раствора и растворов сравнения (а), (Ь) и (д).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, О, Е+Р и О, используя хро¬
матограмму раствора сравнения (б) и хроматограмму,
прилагаемую к ФСО симвастатина для идентифи¬
кации пиков.
Относительное удерживание (по отношению к
симвастатину, время удерживания — около 2,6 мин):
примесь А — около 0,5; примесь Е+Р — около 0,6;
Волновое число (см*1)
Рисунок #1563.-1. Инфракрасный спектр ФСО симвастатина
Симвастатин
901
примесь С — около 0,8; примеси В и С — около 2,4;
примесь О — около 3,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 4,0 между пиком приме¬
си Е и пиком симвастатина.
Предельное содержание примесей:
- сумма примесей Е и Р (не более 1,0 %): на хро¬
матограмме испытуемого раствора площадь пика,
соответствующего примесям Е и Р, не должна превы¬
шать 2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- сумма примесей В су С (не более 0,8 %): на хро¬
матограмме испытуемого раствора площадь пика, со¬
ответствующего примесям В и С, не должна превы¬
шать 1,6-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- примеси А, О, <3 (не более 0,4 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, О и 6, не должны превы¬
шать 0,8 площади основного пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, й, Е, Р и С, не должна превышать 0,2
площади основного пика на хроматограмме раствора
сравнения (Ь);
- сумма примесей, кроме примесей Е и Е (не бо¬
лее 1,0 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пи¬
ков примесей Е и Р, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы {2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 60 °С в глубоком вакууме в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемого
раствора и раствора сравнения (с).
Содержание симвастатина в процентах рассчи¬
тывают с учетом содержания симвастатина в ФСО
симвастатина.
ХРАНЕНИЕ
В защищенном от света месте.
Если субстанция не содержит антиоксидан¬
та — в атмосфере азота в воздухонепроницаемом
контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, 6.
со2н
А. (ЗЯ,5Я)-7-[(15,25,6Я,85,8аЯ)-8-[(2,2-Диметил-
бутаноил)окси]-2,6-диметил-1,2,6,7,8,8а-гексагидрона
фтален-1-ил]-3,5-дигидроксигептановая кислота (тени-
вастатин).
к.
Н* ^ ’ХГ "сн3
В. (15,ЗР,75,85,8аЯ)-8-[2-[(2Я?,4Я)-4-(Ацетилокси)-
6-оксотетрагидро-2Н-пиран-2-ил]этил]-3,7-диметил-
1,2,3,7,8,8а-гексагидронафтален-1-ил-2,2-диметил-
бутират.
о
С. (15,ЗЯ75,85,8аР)-3,7-Диметил-8-[2-[(2Я)-6-оксо-
3,6-дигид ро-2Н-пиран-2-ил]этил]-1,2,3,7,8,8а-гексагидро-
нафтален-1 -ил-2,2-диметилбутират.
О. (2Я,4Я)-2-[2-[(15,25,6Я,85,8аЯ)-8-[(2,2-Диметил-
бутаноил)окси]-2,6-диметил-1,2,6,7,8,8а-гексагидро-
нафтален-1-ил]этил]-6-оксотетрагидро-2Я-пиран-4-ил-
(ЗЯ5Я)-7-[(15,25,6Я85,8аЯ)-8-[(2,2-диметилбутаноил)-
окси]-2,6-диметил-1,2,6,7,8,8а-гексагидронафтален-1-
ил]-3,5-дигидроксигептаноат (димер).
о
Е. Р1 = Р4 = СН3, В2 = КЗ = Н: (1 ЗЖ78,88,8аЯ)-
8-[2-[(2Я, 4Р)-4-Гидрокси-6-оксотетрагидро-2АУ-пиран-
2-ил]этил]-3,7-диметил-1,2,3,7,8,8а-гексагидронафтален-
1- ил-(25)-2-метил бутират (ловастатин).
Р. Р1 = РЗ = Н, Р2 = Р4 = СН3: (15,ЗЯ,75,85,8аЯ)-
8-[2-[(2Я,4Я)-4-Гидрокси-6-оксотетрагидро-2Н-пиран-
2- ил]этил]-3,7-диметил-1,2,3,7,8,8а-гексагидронафтален-
1 -ил-(2Я)-2-метилбутират (эпиловастатин).
С. Р1 = Р2 = СН3, Р2 + Р4 = СН2: (15,ЗЯ,75,85,8аЯ)-
8-[2-[(2Я,4/?)-4-Гидрокси-6-оксотетрагидро-2Н-пиран-
2-ил]этил]-3,7-диметил-1,2,3,7,8,8а-гексагидронафтален-
1 -ил-2,2-диметилбут-3-еноат.
902
Гэсударственная фармакопея Республики Беларусь
07/2016:1265
СОЕВОЕ МАСЛО
ГИДРОГЕНИЗИРОВАННОЕ
Зо/ае о1еит Ьус/годепаХит
80УА-ВЕАЫ ОН, НУОВОвЕЫАТЕО
ОПРЕДЕЛЕНИЕ
Продукт, получаемый очисткой, отбеливанием,
гидрогенизацией и дезодорированием масла, полу¬
ченного из семян <31усте тах (1_.) Мегг. (6. ЫзрШа
(МоепсИ) Мах1т.). Масло состоит главным образом из
триглицеридов пальмитиновой и стеариновой кислот.
ОПИСАНИЕ(СВОЙСТВА)
Белая или почти белая масса или порошок, при
нагревании плавится, переходя в прозрачную бледно-
желтую жидкость.
Практически нерастворимо в воде, легко рас¬
творимо в метиленхлориде, толуоле и, при нагре¬
вании, в петролейном эфире (температура кипения
(65—70) °С), очень мало растворимо в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испыта¬
ние «Температура плавления», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Состав жирных кислот», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Температура плавления (2.2.15). От 66 °С до
72 °С.
Кислотное число (2.5.1). Не более 0,5. 10,0 г ис¬
пытуемого образца растворяют в 50 мл горячей сме¬
си из равных объемов 96 % спирта Р и толуола Р,
предварительно нейтрализованной 0,1 М раствором
калия гидроксида, используя в качестве индикатора
0,5 мл раствора фенолфталеина Р1. Немедленно
титруют горячий раствор.
Перекисное (пероксидное) число (2.5.5, ме¬
тод А). Не более 5,0.
Неомыляемые вещества (2.5.7). Не более
1,0%. Определение проводят из 5,0 г испытуемого
образца.
Щелочные примеси в жирных маслах (2.4.19).
2,0 г испытуемого образца растворяют при осторожном
нагревании в смеси из 1,5 мл 96 % спирта Р и 3 мл то¬
луола Р, прибавляют 0,05 мл раствора 0,4 г/л бромфе-
нолового синего Рв 96% спирте Р. При прибавлении
не более 0,4 мл 0,01 М раствора кислоты хлористо¬
водородной должно появиться желтое окрашивание.
Состав жирных кислот (2.4.22, метод А). Ис¬
пользуют смесь веществ для градуировки, как указано
в таблице 2.4.22.-3.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 25 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(цианопропил)силоксана Р (толщина слоя 0,2 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 0,65 мл/мин;
- деление потока: 1:100;
- температура колонки: 180 °С в течение 20 мин;
- температура блока ввода пробы и детекто¬
ра: 250 °С;
- детектор: пламенно-ионизационный.
Состав фракции жирных кислот испытуемого
образца:
- насыщенные жирные кислоты с длиной цепи
менее С14: не более 0,1 %;
- миристиновая кислота: не более 0,5 %;
- пальмитиновая кислота: от 9,0 % до 16,0 %;
- стеариновая кислота: от 79,0 % до 89,0 %;
- олеиновая кислота и ее изомеры: не более 4,0 %;
- линолевая кислота и ее изомеры: не более 1,0 %;
- линоленовая кислота и ее изомеры: не более 0,2 %;
- арахидоновая кислота: не более 1,0 %;
- бегеновая кислота: не более 1,0 %.
Никель. Не более 0,0001 % (1 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод II).
Испытуемый раствор. 5,0 г испытуемого об¬
разца помещают в предварительно прокаленный и
взвешенный платиновый или кварцевый тигель. Осто¬
рожно нагревают и вводят в образец фитиль, изготов¬
ленный из скрученной беззольной фильтровальной
бумаги. Фитиль поджигают. Когда испытуемый обра¬
зец масла воспламенится, нагрев прекращают. После
сгорания остаток прокаливают в муфельной печи при
температуре около (600±50) °С. Прокаливание про¬
должают до образования белой золы. После охлаж¬
дения остаток растворяют с использованием двух
порций по 2 мл кислоты хлористоводородной разве¬
денной Р и переносят в мерную колбу вместимостью
25 мл, прибавляют 0,3 мл кислоты азотной Р и дово¬
дят водой Р до объема 25,0 мл.
Растворы сравнения. Готовят 3 раствора срав¬
нения, прибавляя к 2,0 мл испытуемого раствора по
1,0 мл, 2,0 мл и 4,0 мл эталонного раствора никеля
(0,2 ррт Щ Р и доводя водой Р до объема 10,0 мл.
Источник излучения: лампа с полым катодом
для определения никеля.
Длина волны: 232 нм.
Атомизатор: графитовая печь.
Газ-носитель: аргон Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1473
СОЕВОЕ МАСЛО
РАФИНИРОВАННОЕ
5о/ае о!еит гаШпаХит
80УА-ВЕАМ ОН, КЕРШЕО
ОПРЕДЕЛЕНИЕ
Жирное масло, полученное из семян <31усте тах
(1_.) Мегг. (6/ус/ле М$р1да (МоепсЬ) Мах1т.) методом
экстракции с последующим рафинированием. Масло
может содержать подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бледно-желтая жидкость.
Практически нерастворимо в 96 % спирте, сме¬
шивается с петролейным эфиром (температура кипе¬
ния (50—70) °С).
Сорбиновая кислота
903
Относительная плотность: около 0,922.
Показатель преломления: около 1,475.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Идентификация жирных масел методом тонкос¬
лойной хроматографии (2.3.2).
Результаты: хроматограмма испытуемого об¬
разца соответствует типичной хроматограмме, пред¬
ставленной на рисунке 2.3.2.-1.
ИСПЫТАНИЯ
- арахидоновая кислота: не более 1,0 %;
- эйкозаноевая кислота: не более 1,0 %;
- бегеновая кислота: не более 1,0 %.
Брассикастерол (2.4.23). Не более 0,3 % в сте-
риновой фракциии масла.
Вода (2.5.32). Не более 0,1 %. Определение про¬
водят из 1,00 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Кислотное число (2.5.1). Не более 0,5. Опреде¬
ление проводят из 10,0 г испытуемого образца.
Перекисное (пероксидное) число (2.5.5, метод
А). Не более 10,0. Не более 5,0, если испытуемая
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения.
Неомыляемые вещества (2.5.7). Не более
1,5%. Определение проводят из 5,0 г испытуемого
образца.
Щелочные примеси в жирных маслах (2.4.19).
Испытуемый образец должен выдерживать испыта¬
ние на щелочные примеси в жирных маслах.
Состав жирных кислот (2.4.22, метод А). Ис¬
пользуют смесь веществ для градуировки, как указано
в таблице 2.4.22.-3.
Состав фракции жирных кислот испытуемого
образца:
- насыщенные жирные кислоты с длиной цепи
менее С14: не более 0,1 %;
- миристиновая кислота: не более 0,2 %;
- пальмитиновая кислота: от 9,0 % до 13,0 %;
- пальмитолеиновая кислота: не более 0,3 %;
- стеариновая кислота: от 2,5 % до 5,0 %;
- олеиновая кислота: от 17,0 % до 30,0 %;
- линолевая кислота: от 48,0 % до 58,0 %;
- линоленовая кислота: от 5,0 % до 11,0 %;
ХРАНЕНИЕ
В заполненном доверху контейнере, в защищен¬
ном от света месте, при температуре не выше 25 °С.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
07/2016:0592
СОРБИНОВАЯ КИСЛОТА
Аадит зогЫсит
ЗОКВ1САСЮ
/С02Н
Н3СГ
С.Н.О, М.м. 112,1
[110-44-1]
ОПРЕДЕЛЕНИЕ
(Е,Е)-Гекса-2,4-диеновая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
Рисунок #0592.-1. Инфракрасный спектр ФСО кислоты сорбиновой
904
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворима в воде, легко растворима в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 132 °С до
136 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
250,0 мл этим же растворителем. 2,0 мл полученного
раствора доводят 0,1 М раствором кислоты хлори¬
стоводородной цо объема 200,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 264 нм.
Удельный показатель поглощения в максимуме:
от 2150 до 2550.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО кислоты сорбиновой #или
спектр, представленный на рисунке #0592.-1#.
й. 0,2 г испытуемого образца растворяют в 2 мл
96 % спирта Р и прибавляют 0,2 мл воды бромной Р.
Раствор обесцвечивается.
ИСПЫТАНИЯ
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в 96 % спирте Р и доводят до объема 25 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Альдегиды. Не более 0,15% в пересчете на
С2Н40.1,0 г испытуемого образца растворяют в смеси
из 30 мл воды Р и 50 мл 2-пропанола Р, доводят до
рН 4 0,1 М раствором кислоты хлористоводородной
или 0,1 М раствором натрия гидроксида и разво¬
дят водой Р до объема 100 мл. К 10 мл полученного
раствора прибавляют 1 мл обесцвеченного раствора
фуксина Р и выдерживают в течение 30 мин. Окра¬
ска раствора должна быть не интенсивнее окраски
эталона, приготовленного параллельно прибавле¬
нием 1 мл обесцвеченного раствора фуксина Р к
смеси из 1,5 мл эталонного раствора ацетальде¬
гида (100 ррт С2Н40) Р, 4 мл 2-пропанола Р и 4,5 мл
воды Р.
Тяжелые металлы (2.4.8, метод В). Не более
0,0010 % (10 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон го¬
товят с использованием 5 мл эталонного раствора
свинца (1 ррт РЬ) Р и 5 мл 96 % спирта Р.
Вода (2.5.12). Не более 1,0 %. Определение про¬
водят из 2,000 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,1000 г испытуемого образца растворяют в
20 мл 96 % спирта Р и титруют 0,1 М раствором на¬
трия гидроксида до розового окрашивания, исполь¬
зуя в качестве индикатора 0,2 мл раствора фенол¬
фталеина Р
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 11,21 мг С6Н802.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0435
СОРБИТОЛ (СОРБИТ)
8огЬНо1ит
80КВ1Т01.
Свн1406 М.м. 182,2
[50-70-4]
ОПРЕДЕЛЕНИЕ
Р-Глюцитол (О-сорбитол).
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определе¬
ние».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
B. 0,5 г испытуемого образца растворяют при на¬
гревании в смеси из 0,5 мл пиридина Р и 5 мл анги¬
дрида уксусного Р. Через 10 мин раствор вливают в
25 мл воды Р и выдерживают в ледяной бане в тече¬
ние 2 ч. Осадок перекристаллизовывают из неболь¬
шого количества 96 % спирта Р и сушат в вакууме.
Температура плавления (2.2.14) полученного остатка:
от 98 °С до 104 °С.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (а). 25 мг ФСО сорбитола
растворяют в воде Р и доводят до объема 10 мл этим
же растворителем.
Раствор сравнения (Ь). 25 мг ФСО маннитола и
25 мг ФСО сорбитола растворяют в воде Р и доводят
до объема 10 мл этим же растворителем.
Сорбитол (сорбит)
905
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р.
Подвижная фаза: вода Р — этилацетат Р —
пропанол Р (10:20:70, об/об/об).
Объем наносимой пробы: 2 мкл.
Фронт подвижной фазы: не менее 17 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
4-аминобензойной кислоты Р; сушат в потоке холод¬
ного воздуха до удаления ацетона; нагревают при
температуре 100 °С в течение 15 мин; охлаждают и
опрыскивают раствором 2 г/л натрия перйодата Р;
сушат в потоке холодного воздуха; нагревают при тем¬
пературе 100 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
О. Удельное оптическое вращение (2.2.7): от +4,0
до +7,0 (в пересчете на безводное вещество). 5,00 г
испытуемого образца растворяют в растворе 6,4 г
динатрия тетрабората Р в 40 мл воды Р, выдержи¬
вают в течение 1 ч при периодическом встряхивании,
доводят водой Р до объема 50,0 мл и, при необходи¬
мости, фильтруют.
ИСПЫТАНИЯ
Раствор 3. 5 г испытуемого образца растворяют
в воде Р и доводят до объема 50 мл этим же раство¬
рителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Электропроводность (2.2.38). Не более
20 мкСм см"1. 20,0 г испытуемого образца растворяют
в воде, свободной от углерода диоксида, Р, полу¬
ченной из воды дистиллированной Р, и доводят до
объема 100,0 мл этим же растворителем. Измеряют
электропроводность полученного раствора при осто¬
рожном перемешивании на магнитной мешалке.
Восстанавливающие сахара. Не более 0,2 % в
пересчете на глюкозу. 5,0 г испытуемого образца раст¬
воряют в 6 мл воды Р при легком нагревании. Раствор
охлаждают, прибавляют 20 мл медно-цитратного
раствора Р и несколько стеклянных шариков. Нагрева¬
ют таким образом, чтобы через 4 мин началось кипе¬
ние, и поддерживают кипение в течение 3 мин. Быстро
охлаждают и прибавляют 100 мл 2,4 % (об/об) раствора
кислоты уксусной ледяной Р и 20,0 мл 0,025 М раст¬
вора йода. При непрерывном перемешивании прибав¬
ляют 25 мл смеси из 6 объемов кислоты хлористово¬
дородной Р и 94 объемов воды Р. После того как осадок
растворится, титруют избыток йода 0,05 М раствором
натрия тиосульфата, используя в качестве индика¬
тора 1 мл раствора крахмала Р, который прибавляют
в конце титрования. На титрование должно пойти не
менее 12,8 мл 0,05 Мраствора натрия тиосульфата.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 5,0 г испытуемого образ¬
ца растворяют в 20 мл воды Р и доводят до объема
100,0 мл этим же растворителем.
Раствор сравнения (а). 0,50 г ФСО сорбито¬
ла растворяют в 2 мл воды Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (Ь). 2,0 мл испытуемого раст¬
вора доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). 5,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 100,0 мл.
Раствор сравнения (д). 0,5 г сорбитола Р и 0,5 г
маннитола Р (примесь А) растворяют в 5 мл воды Р
и доводят до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
7,8 мм, заполненная катионообменной смолой силь¬
ной (кальциевая форма) Р с размером частиц 9 мкм;
- температура: (85±1) °С;
- подвижная фаза: дегазированная вода Р;
- скорость подвижной фазы: 0,5 мл/мин;
- детектор: рефрактометрический, установлен¬
ный при постоянной температуре (нарпимер, 35 °С);
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (с1);
- время хроматографирования: 2-кратное вре¬
мя удерживания сорбитола.
Относительное удерживание (по отношению к
сорбитолу, время удерживания — около 27 мин): при¬
месь С — около 0,6; примесь А — около 0,8; примесь
В — около 1,1.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 2,0 между пиками при¬
меси А и сорбитола.
Предельное содержание примесей:
- любая примесь (не более 2 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кро¬
ме основного, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 3 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
1,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
Свинец (2.4.10). Не более 0,00005% (0,5 ррт).
Испытуемый образец должен выдерживать испыта¬
ние на содержание свинца в сахарах.
Никель (2.4.15). Не более 0,0001 % (1 ррт). Ис¬
пытуемый образец должен выдерживать испытание
на содержание никеля в полиолах. Испытуемый обра¬
зец растворяют в 150,0 мл предписанной смеси раст¬
ворителей.
Вода (2.5.12). Не более 1,5 %. Определение про¬
водят из 1,00 г испытуемого образца. В качестве рас¬
творителя используют смесь формамид Р— метанол
безводый Р (1:2, об/об).
Микробиологическая чистота
Если субстанция предназначена для производ¬
ства лекарственных средств для парентерального
применения:
- ОКА: критерий приемлемости 102 КОЕ/г (2.6.72).
Если субстанция не предназначена для произ¬
водства лекарственных средств для парентерального
применения:
- ОКА: критерий приемлемости 103 КОЕ/г (2.6.12);
- ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12);
906
Государственная фармакопея Республики Беларусь
- ЕзсЬепсЫа соН: отсутствие (2.6.13);
- 8а1топе11а: отсутствие (2.6.13).
Бактериальные эндотоксины (2.6.14). Если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения
без дальнейшей соответствующей процедуры удале¬
ния бактериальных эндотоксинов, то содержание эн¬
дотоксинов должно быть:
- менее 4 МЕ/г для лекарственных средств для
парентерального применения с концентрацией сорби¬
тола менее 100 г/л;
- менее 2,5 МЕ/г для лекарственных средств для
парентерального применения с концентрацией сорби¬
тола 100 г/л и более.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание О-сорбитола в процентах рассчитыва¬
ют с учетом содержания сорбитола в ФСО сорбитола.
МАРКИРОВКА
При необходимости указывают:
- предельное содержание бактериальных эндо¬
токсинов;
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения.
ПРИМЕСИ
А. О-Маннитол.
С. 4-О-а-О-Глюкопиранозил-О-глюцитол (О-мапьти-
тол).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Данный раздел не является обязательным, и
для подтверждения соответствия требованиям
частной статьи проверка указанных характери¬
стик не обязательна. Однако контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для сорбитола, используемого в качестве
наполнителя и связующего вещества при произ¬
водстве таблеток.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошков (2.9.36).
07/2016:0436
СОРБИТОЛА РАСТВОР
КРИСТАЛЛИЗУЮЩИЙСЯ (СОРБИТА
РАСТВОР КРИСТАЛЛИЗУЮЩИЙСЯ)
ЗогЫШит ^шдит спвШНзаЬИе
ЗОКВ1ТОЦ исшю (СКУЗТАШ81ЫС)
ОПРЕДЕЛЕНИЕ
Водный раствор гидрированного частично гидро¬
лизованного крахмала.
Содержание:
- безводное вещество: не менее 68,0 % (м/м) и
не более 72,0 % (м/м);
- О-глюцитол (О-сорбитол; М.м. С6Н1406): не ме¬
нее 92,0 % и не более 101,0 % (в пересчете на безво¬
дное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная сиропообразная жидкость.
Смешивается с водой.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
B. К 7,0 г испытуемого образца прибавляют 40 мл
воды Р, 6,4 г динатрия тетрабората Р, выдержива¬
ют в течение 1 ч при периодическом встряхивании,
доводят водой Р до объема 50,0 мл и, при необходи¬
мости, фильтруют. Угол оптического вращения (2.2.7)
полученного раствора: от 0° до +1,5°.
C. При температуре 25 °С испытуемый образец
представляет собой прозрачную сиропообразную
жидкость.
ИСПЫТАНИЯ
Раствор 5. 7,0 г испытуемого образца доводят
водой Р до объема 50 мл.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Сорбитола раствор некристаллизующийся (сорбита раствор некристаллизующийся)
907
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Электропроводность (2.2.38). Не более
10 мкСм-см"1. Измеряют электропроводность нераз-
веденного испытуемого образца при осторожном пе¬
ремешивании на магнитной мешалке.
Восстанавливающие сахара. Не более 0,2 %
в пересчете на глюкозу. К 5,0 г испытуемого образ¬
ца прибавляют 6 мл воды Р, 20 мл медно-цитрат-
ново раствора Р и несколько стеклянных шариков.
Нагревают таким образом, чтобы через 4 мин на¬
чалось кипение, и поддерживают кипение в течение
3 мин. Быстро охлаждают и прибавляют 100 мл 2,4 %
(об/об) раствора кислоты уксусной ледяной Р и
20.0 мл 0,025 М раствора йода. При непрерывном
встряхивании прибавляют 25 мл смеси из 6 объ¬
емов кислоты хлористоводородной Р и 94 объемов
воды Р. После того как осадок растворится, титруют
избыток йода 0,05 М раствором натрия тиосульфа¬
та, используя в качестве индикатора 1 мл раствора
крахмала Р, который прибавляют в конце титрования.
На титрование должно пойти не менее 12,8 мл 0,05 М
раствора натрия тиосульфата.
Свинец (2.4.10). Не более 0,00005 % (0,5 ррт).
Никель (2.4.15). Не более 0,0001 % (1 ррт).
Вода (2.5.12). Не менее 28,0 % (м/м) и не более
32.0 % (м/м). Определение проводят из 0,1 г испытуе¬
мого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,00 г испытуемого об¬
разца смешивают с 20 мл воды Р и доводят до объ¬
ема 50,0 мл этим же растворителем.
Раствор сравнения (а). 65,0 мг ФСО сорбитола
растворяют в 2 мл воды Р и доводят до объема 5,0 мл
этим же растворителем.
Раствор сравнения (Ь). 65 мг сорбитола Р и
65 мг маннитола Р растворяют в 2 мл воды Р и дово¬
дят до объема 5,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
7,8 мм, заполненная катионообменной смолой силь¬
ной (кальциевая форма) Р с размером частиц 9 мкм;
- температура: (85±1) °С;
- подвижная фаза: дегазированная вода Р;
- скорость подвижной фазы: 0,5 мл/мин;
- детектор: рефрактометрический, установлен¬
ный при постоянной температуре (например, 35 °С);
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания сорбитола.
Относительное удерживание (по отношению к
сорбитолу, время удерживания — около 27 мин): ман-
нитол — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками ман¬
нитола и сорбитола.
Содержание Р-сорбитола в процентах рассчиты¬
вают с учетом содержания сорбитола в ФСО сорби¬
тола.
07/2016:0437
СОРБИТОЛА РАСТВОР
НЕКРИСТАЛЛИЗУЮЩИЙСЯ
(СОРБИТА РАСТВОР
НЕКРИСТАЛЛИЗУЮЩИЙСЯ)
ЗогЬНо1ит Ндшс1ит поп спзШНзаЬНе
80КВ1ТОЦ /./<3(7/0 (ЫОЫ-
СКУ8ТАШ81ЫО)
ОПРЕДЕЛЕНИЕ
Водный раствор гидрированного частично гидро¬
лизованного крахмала.
Содержание:
- безводное вещество: не менее 68,0 % (м/м) и
не более 72,0 % (м/м);
- О-глюцитол (Р-сорбитол; М.м. С6Н1406): не ме¬
нее 72,0 % и не более 92,0 % (в пересчете на безво¬
дное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная сиропообразная жидкость.
Смешивается с водой.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определе¬
ние».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
B. К 7,0 г испытуемого образца прибавляют 40 мл
воды Р, 6,4 г динатрия тетрабората Р, выдержива¬
ют в течение 1 ч при периодическом встряхивании,
доводят водой Р до объема 50,0 мл и, при необходи¬
мости, фильтруют. Угол оптического вращения (2.2.7)
полученного раствора: от +1,5° до +3,5°.
C. При температуре 25 °С испытуемый образец
представляет собой прозрачную сиропообразную
жидкость.
ИСПЫТАНИЯ
Раствор 5. 7,0 г испытуемого образца доводят
водой Р до объема 50 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Электропроводность (2.2.38). Не более
10 мкСм-см-1. Измеряют электропроводность испы¬
туемого образца при осторожном перемешивании на
магнитной мешалке.
Восстанавливающие сахара. Не более 0,2 %
в пересчете на глюкозу. К 5,0 г испытуемого образ¬
ца прибавляют 6 мл воды Р, 20 мл медно-цитрат-
ного раствора Р и несколько стеклянных шариков.
Нагревают таким образом, чтобы через 4 мин на¬
чалось кипение, и поддерживают кипение в течение
3 мин. Быстро охлаждают и прибавляют 100 мл 2,4 %
(об/об) раствора кислоты уксусной ледяной Р и
20,0 мл 0,025 М раствора йода. При непрерывном
перемешивании прибавляют 25 мл смеси из 6 объ¬
емов кислоты хлористоводородной Р и 94 объемов
воды Р. После того как осадок растворится, титруют
908
Гэсударственная фармакопея Республики Беларусь
избыток йода 0,05 М раствором натрия тиосульфа¬
та, используя в качестве индикатора 1 мл раствора
крахмала Р, который прибавляют в конце титрования.
На титрование должно пойти не менее 12,8 мл 0,05 М
раствора натрия тиосульфата.
Восстанавливающие сахара после гидролиза.
Не более 9,3 % в пересчете на глюкозу. К 6,0 г испы¬
туемого образца прибавляют 35 мл воды Р, 40 мл 1 М
раствора кислоты хлористоводородной и несколько
стеклянных шариков. Кипятят в течение 4 ч с обрат¬
ным холодильником. Смесь охлаждают, нейтрализу¬
ют раствором натрия гидроксида разведенным Р,
используя в качестве индикатора 0,2 мл раствора
бромтимолового синего Р1. После охлаждения дово¬
дят водой Р до объема 100,0 мл. К 3,0 мл полученного
раствора прибавляют 5 мл воды Р, 20 мл медно-ци-
тратного раствора Р и несколько стеклянных ша¬
риков. Нагревают таким образом, чтобы через 4 мин
началось кипение, и поддерживают кипение в тече¬
ние 3 мин. Быстро охлаждают и прибавляют 100 мл
2,4 % (об/об) раствора кислоты уксусной ледяной Р
и 20,0 мл 0,025 М раствора йода. При непрерывном
перемешивании прибавляют 25 мл смеси из 6 объ¬
емов кислоты хлористоводородной Р и 94 объемов
воды Р. После того как осадок растворится, титруют
избыток йода 0,05 М раствором натрия тиосульфа¬
та, используя в качестве индикатора 1 мл раствора
крахмала Р, который прибавляют в конце титрования.
На титрование должно пойти не менее 8,0 мл 0,05 М
раствора натрия тиосульфата.
Свинец (2.4.10). Не более 0,00005 % (0,5 ррт).
Никель (2.4.15). Не более 0,0001 % (1 ррт).
Вода (2.5.12). Не менее 28,0 % (м/м) и не более
32,0 % (м/м). Определение проводят из 0,1 г испытуе¬
мого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,00 г испытуемого об¬
разца смешивают с 20 мл воды Р и доводят до объ¬
ема 50,0 мл этим же растворителем.
Раствор сравнения (а). 55,0 мг ФСО сорбитола
растворяют в 2 мл воды Р и доводят до объема 5,0 мл
этим же растворителем.
Раствор сравнения (Ь). 55 мг сорбитола Р и
55 мг маннитола Р растворяют в 2 мл воды Р и дово¬
дят до объема 5,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
7,8 мм, заполненная катионообменной смолой силь¬
ной (кальциевая форма) Р с размером частиц 9 мкм;
- температура: (85±1) °С;
- подвижная фаза: дегазированная вода Р\
- скорость подвижной фазы: 0,5 мл/мин;
- детектор: рефрактометрический, установлен¬
ный при постоянной температуре (например, 35° С);
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания сорбитола.
Относительное удерживание (по отношению к
сорбитолу, время удерживания — около 27 мин): ман-
нитол — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками ман¬
нитола и сорбитола.
Содержание Э-сорбитола в процентах рассчиты¬
вают с учетом содержания сорбитола в ФСО сорби¬
тола.
07/2016:1842
СОСНЫ ОБЫКНОВЕННОЙ МАСЛО
Р/л/ зу^езНз ае1Ьего1еит
РШЕ 8ПУЕ8ТШ8 ОН
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное методом перегон¬
ки с водяным паром из свежей хвои и ветвей Р'шиз
зу1уе$1пз 1_. Может содержать подходящий антиокси¬
дант.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или светло-желтая жид¬
кость с характерным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 мл испытуемого образ¬
ца доводят толуолом Р до объема 10 мл.
Раствор сравнения. 10 мг борнеола Р и 10 мкл
борнилцетата Р растворяют в толуоле Р и доводят
до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: по 10 мкл [по 2 мкл] в
виде полос.
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раство¬
ром анисового альдегида Р, нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин и про¬
сматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые зоны.
Верх хроматографической пластинки
Зона розового цвета (угле¬
водороды)
Борнилацетат: зона корич¬
невого или серо-коричнево¬
го цвета
Зона коричневого или серо¬
коричневого цвета (борни¬
лацетат)
Зона розового цвета
Борнеол: зона коричневого
или серо-коричневого цвета
Скопление зон фиолетово¬
го цвета
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Спирамицин
909
Результаты: характерные пики на хроматограм¬
ме испытуемого раствора по времени удерживания
соответствуют характерным пикам на хроматограмме
раствора сравнения (а).
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,855 до 0,875.
Показатель преломления (2.2.6). От 1,465 до 1,480.
Угол оптического вращения (2.2.7). От -9° до -30°.
Кислотное число (2.5.1). Не более 1,0.
Перекисное (пероксидное) число (2.5.5). Не бо¬
лее 20.
Жирные и минеральные масла (2.8.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
внутренней нормализации.
Испытуемый раствор. Испытуемый образец.
Раствор сравнения (а). 30 мкл а-пинена Р, 10 мг
камфена Р, 20 мкл р-пинена Р, 10 мкл кар-3-ена Р,
10 мкл Р-мирцена Р, 20 мкл лимонена Р, 10 мкл
п-цимена Р, 10 мкл терпинолена Р, 10 мкл борнила-
цетата Р и 10 мкл р-кариофиллена Р растворяют в
1 мл гептана Р.
Раствор сравнения (Ь). 10 мг камфена Р раство¬
ряют в гептане Р и доводят до объема 2 мл этим же
растворителем. 0,1 мл полученного раствора доводят
гептаном Р до объема 1 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м и
внутренним диаметром 0,22 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,2 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
65
10—41
65 -* 220
41—50
220
Блок ввода проб
220
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,2 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения (а); отмечают време¬
на удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками кар-3-
ена и р-мирцена.
Идентификация компонентов: по временам
удерживания компонентов раствора сравнения (а)
идентифицируют компоненты испытуемого раствора.
Пик р-фелландрена элюируется после пика лимонена
с относительным удерживанием около 1,03 по отно¬
шению к лимонену.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- а-пинен: от 32,0 % до 60,0 %;
- камфен: от 0,5 % до 2,0 %;
- Р-пинен: от 5,0 % до 22,0 %;
- кар-3-ен: от 6,0 % до 18,0 %;
-Р-мирцен: от 1,5 % до 10,0 %;
- лимонен: от 7,0 % до 12,0 %;
- Р-фелландрен: не более 2,5 %;
- п-цимен: не более 2,0 %;
- терпинолен: не более 4,0 %;
- борнилацетат: от 1,0 % до 4,0 %;
- р-кариофиллен: от 1,0 % до 6,0 %;
-неучитываемый предел: площадь основного
пика на хроматограмме раствора сравнения (Ь).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:0293
СПИРАМИЦИН
8р1гатустит
ЗР1КАМУС1Ы
н3с
\
Состав
В
Молекулярная
формула
М.м.
Спирамицин I
н
с„н74м2о14
843,1
Спирамицин II
со-сн3
с46н76м2о16
885,1
Спирамицин III
со-сн2-сн3
с46н78м2о15
899,1
ОПРЕДЕЛЕНИЕ
Макролидный антибиотик, который получают вы¬
ращиванием определенных штаммов 81гер(отусез
атЬо^ааепз ил и другими способами. Основным компо¬
нентом является (4Я55,65,7/?,9/?,10/?,11Е,13Е,16/?)-
6-[[3,6-дидезокси-4-0-(2,6-дидезокси-3-С-метил-
а-1_-р17бо-гексопиранозил)-3-(диметиламино)-Р-0-
глюкопиранозил]окси]-4-гидрокси-5-метокси-9,16-
диметил-7-(2-оксоэтил)-10-[[2,3,4,6-тетрадезокси-
4-(диметиламино)-0-эрш77ро-гексопиранозил]окси]-
оксациклогексадека-11,13-диен-2-он (спирамицин I;
М.м. 843). Также присутствуют спирамицин
II (4-О-ацетилспирамицин I) и спирамицин III
(4-О-пропаноилспирамицин I).
Активность: не менее 4100 МЕ/мг (в пересчете
на сухое вещество).
910
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок. Слегка
гигроскопичен.
Мало растворим в воде, легко растворим в аце¬
тоне, в 96 % спирте и в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в уль¬
трафиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,10 г испытуемого
образца растворяют в метаноле Р и доводят до
объема 100,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят метанолом Р до
объема 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 232 нм.
Удельный показатель поглощения в максиму¬
ме: около 340.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 40 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (а). 40 мг ФСО спирами-
цина растворяют в метаноле Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (Ь). 40 мг ФСО эритро¬
мицина А растворяют в метаноле Р и доводят до
объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля О Р.
Подвижная фаза: верхний слой смеси из 4 объ¬
емов 2-пропанола Р, 8 объемов раствора 150 г/л
аммония ацетата Р, предварительно доведенно¬
го раствором натрия гидроксида концентриро¬
ванным Р до рН 9,6, и 9 объемов этилацетата Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р1 и нагревают при тем¬
пературе 110 °С в течение 5 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а). Если на хроматограмме испытуемого
раствора обнаруживаются одно или 2 пятна со зна¬
чениями Рг немного большими, чем для основного
пятна, то эти пятна соответствуют по расположе¬
нию и цвету вторичным пятнам на хроматограмме
раствора сравнения (а) и отличаются от пятен на
хроматограмме раствора сравнения (Ь).
C. 0,5 г испытуемого образца растворяют в
10 мл 0,05 М раствора кислоты серной, прибав¬
ляют 25 мл воды Р, доводят 0,1 М раствором на¬
трия гидроксида до приблизительно рН 8 и дово¬
дят водой Р до объема 50 мл. К 5 мл полученно¬
го раствора прибавляют 2 мл смеси из 1 объема
воды Р и 2 объемов кислоты серной Р. Появляет¬
ся коричневое окрашивание.
ИСПЫТАНИЯ
рН (2.2.3). От 8,5 до 10,5. 0,5 г испытуемого об¬
разца растворяют в 5 мл метанола Р и доводят во¬
дой, свободной от углерода диоксида, Р до объема
100 мл.
Удельное оптическое вращение (2.2.7). От
-80 до -85 (в пересчете на сухое вещество). 1,00 г
испытуемого образца растворяют в 10% (об/об)
растворе кислоты уксусной разведенной Р и до¬
водят до объема 50,0 мл этим же растворителем.
Состав. Жидкостная хроматография (2.2.29),
как указано в испытании «Сопутствующие приме¬
си».
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и раствора сравнения (а).
Содержание в процентах рассчитывают с уче¬
том содержания спирамицинов I, II и III в ФСО спи-
рамицина.
Состав спирамицинов (в пересчете на сухое
вещество):
- спирамицин I: не менее 80,0 %;
- спирамицин II: не более 5,0 %;
- спирамицин III: не более 10,0 %;
- сумма спирамицинов I, II и III: не менее
90,0 %.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием.
Смесь растворителей. Метанол Р — вода Р
(30:70, об/об).
Испытуемый раствор. 25,0 мг испытуемого
образца растворяют в смеси растворителей и до¬
водят до объема 25,0 мл этой же смесью раство¬
рителей.
Раствор сравнения (а). 25,0 мг ФСО спирами-
цина растворяют в смеси растворителей и доводят
до объема 25,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 2,0 мл раствора срав¬
нения (а) доводят смесью растворителей до объ¬
ема 100,0 мл.
Раствор сравнения (с). 5 мг ФСО спирами-
цина растворяют в 15 мл буферного раствора рН
2,2 Р и доводят водой Р до объема 25 мл, затем
нагревают в водяной бане при температуре 60 °С
в течение 5 мин и охлаждают под холодной водой.
Холостой раствор. Смесь растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная полимером органоси¬
ликатным аморфным, с введенными полярными
группами, октадецилсилильным эндкепирован-
ным Р (размер частиц 5 мкм) (гель метилсиликат-
ный, с введенными полярными группами, октаде-
цилсилильный) с размером пор 12,5 нм и содержа¬
нием углерода 15 %;
- температура: 70 °С;
- подвижная фаза: смешивают 5 объемов
раствора 34,8 г/л дикалия гидрофосфата Р, дове¬
денного раствором 27,2 г/л калия дигидрофосфа¬
та Р до рН 6,5, 40 объемов ацетонитрила Р и 55
объемов воды Р\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 232 нм;
- объем вводимой пробы: по 20 мкл холостого
раствора, испытуемого раствора и растворов срав¬
нения (Ь) и (с);
- время хроматографирования: 3-кратное вре¬
мя удерживания спирамицина I.
Идентификация пиков спирамицинов: иденти¬
фицируют пики спирамицинов I, II и III, используя
Спирамицин
911
хроматограмму раствора сравнения (а) и хромато¬
грамму, прилагаемую к ФСО спирамицина.
Относительное удерживание (по отношению
к спирамицину I, время удерживания — от 20 мин
до 30 мин): примесь Р — около 0,41; примесь А —
около 0,45; примесь й — около 0,50; примесь О —
около 0,66; примесь В — около 0,73; примесь Н —
около 0,87; спирамицин II — около 1,4; спирамицин
III — около 2,0; примесь Е — около 2,5.
При необходимости корректируют состав под¬
вижной фазы, изменяя количество ацетонитрила.
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 10,0 между пиками
примеси А и спирамицина I.
Предельное содержание примесей:
- примеси А, В, О, Е, Р, С и Н (не более 2,0 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, О, Е, Р, О и
H, не должны превышать площадь основного пика
на хроматограмме раствора сравнения (Ь);
- любая другая примесь (не более 2,0 %): на
хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пиков примесей А,
В, О, Е, Р, С и Н, не должна превышать площадь
основного пика на хроматограмме раствора срав¬
нения (Ь);
- сумма примесей (не более 10,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика
на хроматограмме раствора сравнения (Ь); не учи¬
тывают пики холостого раствора и спирамицинов
I, II и III.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 3,5 %. 0,500 г испытуемого образца сушат
над фосфора (V) оксидом Р при температуре 80 °С
и давлении не более 0,67 кПа в течение 6 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): С.
A. К1 = Н, К2 = ОН, КЗ = СН2-СНО: (4Р,58,68,
7Р,9Р, 10К, 11Е, 13 Е, 16К)-6-[[3,6-Дидезокси-3-(диметила-
мино)-р-0-глюкопиранозил]окси]-4-гидрокси-5-метокси-
9,16-диметил-7-(2-оксоэтил)-10-[[2,3,4,6-тетрадезокси-
4-(диметиламино)-р-0-эр(япро-гексопиранозил]окси]-
оксациклогексадека-11,13-диен-2-он (неоспирамицин I).
B. К1 = Н, К2 = озил, КЗ = СН2-СН2ОН: (4Я,55,65,
7Я,9Я, 10Я, 11Е, 13Е, 16Я)-6-[[3,6-Дидезокси-4-0-(2,6-
дидезокси-3-С-метил-а-Ь-р1/бо-гексопиранозил)-3-
(диметиламино)-р-0-глюкопиранозил]окси]-4-гидрокси-
7-(2-гидроксиэтил)-5-метокси-9,16-диметил-10-[[2,3,4,6-
тетрадезокси-4-(диметиламино)-р-0-эр1/тро-
гексопиранозил]окси]оксациклогексадека-11,13-диен-
2-он (спирамицин IV).
C. К1 = Н, К2 = озил, КЗ = С(=СН2)-СНО: (4Я,55,
68,7 8 Ж1ОН, 11Е, 13Е, 16Я)-6-[[3,6-Дидезокси-4-0-(2,6-
дидезокси-3-С-метил-а-Ьрб/бо-гексопиранозил)-3-(диме-
тиламино)-Р-0-глюкопиранозил]окси]-7-(1-формилэте-
нил)-4-гидрокси-5-метокси-9,16-диметил-10-[[2,3,4,6-
тетрадезокси-4-(диметиламино)-р-Э-эр1/шро-гексопи-
ранозил]окси]оксациклогексадека-11,13-диен-2-он
(17-метиленспирамицин I).
Е. К1 = Н, К2 = озил, КЗ = СН2-СН3: (АН,55,65,75,
9Н, 10Я, 11Е, 13 Е, 16Я)-6-[[3,6-Дидезокси^-0-(2,6-дидезок-
си-3-С-метил-а-^-рибо-гексопиранозил)-3-(диметила-
мино)-р-0-глюкопиранозил]окси]-7-этил-4-гидрокси-5-
метокси-9,16-диметил-10-[[2,3,4,6-тетрадезокси-4-
(диметиламино)-р-0-эр1/шро-гексопиранозил]окси]-
оксациклогексадека-11,13-диен-2-он (18-дезокси-18-
дигидроспирамицин I, й8РМ).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят количественное определение антибио¬
тиков микробиологическим методом (2.7.2).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, О, Е, Р, С, Н.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
ов. К1 = СО-СН3, К2 = ОН, КЗ = СН2-СНО: (4Я,55,
68,7 Р,9Р, 10Н, 11Е, 13Е, 16Я)-6-[[3,6-Дидезокси-3-(диме-
тиламино)-р-0-глюкопиранозил]окси]-5-метокси-9,16-
диметил-2-оксо-7-(2-оксоэтил)-10-[[2,3,4,6-тетрадезокси-
4-(диметиламино)-Р-0-эрш?ро-гексопиранозил]окси]-
оксациклогексадека-11,13-диен-4-илацетат (неоспира¬
мицин II).
Н. К1 = СО-С2Н5, К2 = ОН, КЗ = СН2-СНО: (4К,55,
65,7К,9К, 10Н, 11Е, 13 Е, 16К)-6-[[3,6-Дидезокси-3-(диме-
тиламино)-р-Э-гл юкопиранозил]окси]-5-метокси-9,16-
диметил-2-оксо-7-(2-оксоэтил)-10-[[2,3,4,6-тетрадезокси-
4-(диметиламино)-р-0-эр1ялро-гексопиранозил]окси]-
оксациклогексадека-11,13-диен-4-илпропаноат (нео¬
спирамицин III).
912
Гэсударственная фармакопея Республики Беларусь
Э. (4/?,55,65,7/?,9Я, 10Я, 11Е, 13Е, 16Я)-6-[[3,6-
Дидезокси^-0-(2,6-дидезокси-3-С-метил-а-Ь-рибо-гексо-
пиранозил)-3-(диметиламино)-Р-0-глюкопиранозил]-
окси]-10-[(2,6-дидезокси-3-С-метил-а-!_-рибо-
гексопиранозил)окси]-4-гидрокси-5-метокси-9,16-
диметил-7-(2-оксоэтил )-оксациклогексадека-11,13-диен-
2-он (спирамицин V).
Р. Спирамицина димер.
07/2016:0688
СПИРОНОЛАКТОН
8ркопо1ас1опит
8Р1К0Ы01.АСТ0ЫЕ
с Н О 5
^24* в32ч/4'*
[52-01-7]
ОПРЕДЕЛЕНИЕ
(2'/?)-7а-(Ацетилсульфанил)-3\4'-дигидро-5'Н-
спиро[андрост-4-ен-17,2'-фуран]-3,5'-дион.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок.
Практически нерастворим в воде, растворим в
96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО спиронолактона.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то испытуе¬
мый образец и ФСО спиронолактона растворяют по
отдельности в минимальном количестве метанола Р
и выпаривают до сухих остатков, которые используют
для получения новых спектров.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метиленхлориде Р и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО спиронолактона
растворяют в метиленхлориде Р и доводят до объ¬
ема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: вода Р — циклогексан Р —
этилацетат Р (1:24:75, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
C. К 10 мг испытуемого образца прибавляют 2 мл
50 % (об/об) раствора кислоты серной Р и перемеши¬
вают. Образуется раствор оранжевого цвета с интен¬
сивной желтовато-зеленой флуоресценцией. Полу¬
ченный раствор осторожно нагревают. Окрашивание
раствора переходит в темно-красное, и выделяется
сероводород, который обнаруживают по почернению
бумаги свинцово-ацетатной Р. Полученный раствор
прибавляют к 10 мл воды Р. Образуется опалесциру¬
ющий раствор желтовато-зеленого цвета (может вы¬
падать осадок).
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От -41
до -49 (в пересчете на сухое вещество). 0,100 г испы¬
туемого образца растворяют в 96% спирте Р и дово¬
дят до объема 10,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Смесь растворителей. Ацетонитрил Р —
вода Р (50:50, об/об).
М.м. 416,6
Спиронолактон
913
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в 2,5 мл тетрагидрофурана Р
и доводят смесью растворителей до объема 25,0 мл.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 100,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора (Ь) доводят смесью растворителей до объема
10.0 мл.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО спиронолактона для проверки пригодности
хроматографической системы (содержит примеси
А, С, О, Е и I) растворяют с использованием ультра¬
звука в 1,0 мл смеси растворителей.
Раствор сравнения (с). 50,0 мг ФСО спироно¬
лактона растворяют в 2,5 мл тетрагидрофурана Р
и доводят смесью растворителей до объема 25,0 мл.
1.0 мл полученного раствора доводят смесью раство¬
рителей до объема 100,0 мл.
Раствор сравнения (д). 5,0 мг ФСО канренона
(примесь Р) растворяют в 2,5 мл тетрагидрофу¬
рана Р и доводят смесью растворителей до объема
25.0 мл. 3,0 мл полученного раствора доводят смесью
растворителей до объема 100,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 3 мкм;
- температура: 40 °С;
- подвижная фаза: ацетонитрил Р — тетраги-
дрофуран Р — метанол Р — вода Р (15:20:425:540,
об/об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (б);
- время хроматографирования: 2,5-кратное вре¬
мя удерживания спиронолактона.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, С, О, Е и I, используя хрома¬
тограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО спиронолактона для проверки
пригодности хроматографической системы, иден¬
тифицируют пик примеси Р, используя хроматограмму
раствора сравнения (б).
Относительное удерживание (по отношению к
спиронолактону, время удерживания — около 26 мин):
примесь А — около 0,95; примесь Р — около 1,2; при¬
месь С — около 1,5; примесь О — около 1,6; примесь
Е — около 1,7; примесь I — около 1,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- коэффициент разделения пиков: не менее 1,5
(Нр — высота пика примеси А относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси А и пик
спиронолактона).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Р — 2,3):
- примесь I (не более 0,5 %): на хроматограмме ис¬
пытуемого раствора (а) площадь пика, соответствующего
примеси I, не должна превышать 5-кратную площадь ос¬
новного пика на хроматограмме раствора сравнения (а);
- примеси Е, Е (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям Е и Р, не должны пре¬
вышать 3-кратную площадь основного пика на хро¬
матограмме раствора сравнения (а);
- примеси А, С (не более 0,2 %): на хромато¬
грамме испытуемого раствора (а) площади пиков,
соответствующих примесям А и С, не должны пре¬
вышать 2-кратную площадь основного пика на хро¬
матограмме раствора сравнения (а);
-примесь О (не более 0,15%): на хромато¬
грамме испытуемого раствора (а) площадь пика,
соответствующего примеси О, не должна превы¬
шать 1,5-кратную площадь основного пика на хро¬
матограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
(а) площадь любого пика, кроме основного и пиков
примесей А, С, О, Е, Р и I, не должна превышать
площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- сумма примесей (не более 0,7 %): на хрома¬
тограмме испытуемого раствора (а) сумма площа¬
дей всех пиков, кроме основного, не должна превы¬
шать 7-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора (а) не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (а).
Свободные меркаптосоединения. К 2,0 г
испытуемого образца прибавляют 20 мл воды Р,
встряхивают в течение 1 мин и фильтруют. К 10 мл
полученного фильтрата прибавляют 0,05 мл 0,01 М
раствора йода и 0,1 мл раствора крахмала Р и
перемешивают. Должно появиться синее окраши¬
вание.
Хром. Не более 0,0050 % (50 ррт). 0,20 г ис¬
пытуемого образца помещают в платиновый ти¬
гель, прибавляют 1 г калия карбоната Р и 0,3 г
калия нитрата Р. Осторожно нагревают до рас¬
плавления и прокаливают при температуре от
600 °С до 650 °С до сгорания углерода. Охлажда¬
ют, полученный остаток растворяют как можно пол¬
нее в 10 мл воды Р при осторожном нагревании,
фильтруют и доводят водой Р до объема 20 мл.
К 10 мл полученного раствора прибавляют 0,5 г мо¬
чевины Р и 14 % (об/об) раствор кислоты серной Р
до кислой реакции среды. После прекращения
выделения пузырьков газа прибавляют 1 мл 14%
(об/об) раствора кислоты серной Р, доводят во¬
дой Р до объема 20 мл и прибавляют 0,5 мл раст¬
вора дифенилкарбазида Р. Окраска полученного
раствора должна быть не интенсивнее окраски
стандартного раствора, приготовленного следую¬
щим образом: 1 мл 14 % (об/об) раствора кислоты
серной Р прибавляют к 0,50 мл свежеприготовлен¬
ного раствора 28,3 мг/л калия дихромата Р, дово¬
дят водой Р до объема 20 мл и прибавляют 0,5 мл
раствора дифенилкарбазида Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
914
Государственная фармакопея Республики Беларусь
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С24Н32045 в процентах рассчитыва¬
ют с учетом содержания спиронолактона в ФСО спи-
ронолактона.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, С, О, Е, Г, I.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В, С, Н.
А. (2'Р)-7а-(Ацетилсульфанил)-5'Н-спиро[андрост-
4-ен-17,2'-фуран]-3,5'-дион (А20-спиронолактон).
В. (27?)-7а-(Ацетилсульфанил)-4-бром-3',4'-ди-
гидро-5'Н-спиро[андрост-4-ен-17,2'-фуран]-3,5'-дион
(4-бромоспиронолактон).
С. (2'Я)-3',4-Дигидро-5'Н-спиро[андрост-4-ен-17,2'-
фуран]-3,5'-дион (алдон).
й. (27?)-7а-(Ацетилдисульфанил)-3',4-дигидро-5'Я-
спиро[андрост-4-ен-17,2-фуран]-3,5-дион (дисульфа-
нилспиронолактон).
Е. (27?)-7р-(Ацетилсульфанил)-3',4'-дигидро-5'Н-
спиро[андрост-4-ен-17,2'-фуран]-3,5’-дион (7Р-спиро-
нолактон).
Р. (2'Я)-3',4'-Дигидро-5'Н-спиро[андрост-4,6-диен-
17,2'-фуран]-3,5-дион (канренон).
С. (2'Я)-7а-(Ацетилсульфанил)-бр-гидрокси-3',4'-
дигидро-5'Н-спиро[андрост-4-ен-17>2'-фуран]-3,5'-дион
(бр-гидроксиспиронолактон).
1. 5-[17а-(Этоксиметил)-17-гидрокси-З-оксоанрост-
4-ен-7а-ил]этантиоат.
Стеариновая кислота
915
07/2016:0753
СТЕАРИЛОВЫЙ СПИРТ
А1со1ю1 з1еагуНсиз
8ТЕАКУ1. А1.С0Н01-
ОПРЕДЕЛЕНИЕ
Смесь твердых спиртов, состоящая преимуще¬
ственно из октадекан-1-ола (С18Н380; М.м. 270,5), жи¬
вотного или растительного происхождения.
Содержание: не менее 95,0 % С18Н380.
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые маслянистые хлопья,
гранулы или масса.
Практически нерастворим в воде, растворим в
96 % спирте. В расплавленном состоянии смешивает¬
ся с жирными маслами, маслом вазелиновым и рас¬
плавленным ланолином.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные как
указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (Ь).
ИСПЫТАНИЯ
Раствор 3. 0,50 г испытуемого образца раство¬
ряют в 20 мл кипящего 96 % спирта Р и охлаждают.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)6.
Температура плавления (2.2.14). От 57 °С до
60 °С.
Кислотное число (2.5.1). Не более 1,0.
Гидроксильное число (2.5.3, метод А). От 197
до 217.
Йодное число (2.5.4, метод А). Не более 2,0.
2,00 г испытуемого образца растворяют в метилен-
хлориде Р, при необходимости подогревая, и доводят
до объема 25 мл этим же растворителем.
Число омыления (2.5.6). Не более 2,0.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28): определение
проводят методом внутренней нормализации.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (а). 50 мг спирта цетилово¬
го Р растворяют в 96% спирте Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (Ь). 50 м г ФСО спирта сте¬
аринового растворяют в 96% спирте Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (с). Смешивают 1 мл раст¬
вора сравнения (а) и 1 мл раствора сравнения (Ь) и
доводят до объема 10 мл 96 % спиртом Р.
Условия хроматографирования:
- колонка длиной 30 м и внутренним диаметром
0,32 мм, покрытая слоем поли(диметил)силоксана Р
(толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—20
150 —► 250
20—40
250
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора и растворов сравнения (Ь) и (с).
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 5,0 между пиком цетило¬
вого спирта и пиком стеарилового спирта.
Рассчитывают процентное содержание С18Н380.
07/2016:1474
СТЕАРИНОВАЯ КИСЛОТА
АсШит з1еапсит
5ТЕАЯ1С АСЮ
ОПРЕДЕЛЕНИЕ
Смесь, состоящая главным образом из кислоты
стеариновой (октадекановой) (С18Н3602; М.м. 284,5) и
кислоты пальмитиновой (гексадекановой) (С1бН3202;
М.м. 256,4). Получают из жиров или масел раститель¬
ного или животного происхождения.
Содержание:
Стеариновая
кислота 50
Стеариновая кислота: не менее 40,0 % и
не более 60,0 %.
Сумма стеариновой и пальмитиновой
кислот: не менее 90,0 %
Стеариновая
кислота 70
Стеариновая кислота: не менее 60,0 % и
не более 80,0 %.
Сумма стеариновой и пальмитиновой
кислот: не менее 90,0 %
Стеариновая
кислота 95
Стеариновая кислота: не менее 90,0 %.
Сумма стеариновой и пальмитиновой
кислот: не менее 96,0 %
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые воскообразные хлопье¬
видные кристаллы, белая или почти белая твердая
масса или белый или желтовато-белый порошок.
Практически нерастворима в воде, растворима в
96 % спирте и петролейном эфире (температура кипе¬
ния (50—70) °С).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Температура замерзания», как описано в разделе
«Испытания».
B. Кислотное число (2.5.1): от 194 до 212. Опре¬
деление проводят из 0,5 г испытуемого образца.
C. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определе¬
ние».
Результаты: основные пики на хроматограмме
испытуемого раствора по времени удерживания соот¬
ветствуют основным пикам на хроматограмме раст¬
вора сравнения.
916
Государственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Цветность (2.2.2, метод I). Испытуемый образец
нагревают до температуры около 75 °С. Окраска по¬
лученной жидкости должна быть не интенсивнее эта¬
лона У(Ж)7 или ВУ(КЖ)7.
Кислотность. 5,0 г испытуемого образца рас¬
плавляют, встряхивают в течение 2 мин с 10 мл горя¬
чей воды, свободной от углерода диоксида, Р, мед¬
ленно охлаждают и фильтруют. К фильтрату прибав¬
ляют 0,05 мл раствора метилового оранжевого Р.
Не должно появляться красное окрашивание.
Йодное число (2.5.4). См. таблицу 1474.-1.
Температура замерзания (2.2.18). См. таблицу
1474.-1.
Таблица 1474.-1.
Тип
Йодное число
Температура
замерзания (°С)
Кислота
стеариновая 50
не более 4,0
53—59
Кислота
стеариновая 70
не более 4,0
57—64
Кислота
стеариновая 95
не более 1,5
64—69
Никель (2.4.31). Не более 0,0001 % (1 ррт).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28): определение
проводят методом внутренней нормализации.
Испытуемый раствор. В конической колбе, осна¬
щенной обратным холодильником, 0,100 г испытуемого
образца растворяют в 5 мл раствора бора фторида в
метаноле Р. Кипятят с обратным холодильником в те¬
чение 10 мин. Прибавляют 4,0 мл гептана Р через от¬
верстие холодильника, кипятят еще 10 мин и охлажда¬
ют. Прибавляют 20 мл насыщенного раствора натрия
хлорида Р Встряхивают и дают слоям разделиться.
Около 2 мл органического слоя удаляют и сушат над
0,2 г натрия сульфата безводного Р. 1,0 мл получен¬
ного раствора доводят гептаном Р до объема 10,0 мл.
Раствор сравнения. Готовят раствор сравнения
таким же образом, как и испытуемый раствор, исполь¬
зуя 50 мг ФСО кислоты пальмитиновой и 50 мг ФСО
кислоты стеариновой вместо испытуемого образца.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром 0,32 мм, покрытая слоем ма-
крогола 20 000 Р (толщина слоя 0,5 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 2,4 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—2
70
2—36
70 -> 240
36—41
240
Блок ввода проб
220
Детектор
260
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению к
метилстеарату): метилпальмитат — около 0,9.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 5,0 между пиками ме-
тилпальмитата и метилстеарата;
- относительное стандартное отклонение: не
более 3,0 % для площадей пиков метилпальмитата
и метилстеарата при 6 повторных вводах пробы; не
более 1,0 % для отношений площадей пиков метил¬
пальмитата к площадям пиков метилстеарата при 6
повторных вводах пробы.
МАРКИРОВКА
Указывают тип стеариновой кислоты (50, 70, 95).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для стеариновой кислоты, используемой в каче¬
стве скользящего вещества при производстве та¬
блеток и капсул.
Распределение частиц по размерам (2.9.31).
Удельная площадь поверхности (2.9.26, метод I).
07/2016:0053
СТРЕПТОМИЦИНА СУЛЬФАТ
81гер1отуап'1 зи№аз
ЗТКЕРТОМУСШ ЗШ.ЕАТЕ
он
• ЗНг304 М.м. 1457
[3810-74-0]
Стрептомицина сульфат
917
ОПРЕДЕЛЕНИЕ
Бис[Л/,Л/'-бис(аминоиминометил)-4-0-[5-дез-
окси-2-0-[2-дезокси-2-(метиламино)-а-1--глюко-
пиранозил]-3-С-формил-а-1-ликсофуранозил]-0-
стрептамина] трисульфат.
Стрептомицина сульфат получают выращивани¬
ем определенных штаммов 81гер(отусез дпзеиз или
любыми другими способами. Могут добавляться ста¬
билизаторы.
Активность: не менее 720 МЕ/мг (в пересчете
на сухое вещество).
ПРОИЗВОДСТВО
При производстве используют методы, позволя¬
ющие уменьшить либо удалить вещества, снижаю¬
щие кровяное давление.
Метод производства должен быть отвалидиро-
ван, чтобы при испытании субстанция отвечала сле¬
дующему требованию.
Аномальная токсичность (2.6.9). Испытуемый
образец должен быть нетоксичным. Тест-доза — 1 мг
испытуемого образца в 0,5 мл воды для инъекций Р
вводят в каждую мышь.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Очень легко растворим в воде, практически не¬
растворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (а). 10 мг ФСО стрепто¬
мицина сульфата растворяют в воде Р и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО канамицина
моносульфата, 10 мг ФСО неомицина сульфата и
10 мг ФСО стрептомицина сульфата растворяют в
воде Р и доводят до объема 10 мл этим же раство¬
рителем.
Пластинка: ТСХ пластинка, покрытая слоем
0,75 мм смеси следующего состава: 0,3 г карбомера Р
смешивают с 240 мл воды Р и выдерживают в тече¬
ние 1 ч при периодическом встряхивании; доводят до
рН 7 последовательными прибавлениями раствора
натрия гидроксида разведенного Р при постоянном
встряхивании и прибавляют 30 г силикагеля Н Р. Пла¬
стинку нагревают при температуре 110 °С в течение
1 ч, охлаждают и немедленно используют.
Подвижная фаза: раствор 70 г/л калия дигидро¬
фосфата Р.
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку опрыскивают смесью из
равных объемов раствора 2 г/л 1,3-дигидроксинаф-
талина Р в 96% спирте Р и раствора 460 г/л кисло¬
ты серной Р. Нагревают при температуре 150 °С в
течение (5—10) мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются три полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
B. (5—10) мг испытуемого образца растворяют в
4 мл воды Р, прибавляют 1 мл 1 М раствора натрия
гидроксида и нагревают на водяной бане в течение
4 мин. Прибавляют незначительный избыток кислоты
хлористоводородной разведенной Р и 0,1 мл раст¬
вора железа (III) хлорида Р1. Появляется фиолетовое
окрашивание.
C. 0,1 испытуемого образца растворяют в 2 мл
воды Р, прибавляют 1 мл раствора а-нафтола Р и
2 мл смеси из равных объемов раствора натрия ги¬
похлорита концентрированного Р и воды Р. Появля¬
ется красное окрашивание.
й. Около 10 мг испытуемого образца растворяют
в воде Р и прибавляют 1 мл 1 М раствора кислоты
хлористоводородной. Нагревают в водяной бане
в течение 2 мин. Прибавляют 2 мл раствора 5 г/л
а-нафтола Р в 1 М растворе натрия гидроксида и
нагревают в водяной бане в течение 1 мин. Появляет¬
ся слабое желтое окрашивание.
Е. Испытуемый образец дает реакции на сульфа¬
ты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 10 мл этим же растворителем.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее третьего эталона шкалы
наиболее подходящего цвета.
Прозрачность (2.2.1). Раствор 5 выдерживают в
защищенном от света месте при температуре около
20 °С в течение 24 ч. Раствор 3 по степени мутности
не должен превышать эталон II.
рН (2.2.3). От 4,5 до 7,0. Измеряют рН раствора 3.
Метанол. Газовая хроматография (2.2.28).
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
25,0 мл этим же растворителем.
Раствор сравнения. 12,0 мг метанола Р дово¬
дят водой Р до объема 100 мл.
Условия хроматографирования:
- колонка длиной (1,5—2,0) м и внутренним диа¬
метром (2—4) мм, заполненная слоем сополимером
этилвинилбензол-дивинилбензола Р (толщина слоя
(150—180) мкм);
- газ-носитель: азот для хроматографии Р\
- скорость газа-носителя: (30—40) мл/мин;
- температура колонки: должна быть постоян¬
ной в диапазоне от 120 °С до 140 °С;
- температура блока ввода проб и детекто¬
ра: должна быть выше, чем температура колонки, по
крайней мере на 50 °С;
- детектор: пламенно-ионизационный.
Предельное содержание метанола (не более
0,3 %); на хроматограмме испытуемого раствора пло¬
щадь пика, соответствующего метанолу, не должна
превышать площадь пика на хроматограмме раст¬
вора сравнения.
Стрептомицин В. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,2 г испытуемого об¬
разца растворяют в свежеприготовленной смеси из
3 объемов кислоты серной Р и 97 объемов метано-
918
Государственная фармакопея Республики Беларусь
лаР\л доводят до объема 5 мл этой же смесью раст¬
ворителей. Нагревают с обратным холодильником в
течение 1 ч, охлаждают, ополаскивают холодильник
метанолом Р и доводят до объема 20 мл этим же
растворителем (раствор 10 г/л).
Раствор сравнения. 36 мг маннозы Р раство¬
ряют в свежеприготовленной смеси из 3 объемов
кислоты серной Р и 97 объемов метанола Р и дово¬
дят до объема 5 мл этой же смесью растворителей.
Нагревают с обратным холодильником в течение 1 ч,
охлаждают, ополаскивают холодильник метанолом Р
и доводят до объема 50 мл этим же растворителем.
5 мл полученного раствора доводят до объема 50 мл
метанолом Р (раствор 0,3 г/л в пересчете на стрепто¬
мицин В; 1 мг маннозы Р соответствует 4,13 мг стреп¬
томицина В).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р.
Подвижная фаза: кислота уксусная ледяная Р—
метанол Р — толуол Р (25:25:50, об/об/об).
Наносимый объем пробы: 10-мкл.
Фронт подвижной фазы: (13—15) см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают свежепри¬
готовленной смесью из равных объемов раствора
2 г/л 1,3-дигидроксинафталина Р в 96% спирте Р и
20 % (об/об) раствора кислоты серной Р. Нагревают
при температуре 110 °С в течение 5 мин.
Предельное содержание стрептомицина В (не
более 3,0 %):
- на хроматограмме испытуемого раствора пят¬
но, соответствующее стрептомицину В, должно быть
не интенсивнее пятна на хроматограмме раствора
сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 1,000 г испытуемого образца сушат при
температуре 60 °С над фосфора (V) оксидом Р при
давлении не более 0,1 кПа в течение 24 ч.
Сульфатная зола (2.4.14). Не более 1,0 %. Опре¬
деление проводят из 1,000 г испытуемого образца.
Сульфаты. Не менее 18,0 % и не более 21,5 %
сульфата (504) (в пересчете на сухое вещество).
0,250 г испытуемого образца растворяют в 100 мл
воды Р и доводят до рН 11 раствором аммиака кон¬
центрированным Р. Прибавляют 10,0 мл 0,1 М раст¬
вора бария хлорида и около 0,5 мг фталеинового
пурпурного Р. Титруют 0,1 М раствором натрия эде-
тата, прибавляя 50 мл 96 % спирта Р, когда окраши¬
вание раствора начинает изменяться, и продолжают
титрование до появления фиолетового окрашивания.
1 мл 0,1 М раствора бария хлорида соответству¬
ет 9,606 мг сульфата (804).
Колориметрическое испытание. Испытуемый
образец и ФСО стрептомицина сульфата сушат при
температуре 60 °С над фосфора (V) оксидом Р при
давлении не более 0,1 кПа в течение 24 ч. 0,100 г вы¬
сушенного испытуемого образца растворяют в воде Р
и доводят до объема 100,0 мл этим же растворите¬
лем. Раствор сравнения готовят таким же образом,
используя 0,100 г высушенного ФСО стрептомицина
сульфата. По 5,0 мл каждого раствора помещают в
две пробирки, а в третью пробирку помещают 5 мл
воды Р. В каждую пробирку прибавляют 5,0 мл 0,2 М
раствора натрия гидроксида и нагревают в водяной
бане точно в течение 10 мин. Охлаждают в ледяной
бане точно в течение 5 мин, прибавляют 3 мл раст¬
вора 15 г/л железа (III) аммония сульфата Р в 0,5 М
растворе кислоты серной, доводят водой Р до объ¬
ема 25,0 мл и перемешивают. Точно через 20 мин по¬
сле прибавления раствора железа аммония сульфата
измеряют оптическую плотность (2.2.25) испытуемого
раствора и раствора сравнения в кювете с толщиной
слоя 2 см при длине волны 525 нм. В качестве ком¬
пенсационного раствора используют раствор, приго¬
товленный из 5 мл воды Р. Оптическая плотность ис¬
пытуемого раствора должна быть не менее 90,0 % от
оптической плотности раствора сравнения.
Бактериальные эндотоксины (2.6.14). Не бо¬
лее 0,25 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без дальнейшей подходящей
процедуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят количественное определение антибио¬
тиков микробиологическим методом (2.7.2).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Стерильная субстанция — в стерильном, возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
07/2016:0248
СУКСАМЕТОНИЯ ХЛОРИД
ЗихатеПюпН сЫопс1ит
ЗиХАМЕТНОИШМ СШОПЮЕ
о сн3
С14Н30С12М2О4 ■ 2НгО М.м. 397,3
[6101-15-1]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 102,0 %
2,2-[бутандиолбис(окси)]бис(А/,А/,Л/-триметилэтанамина)
дихлорид (в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Легко растворим в воде, мало растворим в 96 %
спирте.
Температура плавления: около 160 °С без пред¬
варительной сушки.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО суксаметония хлорида.
B. К 1 мл раствора 8, приготовленного как указано
в разделе «Испытания», прибавляют 9 мл воды Р, 10 мл
Сульпирид
919
кислоты серной разведенной Р и 30 мл раствора ам¬
мония рейнеката Р. Образуется розовый осадок. Вы¬
держивают в течение 30 мин, фильтруют, промывают
водой Р, 96 % спиртом Р и затем эфиром Р и сушат при
температуре 80 °С. Температура плавления (2.2.14) по¬
лученного осадка должна быть от 180 °С до 185 °С.
С. 25 мг испытуемого образца растворяют в 1 мл
воды Р и прибавляют 0,1 мл раствора 10 г/л кобальта
хлорида Р и 0,1 мл раствора калия ферроцианида Р.
Появляется зеленое окрашивание.
О. Около 20 мг испытуемого образца дают реак¬
цию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). 4 мл раствор 3 до¬
водят водой Р до объема 10 мл. Полученный раствор
должен быть бесцветным.
рН (2.2.3). От 4,0 до 5,0. 1 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объема
10 мл.
Холина хлорид. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,4 г испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 0,4 г ФСО суксаметония
хлорида и 2 мг холина хлорида Р растворяют в мета¬
ноле Р и доводят до объема 10 мл этим же раство¬
рителем.
Пластинка: ТСХ пластинка со слоем целлюлозы
для хроматографии Р1.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — бутанол Р (10:40:50, об/об/об).
Смесь встряхивают в течение 10 мин, выдерживают и
используют верхний слой.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии страта.
Высушивание: в потоке воздуха.
Проявление: пластинку опрыскивают раствором
калия йодовисмутата Р.
Пригодность хроматографической системы:
раствор сравнения:
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- холина хлорид (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна, со¬
ответствующего холина хлориду, на хроматограмме
раствора сравнения.
Вода (2.5.12). Не менее 8,0 % и не более 10,0 %.
Определение проводят из 0,30 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
уксусного ангидрида Р. Титруют 0,1 М раствором
кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 18,07 мг С14Н30С12М2О4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:1045
СУЛЬПИРИД
8и1рИс1ит
31Л.Р1МОЕ
С15Н23М3045 М.м. 341,4
[15676-16-1]
ОПРЕДЕЛЕНИЕ
(#5)-Л/-[(1-Этилпирралидин-2-ил)метил]-2-метокси-
5-сульфамоилбензамид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, умеренно рас¬
творим в метаноле, мало растворим в 96 % спирте и
в метиленхлориде. Растворяется в разведенных рас¬
творах минеральных кислот и гидроксидов щелочных
металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 177 °С до
181 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сульпирида #или спектр, пред¬
ставленный на рисунке #1045.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Примесь А».
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: основное пятно на хроматограмме
испытуемого раствора (Ь) соответствует по располо¬
жению и размеру основному пятну на хроматограмме
раствора сравнения (а).
О. В фарфоровую чашку помещают около 1 мг
испытуемого образца, прибавляют 0,5 мл кислоты
серной Р и 0,05 мл раствора формальдегида Р. При
просматривании в ультрафиолетовом свете при длине
волны 365 нм раствор имеет синюю флуоресценцию.
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в кислоте уксусной разведенной Р и доводят до
объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
920
Гэсударственная фармакопея Республики Беларусь
Рисунок #1045.-1. Инфракрасный спектр ФСО сульпирида
Цветность (2.2.2, метод /). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
СОПУТСТВУЮЩИЕ ПРИМЕСИ
Примесь А. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор (а). 0,20 г испытуемого
образца растворяют в метаноле Р, обрабатывают
ультразвуком до полного растворения и доводят до
объема 10,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемо¬
го раствора (а) доводят метанолом Р до объема
10,0мл.
Раствор сравнения (а). 20 мг ФСО сульпири¬
да растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО сульпирида
примеси А растворяют в метаноле Р и доводят до
объема 25,0 мл этим же растворителем. 1,0 мл полу¬
ченного раствора доводят метанолом Р до объема
10,0мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ляГ254 Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — диоксан Р — метанол Р — метилен-
хлорид Р (2:10:14:90, об/об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы, не менее 1/2 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм для
проведения подлинности С, затем опрыскивают рас¬
твором нингидрина Р; нагревают при температуре от
100 °С до 105 °С в течение 15 мин и просматривают
при дневном свете.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора (а) пятно, соответствующее
примеси А, должно быть не интенсивнее соответ¬
ствующего пятна на хроматограмме раствора срав¬
нения (Ь).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 5 мг ФСО сульпирида примеси В растворяют в
подвижной фазе и доводят до объема 50,0 мл этим
же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р (раз¬
мер частиц 5 мкм);
-подвижная фаза: ацетонитрил Р — мета¬
нол Р — раствор, содержащий 6,8 г/л калия диги¬
дрофосфата Р и 1 г/л натрия октансульфоната Р,
предварительно доведенный кислотой фосфорной Р
до рН 3,3 (10:10:80, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 240 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания сульпирида.
Идентификация пиков примесей: идентифици¬
руют пик примеси В, используя хроматограмму раст¬
вора сравнения (Ь).
Сультамициллина тозилат дигидрат
921
Относительное удерживание (по отношению
к сульпириду, время удерживания — около 15 мин):
примесь В — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками при¬
меси В и сульпирида.
Расчет процентного содержания:
- для каждой примеси — используют концентра¬
цию сульпирида в растворе сравнения (а).
Предельное содержание примесей:
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,3 %;
- учитываемый предел: 0,05 %.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
1,0 г испытуемого образца встряхивают с 20 мл
воды Р, фильтруют через стеклянный фильтр (40)
(2.1.2). К 10 мл фильтрата прибавляют 5 мл воды Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца сжигают в кварцевом тигле. К
полученному остатку прибавляют 1 мл 1 М раствора
кислоты хлористоводородной, 3 мл воды Р и 0,1 мл
кислоты азотной Р. Нагревают на водяной бане в
течение около 5 минут. Раствор помещают в пробир¬
ку. Тигель промывают 4 мл воды Р. Промывные воды
собирают в пробирку и доводят водой Р до объема
10 мл.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 80 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 34,14 мг С15Н23М3045.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацев¬
тического использования (2034). Вследствие этого
нет необходимости идентифицировать эти примеси
для доказательства соответствия требованиям. См.
также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): В, С,
О, Е, Р, в.
Н21Ч
Н' N
и энантиомер
СН3
А. [(2Р5)-1 -Этилпирролидин-2-ил]метанамин.
зоат.
зоат.
лота.
B. Р = 0-СН3: Метил-2-метокси-5-сульфамоилбен-
C. Р = 0-С2Н5: Этил-2-метокси-5-сульфамоилбен-
0. Р = ОН: 2-Метокси-5-сульфамоилбензойная кис-
а.
Е. Р = МН2: 2-Метокси-5-сульфамоилбензамид.
Р. 1 -Этил-2-[[(2-метокси-5-сульфамоилбензоил )-
амино]метил]пирролидин-1 -оксид.
о
н2м
N . ^ ,
Н / и энантиомер
^СН3
С. (Р5)-А/-[(1-Этилпирролидин-2-ил)метил]-2-гидро-
кси-5-сульфамоилбензамид.
07/2016:2212
СУЛЬТАМИЦИЛЛИНА ТОЗИЛАТ
ДИГИДРАТ
5иНатюННпИозНаз (ИЬудпсиз
81Л.ТАМ1С1ШЫ ТО8ПАТЕ О/НУОКАТЕ
С25Н30М4О982 ■ С7Н8035 ■ 2Н20 М.м. 803
ОПРЕДЕЛЕНИЕ
[(25,5/?,6/?)-6-[[(2/?)-Аминофенилацетил]амино]-
3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]гептан-
2-карбоксил][(25,5Р)-3,3-диметил-4,4,7-триоксо-4А6-
тиа-1-азабицикло[3.2.0]гептан-2-карбоксил]метилена
4-метилбензенсульфонат дигидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
116. Зак. 1060.
922
Гэсударствённая фармакопея Республики Беларусь
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ПРОИЗВОДСТВО
Необходимо учитывать, что алкилтолуолсульфо-
наты являются генотоксичными соединениями и могут
присутствовать в качестве примесей в сультамицил-
лина тозилате. При разработке процесса производ¬
ства необходимо соблюдать принципы управления
рисками качества, принимая во внимание качество
исходных материалов, мощность производственного
процесса и валидацию. Производитель может исполь¬
зовать метод, описанный в статье 2.5.40. Метил-,
этил- и изпропилтолуолсульфонат в фармацевти¬
ческих субстанциях.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, умеренно рас¬
творим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сультамициллина тозилата
#или спектр, представленный на рисунке #2212.-1#.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+178 до +195 (в пересчете на безводное вещество).
1,000 г испытуемого образца растворяют в диметил-
формамиде Р и доводят до объема 50,0 мл этим же
растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием или хранят
при температуре от 2 °С до 8 °С в течение не
более 6 ч.
Раствор А. Метанол Р1 — ацетонитрил Р1
(20:80, об/об).
Раствор В. 1,56 г натрия дигидрофосфата Р
растворяют в 900 мл воды Р, прибавляют 7,0 мл кис¬
лоты фосфорной Р и доводят водой Р до объема
1000 мл.
Контрольный раствор. Раствор В — раствор А
(30:70, об/об).
Испытуемый раствор. 70,0 мг испытуемого об¬
разца растворяют в 35 мл раствора А, обрабатыва¬
ют ультразвуком в течение около 1 мин, прибавляют
13 мл раствора В, перемешивают, обрабатывают уль¬
тразвуком в течение около 1 мин, доводят раствором
В до объема 50,0 мл и перемешивают.
Раствор сравнения (а). 70,0 мг ФСО сультами¬
циллина тозилата растворяют в 35 мл раствора А,
обрабатывают ультразвуком в течение около 1 мин,
прибавляют 13 мл раствора В, перемешивают, обра¬
батывают ультразвуком в течение около 1 мин, дово¬
дят раствором В до объема 50,0 мл и перемешивают.
Раствор сравнения (Ь). 15 мг испытуемого об¬
разца суспендируют в 20 мл раствора 0,4 г/л натрия
гидроксида Р и обрабатывают ультразвуком в ультра¬
звуковой бане в течение около 5 мин. Прибавляют
20 мл раствора 0,36 г/л кислоты хлористоводород¬
ной Р и доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). 0,200 г испытуемого об¬
разца растворяют в 70,0 мл раствора А, обрабатыва¬
ют ультразвуком в течение около 1 мин, прибавляют
25.0 мл раствора В, перемешивают, обрабатывают
ультразвуком в течение около 1 мин, доводят объем
раствора раствором В до 100,0 мл и перемешивают.
1.0 мл полученного раствора доводят контрольным
раствором до объема 100,0 мл.
Раствор сравнения (ф. 32,3 мг ФСО ампицил¬
лина тригидрата (примесь В) и 7,0 мг ФСО сульбак-
тама (примесь А) растворяют в воде Р и доводят до
объема 1000 мл этим же растворителем.
Рисунок #2212.-1. Инфракрасный спектр ФСО сультамициллина тозилата
Сультамициллина тозилат дигидрат
923
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
3,5 мкм;
- температура: 25 °С;
- подвижная фаза:
- подвижная фаза А: раствор 4,68 г/л натрия
дигидрофосфата Р, доведенного кислотой
фосфорной Р до рН 3,0;
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
95 —30
5 — 70
15—16
30
70
16—16,5
30 — 95
70 — 5
16,5—20
95
5
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: по 5 мкл контрольного
раствора, испытуемого раствора и растворов сравне¬
ния (Ь), (с) и (с1).
Относительное удерживания (по отношению
к сультамициллину, время удерживания — около
9,3 мин): примесь А — около 0,41; пеницилловая кис¬
лота ампициллина — около 0,47; тозилат — около 0,50;
примесь В — около 0,55; примесь С — около 0,94; при¬
месь й — около 1,09; примесь Р — около 1,23; при¬
месь Е — около 1,26; примесь 6 — около 1,42.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками пени-
цилловой кислоты ампициллина и тозилата и не ме¬
нее 2,5 между пиками тозилата и примеси В.
Предельное содержание примесей:
- примесь В (не более 2,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (б);
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с!);
- примеси С, О, Е, Р, О (не более 0,5 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям С, О, Е, Р, С, не должны
превышать 0,5 площади пика сультамициллина на
хроматограмме раствора сравнения (с);
-любая другая примесь (не более 0,5 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного пика и пиков примесей А, В, С, О,
Е, Р, С, не должна превышать 0,5 площади пика сульта¬
мициллина на хроматограмме раствора сравнения (с);
- сумма примесей (не более 4,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 4-крат-
ную площадь пика сультамициллина на хроматограм¬
ме раствора сравнения (с);
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики,
площадь которых составляет менее 0,1 площади пика
сультамициллина на хроматограмме раствора срав¬
нения (с).
Этилацетат. Парофазная газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в 7,0 мл смеси из воды Р и диме-
тилформамида Р (1:99, об/об).
Раствор сравнения. 0,200 г этилацетата Р
растворяют в 240 мл смеси из воды Р и диметилфор-
мамида Р (1:99, об/об) и доводят до объема 250,0 мл
этим же растворителем. 5,0 мл полученного раствора
доводят смесью из воды Р и диметилформамида Р
(1:99, об/об) до объема 7,0 мл.
Флаконы немедленно плотно закрывают рези¬
новыми пробками, покрытыми политетрафторэ¬
тиленом, и обжимают алюминиевыми колпачками.
Встряхивают до получения однородного раствора.
Условия хроматографирования:
- колонка: капиллярная кварцевая длиной 50 м
и внутренним диаметром 0,32 мм, покрытая слоем
поли(диметил)силоксана Р толщиной от 1,8 мкм до
3 мкм;
- газ-носитель: гелий для хроматографии Р;
- линейная скорость газа-носителя: 35 см/с;
- деление потока: 1:5.
- параметры парофазного пробоотборника:
- равновесная температура: 105 °С,
- время достижения равновесия: 45 мин,
- температура линии подачи газовой пробы:
110*0,
- время нахождения под давлением: 30 с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—6
70
6—16
70 - 220
16—18
220
Блок ввода проб
140
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мл.
Относительное удерживание (по отношению к
диметилформамиду, время удерживания — около
14 мин): этилацетат — около 0,7.
Предельное содержание:
- этилацетат: не более 2,0 %
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррлп). 2,0 г испытуемого образца раст¬
воряют в смеси из метанола Р и ацетонитрила Р
(40:60, об/об) и доводят до объема 20,0 мл этой же
смесью растворителей. 12 мл полученного раст¬
вора должны выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием эта¬
лонного раствора свинца (2 ррт РЬ), полученного
путем разведения эталонного раствора свинца
(100 ррт РЬ) смесью из метанола Р и ацетони¬
трила Р (40:60, об/об).
Вода (2.5.12). Не менее 4,0 % и не более 6,0 %.
Определение проводят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
116*. Зак. 1060.
924
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
- объем вводимой пробы: по 5 мкл испытуемого
раствора и раствора сравнения (а).
Содержание сультамициллина тозилата
(С25Н30М4О952 С7Н8О35) рассчитывают в процентах с
учетом содержания сультамициллина тозилата в ФСО
сультамициллина тозилата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Г, 6.
А. (25,5Я)-3,3-Диметил-7-оксо-4-тиа-1-азаби-
цикло[3.2.0]гептан-2-карбоновой кислоты 4,4-диоксид
(сульбактам).
В. (25,5Я,6Я)-6-[[(2Я)-2-Амино-2-фенилацетил]-
амино]-3,3-диметил-7-оксо-4-тиа-1-азабицикло[3.2.0]-
гептан-2-карбоновая кислота (ампициллин)
С. [[(2Я)-Аминофенилацетил]амино][(4Я)-4-
[[[[[(25,5#)-3,3-диметил-4,4,7-триоксо-4А6-тиа-1-
азабицик-ло[3.2.0]гепт-2-ил]карбонил]окси]метокси]-
карбонил]-5,5-диметилтиазолидин-2-ил]уксусная кис¬
лота (пеницилловые кислоты сультамициллина).
О. [(25,5#,6Я)-3,3-Диметил-6-[[(2Я)-[(1 -метил-4-
оксопентилиден)амино]фенилацетил]амино]-7-оксо-4-
тиа-1-азабицикло[3.2.0]гептан-2-карбоксил][(25,5Я)-
3,3-диметил-7-оксо-4-окса-1-азабицикло[3.2.0]гептан-
2-карбоксил]метилен.
Е. Метиленбис[(25,5Я)-3,3-диметил-4,4,7-триоксо-
4Л6-тиа-1 -азабицикло[3.2.0]гептан-2-карбоксилат] (ме¬
тиленовый эфир сульбактама).
Р. [(25|5Я,6Я)-6-[[(2Я)-[[[(25,5Я6Я)-6-[[(2Я)-Ами-
нофенилацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-
азабицикло[3.2.0]гепт-2-ил]карбонил]амино]фенилаце-
тил]амино]-3,3-диметил-7-оксо-4-тиа-1-азабицик-
ло[3.2.0]гептан-2-карбоксил][(25,5Я)-3,3-диметил-4,4,7-
триоксо-4А6-тиа-1-азабицикло[3.2.0]гептан-2-карбоксил]-
метилен (ампициллинсультамициллинамид).
О. [(25,5Я,6/?)-6-[[(2ЯН[[[(2Я)-Аминофенилацетил]-
амино][(45)-4-[[[[[(25,57?)-3,3-диметил-4,4,7-триоксо-4Л6-
тиа-1-азабицикло[3.2.0]гепт-2-ил]карбонил]окси]меток-
си]карбонил]-5,5-диметилтиазолидин-2-ил]ацетил]ами-
но]фенилацетил]амино]-3,3-диметил-7-оксо-4-тиа-1-
азабицикло[3.2.0]гептан-2-карбоксил][(25,57?)-3,3-диме-
тил-4,4,7-триоксо-4А6-тиа-1-азабицикло[3.2.0]гептан-2-
карбоксил]метилен (димер сультамициллина).
07/2016:1476
СУЛЬФАГУАНИДИН
ЗиНадиапШ'тит
5(Л.РА6(7А/УЮ/Л/Е
С7Н10Ы4О25 М.м. 214,3
[57-67-0]
ОПРЕДЕЛЕНИЕ
(4-Аминофенилсульфонил)гуанидин.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый мелкокристаллический
порошок.
Очень мало растворим в воде и в 96 % спирте,
мало растворим в ацетоне, практически нерастворим
Сульфагуанидин
925
в метиленхлориде. Растворяется в разведенных рас¬
творах минеральных кислот.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О, Е.
A. Температура плавления (2.2.14): от 189 °С до
193 °С. Определение проводят из высушенного испы¬
туемого образца.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сульфагуанидина #или спектр,
представленный на рисунке #147б.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
й. 5 мг испытуемого образца растворяют в 10 мл
1 М раствора хлористоводородной кислоты. 1 мл
полученного раствора доводят водой Р до объема
10мл. Полученный раствор без дальнейшего под¬
кисления дает реакцию на первичные ароматические
амины (2.3.1).
Е. 0,1 г испытуемого образца растворяют в 2 мл
воды Р, прибавляют 1 мл раствора а-нафтола Р и
2 мл смеси из равных объемов воды Р и раствора
натрия гипохлорита концентрированного Р. Появ¬
ляется красное окрашивание.
ИСПЫТАНИЯ
Раствор 3. К 2,5 г испытуемого образца прибав¬
ляют 40 мл воды, свободной от углерода диоксида, Р,
нагревают при температуре около 70 °С в течение
5 мин, охлаждают при взбалтывании в ледяной воде
в течение около 15 мин, фильтруют и доводят водой,
свободной от углерода диоксида, Р до объема 50 мл.
Кислотность. К 20 мл раствора 3 прибавляют
0,1мл раствора бромтимолового синего Р1. При
прибавлении не более 0,2 мл 0,1 М раствора натрия
гидроксида окраска раствора должна измениться.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 50 мг испытуемого об¬
разца растворяют'в ацетоне Р и доводят до объема
5 мл этим же растворителем.
Испытуемый раствор (Ь). 2 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО сульфагуани¬
дина растворяют в ацетоне Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят до объема 200 мл ацетоном Р.
Раствор сравнения (с). 5 мл раствора сравнения
(Ь) доводят до объема 10 мл ацетоном Р.
Раствор сравнения (ф. 10 мг сульфаниламида Р
растворяют в испытуемом растворе (Ь) и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля СГ254 Р.
Подвижная фаза: кислота муравьиная безвод¬
ная Р — метанол Р — метиленхлорид Р (10:20:70,
об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (й):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь: на хроматограмме испытуемо¬
го раствора (а) любое пятно, кроме основного, должно
Рисунок #1476.-1. Инфракрасный спектр ФСО сульфагуанидина
117. Зак. 1060.
926
Гэсударственная фармакопея Республики Беларусь
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения (Ь) (0,5 %) и не более одного из таких
пятен может быть интенсивнее пятна на хроматограм¬
ме раствора сравнения (с) (0,25 %).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 8,0 %. 1,000 г сушат при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,175 г испытуемого образца растворяют в 50 мл
кислоты хлористоводородной разведенной Р и ох¬
лаждают раствор на ледяной бане. Проводят опре¬
деление аминного азота в первичной ароматической
аминогруппе (2.5.8). Конечную точку титрования опре¬
деляют электрометрическим методом.
1 мл 0,1 М раствора натрия нитрита соответ¬
ствует 21,42 мг С7Н10М4О25.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
V
'МН,
Н2Ы‘
А. 4-Аминобензолсульфонамид (сульфаниламид).
V
и
л.
N42
Н2М
В. Л/-[(4-Аминофенил)сульфонил]мочевина (суль-
факарбамид).
07/2016:0108
СУЛЬФАМЕТОКСАЗОЛ
8и1?атеиюхаго1ит
5Ш.ЕАМЕГНОХА20[.Е
[723-46-6]
ОПРЕДЕЛЕНИЕ
4-Амино-А/-(5-метилизоксазол-3-ил)бензолсуль-
фонамид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический по¬
рошок.
Рисунок #0108.-1. Инфракрасный спектр ФСО сульфаметоксазола
Сульфаметоксазол
927
Практически нерастворим в воде, легко раство¬
рим в ацетоне, умеренно растворим 96 % спирте. Рас¬
творяется в разведенном растворе натрия гидроксида
и в разведенных кислотах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 169 °С до
172 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сульфаметоксазола #или
спектр, представленный на рисунке #0108.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 3 мл смеси из раствора аммиака
концентрированного Р и метанола Р (2:48, об/об) и
доводят до объема 5 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО сульфаметок¬
сазола растворяют в 3 мл смеси из раствора ам¬
миака концентрированного Р и метанола Р (2:48,
об/об) и доводят до объема 5 мл этой же смесью
растворителей.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: раствор аммиака разведен¬
ный Р1 — вода Р — нитрометан Р — диоксан Р
(3:5:41:51, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: сушат при температуре от 100 °С
до 105 °С.
Проявление: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
О. 5 мг испытуемого образца растворяют в 10 мл
1 М раствора кислоты хлористоводородной. 1 мл
полученного раствора доводят водой Р до объема
10мл. Полученный раствор без дальнейшего под¬
кисления дает реакцию на первичные ароматические
амины (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в смеси из 5 мл раствора натрия гидроксида
разведенного Р и 5 мл воды Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)5, ВУ(КЖ)5
или ОУ(ЗЖ)5.
Кислотность. К 1,25 г тонкоизмельченного ис¬
пытуемого образца прибавляют 25 мл воды Р и на¬
гревают при температуре 70 °С в течение 5 мин.
Охлаждают в ледяной воде в течение около 15 мин
и фильтруют. К 20 мл фильтрата прибавляют 0,1 мл
раствора бромтимолового синего Р1. При при¬
бавлении не более 0,3 мл 0,1 М раствора натрия
гидроксида окрашивание раствора должно изме¬
ниться.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 45 мл подвижной фазы при помо¬
щи ультразвука при температуре около 45 °С в тече¬
ние 10 мин, охлаждают и доводят до объема 50,0 мл
этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 10,0мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (Ь). 1 мг испытуемого образ¬
ца и 1 мг ФСО сульфаметоксазола примеси А раство¬
ряют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (с). 1,0 мг ФСО сульфаме¬
токсазола примеси Р растворяют в подвижной фазе
и доводят до объема 10,0 мл этим же растворителем.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: смешивают 35 объемов ме¬
танола Р2 и 65 объемов раствора 13,6 г/л калия ди¬
гидрофосфата Р, предварительно доведенного до
рН 5,3 раствором 20 г/л калия гидроксида Р\
- скорость подвижной фазы: 0,9 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания сульфаметоксазола.
Относительное удерживание (по отношению
к сульфаметоксазолу, время удерживания — около
10 мин): примесь О — около 0,3; примесь Е — около
0,35; примесь Р — около 0,45; примесь С — около 0,5;
примесь А — около 1,2; примесь В — около 2,0.
Условия хроматографирования: раствор срав¬
нения (Ь):
-разрешение: не менее 3,5 между пиками суль¬
фаметоксазола и примеси А.
Предельное содержание примесей:
- примеси А, В, С, О, Е (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь пиков,
соответствующих примесям А, В, С, О и Е, не должны
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- примесь Р (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси Р, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (с);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С, О, Е
и Р, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,025 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
117*. Зак. 1060.
928
Гэсударственная фармакопея Республики Беларусь
Тяжелые металлы (2.4.8, метод О). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
С. 5-Метилизоксазол-З-амин.
О. Р = ОН: 4-Аминобензолсульфоновая кислота
(сульфаниловая кислота).
Е. Р = МН2:4-Аминобензолсульфонамид (сульфа¬
ниламид).
Проводят определение аминного азота в первич¬
ной ароматической аминогруппе (2.5.8), используя
0,200 г испытуемого образца. Конечную точку титро¬
вания определяют электрометрическим методом.
1 мл 0,1 М раствора натрия нитрита соответ¬
ствует 25,33 мг С^Н^ЫзОзЗ.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
A. Р = СО-СН3: Л/-[4-[(5-Метилизоксазол-3-ил)суль-
фамоил]фенил]ацетамид.
B. Р = 502-С6Н4-л1ЧН2:4-[[(4-Аминофенил)сульфо-
нил]амино]-Л/-(5-метилизоксазол-3-ил)бензолсульфо-
намид.
Р. 4-Амино-А/-(3-метилизоксазол-5-ил)бензолсуль-
фонамид.
07/2016:1571
СУЛЬФАНИЛАМИД
ВиНапИатШит
81И-РАЫИАМЮЕ
Свн,м2023 М.м. 172,2
[63-74-1]
Рисунок #1571.-1. Инфракрасный спектр ФСО сульфаниламида
Сульфацетамид натрия
929
ОПРЕДЕЛЕНИЕ
4-Аминобензолсульфонамид. #Стрептоцид#.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белые или желтовато-белые кристаллы или мел¬
кий порошок.
Мало растворим в воде, легко растворим в аце¬
тоне, умеренно растворим в 96 % спирте, практически
нерастворим в метиленхлориде. Растворяется в рас¬
творах гидроксидов щелочных металлов и разведен¬
ных минеральных кислотах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 164,5 °С
до 166,0 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО сульфаниламида #или спектр,
представленный на рисунке #1571.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (а) обнаруживается пятно, соответствующее
по расположению и размеру основному пятну на хро¬
матограмме раствора сравнения (а).
0. 5 мг испытуемого образца растворяют в 10 мл
1 М раствора кислоты хлористоводородной. 1 мл
полученного раствора доводят водой Р до объема
10мл. Полученный раствор без дальнейшего под¬
кисления дает реакцию на первичные ароматические
амины (2.3.1).
ИСПЫТАНИЯ
Раствор 5. К 2,5 г испытуемого образца при¬
бавляют 50 мл воды, свободной от углерода диок¬
сида, Р, нагревают при температуре 70 °С в течение
около 5 мин, охлаждают в ледяной воде в течение
около 15 мин и фильтруют.
Кислотность. К 20 мл раствора 5 прибавляют
0,1 мл раствора бромтимолового синего Р1. При при¬
бавлении не более 0,2 мл 0,1 М раствора натрия ги¬
дроксида окрашивание раствора должно измениться.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 20 мг испытуемого об¬
разца растворяют в 3 мл смеси из 2 объемов раст¬
вора аммиака концентрированного Р и 48 объемов
метанола Р и доводят до объема 5 мл этой же сме¬
сью растворителей.
Испытуемый раствор (Ь). 0,10 г испытуемого
образца растворяют в 0,5 мл раствора аммиака кон¬
центрированного Р и доводят метанолом Р до объ¬
ема 5 мл (при необходимости аккуратно нагревают до
полного растворения).
Раствор сравнения (а). 20 мг ФСО сульфанила¬
мида растворяют в 3 мл смеси из 2 объемов раст¬
вора аммиака концентрированного Р и 48 объемов
метанола Р и доводят до объема 5 мл этой же сме¬
сью растворителей.
Раствор сравнения (Ь). 1,25 мл испытуемого
раствора (а) доводят смесью из 2 объемов раствора
аммиака концентрированного Р и 48 объемов мета¬
нола Р до объема 50 мл.
Раствор сравнения (с). 20 мг испытуемого об¬
разца и 20 мг ФСО сульфамеразина растворяют в
3 мл смеси из 2 объемов раствора аммиака концен¬
трированного Р и 48 объемов метанола Р и доводят
до объема 5 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР214Р
Подвижная фаза: раствор аммиака разведен¬
ный Р1 — вода Р — нитрометан Р — диоксан Р
(3:5:40:50, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два четко
разделенных пятна.
Предельное содержание примесей:
-любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (Ь) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят определение аминного азота в первич¬
ной ароматической аминогруппе (2.5.8), используя
0,140 г испытуемого образца. Конечную точку титро¬
вания определяют электрометрическим методом.
1 мл 0,1 М раствора натрия нитрита соответ¬
ствует 17,22 мг С6Н8М2023.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0107
СУЛЬФАЦЕТАМИД НАТРИЯ
ЗиНасе^тМит паМсит
5Ш.РАСЕТАМЮЕ ЗОйШМ
Н20
СаН(Ы2Ыа033 • Н20
М.м. 254,2
118. Зак. 1060.
930
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Натрия ацетил[(4-аминофенил)сульфонил]азанид
моногидрат. #Сульфацил натрия*.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или желтовато-белый кристаллический
порошок.
Легко растворим в воде, мало растворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е
Вторая идентификация: А, С, Е, Е
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,1 г испытуемого образ¬
ца растворяют в фосфатном буферном растворе рН
7.0 Р и доводят до объема 100,0 мл этим же раство¬
рителем. 1,0 мл полученного раствора доводят фос¬
фатным буферным раствором рН 7,0 Р до объема
100.0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 255 нм.
Удельный показатель поглощения в максимуме:
от 660 до 720 (в пересчете на безводное вещество).
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО сульфацетамида натрия * или
спектр, представленный на рисунке #0107.-1#.
C. Температура плавления (2.2.14): от 181 °С до
185 °С. 1 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют 6 мл кислоты уксусной разве¬
денной Р и фильтруют. Осадок промывают неболь¬
шим количеством воды Р и высушивают при темпера¬
туре от 100 °С до 105 °С в течение 4 ч.
Е. 1 мг осадка, полученного в идентификации С,
растворяют при нагревании в 1 мл воды Р. Получен¬
ный раствор дает реакцию на первичные ароматиче¬
ские амины (2.3.1) с образованием оранжево-красно¬
го осадка.
Р. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции (а) и (Ь) на натрий
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона <ЗУ(ЗЖ)4.
рН (2.2.3). От 8,0 до 9,5. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием, испытание прово¬
дят с защитой от света.
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО сульфацета¬
мида натрия и 5 мг сульфаниламида Р (примесь А)
растворяют в 1,0 мл подвижной фазы.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: кислота уксусная ледя¬
ная Р— метанол Р — вода для хроматографии Р
(1:10:89, об/об/об);
Рисунок #0107.-1. Инфракрасный спектр ФСО сульфацетамида натрия
Тадалафил
931
- скорость подвижной фазы: 0,8 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 7-кратное вре¬
мя удерживания сульфацетамида.
Относительное удерживание (по отношению к
сульфацетамиду, время удерживания — около 5 мин):
примесь А — около 0,5.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 5,0 между пиками при¬
меси А и сульфацетамида.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—0,5):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
-неучитываемый предел (0,05%): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
2,5 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 25 мл этим же
растворителем. Прибавляют 25 мл кислоты уксусной
разведенной Р, встряхивают в течение 30 мин и филь¬
труют. 15 мл фильтрата должны выдерживать испыта¬
ние на сульфаты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020% (20 ррт). 12 мл фильтрата, полученного в
испытании «Сульфаты», должны выдерживать испы¬
тание на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.72). Не менее 6,0 % и не более 8,0 %.
Определение проводят из 0,200 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.7.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г испытуемого образца растворяют в смеси
из 50 мл воды Р и 20 мл кислоты хлористоводород¬
ной разведенной Р. Полученный раствор охлаждают в
ледяной бане и проводят определение аминного азо¬
та в первичной ароматической аминогруппе (2.5.8).
Конечную точку титрования определяют электроме¬
трическим методом.
1 мл 0,1 М раствора натрия нитрита соответ¬
ствует 23,62 мг С8Н91\12Ма035.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
А. 4-Аминобензолсульфонамид (сульфаниламид).
B. Р = СО-СН3, Р' = Н: Л/-(4-Сульфамоилфенил)-
ацетамид.
C. Р = Р' = СО-СН3: Л/-[[4-(Ацетиламино)фенил]-
сульфонил]ацетамид.
О. 4,4-Сульфонилдианилин (дапсон).
07/2016:2606
ТАДАЛАФИЛ
Та6а!аТ\\ит
ТАОА1-АГП.
С22Н19М304 М.М. 389’4
[171596-29-5]
ОПРЕДЕЛЕНИЕ
(6Р, 72аРу)-6-(1,3-Бензодиоксол-5-ил)-2-метил-
2,3,6,7,12,12а-гексагйдропиразино[1,,2,:1,6]-пиридо-
[3,4-Ь]индол-1,4-дион.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
118*. Зак. 1060.
932
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в диметилсульфоксиде, мало растворим в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Проводят определение подлинности (идентифи¬
кацию) испытаниями А, В или А, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тадалафила #или спектр, пред¬
ставленный на рисунке #2606.-1#.
B. Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Примеси А, В и С», со следующими
изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
размеру соответствует основному пику на хромато¬
грамме раствора сравнения (а).
C. Удельное оптическое вращение (2.2.7): от
+78,0 до +84,0 (в пересчете на сухое вещество).
0,250 г испытуемого образца растворяют в диметил¬
сульфоксиде Р и доводят этим же растворителем до
объема 25,0 мл.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р1 — гек¬
сан Р — 2-пропанол Р1 (20:40:40, об/об/об).
Раствор А. 27 г тетрабутиламмония гидрокси¬
да Р растворяют в метаноле Р и доводят до объема
100,0 мл этим же растворителем.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (а). 25,0 мг ФСО тадалафи¬
ла растворяют в смеси растворителей и доводят до
объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). Для приготовления при¬
меси А /л зНи 25 мг испытуемого образца растворяют
в 40 мл смеси растворителей, прибавляют 1 мл раст¬
вора А, тщательно перемешивают и выдерживают в
течение 20 мин. К полученному раствору прибавляют
1 мл кислоты трифторуксусной Р и доводят смесью
растворителей до объема 100,0 мл.
Раствор сравнения (д). К 1,0 мл испытуемого
раствора прибавляют 1,0 мл раствора сравнения (с)
и доводят смесью растворителей до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем АО для хи¬
ральных разделений Р (размер частиц 10 мкм);
- температура: 30 °С;
-подвижная фаза: гексан Р — 2-пропанол Р1
(50:50, об/об);
- скорость подвижной фазы: 0,75 мл/мин;
-спектрофотометрический детектор, длина
волны 222 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (б);
- время хроматографирования: 2,2-кратное вре¬
мя удерживания тадалафила.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (б).
Рисунок #2606.-1. Инфракрасный спектр ФСО тадалафила
Тадалафил
933
Относительное удерживание (по отношению
к тадалафилу, время удерживания — около 11 мин):
примесь А — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 2,0 между пиками при¬
меси А и тадалафила.
Предельное содержание примесей:
-примесь А (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
1,5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси А, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). При приготовлении раство¬
ров не используется обработка ультразвуком.
Смесь растворителей. Ацетонитрил Р —
2-пропанол Р (50:50, об/об).
Раствор А. 27 г тетрабутиламмония гидрок¬
сида Р растворяют в метаноле Р и доводят этим
же растворителем до объема 100,0 мл.
Испытуемый раствор (а). 40 мг испытуемого
образца растворяют в 50 мл ацетонитрила Р и до¬
водят подвижной фазой А до объема 100,0 мл.
Испытуемый раствор (Ь). 50,0 мг испытуемо¬
го образца растворяют в 50 мл ацетонитрила Р и
доводят подвижной фазой А до объема 100,0 мл. К
10.0 мл полученного раствора прибавляют 25,0 мл
ацетонитрила Р и доводят подвижной фазой А до
объема 50,0 мл.
Раствор сравнения (а). К 1,0 мл испытуемого
раствора (а) прибавляют 50 мл ацетонитрила Р и
доводят подвижной фазой А до объема 100,0 мл.
К 1,0 мл полученного раствора прибавляют 5 мл
ацетонитрила Р и доводят подвижной фазой А до
объема 10,0 мл.
Раствор сравнения (Ь). Для приготовления
примеси А т зНи 4,0 мг испытуемого образца раст¬
воряют в 50 мл смеси растворителей, прибавляют
1 мл раствора А, перемешивают и выдерживают в
течение 40 мин. К полученному раствору прибав¬
ляют 1 мл кислоты трифторуксусной Р и доводят
смесью растворителей до объема 100,0 мл.
Раствор сравнения (с). 1 мл раствора сравне¬
ния (Ь) доводят испытуемым раствором (а) до объ¬
ема 50,0 мл.
Раствор сравнения (д). 50,0 мг ФСО тадала¬
фила растворяют в 50 мл ацетонитрила Р и до¬
водят подвижной фазой А до объема 100,0 мл. К
10.0 мл полученного раствора прибавляют 25,0 мл
ацетонитрила Р и доводят подвижной фазой А до
объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р (размер частиц 5 мкм);
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: 1,0 мл кислоты трифто¬
руксусной Р доводят водой Р до объема 1000 мл;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
85
15
3—30
85-* 5
15 —♦ 95
30—33
5
95
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 285 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (с).
Идентификация пиков примесей: идентифици¬
руют пик суммы примесей А и С, используя хромато¬
грамму раствора сравнения (с).
Относительное удерживание (по отношению
к тадалафилу, время удерживания — около 16 мин):
примеси А и С— около 1,03.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 3,3
(Нр — высота пика суммы примесей А и С относитель¬
но базовой линии; Ну — расстояние между базовой
линией и нижней точкой кривой, разделяющей пик
суммы примесей А и С и пик тадалафила).
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (а); не учитывают
пики примесей А и/или С.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: ацетонитрил Р — подвиж¬
ная фаза А (45:55, об/об);
- скорость подвижной фазы: 1,5 мл/мин;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (б);
- время хроматографирования: 2-кратное вре¬
мя удерживания тадалафила.
Время удерживания: тадалафил — около 4,5 мин.
Содержание С22Н19^04 в процентах рассчитывают
с учетом содержания тадалафила в ФСО тадалафила.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимити¬
руется общим критерием приемлемости для других/
119. Зак. 1060.
934
Гэсударственная фармакопея Республики Беларусь
неспецифицированных примесей и/или общей статьей
Субстанции для фармацевтического использования
(2034). Вследствие этого нет необходимости идентифи¬
цировать эти примеси для доказательства соответствия
требованиям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического использо¬
вания): В, С, О, Е, Е, С, Н, I.
А. (6Я12а5)-6-(1,3-Бензодиоксол-5-ил)-2-метил-
2,3,6,7,12,12а-гексагидропиразино[1 ',2': 1,6]пиридо[3,4-Ь]-
индол-1,4-дион.
В. (65,72а5)-6-(1,3-Бензодиоксол-5-ил)-2-метил-
2,3,6,7,12,12а-гексагидропиразино[1 \2':1,6]пиридо[3,4-Ь]-
индол-1,4-дион.
С. (65,12аЯ)-6-(1,3-Бензодиоксол-5-ил )-2-метил-
2,3,6,7,12,12а-гексагидропиразино[1 ',2':1,6]пиридо[3,4-Ь]-
индол-1,4-дион.
О. (6ЬТ?)-12-(1,3-Бензодиоксол-5-ил)-12а-гидрокси-
8-метил-6а,6Ь,8,9,12,12а-гексагидропиразино[1 ',2': 1,2]-
пирроло[3,4-с]хинолин-6,7,10(5Н)-трион.
Е. (6Я12аЯ 12ЪР)-6-( 1,3-Бензодиоксол-5-ил)-6а-
гидрокси-2-метил-2,3,6а,7,12а,12Ь-гексагидропира-
зино[1',2':1,5]пирроло[3,4нЬ]хинолин-1,4,12(6Н)-трион.
Р. (8а,/?)-6’-(1,3-Бензодиоксол-5-ил)-2'-метил-
г'.З'.в'.ва'-тетрагидро-б'Н-спирор, 1 -бензоксазин-4,7'-
пирроло[1,2-а]пиразин]-1 \2,4'(1 Н)-трион.
О. (12ЬЯ)-6-(1,3-Бензодиоксол-5-ил)-12-гидрокси-
2-метил-2,3,6,12Ь-тетрагидропиразино[1 ',2': 1,5]-
пирроло[3,4нЬ]хинолин-1,4-дион.
Н. (6Я14аЯ)-6-(1,3-Бензодиоксол-5-ил)-2-метил-
2,3,14,14а-тетрагидропиразино[1,2-с/|[1,4]бензодиазонин-
1,4,7,13-(6Н,8Р/)-тетрон.
I. (8а'/?)-6'-(1,3-Бензодиоксол-5-ил)-2'-метил-
2,,3',8,,8а,-тетрагидро-6'Н-спиро[индол-3,7,-пир-
роло[1,2-а]пиразин]-1',2,4'(1Н)-трион.
07/2016:0438
ТАЛЬК
Та1сит
ТА1.С
[14807-96-6]
ОПРЕДЕЛЕНИЕ
Порошкообразный отобранный природный ги¬
дратированный магния силикат. Чистый тальк имеет
формулу (Мд3514О10(ОН)2; М.м. 379,3). Может содер¬
жать различные количества сопутствующих минера-
Тальк
935
лов, среди которых преобладают хлориты (гидратиро¬
ванные силикаты алюминия и магния), магнезит (маг¬
ния карбонат), кальцит (кальция карбонат) и доломит
(кальция и магния карбонат).
ПРОИЗВОДСТВО
Тальк, полученный из природных месторож¬
дений, про которые известно, что они содержат со¬
путствующий асбест, не пригоден для медицинского
применения. Производитель несет ответственность
#(и обязан подтверждать это в сопроводительной до¬
кументации^ за отсутствие в продукте асбеста, что
подтверждается испытанием на амфиболы и серпен¬
тины. Наличие в продукте амфибол и серпентинов
обнаруживается методом рентгеновской дифракции
или методом абсорбционной спектрофотометрии в
инфракрасной области (см. А и В). Если они обнару¬
жены, то с помощью подходящего метода оптической
микроскопии по специфическим морфологическим
признакам асбеста отличают тремолитный и хризоти-
ловый асбест, как описано ниже.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках с калия бромидом Р.
В диапазоне от 740 см-1 до 760 см-1 при исполь¬
зовании детализированной (расширенной) шкалы, по¬
глощение при (758±1) см-1 может указывать на нали¬
чие тремолита или хлорита. Если поглощение сохра¬
няется после прокаливания испытуемого образца при
температуре (850±50) °С в течение не менее 30 мин,
то это указывает на наличие тремолита. В диапазоне
от 600 см-1 до 650 см-1 при использовании детализи¬
рованной (расширенной) шкалы, любая полоса по¬
глощения или плечо может указывать на присутствие
серпентинов.
B. Дифракция рентгеновского излучения.
Приготовление: испытуемый образец помещают
на держатель образца; уплотняют и сглаживают по¬
верхность испытуемого образца с помощью полиро¬
ванного предметного стекла для микроскопии.
Излучение: Си Ка монохромное; 40 кВ, (24—30) мА.
Входная щель: 1°.
Щель детектора: 0,2°.
Скорость гониометра: 1/10° 20/мин.
Диапазон сканирования: 10е—13° 28 и 24°—26° 20.
Образец: не ориентирован.
Результаты: наличие амфибол определяется
по наличию пика дифракции при (10,5±0,1)° 20, на¬
личие серпентинов определяется по наличию пиков
дифракции при (24,3±0,1)° 20 и при (12,1±0,1)° 20.
Если присутствие амфибол и/или серпентинов
определено одним из двух методов, тип асбеста
определяют подходящим методом оптической ми¬
кроскопии.
Присутствие асбеста подтверждают, если выпол¬
няются следующие 2 критерия:
-отношение длины к ширине от 20:1 до 100:1
или более для волокон длиннее 5 мкм;
-способность расщепляться на тончайшие во¬
локна;
и если выполняются хотя бы 2 из следующих 4
критериев:
- параллельные волокна встречаются в форме
пучков;
- концы волокон в пучках истончены;
- волокна в форме тонких игл;
-спутанная масса из индивидуальных волокон
и/или искривленных волокон.
ОПИСАНИЕ(СВОЙСТВА)
Легкий гомогенный порошок белого или почти бе¬
лого цвета, жирный на ощупь (не абразивный).
Практически нерастворим в воде, 96 % спирте,
разведенных растворах кислот и щелочей.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках с калия бромидом Р.
Полосы поглощения: при длинах волн (3677±2) см-1,
(1018±2) см"1 и (669±2) см"1.
B. В платиновом тигле расплавляют смесь, со¬
стоящую из 0,2 г натрия карбоната безводного Р
и 2,0 г калия карбоната Р. К расплавленной массе
прибавляют 0,1 г испытуемого образца и нагрева¬
ют до полного расплавления смеси. Охлаждают
и переносят расплавленную массу в выпаритель¬
ную чашку, содержащую 50 мл горячей воды Р.
Прибавляют кислоту хлористоводородную Р до
прекращения выделения пузырьков. Прибавляют
еще 10 мл кислоты хлористоводородной Р и вы¬
паривают на водяной бане досуха. Охлаждают.
Прибавляют 20 мл воды Р, нагревают до кипения
и фильтруют (остаток используют для проведения
подлинности С). К 5 мл фильтрата прибавляют 1
мл раствора аммиака Р и 1 мл раствора аммония
хлорида Р и фильтруют. К фильтрату прибавляют 1
мл раствора динатрия гидрофосфата Р. Образу¬
ется кристаллический осадок белого цвета.
C. Остаток, полученный в подлинности В, дает
реакцию (а) на силикаты (2.3.1).
ИСПЫТАНИЯ
Раствор 51. 10,0 г испытуемого образца по¬
мещают в коническую колбу с обратным холодиль¬
ником, постепенно, при постоянном перемешива¬
нии, прибавляют 50 мл 0,5 М раствора кислоты
хлористоводородной и нагревают на водяной бане
в течение 30 мин. Охлаждают. Переносят смесь в
лабораторный стакан и отстаивают до осаждения
нерастворенных веществ. Фильтруют надосадоч-
ную жидкость через среднепористый бумажный
фильтр в мерную колбу вместимостью 100 мл, ста¬
раясь по возможности, чтобы осадок оставался в
стакане. Осадок и стакан трижды промывают горя¬
чей водой Р порциями по 10 мл. Фильтр промыва¬
ют 15 мл горячей воды Р, фильтрат охлаждают и
доводят водой Р до объема 100,0 мл.
Раствор 52. Перхлораты в смеси с тяжелы¬
ми металлами взрывоопасны. Примите необхо¬
димые меры предосторожности при проведении
следующих процедур. 0,5 г испытуемого образца
помещают в политетрафторэтиленовую чашку вме¬
стимостью 100 мл, прибавляют 5 мл кислоты хло¬
ристоводородной Р, 5 мл кислоты азотной, сво¬
бодной от свинца, Р и 5 мл кислоты хлорной Р.
Аккуратно перемешивают, затем прибавляют 35 мл
кислоты фтористоводородной Р и медленно вы¬
паривают на горячей плитке досуха. К остатку при¬
бавляют 5 мл кислоты хлористоводородной Р, на¬
крывают часовым стеклом, нагревают до кипения
и охлаждают. Промывают часовое стекло и чашку
119*. Зак. 1060.
936
Гэсударственная фармакопея Республики Беларусь
водой Р. Переносят содержимое в мерную колбу,
промывают чашку водой Р и доводят до объема
50.0 мл этим же растворителем.
Кислотность или щелочность. К 2,5 г испы¬
туемого образца прибавляют 50 мл воды, свобод¬
ной от углерода диоксида, Р и кипятят с обратным
холодильником. Фильтруют под вакуумом. К 10 мл
полученного фильтрата прибавляют 0,1 мл раст¬
вора бромтимолового синего Р1. При прибавле¬
нии не более 0,4 мл 0,01 М раствора кислоты
хлористоводородной должно появиться зеленое
окрашивание. К 10 мл полученного фильтрата при¬
бавляют 0,1 мл раствора фенолфталеина Р1.
При прибавлении не более 0,3 мл 0,01 М раст¬
вора натрия гидроксида должно появиться розо¬
вое окрашивание.
Растворимые в воде вещества. Не более
0,2%. К 10,0 г испытуемого образца прибавляют
50 мл воды, свободной от углерода диоксида, Р,
нагревают до кипения и поддерживают кипение с
обратным холодильником в течение 30 мин. Охлаж¬
дают, фильтруют через среднепористый бумажный
фильтр и доводят водой, свободной от углерода
диоксида, Р до объема 50,0 мл. 25,0 мл фильтрата
выпаривают досуха и нагревают при температуре
105 °С в течение 1 ч. Масса остатка не должна пре¬
вышать 10 мг.
Алюминий. Не более 2,0 %.
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Испытуемый раствор. К 5,0 мл раствора 52 при¬
бавляют 10 мл раствора 25,34 г/л цезия хлорида Р,
10.0 мл кислоты хлористоводородной Р и доводят
водой Р до объема 100,0 мл.
Растворы сравнения. В 4 одинаковые мерные
колбы, содержащие по 10,0 мл кислоты хлористово¬
дородной Р и по 10 мл раствора 25,34 г/л цезия хло¬
рида Р, прибавляют соответственно 5,0 мл, 10,0 мл,
15.0 мл и 20,0 мл эталонного раствора алюминия
(100 рртА1) Р и доводят водой Р до объема 100,0 мл.
Источник излучения: лампа с полым катодом
для определения алюминия.
Длина волны: 309,3 нм.
Генератор атомного пара: пламя закись азо¬
та — ацетилен.
Кальций. Не более 0,9 %.
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Испытуемый раствор. К 5,0 мл раствора 32 при¬
бавляют 10,0 мл кислоты хлористоводородной Р,
10 мл раствора лантана (III) хлорида Р и доводят во¬
дой Р до объема 100,0 мл.
Растворы сравнения. В 4 одинаковые мер¬
ные колбы, содержащие по 10,0 мл кислоты хло¬
ристоводородной Р и по 10 мл раствора лантана
(III) хлорида Р, прибавляют соответственно 1,0 мл,
2.0 мл, 3,0 мл и 5,0 мл эталонного раствора каль¬
ция (100 ррт Са)Р1 и доводят водой Р до объема
100.0 мл.
Источник излучения: лампа с полым катодом
для определения кальция.
Длина волны: 422,7 нм.
Генератор атомного пара: пламя закись азо¬
та — ацетилен.
Железо. Не более 0,25 %.
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. К 2,5 мл раствора 31
прибавляют 50,0 мл 0,5 М раствора кислоты хло¬
ристоводородной и доводят водой Р до объема
100.0 мл.
Растворы сравнения. В 4 одинаковые мерные
колбы, содержащие по 50,0 мл 0,5 М раствора
кислоты хлористоводородной, прибавляют соот¬
ветственно 2,0 мл, 2,5 мл, 3,0 мл и 4,0 мл эталон¬
ного раствора железа (250 ррт Ре) Р и доводят
водой Р до объема 100,0 мл.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Коррекция фона: дейтериевая лампа.
Свинец. Не более 0,0010 % (10 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод Г).
Испытуемый раствор. Используют раствор 51.
Растворы сравнения. В 4 одинаковые мерные
колбы, содержащие по 50,0 мл 0,5 М раствора кис¬
лоты хлористоводородной, прибавляют соответ¬
ственно 5,0 мл, 7,5 мл, 10,0 мл и 12,5 мл эталонного
раствора свинца (10 ррт РЬ) Р1 и доводят водой Р
до объема 100,0 мл.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 217,0 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Магний. От 17,0 % до 19,5 %.
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. 0,5 мл раствора 52 дово¬
дят водой Р до объема 100,0 мл. К 4,0 мл полученного
раствора прибавляют 10,0 мл кислоты хлористово¬
дородной Р, 10 мл раствора лантана (III) хлорида Р
и доводят водой Р до объема 100,0 мл.
Растворы сравнения. В 4 одинаковые мерные
колбы, содержащие по 10,0 мл кислоты хлористово¬
дородной Р и 10 мл раствора лантана (III) хлорида Р,
прибавляют соответственно 2,5 мл, 3,0 мл, 4,0 мл и
5.0 мл эталонного раствора магния (10 ррт Мд) Р1
и доводят водой Р до объема 100,0 мл.
Источник излучения: лампа с полым катодом
для определения магния.
Длина волны: 285,2 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Потеря в массе при прокаливании. Не более
7,0%. 1,00 г испытуемого образца прокаливают до
постоянной массы при температуре от 1050 °С до
1100 °С.
Микробиологическая чистота
Если субстанция предназначена для производ¬
ства лекарственных средств для местного применения:
- ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
Если субстанция предназначена для производства
лекарственных форм для внутреннего применения:
- ОКА: критерий приемлемости 103 КОЕ/г (2.6.12)\
- ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
МАРКИРОВКА
Указывают:
-субстанция предназначена для производства
лекарственных средств для внутреннего или наруж¬
ного применения.
#Таурин
937
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о
характеристиках, которые являются важны¬
ми параметрами, связанными с одной или более
функцией субстанции, используемой в качестве
вспомогательного вещества (см. статью 5.15).
Некоторые из характеристик, описанных в раз¬
деле «Функционально-обусловленные характери¬
стики», могут также присутствовать в части
фармакопейной статьи, обязательной для вы¬
полнения, так как одновременно они являются
и показателями качества. В таком случае в раз¬
деле «Функционально-обусловленные характе¬
ристики» дается перекрестная ссылка на это
испытание в части фармакопейной статьи,
обязательной для выполнения. Контроль данных
характеристик может вносить вклад в качество
лекарственного средства путем достижения по¬
стоянства производственного процесса и улуч¬
шения свойств лекарственного средства при его
использовании. Предлагаемые методы испыта¬
ний являются подходящими для указанных целей,
однако другие методы также могут быть ис¬
пользованы. В случае указания результатов кон¬
кретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть
важны для талька, используемого в качестве
антифрикционного вещества (противоприлипа-
ющего вещества или скользящего вещества) при
производстве таблеток и капсул или в качестве
антиадгезивного вещества при производстве
таблеток, покрытых оболочкой и пленочной обо¬
лочкой.
Распределение частиц по размерам (2.9.31).
Удельная площадь поверхности (2.9.26).
07/2016:1477
ТАНИНОВАЯ КИСЛОТА
Тапп'тит
ТАЫШСАСШ
ОПРЕДЕЛЕНИЕ
Смесь эфиров глюкозы с галловой кислотой и
3-галлоилгалловой кислотой, Нанин*.
ОПИСАНИЕ(СВОЙСТВА)
Желтовато-белый или слегка коричневый аморф¬
ный легкий порошок или блестящие пластины.
Очень легко растворим в воде, легко растворим в
ацетоне, в 96 % спирте и в глицерине (85 %), практи¬
чески нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,1 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», доводят водой Р до объ¬
ема 5 мл. Прибавляют 0,1 мл раствора железа (III)
хлорида Р1. Появляется черновато-синее окрашива¬
ние, переходящее в зеленое при прибавлении 1 мл
кислоты серной разведенной Р.
B. К 1 мл раствора 3 прибавляют 3 мл раствора
1 г/л желатина Р. Смесь становится мутной и образу¬
ется хлопьевидный осадок.
C. 0,1 мл раствора 3 доводят водой Р до объема
5 мл. Прибавляют 0,3 мл раствора бария гидрокси¬
да Р. Образуется зеленовато-синий осадок.
ИСПЫТАНИЯ
Раствор 3. 4,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Декстрины, камеди, соли, сахара. К 2 мл раст¬
вора 3 прибавляют 2 мл 96 % спирта Р. Раствор про¬
зрачный. Прибавляют 1 мл эфира Р. Раствор остается
прозрачным в течение не менее 10 мин.
Смолы. К 5 мл раствора 3 прибавляют 5 мл
воды Р. Смесь должна быть прозрачной (2.2.1) в тече¬
ние не менее 15 мин.
Потеря в массе при высушивании (2.2.32). Не
более 12,0 %. 0,200 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:РБ0011
#ТАУРИН
Таиппит
ТАЧШЫЕ
.зо3н
И2Ы
С2Н7М035 М.м. 125,15
[107-35-7]
ОПРЕДЕЛЕНИЕ
2-Аминоэтансульфоновая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или белые или бесцветные кристаллы.
Растворим в воде, практически нерастворим в
этаноле безводном.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спетрофотометрия в инфракрас¬
ной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО таурина или спектр, представ¬
ленный на рисунке РБ0011.-1.
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
120. Зак. 1060.
938
Гэсударственная фармакопея Республики Беларусь
Рисунок РБ0011.-1. Инфракрасный спектр ФСО таурина
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,1 до 5,6. 1,0 г испытуемого об¬
разца растворяют в воде, свободной от углерода
диоксида, Р и доводят до объема 20 мл этим же
растворителем.
Сопутствующие примеси. Тонкослойная хро¬
матография (2.2.27).
Испытуемый раствор. 1,0 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
50 мл этим же растворителем.
Раствор сравнения. 1,0 мл испытуемого раст¬
вора доводят водой Р до объема 50,0 мл. 1,0 мл
полученного раствора доводят водой Р до объема
10,0 мл.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: кислота уксусная без¬
водная Р — бутанол Р — вода Р — этанол Р
(0,1:10:15:15, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 10 см от
линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром нингидрина Р и высушивают при температуре
от 100 °С до 105 °С в течение 5 мин.
Предельное содержание примесей:
- любая примесь (не более 0,2 %): на хрома¬
тограмме испытуемого раствора кроме основного
пятна допускается только одно дополнительное
пятно, которое должно быть не интенсивнее основ¬
ного пятна на хроматограмме раствора сравнения.
Хлориды (2.4.4). Не более 0,0110% (110 ррт).
Раствор 0,45 г испытуемого образца растворяют в
воде Р и доводят до объема 15 мл этим же раство¬
рителем. Полученный раствор должен выдерживать
испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
1,5 г испытуемого образца растворяют в воде Р и до¬
водят до объема 15,0 мл этим же растворителем. По¬
лученный раствор должен выдерживать испытание на
сульфаты.
Аммония соли (2.4.1, метод А). Не более
0,0200 % (200 ррт). 50 мг испытуемого образца долж¬
ны выдерживать испытание на соли аммония. Эталон
готовят с использованием 0,1 мл эталонного раст¬
вора аммония (100 ррт NН^ Р.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,2 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 50 мл
воды Р, прибавляют 5 мл раствора формальдеги¬
да Р и титруют 0,1 М раствором натрия гидроксида
потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 12,52 мг С2Н7Ы035.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
Тейкопланин
939
07/2016:2358
ТЕЙКОПЛАНИН
Те1сор1ат'пит
ТЕ1СОР1.АМ1Ы
120*. Зак. 1060.
940
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Смесь из гликопептидов, получаемая с помощью
определенных штаммов АсИпор1апез {е'юЬотусеИсиз
зр.; 6 основными компонентами смеси являются тей-
копланин от А^ до А2 5 и тейкопланин А3_г
Является продуктом ферментации.
Активность: не менее 900 МЕ/мг (в пересчете на
безводное и не содержащее натрия хлорид вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтоватый аморфный порошок.
Легко растворим в воде, умеренно растворим
в диметилформамиде, практически нерастворим в
96 % (об/об) спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тейкопланина для иденти¬
фикации #или спектр, представленный на рисунке
#2358.-1#.
B. Просматривают хроматограммы, полученные
в испытании «Состав и сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора основные пики (тейкопланины А^ г А^, А^,
А2 3, А2^ и А^) соответствуют по времени удерживания
и величине основным пикам на хроматограмме раст¬
вора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 0,8 г испытуемого образца раство¬
ряют в 10 мл воды Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод I). Окраска раствора
3 должна быть не интенсивнее эталона ВУ(КЖ)3 или
В(К)4.
рН (2.2.3). От 6,5 до 7,5. 0,50 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят этим же растворителем до объ¬
ема 10,0 мл.
Состав и сопутствующие примеси. Жидкост¬
ная хроматография (2.2.29): испытание проводят ме¬
тодом нормализации.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО тейкоплани¬
на для идентификации растворяют в воде Р и дово¬
дят до объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят водой Р до объема 10,0 мл. 1,0 мл
полученного раствора доводят водой Р до объема
20.0 мл.
Раствор сравнения (с). 50,0 мг ФСО мезити¬
ла оксида растворяют в воде Р и доводят до объема
25.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным эндкепированным для хроматографии Р
(размер частиц 5 мкм);
- подвижная фаза:
- подвижная фаза А: смешивают 900 мл
раствора 3,0 г/л натрия дигидрофосфата
безводного Р, доведенного 7 М раствором
натрия гидроксида до рН 6,0, и 100 мл ацето¬
нитрила Р;
- подвижная фаза В: смешивают 300 мл
раствора 3,0 г/л натрия дигидрофосфата
безводного Р, доведенного 7 М раствором
натрия гидроксида до рН 6,0, и 700 мл ацето¬
нитрила Р;
Рисунок #2358.-1. Инфракрасный спектр ФСО тейкопланина для идентификации
Тейкопланин
941
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
о
ю
т
о
о
о
ю
т
о
30—31
50 —► 10
сл
0
1
СО
о
31—35
10
90
- скорость подвижной фазы: 2,3 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл.
Идентификация: идентифицируют пики групп и
примесей, используя хроматограмму раствора срав¬
нения (а) и хроматограмму, прилагаемую к ФСО тей-
копланина для идентификации.
Относительное удерживание групп и примесей
(по отношению к тейкопланину А2 2):
- группа тейкопланина Ад ^ 0,70;
- группа тейкопланина А2 > 0,70 и < 1,25 и внутри
этой группы:
- тейкопланин А^ = 1;
- группа тейкопланина \л < 1;
- группа тейкопланина 3 > 1 и < 1,12;
- тейкопланин А^ = около 1,12;
- группа тейкопланина А2 5 > 1,12 и < 1,25;
- примеси > 1,25.
Относительное удерживание основных пиков
групп (по отношению к тейкопланину А^, время удер¬
живания — около 18 мин): тейкопланин А^1 — около
0,43; тейкопланин А^ — около 0,93; тейкопланин
А^ — около 1,04; тейкопланин А^ — около 1,12; тей¬
копланин — около 1,14.
Пригодность хроматографической системы:
раствор сравнения (а):
- хроматограмма раствора сравнения (а) должна
соответствовать хроматограмме, прилагаемой к ФСО
тейкопланина для идентификации;
- разрешение: не менее 1,0 между пиками тейко¬
планина А^ и тейкопланина \ 5.
Рассчитывают содержание различных компонен¬
тов в процентах по формулам:
§
группа тейкопланина А, = - - ® - - -- • 100
^ 5а + 0,83 • + $с
тейкопланин ^ 2 =
52
За + 0,83 • + 5С
•100
группа тейкопланина А, , = - --- -■
оа + У,оо • оь + ос
•100
группа тейкопланина Аз 3=
$4
За + 0,83 • 5Ь +5
•100
тейкопланин А^=
34
5а + 0,83 • 5Ь + 5
•100
группа тейкопланина А, = -- 5 _ -100
5а + 0,83 • 5Ь + 5С
группа тейкопланина А3=
За
ь -100
+ 0,83-5Ь+5С
примеси =
Зс
5а + 0,83 • 5Ь
где:
5а — сумма площадей пиков группы тейкоплани¬
на Аз на хроматограмме испытуемого раствора;
5Ь — сумма площадей пиков группы тейкопла¬
нина А3 на хроматограмме испытуемого раствора;
не учитывают пики мезитила оксида;
5с — сумма площадей пиков с относительным
удерживанием более 1,25;
51 — сумма площадей пиков группы тейкопла¬
нина А^ на хроматограмме испытуемого раствора;
52 — площадь пика тейкопланина А2 2 на хро¬
матограмме испытуемого раствора;
53 — сумма площадей пиков группы тейкопла¬
нина А2 з на хроматограмме испытуемого раствора;
34 — площадь пика тейкопланина А2 4 на хро¬
матограмме испытуемого раствора;
35 — сумма площадей пиков группы тейкопла¬
нина А2 5 на хроматограмме испытуемого раствора.
Предельное содержание:
- группа тейкопланина А2: не менее 80,0 %;
- тейкопланин А22: не менее 35,0 % и не бо¬
лее 55,0 %;
- группа тейкопланина А21: не более 20,0 %;
- группа тейкопланина А2г: не более 20,0 %;
- тейкопланин А2 А\ не более 20,0 %;
- группа тейкопланина А25: не более 20,0 %;
- группа тейкопланина А3: не более 15,0 %;
- сумма примесей, кроме мезитила оксида, с
относительным удерживанием более 1,25: не бо¬
лее 5,0 %;
- неучитываемый предел (0,25 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее площади пика тейкоплани¬
на А2 2 на хроматограмме раствора сравнения (Ь).
Хлориды. Не более 5,0 % (в пересчете на на¬
трия хлорид и безводное вещество). 1,000 г испы¬
туемого образца растворяют в 300 мл воды Р, пе¬
ремешивают, подкисляют 2 мл кислоты азотной Р
и титруют 0,1 М раствором серебра нитрата по-
тенциометрически (2.2.20).
1 мл 0,1 М раствора серебра нитрата соот¬
ветствует 5,844 мг №С1.
Тяжелые металлы (2.4.8, метод 6). Не бо¬
лее 0,0020 % (20 ррт). 0,50 г испытуемого образца
должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием 100 мкл
эталонного раствора свинца (100 ррт РЬ) Р. Рас¬
творы фильтруют через мембранный фильтр (но¬
минальный размер пор 0,45 мкм).
Примесь А. Жидкостная хроматография (2.2.29),
как указано в испытании «Состав и сопутствующие
примеси», со следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и раствора сравнения (с).
Относительное удерживание (по отношению
к тейкопланину А2 2, время удерживания — около
18 мин): примесь А— около 0,6.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси А, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (с).
Вода (2.5.12). Не более 15,0 %. Определение
проводят из 0,300 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
942
Гэсударственная фармакопея Республики Беларусь
Бактериальные эндотоксины (2.6.14). Менее
0,31 МЕ/мг.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Проводят количественное определение антибио¬
тиков микробиологическим методом (2.7.2), используя
метод диффузии. В качестве стандартного образца
используют ФСО тейкопланина.
ХРАНЕНИЕ
В защищенном от света месте при температуре
от 2 °С до 8 °С.
ПРИМЕСИ
Специфицированные примеси: А.
СН3 О
Н3С^ СН3
А. 4-Метилпент-3-ен-2-он (мезитила оксид).
07/2016:0298
ТЕОБРОМИН
ТЬеоЬготтит
ТНЕОВРОМ1ЫЕ
С7н8м402 М.м. 180,2
[83-67-0]
ОПРЕДЕЛЕНИЕ
3,7-Диметил-3,7-дигидро-1Н-пурин-2,6-дион.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим в воде и в этаноле, мало
растворим в растворе аммиака. Растворяется в раз¬
веденных растворах гидроксидов щелочных металлов
и минеральных кислотах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО теобромина.
B. Около 20 мг испытуемого образца растворяют
в 2 мл раствора аммиака разведенного Р1, слегка
нагревая, и охлаждают. При прибавлении к получен¬
ному раствору 2 мл раствора серебра нитрата Р2
раствор остается прозрачным. Полученный раствор
кипятят несколько минут, при этом образуется белый
кристаллический осадок.
C. Испытуемый образец дает реакцию на ксан¬
тины (2.3.1).
ИСПЫТАНИЯ
Кислотность. К 0,4 г испытуемого образца
прибавляют 20 мл кипящей воды Р и кипятят в те¬
чение 1 мин, охлаждают и фильтруют. При прибав¬
лении к полученному раствору 0,05 мл раствора
бромтимолового синего Р1 раствор приобретает
желтый или желтовато-зеленый цвет. При прибав¬
лении не более 0,2 мл 0,01 М раствора натрия
гидроксида окраска индикатора должна изменить¬
ся на синюю.
Сопутствующие примеси. Тонкослойная хро¬
матография (2.2.27).
Испытуемый раствор. К 0,2 г мелко измель¬
ченного испытуемого образца прибавляют 10 мл
смеси из 4 объемов метанола Р и 6 объемов
хлороформа Р, нагревают на водяной бане с об¬
ратным холодильником в течение 15 мин при пе¬
риодическом встряхивании, затем охлаждают и
фильтруют.
Раствор сравнения. 5 мг ФСО теобромина
растворяют в смеси из 4 объемов метанола Р и 6
объемов хлороформа Р и доводят этой же смесью
растворителей до объема 50 мл.
Пластинка: ТСХ пластинка со слоем силика-
геля вР254 Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р — ацетон Р — хлороформ Р —
бутанол Р (10:30:30:40, об/об/об/об).
Наносимый объем пробы: 10 мкл испытуемого
раствора и раствора сравнения.
Фронт подвижной фазы: не менее 15 см от
линии страта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хрома¬
тограмме испытуемого раствора любое пятно, кро¬
ме основного, должно быть не интенсивнее пятна
на хроматограмме раствора сравнения.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2 мл эталонно¬
го раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца су¬
шат при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
125 мл кипящей воды Р, охлаждают до темпера¬
туры от 50 °С до 60 °С, прибавляют 25 мл 0,1 М
раствора серебра нитрата и титруют по каплям
0,1 М раствором натрия гидроксида до появления
розовой окраски, используя в качестве индикатора
раствор фенолфталеина Р.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 18,02 мг С7Н8Н402.
Теофиллин
943
07/2016:0299
ТЕОФИЛЛИН
ТЬеорЬуШпит
ТНЕОРНУШЫЕ
С.Н.Ы.О, М.м. 180,2
[58-55-9]
ОПРЕДЕЛЕНИЕ
1,3-Диметил-3,7-дигидро-1 Н-пурин-2,6-дион.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в веще, умеренно растворим в
96% спирте. Растворяется в растворах гидроксидов
щелочных металлов, аммиаке и минеральных кислотах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О, Е.
A. Температура плавления (2.2.14): от 270 °С до
274 °С. Испытуемый образец предварительно сушат
при температуре от 100 °С до 105 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр теофиллина по
Европейской фармакопее #или спектр, представлен¬
ный на рисунке #0299.-1#.
С. 10 мг испытуемого образца нагревают с 1,0 мл
раствора 360 г/л калия гидроксида Р в водяной бане
при температуре 90 °С в течение 3 мин, затем при¬
бавляют 1,0 мл раствора кислоты сульфаниловой
диазотированной Р. Медленно появляется красное
окрашивание. Проводят холостой опыт.
О. Испытуемый образец выдерживает испытание
«Потеря в массе при высушивании», как указано в
разделе «Испытания».
Е. Испытуемый образец дает реакцию на ксанти¬
ны (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют при нагревании в воде, свободной от углерода
диоксида, Р, охлаждают и доводят до объема 75 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность. К 50 мл раствора 5 прибавляют
0,1 мл раствора метилового красного Р. Должно по¬
явиться красное окрашивание. При прибавлении не
более 1,0 мл 0,01 М раствора натрия гидроксида
окрашивание индикатора должно измениться на жел¬
тое.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Рисунок #0299.-1. Инфракрасный спектр теофиллина
944
Гэсударственная фармакопея Республики Беларусь
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг теобромина Р
растворяют в подвижной фазе, прибавляют 5 мл ис¬
пытуемого раствора и доводят до объема 100,0 мл
этим же растворителем. 5 мл полученного раствора
доводят до объема 50 мл подвижной фазой.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 7 мкм;
- подвижная фаза: смешивают 7 объемов аце¬
тонитрила для хроматографии Р и 93 объемов
раствора 1,36 г/л натрия ацетата Р, содержащего
5.0 мл/л кислоты ледяной уксусной Р.
- скорость подвижной фазы: 2,0 мл/мин;
- спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания теофиллина.
Относительное удерживание (по отношению к
теофиллину, время удерживания — около 6 мин): при¬
месь С — около 0,3; примесь В — около 0,4; примесь
О — около 0,5; примесь А — около 2,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 2,0 между пиками тео¬
бромина и теофиллина.
Предельное содержание примесей:
- примеси А, В, С, О (не более 0,1 %); на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В, С и О, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С и О,
не должна превышать площадь основного пика на
хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
100 мл воды Р, прибавляют 20 мл 0,1 М раствора
серебра нитрата и встряхивают. Титруют 0,1 М рас¬
твором натрия гидроксида, используя в качестве ин¬
дикатора 1 мл раствора бромтимолового синего Р1.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 18,02 мг С7Н8М402.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е, Р.
А. 1,3,7-Триметил-3,7-дигидро-1Н-пурин-2,6-дион
(кофеин).
В. 3-Метил-3,7-дигидро-1Н-пурин-2,6-дион.
С. А/-(6-Амино-1,3-диметил-2,4-диоксо-1,2,3,4-
тетрагидропиримидин-5-ил)формамид.
Э. А/-Метил-5-(метиламино)-1 Н-имидазолы-
карбоксамид (теофиллидин).
Е. 1,3-Диметил-7,9-дигидро-1Н-пурин-2,6,8(ЗН)-
трион.
Р. 7-(2-Гидроксиэтил)-1,3-диметил-3,7-дигидро-1Я-
пурин-2,6-дион (этофиллин).
Теофиллин-этилендиамин
945
07/2016:0300
ТЕОФИЛЛИН-ЭТИЛЕНДИАМИН
ТНеорЬуШпит е^ еМу/епесИаттит
ТНЕ0РНУ1.ШЕ-ЕТНУ1-ЕЫЕ01АМ1ЫЕ
С2Н8М2 (07Н8М4О2)2 М.м. 420,4
[317-34-0]
ОПРЕДЕЛЕНИЕ
#Эуфиллин, аминофиллин#.
Содержание:
-теофиллин (С7Н81Ч402; М.м. 180,2): не менее
84,0 % и не более 87,4 % (в пересчете на безводное
вещество);
- этилендиамин (С2Н8Ы2; М.м. 60,1): не менее
13,5 % и не более 15,0 % (в пересчете на безводное
вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок или гра¬
нулы. Гигроскопичен.
Легко растворим в воде (поглощая углекислый газ,
раствор мутнеет), практически нерастворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С, Е.
Вторая идентификация: А, С, О, Е, Е
1,0 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют по каплям при встряхивании
2 мл кислоты хлористоводородной разведенной Р и
фильтруют. Для идентификации А, В, й и Р использу¬
ют осадок, для идентификации С — фильтрат.
A. Температура плавления (2.2.14) осадка, про¬
мытого водой Р и высушенного при температуре
105 °С: от 270 °С до 274 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: осадок промывают водой Р и су¬
шат при температуре 105 °С.
Сравнение: ФСО теофиллина*\лш спектр, пред¬
ставленный на рисунке #0300.-1*.
C. К фильтрату прибавляют 0,2 мл бензоилхлори-
да Р, подщелачивают раствором натрия гидроксида
разведенным Р, интенсивно встряхивают и фильтру¬
ют. Остаток на фильтре промывают 10 мл воды Р,
растворяют в 5 мл горячего 96 % спирта Р и прибав¬
ляют 5 мл воды Р. Температура плавления (2.2.14)
полученного осадка, промытого водой Р и высушен¬
ного при температуре 105 °С: от 248 °С до 252 °С.
O. К 10 мг осадка прибавляют 1,0 мл раствора
360 г/л калия гидроксида Р, нагревают в водяной бане
при температуре 90 °С в течение 3 мин и прибавляют
1,0 мл раствора кислоты сульфаниловой диазоти-
рованной Р. Медленно появляется красное окрашива¬
ние. Проводят контрольный опыт.
Е. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
P. Осадок дает реакцию на ксантины (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 0,5 г испытуемого образца раство¬
ряют при осторожном нагревании в 10 мл воды, сво¬
бодной от углерода диоксида, Р.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Рисунок #0300.-1. Инфракрасный спектр ФСО теофиллина
946
Гэсударственная фармакопея Республики Беларусь
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ОУ(ЗЖ)б.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 47 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг ФСО теобромина
(примесь О) растворяют в подвижной фазе, прибавля¬
ют 5 мл испытуемого раствора и доводят подвижной
фазой до объема 100 мл. 5 мл полученного раствора
доводят подвижной фазой до 50 мл.
Условия хроматографирования:
- колонка 0,25 м и внутренним диаметром 4 мм,
заполненная силикагелем октадецилсилильным для
хроматографии Р с размером частиц 7 мкм;
-подвижная фаза: смешивают 7 объемов аце¬
тонитрила для хроматографии Р и 93 объема
раствора 1,36 г/л натрия ацетата Р, содержащего
0,50 % (об/об) кислоты уксусной ледяной Р\
- скорость подвижной фазы: 2,0 мл/мин;
- спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания теофиллина.
Относительное удерживание (по отношению к
теофиллину, время удерживания — около 6 мин): при¬
месь О — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси О и теофиллина.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- сумма примесей (не более 0,1 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0020 % (20 ррт). 0,500 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 1 мл эталонного
раствора свинца (10 ррт РЬ) Р. После подкисления
буферным раствором рН 3,5 Р выпадает осадок. По¬
лученную смесь доводят водой Р до объема 100 мл.
Осадок полностью растворяется.
Вода (2.5.12). Не более 1,5 %. Определение про¬
водят из 0,50 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этилендиамин. 0,250 г испытуемого образца
растворяют в 30 мл воды Р, прибавляют 0,1 мл раст¬
вора бромкрезолового зеленого Р и титруют 0,1 М
раствором кислоты хлористоводородной до появ¬
ления зеленого окрашивания.
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 3,005 мг С2Н8М2.
Теофиллин. 0,200 г испытуемого образца сушат
при температуре 135 °С до постоянной массы. Оста¬
ток растворяют при нагревании в 100 мл воды Р, ох¬
лаждают, прибавляют 20 мл 0,1 М раствора серебра
нитрата, встряхивают, прибавляют 1 мл раствора
бромтимолового синего Р1 и титруют 0,1 М раство¬
ром натрия гидроксида.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 18,02 мг С7Н8Ы402.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического исполь¬
зования (2034). Вследствие этого нет необходимости
идентифицировать эти примеси для доказательства
соответствия требованиям. См. также статью 5.10.
Контроль примесей в субстанциях для фармацевти¬
ческого использования): А, В, С, Ц Е, Р, 6.
СН3
А. 1,3,7-Триметил-3,7-дигидро-1Я-пурин-2,6-дион
(кафеин).
В. 3-Метил-3,7-дигидро-1 Я-пурин-2,6-дион.
С. Л/-(6-Амино-1,3-диметил-2,4-диоксо-1,2,3,4-
тетрагидропиримидин-5-ил)формамид.
О. А/-Метил-5-(метиламино)-1 Я-имидазол-4-карбо-
ксамид.
Теофиллин-этилендиамин гидрат
947
Е. 1,3-Диметил-7,9-дигидро-1 Н-пурин-2,6,8(ЗН)-
трион.
СН3
Р. 7-(2-Гидроксиметил)-1,3-диметил-3,7-дигидро-
1Н-пурин-2,6-дион (этофиллин).
6. 3,7-Диметил-3,7-дигидро-1Н-пурин-2,6-дион (те¬
обромин).
07/2016:0301
ТЕОФИЛЛИН-ЭТИЛЕНДИАМИН
ГИДРАТ
ТЬеорЬуШпит е? еМу1епесИаттит Нудпсит
ТНЕ0РНУШЫЕ-ЕТНУ1.ЕЫЕ01АМШЕ
НУЭКАТЕ
1,0 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют по каплям при встряхивании
2 мл кислоты хлористоводородной разведен¬
ной Р и фильтруют. Для идентификации А, В, О и Р
используют осадок, для идентификации С — филь¬
трат.
A. Температура плавления (2.2.14) осадка,
промытого водой Р и высушенного при температу¬
ре 105 °С: от 270 °С до 274 °С.
B. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Приготовление: осадок промывают водой Р и
сушат при температуре 105 °С.
Сравнение: ФСО теофиллина #или спектр,
представленный на рисунке #0301.-1#.
C. К фильтрату прибавляют 0,2 мл бензоил-
хлорида Р, подщелачивают раствором натрия
гидроксида разведенным Р, интенсивно встряхи¬
вают и фильтруют. Остаток на фильтре промыва¬
ют 10 мл воды Р, растворяют в 5 мл горячего 96 %
спирта Р и прибавляют 5 мл воды Р. Температура
плавления (2.2.14) полученного осадка, промытого
водой Р и высушенного при температуре 105 °С: от
248 °С до 252 °С.
Р. К 10 мг осадка прибавляют 1,0 мл раствора
360 г/л калия гидроксида Р, нагревают в водяной
бане при температуре 90 °С в течение 3 мин и при¬
бавляют 1,0 мл раствора кислоты сульфаниловой
диазотированной Р. Медленно появляется крас¬
ное окрашивание. Проводят контрольный опыт.
Е. Испытуемый образец выдерживает испыта¬
ние «Вода», как указано в разделе «Испытания».
Р. Осадок дает реакцию на ксантины (2.3.1).
ИСПЫТАНИЯ
хН20
С2Н8Ы2 ■ (С7н8ы402)2 ■ хн20 М.м. 420,4
(безводное вещество)
[72487-55-9]
ОПРЕДЕЛЕНИЕ
#Эуфиллина гидрат, аминофиллина гидрат*
Содержание:
- теофиллин (С7Н8М402; М.м. 180,2): не менее
84,0 % и не более 87,4 % (в пересчете на безводное
вещество);
- этилендиамин (С2Н8Ы2; М.м. 60,1): не менее
13,5 % и не более 15,0 % (в пересчете на безводное
вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок или гра¬
нулы.
Легко растворим в воде (поглощая углекислый
газ, раствор мутнеет), практически нерастворим в
этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С, Е.
Вторая идентификация: А, С, О, Е, Р.
Раствор 5. 0,5 г испытуемого образца раст¬
воряют при осторожном нагревании в 10 мл воды,
свободной от углерода диоксида, Р.
Прозрачность (2.2.1). Раствор 3 по степени
мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона ОУ(ЗЖ)6.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят подвижной фазой до объема
100,0 мл. 1,0 мл полученного раствора доводят
подвижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг ФСО теобро¬
мина (примесь О) растворяют в подвижной фазе,
прибавляют 5 мл испытуемого раствора и доводят
подвижной фазой до объема 100 мл. 5 мл получен¬
ного раствора доводят подвижной фазой до 50 мл.
Условия хроматографирования:
- колонка 0,25 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером ча¬
стиц 7 мкм;
- подвижная фаза: смешивают 7 объемов аце¬
тонитрила для хроматографии Р и 93 объема
раствора 1,36 г/л натрия ацетата Р, содержаще¬
го 0,50 % (об/об) кислоты уксусной ледяной Р;
- скорость подвижной фазы: 2,0 мл/мин;
948
Гэсударственная фармакопея Республики Беларусь
Рисунок #0301 .-1. Инфракрасный спектр ФСО теофиллина
- спектрофотометрический детектор, дли¬
на волны 272 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания теофиллина.
Относительное удерживание (по отношению к
теофиллину, время удерживания — около 6 мин): при¬
месь О — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси О и теофиллина.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
-сумма примесей (не более 0,1 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Н). Не бо¬
лее 0,0020 % (20 ррт). 0,500 г испытуемого об¬
разца должны выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием 1 мл
эталонного раствора свинца (10 ррт РЬ) Р. После
подкисления буферным раствором рН 3,5 Р выпа¬
дает осадок. Полученную смесь доводят водой Р до
объема 100 мл. Осадок полностью растворяется.
Вода (2.5.12). Не менее 3,0 % и не более 8,0 %.
Определение проводят из 0,50 г испытуемого об¬
разца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этилендиамин. 0,250 г испытуемого образ¬
ца растворяют в 30 мл воды Р, прибавляют 0,1 мл
раствора бромкрезолового зеленого Р и титруют
0,1 М раствором кислоты хлористоводородной
до появления зеленого окрашивания.
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 3,005 мг С2Н8М2.
Теофиллин. 0,200 г испытуемого образца су¬
шат при температуре 135 °С до постоянной мас¬
сы. Остаток растворяют при нагревании в 100 мл
воды Р, охлаждают, прибавляют 20 мл 0,1 М раст¬
вора серебра нитрата, встряхивают, прибавляют
1 мл раствора бромтимолового синего Р1 и ти¬
труют 0,1 М раствором натрия гидроксида.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 18,02 мг С7Н8Ы402.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом
контейнере в защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
#Теофиллин-этилендиамин для инъекций
949
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, В, С, О, Е, Е, С.
СН3
А. 1,3,7-Триметил-3,7-дигидро-1Н-пурин-2,6-дион
(кафеин).
В. З-Метил-3,7-дигидро-1 Н-пурин-2,6-дион.
С. Л/-(6-Амино-1,3-диметил-2,4-диоксо-1,2,3,4-
тетрагидропиримидин-5-ил)формамид.
й. Л/-Метил-5-(метиламино)-1 Н-имидазол-4-
карбоксамид.
н3с^
СН3
Е. 1,3-Диметил-7,9-дигидро-1Н-пурин-2,6,8(3/-/)-
трион.
о
1 'он
Нз<ч”ЛгЛ
хх>
О' 'И
сн
Р. 7-(2-Гидроксиметил)-1,3-диметил-3,7-дигидро-
1Н-пурин-2,6-дион (этофиллин).
Р СН3
XX?
о' у
сн-
О. 3,7-Диметил-3,7-дигидро-1Р/-пурин-2,6-дион (те¬
обромин).
07/2016.РБ0012
#ТЕОФИЛЛИН-ЭТИЛЕНДИАМИН
ДЛЯ ИНЪЕКЦИЙ
ТНеорЬуШпит е1 е№у1епесЛаттит рго
т]есИоп'1Ьи$
ТНЕ0РНУШЫЕ-ЕТНУ1-ЕЫЕ01АМШЕ
РОК ШЕСТ10Ы
Рисунок РБ0012.-1. Инфракрасный спектр ФСО теофиллина
950
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Эуфиллин для инъекций, аминофиллин для инъ¬
екций.
Содержание:
-теофиллин (С7Н81\1402; М.м. 180,2): не менее
75.0 % и не более 82,0 % (в пересчете на безводное
вещество);
- этилендиамин (С2Н81Ч2; М.м. 60,1): не менее
18.0 % и не более 22,0 % (в пересчете на безводное
вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок или гра¬
нулы.
Легко растворим в воде (поглощая углекислый
газ, раствор мутнеет), практически нерастворим в
этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С, Е.
Вторая идентификация: А, С, О, Е, Р.
1,0 г испытуемого образца растворяют в 10 мл
воды Р, прибавляют по каплям при встряхивании
2 мл кислоты хлористоводородной разведенной Р и
фильтруют. Для идентификации А, В, О и Р использу¬
ют осадок, для идентификации С — фильтрат.
A. Температура плавления (2.2.14) осадка, про¬
мытого водой Р и высушенного при температуре
105 °С: от 270 °Сдо 274 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: осадок промывают водой Р и су¬
шат при температуре 105 °С.
Сравнение: ФСО теофиллина или спектр, пред¬
ставленный на рисунке РБ0012.-1.
C. К фильтрату прибавляют 0,2 мл бензоилхлори-
да Р, подщелачивают раствором натрия гидроксида
разведенным Р, интенсивно встряхивают и фильтру¬
ют. Остаток на фильтре промывают 10 мл воды Р,
растворяют в 5 мл горячего 96 % спирта Р и прибав¬
ляют 5 мл воды Р. Температура плавления (2.2.14)
полученного осадка, промытого водой Р и высушен¬
ного при температуре 105 °С: от 248 °С до 252 °С.
Э. К 10 мг осадка прибавляют 1,0 мл раствора
360 г/л калия гидроксида Р, нагревают в водяной бане
при температуре 90 °С в течение 3 мин и прибавляют
1.0 мл раствора кислоты сульфаниловой диазот и-
рованной Р. Медленно появляется красное окрашива¬
ние. Проводят контрольный опыт.
Е. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
Р. Осадок дает реакцию на ксантины (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют при встряхивании в 5 мл воды, свободной от
углерода диоксида, Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 8 дол¬
жен быть бесцветным или не интенсивнее эталона
СУ(ЗЖ)7 или У(Ж)7.
рН (2.2.3). От 8,0 до 10,0. 0,4 г испытуемого об¬
разца растворяют, при необходимости слегка подо¬
гревая, в 10 мл воды Р.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 0,2 г испытуемого образ¬
ца растворяют при нагревании в 2 мл воды Р и дово¬
дят метанолом Р до объема 10 мл.
Раствор сравнения. 0,5 мл испытуемого раст¬
вора доводят метанолом Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р254 Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — ацетон Р — хлороформ Р — бута¬
нол Р (10:30:30:40, об/об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения.
Сульфаты (2.4.13). Не более 0,0200%
(200 ррт). 1,0 г испытуемого образца растворяют в
15 мл воды дистиллированной Р, прибавляют 5 мл
кислоты хлористоводородной разведенной Р, интен¬
сивно встряхивают и фильтруют. Фильтр и осадок про¬
мывают водой дистиллированной Р и доводят объем
фильтрата и промывной жидкости до 20 мл этим же
растворителем. 15 мл полученного раствора должны
выдерживать испытание на тяжелые металлы.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 4,5 %. 0,50 г испытуемого
образца растворяют в 20 мл пиридина безводного Р.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Пирогенность (2.6.8). Испытуемый образец дол¬
жен быть апирогенным. Тест-доза —12 мг испытуемо¬
го образца в 0,5 мл раствора 9 г/л натрия хлорида Р
в воде для инъекций Р на 1,0 кг массы тела кролика.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этилендиамин. 0,250 г испытуемого образца
растворяют в 30 мл воды Р, прибавляют 0,1 мл раст¬
вора бромкрезолового зеленого Р и титруют 0,1 М
раствором кислоты хлористоводородной до появ¬
ления зеленого окрашивания.
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 3,005 мг С2Н8М2.
Теофиллин. 0,200 г испытуемого образца сушат
при температуре 135 °С до постоянной массы. Оста¬
ток растворяют при нагревании в 100 мл воды Р, ох¬
лаждают, прибавляют 20 мл 0,1 М раствора серебра
нитрата, встряхивают, прибавляют 1 мл раствора
бромтимолового синего Р1 и титруют 0,1 М раство¬
ром натрия гидроксида.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 18,02 мг С7Н81М402.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Тербииафина гидрохлорид
951
07/2016:1734
ТЕРБИИАФИНА ГИДРОХЛОРИД
ТегЫпаЛп/ ЬубгосЫопдит
ТЕКВ1ЫАЕШЕ НУОКОСШ-ОРиОЕ
С21н25м - НС1 М.м. 327,9
[78628-80-5]
ОПРЕДЕЛЕНИЕ
(2Е)-А/,6,6-Триметил-Л/-(нафтален-1-илметил)
гепт-2-ен-4-ин-1 -амина гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Очень мало растворим или мало растворим в
воде, легко растворим в этаноле и в метаноле, мало
растворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тербииафина гидрохлорида
#или спектр, представленный на рисунке #1734.-1#.
B. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.7). В качестве растворителя используют
этанол Р.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от света.
Смесь растворителей А. Ацетонитрил Р —
вода Р (50:50, об/об).
Смесь растворителей В. Ацетонитрил Р—ме¬
танол Р (40:60, об/об).
Буферный раствор. 2,0 мл триэтиламина Р1
доводят водой Р до объема 950 мл, доводят до рН
7,5 смесью из 5 объемов кислоты уксусной ледя¬
ной Р и 95 объемов воды Р и разводят водой Р до
объема 1000,0 мл.
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в смеси растворителей А и до¬
водят до объема 50,0 мл этой же смесью раство¬
рителей.
Раствор сравнения (а). 5 мг ФСО тербина-
фина для проверки пригодности хроматографи¬
ческой системы (содержит примеси В и Е) раство¬
ряют в 10,0 мл смеси растворителей А.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей А до объ¬
ема 100,0 мл. 1,0 мл полученного раствора доводят
смесью растворителей А до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 3,0 мм, заполненная сферическим силикаге¬
лем октадецилсилильным эндкепированным для
хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: буферный раствор —
смесь растворителей В (30:70, об/об);
-подвижная фаза В: буферный раствор —
смесь растворителей В (5:95, об/об);
Рисунок #1734.-1. Инфракрасный спектр ФСО тербииафина гидрохлорида
952
Гэсударственная фармакопея Республики Беларусь
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-4
100
0
4—25
о
т
о
о
т—
0^ 100
25-30
0
100
- скорость подвижной фазы: 0,8 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В и Е, используя хроматограмму
раствора сравнения (а) и хроматограмму, прилагае¬
мую к ФСО тербинафина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
тербинафину, время удерживания — около 15 мин):
примесь В — около 0,9; примесь Е — около 1,7.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиком приме¬
си В и пиком тербинафина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Е — 0,5):
- примесь В (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь Е (не более 0,05 %): на хромато¬
грамме испытуемого раствора площадь пика, соот¬
ветствующего примеси Е, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В и Е, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
96% спирта Р, прибавляют 5 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 32,79 мг С21Н25Г\1-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, С, й, Р.
А. Л/-Метил-С-(нафтален-1 -ил)метанамин.
В. (22)-А/,6,6-Триметил-Л/-(нафтален-1 -илметил)-
гепт-2-ен-4-ин-1 -амин (щ/с-тербинафин).
н3с СН3
С. (2Е)-Л/,6,6-Триметил-А/-(нафтален-2-илметил)-
гепт-2-ен-4-ин-1 -амин (транс-изотербианфин).
й. (2Е)-Л/,6,6-Триметил-Л/-[(4-метилнафтален-1 -ил)-
метил]гепт-2-ен-4-ин-1 -амин (4-метилтербинафин).
Е. (2Е,4Е>4-(4,4-Диметилпент-2-ин-1 -илиден^А/,/^-
диметил-А/,А/'-бис(нафтален-1-илметил)пент-2-ен-1,5-
диамин.
Р. (22)-А/,6,6-Триметил-Л/-(нафтален-2-илметил)-
гепт-2-ен-4-ин-1 -амин (сщс-изотербинафин).
Терпентинное масло, тип Ртиз ртаз!ег
953
07/2016.РБ0013
«ТЕРПЕНТИННОЕ МАСЛО
ТегеЫпМтае о1еит
Т11КРЕМТШЕ он
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное из древесины или
смолы растений видов Ртиз (Ртасеае) методом пе¬
регонки с водяным паром и очищенное подходящим
методом. Скипидар.
Содержание: не менее 85 % суммы терпенов в
пересчете на а-пинен.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная летучая жидкость с ха¬
рактерным запахом без осадка и воды.
Практически нерастворимо в воде, растворимо в
96 % спирте. Смешивается с эфиром, хлороформом,
петролейным эфиром и жирными маслами.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 3 мл испытуемого образца очень осторожно,
по стенке, не взбалтывая, прибавляют 1 мл раствора
5 г/л п-диметиламинобензальдегида в кислоте сер¬
ной Р. На границе раздела слоев появляется коричне¬
вато-красное кольцо.
B. Газовая хроматография (2.2.28).
Испытуемый раствор. 1,0 мл испытуемого об¬
разца растворяют в гептане Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения. 30 мкл а-пинена Р, 20 мкл
Р-пинена Р, 10 мкл кар-3-ена Р, 10 мкл Р-мирцена Р
растворяют в 1,0 мл гептана Р.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м
и диаметром 0,25 мм, покрытая слоем макрогола
20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,0 мл/мин;
- деление потока: 1:200;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
60
10—80
60 — 200
80—120
200
Блок ввода проб
200
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1,0 мкл.
Порядок элюирования: порядок, указанный при
приготовлении раствора сравнения; отмечают време¬
на удерживания компонентов раствора сравнения.
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками кар-3-
ена и р-мирцена на хроматограмме раствора сравнения.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются не менее четырех пиков,
два из которых — а-пинен и Р-пинен.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,855 до 0,863.
Показатель преломления (2.2.6). От 1,465 до 1,472.
Кислотное число (2.5.1). Не более 0,7.
Жирные и минеральные масла (2.8.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Перекиси. 2 мл испытуемого образца смеши¬
вают с раствором 0,02 г бензидина Р в 2 мл спирта
(95 %, об/об) Р. Не должно появляться красное окра¬
шивание (допускается желтое окрашивание).
Альдегиды. Кусочек натрия гидроксида Р (раз¬
мером около (6—10) мм) помещают в сухую пробир¬
ку, прибавляют 3 мл свежеперегнанного испытуемого
образца и выдерживают в течение 4 ч. Жидкость над
осадком не должна окрашиваться в коричневый или
желто-коричневый цвет.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца доводят 96 % спир¬
том Р до объема 100,0 мл. 1,0 мл полученного раст¬
вора доводят 96 % спиртом Р до объема 50,0 мл.
Измеряют оптическую плотность (2.2.25) полученного
раствора при длине волны 210 нм, используя 96%
спирт Р в качестве компенсационного раствора.
Содержание суммы терпенов в пересчете на
а-пинен в процентах рассчитывают по формуле:
А-5000
237,5 /7? ’
где:
237,5 — удельный показатель поглощения
а-пинена;
А — оптическая плотность испытуемого раствора;
т — масса навески испытуемого образца, г.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом
контейнере в защищенном от света месте при темпе¬
ратуре не выше 15 °С.
07/2016:1627
ТЕРПЕНТИННОЕ МАСЛО,
ТИП Р//У(/5 РШАЗТЕК
ТегеЬШЫпае аеИ1его1еит а Рто р‘таз1го
ТиКРЕЫТШЕ ОН, Р1ЫС18 РША8ТЕР
ТУРЕ
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное из смолы растений
Ртиз ртаз(ег Айоп и/или Ртиз таззотапа Э.Ооп с
помощью перегонки с водяным паром и последующей
ректификации при температуре ниже 180 °С. Может
добавляться подходящий антиоксидант.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или бледно-желтая жид¬
кость с характерным запахом.
Напоминает запах а-пинена и р-пинена.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 мл испытуемого образ¬
ца растворяют в 10 мл толуола Р.
Раствор сравнения. 10 мкл Р-кариофиллена Р
и 10 мкл кариофиллена оксида Р растворяют в 10 мл
толуола Р.
954
Гэсударственная фармакопея Республики Беларусь
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)).
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: Юмкл [или 2 мкл] в
виде полос 10 мм [или 8 мм].
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р, нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин. Просма¬
тривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. Кроме того, на хроматограмме
испытуемого раствора могут присутствовать другие
слабые зоны, расположенные ниже зоны, соответ¬
ствующей кариофиллена оксиду.
Верх хроматографической пластинки
(3-кариофиллен: розовая
зона
Розовая зона
Кариофиллена оксид: розо¬
вая зона
Розовая зона (кариофилле¬
на оксид)
Коричневато-фиолетовая
зона
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
в испытании «Хроматографический профиль».
Результаты: пики на хроматограмме испытуе¬
мого раствора по времени удерживания соответству¬
ют пикам на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,856 до
0,872.
Показатель преломления (2.2.6). От 1,465 до
1,475.
Оптическое вращение (2.2.7). От -40° до -28°.
Кислотное число (2.5.1). Не более 1,0.
Жирные и минеральные масла (2.8.7). Испы¬
туемый образец должен выдерживать испытание на
жирные и минеральные масла.
Хроматографический профиль. Газовая хрома¬
тография (2.2.28): используют метод нормализации.
Испытуемый раствор. 1,0 мл испытуемого об¬
разца растворяют в гептане Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 30 мкл а-пинена Р, 10 мг
камфена Р, 20 мкл р-пинена Р, Юмкл кар-3-ена Р,
10 мкл р-мирцена Р, 20 мкл лимонена Р, 10 мкл лон-
гифолена Р, 10 мкл р-кариофиллена Р и 10 мг карио¬
филлена оксида Р растворяют в 1,0 мл гептана Р.
Раствор сравнения (Ь). 5 мкл Р-кариофиллена Р
растворяют в гептане Р и доводят до объема 10,0 мл
этим же растворителем. 0,1 мл полученного раствора
доводят гептаном Р до объема 1,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м
и диаметром 0,25 мм, покрытая слоем макрогола
20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,0 мл/мин;
- деление потока: 1:200;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
60
10—80
60 200
80—120
200
Блок ввода проб
200
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1,0 мкл.
Порядок элюирования: порядок, указанный при
приготовлении раствора сравнения (а); отмечают вре¬
мена удерживания компонентов раствора сравнения (а).
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками кар-
3-ена и (3-мирцена на хроматограмме раствора срав¬
нения (а).
Используя времена удерживания, полученные из
хроматограммы раствора сравнения (а), на хромато¬
грамме испытуемого раствора определяют располо¬
жение пиков компонентов раствора сравнения (а).
Рассчитывают процентное содержание компо¬
нентов.
Пределы содержания компонентов:
- а-пинен: от 70,0 % до 85,0 %;
- камфен: от 0,5 % до 2,0 %;
- р-пинен: от 5,0 % до 20,0 %;
- кар-3-ен: не более 1,0 %;
- Р-мирцен: от 0,4 % до 1,5 %;
- лимонен: от 1,0 % до 7,0 %;
- лонгифолен: от 0,2 % до 4,0 %;
- р-кариофиллен: от 0,1 % до 3,0 %;
- кариофиллена оксид: не более 1,0 %;
- неучитываемый предел (0,05 %): не учитыва¬
ют пики с площадью менее площади пика на хромато¬
грамме раствора сравнения (Ь).
Остаток после выпаривания (2.8.9). Не более
2,5 %. Выпаривают на водяной бане в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:1373
ТЕСТОСТЕРОН
Тез1оз1егопит
ТЕ8Т08ТЕЕ0ЫЕ
Тестостерон
955
ОПРЕДЕЛЕНИЕ
17р-Гидроксиандрост-4-ен-3-он.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные или желтовато-белые кристаллы.
Практически нерастворим в воде, легко раство¬
рим в 96 % спирте и в метиленхлориде, практически
нерастворим в жирных маслах.
Температура плавления: около 155 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тестостерона.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+106 до +114 (в пересчете на сухое вещество). 0,250 г
испытуемого образца растворяют в этаноле Р и дово¬
дят до объема 25,0 мл этим же растворителем.
Примеси О и Р. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 1 мг станолона Р раст¬
воряют в метаноле Р и доводят до объема 10 мл
этим же растворителем. В 1 мл полученного раствора
растворяют 10 мг ФСО тестостерона для иденти¬
фикации примеси О (тестостерон, содержащий около
1 % примеси й).
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят метанолом Р до объема 100,0 мл.
Раствор сравнения (с). 2,0 мл раствора сравне¬
ния (Ь) доводят до объема 10,0 мл метанолом Р.
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (Ь) доводят до объема 10,0 мл метанолом Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((6—8) мкм).
Предварительная подготовка пластинок (в за¬
щищенном от света месте): 5 г порошкообразного се¬
ребра нитрата Р прибавляют к 100 мл метанола Р.
Суспензию перемешивают в течение 30 мин, филь¬
труют или декантируют суспензию и погружают пла¬
стинку в раствор серебра нитрата на не менее 30 мин.
Сушат при температуре 75 °С в течение 30 мин.
Подготовленная пластинка может храниться в за¬
щищенном от света месте в течение 5—7 дней
Подвижная фаза: кислота уксусная Р — эта¬
нол Р — диоксан Р— метиленхлорид Р (1:2:10:90,
об/об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки в насыщенной камере.
Высушивание: при комнатной температуре в за¬
щищенном от света месте в течение 30 мин.
Проявление: пластинку опрыскивают раствором
200 г/л кислоты толуолсульфоновой Р в этаноле Р и
нагревают при температуре 105 °С в течение 10 мин.
Пластинку просматривают в ультрафиолетовом свете
при длине волны 365 нм.
Пригодность хроматографической системы:
- на хроматограмме раствора сравнения (а) об¬
наруживаются три полностью разделенных пятна;
- фактор подвижности (Р^: примесь О — около
0,5; тестостерон — около 0,65; примесь Р — около 0,7.
Предельное содержание примесей:
- примесь О (не более 0,2 %): на хроматограмме
испытуемого раствора пятно, соответствующее при¬
меси Ь, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (с);
- примесь Р (не более 0,1 %): на хроматограмме
испытуемого раствора пятно, соответствующее при¬
меси Р, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (с!).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО тестостеро¬
на для проверки пригодности хроматографической
системы (содержит примеси С и I) растворяют в 1 мл
метанола Р.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят метанолом Р до объема 20,0 мл. 1,0 мл
полученного раствора доводят метанолом Р до объ¬
ема 10,0 мл.
Раствор сравнения (с). 2,0 мл раствора сравне¬
ния (Ь) доводят до объема 10,0 мл метанолом Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным эндкепированным для хрома¬
тографии Р (размер частиц 5 мкм) с размером пор
15 нм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: вода для хроматогра¬
фии Р — метанол Р (45:55, об/об);
- подвижная фаза В: метанол Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—4
100
0
4—24
о
со
т
о
о
о
чТ
т
о
24—53
60->0
о
о
7
о
53—55
0
100
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению к
тестостерону, время удерживания — около 18 мин):
примесь С — около 0,6; примесь Н — около 0,8; при¬
месь А — около 0,9; примесь I — около 0,95; примесь
С — около 1,2; примесь Е — около 1,7; примесь 3 —
около 2,1; примесь В — около 2,5.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: разделение между пиками приме¬
си I и тестостерона до базовой линии.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси I — 2,9):
-для определения примесей С и I используют
хроматограмму раствора сравнения (а);
- примесь С (не более 0,5 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответ¬
ствующего примеси С, не должна превышать пло-
956
Гэсударственная фармакопея Республики Беларусь
щадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь I (не более 0,2 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси I, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (с);
- примеси А, В, Е, С, Н, Л (не более 0,1 %): для
каждой примеси на хроматограмме испытуемого
раствора площади пиков, соответствующих приме¬
сям А, В, Е, О, Н и 3, не должны превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (с);
-любая другая примесь (не более 0,1 %): на
хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пиков примесей
А, В, Е, О, Н и 3, не должна превышать площадь
основного пика на хроматограмме раствора срав¬
нения (с);
- сумма примесей (не более 0,6 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 1,2-кратную площадь основного пика на хро¬
матограмме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32).
Не более 1,0 %. 0,500 г испытуемого образца су¬
шат при температуре 105 °С в течение 2 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50,0 мг испытуемого образца растворяют в 96%
спирте Р и доводят до объема 100,0 мл этим же
растворителем. 2,0 мл полученного раствора дово¬
дят спиртом Р до объема 100,0 мл. Измеряют опти¬
ческую плотность (2.2.25) полученного раствора при
241 нм.
Содержание С19Н2802 рассчитывают с учетом
удельного показателя поглощения, равного 569.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, 8, С, О, Е, Р, в,
Н, I, и.
А. К1 + К2 = КЗ + К4 = О: Андрост-4-ен-3,17-дион
(андростендион).
С. К1 + К2 = О, КЗ = Н, К4 = ОН: 17а-Гидрокси-
андрост-4-ен-З-он (эпитестостерон).
О. К1 = КЗ = ОН, К2 = К4 = Н: Андрост-4-ен-Зр,17р-
диол (Д4-андростендиол).
Е. К1 + К2 = О, КЗ = 0-СО-СН3, К4 = Н: З-Оксоан-
дрост-4-ен-17р-илацетат (тестостерона ацетат).
В. К = С2Н5: 3-Этоксиандроста-3,5-диен-17-он (ан-
дростендиона этиловый эфир).
и. К = СН3:3-Метоксиандроста-3,5-диен-17-он (ан-
дростендиона метиловый эфир).
Р. 17р-Гидрокси-5а-андростан-3-он (андростано-
лон, станолон).
О. К1 + К2 = О: Андроста-1,4-диен-3,17-дион (ан-
дростадиендион).
Н. К1 = ОН, К2 = Н: 17р-Гидроксиандроста-1,4-
диен-3-он (болденон),
1.17р-Гидроксиандроста-4,6-диен-3-он (Аб-тесто-
стерон).
07/2016:0297
ТЕСТОСТЕРОНА ПРОПИОНАТ
Те$(оз(егот ргорюпаз
ТЕЗТОЗ ТЕЯОЫЕ РКОРЮЫАТЕ
С^Н^О, М.м. 344,5
[57-85-2]
ОПРЕДЕЛЕНИЕ
3-Оксоандрост-4-ен-17р-илпропаноат.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
Тестостерона пропионат
957
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок или бесцветные
кристаллы.
Практически нерастворим в воде, легко раство¬
рим в ацетоне и в 96 % спирте, растворим в жирных
маслах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тестостерона пропионата.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От +84
до +90 (в пересчете на сухое вещество). 0,250 г испы¬
туемого образца растворяют в этаноле Р и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО тестостеро¬
на пропионата для проверки пригодности хромато¬
графической системы (содержит примеси А, В и С)
растворяют в 5,0 мл метанола Р.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
1.0 мл полученного раствора доводят метанолом Р
до объема 10,0 мл.
Раствор сравнения (с). 20,0 мг ФСО тестосте¬
рона пропионата растворяют в 50,0 мл метанола Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: вода Р — метанол Р (20:80,
об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (а) и (Ь);
- время хроматографирования: 2-кратное вре¬
мя удерживания тестостерона пропионата.
Идентификация примесей: идентифицируют
пики примесей А, В и С, используя хроматограмму
раствора сравнения (а) и хроматограмму, прилагае¬
мую к ФСО тестостерона пропионата для проверки
пригодности хроматографической системы.
Относительное удерживание (по отношению к
тестостерона пропионату, время удерживания — око¬
ло 8 мин): примесь С — около 0,4; примесь А — около
0,7; примесь В — около 1,4.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее 3,0
(Нр — высота пика примеси В относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси В и пик
тестостерона пропионата).
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь С (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси С, не должна превышать 2-кратную площадь ос¬
новного пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,7 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 7-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (с).
Содержание С^Н^Оз в процентах рассчитывают
с учетом содержания тестостерона пропионата в ФСО
тестостерона пропионата.
ПРИМЕСИ
Специфицированные примеси: А, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, О, Е.
А. 3-Оксоандрост-4-ен-17р-илацетат (тестостеро¬
на ацетат).
В. 3-Оксоандрост-4-ен-17р-ил-2-метилпропаноат
(тестостерона изобутират).
958
Гэсударственная фармакопея Республики Беларусь
С. 17р-Гидроксиандрост-4-ен-3-он (тестостерон).
й. З-Оксоандроста-1,4-диен-17р-илпропаноат.
Е. 3-Оксоандроста-4,6-диен-17р-илпропаноат.
07/2016:0057
ТЕТРАКАИНА ГИДРОХЛОРИД
Те1гаса'т1 Ьус1госЫопс1ит
ТЕТКАСА1ЫЕ НУОКОСШОКЮЕ
С15Н*А°2 ' НС! М.М. 300,8
[136-47-0]
ОПРЕДЕЛЕНИЕ
2-(Диметиламино)этил-4-(бутиламино)бензоата
гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Слегка гигроскопичен.
Легко растворим в воде, растворим в 96 % спирте.
Температура плавления: около 148 °С; в слу¬
чае присутствия двух разных кристаллических форм
с температурами плавления около 134 °С и около
139 °С: от 134 °Сдо147 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тетракаина гидрохлорида
#или спектр, представленный на рисунке #0057.-1#.
B. К 10 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», прибавляют 1 мл раст¬
вора аммония тиоцианата Р. Образуется белый кри¬
сталлический осадок. Полученный осадок перекри-
сталлизовывают из воды Р и сушат при температуре
80 °С в течение 2 ч. Температура плавления (2.2.14)
полученных кристаллов: около 131 °С.
С. К 5 мг испытуемого образца прибавляют 0,5 мл
азотной кислоты дымящейся Р, выпаривают на во¬
дяной бане досуха, охлаждают, растворяют остаток в
5 мл ацетона Р и прибавляют 1 мл 0,1 М спиртового
раствора калия гидроксида. Появляется фиолетовое
окрашивание.
й. Раствор 3 дает реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Раствор 31. 2 мл раствора 3 доводят водой Р до
объема 10 мл.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 31 должен
быть бесцветным.
рН (2.2.3). От 4,5 до 6,5.1 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объема
10 мл.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием либо хранят их при
температуре от 2 °С до 8 °С.
Смесь растворителей. Ацетонитрил Р —
вода Р (20:80, об/об).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100,0 мл.1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО тетракаина для проверки пригодности хрома¬
тографической системы (содержит примеси А, В и
С) растворяют в 2 мл смеси растворителей.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: 1,36 г калия дигидрофос¬
фата Р растворяют в воде Р, прибавляют 0,5 мл
кислоты фосфорной Р и доводят водой Р до
объема 1000 мл;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
80
20
3—18
с»
0
1
-Ь*
о
20 — 60
18—23
40
60
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 300 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и С, используя хроматограм-
Тетракаина гидрохлорид
959
Рисунок #0057.-1. Инфракрасный спектр ФСО тетракаина гидрохлорида
му раствора сравнения (Ь) и хроматограмму, прилага¬
емую к ФСО тетракаина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
тетракаину, время удерживания — около 8 мин): при¬
месь А — около 0,3; примесь В — около 1,7; примесь
С — около 2,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5 между пиками тетрака¬
ина и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,6; для примеси С — 0,7):
- примесь А (не более 0,05 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (а);
- примеси В, С (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В и С, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В и С, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 12 мл раствора 8 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 30,08 мг С15Н24Ы202-НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. 4-Аминобензойная кислота.
960
Гэсударственная фармакопея Республики Беларусь
С. Метил-4-(бутиламино)бензоат.
07/2016:0211
ТЕТРАЦИКЛИН
Те1гасусНпит
ТЕТКАСУСШЕ
С22Н24М2°8 М.М. 444,4
[60-54-8]
ОПРЕДЕЛЕНИЕ
(45,4а5,5а5,65,12а5)-4-(Диметиламино)-3,6,10,-
12,12а-пентагидрокси-6-метил-1,11 -диоксо-1,4,4а,-
5,5а,6,11,12а-октагидротетрацен-2-карбоксамид.
Получают с использованием штаммов 81гер1оту-
сез аегоГааепз или любым другим способом.
Содержание: не менее 88,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Желтый кристаллический порошок.
Очень мало растворим в воде, растворим в 96 %
спирте и в метаноле, умеренно растворим в ацетоне.
Растворяется в разведенных кислотах и разведенных
растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО тетрациклина
гидрохлорида растворяют в метаноле Р и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО тетрациклина
гидрохлорида, 5 мг демеклоциклина гидрохлорида Р
и 5 мг окситетрациклина гидрохлорида Р в метано¬
ле Р и доводят до объема 10 мл этим же растворите¬
лем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: смешивают 20 объемов аце¬
тонитрила Р, 20 объемов метанола Р и 60 объемов
раствора 63 г/л щавелевой кислоты Р, доведенного
до рН 2 раствором аммиака концентрированным Р.
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме видны три полностью разде¬
ленных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
B. К 2 мг испытуемого образца прибавляют 5 мл
кислоты серной Р. Появляется фиолетово-красное
окрашивание. Полученный раствор прибавляют к 2,5 мл
воды Р. Окраска раствора изменяется на желтую.
C. 10 мг испытуемого образца растворяют в сме¬
си из 1 мл кислоты азотной разведенной Р и 5 мл
воды Р, встряхивают и прибавляют 1 мл раствора
серебра нитрата Р2. Полученный раствор по сте¬
пени мутности не превышает смесь из 1 мл кислоты
азотной разведенной Р, 5 мл воды Р и 1 мл раствора
серебра нитрата Р2.
ИСПЫТАНИЯ
рН (2.2.3). От 3,5 до 6,0. 0,1 г испытуемого образ¬
ца суспендируют в 10 мл воды, свободной от углеро¬
да диоксида, Р.
Удельное оптическое вращение (2.2.7). От
-260 до -280 (в пересчете на сухое вещество). 0,250 г
испытуемого образца растворяют в 0,1 М растворе
кислоты хлористоводородной и доводят до объема
50.0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в 0,01 М растворе кислоты хлори¬
стоводородной и доводят до объема 25,0 мл этим же
растворителем.
Раствор сравнения (а). 25,0 мг ФСО тетраци¬
клина гидрохлорида растворяют в 0,01 М растворе
кислоты хлористоводородной и доводят до объема
25.0 мл этим же растворителем.
Раствор сравнения (Ь). 12,5 мг ФСО 4-эпите-
трациклина гидрохлорида растворяют в 0,01 М рас¬
творе кислоты хлористоводородной и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (с). 10,0 мг ФСО ангидроте-
трациклина гидрохлорида растворяют в 0,01 М рас¬
творе кислоты хлористоводородной и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (д). 10,0 мг ФСО 4-эпианги-
дротетрациклина гидрохлорида растворяют в 0,01 М
растворе кислоты хлористоводородной и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения (е). К 1,0 мл раствора срав¬
нения (а) прибавляют 2,0 мл раствора сравнения (Ь),
5.0 мл раствора сравнения (с!) и доводят 0,01 М рас¬
твором кислоты хлористоводородной до объема
25.0 мл.
Раствор сравнения (I). К 40,0 мл раствора срав¬
нения (Ь) прибавляют 20,0 мл раствора сравнения (с),
Тетризолина гидрохлорид
961
5.0 мл раствора сравнения (й) и доводят 0,01 М рас¬
твором кислоты хлористоводородной до объема
200.0 мл.
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (с) доводят 0,01 М раствором кислоты хлори¬
стоводородной до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сополимером стирол-диви-
нилбензола Р с размером частиц 8 мкм;
- температура: 60 °С;
- подвижная фаза: 80,0 г 2-метил-2-пропанола Р
переносят в мерную колбу вместимостью 1000 мл с
использованием 200 мл воды Р, прибавляют 100 мл
раствора 35 г/л дикалия гидрофосфата Р, доведен¬
ного до рН 9,0 кислотой фосфорной разведенной Р,
200 мл раствора 10 г/л тетрабутиламмония гидро¬
сульфата Р, доведенного до рН 9,0 раствором на¬
трия гидроксида разведенным Р, и 10 мл раствора
40 г/л натрия эдетата Р, доведенного до рН 9,0 рас¬
твором натрия гидроксида разведенным Р, и дово¬
дят водой Р до объема 1000,0 мл;
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (е), (1) и (д).
Пригодность хроматографической системы:
-разрешение: не менее 2,5 между пиками при¬
меси А (первый пик) и тетрациклина (второй пик) на
хроматограмме раствора сравнения (е); не менее
8.0 между пиками тетрациклина и примеси й (третий
пик) на хроматограмме раствора сравнения (е); при
необходимости изменяют концентрацию 2-метил-2-
пропанола в подвижной фазе;
- отношение сигнал/шум: не менее 3 для основ¬
ного пика на хроматограмме раствора сравнения (д);
- фактор асимметрии: не более 1,25 для пика те¬
трациклина на хроматограмме раствора сравнения (е).
Предельное содержание примесей:
- примесь А (не более 5,0 %): на хроматограмме
испытуемого раствора площадь пика примеси А не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения (0;
- примесь В (не более 2,0 %) (элюируется в хво¬
сте основного пика): на хроматограмме испытуемого
раствора площадь пика примеси В не должна превы¬
шать 0,4 площади пика примеси А на хроматограмме
раствора сравнения (Г);
- примесь С (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика примеси С не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения (1)ш,
- примесь О (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика примеси О не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения (0-
Тяжелые металлы (2.4.8, метод С). Не более
0,0050 % (50 ррт). 0,5 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 13,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С^Н^ЫРз рассчитывают в процентах.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
A. Н1 = МН2, Р2 = Н, РЗ = М(СН3)2: (4Я,4а5,5а5,
65,12а5)-4-(Диметиламино)-3,6,10,12,12а-пентагидро-
кси-6-метил-1,11-диоксо-1,4,4а,5,5а,6,11,12а-октагидро-
тетрацен-2-карбоксамид (4-эпитетрациклин).
B. Р1 = СН3, Р2 = 1Ч(СН3)2, КЗ = Н: (45,4а5,5а5,
65,12а5)-2-Ацетил-4-(диметиламино)-3,6,10,12,12а-
пентагидрокси-6-метил-4а,5а,6,12а-тетрагидротерацен-
1,11 (4Н,5Н)-Дион (2-ацетил-2-декарбамоилтетрациклин).
С. Р1 = Ы(СИ3)2, Р2 = Н: (45,4а5,12а5)-4-(Диме-
тиламино)-3,10,11,12а-тетрагидрокси-6-метил-1,12-
диоксо-1,4,4а,5,12,12а-гексагидротетрацен-2-карбо-
ксамид (ангидротетрациклин).
й. Р1 = Н, Р2 = 1М(СН3)2: (4Я,4а5,12а5)-4-(Диме-
тиламино)-3,10,11,12а-тетрагидрокси-6-метил-1,12-
диоксо-1,4,4а,5,12,12а-гексагидротетрацен-2-карбо-
ксамид (4-эпиангидротетрациклин).
07/2016:2101
ТЕТРИЗОЛИНА ГИДРОХЛОРИД
Те(гугоНп1 НуйгосЫопдит
ТЕТЯУЮиЫЕ НУОКОСН1-ОВЮЕ
Спниы2 • НС1 М.м. 236,7
[522-48-5]
ОПРЕДЕЛЕНИЕ
2-[(1/?5)-1,2,3,4-Тетрагидронафтален-1-ил]-4,5-
дигидро-1 Н-имидазола гидрохлорид.
121. Зак. 1060.
962
Гэсударственная фармакопея Республики Беларусь
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворим в воде, в этаноле и в 96 % спир¬
те, практически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тетризолина гидрохлорида
#или спектр, представленный на рисунке #2101.-1#.
B. 50 мг испытуемого образца растворяют в
10 мл воды Р. Полученный раствор дает реакцию (а)
на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 8 должен
быть бесцветным.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. 1,0 г испытуемого образ¬
ца растворяют в смеси 1 М раствор натрия гидрок¬
сида — метанол Р (25:75, об/об) и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 1,0 мл испытуемого раст¬
вора доводят смесью 1 М раствор натрия гидрокси¬
да — метанол Р (25:75, об/об) до объема 100,0 мл.
1,0 мл полученного раствора доводят смесью 1 М
раствор натрия гидроксида — метанол Р (25:75,
об/об) до объема 10,0 мл.
Условия хроматографирования:
- колонка капиллярная кварцевая длиной 25 м
и внутренним диаметром 0,32 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- деление потока: 1:40;
- скорость газа-носителя: 2,5 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—8
160
8—11
160 —► 220
11—15
220
Блок ввода проб
220
Детектор
220
- детектор: пламенно-ионизационный;
- объем водимой пробы: 1 мкл.
Относительное удерживание (по отношению
к тетризолину, время удерживания — около 12 мин):
примесь А — около 0,5.
Пригодность хроматографической системы:
раствор сравнения:
- отношение сигнал/шум: не менее 50 для ос¬
новного пика.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения;
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пика примеси А, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения;
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
Рисунок #2101 .-1. Инфракрасный спектр ФСО тетризолина гидрохлорида
Тиамина гидрохлорид
963
ную площадь основного пика на хроматограмме раст¬
вора сравнения;
-неучитываемый предел (0,05%): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого сушат при темпера¬
туре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в
100 мл смеси кислота уксусная безводная Р — анги¬
дрид уксусный Р (3:7, об/об) и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 23,67 мг С13Н16М2 НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А.
А. (1Я5)-1,2,3,4-Тетрагидронафтален-1-карбони-
трил (сг-цианотетралин).
07/2016:0303
ТИАМИНА ГИДРОХЛОРИД
ТМат/т ЬубгосЫопдит
ТН1АМШЕ НУОКОСШОКЮЕ
С12Н17С1Ы403 • НС1 М.м. 337,3
[67-03-8]
ОПРЕДЕЛЕНИЕ
3-[(4-Амино-2-метилпиримидин-5-ил)метил]-5-(2-
гидроксиэтил)-4-метилтиазолия хлорида гидрохло¬
рид. #Витамин В/.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, растворим в глицерине,
мало растворим в 96 % спирте, практически нераст¬
ворим в гептане.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, С.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тиамина гидрохлорида #или
спектр, представленный на рисунке #0303.-1#.
Рисунок #0303.-1. Инфракрасный спектр ФСО тиамина гидрохлорида
121*. Зак. 1060.
964
Гэсударственная фармакопея Республики Беларусь
B. 20 мг испытуемого образца растворяют в
10 мл воды Р, прибавляют 1 мл кислоты уксусной
разведенной Р и 1,6 мл 1 М раствора натрия гидрок¬
сида, нагревают на водяной бане в течение 30 мин и
охлаждают. Прибавляют 5 мл раствора натрия ги¬
дроксида разведенного Р, 10 мл раствора феррици-
анида калия Р, 10 мл бутанола Р и интенсивно встря¬
хивают в течение 2 мин. Верхний спиртовой слой име¬
ет интенсивную голубую флуоресценцию, особенно
в ультрафиолетовом свете при длине волны 365 нм.
Повторяют испытание, используя 0,9 мл 1 М раст¬
вора натрия гидроксида и 0,2 г натрия сульфита Р
вместо 1,6 мл 1 М раствора натрия гидроксида. Флу¬
оресценция практически не обнаруживается.
C. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). 2,5 мл раствора 3 доводят
водой Р до объема 5 мл. Полученный раствор должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора,
приготовленного как указано в испытании «Прозрач¬
ность», должна быть не интенсивнее эталона У(Ж)7
или <ЗУ(ЗЖ)7.
рН (2.2.3). От 2,7 до 3,3. 2,5 мл раствора 3 дово¬
дят водой Р до объема 10 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Раствор А. К 95 объемам воды Р прибавляют 5
объемов кислоты уксусной ледяной Р и перемешивают.
Испытуемый раствор. 0,35 мг испытуемого об¬
разца растворяют в 15,0 мл раствора А и доводят во¬
дой Р до объема 100,0 мл.
Раствор сравнения (а). 5 мг испытуемого образ¬
ца и 5 мг ФСО тиамина примеси Е растворяют в 4 мл
раствора А и доводят водой Р до объема 25,0 мл.
5,0 мл полученного раствора доводят водой Р до объ¬
ема 25,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят водой Р до объема 50,0 мл. 5,0 мл полу¬
ченного раствора доводят водой Р до объема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецил-
силильным эндкепированным для хроматографии Р
(размер частиц 5 мкм) с удельной площадью поверх¬
ности 350 м2/г и размером пор 10 нм;
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: 3,764 г/л раствор натрия
гексансульфоната Р, доведенный кислотой
фосфорной Р до рН 3,1;
- подвижная фаза В: метанол Р2\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—25
о
г-
т
о
о
со
?
о
25—33
0
1
сл
о
со
0
1
СЛ
о
33—40
50
50
40—45
50 —► 90
50 —► 10
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 248 нм;
- объем вводимой пробы: 25 мкл.
Относительное удерживание (по отношению к
тиамину, время удерживания — около 30 мин): при¬
месь А — около 0,3; примесь В — около 0,9; примесь
С — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,6 между пиками при¬
меси Е и тиамина.
Предельное содержание примесей:
- любая примесь (не более 0,4 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,125 площади основного пика на
хроматограмме раствора сравнения (Ь).
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(2 ррт РЬ) Р.
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,40 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,110 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
уксусного ангидрида Р и немедленно титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20) в течение 2 мин.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора хлорной кислоты соответ¬
ствует 16,86 мг С12Н17С1М4ОЗ НС1.
ХРАНЕНИЕ
В неметаллическом контейнере в защищенном
от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О, Е, Р, 6, Н.
Тимолола малеат
965
Н. (3/?5)-3-[[[(4-Амино-2-метилпиримидин-5-ил)-
метил]тиокарбамоил]сульфанил]-4-оксопентилацетат
(кетодитиокарбамат).
A. ГС1 = СН3, Р2 = 0-803": 3-[(4-Амино-2-метил-
пиримидин-5-ил)метил]-4-метил-5-[2-(сульфонатоокси)-
этил]тиазолий (эфир тиамина сульфата).
B. Н1 = Н, Р2 = ОН: 3-[(4-Аминопиримидин-5-ил)-
метил]-5-(2-гидроксиэтил)-4-метилтиазолий (дезметил-
тиамин).
C. Р1 = СН3, Р2 = С1: 3-[(4-Амино-2-метил-
пиримидин-5-ил)метил]-5-(2-хлорэтил>4-метилтиаэолий
(хлортиамин).
Р. Р1 = С2Н5, К2 = ОН: 3-[(4-Амино-2-этилпи-
римидин-5-ил)метил]-5-(2-гидроксиэтил)-4-метилтиа-
золий (этилтиамин).
О. ГС1 = СН3> Р2 = 0-С0-СН3: 5-[2-(Ацетилокси)-
этил]-3-[(4-амино-2-метилпиримидин-5-ил)метил]-4-
метилтиазолий (ацетилтиамин).
О. X = О: 3-[(4-Амино-2-метилпиримидин-5-ил)ме-
тил]-5-(2-гидроксиэтил)-4-метилтиазол-2(ЗН)-он (ок-
сотиамин).
Е. X = 3: 3-[(4-Амино-2-метилпиримидин-5-ил)ме-
тил]-5-(2-гидроксиэтил)-4-метилтиазол-2(ЗН)-тион
(тиоксотиамин).
07/2016:0572
ТИМОЛОЛА МАЛЕАТ
Г/шо/о// та1еаз
Т1 МО 1-01- МА/.ЕАГЕ
,со2н
оо2н
С13Н24М4035 • С4н404 М.м.°432,5
[26921-17-5]
ОПРЕДЕЛЕНИЕ
(25)-1-[(1,1-Диметилэтил)амино]-3-[[4-(морфолин-
4-ил)-1,2,5-тиадиазол-3-ил]окси]пропан-2-ола (2)-
бутендионат.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
н3сч
о
н
СН3
и энантиомер
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Растворим в воде и в 96 % спирте.
Температура плавления: около 199 °С (с разло¬
жением).
Рисунок #0572.-1. Инфракрасный спектр ФСО тимолола малеата
122. Зак. 1060.
966
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Удельное оптическое вращение (2.2.7): то -5,7
до -6,2. 1,000 г испытуемого образца растворяют в
1 М растворе кислоты хлористоводородной и дово¬
дят до объема 10,0 мл этим же растворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тимолола малеата #или
спектр, представленный на рисунке #0572.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образ¬
ца растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения. 5 мг ФСО тимолола мале¬
ата растворяют в метаноле Р и доводят до объема
5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля 6Р254
Подвижная фаза: аммиака раствор концен¬
трированный Р — метанол Р — метиленхлорид Р
(1:20:80, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают парами
йода в течение 2 ч.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. 0,1 г испытуемого образца растирают со
смесью из 1 мл раствора натрия гидроксида раз¬
веденного Р и 3 мл воды Р. Встряхивают трижды с
эфиром Р порциями по 5 мл. К 0,1 мл водного слоя
прибавляют раствор, содержащий 10 мг резорци¬
на Р в 3 мл кислоты серной Р. Нагревают на водя¬
ной бане в течение 15 мин. Фиолетово-красное окра¬
шивание не появляется. Оставшуюся часть водного
слоя нейтрализуют кислотой серной разведенной Р
и прибавляют 1 мл воды бромной Р. Нагревают
на водяной бане в течение 15 мин, затем нагрева¬
ют до кипения и охлаждают. К 0,2 мл полученного
раствора прибавляют раствор, содержащий 10 мг
резорцина Р в 3 мл кислоты серной Р. Нагревают
на водяной бане в течение 15 мин. Появляется фи¬
олетово-красное окрашивание. Прибавляют 0,2 мл
раствора 100 г/л калия бромида Р и нагревают на
водяной бане в течение 5 мин. Окраска раствора
становится фиолетово-синей.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)8.
рН (2.2.3). От 3,8 до 4,3. Измеряют рН раствора 3.
Энантиомерная чистота. Жидкостная хромато¬
графия (2.2.29). Испытания проводят с защитой от
актиничного света.
Смесь растворителей. Метиленхлорид Р —
2-пропанол Р (10:30, об/об).
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). 30 мг ФСО тимолола
малеата растворяют в смеси растворителей и дово¬
дят до объема 10 мл этой же смесью растворителей.
Раствор сравнения (Ь). 3 мг ФСО (Р)-тимолола
(примесь А) растворяют в смеси растворителей и до¬
водят до объема 10,0 мл этой же смесью раствори¬
телей. 1,0 мл полученного раствора доводят смесью
растворителей до объема 10,0 мл.
Раствор сравнения (с). 1 мл раствора сравнения
(а) доводят смесью растворителей до объема 100 мл.
К 1 мл полученного раствора прибавляют 1 мл раст¬
вора сравнения (Ь).
Раствор сравнения (ф. 1,0 мл испытуемого раст¬
вора доводят смесью растворителей до объема 100,0 мл.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,25 м
и внутренним диаметром 4,6 мм, заполненная сили¬
кагелем ОО для хиральных разделений Р с размером
частиц 5 мкм;
- подвижная фаза: диэтиламин Р — 2-пропа¬
нол Р— гексан Р (2:40:960, об/об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 297 нм;
- объем вводимой пробы: 5 мкл.
При хроматографировании в описанных услови¬
ях примесь А выходит первой.
Пригодность хроматографической системы:
- разрешение: не менее 4,0 между пиками при¬
меси А и (З)-энантиомера на хроматограмме раствора
сравнения (с);
- время удерживания основного пика на хрома¬
тограмме испытуемого раствора должно соответство¬
вать времени удерживания основного пика (соответ¬
ствующего (З)-энантиомеру) на хроматограмме раст¬
вора сравнения (а).
Предельное содержание примесей:
-примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (б).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 20 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО тимолола для проверки пригодности хромато¬
графической системы (содержит примеси В, С, О и Р)
растворяют 1,0 мл подвижной фазы А.
Раствор сравнения (с). 2 мг испытуемого образ¬
ца и 20 мг малеиновой кислоты Р растворяют в 10 мл
ацетонитрила Р. 1 мл полученного раствора поме¬
щают в контейнер из желтого стекла и выпаривают
досуха в потоке азота Р. Открытый контейнер нагре¬
вают при температуре 105 °С в течение 1 ч. Получен¬
ный остаток растворяют в 1,0 мл подвижной фазы А.
Условия хроматографирования:
-колонка длиной 0,150 м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецил-
Тимолола малеат
967
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: смесь из равных объемов
метанола Р и раствора 4,32 г/л натрия октан-
сульфоната Р, предварительно доведенного до
рН 3,0 кислотой уксусной ледяной Р\
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%,об/об)
0—10
97,5
2,5
10—11
97,5 -+ 70
2,5 —> 30 |
11—20
70
30
- скорость подвижной фазы. 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 295 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифи¬
цируют пики примесей В, С, О и Р, используя хрома¬
тограмму раствора сравнения (б) и хроматограмму,
прилагаемую к ФСО тимолола для проверки пригод¬
ности хроматографической системы; идентифици¬
руют пик примеси Е, используя хроматограмму раст¬
вора сравнения (с).
Относительное удерживание (по отношению к
тимололу, время удерживания — около 7,5 мин): ма¬
леиновая кислота — около 0,1; примесь О — около
0,3; примесь Е — около 0,4; примесь В — около 0,7;
примесь Р — около 0,8; примесь С — около 2,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси Р и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 0,6):
- примеси В, С, О, Е, Р (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, С, О, Е и Р, не долж¬
ны превышать 2-кратную площадь основного пика на
хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В, С, й, Е и Р, не должна превышать площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 0,4 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 4-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,5 площади основного пика на хроматограмме
раствора сравнения (а) (0,05 %) и пик, соответствую¬
щий малеиновой кислоте.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 43,25 мг С13Н24М4035 С4Н404.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): О, Н, I, с/.
А. (2Я)-1-[(1,1-Диметилэтил)амино]-3-[[4-(морфо-
лин-4-ил)-1,2,5-тиадиазол-3-ил]окси]пропан-2-ол ((Р)-
тимолол).
он
В. (2Я5)-3-[(1,1 -Диметилэтил)амино]-[[4-(морфолин-
4-ил )-1,2,5-тиадиазол-3-ил]окси]пропан-1 -ол.
С. (2/?5)-Л/-(1,1-Диметилэтил)-2,3-бис[[4-(морфо-
лин-4-ил )-1,2,5-тиадиазол-3-ил]окси]пропан-1-амин.
й. 4-(Морфолин-4-ил)-1,2,5-тиадиазол-З-ол.
122*. Зак. 1060.
968
Государственная фармакопея Республики Беларусь
Е. (22)-4-[(15)-1 -[[(1,1 -Диметилэтил)амино]метил]-
2-[[4-(морфолин-4-ил)-1,2,5-тиадиазол-3-ил]окси]этокси]-
4-оксобут-2-еновая кислота.
/Vе'
\
■Л.
Р. 4-(4-Хлор-1,2,5-тиадиазол-3-ил)морфолин.
0=5;
6. 4-(Морфолин-4-ил)-1,2,5-тиадиазол-3(2Я)-он
1 -оксид.
N-—-0
// 1
н он
X'
СН3
и энантиомер
Н3С СН3
Н. 2-[(2Я5)-3-[(1,1-Диметилэтил)амино]-2-гидро-
ксипропилН-(морфолин-4-ил)-1>2>5-тиадиазол-3(2Н)-он.
I. (2РЗ)-1 -(Этиламино)-3-[[4-(морфолин-4-ил)-1,2,5-
тиадиазол-3-ил]окси]пропан-2-ол.
он он
Н3с.
Н3С Ън3
V
V"
СН3
а"
НдС7 СНз
3.1,1-[1,2,5-Тиадиазол-3,4-диилбис(окси)]бис[(1,1 -
диметилэтил)амино]пропан-2-ол.
07/2016:1161
ТИРОЗИН
Тугозтит
ТУК08ШЕ
[60-18-4]
ОПРЕДЕЛЕНИЕ
(5)-2-Амино-3-(4-гидроксифенил)пропановая
кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень мало растворим в воде, практически не¬
растворим в 96 % спирте. Растворяется в разведен¬
ных минеральных кислотах и в разведенных раство¬
рах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО тирозина.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 1 мл раствора аммиака Р2 и до¬
водят водой Р до объема 50 мл.
Раствор сравнения. 10 мг ФСО тирозина раст¬
воряют в 1 мл раствора аммиака Р2 и доводят во¬
дой Р до объема 50 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: концентрированный раствор
аммиака Р1 — пропанол Р (30:70, об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и высушивают при температуре 105 °С
в течение 15 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. К 50 мг испытуемого образца прибавляют 1 мл
кислоты азотной разведенной Р. В течение 15 мин
появляется темно-красное окрашивание.
Е. Около 30 мг испытуемого образца растворяют
в 2 мл натрия гидроксида раствора разведенного Р,
прибавляют 3 мл свежеприготовленной смеси из рав¬
ных объемов раствора 100 г/л натрия нитрита Р и
раствора, содержащего 0,5 г сульфаниловой кисло¬
ты Р в смеси из 6 мл кислоты хлористоводород¬
ной Р1 и 94 мл воды Р. Появляется оранжево-красное
окрашивание.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в кислоте хлористоводородной разведенной Р
и доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)7.
Удельное оптическое вращение (2.2.7). От
-12,3 до -11,0 (в пересчете на сухое вещество). 1,25 г
испытуемого образца растворяют в смеси из равных
объемов кислоты хлористоводородной разведен-
Тирозин
969
ной Р и воды Р и доводят до объема 25,0 мл этим же
растворителем.
Нингидрин-положительные вещества. Анализ
аминокислот (2.2.56). Для анализа используют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образца, подходящий для применяемого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг фенилаланина Р
(примесь А) растворяют в растворе А и доводят до
объема 100,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
250.0 мл.
Раствор сравнения (с). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (д). 6,0 мл эталонного раст¬
вора аммония (100 ррт А/Н^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (е). 30 мг изолейцина Р и
30 мг лейцина Р растворяют в растворе А и доводят
до объема 50,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
200.0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
вводят в аминокислотный анализатор. Запускают под¬
ходящую программу для определения физиологиче¬
ских аминокислот.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для примеси А — используют концентрацию
примеси А в растворе сравнения (Ь);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию тирозина в растворе сравне¬
ния (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (с); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
- примесь А при длине волны 570 нм: не более
0,5 %;
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,6 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в 3 мл кис¬
лоты азотной разведенной Р и доводят водой Р до
объема 15 мл. Полученный раствор должен выдержи¬
вать испытание на хлориды без дополнительного при¬
бавления азотной кислоты.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
0,5 г испытуемого образца растворяют при осторож¬
ном нагревании в 5 мл кислоты хлористоводородной
разведенной Р и доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (б), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод 6). Не более
0,0010 % (10 ррт). 0,5 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 0,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р. Прибавляют
30 мл кислоты уксусной безводной Р. Титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 18,12 мг СдН^Оз.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
123. Зак. 1060.
970
Гэсударственная фармакопея Республики Беларусь
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей. Вслед¬
ствие этого нет необходимости идентифицировать
эти примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль примесей
в субстанциях для фармацевтического использова¬
ния): В, С.
А. (25)-2-Амино-3-фенилпропановая кислота (фе¬
нилаланин).
н ш2
н2м со2н
В. (25)-2,6-Диаминогексановая кислота (лизин).
Н N42
Н МН2
С02Н
С. 3,3-Дисульфандиилбис[(2/?)-2-аминопропановая
кислота] (цистин).
07/2016:0150
ТИТАНА ДИОКСИД
ТПапи сНохШт
Т1ТАЫШМ йЮХЮЕ
ТЮ2 М.м. 79,9
[13463-67-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 98,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде. Не растворяет¬
ся в разведенных минеральных кислотах, но медлен¬
но растворяется в горячей концентрированной серной
кислоте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. При сильном нагревании испытуемый образец
приобретает бледно-желтое окрашивание, исчезаю¬
щее при охлаждении.
B. К 5 мл раствора 52, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,1 мл раст¬
вора водорода пероксида концентрированного Р. По¬
является оранжево-красное окрашивание.
C. К 5 мл раствора 52 прибавляют 0,5 г цинка Р в
гранулах. Через 45 мин смесь приобретает фиолето¬
во-синее окрашивание.
ИСПЫТАНИЯ
Раствор 51. 20,0 г испытуемого образца встря¬
хивают с 30 мл кислоты хлористоводородной Р в
течение 1 мин. Прибавляют 100 мл воды дистилли¬
рованной Р и нагревают смесь до кипения. Фильтруют
горячую смесь через плотную фильтровальную бума¬
гу до получения прозрачного фильтрата. Промывают
фильтр 60 мл воды дистиллированной Р, фильтрат
и промывные воды объединяют и доводят водой дис¬
тиллированной Р до объема 200 мл.
Раствор 32. 0,500 г (т, г) испытуемого образца
смешивают с 5 г натрия сульфата безводного Р в
колбе с длинным горлом для сжигания вместимостью
300 мл. Прибавляют 10 мл воды Р и перемешивают.
Прибавляют 10 мл кислоты серной Р и кипятят, со¬
блюдая меры предосторожности, до получения про¬
зрачного раствора. Охлаждают, медленно прибавля¬
ют охлажденную смесь из 30 мл воды Р и 10 мл кис¬
лоты серной Р, снова охлаждают и доводят водой Р
до объема 100,0 мл.
Прозрачность (2.2.1). Раствор 52 по степени
мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Раствор 52 должен
быть бесцветным.
Кислотность или щелочность. 5,0 г испытуемо¬
го образца встряхивают с 50 мл воды, свободной от
углерода диоксида, Р, в течение 5 мин. Центрифуги¬
руют или фильтруют до получения прозрачного раст¬
вора. К10 мл полученного раствора прибавляют 0,1 мл
раствора бромтимолового синего Р1. При прибавле¬
нии не более 1,0 мл 0,01 М раствора хлористоводо¬
родной кислоты или 0,01 М раствора натрия гидрок¬
сида окраска раствора должна измениться.
Растворимые в воде вещества. Не более 0,5 %.
К 10,0 г испытуемого образца прибавляют раствор
0,5 г аммония сульфата Р в 150 мл воды Р и кипятят
в течение 5 мин. Охлаждают, доводят водой Р до объ¬
ема 200 мл и фильтруют до получения прозрачного
раствора. 100 мл полученного раствора выпаривают
досуха в предварительно взвешенной чашке для вы¬
паривания и прокаливают. Масса остатка не должна
превышать 25 мг.
Сурьма. Не более 0,0100% (100 ррт). К 10 мл
раствора 52 прибавляют 10 мл кислоты хлористо¬
водородной Р и 10 мл воды Р. При необходимости ох¬
лаждают до температуры 20 °С и прибавляют 0,15 мл
раствора натрия нитрита Р. Через 5 мин прибавля¬
ют 5 мл раствора 10 г/л гидроксиламина гидрохлори¬
да Р и 10 мл свежеприготовленного раствора 0,1 г/л
родамина ВРг тщательно перемешивая после каж¬
дого прибавления. Интенсивно встряхивают с 10,0 мл
толуола Р в течение 1 мин. Дают слоям разделить¬
ся и при необходимости центрифугируют в течение
2 мин. Розовая окраска толуольного слоя должна
быть не интенсивнее окраски толуольного слоя этало¬
на, приготовленного параллельно с использованием
смеси из 5,0 мл эталонного раствора сурьмы (1 ррт
8Ь) Р, 10 мл кислоты хлористоводородной Р и 15 мл
раствора, содержащего 0,5 г натрия сульфата без¬
водного Р и 2 мл кислоты серной Р, вместо смеси из
10 мл раствора 52,10 мл кислоты хлористоводород¬
ной Р и 10 мл воды Р.
Мышьяк (2.4.2, метод А). Не более 0,0005 %
(5 ррт). 0,50 г испытуемого образца помещают в
круглодонную колбу вместимостью 250 мл, осна¬
щенную термометром, воронкой с краном и отво¬
дной трубкой, соединенной с колбой, содержащей
30 мл воды Р. Прибавляют 50 мл воды Р, 0,5 г ги¬
дразина сульфата Р, 0,5 г калия бромида Р и 20 г
натрия хлорида Р. Через воронку прибавляют по
каплям 25 мл кислоты серной Р, нагревают и под¬
держивают температуру жидкости от 110 °С до
115 °С в течение 20 мин. Собирают летучие пары в
Тозилхлорамид натрия
971
колбу, содержащую 30 мл воды Р. Доводят водой Р
до объема 50 мл. 20 мл полученного раствора
должны выдерживать испытание на мышьяк.
Барий. К 10 мл раствора 31 прибавляют 1 мл
кислоты серной разведенной Р Через 30 мин по¬
лученный раствор по степени мутности не должен
превышать смесь из 10 мл раствора 31 и 1 мл
воды дистиллированной Р.
Железо. Не более 0,0200 % (200 ррт). К 8 мл
раствора 52 прибавляют 4 мл воды Р. Перемеши¬
вают и прибавляют 0,05 мл бромной воды Р. Вы¬
держивают в течение 5 мин и удаляют избыток
брома потоком воздуха. Прибавляют 3 мл раст¬
вора калия тиоцианата Р. Окраска полученного
раствора должна быть не интенсивнее окраски
эталона, приготовленного параллельно с исполь¬
зованием смеси из 4 мл эталонного раствора же¬
леза (2 ррт Ре) Р и 8 мл раствора 200 г/л кислоты
серной Р.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). К 10 мл раствора 51 прибавля¬
ют по каплям раствор аммиака концентрирован¬
ный Р до достижения рН 4 и доводят водой Р до
объема 20 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 300 г цинка Р в гранулах (710) прибавля¬
ют 300 мл раствора 20 г/л ртути нитрата Р и
2 мл кислоты азотной Р, встряхивают в течение
10 мин и промывают водой Р. Стеклянную трубку
длиной около 400 мм и диаметром около 20 мм с
крышкой и фильтровальной мембраной заполняют
амальгамированным цинком и через полученную
колонку пропускают 100 мл кислоты серной раз¬
веденной Р и затем 100 мл воды Р, при этом необ¬
ходимо следить, чтобы амальгама была постоян¬
но покрыта жидкостью. Через колонку медленно,
со скоростью около 3 мл/мин, пропускают смесь из
100 мл кислоты серной разведенной Р и 100 мл
воды Р, а затем 100 мл воды Р. Собирают элюат в
коническую колбу вместимостью 500 мл, содержа¬
щую 50,0 мл раствора 150 г/л железа (1/1) аммония
сульфата Р в смеси кислота серная Р — вода Р
(1:3, об/об). Прибавляют 0,1 мл ферроина Р и не¬
замедлительно титруют 0,1 М раствором аммо¬
ния церия нитрата до появления зеленоватого
окрашивания (лг мл). Через колонку медленно,
со скоростью около 3 мл/мин, пропускают смесь
из 50 мл кислоты серной разведенной Р и 50 мл
воды Р, а затем 20,0 мл раствора 52, смесь из
50 мл кислоты серной разведенной Р и 50 мл
воды Р ив конце 100 мл воды Р. Собирают элюат
в коническую колбу вместимостью 500 мл, содер¬
жащую 50,0 мл раствора 150 г/л железа (III) ам¬
мония сульфата Р в смеси кислота серная Р —
вода Р (1:3, об/об). Ополаскивают нижний конец
колонки водой Р, прибавляют 0,1 мл ферроина Р
и незамедлительно титруют 0,1 М раствором ам¬
мония церия нитрата до появления зеленоватого
окрашивания (п2, мл).
Содержание ТЮ2 в процентах рассчитывают по
формуле:
3,99 • (п2 - пл)
т
где:
т — масса испытуемого образца, взятого для
приготовления раствора 52, г.
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомогатель¬
ного вещества (см. статью 5.15). Некоторые из ха¬
рактеристик, описанных в разделе «Функционально-
обусловленные характеристики», могут также при¬
сутствовать в части фармакопейной статьи, обяза¬
тельной для выполнения, так как одновременно они
являются и показателями качества. В таком случае
в разделе «Функционально-обусловленные характери¬
стики» дается перекрестная ссылка на это испыта¬
ние в части фармакопейной статьи, обязательной
для выполнения. Контроль данных характеристик
может вносить вклад в качество лекарственного
средства путем достижения постоянства произ¬
водственного процесса и улучшения свойств лекар¬
ственного средства при его использовании. Предла¬
гаемые методы испытаний являются подходящими
для указанных целей, однако другие методы также
могут быть использованы. В случае указания резуль¬
татов конкретных характеристик требуется указы¬
вать также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для титана диоксида, используемого в качестве
коррегента цвета (пигмента) при производстве
твердых дозированных лекарственных форм для
внутреннего применения и лекарственных средств
для наружного применения.
Распределение частиц по размерам (2.9.31).
07/2016:0381
ТОЗИЛХЛОРАМИД НАТРИЯ
То$у1сЫогатМит паМсит
ТОЗУ1.СН1.0КАМЮЕ ЗСЮШМ
С7Н7СМ№028 • ЗН20 М.м. 281,7
ОПРЕДЕЛЕНИЕ
Л/-Хлор-4-метилбензол-сульфонимидат натрия
тригидрат.
Содержание: не менее 98,0 % и не более 103,0 %
С7Н7С1Н№025 • ЗН20.
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтый кристаллический по¬
рошок.
Легко растворим в воде, растворим в 96 % спирте.
123*. Зак. 1060.
972
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 5, приготовленный как указано в
разделе «Испытания», окрашивает красную лакму¬
совую бумагу Р в синий и затем обесцвечивает ее.
B. К 10 мл раствора 5 прибавляют 10 мл раст¬
вора водорода пероксида разведенного Р. Образу¬
ется белый осадок, который растворяется при на¬
гревании. Горячий раствор фильтруют и охлажда¬
ют. Температура плавления (2.2.14) образовавших¬
ся белых кристаллов, промытых и высушенных при
температуре (100—105) °С: от 137 °С до 140 °С.
C. 1 г испытуемого образца осторожно (из-за
риска мгновенного сгорания) прокаливают. Раст¬
вор полученного остатка в 10 мл воды Р дает реак¬
цию (а) на хлориды (2.3.1).
О. Раствор, приготовленный в подлинности С,
дает реакцию (а) на сульфаты (2.3.1).
Е. Раствор, приготовленный в подлинности С,
дает реакцию (Ь) на натрий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 по степени
мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 8,0 до 10,0. Измеряют рН раст¬
вора 3.
Орто-замещенное соединение. К 2 г испы¬
туемого образца прибавляют 10 мл воды Р, пере¬
мешивают, прибавляют 1 г натрия метабисульфи¬
та Р и нагревают до кипения. Охлаждают до 0 °С,
быстро фильтруют и трижды промывают ледяной
водой Р порциями по 5 мл. Температура плавле¬
ния (2.2.14) осадка, высушенного над фосфора (V)
оксидом Р при давлении не более 600 Па: не ме¬
нее 134 °С.
Вещества, нерастворимые в этаноле. Не бо¬
лее 2 %. 1,00 г испытуемого образца встряхивают
с 20 мл этанола Р в течение 30 мин, фильтруют
через взвешенный фильтр, промывают осадок 5 мл
этанола Р и сушат при температуре от 100 °С до
105 °С. Масса остатка не должна превышать 20 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,125 г испытуемого образца помещают в
колбу с притертой пробкой и растворяют в 100 мл
воды Р. Прибавляют 1 г калия йодида Р и 5 мл
кислоты серной разведенной Р и выдерживают в
течение 3 мин. Титруют 0,1 М раствором натрия
тиосульфата, используя 1 мл раствора крахма¬
ла Р в качестве индикатора.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 14,08 мг С7Н7С1М1Ма023-ЗН20.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
07/2016:0439
а-ТОКОФЕРИЛАЦЕТАТ
М-гас-а-ТосорЬегуНз асе!аз
аИ-гас-а-ТОСОРНЕКУ1. АСЕТАТЕ
С31Н„03 М.м. 472,7
[7695-91-2]
ОПРЕДЕЛЕНИЕ
а11-гас-2,5,7,8-Тетраметил-2-(4,8,12-триметил-
тридецил)-3,4-дигидро-2/-/-1-бензопиран-6-илацетат.
#Витамин Е#.
Содержание: не менее 96,5 % и не более 102,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слегка зеленовато-
желтая вязкая маслянистая жидкость.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, в этаноле и в жирных маслах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Угол оптического вращения (2.2.7): от -0,01°
до +0,01°. 2,50 г испытуемого образца растворяют в
этаноле Р и доводят до объема 25,0 мл этим же раст¬
ворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО а-токоферилацетата #или
спектр, представленный на рисунке #0439.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 2 мл циклогексана Р.
Раствор сравнения. 10 мг ФСО а-токоферил¬
ацетата растворяют в 2 мл циклогексана Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: эфир Р — циклогексан Р
(20:80, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Сопутствующие примеси. Газовая хромато¬
графия (2.2.28): определение проводят методом вну¬
тренней нормализации.
Раствор внутреннего стандарта. 1,0 г сквала-
на Р растворяют в циклогексане Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
а- Токоферилацетат
973
3000
1000
2000 1500
Волновое число (см'1)
Рисунок #0439.-1. Инфракрасный спектр ФСО а-токоферилацетата
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в 10,0 мл внутреннего стандарт¬
ного раствора.
Испытуемый раствор (Ь). 0,100 г испытуемого
образца растворяют в 10 мл циклогексана Р.
Раствор сравнения (а). 0,100 г ФСО а-токофе-
рилацетата растворяют в 10,0 мл внутреннего стан¬
дартного раствора.
Раствор сравнения (Ь). 10 мг испытуемого об¬
разца и 10 мг а-токоферола Р растворяют в цикло¬
гексане Р и доводят до объема 100,0 мл этим же
растворителем.
Раствор сравнения (с). 10 мг ФСО а-токоферил-
ацетата для идентификации пиков (содержит при¬
меси А и В) растворяют в циклогексане Р и доводят до
объема 1 мл этим же растворителем.
Раствор сравнения (д). 1,0 мл испытуемого
раствора (Ь) доводят циклогексаном Р до объема
100,0 мл. 1,0 мл полученного раствора доводят ци¬
клогексаном Р до объема 10,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,25 мм, покрытая слоем поли(диметил)-
силоксана Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
- колонка: 280 °С;
- блок ввода проб и детектор: 290 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора (Ь) и растворов сравнения (а), (Ь), (с) и (с!)
(вводят непосредственно в колонку или через доста¬
точно инертный стеклянный вкладыш (лайнер) с ис¬
пользованием автоматического блока ввода проб или
другим воспроизводимым способом введения проб);
- время хроматографирования: 2-кратное вре¬
мя удерживания а-токоферилацетата.
Идентификация пиков примесей: идентифицируют
пики примесей А и В, используя хроматограмму, прила¬
гаемую к ФСО а-токоферилацетата для идентифика¬
ции пиков, и хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению
к а-токоферилацетату, время удерживания — около
15 мин): сквалан — около 0,4; примесь А— около 0,7;
примесь В — около 0,8; примесь С — около 0,9; при¬
меси О и Е — около 1,05 (элюируются сразу после
а-токоферилацетата).
Пригодность хроматографической системы:
- разрешение: не менее 3,5 между пиками при¬
меси С и а-токоферилацетата на хроматограмме
раствора сравнения (Ь);
- на хроматограмме раствора сравнения (а) пло¬
щадь пика примеси С должна составлять не более
чем 0,2 % от площади пика а-токоферилацетата.
Предельное содержание примесей:
- примесь А: не более 0,5 %;
- примесь В: не более 1,5 %;
- примесь С: не более 0,5 %;
- сумма примесей О и Е: не более 1,0 %;
- любая другая примесь: не более 0,25 %;
- сумма примесей: не более 2,5 %;
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора (Ь) не учитывают пики
с площадью менее площади основного пика на хро¬
матограмме раствора сравнения (с1).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
124. Зак. 1060.
974
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28), как указано в ис¬
пытании «Сопутствующие примеси», со следующими
изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 1 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- фактор асимметрии: не менее 0,6 для основ¬
ного пика.
Содержание С31Н5203 рассчитывают в процен¬
тах с учетом содержания а-токоферилацетата в ФСО
а-токоферилацетата.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
А. а11-гас-я?ранс-2,3,4,6,7-Пентаметил-2-(4,8,12-
триметилтридецил)-2,3-дигид робензофуран-5-илацетат.
В. а11-гас-фУС-2,3,4,6,7-Пентаметил-2-(4,8,12-
триметилтридецил)-2,3-дигидробензофуран-5-илацетат.
С. а11-гас-2,5,7,8-Тетраметил-2-(4,8,12-триметил-
тридецил)-3,4-дигидро-2Н-1-бензопиран-6-ол (аН-гас-
а-токоферол).
0.4-Метокси-2,3,6-триметил-5-[(аН-Я5,Е)-3,7,11,15-
тетраметилгексадек-2-енил]фенилацетат.
Е. (а11-Я5,аН-Е)-2,6,10,14,19,23,27,31 -Октаметил-
дотриаконта-12,14,18-триен.
07/2016:0692
а-ТОКОФЕРОЛ
1'гй-гас-а-ТосорНего1ит
а11-гас-а-ТОСОРНЕК01.
С.зН^О, М.м. 430,7
[10191-41-0]
ОПРЕДЕЛЕНИЕ
а11-гас-2,5,7,8-Тетраметил-2-(4,8,12-триметил-
тридецил)-3,4-дигидро-2Н-1 -бензопиран-6-ол. вита¬
мин Е#.
Содержание: не менее 96,0 % и не более 102,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или желтовато-коричне¬
вая вязкая маслянистая жидкость.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, в этаноле, в метиленхлориде и в жир¬
ных маслах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Угол оптического вращения (2.2.7): от -0,01°
до +0,01°. 2,50 г испытуемого образца растворяют в
этаноле Р и доводят до объема 25,0 мл этим же раст¬
ворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО а-токоферола #или спектр,
представленный на рисунке #0692.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 2 мл циклогексана Р.
Раствор сравнения. 10 мг ФСО а-токоферола
растворяют в 2 мл циклогексана Р.
Пластинка: ТСХ пластинка со слоем силикаге-
пяР264Р.
Подвижная фаза: эфир Р — циклогексан Р
(20:80, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Сопутствующие примеси. Газовая хромато¬
графия (2.2.28): определение проводят методом вну¬
тренней нормализации.
Раствор внутреннего стандарта. 1,0 г сквала-
на Р растворяют в циклогексане Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
а -Токоферол
975
Рисунок #0692.-1. Инфракрасный спектр ФСО а-токоферола
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в 10,0 мл внутреннего стандарт¬
ного раствора.
Испытуемый раствор (Ь). 0,100 г испытуемого
образца растворяют в 10 мл циклогексана Р.
Раствор сравнения (а). 0,100 г ФСО а-токофе-
рола растворяют в 10,0 мл внутреннего стандартного
раствора.
Раствор сравнения (Ь). 10 мг испытуемого об¬
разца и 10 мг а-токоферилацетата Р растворяют в
циклогексане Р и доводят до объема 100,0 мл этим
же растворителем.
Раствор сравнения (с). 10 мг ФСО а-токоферола
для идентификации пиков (содержит примеси А и В)
растворяют в циклогексане Р и доводят до объема
1 мл этим же растворителем.
Раствор сравнения (д). 1,0 мл испытуемого
раствора (Ь) доводят циклогексаном Р до объема
100,0 мл. 1,0 мл полученного раствора доводят ци¬
клогексаном Р цо объема 10,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,25 мм, покрытая слоем поли(диметил)
силоксана Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
- колонка: 280 °С;
- блок ввода проб и детектор: 290 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора (Ь) и растворов сравнения (Ь), (с) и (д);
- время хроматографирования: 2-кратное вре¬
мя удерживания а-токоферола.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму,
прилагаемую к ФСО а-токоферолу для идентифика¬
ции пиков, и хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению к
а-токоферолу, время удерживания — около 9 мин):
сквалан — около 0,5; примесь А— около 0,7; примесь
В — около 0,8; примеси СиЭ — около 1,05 (элюиру¬
ются сразу после а-токоферола).
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,5 между пиками
а-токоферола и а-токоферилацетата.
Предельное содержание примесей:
- примесь А: не более 0,5 %;
- примесь В: не более 1,5 %;
- сумма примесей С и О: не более 1,0 %;
- любая другая примесь: не более 0,25 %;
- сумма примесей: не более 2,5 %;
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора (Ь) не учитывают пики
с площадью менее площади основного пика на хро¬
матограмме раствора сравнения (б).
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28), как указано в ис¬
пытании «Сопутствующие примеси», со следующими
изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 1 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
124*. Зак. 1060.
976
Гэсударственная фармакопея Республики Беларусь
- фактор асимметрии: не менее 0,6 для основ¬
ного пика.
Содержание С29Н50О2 рассчитывают в процентах
с учетом содержания а-токоферола в ФСО а-токо-
ферола
ХРАНЕНИЕ
В атмосфере инертного газа, в защищенном от
света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. а11-гас-л?ранс-2,3,4,6,7-Пентаметил-2-(4,8,12-
триметилтридецил)-2,3-дигидробензофуран-5-ол.
триметилтридецил)-2,3-дигидробензофуран-5-ол.
С. 4-Метокси-2,3,6-триметил-5-[(а11-Я5,Е)-3,7,11,15-
тетраметилгексадек-2-енил]фенол.
О. (а11-Я5,аИ-Е)-2,6,10,14,19,23,27,31 -Октаметил-
дотриаконта-12,14,18-триен.
07/2016:1257
ЯЯ^а-ТОКОФЕРИЛАЦЕТАТ
ЯЯЯ-а- ТосорЬегуНз асе(аз
ЯЯЯ-а-ГОСОРЯЕЯУТ. АСЕТАТЕ
ОПРЕДЕЛЕНИЕ
(2/?)-2,5,7,8-Тетраметил-2-[(4/?,8#)-4,8,12-три-
метилтридецил]-3,4-дигидро-2Н-1-бензопиран-6-
илацетат. #Витамин Е#.
Содержание: не менее 95,0 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слегка зеленовато-
желтая вязкая маслянистая жидкость.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, в этаноле и в жирных маслах, раство¬
рим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Угол оптического вращения (2.2.7): от +0,25°
до +0,35°. 2,50 г испытуемого образца растворяют в
этаноле Р и доводят до объема 25,0 мл этим же раст¬
ворителем.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО а-токоферилацетата #или
спектр, представленный на рисунке #1257.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 10 мг испытуемого об¬
разца растворяют в 2 мл циклогексана Р.
Испытуемый раствор (Ь). В пробирке с при¬
тертой стеклянной пробкой растворяют 10 мг испы¬
туемого образца в 2 мл 2,5 А4 спиртового раствора
кислоты серной Р и нагревают на водяной бане в
течение 5 мин. Охлаждают, прибавляют 2 мл воды Р
и 2 мл циклогексана Р. Встряхивают в течение 1 мин.
Используют верхний слой.
Раствор сравнения (а). 10 мг ФСО а-токоферил-
ацетата растворяют в 2 мл циклогексана Р.
Раствор сравнения (Ь). Готовят аналогич¬
но испытуемому раствору (Ь), используя ФСО
а-токоферилацетата вместо испытуемого образца.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР2В4Р.
Подвижная фаза: эфир Р — циклогексан Р
(20:80, об/об).
Наносимый объем пробы. 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора (а) обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному пятну
на хроматограмме раствора сравнения (а). На хромато¬
граммах испытуемого раствора (Ь) и раствора сравне¬
ния (Ь) допускается два пятна в зависимости от степени
гидролиза: пятно с более высоким значением /?, отно¬
сится к а-токоферилацетату и соответствует пятну на
хроматограмме раствора сравнения (а); пятно с более
низким значением соответствует а-токоферолу.
ИСПЫТАНИЯ
Сопутствующие примеси. Газовая хромато¬
графия (2.2.28): определение проводят методом вну¬
тренней нормализации.
Раствор внутреннего стандарта. 1,0 г сквала-
на Р растворяют в циклогексане Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
ККК-а-Токоферилацетат
977
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в 10,0 мл внутреннего стандарт¬
ного раствора.
Испытуемый раствор (Ь). 0,100 г испытуемого
образца растворяют в 10 мл циклогексана Р.
Раствор сравнения (а). 0,100 г ФСО а-токофе-
рилацетата растворяют в 10,0 мл внутреннего стан¬
дартного раствора.
Раствор сравнения (Ь). 10 мг а-токоферола Р и
10 мг а-токоферилацетата Р растворяют в цикло¬
гексане Р и доводят до объема 100,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,25 мм, покрытая слоем поли(диметил)-
силоксана Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—15
280
Блок ввода проб
290
Детектор
290
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь) (вводят
непосредственно в колонку или через достаточно
инертный стеклянный вкладыш (лайнер) с использо¬
ванием автоматического блока ввода проб или другим
воспроизводимым способом введения проб).
Пригодность хроматографической системы:
- разрешение: не менее 3,5 между пиками
а-токоферола и а-токоферилацетата на хроматограм¬
ме раствора сравнения (Ь);
- на хроматограмме раствора сравнения (а) пло¬
щадь пика а-токоферола должна составлять не более
чем 0,2 % от площади пика а-токоферилацетата.
Предельное содержание примесей:
- сумма примесей: не более 4,0 %;
- неучитываемый предел: 0,1 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28), как указано в ис¬
пытании «Сопутствующие примеси», со следующими
изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 1 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а).
- фактор асимметрии: не менее 0,6 для основ¬
ного пика.
Содержание С31Н5203 рассчитывают в процен¬
тах с учетом содержания а-токоферилацетата в ФСО
а-токоферилацетата.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Использу¬
ют свежеприготовленные растворы.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в 2-пропаноле Р и доводят до объ¬
ема 50,0 мл этим же растворителем. 2,0 мл получен¬
ного раствора доводят 2-пропанолом Р до объема
25,0 мл.
Раствор сравнения. 0,100 г ФСО а-токоферил¬
ацетата растворяют в 2-пропаноле Р и доводят до
объема 50,0 мл этим же растворителем. 2,0 мл полу-
125. Зак. 1060.
978
Государственная фармакопея Республики Беларусь
ченного раствора доводят 2-пропанолом Р до объема
25,0 мл.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,15 м
и внутренним диаметром 2,1 мм, заполненная сили¬
кагелем октилсилильным для хроматографии Р, с
размером частиц 3,5 мкм;
- подвижная фаза: вода Р — метанол Р (2:98,
об/об);
- скорость подвижной фазы: 0,25 мл/мин;
-спектрофотометрический детектор, длина
волны 285 нм;
- объем вводимой пробы: 5 мкл.
Содержание С31Н5203 рассчитывают в процен¬
тах с учетом содержания а-токоферилацетата в ФСО
а-токоферилацетата.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (2Я)-2,5,7,8-Тетраметил-2-[(4Я,8Я)-4,8,12-три-
метилтридецил]-3,4-дигидро-2Н-1-бензопиран-6-ол
(/?/?Я-а-токоферол).
B. Р = Н, Р' = СН3: (2Я)-2,5,8-Триметил-2-[(4Я,8Я)-
4.8.12- триметилтридецил]-3,4-дигидро-2Н-1 -бензопиран-
6-илацетат (ЯЯЯ-Р-токоферилацетат).
C. В = СН3, Я' = Н: (2Я)-2,7,8-Триметил-2-[(4Я,8Я)-
4.8.12- триметилтридецил]-3,4-дигид ро-2Н-1 -бензопиран-
6-илацетат (РРР-у-токоферилацетат).
О. р = р' = н: (2Я)-2,8-Диметил-2-[(4/?,8Я)-4,8,12-
триметилтридецил]-3,4-дигидро-2Н-1-бензопиран-6-
илацетат (РРР-5-токоферилацетат).
07/2016:0304
ТОЛБУТАМИД
То1Ьи1атШит
Т01-ВиТАМЮЕ
С12Н1вМ2°з8 М.М. 270»3
[64-77-7]
ОПРЕДЕЛЕНИЕ
1-Бутил-3-[(4-метилфенил)сульфонил]мочевина.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, растворим в
ацетоне и в 96 % спирте. Растворяется в разведенных
растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: А, В, О.
A. Температура плавления {2.2.14): от 126 °С до
130 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор (а). 25,0 мг испытуемого
образца растворяют в метаноле Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Испытуемый раствор (Ь). 10,0 мл испытуемо¬
го раствора (а) доводят метанолом Р до объема
250.0 мл.
Диапазон длин волн: от 245 нм до 300 нм для ис¬
пытуемого раствора (а); от 220 нм до 235 нм для ис¬
пытуемого раствора (Ь).
Максимумы поглощения: при 258 нм, 263 нм и
275 нм для испытуемого раствора (а); при 228 нм для
испытуемого раствора (Ь).
Плечо: при 268 нм для испытуемого раствора (а).
Удельный показатель поглощения в максимуме:
от 480 до 520 для испытуемого раствора (Ь).
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО толбутамида.
й. К 0,2 г испытуемого образца прибавляют 8 мл
раствора 500 г/л кислоты серной Р, нагревают с об¬
ратным холодильником в течение 30 мин и охлажда¬
ют. Температура плавления (2.2.14) полученных кри¬
сталлов, перекристаллизованных из горячей воды Р
и высушенных при температуре 105 °С: от 135 °С до
140 °С.
ИСПЫТАНИЯ
Раствор 5. 0,2 г испытуемого образца раство¬
ряют в 5 мл раствора натрия гидроксида разведен¬
ного Р и прибавляют 5 мл воды Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,5 до 5,5. К 2,0 г испытуемого об¬
разца прибавляют 50 мл воды, свободной от углеро¬
да диоксида, Р и нагревают при температуре 70 °С в
течение 5 мин. Быстро охлаздают и фильтруют.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед применением.
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 10 мг толуолсульфона-
мида Р (примесь А) и 10 мг толуолсульфонилмоче-
вины Р (примесь В) растворяют в подвижной фазе и
доводят до объема 10,0 мл этим же растворителем.
Толнафтат
979
1.0 мл полученного раствора доводят до объема
100.0 мл подвижной фазой.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: смешивают 35 объемов аце¬
тонитрила Р1 с 65 объемами раствора 1,36 г/л калия
дигидрофосфата Р, доведенного до рН 3,5 кислотой
фосфорной Р.
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор: длина
волны 230 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания толбутамида.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь).
Относительное удерживание (по отношению к
толбутамиду, время удерживания — около 18 мин):
примесь В — около 0,2; примесь А — около 0,3.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 2,0 между пиками при¬
меси А и примеси В.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод В). Не более
0,0010% (Юррт). 1,0 г испытуемого образца раст¬
воряют в смеси вода Р — ацетон Р (15:85, об/об) и
доводят до объема 20 мл этой же смесью раство¬
рителей. 12 мл полученного раствора должны вы¬
держивать испытание на тяжелые металлы. Эталон
готовят с использованием эталонного раствора свин¬
ца (0,5 ррт РЬ), полученного путем разведения эта¬
лонного раствора свинца (100 ррт РЬ) Р смесью
вода Р — ацетон Р (15:85, об/об).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в сме¬
си из 20 мл воды Р и 40 мл 96 % спирта Р и титруют
0,1 М раствором натрия гидроксида, используя в ка¬
честве индикатора 1 мл раствора фенолфталеина Р.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 27,03 мг С12Н18М2035.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С.
С. 1-Азепан-1-ил-3[(4-метилфенил)сульфонил]-
мочевина (толазамид).
07/2016:1158
ТОЛНАФТАТ
То1паЯа(ит
Т01-МАРТАТЕ
С19Н17М05 М.м. 307,4
[2398-96-1]
ОПРЕДЕЛЕНИЕ
0-Нафтален-2-ил-метил(3-метилфенил)тиокар-
бамат.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок.
Практически нерастворим в воде, легко раство¬
рим в ацетоне и в метиленхлориде, очень мало рас¬
творим в 96 % спирте.
125*. Зак. 1060.
980
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО толнафтата #или спектр,
представленный на рисунке #1158.-1#.
ИСПЫТАНИЯ
Примесь й. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 0,400 г испытуемого об¬
разца растворяют в 2 мл метиленхлорида Р и триж¬
ды экстрагируют 0,01 М раствором кислоты хлори¬
стоводородной порциями по 3 мл. Водные извлече¬
ния объединяют и доводят 0,01 М раствором кисло¬
ты хлористоводородной до объема 10,0 мл.
Раствор сравнения (а). 20,0 мг Ы-метил-м-
толуидина Р (примесь Р) растворяют в 50,0 мл ме¬
тиленхлорида Р.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят метиленхлоридом Р до объема
100,0 мл. 2,0 мл полученного раствора трижды экс¬
трагируют 0,01 М раствором кислоты хлористо¬
водородной порциями по 3 мл. Водные извлечения
объединяют и доводят 0,01 М раствором кислоты
хлористоводородной до объема 10,0 мл.
Раствор сравнения (с). 10 мг испытуемого об¬
разца растворяют в 25 мл метанола Р. 2 мл получен¬
ного раствора прибавляют к 2 мл раствора сравнения
(а) и доводят метанолом Р до объема 25 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: трифторуксусная кисло¬
та Р—метанол Р — вода Р (0,1:10:90, об/об/об);
- подвижная фаза В: трифторуксусная кисло¬
та Р — вода Р — метанол Р (0,1:10:90, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
70
30
3—8
70-0
30 — 100
8—20
0
100
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
-объем вводимой пробы: по 100 мкл испытуе¬
мого раствора и раствора сравнения (Ь); 10 мкл раст¬
вора сравнения (с).
Относительное удерживание (по отношению
к толнафтату, время удерживания — около 15 мин):
примесь й — около 0,25.
Пригодность хроматографической системы:
- разрешение: не менее 5,0 между пиками при¬
меси О и толнафтата на хроматограмме раствора
сравнения (с);
- фактор асимметрии: не более 1,9 для пика
примеси О на хроматограмме раствора сравнения (Ь).
Предельное содержание примесей:
- примесь О (не более 20 ррт): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси О, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 5 мл метанола Р и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
1,0 мл полученного раствора доводят метанолом Р
до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО толнафтата
для проверки пригодности хроматографической си¬
стемы (содержит компонент А для проверки разре¬
шения) растворяют в 5 мл метанола Р.
Рисунок #1158.-1. Инфракрасный спектр ФСО толнафтата
Трагакант
981
Условия хроматографирования:
- колонка 0,15 м и внутренним диаметром 4,6 мм,
заполненная силикагелем октадецилсилильным для
хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: трифторуксусная кисло¬
та Р — вода Р—метанол Р (0,1:30:70, об/об/об);
- подвижная фаза В: трифторуксусная кисло¬
та Р — вода Р—метанол Р (0,1:10:90, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—12
100
0
12—30
о
т
о
о
0 — 100
30—33
0
100
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к толнафтату, время удерживания — около 18 мин):
разрешающий компонент А— около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 5,0 между пиком компо¬
нента А для проверки разрешения и пиком толнафтата.
Предельное содержание примесей:
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 60 °С и давлении не более 0,7 кПа в те¬
чение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50,0 мг испытуемого образца растворяют в ме¬
таноле Р и доводят до объема 250,0 мл этим же раст¬
ворителем. 2,0 мл полученного раствора доводят ме¬
танолом Р до объема 50,0 мл. Измеряют оптическую
плотность (2.2.25) полученного раствора в максимуме
при 257 нм.
Содержание С19Н17ЫОЗ рассчитывают с учетом
удельного показателя поглощения, равного 720.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В.
А. Нафтален-2-ол (Р-нафтол).
В. 0,0-Динафтален-2-илтиокарбонат.
СН3
О. Л/,3-Диметиланилин (Л/-метил-/и-толуидин).
07/2016:0532
ТРАГАКАНТ
ТгадасапМа
ТКАСАСАЫТН
[9000-65-1]
ОПРЕДЕЛЕНИЕ
Застывшая на воздухе камедь, истекающая есте¬
ственным путем или получаемая надрезанием ствола
и ветвей Аз1гада!из диттКег 1_аЬШ. и некоторых дру¬
гих видов Аз&ада1из из западной Азии.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Трагакант представляет собой тонкие упло¬
щенные лентовидные белые или бледно-желтые
полупрозрачные полоски длиной около 30 мм, ши¬
риной 10 мм и толщиной до 1 мм, слегка изогнутые,
ороговевшие, с хрупким изломом; на поверхности на¬
блюдается мелкая продольная исчерченность и кон¬
центрические пересекающиеся ребра. Могут также
содержаться кусочки, сходные по форме, но немного
толще, менее прозрачные и менее хрупкие.
B. Испытуемый образец измельчают (355)
(2.9.12). После измельчения представляет собой бе¬
лый или почти белый порошок, образующий вязкий
гель при добавлении воды Р в количестве, превыша¬
ющем массу испытуемого образца примерно в 10 раз.
Просматривают под микроскопом, используя 50 %
(об/об) раствор глицерина Р. В микропрепарате по¬
рошка в смолистой массе должны наблюдаться мно¬
гочисленные слоистые клеточные мембраны, которые
медленно приобретают фиолетовую окраску при об¬
работке раствором цинка хлорида йодированным Р.
Смолистая масса содержит в себе крахмальные зер¬
на, отдельные или в небольших группах, обычно окру¬
глой формы и иногда деформированные, от 4 мкм до
10 мкм в диаметре, изредка до 20 мкм, с центральным
982
Гэсударственная фармакопея Республики Беларусь
ядром, видимым при рассмотрении через перекрещи¬
вающиеся призмы Николя.
С. Просматривают хроматограммы, полученные
в испытании «Гуммиарабик».
Результаты: на хроматограмме испытуемого
раствора обнаруживается 3 зоны, соответствующие
галактозе, арабинозе и ксилозе. Могут присутство¬
вать бледная желтоватая зона на уровне фронта под¬
вижной фазы и серовато-зеленая зона между зонами
галактозы и арабинозы.
Р. 0,5 г измельченного испытуемого образца
(355) (2.9.12) увлажняют 1 мл 96% спирта Р и посте¬
пенно прибавляют при встряхивании 50 мл воды Р до
получения гомогенной слизи. К 5 мл слизи прибавля¬
ют 5 мл воды Р и 2 мл раствора бария гидроксида Р.
Образуется незначительный хлопьевидный осадок.
Нагревают на водяной бане в течение 10 мин. Появ¬
ляется интенсивное желтое окрашивание.
ИСПЫТАНИЯ
Гуммиарабик. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 100 мг измельченного ис¬
пытуемого образца (355) (2.9.12) помещают в толсто¬
стенную центрифужную пробирку и прибавляют 2 мл
раствора 100 г/л трифторуксусной кислоты Р, интен¬
сивно встряхивают до растворения образовавшегося
геля, укупоривают пробирку и нагревают смесь при тем¬
пературе 120 °С в течение 1 ч. Полученный гидролизат
центрифугируют, осторожно переносят прозрачную
надосадочную жидкость в колбу вместимостью 50 мл,
прибавляют 10 мл воды Р и досуха выпаривают раст¬
вор при пониженном давлении досуха. К полученной
прозрачной пленке прибавляют 0,1 мл воды Р и 0,9 мл
метанола Р. Центрифугируют до отделения аморфно¬
го осадка, собирают надосадочную жидкость и, при не¬
обходимости, доводят метанолом Р до объема 1 мл.
Раствор сравнения. 10 мг арабинозы Р, 10 мг га¬
лактозы Р, 10 мг рамнозы Р и 10 мг ксилозы Р раст¬
воряют в 1 мл воды Р и доводят метанолом Р до объ¬
ема 10 мл.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: раствор 16 г/л натрия диги¬
дрофосфата Р — бутанол Р — ацетон Р (10:40:50,
об/об/об).
Наносимый объем пробы: 10 мкл в виде полос.
Фронт подвижной фазы А: не менее 10 см от ли¬
нии старта.
Высушивание А: в потоке теплого воздуха в тече¬
ние нескольких минут.
Фронт подвижной фазы В: не менее 15 см от ли¬
нии старта с использованием той же подвижной фазы.
Высушивание В: при температуре 110 °С в тече¬
ние 10 мин.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р и сушат при температуре
110 °С в течение 10 мин.
Результаты: на хроматограмме раствора срав¬
нения обнаруживаются 4 окрашенные полностью
разделенные зоны в порядке увеличения Рр, соответ¬
ствующие галактозе (серовато-зеленая или зеленая),
арабинозе (желтовато-зеленая), ксилозе (зеленовато¬
серая или желтовато-серая) и рамнозе (желтовато¬
зеленая); на хроматограмме испытуемого раствора
не обнаруживается желтовато-зеленая зона, соответ¬
ствующая зоне рамнозы на хроматограмме раствора
сравнения.
Метилцеллюлоза. Просматривают хромато¬
граммы, полученные в испытании «Гуммиарабик».
Результаты: на хроматограмме испытуемо¬
го раствора не обнаруживается красная зона возле
фронта подвижной фазы.
Камедь стеркулии
A. 0,2 г измельченного испытуемого образца (355)
(2.9.12) помещают в цилиндр с притертой пробкой
вместимостью 10 мл с ценой деления 0,1 мл. Прибав¬
ляют 10 мл спирта (60 %, об/об) Р и встряхивают. Об¬
разовавшийся гель должен занимать не более 1,5 мл.
B. К 1,0 г измельченного испытуемого образца
(355) (2.9.12) прибавляют 100 мл воды Р, встряхивают
и прибавляют 0,1 мл раствора метилового красно¬
го Р. При прибавлении не более 5,0 мл 0,01 М раст¬
вора натрия гидроксида окрашивание индикатора
должно измениться.
Посторонние примеси. Не более 1,0 %.
2.0 г измельченного испытуемого образца (355)
(2.9.12) помещают в круглодонную колбу вместимо¬
стью 250 мл и прибавляют 95 мл метанола Р. Пере¬
мешивают до увлажнения порошка и прибавляют
60 мл кислоты хлористоводородной Р1. Прибавляют
несколько стеклянных шариков диаметром около 4 мм
и нагревают с обратным холодильником на водяной
бане в течение 3 ч при периодическом встряхивании.
Удаляют стеклянные шарики и фильтруют горячую су¬
спензию под вакуумом через стеклянный фильтр (160)
(2.1.2). Колбу промывают небольшим количеством
воды Р и пропускают смыв через фильтр. Промывают
остаток на фильтре около 40 мл метанола Р и сушат
до постоянной массы при температуре 110 °С (около
1 ч). Охлаждают в эксикаторе и взвешивают. Масса
сухого остатка не должна превышать 20 мг.
Время истечения. Не менее 10 с или не менее
50 с, если субстанция применяется для приготовле¬
ния эмульсий.
1.0 г измельченного испытуемого образца (125—250)
(2.9.12) помещают в круглодонную колбу с притертой
стеклянной пробкой вместимостью 1000 мл, прибав¬
ляют 8,0 мл 96 % спирта Р и укупоривают колбу. Су¬
спензию диспергируют на внутренней поверхности
колбы при встряхивании, принимая меры предосто¬
рожности, чтобы не смочить пробку. Открывают кол¬
бу и в один прием прибавляют 72,0 мл воды Р. Уку¬
поривают колбу и интенсивно встряхивают в течение
3 мин. Выдерживают в течение 24 ч и снова интен¬
сивно встряхивают в течение 3 мин. Удаляют пузырь¬
ки воздуха действием вакуума над слизью в течение
5 мин. Слизь переносят в цилиндр вместимостью
50 мл и погружают в нее часть стеклянной трубки дли¬
ной 200 мм и внутренним диаметром 6,0 мм, трубка
должна быть градуирована в диапазоне от 20 мм до
120 мм от нижнего конца; трубка не должна быть про¬
мыта поверхностно-активными веществами. Как толь¬
ко слизь достигнет верхней метки, трубку закрывают
пальцем. Вынимают закрытую трубку, убирают палец
и измеряют секундомером время, необходимое для
достижения мениском нижнего уровня шкалы. Проце¬
дуру выполняют 4 раза и определяют среднее значе¬
ние 3 последних определений.
Общая зола (2.4.16). Не более 4,0 %.
Микробиологическая чистота
- ОКА: критерий приемлемости 104 КОЕ/г (2.6.12);
- ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12);
- ЕзсЬепсЫа соН: отсутствие (2.6.13);
- 8а1топе11а: отсутствие (2.6.13).
Трамадола гидрохлорид
983
МАРКИРОВКА
Указывают, пригодно ли содержимое для приго¬
товления эмульсий.
07/2016:1681
ТРАМАДОЛА ГИДРОХЛОРИД
ТгатадоН ЬуйгосЫопдит
ГЯАШОО/. НУОКОСШ-СтЮЕ
Н3С. ХН3
N
н
он
ОСН3
и энантиомер НС1
С1вН25М02 ■ НС! М.м. 299,8
[36282-47-0]
ОПРЕДЕЛЕНИЕ
(1 Я5,2#5)-2-[(Диметиламино)метил]-1 -(3-метокси-
фенил)циклогексанола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в метаноле, очень мало
растворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14). От 180 °С до
184 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО трамадола.
C. Просматривают хроматограммы, полученные
в испытании «Примесь Е».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
й. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность. К 10 мл раствора 3 прибавляют
0,2 мл раствора метилового красного Р и 0,2 мл
0,01 М раствора кислоты хлористоводородной.
Раствор должен иметь красное окрашивание. При при¬
бавлении не более 0,4 мл 0,01 М раствора натрия
гидроксида должно появиться желтое окрашивание.
Угол оптического вращения (2.2.7). От -0,10°
до +0,10°. Измеряют угол оптического вращения раст¬
вора 3.
Примесь Е. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в метаноле Р и доводят до объ¬
ема 2 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят метанолом Р до объема 10 мл.
Раствор сравнения (а). 25 мг ФСО трамадола
гидрохлорида растворяют в метаноле Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 5 мг ФСО трамадола при¬
меси Е растворяют в 5 мл метанола Р. 1 мл получен¬
ного раствора доводят метанолом Р до объема 10 мл.
Раствор сравнения (с). 5 мг ФСО трамадола
примеси А растворяют в 1 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р254 Р, предварительно промытая метанолом Р.
Подвижная фаза: раствор аммиака концентриро¬
ванный Р — 2-пропанол Р—толуол Р( 1:19:80, об/об/об).
Наносимый объем пробы. 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки, пластинку насыщают раствором аммиака
концентрированным Р в течение 20 мин. Для этого
раствор аммиака концентрированный Р помещают
в один из двух желобов камеры. Непосредственно
перед началом хроматографирования помещают под¬
вижную фазу в другой желоб, пластинку помещают в
хроматографическую камеру, удостоверившись, что
слой силикагеля ориентирован по направлению к
середине камеры #(слой силикагеля должен быть на¬
правлен к желобу с раствором для насыщения)#.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в йодной
камере в течение 1 ч и просматривают в ультрафио¬
летовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- примесь Е (не более 0,2 %): на хроматограмме
испытуемого раствора (а) пятно, соответствующее при¬
меси Е, должно превышать пятно на хроматограмме
раствора сравнения (Ь) по интенсивности и размеру.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,15 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100 мл этим же растворителем.
Раствор сравнения (а). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 10,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100 мл.
Раствор сравнения (Ь). 5 мг ФСО трамадола
примеси А растворяют в 4,0 мл испытуемого раствора
и доводят подвижной фазой до объема 100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, эндкепированным для хроматографии Р с разме¬
ром частиц 5 мкм;
984
Государственная фармакопея Республики Беларусь
-подвижная фаза: 295 объемов ацетонитри¬
ла Р и 705 объемов смеси из 0,2 мл кислоты триф-
торуксусной Р и 100 мл воды Р\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 270 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания трамадола.
Относительное удерживание (по отношению к
трамадолу, время удерживания — около 5 мин): при¬
месь А — около 0,85.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками при¬
меси А и трамадола.
Предельное содержание примесей:
-примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси А, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,4 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,02 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят этим же растворителем до
объема 20 мл. 12 мл полученного раствора должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раствора
свинца (2 ррт РЬ) Р.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,000 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,180 г испытуемого образца растворяют в 25 мл
кислоты уксусной безводной Р, прибавляют 10 мл ан¬
гидрида уксусного Р и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 29,98 мг С16Н25М02 НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
Ат
/ -
ОСН3
Н3С\ /СН3
N
АГ
и энантиомер
ОН
А. (1 Р5,25Р)-2-[(Диметиламино)метил]-1 -(3-
метоксифенил)циклогексанол.
Н3С\ /СНз
N
В. [2-(3-Метоксифенил)циклогекс-1 -енил]-А/,А/-
диметилметанамин.
и энантиомер
С. (1 Р5)-[2-(3-Метоксифенил)циклогекс-2-енил]-
А/,А/-диметилметанамин.
Н3С^ /СН;
и энантиомер
О. (1Р5,2Р5)-2-[(Диметиламино)метил]-1-(3-
гидроксифенил)циклогексанол.
Н3С^ /СН3
N
и энантиомер
Е. (2Я5)-2-[(Диметиламино)метил]циклогексанон.
07/2016:1049
ТРЕОНИН
ТЬгеоптит
ТНПЕОЫШЕ
С.Н.ИО, М.м. 119,1
[72-19-5]
Треонин
985
ОПРЕДЕЛЕНИЕ
(25,3#)-2-Амино-3-гидроксибутановая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Растворим в воде, практически нерастворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО треонина #или спектр, пред¬
ставленный на рисунке #1049.-1*
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, со¬
ответствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения (а).
0.1 мл раствора 2 г/л испытуемого образца сме¬
шивают с 1 мл раствора 20 г/л натрия перйодата Р,
прибавляют 0,2 мл пиперидина Р и 0,1 мл раствора
25 г/л натрия нитропруссида Р. Появляется синее
окрашивание, переходящее через несколько минут в
желтое.
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 5,0 до 6,5. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
-27,6 до -29,0 (в пересчете на сухое вещество). 1,50 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Нингидрин-положительные вещества. Испы¬
тание проводят одним из приведенных ниже методов.*
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образца, подходящий для применяемого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
986
Гэсударственная фармакопея Республики Беларусь
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт Л/Н^ Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (д). 30 мг изолейцина Р (при¬
месь й) и 30 мг лейцина Р растворяют в растворе А и
доводят до объема 50,0 мл этим же растворителем.
1,0 мл полученного раствора доводят раствором А до
объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь) и (с!) вводят в аминокислотный анализатор.
Запускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (с1):
- разрешение: не менее 1,5 между пиками при¬
меси Э и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию треонина в растворе сравне¬
ния (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
- любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для фар¬
мацевтического использования (2034), не применяют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в кислоте хлористоводородной
разведенной Р и доводят до объема 10 мл этим же
растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО треонина
растворяют в 1 % (об/об) растворе кислоты хлори¬
стоводородной Р и доводят до объема 50 мл этим же
растворителем.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (с). 10 мг ФСО треонина
и 10 мг ФСО пролина растворяют в 1 % (об/об) рас¬
творе кислоты хлористоводородной Р и доводят до
объема 25 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром нингидрина Р и высушивают при температуре от
100 °С до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200% (200 ррт).
10 мл раствора 3 доводят водой Рдо 15 мл. Полученный
раствор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
0,5 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 15 мл этим же
растворителем. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато¬
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 0,10 г испытуемого
образца должны выдерживать испытание на аммония
соли. Эталон готовят с использованием 0,2 мл эта¬
лонного раствора аммония (100ррт NН^ Р*
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 0,5 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 0,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 5 мл
кислоты муравьиной безводной Р, прибавляют 30 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 11,91 мгС4Н9М03.
Триамцинолона ацетонид
987
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимити¬
руется общим критерием приемлемости для других/не-
специфицированных примесей. Вследствие этого нет
необходимости идентифицировать эти примеси для
доказательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е.
Н ИНг
Х02Н
A. (25)-2-Амино-3-гидроксибутановая кислота (го¬
мосерин).
V-
Н3С^ТХ)2Н
B. (25)-2-Аминопропановая кислота (аланин).
Н Ь1Н2
нзс\ ЗЧ
со2н
СНз
лин).
С. (25)-2-Амино-3-метилбутановая кислота (ва-
н3с
Н СН3
й. (25,35)-2-Амино-3-метилбутановая кислота (изо¬
лейцин).
Н ИНг
н2ы' ^ ^ "со2н
Е. (25)-2,6-Диаминогексановая кислота (лизин).
07/2016:0533
ТРИАМЦИНОЛОНА АЦЕТОНИД
Тпатсто1оп1 асе(отс1ит
ТШАМС1Ы01-0ЫЕ А СЕТОЫЮЕ
ОН
СНз
СНз
[76-25-5)
М.м. 434,5
ОПРЕДЕЛЕНИЕ
9-Фтор-11 р,21 -дигидрокси-16а, 17-(1 -метилэтил-
идендиокси)прегна-1,4-диен-3,20-дион.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, умеренно рас¬
творим в 96 % спирте.
Обладает полиморфизмом (5.9).
Рисунок. #0533.-1 Инфракрасный спектр ФСО триамцинолона ацетонида
988
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С.
Вторая идентификация: В, О.
A. Абсорбционная спектрофотометрия в ин¬
фракрасной области (2.2.24).
Сравнение: ФСО триамцинолона ацетонида
#или спектр, представленный на рисунке #0533.-1#.
Если спектры, полученные с использовани¬
ем образцов в твердом состоянии, отличаются, то
испытуемый образец и ФСО триамцинолона аце¬
тонида растворяют по отдельности в минималь¬
ном объеме метанола Р, выпаривают до сухих
остатков и готовят диски с солями галогенидов или
эмульсии в масле вазелиновом Р, которые исполь¬
зуют для получения новых спектров.
B. Тонкослойная хроматография (2.2.27). Рас¬
творы готовят непосредственно перед исполь¬
зованием и защищают от света.
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО триамци¬
нолона ацетонида растворяют в метаноле Р и до¬
водят до объема 20 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО триам¬
цинолона гексацетонида растворяют в растворе
сравнения (а) и доводят до объема 10 мл этим же
растворителем.
Пластинка: ТСХ пластинка со слоем подхо¬
дящего силикагеля Р254 Р.
Подвижная фаза: к смеси из 15 объемов эфи¬
ра Р и 77 объемов метиленхлорида Р прибавляют
смесь из 1,2 объема воды Р и 8 объемов метано¬
ла Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: немедленно после хроматогра¬
фирования пластинку просматривают в ультрафи¬
олетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два пол¬
ностью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее
по расположению и размеру основному пятну на
хроматограмме раствора сравнения (а).
C. Просматривают хроматограммы, получен¬
ные как указано в разделе «Количественное опре¬
деление».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хрома¬
тограмме раствора сравнения (с).
0. 5 мг испытуемого образца смешивают с
45 мг магния оксида тяжелого Р и сжигают в ти¬
гле до получения почти белого остатка (обычно ме¬
нее 5 мин). Охлаждают, прибавляют 1 мл воды Р,
0,05 мл раствора фенолфталеина Р1 и около
1 мл кислоты хлористоводородной разведен¬
ной Р до обесцвечивания раствора и фильтруют.
1,0 мл полученного фильтрата прибавляют к све¬
жеприготовленной смеси из 0,1 мл раствора али¬
зарина 8 Р и 0,1 мл раствора цирконила нитра¬
та Р и перемешивают. Выдерживают в течение
5 мин и сравнивают окраску полученного раствора
с окраской контрольного раствора, приготовлен¬
ного аналогично. Окраска испытуемого раствора
желтая, контрольного раствора — красная.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+110 до +117 (в пересчете на безводное веще¬
ство). 0,100 г испытуемого образца растворяют в
96 % спирте Р и доводят до объема 20,0 мл этим
же растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Испытание проводят с за¬
щитой от света.
Испытуемый раствор. 25,0 мг испытуемого
образца растворяют в подвижной фазе В и доводят
до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 5 мг ФСО триамцино¬
лона ацетонида для проверки пригодности хро¬
матографической системы (содержит примеси В
и С) растворяют в подвижной фазе В и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят подвижной фазой В до объема
100,0 мл. 1,0 мл полученного раствора доводят
подвижной фазой В до объема 10,0 мл.
Раствор сравнения (Ь). 25,0 мг ФСО триам¬
цинолона ацетонида растворяют в подвижной
фазе В и доводят до объема 25,0 мл этим же раст¬
ворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р —
вода для хроматографии Р (32:68, об/об);
- подвижная фаза В: вода для хроматогра¬
фии Р — ацетонитрил Р (35:65, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
100
0
20—25
о
0
1
О
0 — 100
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и растворов сравнения (а) и (Ь).
Идентификация пиков примесей: идентифи¬
цируют пики примесей В и С, используя хромато¬
грамму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО триамцинолона ацетонида
для проверки пригодности хроматографической
системы.
Относительное удерживание (по отношению
к триамцинолона ацетонида, время удержива¬
ния — около 16 мин): примесь С — около 0,7; при¬
месь В — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (а):
-разрешение: не менее 2,5 между пиками
примеси С и примеси В.
Предельное содержание примесей:
Триамцинолона ацетонид
989
- примесь В (не более 0,2 %): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси В, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
-примесь С (не более 0,15%): на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси С, не должна превышать
1,5-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей В и С, не должна превышать площадь
основного пика на хроматограмме раствора срав¬
нения (Ь);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 5-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь).
Вода (2.5.12). Не более 2,0 %. Определение
проводят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12,2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Количественное определение проводят с за¬
щитой от света.
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: подвижная фаза А;
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и раствора сравнения (с);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания триамцинолона ацетонида.
Время удерживания: триамцинолона ацето¬
нид — около 16 мин.
Содержание С24Н31Р06 в процентах рассчиты¬
вают с учетом содержания триамцинолона ацето¬
нида в ФСО триамцинолона ацетонида.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, О, Е, Р.
он
А. 9-Фтор-11 р, 16а, 17,21 -тетрагидроксипрегна-1,4-
диен-3,20-дион (триамцинолон).
он
В. 9-Фтор-11 р,21 -дигидрокси-16а, 17-(1 -метил-
этил идендиокси)прегна-1,4,14-триен-3,20-дион (А14-три-
амцинолона ацетонид).
он
С. 9-Фтор-11 р,21,21 -тригидрокси-16а, 17-(1 -метил-
этилидендиокси)прегна-1,4-диен-3,20-дион (триамци¬
нолона ацетонида 21-альдегид гидрат).
он
й. 9-Хлор-11Э,21 -дигидрокси-16а, 17-(1-метил-
этилидендиокси)прегна-1,4-диен-3,20-дион (9а-хлор-
триамцинолона ацетонид).
Е. 9-Фтор-11 р,21 -дигидрокси-16а, 17-(1 -метилэтил-
идендиокси)прегна-4-ен-3,20-дион (1,2-дигидротриам-
цинолона ацетонид).
Р. 9-Фтор-11 р-гидрокси-16а, 17-(1 -метилэтил-
идендиокси)-3,20-диоксопрегна-1,4-диен-21 -ил ацетат
(21-ацетат триамцинолона ацетонида).
990
Гэсударственная фармакопея Республики Беларусь
07/2016:1740
ТРИБЕНОЗИД
ТпЬепозШит
ТШВЕЫОЗЮЕ
он
С29НмОв М.м. 478,6
[10310-32-4]
ОПРЕДЕЛЕНИЕ
Смесь а- и (З-аномеров этил-3,5,6-три-О-бензил-
О-глюкофуранозида.
Содержание: не менее 96,0 % и не более 102,0 %.
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная вязкая жидкость от желтоватого до
бледно-желтого цвета.
Практически нерастворим в воде, очень легко
растворим в ацетоне, в метаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО трибенозида #или спектр,
представленный на рисунке #1740.-1#.
ИСПЫТАНИЯ
Раствор 5. 4,00 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20,0 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,10
при 420 нм. Измеряют оптическую плотность раст¬
вора 5.
Удельное оптическое вращения (2.2.7). От
-31,0 до -40,0. 2,0 мл раствора 5 доводят метано¬
лом Р до объема 20,0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 1,000 г испытуемо¬
го образца растворяют в смеси вода Р — ацетони¬
трил Р (5:95, об/об) и доводят до объема 25,0 мл этой
же смесью растворителей.
Испытуемый раствор (Ь). 50,0 мг испытуемо¬
го образца растворяют в смеси вода Р — ацетони¬
трил Р (5:95, об/об) и доводят до объема 50,0 мл этой
же смесью растворителей.
Раствор сравнения (а). 25,0 мг бензальдегида Р
(примесь С) и 30,0 мг ФСО трибенозида примеси А раст¬
воряют в ацетонитриле Р и доводят до объема 100,0 мл
этим же растворителем. В мерную колбу на 50 мл вно¬
сят 20,0 мл полученного раствора и прибавляют 2,5 мл
воды Р и доводят ацетонитрилом Р до объема 50,0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО трибено¬
зида растворяют в смеси вода Р — ацетонитрил Р
(5:95, об/об) и доводят до объема 50,0 мл этой же
смесью растворителей.
Раствор сравнения (с). 12,0 мг ФСО трибенози¬
да примеси О растворяют в смеси вода Р — ацето¬
нитрил Р (5:95, об/об) и доводят до объема 100,0 мл
этой же смесью растворителей.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
Рисунок #1740.-1. Инфракрасный спектр ФСО трибенозида
Триметазидина дигидрохлорид
991
- подвижная фаза:
- подвижная фаза А: 0,1 % (об/об) раствор кис¬
лоты фосфорной Р;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—40
55 — 10
45 — 90
40—55
10
90
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь) и (с).
Идентификация пиков примесей: идентифици¬
руют пики примесей А и С, используя хроматограмму
раствора сравнения (а); идентифицируют пик примеси
О, используя хроматограмму раствора сравнения (с).
Относительное удерживание (по отношению к
Р-аномеру трибенозида, время удерживания — около
18 мин): а-аномер — около 1,1; примесь С — около
0,2; примесь В — около 0,6; примесь Э — около 0,8;
примесь А — около 1,4.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 3,0 между пиками
а-аномера и р-аномера трибенозида.
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать 1,7-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (а);
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (а); если площадь пика примеси
С на хроматограмме испытуемого раствора превыша¬
ет площадь соответствующего пика на хроматограмме
раствора сравнения (а) (0,25 %), то испытуемый раст¬
вор разбавляют до получения площади пика, равной
или меньшей площади пика на хроматограмме раст¬
вора сравнения (а); при расчете содержания примеси
С учитывают фактор разведения;
- примесь О (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси О, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (с);
- любая другая примесь (не более 0,3 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, С и О, не
должна превышать площадь пика примеси А на хро¬
матограмме раствора сравнения (а);
- сумма примесей (не более 2,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
6,7-кратную площадь пика примеси А на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,17 площади пика примеси А на
хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 5,0 мл раствора 5 доводят мета¬
нолом Р до объема 20,0 мл. 12 мл полученного раст¬
вора должны выдерживать испытание на тяжелые
металлы. Эталон готовят с использованием эталон¬
ного раствора свинца (1 ррт РЬ), полученного путем
разведения эталонного раствора свинца (100 ррт
РЬ) Р метанолом Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (Ь).
Содержание суммы а-аномера и 0-аномера три¬
бенозида рассчитывают в процентах, принимая во
внимание содержание трибенозида в ФСО трибено¬
зида.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в среде
азота.
ПРИМЕСИ
A. Р = СН2-С6Н5:3,5,6-Три-0-бензил-1,2-0-(1-метил-
этилиден)-а-0-глюкофураноза.
B. Р = Н: 3,5-Ди-0-бензил-1,2-0-(1-метилэтилиден)-
а-О-глюкофураноза.
C. С6Н5-СНО: Бензальдегид.
О. С6Н5-СН2-0-СН2-С6Н5: Дибензиловый эфир.
07/2016:1741
ТРИМЕТАЗИДИНА
ДИГИДРОХЛОРИД
Тпте(а21сИп1 сПЬуйгосЫопйит
ТШМЕТА1ЮШЕ 01НУОКОСН1-ОШОЕ
С14Н22М203 ’ 2НС1 М.М. 339,3
[13171-25-0]
ОПРЕДЕЛЕНИЕ
1-(2,3,4-Триметоксибензил)пиперазина дигидрох¬
лорид.
Содержанеие: не менее 98,5 % и не более
101,5 % (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Слегка гигроскопичен.
992
Гэсударственная фармакопея Республики Беларусь
Легко растворим в воде, умеренно растворим в
96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр триметазидина
дигидрохлорида по Европейской Фармакопее #или
спектр, представленный на рисунке #1741.-1*
B. 25 мг испытуемого образца растворяют в 5 мл
воды Р. 2 мл полученного раствора дают реакцию (а)
на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 20,0 мг ФСО тримета¬
зидина для проверки пригодности хроматографиче¬
ской системы растворяют в воде Р и доводят до объ¬
ема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 2,0 мл испытуемого
раствора доводят водой Р до объема 100,0 мл. 5,0 мл
полученного раствора доводят водой Р до объема
100.0 мл.
Раствор сравнения (с). 25,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикагелем
октадецилсилильным для хроматографии Р (размер
частиц 5 мкм) с размером пор 10 нм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: метанол Р — раствор
2,87 г/л натрия гептансульфоната Р, доведен¬
ный до рН 3,0 кислотой фосфорной разведен¬
ной Р, (357:643, об/об);
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—50
со
сл
1
->|
сл
5 —* 25
50—52
75 —► 95
25 —► 5
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 240 нм;
- уравновешивание колонки: не менее 1 ч с соот¬
ношением подвижных фаз А:В (95:5, об/об);
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
триметазидину, время удерживания — около 25 мин):
примесь О — около 0,2; примесь С — около 0,4; при¬
месь Н — около 0,6; примесь А и примесь I — около
0,9; примесь Е — около 0,95; примесь Р — около 1,4;
примесь В — около 1,8.
Пригодность хроматографической системы:
- коэффициент разделения пиков: не менее 3
(Нр — высота пика примеси Е относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси Е и
триметазидина на хроматограмме раствора сравне¬
ния (а));
- отношение сигнал/шум: не менее 10 для основ¬
ного пика на хроматограмме раствора сравнения (с).
Рисунок #1741 .-1. Инфракрасный спектр триметазидина дигидрохлорида
Триметоприм
993
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,55; для примеси С — 0,37; для при¬
меси Р — 0,71):
- примеси А, В, С, О, Е, Р, Н, I (не более 0,1 %): на
хроматограмме испытуемого раствора площадь пика,
соответствующего каждой из примесей, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С, й,
Е, Р, Н и I, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
Примесь О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 22,6 мг пиперазина гидра¬
та Р растворяют в метаноле Р и доводят до объема
100 мл этим же растворителем. 10 мл полученного
раствора доводят метанолом Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — 96% спирт Р (20:80, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: при температуре от 100 °С до
105 °С в течение 30 мин.
Проявление: пластинку опрыскивают реактивом
йодплатината Р и просматривают при дневном свете.
Предельное содержание примесей:
- примесь О (не более 0,1 % в пересчете на без¬
водный пиперазин): на хроматограмме испытуемого
раствора пятно, соответствующее примеси 6, должно
быть не интенсивнее пятна на хроматограмме раст¬
вора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 2,5 %. 1,000 г испытуемого образца сушат над
фосфора (V) оксидом Р при температуре 105 °С и
давлении не превышающем 15 кПа.
Сульфатная зола (2.4.14 А). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в 50 мл
воды Р, прибавляют 1 мл кислоты азотной Р и ти¬
труют 0,1 М раствором серебра нитрата потенцио-
метрически (2.2.20).
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 16,96 мг С14Н221\1203-2НС1.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, О, Н, I.
Я1
А. Р1 = Р4 = Н, Р2 = КЗ = ОСН3:1-(3,4,5-Триметок-
сибензил)пиперазин.
Е. Р1 = КЗ = ОСН3, Р2 = Р4 = Н: 1-(2,4,5-Триметок-
сибензил)пиперазин.
Р. Р1 = Р4 = ОСН3, Р2 = КЗ = Н: 1-(2,4,6-триметок-
сибензил)пиперазин.
ОСН3
В. 1,4-Бис(2,3,4-триметоксибензил)пиперазин.
ОСН3
я.
-ОСН3
осн3
С. Р = СНО: 2,3,4-Триметоксибензальдегид.
О. Р = СН2ОН: (2,3,4-Триметоксифенил)метанол.
1ЧН
ни
С. Пиперазин.
ОСН3
,осн3
^^^ОСНз
H. Р = СООС2Н5: Этил-4-(2,3,4-триметоксибензил)-
пиперазин-1 -карбоксилат.
I. Р = СН3:1-Метил-4-(2,3,4-триметоксибензил)пи-
перазин (Л/-метилтриметазидин).
07/2016:0060
ТРИМЕТОПРИМ
ТптеОюрптит
тшметноршм
СиН,Л04 М.м. 290,3
[738-70-5]
ОПРЕДЕЛЕНИЕ
5-(3,4,5-Триметоксибензил)пиримидин-2,4-диамин.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
994
Гэсударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок.
Очень мало растворим в воде, мало растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 199 °С до
203 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 0,1 М растворе натрия гидрок¬
сида и доводят до объема 100,0 мл этим же раство¬
рителем. 1,0 мл полученного раствора доводят 0,1 М
раствором натрия гидроксида до объема 10,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 287 нм.
Удельный показатель поглощения в максимуме:
от 240 до 250.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО триметоприма *или спектр,
представленный на рисунке #0060.-1#.
0. 25 мг испытуемого образца растворяют (при
необходимости нагревая) в 5 мл 0,005 М раствора
кислоты серной, прибавляют 2 мл раствора 16 г/л
калия перманганата Р в 0,1 М растворе натрия ги¬
дроксида и нагревают до кипения. К горячему раство¬
ру прибавляют 0,4 мл раствора формальдегида Р,
перемешивают, прибавляют 1 мл 0,5 М раствора кис¬
лоты серной, перемешивают, нагревают до кипения,
охлаждают и фильтруют. К полученному фильтрату
прибавляют 2 мл метиленхлорида Р и интенсивно
встряхивают. Органический слой в ультрафиолетовом
свете при длине волны 365 нм имеет флуоресценцию
зеленого цвета.
ИСПЫТАНИЯ
Цветность (2.2.2, метод II). 0,5 г испытуемо¬
го образца растворяют в 10 мл смеси из 1 объема
воды Р, 4,5 объемов метанола Р и 5 объемов мети¬
ленхлорида Р. Окраска полученного раствора должна
быть не интенсивнее эталона ВУ(КЖ)7.
Сопутствующие примеси.
А. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО триметоприма для проверки пригодности хро¬
матографической системы (содержит примесь Е)
растворяют в 1 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,250 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецил-
синильным, деактивированным по отношению к ос¬
нованиям, для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: метанол Р — раствор 1,4 г/л
натрия перхлората Р, доведенный до рН 3,6 кисло¬
той фосфорной Р, (30:70, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 11-кратное вре¬
мя удерживания триметоприма.
Относительное удерживание (по отношению к
триметоприму, время удерживания — около 5 мин):
Рисунок #0060.-1. Инфракрасный спектр ФСО триметоприма
Триметоприм
995
примесь С — около 0,8; примесь Е — около 0,9; при¬
месь А— около 1,5; примесь О — около 2,0; примесь
О — около 2,1; примесь В — около 2,3; примесь 3 —
около 2,7; примесь Р — около 4,0.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,5 между пиками при¬
меси Е и триметоприма.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков, на
соответствующие, поправочные коэффициенты: для
примеси В — 0,43, для примеси Е — 0,53, для при¬
меси 3 — 0,66):
- любая примесь (не более 0,1 %): на хромато¬
грамме испытуемого раствора площадь любого пика,
кроме основного, не должна превышать 0,2 площади
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 0,4
площади основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,04 площади основного пика на хроматограм¬
ме раствора сравнения (а) (0,02 %), и пик примеси Н
(относительное удерживание — около 10,3).
В. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 200,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО триме¬
топрима и 5,0 мг ФСО триметоприма примеси В
растворяют в подвижной фазе и доводят до объема
100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем нитрильным
для хроматографии Р (размер частиц 5 мкм) с удель¬
ной площадью поверхности 350 м2/г и размером пор
10 нм;
- подвижная фаза: 1,14 г натрия гексансульфо-
ната Р растворяют в 600 мл раствора 13,6 г/л калия
дигидрофосфта Р, доводят до рН 3,1 кислотой фос¬
форной Р и смешивают с 400 мл метанола Р;
- скорость подвижной фазы: 0,8 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл;
-время хроматографирования: 6-кратное вре¬
мя удерживания триметоприма.
Относительное удерживание (по отношению к
триметоприму, время удерживания — около 4 мин):
примесь Н — около 1,8; примесь I — около 4,9.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 2,0 между пиками три¬
метоприма и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Н — 0,50, для примеси I — 0,28):
-любая примесь (не более 0,1 %): на хромато¬
грамме испытуемого раствора площадь любого пика,
кроме основного, не должна превышать 0,2 площади
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 0,4
площади основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,04 площади основного пика на хроматограм¬
ме раствора сравнения (а) (0,02 %) и пик примеси В
(относительное удерживание — около 1,3).
Примесь К. Газовая хроматография (2.2.28).
Испытуемый раствор. 0,500 г испытуемого об¬
разца растворяют в 35,0 мл цитратного буферного
раствора рН 5,0 Р, прибавляют 10,0 мл 1,1-димети-
лэтилметилового эфира Р, интенсивно встряхива¬
ют и центрифугируют в течение 10 мин. Используют
верхний слой.
Раствор сравнения. 5,0 мл кислоты хлористо¬
водородной Р доводят водой Р до объема 50,0 мл,
прибавляют 12,5 мг анилина Р (примесь К) и интен¬
сивно встряхивают. 10,0 мкл полученного раствора и
10,0 мл 1,1-диметилэтилметилового эфира Р при¬
бавляют к 35,0 мл цитратного буферного раствора
рН 5,0 Р, интенсивно встряхивают и центрифугируют
в течение 10 мин. Используют верхний слой.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и внутренним диаметром 0,53 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 3 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 12 мл/мин;
- температура: колонка — 80 °С, блок ввода
проб — 230 °С, детектор — 270 °С;
- детектор: термоионизационный, азот- и фос-
фор-селективный;
- объем вводимой пробы: 3 мкл;
- время хроматографирования: 15 мин.
Пригодность хроматографической системы:
раствор сравнения:
- относительное стандартное отклонение:
не более 5,0 % для площадей пиков при 6 повторных
вводах пробы.
Предельное содержание примесей:
-примесь К (не более 5ррт): на хроматограм¬
ме испытуемого раствора площадь пика примеси К не
должна превышать площадь соответствующего пика
на хроматограмме раствора сравнения.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас-
996
Гэсударственная фармакопея Республики Беларусь
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 29,03 мг С14Н18М403.
ПРИМЕСИ
Определяемые жидкостной хроматографией
(A) : А, В, С, О, Е, Р, 6, Н,
Определяемые жидкостной хроматографией
(B) :В,Н,1
Определяемые газовой хроматографией: К.
А. Л/2-Метил-5-(3,4,5-триметоксибензил)пиримидин-
2,4-диамин.
B. П + Р' = О: (2,4-Диаминопиримидин-5-ил)-(3,4,5-
триметоксифенил)метанон.
C. П = ОН, П' = Н: (Р5)-(2,4-Диаминопиримидин-
5-ил)(3,4,5-триметоксифенил)метанол.
й. Р2 = МН2, Р4 = ОН: 2-Амино-5-(3,4,5-триме-
токсибензил)пиримидин-4-ол.
Е. Р2 = ОН, Р4 = МН2: 4-Амино-5-(3,4,5-триметок-
сибензил)пиримидин-2-ол.
Р. КЗ = Вг, П4 = ОСН3: 5-(3-Бром-4,5-диметокси-
бензил)пиримидин-2,4-диамин.
О. РЗ = ОСН3, Р4 = ОС2Н5:5-(4-Этокси-3,5-диметок-
сибензил)пиримидин-2,4-диамин.
Н. Р = СН3: Метил-3,4,5-триметоксибензоат.
Р = Н: 3,4,5-Триметоксибензойная кислота.
ОСН3
I. 3-(Фениламино)-2-(3>4>5-триметоксибензил)проп-
2-еннитрил.
К. Анилин.
07/2016:1272
ТРИПТОФАН
Тгур(орЬапит
ТКУРТОРНАИ
[73-22-3]
ОПРЕДЕЛЕНИЕ
(5)-2-Амино-3-(1 Н-индол-3-ил)пропионовая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический или
аморфный порошок.
Умеренно растворим в воде, мало растворим в
96 % спирте. Растворяется в разведенных растворах
минеральных кислот и растворах гидроксидов щелоч¬
ных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО триптофана #или спектр,
представленный на рисунке #1272.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, размеру и цвету основному пятну
на хроматограмме раствора сравнения (а).
О. 20 мг испытуемого образца растворяют в
10 мл воды Р, прибавляют 5 мл раствора диметила-
минобензальдегида Рб, 2 мл кислоты хлористово¬
дородной Р1 и нагревают на водяной бане. Появляет¬
ся фиолетово-синее окрашивание.
ИСПЫТАНИЯ
Раствор 3. 0,1 г испытуемого образца раство¬
ряют в 1 М растворе кислоты хлористоводородной
и доводят до объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
Триптофан
997
Волновое число (см1)
Рисунок #1272.-1. Инфракрасный спектр ФСО триптофана
Удельное оптическое вращение (2.2.7). От
-30,0 до -33,0 (в пересчете на сухое вещество).
0,25 г испытуемого образца растворяют в воде Р, при
необходимости нагревая на водяной бане, и доводят
до объема 25,0 мл этим же растворителем.
Нингидрин-положительные вещества
Испытание проводят одним из приведенных
ниже методов.#
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости
от чувствительности используемого оборудования.
Концентрации всех растворов могут быть измене¬
ны (при сохранении предписанных соотношений их
концентраций) с условием выполнения требований к
пригодности хроматографической системы, установ¬
ленных в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготов¬
ления образца, подходящий для применяемого при¬
бора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят раствором А до объема 100,0 мл.
2.0 мл полученного раствора доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного
раствора аммония (100 ррт ЫН) Р доводят раство¬
ром А до объема 50,0 мл. 1,0 мл полученного раст¬
вора доводят раствором А до объема 100,0 мл.
Раствор сравнения (д). 30 мг изолейцина Р и
30 мг лейцина Р растворяют в растворе А и доводят
до объема 50,0 мл этим же растворителем. 1,0 мл по¬
лученного раствора доводят раствором А до объема
200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь) и (б) вводят в аминокислотный анализатор.
Запускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентного содержания:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию триптофана в растворе срав¬
нения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
-любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для
фармацевтического использования (2034), не при¬
меняют.
#Тонкослойная хроматография (2.2.27).
Смесь растворителей. Кислота уксусная ледя¬
ная Р — вода Р (50:50, об/об).
998
Государственная фармакопея Республики Беларусь
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в смеси растворителей и дово¬
дят до объема 10 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 50 мл.
Раствор сравнения (а). 10 мг ФСО триптофана
растворяют в смеси растворителей и доводят до объ¬
ема 50 мл этой же смесью растворителей.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят смесью растворителей до объема
20 мл.
Раствор сравнения (с). 10 мг ФСО триптофана
и 10 мг ФСО тирозина растворяют в смеси раство¬
рителей и доводят до объема 25 мл этой же смесью
растворителей.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 °С
до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Примесь А и другие сопутствующие примеси.
Жидкостная хроматография (2.2.29). Эталонный раст¬
вор, испытуемые растворы и растворы сравнения го¬
товят непосредственно перед использованием.
Буферный раствор рН 2,3. 3,90 г натрия диги¬
дрофосфата Р растворяют в 1000 мл воды Р, при¬
бавляют около 700 мл раствора 2,9 г/л кислоты фос¬
форной Р и доводят до рН 2,3 этим же раствором
кислоты.
Смесь растворителей. Ацетонитрил Р —
вода Р (10:90, об/об).
Эталонный раствор. 10,0 мг Ы-ацетилтрип-
тофана Р растворяют в смеси растворителей и до¬
водят до объема 100,0 мл этим же растворителем.
2.0 мл полученного раствора доводят смесью раство¬
рителей до объема 100,0 мл.
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в смеси растворителей и доводят
до объема 10,0 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 0,10 г испытуемого
образца растворяют в эталонном растворе и доводят
до объема 10,0 мл этой же смесью растворителей.
Раствор сравнения (а). Содержимое контейне¬
ра ФСО 1,1'-этилиденбистриптофана (примесь А)
растворяют в 1,0 мл смеси растворителей.
Раствор сравнения (Ь). Содержимое контейне¬
ра ФСО 1,1'-этилиденбистриптофана (примесь А)
растворяют в 1,0 мл эталонного раствора.
Раствор сравнения (с). 0,5 мл раствора срав¬
нения (а) доводят смесью растворителей до объема
5.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным для хроматографии Р с разметом частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — бу¬
ферный раствор рН 2,3 (115:885, об/об);
- подвижная фаза В: ацетонитрил Р — бу¬
ферный раствор рН 2,3 (350:650, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100
0
10—45
100 —► 0
0 —* 100
45—65
0
100
- скорость подвижной фазы: 0,7 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 20 мкл испытуемых
растворов (а) и (Ь) и растворов сравнения (Ь) и (с).
Времена удерживания: триптофан — около
8 мин; Л/-ацетилтриптофан — около 29 мин; примесь
А—около 34 мин.
Пригодность хроматографической системы:
- разрешение: не менее 8,0 между пиками
Л/-ацетилтриптофана и примеси А на хроматограмме
раствора сравнения (Ь); при необходимости изменяют
временную программу градиента (увеличение време¬
ни элюирования подвижной фазой А приводит к увели¬
чению времен удерживания и лучшему разрешению);
- отношение сигнал/шум: не менее 15 для основ¬
ного пика на хроматограмме раствора сравнения (с);
- фактор асимметрии: не более 3,5 для пика
примеси А на хроматограмме раствора сравнения (Ь);
- на хроматограмме испытуемого раствора (а) не
должно обнаруживаться пика со временем удержива¬
ния, соответствующим А/-ацетилтриптофану (в случае
присутствия такого пика необходимо откорректиро¬
вать площадь пика Л/-ацетилтриптофана).
Предельное содержание примесей:
- примесь А (не более 10 ррт): на хроматограм¬
ме испытуемого раствора (Ь) площадь пика, соот¬
ветствующего примеси А, не должна превышать 0,5
площади основного пика на хроматограмме раствора
сравнения (с);
-сумма примесей со временами удерживания
меньшими, чем время удерживания триптофана
(не более 100 ррт): на хроматограмме испытуемо¬
го раствора (Ь) сумма площадей пиков со времена¬
ми удерживания меньшими, чем время удерживания
триптофана, не должна превышать 0,6 площади пика
/У-ацетилтриптофана на хроматограмме раствора
сравнения (Ь);
- сумма примесей со временами удержива¬
ния большими, чем время удерживания трип¬
тофана, и меньшими 1,8 времени удерживания
Ы-ацетилтриптофана (не более 300 ррт): на хрома¬
тограмме испытуемого раствора (Ь) сумма площадей
пиков со временами удерживания большими, чем вре¬
мя удерживания триптофана, и меньшими 1,8 времени
удерживания А/-ацетилтриптофана, не должна превы¬
шать 1,9-кратную площадь пика /У-ацетилтриптофана
на хроматограмме раствора сравнения (Ь);
-неучитываемый предел: на хроматограмме
испытуемого раствора не учитывают пики с площа-
Триптофан
999
дью менее 0,02 площади пика Л/-ацетилтриптофана
на хроматограмме раствора сравнения (Ь) и пик
Л/-ацетилтриптофана.
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в 3 мл кис¬
лоты азотной разведенной Р и доводят водой Р до
объема 15 мл. Полученный раствор без дальнейшего
прибавления кислоты азотной должен выдерживать
испытание на хлориды.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
0,5 г испытуемого образца растворяют в смеси кисло¬
та хлористоводородная разведенной Р — вода дис¬
тиллированная Р (5:25, об/об) и доводят до объема
15 мл этим же растворителем.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хро¬
матограмме испытуемого раствора площадь пика,
соответствующего аммонию, не должна превышать
площадь соответствующего пика на хроматограм¬
ме раствора сравнения (с), учитывая пик, соответ¬
ствующий аммонию на хроматограмме холостого
раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хрома¬
тографии, то испытание «Аммония соли» (2.4.1, ме¬
тод В) проводят следующим образом. 0,10 г испы¬
туемого образца должны выдерживать испытание
на аммония соли. Эталон готовят с использовани¬
ем 0,2 мл эталонного раствора аммония (100 ррт
N14^ Р*
Железо (2.4.9). Не более 0,0020 % (20 ррт).
0,50 г испытуемого образца помещают в делитель¬
ную воронку, растворяют в 10 мл кислоты хлори¬
стоводородной разведенной Р, встряхивают триж¬
ды, каждый раз в течение 3 мин, с метилизобу-
тилкетоном Р1 порциями по 10 мл. Объединенные
органические слои встряхивают с 10 мл воды Р в
течение 3 мин. Водный слой должен выдерживать
испытание на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р, прибавляют 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 20,42 мг С^Н^Ы/^.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следую¬
щие вещества, если они присутствуют в значи¬
тельных количествах, следует определять тем или
иным испытанием, описанным в частной статье.
Их содержание лимитируется общим критерием
приемлемости для других/неспецифицированных
примесей. Вследствие этого нет необходимости
идентифицировать эти примеси для доказатель¬
ства соответствия требованиям. См. также статью
5.10. Контроль примесей в субстанциях для фар¬
мацевтического использования): В, С, О, Е, Р, С,
Н, I, 3, К,
А. 3,3г-[Этилиденбис(1 Н-индол-1,3-диил)]бис[(25)-
2-аминопропановая] кислота (1,1-этил иденбистрипто-
фан).
и эпимер у С*
С02н
В. (5)-2-Амино-3-[(ЗР5)-3-гидрокси-2-оксо-2,3-
дигидро-1Я-индол-3-ил]пропановая кислота (диокси-
индолилаланин).
С. Р = Н: (5)-2-Амино-4-(2-аминофенил)-4-оксо-
бутановая кислота (кинуренин).
Е. Р = СНО: (5)-2-Амино-4-[2-(формиламино)фе-
нил]-4-оксобутановая кислота (А/-формилкинуренин).
О. (5)-2-Амино-3-(5-гидрокси-1 Я-индол-З-илПро¬
пановая кислота (5-гидрокситриптофан).
Н N42
со2н
Р. (5)-2-Амино-3-(фениламино)пропановая кисло¬
та (3-фениламиноаланин).
1000
Гэсударственная фармакопея Республики Беларусь
он
С. (5)-2-Амино-3-(2-гидрокси-1 Я-индол-3-ил)про-
пановая кислота (2-гидрокситриптофан).
H. Я = Н: (ЗЯ5)-1,2,3,4-Тетрагидро-9Н-р-карболин-
3-карбоновая кислота.
I. К = СН3:1-Метил-1,2,3,4-тетрагидро-9Н-р-карбо-
лин-3-карбоновая кислота.
и. Я = СНОН-СН2-ОН: (5)-2-Амино-3-[2-[2,3-диги-
дрокси-1 -(1 Н-индол-3-ил)пропил]-1 Н-индол-3-ил]про-
пановая кислота.
К. Р = И: (5)-2-Амино-3-[2-(1Н-индол-3-илметил)-
1Н-индол-3-ил]пропановая кислота.
Ь. 1 -(1 АУ-Индол-3-илметил)-1,2,3,4-тетрагидро-9Н-
Р-карболин-З-карбоновая кислота.
07/2016:2133
ТРОКСЕРУТИН
ТгохегиНпит
ТКОХЕРШШ
ОПРЕДЕЛЕНИЕ
Смесь О-гидроксиэтилированных производных ру¬
тина, содержащая не менее 80,0 % 2-[3,4-бис(2-гидрокси-
этокси)фенил]-3-[[6-0-(6-дезокси-а-!_-маннопиранозил)-
Р-0-глюкопиранозил]окси]-5-гидрокси-7-(2-гидрокси-
этокси)-4Н-1 -бензопиран-4-она (трис(гидроксиэтил)ру-
тин).
Содержание: не менее 95,0 % и не более 105,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтовато-зеленый кристаллический порошок.
Гигроскопичен.
Легко растворим в воде, мало растворим в 96 %
спирте, практически нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО троксерутина #или спектр,
представленный на рисунке #2133.-1#.
B. Просматривают хроматограммы, полученные
в испытании «Состав».
Результаты, на хроматограмме испытуемого
раствора основной пик по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
ИСПЫТАНИЯ
Состав. Жидкостная хроматография (2.2.29):
определение проводят методом внутренней нормали¬
зации.
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют в подвижной фазе, при необходи¬
мости используют ультразвуковую баню, и доводят
подвижной фазой до объема 10,0 мл.
Раствор сравнения (а). 10,0 мг ФСО троксеру¬
тина растворяют в подвижной фазе, при необходи¬
мости используют ультразвуковую баню, и доводят
подвижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). 1 мл раствора сравнения
(а) доводят подвижной фазой до объема 10 мл. 1 мл
полученного раствора доводят подвижной фазой до
объема 100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: смесь из 20 объемов ацето¬
нитрила Р и 80 объемов раствора 15,6 г/л натрия
дигидрофосфата Р, доведенного до рН 4,4 кислотой
фосфорной разведенной Р или раствором натрия
гидроксида разведенным Р\
- скорость подвижной фазы: 0,5 мл/мин;
- спектрофотометрический детектор, длина
волны 350 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания основного компонента троксерутина
(трис(гидроксиэтил)рутин).
Относительное удерживание (по отношению
к трис(гидроксиэтил)рутину, время удерживания —
около 25 мин): тетракис(гидроксиэтил)рутин —
около 0,5; моно(гидроксиэтил)рутин — около 0,8;
бис(гидроксиэтил)рутин — около 1,1.
Пригодность хроматографической системы:
Троксерутин
1001
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика бис(гидроксиэтил)рутина относи¬
тельно базовой линии; — расстояние между ба¬
зовой линией и нижней точкой кривой, разделяющей
пики бис(гидроксиэтил)рутина и трис(гидроксиэтил)
рутина) на хроматограмме раствора сравнения (а);
- отношение сигнал/шум: не менее 10 для основ¬
ного пика на хроматограмме раствора сравнения (Ь).
Состав:
- основной пик: не менее 80 %;
-любые другие пики: не более 5 %; допускается
присутствие одного пика более 5 %, но менее 10 %;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади основного пика на хроматограмме
раствора сравнения (Ь).
Этиленоксид. Парофазная газовая хроматогра¬
фия (2.2.28).
Испытуемый раствор. К 1,00 г испытуемого об¬
разца во флаконе прибавляют 1,0 мл воды Р и пере¬
мешивают до получения гомогенного раствора. На¬
гревают при температуре 70 °С в течение 45 мин.
Раствор сравнения. К 1,00 г испытуемого об¬
разца во флаконе прибавляют 50 мкл этиленоксида
раствора Р4 и 950 мкл воды Р, плотно укупоривают.
Нагревают при температуре 70 °С в течение 45 мин.
Условия хроматографирования:
- колонка кварцевая капиллярная дли¬
ной 30 м и диаметром 0,32 мм, покрытая слоем
поли(цианопропил) (7) (фенил) (7) (метил) (86)силокса-
на Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость подвижной фазы: 1,1 мл/мин;
- параметры парофазного пробоотборника:
- равновесная температура: 80 °С;
- время достижения равновесия: 45 мин;
- температура передающей линии: 110 °С;
- время приложения избыточного давления: 2 мин;
- время ввода пробы: 12 с;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
40
5—18
40 200
Блок ввода проб
150
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1,0 мл.
Содержание этиленоксида Р в ррт рассчитыва¬
ют по формуле:
(52тг)-(8,т3)’
где:
51 — площадь пика этиленоксида на хроматогра-
ме испытуемого раствора;
$2 — площадь пика этиленоксида на хроматогра-
ме раствора сравнения;
ш1 — масса навески этиленоксида, взятого для
приготовления раствора сравнения, мкг;
т2 — масса навески испытуемого образца, взято¬
го для приготовления испытуемого раствора, г;
Л73 — масса навески испытуемого образца, взято¬
го для приготовления раствора сравнения, г.
Предельное содержание примесей:
- этиленоксид: не более 1 ррт.
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
126. Зак. 1060.
1002
Гэсударственная фармакопея Республики Беларусь
Сульфатная зола (2.4.14). Не более 0,4%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в
100,0 мл воды Р. 10,0 мл полученного раствора дово¬
дят водой Р до объема 100,0 мл. 10,0 мл полученного
раствора доводят водой Р до объема 100,0 мл. Из¬
меряют оптическую плотность раствора (2.2.25) при
350 нм.
Содержание С33Н42019 в процентах рассчитывают
с использованием удельного показателя поглощения,
равного 250.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:1577
ТРОЛАМИН
Тго1аттит
ТК01.АМ1ЫЕ
ОН
СвН15ЫО, М.м. 149,2
[102-71-6]
ОПРЕДЕЛЕНИЕ
2,2',2"-Нитрилотриэтанол. Лриэтаноламин*.
Содержание: не менее 99,0 % (м/м) и не более
103,0 % (м/м) суммы оснований (в пересчете на без¬
водное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная бесцветная или светло-желтая вяз¬
кая жидкость. Очень гигроскопичен.
Смешивается с водой и 96 % спиртом, растворим
в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, В, О.
A. Относительная плотность (2.2.5): от 1,120 до
1,130.
B. Показатель преломления (2.2.6): от 1,482 до
1,485.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
й. К 1 мл испытуемого образца прибавляют
0,3 мл раствора меди (II) сульфата Р. Появляется
голубое окрашивание. Прибавляют 2,5 мл раствора
натрия гидроксида разведенного Р и нагревают до
кипения. Голубое окрашивание не изменяется.
ИСПЫТАНИЯ
Раствор 5. 12 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)6.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28).
Раствор внутреннего стандарта. 5,0 г 3-ами-
нопропанола Р растворяют в воде Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Испытуемый раствор. 10,0 г испытуемого об¬
разца растворяют в воде Р, прибавляют 1,0 мл раст¬
вора внутреннего стандарта и доводят водой Р до
объема 100,0 мл.
Раствор сравнения (а). 1,0 г ФСО троламина
растворяют в водеР и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 0,1 г ФСО троламина
примеси А, 0,5 г ФСО троламина примеси В и 0,1 г
ФСО троламина растворяют в воде Р и доводят до
объема 10,0 мл этим же растворителем. К 1,0 мл
полученного раствора прибавляют 1,0 мл раствора
внутреннего стандарта и доводят водой Р до объема
100,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 25 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)(дифенил)силоксана Р (толщина слоя
0,50 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:35;
- температура:
Время (мин)
Температура (°С)
Колонка
0
60
0—8,5
60 — 230
8,5—14
230
Блок ввода проб
260
Детектор
280
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 2 мкл; при необходи¬
мости вводят контрольный раствор.
Порядок выхода пиков: примесь А, 3-аминопро-
панол, примесь В, троламин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками 3-ами-
нопропанола и примеси А.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора отношение площади пика, со¬
ответствующего примеси А, к площади пика внутрен¬
него стандарта не должно превышать отношение (/?,)
площади пика примеси А к площади пика внутреннего
стандарта на хроматограмме раствора сравнения (Ь);
- примесь В (не более 0,5 %): на хроматограмме
испытуемого раствора отношение площади пика, со¬
ответствующего примеси В, к площади пика внутрен¬
него стандарта не должно превышать отношение (Р2)
площади пика примеси В к площади пика внутреннего
стандарта на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора отношение суммы
Троламин
1003
площадей всех пиков, кроме основного и пика вну¬
треннего стандарта, к площади пика внутреннего
стандарта не должно превышать более чем в 10
раз отношение (Р4) пика троламина к площади пика
внутреннего стандарта на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики, от¬
ношение площадей которых к площади пика внутрен¬
него стандарта менее 0,5 отношения площади пика
троламина к площади пика внутреннего стандарта на
хроматограмме раствора сравнения (Ь).
Примесь С. Гаэовая хроматография (2.2.28).
Смесь растворителей. Ацетонитрил Р — хло¬
роформ Р (10:50, об/об).
Приготовление колонки для твердофазной экс¬
тракции
Колонка А. В стеклянную колонку (длиной 400 мм
и внутренним диаметром 20 мм), снабженную тефло¬
новым краном и стеклянным фильтром (160) (2.1.2),
помещают 3 г натрия сульфата безводного Р, сверху
на него помещают 17 г кизельгура для хроматогра¬
фии Р и 3 г калия карбоната Р. Уплотняют сорбент
аккуратным постукиванием колонки.
Колонка В. Стеклянную колонку (длиной 400 мм
и внутренним диаметром 20 мм), снабженную тефло¬
новым краном и стеклянным фильтром (160) (2.1.2),
заполняют суспензией 25 г силикагеля для хромато¬
графии Р (от 0,063 мм до 0,200 мм) в смеси раство¬
рителей. Для уплотнения сорбента прилагают неболь¬
шое давление. На поверхность сорбента помещают
5 г натрия сульфата безводного Р.
Стандартный раствор (а). 50 мкл А1-нитрозоди-
этаноламина Р (примесь С) растворяют в метано¬
ле Р и доводят до объема 50,0 мл этим же раствори¬
телем. 100 мкл полученного раствора доводят мета¬
нолом Р до объема 100,0 мл.
Стандартный раствор (Ь). 10,0 мл раствора
стандарта (а) доводят метанолом Р до объема
50,0 мл.
Стандартный раствор (с). 50 мг Ы-нитрозоди-
изопропаноламина Р растворяют в метаноле Р и
доводят до объема 50,0 мл этим же растворителем.
100 мкл полученного раствора доводят метанолом Р
до объема 100,0 мл.
Испытуемый раствор. К 2,000 г испытуемо¬
го образца прибавляют 200 мкл метанола Р и 0,5 г
сульфаминовой кислоты Р, растворяют в 8 мл воды
для хроматографии Р и наносят на колонку А. Сосуд
дважды ополаскивают водой для хроматографии Р
порциями по 1,5 мл, промывные воды также наносят
на колонку. Через 15 мин уравновешивания элюируют
с использованием 100 мл этилацетата Р, собирают
элюат в колбу для перегонки вместимостью 250 мл и
выпаривают досуха. Полученный остаток растворяют
в 1 мл смеси растворителей, наносят на колонку В и
выдерживают до осаждения. Колбу дважды опола¬
скивают смесью растворителей порциями по 2 мл,
промывные растворы также наносят на колонку, и
выдерживают до осаждения. Колонку промывают с
помощью 100 мл смеси растворителей и промывной
раствор отбрасывают. Элюируют с использованием
120 мл ацетона Р, элюат собирают в колбу для пере¬
гонки вместимостью 250 мл и выпаривают досуха. По¬
лученный остаток переносят с помощью небольшого
объема ацетона Р в колбу и снова выпаривают досу¬
ха в потоке азота Р. Полученный остаток растворяют
в 100 мкл триметилпентана для хроматографии Р,
прибавляют 100 мкл Ы-метилтриметилсилил-три-
фторацетамида Р и нагревают при температуре
70 °С в течение 1 ч.
Раствор сравнения (а). К 2,000 испытуемого об¬
разца прибавляют 200 мкл стандартного раствора
(Ь) и 0,5 г сульфаминовой кислоты Р и растворяют
в 8 мл воды для хроматографии Р, после чего полу¬
ченный раствор обрабатывают аналогично испытуе¬
мому раствору.
Раствор сравнения (Ь). К 1,0 мл стандартного
раствора (а) прибавляют 4,0 мл стандартного раст¬
вора (с) и перемешивают. 500 мкл полученного раст¬
вора помещают в колбу и выпаривают досуха в по¬
токе азотаР. Полученный остаток растворяют в
200 мкл триметилпентана для хроматографии Р,
прибавляют 200 мкл И-метилтриметилсилил-три-
фторацетамида Р и нагревают при температуре
70 °С в течение 1 ч.
Раствор сравнения (с). В колбу помещают
200 мкл стандартного раствора (Ь) и выпаривают досу¬
ха в потоке азота Р. Полученный остаток растворяют
в 100 мкл триметилпентана для хроматографии Р,
прибавляют 100 мкл И-метилтриметилсилил-три-
фторацетамида Р и нагревают при температуре
70 °С в течение 1 ч.
Контрольный раствор. В стеклянную хромато¬
графическую колбу помещают 200 мкл метанола Р
и выпаривают досуха в потоке азота Р. Получен¬
ный остаток растворяют в 100 мкл триметилпен¬
тана для хроматографии Р, прибавляют 100 мкл
И-метилтриметилсилил-трифторацетамида Р и
нагревают при температуре 70 °С в течение 1 ч.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
диаметром 0,25 мм, покрытая слоем поли(диметил)-
(дифенил)силоксана, деактивированного по отно¬
шению к основаниям, Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 2 мл/мин;
- деление потока: 1:10;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
5—10
180 — 280
280
Блок ввода проб
220
- детектор: хемолюминесцентный:
-двойная горелка плазмы в режиме нитроза-
мина;
- температура горелки: 450 °С;
- скорость потока кислорода: (4,4—5,0) мл/мин;
- объема вводимой пробы: 4 мкл.
Пригодность хроматографической системы:
- разрешение: не менее 1,3 между пиками при¬
меси С и А/-нитрозодиизопропаноламина на хромато¬
грамме раствора сравнения (Ь);
- открываемость: не менее 50 %; разница
между площадью пика примеси С на хроматограмме
раствора сравнения (а) и площадью пика соответству¬
ющего пика на хроматограмме испытуемого раствора
не должна превышать 0,5 площади соответствующего
пика на хроматограмме раствора сравнения (с).
Предельное содержание примесей:
- примесь С (не более 24 ррЬ): на хроматограм¬
ме испытуемого раствора площадь пика примеси С
не должна превышать разницу между площадью пика
126*. Зак. 1060.
1004
Гэсударственная фармакопея Республики Беларусь
примеси С на хроматограмме раствора сравнения (а)
и площадью соответствующего пика на хроматограм¬
ме испытуемого раствора.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 5 мл раствора 5 доводят водой Р
до объема 30 мл. Полученный раствор должен выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Вода (2.5.12). Не более 1,0 %. Определение про¬
водят из 1,000 г испытуемого образца. Навеску испы¬
туемого образца помещают непосредственно в сосуд
с оттитрованным растворителем.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образ¬
ца. Предварительное нагревание на водяной бане не
проводят.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,200 г испытуемого образца растворяют в 75 мл
воды, свободной от углерода диоксида, Р, прибавля¬
ют 0,3 мл раствора метилового красного Р и титру¬
ют 1 М раствором кислоты хлористоводородной.
1 мл 1 М раствора кислоты хлористоводород¬
ной соответствует 0,149 г С6Н15М03.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
А. 2-Аминоэтанол (этаноламин).
В. 2,2-Иминодиэтанол (диэтаноламин).
N0
Ли
С. 2,2-(Нитрозоимино)диэтанол (Л/-нитрозодиэта-
ноламин).
07/2016:1053
ТРОМЕТАМОЛ
ТготеХато/ит
томЕТАМои
НО
ОН
С^ЫОз М.м. 121,1
[77-86-1]
ОПРЕДЕЛЕНИЕ
Аминометилидентри(метанол).
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, умеренно растворим в
96 % спирте, очень мало растворим в этилацетате.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С.
Вторая идентификация: А, В, О.
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», имеет сильнощелочную реакцию
среды (2.2.4).
B. Температура плавления (2.2.14): от 168 °С до
174 °С.
C. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО трометамола.
О. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается пятно, соответствующее
по расположению, цвету и размеру основному пятну
на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 10,0 до 11,5. Измеряют рН свеже¬
приготовленного раствора 3.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,20 г испытуемого
образца растворяют при нагревании в 1 мл воды Р и
доводят метанолом Р до объема 10 мл.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят метанолом Р до объема 10 мл.
Раствор сравнения (а). 20 мг ФСО трометамо¬
ла растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 1 мл испытуемого раст¬
вора (а) доводят метанолом Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р. Перед нанесением растворов пластинку не¬
обходимо промыть метанолом Р.
Подвижная фаза: раствор аммиака разведен¬
ный Р1 — 2-пропанол Р (10:90, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 10 см от ли¬
нии страта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: пластинку опрыскивают раствором
5 г/л калия перманганата Р в растворе 10 г/л натрия
карбоната Р. Через 10 мин пластинку просматривают
при дневном свете.
Предельное содержание примесей:
- любая примесь (не более 1,0 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь).
Трописетрона гидрохлорид
1005
Хлориды (2.4.4). Не более 0,0100 % (100 ррт). К
10 мл раствора 3 прибавляют 2,5 мл кислоты азот¬
ной разведенной Р и доводят водой Р до объема
15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца раство¬
ряют в воде Р. Раствор нейтрализуют кислотой хло¬
ристоводородной Р1 и доводят водой Р до объема
20 мл. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ)Р.
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца растворяют в воде Р и доводят
до объема 10 мл этим же растворителем.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,03 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 20 мл
воды Р, прибавляют 0,2 мл раствора метилового
красного Р и титруют 0,1 М раствором кислоты хло¬
ристоводородной до перехода окраски от желтой до
красной.
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 12,11 мгС4Н11М03.
ПРИМЕСИ
но.
рн
М02
.ОН
А. Нитрометилидентри(метанол).
07/2016:2102
ТРОПИСЕТРОНА ГИДРОХЛОРИД
Тгор'1ве1гоп1 Ьус1гос111опс1ит
ТЕОР18ЕТКОЫ НУОКОСШОКЮЕ
С17Н20М2О2 ’ НС1 М М’ 320,8
[105826-92-4]
ОПРЕДЕЛЕНИЕ
(1 Я,Зг,55)-8-Метил-8-азабицикло[3.2.1 ]окт-3-ил]-
1 Н-индол-З-карбоксилата гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Легко растворим или растворим в воде, умерен¬
но растворим в 96 % спирте, очень мало растворим в
метиленхлориде.
Рисунок #2102.-1. Инфракрасный спектр ФСО трописетрона гидрохлорида
127. Зак. 1060.
1006
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
25,0 мл этим же растворителем. 1,0 мл полученного
раствора доводят метанолом Р до 100,0 мл.
Диапазон длин волн: от 220 нм до 360 нм.
Максимумы поглощения: при 228 нм и при
282 нм.
Отношение оптических плотностей:
^228^282 °Т 1 ,Э Д° 1 ,4.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО трописетрона гидрохлорида
#или спектр, представленный на рисунке #2102.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образ¬
ца растворяют в смеси из равных объемов метано¬
ла Р и метиленхлорида Р и доводят до объема 5 мл
этой же смесью растворителей.
Раствор сравнения. 5 мг ФСО трописетрона
гидрохлорида растворяют в смеси из равных объемов
метанола Р и метиленхлорида Р и доводят до объ¬
ема 5 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метанол Р — метиленхлорид Р
(2:2:30:70, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на холодном воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
Проявление В: сначала пластинку опрыскивают
раствором, приготовленным следующим образом:
0,85 г висмута нитрата основного Р растворяют в
смеси из 10 мл кислоты уксусной Р и 40 мл воды Р; к
5 мл полученного раствора прибавляют 5 мл раствора
400 г/л калия йодида Р и доводят водой Р до объема
100 мл. Затем пластинку опрыскивают раствором пе¬
роксида водорода концентрированным Р.
Результаты В: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения.
О. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,00 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.7). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)7.
Примесь А. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в смеси из равных объемов мета¬
нола Р и метиленхлорида Р и доводят до объема
5.0 мл этой же смесью растворителей.
Раствор сравнения (а). 5,0 мг ФСО тропина
(примесь А) растворяют в смеси из равных объемов
метанола Р и метиленхлорида Р и доводят до объ¬
ема 25,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят смесью из равных объемов метанола Р
и метиленхлорида Р до объема 20,0 мл. К 0,1 мл
полученного раствора прибавляют 1,0 мл раствора
сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге-
ляР294Р.
Подвижная фаза: раствор аммиака Р — мета¬
нол Р — метиленхлорид Р (5:40:60, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку погружают в раствор ка¬
лия йодовисмутата Р1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора пятно, соответствующее при¬
меси А, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (а).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО трописетро¬
на примеси В и 5 мг ФСО этилиндол-3-карбоксилата
растворяют в подвижной фазе А и доводят до объема
20.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят подвижной фазой А до объема
50.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: триэтиламин Р —
ацетонитрил Р — вода Р — метанол Р
(0,3:35:400:565, об/об/об/об);
-подвижная фаза В: триэтиламин Р —
ацетонитрил Р — вода Р — метанол Р
(0,3:100:100:800, об/об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—14
100
0
14—32
100 —> 0
0
1
О
О
32—36
0
100
36—37
о
о
о
о
т
о
о
37—52
100
0
Углерода диоксид
1007
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению к
трописетрону, время удерживания — около 22 мин):
примесь В — около 0,05; этилиндол-3-карбоксилат —
около 0,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4 между пиком примеси
В и пиком этилиндол-3-карбоксилата.
Предельное содержание примесей:
- примесь В (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си В, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,3 %): на хромато¬
грамме испытуемого раствора сумма площадей пи¬
ков, кроме основного, не должна превышать 3-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
М,А/-Диметиланилин. Жидкостная хроматогра¬
фия (2.2.29). Растворы готовят непосредственно
перед использованием.
Испытуемый раствор. 250 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения. 10,0 мг Ы,И-диметил-
анилина Р растворяют в подвижной фазой А и до¬
водят до объема 100,0 мл этим же растворителем.
1,0 мл полученного раствора доводят подвижной фа¬
зой А до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
-подвижная фаза А: триэтиламин Р —
ацетонитрил Р — вода Р — метанол Р
(0,3:35:400:565, об/об/об/об);
- подвижная фаза В: триэтиламин Р —
ацетонитрил Р — вода Р — метанол Р
(0,3:100:100:800, об/об/об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100
0
10—11
о
т
о
о
0
1
о
о
11—30
0
100
30—31
0 — 100
о
т
о
о
31—50
100
0
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 248 нм;
- объем вводимой пробы: 20 мкл.
Предельное содержание примесей:
- А1,Ы-диметиланилин (не более 20 ррт): на хро¬
матограмме испытуемого раствора площадь пика А/,А/-
диметиланилина не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 0,3 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 10 мл
кислоты уксусной безводной Р, прибавляют 70 мл ук¬
сусного ангидрида Р и титруют 0,1 М раствором кис¬
лоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 32,08 мг С17Н20М2О2 НС1.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. (1 ЯЗг,55)-8-Метил-8-азабицикло[3.2.1]октан-3-
ол (тропин).
со2н
В. 1Н-Индол-3-карбоновая кислота.
07/2016:0375
УГЛЕРОДА ДИОКСИД
СагЬопе/ с1юх1с1ит
САКВОЙ о/ох/ое
С02 М.м. 44,01
[124-38-9]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,5 % (об/об) С02 в газо¬
образной фазе.
Данная статья распространяется на углерода ди¬
оксид для медицинского применения.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветный газ.
При температуре 20 °С и давлении 101 кПа 1
объем растворяется в 1 объеме воды.
ПРОИЗВОДСТВО
Проводят испытания газовой фазы.
Если проводят испытание баллона, баллон вы¬
держивают при комнатной температуре в течение
не менее 6 ч перед проведением испытаний. Баллон
держат в вертикальном положении таким образом,
чтобы вентиль был расположен сверху.
127*. Зак. 1060.
1008
Гэсударственная фармакопея Республики Беларусь
Углерода монооксид. Газовая хроматография
(2.2.28).
Испытуемый газ. Испытуемый образец.
Газ сравнения. Смесь, содержащая 5 ррт (об/об)
углерода монооксида Р в азоте Р1.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 2 м и
диаметром 4 мм, заполненная подходящим молеку¬
лярным ситом для хроматографии (0,5 нм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 60 мл/мин;
- температура колонки: 50 °С;
- температура детектора: 130 °С;
- детектор: пламенно-ионизационный детектор
с метанизатором;
- вводимая проба: через петлевой инжектор.
Условия работы прибора и объем вводимой про¬
бы регулируют таким образом, чтобы высота пика
углерода монооксида на хроматограмме газа сравне¬
ния составляла не менее 35 % от полной шкалы реги¬
стрирующего устройства.
Предельное содержание:
- углерода монооксид (не более 5 ррт (об/об)):
на хроматограмме испытуемого газа площадь пика,
соответствующего углерода монооксиду, не должна
превышать площадь основного пика на хроматограм¬
ме газа сравнения.
Азота монооксид и азота диоксид. Не более
2 ррт (об/об) суммарно. С использованием хемилю¬
минесцентного анализатора (2.5.26).
Испытуемый газ. Испытуемый образец.
Газ сравнения (а). Углерода диоксид Р1.
Газ сравнения (Ь). Смесь, содержащая 2 ррт
(об/об) азота монооксида Р в азоте Р1 или в углеро¬
да диоксиде Р1.
Анализаторы калибруют и настраивают чувстви¬
тельность с помощью газов сравнения (а) и (Ь). Изме¬
ряют содержание азота монооксида и азота диоксида
в испытуемом газе.
При использовании в газе сравнения (Ь) азо¬
та вместо углерода диоксида полученный результат
умножают на поправочный фактор для поправки на
эффект гашения отклика анализатора, вызываемого
матричным эффектом углерода диоксида.
Поправочный фактор определяют на известной
стандартной смеси азота монооксида в углерода ди¬
оксиде и сравнивают фактическое содержание с пока¬
заниями анализатора, откалиброванного при помощи
стандартной смеси ЫО/Ы2
„ . Фактическое содержание азота монооксида
Поправочный фактор = ил —.
показания анализатора (азота монооксида)
Общая сера. Не более 1 ррт (об/об). Испытание
проводят с использованием флуоресцентного анали¬
затора в ультрафиолетовой области, после окисления
соединений серы при нагревании при температуре
1000 °С (см. рисунок 0375.-1).
Прибор состоит из следующих частей:
- система, генерирующая ультрафиолетовое из¬
лучение с длиной волны 210 нм, состоящая из УФ-
лампы, коллиматора и селективного светофильтра;
пучок периодически блокируется с помощью прерыва¬
теля, вращающегося с высокой скоростью;
- реакционная камера, через которую пропускают
предварительно профильтрованный испытуемый газ;
- система, детектирующая излучение, испускае¬
мое при длине волны 350 нм, состоящая из селектив¬
ного светофильтра, фотоумножителя и усилителя.
Испытуемый газ. Испытуемый образец.
Газ сравнения (а). Углерода диоксид Р1.
Газ сравнения (Ь). Смесь, содержащая от 0,5 ррт
(об/об) до 2 ррт (об/об) сероводорода Р1 в углерода
диоксиде Р1.
Прибор калибруют и настраивают чувствитель¬
ность при помощи газов сравнения (а) и (Ь). Испыту¬
емый газ пропускают через кварцевую печь, нагретую
до температуры 1000 °С. Кислород Р должен цирку¬
лировать в печи с десятикратной скоростью потока
испытуемого газа. Измеряют содержание серы диок¬
сида в газообразной смеси, выходящей из печи.
прерыватель
□3
источник
ультрафиолетового
излучения
А |
фильтр 210 нм
коллиматор
ВХОДЯЩИЙ выходящий
газ газ
I !
фотоумножитель усилитель
Рисунок 0375.-1. Флуоресцентный анализатор в
ультрафиолетовой области
Вода. Не более 67 ррт (об/об). Определение
проводят на электролитическом гигрометре (2.5.28).
Количественное определение. С использова¬
нием инфракрасного анализатора (2.5.35).
Испытуемый газ. Испытуемый образец. Во избе¬
жание светорассеяния фильтруют.
Газ сравнения (а). Углерода диоксид Р1.
Газ сравнения (Ь). Смесь, состоящая из 5,0%
(об/об) азота Р1 и 95,0 % (об/об) углерода диоксида Р1.
Анализатор калибруют и настраивают чувствитель¬
ность при помощи газов сравнения (а) и (Ь). Измеряют
содержание углерода диоксида в испытуемом газе.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр углерода диокси¬
да по Европейской фармакопее.
B. В испытуемый газ вносят тлеющую деревян¬
ную лучину. Лучина должна потухнуть.
C. Испытуемый газ пропускают через раствор
бария гидроксида Р. Образуется белый осадок, рас¬
творяющийся в кислоте уксусной разведенной Р с
выделением пузырьков.
ИСПЫТАНИЯ
Проводят испытания газовой фазы.
Если проводят испытание баллона, баллон вы¬
держивают при комнатной температуре в течение
не менее 6 ч перед проведением испытаний. Баллон
держат в вертикальном положении таким образом,
чтобы вентиль был расположен сверху.
Углерода монооксид (2.1.6). Не более 5 ррт
(об/об). Определение проводят с помощью индика¬
торной трубки для углерода монооксида.
Уголь активированный
1009
Сероводород (2.1.6). Не более 1 ррт (об/об).
Определение проводят с помощью индикаторной
трубки для сероводорода.
Азота монооксид и азота диоксид (2.1.6). Не
более 2 ррт (об/об). Определение проводят с помо¬
щью индикаторной трубки для азота монооксида и
азота диоксида.
Серы диоксид (2.1.6). Не более 2 ррт (об/об).
Определение проводят с помощью индикаторной
трубки для серы диоксида.
Пары воды (2.1.6). Не более 67 ррт (об/об).
Определение проводят с помощью индикаторной
трубки для паров воды.
ХРАНЕНИЕ
В сжиженном состоянии под давлением в подхо¬
дящих для газов контейнерах, соответствующих уста¬
новленным требованиям.
ПРИМЕСИ
A. N0: Азота монооксид.
B. 1Ч02: Азота диоксид.
C. СО: Углерода монооксид.
О. Общая сера.
Е. Н20: вода.
07/2016:0313
УГОЛЬ АКТИВИРОВАННЫЙ
СагЬо ас1ма1из
СНАПСОАЦ АСТЫАТЕО
ОПРЕДЕЛЕНИЕ
Получают из растительных материалов путем
подходящих процессов карбонизации, обеспечиваю¬
щих высокую поглощающую способность.
ОПИСАНИЕ (СВОЙСТВА)
Черный легкий порошок без комковатости.
Практически нерастворим во всех обычных рас¬
творителях.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец, нагреваемый докрасна,
медленно сгорает без пламени.
B, Испытуемый образец выдерживает испытание
«Адсорбционная способность», как указано в разделе
«Испытания».
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца поме¬
щают в коническую колбу с притертым горлышком
и прибавляют 50 мл кислоты хлористоводород¬
ной разведенной Р. Осторожно кипятят с обратным
холодильником в течение 1 ч, фильтруют и про¬
мывают фильтр кислотой хлористоводородной
разведенной Р. Объединенный фильтрат и про¬
мывные растворы выпаривают досуха на водяной
бане, остаток растворяют в 0,1 М растворе кис¬
лоты хлористоводородной и доводят до объема
50,0 мл этим же растворителем.
Кислотность или щелочность. К 2,0 г испы¬
туемого образца прибавляют 40 мл воды Р и кипя¬
тят в течение 5 мин. Охлаждают, доводят до перво¬
начальной массы водой, свободной от углерода
диоксида, Р и фильтруют. Первые 20 мл фильтра¬
та отбрасывают. К 10 мл фильтрата прибавляют
0,25 мл раствора бромтимолового синего Р1
и 0,25 мл 0,02 М раствора натрия гидроксида.
Раствор синий. При прибавлении не более 0,75 мл
0,02 М раствора кислоты хлористоводородной
должно появиться желтое окрашивание.
Вещества, растворимые в кислоте. Не бо¬
лее 3 %. К 1,0 г испытуемого образца прибавляют
25 мл кислоты азотной разведенной Р и кипятят в
течение 5 мин. Горячий раствор фильтруют через
стеклянный пористый фильтр (10) (2.1.2) и промы¬
вают 10 мл горячей воды Р. Объединенный филь¬
трат и промывные растворы выпаривают досуха на
водяной бане, к остатку прибавляют 1 мл кислоты
хлористоводородной Р, снова выпаривают досуха
и остаток высушивают до постоянной массы при
температуре от 100 °С до 105 °С. Масса остатка не
должна превышать 30 мг.
Окрашенные вещества, растворимые в
щелочи. К 0,25 г испытуемого образца прибав¬
ляют 10 мл раствора натрия гидроксида разве¬
денного Р и кипятят в течение 1 мин. Охлаждают,
фильтруют и объем фильтрата доводят водой Р до
объема 10 мл. Окраска раствора должна быть не
интенсивнее окраски эталона ОУ(ЗЖ)4 (2.2.2, ме¬
тод II).
Вещества, растворимые в 96 % спирте. Не
более 0,5 %. К 2,0 г испытуемого образца прибав¬
ляют 50 мл 96 % спирта Р и кипятят с обратным
холодильником в течение 10 мин. Немедленно
фильтруют, охлаждают и доводят до объема 50 мл
96 % спиртом Р. Окраска фильтрата должна быть
не интенсивнее окраски эталона У(Ж)6 или ВУ(КЖ)6
(2.2.2, метод II). 40 мл фильтрата упаривают досу¬
ха и сушат до постоянной массы при температуре
от 100 °С до 105 °С. Масса остатка не должна пре¬
вышать 8 мг.
Флуоресцирующие вещества. 10,0 г испы¬
туемого образца обрабатывают 100 мл циклогек¬
сана Р1 в течение 2 ч в аппарате для периодиче¬
ской экстракции (аппарате Сокслета). Собирают
жидкость и доводят циклогексаном Р1 до объема
100 мл. Исследуют в ультрафиолетовом свете при
длине волны 365 нм. Флуоресценция раствора не
должна превышать интенсивность флуоресценции
раствора 83 мкг хинина Р в 1000 мл 0,005 М раст¬
вора кислоты серной в тех же условиях.
Сульфиды. 1,0 г испытуемого образца по¬
мещают в коническую колбу и прибавляют 5 мл
кислоты хлористоводородной Р1 и 20 мл воды Р.
Нагревают до кипения. Образующиеся пары не
должны окрашивать в коричневый цвет свинцово¬
ацетатную бумагу Р.
Медь. Не более 0,0025 % (25 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Испытуемый раствор. Раствор 3.
Раствор сравнения. Готовят соответствую¬
щими разведениями эталонного раствора меди
(0,1 % Си) Р 0,1 М раствором кислоты хлористо¬
водородной.
Источник излучения: лампа с полым катодом
для определения меди.
Длина волны: 325,0 нм.
Генератор атомного пара: воздушно-ацети¬
леновое пламя.
Свинец. Не более 0,0010 % (10 ррт).
128. Зак. 1060.
1010
Гэсударственная фармакопея Республики Беларусь
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. Раствор 3.
Раствор сравнения. Готовят соответствующи¬
ми разведениями эталонного раствора свница
(100 ррт РЬ) Р 0,1 М раствором кислоты хлористо¬
водородной.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 283,3 нм; в зависимости от прибо¬
ра может использоваться 217,0 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Цинк. Не более 0,0025 % (25 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод /).
Испытуемый раствор. Раствор 3.
Раствор сравнения. Готовят соответствующими
разведениями эталонного раствора цинка (100 ррт
2п) Р 0,1 М кислотой хлористоводородной.
Источник излучения: лампа с полым катодом
для определения цинка.
Длина волны: 214,0 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Потеря в массе при высушивании (2.2.32). Не
более 15 %. 1,000 г испытуемого образца сушат при
температуре 120 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 5,0%.
Определение проводят из 1,0 г испытуемого образца.
Адсорбционная способность. 0,300 г испыту¬
емого образца помещают в коническую колбу вме¬
стимостью 100 мл со стеклянной притертой пробкой.
Прибавляют 25,0 мл свежеприготовленного раствора
0,5 г феназона Р в 50 мл воды Р. Энергично встря¬
хивают в течение 15 мин. Фильтруют, первые 5 мл
фильтрата отбрасывают. К 10,0 мл фильтрата при¬
бавляют 1,0 г калия бромида Р и 20 мл кислоты хло¬
ристоводородной разведенной Р. Титруют 0,0167 М
раствором калия бромата до исчезновения крас¬
ного окрашивания, используя в качестве индикатора
0,1 мл раствора метилового красного Р. В конце ти¬
трования титруют медленно (1 капля в 15 сек). Про¬
водят контрольный опыт, используя 10,0 мл раствора
феназона.
Количество адсорбированного феназона в 100 г
угля активированного рассчитывают по формуле:
2,353 (а-Ь)
т
где:
а — количество 0,0167 М раствора калия брома¬
та, пошедшее на титрование в контрольном опыте,
мл;
Ь — количество 0,0167 М раствора калия брома¬
та, пошедшее на титрование испытуемого образца,
мл;
т — масса навески испытуемого образца, г.
100 г угля активированного (в пересчете на сухое
вещество) должно адсорбировать не менее 40 г фе¬
назона.
Микробиологическая чистота
- ОКА: критерий приемлемости 103 КОЕ/г (2.6.12);
- ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0590
УКСУСНАЯ КИСЛОТА ЛЕДЯНАЯ
Аас1ит асеИсит д1аа'а1е
АСЕТ1САСЮ, С1.АС/А1.
О
С2н402 М.м. 60,1
[64-19-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % (м/м) и не более
100,5 % (м/м).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллическая масса или прозрачная, бесц¬
ветная, летучая жидкость.
Смешивается с водой, 96 % спиртом и метилен-
хлоридом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 100 г/л испытуемого образца имеет
сильнокислую реакцию (2.2.4).
B. К 0,03 мл испытуемого образца прибавляют
3 мл воды Р и нейтрализуют раствором натрия ги¬
дроксида разведенным Р. Полученный раствор дает
реакцию (Ь) на ацетаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 20 мл испытуемого образца доводят
водой Р до объема 100 мл.
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Испытуемый обра¬
зец должен быть бесцветным.
Температура затвердевания (2.2.18). Не менее
14,8 °С.
Восстанавливающие вещества. 2,0 мл испыту¬
емого образца доводят водой Р до объема 10,0 мл,
прибавляют 0,1 мл 0,02 М раствора калия перманга¬
ната, нагревают на водяной бане в течение 1 мин,
появляется розовое окрашивание.
Хлориды (2.4.4). Не более 25 мг/л. 10 мл раст¬
вора 5 доводят водой Р до объема 15 мл. Полученный
раствор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 50 мг/л. 15 мл раст¬
вора 5 должны выдерживать испытание на сульфаты.
Железо (2.4.9). Не более 0,0005 % (5 ррт).
5,0 мл раствора А, полученного при проведении испы¬
тания «Тяжелые металлы», доводят водой Р до объ¬
ема 10,0мл. Полученный раствор должен выдержи¬
вать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0005 % (5 ррт). Остаток, полученный при проведе¬
нии испытания «Остаток после выпаривания», раство¬
ряют при нагревании двумя порциями по 15 мл воды Р
и доводят до объема 50,0 мл этим же растворителем
(раствор А). 12 мл раствора А должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с исполь¬
зованием эталонного раствора свинца (2 ррт РЬ) Р.
Остаток после выпаривания. Не более
0,01 %. 20 г испытуемого образца выпаривают до¬
суха на водяной бане и сушат при температуре от
100 °С до 105 °С. Масса сухого остатка не должна
превышать 2,0 мг.
Фамотидин
1011
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Точно взвешивают коническую колбу со стеклян¬
ной притертой пробкой, содержащую 25 мл воды Р,
прибавляют 1,0 мл испытуемого образца и снова
точно взвешивают. Прибавляют 0,5 мл раствора фе¬
нолфталеина Р и титруют 1 М раствором натрия
гидроксида.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 60,1 мгС2Н402.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:1012
ФАМОТИДИН
РатоНсИпит
РАМОТЮШЕ
[76824-35-6]
ОПРЕДЕЛЕНИЕ
3-[[[2-[(Диаминометилен)амино]тиазол-4-ил]ме-
тил]сульфанил]-А/-сульфамоилпропанимидамид.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый кристаллический
порошок или кристаллы.
Очень мало растворим в воде, легко растворим
в кислоте уксусной ледяной, очень мало растворим
в этаноле, практически нерастворим в этилацетате.
Растворяется в разведенных минеральных кислотах.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фамотидина #или спектр, пред¬
ставленный на рисунке #1012.-1#.
Если полученные спектры отличаются, то 0,10 г
испытуемого образца и 0,10 г ФСО фамотидина су¬
спендируют по отдельности в 5 мл воды Р, нагрева¬
ют до кипения и охлаждают, потирая стенки пробирки
стеклянной палочкой для инициации кристаллиза¬
ции. Фильтруют, промывают кристаллы 2 мл ледяной
воды Р и сушат при температуре 80 °С и давлении, не
превышающем 670 Па, в течение 1 ч. Сухие остатки
используют для получения новых спектров.
ИСПЫТАНИЯ
Раствор 3. 0,20 г испытуемого образца раство¬
ряют в растворе 50 г/л кислоты хлористоводород¬
ной Р, нагревая при необходимости до 40 °С, и дово¬
дят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)7.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Рисунок #1012.-1. Инфракрасный спектр ФСО фамотидина
128*. Зак. 1060.
1012
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. 12,5 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 10,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 100,0 мл.
Раствор сравнения (Ь). 2,5 мг ФСО фамотидина
примеси О растворяют в метаноле Р и доводят до объ¬
ема 10,0 мл этим же растворителем. К 1,0 мл получен¬
ного раствора прибавляют 0,50 мл испытуемого раст¬
вора и доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (с). 5,0 мг ФСО фамотиди¬
на для проверки пригодности хроматографической
системы (содержит примеси А, В, С, О, Е, Р, О) раст¬
воряют в подвижной фазе А и доводят до объема
10.0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 50 °С;
- подвижная фаза:
- подвижная фаза А: смесь из 6 частей мета¬
нола Р, 94 частей ацетонитрила Р и 900 объ¬
емов раствора 1,882 г/л натрия гексансульфо-
ната Р, доведенного кислотой уксусной Р до
рН 3,5;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
Скорость
подвижной
фазы
(мл/мин)
0—23
о
0
1
со
о>
0-4
1
23—27
96
4
1 —2
21—41
96 — 78
4 — 22
2
47-48
^1
00
1
О
о
О
т
СМ
СМ
2
48—54
100
0
2 — 1
- спектрофотометрический детектор, длина
волны 265 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, Р и О, используя хрома¬
тограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО фамотидина для проверки при¬
годности хроматографической системы; иденти¬
фицируют пик примеси О, используя хроматограмму
раствора сравнения (Ь).
Относительные времена удерживания (по от¬
ношению к фамотидину, время удерживания — около
21 мин): примесь й — около 1,1; примесь С — око¬
ло 1,2; примесь (3 — около 1,4; примесь Р — око¬
ло 1,5; примесь А—около 1,6; примесь В — около 2,0;
примесь Е — около 2,1.
Пригодность хроматографической системы:
- хроматограмма раствора сравнения (с) должна
соответствовать хроматограмме ФСО фамотидина
для проверки пригодности хроматографической си¬
стемы,
- времена удерживания: фамотидин —
(19—23) мин на всех хроматограммах; примесь Е — не
более 48 мин на хроматограмме раствора сравнения (с);
- разрешение: не менее 3,5 между пиками фа¬
мотидина и примеси О на хроматограмме раствора
сравнения (Ь).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А— 1,9; для примеси В — 2,5; для примеси
С — 1,9; для примеси Р — 1,7; для примеси О — 1,4):
- примеси С, О (не более 0,3 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С и О, не должны превышать
3-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примеси А, В, р <3 (не более 0,15 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, В, Р и О, не должны
превышать 1,5-кратную площадь основного пика на
хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Р и С, не должна превышать пло¬
щадь основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей (не более 0,8 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 8-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010 % (10 ррт). 0,5 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 0,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 80 °С и давлении, не превышающем
670 Па, в течение 5 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором хлорной кислоты потенциометрически
(2.2.20).
1 мл 0,1 М раствора хлорной кислоты соответ¬
ствует 16,87 мг С8Н15М70233.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Р, 6.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е, Н, I, 3.
Феназон
1013
А. 3-[[[2-[(Диаминометилиден)амино]тиазол-4-ил]-
метил]суль-фанил]пропанимидамид.
В. 3,5-Бис[2-[[[[диаминометилиден)амино]тиазол-
4-ил]метил]сульфанил]этил]-4Н-1,2,4,6-триатризина
1,1-диоксид.
С. 3-[[[2-[(Диаминометилиден)амино]тиазол-4-ил]-
метил]сульфанил]-Л/-сульфамоилпропанамид.
о
О. 3-[[[2-[(Диаминометилиден)амино]тиазол-4-ил]-
метил]суль-фанил]пропанамид.
Е. 2,2,-1Дисульфандиилбис(метилентиазол-4>2-
диил)]дигуанидин.
Р. 3-[[[2-[(Диаминометилиден)амино]тиазол-4-ил]-
метил]сульфанил]пропановая кислота.
ин
О. А/-Циано-3-[[[2-[(диаминометилиден)амино]-
тиазол-4-ил]метил]сульфанил]пропанимидамид.
N4
Н. [2-[(Диаминометилиден)амино]тиазол-4-ил]ме-
тилкарбамимидотиоат.
I. 3-[[[2-[(Диаминометилиден)амино]тиазол-4-ил]-
метил]сульфинил]-А/-сульфамоилпропанамид.
о
3. Метил 3-[[[2-[(диаминометилиден)амино]тиазол-
4-ил]метил]сульфанил]пропаноат.
07/2016:0421
ФЕНАЗОН
РНепагопит
РНЕЫА20ЫЕ
С^Н^О М.м. 188,2
[60-80-0]
ОПРЕДЕЛЕНИЕ
1,5-Диметил-2-фенил-1,2-дигидро-ЗН-пиразол-З-он.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический
порошок или бесцветные кристаллы.
Очень легко растворим в воде, в 96 % спирте и в
метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Температура плавления (2.2.14): от 109 °С до
113 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО феназона *или спектр,
представленный на рисунке #0421.-1#.
C. К 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 4 мл воды Р и
0,25 мл кислоты серной разведенной Р. Прибавляют
1 мл раствора натрия нитрита Р. Появляется зеле¬
ное окрашивание.
й. К 1 мл раствора 3 прибавляют 4 мл воды Р и
0,5 мл раствора железа (III) хлорида Р2. Появляется
красное окрашивание, исчезающее при прибавлении
кислоты серной разведенной Р.
129. Зак. 1060.
1014
Гэсударственная фармакопея Республики Беларусь
Рисунок #0421 .-1. Инфракрасный спектр ФСО феназона
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора фенолфта¬
леина Р; раствор бесцветный. Прибавляют 0,2 мл
0,01 М раствора натрия гидроксида. Появляется
красное окрашивание. К полученному раствору при¬
бавляют 0,25 мл раствора метилового красного Р
и 0,4 мл 0,01 М раствора кислоты хлористоводо¬
родной. Появляется красное или желтовато-красное
окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО феназона при¬
меси А растворяют в подвижной фазе, прибавляют
10 мл испытуемого раствора и доводят до объема
20.0 мл подвижной фазой. 1,0 мл полученного раст¬
вора доводят до объема 50,0 мл подвижной фазой.
Раствор сравнения (с). 5,0 мг ФСО феназона
примеси А растворяют в подвижной фазе и доводят
до объема 20,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл. 1,0 мл полученного раствора дово¬
дят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 6,0 мм, заполненная сферическим силикагелем
октадецилсилильным для хроматографии Р с раз¬
мером частиц 5 мкм;
- подвижная фаза: 6,8 г калия дигидрофос¬
фата Р растворяют в воде Р и доводят до объема
1000 мл этим же растворителем. Прибавляют 2 мл
триэтиламина Р и доводят до рН 7,0 раствором на¬
трия гидроксида Р. Прибавляют 430 мл метанола Р;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания феназона.
Относительное удерживание (по отношению к
феназону, время удерживания — около 13 мин): при¬
месь А — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками при¬
меси А и феназона.
Предельное содержание примесей:
- примесь А (не более 0,05 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси А, не должна превышать площадь соответству¬
ющего пика на хроматограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,05 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика при¬
меси А, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (а);
-сумма примесей (не более 0,1 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
Фенилаланин
1015
- неучитываемый предел (0,03 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,3 площади основного пика на хро¬
матограмме раствора сравнения (а).
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
1,5 г испытуемого образца растворяют в воде дис¬
тиллированной Р и доводят до объема 15 мл этим же
растворителем. Полученный раствор должен выдер¬
живать испытания на сульфаты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 6 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 20 мл
воды Р. Прибавляют 2 г натрия ацетата Р, 1 мл
уксусной кислоты разведенной Р и 25,0 мл 0,05 М
раствора йода и выдерживают в защищенном от све¬
та месте в течение 30 мин. Прибавляют 25 мл мети-
ленхлорида Р и встряхивают до растворения осадка.
Титруют 0,1 М раствором натрия тиосульфата, ис¬
пользуя в качестве индикатора 1 мл раствора крах¬
мала Р, добавляя его в конце титрования.
Рисунок #0782.-1. Инфракрасный спектр ФСО фенилаланина
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствор йода соответствует 9,41 мг
С.ДДО.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
А. 5-Метил-2-фенил-2,4-дигидро-ЗН-пиразол-3-он.
07/2016:0782
ФЕНИЛАЛАНИН
РМепу1а1аптит
РНЕЫУ1.А1-АЫ1ЫЕ
С9Н11Ы02 М.м. 165,2
[63-91-2]
ОПРЕДЕЛЕНИЕ
(5)-2-Амино-3-фенилпропановая кислота.
Продукт ферментации, экстракт или гидролизат
белка.
129*. Зак. 1060.
1016
Гэсударственная фармакопея Республики Беларусь
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или блестящие белые хлопья.
Умеренно растворим в воде, очень мало раство¬
рим в спирте. Растворяется в разведенных минераль¬
ных кислотах и в разведенных растворах гидроксидов
щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фенилаланина #или спектр,
представленный на рисунке #0782.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру ос¬
новному пятну на хроматограмме раствора сравне¬
ния (а).
й. К 10 мг испытуемого образца прибавляют 0,5 г
калия нитрата Р и 2 мл кислоты серной Р. Нагре¬
вают на водяной бане в течение 20 мин и охлажда¬
ют. Прибавляют 5 мл раствора 50 г/л гидроксилами-
на гидрохлорида Р и выдерживают в ледяной бане в
течение 10 мин. Прибавляют 9 мл раствора натрия
гидроксида концентрированного Р. Появляется окра¬
шивание от фиолетово-красного до фиолетово-корич¬
невого цвета.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в 1 М растворе кислоты хлористоводород¬
ной Р и доводят до объема 10 мл этим же раствори¬
телем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От
-33,0 до -35,5 (в пересчете на сухое вещество). 0,50 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов.#
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образца, подходящий для применяемого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 6,0 мл эталонного раст¬
вора аммония (100 ррт N14) Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (д). 30 мг изолейцина Р и 30 мг
лейцина Р растворяют в растворе А и доводят до объ¬
ема 50,0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят раствором А до объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
(а), (Ь) и (б) вводят в аминокислотный анализатор.
Запускают подходящую программу для определения
физиологических аминокислот.
Пригодность хроматографической системы:
раствор сравнения (б):
-разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию фенилаланина в растворе
сравнения (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (Ь); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
- любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для фар¬
мацевтического использования (2034), не применяют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в смеси равных объемов кисло¬
ты уксусной ледяной Р и воды Р и доводят до объема
10 мл этой же смесью растворителей.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят до объема 50 мл смесью равных
объемов кислоты уксусной ледяной Р и воды Р.
Раствор сравнения (а). 10 мг ФСО фенилала¬
нина растворяют в смеси равных объемов кислоты
уксусной ледяной Р и воды Р и доводят до объема
50 мл этой же смесью растворителей.
Раствор сравнения (Ь). 5 мл испытуемого раст¬
вора (Ь) доводят до объема 20 мл смесью равных
объемов кислоты уксусной ледяной Р и воды Р.
Раствор сравнения (с). 10 мг ФСО фенилалани¬
на и 10 мг ФСО тирозина растворяют в смеси равных
объемов кислоты уксусной ледяной Р и воды Р и до¬
водят до объема 25 мл этой же смесью растворителей.
Пластинка: ТСХпластинка со слоем силикагеля Р.
Фенилэфрин
1017
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре от 100 °С
до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных основных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее основного
пятна на хроматограмме раствора сравнения (Ь).
Хлориды (2.4.4). Не более 0,0200 % (200 ррт).
0,25 г испытуемого образца растворяют в 3 мл кис¬
лоты азотной разведенной Р и доводят водой Р до
объема 15 мл. Полученный раствор должен выдержи¬
вать испытание на хлориды без дополнительного при¬
бавления азотной кислоты.
Сульфаты (2.4.13). Не более 0,0300%
(300 ррт). 0,5 г испытуемого образца растворяют в
смеси, состоящей из кислоты хлористоводородной
разведенной Р и воды дистиллированной Р (5:25,
об/об) и доводят до объема 15 мл этой же смесью
растворителей. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(с) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хромато¬
графии, то испытание «Аммония соли» (2.4.1, метод
В) проводят следующим образом. 50 мг испытуемого
образца должны выдерживать испытание на соли ам-
мония. Эталон готовят с использованием 0,1 мл эта¬
лонного раствора аммония (100 ррт АIН^ Р.#
Железо (2.4.9). Не более 0,0010 % (10 ррт). 1,0 г
испытуемого образца помещают в делительную во¬
ронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 3 мл
кислоты муравьиной безводной Р. Прибавляют
30 мл кислоты уксусной безводной Р. Титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,52 мг СдН^ЫО,.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие ве¬
щества, если они присутствуют в значительных количе¬
ствах, следует определять тем или иным испытанием,
описанным в частной статье. Их содержание лимити¬
руется общим критерием приемлемости для других/не-
специфицированных примесей. Вследствие этого нет
необходимости идентифицировать эти примеси для
доказательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О.
А. (2$)-2-Амино4-метилпентановая кислота (лейцин).
н3с.
В. (25)-2-Амино-4-(метилсульфанил)бутановая
кислота (метионин).
^ н ын2
со2н
С. (25)-2-Амино-3-(4-гидроксифенил)пропановая
кислота (тирозин).
Н 1МН2
со2н
СНз
О. (25)-2-Амино-3-метилбутановая кислота (валин).
07/2016:1035
ФЕНИЛЭФРИН
РНепу1ерМпит
РНЕЫПЕРНРШЕ
[59-42-7]
СН3
М.м. 167,2
130. Зак. 1060.
1018
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
(1/?)-1-(3-Гидроксифенил)-2-(метиламино)этанол.
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде, умеренно растворим в
метаноле, мало растворим в 96 % спирте. Растворя¬
ется в разведенных минеральных кислотах и разве¬
денных растворах гидроксидов щелочных металлов.
Температура плавления: около 174 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фенилэфрина пили спектр,
представленный на рисунке #7035.-7*.
C. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Смесь из равных объ¬
емов метиленхлорида Р и метанольного раствора
кислоты хлористоводородной (1 объем кислоты хло¬
ристоводородной Р доводят метанолом Р до 10 объ¬
емов).
Испытуемый раствор. 0,1 г испытуемого образ¬
ца растворяют в смеси растворителей и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО фенилэфрина
растворяют в смеси растворителей и доводят до объ¬
ема 1 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р —■ метанол Р — метиленхлорид Р
(0,5:25:70, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: на менее 15 см от ли¬
нии старта.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм; пластин¬
ку опрыскивают раствором 1 г/л соли прочного синего
В Р в растворе 50 г/л натрия карбоната Р и просма¬
тривают при дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
0.10 мг испытуемого образца растворяют в 1 мл
1 М растворе кислоты хлористоводородной, при¬
бавляют 0,05 мл раствора меди сульфата Р и 1 мл
раствора 200 г/л натрия гидроксида Р. Появляется
фиолетовое окрашивание. Прибавляют 1 мл эфира Р
и встряхивают. Верхний слой остается бесцветным.
ИСПЫТАНИЯ
Раствор 5. 1 г испытуемого образца растворяют
в 1 М растворе кислоты хлористоводородной и до¬
водят до объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)7.
Удельное оптическое вращение (2.2.7). От -53
до -57 в пересчете на сухое вещество. 1,250 г испыту¬
емого образца растворяют в 1 М растворе кислоты
хлористоводородной и доводят до объема 25,0 мл
этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Рисунок #1035.-1. Инфракрасный спектр ФСО фенилэфрина
Фенилэфрин
1019
Смесь растворителей. Раствор кислоты хло¬
ристоводородной разведенной Р — подвижная фаза
В — подвижная фаза А (5:200:800, об/об/об).
Буферный раствор рН 2,8. 3,25 г натрия октан-
сульфоната моногидрата Р растворяют в 1000 мл
воды Р при перемешивании в течение 30 мин и дово¬
дят кислотой фосфорной разведенной Р до рН 2,8.
Испытуемый раствор. 41,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 2,0 мл полученного раствора доводят сме¬
сью растворителей до объема 100,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
с ФСО фенилэфрина гидрохлорида для идентифи¬
кации пиков (содержит примеси С и Е) растворяют в
2.0 мл смеси растворителей.
Условия хроматографирования:
- колонка длиной 0,055 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 — бу¬
ферный раствор рН 2,8 (10:90, об/об);
-подвижная фаза В: буферный раствор рН
2,8 — ацетонитрил Р1 (10:90, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
93
7
3—13
93 —> 70
о
со
т
13—14
70—>93
30->7
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отноше¬
нию к фенилэфрину, время удерживания — около
2,8 мин): примесь С — около 1,3; примесь Е — око¬
ло 3,6.
Пригодность хроматографической системы:
-фактор асимметрии: не более 1,9 для ос¬
новного пика на хроматограмме испытуемого раст¬
вора;
-коэффициент разделения пиков: не менее
5 (Нр — высота пика примеси С относительно базо¬
вой линии; Ну — расстояние между базовой линией и
нижней точкой кривой, разделяющей пики примеси С
и фенилэфрина) на хроматограмме раствора сравне¬
ния (Ь).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков, на
соответствующие поправочные коэффициенты: для
примеси С — 0,5; для примеси Е — 0,5):
-примеси С, Е (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С и Е, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей С и Е, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образ¬
ца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 60 мл
уксусной кислоты безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 16,72 мг С9Н13М02.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, О.
А. Р = Р' = Н: (1Р)-2-Амино-1-(3-гидроксифенил)-
этанол (норфенилэфрин).
О. Р = СН2-С6Н5, Р' = СН3: (1Р)-2-(Бензилметил-
амино)-1 -(З-гидроксифенил)этанол (бензилфенилэф-
рин).
С. Р = Н: 1-(3-Гидроксифенил)-2-(метиламино)эта-
нон (фенилэфрон).
Е. Р = СН2-С6Н5:2-(Бензилметиламино)-1 -(3-гидро-
ксифенил)этанон (бензилфенилэфрон).
130*. Зак. 1060.
1020
Государственная фармакопея Республики Беларусь
07/2016:0632
ФЕНИЛЭФРИНА ГИДРОХЛОРИД
РЬепу1ер11пп1 Ьус1госЫопс1ит
РНЕМП-ЕРНПШЕ НУОПОСШОПЮЕ
С9Н13М02 - НС1 М.м. 203,7
[61-76-7]
ОПРЕДЕЛЕНИЕ
(1/?)-1-(3-Гидроксифенил)-2-(метиламино)этано-
ла гидрохлорид.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в 96 % спирте.
Температура плавления: около 143 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, С, Е.
Вторая идентификация: А, В, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Температура плавления (2.2.14): от 171 °С до
176 °С. 0,3 г испытуемого образца растворяют в 3 мл
воды Р, прибавляют 1 мл раствора аммиака разве¬
денного Р1 и инициируют кристаллизацию путем тре¬
ния стеклянной палочкой о стенки колбы. Полученные
кристаллы промывают ледяной водой Р и сушат при
температуре 105 °С в течение 2 ч.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО фенилэфрина гидрохлорида
#или спектр, представленный на рисунке #0632.-1#.
й. 10 мг испытуемого образца растворяют в 1 мл
воды Р, прибавляют 0,05 мл раствора 125 г/л меди
сульфата Р и 1 мл раствора 200 г/л натрия гидрок¬
сида Р. Появляется фиолетовое окрашивание. При¬
бавляют 1 мл эфира Р и встряхивают. Верхний слой
остается бесцветным.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,00 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, по¬
лученной из воды дистиллированной Р, и доводят до
объема 100,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К10 мл раствора
3 прибавляют 0,1 мл раствора метилового красного
и 0,2 мл 0,01 М раствора натрия гидроксида. По¬
является желтое окрашивание. При прибавлении не
более 0,4 мл 0,01 М раствора кислоты хлористово¬
дородной должно появиться красное окрашивание.
Удельное оптическое вращение (2.2.7). От -43
до -47 (в пересчете на сухое вещество). Определяют
удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Рисунок #0632.-1. Инфракрасный спектр ФСО фенилэфрина гидрохлорида
Фенилэфрина гидрохлорид
1021
Смесь растворителей. Подвижная фаза В —
подвижная фаза А (20:80, об/об).
Буферный раствор рН 2,8. 3,25 г натрия октан-
сульфоната моногидрата Р растворяют в 1000 мл
воды Р при перемешивании в течение 30 мин и дово¬
дят до рН 2,8 кислотой фосфорной разведенной Р.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 2,0 мл полученного раствора доводят сме¬
сью растворителей до объема 100,0 мл.
Раствор сравнения (Ь). Содержимое контейнера
ФСО фенилэфрина гидрохлорида для идентифика¬
ции пиков (содержит примеси С и Е) растворяют в
2.0 мл смеси растворителей.
Условия хроматографирования:
- колонка длиной 0,055 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 3 мкм;
- температура: 45 °С;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 — бу¬
ферный раствор рН 2,8 (10:90, об/об);
- подвижная фаза В: буферный раствор рН
2,8 — ацетонитрил Р1 (10:90, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
93
7
3—13
о
1^
т
со
О)
о
со
т
г-
13—14
со
О)
т
о
30 —► 7
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
фенилэфрину, время удерживания — около 2,8 мин):
примесь С — около 1,3; примесь Е — около 3,6.
Пригодность хроматографической системы:
- фактор асимметрии: не более 1,9 для основ¬
ного пика на хроматограмме испытуемого раствора;
- коэффициент разделения пиков: не менее 5
(Нр — высота пика примеси С относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пики примеси С и
фенилэфрина на хроматограмме раствора сравне¬
ния (Ь)).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 0,5, для примеси Е — 0,5):
-примеси С, Е (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям С и Е, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей С и Е, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Сульфаты (2.4.13). Не более 0,0500 % (500 ррт).
Раствор 5 должен выдерживать испытание на суль¬
фаты.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
смеси из 0,5 мл 0,1 М раствора кислоты хлори¬
стоводородной и 80 мл 96 % спирта Р и титруют
0,1 М раствором натрия гидроксида этанольным
потенциометрически (2.2.20). Отмечают объем ти-
транта между двумя точками перегиба на кривой
титрования.
1 мл 0,1 М раствора натрия гидроксида эта-
нольного соответствует 20,37 мг С9Н13М02 НС1.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, О.
А. Р = Р' = Н: (1Р)-2-Амино-1-(3-гидроксифенил)-
этанол (норфенилэфрин).
О. Р = СН2-С6Н5, Р' = СН3: (1Р)-2-(Бензил-метил-
амино)-1 -(З-гидроксифенил)этанол (бензилфенилэф-
рин).
С. Р = Н: 1-(3-Гидроксифенил)-2-(метиламино)эта-
нон (фенилэфрон).
Е. Р = СН2-С6Н5: 2-(Бензилметиламино)-1-(3-
гидроксифенил)этанон (бензилфенилэфрон).
1022
Гэсударственная фармакопея Республики Беларусь
07/2016:1357
ФЕНИРАМИНА МАЛЕАТ
РЬеп'1гатт1 та1еаз
РНЕМКАММЕ МА1-ЕАТЕ
>3 и энантиомер
^со2н
ХЮ2Н
С16Н20М2 " С4Н404 М-М. 356,4
[132-20-7]
ОПРЕДЕЛЕНИЕ
(ЗЯ5)-А/,А/-Диметил-3-фенил-3-(пиридин-2-ил)-
пропан-1-амина (2)-бутендиоат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень легко растворим в воде, легко растворим в
96 % спирте, в метаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: С.
Вторая идентификация: А, В, О.
A. Температура плавления (2.2.14): от 106 °С до
109 °С.
B. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в 0,1 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем. 5,0 мл полученного раствора дово¬
дят 0,1 М раствором кислоты хлористоводородной
до объема 50,0 мл.
Диапазон длин волн: от 220 нм до 320 нм.
Максимум поглощения: при 265 нм.
Плечо: при 261 нм.
Удельный показатель поглощения в максимуме:
от 200 до 220.
С. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фенирамина малеата #или
спектру, представленному на рисунке #1357.-1#.
й. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
5,0 мл этим же растворителем.
Раствор сравнения (а). 65 мг кислоты малеино¬
вой Р растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 0,10 г ФСО фенирамина
малеата растворяют в метаноле Р и доводят до объ¬
ема 5,0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: вода Р — кислота муравьи¬
ная безводная Р — метанол Р — диизопропиловый
эфир Р (3:7:20:70, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются два полностью разделен¬
ных пятна: верхнее пятно соответствует по располо¬
жению и размеру пятну на хроматограмме раствора
Рисунок #1357.-1. Инфракрасный спектр ФСО фенирамина малеата
Фенирамина малеат
1023
сравнения (а); нижнее пятно соответствует по распо¬
ложению и размеру нижнему пятну на хроматограмме
раствора сравнения (Ь).
ИСПЫТАНИЯ
Раствор 5. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20,0 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)6.
рН (2.2.3). От 4,5 до 5,5. 0,20 г испытуемого об¬
разца растворяют в 20 мл воды, свободной от угле-
рода диоксида, Р.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Ацетонитрил Р — под¬
вижная фаза А (10:90, об/об).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
Раствор сравнения (а). 10,0 мг ФСО фенирами¬
на примеси А и 10 мг 4-бензилпиридина Р (примесь В)
растворяют в 10,0 мл смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (а) доводят смесью растворителей до объема
50.0 мл.
Раствор сравнения (д). 1,0 мл испытуемого раст¬
вора доводят раствором сравнения (а) до объема
10.0 мл. 1,0 мл полученного раствора доводят смесью
растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,30 м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
10 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 5,056 г/л натрия
гептансульфоната Р, доведенный кислотой
фосфорной разведенной Р до рН 2,5;
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
90
10
2—37
90 —► 62
00
со
т
о
Т"-
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 264 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (а).
Относительное удерживание (по отношению к
фенирамину, время удерживания — около 31 мин):
малеиновая кислота — около 0,1; примесь А — около
0,9; примесь В — около 0,97.
Пригодность хроматографической системы:
раствор сравнения (с!):
- разрешение: не менее 1,5 между пиком приме¬
си В и пиком фенирамина.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хроматограм¬
ме испытуемого раствора площадь пятна, соответ¬
ствующего примеси А, не должна превышать пло¬
щадь соответствующего пика на хроматограмме
раствора сравнения (с);
- примесь В (не более 0,2 %): на хромато¬
грамме испытуемого раствора площадь пятна, со¬
ответствующего примеси В, не должна превышать
2-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А и В, не должна превышать площадь
основного пика на хроматограмме раствора срав¬
нения (Ь);
- сумма примесей: не более 1,0 %;
- неучитываемый предел: на хроматограмме
испытуемого раствора не учитывают пики с площа¬
дью менее 0,5 площади основного пика на хрома¬
тограмме раствора сравнения (Ь) (0,05 %); не учи¬
тывают пик, соответствующий малеиновой кислоте.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 12 мл раствора 3 должны вы¬
держивать испытание на тяжелые металлы. Эта¬
лон готовят с использованием эталонного раст¬
вора свинца (2 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 г испытуемого образца сушат
в вакууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,130 г испытуемого образца растворят в 50 мл
кислоты уксусной безводной Р и титруют 0,1 М рас¬
твором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 17,82 мг С16Н201Ч2-С4Н4О4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В.
А. 2-Бензилпиридин.
В. 4-Бензилпиридин.
1024
Гэсударственная фармакопея Республики Беларусь
07/2016:0201
ФЕНОБАРБИТАЛ
РЬепоЬагЫШит
РНЕЫОВАКВ1ТА1.
С12Н12М203 М.М. 232’2
[50-06-6]
ОПРЕДЕЛЕНИЕ
5-Этил-5-фенил пиримидин-2,4,6(1Н,ЗН,5Н)-
трйон.
Содержание: не менее 99,0 % и не более 101,0 %
в пересчете на сухое вещество.
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень мало растворим в воде, легко растворим в
96 % спирте. Образует водорастворимые соединения
при взаимодействии с гидроксидами и карбонатами
щелочных металлов и аммония.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
А. Температура плавления (2.2.14). Определяют
температуру плавления испытуемого образца. Сме¬
шивают равные части испытуемого образца и ФСО
фенобарбитала и определяют температуру плавле¬
ния смеси. Разница между температурами плавления
(около 176 °С) не должна превышать 2 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фенобарбитала #или спектр,
представленный на рисунке #0201 .-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в 96 % спирте Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО фенобарбита¬
ла растворяют в 96 % спирте Р и доводят до объема
10,0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля (ЗР254 Р.
Подвижная фаза: нижний слой смеси раствор
аммиака концентрированный Р — 96% спирт Р —
метиленхлорид Р (5:15:80, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения.
й. Испытуемый образец дает реакцию на барби¬
тураты (за исключением А/-замещенных) (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в смеси из 4 мл раствора натрия гидроксида
разведенного Р и 6 мл воды Р.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Рисунок #0201 .-1. Инфракрасный спектр ФСО фенобарбитала
Фенобарбитал
1025
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
Кислотность. К 1,0 г испытуемого образца при¬
бавляют 50 мл воды Р и кипятят в течение 2 мин, ох¬
лаждают и фильтруют. К 10 мл фильтрата прибавляют
0,15 мл раствора метилового красного Р. Появляет¬
ся оранжево-желтое окрашивание. При прибавлении
не более 0,1 мл 0,1 М раствора натрия гидроксида
должно появиться желтое окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,125 г испытуемого об¬
разца растворяют в 5 мл метанола Р и доводят под¬
вижной фазой до объема 25,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора смешивают с 20,0 мл метанола Р и доводят под¬
вижной фазой до объема 100,0 мл. 1,0 мл полученно¬
го раствора смешивают с 2,0 мл метанола Р и дово¬
дят подвижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). 5,0 мг ФСО фенобарби¬
тала примеси А и 5,0 мг ФСО фенобарбитала при¬
меси В растворяют в 2,0 мл метанола Р и доводят
подвижной фазой до объема 10,0 мл. 1,0 полученного
раствора смешивают с 20,0 мл метанола Р и доводят
подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: 6,60 г натрия ацетата Р
растворяют в 900 мл воды Р, прибавляют 3 мл кисло¬
ты уксусной ледяной Р, доводят до рН 4,5 кислотой
уксусной ледяной Р и разводят водой Р до объема
1000 мл; 60 объемов полученного раствора смешива¬
ют с 40 объемами метанола Р;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2,1-кратное вре¬
мя удерживания фенобарбитала.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь).
Относительное удерживание (по отношению к
фенобарбиталу, время удерживания — около 14 мин):
примесь А— около 0,2; примесь В — около 0,3.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками при¬
меси А и примеси В.
Предельное содержание примесей:
- примесь А (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать 1,5-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (Ь);
- примесь В (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать 1,5-кратную
площадь соответствующего пика на хроматограмме
раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А и В, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 40 мл
96 % спирта Р, прибавляют 20 мл воды Р и титруют
0,1 М раствором натрия гидроксида потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора натрия гидроксида эта-
нольного соответствует 23,22 мг С12Н12Ы203.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
А. (5/?5)-5-Этил-2,6-диимино-5-фенилтетрагидро-
пиримидин-4-(1 И)- он.
В. (5Я5)-5-Этил-6-имино-5-фенилдигидропири-
мидин-2,4-(1 Н,ЗН)-дион.
С. 5-Метил-5-фенилпиримидин-2,4,6(1 Н,ЗН,5Н)~
трион.
1026
Государственная фармакопея Республики Беларусь
07/2016:0148
ФЕНОКСИМЕТИЛПЕНИЦИЛЛИН
РНепохуте01у1реп1сННпит
РНЕМОХУМЕТНУ1.РЕЫ1С1ШЫ
С1вН1вЫ2058 М.м. 350,4
[87-08-1]
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
О. Около 2 мг испытуемого образца помещают в
пробирку длиной около 150 мм и диаметром 15 мм.
Смачивают 0,05 мл воды Р и прибавляют 2 мл фор¬
мальдегида раствора в серной кислоте Р. Содержи¬
мое пробирки перемешивают вращательными движе¬
ниями; раствор приобретает красновато-коричневую
окраску. Пробирку помещают на водяную баню на
1 мин; появляется темное красновато-коричневое
окрашивание.
ИСПЫТАНИЯ
ОПРЕДЕЛЕНИЕ
(25,5Я,6Я)-3,3-Диметил-7-оксо-6-[(феноксиацетил)-
амино]-4-тиа-1-азабицикло[3.2.0]гептан-2-карбоновая
кислота.
Субстанцию получают выращиванием определен¬
ных штаммов РепюИНит поШит или родственных ор¬
ганизмов на питательных средах, содержащих подхо¬
дящий предшественник, или любым другим способом.
Содержание: не менее 95,0 % и не более 102,0 %
суммы феноксиметилпенициллина и 4-гидроксифе-
ноксиметилпенициллина (в пересчете на безводное
вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Слегка гигроскопичен
Очень мало растворим в воде, растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО феноксиметилпенициллина.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 25 мг испытуемого об¬
разца растворяют в 5 мл ацетона Р.
Раствор сравнения (а). 25 мг ФСО феноксиме¬
тилпенициллина растворяют в 5 мл ацетона Р.
Раствор сравнения (Ь). 25 мг ФСО калия бензил-
пенициллина и 25 мг ФСО калия феноксиметилпени¬
циллина растворяют в 5 мл воды Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: смешивают 30 объемов ацето¬
на Р и 70 объемов раствора 154 г/л аммония ацета¬
та Р, доведенного кислотой уксусной ледяной Р до
рН 5,0.
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в парах
йода до появления пятен и просматривают при днев¬
ном свете.
Пригодность хроматографической системы:
раствор сравнения (Ь):
рН (2.2.3). От 2,4 до 4,0. 50 мг испытуемого об¬
разца суспендируют в 10 мл воды, свободной от
углерода диоксида, Р.
Удельное оптическое вращение (2.2.7). От+186
до +200 (в пересчете на безводное вещество). 0,250 г
испытуемого образца растворяют в бутаноле Р и до¬
водят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. К 250 мл 0,2 М раст¬
вора калия дигидрофосфата Р прибавляют 500 мл
воды Р, доводят раствором 8,4 г/л натрия гидрокси¬
да Р до рН 6,5 и разводят водой Р до объема 1000 мл.
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 50,0 мл этой же смесью растворителей.
Испытуемый раствор (Ь). Готовят непосред¬
ственно перед применением. 80,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 20,0 мл этой же смесью растворителей.
Раствор сравнения (а). 55,0 мг ФСО калия фе¬
ноксиметилпенициллина растворяют в смеси раство¬
рителей и доводят до объема 50,0 мл этой же смесью
растворителей.
Раствор сравнения (Ь). 4,0 мг ФСО калия 4-ги-
дроксифеноксиметилпенициллина растворяют в
смеси растворителей и доводят до объема 10,0 мл
этой же смесью растворителей. 5,0 мл полученного
раствора доводят до объема 100,0 мл смесью раст¬
ворителей.
Раствор сравнения (с). 10 мг ФСО калия фенок¬
симетилпенициллина и 10 мг ФСО натрия бензилпе-
нициллина (примесь А) растворяют в смеси раство¬
рителей и доводят до объема 50 мл этой же смесью
растворителей.
Раствор сравнения (д). 1,0 мл раствора срав¬
нения (а) доводят смесью растворителей до объема
20 мл. 1,0 мл полученного раствора доводят смесью
растворителей до объема 50 мл.
Раствор сравнения (е). 1,0 мл раствора сравне¬
ния (а) доводят до объема 25,0 мл смесью раствори¬
телей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: фосфатный буфер¬
ный раствор рН 3,5 Р — метанол Р — вода Р
(10:30:60, об/об/об);
Феноксиметилпенициллин
1027
- подвижная фаза В: фосфатный буфер¬
ный раствор рН 3,5 Р — вода Р — метанол Р
(10:35:55, об/об/об);
Время (мин)
Подвижная фаза
А
(%, об/об)
Подвижная фаза
В
(%, об/об)
0-1г
60
40
20)
60 — 0
40 — 100
(1, + 20)-(1,+ 35)
0
100
(1,+ 35)-а+50)
0 — 60
о
т
о
о
Хг— время удерживания феноксиметилпенициллина на
хроматограмме раствора сравнения (б).
Если для достижения необходимого разрешения
изменялось соотношение подвижных фаз А и В, то
подвижную фазу полученного состава используют в
начальном времени градиента и для количественного
определения.
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл растворов
сравнения (с), (б) и (е) хроматографируют изократи-
чески, используя соотношение подвижных фаз А и В
начального времени градиента; 20 мкл испытуемого
раствора (Ь) хроматографируют согласно описанной
выше программе градиента; в качестве контрольного
раствора хроматографируют смесь растворителей со¬
гласно описанной выше программе градиента.
Пригодность хроматографической системы:
- разрешение: не менее 6,0 между пиками при¬
меси А и феноксиметилпенициллина на хроматограм¬
ме раствора сравнения (с); при необходимости кор¬
ректируют соотношение подвижных фаз А и В;
- отношение сигнал/шум: не менее 3 для основ¬
ного пика на хроматограмме раствора сравнения (б);
- коэффициент распределения масс: от 5,0 до
7,0 для пика феноксиметилпенициллина (2-й пик) на
хроматограмме раствора сравнения (с).
Предельное содержание примесей:
-любая примесь (не более 1 %): на хроматограм¬
ме испытуемого раствора (Ь) площадь любого пика, кро¬
ме основного, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (е);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора (Ь) не учитывают пик 4-гидрокси-
феноксиметилпенициллина.
4-Гидроксифеноксиметилпенициллин. Жид¬
костная хроматография (2.2.29), как указано в испы¬
тании «Сопутствующие примеси», со следующими
изменениями.
Подвижная фаза: исходный состав смеси под¬
вижных фаз А и В, при необходимости откорректиро¬
ванный.
Вводимая проба: испытуемый раствор (а) и раст¬
вор сравнения (Ь).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
4-гидроксифеноксиметилпенициллина (если необходи¬
мо) — указанный в документах, прилагаемых к ФСО):
- 4-гидроксифеноксиметилпенициллин: не бо¬
лее 4,0 % (в пересчете на безводное вещество).
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,000 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Подвижная фаза: исходный состав смеси под¬
вижных фаз А и В, при необходимости откорректиро¬
ванный.
Вводимая проба: испытуемый раствор (а) и рас¬
творы сравнения (а) и (Ь).
Пригодность системы: раствор сравнения (а):
- относительное стандартное отклонение: не
более 1,0 % при 6 повторных вводах пробы.
Процентное содержание феноксиметилпеницил¬
лина рассчитывают умножением процентного содер¬
жания феноксиметилпенициллина калия на 0,902.
Процентное содержание 4-гидроксифеноксиметил-
пенициллина рассчитывают умножением (если необ¬
ходимо) на поправочный коэффициент, указанный в
документах к ФСО.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
А. (25,5Я,6Я)-3,3-Диметил-7-оксо-6-[(фенилацетил)-
амино]-4-тиа-1-азабицикло[3.2.0]гептан-2-карбоновая
кислота (бензилпенициллин).
В. Феноксиуксусная кислота.
н н
С. (25,5Я6Я)-6-Амино-3,3-диметил-7-оксо-4-тиа-
1-азабицикло[3.2.0]гептан-2-карбоновая кислота (6-ами-
нопенициллановая кислота).
О. (25,5Р,6Р)-3,3-Диметил-7-оксо-6-[[2-(4-гидрокси-
фенокси)ацетил]аминоН-тиа-1-азабицикло[3.2.0]гептан-
2-карбоновая кислота (4-гидроксифеноксиметилпени-
циллин).
Е. Р = С02Н: (45)-2-[Карбокси[(феноксиацетил)-
амино]метил]-5,5-диметилтиазолидин-4-карбоновая
кислота (пенициллоиновые кислоты феноксиметилпе¬
нициллина).
1028
Гэ сударственная фармакопея Республики Беларусь
Р. К = Н: (2К5,45)-5,5-Диметил-2-[[(феноксиацетил)-
амино]метил]тиазолидин-4-карбоновая кислота (пенил-
лоиновые кислоты феноксиметилпенициллина).
07/2016:0631
ФЕНОЛ
РЬепоШт
РНЕЫ01.
С.Н.О М.м. 94,1
[108-95-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветные или бледно-розовые или бледно¬
желтоватые кристаллы или кристаллическая масса.
Расплывается на воздухе.
Растворим в воде, очень легко растворим в 96 %
спирте, глицерине и метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,5 г испытуемого образца растворяют в 2 мл
раствора аммиака концентрированного Р. Испыту¬
емый образец растворяется полностью. Полученный
раствор доводят водой Р до объема около 100 мл. К
2 мл полученного раствора прибавляют 0,05 мл раст¬
вора натрия гипохлорита концентрированного Р.
Появляется синее окрашивание, которое со временем
усиливается.
B. К 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 10 мл воды Р
и 0,1 мл раствора железа (III) хлорида Р1. Появляет¬
ся фиолетовое окрашивание, исчезающее при при¬
бавлении 5 мл 2-пропанола Р.
C. К 1 мл раствора 3 прибавляют 10 мл воды Р и
1 мл бромной воды Р. Образуется белый осадок.
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 15 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона В(К)6.
Кислотность. К 2 мл раствора 3 прибавляют
0,05 мл раствора метилового оранжевого Р. Раст¬
вор должен быть желтым.
Температура затвердевания (2.2.18). Не менее
39,5 °С.
Остаток после выпаривания. Не более 0,05 %.
5,000 г испытуемого образца выпаривают досуха
на водяной бане и высушивают при температуре от
100 °С до 105 °С в течение 1 ч. Масса остатка не
должна превышать 2,5 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца растворяют в
воде Р и доводят до объема 1000,0 мл этим же раст¬
ворителем. 25,0 мл полученного раствора помещают
в колбу с притертой стеклянной пробкой, прибавляют
50,0 мл 0,0167 М раствора бромид-бромата и 5 мл
кислоты хлористоводородной Р. Колбу закрывают
пробкой, выдерживают в течение 30 мин при перио¬
дическом перемешивании и еще 15 мин без переме¬
шивания. Прибавляют 5 мл раствора 200 г/л калия
йодида Р, встряхивают и титруют 0,1 М раствором
натрия тиосульфата до бледно-желтого окрашива¬
ния. Прибавляют 0,5 мл раствора крахмала Р, 10 мл
хлороформа Р и продолжают титровать при интенсив¬
ном встряхивании.
Параллельно проводят контрольный опыт.
1 мл 0,0167 М раствора бромид-бромата соот¬
ветствует 1,569 мг СбНбО.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:1584
ФЕНОЛФТАЛЕИН
РЬепо1рЫЬа1етит
РНЕЫ01.РНТНА1-Е1Ы
ОПРЕДЕЛЕНИЕ
3,3-Бис(4-гидроксифенил)изобензофуран-1 (ЗН)-он.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, растворим в
96 % спирте.
Температура плавления: около 260 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25). 25,0 мг испы¬
туемого образца растворяют в 96% спирте Р и доводят
до объема 100,0 мл этим же растворителем (раствор
А). К 2,0 мл раствора А прибавляют 5,0 мл 1М раст¬
вора кислоты хлористоводородной и доводят 96 %
спиртом Р до объема 50,0 мл (раствор А,). К 10,0 мл
раствора А прибавляют 5,0 мл 1 М раствора кислоты
хлористоводородной и доводят 96 % спиртом Р до
объема 50,0 мл (раствор А^. К 2,0 мл раствора А при¬
бавляют 5,0 мл 1 М раствора натрия гидроксида и до¬
водят 96 % спиртом Р до объема 50,0 мл (раствор В).
Спектр поглощения раствора А1 в области длин волн
Фентанил
1029
от 220 нм до 250 нм имеет максимум при длине волны
229 нм. Удельный показатель поглощения в максиму¬
ме при длине волны 229 нм составляет от 922 до 1018.
Спектр поглощения раствора в области длин волн
от 250 нм до 300 нм имеет максимум при длине волны
276 нм. Удельный показатель поглощения в максиму¬
ме при длине волны 276 нм составляет от 142 до 158.
Спектр поглощения раствора В в области длин волн
от 230 нм до 270 нм имеет максимум при длине волны
249 нм. Удельный показатель поглощения в максимуме
при длине волны 249 нм составляет от 744 до 822.
В. Около 10 мг испытуемого образца растворяют
в 96 % спирте Р и прибавляют 1 мл раствора на¬
трия гидроксида разведенного Р. Появляется крас¬
ное окрашивание. Прибавляют 5 мл кислоты серной
разведенной Р. Раствор обесцвечивается.
ИСПЫТАНИЯ
Раствор 3. К 2,0 г испытуемого образца прибав¬
ляют 40 мл воды дистиллированной Р и нагревают
до кипения. Охлаждают и фильтруют.
Раствор 31. 0,20 г испытуемого образца раство¬
ряют в 5 мл 96 % спирта Р
Прозрачность (2.2.7). Раствор 51 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
51 должна быть не интенсивнее эталона У(Ж)7.
Кислотность или щелочность. К 10 мл раст¬
вора 5 прибавляют 0,15 мл раствора бромтимолово-
го синего Р1. При прибавлении 0,05 мл 0,01 М раст¬
вора кислоты хлористоводородной окраска раствора
должна измениться на желтую. При прибавлении к по¬
лученному раствору 0,10 мл 0,01 М раствора натрия
гидроксида должно появиться синее окрашивание.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 0,5 г испытуемого образ¬
ца растворяют в 96 % спирте Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 1 мл испытуемого раст¬
вора доводят 96 % спиртом Р до объема 10 мл. 5 мл
полученного раствора доводят 96 % спиртом Р до
объема 100 мл.
Раствор сравнения (Ь). 25 мг флуорена Р раст¬
воряют в 96 % спирте Р, прибавляют 0,5 мл испытуе¬
мого раствора и доводят 96 % спиртом Р до объема
10 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: ацетон Р — метиленхлорид Р
(50:50, об/об).
Наносимый объем пробы: по 5 мкл каждого раст¬
вора.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм до и после
обработки парами аммиака.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживается два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (а).
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
10 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
15 мл раствора 5 должны выдерживать испытание на
сульфаты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). К Зг испытуемого образца при¬
бавляют 50 мл кислоты хлористоводородной раз¬
веденной Р, нагревают на водяной бане в течение
5 мин и фильтруют. Фильтрат выпаривают почти до¬
суха, остаток растворяют в 30 мл воды Р. 12 мл полу¬
ченного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
10 мл эталонного раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 5 мл
диметилформамида Р, прибавляют 5 мл раствора
натрия карбоната Р, 10 мл раствора натрия гидро¬
карбоната Р, 35 мл воды Р и 50,0 мл 0,05 М раст¬
вора йода. Прибавляют 10 мл метиленхлорида Р,
20 мл кислоты серной разведенной Р и титруют из¬
быток йода 0,1 М раствором натрия тиосульфата,
используя в качестве индикатора 0,3 мл раствора
крахмала Р, который прибавляют в конце титрования.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
3,979 мгС20Н14О4.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:1210
ФЕНТАНИЛ
Реп1апу!ит
РЕМТАЫ У/.
С22Н2.М20 М М- 336>5
[437-38-7]
ОПРЕДЕЛЕНИЕ
А/-Фенил-А/-[1-(2-фенилэтил)пиперидин-4-ил]про-
панамид.
1030
Гэсударственная фармакопея Республики Беларусь
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в 96 % спирте и в метаноле.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр фентанила по
Европейской Фармакопее.
Если спектр, полученный с использованием об¬
разца в твердом состоянии, отличается, то испытуе¬
мый образец растворяют в минимальном количестве
этанола Р и выпаривают до сухого остатка, который
используют для получения нового спектра.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО фентанила
для проверки пригодности хроматографической си¬
стемы (содержит примеси А, В, С, О и Н) растворяют
в 1,0 мл метанола Р.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
1.0 мл полученного раствора доводят метанолом Р
до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,1 м и внутренним диаметром
3.0 мм, заполненная силикагелем октадецилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 3 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 5 г/л аммония
карбоната Р в смеси тетрагидрофуран Р —
вода Р (10:90, об/об);
- подвижная фаза В: ацетонитрил Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
90 —► 40
о
со
т
о
15—20
40
60
- скорость подвижной фазы: 0,64 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, О и Н, используя хрома¬
тограмму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО фентанила для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению к
фентанилу, время удерживания — около 15 мин): при¬
месь В — около 0,1; примесь А — около 0,3; примесь
С — около 0,9; примесь О — около 1,1; примесь Н —
около 1,2.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,0 между пиками фен¬
танила и примеси О.
Предельное содержание примесей:
- примеси А, В, С, О (не более 0,25 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, В, С, О, не должны
превышать 2,5-кратную площадь основного пика на
хроматограмме раствора сравнения (Ь);
- примесь Н (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси Н, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, й и Н, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): На хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 50 °С.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 50 мл
смеси из 1 объема кислоты уксусной безводной Р и
7 объемов метилэтилкетона Р и титруют 0,1 М рас¬
твором кислоты хлорной, используя в качестве ин¬
дикатора 0,2 мл раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 33,65 мг С^Н^О.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Е, Р, 6.
А. Л/-Фенил-Л/-[цис,транс-1 -оксид-1 -(2-фенилэтил)-
пиперидин-4-ил]пропанамид.
Фибриноген человеческий
1031
B. Р = СО-С2Н5> К' = Н: Л/-Фенил-Л/-(пиперидин-4-
ил)пропанамид.
C. П = СО-СН31 Р' = СН2-СН2-С6Н5: Л/-Фенил-Л/-[1-
(2-фенилэтил)пиперидин-4-ил]ацетамид.
й. Р = Н, Р' = СН2-СН2-СбН5: Л/-Фенил-1 -(2-фенил-
этил)пиперидин-4-амин.
Е. Р = ОНО: Бензальдегид.
Р. Р = 1ЧН2: Анилин (фениламин).
6. Р = 1\1Н-СО-С2Н5: Л/-Фенилпропанамид.
07/2016:0024
ФИБРИНОГЕН ЧЕЛОВЕЧЕСКИЙ
НЬпподвпит Ьитапит
нимАЫ рттыооЕЫ
ОПРЕДЕЛЕНИЕ
Стерильный лиофилизированный препарат бел¬
ковой фракции плазмы, содержащий растворимый
компонент человеческой плазмы, который превра¬
щается в фибрин при прибавлении тромбина. Полу¬
чают из Плазмы человеческой для фракционирования
(0853). Препарат может содержать вспомогательные
вещества, такие как соли, буферы и стабилизаторы.
При растворении в указанном на этикетке объ¬
еме растворителя содержание фибриногена в раство¬
ре должно быть не менее 10 г/л.
ПРОИЗВОДСТВО
Метод изготовления должен обеспечивать це¬
лостность человеческого фибриногена. Метод должен
включать стадию или стадии, обеспечивающие удале¬
ние или инактивацию известных инфекционных аген¬
тов; если при производстве применяются вещества
для инактивации вирусов, то процедура последующей
очистки должна быть валидирована, чтобы показать,
что концентрация этих веществ снижена до приемле¬
мого уровня и остаточные количества не влияют на
безопасность препарата для пациентов.
Специфическая активность (содержание фибри¬
ногена по отношению к содержанию общего белка)
перед прибавлением любого белкового стабилизато¬
ра должна составлять не менее 80 %. Содержание
фибриногена определяют подходящим методом, как
указано в разделе «Количественное определение»;
общее содержание белка определяют подходящим
методом, как указано в испытании «Общий белок» в
Человеческого альбумина растворе. Одновременно
при фракционировании вместе с фибриногеном мо¬
жет быть получен альбумин; в этом случае проводят
специфическое определение альбумина с помощью
подходящего иммунохимического метода (2.7.1) и
найденное количество альбумина вычитают из коли¬
чества общего белка при расчете специфической ак¬
тивности.
Белковую фракцию растворяют в подходящем
растворителе. Антимикробные консерванты или ан¬
тибиотики не добавляют. Раствор пропускают через
фильтр, задерживающий бактерии, расфасовывают в
асептических условиях в конечные контейнеры и не¬
медленно замораживают. Полученный препарат впо¬
следствии лиофильно сушат и контейнеры укупори¬
вают под вакуумом или в атмосфере инертного газа.
ОПИСАНИЕ (СВОЙСТВА)
Белый или бледно-желтый порошок или рыхлая
масса. Гигроскопичен.
Испытуемый препарат восстанавливают, как
указано на этикетке, непосредственно перед опре¬
делением подлинности, проведением испытаний (за
исключением испытаний на растворимость и воду)
и количественного определения.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Должен выдерживать испытание «Количествен¬
ное определение».
ИСПЫТАНИЯ
Растворимость. К содержимому одного контей¬
нера с испытуемым образцом прибавляют указанный
на этикетке объем жидкости при рекомендованной
температуре. Препарат должен растворяться в тече¬
ние 30 мин при температуре от 20 °С до 25 °С, образуя
почти бесцветный слегка опалесцирующий раствор.
рН (2.2.3). От 6,5 до 7,5.
Осмоляльность (2.2.35). Не менее 240 мОсмоль/кг.
Стабильность раствора. Восстановленный
раствор выдерживают при температуре от 20 °С до
25 °С. Не должен образовываться гель в течение
60 мин после восстановления.
Вода. Определяют подходящим методом, таким
как полумикроопределение воды (2.5.72), потеря в
массе при высушивании (2.2.32) или спектрофотоме¬
трия ближнего инфракрасного диапазона (2.2.40); со¬
держание воды должно быть в пределах, утвержден¬
ных компетентным органом.
Стерильность (2.6.1). Восстановленная субстан¬
ция должна выдерживать испытание на стерильность.
Пирогенность (2.6.8) или Бактериальные эн¬
дотоксины (2.6.14).
Фибриноген человеческий должен выдерживать
испытание на пирогенность или, предпочтительнее
и если обосновано и утверждено компетентным ор¬
ганом, валидированное испытание т уИго, такое как
испытание «Бактериальные эндотоксины».
Для испытания «Пирогенность» тест-доза — объ¬
ем, соответствующий не менее 30 мг фибриногена на
1,0 кг массы тела кролика.
При применении испытания «Бактериальные
эндотоксины» испытуемая субстанция должна содер¬
жать менее 0,03 МЕ/мг.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,2 мл восстановленного испытуемого образца
смешивают с 2 мл подходящего буферного раствора
(рН 6,6—6,8), содержащего достаточное количество
тромбина (около 3 МЕ/мл) и кальция (0,05 моль/л):
Выдерживают при температуре 37 °С в течение
20 мин, осадок отделяют центрифугированием
1032
Гэсударственная фармакопея Республики Беларусь
(5000 д, 20 мин) и тщательно промывают раствором
9 г/л натрия хлорида Р. Определяют содержание
азота после минерализации серной кислотой (2.5.9)
и рассчитывают содержание фибриногена (свернув¬
шийся белок) умножением полученного результата
на 6,0. Содержание должно быть не менее 70 % и не
более 130 % от количества фибриногена, указанного
на этикетке.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
МАРКИРОВКА
Указывают:
- количество фибриногена в контейнере;
- название и объем жидкости, используемой для
восстановления.
При необходимости указывают:
- название и количество белкового стабилизато¬
ра в препарате.
07/2016:1781
ФЛУДАРАБИНА ФОСФАТ
РЫагаЫп! рЬозрЬаз
РИЮАКАВМЕ РНОЗРНАТЕ
С10Н13РЫ5О7Р М.м. 365,2
[75607-67-9]
ОПРЕДЕЛЕНИЕ
2-Фтор-9-(5-О-фосфон-р-0-арабинофуранозил)-
9Н-пурин-6-амин.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Мало растворим в воде, легко растворим в ди-
метилформамиде, очень мало растворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО флударабина фосфата #или
спектр, представленный на рисунке #1781 .-1#.
ИСПЫТАНИЯ
Раствор 3. 50 мг испытуемого образца раство¬
ряют с помощью ультразвука в 5,0 мл диметилфор-
мамида Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)5.
Удельное оптическое вращение (2.2.7): от
+10,0 до +14,0 (в пересчете на безводное вещество).
0,100 г испытуемого образца растворяют с помощью
ультразвука в воде Р и доводят до объема 20,0 мл
этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29): определение проводят методом
внутренней нормализации. Растворы готовят непо¬
средственно перед использованием.
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют с помощью ультразвука в 50 мл
воды Р и доводят до объема 100,0 мл этим же раст¬
ворителем.
Раствор сравнения (а). 20 мг ФСО флударабина
фосфата растворяют с помощью ультразвука в 50 мл
воды Р и доводят до объема 100,0 мл этим же раст¬
ворителем.
Раствор сравнения (Ь). 20 мг испытуемого образ¬
ца растворяют с помощью ультразвука в 20 мл 0,1 М
раствора кислоты хлористоводородной, нагрева¬
ют в водяной бане при температуре 80 °С в течение
15 мин, охлаждают до комнатной температуры, пере¬
мешивают и доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравнения
(а) доводят водой Р до объема 100,0 мл. 1,0 мл полу¬
ченного раствора доводят водой Р до объема 20,0 мл.
Раствор сравнения (д). 5 мг ФСО флудараби¬
на для проверки пригодности хроматографической
системы (содержит примеси О, Е и Р) растворяют в
10 мл воды Р с помощью ультразвука и доводят до
объема 25,0 мл этим же растворителем.
Холостой раствор. 0,02 М раствор кислоты
хлористоводородной.
А. Рано элюирующиеся примеси.
Условия хроаматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: метанол Р—раствор 1,36 г/л
калия дигидрофосфата Р (00:940, об/оЩ;,
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длины
волн — 260 нм и 292 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а), (Ь), (с);
- время хроматографирования: 4,5-кратное вре¬
мя удерживания основного пика на хроматограмме
испытуемого раствора.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В, используя хроматограмму
раствора сравнения (Ь) при длине волны 292 нм, от¬
клик при длине волны 292 нм намного выше, чем при
длине волны 260 нм; идентифицируют пик примеси С,
используя хроматограмму, прилагаемую к ФСО флу¬
дарабина фосфата, и хроматограмму раствора срав¬
нения (а) при длине волны 260 нм.
Относительное удерживание (по отношению к
флударабина фосфату, время удерживания — около
9 мин): примесь А — около 0,26; примесь В — около
0,34; примесь С — около 0,42.
Пригодность хроматографической системы:
раствор сравнения (Ь) при 292 нм:
Флударабина фосфат
1033
Рисунок #1781 .-1. Инфракрасный спектр ФСО флударабина фосфата
- разрешение: не менее 2,0 между пиками при¬
меси А и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 4,0; для примеси В — 2,5; для примеси
С — 1,9): при 260 нм:
- примесь А: не более 0,8 %;
- примесь В: не более 0,2 %;
- примесь С: не более 0,4 %;
- неспецифицированные примеси, элюирующие¬
ся перед флударабина фосфатом: не более 0,10 %
для каждой примеси;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади основного пика на хроматограмме
раствора сравнения (с) (0,05 %) и пики, элюирующи¬
еся после пика флударабина фосфата.
В. Поздно элюирующиеся примеси.
Жидкостная хроматография, как указано в испы¬
тании А, со следующими изменениями.
Условия хроматографирования:
- подвижная фаза: метанол Р—раствор 1,36 г/л
калия дигидрофосфата Р (200:800, об/об);
-спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (с) и (б);
- время хроматографирования: 8-кратное вре¬
мя удерживания основного пика на хроматограмме
испытуемого раствора.
Идентификация пиков примесей: идентифици¬
руют пики примесей О, Е, Р, используя хроматограмму
раствора сравнения (б) и хроматограмму, прилагае¬
мую к ФСО флударабина для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
флударабина фосфату, время удерживания — около
2,5 мин): примесь О — около 1,5; примесь Е — около
1,9; примесь Р — около 2,5.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 5,0 между пиками флу¬
дарабина фосфата и примеси Р.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 0,5; для примеси Е — 0,6; для примеси
Р—1,8):
- примесь Е: не более 0,2 %;
- примесь Р: не более 0,2 %;
- примесь О: не более 0,15 %;
-неспецифицированные примеси, элюирующи¬
еся после флударабина фосфата: не более 0,10 %
для каждой примеси;
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади основного пика на хроматограмме
раствора сравнения (с) (0,05 %) и пики, элюирующи¬
еся до пика флударабина фосфата.
Сумма примесей, элюирующихся в испытании А
перед флударабина фосфатом, кроме примесей А, В
и С, и примесей, элюирующихся в испытании В после
флударабина фосфата, кроме примесей О, Ей Р: не
более 0,5 %.
Сумма всех примесей, элюирующихся в испыта¬
нии А перед флударабина фосфатом, и всех приме¬
сей, элюирующихся в испытании В после флудараби¬
на фосфата: не более 2,0 %.
Этанол (2.4.24, система А). Не более 1,0 %.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раство¬
ряют при нагревании в 10 мл воды Р, охлаждают, при¬
бавляют раствор аммиака Р до слабощелочной ре¬
акции по лакмусовой бумаге, доводят до рН 3,0—4,0
1034
Государственная фармакопея Республики Беларусь
кислотой уксусной разведенной Р и разводят водой Р
до объема 20 мл. 12 мл полученного раствора долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием эталонного раст¬
вора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 3,0%. К 0,200 г испы¬
туемого образца (размолотого до очень мелкого по¬
рошка) прибавляют 15 мл метанола безводного Р и
перемешивают на магнитной мешалке в течение 15 с
перед титрованием.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси. Испытание
А», со следующими изменениями.
Испытуемый раствор. 24,0 мг испытуемого об¬
разца растворяют с помощью ультразвука в 50 мл
воды Р и доводят до объема 100,0 мл этим же раство¬
рителем. 25,0 мл полученного раствора доводят под¬
вижной фазой до объема 100,0 мл.
Раствор сравнения. 24,0 мг ФСО флударабина
фосфата растворяют с помрщью ультразвука в 50 мл
воды Р и доводят до объема 100,0 мл этим же раство¬
рителем. 25,0 мл полученного раствора доводят под¬
вижной фазой до объема 100,0 мл.
Условия хроматографирования:
- спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: 10 мкл.
Содержание С10Н13РМ5О7Р рассчитывают с уче¬
том содержания флударабина фосфата в ФСО флу¬
дарабина фосфата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте при температуре от 2 °С до
8 °С.
ПРИМЕСИ
В. 6-Амино-7Н-пурин-2-ол.
С. 9-(3,5-Ди-0-фосфон-р-0-арабинофуранозил)-
2-фтор-9Н-пурин-6-амин.
О. 2-фтор-7Н-пурин-6-амин.
Е. 9-Р-0-Арабинофуранозил-2-фтор-9Н-пурин-6-
амин.
Специфицированные примеси: А, В, С, О, Е, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, Н, I, 3.
Р. 2-Этокси-9-(5-0-фосфон-р-0-арабинофура-
нозил)-9Н-пурин-6-амин.
А. 6-Амино-9-(5-0-фосфон-р-0-арабинофурано-
зил )-9Я-пурин-2-ол.
С. 9-(2-Хлор-2-дезокси-5-0-фосфон-Р-0-арабино-
фуранозил)-2-фтор-9Н-пурин-6-амин.
Пропускание
Флуконазол
1035
Н. 9-(2,5-Ангидро-р-0-арабинофуранозил)-2-фтор-
9Н-пурин-6-амин.
1.9-(5-0-Фосфон-Р-0-арабинофуранозил )-9Н-пурин-
2,6-диамин.
07/2016:2287
ФЛУКОНАЗОЛ
Р1исопаю1ит
РШС0ЫА201-Е
С13Н12Р2М60 М.М. 306»3
[86386-73-4]
ОПРЕДЕЛЕНИЕ
2-(2,4-Дифторфенил)-1,3-бис(1Н-1,2,4-триазол-
1-ил)пропан-2-ол.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
3. 2-Метокси-9-(5-0-фосфон-Р-0-арабинофура-
нозил)-9Н-пурин-6-амин.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Мало растворим в воде, легко растворим в мета¬
ноле, растворим в ацетоне.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО флуконазола #или спектр, пред¬
ставленный на рисунке #2287.-1#.
Рисунок #2287.-1. Инфракрасный спектр ФСО флуконазола
1036
Гэсударственная фармакопея Республики Беларусь
Если спектры, полученные с использованием
образцов в твердом состоянии, отличаются, то ис¬
пытуемый образец и ФСО флуконазола растворяют
по отдельности в минимальном количестве мети-
ленхлорида Р, выпаривают на водяной бане до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе, при необходи¬
мости с помощью ультразвука, и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг ФСО флуконазо¬
ла для идентификации пиков (содержит примесь А)
растворяют в подвижной фазе, при необходимости
с помощью ультразвука, и доводят до объема 10 мл
этим же растворителем.
Раствор сравнения (с). 3,0 мг ФСО флуконазола
примеси В растворяют в подвижной фазе, при необ¬
ходимости с помощью ультразвука, и доводят до объ¬
ема 100,0 мл этим же растворителем.
Раствор сравнения (д). 2,0 мг ФСО флуконазола
примеси С растворяют в подвижной фазе и доводят
до объема 20,0 мл этим же растворителем. К 1,0 мл
полученного раствора прибавляют 1,0 мл испытуемо¬
го раствора и доводят подвижной фазой до объема
10.0 мл.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р1 с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза: ацетонитрил Р — раствор
0,63 г/л аммония формиата Р (14:86, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 260 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания флуконазола.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (Ь) и хроматограмму, прилагаемую к
ФСО флуконазола для идентификации пиков; иден¬
тифицируют пик примеси В, используя хроматограм¬
му раствора сравнения (с); идентифицируют пик при¬
меси С, используя хроматограмму раствора сравне¬
ния (б).
Относительное удерживание (по отношению к
флуконазолу, время удерживания — около 11 мин):
примесь В — около 0,4; примесь А — около 0,5; при¬
месь С — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 3,0 между пиками при¬
меси С и флуконазола.
Предельное содержание примесей:
- примесь А (не более 0,4 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать 0,8 площади основ¬
ного пика на хроматограмме раствора сравнения (а);
- примесь В (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси В, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (с);
-примесь С (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси С, не должна превышать площадь соответству¬
ющего пика на хроматограмме раствора сравнения (с!);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В и С, не должна превышать 0,2 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,6 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
1,2-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод В). Не более
0,0010% (Юррт). 2,0 г испытуемого образца раст¬
воряют в смеси из 15 объемов воды Р и 85 объемов
метанола Р и доводят до объема 20,0 мл этой же
смесью растворителей. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,125 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 15,32 мг С13Н12Р2М60.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
Флуоцинолона ацетонид
1037
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О, Е, Г, С, Н, I.
А. (2Я5)-2-(2,4-Дифторфенил)-1 -(1Н-1,2,4-триазол-
1-ил)-3-(4Н-1,2,4-триазол-4-ил)пропан-2-ол.
В. 2-[2-Фтор-4-(1 Н-1,2,4-триазол-1-ил)фенил]-1,3-
бис(1 Н-1,2,4-триазол-1 -ил)пропан-2-ол.
С. 1,1 '-(1,3-Фенилен)ди-1 Н-1,2,4-триазол.
й. 2-(4-Фторфенил)-1,3-бис(1 Н-1,2,4-триазол-1 -
ил)-пропан-2-ол.
р
р
Е. 1 -(2,4-Дифторфенил)-2-(1 Н-1,2,4-триазол-1 -ил)-
этанон.
и энантиомер
Р. (2/?5)-2-(2,4-Дифторфенил)-3-(1 Н-1,2,4-триазол-
1 -ил)пропан-1,2-диол.
и энантиомер
С. 1-[[(2Я5)-2-(2,4-Дифторфенил)оксиран-2-ил]-
метил]-1 Н-1,2,4-триазол.
и энантиомер
Н. (2Н8)-1 -Бром-2-(2,4-дифторфенил)-3-(1 Н-1,2,4-
триазол-! -ил)пропан-2-ол.
I. 4-Амино-1-[(2/?5)-2-(2,4-дифторфенил)-2-
ги д рокси-3 (1 Н-1,2,4-триазол-1-ил)пропил]-4Н-1,2,4-
триазолий.
07/2016:0494
ФЛУОЦИНОЛОНА АЦЕТОНИД
Р1иосто1от' асе1от'с1ит
РШОС1ЫОЕОЫЕ АСЕТОЫЮЕ
С24Нзор2°б М.м. 452,5
[67-73-2]
ОПРЕДЕЛЕНИЕ
6а,9-Дифтор-11 р,21 -дигидрокси-16а, 17-(1 -метил-
этилидендиокси)прегна-1,4-диен-3,20-дион.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, растворим в
ацетоне и этаноле безводном, практически нераство¬
рим в гептане.
Обладает полиморфизмом (5.9).
1038
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО флуоцинолона ацетонида #или
спектр, представленный на рисунке #0494.-1#.
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то испытуе¬
мый образец и ФСО флуоцинолона ацетонида раст¬
воряют по отдельности в этаноле Р и выпаривают до
сухих остатков, которые используют для получения
новых спектров.
B. Просматривают хроматограммы, полученные
в испытании «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора (Ь) соответствует по времени
удерживания и размеру основному пику на хромато¬
грамме раствора сравнения (б).
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+100 до +104 (в пересчете на сухое вещество). 0,100 г
испытуемого образца растворяют в этаноле Р и дово¬
дят до объема 10,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от света, растворы готовят непосредственно пе¬
ред применением.
Смесь растворителей. Ацетонитрил Р —
вода Р (20:80, об/об).
Буферный раствор. 5,68 г динатрия гидро¬
фосфата безводного Р и 3,63 г калия дигидрофос¬
фата Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем.
Испытуемый раствор (а). 20,0 мг испытуемого
образца растворяют в 20 мл ацетонитрила Р и дово¬
дят водой Р до объема 100,0 мл.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 10,0 мл.
Раствор сравнения (а). 2 мг ФСО флуоцинолона
ацетонида для проверки пригодности хроматогра¬
фической системы (содержит примеси Н, 3 и К) раст¬
воряют в 2 мл ацетонитрила Р и доводят водой Р до
объема 10,0 мл.
Раствор сравнения (Ь). 2 мг ФСО флуоциноло¬
на ацетонида для идентификации пиков (содержит
примеси й и I) растворяют в 2 мл ацетонитрила Р и
доводят водой Р до объема 10,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора (а) доводят смесью растворителей до объема
100,0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Раствор сравнения (д). 20,0 мг ФСО флуоцино¬
лона ацетонида растворяют в 20 мл ацетонитри¬
ла Р и доводят водой Р до объема 100,0 мл. 1,0 мл
полученного раствора доводят смесью растворителей
до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — бу¬
ферный раствор (28:72, об/об);
- подвижная фаза В: вода Р—ацетонитрил Р
(40:60, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
100
0
2-42
о
т
о
о
т—
о
о
7
о
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 238 нм;
- объем вводимой пробы: по 40 мкл испытуемого
раствора (а) и растворов сравнения (а), (Ь), (с).
Рисунок #0494.-1. Инфракрасный спектр ФСО флуоцинолона ацетонида
Флуоцинолона ацетонид
1039
Идентификация пиков примесей: идентифи¬
цируют пики примесей Н, 3 м К, используя хрома¬
тограмму раствора сравнения (а) и хроматограм¬
му, прилагаемую к ФСО флуоцинолона ацетонида
для проверки пригодности хроматографической
системы; идентифицируют пики примесей й и I,
используя хроматограмму раствора сравнения (Ь)
и хроматограмму, прилагаемую к ФСО флуоцино¬
лона ацетонида для идентификации пиков.
Относительное удерживание (по отношению
к флуоцинолона ацетониду, время удерживания —
около 18 мин): примесь О — около 0,8; примесь
Н — около 0,90; примесь I — около 0,94; примесь
3 — около 1,05; примесь К — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициент разделения пиков: не менее
5,0 (Нр — высота пика примеси 3 относительно
базовой линии; Ну — расстояние между базовой
линией и нижней точкой кривой, разделяющей пик
примеси 3 и пик флуоцинолона ацетонида).
Расчет процентного содержания:
- для каждой примеси — используют концен¬
трацию флуоцинолона ацетонида в растворе срав¬
нения (с).
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси К—1,3):
- примесь К: не более 0,3 %;
- примеси О, 3: не более 0,2 % для каждой
примеси;
- примеси Н, I: не более 0,15% для каждой
примеси;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,7 %;
- учитываемый предел: 0,05 %.
Потеря в массе при высушивании (2.2.32).
Не более 1,0 %. 1,000 г испытуемого образца су¬
шат при температуре 105 °С в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы, по 20 мкл испытуе¬
мого раствора (Ь) и раствора сравнения (с!).
Содержание С24Н30Р2О6 в процентах рассчиты¬
вают с учетом содержания флуоцинолона ацетони¬
да в ФСО флуоцинолона ацетонида.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: О, Н, 1, 3, К.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): А, В, С, Е, Р, С, М.
А. 6а,9-Дифтор-11 Р-гидрокси-16а, 17-(1 -метилэтил-
идендиокси)-3,20-диоксопрегна-1,4-диен-21-овая кис¬
лота.
В. 6а,9-Дифтор-11 Р-гидрокси-16а, 17-(1 -метилэтил-
идендиокси)-3-оксоандроста-1,4-диен-17р-карбоновая
кислота.
С. 6а,9-Дифтор-11 р, 16а, 17,21 -тетрагидроксипрегна-
1,4-диен-3,20-дион (флуоцинолон).
н р
О. 6а,9-Дифтор-11 р-гидрокси-16а, 17-(1 -метилэтил-
идендиокси)-3,20-диоксопрегна-1,4-диен-21-аль.
Е. 9,11 Р-Эпокси-6а-фтор-21 -гидрокси-16а, 17-(1 -
метилэтилидендиокси)-9р-прегна-1,4-диен-3,20-дион.
1040
Гэсударственная фармакопея Республики Беларусь
Р. 6а-Фтор-21 -гидрокси-16а, 17-(1 -метилэтил
идендиокси)прегна-4-ен-3,20-дион.
С. 6а-Фтор-11 0-гидрокси-16а, 17-(1 -метилэтил-
идендиокси)-3,20-диоксопрегна-4-ен-21-илацетат.
Н. 9-Фтор-11 (3,21-дигидрокси-16а, 17-(1 -метилэтил-
идендиокси)прегна-1,4-диен-3,20-дион.
I. 6а,9-Дифтор-11 р,21-дигидрокси-16а, 17-(1 -метил-
этилидендиокси)прегна-1,4,14-триен-3,20-дион.
6[3,9-Дифтор-11 р,21 -дигидрокси-16а, 17-(1 -метил¬
этил идендиокси)прегна-1,4-диен-3,20-дион.
К. 4,9-Дифтор-11 р,21 -дигидрокси-16а, 17-(1 -метил-
этилидендиокси)прегна-1,4-диен-3,20-дион.
I. 6а-Хлор-9-фтор-11 р,21 -дигидрокси-16а, 17-(1-
метилэтилидендиокси)прегна-1,4-диен-3,20-дион.
М. 6а,9-Дифтор-11 р-гидрокси-16а,17-(1 -метилэтил-
идендиокси)-3,20-диоксопрегна-1,4-диен-21 -илацетат.
07/2016:1423
ФЛУТАМИД
Р1и1ат1с1ит
РШТАМЮЕ
С11Н11Р3М203 М 276»2
[13311-84-7]
ОПРЕДЕЛЕНИЕ
2-Метил-Л/-[4-нитро-3-(трифторметил)фенил]про-
панамид.
Содержание: не менее 97,5 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Кристаллический порошок бледно-желтого цвета.
Практически нерастворим в воде, легко раство¬
рим в ацетоне и в 96 % спирте, практически нераст¬
ворим в гептане.
Температура плавления: около 112 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО флутамида.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор (а). 20,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 20,0 мл этим же растворителем.
Испытуемый раствор (Ь). 1,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
10,0 мл.
Фолиевая кислота
1041
Раствор сравнения (а). 5,0 мг ФСО флутами-
да для проверки пригодности хроматографической
системы (содержит примеси А, В и С) растворяют в
подвижной фазе и доводят до объема 5,0 мл этим же
растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора (а) доводят подвижной фазой до объема
100.0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 10,0 мл.
Раствор сравнения (с). 20,0 мг ФСО флутамида
растворяют в подвижной фазе и доводят до объема
20.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят подвижной фазой до объема 10,0 мл .
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — вода Р
(50:50, об/об);
- скорость подвижной фазы: 0,5 мл/мин;
-спектрофотометрический детектор, длина
волны 240 нм;
- вводимый объем пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (Ь);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания флутамида.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и С, используя хроматограм¬
му раствора сравнения (а) и хроматограмму, прилага¬
емую к ФСО флутамида для проверки пригодности
хроматографической системы.
Относительное удерживание (по отношению к
флутамиду, время удерживания — около 19 мин): при¬
месь В — около 0,5; примесь А — около 0,6; примесь
С — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
месей В и А.
Расчет процентного содержания:
- для каждой примеси — используют концентра¬
цию флутамида в растворе сравнения (Ь).
Предельное содержание примесей:
- примесь С (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси С, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, В, С: не более 0,2 % для каждой при¬
меси;
- неспецифицированные примеси: не более
0,10 % для каждой примеси;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С^Н^Рз^Оз в процентах рассчитыва¬
ют с учетом содержания флутамида в ФСО флутамида.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О, Е, Р.
A. Р = Н, Р' = 1\Ю2:4-Нитро-3-(трифторметил)ани-
лин.
B. Р = СО-СН3, Р' = Ы02: Л/-[4-Нитро-3-(трифтор-
метил)фенил]ацетамид.
C. Р = СО-СН2-СН3, Р' = Ы02: А/-[4-Нитро-3-(три-
фторметил)фенил]пропанамид.
Э. Р = Р' = Н: 3-(Трифторметил)анилин.
Е. Р = Н: 2-Метил-Л/-[3-(трифторметил)фенил]про-
панамид.
Р. Р = М02:2-Метил-Л/-[2-нитро-5-(трифторметил)-
фенил]пропанамид.
07/2016:0067
ФОЛИЕВАЯ КИСЛОТА
АсШит (оНсит
роисАст
С1аН19М7Ов М.м. 441,4
[59-30-3]
131. Зак. 1060.
т42
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
(25)-2-[[4-[[(2-Амино-4-оксо-1,4-дигидроптери-
дин-6-ил)метил]амино]бензоил]амино]пентандиовая
кислота.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтоватый или оранжевый кристаллический
порошок.
Практически нерастворим в воде и большинстве ор¬
ганических растворителей. Растворяется в разведенных
растворах кислот и гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
A. Удельное оптическое вращение (2.2.7): от +18
до +22 (в пересчете на безводное вещество). 0,25 г
испытуемого образца растворяют в 0,1 М растворе
натрия гидроксида и доводят до объема 25,0 мл этим
же растворителем.
B. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания соот¬
ветствует основному пику на хроматограмме раствора
сравнения (а).
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси из раствора аммиака кон¬
центрированного Р и метанола Р (2:9, об/об) и дово¬
дят до объема 100 мл этой же смесью растворителей.
Раствор сравнения. 50 мг ФСО фолиевой кисло¬
ты растворяют в смеси из раствора аммиака концен¬
трированного Р и метанола Р (2:9, об/об) и доводят
до объема 100 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикагеля 6 Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — пропанол Р — 96% спирт Р (20:20:60,
об/об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению, флуоресценции и размеру основному
пятну на хроматограмме раствора сравнения.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в 5 мл раствора 28,6 г/л натрия
карбоната Р и доводят подвижной фазой до объема
100.0 мл. 2,0 мл полученного раствора доводят под¬
вижной фазой до объема 10,0 мл.
Раствор сравнения (а). 0,100 г ФСО фолиевой
кислоты растворяют в 5 мл раствора 28,6 г/л натрия
карбоната Р и доводят подвижной фазой до объема
100.0 мл. 2,0 мл полученного раствора доводят под¬
вижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). К 20 мг ФСО фолие¬
вой кислоты примеси О прибавляют 5 мл раствора
28.6 г/л натрия карбоната Р, доводят до объема
100.0 мл этим же раствором и перемешивают до пол¬
ного растворения. 1,0 мл полученного раствора сме¬
шивают с 1,0 мл раствора сравнения (а) и доводят
подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (ф. 10,0 мг ФСО фолиевой
кислоты примеси А растворяют в 1 мл раствора
28.6 г/л натрия карбоната Р и доводят подвижной
фазой до объема 100,0 мл. 1,0 мл полученного раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (е). К 12,0 мг ФСО фолие¬
вой кислоты примеси О прибавляют 1 мл раствора
28.6 г/л натрия карбоната Р, доводят до объема
100.0 мл этим же раствором и перемешивают до пол¬
ного растворения. 1,0 мл полученного раствора дово¬
дят подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4.0 мм, заполненная сферическим силикагелем ок-
тилсилильным для хроматографии Р (размер частиц
5 мкм) с удельной площадью поверхности 350 м2/г, раз¬
мером пор 10 нм и содержащим около 12,5 % углерода;
-подвижная фаза: 12 объемов метанола Р
смешивают с 88 объемами раствора, содержащего
11,16 г/л калия дигидрофосфата Р и 5,50 г/л дикалия
гидрофосфата Р\
- скорость подвижной фазы: 0,6 мл/мин;
-спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: по 5 мкл испытуемого
раствора и растворов сравнения (Ь), (с), (с!) и (е);
- время хроматографирования: 3-кратное вре¬
мя удерживания фолиевой кислоты.
Относительное удерживание (по отношению
к фолиевой кислоте, время удерживания — около
8,5 мин): примесь А—около 0,5; примесь В — около 0,6;
примесь С — около 0,9; примесь Е — около 1,27; при¬
месь О — около 1,33; примесь Р — около 2,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками фоли¬
евой кислоты и примеси О.
Предельное содержание примесей:
- примесь О (не более 0,6 %): на хроматограмме
испытуемого раствора площадь пика соответствующе¬
го примеси О, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (е);
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика соответствующе¬
го примеси А, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (с*);
- любая другая примесь (не более 0,5 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А и О, не
должна превышать площадь основного пика на хро¬
матограмме раствора сравнения (с);
- сумма других примесей (не более 1,0 %): на
хроматограмме испытуемого раствора сумма площа¬
дей всех пиков, кроме основного и пиков примесей А
и О, не должна превышать 2-кратную площадь основ¬
ного пика на хроматограмме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (с).
'III
Формальдегида 35 % раствор
1043
Вода {2.5.12). Не менее 5,0 % и не более 8,5 %.
Определение проводят из 0,150 г испытуемого образца.
Сульфатная зола {2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота {2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография {2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы, по 5 мкл испытуемого
раствора и раствора сравнения (а).
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
н ро2н
Н21\Г
'С02Н
А. (25)-2-[(4-Аминобензоил)амино]пентандиовая
кислота (Л/-(4-аминобензоил)-1_-глутаминовая кислота).
о
н2гд
л
ИНг
МН2
В. 2,5,6-Триаминопиримидин-4(1 Н)-он.
С. (25)-2-[[4-[[(2-Амино-4-оксо-1,4-дигидроптери-
дин-7-ил)метил]амино]бензоил]амино]пентандиовая
кислота (изофолиевая кислота).
0.4-[[(2-Амино-4-оксо-1,4-дигидроптеридин-6-ил)-
метил]амино]бензойная кислота (птероевая кислота).
Е. (25)-2-[[4-[Бис[(2-амино-4-оксо-1,4-дигидропте-
ридин-6-ил)метил]амино]бензоил]амино]пентандиовая
кислота (6-птеринилфолиевая кислота).
о
Р. 2-Амино-7-(хлорметил)птеридин-4(1 Н)-он.
07/2016:0826
ФОРМАЛЬДЕГИДА 35 % РАСТВОР
Рогта/с/еМусИ зо1иИо (35 рег сеп1ит)
РОКМА/-ОЕНУОЕ 80ШТ10Ы (35 РЕК
СБЫТ)
[50-00-0]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 34,5 % {м/м) и не более
38,0 % {м/м) формальдегида (СН20; М.м. 30,03).
Содержит метанол в качестве стабилизатора.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
Смешивается с водой и 96 % спиртом.
При хранении может мутнеть.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 мл раствора 3, приготовленного как ука¬
зано в разделе «Испытания», доводят водой Р до
объема 10 мл. К 0,05 мл полученного раствора при¬
бавляют 1 мл раствора 15 г/л хромотроповой кис¬
лоты натриевой соли Р, 2 мл воды Р и 8 мл кисло¬
ты серной Р. В течение 5 мин появляется фиоле¬
тово-синее или фиолетово-красное окрашивание.
B. К 0,1мл раствора 3 прибавляют 10 мл
воды Р, 2 мл приготовленного непосредственно
перед использованием раствора 10 г/л фенилги-
дразина гидрохлорида Р, 1 мл раствора калия
феррицианида Р и 5 мл кислоты хлористоводо¬
родной Р. Появляется интенсивное красное окра¬
шивание.
C. 0,5 мл испытуемого образца смешивают в
пробирке с 2 мл воды Р и 2 мл раствора серебра
нитрата Р2. К полученному раствору прибавляют
раствор аммиака разведенный Р2 до слабощелоч¬
ной реакции и нагревают на водяной бане. Образу¬
ется серый осадок или серебряное зеркало.
О. Испытуемый образец выдерживает требо¬
вания раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 10 мл испытуемого образца при не¬
обходимости фильтруют и доводят водой, свободной
от углерода диоксида, Р до объема 50 мл.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность. К 10 мл раствора 3 прибавляют
1 мл раствора фенолфталеина Р. При прибавлении
не более 0,4 мл 0,1 М раствора натрия гидроксида
должно появиться красное окрашивание.
Метанол. Газовая хроматография {2.2.28).
Раствор внутреннего стандарта. 10 мл этано¬
ла Р1 доводят водой Р до объема 100 мл.
Испытуемый раствор. К 10,0 мл испытуемого
образца прибавляют 10,0 мл раствора внутреннего
стандарта и доводят водой Р до объема 100,0 мл.
131*. Зак. 1060.
1044
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения. К 1,0 мл метанола Р при¬
бавляют 10,0 мл раствора внутреннего стандарта и
доводят водой Р до объема 100,0 мл.
Условия хроматографирования:
-колонка стеклянная длиной (1,5—2,0) м и
внутренним диаметром (2—4) мм, заполненная со¬
полимером этилвинилбензол-дивинилбензола Р
((150—180) мкм);
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: (30—40) мл/мин;
- температура: колонка — 120 °С, блок вво¬
да проб — 150 °С, детектор — 150 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемо¬
го раствора и раствора сравнения.
Пригодность хроматографической системы.
раствор сравнения:
- разрешение: не менее 2,0 между пиками ме¬
танола и этанола.
Предельное содержание примесей:
- метанол: не менее 9,0 % (об/об) и не более
15,0 % (об/об).
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого раствора помещают в
мерную колбу вместимостью 100 мл, содержащую
2,5 мл воды Р и 1 мл раствора натрия гидроксида
разведенного Р, встряхивают и доводят водой Р до
объема 100,0 мл. К 10,0 мл полученного раствора
прибавляют 30,0 мл 0,05 М раствора йода, пере¬
мешивают и прибавляют 10 мл раствора натрия
гидроксида разведенного Р. Выдерживают в те¬
чение 15 мин, прибавляют 25 мл кислоты серной
разведенной Р, 2 мл раствора крахмала Р и титру¬
ют 0,1 М раствором натрия тиосульфата.
1 мл 0,05 М раствора йода соответствует
1,501 мгСН20.
ХРАНЕНИЕ
В защищенном от света месте при температу¬
ре от 15 °С до 25 °С.
07/2016:0004
ФОСФОРНАЯ КИСЛОТА
КОНЦЕНТРИРОВАННАЯ
АсМит р/юзрНопсит сопсеп(га{ит
РНОЗРНОШСАСЮ, СОЫСЕЫТРАТЕО
Н3Р04 М.м. 98,0
[7664-38-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 84,0 % (м/м) и не более
90,0 % (м/м).
ОПИСАНИЕ(СВОЙСТВА)
Прозрачная бесцветная сиропообразная едкая
жидкость.
Смешивается с водой и 96 % спиртом.
При хранении при низких температурах мо¬
жет затвердевать в бесцветную кристаллическую
массу, которая не плавится при температуре ниже
28 °С.
Относительная плотность: около 1,7.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец разводят водой Р. Полу¬
ченный раствор имеет сильнокислую реакцию (2.2.4).
B. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», нейтрализованный раствором
натрия гидроксида разведенным Р, дает реакции (а)
и (Ь) на фосфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца дово¬
дят водой Р до объема 150 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Вещества, осаждаемые аммиаком. К 10 мл
раствора 3 прибавляют 8 мл раствора аммиа¬
ка разведенного Р1. Опалесценция полученного
раствора не должна превышать опалесценцию
смеси из 10 мл раствора 3 и 8 мл воды Р.
Гипофосфористая и фосфористая кисло¬
ты. К 5 мл раствора 3 прибавляют 2 мл раствора
серебра нитрата Р2, нагревают на водяной бане
в течение 5 мин. Внешний вид раствора не должен
изменяться.
Хлориды (2.4.4). Не более 0,0050 % (50 ррт).
15 мл раствора 3 должны выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0100%
(100 ррт). 1,5 г испытуемого образца доводят во¬
дой дистиллированной Р до объема 15 мл. Полу¬
ченный раствор должен выдерживать испытание
на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 7,5 мл раствора 3 должны выдерживать
испытание на мышьяк.
Железо (2.4.9). Не более 0,0050 % (50 ррт).
3 мл раствора 3 доводят водой Р до объема 10 мл.
Полученный раствор должен выдерживать испыта¬
ние на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). К 2,5 г испытуемого образца
прибавляют 4 мл раствора аммиака разведенно¬
го Р1 и доводят водой Р до объема 25 мл. 12 мл
полученного раствора должны выдерживать ис¬
пытание на тяжелые металлы. Эталон готовят с
использованием эталонного раствора свинца
(1 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 1,000 г испытуемого образца прибавляют раст¬
вор 10 г натрия хлорида Р в 30 мл воды Р и титруют
1 М раствором натрия гидроксида, используя в каче¬
стве индикатора раствор фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 49,00 мг Н3Р04.
ХРАНЕНИЕ
В стеклянном контейнере.
Фрамицетина сульфат
1045
07/2016:0005
ФОСФОРНАЯ КИСЛОТА
РАЗВЕДЕННАЯ
Ас1с1ит рЬозрЬопсит с1Ии1ит
РНОЗРНОШС АСЮ, 01ШТЕ
ОПРЕДЕЛЕНИЕ
Содержание: не менее 9,5 % (м/м) и не более
10,5 % (м/м) Н3Р04 (м.м. 98,0).
ПРИГОТОВЛЕНИЕ
К 885 г воды Р прибавляют 115 г кислоты фос¬
форной концентрированной и перемешивают.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец имеет сильнокислую ре¬
акцию (2.2.4).
B. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», нейтрализованный раствором
натрия гидроксида разведенным Р, дает реакции (а)
и (Ь) на фосфаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 86 г испытуемого образца доводят
водой Р до объема 150 мл.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Вещества, осаждаемые аммиаком. К 10 мл
раствора 3 прибавляют 8 мл раствора аммиака раз¬
веденного Р1. Опалесценция полученного раствора
не должна превышать опалесценцию смеси из 10 мл
раствора 3 и 8 мл воды Р.
Гипофосфористая и фосфористая кислоты. К
5 мл раствора 3 прибавляют 2 мл раствора серебра
нитрата Р2 и нагревают на водяной бане в течение
5 мин. Внешний вид раствора не должен изменяться.
Хлориды (2.4.4). Не более 0,0006 % (6 ррт).
15 мл раствора 3 должны выдерживать испытание на
хлориды.
Сульфаты (2.4.13). Не более 0,0010 % (10 ррт).
15 мл испытуемого образца должны выдерживать ис¬
пытание на сульфаты.
Мышьяк (2.4.2, метод А). Не более 0,00002 %
(0,2 ррт). 7,5 мл раствора 5 должны выдерживать ис¬
пытание на мышьяк.
Железо (2.4.9). Не более 0,0006 % (6 ррт). 3 мл
раствора 3 доводят водой Р до объема 10 мл. Получен¬
ный раствор должен вьщерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0001 % (1 ррт). К 20 г испытуемого образца при¬
бавляют 4 мл раствора аммиака разведенного Р1 и
доводят водой Р до объема 25 мл. 12 мл полученного
раствора должны выдерживать испытание на тяже¬
лые металлы. Эталон готовят с использованием сме¬
си из 8 мл эталонного раствора свинца (1 ррт РЬ) Р
и 2 мл воды Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 8,60 г испытуемого образца прибавляют раст¬
вор 10 г натрия хлорида Р в 30 мл воды Р и титруют
1 М раствором натрия гидроксида, используя в каче¬
стве индикатора раствор фенолфталеина Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 49,00 мг Н3Р04.
07/2016:0180
ФРАМИЦЕТИНА СУЛЬФАТ
РгатусеНт зиКаз
РРАМУСЕШ 8Ш-РАТЕ
МН2
С23Н46М6013 ■ хН25 04 М.м. 615 (основание)
[4146-30-9]
ОПРЕДЕЛЕНИЕ
2-Дезокси-4-0-(2,6-диамино-2,6-дидезокси-а-0-
глюкопиранозил)-5-0-[3-0-(2,6-диамино-2,6-дидезокси-
Р-1_-идопиранозил)-р-0-рибофуранозил]-Э-стрептамина
сульфат (неомицин В).
Субстанция продуцируется определенными
штаммами 81гер1отусез ТгасНае или 81гер1отусез
десапз или субстанцию получают другими способами.
Содержание: не менее 630 МЕ/мг (в пересчете на
сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок. Гигроско¬
пичен.
Легко растворим в воде, очень мало растворим
в 96 % спирте, практически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси» #или «При¬
месь С»#.
Результаты:
- основной пик на хроматограмме испытуемого
раствора по времени удерживания соответствует ос¬
новному пику на хроматограмме раствора сравнения
(а) #или на хроматограмме испытуемого раствора
обнаруживается основное пятно, соответствующее
основному пятну на хроматограмме раствора срав¬
нения (а)#;
- испытуемый образец выдерживает испытание
на содержание примеси С.
B. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
ИСПЫТАНИЯ
рН (2.3.3). От 6,0 до 7,0. 0,1 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+52,5 до +55,5 (в пересчете на сухое вещество). 1,00 г
испытуемого образца растворяют в воде Р и доводят
до объема 10,0 мл этим же растворителем.
132. Зак. 1060.
1046
Гэсударственная фармакопея Республики Беларусь
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). Содержимое контейнера
с ФСО фрамицетина сульфата растворяют в под¬
вижной фазе и доводят этим же растворителем до по¬
лучения раствора с концентрацией 0,5 мг/мл.
Раствор сравнения (Ь). 3,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (ф. Содержимое контейне¬
ра ФСО неамина (соответствует 0,5 мг) растворяют в
подвижной фазе и доводят до объема 100,0 мл этим
же растворителем.
Раствор сравнения (е). 10 мг ФСО неомицина
сульфата растворяют в подвижной фазе и доводят
до объема 100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным, деактивированным по отношению к ос¬
нованиям, для хроматографии Р с размером частиц
5 мкм;
- температура: 25 °С;
- подвижная фаза: смешивают 20,0 мл кисло¬
ты трифторуксусной Р, 6,0 мл раствора натрия
гидроксида, свободного от карбонатов, Р и 500 мл
воды Р, выдерживают до установления равновесия,
доводят водой Р до объема 1000 мл и дегазируют;
- скорость подвижной фазы: 0,7 мл/мин;
-постколоночный раствор: раствор натрия
гидроксида, свободного от карбонатов Р, предвари¬
тельно дегазированный и разведенный в 25 раз, ко¬
торый подают безимпульсным потоком в колоночный
элюент с использованием полимерного смешивающе¬
го змеевика объемом 375 мкп;
- скорость постколоночного раствора: 0,5 мл/мин;
- амперометрический детектор, работающий в
импульсном режиме, с золотым рабочим электродом,
хлорсеребряным электродом сравнения и вспомога¬
тельным электродом из нержавеющей стали, с потен¬
циалами детектирования 0,00 В, окисления +0,80 В и
восстановления -0,60 В соответственно, с импульс¬
ной продолжительностью в соответствии с использу¬
емым оборудованием;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 1,5-кратное вре¬
мя удерживания неомицина В.
Относительное удерживание (по отношению к
неомицину В, время удерживания — около 10 мин):
примесь А — около 0,65; примесь С — около 0,9; при¬
месь С— около 1,1.
Пригодность хроматографической системы:
- разрешение: не менее 2,0 между пиками при¬
меси С и неомицина В на хроматограмме раствора
сравнения (е); при необходимости изменяют содер¬
жание раствора натрия гидроксида, свободного от
карбонатов, в подвижной фазе;
- отношение сигнал/шум: не менее 10 для основ¬
ного пика на хроматограмме раствора сравнения (с).
Предельное содержание примесей:
-примесь А (не более 1,0%): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (б) (при рас¬
чете учитывают содержание неамина в ФСО неамина)]
- примесь С (не более 3,0 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси С, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь);
- сумма других примесей (не более 3,0 %): на
хроматограмме испытуемого раствора сумма площа¬
дей всех пиков, кроме основного и пиков примеси А и
С, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (Ь);
-неучитываемый предел (1,0%): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
*Испытания «Примесь А» и «Примесь С» ис¬
пользуются в качестве альтернативных испыта¬
нию «Сопутствующие примеси»*.
#Примесь А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,250 г испытуемого об¬
разца растворяют в водеР и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 0,5 мг ФСО неамина
растворяют в 2,0 мл воды Р.
Раствор сравнения (Ь). 0,5 мл испытуемого раст¬
вора смешивают с 0,5 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со споем силикагеля Н Р.
Подвижная фаза: метиленхлорид Р — раст¬
вор аммиака концентрированный Р — метанол Р
(10:20:30, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 8 см от линии
старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 10 мин.
Проявление: пластинку опрыскивают реакти¬
вом нингидрина и олова (II) хлорида Р и нагревают
при температуре 110 °С в течение 15 мин. Пластинку
снова опрыскивают реактивом нингидрина и олова
(II) хлорида Р и нагревают при температуре 110 °С в
течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- примесь А (не более 1 %): на хроматограмме
испытуемого раствора пятно, соответствующее не-
амину, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (а).
#Примесь С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 40 мг испытуемого образ¬
ца растворяют в воде Р и доводят до объема 5,0 мл
этим же растворителем.
Раствор сравнения (а). 40 мг ФСО фрамицети¬
на сульфата растворяют в воде Р и доводят до объ¬
ема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 30 мг ФСО фрамицети¬
на сульфата растворяют в воде Р и доводят до объ¬
ема 25,0 мл этим же растворителем. 5,0 мл получен¬
ного раствора доводят водой Р до объема 25,0 мл.
Раствор сравнения (с). 40 мг ФСО неомицина
сульфата растворяют в воде Р и доводят до объема
5.0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: метанол Р — раствор 200 г/л
натрия хлорида Р (20:80, об/об).
т
Фруктоза
1047
Наносимый объем пробы: по 5 мкл в виде полос.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 10 мин.
Проявление: пластинку опрыскивают раствором
нингидрина Р1 и сушат при температуре от 100 °С до
105 °С в течение10 мин.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживается пятно со зна¬
чением /уменьшим, чем значение /^основного пятна.
Предельное содержание примесей:
- примесь С (не более 3 %): на хроматограмме
испытуемого раствора пятно, соответствующее не-
омицину С (значение Рг меньшее, чем значение Рг
основного пятна), должно быть не интенсивнее пятна
на хроматограмме раствора сравнения (Ь).
Сульфаты. Не менее 27,0 % и не более 31,0 %
(в пересчете на сухое вещество). 0,250 г испытуемо¬
го образца растворяют в 100 мл воды Р и доводят до
рН 11 раствором аммиака концентрированным Р.
Прибавляют 10,0 мл 0,1 М раствора бария хлорида,
около 0,5 мг фталеинового пурпурного Р и титруют
0,1 М раствором натрия эдетата; в момент изме¬
нения окраски раствора прибавляют 50 мл 96 % спир¬
та Рм продолжают титрование до исчезновения фио¬
летово-синего окрашивания.
1 мл 0,1 М раствора бария хлорида соответству¬
ет 9,606 мг 504.
Потеря в массе при высушивании (2.2.32). Не
более 8,0 %. 1,000 г испытуемого образца сушат над
фосфора (V) оксидом Р при температуре 60 °С и дав¬
лении, не превышающем 0,7 кПа, в течение 3 ч.
Сульфатная зола (2.4.14). Не более 1,0%.
Определение проводят из 1,0 г испытуемого образца.
Стерильность (2.6.1). Если субстанция пред¬
назначена для производства лекарственных средств
для внутриполостного введения без дальнейшей под¬
ходящей процедуры стерилизации, то она должна вы¬
держивать испытание на стерильность.
Бактериальные эндотоксины (2.6.14, метод О).
Менее 1,3 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для внутрипо¬
лостного введения без дальнейшей подходящей про¬
цедуры удаления бактериальных эндотоксинов.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Микробиологический метод (2.7.2). В качестве
стандартного образца используют ФСО фрамицети-
на сульфата.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Если субстанция предназначена для производ¬
ства лекарственных средств для внутриполостного
введения — в стерильном контейнере с контролем
первого вскрытия.
#МАРКИРОВКА
При необходимости указывают:
- субстанция стерильна;
- субстанция не содержит бактериальных эндо¬
токсинов.
ПРИМЕСИ
МН2
A. К1 = Н, К2 = 1ЧН2: 2-Дезокси-4-0-(2,6-диамино-
2,6-дидезокси-а-0-глюкопиранозил)-0-стрептамин (не-
амин или неомицин А-/.Р).
B. К1 = СО-СН3, К2 = 1ЧН2: 3-Л/-Ацетил-2-дезокси-
4-0-(2,6-диамино-2,6-дидезокси-а-0-глюкопиранозил)-
О-стрептамин (3-ацетилнеамин).
О. К1 = Н, К2 = ОН: 4-0-(2-Амино-2-дезокси-а-0-
глюкопиранозил)-2-дезокси-0-стрептамин (паромамин
или неомицин О).
С. К1 = СН2-МН2, К2 = КЗ =Н, К4 = ЫН,: 2-Дезокси-
4- 0-(2,6-диамино-2,6-дидезокси-а-0-глюкопиранозил)-
5- 0-[3-0-(2,6-диамино-2,6-дидезокси-а-0-глюкопи-
ранозил)-Р-0-рибофуранозил]-0-стрептамин (неоми¬
цин С).
Е. К1 = КЗ = Н, К2 = СН2-МН2, К4 = ОН: 4-0-(2-
Амино-2-дезокси-а-0-глюкопиранозил)-2-дезокси-5-0-
[3-0-(2,6-диамино-2,6-дидезокси-р-1_-идопиранозил)-Р-
0-рибофуранозил]-0-стрептамин (паромомицин I или
неомицин Е).
Р. К1 = СН2-МН2, К2 = КЗ = Н, К4 = ОН: 4-0-(2-
Амино-2-дезокси-а-0-глюкопиранозил)-2-дезокси-5-0-
[3-0-(2,6-диамино-2,6-дидезокси-а-0-глюкопиранозил)-
Р-0-рибофуранозил]-0-стрептамин (паромомицин II
или неомицин Р).
О. К1 = Н, К2 = СН2-ЫН2, КЗ = СО-СН3, К4 = ЫН2:
3-Л/-Ацетил-2-дезокси-4-0-(2,6-диамино-2,6-дидезокси-
а-0-глюкопиранозил)-5-0-[3-0-(2,6-диамино-2,6-диде-
зокси-р-1_-идопиранозил)-р-0-рибофуранозил]-0-
стрептамин (неомицин В-ЛР).
07/2016:0188
ФРУКТОЗА
Ргис1озит
РШ1СТ08Е
СвН12Ов М.м. 180,2
[57-48-7]
ОПРЕДЕЛЕНИЕ
О-арабино-Гекс-2-улопираноза.
Субстанция, описанная в данной частной статье,
не всегда подходит для парентерального применения.
132*. Зак. 1060.
1048
Государственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Очень легко растворим в воде, растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(2:3, об/об).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 20 мл этой же смесью растворителей.
Раствор сравнения (а). 10 мг ФСО фруктозы
растворяют в смеси растворителей и доводят до объ¬
ема 20 мл этой же смесью растворителей.
Раствор сравнения (Ь). 10 мг фруктозы Р, 10 мг
глюкозы Р, 10 мг лактозы Р и 10 мг сахарозы Р раст¬
воряют в смеси растворителей и доводят до объема
20 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикагеля С Р.
Подвижная фаза: вода Р—метанол Р—кислота
уксусная безводная Р — этиленхлорид Р (10:15:25:50,
об/об/об/об). Объемы отмеривают точно, так как незна¬
чительный избыток воды вызывает помутнение.
Наносимый объем пробы: 2 мкл; тщательно вы¬
сушивают нанесенные пробы.
Фронт подвижной фазы А: не менее 3/4 высоты
пластинки.
Высушивание А: в потоке теплого воздуха.
Фронт подвижной фазы В: немедленно, не ме¬
нее 3/4 высоты пластинки, после замены подвижной
фазы на новую.
Высушивание В: в потоке теплого воздуха.
Проявление: пластинку опрыскивают раствором
0,5 г тимола Р в смеси из 5 мл кислоты серной Р и
95 мл 96 % спирта Р. Нагревают при температуре
130 °С в течение 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются четыре
полностью разделенных пятна.
Результаты: основное пятно на хроматограмме
испытуемого раствора соответствует по расположе¬
нию, цвету и размеру основному пятну на хромато¬
грамме раствора сравнения (а).
B. 0,1 г испытуемого образца растворяют в 10 мл
воды Р. Прибавляют 3 мл раствора медно-тартрат-
ного Р и нагревают. Образуется красный осадок.
C. К 1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 9 мл воды Р.
К 1 мл полученного раствора прибавляют 5 мл кисло¬
ты хлористоводородной Р и нагревают при темпера¬
туре 70 °С. Появляется коричневое окрашивание.
О. 5 г испытуемого образца растворяют в воде Р
и доводят до объема 10 мл этим же растворителем.
К 0,5 мл полученного раствора прибавляют 0,2 г ре¬
зорцина Р и 9 мл кислоты хлористоводородной раз¬
веденной Р и нагревают на водяной бане в течение
2 мин. Появляется красное окрашивание.
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 100 мл этим же растворителем.
Раствор 31. 5,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). К раствору 31 при¬
бавляют 10 мл воды Р. Полученный раствор должен
быть бесцветным.
Кислотность или щелочность. 6,0 г испытуемо¬
го образца растворяют в 25 мл воды, свободной от
углерода диоксида, Р и прибавляют 0,3 мл раствора
фенолфталеина Р. Раствор бесцветный. При при¬
бавлении не более 0,15 мл 0,1 М раствора натрия
гидроксида должно появиться розовое окрашивание.
Удельное оптическое вращение (2.2.7). От -91
до -93,5 (в пересчете на безводное вещество). 10,0 г
испытуемого образца растворяют в 80 мл воды Р, при¬
бавляют 0,2 мл раствора аммиака разведенного Р1,
выдерживают в течение 30 мин и доводят водой Р до
объема 100,0 мл.
Посторонние сахара. 5,0 г испытуемого образца
растворяют в воде Р и доводят до объема 10 мл этим
же растворителем. К 1 мл полученного раствора при¬
бавляют 9 мл 96 % спирта Р. Опалесценция испытуе¬
мого раствора должна быть не интенсивнее опалесцен¬
ции смеси из 1 мл исходного раствора и 9 мл воды Р.
5-Гид рокси мети л фурфурал ь и родственные
соединения. К 5 мл раствора 3 прибавляют 5 мл
воды Р. Оптическая плотность (2.2.25), измеренная
при длине волны 284 нм, не должна превышать 0,32.
Барий. К 10 мл раствора 3 прибавляют 1 мл
кислоты серной разведенной Р. Опалесценция полу¬
ченного раствора сразу после приготовления и через
1 ч должна быть не интенсивнее опалесценции раст¬
вора, состоящего из 1 мл воды дистиллированной Р
и 10 мл раствора 3.
Свинец (2.4.10). Не более 0,00005 % (0,5 ррт).
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,000 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %. 5,0 г
испытуемого образца растворяют в 10 мл воды Р, при¬
бавляют 2 мл кислоты серной Р, выпаривают досуха
на водяной бане и прокаливают до постоянной массы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:0611
ФТОРУРАЦИЛ
ИиогоигасИит
ршсжоипАСч.
С4Н3РЫ202 М.м.130,1
[51-21-8]
ОПРЕДЕЛЕНИЕ
5-Фторпиримидин-2,4(1 Н,ЗЯ)-дион.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Умеренно растворим в воде, мало растворим в
96 % спирте.
Фторурацил
1049
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фторурацила.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7 или У(Ж)7.
рН (2.2.3). От4,5 до 5,0. Измеряют рН раствора 3.
Примеси Р и О. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в смеси из метанола Р и воды Р
(1:1, об/об) и доводят до объема 10,0 мл этой же сме¬
сью растворителей.
Раствор сравнения (а). 5,0 мг ФСО фторураци¬
ла примеси Р растворяют в смеси из метанола Р и
воды Р( 1:1, об/об) и доводят до объема 200,0 мл этой
же смесью растворителей.
Раствор сравнения (Ь). 20,0 мг мочевины Р
(примесь О) растворяют в метаноле Р и доводят до
объема 10,0 мл этим же растворителем. 1,0 мл полу¬
ченного раствора доводят метанолом Р до объема
100,0 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Я
Подвижная фаза: метанол Р — вода Р—этила-
цетат Р (15:15:70, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление:
- примесь Р: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм;
- примесь О: пластинку опрыскивают смесью из
200 мл раствора 10 г/л диметиламинобензальдеги-
да Р в этаноле Р и 20 мл кислоты хлористоводо¬
родной Р и сушат при температуре 80 °С в течение
(3—4) мин. Пластинку просматривают при дневном
свете (примесь 6 проявляется в виде желтого пятна,
фторурацил не проявляется при опрыскивании).
Пригодность хроматографической системы:
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна после обоих проявлений.
Предельное содержание примесей:
- примесь Р (не более 0,25 %): на хроматограм¬
ме испытуемого раствора пятно, соответствующее
примеси Р, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (а);
- примесь 6 (не более 0,2 %): на хроматограмме
испытуемого раствора пятно, соответствующее при¬
меси 6, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (Ь).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытание проводят с защитой
от света.
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем. 5,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 50,0 мл.
Раствор сравнения (а). 5,0 мг ФСО фторураци¬
ла примеси С растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (с). 5,0 мг ФСО фторураци¬
ла примеси А растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл. 1,0 мл полученного раствора дово¬
дят подвижной фазой до объема 10,0 мл.
Раствор сравнения (д). 5,0 мг ФСО фторураци¬
ла примеси В растворяют в подвижной фазе и дово¬
дят до объема 50,0 мл этим же растворителем. 1,0 мл
полученного раствора доводят подвижной фазой до
объема 100,0 мл. 1,0 мл полученного раствора дово¬
дят подвижной фазой до объема 10,0 мл.
Раствор сравнения (е). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (1). К 1 мл раствора сравне¬
ния (а) прибавляют 1 мл испытуемого раствора и до¬
водят подвижной фазой до объема 10 мл.
Раствор сравнения (д). Содержимое контейнера
ФСО фторурацила смеси примесей (содержит при¬
меси О и Е) растворяют в 1,0 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилильным
для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: раствор 6,805 г/л калия диги¬
дрофосфата Р, доведенный до рН (5,7±0,1) 5 М рас¬
твором калия гидроксида Р\
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 266 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания фторурацила.
Идентификация пиков примесей: идентифици¬
руют пики примесей О и Е, используя хроматограмму
раствора сравнения (д) и хроматограмму, прилагае¬
мую к ФСО фторурацила смеси примесей.
Относительное удерживание (по отношению
к фторурацилу, время удерживания — около 6 мин):
примесь А — около 0,5; примесь В — 0,7; примесь
С — 0,9; примесь О — 1,6; примесь Е — около 1,9.
Пригодность хроматографической системы:
раствор сравнения ({):
- разрешение: не менее 2,0 между пиками при¬
меси С и фторурацила.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси й — 1,5; для примеси Е — 1,3):
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (с);
- примесь В (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (б);
133. Зак. 1060.
1050
Гэсударственная фармакопея Республики Беларусь
- примесь С (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (Ь);
- примеси О, Е (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям О и Е, не должны превышать
площадь основного пика на хроматограмме раствора
сравнения (е);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей А,
В, С, О и Е, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (е);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (е);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (е).
Тяжелые металлы (2.4.8, метод С). Не более
0,0020% (20ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Используют платиновый тигель. Эталон готовят с
использованием 2 мл эталонного раствора свинца
(10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 80 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца
в платиновом тигле.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 80 мл
диметилформамида Р, слегка подогревая. Охлаждают
и титруют 0,1М раствором тетрабутламмония гидрок¬
сида, используя в качестве индикатора 0,25 мл раствора
10 г/л тимолового синего Р в диметилформамиде Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора тетрабутиламония ги¬
дроксида соответствует 13,01 мг С4Н3Н\1202.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, С.
о
А. Пиримидин-2,4,6(1 Н,ЗЯ,5М)-трион (барбитуро¬
вая кислота).
о
л
В. Дигидропиримидин-2,4,5(ЗН)-трион (изобарби-
туровая кислота или 5-гидроксиурацил).
о
о^м
н
С. Пиримидин-2,4(1 Н,ЗН)-дион (урацил).
о
н
О. 5-Метоксипиримидин-2,4(1Н,ЗН)-дион (5-меток-
сиурацил).
ОТ N
Н
X)
Е. 5-Хлоропиримидин-2,4(1Н,ЗН)-дион (5-хлору-
рацил).
л.
н3с 'СГ 'м
Р. 2-Этокси-5-фторопиримидин-4(1Н)-он (2-этокси-
5-фторурацил).
н2ы ж2
О. Карбамид (мочевина).
07/2016:РБ0014
#ФУРАЗОЛИДОН
РигагоИбопит
рцкАгоиооыЕ
о2ы
с,н7и3о5
[67-45-8]
ОПРЕДЕЛЕНИЕ
3-(5-Нитрофурфурилиденамино)оксазолидин-2-он.
Содержание: не менее 97,0 % и не более 103,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Желтый кристаллический порошок.
Очень мало растворим в воде и в 96 % спирте,
практически нерастворим в эфире.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО фуразолидона или спектр,
представленный на рисунке РБ0014.-1.
B. 1 мг испытуемого образца растворяют в 1 мл
диметилформамида Р и прибавляют 0,05 мл 1 М
раствора калия гидроксида спиртового раствора.
Появляется темно-синее окрашивание.
ИСПЫТАНИЯ
рН (2.2.3). От 4,5 до 7,0.1 г испытуемого образца
встряхивают с 100 мл воды, свободной от углерода
диоксида Р в течение 15 мин и фильтруют.
Фуросемид
1051
Рисунок РБ0014.-1. Инфракрасный спектр ФСО фуразолидона
Нитрофурфураля диацетат. Тонкослойная хро¬
матография (2.2.27).
Испытание проводят с защитой от яркого света.
Испытуемый раствор. 50 мг испытуемого образ¬
ца растворяют в 5 мл диметилформамида Р при на¬
гревании на водяной бане в течение нескольких минут,
охлаждают и доводят ацетоном Р до объема 10 мл.
Раствор сравнения. 5,0 мг ФСО нитрофурфу¬
раля диацетата растворяют в смеси из равных объ¬
емов диметилформамида Р и ацетона Р и доводят
до объема 50,0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля С Р.
Подвижная фаза: толуол Р — диоксан Р (95:5,
об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 10 мкл раствора сравнения.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: при температуре 105 °С в течение
5 мин.
Проявление: горячую пластинку опрыскивают
раствором фенилгидразина гидрохлорида Р.
Предельное содержание примесей:
-нитрофурфураля диацетат (не более 1 %):
на хроматограмме испытуемого раствора пятно со¬
ответствующее нитрофураля диацетату должно быть
не интенсивнее соответствующего пятна на хромато¬
грамме раствора сравнения.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от света.
К 80 мг испытуемого образца прибавляют 150 мл
диметилформамида Р, перемешивают до полного
растворения и доводят водой Р до объема 500,0 мл.
5,0 мл полученного раствора доводят водой Р до объ¬
ема 100,0 мл. Измеряют оптическую плотность (2.2.25)
полученного раствора в максимуме при 367 нм.
Содержание С8Н7М3Об рассчитывают с учетом
удельного показателя поглощения, равного 750.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0391
ФУРОСЕМИД
Риго8ет1с1ит
РШЮЗЕМЮЕ
С12Н11С1Ы2058 М.м. 330,7
[54-31-9]
ОПРЕДЕЛЕНИЕ
4-Хлор-2-[(фуран-2-илметил)амино]-5-сульфамоил-
бензойная кислота.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, растворим в аце¬
тоне, умеренно растворим в 96 % спирте, практически
нерастворим в метиленхлориде. Растворяется в разве¬
денных растворах гидроксидов щелочных металлов.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
133*. Зак. 1060.
1052
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. 50 мг испытуемого образ¬
ца растворяют в растворе 4 г/л натрия гидроксида Р
и доводят до объема 100 мл этим же растворителем.
1 мл полученного раствора доводят раствором 4 г/л
натрия гидроксида Р до объема 100 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимум поглощения: при 228 нм, при 270 нм
и при 333 нм.
Отношение оптических плотностей:
Лш/Ляв — от °>52 Д° °>57-
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО фуросемида * или спектр, пред¬
ставленный на рисунке #0391.-1#.
C. 25 мг испытуемого образца растворяют в
10 мл 96% спирта Р. К 5 мл полученного раствора
прибавляют 10 мл воды Р. К 0,2 мл полученного раст¬
вора прибавляют 10 мл кислоты хлористоводород¬
ной разведенной Р и нагревают с обратным холо¬
дильником в течение 15 мин. Охлаждают, прибавляют
18 мл 1 М раствора натрия гидроксида и 1 мл раст¬
вора 5 г/л натрия нитрита Р. Выдерживают в тече¬
ние 3 мин, прибавляют 2 мл раствора 25 г/л кислоты
сульфаминовой Р, перемешивают и прибавляют 1 мл
раствора 5 г/л нафтилэтилендиамина дигидрохло¬
рида Р. Появляется фиолетово-красное окрашивание.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в 0,5 М растворе натрия гидроксида и доводят
до объема 10,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)5.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием, испытание прово¬
дят с защитой от света.
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО фуросемида
примеси А растворяют в подвижной фазе, прибавля-
I
95 |
85 |
80 |
75 |
70 |
65 |
ют 2,0 мл испытуемого раствора и доводят до объема
20.0 мл этим же растворителем. 0,5 мл полученного
раствора доводят подвижной фазой до объема 20,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (с). 2 мг ФСО фуросемида
для идентификации пиков (содержит примеси С и О)
растворяют в 2,0 мл подвижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- подвижная фаза: 2,0 г калия дигидрофос¬
фата Р и 2,5 г цетримида Р растворяют в 700 мл
воды Р, доводят до рН 7,0 раствором аммиака Р и
прибавляют 300 мл пропанола Р;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 238 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания фуросемида.
Идентификация пиков примесей: идентифици¬
руют пик примеси А, используя хроматограмму раст¬
вора сравнения (а); идентифицируют пики примесей
С и Э, используя хроматограмму раствора сравнения
(с) и хроматограмму, прилагаемую к ФСО фуросемида
для идентификации пиков.
Относительное удерживание (по отношению к
фуросемиду, время удерживания — около 9 мин): при¬
месь С — около 0,5; примесь А — около 0,8; примесь
й — около 1,5.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 4 между пиками приме¬
си А и фуросемида.
- отношение сигнал/шум: не менее 40 для основ¬
ного пика на хроматограмме раствора сравнения (Ь);
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
60 |
55 |
50 |
45 |
40 :
Рисунок #0391 .-1. Инфракрасный спектр ФСО фуросемида
Химотрипсин
1053
соответствующие поправочные коэффициенты: для
примеси С — 1,4; для примеси О — 2,0):
- примесь С (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примесь О (не более 0,15 %): на хроматограм¬
ме испытуемого раствора площадь пика, соответству¬
ющего примеси О, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей С и О, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
)Оюриды (2.4.4). Не более 0,0200 % (200 ррт).
К 0,5 г испытуемого образца прибавляют смесь из
0,2 мл кислоты азотной Р и 30 мл воды дистилли¬
рованной Р и встряхивают в течение 5 мин. Выдержи¬
вают в течение 15 мин и фильтруют.
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
К 1,0 г испытуемого образца прибавляют смесь из
0,2 мл кислоты уксусной Р и 30 мл воды дистилли¬
рованной Р и встряхивают в течение 5 мин. Выдержи¬
вают в течение 15 мин и фильтруют.
Тяжелые металлы (2.4.8, метод Н). Не более
0,0020 % (20 ррт).
Смесь растворителей. Вода Р — ацетон Р
(20:80, об/об).
0,25 г испытуемого образца должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием 0,5 мл эталонного раствора свин¬
ца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, Е, Р.
А. 2-Хлор-4-[(фуран-2-илметил)амино]-5-сульфа-
моилбензойная кислота.
С. 2-Амино-4-хлор-5-сульфамоилбензойная кис¬
лота.
й. 2,4-Бис[(фуран-2-илметил)амино]-5-сульфамоил-
бензойная кислота.
Р.4-Хлор-5-сульфамоил-2-[[((2К5)-тетрагидро-
фуран-2-ил)метил]амино]бензойная кислота.
07/2016:0476
ХИМОТРИПСИН
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 40 мл
диметилформамида Р и титруют 0,1 М раствором
натрия гидроксида потенциометрически (2.2.20).
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 33,07 мг С12Н11С1М2053.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
СМуто1гурз/пит
СНУМОТКУР8Ш
[9004-07-3]
ОПРЕДЕЛЕНИЕ
Химотрипсин представляет собой протеолити¬
ческий фермент, получаемый активацией химотрип-
синогена, экстрагируемого из поджелудочной железы
крупного рогатого скота (Воз 1аигиз 1_.). Активность: не
менее 5,0 микрокатал/мг. В растворе при рН около 8
обладает максимальной ферментативной активно¬
стью; активность обратимо ингибируется при рН 3;
при значении рН 3 химотрипсин является наиболее
стабильным.
134. Зак. 1060.
1054
Гэсударственная фармакопея Республики Беларусь
ПРОИЗВОДСТВО
Животные, используемые для получения химо-
трипсина, должны выдерживать требования, предъ¬
являемые к здоровью животных, используемых че¬
ловеком в пищу. Кроме того, используемые ткани не
должны содержать опасные вещества в соответствии
с международным или национальным законодатель¬
ством.
Метод производства должен валидироваться,
чтобы подтвердить, что субстанция выдерживает сле¬
дующее испытание.
Гистамин (2.6.10). Не более 1 мкг (в пересчете
на основание гистамина) на 5 микрокатал химотрип-
синовой активности. Перед проведением испытания
раствор испытуемого образца нагревают на водяной
бане в течение 30 мин.
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический или
аморфный порошок. Аморфная форма является ги¬
гроскопичной.
Умеренно растворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 1 мл раствора 3, приготовленного как указа¬
ла в разделе «Ксштетя», девсдяг вааЪсгРда объ¬
ема 10 мл. В лунке белой пластинки для капельных
реакций смешивают 0,05 мл полученного раствора
и 0,2 мл раствора субстрата. Появляется пурпурное
окрашивание.
Раствор субстрата. К 24,0 мг ацетилтирозина
этилового эфира Р прибавляют 0,2 мл 96 % спир¬
та Р и перемешивают до растворения. Прибавляют
2.0 мл 0,067 М фосфатного буферного раствора рН
7.0 Р и 1 мл смешанного раствора метилового крас¬
ного Р и доводят водой Р до объема 10,0 мл.
B. 0,5 мл раствора 3 доводят водой Р до объема
5 мл и прибавляют 0,10 мл раствора 20 г/л тозил-
фенилаланилхлорметана Р в 96% спирте Р. Дово¬
дят до рН 7,0 и встряхивают в течение 2 ч. В лунке
белой пластинки для капельных реакций смешивают
0,05 мл полученного раствора и 0,2 мл раствора суб¬
страта, приготовленного как указано в подлинности А.
Окрашивание не появляется в течение 3 мин после
смешивания.
ИСПЫТАНИЯ
Раствор 3. 0,10 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 10,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
рН (2.2.3). От 3,0 до 5,0. Измеряют рН раствора 3.
Оптическая плотность (2.2.25). 30,0 мг испы¬
туемого образца растворяют в 0,001 М растворе
кислоты хлористоводородной и доводят до объема
100.0 мл этим же растворителем. Спектр поглощения
испытуемого раствора должен иметь максимум при
281 нм и минимум при 250 нм. Удельный показатель
поглощения: в максимуме — от 18,5 до 22,5; в мини¬
муме — не более 8.
Трипсин
Раствор субстрата. К 98,5 мг тозиларгинина
метилового эфира гидрохлорида Р прибавляют 5 мл
трис(гидроксиметил)аминометан-буферного раст¬
вора рН 8,1 и перемешивают до растворения. К по¬
лученному раствору прибавляют 2,5 мл смешанного
раствора метилового красного Р и доводят водой Р
до объема 25,0 мл.
Испытуемый раствор. В лунке белой пла¬
стинки для капельных реакций смешивают 0,01 мл
трис(гидроксиметил)аминометан-буферного раст¬
вора рН 8,1 Р и 0,1 мл раствора 5 и прибавляют 0,2 мл
раствора субстрата.
Раствор сравнения. Готовят параллельно с ис¬
пытуемым раствором, используя испытуемый обра¬
зец, к которому прибавлено не более 1 % (м/м) БСП
трипсина.
В испытуемом растворе в течение (3—5) мин по¬
сле прибавления раствора субстрата не должно появ¬
ляться окрашивание. В растворе сравнения появляет¬
ся пурпурное окрашивание.
Потеря в массе при высушивании (2.2.32). Не
более 5,0 %. 0,100 г испытуемого образца сушат при
температуре 60 °С и давлении, не превышающем
0,7 кПа, в течение 2 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Активность испытуемого образца определяют,
<ж?р<жгб, с ж
тилтирозина этиловый эфир Р, со скоростью, с
которой БСП химотрипсина гидролизует этот же суб¬
страт в этих же условиях.
Прибор. Используют реакционный сосуд вмести¬
мостью около 30 мл, снабженный:
- устройством, поддерживающим температуру
(25,0±0,1)°С;
-устройством для перемешивания (например,
магнитной мешалкой);
- крышкой с отверстиями для электродов, нако¬
нечника бюретки, трубки для подвода азота и прибав¬
ления реактивов.
Может быть использована автоматическая или
ручная установка для титрования. В последнем слу¬
чае бюретка имеет цену деления 0,005 мл, а рН-
метр — широкую шкалу значений и снабжен стеклян¬
ным и хлорсеребряным электродами или другими
подходящими электродами.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в 0,001 М растворе кислоты хло¬
ристоводородной и доводят до объема 250,0 мл этим
же растворителем.
Раствор сравнения. 25,0 мг БСП химотрипсина
растворяют в 0,001 М растворе кислоты хлористо¬
водородной и доводят до объема 250,0 мл этим же
растворителем.
Растворы хранят при температуре от 0 °С до
5 °С. 1 мл каждого раствора нагревают до температу¬
ры около 25 °С в течение 15 мин и используют 50 мкл
каждого раствора (соответствует около 25 нанока-
тал) для каждого титрования. Титрование проводят
в атмосфере азота. 10,0 мл 0,01 М раствора каль¬
ция хлорида Р помещают в реакционный сосуд и при
перемешивании прибавляют 0,35 мл 0,2 М раствора
ацетилтирозина этилового эфира Р. Когда темпе¬
ратура установится (25,0±0,1) °С (приблизительно че¬
рез 5 мин) доводят точно до рН 8,0 0,02 М раствором
натрия гидроксида. Прибавляют 50 мкл испытуемого
раствора (соответствует около 5 мкг испытуемого об¬
разца) и начинают отсчет времени. Поддерживают рН
8,0, прибавляя 0,02 М раствор натрия гидроксида,
1ВГ
Хинидина сульфат
1055
отмечая прибавленный объем каждые 30 с. Рассчи¬
тывают использованный объем 0,02 М раствора на¬
трия гидроксида в секунду между 30 секундой и 210
секундой титрования. Аналогично проводят титрова¬
ние с использованием раствора сравнения и рассчи¬
тывают использованный объем 0,02 М раствора на¬
трия гидроксида в секунду.
Активность в микрокатал на миллиграмм рассчи¬
тывают по формуле:
лГ-У
тУ*
где:
т — масса испытуемого образца, мг;
т' — масса БСП химотрипсина, мг;
V — объем 0,02 М раствора натрия гидроксида,
израсходованный на титрование испытуемого раст¬
вора в секунду;
V — объем 0,02 М раствора натрия гидрокси¬
да, израсходованный на титрование раствора сравне¬
ния в секунду;
А — активность БСП химотрипсина, в микрока¬
тал на миллиграмм.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищен¬
ном от света месте при температуре от 2 °С до 8 °С.
МАРКИРОВКА
Указывают:
- количество химотрипсина и общую активность
в микрокатал на контейнер;
-для аморфного химотрипсина — «гигроскопичен».
07/2016:0017
ХИНИДИНА СУЛЬФАТ
СЫтсИм зиНав
оитюмЕ зш-рате
[6591-63-5]
• Н2504 • 2Н20
М.м. 783
ОПРЕДЕЛЕНИЕ
Моносульфаты алкалоидов, в пересчете на бис-
[(5)-[(2Я,45,5#)-5-этенил-1-азабицикло[2.2.2]окт-2-
ил](6-метоксихинолин-4-ил)метанола] сульфат диги-
драт.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные шелковистые игольчатые кри¬
сталлы.
Мало растворим в воде, растворим в кипящей воде
и в 96 % спирте, практически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 0,10 г ФСО хинидина суль¬
фата растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля С Р.
Подвижная фаза: диэтиламин Р — эфир Р —
толуол Р (10:24:40, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: дважды; не менее 15 см
от линии старта; после первого хроматографирования
пластинку сушат в потоке воздуха в течение 15 мин.
Высушивание: при температуре 105 °С в течение
30 мин и охлаждают.
Проявление: пластинку опрыскивают реактивом
йодплатината Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
B. 5 мг испытуемого образца растворяют в 5 мл
воды Р и прибавляют 0,2 мл бромной воды Р и 1 мл
раствора аммиака разведенного Р2. Появляется зе¬
леное окрашивание.
C. 0,1 г испытуемого образца растворяют в 3 мл
кислоты серной разведенной Р и доводят водой Р до
объема 100 мл. Полученный раствор просматривают
в ультрафиолетовом свете при длине волны 366 нм.
Обнаруживается интенсивная голубая флуоресцен¬
ция, исчезающая почти полностью при прибавлении
1 мл кислоты хлористоводородной Р.
O. Около 50 мг испытуемого образца растворяют
в 5 мл горячей воды Р, охлаждают, прибавляют 1 мл
раствора серебра нитрата Р1 и перемешивают сте¬
клянной палочкой. Через несколько минут появляется
белый осадок, который растворяется при прибавле¬
нии кислоты азотной разведенной Р.
Е. Испытуемый образец дает реакцию (а) на
сульфаты (2.3.1).
P. Испытуемый образец выдерживает испытание
на рН, как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раство¬
ряют в 0,1 М растворе кислоты хлористоводород¬
ной Р и доводят до объема 25,0 мл этим же раство¬
рителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона СУ(ЗЖ)6.
рН (2.2.3). От 6,0 до 6,8. 0,10 г испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+275 до +290 (в пересчете на сухое вещество). Ис¬
пользуют раствор 3.
Другие хинные алкалоиды. Жидкостная хро¬
матография (2.2.29): определение проводят методом
внутренней нормализации.
134*. Зак. 1060.
1056
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 5 мл подвижной фазы, при не¬
обходимости слегка подогревая, и доводят до объ¬
ема 10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО хинина
сульфата (примесь А) растворяют 5 мл подвижной
фазы, при необходимости слегка подогревая, и до¬
водят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 20 мг ФСО хинидина
сульфата растворяют 5 мл подвижной фазы, при
необходимости слегка подогревая, и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения (с). К 1 мл раствора срав¬
нения (а) прибавляют 1 мл раствора сравнения (Ь).
Раствор сравнения (д). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой до объема
10,0 мл. 1,0 мл полученного раствора доводят под¬
вижной фазой до объема 50,0 мл.
Раствор сравнения (е). 10 мг тиомочевины Р
растворяют в подвижной фазе и доводят до объема
10 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной (0,15—0,25) м и внутренним
диаметром 4,6 мм, заполненная силикагелем ок-
тадецилсилильным для хроматографии Р (размер
частиц (5—10) мкм);
- подвижная фаза: 6,8 г калия дигидрофосфа¬
та Р и 3,0 г гексиламина Р растворяют в 700 мл
воды Р, доводят кислотой фосфорной разведен¬
ной Р до рН 2,8, прибавляют 60 мл ацетонитри¬
ла Р и доводят водой Р до объема 1000 мл;
- скорость подвижной фазы. 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 250 нм для раствора сравнения (е) и
316 нм для всех остальных растворов;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания хинидина.
Идентификация пиков примесей: идентифици¬
руют пики примеси А и дигидрохинина, используя
хроматограмму раствора сравнения (а); иденти¬
фицируют пики хинидина и примеси С, используя
хроматограмму раствора сравнения (Ь); на хрома¬
тограмме раствора сравнения (с) присутствуют 4
пика, соответствующих хинидину, примеси А, при¬
меси С и дигидрохинину, которые идентифицируют
путем сравнения их времен удерживания с соответ¬
ствующими пиками на хроматограммах растворов
сравнения (а) и (Ь).
Относительное удерживание (по отношению к
примеси А): дигидрохинин — около 1,4.
Относительное удерживание (по отношению к
хинидину): примесь С — около 1,5.
Пригодность хроматографической системы:
- разрешение: не менее 3,0 между пиками при¬
меси А и хинидина и не менее 2,0 между пиками
примеси С и примеси А на хроматограмме раствора
сравнения (с);
- отношение сигнал/шум: не менее 4 для ос¬
новного пика на хроматограмме раствора сравне¬
ния (с!);
- коэффициент удерживания: не менее 3,5 и
не более 4,5 для пика примеси А на хроматограм¬
ме раствора сравнения (Ь), определяют с помо¬
щью пика тиомочевины на хроматограмме раствора
сравнения (е); при необходимости корректируют
концентрацию ацетонитрила в подвижной фазе.
Предельное содержание примесей:
- примесь С: не более 15%;
- любая примесь, элюирующаяся раньше хини¬
дина: не более 5 %;
- любая другая примесь: не более 2,5 %;
- неучитываемый предел (0,2 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хро¬
матограмме раствора сравнения (б).
Бор. Не более 0,0005 % (5 ррт). По возможно¬
сти стараются избегать использования стеклян¬
ной посуды.
Испытуемый раствор. 1,00 г испытуемого об¬
разца растворяют в смеси из 0,5 мл кислоты хлори¬
стоводородной Р и 4,0 мл воды Р.
Раствор сравнения. 0,572 г борной кислоты Р
растворяют в воде Р и доводят до объема 1000,0 мл
этим же растворителем. 5,0 мл полученного раст¬
вора доводят водой Р до объема 100,0 мл. К 1,0 мл
полученного раствора прибавляют 3,0 мл воды Р и
0,5 мл кислоты хлористоводородной Р.
Холостой раствор. К 0,5 мл кислоты хлори¬
стоводородной Р прибавляют 4,0 мл воды Р.
К испытуемому, холостому растворам и рас¬
твору сравнения прибавляют по 3,0 мл раствора
100 г/л 2-этилгексан-1,3-диола Р в метиленхлори-
де Р, встряхивают 1 мин и выдерживают в течение
6 мин. К 1,0 мл нижнего слоя прибавляют 2,0 мл
раствора 3,75 г/л куркумина Р в кислоте уксусной
безводной Р и 0,3 мл кислоты серной Р и пере¬
мешивают. Через 20 мин прибавляют 25,0 мл 96 %
спирта Р и перемешивают. Холостой раствор при¬
обретает желтую окраску. Красная окраска испытуе¬
мого раствора должна быть не интенсивнее окраски
раствора сравнения.
Потеря в массе при высушивании (2.2.32).
От 3,0 % до 5,0 %. 1,000 г испытуемого образца су¬
шат при температуре 130 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 20 мл
уксусного ангидрида Р и титруют 0,1 М раствором
кислоты хлорной, используя в качестве индикатора
0,15 мл раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 24,90 мг С40Н48М4О4-Н25О4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (Д)-[(25,45,5Д)-5-Этенил-1 -азабицикло[2.2.2]-
окт-2-ил](6-метоксихинолин-4-ил)метанол (хинин).
Хинина гидрохлорид
1057
В. (5)-[(2Я,45,5Я)-5-Этенил-1-азабицикло[2.2.2]-
окт-2-ил](хинолин-4-ил)метанол (цинхонин).
С. (5)-[(2Я45,5Я)-5-Этил-1 -азабицикло[2.2.2]окт-
2-ил](6-метоксихинолин-4-ил)метанол (дигидрохини-
дин).
07/2016:0018
ХИНИНА ГИДРОХЛОРИД
С/7 /17//71 Нус/госШопс/ит
ОШЫШЕ НУОКОСШ-ОКЮЕ
С20Н24М2°2 ’ НС1 ■ 2Н2° М м- 396>9
[6119-47-7]
ОПРЕДЕЛЕНИЕ
Моногидрохлориды алкалоидов, в пересчете на (РУ
[(25,45,5Я)-5-этенил-1-азабицикло[2.2.2]окт-2-ил](6-ме-
токсихинолин^4-ил)метанола] гидрохлорид дигидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые или бесцветные мелкие
шелковистые игольчатые кристаллы, часто собран¬
ные в кластеры.
Растворим в воде, легко растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 0,10 г ФСО хинина сульфа¬
та растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля С Р.
Подвижная фаза: диэтиламин Р — эфир Р —
толуол Р (10:24:40, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: дважды; не менее 15 см
от линии старта; после первого хроматографирования
пластинку сушат в потоке воздуха в течение 15 мин.
Высушивание: при температуре 105 °С в течение
30 мин и охлаждают.
Проявление: пластинку опрыскивают реактивом
йодплатината Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
B. Около 10 мг испытуемого образца растворяют
в воде Р и доводят до объема 10 мл этим же раство¬
рителем. К 5 мл полученного раствора прибавляют
0,2 мл бромной воды Р и 1 мл раствора аммиака раз¬
веденного Р2. Появляется зеленое окрашивание.
C. 0,1 г испытуемого образца растворяют в 3 мл
кислоты серной разведенной Р и доводят до объема
100 мл водой Р. Полученный раствор просматривают
в ультрафиолетовом свете при длине волны 366 нм.
Обнаруживается интенсивная голубая флуоресцен¬
ция, исчезающая почти полностью при добавлении
1 мл кислоты хлористоводородной Р.
й. Испытуемый образец дает реакции на хлори¬
ды (2.3.1).
Е. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 6,0 до 6,8. 10 мл раствора 3 до¬
водят водой, свободной от углерода диоксида, Р до
объема 20 мл.
Удельное оптическое вращение (2.2.7). От
-245 до -258 (в пересчете на сухое вещество). 0,500 г
испытуемого образца растворяют в 0,1 М растворе
кислоты хористоводородной Р и доводят до объема
25.0 мл этим же растворителем.
Другие хинные алкалоиды. Жидкостная хро¬
матография (2.2.29): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 5 мл подвижной фазы, при необ¬
ходимости слегка подогревая, и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО хинина суль¬
фата растворяют 5 мл подвижной фазы, при необ¬
ходимости слегка подогревая, и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 20 мг ФСО хинидина
сульфата (примесь А) растворяют 5 мл подвижной
фазы, при необходимости слегка подогревая, и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (с). К 1 мл раствора сравне¬
ния (а) прибавляют 1 мл раствора сравнения (Ь).
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 10,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 50,0 мл.
135. Зак. 1060.
1058
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (е). 10 мг тиомочевиныР
растворяют в подвижной фазе и доводят до объема
10 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной (0,15—0,25) м и внутренним
диаметром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р (размер частиц
(5—10) мкм);
- подвижная фаза: 6,8 г калия дигидрофосфа¬
та Р и 3,0 г гексиламина Р растворяют в 700 мл
воды Р, доводят кислотой фосфорной разведен¬
ной Р до рН 2,8, прибавляют 60 мл ацетонитрила Р
и доводят водой Р до объема 1000 мл;
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 250 нм для раствора сравнения (е) и 316 нм
для всех остальных растворов;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания хинина.
Идентификация пиков примесей: идентифи¬
цируют пики хинина и примеси С, используя хрома¬
тограмму раствора сравнения (а); идентифицируют
пики примеси А и дигидрохинидина, используя хрома¬
тограмму раствора сравнения (Ь);на хроматограмме
раствора сравнения (с) присутствует 4 пика, соответ¬
ствующих хинину, примеси А, примеси С и дигидрохи-
нидину, которые идентифицируют путем сравнения их
времен удерживания с соответствующими пиками на
хроматограммах растворов сравнения (а) и (Ь).
Относительное удерживание (по отношению к
хинину): примесь С — около 1,4.
Относительное удерживание (по отношению к
примеси А): дигидрохинидин — около 1,5.
Пригодность хроматографической системы:
- разрешение: не менее 3,0 между пиками хинина и
примеси А и не менее 2,0 между пиками дигидрохиниди¬
на и хинина на хроматограмме раствора сравнения (с);
- отношение сигнал/шум: не менее 4 для основ¬
ного пика на хроматограмме раствора сравнения (б);
- коэффициент удерживания: не менее 3,5 и не
более 4,5 для пика примеси А на хроматограмме раст¬
вора сравнения (Ь), *я< определяют с помощью пика
тиомочевины на хроматограмме раствора сравнения
(е); при необходимости корректируют концентрацию
ацетонитрила в подвижной фазе.
Предельное содержание примесей:
- примесь С: не более 10 %;
- любая примесь, элюирующаяся раньше хини¬
на: не более 5 %;
- любая другая примесь: не более 2,5 %;
- неучитываемый предел (0,2 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (б).
Сульфаты (2.4.13). Не более 0,05% (500 ррт).
Раствор 5 должен выдерживать испытание на сульфаты.
Барий. К 15 мл раствора 5 прибавляют 1 мл кис¬
лоты серной разведенной Р и выдерживают в течение
15 мин. Опалесценция полученного раствора должна
быть не интенсивнее опалесценции смеси, состоящей
из 15 мл раствора 3 и 1 мл воды дистиллированной Р.
Потеря в массе при высушивании (2.2.32). От
6,0 % до 10,0 %. 1,000 г испытуемого образца сушат
при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 50 мл
96 % спирта Р и прибавляют 5,0 мл 0,01 М кислоты
хлористоводородной. Титруют 0,1 М раствором на¬
трия гидроксида потенциометрически (2.2.20). Отме¬
чают объем титранта между двумя точками перегиба
на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 36,09 мг С20Н24М2О2 НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (3)-[(2/?,45,5/?)-5-Этенил-1-азабицикло[2.2.2]-
окт-2-ил](6-метоксихинолин-4-ил)метанол (хинидин).
В. (#)-[(2$,4$,5Я)-5-Этенил-1-азабицикло[2.2.2]-
окт-2-ил](хинолин-4-ил)метанол (цинхонидин).
С. (Я?)-[(25,45,5Я)-5-Этил-1-азабицикло[2.2.2]окт-
2-ил](6-метоксихинолин-4-ил)метанол (дигидрохинин).
07/2016:0019
ХИНИНА СУЛЬФАТ
СЛ/л/л/ зиНаз
ОС//Л//А/Е 51Л.РАТЕ
С^Н^О, • Н25 04 ■ 2Н20 М.м. 783
[6119-70-6]
Хинина сульфат
1059
ОПРЕДЕЛЕНИЕ
Моносульфаты алкалоидов, в пересчете на бис[(/?)-
[(2#,45,5Я)-5-этенил-1-азабицикло[2.2.2]окт-2-ил]-
(6-метоксихинолин-4-ил)метанола] сульфат дигидрат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или мелкие бесцветные игольчатые кристаллы.
Мало растворим в воде, умеренно растворим в
кипящей воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 0,10 г ФСО хинина сульфа¬
та растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: диэтиламин Р — эфир Р —
толуол Р (10:24:40, об/об/об).
Наносимый объем пробы. 5 мкл.
Фронт подвижной фазы: дважды; не менее 15 см
от линии старта; после первого хроматографирования
пластинку сушат в потоке воздуха в течение 15 мин.
Высушивание: при температуре 105 °С в течение
30 мин и охлаждают.
Проявление: пластинку опрыскивают реактивом
йодплатината Р.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
B. Около 5 мг испытуемого образца растворяют
в воде Р и доводят до объема 5 мл этим же раство¬
рителем. Прибавляют 0,2 мл бромной воды Р и 1 мл
раствора аммиака разведенного Р2. Появляется зе¬
леное окрашивание.
C. 0,1 г испытуемого образца растворяют в 3 мл
кислоты серной разведенной Р и доводят до объема
100 мл водой Р. Полученный раствор просматривают
в ультрафиолетовом свете при длине волны 366 нм.
Обнаруживается интенсивная голубая флуоресцен¬
ция, исчезающая почти полностью при добавлении
1 мл кислоты хлористоводородной Р.
й. Около 45 мг испытуемого образца растворяют
в 5 мл кислоты хлористоводородной разведенной Р.
Полученный раствор дает реакцию (а) на сульфаты
(2.3.1).
Е. Испытуемый образец выдерживает испытание
«рН», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 0,500 г испытуемого образца раство¬
ряют в 0,1 М растворе кислоты хлористоводород¬
ной Р и доводят до объема 25,0 мл этим же раство¬
рителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона <ЗУ(ЗЖ)6.
рН (2.2.3). От 5,7 до 6,6. Суспензия 10 г/л испы¬
туемого образца в воде Р должна выдерживать испы¬
тание на рН.
Удельное оптическое вращение (2.2.7). От
-237 до -245 (в пересчете на сухое вещество). Прово¬
дят испытание раствора 3.
Другие хинные алкалоиды. Жидкостная хро¬
матография (2.2.29): определение проводят методом
внутренней нормализации.
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 5 мл подвижной фазы, при необ¬
ходимости слегка подогревая, и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО хинина суль¬
фата растворяют 5 мл подвижной фазы, при необ¬
ходимости слегка подогревая, и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 20 мг ФСО хинидина
сульфата (примесь А) растворяют 5 мл подвижной
фазы, при необходимости слегка подогревая, и дово¬
дят до объема 10 мл этим же растворителем.
Раствор сравнения (с). К 1 мл раствора сравне¬
ния (а) прибавляют 1 мл раствора сравнения (Ь).
Раствор сравнения (д). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 10,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 50,0 мл.
Раствор сравнения (е). 10 мг тиомочевины Р
растворяют в подвижной фазе и доводят до объема
10 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной (0,15—0,25) м и внутренним
диаметром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р (размер частиц
(5—10)мкм);
- подвижная фаза: 6,8 г калия дигидрофосфа¬
та Р и 3,0 г гексиламина Р растворяют в 700 мл
воды Р, доводят кислотой фосфорной разведен¬
ной Р до рН 2,8, прибавляют 60 мл ацетонитрила Р
и доводят водой Р до объема 1000 мл;
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 250 нм для раствора сравнения (е) и 316 нм
для всех остальных растворов;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания хинина.
Идентификация пиков примесей: идентифи¬
цируют пики хинина и примеси С, используя хрома¬
тограмму раствора сравнения (а); идентифицируют
пики примеси А и дигидрохинидина, используя хрома¬
тограмму раствора сравнения (Ь);на хроматограмме
раствора сравнения (с) присутствует 4 пика, соответ¬
ствующих хинину, примеси А, примеси С и дигидрохи-
нидину, которые идентифицируют путем сравнения их
времен удерживания с соответствующими пиками на
хроматограммах растворов сравнения (а) и (Ь).
Относительное удерживание (по отношению к
хинину): примесь С — около 1,4.
Относительное удерживание (по отношению к
примеси А): дигидрохинидин — около 1,5.
Пригодность хроматографической системы:
- разрешение: не менее 3,0 между пиками хини¬
на и примеси А и не менее 2,0 между пиками диги¬
дрохинидина и хинина на хроматограмме раствора
сравнения (с);
- отношение сигнал/шум: не менее 4 для основ¬
ного пика на хроматограмме раствора сравнения (с1);
- коэффициент удерживания: не менее 3,5 и не
более 4,5 для пика примеси А на хроматограмме раст-
135*. Зак. 1060.
1060
Гэсударственная фармакопея Республики Беларусь
вора сравнения (Ь), 1Я> определяют с помощью пика
тиомочевины на хроматограмме раствора сравнения
(е); при необходимости корректируют концентрацию
ацетонитрила в подвижной фазе.
Предельное содержание примесей:
- примесь С: не более 10 %;
- любая примесь, элюирующаяся раньше хини¬
на: не более 5 %;
- любая другая примесь: не более 2,5 %;
- неучитываемый предел (0,2 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (б).
Потеря в массе при высушивании (2.2.32). От
3,0% до 5,0%. 1,000 г испытуемого образца сушат
при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в сме¬
си, состоящей из 10 мл хлороформа Р и 20 мл уксус¬
ного ангидрида Р, и титруют 0,1 М раствором кисло¬
ты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 24,90 мг С40Н481Ч4О4-Н25О4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (5)-[(2Я,45,5Я)-5-Этенил-1 -азабицикло[2.2.2]-
окт-2-ил](6-метоксихинолин-4-ил)метанол (хинидин).
В. (Я)-[(25,45,5Я)-5-Этенил-1-азабицикло[2.2.2]-
окт-2-ил](хинолин-4-ил)метанол (цинхонидин).
С. (Р)-[(28А5,бР)-5-Эт]АП-1-азаб]лиу\кло[2.2.2]окт-
2-ил](6-метоксихинолин-4-ил)метанол (дигидрохинин).
07/2016:0265
ХЛОРАЛГИДРАТ
СЫогаИ Ьудгаз
СЫ.ОКА1. НУйКАТЕ
С.Н,С1,02 М.м. 165,4
[302-17-0]
ОПРЕДЕЛЕНИЕ
2,2,2-Трихлорэтан-1,1-диол.
Содержание: не менее 98,5 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветные прозрачные кристаллы.
Очень легко растворим в воде, легко растворим
в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. К 10 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», прибавляют 2 мл раствора
натрия гидроксида разведенного Р. Раствор мутнеет,
при нагревании появляется запах хлороформа.
B. К 1 мл раствора 3 прибавляют 2 мл раствора
натрия сульфида Р. Появляется желтое окрашива¬
ние быстро переходящее в красновато-коричневое.
При выдерживании в течение небольшого промежут¬
ка времени может образоваться красный осадок.
ИСПЫТАНИЯ
Раствор 5. 3,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 30 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 3,5 до 5,5. Измеряют рН раствора 3.
Хлоралалкоголят. К 1,0 г испытуемого образ¬
ца прибавляют 10 мл раствора натрия гидроксида
разведенного Р и нагревают. Надосадочный раствор
фильтруют, к фильтрату прибавляют по каплям 0,05 М
раствор йода до появления желтого окрашивания.
Выдерживают в течение 1 ч. Не должен образовы¬
ваться осадок.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
5 мл раствора 3 доводят водой Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более
0,0020 % (20 ррт). 10 мл раствора 3 доводят водой Р
до объема 20 мл. Полученный раствор должен выдер¬
живать испытание на тяжелые металлы. Эталон гото¬
вят с использованием эталонного раствора свинца
(1 ррт РЬ) Р.
Нелетучий остаток. Не более 0,1 %. 2,000 г ис¬
пытуемого образца выпаривают на водяной бане.
Масса полученного остатка не должна превышать 2 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
4,000 г испытуемого образца растворяют в 10 мл
воды Р и прибавляют 40,0 мл 1 М раствора натрия
Хлорамфеникол
1061
гидроксида. Выдерживают точно 2 мин и титруют
0,5 М раствором кислоты серной, используя в каче¬
стве индикатора 0,1 мл раствора фенолфталеина Р.
Нейтрализованный раствор титруют 0,1 М раствором
серебра нитрата, используя в качестве индикатора
0,2 мл раствора калия хромата Р. Объем 1 М раст¬
вора натрия гидроксида, израсходованного на титро¬
вание, рассчитывают путем вычитания объема 0,5 М
раствора кислоты серной, израсходованного в 1-м
титровании, и двух пятнадцатых объема 0,1 М раст¬
вора серебра нитрата, израсходованного во 2-м ти¬
тровании, из объема 1 М раствора натрия гидрокси¬
да, прибавленного до титрования.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 0,1654 г С2Н3С1302.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере #в защи¬
щенном от света месте#.
07/2016:0071
ХЛОРАМФЕНИКОЛ
СЫогатр1пеп1со1ит
СНШКАМРНЕЫ1С01.
[56-75-7]
ОПРЕДЕЛЕНИЕ
2,2-Дихлор-Л/-[(1#,2Я)-2-гидрокси-1-(гидроксиме-
тил)-2-(4-нитрофенил)этил]ацетамид. #Левомицетин#.
Субстанция продуцируется некоторыми штам¬
мами 31гер1отусе8 уепегие!ае в подходящей среде.
Обычно получают синтетическим путем.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый, серовато-белый или желтовато-белый
мелкий кристаллический порошок или мелкие кри¬
сталлы, игольчатые или вытянутые пластинки.
Мало растворим в воде, легко растворим в 96 %
спирте и в пропиленгликоле.
Раствор в этаноле является правовращающим, а
раствор в этилацетате — левовращающим.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О, Е.
A. Температура плавления (2.2.14): от 149 °С до
153 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО хлорамфеникола #или спектр,
представленный на рисунке #0071.-1#.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме с 1 мкл испыту¬
емого раствора обнаруживается основное пятно, со¬
ответствующее по расположению и размеру основно¬
му пятну на хроматограмме раствора сравнения (а).
О. 10 мг испытуемого образца растворяют в 1 мл
спирта (50 %, об/об) Р, прибавляют 3 мл раствора
10 г/л кальция хлорида Р, 50 мг порошка цинка Р и
нагревают на водяной бане в течение 10 мин. Горячий
1062
Гэсударственная фармакопея Республики Беларусь
раствор фильтруют и охлаждают. Прибавляют 0,1 мл
бензоилхлорида Р и встряхивают в течение 1 мин.
Прибавляют 0,5 мл раствора железа (III) хлорида Р1
и 2 мл хлороформа Р и встряхивают. В водном слое
появляется окраска от светлой фиолетово-красной до
красно-фиолетовой.
Е. 50 мг испытуемого образца помещают в фар¬
форовый тигель, прибавляют 0,5 г натрия карбоната
безводного Р, нагревают на открытом огне в течение
10 мин и охлаждают. Осадок смывают 5 мл кислоты
азотной разведенной Р и фильтруют. К 1 мл филь¬
трата прибавляют 1 мл воды Р. Полученный раствор
дает реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Кислотность или щелочность. К 0,1 г испыту¬
емого образца прибавляют 20 мл воды, свободной
от углерода диоксида, Р, встряхивают и прибавляют
0,1 мл раствора бромтимолового синего Р1. При при¬
бавлении не более 0,1 мл 0,02 М раствора кислоты
хлористоводородной или 0,02 М раствора натрия ги¬
дроксида окраска раствора должна измениться.
Удельное оптическое вращение (2.2.7). От
+18,5 до +20,5. 1,50 г испытуемого образца раство¬
ряют в этаноле Р и доводят до объема 25,0 мл этим
же растворителем.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 0,10 г ФСО хлорамфени-
кола растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 0,5 мл раствора сравне¬
ния (а) доводят ацетоном Р до объема 100 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
0=254 Р-
Подвижная фаза: вода Р — метанол Р — хлоро¬
форм Р (1:10:90, об/об/об).
Наносимый объем пробы: по 1 мкл испытуемого
раствора и раствора сравнения (а) и по 20 мкл испы¬
туемого раствора и раствора сравнения (Ь).
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме с 20 мкл испытуемого раствора любое пятно,
кроме основного, должно быть не интенсивнее основ¬
ного пятна на хроматограмме раствора сравнения (Ь).
Хлориды (2.2.4). Не более 0,0100 % (100 ррт). К
1,00 г испытуемого образца прибавляют 20 мл воды Р
и 10 мл кислоты азотной Р и встряхивают в тече¬
ние 5 мин. Фильтруют через фильтровальную бума¬
гу, предварительно промытую водой Р, порциями по
5 мл до тех пор, пока 5 мл фильтрата не перестанут
опалесцировать после прибавления 0,1 мл кислоты
азотной Р и 0,1 мл раствора серебра нитрата Р1.
15 мл полученного фильтрата должны выдерживать
испытание на хлориды.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 2,0 г испытуемого образца.
Пирогенность (2.6.8). Субстанция должна вы¬
держивать испытание на пирогенны, если предназна¬
чена для производства лекарственных средств для
парентерального применения без последующей про¬
цедуры удаления пирогенов. Готовят раствор 2 мг/мл
испытуемого образца. Тест-доза — 2,5 мл полученно¬
го раствора на 1 кг массы тела кролика.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в
воде Р и доводят до объема 500,0 мл этим же раст¬
ворителем. 10,0 мл полученного раствора доводят
водой Р до объема 100,0 мл. Измеряют оптическую
плотность (2.2.25) полученного раствора в максимуме
при 278 нм.
Содержание С1(1Н12С12М205 рассчитывают с уче¬
том удельного показателя поглощения, равного 297.
ХРАНЕНИЕ
В защищенном от света месте.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
07/2016:0473
ХЛОРАМФЕНИКОЛА ПАЛЬМИТАТ
СЫогатрЬепюоН ра1тНаз
СНЮРАМРНЕШСО/. РАШ1ТАТЕ
С27Н42С12Ы2Ов М.м. 561,6
[530-43-8]
ОПРЕДЕЛЕНИЕ
(2Я,ЗЯ)-2-[(Дихлорацетил)амино]-3-гидрокси-3-
(4-нитрофенил)пропилгексадеканоат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Мелкий, маслянистый порошок белого или почти
белого цвета.
Практически нерастворим в воде, легко раство¬
рим в ацетоне, умеренно растворим в 96 % спирте,
очень мало растворим в гексане.
Температура плавления: от 87 °С до 95 °С.
Обладает полиморфизмом (5.9). Термодинами¬
чески устойчивая форма обладает низкой биодоступ¬
ностью при пероральном применении.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в смеси из 1 мл 1 М раствора на-
Хлорамфеникола пальмитат
1063
трия гидроксида и 5 мл ацетона Р и выдерживают
в течение 30 мин. Прибавляют 1,1 мл 1 М раствора
кислоты хлористоводородной и 3 мл ацетона Р.
Раствор сравнения (а). 10 мг ФСО хлорамфени¬
кола растворяют в ацетоне Р и доводят до объема
5 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг кислоты пальми¬
тиновой Р растворяют в ацетоне Р и доводят до объ¬
ема 5 мл этим же растворителем.
Раствор сравнения (с). 10 мг испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р.
Подвижная фаза: раствор 100 г/л аммония аце¬
тата Р — 96% спирт Р (30:70, об/об).
Наносимый объем пробы: по 4 мкл каждого раст¬
вора.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором,
содержащим 0,2 г/л дихлорфлуоресцеина Р и 0,1 г/л
родамина В Р в 96% спирте Р, сушат на воздухе и
посматривают в ультрафиолетовом свете при длине
волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются три пятна, соответствую¬
щие по расположению основным пятнам на хромато¬
граммах растворов сравнения (а), (Ь) и (с).
B. 0,2 г испытуемого образца растворяют в 2 мл
пиридина Р, прибавляют 2 мл раствора 100 г/л калия
гидроксида Р и нагревают на водяной бане. Появля¬
ется красное окрашивание.
C. Около 10 мг испытуемого образца растворяют
в 5 мл 96 % спирта Р, прибавляют 4,5 мл кислоты
серной разведенной Р и 50 м г цинка порошка Р, вы¬
держивают в течение 10 мин и, при необходимости,
отделяют надосадочную жидкость или фильтруют.
Полученный раствор охлаждают в ледяной воде,
прибавляют 0,5 мл раствора натрия нитрита Р.
Выдерживают в течение 2 мин и прибавляют 1 г мо¬
чевины Р, 2 мл раствора натрия гидроксида концен¬
трированного Р, 1 мл раствора р-нафтола Р. Появ¬
ляется красное окрашивание.
ИСПЫТАНИЯ
Кислотность. 1,0 г испытуемого образца раст¬
воряют в 5 мл смеси из равных объемов 96 % спир¬
та Р и эфира Р, нагревают до температуры 35 °С и
прибавляют 0,2 мл раствора фенолфталеина Р. При
прибавлении не более 0,4 мл 0,1 М раствора натрия
гидроксида окраска раствора должна измениться на
розовую и сохраняться в течение 30 с.
Удельное оптическое вращение (2.2.7). От
+22,5 до +25,5. 1,25 г испытуемого образца раство¬
ряют в этаноле безводном Р и доводят до объема
25,0 мл этим же растворителем.
Свободный хлорамфеникол. Не более
450 ррт. 1,0 г испытуемого образца растворяют в
80 мл ксилола Р при слабом нагревании, охлаждают
и трижды встряхивают с порциями воды Р, каждая
по 15мл. Объединенные водные экстракты доводят
водой Р до объема 50 мл и встряхивают с 10 мл то¬
луола Р. Отделяют и отбрасывают слой с толуолом.
Часть водного слоя центрифугируют и измеряют оп¬
тическую плотность А (2.2.25) в максимуме при длине
волны 278 нм, используя в качестве компенсационно¬
го раствора контрольный раствор с оптической плот¬
ностью не более 0,05.
Содержание свободного хлорамфеникола в ча¬
стях на миллион частей (ррт) рассчитывают по фор¬
муле:
Л-104
5,96 '
Сопутствующие примеси. Тонкослойная хрома¬
тография* (2.2.27).
Испытуемый раствор. 0,1 г испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 20 мг ФСО хлорамфени¬
кола пальмитата изомера растворяют в ацетоне Р
и доводят до объема 10 мл этим же растворителем.
1 мл полученного раствора доводят ацетоном Р до
объема 10 мл.
Раствор сравнения (Ь). 20 мг ФСО хлорамфени¬
кола дипальмитата растворяют в ацетоне Р и дово¬
дят до объема 10 мл этим же растворителем. 1 мл по¬
лученного раствора доводят ацетоном Р до объема
10 мл.
Раствор сравнения (с). 5 мг ФСО хлорамфени¬
кола растворяют в ацетоне Р и доводят до объема
10 мл этим же растворителем. 1 мл полученного раст¬
вора доводят ацетоном Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикагеля
Подвижная фаза: метанол Р — хлороформ Р —
циклогексан Р (10:40:50, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Предельное содержание примесей:
- хлорамфеникола пальмитата изомер и хло¬
рамфеникола дипальмитат (не более 2,0 %): на
хроматограмме испытуемого раствора пятна, соот¬
ветствующие хлорамфеникола пальмитата изомеру
и хлорамфеникола дипальмитату, должны быть не
интенсивнее соответствующих пятен на хроматограм¬
мах растворов сравнения (а) и (Ь);
- любая другая примесь (не более 0,5 %): на хро¬
матограмме испытуемого раствора любое пятно, кро¬
ме основного и пятен, соответствующих хлорамфени¬
кола пальмитата изомеру и хлорамфеникола дипаль¬
митату, должно быть не интенсивнее основного пятна
на хроматограмме раствора сравнения (с).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат над
фосфора (V) оксидом Р при температуре 80 °С и дав¬
лении, не превышающем 0,1 кПа, в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
90,0 мг испытуемого образца растворяют в 96%
спирте Р и доводят до объема 100,0 мл этим же раст¬
ворителем. 10,0 мл полученного раствора доводят
96 % спиртом Р до объема 250,0 мл. Измеряют опти-
1064
Гэсударственная фармакопея Республики Беларусь
ческую плотность (2.2.25) полученного раствора при
длине волны 271 нм.
Содержание С27Н42С121Ч206 рассчитывают с уче¬
том удельного показателя поглощения, равного 178.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (1 Р,2Р)-2-[(Дихлорацетил)амино]-3-гидрокси-1 -
(4-нитрофенил)пропилгексадеканоат (изомер хлорам-
феникола пальмитата).
В. (1Р,2Р)-2-[(Дихлорацетил)амино]-1-(4-нитрофе-
нил)пропан-1,3-диилбисгексадеканоат (хлорамфени-
кола пальмитат).
07/2016:0382
ХЛОРБУТАНОЛ БЕЗВОДНЫЙ
СШогоЬи1апо1ит апЪудпсит
сн1-01ЮвитАыо1., Аынгокоиз
н3с он
Н3С^Ч'''СС13
С4Н7С1,0 М.м. 177,5
[57-15-8]
ОПРЕДЕЛЕНИЕ
1,1,1 -Трихлор-2-метилпропан-2-ол.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Легко сублимируется.
Мало растворим в воде, очень легко растворим в
96 % спирте, растворим в глицерине (85 %).
Температура плавления: около 95 °С (без пред¬
варительного высушивания).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Около 20 мг испытуемого образца прибавляют
к смеси из 1 мл пиридина Р и 2 мл раствора натрия
гидроксида концентрированного Р. Нагревают в во¬
дяной бане, встряхивают и выдерживают. В пиридино¬
вом слое появляется красное окрашивание.
B. Около 20 мг испытуемого образца прибавляют
к 5 мл раствора серебра нитрата аммиачного Р и
слегка нагревают. Образуется черный осадок.
С. К 20 мг испытуемого образца прибавляют 3 мл
1 М раствора натрия гидроксида и встряхивают до
растворения. Прибавляют 5 мл воды Р и затем мед¬
ленно прибавляют 2 мл раствора калия йодида йоди¬
рованного Р. Образуется желтоватый осадок.
й. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 3. 5 г испытуемого образца растворяют
в 96% спирте Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)5.
Кислотность. К 4 мл раствора 5 прибавляют
15 мл 96% спирта Р и 0,1 мл раствора бромтимо-
лового синего Р1. При прибавлении не более 1,0 мл
0,01 М раствора натрия гидроксида окрашивание
индикатора должно измениться на синее.
Хлориды (2.4.4). Не более 0,0300 % (300 ррт).
0,17 г испытуемого образца растворяют в 5 мл 96 %
спирта Р и доводят водой Р до объема 15 мл. При
приготовлении эталона вместо 5 мл воды Р использу¬
ют 5 мл 96 % спирта Р.
Вода (2.5.12). Не более 1,0 %. Определение про¬
водят из 2,00 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 20 мл
96 % спирта Р. Прибавляют 10 мл раствора натрия
гидроксида разведенного Р, нагревают в водяной
бане в течение 5 мин и охлаждают. Прибавляют 20 мл
кислоты азотной разведенной Р, 25,0 мл 0,1 М раст¬
вора серебра нитрата и 2 мл дибутилфталата Р и
энергично встряхивают. Прибавляют 2 мл раствора
железа (!!!) аммония сульфата Р2 и титруют 0,1 М
раствором аммония тиоцианата Р до появления
оранжевого окрашивания.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,92 мг С4Н7С130.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0383
ХЛОРБУТАНОЛ ГЕМИГИДРАТ
СЫогоЬи(апо1ит ^етШудп'сит
сниоковитАысп_ немшуокате
н3с он
3/ 0,5 Н20
Н3СГ ХС13
С4Н7С1,0 ■ 0,5 Н20 М.м. 186,5
[6001-64-5]
ОПРЕДЕЛЕНИЕ
1,1,1 -Трихлор-2-метилпропан-2-ол гемигидрат.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на безводное вещество).
Хлоргексидина диацетат
1065
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы. Легко сублимируется.
Мало растворим в воде, очень легко растворим в
96 % спирте, растворим в глицерине (85 %).
Температура плавления: около 78 °С (без пред¬
варительной сушки).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Около 20 мг испытуемого образца прибавляют
к смеси из 1 мл пиридина Р и 2 мл раствора натрия
гидроксида концентрированного Р. Нагревают на во¬
дяной бане, встряхивают и выдерживают. В пиридино¬
вом слое появляется красное окрашивание.
B. Около 20 мг испытуемого образца прибавляют
к 5 мл раствора серебра нитрата аммиачного Р и
слегка нагревают. Образуется черный осадок.
C. К 20 мг испытуемого образца прибавляют 3 мл
1 М раствора натрия гидроксида и встряхивают до
растворения. Прибавляют 5 мл воды Р и затем мед¬
ленно прибавляют 2 мл раствора калия йодида йоди¬
рованного Р. Образуется желтоватый осадок.
О. Испытуемый образец выдерживает испытание
«Вода», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Раствор 5. 5 г испытуемого образца растворяют
в 96 % спирте Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)5.
Кислотность. К 4 мл раствора 3 прибавляют
15 мл 96% спирта Р и 0,1 мл раствора бром тимо¬
лового синего Р1. При прибавлении не более 1,0 мл
0,01 М раствора натрия гидроксида окрашивание
индикатора должно измениться на синее.
Хлориды (2.4.4). Не более 0,0100 % (100 ррт).
К 1 мл раствора 3 прибавляют 4 мл 96% спирта Р и
доводят водой Р до объема 15 мл. При приготовлении
эталона вместо 5 мл воды Р используют 5 мл 96%
спирта Р.
Вода (2.5.12). Не менее 4,5 % и не более 5,5 %.
Определение проводят из 0,300 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 20 мл
96 % спирта Р. Прибавляют 10 мл раствора натрия
гидроксида разведенного Р, нагревают в водяной
бане в течение 5 мин и охлаждают. Прибавляют 20 мл
кислоты азотной разведенной Р, 25,0 мл 0,1 М раст¬
вора серебра нитрата и 2 мл дибутилфталата Р и
энергично встряхивают. Прибавляют 2 мл раствора
железа (III) аммония сульфата Р2 и титруют 0,1 М
раствором аммония тиоцианата Р до появления
оранжевого окрашивания.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 5,92 мг С4Н7С1эО.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:0657
ХЛОРГЕКСИДИНА ДИАЦЕТАТ
СЫогНех1сНт' сНасеЫз
СН1.ОКНЕХЮШЕ Р1АСЕТАТЕ
[56-95-1]
ОПРЕДЕЛЕНИЕ
1,1 '-(Гексан-1,6-диил)бис[5-(4-хлорфенил)бигуа-
нида] диацетат.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый микрокристаллический
порошок.
Умеренно растворим в воде, растворим в 96 %
спирте, мало растворим в глицерине и пропиленгли-
коле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО хлоргексидина диацетата.
B. Около 5 мг испытуемого образца растворяют
в 5 мл теплого раствора 10 г/л цетримида Р и при¬
бавляют 1 мл раствора натрия гидроксида концен¬
трированного Р и 1 мл воды бромной Р. Появляется
темно-красное окрашивание.
C. 0,3 г испытуемого образца растворяют в 10 мл
смеси из равных объемов кислоты хлористоводо¬
родной Р и воды Р. К полученному раствору прибав¬
ляют 40 мл воды Р, фильтруют при необходимости
и охлаждают в ледяной бане. Полученный раствор
подщелачивают по бумаге титанового желтого Р,
прибавляя по каплям при перемешивании раствор
натрия гидроксида концентрированный Р, и при¬
бавляют 1 мл избытка. Фильтруют, осадок промывают
водой Р до отсутствия щелочи в промывных водах,
перекристаллизовывают из спирта (70 %, об/об) Р и
сушат при температуре от 100 °С до 105 °С. Темпера¬
тура плавления (2.2.14) полученного осадка от 132 °С
до 136 °С.
О. Испытуемый образец дает реакцию (а) на аце¬
таты (2.3.1).
ИСПЫТАНИЯ
Примесь Р (хлоранилин). Не более 0,0500 %
(500 ррт).
Испытуемый раствор. 0,20 г испытуемого об¬
разца растворяют в 25 мл воды Р, встряхивая при не¬
обходимости. К полученному раствору прибавляют 1 мл
кислоты хлористоводородной Р и доводят водой Р до
\
1066
Гэсударственная фармакопея Республики Беларусь
объема 30 мл. Быстро прибавляют, тщательно пере¬
мешивая после каждого прибавления: 5 мл раствора
103 г/л кислоты хлористоводородной Р, 0,35 мл раст¬
вора натрия нитрита Р, 2 мл раствора 50 г/л аммония
сульфамата Р, 5 мл раствора 1,0 г/л нафтилэтилен-
диамина дигидрохлорида Р и 1 мл 96 % спирта Р, коли¬
чественно переносят в мерную колбу, доводят водой Р
до объема 50,0 мл и выдерживают в течение 30 мин.
Растворы сравнения. Готовят растворы сравне¬
ния, содержащие соответственно 50 ррт, 100 ррт,
200 ррт, 500 ррт и 600 ррт хлоранилина Р (при¬
месь Р) в испытуемом образце, следующим образом:
1,0 мл, 2,0 мл, 4,0 мл, 10,0 мл и 12,0 мл раствора
0,010 г/л хлоранилина Р (примесь Р) в кислоте хло¬
ристоводородной разведенной Р доводят водой Р
до объема 20 мл. Затем прибавляют 10 мл воды Р
Быстро прибавляют, тщательно перемешивая после
каждого прибавления: 5 мл раствора 103 г/л кислоты
хлористоводородной Р, 0,35 мл раствора натрия
нитрита Р, 2 мл раствора 50 г/л аммония сульфама¬
та Р, 5 мл раствора 1 г/л нафтилэтилендиамина ди¬
гидрохлорида Р и 1 мл 96 % спирта Р; количественно
переносят в мерную колбу, доводят водой Р до объ¬
ема 50,0 мл и выдерживают в течение 30 мин.
Измеряют оптическую плотность (2.2.25) каждого
раствора сравнения при длине волны 556 нм и строят
градуировочный график.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при длине волны 556 нм. Определяют
концентрацию хлоранилина, используя градуировоч¬
ный график.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы хранят при темпера¬
туре, не превышающей 12 °С.
Испытуемый раствор. 0,140 г испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). Содержимое контейнера
с ФСО хлоргексидина для проверки пригодности хро¬
матографической системы (содержит примеси А, В,
Н, I, К, N и О) растворяют в 1,0 мл подвижной фазы А.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
-подвижная фаза А: 0,1% (об/об) раствор
кислоты трифторуксусной Р в ацетонитри¬
ле Р — 0,1 % (об/об) раствор кислоты трифто¬
руксусной Р в воде Р (20:80, об/об);
-подвижная фаза В: 0,1% (об/об) раствор
кислоты трифторуксусной Р в воде Р — 0,1 %
(об/об) раствор кислоты трифторуксусной Р в
ацетонитриле Р (10:90, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
100
0
2—32
100 — 80
0 — 20
32—37
80
20
37-47
80 — 70
20 — 30
47—54
70
30
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, Н, I, К, N и О, используя хро¬
матограмму раствора сравнения (а) и хроматограмму,
прилагаемую к ФСО хлоргексидина для проверки при¬
годности хроматографической системы.
Относительное удерживание (по отношению к
хлоргексидину, время удерживания — около 35 мин):
примесь N — около 0,35; примесь В — около 0,36;
примесь А — около 0,6; примесь Н — около 0,85; при¬
месь О — около 0,90; примесь I — около 0,91; при¬
месь К — около 1,4.
Пригодность хроматографической системы:
раствор сравнения (а):
- коэффициёнт разделения пиков: не менее 2,0
(Нр — высота пика примеси В относительно базовой
линии; Ну — расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси В и пик
примеси !Ч).
Предельное содержание примесей:
- сумма примесей I и О (не более 0,4 %): на хро¬
матограмме испытуемого раствора сумма площадей
пиков примесей I и О не должна превышать 0,4-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- примесь К (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси К, не должна превышать 0,3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, Н и N (не более 0,15 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, Н и 14, не должны превы¬
шать 0,15-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, Н, I, К, N и О, не должна превышать 0,1-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- сумма примесей (не более 0,8 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
0,8-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,05 площади основного пика на хроматограм¬
ме раствора сравнения (Ь) (0,05 %).
Потеря в массе при высушивании (2.2.32). Не
более 3,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,15%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,140 г испытуемого образца растворяют в
100 мл кислоты уксусной безводной Р и титруют
0,1 М раствором кислоты хлорной потенциометри-
чески (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 15,64 мг С22Н30С121М10*2С2Н4О2.
I
Хлоргексидина диглюконата раствор
1067
ПРИМЕСИ
Специфицированные примеси: А, Н, I, К, Л/, О, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, Е, Р, С, М.
А. 1 -(4-Хлорфенил)-5-[6-[(цианокарбамимидоил)-
амино]гексил]бигуанид.
В. 1 -[[6-[[[(4-Хлорфенил)карбамимидоил]карбами-
мидоил]амино]гексил]карбамимидоил]мочевина.
Е. 1-(4-Хлорфенил)гуанидин.
Р. 1-(4-Хлорфенил)мочевина.
С. 1 -(6-Аминогексил)-5-(4-хлорфенил)бигуанид.
H. 1,1 -[Иминобис(карбонимидоилиминогексан-6,1-
диил)]бис[5-(4-хлорфенил)бигуанид].
I. Структура не установлена.
К. 1 -(4-Хлорфенил)-3-[[6-[[[(4-хлорфенил)карба-
мимидоил]карбамимидоил]амино]гексил]карбамими-
доил]мочевина.
М. 5-(4-Хлорфенил}-5'-фенил-1,1 '-(гексан-1,6-диил)-
дибигуанид.
N. 1-[6-(Карбамимидоиламино)гексил]-5-(4-
хлорфенил)бигуанид.
О. 5-(2-Хлорфенил)-5,-(4-хлорфенил)-1,1'-(гексан-
1,6-диил)дибигуанид.
С!\
Р. 4-Хлоранилин.
мн2
07/2016:0658
ХЛОРГЕКСИДИНА ДИГЛЮКОНАТА
РАСТВОР
С/7/ог/7ех/с//п/ сНд1исопаНз 8о1иИо
СШ-ОКНЕХЮШЕ 01СШС0ИАТЕ
80ШТЮЫ
1068
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
Водный раствор 1,1'-(гексан-1,6-диил)бис[5-(4-
хлорфенил)бигуанида] ди-Э-глюконата.
Содержание: не менее 190 г/л и не более 210 г/л.
ОПИСАНИЕ (СВОЙСТВА)
Почти бесцветная или бледно-желтоватая жидкость.
Смешивается с водой, с не более 3 частями
ацетона и с не более 5 частями 96 % спирта.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: к 1 мл испытуемого образца
прибавляют 40 мл воды Р, охлаждают в ледяной
бане, подщелачивают при перемешивании по каплям
растворомнатриягидроксидаконцентрированным Р
с использованием бумаги титанового желтого Р и
прибавляют еще 1 мл раствора натрия гидроксида
концентрированного Р. Фильтруют, осадок промыва¬
ют водой Р до исчезновения щелочной реакции
промывной воды, перекристаллизовывают из спирта
(70 %, об/об) Р и сушат при температуре от 100 °С до
105 °С. Используют сухой остаток.
Сравнение: ФСО хлоргексидина #или спектр,
представленный на рисунке #0658.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10,0 мл испытуемого об¬
разца доводят водой Р до объема 50 мл.
Раствор сравнения. 25 мг ФСО кальция глюко¬
ната растворяют в 1 мл воды Р.
Пластинка: ТСХпластинка со слоем силикагеля Р.
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетат Р — вода Р — 96%
спирт Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 1/2 высоты
пластинки.
Высушивание: при температуре 100 °С в течение
20 мин и охлаждают.
Проявление: пластинку опрыскивают раствором
25 г/л аммония молибдата Р и 10 г/л церия (IV) суль¬
фата Р в кислоте серной разведенной Р и нагревают
при температуре 110 °С в течение 10 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
С. К 1 мл испытуемого образца прибавляют
40 мл воды Р, охлаждают в ледяной бане, подщела¬
чивают при перемешивании по каплям раствором на¬
трия гидроксида концентрированным Р с использо¬
ванием бумаги титанового желтого Р и прибавляют
еще 1 мл раствора натрия гидроксида концентри¬
рованного Р. Фильтруют, осадок промывают водой Р
до исчезновения щелочной реакции промывной воды,
перекристаллизовывают из спирта (70 %, об/об) Р и
сушат при температуре от 100 °С до 105 °С. Темпе¬
ратура плавления (2.2.14) полученного остатка: от
132 °С до 136 °С.
О. К 0,05 мл испытуемого образца прибавляют
5 мл раствора 10 г/л цетримида Р, 1 мл раствора
натрия гидроксида концентрированного Р и 1 мл
бромной воды Р. Появляется интенсивное красное
окрашивание.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 1,06 до
1,07.
рН (2.2.3). От 5,5 до 7,0. 5,0 мл испытуемого об¬
разца доводят водой, свободной от углерода диокси¬
да, Р до объема 100 мл.
Рисунок #0658.-1. Инфракрасный спектр ФСО хлоргексидина
Хлоргексидина диглюконата раствор
1069
Примесь Р (хлоранилин). Не более 0,0500 %
(500 ррт) в пересчете на раствор хлоргексидина ди-
глюконата,
Испытуемый раствор. 0,20 г испытуемого об¬
разца доводят водой Р до объема 30 мл. Прибав¬
ляют быстро, перемешивая после каждого прибав¬
ления: 5 мл раствора 103 г/л кислоты хлористово¬
дородной Р, 0,35 мл раствора натрия нитрита Р,
2 мл раствора 50 г/л аммония сульфамата Р, 5 мл
раствора 1 г/л нафтилэтилендиамина дигидрох¬
лорида Р, 1 мл 96 % спирта Р. Переносят количе¬
ственно в мерную колбу, доводят водой Р до объема
50.0 мл и выдерживают в течение 30 мин.
Растворы сравнения. Растворы сравнения с
концентрацией хлоранилина Р (примесь Р) в испы¬
туемом образце 50 ррт, 100 ррт, 200 ррт, 500 ррт
и 600 ррт соответственно готовят следующим обра¬
зом: 1,0 мл, 2,0 мл, 4,0 мл, 10,0 мл и 12,0 мл раст¬
вора 0,010 г/л хлорамина Р (примесь Р) в кислоте
хлористоводородной разведенной Р доводят во¬
дой Р до объема 20 мл. Затем прибавляют 10 мл
воды Р. Прибавляют быстро, перемешивая после
каждого прибавления: 5 мл раствора 103 г/л кис¬
лоты хлористоводородной Р, 0,35 мл раствора
натрия нитрита Р, 2 мл раствора 50 г/л аммония
сульфамата Р, 5 мл раствора 1 г/л нафтилэтилен¬
диамина дигидрохлорида Р, 1 мл 96 % спирта Р.
Каждые раствор переносят количественно в мерную
колбу, доводят водой Р до объема 50,0 мл и выдер¬
живают в течение 30 мин.
Измеряют оптическую плотность (2.2.25) каж¬
дого раствора сравнения при длине волны 556 нм и
строят калибровочную кривую.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора при длине волны 556 нм. Опреде¬
ляют концентрацию хлоранилина с помощью кали¬
бровочной кривой.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы хранят при темпера¬
туре, не превышающей 12 °С.
Испытуемый раствор. 1,0 мл испытуемого
образца доводят подвижной фазой А до объема
100.0 мл.
Раствор сравнения (а). Содержимое контейне¬
ра с ФСО хлоргексидина для проверки пригодности
хроматографической системы (содержит примеси
А, В, Р, С, Н, I, Л, К, Ц N и О) растворяют в 1,0 мл
подвижной фазы А.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят подвижной фазой А до объема
100.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным эндкепированным для хроматогра¬
фии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: смесь 0,1 % (об/об) раст¬
вор трифторуксусной кислоты Р в ацетони¬
триле Р — 0,1 % (об/об) раствор трифторук¬
сусной кислоты Р в воде Р (20:80, об/об)]
- подвижная фаза В: смесь 0,1 % (об/об) раст¬
вор трифторуксусной кислоты Р в воде Р —
0,1 % (об/об) раствор трифторуксусной кисло¬
ты Р в ацетонитриле Р (10:90, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
100
0
2—32
100 —> 80
о
ом
т
о
32—37
80
20
37-47
о
1^
т
о
00
о
со
т
о
ом
47—54
70
30
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Идентификация пиков примесей: идентифи¬
цируют пики примесей А, В, Р, С, Н, I, 3, К, Ц N и О,
используя хроматограмму раствора сравнения (а) и
хроматограмму, прилагаемую к ФСО хлоргексидина
для проверки пригодности хроматографической си¬
стемы.
Относительное удерживание (по отношению к
хлоргексидину, время удерживания — около 35 мин):
примесь I — около 0,23; примесь О — около 0,24; при¬
месь (3 — около 0,25; примесь N — около 0,35; при¬
месь В — около 0,36; примесь Р — около 0,5; примесь
А— около 0,6; примесь Н — около 0,85; примесь О —
около 0,90; примесь I — около 0,91; примесь Л — око¬
ло 0,96; примесь К — около 1,4.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 3,0 между пиками при¬
месей 1_ и <3;
- коэффициент разделения пиков: не менее 2,0
(Нр — высота пика примеси В относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси В и пик
примеси N1).
Предельное содержание примесей:
- примесь N (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси N. не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (Ь);
- примесь Н (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси Н, не должна превышать 0,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси А, ^ и К (не более 0,4 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, 3 и К, не должны превы¬
шать 0,4-кратную площадь основного пика на хрома¬
тограмме раствора сравнения (Ь);
- сумма примесей I и О (не более 0,4 %): на хро¬
матограмме испытуемого раствора сумма площадей
пиков примесей I и О не должна превышать 0,4-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- примесь О (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси С, не должна превышать 0,3-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- примеси В, Р, /_ и О (не более 0,2 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, Р, I. и О, не должны
превышать 0,2-кратную площадь основного пика на
хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
1070
Гэсударственная фармакопея Республики Беларусь
площадь любого пика, кроме основного и пиков при¬
месей А, В, Р, 6, Н, I, Л, К, Ц N О и О, не должна пре¬
вышать 0,1 -кратную площадь основного пика на хро¬
матограмме раствора сравнения (Ь);
- сумма примесей (не более 3,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (Ь).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют относительную плотность (2.2.5)
испытуемого образца. К 1,00 г испытуемого образца
прибавляют 50 мл кислоты уксусной безводной Р и
титруют 0,1 М раствором кислоты хлорной потенци-
ометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 22,44 мг С22Н30С12М10-2С6Н12Ог
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, Р, О, Н, I, 3,
К, и А/, О, Р, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): Е, М.
А. 1 -(4-Хлорфенил)-5-[6-[(цианокарбамимидоил)-
амино]гексил]бигуанид.
В. 1 -[[6-[[[(4-Хлорфенил)карбамимидоил]карбами-
мидоил]амино]гексил]карбамимидоил]мочевина.
Е. 1-(4-Хлорфенил)гуанидин.
Р. 1-(4-Хлорфенил)мочевина.
6.1-(6-Аминогексил)-5-(4-хлорфенил)бигуанид.
Н. 1,1 '-[Иминобис(карбонимидоил иминогексан-6,1 -
диил)]бис[5-(4-хлорфенил)бигуанид].
С. Структура не установлена.
3. 1 -(4-Хлорфенил)-5-[6-[[4-[(4-хлорфенил)амино]>
6-[(15,2/?,ЗР,4Р)-1,2,3,4,5-пентагидроксипентил]-1,3,5-
триазин-2-ил]амино]гексил]бигуанид].
К. 1 -(4-Хлорфенил )-3-[[6-[[[(4-хлорфенил )карба-
мимидоил]амино]гексил]карбамимидоил]мочевина.
(5Я,65)-2-[(4-Хлорфенил)амино]-5-гидрокси-6-
[(1Я 2Я)-1,2,3-тригидроксипропил]-5,6-дигидро-4Н-1,3-
оксазин-4-он.
Хлористоводородная кислота концентрированная
1071
М. 5-(4-Хлорфенил)-5Ч1>енил-1,1'-(гексан-1,6-диил)-
дибигуанид.
N. 1 -[6-(Карбамимидоиламино)гексил]-5-(4-
хлорфенил)бигуанид.
О. 5-(2-Хлорфенил)-5'-(4-хлорфенил)-1,1 '-(гекеан-
1,6-диил)дибигуанид.
Р. 4-Хлоранилин.
О. Структура не установлена.
07/2016:0002
ХЛОРИСТОВОДОРОДНАЯ КИСЛОТА
КОНЦЕНТРИРОВАННАЯ
Ас1с1ит Ьус1госЫопс1ит сопсеп1га1ит
НУОКОСШ. ОШ С А ею, соысеыткатео
НС1 М.м. 36,46
[7647-01-0]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 35,0 % (м/м) и не более
39,0 % (м/м).
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная дымящая жидкость.
Смешивается с водой.
Относительная плотность: около 1,18.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец разводят водой Р. Полу¬
ченный раствор имеет сильнокислую реакцию (2.2.4).
B. Испытуемый образец дает реакции на хлори¬
ды (2.3.1).
С. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 5. К 2 мл испытуемого образца при¬
бавляют 8 мл воды Р.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Свободный хлор. Не более 0,0004 % (4 ррт).
К 15 мл испытуемого образца прибавляют 100 мл
воды, свободной от углерода диоксида, Р, 1 мл
раствора 100 г/л калия йодида Р и 0,5 мл раст¬
вора крахмала, свободного от йодидов, Р. Полу¬
ченный раствор выдерживают в защищенном от
света месте в течение 2 мин. Голубое окрашива¬
ние раствора должно исчезнуть при прибавлении
0,2 мл 0,01 М раствора натрия тиосульфата.
Сульфаты (2.4.13). Не более 0,0020 %
(20 ррт). К 6,4 мл испытуемого образца прибав¬
ляют 10 мг натрия гидрокарбоната Р и выпари¬
вают досуха на водяной бане. Остаток растворяют
в 15 мл воды дистиллированной Р. Полученный
раствор должен выдерживать испытание на суль¬
фаты.
#Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 4,2 мл испытуемого образца доводят во¬
дой Р до объема 10 мл. 1 мл полученного раствора
должен выдерживать испытание на мышьяк.
Шепезо (2.4.9). Не более 0,00015 % (1,5 ррт).
6,7 мл испытуемого образца доводят водой Р до
объема 10 мл. Полученный раствор должен выдер¬
живать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не более
0,0002 % (2 ррт). Остаток, полученный в испыта¬
нии «Остаток после выпаривания», растворяют
в 1 мл кислоты хлористоводородной разведен¬
ной Р и доводят водой Р до объема 25 мл. 5 мл
полученного раствора доводят водой Р до объема
20 мл. 12 мл полученного раствора должны выдер¬
живать испытание на тяжелые металлы. Эталон
готовят с использованием эталонного раствора
свинца (2 ррт РЬ) Р.
Остаток после выпаривания. Не более
0,01 %. 100,0 г испытуемого образца выпаривают
досуха на водяной бане и сушат при температуре
от 100 °С до 105 °С. Масса сухого остатка не долж¬
на превышать 10 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Точно взвешивают колбу, закрытую притертой
стеклянной пробкой, в которой содержится 30 мл
воды Р. Прибавляют 1,5 мл испытуемого образца
и снова взвешивают. Полученный раствор титруют
1 М раствором натрия гидроксида, используя в
качестве индикатора раствор метилового красно¬
го Р.
1 мл 1 М раствора натрия гидроксида соот¬
ветствует 36,46 мг НС1.
ХРАНЕНИЕ
В закрытом контейнере из стекла или друго¬
го инертного материала при температуре не выше
30 °С.
1072
Гэсударственная фармакопея Республики Беларусь
07/2016:0003
ХЛОРИСТОВОДОРОДНАЯ КИСЛОТА
РАЗВЕДЕННАЯ
Ас Мит Ьус/госЫогМит сНЫит
НУОПОСШ-ОМС АСЮ, й/ШТЕ
ОПРЕДЕЛЕНИЕ
Содержание: не менее 9,5 % (м/м) и не более
10,5 % (м/м) НС! (М.м. 36,46).
ПРИГОТОВЛЕНИЕ
К 726 г воды Р прибавляют 274 г кислоты хлори¬
стоводородной концентрированной и перемешивают.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец имеет сильнокислую ре¬
акцию (2.2.4).
B. Испытуемый образец дает реакции на хлори¬
ды (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Испытуемый обра¬
зец должен быть бесцветным.
Свободный хлор. Не более 0,0001 % (1 ррт). К
60 мл испытуемого образца прибавляют 50 мл воды,
свободной от углерода диоксида, Р, 1 мл раствора
100 г/л калия йодида Р и 0,5 мл раствора крахмала,
свободного от йодидов, Р. Полученный раствор вы¬
держивают в защищенном от света месте в течение
2 мин. Голубое окрашивание раствора должно ис¬
чезнуть при прибавлении 0,2 мл 0,01 М раствора на¬
трия тиосульфата.
Сульфаты (2.4.13). Не более 0,0005 % (5 ррт). К
26 мл испытуемого образца прибавляют 10 мг натрия
гидрокарбоната Р и выпаривают досуха на водяной
бане. Остаток растворяют в 15 мл воды дистиллиро¬
ванной Р. Полученный раствор должен выдерживать
испытание на сульфаты.
Тяжелые металлы (2.4.8, метод А). Не более
0,0002 % (2 ррт). Остаток, полученный в испытании
«Остаток после выпаривания», растворяют в 1 мл
кислоты хлористоводородной разведенной Р и дово¬
дят водой Р до объема 25 мл. 5 мл полученного раст¬
вора доводят водой Р до объема 20 мл. 12 мл раст¬
вора должны выдерживать испытание на тяжелые ме¬
таллы. Эталон готовят с использованием эталонного
раствора свинца (2 ррт РЬ) Р.
Остаток после выпаривания. Не более 0,01 %.
100,0 г испытуемого образца выпаривают досуха на
водяной бане и сушат при температуре от 100 °С до
105 °С. Масса сухого остатка не должна превышать
10 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 6,00 г испытуемого образца прибавляют 30 мл
воды Р. Полученный раствор титруют 1 М раствором
натрия гидроксида, используя в качестве индикатора
раствор метилового красного Р.
1 мл 1 М раствора натрия гидроксида соответ¬
ствует 36,46 мг НС1.
07/2016:0384
ХЛОРКРЕЗОЛ
СЫогосгезо/ит
СЖ.ОКОСКЕ501.
С7Н7СЮ М.м. 142,6
[59-50-7]
ОПРЕДЕЛЕНИЕ
4-Хлор-З-метилфенол.
Содержание: не менее 98,0 % и не более 101,0%.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок, или плотная кристаллическая масса в виде гра¬
нул, или бесцветные или белые кристаллы.
Мало растворим в воде, очень легко растворим в
96 % спирте, легко растворим в жирных маслах. Рас¬
творяется в растворах гидроксидов щелочных метал¬
лов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14). От 64 °С до
67 °С.
B. К 0,1 г испытуемого образца прибавляют
0,2 мл бензоилхлорида Р, 0,5 мл раствора натрия
гидроксида разведенного Р и энергично встряхива¬
ют до образования кристаллического осадка белого
цвета, прибавляют 5 мл воды Р и фильтруют. Осадок
перекристаллизовывают из 5 мл метанола Р и су¬
шат при температуре 70 °С. Температура плавления
(2.2.14) полученных кристаллов: от 85 °С до 88 °С.
C. К 5 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 0,1 мл раст¬
вора железа хлорида Р1. Появляется голубоватое
окрашивание.
ИСПЫТАНИЯ
Раствор 3. К 3,0 г мелкоизмельченного испытуе¬
мого образца прибавляют 60 мл воды, свободной от
углерода диоксида, Р, встряхивают в течение 2 мин и
фильтруют.
Раствор 51. 1,25 г испытуемого образца раст¬
воряют в 96% спирте Р и доводят до объема 25 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
31 должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность. К 10 мл раствора 3 прибавляют
0,1 мл раствора метилового красного Р При при¬
бавлении не более 0,2 мл 0,01 М раствора натрия
гидроксида должно появиться чистое желтое окраши¬
вание.
Сопутствующие примеси. Газовая хроматогра¬
фия (2.2.28): испытание проводят методом нормали¬
зации.
Испытуемый раствор. 1,0 г испытуемого об¬
разца растворяют в ацетоне Р и доводят до объема
100 мл этим же растворителем.
#Хлороформ
1073
Раствор сравнения. 1,0 мл испытуемого раст¬
вора доводят ацетоном Р до объема 100,0 мл. 5,0 мл
полученного раствора доводят ацетоном Р до объ¬
ема 100,0 мл.
Условия хроматографирования:
- колонка стеклянная длиной 1,80 м и внутрен¬
ним диаметром (3—4) мм, заполненная диатомитом
силанизированным для газовой хроматографии Р,
импрегнированным (3—5) % (м/м) полиметилфенил-
силоксана Р;
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 30 мл/мин;
- температура колонки: 125 °С;
- температура блока ввода проб: 210 °С;
- температура детектора: 230 °С;
- детектор*, лламеппо-иониэациоппый*,
- время хроматографирования: 3-кратное вре¬
мя удерживания хлорокрезола.
Время удерживания: хлорокрезол — около 8 мин.
Предельное содержание примесей:
- неспецифицированные примеси: не более 0,10 %
- сумма примесей: не более 1 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения.
Нелетучие вещества. Не более 0,1 %. 2,0 г испы¬
туемого образца выпаривают на водяной бане досуха
и остаток сушат при температуре от 100 °С до 105 °С.
Масса сухого остатка не должна превышать 2 мг.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
70,0 мг испытуемого образца помещают в колбу
с притертой стеклянной пробкой, растворяют в 30 мл
кислоты уксусной ледяной Р, прибавляют 25,0 мл
0,0167 М раствора калия бромата, 20 мл раствора
150 г/л калия бромида Р, 10 мл кислоты хлористово¬
дородной Р и выдерживают в защищенном от света
месте в течение 15 мин. К полученному раствору при¬
бавляют 1 г калия йодида Р, 100 мл воды Р и титру¬
ют, энергично встряхивая, 0,1 М раствором натрия
тиосульфата, используя в качестве индикатора 1 мл
раствора крахмала Р, который прибавляют в конце
титрования.
Параллельно проводят контрольный опыт.
1 мл 0,0167 М раствора калия бромата соответ¬
ствует 3,565 г С7Н7СЮ.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:РБ0015
#ХЛОРОФОРМ
СЫого^огт'ют
сниопоропм
СНС1, М.м. 119,4
[67-66-3]
ОПРЕДЕЛЕНИЕ
Трихлорметан.
В качестве стабилизатора содержит не менее
0,4 % (м/м) и не более 1,0 % (м/м) этанола.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная тяжелая подвижная ле¬
тучая жидкость с характерным запахом.
Мало растворим в воде, смешивается с 96 %
спиртом, не смешивается с глицерином.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Температура кипения», как указано в разделе «Ис¬
пытания».
ИСПЫТАНИЯ
Раствор $. А 5 мл испытуемого образца встряхи¬
вают с 30 мл воды Р. Используют водный слой.
Кислотность. К 10 мл раствора 3 прибавляют
4 капли раствора бромфенолового синего Р1. Долж¬
но появиться сине-фиолетовое окрашивание.
Посторонний запах. 20 мл испытуемого образца
выливают на чистую, не имеющую запаха фильтро¬
вальную бумагу, сложенную вчетверо, и помещают в
чашку Петри, находящуюся на водяной бане при тем¬
пературе 50 °С. После испарения хлороформа бумага
не должна иметь постороннего запаха.
Относительная плотность (2.2.5). От 1,475 до
1,481.
Температура кипения (2.2.12). От 59,5 °С до
62,0 °С.
Органические примеси. 15 мл испытуемого об¬
разца помещают в колбу со шлифом, предварительно
сполоснутую дважды испытуемым образцом и дваж¬
ды кислотой серной Р, прибавляют 10 мл кислоты
серной Р, встряхивают в течение (15—20) с и выдер¬
живают в защищенном от света месте при темпера¬
туре (15±2) °С в течение 1 ч. Слой серной кислоты не
должен окрашиваться.
Нелетучий остаток. Не более 0,002 %. 50,0 г ис¬
пытуемого образца выпаривают на водяной бане до¬
суха и сушат при температуре от 100 °С до 105 °С.
Масса полученного остатка не должна превышать 1 мг.
Этанол. Не менее 0,4 % (м/м) и не более 1,0 %
(м/м). 1,00 г испытуемого образца (т, г) помещают в
колбу со шлифом, прибавляют 15,0 мл нитрохромо-
вого реактива Р, колбу закрывают, энергично встря¬
хивают в течение 2 мин и выдерживают в течение
15 мин. Прибавляют 100 мл воды Р и 5 мл раствора
200 г/л калия йодида Р. Через 2 мин титруют 0,1 М
раствором натрия тиосульфата до появления
светло-зеленого окрашивания (п^, мл 0,1 Мраствора
натрия тиосульфата), используя в качестве индика¬
тора 1 мл раствора крахмала Р.
Параллельно проводят контрольный опыт (п2, мл
0,1 М раствора натрия тиосульфата).
Содержание этанола в процентах рассчитывают
по формуле:
(п2 - л1) • 0,115
т
Вода. 10 мл испытуемого образца охлаждают до
температуры от -3 °С до -4 °С. Не должно появлять¬
ся мути.
Свободный хлор. К 10 мл раствора 3 прибавля¬
ют 0,5 мл раствора калия йодида Р и 0,5 мл раст¬
вора крахмала Р. Не должно появляться синее окра¬
шивание.
1074
Гэсударственная фармакопея Республики Беларусь
Хлориды. 5 мл раствора 3 доводят водой Р до
объема 10 мл. Полученный раствор не должен давать
реакции на хлориды (2.3.1).
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом стеклянном контейне¬
ре в защищенном от света месте при температуре от
8°С до 15 °С.
07/2016:0546
ХЛОРТАЛИДОН
СЫог1аНс1опит
стортАиооыЕ
С14Н11С1М2043 М.м. 338,8
[77-36-1]
ОПРЕДЕЛЕНИЕ
2-Хпор-5-[(1Я5)-1-гидрокси-3-оксо-2,3-дигидро-
1 Н-изоиндол-1 -ил]бензолсульфонамид.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или желтовато-белый порошок.
Очень мало растворим в воде, растворим в аце¬
тоне и в метаноле, практически нерастворим в мети-
ленхлориде. Растворяется в разведенных растворах
гидроксидов щелочных металлов.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО хлорталидона #или спектр,
представленный на рисунке #0546.-1
Если спектры, полученные с использованием об¬
разцов в твердом состоянии, отличаются, то испытуе¬
мый образец и ФСО хлорталидона растворяют по от¬
дельности в метаноле Р и выпаривают до сухих остат¬
ков, которые используют для получения новых спектров.
ИСПЫТАНИЯ
Кислотность. 1,0 г испытуемого образца раст¬
воряют при нагревании в смеси из 25 мл ацетона Р
и 25 мл воды, свободной от углерода диоксида, Р,
охлаждают и титруют 0,1 М раствором натрия ги¬
дроксида потенциометрически (2.2.20). Объем 0,1 М
раствора натрия гидроксида, пошедший на титрова¬
ние, не должен превышать 0,75 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Смешивают 2 объема
раствора 2 г/л натрия гидроксида Р, 48 объемов под¬
вижной фазы В и 50 объемов подвижной фазы А.
Испытуемый раствор (а). 50,0 мг испытуемого
образца растворяют в смеси растворителей и доводят
до объема 50,0 мл этим же растворителем.
Испытуемый раствор (Ь). 10,0 мл испытуемого
раствора (а) доводят смесью растворителей до объ¬
ема 100,0 мл.
Раствор сравнения (а). 1,0 мл испытуемого
раствора (а) доводят смесью растворителей до
Хлорталидон
1075
объема 100,0 мл. 1,0 мл полученного раствора до¬
водят смесью растворителей до объема 10,0 мл.
Раствор сравнения (Ь). Содержимое контей¬
нера ФСО хлорталидона для идентификации пи¬
ков (содержит примеси В, <3 и ч!) растворяют в 1 мл
смеси растворителей.
Раствор сравнения (с). 50,0 мг ФСО хлорта¬
лидона растворяют в смеси растворителей и до¬
водят до объема 50,0 мл этим же растворителем.
10,0 мл полученного раствора доводят смесью
растворителей до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: 1,32 г аммония фосфа¬
та Р растворяют в 900 мл воды Р, доводят до
рН 5,5 кислотой фосфорной разведенной Р и
разводят водой Р до объема 1000 мл;
- подвижная фаза В: метанол Р2\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—16
65
35
16—21
65 — 50
35 — 50
21—35
50
50
35—45
50 — 65
50 — 35
- скорость подвижной фазы: 1,4 мл/мин;
-спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и растворов сравнения (а) и (Ь).
Идентификация пиков примесей: идентифи¬
цируют пики примесей В, С и 3, используя хрома¬
тограмму раствора сравнения (Ь) и хроматограмму,
прилагаемую к ФСО хлорталидона для идентифи¬
кации пиков.
Относительное удерживание (по отношению к
хлорталидону, время удерживания — около 7 мин):
примесь В — около 0,7; примесь ^ — около 0,9; при¬
месь 6 — около 6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,5 между пиками при¬
меси ^ и хлорталидона.
Предельное содержание примесей:
- примесь В (не более 0,7 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси В, не должна превышать 7-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь ^ (не более 0,3 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси ^ не должна превышать 3-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- примесь С (не более 0,2 %): на хроматограмме
испытуемого раствора (а) площадь пика, соответству¬
ющего примеси С, не должна превышать 2-кратную
площадь основного пика на хроматограмме раствора
сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора (а)
площадь любого пика, кроме основного и пиков при¬
месей В, О и 3, не должна превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,2 %): на хромато¬
грамме испытуемого раствора (а) сумма площадей
всех пиков, кроме основного, не должна превышать
12-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора (а) не учитывают пики с
площадью менее 0,5 площади основного пика на хро¬
матограмме раствора сравнения (а).
Хлориды (2.2.4). Не более 0,0350 % (350 ррт).
O, 3 г испытуемого образца растирают в мелкий по¬
рошок, прибавляют 30 мл воды Р, встряхивают в те¬
чение 5 мин и фильтруют. 15 мл фильтрата должны
выдерживать испытание на хлориды. Эталон готовят
с использованием 10 мл эталонного раствора хло¬
рида (5 ррт С1) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (с).
Содержание С14Н11С1Ы2043 в процентах рассчи¬
тывают с учетом содержания хлорталидона в ФСО
хлорталидона.
ПРИМЕСИ
Специфицированные примеси: В, 6, 3.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содер¬
жание лимитируется общим критерием приемле¬
мости для других/неспецифицированных примесей
и/или общей статьей Субстанции для фармацевти¬
ческого использования (2034). Вследствие этого нет
необходимости идентифицировать эти примеси для
доказательства соответствия требованиям. См. так¬
же статью 5.10. Контроль примесей в субстанциях
для фармацевтического использования): А, С, О, Е,
P, Н, I.
оч о
у
/О.
о к
ТТ1
С1^^
A. Р = Н, Р' = ОН: 2-(4-Хлор-3-сульфобензоил)бен-
зойная кислота.
B. Р = Н, Р' = 1ЧН2:2-(4-Хлор-3-сульфамоилбензоил)
бензойная кислота.
C. Р = СН2-СН3, Р' = ЫН2: Этил-2-(4-хлор-3-сульфа-
моилбензоил)бензоат.
I. Р = СН(СН3)2, Р' = МН2: 1 -Метилэтил-2-(4-хлор-
3-сульфамоилбензоил)бензоат.
1076
Гэсударственная фармакопея Республики Беларусь
й. К = ОС2Н5, К' = 302-1МН2:2-Хлор-5-[(1Я5)-1-эток-
си-З-оксо-2,3-дигидро-1 Н-изоиндол-1 -ил]бензолсуль-
фонамид.
Е. В = Н, К' = 302-МН2:2-Хлор-5-[(1Я5)-3-оксо-2,3-
дигидро-1 Н-изоиндол-1 -ил]бензолсул ьфонамид.
<3. К = ОН, К' = С1: (3/?5)-3-(3,4-Дихлорфенил)-3-
гидрокси-2,3-дигидро-1 Н-изоиндол-1 -он.
Н. К = ОСН(СН3)2, К' = 502-[\1Н2: 2-Хлор-5-[(1Я5)-
1 -(1 -метилэтокси)-3-оксо-2,3-дигидро-1 Н-изоиндол-1 -
ил]бензолсульфонамид.
Р. Бис[2-хлор-5-(1-гидрокси-3-оксо-2,3-дигидро-1Н-
изоиндол-1-ил)бензолсульфонил]амин.
*!. Структура не установлена.
07/2016:0386
ХЛОРФЕНАМИНА МАЛЕАТ
СЫогрНепат1т та1еаз
СН1-ОКРНЕЫАМШЕ МАБЕАТЕ
ОПРЕДЕЛЕНИЕ
(3/?5)-3-(4-Хлорфенил)-Л/,Л/-диметил-3-(пиридин-
2-ил)пропан-1-амина гидро-(2)-бутендиоат. #Хлорфе-
нирамина малеат*.
Содержание: не менее 98,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Легко растворим в воде, растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14): от 130 °С до
135 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО хлорфенамина малеата #или
спектр, представленный на рисунке #0386.-1#.
C. Испытуемый образец выдерживает испытание
«Угол оптического вращения», как указано в разделе
«Испытания».
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20,0 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)6.
Угол оптического вращения (2.2.7). От -0,10° до
+0,10°. Измеряют оптическое вращение раствора 8.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 0,5 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (с). 5 мг ФСО хлорфенамина
малеата примеси С растворяют в 5 мл испытуемо¬
го раствора и доводят подвижной фазой до объема
50,0 мл. 2 мл полученного раствора доводят подвиж¬
ной фазой до объема 20 мл.
Раствор сравнения (д). 5 мг 2,2-дипиридилами-
на Р (примесь В) растворяют в подвижной фазе и до¬
водят до объема 100 мл этим же растворителем.
Раствор сравнения (е). Содержимое контейнера
ФСО хлорфенамина малеата примеси А растворяют
в 2 мл испытуемого раствора и обрабатывают ультра¬
звуком в течение 5 мин.
Условия хроматографирования:
- колонка длиной 0,30 м и внутренним диаме¬
тром 3,9 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
10 мкм;
- подвижная фаза: 20 объемов ацетонитрила Р
смешивают с 80 объемами раствора 8,57 г/л аммония
дигидрофосфата Р, предварительно доведенного до
рН 3,0 кислотой фосфорной Р\
- скорость подвижной фазы: 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3,5-кратное вре¬
мя удерживания хлорфенамина.
Относительное удерживание (по отношению к
хлорфенамину, время удерживания — около 11 мин):
малеиновая кислота — около 0,2; примесь А — около
0,3; примесь В — около 0,4; примесь С — около 0,9;
примесь О — около 3,0.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 1,5 между пиками при¬
меси С и хлорфенамина.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А — 1,5; для примеси В — 1,4):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси А, не должна превышать 0,4 площади
основного пика на хроматограмме раствора сравне¬
ния (а);
- примеси В, С, О (не более 0,1 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям В, С и О, не должны превы-
Хлорфенамина малеат
1077
Рисунок #0386.-1. Инфракрасный спектр ФСО хлорфенамина малеата
шать 0,2 площади основного пика на хроматограмме
раствора сравнения (а);
- любая другая примесь (не более 0,10%): на
хроматограмме испытуемого раствора площадь лю¬
бого пика, кроме основного и пиков примесей А, В, С и
О, не должна превышать 0,2 площади основного пика
на хроматограмме раствора сравнения (а);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади основного пика на хроматограмме
раствора сравнения (Ь) (0,05 %), а также пики подвиж¬
ной фазы и малеиновой кислоты.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытания на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворят в 25 мл
кислоты уксусной безводной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 19,54 мг С16Н19С1М2-С4Н404.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
А. 2-(4-Хлорфенил)-4-(диметиламино)-2-[2-(диме-
тиламино)этил]бутаннитрил.
н
^ ж
В. Л/-(Пиридин-2-ил)пиридин-2-амин (2,2-дипири-
диламин).
и энантиомер
С. (ЗЯ5)-3-(4-Хлорофенил)-А/-метил-3-(пиридин-
2-ил)пропан-1 -амин.
и энантиомер
О. (2/?5)-2-(4-Хлорофенил)-4-(диметиламино)-2-
(пиридин-2-ил)бутаннитрил.
1078
Гэсударственная фармакопея Республики Беларусь
07/2016:0072
ХОЛЕКАЛЫДИФЕРОЛ
С1ю1еса1сИего1ит
СН01-ЕСА1-С1РЕ1Ю1.
С27Н..О М.м. 384,6
[67-97-0]
ОПРЕДЕЛЕНИЕ
(52,7Е)-9,10-Секохолеста-5,7,10(19)-триен-3|3-ол.
Содержание: не менее 97,0 % и не более 102,0 %.
В растворе в зависимости от температуры и вре¬
мени имеет место обратимая изомеризация в прехо-
лекальциферол. Активность обуславливается обоими
компонентами (см. раздел «Количественное опреде¬
ление»).
1 мг холекальфицерола соответствует 40 000 МЕ
антирахитической активности (витамин й) у крыс.
ОПИСАНИЕ (СВОЙСТВА)
Белые или почти белые кристаллы.
Практически нерастворим в воде, легко раство¬
рим в 96 % спирте, растворим в триметилпентане и в
жирных маслах.
Чувствителен к воздуху, нагреванию и свету. Рас¬
творы нестабильны без использования антиоксидан¬
тов и поэтому должны использоваться немедленно.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО холекальциферола.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+105 до +112. 0,200 г испытуемого образца быстро
растворяют в спирте, свободном от альдегидов, Р
без нагревания и доводят до объема 25,0 мл этим
же растворителем. Испытание проводят в течение
30 мин после приготовления раствора.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием, избегая воздей¬
ствия актиничного света и воздуха.
Испытуемый раствор. 10,0 мг испытуемого образ¬
ца растворяют в триметилпентане Р без нагревания
и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО холекаль¬
циферола растворяют в триметилпентане Р без на¬
гревания и доводят до объема 10,0 мл этим же раст¬
ворителем.
Раствор сравнения (Ь). 1,0 мл ФСО холекаль¬
циферола для проверки пригодности хроматогра¬
фической системы (содержит примесь А) доводят
подвижной фазой до объема 5,0 мл. Нагревают на
водяной бане при температуре 90 °С с обратным хо¬
лодильником в течение 45 мин и охлаждают (образо¬
вание прехолекальциферола).
Раствор сравнения (с). 10,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 100,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем для хромато¬
графии Р с размером частиц 5 мкм;
- подвижная фаза: пентанол Р — гексан Р
(0,3:99,7, об/об);
- скорость подвижной фазы: 2 мл/мин;
- спектрофотометрический детектор, длина
волны 265 нм;
- объем вводимой пробы: по 5 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 2-кратное вре¬
мя удерживания холекальциферола.
Относительное удерживание (по отношению
к холекальциферолу, время удерживания — около
19 мин): прехолекальциферол — около 0,5; примесь
А — около 0,6.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками пре¬
холекальциферола и примеси А.
Предельное содержание примесей:
- примесь А (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пика приме¬
си А, не должна превышать площадь основного пика
на хроматограмме раствора сравнения (с);
- сумма примесей (не более 1,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
10-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,5 площади основного пика на
хроматограмме раствора сравнения (с); не учитывают
пик прехолекальциферола.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- вводимая проба: испытуемый раствор и раст¬
вор сравнения (а).
Содержание С^Н^О в процентах рассчитывают
с учетом содержания холекальциферола в ФСО холе¬
кальциферола.
Холестерин
1079
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в атмос¬
фере азота, в защищенном от света месте, при темпе¬
ратуре от 2 °С до 8 °С.
Содержимое открытого контейнера должно ис¬
пользоваться немедленно.
ПРИМЕСИ
Специфицированная примесь: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О, Е.
А. (5Е,7Е)-9,10-Секохолеста-5,7,10(19)-триен-Зр-
ол (транс-холекальциферол, транс-витамин 03).
В. Холеста-5,7-диен-3(3-ол (7,8-дидегидрохолесте-
рин, провитамин 03).
С. 9р,10а-Холеста-5,7-диен-Зр-ол (люмистерин3).
О. (6Е)-9,10-Секохолеста-5(10),6,8(14)-триен-Зр-
ол (изо-тахистерин3).
Е. (6Е)-9,10-Секохолеста-5(10),6,8-триен-Зр-ол (та-
хистерин3).
07/2016:0993
ХОЛЕСТЕРИН
СЬо1ез1его1ит
СН01.Е8ТЕ1Ю1.
С^Н^О М.м. 386,7
[57-88-5]
ОПРЕДЕЛЕНИЕ
Хол ест-5-ен-ЗР-ол.
Содержание:
- холестерин: не менее 95,0 % (в пересчете на
сухое вещество);
- сумма стеринов: не менее 97,0 % и не более
103,0 % (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Практически нерастворим в воде, умеренно рас¬
творим в ацетоне и в 96 % спирте.
Чувствителен к свету.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14): от 147 °С до
150 °С.
B. Тонкослойная хроматография (2.2.27). Рас¬
творы готовят непосредственно перед использо¬
ванием.
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в этиленхлориде Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО холестерина
растворяют в этиленхлориде Р и доводят до объема
5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
1080
Гэсударственная фармакопея Республики Беларусь
Подвижная фаза, этилацетат Р — толуол Р
(33:66, об/об).
Наносимый объем пробы: 20 мкл.
Фронт подвижной фазы: немедленно, в защи¬
щенном от света месте, не менее 15 см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают 3 раза
раствором мышьяка трихлорида Р, просматрива¬
ют в течение (3—4) мин.
Результаты, на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения.
С. Около 5 мг испытуемого образца раство¬
ряют в 2 мл метиленхлорида Р. Прибавляют 1 мл
уксусного ангидрида Р, 0,01 мл кислоты серной Р
и встряхивают. Появляется розовое окрашивание,
которое быстро переходит в красное, затем в голу¬
бое и наконец становится ярко-зеленым.
ИСПЫТАНИЯ
Растворимость в 96 % спирте. 0,5 г испытуе¬
мого образца помещают в колбу с пробкой и раст¬
воряют в 50 мл 96 % спирта Р при температуре
50 °С. Выдерживают в течение 2 ч. Не должен вы¬
падать осадок и раствор не должен мутнеть.
Кислотность. 1,0 г испытуемого образца
растворяют в 10 мл эфира Р, прибавляют 10,0 мл
0,1 М раствора натрия гидроксида и встряхивают
в течение 1 мин. Осторожно нагревают для удале¬
ния эфира и затем кипятят в течение 5 мин. Охлаж¬
дают, прибавляют 10 мл воды Р и 0,1 мл раствора
фенолфталеина Р в качестве индикатора и титру¬
ют 0,1 М раствором кислоты хлористоводород¬
ной до момента исчезновения розового окрашива¬
ния, энергично перемешивая во время титрования.
Выполняют контрольный опыт. Разность между
объемами 0,1 М раствора кислоты хлористово¬
дородной, необходимой для изменения окраски
индикатора в контрольном опыте и с испытуемым
раствором не должна быть более 0,3 мл.
Потеря в массе при высушивании (2.2.32).
Не более 0,3%. 1,000 г испытуемого образца
сушат в вакууме при температуре 60 °С в тече¬
ние 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 0,100 г
ФСО прегненолона изобутирата растворяют в
гептане Р и доводят до объема 100,0 мл этим же
растворителем.
Испытуемый раствор. 25,0 мг испытуемого
образца растворяют в растворе внутреннего стан¬
дарта и доводят до объема 25,0 мл этим же раст¬
ворителем.
Раствор сравнения. 25,0 мг ФСО холестери¬
на растворяют в растворе внутреннего стандарта
и доводят до объема 25,0 мл этим же растворите¬
лем.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной
30 м и внутренним диаметром 0,25 мм, покрытая
слоем поли(диметил)силоксана Р (толщина слоя
0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 2 мл/мин;
- деление потока: 1:25;
- температура: колонка — 275 °С, блок вво¬
да проб — 285 °С, детектор — 300 °С;
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1,0 мкл.
Пригодность хроматографической системы:
раствор сравнения:
-разрешение: не менее 10,0 между пиками
прегненолона изобутирата и холестерина;
- фактор асимметрии: не менее 0,6 для пика
холестерина.
Процентное содержание холестерина рассчи¬
тывают с учетом заявленного содержания в ФСО
холестерина. Процентное содержание суммы сте-
ринов рассчитывают, суммируя содержание холе¬
стерина и содержание других веществ со време¬
нем удерживания менее или равным 1,5-кратному
времени удерживания холестерина. Не учитывают
пики внутреннего стандарта и растворителя.
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
На этикетке должен быть указан источник мате¬
риала для производства холестерина (например, го¬
ловной и спинной мозг крупного рогатого скота, лано¬
лин или куриные яйца).
ПРИМЕСИ
А. 5а-Холест-7-ен-30-ол (латостерин).
С. 5а-Холеста-7,24-диен-30-ол.
Хондроитина натрия сульфат
1081
07/2016:2064
ХОНДРОИТИНА НАТРИЯ СУЛЬФАТ
С1юпс1гоШп1 па\п\ зиКаз
СНОЫОКОПт 8Ш.РАТЕ зоошм
Н20(С14Н19ММа20143)х
ОПРЕДЕЛЕНИЕ
Природный сополимер, образованный в ос¬
новном двумя дисахаридами: натриевых солей
[4)-((3-0-глюкопиранозилуроновой кислоты)-(1 —►З)-
[2-(ацетиламино)-2-дезокси-р-0-галактопиранозил-4-
сульфата]-(1 —►] и [4)-(Р-0-глюкопиранозилуроновой
кислоты)-(1—►3)-[2-(ацетиламино)-2-деокси-р-0-галак-
топиранозил-6-сульфата]-(1—►]. При полном гидроли¬
зе образуются Б-галактозамин, О-глюкуроновая кис¬
лота, уксусная кислота и серная кислота. Получают из
хрящевой ткани наземных и морских видов животных.
В зависимости от вида животного, являющегося ис¬
точником хондроитина натрия сульфата, могут быть
различные соотношения 4-сульфатных и 6-сульфат-
ных групп.
Содержание: от 95 % до 105 % (в пересчете на
сухое вещество).
ПРОИЗВОДСТВО
Животные, из которых получают хондроитина на¬
трия сульфат, должны выдерживать требования к жи¬
вотным для потребления человеком.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Гигроскопичен.
Легко растворим в воде, практически нераство¬
рим в ацетоне и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках с калия бромидом Р.
Сравнение: для хондроитина натрия суль¬
фата, полученного из хрящевых тканей наземных
животных, используют ФСО хондроитина натрия
сульфата; для хондроитина натрия сульфата, по¬
лученного из хрящевых тканей морских животных,
используют ФСО хондроитина натрия сульфата
(морской).
B. Раствор 31, приготовленный как указано в
разделе «Испытания», дает реакцию (Ь) на натрий
(2.3.1).
C. Просматривают электрофореграммы, полу¬
ченные в испытании «Сопутствующие примеси».
Результаты: на электрофореграмме ис¬
пытуемого раствора обнаруживается основная
полоса, соответствующая по расположению ос¬
новной полосе на электрофореграмме раствора
сравнения (а).
ИСПЫТАНИЯ
Раствор 31. 2,500 г испытуемого образца
растворяют в 50,0 мл воды, свободной от углеро¬
да диоксида, Р.
Раствор 32. 1,0 мл раствора 31 доводят до
объема 10,0 мл водой Р.
рН (2.2.3). От 5,5 до 7,5. Измеряют рН раст¬
вора 31.
Удельное оптическое вращение (2.2.7). От
-20 до -30 (для хондроитина натрия сульфата из
хрящевых тканей наземных животных) или от -12
до -19 (для хондроитина натрия сульфата из хря¬
щевых тканей морских животных) (в пересчете на
сухое вещество). Используют раствор 31.
Характеристическая вязкость. От 0,01 м3/кг
до 0,15 м3/кг.
Испытуемый раствор (а). Взвешивают 5,000 г
(т0р) испытуемого образца и прибавляют около
80 мл раствора 11,7 г/л натрия хлорида Р ком¬
натной температуры. Растворяют, встряхивая, при
комнатной температуре в течение 30 мин. Доводят
раствором 11,7 г/л натрия хлорида Р до объема
100,0 мл. Фильтруют через мембранный фильтр с
размером пор 0,45 мкм, отбрасывая первые 10 мл
фильтрата. Концентрация испытуемого раствора
(а) является только оценочной и ее необходимо от¬
корректировать после первоначального определе¬
ния вязкости испытуемого раствора (а).
Испытуемый раствор (Ь). К 15,0 мл испыту¬
емого раствора (а) прибавляют 5,0 мл раствора
11.7 г/л натрия хлорида Р.
Испытуемый раствор (с). К 10,0 мл испыту¬
емого раствора (а) прибавляют 10,0 мл раствора
11.7 г/л натрия хлорида Р.
Испытуемый раствор (д). К 5,0 мл испыту¬
емого раствора (а) прибавляют 15,0 мл раствора
11.7 г/л натрия хлорида Р.
Определяют время вытекания (2.2.9) для раст¬
вора 11,7 г/л натрия хлорида Р (/0) и времена вы¬
текания 4 испытуемых растворов (/.,, /2, /3 и /4) при
температуре (25,00±0,03) °С. Используют подходя¬
щий вискозиметр с висячим уровнем (характери¬
стики: номинальная постоянная вискозиметра —
около 0,005 мм2/с2, диапазон кинематической вяз¬
кости— (1—5) мм2/с2, внутренний диаметр трубки
Р— 0,53 мм, объем расширения С — 5,6 мл, вну¬
тренний диаметр трубки N — (2,8—3,2) мм) с во¬
ронкообразным нижним капиллярным концом. Для
всех измерений используют один и тот же вискози¬
метр; все значения времени вытекания жидкости
определяют трижды.
Пригодность:
- результаты не должны отличаться более чем
на 0,35 % от среднего значения;
- время вытекания должно быть не менее
1,6 /0 и не более 1,8/0.
Если данные условия не выполняются, то кон¬
центрацию испытуемого раствора (а) корректируют
и испытание повторяют.
Расчет относительной вязкости.
Поскольку плотности растворов хондроити¬
на натрия сульфата и растворителя практически
одинаковы, то относительные вязкости #г/ (обозна¬
чаемые (7г1, цт2, цт3 и г]т4) могут быть рассчитаны из
отношения времен вытекания соответствующих
растворов /. (обозначаемых 12, /3 и /4) ко времени
вытекания растворителя с учетом поправочного
136. Зак. 1060.
1082
Гэсударственная фармакопея Республики Беларусь
коэффициента на изменение кинетической энергии
для капилляра (В = 30 800 с3), как показано ниже:
В
В •
Расчет концентраций.
Рассчитывают концентрацию с1 (выраженную в
кг/м3) хондроитина натрия сульфата в испытуемом
растворе (а) по следующей формуле:
х 100-Л
т°р ’ 100 ' 100
10,
где:
х—содержание хондроитина натрия сульфата, най¬
денное в испытании «Количественное определение», %;
/? — потеря в массе при высушивании, %.
Рассчитывают концентрацию с2 (кг/м3) хондрои¬
тина натрия сульфата в испытуемом растворе (Ь), ис¬
пользуя следующую формулу:
с1 • 0,75.
Рассчитывают концентрацию с3 (кг/м3) хондрои¬
тина натрия сульфата в испытуемом растворе (с), ис¬
пользуя следующую формулу:
сг 0,50.
Рассчитывают концентрацию с4 (выраженную в
кг/м3) хондроитина натрия сульфата в испытуемом
растворе (с1), используя следующую формулу:
с1 • 0,25.
Расчет характеристической вязкости.
Удельную вязкость г)5. испытуемого раствора
(обозначенные как о81, П%2> Я8зи 0^) определяют по зна¬
чениям относительной вязкости цг. (обозначенные пг1,
Оа’ Ягз и 0г4)> используя следующую формулу:
Пп~ 1-
Характеристическую вязкость [/7], определенную
как
[г]]= Нт[
с-»о1 с
рассчитывают методом линейной регрессии наи¬
меньших квадратов, используя следующую формулу:
п*=сгкн+[ч1
с,
где:
с. — концентрация испытуемого вещества, кг/м3;
кИ — постоянная Хаггинса.
Сопутствующие примеси. Электрофорез (2.2.31).
Буферный раствор А (0,1 М раствор бария
ацетата рН 5,0). 25,54 г бария ацетата Р раство¬
ряют в 900 мл воды Р, доводят кислотой уксусной
ледяной Р до рН 5,0 и разводят водой Р до объема
1000.0 мл.
Буферный раствор В (1 М раствор бария аце¬
тата рН 5,0). 255,43 г бария ацетата Р раство¬
ряют в 900 мл воды Р, доводят кислотой уксусной
ледяной Р до рН 5,0 и разводят водой Р до объема
1000.0 мл.
Окрашивающий раствор. 1,0 г толуидиново-
го синего Р и 2,0 г натрия хлорида Р растворяют в
1000 мл 0,01 М раствора кислоты хлористоводо¬
родной и фильтруют.
Испытуемый раствор. Готовят раствор 30 мг/мл
испытуемого образца в воде Р.
Раствор сравнения (а). Готовят раствор 30 мг/мл
ФСО хондроитина натрия сульфата в воде Р.
Раствор сравнения (Ь). 2,0 мл раствора сравне¬
ния (а) доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). Смешивают равные объ¬
емы раствора сравнения (Ь) и воды Р.
Методика. Основание прибора для электрофо¬
реза охлаждают до 10 °С. Предварительно уравно¬
вешивают агарозный гель в буферном растворе А в
течение 1 мин. Избыток жидкости осторожно слива¬
ют. Высушивают гель около 5 мин. В каждый резер¬
вуар прибора для электрофореза помещают 400 мл
буферного раствора В. В каналы агарозного геля
вносят по 1 мкл каждого раствора. На охлажденную
пластинку прибора для электрофореза пипеткой на¬
носят несколько миллилитров 50 % (об/об) раствора
глицерина Р и помещают гель на середину керами¬
ческой пластинки. Помещают фитиль, пропитанный
буферным раствором В, на положительно и отри¬
цательно заряженные стороны агарозного геля. Не¬
обходимо убедиться, что имеется хороший контакт
между буфером для электрофореза и агарозным ге¬
лем. Электрофорез проводят в следующих условиях:
75 мА/гель при напряжении (100—150) В (максимум
(300—400) В) для геля размером около 12 см х 10 см.
Проводят электрофорез в течение 12 мин. Гель по¬
мещают на 2 мин в смесь, состоящую из этанола
безводного Р и буферного раствора А (10:90, об/об).
Проводят электрофорез в течение 20 мин. Гель поме¬
щают на 2 мин в смесь, состоящую из этанола безво¬
дного Р и буферного раствора А (30:70, об/об). Прово¬
дят электрофорез в течение 20 мин. Гель окрашивают
окрашивающим раствором в течение 10 мин. Краску
удаляют с геля в течение 15 мин в потоке водопрово¬
дной воды, затем в течение (10—15) мин водой Р пока
не станет видимой полоса на электрофореграмме
раствора сравнения (с). Гель высушивают.
Пригодность системы:
- на электрофореграмме раствора сравнения (с)
обнаруживается полоса;
- полоса на электрофореграмме раствора срав¬
нения (Ь) четко видна и соответствует по расположе¬
нию полосе на электрофореграмме раствора сравне¬
ния (а).
Результаты (не более 2 %): любая вторичная
полоса на электрофореграмме испытуемого раствора
не должна быть интенсивнее полосы на электрофоре¬
грамме раствора сравнения (Ь).
Общий белок (2.5.33, метод 2). Не более 3,0 %
(в пересчете на сухое вещество).
Испытуемый раствор. 1,0 мл раствора 81 дово¬
дят 0,1 М раствором натрия гидроксида до объема
50.0 мл.
Стандартные растворы. Около 0,100 г альбу¬
мина бычьего Р (точная навеска) растворяют в 0,1 М
растворе натрия гидроксида и доводят до объема
50.0 мл этим же растворителем. Все дополнительные
разведения проводят с использованием 0,1 М раст¬
вора натрия гидроксида.
Хлориды (2.4.4). Не более 0,5 %. 1 мл раствора
82 доводят водой Р до объема 15 мл. Кислоту азот¬
ную разведенную не прибавляют. Эталон готовят с
использованием 5 мл эталонного раствора хлори¬
да (5 ррт С1) Р и 10 мл воды Р. Полученный раствор
должен выдерживать испытания на хлориды.
Целекоксиб
1083
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 12,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 4 ч.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
81арНу1ососсиз аигеиз: отсутствие (2.6.13).
Рзеидотопаз аегид/поза: отсутствие (2.6.13).
ЕзсЬепсЫа со1г. отсутствие (2.6.13).
За!топеИа: отсутствие (2.6.13).
Грамотрицательные бактерии, устойчивые к жел¬
чи: отсутствие (2.6.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор (а). 0,100 г (т,) испытуе¬
мого образца, растворяют в воде Р и доводят до объ¬
ема 100,0 мл этим же растворителем.
Испытуемый раствор (Ь). 5,0 мл испытуемого
раствора (а) доводят водой Р до объема 50,0 мл.
Раствор сравнения (а). 0,100 г (т0) ФСО хондро-
итина натрия сульфата, предварительно высушен¬
ного, как указано в испытании «Потеря в массе при
высушивании», растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят водой Р до объема 50,0 мл.
Раствор титранта (а). Взвешивают 4,000 г це-
тилпиридиния хлорида моногидрата Р и доводят до
объема 1000 мл водой Р.
Раствор титранта (Ь). Взвешивают 1,000 г це-
тилпиридиния хлорида моногидрата Р и доводят до
объема 1000 мл водой Р.
Проводят визуальное или фотометрическое ти¬
трование следующим образом.
Визуальное титрование. 40,0 мл раствора
сравнения (а) и 40,0 мл испытуемого раствора (а)
титруют раствором титранта (а). Раствор становится
мутным. В конечной точке жидкость становится про¬
зрачной с почти белыми взвешенными частицами.
Частицы становятся более заметными, если перед
началом титрования прибавить 0,1 мл 1 % метило¬
вого синего Р. Взвешенные частицы более заметны
на синем фоне.
Фотометрическое титрование. 50,0 мл раст¬
вора сравнения (Ь) и 50,0 мл испытуемого раствора
(Ь) титруют раствором титранта (Ь). Для определения
конечной точки используют подходящий автоматиче¬
ский титратор, оборудованный фототродом при под¬
ходящей длине волны (не является критической) в
видимой области.
Содержание хондроитина натрия сульфата в
процентах рассчитывают по формуле:
У,т0 Ю0
V^Щ 100-1 ’
где:
у0 — объем подходящего раствора титранта при
титровании соответствующего раствора сравнения, мл;
у, — объем подходящего раствора титранта при ти¬
тровании соответствующего испытуемого раствора, мл;
Н — потеря в массе при высушивании испытуе¬
мого образца, %;
I — содержание Н20(С14Н19М№20143)Х в ФСО
хондроитина натрия сульфата, %.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере, в защи¬
щенном от света месте.
МАРКИРОВКА
На этикетке указывают источник вещества (из хря¬
щевой ткани морских или наземных видов животных).
07/2016:2591
ЦЕЛЕКОКСИБ
Се1есох1Ьит
СЕ1.ЕСОХ1В
с 17Н р N О 5 М.м. 381,4
[169590-42-5]
ОПРЕДЕЛЕНИЕ
4-[5-(4-Метилфенил)-3-(трифторметил)-1Н-пиразол-
1 -ил]бензолсульфонамид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический или
аморфный порошок.
Практически нерастворим в воде, от легко рас¬
творим до растворим в этаноле, растворим в мети-
ленхлориде.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО целекоксиба #или спектр, пред¬
ставленный на рисунке #2591.-1#.
Если полученные спектры отличаются, то испы¬
туемый образец и ФСО целекоксиба растворяют по
отдельности в 2-пропаноле Р и выпаривают до сухих
остатков, которые используют для получения новых
спектров.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Смесь растворителей. Вода Р — метанол Р2
(25:75, об/об).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в смеси растворителей и доводят
до объема 100,0 мл этой же смесью растворителей.
136*. Зак. 1060.
1084
Гэсударственная фармакопея Республики Беларусь
Рисунок #2591 .-1. Инфракрасный спектр ФСО целекоксиба
Раствор сравнения (а). 50,0 мг ФСО целекокси¬
ба растворяют в смеси растворителей и доводят до
объема 100,0 мл этой же смесью растворителей.
Раствор сравнения (Ь). 3 мг ФСО целекоксиба
примеси Л и 3 мг ФСО целекоксиба примеси В раст¬
воряют в смеси растворителей и доводят до объема
50.0 мл этой же смесью растворителей. 1,0 мл полу¬
ченного раствора доводят раствором сравнения (а)
до объема 25,0 мл.
Раствор сравнения (с). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема
100.0 мл. 1,0 мл полученного раствора доводят сме¬
сью растворителей до объема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилсилиль-
ным эндкепированным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 60 °С;
-подвижная фаза: ацетонитрил Р1 — мета¬
нол Р2 — раствор 2,7 г/л калия дигидрофосфата Р,
предварительно доведенный до рН 3,0 кислотой
фосфорной Р (10:30:60, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
-уравновешивание: подвижная фаза в течение
не менее 1 ч;
- объем вводимой пробы: по 25 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 1,5-кратное вре¬
мя удерживания целекоксиба.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и В используя хроматограмму
раствора сравнения (Ь).
Относительное удерживание (по отношению
к целекоксибу, время удерживания — около 27 мин):
примесь А — около 0,9; примесь В — около 1,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси А и целекоксиба и не менее 1,8 пиками целекок¬
сиба и примеси В.
Расчет содержания примесей: для всех приме¬
сей используют концентрацию целекоксиба в раство¬
ре сравнения (с).
Предельное содержание примесей:
- примесь А: не более 0,4 %;
- неспецифицированные примеси: не более 0,10 %;
- сумма примесей: не более 0,5 %;
- учитываемый предел: 0,05 %.
Тяжелые металлы (2.4.8, метод Н). Не более
0,0020 % (20 ррт).
Смесь растворителей. Вода Р — ацетон Р
(15:85, об/об).
0,50 г испытуемого образца должны выдержи¬
вать испытание на тяжелые металлы. Эталон готовят
с использованием 1 мл эталонного раствора свинца
(ЮрртРЬ)Р.
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 0,400 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца
в платиновом тигле.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 25 мкл испытуемого
раствора и раствора сравнения (а).
Содержание С17Н14Р31\13028 в процентах рассчи¬
тывают с учетом содержания целекоксиба в ФСО це¬
лекоксиба.
Целлюлоза микрокристаллическая
1085
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В.
А. 4-[5-(3-Метилфенил)-3-(трифторметил)-1 Н-
пиразол-1 -ил]бензолсульфонамид.
В. 4-[3-(4-Метилфенил)-5-(трифторметил)-1 Н-
пиразол-1 -ил]бензолсульфонамид.
07/2016:0316
ЦЕЛЛЮЛОЗА
МИКРОКРИСТАЛЛИЧЕСКАЯ
СеНи/озит т1сгоспзШНпит
СЕ1-ИЛ-08Е, М1СКОСКУ8ТАШЫЕ
виде пульпы из растительного волокнистого материа¬
ла) минеральными кислотами.
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый мелкий или гранулиро¬
ванный порошок.
Практически нерастворим в воде, ацетоне, этано¬
ле безводном, толуоле, разведенных кислотах и рас¬
творе 50 г/л натрия гидроксида.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Около 10 мг испытуемого образца помещают
на часовое стекло и диспергируют в 2 мл раствора
цинка хлорида йодированного Р. Испытуемый обра¬
зец приобретает фиолетово-голубую окраску.
B. Степень полимеризации не более 350. 1,300 г
испытуемого образца помещают в коническую кол¬
бу емкостью 125 мл, прибавляют 25,0 мл воды Р и
25,0 мл раствора меди-этилендиамина щелочного Р.
Немедленно пропускают азот Р, закрывают пробкой
и встряхивают до полного растворения. Необходи¬
мый объем полученного раствора помещают в под¬
ходящий капиллярный вискозиметр (2.2.9). Раствор
термостатируют при температуре (25±0,1)°С не ме¬
нее 5 мин. Отмечают время вытекания (/.,) в секундах
между двумя отметками вискозиметра. Рассчитывают
кинематическую вязкость (и,) раствора по формуле:
где:
/с, — постоянная вискозиметра.
Смешивают одинаковые объемы раствора меди-
этилендиамина щелочного Р и воды Р и измеряют
время вытекания (?2), используя подходящий
капиллярный вискозиметр. Рассчитывают кинемати¬
ческую вязкость (о2) растворителя по формуле:
и2 — ^2 * ^2 ’
где:
к2 — постоянная вискозиметра.
Рассчитывают относительную вязкость (потн)
испытуемого образца по формуле:
Ол
Потн = —■
и2
Определяют истинную вязкость (1п]ист) по таблице
0316.-1, используя интерполяцию.
Рассчитывают степень полимеризации (Р) по
формуле:
ОПРЕДЕЛЕНИЕ
Очищенная частично деполимеризованная цел¬
люлоза, полученная обработкой альфа-целлюлозы (в
р 95-Ыии.
/77[(100-Ь)/100]’
где:
т — масса испытуемого образца, г;
Ь — потеря в массе при высушивании, %.
ИСПЫТАНИЯ
Растворимость. 50 мг испытуемого образца
растворяют в 10 мл раствора меди тетрааммиаката
аммиачного Р. Растворяется полностью без остатка.
рН (2.2.3). От 5,0 до 7,5. 5 г испытуемого образ¬
ца встряхивают с 40 мл воды, свободной от углерода
диоксида, Р, в течение 20 мин и центрифугируют. Из¬
меряют рН надосадочной жидкости.
Электропроводность (2.2.38). Электропровод¬
ность испытуемого раствора не должна превышать
электропроводность воды более чем на 75 мкСм см"1.
137. Зак. 1060.
1086
Гэсударственная фармакопея Республики Беларусь
Таблица знамений истинной вязкости
Таблица 0316.-1.
Истинная вязкость 1п]ист
при различных значениях относительной вязкости [/у]отн
И*™
^отн
0,00
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
1,1
0,098
0,106
0,115
0,125
0,134
0,143
0,152
0,161
0,170
0,180
1,2
0,189
0,198
0,207
0,216
0,225
0,233
0,242
0,250
0,259
0,268
1,3
0,276
0,285
0,293
0,302
0,310
0,318
0,326
0,334
0,342
0,350
1,4
0,358
0,367
0,375
0,383
0,391
0,399
0,407
0,414
0,422
0,430
1,5
0,437
0,445
0,453
0,460
0,468
0,476
0,484
0,491
0,499
0,507
1,6
0,515
0,522
0,529
0,536
0,544
0,551
0,558
0,566
0,573
0,580
1,7
0,587
0,595
0,602
0,608
0,615
0,622
0,629
0,636
0,642
0,649
1,8
0,656
0,663
0,670
0,677
0,683
0,690
0,697
0,704
0,710
0,717
1,9
0,723
0,730
0,736
0,743
0,749
0,756
0,762
0,769
0,775
0,782
2,0
0,788
0,795
0,802
0,809
0,815
0,821
0,827
0,833
0,840
0,846
2,1
0,852
0,858
0,864
0,870
0,876
0,882
0,888
0,894
0,900
0,906
2,2
0,912
0,918
0,924
0,929
0,935
0,941
0,948
0,953
0,959
0,965
2,3
0,971
0,976
0,983
0,988
0,994
1,000
1,006
1,011
1,017
1,022
2,4
1,028
1,033
1,039
1,044
1,050
1,056
1,061
1,067
1,072
1,078
2,5
1,083
1,089
1,094
1,100
1,105
1,111
1,116
1,121
1,126
1,131
2,6
1,137
1,142
1,147
1,153
1,158
1,163
1,169
1,174
1,179
1,184
2,7
1,190
1,195
1,200
1,205
1,210
1,215
1,220
1,225
1,230
1,235
2,8
1,240
1,245
1,250
1,255
1,260
1,265
1,270
1,275
1,280
1,285
2,9
1,290
1,295
1,300
1,305
1,310
1,314
1,319
1,324
1,329
1,333
3,0
1,338
1,343
1,348
1,352
1,357
1,362
1,367
1,371
1,376
1,381
3,1
1,386
1,390
1,395
1,400
1,405
1,409
1,414
1,418
1,423
1,427
3,2
1,432
1,436
1,441
1,446
1,450
1,455
1,459
1,464
1,468
1,473
3,3
1,477
1,482
1,486
1,491
1,496
1,500
1,504
1,508
1,513
1,517
3,4
1,521
1,525
1,529
1,533
1,537
1,542
1,546
1,550
1,554
1,558
3,5
1,562
1,566
1,570
1,575
1,579
1,583
1,587
1,591
1,595
1,600
3,6
1,604
1,608
1,612
1,617
1,621
1,625
1,629
1,633
1,637
1,642
3,7
1,646
1,650
1,654
1,658
1,662
1,666
1,671
1,675
1,679
1,683
3,8
1,687
1,691
1,695
1,700
1,704
1,708
1,712
1,715
1,719
1,723
3,9
1,727
1,731
1,735
1,739
1,742
1,746
1,750
1,754
1,758
1,762
4,0
1,765
1,769
1,773
1,777
1,781
1,785
1,789
1,792
1,796
1,800
4,1
1,804
1,808
1,811
1,815
1,819
1,822
1,826
1,830
1,833
1,837
4,2
1,841
1,845
1,848
1,852
1,856
1,859
1,863
1,867
1,870
1,874
4,3
1,878
1,882
1,885
1,889
1,893
1,896
1,900
1,904
1,907
1,911
4,4
1,914
1,918
1,921
1,925
1,929
1,932
1,936
1,939
1,943
1,946
4,5
1,950
1,954
1,957
1,961
1,964
1,968
1,971
1,975
1,979
1,982
4,6
1,986
1,989
1,993
1,996
2,000
2,003
2,007
2,010
2,013
2,017
4,7
2,020
2,023
2,027
2,030
2,033
2,037
2,040
2,043
2,047
2,050
4,8
2,053
2,057
2,060
2,063
2,067
2,070
2,073
2,077
2,080
2,083
4,9
2,087
2,090
2,093
2,097
2,100
2,103
2,107
2,110
2,113
2,116
5,0
2,119
2,122
2,125
2,129
2,132
2,135
2,139
2,142
2,145
2,148
5,1
2,151
2,154
2,158
2,160
2,164
2,167
2,170
2,173
2,176
2,180
5,2
2,183
2,186
2,190
2,192
2,195
2,197
2,200
2,203
2,206
2,209
5,3
2,212
2,215
2,218
2,221
2,224
2,227
2,230
2,233
2,236
2,240
5,4
2,243
2,246
2,249
2,252
2,255
2,258
2,261
2,264
2,267
2,270
5,5
2,273
2,276
2,279
2,282
2,285
2,288
2,291
2,294
2,297
2,300
5,6
2,303
2,306
2,309
2,312
2,315
2,318
2,320
2,324
2,326
2,329
5,7
2,332
2,335
2,338
2,341
2,344
2,347
2,350
2,353
2,355
2,358
5,8
2,361
2,364
2,367
2,370
2,373
2,376
2,379
2,382
2,384
2,387
5,9
2,390
2,393
2,396
2,400
2,403
2,405
2,408
2,411
2,414
2,417
Целлюлоза микрокристаллическая
1087
Истинная вязкость 1п]ист
при различных значениях относительной вязкости [/?]
отн
0,00
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
6,0
2,419
2,422
2,425
2,428
2,431
2,433
2,436
2,439
2,442
2,444
6,1
2,447
2,450
2,453
2,456
2,458
2,461
2,464
2,467
2,470
2,472
6,2
2,475
2,478
2,481
2,483
2,486
2,489
2,492
2,494
2,497
2,500
6,3
2,503
2,505
2,508
2,511
2,513
2,516
2,518
2,521
2,524
2,526
6,4
2,529
2,532
2,534
2,537
2,540
2,542
2,545
2,547
2,550
2,553
6.5
2,555
2558
2,561
2,563
2,566
2,568
2,571
2,574
2,576
2,579
6.6
2.581
2584
2,587
2,590
2,592
2,595
2,597
2,600
2,603
2,605
6,7
2608
2610
2,613
2,615
2,618
2,620
2,623
2,625
2,627
2,630
€*
2633
2635
2,637
2,640
2,643
2,645
2,648
2,650
2,653
2,655
5..3
2558
2660
2,663
2,665
2,668
2,670
2,673
2,675
2,678
2,680
е' .и
2,683
2,685
2,687
2,690
2,693
2,695
2,698
2,700
2,702
2,705
7.1
2,707
2,710
2,712
2,714
2,717
2,719
2,721
2,724
2,726
2,729
12
2,731
2,733
2,736
2,738
2,740
2,743
2,745
2,748
2,750
2,752
7,3
2,755
2,757
2,760
2,762
2,764
2,767
2,769
2,771
2,774
2,776
7,4
2,779
2,781
2,783
2,786
2,788
2,790
2,793
2,795
2,798
2,800
7.5
2,802
2,805
2,807
2,809
2,812
2,814
2,816
2,819
2,821
2,823
7,6
2,826
2,828
2,830
2,833
2,835
2,837
2,840
2,842
2,844
2,847
7,7
2,849
2,851
2,854
2,856
2,858
2,860
2,863
2,865
2,868
2,870
7,8
2,873
2,875
2,877
2,879
2,881
2,884
2,887
2,889
2,891
2,893
7.9
2895
2898
2,900
2,902
2,905
2,907
2,909
2,911
2,913
2,915
2918
2920
2,922
2,924
2,926
2,928
2,931
2,933
2,935
2,937
* *
2939
2942
2,944
2,946
2,948
2,950
2,952
2,955
2,957
2,959
82
2.961
2963
2,966
2,968
2,970
2,972
2,974
2,976
2,979
2,981
8.3
2.983
2,985
2,987
2,990
2,992
2,994
2,996
2,998
3,000
3,002
8,4
3,004
3,006
3,008
3,010
3,012
3,015
3,017
3,019
3,021
3,023
8,5
3,025
3,027
3,029
3,031
3,033
3,035
3,037
3,040
3,042
3,044
8,6
3,046
3,048
3,050
3,052
3,054
3,056
3,058
3,060
3,062
3,064
8,7
3,067
3,069
3,071
3,073
3,075
3,077
3,079
3,081
3,083
3,085
8,8
3,087
3,089
3,092
3,094
3,096
3,098
3,100
3,102
3,104
3,106
8,9
3,108
3,110
3,112
3,114
3,116
3,118
3,120
3,122
3,124
3,126
9,0
3,128
3,130
3,132
3,134
3,136
3,138
3,140
3,142
3,144
3,146
5 «
3,148
3,150
3,152
3,154
3,156
3,158
3,160
3,162
3,164
3,166
92
3,168
3,170
3,172
3,174
3,176
3,178
3,180
3,182
3,184
3,186
9,3
3,188
3,190
3,192
3,194
3,196
3,198
3,200
3,202
3,204
3,206
9,4
3,208
3,210
3,212
3,214
3,215
3,217
3,219
3,221
3,223
3,225
9,5
3,227
3,229
3,231
3,233
3,235
3,237
3,239
3,241
3,242
3,244
9,6
3,246
3,248
3,250
3,252
3,254
3,256
3,258
3,260
3,262
3,264
9,7
3,266
3,268
3,269
3,271
3,273
3,275
3,277
3,279
3,281
3,283
9,8
3,285
3,287
3,289
3,291
3,293
3,295
3,297
3,298
3,300
3,302
9.9
3,304
3,305
3,307
3,309
3,311
3,313
3,316
3,318
3,320
3,321
Истинная вязкость [п]ист
при различных значениях относительной вязкости [/?'
отн
№1ист
Пшт
ад
0,1
0.2
0,3
0,4
0,5
0,6
0.7
0,8
0,9
10
3.32
3.34
3,36
3,37
3,39
3,41
3,43
3,45
3,46
3,48
11
3.50
3,52
3,53
3,55
3,56
3,58
3,60
3,61
3,63
3,64
< 12
3,66
3.68
3,69
3,71
3,72
3,74
3,76
3,77
3,79
3,80
13
3,80
3,83
3,85
3,86
3,88
3,89
3,90
3,92
3,93
3,95
14
3,96
3,97
3,99
4,00
4,02
4,03
4,04
4,06
4,07
4,09
15
4,10
4,11
4,13
4,14
4,15
4,17
4,18
4,19
4,20
4,22
16
4,23
4,24
4,25
4,27
4,28
4,29
4,30
4,31
4,33
4,34
17
4,35
4,36
4,37
4,38
4,39
4,41
4,42
4,43
4,44
4,45
18
4,46
4,47
4,48
4,49
4,50
4,52
4,53
4,54
4,55
4,56
19
4,57
4,58
4,59
4,60
4,61
4,62
4,63
4,64
4,65
4,66
137*. Зак. 1060.
1088
Гэсударственная фармакопея Республики Беларусь
В качестве испытуемого раствора используют надо-
садочную жидкость, полученную в испытании «рН».
После установления стабильных показаний измеряют
электропроводность надосадочной жидкости. Изме¬
ряют электропроводность воды, которая использова¬
лась при приготовлении испытуемого раствора.
Вещества, извлекаемые эфиром. Не более
0,05 %. 10,0 г испытуемого образца помещают в хро¬
матографическую колонку с внутренним диаметром
около 20 мм. Пропускают через колонку 50 мл эфира,
свободного от пероксидов, Р. Элюат выпаривают до¬
суха. Остаток сушат при температуре 105 °С в тече¬
ние 30 мин, охлаждают в эксикаторе и взвешивают.
Параллельно проводят контрольный опыт. Разница
между массой остатка и массой, полученной в ходе
контрольного опыта, не должна превышать 5 мг.
Растворимые в воде вещества. Не более
0,25 %. 5,0 г испытуемого образца встряхивают с
80 мл воды Р в течение 10 мин, фильтруют через бу¬
мажный фильтр под вакуумом во взвешенную колбу,
выпаривают досуха на водяной бане, не допуская об¬
угливания. Остаток сушат при температуре 105 °С в
течение 1 ч, выдерживают в эксикаторе и взвешивают.
Параллельно проводят контрольный опыт. Разница
между массой остатка и массой, полученной в ходе
контрольного опыта, не должна превышать 12,5 мг.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
ЕзсЬепсЫа соН: отсутствие (2.6.13).
Рзеидотопаз аегидтоза: отсутствие (2.6.13).
81арЬу1ососсиз аигеиз: отсутствие (2.6.13).
8а1топе11а: отсутствие (2.6.13).
ФУНКЦИОНАЛЬНО-ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
В данном разделе приведена информация о ха¬
рактеристиках, которые являются важными пара¬
метрами, связанными с одной или более функцией
субстанции, используемой в качестве вспомога¬
тельного вещества (см. статью 5.15). Данный раз¬
дел не является обязательным, и для подтвержде¬
ния соответствия требованиям частной статьи
проверка указанных характеристик не обязательна.
Однако контроль данных характеристик может
вносить вклад в качество лекарственного средства
путем достижения постоянства производственно¬
го процесса и улучшения свойств лекарственного
средства при его использовании. Предлагаемые ме¬
тоды испытаний являются подходящими для ука¬
занных целей, однако другие методы также могут
быть использованы. В случае указания результатов
конкретных характеристик требуется указывать
также и методы проведения испытаний.
Следующие характеристики могут быть важ¬
ны для микрокристаллической целлюлозы, использу¬
емой в качестве связующего вещества, разбавите¬
ля или разрыхлителя.
Распределение частиц по размерам (2.9.31
или 2.9.38).
Текучесть порошка (2.9.36).
07/2016:0540
ЦЕТИЛОВЫЙ СПИРТ
А1со1ю1 се(уНсиз
СЕТП. А1.СОН01.
ОПРЕДЕЛЕНИЕ
Смесь твердых спиртов, состоящая в основном
из гексадекан-1-ола (С^Н^О; М.м. 242,4) животного
или растительного происхождения.
Содержание: не менее 95,0 % С^Н^О.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая воскообразная масса, по¬
рошок, хлопья или гранулы.
Практически нерастворим в воде, легко раство¬
рим или умеренно растворим в 96 % спирте; в рас¬
плавленном состоянии смешивается с маслами рас¬
тительного и животного происхождения, маслом вазе¬
линовым и расплавленным ланолином.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные как
указано в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 0,50 г испытуемого образца раство¬
ряют в 20 мл кипящего 96 % спирта Р и охлаждают.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона В(К)6.
Температура плавления (2.2.14). От 46 °С до
52 °С.
Кислотное число (2.5.1). Не более 1,0.
Гидроксильное число (2.5.3, метод А). От 218
до 238.
Йодное число (2.5.4, метод А). Не более 2,0.
2,00 г испытуемого образца растворяют в метилен-
хлориде Р и доводят до объема 25 мл этим же раст¬
ворителем.
Число омыления (2.5.6). Не более 2,0.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28): определение
проводят методом нормализации.
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в 96% спирте Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
Раствор сравнения (а). 50 мг ФСО цетилового
спирта растворяют в 96 % спирте Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 50 мг ФСО стеариново¬
го спирта растворяют в 96% спирте Р и доводят до
объема 10 мл этим же растворителем.
Цетилпальмитат
1089
Раствор сравнения (с). Смешивают 1 мл раст¬
вора сравнения (а) и 1 мл раствора сравнения (Ь) и
доводят 96 % спиртом Р до объема 10 мл.
Условия хроматографирования:
- колонка длиной 30 м и внутренним диаметром
0,32 мм, покрытая слоем поли(диметил)силоксана Р
(толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—20
150 — 250
20—40
250
. Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: по 1 мкл испытуемого
раствора и растворов сравнения (а) и (с).
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 5,0 между пиками цети¬
лового спирта и стеарилового спирта.
Рассчитывают процентное содержание С^Н^О.
07/2016:1906
ЦЕТИЛПАЛЬМИТАТ
Се1уЧз ра1тПаз
СЕТП РА1-М1ТАТЕ
ОПРЕДЕЛЕНИЕ
Смесь С14—С18 эфиров лауриловой (додекано-
вой), миристиновой (тетрадекановой), пальмитиновой
(гексадекановой) и стеариновой (октадекановой) кис¬
лот («воск цетиловых эфиров»).
Содержание (в пересчете на гексадецилгексаде-
каноат): не менее 10,0 % и не более 20,0 % для це-
тилпальмитата 15; не менее 60,0 % и не более 70,0 %
для цетилпальмитата 65; не менее 90,0 % для цетил-
пальмитата 95.
СВОЙСТВА (ОПИСАНИЕ)
Белые или почти белые воскообразные пласти¬
ны, хлопья или порошок.
Практически нерастворим в воде, растворим в
кипящем этаноле безводном и метиленхлориде, мало
растворим в петролейном эфире, практически нераст¬
ворим в этаноле безводном.
Температура плавления: около 45 °С для цетип-
пальмитата 15 и цетилпальмитата 65 и около 52 °С
для цетилпальмитата 95.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение», и на
хроматограмме испытуемого раствора обнаружива¬
ются основные характерные пики.
B. Испытуемый образец выдерживает испытание
«Число омыления», как указано в разделе «Испытания».
ИСПЫТАНИЯ
Цветность (2.2.2, Метод II). 4,0 г испытуемого
образца растворяют в метиленхлориде Р и доводят
до объема 20 мл этим же растворителем. Окраска
полученного раствора должна быть не интенсивнее
эталона У(Ж)6.
Кислотное число (2.5.1). Не более 4,0. 10,0 г
испытуемого образца растворяют в 50 мл указанной
смеси растворителей при нагревании с обратным хо¬
лодильником на водяной бане в течение 5 мин.
Гидроксильное число (2.5.3, метод А). Не бо¬
лее 20,0.
Йодное число (2.5.4, метод А). Не более 2,0.
Число омыления (2.5.6). От 105 до 120. Нагре¬
вают с обратным холодильником в течение 2 ч.
Щелочность. 2,0 г испытуемого образца раство¬
ряют при слабом нагревании в смеси из 1,5 мл 96 %
спирта Р и 3 мл толуола Р. Прибавляют 0,05 мл
раствора 0,4 г/л бромфенолового синего Р в 96%
спирте Р. При прибавлении не более 0,4 мл 0,01 М
раствора кислоты хлористоводородной должно по¬
явиться желтое окрашивание.
Никель (2.4.31). Не более 0,0001 % (1 ррт).
Вода (2.5.12). Не более 0,3 %. Определение про¬
водят из 1,0 г испытуемого образца. В качестве рас¬
творителя используют смесь равных объемов мета¬
нола безводного Р и метиленхлориде Р.
Общая зола (2.4.16). Не более 0,2 %. Определе¬
ние проводят из 1,0 г испытуемого образца.
^Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28): определение
проводят методом нормализации.
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в гексане Р и доводят до объема
20,0 мл этим же растворителем.
Раствор сравнения (а). 20,0 мг ФСО цетилпаль¬
митата 95 растворяют в гексане Р и доводят до объ¬
ема 20,0 мл этим же растворителем.
Раствор сравнения (Ь). 20,0 мг ФСО цетилпаль¬
митата 15 растворяют в гексане Р и доводят до объ¬
ема 20,0 мл этим же растворителем.
Условия хроматографирования:
-колонка из нержавеющей стали длиной 10м
и внутренним диаметром 0,53 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 2,65 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 6,5 мл/мин;
- деление потока: 1:10;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
100 — 300
10—15
300
Блок ввода проб
350
Детектор
350
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Относительное удерживание (по отношению
к цетилпальмититу, время удерживания — около 9
мин): цетиловый спирт — около 0,3; пальмитиновая
кислота — около 0,4; лауриловый эфир — около
0,8; миристиновый эфир — около 0,9; стеариновый
эфир — около 1,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками цетил¬
пальмитата и цетилстеарата.
138. Зак. 1060.
1090
Гэсударственная фармакопея Республики Беларусь
ХРАНЕНИЕ
При температуре не выше 25 °С.
МАРКИРОВКА
Указывают тип цетилпальмитата.
07/2016:0379
ЦЕТИЛПИРИДИНИЯ ХЛОРИД
Се1у1рупс1тН сЫопс1ит
СЕТПРУШ01ЫШМ СШОКЮЕ
С21Н38С1Ы ■ Н20 М.м. 358,0
[6004-24-6]
ОПРЕДЕЛЕНИЕ
1-Гексадецилпиридиния хлорид.
Содержание: не менее 96,0 % и не более 101,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый слегка мыльный на
ощупь порошок.
Растворим в воде и в 96 % спирте.
Водный раствор дает обильную пену при встря¬
хивании.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
А. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем. 5,0 мл полученного
раствора доводят водой Р до объема 100,0 мл.
Диапазон длин волн: от 240 нм до 300 нм.
Максимум поглощения: при 259 нм.
Плечи: при около 254 нм и при около 265 нм.
Удельный показатель поглощения в максимуме
(в пересчете на безводное вещество): от 126 до 134.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цетилпиридиния хлорида #или
спектр, представленный на рисунке #0379.-1#.
C. К 5 мл раствора натрия гидроксида разве¬
денного Р прибавляют 0,1 мл раствора бромфеноло-
вого синего Р1 и 5 мл хлороформа Р и встряхивают.
Хлороформный слой бесцветный. Прибавляют 0,1 мл
раствора 5, приготовленного как указано в разделе
«Испытания», и встряхивают. Слой хлороформа окра¬
шивается в синий цвет.
й. Раствор 5 дает реакцию (а) на хлориды (2.3.7).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида Р, и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.7). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Раствор 5 должен
быть бесцветным.
Кислотность. К 50 мл раствора 3 прибавляют
0,1 мл раствора фенолфталеина Р. При прибавле¬
нии не более 2,5 мл 0,02 М раствора натрия гидрок¬
сида окраска индикатора должна измениться.
Амины и соли аминов. 5,0 г испытуемого об¬
разца растворяют при нагревании в 20 мл смеси из
3 объемов 7 М раствора кислоты хлористводород-
Рисунок #0379.-1. Инфракрасный спектр ФСО цетилпиридиния хлорида
Цетиризина дигидрохлорид
1091
ной и 97 объемов метанола Р и прибавляют 100 мл
2-пропанола Р. Через раствор медленно пропуска¬
ют струю азотаР. Постепенно прибавляют 12,0 мл
0,1 М раствора тетрабутиламмония гидроксида
и строят кривую потенциометрического титрования
(2.2.20). Если кривая имеет 2 точки перегиба, то объ¬
ем титранта, прибавленный между 2-мя точками,
должен быть не более 5,0 мл. Если кривая не имеет
точек перегиба, то испытуемый образец не выдержи¬
вает испытание. Если кривая тлеет одну точку пере¬
гиба, то испытание повторяют, но прибавляют 3,0 мл
раствора 25,0 г/л дшлетипдециламина Р в 2-пропа¬
ноле Р перед титрованием. Если кривая титрования
после прибавления 12.0 мл титранта имеет только
одну точку перегиба, то испытуемый образец не вы¬
держивает испытание.
Вода (2.5.12). Не менее 4,5 % и не более 5,5 %.
Определение проводят из 0,300 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,00 г испытуемого образца растворяют в воде Р
и доводят до объема 100,0 мл этим же растворите¬
лем. 25,0 мл полученного раствора переносят в де¬
лительную воронку, прибавляют 25 мл хлорофор¬
ма Р, 10 мл 0,1 М раствора натрия гидроксида и
10,0 мл свежеприготовленного раствора 50 г/л калия
йодида Р, энергично встряхивают и после разделе¬
ния отбрасывают слой хлороформа. Водный слой
встряхивают с хлороформом Р трижды порциями по
10 мл и отбрасывают слои хлороформа. К водному
слою прибавляют 40 мл кислоты хлористоводород¬
ной Р, охлаждают и титруют 0,05 М раствором калия
йодата до почти полного исчезновения темно-ко¬
ричневой окраски. Прибавляют 2 мл хлороформа Р
и продолжают титрование, интенсивно встряхивая,
до неизменной окраски слоя хлороформа. Проводят
контрольный опыт для смеси из 10,0 мл свежепри¬
готовленного раствора 50 г/л калия йодида Р, 20 мл
воды Р и 40 мл кислоты хлористоводородной Р.
1 мл 0,05 М раствора калия йодата соответ¬
ствует 34,0 мг С21Н38С1М.
07/2016:1084
ЦЕТИРИЗИНА ДИГИДРОХЛОРИД
СеНпят сИМудгосЫоп'дит
СЕТ1Ш21ЫЕ 0/НУ0Я0СМ.0Я/0Е
.со2н
• 2 НС1
и энантиомер
М.м. 461,8
С21Н25С1М203
[83881-52-1]
2НС!
ОПРЕДЕЛЕНИЕ
(Я5)-2-[2-[4-[(4-Хлорфенил)фенилметил]пиперазин-
1-ил]этокси]уксусной кислоты дигидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок.
138*. Зак. 1060.
1092
Гэсударственная фармакопея Республики Беларусь
Легко растворим в воде, практически нераство¬
рим в ацетоне и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, О.
Вторая идентификация: А, С, О.
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 50 мл раствора 10,3 г/л кислоты
хлористоводородной Р и доводят до объема 100,0 мл
этим же растворителем. 10,0 мл полученного раст¬
вора доводят раствором 10,3 г/л кислоты хлористо¬
водородной Р до объема 100,0 мл.
Диапазон длин волн: от 210 нм до 350 нм.
Максимум поглощения: при 231 нм.
Удельный показатель поглощения в максимуме:
от 359 до 381.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цетиризина дигидрохлорида
#или спектр, представленный на рисунке #1084.-1#.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор: 10 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения (а). 10 мг ФСО цетиризина
дигидрохлорида растворяют в воде Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО хлорфенами-
на малеата растворяют в воде Р и доводят до объ¬
ема 5 мл этим же растворителем. К 1 мл полученного
раствора прибавляют 1 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем силикаге-
ля вР254 Р.
Подвижная фаза: раствор аммиака Р — мета¬
нол Р — метиленхлорид Р( 1:10:90, об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается пятно, соответствующее по
расположению и размеру основному пятну на хрома¬
тограмме раствора сравнения (а).
й. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ВУ(КЖ)7.
рН (2.2.3). От 1,2 до 1,8. Измеряют рН раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 2 мг ФСО цетиризина
дигидрохлорида и 2 мг ФСО цетиризина примеси А
растворяют в подвижной фазе и доводят до объема
10.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят подвижной фазой до объема
100.0 мл.
Раствор сравнения (Ь). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
5.0 мл полученного раствора доводят подвижной фа¬
зой до объема 100,0 мл.
Раствор сравнения (с). Содержимое контейнера
с ФСО цетризина для идентификации пиков (содер¬
жит примеси В, С, О, Е и Р) растворяют в 5,0 мл под¬
вижной фазы.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем для хромато¬
графии Р с размером частиц 5 мкм;
- подвижная фаза: кислота серная разве¬
денная Р — вода Р — ацетонитрил Р (0,4:6,6:93,
об/об/об);
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания цетиризина.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, О-, Е и Р, используя хрома¬
тограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО цетризина для идентификации
пиков; идентифицируют пик примеси А, используя
хроматограмму раствора сравнения (а).
Относительное удерживание (по отношению к
цетризину, время удерживания — около 10 мин): при¬
месь О — около 0,6; примесь В — около 0,8; примесь
С — около 0,9; примесь Е — около 1,2; примесь Р —
около 1,37; примесь А—около 1,42.
Пригодность хроматографической системы:
раствор сравнения (с):
- коэффициент разделения пиков: не менее 5,0
(Нр — высота пика примеси С относительно базовой
линии; Ну— расстояние между базовой линией и ниж¬
ней точкой кривой, разделяющей пик примеси С и пик
цетиризина).
Предельное содержание примесей (для расче¬
та содержания примесей умножают площади пиков
на соответствующие поправочные коэффициенты:
для примеси А — 0,7; для примеси С — 1,9; для при¬
меси О — 0,6; для примеси Е — 1,3; для примеси
Р — 1,9):
- примеси А, В, С, О, Е, Р (не более 0,15 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, С, О, Е и Р,
не должны превышать 0,75 площади основного пика
на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков
примесей А, В, С, О, Е, Р, не должна превышать 0,5
площади основного пика на хроматограмме раст¬
вора сравнения (Ь);
- сумма примесей (не более 0,3 %); на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
1,5-кратной площади основного пика на хромато¬
грамме раствора сравнения (Ь);
Цетостеариловый спирт
1093
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 70 мл
смеси из воды Р и ацетона Р (30:70, об/об). Титруют
0,1 М раствором натрия гидроксида потенциометри-
чески (2.2.20) до второго скачка титрования.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 15,39 мг С21Н25С1М203-2НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): С.
А. (/?$)-1 -[(4-Хлорфенил)фенилметил]пиперазин.
В. (Я5)-2-[4-[(4-Хлорфенил)фенилметил]пиперазин-
1-ил]уксусная кислота.
С. (Я5)-2-[2-[4-[(2-Хлорфенил)фенилметил]пипе-
разин-1 -ил]этокси]уксусная кислота.
0.1,4-бис[(4-Хлорфенил)фенилметил]пиперазин.
Е. (Я5)-2-[2-[2-[4-[(4-Хлорфенил)фенилметил]пи-
перазин-1-ил]этокси]этокси]уксусная кислота (этокси-
цетиризин).
Р. [2-[4-(Дифенилметил)пиперазин-1 -ил]этокси]ук-
сусная кислота.
О. 2-[4-[(/?5)-(4-Хлорфенил)фенилметил]пипе-
разин-1 -ил]этанол.
07/2016:0702
ЦЕТОСТЕАРИЛОВЫЙ СПИРТ
А1со1ю1 се1уИсиз е1 з1еагуНсиз
СЕТ08ТЕАКП. А^СОНО^
ОПРЕДЕЛЕНИЕ
Смесь твердых алифатических спиртов, состо¬
ящая в основном из октадекан-1-ола (стеариновый
спирт; С18Н380; М.м. 270,5) и гексадекан-1-ола (цети¬
ловый спирт; С^Н^О; М.м. 242,4) животного или рас¬
тительного происхождения.
Содержание:
- стеариновый спирт: не менее 40,0 %;
-сумма стеаринового спирта и цетилового
спирта: не менее 90,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белая или бледно-желтая воскообразная масса,
пластинки, хлопья или гранулы.
Практически нерастворим в воде, растворим в
96 % спирте и в петролейном эфире. В расплавлен-
139. Зак. 1060.
1094
Гэсударственная фармакопея Республики Беларусь
ном виде смешивается с жирными маслами, маслом
вазелиновым и расплавленным ланолином.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Просматривают хроматограммы, полученные как
указано в разделе «Количественное определение».
Результаты: два основных пика на хромато¬
грамме испытуемого раствора по времени удержива¬
ния соответствует основным пикам на хроматограмме
раствора сравнения.
ИСПЫТАНИЯ
Раствор 3. 0,50 г испытуемого образца раство¬
ряют в 20 мл кипящего 96 % спирта Р и охлаждают.
Прозрачность {2.2.1). Раствор 3 должен быть
прозрачным.
Цветность {2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона В(К)6.
Температура плавления {2.2.14). От 49 °С до
56 °С.
Кислотное число {2.5.1). Не более 1,0.
Гидроксильное число {2.5.3, метод А). От
208 до 228.
Йодное число {2.5.4, метод А). Не более 2,0.
2,00 г испытуемого образца растворяют в метилен-
хлориде Р и доводят до объема 25 мл этим же раст¬
ворителем.
Число омыления {2.5.6). Не более 2,0.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28): определение
проводят методом внутренней нормализации.
Испытуемый раствор. 0,100 г испытуемого
образца растворяют в 96 % спирте Р и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения. 60 мг ФСО спирта цети¬
лового и 40 мг ФСО спирта стеаринового раство¬
ряют в 96 % спирте Р и доводят до объема 10 мл
этим же растворителем. 1 мл полученного раст¬
вора доводят 96 % спиртом Р до объема 10 мл.
Условия хроматографирования:
- колонка длиной 30 м и внутренним диаме¬
тром 0,32 мм, покрытая слоем поли(диметил)си-
локсана Р (толщина слоя 1 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость потока: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—20
150 — 250
20-40
250
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 5,0 между пиками цети¬
лового спирта и стеарилового спирта.
Рассчитывают процентное содержание С^Н^О и
с18н38о.
07/2016:0801
ЦЕТОСТЕАРИЛОВЫЙ СПИРТ
(ТИП А) ЭМУЛЬСИОННЫЙ
А1со1ю1 се!уНсиз е/ з!еагуНсиз ети1зШсапз А
СЕТ08ТЕАКУ1. А1-СОН01- (ТУРЕ А),
ЕМи1-$1РУШв
ОПРЕДЕЛЕНИЕ
Смесь цетостеарилового спирта и натрия це-
тостеарилсульфата. Может содержать подходящий
буфер.
Содержание:
- спирт цетостеариловый: не менее 80,0 % (в
пересчете на безводное вещество);
- натрия цетостеарилсульфат: не менее 7,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белая или бледно-желтая воскообразная масса,
пластины, хлопья или гранулы.
Растворим в горячей воде с образованием опа¬
лесцирующего раствора, практически нерастворим в
холодной воде, мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С, О.
Вторая идентификация: А, С, О.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,1 г испытуемого об¬
разца растворяют в 10 мл триметилпентана Р при
нагревании на водяной бане. Встряхивают с 2 мл
спирта (70 %, об/об) Р и выдерживают до разделения
слоев. Нижний слой используют в качестве испытуе¬
мого раствора (Ь). 1 мл верхнего слоя доводят триме-
тилпентаном Р до объема 8 мл.
Испытуемый раствор (Ь). Используют нижний
слой, полученный при приготовлении испытуемого
раствора (а).
Раствор сравнения (а). 24 мг ФСО цетилового
спирта Р и 16 мг ФСО стеарилового спирта раство¬
ряют в 10 мл триметилпентана Р.
Раствор сравнения (Ь). 20 мг натрия цетосте-
арилсульфата Р растворяют в 10 мл спирта (70 %,
об/об) Р при нагревании на водяной бане.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: вода Р — ацетон Р — мета¬
нол Р (20:40:40, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
50 г/л фосфорномолибденовой кислоты Р в 96%
спирте Р; нагревают при температуре 120 °С до по¬
явления пятен (около 5 мин).
Результаты:
-на хроматограмме испытуемого раствора (а)
обнаруживаются 2 основных пятна, соответствующих
по расположению и цвету основным пятнам на хрома¬
тограмме раствора сравнения (а);
-на хроматограмме испытуемого раствора (Ь)
обнаруживаются 2 пятна, соответствующих по распо¬
ложению и цвету основным пятнам на хроматограмме
раствора сравнения (Ь).
Цетостеариловый спирт (тип А) эмульсионный
1095
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение
цетостеарилового спирта».
Результаты: 2 основных пика на хроматограм¬
ме испытуемого раствора по времени удерживания
соответствуют 2 основным пикам на хроматограммах
растворов сравнения (а) и (Ь).
C. Испытуемый образец окрашивает бесцветное
пламя в желтый цвет.
й. К 0,3 г испытуемого образца прибавляют
20 мл этанола безводного Р и нагревают до кипения
на водяной бане при перемешивании. Немедленно
фильтруют смесь, выпаривают досуха и прибавляют
к остатку 7 мл воды Р. К 1 мл полученного раствора
прибавляют 0,1 мл раствора 1 г/л метиленового си¬
него Р. 2 мл кислоты серной разведенной Р и 2 мл
метиленхлорида Р и встряхивают. Нижний слой окра¬
шивается в синий цвет.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 2,0.
Йодное число (2.5.4, метод А). Не более 3,0.
2,00 г испытуемого образца растворяют в 25 мл ме¬
тиленхлорида Р.
Число омыления (2.5.6). Не более 2,0.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 2,50 г испытуемого образца.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Цетостеариловый спирт. Газовая хроматогра¬
фия (2.2.28).
Раствор внутреннего стандарта. 0,200 г ФСО
1-нонадеканола растворяют в этаноле Р и доводят
до объема 100,0 мл этим же растворителем.
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в 25,0 мл раствора внутреннего
стандарта, прибавляют 25 мл воды Р и встряхивают
4 раза с пентаном Р порциями по 25 мл, прибавляя на¬
трия хлорид Р при необходимости, для лучшего разде¬
ления слоев. Объединяют верхние слои. Дважды про¬
мывают водой Р порциями по 30 мл, высушивают над
натрия сульфатом безводным Р и фильтруют.
Раствор сравнения (а). 0,100 г ФСО цетилового
спирта растворяют в 25,0 мл раствора внутреннего
стандарта, прибавляют 25 мл воды Р и встряхивают
4 раза с пентаном Р порциями по 25 мл, прибавляя на¬
трия хлорид Р при необходимости, для лучшего разде¬
ления слоев. Объединяют верхние слои. Дважды про¬
мывают водой Р порциями по 30 мл, высушивают над
натрия сульфатом безводным Р и фильтруют.
Раствор сравнения (Ь). 0,100 г ФСО стеарино¬
вого спирта растворяют в 25,0 мл раствора внутрен¬
него стандарта, прибавляют 25 мл воды Р и встряхи¬
вают 4 раза с пентаном Р порциями по 25 мл, при¬
бавляя натрия хлорид Р при необходимости, для
лучшего разделения слоев. Объединяют верхние
слои. Дважды промывают водой Р порциями по 30 мл,
высушивают над натрия сульфатом безводным Р и
фильтруют.
Условия хроматографирования:
-колонка кварцевая капиллярная длиной 25 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р;
- скорость потока: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—20
150 —► 250
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: спирт цетиловый, спирт
стеариновый и 1-нонадеканол.
Процентное содержание спирта цетилового в ис¬
пытуемом образце рассчитывают с учетом содержа¬
ния спирта цетилового в ФСО спирта цетилового по
формуле:
А,
А2 тх,у
А А .у
1
• — •100,
т
где:
Ах — площадь пика спирта цетилового на хрома¬
тограмме испытуемого раствора;
Аху— площадь пика ФСО спирта цетилового на
хроматограмме раствора сравнения (а);
А1 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора;
А2 — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (а);
т — масса навески испытуемого образца, ис¬
пользованной для приготовления испытуемого раст¬
вора, мг;
тху — масса навески ФСО спирта цетилового,
использованной для приготовления раствора сравне¬
ния (а), мг.
Процентное содержание спирта стеарилового в
испытуемом образце рассчитывают с учетом содер¬
жания спирта стеарилового в ФСО спирта стеарило¬
вого по формуле:
А-
4^.1.100,
А А ,у т
где:
Аг — площадь пика спирта стеарилового на хро¬
матограмме испытуемого раствора;
Ау— площадь пика ФСО спирта стеарилового
на хроматограмме раствора сравнения (Ь);
А1 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора;
Аъ — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (Ь);
т — масса навески испытуемого образца, ис¬
пользованной для приготовления испытуемого раст¬
вора, мг;
тху — масса навески ФСО спирта стеарило¬
вого, использованной для приготовления раствора
сравнения (Ь), мг.
Процентное содержание цетостеарилового спир¬
та соответствует суммарному процентному содержа¬
нт* спирта цетилового и спирта стеарилового.
Натрия цетостеарилсульфат. 0,300 г испыту¬
емого образца диспергируют в 25 мл метиленхло¬
рида Р. Прибавляют 50 мл воды Р и 10 мл раствора
димидия бромида и сульфанового синего смешанно¬
го Р. Титруют 0,004 М раствором бензэтония хлори¬
да до изменения окраски нижнего слоя от розовой до
серой. При титровании используют ультразвук и на¬
гревание; перед каждым прибавлением титрованного
раствора дают слоям разделиться.
139*. Зак. 1060.
1096
Гэсударственная фармакопея Республики Беларусь
1 мл 0,004 М раствора бензэтония хлорида со¬
ответствует 1,434 мг натрия цетостеарилсульфата.
МАРКИРОВКА
При необходимости указывают название и кон¬
центрацию добавленного буфера.
07/2016:0802
ЦЕТОСТЕАРИЛОВЫЙ СПИРТ
(ТИП В) ЭМУЛЬСИОННЫЙ
А1со1ю1 се1уИсиз е1 з1еагуНсиз ети!зШсапз В
СЕТ08ТЕАКУ11А1-СОН01. (ТУРЕ В),
ЕМ1Л.31РУ1ЫО
ОПРЕДЕЛЕНИЕ
Смесь цетостеарилового спирта и натрия лау-
рилсульфата. Может содержать подходящий буфер.
Содержание:
- спирт цетостеариловый: не менее 80,0 % (в
пересчете на безводное вещество);
- натрия лаурилсульфат: не менее 7,0 % (в пе¬
ресчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белая или бледно-желтая воскообразная масса,
пластины, хлопья или гранулы.
Растворим в горячей воде с образованием опа¬
лесцирующего раствора, практически нерастворим в
холодной воде, мало растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, С, О.
Вторая идентификация: А, С, О.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,1 г испытуемого об¬
разца растворяют в 10 мл триметилпентана Р при
нагревании на водяной бане. Встряхивают с 2 мл
спирта (70 %, об/об) Р и выдерживают до разделения
слоев. Нижний слой используют в качестве испытуе¬
мого раствора (Ь). 1 мл верхнего слоя доводят триме-
тилпентаном Р до объема 8 мл.
Испытуемый раствор (Ь). Используют нижний
слой, полученный при приготовлении испытуемого
раствора (а).
Раствор сравнения (а). 24 мг ФСО цетилового
спирта Р и 16 мг ФСО стеаринового спирта раство¬
ряют в 10 мл триметилпентана Р.
Раствор сравнения (Ь). 20 мг ФСО натрия ла-
урилсульфата растворяют в 10 мл спирта (70%,
об/об) Р при нагревании на водяной бане.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля октадецилсилильного Р254 Р.
Подвижная фаза: вода Р — ацетон Р — мета¬
нол Р (20:40:40, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
50 г/л фосфорномолибденовой кислоты Р в 96%
спирте Р; нагревают при температуре 120 °С до по¬
явления пятен (около 5 мин).
Результаты:
-на хроматограмме испытуемого раствора (а)
обнаруживаются 2 основных пятна, соответствующих
по расположению и цвету основным пятнам на хрома¬
тограмме раствора сравнения (а);
- на хроматограмме испытуемого раствора (Ь)
одно из пятен соответствует по расположению и цвету
основному пятну на хроматограмме раствора сравне¬
ния (Ь).
B. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение
цетостеарилового спирта».
Результаты: 2 основных пика на хроматограм¬
ме испытуемого раствора по времени удерживания
соответствуют 2 основным пикам на хроматограммах
растворов сравнения (а) и (Ь).
C. Испытуемый образец окрашивает бесцветное
пламя в желтый цвет.
О. К 0,3 г испытуемого образца прибавляют
20 мл этанола безводного Р и нагревают до кипения
на водяной бане при перемешивании. Немедленно
фильтруют смесь, выпаривают досуха и прибавляют
к остатку 7 мл воды Р. К 1 мл полученного раствора
прибавляют 0,1 мл раствора 1 г/л метиленового си¬
него Р, 2 мл кислоты серной разведенной Р и 2 мл
метиленхлорида Р и встряхивают. Нижний слой окра¬
шивается в синий цвет.
ИСПЫТАНИЯ
Кислотное число (2.5.1). Не более 2,0.
Йодное число (2.5.4, метод А). Не более 3,0.
2,00 г испытуемого образца растворяют в 25 мл ме¬
тиленхлорида Р.
Число омыления (2.5.6). Не более 2,0.
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 2,50 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Цетостеариловый спирт. Газовая хроматогра¬
фия (2.2.28).
Раствор внутреннего стандарта. 0,200 г ФСО
1-нонадеканола растворяют в этаноле безводном Р и
доводят до объема 100,0 мл этим же растворителем.
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в 25,0 мл раствора внутреннего
стандарта, прибавляют 25 мл воды Р и встряхивают 4
раза с пентаном Р порциями по 25 мл, прибавляя на¬
трия хлорид Р, при необходимости, для лучшего раз¬
деления слоев. Объединяют верхние слои. Дважды
промывают водой Р порциями по 30 мл, высушивают
над натрия сульфатом безводным Р и фильтруют.
Раствор сравнения (а). 0,100 г ФСО цетилового
спирта растворяют в 25,0 мл раствора внутреннего
стандарта, прибавляют 25 мл воды Р и встряхивают 4
раза с пентаном Р порциями по 25 мл, добавляя на¬
трия хлорид Р, при необходимости, для лучшего раз¬
деления слоев. Объединяют верхние слои. Дважды
промывают водой Р порциями по 30 мл, высушивают
над натрия сульфатом безводным Р и фильтруют.
Раствор сравнения (Ь). 0,100 г ФСО стеарино¬
вого спирта растворяют в 25,0 мл раствора внутрен¬
него стандарта, прибавляют 25 мл воды Р и встря¬
хивают 4 раза с пентаном Р порциями по 25 мл,
добавляя натрия хлорид Р, при необходимости, для
лучшего разделения слоев. Объединяют верхние
слои. Дважды промывают водой Р порциями по 30 мл,
высушивают над натрия сульфатом безводным Р и
фильтруют.
Цетримид
1097
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 25 м
и внутренним диаметром 0,25 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость потока: 1 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—20
150 —> 250
Блок ввода проб
250
Детектор
250
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: спирт цетиловый, спирт
стеариловый и 1-нонадеканол.
Процентное содержание спирта цетилового в ис¬
пытуемом образце рассчитывают с учетом содержа¬
ния спирта цетилового в ФСО спирта цетилового по
формуле:
А
о,
А А,у т
где:
— площадь пика спирта цетилового на хрома¬
тограмме испытуемого раствора;
Аху— площадь пика ФСО цетилового спирта на
хроматограмме раствора сравнения (а);
А1 — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора;
А2 — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (а);
т — масса навески испытуемого образца, ис¬
пользованной для приготовления испытуемого раст¬
вора, мг;
тху — масса навески ФСО цетилового спирта,
использованной для приготовления раствора сравне¬
ния (а), мг.
Процентное содержание спирта стеарилового в
испытуемом образце рассчитывают с учетом содер¬
жания спирта стеарилового в ФСО спирта стеарило¬
вого по формуле:
АА. ^.1.100,
А А,у т
где:
А2 — площадь пика спирта стеарилового на хро¬
матограмме испытуемого раствора;
А — площадь пика ФСО спирта стеарилового
на хроматограмме раствора сравнения (Ь);
А — площадь пика внутреннего стандарта на
хроматограмме испытуемого раствора;
А3 — площадь пика внутреннего стандарта на
хроматограмме раствора сравнения (Ь);
т — масса навески испытуемого образца, ис¬
пользованной для приготовления испытуемого раст¬
вора, мг;
тху — масса навески ФСО спирта стеарило¬
вого, использованной для приготовления раствора
сравнения (Ь), мг.
Процентное содержание цетостеарилового спир¬
та соответствует суммарному процентному содержа¬
нию спирта цетилового и спирта стеарилового.
Натрия лаурилсульфат. 0,300 г испытуемого
образца диспергируют в 25 мл метиленхлорида Р.
Прибавляют 50 мл воды Р и 10 мл раствора дими¬
дия бромида и сульфанового синего смешанного Р.
Титруют 0,004 М раствором бензэтония хлорида до
изменения окраски нижнего слоя от розовбй до серой.
При титровании используют ультразвук и нагревание;
перед каждым прибавлением титрованного раствора
дают слоям разделиться.
1 мл 0,004 М раствора бензэтония хлорида со¬
ответствует 1,154 мг натрия лаурилсульфата.
МАРКИРОВКА
При необходимости указывают название и кон¬
центрацию добавленного буфера.
07/2016:0378
ЦЕТРИМИД
СеМтМит
СЕТШМЮЕ
Н3С СНз
\+/
Вг
СНз
ОПРЕДЕЛЕНИЕ
Цетримид состоит из триметилтетрадециламмо-
ния бромида и может содержать небольшие количества
додецил- и гексадецилтриметиламмония бромидов.
Содержание: не менее 96,0 % и не более 101,0 %
алкилтриметиламмония бромидов, в пересчете на
С^НззВгЫ (М.м. 336,4) (в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый объемистый сыпучий по¬
рошок.
Легко растворим в воде и в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,25 г испытуемого образца растворяют в 96%
спирте Р и доводят до объема 25,0 мл этим же раст¬
ворителем. Оптическая плотность (2.2.25) получен¬
ного раствора, измеренная в диапазоне длин волн от
260 нм до 280 нм, составляет не более 0,05.
B. 5 мг испытуемого образца растворяют в 5 мл
буферного раствора рН 8,0 Р. Прибавляют 10 мг
калия феррицианида Р. Образуется желтый осадок.
Параллельно готовят контрольный раствор без испы¬
туемого образца: наблюдается желтое окрашивание
раствора без образования осадка.
C. Раствор 5, приготовленный как указано в раз¬
деле «Испытания», при встряхивании образует обиль¬
ную пену.
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,10 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения. 0,10 г ФСО триметилте-
традециламмония бромида растворяют в воде Р и
доводят до объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля силанизированного Р254 Р.
Подвижная фаза: ацетон Р— раствор 270 г/л на¬
трия ацетата Р — метанол Р (20:35:45; об/об/об).
Наносимый объем пробы: 1 мкл.
Фронт подвижной фазы: не менее 12 см от ли¬
нии страта.
140. Зак. 1060.
1098
Гэсударственная фармакопея Республики Беларусь
Высушивание: в потоке горячего воздуха.
Проявление: охлаждают; выдерживают в парах
йода и просматривают в дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
Е. Испытуемый образец дает реакцию (а) на бро¬
миды (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 2,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 50 мл раст¬
вора 8 прибавляют 0,1 мл раствора бромкрезолово-
го пурпурового Р. При прибавлении не более 0,1 мл
0,1 М раствора кислоты хлористоводородной или
0,1 М раствора натрия гидроксида окрашивание
раствора должно измениться.
Амины и соли аминов. 5,0 г испытуемого образ¬
ца растворяют в 30 мл смеси из 1 М раствора кисло¬
ты хлористоводородной и метанола Р (1:99, об/об),
прибавляют 100 мл 2-пропанола Р. Через раствор
медленно пропускают азотР. Постепенно прибав¬
ляют 15,0 мл 0,1 М раствора тетрабутиламмония
гидроксида и строят кривую потенциометрического
титрования (2.2.20). Если на кривой титрования об¬
наруживаются 2 скачка потенциала, объем титранта
между двумя точками перегиба на кривой титрования
не должен превышать 2,0 мл.
Потеря в массе при высушивании (2.2.32). Не
более 2,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 2 ч.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г испытуемого образца растворяют в
воде Р и доводят до объема 100,0 мл этим же раст¬
ворителем. 25,0 мл полученного раствора переносят
в делительную воронку, прибавляют 25 мл хлорофор¬
ма Р, 10 мл 0,1 М раствора натрия гидроксида Р и
10.0 мл свежеприготовленного раствора 50 г/л калия
йодида Р. Встряхивают, выдерживают для разделе¬
ния слоев и отделяют хлороформный слой. Водный
слой встряхивают с тремя порциями хлороформа Р,
каждая по 10 мл, и отделяют хлороформные слои.
Прибавляют 40 мл кислоты хлористоводородной Р,
охлаждают и титруют 0,05 М раствором калия йода¬
та до почти полного исчезновения темно-коричне¬
вого окрашивания. Прибавляют 2 мл хлороформа Р
и продолжают титровать, энергично встряхивая, до
момента, когда окрашивание хлороформного слоя пе¬
рестанет изменяться. Выполняют контрольный опыт,
используя в качестве раствора смесь, состоящую из
10.0 мл свежеприготовленного раствора 50 г/л калия
йодида Р, 20 мл воды Р и 40 мл кислоты хлористо¬
водородной Р.
1 мл 0,05 М раствора калия йодата соответ¬
ствует 33,64 мг С^Н^ВгМ.
07/2016:0988
ЦЕФАЗОЛИН НАТРИЯ
СеТаюНпит па1псит
СЕРА20Ш 800ШМ
[27164-46-1]
ОПРЕДЕЛЕНИЕ
Натрия (6Р, 7 Р)-3-[[(5-мети л-1,3,4-тиадиазол-2-ил)-
сульфанил]метил]-8-оксо-7-[( 1 Н-тетразол-1 -илацетил )-
амино]-5-тиа-1-азабицикло[4.2.0]окг-2-ен-2-карбоксилат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый порошок. Очень гигро¬
скопичен.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: 0,150 г испытуемого образца
растворяют в 5 мл воды Р, прибавляют 0,5 мл кисло¬
ты уксусной разведенной Р, взбалтывают и выдер¬
живают в ледяной бане в течение 10 мин. Фильтруют
и осадок на фильтре промывают (1—2) мл воды Р.
Осадок растворяют в смеси вода Р — ацетон Р (1:9,
об/об). Растворитель упаривают почти досуха и высу¬
шивают при температуре 60 °С в течение 30 мин.
Сравнение: ФСО цефазолина #или спектр, пред¬
ставленный на рисунке #0988.-1#.
B. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 2,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,15 при
430 нм. Измеряют оптическую плотность раствора 8.
рН (2.2.3). От 4,0 до 6,0. Измеряют рН раствора 8.
Удельное оптическое вращение (2.2.7). От -15
до -24 (в пересчете на безводное вещество). 1,25 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в водеР и доводят до объема
100,0 мл этим же растворителем. 2,0 мл полученного
Цефазолин натрия
1099
Рисунок #0988.-1. Инфракрасный спектр ФСО цефазолина
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 5 мкл.
Пригодность хроматографической системы.
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками цефа¬
золина и примеси 1_ (см. рисунок 0988.-2).
Предельное содержание примесей:
- любая примесь (не более 1,0 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 3,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
3,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (а).
/У,А/-Диметиланилин (2.4.26, метод В). Не бо¬
лее 0,0020 % (20 ррт).
Вода (2.5.12). Не более 6,0 %. Определение про¬
водят из 0,300 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,15 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без последующей процедуры удале¬
ния бактериальных эндотоксинов.
#Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным. Тест-доза — 50 мг испы¬
туемого образца в 1 мл воды для инъекций Р на 1,0 кг
массы тела кролика.
Испытание «Пирогенность» проводят в качестве
альтернативного испытанию «Бактериальные эндо¬
токсины».
раствора доводят раствором натрия гидрокарбона¬
та Рдо 100,0 мл.
Диапазон длин волн: от 220 нм до 350 нм.
Максимумы поглощения: при 272 нм.
Удельный показатель поглощения в максимуме:
от 260 до 300 (в пересчете на безводное вещество).
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (Ь). 20 мг испытуемого об¬
разца растворяют в 10 мл раствора 2 г/л натрия ги¬
дроксида Р и выдерживают в течение (15—30) мин.
1.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 20 мл.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаметром
4.0 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- температура: 45 °С;
- подвижная фаза:
-подвижная фаза А: раствор, содержащий
14,54 г/л динатрия гидрофосфата Р и 3,53 г/л
калия дигидрофосфата Р;
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—2
98
2
2-4
98—>85
2—15
4—10
85—>60
15 — 40
10—11,5
60 —> 35
40 — 65
11,5—12
35
65
12—15
35 — 98
65 — 2
15—21
98
2
140*. Зак. 1060.
1100
Гэсударственная фармакопея Республики Беларусь
1. Примесь и. 3. Структура не установлена. 5. Примесь I.
2. Примесь Е. 4. Цефазолин.
Рисунок 0988.-2. Хроматограмма для испытания
«Сопутствующие примеси» цефазолина натрия: раствор сравнения (Ь)
#Аномальная токсичность (2.6.9). Испытуе¬
мый образец должен быть нетоксичным. Тест-доза —
25 мг испытуемого образца в 0,5 мл воды для инъек¬
ций Р, внутривенно. Срок наблюдения — 48 ч.
#Стерильность (2.6.1). Цефазолин натрия в
условиях испытания обладает антимикробным дей¬
ствием. Для устранения антимикробного действия
посев на питательные среды проводят методом мем¬
бранной фильтрации. Содержание фермента пени-
циллиназы в промывной жидкости — 2000 ЕД/мл.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг ФСО цефазолина
растворяют в подвижной фазе и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО цефуроксима
натрия растворяют в 10,0 мл раствора сравнения (а)
и доводят подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
-подвижная фаза: смесь из 10 объемов аце¬
тонитрила Р и 90 объемов раствора, содержащего
2,77 г/л динатрия гидрофосфата Р и 1,86 г/л кисло¬
ты лимонной Р\
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 270 нм;
- объем вводимой пробы: 20 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками цефа¬
золина и цефуроксима.
Содержание цефазолина натрия рассчитывают в
процентах умножая процентное содержание цефазо¬
лина на 1,048.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): А, В, С, О, Е, О,
Н, I, 3, К,
А. (6/?,7Я)-7-Амино-3-[[(5-метил-1,3,4-тиадиазол-
2-ил)сульфанил]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]-
окт-2-ен-2-карбоновая кислота.
В. (6#,7Я)-7-[(2,2-Диметилпропаноил)амино]-3-
[[(5-метил-1,3,4-тиадиазол-2-ил)сульфанил]метил]-8-
оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая
кислота.
Цефаклор
1101
С. (6Я7Д)-3-Метил-8-оксо-7-[(1 Н-тетразол-1 -ил-
ацетил )амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоновая кислота.
О. (6/?,7/?)-3-[(Ацетилокси)метил]-8-оксо-7-[(1Н-
тетразол-1 -илацетил)амино]-5-тиа-1 -азабицикло[4.2.0]-
окт-2-ен-2-карбоновая кислота.
зн
Е. 5-Метил-1,3,4-тиодиазол-2-тиол (ММТй).
(3. (5аЯ,6Я)-6-[(1Н-Тетразол-1-илацетил)амино]-
5а,б-дигидро-ЗН,7Н-азето[2,1-Ь]фуро[3,4-сУ][1,3]тиазин-
1,7(4Н)-дион.
Н. (6/?,7Я)-3-[(Ацетилокси)метил]-7-амино-8-оксо-
5-тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоновая кисло¬
та (7-АЦК).
I. 2-[Карбокси[(1 Н-тетразол-1-илацетил)амино]-
метил]-5-[[(5-метил-1,3,4-тиодиазол-2-ил)сульфанил]-
метил]-5,6-дигидро-2Н-1,3-тиазин-4-карбоновая кисло¬
та (цефазолоевая кислота).
*}. 2-[Карбокси[(1 Н-тетразол-1-илацетил)амино]-
метил]-5-метилиден-5,6-дигидро-2Н-1,3-тиазин-4-кар-
боновая кислота.
Н3с
К. (6/?,7Я)-3-[[(5-Метил-1,3,4-тиадиазол-2-ил)суль-
фанил]метил]-8-оксо-7-[(1 Н-тетразол-1 -илацетил)амино]-
5-тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоксамид (це-
фазолинамид).
I- (6Н,75)-3-[[(5-Метил-1,3,4-тиадиазол-2-ил)суль-
фанил]метил]-8-оксо-7-[(1 Н-тетразол -1 -илацетил)ами-
но]-5-тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоновая кис¬
лота.
07/2016:0986
ЦЕФАКЛОР
Се/ас/ошш
СЕРАС1-ОЯ
Н N42
о
С Н СМ О 3 ■
^15 1Г З Г
[70356-03-5]
н2о
ОПРЕДЕЛЕНИЕ
(6Я,7Я)-7-[[(2Я)-2-Амино-2-фенилацетил]амино]-
3-хлор-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоновая кислота моногидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтый порошок.
Мало растворим в воде, практически нераство¬
рим в метаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цефаклора * или спектр, пред¬
ставленный на рисунке #0986.-1#.
ИСПЫТАНИЯ
рН (2.2.3). От 3,0 до 4,5. 0,250 г испытуемого об¬
разца суспендируют в воде, свободной от углерода
диоксида, Р и доводят до объема 10 мл этим же раст¬
ворителем.
1102
Государственная фармакопея Республики Беларусь
Рисунок #0986.-1. Инфракрасный спектр ФСО цефаклора
Удельное оптическое вращение (2.2.7). От
+101 до +111 (в пересчете на безводное вещество).
0,250 г испытуемого образца растворяют в растворе
10 г/л кислоты хлористоводородной Р и доводят до
объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 10,0 мл раствора 2,7 г/л натрия
дигидрофосфата Р, доведенного до рН 2,5 кислотой
фосфорной Р.
Раствор сравнения (а). 2,5 мг ФСО цефаклора и
5,0 мг ФСО дельта-3-цефаклора (примесь О) раство¬
ряют в 100,0 мл раствора 2,7 г/л натрия дигидрофос¬
фата Р, доведенного до рН 2,5 кислотой фосфор¬
ной Р
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят до объема 100,0 мл раствором 2,7 г/л
натрия дигидрофосфата Р, доведенного до рН 2,5
кислотой фосфорной Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
пильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: раствор 7,8 г/л натрия
дигидрофосфата, доведенный до рН 4,0 кисло¬
той фосфорной Р\
- подвижная фаза В: ацетонитрил Р — под¬
вижная фаза А (450:550, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—30
95 — 75
5 — 25
30—45
75 — 0
25—100
45—55
0
100
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем водимой пробы: 20 мкл.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: менее 2 между пиками цефаклора
и примеси 0\
-фактор асимметрии: не более 1,2 для пика
цефаклора; при необходимости, изменяют концентра¬
цию ацетонитрила в подвижной фазе.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора площадь любого пика,
кроме основного, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (Ь);
- сумма примесей (не более 2 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод С). Не более
0,0030 % (30 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 3 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 3,0 % и не более 6,5 %.
Определение проводят из 0,200 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Цефалексин моногидрат
1103
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 15,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 15,0 мг ФСО цефаклора
растворяют в подвижной фазе и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (Ь). 3,0 мг ФСО цефаклора и
3.0 мг ФСО дельта-3-цефаклора (примесь й) раство¬
ряют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
синильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: 220 мл метанола Р прибав¬
ляют к смеси из 780 мл воды Р, 10 мл триэтилами-
на Р и 1 г натрия гептансульфоната Р\ доводят до
РН 2,5 кислотой фосфорной Р;
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 265 нм;
- объем вводимой пробы: 20 мкл.
Пригодность хроматографической системы:
-разрешение: не менее 2,5 между пиками це¬
факлора и примеси О на хроматограмме раствора
сравнения (Ь); при необходимости изменяют концен¬
трацию метанола в подвижной фазе;
- фактор асимметрии: не более 1,5 для пика
цефаклора на хроматограмме раствора сравне¬
ния (Ь);
- относительное стандартное отклонение: не
более 1,0 % для площадей пиков цефаклора на хро¬
матограммах раствора сравнения (а) при 6 повторных
вводах пробы.
ПРИМЕСИ
Н ИНг
|^^р^со2н
А. (2Р)-2-Амино-2-фенилуксусная кислота (фенил-
глицин).
со,н
у-^ 'С|
н н
В. (6Р,7/?)-7-Амино-3-хлор-8-оксо-5-тиа-1 -азаби-
цикло[4.2.0]окт-2-ен-2-карбоновая кислота.
С. (6Р,7Р)-7-[[(25)-2-Амино-2-фенилацетил]амино]-
3-хлор-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбо-
новая кислота.
и эпимер у С*
О. (2Р,Ж,7ПУ и (25,6Я,7Я)-7-[[(2Я)-2-Амино-2-фе-
нилацетил]амино]-3-хлор-8-оксо-5-тиа-1-азабици-
кло[4.2.0]окт-3-ен-2-карбоновая кислота (дельта-3-
цефаклор).
Е. 2-[[(2Я)-2-Амино-2-фенилацетил]амино]-2-(5-
хлор-4-оксо-3,4-дигидро-2Н-1,3-тиазин-2-ил)уксусная
кислота.
Р. З-Фенилпиразин-2-ол.
0. (2Я,6Я,7Я)- и (25,6Я,7Я)-7-[[(2Я)-2-Амино-2-
фенилацетил]амино]-3-метилен-8-оксо-5-тиа-1-азабици-
кло[4.2.0]октан-2-карбоновая кислота (изоцефалексин).
Н. (6/?,7/?)-7-[[(2/?)-2-[[(2/?)-2-Амино-2-фенилацетил]-
амино]-2-фенилацетил]амино]-3-хлор-8-оксо-5-тиа-1-
азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (А/-фе-
нилглицилцефаклор).
07/2016:0708
ЦЕФАЛЕКСИН МОНОГИДРАТ
СеШех'тит топоНубпсит
СЕРА1.ЕХШ МОЫОНУйКАТЕ
Н 1ЧН2
С16Н17М3045 • Н20
[23325-78-2]
1104
Гэсударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ
(6Я,7/?)-7-[[(2#)-2-Амино-2-фенилацетил]амино]-
3-метил-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоновая кислота моногидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Умеренно растворим в воде, практически нераст¬
ворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цефалексина моногидрата
#или спектр, представленный на рисунке #0708.-1#.
ИСПЫТАНИЯ
рН (2.2.3). От 4,0 до 5,5. 50 мг испытуемого об¬
разца растворяют в воде, свободной от углерода ди¬
оксида, Р и доводят до объема 10 мл этим же раство¬
рителем.
Удельное оптическое вращение (2.2.7). От
+149 до +158 (в пересчете на безводное вещество).
0,125 г испытуемого образца растворяют во фталат-
ном буферном растворе рН 4,4 Р и доводят до объ¬
ема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе А и доводят до
объема 50,0 мл этим же растворителем.
Растворсравнения(а)^0,0 тй-фенилглицина Р
растворяют в подвижной фазе А и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (Ь). 10,0 мг ФСО кислоты
7-аминодезацетоксицефалоспорановой раство¬
ряют в фосфатном буферном растворе рН 7,0 Р5
и доводят подвижной фазой А до объема 10,0 мл.
Раствор сравнения (с). 1,0 мл раствора срав¬
нения (а) и 1,0 мл раствора сравнения (Ь) доводят
подвижной фазой А до объема 100,0 мл.
Раствор сравнения (д). 10 мг диметилфор-
мамида Р и 10 мг диметилацетамида Р раство¬
ряют в подвижной фазе А и доводят до объема
10.0 мл этим же растворителем. 1,0 мл полученно¬
го раствора доводят подвижной фазой А до объема
100.0 мл.
Раствор сравнения (е). 1,0 мл раствора срав¬
нения (с) доводят подвижной фазой А до объема
20.0 мл.
Раствор сравнения (7). 10 мг ФСО цефотак-
сима натрия растворяют в подвижной фазе А и до¬
водят до объема 10,0 мл этим же растворителем.
К 1,0 мл полученного раствора прибавляют 1,0 мл
испытуемого раствора и доводят подвижной фазой
А до объема 100 мл.
Условия хроматографирования:
- колонка длиной 0,10 м и внутренним диаме¬
тром 4,6 мм, заполненная сферическим силикаге¬
лем октадецилсилильным для хроматографии Р
с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: фосфатный буферный
раствор рН 5,0 Р\
- подвижная фаза В: метанол Р2\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—1
98
2
1—20
о
г-
т
со
О)
2 —► 30
Рисунок #0708.-1. Инфракрасный спектр ФСО цефалексина моногидрата
Цефалексин моногидрат
1105
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 220 нм;
- объем вводимой пробы: по 20 мкл испытуе¬
мого раствора и растворов сравнения (с), (с!), (е)
и(0-
Пригодность хроматографической системы:
-разрешение: не менее 2,0 между пиками
примесей А и В на хроматограмме раствора срав¬
нения (с) и не менее 1,5 между пиком цефалексина
и пиком цефотаксима на хроматограмме раствора
сравнения (Г).
Предельное содержание примесей:
- примесь В (не более 1,0 %); на хромато¬
грамме испытуемого раствора площадь пика, со¬
ответствующего примеси В, не должна превышать
площадь второго пика на хроматограмме раствора
сравнения (с);
-любая другая примесь (не более 1,0 %): на
хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пика примеси В,
не должна превышать площадь первого пика на
хроматограмме раствора сравнения (с);
- сумма примесей (не более 3,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать 3-кратную площадь первого пика на хромато¬
грамме раствора сравнения (с);
- неучитываемый предел: на хроматограмме
испытуемого раствора не учитывают пики диме-
тилформамида и диметилацетамида и пики с пло¬
щадью менее площади второго пика на хромато¬
грамме раствора сравнения (е) (0,05 %).
Л/,А/-Диметиланилин (2.4.26, метод В). Не бо¬
лее 0,0020 % (20 ррт).
Вода (2.5.12). От 4,0 % до 8,0 %. Определение
проводят из 0,300 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого об¬
разца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем.
Раствор сравнения (а). 50,0 мг ФСО цефалек¬
сина моногидрата растворяют в воде Р и доводят
до объема 100,0 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО цефрадина
растворяют в 20 мл раствора сравнения (а) и дово¬
дят водой Р до объема 100 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза: метанол Р — ацетони¬
трил Р — раствор 13,6 г/л калия дигидрофосфа¬
та Р— вода Р (2:5:10:83, об/об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 254 нм;
- объем вводимой пробы: 20 мкл.
Пригодность хроматографической системы:
раствора сравнения (Ь):
- разрешение: не менее 4,0 между пиками цефа¬
лексина и цефрадина.
Содержание цефалексина моногидрата рассчи¬
тывают в процентах.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (2Я)-2-Амино-2-фенилуксусная кислота (О-фе-
нилглицин).
В. (6/?,7/?)-7-Амино-3-метил-8-оксо-5-тиа-1 -аза-
бицикло[4.2.0]окт-2-ен-2-карбоновая кислота (7-амино-
дезацетоксицефалоспорановая кислота, 7-аЪсА).
С. (6/?,7Я)-7-[[(2/?)-2-[[(2/?)-2-Амино-2-фенил-
ацетил]амино]-2-фенилацетил]амино]-3-метил-8-оксо-
5-тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоновая кис¬
лота.
н3с он
Р. 3-Гидрокси-4-метилтиофен-2(5Н)-он.
Е. (6Я,7Я)-7-[(2,2-Диметилпропаноил)амино]-3-
метил-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоновая кислота (7-АйСА пиваламид).
и эпимер при С*
Р. (2Я5,6#,7Я)-7-[[(2Я)-2-Амино-2-фенилацетил]-
амино]-3-метил-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-3-
ен-2-карбоновая кислота (дельта-2-цефалексин).
1106
Гэсударственная фармакопея Республики Беларусь
07/2016:2126
ЦЕФЕПИМА ДИГИДРОХЛОРИД
МОНОГИДРАТ
Се/ер/ш/ сИНус1госМопс1ит топоЬубпсит
СЕРЕР1МЕ О/НУОЛОСЖ-ОК/ОЕ
МОЛЮЯУОЯАГЕ
ОПРЕДЕЛЕНИЕ
(6/?,7/?)-7-[[(22)-(2-Аминотиазол-4-ил)(метоксии-
мино)ацетил]амино]-3-[(1-метилпирролидинио)метил]-
8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилата
дигидрохлорид моногидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 97,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легко растворим в воде и в метаноле, практиче¬
ски нерастворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цефепима дигидрохлорида мо¬
ногидрата #или спектр, представленный на рисунке
#2126.-1#.
В. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 20 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод //). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)3.
Удельное оптическое вращение (2.2.7). От +40
до +45 (в пересчете на безводное вещество). 0,250 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Примесь С. Жидкостная хроматография (2.2.29).
Растворы готовят непосредственно перед исполь¬
зованием.
Испытуемый раствор. 0,100 г испытуемого образ¬
ца растворяют в 0,01 М растворе кислоты азотной и
доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 0,250 г А1-метилпир-
ролидина Р (примесь С) доводят водой Р до объема
100,0 мл. 2,0 мл полученного раствора доводят 0,01 М
раствором кислоты азотной до объема 100,0 мл.
Раствор сравнения (Ь). 0,250 г пирролидина Р
доводят 0,01 М раствором кислоты азотной до
объема 100 мл. 2 мл полученного раствора дово¬
дят 0,01 М раствором кислоты азотной до объема
100 мл. Смешивают 5 мл полученного раствора и
5 мл раствора сравнения (а).
Условия хроматографирования:
- колонка длиной 0,05 м и внутренним диаме¬
тром 4,6 мм, заполненная катионообменной смолой
сильной Р с размером частиц 5 мкм;
Рисунок #2126.-1. Инфракрасный спектр ФСО цефепима дигидрохлорида моногидрата
Цефепима дигидрохлорид моногидрат
1107
-подвижная фаза: ацетонитрил Р — 0,01 М
раствор кислоты азотной (1:100, об/об); фильтруют
через фильтр с размером пор 0,2 мкм;
- скорость подвижной фазы: 1 мл/мин;
- кондуктометрический детектор;
- объем вводимой пробы. 100 мкл;
- время хроматографирования: 1,1-кратное вре¬
мя удерживания цефепима.
Время удерживания: цефепим — около 50 мин,
элюируется широким пиком.
Пригодность хроматографической системы:
- фактор асимметрии: не более 2,5 для пика
примеси С на хроматограмме раствора сравнения (а);
- относительное стандартное отклонение: не
более 5,0 % при 6 повторных вводах раствора сравне¬
ния (а);
-коэффициент разделения пиков: не менее 3
между пиками пирролидина и примеси С на хромато¬
грамме раствора сравнения (Ь).
Рассчитывают содержание примеси О в процен¬
тах в испытуемом растворе, используя раствор срав¬
нения (а).
Предельное содержание примесей:
- примесь 6: не более 0,5 %.
Сопутствующие примеси. Жидкостная хромато¬
графия (2.2.29). Растворы готовят непосредственно
перед применением или хранят охлажденными при
температуре от 4 °С до 8 °С в течение не более 12 ч.
Испытуемый раствор. 70,0 мг испытуемого об¬
разца растворяют в подвижной фазе А, доводят до
объема 50,0 мл этим же растворителем, обрабатывают
ультразвуком в течение 30 с и перемешивают в тече¬
ние около 5 мин.
Раствор сравнения (а). 70,0 мг ФСО цефепима
дигидрохлорида моногидрата растворяют в подвиж¬
ной фазе А, доводят этим же растворителем до объема
50.0 мл, обрабатывают ультразвуком в течение 30 с и
перемешивают в течение около 5 мин.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой А до объема 10,0 мл.
2.0 мл полученного раствора доводят подвижной фа¬
зой А до объема 100,0 мл.
Раствор сравнения (с). 7 мг ФСО цефепима диги¬
дрохлорида моногидрата для проверки пригодности
хроматографической системы (содержит примеси А,
В и Р) растворяют в подвижной фазе А и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения (ф. 2 мг ФСО цефепима при¬
меси Е растворяют в подвижной фазе А и доводят до
объема 25,0 мл этим же растворителем. 1,0 мл полу¬
ченного раствора доводят подвижной фазой А до объ¬
ема 10,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным эндкепированным для хроматографии Р с разме¬
ром частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р — раст¬
вор 0,68 г/л калия дигидрофосфата Р, доведен¬
ный 0,5 М раствором калия гидроксида до рН 5,0
(10:90, об/об);
- подвижная фаза В: ацетонитрил Р — раст¬
вор 0,68 г/л калия дигидрофосфата Р, доведен¬
ный 0,5 М раствором калия гидроксида до рН 5,0
(50:50, об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100
0
10—30
о
0
1
сл
о
0 —► 50
30—35
50
50
35—36
50 —► 100
сл
0
1
О
36-45
100
0
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (с1).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В и Р, используя хроматограм¬
му раствора сравнения (с) и хроматограмму, прилага¬
емую к ФСО цефепима дигидрохлорида моногидра¬
та для проверки пригодности хроматографической
системы; идентифицируют пик примеси Е используя
хроматограмму раствора сравнения (с1).
Относительное удерживание (по отношению к
цефепиму, время удерживания — около 7 мин): при¬
месь Е — около 0,4; примесь Р — около 0,8; примесь
А— около 2,5; примесь В — около 4,1.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 1,5 между пиками при¬
меси Р и цефепима.
Предельное содержание примесей (для расчета со¬
держания примесей умножают площади пиков на соот¬
ветствующие поправочные коэффициенты: для примеси
А— 1,4; для примеси В —1,4; для примеси Е — 1,8):
- примесь А (не более 0,3 %): на хроматограмме ис¬
пытуемого раствора площадь пика, соответствующего
примеси А, не должна превышать 1,5-кратную площадь
основного пика на хроматограмме раствора сравнения (Ь);
- примеси ВиР (не более 0,2 %): на хроматограмме
испытуемого раствора площади пиков, соответствующих
примесям В и Р, не должны превышать площадь основ¬
ного пика на хроматограмме раствора сравнения (Ь);
-примесь Е (не более 0,1 %): на хроматограмме
испытуемого раствора площадь пика, соответствующего
примеси Е, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей А,
В, Е и Р, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь);
-сумма примесей (не более 1,0%): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-кратную
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (Ь).
Вода (2.5.12). Не менее 3,0 % и не более 4,5 %.
Определение проводят из 0,400 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
0,04 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
1108
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- подвижная фаза: подвижная фаза А;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а);
- время хроматографирования: 1,4-кратное вре¬
мя удерживания цефепима.
Содержание С19Н241Ч60552-2НС1 в процентах рас¬
считывают с учетом содержания цефепима дигидрох¬
лорида в ФСО цефепима дигидрохлорида моноги¬
драта.
ХРАНЕНИЕ
В защищенном от света месте. Если субстанция
стерильная, ее хранят в стерильном воздухонепрони¬
цаемом контейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, Е, Р, О.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, О.
А. (6/?,7/?)-7-[[(2Е)-(2-Аминотиазол-4-ил)(метокси-
имино)ацетил]амино]-3-[(1-метилпирролидинио)метил]-
8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат
(ангш-цефепим).
В. (6Я,7Я?)-7-[[(22)-[2-[[(22)-(2-Аминотиазол-4-ил)-
(метоксиимино)ацетил]амино]тиазол-4-ил](метоксии-
мино)ацетил]амино]-3-[(1-метилпирролидинио)метил]-
8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат.
С. (22)-2-(2-Аминотиазол-4-ил)-А/-(формилметил)-
2-(метоксиимино)ацетамид.
СН3
со2н
Н2И-—^
V
О. (22)-(2-Аминотиазол-4-ил)(метоксиимино)уксус¬
ная кислота.
Н2М"
Е. (6/?,7Я)-7-Амино-3-[(1 -метилпирролидинио)ме-
тил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-кар-
боксилат.
Р. (6/?,7А?)-7-[[[(6/?,7/?)-7-[[(27)-(2-Аминотиазол-4-
ил)(метоксиимино)ацетил]амино]-3-[(1-метилпир-
ролидинио)метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-
2-ен-2-ил]карбонил]амино]-3-[(1-метилпирролидинио)-
метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоксилат.
С. 1-Метилпирролидин (А/-метилпирролидин).
07/2016:1404
ЦЕФОПЕРАЗОН НАТРИЯ
СеТорегагопит паМсит
СЕР0РЕКА20МЕ 800ШМ
С25Н26М9Ыа0852 М.м. 668
[62893-20-3]
ОПРЕДЕЛЕНИЕ
Натрия (6Я,7Я)-7-[[(2Я)-2-[[(4-этил-2,3-диоксопи-
перазин-1-ил)карбонил]амино]-2-(4-гидроксифенил)-
ацетил]амино]-3-[[(1 -метил-1 Н- тетразол-5-ил )сульфа-
нил]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоксилат.
Цефоперазон натрия
1109
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок. Гигро¬
скопичен.
Легко растворим в воде, растворим в метаноле,
мало растворим в 96 % спирте.
В кристаллической форме обладает полимор¬
физмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: растворяют в метаноле Р, вы¬
паривают досуха и используют сухой остаток для по¬
лучения спектра.
Сравнение: ФСО цефоперазона натрия #или
спектр, представленный на рисунке #1404.-1#.
B. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора (а) по времени удерживания и
величине соответствует основному пику на хромато¬
грамме раствора сравнения (а).
C. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 25,0 мл этим же
растворителем.
Прозрачность (2.2.7). Раствор 3 должен быть
прозрачным.
Оптическая плотность (2.2.25). Не более 0,15
при 430 нм. Измеряют оптическую плотность раствора 3.
рН (2.2.3). От 4,5 до 6,5. 2,5 г испытуемого образ¬
ца растворяют в воде, свободной от углерода диок¬
сида, Р и доводят до объема 10 мл раствора этим же
растворителем.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор (а). 25,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 250,0 мл этим же растворителем.
Испытуемый раствор (Ь). 25,0 мг испытуемого
образца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 25,0 мг ФСО цефопе¬
разона дигидрата растворяют в подвижной фазе
и доводят до объема 250,0 мл этим же раствори¬
телем.
Раствор сравнения (Ь). 5,0 мл раствора сравне¬
ния (а) доводят подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: смесь из 884 объемов воды Р,
110 объемов ацетонитрила Р, 3,5 объема раствора
60 г/л кислоты уксусной Р, 2,5 объема раствора, при¬
готовленного следующим образом: 14 мл триэтила-
мина Р и 5,7 мл кислоты уксусной ледяной Р доводят
водой Р до объема 100 мл;
- скорость подвижной фазы: 1 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора (Ь) и раствора сравнения (а) и (Ь);
- время хроматографирования: 2,5-кратное вре¬
мя удерживания цефоперазона.
Рисунок #1404.-1. Инфракрасный спектр ФСО цефоперазона натрия
1110
Гэсударственная фармакопея Республики Беларусь
Время удерживания: цефоперазон — около
15 мин.
Пригодность хроматографической системы:
раствор сравнения (а):
- число теоретических тарелок: не менее
5000 для основного пика; при необходимости изме¬
няют концентрацию ацетонитрила Р в подвижной
фазе;
- фактор асимметрии: не более 1,6 для основ¬
ного пика; при необходимости изменяют концентра¬
цию ацетонитрила Р в подвижной фазе.
Предельное содержание примесей:
- любая примесь (не более 1,5 %): на хрома¬
тограмме испытуемого раствора (Ь) площадь лю¬
бого пика, кроме основного, не должна превышать
1.5- кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- сумма примесей (не более 4,5 %): на хромато¬
грамме испытуемого раствора (Ь) сумма площадей
всех пиков, кроме основного, не должна превышать
4.5- кратную площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора (Ь) не учитывают пики
с площадью менее 0,1 площади основного пика на
хроматограмме раствора сравнения (Ь).
Ацетон (2.4.24, система В). Не более 2,0 %.
Исходный раствор испытуемого образца.
0,500 г испытуемого образца растворяют в воде Р и
доводят до объема 10,0 мл этим же растворителем.
Раствор остаточного растворителя. 0,350 г
ацетона Р растворяют в воде Р и доводят до объ¬
ема 100,0 мл этим же растворителем. 10,0 мл по¬
лученного раствора доводят водой Р до объема
100,0 мл.
Готовят 4 флакона, как указано в таблице ниже:
№
флакона
Исходный
раствор ис¬
пытуемого
образца, мл
Раствор оста¬
точного рас¬
творителя,
мл
Вода Р,
мл
1
1,0
0
4,0
2
1,0
1,0
3,0
3
1,0
2,0
2,0
4
1,0
3,0
1,0
Могут быть использованы следующие условия
проведения парофазного анализа:
- время уравновешивания: 15 мин;
-температура линии подачи газовой пробы:
110 °С;
- температура колонки: 40 °С в течение 10 мин.
Тяжелые металлы (2.4.8, метод С). Не более
0,0005 % (5 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 5,0 %. Определение про¬
водят из 0,200 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,20 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
#Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным. Тест-доза — 10 мг испы¬
туемого образца в 1 мл воды для инъекций Р на 1 кг
массы тела кролика.
Испытание «Пирогенность» проводят в качестве
альтернативного испытанию «Бактериальные эндо¬
токсины».
#Стерильность (2.6.1) Испытуемый образец в
условиях испытания обладает антимикробным дей¬
ствием. Для устранения антимикробного действия
посев на питательные среды проводят методом мем¬
бранной фильтрации.
#Аномальная токсичность (2.6.9). Испытуемый
образец должен быть нетоксичным. Тест-доза — 20 мг
испытуемого образца в 0,5 мл воды для инъекций Р,
внутривенно. Срок наблюдения — 48 ч.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора (а) и раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение: не
более 1,0 % для площади пика цефоперазона при 6
повторных вводах пробы.
Содержание цефоперазона натрия рассчитыва¬
ют в процентах умножая процентное содержание це¬
фоперазона на 1,034.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищен¬
ном от света месте при температуре от 2 °С до 8 °С.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
разин-1-ил)карбонил]амино]-2-(4-гидроксифенил)аце-
тил]амино]-5а,6-дигидро-ЗН,7Н-азето[2,1 -Ь]фуро[3,4-д]-
[1,3]тиазин-1,7(4Н)-дион.
1-ил)карбонил]амино]-2-(4-гидроксифенил)ацетил]-
амино]-3-[(4-метил-5-тиоксо-4,5-дигидро-1Н-тетразол-
1 -ил)метил]-8-оксо-5-тиа-1 -азабицико[4.2.0]окт-2-ен-2-
карбоновая кислота.
Цефотаксим натрия
1111
зн
07/2016:0989
\
^-СНз
/
С. 1-Метил-1 Н-тетразол-5-тиол.
й. (6Я,7#)-7-Амино-8-оксо-3-[(1Н-1,2,3-тиазол-4-
илсульфанил)метил]-5-тиа-1 -азабицикло14.2.0]окг-2-
ен-2-карбоновая кислота (7-ТАЦК, 7-ТАСА).
со,н о
н н
Е. (6#,7#)-3-[(Ацетокси)метил]-7-амино-8-оксо-5-
тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота
(7-АЦК, 7-АСА).
Р. (6Я,75)-7-[[(2Я)-2-[[(4-Этил-2,3-диоксопиперазин-
1-ил)карбонил]амино]-2-(4-гидроксифенил)ацетил]-
амино]-3-[[(1-метил-1 Н-тетразол-5-ил)сульфанил]метил]-
8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая
кислота.
ЦЕФОТАКСИМ НАТРИЯ
СеМах1тит паМсит
СЕРОТАХ1МЕ ЗОйШМ
С1вН16Ы5Ыа0782 М.м. 477,4
[64485-93-4]
ОПРЕДЕЛЕНИЕ
Натрия (6/?,7/?)-3-[(ацетилокси)метил]-7-[[(22)-2-(2-
аминотиазол-4-ил)-2-(метоксиимино)ацетил]амино]-8-
оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый порошок. Гигро¬
скопичен.
Легко растворим в воде, умеренно растворим в
метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цефотаксима натрия #или
спектр, представленный на рисунке #0989.-1#.
Рисунок #0989.-1. Инфракрасный спектр ФСО цефотаксима натрия
1112
Гэсударственная фармакопея Республики Беларусь
В. Испытуемый образец дает реакцию (а) на на¬
трий {2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным. К 10 мл раствора 3 прибавляют 1 мл
кислоты уксусной ледяной Р и немедленно проводят
испытание. Полученный раствор должен быть про¬
зрачным.
рН (2.2.3). От 4,5 до 6,5. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
+58,0 до +64,0 (в пересчете на безводное вещество).
0,100 г испытуемого образца растворяют в воде Р и
доводят до объема 10,0 мл этим же растворителем.
Оптическая плотность (2.2.25). Не более 0,40 при
430 нм. Измеряют оптическую плотность раствора 3.
Удельный показатель поглощения (2.2.25). От
360 до 390 в максимуме при 235 нм (в пересчете на
безводное вещество). 20,0 мг испытуемого образца
растворяют в воде Р и доводят до объема 100,0 мл
этим же растворителем. 10,0 мл полученного раст¬
вора доводят водой Р до 100,0 мл.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29). Растворы готовят непосред¬
ственно перед использованием.
Раствор А. Подвижная фаза В — подвижная
фаза А (14:86, об/об).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 8,0 мг ФСО цефотакси-
ма кислоты растворяют в растворе А и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл.
Раствор сравнения (с). К 4,0 мл испытуемого
раствора прибавляют 1,0 мл кислоты хлористово¬
дородной разведенной Р, нагревают при температуре
40 °С в течение 2 ч и прибавляют 5,0 мл фосфатного
буферного раствора рН 6,6 Р и 1,0 мл раствора на¬
трия гидроксида разведенного Р.
Раствор сравнения (д). 4 мг ФСО цефотаксима
для идентификации пиков (содержит примеси А, В, С,
Е и Р) растворяют в 5 мл раствора А.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаметром
3,9 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: раствор 7,1 г/л динатрия
гидрофосфата Р, доведенный до рН 6,25 кисло¬
той фосфорной Р]
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—7
86
14
7—9
86—>82
14 18
9—16
82
18
16—45
82 —> 60
00
1
о
45—50
60
40
50—55
со
00
т
о
со
40->14
55—60
86
14
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 235 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (б).
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, С, Е и Р, используя хромато¬
грамму раствора сравнения (б) и хроматограмму, при¬
лагаемую к ФСО цефотаксима для идентификации
пиков.
Относительное удерживание (по отношению к
цефотаксиму, время удерживания — около 13 мин):
примесь В — около 0,3; примесь А — около 0,5; при¬
месь Е — около 0,6; примесь С — около 1,9; примесь
О — около 2,3; примесь Р — около 2,4; примесь 6 —
около 3,1.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 3,5 между пиками при¬
меси Е и цефотаксима;
- фактор асимметрии: не более 2,0 для пика
цефотаксима.
Предельное содержание примесей:
- примеси А, В, С, О, Е, Р (не более 1,0 %): на
хроматограмме испытуемого раствора площади пи¬
ков, соответствующих примесям А, В, С, О, Е и Р, не
должны превышать площадь основного пика на хро¬
матограмме раствора сравнения (Ь);
- любая другая примесь (не более 0,2 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С, О,
Е и Р, не должна превышать 0,2 площади основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 3,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 3-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,05 площади основного пика на
хроматограмме раствора сравнения (Ь).
Этанол (2.4.24, система А). Не более 1,0 %.
М,М-Диметиланилин (2.4.26, метод В). Не бо¬
лее 0,0020 % (20 ррт).
2-Этилгексановая кислота (2.4.28). Не более
0,5 % (м/м).
Вода (2.5.12). Не более 3,0 %. Определение про¬
водят из 0,300 г испытуемого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,05 МЕ/мг, если субстанция предназначена для
производства лекарственных средств для паренте¬
рального применения без дальнейшей подходящей
процедуры удаления бактериальных эндотоксинов.
#Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным. Тест-доза — 50 мг испы¬
туемого образца в 1 мл воды для инъекций Р на 1,0 кг
массы тела кролика.
Испытание «Пирогенность» проводят в качестве
альтернативного испытанию «Бактериальные эндо¬
токсины».
#Стерильность (2.6.1). Цефотаксим натрия в ус¬
ловиях испытания обладает антимикробным действи¬
ем. Для устранения антимикробного действия посев
на питательные среды проводят методом мембран¬
ной фильтрации.
Цефтазидим пентагидрат
1113
#Аномальная токсичность (2.6.9). Испытуемый
образец должен быть нетоксичным. Тест-доза — 40 мг
испытуемого образца в 0,5 мл воды для инъекций Р,
внутривенно. Срок наблюдения — 48 ч.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание цефотаксима натрия рассчитывают
в процентах, умножая процентное содержание цефо¬
таксима на 1,048.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): О.
A. К = К' = Н: (6Р,7Р)-7-[[(22)-2-(2-Аминотиазол-
4- ил)-2-метоксиимино)ацетил]амино]-3-метил-8-оксо-
5- тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоновая кисло¬
та (дезацетоксицефотаксим).
B. Р = ОН, Р' = Н: (6/?,7Я)-7-[[(22)-2-(2-Аминотиа-
зол-4-ил)-2-метоксиимино)ацетил]амино]-3-(гидро-
ксиметил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-
карбоновая кислота (дезацетилцефотаксим).
C. Р = 0-С0-СН3, Р' = СНО: (6Р,7Р)-3-[(Ацетилокси)-
метил]-7-[[(22)-2-[2-(формиламино)тиазол-4-ил]-2(ме-
токсиимино)ацетил]амино]-8-оксо-5-тиа-1-азабици-
кло[4.2.0]окт-2-ен-2-карбоновая кислота (Л/-формил-
цефотаксим).
О. (6Р,7Р)-3-[(Ацетилокси)метил]-7-[[(2Е)-2-(2-
аминотиазол-4-ил)-2-(метоксиимино)ацетил]амино]-8-
оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая
кислота (Е-цефотаксим).
Е. (5аР,6Р)-6-[[(22)-2-(2-Аминотиазол-4-ил)-2-мето-
ксиимино)ацетил]амино]-5а,6-дигидро-ЗН,7Н-азето[2,1 -Ь]-
фуро[3,4-с/][1,3]тиазин-1,7(4Н)-дион (дезацетилцефо-
таксима лактон).
Р. (6Р,7Р)-3-[(Ацетилокси)метил]-7-[[(22)-2-[2-
[[[(6Е,7/?)-7-[[(22)-2-(2-аминотиазол-4-ил)-2-(мето-
ксиимино)ацетил]амино]-2-карбокси-8-оксо-5-тиа-1-
азабицикло[4.2.0]окт-2-ен-2-ил]мелтил]амино]тиазол-
4-ил]-2-(метоксиимино)ацетил]амино]-8-оксо-5-тиа-1-
азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (ди¬
мер цефотаксима).
С.(6Р,7Р)-3-[(Ацетилокси)метил]-7-[[(22)-2-[2-[[(22)-
2-(2-аминотиазол-4-ил)-2-(метоксиимино)ацетил]ами-
но]тиазол-4-ил]-2-(метоксиимино)ацетил]амино]-8-оксо-
5-тиа-1 -азабицикло[4.2.0]окт-2-ен-2-карбоновая кисло¬
та (АТА цефотаксим).
07/2016:1405
ЦЕФТАЗИДИМ ПЕНТАГИДРАТ
СеПаясНтит реп1аЬус1псит
СЕРТА1Ю1МЕ РЕЫТАНУОРАТЕ
• 5Н20 М.м. 637
[78439-06-2]
ОПРЕДЕЛЕНИЕ
(6/?,7/?)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-[(1-карбо-
кси-1-метилэтокси)имино]ацетил]амино]-8-оксо-3-
1114
Гэсударственная фармакопея Республики Беларусь
[(пиридин-1 -иум-1 -ил)метил]-5-тиа-1 -азабицикло[4.2.0]-
окт-2-ен-2-карбоксилат пентагидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 95,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Мало растворим в воде и метаноле, практически
нерастворим в ацетоне и 96 % спирте. Растворяется в
растворах кислот и гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цефтазидима #или спектр,
представленный на рисунке #1405.-1#.
ИСПЫТАНИЯ
Раствор 3. 0,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 8 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 8 должен
быть бесцветным.
рН (2.2.3). От 3,0 до 4,0. Измеряют рН раствора 8.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,150 г испытуемого об¬
разца суспендируют в 5 мл ацетонитрила Р, раство¬
ряют, прибавляя воду Р, и доводят водой Р до объема
100 мл.
Раствор сравнения (а). К 1,0 мл испытуемого
раствора прибавляют 5,0 мл ацетонитрила Р и дово¬
дят водой Р до объема 100,0 мл. 1,0 мл полученного
раствора доводят водой Р до объема 5,0 мл.
Раствор сравнения (Ь). Для приготовления при¬
меси В 1п зНи 5 мл испытуемого раствора выдержи¬
вают в ультрафиолетовом свете при длине волны
254 нм в течение около 24 ч.
Раствор сравнения (с). 3 мг ФСО цефтазидима
для идентификации пиков (содержит примеси А и О)
суспендируют в 0,5 мл ацетонитрила Р, растворяют,
прибавляя воду Р, и доводят водой Р до объема 2 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 40 °С;
- подвижная фаза:
-подвижная фаза А: раствор, содержащий
3,6 г/л динатрия гидрофосфата Р и 1,4 г/л калия
дигидрофосфата Р, доведенный 10% (об/об)
раствором кислоты фосфорной Р до рН 3,4;
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р]
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-^
96-* 89
4 —* 11
4—5
89
11
5—8
89-* 84
11 —* 16
8—11
84-* 80
16 -* 20
11—15
00
0
1
сл
о
20 — 50
15—18
50 —* 20
сл
0
1
00
о
18—22
20
80
- скорость подвижной фазы: 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к цефтазидиму, время удерживания — около 8 мин):
Рисунок #1405.-1. Инфракрасный спектр ФСО цефтазидима
Цефтазидим пентагидрат
1115
примесь Р — около 0,4; примесь С — около 0,8; при¬
месь А — около 0,9; примесь В — около 1,4.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и С, используя хроматограмму
раствора сравнения (с) и хроматограмму, прилагае¬
мую к ФСО цефтазидима для идентификации пиков;
идентифицируют пик примеси В, используя хромато¬
грамму раствора сравнения (Ь).
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 4,0 между пиками при¬
меси А и цефтазидима.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 3,0):
- примеси А, В и О (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В и О, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В и О, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а); не учитывают
пик примеси Р.
Примесь Р. Жидкостная хроматография (2.2.29).
Растворы готовят непосредственно перед исполь¬
зованием.
Фосфатный буферный раствор. Готовят 10%
(об/об) раствор фосфатного буферного раствора
рН 7,0 Р4.
Испытуемый раствор. 0,500 г испытуемого об¬
разца растворяют в фосфатном буферном растворе и
доводят до объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,00 г пиридина Р раст¬
воряют в воде Р и доводят этим же растворителем до
объема 100,0 мл. 5,0 мл полученного раствора дово¬
дят водой Р до объема 200,0 мл. 1,0 мл полученного
раствора доводят фосфатным буферным раствором
до объема 100,0 мл.
Раствор сравнения (Ь). 1 мл испытуемого раст¬
вора доводят фосфатным буферным раствором до
объема 200 мл. К 1 мл полученного раствора прибав¬
ляют 20 мл раствора сравнения (а) и доводят фос¬
фатным буферным раствором до объема 200 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: раствор 28,8 г/л аммония ди¬
гидрофосфата Р, предварительно доведенный рас¬
твором аммиака Р до рН 7,0 — ацетонитрил Р —
вода Р (8:24:68, об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 255 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 7,0 между пиками цеф¬
тазидима и примеси Р.
Предельное содержание примесей:
- примесь Р (не более 0,0500 % (500 ррт)): на
хроматограмме испытуемого раствора площадь пика,
соответствующего примеси Р, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (а).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 2,0 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 13,0% и не более
15,0 %. Определение проводят из 0,100 г испытуемо¬
го образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
0,10 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 25,0 мг ФСО цефтази¬
дима растворяют в подвижной фазе и доводят до объ¬
ема 25,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО цефтазиди¬
ма для идентификации пиков (содержит примеси А и
О) растворяют в подвижной фазе и доводят до объ¬
ема 5,0 мл этим же растворителем.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем гексилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: 4,3 г динатрия гидрофосфата Р
и 2,7 г калия дигидрофосфата Р растворяют в 980 мл
воды Р, затем прибавляют 20 мл ацетонитрила Р,
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 245 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 6 мин.
Относительное удерживание (по отношению к
цефтазидиму, время удерживания — около 4,5 мин):
примесь А— около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси А и цефтазидима.
Содержание С22Н221\160752 рассчитывают с учетом
содержания цефтазидима в ФСО цефтазидима.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере. Если суб¬
станция стерильная, ее хранят в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
1116
Гэсударственная фармакопея Республики Беларусь
ПРИМЕСИ
Специфицированные примеси: А, В, Р, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, Е, Н.
А. (2Я5,6Я,7Я)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-
[(1 -карбокси-1 -метилэтокси)имино]ацетил]амино]-8-
оксо-3-[(пиридин-1 -иум-1 -ил)метил]-5-тиа-1 -азабици-
кло[4.2.0]окт-3-ен-2-карбоксилат (Д-2-цефтазидим).
В. (6Я,7Я)-7-[[(2Е)-2-(2-Аминотиазол-4-ил)-2-[(1 -
карбокси-1-метилэтокси)имино]ацетил]амино]-8-оксо-
3-[(пиридин-1 -иум-1 -ил)метил]-5-тиа-1 -азабицикло[4.2.0]-
окт-2-ен-2-карбоксилат.
С. (6Я7Я)-7-Амино-8-оксо-3-[(пиридин-1 -иум-1 -ил)-
метил]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат.
Е. (6Я,7Я)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-[[2-
(1,1 -диметилэтокси)-1,1 -диметил-2-оксоэтокси]имино]-
ацетил]амино]-8-оксо-3-[(пиридин-1 -иум-1 -ил )метил]-
5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат.
Р. Пиридин.
С. 2-[[[(12)-1-(2-Аминотиазол-4-ил)-2-[(оксоэтил)-
амино]-2-оксоэтилиден]амино]окси]-2-метилпропановая
кислота.
Н. (6Я,7Я)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-[(2-
метокси-1,1 -диметил-2-оксоэтокси)имино]ацетил]амино]-
8-оксо-3-[(пиридин-1 -иум-1 -ил )метил]-5-тиа-1 -азабици-
кло[4.2.0]окт-2-ен-2-карбоксилат.
07/2016:2344
ЦЕФТАЗИДИМ ПЕНТАГИДРАТ
С НАТРИЯ КАРБОНАТОМ
ДЛЯ ИНЪЕКЦИЙ
СеПаясНтит реМаЬудпсит е1 па1т сагЬопаз
ад 1п1еЫаЫ1е
СЕРТА2Ю1МЕ РЕЫТАНУОЯАТЕ МТН
ЗОйШМ САЯВОЫАТЕ РОЯ ШЕСТЮЫ
ОПРЕДЕЛЕНИЕ
Стерильная смесь цефтазидима пентагидрата и
натрия карбоната безводного.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание:
- цефтазидим: не менее 93,0 % и не более
105,0 % (в пересчете на сухое и не содержащее кар¬
бонат вещество);
- натрия карбонат: не менее 8,0 % и не более
10,0%.
ОПИСАНИЕ (СВОЙСТВА)
Белый или бледно-желтый кристаллический по¬
рошок.
Легко растворим в воде и в метаноле, практиче¬
ски нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
в разделе «Количественное определение».
Результаты: основной пик на хроматограмме
испытуемого раствора по времени удерживания со¬
ответствует основному пику на хроматограмме раст¬
вора сравнения (а).
B. Испытуемый образец дает реакцию (а) на кар¬
бонаты (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,60 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цефтазидим пентагидрат с натрия карбонатом
1117
Оптическая плотность (2.2.25). Не более 0,50
при длине волны 425 нм.
рН (2.2.3). От 5,0 до 7,5. Измеряют рН раствора 5.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,150 г испытуемого образ¬
ца суспендируют в 5 мл ацетонитрила Р, растворяют,
прибавляя воду Р, и доводят водой Р до объема 100 мл.
Раствор сравнения (а). К 1,0 мл испытуемого
раствора прибавляют 5,0 мл ацетонитрила Р и дово¬
дят водой Р до объема 100,0 мл. 1,0 мл полученного
раствора доводят водой Р до объема 5,0 мл.
Раствор сравнения (Ь). Для приготовления при¬
меси В т зНи 5 мл испытуемого раствора выдержи¬
вают в ультрафиолетовом свете при длине волны
254 нм в течение около 24 ч.
Раствор сравнения (с). 3 мг ФСО цефтазидима
для идентификации пиков (содержит примеси А и О)
суспендируют в 0,5 мл ацетонитрила Р, растворяют,
прибавляя воду Р и доводят водой Р до объема 2 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза:
- подвижная фаза А: раствор, содержащий
3,6 г/л динатрия гидрофосфата Р и 1,4 г/л калия
дигидрофосфата Р, доведенный 10% (об/об)
раствором кислоты фосфорной Р до рН 3,4;
- подвижная фаза В: ацетонитрил для хрома¬
тографии Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0-4
О)
00
т
со
О)
4 — 11
4—5
89
11
5—8
89 —* 84
11 — 16
8—11
84 —80
16 — 20
11—15
80 — 50
20 — 50
15—18
50—>20
сл
0
1
00
о
18—22
20
80
- скорость подвижной фазы. 1,3 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению
к цефтазидиму, время удерживания — около 8 мин):
примесь Р — около 0,4; примесь О — около 0,8; при¬
месь А — около 0,9; примесь В — около 1,4.
Идентификация пиков примесей: идентифици¬
руют пики примесей А и О, используя хроматограмму
раствора сравнения (с) и хроматограмму, прилагае¬
мую к ФСО цефтазидима для идентификации пиков;
идентифицируют пик примеси В, используя хромато¬
грамму раствора сравнения (Ь).
Пригодность хроматографической системы.
раствор сравнения (с):
- разрешение: не менее 4,0 между пиками при¬
меси А и цефтазидима.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 3,0):
- примеси А, В и О (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям А, В и О, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В и С, не должна превышать 0,5 площади основного
пика на хроматограмме раствора сравнения (а);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 5-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а); не учитывают
пик примеси Р.
Примесь Р. Жидкостная хроматография (2.2.29).
Растворы готовят непосредственно перед исполь¬
зованием.
Фосфатный буферный раствор. Готовят 10%
(об/об) раствор фосфатного буферного раствора
рН 7,0 Р4.
Испытуемый раствор. 0,500 г испытуемого об¬
разца растворяют в фосфатном буферном растворе и
доводят до объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,00 г пиридина Р раст¬
воряют в воде Р и доводят до объема 100,0 мл этим
же растворителем. 5,0 мл полученного раствора дово¬
дят водой Р до объема 200,0 мл. 1,0 мл полученного
раствора доводят фосфатным буферным раствором
до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят фосфатным буферным раствором
до объема 200,0 мл. К 1,0 мл полученного раствора
прибавляют 20,0 мл раствора сравнения (а) и дово¬
дят фосфатным буферным раствором до объема
200,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: раствор 28,8 г/л аммония ди¬
гидрофосфата Р, предварительно доведенный рас¬
твором аммиака Р до рН 7,0 — ацетонитрил Р —
вода Р (8:24:68, об/об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 255 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 10 мин.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 7,0 между пиками цеф¬
тазидима и примеси Р.
Предельное содержание примесей:
- примесь Р (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси Р, не должна превышать 6-кратную
площадь основного пика на хроматограмме раствора
сравнения (а).
Потеря в массе при высушивании (2.2.32). Не
более 13,5 %. 0,300 г испытуемого образца сушат при
температуре 25 °С и давлении не более 0,67 кПа в те¬
чение 4 ч, затем нагревают остаток при температуре
100 °С и давлении не более 0,67 кПа в течение 3 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
1118
Гэсударственная фармакопея Республики Беларусь
• Бактериальные эндотоксины (2.6.14). Менее
0,10 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 25,0 мг ФСО цефтази-
дима растворяют в подвижной фазе и доводят до объ¬
ема 25,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО цефтазиди-
ма для идентификации пиков (содержит примеси А и
(3) растворяют в подвижной фазе и доводят до объ¬
ема 5,0 мл этим же растворителем.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем гексилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: 4,3 т динатрия гидрофосфа¬
та Р и 2,7 г калия дигидрофосфата Р растворяют
в 980 мл воды Р, затем прибавляют 20 мл ацетони¬
трила Р;
- скорость подвижной фазы: 2 мл/мин;
- спектрофотометрический детектор, длина
волны245нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 6 мин.
Относительное удерживание (по отношению к
цефтазидиму, время удерживания — около 4,5 мин):
примесь А — около 0,7.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между пиками при¬
меси А цефтазидима.
Содержание С22Н221Ч60732 рассчитывают с учетом
содержания цефтазидима в ФСО цефтазидима.
Натрия карбонат. Атомно-абсорбционная спек¬
трометрия (2.2.23, метод I).
Буферный раствор цезия хлорида. К 12,7 г цезия
хлорида Р прибавляют 500 мл воды Р и 86 мл кисло¬
ты хлористоводородной Р и доводят водой Р до объ¬
ема 1000,0 мл.
Эталонный раствор натрия (1000 мг/л). 3,70 г
натрия нитрата Р растворяют в воде Р и доводят до
объема 500 мл этим же растворителем, прибавляют
48,5 г кислоты азотной Р и доводят водой Р до объ¬
ема 1000 мл.
Испытуемый раствор. 650,0 мг испытуемого
образца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем. К 10,0 мл получен¬
ного раствора прибавляют 5,0 мл буферного раствора
цезия хлорида и доводят водой Р до объема 50,0 мл.
Раствор сравнения. По 20,0 мл буферного раст¬
вора цезия хлорида помещают в 4 идентичные кол¬
бы, затем прибавляют соответственно 0 мл, 5,00 мл,
10.00 мл, 15,00 мл эталонного раствора натрия
(1000 мг/л) и доводят водой Р до объема 200,0 мл.
Источник излучения: лампа с полым катодом
для определения натрия.
Длина волны: от 330,2 нм до 330,3 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Рассчитывают содержание натрия карбоната в
процентах.
ХРАНЕНИЕ
В стерильном воздухонепроницаемом контейне¬
ре с контролем первого вскрытия в защищенном от
света и влажности месте.
МАРКИРОВКА
Указывают содержание цефтазидима в процен¬
тах (м/м).
ПРИМЕСИ
Специфицированные примеси: А, В, Р, С.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С, Е, Н.
А. (2Я5,6Я,7#)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-
[(1-карбокси-1-метилэтокси)имино]ацетил]амино]-8-
оксо-3-[(пиридин-1 -иум-1 -ил)метил]-5-тиа-1 -азабици-
кло[4.2.0]окт-3-ен-2-карбоксилат (А-2-цефтазидим).
В. (6Я,7/?)-7-[[(2Е)-2-(2-Аминотиазол-4-ил)-2-[(1-
карбокси-1-метилэтокси)имино]ацетил]амино]-8-оксо-
3-[(пиридин-1 -иум-1 -ил)метил]-5-тиа-1 -азабицикло[4.2.0]-
окт-2-ен-2-карбоксилат.
С. (6Я7К)-7-Амино-8-оксо-3-[(пиридин-1-иум-1 -ил)-
метил]-5-тиа-1-азабицикло[4.2.0]окг-2-ен-2-карбоксилат.
Е. (6/?,7Я)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-[[2-
(1,1-диметилэтокси)-1,1-диметил-2-оксоэтокси]имино]-
ацетил]амино]-8-оксо-3-[(пиридин-1-иум-1-ил)метил]-
5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат.
Цефтриаксон натрия
1119
Р. Пиридин.
6. 2-[[[( 12)-1 -(2-Аминотиазол-4-ил)-2-[(оксоэтил)-
амино]-2-оксоэтилиден]амино]окси]-2-метилпропановая
кислота.
Н. (6Я,7Я)-7-[[(22)-2-(2-Аминотиазол-4-ил)-2-[(2-
метокси-1,1 -диметил-2-оксоэтокси)имино]ацетил]амино]-
8-оксо-3-[(пиридин-1-иум-1-ил)метил]-5-тиа-1-азабици-
кло[4.2.0]окт-2-ен-2-карбоксилат.
07/2016:0991
ЦЕФТРИАКСОН НАТРИЯ
СеПпахопит паМсит
СЕРТШАХОЫЕ 20ВШМ
ОПРЕДЕЛЕНИЕ
Динатрия (6/?,7Я)-7-[[(22)-(2-аминотиазол-4-ил)
(метоксиимино)ацетил]амино]-3-[[(2-метил-6-оксидо-5-
оксо-2,5-дигидро-1,2,4-тиазин-3-ил)сульфанил]метил]-
8-оксо-5-тиа-1 -азабицикло[4.2.0] окт-2-ен-2-карбоксилат
3,5-гидрат.
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Почти белый или желтоватый кристаллический
порошок. Слегка гигроскопичен.
Легко растворим в воде, умеренно растворим в
метаноле, очень мало растворим в этаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цефтриаксона натрия #или
спектр, представленный на рисунке #0991 .-1#.
В. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 2,40 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20,0 мл этим же растворителем.
Раствор 31. 2 мл раствора 5 доводят водой Р до
объема 20 мл.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2). Окраска раствора 31 должна
быть не интенсивнее эталона У(Ж)5 или ВУ(КЖ)5.
рН (2.2.3). От 6,0 до 8,0. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От
-155 до “170 (в пересчете на безводное вещество).
0,250 г испытуемого образца растворяют в воде Р и
доводят до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 30,0 мг ФСО цефтриак¬
сона натрия растворяют в подвижной фазе и доводят
до объема 100,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО цефтриак¬
сона натрия и 5,0 мг ФСО цефтриаксона примеси А
растворяют в подвижной фазе и доводят до объема
100,0 мл этим же растворителем.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза: 2,0 г тетрадециламмония
бромида Р и 2,0 г тетрагептиламмония бромида Р
растворяют в смеси из 440 мл воды Р, 55 мл 0,067 М
фосфатного буферного раствора рН 7,0 Р, 5,0 мл
цитратного буферного раствора рН 5,0 (20,17 г лимон¬
ной кислоты Р растворяют в 800 мл воды Р, доводят
до рН 5,0 раствором натрия гидроксида концентри¬
рованным Р и разводят водой Р до объема 1000,0 мл)
и 500 мл ацетонитрила Р;
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 2-кратное вре¬
мя удерживания цефтриаксона.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 3,0 между пиками цеф¬
триаксона и примеси А.
Предельное содержание примесей:
- любая примесь (не более 1,0 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с);
- сумма примесей (не более 4,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 4-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
1120
Гэсударственная фармакопея Республики Беларусь
Рисунок #0991 .-1. Инфракрасный спектр ФСО цефтриаксона натрия
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,1 площади основного пика на хро¬
матограмме раствора сравнения (с).
№,№Диметиланилин (2.4.26, метод В). Не бо¬
лее 0,0020 % (20 ррпл).
2-Этилгексановая кислота (2.4.28). Не более
0,8 % (м/м).
Вода (2.5.12). Не менее 8,0% и не более
11,0 %. Определение проводят из 0,100 г испытуе¬
мого образца.
Бактериальные эндотоксины (2.6.14). Менее
0,08 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
#Пирогенность (2.6.8). Испытуемый образец
должен быть апирогенным. Тест-доза — 40 мг испы¬
туемого образца в 1 мл воды для инъекций Р на 1,0 кг
массы тела кролика.
Испытание «Пирогенность» проводят в качестве
альтернативного испытанию «Бактериальные эндо¬
токсины».
#Стерильность (2.6.1). Цефтриаксон натрия в
условиях испытания обладает антимикробным дей¬
ствием. Для устранения антимикробного действия
посев на питательные среды проводят методом мем¬
бранной фильтрации.
#Аномальная токсичность (2.6.9). Испытуемый
образец должен быть нетоксичным. Тест-доза — 30 мг
испытуемого образца в 0,5 мл воды для инъекций Р,
внутривенно. Срок наблюдения — 48 ч.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание цефтриаксона натрия рассчитывают
в процентах, учитывая содержание С18Н16М8Ыа20783 в
ФСО цефтриаксона натрия.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Стерильная субстанция — в стерильном возду¬
хонепроницаемом контейнере с контролем первого
вскрытия.
ПРИМЕСИ
А. (6Я,7#)-7-[[(2Е)-(2-Аминотиазол-4-ил)(метокси-
имино)ацетил]амино]-3-[[(2-метил-5,6-диоксо-1,2,5,6-
тетрагидро-1,2,4-триазин-3-ил)сульфанил]метил]-8-
оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая
кислота ((Е)-изомер).
В. (5аЯ,6Я)-6-[[(22)-(2-Аминотиазол-4-ил)(меток-
сиимино)ацетил]амино]-5а,6-дигидро-ЗЯ,7Н-азето[2,1 -Ь]-
фуро[3,4-</][1,3]тиазин-1,7(4Я)-дион.
Цианокобаламин
1121
нА
Т
СН3
С. 2-Метил-3-сульфанил-1,2-дигидро-1,2,4-триазин-
5,6-дион.
О. 5-Бензотиазол-2-ил-(22)-(2-аминотиазол-4-ил)-
(метоксиимино)тиоацетат.
Е. (6#,7#)-7-Амино-3-[[(2-метил-5,6-диоксо-1,2,5,6-
тетрагидро-1,2,4-триазин-3-ил)сульфанил]метил]-8-
оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая
кислота.
07/2016:0547
ЦИАНОКОБАЛАМИН
СуапосоЬа1аттит
СУАЫОСОВА1-АМ1Ы
ОПРЕДЕЛЕНИЕ
а-(5,6-Диметилбензимидазол-1 -ил)кобамида ци¬
анид. #Витамин В12#.
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на сухое вещество).
Требования данной статьи применяются к циано-
кобаламину, получаемому путем ферментации.
ОПИСАНИЕ (СВОЙСТВА)
Темно-красный кристаллический порошок или
темно-красные кристаллы. Безводная субстанция
очень гигроскопична.
Умеренно растворим в воде и 96 % спирте, прак¬
тически нерастворим в ацетоне.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в уль¬
трафиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 2,5 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема
100.0 мл этим же растворителем.
Диапазон длин волн: от 260 нм до 610 нм.
Максимумы поглощения: при 278 нм, при
361 нм, а также в диапазоне от 547 нм до 559 нм.
Отношение оптических плотностей:
- Аз61/А547-559 — от 3,15 до 3,45;
- Азв1/А278 — от 1,70 до 1,90.
B. Тонкослойная хроматография (2.2.27). Ис¬
пытания проводят с защитой от света.
Испытуемый раствор. 2 мг испытуемого об¬
разца растворяют в 1 мл смеси 96 % спирт Р —
вода Р (1:1, об/об).
Раствор сравнения. 2 мг ФСО цианокобала-
мина растворяют в 1 мл смеси 96 % спирт Р —
вода Р (1:1, об/об).
Пластинка: ТСХ пластинка со слоем силика¬
геля О Р.
Подвижная фаза: раствор аммиака разведен¬
ный Р1 ■— метанол Р — метиленхлорид Р (9:30:45,
об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: камеру не насыщают
парами подвижной фазы, не менее 2/3 высоты пла¬
стинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают при
дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соот¬
ветствующее по расположению, цвету и размеру
основному пятну на хроматограмме раствора срав¬
нения.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого
образца растворяют в подвижной фазе и доводят
до объема 10,0 мл этим же растворителем. Исполь¬
зуют в течение 1 ч.
Раствор сравнения (а). 3,0 мл испытуемо¬
го раствора доводят подвижной фазой до объема
100.0 мл. Используют в течение 1 ч.
Раствор сравнения (Ь). 5,0 мл испытуемо¬
го раствора доводят подвижной фазой до объема
50.0 мл. 1,0 мл полученного раствора доводят под-
141. Зак. 1060.
1122
Гэсударственная фармакопея Республики Беларусь
вижной фазой до объема 100,0 мл. Используют в
течение 1 ч.
Раствор сравнения (с). 25 мг испытуемого об¬
разца растворяют в 10 мл воды Р, при необходимо¬
сти нагревая. Охлаждают, прибавляют 5 мл раст¬
вора 1,0 г/л хлорамина Р и 0,5 мл 0,05 М раствора
кислоты хлористоводородной, доводят водой Р
до объема 25 мл, перемешивают и выдерживают в
течение 5 мин. 1 мл полученного раствора доводят
подвижной фазой до объема 10 мл и используют
немедленно.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4 мм, заполненная силикагелем октилси-
лильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: смесь из 26,5 объемов ме¬
танола Р и 73,5 объемов раствора 10 г/л динатрия
гидрофосфата Р, доведенного до рН 3,5 кислотой
фосфорной Р (раствор годен в течение 2 дней);
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 361 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания цианокобаламина.
Пригодность хроматографической системы:
- на хроматограмме раствора сравнения (с)
должно обнаруживаться два основных пика;
- разрешение: не менее 2,5 между двумя ос¬
новными пиками на хроматограмме раствора (с);
- отношение сигнал/шум: не менее 5 для ос¬
новного пика на хроматограмме раствора сравне¬
ния (Ь).
Предельное содержание примесей:
- сумма примесей (не более 3 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раст¬
вора сравнения (а);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32).
Не более 12,0%. 40,00 мг испытуемого образца
сушат в вакууме при температуре 105 °С в тече¬
ние 2 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
100,0 мг испытуемого образца растворяют в
воде Р и доводят до объема 500,0 мл этим же раст¬
ворителем. 25,0 мл полученного раствора доводят
водой Р до объема 200,0 мл. Измеряют оптическую
плотность (2.2.25) полученного раствора в максимуме
при длине волны 361 нм.
Содержание Сб3Н88СоМ14014Р рассчитывают с
учетом удельного показателя поглощения, равно¬
го 207.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
07/2016:0711
ЦИКЛОФОСФАМИД
Сус1орМозрНат1с1ит
СУС1-ОРНОЗРНАМЮЕ
О ,
\
о
/
1
и энантиомер
О
сч
X
1 \
\
,С12М202РН20
С1
М.м. 279,1
И 9-2]
ОПРЕДЕЛЕНИЕ
(2Я5)-А/,А/-Бис(2-хлорэтил)тетрагидро-2Н-1,3,2-
оксазафосфорин-2-амина 2-оксид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде, легко растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А, С, О.
A. Определяют температуру плавления (2.2.14)
испытуемого образца. Смешивают равные количе¬
ства испытуемого образца и ФСО циклофосфамида
и определяют температуру плавления полученной
смеси. Разница между полученными значениями тем¬
пературы плавления (около 51 °С) не должна превы¬
шать 2 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО циклофосфамида.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси». На хрома¬
тограмме испытуемого раствора (Ь) обнаруживается
основное пятно, соответствующее по расположению,
цвету и размеру основному пятну на хроматограмме
раствора сравнения (а).
О. 0,1 г испытуемого образца растворяют в 10 мл
воды Р и прибавляют 5 мл раствора серебра нитра¬
та Р1. Раствор прозрачный. При кипячении образует¬
ся белый осадок, растворимый в растворе аммиака
концентрированном Р и выпадающий снова при при¬
бавлении кислоты азотной разведенной Р.
ИСПЫТАНИЯ
Раствор 3. 0,50 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 4,0 до 6,0. Измеряют рН раствора 3
непосредственно после приготовления.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого об¬
разца растворяют в 96 % спирте Р и доводят до объ¬
ема 5 мл этим же растворителем.
Циластатин натрия
1123
Испытуемый раствор (Ь). 1 мл испытуемо¬
го раствора (а) доводят 96 % спиртом Р до объема
10 мл.
Раствор сравнения (а). 10 мг ФСО циклофосфа-
мида растворяют в 96% спирте Р и доводят до объ¬
ема 5 мл этим же растворителем.
Раствор сравнения (Ь). 0,1 мл испытуемого раст¬
вора (а) доводят 96 % спиртом Р до объема 10 мл.
Пластинка: ТСХ пластинка со споем силикагеля 6 Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — ацетон Р — вода Р — метилэтилкетон Р
(2:4:12:80, о&об/об/об).
Наносимый объем пробы. 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: в потоке теплого воздуха и нагре¬
вают при температуре 110 °С в течение 10 мин.
Обработка хлором: на дно хроматографической
камеры помещают выпарительную чашку, содержа¬
щую смесь из равных объемов кислоты хлористо¬
водородной Р и раствора 50 г/л калия пермангана¬
та Р. Горячую пластинку выдерживают закрытой
камере в парах хлора в течение 2 мин. Затем пла¬
стинку обрабатывают струей холодного воздуха до
удаления избытка хлора, при этом пластинка ниже
точек нанесения растворов не должна окрашиваться
в синий цвет от прибавления капли раствора калия
йодида и крахмала Р (допускается появление только
очень незначительного бледно-синего окрашивания).
Необходимо избегать длительной обработки холод¬
ным воздухом.
Проявление: пластинку опрыскивают раствором
калия йодида и крахмала Р и выдерживают в течение
5 мин.
Предельное содержание примесей:
-любая примесь (не более 1,0 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора (а) не учитывают пятна на линии
старта.
Хлориды (2.4.4). Не более 0,0330 % (330 ррт).
0,15 г испытуемого образца растворяют в воде Р и до¬
водят до объема 15 мл этим же растворителем. Све¬
жеприготовленный раствор должен выдерживать ис¬
пытание на хлориды.
Фосфаты (2.4.11). Не более 0,0100 % (100 ррт).
0,10 г испытуемого образца растворяют в воде Р и до¬
водят до объема 100 мл этим же растворителем. По¬
лученный раствор должен выдерживать испытание на
фосфаты.
Тяжелые металлы (2.4.8, метод С). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не менее 6,0% и не более 7,0 %.
Определение проводят из 0,300 г испытуемого образца.
#Пирогенность (2.6.8). Если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения без последующей
процедуры удаления пирогенов, испытуемый образец
должен быть апирогенным. Тест-доза — 50 мг испы¬
туемого образца в 4 мл раствора 9 г/л натрия хлори¬
да Р на 1,0 кг массы тела кролика за 60 с. Срок на¬
блюдения — 72 ч.
#Стерильность (2.6.1). Если субстанция пред¬
назначена для производства лекарственных средств
для парентерального применения без последующей
процедуры стерилизации, испытуемый образец дол¬
жен выдерживать испытание на стерильность мето¬
дом мембранной фильтрации.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 50 мл
раствора 1 г/л натрия гидроксида Р в этиленглико¬
ле Р и кипятят с обратным холодильником в течение
30 мин. Охлаждают, холодильник промывают 25 мл
воды Р, прибавляют 75 мл 2-пропанола Р, 15 мл кис¬
лоты азотной разведенной Р, 10,0 мл 0,1 М раст¬
вора серебра нитрата, 2,0 мл раствора железа (III)
аммония сульфата Р2 и титруют 0,1 М раствором
тиоцианата.
1 мл 0,1 М раствора серебра нитрата соответ¬
ствует 13,05 мг С7Н15С12М202Р.
#ХРАНЕНИЕ
Если субстанция стерильная, ее хранят в сте¬
рильном воздухонепроницаемом контейнере с кон¬
тролем первого вскрытия.
07/2016:1408
ЦИЛАСТАТИН НАТРИЯ
СНазШтит паМсит
СН-АЗТАТ1Ы ВООШМ
С16Н25М2Ыа053 М.м. 380,4
[81129-83-1]
ОПРЕДЕЛЕНИЕ
Натрия (2)-7-[[(2/?)-2-амино-2-карбоксиэтил]суль-
фанил]-2-[[[(15)-2,2-диметилциклопропил]карбонил]-
амино]гепт-2-еноат.
Содержание: не менее 98,0 % и не более 101,5 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или светло-желтый аморфный порошок.
Гигроскопичен.
Очень легко растворим в воде и в метаноле, мало
растворим в этаноле, очень мало растворим в диме-
тилсульфоксиде, практически нерастворим в ацетоне
и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО циластатина натрия.
C. Испытуемый образец дает реакцию (а) на на¬
трий (2.3.1).
141*. Зак. 1060.
1124
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона У(Ж)6.
рН (2.2.3). От 6,5 до 7,5. Измеряют рН раствора 5.
Удельное оптическое вращение (2.2.7). От
+41,5 до +44,5 (в пересчете на безводное вещество).
0,250 г испытуемого образца растворяют в смеси
из кислоты хлористоводородной Р и метанола Р
(1:120, об/об) и доводят до объема 25,0 мл этой же
смесью растворителей.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Растворы готовят непосредственно
перед применением.
Испытуемый раствор. 32 мг испытуемого образ¬
ца растворяют в воде Р и доводят до объема 20,0 мл
этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят водой Р до объема 100,0 мл. 1,0 мл
полученного раствора доводят водой Р до объема
10.0 мл.
Раствор сравнения (Ь). 3 мг ФСО циластатина
для проверки пригодности хроматографической си¬
стемы 1 (содержит примеси А, В, Е, Р, О (эпимер 2)
и Н) растворяют в воде Р и доводят до объема 2,0 мл
этим же растворителем.
Раствор сравнения (с). 3 мг ФСО циластатина
для проверки пригодности хроматографической си¬
стемы 2 (содержит примеси С и О (эпимер 1)) раст¬
воряют в воде Р и доводят до объема 2,0 мл этим же
растворителем.
Раствор сравнения (д). 32 мг мезитилоксида Р
(примесь О) растворяют в 100,0 мл воды Р. 1,0 мл
полученного раствора доводят водой Р до объема
50.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем для хрома¬
тографии, совместимым с подвижными фазами со
100% содержанием воды, октадецилсилильным,
эндкепированным Р с размером частиц 5 мкм;
- температура: 50 °С;
- подвижная фаза:
- подвижная фаза А: фосфатный буферный
раствор рН 3,25 Р;
-подвижная фаза В: ацетонитрил Р1, фос¬
фатный буферный раствор рН 3,25 Р (50:50,
об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
100
0
3—28
о
о
СО
о
0 — 10
28—38
90
10
38—63
со
о
4
сл
о
0
1
сл
о
63—78
50 —► 30
сл
о
4
■^1
О
78—88
30
70
- скорость подвижной фазы: 2,0 мл/мин;
-спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей А, В, Е, Р, 6 (эпимер 2) и Н, ис¬
пользуя хроматограмму раствора сравнения (Ь) и хро¬
матограмму, прилагаемую к ФСО циластатина для
проверки пригодности хроматографической систе¬
мы 1\ идентифицируют пики примесей С и С (эпимер
1), используя хроматограмму раствора сравнения (с)
и хроматограмму, прилагаемую к ФСО циластатина
для проверки пригодности хроматографической си¬
стемы 2; идентифицируют пик примеси О, используя
хроматограмму раствора сравнения (б).
Относительное удерживание (по отношению к
циластатину, время удерживания — около 50 мин):
примесь Е — около 0,2; примесь А (эпимер 1) — око¬
ло 0,60; примесь А (эпимер 2) — около 0,62; примесь
О — около 0,9; примесь Р — около 0,98; примесь 6
(эпимер 1) — около 1,02; примесь С (эпимер 2) — око¬
ло 1,05; примесь Н — около 1,06; примесь В — около
1,17; примесь С — около 1,23.
Пригодность хроматографической системы:
- коэффициент разделения пиков: не менее 10
(Нр — высота пика примеси Р относительно базовой
линии; — расстояние между базовой линией и
нижней точкой кривой, разделяющей пик примеси Р
и пик циластатина на хроматограмме раствора срав¬
нения (Ь));
- коэффициент разделения пиков: не менее 2,5
(Нр — высота пика примеси 6 (эпимер 1) относитель¬
но базовой линии; Ну — расстояние между базовой
линией и нижней точкой кривой, разделяющей пик
примеси С (эпимер 1) и пик циластатина на хромато¬
грамме раствора сравнения (с)).
Расчет процентного содержания:
-для всех примесей — используют концентра¬
цию циластатина в растворе сравнения (а).
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси С — 1,3; для примеси Е — 3,3; для примеси
О (эпимер 1) — 3,3; для примеси (3 (эпимер 2) — 1,6):
- примеси А (сумма всех эпимеров): не более 0,5 %;
- примесь С (сумма всех эпимеров): не более 0,4 %;
- примесь Е (сумма всех эпимеров): не более 0,3 %;
- примеси В, Р, Н: не более 0,1 % для каждой при¬
меси;
- примесь О: не более 0,1 % для каждого эпимера;
- неспецифицированные примеси: не более
0,05 % для каждой примеси;
- сумма примесей: не более 1,0 %;
-учитываемый предел: 0,03%; не учитывают
пики, соответствующие пику примеси О на хромато¬
грамме раствора сравнения (б).
Примесь О, ацетон и метанол. Газовая хрома¬
тография (2.2.28).
Раствор внутреннего стандарта. 0,5 мл про¬
панола Р растворяют в воде Р и доводят до объема
1000 мл этим же растворителем.
Испытуемый раствор. 0,200 г испытуемого об¬
разца растворяют в воде Р, прибавляют 2,0 мл раст¬
вора внутреннего стандарта и доводят водой Р до
объема 10,0 мл.
Раствор сравнения. 2,0 мл ацетона Р, 0,5 мл
метанола Р и 0,5 мл мезитилоксида Р (примесь й)
растворяют в воде Р и доводят до объема 1000 мл
этим же растворителем. К 2,0 мл полученного раст¬
вора прибавляют 2,0 мл раствора внутреннего стан¬
дарта и доводят водой Р до объема 10,0 мл. Получен¬
ный раствор содержит 316 мкг ацетона, 79 мкг метано¬
ла и 86 мкг примеси О в 1 мл.
Циластатин натрия
1125
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м и
внутренним диаметром около 0,53 мм, покрытая сло¬
ем макрогола 20 000 Р (толщина слоя 1,0 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 9 мл/мин;
- температура:
Время (мин)
Температура (°С)
Колонка
0—2,5
50
2,5—5
50 —* 70
5—5,5 !
! 70
Блок ввода проб
! 160
Детектор
| 220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Содержание ацетона, метанола и примеси О в
процентах рассчитывают по формуле:
Я/
где:
С — концентрация растворителя в растворе
сравнения, мкг/мл;
IN — количество циластатина натрия в испытуе¬
мом растворе, мг;
Ри — отношение площади пика растворителя к
площади пика пропанола на хроматограмме испыту¬
емого раствора;
Р$ — отношение площади пика растворителя к
площади пика пропанола на хроматограмме раствора
сравнения.
Предельное содержание примесей:
- ацетон: не более 1,0 % (м/м)\
- метанол: не более 0,5 % (м/м);
- примесь О: не более 0,4 % (м/м).
Тяжелые металлы (2.4.8, метод Н). Не более
0,0010 % (10 ррт). 1,0 г испытуемого образца должен
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 1 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Вода (2.5.12). Не более 2,0 %. Определение про¬
водят из 0,500 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
Бактериальные эндотоксины (2.6.14). Менее
0,17 МЕ/мг, если субстанция предназначена для про¬
изводства лекарственных средств для парентераль¬
ного применения без дальнейшей подходящей проце¬
дуры удаления бактериальных эндотоксинов.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в 30 мл
метанола Р и прибавляют 5 мл воды Р. Полученный
раствор доводят до рН около 3,0 0,1 М раствором
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Наблюдаются три скачка потенциала. Отме¬
чают объем, прибавленный между 1-й и 3-й точками
эквивалентности.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 19,02 мг С16Н25Ы2Ма053.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере при темпе¬
ратуре от 2 °С до 8 °С. Если субстанция стерильная,
ее хранят в стерильном воздухонепроницаемом кон¬
тейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Р, 6, Н.
но2с^ ^со2н
Н N42 о
и эпимер при 3
А. (2>7-[(Я5)-[(2Я)-2-Амино-2-карбоксиэтил]суль-
финил]-2-[[[(15)-2,2-диметилциклопропил]карбонил]-
амино]гепт-2-еноевая кислота.
и эпимер при С*
в. (2)-7-[[(2Я)-2-Карбокси-2-[[(1 Я5)-1 -Метил-З-оксо-
бутил]амино]этил]сульфанил]-2-[[[(15)-2,2-диметил-
циклопропил]карбонил]амино]гепт-2-еноевая кислота.
со2н
н3с
С. (2)-7-[[(2/?)-2-Карбокси-2-[(1,1-диметил-З-оксо-
бутил)амино]этил]сульфанил]-2-[[[(15)-2,2-диметил-
циклопропил]карбонил]амино]гепт-2-еноевая кислота.
сн3
н3с
сн3
0.4-Метилпент-3-ен-2-он (мезитилоксид).
Н°2С\ /С02Н
Е. 7-[[(2Я)-2-Амино-2-карбоксиэтил]сульфанил]-2-
оксогептановая кислота.
Р. (2)-7-[[(2Я)-2-Амино-2-карбоксиэтил]сульфанил]-
2-[(2,3-диметилбут-3-еноил)амино]гепт-2-этил]суль-
фанил]-2-[[[(15)-2,2-диметилциклопропил]карбонил]-
амино]гепт-2-еноевая кислота.
и эпимер при С*
(3. (Е)-(2Я$)-7-[[(2Я)-2-Амино-2-карбоксиэтил]суль-
фанил]-2-[[[(15)-2,2-диметилциклопропил]карбонил]-
амино]гепт-3-еноевая кислота.
142. Зак. 1060.
1126
Гэсударственная фармакопея Республики Беларусь
Н. (2)-7-[(2-Аминоэтил)сульфанил]-2-[[[(15)-2,2-
диметилциклопропил]карбонил]амино]гепт-2-еноевая
кислота.
07/2016:1482
ЦИНКА АЦЕТАТ ДИГИДРАТ
2//7С/ асе1аз сНЬудпсиз
ШСАСЕТАТЕ 01НУ0РАТЕ
С4Н6042п ■ 2Н20 М.м. 219,5
[5970-45-6]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %
С4Н6042п-2Н20.
ОПИСАНИЕ(СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или хлопья.
Легко растворим в воде, растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец дает реакцию (а) на аце¬
таты (2.3.1).
B. Испытуемый образец дает реакцию (а) на цинк
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 10,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р, при¬
готовленной из воды дистиллированной Р, и доводят
до объема 100 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод И). Раствор 5 должен
быть бесцветным.
рН (2.2.3). От 5,8 до 7,0. 10 мл раствора 3 до¬
водят водой, свободной от углерода диоксида, Р до
объема 20 мл. Измеряют рН полученного раствора.
Восстанавливающие вещества. К 10 мл раст¬
вора 3 прибавляют 90 мл воды Р, 5 мл кислоты
серной разведенной Р, 1,5 мл раствора 0,3 г/л калия
перманганата Р и кипятят в течение 5 минут. Раствор
должен сохранять розовое окрашивание.
Хлориды (2.4.4). Не более 0,0050 % (50 ррт).
10 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
15 мл раствора 3 должны выдерживать испытание на
сульфаты.
Алюминий. Не более 0,0005 % (5 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. 2,50 г испытуемого об¬
разца растворяют в 20 мл раствора 200 г/л кислоты
азотной, свободной от свинца и кадмия, Р и доводят
до объема 25,0 мл этим же растворителем.
Растворы сравнения. Растворы сравнения го¬
товят соответствующими разведениями эталонного
раствора алюминия (200 ррт А1) Р раствором 200 г/л
кислоты азотной, свободной от свинца и кадмия, Р.
Источник излучения: лампа с полым катодом
для определения алюминия.
Длина волны: 309,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя либо пламя ацетилен — закись азота.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррт). 0,5 г испытуемого образца должны выдержи¬
вать испытание на мышьяк.
Кадмий. Не более 0,0002 % (2 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. Используют раствор, при¬
готовленный как указано в испытании «Алюминий».
Растворы сравнения. Растворы сравнения го¬
товят соответствующими разведениями эталонного
раствора кадмия (0,1 % Сф Р раствором 200 г/л кис¬
лоты азотной, свободной от свинца и кадмия, Р.
Источник излучения: лампа с полым катодом
для определения кадмия.
Длина волны: 228,8 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Медь. Не более 0,0050 % (50 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. Используют раствор,
приготовленный как указано в испытании «Железо».
Растворы сравнения. Растворы сравнения го¬
товят соответствующими разведениями эталонного
раствора меди (10 ррт Си) Р раствором 200 г/л кис¬
лоты азотной, свободной от свинца и кадмия, Р.
Источник излучения: лампа с полым катодом
для определения меди.
Длина волны: 324,8 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Железо. Не более 0,0050 % (50 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. 1,25 г испытуемого об¬
разца растворяют в 20 мл раствора 200 г/л кислоты
азотной, свободной от свинца и кадмия, Р и доводят
до объема 25,0 мл этим же растворителем.
Растворы сравнения. Растворы сравнения го¬
товят соответствующими разведениями эталонного
раствора железа (20 ррт Ре) Р раствором 200 г/л
кислоты азотной, свободной от свинца и кадмия, Р.
Источник излучения: лампа с полым катодом
для определения железа.
Длина волны: 248,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Свинец. Не более 0,0010 %. (10 ррт).
Атомно-абсорбционная спектрометрия (2.2.23,
метод I).
Испытуемый раствор. 5,00 г испытуемого об¬
разца растворяют в 20 мл раствора 200 г/л кислоты
азотной, свободной от свинца и кадмия, Р и доводят
до объема 25,0 мл этим же растворителем.
Растворы сравнения. Растворы сравнения го¬
товят соответствующими разведениями эталонного
раствора свинца (0,1 % РЬ) Р раствором 200 г/л кис¬
лоты азотной, свободной от свинца и кадмия, Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Цинка глюконат
1127
Длина волны: 283,3 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 5 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое определение цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 21,95 мг С4Н6042п-2Н20.
ХРАНЕНИЕ
В неметаллическом контейнере.
07/2016:2164
ЦИНКА ГЛЮКОНАТ
2/пс/ д1исопаз
21N С вШСОЫАТЕ
С12Нг22п014 • хН20 М.м. 455,7
(безводное вещество)
ОПРЕДЕЛЕНИЕ
Цинка О-гпюконат безводный или гидратированный.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок. Гигроскопичен.
Растворим в воде, практически нерастворим в
этаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в 1 мл воды Р.
Раствор сравнения. 20 мг ФСО кальция глюко¬
ната растворяют в 1 мл воды Р, нагревая при необхо¬
димости в водяной бане при температуре 60 °С.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: раствор аммиака концен¬
трированный Р — этилацетат Р — вода Р — 96%
спирт Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: 1 мкп.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: при температуре от 100 °С до
105 °С в течение 20 мин, затем охлаждают до комнат¬
ной температуры.
Проявление: пластинку опрыскивают раствором,
содержащим 25 г/л аммония молибдата Р и 10 г/л
церия сульфата Р в кислоте серной разведенной Р,
и нагревают при температуре от 100 °С до 105 °С в
течение около 10 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
В. 0,1 г испытуемого образца растворяют в 5 мл
воды Р, прибавляют 0,5 мл раствора калия ферроци¬
анида Р. Образуется белый осадок, который не рас¬
творяется при прибавлении 5 мл кислоты хлористо¬
водородной Р.
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 по степени мут¬
ности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)6.
Сахароза и восстанавливающие сахара. 0,5 г
испытуемого образца растворяют в смеси из 2 мл
кислоты хлористоводородной Р1 и 10 мл воды Р.
Кипятят в течение 5 мин, охлаждают, прибавляют
10 мл раствора натрия карбоната Р, выдерживают
в течение 10 мин, доводят водой Р до объема 25 мл и
фильтруют. К 5 мл фильтрата прибавляют 2 мл раст¬
вора медно-тартратного Р, кипятят в течение 1 мин
и выдерживают в течение 2 мин. Не должен образо¬
вываться красный осадок.
Хлориды (2.4.4). Не более 0,0500 % (500 ррт).
5 мл раствора 5 доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,0500%
(500 ррт). 2,0 г испытуемого образца растворяют в
смеси из 10 мл кислоты уксусной Р и 90 мл воды дис¬
тиллированной Р.
Кадмий. Атомно-абсорбционная спектрометрия
(2.2.23, метод II). Не более 0,0002 % (2 ррт).
Испытуемый раствор. 5,00 г испытуемого об¬
разца растворяют с помощью ультразвука в 20 мл
воды дистиллированной деионизированной Р и дово¬
дят этим же растворителем до объема 25,0 мл.
Растворы сравнения: готовят разбавлением
эталонного раствора кадмия (0,1 % Сд) водой дис¬
тиллированной деионизированной Р.
Источник излучения: лампа с полым катодом
для определения кадмия.
Длина волны: 228,8 нм.
Атомизатор: воздушно-ацетиленовое пламя.
Тяжелые металлы (2.4.8, метод А). Не более
0,0010% (10 ррт). 2,0 г испытуемого образца раст¬
воряют в 20 мл воды Р, нагревая в водяной бане
при температуре 60 °С. 12 мл полученного раствора
должны выдерживать испытание на тяжелые метал¬
лы. Эталон готовят с использованием эталонного
раствора свинца (1 ррт РЬ) Р.
Вода (2.5.32). Не более 12,0%. Определение
проводят из 80,0 мг испытуемого образца.
Микробиологическая чистота
ОКА: критерий приемлемости 103 КОЕ/г (2.6.12).
ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 г испытуемого образца растворяют в 5 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое титрование цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 45,57 мг С12Н222п014.
142*. Зак. 1060.
1128
Гэсударственная фармакопея Республики Беларусь
ХРАНЕНИЕ
В неметаллическом воздухонепроницаемом кон¬
тейнере.
07/2016:0252
ЦИНКА ОКСИД
2\пс\ ох1'с1ит
ШС ОХЮЕ
2пО М.м. 81,4
[1314-13-2]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на прокаленное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Мягкий белый или бледно желтовато-белый
аморфный порошок, не содержащий твердых частиц.
#Поглощает из воздуха диоксид углерода.#
Практически нерастворим в воде и 96 % спирте.
Растворяется в разведенных растворах минеральных
кислот, #в уксусной кислоте, в растворах гидроксидов
щелочных металлов#.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец желтеет при сильном на¬
гревании. Желтая окраска исчезает при охлаждении.
B. 0,1 г испытуемого образца растворяют в 1,5 мл
кислоты хлористоводородной разведенной Р и до¬
водят водой Р до объема 5 мл. Полученный раствор
дает реакцию на цинк (2.3.7).
ИСПЫТАНИЯ
Щелочность. 1,0 г испытуемого образца встря¬
хивают с 10 мл кипящей воды Р, прибавляют 0,1 мл
раствора фенолфталеина Р и фильтруют. Если
фильтрат окрашен в красный цвет, то при прибавлении
не более 0,3 мл 0,1 М раствора кислоты хлористо¬
водородной окраска фильтрата должна измениться.
Карбонаты и вещества, нерастворимые в кис¬
лотах. 1,0 г испытуемого образца растворяют в 15 мл
кислоты хлористоводородной разведенной Р. При
растворении не должно выделяться пузырьков газа.
Полученный раствор по степени мутности не должен
превышать эталон II (2.2.1) и должен быть бесцвет¬
ным (2.2.2, метод II).
Мышьяк (2.4.2, метод А). Не более 0,0005 %
(5 ррт). Определение проводят из 0,2 г испытуемого
образца.
Кадмий. Не более 0,0010 % (10 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 2,0 г испытуемого об¬
разца растворяют в 14 мл смеси из равных объемов
воды Р и кислоты азотной, свободной от кадмия и
свинца, Р, кипятят в течение 1 мин, охлаждают и до¬
водят водой Р до объема 100,0 мл.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора кадмия
(0,1 % Сф Р 3,5 % (об/об) раствором кислоты азот¬
ной, свободной от кадмия и свинца, Р.
Источник излучения: лампа с полым катодом
для определения кадмия.
Длина волны: 228,8 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое или воздушно-пропановое пламя.
Железо (2.4.9). Не более 0,0200 % (200 ррт).
50 мг испытуемого образца растворяют в 1 мл кисло¬
ты хлористоводородной разведенной Р и доводят
водой Р до объема 10 мл. Полученный раствор дол¬
жен выдерживать испытание на железо. Используют
0,5 мл кислоты тиогиколиевой Р.
Свинец. Не более 0,0050 % (50 ррт). Атомно-аб-
сорбционная спектрометрия (2.2.23, метод II).
Испытуемый раствор. 5,0 г испытуемого об¬
разца растворяют в 24 мл смеси из равных объемов
воды Р и кислоты азотной, свободной от кадмия и
свинца, Р, кипятят в течение 1 мин, охлаждают и до¬
водят водой Р до объема 100,0 мл.
Растворы сравнения. Готовят соответствую¬
щими разведениями эталонного раствора свинца
(0,1 % РЬ) Р 3,5 % (об/об) раствором кислоты азот¬
ной, свободной от кадмия и свинца, Р.
Источник излучения: лампа с полым катодом
для определения свинца.
Длина волны: 283,3 нм или 217,0 нм (в зависимо¬
сти от оборудования).
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Потеря в массе при прокаливании. Не более
1,0 %. 1,00 г испытуемого образца прокаливают при
температуре (500±50) °С до постоянной массы.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 10 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое определение цинка (2.5.11).
1 мл 0,7 М раствора натрия эдетата соответ¬
ствует 8,14 мг 2пО.
#ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
07/2016:1683
ЦИНКА СУЛЬФАТ ГЕКСАГИДРАТ
2/лс/ зиНаз ЬехаЬубпсиз
2ШС ЗШ-ГАТЕ НЕХАНУйКАТЕ
2п304 ■ 6Н20 М.м. 269,5
[13986-24-8]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 104,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бездетные прозрачные кристаллы. Выветри¬
вается на воздухе.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в
разделе «Испытания», дает реакции на сульфаты
(2.3.1).
B. Раствор 3 дает реакцию на цинк (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
Цинка сульфат моногидрат
1129
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,4 до 5,6. Измеряют рН раствора 3.
Хлориды (2.4.4). Не более 0,0300 % (300 ррт).
3,3 мл раствора 5 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Железо (2.4.9). Не более 0,0100% (100 ррт).
2 мл раствора 3 доводят водой Р до объема 10 мл. По¬
лученный раствор должен выдерживать испытание на
железо. Используют 0,5 мл кислоты тиогликолевой Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 5 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое определение цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 26,95 мг 2п304-6Н20.
ХРАНЕНИЕ
В неметаллическом воздухонепроницаемом кон¬
тейнере.
07/2016:0111
ЦИНКА СУЛЬФАТ ГЕПТАГИДРАТ
7апс\ зи1?аз Мер(аМус1псиз
2ШС ЗШ-РАТЕ НЕРТАНУОНАТЕ
2п504 - 7Н20 М.м. 287,5
[7446-20-0]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 104,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцетные прозрачные кристаллы. Выветри¬
вается на воздухе.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции на сульфаты (2.3.1).
B. Раствор 3 дает реакцию на цинк (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,4 до 5,6. Измеряют рН раствора 3.
Хлориды (2.4.4). Не более 0,0300 % (300 ррт).
3,3 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Железо (2.4.9). Не более 0,0100% (100 ррт).
2 мл раствора 3 доводят водой Р до объема 10 мл.
Полученный раствор должен выдерживать испытание
на железо. Используют 0,5 мл кислоты тиогликоле¬
вой Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 5 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое определение цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 28,75 мг 2п304*7Н20.
ХРАНЕНИЕ
В неметаллическом воздухонепроницаемом кон¬
тейнере.
07/2016:2159
ЦИНКА СУЛЬФАТ МОНОГИДРАТ
2/лс/ зиКаз топоЬудпсиз
ШС 81Л.РАТЕ МОЫОНУОНАТЕ
2п504 ■ НгО М.м. 179,5
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,0 % и не более 101,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцетные прозрачные кристаллы.
Очень легко растворим в воде, практически не¬
растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Раствор 3, приготовленный как указано в раз¬
деле «Испытания», дает реакции на сульфаты (2.3.1).
B. Раствор 3 дает реакцию на цинк (2.3.1).
C. Испытуемый образец выдерживает требова¬
ния раздела «Количественное определение».
ИСПЫТАНИЯ
Раствор 3. 2,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 50 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 4,4 до 5,6. Измеряют рН раствора 3.
Хлориды (2.4.4). Не более 0,0300 % (300 ррт).
3,3 мл раствора 3 доводят водой Р до объема 15 мл.
Полученный раствор должен выдерживать испытание
на хлориды.
Железо (2.4.9). Не более 0,0100% (100 ррт).
2 мл раствора 3 доводят водой Р до объема 10 мл.
Полученный раствор должен выдерживать испытание
на железо. Используют 0,5 мл кислоты тиогликоле¬
вой Р.
143. Зак. 1060.
1130
Гэсударственная фармакопея Республики Беларусь
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,160 г испытуемого образца растворяют в 5 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое определение цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 17,95 мг 2п304 Н20.
ХРАНЕНИЕ
В неметаллическом контейнере.
07/2016:0110
ЦИНКА ХЛОРИД
2/лс/ сЫогМит
2МС СНШНЮЕ
2пС12 М.м. 136,3
[7646-85-7]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 95,0 % и не более 100,5 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или белые или почти белые кусочки в форме па¬
лочек. Расплывается на воздухе.
Очень легко растворим в воде, легко растворим в
96 % спирте и глицерине.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. 0,5 г испытуемого образца растворяют в кис¬
лоте азотной разведенной Р и доводят до объема
10 мл этим же растворителем. Полученный раствор
дает реакцию (а) на хлориды (2.3.1).
B. 5 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», дают реакцию (а) на цинк
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. К 2,0 г испытуемого образца прибав¬
ляют 38 мл воды, свободной от углерода диоксида, Р,
приготовленной из воды дистиллированной Р, и при¬
бавляют по каплям кислоту хлористоводородную раз¬
веденную Р до полного растворения. Доводят водой,
свободной от углерода диоксида, Р, приготовленной
из воды дистиллированной Р, до объема 40 мл.
рН (2.2.3). От 4,6 до 5,5.1,0 г испытуемого образца
растворяют в 9 мл воды, свободной от углерода ди¬
оксида, Р, допускается небольшая мутность раствора.
Оксихлориды. 10,0 г испытуемого образца раст¬
воряют в 10 мл воды, свободной от углерода диок¬
сида, Р. Полученный раствор по степени мутности
не должен превышать эталон II (2.2.1). К 1,5 мл полу¬
ченного раствора прибавляют 7,5 мл 96 % спирта Р.
Раствор может помутнеть в течение 10 мин. При при¬
бавлении 0,2 мл кислоты хлористоводородной раз¬
веденной Р мутность должна исчезать.
Сульфаты (2.4.13). Не более 0,0200 % (200 ррт).
5 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты. Эталон готовят с ис¬
пользованием 5 мл эталонного раствора сульфата
(10 ррт 80^ Р и 10 мл воды дистиллированной Р.
Алюминий, кальций, тяжелые металлы, же¬
лезо, магний. К 8 мл раствора 3 прибавляют 2 мл
раствора аммиака концентрированного Р и встряхи¬
вают. Полученный раствор должен быть прозрачным
(2.2.1) и бесцветным (2.2.2, метод II). Прибавляют
1 мл раствора динатрия гидрофосфата Р. Раствор
должен оставаться прозрачным в течение 5 минут.
Прибавляют 0,2 мл раствора натрия сульфида Р.
Образуется белый осадок, надосадочная жидкость
остается бесцветной.
Аммония соли (2.4.1). Не более 0,0400%
(400 ррт). 0,5 мл раствора 3 доводят водой Р до объ¬
ема 15 мл. Полученный раствор должен выдерживать
испытание на соли аммония.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в 5 мл
кислоты уксусной разведенной Р и проводят ком¬
плексометрическое определение цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответ¬
ствует 13,63 мг2пС12.
ХРАНЕНИЕ
В неметаллическом контейнере.
07/2016:0816
ЦИННАРИЗИН
Стпап21пит
СМЫАШШЕ
ги
кк
С2вН28К12 М.м. 368,5
[298-57-7]
ОПРЕДЕЛЕНИЕ
(Е)-1-(Дифенилметил)-4-(3-фенилпроп-2-енил)
пиперазин.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко раство¬
рим в метиленхлориде, растворим в ацетоне, мало
растворим в 96 % спирте и в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О
A. Температура плавления (2.2.14): от 118 °С до
122 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
1131
ч
Циннаризин
Сравнение: ФСО циннаризина #или спектр, пред¬
ставленный на рисунке #0816.-1#.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
20 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО циннаризина
растворяют в метаноле Р и доводят до объема 20 мл
этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО циннаризина
и 10 мг ФСО фпунариэшв дигидрохлорида раство¬
ряют в метаноле Р и доводят до объема 20 мл этим
же растворителем.
Плаотма: ТСХ пластинка со споем силикаге¬
ля октадеципсилильного Р.
Подвижная фаза: раствор 58,4 г/л натрия хлори¬
да Р — метанол Р — ацетон Р (20:30:50, об/об/об)
(камеру не насыщают парами подвижной фазы).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два четко
разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
О. 0,2 г кислоты лимонной безводной Р раство¬
ряют в 10 мл уксусного ангидрида Р при нагревании
в водяной бане при температуре 80 °С, выдерживают
при этой температуре в течение 10 мин и прибавляют
около 20 мг испытуемого образца. Появляется крас¬
новато-фиолетовое окрашивание.
ИСПЫТАНИЯ
Раствор 5. 0,5 г испытуемого образца раство¬
ряют в метиленхлориде Р и доводят до объема 20 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)Г
Кислотность или щелочность. 0,5 г испытуемо¬
го образца суспендируют в 15 мл воды Р и кипятят в
течение 2 мин. Охлахщают и фильтруют. Фильтрат до¬
водят водой, свободной от углерода диоксида, Р до
объема 20 мл. К 10 мл полученного раствора прибав¬
ляют 0,1 мл раствора фенолфталеина Р. При при¬
бавлении не более 0,25 мл 0,01 М раствора натрия
гидроксида должно появиться розовое окрашивание. К
оставшимся 10 мл раствора прибавляют 0,1 мл раст¬
вора метилового красного Р. При прибавлении не бо¬
лее 0,25 мл 0,01 М раствора кислоты хлористоводо¬
родной должно появиться красное окрашивание.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 12,5 мг ФСО циннаризи¬
на и 15,0 мг ФСО флунаризина дигидрохлорида раст¬
воряют в метаноле Р и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят метанолом Р до объема 20,0 мл.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят метанолом Р до объема 100,0 мл.
5.0 мл полученного раствора доводят метанолом Р
до объема 20,0 мл.
Условия хроматографирования:
- колонка длиной 0,1 м и внутренним диаметром
4.0 мм, заполненная силикагелем октадецилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 3 мкм;
Рисунок #0816.-1. Инфракрасный спектр ФСО циннаризина
143*. Зак. 1060.
1132
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза:
-подвижная фаза А: раствор 10 г/л аммония
ацетата Р;
- подвижная фаза В: 0,2 % (об/об) раствор кис¬
лоты уксусной ледяной Р в ацетонитриле Р1.
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—20
75 —> 10
о
О)
т
ю
см
20—25
10
90
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл.
Относительное удерживание (по отношению к
цийнаризину, время удерживания — около 11 мин):
примесь А — около 0,4; флунаризин — около 1,05;
примесь В — около 1,1; примесь С — около 1,2; при¬
месь О — около 1,6; примесь Е — около 1,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 5,0 между пиками цин-
наризина и флунаризина.
Предельное содержание примесей:
- примеси А, В, С, О, Е (не более 0,25 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям А, В, С, О и Е, не должна
превышать площадь основного пика на хроматограм¬
ме раствора сравнения (Ь);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
А, В, С, Э и Е, не должна превышать 0,4 площади ос¬
новного пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 0,5 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат-
ную площадь основного пика на хроматограмме раст¬
вора сравнения (Ь);
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод В). Не более
0,0020 % (20 ррт). 1,0 г испытуемого образца раство¬
ряют в смеси вода Р — ацетон Р (15:85, об/об), при¬
бавляя кислоту хлористоводородную разведенную Р
до полного растворения, и доводят смесью вода Р —
ацетон Р (15:85, об/об) до объема 20 мл. 12 мл полу¬
ченного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
10 мл эталонного раствора свинца (1 ррт РЬ), полу¬
ченного при разведении эталонного раствора свинца
(100 ррт РЬ) Р смесью вода Р—ацетон Р (15:85, об/об).
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 60 °С в течение 4 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
смеси кислота уксусная безводная Р — метилэ-
тилкетон Р (1:7, об/об) и титруют 0,1 М раствором
кислоты хлорной, используя в качестве индикатора
0,2 мл раствора нафтолбензеина Р.
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 18,43 мг С26Н28М2.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
с6н5
А. 1-(Дифенилметил)пиперазин.
С6Н5
В. (2)-1-(Дифенилметил)-4-(3-фенилпроп-2-енил)-
пиперазин.
С6Н5
С. (4-(Дифенилметил)-1,1-бис[(Е)-3-фенилпроп-2-
енил]пиперазиния хлорид.
с6н5
О. 1-(Дифенилметил)-4-[(1/?5,ЗЕ)-4-фенил-1-[(Е)-
2фенилэтил]бут-3-енил]пиперазин.
С6Н5
Е. 1,4-Бис(дифенилметил)пиперазин.
07/2016:1089
ЦИПРОФЛОКСАЦИН
аргоЛохастит
С1РКОР1- ОХА С1Ы
С17Н13РМ303 М.м. 331,4
[85721-33-1]
Ципрофлоксацин
1133
ОПРЕДЕЛЕНИЕ
1 -Циклопропил-6-фтор-4-оксо-7-(пиперазин-1 -
ил)-1,4-дигидрохинолин-З-карбоновая кислота.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Почти белый или бледно-желтый кристалличе¬
ский порошок. Слегка гигроскопичен.
Практически нерастворим в воде, очень мало
растворим в этаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ципрофлоксацин а.
ИСПЫТАНИЯ
Раствор 5. 0,25 г испытуемого образца раство¬
ряют в 0,1 М растворе кислоты хлористоводородной
и доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 3
должна быть не интенсивнее эталона ОУ(ЗЖ)5.
Примесь А. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 50 мг испытуемого образ¬
ца растворяют в растворе аммиака разведенном Р1
и доводят до объема 5 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО ципрофлоксаци-
на примеси А растворяют в смеси из 0,1 мл раствора
аммиака разведенного Р1 и 90 мл воды Р и доводят
водой Р до объема 100 мл. 2 мл полученного раст¬
вора доводят водой Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ля р214 Р.
Наносимый объем пробы: 5 мкл.
Пластинку помещают в хроматографическую ка¬
меру, на дне которой находится выпарительная чашка
с 50 мл раствора аммиака концентрированного Р,
камеру закрывают крышкой и выдерживают пластинку
в парах аммиака в течение 15 мин. Пластинку поме¬
щают во вторую хроматографическую камеру с под¬
вижной фазой и проводят хроматографирование.
Подвижная фаза: ацетонитрил Р — раствор
аммиака концентрированный Р — метанол Р — ме-
тиленхлорид Р (10:20:40:40, об/об/об/об).
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора пятно, соответствующее при¬
меси А, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. К 25,0 мг испытуемо¬
го образца прибавляют 0,2 мл кислоты фосфорной
разведенной Р, доводят подвижной фазой до объема
50,0 мл и обрабатывают ультразвуком до получения
прозрачного раствора.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 5,0 мл.
Раствор сравнения (Ь). 2,5 мг ФСО ципрофлок-
сацина гидрохлорида для идентификации пиков (со¬
держит примеси В, С, О и Е) растворяют в подвижной
фазе и доводят до объема 5,0 мл этим же раствори¬
телем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем эндкепиро-
ванным октадецилсилильным, деактивированным
по отношению к основаниям, для хроматографии Р
с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: ацетонитрил Р — раствор
2,45 г/л кислоты фосфорной Р, доведенный до рН
3.0 триэтиламином Р, (13:87, об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 278 нм;
- объем вводимой пробы: 50 мкл;
- время хроматографирования: 2-кратное вре¬
мя удерживания ципрофлоксацина.
Идентификация примесей: идентифицируют
пики примесей В, С, Р и Е, используя хроматограмму
раствора сравнения (Ь) и хроматограмму, прилага¬
емую к ФСО ципрофлоксацина для идентификации
пиков.
Относительное удерживание (по отношению
к ципрофлоксацину, время удерживания — около
9 мин): примесь Е — около 0,4; примесь В — около
0,6; примесь С — около 0,7; примесь О — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-разрешение: не менее 1,3 между пиками при¬
меси В и примеси С.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,7; для примеси С — 0,6; для примеси
О — 1,4; для примеси Е — 6,7):
- примеси В, С, О, Е (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям В, С, й и Е, не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей В, С, й и Е, не должна превышать 0,5 площади
основного пика на хроматограмме раствора сравне¬
ния (а);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Тяжелые металлы (2.4.8, метод Е). Не более
0,0020 % (20 ррт). 0,5 г испытуемого образца раст¬
воряют в кислоте уксусной разведенной Р и доводят
до объема 30 мл этим же растворителем. Прибавля¬
ют 2 мл воды Р вместо 2 мл буферного раствора рН
3,5 Р. Полученный фильтрат должен выдерживать ис-
144. Зак. 1060.
1134
Гэсударственная фармакопея Республики Беларусь
пытание на тяжелые металлы. Эталон готовят с ис¬
пользованием 10 мл эталонного раствора свинца
(1 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат в ва¬
кууме при температуре 120 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Используют платиновый тигель.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
О. 7-Хлор-1 -циклопропил-4-оксо-6-(пиперазин-1 -
ил)-1,4-дигидрохинолин-З-карбоновая кислота.
07/2016:0888
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в 80 мл
уксусной кислоты ледяной Р и титруют 0,1 М раство¬
ром кислоты хлорной потенциометрически (2.2.20).
1 мп 0,1 М раствора кислоты хлорной соответ¬
ствует 33,14 мг С17Н18РМ303.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Р.
А. Р = С1: 7-Хлор-1-циклопропил-6-фтор-4-оксо-
1,4-дигидрохинолин-З-карбоновая кислота (фторхино-
лоновая кислота).
С. Р = МН-[СН2]2-ЫН2: 7-[(2-Аминоэтил)амино]-1-
циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-З-
карбоновая кислота (соединение этилендиамина).
В. Р = С02Н, Р' = Н: 1-Циклопропил-4-оксо-7-
(пиперазин-1 -ил)-1,4-дигидрохинолин-З-карбоновая
кислота (соединение, не содержащее фтор).
Е. Р = Н, Р' = Р: 1-Циклопропил-6-фтор-7-
(пиперазин-1 -ил)хинолин-4(1 И)-он (декарбоксилиро-
ванное соединение).
Р. Р = С02Н, Р' = ОН: 1-Циклопропил-6-гидрокси-
4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-
карбоновая кислота.
ЦИПРОФЛОКСАЦИНА
ГИДРОХЛОРИД
аргоПохаат Ну4госЫопс1ит
С1РКОР1. ОХА С1И НУОРОСНШНЮЕ
С17Н„РН,0, ■ НС1 • хН20 М.м. 367,8
(безводное вещество)
ОПРЕДЕЛЕНИЕ
1 -Циклопропил-6-фтор-4-оксо-7-(пиперазин-1 -ил)-
1,4-дигидрохинолин-З-карбоновой кислоты гидрохло¬
рид. Содержит переменное количество воды.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Бледно-желтый кристаллический порошок. Слег¬
ка гигроскопичен.
Растворим в воде, мало растворим в метаноле,
очень мало растворим в этаноле безводном, практи¬
чески нерастворим в ацетоне, этилацетате и метилен-
хлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО ципрофлоксацина гидрохлори¬
да #или спектр, представленный на рисунке #0888.-1#.
B. 0,1 г испытуемого образца дает реакцию (Ь) на
хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 8. 0,5 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). 10 мл раствора 8 доводят
водой, свободной от углерода диоксида, Р до объема
20 мл. Полученный раствор должен быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора,
приготовленного для испытания «Прозрачность»,
должна быть не интенсивнее эталона <ЗУ(ЗЖ)5.
рН (2.2.3). От 3,5 до 4,5. Измеряют рН раствора 8.
Примесь А. Не более 0,2 %. Тонкослойная хро¬
матография (2.2.27).
Ципрофлоксацина гидрохлорид
1135
Рисунок #0888.-1. Инфракрасный спектр ФСО ципрофлоксацина гидрохлорида
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Раствор сравнения. 10 мг ФСО ципрофлоксаци¬
на примеси А растворяют в смеси из 0,1 мл раствора
аммиака разведенного Р1 и 90 мл воды Р и доводят
водой Р до объема 100 мл. 2 мл полученного раст¬
вора доводят водой Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР^Р.
Пластинку помещают в хроматографическую ка¬
меру, на дне которой находится выпарительная чашка
с 50 мл раствора аммиака концентрированного Р,
камеру закрывают крышкой и выдерживают пластинку
в парах аммиака в течение 15 мин. Пластинку поме¬
щают во вторую хроматографическую камеру с под¬
вижной фазой и проводят хроматографирование.
Подвижная фаза: ацетонитрил Р — раствор
аммиака концентрированный Р — метанол Р — ме-
тиленхлорид Р (10:20:40:40, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 3/4 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Предельное содержание примесей:
-примесь А: на хроматограмме испытуемого
раствора пятно, соответствующее примеси А, должно
быть не интенсивнее основного пятна на хромато¬
грамме раствора сравнения.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 25,0 мг ФСО ципрофлок¬
сацина гидрохлорида растворяют в подвижной фазе
и доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 2,5 мг ФСО ципрофлок¬
сацина гидрохлорида для идентификации пиков (со¬
держит примеси В, С, Р и Е) растворяют в подвижной
фазе и доводят до объема 5,0 мл этим же раствори¬
телем.
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 50,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,25м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем эндкепиро-
ванным октадецилсилильным, деактивированным
по отношению к основаниям, для хроматографии Р
с размером частиц 5 мкм;
- температура: 40 °С;
- подвижная фаза: ацетонитрил Р — раствор
2,45 г/л кислоты фосфорной Р, доведенный три-
этиламином Р до рН 3,0, (13:87, об/об);
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 278 нм;
- объем вводимой пробы: по 50 мкл испытуемого
раствора и растворов сравнения (Ь) и (с);
- время хроматографирования: 2,3-кратное вре¬
мя удерживания ципрофлоксацина.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, Р и Е, используя хромато¬
грамму раствора сравнения (Ь) и хроматограмму, при¬
лагаемую к ФСО ципрофлоксацина гидрохлорида для
идентификации пиков.
Относительное удерживание (по отношению
к ципрофлоксацину, время удерживания — около
9 мин): примесь Е — около 0,4; примесь В — около
0,6; примесь С — около 0,7; примесь Р — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (Ь):
144*. Зак. 1060.
1136
Гэсударственная фармакопея Республики Беларусь
-разрешение: не менее 1,3 между пиками при¬
месей В и С.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси В — 0,7; для примеси С — 0,6; для примеси
О — 1,4; для примеси Е — 6,7):
- примесь Е (не более 0,3 %): на хроматограмме
испытуемого раствора площадь пика, соответствую¬
щего примеси Е, не должна превышать 1,5-кратную
площадь основного пика на хроматограмме раствора
сравнения (с);
- примеси В, С, О (не более 0,2 %): на хрома¬
тограмме испытуемого раствора площади пиков, со¬
ответствующих примесям В, С и О , не должны пре¬
вышать площадь основного пика на хроматограмме
раствора сравнения (с);
- неспецифицированные примеси (не более
0,10 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пиков примесей
В, С, Р и Е, не должна превышать 0,5 площади основ¬
ного пика на хроматограмме раствора сравнения (с);
- сумма примесей (не более 0,5 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
2,5-кратную площадь основного пика на хроматограм¬
ме раствора сравнения (с);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (с) (0,05%).
Тяжелые металлы (2.4.8, метод Е). Не более
0,0020 % (20 ррт). 0,25 г испытуемого образца раст¬
воряют в воде Р, доводят до объема 30 мл этим же
растворителем и проводят предфильтрацию. Полу¬
ченный фильтрат должен выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
5 мл эталонного раствора свинца (1 ррт РЬ) Р.
Вода (2.5.12). Не более 6,7 %. Определение про¬
водят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
Используют платиновый тигель.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Рассчитывают процентное содержание
С17Н18НЧ303 НС1, принимая во внимание содержание
ципрофлоксацина гидрохлорида в ФСО ципрофлок-
сацина гидрохлорида.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей и/или об¬
щей статьей Субстанции для фармацевтического
использования (2034). Вследствие этого нет необ¬
ходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): Р.
А. 7-Хлор-1-циклопропил-6-фтор-4-оксо-1,4-
дигидрохинолин-3-карбоновая кислота (фторхиноло-
новая кислота).
В. 1-Циклопропил-4-оксо-7-(пиперазин-1-ил)-1,4-
дигидрохинолин-3-карбоновая кислота (соединение,
не содержащее фтор).
С. 7-[(2-Аминоэтил)амино]-1 -циклопропил-6-фтор-
4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (со¬
единение этилендиамина).
Р. 7-Хлор-1-циклопропил-4-оксо-6-(пиперазин-1-
ил)-1,4-дигидрохинолин-З-карбоновая кислота.
Е. 1 -Циклопропил-6-фтор-7-(пиперазин-1 -ил )хи-
нолин-4(1Н)-он (декарбоксилированное соединение).
Р. 1 -Циклопропил-6-гидрокси-4-оксо-7-(пиперазин-
1-ил)-1,4-дигидрохинолин-З-карбоновая кислота.
Цисплатин
1137
07/2016:0599
ЦИСПЛАТИН
азр1аИпит
С13Р1ЛТ1Ы
С1\ ЖЫН3
СГ ЫН3
РЮ12(ЫН3)2 М.м. 300,0
[15663-27-1]
ОПРЕДЕЛЕНИЕ
цис-Диаминдихлорплатина (II).
Содержание: не менее 97,0 % и не более 102,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Желтый порошок или желтые или оранжево-жел¬
тые кристаллы.
Мало растворим в воде, умеренно растворим
в диметилформамиде, практически нерастворим в
96 % спирте.
Идентификацию В и испытания (кроме испыта¬
ния «Серебро») проводят с защитой от света.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: В, С.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО цисплатина #или спектр, пред¬
ставленный на рисунке #0599.-1#.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 мл раствора 32, полу¬
ченного как указано в разделе «Испытания», доводят
диметилформамидом Р до объема 10 мл.
Раствор сравнения. 10 мг ФСО цисплатина
растворяют в 5 мл диметилформамида Р.
Пластинка: ТСХ пластинка со слоем целлюлозы
для хроматографии Р1.
Активирование пластинки: пластинку нагрева¬
ют при температуре 150 °С в течение 1 ч.
Подвижная фаза: ацетон Р — диметилформа-
мид Р (10:90, об/об).
Наносимый объем пробы: 2 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
50 г/л олова (II) хлорида Р в смеси из равных объе¬
мов кислоты хлористоводородной разведенной Р и
воды Р и выдерживают в течение 1 ч.
Результат: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
С. 2 мл раствора натрия гидроксида разведен¬
ного Р помещают в стеклянную выпарительную чашку,
прибавляют 50 мг испытуемого образца и выпаривают
досуха. Полученный остаток растворяют в смеси из
0,5 мл кислоты азотной Р и 1,5 мл кислоты хлори¬
стоводородной Р и выпаривают досуха. Полученный
остаток оранжевого цвета. Остаток растворяют в 0,5 мл
воды Р и прибавляют 0,5 мл раствора аммония хлори¬
да Р. Образуется желтый кристаллический осадок.
ИСПЫТАНИЯ
Раствор 31. 25 мг испытуемого образца раство¬
ряют в растворе 9 г/л натрия хлорида Р в воде, сво¬
бодной от углерода диоксида, Р и доводят до объема
25 мл этим же растворителем.
Раствор 52. 0,20 г испытуемого образца раст¬
воряют в диметилфомамиде Р и доводят до объема
10 мл этим же растворителем.
Рисунок #0599.-1. Инфракрасный спектр ФСО цисплатина
145. Зак. 1060.
1138
Гэсударственная фармакопея Республики Беларусь
Прозрачность (2.2.1). Растворы 31 и 32 должны
быть прозрачными.
Цветность (2.2.2, метод II). Окраска раствора
31 должна быть не интенсивнее эталона ОУ(ЗЖ)5.
рН (2.2.3). От 4,5 до 6,0. Измеряют рН раствора
31 немедленно после приготовления.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29). Испытания проводят с защитой
от света. Растворы, содержащие платину, не на¬
гревают и не подвергают воздействию ультразву¬
ка. Все растворы необходимо использовать в тече¬
ние 4 ч после приготовления.
Испытуемый раствор. 25,0 мг испытуемого об¬
разца растворяют в растворе 9,0 г/л натрия хлорида Р
и доводят до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 25,0 мл ФСО цисплати-
на растворяют в растворе 9,0 г/л натрия хлорида Р и
доводят до объема 25,0 мл этим же растворителем.
Раствор сравнения (Ь). 5,0 мг ФСО цисплатина
примеси А растворяют в растворе 9,0 г/л натрия хло¬
рида Р и доводят до объема 50,0 мл этим же раство¬
рителем.
Раствор сравнения (с). 5,6 мг ФСО цисплатина
примеси В растворяют в растворе 9,0 г/л натрия хло¬
рида Р и доводят до объема 100,0 мл этим же раст¬
ворителем.
Раствор сравнения (ф. К 0,05 мл испытуемого
раствора прибавляют 5,0 мл раствора сравнения (Ь),
5.0 мл раствора сравнения (с) и доводят раствором
9.0 г/л натрия хлорида Р до объема 25,0 мл.
Раствор сравнения (е). 5,0 мл раствора сравне¬
ния (с!) доводят раствором 9,0 г/л натрия хлорида Р
до объема 20,0 мл.
Холостой раствор. Раствор 9,0 г/л натрия хло¬
рида Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,0 мм, заполненная силикагелем октилсилиль-
ным, деактивированным по отношению к основани¬
ям, для хроматографии Р с размером частиц 4 мкм;
- температура: 30 °С;
- подвижная фаза: 1,08 г натрия октансульфо-
натаР, 1,70 г тетрабутиламмония гидросульфа¬
та Р и 2,72 г калия дигидрофосфата Р растворяют в
950 мл воде для хроматографии Р, доводят до рН 5,9
1 М раствором натрия гидроксида и разводят водой
для хроматографии Р до объема 1000 мл;
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 210 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора, растворов сравнения (с!) и (е) и холостого
раствора;
- время хроматографирования: 7-кратное вре¬
мя удерживания цисплатина.
Пик неудерживаемого компонента — последний
пик из группы пиков реакции на ввод пробы на хрома¬
тограмме холостого раствора.
Идентификация пика аквакомплекса цисплати-.
на: для идентификации пика аквакомплекса цисплати¬
на используют хроматограмму раствора сравнения (а)
и хроматограмму, прилагаемую к ФСО цисплатина.
Относительное удерживание (по отношению к
цисплатину, время удерживания — около 3,8 мин): пик
неудерживаемого компонента — около 0,5; примесь
А— около 0,6; примесь В — около 0,7; аквакомплекс
цисплатина — около 1,2.
Пригодность хроматографической системы:
раствор сравнения (с1):
- разрешение: не менее 2,5 между пиками при¬
меси А и примеси В; пики неудерживаемого компонен¬
та и примеси А должны разделяться.
Предельное содержание примесей:
- примесь А (не более 2,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси А, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (д);
- примесь В (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответству¬
ющего примеси В, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (б);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков приме¬
сей А и В, не должна превышать 0,5 площади пика ци¬
сплатина на хроматограмме раствора сравнения (с));
- сумма примесей, кроме примесей А и В( не бо¬
лее 0,5 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пиков
примесей А и В, не должна превышать 2,5-кратную
площадь пика цисплатина на хроматограмме раст¬
вора сравнения (б);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее площади пика цисплатина на хроматограмме
раствора сравнения (е) (0,05 %) и пик аквакомплекса
цисплатина.
Серебро. Не более 0,0250 % (250 ррт). Атомно¬
абсорбционная спектрометрия (2.2.23, метод I).
Испытуемый раствор. 0,100 г испытуемого об¬
разца растворяют в 15 мл кислоты азотной Р при на¬
гревании до температуры 80 °С, охлаждают и доводят
водой Р до объема 25,0 мл.
Раствор сравнения. К подходящему объему
(от 10 мл до 30 мл) эталонного раствора серебра
(5 ррт Ад) Р прибавляют 50 мл кислоты азотной Р и
доводят водой Р до объема 100,0 мл.
Источник излучения: лампа с полым катодом
для определения серебра, предпочтительнее исполь¬
зовать ширину щели 0,5 нм.
Длина волны: 328 нм.
Генератор атомного пара: воздушно-ацетиле¬
новое пламя.
Параллельно проводят контрольный опыт.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (а).
Содержание РЮ12(МН3)2 рассчитывают в процен¬
тах по сумме площадей пиков цисплатина и акваком¬
плекса цисплатина с учетом содержания цисплатина
в ФСО цисплатина.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
Цистеина гидрохлорид моногидрат
1139
ПРИМЕСИ
Специфицированные примеси: А, В.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): С.
Ск ЫИ3
р\
Нз^ ^С1
A. шранс-Диаминдихлорплатина (II) (трансплатин).
■ ск жмн3' ■
СГ С1
B. Аминтрихлорплатинат (-).
ск ^сМ2-
СГ С1
Сравнение: ФСО цистеина гидрохлорида моно¬
гидрата #или спектр, представленный на рисунке
#0895.-1#.
С. Просматривают хроматограммы, полученные
в испытании «Нингидрин-положительные вещества.
#Тонкослойная хроматография (2.2.27)».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
О. Около 5 мг испытуемого образца растворяют
в 1 мл раствора натрия гидроксида разведенного Р,
прибавляют 1 мл раствора 30 г/л натрия нитропрус-
сида Р. Появляется интенсивное фиолетовое окраши¬
вание, переходящее в коричневато-красное, а затем в
оранжевое. Прибавляют 1 мл кислоты хлористово¬
дородной Р. Появляется зеленое окрашивание.
Е. Около 50 мг испытуемого образца растворяют
в 5 мл воды Р, нагревают до температуры около 60 °С
на водяной бане и осторожно по каплям прибавляют
5 мл раствора водорода пероксида концентрирован¬
ного Р. Нагревают водяную баню до кипения и выдер¬
живают на водяной бане в течение 1 ч. Охлаждают до
комнатной температуры и доводят водой Р до объема
10 мл. 2 мл полученного раствора дает реакцию (а) на
хлориды (2.3.1).
С. Тетрахлорплатинат (2-).
07/2016:0895
ЦИСТЕИНА ГИДРОХЛОРИД
МОНОГИДРАТ
Суз/е/У?/ НудгосЫоп’с1ит топоЬудпсит
СУЗТЕ1ЫЕ НУОКОСШСЖЮЕ
МОЫОНУОПАТЕ
н мн2
НЗ. ЗС. * НС| * Н20
со2н
С3Н7Ы025 ■ НС1 ■ н20 М.м. 175,6
[7048-04-6]
ОПРЕДЕЛЕНИЕ
(2Р)-2-Амино-3-сульфанилпропановой кислоты
гидрохлорид моногидрат.
Продукт ферментации, экстракт или гидролизат
белка.
Содержание: не менее 98,5 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, мало растворим в 96 %
спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, Е.
Вторая идентификация: А, С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
ИСПЫТАНИЯ
Раствор 5. 2,5 испытуемого образца растворяют
в воде дистиллированной Р и доводят до объема
50 мл этим же растворителем.
Раствор 51. 10 мл раствора 5 доводят водой Р
до объема 20 мл.
Прозрачность (2.2.1). Раствор 31 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
31 должна быть не интенсивнее эталона ВУ(КЖ)6.
Удельное оптическое вращение (2.2.7). От +5,5
до +7,0 (в пересчете на сухое вещество). 2,00 г ис¬
пытуемого образца растворяют в кислоте хлористо¬
водородной Р1 и доводят до объема 25,0 мл этим же
растворителем.
Нингидрин-положительные вещества.
Испытание проводят одним из приведенных
ниже методов.#
Анализ аминокислот (2.2.56). Для анализа ис¬
пользуют Метод 1. Растворы готовят непосред¬
ственно перед применением.
Концентрации испытуемого раствора и раство¬
ров сравнения могут быть изменены в зависимости от
чувствительности используемого оборудования. Кон¬
центрации всех растворов могут быть изменены (при
сохранении предписанных соотношений их концен¬
траций) с условием выполнения требований к пригод¬
ности хроматографической системы, установленных
в общей статье 2.2.46.
Раствор А. Кислота хлористоводородная раз¬
веденная Р1 или буферный раствор для приготовле¬
ния образца, подходящий для применяемого прибора.
Испытуемый раствор. 30,0 мг испытуемого об¬
разца растворяют в растворе А и доводят до объема
50,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят раствором А до объема 100,0 мл. 2,0 мл
полученного раствора доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 30,0 мг /_-цистина Р (при¬
месь А) растворяют в растворе А и доводят до объема
145*. Зак. 1060.
1140
Гэсударственная фармакопея Республики Беларусь
Волновое число (см'1)
Рисунок #0895.-1. Инфракрасный спектр ФСО цистеина гидрохлорида моногидрата
100,0 мл этим же растворителем. 1,0 мл полученного
раствора доводят раствором А до объема 250,0 мл.
Раствор сравнения (с). 30,0 мг пролина Р раст¬
воряют в растворе А и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора
доводят раствором А до объема 250,0 мл.
Раствор сравнения (д). 6,0 мл эталонного раст¬
вора аммония (100 ррт Р доводят раствором А
до объема 50,0 мл. 1,0 мл полученного раствора до¬
водят раствором А до объема 100,0 мл.
Раствор сравнения (е). 30 мг изолейцина Р и 30 мг
лейцина Р растворяют в растворе А и доводят этим же
растворителем до объема 50,0 мл. 1,0 мл полученного
раствора доводят раствором А до объема 200,0 мл.
Холостой раствор. Раствор А.
Подходящие равные количества испытуемого
раствора, холостого раствора и растворов сравнения
вводят в аминокислотный анализатор. Запускают под¬
ходящую программу для определения физиологиче¬
ских аминокислот.
Пригодность хроматографической системы:
раствор сравнения (е):
- разрешение: не менее 1,5 между пиками изо¬
лейцина и лейцина.
Расчет процентных содержаний:
-для примеси А — используют концентрацию
примеси А в растворе сравнения (Ь);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 570 нм — ис¬
пользуют концентрацию цистеина в растворе сравне¬
ния (а);
-для любого нингидрин-положительного веще¬
ства, определяемого при длине волны 440 нм — ис¬
пользуют концентрацию пролина в растворе сравне¬
ния (с); если пик выше учитываемого предела при
обеих длинах волн, для расчетов используют резуль¬
тат, полученный при длине волны 570 нм.
Предельное содержание примесей:
- примесь А при длине волны 570 нм: не более
0,5 %;
- любое нингидрин-положительное вещество:
не более 0,2 % для каждой примеси;
- сумма примесей: не более 1,0 %;
- учитываемый предел: 0,05 %.
Пределы, указанные в разделе «Сопутствующие
примеси» (таблица 2034.-1) статьи Субстанции для фар¬
мацевтического использования (2034), не применяют.
#Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,20 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 10 мл
этим же растворителем. К 5 мл полученного раствора
прибавляют 5 мл раствора 40 г/л И-этилмалеимида в
96 % спирте Р и выдерживают в течение 5 мин.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 20 мг ФСО цистеина ги¬
дрохлорида моногидрата растворяют в воде Р и до¬
водят до объема 10 мл этим же растворителем. При¬
бавляют 10 мл раствора 40 г/л И-этилмалеимида в
96 % спирте Р и выдерживают в течение 5 мин.
Раствор сравнения (Ь). 2 мл раствора сравнения
(а) доводят водой Р до объема 10 мл.
Раствор сравнения (с). 5 мл испытуемого раст¬
вора (Ь) доводят водой Р до объема 20 мл.
Раствор сравнения (д). 10 мг ФСО тирозина
растворяют в 10 мл раствора сравнения (а) и доводят
водой Р до объема 25 мл.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р— бутанол Р (20:20:60, об/об/об).
Наносимый объем пробы: по 5 мкл испытуемых
растворов (а) и (Ь) и растворов сравнения (Ь), (с) и (б).
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Цитарабин
1141
Высушивание: при температуре 80 °С в течение
30 мин.
Проявление: пластинку опрыскивают раство¬
ром нингидрина Р и высушивают при температуре от
100 °С до 105 °С в течение 15 мин.
Пригодность хроматографической системы:
раствор сравнения (б):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (с).
Сульфаты (2.4.13). Не более 0,0300 % (300 ррт).
10 мл раствора 3 доводят водой дистиллированной Р
до объема 15 мл. Полученный раствор должен выдер¬
живать испытание на сульфаты.
Аммония соли. Не более 0,02 %.
Анализ аминокислот (2.2.56), как указано в ис¬
пытании «Нингидрин-положительные вещества», со
следующими изменениями.
Подходящие равные количества испытуемого
раствора, холостого раствора и раствора сравнения
(б) вводят в аминокислотный анализатор.
Предельное содержание примесей:
- аммоний при длине волны 570 нм: на хромато¬
грамме испытуемого раствора площадь пика, соответ¬
ствующего аммонию, не должна превышать площадь
соответствующего пика на хроматограмме раствора
сравнения (б), учитывая пик, соответствующий аммо¬
нию на хроматограмме холостого раствора.
#Если испытание «Нингидрин-положительные
вещества» проводят методом тонкослойной хрома¬
тографии, то испытание «Аммония соли» (2.4.1, ме¬
тод В) проводят следующим образом. 50 мг испытуе¬
мого образца должны выдерживать испытание на ам¬
мония соли. Эталон готовят с использованием 0,1 мл
эталонного раствора аммония (100 ррт N14) Р *.
Железо (2.4.9). Не более 0,0020 % (20 ррт).
0,50 г испытуемого образца помещают в делительную
воронку, растворяют в 10 мл кислоты хлористоводо¬
родной разведенной Р, встряхивают трижды, каждый
раз в течение 3 мин, с метилизобутилкетоном Р1
порциями по 10 мл. Объединенные органические слои
встряхивают с 10 мл воды Р в течение 3 мин. Водный
слой должен выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод 6). Не более
0,0010 % (10 ррт). 0,5 г испытуемого образца долж¬
ны выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 0,5 мл эталонного
раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 8,0% и не более 12,0%. 1,000 г испытуемо¬
го образца сушат при давлении, не превышающем
0,7 кПа, в течение 24 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца и 4 г калия йодида Р
помещают в колбу со шлифом, растворяют в 20 мл
воды Р, охлаждают в ледяной бане, прибавляют 3 мл
кислоты хлористоводородной Р1 и 25,0 мл 0,05 М
раствора йода. Колбу закрывают пробкой и выдержи¬
вают в защищенном от света месте в течение 20 мин.
Титруют 0,1 М раствором тиосульфата натрия, ис¬
пользуя в качестве индикатора 3 мл раствора крах¬
мала Р, который прибавляют в конце титрования.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
15,76 мг С3Н71Ч023-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных ко¬
личествах, следует определять тем или иным испы¬
танием, описанным в частной статье. Их содержание
лимитируется общим критерием приемлемости для
других/неспецифицированных примесей. Вследствие
этого нет необходимости идентифицировать эти при¬
меси для доказательства соответствия требованиям.
См. также статью 5.10. Контроль примесей в субстан¬
циях для фармацевтического использования): В.
Н ИНг
НО;
Н" ЫН2
со2н
А. (2Я,2'/?)-3,3'-дисульфандиилбис(2-аминопро-
пановая кислота) (цистин).
В. (25)-2-амино-3-гидроксипропановая кислота (се¬
рин).
07/2016:0760
ЦИТАРАБИН
Су1агаЫпит
СУТАКАВШЕ
С9Н13Ы,05 М.М. 243.2
[147-94-4]
ОПРЕДЕЛЕНИЕ
4-Амино-1-р-0-арабинофуранозилпиримидин-
2(1Н)-он.
Содержание: не менее 99,0 % и не более 100,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
1142
Государственная фармакопея Республики Беларусь
Легко растворим в воде, очень мало растворим в
96 % спирте и в метиленхлориде.
Температура плавления: около 215 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 20,0 мг испытуемого об¬
разца растворяют в 0,1 М растворе кислоты хлори¬
стоводородной и доводят до объема 100,0 мл этим
же растворителем. 5,0 мл полученного раствора дово¬
дят 0,1 М раствором кислоты хлористоводородной
до объема 100,0 мл.
Диапазон длин волн: от 230 нм до 350 нм.
Максимум поглощения: при 281 нм.
Удельный показатель поглощения в максимуме:
от 540 до 570.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО цитарабина.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси», в ультрафи¬
олетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуемого
раствора (Ь) основное пятно соответствует по распо¬
ложению и размеру основному пятну на хроматограм¬
ме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 3. 1,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 10 мл этим же
растворителем.
Прозрачность (2.2.7). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод //). Окраска раствора 3
должна быть не интенсивнее эталона У(Ж)5.
Удельное оптическое вращение (2.2.7). От
+154 до +160 (в пересчете на сухое вещество). 0,250 г
испытуемого образца растворяют в воде Р и доводят
до объема 25,0 мл этим же растворителем.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,25 г испытуемого об¬
разца растворяют в воде Р и доводят до объема 5 мл
этим же растворителем.
Испытуемый раствор (Ь). 2 мл испытуемого
раствора (а) доводят водой Р до объема 50 мл.
Раствор сравнения (а). 10 мг ФСО цитарабина
растворяют в воде Р и доводят до объема 5 мл этим
же растворителем.
Раствор сравнения (Ь). 0,5 мл испытуемого раст¬
вора (а) доводят водой Р до объема 100 мл.
Раствор сравнения (с). 20 мг уридина Р и 20 мг
ФСО урациларабинозида растворяют в метаноле Р
и доводят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ вР254 Р•
Подвижная фаза: вода Р — ацетон Р — метил-
этилкетон Р (15:20:65, об/об/об).
Наносимый объем пробы: по 5 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Предельное содержание примесей (0,5 %): на
хроматограмме испытуемого раствора (а) любое пят¬
но, кроме основного, должно быть не интенсивнее
пятна на хроматограмме раствора сравнения (Ь).
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 0,250 г испытуемого образца сушат над
фосфора (V) оксидом Р при температуре 60 °С и дав¬
лении от 0,2 кПа до 0,7 кПа в течение 3 ч.
Сульфатная зола (2.4.74). Не более 0,5%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.7.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в 60 мл
кислоты уксусной безводной Р, нагревая при необхо¬
димости, и титруют 0,1 М раствором кислоты хлор¬
ной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ¬
ствует 24,32 мг С9Н13М305.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте.
ПРИМЕСИ
A. Р = ОН, Р' = Н: 1-р-О-Арабинофуранозилпири-
мидин-2,4(1 Н,ЗН)-дион (урациларабонозид).
B. Р = Н, Р' = ОН: 1-(ЗЧЭ-Рибофуранозилпиримидин-
2,4(1 Н,ЗН)-дион (уридин).
07/2016:1837
ЧАЙНОГО ДЕРЕВА МАСЛО
МеЫеисае аеМего1еит
ТЕА ТНЕЕ ОН
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное из листьев и верхних
побегов МеШеиса аНегпНоНа (Мак1еп апд Ве1сИ) СНее1,
М. НпапИоНа ЗтИИ, М. сИззШЛога Р. МиеПег и/или других
видов МеШеиса методом перегонки с водяным паром.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная, подвижная, бесцветная или бледно-
желтого цвета жидкость с характерным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,1 мл испытуемого об¬
разца растворяют в 5 мл гептана Р.
Шалфея мускатного масло
1143
Раствор сравнения. 30 мкл цинеола Я, 60 мкл
терпинен-4-ола Р и 10 мг а-терпинеола Р раство¬
ряют в 10 мл гептана Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — гептан Р
(20:80, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р, нагревают при темпе¬
ратуре от 100 °С до 105 °С в течение (5—10) мин
до появления пятен. Просматривают при дневном
свете.
Результаты: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения
и испытуемого раствора. На хроматограмме испы¬
туемого раствора могут обнаруживаться и другие
зоны.
Верх хроматографической пластинки
Цинеол: зона фиолетово-ко¬
ричневого цвета
Терпинен-4-ол: зона корич¬
невато-фиолетового цвета
а-Терпинеол: зона фиолето¬
вого или коричневато-фио¬
летового цвета
Зона фиолетово-коричнево¬
го цвета, менее интенсивная
(цинеол)
Зона коричневато-фиолето¬
вого цвета (терпинен-4-ол)
Зона фиолетового или ко¬
ричневато-фиолетового
цвета (а-терпинеол)
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
как указано в испытании «Хроматографический про¬
филь».
Результаты: на хроматограмме испытуемого
раствора обнаруживаются характерные пики, соот¬
ветствующие по времени удерживания характерным
пикам на хроматограмме раствора сравнения,
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,885до 0,906.
Показатель преломления (2.2.6). От 1,475 до 1,482.
Угол оптического вращения (2.2.7). От +5° до +15°.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
нормализации.
Испытуемый раствор. 0,15 мл испытуемого об¬
разца растворяют в 10 мл гексана Р.
Раствор сравнения. 5 мкл а-пинена Р, 5 мкл
сабинена Я, 15 мкл а-терпинена Р, 5 мкл лимо¬
нена Р, 5 мкл цинеола Р, 30 мкл у-терпинена Я,
5 мкл п-цимена Я, 5 мкл терпинолена Я, 60 мкл
терпинен-4-ола Я, 5 мкл аромадендрена Я и 5 мкл
а-терпинеола Р растворяют в 10 мл гексана Р.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной от 30 м
(толщина слоя может быть 1 мкм) до 60 м (толщина
слоя может быть 0,2 мкм) и внутренним диаметром
от 0,25 мм до 0,53 мм, покрытая слоем макрогола
20 000 Р;
- газ-носитель: гелий для хроматографии Я;
- скорость газа-носителя: 1,3 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—1
50
1—37
50 -► 230
37—45
230
Блок ввода проб
240
Детектор
240
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения; отмечают времена
удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 2,7 между пиками
терпинен-4-ола и аромадендрена.
По временам удерживания компонентов раст¬
вора сравнения определяют состав испытуемого
раствора. Не учитывают пик гексана.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- а-пинен: от 1,0 % до 6,0 %;
- сабинен: не более 3,5 %;
- а-терпинен: от 5,0 % до 13,0 %;
- лимонен: от 0,5 % до 4,0 %;
- цинеол: не более 15,0 %;
- у-терпинен: от 10,0 % до 28,0 %;
- п-цимен: от 0,5 % до 12,0 %;
- терпинолен: от 1,5 % до 5,0 %;
- терпинен-4-ол: не менее 30,0 %;
- аромадендрен: не более 7,0 %;
- а-терпинеол: от 1,5 % до 8,0 %.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:1850
ШАЛФЕЯ МУСКАТНОГО МАСЛО
8аМае зс1агеае аеШего/еит
С/.АЯУ5ДСЕ О//.
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное из свежих или высу¬
шенных соцветий 8аМа зс1агеа 1_. методом перегонки
с водяным паром.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная или коричневато-желтая жидкость
(обычно светло-желтая) с характерным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 мл испытуемого об¬
разца растворяют в толуоле Я и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 60 мкл линалола Я, 200 мкл
линалилацетата Я и 60 мкл а-терпинеола Я раство¬
ряют в толуоле Я и доводят до объема 10 мл этим же
растворителем.
1144
Гэсударственная фармакопея Республики Беларусь
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 5 мкл испытуемого об¬
разца и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы, не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С
до 105 °С в течение (5—10) мин; просматривают при
дневном свете в течение 5 мин.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора могут обнаруживаться и другие слабые
зоны.
Верх хроматографической пластинки
а-Терпинеол: зона темно-
фиолетового цвета
Зона темно-фиолетового цвета
Линалилацетат: зона тем¬
но-фиолетового цвета
Зона темно-фиолетового цвета
Линалол: зона темно-фи¬
олетового цвета
Зона темно-фиолетового цвета
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
как указано в испытании «Хроматографический про¬
филь».
Результаты: на хроматограмме испытуемого
раствора пять пиков по положению соответствуют
пяти пикам на хроматограмме раствора сравнения.
Два пика, соответствующие а- и (3-туйону, на хрома¬
тограмме испытуемого раствора могут отсутствовать.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). От 0,890 до 0,908.
Показатель преломления (2.2.6). От 1,456 до 1,466.
Угол оптического вращения (2.2.7). От -26° до -10°.
Кислотное число (2.5.1). Не более 1,0.
Хроматографический профиль. Газовая хро¬
матография (2.2.28): определение проводят методом
нормализации.
Испытуемый раствор. Испытуемый образец.
Раствор сравнения. К 1 г гексана Р прибавляют
5 мкл туйона Р, 5 мкл линалола Р, 100 мкл линали-
лацетата Р, 10 мкл а-терпинеола Р и 25 мг (±20 %)
склареола Р и тщательно перемешивают путем встря¬
хивания.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной от 30 м
(толщина слоя может быть 1 мкм) до 60 м (толщина
слоя может быть 0,2 мкм) и внутренним диаметром
от 0,25 мм до 0,53 мм, покрытая слоем макрогола
20 000 Р;
- газ-носитель: гелий для хроматографии Р;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—10
60
10—75
60 —>190
75—120
190
Блок ввода проб
220
Детектор
240
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,2 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения; отмечают времена
удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,5 между пиками лина¬
лола и линалилацетата.
По временам удерживания компонентов раст¬
вора сравнения определяют состав испытуемого
раствора (не учитывают пик гексана). Туйон Р пред¬
ставляет собой смесь а- и р-туйона. а-Туйон в описан¬
ных выше условиях элюируется раньше (3-туйона.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Также определяют содержание гермакрена-й в
процентах. Пик гермакрена-0 на хроматограмме ис¬
пытуемого раствора идентифицируют по относитель¬
ному удерживанию, которое равно 1,23 относительно
времени удерживания пика линалола.
Содержание:
-а-и р-туйон: не более 0,2 %;
- линалол: от 6,5 % до 24 %;
- линалилацетат: от 56 % до 78 %;
- а-терпинеол: не более 5,0 %;
- гермакрен-О: от 1,0 % до 12 %;
- склареол: от 0,4 % до 2,6 %.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:1149
ШЕЛЛАК
1-асса
ЗНЕЦ.АС
ОПРЕДЕЛЕНИЕ
Очищенный продукт, полученный из смолистой
секреции женской особи насекомого Кета 1асса (Кегг)
ЫпсПпдег (/.асс//ег /асса Кегг). В зависимости от об¬
работки необработанного сырья (сырцовый шеллак)
различают четыре вида шеллака: восксодержащий,
отбеленный, депарафинизированный и отбеленный
депарафинизированный.
Восксодержащий шеллак получают из сырцового
шеллака; очищают фильтрацией расплавленного сы¬
рья и/или горячей экстракцией с использованием под¬
ходящего растворителя.
Отбеленный шеллак получают из сырцового шел¬
лака обработкой натрия гипохлоритом после раство¬
рения в подходящем растворе щелочи, осаждением
разведенной кислотой и высушиванием.
Депарафинизированный шеллак получают из
восксодержащего или сырцового шеллака обработ¬
кой подходящим растворителем и удалением нерас-
творенного воска фильтрацией.
Отбеленный депарафинизированный шеллак по¬
лучают из восксодержащего или сырцового шеллака
обработкой натрия гипохлоритом после растворения
в подходящем растворе щелочи; нерастворенный
воск удаляют фильтрацией. Осаждают разведенной
кислотой и высушивают.
Шеллак
1145
ОПИСАНИЕ (СВОЙСТВА)
Коричневато-оранжевые или желтые блестящие
полупрозрачные твердые или хрупкие относительно
тонкие хлопья (восксодержащий и депарафинизиро¬
ванный шеллак) или кремово-белый или коричневато-
желтый порошок (отбеленный и отбеленный депара¬
финизированный шеллак).
Практически нерастворим в воде, с этанолом
безводным образует более или менее опалесцирую¬
щий раствор (восксодержащий и отбеленный шеллак)
или прозрачный раствор (депарафинизированный и
отбеленный депарафинизированный шеллак). При
нагревании умеренно растворим или растворим в ще¬
лочных растворах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,25 г порошкообразного
испытуемого образца (500) (2.9.12) нагревают на во¬
дяной бане с 2 мл раствора натрия гидроксида раз¬
веденного Р в течение 5 мин. Охлаждают, прибавляют
5 мл этилацетата Р и медленно, при перемешива¬
нии, 2 мл кислоты уксусной разведенной Р. Встряхи¬
вают и фильтруют верхний слой через натрия суль¬
фат безводный Р.
Раствор сравнения. 6,0 мг кислоты алеурино-
вой Р растворяют в 1,0 мл метанола Р, при необхо¬
димости слегка подогревая.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯР214Р.
Подвижная фаза: кислота уксусная Р — ме¬
танол Р — метиленхлорид Р — этилацетатР
(1:8:32:60, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде
полос.
Фронт подвижной фазы: дважды не менее 15 см
от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р, нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин и просма¬
тривают при дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются несколько окрашенных
зон, одна из которых по расположению и цвету соот¬
ветствует зоне на хроматограмме раствора сравне¬
ния. Выше этой зоны на хроматограмме испытуемо¬
го раствора обнаруживается зона розового цвета, а
ниже — несколько зон фиолетового цвета. Ниже зоны,
соответствующей алеуриновой кислоте, обнаружива¬
ется зона светло-синего цвета (шеллоевая кислота)
в сопровождении зон такого же цвета, но меньшей
интенсивности. Могут обнаруживаться и другие блед¬
ные серые и фиолетовые зоны.
B. Просматривают хроматограмму, полученную
как указано в испытании «Канифоль».
Результаты: на хроматограмме испытуемого
раствора восксодержащего шеллака обнаруживается
более или менее интенсивная зона голубовато-серого
цвета, находящаяся непосредственно над зоной ти-
молфталеина на хроматограмме раствора сравнения.
На хроматограмме испытуемого раствора депарафи¬
низированного шеллака эти зоны, расположенные не¬
посредственно над зоной тимолфталеина на хромато¬
грамме раствора сравнения, не обнаруживаются.
ИСПЫТАНИЯ
Кислотное число (2.5.1). От 65 до 95 (в пере¬
счете на сухое вещество). Определение проводят из
1.00 г грубоизмельченного испытуемого образца. Ко¬
нечную точку титрования определяют потенциометри-
чески (2.2.20).
Канифоль. Тонкослойная хроматография
(2.2.27), как указано в разделе «Подлинность» для ис¬
пытания А, со следующими изменениями.
Испытуемый раствор. 50 мг порошкообразного
испытуемого образца (500) (2.9.12) растворяют при
нагревании в смеси из 0,5 мл метиленхлорида Р и
0,5 мл метанола Р.
Раствор сравнения. 2,0 мг тимолфталеина Р
растворяют в 1,0 мл метанола Р.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм и отмеча¬
ют зоны поглощения на хроматограмме испытуемого
раствора, значения РР которых близки со значением
Рр зоны поглощения тимолфталеина на хроматограм¬
ме раствора сравнения; пластинку опрыскивают рас¬
твором анисового альдегида Р, нагревают при тем¬
пературе от 100 °С до 105 °С в течение (5—10) мин и
просматривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается основная зона красновато-фи¬
олетового цвета (тимолфталеин). На хроматограмме
испытуемого раствора ни одна из зон поглощения,
значения РР которых близки со значением РР зоны
поглощения тимолфталеина на хроматограмме раст¬
вора сравнения, не должны иметь более или менее
интенсивный фиолетовый или коричневатый цвет
(канифоль). Не учитывают слабые зоны фиолетово¬
го цвета, обнаруживаемые на указанном уровне, ко¬
торые не обнаруживались как зоны поглощения при
просматривании в ультрафиолетовом свете до опры¬
скивания и нагревания.
Мышьяк (2.4.2, метод А). Не более 0,0003 %
(3 ррт). 0,33 г испытуемого образца и 5 мл кислоты
серной Р помещают в колбу для сжигания. Осторожно
прибавляют несколько миллилитров раствора водо¬
рода пероксида концентрированного Р, нагревают
до кипения и кипятят до получения прозрачного бесц¬
ветного раствора. Продолжают нагревание для удале¬
ния воды и, насколько это возможно, серной кисло¬
ты и доводят водой Р до объема 25 мл. Полученный
раствор должен выдерживать испытание на мышьяк.
Тяжелые металлы (2.4.8, метод О). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 2,0 % для неотбеленного шеллака и не более
6.0 % для отбеленного шеллака. 1,000 г порошкоо¬
бразного испытуемого образца (500) (2.9.12) сушат
при температуре от 40 °С до 45 °С в течение 24 ч.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте. Отбеленный шел¬
лак и отбеленный депарафинизированный шеллак
хранят при температуре не выше 15 °С.
МАРКИРОВКА
Указывают тип шеллака.
1146
Гэсударственная фармакопея Республики Беларусь
07/2016:0390
ЭВКАЛИПТОВОЕ МАСЛО
Еиса1урИ ае0юго1еит
Е1/СА/.УР7Ч/5 0/1.
ОПРЕДЕЛЕНИЕ
Эфирное масло, полученное методом перегонки
с водяным паром с последующей ректификацией из
свежих листьев или свежих концевых веточек разных
видов Еиса1ур1из, содержащих 1,8-цинеол. Использу¬
ют преимущественно виды Еиса1ур(из д!оЬи!из 1_аЬШ.,
Еиса1ур(и8 ро1уЬгас1еа Р.Т. Вакег или Еиса1ур1из зтПНН
П.Т. Вакег.
ОПИСАНИЕ(СВОЙСТВА)
Бесцветная или светло-желтая жидкость с запа¬
хом, напоминающим запах 1,8-цинеола.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В.
Вторая идентификация: А.
А. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,1 г испытуемого об¬
разца растворяют в толуоле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 20 мкл а-терпинеола Р и
50 мкл цинеола Р растворяют в толуоле Р и доводят
до объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: этилацетат Р — толуол Р
(10:90, об/об).
Наносимый объем пробы: по 10 мкл [или по
2 мкл] в виде полос длиной 10 мм [или 6 мм].
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин и просма¬
тривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора могут обнаруживаться и другие слабые
зоны около фронта подвижной фазы и на уровне
а-терпинеола.
Верх хроматографической пластинки
1,8-Цинеол: фиолетово-ко¬
ричневая зона
Интенсивная фиолетово-ко¬
ричневая зона (1,8-цинеол)
а-Терпинеол: фиолетово-ко¬
ричневая зона
Раствор сравнения
Испытуемый раствор
В. Просматривают хроматограммы, полученные
как указано в испытании «Хроматографический про¬
филь».
Результаты: характерные пики а-пинена,
Р-пинена, а-фелландрена, лимонена и 1,8-цинеола
на хроматограмме испытуемого раствора соответ¬
ствуют по времени удерживания пикам на хромато¬
грамме раствора сравнения (а). На хроматограмме
испытуемого раствора могут обнаруживаться саби-
нен и камфора.
ИСПЫТАНИЯ
Относительная платность (2.2.5). От 0,906 до 0,927.
Показатель преломления (2.2.6). От 1,458 до 1,470.
Угол оптического вращения (2.2.7). От 0° до +10°.
Растворимость в спирте (2.8.10). Испытуемый
образец растворим в 5 объемах спирта (70 %, об/об) Р.
Альдегиды. 10 мл испытуемого образца поме¬
щают в пробирку длиной 150 мм, диаметром 25 мм,
с притертой стеклянной пробкой и прибавляют 5 мл
толуола Р и 4 мл раствора гидроксиламина спир¬
тового Р. Интенсивно встряхивают и немедленно ти¬
труют 0,5 М раствором калия гидроксида в спирте
(60 %, об/об) до перехода окраски от красной к жел¬
той. Продолжают титрование при постоянном встря¬
хивании до тех пор, пока чисто-желтое окрашивание
нижнего слоя не будет устойчивым в течение 2 мин
после встряхивания и разделения слоев. Реакция
завершается приблизительно через 15 мин. Прово¬
дят титрование новой порции 10 мл испытуемого об¬
разца. Для определения точки конца титрования ис¬
пользуют цвет оттитрованной жидкости предыдуще¬
го определения, к которой добавлено дополнительно
0,5 мл 0,5 М раствора калия гидроксида в спирте
(60 %, об/об). На второе титрование должно пойти
не более 2,0 мл 0,5 М раствора гидроксида калия в
спирте (60 %, об/об).
Хроматографический профиль. Газовая хро¬
матография (2.2.28): испытание проводят методом
нормализации.
Испытуемый раствор. 200 мкл испытуемого об¬
разца растворяют в гептане Р и доводят до объема
10,0 мл этим же растворителем.
Раствор сравнения (а). 10 мкл а-пинена Р, 5 мкл
Р-пинена Р, 5 мкл сабинена Р, 5 мкл а-фелландрена Р,
10 мкл лимонена Р, 50 мкл цинеола Р и 5 мг камфо¬
ры Р растворяют в гептане Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 5 мкл лимонена Р раст¬
воряют в гептане Р и доводят до объема 50,0 мл
этим же растворителем. 0,5 мл полученного раствора
доводят гептаном Р до объема 5,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м и
внутренним диаметром около 0,25 мм, покрытая сло¬
ем макрогола 20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:50;
- температура:
Время (мин)
Температура (°С)
Колонка
0—5
60
5—33
60 — 200
33—38
200
Блок ввода проб
220
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения (а); отмечают време¬
на удерживания всех веществ.
Эметина гидрохлорид пентагидрат
1147
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками лимо¬
нена и цинеола.
Идентификация компонентов: по временам
удерживания компонентов раствора сравнения (а)
определяют состав испытуемого раствора.
Рассчитывают содержание каждого из компонен¬
тов в процентах.
Содержание:
- а-пинен: от 0,05 % до 10,0 %;
- Р-пинен: от 0,05 % до 1,5 %;
- сабинен: не более 0,3 %;
- а-фелландрен: от 0,05 % до 1,5 %;
- лимонен: от 0,05 % до 15,0 %;
- 1,8-цинеол: не менее 70,0 %;
- камфора: не более 0,1 %;
- неучитываемый предел (0,05 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (Ь).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
При температуре не выше 25 °С.
07/2016:0081
ЭМЕТИНА ГИДРОХЛОРИД
ПЕНТАГИДРАТ
ЕтеИп/ Ьус/госЫгогМит реМаЬуйпсит
ЕМЕТШЕ НУОКОСНШКЮЕ
РЕЫТАНУОЕАТЕ
ОСН3
ОСН3
2НС1 5Н20
Н
С^Н^О, ■ 2НС1 ■ 5Н20 М.м. 644
ОПРЕДЕЛЕНИЕ
(25,ЗК, 11 Ь5)-2-[[(1 Я)-6,7-Диметокси-1,2,3,4-тетра-
гидроизохинолин-1 -ил]метил]-3-этил-9,10-диметокси-
1,3,4,6,7,11Ь-гексагидро-2Н-бензо[а]хинолизина диги¬
дрохлорид.
Содержание: не менее 98,0 % и не более 102,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый кристаллический
порошок.
Легко растворим в воде и в спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, Е.
Вторая идентификация: В, С, О, Е.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО эметина гидрохлорида.
B. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси», в ультрафи¬
олетовом свете при длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, флуоресценции и разме¬
ру пятну на хроматограмме раствора сравнения (а).
C. Около 10 мг испытуемого образца растворяют
в 2 мл раствора водорода пероксида разведенного Р,
прибавляют 1 мл кислоты хлористоводородной Р и
нагревают. Появляется оранжевое окрашивание.
Э. Около 5 мг испытуемого образца распределя¬
ют на поверхности 1 мл сульфомолибденового реак¬
тива Р2. Появляется ярко-зеленое окрашивание.
Е. Испытуемый образец дает реакцию (а) на хло¬
риды (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 1,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и до¬
водят до объема 25 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора
3 должна быть не интенсивнее эталона У(Ж)5 или
ВУ(КЖ)5.
рН (2.2.3). От 4,0 до 6,0.4 мл раствора 3 доводят
водой, свободной от углерода диоксида, Р до объ¬
ема 10 мл.
Удельное оптическое вращение (2.2.7). От +16
до +19 (в пересчете на сухое вещество). Навеску ис¬
пытуемого образца, соответствующую 1,250 г сухого
вещества, растворяют в воде Р и доводят до объема
25,0 мл этим же растворителем.
Сопутствующие примеси. Тонкослойная хро¬
матография (2.2.27). Растворы готовят непосред¬
ственно перед использованием.
Испытуемый раствор. 50 мг испытуемого об¬
разца растворяют в метаноле Р, содержащем 1 %
(об/об) раствора аммиака разведенного Р2, и дово¬
дят до объема 100 мл этим же растворителем.
Раствор сравнения (а). 50 мг ФСО эметина ги¬
дрохлорида растворяют в метаноле Р, содержащем
1 % (об/об) раствора аммиака разведенного Р2, и до¬
водят до объема 100 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО изоэметина
гидробромида растворяют в метаноле Р, содержа¬
щем 1 % (об/об) раствора аммиака разведенного Р2,
и доводят до объема 100 мл этим же растворителем.
5 мл полученного раствора доводят до объема 50 мл
метанолом Р, содержащем 1 % (об/об) раствора ам¬
миака разведенного Р2.
Раствор сравнения (с). 10 мг ФСО цефаэлина
гидрохлорида растворяют в метаноле Р, содержа¬
щем 1 % (об/об) раствора аммиака разведенного Р2,
и доводят до объема 100 мл этим же растворителем.
5 мл полученного раствора доводят до объема 50 мл
метанолом Р, содержащем 1 % (об/об) раствора ам¬
миака разведенного Р2.
Раствор сравнения (д). 1 мл раствора сравнения
(а) доводят до объема 100 мл метанолом Р, содержа¬
щем 1 % (об/об) раствора аммиака разведенного Р2.
Раствор сравнения (е). К 1 мл раствора срав¬
нения (а) прибавляют 1 мл раствора сравнения (Ь) и
1 мл раствора сравнения (с).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
1148
Гэсударственная фармакопея Республики Беларусь
Подвижная фаза: диэтиламин Р — вода Р — ме¬
танол Р—этиленгликоля монометиловый эфир Р —
хлороформ Р (0,5:2:5:20:100, об/об/об/об/об).
Наносимый объем пробы: по 10 мкл испытуемо¬
го раствора и растворов сравнения (а), (Ь), (с), (с!) и
30 мкл раствора сравнения (е).
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе до полного испарения
подвижной фазы.
Проявление: пластинку опрыскивают раствором
йода в хлороформе Р в хорошо вентилируемом вы¬
тяжном шкафу и нагревают при температуре 60 °С в
течение 15 мин. Просматривают в ультрафиолетовом
свете при длине волны 365 нм.
Пригодность хроматографической системы:
раствор сравнения (е):
- на хроматограмме должны обнаруживаться три
полностью разделенных пятна.
Предельное содержание примесей:
- изоэметин (не более 2,0 %): на хроматограмме
испытуемого раствора пятно, соответствующее изоэ¬
метину, должно быть не интенсивнее пятна на хрома¬
тограмме раствора сравнения (Ь);
- цефаэлин (не более 2,0 %): на хроматограмме
испытуемого раствора пятно, соответствующее цефа-
элину, должно быть не интенсивнее пятна на хромато¬
грамме раствора сравнения (с);
- любая другая примесь (не более 1,0 %): на
хроматограмме испытуемого раствора любое пятно,
кроме основного и пятен изоэметина и цефаэлина,
должно быть не интенсивнее пятна на хроматограмме
раствора сравнения (б).
Потеря в массе при высушивании (2.2.32). Не
менее 11,0 % и не более 15,0 %. 1,00 г испытуемого
образца сушат при температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют в сме¬
си, состоящей из 5,0 мл 0,01 М раствора кислоты
хлористоводородной и 50 мл 96 % спирта Р, и ти¬
труют 0,1 М раствором натрия гидроксида потенцио-
метрически (2.2.20). Отмечают объем титранта между
двумя точками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 27,68 мг С29Н40М2О4-2НС1.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:РБ0030
#ЭМУЛЬГАТОР № 1
Ети1депз № 1
ЕМШ-31РУ1ЫС АвЕЫТ № 1
ОПРЕДЕЛЕНИЕ
Смесь спиртов синтетических первичных жир¬
ных фракций С16—С20 или С1б—С18 с натриевой солью
сульфоэфиров таких же спиртов.
Содержание: не менее 92,0 % суммы цетилового
спирта (С^Н^О) и стеаринового спирта (С18Н380).
ОПИСАНИЕ (СВОЙСТВА)
Твердая масса в виде плиток или чешуек, жирная
на ощупь, от белого до белого с желтоватым оттенком
цвета, со специфическим запахом.
Умеренно растворим в 96 % спирте, мало раство¬
рим в ацетоне и в хлороформе, очень мало раство¬
рим в гексане, практически нерастворим в воде. До¬
пускается опалесценция полученных растворов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Просматривают хроматограммы, полученные
как указано в разделе «Количественное определение».
Результаты: пики, соответствующие спиртам С16
(цетиловому) и С18 (стеариловому) на хроматограмме
испытуемого раствора по времени удерживания соот¬
ветствуют пикам спиртов С16 (цетиловому) и С18 (сте¬
ариловому) на хроматограмме раствора сравнения.
B. 5 г испытуемого образца помещают в кониче¬
скую колбу вместимостью 250 мл и прибавляют 60 мл
кислоты хлористоводородной разбавленной Р. Кол¬
бу закрывают воронкой и кипятят до получения про¬
зрачного верхнего слоя (около 1 ч), после чего охлаж¬
дают, ставя на лед. На поверхности жидкости образу¬
ется твердая масса со специфическим запахом.
C. 5 мл жидкости, полученной в испытании В,
дают реакцию (а) на сульфаты (2.3.1).
ИСПЫТАНИЯ
Температура каплепадения (2.2.17). От 50 °С
до 62 °С.
Эмульгирующая способность. 3,50 г испытуе¬
мого образца, предварительно растертого в фарфо¬
ровой ступке, помещают в стеклянный стакан вмести¬
мостью 100 мл и прибавляют 46,5 мл воды, свободной
от углерода диоксида, Р. Стакан нагревают на водя¬
ной бане до температуры от 75 °С до 80 °С, тщатель¬
но перемешивая смесь стеклянной палочкой до обра¬
зования однородной массы.
Содержимое стакана охлаждают до комнатной
температуры при постоянном перемешивании. Долж¬
на образоваться белая, однородная эмульсия (пробу
оставляют для проведения испытаний «рН» и «Термо¬
стабильность»).
рН (2.2.3). От 7,5 до 8,5. Измеряют рН эмульсии,
полученной в испытании «Эмульгирующая способ¬
ность».
Термостабильность. 30 г эмульсии, полученной
в испытании «Эмульгирующая способность», нагре¬
вают в закрытом бюксе в термостате при температуре
45 °С в течение 6 ч. Эмульсия не должна расслаи¬
ваться.
Гидроксильное число (2.5.3, метод А). От 200
до 220.
Йодное число (2.5.4). Не более 1,0. Для раст¬
ворения навески испытуемого образца используют
10 мл хлороформа Р.
Эфирное число. От 4 до 10. 5,00 г испытуемого
образца, предварительно растертого в фарфоровой
ступке, помещают в коническую колбу вместимостью
250 мл, прибавляют 25 мл воды Р, нагревают на водя¬
ной бане до расплавления, прибавляют 0,25 мл раст¬
вора фенолфталеина Р1 и нейтрализуют 0,1 М рас¬
твором натрия гидроксида до слабо розовой окраски
или 0,1 М раствором кислоты хлористоводородной
до обесцвечивания пробы. Затем в колбу прибавляют
#Эмульгатор № 1
1149
25 мл 0,5 М раствора кислоты хлористоводородной
и осторожно кипятят в колбе с обратным холодильни¬
ком до получения прозрачного верхнего слоя (около
2 ч). Затем холодильник промывают 25 мл горячей
воды Р и содержимое колбы титруют 0,5 М раство¬
ром натрия гидроксида до розовой окраски, исполь¬
зуя 0,25 мл раствора фенолфталеина Р1 в качестве
индикатора.
Параллельно проводят контрольный опыт.
Эфирное число рассчитывают по формуле:
(У, -Уг)'28,05
т
где:
V, — объем 0,5 М раствора натрия гидроксида,
пошедший на титрование испытуемого раствора, мл;
\/2 — объем 0,5 М раствора натрия гидроксида,
пошедший на титрование в контрольном опыте, мл;
т — масса навески субстанции, г;
28,05 — количество калия гидроксида, соответ¬
ствующее 1 мл 0,5 М раствора натрия гидроксида, мг.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Газовая хроматография (2.2.28).
Раствор внутреннего стандарта. 1,5 г лаури-
лового спирта Р растворяют в 100 мл хлороформа Р,
доводят до объема 200,0 мл этим же растворителем
и перемешивают. Срок годности полученного раст¬
вора —10 сут.
Испытуемый раствор. 0,500 г испытуемого об¬
разца, предварительно растертого в фарфоровой
ступке, помещают в коническую колбу вместимо¬
стью 100 мл, прибавляют 20 мл 0,5 М раствора кис¬
лоты хлористоводородной и осторожно кипятят с
обратным холодильником до получения прозрачного
верхнего слоя (около 2 ч). Затем содержимое колбы
охлаждают до комнатной температуры, холодильник
промывают 15 мл теплого хлороформа Р, в колбу
прибавляют 10,0 мл раствора внутреннего стандар¬
та, переносят содержимое колбы в делительную во¬
ронку и встряхивают в течение 2 мин. После разде¬
ления слоев нижний хлороформный слой отделяют
и фильтруют в мерную колбу вместимостью 50 мл
через вату с 2 г натрия сульфата безводного Р,
предварительно смоченного хлороформом Р. Холо¬
дильник и коническую колбу промывают 10 мл те¬
плого хлороформа Р, помещают в ту же делитель¬
ную воронку и экстрагируют при встряхивании в те¬
чение 2 мин. Хлороформный слой отделяют и филь¬
труют в ту же мерную колбу. Процедуру промывания
холодильника и колбы и экстракцию повторяют еще
раз, используя 10 мл хлороформа Р. Затем филь¬
трат доводят хлороформом Р до объема 50,0 мл и
перемешивают. Используют свежеприготовленный
раствор.
Раствор сравнения. 0,180 г ФСО цетилового
спирта и 0,300 г ФСО стеаринового спирта раст¬
воряют в 20 мл хлороформа Р, прибавляют 10,0 мл
раствора внутреннего стандарта, доводят хлорофор¬
мом Р до объема 50,0 мл и перемешивают. Использу¬
ют свежеприготовленный раствор.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и диаметром 0,32 мм, покрытая слоем 5 % полиме-
тилфенилсилоксана Р (толщина слоя 0,25 мкм) или
аналогичная;
- газ-носитель: гелий для хроматографии Р\
- скорость газа-носителя: 8 мл/мин;
- деление потока: 1:50;
- температура:
Время
(мин)
Темпера-
тура («С)
Ско¬
рость
нагрева
(°С/мин)
Режим
Колонка
0—1,6
150—И 90
25
линейный гра¬
диент
1,6—3,6
190
—
изотермиче¬
ский
3,6—9,7
190 — 300
35
линейный гра¬
диент
9,7—10,7
300
—
изотермиче¬
ский
Блок
ввода
проб
270
Детектор
350
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: цетиловый спирт, стеари-
ловый спирт.
Пригодность хроматографической системы:
раствор сравнения:
- число теоретических тарелок: не менее
15 000, рассчитывают по пику цетилового спирта;
- разрешение: не менее 2,0 между пиками цети¬
лового спирта и стеарилового спирта;
- относительное стандартное отклонение: не
более 2,0 %, рассчитанные для отношений площадей
пиков цетилового спирта к площадям пиков внутрен¬
него стандарта и для отношений площадей пиков
стеарилового спирта к площадям пиков внутреннего
стандарта при 5 повторных вводах.
Содержание цетилового спирта и стеарилового
спирта в процентах расчитывают по формуле:
В, • тЫ ' Р,
Вош-т ’
где:
В} — средние значения отношений площадей пи¬
ков цетилового спирта или стеарилового спирта к пло¬
щадям пиков внутреннего стандарта, вычисленные из
хроматограмм испытуемого раствора;
Во.—средние значения отношений площадей пи¬
ков цетилового спирта или стеарилового спирта к пло¬
щадям пиков внутреннего стандарта, вычисленные из
хроматограмм раствора сравнения;
то1 — масса навесок цетилового спирта или стеа¬
рилового спирта, использованных для приготовления
раствора сравнения, г;
т — масса навески испытуемого образца, г;
Р. — содержание цетилового спирта или стеари¬
лового спирта в образце, взятом для приготовления
раствора сравнения, %.
Рассчитывают суммарное содержание С^Н^О
(цетилового спирта) и С^Н^О (стеарилового спирта)
в эмульгаторе № 1 в процентах.
ХРАНЕНИЕ
В защищенном от влаги и света месте при темпе¬
ратуре от 15 °С до 25 °С.
1150
Гэсударственная фармакопея Республики Беларусь
07/2016:1420 ИСПЫТАНИЯ
ЭНАЛАПРИЛА МАЛЕАТ
Епа1арпН та1еаз
ЕЛ/Д1АРЯ//. МА1.ЕАТЕ
со2н
со2н
С20Н28М2О5 ’ С4Н404 М.М. 492>5
[76095-16-4]
ОПРЕДЕЛЕНИЕ
(25)-1-[(25)-2[[(15)-1-(Этоксикарбонил )-3-фенил-
пропил]амино]пропаноил]пирролидин-2-карбоновой
кислоты (2)-бутендиоат.
Содержание: не менее 98,5 % и не более 101,5 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Умеренно растворим в воде, легко растворим в
метаноле, практически нерастворим в метиленхлори-
де. Растворяется в разведенных растворах гидрокси¬
дов щелочных металлов.
Температура плавления: около 144 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО эналаприла малеата #или
спектр, представленный на рисунке #1420.-1#.
Раствор 5. 0,25 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
рН (2.2.3). От 2,4 до 2,9. Измеряют рН раствора 3.
Удельное оптическое вращение (2.2.7). От -48
до -51 (в пересчете на сухое вещество). Определяют
удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Буферный раствор А. 2,8 г натрия дигидрофос¬
фата моногидрата Р растворяют в 950 мл воды Р,
доводят до рН 2,5 кислотой фосфорной Р и разводят
водой Р до объема 1000 мл.
Буферный раствор В. 2,8 г натрия дигидрофос¬
фата моногидрата Р растворяют в 950 мл воды Р, до¬
водят до рН 6,8 раствором натрия гидроксида концен¬
трированным Р и разводят водой Р до объема 1000 мл.
Смесь для растворения. Смешивают 50 мл аце¬
тонитрила Р1 и 950 мл буферного раствора А.
Испытуемый раствор. 30 мг испытуемого об¬
разца растворяют в смеси для растворения и доводят
до объема 100,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят смесью для растворения до объема
100,0 мл.
Раствор сравнения (Ь). 3,0 мг ФСО эналаприла
для проверки пригодности хроматографической
системы (содержит примесь А) растворяют в смеси
для растворения и доводят до объема 10,0 мл этой же
смесью для растворения.
Раствор сравнения (с). Содержимое контейнера
с ФСО эналаприла смеси примесей (примеси В, С, Э,
Е и Н) растворяют в 1,0 мл смеси для растворения.
Рисунок #1420.-1. Инфракрасный спектр ФСО эналаприла малеата
Эналаприла малеат
1151
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,1 мм, заполненная сополимером стирол-диви-
нилбензола Р с размером частиц 5 мкм;
- температура: 70 °С;
- подвижная фаза:
- подвижная фаза А: смешивают 50 мл ацето-
нитрила Р1 и 950 мл буферного раствора В;
- подвижная фаза В: смешивают 340 мл бу¬
ферного раствора В и 660 мл ацетонитрила Р1\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—20
95 —► 40
сл
1
О)
о
20—25
40
60
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: 50 мкл.
Идентификация пиков примесей: идентифици¬
руют пики примесей В, С, й, Е и Н, используя хрома¬
тограмму раствора сравнения (с) и хроматограмму,
прилагаемую к ФСО эналаприла смеси примесей;
идентифицируют пик примеси А, используя хромато¬
грамму раствора сравнения (Ь).
Относительное удерживание (по отношению
к эналаприлу, время удерживания — около 11 мин):
примесь С — около 0,2; примесь В — около 0,8; при¬
месь А— около 1,1; примесь Н — около 1,3; примесь
Е — около 1,5; примесь О — около 2,1.
Пригодность хроматографической системы:
раствор сравнения (Ь):
-коэффициент разделения пиков: не менее 10
(Нр — высота пика примеси А относительно базовой
ли-нии; Ну — расстояние между базовой линией и
нижней точкой кривой, разделяющей пики примеси А
и эналаприла).
Предельное содержание примесей:
- примесь А (не более 1,0 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- примеси В, С, О, Е, Н (не более 0,3 %): на хро¬
матограмме испытуемого раствора площади пиков,
соответствующих примесям В, С, О, Е, Н, не должны
превышать 0,3 площади основного пика на хромато¬
грамме раствора сравнения (а);
- неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раствора
площадь любого пика, кроме основного и пиков при¬
месей А, В, С, О, Е и Н, не должна превышать 0,1
площади основного пика на хроматограмме раствора
сравнения (а);
- сумма примесей кроме примеси А (не более
1,0 %): на хроматограмме испытуемого раствора
сумма площадей всех пиков, кроме основного и пика
примеси А, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (а);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора не учитывают пики с площадью
менее 0,05 площади основного пика на хроматограм¬
ме раствора сравнения (а) (0,05 %) и пик, соответству¬
ющий малеиновой кислоте.
Тяжелые металлы (2.4.8, метод С). Не более
0,0010 % (10 ррт). 2,0 г испытуемого образца должны
выдерживать испытание на тяжелые металлы. Эта¬
лон готовят с использованием 2 мл эталонного раст¬
вора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 г испытуемого образца сушат при
температуре 105 °С в течение 3 ч.
Сульфатная зола (2.4.74). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.72, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в воде,
свободной от углерода диоксида, Р, доводят до объ¬
ема 30 мл этим же растворителем и титруют 0,1 М
раствором натрия гидроксида потенциометрически
(2.2.20). Титруют до второй точки перегиба на кривой
титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 16,42 мг С20Н28Ы2О5 О4Н4О4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Н.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.70. Контроль примесей в субстанциях для
фармацевтического использования): Р, 6,1.
А. (25)-1 -[(25)-2-[[(1 Е)-1-(Этоксикарбонил)-3-фенил-
пропил]амино]пропаноил]пирролидин-2-карбоновая
кислота.
В. (25)-2-[[(15)-1-(Этоксикарбонил )-3-фенилпропил]-
амино]пропионовая кислота.
С. Р = Н: (25)-1 -[(25)-2-[[(15)-1 -Карбокси-З-фенил-
пропил]амино]пропаноил]пирролидин-2-карбоновая
кислота.
Е. Р = СН2-СН2-СбН5: (25)-1-[(25)-2-[[(15)-3-Фенил-
1- [(2-фенилэтокси)карбонил]пропил]амино]пропаноил]-
пирролидин-2-карбоновая кислота.
Р. Р = С4Н9: (25)-1-[(25)-2-[[(15)-1-(Бутоксикар-
бонил)-3-фенилпропил]амино]пропаноил]пирролидин-
2- карбоновая кислота.
1152
Гэсударственная фармакопея Республики Беларусь
Э. Этиловый эфир (2Э)-2-[(35,8а5)-3-метил-1,4-
диоксо-октагидропирроло[1,2-а]пиразин-2-ил]-4-фе-
нилбутановой кислоты.
(3. (25)-2-[[(15)-3-Циклогексил-1-(этоксикарбонил)-
пропил]амино]пропионовая кислота.
Н. (23)-1-[(25)-2-[[(1 $)-3-Циклогексил-1 -(этоксикар¬
бонил )пропил]амино]пропаноил]пирролидин-2-карбо-
новая кислота.
I. 1Н-Имидазол.
07/2016:0082
ЭРГОКАЛЬЦИФЕРОЛ
Егдоса1сЯего1ит
ЕП60СА1-С1РЕП01.
С^Н^О М.м. 396,7
[50-14-6]
ОПРЕДЕЛЕНИЕ
(52,7Е,22Е)-9> 1О-Секоэргоста-5,7,10(19),22-тетраен-
Зр-ол. #Витамин О*.
Содержание: не менее 97,0 % и не более 103,0 %.
1 мг эргокальциферола соответствует 40 000 МЕ
антирахитной активности (витамин Э) на крысах.
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтоватый кристаллический
порошок или белые или почти белые кристаллы.
Практически нерастворим в воде, легко раство¬
рим в 96 % спирте, растворим в жирных маслах.
Эргокальциферол чувствителен к воздуху, на¬
греванию и свету. Растворы в летучих растворителях
являются нестабильными и должны использоваться
немедленно.
В растворах в зависимости от температуры и
времени может происходить обратимая изомериза¬
ция в пре-эргокальциферол. Активность субстанции
обусловлена обоими компонентами.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО эргокальциферола #или спектр,
представленный на рисунке #0082.-1#.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От
+103 до +107. 0,200 г испытуемого образца быстро и
без нагревания растворяют в 96% спирте, свобод¬
ном от альдегидов, Р и доводят до объема 25,0 мл
этим же растворителем. Полученный раствор исполь¬
зуют в течение 30 мин после приготовления.
Восстанавливающие вещества. Не более
0,0020 % (20 ррт).
Испытуемый раствор. 0,1 г испытуемого образ¬
ца растворяют в 96% спирте, свободном от альде¬
гидов, Р и доводят до объема 10,0 мл этим же раство¬
рителем. Прибавляют 0,5 мл раствора 5 г/л тетразо-
лиевого синего Р в 96% спирте, свободном от аль¬
дегидов, Р, 0,5 мл раствора тетраметиламмония
гидроксида разведенного Р, выдерживают в течение
5 мин (точно) и прибавляют 1,0 мл кислоты уксусной
ледяной Р.
Раствор сравнения. К 10,0 мл раствора
0,2 мкг/мл гидрохинона Р в 96% спирте, свободном
от альдегидов, Р прибавляют 0,5 мл раствора 5 г/л
тетразолиевого синего Р в 96% спирте, свободном
от альдегидов, Р, 0,5 мл раствора тетраметилам¬
мония гидроксида разведенного Р, выдерживают в
течение 5 мин (точно) и прибавляют 1,0 мл кислоты
уксусной ледяной Р.
Компенсационный раствор. К 10,0 мл 96 % спир¬
та, свободного от альдегидов, Р прибавляют 0,5 мл
раствора 5 г/л тетразолиевого синего Рв96% спир¬
те, свободном от альдегидов, Р, 0,5 мл раствора
тетраметиламмония гидроксида разведенного Р,
выдерживают в течение 5 мин (точно) и прибавляют
1,0 мл кислоты уксусной ледяной Р.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора и раствора сравнения при длине вол¬
ны 525 нм. Оптическая плотность испытуемого раст¬
вора не должна превышать оптическую плотность
раствора сравнения.
Эргостерол. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. 0,25 г испытуемого об¬
разца растворяют в этиленхлориде Р, содержащем
10 г/л сквалана Р и 0,1 г/л бутилгидрокситолуола Р,
и доводят до объема 5 мл этим же растворителем.
Раствор готовят непосредственно перед использова¬
нием.
Эргокальциферол
1153
Рисунок #0082.-1. Инфракрасный спектр ФСО эргокальциферола
Раствор сравнения (а). 0,10 г ФСО эргокаль¬
циферола растворяют в этиленхлориде Р, содер¬
жащем 10 г/л сквалана Р и 0,1 г/л бутилгидрок-
ситолуола Р, и доводят до объема 2 мл этим же
растворителем. Раствор готовят непосредственно
перед использованием.
Раствор сравнения (Ь). 5 мг ФСО эргостеро-
ла растворяют в этиленхлориде Р, содержащем
10 г/л сквалана Р и 0,1 г/л бутилгидрокситолуо-
ла Р, и доводят до объема 50 мл этим же раство¬
рителем. Раствор готовят непосредственно перед
использованием.
Раствор сравнения (с). Смешивают равные
объемы раствора сравнения (а) и раствора срав¬
нения (Ь). Раствор готовят непосредственно перед
использованием.
Пластинка: ТСХ пластинка со слоем силика¬
геля О Р.
Подвижная фаза: смесь из равных объемов
циклогексана Р и эфира, свободного от перокси¬
дов, Р, содержащая 0,1 г/л бутилгидрокситолуо-
ла Р.
Наносимый объем пробы: по 10 мкл испыту¬
емого раствора и растворов сравнения (а) и (Ь) и
20 мкл раствора сравнения (с).
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта. Хроматографируют немедленно после
нанесения проб с защитой от света.
Высушивание: на воздухе.
Проявление: пластинку трижды опрыскивают
раствором сурьмы (III) хлорида Р1. Просматрива¬
ют при дневном свете через (3—4) мин после опры¬
скивания.
Пригодность хроматографической системы:
раствор сравнения (с):
- на хроматограмме обнаруживаются два пол¬
ностью разделенных пятна.
Предельное содержание примесей:
- эргостерол (не более 0,2 %): на хромато¬
грамме испытуемого раствора обнаруживается ос¬
новное пятно изначально оранжево-желтого цвета,
которое постепенно окрашивается в коричневый
цвет; на хроматограмме испытуемого раствора пят¬
но фиолетового цвета (эргостерол), которое мед¬
ленно проявляется непосредственно ниже основ¬
ного пятна, должно быть не интенсивнее пятна на
хроматограмме раствора сравнения (Ь); на хрома¬
тограмме испытуемого раствора не должны обна¬
руживаться пятна, не соответствующие пятнам на
хроматограммах растворов сравнения (а) и (Ь).
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требо¬
вания статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Ипытание
проводят как можно быстро с защитой от воздей¬
ствия актиничного света и воздуха.
Испытуемый раствор. 10,0 мг испытуемого об¬
разца растворяют без нагревания в 10,0 мл толуо¬
ла Р и доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (а). 10,0 мг ФСО эргокальци¬
ферола растворяют без нагревания в 10,0 мл толуо¬
ла Р и доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (Ь). 1,0 мл ФСО холекальци-
ферола для проверки пригодности хроматографи¬
ческой системы доводят подвижной фазой до объ¬
ема 5,0 мл, нагревают с обратным холодильником на
водяной бане при температуре 90 °С в течение 45 мин
и охлаждают.
Условия хроматографирования:
- колонка из нержавеющей стали длиной 0,25 м
и внутренним диаметром 4,6 мм, заполненная подхо¬
дящим силикагелем с размером частиц 5 мкм;
1154
Гэсударственная фармакопея Республики Беларусь
- подвижная фаза: пентанол Р — гексан Р
(3:997, об/об);
- скорость подвижной фазы: 2 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: подходящий и доста¬
точный объем.
Относительное удерживание (по отношению к
холекальциферолу): пре-холекалыдиферол — около
0,4; транс-холекальциферол — около 0,5.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- относительное стандартное отклонение: не
более 1,0 % для площадей пиков холекальциферола
при 6 повторных вводах пробы;
- разрешение: не менее 1,0 между пиками пре-
холекальциферола и транс-холекальциферола. При
необходимости изменяют соотношение компонентов
подвижной фазы или скорость подвижной фазы.
Содержание эргокальциферола в процентах рас¬
считывают по формуле:
где:
т — масса навески испытуемого образца в ис¬
пытуемом растворе, мг;
т' — масса навески ФСО эргокальциферола в
растворе сравнения (а), мг;
50 — площадь (высота) пика эргокальциферола
на хроматограмме испытуемого раствора;
— площадь (высота) пика эргокальциферола
на хроматограмме раствора сравнения (а).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в атмос¬
фере азота в защищенном от света месте при темпе¬
ратуре от 2 °С до 8 °С.
Содержимое контейнера используют немедлен¬
но после вскрытия.
ПРИМЕСИ
А. (5Е,7Е,22Е)-9,10-Секоэргоста-5,7,10(19),22-
тетраен-ЗР-ол (транс-витамин 02).
В. (22Е)-Эргоста-5,7,22-триен-Зр-ол (эргостерол).
С. (9р, 10а,22Е)-Эргоста-5,7,22-триен-Зр-ол (лю-
мистерол2).
О. (6Е,22Е)-9,10-Секоэргоста-5(10),6,8(14),22-
тетраен-Зр-ол (изо-тахистерол2).
Е. (6Е,22Е)-9,10-Секоэргоста-5(10),6,8,22-тетраен-
Зр-ол (тахистерол2).
07/2016:1803
ЭРИТРИТОЛ
Егу1М1о1ит
ЕКУТНК1Т01-
С4Н10О4 М.м. 122,1
[149-32-6]
ОПРЕДЕЛЕНИЕ
(2Е,35)-Бутан-1,2,3,4-тетрол (мезо-эритритол).
Содержание: не менее 96,0 % и не более 102,0 %
(в пересчете на безводное вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или сыпучие гранулы.
Легко растворим в воде, очень мало растворим
в 96 % спирте.
Эритритол
1155
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Температура плавления (2.2.14): от 119 °Сдо 122 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО эритритола.
ИСПЫТАНИЯ
Раствор 3. 5,0 г испытуемого образца раство¬
ряют в воде Р и доводят до объема 50 мл этим же
растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Электропроводность (2.2.38). Не более 20 мкСм-см*1.
20,0 г испытуемого образца растворяют в воде,
свободной от углерода диоксида, Р, приготовленной
из воды дистиллированной Р, и доводят до объема
100.0 мл этим же растворителем. Измеряют электро¬
проводность полученного раствора при осторожном
помешивании магнитной мешалкой.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 0,50 г испытуемого об¬
разца растворяют в воде Р и доводят до объема
10.0 мл этим же растворителем.
Раствор сравнения (а). 0,50 г ФСО эритрито¬
ла растворяют в воде Р и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (Ь). 2,0 мл испытуемого раст¬
вора доводят водой Р до объема 100,0 мл.
Раствор сравнения (с). 5,0 мл раствора сравне¬
ния (Ь) доводят водой Р до объема 100,0 мл.
Раствор сравнения (д). 1,0 г эритритола Р и
1.0 г глицерина Р растворяют в воде Р и доводят до
объема 20,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,3 м и внутренним диаметром
7,8 мм, заполненная катионообменной смолой Р с
размером частиц 9 мкм;
- температура: 70 °С;
- подвижная фаза: 0,01 % (об/об) раствор кисло¬
ты серной Р;
- скорость подвижной фазы: 0,8 мл/мин;
- детектор: рефрактометр при постоянной тем¬
пературе;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (Ь), (с) и (б);
- время хроматографирования: 3-кратное вре¬
мя удерживания эритритола.
Относительное удерживание (по отношению к
эритритолу, время удерживания — около 11 мин): при¬
месь А — около 0,77; примесь В — около 0,90; при¬
месь С — около 0,94; примесь й — около 1,10.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 2 между пиками эритри¬
тола и примеси О.
Предельное содержание примесей:
- любая примесь (не более 2,0 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (Ь);
- сумма примесей (не более 2,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора
сравнения (Ь);
- неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с.
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (с).
Свинец (2.4.10). Не более 0,00005 % (0,5 ррт).
Вода (2.5.12). Не более 0,5 %. Определение про¬
водят из 1,00 г испытуемого образца.
Микробиологическая чистота
' Если субстанция предназначена для производ¬
ства лекарственных средств для парентерального
применения:
- ОКА: критерий приемлемости 102 КОЕ/г (2.6.12).
Если субстанция не предназначена для произ¬
водства лекарственных средств для парентерального
применения:
- ОКА: критерий приемлемости 103 КОЕ/г (2.6.12);
- ОКГ: критерий приемлемости 102 КОЕ/г (2.6.12);
- ЕвсЬепсЫа соН: отсутствие (2.6.13);
- 8а1топе11а: отсутствие (2.6.13).
Бактериальные эндотоксины (2.6.14). Если
субстанция предназначена для производства лекар¬
ственных средств для парентерального применения
без дальнейшей подходящей процедуры удаления
бактериальных эндотоксинов:
- менее 4 МЕ/г для лекарственных средств для
парентерального применения с содержанием эритри¬
тола 100 г/л или менее;
- менее 2,5 МЕ/г для лекарственных средств для
парентерального применения с содержанием эритри¬
тола более 100 г/л.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 20 мкл испытуемого
раствора и раствора сравнения (а).
Содержание эритритола в процентах рассчи¬
тывают с использованием хроматограммы раствора
сравнения (а) и с учетом содержания эритритола в
ФСО эритритола.
МАРКИРОВКА
При необходимости указывают:
- субстанция пригодна для производства лекар¬
ственных средств для парентерального применения
ПРИМЕСИ
А. 4-О-а-О-Глюкопиранозил-О-глюцитол (О-мальти-
тол).
но н
В. О-Глюцитол (й-сорбитол).
1156
Гэсударственная фармакопея Республики Беларусь
С. (2/?,35,45)-Пентан-1,2,3,4,5-пентол (мезо-ри-
битол).
Э. Пропан-1,2,3-триол (глицерин).
07/2016:0179
ЭРИТРОМИЦИН
Егу1Ьготус'тит
ЕКУТНКОМУСШ
Эритро¬
мицин
Молекулярная
формула
М.м.
К1
К2
А
с37н67ко13
734
ОН
сн3
В
с37н65мо12
718
н
сн3
С
«УУЮ,,
720
он
н
ОПРЕДЕЛЕНИЕ
Смесь макролидных антибиотиков, продуцируе¬
мых штаммом 8&ер{отусез егу1Ьгеиз, основным ком¬
понентом которой является (3/?,45,55,6Я,7/?,9/?,11Я,-
12Я, 135,14/?)-4-[(2,6-дидезокси-3-С-метил-3-0-метил-
а-1_-рибо-гексопиранозил)окси]-14-этил-7,12,13-
тригидрокси-3,5,7,9,11,13-гексаметил-6-[(3,4,6-
тридезокси-З-диметиламино-Р-О-шуло-гексопиранозил)-
окси]оксациклотетрадекан-2,10-дион (эритромицин А).
Содержание:
- суммарное содержание эритромицина А, эри¬
тромицина В и эритромицина С: не менее 93,0 % и
не более 102,0% (в пересчете на безводное веще¬
ство);
- эритромицин 8: не более 5,0 %;
- эритромицин С: не более 5,0 %.
#1 мкг эритромицина А соответствует специфиче¬
ской активности 1 ЕД.#
ОПИСАНИЕ (СВОЙСТВА)
Белый или слегка желтый порошок или бесцвет¬
ные или слегка желтые кристаллы. Слегка гигроско¬
пичен.
Мало растворим в воде (растворимость умень¬
шается при увеличении температуры), легко раство¬
рим в 96 % спирте, растворим в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: Б, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО эритромицина А #или спектр,
представленный на рисунке #0179.-1#. Не учитывают
полосы в диапазоне от 1980 см1 до 2050 см-1.
Если полученные спектры отличаются, то по
50 мг испытуемого образца и ФСО эритромицина А
растворяют по отдельности в 1,0 метиленхлорида Р,
высушивают при температуре 60 °С и давлении, не
превышающем 670 Па, в течение 3 ч. Остатки исполь¬
зуют для получения новых спектров.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 10 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (а). 10 мг ФСО эритромици¬
на А растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения (Ь). 20 мг ФСО спирамицина
растворяют в метаноле Р и доводят до объема 10 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: смешивают 4 объема 2-пропа-
нола Р, 8 объемов раствора 150 г/л аммония ацета¬
та Р, доведенного до рН 9,6 раствором аммиака Р, и
9 объемов этилацетата Р. Смесь выдерживают до
разделения и используют верхний слой.
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р1 и нагревают при температу¬
ре 110 °С в течение 5 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения
(а); обнаруживающиеся пятна отличаются по распо¬
ложению и цвету от пятен на хроматограмме раствора
сравнения (Ь).
C. К 5 мг испытуемого образца прибавляют 5 мл
раствора 0,2 г/л ксантгидрола Р в смеси из 1 объема
кислоты хлористоводородной Р и 99 объемов кисло¬
ты уксусной Р и нагревают на водяной бане. Появля¬
ется красное окрашивание.
О. 10 мг испытуемого образца растворяют в
5 мл кислоты хлористоводородной Р1 и выдержи¬
вают в течение (10—20) мин. Появляется желтое
окрашивание.
ИСПЫТАНИЯ
Удельное оптическое вращение (2.2.7). От -71
до -78 (в пересчете на безводное вещество). 1,00 г
испытуемого образца растворяют в этаноле Р и дово¬
дят до объема 50,0 мл этим же растворителем. Опре¬
деление проводят не менее чем через 30 мин после
приготовления раствора.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 40,0 мг испытуемого об¬
разца растворяют в смеси из 1 объема метанола Р
и 3 объемов фосфатного буферного раствора рН
Эритромицин
1157
Рисунок #0179.-1. Инфракрасный спектр ФСО эритромицина А
7.0 Р1 и доводят до объема 10,0 мл этой же смесью
растворителей.
Раствор сравнения (а). 40,0 мг ФСО эритроми¬
цина А растворяют в смеси из 1 объема метанола Р
и 3 объемов фосфатного буферного раствора рН
7.0 Р1 и доводят до объема 10,0 мл этой же смесью
растворителей.
Раствор сравнения (Ь). 10,0 мг ФСО эритроми¬
цина В и 10,0 мг ФСО эритромицина С растворяют
в смеси из 1 объема метанола Р и 3 объемов фос¬
фатного буферного раствора рН 7,0 Р1 и доводят
до объема 50,0 мл этой же смесью растворителей.
Раствор сравнения (с). 5 мг ФСО И-деметил-
эритромицина А растворяют в растворе сравнения (Ь),
прибавляют 1,0 мл раствора сравнения (а) и доводят
раствором сравнения (Ь) до объема 25 мл.
Раствор сравнения (д). 3,0 мл раствора срав¬
нения (а) доводят смесью из 1 объема метанола Р
и 3 объемов фосфатного буферного раствора рН
7.0 Р1 до объема 100,0 мл.
Раствор сравнения (е). 40 мг ФСО эритроми¬
цина А помещают в стеклянную пробирку таким об¬
разом, чтобы образовался слой толщиной не более
1 мм. Нагревают при температуре 130 °С в течение
4 ч. Охлаждают и растворяют в смеси из 1 объема
метанола Р и 3 объемов фосфатного буферного
раствора рН 7,0 Р1 и доводят до объема 10 мл этой
же смесью растворителей.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная сополимером стирол-дивинилбен-
зола Р с размером частиц 8 мкм (размер пор 100 нм);
- температура: 70 °С (для колонки и не менее
1/3 части капилляра, идущего к колонке);
- подвижная фаза: к 50 мл раствора 35 г/л дика¬
лия гидрофосфата Р, доведенного до рН 9,0±0,05
кислотой фосфорной разведенной Р, прибавля¬
ют 400 мл воды Р, 165 мл 2-метил-2-пропанола Р,
30 мл ацетонитрила Р и доводят водой Р до объема
1000 мл;
- скорость подвижной фазы: 2,0 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: по 100 мкл испытуемо¬
го раствора и растворов сравнения (с), (б) и (е);
- время хроматографирования: 5-кратное вре¬
мя удерживания эритромицина А.
Относительное удерживание (по отношению к
эритромицину А, время удерживания — около 15 мин):
примесь А — около 0,3; примесь В — около 0,45; эри¬
тромицин С — около 0,5; примесь С — около 0,9; при¬
месь Э — около 1,4; примесь Р — около 1,5; эритроми¬
цин В — около 1,8; примесь Е — около 4,3.
Пригодность хроматографической системы:
раствор сравнения (с):
-разрешение: не менее 0,8 между пиками при¬
меси В и эритромицина С и не менее 5,5 между пика¬
ми примеси В и эритромицина А; при необходимости
увеличивают концентрацию 2-метил-2-пропанола в
подвижной фазе либо снижают скорость подвижной
фазы до 1,5 мл/мин или 1,0 мл/мин.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Е — 0,09; для примеси Р — 0,15):
- любая примесь (не более 3,0 %): на хроматограм¬
ме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать площадь основного
пика на хроматограмме раствора сравнения (с1);
- сумма примесей (не более 7,0 %): на хрома¬
тограмме испытуемого раствора сумма площадей
пиков, соответствующих всем примесям, не должна
превышать 2,3-кратную площадь основного пика на
хроматограмме раствора сравнения (б);
1158
Гэсударственная фармакопея Республики Беларусь
- неучитываемый предел: на хроматограмме
испытуемого раствора не учитывают пики с площа¬
дью менее 0,02 площади основного пика на хрома¬
тограмме раствора сравнения (с!) (0,06 %), а также
пики эритромицина В и эритромицина С.
Тиоцианат. Не более 0,3 %. Растворы гото¬
вят непосредственно перед использованием и за¬
щищают от воздействия актиничного света.
Компенсационный раствор. 1,0 мл раствора
90 г/л железа (III) хлорида Р доводят метанолом Р
до объема 50,0 мл.
Испытуемый раствор. 0,100 г (т, г) испыту¬
емого образца растворяют в 20 мл метанола Р,
прибавляют 1,0 мл раствора 90 г/л железа (III) хло¬
рида Р и доводят метанолом Р до объема 50,0 мл.
Гэтовят отдельно два раствора сравнения.
Раствор сравнения. 0,100 г калия тиоциана¬
та Р, предварительно высушенного при темпера¬
туре 105 °С в течение 1 ч, растворяют в метано¬
ле Р и доводят до объема 50,0 мл этим же раство¬
рителем. 5,0 мл полученного раствора доводят ме¬
танолом Р до объема 50,0 мл. К 5,0 мл получен¬
ного раствора прибавляют 1,0 мл раствора 90 г/л
железа (III) хлорида Р и доводят метанолом Р до
объема 50,0 мл.
Измеряют оптическую плотность (2.2.25) рас¬
творов сравнения (А,, А2) и испытуемого раствора
(А) в максимуме (около 492 нм).
Коэффициент пригодности (5) рассчитывают
по формуле:
С. /ту А
тЛА2'
где:
ш1> ^2 — массы навесок калия тиоцианата, ис¬
пользованных при приготовлении соответствующих
растворов сравнения.
Пригодность системы:
- коэффициент пригодности 5: не менее 0,985
и не более 1,015.
Содержание тиоцианата в процентах рассчиты¬
вают по формуле:
А • 58,08 - 0,5
т 97,18
гт1 + т1Л
А\ ^2 у
где:
58,08 — относительная молекулярная масса
тиоцианатной группы;
97,18 — относительная молекулярная масса
калия тиоцианата
Вода (2.5.12). Не более 6,5 %. Определение
проводят из 0,200 г испытуемого образца. В ка¬
честве растворителя используют раствор 100 г/л
имидазола Р в метаноле безводном Р.
Сульфатная зола (2.4.14). Не более 0,2%.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 100 мкл испытуемо¬
го раствора и растворов сравнения (а) и (Ь).
Пригодность хроматографической системы:
раствор сравнения (а):
- фактор асимметрии: не более 5;
- относительное стандартное отклонение:
не более 1,2 % для площадей пиков при 6 повтор¬
ных вводах пробы.
Содержание эритромицина А в процентах рас¬
считывают с использованием хроматограммы раст¬
вора сравнения (а). Содержание эритромицина В и
эритромицина С в процентах рассчитывают с исполь¬
зованием хроматограммы раствора сравнения (Ь).
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
А. (3/?,45,55,67?,7Я,97?, 11/?,127?,135,147?)-4-[(2,6-
Дидезокси-З-С-метил-З-О-метил-а-Ь-рибо-гексо-
пиранозил)окси]-14-этил-7,12,13-тригидрокси-3-
(гидрокси-метил)-5,7,9,11,13-пентаметил-6-[[3,4,6-
тридезокси-3-(диметиламино)-р-0-ксш70-гексопира-
нозил]окси]-оксациклотетрадекан-2,10-дион (эритро¬
мицин Р).
В. (37?,45,55,6/?,77?,97?, 11Я127?, 135,147?)-4-[(2,6-
Дидезокси-З-С-метил-З-О-метил-а-Ьрибо-гексопира-
нозил )окси]-14-этил-7,12,13-тригидрокси-3,5,7,9,11,13-
гексаметил-6-[[3,4,6-тридезокси-3-(метиламино)-р-
0-кс1шо-гексопиранозил]окси]оксациклотетрадекан-
2,10-дион (3"-/\/-деметилэритромицин А).
Эритромицина эстолат
1159
С. (гМаЯ^'Я.б'В.б'З^ Д,85,9Я, 10/?, 12Я, 1 4Я, 1
5Я, 1 63)-7-Этил-5',8,9, 14-тетрагидрокси-4'-метокси-
4',6',8,10,12,14,16-гептаметил-15-[[3,4,6-тридезокси-
3-(диметиламино)-р-0-ксш70-гексопиранозил]окси]-
гексадекагидроспиро[5Н,11Н-1,3-диоксино[5,4-с]-
оксацикло-тетрадецин-2,2'-пиран]-5,11 -дион (эри¬
тромицин Е).
О. (15,2Я,ЗЯ,45,5Я,8Я,95,105,11Я,12Я,14Я)-9-
[(2,6-Дидезокси-3-С-метил-3-0-метил-а-1_-р1/бо-гексо-
пиранозил )окси]-5-этил-3-гидрокси-2,4,8,10,12,14-
гексаметил-11-[[3,4,6-тридезокси-3-(диметиламино)-
Р-0-ксш70-гексопиранозил]окси]-6,15,16-триоксатри-
цикло[10.2.1.11>4]гексадекан-7-он (ангидроэритроми-
цин А).
СН3
Е. (27?,3/?,45,5Я?,8Я,95,105,11/?,12Я)-9-[(2,6-
Дидезокси-З-С-метил-З-О-метил-а-Ь-рыбо-гексо-
пиранозил)окси]-5-этил-3,4-дигидрокси-2,4,8,10,-
12,14-гексаметил-11 -[[3,4,6-тридезокси-3-(диме-
тиламино)-Р-0-/ссало-гексопиранозил]окси]-6,15-
диоксабицикло[10.2.1]пентадец-1(14)-ен-7-он (эри¬
тромицина А еноловый эфир).
Р. (2/?,ЗЯ,6Я,75,85,9Я,10Я)-7-[(2,6-Дидезокси-
3-С-метил-3-0-метил-а-Ь-р1/бо-гексопиранозил)ок-
си]-3-[(1Я,2Я)-1,2-дигидрокси-1-метилбутил]-2,6,8,-
10,12-пентаметил-9-[[3,4,6-тридезокси-3-(диметил-
амино)-р~0-/сс^ло-гексопиранозил]окси]-4,13-диокса-
бицикло[8.2.1]тридец-1(12)-ен-5-он (псевдоэритро¬
мицина А еноловый эфир).
07/2016:0552
ЭРИТРОМИЦИНА ЭСТОЛАТ
Егу01готус1П1 ез1о1аз
ЕКУТНЕОМУСШ Е8Т01.АТЕ
о
Эритроми¬
цин (эсто¬
лат)
Молекулярная
формула
М.м.
К1
К2
А
с52н97мо18з
1056
ОН
сн3
В
с52н97ыо17з
1040
н
сн3
С
СяН^З
1042
он
н
ОПРЕДЕЛЕНИЕ
Основной компонент: (ЗЯ,45,55,6Я,7Я,9Я,11Я,-
12/?, 135,14/?)-4-[(2,6-дидезокси-3-С-метил-3-0-метил-
а-1_-р1;бо-гексопиранозил)окси]-14-этил-7,12,13-
тригидрокси-3,5-7,9,11,13-гексаметил-6-[[3,4,6-триде-
зокси-3-(диметиламино)-2-0-пропионил-Р-0-кс1/ло-
гексопиранозил]окси]оксациклотетрадекан-2,10-диона
додецилсульфат (эритромицина А 2"-пропионата до-
децилсульфат).
Полусинтетический продукт, полученный из про¬
дукта ферментации.
Содержание:
- эритромицина эстолат: не менее 86,0 % и не
более 102,0 % (в пересчете на безводное вещество);
- эритромицин В: не более 5,0 % (в пересчете на
безводное вещество);
- эритромицин С: не более 5,0 % (в пересчете
на безводное вещество).
ОПИСАНИЕ
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легко раство¬
рим в 96 % спирте, растворим в ацетоне. Практически
не растворяется в кислоте хлористоводородной раз¬
веденной.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО эритромицина эстолата #или
спектр, представленный на рисунке #0552.-1#.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Гидролизный раствор. Раствор 20 г/л дикалия
гидрофосфата Р, доведенный до рН 8,0 кислотой
фосфорной Р.
1160
Гэсударственная фармакопея Республики Беларусь
Рисунок #0552.-1. Инфракрасный спектр ФСО эритромицина эстолата
Испытуемый раствор. 0,150 г испытуемого об¬
разца растворяют в 25 мл метанола Р, прибавляют
20 мл гидролизного раствора, перемешивают и вы¬
держивают при комнатной температуре в течение не
менее 12 ч. Доводят гидролизным раствором до объ¬
ема 50,0 мл.
Раствор сравнения (а). 40,0 мг ФСО эритроми¬
цина А растворяют в 10 мл метанола Р и доводят ги¬
дролизным раствором до объема 20,0 мл.
Раствор сравнения (Ь). 10,0 мг ФСО эритроми¬
цина В и 10,0 мг ФСО эритромицина С растворяют
в 50,0 мл метанола Р, прибавляют 5,0 мл раствора
сравнения (а) и доводят гидролизным раствором до
объема 100,0 мл.
Раствор сравнения (с). 2 мг ФСО И-деметилэри-
тромицинаА растворяют в 20 мл раствора сравнения (Ь).
Раствор сравнения (ф. 3,0 мл раствора сравне¬
ния (а) доводят смесью из равных объемов метано¬
ла Р и гидролизного раствора до объема 100,0 мл.
Раствор сравнения (е). 40 мг ФСО эритроми¬
цина А, предварительно нагретого при температуре
130 °С в течение 3 ч, растворяют в 10 мл метанола Р
и доводят гидролизным раствором до объема 20 мл
(приготовление т $Ни примесей Е и Р).
Раствор сравнения (I). 2 мг ФСО эритромици¬
на А растворяют в 10 мл 0,01 М раствора кислоты
хлористоводородной, выдерживают при комнатной
температуре в течение 30 мин и доводят гидролиз¬
ным раствором до объема 20 мл (приготовление т
зПи примеси й).
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная сополимером стирол-диви-
нилбензола Р (размер частиц 8 мкм) с размером пор
100 нм;
- температура: 70 °С (для колонки и не менее
1/3 части капилляра, идущего к колонке);
- подвижная фаза: к 50 мл раствора 35 г/л дика¬
лия гидрофосфата Р, доведенного до рН 9,0±0,05 кис¬
лотой фосфорной разведенной Р, прибавляют 400 мл
воды Р, 165 мл 2-метил-2-пропанола Р, 30 мл ацето¬
нитрила Р и доводят водой Р до объема 1000 мл;
- скорость подвижной фазы: 2,0 мл/мин;
-спектрофотометрический детектор, длина
волны 215 нм;
- объем вводимой пробы: по 200 мкл испытуемо¬
го раствора и растворов сравнения (с), (с!), (е) и (0;
- время хроматографирования: 5-кратное вре¬
мя удерживания эритромицина А; интегрирование на¬
чинают после гидролизного пика.
Идентификация пиков примесей: идентифици¬
руют пики примесей Е и Р, используя хроматограмму
раствора сравнения (е).
Относительное удерживание (по отношению
к эритромицину А, время удерживания — около
15 мин): гидролизный пик — менее 0,3; примесь А —
около 0,3; примесь В — около 0,45; эритромицин С —
около 0,5; примесь С — около 0,9; примесь О — около
1,3; примесь О — около 1,4; примесь Р — около 1,5;
эритромицин В — около 1,8; примесь Е — около 4,3.
Пригодность хроматографической системы:
раствор сравнения (с):
- разрешение: не менее 0,8 между пиками при¬
меси В и эритромицина С; не менее 5,5 между пиками
примеси В и эритромицина А.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси Е — 0,09; для примеси Р — 0,15; для при¬
меси (3 — 0,14):
- примеси А, В, С, О, Е, Р, О (не более 3,0 %):
на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям А, В, С, О, Е, Р и
О, не должны превышать площадь основного пика на
хроматограмме раствора сравнения (б);
Эритромицина эстолат
1161
- любая другая примесь (не более 0,2 %): на
хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пиков примесей А,
В, С, О, Е, Р и С, не должна превышать 0,067 пло¬
щади основного пика на хроматограмме раствора
сравнения (с!);
- сумма примесей (не более 5,0 %): на хро¬
матограмме испытуемого раствора сумма площа¬
дей всех пиков, кроме основного, не должна пре¬
вышать 1,67-кратную площадь основного пика на
хроматограмме раствора сравнения (с1);
- неучитываемый предел (0,06 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,02 площади основного
пика на хроматограмме раствора сравнения (с1).
Свободный эритромицин. Жидкостная хро¬
матография (2.2.29). Растворы готовят непо¬
средственно перед использованием.
Испытуемый раствор. 0,250 г испытуемого
образца растворяют в подвижной фазе и доводят
до объема 50,0 мл этим же растворителем.
Раствор сравнения. 75,0 мг ФСО эритроми¬
цина А растворяют в подвижной фазе и доводят до
объема 50,0 мл этим же растворителем. 5,0 мл по¬
лученного раствора доводят ацетонитрилом Р до
объема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: смешивают 35 объемов
ацетонитрила Р1 и 65 объемов раствора, содер¬
жащего 3,4 г/л калия дигидрофосфата Р и 2,75 г/л
триэтиламина Р, доведенного до рН 3,0 кисло¬
той фосфорной разведенной Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 195 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: для раст¬
вора сравнения — 2-кратное время удерживания
эритромицина А; для испытуемого раствора —
4,5-кратное время удерживания первого пика эри¬
тромицина пропионата.
Времена удерживания: эритромицин А — око¬
ло 5 мин; первый пик эритромицина пропионата —
около 10 мин.
Предельное содержание примесей:
-свободный эритромицин: на хроматограмме
испытуемого раствора площадь пика свободного
эритромицина не должна превышать площадь ос¬
новного пика на хроматограмме раствора сравнения.
Додецилсульфат. Не менее 23,0 % и не бо¬
лее 25,5 % С12Н26043 (в пересчете на безводное
вещество). 0,500 г испытуемого образца раство¬
ряют в 25 мл диметилформамида Р и титруют
0,1 М раствором натрия метилата, используя в
качестве индикатора 0,05 мл раствора 3 г/л тимо¬
лового синего Р в метаноле Р.
1 мп 0,1 М раствора натрия метилата соот¬
ветствует 26,64 мг С12Н26043.
Вода (2.5.12). Не более 4,0%. Определение
проводят из 0,300 г испытуемого образца. В ка¬
честве растворителя используют раствор 100 г/л
имидазола Р в метаноле безводном Р.
Сульфатная зола (2.4.14). Не более 0,5%.
Определение проводят из 0,5 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как ука¬
зано в испытании «Сопутствующие примеси», со
следующими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 200 мкл испытуе¬
мого раствора и растворов сравнения (а) и (Ь).
Пригодность хроматографической системы:
раствор сравнения (а):
- относительное стандартное отклонение:
не более 1,2 % для площадей пиков при 6 повтор¬
ных вводах пробы.
Содержание эритромицина А рассчитывают
в процентах, используя хроматограмму раствора
сравнения (а). Содержание эритромицина А эсто-
лата рассчитывают, умножая процентное содержа¬
ние эритромицина А на 1,4387.
Содержание эритромицина В и эритромицина
С рассчитывают в процентах, используя хромато¬
грамму раствора сравнения (Ь). Содержание эри¬
тромицина В эстолата и эритромицина С эстолата
рассчитывают, умножая их процентное содержание
на 1,4387.
Содержание эритромицина эстолата рассчи¬
тывают путем суммирования содержания эритро-
мицинов А, В и С эстолатов.#
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, О, Е, Е, С.
A. Р1 = ОН, Н2 = СН3: Эритромицин Р.
B. Р1 = Р2 = Н: Л/-Деметилэритромицин А.
О. Р1 = Н, Р2 = СО-С2Н5: Л/-Деметил-Л/-пропано-
илэритромицин А.
С. Эритромицин Е.
146. Зак. 1060.
Пропускание
1162
Гэсударственная фармакопея Республики Беларусь
Р. Ангидроэритромицин А.
07/2016:1591
ЭТАКРИДИНА ЛАКТАТ
МОНОГИДРАТ
ЕМаспсИм 1ас(аз топоМус/псиз
ЕТНАСМОШЕ 1.АСТАТЕ
МОИОНУОПАТЕ
С15н15ыз0 ■ Сзнб03 - н20 М.м. 361,4
[6402-23-9]
ОПРЕДЕЛЕНИЕ
7-Этоксиакридин-3,9-диамина (2/?5)-2-гидрокси-
пропаноат.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
Е. Эритромицина А еноловый эфир.
Р. Псевдоэритромицина А еноловый эфир.
ОПИСАНИЕ (СВОЙСТВА)
Желтый кристаллический порошок.
Умеренно растворим в воде, очень мало раство¬
рим в 96 % спирте, практически нерастворим в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А.
Вторая идентификация: В, С, О.
А. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО этакридина лактата моно¬
гидрата #или спектр, представленный на рисунке
#1561 .-1#.
Рисунок #1561 .-1. Инфракрасный спектр ФСО этакридина лактата моногидрата
Этанол безводный
1163
B. К 0,1 мл раствора 3, приготовленного как
указано в разделе «Испытания», прибавляют 100 мл
воды Р. Появляются зеленовато-желтое окрашива¬
ние и интенсивная зеленая флуоресценция в уль¬
трафиолетовом свете при длине волны 365 нм. При¬
бавляют 5 мл 1 М раствора кислоты хлористово¬
дородной. Флуоресценция сохраняется.
C. К 0,5 мл раствора 3 прибавляют 1,0 мл
воды Р, 0,1 мл раствора 10 г/л кобальта хлори¬
да Р и 0,1 мл раствора 50 г/л калия ферроциани¬
да Р. Появляется зеленое окрашивание.
О. К 50 мл раствора 3 прибавляют 10 мл раст¬
вора натрия гидроксида разведенного Р и филь¬
труют. К 5 мл полученного фильтрата прибавляют
1 мл кислоты серной разведенной Р. 5 мл полу¬
ченного раствора дают реакцию на лактаты (2.3.1).
ИСПЫТАНИЯ
Раствор 3. 2,0 г испытуемого образца раство¬
ряют в воде, свободной от углерода диоксида, Р
и доводят до объема 100,0 мл этим же раствори¬
телем.
рН (2.2.3). От 5,5 до 7,0. Измеряют рН раст¬
вора 3.
Сопутствующие примеси. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого
образца растворяют в подвижной фазе и доводят
до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят подвижной фазой до объема
100.0 мл.
Раствор сравнения (Ь). 1,0 мл раствора срав¬
нения (а) доводят подвижной фазой до объема
10.0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза: 1,0 г натрия октансуль-
фоната Р растворяют в смеси из 300 мл ацетони¬
трила Р и 700 мл фосфатного буферного раст¬
вора рН 2,8 Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 268 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 3-кратное вре¬
мя удерживания этакридина.
Время удерживания: этакридин — около
15 мин.
Предельное содержание примесей:
-любая примесь (не более 0,3 %): на хрома¬
тограмме испытуемого раствора площадь любо¬
го пика, кроме основного, не должна превышать
3-кратную площадь основного пика на хромато¬
грамме раствора сравнения (Ь);
- сумма примесей (не более 1 %): на хрома¬
тограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превы¬
шать площадь основного пика на хроматограмме
раствора сравнения (а);
- неучитываемый предел (0,05 %): на хро¬
матограмме испытуемого раствора не учитывают
пики с площадью менее 0,5 площади основного
пика на хроматограмме раствора сравнения (Ь).
Тяжелые металлы (2.4.8, метод Р). Не более
0,0050 % (50 ррт). 1,0 г испытуемого образца дол¬
жен выдерживать испытание на тяжелые металлы.
Эталон готовят с использованием 5,0 мл эталон¬
ного раствора свинца (10 ррт РЬ) Р.
Потеря в массе при высушивании (2.2.32). Не
менее 4,5 % и не более 5,5 %. 1,000 г испытуемого об¬
разца сушат в вакууме при температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,270 г испытуемого образца растворяют в
5,0 мл кислоты муравьиной безводной Р, прибавля¬
ют 60,0 мл уксусного ангидрида Р и титруют 0,1 М
раствором кислоты хлорной потенциометрически
(2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответ-
ствует 34,34 мг С15Н15М3ОС3Н603.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
о
А. 6-Амино-2-этоксиакридин-9(10Н)-он.
B. Р = С1: 6-Хлор-2-этоксиакридин-9-амин.
C. Р = 0-СН2-СН2-0Н: 2-[(9-Амино-7-этоксиакридин-
3-ил)окси]этанол.
07/2016:1318
ЭТАНОЛ БЕЗВОДНЫЙ
Е1Напо1ит апЬус/псит
ЕТНАЫОЦ >ШНУОЯО(75
НзО^^ОН
02Н6О М.м. 46,07
[64-17-5]
ОПРЕДЕЛЕНИЕ
Содержание: не менее 99,5 % (об/об) С2НбО
(99,2 % (м/м)) при температуре 20 °С, рассчитанное
по относительной плотности с помощью алкоголеме¬
трических таблиц (5.5).
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная, прозрачная, летучая, легковоспла¬
меняющаяся жидкость. Гигроскопичен.
Смешивается с водой и метиленхлоридом.
Горит голубым бездымным пламенем.
Температура кипения: около 78 °С.
146*. Зак. 1060.
1164
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр этанола безво¬
дного по Европейской Фармакопее.
C. 0,1 мл испытуемого образца смешивают в про¬
бирке с 1 мл раствора 10 г/л калия перманганата Р
и 0,2 мл кислоты серной разведенной Р. Немедлен¬
но накрывают фильтровальной бумагой, смоченной
свежеприготовленным раствором 0,1 г натрия ни-
тропруссида Р и 0,5 г пиперазина гидрата Р в 5 мл
воды Р. Через несколько минут бумага окрашивается
в интенсивный синий цвет, который бледнеет через
(10—15) мин.
й. К 0,5 мл испытуемого образца прибавляют
5 мл воды Р, 2 мл раствора натрия гидроксида раз¬
веденного Р, затем медленно прибавляют 2 мл 0,05 М
раствора йода Р. В течение 30 мин образуется жел¬
тый осадок.
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец
должен быть прозрачным при сравнении с водой Р.
1.0 мл испытуемого образца доводят водой Р до объ¬
ема 20 мл. Выдерживают в течение 5 мин, раствор
остается прозрачным (2.2.1) по сравнению с водой Р.
Цветность (2.2.2, метод II). Испытуемый раст¬
вор должен быть бесцветным при сравнении с во¬
дой Р.
Кислотность или щелочность. К 20 мл испыту¬
емого образца прибавляют 20 мл воды, свободной от
углерода диоксида, Р и 0,1 мл раствора фенолфта¬
леина Р. Раствор остается бесцветным. Прибавляют
1.0 мл 0,01 М раствора натрия гидроксида. Должно
появиться розовое окрашивание (0,0030 % (30 ррт) в
пересчете на уксусную кислоту).
Относительная плотность (2.2.5). От 0,790 до
0,793.
Оптическая плотность (2.2.25). Не более 0,40
при длине волны 240 нм, не более 0,30 в диапазоне
длин волн от 250 нм до 260 нм и не более 0,10 в диа¬
пазоне длин волн от 270 нм до 340 нм. Спектр погло¬
щения представляет собой непрерывно нисходящую
кривую без видимых пиков и выступов.
Определение проводят в кюветах с толщиной слоя
5 см в диапазоне длин волн от 235 нм до 340 нм, исполь¬
зуя воду Р в качестве компенсационного раствора.
Летучие примеси. Газовая хроматография
(2.2.28).
Испытуемый раствор (а). Испытуемый образец.
Испытуемый раствор (Ь). К 500,0 мл испытуе¬
мого образца прибавляют 150 мкл 4-метилпентан-
2-ола Р.
Раствор сравнения (а). 100 мкл метанола без¬
водного Р доводят испытуемым образцом до объема
50.0 мл. 5,0 мл полученного раствора доводят испы¬
туемым образцом до объема 50,0 мл.
Раствор сравнения (Ь). 50 мкл метанола без¬
водного Р и 50 мкл ацетальдегида Р доводят испы¬
туемым образцом до объема 50,0 мл. 100 мкл полу¬
ченного раствора доводят испытуемым образцом до
объема 10,0 мл.
Раствор сравнения (с). 150 мкл ацеталя Р до¬
водят испытуемым образцом до объема 50,0 мл.
100 мкл полученного раствора доводят испытуемым
образцом до объема 10,0 мл.
Раствор сравнения (д). 100 мкл бензола Р до¬
водят испытуемым образцом до объема 100,0 мл.
100 мкл полученного раствора доводят испытуемым
образцом до объема 50,0 мл.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 30 м
и внутренним диаметром 0,32 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 1,8 мкм);
- газ-носитель: гелий для хроматографии Р\
- линейная скорость: 35 см/с;
- деление потока: 1:20;
- температура:
Время (мин)
Температура (°С)
Колонка
0—12
40
12—32
40 — 240
32—42
240
Блок ввода проб
200
Детектор
280
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между первым пиком
(ацетальдегид) и вторым пиком (метанол).
Предельное содержание примесей:
- метанол (не более 0,0200 % (200 ррт) (об/об)):
на хроматограмме испытуемого раствора (а) площадь
пика, соответствующего метанолу, должна быть не
более половины площади соответствующего пика на
хроматограмме раствора сравнения (а);
- сумма содержания ацетальдегида и ацеталя:
не более 0,0010% (10 ррт) (об/об), в пересчете на
ацетальдегид.
Суммарное содержание ацетальдегида и ацета¬
ля в частях на миллион (об/об) рассчитывают по фор¬
муле:
10 Ае 30Се 44,05
АТ-АЕ+СГ-СЕ‘118,2’
где:
Ае — площадь пика ацетальдегида на хромато¬
грамме испытуемого раствора (а);
АТ — площадь пика ацетальдегида на хромато¬
грамме раствора сравнения (Ь);
СЕ — площадь пика ацеталя на хроматограмме
испытуемого раствора (а);
Ст — площадь пика ацеталя на хроматограмме
раствора сравнения (с);
44,05 — молекулярная масса ацетальдегида;
118,2 — молекулярная масса ацеталя.
- бензол (не более 0,0002 % (2 ррт) (об/об)).
Содержание бензола в частях на миллион (об/об)
рассчитывают по следующей формуле:
2 ВЕ
ВТ-ВЕ ’
где:
ВЕ — площадь пика бензола на хроматограмме
испытуемого раствора (а);
Вт — площадь пика бензола на хроматограмме
раствора сравнения (д).
Этилморфина гидрохлорид
1165
При необходимости подлинность бензола мо¬
жет быть подтверждена при помощи другой подхо¬
дящей хроматографической системы (с неподвиж¬
ной фазой с другой полярностью);
- сумма других примесей (не более 0,0300 %
(300 ррт)): на хроматограмме испытуемого раст¬
вора (Ь) сумма площадей всех пиков, кроме ос¬
новного пика и пиков метанола, ацетальдегида,
ацеталя, бензола и 4-метилпентан-2-ола, не долж¬
на превышать площадь пика, соответствующего
4-метилпентан-2-олу на хроматограмме испытуе¬
мого раствора (Ь);
- неучитываемый предел (0,0009 % (9 ррт)):
на хроматограмме испытуемого раствора (Ь) не
учитывают пики с площадью менее 0,03 площади
пика, соответствующего 4-метилпентан-2-олу на
хроматограмме испытуемого раствора (Ь).
Остаток при выпаривании. Не более
0,0025 % (25 ррт) (м/об). 100 мл испытуемого об¬
разца выпаривают досуха на водяной бане и сушат
при температуре от 100 °С до 105 °С в течение 1 ч.
Масса остатка не должна превышать 2,5 мг.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
Н3СК
СНз
СН3
О
з
С. Пропан-2-он (ацетон).
07/2016:0491
ЭТИЛМОРФИНА ГИДРОХЛОРИД
ЕН1у1тогрЫт Ьус1гос111опс1ит
ЕТНУ1.МОКРН1МЕ НУОРОСШОРШЕ
/СН3
Н N
• НС1 ■ 2Н20
Н
и ~ н ОН
С^Н^ЫОз ■ НС1 • 2НгО М.м. 385,9
ОПРЕДЕЛЕНИЕ
7,8-Дидегидро-4,5а-эпокси-3-этокси-17-метил-
морфинан-6а-ола гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на безводное вещество).
А.
1,1-Диэтоксиэтан (ацеталь).
о
В. Ацетальдегид.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок.
Растворим в воде и в 96 % спирте, практически
нерастворим в циклогексане.
Рисунок #0491 .-1. Спектр этилморфина гидрохлорида
147. Зак. 1060.
1166
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, О.
Вторая идентификация: В, С, О.
A. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр этилморфи-
на гидрохлорида по Европейской Фармакопее #или
спектр, представленный на рисунке #0491.-1#.
B. 0,5 г испытуемого образца помещают в про¬
бирку, растворяют в 6 мл воды Р и прибавляют 15 мл
0,1 М раствора натрия гидроксида. Потирают вну¬
тренние стенки пробирки стеклянной палочкой. Об¬
разуется белый кристаллический осадок. Осадок со¬
бирают, промывают и растворяют в 20 мл воды Р, на¬
гретой до температуры 80 °С. Фильтруют и охлаждают
в ледяной бане. Полученный остаток сушат в вакууме
в течение 12 ч. Температура плавления (2.2.14) полу¬
ченных кристаллов: от 85 °С до 89 °С.
C. К 10 мг испытуемого образца прибавляют 1 мл
кислоты серной Р, 0,05 мл раствора железа (III) хло¬
рида Р2 и нагревают на водяной бане. Появляется си¬
нее окрашивание. Прибавляют 0,05 мл кислоты азот¬
ной Р. Окрашивание раствора изменяется на красное.
О. Раствор 5, приготовленный как указано в разде¬
ле «Испытания», дает реакцию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор 5. 0,500 г испытуемого образца раст¬
воряют в воде, свободной от углерода диоксида, Р и
доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 5
должна быть не интенсивнее эталона ВУ(КЖ)6.
Кислотность или щелочность. К 10 мл раствора
3 прибавляют 0,05 мл раствора метилового красно¬
го Р и 0,2 мл 0,02 М раствора кислоты хлористоводо¬
родной. Должно появиться красное окрашивание. При
прибавлении не более 0,4 мл 0,02 М раствора натрия
гидроксида должно появиться желтое окрашивание.
Удельное оптическое вращение (2.2.7). От -102
до -105 (в пересчете на безводное вещество). Опре¬
деляют удельное оптическое вращение раствора 3.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 20,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 25,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 20,0 мл.
Раствор сравнения (Ь). 12,5 мг кодеина Р раст¬
воряют в подвижной фазе и доводят до объема 5,0 мл
этим же растворителем.
Раствор сравнения (с). 0,5 мл раствора сравне¬
ния (Ь) доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (ф. К 1,0 мл испытуемого
раствора прибавляют 1,0 мл раствора сравнения (Ь) и
доводят подвижной фазой до объема 50,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: к смеси из 12,5 мл кислоты
уксусной ледяной Р и 5 мл 20 % (об/об) раствора
триэтиламина Р в смеси из равных объемов мета¬
нола Р и воды Р прибавляют 1,25 г натрия гептан-
сульфоната Р и доводят водой Р до объема 1000 мл.
К 550 мл полученного раствора прибавляют 450 мл
метанола Р;
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 230 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: 4-кратное вре¬
мя удерживания этилморфина.
Относительное удерживание (по отношению к
этилморфину, время удерживания — около 6,2 мин):
примесь В — около 0,7; примесь С — около 0,8; при¬
месь О — около 1,3; примесь А — около 2,5.
Пригодность хроматографической системы:
раствор сравнения (д):
-разрешение: не менее 5 между пиками этил¬
морфина и примеси С.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси О — 0,4):
- примеси А, В, О (не более 0,2 %): на хромато¬
грамме испытуемого раствора площади пиков, соот¬
ветствующих примесям А, В и О, не должны превы¬
шать площадь основного пика на хроматограмме
раствора сравнения (а);
- примесь С (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси С, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (с);
- любая другая примесь (не более 0,1 %): на хро¬
матограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А, В, С и О,
не должна превышать 0,5 площади основного пика на
хроматограмме раствора сравнения (а);
- сумма примесей, кроме примеси С (не более
0,5 %): на хроматограмме испытуемого раствора сум¬
ма площадей всех пиков, кроме основного и пика при¬
меси С, не должна превышать 2,5-кратную площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Вода (2.5.12). Не менее 8,0 % и не более 10,0 %.
Определение проводят из 0,250 г испытуемого образца.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют в смеси
из 5 мл 0,01 М раствора кислоты хлористоводород¬
ной и 30 мл 96 % спирта Р и титруют 0,1 М раство¬
ром натрия гидроксида потенциометрически (2.2.20).
Отмечают объем титранта между двумя точками пе¬
региба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 34,99 мг С19Н23М03*НС1.
ХРАНЕНИЕ
В защищенном от света месте.
Этиловый спирт 96 %
1167
Таблица РБ0032.-1.
Содержание спирта этилового
95%
90%
80%
70%
60%
40%
Плотность
0,8114—0,8075
0,8292—0,8259
0,8593—0,8565
0,8855—0,8830
0,9092—0,9069
0,9487—0,9473
Содержание, % (об/об)
95—96
90—91
00
0
1
00
70—71
60—61
39,5—40,5
ПРИМЕСИ
Специфицированные примеси: А, В, С, О.
A. Я = К' = С2Н5: 7,8-Дидегидро-4,5а-эпокси-3,6а-
диэтокси-17-метил морфинан.
B. Я = Я' = И: 7,8-Дидегидро-4,5а-эпокси-17-ме-
тилморфинан-3,6а-диол (морфин).
C. Р = СН3, Р' = Н: 7,8-Дидегидро-4,5а-эпокси-3-
й. 7,8-Дидегидро-4,5а-эпокси-3-этокси-17-ме-
тилморфинан-6-он (этилморфинон).
07/2016: РБ0032
#ЭТИЛОВЫЙ СПИРТ
95 %, 90 %, 80 %, 70 %, 60 %, 40 %
8р1'гНи$ аеМуНсиз
95 %, 90 %, 80 %, 70 %, 60 %, 40 %
ЕТНАМ01195 % 90 %, 80 % 70 % 60 % 40 %
ОПРЕДЕЛЕНИЕ
Спирт этиловый 95 %, 90 %, 80 %, 70 %, 60 % и
40 % представляет собой спирто-водный раствор.
ПРИГОТОВЛЕНИЕ
Смешивают необходимое количество Этилово¬
го спирта 96% (1317) с необходимым количеством
Воды очищенной (0008) в соответствии с алкоголеме¬
трическими таблицами (5.5).
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость с характерным
спиртовым запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 0,5 мл испытуемого образца прибавляют 5 мл
воды Р, 2 мл разведенного раствора натрия гидрок¬
сида Р и затем медленно 2 мл 0,05 М раствора йода.
Через 30 мин образуется желтый осадок.
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным.
Цветность (2.2.2, метод II). Испытуемый обра¬
зец должен быть бесцветным.
Плотность (2.2.5). Плотность и ее соответствие
содержанию спирта этилового (об/об) представлена в
таблице: РБ0032.-1.
Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
07/2016:1317
ЭТИЛОВЫЙ СПИРТ 96 %
ЕМапо1ит (96 рег сеМит)
(#8р1гПиз аеМуНсиз 96 %)
ЕТНАЫ01. (96 РЕК СЕЫТ)
ОПРЕДЕЛЕНИЕ
Содержание:
-этанол (С2Н60; М.м. 46,07): не менее 95,1 %
(об/об) (92,6 % (м/м)) и не более 96,9 % (об/об) (95,2 %
(м/м)) при температуре 20 °С, рассчитывают по отно¬
сительной плотности с использованием алкоголеме¬
трических таблиц (5.5);
- вода.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная, прозрачная, летучая, легковоспла¬
меняющаяся жидкость. Гигроскопична.
Смешивается с водой и метиленхлоридом. Горит
голубым, бездымным пламенем.
Температура кипения: около 78 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С, О.
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: эталонный спектр этилового спир¬
та 96% по Европейской Фармакопее #или спектр,
представленный на рисунке #1317.-1#.
C. 0,1 мл испытуемого образца смешивают в про¬
бирке с 1 мл раствора 10 г/л калия перманганата Р и
0,2 мл кислоты серной разведенной Р. Пробирку сра¬
зу накрывают фильтровальной бумагой, смоченной в
свежеприготовленном растворе, содержащем 0,1 г на¬
трия нитропруссида Р и 0,5 г пиперазина гидрата Р
в 5 мл воды Р. Через несколько минут бумага окраши¬
вается в интенсивный синий цвет, который бледнеет
через (10—15) мин.
О. К 0,5 мл испытуемого образца прибавляют
5 мл воды Р, 2 мл раствора натрия гидроксида раз¬
веденного Р и затем медленно 2 мл 0,05 М раствора
йода. Через 30 мин образуется желтый осадок.
ИСПЫТАНИЯ
Прозрачность (2.2.1). Испытуемый образец дол¬
жен быть прозрачным при сравнении с водой Р. Затем
1,0 мл испытуемого образца доводят водой Р до объ¬
ема 20 мл. Полученный раствор через 5 мин должен
быть прозрачным при сравнении с водой Р.
Цветность (2.2.2, метод II). Испытуемый образец
должен быть бесцветным при сравнении с водой Р.
147*. Зак. 1060.
1168
Гэсударственная фармакопея Республики Беларусь
Рисунок #1317.-1. Инфракрасный спектр этилового спирта 96%
Кислотность или щелочность. К 20 мл испыту¬
емого образца прибавляют 20 мл воды, свободной от
углерода диоксида, Р и 0,1 мл раствора фенолфта¬
леина Р. Раствор остается бесцветным. К полученно¬
му раствору прибавляют 1,0 мл 0,01 М раствора на¬
трия гидроксида Р. Появляется розовое окрашивание
(0,0030 % (30 ррт) в пересчете на уксусную кислоту).
Относительная плотность (2.2.5). От0,805 до 0,812.
Оптическая плотность (2.2.25). Оптическая
плотность должна быть не более 0,40 при длине
волны 240 нм, не более 0,30 в диапазоне от 250 нм
до 260 нм и не более 0,10 в диапазоне от 270 нм до
340 нм. Спектр поглощения представляет собой не¬
прерывно нисходящую кривую без видимых пиков и
выступов.
Определение проводят в диапазоне длин волн от
235 до 340 нм, в кювете толщиной 5 см с использова¬
нием воды Р в качестве компенсационной жидкости.
Летучие примеси. Газовая хроматография
(2.2.28).
Испытуемый раствор (а). Испытуемый образец.
Испытуемый раствор (Ь). 150 мкл 4-метил-
пентан-2-ола Р прибавляют к 500,0 мл испытуемого
образца.
Раствор сравнения (а). 100 мкл метанола без¬
водного Р доводят испытуемым образцом до объема
50,0 мл. 5,0 мл полученного раствора доводят испы¬
туемым образцом до объема 50,0 мл.
Раствор сравнения (Ь). 50 мкл метанола без¬
водного Р и 50 мкл ацетальдегида Р доводят испы¬
туемым образцом до объема 50,0 мл. 100 мкл полу¬
ченного раствора доводят испытуемым образцом до
объема 10,0 мл.
Раствор сравнения (с). 150 мкл ацеталя Р до¬
водят испытуемым образцом до объема 50,0 мл.
100 мкл полученного раствора доводят испытуемым
образцом до объема 10,0 мл.
Раствор сравнения (д). 100 мкл бензола Р до¬
водят испытуемым образцом до объема 100,0 мл.
100 мкл полученного раствора доводят испытуемым
образцом до объема 50,0 мл.
Условия хроматографирования;
- колонка кварцевая капиллярная длиной 30 м
и внутренним диаметром 0,32 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силоксана Р
(толщина слоя 1,8 мкм);
- газ-носитель: гелий для хроматографии Р;
- линейная скорость: 35 см/с;
- деление потока: 1:20;
- температура:
Время (мин)
Температура (°С)
Колонка
0—12
40
12—32
40 -> 240
32—42
240
Блок ввода проб
200
Детектор
280
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 1,5 между первым (аце¬
тальдегид) и вторым (метанол) пиками.
Предельное содержание примесей:
- метанол (не более 0,0200 % (об/об) (200 ррт,
об/об)): на хроматограмме испытуемого раствора (а)
площадь пика, соответствующего метанолу, не долж¬
на превышать 0,5 площади соответствующего пика на
хроматограмме раствора сравнения (а);
- ацетальдегид + ацеталь: не более 0,0010 %
(об/об) (10 ррт, об/об) в пересчете на ацетальдегид.
Суммарное содержание ацетальдегида и ацета¬
ля в частях на миллион (об/об) рассчитывают по фор¬
муле:
Этиловый спирт 96 %
1169
10 Ае 30 СЕ 44,05
АТ-АЕ + СТ-СЕ' 118,2’
где:
Ае — площадь пика ацетальдегида на хромато¬
грамме испытуемого раствора (а);
Ат — площадь пика ацетальдегида на хромато¬
грамме раствора сравнения (Ь);
СЕ — площадь пика ацеталя на хроматограмме
испытуемого раствора (а);
Ст — площадь пика ацеталя на хроматограмме
раствора сравнения (с);
44,05 — молекулярная масса ацетальдегида;
118,2 — молекулярная масса ацеталя.
- бензол: не более 0,0002 % (об/об) (2 ррт, об/об).
Содержание бензола в частях на миллион (об/об)
рассчитывают по формуле:
2Ве
вт~вЕ9
где:
ВЕ — площадь пика бензола на хроматограмме
испытуемого раствора (а);
Вт— площадь пика бензола на хроматограмме
раствора сравнения (6).
При необходимости подлинность бензола может
быть подтверждена использованием другой подхо¬
дящей хроматографической системы (неподвижной
фазы другой полярности).
-сумма других примесей (не более 0,0300%
(300 ррт)): на хроматограмме испытуемого раствора (Ь)
сумма площадей всех пиков, кроме основного и пиков
метанола, ацетальдегида, ацеталя и бензола, не долж¬
на превышать площади пика 4-метилпентан-2-ола;
- неучитываемый предел (0,0009 % (9 ррт)):
не учитывают пики с площадью менее 0,03 площади
пика 4-метилпентан-2-ола на хроматограмме испыту¬
емого раствора (Ь).
Для информации представлена хроматограмма
на рисунке #1317.-2.
#Летучие примеси. Газовая хроматография
(2.2.28).
Испытуемый раствор (а). Испытуемый образец.
Градуировку хроматографа выполняют, исполь¬
зуя не менее трех готовых аттестованных градуиро¬
вочных смесей, соответствующих диапазону измеряе¬
мых концентраций следующего состава:
1) 0,012% спирта метилового и по (8—9) мг/л
ацетальдегида, метилацетата, этилацетата, 2-пропа¬
нола, 1-пропанола, спирта изобутилового, 1-бутанола,
спирта изоамилового;
2) 0,006 % спирта метилового и по (4—5) мг/л
ацетальдегида, метилацетата, этилацетата, 2-пропа¬
нола, 1-пропанола, спирта изобутилового, 1-бутанола,
спирта изоамилового;
3) 0,003 % спирта метилового и по (0,8—2) мг/л
уксусного альдегида, метилацетата, этилацетата,
2-пропанола, 1-пропанола, спирта изобутилового,
1-бутанола, спирта изоамилового.
Условия хроматографирования:
- колонка капиллярная длиной 60 м и внутрен¬
ним диаметром 0,53 мм, покрытая слоем нитроте-
рефталевой кислоты, модифицированной полиэти¬
ленгликолем Р (толщина слоя 1 мкм);
1. Ацетальдегид
2. Метанол
3. Этанол
4. Ацетон
5. Пропанол-2
6. Бутанон-2
8. Бутанол-2
9. Циклогексан
10. Бензол
11. 2-Метил-1-пропанол
12. Бутанол
13. Ацеталь
14. Метилизобутилкетон
15. Пентанол
16. Фурфурол
17. Октанол.
Рисунок #1317.-2. Хроматограмма определения летучих примесей
148. Зак. 1060.
1170
Гэсударственная фармакопея Республики Беларусь
-газ-носитель: гелий для хроматографии Р
или азот для хроматографии Р\
- скорость газа-носителя: 4,9 мл/мин;
- деление потока: 1:20;
- температура:
Время (мин)
Температура (°С)
Колонка
0—9
73
9—18,4
73-120
18,4—25
120
Блок ввода проб
200
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Порядок выхода пиков: уксусный альдегид, ме-
тилацетат, этилацетат, спирт метиловый, 2-пропанол,
спирт этиловый, 1-пропанол, спирт изобутиловый,
1-бутанол, спирт изоамиловый.
Пригодность хроматографической системы:
градуировочная смесь:
- эффективность хроматографической колон¬
ки: не менее 2000 теоретических тарелок, рассчитан¬
ная по пикам уксусного альдегида, спирта метилово¬
го, метилацетата, этилацетата, 2-пропанола на хрома¬
тограммах стандартных образцов примесей в спирте
этиловом; не менее 10 000, рассчитанная по пикам
1 -пропанола, спирта изобутилового, 1-бутанола, спир¬
та изоамилового;
- разрешение: не менее 1,0 между пиком опре¬
деляемого компонента примеси и его ближайшими,
соседними пиками;
- относительное стандартное отклонение: не
более 15 %, рассчитанное для площадей пиков опре¬
деляемых примесей.
Для пересчета на безводный спирт результаты
умножают на коэффициент П, который рассчитывают
по формуле:
/7 = 100 :Р,
где:
Р — объемная доля спирта этилового в испытуе¬
мом образце, %;
100 — объемная доля безводного спирта, %.
Предельное содержание примесей:
- сложные эфиры: не более 10 мг/л, в пересчете
на безводный спирт (рассчитывают как сумму концен¬
траций метилацетата и этилацетата);
- сивушное масло: не более 6 мг/л, в пересчете
на безводный спирт (рассчитывают как сумму концен¬
траций 2-пропанола, 1-пропанола, спирта изобутило¬
вого, 1-бутанола и спирта изоамилового);
- ацетальдегид: не более 2 мг/л, в пересчете на
безводный спирт;
- метанол: не более 0,03 % (об/об), в пересчете
на безводный спирт.
Для информации представлена хроматограмма
на рисунке #1317.-3.
Остаток после выпаривания. Не более
0,0025% (м/об) (25ррт, м/об). 100 мл испытуемого
образца выпаривают досуха на водяной бане и сушат
при температуре от 100 °С до 105 °С в течение 1 ч.
Масса остатка не должна превышать 2,5 мг.
#Тяжелые металлы (2.4.8, метод А). Не более
0,0002 % (2 ррт). Сухой остаток, полученный в испы¬
тании на «Остаток после выпаривания», растворяют
в 1 мл 1 М раствора кислоты хлористоводородной,
количественно переносят в колбу и доводят водой Р
до объема 100,0 мл, перемешивают. 12 мл получен¬
ного раствора должны выдерживать испытание на
тяжелые металлы. Эталон готовят с использованием
10 мл эталонного раствора свинца (2 ррт РЬ) Р.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
СН3
А. 1,1-Диэтоксиэтан (ацеталь).
В. Ацетальдегид.
Рисунок #1317.-3. Хроматограмма определения летучих примесей
Этиловый спирт 96 %
1171
А„
Н3С хн3
С. Пропан-2-он (ацетон).
О. Бензол.
Е. Циклогексан.
Р. Метанол.
н3с—он
н3с.
сн3
СВ. Бутан-2-он (метилэтилкетон).
сн3 о
н3с" ^ хн3
Н. 4-Метилпентан-2-он (метилизобутилкетон).
юн
н3с"
I. Пропан-1-ол (пропанол).
он
н3с сн3
и. Пропан-2-ол (изопропиловый спирт).
Н3С^ /ОН
К. Бутан-1-ол (бутанол).
он
Н3а
I.. Бутан-2-ол.
сн3
Н3С'
сн3
.он
М. 2-Метилпропан-1-ол (изобутанол).
о
N. Фуран-2-карбальдегид (фурфураль).
н3с он
НзСГХНз
O. 2-Метилпропан-2-ол (1,1-диметилэтиловый
спирт).
н3с он
хн3
Р. 2-Метилбутан-2-ол.
он
н3с
хсн3
О. Пентан-2-ол.
Р. Пентан-1-ол (пентанол).
3. Гексан-1-ол (гексанол).
Н3С‘
Т. Гептан-2-ол
н3с
II. Гексан-2-ол.
н3с^
V. Гексан-З-ол.
О. Бензол.
Е. Циклогексан.
Р. Метанол.
н3с—он
Н3с.
сн3
С. Бутан-2-он (метилэтилкетон).
СН3 О
н3с
сн3
Н. 4-Метилпентан-2-он (метилизобутилкетон).
/ОН
н3с"
I. Пропан-1-ол (пропанол).
он
Н3С хн3
Л. Пропан-2-ол (изопропиловый спирт).
К. Бутан-1-ол (бутанол).
он
Н3С^
Бутан-2-ол.
СН3
Н3С'
сн3
он
M. 2-Метил пропан-1-ол (изобутанол).
V.'
N. Фуран-2-карбальдегид (фурфураль).
148*. Зак. 1060.
1172
Гэсударственная фармакопея Республики Беларусь
н3с он
Н3С^
он.
О. 2-Метилпропан-2-ол (1,1-диметилэтиловый
спирт).
н3с.
н3с он
сн3
Р. 2-Метилбутан-2-ол.
он
н3с
сн
3
О. Пентан-2-ол.
н3<
Я. Пентан-1-ол (пентанол).
5. Гексан-1-ол (гексанол).
н3с
Т. Гептан-2-ол.
он
н3с
I). Гексан-2-ол.
Н3С
сн3
он
V. Гексан-З-ол.
07/2016:0900
ЭТИЛПАРАГИДРОКСИБЕНЗОАТ
ЕИпуИз рагаЬубгохуЪепгоаз
ЕТНП РАЙАНУОРЮХУВЕИЮАТЕ
ОПРЕДЕЛЕНИЕ
Этил-4-гидроксибензоат.
Содержание: не менее 98,0 % и не более 102,0 %.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Очень мало растворим в воде, легко растворим в
96 % спирте и в метаноле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В.
Вторая идентификация: А, С.
А. Температура плавления (2.2.14): от 115 °С до
118 °С.
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО этилпарагидроксибензоата.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). 0,10 г испытуемого
образца растворяют в ацетоне Р и доводят этим же
растворителем до объема 10 мл.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят ацетоном Р до объема 10 мл.
Раствор сравнения (а). 10 мг ФСО этилпараги¬
дроксибензоата растворяют в ацетоне Р и доводят
до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 10 мг ФСО метилпара-
гидроксибензоата растворяют в 1 мл испытуемого
раствора (а) и доводят ацетоном Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем октаде-
цилсилильного силикагеля Р254 Р
Подвижная фаза: кислота ледяная уксусная Р —
вода Р — метанол Р (1:30:70, об/об/об).
Наносимый объем пробы: 2 мкл испытуемого
раствора (Ь) и растворов сравнения (а) и (Ь).
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультра¬
фиолетовом свете при длине волны 254 нм.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- на хроматограмме обнаруживаются два полно¬
стью разделенных пятна.
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соот¬
ветствующее по расположению и размеру основному
пятну на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Раствор 5. 1,0 г испытуемого образца раство¬
ряют в 96% спирте Р и доводят до объема 10 мл
этим же растворителем.
Прозрачность (2.2.1). Раствор 5 должен быть
прозрачным.
Цветность (2.2.2, метод II). Окраска раствора 8
должна быть не интенсивнее эталона ВУ(КЖ)б.
Кислотность. К 2 мл раствора 8 прибавляют
3 мл 96 % спирта Р, 5 мл воды, свободной от угле¬
рода диоксида, Р и 0,1 мл раствора бромкрезолового
зеленого Р. При прибавлении не более 0,1 мл раст¬
вора 0,1 М натрия гидроксида окраска индикатора
должна измениться на зеленую.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 50,0 мг испытуемого об¬
разца растворяют в 2,5 мл метанола Р и доводят
подвижной фазой до объема 50,0 мл. 10,0 мл полу¬
ченного раствора доводят подвижной фазой до объ¬
ема 100,0 мл.
Раствор сравнения (а). 5 мг 4-гидроксибензой-
ной кислоты Р (примесь А), 5 мг метилпарагидрокси-
бензоата Р (примесь В) и 5 мг испытуемого образца
растворяют в подвижной фазе и доводят до объема
100.0 мл этим же растворителем. 1,0 мл полученного
раствора доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (Ь). 50,0 мг ФСО этилпара¬
гидроксибензоата Р растворяют в 2,5 мл метано¬
ла Р и доводят подвижной фазой до объема 50,0 мл.
10.0 мл полученного раствора доводят подвижной
фазой до объема 100,0 мл.
Эфедрина гидрохлорид
1173
Раствор сравнения (с). 1,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 20,0 мл.
1,0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Условия хроматографирования:
-колонка длиной 0,15м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: раствор 6,8 г/л калия диги¬
дрофосфата Р — метанол Р (35:65, об/об);
- скорость подвижной фазы: 1,3 мл/мин;
-спектрофотометрический детектор, длина
волны 272 нм;
- объем вводимой пробы: по 10 мкл испытуемого
раствора и растворов сравнения (а) и (с);
- время хроматографирования: 4-кратное вре¬
мя удерживания этилпарагидроксибензоата.
Относительное удерживание (по отношению
к этилпарагидроксибензоату, время удерживания —
около 3,0 мин): примесь А — около 0,5; примесь В —
около 0,8.
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 2,0 между пиками при¬
меси В и этилпарагидроксибензоата.
Предельное содержание примесей (для расчета
содержания примеси умножают площадь пика на по¬
правочный коэффициент: для примеси А — 1,4):
- примесь А (не более 0,5 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (с);
- неспецифицированные примеси (не более
0,5 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси
А, не должна превышать площадь основного пика на
хроматограмме раствора сравнения (с);
- сумма примесей (не более 1,0 %): на хромато¬
грамме испытуемого раствора сумма площадей всех
пиков, кроме основного, не должна превышать 2-крат¬
ную площадь основного пика на хроматограмме раст¬
вора сравнения (с);
-неучитываемый предел (0,1 %): на хромато¬
грамме испытуемого раствора не учитывают пики с
площадью менее 0,2 площади основного пика на хро¬
матограмме раствора сравнения (с).
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29), как указано
в испытании «Сопутствующие примеси», со следую¬
щими изменениями.
Условия хроматографирования:
- объем вводимой пробы: по 10 мкл испытуемого
раствора и раствора сравнения (Ь).
Содержание С9Н10О3 в процентах рассчитывают с
учетом содержания этилпарагидроксибензоата в ФСО
этилпарагидроксибензоата.
ПРИМЕСИ
Специфицированная примесь: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным ис¬
пытанием, описанным в частной статье. Их содержа¬
ние лимитируется общим критерием приемлемости
для других/неспецифицированных примесей и/или
общей статьей Субстанции для фармацевтическо¬
го использования (2034). Вследствие этого нет не¬
обходимости идентифицировать эти примеси для до¬
казательства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для
фармацевтического использования): В, С, О.
А. 4-Гидроксибензойная кислота.
В. Метил-4-гидроксибензоат (метилпарагидрокси-
бензоат).
С. Пропил-4-гидроксибензоат (пропилпарагидрок-
сибензоат).
й. Бутил-4-гидроксибензоат (бутилпарагидрокси-
бензоат).
07/2016:0487
ЭФЕДРИНА ГИДРОХЛОРИД
ЕрНеДпп! ЬудгосЫопс/ит
ЕРНЕОРШЕ НУОКОСШ-ОРЮЕ
С10Н15ЫО ■ НС1 М.м. 201,7
[50-98-6]
ОПРЕДЕЛЕНИЕ
(1/?,25)-2-(Метиламино)-1-фенилпропан-1-ола
гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, растворим в 96 % спирте.
Температура плавления: около 219 °С.
149. Зак. 1060.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
A. Испытуемый образец выдерживает испытание
«Удельное оптическое вращение», как указано в раз¬
деле «Испытания».
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Сравнение: ФСО эфедрина гидрохлорида #или
спектр, представленный на рисунке #0487.-1*
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мг испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Раствор сравнения. 10 мг ФСО эфедрина ги¬
дрохлорида растворяют в метаноле Р и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: метиленхлорид Р — раст¬
вор аммиака концентрированный Р — 2-пропанол Р
(5:15:80, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты
пластинки.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 110 °С в
течение 5 мин.
Результаты: на хроматограмме испытуемого
раствора обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения.
О. К 0,1 мл раствора 3, приготовленного как указа¬
но в разделе «Испытания», прибавляют 1 мл воды Р,
0,2 мл раствора меди сульфата Р и 1 мл раствора
натрия гидроксида концентрированного Р. Появля¬
ется фиолетовое окрашивание. Прибавляют 2 мл ме¬
тиленхлорида Р и встряхивают. В нижнем (органиче¬
ском) слое появляется темно-серое окрашивание, а в
верхнем (водном) слое — синее окрашивание.
Е. К 5 мл раствора 3 прибавляют 5 мл воды Р.
Полученный раствор дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,00 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора метилового
красного Р и 0,2 мл 0,01 М раствора натрия гидрок¬
сида. Должно появиться желтое окрашивание. При
прибавлении не более 0,4 мл 0,01 М раствора кисло¬
ты хлористоводородной должно появиться красное
окрашивание.
Удельное оптическое вращения (2.2.7). От
-33,5 до -35,5 (в пересчете на сухое вещество).
12,5 мл раствора 3 доводят водой Р до объема
25.0 мл.
Сопутствующие примеси. Жидкостная хрома¬
тография (2.2.29).
Испытуемый раствор. 75 мг испытуемого об¬
разца растворяют в подвижной фазе и доводят до
объема 10 мл этим же растворителем.
Раствор сравнения (а). 2,0 мл испытуемого раст¬
вора доводят подвижной фазой до объема 100,0 мл.
1.0 мл полученного раствора доводят подвижной фа¬
зой до объема 10,0 мл.
Раствор сравнения (Ь). 5 мг испытуемого образ¬
ца и 5 мг ФСО псевдоэфедрина гидрохлорида раст-
Л 3000 * ----- '1500 " * 1000
Волновое число (см‘1)
Рисунок. #0487.-1 Инфракрасный спектр ФСО эфедрина гидрохлорида
Эфедрина гидрохлорид рацемический
1175
воряют в подвижной фазе и доводят до объема 50 мл
этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,15 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем фенилсилиль-
ным для хроматографии Р с размером частиц 3 мкм;
- подвижная фаза: метанол Р— раствор 11,6 г/л
аммония ацетата Р, доведенного кислотой уксус¬
ной ледяной Р до рН 4,0, (6:94, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 257 нм;
- объем вводимой пробы: 20 мкл;
- время хроматографирования: 2,5-кратное вре¬
мя удерживания эфедрина.
Относительное удерживание (по отношению к
эфедрину, время удерживания — около 8 мин): при¬
месь В — около 1,1; примесь А— около 1,4.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 2,0 между пиками эфе¬
дрина и примеси В.
Предельное содержание примесей (для расчета
содержания примесей умножают площади пиков на
соответствующие поправочные коэффициенты: для
примеси А—0,4):
- примесь А (не более 0,2 %): на хроматограмме
испытуемого раствора площадь пика, соответствующе¬
го примеси А, не должна превышать площадь основно¬
го пика на хроматограмме раствора сравнения (а);
- неспецифицированные примеси (не более
0,1 %): на хроматограмме испытуемого раствора пло¬
щадь любого пика, кроме основного и пика примеси А,
не должна превышать 0,5 площади основного пика на
хроматограмме раствора сравнения (а);
- сумма примесей, кроме примеси А (не более
0,5 %): на хроматограмме испытуемого раствора сум¬
ма площадей всех пиков, кроме основного и пика при¬
меси А, не должна превышать 2,5-кратную площадь
основного пика на хроматограмме раствора сравне¬
ния (а);
- неучитываемый предел (0,05 %): на хрома¬
тограмме испытуемого раствора не учитывают пики
с площадью менее 0,25 площади основного пика на
хроматограмме раствора сравнения (а).
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
Используют раствор 5.
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ПРИМЕСИ
Специфицированные примеси: А.
Другие обнаруживаемые примеси (следующие
вещества, если они присутствуют в значительных
количествах, следует определять тем или иным
испытанием, описанным в частной статье. Их со¬
держание лимитируется общим критерием прием¬
лемости для других/неспецифицированных приме¬
сей и/или общей статьей Субстанции для фарма¬
цевтического использования (2034). Вследствие
этого нет необходимости идентифицировать эти
примеси для доказательства соответствия требо¬
ваниям. См. также статью 5.10. Контроль приме¬
сей в субстанциях для фармацевтического ис¬
пользования): В.
А. (-)-(1Р)-1-Гидрокси-1-фенилпропан-2-он.
В. (15,25)-2-(Метиламино)1-фенилпропан-1-ол
(псевдоэфедрин).
07/2016:0715
ЭФЕДРИНА ГИДРОХЛОРИД
РАЦЕМИЧЕСКИЙ
ЕрЬвдпп1 гасетю НудгосЫопс1ит
ЕРНЕйРШЕ НУОРОСШОРЮЕ,
РАСЕ М/С
н ОН
н сн3
и энантиомер • НС1
С10Н15МО ■ НС1 М.м. 201,7
[134-71-4]
ОПРЕДЕЛЕНИЕ
(1 Р5,2$Р)-2-(Метиламино)-1 -фенил пропан-1 -ола
гидрохлорид.
Содержание: не менее 99,0 % и не более 101,0 %
(в пересчете на сухое вещество).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в 50 мл
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 20,17 мг С10Н15МО*НС1.
ХРАНЕНИЕ
В защищенном от света месте.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический поро¬
шок или бесцветные кристаллы.
Легко растворим в воде, растворим в 96 % спирте.
Температура плавления: около 188 °С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, Е.
Вторая идентификация: А, С, О, Е.
А. Испытуемый образец выдерживает испытание
«Угол оптического вращения», как указано в разделе
«Испытания».
149*. Зак. 1060.
1176
Гэсударственная фармакопея Республики Беларусь
B. Абсорбционная спектрофотометрия в инфра¬
красной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО эфедрина гидрохлорида раце¬
мического #или спектр, представленный на рисунке
#0715.-1*.
C. Просматривают хроматограммы, полученные
в испытании «Сопутствующие примеси».
Результаты: на хроматограмме испытуемого
раствора (Ь) обнаруживается основное пятно, соответ¬
ствующее по расположению, цвету и размеру основ¬
ному пятну на хроматограмме раствора сравнения (а).
О. К 0,1 мл раствора 5, приготовленного как
указано в разделе «Испытания», прибавляют 1 мл
воды Р, 0,2 мл раствора меди сульфата Р и 1 мл
раствора натрия гидроксида концентрированно¬
го Р. Появляется фиолетовое окрашивание. Прибав¬
ляют 2 мл эфира Р и встряхивают. В эфирном слое
появляется красно-фиолетовое окрашивание, а в во¬
дном слое — синее окрашивание.
Е. К 5 мл раствора 3 прибавляют 5 мл воды Р.
Полученный раствор дает реакцию (а) на хлориды
(2.3.1).
ИСПЫТАНИЯ
Раствор 5. 5,00 г испытуемого образца раство¬
ряют в воде дистиллированной Р и доводят до объ¬
ема 50,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор 3 должен быть
прозрачным.
Цветность (2.2.2, метод II). Раствор 3 должен
быть бесцветным.
Кислотность или щелочность. К 10 мл раст¬
вора 3 прибавляют 0,1 мл раствора метилового
красного Р и 0,1 мл 0,01 М раствора натрия гидрок¬
сида. Должно появиться желтое окрашивание. При
прибавлении не более 0,2 мл 0,01 М раствора кисло¬
ты хлористоводородной должно появиться красное
окрашивание.
Угол оптического вращения (2.2.7). От +0,2° до
-0,2°. Измеряют угол оптического вращения раствора 3.
Сопутствующие примеси. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор (а). 0,20 г испытуемого об¬
разца растворяют в метаноле Р и доводят до объема
10 мл этим же растворителем.
Испытуемый раствор (Ь). 1 мл испытуемого
раствора (а) доводят метанолом Р до объема 10 мл.
Раствор сравнения (а). 20 мг ФСО эфедрина ги¬
дрохлорида рацемического растворяют в метаноле Р
и доводят до объема 10 мл этим же растворителем.
Раствор сравнения (Ь). 1 мл испытуемого раст¬
вора (а) доводят метанолом Р до объема 200 мл.
Пластинка: ТСХ пластинка со слоем силикагеля С Р.
Подвижная фаза: хлороформ Р — раствор
аммиака концентрированный Р — 2-пропанол Р
(5:15:80, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидрина Р и нагревают при температуре 110 °С в
течение 5 мин.
Предельное содержание примесей:
- любая примесь (не более 0,5 %): на хромато¬
грамме испытуемого раствора (а) любое пятно, кроме
основного, должно быть не интенсивнее пятна на хро¬
матограмме раствора сравнения (Ь);
- неучитываемый предел: на хроматограмме ис¬
пытуемого раствора (а) не учитывают пятна, которые
светлее фона.
Сульфаты (2.4.13). Не более 0,0100 % (100 ррт).
15 мл раствора 3 должны выдерживать испытание на
сульфаты.
Рисунок #0715.-1. Инфракрасный спектр ФСО эфедрина гидрохлорида рацемического
Эфир анестезирующий
1177
Потеря в массе при высушивании (2.2.32). Не
более 0,5 %. 1,000 г испытуемого образца сушат при
температуре 105 °С.
Сульфатная зола (2.4.14). Не более 0,1 %.
Определение проводят из 1,0 г испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,170 г испытуемого образца растворяют в 30 мл
96 % спирта Р, прибавляют 5,0 мл 0,01 М раствора
кислоты хлористоводородной и титруют 0,1 М рас¬
твором натрия гидроксида потенциометрически
(2.2.20). Отмечают объем титранта между двумя точ¬
ками перегиба на кривой титрования.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 20,17 мг С10Н151МО-НС1.
ХРАНЕНИЕ
В защищенном от света месте.
07/2016:0650
ЭФИР
АеОпег
ЕТНЕН
С4Н10О М.м. 74,1
[60-29-7]
ОПРЕДЕЛЕНИЕ
Диэтиловый эфир.
Может содержать соответствующий нелетучий
антиоксидант в соответствующей концентрации.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость, летучая.
Растворим в воде, смешивается с 96 % спиртом,
метиленхлоридом и жирными маслами.
Легко воспламеняется.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Температурные пределы перегонки», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Кислотность. К 20 мл 96 % спирта Р прибав¬
ляют 0,25 мл раствора бромтимолового синего Р1
и по каплям 0,02 М раствор натрия гидроксида до
окрашивания раствора в синий цвет, устойчивый в те¬
чение 30 с. Прибавляют 25 мл испытуемого образца,
встряхивают и прибавляют по каплям 0,02 М раствор
натрия гидроксида до повторного окрашивания раст¬
вора в синий цвет, устойчивый в течение 30 с. При
этом должно быть израсходовано не более 0,4 мл
0,02 М раствора натрия гидроксида.
Относительная плотность (2.2.5). От 0,714 до
0,716.
Температурные пределы перегонки (2.2.11).
От 34,0 °С до 35,0 °С. Испытание проводят при со¬
ответствии испытуемого образца требованиям
испытания «Пероксиды». Испытание проводят с ис¬
пользованием подходящего прибора для нагревания,
соблюдая меры предосторожности, чтобы избежать
прямого нагревания колбы выше уровня жидкости.
Альдегиды. 10,0 мл испытуемого образца по¬
мещают в цилиндр с притертой стеклянной пробкой,
прибавляют 1 мл щелочного раствора калия те-
трайодмеркурата Р, встряхивают в течение Юс и
выдерживают в течение 5 мин в защищенном от света
месте. Нижний опалесцирующий слой может иметь
желтый или красновато-коричневый цвет, но не серый
или черный.
Пероксиды. В цилиндр с притертой стеклянной
пробкой вместимостью 12 мл и диаметром около
15 мм помещают 8 мл раствора крахмала с калия
йодидом Р и заполняют испытуемым образцом до
объема 12 мл, перемешивают и выдерживают в за¬
щищенном от света месте в течение 5 мин. Не должно
появляться окрашивание.
Нелетучие вещества. Не более 20 мг/л. Испы¬
тание проводят при соответствии испытуемого
образца требованиям испытания «Пероксиды».
50 мл испытуемого образца выпаривают на водяной
бане досуха и высушивают при температуре от 100 °С
до 105 °С. Масса сухого остатка не должна превы¬
шать 1 мг.
Вещества с посторонним запахом. Бумажный
фильтр диаметром 80 мм смачивают 5 мл испытуемо¬
го образца и оставляют до полного испарения. Сразу
после испарения не должно обнаруживаться посто¬
ронних запахов.
Вода (2.5.12). Не более 2 г/л. Определение про¬
водят из 20 мл испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте #при температуре от 8 °С до
15 °С#.
#МАРКИРОВКА
Указывают название и концентрацию добавлен¬
ного нелетучего антиоксиданта.
07/2016:0367
ЭФИР АНЕСТЕЗИРУЮЩИЙ
АеМег апаезНюНсиз
ЕТНЕК, АМАЕЗТНЕТ1С
С4н1()0 М.м. 74,1
[60-29-7]
ОПРЕДЕЛЕНИЕ
Диэтиловый эфир.
Может содержать соответствующий нелетучий
антиоксидант в соответствующей концентрации.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость, летучая, легко
подвижная.
150. Зак. 1060.
1178
Государственная фармакопея Республики Беларусь
Растворим в 15 частях воды, смешивается с 96 %
спиртом и жирными маслами.
Легко воспламеняется.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Испытуемый образец выдерживает испытание
«Относительная плотность», как указано в разделе
«Испытания».
B. Испытуемый образец выдерживает испытание
«Температурные пределы перегонки», как указано в
разделе «Испытания».
ИСПЫТАНИЯ
Кислотность. К 20 мл 96 % спирта Р прибав¬
ляют 0,25 мл раствора бромтимолового синего Р1
и по каплям 0,02 М раствор натрия гидроксида до
окрашивания раствора в синий цвет, устойчивый в те¬
чение 30 с. Прибавляют 25 мл испытуемого образца,
встряхивают и прибавляют по каплям 0,02 М раствор
натрия гидроксида до повторного окрашивания раст¬
вора в синий цвет, устойчивый в течение 30 с. При
этом должно быть израсходовано не более 0,4 мл
0,02 М раствора натрия гидроксида.
Относительная плотность (2.2.5). От 0,714 до
0,716.
Температурные пределы перегонки (2.2.11).
От 34,0 °С до 35,0 °С. Испытание проводят при со-
ответствии испытуемого образца требованиям
испытания «Пероксиды». Испытание проводят с ис¬
пользованием подходящего прибора для нагревания,
соблюдая меры предосторожности, чтобы избежать
прямого нагревания колбы выше уровня жидкости.
Ацетон и альдегиды. 10,0 мл испытуемого об¬
разца помещают в цилиндр с притертой стеклянной
пробкой, прибавляют 1 мл щелочного раствора ка¬
лия тетрайодмеркурата Р, встряхивают в течение
10 с и выдерживают в течение 5 мин в защищенном от
света месте. Нижний слой должен приобрести только
легкую опалесценцию.
Если испытуемый образец не выдерживает дан¬
ное испытание, но соответствует требованиям ис¬
пытания «Пероксиды», то 40 мл испытуемого образ¬
ца перегоняют до тех пор, пока не останется только
5 мл. Дистиллят собирают в колбу-приемник, поме¬
щенную в ледяную баню. 10,0 мл полученного дис¬
тиллята должны выдерживать испытание на «Ацетон
и альдегиды».
Пероксиды. В цилиндр с притертой стеклянной
пробкой вместимостью 12 мл и диаметром около
15 мм помещают 8 мл раствора крахмала с калия йо¬
дидом Р и заполняют испытуемым образцом до объ¬
ема 12 мл, интенсивно встряхивают и выдерживают
в защищенном от света месте в течение 30 мин. Не
должно появляться окрашивание.
Нелетучие вещества. Не более 20 мг/л. Испы¬
тание проводят при соответствии испытуемого
образца требованиям испытания «Пероксиды».
50 мл испытуемого образца выпаривают на водяной
бане досуха и высушивают при температуре от 100 °С
до 105 °С. Масса сухого остатка не должна превы¬
шать 1 мг.
Вещества с посторонним запахом. Бумажный
фильтр диаметром 80 мм смачивают 5 мл испытуемо¬
го образца и оставляют до полного испарения. Сразу
после испарения не должно обнаруживаться посто¬
ронних запахов.
Вода (2.5.12). Не более 2 г/л. Определение про¬
водят из 20 мл испытуемого образца.
#Микробиологическая чистота (2.6.12, 2.6.13).
Испытуемый образец должен выдерживать требова¬
ния статьи 5.1.4.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защи¬
щенном от света месте #при температуре от 8 °С до
15 °С#. Содержимое частично наполненного контей¬
нера может быстро испортиться.
#МАРКИРОВКА
Указывают название и концентрацию добавлен¬
ного нелетучего антиоксиданта.
Аира корневища
1179
ЧАСТНЫЕ ФАРМАКОПЕЙНЫЕ
СТАТЬИ НА ЛЕКАРСТВЕННОЕ
РАСТИТЕЛЬНОЕ СЫРЬЕ
Частные фармакопейные статьи на лекар¬
ственное растительное сырье (ЛРС) приведены в
алфавитном порядке по названиям ЛРС на русском
языке. Название ЛРС на английском и латинском язы¬
ках приведено в соответствии с названием ЛРС Ев¬
ропейской Фармакопеи в единственном числе. ЛРС,
на которое отсутствует частная фармакопейная
статья Европейской Фармакопеи, обозначено знач¬
ком «*» около наименования на английском языке.
Статьи и/или отдельные испытания, полно¬
стью соответствующие требованиям Европейской
Фармакопеи, обозначены значком «(И)».
В частных статьях Фармакопеи приведены
требования к цельному лекарственному расти¬
тельному сырью, допускаемому к переработке, если
иного не указано в статье. Общие требования, в
том числе значения и допустимые нормы при сито¬
вом анализе, к измельченному ЛРС описаны в общей
статье «Сборы», если не указано иное в частных
фармакопейных статьях.
Степень измельчения ЛРС, необходимая для
проведения испытаний, указывается в скобках. На¬
пример, измельченное сырье с частицами, проходя¬
щими сквозь сито с размером стороны отверстия
355 мкм, обозначено как измельченное сырье (355).
07/2016:РБ0033
АИРА КОРНЕВИЩА
Асоп са1ат1 гЫгота
ЗШЕЕТ Я.АС КН120МЕ*
ОПРЕДЕЛЕНИЕ
Собранные осенью или ранней весной, отмытые
от земли, освобожденные от корней, остатков листьев
и стеблей, высушенные корневища многолетнего тра¬
вянистого растения Асогиз са/атиз 1_.
Содержание:
-цельное сырье: не менее 20 мл/кг эфирного
масла (в пересчете на сухое сырье);
- измельченное сырье: не менее 15 мл/кг эфир¬
ного масла (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корневища или
куски корневищ длиной до 30 см и толщиной до 2 см,
цилиндрические, слегка сплюснутые и изогнутые,
преимущественно продольно-разрезанные. На верх¬
ней стороне корневища видны треугольные широкие
рубцы от отмерших листьев, на нижней — многочис¬
ленные мелкие круглые коричневые ямки, следы уда¬
ленных корней, которые расположены зигзагообраз¬
но; излом неровный, зернистый. Снаружи корневища
желтовато-коричневого или красновато-коричневого
цвета, иногда с серовато-зеленым оттенком, на из¬
ломе — беловато-розовые, иногда желтоватые или
зеленоватые. На поперечном разрезе можно увидеть
в форме круга темную эндодерму, при увеличении —
мелкие точки. Запах сильный, ароматный.
B. Микроскопия (2.8.23). При просматрива¬
нии поперечного среза корневища видна покровная
ткань— эпидермис, состоящий из узких, вытянутых
вдоль корневища некрупных клеток. Основная ткань
состоит из округлых клеток паренхимы, которые от¬
делены друг от друга широкими межклеточными про¬
странствами (аэренхима). Аэренхима содержит или
мелкие крахмальные зерна ((2—8) мкм), или клетки
с коричневым содержимым, которое окрашивается в
красный цвет при обработке свежеприготовленным
раствором 20 г/л ванилина Р в кислоте хлористово¬
дородной Р. Несколько более крупные секреторные
клетки, чем клетки окружающей паренхимы, содержат
сильно просвечивающее желтоватое эфирное масло.
В коре находятся коллатеральные проводящие пучки
с волокнистой, кристаллоносной обкладкой. Эндодер¬
ма выражена не слишком четко, в центральном ци¬
линдре сосудистые пучки без волокнистых и кристал¬
лических обкладок.
C. 1,0 г измельченного сырья (355) (2.9.12) поме¬
щают в колбу вместимостью 50 мл, прибавляют 10 мл
96 % спирта Р и доводят до кипения на водяной
бане, охлаждают и фильтруют (раствор А). К 1,2 мл
раствора А прибавляют 2 мл спирта (90 %, об/об) Р
и 0,2 мл раствора железа (III) хлорида Р. Появляется
серовато-зеленое окрашивание.
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Используют раствор А,
приготовленный как указано выше.
Раствор сравнения. 10 мг тимола Р и 15 мг ане¬
тола Р растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: этилацетат Р — гексан Р
(10:90, об/об).
Наносимый объем пробы: 30 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 105 °С до 110 °С в течение (8—10) мин. Просматри¬
вают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается зона оранжевого цвета (тимол)
в средней части хроматограммы и зона фиолетового
цвета (анетол) в верхней трети хроматограммы. На
хроматограмме испытуемого раствора непосред¬
ственно ниже зоны тимола на хроматограмме раст¬
вора сравнения зона серого и две зоны фиолетового
цвета; на уровне зоны анетола — зоны фиолетового
цвета, ниже которых расположена зона розового цве¬
та. На хроматограмме испытуемого раствора могут
обнаруживаться и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корневища, побуревшие в изломе, —
не более 5 %; корневища, плохо очищенные от кор¬
ней и остатков листьев, — не более 5 %. Органиче¬
ские примеси: не более 1 %. Минеральные примеси:
не более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 6,0 %.
150*. Зак. 1060.
1180
Государственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод В или О). Ис¬
пользуют 10,0 г измельченного сырья (1400) (2.9.12).
Перегонку проводят в течение 90 мин.
07/2016:1126
АЛТЕЯ КОРНИ
АШнаеае гасНх
МАР8НМАИОШ НООТ
ОПРЕДЕЛЕНИЕ
Собранные осенью или весной, тщательно
очищенные от земли и пробкового слоя и высу¬
шенные боковые и неодревесневшие стержневые
корни многолетних травянистых растений АННаеа
оШапаНз 1_. и АИЬаеа агтеп/аса Теп.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корни, очи¬
щенные от пробки, почти цилиндрической формы
или расщепленные вдоль на 2—4 части, слегка
суживающиеся к концу, длиной (10—35). см и тол¬
щиной до 2 см. Поверхность корня продольно-бо¬
роздчатая с отслаивающимися длинными, мягкими
лубяными волокнами и темными точками — следа¬
ми отпавших или отрезанных тонких корней. Излом
в центре зернисто-шероховатый, снаружи волокни¬
стый. Цвет корня снаружи и в изломе белый, жел¬
товато-белый (АННаеа оТПстаИз) или сероватый
(АИЬаеа агтетаса). Запах слабый, своеобразный.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.12). Порошок имеет
серовато-коричневый (неочищенный корень) или
беловатый (очищенный корень) цвет. Просматри¬
вают под микроскопом с использованием раст¬
вора хлоралгидрата Р. В порошке обнаруживают¬
ся следующие диагностические признаки (рисунок
1126.-1): фрагменты бесцветных, преимуществен¬
но неодревесневших, толстостенных волокон [С,
О, М] с расщепленными или остроконечными кон¬
цами [О], иногда с прилегающими паренхиматозны¬
ми клетками лучей сердцевины [М], или сгруппи¬
рованные фрагменты волокон [С]; фрагменты со¬
судов, окантованных порами, или с сетчатыми или
лестничными утолщениями [О, Н]; друзы кальция
оксалата размером около (20—35) мкм, преиму¬
щественно (25—30) мкм, расположенные отдельно
[К] или включенные в паренхиматозные клетки [В];
фрагменты паренхимы [Е] с клетками, содержащи¬
ми слизь [Еа, Р]; фрагменты корки с тонкостенны¬
ми, слоистыми клетками (вид с поверхности [А] и
вид в поперечном срезе [1_]) (неочищенный корень).
Просматривают под микроскопом с использовани¬
ем раствора рутения красного Р. В порошке об¬
наруживаются группы клеток паренхимы, содер¬
жащей слизь, окрашивающей в оранжево-красный
цвет. Просматривают под микроскопом с использо¬
ванием воды Р. В порошке обнаруживаются мно¬
гочисленные зерна крахмала [3], размером около
(3—25) мкм, иногда с продольными ядрами. Зерна
крахмала преимущественно одинарные ^а], неко¬
торые из них 2—4-составные [ЗЬ].
Рисунок 1126.-1. Микроскопические признаки
корней алтея
С. Срез корня или измельченное сырье (355)
(2.9.12) смачивают раствором аммиака разведен¬
ным Р1 или раствором натрия гидроксида разведен¬
ным Р. Появляется желтое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: деревянистые корни — не более 3 %;
корни, плохо очищенные от пробки, — не более 3 %.
Органические примеси: не более 0,5 %. Минеральные
примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
(§) Коэффициент набухания (2.8.4). Не менее
10. Используют измельченное сырье (710) (2.9.12).
07/2016: РБ0034
АРАЛИИ МАНЬЧЖУРСКОЙ КОРНИ
АгаНае тапдвЬипсае гасНх
МРАЫЕЗЕ АЫвЕИСА ТРЕЕ НО ОТ*
ОПРЕДЕЛЕНИЕ
Собранные весной или поздней осенью, тща¬
тельно очищенные от земли, разрубленные на куски
и высушенные корни дерева АгаНа е1а1а (М|ц.) Веет.
(А. тапдзЬипса Рирг. е1 Мах1т.).
Содержание: не менее 5,0 % суммы аралозидов
в пересчете на аммонийную соль аралозидов А, В и С
с усредненной молекулярной массой (в пересчете на
сухое сырье).
Арники цветки
1181
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
продольно-расщепленные куски корней длиной
до 8 см и диаметром до 3 см, с немногочислен¬
ными мелкими боковыми корнями. Корни легкие,
продольно-морщинистые, с сильно шелушащейся
пробкой. Кора тонкая, легко отделяется от древе¬
сины. Излом корня занозистый. Цвет корней сна¬
ружи коричневато-серый, на изломе беловато- или
желтовато-серый. Запах ароматный.
B. Микроскопия (2.8.23). На поперечном сре¬
зе корня виден слой сильно шелушащейся пробки.
Кора состоит из клеток паренхимы с тонкими стен¬
ками, среди которых концентрическими поясами
расположены секреторные каналы диаметром от
7 м«м до 20 мкм. Паренхимные клетки вокруг се¬
креторных каналов и клетки сердцевинных лучей
заполнены крахмальными зернами. Крахмальные
зерна простые и 2—8-сложные. В наружной части
коры встречаются друзы оксалата кальция. Кора
отделяется от древесины узким слоем камбия.
Древесина кольцесосудистая. Сердцевинные лучи
одно-, пятирядные.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (5600) (2.9.12) прибавляют 20 мл метано¬
ла Р и кипятят с обратным холодильником в водя¬
ной бане при температуре от 80 °С до 85 °С в тече¬
ние 1 ч. Охлаждают и центрифугируют. Собирают
надосадочную жидкость.
Раствор сравнения. 60 мг сапарала Р раство¬
ряют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силика¬
геля С Р.
Подвижная фаза: хлороформ Р — мета¬
нол Р — вода Р (61:32:7, об/об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде по¬
лос.
Фронт подвижной фазы: не менее 18 см от
линии старта.
Высушивание: на воздухе в течение 10 мин.
Проявление: пластинку опрыскивают раство¬
ром 200 г/л кислоты серной Р и нагревают при
температуре 105 °С в течение 10 мин. Просматри¬
вают при дневном свете.
Результаты: на хроматограмме раствора
сравнения обнаруживаются три основные зоны
темно-красного цвета (аралозиды). На хромато¬
грамме испытуемого раствора обнаруживаются
зоны, соответствующие по расположению и цвету
основным зонам на хроматограмме раствора срав¬
нения. На хроматограмме испытуемого раствора
могут обнаруживаться и другие зоны темно-крас¬
ного или другого цвета.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: куски корней длиной более 8 см — не
более 15 %; куски корней диаметром более 3 см — не
более 15 %; корни, почерневшие в изломе, — не бо¬
лее 4 %. Органические примеси: не более 1 %. Мине¬
ральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 7,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,000 г измельченного сырья (710) (2.9.12) по¬
мещают в патрон из фильтровальной бумаги и опу¬
скают в экстрактор аппарата Сокслета с рабочим
объемом (150—200) мл. В колбу-приемник прибав¬
ляют 250 мл метанола Р, 70 мл раствора 500 г/л
кислоты серной Р и экстрагируют в водяной бане
в течение 7 ч. К полученному в колбе-приемнике
раствору прибавляют равное количество воды Р
и охлаждают под струей холодной воды в течение
10 мин. Выпавший осадок отфильтровывают через
стеклянный фильтр (ПОР 16) диаметром не менее
50 мм. Первую порцию фильтруют без вакуума, за¬
тем, когда отделение фильтрата почти прекратится,
фильтруют оставшуюся часть под вакуумом. Осадок
на фильтре промывают водой Р (около 1000 мл), пе¬
ремешивая его 2—3 раза, до значения рН использу¬
емой воды Р и затем подсушивают, не выключая ва¬
куума. Осадок количественно переносят с помощью
50 мл горячей смеси метанол Р — 2-бутанол Р1
(1:1,5, об/об) в стакан вместимостью 100 мл.
Титруют потенциометрически (2.2.20) 0,1 М
раствором натрия гидроксида в смеси из мета¬
нола и бензола с использованием стеклянного ин¬
дикаторного электрода. При титровании отмечают
количество титранта, израсходованного на доведе¬
ние рН испытуемого раствора до 7,0.
Параллельно проводят контрольный опыт.
Содержание суммы аралозидов в пересчете
на аммонийную соль аралозидов А, В и С с усред¬
ненной молекулярной массой в процентах рассчи¬
тывают по формуле:
0,10422 - (У - У, - У2) • 100
т
где:
0,10422 — количество аммонийных солей арало¬
зидов, соответствующее 1 мл 0,1 М раствора гидрок¬
сида натрия в смеси из метанола и бензола, г;
V— объем титранта, израсходованного на титро¬
вание испытуемого раствора, мл;
V, — объем титранта, израсходованного на до¬
ведение рН испытуемого раствора до 7,0, мл;
\/2 — объем титранта, израсходованного в кон¬
трольном опыте, мл;
т — масса навески испытуемого сырья, г.
07/2016:1391
АРНИКИ ЦВЕТКИ
Ат ‘\сае Лоз
АКЫ1СА Р1-01/УЕЯ
ОПРЕДЕЛЕНИЕ
Собранные в начале цветения и высушенные
цветки многолетнего травянистого растения Агтса
топ1апа 1_., Агтса IоНоза ЫиИ. и Агтса 8сЬат18зотз
Ьесе.
Содержание: не менее 0,40 % (м/м) суммы се-
сквитерпеновых лактонов в пересчете на дигидроге-
леналина тиглат (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Сырье пред¬
ставляет собой отдельные краевые ложноязыч-
1182
Государственная фармакопея Республики Беларусь
ковые и трубчатые цветки, семянки с хохолком,
цветоложа распавшихся соцветий, реже цель¬
ные корзинки. Ложноязычковые цветки длиной до
2,5 см с трехзубчатым отгибом, трубчатые — дли¬
ной до 1,5 см пятичленные; окраска цветков от
оранжево-желтой до светло-оранжево-желтой.
Цветоложе соцветия слегка выпуклое, ямчатое, с
короткими щетинистыми волосками вокруг ямок.
Корзинки диаметром (2—6) см (с краевыми цветка¬
ми) и (1,2—3,2) см (без краевых цветков) с остат¬
ками цветоносов длиной до 3 см или без них. Се¬
мянки продолговатые светло-желто-коричневого
цвета с однорядным хохолком из желтоватых, не¬
ветвистых, тонких щетинок длиной до 1 см. Запах
слабый, ароматный.
(§*)В. Микроскопия (2.8.23). Разделяют голов¬
ку на различные части. Исследуют измельченное
сырье (355) (2.9.12). Просматривают под микро¬
скопом с использованием раствора хлоралги¬
драта Р. В порошке обнаруживаются следующие
диагностические признаки (рисунок 1391.-1): эпи¬
дермис прицветника обертки соцветия [Ц М, О, О]
имеет устьица [1_Ь, Оа, Оа] и трихомы, наиболее
многочисленные на внешней (абаксиальной) по¬
верхности. Обнаруживается несколько различных
типов трихом: однорядные многоклеточные покры¬
вающие трихомы, особенно многочисленные по
краям прицветника, длина которых варьирует от
50 мкм до 500 мкм, целые [1_а] или фрагментиро¬
ванные [Р]; секреторные трихомы с одно- и двух¬
рядными многоклеточными ножками и многокле¬
точными сферическим головками, длиной около
300 мкм, многочисленные на внешней поверхности
прицветника [ОЬ]; секреторные трихомы с много¬
клеточными ножками и многоклеточными сфериче¬
ским головками, длиной около 80 мкм, многочис¬
ленные на внутренней поверхности прицветника
(вид с поверхности [ОЬ] или вид сбоку [Ма]). Эпи¬
дермис язычкового венчика [С, О, Н, ^ состоит из
дольчатых или продолговатых клеток, покрытых
бороздчатой кутикулой [Оа], и нескольких устьиц
и трихом различного типа: покрывающие трихомы
с очень острыми концами, длина которых может
превышать 500 мкм, состоящие из 1—3 прокси¬
мальных толстостенных клеток и 2—4 дистальных
тонкостенных клеток [С, НЬ]; секреторные трихомы
с двухрядными многоклеточными головками (вид с
поверхности [<ЗЬ] или вид сбоку ^а]); секреторные
трихомы с многоклеточными ножками и многокле¬
точными сферическим головками [К]. Язычок за¬
канчивается в сферических сосочковидных клетках
[На]. Фрагменты эпидермиса завязи [А, В, Э] по¬
крыты трихомами двух типов: секреторными три¬
хомами с короткими ножками и многоклеточными
сферическими головками (вид с поверхности [Аа]
или вид сбоку [Эа]); сдвоенными покрывающими
трихомами, обычно состоящими из двух объеди¬
ненных в продольном направлении клеток, с общи¬
ми ямчатыми стенками (вид с поверхности [АЬ] или
вид сбоку [Ва]); их концы острые и иногда расще¬
пленные. Эпидермис чашечки состоит из продол¬
говатых клеток, имеющих короткие одноклеточные
покрывающие трихомы, направленные к верхнему
концу щетины [Е]. Зерна пыльцы диаметром около
30 мкм имеют сферическую форму, колючую экзи-
ну и 3 зародышевые поры [Р, N1].
Рисунок 1391.-1. Микроскопические признаки
цветков арники
®С. Просматривают хроматограмму, получен¬
ную как указано в испытании «Са1епди1а оЛ1апаНз I. —
Не1его1Ьеса юи1о1с1ез Сазз».
Результаты: на хроматограмме испытуемого
раствора в средней части обнаруживается флуо¬
ресцирующая зона синего цвета, соответствующая
Арники цветки
1183
зоне хлорогеновой кислоты на хроматограмме
раствора сравнения; над этой зоной — три флуо¬
ресцирующие зоны желтовато-коричневого или
оранжево-желтого цвета; над этими тремя зона¬
ми — флуоресцирующая зона зеленовато-желтого
цвета, соответствующая астрагалину. Зона, рас¬
положенная ниже зоны астрагалина, соответству¬
ет изокверцитрозиду; зона, расположенная непо¬
средственно под ней, соответствует лютеолин-7-
глюкозиду. На хроматограмме испытуемого раст¬
вора также обнаруживается флуоресцирующая
зона зеленовато-синего цвета, расположенная
ниже зоны кофейной кислоты на хроматограмме
раствора сравнения.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: частицы, проходящие сквозь сито (2000)
(2.9.12) , — не более 6 %. Органические примеси: не
более 2 %. Минеральные примеси: не более 1 %.
® Са/епс/и/а оТПстаПз Ь. — Не(его1Неса
ти1оШез Са$$. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. К 2,00 г измельченного
сырья (710) (2.9.12) прибавляют 10 мл метанола Р,
нагревают при перемешивании в водяной бане при
60 °С в течение 5 мин, охлаждают и фильтруют.
Раствор сравнения. 2,0 мг кофейной кисло¬
ты Р, 2,0 мг хлорогеновой кислоты Р и 5,0 мг рути¬
на Р растворяют в метаноле Р и доводят до объема
30 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безвод¬
ная Р — вода Р — метилэтилкетон Р — этилаце-
тат Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: по 15 мкл в виде по¬
лос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе в течение нескольких
минут.
Проявление: пластинку опрыскивают раствором
10 г/л дифенилборной кислоты аминоэтилового
эфира Р в метаноле Р и затем раствором 50 г/л ма-
крогола 400 Р в метаноле Р. Нагревают при темпера¬
туре от 100 °С до 105 °С в течение 5 мин. Высушива¬
ют на воздухе и просматривают в ультрафиолетовом
свете при 365 нм.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается в нижней части флуоресциру¬
ющая зона оранжево-желтого цвета (рутин), в сред¬
ней части — флуоресцирующая зона (хлорогеновая
кислота), в верхней части — флуоресцирующая зона
светло-синеватого цвета (кофейная кислота). На хро¬
матограмме испытуемого раствора не должны обна¬
руживаться ни флуоресцирующая зона оранжево¬
желтого цвета, соответствующая рутину на хромато¬
грамме раствора сравнения, ни зона, расположенная
ниже зоны, соответствующей рутину.
® Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 9,0% (Агпюа
топ1апа 1_., Агпюа ТоНова Ыий.) и не более 12,0%
(Атюа зс11ат1880П181_езз.).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Жидкостная хроматография (2.2.29).
Раствор внутреннего стандарта. Непосред¬
ственно перед использованием 0,010 г ФСО сан¬
тонина растворяют в 10,0 мл метанола Р.
Испытуемый раствор. 1,00 г измельченно¬
го сырья (355) (2.9.12) помещают в круглодонную
колбу вместимостью 250 мл и прибавляют 50 мл
смеси из равных объемов метанола Р и воды Р.
Нагревают с обратным холодильником в водяной
бане при температуре от 50 °С до 60 °С при ча¬
стом встряхивании в течение 30 мин, охлаждают
и фильтруют через бумажный фильтр. Помещают
бумажный фильтр, порезанный на кусочки, в кру¬
глодонную колбу к остатком, прибавляют 50 мл
смеси из равных объемов метанола Р и воды Р и
нагревают с обратным холодильником в водяной
бане при температуре от 50 °С до 60 °С при ча¬
стом встряхивании в течение 30 мин. Экстракцию
повторяют дважды. К полученному фильтрату при¬
бавляют 3,00 мл раствора внутреннего стандарта
и упаривают до 18 мл при пониженном давлении.
Ополаскивают круглодонную колбу водой Р и до¬
водят до объема 20,0 мл этим же растворителем.
Переносят раствор в хроматографическую колонку
длиной около 0,15 м и внутренним диаметром око¬
ло 30 мм, заполненную 15 г кизельгура для хро¬
матографии Р. Выдерживают в течение 20 мин,
промывают 200 мл смеси из равных объемов эти-
лацетата Р и метиленхлорида Р. Элюат выпари¬
вают досуха в круглодонной колбе вместимостью
250 мл. Остаток растворяют в 10,0 мл метанола Р
и прибавляют 10,0 мл воды Р. Прибавляют 7,0 г
нейтрального оксида алюминия Р, встряхивают в
течение 120 с, центрифугируют при 5000 д в тече¬
ние 10 мин и фильтруют через бумажный фильтр.
10,0 мл фильтрата выпаривают досуха. Остаток
растворяют в 3,0 мл смеси из равных объемов ме¬
танола Р и воды Р и фильтруют.
Условия хроматографирования:
- колонка длиной 0,12 м и внутренним диа¬
метром 4 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 4 мкм;
- подвижная фаза:
- подвижная фаза А: вода Р\
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—3
62
38
3—20
62 — 55
38 — 45
20—30
55
45
30—55
55 — 45
45—55
55—57
45— 0
55—100
57—70
0
100
70—90
62
38
- скорость подвижной фазы: 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 225 нм;
- объем вводимой пробы: 20 мкл.
Содержание суммы сесквитерпеновых лактонов
в пересчете на дигидрогеленалина тиглат в процентах
рассчитывают по формуле:
1184
Гэсударственная фармакопея Республики Беларусь
5^-С-У •1,187-100
/77 -1000
где:
— площадь всех пиков, соответствующих
сесквитерпеновым лактонам, элюирующихся после
пика сантонина на хроматограмме испытуемого
раствора;
— площадь пика сантонина на хроматограм¬
ме испытуемого раствора;
/л — масса навески испытуемого сырья, г;
С — концентрация сантонина в растворе вну¬
треннего стандарта, мг/мл;
V — объем раствора внутреннего стандарта,
используемый для получения испытуемого раст¬
вора, мл;
1,187 — коэффициент пропорциональности
для пиков дигидрогеленалина тиглата и сантонина.
07/2016:РБ0035
БАГУЛЬНИКА БОЛОТНОГО ПОБЕГИ
/.ес//' ра1изМз согтиз
СКУ8ТА1. ТЕА 1-ЕйШЛ*
ОПРЕДЕЛЕНИЕ
Собранные в фазу созревания плодов и высу¬
шенные олиственные побеги текущего года вечно¬
зеленого кустарника 1.ес1ит ра1и8(ге 1_.
Содержание: не менее 1 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь оли-
ственных побегов, листьев и небольшого количе¬
ства плодов. Листья очередные, на коротких че¬
решках, кожистые, линейно-продолговатые или
продолговато-эллиптические, цельнокрайние, дли¬
ной (15—45) мм, шириной (1—5) мм, с завернуты¬
ми вниз краями; с верхней стороны темно-зеленые,
блестящие; с нижней стороны покрыты густым
оранжево-коричневым войлочным опушением.
Стебли цилиндрические с оранжево-коричневым
войлочным опушением. Плод — многосемянная
продолговатая коробочка длиной (3—8) мм, желе¬
зисто-опушенная, раскрывающаяся при созрева¬
нии снизу вверх пятью створками. Запах резкий,
специфический.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны (рисунок РБ0035.-1):
клетки эпидермиса с обеих сторон листа — мелкие
с тонкими или четковидно-утолщенными стенками,
над жилками — с прямыми [А]. Устьица только на
нижней стороне, с 4—8 околоустьичными клетками
(аномоцитный тип). Верхняя сторона листа покрыта
толстой кутикулой; волоски встречаются редко. Ниж¬
няя сторона густо опушена волосками трех типов:
длинными, многоклеточными лентовидными, изви¬
листыми и перекрученными волосками, состоящими
из двух рядов клеток, с красно-коричневым содер¬
жимым [В1]; мелкими одноклеточными волосками с
толстой оболочкой, покрытой бородавчатой кутику¬
лой [В2]; головчатыми волосками на одно- или много¬
клеточной ножке с многоклеточной круглой головкой,
содержащей маслянистые капли [ВЗ]. Эфиромаслич¬
ные железки встречаются на обеих сторонах листа,
но больше на нижней; они состоят из крупной округ¬
ло-приплюснутой головки, образованной клетками
двух типов: 6—10 мелких округлых клеток, располо¬
женных у основания железки, и 10—12 крупных, поч¬
ти плоских клеток, образующих купол над первыми;
ножка железки короткая, двухрядная, из нескольких
мелких клеток [С]. Мезофилл листа характеризует¬
ся ярко выраженной аэренхимой и содержит друзы
оксалата кальция, реже одиночные призматические
кристаллы и их сростки [О].
Рисунок РБ0035.-1. Микроскопические признаки по¬
бегов багульника болотного
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 20 мкл масла, получен¬
ного при количественном определении, прибавляют
1 мл толуола Р.
Раствор сравнения. 5 мг тимола Р и 10 мг мен¬
тола Р растворяют в 10 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 15 мкл испытуемого
раствора и 20 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 13 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р, нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин. Просма¬
тривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживаются синяя зона (ментол) в нижней
части и над ней розовая зона (тимол). На хромато-
Барбариса обыкновенного корни
1185
грамме испытуемого раствора обнаруживается зона
от фиолетового до красновато-фиолетового цвета не¬
много выше пятна ментола на хроматограмме раст¬
вора сравнения (ледол), а также зона от фиолетового
до красновато-фиолетового цвета немного выше пят¬
на тимола на хроматограмме раствора сравнения (па-
люстрол). На хроматограмме испытуемого раствора
могут обнаруживаться и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: серовато-коричневые стебли — не бо¬
лее 10 %. Органические примеси: не более 1 %. Ми¬
неральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 4,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют содержание эфирного масла (2.8.12,
метод С). 30,0 г измельченного сырья (2000) (2.9.12)
помещают в круглодонную колбу вместимостью
1000 мл и прибавляют 400 мл воды Р. Время перегон¬
ки — 4 ч, после чего охлаждение холодильника пре¬
кращают, чтобы закристаллизовавшаяся часть эфир¬
ного масла на стенках холодильника расплавилась и
опустилась в приемник. Затем немедленно включают
подачу воды в холодильник во избежание нагревания
приемника.
07/2016:РБ0036
БАДАНА КОРНЕВИЩА
Вегдеп/ае сгаззЯоНае гЫгота
1.ЕАТНЕК ВЕКвЕША КООТ*
ОПРЕДЕЛЕНИЕ
Собранные в июне—июле, освобожденные от
земли, корней и надземных частей, разрезанные на
куски и высушенные корневища многолетнего травя¬
нистого растения Вегдета сгаззНоНа (1_.) РгКзсЬ.
Содержание: не менее 20,0 % дубильных ве¬
ществ в пересчете на танин (в пересчете на сухое
сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Куски корневищ
цилиндрической формы длиной до 20 см, толщиной
от 1 см до 3,5 см, имеющие на поверхности чешуе¬
видные остатки черешков листьев и округлые следы
корней. Цвет корневища и чешуек, покрывающих кор¬
невище, темно-коричневый или почти черный. На из¬
ломе корневище зернистое, светло-розовое или свет¬
ло-коричневое. Запах отсутствует.
B. Микроскопия (2.8.23). При просматрива¬
нии поперечного среза видно, что корневище имеет
пучковый тип строения. Покровная ткань состоит из
4—5 рядов клеток пробки. Проводящие пучки откры¬
тые коллатеральные, расположены кольцом. Парен¬
хима коры, сердцевинных лучей и сердцевины со¬
стоит из крупных тонкостенных клеток, заполненных
крахмальными зернами и друзами оксалата кальция.
Крахмальные зерна простые, округлые, диаметром от
7 мкм до 25 мкм.
С. Срез корневища смачивают раствором 10 г/л
железа (III) аммония сульфата Р или железа (III) хло¬
рида Р. Появляется черно-синее окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни, надземные части, в том числе
отделенные при анализе, — не более 1 %. Органиче¬
ские примеси: не более 1 %. Минеральные примеси:
не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 4,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г измельченного сырья (2000) (2.9.12) поме¬
щают в коническую колбу со шлифом вместимостью
500 мл, прибавляют 250 мл кипящей воды Р и кипя¬
тят с обратным холодильником в течение 30 мин при
периодическом перемешивании. Охлаждают до ком¬
натной температуры, процеживают и доводят водой Р
до объема 250,0 мл. 25,0 мл полученного извлечения
помещают в коническую колбу вместимостью 750 мл,
прибавляют 500 мл воды Р, 25,0 мл раствора индиго-
сульфокислоты (0,25 мг индигокармина Р растворяют
в 6,5 мл кислоты серной Р, прибавляют 6,5 мл кис¬
лоты серной Р, доводят водой Р до объема 250 мл и
фильтруют). Титруют при постоянном перемешивании
0,02 М раствором калия перманганата до желтого
окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07/2016:РБ0037
БАРБАРИСА ОБЫКНОВЕННОГО
КОРНИ
ВегЬепсИз уи/дапз гасИх
ВАКВЕККУ КООТ*
ОПРЕДЕЛЕНИЕ
Собранные ранней весной (до начала распуска¬
ния почек) или осенью (после созревания плодов),
тщательно очищенные от земли и высушенные корни
ВегЬепз VиIда^^з 1_.
Содержание: не менее 0,6 % берберина в пере¬
счете на берберина бисульфат (С20Н181ЧО4-Н5О4, М.м.
433,4) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Куски деревяни¬
стых корней длиной от 2 см до 20 см, толщиной до
6 см, цельные, почти цилиндрические, прямые или
изогнутые, часто разветвленные, продольно-морщини¬
стые, встречаются куски корней расщепленные вдоль;
излом грубоволокнистый. Цвет корней снаружи серо¬
вато-коричневый или темно-коричневый, на изломе зе¬
леновато-желтый. Запах слабый, своеобразный.
1186
Гэсударственная фармакопея Республики Беларусь
B. Микроскопия (2.8.23). На поперечном срезе
видны: узкая кора и широкая древесина; лубяные
одревесневшие волокна расположены группами или
встречаются одиночно. Вблизи сердцевинных лучей
и в лучах встречаются одиночно или расположенные
группами овальные или четырехугольные камени¬
стые клетки. В клетках сердцевинных лучей встреча¬
ются одиночные призматические кристаллы оксалата
кальция.
C. Срез корня смачивают кислотой азотной Р.
Появляется красновато-коричневое окрашивание.
О. Срез корня смачивают кислотой серной Р.
Появляется оранжево-красное окрашивание, которое
при нагревании переходит в серовато-зеленое.
Е. Срез корня смачивают раствором водорода
пероксида разведенным Р. Появляется фиолетовое
окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни, почерневшие в изломе, — не
более 5 %. Органические примеси: не более 1 %. Ми¬
неральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (710) (2.9.12) по¬
мещают в колбу вместимостью 25 мл, прибавляют
10,0 мл 96 % спирта Р, взвешивают с точностью до
0,01 г и нагревают с обратным холодильником на во¬
дяной бане при температуре от 90 °С до 95 °С в тече¬
ние 30 мин. Охлаждают и, если необходимо, доводят
до первоначальной массы 96 % спиртом Р. Отста¬
ивают до оседания частиц, при необходимости ис¬
пользуют центрифугирование. (0,05—0,10) мл надо-
садочной жидкости (Уг мл) наносят на линию старта
ТСХ пластинки со слоем подходящего силикагеля Р.
На ту же пластинку в качестве свидетеля наносят
0,02 мл раствора 5 г/л берберина бисульфата Р в
96 % спирте Р Пластинку с нанесенными проба¬
ми высушивают на воздухе и хроматографируют с
использованием подвижной фазы хлороформ Р —
96 % спирт Р — раствор аммиака концентриро¬
ванный Р (3:3:1, об/об/об). Когда фронт подвижной
фазы пройдет (10—14) см от линии старта, пластин¬
ку вынимают из камеры и сушат на воздухе в течение
(1-2)ч.
Испытуемый раствор. На хроматограмме ис¬
пытуемого раствора участок сорбента с пятном,
расположенным на уровне свидетеля берберина и
соответствующий ему по величине и характеру
флуоресценции в ультрафиолетовом свете, снима¬
ют скальпелем, элюируют 0,05 М раствором серной
кислоты (\/2, мл) при нагревании на водяной бане в
течение (5—10) мин. Охлаждают и центрифугируют
при (5000—10 000) об/мин в течение (15—20) мин.
Используют надосадочную жидкость.
Компенсационный раствор. Готовят аналогично
испытуемому раствору, используя участок сорбента с
чистого участка той же пластинки такой же площади.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора при 345 нм.
Содержание берберина в пересчете на бербери¬
на бисульфат в процентах рассчитывают по формуле:
А Л/2 10
^•^ 646 ’
где:
646 — удельный показатель поглощения бербе¬
рина бисульфата;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0038
БАРБАРИСА ОБЫКНОВЕННОГО
ЛИСТЬЯ
ВегЬепсИз VиIда^^з Ышт
ВАПВЕГЖУ 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в фазу бутонизации и цветения и вы¬
сушенные листья кустарника ВегЬепз уи1дапз 1_.
Содержание: не менее 0,15 % суммы алкалоидов
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные ли¬
стья длиной (2—7) см и шириной (1—4) см, с кли¬
новидным основанием и округлой верхушкой, тон¬
кие, с обеих сторон покрытые восковым налетом;
по краю мелкопильчатые, зубцы листа вытянуты
в мягкую иголочку. Жилкование перисто-сетчатое,
главная жилка слегка напоминает ломаную линию.
Черешок голый, желобчатый, в верхней части слег¬
ка крылатый. Цвет листьев с верхней стороны тем¬
но-зеленый, матовый, с нижней — светлый. Запах
своеобразный.
B. Микроскопия (2.8.23). При рассмотрении
листа с поверхности у молодых тонких листьев
клетки эпидермиса сильно извилистые. У старых
кожистых листьев эпидермис верхней и нижней сто¬
рон имеет четковидно-утолщенные стенки клеток.
Клетки эпидермиса по краю листа, особенно над
зубчиками, отличаются более мелкими размерами и
довольно толстыми стенками, по краю зубчика они
образуют пирамидальные выросты. Устьица аномо-
цитного типа, располагаются только на нижнем эпи¬
дермисе. Волоски и кристаллы отсутствуют.
C. К 0,5 г измельченного сырья (355) (2.9.12)
прибавляют 5 мл кислоты уксусной разведен¬
ной Р, нагревают при перемешивании и фильтруют.
К фильтрату прибавляют раствор 10 г/л кислоты
кремневольфрамовой Р Появляется муть, перехо¬
дящая в хлопьевидный осадок желтовато-зеленого
цвета.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: частицы сырья, проходящие сквозь сито
(2000) (2.9.12), — не более 5 %; листья, утратившие
нормальную окраску, — не более 4 %; другие части
растения — не более 2 %. Органические примеси: не
более 2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
Бегонии листья
1187
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,00 г измельченного сырья (710) (2.9.12) по¬
мещают в колбу вместимостью 250 мл, прибавляют
150 мл эфира Р, 7 мл раствора аммиака разведенно¬
го Р1 и встряхивают в течение 1 ч. Эфирное извле¬
чение быстро процеживают через ватный тампон в
колбу вместимостью 200 мл, прикрывая воронку часо¬
вым стеклом. К фильтрату прибавляют 5 мл воды Р,
встряхивают и оставляют до просветления эфирного
слоя. 90 мл эфирного извлечения с помощью мерного
цилиндра помещают в делительную воронку вмести¬
мостью 200 мл. Мерный цилиндр дважды ополаскива¬
ют эфиром Р порциями по 10 мл, которые присоеди¬
няют к отмеренному эфирному извлечению. Эфирное
извлечение встряхивают с раствором 10 г/л кислоты
хлористоводородной Р последовательно порциями
по 20 мл, 15 мл и 10 мл до полного извлечения ал¬
калоидов (проба с реактивом Майера Р), каждый
раз фильтруя через смоченный водой Р бумажный
фильтр диаметром 5 см во вторую делительную во¬
ронку вместимостью 200 мл. Фильтр промывают
дважды раствором 10 г/л кислоты хлористоводо¬
родной Р порциями по 5 мл, присоединяя промывную
жидкость к общему кислотному извлечению. Кислот¬
ное извлечение подщелачивают раствором аммиака
разведенным Р до щелочной реакции по раствору
фенолфталеина Р и трижды встряхивают по 3 мин с
хлороформом Р последовательно порциями по 20 мл,
15 мл и 10 мл. Хлороформные извлечения фильтруют
в предварительно высушенную при температуре от
90 °С до 100 °С и взвешенную с точностью до 0,001 г
круглодонную колбу вместимостью 100 мл через бу¬
мажный фильтр с (4—5) г натрия сульфата безво¬
дного Р, смоченного хлороформом Р. Фильтр дважды
промывают хлороформом Р порциями по 5 мл. Хло¬
роформ выпаривают досуха на водяной бане в ваку¬
уме. Полученный остаток сушат при температуре от
90 °С до 100 °С в течение (30—45) мин, охлаждают и
взвешивают.
Содержание суммы алкалоидов в процентах рас¬
считывают по формуле:
а-100
т
где:
а — масса сухого остатка, г;
т — масса сырья, соответствующая отмеренно¬
му объему эфирного извлечения, г.
07/2016: РБ0039
БЕГОНИИ ЛИСТЬЯ
Ведома IЪНит
Е1.ЕРАНГ8-ЕАК 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в фазу цветения, высушенные листья
многолетнего растения Ведома егу1горЬуИа Ыеит.
Содержание: не менее 0,1 % суммы антоцианов
в пересчете на цианидин-3,5-дигликозид (в пересчете
на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Листовая пла¬
стинка цельная, широкоовальная, с сердцевидным
основанием, со слегка волнистым расставлено-зуб-
чатым краем. Длина листовой пластинки (9—10) см,
ширина — (12—15) см. Верхняя сторона листа темно¬
зеленая, блестящая, голая; нижняя — темно-красная
с зелеными жилками, покрыта тонкими красными во¬
лосками. Жилки без опушения, с красными штрихами.
Запах слабый.
B. Микроскопия (2.8.23). Верхняя часть эпидер¬
мы листа без устьиц, образована относительно мел¬
кими бесцветными паренхимными 4—9- (чаще 4—5-)
угольными клетками с прямыми стенками. Клетки
верхней эпидермы уплощены. Под эпидермой рас¬
полагается верхний слой полупрозрачных крупных
паренхимных клеток. На поперечных срезах листа
видно, что он образован чаще двумя, иногда тремя
рядами клеток, которые располагаются один над дру¬
гим. Клетки нижнего ряда по высоте чаще в полтора
раза превышают верхние. Нижняя эпидерма листа,
как и верхняя, также образована однотипными, бесц¬
ветными паренхимными клетками с прямыми стенка¬
ми. Имеются многочисленные устьица, волоски. Ос¬
новные клетки нижней эпидермы многоугольные. Они
несколько мельче клеток верхней эпидермы. Устьица
овальные, окружены тремя околоустьичными клетка¬
ми (анизоцитный тип) (2.8.3). Устьица располагаются
обычно одиночно, реже попарно. На нижней стороне
листа видны радиально расположенные неокрашен¬
ные жилки (проводящие пучки).
C. 1,0 мл раствора 5, приготовленного как указа¬
но в разделе «Испытания», доводят спиртом (70 %,
об/об) Р до объема 100,0 мл. Спектр поглощения
(2.2.25) полученного раствора в области от 220 до
340 нм имеет максимум при (275±5) нм.
О. К 1 мл раствора 5 прибавляют 5 мл спирта
(70 %, об/об) Р, перемешивают и прибавляют не¬
сколько капель раствора 100 г/л натрия гидрокси¬
да Р. Появляется коричневато-зеленое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Раствор 5. 1,0 г измельченного сырья (2000)
(2.9.12) помещают в колбу вместимостью 100 мл, при¬
бавляют 20 мл спирта (70 %, об/об) Р, выдерживают
в закрытой колбе в течение 30 мин и нагревают с об¬
ратным холодильником на водяной бане при темпера¬
туре от 80 °С до 90 °С в течение 30 мин. Охлаждают
и фильтруют.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие и побуревшие листья —
не более 5 %; другие части растения (черешки и цве¬
тоносы) — не более 5 %. Органические примеси: не
более 1 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 20,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (2000) (2.9.12) по¬
мещают в колбу вместимостью 100 мл, прибавляют
25 мл раствора 10 г/л кислоты хлористоводород¬
ной Р и выдерживают в водяной бане при температу¬
ре от 50 °С до 55 °С в течение 20 мин. Процеживают
через ватный тампон в колбу вместимостью 150 мл.
Ватный тампон помещают в колбу с остатком сырья,
прибавляют 25 мл раствора 10 г/л кислоты хлористо¬
водородной Р, смывая частицы сырья с воронки в кол¬
бу, и выдерживают в водяной бане при температуре от
50 °С до 55 °С в течение 20 мин. Процеживают через
1188
Гэсударственная фармакопея Республики Беларусь
ватный тампон в ту же колбу вместимостью 150 мл,
прибавляют 30 мл 96 % спирта Р, перемешивают, ох¬
лаждают до комнатной температуры и фильтруют че¬
рез бумажный фильтр в мерную колбу вместимостью
100 мл, промывая колбу и фильтр (10—15) мл раст¬
вора 10 г/л кислоты хлористоводородной Р. Доводят
раствором 10 г/л кислоты хлористоводородной Р до
объема 100,0 мл.
Измеряют оптическую плотность (2.2.25) раст¬
вора при 510 нм, используя раствор 10 г/л кислоты
хлористоводородной Р в качестве компенсационного
раствора.
Содержание суммы антоцианов в пересчете на
цианидин-3,5-дигликозид в процентах рассчитывают
по формуле:
АЮ0
т-453 ’
где:
453 — удельный показатель поглощения циани-
дин-3,5-дигликозида;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0040
БЕЛЕНЫ ЧЕРНОЙ ЛИСТЬЯ
Нуовсуат ’1 п1дп IоНит
НЕЫВАЫЕ1-ЕАЕ*
ОПРЕДЕЛЕНИЕ
Собранные в течение лета и высушенные при¬
корневые и стеблевые листья двухлетнего травяни¬
стого растения Нуозсуатиз п/дег 1_.
Содержание: не менее 0,05 % суммы алкалоидов
в пересчете на гиосциамин (С17Н23М03; М.м. 289,4)
(в пересчете на сухое сырье).
Основными составляющими алкалоидов явля¬
ются гиосциамин вместе с небольшим количеством
гиосцина (скополамина).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
частично измельченные листья продолговато-яйце¬
видной, яйцевидной или эллиптической формы, пери¬
столопастные или цельные с неравномерно-зубчатым
краем. Прикорневые листья с длинным черешком,
с обеих сторон покрыты густыми, длинными, мягки¬
ми волосками; стеблевые — без черешков, менее
опушены, волоски располагаются преимущественно
по жилкам и краю пластинки листа. Длина листьев
(5—20) см, ширина (3—10) см. Срединная жилка бе¬
ловатая, плоская, сильно расширяется к основанию.
Цвет листьев серовато-зеленый. Запах слабый, свое¬
образный, усиливающийся при увлажнении.
B. Микроскопия (2.8.23). При просматрива¬
нии листа с поверхности видны клетки эпидермиса
с верхней стороны с мало извилистыми стенками, с
нижней — с более извилистыми. Устьица многочис¬
ленные с обеих сторон листа, окружены 3 (реже 4)
околоустьичными клетками, из которых одна обыч¬
но мельче других (анизоцитный тип) (2.8.3). Волоски
многочисленные, двух типов: простые и головчатые.
Простые волоски тонкостенные, одни из них 2—3-кле-
точные, небольшие, другие — многоклеточные, очень
крупные. Головчатые волоски с длинной многоклеточ¬
ной ножкой и 4—8-клеточной (реже 1—2-клеточной)
железистой головкой. В мезофилле листа содержат¬
ся одиночные призматические кристаллы оксалата
кальция; нередко встречаются кристаллы в виде кре¬
стообразных сростков или тупоконечных друз. В круп¬
ных жилках имеются удлиненно-овальные клетки,
заполненные кристаллическим песком. В молодых
листьях содержатся только мелкие, едва заметные
призматические кристаллы, расположенные вблизи
жилок.
С. 1 г измельченного сырья (180) (2.9.12) встря¬
хивают с 10 мл 0,05 М раствора серной кислоты в
течение 2 мин и фильтруют. К фильтрату прибавля¬
ют 1 мл раствора аммиака концентрированного Р,
5 мл воды Р и осторожно встряхивают с 15 мл эфира,
свободного от пероксидов, Р, избегая образования
эмульсии. Эфирный слой отделяют, фильтруют через
фильтр с безводным сульфатом натрия Р в фарфо¬
ровую чашку и эфир выпаривают досуха. К остатку
прибавляют 0,5 мл кислоты азотной дымящейся Р
и выпаривают досуха на водяной бане. Прибавляют
10 мл ацетона Р и, по каплям, раствор 30 г/л калия
гидроксида Р в 96% спирте Р. Появляется темно-
фиолетовое окрашивание.
й. Просматривают хроматограмму, полученную в
испытании «Хроматография».
Результаты: основные зоны на хроматограмме
испытуемого раствора по расположению, цвету и раз¬
меру соответствуют основным зонам на хроматограм¬
ме такого же объема раствора сравнения.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Хроматография. Тонкослойная хроматография
(2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (180) (2.9.12) прибавляют 15 мл 0,05 М раст¬
вора серной кислоты, встряхивают в течение 15 мин
и фильтруют. Фильтр промывают 0,05 М раствором
серной кислоты до получения 20 мл фильтрата. К
фильтрату прибавляют 1 мл раствора аммиака кон¬
центрированного Р и встряхивают с двумя объемами
эфира, свободного от пероксидов, Р, порциями по
10мл. При необходимости слои разделяют при по¬
мощи центрифугирования. Объединенные эфирные
слои высушивают натрия сульфатом безводным Р,
фильтруют и выпаривают досуха на водяной бане.
Остаток растворяют в 0,5 мл метанола Р.
Раствор сравнения. 50 мг гиосциамина сульфа¬
та Р растворяют в 10 мл метанола Р. 15 мг гиосци¬
на гидробромида Р растворяют в 10 мл метанола Р.
К 2 мл раствора гиосцина гидробромида прибавляют
4 мл раствора гиосциамина сульфата и доводят ме¬
танолом Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — вода Р — ацетон Р (3:7:90, об/об/об).
Наносимый объем пробы: по 10 мкл и по 20 мкл
каждого раствора в виде полос длиной 20 мм и шири¬
ной 3 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 15 мин и охлаждают на воздухе.
Проявление А: опрыскивают пластинку раство¬
ром калия йодвисмутата разведенным Р до появле¬
ния оранжевых или коричневых зон на желтом фоне.
Белладонны листья (красавки листья)
1189
Результаты А: зоны на хроматограммах испыту¬
емого раствора соответствуют зонам на хроматограм¬
мах раствора сравнения по расположению (гиосциа-
мин в нижней трети и гиосцин в верхней трети хро¬
матограммы) и цвету. Могут наблюдаться слабые вто¬
ростепенные зоны в средней части хроматограммы
20 мкл испытуемого раствора или около линии старта
на хроматограмме 10 мкл испытуемого раствора.
Проявление В: пластинку опрыскивают раство¬
ром натрия нитрита Р до тех пор, пока подложка не
станет видимой через слой сорбента. Просматривают
через 15 мин при дневном свете.
Результаты В: зоны на хроматограммах раст¬
вора сравнения и испытуемого раствора, соответ¬
ствующие гмосциамину, изменяют цвет с коричневого
на красновато-коричневый, но не на серовато-синий
(атропин); все второстепенные зоны исчезают.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: пожелтевшие, побуревшие, почернев¬
шие листья — не более 3 %; другие части растения
(стебли, цветки, плоды) — не более 5 %. Органиче¬
ские примеси: не более 1 %. Минеральные примеси:
не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья (355)
{2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 20,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50 г испытуемого сырья полностью измельчают
(180) (2.9.12). Измельченное сырье используют для
определения потери в массе при высушивании и сум¬
мы алкалоидов.
a) Определяют потерю в массе при высушивании
(2.2.32). 2,000 г измельченного сырья сушат при тем¬
пературе 105 °С.
b) 10,00 г измельченного сырья смачивают экс¬
тракционной смесью раствор аммиака Р — 96%
спирт Р — эфир, свободный от пероксидов, Р (1:2:6,
об/об/об) и тщательно перемешивают. Смесь пере¬
носят в подходящий перколятор, применяя, если не¬
обходимо, экстракционную смесь. Настаивают в тече¬
ние 4 ч, затем перколируют смесью из хлороформа Р
и эфира, свободного от пероксидов, Р (1:3, об/об) до
полного извлечения алкалоидов. Отбирают несколь¬
ко миллилитров жидкости, вытекающей из перколя-
тора, высушивают досуха, сухой остаток растворяют
в 0,25 М растворе серной кислоты и проверяют от¬
сутствие алкалоидов с помощью раствора тетра-
йодмеркурата калия Р. Перколят концентрируют до
объема около 50 мл, отгоняя растворитель на водя¬
ной бане, затем количественно переносят в делитель¬
ную вороту, используя эфир, свободный от перок¬
сидов, Р. Прибавляют эфир, свободный от перокси¬
дов, Р в количестве, не менее чем в 2,1 раза превы¬
шающем объем перколята, до получения раствора с
плотностью значительно меньшей, чем у воды. Раст¬
вор встряхивают, избегая нагрева, трижды с 0,25 М
раствором серной кислоты порциями по 20 мл. Слои
разделяют, при необходимости используют центри¬
фугирование. Кислотный слой переносят во вторую
делительную воронку, подщелачивают раствором
аммиака Р и экстрагируют три раза хлороформом Р
порциями по 30 мл. К объединенным хлороформным
экстрактам прибавляют 4 г натрия сульфата без¬
водного Р и выдерживают в течение 30 мин, перио¬
дически взбалтывая. Затем хлороформный экстракт
сливают, сульфат натрия трижды промывают хлоро¬
формом Р порциями по 10 мл и промывную жидкость
объединяют с экстрактом. Растворитель выпаривают
досуха на водяной бане. Остаток нагревают при тем¬
пературе от 100 °С до 105 °С в течение 15 мин. Сухой
остаток растворяют в нескольких миллилитрах хлоро¬
форма Р, прибавляют 20,0 мл 0,01 М раствора сер¬
ной кислоты и удаляют хлороформ выпариванием
на водяной бане. После охлаждения избыток кислоты
титруют 0,02 М раствором натрия гидроксида, ис¬
пользуя в качестве индикатора смешанный раствор
метилового красного Р.
Содержание суммы алкалоидов в пересчете на
гиосциамин в процентах рассчитывают по формуле:
57,88 • (20 - п)
(100-(У) т ’
где:
д— потеря в массе при высушивании измельчен¬
ного сырья (180) (2.9.12), %;
п — с&ъем 0,02 М раствора натрия гидроксида,
пошедшего на титрование, мл;
т — масса навески испытуемого сырья, г.
07/2016:0221
БЕЛЛАДОННЫ ЛИСТЬЯ
(КРАСАВКИ ЛИСТЬЯ)
ВеНадоппае Шит
ВЕНАООЫЫА ГЕАР
ОПРЕДЕЛЕНИЕ
Собранные в фазу начала бутонизации до массо¬
вого плодоношения и высушенные листья или смесь
из высушенных листьев и цветущих верхушек побе¬
гов, иногда с плодами А(гора ЬеНадоппа 1_.
Содержание: не менее 0,30 % суммы алкалоидов
в пересчете на гиосциамин (С17Н23Ы03; М.м. 289,4)
(в пересчете на сухое сырье).
Основными составляющими алкалоидов явля¬
ются гиосциамин вместе с небольшим количеством
гиосцина (скополамина).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Листья от зелено¬
го до коричнево-зеленого цвета, снизу более светлые,
часто скрученные и измельченные, эллиптической,
яйцевидной или продолговато-яйцевидной формы, к
верхушке заостренные, цельнокрайние, к основанию
суживающиеся в короткий черешок, тонкие, длиной
до 20 см и шириной до 10 см. Цветоносные побеги
со сплющенными стеблями и попарно сближенными
листьями. В паре один из листьев крупнее другого. В
пазухах листьев встречаются цветки, иногда плоды.
Цветки со спайнолистной чашечкой и колокольчатым
венчиком. Плоды шарообразные ягоды, от зеленого
до черно-коричневого цвета с неопадающей чашеч¬
кой и широко раскрытыми чашелистиками. Запах сла¬
бый, своеобразный.
®)В. Микроскопия (2.8.23}. Исследуют измель¬
ченное сырье (355) (2.9.12). Порошок имеет темно¬
зеленый цвет. Просматривают под микроскопом с
использованием раствора хлоралгидрата Р. В по-
1190
Гэсударственная фармакопея Республики Беларусь
рошке обнаруживаются следующие диагностические
признаки (рисунок 0221.-1): фрагменты пластины, в
которых видны эпидермальные клетки со стенками
волнистого вида с бороздчатой кутикулой [А, С] и
часть расположенной внизу палисадной паренхимы
[Аа], соединенной с верхним эпидермисом [А]; много¬
численные устьица [Са] анизоцитного типа и также
несколько аномоцитного типа (2.8.3) расположены
наиболее часто на нижнем эпидермисе [С]; много¬
клеточные, однорядные покрывающие трихомы с
гладкой кутикулой [Р], железистые трихомы с одно¬
клеточными головками и многоклеточными, одноряд¬
ными ножками [О] или с многоклеточными головками
и одноклеточными ножками [В]; клетки паренхимы,
включая закругленные клетки, некоторые из которых
содержат микросфеноидальные кристаллы кальция
оксалата [Е]; кольцеобразно и спирально утолщен¬
ные сосуды [К]. В измельченном растительном сырье
могут также обнаруживаться волокна и сетчато утол¬
щенные сосуды стеблей; субсфероидальные зерна
пыльцы диаметром (40—50) мм с 3 зародышевыми
порами, 3 бороздками и в значительной степени ям-
чатой экзиной [Н]; фрагменты венчика с бородавча¬
тым эпидермисом [*Ц, или имеющего многочисленные
покрывающие или железистые трихомы вышеописан¬
ных типов [!_]; фрагменты коричневато-желтой тесты,
состоящей из скпереидных клеток неправильной
формы.
Рисунок 0221 .-1. Микроскопические признаки
листьев белладонны
®С. 1 г измельченного сырья (180) (2.9.12)
встряхивают с 10 мл 0,05 М раствора серной кисло¬
ты в течение 2 мин и фильтруют. К фильтрату прибав¬
ляют 1 мл раствора аммиака концентрированного Р,
5 мл воды Р и осторожно встряхивают с 15 мл эфира,
свободного от пероксидов, Р, избегая образования
эмульсии. Эфирный слой отделяют, фильтруют через
фильтр с безводным сульфатом натрия Р в фарфо¬
ровую чашку и эфир выпаривают досуха. К остатку
прибавляют 0,5 мл кислоты азотной дымящейся Р
и выпаривают досуха на водяной бане. Прибавляют
10 мл ацетона Р и, по каплям, раствор 30 г/л калия
гидроксида Р в 96% спирте Р. Появляется темно-
фиолетовое окрашивание.
Просматривают хроматограмму, получен¬
ную в испытании «Хроматография».
Результаты: основные зоны на хроматограмме
испытуемого раствора по расположению, цвету и раз¬
меру соответствуют основным зонам на хроматограм¬
ме такого же объема раствора сравнения.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
(*») Хроматография. Тонкослойная хроматогра¬
фия (2.2.27).
Испытуемый раствор. К 0,6 г измельченного
сырья (180) (2.9.12) прибавляют 15 мл 0,05 М раст¬
вора серной кислоты, встряхивают в течение 15 мин
и фильтруют. Фильтр промывают 0,05 М раствором
серной кислоты до получения 20 мл фильтрата. К
фильтрату прибавляют 1 мл раствора аммиака кон¬
центрированного Р и встряхивают с двумя объемами
эфира, свободного от пероксидов, Р, порциями по
10мл. При необходимости слои разделяют при по¬
мощи центрифугирования. Объединенные эфирные
слои высушивают натрия сульфатом безводным Р,
фильтруют и выпаривают досуха на водяной бане.
Остаток растворяют в 0,5 мл метанола Р.
Раствор сравнения. 50 мг гиосциамина сульфа¬
та Р растворяют в 9 мл метанола Р. 15 мг гиосцина
гидробромида Р растворяют в 10 мл метанола Р.
1,8 мл раствора гиосцина гидробромида смешивают
с 8 мл раствора гиосциамина сульфата.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — вода Р — ацетон Р (3:7:90, об/об/об).
Наносимый объем пробы: по 10 мкл и по 20 мкл
каждого раствора в виде полос длиной 20 мм и шири¬
ной 3 мм, оставив 1 см между полосами.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 15 мин, затем охлаждают на воздухе.
Проявление А: пластинку опрыскивают раство¬
ром калия йодвисмутата Р2, используя около 10 мл
на квадратную пластинку со стороной 200 мм до по¬
явления оранжевых или коричневых зон на желтом
фоне. Просматривают при дневном свете.
Результаты А: зоны на хроматограммах испыту¬
емого раствора соответствуют зонам на хроматограм¬
мах раствора сравнения по расположению (гиосциа-
мин в нижней трети и гиосцин в верхней трети хро¬
матограммы) и цвету. Размер зон на хроматограммах
испытуемого раствора не меньше размера соответ¬
ствующих зон на хроматограммах такого же объема
раствора сравнения. Могут наблюдаться слабые вто¬
ростепенные зоны в средней части хроматограммы
20 мкл испытуемого раствора или около линии старта
на хроматограмме 10 мкл испытуемого раствора.
Проявление В: пластинку опрыскивают раство¬
ром натрия нитрита Р до тех пор, пока подложка не
станет видимой через слой сорбента. Просматривают
через 15 мин при дневном свете.
Результаты В: зоны на хроматограммах раст¬
вора сравнения и испытуемого раствора, соответ¬
ствующие гиосциамину, изменяют цвет с коричневого
Березы листья
1191
на красновато-коричневый, но не на серовато-синий
(атропин); все второстепенные зоны исчезают.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли диаметром более 5 мм — не
более 3 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
@ Общая зола (2.4.16). Не более 16,0 %.
Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
I* 50 г испытуемого сырья полностью измель¬
чают (180) (2.9.12). Измельченное сырье используют
для определения потери в массе при высушивании и
суммы алкалоидов.
a) Определяют потерю в массе при высушивании
(2.2.32). 2,000 г измельченного сырья сушат при тем¬
пературе от 100 °С до 105 °С.
b) 10,00 г измельченного сырья смачивают экс¬
тракционной смесью из 5 мл раствора аммиака Р,
10 мл 96 % спирта Р и 30 мл эфира, свободного от
пероксидов, Р и тщательно перемешивают. Смесь
переносят в подходящий перколятор, применяя,
если необходимо, экстракционную смесь. Настаи¬
вают в течение 4 ч, затем перколируют смесью из
хлороформа Р и эфира, свободного от перокси¬
дов, Р (1:3, об/об) до полного извлечения алкалои¬
дов. Отбирают несколько миллилитров жидкости,
вытекающей из перколятора, высушивают досуха,
сухой остаток растворяют в 0,25 М растворе сер¬
ной кислоты и проверяют отсутствие алкалоидов с
помощью раствора тетрайодмеркурата калия Р.
Перколят концентрируют до объема около 50 мл, от¬
гоняя растворитель на водяной бане, затем количе¬
ственно переносят в делительную воронку, исполь¬
зуя эфир, свободный от пероксидов, Р. Прибавляют
эфир, свободный от пероксидов, Р в количестве, не
менее чем в 2,1 раза превышающем объем перко-
лята, до получения раствора с плотностью значи¬
тельно меньшей, чем у воды. Раствор встряхивают,
избегая нагрева, трижды с 0,25 М раствором сер¬
ной кислоты порциями по 20 мл. Слои разделяют,
при необходимости используют центрифугирование.
Кислотный слой переносят во вторую делительную
воронку, подщелачивают раствором аммиака Р и
экстрагируют три раза хлороформом Р порциями
по 30 мл. К объединенным хлороформным экстрак¬
там прибавляют 4 г натрия сульфата безводного Р
и выдерживают в течение 30 мин, периодически
взбалтывая. Затем хлороформный экстракт слива¬
ют, сульфат натрия трижды промывают хлорофор¬
мом Р порциями по 10 мл, и объединяют с экстрак¬
том. Растворитель выпаривают досуха на водяной
бане. Остаток нагревают при температуре от 100 °С
до 105 °С в течение 15 мин. Сухой остаток раство¬
ряют в нескольких миллилитрах хлороформа Р, при¬
бавляют 20,0 мл 0,01 М раствора серной кислоты
и удаляют хлороформ выпариванием на водяной
бане. После охлаждения избыток кислоты титруют
0,02 М раствором натрия гидроксида, используя в
качестве индикатора смешанный раствор метило¬
вого красного Р.
Содержание суммы алкалоидов в пересчете на
гиосциамин в процентах рассчитывают по формуле:
57,88-(20 -п)
(100 — сУ) * /77 ’
где:
с/— потеря в массе при высушивании измельчен¬
ного сырья (180) (2.9.12), %;
п — объем 0,02 М раствора натрия гидроксида,
пошедшего на титрование, мл;
т — масса навески испытуемого сырья, г
07/2016:1174
<§> БЕРЕЗЫ ЛИСТЬЯ
Ве1и1ае ТоНит
В1КСН 1-ЕАР
ОПРЕДЕЛЕНИЕ
Цельные или фрагментированные высушенные
листья Ве1и!а репди1а Ро1И и/или ВеЫа риЬезсепз
ЕИгИ., а так же их гибридов.
Содержание: не менее 1,5 % флавоноидов в пе¬
ресчете на гиперозид (С21Н20О12; М.м. 464,4) (в пере¬
счете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Верхняя поверх¬
ность листьев обоих видов имеет темно-зеленый цвет,
нижняя — зеленовато-серый; на листьях наблюдает¬
ся характерное плотное сетчатое жилкование. Жилки
светло-коричневые или почти белые.
Листья В. репди!а гладкие с близко расположен¬
ными железистыми порами на обеих поверхностях.
Листья длиной (3—7) см и шириной (2—5) см; чере¬
шок длинный, край листа дважды пильчатый, пластин¬
ка листа по форме треугольная или ромбовидная с
широким клиновидным или почти усеченным основа¬
нием. Угол с каждой стороны неокругленный или слег¬
ка округленный, верхушка длинная и заостренная.
Листья В. риЬезсепз имеют небольшое количе¬
ство железистых волосков, слегка опушены на обеих
поверхностях. На нижней стороне в точках разветвле¬
ния жилок находятся небольшие пучки желтовато-се¬
рых волосков. Листья В. риЬезсепз немного меньше,
овальные или ромбические и более округлые. Пиль-
чатость более грубая и более регулярная. Верхушка
листа не длинная и не заостренная.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Порошок имеет зеленовато¬
серый цвет. Исследуют под микроскопом, используя
раствор хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки (рису¬
нок 1174.-1): многочисленные фрагменты листовой
пластинки (вид с поверхности) с прямостенными
клетками эпидермиса верхней стороны листа с ниже-
расположенной палисадной паренхимой [Е] и клетка¬
ми эпидермиса нижней стороны листа с устьицами
аномоцитного типа (2.8.3) [О]; большие свободные
железистые волоски размером обычно от 100 мкм
до 120 мкм рЭ]; фрагменты листовой пластинки в по¬
перечном срезе [В], на котором видны железистые
волоски на эпидермисе [Ва], неоднородный, ассиме-
тричный мезофилл, содержащий друзы кристаллов
[ВЬ] и призматические кристаллы [Вс] оксалата каль¬
ция; фрагменты пористой паренхимы [А] с кристал¬
лоносной обкладкой [Аа] и клетками, содержащими
друзы кристаллов оксалата кальция [АЬ]; фрагменты
1192
Гэсударственная фармакопея Республики Беларусь
сосудов и волокон склеренхимы [С]. Если лекарствен¬
ное растительное сырье содержит В. риЬезсепз, в
порошке обнаруживаются одноклеточные покровные
волоски с очень толстыми стенками, длиной около
(80—600) мкм, обычно (100—200) мкм, многочислен¬
ные на краевой части листовой пластинки [Р] или на
эпидермисе (вид с поверхности [Н]).
Рисунок 1174.-1. Микроскопические признаки
листьев березы
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл метанола Р,
встряхивают и нагревают на водяной бане при 60 °С в
течение 5 мин. Охлаждают и фильтруют.
Раствор сравнения. 1 мг кофейной кислоты Р,
1 мг хлорогеновой кислоты Р, 2,5 мг гиперозида Р и
2,5 мг рутина Р растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р —метилэтилкетон Р — этилаце-
тат Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы, по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку обрабатывают раство¬
ром 10 г/л дифенилборной кислоты аминоэтилово¬
го эфира Р в метаноле Р и затем раствором 50 г/л
макрогола 400 Р в метаноле Р. Сушат на воздухе в
течение 30 мин и просматривают в ультрафиолетовом
свете при длине волны 365 нм.
Результаты: на хроматограмме раствора срав¬
нения в нижней половине обнаруживаются три флу¬
оресцирующие зоны (в порядке увеличения значения
Рг): желтовато-коричневого цвета (рутин), светло-си¬
него цвета (хлорогеновая кислота) и желтовато-ко¬
ричневого цвета (гиперозид); в верхней трети — флуо¬
ресцирующая зона светло-синего цвета (кофейная
кислота). На хроматограмме испытуемого раствора
обнаруживаются три флуоресцирующие зоны, соот¬
ветствующие зонам рутина, хлорогеновой кислоты и
гиперозида на хроматограмме раствора сравнения.
Зона, соответствующая рутину, имеет очень слабую
флуоресценцию, а зона, соответствующая гиперози-
ду, — интенсивную флуоресценцию. Также наблюда¬
ются другие желтовато-коричневые зоны со слабой
флуоресценцией между зонами, соответствующими
кофейной кислоте и хлорогеновой кислоте на хрома¬
тограмме раствора сравнения. Около фронта раст¬
ворителей обнаруживается флуоресцирующая зона
красного цвета, соответсвующая хлорофиллам. На
хроматограмме испытуемого раствора между этой
зоной и зоной, соответствующей кофейной кислоте
на хроматограмме раствора сравнения, обнаружи¬
вается коричневато-желтая зона, соответствующая
кверцетину.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: фрагменты сережек — не более 3 %.
Другие допустимые примеси: не более 3 %.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Исходный раствор. 0,200 г измельченного сырья
(355) (2.9.12) помещают в круглодонную колбу вме¬
стимостью 100 мл, прибавляют 1 мл раствора 5 г/л
гексаметилентетрамина Р, 20 мл ацетона Р и 2 мл
кислоты хлористоводородной Р1. Кипятят с обрат¬
ным холодильником в течение 30 мин, после чего из¬
влечение процеживают через ватный тампон в колбу
вместимостью 100 мл. Ватный тампон помещают в
круглодонную колбу с остатком и дважды экстраги¬
руют, кипятя с обратным холодильником в течение
10 мин, каждый раз прибавляя 20 мл ацетона Р в ка¬
честве экстрагента. Охлаждают и процеживают через
ватный тампон. Объединенные ацетоновые извлече¬
ния фильтруют через бумажный фильтр в мерную кол¬
бу вместимостью 100 мл, ополаскивают круглодонную
колбу ацетоном Р и фильтруют через тот же фильтр,
после чего промывают фильтр ацетоном Р. Объем
раствора доводят ацетоном Р до объема 100,0 мл.
20,0 мл полученного раствора помещают в делитель¬
ную воронку, прибавляют 20 мл воды Р и экстраги¬
руют этилацетатом Р порцией 15 мл, затем еще
трижды порциями по 10 мл. Этилацетатные извлече¬
ния объединяют, помещают в делительную воронку и
дважды промывают водой Р порциями по 50 мл. Ор¬
ганический слой фильтруют через бумажный фильтр с
10 г натрия сульфата безводного Р в мерную колбу
вместимостью 50 мл и доводят этилацетатом Р до
объема 50,0 мл.
Испытуемый раствор. К 10,0 мл исходного раст¬
вора прибавляют 1 мл реактива алюминия хлорида Р
и доводят 5 % (об/об) раствором кислоты уксусной
ледяной Р в метаноле Р до объема 25,0 мл.
Компенсационный раствор. 10,0 мл исходного
раствора доводят 5 % (об/об) раствором кислоты ук¬
сусной ледяной Р в метаноле Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
растворов (2.2.25) при 425 нм.
Бессмертника песчаного цветки
1193
Содержание флавоноидов в пересчете на гипе-
розид в процентах рассчитывают по формуле:
Л-625
ш- 500 ’
500 — удельный показатель поглощения гиперо-
зида;
К — ощшш \йК\-
вора;
/л — масса навески испытуемого сырья, г.
07/2016: РБ0041
БЕРЕЗЫ ПОЧКИ
ВеШае детта
втсн вао*
ОПРЕДЕЛЕНИЕ
Собранные до распускания и высушенные почки
ВеМа репди1а Яо1Н и/или ВеЮ1а риЬезсепз ЕМгЬ.
Содержание: не менее 2 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Почки удлинен¬
но-конические, заостренные или притупленные, часто
клейкие. Чешуйки расположены черепицеобразно,
плотно прижаты по краям, слегка реснитчатые (ниж¬
ние короче верхних и иногда с несколько отстающи¬
ми кончиками); длина (3—7) мм, ширина от 1,5 мм до
3 мм. Цвет коричневый, у основания иногда зеленова¬
тый. Запах бальзамический.
B. Микроскопия (2.8.23). При просматривании
чешуи почки с поверхности видны клетки эпидерми¬
са, слегка вытянутые с прямыми, местами четковид-
но-утолщенными стенками. Устьица на наружном
эпидермисе аномоцитного типа, расположены в углу¬
блении в виде воронки. Замыкающие клетки устьица
в 2—3 раза крупнее эпидермальных. По краю чешуи и
жилкам встречаются простые одноклеточные волоски
с коричневым содержимым и бородавчатой поверх¬
ностью. В мезофилле видны многочисленные друзы
оксалата кальция. При просматривании листового за¬
чатка с поверхности видны крупные коричневые же¬
лезки; на зубчиках форму конуса, на поверхности ли¬
сточка — в виде гриба. Железки состоят из округлых
или слегка продольно-вытянутых внутренних клеток,
заполненных коричневым содержимым, и радиально¬
вытянутых прозрачных наружных клеток.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (веточки, в том
числе отделенные от почек при анализе, сережки и
другие) — не более 8 %; почки, тронувшиеся в рост
и слегка распустившиеся, — не более 2 %. Органиче¬
ские примеси: не более 1 %. Минеральные примеси:
не более 0,5 %
Потеря в массе при высушивании (2.2.32). Не
более 10,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 4,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,7 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют содержание эфирного масла (2.8. 12,
метод В). 20,0 г измельченного сырья (2000) (2.9.12)
помещают в колбу вместимостью 1000 мл, прибавля¬
ют 400 мл воды Р в качестве жидкости для перегонки.
Время перегонки 2 ч.
07/2016: РБ0042
БЕССМЕРТНИКА ПЕСЧАНОГО
ЦВЕТКИ
НеНсЬгузI агепагИ Лоз
ЗАЫОУ ЕУЕ^АЗТШв Р/.ОУУЕЯ*
ОПРЕДЕЛЕНИЕ
Собранные до распускания цветков и высушен¬
ные корзинки многолетнего травянистого растения
НеНсЬгузит агепапит (1_.) МоепсН.
Содержание: не менее 2,5 % суммы флавонои¬
дов в пересчете на рутин (С27Н30О16*ЗН2О; М.м. 665)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корзинки ша¬
ровидные, одиночные или по несколько вместе на
коротких шерстисто-войлочных цветоносах длиной
до 1 см, диаметром около 7 мм. Корзинки состо¬
ят из многочисленных цветков, расположенных на
голом цветоложе, окруженных многочисленными,
неплотно прижатыми листочками обвертки. Все
цветки трубчатые, пятизубчатые, обоеполые, с хо¬
холком. Листочки обвертки вогнутые, сухие, плен¬
чатые, блестящие, наружные — яйцевидные, сред¬
ние — лопатчатые, удлиненные, внутренние — уз¬
кие, линейные. Цвет обвертки зеленовато-желтый,
цветков — зеленовато-желтый или оранжевый. За¬
пах слабый, ароматный.
B. Микроскопия (2.8.23). При просматривании
листочков обвертки с поверхности виден эпидермис
из слегка вытянутых пористых клеток, в суженной
части листочка — множество простых бичевидных
волосков с несколькими короткими базальными и
одной длинной конечной клетками и эфиромаслич¬
ных овальных двухрядных, многоярусных железок,
состоящих из 8—12 клеток. При просматривании
цветка с поверхности видна овальная завязь с мно¬
гочисленными вздутыми волосками и ее кольцевое
основание из четырехугольных толстостенных кле¬
ток. На верхушке завязи виден хохолок, состоящий
из тонких щетинок, сросшихся друг с другом у ос¬
нования. Зубцы венчика с неровными и бахромча¬
тыми краями. На венчике множество головчатых
волосков с одноклеточной головкой на 12—14-кле¬
точной ножке.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (1400) (2.9.12) прибавляют 10 мл метанола Р,
нагревают при перемешивании в водяной бане при
60 °С в течение 5 мин, охлаждают и фильтруют.
Раствор сравнения. 2 мг хлорогеновой кисло¬
ты Р и 5 мг кверцетина дигидрата Р растворяют в
метаноле Р и доводят до объема 20 мл этим же раст¬
ворителем.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
1194
Гэсударственная фармакопея Республики Беларусь
Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат (11:11:27:100, об/об/об/об).
Наносимый объем пробы: 30 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: пластинку опрыскивают раствором
10 г/л дифенилборной кислоты аминоэтилового
эфира Р в метаноле Р и затем раствором 50 г/л ма-
крогола 400 Р в метаноле Р. Высушивают на воздухе
в течение 30 мин и просматривают в ультрафиолето¬
вом свете при 365 нм.
Результаты: на хроматограмме раствора срав¬
нения в нижней половине обнаруживается флуорес¬
цирующая зона светло-синего цвета (хлорогеновая
кислота); в верхней трети обнаруживается зона жел¬
товато-коричневого цвета (кверцетин). На хромато¬
грамме испытуемого раствора обнаруживаются флуо¬
ресцирующие зоны, соответствующие зонам хло-
рогеновой кислоты и кверцетина на хроматограмме
раствора сравнения. На хроматограмме испытуемого
раствора обнаруживаются и другие флуоресцирую¬
щие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: соцветия с остатками стеблей длиной
свыше 1 см — не более 5 %; остатки корзинок (цве¬
толожа с обвертками) — не более 5 %; измельченные
частицы, проходящие сквозь сито (1400) (2.9.12), —
не более 5 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (1400) (2.9.12) по¬
мещают в коническую колбу вместимостью 250 мл,
прибавляют 100 мл 96% спирта Р и нагревают с
обратным холодильником на водяной бане в тече¬
ние 30 мин. Охлаждают до комнатной температуры и
фильтруют через бумажный фильтр, предварительно
смоченный 96 % спиртом Р. Экстракцию указанным
выше способом повторяют еще три раза, исполь¬
зуя каждый раз по 50 мл 96 % спирта Р. Фильтраты
объединяют и доводят 96 % спиртом Р до объема
250,0 мл (раствор А).
Испытуемый раствор. К 2,0 мл раствора А при¬
бавляют 1,0 мл раствора 20 г/л алюминия хлорида Р
в 96% спирте Р, 5 капель кислоты хлористоводо¬
родной разведенной Р и доводят 96 % спиртом Р до
объема 25,0 мл.
Раствор сравнения. 0,0500 г рутина Р, предва¬
рительно высушенного при температуре от 130 °С до
135 °С в течение 3 ч, растворяют в 80 мл 96 % спир¬
та Р при нагревании на водяной бане, охлаждают и
доводят до объема 100,0 мл этим же растворителем
(раствор В). К 1,0 мл раствора В прибавляют 1,0 мл
раствора 20 г/л алюминия хлорида Рв96% спирте Р,
5 капель кислоты хлористоводородной разведен¬
ной Р и доводят 96 % спиртом Р до объема 25,0 мл.
Компенсационный раствор (а). К 2,0 мл раст¬
вора А прибавляют 5 капель кислоты хлористоводо¬
родной разведенной Р и доводят 96 % спиртом Р до
объема 25,0 мл.
Компенсационный раствор (Ь). К 1,0 мл раст¬
вора В прибавляют 5 капель кислоты хлористоводо¬
родной разведенной Р и доводят 96 % спиртом Р до
объема 25,0 мл.
Через 40 минут измеряют оптическую плотность
(2.2.25) испытуемого раствора и раствора сравнения
при 411 нм, используя компенсационные растворы (а)
и (Ь) соответственно.
Содержание суммы флавоноидов в пересчете на
рутин в процентах рассчитывают по формуле:
АлуР.1,25
А0т
где:
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески рутина Р, г;
07/2016:1432
<@> БОЯРЫШНИКА ЛИСТЬЯ
И ЦВЕТКИ
С^а^аед^ ТоНит сит Логе
НАШНОКЫ 1.ЕАРАЫО Я.01УЕЯ
ОПРЕДЕЛЕНИЕ
Целые или резаные высушенные цветоносные
ветви СгаХаедиз топодупа иасц. (ипс!т), С. /аемда/а
(Р01Г.) ЭС. (синонимы: С. охуасапНюШез ТИиШ.;
С. охуасапОпа аис!.) или их гибридов или, значительно
реже, прочих европейских видов СгаХаедиз, в том чис¬
ле С. репХадупа \Л/а1с1з*. е! К\Х. ех №114., С. т'дга \Л/а1с1з1.
еХ КП. и С. ахагоШз !_.
Содержание: не менее 1,5 % суммы флавонои¬
дов в пересчете на гиперозид (С21Н20О12; М.м. 464,4)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Стебли темно-ко¬
ричневого цвета, деревянистые, диаметром от 1 мм
до 2,5 мм, с чередующимися черешчатыми листьями
с мелкими, часто опадающими прилистниками и щит¬
ками многочисленных мелких белых цветков. Листья
более или менее глубоко разделены на лопасти со
слабозубчатыми или почти ровными краями; листья
С. /аемда/а перистолопастные или перисторассечен-
ные с 3, 5 или 7 тупоконечными лопастями, листья
С. топодупа перисторассеченные с 3 или 5 остры¬
ми лопастями; верхняя поверхность темно-зелено¬
го или коричневато-зеленого цвета, нижняя поверх¬
ность — светлее, серовато-зеленая, с выступающим
плотным сетчатым жилкованием. Листья С. 1аеу1да1а,
С. топодупа и С. реп!адупа гладкие или с редкими
волосками; листья С. агагоШз и С. л/дга густо опуше¬
ны. Цветки имеют коричневато-зеленую трубчатую
чашечку, состоящую из пяти раздельных завернутых
чашелистиков, венчик, состоящий из пяти раздельных
желтовато-белых или коричневатых закругленных или
широко яйцевидных и коротких ноготковидных лелеет-
Боярышника листья и цветки
1195
ков и многочисленных тычинок. Завязь, сросшаяся с
чашечкой, состоит из 1—5 плодолистиков, каждый из
которых имеет длинный столбик и содержит одиноч¬
ную семяпочку; С. топодупа имеет один плодолистик,
С. /аемда^а — два или три, С. агаго1из — два или три
или иногда только один, С. реп1адупа — пять или ино¬
гда четыре.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное лекарственное растительное сырье (355) (2.9.12).
Порошок желтовато-зеленого цвета. Исследуют под
микроскопом, используя раствор хлоралгидрата Р.
В порошке обнаруживаются следующие диагности¬
ческие признаки: одноклеточные покровные волоски,
обычно с толстой стенкой и широким просветом, почти
прямые или слегка изогнутые, ямчатые у основания;
фрагменты эпидермиса листа с клетками, которые
имеют извилистые или многоугольные антиклиналь¬
ные стенки, и крупными устьицами аномоцитного типа
(2.8.3), окруженными 4—7 клетками; паренхиматоз¬
ные клетки мезофилла, содержащие друзы кристал¬
лов оксалата кальция размером обычно (10—20) мкм,
которые связаны с жилками, содержащими группы
мелких призматических кристаллов; фрагменты ле¬
пестков с закругленными многоугольными сильно бо¬
родавчатыми толстостенными клетками эпидермиса,
кутикула которых имеет явственно заметную волно¬
образную бороздчатость; фрагменты пыльников, в
которых виден эндотеций с изогнутым и равномерно
утолщенным краем; фрагменты стеблей, содержащие
клетки колленхимы, разделенные ямчатые сосуды и
группы лигнифицированных волокон склеренхимы
с узкими просветами; многочисленные сферические
или эллиптические, или треугольные зерна пыльцы
диаметром до 45 мкм с тремя зародышевыми порами
и слегка зернистой экзиной.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
лекарственного растительного сырья (355) (2.9.12)
прибавляют 10 мл метанола Р и нагревают с обрат¬
ным холодильником в водяной бане при температуре
65 °С в течение 5 мин. Охлаждают и фильтруют.
Раствор сравнения. 1,0 мг хлорогеновой кисло¬
ты Р и 2,5 мг гиперозида Р растворяют в 10 мл ме¬
танола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метилэтилкетон Р — этилаце-
тат Р (10:10:30:50, об/б/об/об).
Наносимый объем пробы: по 20 мкл в виде по¬
лос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: горячую пластинку опрыскивают
раствором 10 г/л аминоэтилового эфира дифенил-
борной кислоты Р в метаноле Р и затем раствором
50 г/л макрогола 400 Р в метаноле Р. Пластинку
высушивают на воздухе в течение 30 мин и просма¬
тривают в ультрафиолетовом свете при длине волны
365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие флуоресци¬
рующие зоны.
Верх хроматографической пластинки
Гиперозид: флуоресцирую¬
щая зона желтовато-оран¬
жевого цвета
Хлорогеновая кислота: флу¬
оресцирующая зона светло-
синего цвета
Флуоресцирующая зона
желтовато-зеленого цвета
(витексин)
Флуоресцирующая зона
желтовато-оранжевого
цвета (гиперозид)
Флуоресцирующая зона
светло-синего цвета (хлоро¬
геновая кислота)
Флуоресцирующая зона
желтовато-зеленого цвета
(витексин-2"-рамнозид)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: одревесневшие побеги диаметром бо¬
лее 2,5 мм — не более 8 %. Другие допустимые при¬
меси: не более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 1,000 г измельченного лекарственного
растительного сырья (355) (2.9.12) сушат при темпе¬
ратуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Исходный раствор. 0,400 г измельченного ле¬
карственного растительного сырья (250) (2.9.12) по¬
мещают в колбу вместимостью 200 мл, прибавляют
40 мл спирта (60 %, об/об) Р. Нагревают в водяной
бане при температуре 60 °С в течение 10 мин при
частом встряхивании. Охлаждают, фильтруют че¬
рез ватный тампон в мерную колбу вместимостью
100 мл. Ватный тампон с остатком лекарственно¬
го растительного сырья переносят обратно в колбу
вместимостью 200 мл, прибавляют 40 мл спирта
(60 %, об/об) Р и вновь нагревают в водяной бане
при температуре 60 °С в течение 10 мин при ча¬
стом встряхивании. Охлаждают и фильтруют в ту
же мерную колбу вместимостью 100 мл. Ополаски¬
вают спиртом (60 %, об/об) Р колбу вместимостью
200 мл, фильтруют и переносят в ту же мерную кол¬
бу вместимостью 100 мл. Доводят спиртом (60%,
об/об) Р до объема 100,0 мл и фильтруют.
Испытуемый раствор. 5,0 мл исходного раст¬
вора помещают в круглодонную колбу и выпаривают
досуха при пониженном давлении. Полученный оста¬
ток растворяют в 8 мл смеси метанола Р и кислоты
уксусной безводной Р (10:100, об/об) и переносят в
мерную колбу вместимостью 25 мл. Круглодонную
колбу ополаскивают 3 мл смеси метанола Р и кисло¬
ты уксусной безводной Р (10:100, об/об) и переносят
в ту же мерную колбу вместимостью 25 мл. Прибавля¬
ют 10,0 мл раствора, содержащего 25,0 г/л кислоты
борной Р и 20,0 г/л кислоты щавелевой Р в кислоте
муравьиной безводной Р и доводят кислотой уксус¬
ной безводной Р до объема 25,0 мл.
Компенсационный раствор. 5,0 мл исходного
раствора помещают в круглодонную колбу и выпа¬
ривают досуха при пониженном давлении. Получен¬
ный остаток растворяют в 8 мл смеси из метанола Р
и кислоты уксусной безводной Р (10:100, об/об) и
переносят в мерную колбу вместимостью 25 мл. Кру¬
глодонную колбу ополаскивают 3 мл смеси из ме-
1196
Гэсударственная фармакопея Республики Беларусь
танола Р и кислоты уксусной безводной Р (10:100,
об/об) и переносят в ту же мерную колбу вместимо¬
стью 25 мл. Прибавляют 10,0 мл кислоты муравьи¬
ной безводной Р и доводят кислотой уксусной безво¬
дной Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
растворов (2.2.25) при 410 нм.
Содержание суммы флавоноидов в пересчете на
гиперозид в процентах рассчитывают по формуле:
А 500
т-405’
где:
405 — удельный показатель поглощения гиперо-
зида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0043
БОЯРЫШНИКА ЛИСТЬЯ
СгаГаед/ ГоНит
НАШНОИЫ 1.ЕАГ*
ОПРЕДЕЛЕНИЕ
Собранные во время цветения и высушенные ли¬
стья кустарников или небольших деревьев Сга(аедиз
запдшпеа РаН. и С. 1аеПда1а (Ро1г) ОС. (С. охуасапОпа
зепзи Ро]агк.).
Содержание:
- сумма флавоноидов: не менее 0,25 % в пере¬
счете на рутин (С27Н30О16-ЗН2О; М.м. 665) (в пересчете
на сухое сырье);
- сумма процианидинов: не менее 5,0 % в пере¬
счете на цианидина хлорид (С^Н^СЮ^ М.м. 322,7)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь цельных
или частично изломанных листьев. Листья в очер¬
тании яйцевидные, обратнояйцевидные или яйце¬
видно-ромбические; у Сга1аедиз запдшпеа с острой
вершиной и клиновидным основанием, у Сга1аедиз
охуасапИпа — с притупленной вершиной. Листья ло¬
пастные, с 3—7 (реже 1—2) неглубокими городчато-
зубчатыми или пильчатыми лопастями. У Сга1аедиз
запдшпеа листья негусто-волосистые с обеих сторон,
сверху темно-зеленые, снизу — светлее; черешки
желобчатые, голые или негусто-волосистые. Длина
листовой пластинки (2—10) см, ширина (2,5—5) см,
длина черешка до 5 см. У Сга1аедиз охуасапХЬа ли¬
стья ярко-зеленые с более светлой нижней стороной,
голые, иногда снизу с бородками из волосков в углах
жилок. Длина листовой пластинки до 6 см, ширина до
5 см, длина черешка до 3 см. Запах слабый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны: нижний эпидермис листа
с сильно извилистыми тонкими стенками; многочис¬
ленные устьица аномоцитного типа, распложенные
неравномерно и беспорядочно среди эпидермаль¬
ных клеток. Форма устьиц овальная, длина пример¬
но в полтора раза превышает ширину. Над жилкой
эпидермис состоит из узких продолговатых клеток,
плотно прилегающих друг к другу. Клеточная стенка
толще, чем у межжилкового эпидермиса и без харак¬
терной извилистости. Волоски на нижнем и верхнем
эпидермисе простые, одноклеточные, длинные, за¬
остренные. На верхнем эпидермисе устьиц нет. Эпи¬
дермальные клетки различной формы и размеров.
Клеточная стенка слабоизвилистая. Волосков мень¬
ше, чем на нижнем эпидермисе. При повреждениях
сосудисто-волокнистых пучков могут быть видны спи¬
ральные утолщения стенок спиральных сосудов. На
поперечном срезе через главную жилку листа в па¬
ренхиме видны одиночные, редко разбросанные кри¬
сталлы оксалата кальция. В периферических слоях
видны коричневые включения каротиноидов.
С. К 1 мл раствора А, приготовленного как указа¬
но в разделе «Количественное определение. Опреде¬
ление содержания суммы флавоноидов», прибавля¬
ют 57 капель раствора 36,2 г/л алюминия хлорида Р в
спирте (70 %, об/об) Р и нагревают на водяной бане
в течение (3—5) мин. Появляется зеленовато-желтое
окрашивание.
Б. К 0,2 мл раствора В, приготовленного как
указано в разделе «Количественное определение.
Определение содержания суммы процианидинов в
пересчете на цианидина хлорид», прибавляют 0,9 мл
спирта (70 %, об/об) Р, 6 мл 5 % (об/об) раствора кис¬
лоты хлористоводородной Р в бутаноле Р, 0,2 мл
раствора железа (III) аммония сульфата Р7 и нагре¬
вают в водяной бане в течение (20—30) мин. Появля¬
ется красное окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: листья, изменившие окраску, — не бо¬
лее 5 %; другие части растения (стебли, цветки и дру¬
гие) — не более 3 %. Органические примеси: не более
3 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания суммы флавоно¬
идов в пересчете на рутин. 2,000 г измельченного
сырья (250) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 250 мл, прибавляют 80 мл спирта
(70 %, об/об) Р и кипятят с обратным холодильником
на водяной бане в течение 1 ч. Выдерживают в течение
2 ч и фильтруют в мерную колбу вместимостью 100 мл.
Круглодонную колбу и фильтр промывают 20 мл спир¬
та (70 %, об/об) Р в ту же мерную колбу и доводят до
объема 100,0 мл этим же растворителем (раствор А).
Буферный раствор 8. К 10 мл 7 М раствора на¬
трия гидроксида прибавляют 57 мл раствора 60 г/л
кислоты уксусной ледяной Р и доводят водой Р до
объема 100,0 мл.
Стандартный раствор. 0,0500 г рутина Р,
предварительно высушенного при температуре от
130 °С до 135 °С в течение 3 ч, растворяют в спирте
(70 %, об/об) Р при нагревании на водяной бане, ох¬
лаждают и доводят до объема 100,0 мл этим же раст¬
ворителем.
Испытуемый раствор. 5,0 мл раствора А поме¬
щают в мерную колбу вместимостью 25 мл, прибавля¬
ют 6 мл раствора 36,2 г/л алюминия хлорида Р в спир¬
те (70 %, об/об) Р и выдерживают в водяной бане в
Боярышника плоды
1197
течение 3 мин. Быстро охлаждают до комнатной тем¬
пературы, прибавляют 2 мл буферного раствора 5 и
доводят спиртом (70 %, об/об) Р до объема 25,0 мл.
Раствор сравнения. Готовят одновременно с ис¬
пытуемым раствором. 1,0 мл стандартного раствора
помещают в мерную колбу вместимостью 25 мл, при¬
бавляют 6 мл раствора 36,2 г/л алюминия хлорида Р в
спирте (70 %, об/об) Р и выдерживают в водяной бане
в течение 3 мин. Быстро охлаждают до комнатной тем¬
пературы, прибавляют 2 мл буферного раствора 3 и
доводят спиртом (70 %, об/об) Р до объема 25,0 мл.
Компенсационный раствор (а). 5,0 мл раствора
А помещают в мерную колбу вместимостью 25 мл,
прибавляют 2 мл буферного раствора 3 и доводят
спиртом (70 %, об/об) Р до объема 25,0 мл.
Компенсационный раствор (Ь). 1,0 мл стандарт¬
ного раствора помещают в мерную колбу вместимо¬
стью 25 мл, прибавляют 2 мл буферного раствора 3 и
доводят спиртом (70 %, об/об) Р до объема 25,0 мл.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора и раствора сравнения при 409 нм,
используя компенсационные растворы (а) и (Ь) соот¬
ветственно.
Содержание суммы флавоноидов в пересчете на
рутин в процентах рассчитывают по формуле:
А • т0 • 20
А0 т ’
где:
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески рутина Р, г.
Определение содержания суммы проциани-
динов в пересчете на цианидина хлорид. 1,000 г
измельченного сырья (250) (2.9.12) помещают в кру¬
глодонную колбу вместимостью 100 мл, прибавляют
20,0 мл спирта (70 %, об/об) Р, закрывают пробкой и
взвешивают с точностью до 0,01 г. Кипятят с обратным
холодильником на водяной бане в течение 30 мин, ох¬
лаждают и доводят спиртом (70 %, об/об) Р до перво¬
начальной массы. Полученный раствор центрифуги¬
руют при 3000 об/мин в течение 10 мин и собирают
надосадочную жидкость (раствор В).
Испытуемый раствор. 0,1 мл раствора В по¬
мещают в круглодонную колбу вместимостью 50 мл,
прибавляют 0,9 мл спирта (70 %, об/об) Р, 6 мл 5 %
(об/об) раствора кислоты хлористоводородной Р в
бутаноле Р, 0,2 мл раствора железа (III) аммония
сульфата Р7 и нагревают в водяной бане в течение
50 мин. Охлаждают.
Компенсационный раствор. К 1,0 мл спирта
(70 %, об/об) Р прибавляют 6 мл 5 % (об/об) раст¬
вора кислоты хлористоводородной Р в бутаноле Р
и 0,2 мл раствора железа (III) аммония сульфата Р7.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 550 нм.
Содержание суммы процианидинов в пересчете
на цианидина хлорид в процентах рассчитываю по
формуле:
А 1440
т -136 ’
где:
136 — удельный показатель поглощения продук¬
та реакции цианидина хлорида с железа (III) аммония
сульфатом;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1220
БОЯРЫШНИКА ПЛОДЫ
Сга(аед1 кис1из
НАШТНОКЫ ВЕКШЕ8
ОПРЕДЕЛЕНИЕ
Собранные в фазу полного созревания и высушен¬
ные плоды кустарников или небольших деревьев раз¬
личных видов: Сга1аедиз запдитеа Ра11.; С. 1аемда1а
(Ро1г) РС. (зуп.: С. охуасапИпа зепзи Ро]агк.); С. кого/коуИ
1_. Непгу; С. аНаюа (Ьоиб.) Ьалде; С. сЫогосагра 1_еппе
е* С. КосИ; С. баНипса КоеИпе ех ЗсИпе1с1.; С. топодупа
Засц.; С. а1етапп1епз1з С1п.; С. опеп1оЬаШса Ст.;
С. сип/1зера1а ипбт.; С. х сигот'са С1п.; С. х с1ипепз1з
Ст.
Содержание:
- не менее 0,06 % процианидинов в пересчете на
цианидина хлорид (С15Н10Об НС1; М.м. 322,7) (в пере¬
счете на сухое сырье);
- или не менее 0,06 % суммы флавоноидов в пе¬
ресчете на гиперозид (С21Н20О12; М.м. 464,4) (в пере¬
счете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Плоды яблоко¬
образные, от почти шаровидной до эллипсоидальной
формы, твердые, морщинистые, длиной (5—13) мм,
шириной (4—10) мм, сверху с кольцевой оторочкой,
образованной ссохшимися чашелистиками. В желто¬
ватой мякоти плода находятся 1—5 деревянистых ко¬
сточек, имеющих неправильную треугольную, оваль¬
ную или сжатую с боков форму. Поверхность косто¬
чек ямчато-морщинистая или бороздчатая по спинке.
Цвет плодов от желто-оранжевого и темно-красного
до коричневато-красного, коричневого или черного,
иногда с беловатым налетом выкристаллизовавшего¬
ся сахара.
@> В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Порошок имеет серовато¬
красный цвет. Порошок исследуют под микроскопом,
используя раствор хлоралгидрата Р. Порошок об¬
ладает следующими диагностическими признаками
(рисунок 1220.-1): покрывающие трихомы [Р] изну¬
три диска, длинные, одноклеточные, часто изогну¬
тые, суживающиеся до определенного положения,
с сильно утолщенными и одревесневшими стенка¬
ми; фрагменты красного наружного слоя цветоложа
(вид поверхности [О]); фрагменты внутренних слоев
цветоложа [А], несколько клеток, содержащих друзы
кристаллов [Аа] или призматические кристаллы [АЬ]
кальция оксалата; иногда встречаются фрагменты [3, К],
включающие группы склереид [Ка] и пучки сосудов
[ба, КЬ], объединенные с рядами клеток, содержащих
призматические кристаллы кальция оксалата [Лэ, Кс];
фрагменты перикарпия [В], состоящие из паренхимы,
включающей несколько клеток, содержащих друзы
кристаллов кальция оксалата [Ва] и группы склере¬
ид различных размеров с многочисленными ямками
[ВЬ]; толстостенные склереиды [Е, Н], некоторые из
которых бороздчатые [Е], а некоторые имеют заметно
1198
Гэсударстеенная фармакопея Республики Беларусь
разветвленные каналы [Н]; несколько фрагментов се¬
менной кожуры [С], имеющей наружный слой, состо¬
ящий из гексагональных, слизистых клеток [Са], ниже
которых расположен желтовато-коричневый пигмент¬
ный слой, содержащий многочисленные призмати¬
ческие кристаллы кальция оксалата [СЬ]; паренхима
эндосперма и семядоли, состоящая из клеток, содер¬
жащих зерна алейрона и гранулы зафиксированного
масла [О].
Рисунок 1220.-1. Микроскопические признаки
плодов боярышника
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р и
нагревают на водяной бане при 65 °С в течение 5 мин
при частом встряхивании. Охлаждают до комнатной
температуры и фильтруют. Фильтрат доводят мета¬
нолом Р до объема 10 мл.
Раствор сравнения. 2 мг хлорогеновой кисло¬
ты Р, 2 мг кофейной кислоты Р, 5 мг гиперозида Р и
5 мг рутина Р растворяют в 20 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метилэтилкетон Р — этилаце-
тат Р (10:10:30:50 об/об/об/об).
Наносимый объем пробы: 30 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: горячую пластинку опрыскивают
раствором 10 г/л дифенилборной кислоты аминоэ¬
тилового эфира Р в метаноле Р и затем раствором
50 г/л макрогола 400 Р в метаноле Р. Пластинку вы¬
сушивают на воздухе в течение 30 мин и просматри¬
вают в ультрафиолетовом свете при длине волны
365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора обнаруживаются и другие флуоресцирую¬
щие зоны.
Верх хроматографической пластинки
Кофейная кислота: флуо¬
ресцирующая зона светло-
синего цвета
Флуоресцирующие зоны
светло-синего цвета
Флуоресцирующая зона
светло-синего цвета
Флуоресцирующие зоны
светло-синего цвета
Гиперозид: флуоресцирую¬
щая зона желтовато-корич¬
невого цвета
Хлорогеновая кислота: флуо¬
ресцирующая зона светло-
синего цвета
Рутин: флуоресцирующая
зона желтовато-коричнево¬
го цвета
Две слабых флуоресциру¬
ющих зоны красноватого
цвета
Флуоресцирующая зона
желтовато-коричневого
цвета
Флуоресцирующая зона
светло-синего цвета
Слабая флуоресцирующая
зона красноватого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2).Несырьевые части
растения: подгоревшие плоды — не более 2 %; пло¬
ды недозрелые (буровато-зеленые) — не более 1 %;
плоды, поврежденные вредителями, дробленные, от¬
дельные косточки, плодоножки, в том числе отделен¬
ные при анализе, — не более 5 %. Органические при¬
меси: не более 1 %. Минеральные примеси: не более
0,5 %.
Потеря в массе при высушивании (2.2.32) Не
более 14,0 %. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
@> Общая зола (2.4.16). Не более 5,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
@> Определение содержания процианидинов
в пересчете на цианидина хлорид. К 2,50 г измель¬
ченного сырья (355) (2.9.12) прибавляют 30 мл спир¬
та (70 %, об/об) Р, нагревают с обратным холодиль¬
ником в течение 30 мин и фильтруют. Остаток про¬
мывают 10,0 мл спирта (70 %, об/об) Р. К фильтрату
прибавляют 15,0 мл кислоты хлористоводород¬
ной Р1 и 10,0 мл воды Р и нагревают с обратным хо¬
лодильником в течение 80 мин, после чего охлаждают,
фильтруют и промывают остаток спиртом (70 %, об/
об) Р до прекращения окрашивания фильтрата. Объ¬
ем фильтрата доводят спиртом (70 %, об/об) Р до
250,0 мл. 50,0 мл полученного раствора выпаривают
в круглодонной колбе до объема приблизительно 3 мл
и переносят в делительную воронку. Ополаскивают
круглодонную колбу последовательно 10 мл и 5 мл
воды Р и переносят в делительную воронку. Полу¬
ченный раствор встряхивают трижды с бутанолом Р
Боярышника цветки
1199
порциями по 15 мл. Органические слои объединяют и
доводят бутанолом Р до объема 100,0 мл.
Измеряют оптическую плотность (2.2.25) раст¬
вора при 555 нм.
Содержание процианидинов в пересчете на ци-
анидина хлорид в процентах рассчитывают по фор¬
муле:
Д-500
/77 -1200 ’
где:
1200 — удельный показатель поглощения циани-
дина хлорида;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
Определение содержания флавоноидов в
пересчете на гиперозид. 5,000 г измельченного сы¬
рья (2000) (2.9.12) помещают в круглодонную колбу со
шлифом вместимостью 100 мл, прибавляют 50,0 мл
96 % спирта Р, взвешивают с точностью до 0,01 г и
нагревают на кипящей водяной бане в течение 1 ч.
Колбу с содержимым охлаждают до комнатной тем¬
пературы, взвешивают и доводят массу колбы до
первоначальной 96 % спиртом Р. Полученный раст¬
вор процеживают через ватный тампон толщиной не
более 0,5 см, отбрасывая первые 15 мл фильтрата;
25.0 мл фильтрата переносят в круглодонную колбу
вместимостью 50 мл и выпаривают досуха в вакууме.
Сухой остаток обрабатывают дважды горячим рас¬
твором 100 г/л натрия хлорида Р порциями по 10 мл,
каждый раз нагревая содержимое колбы на водяной
бане в течение 2 мин. Раствор охлаждают, процежи¬
вают через ватный тампон, смоченный водой Р, на
колонку, заполненную полиамидом для колоночной
хроматографии Р [1,0 г полиамида для колоночной
хроматографии Р помещают в стаканчик вместимо¬
стью 50 мл, прибавляют 30 мл воды Р, перемешивают
и выливают через воронку в колонку диаметром 1,5 см
и высотой 25 см. В нижнюю часть колонки предвари¬
тельно помещают небольшой ватный тампон, смочен¬
ный водой Р. Колонку заполняют при открытом кране.
Элюирование проводят со скоростью 4 мл/мин, не
допуская обнажения поверхности сорбента. Толщина
слоя жидкости над сорбентом должна быть не менее
(4—5) мм]. Колонку промывают 30,0 мл воды Р, из них
10 мл используют для промывания фильтра, который
после этого убирают. Когда над сорбентом останется
слой жидкости толщиной (7—10) мм, водный элюат
отбрасывают. Элюирование суммы флавоноидов про¬
водят 25 мл 96 % спирта Р, который прибавляют в ко¬
лонку постепенно, порциями по 5 мл. Первые порции
элюата (бесцветные и прозрачные) собирают в граду¬
ированную пробирку вместимостью 10 мл, диаметром
около 1 см. Когда элюат приобретет окраску и обьем
окрашенного элюата в пробирке достигнет 1 мл, мер¬
ную пробирку убирают (граница раздела бесцветно¬
го водного и окрашенного спиртового слоев элюата
в пробирке хорошо различима визуально). Элюат из
пробирки отбрасывают. Последующие порции элю¬
ата собирают в мерную колбу вместимостью 25 мл.
Объем элюата в колбе доводят 96 % спиртом Р до
25.0 мл (раствор А).
Испытуемый раствор. 2,0 мл раствора А дово¬
дят 96 % спиртом Р до объема 10,0 мл.
Раствор сравнения. 2,0 мл раствора 1 г/л ФСО
гиперозида в 96 % спирте Р помещают в круглодон¬
ную колбу вместимостью 50 мл со шлифом и выпа¬
ривают досуха под вакуумом. Остаток в колбе обра¬
батывают дважды горячим раствором 100 г/л натрия
хлорида Р порциями по 10 мл, каждый раз нагревая
содержимое колбы на водяной бане в течение 2 мин,
и сливают раствор на вновь приготовленную колонку,
заполненную полиамидом для колоночной хромато¬
графии Р, процеживая через ватный тампон, смочен¬
ный водой Р. Элюат для измерения оптической плот¬
ности стандартного образца гиперозида получают
аналогично элюату суммы флавоноидов.
Компенсационный раствор. Получают аналогич¬
но элюату суммы флавоноидов путем пропускания
25.0 мл 96 % спирта Р через вновь приготовленную
колонку, заполненную полиамидом для колоночной
хроматографии Р в мерную колбу вместимостью
25 мл. Объем элюата доводят 96 % спиртом Р до
25.0 мл.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора и раствора сравнения при 365 нм.
Содержание суммы флавоноидов в пересчете на
гиперозид в процентах рассчитывают по формуле:
Ат0Р- 0,08
А0-т
где:
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески ФСО гиперозида, г;
Р — содержание гиперозида в ФСО, %.
07/2016:РБ0045
БОЯРЫШНИКА ЦВЕТКИ
Сгайед/ Лоз
НАМТНОПЫ Р/.ОИ/ЕК*
ОПРЕДЕЛЕНИЕ
Собранные в начале цветения и высушенные со¬
цветия кустарников или небольших деревьев: Сга1аедиз
запдшпеа Ра11.; С. /аемда/а (Ро1г) ОС. (зуп.: С. охуасапХЪа
зепзи Ро]агк.); С. кого1кот 1_. Непгу; С. аНаюа (1_ои<±)
1_апде; С. сЫогосагра 1_еппе е! С. КосН; С. аНаюа (1_оис1.)
1_апде; С. даЬипса КоеИпе ех 5сИпек1.; С. топодупа
*}асц.\ С. а1етапп1епз'13 Сю.; С. реп1адупа \Л/а1сЫ. е1 КК.;
С. опеп1оЬаШса Сю.; С. сигУ1зера1а Ыпс1т.; С. х сигопюа
Ст.; С. х с1ипепз1з Ст.
Содержание: не менее 0,5 % гиперозида
(^21Н2о^12* М.м. 464,4) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Смесь цельных
щитковидных, реже зонтиковидных соцветий и их
частей — отдельных цветков, бутонов, цветоножек,
лепестков, тычинок и пыльников. Цветки правиль¬
ные, с двойным околоцветником, состоящим из пяти
продолговато-треугольных, треугольных или узких
ланцетных зеленоватых чашелистиков и пяти оваль¬
ных коричневато- или желтовато-белых лепестков;
тычинок до 20, с красными пыльниками, столбиков
от 1 до 5; цветоножки обычно голые или слабо опу¬
шенные, длиной до 35 мм. Диаметр распустивших¬
ся цветков (10—15) мм, бутонов — (3—4) мм. Запах
слабый, своеобразный.
1200
Гэсударственная фармакопея Республики Беларусь
B. Микроскопия (2.8.23). При просматривании ча¬
шелистиков и лепестков с поверхности видны клетки
эпидермиса, имеющие с наружной стороны прямые или
слабо извилистые стенки и складчатую кутикулу. Устьи¬
ца крупные, редкие, аномоцитного типа (2.8.3), располо¬
женные на чашелистиках с наружной стороны. Клетки
внутреннего эпидермиса лепестков имеют сосочковид¬
ные выросты. По краю чашелистиков расположены мно¬
гоклеточные шаровидные железки (сидячие и на много¬
клеточных ножках) с желтовато-коричневым содержи¬
мым; на поверхности — многочисленные простые, од¬
ноклеточные волоски с толстыми стенками, гладкие, на
верхушке заостренные, прямые или слабо изогнутые, у
основания слегка расширенные и окруженные розеткой
из пяти эпидермальных клеток. В мезофилле чашели¬
стиков и завязи имеются друзы, изредка встречаются
призматические кристаллы оксалата кальция.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (355) (2.9.12) прибавляют 5 мл 96 % спирта Р и
кипятят с обратным холодильником на водяной бане в
течение 15 мин. Охлаждают, собирают надосадочную
жидкость.
Раствор сравнения. 5 мг гиперозида Р раство¬
ряют в 10 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: хлороформ Р — метанол Р
(80:20, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе в течение 2 мин.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается зона темно-коричневого
цвета на уровне зоны гиперозида на хроматограмме
раствора сравнения.
Проявление В: пластинку опрыскивают раство¬
ром 50 г/л алюминия хлорида Р в 96% спирте Р, на¬
гревают при температуре от 100 °С до 105 °С в тече¬
ние (2—3) мин и просматривают в ультрафиолетовом
свете при 365 нм и при дневном свете.
Результаты В: в ультрафиолетовом свете зоны,
соответствующие гиперозиду, приобретают желто-зе¬
леную флуоресценцию, при дневном свете — ярко-
желтую окраску.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (веточки, ли¬
стья) — не более 6 %. Органические примеси: не бо¬
лее 0,5 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор. 2,000 г измельченного сы¬
рья (2000) (2.9.12) помещают в колбу вместимостью
250 мл, прибавляют 100 мл 96 % спирта Р, взвешива¬
ют с точностью до 0,01 г и нагревают с обратным хо¬
лодильником на водяной бане в течение 1 ч. Охлажда¬
ют до комнатной температуры, взвешивают и доводят
массу колбы до первоначальной 96 % спиртом Р. Про¬
цеживают через ватный тампон толщиной не более 0,5
см, отбрасывая первые 30 мл фильтрата. 50,0 мл филь¬
трата помещают в круглодонную колбу со шлифом вме¬
стимостью 100 мл и упаривают в вакууме до объема
(2—3) мл. Доводят 96 % спиртом Р до объема 10,0 мл,
перемешивают, дают осесть образовавшемуся аморф¬
ному осадку и собирают надосадочную жидкость.
Раствор сравнения. 0,050 г гиперозида Р, высу¬
шенного при температуре от (105±2) °С, помещают в
колбу со шлифом вместимостью 100 мл, прибавляют
40 мл 96 % спирта Р и нагревают с обратным холо¬
дильником на водяной бане до полного растворения
кристаллов. Охлаждают и доводят 96 % спиртом Р
до объема 50,0 мл.
На линию старта ТСХ пластинки со слоем си¬
ликагеля Р наносят по 80 мкл раствора сравнения и
испытуемого раствора в виде полос длиной 5 см на
расстоянии 1,5 см от края. Пластинку с нанесенными
пробами высушивают на воздухе в течение 5 мин и
хроматографируют с использованием подвижной фазы
хлороформ Р — метанол Р (80:20, об/об) (камеру не
насыщают парами подвижной фазы). Когда фронт под¬
вижной фазы пройдет около 13 см, пластинку вынима¬
ют из камеры, высушивают на воздухе в течение 2 мин
и повторно хроматографируют в описанных выше ус¬
ловиях. Пластинку высушивают на воздухе в течение
2 мин и просматривают в ультрафиолетовом свете при
длине волны 365 нм. Отмечают зоны гиперозида на
хроматограммах раствора сравнения и испытуемого
раствора. Вырезают участки пластинки с зонами, а так¬
же чистый участок равной площади этой же пластинки
для контрольного опыта, разрезают каждый на кусочки
размером (0,3—0,5) см, помещают в колбы со шлифа¬
ми вместимостью 50 мл, прибавляют по 10,0 мл смеси
из диоксана Р и воды Р (50:50, об/об), закрывают проб¬
ками и встряхивают в течение 60 мин. Содержимое
колб переносят в центрифужные пробирки и центри¬
фугируют со скоростью 1000 об/мин в течение 5 мин.
Измеряют оптическую плотность (2.2.25) элюа-
тов испытуемого раствора и раствора сравнения при
365 нм, используя элюат контрольного опыта в каче¬
стве компенсационного раствора.
Содержание гиперозида в процентах рассчиты¬
вают по формуле:
А * т0 • 40
А0 т '
где:
А — оптическая плотность элюата испытуемого
раствора;
А0 — оптическая плотность элюата раствора
сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески гиперозида Р, г.
07/2016: РБ0046
БРУСНИКИ ЛИСТЬЯ
УасстН чШз-Шаеае ТоНит
СОМВЕВКУ 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные до начала цветения или после созре¬
вания плодов, высушенные листья многолетнего веч¬
нозеленого кустарничка Уасстшт уШзчдаеа 1~
Содержание арбутина (С12Н1607, М.м. 272,3) (в
пересчете на сухое сырье):
Брусники листья
1201
- не менее 4,5 %, определенное методом титро¬
вания (#2.2.90);
- или не менее 5,0 %, определенное методом
жидкостной хроматографии (2.2.29).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья коротко¬
черешковые, кожистые, эллиптические или обратно¬
яйцевидные, на верхушке притупленные или слабо¬
выемчатые с цельными или слегка зазубренными,
завернутыми вниз краями, длиной от 7 мм до 30 мм,
шириной от 5 мм до 15 мм. Листья сверху темно-зеле¬
ные, снизу светло-зеленые с ясно заметными темно-
коричневыми точками (железками).
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны слегка извилистые стенки
клеток верхнего и нижнего эпидермиса. Устьица толь¬
ко на нижнем эпидермисе мелкие, окружены двумя
околоустьичными клетками, расположенными парал¬
лельно устьичной щели (парацитный тип) (2.8.3). На
нижней стороне листа имеются железки, состоящие
из многоклеточной ножки, постепенно переходящей
в овальную многоклеточную головку с коричневым
содержимым. На верхней стороне листа по жилкам
встречаются редкие одноклеточные прямые или ду¬
говидно изогнутые волоски с толстыми стенками и
гладкой или слабобородавчатой поверхностью. В ме¬
зофилле содержатся редкие одиночные кристаллы
оксалата кальция в виде призм.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (355) (2.9.12) помещают в колбу, прибавляют 5 мл
смеси из метанола Р и воды Р (50:50, об/об) и кипятят
с обратным холодильником в водяной бане в течение
10 мин. Горячее извлечение фильтруют. Фильтр и кол¬
бу промывают смесью из метанола Р и воды Р (50:50,
об/об), объединяют промывные воды с фильтратом
и доводят смесью из метанола Р и воды Р (50:50,
об/об) до объема 5 мл.
Раствор сравнения. 2,5 мг арбутина Р раство¬
ряют в 5 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р.
Подвижная фаза: этилацетат Р — кислота му¬
равьиная безводная Р — вода Р (44:3:3, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: пластинку опрыскивают раствором
10 г/л 4-аминопиразолона Р, затем раствором 20 г/л
калия феррицианида Р и проявляют в парах аммиака.
Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Арбутин: зона красного
цвета
Зона красного цвета (пирозид)
Зона красного цвета (арбутин)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: листья, побуревшие и почерневшие с
обеих сторон, — не более 7 %; другие части расте¬
ния — не более 1 %. Органические примеси: не более
1 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 7,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания арбутина мето¬
дом титрования. 0,500 г измельченного сырья (710)
(2.9.12) помещают в колбу вместимостью 100 мл,
прибавляют 50 мл воды Р и нагревают, поддерживая
слабое кипение, в течение 30 мин. Горячее извлече¬
ние фильтруют в мерную колбу вместимостью 100 мл
через бумажный фильтр, избегая попадания частиц
сырья на фильтр. В колбу с сырьем повторно при¬
бавляют 25 мл воды Р и кипятят в течение 20 мин. Го-
рячее извлечение вместе с сырьем переносят на тот
же фильтр и остаток на фильтре дважды промывают
горячей водой Р порциями по 10 мл. К фильтрату при¬
бавляют 3 мл раствора свинца (II) ацетата основ¬
ного Р, перемешивают, охлаждают и доводят объем
фильтрата водой Р до 100,0 мл. Колбу помещают в
водяную баню и выдерживают до полной коагуляции
осадка. Горячую жидкость полностью отфильтро¬
вывают в колбу через бумажный фильтр, прикрывая
воронку часовым стеклом. Охлаждают, к фильтра¬
ту прибавляют 1 мл кислоты серной Р, колбу взве¬
шивают с точностью до 0,01 г и кипятят с обратным
холодильником в течение 1,5 ч, поддерживая равно¬
мерное и слабое кипение. Охлаждают до комнатной
температуры, взвешивают и доводят массу колбы
до первоначальной водой Р и полностью отфильтро¬
вывают в колбу вместимостью 250 мл через бумаж¬
ный фильтр. К фильтрату прибавляют 0,1 г порошка
цинка Р и встряхивают в течение 5 мин. Жидкость
нейтрализуют натрия гидрокарбонатом Р (около
(1—2) г) по красной лакмусовой бумаге Р, прибавляют
еще 2 г натрия гидрокарбоната Р и после его раст¬
ворения фильтруют в колбу через бумажный фильтр.
50,0 мл фильтрата переносят в плоскодонную
колбу вместимостью 500 мл, прибавляют 200 мл
воды Р и немедленно титруют из микро- или полуми-
кробюретки 0,05 М раствором йода до появления си¬
него окрашивания, не исчезающего в течение 1 мин,
используя в качестве индикатора раствор крахмала,
свободный от йодидов, Р.
1 мл 0,05 М раствора йода соответствует
13,61 мг арбутина (С12Н1607).
Определение содержания арбутина методом
жидкостной хроматографии.
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,000 г измельченного
сырья (710) (2.9.12) помещают в круглодонную колбу
вместимостью 250 мл, прибавляют 50,0 мл воды Р,
взвешивают с точностью до 0,01 г и нагревают с об¬
ратным холодильником в водяной бане в течение
45 мин. Охлаждают, доводят массу до первоначаль¬
ной и центрифугируют. Надосадочную жидкость филь¬
труют через мембранный фильтр с номинальным раз¬
мером пор 0,45 мкм.
151. Зак. 1060.
1202
Гэсударственная фармакопея Республики Беларусь
Раствор сравнения (а). 50,0 мг ФСО арбутина
растворяют в 50,0 мл воды Р.
Раствор сравнения (Ь). 2 мг гидрохинона Р раст¬
воряют в воде Р, прибавляют 2,0 мл раствора сравне¬
ния (а) и доводят водой Р до объема 25,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 30 °С;
- подвижная фаза:
- подвижная фаза А: 0,01 М раствор дигидро¬
фосфата калия, доведенный до рН 3,0±0,2 кис¬
лотой фосфорной Р\
- подвижная фаза В: ацетонитрил Р;
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—20
95 — 80
5 — 20
20—20,1
00
0
1
СО
сл
20 — 5
20,1—23
95
5
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 10 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 8,0 между пиком арбути¬
на и гидрохинона.
Рисунок РБ0046-1. Хроматограмма испытуемого
раствора
Содержание арбутина в процентах рассчитыва¬
ют по формуле:
8, т2 Р
32т,
где:
— площадь пика арбутина на хроматограмме
испытуемого раствора;
52 — площадь пика арбутина на хроматограмме
раствора сравнения;
ш1 —масса навески испытуемого сырья, г;
т2 — масса навески ФСО арбутина, исполь¬
зованного для приготовления раствора сравнения
(а), г;
Р — содержание арбутина в стандартном образ¬
це, %.
07/2016:1217
БУЗИНЫ ЧЕРНОЙ ЦВЕТКИ
8атЬиа Поз
Е1.0Е/? Р/.01УЕЯ
ОПРЕДЕЛЕНИЕ
Собранные в период цветения, высушенные
цветки и бутоны кустарника ЗатЬисиз Ыдга 1_.
Содержание:
- не менее 0,80 % флавоноидов в пересчете на
изокверцитрозид (С21Н20О12; М.м. 464,4) (в пересчете
на сухое сырье);
- или не менее 2,0 % флавоноидов в пересчете
на рутин (С27Н30О16; М.м. 611) (в пересчете на сухое
сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
®А. Внешние признаки (2.8.23). Цветки диа¬
метром около 5 мм с тремя маленькими прицвет¬
никами, видимыми при увеличении, может при¬
сутствовать цветоножка. Пятизубчатая чашечка
слабозаметная, венчик светло-желтого цвета с пя¬
тью широкоовальными лепестками, сросшимися у
основания с образованием трубочки. Тычиночные
нити пяти желтых тычинок чередуются с лепест¬
ками. Венчик часто отдельный или вместе с при¬
росшими у основания тычинками. Завязь нижняя с
коротким столбиком с тремя тупоконечными рыль¬
цами.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.12). Цвет зеленовато-
желтый. Просматривают под микроскопом с исполь¬
зованием раствора хлоралгидрата Р. В порошке
обнаруживаются следующие диагностические при¬
знаки (рисунок 1217.-1): многочисленные сфериче¬
ские или иногда эллипсоидальные зерна пыльцы
диаметром около 30 мкм с тремя зародышевыми
порами и очень мелко ямчатой экзиной [СЗ]; клетки
нижнего эпидермиса чашелистиков, часто с капля¬
ми масла, покрытые бороздчатой кутикулой при
просматривании с поверхности [А]; редкие фраг¬
менты краев чашелистиков с одноклеточными кра¬
евыми зубчиками в поперечном разрезе [Е]; фраг¬
менты лепестков с многочисленными маленькими
каплями эфирного масла [Н]; фрагменты верхнего
эпидермиса чашелистиков [В] или лепестков [Р]
при просматривании с поверхности со слабо и не¬
регулярно утолщенными стенками [Ва, Ра], аномо-
цитными устьицами (2.8.3) [ВЬ, РЬ] и бороздчатой
кутикулой; мезофильные клетки лепестков и ча¬
шелистиков с идиобластами, содержащими много¬
численные микроклиновидные кристаллы оксалата
кальция [Вс]; фрагменты пыльников в поперечном
разрезе [С] и при просматривании с поверхности
[О] с внешним слоем [Са] и клетками волокнистого
слоя [СЬ, Сс, О].
Бузины черной цветки
1203
Рисунок 1217.-1. Микроскопические признаки
цветков бузины
® С. Испытуемый раствор. К 0,5 г измель¬
ченного сырья (355) (2.9.12) прибавляют 5 мл ме¬
танола Р, обрабатывают ультразвуком в течение
10 мин и центрифугируют в течение 5 мин.
Раствор сравнения. 1 мг кофейной кисло¬
ты Р, 1 мг хлорогеновой кислоты Р, 2,5 мг гипе-
розида Р и 2,5 мг рутина Р растворяют в 10 мл
метанола Р
Пластинка: ТСХ пластинка со слоем силика¬
геля Р ((2—10) мкм).
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метилэтилкетон Р — этила-
цетат Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: по 4 мкл в виде по¬
лос длиной 8 мм.
Фронт подвижной фазы: не менее 6 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку нагревают в течение
5 мин при температуре 100 °С и обрабатывают рас¬
твором 1 г/л дифенилборной кислоты аминоэти¬
лового эфира Р в этилацетате Р и затем раство¬
ром 5 г/л макрогола 400 Р в метиленхлориде Р;
сушат пластинку на воздухе в течение 30 мин. Про¬
сматривают при дневном свете (результаты А) и в
ультрафиолетовом свете при длине волны 365 нм
(результаты В).
Результаты А: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения
и испытуемого раствора. На хроматограмме испы¬
туемого раствора могут обнаруживаться и другие
бледные зоны.
Верх хроматографической пластинки
Гиперозид: зона темно-
оранжевого цвета
Зона оранжевого цвета
Рутин: зона темно-желто¬
го цвета
Зона темно-желтого цвета
Раствор сравнения
Испытуемый раствор
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие бледные
зоны.
Верх хроматографической пластинки
Кофейная кислота: флуо¬
ресцирующая зона голубо¬
го цвета
Интенсивная флуоресциру¬
ющая зона светло-голубо¬
го цвета
2 флуоресцирующих зоны
светло-голубого цвета
Гиперозид: флуоресцирую¬
щая зона оранжевого цвета
Хлорогеновая кислота: флу¬
оресцирующая зона светло-
голубого цвета
Флуоресцирующая зона
оранжевого цвета
Интенсивная флуоресциру¬
ющая зона светло-голубо¬
го цвета
Рутин: флуоресцирующая
зона оранжевого цвета
Флуоресцирующая зона
оранжевого цвета
Раствор сравнения
Испытуемый раствор
ЧИСЛОВЫЕ ПОКАЗАТЕЛИ
(^Допустимые примеси (2.8.2). Несырьевые
части растения: побуревшие цветки — не более 15 %.
Фрагменты крупных цветоносов и другие посторонние
примеси: не более 8 %. Определение проводят из 10 г
испытуемого сырья.
ЗатЬисиз еЬи1из Ь. Просматривают хрома¬
тограммы, полученные в подлинности С.
Результаты В: на хроматограмме испытуемого
раствора не должна обнаруживаться флуоресциру¬
ющая зона зеленовато-белого цвета, расположенная
выше зоны хлорогеновой кислоты на хроматограмме
раствора сравнения; на хроматограмме испытуемого
раствора не должна обнаруживаться флуоресциру¬
ющая зона зеленого цвета непосредственно ниже
флуоресцирующей зоны оранжевого цвета на хрома¬
тограмме раствора сравнения.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 10,0 %.
151*. Зак. 1060.
1204
Гэсударственная фармакопея Республики Беларусь
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания флавоноидов в
пересчете на изокверцитрозид.
Исходный раствор. 0,600 г измельченного сы¬
рья (355) (2.9.12) помещают в круглодонную колбу
вместимостью 100 мл, прибавляют 1 мл раствора
5 г/л гексаметилентетрамина Р, 20 мл ацетона Р
и 2 мл кислоты хлористоводородной Р1. Кипятят с
обратным холодильником в течение 30 мин, после
чего извлечение процеживают через ватный тампон
в колбу. Ватный тампон помещают в круглодонную
колбу с остатком сырья и дважды кипятят с обрат¬
ным холодильником в течение 10 мин, каждый раз
прибавляя 20 мл ацетона Р в качестве экстрагента.
Охлаждают и процеживают через ватный тампон.
Объединенные ацетоновые извлечения фильтруют
через бумажный фильтр в мерную колбу вмести¬
мостью 100 мл, ополаскивают колбу ацетоном Р и
фильтруют через тот же фильтр, после чего промы¬
вают фильтр ацетоном Р. Объем раствора доводят
ацетоном Р до 100,0 мл. 20,0 мл полученного раст¬
вора помещают в делительную воронку, прибавля¬
ют 20 мл воды Р и экстрагируют этилацетатом Р
порцией 15 мл, затем еще трижды порциями по
10 мл. Этилацетатные извлечения объединяют, по¬
мещают в делительную воронку и дважды промыва¬
ют водой Р порциями по 50 мл. Органический слой
фильтруют через бумажный фильтр с 10 г натрия
сульфата безводного Р в мерную колбу вместимо¬
стью 50 мл и доводят этилацетатом Р до объема
50,0 мл.
Испытуемый раствор. К 10,0 мл исходного
раствора прибавляют 1 мл реактива алюминия хло¬
рида Р и доводят 5 % (об/об) раствором кислоты ук¬
сусной ледяной Р в метаноле Р до объема 25,0 мл.
Компенсационный раствор. 10,0 мл раствора
А доводят 5 % (об/об) раствором кислоты уксусной
ледяной Р в метаноле Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 425 нм.
Содержание флавоноидов в пересчете на изо¬
кверцитрозид в процентах рассчитывают по фор¬
муле:
А- 625
т■500’
где:
500 — удельный показатель поглощения изо-
кверцитрозида;
А — оптическая плотность испытуемого раст¬
вора;
/77 — масса навески испытуемого сырья, г.
Определение содержания суммы флавонои¬
дов в пересчете на рутин.
Исходный раствор. 1,000 г измельченного сы¬
рья (710) (2.9.12) помещают в колбу со шлифом вме¬
стимостью 250 мл, прибавляют 100 мл спирта (70 %,
об/об) Р, взвешивают с точностью до 0,01 г и нагрева¬
ют с обратным холодильником на водяной бане в те¬
чение 30 мин, периодически встряхивая для смывания
частиц со стенок колбы. Охлаждают до комнатной тем¬
пературы, доводят массу колбы до первоначальной
спиртом (70 %, об/об) Р и фильтруют через бумажный
фильтр, отбрасывая первые 10 мл фильтрата.
Испытуемый раствор. К 2,0 мл раствора А
прибавляют 15 мл 96 % спирта Р, 0,5 мл кислоты
уксусной разведенной Р, 3 мл раствора 50 г/л алю¬
миния хлорида Р в спирте (70 %, об/об) Р, 2 мл
раствора 50 г/л гексаметилентетрамина Р и дово¬
дят водой Р до объема 25,0 мл.
Компенсационный раствор. К 2,0 мл раствора
А прибавляют 15 мл 96 % спирта Р, 0,5 мл кисло¬
ты уксусной разведенной Р, 2 мл раствора 50 г/л
гексаметилентетрамина Р и доводят водой Р до
объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 407 нм.
Содержание суммы флавоноидов в пересчете
на рутин в процентах рассчитывают по формуле:
А-1250
т-220’
где:
220 — удельный показатель поглощения ком¬
плекса рутина с алюминия хлоридом;
А—оптическая плотность испытуемого раствора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0031
ВАЛЕРИАНЫ КОРНЕВИЩА
С КОРНЯМИ
Уа/епапае гЫгота сит гасЛаЬиз
УА/.ЕЯ/>Ш ЯООГ
ОПРЕДЕЛЕНИЕ
Высушенные целые, фрагментированные или
измельченные подземные части растения Уа\епапа
оШс/паНз Ь. 8.1., включая корневища с корнями и сто¬
лонами.
Содержание:
- цельное и фрагментированное сырье:
-не менее 0,17% (м/м) суммы сесквитерпе-
новых кислот в пересчете на валереновую кис¬
лоту (015Н22О2, М.м. 234,3) (в пересчете на сухое
сырье);
- или не менее 0,70 % валепотриатов в пере¬
счете на пирилиевую соль валтрата (в пересчете
на сухое сырье) и не менее 2,0 % суммы сложных
эфиров в пересчете на этиловый эфир валере-
новой кислоты (017Н26О2; М.м. 262,4) (в пересчете
на сухое сырье);
- измельченное сырье:
-не менее 0,10 % (м/м) суммы сесквитерпе-
новых кислот в пересчете на валереновую кис¬
лоту (С15Н2202> М.м. 234,3) (в пересчете на сухое
сырье);
- или не менее 0,70 % валепотриатов в пере¬
счете на пирилиевую соль валтрата (в пересчете
на сухое сырье) и не менее 2,0 % суммы сложных
эфиров в пересчете на этиловый эфир валере-
новой кислоты (С17Н2602; М.м. 262,4) (в пересчете
на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Целые и фраг¬
ментированные подземные части растения пред¬
ставляют собой корневища от желтовато-серого до
Валерианы корневища с корнями
1205
бледного коричневато-серого цвета, от конической
до цилиндрической формы, длиной приблизительно
до 50 мм и диаметром 30 мм, основание удлинен¬
ное или сжатое, обычно полностью покрытое много¬
численными корнями. На верхушке обычно имеется
чашеобразный рубец от надземных частей; иногда
присутствуют основания стеблей. На продольном
разрезе в центре сердцевины видна полость с по¬
перечными перегородками. Корни многочисленные,
почти цилиндрической формы, цвет такой же, как у
корневищ, диаметром от 1 мм до 3 мм, длина ино¬
гда превышает 100 мм. Присутствует небольшое ко¬
личество нитевидных хрупких вторичных корней. На
столонах имеются выступающие узлы, разделенные
бороздчатыми междоузлиями длиной от 20 мм до
50 мм, имеющими волокнистый излом.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет серовато-коричневый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки: клетки,
содержащие бледно-коричневую смолу или капле¬
видные включения эфирного масла; группы неболь¬
ших прямоугольных склереид с толстыми стенками;
редкие группы более крупных склереид основания
стебля с более тонкими клеточными стенками; лиг-
нифицированные сосуды с сетчатыми утолщениями,
встречающиеся группами или по отдельности; от¬
дельные фрагменты клеток коры и эпидермических
клеток, на некоторых имеются корневые волоски;
редкие фрагменты пробки.
При использовании 50 % (об/об) раствора гли¬
церина Р в измельченном сырье (355) (2.9.12) вид¬
ны многочисленные крахмальные зерна, в основном
сложные, содержащие до 4—6 составляющих, но ча¬
сто распадающиеся на отдельные гранулы округлой
или неправильной формы диаметром до 15 мкм;
большинство гранул имеют неотчетливые трещинки
или радиальный центр наслоения.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 г измельченного сырья
(355) (2.9.12) суспендируют в 10 мл метанола Р и
обрабатывают ультразвуком в течение 10 мин. На-
досадочную жидкость фильтруют через мембранный
фильтр с размером пор 0,45 мкм. Используют филь¬
трат.
Раствор сравнения. 5 мг кислоты ацетоксива-
лереновой Р и 5 мг кислоты валереновой Р раство¬
ряют в 20 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р ((2—10) мкм)].
Подвижная фаза: кислота уксусная ледя¬
ная Р— этилацетат Р — циклогексан Р (2:38:60,
об/об/об).
Наносимый объем пробы: по 20 мкл [или 5 мкл]
в виде полос длиной 10 мм [или 8 мм].
Фронт подвижной фазы: не менее 10 см [6 см]
от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р. Нагревают при темпе¬
ратуре от 100 °С до 105 °С в течение (5—10) мин.
Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны фио¬
летового цвета.
Верх хроматографической пластинки
Валереновая кислота: зона
фиолетового цвета
Ацетоксивалереновая кис¬
лота: зона фиолетового
цвета
Зона фиолетового цвета
(валереновая кислота)
Зона фиолетового цвета
(ацетоксивалереновая кис¬
лота)
Две слабые или очень
слабые зоны фиолетово¬
го цвета
Раствор сравнения
Испытуемый раствор
й. 10 мл раствора А, приготовленного как ука¬
зано в разделе «Количественное определение со¬
держания валепотриатов в пересчете на пирилиевую
соль валтрата», выпаривают под вакуумом на водя¬
ной бане при температуре от 70 °С до 80 °С. К полу¬
ченному остатку прибавляют 5 мл кислоты уксусной
ледяной Р и 5 мл кислоты хлористоводородной Р1.
Появляется желто-зеленое окрашивание, постепенно
переходящее в интенсивное синее окрашивание.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: остатки листьев и стеблей, в том чис¬
ле и отделенные при анализе, а также старые от¬
мершие корневища — не более 5 %. Органические
примеси: не более 2 %. Минеральные примеси: не
более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 15,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 13,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания суммы сескви-
терпеновых кислот. Жидкостная хроматография
(2.2.29).
Испытуемый раствор. 1,50 г измельченного
сырья (710) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 100 мл со шлифом, прибавляют
20 мл метанола Р1, перемешивают и нагревают на
водяной бане с обратным холодильником в течение
30 мин. Охлаждают и фильтруют. Фильтр с остатком
помещают в ту же круглодонную колбу вместимостью
100 мл, прибавляют 20 мл метанола Р1 и нагревают
на водяной бане с обратным холодильником в тече¬
ние 15 мин. Охлаждают и фильтруют. Фильтраты объ¬
единяют и доводят метанолом Р1 до объема 50,0 мл,
ополаскивая круглодонную колбу и фильтр этим же
растворителем.
Раствор сравнения. Количество ФСО сухого
экстракта валерианы стандартизированного, со¬
ответствующее 1,0 мг валереновой кислоты, раство¬
ряют в метаноле Р1 и доводят до объема 10,0 мл
этим же растворителем. Обрабатывают ультразвуком
и фильтруют через мембранный фильтр с размером
пор 0,45 мкм.
152. Зак. 1060.
1206
Гэсударственная фармакопея Республики Беларусь
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: ацетонитрил Р1 —
раствор 5 г/л фосфорной кислоты Р (20:80,
об/об);
- подвижная фаза В: раствор 5 г/л фосфор¬
ной кислоты Р1 — ацетонитрил Р (20:80,
об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
55
45
5—18
55 — 20
45-80
18—20
20
80
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 220 нм;
- объем вводимой пробы: 20 мкл.
Идентификация пиков: для идентификации
пиков ацетоксивалереновой кислоты и валерено-
вой кислоты на хроматограмме раствора сравне¬
ния используют хроматограмму, прилагаемую к
ФСО сухого экстракта валерианы стандартизи¬
рованного.
Пригодность хроматографической системы:
раствор сравнения:
- относительное время удерживания (по от¬
ношению к валереновой кислоте, время удержива¬
ния — около 19 мин): ацетоксивалереновая кисло¬
та — около 0,5.
Содержание суммы сесквитерпеновых кислот
в пересчете на валереновую кислоту в процентах
рассчитывают по формуле:
(3,+32)т2 -Р-5
8,171,
где:
51 — площадь пика ацетоксивалереновой кисло¬
ты на хроматограмме испытуемого раствора;
$2 — площадь пика валереновой кислоты на хро¬
матограмме испытуемого раствора;
$3 — площадь пика валереновой кислоты на хро¬
матограмме раствора сравнения;
т^ — масса навески испытуемого сырья, г;
т2 — масса навески ФСО сухого экстракта ва¬
лерианы стандартизированного, г;
Р — содержание валереновой кислоты в ФСО су¬
хого экстракта валерианы стандартизированного, %.
Определение содержания валепотриатов в
пересчете на пирилиевую соль валтрата. К 2,000 г
измельченного сырья (710) (2.9.12) прибавляют
200 мл смеси спирт (95 %, об/об) Р — хлороформ Р
(5:95, об/об) и встряхивают в течение 1 ч. Содержи¬
мое колбы фильтруют через бумажный фильтр «си¬
няя лента» в мерную колбу вместимостью 250 мл,
промывая остаток на фильтре смесью спирт (95 %,
об/об) Р — хлороформ Р (5:95, об/об) порциями по
20 мл, 20 мл и 10 мл и присоединяя промывную жид¬
кость к фильтрату в мерной колбе, и доводят до объ¬
ема 250,0 мл этим же растворителем (раствор А).
Испытуемый раствор. 50,0 мл раствора (А) вы¬
паривают досуха под вакуумом при остаточном дав¬
лении (20—40) мм рт. ст. при температуре не выше
45 °С. К полученному остатку прибавляют 50,0 мл
смеси кислота хлористоводородная Р — кислота
уксусная ледяная Р (36:25, об/об), встряхивают в те¬
чение 20 мин и выдерживают в течение (16—18) ч.
Полученный раствор фильтруют через бумажный
фильтр «синяя лента».
Компенсационный раствор. Кислота хлористо¬
водородная Р — кислота уксусная ледяная Р (36:25,
об/об).
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 597 нм.
Содержание валепотриатов в пересчете на пири¬
лиевую соль валтрата в процентах рассчитывают по
формуле:
А- 250
т-91,1 '
где:
91,1 — удельный показатель поглощения пири-
лиевой соли валтрата;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Определение содержания суммы сложных
эфиров в пересчете на этиловый эфир валере¬
новой кислоты. 5,000 г измельченного сырья (500)
помещают в коническую колбу со шлифом вмести¬
мостью 100 мл, прибавляют 50 мл смеси хлоро¬
форм Р — 96% спирт Р (5:1, об/об) и встряхивают в
течение 45 мин. Фильтруют через бумажный фильтр,
смоченный смесью хлороформ Р — 96% спирт Р
(5:1, об/об), в мерную колбу вместимостью 100 мл,
избегая попадания частиц сырья на фильтр. В кол¬
бу с остатком сырья прибавляют 40 мл смеси хлоро¬
форм Р — 96% спирт Р (5:1, об/об) и встряхивают в
течение 15 мин. Фильтруют в ту же мерную колбу и
доводят смесью хлороформ Р — 96% спирт Р (5:1,
об/об) до объема 100,0 мл (раствор А).
Испытуемый раствор. 5,0 мл раствора А по¬
мещают в круглодонную колбу вместимостью 50 мл
и выпаривают в вакууме при температуре от 40 °С
до 50 °С досуха. К полученному сухому остатку при¬
бавляют 5,0 мл гидроксиламина раствора щелочно¬
го РЗ, выдерживают в течение 20 мин, прибавляют
10,0 мл 1 М раствора хлористоводородной кисло¬
ты, 5,0 мл раствора 10 г/л железа (III) хлорида Р в
0,1 М растворе хлористоводородной кислоты, пере¬
мешивают и фильтруют через бумажный фильтр, смо¬
ченный водой Р.
Компенсационный раствор. К 5,0 мл гидрокси¬
ламина раствора щелочного РЗ прибавляют 10,0 мл
1 М раствора хлористоводородной кислоты и 5,0 мл
раствора 10 г/л железа (III) хлорида Рв0,1 М раство¬
ре хлористоводородной кислоты.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 512 нм.
Содержание суммы сложных эфиров в пересчете
на этиловый эфир валереновой кислоты в процентах
рассчитывают по формуле:
А -400
т -10,5 ’
где:
10,5 — удельный показатель поглощения гидрок-
самата валереновой кислоты;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Вахты трехлистной листья
1207
07/2016:РБ0047
ВАСИЛЬКА СИНЕГО ЦВЕТКИ
Сеп1аигеае суап\ Лоз
ВШЕ СОМН-ОМЕК*
ОПРЕДЕЛЕНИЕ
Собранные в период цветения и высушенные
краевые и срединные цветки одно- двухлетнего тра¬
вянистого растения Сеп(аигеа суапиз 1_.
Содержание: не менее 0,6 % суммы антоцианов
в пересчете на цианидин-3,5-дигликозид (в пересчете
на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь краевых
и срединных цветков. Краевые цветки бесполые, во¬
ронковидные, длиной до 2 см, венчиковидные, не¬
правильной формы, с 5—8 глубоко надрезанными
ланцетовидными долями отгиба и трубчатым осно¬
ванием до 6 мм длиной. Срединные — обоеполые,
трубчатые, длиной около 1 см, оканчивающиеся пя¬
тью прямыми зубцами, от середины к основанию рез¬
ко суженные. Тычинок пять, со свободными шерсти¬
стыми нитями и сросшимися пыльниками. Пестик с
нижней завязью. Цвет краевых цветков синий, у осно¬
вания бесцветный; срединных — сине-фиолетовый.
Запах слабый.
B. Микроскопия (2.8.23). Клетки эпидермиса
краевых цветков с обеих сторон вытянутые, с зао¬
стренными концами и извилистыми стенками. В труб¬
чатой части цветка стенки клеток прямые или слабо
волнистые. В тканях трубочки содержатся многочис¬
ленные призматические кристаллы оксалата кальция.
Эпидермис трубчатых цветков имеет аналогичную
структуру, но с более мелкими клетками. Встречаются
зерна пыльцы овальной формы.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: цветочные корзинки — не более 1 %;
цветки, потерявшие естественную окраску, — не бо¬
лее 10 %. Органические примеси: не более 0,5 %. Ми¬
неральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Растворитель 3. 20,0 мл кислоты хлористово¬
дородной Р1 доводят водой Р до объема 500,0 мл.
Испытуемый раствор. 0,3 г измельченного сырья
(710) (2.9.12) помещают в колбу вместимостью 250 мл,
прибавляют 100 мл растворителя 3, выдерживают в
водяной бане при температуре от 40 °С до 45 °С в те¬
чение 15 мин. Извлечение процеживают через ватный
тампон в мерную колбу вместимостью 250 мл. Ватный
тампон с сырьем снова помещают в колбу, прибавляют
100 мл растворителя 3, предварительно смывая части¬
цы сырья с воронки в колбу, и повторяют экстрагиро¬
вание указанным выше способом. Содержимое колбы
процеживают через ватный тампон в ту же мерную кол¬
бу. Сырье на ватном тампоне промывают 40 мл раство¬
рителя 3. После охлаждения доводят объем извлече¬
ния растворителем 3 до 250,0 мл. Полученное извле¬
чение фильтруют через бумажный фильтр, отбрасывая
первые 10 мл фильтрата.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 510 нм, используя растворитель 3
в качестве компенсационного раствора.
Содержание суммы антоцианов в пересчете на
цианидин-3,5-дигликозид в процентах рассчитывают
по формуле:
А 250
453 • т '
где:
453 — удельный показатель поглощения
цианидин-3,5-дигликозида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1605
ВАХТЫ ТРЕХЛИСТНОЙ ЛИСТЬЯ
МепуапШсИ5 МТоШае IЬНит
ВОвВЕАЫ/.ЕАР
ОПРЕДЕЛЕНИЕ
Высушенные цельные или фрагментированные
листья МепуапМез МоНа!а 1_.
ОПИСАНИЕ
Очень горький и стойкий вкус.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
@) А. Внешние признаки (2.8.23). Листья с длин¬
ным черешком, трехлистные с длинной обверткой у
основания; черешок диаметром до 5 мм с выражен¬
ной продольной бороздчатостью. Листовая пластинка
разделена на три равных сидячих листочка обратно¬
яйцевидной формы длиной до 10 см и шириной до
5 см с цельным, иногда извилистым краем, с корич¬
неватыми или красноватыми гидатодами и лопато¬
видным основанием; листья гладкие с темно-зеленой
верхней поверхностью и бледно-зеленой нижней, с
широкой беловатой мелкобороздчатой выступающей
главной жилкой.
О») В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет желтовато-зеленый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки: фрагмен¬
ты верхнего эпидермиса с многогранными клетками и
тонкие волнистые стенки; фрагменты нижнего эпидер¬
миса с извилистыми стенками; аномоцитные устьица
(2.8.3) на обеих сторонах листа, прилегающие клет¬
ки с радиальной бороздчатостью; клетки эпидерми¬
са жилок прямостенные с бородавчатые; фрагменты
паренхимы мезофилла с большими межклеточными
пространствами (аэренхима); клетки неправильной
формы с редкими склереидами; фрагменты спираль¬
ных или кольцеобразных сосудов.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл метанола Р и на¬
гревают в водяной бане при температуре 60 °С в тече¬
ние 5 мин при перемешивании. Охлаждают, фильтру¬
ют и выпаривают досуха при пониженном давлении
152*. Зак. 1060.
1208
Гэсударственная фармакопея Республики Беларусь
при температуре 60 °С. Остаток растворяют в 2,0 мл
метанола Р.
Раствор сравнения. 5 мг логанина Р растворяют
в 15 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: вода Р — метанол Р — этила-
цетат Р (8:15:77, об/об/об).
Наносимый объем пробы: по 30 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С до
105 °С в течение 10 мин. Просматривают при дневном
свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора обнаруживаются и другие зоны.
Верх хроматографической пластинки
Логанин: зона серовато-фи¬
олетового цвета
Зона фиолетового цвета
Интенсивная зона синего
цвета
Зона от фиолетового до се¬
ровато-фиолетового цвета
Зона от серого до серовато-
синего цвета
Зона коричневатого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: листья с черешками длиннее 3 см —
не более 8 %; отдельные черешки — не более 3 %;
почерневшие и побуревшие листья — не более 5 %.
Органические примеси: не более 1 %. Минеральные
примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
<§> Показатель горечи (2.8.15). Не менее 3000.
07/2016:1828
(©ГИНКГО листья
Откдотз Шит
СШКОО 1.ЕАР
ОПРЕДЕЛЕНИЕ
Целые или фрагментированные высушенные ли¬
стья Сткдо ЬНоЬа 1_.
Содержание: не менее 0,5 % флавоноидов в пе¬
ресчете на флавоновые гликозиды (М.м. 757) (в пере¬
счете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Лист сероватый,
желтовато-зеленый или желтовато-коричневый. Верх¬
няя поверхность немного темнее нижней. Черешки
длиной около (4—9) см. Листовая пластинка шириной
около (4—10) см, веерообразная, обычно двухло¬
пастная или иногда неразделенная. Обе поверхности
гладкие, жилкование дихотомическое, жилки рас¬
ходятся по радиусу от основания; они одинаково за¬
метны на обеих поверхностях листа. Дистальный край
неравномерно и в разной степени рассечен, несимме¬
трично двухлопастной или выемчатый. Латеральные
края цельные и постепенно сужаются по направлению
к основанию.
В. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет сероватый, желтовато¬
зеленый или желтовато-коричневый. Просматривают
под микроскопом с использованием раствора хло¬
ралгидрата Р. В порошке обнаруживаются следую¬
щие диагностические признаки (рисунок 1828.-1): фраг¬
менты листовой пластинки неправильной формы
[А, В, О, Е] с верхним эпидермисом (вид с поверхности
Р] и поперечный срез [Е]), состоящим из удлиненных
клеток с неравномерно извилистыми стенками ра],
часто с прилегающей палисадной паренхимой РЬ],
и нижним эпидермисом (вид с поверхности [А] и по¬
перечный срез [В]), состоящим из небольших клеток с
мелко исчерченной кутикулой и небольшими сосочка¬
ми у каждой [Аа] и глубоко погруженного устьичного
комплекса [АЬ] шириной около 60 мкм из 6—8 вспо¬
могательных клеток; фрагменты сосудистой ткани из
черешка и жилок [С] с ксилемой [Са] и паренхимой,
отдельные клетки содержат многочисленные друзы
кристаллов кальция оксалата разных размеров [СЬ].
25 ут
Рисунок 1828.-1. Микроскопические признаки
листьев гинкго
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 2,0 г измельченного
сырья (710) (2.9.12) прибавляют 10 мл метанола Р и
нагревают в водяной бане при температуре 65 °С в
течение 10 мин, часто встряхивая. Охлаждают до ком¬
натной температуры и фильтруют.
Гинкго листья
1209
Раствор сравнения. 1,0 мг хлорогеновой кисло¬
ты Р и 3,0 мг рутина Р растворяют в 20 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р
Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат Р (7,5:7,5:17,5:67,5, об/об/об/об).
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 17 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: горячую пластинку опрыскивают
раствором 10 г/л дифенилборной кислоты амино¬
этилового эфира Р в метаноле Р и затем таким же
объемом раствора 50 г/л макрогола 400 Р в мета¬
ноле Р. Пластинку высушивают на воздухе в течение
около 30 мин и просматривают в ультрафиолетовом
свете при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться другие слабые флуо¬
ресцирующие зоны.
Верх хроматографической пластинки
Хлорогеновая кислота: флу¬
оресцирующая зона светло-
синего цвета
Рутин: флуоресцирующая
зона желтовато-коричнево¬
го цвета
Флуоресцирующая зона
желтовато-коричневого
цвета
Флуоресцирующая зона зе¬
леного цвета
Две флуоресцирующие
зоны желтовато-коричнево¬
го цвета
Интенсивная флуоресциру¬
ющая зона светло-синего
цвета, иногда перекрываю¬
щаяся с флуоресцирующей
зоной зеленовато-коричне¬
вого цвета
Флуоресцирующая зона зе¬
леного цвета
Две флуоресцирующие
зоны желтовато-коричнево¬
го цвета
Флуоресцирующая зона зе¬
леного цвета
Флуоресцирующая зона
желтовато-коричневого
цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побеги — не более 5 %. Другие допу¬
стимые примеси: не более 2 %.
Потеря в массе при высушивании (2.2.32).
Не более 11,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 11,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Флавоноиды. Жидкостная хроматография
(2.2.29).
Испытуемый раствор. К 2,500 г измельченного сы¬
рья (710) (2.9.12) прибавляют 50 мл 60 % (об/об) раст¬
вора ацетона Р, нагревают с обратным холодильником
в течение 30 мин и фильтруют. Повторяют экстракцию,
используя 40 мл 60 % (об/об) раствора ацетона Р, и
фильтруют. Фильтраты объединяют и доводят 60 %
(об/об) раствором ацетона Р до объема 100,0 мл.
50,0 мл полученного раствора упаривают до удале¬
ния ацетона и переносят в мерную колбу вместимо¬
стью 50 мл с помощью 30 мл метанола Р, прибавля¬
ют 4,4 мл кислоты хлористоводородной Р1, доводят
водой Р до объема 50,0 мл и центрифугируют. 10 мл
надосадочной жидкости помещают в сосуд из темно¬
го стекла вместимостью 10 мл, закрывают резиновой
пробкой с алюминиевым колпачком и нагревают на во¬
дяной бане в течение 25 мин. Охлаждают до комнатной
температуры.
Раствор сравнения. 10,0 мг кверцетина диги¬
драта Р растворяют в 20 мл метанола Р. Прибавля¬
ют 15,0 мл кислоты хлористоводородной разведен¬
ной Р и 5 мл воды Р и доводят метанолом Р до объ¬
ема 50,0 мл.
Условия хроматографирования:
-колонка длиной 0,125 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц
5 мкм;
- температура: 25 °С;
- подвижная фаза:
- подвижная фаза А: раствор 0,3 г/л кислоты
фосфорной Р, доведенный до рН 2,0;
- подвижная фаза В: метанол Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—1
60
40
1—20
60 —»45
40 —► 55
20—21
45-»0
55-» 100
21—25
0
100
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 370 нм;
- объем вводимой пробы: 10 мкл.
Относительные времена удерживания (по от¬
ношению к кверцетину, время удерживания — около
12,5 мин): кемпферол — около 1,4; изорамнетин —
около 1,5.
Пригодность хроматографической системы:
- разрешение: не менее 1,5 между пиками кемп-
ферола и изорамнетина.
Пики, элюируемые до пика кверцетина или после
пика изорамнетина, на хроматограмме испытуемого
раствора не учитывают.
Содержание флавоноидов в пересчете на фла-
воновые гликозиды в процентах рассчитывают по
формуле:
2 Е, /т? ! 2 ,514 р
Р2т2
где:
Р^ — сумма площадей всех учитываемых пиков
на хроматограмме испытуемого раствора;
Р2 — площадь пика кверцетина на хроматограм¬
ме раствора сравнения;
/Т71 — масса навески кверцетина, используемого
для приготовления раствора сравнения, г;
т2 — масса навески сырья, используемого для
приготовления испытуемого раствора, г;
р — содержание безводного кверцетина в квер¬
цетине дигидрате Р, %.
1210
Гэсударственная фармакопея Республики Беларусь
07/2016:0392
<§> ГОРЕЧАВКИ КОРНИ
СепИапае гасИх
ОЕЫТ1АЫ ПООТ
ОПРЕДЕЛЕНИЕ
Высушенные фрагментированные подземные
части СепИапа Меа 1_.
ОПИСАНИЕ (СВОЙСТВА)
Имеют сильный и устойчивый горький вкус.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Представляют
собой одиночные или ветвистые части корней суб¬
цилиндрической формы различной длины (обычно
(5—15) см), диаметром обычно (5—40) мм. Поверх¬
ность желтовато-коричневая или серовато-коричне¬
вая, цвет на изломе желтоватый или красновато-жел¬
тый, но не красновато-коричневый. Корень продоль¬
но морщинистый с редкими следами от отростков.
Ответвления корневища часто имеют терминальную
почку и всегда окружены близко расположенными
рубцами от листьев. Корневище и корни в сухом виде
хрупкие и легко ломаются, но они быстро поглощают
влагу и становятся гибкими. На ровном поперечном
срезе видна кора, занимающая около одной четвер¬
той радиуса, отделенная четко заметным камбием от
неотчетливо радиально-симметричной, в основном,
паренхиматозной ксилемы.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет светло-коричне¬
вый или желтовато-коричневый. Просматривают
под микроскопом с использованием раствора хло¬
ралгидрата Р. В порошке обнаруживаются следу¬
ющие диагностические признаки (рисунок 0392.-1):
фрагменты пробки с многогранными, тонкостенны¬
ми, желтовато-коричневыми клетками (вид с поверх¬
ности [Е]); фрагменты дермальной ткани (попереч¬
ный срез [С]), состоящей из тонкостенных, желтова¬
то-коричневых клеток пробки [Са] и толстостенных
клеток колленхимы (феллодермы) [СЬ]; фрагменты
паренхимы (продольный срез [В], поперечный срез
[О]) с клетками с умеренно утолщенными стенками,
содержащими капельки масла [Ва, йа], небольшие
призматические кристаллы [ВЬ, ОЬ] и мельчайшие
иглы кальция оксалата [Вс, Ос]; отдельные фраг¬
менты одревесневших сосудов со спиралевидным
[Н] или сетчатым [6] утолщением и имеющих диа¬
метр до 80 мкм; фрагменты ксилемы (продольный
срез [А], поперечный срез [Р]), состоящие из сосу¬
дов [Аа, Ра] и клеток паренхимы с умеренно утол¬
щенными стенками, содержащих капельки масла
[АЬ, РЬ].
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 25 мл метанола Р,
встряхивают в течение 15 мин и фильтруют. Филь¬
трат выпаривают досуха при пониженном давлении
при температуре не выше 50 °С. Остаток раство¬
ряют в небольших порциях метанола Р до полу¬
чения 5 мл раствора, который может содержать
осадок.
Раствор сравнения. 5 мг гиперозида Р и 5 мг
феназона Р растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯР2$4Р.
Наносимый объем пробы: по 20 мкл в виде полос.
Подвижная фаза: вода Р — кислота муравьиная
безводная Р — этилформиат Р (4:8:88, об/об/об).
Фронт подвижной фазы: в ненасыщенной хрома¬
тографической камере не менее 8 см от линии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Феназон: зона поглощения
Отчетливая зона поглоще¬
ния
Зона слабого поглощения
(амарогентин)
Гиперозид: зона поглощения
Отчетливая зона поглоще¬
ния (гентиопикрозид)
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку обрабатывают рас¬
твором 100 г/л калия гидроксида Р в метаноле Р
и затем свежеприготовленным раствором 2 г/л
соли прочного синего В Р в смеси из этанола Р и
воды Р (50:50, об/об). Просматривают при дневном
свете.
Результаты В: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения
и испытуемого раствора. На хроматограмме ис¬
пытуемого раствора могут обнаруживаться другие
зоны.
Гэрицвета весеннего трава
1211
Верх хроматографической пластинки
Отчетливая зона темно-фи¬
олетового цвета
Зона фиолетово-красного
цвета (амарогентин)
Гиперозид: зона коричнева¬
то-красного цвета
Слабая зона светло-корич¬
невого цвета (гентиопикро-
зид)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Другие виды ОепИапа. Просматривают хрома¬
тограммы, полученные в проявлении В испытания С
на подлинность.
Результаты: на хроматограмме испытуемого
раствора не должны обнаруживаться зоны фиолето¬
вого цвета непосредственно над зоной амарогентина.
Общая зола (2.4.16). Не более 6,0 %.
Показатель горечи (2.8.15). Не менее 10 000.
Вещества, извлекаемые водой. Не менее 33 %.
К 5,0 г измельченного лекарственного растительно¬
го сырья (710) (2.9.12) прибавляют 200 мл кипящей
воды Р, выдерживают в течение 10 мин при периоди¬
ческом встряхивании, охлаждают, доводят водой Р до
объема 200,0 мл и фильтруют. 20,0 мл полученного
фильтрата выпаривают досуха на водяной бане, оста¬
ток сушат при температуре от 100 °С до 105 °С. Масса
остатка должна составлять не менее 0,165 г.
07/2016: РБ0048
ГОРИЦВЕТА ВЕСЕННЕГО ТРАВА
Аёоп 'кНз уегпаНз НегЬа
АООЛ//5 НЕЯВ*
ОПРЕДЕЛЕНИЕ
Собранная в период цветения до начала осыпа¬
ния плодов и высушенная трава многолетнего травя¬
нистого растения Адотз уегпаНз 1_.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные или ча¬
стично измельченные олиственные стебли с цветка¬
ми или без них, реже с бутонами или плодами разной
степени развития, иногда частично осыпавшимися.
Стебли, срезанные выше коричневых низовых че¬
шуевидных листьев, длиной (10—35) см, толщиной
до 0,4 см, простые или маловетвистые. Листья оче¬
редные, сидячие, полустеблеобъемлющие, в общем
очертании округлые или широкоовальные, папьчато-
рассеченные на пять долей, из которых две нижних —
перисторассеченные, три верхних — дваждыпери-
сторассеченные; доли листьев линейные, у верхуш¬
ки шиловидно-заостренные, цельнокрайние, длиной
(0,5—2) см, шириной (0,5—1) мм. Листья по отцвета¬
нии жестковатые. Цветки одиночные на верхушке сте¬
блей, правильные, около 3,5 см в поперечнике, сво¬
боднолепестные, с 5—8 чашелистиками, с 15—20 ле¬
пестками, с многочисленными тычинками и пестика¬
ми. Чашелистики яйцевидные, вверху притупленные,
с редкими зубцами, опушенные, длиной (12—20) мм,
шириной около 12 мм, легко опадающие. Лепестки
продолговато-эллиптические, на верхушке суженные,
зазубренные. Плод сборный, овальный, состоит из
многочисленных сухих орешков, сидящих на цилин¬
дрическом буроватом цветоложе. Орешки длиной от
3,5 мм до 5,5 мм, шириной около 3 мм, овальные, с
коротким крючкообразно загнутым столбиком, морщи¬
нисто-ячеистые, опушенные. Цвет стеблей и листьев
зеленый, цветков — желтый, плодов — серовато-зе¬
леный. Запах слабый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности с обеих сторон видны крупные
клетки эпидермиса с сильно извилистыми стенками,
несколько вытянуты по длине дольки. Клетки верхне¬
го эпидермиса иногда имеют четковидные утолщения.
Кутикула с ясно выраженной продольной, волнистой
складчатостью. Устьица только на нижней стороне,
крупные, овальные, слегка выступающие над поверх¬
ностью листа, окружены 4—5 клетками эпидермиса и
ориентированы вдоль пластинки листа (аномоцитный
тип) (2.8.3). По краю долек листа и у основания изред¬
ка встречаются одноклеточные волоски двух типов:
длинные, лентовидные с закругленной верхушкой,
суженные у основания; короткие булавовидные воло¬
ски, резко суженные у места прикрепления. Все воло¬
ски со спирально-складчатой кутикулой, прикреплены
к очень маленькой округлой клетке эпидермиса.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 20 мл спирта (50 %,
об/об) Р, 10 мл раствора 100 г/л свинца ацетата Р
и нагревают с обратным холодильником на водяной
бане в течение 2 мин. Охлаждают, центрифугируют.
Надосадочную жидкость дважды встряхивают с хло¬
роформом Р порциями по 15 мл, затем дважды встря¬
хивают со смесью из хлороформа Р и метанола Р
(50:50, об/об) порциями по 15 мл. При необходимости
слои разделяют центрифугированием. К объединен¬
ным хлороформным слоям прибавляют 1 г полиами¬
да для колоночной хроматографии Р, перемешива¬
ют, фильтруют через бумажный фильтр с 2 г натрия
сульфата безводного Р и выпаривают досуха. Оста¬
ток растворяют в 1 мл смеси из хлороформа Р и ме¬
танола Р (50:50, об/об).
Раствор сравнения. 5 мг дигитоксина Р раст¬
воряют в 1 мл смеси из хлороформа Р и метанола Р
(50:50, об/об).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля в Р.
Подвижная фаза: хлороформ Р — толуол Р —
метанол Р (80:10:10, об/об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С до
105 °С в течение 5 мин. Просматривают при дневном
свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается зона серо-фиолетового цвета
со значением 0,38—0,42 (дигитоксин). На хромато¬
грамме испытуемого раствора обнаруживается зона
зеленого цвета (цимарин) расположенная немного
выше зоны дигитоксина на хроматограмме раствора
сравнения. На хроматограмме испытуемого раствора
обнаруживаются и другие зоны.
153*. Зак. 1060.
1212
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые части
растения: побуревшие части растения — не более 3 %;
растения со стеблями, имеющими коричневые чешуй¬
чатые листья, — не более 2 %. Органические примеси:
не более 2 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
07/2016:2384
ГОРЦА ЗМЕИНОГО КОРНЕВИЩА
(ЗМЕЕВИКА КОРНЕВИЩА)
Ро1удом Ыз1ог1ае гЫгота (ЕНз1ог1ае гЫгота)
В18ТОПТ КН120МЕ
ОПРЕДЕЛЕНИЕ
Собранные после отцветания, очищенные от кор¬
ней, остатков листьев и стеблей, отмытые от земли
и высушенные корневища многолетних травянистых
растений Регз1сапа Ыз1ог1а (1_.) Затр. (син. Ро1удопит
Ыз1ог1а 1_.).
Содержание:
- не менее 3,0 % дубильных веществ в пересче¬
те на пирогаллол (С6Нб03; М.м. 126,1) (в пересчете на
сухое сырье);
- или не менее 15,0 % дубильных веществ в пе¬
ресчете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корневище твер¬
дое, змеевидноизогнутое, несколько сплюснутое,
с поперечными кольчатыми утолщениями и следа¬
ми обрезанных корней. Длина корневища обычно
(3—13) см, толщина (1,5—2,5) см. Могут встречаться
фрагменты корневища диаметром около 0,3 см и дли¬
ной до 1 см, остатки корней длиной не более 1 см и
диаметром около 1 мм. Цвет пробки темный, красно¬
вато-коричневый; на изломе — розоватый или корич¬
невато-розовый, излом ровный.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Порошок имеет краснова¬
то-коричневый цвет. Просматривают под микроскопом
с использованием раствора хлоралгидрата Р. В по¬
рошке обнаруживаются следующие диагностические
признаки (рисунок 2384.-1): очень многочисленные
друзы кальция оксалата, диаметром (15—65) мкм,
расположенные отдельно [С] или включенные в клет¬
ки паренхимы [Оа]\ изредка встречаются фрагменты
корки (вид сбоку [В] или вид с поверхности [Н]); со¬
судистые пучки (вид продольного среза [Е] или вид
поперечного среза [3]), включая небольшие ямчатые
сосуды [Еа, За], с прилегающими мелкоямчатыми,
толстостенными волокнами [ЕЬ, Лэ]; отдельно рас¬
положенные фрагменты сосудов [С]; отдельно рас¬
положенные волокна [Р]; фрагменты паренхимы [О] с
округлыми клетками со слегка утолщенными стенка¬
ми; фрагменты колленхимы [К]. Просматривают под
микроскопом с использованием 50 % (об/об) раствора
глицерина Р. В порошке обнаруживаются округлые
или яйцевидные простые зерна крахмала диаметром
около (5—12) мкм, расположенные отдельно или
включенные в клетки паренхимы [А].
Рисунок 2384.-1. Микроскопические признаки
корневищ горца змеиного
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл смеси из равных
объемов метанола Р и воды Р, нагревают на водяной
бане при температуре около 65 °С в течение 30 мин и
фильтруют.
Раствор сравнения. 5 мг фруктозы Р и 5 мг ка-
техина Р растворяют в 5 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((2—10) мкм).
Подвижная фаза: вода Р — кислота муравьиная
безводная Р — этилацетат Р (5:10:85, об/об/об/об).
Наносимый объем пробы: по 2 мкл в виде полос.
Фронт подвижной фазы: не менее 7 см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раство¬
ром анисового альдегида Р и нагревают при темпера¬
туре от 100 °С до 105 °С в течение 5 мин. Пластинку
просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора обнаруживаются и другие слабые зоны.
Верх хроматографической пластинки
Катехин: флуоресцирующая
зона коричневого цвета
Зона коричневого цвета (ка¬
техин)
Зона коричневого цвета
Зона фиолетового цвета
Зона коричневого цвета
Зона оранжевого цвета
Фруктоза: зона зеленого
цвета
Зона зеленого цвета (фрук¬
тоза)
Раствор сравнения
Испытуемый раствор
*
Горца перечного трава (водяного перца трава)
1213
О. К 2 г измельченного сырья (1000) (2.9.12)
прибавляют 20 мл воды Р, кипятят с обратным хо¬
лодильником в течение 10 мин и фильтруют. К 1 мл
полученного раствора прибавляют 2—3 капли раст¬
вора железа (III) аммония сульфата Р5. Появляется
черно-синее окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корневища, почерневшие в изломе, —
не более 10 %; остатки листьев и стеблей, в том числе
отделенные при анализе, — не более 1 %. Органиче¬
ские примеси: не более 0,5 %. Минеральные примеси:
не более 1 %.
Се®) Рапз ро!урНу11а Зт. или Рапз циадгНоНа 1_.
Микроскопия (2.8.23). Просматривают под микроско¬
пом с использованием раствора хлоралгидрата Р.
Присутствие рафид кальция оксалата в свободном
виде или в пучках свидетельствует о загрязнении кор¬
невищами Р. ро1урЬу11а 5т. маг. уиппапепз1в (РгапсИ.)
Напс1.-Ма22:. или Р. ро!урЬу11а 5т. мах. сЫпелз/з
(РгапсЬ.) Н.Нага или Р. чиадпТоНа 1_.
< ЕФ.- Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 9,0 %.
@) Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Определение содержания дубильных ве¬
ществ в пересчете на пирогаллол (2.8.14). Исполь¬
зуют 1,000 г измельченного сырья (180) (2.9.12).
Определение содержания дубильных ве¬
ществ в пересчете на танин. 2,000 г измельченного
сырья (2000) (2.9.12) помещают в коническую колбу
со шлифом вместимостью 500 мл, прибавляют 250 мл
кипящей воды Р и кипятят с обратным холодильником
в течение 30 мин при периодическом перемешива¬
нии. Затем охлаждают до комнатной температуры,
процеживают и доводят водой Р до объема 250,0 мл.
25,0 мл полученного извлечения помещают в ко¬
ническую колбу вместимостью 750 мл, прибавляют
500 мл воды Р, 25,0 мл раствора индигосульфокисло-
ты (0,25 мг индигокармина Р растворяют в 6,5 мл кис¬
лоты серной Р, прибавляют 6,5 мл кислоты серной Р,
доводят водой Р до объема 250 мл и фильтруют). Ти¬
труют при постоянном перемешивании 0,02 М раство¬
ром калия перманганата до желтого окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07/2016: РБ0049
ГОРЦА ПЕРЕЧНОГО ТРАВА
(ВОДЯНОГО ПЕРЦА ТРАВА)
Ро1удоп1 Ьус1гор1репз ЬегЬа
И(АТЕВ Р1РЕР НЕРВ*
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная тра¬
ва однолетнего травянистого растения Ро1удопит
Ьудгор!рег 1_.
Содержание: не менее 0,50 % суммы флавонои-
дов в пересчете на кверцетин (С15Н10О7; М.м. 302,2) (в
пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
частично измельченные цветоносные олиственные
побеги длиной до 45 см без грубых нижних частей, с
плодами разной степени зрелости. Стебли цилиндри¬
ческие со вздутыми узлами. Листья очередные, корот¬
кочерешковые, продолговато-ланцетные, заостренные
или туповатые, цельнокрайние, голые, длиной до 9 см,
шириной до 1,8 см. У основания черешков находятся
два прилистника, сросшиеся в пленчатые стеблеобъ¬
емлющие цилиндрические раструбы длиной до 1,5 см.
Поверхность раструбов голая, верхний край с коротки¬
ми (около 2 мм) щетинками. Соцветия — тонкие пре¬
рывистые кисти длиной до 6 см, цветки на коротких
цветоножках. Околоцветник венчиковидный с А—5 ту¬
поватыми долями, длиной (3—4) мм, покрытыми мно¬
гочисленными бурыми точками (вместилища). Тычинок
6, реже 8, пестик с верхней одногнездной завязью и
2—3 столбиками. Плоды — яйцевидно-эллиптические
орешки, с одной стороны плоские, с другой — выпу¬
клые, заключенные в остающийся околоцветник. Цвет
стеблей зеленый или красноватый, листьев—зеленый,
раструбов — красноватый, цветков — зеленоватый или
розоватый, плодов — черный. Запах отсутствует.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны: клетки эпидермиса с из¬
вилистыми стенками; устьица с обеих сторон листа,
окружены 2—4 околоустьичными клетками (аномо-
цитный тип) (2.8.3). На поверхности имеются мелкие
бесцветные или светло-коричневые железки, состо¬
ящие из 2—4 клеток. По краю пластинки и по жилке
с нижней стороны листа расположены конусовидные
пучковые волоски, сросшиеся из нескольких кле¬
ток. В мезофилле листа многочисленные крупные
остроконечные друзы оксалата кальция и крупные
округлые или овальные схизогенные вместилища с
содержимым светло-коричневого, коричневого или
золотисто-желтого цвета. Наиболее важным диагно¬
стическим признаком, позволяющим отличить сырье
Р Ьудгор!рег от близких видов, является наличие по¬
груженных вместилищ в паренхиме всех надземных
органов: листа, стебля, околоцветника и раструба. Из
других видов горцев вместилища встречаются у горца
мягкого только в мезофилле листа.
C. К 1 г измельченного сырья (710) (2.9.12) при¬
бавляют 20 мл воды Р, кипятят в течение 5 мин и
фильтруют. К 5 мл фильтрата прибавляют 3 мл раст¬
вора 10 г/л алюминия хлорида Р в спирте (95%,
об/об) Р. Появляется желто-зеленое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побуревшие, почерневшие и пожелтев¬
шие части растения — не более 5 %. Органические
примеси: не более 3 %. Минеральные примеси: не
более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 8,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (710) (2.9.12) по¬
мещают в колбу со шлифом вместимостью 150 мл,
154. Зак. 1060.
1214
Гэсударственная фармакопея Республики Беларусь
прибавляют 30 мл 1 % (об/об) раствора кислоты хло¬
ристоводородной Р в спирте (90 %, об/об) Р и нагре¬
вают с обратным холодильником на водяной бане в
течение 30 мин. Охлаждают до комнатной темпера¬
туры и фильтруют через бумажный фильтр в мерную
колбу вместимостью 100 мл. Экстракцию повторяют
еще 1 раз, как указано выше, затем 1 раз с исполь¬
зованием спирта (90 %, об/об) Р в течение 30 мин.
Извлечения фильтруют через тот же фильтр в ту же
мерную колбу, промывают фильтр спиртом (90 %,
об/об) Р и доводят спиртом (90 %, об/об) Р до объ¬
ема 100,0 мл (раствор А).
Испытуемый раствор. К 2,0 мл раствора А при¬
бавляют 1,0 мл раствора 10 г/л алюминия хлорида
в спирте (95 %, об/об) Р и доводят спиртом (95 %,
об/об) Р до объема 25,0 мл.
Компенсационный раствор. 2,0 мл раствора А
доводят спиртом (95 %, об/об) Р до объема 25,0 мл.
Через 20 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 430 нм.
Содержание суммы флавоноидов в пересчете на
кверцетин в процентах рассчитывают по формуле:
А-1250
ш -764,6 ’
где:
764,6 — удельный показатель поглощения ком¬
плекса кверцетина с алюминия хлоридом при 430 нм;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0050
ГОРЦА ПОЧЕЧУЙНОГО ТРАВА
Ро1удоп! регзюапае ЬегЬа
8РОТТЕО ^А^VЗТН^МВ*
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная тра¬
ва однолетнего травянистого растения Ро1удопит
регзюапа I.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные или
частично измельченные цветоносные олиственные
побеги длиной до 40 см без грубых нижних частей, с
плодами разной степени зрелости. Стебли ветвистые
или простые, продольно-бороздчатые, с вздутыми
узлами. Листья очередные, короткочерешковые, лан¬
цетные, длинно-заостренные с клиновидным основа¬
нием, на верхней стороне с темным пятном или без
него, цельнокрайние, длиной до 16 см, шириной до
2,5 см. Находящиеся при основании черешков ли¬
стьев пленчатые раструбы покрыты прижатыми воло¬
сками и плотно охватывают стебли, по верхнему краю
с ресничками длиной от 0,2 мм до 4,5 мм. Соцветия
верхушечные, густые колосовидные кисти. Цветки
мелкие, с простым глубоко 4-5-рассеченным около¬
цветником, длиной около (2—3,5) мм. Доли около¬
цветника и цветонос с единичными железками (уве¬
личение 10*). Плоды трехгранные, чечевицеобраз¬
ные или плоские с одной или с обеих сторон, орешки
длиной (2,2—2,9) мм, шириной (1,6—2) мм, блестя¬
щие, черные или темно-коричневые. Цвет стеблей
зеленый, иногда с коричневатым оттенком; листьев с
верхней стороны зеленый, с нижней — серовато-зе¬
леный; околоцветника — розовый, реже белый, при
основании зеленоватый.
В. Микроскопия (2.8.23). При просматривании
листа с поверхности видны клетки верхнего эпидер¬
миса с прямыми стенками, нижнего — с извилисты¬
ми. Устьица с 2—4 околоустьичными клетками, ино¬
гда они окружены 2 клетками, расположенными вдоль
устьичной щели (аномоцитный тип) (2.8.3). На обеих
поверхностях листа имеются железки на 2—4-кле¬
точной ножке с головкой из 8 (12—16) клеток, реже
с 2—4-клеточной головкой с красновато-коричневым
содержимым или бесцветные. По всей пластинке ли¬
ста и по краю встречаются пучковые волоски, образо¬
ванные 2—5 сросшимися клетками, которые на вер¬
хушке волоска часто слегка расходятся. В мезофилле
листа крупные друзы оксалата кальция. На эпидерми¬
се стебля и раструба, кроме вышеперечисленных при¬
знаков, встречаются пленчатые волоски, состоящие
из нескольких рядов клеток и имеющие 2-клеточное
основание. В ткани околоцветника — призматические
кристаллы оксалата кальция.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побуревшие, почерневшие, пожелтев¬
шие части растения — не более 10 %. Органические
примеси: не более 3 %. Минеральной примеси: не бо¬
лее 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не болееЮ.О %.
07/2016:1885
ГОРЦА ПТИЧЬЕГО ТРАВА
(СПОРЫША ТРАВА)
Ро1удот амси1апз ЬегЬа
КЫОТ6КА88
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная тра¬
ва однолетнего травянистого растения Ро1удопит
аПси1аге 1_. з.1.
Содержание:
- не менее 0,30 % флавоноидов в пересчете на
гиперозид (С21Н20О12; М.м. 464,4) (в пересчете на су¬
хое сырье);
- или не менее 0,5 % флавоноидов в пересчете
на авикулярин (С^Н^О^; М.м. 434,4) (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Стебель тол¬
щиной (0,5—2) мм, разветвленный, коленчатый, ци¬
линдрический или слегка угловатый с продольной
бороздчатостью. Листья сидячие или короткочерешко¬
вые, гладкие, цельнокрайние, сильно различающиеся
по форме и размеру. Прилистники, сросшиеся в рас¬
труб, рассечены и имеют серебристую окраску. Не¬
большие пазушные цветки имеют 5 зеленовато-белых
околоцветниковых сегмента, верхушки которых зача¬
стую имеют красный цвет. Плоды сухие, нераскрыва-
Гэрца птичьего трава (спорыша трава)
1215
ющиеся, размером (2—4) мм, коричневого или черного
цвета, треугольные, обычно с точками или бороздками.
@>В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет порошка зеленова¬
то-коричневый. Просматривают под микроскопом с
использованием раствора хлоралгидрата Р. В по¬
рошке обнаруживаются следующие диагностические
признаки (рисунок 1885.-1): фрагменты нижнего [А] и
верхнего [6] эпидермиса листьев с исчерченной кути¬
кулой и анизоцитными устьицами (2.8.3) [Аа, 0а]\ мно¬
гоугольные клетки верхнего эпидермиса [О] со слегка
утолщенными четковидными стенками, часто вместе
с палисадной паренхимой [ОЬ]; клетки нижнего эпи¬
дермиса [А] с тонкими извилистыми стенками; фраг¬
менты краев пластинки листа с неправильными клет¬
ками у]; фрагменты паренхимы [О] с многочисленны¬
ми клетками, содержащими друзы оксалата кальция,
некоторые очень большие [Оа], часто вместе с сосу¬
дами [ОЬ]; группы волокон [В, С] с толстыми стенками
[Ва, СЬ] гиподермы стебля с сопутствующим эпидер¬
мисом [Са] или паренхимой, состоящей из клеток, со¬
держащих друзы оксалата кальция [ВЬ]; фрагменты
раструба [Е] с вытянутыми тонкостенными клетками
[Еа], вдоль каждого из которых идут очень вытянутые
волокна [ЕЬ]; шарообразные зерна пыльцы с гладкой
экзиной и тремя зародышевыми порами [Н]; редкие
коричневые фрагменты экзокарпия, состоящего из
клеток с толстыми извилистыми клетками [Р].
Рисунок 1885.-1. Микроскопические признаки
травы горца птичьего
@> С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл метанола Р и на¬
гревают с обратным холодильником в водяной бане в
течение 10 мин. Охлаждают и фильтруют.
Раствор сравнения. 1 мг кофейной кислоты Р,
2,5 мг гиперозида Р и 1 мг хлорогеновой кислоты Р
растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат Р (7:7:14:72, об/об/об/об).
Наносимый объем пробы: по 20 мкл в виде по¬
лос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: пластинку обрабатывают раство¬
ром 10 г/л дифенилборной кислоты аминоэтилового
эфира Р в метаноле Р и затем раствором 50 г/л ма-
крогола 400 Р в метаноле Р. Пластинку высушивают
на воздухе в течение 30 мин и просматривают в уль¬
трафиолетовом свете при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора обнаруживаются и другие флуоресцирую¬
щие зоны.
Верх хроматографической пластинки
Кофейная кислота: флуо¬
ресцирующая зона светло-
голубого цвета
Гиперозид: флуоресцирую¬
щая зона желтовато-корич¬
невого цвета
Хлорогеновая кислота: флу¬
оресцирующая зона светло-
голубого цвета
Одна или две флуоресциру¬
ющие зоны голубого цвета
(кофейная кислота)
Одна или две флуоресциру¬
ющие зоны желтовато-зеле¬
ного цвета
Флуоресцирующая зона
желтого цвета
Флуоресцирующая зона
желтовато-коричневого
цвета
флуоресцирующая зона
светло-голубого цвета (хло¬
рогеновая кислота)
Флуоресцирующая зона
желтовато-коричневого
цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побуревшие и почерневшие части рас¬
тения — не более 3 %; корни — не более 2 %. Орга¬
нические примеси: не более 2 %. Минеральные при¬
меси: не более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 13,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
®> Определение содержания флавоноидов в
пересчете на гиперозид.
Исходный раствор. 0,800 г измельченного сы¬
рья (355) (2.9.12) помещают в круглодонную колбу
вместимостью 100 мл, прибавляют 1мл раствора
5 г/л гексаметилентетрамина Р, 20 мл ацетона Р и
2 мл кислоты хлористоводородной Р1. Кипятят с об¬
ратным холодильником в течение 30 мин, после чего
154*. Зак. 1060.
1216
Гэсударственная фармакопея Республики Беларусь
извлечение процеживают через ватный тампон в кол¬
бу. Ватный тампон помещают в круглодонную колбу с
остатком сырья и дважды кипятят с обратным холо¬
дильником в течение 10 мин, каждый раз прибавляя
20 мл ацетона Р в качестве экстрагента. Охлаждают
и процеживают через ватный тампон. Объединенные
ацетоновые извлечения фильтруют через бумажный
фильтр в мерную колбу вместимостью 100 мл, опола¬
скивают колбу ацетоном Р и фильтруют через тот же
фильтр, после чего промывают фильтр ацетоном Р.
Объем раствора доводят до 100,0 мл ацетоном Р.
20.0 мл полученного раствора помещают в делитель¬
ную воронку, прибавляют 20 мл воды Р и экстраги¬
руют этилацетатом Р порцией 15мл, затем еще
трижды порциями по 10 мл. Этилацетатные извлече¬
ния объединяют, помещают в делительную воронку и
дважды промывают водой Р порциями по 50 мл. Ор¬
ганический слой фильтруют через бумажный фильтр с
10 г натрия сульфата безводного Р в мерную колбу,
вместимостью 50 мл и доводят этилацетатом Р до
объема 50,0 мл.
Испытуемый раствор. К 10,0 мл исходного раст¬
вора прибавляют 1 мл реактива алюминия хлорида Р
и доводят 5 % (об/об) раствором кислоты уксусной
ледяной Р в метаноле Р до объема 25,0 мл.
Компенсационный раствор. 10,0 мл исходного
раствора доводят 5 % (об/об) раствором кислоты ук¬
сусной ледяной Р в метаноле Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 425 нм.
Содержание флавоноидов в пересчете на гипе-
розид в процентах рассчитывают по формуле:
А- 625
т-500 *
где:
500 — удельный показатель поглощения гиперо-
зида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Определение содержания суммы флавонои¬
дов в пересчете на авикулярин.
Исходный раствор. 1,000 г измельченного сырья
(710) (2.9.12) помещают в колбу вместимостью 150 мл,
прибавляют 30 мл спирта (70 %, об/об) Р. Нагревают
с обратным холодильником в водяной бане в течение
30 мин, периодически встряхивая для смывания ча¬
стиц сырья со стенок колбы. Охлаждают и фильтруют
через бумажный фильтр в мерную колбу вместимо¬
стью 100 мл так, чтобы частицы сырья не попадали на
фильтр. Экстракцию повторяют дважды в описанных
выше условиях, фильтруя извлечения в ту же мерную
колбу. Фильтр промывают спиртом (70 %, об/об) Р и
доводят до объема 100,0 мл этим же растворителем.
Испытуемый раствор. 4,0 мл исходного раст¬
вора и 2,0 мл раствора 20 г/л алюминия хлорида Р в
96 % спирте Р доводят 96 % спиртом Р до объема
25.0 мл.
Компенсационный раствор. 4,0 мл исходного
раствора и 1 каплю кислоты хлористоводородной
разведенной Р доводят 96 % спиртом Р до объема
25.0 мл
Через 20 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 410 нм.
Содержание суммы флавоноидов в пересчете на
авикулярин в процентах рассчитывают по формуле:
А•2500
т-330 ’
где:
330 — удельный показатель поглощения ком¬
плекса авикулярина с алюминия хлоридом;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0051
ДЕВЯСИЛА КОРНЕВИЩА И КОРНИ
1пи1ае Ье1епН гЫгота е* гасЛх
Е1.ЕСАМРАЫЕ РООТ*
ОПРЕДЕЛЕНИЕ
Собранные осенью и высушенные корневища
и корни многолетнего травянистого растения 1пи1а
Ье1епшт 1_.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корневища и
корни цилиндрические, большей частью продольно-
расщепленные, снаружи продольно-мелкоморщини-
стые, длиной от 2 см до 20 см, толщиной — от 0,5 см
до 3 см, твердые, в изломе слабозернистые, с за¬
метными коричневатыми блестящими точечками —
вместилищами с эфирным маслом (при увеличении
10*). Цвет снаружи серовато-коричневый, на изло¬
ме — желтовато-белый или желтовато-серый. Запах
ароматный.
B. Микроскопия (2.8.23). На поперечном сре¬
зе корня видна многорядная серовато-коричневая
пробка, кора и древесина. Паренхима коры состо¬
ит из крупных клеток, содержащих инулин в виде
бесформенных, бесцветных, сильно преломляющих
свет «глыбок» (смотреть препарат без нагревания!).
Во вторичной коре заметны участки луба в виде
мелких клеток, расположенных небольшими группа¬
ми. Линия камбия четкая. В древесине видны круп¬
ные сосуды, особенно близ камбия, расположенные
группами. В коре и древесине корня имеются круп¬
ные схизогенные вместилища со смолой и эфирным
маслом. Они округлые или овальные, с хорошо за¬
метным слоем выделительных клеток. После окра¬
ски раствором Судана IIIР капли смолистого содер¬
жимого во вместилищах приобретают яркий оранже¬
во-красный цвет.
C. Срез корневища смачивают 2—3 каплями
раствора йода Р1. Синее окрашивание не появляет¬
ся (отсутствие крахмала).
О. Срез корневища смачивают 2—3 каплями
раствора а-нафтола Р1 или раствора тимола Р и
1 каплей серной кислоты Р. Появляется красно-фио¬
летовое или оранжево-красное окрашивание соответ¬
ственно (инулин).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: дряблые корневища и корни, основания
стеблей и другие части девясила — не более 5 %; кор¬
невища и корни, потемневшие в изломе, — не более
5 %; куски корней длиной менее 2 см — не более 5 %.
Органические примеси: не более 0,5 %. Минеральные
примеси: не более 1 %.
Девясила цветки
1217
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 4,0 %.
07/2016:РБ0052
ДЕВЯСИЛА ЦВЕТКИ
1пи1ае Ье1епН Лоз
Е1.ЕСАМРАЫЕ Я.ОИГЕК*
ОПРЕДЕЛЕНИЕ
Собранные в начале цветения и высушенные
цветочные корзинки многолетнего травянистого рас¬
тения 1пи1а Ыепшт Ь.
Содержание: не менее 3,0 % суммы фенольных
соединений в пересчете на кверцетин (С15Н10О7; М.м.
302,2) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные и рас¬
павшиеся цветочные корзинки, отдельные ложноязыч¬
ковые и трубчатые цветки, семянки с хохолком, цвето¬
ложа распавшихся соцветий. Корзинки диаметром до
6 см. Краевые цветки ложноязычковые, пестичные,
длиной до 3,5 см; трубка венчика длиной (6—7) мм,
на одну треть короче хохолка, язычки остротрехзуб¬
чатые, длиной (2,6—3,0) см, шириной (1,0—1,5) см.
Срединные цветки трубчатые, обоеполые, многочис¬
ленные, с пятью зубцами, длиной до 1,5 см, немного
длиннее хохолков или равны им. Тычинок 5; пыльники,
сросшиеся в трубку. Пестик с нижней завязью, длин¬
ным, тонким столбиком и двумя рыльцами. Плод че¬
тырехгранная коричневая семянка, длиной (4—5) мм,
с хохолком вдвое длиннее семянки. Обертка с много¬
численными черепитчатыми листочками; внутренние
листочки обертки кверху расширенные, тупые, на¬
ружные — более короткие, яйцевидные, травянистые,
серо-войлочные. Цвет цветков от светло-желтого до
желтого, листочков обертки от зеленого до коричне¬
вато-зеленого, семянок от светло-зеленого до светло-
коричневого. Запах слабый, ароматный.
B. Микроскопия (2.8.23). При просматривании
ложноязычковых цветков с поверхности видны уд¬
линенные клетки эпидермиса с прямыми стенками
и округлыми хромопластами. Клетки эпидермиса
трубчатых цветков продольно-вытянутые с прямы¬
ми стенками, по краю зубчиков сосочковидные. На
поверхности ложноязычковых и трубчатых цветков
имеются многочисленные эфирномасличные же¬
лезки. Железки многоклеточные, их выделительные
клетки расположены в один или два ряда (вид сбоку),
при просматривании сверху железки видны в виде
овальных образований с поперечной перегородкой.
На верхушке завязи ложноязычковых и трубчатых
цветков виден хохолок, состоящий из тонких щети¬
нок, сросшихся друг с другом у основания. Пыльце¬
вые зерна округлые или трехлопастные, трехборозд-
но-поровые, с шиповатой экзиной. Эпидермис вну¬
треннего листочка обертки состоит из узких и сильно
вытянутых прямостенных клеток, в верхней части —
из многоугольных и слегка вытянутых клеток, стенки
которых пронизаны порами. Клетки эпидермиса вну¬
тренней стороны наружного листочка обертки много¬
угольные, слегка вытянутые, прямостенные, прони¬
занные порами, без устьиц, наружной стороны — с
прямыми или слабоизвилистыми стенками и устьи¬
цами, окруженными 4—7 околоустьичными клетками
(аномоцитный тип) (2.8.3). Над жилкой клетки эпи¬
дермиса многоугольные вытянутые. Волоски много¬
численные, разнообразные по строению. По краю
внутреннего листочка обертки встречаются простые
одноклеточные тонко- или толстостенные волоски,
представляющие собой выросты клеток эпидермиса,
нередко сросшиеся по нескольку, по верхнему краю
волоски с более округлым основанием и более вытя¬
нутой верхушкой. Простые многоклеточные волоски,
состоящие из одной или нескольких коротких клеток
с тонкими, реже утолщенными оболочками и прямой
или извилистой конечной клетки различной длины
с толстыми стенками и заостренным концом, густо
покрывают всю поверхность листочков обертки, осо¬
бенно наружного листочка и его край, образуя вой¬
лочное опушение. Простые короткие одноклеточные
волоски с расширенным основанием, встречаются по
всей поверхности листочков обертки. При отпадении
волосков на месте их прикрепления остаются кру¬
глые валики. Эфирномасличные железки на листоч¬
ках обертки такого же строения, как и на цветках. На
поверхности ложноязычковых и трубчатых цветков
волоски встречаются редко.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,00 г измельченного
сырья (355) (2.9.12) прибавляют 20 мл 96 % спирта Р
и нагревают на водяной бане с обратным холодильни¬
ком в течение 30 мин. Охлаждают и фильтруют через
бумажный фильтр.
Раствор сравнения. 25 мг кверцетина Р раство¬
ряют в 25 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: толуол Р — этилацетат Р —
кислота уксусная ледяная Р (36:12:5, об/об/об).
Наносимый объем пробы. 30 мкл испытуемого
раствора и 5 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 10 мин, затем охлаждают на воздухе.
Проявление: пластинку опрыскивают раствором
20 г/л алюминия хлорида Ра 96% спирте Р. Просма¬
тривают в ультрафиолетовом свете при длине волны
365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Кверцетин: флуоресцирую¬
щая зона желтого цвета
Флуоресцирующая зона фи¬
олетового цвета
Флуоресцирующая зона
желтого цвета (кверцетин)
Флуоресцирующая зона
желто-коричневого цвета
Раствор сравнения
Испытуемый раствор
О. На хроматограмме испытуемого раствора,
полученного как указано в разделе «Количественное
определение», обнаруживаются пики 1, 2, 3, 4, 5 (ри¬
сунок РБ0052.-1).
155. Зак. 1060.
1218
Государственная фармакопея Республики Беларусь
1,2,3, 5. Фенольные соединения. 4. Кверцетин.
Рисунок РБ0052.-1. Хроматограмма испытуемого раствора
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: кусочки стеблей, цветоносов и ли¬
стьев — не более 3 %. Органические примеси: не бо¬
лее 2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,000 г измельченного
сырья (355) (2.9.12) помещают в круглодонную кол¬
бу со шлифом вместимостью 250 мл, прибавляют
40,0 мл спирта (60 %, об/об) Р, взвешивают с точно¬
стью до 0,01 г и нагревают с обратным холодильни¬
ком на водяной бане в течение 30 мин. Охлаждают,
доводят массу до первоначальной спиртом (60 %,
об/об) Р и центрифугируют в течение 5 мин со ско¬
ростью 3000 об/мин. Надосадочную жидкость филь¬
труют через мембранный фильтр с диаметром пор
0,45 мкм.
Раствор сравнения. 10,0 мг кверцетина диги¬
драта Р (с учетом содержания безводного кверце¬
тина в кверцетине дигидрате Р) растворяют в 96%
спирте Р и доводят до объема 100,0 мл этим же раст¬
ворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним ди¬
аметром 4,6 мм, заполненная силикагелем
октадецилсилильным для хроматографии Р с раз¬
мером частиц 5 мкм;
- температура: 30 °С;
-подвижная фаза: ацетонитрил Р — 0,01 М
раствор калия дигидрофосфата Р, доведенный кис¬
лотой фосфорной Р до рН 3,0±0,2 (20:80, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 340 нм;
- объем вводимой пробы: 10 мкл.
Содержание суммы фенольных соединений в
пересчете на кверцетин в процентах рассчитывают по
формуле:
А, т2 0,16
Аг'Щ '
где:
А1 — сумма площадей пиков 1, 2, 3, 4, 5 на хро¬
матограмме испытуемого раствора (см. рисунок
РБ0052.-1);
А2 — площадь пика кверцетина на хроматограм¬
ме раствора сравнения;
А7?1 — масса навески испытуемого сырья, г;
т2 — масса навески кверцетина дигидрата Р
(в пересчете на содержание безводного кверцетина в
кверцетине дигидрате Р), мг.
07/2016:2120
ДОННИКА ТРАВА
МеШоН ЬегЬа
МЕШОТ
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная трава
двулетнего травянистого растения МеН1о1из о^стаНз
(Ь.) !_ат. и МеНЫиз аШзз1тиз Пии!.
Содержание: не менее 0,3 % кумарина (С9Н602;
М.м. 146,1) (в пересчете на сухое сырье).
Донника трава
1219
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные об¬
лиственные цветущие верхушки и боковые веточки
растений со стеблем длиной до 30 см и диаметром
до 3 мм. Прилистники ланцетные или шиловидные,
цельнокрайние, редко, у самых нижних листьев, с
1—2 зубчиками. Нижние листья обратнояйцевидные,
верхние — продолговатые или ланцетные, по краю
с обеих сторон с 10—13 неровными зубчиками у М.
оШс'таНз и с 8—20 зубчиками у М. аШзз1тиз. Цветки
мотыльковые, мелкие, длиной от 5 мм до 7 мм. Ча¬
шечка колокольчатая, пятизубчатая, остающаяся при
плоде, голая у донника лекарственного и опушенная
у донника рослого. Иногда встречаются в незначи¬
тельном количестве мелкие незрелые плоды — бобы
длиной от 3 мм до 5 мм, неясносетчатые или попереч¬
но-морщинистые, голые или покрытые редкими воло¬
сками. Семя одно, реже два. Цвет стеблей, чашечек и
плодов — зеленый, венчиков — желтый. Запах аро¬
матный, свежего сена (кумарин).
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Порошок имеет желтова¬
то-зеленый цвет. Просматривают под микроскопом с
использованием раствора хлоралгидрата Р. В по¬
рошке обнаруживаются следующие диагностические
признаки (рисунок 2120.-1): фрагменты пластины ли¬
ста (вид поверхности [О]), имеющие неравномерно
утолщенные, слегка волнистые клетки эпидермиса;
многочисленные устьица [ОЬ] преимущественно ано-
моцитного типа (2.8.3) с 3—6 дочерними клетками
Ра] и часто с расположенной внизу палисадной па¬
ренхимой рс]; однорядные покрывающие трихомы с
2 короткими, гладкостенными базальными клетками,
изогнутыми под прямым углом, с толстой стенкой и
бородавчатой кутикулой [А, В]; иногда встречающиеся
железистые трихомы с короткой 2-, 3-клеточной нож¬
кой и яйцевидной двухрядной головкой с 4 нечетки¬
ми клетками [Н]; фрагменты лепестков, состоящие из
клеток с бородавчатыми стенками [М]; фрагменты со¬
судистой ткани стебля [Р, О], включая большие сосу¬
ды [О], иногда соединенные с неодревесневшими раз¬
деленными перегородкой волокнами [Ра] и оберткой
клеток паренхимы, содержащих призматические кри¬
сталлы кальция оксалата [РЬ]; фрагменты мезофилла
У], включающие несколько клеток, которые могут ино¬
гда содержать друзы кальция оксалата ^!а]; фрагмен¬
ты эпидермиса стебля с продолговатыми клетками с
прямыми стенками и устьицами аномоцитного типа
[Ц (2.8.3); фрагменты волокнистого слоя пыльников
(вид с поверхности [Е] и вид поперечного среза [К]);
сферические или яйцевидные зерна пыльцы длиной
около 25 мкм с 3 зародышевыми порами и гладкой эк-
зиной [С].
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,3 г измельченного сы¬
рья (355) (2.9.12) прибавляют Змл метанола Р, на¬
гревают на водяной бане при температуре 100 °С в
течение 1 мин и фильтруют.
Раствор сравнения. 50 м г ФСО кумарина и
20 мг о-кумаровой кислоты Р растворяют в 50 мл
метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: верхний слой смеси кисло¬
та уксусная разведенная Р — эфир Р — толуол Р
(10:50:50, об/об/об).
м "
Рисунок 2120.-1. Микроскопические признаки
травы донника
Наносимый объем пробы: по 25 мкл [или по
3 мкл] в виде полос длиной 10 мм [или 8 мм].
Фронт подвижной фазы: не менее 12 см [или
б см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают 2 Мраство¬
ром калия гидроксида спиртовым Р и просматривают
в ультрафиолетовом свете при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться другие флуоресциру¬
ющие зоны разных цветов.
Верх хроматографической пластинки
Кумарин: флуоресцирую¬
щая зона зеленовато-желто¬
го цвета
Флуоресцирующая зона зе¬
леновато-желтого цвета (ку¬
марин)
о-Кумаровая кислота: флуо¬
ресцирующая зона зелено¬
вато-желтого цвета
Флуоресцирующая зона
синего цвета
Флуоресцирующая зона зе¬
леновато-желтого цвета
(о-кумаровая кислота)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли диаметром свыше 3 мм — не
более 2 %; частицы, проходящие сквозь сито (500)
(2.9.12) , — не более 5 %; пожелтевшие, побуревшие
и почерневшие части растения — не более 2 %. Орга¬
нические примеси: не более 1 %. Минеральные при¬
меси: не более 0,5 %.
® Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 10,0 %.
155*. Зак. 1060.
1220
Гэсударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Жидкостная хроматография (2.2.29).
Испытуемый раствор. 50 г испытуемого сырья
полностью измельчают (500) (2.9.12). К 5,00 г измель¬
ченного сырья прибавляют 90 мл метанола Р, ки¬
пятят с обратным холодильником в течение 30 мин,
охлаждают и фильтруют под вакуумом через тампон
из стекловолокна. К остатку вместе с измельченным
тампоном прибавляют 90 мл метанола Р и повторяют
экстракцию. Фильтраты объединяют и доводят мета¬
нолом Р до объема 250,0 мл.
Раствор сравнения. 25,0 мг ФСО кумарина раст¬
воряют в метаноле Р и доводят до объема 250,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: ацетонитрил Р — раствор
5 г/л кислоты фосфорной Р (22:78, об/об);
- скорость подвижной фазы: 1,7 мл/мин;
- спектрофотометрический детектор, длина
волны 275 нм;
- объем вводимой пробы: 20 мкп.
Пригодность хроматографической системы:
- время удерживания: кумарин — около 7,8 мин.
Содержание кумарина рассчитывают в процен¬
тах по формуле:
А, • т2 • Р
А2 • т^
где:
Л1 — площадь пика кумарина на хроматограмме
испытуемого раствора;
А2 — площадь пика кумарина на хроматограмме
раствора сравнения;
/л1 — масса навески испытуемого сырья, г;
т2 — масса навески ФСО кумарина, г;
Р — содержание кумарина в ФСО кумарина, %.
07/2016:1887
ДУБА КОРА
Оиегсиз сог1ех
ОАК ВАЯК
ОПРЕДЕЛЕНИЕ
Измельченная и высушенная кора поросли,
тонких стволов и молодых ветвей Оиегсиз гоЬиг Ь.,
О. ре/гаеа (Май.) ЫеЫ. (зуп.: О. зеззШЛога ЗаПзЬ.) и
О. риЬезсепз \М11с1.
Содержание:
- не менее 3,0 % дубильных веществ в пересче¬
те на пирогаллол (С6Н603; М.м. 126,1) (в пересчете на
сухое сырье);
- или не менее 8,0 % дубильных веществ в пере¬
счете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Трубчатые, жело¬
боватые или в виде узких полосок различной длины
кусочки, толщиной около (2—3) мм (до 6 мм). Наруж¬
ная поверхность довольно гладкая, с редкими трещин¬
ками. Внутренняя поверхность матовая с невысокими
продольными бороздками шириной (0,5—1) мм. На из¬
ломе наружная кора зернистая, ровная, внутренняя —
сильноволокнистая, занозистая. Цвет коры снаружи
светло-коричневый или светло-серый, серебристый,
внутри — коричневый, красновато-коричневый или
желтовато-коричневый. Запах слабый, своеобразный,
усиливающийся при смачивании коры водой.
В. Микроскопия (2.8.23). На поперечном срезе
виден коричневый пробковый слой из многочислен¬
ных рядов клеток. В наружной коре находятся друзы
оксалата кальция, группы каменистых клеток и на
некотором расстоянии от пробки тангентально рас¬
положенный механический пояс, состоящий из чере¬
дующихся групп лубяных волокон и каменистых кле¬
ток. В наружной коре по направлению от пояса внутрь
разбросаны группы волокон и каменистых клеток.
Некоторые клетки паренхимы содержат флобафе-
ны в виде включений красно-коричневого цвета. Во
внутренней коре многочисленные, тангентально вы¬
тянутые группы лубяных волокон с кристаллоносной
обкладкой, расположены параллельными концентри¬
ческими поясами. Между группами волокон проходят
однорядные сердцевинные лучи, реже встречаются
более широкие лучи, которые близ камбия содержат
группы каменистых клеток, что обуславливает при вы¬
сыхании образование продольных ребер, видимых на
внутренней поверхности.
® С. К 1 г измельченного сырья (710) (2.9.12)
прибавляют 10 мл спирта (30 %, об/об) Р и нагрева¬
ют на водяной бане с обратным холодильником в те¬
чение 30 мин. Охлаждают и фильтруют. К 1 мл филь¬
трата прибавляют 2 мл раствора 10 г/л ванилина Р в
кислоте хлористоводородной Р. Появляется красное
окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: куски коры, потемневшие с внутренней
поверхности, — не более 5 %; куски коры толщиной
более 6 мм — не более 5 %. Органические примеси:
не более 1 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 15,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
(^Определение содержания дубильных ве¬
ществ в пересчете на пирогаллол (2.8.14). Исполь¬
зуют 0,700 г измельченного сырья (710) (2.9.12).
Определение содержания дубильных веществ
в пересчете на танин. 2,000 г измельченного сырья
(2000) помещают в коническую колбу со шлифом
вместимостью 500 мл, прибавляют 250 мл кипящей
воды Р и кипятят с обратным холодильником в тече¬
ние 30 мин при периодическом перемешивании. За¬
тем охлаждают до комнатной температуры, процежи¬
вают и доводят водой Р до объема 250,0 мл.
25,0 мл полученного извлечения помещают в ко¬
ническую колбу вместимостью 750 мл, прибавляют
500 мл воды Р, 25,0 мл раствора индигосульфокисло-
ты (0,25 мг индигокармина Р растворяют в 6,5 мл кис¬
лоты серной Р, прибавляют 6,5 мл кислоты серной Р,
доводят водой Р до объема 250 мл и фильтруют). Ти¬
труют при постоянном перемешивании 0,02 Мраство¬
ром калия перманганата до желтого окрашивания.
Дурмана листья
1221
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07/2016:0246
ДУРМАНА ЛИСТЬЯ
3(гатопи 1оНит
ЗТКАМОЫШМ 1.ЕАР
ОПРЕДЕЛЕНИЕ
Высушенные листья или смесь из высушенных
листьев и цветущих верхушек побегов, иногда с пло¬
дами Оа{ига вкатопшт 1_. и его разновидностей.
Содержание: не менее 0,25 % суммы алкало¬
идов, в пересчете на гиосциамин (С17Н23М03; М.м.
289,4) (в пересчете на сухое сырье). Основными со¬
ставляющими алкалоидов являются гиосциамин вме¬
сте с непостоянным количеством гиосцина (скопола-
мина).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Листья от
темно-коричневато-зеленых до темно-серовато-зе-
леных с короткими черешками, часто сильно скру¬
ченные и смятые при высушивании, тонкие и лом¬
кие, яйцевидной или треугольно-яйцевидной формы,
крупновыемчато-зубчатые с заостренной вершиной
и часто неравнобоким основанием. Молодые ли¬
стья по жилкам опушенные, зрелые листья почти
голые. Стебли зеленые или красновато-зеленые,
прямые, изогнутые или искривленные, продольно
морщинистые, иногда поперечно морщинистые, ди¬
хотомически ветвящиеся, с единственным цветком
или незрелым плодом в развилке. Цветы на корот¬
ком цветоносе, имеют сростнолепестную чашечку с
пятью лепестками и воронкообразным коричневато¬
белым или красноватым венчиком. Плод — коробоч¬
ка, обычно покрытая многочисленными короткими,
жесткими шипами, раскрывается четырьмя створка¬
ми; семена сплюснутые, от коричневого до черного
цвета с мелкоямчатой кожурой.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет серовато-зеленый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки (рисунок
0246.-1): фрагменты верхнего [А] и нижнего [С] эпи¬
дермиса листовой пластинки (при просматривании с
поверхности), представляющего собой клетки со сла¬
бо волнистыми антиклинальными стенками и гладкой
кутикулой, сопровождаемого палисадной [Аа] и губ¬
чатой [Са] паренхимой; анизоцитные [Ас, СЬ] и ано-
моцитные [АЬ] устьица чаще встречаются в нижнем
эпидермисе; фрагменты покровных конических [Е] од¬
норядных 3—5-клеточных волосков с бородавчатыми
стенками, иногда со спавшимися стенками [Еа]; корот¬
кие булавовидные железистые волоски с 2—7-клеточ-
ной головкой при просматривании сбоку [В]; дорсовен-
тральный мезофилл в поперечном разрезе [Р] с одним
рядом палисадных клеток [Ра] и губчатой паренхимой
[РЬ], содержащей друзы оксалата кальция [Рс]; фраг¬
менты губчатой паренхимы [О], некоторые клетки ко¬
торой содержат малые друзы оксалата кальция [РЬ],
с кольцеобразно или спиралеобразно утолщенными
сосудами [Оа] при просматривании с поверхности. В
порошке могут также обнаруживаться: волокна и сет¬
чато-утолщенные сосуды стебля; сферические зерна
пыльцы диаметром около (60—80) мкм с тремя заро¬
дышевыми порами и почти гладкой экзиной [О]; фраг¬
менты венчика [Н] с клетками с волнистыми стенками
[На] и нижележащим мезофиллом [НЬ] с некоторыми
клетками, содержащими призмы [Нс] или друзы [Нб]
оксалата кальция; фрагменты семян, содержащих
желтовато-коричневые извилистые толстостенные
склереиды у] семенной кожуры и редкие призмати¬
ческие и микросфеноидальные кристаллы оксалата
кальция.
Рисунок 0246.-1. Микроскопические признаки
листьев дурмана
® С. Просматривают хроматограммы, получен¬
ные в испытании «Хроматография».
Результаты: основные зоны на хроматограмме
испытуемого раствора по расположению, цвету и раз¬
меру соответствуют основным зонам на хроматограм¬
ме такого же объема раствора сравнения.
®0. 1 г измельченного сырья (180) (2.9.12)
встряхивают с 10 мл 0,05 М раствора серной кисло¬
ты в течение 2 мин и фильтруют. К фильтрату при¬
бавляют 1 мл раствора аммиака концентрированно¬
го Р, 5 мл воды Р и осторожно встряхивают с 15 мл
эфира, свободного от пероксидов, Р, избегая обра¬
зования эмульсии. Эфирный слой отделяют, фильтру¬
ют через фильтр с натрия сульфатом безводным Р
в фарфоровую чашку и эфир выпаривают досуха. К
остатку прибавляют 0,5 мл кислоты азотной Р и вы¬
паривают досуха на водяной бане. Прибавляют 10 мл
ацетона Р и, по каплям, раствор 30 г/л калия гидро¬
ксида Р в 96% спирте Р. Появляется темно-фиоле¬
товое окрашивание.
1222
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
(*>) Хроматография. Тонкослойная хроматогра¬
фия (2.2.27).
Испытуемый раствор. К 1,0 г измельченно¬
го сырья (180) (2.9.12) прибавляют 10 мл 0,05 М
раствора серной кислоты, встряхивают в тече¬
ние 15 мин и фильтруют. Фильтр промывают 0,05 М
раствором серной кислоты до получения 25 мл
фильтрата. К фильтрату прибавляют 1 мл раствора
аммиака концентрированного Р и встряхивают с
двумя объемами эфира, свободного от перокси¬
дов, Р, порциями по 10 мл. При необходимости слои
разделяют при помощи центрифугирования. Объ¬
единенные эфирные слои высушивают безводным
сульфатом натрия Р, фильтруют и выпаривают до¬
суха на водяной бане. Остаток растворяют в 0,5 мл
метанола Р.
Раствор сравнения. 50 мг гиосциамина суль¬
фата Р растворяют в 9 мл метанола Р. 15 мг гиос-
цина гидробромида Р растворяют в 10 мл метано¬
ла Р. 3,8 мл раствора гиосциамина сульфата сме¬
шивают с 4,2 мл раствора гиосцина гидробромида и
доводят метанолом Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р— вода Р— ацетон Р (3:7:90, об/об/об).
Наносимый объем пробы: по 10 мкл и по 20 мкл
каждого раствора в виде полос длиной 20 мм и шири¬
ной Змм, оставив 1 см между полосами.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 15 мин, затем охлаждают на воздухе.
Проявление А: пластинку опрыскивают раство¬
ром калия йодвисмутата Р2, используя около 10 мл
на квадратную пластинку со стороной 200 мм до по¬
явления оранжевых или коричневых зон на желтом
фоне. Просматривают при дневном свете.
Результаты А: зоны на хроматограммах испы¬
туемого раствора соответствуют зонам на хромато¬
граммах раствора сравнения по их расположению
(гиосциамин в нижней трети и гиосцин в верхней
трети хроматограммы) и цвету. Размер зон на хрома¬
тограммах испытуемого раствора не меньше разме¬
ра соответствующих зон на хроматограммах такого
же объема раствора сравнения. Могут наблюдаться
слабые второстепенные зоны в средней части хро¬
матограммы 20 мкл испытуемого раствора или около
линии старта на хроматограмме 10 мкл испытуемого
раствора.
Проявление В: пластинку опрыскивают раство¬
ром натрия нитрита Р до тех пор, пока подложка не
станет видимой через слой сорбента. Просматривают
через 15 мин при дневном свете.
Результаты В: зоны на хроматограммах раст¬
вора сравнения и испытуемого раствора, соответ¬
ствующие гиосциамину, изменяют цвет с коричневого
на красновато-коричневый, но не на серовато-синий
(атропин), все второстепенные зоны исчезают.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли диаметром более 5 мм — не
более 3 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 20,0 %.
<§) Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® а) Определяют потерю в массе при высуши¬
вании (2.2.32). 2,000 г измельченного сырья (180)
(2.9.12) сушат при температуре 105 °С.
Ь) 10,00 г измельченного сырья (180) (2.9.12)
смачивают смесью из 5 мл раствора аммиака Р,
10 мл 96 % спирта Р и 30 мл эфира, свободного от
пероксидов, Р и тщательно перемешивают. Смесь
переносят в подходящий перколятор, применяя,
если необходимо, экстракционную смесь. Настаи¬
вают в течение 4 ч, затем перколируют смесью из
хлороформа Р и эфира, свободного от перокси¬
дов, Р (1:3, об/об) до полного извлечения алкалои¬
дов. Отбирают несколько миллилитров жидкости,
вытекающей из перколятора, высушивают досуха,
сухой остаток растворяют в 0,25 М растворе сер¬
ной кислоты и проверяют отсутствие алкалоидов с
помощью раствора тетрайодмеркурата калия Р.
Перколят концентрируют до объема около 50 мл, от¬
гоняя растворитель на водяной бане, затем количе¬
ственно переносят в делительную воронку, исполь¬
зуя эфир, свободный от пероксидов, Р. Прибавляют
эфир, свободный от пероксидов, Р в количестве, не
менее чем в 2,1 раза превышающем объем перколя-
та, до получения раствора с плотностью значительно
меньшей, чем у воды. Раствор встряхивают не менее
трех раз с 0,25 М раствором серной кислоты, пор¬
циями по 20 мл. Слои разделяют, при необходимо¬
сти используют центрифугирование. Кислотный слой
переносят во вторую делительную воронку, подще¬
лачивают раствором аммиака Р и экстрагируют три
раза хлороформом Р, порциями по 30 мл. К объ¬
единенным хлороформным экстрактам прибавляют
4 г натрия сульфата безводного Р и выдерживают
в течение 30 мин, периодически взбалтывая. Затем
хлороформный экстракт сливают, сульфат натрия
трижды промывают хлороформом Р, порциями по
10 мл, и объединяют с экстрактом. Растворитель вы¬
паривают досуха на водяной бане. Остаток нагрева¬
ют при температуре от 100 °С до 105 °С в течение
15 мин. Сухой остаток растворяют в нескольких мил¬
лилитрах хлороформа Р, прибавляют 20,0 мл 0,01 М
раствора серной кислоты и удаляют хлороформ
выпариванием на водяной бане. После охлаждения
избыток кислоты титруют 0,02 М раствором натрия
гидроксида, используя в качестве индикатора сме¬
шанный раствор метилового красного Р.
Содержание суммы алкалоидов в пересчете на
гиосциамин в процентах рассчитывают по формуле:
57,88 (20 -п)
(ЮО-сУ) ш ’
где:
д— потеря в массе при высушивании измельчен¬
ного испытуемого сырья, %;
п — объем 0,02 М раствора натрия гидроксида,
пошедшего на титрование, мл;
т — масса навески измельченного испытуемого
сырья, г.
ХРАНЕНИЕ
® В защищенном от влаги месте.
Душицы трава
1223
07/2016:1880
ДУШИЦЫ ТРАВА
Оп'дат ЛегЬа
ОКЕвАЫО
ОПРЕДЕЛЕНИЕ
Собранная во время цветения и высушенная
трава многолетнего травянистого растения Опдапит
опИез 1_. или Опдапит уи1даге 1_. подв. Ыг1ит (Ыпк)
1е1зуу. или смеси этих видов.
Содержание:
- эфирного масла:
- цельное сырье: не менее 1 мл/кг (в пересчете
на сухое сырье);
- измельченное сырье: не менее 0,8 мл/кг (в
пересчете на сухое сырье);
- суммы флавоноидов в пересчете на лютеолин
(С15Н10Об; М.м. 286,2): не менее 1,0 % (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). О.опИез. Ли¬
стья желтовато-зеленого цвета, обычно (4—22) мм
длиной и (3—14) мм шириной, бесчерешковые или
имеют длинный или короткий черешок. Пластина яй¬
цевидной, эллиптической или овально-ланцетовид¬
ной формы. Края цельные или зубчатые, верхушка
остроконечная или тупоконечная. Прожилки желто¬
ватого цвета, хорошо заметные на адаксиальной по¬
верхности. Цветки одиночные или видны в виде раз¬
дробленных частей щитка. Чашечка подобна прицвет¬
нику и незаметна. Венчик белого цвета, на верхушке
соцветия либо присутствуют отдельные цветки, либо
они незаметны. Прицветники перекрываются и имеют
зеленую окраску, как у листьев. Лекарственное расти¬
тельное сырье содержит желтоватые или желтовато-
коричневые части стебля.
0.уи1даге (подв. /7Мит). Листья зеленого цвета,
обычно (3—28) мм длиной и (2,5—19) мм шириной.
Черешковые или бесчерешковые. Пластина яйцевид¬
ной или овально-элиптической формы. Края цельные
или зубчатые, верхушка остроконечная или тупоко¬
нечная. Цветки редкие, обнаруживаются в виде раз¬
дробленных частей щитков. Прицветники перекрыва¬
ются и имеют зеленовато-желтую окраску. Чашечка
подобна прицветнику и незаметна. Венчик белого
цвета слегка заметный или незаметный на верхушке
соцветия.
® В. Микроскопия (2.8.23). Измельчают до
порошкообразного состояния (710) (2.9.12). Поро¬
шок имеет зеленый (О. уи1даге) или желтовато-зе¬
леный (О. опИез) цвет. Порошок исследуют под ми¬
кроскопом, используя раствор хлоралгидрата Р
(рисунок 1880.-1).
Порошок О. опИез имеет фрагменты эпидермиса
листьев [А, О, 6], состоящего из клеток с извилисты¬
ми стенками, диацитных устьиц (2.8.3) [Са], покры¬
вающих трихом и железистых трихом; присутствуют
2 типа железистых трихом: некоторые трихомы ла-
миацетного типа с 8—16 клетками (вид с поверхно¬
сти [йа]) и трихомы очень распространенного типа
с одноклеточной головкой и одно- [Ос], двух- [Н] или
трехклеточной ножкой; покрывающие трихомы имеют
гладкие, толстые стенки; некоторые из них являются
многоклеточными [В, (ВЬ], часто раздробленными [Аа]
и содержат призматические кристаллы кальция окса¬
лата, в то время как все остальные являются редкими,
одноклеточными и коническими [С]; на эпидермисе
[<3с1, <3е] видны рубцы от покрывающих и грандуляр-
ных трихом; часто встречаются зерна пыльцы с глад¬
кой экзиной.
Порошок О. уи1даге подв. НШт имеет фрагмен¬
ты верхнего эпидермиса с клетками с извилистыми,
четковидными стенками, с прилегающей палисадной
паренхимой у]; фрагменты нижнего эпидермиса [14],
состоящего из клеток с тонко и неравномерно утол¬
щенными стенками, диацитных устьиц (2.8.3) [№],
покрывающих трихом и железистых трихом; присут¬
ствуют 2 типа железистых трихом: некоторые трихомы
ламиацетного типа с 12 клетками (вид с поверхности
[N6]) и трихомы редкого типа с одноклеточной голов¬
кой [N0] и двух- или трехклеточной ножкой; покрыва¬
ющие трихомы имеют толстые, бородавчатые стенки
и содержат мелкие иглы кальция оксалата; некоторые
из них конические, многоклеточные и зубчатые [Ц М],
в то время как все остальные являются редкими, од¬
ноклеточными [К]; иногда присутствуют зерна пыльцы
с гладкой экзиной [Е, Р].
Рисунок 1880.-1. Микроскопические признаки
травы душицы
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (355) (2.9.12) прибавляют 5 мл метиленхлорида Р,
встряхивают в течение 3 мин и фильтруют через бу¬
мажный фильтр с 2 г натрия сульфата безводного Р.
Раствор сравнения. 1 мг тимола Р и 10 мкл кар-
вакрола Р растворяют в 10 мл метиленхлорида Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: метиленхлорид Р.
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
1224
Гэсударственная фармакопея Республики Беларусь
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р, используя 10 мл на квадрат¬
ную пластинку со стороной 200 мм, и нагревают в те¬
чение 10 мин при температуре от 100 °С до 105 °С.
Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора в нижней трети и в верхней части могут об¬
наруживаться и другие зоны.
Верх хроматографической пластинки
Зона синевато-фиолетово¬
го цвета
Тимол: зона розового цвета
Карвакрол: зона бледно-фи¬
олетового цвета
Зона бледно-зеленого цвета
Зона розового цвета (тимол)
Зона бледно-фиолетового
цвета (карвакрол)
Зона бледно-фиолетового
цвета
Зона серого цвета
Зона бледно-зеленого цвета
Зона синевато-фиолетово¬
го цвета
Интенсивная зона коричне¬
вого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие и побуревшие части рас¬
тения — не более 7 %; кусочки стеблей и боковых
веточек, в том числе отделенные при анализе, — не
более 40 %. Органические примеси: не более 1 %.
Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%, 3,000 г измельченного сырья (2000)
(2.9.72) сушат при температуре 105 °С.
(§) Общая зола (2.4.16). Не более 15,0 %.
® Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.72, метод А или С). 25,0 г
измельченного сырья (2000) (2.9.72) помещают в колбу
вместимостью 1000 мл, прибавляют 400 мл воды Р в
качестве жидкости для перегонки. Время перегонки 2 ч.
Определение содержания суммы флавонои-
дов в пересчете на лютеолин.
Исходный раствор. 1,000 г измельченного сырья
(710) (2.9.12) помещают в коническую колбу со шли¬
фом, прибавляют 50,0 мл спирта (60 %, об/об) Р. Кол¬
бу закрывают, встряхивают и взвешивают с точностью
до 0,01 г и кипятят с обратным холодильником в водя¬
ной бане в течение 1,5 ч. Затем колбу с содержимым
охлаждают до комнатной температуры, взвешивают
и доводят массу колбы до первоначальной спиртом
(60 %, об/об) Р. Извлечение фильтруют через бумаж¬
ный фильтр, отбрасывая первые 10 мл фильтрата.
Испытуемый раствор. К 1,0 мл исходного раст¬
вора прибавляют 4,0 мл раствора 20 г/л алюминия
хлорида Рв96% спирте Р и доводят спиртом (60 %,
об/об) Р до объема 50,0 мл.
Компенсационный раствор. К 1,0 мл исходного
раствора прибавляют 0,1 мл кислоты уксусной ледя¬
ной Р и доводят спиртом (60 %, об/об) Р до объема
50,0 мл.
Через 30 мин измеряют оптическую плотность
растворов (2.2.25) при 400 нм.
Содержание суммы флавоноидов в пересчете на
лютеолин в процентах рассчитывают по формуле:
А 2500
549,41- т ’
где:
549,41 — удельный показатель поглощения ком¬
плекса лютеолина с алюминия хлоридом;
А — оптическая плотность испытуемого раст¬
вора;
/л — масса навески испытуемого сырья, г.
07/2016:1857
@)ДЯГИЛЯ КОРНИ
АпдеНсае агсЬапдеНсае гасИх
АЫОЕЫСА АКСНАЫОЕиСА ЯООГ
ОПРЕДЕЛЕНИЕ
Цельные или резаные, тщательно высушенные
корневища и корни АпдеНса агсЬапдеИса 1_. (синоним:
А. оШсшаНз НоЯтп.).
Содержание: не менее 2,0 мл/кг эфирного масла
(в пересчете на сухое сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеют горький вкус.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корневище име¬
ет серовато-коричневый или красновато-коричневый
цвет и поперечные кольцеобразные утолщения. От
основания отходят серовато-коричневые или крас¬
новато-коричневые цилиндрические продольно-бо-
розчатые, изредка разветвленные корни, часто с не¬
полностью охватывающими поперечными выступами.
Верхняя часть иногда имеет остатки стебля и основа¬
ний листьев. Излом неровный. На изломе видна се¬
ровато-белая, рыхлая, отчетливо радиально-симме¬
тричная кора, в которой заметны секреторные каналы
в виде коричневых пятен, и ярко-желтая или серова¬
то-желтая древесина, которая в корневище окружает
сероватую или коричневато-белую сердцевину.
B. Микроскопия (2.8.23). При просматривании
измельченного сырья (355) (2.9.12) (коричневато-бе¬
лого цвета) с использованием раствора хлоралги¬
драта Р видны (рисунок 1857.-1): фрагменты коры,
состоящие из нескольких слоев тонкостенных серо¬
вато-коричневых или красновато-коричневых клеток
(вид поверхности [С] и поперечный срез [Е]); большие
желтовато-коричневые секреторные цельные или
фрагментированные каналы (поперечный срез [А] или
продольный срез [Р]); фрагменты сердцевинных лу¬
чей толщиной в 2—4 клетки [0]\ фрагменты ксилемы
[В], состоящие из одревесневших сосудов с сетчатым
утолщением [Ва], расположенным по отдельности или
в небольших группах, и неодревесневшей паренхимы,
в которой некоторые клетки, соединенные с сосуда¬
ми, колленхиматозно утолщены. При просматривании
Дягиля корни
1225
измельченного сырья с использованием 50 % (об/об)
раствора глицерина Р видны многочисленные про¬
стые крахмальные зерна диаметром (2—4) мкм, от¬
дельные или включенные в клетки паренхимы [О].
Рисунок 1857.-1. Микроскопические признаки
корней дягиля
С. Просматривают хроматограммы, полу¬
ченные, как указано в испытании «Другие виды
АпдеИса, 1.еУ1зНсит и ЫдизИсит».
Результаты А: ниже приведена последова¬
тельность зон хроматограмм раствора сравнения и
испытуемого раствора. На хроматограмме испытуе¬
мого раствора могут обнаруживаться и другие сла¬
бые флуоресцирующие зоны.
Верх хроматографической пластинки
(2)-Лигустилид: флуоресци¬
рующая зона голубовато-бе¬
лого цвета
Остол: флуоресцирующая
зона синего цвета
Императорин: флуоресциру¬
ющая зона беловатого цвета
Флуоресцирующая зона
синего цвета
Флуоресцирующая зона бе¬
ловатого цвета
Флуоресцирующая зона
синего цвета
3 флуоресцирующие зоны
синего цвета
Раствор сравнения
Испытуемый раствор
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые флу¬
оресцирующие зоны.
Верх хроматографической пластинки
(2)-Лигустилид: флуоресци¬
рующая зона синего цвета
Остол: зона поглощения
Императорин: зона погло¬
щения
Зона поглощения
Зона поглощения
Зона поглощения
Несколько зон поглощения
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Другие виды АпдеНса, 1~еУ1вИсит и ЫдизИсит.
Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1 г свежеизмельченно-
го сырья (355) (2.9.12) прибавляют 4 мл гептана Р,
закрывают и обрабатывают ультразвуком в течение
5 мин. Смесь центрифугируют и используют надоса-
дочную жидкость.
Раствор сравнения. 1 мг императорина Р, 1 мг
(2)-лигустилида Р и 1 мг остола Р растворяют 10 мл
метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((2—10) мкм).
Подвижная фаза: кислота уксусная ледяная Р —
этилацетат Р — толуол Р (1:10:90, об/об/об).
Наносимый объем пробы: по 4 мкл в виде полос
длиной 8 мм.
Фронт подвижной фазы: не менее 6 см от линии
старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты А: на хроматограмме испытуемого
раствора не должна обнаруживаться зона, соответ¬
ствующая по расположению зоне (2)-лигустилида на
хроматограмме раствора сравнения.
Проявление В: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты В: на хроматограмме испытуемого
раствора не должна обнаруживаться зона, соответ¬
ствующая по расположению или находящаяся непо¬
средственно под зоной (7)-лигустилида на хромато¬
грамме раствора сравнения.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: основания листьев и основания сте¬
блей — не более 5 %; частицы, потерявшие есте¬
ственную окраску, — не более 5 %. Другие допусти¬
мые примеси: и не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 40,0 г испыту¬
емого сырья, измельченного непосредственно перед
испытанием (500) (2.9.12), помещают в круглодонную
колбу вместимостью 2 л, прибавляют 10 капель вазе¬
линового масла Р и 500 мл воды Р в качестве жид¬
кости для перегонки. 0,50 мл ксилола Р помещают в
градуированную трубку. Перегонку проводят со скоро¬
стью (2—3) мл/мин в течение 4 ч.
1226
Гэсударственная фармакопея Республики Беларусь
07/2016:1523
ЖЕНЬШЕНЯ КОРНИ
Отзепд гасИх
01Ы8ЕЫС
ОПРЕДЕЛЕНИЕ
Цельные или разрезанные на части высушенные
корни (белый женьшень) или обработанные паром и
затем высушенные корни (красный женьшень) Рапах
дтзепд С. А. Меуег.
Содержание:
- не менее 0,40 % суммы панаксозидов Кд1
(С42Н72°14-2Н2°; Мм• 837) ” *Ь1 (С^О^ЗН.О;
М.м. 1163) (в пересчете на сухое сырье);
- или не менее 1,0 % суммы панаксозидов
в пересчете на панаксозид РЫ (С^Н^О^-ЗНзО;
М.м. 1163) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корни стержне¬
вые, большей частью цилиндрические, с ответвлени¬
ями. Могут быть изогнутыми. Длина корней до 20 см,
диаметр до 2,5 см. Поверхность от бледно-желтого до
белого с желтоватым оттенком цвета (белый женьшень)
или коричневато-красного цвета (красный женьшень), с
продольными морщинами. В верхней части могут быть
остатки побегов в виде короны. Излом ровный. На попе¬
речном срезе видна широкая наружная зона с рассеян¬
ными смоляными каналами оранжево-красного цвета и
тонколучевая внутренняя зона. В нижней части белого
женьшеня имеются многочисленные мелкие корешки,
которые обычно отсутствуют в красном женьшене.
® В. Микроскопия (2.8.23). При просматривании
измельченного сырья (355) (2.9.12) (светло-желтого
цвета) с использованием раствора хлоралгидрата Р
видны следующие диагностические признаки (рисунок
1523.-1): многочисленные фрагменты паренхиматоз¬
ных клеток [А, Е] с тонкими [Еа] или слегка утолщен¬
ными [Аа] стенками, в некоторых клетках обнаружива¬
ются друзы оксалата кальция [АЬ]; фрагменты крупных
секреторных каналов [ЕЬ], содержащих смолу желто¬
вато-коричневого цвета [Ес]; нелигнифицированные
трахеиды и частично лигнифицированные сосуды со
спиральной или сетчатой утолщенностью, изолирован¬
ные [)]; фрагменты ксилемы (вид продольного среза
[С], вид поперечного среза [Р]), состоящие из сосу¬
дов [Са, Ра] и тонкостенных паренхиматозных клеток
[СЬ, РЬ]; рассеянные друзы оксалата кальция р, О];
фрагменты коры (вид с поверхности [В], вид поперечно¬
го среза [Н]), часто представляющие собой феллодерму
со слегка утолщенными стенками [На] и наружным сло¬
ем корковой паренхимы [НЬ]. При использовании 50 %
(об/об) раствора глицерина Р видны многочисленные
крахмальные зерна [К], простые или сложные, диаме¬
тром от 1 мкм до 10 мкм. В красном женьшене крах¬
мальные зерна часто деформированы и разрушены под
действием пара или могут полностью отсутствовать.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл 70% (об/об)
раствора метанола Р. Кипятят с обратным холодиль¬
ником в течение 15 мин. Охлаждают, фильтруют и до¬
водят метанолом Р до объема 10,0 мл.
Раствор сравнения. 5,0 мг эсцина Р и 5,0 мг ар¬
бутина Р растворяют в 1 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Рисунок 1523.-1. Микроскопические
признаки корня женьшеня
Подвижная фаза: этилацетат Р — вода Р —
бутанол Р (25:50:100, об/об/об), используют верхний
слой после отстаивания в течение 10 мин.
Наносимый объем пробы: по 20 мкл [или по
4 мкл] в виде полос.
Фронт подвижной фазы: не менее 10 см [или
5 см] от линии старта в ненасыщенной хроматографи¬
ческой камере.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р. Нагревают при температуре
от 105 °С до 110 °С в течение (5—10) мин. Просматри¬
вают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Арбутин: зона коричнево¬
го цвета
Зона фиолетового цвета
(панаксозиды Кд1+Кд2)
Слабая зона фиолетового
цвета (панаксозид КГ)
Зона фиолетового цвета
(панаксозид Ке)
Зона фиолетового цвета
(панаксозид Кс1)
Слабая зона фиолетового
цвета
Эсцин: зона серого цвета
Зона фиолетового цвета
(панаксозид Кс)
Зона фиолетового цвета
(панаксозиды КЫ+КЬ2)
Раствор сравнения
Испытуемый раствор
Женьшеня корни
1227
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Сумма допусти¬
мых примесей: не более 2 %.
(§) Рапах дшлдие/о/шт. Просматривают хро¬
матограмму, полученную в разделе «Количествен¬
ное определение. Определение содержания суммы
панаксозидов Рд1 и РЫ». В случае подмены корней
Рапах д'тзепд на корни Рапах дитциеМит на хрома¬
тограмме испытуемого раствора отсутствует пик, со¬
ответствующий панаксозиду Я* (рисунок 1523.-2).
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 7,0 %.
® Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Определение содержания суммы панаксо¬
зидов Рд1 и ВЫ.
Жидкостная хроматография (2.2.29).
Испытуемый раствор. Измельчают около 50 г
испытуемого сырья (355) (2.9.12). 1,00 г измельчен¬
ного сырья помещают в круглодонную колбу вмести¬
мостью 250 мл, прибавляют 70 мл 50 % (об/об) раст¬
вора метанола Р, добавляют несколько кусочков
пемзы и кипятят с обратным холодильником на водя¬
ной бане в течение 1 ч. Охлаждают, центрифугируют
и собирают надосадочную жидкость. Остаток пере¬
носят в ту же круглодонную колбу и повторяют экс¬
тракцию, как указано выше. Надосадочные жидкости
объединяют и выпаривают досуха при пониженном
давлении при температуре не выше 60 °С. Остаток
растворяют в 20,0 мл смеси из ацетонитрила Р
и воды Р (20:80, об/об). 2,0 мл полученного раст¬
вора разводят смесью из ацетонитрила Р и воды Р
(20:80, об/об) до объема 10,0 мл и перед введением
пробы фильтруют через мембранный фильтр с диа¬
метром пор 0,45 мкм.
Раствор сравнения. 3,0 мг панаксозида Рд1 Р,
3.0 мг панаксозида Ре Р, 3,0 мг панаксозида Р(Р и
3.0 мг панаксозида РЫ Р растворяют в метаноле Р
и доводят до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,125 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером ча¬
стиц 5 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: вода Р, доведенная кис¬
лотой фосфорной Р до рН 2;
- подвижная фаза В: ацетонитрил Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—8
80
20
8—40
00
0
1
05
О
о
т
о
см
40-45
60 —* 40
40 — 60
45-47
40 — 0
О)
0
1
о
о
47—52
0
100
52—55
0 — 80
100 — 20
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 203 нм;
- время уравновешивания системы: 20 мин пе¬
ред каждым вводом;
- объем вводимой пробы: 20 мкл.
Порядок выхода пиков: в порядке перечисления
составляющих раствора сравнения; отмечают их вре¬
мена удерживания.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,0 между пиками панак¬
созида Рд1 и панаксозида Ре.
Определяют расположение пиков, соответствую¬
щих панаксозиду РЫ и панаксозиду Рд1, на хромато¬
грамме испытуемого раствора.
Содержание панаксозида Рд1 и панаксозида РЫ
в процентах рассчитывают по формуле:
‘^2 *^1 ^ ^2 -Л13 ‘РI
53 • ш1 -100 34 •Л71 -100 ’
где:
51 — площадь пика панаксозида РЫ на хромато¬
грамме испытуемого раствора;
$2 — площадь пика панаксозида Рд1 на хромато¬
грамме испытуемого раствора;
53 — площадь пика панаксозида РЫ на хромато¬
грамме раствора сравнения;
54 — площадь пика панаксозида Рд1 на хромато¬
грамме раствора сравнения;
1. Панаксозид Рд1. 4. Панаксозид РЫ. 7. Панаксозид Р<±
2. Панаксозид Ре. 5. Панаксозид Рс.
3. Панаксозид НГ. 6. Панаксозид РЬ2.
Рисунок 1523.-2. Хроматограмма испытуемого раствора
1228
Гэсударственная фармакопея Республики Беларусь
а7?1 — масса навески испытуемого сырья, г;
т2 — масса навески панаксозида ЯМ в растворе
сравнения, мг;
т3 — масса навески панаксозида Рд1 в растворе
сравнения, мг;
Р, — содержание панаксозида КМ в реактиве, %;
Р2 — содержание панаксозида Рд1 в реактиве, %.
Определение содержания суммы панаксози-
дов в пересчете на панаксозид РЫ.
Исходный раствор. 1,000 г измельченного сырья
(710) (2.9.12) помещают в колбу со шлифом вмести¬
мостью 250 мл, прибавляют 50,0 мл спирта (40 %,
об/об) Р, взвешивают с точностью до 0,01 г и нагре¬
вают с обратным холодильником в водяной бане в
течение 60 мин. Охлаждают, доводят массу до перво¬
начальной спирта (40 %, об/об) Р и фильтруют.
Испытуемый раствор. 2,0 мл исходного раст¬
вора помещают в колбу со шлифом вместимостью
50 мл, прибавляют 1,0 мл раствора 50 г/л кислоты
фосфорномолибденовой Р в 96% спирте Р, 10 мл
воды Р и нагревают с обратным холодильником в
водяной бане в течение 2 ч. Быстро охлаждают до
комнатной температуры. Полученный раствор коли¬
чественно переносят в мерную колбу вместимостью
50 мл, используя воду Р, и доводят до объема 50,0 мл
этим же растворителем.
Компенсационный раствор. 2,0 мл спирта
(40 %, об/об) Р помещают в колбу со шлифом вмести¬
мостью 50 мл, прибавляют 1,0 мл раствора 50 г/л кис¬
лоты фосфорномолибденовой Р в 96% спирте Р,
10 мл воды Р и нагревают с обратным холодильником
в водяной бане в течение 2 ч. Быстро охлаждают до
комнатной температуры. Полученный раствор коли¬
чественно переносят в мерную колбу вместимостью
50 мл, используя воду Р, и доводят до объема 50,0 мл
этим же растворителем.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 730 нм.
Содержание суммы панаксозидов в пересчете на
панаксозид РЫ в процентах рассчитывают по формуле:
Д-1250
/г? * 193 ’
где:
193 — удельный показатель поглощения продук¬
тов реакции панаксозида РЫ с кислотой фосфорно¬
молибденовой;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0053
ЖОСТЕРА СЛАБИТЕЛЬНОГО
ПЛОДЫ
КЬатп! саМагИсае 1гис1из
соммоы висктнсжы грилт*
ОПРЕДЕЛЕНИЕ
Собранные осенью зрелые и высушенные плоды
кустарника РЬатпиз саМа/Иса 1_.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Плоды — окру¬
глые костянки с блестящей морщинистой поверхно¬
стью, диаметром (5—8) мм, с небольшим малоза¬
метным остатком столбика и с сохранившейся плодо¬
ножкой или углублением на месте ее отрыва. Мякоть
бурая, с 3—4 (реже 2) темно-бурыми косточками с
твердой кожурой, трехгранной или яйцевидной фор¬
мы. Цвет плодов почти черный. Запах слабый, непри¬
ятный.
B. Микроскопия (2.8.23). При просматривании
в поперечном срезе в паренхимной ткани видны: со-
судисто-проводящие пучки, секреторные вместили¬
ща и друзы оксалата кальция. Эндокарп состоит из
кристаллоносных клеток, склереид и склеренхимы.
Семенная кожура также содержит извилисто- и тол¬
стостенные склереиды.
C. К 1 г измельченного сырья (710) (2.9.12) при¬
бавляют 10 мл раствора 100 г/л натрия гидрокси¬
да Р, кипятят в течение нескольких минут, прибавляют
10 мл воды Р и фильтруют. При охлаждении фильтрат
подкисляют кислотой хлористоводородной разве¬
денной Р до слабокислой реакции. К полученному
раствору прибавляют 10 мл эфира Р и встряхивают.
Эфирный слой окрашивается в желтый цвет. 5 мл
эфирного извлечения встряхивают с 5 мл раствора
аммиака разведенного Р1. Водный слой окрашива¬
ется в темно-красный цвет, а эфирный слой остается
окрашенным в желтый цвет.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: недозрелые плоды — не более 4 %;
подгоревшие плоды — не более 5 %. Органические
примеси: не более 2 %. Минеральные примеси: не бо¬
лее 0,5 %.
Примечание. Не допускается примесь плодов
крушины ольховидной (Ргапди1а а1пиз МН1.) — черные,
неблестящие, шарообразные костянки, содержащие 2
(3) чечевицеобразные косточки с клювовидным хря¬
щеватым выростом.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 4,0 %.
07/2016:1438
ЗВЕРОБОЯ ТРАВА
Нурепс/ МегЬа
8Т. ЛЭНЫ’З IУОЯ7"
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения высушенная цель¬
ная или фрагментированная трава многолетних тра¬
вяных растений Нурепсит рег?ога№т и Нурепсит
тасиЫит Сгап*2 (Н. циадгапди1ит 1_.).
Содержание:
- не менее 0,08 % суммы гиперицинов в пересче¬
те на гиперецин (С30Н16О8; М.м. 504,4) (в пересчете на
сухое сырье);
-- или не менее 1,5 % суммы флавоноидов в пе¬
ресчете на рутин (С27Н30О16; М.м. 611) (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Верхние части
стеблей с листьями, цветками, бутонами и недозре-
Зверобоя трава
1229
лыми плодами. Стебли полые, цилиндрические, дли¬
ной до 30 см, с двумя (Н. регГога1ит) или четырьмя
(Н. таси1а1ит) продольными ребрами. Листья супро¬
тивные, сидячие, продолговатые или продолговато¬
овальные, цельнокрайние, голые, до 3,5 см, шириной
до 1,4 см. У Нурепсит рег(ога1ит листья с много¬
численными просвечивающимися вместилищами в
виде светлых точек. Цветки многочисленные около
(1—1,5) см в диаметре, собраны в щитковидную ме¬
телку. Чашечка сростнолистная, глубокопятираздель¬
ная, чашелистики ланцетовидные, тонко заостренные
(Н. регТогаШт) или продолговато-овальные с при¬
тупленной верхушкой (Н. тасиШит). Венчик раз¬
дельнолепестной, в 2—3 раза длиннее чашечки, ле¬
пестков пять. Тычинки многочисленные, сросшиеся у
основания нитями в три пучка. Плод — трехгнездная
многосемянная коробочка. Цвет стеблей — от зеле¬
новато-желтого до серовато-зеленого, иногда розова¬
то-фиолетовый; листьев — от серовато-зеленого до
темно-зеленого; лепестков — ярко-желтый или жел¬
тый с черными точками, хорошо заметными под лу¬
пой; плодов зеленовато-коричневый. Запах слабый,
своеобразный.
В. Микроскопия (2.8.23). При просматривании
листа с поверхности видны клетки эпидермиса с из¬
вилистыми стенками, имеющими четковидные утол¬
щения. Устьица аномоцитного или парацитного типа
(2.8.3), расположены только на нижней стороне листа.
Встречаются вместилища двух типов: пигментиро¬
ванные вместилища овальной формы, содержащие
красновато-фиолетовый пигмент, расположены в ос¬
новном по краю листа; бесцветные просвечивающи¬
еся вместилища (Н. регГога(ит) встречаются по всей
пластинке листа, вдоль жилок они продольно вытя¬
нуты, у Нурепсит тасиШит встречаются редко или
отсутствуют.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (500) (2.9.12) прибавляют 10 мл метанола Р, вы¬
держивают в водяной бане при температуре 60 °С в
течение 10 мин при перемешивании и фильтруют.
Раствор сравнения. 5 мг рутина Р и 5 мг гипе-
розида Р растворяют в метаноле Р и доводят объем
до 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безвод¬
ная Р — вода Р — этилацетат Р (6:9:90, об/об).
Наносимый объем пробы: 10 мкл испытуемого
раствора и 5 мкл раствора сравнения в виде полос
длиной 10 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С в течение 10 мин.
Проявление: пластинку обрабатывают раство¬
ром 10 г/л дифенилборной кислоты аминоэтилового
эфира Р в метаноле Р и затем раствором 50 г/л ма-
крогола 400 Р в метаноле Р. Через 30 мин пластинку
просматривают в ультрафиолетовом свете при длине
волны 365 нм.
Результаты: на хроматограмме раствора срав¬
нения в нижней трети обнаруживаются флуоресциру¬
ющая зона (рутин) и расположенная над ней флуорес¬
цирующая зона (гиперозид), обе зоны желто-оранже¬
вого цвета. На хроматограмме испытуемого раствора
в нижней трети обнаруживаются красно-оранжевые
флуоресцирующие зоны, соответствующие рутину и
гиперозиду, в нижней части верхней трети флуоресци¬
рующая зона (псевдогиперицин) и над ней флуорес¬
цирующая зона (гиперицин), обе зоны красного цвета.
На хроматограмме испытуемого раствора могут обна¬
руживаться и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли — не более 50 %. Органические
примеси: не более 1 %. Минеральные примеси: не бо¬
лее 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
(^Определение содержания суммы гипери-
цинов в пересчете на гиперицин.
Испытуемый раствор. 0,800 г измельченного
сырья (500) (2.9.12) помещают в круглодонную колбу
вместимостью 100 мл с магнитной мешалкой и при¬
бавляют 60 мл смеси из воды Р и тетрагидрофура-
на Р (20:80, об/об). Смесь кипятят с обратным холо¬
дильником в водяной бане при температуре 70 °С в
течение 30 мин, затем центрифугируют при 700 д в
течение 2 мин. Надосадочную жидкость помещают
в колбу вместимостью 250 мл. Остаток помещают в
ту же круглодонную колбу объемом 100 мл, прибав¬
ляют 60 мл смеси из воды Р и тетрагидрофурана Р
(20:80, об/об), нагревают с обратным холодильником
в течение 30 мин и центрифугируют при 700 д в те¬
чение 2 мин. Надосадочную жидкость помещают в
колбу с первым извлечением и выпаривают досуха.
Остаток после выпаривания с помощью ультразвука
растворяют в 15 мл метанола Р, переносят в мерную
колбу вместимостью 25 мл, в которую прибавляют ме¬
танол Р после промывания колбы объемом 250 мл, и
доводят до объема 25,0 мл этим же растворителем.
Центрифугируют и фильтруют 10 мл надосадочной
жидкости через мембранный фильтр (0,2 мкм), отбра¬
сывая первые 2 мл фильтрата. 5,0 мл фильтрата до¬
водят метанолом Р до объема 25,0 мл.
Компенсационный раствор. Метанол Р.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 590 нм. Содержание гиперицинов
в пересчете на гиперицин в процентах рассчитывают
по формуле:
А-125
т-870 ’
где:
870 — удельный показатель поглощения гипери-
цина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Определение содержания суммы флавонои-
дов в пересчете на рутин.
Исходный раствор. 1,000 г измельченного сырья
(710) помещают в колбу вместимостью 150 мл, при¬
бавляют 30 мл спирта (50 %, об/об) Р. Нагревают с
обратным холодильником в водяной бане в течение
30 мин, периодически встряхивая для смывания ча-
1230
Гэсударственная фармакопея Республики Беларусь
стиц сырья со стенок колбы. Горячее извлечение
процеживают через ватный тампон в мерную колбу
вместимостью 100 мл так, чтобы частицы сырья не
попадали на фильтр. Ватный тампон помещают в кол¬
бу для экстрагирования и прибавляют 30 мл спирта
(50 %, об/об) Р. Экстракцию повторяют еще дважды в
описанных выше условиях, процеживая извлечение в
ту же мерную колбу. Охлаждают и доводят спиртом
(50 %, об/об) Р до объема 100,0 мл.
Испытуемый раствор. 1,0 мл исходного раст¬
вора и 1,0 мл раствора 20 г/л алюминия хлорида Р в
96 % спирте Р доводят 96 % спиртом Р до объема
25,0 мл.
Раствор сравнения. 0,0500 г рутина Р, предва¬
рительно высушенного при температуре от 130 °С до
135 °С в течение 3 ч, растворяют в 96 % спирте Р при
нагревании на водяной бане, охлаждают и доводят
до объема 100,0 мл этим же растворителем. К 1,0 мл
полученного раствора прибавляют 1,0 мл раствора
20 г/л алюминия хлорида Р в 96% спирте Р и дово¬
дят 96 % спиртом Р до объема 25,0 мл.
Компенсационный раствор. 1,0 мл исходного
раствора и 1 каплю кислоты уксусной разведенной Р
доводят 96 % спиртом Р до объема 25,0 мл.
Через 40 мин измеряют оптическую плотность
(2.2.25) растворов при 415 нм.
Содержание суммы флавоноидов в пересчете на
рутин в процентах рассчитывают по формуле:
А т0 -100
А0т
где:
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески рутина Р, г.
07/2016: РБ0054
ЗЕМЛЯНИКИ ЛЕСНОЙ ЛИСТЬЯ
Ргадапае уезсае Шит
8ТВА\МВЕГЖУ 1-ЕАГ*
ОПРЕДЕЛЕНИЕ
Собранные в фазу цветения и высушенные ли¬
стья дикорастущего многолетнего травянистого рас¬
тения Ргадапа уезса 1_.
Содержание: не менее 4,0 % суммы феноль¬
ных соединений в пересчете на галловую кислоту
(С7Нб05 ■ Н20; М.м. 188,1) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные или
частично измельченные листья с остатками череш¬
ков длиной до 1 см. Листья сложные, состоят из трех
почти сидячих листочков длиной (1,5—6) см, шириной
(1,6—4,5) см. Средний листочек яйцевидной или ром¬
бической формы, боковые — косо-яйцевидные. Края
листочков зубчатые. Зубцы треугольные или округло¬
треугольные с заостренной верхушкой. Конечный зу¬
бец на верхушке листочка несколько меньше по раз¬
мерам соседних зубцов. Верхняя сторона листочка с
редкими волосками и слегка вдавленными жилками,
нижняя с довольно густым опушением и выпуклыми
центральной и боковыми жилками первого порядка.
Цвет листьев сверху зеленый или темно-зеленый,
снизу — сероватый или серовато-зеленый.
B. Микроскопия (2.8.23). При просматрива¬
нии препарата листа под микроскопом видны пря¬
мостенные клетки верхнего эпидермиса, местами с
четковидным утолщением, и извилистостенные клет¬
ки — нижнего. Устьица многочисленные, овальные,
погруженные, окружены 3—6 клетками эпидермиса
(аномоцитный тип) (2.8.3). На обеих сторонах листа,
а также по жилкам и вблизи их встречаются простые
и головчатые волоски. Простые волоски длинные,
одноклеточные, толсто- и тонкостенные, остроко¬
нечные с расширенным основанием. Головчатые же¬
лезистые волоски состоят из 2—3-клеточной (реже
1- и 4-клеточной) ножки и одноклеточной овальной
или шаровидной головки. Клетки эпидермиса вокруг
основания волосков образуют розетку. В мезофилле
листа, главной жилки, черешочков и черешков встре¬
чаются друзы и ромбические кристаллы оксалата
кальция.
C. К 1 мл раствора А, полученного как указано в
разделе «Количественное определение», в фарфоро¬
вой чашке прибавляют 2 капли раствора 10 г/л железа
(III) хлорида Рв96% спирте Р. Появляется сине-фи¬
олетовое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: листья, побуревшие или почернев¬
шие, — не более 2 %; листья с остатками черешка
длиной более 1 см — не более 5 %; цветоносные
стебли, плоды и другие части растения — не более
5 %. Органические примеси: не более 1 %. Минераль¬
ные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16) Не более 12,0%.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0%.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
В колбу со шлифом вместимостью 100 мл по¬
мещают 0,500 г измельченного сырья (2000) (2.9.12),
прибавляют 50 мл спирта (80 %, об/об) Р, закрывают
пробкой, взвешивают с точностью до 0,01 г и нагре¬
вают с обратным холодильником на водяной бане в
течение 45 мин. Затем колбу с содержимым охлажда¬
ют до комнатной температуры, взвешивают вместе с
пробкой и доводят массу колбы до первоначальной
спиртом (80 %, об/об) Р. Извлечение фильтруют, от¬
брасывая первые 5 мл фильтрата (раствор А).
Испытуемый раствор. К 0,10 мл раствора А
прибавляют 0,50 мл реактива фосфорномолибдено-
во-вольфрамового Р, 2,5 мл насыщенного раствора
натрия карбоната Р и доводят водой Р до объема
25,0 мл.
Компенсационный раствор. К 0,50 мл реактива
фосфорномолибденово-вольфрамового Р прибавля¬
ют 2,5 мл насыщенного раствора натрия карбона¬
та Р и доводят водой Р до объема 25,0 мл.
Через 60 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 760 нм.
Содержание суммы фенольных соединений в
пересчете на галловую кислоту в процентах рассчи¬
тывают по формуле:
Золототысячника трава
1231
Л-12500
т • 850
где:
850 — удельный показатель поглощения продук¬
тов взаимодействия галловой кислоты с реактивом
фосфорномолибденово-вольфрамовым;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0055
ЗЕМЛЯНИКИ ЛЕСНОЙ ПЛОДЫ
Ргадапае уезсае 1гис1из
ЗТКАМВЕККУ РКШТ*
ОПРЕДЕЛЕНИЕ
Собранные в фазу полного созревания и высу¬
шенные плоды дикорастущего многолетнего травяни¬
стого растения Ргадапа Vезса Ь.
Содержание: не менее 3,0 % фенольных соеди¬
нений в пересчете на галловую кислоту (С7Нб05 • Н20;
М.м. 188,1) (в пересчете на сухое сырье).
A. Внешние признаки (2.8.23). Цельные, очи¬
щенные от чашелистиков и плодоножек, ложные
плоды разнообразной формы — от яйцевидной или
овальной до конусообразной, длиной от 6 мм до
10 мм и шириной от 3 мм до 7 мм с многочисленны¬
ми погруженными до половины в мякоть продолго¬
вато-коническими, сухими, желтоватыми плодика¬
ми-орешками. Цвет плодов от оранжево-красного до
ярко- и темно-красного. Запах ароматный. Вкус кис¬
ловато-сладкий.
B. Микроскопия (2.8.23). При просматрива¬
нии с поверхности видны многоугольные клетки
эпидермиса с прямыми стенками и редкими окру¬
глыми устьицами, окруженными 5—7 клетками
(аномоцитный тип) (2.8.3). На поверхности эпидер¬
миса встречаются волоски — простые одноклеточ¬
ные мелкие тонкостенные извилистые, часто нахо¬
дящиеся по 2—3 вместе, крупные толстостенные
с узкой полостью и 2—3-клеточные. Поверхность
плода покрыта многочисленными ворсинками, рас¬
положенными по разросшемуся цветоложу. Эпи¬
дермис кожуры орешка состоит из многоугольных
клеток, местами имеющих четковидные утолще¬
ния. Механический слой кожуры орешка представ¬
лен вытянутыми взаимоперекрещивающимися во¬
локнами с призматическими кристаллами оксалата
кальция.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие и пригоревшие плоды —
не более 8 %; недозрелые плоды — не более 5 %;
плоды с плодоножками — не более 3 %; отделивши¬
еся семена — не более 5 %; листья и стебли — не
более 0,1 %. Органические примеси: не более 1 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Зола общая (2.4.16). Не более 4,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
В колбу со шлифом вместимостью 100 мл по¬
мещают 0,500 г измельченного сырья (1000) (2.9.12),
прибавляют 50 мл спирта (50 %, об/об) Р, закрывают
пробкой, взвешивают с точностью до 0,01 г и нагре¬
вают с обратным холодильником на водяной бане в
течение 60 мин. Затем колбу с содержимым охлажда¬
ют до комнатной температуры, взвешивают вместе с
пробкой и доводят массу колбы до первоначальной
спиртом (50 %, об/об) Р. Извлечение фильтруют, от¬
брасывая первые 5 мл фильтрата (раствор А).
Испытуемый раствор. К 0,10 мл раствора А
прибавляют 0,50 мл реактива фосфорномолибдено-
во-вольфрамового Р, 2,5 мл насыщенного раствора
натрия карбоната Р и доводят водой Р до объема
25,0 мл закрывают пробкой, перемешивают и нагре¬
вают в водяной бане при температуре 60 °С в течение
2 мин.
Компенсационный раствор. К 0,50 мл реактива
фосфорномолибденово-вольфрамового Р прибавля¬
ют 2,5 мл насыщенного раствора натрия карбона¬
та Р и доводят водой Р до объема 25,0 мл.
Через 60 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 760.
Содержание суммы фенольных соединений в
пересчете на галловую кислоту в процентах рассчи¬
тывают по формуле:
А-12500
ш-850 ’
где:
850 — удельный показатель поглощения продук¬
тов взаимодействия галловой кислоты с реактивом
фосфорномолибденово-вольфрамовым;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1301
ЗОЛОТОТЫСЯЧНИКА ТРАВА
СеМаигН ЬегЬа
СЕЫТА1ЮУ
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная тра¬
ва травянистых растений СеМаипит егу1Ьгаеа Ра1п,
включая С. та]из (Н. е\ Ь.) 2еГСпег, С. зиЯгиИсозит
(СпзеЬ.) Яопп. (зуп.: Егу01гаеа сеМаипит Регзооп;
С. итЬе11а(ит ОШЬег*; С. ттиз СВагз.).
ОПИСАНИЕ (СВОЙСТВА)
(!*) Вкус горький.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
(§)А. Внешние признаки (2.8.23). Стебель по¬
лый цилиндрический от зеленого до темно-коричне¬
вого цвета, с продольными ребрами, ветвится только
в верхней части. Листья сидячие, цельные, располо¬
женные крестообразно, листовая пластинка от оваль¬
ной до ланцетовидной формы, длиной до 3 см. Обе
поверхности голые от зеленого до коричневато-зеле¬
ного цвета. Соцветия щитковидные. Трубчатая чашеч¬
ка зеленого цвета с 5 ланцетовидными заостренными
зубчиками. Венчик состоит из беловатой трубки, де-
1232
Гэсударственная фармакопея Республики Беларусь
лящейся на 5 вытянутых ланцетовидных долей от ро¬
зового до красного цвета, длина (5—8) мм. 5 тычинок
присоединены к верхней части трубки венчика. За¬
вязь верхняя с коротким пестиком, широким раздво¬
енным рыльцем и многочисленными семяпочками.
Часто встречаются цилиндрические коробочки дли¬
ной (7—10) мм с маленьким коричневыми семенами
с заметной неровностью.
В. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). При просматривании листа с
поверхности (рисунок #1301.-1) видны клетки эпидер¬
миса обеих сторон с извилистыми стенками и склад¬
чатой кутикулой. Клетки эпидермиса нижней стороны
листа отличаются меньшими размерами и более из¬
вилистыми стенками [А]. Устьица с обеих сторон ли¬
ста анизоцитного типа (2.8.3), в большем количестве
на нижней, окружены 2—3 околоустьичными клетка¬
ми, на нижней стороне листа С. ри1с1ю11ит встреча¬
ются устьица диацитного типа [2.8.3]. По краю пла¬
стинки 1—2 ряда клеток эпидермиса имеют толстые
оболочки и слегка вытянуты в сосочек [В]. В клетках
мезофилла листа видны мелкие одиночные призма¬
тические кристаллы оксалата кальция, иногда встре¬
чаются крестообразносросшиеся кристаллы и реже
мелкие друзы [С]. Эпидермис чашечки с прямыми
стенками [Р], венчика — с тупыми стенками и склад¬
чатой кутикулой [О]. Трехпоровая желтая пыльца диа¬
метром около 30 мкм [Е].
Рисунок #1301 .-1. Микроскопические признаки
травы золототысячника
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (335) (2.9.12) прибавляют 25 мл метанола Р,
встряхивают в течение 15 мин и фильтруют. Фильтрат
выпаривают досуха при пониженном давлении при
температуре не выше 50 °С. К остатку прибавляют
метанол Р до получения 5 мл раствора (может со¬
держать осадок).
Раствор сравнения. 1 мг рутина Р и 1 мг свер-
тиамарина Р растворяют в 1 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р245 Р ((2—10) мкм)].
Подвижная фаза: кислота муравьиная без¬
водная Р — вода Р — этилформиат Р (8:4:88,
об/об/об).
Наносимый объем пробы: по Юмкл [или по
5 мкл] в виде полос.
Фронт подвижной фазы: не менее 12 см [или
6 см] от линии старта в ненасыщенной хроматографи¬
ческой камере.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при 254 нм.
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны с мень¬
шей интенсивностью поглощения.
Верх хроматографической пластинки
Свертиамарин: зона погло¬
щения
Рутин: зона поглощения
Зона выраженного поглоще¬
ния (свертиамарин)
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р, нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин и про¬
сматривают при дневном свете.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и
испытуемого раствора. На хроматограмме испы¬
туемого раствора могут обнаруживаться и другие
менее интенсивные зоны.
Верх хроматографической пластинки
Свертиамарин: зона корич¬
невого цвета
Рутин: зона желтого цвета
Зона коричневого цвета
(свертиамарин)
Зона коричневато-серого
цвета
Зона желтого цвета
Зона серого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни, в том числе отделенные при
анализе, — не более 2 %. Органические примеси: не
более 1 %. Минеральные примеси: не более 1 %.
® Показатель горечи (2.8.15). Не менее 2000.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 7,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,5 %.
Ивы кора
1233
07/2016:1583
ИВЫ КОРА
ЗаНа'з сог1ех
И/ШОЮВАКК
ОПРЕДЕЛЕНИЕ
Высушенные цельные или фрагментированные
куски коры молодых ветвей и цельные части побегов
текущего года различных видов рода ЗаНх, включая
5. ригригеа Ь., 5. а/Ьа 1_., 5. ГгадШз 1_. и 5. дарЬпоШез УШ.
Содержание: не менее 1,5 % производных сали¬
цина в пересчете на салицин (С13Н1807; М.м. 286,3)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
СЕ) А. Внешние признаки (2.8.23). Кора толщи¬
ной (1—2) мм в виде гибких продолговатых трубчатых
или изогнутых кусков. Внешняя поверхность гладкая
или продольно слегка морщинистая серовато-жел¬
того или коричневато-серого цвета. Внутренняя по¬
верхность гладкая или продольно мелкоисчерченная
и в зависимости от вида белого, бледно-желтого или
красновато-коричневого цвета. Излом мелкощети¬
нистый на внешней стороне и грубоволокнистый на
внутренней стороне. Диаметр побегов текущего года
не превышает 10 мм. Древесина белого или бледно-
желтого цвета.
СЕ) В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет бледно-желтый, зе¬
леновато-желтый или светло-коричневый. Просматри¬
вают под микроскопом с использованием раствора
хлоралгидрата Р. В порошке обнаруживаются сле¬
дующие диагностические признаки (рисунок 1583.-1):
пучки [В, С] тонких волокон [Ва, Са] длиной до 600 мкм
с очень толстыми стенками и кристаллической обклад¬
кой, состоящей из призм оксалата кальция [ВЬ, СЬ]; па¬
ренхиматозные клетки коры [О, 3] с толстыми ямчаты-
ми и очень четковидными стенками [йа], содержащие
крупные друзы оксалата кальция [Са, За]; некоторые
клетки паренхимы являются колленхиматозными [С];
однорядные сердцевинные лучи в поперечном разрезе
РЬ]; утолщенные клетки пробки при просматривании с
поверхности [Р]; многочисленные рассеянные призмы
[Е] и друзы [А] оксалата кальция; могут обнаруживать¬
ся фрагменты коричневатой колленхимы почек. Кроме
этого при просматривании побегов обнаруживаются:
фрагменты древесины [Н], включающей в себя лигни-
фицированные волокна [На] и сосуды [НЬ]; иногда в со¬
провождении сердцевинных лучей [Нс].
(Е) С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор (а). К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р.
Нагревают на водяной бане при температуре около
50 °С в течение 10 мин при частом встряхивании. Ох¬
лаждают и фильтруют.
Испытуемый раствор (Ь). К 5,0 мл испытуемого
раствора (а) прибавляют 1,0 мл раствора 50 г/л на¬
трия карбоната безводного Р и нагревают в водяной
бане при температуре около 60 °С в течение 10 мин.
Охлаждают и при необходимости фильтруют.
Раствор сравнения. 2 мг салицина Р и 2 мг хлоро-
геновой кислоты Р растворяют в 1,0 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Рисунок 1583.-1. Микроскопические признаки
коры ивы
Подвижная фаза: вода Р— метанол Р— этила-
цетат Р (8:15:77, об/об/об).
Наносимый объем пробы: по 10 мкл [или 2 мкл]
в виде полос.
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку обрабатывают смесью
серная кислота Р — метанол Р (5:95, об/об) и на¬
гревают при температуре от 100 °С до 105 °С в те¬
чение 5 мин. Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемых растворов (а) и (Ь). На хроматограмме
испытуемых растворов (а) и (Ь) могут обнаружи¬
ваться и другие зоны.
Верх хроматографической пластинки
Могут обнаружи¬
ваться несколь¬
ко зон краснова¬
то-фиолетового
цвета
Слабая зона крас¬
новато-фиолето¬
вого цвета (сали¬
цин)
Салицин: зона
красновато-фио¬
летового цвета
Зона краснова¬
то-фиолетового
цвета (салицин)
Хлорогеновая кис¬
лота: зона корич¬
невого цвета
Раствор
сравнения
Испытуемый
раствор (а)
Испытуемый
раствор (Ь)
1234
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побеги диаметром более 10 мм — не
более 3 %; куски коры толщиной более 2 мм — не бо¬
лее 2 %. Органические примеси: не более 1 %. Мине¬
ральные примеси: не более 0,1 %.
® Потеря в массе при высушивании (2.2.32).
Не более 11,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
@> Жидкостная хроматография (2.2.29).
Испытуемый раствор. К 1,000 г измельченного
сырья (355) (2.9.12) прибавляют 40 мл метанола Р и
40,0 мл раствора 4,2 г/л натрия гидроксида Р и на¬
гревают в водяной бане при температуре около 60 °С
с обратным холодильником при частом встряхивании
в течение около 1 ч. После охлаждения прибавля¬
ют 4,0 мл раствора 103,0 г/л кислоты хлористово¬
дородной Р. Суспензию фильтруют в мерную колбу
вместимостью 100 мл, промывают и доводят до объ¬
ема 100,0 мл смесью равных объемов метанола Р и
воды Р. Фильтруют через мембранный фильтр с раз¬
мером пор 0,45 мкм.
Раствор сравнения. 5,0 мг пицеина Р раство¬
ряют в 25,0 мл смеси вода Р — метанол Р (20:80,
об/об) (раствор А). 15,0 мг ФСО салицина растворяют
в 25 мл смеси вода Р — метанол Р (20:80, об/об),
прибавляют 5,0 мл раствора А и доводят водой Р до
объема 50,0 мл.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
3 мкм;
- подвижная фаза:
- подвижная фаза А: тетрагидрофуран Р —
0,5 % (об/об) раствор кислоты фосфорной Р
(1,8:98,2, об/об);
- подвижная фаза В: тетрагидрофуран Р\
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—15
100
0
15—17
о
0
1
со
о
0 — 10
17—23
90
10
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 270 нм;
- объем вводимой пробы: 10 мкл.
Время удерживания: салицин — около 6,4 мин;
пицеин — около 7,7 мин.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,5 между пиками сали¬
цина и пицеина.
Содержание производных салицина в пересчете
на салицин в процентах рассчитывают по формуле:
51гт2 Р 2
32т,
где:
51 — площадь пика салицина на хроматограмме
испытуемого раствора;
52 — площадь пика салицина на хроматограмме
раствора сравнения;
Л71 — масса навески испытуемого сырья, г;
т2 — масса навески ФСО салицина, г;
Р — процентное содержание салицина в ФСО
салицина.
07/2016:1522
<§> ИМБИРЯ КОРНЕВИЩА
Ипд1Ьепз гЫгота
ОШ С ЕЙ
ОПРЕДЕЛЕНИЕ
Высушенное цельное или резаное корневище
Дпд1Ьег оЯ1ста1е Козсое с пробкой, удаленной полно¬
стью или только с широких плоских поверхностей.
Содержание: не менее 15 мл/кг эфирного масла
(в пересчете на безводное сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеют характерный ароматный запах, пряный и
жгучий вкус.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корневище сжа¬
то с боков, имеет короткие, уплощенные, обратнояй¬
цевидные, искривленные разветвления на верхней
стороне, на верхушке каждого из них иногда имеется
глубокий рубец; цельные корневища длиной около
(5—10) см, шириной (1,5—3) см или 4 см и толщиной
(1—1,5) см, иногда разделенные продольно. На очи¬
щенном корневище со светло-коричневой наружной
поверхностью наблюдается продольная бороздча-
тость и редкие свободные волокна; внешняя поверх¬
ность неочищенного корневища имеет цвет от блед¬
но- до темно-коричневого и более или менее покрыта
пробкой, которая имеет заметные узкие продольные и
поперечные ребра; пробка легко счищается с боковых
поверхностей, но сохраняется между ответвлениями.
Излом — короткий и крахмалистый с выступающими
волокнами. На гладком поперечном срезе наблюдает¬
ся узкая кора, отделенная эндодермой от значительно
более широкой стелы; в ней наблюдаются многочис¬
ленные разбросанные сосудисто-волокнистые пучки
и многочисленные разбросанные масляные клетки
с желтым содержимым. Кроме того, неочищенное
корневище имеет наружный слой темно-коричневой
пробки.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет бледно-желтый или
коричневатый. Просматривают под микроскопом с
использованием раствора хлоралгидрата Р. В по¬
рошке обнаруживаются следующие диагностические
признаки (рисунок 1522.-1): группы больших тонко¬
стенных разделенных перегородками волокон с од¬
ной часто пильчатой стенкой [С, О, 6]; фрагменты
[К], содержащие сетчатые сосуды [Ка], часто вместе
с тонкостенными клетками, содержащими коричне¬
вый пигмент [КЬ], и амилопластами паренхимы [Кс];
многочисленные изолированные, достаточно круп¬
ные, сетчатые сосуды [Н, Ц; многочисленная тонко¬
стенная основная паренхима У, М], некоторые клетки
которой содержат коричневую смолу [За]] фрагменты
коричневой пробки обычно видны на поверхности [Р],
но иногда на поперечном срезе [Е]. При просматри¬
вании измельченного сырья с использованием 50 %
Исландского мха слоевища
1235
(об/об) раствора глицерина Р видны: многочисленные
простые, уплощенные, продолговатые или овальные
или нерегулярные зерна крахмала, длиной до около
50 мкм и шириной 25 мкм, с небольшим ядром в виде
точки, расположенным в более узком конце; иногда
зерна имеют слабую, поперечную исчерченность и
могут быть несвязанными [А], объединенными в агло¬
мераты [В] или включенными в клетки паренхимы [Кс].
Рисунок 1522.-1. Микроскопические признаки
корневищ имбиря
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (710) (2.9.12) прибавляют 5 мл метанола Р,
встряхивают в течение 15 мин и фильтруют.
Раствор сравнения. Юмкл цитраля Р и 10 мг
резорцина Р растворяют в 10 мл метанола Р. Раст¬
вор готовят непосредственно перед использованием.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: гексан Р—эфир Р (40:60, об/об).
Наносимый объем пробы: 20 мкл, в виде полос.
Фронт подвижной фазы: в ненасыщенной хро¬
матографической камере не менее 15 см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
10 г/л ванилина Р в кислоте серной Р, нагревают при
температуре от 100 °С до 105 °С в течение 10 мин.
Просматривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения в нижней половине обнаруживается интенсив¬
ная зона красного цвета (резорцин) и в верхней по¬
ловине — две зоны фиолетового цвета (цитраль); на
хроматограмме испытуемого раствора ниже зоны,
соответствующей резорцину на хроматограмме раст¬
вора сравнения, обнаруживаются две интенсивные
зоны фиолетового цвета (гингеролы), в средней части
между зонами, соответствующими резорцину и цитра-
лю на хроматограмме раствора сравнения, обнаружи¬
ваются две другие менее интенсивные зоны фиоле¬
тового цвета (шагаолы); могут обнаруживаться другие
зоны.
ИСПЫТАНИЯ
Вода (2.2.13). Не более 100 мл/кг. Определение
проводят методом отгонки из 20,0 г измельченного сы¬
рья (710) (2.9.12).
Общая зола (2.4.16). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 20,0 г испы¬
туемого сырья, грубо измельченного непосредственно
перед испытанием, помещают в круглодонную колбу
вместимостью 1000 мл, прибавляют 10 капель вазе¬
линового масла Р или другого антивспенивателя и
500 мл воды Р в качестве жидкости для перегонки. В
градуированную трубку помещают 0,5 мл ксилола Р.
Перегонку проводят со скоростью (2—3) мл/мин в те¬
чение 4 ч.
07/2016:1439
(й) ИСЛАНДСКОГО МХА
СЛОЕВИЩА
ЫсЬеп 1з1апсИсиз
/СЕ/-ДЛЮ ЛЮ55
ОПРЕДЕЛЕНИЕ
Цельные или резаные высушенные слоевища
СеХгапа 1з1апсНса (1_.) АсНапиз з.1.
ПОДЛИНННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Слоевища дли¬
ной до 15 см нерегулярно-дихотомическое, состоит
из гладких желобовидных или почти плоских жест¬
ких ломких лент шириной (0,3—1,5) см и толщиной
около 0,5 мм, иногда зубчатые с реснитчатым краем
(пикнидиями). Верхняя поверхность зеленоватого или
зеленовато-коричневого цвета, нижняя поверхность
серовато-белого или светло-коричневого цвета с бе¬
ловатыми вдавленными пятнами (так называемыми
дыхательными полостями). Очень редко на верхуш¬
ках концевых лопастей встречаются коричневые дис¬
кообразные апотеции.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет серовато-коричневый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки: много¬
численные фрагменты псевдопаренхимы, состоящей
из толстостенных гифов с узкими просветами корко¬
вого слоя и гифов с широкими просветами прилега¬
ющего слоя, состоящего из свободно переплетенных
гифов, в котором в сердцевинной зоне располагаются
зеленоватые или коричневатые клетки водорослей
диаметром до 15 мкм; редкие краевые фрагмены
слоевища с трубкообразными или цилиндрически¬
ми спермогониями шириной до 160 мкм и длиной до
400 мкм.
C. К 1,0 г измельченного сырья (355) (2.9.12) при¬
бавляют 10 мл воды Р и кипятят в течение (2—3) мин.
После охлаждения полученного серовато-коричнево¬
го раствора образуется гель, приобретающий синее
окрашивание при прибавлении раствора йода Р.
й. Тонкослойная хроматография (2.2.27).
1236
Гэсударственная фармакопея Республики Беларусь
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (355) (2.9.12) прибавляют 5 мл ацетона Р и на¬
гревают с обратным холодильником в водяной бане в
течение (2—3) мин. Охлаждают и фильтруют.
Раствор сравнения. 5 мг анетола Р и 5 мг ко¬
фейной кислоты Р растворяют в 2 мл ацетона Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: ацетон Р — метанол Р — кис¬
лота муравьиная безводная Р — толуол Р (5:5:10:80,
об/об/об/об).
Наносимый объем пробы: 20 мкл [или 4 мкл] ис¬
пытуемого раствора и 10 мкл [или 2 мкл] раствора
сравнения в виде полос.
Фронт подвижной фазы: не менее 10 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин. Просма¬
тривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые зоны.
Верх хроматографической пластинки
Анетол: зона от синего
до синевато-фиолетово¬
го цвета
Зона серовато-синего цвета
2 слабых зоны серовато-сине¬
го цвета
Слабая зона от серовато-ко¬
ричневого до серого цвета
Кофейная кислота: зона
серовато-синего цвета
Зона серовато-фиолетового
цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Сумма допусти¬
мых примесей: не более 5 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 3,0 %.
Коэффициент набухания (2.8.4). Не менее 4,5.
Используют измельченное сырье (355) (2.9.12).
07/2016:1297
КАЛЕНДУЛЫ ЦВЕТКИ
(НОГОТКОВ ЦВЕТКИ)
Са1епс1и1ае Поз
САТЕЫОШ-А Р/.ОУУЕЯ
ОПРЕДЕЛЕНИЕ
Собранные в начале распускания трубчатых
цветков и высушенные цветочные корзинки растения
Са1епс1и1а оШапаНз I.
Содержание:
- не менее 0,4 % суммы флавоноидов в пересче¬
те на гиперозид (С21Н20О12; М.м. 464,4) (в пересчете на
сухое сырье);
- или не менее 0,6 % суммы флавоноидов в пе¬
ресчете на рутин (С27Н30О16; М.м. 611) (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
частично осыпавшиеся корзинки диаметром до 5 см,
без цветоносов или с остатками цветоносов длиной
не более 3 см. Обвертка серо-зеленая, одно- двух¬
рядная; листочки ее линейные, заостренные, густоо-
пушенные. Цветоложе слегка выпуклое, голое. Крае¬
вые цветки язычковые, длиной (15—28) мм, шириной
(3—5) мм с изогнутой короткой опушенной трубкой,
трехзубчатым отгибом, вдвое превышающим обверт¬
ку, и 4—5 жилками. Цветки расположены в 2—3 ряда
у немахровых и в 10—15 рядов у махровых форм. Пе¬
стик с изогнутой нижней одногнездной завязью, тон¬
ким столбиком и двухлопастным рыльцем. Средин¬
ные цветки трубчатые с пятизубчатым венчиком. Цвет
краевых цветков красновато-оранжевый, оранжевый,
ярко- или бледно-желтый; срединных — оранжевый,
желтовато-коричневый или желтый.
B. Микроскопия (2.8.23). При просматривании
язычковых цветков с поверхности видны удлиненные
клетки эпидермиса с оранжевыми округлыми хромато-
пластами; на зубчиках эпидермис с сосочками, иногда
с устьицами; трубка венчика густо опушена простыми
и железистыми одно-, двухрядными волосками; завязь
также опушена: с выпуклой стороны железистыми, по
краям вогнутой стороны — простыми двухрядными во¬
лосками. Головка железистых волосков состоит из 2,4
или 8 клеток. Эпидермис трубчатых цветков такой же,
как у язычковых, но у зубчиков он с более вытянутыми
сосочками; нижняя часть трубки венчика и завязь гу¬
сто опушены одно-, двухрядными железистыми, реже
двухрядными простыми волосками. Складчатость
кутикулы, обычно маскируемая хромопластами, про¬
сматривается только на отдельных участках. Пыльца
округлая, шиповатая. Эпидермис листочков обвертки
по краю представлен удлиненными клетками с прямы¬
ми стенками, в средней части — извилистыми стен¬
ками и устьицами; листочки обвертки густо опушены:
по краю —* длинными одно- двухрядными простыми,
двухрядными железистыми и ветвистыми волосками;
в средней части — только железистыми клетками.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (500) (2.9.12) прибавляют 10 мл метанола Р и на¬
гревают на водяной бане с обратным холодильником
в течение 10 мин. Охлаждают и фильтруют.
Раствор сравнения. 1,0 мг кислоты кофей¬
ной Р, 1,0 мг кислоты хлорогеновой Р и 2,5 мг рути¬
на Р растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безвод¬
ная Р— вода Р — этилацетат Р (10:10:80, об/об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Календулы цветки (ноготков цветки)
1237
Проявление: горячую пластинку опрыскивают
раствором 10 г/л дифенилборной кислоты аминоэ¬
тилового эфира Р в метаноле Р и затем раствором
50 г/л макрогола 400 Р в метаноле Р. Пластинку вы¬
сушивают на воздухе в течение 30 мин и просматрива¬
ют в ультрафиолетовом свете при длине волны 365 нм.
Результаты: на хроматограмме раствора срав¬
нения обнаруживаются: в нижней части флуоресци¬
рующая зона желтовато-коричневого цвета (рутин),
в средней части светлая флуоресцирующая зона го¬
лубоватого цвета (хлорогеновая кислота) и в верхней
части светлая флуоресцирующая зона голубоватого
цвета (кофейная кислота). На хроматограмме испыту¬
емого раствора обнаруживаются: флуоресцирующая
зона желтовато-коричневого цвета, соответствующая
рутину на хроматограмме раствора сравнения, ниже
ее и непосредственно над ней флуоресцирующие зоны
желтовато-зеленого цвета; светлая флуоресцирующая
зона голубоватого цвета, соответствующая хлорогено-
вой кислоте на хроматограмме раствора сравнения,
и флуоресцирующая зона желтовато-зеленого цвета
над ней; светлая флуоресцирующая зона голубоватого
цвета немного ниже зоны, соответствующей кофейной
кислоте на хроматограмме раствора сравнения. На
хроматограмме испытуемого раствора могут обнару¬
живаться и другие флуоресцирующие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: остатки цветоносов, в том числе от¬
деленные от корзинок при анализе, — не более 6 %;
корзинки с полностью осыпавшимися язычковыми и
трубчатыми цветками (цветоложе с обвертками) — не
более 20 %; побуревшие корзинки — не более 3 %;
другие части растения (кусочки стеблей и листьев) —
не более 3 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 11,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
■® Определение содержания суммы флаво-
ноидов в пересчете на гиперозид.
Исходный раствор. 0,800 г измельченного сы¬
рья (500) (2.9.12) помещают в круглодонную колбу
вместимостью 100 мл, прибавляют 1 мл раствора
5 г/л гексаметилентетрамина Р, 20 мл ацетона Р и
7 мл кислоты хлористоводородной Р1. Кипятят с об¬
ратным холодильником в течение 30 мин, после чего
извлечение процеживают через ватный тампон в кол¬
бу. Ватный тампон помещают в круглодонную колбу с
остатком сырья и дважды кипятят с обратным холо¬
дильником в течение 10 мин, каждый раз прибавляя
20 мл ацетона Р в качестве экстрагента. Охлаждают
и процеживают через ватный тампон. Объединенные
ацетоновые извлечения фильтруют через бумажный
фильтр в мерную колбу вместимостью 100 мл, опола¬
скивают круглодонную колбу ацетоном Р и фильтру¬
ют через тот же фильтр, после чего фильтр промыва¬
ют ацетоном Р. Объем раствора доводят до 100,0 мл
ацетоном Р. 20,0 мл полученного раствора помеща¬
ют в делительную воронку, прибавляют 20 мл воды Р
и экстрагируют этилацетатом Р порцией 15 мл, за¬
тем еще трижды порциями по 10 мл. Этилацетатные
извлечения объединяют, помещают в делительную
воронку и дважды промывают водой Р порциями по
50 мл. Органический слой фильтруют через бумаж¬
ный фильтр с 10 г натрия сульфата безводного Р в
мерную колбу, вместимостью 50 мл и доводят этила¬
цетатом Р до объема 50,0 мл.
Испытуемый раствор. К 10,0 мл исходного раст¬
вора прибавляют 1 мл реактива алюминия хлорида Р
и доводят 5 % (об/об) раствором кислоты уксусной
ледяной Р в метаноле Р до объема 25,0 мл.
Компенсационный раствор. 10,0 мл исходного
раствора доводят 5 % (об/об) раствором кислоты ук¬
сусной ледяной Р в метаноле Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 425 нм.
Содержание флавоноидов в пересчете на гипе¬
розид в процентах рассчитывают по формуле:
А- 625
т-500 ’
где:
500 — удельный показатель поглощения гиперо-
зида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Определение содержания суммы флавонои¬
дов в пересчете на рутин.
Исходный раствор. 1,000 г измельченного сы¬
рья (710) (2.9.12) помещают в колбу вместимостью
150 мл, прибавляют 30 мл спирта (50 %, об/об) Р и
нагревают с обратным холодильником в водяной бане
в течение 30 мин, периодически встряхивая для смы¬
вания частиц сырья со стенок. Горячее извлечение
процеживают через ватный тампон в мерную колбу
вместимостью 100 мл так, чтобы частицы сырья не
попадали на фильтр. Ватный тампон помещают в
колбу с остатком сырья и прибавляют 30 мл спирта
(50 %, об/об) Р. Экстракцию повторяют еще дважды в
описанных выше условиях, процеживая извлечение в
ту же мерную колбу. Охлаждают и доводят спиртом
(50 %, об/об) Р до объема 100,0 мл.
Испытуемый раствор. К 1,0 мл исходного раст¬
вора прибавляют 10 мл спирта (70%, об/об) Р и
2,0 мл раствора 50 г/л алюминия хлорида Р в спирте
(70 %, об/об) Р. Через 10 мин прибавляют 2,0 мл 5 %
(об/об) раствора кислоты уксусной Р в спирте (70 %,
об/об) Р и доводят спиртом (70 %, об/об) Р до объ¬
ема 25,0 мл.
Компенсационный раствор. К 1,0 мл исходного
раствора прибавляют 10 мл спирта (70%, об/об) Р
и 2,0 мл 5 % (об/об) раствора кислоты уксусной Р
в спирте (70 %, об/об) Р и доводят спиртом (70 %,
об/об) Р до объема 25,0 мл.
Через 1 ч измеряют оптическую плотность
(2.2.25) испытуемого раствора при 408 нм.
Содержание суммы флавоноидов в пересчете на
рутин в процентах рассчитывают по формуле:
А-2500
т-220 '
где:
220 — удельный показатель поглощения ком¬
плекса рутина с алюминия хлоридом;
А — оптическая плотность испытуемого раст¬
вора;
/77 — масса навески испытуемого сырья, г.
1238
Государственная фармакопея Республики Беларусь
07/2016:РБ0057
КАЛИНЫ КОРА
УФигт соЛех
VIвиры им в апк*
ОПРЕДЕЛЕНИЕ
Собранная ранней весной кора стволов и ветвей
кустарника или небольшого дерева УФитит ори1из 1_.
Содержание: не менее 4,0 % дубильных веществ
в пересчете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Трубчатые, же¬
лобоватые или плоские куски коры различной длины,
толщиной около 2 мм. Наружная поверхность коры
морщинистая, коричневато-серая или зеленовато-се¬
рая с мелкими чечевичками. Внутренняя поверхность
гладкая, светло- или коричневато-желтая с мелкими
красноватыми пятнышками и полосками. Излом коры
мелкозернистый.
B. Микроскопия (2.8.23). На поперечном срезе
виден коричневый многорядный пробковый слой. На
границе первичной и вторичной коры одиночно или
небольшими группами (по 2—4) расположены лубя¬
ные волокна. Стенки лубяных волокон толстые, сло¬
истые, неодревесневшие, пронизаны тончайшими
порами. Во вторичной коре видны одно-, двухрядные
сердцевинные лучи и крупные, одревесневшие каме¬
нистые клетки желтого цвета с сильно утолщенными,
слоистыми стенками, пронизанными многочисленны¬
ми порами. Каменистые клетки расположены неболь¬
шими (по 2—6) тангентально-вытянутыми группами,
реже одиночно. В паренхиме коры, особенно первич¬
ной, видны многочисленные крупные и мелкие друзы
оксалата кальция.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (710) (2.9.12) прибавляют 10 мл 96 % спирта Р и
выдерживают в течение 20 мин при комнатной тем¬
пературе. Полученное извлечение фильтруют через
бумажный фильтр и упаривают в вакууме до объема
(1—1,5) мл.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: хлороформ Р — метанол Р
(90:10, об/об).
Наносимый объем пробы: 100 мкл в виде полосы.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором,
полученным при растворении 1,0 г диметиламино-
бензальдегида Р в смеси из 5 мл кислоты хлористо¬
водородной Р и 95 мл 96 % спирта Р, и выдерживают
при температуре 110 °С в течение (5—8) мин.
Результаты: на хроматограмме обнаруживают¬
ся 5—9 зон сине-зеленого цвета (иридоиды) и 2—3
зоны розовато-красного цвета (катехины).
Р. Внутреннюю поверхность коры смачивают
раствором железа (III) аммония сульфата Р5. Появ¬
ляется черно-зеленое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: куски коры, потемневшие с внутренней
поверхности, — не более 5 %; куски коры с остатками
древесины и веточек — не более 2 %. Органические
примеси: не более 1,5%. Минеральные примеси: не
более 0,5 %.
Вещества, извлекаемые 50 % (об/об) спиртом.
Не менее 18,0 % (в сухом сырье).
1.000 г измельченного сырья (1000) (2.9.12) по¬
мещают в коническую колбу вместимостью 250 мл,
прибавляют 50,0 мл спирта (50 %, об/об) Р, закры¬
вают колбу пробкой, взвешивают с точностью 0,01 г
и выдерживают в течение 1 ч. Затем колбу присо¬
единяют к обратному холодильнику и нагревают,
поддерживая слабое кипение, в течение 2 ч. После
охлаждения колбу с содержимым вновь закрыва¬
ют той же пробкой, взвешивают и потерю в массе
восполняют спиртом (50 %, об/об) Р. Содержимое
колбы тщательно взбалтывают и фильтруют через
сухой бумажный фильтр. 25,0 мл полученного филь¬
трата переносят в предварительно высушенную при
температуре от 100 °С до 105 °С и взвешенную вы¬
парительную чашку, выпаривают на водяной бане
досуха и сушат при температуре от 100 °С до 105 °С
до постоянной массы.
Проводят два параллельных испытания. За окон¬
чательный ответ принимают среднее арифметиче¬
ское двух параллельных испытаний.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2.000 г измельченного сырья (2000) (2.9.12) по¬
мещают в коническую колбу со шлифом вместимо¬
стью 500 мл, прибавляют 250 мл кипящей воды Р
и кипятят с обратным холодильником в течение
30 мин при периодическом перемешивании. Затем
охлаждают до комнатной температуры, процежива¬
ют и доводят водой Р до объема 250,0 мл. 25,0 мл
полученного извлечения помещают в коническую
колбу вместимостью 750 мл, прибавляют 500 мл
воды Р, 25,0 мл раствора индигосульфокислоты
(0,25 мг индигокармина Р растворяют в 6,5 мл кис¬
лоты серной Р, прибавляют 6,5 мл кислоты сер¬
ной Р, доводят водой Р до объема 250 мл и филь¬
труют). Титруют при постоянном перемешивании
0,02 М раствором калия перманганата до желтого
окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 г дубильных веществ в пересчете
на танин.
07/2016:РБ0008
КАШТАНА КОНСКОГО СЕМЕНА
АезсиН Ырросаз1ап1 зетеп
(Н1рросаз1ат зетеп)
НОКЗЕ-СНЕЗТЫиТ*
ОПРЕДЕЛЕНИЕ
Собранные и высушенные зрелые семена расте¬
ния Аезси1из Ырросаз1апит 1_.
Содержание: не менее 3,0 % суммы тритерпе-
новых сапонинов в пересчете на эсцин (С^Н^О^;
М.м. 1101) (в пересчете на сухое сырье).
Каштана конского семена
1239
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Семена не¬
правильной шаровидной формы диаметром от 2 см
до 4 см, слегка сплюснутые, бугристые, покрыты
гладкой, блестящей, жесткой, темно-коричневой
кожурой с большим серым пятном при основании.
B. Микроскопия (2.8.23). На поперечном сре¬
зе видно, что верхний эпидермис состоит из пали¬
садных клеток, а основная часть семенной кожу¬
ры представлена паренхимной тканью из клеток с
утолщенными стенками, пронзенными поровыми
канальцами. Наружные ряды из плотно сомкнутых
клеток, более глубокие — сложены рыхло с круп¬
ными межклетниками различной формы. По на¬
правлению к зародышу клетки паренхимы мельча¬
ют и спадаются, образуя слой сдавленных клеток,
в котором встречаются проводящие пучки. Глубже
располагается слой из 4—5 рядов крупных про¬
долговатых клеток с тонкими стенками. Эпидермис
семядолей состоит из мелких клеток, ткань семя¬
долей — из многоугольных плотно сомкнутых па¬
ренхимных клеток, содержащих капельки жирного
масла и крахмальные зерна неправильно груше¬
видной формы разного размера, простые и двух- и
трехсложные.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (500) (2.9.12) прибавляют 10 мл спирта (70 %,
об/об) Р, кипятят с обратным холодильником в тече¬
ние 10 мин, охлаждают и фильтруют.
Раствор сравнения. 10 мг эсцина Р растворяют
в 10 мл спирта (70 %, об/об) Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля вР254 Р.
Подвижная фаза: верхний слой смеси кислота
уксусная ледяная Р — вода Р — бутанол Р (10:40:50,
об/об/об).
Наносимый объем пробы: 20 мкп испытуемого
раствора и 10 мкл раствора сравнения в виде полос
длиной 20 мм.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуе¬
мого раствора в нижней половине обнаруживается
зона поглощения, соответствующая по расположе¬
нию зоне эсцина на хроматограмме раствора срав¬
нения.
Проявление В: пластинку опрыскивают рас¬
твором анисового альдегида Р, используя 10 мл
на квадратную пластинку со стороной 200 мм, и
нагревают в течение (5—10) мин при температуре
от 100 °С до 105 °С. Просматривают при дневном
свете.
Результаты В: на хроматограмме испытуе¬
мого раствора обнаруживается сине-фиолетовая
зона, соответствующая по цвету и расположению
зоне эсцина на хроматограмме раствора сравне¬
ния. Кроме того, на хроматограмме испытуемого
раствора обнаруживаются несколько более узких и
слабых полос от коричневого до коричневато-крас¬
ного цвета; в нижней части обнаруживается отчет¬
ливая зона коричнево-серого цвета; немного ниже
этой зоны обнаруживается зона коричневого цвета.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Не более 2 %.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 4,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (500) (2.9.12) по¬
мещают в колбу вместимостью 250 мл со шлифом,
прибавляют 100,0 мл 65% (об/об) раствора мета¬
нола Р. Колбу с содержимым взвешивают с точно¬
стью до 0,1 г и кипятят с обратным холодильником
в водяной бане в течение 30 мин. Охлаждают, при
необходимости доводят до первоначальной массы с
помощью 65 % (об/об) раствора метанола Р и филь¬
труют. 30,0 мл фильтрата помещают в круглодонную
колбу вместимостью 100 мл и выпаривают в вакууме
досуха. Сухой остаток растворяют в 20,0 мл 0,1 М
раствора кислоты хлористоводородной, перено¬
сят в делительную воронку вместимостью 250 мл,
колбу для выпаривания дважды промывают 0,1 М
раствором кислоты хлористоводородной порция¬
ми по 5,0 мл и промывные воды прибавляют в ту же
делительную воронку. В делительную воронку при¬
бавляют 20 мл пропанола Р и 50 мл хлороформа Р
и интенсивно встряхивают в течение 2 мин. После
расслоения нижний слой отбрасывают, верхний слой
повторно встряхивают с нижним слоем, полученным
в результате интенсивного встряхивания в течение
2 мин смеси из 30,0 мп 0,1 М раствора кислоты
хлористоводородной, 20 мл пропанола Р и 50 мл
хлороформа Р. Объединенные хлороформные из¬
влечения (нижние слои) помещают в круглодонную
колбу и выпаривают в вакууме досуха. Полученный
сухой остаток дважды промывают эфиром, свобод¬
ным от пероксидов, Р порциями по 10 мл. Эфирные
извлечения объединяют, фильтруют и фильтр про¬
мывают 10 мл эфира, свободного от пероксидов, Р.
Фильтрат отбрасывают. Фильтр и круглодонную кол¬
бу сушат до исчезновения запаха эфира. Остаток в
колбе трижды промывают кислотой уксусной безво¬
дной Р порциями по 10 мл и фильтруют полученные
растворы через ранее использованный и высушен¬
ный фильтр в мерную колбу вместимостью 50 мл. За¬
тем круглодонную колбу промывают еще небольшим
количеством кислоты уксусной безводной Р. Про¬
мывную жидкость фильтруют в ту же мерную колбу
и промывают фильтр. Объединенные фильтраты
доводят кислотой уксусной безводной Р до объема
50,0 мл (раствор А).
Испытуемый раствор. 5,0 мл раствора А дово¬
дят реактивом железа (III) хлорида и кислоты уксус¬
ной Р до объема 25,0 мл. Выдерживают в водяной
бане при температуре 60 °С в течение 25 мин при пе¬
риодическом перемешивании и охлаждают под стру¬
ей холодной воды до комнатной температуры.
Компенсационный раствор. 5,0 мл кислоты ук¬
сусной безводной Р помещают в мерную колбу вме¬
стимостью 25 мл и доводят реактивом железа (III)
хлорида и кислоты уксусной Р до объема 25,0 мл.
Выдерживают в водяной бане при температуре 60 °С
в течение 25 мин при периодическом перемешивании
и охлаждают под струей холодной воды до комнатной
температуры.
1240
Гэсударственная фармакопея Республики Беларусь
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 540 нм.
Содержание тритерпеновых сапонинов в пе¬
ресчете на эсцин в процентах рассчитывают по
формуле:
А • 833,3
т-60 ’
где:
60 — удельный показатель поглощения продукта
реакции эсцина и реактива железа (III) хлорида и кис¬
лоты уксусной Р\
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1304
КОРИАНДРА ПЛОДЫ
Сопапс1п (ги&из
СОШАЫОЕК
ОПРЕДЕЛЕНИЕ
Собранные в фазу полной зрелости и высушен¬
ные плоды (вислоплодники) однолетнего травянисто¬
го растения Сопапдгит затмит 1_.
Содержание: не менее 3 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Плод ко¬
ричневого или светло-коричневого цвета более или
менее сферической формы диаметром (1,5—5) мм
или овальной формы длиной (2—6) мм. Состоит
из нераспадающегося вислоплодника с обычно
прочно соединенными мерикарпиями. Плод глад¬
кий с 10 волнистыми слегка возвышающимися пер¬
вичными ребрами и 8 прямыми более заметными
вторичными ребрами. Мерикарпии вогнуты на вну¬
тренней стороне. Верхушку венчает стилоподий,
также может присутствовать небольшой фрагмент
плодоножки.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) {2.9.12). Цвет коричневый.
Просматривают под микроскопом с использовани¬
ем раствора хлоралгидрата Р. В порошке обна¬
руживаются следующие диагностические признаки
(рисунок 1304.-1): многочисленные капельки мас¬
ла [В]; фрагменты эндосперма [А] с маленькими
толстостенными клетками правильной формы, со¬
держащими микророзетки [Аа] и микрокристаллы
оксалата кальция, а также капельки масла [АЬ];
фрагменты эндокарпия (вид с поверхности [С, 3],
поперечный срез [Н]) с очень узкими клетками, рас¬
положенными паркетообразно [Са, На], и обычно
связанными со слоем тонкостенных [СЬ, НЬ] или
толстостенных ^а] прямоугольных склереид ме-
зокарпия; фрагменты склеренхиматозного слоя
мезокарпия [О] с короткими сильно утолщенными
ямчатыми веретенообразными клетками, распо¬
ложенными приблизительно под прямым углом к
клеткам прилегающих слоев; фрагменты паренхи¬
мы мезокарпия (поперечный срез [Е]) с маленьки¬
ми клетками со слабо утолщенными клетками [Еа],
остатками выделительных каналов [ЕЬ] и склереи-
дами [Ес]; фрагменты эпикарпия (вид с поверхно¬
сти [Р]) с тонкостенными многоугольными клетка¬
ми, некоторые из них содержат маленькие призмы
оксалата кальция [Ра]; редкие фрагменты выдели¬
тельных каналов с коричневыми клетками (вид с
поверхности [й]); редкие фрагменты сосудистых
пучков [К].
Рисунок 1304.-1. Микроскопические признаки
плодов кориандра
(§) С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г свежеизмельчен-
ного сырья (355) (2.9.12) прибавляют 5 мл метано¬
ла Р, обрабатывают ультразвуком в течение 10 мин и
центрифугируют или фильтруют; используют надоса-
дочную жидкость или фильтрат.
Раствор сравнения. 10 мкл линалола Р и 2 мкл
геранилацетата Р растворяют в 1,0 мл толуола Р.
Пластинка: ТСХ пластинка со слоем
силикагеляР254 Р ((5—40) мкм) [или ТСХ пластинка
со слоем силикагеляР254 Р ((2—10) мкм)].
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Нанесение: по 10 мкл [или 2 мкл] в виде полос
длиной 15 мм [или 8 мм].
Фронт подвижной фазы: не менее 10 см [или
6 см] от линии старта.
Высушивание: на воздухе в течение 5 мин.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р, нагревают при температуре
от 100 °С до 105 °С в течение 5 мин и просматривают
при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора могут обнаруживаться и другие слабые
зоны.
Крапивы листья
1241
Верх хроматографической пластинки
Зона синевато-фиолетового
цвета
Геранилацетат: зона
фиолетово-синего цвета
Линалол: интенсивная зона
фиолетового цвета
Зона фиолетового цвета
Зона
фиолетово-синего цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: поврежденные и недоразвитые пло¬
ды — не более 3 %; эфирномасличные примеси (ду¬
шисты^ плоды и семена других видов) — не более
1 %. Органические примеси: не более 1 %. Минераль¬
ные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
@> Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Эфирное масло (2.8.12, метод А). 30,0 г
грубоизмельченного непосредственно перед исполь¬
зованием сырья помещают в круглодонную колбу
вместимостью 500 мл, прибавляют 200 мл воды Р. В
градуированную трубку помещают 0,5 мл ксилола Р.
Перегонку проводят со скоростью (2—3) мл/мин в те¬
чение 2 ч.
07/2016:1897
КРАПИВЫ ЛИСТЬЯ
Ш/сае IЬНит
А1ЕТПЕ 1.ЕАР
ОПРЕДЕЛЕНИЕ
Собранные во время цветения, высушенные
цельные или измельченные листья многолетнего тра¬
вянистого растения Ш/са (У/о/са 1_. или Ш/са игепиз 1_.
или смесь этих видов.
Содержание:
- не менее 0,3 % суммы кофеоилмаловой и хло-
рогеновой кислот в пересчете на хлорогеновую кисло¬
ту (С16Н1809; М.м. 354,3) (в пересчете на сухое сырье);
- или не менее 0,8 % суммы полифенольных
соединений в пересчете на пирогаллол (С6Н603;
М.м. 126,1) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, С.
Вторая идентификация: А, В, О.
® А. Внешние признаки (2.8.23). Листья темно¬
зеленого, темно-серовато-зеленого или коричневато¬
зеленого цвета на верхней поверхности и более блед¬
ные на нижней поверхности; рассеянные жгучие во¬
лоски встречаются на обеих поверхностях, так же как
мелкие покровные волоски, которые более многочис¬
ленны по краям и вдоль жилок на нижней стороне ли¬
ста. Листовая пластинка сильно сморщенная, оваль¬
ная или продолговатая, длиной до 100 мм и шириной
до 50 мм с крупнопильчатым краем и сердцевидная
в основании. Жилкование сетчатое и отчетливо за¬
метное на нижней стороне листа. Черешок зеленый
или коричневато-зеленый, округлый или уплощенный,
шириной около 1 мм, продольно бороздчатый и скру¬
ченный; имеет жгучие и покровные волоски.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет зеленый или серо¬
вато-зеленый. Просматривают под микроскопом с ис¬
пользованием раствора хлоралгидрата Р. В порош¬
ке обнаруживаются следующие диагностические при¬
знаки (рисунок 1897.-1): фрагменты одноклеточных
жгучих волосков [А, В, С] длиной до 2 мм, выходящих
из приподнятых многоклеточных оснований [Са] и со¬
стоящих из вытянутых конических клеток с легко об¬
ламывающейся жгучей головкой; мелкие железистые
трихомы [Р] ((35—65) мкм) с одно- либо двухклеточ¬
ной ножкой и двух- или четырехклеточной головкой,
отдельные [Ра] или на фрагментах эпидермиса [РЬ];
фрагменты верхнего эпидермиса листьев при просма¬
тривании с поверхности [<3] или на поперечном срезе
[О] со слегка извилистыми клетками [Оа, 6с], одно¬
клеточными прямыми или слегка изогнутыми покров¬
ными волосками, вытянутыми у основания, длиной до
700 мкм [йс, Оа] и многочисленными крупными цисто-
литами [ОЬ, Еа, <ЗЬ], пустыми или содержащими плот¬
ную зернистую массу карбоната кальция; палисадная
паренхима при просматривании с поверхности [Е] с
округлыми клетками [ЕЬ], окруженными цистолитами
[Еа], или при просматривании на поперечном срезе
[Об]; фрагменты нижнего эпидермиса с извилистыми
или волнистыми клетками [Н], аномоцитными [На] или
анизоцитными [НЬ] устьицами (2.8.3), сопровождае-
Рисунок 1897.-1. Микроскопические признаки
листьев крапивы
156. Зак. 1060.
1242
Государственная фармакопея Республики Беларусь
мыми губчатым мезофиллом при просматривании с
поверхности [Нс] и на поперечном срезе [Ре], содер¬
жащим мелкие друзы оксалата кальция при просма¬
тривании с поверхности [Ид] и на поперечном срезе
[Ш]; редкие мелкие группы сосудов с паренхимой, со¬
держащей друзы оксалата кальция [д].
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл метанола Р, ки¬
пятят с обратным холодильником в течение 15 мин,
охлаждают и фильтруют. Фильтрат выпаривают в ва¬
кууме досуха при температуре не выше 40 °С. Оста¬
ток растворяют в 2 мл метанола Р.
Раствор сравнения. 2 мг хлорогеновой кисло¬
ты Р и 1 мг скополетина Р растворяют в 20 мл ме¬
танола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метанол Р — этилацетат Р
(2,5:4:4:50, об/об/об/об).
Наносимый объем пробы: по 10 мкл [или 4 мкл] в
виде полос длиной 10 мм [или 8 мм].
Фронт подвижной фазы: не менее 8 см [или 6 см]
от линии старта.
Высушивание: на воздухе.
Проявление: пластинку нагревают при 100 °С в
течение 5 мин. Горячую пластинку опрыскивают рас¬
твором 10 г/л дифенилборной кислоты аминоэтило¬
вого эфира Р в метаноле Р. Просматривают в уль¬
трафиолетовом свете при 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора в нижней половине могут обнаруживаться
флуоресцирующие зоны бледно-синего или желтого
цвета.
Верх хроматографической пластинки
Скополетин: флуоресцирую¬
щая зона синего цвета
Две зоны красного цвета
Флуоресцирующая зона си¬
него цвета (скополетин)
Флуоресцирующая зона си¬
него цвета
Хлорогеновая кислота: флу¬
оресцирующая зона голубо¬
го цвета
Флуоресцирующая зона го¬
лубого цвета (хлорогеновая
кислота)
Зона коричневато-желтого
цвета
Раствор сравнения
Испытуемый раствор
О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1,0 г измельченного сы¬
рья (250) (2.9.12) встряхивают с 10 мл гексана Р в те¬
чение 3 ч и фильтруют через бумажный фильтр. Филь¬
трат выпаривают досуха при температуре не выше
45 °С. Остаток растворяют в 1 мл гексана Р.
Раствор сравнения. 10 мг фитоменадиона Р
растворяют в 20 мл гексана Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: бензол Р — петролейный
эфир Р (50:50, об/об).
Наносимый объем пробы: 50 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку выдерживают в ультра¬
фиолетовом свете с длиной волны 365 нм в течение
10 мин. Просматривают в ультрафиолетовом свете
при 365 нм.
Результат: на хроматограмме раствора срав¬
нения в нижней половине проявляется зона желто-
зеленого цвета (фитоменадион). На хроматограмме
испытуемого раствора проявляется желто-зеленая
зона, соответствующая зоне фитоменадиона на хро¬
матограмме испытуемого раствора. На хроматограм¬
ме испытуемого раствора могут обнаруживаться и
другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения — не более 5 %;
почерневшие и побуревшие части растения — не бо¬
лее 5 %. Органические примеси: не более 2 %. Мине¬
ральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 20,0 %.
(§) Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
(^Определение содержания суммы кофео-
ил маловой и хлорогеновой кислот.
Жидкостная хроматография (2.2.29).
Испытуемый раствор. К 0,200 г измельченного
сырья (355) (2.9.12) прибавляют 25,0 мл 40 % (об/об)
раствора метанола Р, выдерживают в ультразвуко¬
вой бане при температуре 40 °С в течение 30 мин и
фильтруют.
Раствор сравнения. 10,0 мг ФСО хлорогеновой
кислоты растворяют в 100,0 мл 40 % (об/об) раст¬
вора метанола Р. 5,0 мл полученного раствора до¬
водят 40 % (об/об) раствором метанола Р до объема
25,0 мл.
Условия хроматографирования:
- предколонка длиной 4 мм и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
-колонка длиной 0,125м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- температура: 25 °С;
- подвижная фаза:
- подвижная фаза А: метанол Р — вода Р, до¬
веденная кислотой фосфорной разведенной Р
до рН 2,0 (15:85, об/об).
- подвижная фаза В: метанол Р.
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—1
100
0
1—25
100 — 85
0 — 15
25—35
85
15
35—36
о
т
ю
со
о
о
7
ю
Кровохлебки корни
1243
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, дли¬
на волны 330 нм;
- объем вводимой пробы: 20 мкл.
Относительные времена удерживания (по от¬
ношению к хлорогеновой кислоте, время удержи¬
вания около 13 мин): кофеоилмаловая кислота-
около 2,2.
Содержание хлорогеновой кислоты или кофе-
оилмаловой кислоты в процентах рассчитывают по
формуле:
51 т2Р
32 •ш1 -20 ’
где:
51 — сумма площадей пиков хлорогеновой
кислоты и кофеоилмаловой кислоты на хромато¬
грамме испытуемого раствора;
52 — площадь пика хлорогеновой кислоты на
хроматограмме раствора сравнения;
/т?1 — масса навески испытуемого сырья, г;
т2 — масса навески ФСО хлорогеновой кис¬
лоты, г;
Р— содержание хлорогеновой кислоты в ФСО
хлорогеновой кислоты, %.
Определение содержания суммы полифеноль-
ных соединений.
Испытуемый раствор. 2,500 г измельченно¬
го сырья (500) {2.9.12) помещают в круглодонную
колбу вместимостью 250 мл, прибавляют 150 мл
кипящей воды Р, нагревают с обратным холодиль¬
ником в водяной бане в течение 15 мин. Затем
колбу охлаждают при комнатной температуре в
течение 45 мин, содержимое количественно пере¬
носят в мерную колбу и доводят водой Р до объ¬
ема 250,0 мл. Выдерживают до оседания частиц
и фильтруют через бумажный фильтр, отбрасы¬
вая первые 50 мл фильтрата. 5,0 мл полученного
фильтрата разводят водой Р до объема 25,0 мл.
К 2,0 мл полученного раствора прибавляют 1,0 мл
реактива фосфорномолибденово-вольфрамово-
го Р, 10,0 мл воды Р и доводят раствором 290 г/л
натрия карбоната Р в воде Р до объема 25,0 мл.
Раствор сравнения. 50,0 мг пирогаллола Р
растворяют в воде Р и доводят до объема 100,0 мл
этим же растворителем. 5,0 мл полученного раст¬
вора разводят водой Р до объема 100,0 мл. К
2,0 мл полученного раствора прибавляют 1,0 мл
реактива фосфорномолибденово-вольфрамово-
гоР, 10,0 мл воды и доводят раствором 290 г/л
натрия карбоната Р в воде Р до объема 25,0 мл.
Компенсационный раствор. Вода Р.
Через 30 мин измеряют оптическую плотность
(2.2.25) растворов при 760 нм.
Содержание суммы полифенольных соедине¬
ний в пересчете на пирогаллол в процентах рас¬
считывают по формуле:
А, • т2 • 62,5
А2 • /т?1
где:
А1 — оптическая плотность испытуемого раст¬
вора;
А2 — оптическая плотность раствора сравнения;
т1 — масса навески испытуемого сырья, г;
т2 — масса навески пирогаллола Р, г.
07/2016:2385
КРОВОХЛЕБКИ КОРНИ
ЗапдшзогЬае гасИх
ЗАМОШЗОВВА КООТ
ОПРЕДЕЛЕНИЕ
Цельные или разрезанные на куски высушенные
одревесневшие корневища и корни многолетнего тра¬
вянистого растения ЗапдшзогЬа оШстаИз 1_.
Содержание:
- не менее 5,0 % дубильных веществ в пересче¬
те на пирогаллол (С6Н603; М.м. 126,1) (в пересчете на
сухое сырье);
- или не менее 14 % дубильных веществ в пере¬
счете на танин (в пересчете на сухое сырье).
ОПИСАНИЕ
® Придаточные корни длиной (5—25) см и диа¬
метром до 2 см.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Цельное сы¬
рье состоит из толстых коротких корневищ, зачастую
разветвленных, веретенообразной или цилиндриче¬
ской формы, а также из придаточных корней с крас¬
новато-коричневой или черновато-коричневой по¬
верхностью, с продольной бороздчатостью, иногда с
поперечными трещинами, с видимыми местами при¬
соединения отпавших корешков.
Также могут обнаруживаться более или менее
цилиндрические фрагменты длиной 2 см или диски
эллиптической или неправильной формы. Излом
светлый и очень волокнистый.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет светло-желтова¬
то-коричневый. Просматривают под микроскопом с
использованием раствора хлоралгидрата Р. В по¬
рошке обнаруживаются следующие диагностические
признаки: многочисленные целые или фрагменти¬
рованные волокна флоэмы, обычно отдельные, уз¬
кие, длиной иногда более 500 мкм, часто с грубыми
стенками; отдельные или находящиеся внутри клеток
паренхимы друзы оксалата кальция; небольшое коли¬
чество сетчатых лигнифицированных сосудов; редкие
фрагменты пробки.
При использовании 50 % (об/об) раствора гли¬
церина Р видны: округлые или яйцевидные крах¬
мальные зерна, отдельные или группами по 2—4;
диаметр отдельных зерен может достигать 30 мкм;
некоторые крахмальные зерна обнаруживаются так¬
же в паренхимных клетках и в клетках сердцевинных
лучей.
®>С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 2,0 г измельченно¬
го сырья (355) (2.9.12) прибавляют 50 мл воды Р,
кипятят с обратным холодильником в течение
30 мин, охлаждают и центрифугируют в течение
10 мин. Надосадочную жидкость встряхивают с
двумя порциями по 15 мл диизопропилового эфи¬
ра Р, насыщенного кислотой хлористоводород¬
ной Р. Объединенные эфирные слои выпаривают
досуха, остаток растворяют в 1,0 мл метанола Р и
фильтруют через полипропиленовый фильтр (раз¬
мер пор 0,45 мкм).
Раствор сравнения. 5 мг галловой кислоты Р и
20 мг резорцина Р растворяют в 20 мл метанола Р.
156*. Зак. 1060.
1244
Государственная фармакопея Республики Беларусь
I
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р254 Р ((2—10) мкм)].
Подвижная фаза: кислота муравьиная безво¬
дная Р — этилацетат Р — толуол Р (10:30:60,
об/об/об).
Наносимый объем пробы, по 10 мкл [или 4 мкл]
в виде полос.
Фронт подвижной фазы: не менее 10 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые зоны
поглощения.
Верх хроматографической пластинки
Зона поглощения
Резорцин: зона поглощения
Зона поглощения
Галловая кислота: зона
поглощения
Зона поглощения (галловая
кислота)
Зона поглощения
Зона поглощения
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку опрыскивают раство¬
ром 10 г/л железа (III) хлорида Р в этаноле Р и нагре¬
вают при температуре от 100 °С до 105 °С в течение
15 мин. Просматривают при дневном свете.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора могут обнаруживаться и другие слабые
зоны.
Верх хроматографической пластинки
Резорцин: зона коричневого
цвета
Зона черновато-синего
цвета
Галловая кислота: зона
черновато-синего цвета
Зона черновато-синего
цвета (галловая кислота)
Зона черновато-синего
цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корневища и корни, почерневшие и по¬
буревшие в изломе, — не более 10 %; частицы, про¬
ходящие сквозь сито (2000) (2.9.12), — не более 5 %.
Органические примеси: не более 1 %. Минеральные
примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
(§> Определение содержания дубильных ве¬
ществ в пересчете на пирогаллол (2.8.14). Исполь¬
зуют 0,500 г измельченного сырья (180) (2.9.12).
Определение содержания дубильных веществ
в пересчете на танин. 1,500 г измельченного сырья
(355) (2.9.12) помещают в коническую колбу со шли¬
фом вместимостью 500 мл, прибавляют 250 мл кипя¬
щей воды Р и кипятят с обратным холодильником в
течение 30 мин при периодическом перемешивании.
Затем охлаждают до комнатной температуры, проце¬
живают и доводят водой Р до объема 250,0 мл.
25,0 мл полученного извлечения помещают в ко¬
ническую колбу вместимостью 750 мл, прибавляют
500 мл воды Р, 25,0 мл раствора индигосульфокис-
лоты (0,25 мг индигокармина Р растворяют в 6,5 мл
кислоты серной Р, прибавляют 6,5 мл кислоты сер¬
ной Р, доводят водой Р до объема 250 мл и фильтру¬
ют). Титруют при постоянном перемешивании 0,02 М
раствором калия перманганата до желтого окраши¬
вания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07:2016:0025
КРУШИНЫ КОРА
Ргапди1ае сог1ех
РКАЫВ1Л.А В АПК
ОПРЕДЕЛЕНИЕ
Сухая цельная или разрезанная на куски кора
стволов и ветвей РЬатпиз ?гапди1а 1_. (Ргапди1а а1пи$
МШег).
Содержание: не менее 7,0 % глюкофрангули-
нов, в пересчете на глюкофрангулин А (С27Н30014; М.м.
578,5) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Кора пред¬
ставляет собой искривленные, почти плоские или
перекрученные, трубчатые или желобоватые куски
коры, толщиной обычно от 0,5 мм до 2 мм, различной
длины и ширины. Внешняя поверхность коры серо¬
вато-коричневая или темно-коричневая, продольно¬
морщинистая, часто с многочисленными сероваты¬
ми, поперечно-вытянутыми чечевичками или серыми
пятнами; при легком соскабливании наружной части
пробки обнаруживается темно-красный слой. Вну¬
тренняя поверхность коры оранжево-коричневая или
красновато-коричневая, гладкая, с тонкой продольной
бороздчатостью; краснеет при прибавлении щелочей.
Излом хрупкий, волокнистый во внутренней части.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет желтоватый или
красновато-коричневый. Просматривают под микро¬
скопом с использованием раствора хлоралгидра¬
та Р. В порошке обнаруживаются следующие диагно¬
стические признаки (рисунок 0025.-1): многочислен¬
ные волокна флоэмы на поперечном срезе [О] или на
продольном срезе [К], частично лигнифицированные,
в группах [Оа, Ка] с кристаллоносной обкладкой, со¬
держащей призмы оксалата кальция рь, КЬ], иногда с
сердцевинными лучами рс]; красновато-коричневые
фрагменты пробки [Н]; фрагменты флоэмы паренхи-
1
Крушины кора
1245
мы на продольном срезе [<3], содержащие друзы ок¬
салата кальция [А, Е], или на тангенциальном срезе
[С], с сердцевинными лучами [Са] ил клетки, содержа¬
щие друзы оксалата кальция [СЬ]; немногочисленные
фрагменты колленхимы [Р]; отдельные друзы [В] и
призмы у] оксалата кальция.
Рисунок 0025.-1. Микроскопические признаки
коры крушины
® С. Просматривают при дневном свете хрома¬
тограмму, полученную в испытании А «Другие виды
РЬатпиз\ антроны» в ультрафиолетовом свете при
длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора в нижней трети обнаруживаются две зоны
оранжево-коричневого цвета (глюкофрангулины); в
верхней трети — от двух до четырех зон красного цве¬
та (франгулины (не всегда четко разделяются), и над
ними франгулоэмодин).
® О. К 50 мг измельченного сырья (180) (2.9.12)
прибавляют 25 мл кислоты хлористоводородной раз¬
веденной Р и нагревают на водяной бане в течение
15 мин. Охлаждают, встряхивают с 20 мл эфира Р и от¬
брасывают водный слой. Эфирный слой встряхивают
с 10 мл раствора аммиака разведенного Р1. Водный
слой окрашивается в красновато-фиолетовый цвет.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
® Другие виды КНатпиз; антроны. Тонко¬
слойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (180) (2.9.12) прибавляют 5 мл спирта (70 %,
об/об) Р и нагревают до кипения. Охлаждают и цен¬
трифугируют. Раствор немедленно сливают и исполь¬
зуют в течение 30 мин.
Раствор сравнения. 20 мг барбалоина Р раство¬
ряют в спирте (70 %, об/об) Р и доводят до объема
10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р (2 пластинки).
Подвижная фаза: вода Р — метанол Р — этила-
цетат Р (13:17:100, об/об/об).
Испытание А.
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе в течение 5 мин.
Проявление: пластинку опрыскивают раствором
50 г/л калия гидроксида Р в спирте (50 %, об/об) Р
и нагревают при температуре от 100 °С до 105 °С в
течение 15 мин. Просматривают в ультрафиолетовом
свете при длине волны 365 нм.
Результаты: на хроматограмме раствора срав¬
нения в центральной части обнаруживается зона
коричневато-желтого цвета (барбалоин). На хрома¬
тограмме испытуемого раствора не должны обнару¬
живаться ни зоны с интенсивной желтой флуоресцен¬
цией, ни флуоресцирующая зона от оранжевого до
красноватого цвета, соответствующие барбалоину на
хроматограмме раствора сравнения.
Испытание В.
Наносимый объем пробы: 10 мкл испытуемого
раствора в виде полосы.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе в течение не более
5 мин.
Проявление: пластинку немедленно опрыскива¬
ют раствором 5 г/л нитротетразолиевого синего Р в
метаноле Р и немедленно просматривают при днев¬
ном свете.
Результаты: не должны обнаруживаться фио¬
летовые или серовато-синие зоны.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: куски коры, покрытые кустистыми ли¬
шайниками, — не более 1 %; куски коры с остатками
древесины — не более 1 %; куски коры толще 2 мм —
не более 3 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
® Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Испытание проводят с защитой от яркого
света.
0,250 г измельченного сырья (180) (2.9.12) поме¬
щают в круглодонную колбу со шлифом, прибавляют
25.0 мл 70 % (об/об) раствора метанола Р, переме¬
шивают и взвешивают. Нагревают с обратным холо¬
дильником в водяной бане в течение 15 мин. Охлаж¬
дают, взвешивают, доводят до первоначальной массы
70 % (об/об) раствором метанола Р и фильтруют.
5.0 мл фильтрата помещают в делительную воронку,
прибавляют 50 мл воды Р и 0,1 мл кислоты хлори¬
стоводородной Р и встряхивают пять раз с петро-
лейным эфиром Р порциями по 20 мл. Выдерживают
до разделения слоев и сливают нижний водный слой
в мерную колбу вместимостью 100 мл. Органические
слои объединяют и отмывают дважды водой Р пор¬
циями по 15 мл. Эту воду используют для промывки
делительной воронки и прибавляют к водному раство¬
ру в мерной колбе. Прибавляют 5 мл раствора 50 г/л
натрия карбоната Р и доводят водой Р до объема
100.0 мл. Органический слой отбрасывают. 40,0 мл
водного раствора переносят в круглодонную колбу
157. Зак. 1060.
1246
Гэсударственная фармакопея Республики Беларусь
вместимостью 200 мл со шлифом, прибавляют 20 мл
раствора 200 г/л железа (III) хлорида Р и нагревают
с обратным холодильником в водяной бане в тече¬
ние 20 мин. Уровень воды в бане должен быть выше
уровня раствора в колбе. Прибавляют 2 мл кислоты
хлористоводородной Р и продолжают нагревание
еще 20 мин, интенсивно встряхивая, до растворения
осадка. Охлаждают, переносят смесь в делительную
воронку и встряхивают с тремя объемами, каждый
раз по 25 мл, эфира Р, предварительно использован¬
ного для ополаскивания колбы. Объединенные эфир¬
ные экстракты отмывают дважды водой Р порциями
по 15 мл. Эфирный слой переносят в мерную колбу
и доводят эфиром Р до объема 100,0 мл. 20,0 мл по¬
лученного раствора осторожно выпаривают досуха.
Остаток растворяют в 10,0 мл раствора 5 г/л магния
ацетата Р в метаноле Р.
Измеряют оптическую плотность (2.2.25) раст¬
вора при 515 нм, используя метанол Р в качестве
компенсационного раствора.
Содержание глюкофрангулинов в пересчете на
глюкофрангулин А в процентах рассчитывают по фор¬
муле:
А-625
т-204 ’
где:
204 — удельный показатель поглощения глю-
кофрангулина А;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0058
КУКУРУЗЫ СТОЛБИКИ
С РЫЛЬЦАМИ
Теае таусИз з1уН сит зИдтаНз
МАНЕ 5 ГУТЕ*
ОПРЕДЕЛЕНИЕ
Собранные в период созревания початков и вы¬
сушенные столбики с рыльцами однолетнего травяни¬
стого растения 2еа тауз Ь.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Мягкие шелкови¬
стые нити (столбики), собранные пучками или частич¬
но перепутанные, на верхушке которых находятся
двухлопастные рыльца. Столбики несколько искрив¬
ленные, плоские, шириной (0,1—0,15) мм; длиной от
0,5 см до 20 см, рыльца короткие, длиной от 0,4 мм
до 3 мм. Часто встречаются столбики без рылец. Цвет
коричневый, коричнево-красный, светло-желтый.
B. Микроскопия (2.8.23). При просматривании
с поверхности столбиков с рыльцами видны клетки
эпидермиса удлиненной формы с прямыми стенками.
На эпидермисе расположены редкие простые волоски
двух типов: продольноспаянные многоклеточные во¬
лоски длиной (0,2—0,8) мм с заостренной или кони¬
ческой верхушкой, состоящие из 2—3 ярусов клеток в
длину, и многоклеточные тонкостенные, изогнутые. В
паренхиме двух узких сторон столбиков и рылец про¬
ходят два параллельных проводящих пучка с хорошо
заметными спиральными сосудами. На рыльце замет¬
ны многоклеточные ворсинки.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие столбики с рыльцами —
не более 3 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 7,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,5 %.
Вещества, извлекаемые 70 % (об/об) спиртом.
Не менее 15,0 % (в сухом сырье).
1,000 г измельченного сырья (1400) (2.9.12) поме¬
щают в коническую колбу вместимостью 250 мл, при¬
бавляют 50,0 мл спирта (70 %, об/об) Р, закрывают
колбу пробкой, взвешивают с точностью 0,01 г и вы¬
держивают в течение 1 ч. Затем колбу присоединяют
к обратному холодильнику и нагревают, поддерживая
слабое кипение, в течение 2 ч. После охлаждения кол¬
бу с содержимым вновь закрывают той же пробкой,
взвешивают и потерю в массе восполняют спиртом
(70 %, об/об) Р. Содержимое колбы тщательно взбал¬
тывают и фильтруют через сухой бумажный фильтр.
25,0 мл полученного фильтрата переносят в предва¬
рительно высушенную при температуре от 105 °С и
взвешенную выпарительную чашку, выпаривают на
водяной бане досуха и сушат при температуре 105 °С
до постоянной массы.
Проводят два параллельных испытания. За окон¬
чательный ответ принимают среднее арифметиче¬
ское двух параллельных испытаний.
07/2016:1868
ОПРЕДЕЛЕНИЕ
Цельные или измельченные высушенные цве¬
тущие верхушки РШрепс1и1а и/тапа (1_.) Мах1т. (зуп.:
Зр/гаеа и1тапа 1_.).
Содержание: не менее 1 мл/кг эфирного масла (в
пересчете на сухое сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеет ароматный запах метилсалицилата после
измельчения.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Стебель диаме¬
тром до 5 мм, зеленовато-коричневого цвета, жесткий,
коленчатый, полый за исключением верхушки, с пра¬
вильными прямыми продольными бороздами. Лист
черешчатый, сложный, непарноперистый, с двумя
красновато-коричневыми заостренными прилистни¬
ками. Состоит из 3—9 пар неравномерно пильчатых
листочков, некоторые из них маленькие и веерообраз¬
ные. Листочки на верхней стороне темно-зеленые и
гладкие, на нижней — опушенные и более светлые,
иногда серебристые. Конечный листочек крупнее
остальных, разделен на три части. На нижней поверх¬
ности жилки хорошо заметные, коричневого цвета.
® ЛАБАЗНИКА ВЯЗОЛИСТНОГО
ТРАВА
РШрепс!и1ае и1тапае НегЬа
МЕАООШМЕЕТ
1
Лабазника вязолистного трава
1247
Соцветия сложные и состоят из очень многочислен-
шх цветков, объединенных в неравномерные полу-
эонтачные метелки. Цветки кремовато-белые, диаме-
~тюу от 3 мм до 6 мм; чашечка состоит из пяти тем-
•о-зеленых завернутых волосистых чашелистиков,
сросшихся в основании в вогнутое цветоложе; пять
несросшихся лепестков, которые легко отделяются,
бледно-желтого цвета, обратнояйцевидных и заметно
сужающихся к основанию; многочисленные тычинки с
закругленными пыльниками, простирающиеся за ле¬
пестки; гинецей состоит из около А—б плодолистиков,
каждый из которых имеет короткий столбик и шаро¬
видное рыльце; плодолистики совместно винтообраз¬
но закручиваются, образуя спирально закрученные
плоды желтовато-коричневого цвета. Часто встреча¬
ются нераскрывшиеся цветочные бутоны. Если встре¬
чаются плоды, они имеют спирально закрученную
форму и содержат коричневатые семена.
B. Микроскопия (2.8.23). При просматривании
измельченного сырья (355) (2.9.12) (зеленого или жел¬
товато-зеленого цвета) с использованием раствора
хлоралгидрата Р видны (рисунок 1868.-1): фрагменты
эпидермиса листьев и чашелистиков [С, Е, Р] с клет¬
ками извилистой или волнистой формы [Са, Еа, Ра],
короткими толстостенными конусообразными покров¬
ными волосками, утолщенными в основании (вид с
поверхности [ЕЬ] и вид сбоку [3]), одноклеточными по¬
кровными волосками, тонкостенными, очень длинными
и волнистыми с заостренными концами (вид с поверх¬
ности [Рс] и вид сбоку [А]) или их рубцы (волнистый во¬
лосок [Рс1], конусообразный волосок [Ре]) и редкие бу¬
лавовидные железистые волоски с однорядной ножкой
из 1—3 клеток ([Еб] и [6] соответственно), многоклеточ¬
ной головкой и насыщенным коричневым содержимым;
фрагменты верхнего эпидермиса часто с прилегающей
палисадной паренхимой [СЬ], включающей несколько
гипертрофированных клеток, содержащих друзы кри¬
сталлов оксалата кальция [Сс]; фрагменты нижнего
эпидермиса с устьицами аномоцитного типа [Ес, РЬ]
(2.8.3), иногда с прилегающей пористой паренхимой
[Р1] с несколькими клетками, содержащими друзы кри¬
сталлов оксалата кальция [Рд]; фрагменты лепестков
[Н] с тонкостенными клетками эпидермиса, некоторые
имеют закругленные сосочки [На]; многочисленные
шарообразные зерна пыльцы с тремя порами и слегка
впалой экзиной [ВЬ]; фрагменты пыльника [В, О], во¬
локнистый слой которого имеет характерные утолще¬
ния (вид с поверхности [й] и вид сбоку [Ва]); фрагменты
завязи [К] с эпидермисом, образующим устьица [Ка], и
с паренхимой, содержащей призматические кристаллы
оксалата кальция [КЬ]; фрагменты проводящей ткани
[Ц с кольцеобразными, спиральными или ямчатыми
сосудами листьев и стеблей.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Раствор эфирного масла
в ксилоле, полученный как указано в разделе «Коли¬
чественное определение».
Раствор сравнения. 0,1 мл метилсалицилата Р
и 0,1 мл салицилового альдедгида Р растворяют в
ксилоле Р и доводят до объема 5 мл этим же раство¬
рителем.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: гексан Р — толуол Р (50:50,
об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Рисунок 1868.-1. Микроскопические признаки
травы лабазника вязолистного
Высушивание: на воздухе.
Проявление: пластинку опрыскивают 3 мл раст¬
вора железа (III) хлорида РЗ. Просматривают при
дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Метилсалицилат: зона
фиолетово-коричневого
цвета
Салициловый альдегид:
зона фиолетово¬
коричневого цвета
Зона фиолетово¬
коричневого цвета
(метилсалицилат)
Зона фиолетово¬
коричневого цвета
(салициловый альдегид)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые части
растения: стебли диаметром более 5 мм — не более
5,0 %. Другие допустимые примеси: не более 2,0 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 7,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 50,0 г реза¬
ного сырья помещают в колбу вместимостью 1000 мл,
прибавляют 300 мл кислоты хлористоводородной
разведенной Р в качестве жидкости для перегонки. В
градуированную трубку помещают 0,5 мл ксилола Р.
Перегонку проводят со скоростью (2—3) мл/мин в те¬
чение 2 ч.
157*. Зак. 1060
1248
Гэсударственная фармакопея Республики Беларусь
07/2016:РБ0059
ЛАБАЗНИКА ВЯЗОЛИСТНОГО
ЦВЕТКИ
РШрепс1и1ае и1тапае Лоз
МЕАООШМЕЕТ Р1-ОШЕК*
ОПРЕДЕЛЕНИЕ
Собранные в фазу цветения и высушенные
соцветия многолетнего травянистого растения
РШрепди1а и1тапа (Ь.) Мах1т. (зуп.: 5р/гаеа и1тапа
Содержание:
- не менее 6,0 % суммы флавоноидов в пересче¬
те на спиреозид (С21Н20О12; М.м. 464,4) (в пересчете на
сухое сырье);
- или не менее 5,0 % спиреозида (С21Н20О12;
М.м. 464,4) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь цветков
и их частей, бутонов, недоразвитых плодиков, цве¬
тоножек и тонких (до 1 мм) веточек соцветий. Цвет¬
ки правильные, пятичленные, диаметром (6—8) мм.
Чашечка пяти лопастная, с отогнутыми вниз треуголь¬
но-яйцевидными долями, снаружи слабовойлочная.
Цветоножки и веточки соцветия более или менее гу-
стоопушенные. Венчик раздельнолепестный, в 2—2,5
раза длиннее чашечки. Лепестки обратнояйцевидные,
с длинным ноготком, по краю слегка волнистые, с во¬
гнутой морщинистой поверхностью, с обеих сторон го¬
лые. Тычинки многочисленные, свободные, длиннее
лепестков, отогнутые и одинаковые по длине. Недо¬
развитые плодики — нераскрывающиеся винтообраз¬
но скрученные листовки длиной до 8 мм, одиночные
или по нескольку штук вместе с чашечкой. Цвет ле¬
пестков и бутонов желтовато-белый, чашечек, цвето¬
ножек и веточек — темно-зеленый, плодиков — корич¬
невато-зеленый. Запах медовый.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет от желтого до желто-
зеленого. В порошке обнаруживаются следующие
диагностические признаки (рисунок РБ0059.-1): фраг¬
менты чашелистиков [А, В] с устьицами аномоцитного
типа [Аа, С] (2.8.3) и включениями оксалата кальция
[АЬ, Вс]; клетки эпидермиса прозенхимной [Ас] и па¬
ренхимной формы [Вб]; многочисленные одноклеточ¬
ные извилистые волоски [ВЬ, О]] фрагменты завязи:
отдельные элементы кристаллического песка в клет¬
ках [Е]; отдельный простой пестик [Р], в котором хоро¬
шо просматриваются рыльце [РЬ] с пыльцевыми зер¬
нами [Рс], столбик [Ра], семязачаток [Рб]; фрагменты
венчика [6—I]: паренхимные клетки эпидермиса [Сб],
простые одноклеточные волоски [<3а], включения ок¬
салата кальция [<ЗЬ, На], а также видны отдельные
фрагменты проводящих элементов ксилемы [I]; фраг¬
менты пыльника: вид сбоку []], вид сверху [К], много¬
численные пыльцевые зерна [Ц с неплотно сомкнутой
экзиной [1а] и тремя порами [Щ.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Цветки: не менее
50 %. Органические примеси: не более 1 %. Мине¬
ральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
с н I
Рисунок РБ0059.-1. Микроскопические признаки
цветков лабазника вязолистного
Общая зола (2.4.16). Не более 6,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания флавоноидов в
пересчете на спиреозид. 1,000 г измельченного сы¬
рья (180) (2.9.12) помещают в колбу вместимостью
250 мл, прибавляют 50,0 мл 96 % спирта Р. Колбу с
содержимым взвешивают с точностью до 0,01 г и на¬
гревают с обратным холодильником на водяной бане
в течение 5 ч. Охлаждают до комнатной температуры,
взвешивают, доводят массу до первоначальной 96 %
спиртом Р и перемешивают. Выдерживают в течение
30 мин. 0,50 мл надосадочной жидкости доводят рас¬
твором 6,6 г/л алюминия хлорида Р в метаноле Р до
объема 100,0 мл.
Измеряют оптическую плотность (2.2.25) раст¬
вора при 420 нм, используя раствор 6,6 г/л алюминия
хлорида Р в метаноле Р в качестве компенсационно¬
го раствора.
Содержание суммы флавоноидов в пересчете на
спиреозид в процентах рассчитывают по формуле:
А -10000
т•650 ’
где;
650 — удельный показатель поглощения, соот¬
ветствующий спиреозиду (удельный показатель по¬
глощения комплекса кверцетина с алюминия хлори¬
дом с учетом поправочного коэффициента);
Лаванды цветки
1249
1. Спиреозид.
Рисунок РБ0059-1. Хроматограмма испытуемого раствора
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
Определение содержания спиреозида. Жид¬
костная хроматография (2.2.29).
Испытуемый раствор. 0,500 г измельченного
сырья (180) (2.9.12) помещают в колбу вместимостью
100 мл, прибавляют 25,0 мл спирта (40 %, об/об) Р.
Колбу с содержимым взвешивают с точностью до
0,01 г и нагревают с обратным холодильником на во¬
дяной бане в течение 2 ч, периодически перемеши¬
вая. Охлаждают, доводят массу до первоначальной
спиртом (40 %, об/об) Р, перемешивают и центри¬
фугируют. Надосадочную жидкость фильтруют через
мембранный фильтр с диаметром пор 0,45 мкм.
Раствор сравнения. 15,0 мг спиреозида Р раст¬
воряют в 25,0 мл спирта (40 %, об/об) Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: ацетонитрил Р — 0,01 М
раствор дигидрофосфата калия, доведенный до рН
3,0±0,2 кислотой фосфорной Р (20:80, об/об);
- спектрофотометрический детектор, длина
волны 360 нм;
- объем вводимой пробы: 10 мкл;
- время хроматографирования: не менее 40 мин.
Пригодность хроматографической системы:
раствор сравнения:
- фактор удерживания: не менее 6 для пика спи¬
реозида.
Содержание спиреозидав процентах рассчиты¬
вают по формуле:
3,-т2-Р
32-т, ’
где
— площадь пика спиреозида на хроматограм¬
ме испытуемого раствора (см. рисунок РБ0059.-1);
32 — площадь пика спиреозида на хроматограм¬
ме раствора сравнения;
т1 — масса навески испытуемого сырья, г;
т2 — масса навески спиреозида Р, г;
Р — содержание спиреозида в реактиве, %.
07/2016:1534
@ЛАВАНДЫ ЦВЕТКИ
1-ауапс1и1ае Лоз
1-А\/ЕЫОЕК Р/.01УЕЯ
ОПРЕДЕЛЕНИЕ
Высушенные цветки 1„ауапди1а апдизШоНа МШ.
(/.. оТЛстаНа СНа1х).
Содержание: не менее 13 мл/кг эфирного масла
(в пересчете на безводное сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеют сильный ароматный запах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, О.
Вторая идентификация: А, В, С.
A. Внешние признаки (2.8.23). Цветок имеет
короткую цветоножку и состоит из голубовато-серой
трубчатой чашечки, дистально разделенной на четы¬
ре очень коротких зубчика и маленький закругленный
чашелистик, синего двугубого венчика с верхней раз¬
двоенной губой и нижней трехлопастной губой и четы¬
рех двусильных тычинок с яйцевидными пыльниками.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет голубовато-серый. При
просматривании порошка с использованием раст¬
вора хлоралгидрата Р видны (рисунок 1534.-1): по¬
кровные волоски, раздваивающиеся на одном или бо¬
лее уровнях [С, Ц; секреторные трихомы с короткими
ножками и 8-клеточными головками, характерными
для типа 1.ат1асеае (вид сбоку [Н], вид с поверхности
158. Зак. 1060.
1250
Гэсударственная фармакопея Республики Беларусь
[М]); железистые волоски с одноклеточными [О] или
многоклеточными [К] ножками и одноклеточными го¬
ловками; железистые волоски с длинными неровными
ножками и одноклеточными головками, отделенными
от ножки промежуточной клеткой с гладкой кутикулой,
некоторые волоски имеют венчик из мелких сферои¬
дальных бугорков непосредственно под местом при¬
соединения промежуточной клетки к ножке [С]; фраг¬
менты бородавчатого эпидермиса внутренней по¬
верхности лепестков (вид с поверхности Щ, вид сбоку
[Р]); фрагменты эпидермиса чашечки с извилисто¬
стенными клетками и содержащими призматические
кристаллы оксалата кальция [О]; сферические зерна
пыльцы диаметром около 45 мкм, с экзиной с шестью
щелевидными зародышевыми порами и шестью лен¬
товидными пазухами, расходящимися из полюсов
[А, О, Е, Р]; изредка встречающиеся фрагменты эпи¬
дермиса листьев с устьицами преимущественно диа-
цитного типа [В] (2.8.3); фрагменты сосудистой ткани
со спиральными сосудами, входящими в паренхиму с
несколькими клетками, содержащими небольшие дру¬
зы кристаллов оксалата кальция [Ы].
Рисунок 1534.-1. Микроскопические признаки
цветков лаванды
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного ле¬
карственного растительного сырья (355) (2.9.12) при¬
бавляют 5 мл гексана Р, встряхивают в течение 5 мин
и фильтруют.
Раствор сравнения. 10 мкл линалола Р и 10 мкл
линалилацетата Р растворяют в 5 мл гексана Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р, нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин и просма¬
тривают при дневном свете.
Результаты: на хроматограмме раствора сравне¬
ния в нижней трети обнаруживается зона серовато-си¬
него цвета (линалол) и в средней трети зона серовато-
синего цвета (линалилацетат). На хроматограмме ис¬
пытуемого раствора обнаруживаются зоны линалола и
линалилацетата и зона красновато-фиолетового цвета
(эпоксидигидрокариофиллен) в средней части между
этими зонами. Также обнаруживаются другие зоны.
О. Просматривают хроматограммы, полученные в
испытании «Другие виды и разновидности лаванды».
Результаты: на хроматограмме испытуемого
раствора 5 основных пиков соответствуют по време¬
ни удерживания соответствующим пикам на хрома¬
тограмме раствора сравнения. Среди них преимуще¬
ственно пики линалола и линалилацетата.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли — не более 3 %. Другие допу¬
стимые примеси: не более 2 %.
Другие виды и разновидности лаванды. Газо¬
вая хроматография (2.2.28): испытание проводят ме¬
тодом нормализации.
Испытуемый раствор. 0,2 мл смеси из эфирно¬
го масла и ксилола, полученной как указано в разделе
«Количественное определение», доводят гексаном Р
до объема 5 мл, прибавляют 1 г натрия сульфата
безводного Р, встряхивают и используют надосадоч-
ную жидкость.
Раствор сравнения. 0,1 г лимонена Р, 0,2 г цине-
ола Р, 0,05 г камфоры Р, 0,4 г линалола Р, 0,6 г лина¬
лилацетата Р и 0,2 г а-терпинеола Р растворяют в
100 мл гексана Р.
Условия хроматографирования:
- колонка кварцевая капиллярная длиной 60 м и
внутренним диаметром 0,25 мм, покрытая слоем ма-
крогола 20 000 Р;
- газ-носитель: гелий для хроматографии Р;
- скорость газа-носителя: 1,5 мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0 — 15
70
15 — 70
о
со
7
о
1^
Блок ввода проб
220
Детектор
220
- детектор: пламенно-ионизационный;
- объем вводимой пробы: равные объемы каждо¬
го раствора.
Порядок выхода пиков: порядок выхода пиков со¬
ответствует порядку перечисления веществ при при¬
готовлении раствора сравнения; отмечают времена
удерживания всех веществ.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,5 между пиками лимо¬
нена и цинеола;
-количество теоретических тарелок: не ме¬
нее 30 000 для пика лимонена при температуре
110 °С.
Используя времена удерживания для пиков на
хроматограмме раствора сравнения, на хроматограм-
Ламинарии слоевища (морская капуста)
1251
ме испытуемого раствора определяют пики шести
компонентов раствора сравнения. Пики гексана и кси¬
лола не учитывают.
Предельное содержание примесей:
- камфора: не более 1 %.
Вода (2.2.73). Не более 100 мл/кг. Определение
проводят из 20,0 г испытуемого сырья.
Общая зола (2.4.16). Не более 9,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 20,0 г лекар¬
ственного растительного сырья помещают в кругло¬
донную колбу вместимостью 1000 мл, прибавляют
500 мл воды Р в качестве жидкости для перегонки. В
градуированную трубку помещают 0,5 мл ксилола Р.
Перегонку проводят со скоростью (2—3) мл/мин в те¬
чение 2 ч.
07/2016: РБ0060
ЛАМИНАРИИ СЛОЕВИЩА
(МОРСКАЯ КАПУСТА)
1-ат 'тапае Ма11ота
I. АММАН!А*
ОПРЕДЕЛЕНИЕ
Собранные с июня по октябрь и высушенные сло¬
евища бурых морских водорослей 1-ат1папа ]ароп1са
АгезсИ. и зассЬагта (1_.) 1ат.
Содержание:
- не менее 0,1 % йода (в пересчете на сухое сы-
рье);
- не менее 8,0 % полисахаридов (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цвет цельных
слоевищ от светло-коричневато-зеленого до темно-
коричневато-зеленого, зеленовато-коричневый, крас¬
но-коричневый, иногда зеленовато-черный; снаружи
слоевища покрыты белым налетом солей. Допускает¬
ся наличие пластин с разрывами по краям и середине.
Слоевища /_ат'тапа ]ароп'\са — плотные, кожистые,
лентообразные пластины, сложенные по длине, без
стволиков или куски пластин длиной не менее 15 см,
шириной не менее 7 см. Толщина пластин не менее
0,3 мм. Края пластин цельные, волнистые. Слоевища
1-аттапа 8асс11аппа — плотные, кожистые, морщи¬
нистые лентовидные пластины без стволиков или их
куски длиной не менее 10 см, шириной не менее 5 см.
Толщина пластин не менее 0,3 мм. Края пластин вол¬
нистые. Запах слоевищ своеобразный.
B. Микроскопия (2.8.23). При просматривании
слоевищ с поверхности виден эпидермис, состоящий
из мелких, почти квадратных клеток с толстыми стен¬
ками, сквозь которые просвечивают многочисленные
округлые слизистые вместилища.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Песок. Не более 0,2 %. 20 г измельченного сы¬
рья (710) (2.9.12) помещают в стакан вместимостью
150 мл, прибавляют 60 мл раствора 100 г/л кислоты
хлористоводородной Р и нагревают до кипения, не¬
прерывно перемешивая до прекращения вспучивания
массы. Стакан накрывают часовым стеклом и кипя¬
тят в течение 15 мин. Прибавляют водуР доверху,
интенсивно перемешивают содержимое стеклянной
палочкой и выдерживают в течение (3—5) мин. Че¬
рез пипетку вместимостью (5—10) мл, погруженную
в стакан на расстояние (1—2) см от дна, пропускают
водуР со скоростью (160—170) мл/мин (слив воды
должен происходить через край стакана) в течение
20 мин. Собирают надосадочную жидкость. Для уда¬
ления оставшихся частиц сырья к осадку в стакане
прибавляют (25—30) мл насыщенного раствора на¬
трия хлорида Р, перемешивают, выдерживают до
оседания песка на дно и осторожно сливают жидкость
вместе со взвешенными частицами сырья. Обработку
насыщенным раствором натрия хлорида Р повторя¬
ют 3—4 раза до полного удаления взвешенных ча¬
стиц сырья. Осадок в стакане промывают 2—3 раза
водой Р и количественно переносят на беззольный
фильтр. Осадок вместе с фильтром сжигают в пред¬
варительно прокаленном и взвешенном тигле и про¬
каливают в течение 15 мин при температуре от 550 °С
до 650 °С. Охлаждают и взвешивают. Масса остатка
должна составлять не более 40 мг.
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: слоевища с пожелтевшими краями — не
более 10 %; слоевища толщиной менее 0,3 мм — не
более 15 %. Органические примеси: не допускаются.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 15,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 40 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания йода. 60 мг измель¬
ченного сырья (500) (2.9.12) сжигают в колбе с кис¬
лородом (2.5.10). Используют колбу вместимостью
1000 мл, в качестве поглощающей жидкости — 2 мл
0,1 М раствора натрия гидроксида и 50 мл воды Р.
После сжигания колбу интенсивно встряхивают в те¬
чение 3 мин. Прибавляют 100мл воды Р, промывая
шлиф, держатель и внутренние стенки колбы. При¬
бавляют по каплям до появления слабо-желтого окра¬
шивания смесь из 25 мл раствора 100 г/л калия аце¬
тата Р в кислоте уксусной ледяной Р и 15 капель
брома Р. Выдерживают в течение 2 мин, прибавляют
по каплям 85 % (м/м) раствор кислоты муравьиной
безводной Р до обесцвечивания раствора, выдержи¬
вают в течение 1 мин и прибавляют 3 мл раствора
30 г/л кислоты сульфаминовой Р. Перемешивают в
течение 3 мин, прибавляют 1 г калия йодида Р и ти¬
труют 0,005 М раствором натрия тиосульфата, ис¬
пользуя в качестве индикатора раствор крахмала,
свободный от йодидов, Р.
1 мл 0,005 М раствора натрия тиосульфата
соответствует 0,1058 мг йода.
Определение содержания полисахаридов.
10.00 г измельченного сырья (2000) (2.9.12) помеща¬
ют в колбу вместимостью 500 мл, прибавляют 100 мл
воды Р и кипятят с обратным холодильником при
периодическом перемешивании в течение 30 мин.
Оставляют до оседания частиц и процеживают надо¬
садочную жидкость через ватный тампон в мерную
колбу вместимостью 500 мл. Экстракцию повторяют
еще четыре раза с использованием воды Р порциями
по 100 мл. Водные извлечения процеживают в ту же
мерную колбу и доводят водой Р до объема 500,0 мл.
25.0 мл полученного раствора помещают в центри-
158*. Зак. 1060.
1252
Гэсударственная фармакопея Республики Беларусь
фужную пробирку, прибавляют 25 мл 96 % спирта Р,
перемешивают, подогревают в водяной бане при тем¬
пературе 30 °С в течение 5 мин. Через 30 мин содер¬
жимое центрифугируют при 5000 об/мин в течение
30 мин. Надосадочную жидкость фильтруют под ваку¬
умом через высушенный при температуре 105 °С до
постоянной массы стеклянный фильтр (16) диаметром
40 мм. Осадок количественно переносят на фильтр с
помощью смеси из 96 % спирта Р и воды Р (50:50,
об/об) и промывают 10 мл 96% спирта Р. Фильтр с
осадком высушивают сначала на воздухе, затем при
температуре 105 °С до постоянной массы.
Содержание полисахаридов в процентах рассчи¬
тывают по формуле:
(т2 - /л.,)-2000
т
где:
/7)1 — масса фильтра, г;
т2 — масса фильтра с осадком, г;
т — масса навески испытуемого сырья, г.
07/2016:РБ0061
ЛАНДЫША ЛИСТЬЯ
СопуаНапае Ш‘\ит
ШУ-ОР-ТНЕ-УАИ-ЕУ 1.ЕАЕ*
ОПРЕДЕЛЕНИЕ
Собранные до цветения и в начале цветения и
высушенные листья многолетних травянистых рас¬
тений СопуаНапа та]аНз 1_., СопVаIIа^^а 1гапзсаисаз1са
1Лкт ех СгоззИ. и СопуаНапа /се/з/се/ 1Мд.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья цельные,
реже изломанные, эллиптической или ланцетовидной
формы с заостренной верхушкой, суживающиеся у
основания и постепенно переходящие в длинные вла¬
галища; отдельные или соединенные по два или три.
Край листа цельный, жилкование дугонервное. Листо¬
вая пластинка тонкая, ломкая, с голой, слегка блестя¬
щей поверхностью. Длина листьев до 20 см, ширина
до 8 см. Цвет листьев зеленый, реже — коричневато¬
зеленый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности с обеих сторон видны вытянутые
по длине листа клетки эпидермиса с прямыми стенка¬
ми. Устьица погруженные, округлые, ориентированы
по длине листа, окружены четырьмя клетками эпидер¬
миса. Под верхним эпидермисом видны клетки пали¬
садной ткани, вытянутые по ширине листа. Губчатая
ткань рыхлая и состоит из разветвленных клеток, вы¬
тянутых по ширине листа. В отдельных клетках мезо¬
филла видны пучки тонких рафид и крупные игольча¬
тые кристаллы оксалата кальция.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 20 мл спирта (50 %,
об/об) Р, 10 мл раствора 100 г/л свинца ацетата Р
и нагревают с обратным холодильником на водяной
бане в течение 2 мин. Охлаждают, центрифугируют.
Надосадочную жидкость дважды встряхивают с хло¬
роформом Р порциями по 15 мл, затем дважды встря¬
хивают со смесью из хлороформа Р и метанола Р
(50:50, об/об) порциями по 15 мл. При необходимости
слои разделяют центрифугированием. Хлороформ¬
ные слои объединяют, фильтруют через бумажный
фильтр с 2 г натрия сульфата безводного Р и вы¬
паривают досуха. Остаток растворяют в 1 мл смеси из
хлороформа Р и метанола Р (50:50, об/об).
Раствор сравнения. 5 мг конвалатоксина Р
растворяют в 1 мл смеси из хлороформа Р и метано¬
ла Р (50:50, об/об).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля в Р.
Подвижная фаза: хлороформ Р — метанол Р —
вода Р (80:18:2, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С до
105 °С в течение 5 мин. Просматривают при дневном
свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается зона зеленого цвета со зна¬
чением /?, 0,38—0,48 (конвалатоксин). На хромато¬
грамме испытуемого раствора обнаруживается зона,
соответствующая по расположению и цвету зоне кон¬
валатоксина на хроматограмме раствора сравнения.
На хроматограмме испытуемого раствора обнаружи¬
ваются и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: частицы, проходящие сквозь сито (2000)
(2.9.12) , — не более 3 %; пожелтевшие и побуревшие
листья — не более 5 %. Органические примеси: не бо¬
лее 1 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
07/2016: РБ0062
ЛАНДЫША ТРАВА
СопуаНапае ЬегЬа
иЦУ-ОЕ-ТНЕ-УАЦ-ЕУ НЕРВ*
ОПРЕДЕЛЕНИЕ
Собранная в период цветения и высушенная
трава многолетних травянистых растений СопуаНапа
/7?ауа//з1-., СопуаНапа 1гапзсаисаз1са 1Лк1П ех ОгоззИ. и
СопVаIIапа /се/з/се/ М1д.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Смесь цельных,
реже изломанных листьев, соцветий с цветоносами,
отдельных цветков и кусочков цветоносов. Листья эл¬
липтической или ланцетовидной формы с заострен¬
ной верхушкой, суживающиеся у основания и посте¬
пенно переходящие в длинные замкнутые влагалища,
отдельные или охватывающие друг друга по два или
три. Край листа цельный, жилкование дугонервное.
Лист тонкий, ломкий, с голой и слегка блестящей по¬
верхностью. Длина листьев до 20 см, ширина до 8 см.
Соцветие — односторонняя рыхлая кисть из 3—12 (до
20) желтоватых цветков на ребристом голом цветоно¬
се длиной до 20 см и толщиной до 1,5 мм. Цветки обо¬
еполые с венчиковидным колокольчатым околоцвет-
Ландыша цветки
1253
ником, сростнолепестные, с шестью короткими ото¬
гнутыми зубчиками, на коротких цветоножках, с плен¬
чатыми линейными прицветниками. Цвет листьев
зеленый, реже коричневато-зеленый, цветков — жел¬
товатый, цветоносов — светло-зеленый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности с обеих сторон видны вытянутые
по длине листа клетки эпидермиса с прямыми стен¬
ками. Устьица погруженные, округлые, ориентиро¬
ваны по длине листа, окружены четырьмя клетками
эпидермиса. Под верхним эпидермисом видны клетки
палисадной ткани, вытянутые по ширине листа. Губ¬
чатая ткань рыхлая и состоит из разветвленных кле¬
ток, вытянутых по ширине листа. В отдельных клет¬
ках мезофилла видны пучки тонких рафид и крупные
игольчатые кристаллы оксалата кальция. При про¬
сматривании венчика с поверхности с обеих сторон
видны слегка вытянутые по оси многоугольные клетки
эпидермиса с тонкими прямыми стенками и нежной
складчатостью кутикулы. Устьица погруженные, окру¬
глые, ориентированы по длине околоцветника, окру¬
жены 4—5 клетками эпидермиса. Эпидермис зубчика
с сосочковидными выростами. В ткани околоцветника
видны тонкие рафиды оксалата кальция, встречаются
крупные игольчатые кристаллы; пыльца шаровидной
формы с гладкой поверхностью.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 20 мл спирта (50 %,
об/об) Р, 10 мл раствора 100 г/л свинца ацетата Р
и нагревают с обратным холодильником на водяной
бане в течение 2 мин. Охлаждают, центрифугируют.
Надосадочную жидкость дважды встряхивают с хло¬
роформом Р порциями по 15 мл, затем дважды встря¬
хивают со смесью из хлороформа Р и метанола Р
(50:50, об/об) порциями по 15 мл. При необходимости
слои разделяют центрифугированием. Хлороформ¬
ные слои объединяют, фильтруют через бумажный
фильтр с 2 г натрия сульфата безводного Р и вы¬
паривают досуха. Остаток растворяют в 1 мл смеси из
хлороформа Р и метанола Р (50:50, об/об).
Раствор сравнения. 5 мг конвалатоксина Р
растворяют в 1 мл смеси из хлороформа Р и метано¬
ла Р (50:50, об/об).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля О Р.
Подвижная фаза: хлороформ Р — метанол Р —
вода Р (80:18:2, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С до
105 °С в течение 5 мин. Просматривают при дневном
свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается зона зеленого цвета со зна¬
чением /?, 0,38—0,48 (конвалатоксин). На хромато¬
грамме испытуемого раствора обнаруживается зона,
соответствующая по расположению и цвету зоне кон¬
валатоксина на хроматограмме раствора сравнения.
На хроматограмме испытуемого раствора обнаружи¬
ваются и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Соцветия: не ме¬
нее 5 %. Несырьевые части растения: частицы, про¬
ходящие сквозь сито (2000) (2.9.12), — не более 3 %;
пожелтевшие и побуревшие листья и побуревшие
цветки — не более 5 %. Органические примеси: не бо¬
лее 1 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 9,0 %.
07/2016:РБ0063
ЛАНДЫША ЦВЕТКИ
СопVаIIа^^ае Лоз
ШУ-ОЕ-ТНЕ-УАИ-ЕУ РШМЕП*
ОПРЕДЕЛЕНИЕ
Собранные в период цветения и высушенные
цветки многолетних травянистых растений СопуаНапа
та/аНзСопуаИапа Капзсаисазюа 1Лкт ех ОгоззН. и
СопVаIIапа ке1зке'1 М1д.
ПОДЛИННОСТЬ(ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь соцветий с
остатками цветоносов длиной до 20 см, цветков и ино¬
гда кусочков цветоносов. Цветонос ребристый, голый,
толщиной до 1,5 мм, с односторонней рыхлой кистью
из 3—12 (до 20) желтоватых цветков. Цветки обоепо¬
лые с венчиковидным колокольчатым околоцветни¬
ком, сростнолепестные, с шестью короткими отогну¬
тыми зубчиками, на коротких цветоножках, с пленча¬
тыми линейными прицветниками. Тычинок шесть, на
коротких нитях, прикрепленных к основанию около¬
цветника; завязь верхняя, трехгнездная, столбик с
расширенным трехлопастным рыльцем. Цвет цвето¬
носов светло-зеленый, цветков — желтоватый.
B. Микроскопия (2.8.23). При просматривании
венчика с поверхности с обеих сторон видны слегка
вытянутые по оси многоугольные клетки эпидермиса
с тонкими прямыми стенками и нежной складчатостью
кутикулы. Устьица погруженные, округлые, ориенти¬
рованы по длине околоцветника, окружены 4—5 клет¬
ками эпидермиса. Эпидермис зубчика с сосочковид¬
ными выростами. В ткани околоцветника видны тон¬
кие рафиды оксалата кальция, встречаются крупные
игольчатые кристаллы; пыльца шаровидной формы с
гладкой поверхностью.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 20 мл спирта (50 %,
об/об) Р, 10 мл раствора 100 г/л свинца ацетата Р
и нагревают с обратным холодильником на водяной
бане в течение 2 мин. Охлаждают, центрифугируют.
Надосадочную жидкость дважды встряхивают с хло¬
роформом Р порциями по 15 мл, затем дважды встря¬
хивают со смесью из хлороформа Р и метанола Р
(50:50, об/об) порциями по 15 мл. При необходимости
слои разделяют центрифугированием. Хлороформ¬
ные слои объединяют, фильтруют через бумажный
фильтр с 2 г натрия сульфата безводного Р и вы¬
паривают досуха. Остаток растворяют в 1 мл смеси из
хлороформа Р и метанола Р (50:50, об/об).
Раствор сравнения. 5 мг конвалатоксина Р
растворяют в 1 мл смеси из хлороформа Р и метано¬
ла Р (50:50, об/об).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля С Р.
159. Зак. 1060.
1254
Гэсударственная фармакопея Республики Беларусь
Подвижная фаза: хлороформ Р — метанол Р —
вода Р (80:18:2, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С до
105 °С в течение 5 мин. Просматривают при дневном
свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается зона зеленого цвета со зна¬
чением Я, 0,38—0,48 (конвалатоксин). На хромато¬
грамме испытуемого раствора обнаруживается зона,
соответствующая по расположению и цвету зоне кон-
валатоксина на хроматограмме раствора сравнения.
На хроматограмме испытуемого раствора обнаружи¬
ваются и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: соцветия с побуревшими цветками —
не более 5 %; отдельные цветоносы — не более 1 %.
Органические примеси: не более 0,5 %. Минеральные
примеси: не более 0,3 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
07/2016: РБ0064
ЛАПЧАТКИ БЕЛОЙ ТРАВА
Ро^еп^^IIае а\Ьае ЬегЬа
МН1ТЕ СШСШЕРОИ*
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная трава
многолетнего травянистого растения Ро1епШа а\Ьа 1_.
Содержание: не менее 1,8 % флавоноидов в пе¬
ресчете на рутин (С27Н30О16; М.м. 611) (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
частично измельченные олиственные цветоносные
стебли длиной до 20 см. Листья чешуевидные, при¬
корневые, на длинных черешках, пальчатосложные,
состоят из 5 обратно-ланцетных листочков, сверху
темно-зеленые, снизу шелковистые с темно-коричне¬
выми прилистниками. Стеблевые листья небольшие,
чешуевидные, в количестве 1—2, с маленькими яй¬
цевидно-ланцетными прилистниками. Все растение
покрыто прижатыми шелковистыми серебристыми во¬
лосками. Цветки белые, диаметром до 3 см, на длин¬
ных цветоносах, собраны по 2—5 в верхушечные по-
лузонтики. Венчик из 5 свободных лепестков, чашечка
с подчашием опушены, пятинадрезанные. Тычинок и
пестиков много.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны: клетки эпидермиса много¬
угольные в очертании, с прямыми стенками; устьица
только на нижней стороне листа с 4—7 околоустьич-
ными клетками (аномоцитный тип) (2.8.3); на нижней
стороне листа — многочисленные длинные простые
волоски с одноклеточной ножкой и двухклеточной го¬
ловкой и головчатые волоски с многоклеточной нож¬
кой и одноклеточной головкой; в клетках мезофилла
многочисленные друзы оксалата кальция.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (250) (2.9.12) прибавляют 30 мл спирта (40 %,
об/об) Р, нагревают с обратным холодильником на во¬
дяной бане в течение 30 мин и фильтруют.
Раствор сравнения. 5 мг рутина Р растворяют в
10 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: бутанол Р — кислота уксус¬
ная ледяная Р — вода Р (4:1:2, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе в течение (10—15) мин.
Проявление: пластинку обрабатывают раствором
30 г/л алюминия хлорида Р в 96% спирте Р, нагре¬
вают при температуре от 100 °С до 105 °С в течение
10 мин и просматривают в ультрафиолетовом свете
при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Рутин: флуоресцирующая
зона желто-зеленого цвета
Флуоресцирующая зона
синего цвета
Флуоресцирующая зона
желто-зеленого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие и побуревшие части рас¬
тения — не более 7 %; кусочки стеблей и боковых
веточек, в том числе отделенные при анализе, — не
более 40 %. Органические примеси: не более 1 %.
Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16) Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (250) (2.9.12) поме¬
щают в колбу вместимостью 50 мл со шлифом и при¬
бавляют 30,0 мл спирта (40 %, об/об) Р. Колбу закры¬
вают, взвешивают с точностью до 0,01 г и кипятят с
обратным холодильником на водяной бане в течение
45 мин. Затем колбу с содержимым охлаждают и те¬
чение 30 мин, взвешивают, доводят массу колбы до
первоначальной спиртом (40 %, об/об) Р и фильтру¬
ют (раствор А).
Испытуемый раствор. К 1,0 мл раствора А при¬
бавляют 2,0 мл раствора 30 г/л алюминия хлорида Р в
96 % спирте Р и доводят спиртом (95 %, об/об) Р до
объема 25,0 мл.
Раствор сравнения. 0,050 г ФСО рутозида три-
гидрата (с учетом заявленного содержания рутозида
в ФСО) растворяют при нагревании на водяной бане
Лапчатки корневища
1255
в 50 мл спирта (40 %, об/об) Р, охлаждают и доводят
спиртом (40 %, об/об) Р до объема 100,0 мл (раствор
В). К 1,0 мл раствора В прибавляют 2,0 мл раствора
30 г/л алюминия хлорида Р в 96% спирте Р и дово¬
дят спиртом (95 %, об/об) Р до объема 25,0 мл.
Компенсационный раствор (а). К 1,0 мл раст¬
вора А прибавляют 2—3 капли раствора кислоты
уксусной Р и доводят спиртом (95 %, об/об) Р до объ¬
ема 25,0 мл.
Компенсационный раствор (Ь). К 1,0 мл раст¬
вора В прибавляют 2—3 капли раствора кислоты
уксусной Р и доводят спиртом (95 %, об/об) Р до объ¬
ема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора и раствора сравнения
при 412 нм, используя компенсационный раствор (а) и
компенсационный раствор (Ь) соответственно.
Содержание суммы флавоноидов в пересчете на
рутин в процентах рассчитывают по формуле:
АпV 30
А0т
где:
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески ФСО рутозида тригидрата
(с учетом заявленного содержания рутозида в ФСО), г.
07/2016:1478
ЛАПЧАТКИ КОРНЕВИЩА
ТогтепИНае гЫгота
ТОШЕМТИ.
ОПРЕДЕЛЕНИЕ
Собранные в период цветения, цельные или из¬
мельченные, очищенные от корней, остатков листьев
и стеблей, отмытые от земли и высушенные корне¬
вища многолетнего травянистого растения Ро^еп^^IIа
егес1а (1_.) РаеизсИ. (зуп.: Р /огтепИИа 31окез).
Содержание:
- не менее 7 % дубильных веществ в пересчете
на пирогаллол (С6Н603; М.м. 126,1) (в пересчете на су¬
хое сырье);
- или не менее 20 % дубильных веществ в пере¬
счете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
®А. Внешние признаки (2.8.23). Корневища
цилиндрические веретенообразные, с очень непра¬
вильной формой, часто образующие сплетенные,
спутанные клубни, длиной до 10 см и толщиной
(1—2) см, очень твердые и почти не разветвленные.
Поверхность от коричневого до красновато-коричне¬
вого цвета, морщинистая, с остатками корней и по¬
перечно-вытянутыми вдавленными беловатыми сле¬
дами от стеблей. На верхушке корневищ могут при¬
сутствовать остатки многочисленных надземных сте¬
блей. Разлом хрупкий и зернистый, от темно-красного
до коричневато-желтого цвета.
<Е) В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет красновато-корич¬
невый. Просматривают под микроскопом с использо¬
ванием раствора хлоралгидрата Р. В порошке обна¬
руживаются следующие диагностические признаки:
крупнозубчатые друзы оксалата кальция диаметром
до 60 мкм; фрагменты тонкостенной паренхимы, со¬
держащей красновато-коричневый танин; пучки тон¬
ких точечных сосудов с латеральными порами; тол¬
стостенная и ямчатая многоугольная паренхима; пуч¬
ки и фрагменты склеренхиматозных толстостенных
волокон; редкие фрагменты пробки с тонкостенными
коричневыми слоистыми клетками.
При использовании 50 % (об/об) раствора гли¬
церина Р видны многочисленные крахмальные зерна
округлой и эллиптической формы размером до 20 мкм.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл воды Р, встряхи¬
вают в течение 10 мин и фильтруют. Фильтрат дважды
экстрагируют этилацетатом Р порциями по 10мл.
Объединенные верхние слои фильтруют через бу¬
мажный фильтр с 6 г натрия сульфата безводного Р
и выпаривают досуха в вакууме. Остаток растворяют
в 1,0 мл этилацетата Р.
Раствор сравнения. 1,0 мг катехина Р раство¬
ряют в 1,0 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р —
эфир Р — гексан Р — этилацетат Р (20:20:20:40,
об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе, в течение (10—15) мин.
Проявление: пластинку опрыскивают свежепри¬
готовленным раствором 5 г/л соли прочного синего
В Р. Появляются зоны красноватого цвета. Пластин¬
ку выдерживают в парах аммиака. Зоны приобретают
более интенсивное красновато-коричневое окрашива¬
ние. Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испыту¬
емого раствора. На хроматограмме испытуемого раст¬
вора обнаруживаются и другие более слабые зоны.
Верх хроматографической пластинки
Катехин: интенсивная зона
красновато-коричневого
цвета
Более интенсивная зона
красновато-коричневого
цвета (катехин)
Менее интенсивная зона
Интенсивная зона
Менее интенсивные зоны
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корневища, почерневшие в изломе, —
не более 3 %; корни и надземные части растения, в
том числе отделенные при анализе, — не более 3 %.
Другие допустимые примеси: не более 2 %.
(§•) Кадмий (2.4.27). Не более 0,00020 %
(2,0 ррт).
@> Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 5,0 %.
159*. Зак. 1060.
1256
Государственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Определение содержания дубильных ве¬
ществ в пересчете на пирогаллол (2.8.14). Исполь¬
зуют 0,500 г измельченного сырья (180) (2.9.12).
Определение содержания дубильных ве¬
ществ в пересчете на танин. 2,000 г измельченного
сырья (2000) (2.9.12) помещают в коническую кол¬
бу со шлифом вместимостью 500 мл, прибавляют
250 мл кипящей воды Р и кипятят с обратным хо¬
лодильником в течение 30 мин при периодическом
перемешивании. Затем охлаждают до комнатной
температуры, процеживают и доводят водой Р до
объема 250,0 мл.
25,0 мл полученного извлечения помещают в
коническую колбу вместимостью 750 мл, прибав¬
ляют 500 мл воды Р, 25,0 мл раствора индигосуль-
фокислоты (0,25 мг индигокармина Р растворяют в
6,5 мл кислоты серной Р, прибавляют 6,5 мл кисло¬
ты серной Р, доводят водой Р до объема 250 мл и
фильтруют). Титруют при постоянном перемешива¬
нии 0,02 М раствором калия перманганата до зо¬
лотисто-желтого окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07/2016:РБ0065
ЛЕВЗЕИ САФЛОРОВИДНОЙ
КОРНЕВИЩА С КОРНЯМИ
КЬаропИа саг01ато1с1ез гЫгота сит
гасИаЬиз
1-ЕШЕА КООТ*
ОПРЕДЕЛЕНИЕ
Собранные осенью, очищенные от остатков над¬
земных частей и от земли, промытые и высушенные
корневища с корнями многолетнего травянистого
растения РЬаропИсит саМатоШез (Ш\\д) Н]1п (зуп.:
Ьеитеа саг01ато1дез (\Л/Шс1) ОС).
Содержание: не менее 0,1 % 20-гидроксиэкдизо-
на (С^Н^О,,; М.м. 480,6) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корневища
цельные или разрезанные, деревянистые, иногда с
остатками стеблей длиной до 1 см, цилиндрические,
многоглавые, разветвленные, снаружи неравномерно¬
морщинистые, в изломе неровные, с многочислен¬
ными тонкими ветвящимися придаточными корнями.
Корни упругие, мелкобороздчатые. Толщина корне¬
вищ до 3 см, длина корней — до 36 см. Цвет корне¬
вищ и корней снаружи от коричневого до почти чер¬
ного, в изломе — бледно-желтый; часто встречаются
корни с мелкими участками, лишенными коры. Запах
слабый, своеобразный.
B. Микроскопия (2.8.23). На поперечном сре¬
зе корня заметен широкий участок первичной коры.
Эпидермис однослойный. Клетки эндодермы танген-
тально-вытянутые, утолщенные; вблизи эндодермы
расположены крупные секреторные вместилища
в виде прерывистого пояса. Они состоят из 4—5
крупных выделительных клеток, часто с мелкими ка¬
плями смолистого содержимого (реакция с раство¬
ром Судана IIIР). Молодые корни имеют первичное
строение, старые корни — вторичное. В клетках
лубяной паренхимы содержится инулин; изредка
встречаются одиночные призматические кристал¬
лы и друзы оксалата кальция. Древесина состоит
из множества утолщенных, одревесневших волокон
(либриформ) и немногочисленных узких сосудов и
трахеид. На поперечном срезе корневища видны:
небольшие группы каменистых клеток; крупные се¬
креторные вместилища с содержимым оранжевого
цвета; в древесине — сосуды, либриформ; в сердце-
вине — редкие вместилища; в паренхиме — инулин
и друзы оксалата кальция.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5,0 г измельченного сы¬
рья (710) (2.9.12) помещают в колбу вместимостью
100 мл со шлифом, прибавляют 50 мл спирта (70 %,
об/об) Р, закрывают пробкой и выдерживают при пе¬
риодическом перемешивании в течение 24 ч. Филь¬
труют через бумажный фильтр и доводят спиртом
(70 %, об/об) Р до объема 50 мл. Фильтруют через
фильтр с диаметром пор 0,45 мкм.
Раствор сравнения. 1 мг 20-гидроксиэкдизона Р
растворяют в 10 мл спирта (70 %, об/об) Р.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: толуол Р — ацетон Р — эта¬
нол безводный Р — раствор аммиака концентриро¬
ванный Р (100:140:32:9, об/об/об/об).
Наносимый объем пробы: по 5 мкл в виде полос.
Фронт подвижной фазы: не менее 14 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре от 100 °С до
105 °С. Просматривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается основная зона серовато-тем¬
но-красного цвета (20-гидроксиэкдизон). На хромато¬
грамме испытуемого раствора обнаруживается зона,
соответствующая по расположению и цвету зоне
20-гидроксиэкдизона на хроматограмме раствора
сравнения. На хроматограмме испытуемого раствора
обнаруживаются также две или три зоны розового и
несколько зон коричневого или коричневато-серого
цвета.
Б. К измельченному сырью, полученному пу¬
тем соскабливания поперечного излома корневи¬
ща или корня, прибавляют 1—2 капли раствора
а-нафтола Р1 и 1 каплю кислоты серной Р. Появ¬
ляется красновато-фиолетовое окрашивание (при ис¬
пользовании раствора резорцина Р вместо раствора
а-нафтола Р1 появляется красное окрашивание, при
использовании раствора тимола Р— розовато-крас¬
ное окрашивание).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Крахмал. К измельченному сырью, полученному
путем соскабливания поперечного излома корневища
или корня, прибавляют 2—3 капли раствора йода Р1.
Не должно появляться синее окрашивание.
Допустимые примеси (2.8.2). Несырьевые
части растения: остатки стеблей, в том числе отде¬
ленные при анализе, — не более 2 %. Органические
примеси: не более 1 %. Минеральные примеси: не
более 4 %.
Левзеи сафлоровидной листья
1257
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 9,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор. 15,0 г измельченного
сырья (710) (2.9.12) помещают в патрон из фильтро¬
вальной бумаги и помещают в экстрактор аппарата
Сокслета, рабочий объем которого около 150 мл.
Экстрагируют (180—200) мл метанола Р в течение
7 ч (25—30 сливов). Извлечение упаривают в ваку¬
уме при температуре 40 °С до объема около 5 мл,
количественно переносят в мерную колбу вмести¬
мостью 25 мл, используя метанол Р, и доводят до
объема 25,0 мл этим же растворителем.
Раствор сравнения. 16,8 мг 20-гидроксиэкди-
зона Р растворяют в метаноле Р и доводят до объ¬
ема 10,0 мл этим же растворителем.
ТСХ пластинку со стеклянной подложкой, раз¬
мером 20X20 см, с закрепленным слоем силикаге¬
ля Р делят на пять равных полос. На линию старта
первой и второй полос наносят по 150 мкл испы¬
туемого раствора в виде полос длиной 20 мм, на
третей и четвертой — по 150 мкл раствора сравне¬
ния, пятая полоса используется для приготовления
компенсационного раствора. Пластинку с нанесен¬
ными пробами сушат на воздухе в течение 20 мин
и помещают в камеру со смесью растворителей
хлороформ Р — метанол Р— ацетон Р (6:2:1,
об/об/об) (насыщают хроматографическую камеру
парами растворителя в течение не менее 30 мин).
Когда фронт подвижной фазы пройдет не менее
17 см от линии старта, пластинку вынимают из ка¬
меры и сушат на воздухе в течение 1 ч. Первую и
третью полосы пластинки опрыскивают раствором
ванилина в серной кислоте Р. На второй и четвер¬
той полосах отмечают зоны 20-гидроксиэкдизона,
расположенные на уровне проявленных зон на пер¬
вой и третьей полосах.
Слои сорбента с отмеченных зон на второй и
четвертой полосах количественно переносят в две
колбы вместимостью 100 мл со шлифом, прибав¬
ляют по 15,0 мл метанола Р, закрывают пробкой и
встряхивают в течение 4 ч. Фильтруют элюаты через
стеклянный фильтр (16).
Измеряют оптическую плотность (2.2.25) элю-
атов испытуемого раствора и раствора сравнения
при 242 нм, используя элюат с пятой полосы в каче¬
стве компенсационного раствора.
Содержание 20-гидроксиэкдизона в процентах
рассчитывают по формуле:
А-т0- 0,0025 Р
А) т
где:
А — оптическая плотность элюата испытуемого
раствора;
А0 — оптическая плотность элюата раствора
сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески 20-гидроксиэкдизона, мг;
Р — содержание 20-гидроксиэкдизона в реакти¬
ве, %.
07/2016: РБ0066
ЛЕВЗЕИ САФЛОРОВИДНОЙ
ЛИСТЬЯ
КНаропИа саЛЬатоШез ЬЬНит
1-Е1ЛЕА 1-ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в августе и высушенные листья
РЬаропНсит саАЬатоШез 0Л/Ш<±) 1Ц1п (зуп.: 1.еигеа
саПЬатоШез (\Л/Шс1) РС).
Содержание: не менее 0,1 % 20-гидроксиэкдизо-
на (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья паути¬
нисто-опушенные либо голые. Листья двух видов:
черешковые, длиной (8—60) см и шириной до 30 см,
глубоко перисто-раздельные, с более крупной конеч¬
ной долей и 5—6 парами боковых яйцевидных, за¬
остренных по краю зубчатых долей; либо листья без
черешка, перисто-лапчатые, с сетчатым жилковани¬
ем; с нижней стороны хорошо заметна центральная
жилка светло-зеленого цвета. Цвет листьев с обеих
сторон зеленый.
B. Микроскопия (2.8.23). При просматривании
листа (рисунок РБ0066.-1) с верхней стороны эпи¬
дермиса [О] видны клетки с извилистыми округлыми
стенками [Оа], а также устьица [йЬ] (2.8.3). Клетки
нижнего эпидермиса [С] многоугольные, с прямыми
изломанными стенками, более крупные, 8—12-гран¬
ные [СЬ]. Устьица на обеих сторонах листа окружены
4—6 клетками эпидермиса [Са, йЬ], на нижней сторо¬
не их больше (аномоцитный тип) (2.8.3). Волоски про¬
стые, многоклеточные [А], расположены по всей по¬
верхности листа, по краю, а также вдоль центральной
и боковых жилок [ВЬ]. В паренхиме листа [В] заметны
скопления многоугольных клеток [Ва], а также вмести¬
лища [Вс].
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1,0 г измельченного сы¬
рья (355) (2.9.12) помещают в колбу вместимостью
25 мл со шлифом, прибавляют 10 мл спирта (70 %,
об/об) Р и настаивают при периодическом перемеши¬
вании в течение суток. Фильтруют через бумажный
фильтр и доводят спиртом (70 %, об/об) Р до объема
10 мл.
Раствор сравнения. 1 мг 20-гидроксиэкдизона Р
растворяют в 10 мл спирта (70 %, об/об) Р.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: толуол Р — ацетон Р — эта¬
нол Р — раствор аммиака концентрированный Р
(100:140:32:9, об/об/об/об).
Наносимый объем пробы: по 5 мкл в виде полос.
Фронт подвижной фазы: не менее 14 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают реактивом
ванилина Р и нагревают при температуре 105 °С. Про¬
сматривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается основная зона серовато-темно¬
красного цвета (20-гидроксиэкдизон). На хроматограм¬
ме испытуемого раствора обнаруживается зона, соот¬
ветствующая по расположению и цвету зоне 20-гидрок-
г
1258
Гэсударственная фармакопея Республики Беларусь
I
Рисунок РБ0066.-1. Микроскопические признаки
листьев левзеи сафлоровидной
сиэкдизона на хроматограмме раствора сравнения. На
хроматограмме испытуемого раствора обнаруживают¬
ся также две или три зоны розового и несколько зон
коричневого или коричневато-серого цвета.
й. 3 мл испытуемого раствора, приготовленного
как указано в подлинности С, упаривают в пробирке
до сухого остатка и прибавляют 1 мл свежеприготов¬
ленного раствора 50 г/л ацетилхлорида Р в кислоте
уксусной ледяной Р. Через (15—20) мин появляется
коричневато-красное окрашивание. При прибавлении
3 мл раствора 50 г/л натрия гидроксида Р окраска
переходит в светло-желтую.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: листья, утратившие окраску, присущую
данному виду (побуревшие, почерневшие, выцветшие
и другие), — не более 15 %. Органические примеси:
не более 1 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 15,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 7,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 10,00 г измельченного
сырья (355) (2.9.12) помещают в колбу вместимостью
200 мл со шлифом, прибавляют 100 мл спирта (70 %,
об/об) Р и настаивают при периодическом перемеши¬
вании в течение суток. Фильтруют через бумажный
фильтр и доводят спиртом (70 %, об/об) Р до объема
100,0 мл.
Раствор сравнения. 5,0 мг 20-гидроксиэкдизо-
на Р растворяют в 50,0 мл спирта (70 %, об/об) Р.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 5 мм, заполненная силикагелем октадецилси-
лильным, деактивированным по отношению к ос¬
нованиям, для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: вода Р — ацетонитрил для
хроматографии Р (83:17, об/об);
- скорость подвижной фазы: 0,5 мл/мин;
- температура: 30 °С;
-спектрофотометрический детектор, длина
волны 247 нм;
- объем вводимой пробы: 10 мкл.
Содержание 20-гидроксиэкдизона в процентах
рассчитывают по формуле:
8т0Р
$0т-500 ’
где:
5 — площадь пика 20-гидроксиэкдизона на хро¬
матограмме испытуемого раствора;
50 — площадь пика 20-гидроксиэкдизона на хро¬
матограмме раствора сравнения;
т0 — масса навески 20-гидроксиэкдизона Р, мг;
т — масса навески испытуемого сырья, г;
Р — содержание 20-гидроксиэкдизона в реакти¬
ве, %.
07/2016:2428
(|*)ЛИМОННИКА плоды
8сЫзапс1гае сЫпепз/з 1гис1из
8СН18АЫОКА РРШ1Т
ОПРЕДЕЛЕНИЕ
Цельные, высушенные или обработанные паром
и высушенные, зрелые плоды ЗсЫзапдга сЫпепв/з
(Тигсг.) ВаШ.
Содержание: не менее 0,40 % схизандрина
(С24Н32О7; М.м. 432,5) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Ягоды имеют
более или менее сферическую форму и диаметр до
8 мм; внешняя поверхность красная, красновато-ко¬
ричневая или черноватая, иногда покрытая белова¬
тым инеем; околоплодник сильно высохший; имеются
1—2 почкообразных, желтовато-коричневых блестя¬
щих семени с тонкой семенной оболочкой.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Порошок красновато-
коричневого цвета. Просматривают под микроско¬
пом с использованием раствора хлоралгидрата Р.
Порошок обладает следующими диагностическими
признаками (рисунок 2428.-1): красновато-корич¬
невые фрагменты околоплодника, состоящие из
одного слоя тонкостенных клеток эпикарпия (вид с
поверхности [А]), покрытого бороздчатой кутикулой
[Аа], с прилегающими редкими клетками с эфирным
маслом [АЬ], кутикула, расположенная над клетками
с эфирным маслом не является бороздчатой; фраг¬
менты мезокарпия [С], состоящие из нескольких
слоев клеток яйцевидной формы, более или менее
уплощенных; фрагменты семенной кожуры [В, Е] с
внешним слоем, состоящим из многогранных скле-
реид с диаметром около (11—30) мкм (вид с поверх¬
ности [Ва]), или расположенным в виде частокола
(вид с поверхности [Еа]) с толстыми, одревесневев-
шими, мелкобороздчатыми стенками и небольшой
полостью с красновато-коричневым или чернова¬
тым содержимым с внутренними слоями семенной
кожуры, состоящими из склереид с желтоватыми
или слегка красноватыми стенками, расположенных
отдельно, небольшими группами или совместно с
внешними склереидами, диаметром около 80 мкм, с
одревесневевшими, утолщенными ямчатыми стен¬
ками с заметными бороздками и большой полостью
[ВЬ, ЕЬ]; фрагменты преимущественно бесцветного
Лимонника плоды
1259
эндосперма [Р], состоящие из многогранных кле¬
ток, содержащих капельки масла [йа] и алейроно¬
вые зерна. Исследуют под микроскопом, используя
50 % (об/об) раствор глицерина Р. В порошке обна¬
руживаются паренхиматозные клетки мезокарпия,
содержащие многочисленные небольшие, круглые
или в небольшой степени многогранные зерна крах¬
мала, и иногда отдельные круглые или в небольшой
степени многогранные зерна крахмала, простые
или объединенные в 2—5 зерен [Р]; часто наблю¬
дается ядро.
Рисунок 2428.-1. Микроскопические признаки
плодов лимонника
С. Просматривают хроматограммы, полученные,
как указано в испытании «ЗсЫзапдга зрЬепапНюга».
Результаты А: ниже приведена последователь¬
ность зон поглощения хроматограмм раствора срав¬
нения и испытуемого раствора. На хроматограмме ис¬
пытуемого раствора могут обнаруживаться и другие
слабые зоны поглощения.
Верх хроматографической пластинки
у-Схизандрин: зона
поглощения
Зона поглощения
(у-схизандрин)
Слабая зона поглощения
Схизандрин:зона
поглощения
Зона поглощения
(схизандрин)
Раствор сравнения
Испытуемый раствор
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора могут обнаруживаться и другие слабые
зоны.
Верх хроматографической пластинки
у-Схизандрин: зона
коричневого цвета
Зона коричневого цвета
(у-схизандрин)
Схизандрин: интенсивная
зона коричневато-зеленого
цвета
Интенсивная зона
коричневато-зеленого цвета
(схизандрин)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
ЗсЫзапс1га зрЬепапИтга. Тонкослойная хрома¬
тография (2.2.27).
Испытуемый раствор. К 2,5 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл метанола Р. Экс¬
трагируют при температуре 25 °С в ультразвуковой
бане в течение 5 мин и центрифугируют.
Раствор сравнения. 5 мг схизандрина Р и 5 мг
у-схизандрина Р растворяют в 5 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р254 Р ((2—10) мкм)].
Подвижная фаза: уксусная кислота Р — этила-
цетатР— толуол Р (2:22:46, об/об/об).
Наносимый объем пробы. 5 мкл [или 2 мкл], в
виде полос 10 мм [или 6 мм].
Фронт подвижной фазы: не менее 10 см [или
7см] от линии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 254 нм.
Проявление В: пластинку опрыскивают раство¬
ром 100 г/л кислоты серной Р в метаноле Р и нагре¬
вают при температуре 120 °С в течение 7 мин. Про¬
сматривают при дневном свете.
Результаты В: На хроматограмме испытуемого
раствора обнаруживаются зона схизандрина и зона
у-схизандрина; на хроматограмме в нижней трети не
должна обнаруживаться интенсивная зона фиолето¬
во-розового цвета.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,250 г измельченного
сырья (355) (2.9.12) помещают в коническую колбу
вместимостью 250 мл, прибавляют 90 мл метанола Р
и обрабатывают ультразвуком в течение 30 мин. По¬
лученный раствор фильтруют в мерную колбу, при¬
бавляют, промывая фильтр, 10 мл метанола Р и до¬
водят до объема 100,0 мл этим же растворителем.
Раствор сравнения. 5,0 мг ФСО схизандри¬
на растворяют в метаноле Р и доводят до объема
100,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным эндкепированным для хроматографии Р\
- температура: 25 °С;
- подвижная фаза:
- подвижная фаза А: вода Р — метанол Р
(35:65, об/об);
- подвижная фаза В: метанол Р;
1260
Гэсударственная фармакопея Республики Беларусь
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—10
100
0
10—16
100—>58
0 —> 42
16—26
58
42
- скорость подвижной фазы: 1 мл/мин;
- спектрофотометрический детектор, длина
волны 250 нм;
- объем вводимой пробы: 10 мкл.
Время удерживания: схизандрин — около 8 мин.
Пригодность хроматографической системы:
- число теоретических тарелок, не менее 5000
для пика схизандрина на хроматограмме раствора
сравнения.
Содержание схизандрина в процентах рассчиты¬
вают по формуле:
А-пур
А • Г7?1
где:
А — площадь пика схизандрина на хроматограм¬
ме испытуемого раствора;
А2 — площадь пика схизандрина на хроматограм¬
ме раствора сравнения;
/л1 — масса навески лекарственного раститель¬
ного сырья, использованной для приготовления испы¬
туемого раствора, г;
т2 — масса навески ФСО схизандрина, использо¬
ванной для приготовления раствора сравнения, г;
р — содержание схизандрина в ФСО схизандри¬
на, %.
07/2016:РБ0067
ЛИМОННИКА СЕМЕНА
БсЫзапдгае сЫпепз1сИз зетеп
8СН11АЫОКА 5ЕЕО*
ОПРЕДЕЛЕНИЕ
Зрелые, освобожденные от околоплодников и
высушенные семена деревянистой лианы ЗсЫзапс1га
с/1теп818 (Тигсг.) ВаШ.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Семена округло¬
почковидной формы, на вогнутой стороне с замет¬
ным темно-серым рубчиком, расположенным поперек
семени. Длина от 3 мм до 5 мм, ширина от 2 мм до
4,5 мм, толщина от 1,5 мм до 2,5 мм. Поверхность
гладкая, блестящая, желтовато-коричневого цвета.
Семена состоят из твердой хрупкой кожуры и плотно¬
го ядра, которое у недоразвитых семян может отсут¬
ствовать. Кожура легко ломается и свободно отстает
от ядра. Ядро подковообразной формы, серовато¬
желтое, один конец конусовидно заостренный, другой
округлый. На выпуклой стороне ядра семени прохо¬
дит светло-коричневая бороздка. Основную массу
ядра семени составляет эндосперм. В заостренном
конце верхушки (в эндосперме) лежит небольшой за¬
родыш, заметный при увеличении. Запах при растира¬
нии сильный, специфический.
B. Микроскопия (2.8.23). На поперечном сре¬
зе семени видна семенная кожура, состоящая из
нескольких слоев: эпидермальный слой представ¬
лен крупными радиально вытянутыми клетками,
с утолщенными одревесневшими темно-желтыми
оболочками, пронизанными порами. Под ним рас¬
положен склеренхимный слой, состоящий из 4—6
рядов сильно одревесневших каменистых клеток.
Далее лежит слой спавшихся клеток, а за ним один
ряд очень крупных тонкостенных четырехугольных
клеток, содержащих маслянистые включения в
виде капель зеленовато-желтого цвета. Самый вну¬
тренний слой семенной кожуры — бесструктурная
спавшаяся тонкостенная ткань. Эндосперм семени
состоит из небольших многоугольных клеток, со¬
держащих капли жирного масла и мелкие алейро¬
новые зерна.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (мякоть плода,
веточки) — не более 3 %; поврежденные семена — не
более 5 %. Органические примеси: не более 1 %. Ми¬
неральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 3,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
07/2016:0957
ЛИПЫ ЦВЕТКИ
ТШае Лоз
ЫМЕРЬОМЕК
ОПРЕДЕЛЕНИЕ
Собранные во время цветения и высушенные
соцветия деревьев ТШа согда!а МШег, ТШа р1а1урМу11о8
8сор. и ТШа х Vи^дап8 Неупе или смеси этих видов.
Содержание: не менее 0,5 % суммы флавонои-
дов в пересчете на кверцетин (С15Н10О7; М.м. 302,2)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Соцветия щит¬
ковидные, состоят из 5—15 (Т. согс!а(а) или 2—9
(Т. рШурНуНоз) цветков на удлиненных цветоножках,
сидящих на общем цветоносе, сросшемся в нижней
части с главной жилкой прицветного листа. Цветки
правильные, диаметром от 1 см до 1,5 см. Чашечка из
пяти продолготовато-яйцевидных чашелистиков, густо
опушенных по краю и с внутренней стороны. Венчик
из пяти свободных яйцевидных лепестков, длиннее
чашечки. Тычинки многочисленные, с двумя желты¬
ми пыльниками на длинных нитях, сросшихся в пять
пучков. Пестик один с верхней шаровидной завязью,
густо покрытой пушистыми волосками. Встречаются
цветочные бутоны и незрелые плоды — шаровидные
сильно опушенные орешки до 2 мм в диаметре. При-
цветный лист пленчатый, с густой сетью жилок, дли¬
ной до 6 см и шириной до 1,5 см, продолговато-эллип¬
тической формы с притупленной верхушкой, в нижней
половине сросшийся по главной жилке с цветоносом.
Цвет лепестков беловато-желтый, чашелистиков —
зеленовато- или желтовато-серый, прицветных ли¬
стьев — светло-желтый или зеленовато-желтый. За¬
пах слабый, ароматный.
Липы цветки
1261
В. Микроскопия (2.8.23). При просматривании
прицветного листа с поверхности видны слегка из¬
вилистые клетки эпидермиса с обеих сторон листа.
Устьица только на нижней стороне, овальные, с 4—6
околоустьичными клетками (аномоцитный тип) (2.8.3).
Волоски встречаются преимущественно в средней
части прицветного листа, вблизи места срастания
его с цветоносом. Волоски двух типов: головчатые —
с многоклеточной овальной головкой на короткой
1—3-клеточной ножке и звездчато-лучистые, состоя¬
щие из 3—7 длинных извилистых клеток, сросшихся
основаниями. Мезофилл очень рыхлый, типа аэрен¬
химы, с друзами, реже призматическими кристаллами
оксалата кальция, особенно многочисленными вбли¬
зи жилок. Лепестки и чашелистики характеризуются
наличием друз оксалата кальция и таких же волосков,
как и на прицветном листе. Кроме того, у основания
чашелистиков, с внутренней стороны, расположены
длинные прямые волоски, состоящие из двух парал¬
лельных клеток, сросшихся основаниями, на лепест¬
ках — вильчатые волоски из двух извилистых клеток,
сросшихся основанием. В лепестках хорошо видны
крупные вместилища со слизью.
@> С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р и
нагревают в водяной бане при температуре 65 °С в
течение 5 мин. Охлаждают и фильтруют.
Раствор сравнения. 2,0 мг кислоты кофей¬
ной Р, 5 мг гиперозида Р и 5 мг рутина Р растворяют
в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метилэтилкетон Р — этилаце-
тат Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: горячую пластинку опрыскивают
раствором 10 г/л дифенилборной кислоты аминоэ¬
тилового эфира Р в метаноле Р и затем раствором
50 г/л макрогола 400 Р в метаноле Р. Пластинку вы¬
сушивают на воздухе в течение 30 мин и просматри¬
вают в ультрафиолетовом свете при длине волны
365 нм.
Результаты: на хроматограмме раствора срав¬
нения обнаруживается флуоресцирующие зоны (в
порядке увеличения значения /?г): от желтовато-оран¬
жевого до коричневато-оранжевого цвета (рутин и
гиперозид), зеленовато-синего цвета (кофейная кис¬
лота). На хроматограмме испытуемого раствора об¬
наруживается основная флуоресцирующая зона от ко¬
ричневато-желтого до оранжевого цвета, расположен¬
ная над зоной гиперозида на хроматограмме раствора
сравнения (при просматривании при дневном свете
она также обнаруживается как основная зона); флуо¬
ресцирующая зона коричневато-желтого цвета на
уровне зоны рутина на хроматограмме раствора срав¬
нения; ниже этой зоны могут обнаруживаться две флу¬
оресцирующие зоны желтого цвета; обнаруживаются
флуоресцирующие зоны оранжевого и желтого цвета
между зонами рутина и гиперозида на хроматограмме
раствора сравнения; до пяти флуоресцирующих зон
от желтого до оранжевого цвета между зонами гипе¬
розида и кофейной кислоты на хроматограмме раст¬
вора сравнения; обнаруживается флуоресцирующая
зона синего цвета непосредственно под зоной кофей¬
ной кислоты на хроматограмме раствора сравнения.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: соцветия с прицветниками и отдельные
прицветники, поврежденные вредителями и поражен¬
ные ржавчиной, — не более 2 %; побуревшие и потем¬
невшие части соцветия — не более 4 %; другие части
растения (листья и побеги) — не более 1 %; соцветия,
полностью отцветшие, с плодами — не более 2 %; из¬
мельченные частицы, проходящие сквозь сито (2000)
(2.9.72) , — не более 3%; осыпь отдельных цветков
или соцветий без прицветников — не более 15 %. Ор¬
ганические примеси: не более 0,3 %. Минеральные
примеси: не более 0,1 %.
Не допускается присутствие соцветий с прицвет¬
ником, у которого на верхней стороне имеются звезд¬
чатые волоски с 5—8 лучами, и цветками с явным
двойным венчиком, образованным превращением
пяти тычинок в лепестко-подобные стаминодии, и с
не лопастным и не зубчатым пестиком. Шестичлен¬
ные цветки обнаруживаются редко (77//а атепсапа Ь.,
7/7/а 1отеп1оза МоепсИ).
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.72) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 8,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Исходный раствор. 1,000 г измельченного сы¬
рья (355) (2.9.72) помещают в круглодонную колбу
со шлифом, прибавляют 20 мл ацетона Р, 2 мл кис¬
лоты хлористоводородной Р1, 1 мл раствора 5 г/л
гексаметилентетрамина Р и нагревают с обратным
холодильником в водяной бане в течение 30 мин. Ох¬
лаждают, процеживают через ватный тампон в мерную
колбу вместимостью 100 мл. Ватный тампон вместе
с частицами сырья помещают в ту же круглодонную
колбу, прибавляют 20 мл ацетона Р и нагревают с
обратным холодильником в водяной бане в течение
10 мин. Охлаждают, процеживают в ту же мерную кол¬
бу вместимостью 100 мл. Ватный тампон вместе с ча¬
стицами сырья помещают в ту же круглодонную колбу,
повторяют экстракцию и процеживают в ту же мерную
колбу. Доводят объем фильтрата ацетоном Р до объ¬
ема 100,0 мл. 20,0 мл полученного раствора помеща¬
ют в делительную воронку, прибавляют 20 мл воды Р
и встряхивают с 15 мл этилацетата Р. Повторяют
экстракцию еще трижды с этим же растворителем пор¬
циями по 10мл. Этилацетатные слои объединяют и
промывают дважды водой Р порциями по 40 мл. Эти-
лацетатное извлечение фильтруют через бумажный
фильтр с натрия сульфатом безводным Р в мерную
колбу вместимостью 50 мл. Доводят объем получен¬
ного раствора этилацетатом Р до объема 50,0 мл.
Испытуемый раствор. К 10,0 мл исходного раст¬
вора прибавляют 2 мл реактива алюминия хлорида Р
и доводят 5 % (об/об) раствором кислоты уксусной
ледяной Р в метаноле Р до объема 25,0 мл.
Компенсационный раствор. 10,0 мл исходного
раствора разводят 5 % (об/об) раствором кислоты
уксусной ледяной Р в метаноле Р до объема 25,0 мл.
Через 45 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 425 нм.
1262
Государственная фармакопея Республики Беларусь
Содержание флавоноидов в пересчете на квер¬
цетин в процентах рассчитывают по формуле:
Д-625
/л-714 ’
где:
714 — удельный показатель поглощения кверце¬
тина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:0095
ЛЬНА СЕМЕНА
Им зетеп
Ш8ЕЕО
ОПРЕДЕЛЕНИЕ
Зрелые и высушенные семена травянистого рас¬
тения Ипит изНаИз51тит I.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Семена
сплюснутые, удлиненной яйцевидной формы, дли¬
ной от 4 мм до 6 мм, шириной от 2 мм до 3 мм и тол¬
щиной от 1,5 мм до 2 мм; округлые с одного конца и
заостренные с другого, с ясно заметным семенным
рубчиком. Семенная кожура от темно-красновато¬
коричневого до желтого цвета, гладкая и блестящая,
при увеличении — мелко-выемчатая. Под кожурой
располагается узкий, беловатый эндосперм и заро¬
дыш, состоящий из двух больших плоских желтова¬
тых и маслянистых семядолей; корешок направлен к
семенному рубчику.
(§) В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Порошок маслянистый на
ощупь. Просматривают под микроскопом с использо¬
ванием раствора хлоралгидрата Р. В порошке об¬
наруживаются следующие диагностические признаки
(рисунок 0095.-1): фрагменты внешней семенной ко¬
журы [А, В] с клетками многоугольными при просма¬
тривании с поверхности [Аа] или узкими при просма¬
тривании на поперечном срезе [Ва], заполненными
слизью [ВЬ]; фрагменты колленхиматозно утолщенно¬
го суб-эпидермального слоя (вид на поперечном сре¬
зе [Вс] или с поверхности [АЬ]) с округлыми клетками
с межклеточным пространством треугольной формы,
слой часто прикрепленн к склеренхиматозному слою,
состоящему из вытянутых клеток с утолщенными и
ямчатыми стенками [Са], некоторые с сильно утол¬
щенными и ямчатыми клетками [6]; фрагменты при
просматривании с поверхности [С], состоящие из ги¬
алинового слоя с тонкостенными клетками [СЬ], ча¬
сто остающегося прикрепленным к слою вытянутых
склереид и пересекающего их по примерно прямыми
углами [Са]; фрагменты внутренней семенной кожу¬
ры при просматривании с поверхности [О], состоящих
из средне утолщенных многоугольных клеток, запол¬
ненных коричнево-оранжевым пигментом; маленькие
многогранные массы пигмента [Н]; многочисленные
фрагменты паренхимы семенной кожуры с крупны¬
ми слегка и правильно утолщенными клетками при
просматривании с поверхности у, Ц; паренхима эн¬
досперма [К] и семядолей [Е], содержащая алейроно¬
вые зерна и капельки масла; очень многочисленные
отдельные капельки масла [Р].
ВЬ
Рисунок 0095.-1. Микроскопические признаки
семян льна
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: части коробочек, плодоножек, битых се¬
мян — не более 1 %. Органические примеси: не более
2 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0%. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 6,0 %.
® Коэффициент набухания (2.8.4). Не менее 4.
07/2016:1233
ЛЮБИСТКА КОРНИ
1-еУ18Иа гасИх
^ОVА6Е КООГ
ОПРЕДЕЛЕНИЕ
Цельные или измельченные высушенные корне¬
вища и корни Ьеу‘1зИсит оЯ1ста1е Коей.
Содержание:
- цельное сырье: не менее 4,0 мл/кг эфирного
масла (в пересчете на сухое сырье);
- измельченное сырье: не менее 3,0 мл/кг эфир¬
ного масла (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корневище и
крупные корни часто продольно расщеплены. Кор¬
невище короткое, диаметром до 5 см, светлого серо¬
вато-коричневого или желтовато-коричневого цвета,
простое или с несколькими бугорками; корни мало
Любистка корни
1263
разветвленные такого же цвета, что и корневище;
толщиной обычно до 1,5 см и длиной до около 25 см;
излом обычно гладкий с очень широкой желтовато¬
белой корой и узкой коричневато-желтой древесиной.
B. Микроскопия (2.8.23). При просматривании
измельченного сырья (355) (2.9.12) (коричневато-жел¬
того цвета) с использованием раствора хлоралгидра¬
та Р видны: клетки пробки, многоугольные или шаро¬
образные при просматривании с поверхности с содер¬
жимым коричневого цвета; многочисленные клетки
паренхимы, главным образом тонкостенные и шароо¬
бразной формы, некоторые с более толстыми стенка¬
ми; группы небольших, сетчато-утолщенных сосудов,
расположенные в мелкоклеточной, неодревесневшей
паренхиме; фрагменты более крупных сосудов с сет¬
чатым утолщением, диаметром до 125 мкм; фрагмен¬
ты секреторных каналов шириной до 180 мкм. При
просматривании измельченного сырья с использова¬
нием 50 % (об/об) раствора глицерина Р видны: зер¬
на крахмала, простые, шарообразной или яйцевидной
формы, размером до около 12 мкм и многочисленные
более крупные сложные зерна, многие с несколькими
компонентами.
C. Тонкослойная хроматография (2.2.27).
Просматривают хроматограммы, полученные как
указано в испытании «Виды АпдеНса и ЫдизИсит».
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испыту¬
емого раствора. На хроматограмме испытуемого раст¬
вора могут обнаруживаться и другие слабые флуо¬
ресцирующие зоны.
Верх хроматографической пластинки
(2)-Лигустилид:
флуоресцирующая зона
голубовато-белого цвета
Флуоресцирующая зона
голубовато-белого цвета
Остол: флуоресцирующая
зона синего цвета
Императорин:
флуоресцирующая зона
беловатого цвета
Слабая флуоресцирующая
зона беловатого цвета
Слабая флуоресцирующая
зона беловатого цвета
Раствор сравнения
Испытуемый раствор
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые зоны
поглощения.
Верх хроматографической пластинки
(2)-Лигустилид:
флуоресцирующая зона
синеватого цвета
Флуоресцирующая зона
синеватого цвета
Остол: зона поглощения
Императорин: зона
поглощения
Слабая зона поглощения
Раствор сравнения
Испытуемый раствор
Результаты С: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора могут обнаруживаться и другие слабые
зоны.
Верх хроматографической пластинки
(2)-Лигустилид: зона серого
цвета
2 отчетливые зоны
красноватого цвета
Зона серого цвета
Остол: зона фиолетового
цвета
Императорин: зона серого
цвета
2 зоны пурпурного цвета
Отчетливая зона
коричневого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Виды АпдеНса и ЫдизИсит. Тонкослойная хро¬
матография (2.2.27).
Испытуемый раствор. К1 г свежеизмельченного
сырья (355) (2.9.12) прибавляют 4 мл гептана Р и об¬
рабатывают ультразвуком в течение 5 мин. Смесь цен¬
трифугируют и используют надосадочную жидкость.
Раствор сравнения. 1 мг императорина Р, 1 мг
(1)-лигустилида Р и 1 мг остола Р растворяют в
10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((2—Щ мкм).
Подвижная фаза: кислота уксусная ледяная Р —
этилацетат Р — толуол Р (1:10:90, об/об/об).
Наносимый объем пробы: по 4 мкл в виде полос
8 мм.
Фронт подвижной фазы: не менее 6 см от линии
старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты А: на хроматограмме испытуемого
раствора не должна обнаруживаться флуоресциру¬
ющая зона синего цвета, расположенная непосред¬
ственно под или над зоной императорина на хромато¬
грамме раствора сравнения.
Проявление В: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты В: на хроматограмме испытуемого
раствора не должна обнаруживаться зона, соответ¬
ствующая по расположению императорину или распо¬
ложенная непосредственно под зоной императорина
на хроматограмме раствора сравнения.
Проявление С: пластинку обрабатывают раство¬
ром 20 мл кислоты серной Р в 180 мл охлажденного
на льду метанола Р\ нагревают при температуре от
100 °С до 105 °С в течение 5 мин и просматривают
при дневном свете.
Результаты С: на хроматограмме испытуемого
раствора не должна обнаруживаться зона пурпурного
цвета между 2 зонами красноватого цвета в верхней
части хроматограммы и зоной (2)-лигустилида на хро¬
матограмме раствора сравнения; на хроматограмме
испытуемого раствора не должна обнаруживаться
зона пурпурного цвета между зонами (7)-лигустилида
и остола на хроматограмме раствора сравнения.
Допустимые примеси (2.8.2). Не более 3 %.
Определение проводят из 50 г испытуемого сырья.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
1264
Гэсударственная фармакопея Республики Беларусь
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в кислоте хлористово¬
дородной (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 40,0 г сырья,
измельченного (500) (2.9.12) непосредственно перед
определением, помещают в колбу вместимостью
2 л, прибавляют 10 капель масла вазелинового Р,
500 мл воды Р в качестве жидкости для перегон¬
ки. В градуированную трубку помещают 0,50 мл
ксилола Р. Перегонку проводят со скоростью
(2—3) мл/мин в течение 4 ч.
07/2016: РБ0068
МАКЛЕЙИ ЛИСТЬЯ
Мас1еауае Шит
МАС1.ЕАУА 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в период от бутонизации до цвете¬
ния и высушенные листья многолетних травянистых
растений Мас1еауа согда1а (\Л/Шс1.) Н. Вг. и Мас1еауа
тюгосагра (Мах1т.) Реббе.
Содержание: не менее 0,5 % суммы бисульфатов
сангвинарина и хелеритрина в пересчете на сангвири-
трин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь цельных
или частично измельченных листьев. Листья сердце¬
видной формы, 5—7-раздельные, длиной до 25 см,
сверху голые, от коричневато-зеленого до коричнева¬
то-желтого или серовато-зеленого цвета, снизу густо
опушенные, серого или желтовато-серого цвета. Ку¬
сочки черешков листьев длиной до 12 см и шириной
до 1 см, неправильно цилиндрической, у основания —
подковообразной формы, иногда сплюснутые, от жел¬
товато-серого до коричневато-серого цвета, иногда с
восковым налетом.
B. Микроскопия (2.8.23). При просматривании
листа или фрагментов листовой пластинки с поверх¬
ности видно, что клетки верхнего эпидермиса имеют
многоугольную форму и почти прямые стенки; клетки
нижнего эпидермиса со слабо извилистыми стенками.
Устьица многочисленные, овальной формы, погружен¬
ные с 5—6 околоустьичными клетками (аномоцитный
тип) (2.8.3), расположены только на нижней стороне
листа. Волоски простые, многоклеточные, гладкие,
прямые или слабоизогнутые, встречаются на нижней
стороне листа, чаще по жилкам. Изредка встречаются
многоклеточные волоски с основанием из двух рядов
клеток. В мезофилле листа вдоль проводящих пучков
видны млечники с зернистым содержанием оранжево¬
коричневого цвета.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 г измельченного сырья
(500) (2.9.12) помещают в коническую колбу вмести¬
мостью 50 мл, смачивают 1 мл раствора аммиака
разведенного Р1, тщательно перемешивают, прибав¬
ляют 15 мл хлороформа Р, настаивают при перио¬
дическом взбалтывании в закрытой колбе в течение
10 мин и фильтруют.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: толуол Р — 96% спирт Р —
раствор аммиака концентрированный Р (10:2:0,05,
об/об/об).
Наносимый объем пробы: 20 мкл в виде полосы.
Фронт подвижной фазы: не менее 15 см от
линии старта.
Высушивание: на воздухе.
Проявление: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются зоны оранжевого цвета
с Рг около 0,8 (сангвинарин) и желтого цвета с
около 0,7 (хелеритрин); могут обнаруживаться и
другие слабые зоны.
Э. 5 мл испытуемого раствора, приготовленно¬
го как указано в подлинности С, помещают в про¬
бирку со шлифом, прибавляют 1 мл 10% (об/об)
раствора кислоты серной Р, закрывают пробкой и
взбалтывают в течение 3 мин. При стоянии в во¬
дном слое выпадает осадок оранжевого цвета (би¬
сульфаты сангвинарина и хелеритрина).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (стебли, цветки,
плоды) — не более 5 %. Органические примеси: не
более 1 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 13,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г измельченного сырья (500) (2.9.12)
помещают в коническую плоскодонную колбу со
шлифом вместимостью 50 мл, прибавляют 0,5 мл
раствора аммиака концентрированного Р, тща¬
тельно перемешивают стеклянной палочкой до по¬
лучения однородной увлажненной массы. Закры¬
вают колбу пробкой и выдерживают при комнатной
температуре в течение 30 мин. Затем в колбу при¬
бавляют 25 мл хлороформа Р, закрывают пробкой
и выдерживают при комнатной температуре в те¬
чение 15 ч. По истечении указанного промежутка
времени содержимое колбы перемешивают в тече¬
ние 15 мин и фильтруют через стеклянный фильтр
(ПОР 100) в вакууме в круглодонную колбу вмести¬
мостью 50 мл. Коническую колбу и осадок на филь¬
тре промывают хлороформом Р дважды порциями
по 10 мл, объединяя с фильтратом в круглодонной
колбе. Растворитель отгоняют досуха в вакууме на
водяной бане при температуре 50 °С. Остаток раст¬
воряют в 1 мл хлороформа Р, прибавляют 25 мл
раствора 50 г/л кислоты уксусной ледяной Р, за¬
крывают колбу пробкой и интенсивно встряхивают
в течение (1—2) мин. Полученную эмульсию пере¬
носят в пластиковые центрифужные пробирки с
пробками и центрифугируют при 5000 д в течение
(10—15) мин. 5,0 мл надосадочной жидкости дово¬
дят раствором 50 г/л кислоты уксусной ледяной Р
до объема 25,0 мл.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при 452 нм, используя раствор
50 г/л кислоты уксусной ледяной Р в качестве ком¬
пенсационного раствора.
Малины листья
1265
Содержание суммы бисульфатов сангвинари-
на и хелеритрина в пересчете на сангвиритрин в
процентах рассчитывают по формуле:
А-125
т-106 '
где:
106 — удельный показатель поглощения сангви-
ритрина;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0007
МАЛИНЫ листья
КиЫ Мае/1ЬНит
КА8РВЕНКУ 1.ЕАР
ОПРЕДЕЛЕНИЕ
Цельные или измельченные высушенные листья
кустарника РиЬиз Шаеиз 1_.
Содержание: не менее 3,0 % дубильных веществ
в пересчете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
частично изломанные непарноперистые листья,
тройчатые или с 5—7 листочками. Боковые листоч¬
ки супротивные, почти сидячие, конечный листочек
более крупный, черешковый. Листочки продолго¬
вато-яйцевидной формы, с округлым или сердце¬
видным основанием, заостренной верхушкой и не¬
равномерно-пильчатым краем, сверху голые, снизу
густо опушенные, иногда со щетинками у основа¬
ния главной жилки. Жилкование перисто-краебеж-
ное с выступающей центральной жилкой и отходя¬
щими от нее под одним углом крупными жилками
первого порядка. Длина листа (9—12) см, ширина
(1,5—9) см. Рахисы длиной (2—7) см, цилиндриче¬
ские, с небольшими загнутыми щетинками, продоль¬
ной бороздкой на верхней стороне рахиса и парными
приросшими прилистниками при основании. Цвет ли¬
стьев с верхней стороны зеленый, с нижней — серова¬
то-белый, рахисов — светло-зеленый. Запах слабый.
B. Микроскопия (2.8.23). При рассмотре¬
нии препаратов листа под микроскопом (рисунок
РБ0007.1) видны извилистостенные иногда с четко¬
видным утолщением клетки верхнего эпидермиса [А];
клетки нижнего эпидермиса тонкостенные, с более
извилистыми стенками и гладкой кутикулой [В]. На
нижней стороне листа видны основания волосков [Ва]
и многочисленные устьица [ВЬ], которые окружены
5—7 клетками эпидермиса (аномоцитный тип)
(2.8.3). Волоски простые [Аа] и головчатые (реже).
Простые волоски двух типов: на нижней стороне ли¬
ста многочисленные волоски одноклеточные, тол¬
стостенные, тупоконечные, нелигнифицированные,
с гладким основанием, извитые и изогнутые, обра¬
зующие войлочное опушение [С]; на верхней сторо¬
не листа часто встречаются одноклеточные, кониче¬
ские, толстостенные, лигнифицированные волоски
до 500 мкм длины. По главной жилке и крупным
жилкам изредка встречаются головчатые волоски с
многоклеточной двухрядной ножкой и многоклеточ¬
ной головкой. Мезофилл возле главной жилки со¬
держит друзы кальция оксалата ((30—45) мкм) [АЬ].
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл спирта (95 %,
об/об) Р, нагревают с обратным холодильником на во¬
дяной бане в течение (10—15) мин, охлаждают, филь¬
труют и выпаривают до объема 2 мл.
Раствор сравнения. 2,5 мг рутина Р раство¬
ряют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силика¬
геля 6Р254 Р.
Подвижная фаза: этилацетат Р — муравьи¬
ная кислота безводная Р — уксусная кислота ле¬
дяная Р—вода Р (100:11:11:27, об/об/об/об).
Рисунок РБ0007.-1. Микроскопические признаки
листьев малины
1266
Гэсударственная фармакопея Республики Беларусь
Наносимый объем пробы: по 10 мкл в виде по¬
лос длиной 20 мм.
Фронт подвижной фазы: не менее 13 см от
линии старта.
Высушивание: на воздухе в течение 30 мин.
Проявление: пластинку опрыскивают раствором
20 г/л алюминия хлорида Рв96% спирте Р, высуши¬
вают в сушильном шкафу при температуре 105 °С в
течение (2—3) мин и просматривают в ультрафиоле¬
товом свете при длине волны 365 нм.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются флуоресцирующие зоны
желто-зеленого цвета со значениями около 1,6,1,5,
1,4, 0,85 и 0,8 относительно зоны рутина на хромато¬
грамме раствора сравнения. Допускается наличие
слабых флуоресцирующих зон голубого цвета.
й. К 0,1 г измельченного сырья (500) (2.9.12) при¬
бавляют 10 мл воды Р, кипятят в течение (2—3) мин,
охлаждают и фильтруют. К 1 мл фильтрата прибавля¬
ют 2—3 капли свежеприготовленного раствора 10 г/л
железа (III) аммония сульфата Р. Появляются черно¬
синее окрашивание и осадок.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (цветоносные
побеги, соцветия, недозрелые плоды) — не более
3,0 %; пожелтевшие и потемневшие листья — не бо¬
лее 4,0 %. Органические примеси: не более 2 %. Ми¬
неральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2.000 г измельченного сырья (180) (2.9.12) поме¬
щают в круглодонную колбу со шлифом вместимостью
250 мл, прибавляют 150 мл воды Р, нагревают на во¬
дяной бане в течение 30 мин, охлаждают под проточ¬
ной водой и количественно переносят в мерную колбу
вместимостью 250 мл. Ополаскивают круглодонную
колбу и сливают промывные воды в мерную колбу, по¬
сле чего доводят водой Р до объема 250,0 мл. Дают
осесть твердым частичкам и фильтруют жидкость че¬
рез бумажный фильтр диаметром 125 мм, отбрасывая
первые 50 мл фильтрата.
25.0 мл фильтрата помещают в коническую кол¬
бу вместимостью 750 мл, прибавляют 500 мл воды Р,
25 мл раствора индигокармина Р в кислоте серной Р
(1 г индигокармина Р растворяют в 25 мл кислоты
серной Р, затем прибавляют еще 25 мл кислоты сер¬
ной Р, осторожно доводят водой Р до объемаЮОО мл
и перемешивают) и титруют при постоянном переме¬
шивании 0,02 М раствором калия перманганата до
золотисто-желтого окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07/2016:РБ0069
МАЛИНЫ ПЛОДЫ
НиЫ Мае/ Тгис1из
КА8РВЕППУ РКШТ*
ОПРЕДЕЛЕНИЕ
Собранные в период созревания, освобожден¬
ные от цветоножек и конусовидного цветоложа, высу¬
шенные плоды РиЬиз Шаеиз 1_.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Плоды сбор¬
ные — сложные костянки округлой или конусовидной
формы, состоящие из большого числа (около 30—60)
отдельно сросшихся между собой костянок. Они обра¬
зуют полый конус с округлой верхушкой диаметром от
7,5 мм до 12 мм. Отдельные костянки мелкие, смор¬
щенные, шаровидные или эллипсовидные, опушен¬
ные, внутри с косточкой, имеющей ямчатую поверх¬
ность. Цвет плодов с поверхности от серовато-крас¬
ного до коричневато-красного, мякоти — розоватый,
косточек — темно-желтый. Запах специфический,
приятный.
B. Микроскопия (2.8.23). При просматривании
плода-костянки с поверхности видны многоугольные
клетки эпидермиса, имеющие очень тонкие стенки.
Железистые волоски встречаются по всей поверх¬
ности. Они имеют короткую одноклеточную ножку и
овальную двуклеточную, реже шаровидную однокле¬
точную головку, содержимое которой окрашивается
раствором Судана IIIР в оранжевый цвет. Многочис¬
ленные простые волоски одноклеточные, очень тон¬
костенные, прикреплены к маленькой круглой клетке
кожицы. Встречаются цельные, чаще обломанные
пестики с рыльцем. Клетки паренхимы мякоти круп¬
ные, тонкостенные, содержат мелкие друзы оксалата
кальция. Механическая ткань околоплодника костян¬
ки состоит из каменистых клеток, располагающихся
пластами.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие плоды — не более 8 %;
плоды, слипшиеся в комки, — не более 4 %; плоды
с неотделенными цветоножками и цветоложами — не
более 2 %; другие части растения (листья и стебли) —
не более 0,5 %; измельченные частицы плодов, про¬
ходящие сквозь сито (1400) (2.9.12), — не более 4 %.
Органические примеси: не более 0,5 %. Минеральные
примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 15,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 3,5 %.
07/2016:РБ0070
МАРЕНЫ КОРНЕВИЩА И КОРНИ
НиЫае гШгота е^ гасНх
АТАООЕЯ КООГ*
ОПРЕДЕЛЕНИЕ
Собранные весной в начале вегетации или осе¬
нью в период плодоношения, тщательно очищенные
Марены корневища и корни
1267
от земли и высушенные корневища и корни много¬
летних травянистых растений РиЫа Ипс1огит 1_. или
РиЫа /Ъепса (Р15И. ех ОС). С. КосИ.
Содержание: не менее 3,0 % связанных произво¬
дных антрацена (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корневища и
корни продольно-морщинистые, цилиндрические,
различной длины, толщиной от 2 мм до 18 мм,
обычно с отслаивающейся шелушащейся проб¬
кой. У корневищ в центре обычно имеется полость.
Цвет корневищ и корней снаружи красновато-ко¬
ричневый, на изломе видна красновато-коричне¬
вая кора и оранжево-красная древесина.
B. Микроскопия (2.8.23). На поперечном сре¬
зе корня (или корневища) видно, что пробка со¬
стоит из клеток с очень тонкими оболочками. В не¬
которых клетках корковой паренхимы содержатся
рафиды оксалата кальция. Линия камбия узкая.
Древесина нелучистая. Сосуды древесины распо¬
ложены группами, клетки древесной паренхимы —
радиальными рядами. Все элементы древесины
одревесневшие. В корневище центральная часть
занята крупными клетками сердцевины с утолщен¬
ными пористыми стенками; встречаются рафиды
оксалата кальция.
C. К 50 мг измельченного сырья (180) (2.9.12)
прибавляют 25 мл кислоты хлористоводородной
разведенной Р и нагревают смесь на водяной бане
в течение 15 мин. Охлаждают, встряхивают с 20 мл
эфира Р и отбрасывают водный слой. Эфирный
слой встряхивают с 10 мл раствора аммиака раз¬
веденного Р1. Водный слой приобретает цвет от
красного до фиолетового.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: другие части растения (стебли,
листья и другие) — не более 1,5 %. Органические
примеси: не более 1 %. Минеральные примеси: не
более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0%. 2,000 г измельченного сырья
(2000) (2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Построение градуировочного графика. 50,0 г
кобальта хлорида Р, высушенного в эксикаторе
до постоянной массы, растворяют в 250 мл воды Р,
прибавляют 1 мл хлористоводородной кислоты Р
и доводят водой Р до объема 500,0 мл. Полученный
раствор является исходным (№ 10). Из полученно¬
го раствора готовят серию разведенных растворов
(№ 1, 2, 3, 4, 5, 6, 7, 8 и 9), содержащих кобальта
хлорида (СоС12-6Н20) соответственно 0,010; 0,020;
0,030; 0,040; 0,050; 0,060; 0,070; 0,080 и 0,090 г в
1 мл, и измеряют оптические плотности (2.2.25) по¬
лученных растворов при 530 нм, используя воду Р
в качестве компенсационного раствора. Для по¬
строения градуировочного графика на оси абсцисс
откладывают концентрацию раствора, а на оси ор¬
динат — их оптическую плотность. При этом кон¬
центрации растворов кобальта хлорида выражают
в соответствующих концентрациях производных ан¬
трацена, пользуясь таблицей РБ0070.-1.
Таблица РБ0070.-1.
№
п/п
Концентрация кобальта
хлорида (СоС12-6Н20),
г/мл
Концентрация произво¬
дных антрацена, мг/мл
1
0,010
0,0027
2
0,020
0,0060
3
0,030
0,0088
4
0,040
0,0117
5
0,050
0,0146
6
0,060
0,0176
7
0,070
0,0204
8
0,080
0,0233
9
0,090
0,0260
10
0,100
0,0281
Определение суммы производных антрацена.
0,100 г измельченного сырья (250) (2.9.12) по¬
мещают в колбу вместимостью 200 мл, прибавляют
7,5 мл кислоты уксусной ледяной Р, 1 мл кислоты
хлористоводородной Р и нагревают с обратным хо¬
лодильником в водяной бане в течение 15 мин при
периодическом встряхивании вращательным дви¬
жением, смывая появляющийся на стенках колбы
осадок. Охлаждают, прибавляют 30 мл эфира Р и
кипятят с обратным холодильником на водяной бане
при температуре 35 °С в течение 15 мин. Охлажда¬
ют, процеживают через ватный тампон в делитель¬
ную воронку вместимостью 250 мл. Ватный тампон
переносят в колбу и повторяют экстракцию дважды
с эфиром Р, порциями по 30 мл. Охлаждают, проце¬
живают в ту же делительную воронку вместимостью
250 мл. Эфирное извлечение промывают дважды во¬
дой Р порциями по 20 мл. Водный слой отбрасывают.
Осторожно, по стенкам, прибавляют 15 мл раствора
300 г/л натрия гидроксида Р, 25 мл щелочно-аммиач¬
ного раствора Р и осторожно взбалтывают в течение
5 мин, охлаждая воронку под струей холодной воды.
После полного расслоения прозрачный красный ниж¬
ний слой, не фильтруя, сливают в мерную колбу вме¬
стимостью 500 мл, а эфирный слой обрабатывают
порциями по 25 мл щелочно-аммиачного раствора Р
до прекращения окрашивания жидкости и сливают в
ту же мерную колбу. Прибавляют 3 капли раствора
водорода пероксида концентрированного Р и дово¬
дят щелочно-аммиачным раствором Р до объема
500,0 мл.
Через 10 мин измеряют оптическую плотность
(2.2.25) при 530 нм в кювете с толщиной слоя 5 мм,
используя щелочно-аммиачный раствор Р в качестве
компенсационного раствора.
Концентрацию производных антрацена в раство¬
ре (мг/мл) определяют по градуировочному графику.
Содержание суммы производных антрацена (X) в
процентах рассчитывают по формуле:
х-°*°
т
где:
С — концентрация производных антрацена,
определенная по градуировочному графику, мг/мл;
т — масса навески испытуемого сырья, г.
Определение свободных производных антрацена.
0,100 г измельченного сырья (250) (2.9.12) по¬
мещают в колбу вместимостью 300 мл, прибавляют
50 мл эфира Р и кипятят с обратным холодильником
на водяной бане при температуре 35 °С в течение
1268
Гэсударственная фармакопея Республики Беларусь
10 мин. Охлаждают, процеживают через ватный там¬
пон в делительную воронку вместимостью 300 мл.
Ватный тампон переносят обратно в колбу и повто¬
ряют экстракцию дважды с эфиром Р порциями по
50 мл. Охлаждают, процеживают в ту же делительную
воронку вместимостью 300 мл. Прибавляют 20 мл ще¬
лочно-аммиачного раствора Р и осторожно взбалты¬
вают в течение 5 мин, охлаждая воронку под струей
холодной воды. После полного расслоения нижний
слой, не фильтруя, сливают в мерную колбу вмести¬
мостью 100 мл, а эфирный слой трижды обрабаты¬
вают щелочно-аммиачным раствором Р порциями по
20 мл и сливают в ту же мерную колбу. Прибавляют
1 каплю раствора водорода пероксида концентриро¬
ванного Р и доводят до объема 100,0 мл щелочно-ам¬
миачным раствором Р.
Через 10 мин измеряют оптическую плотность
(2.2.25) при 530 нм в кювете с толщиной слоя 5 мм,
используя щелочно-аммиачный раствор Р в качестве
компенсационного раствора.
Концентрацию производных антрацена в раство¬
ре (мг/мл) определяют по градуировочному графику.
Содержание свободных производных антрацена
(X,) в процентах рассчитывают по формуле:
х1=^,
т
где:
С — концентрация производных антрацена,
определенная по градуировочному графику, мг/мл;
т — масса навески испытуемого сырья, г.
Определение связанных производных антрацена.
Содержание связанных производных антрацена
(Х2) в процентах рассчитывают по формуле:
Х2 =Х-ХГ
07/2016:РБ0071
МАТЬ-И-МАЧЕХИ ЛИСТЬЯ
ТиззИадт/з IагТагае ГоНит
СОИ8РООТ 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в первой половине лета и высушен¬
ные листья дикорастущего многолетнего травянисто¬
го растения ТиззНадо !аг(ага 1_.
Содержание: не менее 4,0 % полисахаридов (в
пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь цельных
или частично измельченных листьев. Листья округло¬
сердцевидные, по кругу выемчатые и неравномерно
редко- и мелкозубчатые, сверху голые, снизу белово¬
йлочные от обилия спутанных длинных волосков. Че¬
решки тонкие, сверху желобоватые, часто с сохранив¬
шимся войлочным опушением. Длина листовой пла¬
стинки обычно (8—15) см, ширина около 10 см, длина
черешка около 5 см. Листья не должны быть слишком
молодыми, то есть не должны иметь густого опушения
на верхней стороне. Цвет листьев с верхней стороны
зеленый, с нижней — беловато-серый.
B. Микроскопия (2.8.23). При просматривании
верхней стороны листа с поверхности видно, что
эпидермис состоит из крупных многоугольных клеток
с прямыми, нередко четковидно-утолщенными боко¬
выми стенками. Над жилками эпидермальные клетки
вытянуты, остальные — изодиаметрические. Кутику¬
ла толстая, морщинисто-складчатая, над жилками
продольно-складчатая. Клетки нижнего эпидермиса
мелкие с сильно извилистыми стенками. Кутикула
тонкая, морщинисто-складчатая, над жилками про¬
дольно-складчатая. Над воздухоносными полостя¬
ми эпидермис приподнят, здесь расположены 1—2
устьица. Устьица крупные, овальные, аномоцитного
типа (2.8.3). На верхней стороне листа устьица встре¬
чаются редко, имеют 4—5 околоустьичных клеток; на
нижней — многочисленные с 7—9 околоустьичными
клетками, расположенными радиально. На обеих сто¬
ронах листа кутикула образует вокруг устьиц радиаль¬
ную складчатость. Верхняя сторона листа почти го¬
лая, нижняя — покрыта многочисленными простыми
волосками. Волоски состоят из короткого основания,
образованного 3—6 небольшими клетками и длинной
конечной, шнуровидной, сильно извилистой клетки.
Волоски переплетаются между собой. Губчатая ткань
имеет характер аэренхимы — ее клетки расположены
однорядными цепочками, образующими крупные воз¬
духоносные полости.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побуревшие листья и листья с бурыми
пятнами ржавчины — не более 8 %. Органические
примеси: не более 2 %. Минеральные примеси: не
более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 20,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,00 г измельченного сырья (1400) (2.9.12) по¬
мещают в колбу вместимостью 500 мл, прибавляют
100 мл воды Р и кипятят с обратным холодильни¬
ком при периодическом перемешивании в течение
30 мин. Оставляют до оседания частиц и собирают
надосадочную жидкость. Экстракцию повторяют еще
четыре раза с использованием воды Р порциями по
100 мл. Водные извлечения объединяют и центрифу¬
гируют при 5000 об/мин в течение 10 мин. Всю надо¬
садочную жидкость процеживают через ватный там¬
пон и доводят водой Р до объема 500,0 мл. 25,0 мл
полученного раствора помещают в центрифужную
пробирку, прибавляют 75 мл 96 % спирта Р, пере¬
мешивают, подогревают в водяной бане при 60 °С в
течение 5 мин. Через 30 мин содержимое центри¬
фугируют при 5000 об/мин в течение 30 мин. Надо¬
садочную жидкость фильтруют под вакуумом через
высушенный при температуре от 100 °С до 105 °С до
постоянной массы стеклянный фильтр (ПОР 16) диа¬
метром 40 мм. Осадок количественно переносят на
фильтр и последовательно промывают 15 мл смеси
из 96 % спирта Р и воды Р (3:1, об/об), 10 мл ацето¬
на Р и 10 мл этилацетата Р. Фильтр с осадком вы¬
сушивают сначала на воздухе, затем при температуре
от 100 °С до 105 °С до постоянной массы.
Содержание полисахаридов в процентах рассчи¬
тывают по формуле:
Мелиссы листья
1269
(т2 -т,) 2000
т
где:
т1 — масса фильтра, г;
т2 — масса фильтра с осадком, г;
т — масса навески испытуемого сырья, г.
07/2016:1447
<&> МЕЛИССЫ листья
МеНззае IЬНит
МЕЫ88А 1ЕАЕ
ОПРЕДЕЛЕНИЕ
Высушенные листья МеНзза оШс'таИз 1_.
Содержание: не менее 1,0 % розмариновой кисло¬
ты (С18Н1608; М.м. 360,3) (в пересчете на сухое сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеют запах, напоминающий запах лимона.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья имеют
черешки различной длины; листовая пластинка ши¬
рокоовальная длиной до 8 см и шириной до 5 см,
остроконечная на вершине и от закругленной до серд¬
цевидной в основании; края листа от городчатого до
пильчатого. Верхняя сторона интенсивного зелено¬
го цвета, нижняя сторона — более бледная зеленая
с заметной главной жилкой и рельефным сетчатым
жилкованием; на верхней поверхности и вдоль жилок
на нижней мелкопятнистой поверхности листьев име¬
ются отдельные волоски.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет зеленоватый. Просма¬
тривают под микроскопом с использованием раст¬
вора хлоралгидрата Р. В порошке обнаруживают¬
ся следующие диагностические признаки (рисунок
1447.-1): видимые с поверхности фрагменты верхнего
эпидермиса с извилистостенными клетками [А, В, С],
иногда с прилегающей палисадной паренхимой [Аа];
фрагменты нижнего эпидермиса [О] с диацитными
устьицами [Об] (2.8.3); короткие прямые одноклеточ¬
ные конические покровные волоски с мелкоборозд¬
чатой кутикулой, отдельные [Е] или прикрепленные
к эпидермису [Иа]; многоклеточные, однорядные по¬
кровные волоски с заостренными концами и толсты¬
ми бородавчатыми кутикулами [С]; восьмиклеточные
железистые волоски пластинчатого типа (при просма¬
тривании с поверхности [6а]); железистые волоски
с 1—3-клеточными ножками и одноклеточными или
реже двуклеточными головками (при просматривании
с поверхности [Ва] или на поперечном срезе [Р]).
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 2,0 г измельченного сы¬
рья (355) (2.9.12) помещают в круглодонную колбу
вместимостью 250 мл, прибавляют 100 мл воды Р.
Осуществляют перегонку в течение 1 ч в приборе для
определения эфирного масла в лекарственном рас¬
тительном сырье (2.8.12, метод А), поместив в граду¬
ированную трубку 0,5 мл ксилола Р. После перегонки
органическую фазу переносят в мерную колбу вме¬
стимостью 1 мл, промывая градуированную трубку
прибора небольшими порциями ксилола Р, и доводят
этим же растворителем до объема 1,0 мл.
Рисунок 1447.-1. Микроскопические признаки
листьев мелиссы
Раствор сравнения. 1,0 мкл цитронеллаля Р и
10,0 мкл цитраля Р (состоит из нераля и гераниаля)
растворяют в 25 мл ксилола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: этилацетат Р — гексан Р
(10:90, об/об).
Наносимый объем пробы: 20 мкл [или 4 мкл], в
виде полос.
Фронт подвижной фазы: в ненасыщенной хро¬
матографической камере не менее 15 см [или 6 см]
от линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С в течение (10—15) мин. Просма¬
тривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Цитронеллаль: зона серого
или серовато-фиолетового
цвета на границе между
верхней и средней третями
Зона серого или серовато¬
фиолетового цвета
(цитронеллаль) на границе
между верхней и средней
третями
Зона красновато¬
фиолетового цвета
Цитраль: 2 зоны серовато¬
фиолетового или синевато¬
фиолетового цвета на
границе между средней и
нижней третями
2 зоны серовато¬
фиолетового или синевато¬
фиолетового цвета
(цитраль) на границе между
средней и нижней третями
Раствор сравнения
Испытуемый раствор
1270
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли диаметром более 1 мм — не бо¬
лее 10 %. Другие допустимые примеси: не более 2 %.
Определение проводят из 20 г испытуемого сырья.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 12,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. Используют колбы из
темного стекла. 0,100 г измельченного сырья (355)
(2.9.12) диспергируют в 90 мл спирта (50 %, об/об) Р.
Кипятят в водяной бане с обратным холодильником
в течение 30 мин, охлаждают и фильтруют в мерную
колбу вместимостью 100 мл. Колбу и фильтр промы¬
вают 10 мл спирта (50 %, об/об) Р и доводят до объе¬
ма 100,0 мл этим же растворителем. Фильтруют через
фильтр с размером пор 0,45 мкм.
Раствор сравнения (а). 20,0 мг ФСО розмари¬
новой кислоты растворяют в спирте (50 %, об/об) Р
и доводят до объема 100,0 мл этим же растворите¬
лем. 20,0 мл полученного раствора доводят спиртом
(50 %, об/об) Рдо объема 100,0 мл.
Раствор сравнения (Ь). 5,0 мг феруловой кисло¬
ты Р растворяют в растворе сравнения (а) и доводят
до объема 50,0 мл этим же раствором.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: кислота фосфорная Р —
ацетонитрил Р — вода Р( 1:19:80, об/об/об);
- подвижная фаза В: кислота фосфорная Р —
метанол Р — ацетонитрил Р (1:40:59, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—20
100—>55
0 —> 45
20—25
55 ->0
45-> 100
25—30
0—>100
100->0
- скорость подвижной фазы. 1,2 мл/мин;
-спектрофотометрический детектор, длина
волны 330 нм;
- объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению к
розмариновой кислоте, время удерживания — около
11 мин): феруловая кислота — около 0,8.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиками феру¬
ловой кислоты и розмариновой кислоты.
Содержание розмариновой кислоты в процентах
рассчитывают по формуле:
А, т2 р-0,2
Аг-тл
где:
А1 — площадь пика розмариновой кислоты на
хроматограмме испытуемого раствора;
А2 — площадь пика розмариновой кислоты на
хроматограмме раствора сравнения (а);
ш1 — масса навески испытуемого сырья, исполь¬
зованной для приготовления испытуемого раствора, г;
т2 — масса навески ФСО розмариновой кисло¬
ты, использованной для приготовления раствора
сравнения (а), г;
р — содержание розмариновой кислоты в ФСО
розмариновой кислоты, %.
07/2016:РБ0072
МЕЛИССЫ ТРАВА
МеНззае ЬегЬа
МЕИЗЗА НЕКВ*
ОПРЕДЕЛЕНИЕ
Собранная в фазу бутонизации и цветения, вы¬
сушенная трава многолетнего травянистого растения
МеНзза оШтаНз 1_.
Содержание: не менее 4,0 % суммы гидроксико-
ричных кислот в пересчете на розмариновую кислоту
(С18Н16Ов; М.м. 360,3) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Олиственные
верхние части стеблей длиной до 35 см с цветками
и листьями и их кусочки, а также цельные или из¬
мельченные листья, соцветия и отдельные цветки.
Стебель ветвистый, четырехгранный, продольно-же¬
лобоватый, с рыхлой серовато-белой сердцевиной;
листья супротивные, черешковые, яйцевидные, го-
родчато-пильчатые по краю, длиной до 8 см и ши¬
риной до 5 см, по всей пластинке листа располо¬
жены эфирномасличные железки; цветки мелкие,
сидячие, собранные по 2—10 в ложные мутовки в
пазухах верхних листьев; прицветники эллиптиче¬
ские, заостренные или продолговатые; чашечка ко¬
роткотрубчатая, колокольчатая, двугубая, верхняя
губа трехзубчатая, нижняя — двухзубчатая; венчик
двугубый, пятилепестной, сросшийся в трубочку, в
полтора-два раза длиннее чашечки; тычинок четыре,
две из которых короче других; пестик с верхней че¬
тырехраздельной завязью и длинным двухраздель¬
ным столбиком. Стебли, листья и чашечки слегка
опушены беловатыми железистыми волосками. Цвет
листьев от серовато-зеленого до зеленого, иногда зе¬
леновато-коричневый; стеблей — от светло-зеленого
до серовато-зеленого; цветков — желтовато-белый,
розоватый или розовато-фиолетовый. Запах слабый,
ароматный.
B. Микроскопия (2.8.23). Видны: клетки верх¬
него эпидермиса с извилистыми боковыми стенка¬
ми, нижнего — более мелкие и извилистые; устьица
диацитного типа (2.8.3), расположенные на обеих
сторонах листа; эфирномасличные железки округлой
формы, расположены преимущественно на нижней
стороне листа и состоят из 8 (реже из 6) радиаль¬
но расположенных выделительных клеток. Волоски
встречаются трех типов: простые длинные многокле¬
точные и мелкие одноклеточные сосочковидные во¬
лоски с гладкой или бородавчатой кутикулой, голов¬
чатые волоски на одноклеточной, реже на длинной
1—3-клеточной ножке с шаровидной одноклеточной
головкой. Простые волоски расположены в основном
вдоль жилок и по краю листа; у многоклеточных про¬
стых волосков часто одна или более клеток спавшие¬
ся. Клетки эпидермиса венчика с сильно извилисты¬
ми стенками. Для венчика характерны также волоски
шлангообразной или пальцевидной формы, 1—3-кле-
Многоколосника морщинистого трава (лофанта трава)
1271
точные, покрытые штриховой кутикулой. Фрагменты
стебля, чашечки и венчика несут те же трихомы, что
и листья.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 2,0 г измельченного сы¬
рья (355) (2.9.12) помещают в круглодонную колбу
вместимостью 250 мл, прибавляют 100 мл воды Р и
нагревают в течение 1 ч в приборе для определения
эфирного масла (2.8.12, метод А), предварительно
поместив в приемник 0,5 мл ксилола Р. После пере¬
гонки органическую фазу переносят в мерную колбу
вместимостью 1 мл, промывая приемник небольшими
порциями ксилола Р, и доводят до 1 мл этим же раст¬
ворителем.
Раствор сравнения. Юмкл цитраля Р раство¬
ряют в 25 мл ксилола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р
Подвижная фаза: этилацетат Р — гексан Р
(10:90, об/об).
Наносимый объем пробы: 10 мкл раствора срав¬
нения и 30 мкл испытуемого раствора в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альдегида Р и нагревают при температуре
от 100 °С до 105 °С в течение 10 мин. Просматривают
при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Цитраль: двойная зона от
серовато-фиолетового до
синевато-фиолетового цвета
Зона красновато¬
фиолетового цвета
(эпоксикариофиллен)
Двойная зона от серовато¬
фиолетового до синевато¬
фиолетового цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли, в том числе отделенные при
анализе, — не более 50 %. Органические примеси: не
более 2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Зола общая (2.4.16). Не более 14,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,200 г измельченного сырья (355) (2.9.12) при¬
бавляют 190 мл спирта (60%, об/об) Р. Кипятят с
обратным холодильником в водяной бане в течение
30 мин. Охлаждают и фильтруют в мерную колбу вме¬
стимостью 200 мл. Промывают фильтр 10 мл спирта
(50 %, об/об) Р в ту же колбу. Доводят спиртом (50 %,
об/об) Р до объема 200,0 мл (раствор А).
Испытуемый раствор. К 1,0 мл раствора А в
пробирке прибавляют 2 мл 0,5 М раствора хлористо¬
водородной кислоты, 2 мл раствора полученного при
растворении 10 г натрия молибдата Р и 10 г натрия
нитрита Р в 100 мл воды Р. Затем прибавляют 2 мл
раствора натрия гидроксида разведенного Р и дово¬
дят водой Р до объема 10,0 мл.
Компенсационный раствор. К 1,0 мл раствора А
в другой пробирке прибавляют 2 мл 0,5 М раствора
хлористоводородной кислоты, 2 мл раствора на¬
трия гидроксида разведенного Р и доводят водой Р
до объема 10,0 мл.
Немедленно измеряют оптическую плотность
(2.2.25) испытуемого раствора при 505 нм.
Содержание суммы гидроксикоричных кислот в
пересчете на розмариновую кислоту в процентах рас¬
считывают по формуле:
А 2000
т-400 ’
где:
400 — удельный показатель поглощения розма¬
риновой кислоты;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0073
МНОГОКОЛОСНИКА
МОРЩИНИСТОГО ТРАВА
(ЛОФАНТА ТРАВА)
Ада&асЬе гидоза НегЬа
01АЫТ-НУЗЗОР*
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная трава
Адаз1асЬе гидоза Упс11.
Содержание: не менее 4,0 % суммы флавонои-
дов в пересчете на лютеолин (С15Н10Об; М.м. 286,2)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или ча¬
стично измельченные облиственные цветоносные по¬
беги длиной от 25 см до 45 см. Стебли четырехгран¬
ные, зеленые, коротко опушенные. Листья супротив¬
ные, длинночерешковые, яйцевидные, часто с серд¬
цевидным основанием, к верхушке заостренные, край
крупнопильчатый. Длина листовой пластинки (3—9) см,
ширина (2—6) см, наиболее широкая часть расположе¬
на ближе к основанию листовой пластинки; длина че¬
решка до 3,5 см. Цвет листьев сверху темно-зеленый,
снизу светло-зеленый из-за густых коротких прижатых
волосков. Цветки собраны в ложные мутовки, объ¬
единенные в верхушечные колосовидные соцветия.
Чашечка длиной (5—8) мм, неровнозубчатая, фиоле¬
тово-красного цвета. Венчик длиной от 7 мм до 10 мм,
двугубый, более светлого цвета. Запах ароматный.
B. Микроскопия (2.8.23). Клетки верхнего эпи¬
дермиса листа со слабо извилистыми стенками,
обычно без устьиц. Имеются короткие 1—2-клеточ¬
ные простые волоски; вдоль жилок часто встречаются
головчатые волоски с двухклеточной головкой, распо¬
ложенные на короткой одноклеточной ножке. Клетки
нижнего эпидермиса с сильно извилистыми стенками.
Устьица многочисленные, как правило окружены дву¬
мя околоустьичными клетками эпидермиса, располо¬
женными перпендикулярно устьичной щели. Встре¬
чаются также устьица, окруженные большим числом
1272
Государственная фармакопея Республики Беларусь
(до 5) клеток эпидермиса. Многочисленные волоски
расположены по жилкам и по всей нижней поверхно¬
сти листовой пластинки. Волоски простые, чаще изо¬
гнутые, 1—3-клеточные, стенки нижней клетки глад¬
кие, стенки остальных — мелкобородавчатые.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 20 мл раствора А, при¬
готовленного как указано в разделе «Количествен¬
ное определение», упаривают в вакууме до объема
(2—3) мл при температуре 40 °С.
Раствор сравнения. 10 мг рутина Р и 10 мг
кверцетина дигидрата Р растворяют в 10 мл спирта
(70 %, об/об) Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: бутанол Р — кислота уксус¬
ная ледяная Р — вода Р (5:1:4, об/об/об).
Наносимый объем пробы: 30 мкл испытуемого
раствора и 20 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе в течение (10—15) мин,
затем при температуре 105 °С до исчезновения запа¬
ха растворителей.
Проявление: пластинку обрабатывают раствором
50 г/л алюминия хлорида Р в 96% спирте Р и нагре¬
вают при температуре от 100 °С до 105 °С в течение
5 мин. Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Кверцетин: зона желто-
зеленого цвета
Зона желто-зеленого цвета
Рутин: зона желто-зеленого
цвета
Окрашенная зона
Зона желто-зеленого цвета
Окрашенная зона
Раствор сравнения
Испытуемый раствор
й. К 0,5 мл раствора А, приготовленного как ука¬
зано в разделе «Количественное определение», при¬
бавляют 1 мл 96 % спирта Р, перемешивают и осто¬
рожно, по стенке прибавляют 1 мл раствора 10 г/л
диметиламинобензальдегида Р в кислоте серной Р.
На границе смешивания растворов появляется корич¬
нево-красное окрашивание (терпеноиды).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие и побуревшие части тра¬
вы — не более 5 %. Органические примеси: не более
1 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 17,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (2000) (2.9.12) по¬
мещают в колбу вместимостью 100 мл, прибавляют
30 мл спирта (70 %, об/об) Р и нагревают с обрат¬
ным холодильником в водяной бане при температу¬
ре от 80 °С до 90 °С в течение 30 мин. Охлаждают до
комнатной температуры и фильтруют надосадочную
жидкость через бумажный фильтр в мерную колбу
вместимостью 100 мл. Экстракцию повторяют дваж¬
ды спиртом (70 %, об/об) Р порциями по 30 мл. По¬
лученные извлечения охлаждают, фильтруют в ту же
мерную колбу и доводят спиртом (70 %, об/об) Р до
объема 100,0 мл (раствор А).
Испытуемый раствор. 1,0 мл раствора А разво¬
дят спиртом (70 %, об/об) Р до объема 100,0 мл.
Компенсационный раствор. Спирт (70 %, об/об) Р.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 330 нм.
Содержание суммы флавоноидов в пересчете на
лютеолин в процентах рассчитывают по формуле:
А-10000
/77 • 560,8 ’
где:
560,8 — удельный показатель поглощения люте-
олина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1532
@> МОЖЖЕВЕЛЬНИКА ПЛОДЫ
Лш/рег/ да1Ьи!из
Л1Ы1РЕК
ОПРЕДЕЛЕНИЕ
Высушенные зрелые плоды (шишко-ягоды)
^ип^ре^из соттитз 1_.
Содержание: не менее 10,0 мл/кг эфирного мас¬
ла (в пересчете на безводное сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеют сильный ароматный запах, особенно при
дроблении.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Плоды ша¬
ровидной формы диаметром до 10 мм фиолето¬
во-коричневого или черновато-коричневого цвета
часто с синеватым оттенком. Шишко-ягода состо¬
ит из трех мясистых чешуек. На верхушке имеется
трехлучевая сомкнутая щель и три не очень четко
выраженных выроста. Остаток плодоножки часто
соединен с основанием. Мякоть рыхлая и коричне¬
ватая. Содержит три, реже два, мелких продолго¬
ватых очень плотных семени, которые имеют три
острые грани, слегка закруглены к основанию и
имеют заостренную верхушку. Семена соединяют¬
ся с частями шишко-ягоды, содержащими мякоть,
внизу наружной стороны их основания. На внеш¬
ней поверхности семян лежат крупные овальные
масляные железы, содержащие липкую смолу.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет коричневый.
Просматривают под микроскопом с использовани¬
ем раствора хлоралгидрата Р. В порошке обна¬
руживаются следующие диагностические призна¬
ки: фрагменты эпидермиса стенок шишко-ягоды,
содержащие клетки с толстыми пористыми бесц¬
ветными стенками и коричневым железистым со¬
держимым, иногда устьица аномоцитного типа
(2.8.3); фрагменты трехлучевой верхушечной щели
шишко-ягоды с пространствами и клетками эпидер¬
миса, соединенного сосочковидными выростами;
Мяты перечной листья
1273
фрагменты гиподермы с колленхимными утолщен¬
ными клетками; фрагменты мезокарпия, состоя¬
щего из больших тонкостенных клеток паренхимы,
обычно округлых, с большими межклеточными про¬
странствами, и большими, неправильной формы,
обычно с редкими углублениями идиобластами
желтого цвета (клетки-вместилища); фрагменты
схизогенных масличных клеток; фрагменты семен¬
ной кожуры с толстостенными, ямчатыми, бесц¬
ветными склереидами, содержащими один или
несколько призматических кристаллов оксалата
кальция; фрагменты эндосперма и зародышевой
ткани с тонкостенными клетками, содержащими
жирные масла и алейроновые зерна.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Смесь эфирного мас¬
ла и ксилола, полученную как указано в разделе
«Количественное определение», доводят гекса¬
ном Р до объема 5,0 мл.
Раствор сравнения. 4,0 мг гвайазулена Р и
50 мкл цинеола Р растворяют в 10 мл гексана Р.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде по¬
лос.
Фронт подвижной фазы: не менее 15 см от
линии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают рас¬
твором анисового альдегида Р и нагревают
при температуре от 100 °С до 105 °С в течение
(5—10) мин. Просматривают при дневном свете.
Результаты: на хроматограмме раствора
сравнения в верхней половине обнаруживается
зона красного цвета (гвайазулен), в нижней поло¬
вине — зона коричневато-фиолетового или серова¬
то-фиолетового цвета (цинеол). На хроматограмме
испытуемого раствора обнаруживается интенсив¬
ная зона фиолетового цвета (моно- и сесквитерпе-
ны), соответствующая по расположению зоне гвай¬
азулена на хроматограмме раствора сравнения;
зона красновато-фиолетового цвета немного выше
зоны, соответствующей цинеолу на хроматограмме
раствора сравнения; зона серовато-фиолетового
цвета (терпинен-4-ол) немного ниже зоны, соот¬
ветствующей цинеолу на хроматограмме раствора
сравнения, и непосредственно под ней — зона си¬
него цвета. На хроматограмме раствора сравнения
может обнаруживаться слабая зона фиолетового
цвета, соответствующая по расположению зоне
цинеола, а также другие зоны.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: неспелых или изменивших окраску
шишко-ягод — не более 5 %. Другие допустимые при¬
меси: не более 2 %.
Вода (2.2.13). Не более 120 мл/кг. Определение
проводят из 20,0 г дробленого сырья.
Общая зола (2.4.16). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 20,0 г сы¬
рья, измельченного с использованием подходящей
мельницы до грубого порошка непосредствен¬
но перед испытанием, помещают в круглодонную
колбу вместимостью 500 мл и прибавляют 200 мл
воды Р в качестве жидкости для перегонки. В гра¬
дуированную трубку помещают 0,5 мл ксилола Р.
Перегонку проводят со скоростью (3—4) мл/мин в
течение 90 мин.
07/2016:0406
МЯТЫ ПЕРЕЧНОЙ ЛИСТЬЯ
МепМае р/регНае IоЧит
РЕРРЕРМШТ 1.ЕАР
ОПРЕДЕЛЕНИЕ
Собранные в фазу цветения, высушенные и об¬
молоченные, цельные или измельченные листья мно¬
голетнего травянистого растения МепМа р/регНа 1_.
Содержание:
-цельное сырье: не менее 12 мл/кг эфирного
масла (в пересчете на сухое сырье);
- измельченное сырье: не менее 9 мл/кг эфирно¬
го масла (в пересчете на сухое сырье).
ОПИСАНИЕ
Обладает характерным и проникающим запахом.
Обладает характерным ароматным вкусом.
Листья мяты перечной имеют цвет от зеленого до
коричневато-зеленого, с коричневато-фиолетовыми
жилками у некоторых сортов. Черешки зеленые или
коричневато-зеленые.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Листья тонкие,
хрупкие. Пластинка цельного листа имеет длину от
3 см до 9 см и ширину от 1 см до 3 см, продолговато¬
овальная, эллиптическая. Край пластинки неравно¬
пильчатый с острыми зубцами. Верхушка листа за¬
круглена или притуплена, основание морщинистое у
черешка, округлое или сердцевидное. Поверхность
листа голая, лишь снизу по жилкам при увеличении
заметны редкие, прижатые волоски и по всей пластин¬
ке листа — блестящие ярко-желтые или более тем¬
ные железки.
®> В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.72). Цвет коричне¬
вато-зеленый. Просматривают под микроскопом
с использованием раствора хлоралгидрата Р.
В порошке обнаруживаются следующие диагно¬
стические признаки (рисунок 0406.-1): фрагменты
эпидермиса, несущие покровные и железистые во¬
лоски; эпидермис верхней стороны листа при про¬
сматривании с поверхности [В, Н] с клетками с из¬
вилисто-волнистыми стенками [На] и исчерченной
кутикулой над жилками [В], вместе с палисадной
паренхимой [НЬ]; эпидермис нижней стороны ли¬
ста [С] с устьицами диацитного типа [Са] (2.8.3);
простые покровные волоски обычно фрагментиро¬
ваны, вытянуты, с 3—8 клетками и с исчерченной
кутикулой [А, Е]; железистые волоски двух типов:
а) одноклеточная ножка с маленькой округлой од¬
ноклеточной головкой диаметром (15—25) мкм при
просматривании с поверхности [Ва, СЬ] или на по¬
перечном срезе [О]; Ь) одноклеточная ножка с вытя¬
нутой овальной головкой диаметром (55—70) мкм,
состоящей из 8 радиально расположенных клеток
160. Зак. 1060.
1274
Гэсударственная фармакопея Республики Беларусь
при просматривании с поверхности [ВЬ] или на по¬
перечном срезе [Са]; фрагменты около краевых
частей пластинки листа [Р] с изодиаметрическими
клетками, антиклинальные стенки которых более
или менее прямые и четковидные [Ра], и короткими
коническими одноклеточными или двуклеточными
покровными волосками [РЬ]; фрагменты дорсовен-
трального мезофилла при просматривании в по¬
перечном разрезе [СВ] с единственным палисадным
слоем [Сс] и 4—6 слоями губчатой паренхимы [ОЬ].
Под кутикулой выделительных клеток могут обна¬
руживаться желтоватые кристаллы ментола.
Рисунок 0406.-1. Микроскопические признаки
листьев мяты перечной
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,2 г свежеизмель-
ченного сырья прибавляют 2 мл метиленхлори-
да Р, встряхивают в течение нескольких минут и
фильтруют. Фильтрат выпаривают досуха при тем¬
пературе около 40 °С, остаток растворяют в 0,1 мл
толуола Р.
Раствор сравнения. 50 мг ментола Р, 20 мкл ци-
неола Р, 10 мг тимола Р и 10 мкл ментилацетата Р
растворяют в толуоле Р и доводят до объема 10 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге-
ля вР254 Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 10 мкл раствора срав¬
нения и 20 мкл испытуемого раствора в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе до удаления раство¬
рителей.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые зоны
поглощения.
Верх хроматографической пластинки
Тимол: зона поглощения
Могут обнаруживаться зоны
поглощения (карвон, пуле-
гон)
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р и нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин. Про¬
сматривают при дневном свете.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие слабые зоны.
Верх хроматографической пластинки
Интенсивная зона фиоле¬
тово-красного цвета (около
фронта растворителя) (угле¬
водороды)
Ментилацетат: зона фиоле¬
тово-синего цвета
Тимол: зона поглощения
Цинеол: зона фиолетово-си¬
него или коричневого цвета
Зона фиолетово-синего цве¬
та (ментилацетат)
Зона зеленовато-синего
цвета (ментон)
Могут обнаруживаться зоны
светло-розового или серо¬
вато-голубого или серова¬
то-зеленого цвета (карвон,
пулегон, изоментон)
Зона бледного фиолетово¬
синего или коричневого цве¬
та (цинеол)
Ментол: интенсивная зона
синего или фиолетового
цвета
Интенсивная зона синего
или фиолетового цвета
(ментол)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли — не более 10 %; почерневшие
листья — не более 5 %. Органические примеси: не бо¬
лее 3 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 14,0 %
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
@> Эфирное масло (2.8.12, метод А). 20,0 г раз¬
мельченного испытуемого сырья помещают в колбу
вместимостью 500 мл и прибавляют 200 мл воды Р. В
градуированную трубку помещают 0,50 мл ксилола Р.
Перегонку проводят со скоростью (3—4) мл/мин в те¬
чение 2 ч.
Наперстянки пурпурной листья
1275
07/2016:0117
НАПЕРСТЯНКИ ПУРПУРНОЙ
ЛИСТЬЯ
О/дПаНз ригригеае ТоНит
01в1ТАиЗ /.ЕАР
ОПРЕДЕЛЕНИЕ
Высушенные листья двухлетнего травянистого
растения 0/д//а//$ ригригеа 1_.
Содержание: не менее 0,3 % суммы сердечных
гликозидов в пересчете на дигитоксин (М.м. 765)
(в пересчете на сухое сырье).
®ОПИСАНИЕ
Обладает слабым, но характерным запахом.
Цельные листья длиной около (10—40) см и
шириной (4—15) см. Листовая пластинка овально¬
ланцетовидная или широко овальная. Длина кры¬
латого черешка составляет от 1/4 длины до равной
длине листовой пластинки.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
(О) А. Внешние признаки (2.8.23). Лист хрупкий
и зачастую поломан. Верхняя поверхность зеленая,
нижняя — серовато-зеленая. Верхушка листа подо¬
страя, край листа неравномерно-городчатый, зубча¬
тый или пильчатый. Основание сбегающее. Жилко¬
вание перистое, боковые жилки заметные, особенно
на нижней стороне листа, отходят от главной жилки
под углом около 45°, у края листа — сетчатонервные;
прожилки оканчиваются в каждом зубчике края листа,
нижние жилки спускаются к крылатому черешку. Верх¬
няя сторона листа морщинистая и опушена; нижняя
сторона листа сильно опушена и на ней видна сетка
выступающих жилок.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.12). Просматривают
под микроскопом с использованием раствора хло¬
ралгидрата Р. В порошке обнаруживаются следу¬
ющие диагностические признаки (рисунок 0117.-1):
фрагменты верхнего эпидермиса при просматри¬
вании с поверхности [К, Ц с клетками с гладкой
кутикулой и антиклинальными стенками, которые
слегка утолщенные, прямые или слегка извили¬
стые, со слабовыраженными четковидными утол¬
щениями и ямчатостью [Ьа], иногда обнаруживают¬
ся места прикрепления покровных волосков [Ка],
фрагменты верхнего эпидермиса с нижележащей
палисадной паренхимой [Щ; фрагменты нижнего
эпидермиса при просматривании с поверхности
[О] с заметно извилистыми клетками и устьицами
аномоцитного типа (2.8.3) [6а]; волоски двух ти¬
пов: а) простые покровные волоски с притупленны¬
ми верхушками, обычно состоящие из 3—5 клеток
[Н, 3], часто с одной или более спавшимися клетка¬
ми [За], стенки в основном мелкобородавчатые или
мелкоисчерченные; Ь) железистые волоски обычно
с одноклеточной [С, О], иногда с многоклеточной
однорядной [А, В, Е] ножкой и одноклеточной го¬
ловкой [А, В, С, Е], двуклеточной головкой при про¬
сматривании сбоку [й] или с поверхности [Р] или,
в исключительных случаях, с четырехклеточной
головкой.
Рисунок 0117.-1. Микроскопические признаки
листьев наперстянки пурпурной
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К1,0 г измельченного сы¬
рья (180) (2.9.12) прибавляют смесь из 20 мл спирта
(50 %, об/об) Р и 10 мл раствора свинца ацетата Р.
Кипятят в течение 2 мин. Охлаждают и центрифуги¬
руют. Встряхивают надосадочную жидкость дважды
с хлороформом Р, порциями по 15 мл; при необходи¬
мости слои разделяют центрифугированием. Хлоро¬
формные слои объединяют и высушивают с помощью
натрия сульфата безводного Р и фильтруют. 10 мл
полученного раствора выпаривают досуха на водяной
бане, остаток растворяют в 1 мл смеси из равных объ¬
емов хлороформа Р и метанола Р.
Раствор сравнения. 5 мг ФСО пурпуреагликози-
да А, 2 мг ФСО пурпуреагликозида В, 5 мг дигитокси-
на Р и 2 мг гитоксина Р растворяют в смеси из рав¬
ных объемов хлороформа Р и метанола Р и доводят
до 10 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля 6 Р.
Подвижная фаза: вода Р — метанол Р—этила-
цетат Р (7,5:10:75, об/об/об).
Наносимый объем пробы: по 20 мкл в виде полос
длиной 20 мм и шириной 3 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: до удаления растворителей.
Проявление: пластинку обрабатывают смесью из
2 объемов раствора 10 г/л хлорамина Р и 8 объемов
раствора 250 г/л кислоты трихлоруксусной Р в 96%
спирте Р и нагревают при температуре от 100 °С до
105 °С в течение 10 мин. Просматривают в ультрафи¬
олетовом свете при длине волны 365 нм.
Результаты: на хроматограмме раствора
сравнения в нижней части обнаруживается флуо¬
ресцирующая зона светло-голубого цвета (пурпу-
160*. Зак. 1060.
1276
Гэсударственная фармакопея Республики Беларусь
реагликозид В) и непосредственно выше — флу¬
оресцирующая зона коричневато-желтого цвета
(пурпуреагликозид А); в средней части обнаружи¬
вается флуоресцирующая зона светло-голубого
цвета (гитоксин) и над ней — флуоресцирующая
зона коричневато-желтого цвета (дигитоксин). На
хроматограмме испытуемого раствора обнаружи¬
ваются зоны, соответствующие по расположению,
цвету и величине зонам на хроматограмме раст¬
вора сравнения. На хроматограмме испытуемого
раствора могут обнаруживаться и другие флуорес¬
цирующие зоны.
® О. 5 мл хлороформного извлечения, полу¬
ченного как указано в идентификации С, выпарива¬
ют досуха на водяной бане. К остатку прибавляют
2 мл раствора кислоты динитробензойной Р и
1 мл 1 М раствора гидроксида натрия. В течение
5 мин появляется красновато-фиолетовое окраши¬
вание.
@> Е. 5 мл хлороформного извлечения, полу¬
ченного как указано в идентификации С, выпарива¬
ют досуха на водяной бане. К остатку прибавляют
3 мл раствора ксантгидрола Р и нагревают на во¬
дяной бане в течение 3 мин. Появляется красное
окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: потемневшие или пожелтевшие ли¬
стья — не более 1 %; другие части растения (стебель,
цветки и плоды) — не более 1 %; измельченные ли¬
стья, проходящие сквозь сито (1400) (2.9.12), — не
более 2 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Б/дИаНз 1апа(а ЕНгН. Присутствие листьев с
малым количеством или с отсутствующими волоска¬
ми и с параллельным жилкованием, или присутствие
клеток нижнего эпидермиса с четковидными антикли¬
нальными стенками и клеток верхнего эпидермиса с
многочисленными устьицами указывает на подмену
Б/дИаНз 1апа!а ЕНгН.
® Потеря в массе при высушивании (2.2.32).
Не более 6,0%. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 12,0 %.
еф Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Испытуемый раствор. 0,250 г измельченного
сырья (180) (2.9.12) встряхивают с 50,0 мл воды Р в
течение 1 ч. Прибавляют 5,0 мл раствора 150 г/л аце¬
тата свинца Р, перемешивают и через несколько ми¬
нут прибавляют 7,5 мл раствора 40 г/л динатрия ги¬
дрофосфата Р. Фильтруют через складчатый бумаж¬
ный фильтр. К 50,0 мл фильтрата прибавляют 5 мл
раствора кислоты хлористоводородной (150 г/л НС1)
и нагревают с обратным холодильником на водяной
бане в течение 1 ч. Переносят в делительную воронку,
дважды ополаскивая колбу водой Р порциями по 5 мл
и встряхивают трижды с хлороформом Р порциями
по 25 мл. Хлороформные слои объединяют, высуши¬
вают с помощью безводного сульфата натрия Р и
доводят хлороформом Р до объема 100,0 мл. 40,0 мл
полученного раствора выпаривают досуха. Остаток
растворяют в 7 мл спирта (50 %, об/об) Р, прибавля¬
ют 2 мл раствора кислоты динитробензойной Р и
1 мл 1 М раствора натрия гидроксида.
Раствор сравнения. Готовят одновременно с
испытуемым раствором. 50,0 мг ФСО дигитоксина
растворяют в 96% спирте Р и доводят до объема
50.0 мл этим же растворителем. 5,0 мл полученно¬
го раствора доводят 96 % спиртом Р до объема
50.0 мл. К 5,0 мл полученного раствора прибавляют
25 мл воды Р и 3 мл раствора кислоты хлористово¬
дородной (150 г/л НС1). Нагревают с обратным холо¬
дильником на водяной бане в течение 1 ч. Переносят
в делительную воронку, дважды ополаскивая колбу
водой Р порциями по 5 мл и встряхивают трижды с
хлороформом Р порциями по 25 мл. Хлороформные
слои объединяют, высушивают с помощью натрия
сульфата безводного Р и доводят хлороформом Р до
объема 100,0 мл. 40,0 мл полученного раствора вы¬
паривают досуха. Остаток растворяют в 7 мл спирта
(50 %, об/об) Р, прибавляют 2 мл раствора кислоты
динитробензойной Р и 1 мл 1 М раствора натрия
гидроксида.
Измеряют оптическую плотность (2.2.25) рас¬
творов при 540 нм несколько раз в течение первых
12 мин, пока не будет достигнуто максимальное зна¬
чение, используя в качестве компенсационного раст¬
вора смесь из 7 мл спирта (50 %, об/об) Р, 2 мл
раствора кислоты динитробензойной Р и 1 мл 1 М
раствора натрия гидроксида.
Содержание сердечных гликозидов в пересчете
на дигитоксин в процентах рассчитывают исходя из
измеренных оптических плотностей и концентраций
растворов.
07/2016:РБ0074
ОБЛЕПИХИ ПЛОДЫ СВЕЖИЕ
Н/'ррорЬаез гатпо1с1ез 1гис1из гесепз
8ЕА висктнокм рки/г, РЯЕ5Н*
ОПРЕДЕЛЕНИЕ
Свежие или замороженные зрелые плоды
Н(ррорНае гатло/дез1_.
Содержание: не менее 0,010% каротиноидов в
пересчете на (3-каротин (С40Н5б; М.м. 536,9) (в пере¬
счете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Плоды — ко¬
стянки, сочные, с одной косточкой, от шарообразной
до удлиненно-эллипсовидной формы, с плодоножкой
или без нее. Длина плодов от 4 мм до 12 мм. Цвет от
желтого до темно-оранжевого. Запах слабый, напоми¬
нающий запах ананаса.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: недозрелые плоды — не более 11 %;
мятые плоды (при условии сохранения сока из этих
плодов) — не более 35 %; поврежденные вредите¬
лями плоды — не более 2 %; веточки и другие части
растения — не более 1 %. Органические примеси:
не более 1 %. Минеральные примеси: не более
0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 87 %. 2,000 г измельченного сырья сушат при
температуре 105 °С.
Общая зола (2.4.16). Не более 1,0 %.
Одуванчика лекарственного корни
1277
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г испытуемого сырья растирают в фарфо¬
ровой ступке с небольшим количеством натрия суль¬
фата безводного Р до получения однородной мас¬
сы. Содержимое ступки количественно переносят в
коническую колбу вместимостью 250 мл, прибавляют
50 мл петролейного эфира Р и встряхивают в течение
30 мин. Собирают надосадочную жидкость, к остатку в
колбе прибавляют 20 мл петролейного эфира Р. Экс¬
тракцию повторяют до тех пор, пока растворитель не
перестанет окрашиваться. Извлечения объединяют,
высушивают безводным сульфатом натрия Р до ис¬
чезновения мутности раствора и фильтруют в мерную
колбу вместимостью 250 мл. Сульфат натрия промы¬
вают трижды петролейным эфиром Р порциями по
10 мл, фильтруя в ту же мерную колбу, и доводят до
объема 250,0 мл этим же растворителем (раствор А).
Испытуемый раствор. 100,0 мл раствора А упа¬
ривают при температуре от 40 °С до 50 °С в вакууме
до объема 20 мл, количественно переносят в мерную
колбу вместимостью 25 мл, используя петролейный
эфир Р, и доводят до объема 25,0 мл этим же раст¬
ворителем.
Раствор сравнения. 0,3600 г калия дихрома¬
та Р, предварительно высушенного при температуре
130 °С до постоянной массы, растворяют в воде Р и
доводят до объема 1000,0 мл этим же растворителем.
Компенсационный раствор. Петролейный
эфир Р.
Измеряют оптическую плотность (2.2.25) раство¬
ров при 450 нм в кюветах с толщиной слоя 5 мм.
Содержание суммы каротиноидов в пересчете на
Р-каротин в процентах рассчитывают по формуле:
Л Л70 -0,00578-6,25
А0т
где:
0,00578 — коэффициент пересчета оптической
плотности раствора 3,6 г/л калия дихромата Р в со¬
держание р-каротина (г/л);
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески калия дихромата Р, г.
ХРАНЕНИЕ
Замороженное сырье — при температуре не
выше -18°С.
07/2016:1852
ОДУВАНЧИКА ЛЕКАРСТВЕННОГО
КОРНИ
Тагахаа оШстаНз гасИх
ОАЫОЕЫОЫ коот
ОПРЕДЕЛЕНИЕ
Собранные осенью, очищенные от корневой
шейки, отмытые от земли и высушенные корни много¬
летнего травянистого растения Тагахасит о1Пста1е
Р-НЛМдд.
Содержание: не менее 0,3 % фенолкарбоновых
кислот в пересчете на кофейную кислоту (С9Н704;
М.м. 180,2) (в пересчете на сухое сырье).
ОПИСАНИЕ (СВОЙСТВА)
® Сырье имеет горький вкус.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корни стержне¬
вые, маловетвистые, цельные или изломанные, дли¬
ной (2—15) см, толщиной (0,3—-3) см, продольно-мор¬
щинистые, иногда спирально-перекрученные, плот¬
ные, хрупкие. Излом неровный. В центре корня видна
небольшая желтая древесина, окруженная широкой
серовато-белой корой, в которой заметны (при увели¬
чении 10х) коричневатые, концентрические тонкие по¬
яса млечников. Цвет снаружи от светло-коричневого
до темно-коричневого.
® В. Микроскопия (2.8.23). Измельчают до по¬
рошкообразного состояния (355) (2.9.12). Порошок
имеет желтовато-коричневый цвет. Просматривают
под микроскопом с использованием раствора хлорал¬
гидрата Р. В порошке обнаруживаются следующие
диагностические признаки (рисунок 1852.-1): фрагмен¬
ты корки коричневого или красновато-коричневого цве¬
та (вид с поверхности [6] и вид поперечного среза [С]) с
уплощенными, тонкостенными клетками [Са], иногда с
прилегающей паренхимой [СЬ]; сетчатые одревеснев¬
шие сосуды [Е, 3, М]; фрагменты паренхимы [А, О, К, Ц,
некоторые из которых содержат разветвленные, содер¬
жащие латекс сосуды (продольный срез [Ка] и попереч¬
ный срез [Ра]); зернистое содержимое сосудов, содер¬
жащих латекс [В, Н]. Просматривают под микроскопом
с использованием глицерина Р. В порошке обнару¬
живаются многочисленные несимметричные, угловые
фрагменты инулина, расположенные отдельно [Р] или
включенные в клетки паренхимы [!_а].
Рисунок 2120.-1. Микроскопические признаки
корней одуванчика
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 2,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р,
161. Зак. 1060.
1278
Гэсударственная фармакопея Республики Беларусь
нагревают на водяной бане при температуре 60 °С в
течение 5 мин и фильтруют через бумажный фильтр.
Раствор сравнения. 0,5 мг хлорогеновой кисло¬
ты Р растворяют в 5 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: хлороформ Р — кислота ук¬
сусная ледяная Р — вода Р (50:42:8, об/об/об).
Наносимый объем пробы: Юмкл испытуемого
раствора и 5 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
20 г/л алюминия хлорида Р в 96% спирте Р. Просма¬
тривают в ультрафиолетовом свете при 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Флуоресцирующая зона
серовато-синего цвета
(кофейная кислота)
Хлорогеновая кислота:
флуоресцирующая зона
серовато-синего цвета
Флуоресцирующая зона
серовато-желтого цвета
(цикориевая кислота)
Флуоресцирующая зона
серовато-синего цвета
(хлорогеновая кислота)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни, плохо очищенные от корневых
шеек и черешков листьев, — не более 4 %; дряблые
корни — не более 2 %; корни, побуревшие в изло¬
ме, — не более 10 %. Органические примеси: не бо¬
лее 0,5 %. Минеральные примеси: не более 2 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 10,0 %.
® Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 3,0 %.
® Показатель горечи (2.8.15). Не менее 100.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Исходный раствор. 1,000 г измельченного сырья
(355) (2.9.12) помещают в колбу со шлифом вместимо¬
стью 100 мл, прибавляют 25 мл воды Р и встряхивают
в течение 30 мин. Водное извлечение процеживают
через ватный тампон в мерную колбу вместимостью
50 мл. Вату помещают в колбу с остатком сырья и экс¬
тракцию повторяют еще раз, прибавляя 25 мл воды Р.
Извлечения объединяют и доводят водой Р до объема
50,0 мл.
Испытуемый раствор. В мерную колбу вмести¬
мостью 10 мл помещают в следующей последова¬
тельности: 5 мл водыР, 1,0 мл исходного раствора,
1 мл 0,5 М раствора хлористоводородной кисло¬
ты, 1 мл раствора, полученного при растворении
10 г натрия молибдата Р и 10 г натрия нитрита Р
в 100мл водыР. Затем прибавляют 1 мл раствора
40 г/л натрия гидроксида Р и доводят водой Р до
объема 10,0 мл.
Компенсационный раствор. В мерную колбу
вместимостью 10 мл помещают в следующей после¬
довательности: 5 мл воды Р, 1 мл 0,5 М раствора
хлористоводородной кислоты, 1 мл раствора, полу¬
ченного при растворении 10 г натрия молибдата Р и
10 г натрия нитрита Р в 100 мл воды Р. Затем при¬
бавляют 1 мл раствора 40 г/л натрия гидроксида Р и
доводят водой Р до объема 10,0 мл.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 490 нм.
Содержание фенолкарбоновых кислот в пере¬
счете на кофейную кислоту в процентах рассчитыва¬
ют по формуле:
А 500
т-285’
где:
285 — удельный показатель поглощения кофей¬
ной кислоты;
А—оптическаяплотностьиспытуемогораствора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0075
ОЛЬХИ СЕРОЙ ЛИСТЬЯ
А1т ’тсапае ТоИит
вПЕУА1-ОЕП 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в течение лета, высушенные и обмо¬
лоченные листья А1пиз тсапа (1_.) Саегёп.
Содержание: не менее 1,0 % гиперозида
(^21^20^12» М.м. 464,4) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья яйцевид¬
ные или широкоэллиптические, овально-ланцето¬
видные, на верхушке клиновидно суженные, иногда
несколько заостренные, реже притупленные, с окру¬
глым или ширококлиновидным основанием. По краю
остродвоякопильчатые. Жилкование перистое, ко¬
личество жилок от 7 до 11 (реже 13). Длина листа от
4 см до 12 см, ширина от 3 см до 7 см, черешок листа
до 2 см. Цвет листьев сверху зеленый, снизу — серо¬
зеленый, с опушением, особенно по жилкам. Запах
слабый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны клетки эпидермиса. Верх¬
ний эпидермис состоит из крупных клеток, в очертании
многоугольных; с нижней стороны клетки эпидермиса
с извилистым контуром. Устьица преимущественно
на нижней стороне, овальные, окружены 4—9 клет¬
ками эпидермиса (аномоцитный тип) (2.8.3). По всей
поверхности листа встречаются простые однокле¬
точные тонкостенные волоски, иногда с коричневым
содержимым; на нижней стороне листа встречаются
простые толстостенные волоски. В мезофилле листа
характерны многочисленные, хорошо сформирован¬
ные остроконечные друзы оксалата кальция. На обе¬
их поверхностях листа имеются железки на короткой
ножке с головкой из пяти клеток с коричневым содер¬
жимым. Жилки листа сопровождаются секреторными
включениями, заполненными коричневым содержи¬
мым, которое окрашивается раствором Судана IIIР в
Ольхи серой листья
1279
красный цвет. Секреторные включения с коричневым
содержимым находятся и в мезофилле листа.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 60 мл спирта (70 %,
об/об) Р и кипятят с обратным холодильником в те¬
чение 40 мин. Охлаждают, центрифугируют со скоро¬
стью 3000 об/мин в течение 5 мин и собирают надо-
садочную жидкость.
Раствор сравнения. 1,0 мг гиперозида Р раство¬
ряют в 1 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем целлюлозы
для хроматографии Р.
Подвижная фаза: кислота уксусная Р. Камеру
насыщают парами подвижной фазы в течение 30 мин.
Наносимый объем пробы, по 5 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
20 г/л алюминия хлорида Р в 96% спирте Р и про¬
сматривают в ультрафиолетовом свете при длине
волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Зона голубого цвета
Зона голубого цвета
Гиперозид: зона желтого
цвета
Зона желтого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: потемневшие и побуревшие ли¬
стья — не более 3 %. Органические примеси: не
более 3 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0%. 2,000 г измельченного сырья
(2000) (2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 6,0 %.
Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,00 г измельченного
сырья (355) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 250 мл, прибавляют 60 мл спирта
(60 %, об/об) Р, взвешивают с точностью до 0,01 г и
кипятят с обратным холодильником в водяной бане
в течение 40 мин. Охлаждают, доводят массу до
первоначальной спиртом (60 %, об/об) Р и центри¬
фугируют. Надосадочную жидкость фильтруют че¬
рез мембранный фильтр с диаметром пор 0,45 мкм.
Раствор сравнения. 7,5 мг гиперозида Р,
5,0 мг эллаговой кислоты Р растворяют в метано¬
ле и доводят до объема 25,0 мл этим же раство¬
рителем.
Условия хроматографирования:
-колонка длиной 0,150 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- температура: 20 °С;
- подвижная фаза:
- подвижная фаза А: вода Р — кислота ук¬
сусная ледяная Р (97:3, об/об);
- подвижная фаза В: метанол Р\
161*. Зак. 1060.
1280
Гэсударственная фармакопея Республики Беларусь
Время
(мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
Скорость
подвижной фазы
(мл/мин)
0—10
100 — 90
0 — 10
0,9 — 1,0
10—40
о
со
т
о
а
10 — 70
1,0
40—44
о
о
7
о
со
70 — 0
1,0 —0,9
44—47
100
0
0,9
-спектрофотометрический детектор, длина
волны 360 нм;
- время уравновешивания системы: 20 мин пе¬
ред каждым вводом;
- объем вводимой пробы: 20 мкл.
Порядок выхода пиков: гиперозид, эллаговая кис¬
лота (рисунок РБ0075.-1).
Пригодность хроматографической системы:
раствор сравнения:
- разрешение между пиками гиперозида и элла-
говой кислоты не менее 1,5.
Содержание гиперозида в процентах рассчиты¬
вают по формуле:
5С-6
51 т
где:
3 — площадь пика гиперозида на хроматограмме
испытуемого раствора;
31 — площадь пика гиперозида на хроматограм¬
ме раствора сравнения;
т — масса навески испытуемого сырья, г;
С — содержание гиперозида в растворе сравне¬
ния, мг/мл.
07/2016:РБ0076
ольхи соплодия
А1п11гис1из
А/.ОЕЯ РКШТ*
ОПРЕДЕЛЕНИЕ
Собранные поздней осенью и зимой, высу¬
шенные соплодия А1пиз тсапа (Ь.) МоепсН и А1пиз
дЫтоза (1_.) Саегит
Содержание: не менее 10 % дубильных веществ
в пересчете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Яйцевидные
или продолговатые соплодия ольхи («шишки»),
расположенные по несколько штук на общей пло¬
доножке или одиночные, с плодоножками либо без
них, чешуйки и плоды. На твердой оси соплодия
расположены многочисленные веерообразные че¬
шуйки с утолщенным, слегка лопастным наружным
краем. В пазухах чешуек находятся односеменные
двукрылые сплюснутые плоды-орешки. Длина об¬
щей плодоножки до нижнего соплодия до 15 мм,
длина соплодий до 20 мм, диаметр до 13 мм. Цвет
соплодий и веточек темно-красновато-коричневый
или темно-коричневый.
B. Микроскопия (2.8.23). На поперечном сре¬
зе оси соплодия располагаются 5—6 сосудисто¬
волокнистых коллатеральных пучков, у основания
которых находится многоклеточная перимедуляр-
ная зона. Флоэма деформирована; над флоэмой
располагается механическая ткань, состоящая из
крупных или продолговатых клеток. На поперечном
срезе чешуйки в средней части видно 5 сосудисто¬
волокнистых коллатеральных пучков, состоящих
из ксилемы, тонкого слоя деформированной флоэ¬
мы и 3—5 рядов склеренхимы, расположенных по
обеим сторонам пучка. Чешуйки покрыты эпидер¬
мисом с кутикулой, более толстой на внешней сто¬
роне соплодий.
С. 2 г измельченного сырья (1000) (2.9.12) по¬
мещают в колбу, прибавляют 20 мл воды Р и кипя¬
тят с обратным холодильником в течение 30 мин.
Горячее извлечение фильтруют. К 2 мл полученно¬
го раствора прибавляют 2 капли раствора железа
(III) аммония сульфата Р5. Появляется черно-си¬
нее окрашивание, быстро переходящее в черное.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: веточки и отделившиеся плодо¬
ножки — не более 1 %; соплодия с длиной общей
плодоножки свыше 15 мм — не более 3 %; измель¬
ченные частицы, проходящие сквозь сито (710)
(2.9.12), — не более 3 %. Органические примеси: не
более 0,5 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 2,000 г измельченного сырья
(2000) (2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 3,5 %.
Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2.000 г измельченного сырья (2000) (2.9.12)
помещают в коническую колбу со шлифом вме¬
стимостью 500 мл, прибавляют 250 мл кипящей
воды Р и кипятят с обратным холодильником в те¬
чение 30 мин при периодическом перемешивании.
Охлаждают до комнатной температуры, процежи¬
вают и доводят водой Р до объема 250,0 мл.
25.0 мл полученного извлечения помещают в
коническую колбу вместимостью 750 мл, прибав¬
ляют 500 мл воды Р, 25,0 мл раствора индигосуль-
фокислоты (0,25 мг индигокармина Р растворяют в
6,5 мл кислоты серной Р, прибавляют 6,5 мл кис¬
лоты серной Р, доводят водой Р до объема 250 мл
и фильтруют). Титруют при постоянном перемеши¬
вании 0,02 М раствором калия перманганата до
желтого окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата
соответствует 4,157 мг дубильных веществ в пере¬
счете на танин.
07/2016:РБ0077
ОЛЬХИ ЧЕРНОЙ листья
А1т дШНпозае Шит
В1.АСК А1.0ЕК 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в течение лета, высушенные и обмо¬
лоченные листья А1пиз д/иИпоза (I.) Саег1п.
Ольхи черной листья
1281
Содержание:
- не менее 5,0 % суммы фенольных соеди¬
нений в пересчете на эллаговую кислоту (С14Н608;
М.м. 302,2) (в пересчете на сухое сырье);
- не менее 0,5 % эллаговой кислоты (С14Н608;
М.м. 302,2) и не мене 0,3 % гиперозида (С21Н20О12;
М.м. 464,4) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья че¬
решковые, широко-обратнояйцевидные или почти
округлые с цельнокрайним клиновидным основа¬
нием; на верхушке притупленные или выемчатые,
с городчато-пильчатым краем. Молодые листья
очень клейкие. Жилкование перистое, количество
жилок 5—7 (реже 9). Длина листа от 2,3 см до
12 см, ширина от 2,5 см до 6 см, длина черешка
от 0,7 см до 3 см. Листья сверху темно-зеленые,
блестящие, снизу светло-зеленые, почти голые
(с редкими волосками по жилкам). Запах слабый.
B. Микроскопия (2.8.23). При просматрива¬
нии листа с поверхности видны: многоугольные
клетки верхнего эпидермиса с незначительно утол¬
щенными оболочками, нижнего — более мелкие
с сильно извилистым контуром; устьица овальные,
преимущественно на нижней стороне, окружены
4—9 клетками (аномоцитный тип) (2.8.3); много¬
численные волоски на обеих сторонах листа (на
нижней стороне их больше); волоски простые,
одноклеточные, с заостренной верхушкой, иногда
с коричневым содержимым, расположенные, в ос¬
новном, по крупным жилкам, особенно с нижней
стороны; на обеих сторонах листа железки на ко¬
роткой ножке с головкой из пяти клеток с коричне¬
вым содержимым. В мезофилле листа много друз
оксалата кальция. Жилки листа сопровождаются
секреторными включениями, заполненными корич¬
невым зернистым или маслянистым содержимым,
которые окрашиваются раствором Судана III Р в
красный цвет.
C. Тонкослойная хроматография (2.2.21).
Испытуемый раствор. К 0,5 г измельченно¬
го сырья (355) (2.9.12) прибавляют 30 мл спирта
(60 %, об/об) Р и кипятят с обратным холодильни¬
ком в течение 40 мин. Охлаждают, центрифугируют
при 3000 об/мин в течение 5 мин и собирают над-
осадочную жидкость.
Раствор сравнения. 1,0 мг эллаговой кисло¬
ты Р, 1,0 мг гиперозида Р и 1 мг рутина Р раство¬
ряют в 1 мл метанола Р.
Пластинка: ТСХ пластинка со слоем целлюло¬
зы для хроматографии Р
Подвижная фаза: 2-пропанол Р — кислота
муравьиная безводная Р — вода Р (2:5:5, об/об/об).
Камеру насыщают парами подвижной фазы в тече¬
ние 30 мин.
Наносимый объем пробы: по 5 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от
линии старта.
Высушивание: на воздухе.
Прояление: пластинку опрыскивают раство¬
ром 20 г/л алюминия хлорида Р в 96 % спирте Р
и просматривают в ультрафиолетовом свете при
длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора.
Верх хроматографической пластинки
Зона голубого цвета
Рутин: зона желтого цвета
Гиперозид: зона желтого
цвета
Зона желтого цвета
Зона желтого цвета
Эллаговая кислота: зона
фиолетового цвета
Зона фиолетового цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: потемневшие и побуревшие листья —
не более 3 %. Органические примеси: не более 3 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания суммы фенольных
соединений в пересчете на эллаговую кислоту.
0,500 г измельченного сырья (355) (2.9.12) помещают в
круглодонную колбу вместимостью 100 мл, прибавляют
30 мл спирта (60 %, об/об) Р, взвешивают с точностью
до 0,01 г и кипятят с обратным холодильником в водя¬
ной бане в течение 40 мин при периодическом встряхи¬
вании для смывания частиц сырья со стенок колбы. Ох¬
лаждают, доводят массу до первоначальной спиртом
(60 %, об/об) Р и центрифугируют при 3000 об/мин в
течение 5 мин. К 10,0 мл надосадочной жидкости при¬
бавляют 10,0 мл спирта (60 %, об/об) Р (раствор А).
Испытуемый раствор. К 0,1 мл раствора А при¬
бавляют 0,5 мл реактива фосфорномолибденово¬
вольфрамового Р, 10 мл воды Р и доводят до объема
25,0 мл раствором 150 г/л натрия карбоната безво¬
дного Р.
Компенсационный раствор. К 0,5 мл реактива
фосфорномолибденово-вольфрамового Р прибавля¬
ют 10 мл воды Р и доводят до объема 25,0 мл раство¬
ром 150 г/л натрия карбоната безводного Р.
Через 40 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 760 нм.
Концентрацию суммы фенольных соединений в
испытуемом растворе (мг/мл) в пересчете на эллаго¬
вую кислоту определяют по градуировочному графику.
Содержание суммы фенольных соединений в
пересчете на эллаговую кислоту в процентах рассчи¬
тывают по формуле:
С • 1500
т
где:
С — содержание фенольных соединений в пере¬
счете на эллаговую кислоту, найденное по калибро¬
вочному графику, мг/мл;
т — масса навески испытуемого сырья, г.
Построение градуировочного графика. 12,5 мг
эллаговой кислоты Р, в пересчете на сухое веще¬
ство, растворяют 10 мл метанола Р и доводят до
объема 25,0 мл этим же растворителем. В пять мер¬
ных колб вместимостью 25 мл помещают по 0,1; 0,2;
0,3; 0,4; и 0,5 мл полученного раствора соответствен-
162. Зак. 1060.
1282
Гэсударственная фармакопея Республики Беларусь
но, прибавляют по 0,5 мл реактива фосфорномолиб-
деново-вольфрамового Р, по 10 мл воды Р и доводят
до объема 25,0 мл раствором 150 г/л натрия карбо¬
ната безводного Р. Для построения градуировочного
графика на оси абсцисс откладывают концентрацию
раствора (мг/мл), а на оси ординат — их оптическую'
плотность.
Определение содержания эллаговой кислоты
и гиперозида. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,00 г измельченного
сырья (355) помещают в круглодонную колбу вме¬
стимостью 250 мл, прибавляют 60 мл спирта (60 %,
об/об) Р, взвешивают с точностью до 0,01 г и кипятят
с обратным холодильником в водяной бане в течение
40 мин. Охлаждают, доводят массу до первоначаль¬
ной спиртом (60%, об/об) Р и центрифугируют. На-
досадочную жидкость фильтруют через мембранный
фильтр с диаметром пор 0,45 мкм.
Раствор сравнения. 2,5 мг гиперозида Р, 5,0 мг
эллаговой кислоты Р растворяют в метаноле и дово¬
дят до объема 25,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,150 м и внутренним диаметром
4,6 мм, заполненная силикагелем октадецилсилиль-
ным для хроматографии Р с размером частиц 5 мкм;
- температура: 20 °С;
- подвижная фаза:
- подвижная фаза А: вода Р — кислота уксус¬
ная ледяная Р (97:3, об/об);
- подвижная фаза В: метанол Р;
Время (мин)
Подвижная
фаза А
(%, об/об)
Подвижная
фаза В
(%, об/об)
Скорость
подвижной
фазы
(мл/мин)
0—10
100 — 90
0 —> 10
0,9 —>1,0
10—40
о
со
т
о
О)
о
г*.
т
о
1,0
40—44
о
о
т
о
со
о
т
о
1,0 —0,9
44—47
100
0
0,9
- спектрофотометрический детектор, дли¬
на волны 360 нм;
- время уравновешивания системы: 20 мин
перед каждым вводом;
- объем вводимой пробы: 20 мкл.
Порядок выхода пиков: гиперозид, эллаговая
кислота (рисунок РБ0077.-1).
Пригодность хроматографической системы:
раствор сравнения:
- разрешение между пиками гиперозида и эл¬
лаговой кислоты не менее 1,5.
Содержание гиперозида в процентах рассчи¬
тывают по формуле:
5,-С-6
32т
где:
31 — площадь пика гиперозида на хромато¬
грамме испытуемого раствора;
52 — площадь пика гиперозида на хромато¬
грамме раствора сравнения;
т — масса навески испытуемого сырья, г;
С — содержание гиперозида в растворе срав¬
нения, мг/мл.
Содержание эллаговой кислоты в процентах
рассчитывают по формуле:
УС-6
54 ■ т
где:
53 — площадь пика эллаговой кислоты на хро¬
матограмме испытуемого раствора;
$4 — площадь пика эллаговой кислоты на хро¬
матограмме раствора сравнения;
т — масса навески испытуемого сырья, г;
С1 — содержание эллаговой кислоты в раство¬
ре сравнения, мг/мл.
1. Гиперозид. 2. Эллаговая кислота.
Рисунок РБ0077.-1. Хроматограмма испытуемого раствора
Ортосифона тычиночного листья (почечного чая листья)
1283
07/2016:1229
ОРТОСИФОНА ТЫЧИНОЧНОГО
ЛИСТЬЯ (ПОЧЕЧНОГО ЧАЯ
ЛИСТЬЯ)
ОгОюз1рНоп18 (оНит
МУА ТЕА
ОПРЕДЕЛЕНИЕ
Собранные в течение вегетации и высушенные
листья и верхушки побегов ОгИю&рЬоп з(ат1пеиз
Веп1Ь. (О. апзШиз Мац.; О. зр‘юа(из Вак.).
Содержание: не менее 0,05 % синенсети-
на (С20Н20О7; М.м. 372,4) (в пересчете на сухое
сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
@>А. Внешние признаки (2.8.23). Листья
хрупкие, длиной до 7,5 см и шириной до 2,5 см.
Черешок короткий. Листовая пластинка овальной
или ланцетовидной формы с острой верхушкой и
клиновидным основанием. Нижняя поверхность
листьев светлая серовато-зеленого цвета, верхняя
поверхность — темно-зеленая или коричневато¬
зеленая. Жилкование перистое с небольшим ко¬
личеством вторичных жилок. При просматривании
при увеличении (10*) видно, что вторичные жилки
идут параллельно главной жилке, а потом расхо¬
дятся под острым углом. Край листовой пластинки
неравномерно- и крупнозубчатый, иногда город-
чатый, нижняя сторона слегка скручена. Черешки
тонкие, четырехугольные, длиной (4—8) мм и, как и
первичное жилкование, обычно фиолетового цве¬
та. Обнаруживаются редко встречающиеся кисте¬
видные соцветия голубовато-белых или фиолето¬
вых, еще не распустившихся цветков.
@> В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.12). Цвет темно-зе¬
леный. Просматривают под микроскопом с исполь¬
зованием раствора хлоралгидрата Р. В порошке
обнаруживаются следующие диагностические при¬
знаки (рисунок 1229.-1): фрагменты эпидермиса
с клетками с извилистым очертанием, несущие
одноклеточные или двухклеточные конические по¬
кровные волоски и простые членистые волоски
длиной до 450 мкм, состоящие из 3—8 клеток с
толстыми ямчатыми стенками; головчатые волоски
с одноклеточными или двухклеточными головками;
секреторные волоски с одноклеточными ножками и
обычно с четырехклеточными головками; устьица
диацитного типа (2.8.3), более многочисленные на
нижнем эпидермисе.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1 г измельченного
сырья (710) (2.9.12) прибавляют 10 мл метанола Р
и нагревают в водяной бане при температуре 60 °С
в течение 5 мин при встряхивании. Охлаждают и
фильтруют.
Раствор сравнения. 1мг синенсетина Р раст¬
воряют в метаноле Р и доводят до объема 20 мл
этим же растворителем.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: метанол Р — этилаце-
тэт Р — толуол Р (5:40:55, об/об/об).
A. Листовая пластинка при просматривании в по¬
перечном разрезе с секреторным волоском с четырех¬
клеточной головкой (Аа) и головчатым волоском с одно¬
клеточной головкой (АЬ).
B. Верхний эпидермис при просматривании с по¬
верхности с головчатым волоском с двухклеточной го¬
ловкой (Ва) и нижерасположенной палисадной паренхи¬
мой (ВЬ).
С и Е. Членистые покровные волоски (обычно обна¬
руживаются только фрагменты).
Р. Нижний эпидермис при просматривании с по¬
верхности с устьицами диацитного типа (Оа) и секретор¬
ным волоском с четырехклеточной головкой (ОЬ).
Р. Край листовой пластинки.
6. Выделительный волосок.
Н. Нижний эпидермис при просматривании с по¬
верхности с устьицами диацитного типа (На), головча¬
тым волоском с одноклеточной головкой (НЬ) и много¬
клеточным покровным волоском (Нс).
Л. Покровные волоски на краю листовой пластинки.
Рисунок 1229.-1. Микроскопические признаки
листьев ортосифона тычиночного
(почечного чая)
Наносимый объем пробы: по 10 мкл в виде
полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора в нижней трети и около фронта подвижной
фазы обнаруживаются флуоресцирующие зоны крас¬
ного цвета.
162*. Зак. 1060.
1284
Гэсударственная фармакопея Республики Беларусь
Верх хроматографической пластинки
Одна или две
флуоресцирующих зоны от
синего до фиолетово-синего
цвета
Синенсетин: интенсивная
флуоресцирующая зона
светло-голубого цвета
Основная
флуоресцирующая зона
голубого цвета (синенсетин)
Две флуоресцирующие зоны
голубоватого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли диаметром более 1 мм — не
более 5 %; почерневшие с обеих сторон листья — не
более 2 %. Органические примеси: не более 1 %. Ми¬
неральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 11,0%. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 12,5 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
(§) Жидкостная хроматография (2.2.29).
Испытуемый раствор. К 2,5 г измельченного сы¬
рья (355) (2.9.12) прибавляют 100 мл метиленхлори-
да Р и нагревают на водяной бане в течение 30 мин
при перемешивании. Фильтруют. Экстракцию повторя¬
ют дважды. Фильтраты объединяют и выпаривают при
пониженном давлении досуха. Остаток растворяют в
25.0 мл подвижной фазы, при необходимости исполь¬
зуют ультразвуковую баню. Фильтруют через нитро¬
целлюлозный фильтр с диаметром пор 0,45 мкм.
Раствор сравнения. 5 мг синенсетина Р раст¬
воряют в 80 мл подвижной фазы, при необходимости
используя ультразвуковую баню, и доводят до объема
100.0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: тетрагидрофуран Р — кис¬
лота уксусная Р — вода Р — метанол Р (5:8:42:45,
об/об/об/об);
- скорость подвижной фазы: 0,5 мл/мин;
- спектрофотометрический детектор, длина
волны 258 нм.
- объем вводимой пробы: 20 мкл.
Содержание синенсетина в процентах рассчиты¬
вают по формуле:
т2 • 51 • 25
тг52 '
где:
51 — площадь пика синенсетина на хроматограм¬
ме испытуемого раствора;
32 — площадь пика синенсетина на хроматограм¬
ме раствора сравнения;
/т?1 — масса навески испытуемого сырья, г;
т2 — масса навески синенсетина Р, г.
07/2016:1459
ПАССИФЛОРЫ ТРАВА
РаззШогае ЬегЬа
РА38ЮЫ Р1.0МЕР
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и/или начала плодо¬
ношения, измельченная и высушенная трава много¬
летнего растения РаззШога тсагпа1а 1_.
Содержание: не менее 1,5% суммы флавонои-
дов в пересчете на витексин (С21Н20О10; М.м. 432,4)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Стебель зеле¬
ного, или зеленовато-серого, или коричневого цвета,
деревянистый, полый, продольно исчерчен, гладкий
или очень слабо опушен, диаметром обычно менее
8 мм. Листья зеленого или зеленовато-коричневого
цвета, очередные, мелкозубчатые, опушенные, глубо¬
кораздельные на три острых доли, средняя из кото¬
рых самая крупная. Главная жилка на много больше
выступает на нижней стороне листа. Черешок опу¬
шен и имеет два темных нектарника около листовой
пластинки. Усики многочисленны, выходят из пазух
листьев; усики мелкие, гладкие, круглые, оканчива¬
ются цилиндрическими спиральками. Лучеобразные
цветки, при их наличии, имеют 3 мелких прицветника
и венчик, состоящий из 5 белых вытянутых лепестка
с несколькими рядами нитевидных лепестковидных
отростков. Могут обнаруживаться зеленоватые или
коричневатые плоды овальной формы или расплю¬
щенные; плоды содержат несколько сплюснутых ко¬
ричневато-желтых ямчатых семян.
@ В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет светло-зеленый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружи¬
ваются следующие диагностические признаки: фраг¬
менты эпидермиса листа с извилистыми стенками и
устьицами аномоцитного типа (2.8.3); многочислен¬
ные отдельные или расположенные вдоль жилок дру¬
зы оксалата кальция; большое количество отдельных
или сгруппированных волокон из стебля, ассоцииро¬
ванных с ямчатыми сосудами и трахеидами; простые
волоски с 1—3 тонкостенными клетками прямые или
слегка искривлены, оканчиваются в точке или ино¬
гда в крючке. Кроме того, если присутствуют цветки,
в порошке обнаруживается бородавчатый эпидермис
лепестков и придатков, зерна пыльцы с сетчатой эк-
зиной; если присутствуют зрелые плоды, в порошке
обнаруживаются разбросанные таниновые клетки и
коричневато-желтые ямчатые фрагменты семенной
кожуры.
® С. Просматривают хроматограммы, получен¬
ные в испытании «Другие виды РаззШога».
На хроматограмме испытуемого раствора ниже
зоны рутина на хроматограмме раствора сравнения
обнаруживается зона с интенсивной флуоресцен¬
цией желтого цвета, над ней — флуоресцирующая
зона зеленого цвета (дигликозилфлавон); ниже зоны
гиперозида на хроматограмме раствора сравнения
обнаруживается флуоресцирующая зона желтого
цвета (изоориентин), над ней — флуоресцирующая
зона зеленого цвета (изовитексин); над зоной типе-
Пастушьей сумки трава
1285
розида на хроматограмме раствора сравнения об¬
наруживается флуоресцирующая зона коричнева¬
то-желтого цвета (ориентин), над ней — флуоресци¬
рующая зона зеленого цвета (витексин). Последние
две зоны могут отсутствовать. На хроматограмме
испытуемого раствора могут обнаруживаться и дру¬
гие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: незрелые плоды — не более 6 %; части
стеблей — не более 60 %. Органические примеси: не
более 2 %. Минеральные примеси: не более 1 %.
® Другие виды РаззШога. Тонкослойная хро¬
матография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.72) прибавляют 5 мл метанола Р и
кипятят с обратным холодильником в течение 10 мин.
Охлаждают и фильтруют.
Раствор сравнения. 2,0 мг рутина Р и 2,0 мг ги-
перозида Р растворяют при нагревании в 10 мл мета¬
нола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метилэтилкетон Р — этилаце-
тат Р (10:10:30:50, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
10 г/л дифенилборной кислоты аминоэтилового
эфира Р в метаноле Р и затем раствором 50 г/л ма-
крогола 400 Р в метаноле Р. Через 30 мин пластинку
просматривают в ультрафиолетовом свете при длине
волны 365 нм.
Результаты: на хроматограмме раствора
сравнения в нижней трети обнаруживается флуо¬
ресцирующая зона желтовато-коричневого цвета
(рутин) и в центальной части — флуоресцирую¬
щая зона желтовато-коричневого цвета (гиперо-
зид). На хроматограмме испытуемого раствора не
должны обнаруживаться флуоресцирующие зоны
зеленовато-желтого или оранжево-желтого цвета
между зонами дигликозилфлавона и изоориентина
(Р. соеги1еа, Р. едиНз).
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 13,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Исходный раствор. 0,200 г измельченного
сырья (250) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 100 мл, прибавляют 40 мл спирта
(60 %, об/об) Р и нагревают с обратным холодиль¬
ником в водяной бане при температуре 60 °С в те¬
чение 30 мин при частом перемешивании. Охлаж¬
дают, процеживают через ватный тампон в колбу
вместимостью 100 мл. Ватный тампон с остатками
сырья переносят обратно в круглодонную колбу,
прибавляют 40 мл спирта (60 %, об/об) Р и нагре¬
вают с обратным холодильником в водяной бане
при температуре 60 °С в течение 10 мин. Охлаж¬
дают, фильтруют через бумажный фильтр вместе с
первым фильтратом в мерную колбу вместимостью
100 мл. Ополаскивают круглодонную колбу, колбу
вместимостью 100 мл и фильтр спиртом (60%,
об/об) Р и доводят до объема 100,0 мл этим же
растворителем.
Испытуемый раствор. 5,0 мл исходного раст¬
вора помещают в круглодонную колбу и выпари¬
вают в вакууме. Остаток растворяют в 10 мл сме¬
си из метанола Р и кислоты уксусной ледяной Р
(10:100, об/об), прибавляют 10 мл раствора, содер¬
жащего 25 г/л кислоты борной Р и 20 г/л кислоты
щавелевой Р в кислоте муравьиной безводной Р, и
доводят кислотой уксусной безводной Р до объема
25,0 мл.
Компенсационный раствор. 5,0 мл исходного
раствора помещают в круглодонную колбу и выпа¬
ривают в вакууме. Остаток растворяют в 10 мл сме¬
си из метанола Р и кислоты уксусной ледяной Р
(10:100, об/об), прибавляют 10 мл кислоты мура¬
вьиной безводной Р и доводят кислотой уксусной
безводной Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 401 нм.
Содержание суммы флавоноидов в пересче¬
те на витексин в процентах рассчитывают по фор¬
муле:
А-500
т-628 ’
где:
628 — удельный показатель поглощения витек-
сина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0078
ПАСТУШЬЕЙ СУМКИ ТРАВА
Вигзае раз(опз ЬегЬа
ЗНЕРНЕРО’З РИВЗЕ НЕРВ*
ОПРЕДЕЛЕНИЕ
Собранная в фазы цветения и начала плодо¬
ношения (до побурения плодов) и высушенная над¬
земная часть однолетнего растения СарзеПа Ьигза-
раз(опз 1_.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Олиственные
стебли длиной до 40 см, простые или ветвистые с
ребристой поверхностью, голые или в нижней части
слабоопушенные, с цветками или незрелыми плода¬
ми на вытянутых кистевидных соцветиях, часто с ро¬
зетками прикорневых листьев. Прикорневые листья
продолговато-ланцетные, черешковые, перистораз¬
дельные с острыми треугольными струговидно-вы-
емчатыми, цельнокрайними или зубчатыми долями;
стеблевые — очередные, сидячие, продолговато-
ланцентые цельнокрайние или выемчато-зубчатые;
верхние — почти линейные со стреловидным основа¬
нием. Цветки мелкие, правильные, раздельнолепест¬
ные. Чашечка из четырех продолговато-яйцевидных,
зеленых чашелистиков. Венчик из четырех обратно¬
яйцевидных лепестков. Тычинок шесть, из которых
четыре длиннее остальных. Плоды — стручочки, об-
1286
Гэсударственная фармакопея Республики Беларусь
ратнотреугольно-сердцевидные, на верхушке слегка
выемчатые, сплюснутые, с двумя раскрывающимися
створками. Цвет стеблей, листьев и плодов зеленый,
цветков — беловатый.
В. Микроскопия (2.8.23). При просматривании
листа с поверхности видны мелкие клетки эпидермиса
с тонкими стенками, с верхней стороны слегка изви¬
листые в очертании, с нижней — сильно извилистые.
Устьица с обеих сторон, на нижней стороне их боль¬
ше, мелкие, окружены тремя клетками эпидермиса, из
которых одна значительно мельче двух других (ани-
зоцитный тип) (2.8.3). На обеих сторонах листа много
одноклеточных волосков: разветвленные волоски трех,
шести- и реже семиконечные с грубобородавчатой по¬
верхностью, лучи волоска прижаты к поверхности ли¬
ста; простые волоски крупные, одноклеточные, реже
двух- и трехклеточные, с широким основанием и узким,
заостренным концом, поверхность гладкая или слегка
бородавчатая; двухконечные вильчатые волоски с лу¬
чами, приподнимающимися над поверхностью листа,
встречаются редко. Эпидермис стебля состоит из кле¬
ток с прямыми стенками, сильно удлиненными по про¬
дольной оси стебля; имеются устьица. На поперечном
срезе стебля видны: снаружи эпидермис, к нему при¬
мыкает коровая паренхима, затем следует сплошное
механическое кольцо, в котором выделяются проводя¬
щие пучки, состоящие из ксилемы, флоэмы и сопро¬
вождающей группы склереид. Сердцевина большая,
заполненная тонкостенной паренхимой, местами разо¬
рванной. Пыльца гладкая, круглая, с тремя порами.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни (в том числе отделенные при ана¬
лизе), части растения, пораженные мучнистой росой,
и пожелтевшие листья — не более 3 %; частицы, про¬
ходящие сквозь сито (710) (2.9.12), — не более 2 %.
Органические примеси: не более 2 %. Минеральные
примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
Вещества, извлекаемые спиртом (70 %, об/об).
Не менее 10 %.
1,000 г измельченного сырья (1400) (2.9.12) поме¬
щают в коническую колбу вместимостью 250 мл, при¬
бавляют 50,0 мл спирта (70 %, об/об) Р, закрывают
колбу пробкой, взвешивают с точностью 0,01 г и вы¬
держивают в течение 1 ч. Затем колбу присоединяют
к обратному холодильнику и нагревают, поддерживая
слабое кипение, в течение 2 ч. После охлаждения кол¬
бу с содержимым вновь закрывают той же пробкой,
взвешивают и потерю в массе восполняют спиртом
(70 %, об/об) Р. Содержимое колбы тщательно взбал¬
тывают и фильтруют через сухой бумажный фильтр.
25,0 мл полученного фильтрата переносят в предва¬
рительно высушенную при температуре от 100 °С до
105 °С и взвешенную выпарительную чашку, выпари¬
вают на водяной бане досуха и сушат при температу¬
ре от 100 °С до 105 °С до постоянной массы.
Проводят два параллельных испытания. За окон¬
чательный ответ принимают среднее арифметиче¬
ское двух параллельных испытаний.
07/2016:1364
<§> ПЕРВОЦВЕТА КОРНИ
Рпти1ае гасЛх
РШМ1ЛА ЯООТ
ОПРЕДЕЛЕНИЕ
Цельные или резанные высушенные корневища
и корни Рпти1а Vе^^з I. или Рпти1а еШюг НШ.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Корневище
неравномерно бугристое, серовато-коричневого
цвета, прямое или слегка изогнутое, длиной около
(1—5) см и толщиной около (2—4) мм. На верхуш¬
ке корневища часто имеются остатки стеблей и ли¬
стьев. От корневищ отходят многочисленные хруп¬
кие корни толщиной около 1 мм и длиной обычно
(6—8) см. Цвет корня Р. е1аИог светло-коричневый
или красновато-коричневый, корня Р. Vе^^з— свет¬
ло-желтый или желтовато-белый. Излом — глад¬
кий.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет серовато-корич¬
невый. Просматривают под микроскопом с исполь¬
зованием раствора хлоралгидрата Р. В порошке
обнаруживаются следующие диагностические при¬
знаки (рисунок 1364.-1): фрагменты паренхимы
коры корня или корневища и сердцевины корневи¬
ща [Э, Н], состоящие из округлых или яйцевидных
клеток с неравномерно утолщенными и ямчатыми
стенками; коричневатые фрагменты поверхност¬
ной ткани корня с всасывающими волосками [С];
желтые или коричневатые фрагменты эпидермиса
корневища, покрытого бороздчатой кутикулой при
просматривании с поверхности [А] или на попереч¬
ном срезе [Р] с прилегающей паренхимой коры [Ра];
сетчатые сосуды [В] иногда со спиральными сосу¬
дами [3]; группы больших, сильноямчатых желто¬
вато-зеленых склереидов сердцевинной паренхи¬
мы корневища [Е], характерные для Р. е!аИог. При
просматривании измельченного сырья с использо¬
ванием 50 % (об/об) раствора глицерина Р видны
простые или сложные зерна крахмала различной
формы и размера [О].
C. Тонкослойная хроматография (2.2.27),
как указано в испытании «Корень Утсе1ох1сит
ЫгипсИпапа МесЛк.», со следующими изменениями.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р, нагревают при темпе¬
ратуре от 100 °С до 105 °С в течение (5—10) мин и
просматривают при дневном свете.
Результаты: на хроматограмме раствора
сравнения обнаруживается основная зона синева¬
то-фиолетового цвета (эсцин), расположенная на
границе между нижней и средней третями. На хро¬
матограмме испытуемого раствора обнаруживают¬
ся одна или две интенсивные зоны темно-фиоле¬
тового цвета, расположенные немного ниже зоны
эсцина на хроматограмме раствора сравнения. На
хроматограмме испытуемого раствора могут об¬
наруживаться и другие зоны бледно-фиолетового,
желтоватого или коричневато-зеленого цвета.
ИСПЫТАНИЯ
Корни \Гтсе1ох'1сит ЫгипсЛпапа МесЛк. Тонко¬
слойная хроматография (2.2.27).
Пижмы цветки
1287
Рисунок 1364.-1. Микроскопические признаки
корней первоцвета
Испытуемый раствор. К 1,0 г измельченного
сырья (500) (2.9.12) прибавляют 10 мл спирта (70 %,
об/об) Р, нагревают с обратным холодильником в те¬
чение 15 мин, охлаждают и фильтруют.
Раствор сравнения. 10 мг эсцина Р растворяют
в 1,0 мл спирта (70 %, об/об) Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР2В4Р.
Подвижная фаза: верхний слой смеси кислота
уксусная ледяная Р — вода Р — бутанол Р (10:40:50,
об/об/об).
Наносимый объем пробы: по 20 мкл в виде
полос.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограммах раствора
сравнения и испытуемого раствора обнаруживается
зона поглощения (эсцин), расположенная на границе
между нижней и средней третями. Отмечают эту зону.
Проявление В: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты В: на хроматограмме испытуемого
раствора не должны обнаруживаться флуоресцирую¬
щие зоны светло-синего или зеленоватого цвета ниже
основной зоны эсцина на хроматограмме раствора
сравнения.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 9,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
07/2016:РБ0079
ПИЖМЫ ЦВЕТКИ
ТапасеИ уи!дапз Лоз
ТАЫ8У Р1-ОШЕК*
ОПРЕДЕЛЕНИЕ
Собранные в начале цветения и высушенные со¬
цветия (цветки) многолетнего травянистого растения
Тапасе1ит уи1даге Ь.
Содержание: не менее 2,5 % суммы флавонои-
дов и фенолкарбоновых кислот в пересчете на лю-
теолин (С15Н10О6; М.м. 286,2) (в пересчете на сухое
сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Части сложного
щитковидного соцветия и отдельные цветочные кор¬
зинки. Корзинки полушаровидной формы с вдавлен¬
ной серединой, диаметром (6—8) мм, состоят из мел¬
ких трубчатых цветков: краевых — пестичных, средин¬
ных — обоеполых. Цветоложе голое, неполое, слегка
выпуклое, окружено обверткой из черепитчато распо¬
ложенных ланцетных с пленчатым краем листочков.
Цветоносы бороздчатые, голые, реже слабо опушен¬
ные. Цвет цветков желтый, листочков обвертки — ко¬
ричневато-зеленый, цветоносов — светло-зеленый.
B. Микроскопия (2.8.23). При просматривании
листочка обвертки с поверхности видна центральная
жилка, сопровождающаяся секреторными ходами.
Клетки эпидермиса с наружной стороны листочка
крупные, с прямыми или слегка извилистыми стен¬
ками, заметна складчатость кутикулы. Клетки эпи¬
дермиса с внутренней стороны узкие и сильно вы¬
тянутые. Устьица и волоски встречаются только с на¬
ружной стороны листочка обвертки и сосредоточены
главным образом по центральной жилке и по краю.
Устьица окружены 4—6 околоустьичными клетками
(аномоцитный тип) (2.8.3). Волоски многоклеточные,
бичевидные, конечная клетка очень длинная, пере¬
крученная и часто обломанная. Клетки эпидермиса
венчика многоугольные, тонкостенные, некоторые из
них имеют четковидные утолщения. На поверхности
цветков имеются эфиромасличные железки, наибо¬
лее густо расположенные на завязи и у основания
трубочки венчика. Железки четырех-, шестиклеточ¬
ные, двухрядные, двух- трехярусные. В мезофилле и
клетках эпидермиса венчика встречаются друзы окса¬
лата кальция, сосредоточенные в местах срастания
лепестков и на границе венчика и завязи. На поверх¬
ности листочка обвертки железки встречаются редко.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р и
нагревают в водяной бане при температуре 65 °С в
течение 10 мин. Охлаждают и фильтруют.
Раствор сравнения. 1 мг кофейной кислоты Р,
1 мг хлорогеновой кислоты Р и 2,5 мг кверцетина
дигидрата Р растворяют в 10 мл метанола Р.
Пластинка : ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат Р (7:7:14:72, об/об/об/об).
Наносимый объем пробы: по 30 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
1288
Гэсударственная фармакопея Республики Беларусь
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: горячую пластинку опрыскивают
раствором 10 г/л дифенилборной кислоты аминоэ¬
тилового эфира Р в метаноле Р и раствором 50 г/л
макрогола 400 Р в метаноле Р. Пластинку высуши¬
вают на воздухе в течение около 30 мин и просма¬
тривают в ультрафиолетовом свете при длине волны
365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие флуоресци¬
рующие зоны.
Верх хроматографической пластинки
Кверцетин:
флуоресцирующая зона
желтовато-коричневого
цвета
Кофейная кислота:
флуоресцирующая зона
светло-синего цвета
Хлорогеновая кислота:
флуоресцирующая зона
светло-синего цвета
Раствор сравнения
Флуоресцирующая зона
желто-коричневого цвета
Одна или две
флуоресцирующие зоны
светло-синего цвета
Флуоресцирующая зона
желтовато-коричневого
цвета
Флуоресцирующая зона
желтовато-коричневого
цвета
Флуоресцирующая зона
светло-синего цвета
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: другие части растения (стебли, ли¬
стья) — не более 40 %; почерневшие и побуревшие
корзинки — не более 8 %. Органические примеси: не
более 1 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 9,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2.000 г измельченного сырья (710) (2.9.12) поме¬
щают в плоскодонную колбу вместимостью 300 мл со
шлифом и прибавляют 200 мл 96 % спирта Р. Колбу
закрывают и взвешивают с точностью до 0,01 г и на¬
гревают с обратным холодильником на водяной бане
в течение 3,5 ч. Колбу с содержимым охлаждают до
комнатной температуры, взвешивают и доводят массу
колбы до первоначальной 96 % спиртом Р. Извлече¬
ние фильтруют через бумажный фильтр, отбрасывая
первые 20 мл фильтрата.
50.0 мл фильтрата переносят в круглодонную
колбу вместимостью 250 мл и выпаривают в ваку¬
уме досуха. Сухой остаток в колбе промывают три
раза по 20 мл 1,2-дихлорэтаном Р, насыщенным
водой Р. Затем содержимое колбы количественно
переносят в мерную колбу вместимостью 100 мл
с помощью буферного раствора рН 9,0 Р1 четы¬
ре раза порциями по 20 мл и доводят до объема
100,0 мл этим же растворителем. Содержимое кол¬
бы переносят в делительную воронку вместимо¬
стью 250 мл и очищают 1,2-дихлорэтаном Р че¬
тыре раза порциями по 20 мл. 1,0 мл очищенного
раствора разводят буферным раствором рН 9,0 Р1
до объема 25,0 мл.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при длине волны 310 нм, используя
в качестве компенсационного раствора буферный
раствор рН 9,0 Р1.
Содержание суммы флавоноидов и фенолкарбо-
новых кислот в пересчете на лютеолин в процентах
рассчитывают по формуле:
А -10000
/77 • 338,2 ’
где:
338,2 — удельный показатель поглощения люте-
олина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0080
ПИОНА УКЛОНЯЮЩЕГОСЯ
КОРНЕВИЩА И КОРНИ
Раеоп/ае апота1ае гЫгота е1 гад'\х
РЕОЫУ КООТ*
ОПРЕДЕЛЕНИЕ
Собранные в период цветения, очищенные от
земли, отмытые, разрезанные на куски и высушенные
корневища и корни многолетнего растения Раеоп/а
апота1а 1_.
Содержание: не менее 3,5 % суммы иридоидов
в пересчете на пеонифлорин (С^Н^О^; М.м. 480,5)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Куски корневищ
и корней различной формы длиной от 1 см до 9 см,
толщиной от 0,2 см до 1,5 см. Снаружи от желтовато-
коричневого до темно-коричневого цвета, продольно¬
морщинистые. Излом неровный, белого или белова¬
то-желтоватого цвета, по краю иногда красновато-фи¬
олетовый. На поперечном разрезе или изломе видны:
снаружи тонкий слой перидермы; белый слой коры;
резко выступающие желтоватые клиновидные участ¬
ки древесины и светлые сердцевинные лучи. Запах
сильный, своеобразный.
B. Микроскопия (2.8.23). При просматривании
поперечного среза корня видно, что покровная ткань
представлена 3—6 рядами небольших клеток пробки.
Под пробкой лежат крупные тангентально-вытянутые
клетки, имеющие значительное утолщение танген-
тальной стенки оболочки и в углах (уголково-пластин¬
чатая колленхима). Кора состоит из округло-овальных
клеток паренхимы, среди которых встречаются тяжи
клеток с неравномерноутолщенными оболочками,
пронизанными овальными порами. Линия камбия чет¬
кая. Флоэма и ксилема расположены в виде узких ра¬
диальных секторов, разделенных широкими сердце¬
винными лучами. Клетки флоэмной паренхимы имеют
уголковые утолщения оболочек. Ксилема состоит из
сосудов, расположенных группами и радиальными ря¬
дами, примыкающих к ним узких трахеид и древесной
паренхимы. В сердцевинных лучах часто встречаются
Пиона уклоняющегося трава
1289
участки клеток с утолщенными оболочками, пронизан¬
ными овальными порами. В клетках паренхимы со¬
держатся: округлые, овальные, удлиненно овальные
зерна крахмала часто с усеченным концом, диаме¬
тром от 3 мкм до 30 мкм; оксалат кальция в виде друз
или сростков неопределенной формы и скоплений
мелких кристаллов.
С. К 2 мл раствора А, приготовленного как ука¬
зано в разделе «Количественное определение», при¬
бавляют 2 мл раствора 20 г/л кислоты хлористово¬
дородной Р и нагревают в течение 5 мин. Появляется
характерный запах. При прибавлении к полученному
раствору 2 мл раствора 30 г/л железа (III) хлорида Р
появляется фиолетовое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корневища с остатками стеблей длин¬
ной до 3 см — не более 10 %. Органические примеси:
не более 0,5 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,000 г измельченного сырья (1400) (2.9.12) по¬
мещают в круглодонную колбу вместимостью 100 мл,
прибавляют 60 мл спирта (40 %, об/об) Р и нагрева¬
ют с обратным холодильником в водяной бане в тече¬
ние 45 мин. Охлаждают и фильтруют в мерную колбу
вместимостью 100 мл. К остатку сырья в круглодон¬
ной колбе прибавляют 40 мл спирта (40 %, об/об) Р и
нагревают с обратным холодильником в водяной бане
в течение 15 мин. Охлаждают. Фильтруют, промывая
круглодонную колбу и остаток на фильтре 5 мл спир¬
та (40 %, об/об) Р, в ту же мерную колбу. Доводят
до объема 100,0 мл спиртом (40 %, об/об) Р. 10 мл
полученного раствора пропускают через стеклянную
колонку диаметром 10 мм, заполненную 5 г алюминия
оксида нейтрального Р (раствор А).
Испытуемый раствор. К 5,0 мл раствора А при¬
бавляют 5,0 мл раствора гидроксиламина щелочно¬
го РЗ, выдерживают в течение 20 мин, прибавляют
10.0 мл 1 М раствора хлористоводородной кислоты
и 5,0 мл раствора 10 г/л железа (III) хлорида Рв0,1 М
раствора хлористоводородной кислоты. Переме¬
шивают и фильтруют через предварительно смочен¬
ный водой Р бумажный фильтр.
Компенсационный раствор. К 5,0 мл воды Р при¬
бавляют 5,0 мл раствора гидроксиламина щелочно¬
го РЗ, выдерживают в течение 20 мин, прибавляют
10.0 мл 1 М раствора хлористоводородной кислоты
и 5,0 мл раствора 10 г/л железа (III) хлорида Рв0,1 М
растворе хлористоводородной кислоты.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 512 нм.
Содержание суммы иридоидов в пересчете на
пеонифлорин в процентах рассчитывают по формуле:
А 500
т-16,2 '
где:
16,2 — удельный показатель поглощения продук¬
тов реакции пеонифлорина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:РБ0081
ПИОНА УКЛОНЯЮЩЕГОСЯ ТРАВА
Раеотае апота1ае ЬегЬа
РЕОЫУНЕКВ*
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная надзем¬
ная часть многолетнего растения Раеол/а апота!а 1_.
Содержание: не менее 2,5 % суммы иридоидов
в пересчете на пеонифлорин (С^Н^О^; М.м. 480,5)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь стеблей,
листьев, цветков, бутонов и незрелых плодов различ¬
ной степени развития. Стебли бороздчатые или ре¬
бристые, голые, длиной до 35 см и толщиной до 2 см,
внизу с рыхлой сердцевиной, или полые, вверху плот¬
ные. Листья рассеченные, очередные, голые, сильно
сморщенные; пластинка листа длиной от 3 см до 13
см, с длинным черешком. Сегменты глубокотройча¬
то- или перисторассеченные, средние сегменты трех¬
лопастные, боковые — ланцетные, цельнокрайние.
Цветки крупные, диаметром от 8 см до 13 см, чашечка
из пяти неодинаковых зеленых листочков, лепестков
пять и более. Тычинки многочисленные, пестиков от
трех до пяти, сидящих на диске. Плод состоит из 3—5
листовок. Цвет стеблей коричневато-зеленый, листья
с верхней стороны темно-зеленые, с нижней — свет¬
ло-зеленые; лепестки красные или красновато-корич¬
неватые. Запах слабый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны: клетки эпидермиса с обе¬
их сторон листа с сильно извилистыми антиклиналь¬
ными стенками; многочисленные устьица аномоцит-
ного типа (2.8.3) с 4—6 околоустьичными клетками
только на нижней стороне листа; клетки верхнего эпи¬
дермиса местами имеющего четковидно-утолщенные
оболочки, особенно часто над жилками и по краю сег¬
ментов листа. Над главной жилкой сегмента листа с
верхней стороны имеются многочисленные короткие
волоски, расположенные под острым углом к поверх¬
ности листа. Основание волоска слегка расширенное,
верхушка суженная, но не заостренная и всегда на¬
правлена к верхушке доли листа. Волоски однокле¬
точные, редко двухклеточные, имеющие довольно
толстую оболочку, зернистое содержимое, покрытые
продольноскладчатой кутикулой. С нижней стороны
листа видна аэренхима. С верхней стороны листа в
клетках мезофила видны скопления мелких кристал¬
лов оксалата кальция.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли с остатками корневищ — не бо¬
лее 20 %. Органические примеси: не более 2 %. Ми¬
неральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 7,0 %.
1290
Гэсударственная фармакопея Республики Беларусь
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
3,000 г измельченного сырья (1400) (2.9.12) по¬
мещают в круглодонную колбу вместимостью 100 мл,
прибавляют 60 мл спирта (40 %, об/об) Р и нагрева¬
ют с обратным холодильником в водяной бане в тече¬
ние 45 мин. Охлаждают и фильтруют в мерную колбу
вместимостью 100 мл. К остатку сырья в круглодон¬
ной колбе прибавляют 40 мл спирта (40 %, об/об) Р и
нагревают с обратным холодильником в водяной бане
в течение 15 мин. Охлаждают. Фильтруют, промывая
круглодонную колбу и остаток на фильтре 5 мл спир¬
та (40 %, об/об) Р, в ту же мерную колбу. Доводят
до объема 100,0 мл спиртом (40 %, об/об) Р. 10 мл
полученного раствора пропускают через стеклянную
колонку диаметром 10 мм, заполненную 5 г алюминия
оксида нейтрального Р (раствор А).
Испытуемый раствор. К 5,0 мл раствора А при¬
бавляют 5,0 мл раствора гидроксиламина щелочно¬
го РЗ, выдерживают в течение 20 мин, прибавляют
10.0 мл 1 М раствора хлористоводородной кислоты
и 5,0 мл раствора 10 г/л железа (III) хлорида Рв0,1 М
раствора хлористоводородной кислоты. Переме¬
шивают и фильтруют через предварительно смочен¬
ный водой Р бумажный фильтр.
Компенсационный раствор. К 5,0 мл воды Р при¬
бавляют 5,0 мл раствора гидроксиламина щелочно¬
го РЗ, выдерживают в течение 20 мин, прибавляют
10.0 мл 1 М раствора хлористоводородной кислоты
и 5,0 мл раствора 10 г/л железа (!!!) хлорида Рв0,1 М
растворе хлористоводородной кислоты.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 512 нм.
Содержание суммы иридоидов в пересчете на
пеонифлорин в процентах рассчитывают по формуле:
А -500
/77 • 16,2 ’
где:
16,2 — удельный показатель поглощения продук¬
тов реакции пеонифлорина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016: РБ0082
ПОДОРОЖНИКА БОЛЬШОГО
ЛИСТЬЯ
Р1ап1ад1П18 тарпз Шит
СОММОЫ Р1.АЫТАШ 1.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные во время цветения и высушенные
листья многолетнего травянистого растения Р1ап!адо
та]ог 1_.
Содержание: не менее 12,0 % полисахаридов (в
пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные или
частично измельченные листья, широкояйцевидные
или широкоэллиптические, цельнокрайние или слегка
зубчатые, с 3—9 продольными дугообразными жилка¬
ми, суженные в широкий черешок различной длины. В
месте обрыва черешка видны длинные остатки тем¬
ных нитевидных жилок. Длина листьев с черешком до
24 см, ширина от 3 см до 11 см. Цвет зеленый или
коричневато-зеленый.
B. Микроскопия (2.8.23). Видны: клетки верхне¬
го эпидермиса — многоугольные с прямыми стенка¬
ми, нижнего — со слабоизвилистыми стенками. Кути¬
кула местами образует складки. Устьица имеются на
обеих сторонах листа, преимущественно на нижней,
округлые, окружены 3—4 клетками эпидермиса (ано-
моцитный тип) (2.8.3). Волоски простые и головчатые.
Простые волоски с расширенным основанием, много¬
клеточные, гладкие. Головчатые волоски двух типов:
на одноклеточной ножке с удлиненной двухклеточной
головкой, реже встречаются головчатые волоски на
многоклеточной ножке с шарообразной или овальной
одноклеточной головкой. В местах прикрепления во¬
лосков клетки эпидермиса образуют розетку.
C. К 10 мл извлечения, полученного как указано в
разделе «Количественное определение», прибавляют
30 мл 96 % спирта Р и перемешивают. Появляются
хлопьевидные сгустки, выпадающие в осадок при сто¬
янии (полисахариды). Раствор с осадком фильтруют
через стеклянный фильтр (16) и собирают осадок с
фильтра. Осадок растворяют в 1 мл 0,1 М раствора
натрия гидрооксида, прибавляют 0,25 мл раствора
5 г/л карбазола Р в 96% спирте, свободном от аль¬
дегидов, Р, 5 мл кислоты серной Р и перемешивают.
Нагревают на водяной бане в течение 10 мин. Появ¬
ляется красно-фиолетовое окрашивание (галактуро-
новая кислота).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побуревшие и почерневшие листья —
не более 5 %; цветочные стрелки — не более 1 %.
Органические примеси: не более 1 %. Минеральные
примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 20,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,00 г измельченного сырья (500) (2.9.12) по¬
мещают в колбу вместимостью 500 мл, прибавляют
100 мл воды Р и кипятят с обратным холодильни¬
ком при периодическом перемешивании в течение
30 мин. Оставляют до оседания частиц и собирают
надосадочную жидкость. Экстракцию повторяют еще
четыре раза с использованием воды Р порциями по
100 мл. Водные извлечения объединяют и центрифу¬
гируют при 5000 об/мин в течение 10 мин. Всю надо¬
садочную жидкость процеживают через ватный там¬
пон и доводят водой Р до объема 500,0 мл. 25,0 мл
полученного раствора помещают в центрифужную
пробирку, прибавляют 75 мл 96 % спирта Р, пере¬
мешивают, подогревают в водяной бане при 60 °С в
течение 5 мин. Через 30 мин содержимое центри¬
фугируют при 5000 об/мин в течение 30 мин. Надо¬
садочную жидкость фильтруют под вакуумом через
высушенный при температуре от 100 °С до 105 °С до
постоянной массы стеклянный фильтр (ПОР 16) диа¬
метром 40 мм. Осадок количественно переносят на
Подорожника ланцетного листья
1291
фильтр и последовательно промывают 15 мл смеси
из 96 % спирта Р и воды Р (3:1, об/об), 10 мл ацето¬
на Р и 10 мл этилацетата Р. Фильтр с осадком вы¬
сушивают сначала на воздухе, затем при температуре
от 100 °С до 105 °С до постоянной массы.
Содержание полисахаридов в процентах рассчи¬
тывают по формуле:
(т2 -лт,)* 2000
т
где:
/771 — масса фильтра, г;
т2 — масса фильтра с осадком, г;
т — масса навески испытуемого сырья, г.
07/2016:1884
® ПОДОРОЖНИКА ЛАНЦЕТНОГО
ЛИСТЬЯ
Р1ап1ад'1П131апсеоШае Шит
ШВШОКТ Р1.АЫТА1Ы
ОПРЕДЕЛЕНИЕ
Целые или фрагментированные, высушенные
листья и стебель Р1ап1адо 1апсео1а1а 1_. 8.1.
Содержание: не менее 1,5 % суммы произво¬
дных о-дигидроксикоричной кислоты в пересчете на
актеозид (С29Н36015; М.м. 624,6) (в пересчете на сухое
вещество).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки. Листья длиной до 30 см
и шириной до 4 см, от желтовато-зеленого до корич¬
невато-зеленого цвета с выпуклыми беловато-зеле¬
ными почти параллельными жилками на абаксиаль-
ный поверхности. Лист состоит из ланцетовидной
листовой пластинки, сужающейся у основания в же¬
лобчатый черешок. Край нечетко зубчатый и часто
волнистый. Имеет 3, 5 или 7 основных жилок прибли¬
зительно одинаковых по длине и идущих почти па¬
раллельно. Волоски могут почти отсутствовать, быть
редко размещенными или иногда густыми, особенно
на нижней поверхности и над жилками. Стебель ко¬
ричневато-зеленый, более длинный, чем листья, с
диаметром (3—4) мм, имеющий глубокие желобки в
продольном направлении с 5—7 заметными жилка¬
ми. Поверхность обычно покрыта тонкими волосками.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет желтовато-зеле¬
ный. Просматривают под микроскопом с использова¬
нием раствора хлоралгидрата Р. В порошке обна¬
руживаются следующие диагностические признаки
(рисунок 1884.-1): фрагменты эпидермиса, состоя¬
щие из клеток с нерегулярными извилистыми анти¬
клинальными стенками, фрагменты верхнего эпи¬
дермиса листовой пластинки (вид с поверхности [Н]
и поперечный срез [й]) с прилегающей столбчатой
паренхимой [Оа, На] и фрагменты нижнего эпидер¬
миса (вид с поверхности [О]) имеют устьица (2.8.3)
преимущественно диацитного типа [Са] и иногда
аномоцитного типа [СЬ]; многоклеточные одноряд¬
ные конические покрывающие трихомы, являющие¬
ся очень типичными, целые [С] или преимуществен¬
но фрагментированные [А] с базальной клеткой
большего по сравнению с другими эпидермальны¬
ми клетками размера, за которой следует короткая
клетка, поддерживающая 2 или более продолго¬
ватые клетки с узкой и изменяющейся полостью,
перекрываемой через интервалы, соответствующие
небольшим набуханиям в трихоме и создающие
сочленение, конечная клетка имеет остроконечную
верхушку и нитевидную полость; железистые трихо¬
мы имеют одноклеточную цилиндрическую ножку и
многоклеточную продолговатую коническую головку,
состоящую из нескольких рядов небольших клеток,
и одиночную конечную клетку [В, Ос]; плотные груп¬
пы одервенелой сосудисто-волокнистой ткани с уз¬
кими спиральными и кольцеобразными утолщенны¬
ми сосудами и небольшими умеренно утолщенными
волокнами [Р]; фрагменты ствола [Е] с клетками с
утолщенными стенками и грубо заостренной кутику¬
лой, устьицами [Ес], многоклеточными однорядными
покрывающими трихомами [ЕЬ] и железистыми три¬
хомами [Еа] ранее описанного типа.
Рисунок 1884.-1 Микроскопические признаки
листьев подорожника ланцетного
С. Просматривают хроматограммы, полученные
как указано в испытании «Листья 01дИаНз 1апа(а».
Результаты А\ ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Актеозид: зона желтого
цвета
Зона желтого цвета
(актеозид)
Аукубин: зона синего цвета
Зона синего цвета (аукубин)
Раствор сравнения
Испытуемый раствор
1292
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Листья 0/д/Та//5 1апа(а. Тонкослойная хромато¬
графия (2.2.27).
Смесь растворителей. Вода Р — метанол Р
(30:70, об/об).
Испытуемый раствор. Используют свежепри¬
готовленный раствор. 1 г измельченного лекарствен¬
ного растительного сырья (355) (2.9.12) помещают в
колбу вместимостью 25 мл, прибавляют 10 мл смеси
растворителей и встряхивают в течение 30 мин. Филь¬
труют, ополаскивают колбу и фильтруют с использова¬
нием двух порций смеси растворителей по 5 мл. Дово¬
дят смесью растворителей до объема 25 мл.
Раствор сравнения. 1 мг актеозида Р и 1 мг
аукубина Р растворяют в 1 мл смеси растворителей.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯР254 Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат Р (11:11:27:100, об/об/об/об).
Наносимый объем пробы: 10 мкл, в виде полос.
Фронт подвижной фазы: не менее 8 см от линии
старта; нагревают незамедлительно после хромато¬
графирования при температуре около 120 °С в тече¬
ние (5—10) мин.
Проявление А: пластинку просматривают при
дневном свете.
Проявление В: пластинку просматривают в уль¬
трафиолетовом свете при длине волны 365 нм.
Результаты В: на хроматограмме испытуемо¬
го раствора не должна обнаруживаться яркая синяя
флуоресцирующая зона, расположенная ниже крас¬
новато-коричневатой флуоресцирующей зоны, соот¬
ветствующей аукубину на хроматограмме раствора
сравнения.
Допустимые примеси (2.8.2). Листья различно¬
го цвета — не более 5 %; другие допустимые приме¬
си — не более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 10,0 %. 1,000 г измельченного лекарственного
растительного сырья (355) (2.9.12) сушат при темпе¬
ратуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 14,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Исходный раствор. 1,000 г измельченного лекар¬
ственного растительного сырья (355) (2.9.12) помещают
в колбу и прибавляют 90 мл спирта (50 %, об/об) Р и
кипятят в водяной бане с обратным холодильником в
течение 30 мин. Охлаждают и фильтруют в мерную кол¬
бу вместимостью 100 мл. Ополаскивают колбу и филь¬
труют с использованием 10 мл спирта (50 %, об/об) Р.
Объединяют фильтрат и промывные воды и доводят
спиртом (50 %, об/об) Р до объема 100,0 мл.
Испытуемый раствор. В мерную колбу вмести¬
мостью 10 мл прибавляют, перемешивая после каж¬
дого прибавления, 1,0 мл исходного раствора, 2 мл
0,5 М раствора кислоты хлористоводородной, 2 мл
раствора, приготовленного растворением 10 г натрия
нитрита Р и 10 г натрия молибдата Р в 100 мл
воды Р, и 2 мл раствора натрия гидроксида раз¬
веденного Р. Полученный раствор доводят водой Р
до объема 10,0мл. Незамедлительно измеряют оп¬
тическую плотность (2.2.25) испытуемого раствора
при длине волны 525 нм, используя в качестве ком¬
пенсационной жидкости раствор, приготовленный
следующим образом: в мерную колбу вместимостью
10 мл прибавляют 1,0 мл исходного раствора, 2 мл
0,5 М раствора кислоты хлористоводородной и 2 мл
раствора натрия гидроксида разведенного Р и дово¬
дят водой Р до объема 10,0 мл.
Сумму производных о-дигидроксикоричной кис¬
лоты в пересчете на актеозид в процентах рассчиты¬
вают, учитывая, что удельный показатель поглощения
для актеозида при длине волны 525 нм равен 185, по
формуле:
А-1000
185 /7? ’
где:
А — оптическая плотность испытуемого раствора
при длине волны 525 нм;
т — масса навески испытуемого образца, г.
07/2016:1380
<§> ПОЛЫНИ ГОРЬКОЙ ТРАВА
АЬзШН ЬегЬа
и/оялтооо
ОПРЕДЕЛЕНИЕ
Прикорневые листья или слегка покрытые ли¬
стьями цветущие верхушки или смесь высушенных
цельных или измельченных этих частей Аг/ет/з/з
аЬз'т1Ыит I.
Содержание: не менее 2 мл/кг эфирного масла (в
пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья серова¬
тые или зеленоватые, густо опушенные на обеих по¬
верхностях. Прикорневые листья с длинными череш¬
ками имеют треугольные или овальные дважды или
трижды перисторассеченные листовые пластинки
с закругленными или ланцетовидными сегментами.
Стеблевые листья менее сегментированы, верху¬
шечные листья ланцетовидной формы. Цветоносный
стебель зеленовато-серый, опушенный, диаметром
до 2,5 мм и обычно с 5 сглаженными продольными
бороздками. Головки соцветий собраны в свободные,
пазушные метелки, расположенные на уровне ланце¬
товидных или слегка перисторассеченных листьев;
они имеют сферическую или уплощенную полусфе¬
рическую форму, диаметр от 2 мм до 4 мм и состо¬
ят из серой опушенной обвертки с наружными про¬
дольными прицветниками и внутренними — оваль¬
ной формы с тупой верхушкой и пленчатыми краями,
цветоложа с очень длинным верхним прицветником
длиной до 1 мм или более, многочисленными желты¬
ми, трубчатыми, гермафродитными цветками длиной
около 2 мм и несколькими желтыми краевыми языч¬
ковыми цветками.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет зеленовато-серый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки (рисунок
1380.-1): многочисленные Т-образные волоски [А] с
короткой однорядной ножкой, состоящей из 1—5 не¬
больших клеток, перпендикулярно покрытой очень
длинной волнообразной конечной клеткой с за¬
остренными концами; фрагменты эпидермиса (вид
с поверхности [О]) с извилистыми или волнообраз-
Пустырника листья
1293
ными стенками, устьица аномоцитного типа (2.8.3)
[Оа], покровные волоски [йЬ] и железистые волоски,
содержащие масло [Ос] либо не содержащие масло
[Од], каждый с короткой, двурядной двуклеточной
ножкой и двурядной головкой с 2—4 клетками; сво¬
бодные железистые волоски (вид сбоку [С]); фраг¬
менты венчиков из трубчатых и язычковых цветков,
некоторые содержат небольшие друзы кристаллов
оксалата кальция [Н]; многочисленные верхние при¬
цветники цветков, каждый из которых состоит из
небольшой клетки, формирующей ножку, и очень
длинной цилиндрической и тонкостенной конечной
клетки длиной около (1—1,5) мм, цельной [Е] или
ограниченной дистальной частью [В]; шарообразные
зерна пыльцы, диаметром около 30 мкм, с 3 порами
и мелко бородавчатой экзиной [О]; фрагменты сосу¬
дистой ткани листьев [Р] или стеблей у], состоящей
из сосудов со спиральными или кольцеобразными
утолщениями [Ра] или с окаймленными порами [да],
волокнами [РЬ, ЗЬ] и паренхиматозными клетками с
ямчатыми, среднеутолщенными стенками [Рс, Зс].
Рисунок 1380.-1. Микроскопические признаки
травы полыни горькой
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 2 г измельченного сы¬
рья (355) (2.9.12) помещают в 50 мл кипящей воды Р
и выдерживают в течение 5 мин, встряхивая колбу
несколько раз. После охлаждения прибавляют 5 мл
раствора 100 г/л свинца (II) ацетата Р, перемешива¬
ют и фильтруют. Колбу и остаток на фильтре промы¬
вают 20 мл воды Р. Фильтрат встряхивают с 50 мл ме-
тиленхлорида Р. Отделяют органический слой, высу¬
шивают с использованием натрия сульфата безво¬
дного Р, фильтруют и выпаривают досуха на водяной
бане. Остаток растворяют в 0,5 мл 96 % спирта Р.
Раствор сравнения. 2 мг метилового красно¬
го Р и 2 мг резорцина Р растворяют в 10,0 мл мета¬
нола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: ацетон Р — кислота уксус¬
ная ледяная Р — толуол Р — метиленхлорид Р
(10:10:30:50, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: пластинку опрыскивают раство¬
ром уксусного ангидрида — кислоты серной Р. Про¬
сматривают при дневном свете.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается зона синего цвета, соответ¬
ствующая артабсину и расположенная немного выше
зоны красного цвета, соответствующей метиловому
красному, на хроматограмме раствора сравнения.
Проявление В: пластинку нагревают при темпе¬
ратуре от 100 °С до 105 °С в течение 5 мин. Просма¬
тривают при дневном свете.
Результаты В: на хроматограмме раствора
сравнения в средней трети обнаруживается зона
красного цвета, соответствующая метиловому красно¬
му, и ниже ее — зона светло-розового цвета, соответ¬
ствующая резорцину. На хроматограмме испытуемого
раствора обнаруживается интенсивная зона красного
или коричневато-красного цвета, соответствующая
абсинтину, со значением Рр, подобным значению РР
для зоны резорцина на хроматограмме равствора
сравнения. На хроматограмме испытуемого раствора
обнаруживаются другие зоны, менее интенсивные,
чем зона абсинтина.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли диаметром более 4 мм — не бо¬
лее 5 %. Другие допустимые примеси: не более 2 %.
Показатель горечи (2.8.15). Не менее 10 000.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в кислоте хлористово¬
дородной (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 50,0 г резано¬
го сырья помещают в круглодонную колбу вместимо¬
стью 1000 мл и прибавляют 500 мл воды Р в качестве
жидкости для перегонки. В градуированную трубку
помещают 0,5 мл ксилола Р. Перегонку проводят со
скоростью (2—3) мл/мин в течение не менее 3 ч.
07/2016: РБ0083
ПУСТЫРНИКА листья
1-еопип 1оНит
МОТНЕШОКТ1-ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные в фазу начала цветения и высушен¬
ные цельные или измельченные листья многолетнего
травянистого растения 1-еопигиз сагсИаса I. и [.еопигиз
ри1прие1оЬа(из СШЬ. (/.. ППозиз РезТ. ех 0’11пл).
Содержание: не менее 0,4 % суммы иридоидов в
пересчете на гарпагида ацетат (С^Н^О^; М.м. 406,4)
(в пересчете на сухое сырье).
1294
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Стеблевые ли¬
стья или листья соцветий цельные или частично из¬
мельченные, скрученные, с обеих сторон опушенные
волосками, с черешками или без них. Стеблевые
листья в очертании яйцевидные или округлые, трех¬
пятилопастные или раздельные, длиной до 14 см, ши¬
риной до 10 см. Листья соцветий в очертании эллип¬
тические или продолговато-ромбические, в основании
яйцевидные или клиновидные, трехлопастные или
ланцетовидные, зубчатые или цельнокрайние. Цвет
от серо-зеленого до темно-зеленого.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности с обеих сторон видны клет¬
ки эпидермиса с тонкими извилистыми боковыми
стенками, особенно на нижней стороне. Устьица
многочисленные, расположены преимущественно
на нижнем эпидермисе, окружены 3—4 (реже 2)
околоустьичными клетками (аномоцитный тип)
(2.8.3). Железки на короткой ножке с 4—6 (реже 8)
радиально расположенными выделительными
клетками. Волоски двух типов: многочисленные
многоклеточные грубобородавчатые, расширен¬
ные в местах соединения клеток; мелкие головча¬
тые волоски на одно-двухклеточной короткой ножке
с округлой головкой, состоящей из 1—2 клеток.
Видны места прикрепления многоклеточных грубо¬
бородавчатых волосков.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Используют раствора А,
приготовленный как указано в разделе «Количествен¬
ное определение».
Пластинка: ТСХ пластинка со слоем силикагеля 6 Р.
Подвижная фаза: хлороформ Р — метанол Р —
вода Р (80:2:0,1, об/об/об).
Наносимый объем пробы: 50 мкп в виде полосы.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром диметиламинобензальдегида Р8. Нагревают при
температуре от 100 °С до 105 °С в течение 5 мин до
проявления зон. Просматривают при дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается не менее трех зон фиолето¬
во-красного цвета со значениями Я, около 0,4 (гарпа-
гид), около 0,6 (аюгозид) и около 0,8 (гарпагида ацетат).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые части
растения: побуревшие, почерневшие и пожелтевшие
листья — не более 5 %; другие части растения (стеб¬
ли, цветки) — не более 5 %. Органические примеси: не
более 2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 13,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,000 г измельченного сырья (250) (2.9.12) по¬
мещают в круглодонную колбу со шлифом вмести¬
мостью 250 мл, прибавляют 80 мл смеси из спирта
(40 %, об/об) Р и нагревают с обратным холодиль¬
ником на водяной бане в течение 45 мин. Охлажда¬
ют и процеживают через ватный тампон в мерную
колбу вместимостью 100 мл. Ватный тампон поме¬
щают в колбу с остатком сырья, прибавляют 40 мл
спирта (40 %, об/об) Р и нагревают с обратным хо¬
лодильником на водяной бане в течение 15 мин. Ох¬
лаждают, процеживают в ту же мерную колбу и до¬
водят спиртом (40 %, об/об) Р до объема 100,0 мл.
10,0 мл полученного раствора упаривают до объ¬
ема около 5 мл, количественно переносят в мер¬
ную колбу вместимостью 10 мл, используя воду Р, и
доводят до объема 10,0 мл этим же растворителем.
Полученный раствор пропускают через стеклянную
колонку диаметром 10 мм, заполненную 3 г алюми¬
ния оксида нейтрального Р (раствор А).
Испытуемый раствор. К 5,0 мл раствора А
прибавляют 5,0 мл раствора гидроксиламина ще¬
лочного РЗ. Выдерживают в течение 5 мин, при¬
бавляют 10,0 мл 1 М раствора хлористоводород¬
ной кислоты, 5,0 мл раствора 10 г/л железа (III)
хлорида Р в 0,1 М раствора хлористоводородной
кислоты и перемешивают.
Компенсационный раствор. К 5,0 мл воды Р,
прибавляют 5,0 мл раствора гидроксиламина ще¬
лочного РЗ, 10,0 мл 1 М раствора хлористоводо¬
родной кислоты и 5,0 мл раствора 10 г/л железа
(!!!) хлорида Рв0,1 М растворе хлористоводород¬
ной кислоты.
Измеряют оптическую плотность (2.2.25) ис¬
пытуемого раствора при 512 нм.
Содержание суммы иридоидов в пересчете
на гарпагида ацетат в процентах рассчитывают по
формуле:
А 500
т- 56,03 ’
где:
56,03 — удельный показатель поглощения про¬
дуктов реакции гарпагида ацетата;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1833
ПУСТЫРНИКА ТРАВА
1-еопип ЬегЬа
МОТНЕПЖОКТ
ОПРЕДЕЛЕНИЕ
Собранная в фазу начала цветения и высу¬
шенная трава многолетнего травянистого растения
Ьеопигиз сагсИаса 1_. и 1-еопигиз дсшдие/оЬа/из СШЬ.
Содержание:
- не менее 0,2 % флавоноидов в пересчете на ги-
перозид (С21Н20О12; М.м. 464,4) (в пересчете на сухое
сырье);
- или не менее 0,3 % суммы иридоидов в пере¬
счете на гарпагида ацетат (С^Н^О^; М.м. 406,4)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
@> А. Внешние признаки (2.8.23). Кусочки сте¬
бля покрыты волосками, продольно полосатые, че¬
тырехугольные, полые, шириной до около 10 мм; на
стеблях расположены супротивные перекрещива¬
ющиеся черешковые листья, в пазухах верхних ли-
Пустырника трава
1295
стьев расположены около 6—12 маленьких цветков,
собранных в сидячие мутовки, образующие длинный
облиственный колос. Нижние листья овально-окру¬
глые, пальчатораздельные на 3—5, реже на 7 до¬
лей, доли неравномерно-зубчатые. Верхние листья
цельные или трехнадрезанные ланцетовидные с
пильчатым краем, в основании клинообразные. Верх¬
няя поверхность листьев зеленая с разбросанными
волосками, нижняя поверхность — более бледного
зеленого цвета, густо опушенная, с выступающим
пальчатым и сетчатым жилкованием. Цветки имеют
воронкообразную чашечку длиной (3—5) мм с 5 жест¬
кими отогнутыми назад зубчиками; двугубый венчик
с верхней губой розового цвета, опушенной на внеш¬
ней стороне, и белой нижней губой с красновато-фио¬
летовыми пятнами; 4 густоопушенных тычинки.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет зеленый. Просма¬
тривают под микроскопом с использованием раст¬
вора хлоралгидрата Р. В порошке обнаруживают¬
ся следующие диагностические признаки (рисунок
1833.-1): многочисленные покровные волоски [А],
цельные [Аа] или фрагментированные [АЬ], простые,
с бородавчатыми стенками, состоящие из 2—8 кле¬
ток с небольшим утолщением в основании, длиной
до 1500 мкм; фрагменты верхнего эпидермиса при
просматривании с поверхности [В] с клетками с пря¬
мыми или извилистыми антиклинальными стенками
[Ва], часто сопровождаемые палисадной паренхи¬
мой [ВЬ]; фрагменты нижнего эпидермиса при про¬
сматривании с поверхности [С] с клетками с извили¬
стыми антиклинальными стенками [Са], с устьицами
диацитного типа (2.8.3) [СЬ], с железистыми трихома¬
ми с короткой одноклеточной ножкой и шарообразной
головкой, состоящей из 8—16 клеток [Сс], с желези¬
стыми трихомами с одно- или двухклеточной ножкой
и двух- или четырехклеточной головкой [Сс1] и иногда
с покровными волосками; фрагменты листовой пла¬
стинки при просматривании в поперечном разрезе
Р], состоящие из эпидермиса, несущего желези¬
стые трихомы с шарообразной головкой, состоящей
из 8—16 клеток [йа], или с 2—4-кпеточной головкой
[ОЬ], из однослойного палисадного мезофилла, зани¬
мающего почти половину поперечного сечения [Ос],
и рыхлой губчатой паренхимы [Рс!]; фрагменты ча¬
шечки [О] с эпидермисом, состоящим из многоуголь¬
ных клеток, с одно- или двухклеточными конически¬
ми покровными волосками со стенками, покрытыми
шипами, часто вместе с веретенообразными клетка¬
ми мезофилла с толстыми стенками и содержащими
мелкие призматические кристаллы оксалата кальция
[<ЗЬ]; отдельные железистые волоски [Н] либо с мно¬
гоклеточной ножкой и одноклеточной головкой (воло¬
ски пыльников) [На], либо с одно- или двухклеточной
ножкой и 2—4-клеточной головкой [НЬ]; сферические
зерна пыльцы диаметром около (25—30) мкм с тре¬
мя порами и тремя бороздками и с гладкой экзиной
[Е]; толстостенные лигнифицированные волокна [Р];
фрагменты стебля со спиральными и кольчатыми
сосудами [К]; редкие фрагменты перикарпа [3], со¬
стоящего из дольчатых клеток с толстыми ямчатыми
стенками, каждая из которых содержит по одному
призматическому кристаллу оксалата кальция у а].
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (355) (2.9.12) прибавляют 5 мл метанола Р и
нагревают на водяной бане при температуре 65 °С в
Рисунок 1833.-1. Микроскопические признаки
травы пустырника
течение 5 мин при встряхивании. Охлаждают и филь¬
труют.
Раствор сравнения. 5 мг нафтола желтого 8 Р и
2,0 мг каталпола Р растворяют в 5,0 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р— этилацетат Р (20:20:60, об/об/об).
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раство¬
ром диметиламинобензальдегида Р2, используя око¬
ло 5 мл на квадратную пластинку со стороной 200 мм.
Нагревают при температуре от 100 °С до 105 °С в те¬
чение 10 мин до проявления зон. Просматривают при
дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться другие слабые зоны
серовато-синего цвета.
Верх хроматографической пластинки
Широкая зона белого цвета
Зона серовато-синего цвета
(иридоид)
Нафтол желтый 8:
интенсивная зона желтого
цвета
Катал пол: зона серовато-
синего цвета
Одна или две зоны
серовато-синего цвета
(иридоид)
Раствор сравнения
Испытуемый раствор
1296
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: побуревшие и почерневшие части
растения — не более 7 %; стебли, в том числе отде¬
ленные при анализе — не более 46 %. Органические
примеси: не более 3 %. Минеральные примеси: не
более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
{2.9.12) сушат при температуре 105 °С.
® Общая зола {2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте {2.8.1). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
®> Определение содержания флавоноидов в
пересчете на изокверцитрозид.
Исходный раствор. 1,00 г измельченного сы¬
рья (355) {2.9.12) помещают в круглодонную колбу
вместимостью 100 мл, прибавляют 1 мл раствора
5 г/л гексаметилентетрамина Р, 20 мл ацетона Р
и 2 мл кислоты хлористоводородной Р1. Кипятят с
обратным холодильником в течение 30 мин, после
чего извлечение процеживают через ватный тампон
в колбу. Ватный тампон помещают в круглодонную
колбу с остатком сырья и дважды кипятят с обрат¬
ным холодильником в течение 10 мин, каждый раз
прибавляя 20 мл ацетона Р в качестве экстрагента.
Охлаждают и процеживают через ватный тампон.
Объединенные ацетоновые извлечения фильтруют
через бумажный фильтр в мерную колбу вмести¬
мостью 100 мл, ополаскивают круглодонную колбу
ацетоном Р и фильтруют через тот же фильтр, по¬
сле чего промывают фильтр ацетоном Р. Объем
раствора доводят до 100,0 мл ацетоном Р. 20,0 мл
полученного раствора помещают в делительную
воронку, прибавляют 20 мл воды Р и экстрагируют
этилацетатом Р порцией 15 мл, затем еще триж¬
ды порциями по 10 мл. Этилацетатные извлечения
объединяют, помещают в делительную воронку и
дважды промывают водой Р порциями по 50 мл.
Органический слой фильтруют через бумажный
фильтр с 10 г натрия сульфата безводного Р в
мерную колбу, вместимостью 50 мл и доводят эти¬
лацетатом Р до объема 50,0 мл.
Испытуемый раствор. К 10,0 мл исходного
раствора прибавляют 1 мл реактива алюминия
хлорида Р и доводят 5 % {об/об) раствором кисло¬
ты уксусной ледяной Р в метаноле Р до объема
25.0 мл.
Компенсационный раствор. 10,0 мл исходно¬
го раствора доводят 5 % {об/об) раствором кисло¬
ты уксусной ледяной Р в метаноле Р до объема
25.0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 425 нм.
Содержание флавоноидов в пересчете на гипе-
розид в процентах рассчитывают по формуле:
А 625
т•500 ’
где:
500 — удельный показатель поглощения гиперо-
зида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Определение содержания суммы иридоидов
в пересчете на гарпагида ацетат. 5,000 г измельчен¬
ного сырья (250) (2.9.72) помещают в круглодонную
колбу со шлифом вместимостью 250 мл, прибавляют
80 мл смеси из спирта (40 %, об/об) Р и нагревают
с обратным холодильником на водяной бане в тече¬
ние 45 мин. Охлаждают и процеживают через ватный
тампон в мерную колбу вместимостью 100 мл. Ват¬
ный тампон помещают в колбу с остатком сырья, при¬
бавляют 40 мл спирта (40 %, об/об) Р и нагревают с
обратным холодильником на водяной бане в течение
15 мин. Охлаждают, процеживают в ту же мерную
колбу и доводят спиртом (40 %, об/об) Р до объема
100.0 мл. 10,0 мл полученного раствора упаривают до
объема около 5 мл, количественно переносят в мер¬
ную колбу вместимостью 10 мл, используя водуР, и
доводят до объема 10,0 мл этим же растворителем.
Полученный раствор пропускают через стеклянную
колонку диаметром 10 мм, заполненную 3 г алюминия
оксида нейтрального Р (раствор А).
Испытуемый раствор. К 5,0 мл раствора А при¬
бавляют 5,0 мл раствора гидроксиламина щелоч¬
ного РЗ. Выдерживают в течение 5 мин, прибавляют
10.0 мл 7 М раствора хлористоводородной кисло¬
ты, 5,0 мл раствора 10 г/л железа (III) хлорида Р в
0,1 М раствора хлористоводородной кислоты и пе¬
ремешивают.
Компенсационный раствор. К 5,0 мл воды Р,
прибавляют 5,0 мл раствора гидроксиламина щелоч¬
ного РЗ, 10,0 мл 7 М раствора хлористоводородной
кислоты и 5,0 мл раствора 10 г/л железа (III) хлори¬
да Р в 0,1 М растворе хлористоводородной кислоты.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 512 нм.
Содержание суммы иридоидов в пересчете на
гарпагида ацетат в процентах рассчитывают по фор¬
муле:
А >500
т • 56,03 ’
где:
56,03 — удельный показатель поглощения про¬
дуктов реакции гарпагида ацетата;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1860
РАСТОРОПШИ ПЛОДЫ
ЗНуЫ тапап/ 1гис1из
МИК-ТН18ПЕ РРШ1Т
ОПРЕДЕЛЕНИЕ
Собранные и высушенные плоды однолетнего
травянистого растения ЗИуЬит тапапит (I.) Саег1пег.
Содержание:
- не менее 1,5 % силимарина в пересчете на си-
либинин (С25Н22О10; М.м. 482,4) (в пересчете на сухое
сырье);
- или не менее 2,7 % суммы флаволигнанов в пе¬
ресчете на силимарин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Семянка
сильно сплюснутая, вытянутой обратно-яйцевид-
Расторопши плоды
1297
ной формы, длиной около (6—8) мм, шириной 3 мм
и толщиной 1,5 мм; внешняя поверхность гладкая и
блестящая серого или бледно-коричневого основно¬
го цвета с продольной темно-коричневой бороздча-
тостью, в результате чего семянка приобретает блед¬
но-серый или коричневый цвет; плод конусообраз¬
ный у основания и с венцом у верхушки с блестящей
бледно-желтым ободком высотой около 1 мм, окру-
жающим~остатки столбика. При поперечном разрезе
плода обнаруживается узкая коричневая внешняя
часть и две больших плотных белых маслянистых
семядоли.
@>В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.72). Цвет коричневато-жел¬
тый с более темными вкраплениями. Просматривают
под микроскопом с использованием раствора хло¬
ралгидрата Р. В порошке обнаруживаются следую¬
щие диагностические признаки: фрагменты эпикарпа,
состоящего из бесцветных клеток, многоугольных при
просматривании с поверхности, просвет, в зависи¬
мости от ориентации, довольно широкий или в виде
маленькой щели; группы паренхиматозных клеток
пигментного слоя, некоторые из них содержат окра¬
шенное вещество ярко-красного цвета; очень много¬
численные группы крупных склереид семенной ко¬
журы с ярко-желтыми ямчатыми стенками и узкими
просветами; редкие фрагменты мелкоклеточной па¬
ренхимы с ямчатыми и четковидными стенками; мно¬
гочисленные тонкостенные паренхиматозные клетки
семядолей, содержащие капельки масла и рассеян¬
ные друзы оксалата кальция; небольшое количество
более крупных призматических кристаллов оксалата
кальция.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (500) (2.9.12) прибавляют 10 мл метанола Р, на¬
гревают с обратным холодильником в водяной бане
при температуре 70 °С в течение 5 мин, охлаждают
и фильтруют. Фильтрат выпаривают досуха, остаток
растворяют в 1,0 мл метанола Р.
Раствор сравнения. 2 мг силибинина Р и 5 мг
таксифолина Р растворяют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — ацетон Р — метиленхлорид Р (8,5:16,5:75,
об/об/об).
Наносимый объем пробы: 30 мкл испытуемого
раствора и 10 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Проявление: теплую пластинку обрабатывают
раствором 10 г/л дифенилборной кислоты аминоэ¬
тилового эфира Р в метаноле Р и затем раствором
50 г/л макрогола 400 Р в метаноле Р. Через 30 мин
пластинку просматривают в ультрафиолетовом свете
при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться другие флуоресциру¬
ющие зоны оранжевого и желтовато-зеленого цвета
между зонами силибинина и таксифолина.
Верх хроматографической пластинки
Силибинин:
флуоресцирующая зона
желтовато-зеленого цвета
Флуоресцирующая зона
желтовато-зеленого цвета
(силибинин)
Таксифолин:
флуоресцирующая зона
оранжевого цвета
Флуоресцирующая
зона оранжевого цвета
(таксифолин)
Флуоресцирующая зона
желтовато-зеленого цвета
(силикристин)
Флуоресцирующая зона
голубого цвета (линия
старта)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: другие части растения (стебли, ли¬
стья) — не более 2 %. Органические примеси: не бо¬
лее 2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (500)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
® Общая зола (2.4.16). Не более 8,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
@ Определение содержания силимарина в
пересчете на силибинин. Жидкостная хроматогра¬
фия (2.2.29).
Испытуемый раствор. 5,00 г измельченного сы¬
рья (500) (2.9.12) помещают в аппарат для экстрак¬
ции, прибавляют 100 мл петролейного эфира Р и на¬
гревают в водяной бане в течение 8 ч. Обезжиренный
остаток сырья охлаждают до комнатной температуры
и помещают в аппарат для экстракции и экстрагиру¬
ют 100 мл метанола Р в водяной бане в течение 5 ч.
Метанольный экстракт упаривают в вакууме до объ¬
ема около 30 мл, фильтруют в мерную колбу вмести¬
мостью 50 мл, ополаскивая аппарат для экстракции и
фильтр метанолом Р, и доводят до объема 50,0 мл
этим же растворителем. 5,0 мл полученного раствора
доводят метанолом Р до объема 50,0 мл.
Раствор сравнения. Навеску ФСО сухого экс¬
тракта расторопши стандартизованного, соответ¬
ствующую 10,0 мг силибинина, растворяют в метано¬
ле Р и доводят до объема 100,0 мл этим же раство¬
рителем.
Условия хроматографирования:
-колонка длиной 0,125м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным эндкепированным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: кислота фосфорная Р —
метанол Р — вода Р (0,5:35:65, об/об/об);
- подвижная фаза В: кислота фосфорная Р —
метанол Р — вода Р (0,5:50:50, об/об/об);
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—28
100 — 0
0—100
28—35
0
100
35—36
0—100
100-0
36—51
100
0
- скорость подвижной фазы: 0,8 мл/мин;
- спектрофотометрический детектор, длина
волны 288 нм;
1298
Гэсударственная фармакопея Республики Беларусь
- объем вводимой пробы: 10 мкл.
Время удерживания: силибинин В — около
30 мин; при необходимости изменяют временную про¬
грамму градиента.
Идентификация пиков: идентифицируют пики
силикристина, силидианина, силибинина А, силибини-
на В, изосилибинина А и изосилибинина В, используя
хроматограмму, прилагаемую к ФСО сухого экстрак¬
та расторопши стандартизованного. На хромато¬
грамме испытуемого раствора пик силидианина мо¬
жет иметь различную величину, может отсутствовать
или быть основным пиком.
Пригодность хроматографической системы:
раствор сравнения:
- разрешение: не менее 1,8 между пиками сили¬
бинина А и силибинина В;
-хроматограмма должна соответствовать хро¬
матограмме, прилагаемой к ФСО сухого экстракта
расторопши стандартизованного.
Определяют площади пиков, соответствующих
силикристину, силидианину, силибинину А, силибини-
ну В, изосилибинину А и изосилибинину В.
Содержание силимарина в пересчете на силиби¬
нин в процентах рассчитывают по формуле:
(А, + А2 +А3+ Ал + А6 + Ар) • т, -Р • 5
(А7 + А8) • т2
где:
А,, А2, Аъ, Ал, А5, А6 — площади пиков соответству¬
ющих силикристину, силидианину, силибинину А, си¬
либинину В, изосилибинину А и изосилибинину В соот¬
ветственно на хроматограмме испытуемого раствора;
Аг Ав — площади пиков соответствующих сили¬
бинину А и силибинину В соответственно на хромато¬
грамме раствора сравнения;
ш1 — масса навески ФСО сухого экстракта рас¬
торопши стандартизованного, взятого для приго¬
товления раствора сравнения, г;
т2 — масса навески испытуемого сырья, г;
Р — суммарное содержание силибинина А и си¬
либинина В в ФСО сухого экстракта расторопши
стандартизованного, %.
Определение содержания суммы флаволиг-
нанов в пересчете на силимарин.
Испытуемый раствор. 0,500 г измельченного сы¬
рья (710) (2.9.12) помещают в коблу со шлифом вме¬
стимостью 100 мл, прибавляют 25 мл 96 % спирта Р
и нагревают с обратным холодильником на водяной
бане в течение 60 мин. Содержимое колбы охлажда¬
ют, фильтруют через бумажный фильтр в мерную кол¬
бу вместимостью 25 мл и доводят 96 % спиртом Р до
объема 25,0 мл. 1,0 мл полученного раствора доводят
96 % спиртом Р до объема 50,0 мл.
Компенсационный раствор. 96 % спирт Р.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 289 нм.
Содержание суммы флаволигнанов в пересчете
на силимарин в процентах рассчитывают по формуле:
А-1250
/77 -444 ’
где:
444 — удельный показатель поглощения силима¬
рина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:0291
РЕВЕНЯ КОРНИ
Я?Ле/ гасИх
КНШВАКВ
ОПРЕДЕЛЕНИЕ
Собранные осенью или ранней весной в возрас¬
те не менее 3 лет, очищенные от гнилых частей, от¬
мытые от земли, разрезанные на части и высушенные
корни и корневища растения ПЬеит ра1та(ит Ь. уаг
1апдиНсит Мах1т. или РЬеит о1Пста1е ВаШоп.
Содержание:
- не менее 2,2 % суммы гидроксиантраценпро-
изводных в пересчете на реин (С15Н806; М.м. 284,2)
(в пересчете на сухое сырье);
- или не менее 2,0 % производных антрацена в
пересчете на истизин (С14Н804; М.м. 240,2) (в пересче¬
те на сухое сырье).
ОПИСАНИЕ
Обладает характерным ароматным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Куски корней и
корневищ различной формы длиной до 25 см, толщи¬
ной до 5 см. Крупные куски корней цилиндрические
или конусовидные, слегка изогнутые, с продольно¬
морщинистой поверхностью. Куски корневищ встреча¬
ются редко, поверхность их поперечно-морщинистая.
Цвет с поверхности темно-коричневый, на изломе —
желто-коричневый или оранжево-коричневый; свежий
излом зернистый, сероватый, с оранжевыми или розо¬
ватыми прожилками.
@> В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет коричневато-жел¬
тый. Просматривают под микроскопом с использова¬
нием раствора хлоралгидрата Р. В порошке обна¬
руживаются следующие диагностические признаки:
крупные друзы оксалата кальция, размер которых
может превышать 100 мкм, а также их фрагменты;
сетчатые нелигнифицированные сосуды размером до
175 мкм. Многочисленные группы округлых или много¬
гранных тонкостенных клеток паренхимы. Склереиды
и волокна отсутствуют. При просматривании порошка
с использованием 50 % (об/об) раствора глицерина Р
обнаруживаются простые округлые или сложные (от 2
до 4) крахмальные зерна со звездоподобным ядром.
(§) С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мг измельченного сы¬
рья (180) (2.9.12) нагревают в водяной бане в течение
15 мин со смесью из 1 мл кислоты хлористоводо¬
родной Р и 30 мл воды Р. Охлаждают и встряхивают с
25 мл эфира Р. Эфирный слой сушат с использовани¬
ем натрия сульфата безводного Р и фильтруют. Вы¬
паривают эфирный слой досуха, остаток растворяют
в 0,5 мл эфира Р
Раствор сравнения. 5 мг эмодина Р растворяют
в 5 мл эфира Р.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — этилацетат Р — петролейный эфир Р
(1:25:75, об/об/об).
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Ревеня корни
1299
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты А: на хроматограмме раствора
сравнения в центральной части обнаруживается
флуоресцирующая зона оранжевого цвета (эмодин).
На хроматограмме испытуемого раствора обнару¬
живаются: зона, соответствующая эмодину; выше
зоны эмодина обнаруживаются две зоны с похожей
флуоресценцией (фисцион и хризофанол, в порядке
уменьшения значений /?р); ниже эмодина обнаружи¬
ваются две зоны с похожей флуоресценцией (реин и
алоэ-эмодин, в порядке уменьшения значения Рр).
Проявление В: пластинку опрыскивают раство¬
ром 100 г/л калия гидроксида Р в метаноле Р.
Результаты В: все зоны приобретают окраску от
красного до фиолетового цвета.
® О. К 50 мг измельченного сырья (180) (2.9.12)
прибавляют 25 мл кислоты хлористоводородной
разведенной Р и нагревают смесь на водяной бане
в течение 15 мин. Охлаждают, встряхивают с 20 мл
эфира Р и отбрасывают водный слой. Эфирный слой
встряхивают с 10 мл раствора аммиака разведенно¬
го Р1. Водный слой приобретает цвет от красного до
фиолетового.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни, почерневшие в изломе, — не
более 5 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
® КЬеит гЬаропЛсит. Тонкослойная хромато¬
графия (2.2.27).
Испытуемый раствор. К 0,2 г измельченного
сырья (180) (2.9.12) прибавляют 2 мл метанола Р и
кипятят с обратным холодильником в течение 5 мин.
Охлаждают и фильтруют.
Раствор сравнения. 10 мг рапонтицина Р раст¬
воряют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля 6 Р.
Подвижная фаза: метанол Р — метиленхло-
рид Р (20:80, об/об).
Наносимый объем пробы: по 20 мкл в виде полос
длиной не более 20 мм и шириной не более 3 мм.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
фосфорномолибденовой кислоты Р.
Результаты: на хроматограмме испытуемого
раствора возле линии старта не должна обнаружи¬
ваться синяя зона, соответствующая зоне рапонтици¬
на на хроматограмме раствора сравнения.
® Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (180)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводород¬
ной кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
(^Определение содержания суммы гидрок-
сиантраценпроизводных в пересчете на реин. Ис¬
пытание проводят с защитой от яркого света.
0,100 г измельченного сырья (180) (2.9.12) по¬
мещают в колбу вместимостью 100 мл. Прибавляют
30,0 мл воды Р, перемешивают и взвешивают. Нагре¬
вают в водяной бане с обратным холодильником в те¬
чение 15 мин. Охлаждают, прибавляют 50 мг натрия
гидрокарбоната Р, взвешивают и доводят водой Р
до первоначальной массы. Центрифугируют. 10,0 мл
надосадочной жидкости переносят в круглодонную
колбу со шлифом вместимостью 100 мл. Прибавляют
20 мл раствора железа (III) хлорида Р1 и перемеши¬
вают. Нагревают с обратным холодильником на водя¬
ной бане в течение 20 мин, прибавляют 1 мл кислоты
хлористоводородной Р и нагревают еще в течение 20
мин, периодически встряхивая. Охлаждают, перено¬
сят в делительную воронку, ополаскивая колбу эфи¬
ром Р, и встряхивают с этим же растворителем три
раза порциями по 25 мл. Объединяют эфирные из¬
влечения и промывают водой Р дважды порциями по
15 мл. Эфирный слой процеживают через ватный там¬
пон и доводят до объема 100,0 мл эфиром Р. 10,0 мл
полученного раствора выпаривают на водяной бане
досуха. Остаток растворяют в 10,0 мл раствора 5 г/л
магния ацетата Р в метаноле Р.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при 515 нм, используя метанол Р в
качестве компенсационного раствора.
Содержание суммы гидроксиантраценпроизвод-
ных в пересчете на реин в процентах рассчитывают
по формуле:
А- 300
т-468 ’
где:
468 — удельный показатель поглощения реина,
рассчитанный исходя из удельного поглощения бар-
балоина;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
Определение содержания производных ан¬
трацена в пересчете на истизин. Испытание прово¬
дят с защитой от яркого света.
0,0500 г измельченного сырья (180) (2.9.12) по¬
мещают в колбу вместимостью 100 мл, прибавляют
7,5 мл кислоты уксусной ледяной Р и нагревают с
обратным холодильником в водяной бане в течение
15 мин. Охлаждают, прибавляют 30 мл эфира Р и
кипятят в водяной бане еще в течение 15 мин. Ох¬
лаждают, процеживают через ватный тампон в дели¬
тельную воронку вместимостью 300 мл и промывают
20 мл эфира Р. Ватный тампон переносят в колбу с
остатком сырья, прибавляют 30 мл эфира Р и кипятят
в водяной бане в течение 10 мин. Охлаждают и про¬
цеживают через ватный тампон в ту же делительную
воронку. Колбу дважды ополаскивают эфиром Р пор¬
циями по 10 мл и процеживают через тот же ватный
тампон. К объединенным извлечениям осторожно,
по стенкам, прибавляют 100 мл щелочно-аммиачно¬
го раствора Р и осторожно взбалтывают в течение
(5—7) мин, охлаждая воронку под струей холодной
воды. После полного расслоения прозрачный красный
нижний слой, не фильтруя, сливают в мерную колбу
вместимостью 250 мл, а эфирный слой обрабатывают
порциями по 20 мл щелочно-аммиачного раствора Р
до прекращения окрашивания жидкости, сливают в ту
же мерную колбу и доводят до объема 250,0 мл ще¬
лочно-аммиачным раствором Р. 25,0 мл полученного
раствора помещают в коническую колбу вместимо¬
стью 100 мл и нагревают в течение 15 мин на водяной
бане при перемешивании. Охлаждают. Раствор коли¬
чественно переносят в мерную колбу вместимостью
25 мл и доводят водой Р до объема 25,0 мл.
163*. Зак. 1060.
1300
Гэсударственная фармакопея Республики Беларусь
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при 530 нм, используя водуР в ка¬
честве компенсационного раствора.
Концентрацию производных антрацена в раство¬
ре (мг/мл) в пересчете на истизин определяют по гра¬
дуировочному графику.
Содержание производных антрацена в пересче¬
те на истизин в процентах рассчитывают по формуле:
С-25
т ’
где:
С — концентрация производных антрацена опре¬
деленная по градуировочному графику, мг/мл;
т — масса навески испытуемого сырья, г.
Построение градуировочного графика. 50,00 г
кобальта хлорида Р, высушенного в эксикаторе до
постоянной массы, растворяют в 250 мл воды Р, при¬
бавляют 1 мл кислоты хлористоводородной Р и до¬
водят водой Р до объема 500,0 мл. Из полученного
раствора готовят серию разведенных растворов (№ 1,
2, 3, 4, 5, 6, 7, 8 и 9), содержащих кобальта хлори¬
да (СоС12-6Н20) соответственно 0,010; 0,015; 0,020;
0,025; 0,030; 0,035; 0,040; 0,045; 0,050 г в 1 мл, и из¬
меряют оптические плотности (2.2.25) полученных
растворов при 530 нм, используя водуР в качестве
компенсационного раствора. Для построения гра¬
дуировочного графика на оси абсцисс откладывают
концентрации растворов, а на оси ординат — их оп¬
тические плотности. При этом концентрации раство¬
ров кобальта хлорида выражают в соответствующих
концентрациях производных антрацена (истизина),
пользуясь таблицей #0291 .-1.
Таблица #0291.-1.
№
п/п
Концентрация кобальта
хлорида (СоС12*6Н20),
г/мл
Концентрация
производных
антрацена в пересчете
на истизин, мг/мл
1
0,010
0,0036
2
0,015
0,0054
3
0,020
0,0072
4
0,025
0,0090
5
0,030
0,0108
6
0,035
0,0126
7
0,040
0,0144
8
0,045
0,0162
9
0,050
0,0180
07/2016:1587
РЕПЕШКА ТРАВА
Адптоп1ае ЬегЬа
Автмоыу
ОПРЕДЕЛЕНИЕ
Высушенные цветущие верхушки Адптоп1а
еираЪпа 1_.
Содержание: не менее 2,0 % дубильных веществ
в пересчете на пирогаллол (С6Н603; М.м. 126,1) (в пе¬
ресчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Стебель зе¬
леного или чаще красноватого цвета, цилиндри¬
ческий с редким ветвлением. Стебель покрыт
длинными, прямыми или спутанными волосками.
Листья непарноперистые, с 3 или 6 противопостав¬
ленными парами листочков, с 2 или 3 более мел¬
кими листочками между ними. Листочки от глубоко
зубчатых до пильчатых, верхняя поверхность тем¬
но-зеленая, нижняя — сероватая и густо опушена.
Цветки маленькие, формируют верхушечный ко¬
лос; пятичленные, расположены в пазухах опушен¬
ных прицветников, чашечки плотно окружены мно¬
гочисленными крючкообразными колосками, кото¬
рые расположены по краю опушенного цветоложа.
Лепестки свободные, желтого цвета, опадающие.
Плодоносное, обратно-конусовидное цветоложе с
глубокими бороздками и изогнутыми щетинками,
обычно находится в основании соцветия.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет желтовато-зеле¬
ный или серый. Просматривают под микроскопом с
использованием раствора хлоралгидрата Р. В по¬
рошке обнаруживаются следующие диагностиче¬
ские признаки (рисунок 1587.-1): многочисленные
прямые или изогнутые одноклеточные длинные
толстостенные (около 500 мкм) покровные воло¬
ски [АЬ, Са, Р], мелко бородавчатые и иногда спи¬
рально изогнутые, часто фрагментированные [Р];
фрагменты эпидермиса стеблей [А] с устьицами
[Аа], покровными волосками [АЬ] и железистыми
волосками [Ас]; фрагменты верхнего эпидермиса
листьев (вид с поверхности [С]) с прямостенными
клетками, покровными волосками [Са] и с приле¬
жащей палисадной паренхимой [СЬ], некоторые
клетки которой содержат призматические кристал¬
лы оксалата кальция [Сс]; фрагменты нижнего
эпидермиса листьев (вид с поверхности ^]) с вол¬
нообразными стенками и многочисленными устьи¬
цами ^а], преимущественно аномоцитного типа
(2.8.3), изредка анизоцитного типа и железистыми
волосками ^Ь]; зерна пыльцы от яйцевидной до
субсферической формы с тремя порами и гладкой
экзиной [0]\ железистые волоски с многоклеточ¬
ной однорядной ножкой и головкой из 1—4 клеток
[В, ч1Ь]; фрагменты стеблей [Н] с пучками волокон
[На] и клетками паренхимы, некоторые из них со¬
держат друзы кристаллов кальция оксалата [НЬ];
небольшие спиралевидные сосуды листочков [О];
фрагменты крупных спиралевидных или с окайм¬
ленными порами сосудов стебля [Е].
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 2,0 г измельченно¬
го сырья (355) (2.9.12) прибавляют 20 мл мета¬
нола Р, нагревают, встряхивая, при температуре
40 °С в течение 10 мин и фильтруют.
Раствор сравнения. 1,0 мг изокверцитрози-
да Р и 1,0 мг рутина Р растворяют в 2 мл мета¬
нола Р.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: кислота муравьиная без¬
водная Р — вода Р — этилацетат Р (10:10:80,
об/об/об).
Наносимый объем пробы: по 10 мкл в виде
полос.
Фронт подвижной фазы: не менее 12 см от
линии старта.
Высушивание: при температуре от 100 °С до
105 °С.
Родиолы розовой корневища и корни
1301
травы репешка
Проявление: горячую пластинку опрыскивают
раствором 10 г/л дифенилборной кислоты ами¬
ноэтилового эфира Р в метаноле Р и затем рас¬
твором 50 г/л макрогола 400 Р в метаноле Р. Пла¬
стинку высушивают на воздухе в течение 30 мин
и просматривают в ультрафиолетовом свете при
длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматограс
рической пластинки
Изокверцитрозид:
флуоресцирующая зона
оранжевого цвета
Рутин: флуоресцирующая
зона оранжевого цвета
Может обнаруживаться
флуоресцирующая
зона оранжевого цвета
(кверцитрозид)
Флуоресцирующая
зона оранжевого цвета
(изокверцитрозид)
Флуоресцирующая
зона оранжевого цвета
(гиперозид)
Флуоресцирующая зона
оранжевого цвета (рутин)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Потеря в массе при высушивании (2.2.32).
Не более 10,0%. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Дубильные вещества (2.8.14). Определе¬
ние проводят из 1,000 г измельченного сырья (180)
(2.9.12) .
07/2016: РБ0084
РОДИОЛЫ РОЗОВОЙ
КОРНЕВИЩА И КОРНИ
/?ос//о/ае гозеае гЫгота е( гасИх
К08ЕМОКТ КООТ*
ОПРЕДЕЛЕНИЕ
Собранные в фазу цветения и плодоношения,
очищенные и отмытые от земли, разрезанные на ку¬
ски и высушенные корневища и корни многолетнего
травянистого растения Родю1а гозеа Ь.
Содержание: не менее 0,8 % салидрозида
(С14Н20О7; М.м. 300,3) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Куски корневищ
и корней различной формы. Куски корневищ длиной
до 9 см, толщиной (2—5) см, твердые, морщинистые,
со следами отмерших стеблей и остатками чешуевид¬
ных листьев. От корневищ отходят немногочисленные
корни длиной (2—9) см, толщиной от 0,5 см до 1 см.
Поверхность корневищ и корней блестящая, серова¬
то-коричневого цвета; при отслаивании пробки обна¬
руживается желтый слой. Цвет на изломе розовато¬
коричневый или светло-коричневый.
B. Микроскопия (2.8.23). На поперечном срезе
корневища видна слоистая перидерма. Корневище
имеет пучковый тип строения. Проводящие пучки
открытые, коллатеральные, веретеновидные, рас¬
положены кольцом, ориентированы к периферии
корневища флоэмой и к центру — ксилемой. Воз¬
можно наличие второго кольца более мелких про¬
водящих пучков, в которых флоэма ориентирована
к центру, а ксилема — к периферии. Паренхима
корневища состоит из крупных клеток, заполненных
крахмальными зернами — простыми, округлыми
или овальными, диаметром от 5 мкм до 20 мкм.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (1400) (2.9.12) прибавляют 10 мл метанола Р. На¬
гревают с обратным холодильником на водяной бане
при температуре 65 °С в течение 20 мин. Охлаждают
и фильтруют.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля Р254 Р, предварительно активированная
посредством нагревания при температуре 110 °С в те¬
чение 1 ч.
Подвижная фаза: хлороформ Р — метанол Р —
вода Р (26:14:3, об/об/об). Камеру насыщают не ме¬
нее 24 ч.
Наносимый объем пробы: 2 мкл в виде полосы.
Фронт подвижной фазы: не менее 13 см от ли¬
нии старта.
Высушивание: на воздухе в течение 5 мин.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается основная зона фиолетово¬
го цвета с значением /?, около 0,4 (розавин). На хрома¬
тограмме испытуемого раствора могут обнаруживать¬
ся и другие зоны.
Проявление В: пластинку опрыскивают раствором
100 г/л натрия карбоната Р и нагревают в течение
2 мин при температуре 110 °С. Затем опрыскивают диа¬
зореактивом Р1 и нагревают в течение 2 мин при тем¬
пературе 110 °С. Просматривают при дневном свете.
164. Зак. 1060.
1302
Гэсударственная фармакопея Республики Беларусь
Результаты В: на хроматограмме испытуе¬
мого раствора обнаруживается зона красноватого
цвета со значением /?, около 0,42 (салидрозид). На
хроматограмме испытуемого раствора могут обна¬
руживаться и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: листья, стебли, в том числе от¬
деленные при анализе — не более 4 %. Органи¬
ческие примеси: не более 1 %. Минеральные при¬
меси: не более 3 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 9,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,500 г измельченного сырья (1400) (2.9.12) по¬
мещают в колбу вместимостью 100 мл, прибавляют
10 мл воды Р и нагревают с обратным холодильником
в водяной бане в течение 15 мин. Фильтруют через бу¬
мажный фильтр в мерную колбу вместимостью 50 мл,
избегая попадания частиц сырья на фильтр. Экстрак¬
цию повторяют трижды с водой Р порциями по 10 мл,
нагревая в течение 10 мин, и фильтруют в ту же мер¬
ную колбу. Охлаждают. К фильтрату прибавляют 6 мл
раствора свинца (II) ацетата Р и 2 мл насыщенного
раствора натрия сульфата Р, перемешивают и до¬
водят водой Р до объема 50,0 мл. Фильтруют через бу¬
мажный фильтр, отбрасывая первые 15 мл фильтрата.
К 5,0 мл полученного фильтрата прибавляют 2,5 мл
раствора 20 г/л натрия карбоната Р, 2,5 мл диазоре¬
актива Р1 и доводят водой Р до объема 25,0 мл.
Через 5 мин измеряют оптическую плотность
(2.2.25) раствора при 486 нм, используя воду Р в каче¬
стве компенсационного раствора.
Содержание салидрозида в процентах рассчиты¬
вают по формуле:
А- 250
т-253 ’
где:
253 — удельный показатель поглощения сали¬
дрозида;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
07/2016:0404
РОМАШКИ ЦВЕТКИ
МаМсапае Лоз
МАТШСАША П-ОЖЕК
ОПРЕДЕЛЕНИЕ
Собранные в начале цветения и высушен¬
ные цветки (цветочные корзинки) МаШапа гесиШа 1_.
(СЬатотШа гесиШа (1_.) КаизсИег1, МаНсапа сЬато-
т'Ша I-.).
Содержание: не менее 3 мл/кг синего эфирного
масла (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Головки де¬
лятся на обвертку, состоящую из большого количества
прицветников, расположенных в 1—3 ряда; вытяну¬
то-коническое цветоложе, иногда полусферического
(молодая головка); 12—20 краевых язычковых цветка
с белым язычком; несколько десятков центральных
желтых трубчатых цветков. Прицветники обвертки
овальной или ланцетовидной формы с коричневато¬
серым пленчатым краем. Цветоложе полое, без че¬
шуек. Венчик язычковых цветков представляет собой
коричневато-желтую трубку в основании, вытягива¬
ющуюся с образованием белого овально-вытянутого
язычка. Завязь нижняя темно-коричневая, овальной
или сферической формы, с длинным столбиком и
раздвоенным рыльцем. Трубчатые цветки желтые с
трубчатым пятизубчиковым венчиком, пятью сростно-
пыльниковыми налепестными тычинками и гинецеем,
подобным на гинецей язычковых цветков.
® В. Микроскопия (2.8.23). Головку разделя¬
ют на составляющие. Просматривают под микроско¬
пом с использованием раствора хлоралгидрата Р.
Края прицветников состоят из тонкостенных клеток,
центральная часть — из вытянутых склереид с ред¬
кими устьицами (2.8.3). На внутреннем эпидерми¬
се венчика язычковых цветков при просматривании
с поверхности видны тонкостенные многоугольные
мелкобугорчатые клетки, клетки внешнего эпидерми¬
са заметно извилистые и сильно исчерчены; венчик
трубчатых цветков с продольно вытянутыми клетка¬
ми эпидермиса с мелкими группами выростов около
верхушек долей. На внешней стороне прицветников и
внешней стороне венчиков обоих типов цветков обна¬
руживаются железистые волоски, каждый из которых
состоит из короткой ножки и 2—3-ярусной головки с 2
клетками в каждом ярусе. Завязи имеют уплотненное
кольцо в основании, стенка состоит из вертикальных
полос тонкостенных продольно вытянутых клеток с
многочисленными железистыми волосками, чередую¬
щихся с веретенообразными группами мелких ради¬
ально вытянутых клеток, содержащих слизь. Клетки
в верхушках рыльца вытянуты, образуя округлые вы¬
росты. Многочисленные мелкие друзы оксалата каль¬
ция обнаруживаются во внутренних тканях завязей и
долей пыльников. Зерна пыльцы от сферической до
треугольной формы диаметром около 30 мкм с тремя
порами и с покрытой шипами экзиной.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 50 мкл эфирного масла,
полученного при количественном определении, раз¬
водят в 1 мл ксилола Р.
Раствор сравнения. 2 мкл хамазулена Р, 5 мкл
(-)-а-бисаболола Р и 10 мг борнилацетата Р раство¬
ряют в 5 мл толуола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раство¬
ром анисового альдегида Р и нагревают в течение
(5—10) мин при температуре от 100 °С до 105 °С.
Просматривают немедленно при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны
Рудбекии шершавой цветки
1303
Верх хроматографической пластинки
Хамазулен: зона красного
или красно-фиолетового
цвета
Одна или две зоны синего
или сине-фиолетового цвета
Зона красного или красно¬
фиолетового цвета
(хамазулен)
Борнилацетат: зона
желтовато-коричневого
цвета
Зона коричневого цвета (ен-
ин-дициклоэфир)
(-)-а-Бисаболол: зона
красновато-фиолетового
или синевато-фиолетового
цвета
Зона красновато¬
фиолетового или синевато¬
фиолетового цвета
((-)-а-бисаболол)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: листья, стебли, корзинки с остатками
цветоносов длиннее 3 см — не более 9 %; почернев¬
шие и побуревшие корзинки — не более 5 %. Органи¬
ческие примеси: части других неядовитых растений и
корзинки других видов ромашки — не более 3 %. Ми¬
неральные примеси: не более 0,5 %.
Примечание. К органической примеси относят со¬
цветия растений, похожих по внешнему виду на ро¬
машку аптечную, но не являющихся лекарственными:
ромашки непахучей — МаМсапа /подога 1_., которая в
отличие от ромашки аптечной имеет сплошное цвето¬
ложе и более крупные корзинки (до 12 мм), пупавки
полевой — АпМет'/з ап/епз/з 1_., имеющей пленчатое
цветоложе, и пупавки собачей —Апдют/з со1и1а 1_., у
которой цветоложе пленчатое только сверху.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 13,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 30 г цельного
сырья помещают в колбу вместимостью 1000 мл, при¬
бавляют 500 мл воды Р в качестве жидкости для пе¬
регонки. В градуированную трубку помещают 0,50 мл
ксилола Р и перегоняют со скоростью (3—4) мл/мин в
течение 4 ч. За несколько минут до истечения време¬
ни перегонки выключают подачу воды в холодильник,
продолжая перегонку, пока синие летучие компоненты
не достигнут нижнего конца холодильника. Немедлен¬
но включают подачу воды в холодильник во избежа¬
ние нагревания приемника. Через 10 мин останавли¬
вают перегонку.
07/2016: РБ0099
РУДБЕКИИ ШЕРШАВОЙ ЦВЕТКИ
КидЬеск/а Ыг1а Логез
В1.АСК-ЕУЕО ЗС/ЗАЫ Н-ОЪУЕЯ*
ОПРЕДЕЛЕНИЕ
Собранные в начале цветения и высушенные
цветочные корзинки многолетнего травянистого рас¬
тения РидЬеск/а Ша 1_.
Содержание:
- не менее 2,5 % суммы фенольных соединений
в пересчете на патулетрин (С21Н20О14; М.м. 496,4) в су¬
хом сырье;
- не менее 0,3 % патулетрина (С21Н20О14; М.м. 496,4)
в сухом сырье.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки. Цельные и распавшиеся
цветочные корзинки, отдельные ложноязычковые и
трубчатые цветки, ложа распавшихся соцветий, ли¬
сточки обвертки, семянки. Корзинки-соцветия круп¬
ные, шаровидной, полушаровидной или конической
формы, диаметром (8—15) см, без цветоноса или с
остатками цветоноса длиной не более 3 см. Краевые
цветки ложноязычковые, длиной (3—7) см, шириной
(0,5—2) см, язычки остротрехзубчатые. Срединные
цветки трубчатые, обоеполые, длиной не более 8 мм,
шириной (1—3) мм, отгиб из пяти зубчиков. На вер¬
хушках зубчиков трубчатых цветков просматриваются
хохолки. Тычинок 5, нити тычинок свободные, пыль¬
ники слипаются между собой боковыми сторонами и
образуют пыльниковую трубку. Длинный, тонкий стол¬
бик пестика заканчивается двумя рыльцами, завязь
нижняя. Ложе соцветия — расширенное окончание
оси цветоноса, основа соцветия — корзинки. Ложе
соцветия голое, по форме выпуклое, коническое или
цилиндрическое. Обвертка состоит из двух рядов че-
репитчато расположенных немногочисленных листоч¬
ков. Наружные листочки обвертки длиной до 2 см,
яйцевидные, с заостренным концом, травянистые,
серовойлочные; внутренние — кверху расширенные,
тупые. Плод — четырехгранная, слегка сжатая семян¬
ка, не более 2,5 мм длиной. Цвет ложноязычковых
цветков от светло-желтого до оранжевого, часто при
основании ложноязычкового цветка расположены
темные пятна. Цвет трубчатых цветков от бурого до
черно-бурого. Цвет листочков обвертки от зеленого
до буровато-зеленого с серовойлочным опушением.
Цвет семянки от коричнево-бурого до черного. Запах
слабый, ароматный.
B. Микроскопия (2.8.23). При просматривании
ложноязычковых цветков с поверхности видны много¬
угольные изодиаметричные клетки эпидермы с прямы¬
ми стенками [1]. Клетки эпидермы трубчатого цветка
прямоугольной формы с прямыми стенками [2]. По
краю зубчиков ложноязычкового цветка сосочковидные
выросты клеток эпидермы [3]. Волоски густо покрыва¬
ют всю поверхность внутренней стороны пластинки
ложноязычкового цветка и ее край, образуя войлоч¬
ное опушение. Волоски многочисленные, среди них
встречаются длинные конусовидные простые волоски,
состоящие из трех/четырех удлиненных клеток с утол¬
щенными, реже тонкими оболочками и прямой конеч¬
ной клетки различной длины с заостренным концом [4].
На поверхности ложноязычковых цветков встречаются
железистые волоски, состоящие из одно- или двухкле¬
точной ножки и одноклеточной овальной головки, за¬
полненной желтовато-оранжевым содержимым [5]. На
верхушках зубчиков трубчатых цветков видны хохолки,
состоящие из двух типов простых волосков: волоски
с тонкой игловидной конечной клеткой [а] и волоски с
конусовидной конечной клеткой [Ь], оба типа волосков
состоят из трех/пяти клеток с прямыми стенками [6].
На поверхности трубчатых цветков обнаружены ко¬
нусовидные многоклеточные простые волоски, состо¬
ящие из нескольких коротких клеток с тонкими, реже
164*. Зак. 1060.
1304
Гэсударственная фармакопея Республики Беларусь
утолщенными стенками и удлиненной конечной клетки
с заостренным концом [7]. На поверхности ложноя¬
зычковых и трубчатых цветков просматриваются эфи¬
ромасличные железки. Железки многоклеточные, со¬
стоящие из 10—12 клеток, расположенных в два ряда
([8], вид сбоку), при просматривании сверху железки
видны в виде овальных образований с поперечной
перегородкой [9]. Пыльцевые зерна округлые, с ши¬
поватой экзиной [10]. Эпидерма внутренней стороны
листочков обвертки состоит из многоугольных, слегка
вытянутых клеток с прямыми стенками [с], над жилкой
клетки эпидермы многоугольные, сильно вытянуты [с!]
[11]. Эпидерма наружной стороны листочка обвертки
представлена клетками с сильноизвилистыми стенка¬
ми и устьицами, которые были окружены 4—6 околоу-
стьичными клетками (аномоцитный тип) [12] (2.8.3). На
поверхности и по краю листочков обвертки встречают¬
ся тонкие игловидные простые волоски, состоящие из
трех-четырех удлиненных клеток с утолщенными, реже
тонкими оболочками, клетка при основании волоска
укорочена [13]. Клетки вокруг основания волосков рас¬
положены радиально (базальные клетки волоска) и об¬
разуют розетку. При отпадении волосков на месте их
прикрепления остаются круглые валики [14]. Волоски
чаще встречаются на внутренней стороне листочков
обвертки и образуют жесткое опушение.
Рисунок РБ0099.-1. Микроскопические признаки
цветков рудбекии шершавой
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,50 г измельченного
сырья (2000) прибавляют 10 мл метанола Р. Кипятят
на водяной бане с обратным холодильником в тече¬
ние 30 мин. Охлаждают, центрифугируют при 5000 д в
течение 5 минут и собирают надосадочную жидкость.
Раствор сравнения. 10 мг патулетрина Р раст¬
воряют в 10 мл метанола Р, обрабатывают ультра¬
звуком в течение 5 мин.
Пластинка: ТСХ пластинка со слоем целлюлозы Р.
Подвижная фаза: этилацетат Р — толуол Р —
кислота уксусная ледяная Р — вода Р (10:1:10:1,
об/об/об/об).
Наносимый объем пробы: в виде полос длиной
20 мм и шириной 5 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Проявление: пластинку опрыскивают раствором
20 г/л алюминия хлорида Р в 96% спирте. Просма¬
тривают в ультрафиолетовом свете при длине волны
365 нм.
Высушивание: при температуре от 100 °С до
105 °С в течение 3 мин, затем охлаждают на воздухе.
Результаты, ниже приведена последователь¬
ность расположения зон хроматограмм раствора
сравнения и испытуемого раствора. На хроматограм¬
ме испытуемого раствора могут обнаруживаться дру¬
гие слабо флуоресцирующие зоны желтого и голубого
цветов.
Верх хроматографической пластинки
Патулетрин: интенсивно
флуоресцирующая зона
желто-зеленого цвета
Флуоресцирующая зона
красного цвета
Интенсивно
флуоресцирующая зона
желто-зеленого цвета
(патулетрин)
Флуоресцирующая зона
желтого цвета
Слабо флуоресцирующая
зона желтого цвета
Раствор сравнения
Испытуемый раствор
Э. На хроматограмме испытуемого раствора, по¬
лученного в разделе «Количественное определение.
Определение содержания патулетрина», обнаружива¬
ется пик патулетрина (рисунок РБ0099.-2).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: кусочки стеблей, цветоносов и ли¬
стьев — не более 3 %. Органические примеси: не бо¬
лее 2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000) су¬
шат при температуре 105 °С.
Общая зола (2.4.16). Не более 14,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержание суммы фенольных
соединений в пересчете на патулетрин. 1,000 г ис¬
пытуемого сырья (2000) помещают в круглодонную
колбу со шлифом вместимостью 100 мл и прибав¬
ляют 50,0 мл спирта Р (50 %, об/об). Взвешивают с
точностью до 0,01 г, нагревают на водяной бане при
температуре 60 °С с обратным холодильником в те¬
чение 30 мин. Колбу с содержимым охлаждают до
комнатной температуры и доводят массу спиртом Р
(50%, об/об) до первоначальной. Центрифугируют
при 5000 д в течение 5 мин и собирают надосадочную
жидкость (раствор А).
Испытуемый раствор. К 1,0 мл раствора А при¬
бавляют 1,0 мл реактива фосфорномолибденово¬
вольфрамового Р и доводят до объема 10,0 мл раство¬
ром 100 г/л натрия карбоната безводного Р.
Построение градуировочного графика. 12,5 мг
патулетрина Р растворяют в 10 мл спирта Р (50 %,
об/об) и доводят до объема 25,0 мл этим же раствори¬
телем. В пять мерных колб вместимостью 25 мл поме¬
щают 0,1; 0,2; 0,3; 0,4 и 0,5 мл полученного раствора
соответственно, прибавляют 1,0 мл реактива фосфор-
номолибденово-вольфрамового Р и доводят до объ¬
ема 25,0 мл раствором 100 г/л натрия карбоната без¬
водного Р. Для построения градуировочного графика
на оси абсцисс откладывают концентрацию раствора
(мг/мл), а на оси ординат — их оптическую плотность.
Рябины плоды
1305
Компенсационный раствор. К 1,0 мл спирта Р
(50 %, об/об) прибавляют 1,0 мл реактива фосфор-
номолибденово-вольфрамового Р доводят до объема
10,0 мл раствором 100 г/л натрия карбоната безво¬
дного Р.
Через 40 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 760 нм.
Содержание суммы фенольных соединений в
пересчете на патулетрин в процентах рассчитывают
по формуле:
С -500
т ’
где:
С — содержание фенольных соединений в пере¬
счете на патулетрин, найденное по градуировочному
графику, мг/мл;
т — масса навески испытуемого сырья, г.
Определение содержания патулетрина. Жид¬
костная хроматография (2.2.29).
Испытуемый раствор. 1,000 г испытуемого сы¬
рья (2000) помещают в круглодонную колбу со шли¬
фом вместимостью 100 мл и прибавляют 50,0 мл
спирта Р (50 %, об/об). Взвешивают с точностью до
0,01 г, нагревают на водяной бане при температуре
60 °С с обратным холодильником в течение 30 мин.
Колбу с содержимым охлаждают до комнатной темпе¬
ратуры и доводят массу спиртом Р (50 %, об/об) до
первоначальной. Центрифугируют при 5000 д в тече¬
ние 5 мин, надосадочную жидкость фильтруют через
мембранный фильтр с диаметром пор 0,45 мкм.
Раствор сравнения. 50,0 мг патулетрина Р
растворяют в спирте Р (50 %, об/об) и доводят до
объема 200,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октильным
для хроматографии Р с размером частиц 5 мкм;
- температура: 30 °С;
- подвижная фаза: ацетонитрил Р — 0,01 М
раствор калия дигидрофосфата Р, доведенный кис¬
лотой фосфорной Р до рН 3,0±0,2 (20:80, об/об);
- скорость подвижной фазы: 1,0 мл/мин;
-спектрофотометрический детектор, длина
волны 360 нм;
- объем вводимой пробы: 10 мкл.
Содержание патулетрина в процентах рассчиты¬
вают по формуле:
5гС-5
32т ’
где:
51 — площадь пика патулетрина на хроматограм¬
ме испытуемого раствора;
52 — площадь пика патулетрина на хроматограм¬
ме раствора сравнения;
т — масса навески испытуемого сырья, г;
С — содержание патулетрина Р в растворе
сравнения, мг/мл.
07/2016:РБ0085
РЯБИНЫ ПЛОДЫ
8огЫ аисирапае ?гис1из
КОШАЫВЕКВУ*
ОПРЕДЕЛЕНИЕ
Собранные в период полного созревания и вы¬
сушенные плоды дерева (реже кустарника) ЗогЬиз
аисирапа 1_.
Содержание:
- не менее 0,01 % кислоты аскорбиновой (в пере¬
счете на сухое сырье);
- не менее 0,003 % каротиноидов в пересчете на
р-каротин (в пересчете на сухое сырье).
1. Патулетрин.
Рисунок РБ0099.-2. Хроматограмма испытуемого раствора (определение содержания патулетрина)
165. Зак. 1060.
1306
Гэсударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Плоды яблокоо¬
бразные, без плодоножек, 2—5-гнездные, округлые или
овально-округлые, в поперечнике до 9 мм, блестящие,
сильноморщинистые, на верхушке с остающейся ча¬
шечкой из пяти малозаметных смыкающихся зубчиков.
В мякоти плода находятся 2—7 слегка серповидно¬
изогнутых, продолговатых, с острыми концами, гладких
красновато-коричневых семян. Цвет плодов краснова¬
то- или желтовато-оранжевый, коричневато-красный.
На поперечном разрезе плода (при увеличении 10*)
видно 2—5 семенных гнезд. Стенки гнезд хрящеватые,
твердые, сросшиеся с мякотью. Внутри каждого гнезда
находятся 1—2 семени с красновато-коричневой твер¬
дой семенной кожурой и белым семенным ядром. Мя¬
коть плода рыхлая, мясистая, сверху покрыта кожицей.
Запах слабый, своеобразный.
B. Микроскопия (2.8.23). При просматривании
эпидермы плода с поверхности видны клетки «оконча-
того» типа, в очертании многоугольные, разновеликие
с неразличимой границей между соседними стенка¬
ми, содержащие капли жирного масла желтого цвета.
Мезокарпий состоит из тонкостенных паренхимных
клеток, разнообразных по форме и величине, с много¬
численными оранжево-желтыми хромопластами (с
преобладанием веретеновидных), содержащими кри¬
сталлы каротина. В темном поле поляризационного
микроскопа кристаллы каротина светятся зеленым
или красным цветом в зависимости от их положе¬
ния. Изредка встречаются проводящие пучки, ксиле¬
ма которых состоит из узких спиральных сосудов. В
более глубоких слоях мезокарпия, особенно вблизи
эндокарпия, рассеяны одиночные или группы камени¬
стых клеток. В мезокарпии встречаются друзы окса¬
лата кальция. Эндокарпий тонкий, плотный, состоит
из склеренхимной ткани и слабо развитых парен¬
химных клеток, содержащих одиночные кристаллы и
друзы оксалата кальция. На наружной стороне пере¬
городки между гнездами по 1—2 и более расположе¬
ны длинные простые одноклеточные волоски более
или менее извитые. Клетки эпидермы кожуры семени
многоугольные, прямостенные с неравномерно утол¬
щенными внутренними стенками, что создает харак¬
терную картину эпидермы с поверхности при низкой
установке микрометрического винта. На поперечном
срезе кожуры семени видны четырехугольные клетки
эпидермы, 3—4 ряда скперенхимных клеток, 1 ряд
более тонкостенных клеток и слой спавшихся клеток.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 г измельченных пло¬
дов (2000) (2.9.12) помещают в коническую колбу со
шлифом вместимостью 25 мл, прибавляют 5 мл хло¬
роформа Р, закрывают пробкой и перемешивают в те¬
чение 1,5 ч. Фильтруют через бумажный фильтр.
Раствор сравнения. 10 мг р-каротина Р раство¬
ряют в 10 мл хлороформа Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: циклогексан Р — эфир Р
(80:20, об/об).
Наносимый объем пробы: 50 мкл испытуемого
раствора и 20 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 9 см от линии
старта.
Высушивание: на воздухе.
Проявление: просматривают при дневном свете.
Результаты: на хроматограмме испытуемого
раствора обнаруживается зона желтого цвета, соответ¬
ствующая зоне желтого цвета (р-каротин) на хромато¬
грамме раствора сравнения. На хроматограмме испы¬
туемого раствора могут обнаруживаться и другие зоны.
й. 1 г измельченного сырья (2000) (2.9.12) рас¬
тирают в ступке с 5 мл раствора 5 г/л кислоты хлори¬
стоводородной Р, затем прибавляют 20 мл раствора
5 г/л кислоты хлористоводородной Р, выдерживают
10 мин, перемешивают и фильтруют через бумаж¬
ный фильтр. К 5 мл фильтрата прибавляют по каплям
раствор 0,6 г/л дихлорфенолиндофенола натриевой
соли Р. Синее окрашивание раствора реактива пере¬
ходит в розовое.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие и пригоревшие плоды —
не более 3 %; недозрелые плоды (светло-желтые,
желтые) — не более 2 %; плодоножки, веточки, ли¬
стья — не более 0,5 %; плоды с плодоножками — не
более 3 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,2 %.
Потеря в массе при высушивании (2.2.32). Не
более 18,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С в течение 3 ч.
Общая зола (2.4.16). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания кислоты аскорби¬
новой. 2,000 г измельченного сырья (2000) (2.9.12)
растирают в ступке с 5 мл раствора 5 г/л кислоты
хлористоводородной Р, количественно переносят
при помощи 40 мл раствора 5 г/л кислоты хлористо¬
водородной Р в мерную колбу вместимостью 50 мл и
доводят до объема 50,0 мл этим же растворителем,
выдерживают 10 мин, перемешивают и фильтруют
через бумажный фильтр, отбрасывая первые 15 мл
фильтрата. 5,0 мл полученного раствора помещают
в коническую колбу, прибавляют 30 мл воды Р и ти¬
труют из микробюретки титрованным раствором
дихлорфенолиндофенола Р до появления светло-ро¬
зовой окраски, не исчезающей в течение (15—20) с.
1 мл титрованного раствора дихлорфенолин¬
дофенола Р соответствует 0,1 мг кислоты аскорбино-
вой (С6Н806).
Определение содержания каротиноидов.
2,000 г измельченного сырья (2000) (2.9.12) помеща¬
ют в коническую колбу вместимостью 250 мл, при¬
бавляют 50 мл 96 % спирта Р и встряхивают в тече¬
ние 30 мин. Извлечение фильтруют через бумажный
фильтр, отбрасывая первые 15 мл фильтрата.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при 450 нм, используя 96 % спирт Р
в качестве компенсационного раствора.
Содержание суммы каротиноидов в пересчете на
р-каротин в процентах рассчитывают по формуле:
А 50
т-250 ’
где:
250 — удельный показатель поглощения р-каро-
тина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Сабельника болотного корневища с корнями
1307
07/2016:РБ0086
САБЕЛЬНИКА БОЛОТНОГО
КОРНЕВИЩА С КОРНЯМИ
Сотап ра/изМз гЫгота сит гасИаЬиз
СОИ/ВЕККУЯООГ*
ОПРЕДЕЛЕНИЕ
Собранные осенью, очищенные от остатков над¬
земных частей и от земли, промытые и высушенные
корневища с корнями и укоренившиеся стебли много¬
летнего травянистого растения Сотагит ра1и81пз 1_.
Содержание: не менее 2,0 % суммы проан-
тоцианидинов в пересчете на цианидина хлорид
(С15Н10О6 НС1; М.м. 322,7) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь полых
кусков корневищ с корнями и кусков укоренившихся
стеблей с остатками оснований черешков листьев,
длиной до 30 см и диаметром (3—7) мм, с четко вы¬
раженными узлами, прямые или слегка изогнутые,
иногда разветвленные. Поверхность продольно-мор¬
щинистая матовая (у корневищ) или блестящая (у уко¬
ренившихся стеблей), с участками отслоившейся тем¬
но-коричневой первичной коры, со светло-коричне¬
выми или темно-коричневыми остатками оснований
стеблеобъемлющих черешков листьев и почечных
чешуй в междоузлиях и остатками нитевидных корней
в узлах корневищ. Цвет корневищ и укоренившихся
стеблей с поверхности от светло-коричневого, почти
черного, излом неровный, желтовато-белый, иногда
зеленоватый.
B. Микроскопия (2.8.23). На поперечном срезе
корневищ и укоренившихся стеблей видно вторичное
непучковое строение. Снаружи корневищ перидерма
из 7—8 радиальных рядов клеток пробки. За пери¬
дермой следует вторичная кора, узкая зона камбия
и широкий слой древесины с заметно выраженными
осенними и весенними элементами вторичной ксиле¬
мы, состоящей из крупных и мелких сосудов, трахеид,
древесной паренхимы. Сердцевинные лучи узкие, од¬
норядные. На поперечном срезе укоренившихся сте¬
блей видны узкий слой перидермы, первичная кора
из паренхимных клеток с крупными межклетниками
(аэренхима), иногда с крупными друзами оксалата
кальция. Перимедулярная зона сердцевины корне¬
вищ и укоренившихся стеблей состоит из толстостен¬
ных клеток, окрашенных в темно-коричневый цвет,
иногда содержащих многочисленные включения фи¬
олетово-красного цвета.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,50 г измельченного
сырья (180) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 50 мл, прибавляют 20 мл спирта
(70 %, об/об) Р и нагревают с обратным холодильни¬
ком на водяной бане в течение 30 мин. Охлаждают и
фильтруют.
Раствор сравнения. 5 мг катехина Р раство¬
ряют в 20 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — мета¬
нол Р — вода Р — кислота муравьиная безводная Р
(85:3:3,5:4, об/об/об/об).
Наносимый объем пробы, по 5 мкл в виде полос
длиной 20 мм и шириной около 3 мм.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: при температуре 105 °С в течение
5 мин, затем охлаждают на воздухе.
Проявление: пластинку опрыскивают раствором
1 г/л ванилина Рв96% спирте Р, затем погружают на
(1—2) мин в раствор 10 г/л кислоты серной Р в 96%
спирте Р и выдерживают при температуре 105 °С в
течение 5 мин. Просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Катехин: зона красного
цвета
Зона красного цвета
(катехин)
Зона красного цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: листья и другие части растения, в том
числе отделенные при анализе — не более 10 %. Ор¬
ганические примеси: не более 3 %. Минеральные при¬
меси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 6,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Реактив 3. 0,5 г железа (III) аммония сульфа¬
та Р растворяют в 25 мл 1 М растворе хлористово¬
дородной кислоты.
0,500 г измельченного сырья (250) (2.9.12) по¬
мещают в круглодонную колбу вместимостью 100 мл,
прибавляют 20,0 мл спирта (70 %, об/об) Р, закрыва¬
ют пробкой, взвешивают с точностью до 0,01 г и на¬
гревают с обратным холодильником на водяной бане
при температуре 80 °С в течение 20 мин. Охлаждают
до комнатной температуры, взвешивают и доводят
массу до первоначальной спиртом (70 %, об/об) Р.
Содержимое колбы центрифугируют при (2000—3000)
об/мин в течение (10—15) мин и собирают надосадоч-
ную жидкость (раствор А).
Испытуемый раствор. 0,10 мл раствора А поме¬
щают в круглодонную колбу вместимостью 50 мл, при¬
бавляют 0,9 мл спирта (70 %, об/об) Р, 0,2 мл реакти¬
ва 5, 6,0 мл 5 % (об/об) раствора кислоты хлористо¬
водородной Р1 в бутаноле Р, нагревают с обратным
холодильником на водяной бане при температуре от
80 °С до 95 °С в течение 60 мин и охлаждают до ком¬
натной температуры.
Компенсационный раствор. 0,10 мл раствора
А помещают в круглодонную колбу вместимостью
50 мл, прибавляют 0,9 мл спирта (70 %, об/об) Р,
0,2 мл реактива 5 и 6,0 мл 5 % (об/об) раствора кис¬
лоты хлористоводородной Р1 в бутаноле Р.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 550 нм.
165*. Зак. 1060.
1308
Гэсударственная фармакопея Республики Беларусь
Содержание суммы проантоцианидинов в пере¬
счете на цианидина хлорид в процентах рассчитыва¬
ют по формуле:
А 1440
т-352 ’
где:
352 — удельный показатель поглощения циани¬
дина хлорида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:0206
(§)СЕННЫ ЛИСТЬЯ
8еппае (оНит
ЗЕЫЫА 1.ЕАР
ОПРЕДЕЛЕНИЕ
Высушенные листочки Сазз1а зеппа 1_.
(С. асиШоНа ОеП1е), известной как александрийская
или хартумская сенна, или Сазз1а апдизШоНа УаЫ, из¬
вестной как тинивельская сенна, или смесь листочков
обоих видов.
Содержание: не менее 2,5 % гидроксиантрацено-
вых гликозидов в пересчете на сеннозид В (С42Н38О20;
М.м. 863) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листочки
С. зеппа серовато-зеленого или коричневато-зелено¬
го цвета, тонкие, хрупкие, ланцетовидные, остроко¬
нечные, асимметричные в основании, длиной обычно
(15—40) мм и шириной (5—15) мм, максимально ши¬
рокие в области немного ниже центра листа; листовая
пластинка слегка волнистая с обеих сторон, покрыта
мелкими короткими волосками. Перистое жилкование
заметно в большей степени на нижней стороне листа,
боковые жилки отходят от центральной под углом око¬
ло 60° и соединяются между собой дугами около края
листа.
Устьичный коэффициент (2.8.3): 10—12,5—15.
Листочки С. апдизШоНа желтовато-зеленого или
коричневато-зеленого цвета, вытянутой ланцето¬
видной формы, слегка асимметричные в основании,
длиной обычно (20—50) мм и шириной (7—20) мм в
центре. Обе поверхности гладкие с очень небольшим
количеством коротких волосков и часто с поперечны¬
ми или косыми линиями.
Устьичный коэффициент (2.8.3): 14—17,5—20.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Порошок имеет зеленый или
желтовато-зеленый цвет. Просматривают под микро¬
скопом с использованием раствора хлоралгидра¬
та Р. В порошке обнаруживаются следующие диа¬
гностические признаки (рисунок 0206.-1): фрагменты
эпидермиса (С. апдизМоНа: [А, В], С. зеппа: [3, К]) с
полигональными клетками [Аа, Ка], устьицами пара-
цитного типа [АЬ, Ас, Ва, За, КЬ] (2.8.3) и покрываю¬
щими одноклеточными волосками конической формы
с бородавчатыми стенками (вид с поверхности [Аб],
вид сбоку (С. зеппа: [О])) или рубцами [ВЬ, Лэ], ча¬
сто вместе с прилегающей палисадной паренхимой
[Ае, Зс]; отдельные, фрагментированные покрываю¬
щие волоски [Е]; волокна [Р] с кристаллической об¬
верткой призматических кристаллов кальция оксалата
[Ра]; отедльные призматические кристаллы оксалата
кальция [0]\ отдельные друзы кальция оксалата [Н];
фрагменты срединной паренхимы листа [С] с клетака-
ми, содержащими друзы кальция оксалата [Са], часто
вместе с палисадной паренхимой [СЬ] и сосудистыми
пучками [Сс].
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (180) (2.9.12) прибавляют 5 мл смеси из равных
объемов 96 % спирта Р и воды Р и нагревают до ки¬
пения. Центрифугируют и используют надосадочную
жидкость.
Раствор сравнения. 10 мг ФСО экстракта сен-
ны растворяют в 1 мл смеси из равных объемов 96 %
спирта Р и воды Р (раствор может содержать неболь¬
шой осадок).
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная Р—
вода Р — этилацетат Р — пропанол Р (1:30:40:40,
об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос
длиной 20 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают 20 %
(об/об) раствором кислоты азотной Р и нагревают
при температуре 120 °С в течение 10 мин. Охлаждают
и опрыскивают раствором 50 г/л калия гидроксида Р в
спирте (50 %, об/об) Р до проявления зон.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются основные зоны, соответ¬
ствующие по расположению (сеннозиды В, А, О и С
в порядке увеличения значения Я,), цвету и размеру
основным зонам на хроматограмме раствора сравне-
Сенны листья с плодами
1309
ния. Между зонами, соответствующими сеннозидам
й и С, может обнаруживаться зона красного цвета
(реин-8-глюкозид).
О. Около 25 мг измельченного сырья (180)
(2.9.12) помещают в коническую колбу, прибавляют
50 мл воды Р и 2 мл кислоты хлористоводородной Р
и нагревают в водяной бане в течение 15 мин. Охлаж¬
дают и встряхивают с 40 мл эфира Р. Эфирный слой
отделяют, высушивают с использованием натрия
сульфата безводного Р. 5 мл полученного раствора
выпаривают досуха и к охлажденному остатку прибав¬
ляют 5 мл раствора аммиака разведенного Р1. Про¬
является желтое или оранжевое окрашивание. Нагре¬
вают на водяной бане в течение 2 мин. Появляется
красновато-фиолетовое окрашивание.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: не более 3 %. Посторонние примеси: не
более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от яркого
света.
0,150 г измельченного сырья (180) (2.9.12) по¬
мещают в колбу вместимостью 100 мл. Прибавляют
30,0 мл воды Р, перемешивают, взвешивают и поме¬
щают в водяную баню. Нагревают с обратным холо¬
дильником в течение 15 мин. Охлаждают, взвешивают
и доводят водой Р до первоначальной массы. Цен¬
трифугируют и переносят 20,0 мл надосадочной жид¬
кости в делительную воронку вместимостью 150 мл.
Прибавляют 0,1 мл кислоты хлористоводородной
разведенной Р и трижды встряхивают с хлорофор¬
мом Р порциями по 15 мл. Отделяют и отбрасывают
хлороформный слой. К водному слою прибавляют
0,10 г натрия гидрокарбоната Р, встряхивают в те¬
чение 3 мин и центрифугируют. 10,0 мл надосадочной
жидкости помещают в круглодонную колбу вместимо¬
стью 100 мл со шлифом, прибавляют 20 мл раствора
железа (III) хлорида Р1 и перемешивают. Нагревают
с обратным холодильником в водяной бане в тече¬
ние 20 мин. Уровень воды в бане должен быть выше
уровня раствора в колбе. Прибавляют 3 мл кислоты
хлористоводородной Р и нагревают еще в течение
20 мин, часто встряхивая, до растворения осадка.
Охлаждают, переносят смесь в делительную воронку
и трижды встряхивают с эфиром Р, предварительно
использованным для ополаскивания колбы, порциями
по 25 мл. Объединяют 3 эфирных слоя и промывают
дважды водой Р порциями по 15 мл. Переносят объ¬
единенный эфирный слой в мерную колбу и доводят
эфиром Р до объема 100,0 мл. 10,0 мл полученно¬
го раствора осторожно выпаривают досуха. Остаток
растворяют в 10,0 мл раствора 5 г/л магния аце¬
тата Р в метаноле Р. Измеряют оптическую плот¬
ность (2.2.25) полученного раствора при длине волны
515 нм, используя метанол Р в качестве компенсаци¬
онного раствора.
Содержание гидроксиантраценовых гликозидов в
пересчете на сеннозид В в процентах рассчитывают
по формуле:
А-300
т-240 ’
где:
240 — удельный показатель поглощения сенно-
зида В;
А — оптическая плотность при длине волны
515 нм;
т — масса навески испытуемого сырья, г.
ХРАНЕНИЕ
В защищенном от влаги месте.
07/2016:РБ0087
СЕННЫ ЛИСТЬЯ С ПЛОДАМИ
8еппае IЬНит сит Iгис1из
5ЕЫЫА /.ЕАЕ 1У/777 Р005*
ОПРЕДЕЛЕНИЕ
Собранные в фазу цветения и плодоношения,
высушенные и обмолоченные листья кустарника
Сазз/а зеппа 1_. (зуп.: С. асиШоНа РеЖе).
Содержание: не менее 1,35 % суммы агликонов
антраценового ряда в пересчете на хризофановую
кислоту (С15Н10О; М.м. 254,25) (в пересчете на сухое
сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Отдельные ли¬
сточки и черешки сложного парноперистого листа,
цельные или частично измельченные, кусочки тонких
травянистых стеблей, бутоны, цветки и незрелые пло¬
ды. Листочки удлиненно-ланцетовидные или ланцето¬
овальные, заостренные к верхушке, наиболее широ¬
кие в средней части, у основания неравнобокие, тон¬
кие, ломкие, цельнокрайние, с очень коротким череш¬
ком. Вторичные жилки, ясно заметные с обеих сторон,
отходят под острым углом от главной жилки и соеди¬
няются между собой дугами, идущими параллельно
краю листочка. Длина листочка (1—4) см, ширина
(0,4—1,5) см, обе поверхности покрыты короткими
волосками. Цвет листочков с обеих сторон серовато-
зеленый или с верхней стороны желтовато-зеленый,
матовый. Плод — боб, плоский, кожистый, слабо изо¬
гнутый, длиной (3—5) см, шириной (1,5—2) см. Цвет
плодов зеленовато-коричневый с темными очертани¬
ями семенных камер. Цвет бутонов и цветков желтый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны клетки эпидермиса с
многоугольными прямыми стенками. Клетки, находя¬
щиеся у основания волоска, располагаясь радиально,
образуют угловатую 6—10-лучевую розетку. Волоски
короткие, простые, часто согнутые, одноклеточные, с
толстыми стенками и грубобородавчатой поверхно¬
стью. Волоски часто опадают и в центре розетки ви¬
ден округлый валик. Устьица окружены 2—3, реже 4
клетками эпидермиса (аномоцитный тип) (2.8.3), рас¬
положены с обеих сторон листа. В мезофилле видны
друзы оксалата кальция. Главные и более крупные
боковые жилки листа окружены кристаллоносной об¬
кладкой.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (180) (2.9.12) прибавляют 5 мл смеси из рав¬
ных объемов 96 % спирта Р и воды Р и нагревают
166. Зак. 1060.
1310
Гэсударственная фармакопея Республики Беларусь
до кипения. Центрифугируют и используют надоса-
дочную жидкость.
Раствор сравнения. 10 мг ФСО экстракта
сенны растворяют в 1 мл смеси из равных объемов
96 % спирта Р и воды Р (раствор может содержать
небольшой осадок).
Пластинка: ТСХ пластинка со слоем силика¬
геля О Р.
Подвижная фаза: кислота уксусная ледя¬
ная Р — вода Р — этилацетат Р — пропанол Р
(1:30:40:40, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде по¬
лос длиной 20 мм и шириной 2 мм.
Фронт подвижной фазы: не менее 10 см от
линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают 20 %
(об/об) раствором кислоты азотной Р и нагревают
при температуре 120 °С в течение 10 мин. Охлаж¬
дают и опрыскивают раствором 50 г/л калия гидрок¬
сида Р в спирте (50 %, об/об) Р до проявления зон.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются основные зоны, соот¬
ветствующие по расположению (сеннозиды В, А,
О и С в порядке увеличения значения РР), цвету и
размеру основным зонам на хроматограмме раст¬
вора сравнения. Между зонами, соответствующи¬
ми сеннозидам О и С, может обнаруживаться зона
красного цвета (реин-8-глюкозид). На хромато¬
грамме испытуемого раствора зоны, соответствую¬
щие сеннозидам О и С, неинтенсивные.
О. Около 25 мг измельченного сырья (180)
(2.9.12) помещают в коническую колбу, прибавля¬
ют 50 мл воды Р и 2 мл кислоты хлористоводо¬
родной Р и нагревают в водяной бане в течение
15 мин. Охлаждают и встряхивают с 40 мл эфи¬
ра Р. Эфирный слой отделяют, высушивают с ис¬
пользованием натрия сульфата безводного Р.
5 мл полученного раствора выпаривают досуха и к
охлажденному остатку прибавляют 5 мл раствора
аммиака разведенного Р1. Проявляется желтое
или оранжевое окрашивание. Нагревают на водя¬
ной бане в течение 2 мин. Появляется красновато¬
фиолетовое окрашивание.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Листочки и пло¬
ды — не менее 60 %. Несырьевые части растения:
кусочки стеблей толще 2 мм — не более 3 %; побу¬
ревшие, почерневшие листочки — не более 3 %. Ор¬
ганические примеси: не более 3 %. Минеральные при¬
меси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор. 0,400 г измельченного
сырья (710) (2.9.12) помещают в колбу вместимо¬
стью 200 мл, прибавляют 100,0 мл воды Р, взвеши¬
вают с точностью до 0,01 г и нагревают с обратным
холодильником в водяной бане в течение 20 мин при
перемешивании. Уровень воды в бане должен быть
выше уровня раствора в колбе. Охлаждают и дово¬
дят водой Р до первоначальной массы. Выдержива¬
ют в течение 10 мин и фильтруют через бумажный
фильтр. 25,0 мл фильтрата переносят в делительную
воронку вместимостью 100 мл и встряхивают дваж¬
ды с эфиром Р в течение 2 мин порциями по 40 мл
и 20 мл. Водный слой переносят в колбу со шлифом
вместимостью 150 мл. Эфирные извлечения объ¬
единяют и дважды промывают водой Р порциями
по 10 мл. Воду помещают в ту же колбу со шлифом
вместимостью 150 мл. Эфирные слои отбрасывают.
Объединенные водные извлечения нагревают на во¬
дяной бане до исчезновения запаха эфира. Прибав¬
ляют 0,1 г натрия гидрокарбоната Р, 10 мл раст¬
вора железа (III) хлорида Р1 и нагревают с обратным
холодильником в водяной бане при перемешивании
в течении 20 мин. Прибавляют 5 мл 50 % (м/м) раст¬
вора кислоты серной Р и нагревают еще в течение
30 мин. Охлаждают, полученный раствор переносят
в делительную воронку вместимостью 300 мл. Колбу
вместимостью 150 мл ополаскивают 20 мл воды Р,
75 мл эфира Р. Промывную воду и эфир присоеди¬
няют к раствору в делительной воронке и встряхива¬
ют в течение 5 мин. Эфирный слой отделяют (тем¬
ные хлопья оставляют в водном слое) и фильтруют
через стеклянный фильтр (ПОР 100) в делительную
воронку вместимостью 500 мл. Водный слой дважды
встряхивают с эфиром Р порциями по 30 мл и 20 мл.
Эфирные извлечения объединяют и фильтруют че¬
рез тот же стеклянный фильтр (100) в ту же дели¬
тельную воронку вместимостью 500 мл и дважды
промывают водой Р порциями по 30 мл. К эфирному
извлечению прибавляют 100 мл щелочно-аммиачно¬
го раствора Р и осторожно встряхивают в течение
5 мин. Прозрачный водный слой сливают в мерную
колбу вместимостью 250 мл (хлопья промежуточно¬
го слоя оставляют в воронке). К эфирному извлече¬
нию прибавляют 20 мл воды Р и 3 мл кислоты хло¬
ристоводородной Р. Воронку охлаждают под струей
воды, встряхивают в течение 2 мин и сливают во¬
дный слой в ту же мерную колбу. Эфирное извлече¬
ние еще раз встряхивают с 50 мл щелочно-аммиач¬
ного раствора Р в течение 2 мин и сливают водный
слой в ту же мерную колбу. Объединенные водные
извлечения доводят щелочно-аммиачным раство¬
ром Р до объема 250,0 мл.
Раствор сравнения. К 1,500 г кобальта хлори¬
да Ру высушенного до постоянной массы в эксикато¬
ре, прибавляют 20 мл воды Р, 0,1 мл кислоты хло¬
ристоводородной Р1 и доводят водой Р до объема
100,0 мл.
Через 15 мин измеряют оптическую плотность
(2.2.25) раствора при 523 нм, используя щелочно¬
аммиачный раствор Р в качестве компенсационного
раствора.
Содержание суммы агликонов антраценового
ряда в пересчете на хризофановую кислоту в процен¬
тах рассчитывают по формуле:
А т0 0,43
А0т
где:
0,43 — коэффициент пересчета оптической плот¬
ности раствора 15 г/л кобальта хлорида Р в содержа¬
ние хризофановой кислоты (г) в 1 л щелочно-аммиач¬
ного раствора;
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т — масса навески испытуемого сырья, г;
т0 — масса навески кобальта хлорида Р, г.
Сенны остролистной плоды
1311
07/2016:0207
<§)СЕННЫ
ОСТРОЛИСТНОЙ плоды
8еппае /гис(из асиШоНае
ЗЕЫЫА РОС5, А1.ЕХАЫОШАЫ
ОПРЕДЕЛЕНИЕ
Высушенные плоды Сазз/а зеппа 1_. (С. асиШоНа
Ое\\\е).
Содержание: не менее 3,4 % гидроксиантрацено-
вых гликозидов в пересчете на сеннозид В (С42Н38О20;
М.м. 863) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Сплющенные,
почковидные плоды зеленого или зеленовато-ко¬
ричневого цвета с коричневыми пятнами, соответ¬
ствующими по расположению семенам, обычно от
40 мм до 50 мм длиной и не менее 20 мм шириной.
На одном конце имеется отметка от столбика, на
другом — короткая плодоножка. Плод содержит
6—7 сплющенных семян обратнояйцевидной фор¬
мы зеленого или бледно-коричневого цвета, по
всей семенной кожуре которых наблюдается сетка
из выступающих ребрышек.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет коричневый. Про¬
сматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружи¬
ваются следующие диагностические признаки (ри¬
сунок 0207.-1): фрагменты эпикарпия (вид поверх¬
ности [В, К]) с многоугольными клетками, устьица
аномоцитного [Ва] или парацитного [ВЬ, Ка] типа
(2.8.3), покрывающие волоски [КЬ] или рубцы от них
[Вс]; отдельные конические бородавчатые волоски,
обычно искривленные [С]; фрагменты мезокарпия
(вид поверхности [Э]) в виде двух перекрестных
слоев волокон ра] с кристаллоносной обкладкой,
состоящей из призматических кристаллов оксала¬
та кальция [ОЬ] и иногда клетки эндокарпия рс];
фрагменты наружного слоя семенной кожуры (вид
поперечного среза ^]), покрытого тонкой кутикулой
[За] с палисадными клетками длиной около 50 мкм,
с тонкими стенками и редуцированной полостью
[Лэ] с прилегающей гиподермой с колонноподоб¬
ными клетками ^с]; палисадные клетки семенной
кожуры (вид поверхости [N1) и фрагменты гиподер¬
мы семени, формирующие кольца (вид поверхно¬
сти [А]); фрагменты семядоли (вид поверхности
[Р], вид поперечного среза [Е]), состоящие из не¬
больших клеток эпидермиса [Еа, Ра] и палисадной
ткани [ЕЬ, РЬ]; отдельные друзы и призматические
кристаллы оксалата кальция ре, Са] или включен¬
ные в паренхиму [6]; фрагменты сосудистых пуч¬
ков [Ц со спиралевиными сосудами [1_а] и и волок¬
нами с умерено уплотненными ямчатыми стенками
[1_Ь]; склереиды [М, О] и волокна [Н] с прилегающей
кристаллической обверткой оксалата кальция [На]
из плодоножки.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (180) (2.9.12) прибавляют 5 мл смеси из рав¬
ных объемов 96 % спирта Р и воды Р и нагревают
до кипения. Центрифугируют и используют надоса-
дочную жидкость.
Рисунок 0207.-1. Микроскопические признаки
плодов сены остролистной
Раствор сравнения. 10 мг ФСО экстракта
сенны растворяют в 1 мл смеси из равных объемов
96 % спирта Р и воды Р (раствор может содержать
небольшой осадок).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: кислота уксусная ледяная Р —
вода Р — этилацетат Р — пропанол Р (1:30:40:40,
об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос
длиной 20 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают 20 %
(об/об) раствором кислоты азотной Р и нагревают
при температуре 120 °С в течение 10 мин. Охлаждают
и обрабатывают раствором 50 г/л калия гидроксида Р
в спирте (50 %, об/об) Р до проявления зон.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются основные зоны, соответ¬
ствующие по расположению (сеннозиды В, А, О и С
в порядке увеличения значения Рр), цвету и размеру
основным зонам на хроматограмме раствора сравне¬
ния. Между зонами, соответствующими сеннозидам
й и С, может обнаруживаться зона красного цвета
(реин-8-глюкозид). На хроматограмме испытуемого
раствора зоны, соответствующие сеннозидам й и С,
неинтенсивные.
О. Около 25 мг измельченного сырья (180)
(2.9.12) помещают в коническую колбу, прибавляют
50 мл воды Р и 2 мл кислоты хлористоводород¬
ной Р и нагревают в водяной бане в течение 15 мин.
Охлаждают и встряхивают с 40 мл эфира Р. Эфир¬
ный слой отделяют, высушивают с использованием
натрия сульфата безводного Р. 5 мл полученно¬
го раствора выпаривают досуха и к охлажденному
166*. Зак. 1060.
1312
Гэсударственная фармакопея Республики Беларусь
остатку прибавляют 5 мл раствора аммиака раз-
веденного Р1. Проявляется желтое или оранже¬
вое окрашивание. Нагревают на водяной бане в
течение 2 мин. Появляется красновато-фиолетовое
окрашивание.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 9,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от яркого света.
0,150 г измельченного сырья (180) (2.9.12) по¬
мещают в колбу вместимостью 100 мл. Прибавляют
30.0 мл воды Р, перемешивают, взвешивают и по¬
мещают в водяную баню. Нагревают с обратным хо¬
лодильником в течение 15 мин. Охлаждают, взвеши¬
вают и доводят водой Р до первоначальной массы.
Центрифугируют и переносят 20,0 мл надосадочной
жидкости в делительную воронку вместимостью
150 мл. Прибавляют 0,1мл кислоты хлористово¬
дородной разведенной Р и трижды встряхивают с
хлороформом Р порциями по 15 мл. Отделяют и от¬
брасывают хлороформный слой. К водному слою
прибавляют 0,10 г натрия гидрокарбоната Р, встря¬
хивают в течение 3 мин и центрифугируют. 10,0 мл
надосадочной жидкости помещают в круглодонную
колбу вместимостью 100 мл со шлифом, прибавляют
20 мл раствора железа (III) хлорида Р1 и перемеши¬
вают. Нагревают с обратным холодильником в водя¬
ной бане в течение 20 мин. Уровень воды в бане дол¬
жен быть выше уровня раствора в колбе. Прибавляют
3 мл кислоты хлористоводородной Р и нагревают
еще в течение 20 мин, часто встряхивая, до раство¬
рения осадка. Охлаждают, переносят смесь в дели¬
тельную воронку и трижды встряхивают с эфиром Р,
предварительно использованным для ополаскивания
колбы, порциями по 25 мл. Объединяют 3 эфирных
слоя и промывают дважды водой Р порциями по
15мл. Переносят объединенное эфирное извлече¬
ние в мерную колбу и доводят эфиром Р до объема
100.0 мл. 10,0 мл полученного раствора осторожно
выпаривают досуха. Остаток растворяют в 10,0 мл
раствора 5 г/л магния ацетата Р в метаноле Р. Из¬
меряют оптическую плотность (2.2.25) полученного
раствора при длине волны 515 нм, используя мета¬
нол Р в качестве компенсационного раствора.
Содержание гидроксиантраценовых гликозидов в
пересчете на сеннозид В в процентах рассчитывают
по формуле:
А -300
т-240 ’
где:
240 — удельный показатель поглощения сенно-
зида В;
А — оптическая плотность при длине волны
515 нм;
т — масса навески испытуемого сырья, г.
ХРАНЕНИЕ
В защищенном от влаги месте.
07/2016:0208
@) СЕННЫ УЗКОЛИСТНОЙ плоды
8еппае Iгис1из апдизШоНае
ЗЕЫЫА РООЗ, ТШЫЕУЕНУ
ОПРЕДЕЛЕНИЕ
Высушенные плоды Сазз/'а апдизШоНа УаЫ.
Содержание: не менее 2,2 % гидроксиантрацено¬
вых гликозидов в пересчете на сеннозид В (С42Н38О20;
М.м. 863) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Сплющенные,
слегка почковидные плоды желтовато-коричневого
или коричневого цвета с темно-коричневыми пят¬
нами, соответствующими по расположению семе¬
нам, обычно (35—60) мм длиной и (14—18) мм ши¬
риной. На одном конце имеется отметка от столби¬
ка, на другом — короткая плодоножка. Плод содер¬
жит 5—8 сплющенных семян обратнояйцевидной
формы зеленого или бледно-коричневого цвета с
неполными волнистыми поперечными ребрышками
на семенной кожуре.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет коричневый. Про¬
сматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружи¬
ваются следующие диагностические признаки (ри¬
сунок 0208.-1): фрагменты эпикарпия (вид поверх¬
ности [В, К]) с многоугольными клетками, устьица
аномоцитного [Ва] или парацитного [ВЬ, Ка] типа
(2.8.3), покрывающие волоски [КЬ] или рубцы от них
[Вс]; отдельные конические бородавчатые волоски,
обычно искривленные [С]; фрагменты мезокарпия
(вид поверхности [й]) в виде двух перекрестных
слоев волокон [Оа] с кристаллоносной обкладкой,
состоящей из призматических кристаллов оксала¬
та кальция рЬ] и иногда клетки эндокарпия рс];
фрагменты наружного слоя семенной кожуры (вид
поперечного среза [3]), покрытого тонкой кутикулой
^а] с палисадными клетками длиной около 50 мкм,
с тонкими стенками и редуцированной полостью
[Лэ] с прилегающей гиподермой с колонноподоб¬
ными клетками [Зс]; палисадные клетки семенной
кожуры (вид поверхости [Ы]) и фрагменты гиподер¬
мы семени, формирующие кольца (вид поверхно¬
сти [А]); фрагменты семядоли (вид поверхности
[Р], вид поперечного среза [Е]), состоящие из не¬
больших клеток эпидермиса [Еа, Ра] и палисадной
ткани [ЕЬ, РЬ]; отдельные друзы и призматические
кристаллы оксалата кальция [Ое, Са] или включен¬
ные в паренхиму [О]; фрагменты сосудистых пуч¬
ков [1_] со спиралевиными сосудами [1_а] и и волок¬
нами с умерено уплотненными ямчатыми стенками
[1_Ь]; склереиды [М, О] и волокна [И] с прилегающей
кристаллической обверткой оксалата кальция [На]
из цветоножки плода.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (180) (2.9.12) прибавляют 5 мл смеси из рав¬
ных объемов 96 % спирта Р и воды Р и нагревают
до кипения. Центрифугируют и используют надоса-
дочную жидкость.
Раствор сравнения. 10 мг ФСО экстракта сен-
ны растворяют в 1 мл смеси из равных объемов 96 %
Сенны узколистной плоды
1313
Рисунок 0208.-1. Микроскопические признаки
плодов сены узколистной
спирта Р и воды Р (раствор может содержать неболь¬
шой осадок).
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: кислота уксусная ледя¬
ная Р — вода Р — этилацетат Р — пропанол Р
(1:30:40:40, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде по¬
лос длиной 20 мм.
Фронт подвижной фазы: не менее 10 см от
линии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают 20 %
(об/об) раствором кислоты азотной Р и нагрева¬
ют при температуре 120 °С в течение 10 мин. Ох¬
лаждают и обрабатывают раствором 50 г/л калия
гидроксида Р в спирте (50 %, об/об) Р до прояв¬
ления зон.
Результаты: на хроматограмме испытуемого
раствора обнаруживаются основные зоны, соот¬
ветствующие по расположению (сеннозиды В, А,
О и С в порядке увеличения значения ПР), цвету и
размеру основным зонам на хроматограмме раст¬
вора сравнения. Между зонами, соответствующи¬
ми сеннозидам Э и С, может обнаруживаться зона
красного цвета (реин-8-глюкозид). На хромато¬
грамме испытуемого раствора зоны, соответствую¬
щие сеннозидам й и С, неинтенсивные.
О. Около 25 мг измельченного сырья (180)
(2.9.12) помещают в коническую колбу, прибавля¬
ют 50 мл воды Р и 2 мл кислоты хлористоводо¬
родной Р и нагревают в водяной бане в течение
15 мин. Охлаждают и встряхивают с 40 мл эфи¬
ра Р. Эфирный слой отделяют, высушивают с ис¬
пользованием натрия сульфата безводного Р.
5 мл полученного раствора выпаривают досуха и к
охлажденному остатку прибавляют 5 мл раствора
аммиака разведенного Р1. Проявляется желтое
или оранжевое окрашивание. Нагревают на водя¬
ной бане в течение 2 мин. Появляется красновато¬
фиолетовое окрашивание.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 12,0 %. 1,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 9,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытание проводят с защитой от яркого света.
0,150 г измельченного сырья (180) (2.9.12) по¬
мещают в колбу вместимостью 100 мл. Прибавля¬
ют 30,0 мл воды Р, перемешивают, взвешивают
и помещают в водяную баню. Нагревают с обрат¬
ным холодильником в течение 15 мин. Охлаждают,
взвешивают и доводят водой Р до первоначальной
массы. Центрифугируют и переносят 20,0 мл надо-
садочной жидкости в делительную воронку вмести¬
мостью 150 мл. Прибавляют 0,1 мл кислоты хлори¬
стоводородной разведенной Р и трижды встряхива¬
ют с хлороформом Р порциями по 15 мл. Отделяют
и отбрасывают хлороформный слой. К водному
слою прибавляют 0,10 г натрия гидрокарбоната Р,
встряхивают в течение 3 мин и центрифугируют.
10,0 мл надосадочной жидкости помещают в кру¬
гл одонную колбу вместимостью 100 мл со шлифом,
прибавляют 20 мл раствора железа (III) хлорида Р1
и перемешивают. Нагревают с обратным холодиль¬
ником в водяной бане в течение 20 мин. Уровень
воды в бане должен быть выше уровня раствора в
колбе. Прибавляют 3 мл кислоты хлористоводо¬
родной Р и нагревают еще в течение 20 мин, часто
встряхивая, до растворения осадка. Охлаждают,
переносят смесь в делительную воронку и трижды
встряхивают с эфиром Р, предварительно исполь¬
зованным для ополаскивания колбы, порциями по
25 мл. Объединяют 3 эфирных слоя и промывают
дважды водой Р порциями по 15мл. Переносят
объединенное эфирное извлечение в мерную кол¬
бу и доводят эфиром Р до объема 100,0 мл. 10,0 мл
полученного раствора осторожно выпаривают до¬
суха. Остаток растворяют в 10,0 мл раствора 5 г/л
магния ацетата Р в метаноле Р. Измеряют оп¬
тическую плотность (2.2.25) полученного раствора
при длине волны 515 нм, используя метанол Р в
качестве компенсационного раствора.
Содержание гидроксиантраценовых гпикозидов в
пересчете на сеннозид В в процентах рассчитывают
по формуле:
А 300
т 240
где:
240 — удельный показатель поглощения сенно-
зида В;
А — оптическая плотность при длине волны
515 нм;
т — масса навески испытуемого сырья, г.
ХРАНЕНИЕ
В защищенном от влага мест.
1314
Гэсударственная фармакопея Республики Беларусь
07/2016.РБ0088
СИНЮХИ КОРНЕВИЩА С КОРНЯМИ
Ро1етопН соеги1е/ гЫюта сит габюЬиз
С НЕ АТ \/А1-ЕШАМ КООТ*
ОПРЕДЕЛЕНИЕ
Собранные ранней весной или осенью, быстро
отмытые от земли и высушенные корневища с корня¬
ми многолетнего травянистого растения Ро1етопшт
соет1еит 1_.
Содержание: не менее 8,0 % суммы тритерпено-
вых сапонинов в пересчете на р-эсцин (М.м. 1101,2)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
разрезанные вдоль корневища с корнями. Корневи¬
ща горизонтальные, прямые или слегка изогнутые,
иногда ветвящиеся, с многочисленными придаточ¬
ными корнями; длина корневищ от 0,5 см до 5 см,
толщина — от 0,3 см до 2 см. Поверхность корневищ
морщинистая, излом ровный или зернистый. В цен¬
тре часто имеется полость вследствие разрушения
сердцевины. Корни тонкие, длиной от 7 см до 35 см,
толщиной (1—2) мм, мелкие, шероховатые, цилин¬
дрические, узловатые, ломкие. Цвет корневищ с по¬
верхности серовато-коричневый, на изломе — жел¬
товато-белый или белый. Корни снаружи желтые, на
изломе — белые.
B. Микроскопия (2.8.23). На поперечном срезе
корневища видно, что покровная ткань состоит из
1—2 слоев клеток эпидермиса с опробковевшими
оболочками. Первичная кора представлена относи¬
тельно крупными паренхимными клетками с меж¬
клетниками треугольной формы. Заметно кольцо
эндодермы, состоящее из тонкостенных, тангенталь-
но-вытянутых клеток. Во вторичной коре находятся
участки флоэмы с неясными границами; паренхима
коры содержит простые и сложные крахмальные
зерна. Камбий слабо выражен. Сердцевинные лучи
одно- и двухрядные. В древесине сосуды распола¬
гаются одиночно или радиальными группами. Серд¬
цевина состоит из крупных паренхимных клеток, за¬
полненных крахмалом.
На поперечном срезе корня видна покровная
ткань, состоящая из 1—2 слоев округлых клеток эпи¬
дермиса с тонкими опробковевшими оболочками.
Первичная кора состоит из крупных, тангентально-
вытянутых клеток с неравномерно утолщенными
оболочками. Эндодерма хорошо выражена, клеточ¬
ные оболочки ее окрашиваются раствором Судана
IIIР в оранжево-красный цвет. Вторичная кора значи¬
тельно уже первичной и состоит из мелких клеток —
проводящих элементов луба и более крупных клеток
лубяной паренхимы. Камбиальная зона слабо выра¬
жена. В древесине корня сосуды разного диаметра,
располагаются без особого порядка, сердцевинные
лучи незаметны. В паренхимных клетках коры и дре¬
весины содержатся капли жирного масла; изредка
встречаются мелкие крахмальные зерна.
C. 2,0 г измельченного сырья (5600) (2.9.12) по¬
мещают в колбу вместимостью 100 мл, прибавляют
50 мл воды Р и нагревают в водяной бане в течение
10 мин при перемешивании. Охлаждают и фильтру¬
ют. 5 мл полученного раствора сильно встряхивают.
Образуется обильная и стойкая пена.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корневища с остатками стеблей длиной
свыше 1 см — не более 5 %; корневища, побурев¬
шие в изломе, — не более 3 %; частицы, проходящие
сквозь сито (710) (2.9.12), — не более 5 %. Органиче¬
ские примеси: не более 1 %. Минеральные примеси:
не более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 13,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,000 г измельченного сырья (710) (2.9.12) по¬
мещают в круглодонную колбу со шлифом вмести¬
мостью 250 мл, прибавляют 50 мл хлороформа Р
и нагревают с обратным холодильником на водя¬
ной бане в течение 30 мин. Горячее хлороформное
извлечение процеживают через ватный тампон,
избегая попадания на него частиц сырья. Ватный
тампон переносят в колбу с сырьем, прибавляют
50 мл хлороформа Р и повторно экстрагируют на
водяной бане в течение 30 мин. Горячее хлоро¬
формное извлечение процеживают через ватный
тампон, избегая попадания на него частиц сырья.
Хлороформные извлечения отбрасывают. Ватный
тампон помещают в колбу с сырьем, прибавляют
50 мл спирта (40 %, об/об) Р и нагревают с об¬
ратным холодильником в водяной бане в течение
30 мин. Процеживают через ватный тампон, из¬
бегая попадания на него частиц сырья, в мерную
колбу вместимостью 100 мл. Ватный тампон поме¬
щают в колбу с сырьем, прибавляют 50 мл спирта
(40 %, об/об) Р и нагревают с обратным холодиль¬
ником в водяной бане в течение 30 мин. Процежи¬
вают через ватный тампон в ту же мерную колбу.
Охлаждают и доводят спиртом (40 %, об/об) Р до
объема 100,0 мл. 1,0 мл полученного раствора до¬
водят водой Р до объема 25,0 мл (раствор А).
Испытуемый раствор. 2,0 мл раствора А по¬
мещают в полипропиленовый патрон с внутренним
диаметром 10 мм, заполненный 0,2 г силикагеля
гексадецилсилильного для хроматографии (па¬
трон с сорбентом предварительно выдерживают
с 0,3 мл 96 % спирта Р в течение 15 мин и про¬
мывают 5 мл воды Р). Промывают 5 мл воды Р со
скоростью 1 мл/мин. Водный элюат отбрасывают.
Через патрон дважды пропускают 96 % спирт Р
порциями по 0,5 мл. Элюаты объединяют и выпа¬
ривают досуха. К остатку прибавляют 10 мл кисло¬
ты серной Р. Выдерживают при температуре 70 °С
в течение 60 мин и охлаждают.
Компенсационный раствор, полипропилено¬
вый патрон с внутренним диаметром 10 мм, за¬
полненный 0,2 г силикагеля гексадецилсилильного
для хроматографии (патрон с сорбентом предва¬
рительно выдерживают с 0,3 мл 96 % спирта Р в
течение 15 мин и промывают 5 мл воды Р). Промы¬
вают 5 мл воды Р со скоростью 1 мл/мин. Водный
элюат отбрасывают. Через патрон пропускают 1 мл
96 % спирта Р. Элюат выпаривают досуха. При¬
бавляют 10 мл кислоты серной Р. Выдерживают
при температуре 70 °С в течение 60 мин и охлаж¬
дают.
Солодки корень (лакричный корень)
1315
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 321 нм.
Содержание суммы тритерпеновых сапонинов в
пересчете на (3-эсцин в процентах рассчитывают по
формуле:
А >12500
т-421 ’
где:
421 — удельный показатель поглощения продук¬
та взаимодействия р-эсцина с серной кислотой;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:0277
СОЛОДКИ КОРЕНЬ
(ЛАКРИЧНЫЙ КОРЕНЬ)
(3/усуггЫгае гасИх (ЩитИве гасИх)
испготсЕ поот
ОПРЕДЕЛЕНИЕ
Высушенные, очищенные или не очищенные
от пробки, цельные или измельченные корни и под¬
земные побеги многолетних травянистых растений
С1усуггЫга д!аЬга 1_. и/или С1усуггЫга ШаХа Ва1. и/или
СХусуггШа ига1епз1з НзсЬ.
Содержание: глицирризиновой кислоты
(^42Н62^16» М.м. 823) (в пересчете на сухое сырье):
- не менее 6,0 % при определении методом спек¬
трофотометрии (2.2.25);
- или не менее 4,0 % при определении методом
жидкостной хроматографии (2.2.29).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Корень с не¬
большим количеством ответвлений. Кора коричневого
или коричневато-серого цвета продольно-морщини¬
стая со следами боковых корней. Цилиндрические
столоны имеют диаметр (1—2) см, внешний вид по¬
добен внешнему виду корней, кроме того дополни¬
тельно обнаруживаются редкие мелкие почки. Излом
корня и столона зернистый и волокнистый. Пробко¬
вый слой тонкий; зона вторичной флоэмы широкая,
светло-желтого цвета с радиальной бороздчатостью.
Цилиндрическая желтая ксилема плотная, с ради¬
ально-симметричной структурой. Столон имеет цен¬
тральную сердцевину, которая отсутствует у корней. У
очищенных корней наружная часть коры отсутствует.
<®> В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет светло-желтый
или бледно-сероватый. Просматривают под микро¬
скопом с использованием раствора хлоралгидра¬
та Р. В порошке обнаруживаются следующие диа¬
гностические признаки: фрагменты желтых толсто¬
стенных волокон длиной (700—1200) мкм и шириной
(10—20) мкм с точкообразым просветом, часто с
кристаллической обкладкой, содержащей призмы
оксалата кальция длиной (10—35) мкм и шириной
(2—5) мкм; стенки сосудов желтые, толщиной
(5—10) мкм, лигнифицированные, с большим количе¬
ством окаймленных пор со щелевидными отверсти¬
ями; фрагменты пробки, состоящие из тонкостенных
клеток, а также отдельные призматические кристаллы
оксалата кальция и фрагменты паренхимной ткани.
У очищенных корней фрагменты пробки отсутству¬
ют. Порошок просматривают под микроскопом с ис¬
пользованием смеси равных объемов глицерина Р и
воды Р. В порошке обнаруживаются следующие диа¬
гностические признаки: простые круглые или оваль¬
ные зерна крахмала диаметром (2—20) мкм.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,50 г измельченного
сырья (180) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 50 мл, прибавляют 16,0 мл воды Р,
4,0 мл кислоты хлористоводородной Р1 и нагревают
с обратным холодильником на водяной бане в тече¬
ние 30 мин. Охлаждают и фильтруют. Круглодонную
колбу и фильтр сушат при температуре 105 °С в тече¬
ние 60 мин. Фильтр помещают в круглодонную колбу,
прибавляют 20,0 мл эфира Р и нагревают с обрат¬
ным холодильником в водяной бане при температуре
40 °С в течение 5 мин. Охлаждают и фильтруют. Филь¬
трат выпаривают досуха, остаток растворяют в 5,0 мл
эфира Р.
Раствор сравнения. 5,0 мг глицирретиновой
кислоты Р и 5,0 мг тимола Р растворяют в 5,0 мл
эфира Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: раствор аммиака концентри¬
рованный Р — вода Р — 96% спирт Р — этилаце-
тат Р (1:9:25:65, об/об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы, не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе в течение 5 мин.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограммах испытуемого
раствора и раствора сравнения в нижней половине
обнаруживается зона поглощения (глицирретиновая
кислота).
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р и нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин. Про¬
сматривают при дневном свете.
Результаты В: на хроматограмме раствора
сравнения в нижней половине обнаруживается зона
фиолетового цвета (глицирретиновая кислота) и в
верхней трети — зона красного цвета (тимол). На
хроматограмме испытуемого раствора в нижней по¬
ловине обнаруживается зона фиолетового цвета со¬
ответствующая зоне глицирретиновой кислоты на
хроматограмме раствора сравнения; в верхней трети
обнаруживается зона желтого цвета (изоликвиридиге-
нин) ниже зоны тимола на хроматограмме раствора
сравнения. На хроматограмме испытуемого раствора
могут обнаруживаться и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Неочищенное сырье
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: корни, дряблые в изломе, желто-корич¬
невые и остатки стеблей — не более 4 %. Органиче¬
ские примеси: не более 1 %. Минеральные примеси:
не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 8,0 %.
1316
Гэсударственная фармакопея Республики Беларусь
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,5 %.
@> Охратоксин А (2.8.22). Не более 20 мкг/кг.
Очищенное сырье
Допустимые примеси (2.8.2). Несырьевые части
растения: корни, плохо очищенные от пробки (с остат¬
ками более трех участков темно-коричневой пробки
на одном куске или поперечнике остатков пробки бо¬
лее 10 мм) — не более 15 %; корни, потемневшие и
побуревшие с поверхности, но светло-желтые в изло¬
ме — не более 20 %. Органические примеси: не более
0,5 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 6,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
@ Охратоксин А (2.8.22). Не более 20 мкг/кг.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях (2.2.25). 2,000 г
измельченного сырья (180) (2.9.12) помещают в кол¬
бу вместимостью 150 мл, прибавляют 20 мл смеси
из кислоты азотной Р и ацетона Р (1:20, об/об) и
встряхивают в течение 1 ч. Извлечение фильтруют в
мерную колбу вместимостью 100 мл. Остаток в колбе
промывают 10 мл ацетона Р и фильтруют через тот
же фильтр. В колбу с остатком прибавляют 20 мл аце¬
тона Р, которым одновременно смывают остатки сы¬
рья с фильтра и кипятят с обратным холодильником на
водяной бане в течение 5 мин. Извлечение фильтруют
через тот же фильтр в ту же мерную колбу. Экстракцию
горячим ацетоном повторяют еще два раза. Остатки
сырья промывают ацетоном Р до объема жидкости
в мерной колбе 100,0 мл. Содержимое мерной кол¬
бы количественно переносят в стакан вместимостью
200 мл, ополаскивая 40 мл 96 % спирта Р. Прибавля¬
ют по каплям при интенсивном перемешивании раст¬
вор аммиака концентрированный Р до появления
обильного светло-желтого творожистого осадка (до
значения рН от 8,3 до 8,6). Маточную жидкость вместе
с осадком фильтруют через бумажный фильтр под ва¬
куумом. Стакан и фильтр с осадком промывают 50 мл
ацетона Р порциями по (10—15) мл. Осадок с филь¬
тром переносят в стакан, в котором производилось
осаждение, прибавляют 50 мл воды Р и перемешива¬
ют. Полученный раствор количественно переносят в
мерную колбу вместимостью 250 мл. Фильтр несколь¬
ко раз промывают небольшими порциями воды Р, при¬
бавляя их к раствору в мерной колбе. Доводят водой Р
до объема 250,0 мл. 30,0 мл полученного раствора
разводят водой Р до объема 500,0 мл.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при 258 нм, используя воду Р в ка¬
честве компенсационного раствора.
Содержание глицирризиновой кислоты в процен¬
тах рассчитывают по формуле:
А-823-417
А77 11000 ’
где:
11000 — молярный показатель поглощения гли¬
цирризиновой кислоты;
823 — молекулярная масса глицирризиновой
кислоты;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
® Жидкостная хроматография (2.2.29).
Испытуемый раствор. 1,000 г измельченного
сырья (180) (2.9.12) помещают в коническую колбу
вместимостью 150 мл, прибавляют 100,0 мл раствора
8 г/л раствора аммиака Р и обрабатывают в ультра¬
звуковой бане в течение 30 мин. Часть полученного
раствора центрифугируют и 1 мл надосадочной жид¬
кости доводят раствором 8 г/л раствора аммиака Р
до объема 5,0 мл. Полученный раствор фильтруют
через мембранный фильтр (номинальный размер пор
0,45 мкм); используют фильтрат в качестве испытуе¬
мого раствора.
Раствор А. 0,130 г ФСО моноамминия глицир-
ризата растворяют в растворе 8 г/л раствора амми¬
ака Р и доводят до объема 100,0 мл этим же раство¬
рителем.
Раствор сравнения (а). 5,0 мл раствора А дово¬
дят раствором 8 г/л раствора аммиака Р до объема
100.0 мл.
Раствор сравнения (Ь). 10,0 мл раствора А дово¬
дят раствором 8 г/л раствора аммиака Р до объема
100.0 мл.
Раствор сравнения (с). 15,0 мл раствора А дово¬
дят раствором 8 г/л раствора аммиака Р до объема
100.0 мл.
Условия хроматографирования:
-колонка длиной 0,10м и внутренним диаме¬
тром 4 мм, заполненная силикагелем октадецилси-
лильным для хроматографии Р с размером частиц
5 мкм;
- подвижная фаза: кислота уксусная ледя¬
ная Р — ацетонитрил Р — вода Р (6:30:64, об/об/об);
- скорость подвижной фазы: 1,5 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
- объем вводимой пробы: 10 мкл.
Строят градуировочный график, указывая на оси
абсцисс массы моноаммония глицирризата, в грам¬
мах, на оси ординат — соответствующие площади
пиков.
Определяют и интегрируют пик 18Р-глицирризино-
вой кислоты на хроматограмме испытуемого раст¬
вора, используя соответствующие времена удержива¬
ния и площади пиков на хроматограммах растворов
сравнения.
Содержание 18р-глицирризиновой кислоты в
процентах рассчитывают по формуле:
где:
А — масса моноамминия глицирризата в испытуе¬
мом растворе, найденная по градуировочному графику, г;
В — содержание моноамминия глицирризата в
ФСО моноамминия глицирризата;
т — масса испытуемого сырья, использованная
для приготовления испытуемого раствора, г;
823 — молекулярная масса 18р-глицирризиновой
кислоты;
840 — молекулярная масса моноамминия гли¬
цирризата (без учета кристаллизационной воды).
МАРКИРОВКА
На упаковке указывают, что сырье очищенное
(или неочищенное).
Сушеницы топяной трава
1317
07/2016:РБ0089
СОСНЫ почки
Р/т детта
рше всю*
ОПРЕДЕЛЕНИЕ
Собранные до распускания и высушенные почки
Р'тиз зНуез(пз 1_.
Содержание: не менее 3 мл/кг эфирного масла
(в пересчете на сухое сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеют характерный ароматный, смолистый запах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Почки (укорочен¬
ные верхушечные побеги) длиной (1—4) см, одиноч¬
ные или по несколько штук в мутовках, окружающих
более крупную центральную почку, без стебля или с
остатком стебля, длиной не более 3 мм. Поверхность
почек покрыта сухими, спирально расположенными
ланцетовидными, заостренными бахромчатыми че¬
шуйками, склеенными между собой выступающей
смолой. Цвет снаружи розовато-коричневый, в изло¬
ме зеленый или коричневый.
B. Микроскопия (2.8.23). При просматривании
чешуи почки с поверхности видны: в центральной ча¬
сти трахеиды со щелевидными порами и заостренны¬
ми концами и два смоляных хода, идущих от основа¬
ния чешуйки до ее верхушки; в периферической части
вытянутые паренхимные клетки, концы которых часто
отогнуты к основанию чешуйки, иногда они заканчи¬
ваются свободно и образуют бахромчатость края че¬
шуйки.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: хвоя — не более 0,5 %; почки со стеблем
длиной более 3 мм и переросшие — не более 10 %;
частицы, проходящие сквозь сито (2800) (2.9.12), —
не более 5 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0 %. 2,000 г крупноизмельченного сырья су¬
шат при температуре 105 °С.
Общая зола (2.4.16). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют содержание эфирного масла (2.8.12,
метод В). 20,0 г крупноизмельченного сырья (без про¬
сеивания) помещают в колбу вместимостью 1000 мл,
прибавляют 400 мл воды Р в качестве жидкости для
перегонки. Время перегонки 90 мин.
07/2016:РБ0090
СУШЕНИЦЫ ТОПЯНОЙ ТРАВА
ОпарЬаШ иИдтоз! ЬегЬа
МАКЗН СДОШЕЕО НЕПВ*
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения и высушенная тра¬
ва с корнями однолетнего травянистого растения
СпарЫшт иНдтозит 1_. з.1.
Содержание: не менее 0,2 % суммы флаво-
ноидов в пересчете на гнафалозид А (С32Н30О15;
М.м. 654,6) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или
частично измельченные олиственные стебли длиной
до 30 см с серовато-белым войлочным опушением.
Корни тонкие стержневые, ветвистые. Стебли тон¬
кие, цилиндрические, обычно от основания распро¬
стерто-ветвистые. Листья длиной (0,5—3,5) см, ши¬
риной (0,1—0,4) см, очередные, короткочерешковые,
линейно-продолговатые, с туповатой верхушкой и
выдающейся срединной жилкой. Соцветие состоит
обычно из нескольких яйцевидных мелких корзинок
длиной (0,3—0,4) см, плотно скученных клубочками
на верхушках побегов и окруженных лучисторасходя-
щимися листьями, превышающими клубочки соцветий.
Обвертка корзинки состоит из 2—3 рядов черепитчато¬
расположенных темно-коричневых листочков; наруж¬
ные листочки яйцевидные, при основании войлочные,
в верхней половине голые, блестящие; внутренние —
продолговато-яйцевидные, заостренные, голые. Цвет¬
ки мелкие, желтоватые, трубчатые, пятизубчатые. Пло¬
ды — семянки с хохолком из 10 отдельных волосков.
Корни стержневые, ветвистые. Цвет зеленовато-серый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны клетки эпидермиса, с обе¬
их сторон более или менее вытянутые по длине ли¬
ста. Клетки эпидермиса верхней стороны со слегка
извилистыми стенками, с нижней — сильно извили¬
стые. Устьица крупные, овальные, погруженные, окру¬
жены 4—5 клетками эпидермиса и ориентированы
по длине листа (аномоцитный тип) (2.8.3): на нижней
стороне их значительно больше. На обеих сторонах
листа встречаются многочисленные простые воло¬
ски с тонкими стенками с 1—3 базальными клетками
и длинной извилистой конечной клеткой. Встречают¬
ся головчатые волоски, состоящие из одноклеточной
ножки и многоклеточной удлиненно-овальной головки;
клетки головки располагаются в один или два ряда.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Органические
примеси: не более 2 %. Минеральные примеси: не
более 2 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 20,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,000 г измельченного сырья (2000) (2.9.12) по¬
мещают в патрон из фильтровальной бумаги и экстра¬
гируют 96 % спиртом Р в аппарате Сокслета с рабо¬
чим объемом 150 мл на водяной бане в течение 5 ч
(20—25 сливов). Извлечение выпаривают по частям
досуха в водяной бане в круглодонной колбе вмести¬
мостью 100 мл.
К сухому остатку в колбе прибавляют 5 мл раст¬
вора 100 г/л натрия хлорида Р. Колбу нагревают
на водяной бане в течение 2 мин, растирая осадок
стеклянной палочкой до растворения. Охлаждают и
количественно переносят на колонку, заполненную
полиамидом для колоночной хроматографии Р (ис¬
пользуют подходящие готовые колонки для твердо-
1318
Гэсударственная фармакопея Республики Беларусь
фазной экстракции или подготавливают колонку сле¬
дующим образом: 1,5 г полиамида для колоночной
хроматографии Р помещают в стаканчик вместимо¬
стью 50 мл, прибавляют 30 мл воды Р, перемешива¬
ют и выливают через воронку в колонку диаметром
1,2 см и высотой 25 см. В нижнюю часть колонки
предварительно помещают небольшой ватный там¬
пон, смоченный водой Р. Колонку заполняют при от¬
крытом кране. Элюирование проводят со скоростью
4 мл/мин, не допуская обнажения поверхности сор¬
бента). Элюат повторно пропускают через колонку.
Слив производят через воронку с ватным тампоном,
предварительно смоченным водой. Колонку промы¬
вают 10 мл воды Р, затем убирают ватный тампон и
промывают колонку еще 20 мл воды Р. Водный элю¬
ат отбрасывают. Флавоноиды элюируют 50 мл 96 %
спирта Р со скоростью 4 мл/мин. Когда зона темно¬
желтого цвета (обнаруживается в ультрафиолетовом
свете при длине волны 365 нм) подойдет к нижней
части сорбента, элюат собирают в мерную колбу вме¬
стимостью 50 мл и доводят 96 % спиртом Р до объ¬
ема 50,0 мл.
Испытуемый раствор. 1,0 мл извлечения дово¬
дят 96 % спиртом Р до объема 25,0 мл.
Раствор сравнения. 0,0300 г дихромата ка¬
лия Р, высушенного до постоянной массы, раство¬
ряют в 1000, 0 мл 0,005 М раствора серной кислоты.
Компенсационный раствор (а). 50 мл 96 % спир¬
та Р пропускают через колонку, заполненную поли¬
амидом для колоночной хроматографии Р. Доводят
объем элюата 96 % спиртом Р до 50,0 мл.
Компенсационный раствор (Ь). 0,005 М раствор
серной кислоты.
Измеряют оптическую плотность (2.2.25) испы¬
туемого раствора и раствора сравнения при 338 нм,
используя компенсационный раствор (а) и компенса¬
ционный раствор (Ь) соответственно.
Содержание суммы флавоноидов в пересчете на
гнафалозид А в процентах рассчитывают по формуле:
Ат0- 0,2020 • 250 • 1,03
А0т
где:
0,2020 — коэффициент пересчета дихромата ка¬
лия на гнафалозид А;
1,03 — поправочный коэффициент на неполное
элюирование гнафалозида А с полиамидного сорбента;
А — оптическая плотность испытуемого раст¬
вора;
А0 — оптическая плотность раствора сравнения;
т0 — масса навески дихромата калия, г;
т — масса навески испытуемого сырья, г.
07/2016: РБ0091
ТЕРМОПСИСА ЛАНЦЕТНОГО ТРАВА
ТЬегторз'кНз 1апсео1а1ае ЬегЬа
1-АЫСЕ1.ЕАР ТНЕРМОР313 НЕРВ*
ОПРЕДЕЛЕНИЕ
Собранная в начале цветения до появления пло¬
дов и высушенная трава многолетнего травянистого
растения ТНегторз1з IапсеоШа Р. Вг.
Содержание: не менее 1,5 % суммы алкалои¬
дов в пересчете на термопсин (С15Н20Ы2О; М.м. 244,3)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Цельные или ча¬
стично измельченные стебли с листьями и цветками.
Стебли простые или ветвистые, бороздчатые, слабоо-
пушенные, длиной до 30 см. Листья очередные, трой¬
чатые на коротких черешках ((4—7) мм), с продолго¬
ватыми или продолговато-ланцетными листочками
длиной от 30 мм до 60 мм, шириной от 5 мм до 12 мм.
Сверху почти голые, снизу покрытые прижатыми во¬
лосками. Прилистники ланцетовидные, почти вдвое
короче дольки листа, опушены прижатыми волосками.
Цветки собраны мутовками в негустую верхушечную
кисть. Чашечка колокольчатая, пятизубчатая с нерав¬
ными по длине зубцами, опушена прижатыми волоска¬
ми. Венчик мотыльковый, длиной (25—28) мм, верхний
лепесток (флаг) с почти округлым отгибом, на верхуш¬
ке с глубоким и узким вырезом; два боковых лепестка
(крылья) лишь немного короче флага; нижние сросши¬
еся лепестки (лодочка) в 1,5—2 раза шире крыльев.
Тычинок 10, все свободные; пестик 1 с длинным стол¬
биком и шелковисто-опушенной завязью. Цвет стеблей
и листьев серовато-зеленый, цветков — желтый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности видны многоугольные клетки
верхнего эпидермиса со слабоизвилистыми стенками,
нижнего — с более извилистыми. Местами, особенно
на верхнем эпидермисе, стенки клеток имеют четко¬
видные утолщения. Устьица овальные, окружены 3—5
околоустьичными клетками (аномоцитный тип) (2.8.3),
погруженные, преобладают на нижней стороне листа.
Волоски многочисленные, двухклеточные и состоят из
короткой базальной клетки и длинной терминальной,
прижатой к поверхности листа. У одних волосков тер¬
минальная клетка длинная, с толстой, снаружи круп¬
нобугристой поверхностью, у других она несколько
короче с тонкой оболочкой и гладкой поверхностью.
Вокруг места прикрепления волоска клетки эпидерми¬
са с почти прямыми стенками, расположены лучисто,
образуя розетку. Если волосок отпал, то в центре ро¬
зетки виден крупный валик. При просветлении листа
раствором хлоралгидрата Р в клетках эпидермиса
видны многочисленные сферокристаллы фенологли-
козида, легко растворимые в растворах гидроксидов
щелочных металлов. В порошке встречаются обрывки
эпидермиса с устьицами, розетками и иногда сферо-
кристаллами, многочисленные волоски, обрывки па¬
ренхимы и сосудов.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: плоды — не более 1 %; побуревшие ча¬
сти травы, корни (в том числе отделенные при анали¬
зе) — не более 4 %. Органические примеси: не более
2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 8,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,00 г измельченного сырья (710) (2.9.12) по¬
мещают в колбу вместимостью 250 мл, прибавляют
100,0 мл хлороформа Р и 5 мл раствора аммиака кон¬
центрированного Р, закрывают пробкой и встряхивают
в течение 2 ч или выдерживают при комнатной темпе¬
ратуре в течение 15 ч и встряхивают в течение 30 мин.
Хлороформное извлечение фильтруют через ватный
Тимьяна трава
1319
тампон. 50,0 мл фильтрата выпаривают досуха. К по¬
лученному остатку прибавляют 2,0 мл 0,1 М раствора
натрия гидроксида и растирают стеклянной палочкой
до полного исчезновения комочков, затем прибавляют
8.0 мл воды Р и перемешивают в течение (2—3) мин.
К полученной смеси прибавляют 10,0 мл 0,1 М раст¬
вора кислоты хлористоводородной, осторожно пере¬
мешивают, выдерживают в течение (8—10) мин, встря¬
хивают в течение (8—10) мин и фильтруют через трой¬
ной бумажный складчатый фильтр диаметром 7 см. К
10.0 мл фильтрата прибавляют 10 мл воды Р, 2—3 кап¬
ли раствора метилового красного Р и оттитровывают
избыток кислоты 0,1 М раствором натрия гидроксида
до появления желтого окрашивания.
Параллельно проводят контрольный опыт, ис¬
пользуя раствор, состоящий из 1 мл 0,1 М раствора
натрия гидроксида, 4,0 мл воды Р, 5,0 мл 0,2 М раст¬
вора кислоты хлористоводородной и 2—3 капель
раствора метилового красного Р.
1 мл 0,1 М раствора кислоты хлористоводо¬
родной соответствует 24,4 мг суммы алкалоидов в
пересчете на термопсин.
07/2016:0865
@> ТИМЬЯНА ТРАВА
ТНут/ ЬегЬа
ТНУМЕ
ОПРЕДЕЛЕНИЕ
Собранная во время цветения, высушенная и
обмолоченная трава ТМутиз уи1дапз 1_. или ТМутиз
гуд'/з 1_. или смесь обоих видов.
Содержание:
- эфирное масло: не менее 12 мл/кг (в пересчете
на безводное сырье);
- суммарное содержание тимола и карвакрола
(оба С10Н14О; М.м. 150,2): не менее 40 % в эфирном
масле.
ОПИСАНИЕ
Запах сильный, ароматный, подобен тимолу.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Листья ТОутиз
VиIда^^з обычно длиной от4ммдо12мми шириной до
3 мм; сидячие или с очень коротким черешком. Листо¬
вая пластинка жесткая, цельная, ланцетная до оваль¬
ной, с отчетливо завернутыми вниз краями, опушены
с обеих сторон от серого до зеленовато-серого цвета;
главная жилка вдавлена с верхней стороны и силь¬
но выступает с нижней стороны. Чашечка трубчатая,
зеленая, часто с фиолетовыми точками; на конце на¬
ходятся две губы, верхняя из которых изогнута назад
и оканчивается тремя долями, нижняя — длиннее и
имеет два опушенных зубца. После цветения чашечка
закрывается своеобразной короной из длинных, жест¬
ких волосков. Венчик в два раза длиннее чашечки,
обычно коричневатый после сушки, слегка двугубый.
Листья ТЬутиз гуд/з обычно длиной от 1,7 мм до
6,5 мм и шириной от 0,4 мм до 1,2 мм; листовая пла¬
стинка от игольчатой до линейно-ланцетной формы с
отчетливо завернутыми вниз краями. Обе поверхно¬
сти листовой пластинки от зеленого до зеленовато-се¬
рого цвета; главная жилка иногда фиолетовая; по кра¬
ям, особенно у основания, с длинными белыми воло¬
сками. Высушенные цветки такие же, как у Т. VиIдапз.
В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет порошка обоих ви¬
дов серовато-зеленый или зеленовато-коричневый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки (рисунок
0865.-1 и рисунок 0865.-2): фрагменты внешнего эпи¬
дермиса венчика при просматривании с поверхности
[А, С, Р], состоящего из клеток с волнистыми и слег¬
ка утолщенными [Рс] или неутолщенными [Ас] стен¬
ками, многочисленными простыми многоклеточными
покровными волосками, зачастую с одной спавшейся
клеткой [Аа], железистыми волосками с одноклеточной
головкой и одноклеточной [Са, РЬ] или многоклеточной
[АЬ] ножкой, устьицами диацитного типа (2.8.3) [Ра] и
железистыми волосками, состоящими из 12 клеток [□];
изодиаметрические со слегка утолщенными стенками
клетки эпидермиса основания венчика [С]; относи¬
тельно редкие сферические и гладкие зерна пыльцы
диаметром около 35 мкм [В] с 6 щелевидными заро¬
дышевыми порами; порошок Т. гуд/з также содержит
многочисленные толстые пучки волокон из главных жи¬
лок и из фрагментов стеблей; эпидермис листьев при
просматривании с поверхности [О, К] имеет клетки с
антиклинальными извилистыми и четковидными стен¬
ками [6а, Ка] и устьица диацитного типа (2.8.3) [6Ь];
многочисленные железистые волоски, состоящие из 12
выделительных клеток, кутикула над которыми обычно
приподнята секретом с образованием сферического
или яйцевидного пузыревидного покрытия [КЬ]; желе¬
зистые волоски с одноклеточной ножкой и шаровидной
или яйцевидной головкой [Кс]; у обоих видов эпидер¬
мис верхней стороны листа несет покровные волоски в
форме заостренных зубов с бородавчатыми стенками
[Ос] и обычно сопровождается расположенной ниже
Рисунок 0865.-1. Микроскопические признаки
травы тимьяна
1320
Гэ сударственная фармакопея Республики Беларусь
палисадной паренхимой [<3с1, Кс!]; эпидермис нижней
стороны листа при просматривании в поперечном раз¬
резе [Н, Ц имеет покровные волоски различных типов:
одноклеточные прямые или слегка искривленные [На,
1а]; двухклеточные или трехклеточные сочлененные
и чаще всего коленчато изогнутые [НЬ, и] (Т. VиIдапз)^,
двухклеточные или трехклеточные более или менее
прямые [14] или очень крупные многоклеточные [М] в
основании листовой пластинки (Т. гуд1з)\ фрагменты
чашечки, покрытые многочисленными простыми воло¬
сками, состоящими из 5—6 клеток и со слабоисчерчен-
ной кутикулой при просматривании с поверхности [Е].
Рисунок 0865.-2. Микроскопические признаки
травы тимьяна
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (355) (2.9.12) прибавляют 5 мл метанола Р, об¬
рабатывают ультразвуком в течение 10 мин и центри¬
фугируют или фильтруют; используют фильтрат или
надосадочную жидкость.
Раствор сравнения. 1 мг рутина Р и 1 мг кисло¬
ты розмариновой Р растворяют в 5 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р254 Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р254 Р ((2—10) мкм)]
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — этилацетат Р( 1:1:15, об/об/об).
Наносимый объем пробы: по 20 мкл [или по
5 мкл] в виде полос длиной 20 мм [или 8 мм].
Фронт подвижной фазы, не менее 15 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку нагревают при темпера¬
туре 100 °С в течение 3 мин, горячую пластинку обра¬
батывают раствором 5 г/л дифенилборной кислоты
аминоэтилового эфира Р в этилацетате Р, затем
раствором 50 г/л макрогола 400 Р в метиленхлори-
де Р\ просматривают в ультрафиолетовом свете при
длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. Кроме того, на хроматограмме ис¬
пытуемого раствора могут обнаруживаться и другие
тусклые флуоресцирующие зоны.
Верх хроматографической пластинки
Розмариновая кислота:
флуоресцирующая зона
голубого цвета
Две флуоресцирующие
зоны красного цвета
Флуоресцирующая
зона голубого цвета
(розмариновая кислота)
Одна или две
флуоресцирующие зоны
голубого цвета
Рутин: флуоресцирующая
зона оранжево-желтого
цвета
Две флуоресцирующие
зоны желтого или
оранжевого цвета
Может обнаруживаться
флуоресцирующая зона
зеленого цвета
Раствор сравнения
Испытуемый раствор
О. Просматривают хроматограммы, полученные
в разделе «Количественное определение. Определе¬
ние содержания тимола и карвакрола».
Результаты: на хроматограмме испытуемого
раствора характерные пики по времени удерживания
соответствуют характерным пикам на хроматограмме
раствора сравнения (а).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли толщиной более 1 мм и длиной
более 15 мм — не более 10%. Другие примеси: не
более 2 %.
ТНутиз зегрШит 1_. Фальсификация Т. зегрШит 1_.
выявляется путем обнаружения листьев с длинными во¬
лосками в основании и слабоопушенных других частей.
Вода (2.2.13). Не более 100 мл/кг. Определение
проводят из 20,0 г измельченного сырья (355) (2.9.12).
Общая зола (2.4.16). Не более 15,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 30,0 г ис¬
пытуемого сырья помещают в колбу вместимостью
1000 мл, прибавляют 400 мл воды Р в качестве жид¬
кости для перегонки. Перегоняют без использования
ксилола Р со скоростью (2—3) мл/мин в течение 2 ч.
Определение содержания тимола и карва¬
крола. Газовая хроматография (2.2.28): определение
проводят методом внутренней нормализации.
Испытуемый раствор. Полученное при количе¬
ственном определении эфирное масло фильтруют че¬
рез бумажный фильтр с небольшим количеством без¬
водного сульфата натрия Р и доводят гексаном Р до
объема 5,0 мл, ополаскивая прибор для определения
эфирного масла и фильтр с сульфатом натрия этим
же растворителем. Далее объем фильтрованного
раствора, соответствующий 100 мкл эфирного масла,
доводят гептаном Р до объема 5,0 мл.
Тмина плоды
1321
Раствор сравнения (а). 0,20 г тимола Р и 50 мг
карвакрола Р растворяют в гексане Р и доводят до
объема 5,0 мл этим же растворителем.
Раствор сравнения (Ь). 10 мкл карвакрола Р дово¬
дят гептаном Р до объема 10,0 мл. 100 мкл получен¬
ного раствора доводят гептаном Р до объема 10,0 мл.
Условия хроматографирования:
-колонка кварцевая капиллярная длиной
(30—60) м и диаметром 0,25 мм, покрытая слоем ма¬
хрогола 20 000 Р (толщина слоя 0,25 мкм);
- газ-носитель: азот для хроматографии Р или
гелий для хроматографии Р\
- скорость газа-носителя: (1—2) мл/мин;
- деление потока: 1:100;
- температура:
Время (мин)
Температура (°С)
Колонка
0—45
40 — 220
Блок ввода проб
190
Детектор
210
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 0,2 мкл.
Порядок элюирования: порядок, как указано в
составе раствора сравнения (а); отмечают времена
удерживания компонентов раствора сравнения (а).
Пригодность хроматографической системы:
раствор сравнения (а):
- разрешение: не менее 1,5 между пиками тимо¬
ла и карвакрола.
По временам удерживания компонентов раст¬
вора сравнения (а) определяют состав испытуемого
раствора.
Рассчитывают процентное содержание тимола и
карвакрола. Не учитывают пик растворителя и пики с
площадью менее площади основного пика на хрома¬
тограмме раствора сравнения (Ь) (0,05 %).
07/2016:1080
ТМИНА ПЛОДЫ
Сам Iгис1из
САКАШАУ РКШТ
ОПРЕДЕЛЕНИЕ
Зрелые и высушенные плоды двухлетнего травя¬
нистого растения Сагит сам 1_.
Содержание: не менее 20 мл/кг эфирного масла
(в пересчете на безводное сырье).
ОПИСАНИЕ (СВОЙСТВА)
Запах напоминает запах карвона.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Плод — вис¬
лоплодник почти цилиндрической формы длиной
обычно (3—6,5) мм и шириной (1—1,5) мм. Мерикар-
пии, обычно отдельные, серовато-коричневого или
коричневого цвета, гладкие, в основном серповидной
формы, с заостренными обоими концами. Каждый ме-
рикарпий имеет по 5 выступающих узких ребрышек.
Поперечный разрез при просматривании имеет почти
правильную пятиугольную форму, с использованием
увеличительного стекла видны четыре эфиромаслич¬
ных канальца на выпуклой стороне и два на плоской.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет желтовато-корич¬
невый. Просматривают под микроскопом с исполь¬
зованием раствора хлоралгидрата Р. В порошке
обнаруживаются следующие диагностические при¬
знаки: фрагменты, состоящие из желтовато-корич¬
невых или коричневых тонкостенных многоугольных
выделительных клеток, часто сопровождаемых слоем
тонкостенных поперечно вытянутых клеток шириной
(8—12) мкм; фрагменты эпикарпа с толстостенны¬
ми клетками и редкими устьицами аномоцитного типа
(2.8.3); многочисленные фрагменты эндосперма, со¬
держащего алейроновые зерна, капельки жирного мас¬
ла и микрокристаллы оксалаты кальция, собранные в
розетки; спиральные сосуды вместе со склеренхимны-
ми волокнами; редкие пучки волокон из плодоноса; мо¬
гут обнаруживаться группы склереид мезокарпа прямо¬
угольной или почти прямоугольной формы со средне
утолщенными и ямчатыми стенками.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (710) (2.9.12) прибавляют 5,0 мл этилацета-
та Р, встряхивают в течение (2—3) мин и фильтруют
через бумажный фильтр с 2 г натрия сульфата без¬
водного Р.
Раствор сравнения. 2 мкл карвона Р и 5 мкл
оливкового масла Р растворяют в 1,0 мл этилаце-
тэта Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ЛЯ Р254 Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: 10 мкл раствора срав¬
нения и 20 мкл испытуемого раствора в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограммах испытуемого
раствора и раствора сравнения в центральной части
обнаруживаются зоны поглощения (карвон).
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р и нагревают при темпе¬
ратуре от 100 °С до 105 °С в течение (2—4) мин. Про¬
сматривают при дневном свете.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Слабая зона фиолетового
цвета (терпеновые
углеводороды)
Триглицериды оливкового
масла: зона фиолетового
цвета
Карвон: интенсивная зона
оранжево-коричневого
цвета
Зона фиолетового цвета
(триглицериды)
Интенсивная зона
оранжево-коричневого
цвета (карвон)
Несколько слабых зон
в основном фиолетово¬
сероватого и коричневатого
цвета
Раствор сравнения
Испытуемый раствор
1322
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: поврежденные, недоразвитые плоды
тмина и другие части растения — не более 2 %. Орга¬
нические примеси: не более 2 %. Минеральные при¬
меси: не более 0,5 %.
®Вода (2.2.13). Не более 100 мл/кг. Опреде¬
ление проводят из 10,0 г измельченного сырья (710)
(2.9.12).
Общая зола (2.4.16). Не более 8,0 %.
Зола, нерастворимая в кислоте хлористово¬
дородной (2.8.1). Не более 1,5 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 10,0 г измель¬
ченного непосредственно перед проведением испы¬
тания сырья (710) (2.9.12) помещают в колбу вмести¬
мостью 500 мл, прибавляют 200 мл воды Р в качестве
жидкости для перегонки. В градуированную трубку
помещают 0,50 мл ксилола Р. Перегонку проводят со
скоростью (2—3) мл/мин в течение 90 мин.
07/2016:1054
ТОЛОКНЯНКИ листья
Шае иг811ЬНит
ВЕАКВЕККУ1-ЕАР
ОПРЕДЕЛЕНИЕ
Собранные весной до и в начале цветения или
осенью с начала созревания плодов до появления
снежного покрова, высушенные листья вечнозеленого
кустарничка Аг&08(арЬу1о8 иVа-и^8^ (1_.) Зргепд.
Содержание: арбутина (С12Н1607; М.м. 272,3)
(в пересчете на сухое сырье):
- не менее 6,0 %, определенное методом титро¬
вания (#2.2.90);
- или не менее 7,0 %, определенное методом
жидкостной хроматографии (2.2.29).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Листья с верхней
стороны блестящие, темно-зеленые, с нижней сто¬
роны светлее, матовые, длиной от 7 мм до 30 мм и
шириной от 5 мм до 12 мм. Листовая пластинка об¬
ратнояйцевидной или удлиненно-овальной формы, с
гладким, слегка завернутым к нижней стороне краем,
сужающаяся в основании к короткому черешку, на
верхушке закругленная, иногда с небольшой выемкой.
Листовая пластинка толстая, кожистая. Жилкование
перистое, с хорошо заметным с обеих сторон листо¬
вой пластинки сетчатым рисунком. Листья с верхней
стороны с ясно заметными вдавленными жилками,
шероховатые на ощупь. Только молодые листья име¬
ют опушенные края. Зрелые листья голые.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.12). Цвет зеленый,
зеленовато-серый или желтовато-зеленый. Просма¬
тривают под микроскопом с использованием раст¬
вора хлоралгидрата Р. В порошке обнаруживают¬
ся следующие диагностические признаки (рисунок
1054.-1): фрагменты верхнего эпидермиса при про¬
сматривании с поверхности [А] с толстостенными
и нерегулярноямчатыми многоугольными клетками
[Аа], обычно вместе с палисадной паренхимой [АЬ];
фрагменты верхнего эпидермиса на поперечном
срезе [О] с прямостенными клетками [6а], покры¬
тыми толстой гладкой кутикулой [6Ь], сопровожда¬
емыми палисадной паренхимой [6с], состоящей из
трех или четырех слоев клеток неравной длины,
некоторые из которых содержат многочисленные
призмы оксалата кальция [66]; фрагменты нижне¬
го эпидермиса при просматривании с поверхности
[В, Е] с устьицами аномоцитного типа [Ва] (2.8.3),
окруженными 5—11 клетками, с местами прикре¬
пления оснований волосков [Еа], с губчатой парен¬
химой [ЕЬ]; группы лигнифицированных волокон
перицикпа [0]\ фрагменты сосудистой системы [Р],
состоящей из пористых сосудов [Ра] и волокон [РЬ],
сопровождаемых рядами клеток, содержащих при¬
змы оксалата кальция [Рс]; в паренхимных клетках
обнаруживаются капельки масла; иногда встреча¬
ются фрагменты конических одноклеточных покров¬
ных волосков [С].
Рисунок 1054.-1. Микроскопические признаки
листьев толокнянки
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного
сырья (355) (2.9.12) прибавляют 5 мл смеси из рав¬
ных объемов метанола Р и воды Р и нагревают с
обратным холодильником в течение 10 мин. Горячее
извлечение фильтруют. Фильтр промывают смесью из
равных объемов метанола Р и воды Р и доводят до
объема 5 мл этим же растворителем.
Раствор сравнения. 50 мг арбутина Р и 25 мг
галловой кислоты Р растворяют в метаноле Р и до¬
водят до объема 20,0 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: кислота муравьиная безво¬
дная Р— вода Р— этилацетат Р (6:6:88, об/об/об).
Наносимый объем пробы: по Юмкл [или по
2 мкл] в виде полос длиной 15 мм [или 8 мм].
Толокнянки листья
1323
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: при температуре от 105 °С до
110 °С до исчезновения запаха растворителей.
Проявление: пластинку обрабатывают раствором
10 г/л дихлорхинонхлоримида Р в метаноле Я Затем
обрабатывают раствором 20 г/л натрия карбоната
безводного Я. Просматривают при дневном свете.
Результат: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны: две
или три зоны синего цвета и несколько зон коричнево¬
го или коричневато-серого цвета.
Верх хроматографической пластинки
Галловая кислота: зона
коричневатого цвета
Зона коричневатого цвета
Коричневая зона
Арбутин: зона синего цвета
Интенсивная зона синего
цвета (арбутин)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (веточки, пло¬
ды) — не более 4 %; побуревшие и потемневшие с
обеих сторон листья — не более 3 %. Органические
примеси: не более 0,5 %. Минеральные примеси: не
более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 4,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания арбутина мето¬
дом титрования. 0,500 г измельченного сырья (710)
(2.9.12) помещают в колбу вместимостью 100 мл, при¬
бавляют 50 мл воды Я и нагревают с обратным холо¬
дильником, поддерживая слабое кипение, в течение
30 мин. Горячее извлечение фильтруют в мерную
колбу вместимостью 100 мл через бумажный фильтр,
избегая попадания частиц сырья на фильтр. В колбу
с сырьем повторно прибавляют 25 мл воды Я и ки¬
пятят в течение 20 мин. Горячее извлечение вместе
с сырьем переносят на тот же фильтр и остаток на
фильтре дважды промывают горячей водой Я порция¬
ми по 10 мл. К фильтрату прибавляют 3 мл раствора
свинца (II) ацетата основного Я, перемешивают, ох¬
лаждают и доводят водой Я до объем 100,0 мл. Колбу
помещают в водяную баню и выдерживают до полной
коагуляции осадка. Горячую жидкость полностью от¬
фильтровывают в колбу через бумажный фильтр,
прикрывая воронку часовым стеклом. Охлаждают,
к фильтрату прибавляют 1 мл кислоты серной Я,
колбу взвешивают с точностью до 0,01 г и кипятят с
обратным холодильником в течение 1,5 ч, поддер¬
живая равномерное и слабое кипение. Охлаждают
до комнатной температуры, взвешивают и доводят
массу колбы до первоначальной водой Я и полностью
отфильтровывают через бумажный фильтр в колбу
вместимостью 250 мл. К фильтрату прибавляют 0,1 г
порошка цинка Я и встряхивают в течение 5 мин.
Жидкость нейтрализуют натрия гидрокарбонатом Я
(около (1—2) г) по красной лакмусовой бумаге Я, при¬
бавляют еще 2 г натрия гидрокарбоната Р и после
его растворения фильтруют через бумажный фильтр.
50,0 мл фильтрата переносят в плоскодонную
колбу вместимостью 500 мл, прибавляют 200 мл
воды Я и немедленно титруют из микро- или полуми-
кробюретки 0,05 М раствором йода до появления си¬
него окрашивания, не исчезающего в течение 1 мин,
используя в качестве индикатора раствор крахмала,
свободный от йодидов, Я.
1 мл 0,05 М раствора йода соответствует
13,61 мг арбутина (С12Н1б07).
® Определение содержания арбутина мето¬
дом жидкостной хроматографии. Жидкостная хро¬
матография (2.2.29).
Испытуемый раствор. 0,800 г измельченного
сырья (250) (2.9.12) помещают в колбу со шлифом
вместимостью 100 мл, прибавляют 20 мл воды Я и на¬
гревают с обратным холодильником на водяной бане
в течение 30 мин. Охлаждают, фильтруют через хлоп¬
ковый ватный тампон. Тампон помещают к остатку сы¬
рья в колбе, прибавляют 20 мл воды Я и нагревают с
обратным холодильником на водяной бане в течение
30 мин. Охлаждают и фильтруют через фильтроваль¬
ную бумагу, отбрасывая первые 10 мл фильтрата.
Раствор сравнения (а). 50,0 мг ФСО арбутина
растворяют в подвижной фазе и доводят до объема
50.0 мл этим же растворителем.
Раствор сравнения (Ь). 2,5 мг гидрохинона Я
растворяют в подвижной фазе и доводят до объема
10.0 мл этим же растворителем. К 5,0 мл полученного
раствора прибавляют 2,5 мл раствора сравнения (а)
и доводят до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаметром
4 мм, заполненная силикагелем октадецилсилиль-
ным деактивированным по отношению к основаниям
для хроматографии Я с размером частиц 5 мкм;
- подвижная фаза: метанол Я — вода Я (10:90,
об/об);
- скорость подвижной фазы: 1,2 мл/мин;
- спектрофотометрический детектор, длина
волны 280 нм;
- объем вводимой пробы: 20 мкл.
Пригодность хроматографической системы:
раствор сравнения (Ь):
- разрешение: не менее 4,0 между пиком арбути¬
на и гидрохинона.
Содержание арбутина в процентах рассчитыва¬
ют по формуле:
51 • ш2 • Р
52т, 1
где:
5,, — площадь пика арбутина на хроматограмме
испытуемого раствора;
52 — площадь пика арбутина на хроматограмме
раствора сравнения;
т1 — масса навески испытуемого сырья, г;
т2 — масса навески ФСО арбутина, использо¬
ванного для приготовления раствора сравнения (а), г;
Я — содержание арбутина в стандартном образ¬
це, %.
1324
Гэсударственная фармакопея Республики Беларусь
07/2016:РБ0092
ТЫКВЫ СЕМЕНА
СисигЬПае зетеп
Р11МРКШ 8ЕЕО*
ОПРЕДЕЛЕНИЕ
Зрелые, очищенные от остатков мякоти око¬
лоплодника и высушенные семена однолетних
растений СисигЬИа реро 1_., С. шах/ша ОисН. и
С. то$с11а1а (РисИ.) Ро1г. или смеси этих видов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Семена эл-
липтические, плотные, слегка суженные с одной
стороны, окаймленные по краю ободком. Поверх¬
ность семян глянцевая или матовая, гладкая или
слегка шероховатая. Кожура семени состоит из
двух частей: деревянистой, легко отделяемой и
внутренней — пленчатой, плотно прилегающей
к зародышу; иногда деревянистая кожура отсут¬
ствует. Зародыш состоит из двух желтовато-белых
семядолей и небольшого корешка. Длина семени
от 1,5 см до 2,5 см, ширина от 0,8 см до 1,4 см.
Цвет семян белый, белый с желтоватым или се¬
роватым оттенком, реже зеленовато-серый или
желтый.
B. Микроскопия (2.8.23). На поперечном
срезе семени тыквы видны: семенная кожура,
алейроновый слой (недоразвитый эндосперм) и
семядоли зародыша. В семенной кожуре эпидер¬
мис представлен крупными палисадными клетка¬
ми с утолщенными и, как правило, волнистыми
боковыми стенками и почти всегда разрушенной
наружной стенкой. Под эпидермисом расположе¬
на мощная склеренхима, в которой различаются
три слоя. Наружная часть склеренхимы состоит
из 5—7 рядов плотно сомкнутых клеток с много¬
численными порами. Срединная часть склерен¬
химы представлена одним слоем очень крупных
округлочетырехугольных клеток с толстой сло¬
истой оболочкой и узкими порами. Внутренняя
часть склеренхимы в зависимости от вида тыквы
содержит от двух до шести рядов клеток звездча¬
той формы, которые образуют крупные межклет¬
ники. К внутренней части склеренхимы примы¬
кает несколько слоев тонкостенных сдавленных
клеток. Алейроновый слой представлен одним
рядом небольших изодиаметрических клеток, гу¬
сто заполненных алейроновыми зернами. В клет¬
ках семядолей хорошо различим эпидермальный
слой из мелких, овальных клеток; далее следуют
клетки палисадного слоя. Все они густо запол¬
нены алейроновыми зернами и каплями жирного
масла.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: фрагменты околоплодника в виде
отделившихся пленок и остатков сухой мякоти — не
более 0,2 %; пустые и поврежденные семена — не
более 2 %. Органические примеси: не более 0,5 %.
Минеральные примеси: не более 0,1 %.
Потеря в массе при высушивании (2.2.32).
Не более 13,0%. 2,000 г измельченного сырья
(2000) (2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
07/2016:1382
ТЫСЯЧЕЛИСТНИКА ТРАВА
МШеТоШ МегЬа
УАККОШ
ОПРЕДЕЛЕНИЕ
Трава тысячелистника состоит из цельных или
измельченных высушенных цветущих верхних частей
АсЫНеа тШеМшт 1_.
Содержание: не менее 1 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные или
частично измельченные цветоносные побеги. Стеб¬
ли округлые, опушенные, с очередными листьями,
длиной до 15 см. Листья длиной до 10 см, шириной
до 3 см, продолговатые, дваждыперисторассечен-
ные на ланцетные или линейные доли. Корзинки
продолговато-яйцевидные, длиной (3—4) мм, ши¬
риной (1,5—3) мм, в щитковидных соцветиях или
одиночные. Обвертки корзинок из черепитчатых
продолговато-яйцевидных листочков с перепон¬
чатыми буроватыми краями. Цветоложе корзинок
с пленчатыми прицветниками. Краевые цветки пе¬
стичные. Средние цветки трубчатые обоеполые.
Цвет стеблей и листьев серовато-зеленый, крае¬
вых цветков — белый, реже розовый, срединных —
желтоватый.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.12). Цвет зеленый
или серовато-зеленый. Просматривают под микро¬
скопом с использованием раствора хлоралги¬
драта Р. В порошке обнаруживаются следующие
диагностические признаки (рисунок 1382.-1): фраг¬
менты эпидермиса стебля при просматривании с
поверхности [К] с клетками с гладкой кутикулой и
устьицами аномоцитного типа (2.8.3); фрагменты
эпидермиса листьев и прицветников при просма¬
тривании с поверхности [В] с извилистыми клетка¬
ми и нерегулярно утолщенными стенками, мелко
исчерченной кутикулой и устьицами аномоцитного
типа (2.8.3); очень редкие железистые волоски с ко¬
роткой ножкой и головкой, образованной двумя ря¬
дами по 3—5 клеток, заключенных в пузыреподоб¬
ную мембрану [Н]; простые цельные или фрагмен¬
тированные покровные волоски [А], состоящие из
4—6 мелких более или менее изодиаметрических
находящихся в основании клеток и толстостенной,
часто слегка извилистой терминальной клетки,
длиной от 400 мкм до свыше 1000 мкм; фрагменты
язычкового венчика с сосочковыми эпидермаль¬
ными клетками [О]; фрагменты трубчатого венчика
с извилистыми клетками эпидермиса, покрытыми
тонкой исчерченной кутикулой при просматрива¬
нии с поверхности [Р]; мелкоклеточная паренхима
венчика трубчатых цветков, содержащая друзы
оксалата кальция [Е]; группы лигнифицированных
и ямчатых клеток прицветников [С]; сферические
зерна пыльцы диаметром около 30 мкм с тремя за¬
родышевыми порами и экзиной, покрытой шипами
[С]; группы склеренхимных волокон и мелких сосу¬
дов стебля со спиральным или кольцевым утолще¬
нием [<}].
Укропа пахучего плоды
1325
Рисунок 1382.-1. Микроскопические признаки
травы тысячелистника
® С. К 2,0 г измельченного сырья (710) (2.9,12)
прибавляют 25 мл этилацетата Р, встряхивают в те¬
чение 5 мин и фильтруют. Выпаривают досуха на во¬
дяной бане и остаток растворяют в 0,5 мл толуола Р
(раствор А). К 0,1 мл полученного раствора прибавля¬
ют 2,5 мл раствора диметиламинобензальдегида Р8
и нагревают на водяной бане в течение 2 мин. Охлаж¬
дают, прибавляют 5 мл петролейного эфира Р и ин¬
тенсивно встряхивают. Водный раствор приобретает
голубое или зеленовато-голубое окрашивание.
® О. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. Используют раствор А,
приготовленный как указано в подлинности С.
Раствор сравнения. 10 мг цинеола Р и 10 мг
гвайазулена Р растворяют в 20 мл толуола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раство¬
ром анисового альдегида Р, выдерживают при тем¬
пературе от 100 °С до 105 °С в течение (5—10) мин.
Просматривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения в верхней части обнаруживается красная зона
(гвайазулен), а в центральной части синяя или серо¬
вато-синяя зона (цинеол). На хроматограмме испы¬
туемого раствора обнаруживаются: фиолетовая зона
немного выше зоны гвайазулена на хроматограмме
раствора сравнения; ниже этой зоны красновато-фи¬
олетовая зона; ниже которой расположены одна или
две не полностью разделенные серовато-фиолетовые
или сероватые зоны (которые изменяют цвет на зеле¬
новато-серый в течение нескольких часов) и красно¬
вато-фиолетовая зона немного выше зоны цинеола
на хроматограмме раствора сравнения. На хромато¬
грамме испытуемого раствора могут обнаруживаться
и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: пожелтевшие, побуревшие и почер¬
невшие части травы — не более 1 %, стебли толще
3 мм — не более 3 %. Органические примеси: не бо¬
лее 0,5 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 15,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 20,0 г из¬
мельченного сырья помещают в круглодонную колбу
вместимостью 1000 мл, прибавляют 500 мл смеси
из воды Р и этиленгликоля Р (1:9, об/об) в качестве
жидкости для перегонки. В градуированную трубку по¬
мещают 0,50 мл 1,2,4-триметилбензола Р и перего¬
няют со скоростью (3—4) мл/мин в течение 4 ч.
За несколько минут до истечения времени пере¬
гонки выключают подачу воды в холодильник, про¬
должая перегонку, пока синие летучие компоненты не
достигнут нижнего конца холодильника. Немедленно
включают подачу воды в холодильник во избежание
нагревания приемника. Через 10 мин останавливают
перегонку.
07/2016:РБ0093
УКРОПА ПАХУЧЕГО ПЛОДЫ
Апе1Ы джео/епНз 1гис1из
йШ РИШТ*
ОПРЕДЕЛЕНИЕ
Зрелые и высушенные плоды однолетнего травя¬
нистого растения АпеОпит джео1епз Ь.
Содержание: не менее 20 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Отдельные по-
луплодики (мерикарпии), реже цельные плоды (вис¬
лоплодники) длиной (3—7) мм, шириной (1,5—4) мм.
Мерикарпий широкоэллиптический, слабовыпуклый
на спинной стороне и плоский — на внутренней. Каж¬
дый мерикарпий с тремя нитевидными спинными ре¬
брами и двумя плоскими крыловидными боковыми.
Цвет плодов зеленовато-коричневый или коричневый,
ребер — желто-коричневый. Запах характерный.
B. Микроскопия (2.8.23). На поперечном срезе
мерикарпия видны тангентально вытянутые клетки
эпидермиса с толстыми стенками. Мезокарпий со¬
стоит из паренхимных клеток с тонкими или слегка
утолщенными стенками, особенно в разросшихся бо¬
ковых ребрышках. В ребрышках расположены про¬
водящие пучки с группами механических волокон. В
ложбинках находятся эфиромасличные канальцы:
четыре на выпуклой стороне, два — на плоской.
1326
Гэсударственная фармакопея Республики Беларусь
Канальцы различных размеров, септированные (с
поперечными перегородками), с коричневыми вы¬
делительными клетками. Эндокарпий плотно срос¬
шийся с семенной кожурой. Эндосперм состоит из
многоугольных клеток, заполненных алейроновыми
зернами, каплями жирного масла, мелкими друзами
оксалата кальция.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (стебли и цвет¬
ки) — не более 1 %. Органические примеси: не более
2 %. Минеральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 12,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод В). 10,00 г испы¬
туемого сырья измельчают в ступке в течение 2 мин с
прибавлением 3 г битого стекла или фарфора, пред¬
варительно отсеянного от пыли сквозь сито (250)
(2.9.12) . Измельченную массу количественно пере¬
носят в колбу для перегонки. Перегонку проводят в
течение 2,5 ч.
07/2016: РБ0094
ФАСОЛИ СТВОРКИ
РЪазеоН \/и!дапз уэ1уэ
НАШСОТРООЗ*
ОПРЕДЕЛЕНИЕ
Высушенные створки зрелых плодов РНазео/из
VиIда^^з 1_.
Содержание: не менее 0,01 % фенолкарбоно-
вых кислот в пересчете на кофейную кислоту (С9Н804;
М.м. 180,2) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Удлиненные,
часто спиралевидно скрученные створки плодов, ча¬
стично изломанные, желобчатые или прямые. Сна¬
ружи поверхность створок гладкая, иногда слегка
морщинистая, матовая, от светло-желтого до желтого
цвета, изредка видны полоски или пятна коричневого
или фиолетового цвета. Внутренняя поверхность бле¬
стящая, белая или желтовато-белая.
B. Микроскопия (2.8.23). Видны: экзокарпий,
который снаружи состоит из прямостенных клеток
эпидермиса со складчатостью кутикулы и с многочис¬
ленными местами прикрепления оснований опавших
волосков в виде радиальных розеток; под экзокарпи-
ем — 2—3 слоя веретеновидно вытянутых склерен-
химных волокон с сильно утолщенными стенками;
внутренняя поверхность створок плодов — эндокар¬
пий — состоит из 2—6 рядов мелких одревесневших
склеренхимных волокон.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 5,0 г измельченного сы¬
рья (355) (2.9.12) прибавляют 10 мл смеси из метано¬
ла Р и воды Р (50:50, об/об), выдерживают в течении
30 мин при перемешивании и фильтруют.
Раствор сравнения. 3 мг аргинина Р растворяют
в 10 мл смеси из метанола Р и воды Р (50:50, об/об).
Пластинка: ТСХ пластинка со слоем силикагеля
С Р.
Подвижная фаза: бутанол Р — кислота уксус¬
ная ледяная Р — вода Р (40:10:10, об/об/об).
Наносимый объем пробы: 4 мкл раствора срав¬
нения и 15 мкл испытуемого раствора в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
нингидринаР1 и нагревают при температуре 105 °С
в течение 5 мин. Просматривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения в нижней трети обнаруживается основная зона
(аргинин). На хроматограмме испытуемого раствора
обнаруживается зона, соответствующая по распо¬
ложению и цвету зоне аргинина на хроматограмме
раствора сравнения. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: почерневшие с обеих сторон створки
плодов — не более 8 %; другие части растения — не
более 3 %. Органические примеси: не более 2 %. Ми¬
неральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32).
Не более 15,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 10,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 0,8 %.
Вещества, извлекаемые водой. Не менее 15 %.
1,000 г измельченного сырья (1400) (2.9.12) помещают
в коническую колбу вместимостью 250 мл, прибавля¬
ют 50,0 мл воды Р, закрывают колбу пробкой, взвеши¬
вают с точностью 0,01 г и выдерживают в течение 1 ч.
Затем колбу присоединяют к обратному холодильнику
и нагревают, поддерживая слабое кипение, в течение
2 ч. После охлаждения колбу с содержимым вновь
закрывают той же пробкой, взвешивают и потерю в
массе восполняют водой Р. Содержимое колбы тща¬
тельно взбалтывают и фильтруют через сухой бумаж¬
ный фильтр. 25,0 мл полученного фильтрата перено¬
сят в предварительно высушенную при температуре
от 100 °С до 105 °С и взвешенную выпарительную
чашку, выпаривают на водяной бане досуха и сушат
при температуре от 100 °С до 105 °С до постоянной
массы.
Проводят два параллельных испытания. За окон¬
чательный результат принимают среднее арифмети¬
ческое двух параллельных испытаний.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,00 г измельченного сырья (710) (2.9.12) по¬
мещают в круглодонную колбу вместимостью 250 мл,
прибавляют 50 мл метанола Р, нагревают с обрат¬
ным холодильником на водяной бане в течение 30 мин
и фильтруют. Дважды повторяют экстракцию метано¬
лом Р порциями по 50 мл и фильтруют. Извлечения
объединяют и выпаривают досуха при пониженном
давлении. Остаток растворяют в 20 мл горячей воды Р
и выдерживают в холодильнике в течение 12 ч. Полу¬
ченный раствор фильтруют через бумажный фильтр в
мерную колбу вместимостью 25 мл и доводят водой Р
до объема 25,0 мл (раствор А).
Фенхеля горького плоды
1327
Испытуемый раствор. К 1,0 мл раствора А при¬
бавляют 5 мл воды Р, 1 мл 0,5 М раствора хлористо¬
водородной кислоты, 1 мл раствора, полученного при
растворении 10 г натрия молибдата Р и 10 г натрия
нитрита Р в 100 мл воды Р, 1 мл раствора 40 г/л
натрия гидроксида Р и доводят водой Р до объема
10,0 мл.
Компенсационный раствор. К 5 мл воды Р при¬
бавляют 1 мл 0,5 М раствора хлористоводородной
кислоты, 1 мл раствора, полученного при растворе¬
нии 10 г натрия молибдата Р и 10 г натрия нитри¬
та Р в 100 мл воды Р, 1 мл раствора 40 г/л натрия
гидроксида Р и доводят водой Р до объема 10,0 мл.
Измеряют оптическую плотность (2.2.25) испыту¬
емого раствора при 490 нм.
Содержание фенолкарбоновых кислот в пере¬
счете на кофейную кислоту в процентах рассчитыва¬
ют по формуле:
А 500
т-285 ’
где:
285 — удельный показатель поглощения кофей¬
ной кислоты;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:0824
(§) ФЕНХЕЛЯ ГОРЬКОГО ПЛОДЫ
Роет'сиН этап &ис1из
РЕЫЫЕЦ В1ТТЕК
ОПРЕДЕЛЕНИЕ
Высушенные вислоплодники и мерикарпии
Роепюи1ит уи1даге МШ. подвид уи1даге уаг. уи1даге.
Содержание:
- эфирное масло: не менее 40 мл/кг (в пересчете
на безводное сырье);
- анетол: не менее 60,0 % в эфирном масле;
- фенхон: не менее 15,0 % в эфирном масле.
ОПИСАНИЕ (СВОЙСТВА)
Имеют зеленовато-коричневый, коричневый или
зеленый цвет.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Плод фенхе¬
ля горького — вислоплодник почти цилиндрической
формы с закругленным основанием и сужающейся
верхушкой, заканчивающейся большим стилоподием.
Длина плода обычно составляет от 3 мм до 12 мм,
ширина от 3 мм до 4 мм. Мерикарпии, обычно несрос-
шиеся, имеют гладкую поверхность. Каэедый мерикар-
пий имеет 5 слегка наклоненных в сторону ребер. На
поперечном срезе плода при просматривании под лу¬
пой наблюдается 4 канальца эфирного масла на дор¬
сальной поверхности и 2 — на комиссуральной.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет серовато-коричневый
или серовато-желтый. Просматривают под микроско¬
пом с использованием раствора хлоралгидрата Р. В
порошке обнаруживаются следующие диагностиче¬
ские признаки (рисунок 0824.-1): желтые фрагменты
широких выделительных каналов, часто состоящие
из многоугольных секреторных клеток с желтовато-ко¬
ричневыми стенками [О, Н]; сетчатая паренхима мезо-
карпия [В]; многочисленные пучки волокон [С] ребер
[Оа] плода, часто с прилежащими узкими спиральны¬
ми сосудами [<ЗЬ]; очень многочисленные фрагменты
[Р] эндосперма, содержащие алейроновые зерна [РЬ]
и очень мелкие друзы кристаллов оксалата кальция
[Ра]; некоторое количество пучков волокон цветоло¬
жа [Е]; фрагменты эндокарпия (вид с поверхности
[А, К]), состоящие из тонкостенных поперечно-вытя¬
нутых клеток шириной от 2 мкм до 9 мкм, расположен¬
ных паркетообразно, иногда с прилежащим внутрен¬
ним слоем мезокарпия [Аа]; фрагменты эпикарпия с
устьицами с прилежащими капельками эфирного мас¬
ла [С]; очень многочисленные капельки масла [3].
Рисунок 0824.-1. Микроскопические признаки
плодов фенхеля горького
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,3 г свежеизмельченно-
го сырья (1400) (2.9.12) встряхивают с 5,0 мл мети-
ленхлорида Р в течение 15 мин. Фильтруют и осто¬
рожно выпаривают фильтрат досуха на водяной бане
при температуре 60 °С. Остаток после выпаривания
растворяют в 0,5 мл толуола Р.
Раствор сравнения. 50 мкл анетола Р и 10 мкл
фенхона Р растворяют в 5,0 мл гексана Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля вР254 Р
Подвижная фаза: гексан Р — толуол Р (20:80,
об/об).
Наносимый объем пробы: по 10 мкл в виде полос
длиной 20 мм и шириной 3 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограмме раствора
сравнения и испытуемого раствора в центральной ча¬
сти обнаруживается зона поглощения, соответствую¬
щая анетолу.
1328
Гэсударственная фармакопея Республики Беларусь
Проявление В: пластинку опрыскивают кислотой
серной Р и нагревают при температуре 140 °С в тече¬
ние (5—10) мин до проявления зоны желтого цвета,
соответствующей фенхону, в нижней трети хромато¬
граммы.
Результаты В: на хроматограмме раствора
сравнения и испытуемого раствора в центральной ча¬
сти обнаруживается зона фиолетового цвета, соответ¬
ствующая анетолу. На хроматограмме испытуемого
раствора в верхней трети обнаруживается также зона
красновато-коричневого цвета (терпены).
ИСПЫТАНИЯ
Эстрагол. Газовая хроматография (2.2.28): испы¬
тание проводят методом нормализации.
Испытуемый раствор. Смесь эфирного масла и
ксилола Р, полученную как указано в разделе «Коли¬
чественное определение эфирного масла», доводят
ксилолом Р до объема 5,0 мл, промывая прибор для
определения эфирного масла.
Раствор сравнения. 5 мг эстрагола Р раство¬
ряют в 0,5 мл ксилола Р.
Условия хроматографирования:
- колонка длиной от 30 м до 60 м и внутрен¬
ним диаметром 0,3 мм, покрытая слоем макрагола
20 000 Р;
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 0,40 мл/мин;
- деление потока: 1:200;
- температура:
Время (мин)
Температура (°С)
Колонка
0—4
60
4—26
60170
26-41
170
Блок ввода проб
220
Детектор
270
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Предельное содержание примесей:
- эстрагол: не более 5,0 % в эфирном масле,
полученном как указано в разделе «Количественное
определение».
Допустимые примеси (2.8.2). Другие части рас¬
тения: плодоножки — не более 1,5%. Другие допусти¬
мые примеси 1,5 %.
Вода (2.2.13). Не более 100 мл/кг. Определение
проводят из 20,0 г измельченного сырья (710) (2.9.12).
Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 5,0 г испы¬
туемого сырья, измельченного до грубого порошка
(1400) (2.9.12) непосредственно перед проведением
испытания, помещают в круглодонную колбу вмести¬
мостью 500 мл, прибавляют 200 мл воды Р в качестве
жидкости для перегонки. В градуированную трубку по¬
мещают 0,50 мл ксилола Р и перегоняют со скоростью
(2—3) мл/мин в течение 2 ч.
Анетол и фенхон. Газовая хроматография
(2.2.28), как указано в испытании «Эстрагол», со сле¬
дующими изменениями.
Раствор сравнения. 5 мг фенхона Р и 5 мг ане¬
тола Р растворяют в 0,5 мл ксилола Р.
Порядок элюирования: порядок, как указано в со¬
ставе раствора сравнения; отмечают времена удер¬
живания компонентов раствора сравнения.
ХРАНЕНИЕ
В защищенном от влаги месте.
07/2016:0825
® ФЕНХЕЛЯ СЛАДКОГО ПЛОДЫ
Роеп1сиН с1и1аз IгисШ
ЕЕЫЫЕЦ 5МЕЕТ
ОПРЕДЕЛЕНИЕ
Высушенные вислоплодники и мерикарпии
Роетси1ит VиIда^е МШ. подвид уи1даге уаг. ди1се (МШ.)
ВаИ.&ТгаЬ.
Содержание:
- эфирное масло: не менее 20 мл/кг (в пересчете
на безводное сырье);
- анетол: не менее 80,0 % в эфирном масле.
ОПИСАНИЕ (СВОЙСТВА)
Имеют бледно-зеленый или бледный желтовато-
коричневый цвет.
ПОДЛИННОСТЬ(ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Плод фенхеля
сладкого — вислоплодник почти цилиндрической
формы с закругленным основанием и сужающейся
верхушкой, заканчивающейся большим стилопо-
дием. Длина плода обычно составляет (3—12) мм,
ширина от 3 мм до 4 мм. Мерикарпии, обычно не-
сросшиеся, имеют гладкую поверхность. Каждый
мерикарпий имеет 5 слегка наклоненных в сторону
выступающих ребер. На поперечном срезе плода
при просматривании под лупой наблюдается 4 ка¬
нальца эфирного масла на дорсальной поверхно¬
сти и 2 — на комиссуральной.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет серовато-корич¬
невый или серовато-желтый. Просматривают под
микроскопом с использованием раствора хлорал¬
гидрата Р. В порошке обнаруживаются следую¬
щие диагностические признаки (рисунок 0825.-1):
желтые фрагменты широких выделительных ка¬
налов, часто состоящие из многоугольных секре¬
торных клеток с желтовато-коричневыми стенками
[Э, Н]; сетчатая паренхима мезокарпия [В]; много¬
численные пучки волокон [С] ребер [<3а] плода,
часто с прилежащими узкими спиральными сосуда¬
ми [ОЬ]; очень многочисленные фрагменты [Р] эн¬
досперма, содержащие алейроновые зерна [РЬ] и
очень мелкие друзы кристаллов оксалата кальция
[Ра]; некоторое количество пучков волокон цветоло¬
жа [Е]; фрагменты эндокарпия (вид с поверхности
[К, А]), состоящие из тонкостенных поперечно-вытя¬
нутых клеток шириной (2—9) мкм, расположенных
паркетообразно, иногда с прилежащим внутренним
слоем мезокарпия [Аа]; фрагменты эпикарпия с
устьицами с прилежащими капельками масла [С];
очень многочисленные капельки масла ^].
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,3 г свежеизмельченно-
го сырья (1400) (2.9.12) встряхивают с 5,0 мл мети-
ленхлорида Р в течение 15 мин. Фильтруют и осто¬
рожно выпаривают фильтрат досуха на водяной бане
при температуре 60 °С. Остаток после выпаривания
растворяют в 0,5 мл толуола Р
Фиалки трава
1329
Рисунок 0825.-1. Микроскопические признаки
плодов фенхеля сладкого
Раствор сравнения. 60 мкл анетола Р раство¬
ряют в 5,0 мл гексана Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля вР254 Р.
Подвижная фаза: гексан Р — толуол Р (20:80,
об/об).
Наносимый объем пробы: по 10 мкл в виде полос
длиной 20 мм и шириной 3 мм.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограмме раствора
сравнения и испытуемого раствора в центральной ча¬
сти обнаруживается зона поглощения, соответствую¬
щая анетолу.
Проявление В: пластинку опрыскивают кислотой
серной Р и нагревают при температуре 140 °С в тече¬
ние 5 мин. Просматривают при дневном свете.
Результаты В: на хроматограмме раствора
сравнения и испытуемого раствора в центральной ча¬
сти обнаруживается зона фиолетового цвета, соответ¬
ствующая анетолу. На хроматограмме испытуемого
раствора в верхней трети обнаруживается также зона
красновато-коричневого цвета (терпены).
ИСПЫТАНИЯ
Эстрагол и фенхон. Газовая хроматография
(2.2.28): испытание проводят методом внутренней
нормализации.
Испытуемый раствор. Смесь эфирного масла и
ксилола Р, полученную как указано в разделе «Коли¬
чественное определение эфирного масла», доводят
ксилолом Р до объема 5,0 мл, промывая прибор для
определения эфирного масла.
Раствор сравнения. 5 мг эстрагола Р и 5 мг
фенхона Р растворяют в 0,5 мл ксилола Р.
Условия хроматографирования:
- колонка длиной от 30 м до 60 м и внутрен¬
ним диаметром 0,3 мм, покрытая слоем макрагола
20 000 Р\
- газ-носитель: азот для хроматографии Р;
- скорость газа-носителя: 0,40 мл/мин;
- деление потока: 1:200;
- температура:
Время (мин)
Температура (°С)
Колонка
0—4
60
4—26
60-* 170
26-41
170
Блок ввода проб
220
Детектор
270
- детектор: пламенно-ионизационный;
- объем вводимой пробы: 1 мкл.
Предельное содержание примесей:
- эстрагол: не более 10,0 % в эфирном масле;
- фенхон: не более 7,5 % в эфирном масле.
Допустимые примеси (2.8.2). Другие части рас¬
тения: плодоножки — не более 1,5 %. Другие допусти¬
мые примеси 1,5 %.
Вода (2.2.13). Не более 80 мл/кг. Определение
проводят из 20,0 г измельченного сырья (710) (2.9.12).
Общая зола (2.4.16). Не более 10,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 10,0 г испы¬
туемого сырья, измельченного до грубого порошка
(1400) (2.9.12) непосредственно перед проведением
испытания, помещают в круглодонную колбу вмести¬
мостью 500 мл, прибавляют 200 мл воды Р в качестве
жидкости для перегонки. В градуированную трубку по¬
мещают 0,50 мл ксилола Р и перегоняют со скоростью
(2—3) мл/мин в течение 2 ч.
Анетол. Газовая хроматография (2.2.28), как ука¬
зано в испытании «Эстрагол и фенхон», со следующи¬
ми изменениями.
Раствор сравнения. 5 мг анетола Р растворяют
в 0,5 мл ксилола Р.
ХРАНЕНИЕ
В защищенном от влаги месте.
07/2016:1855
ФИАЛКИ ТРАВА
1/ю/ае ЬегЬа сит Логе
ИПЕО РАМЗУ
(РЮМЕРШв АЕШАЕ РАРТЗ)
ОПРЕДЕЛЕНИЕ
Собранная в фазу массового цветения и высу¬
шенная трава одно- или двулетнего травянистого рас¬
тения У/о/а /г/со/ог 1_. и/или V. ап/епз1з Мигг.
Содержание: не менее 1,5 % суммы флавонои-
дов в пересчете на виолантин (С27Н30О14, М.м. 578,5)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Стебель
угловатый и полый. Листья овальные, черешко¬
вые с сердцевидным основанием, или вытянутые
и тупоконечные, с лировидными прилистниками,
разделенные посередине. Цветки с длинной цвето¬
ножкой, зигоморфные, с пятью овальными ланце¬
товидными чашелистиками, с придатками, направ¬
ленными наружу и с 5 лепестками, нижний из кото-
167. Зак. 1060.
1330
Государственная фармакопея Республики Беларусь
рых имеет ноготок; у У/о/а ап/епз1з лепестки короче
чашечки, нижний лепесток кремового цвета с чер¬
ными линиями, 4 верхних лепестка могут быть кре¬
мового или фиолетово-синего цвета; у У/о/а /г/со/ог
лепестки длиннее чашечки, фиолетового цвета, с
большим или меньшим желтым оттенком. Андро-
цей, состоящий из 5 тычинок, имеет наверху мем¬
бранный связующий придаток с двумя ноготками.
Трехкамерная завязь имеет короткий столбик и
шаровидное рыльце. Плоды — ладьевидные трех¬
дольные коробочки желтовато-коричневого цвета
длиной от 5 мм до 10 мм. Семена бледно-желтого
цвета, грушевидной формы, длиной около 1 мм, с
присеменником.
В. Микроскопия (2.8.23). При просматрива¬
нии листа с поверхности (рисунок #1855.-1) у обоих
видов фиалки видны клетки эпидермиса, с нижней
стороны более извилистые, чем с верхней; устьица
аномоцитного типа (2.8.3), располагаются с обеих
сторон и окружены 3-4 клетками эпидермиса [А].
Простые волоски нежнобородавчатые, с толстыми
стенками и заостренным концом, располагаются
преимущественно на жилках и по краю листа [А1].
Железистые волоски с многоклеточной головкой на
широкой многоклеточной ножке, встречаются толь¬
ко по краю листа с углублениями между зубцами
и на концах зубцов [В]. В мезофилле листа видны
многочисленные крупные друзы оксалата кальция.
Клетки эпидермиса лепестков имеют сосочковид¬
ные выросты [С1, С2]. На эпидермисе средних и
нижних лепестков (у основания) располагаются
длинные одноклеточные тупоконечные волоски
с тонкими стенками р]. На эпидермисе нижнего
лепестка при входе в шпорец видны извилистые
длинные одноклеточные бугорчатые волоски [Е].
В паренхиме нижней части лепестков встречаются
друзы оксалата кальция.
При измельчении сырья (355) (2.9.12) и про¬
сматривании под микроскопом с использованием
раствора хлоралгидрата Р видны: фрагменты
эпидермиса с волнообразными стенками при про¬
сматривании с поверхности с устьицами аномо¬
цитного типа (2.8.3); конические одноклеточные
покровные волоски более широкие у основания и
заостренные на конце с исчерченной кутикулой;
железистые волоски с многоклеточной головкой
и короткой многоклеточной ножкой в зазубринах
края листовой пластинки; друзы оксалата кальция,
иногда включенные в паренхиму; фрагменты вен¬
чика с извилистостенными эпидермальными клет¬
ками, в срединной части клетки эпидермиса сосоч¬
ковые, некоторые вытянуты до колбовидных или
бутылковидных, эпидермис основания лепестков
несет покровные волоски длиной до 300 мкм с ха¬
рактерными бугорчатыми утолщениями; сфериче¬
ские или многогранные зерна пыльцы диаметром
от 60 мкм до 80 мкм с мелкоямчатой экзиной и с
5 (У/о/а ап/епз/з) или 4 (У/о/а /г/со/ог) порами; ред¬
кие фрагменты спиральных или сетчатых сосудов
и группы волокон стебля.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 2,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл спирта (70 %,
об/об) Р. Нагревают в водяной бане при температуре
.65 °С в течение 5 мин при перемешивании. Охлажда¬
ют и фильтруют.
Раствор сравнения. 2,5 мг рутина Р, 2,5 мг ги-
перозида Р и 1,0 мг кофейной кислоты Р растворяют
в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
*Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат Р (11:11:27:100, об/об/об/об).#
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 12 см от ли¬
нии старта.
Высушивание: при температуре от 100 °С до 105 °С.
Проявление: горячую пластинку опрыскивают
раствором 10 г/л аминоэтилового эфира дифенил-
борной кислоты Р в метаноле Р и затем раствором
50 г/л макрогола 400 Р в метаноле Р. Через 30 мин
пластинку просматривают в ультрафиолетовом свете
при длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие флуоресци¬
рующие зоны.
Верх хроматографической пластинки
Кофейная кислота:
флуоресцирующая зона
от зеленовато-голубого до
светло-голубого цвета
Флуоресцирующая зона
синего цвета
Гиперозид:
флуоресцирующая зона
желтовато-коричневого
цвета
Флуоресцирующая зона
желтовато-зеленого цвета
Рутин: флуоресцирующая
зона желтовато-коричневого
цвета
Интенсивная
флуоресцирующая зона
желтовато-коричневого
цвета (рутин)
Флуоресцирующая зона
желтовато-зеленого цвета
Флуоресцирующая зона
желтовато-зеленого цвета
Флуоресцирующая зона
желтовато-зеленого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: пожелтевшие листья и стебли — не бо¬
лее 7 %; другие части растения (плоды, створки пло¬
ды, корни, в том числе отделенные при анализе) — не
более 3 %. Органические примеси: не более 3 %. Ми¬
неральные примеси: не более 1 %.
@> Коэффициент набухания (2.8.4). Не менее
9. Исследуют измельченное сырье (355) (2.9.12).
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 15,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
Фукус
1331
Рисунок #1855.-1. Микроскопические признаки
травы фиалки
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
@> Исходный раствор. 0,300 г измельченного сы¬
рья (250) (2.9.12) помещают в колбу вместимостью
200 мл, прибавляют 40 мл спирта (60 %, об/об) Р и
нагревают в водяной бане при температуре 60 °С в
течение 10 мин при частом перемешивании. Охлаж¬
дают и процеживают через ватный тампон в мерную
колбу вместимостью 100 мл. Ватный тампон с остат¬
ками сырья переносят обратно в колбу вместимостью
200 мл, прибавляют 40 мл спирта (60 %, об/об) Р и
нагревают в водяной бане при температуре 60 °С в
течение 10 мин при перемешивании. Охлаждают и
процеживают в ту же мерную колбу вместимостью
100 мл. Ополаскивают колбу вместимостью 200мп
спиртом (60 %, об/об) Р, процеживают и переносят в
ту же мерную колбу вместимостью 100 мл. Доводят
спиртом (60 %, об/об) Р до объема 100,0 мл и филь¬
труют.
Испытуемый раствор. 5,0 мл исходного раст¬
вора помещают в круглодонную колбу и выпаривают
досуха при пониженном давлении. Остаток после вы¬
паривания растворяют в 8 мл смеси из метанола Р и
кислоты уксусной ледяной Р (10:100, об/об) и пере¬
носят в мерную колбу вместимостью 25 мл. Опола¬
скивают круглодонную колбу 3 мл смеси из метано¬
ла Р и кислоты уксусной ледяной Р (10:100, об/об) и
переносят в ту же мерную колбу вместимостью 25 мл.
Прибавляют 10,0 мл раствора, содержащего 25,0 г/л
кислоты борной Р и 20,0 г/л кислоты щавелевой Р в
кислоте муравьиной безводной Р, и доводят кисло¬
той уксусной безводной Р до объема 25,0 мл.
Компенсационный раствор. 5,0 мл исходного
раствора помещают в круглодонную колбу и выпари¬
вают досуха в вакуумом. Остаток после выпаривания
растворяют в 8 мл смеси из метанола Р и кислоты
уксусной ледяной Р (10:100, об/об) и переносят в мер¬
ную колбу вместимостью 25 мл. Ополаскивают кру¬
глодонную колбу 3 мл смеси из метанола Р и кисло¬
ты уксусной ледяной Р (10:100, об/об) и переносят в
туже мерную колбу вместимостью 25 мл. Прибавляют
10,0 мл муравьиной кислоты безводной Р и доводят
кислотой уксусной безводной Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 405 нм.
Содержание суммы флавоноидов в пересчете на
виолантин в процентах рассчитывают по формуле:
А- 500
/77-400 ’
где:
400 — удельный показатель поглощения виолан-
тина;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1423
<Е> ФУКУС
Рисиз уе/ АзсорНуНит
КЕ1-Р
ОПРЕДЕЛЕНИЕ
Фрагменты высушенных слоевищ Рисиз
уезюи1озиз или Р зегга(из или АзсорНуНит
подозит Ье чМз.
Содержание: не менее 0,03 % и не более 0,2 %
общего содержания йода (А.м. 126,9) (в пересчете на
сухое сырье).
ОПИСАНИЕ (СВОЙСТВА)
Имеет соленый и слизистый вкус и неприятный
морской запах.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Состоит из фраг¬
ментов слоевищ роговидной консистенции от черно¬
коричневого до зеленовато-коричневого цвета, иногда
покрытых беловатым налетом. Слоевище состоит из
лентовидных полотен, дихотомического ветвления, с
выступающими центральными ребрами (псевдожил¬
ками). Для вида Р. Vез^сиIозиз типично сильноразвет-
вленное полотно с гладкими краями, на котором ино¬
гда располагаются яйцевидные одиночные или пар¬
ные воздушные пузырьки. Концы отдельных ветвей
имеют яйцевидную форму и слегка расширены. На
них располагаются многочисленные репродуктивные
органы (вместилища). Вид Р. зегга1из имеет сильно-
разветвленное полотно с пильчатым краем и без воз¬
душных пузырьков, ветви с концептакулами в мень¬
шей степени раздуты. Слоевище вида А. тодозит
имеет беспорядочное ветвление с одиночными яйце¬
видными воздушными пузырьками, псевдожилка от¬
сутствует, концептакулы серповидной формы распо¬
лагаются на концах небольших ветвей.
B. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет зеленовато-корич¬
невый. Просматривают под микроскопом с исполь¬
зованием раствора хлоралгидрата Р. В порошке
167*. Зак. 1060.
1332
Гэсударственная фармакопея Республики Беларусь
обнаруживаются следующие диагностические при¬
знаки: фрагменты эпителиальных тканей, состоя¬
щие из правильных изодиаметрических клеток с ко¬
ричневым содержимым, и фрагменты глубоко рас¬
положенных тканей с бесцветными удлиненными
клетками, собранными в длинные волокна с боль¬
шим количеством слизи между ними. Иногда видны
толстостенные клетки псевдожилок, расположенные
рядами и плотно прилегающими друг к другу груп¬
пами.
С. К 1 г измельченного сырья (355) (2.9.12) при¬
бавляют 20 мл 2 % (об/об) раствора кислоты хлори¬
стоводородной Р. Интенсивно встряхивают и филь¬
труют. Остаток промывают 10 мл воды Р и фильтру¬
ют. К остатку прибавляют 10 мл раствора 200 г/л
натрия карбоната Р. Встряхивают и центрифуги¬
руют. Собирают надосадочную жидкость, доводят
кислотой серной Р до рН 1,5. Медленно образуется
белый хлопьевидный осадок.
ИСПЫТАНИЯ
Мышьяк (2.4.27). Не более 0,0090 % (90 ррт).
Кадмий (2.4.27). Не более 0,0004 % (4 ррт).
Свинец (2.4.27). Не более 0,0005 % (5 ррт).
Ртуть (2.4.27). Не более 0,00001 % (0,1 ррт).
Коэффициент набухания (2.8.4). Не менее 6.
Потеря в массе при высушивании (2.2.32). Не
более 15,0 %. 1,000 г испытуемого сырья сушат при
температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 24 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Общий йод. К1,000 г измельченного сырья (355)
в высоком кварцевом тигле прибавляют 5 мл воды Р
и 5 г калия гидроксида Р. Перемешивают магниевой
палочкой и нагревают на водяной бане. Прибавляют
1 г калия карбоната Р. Перемешивают и вместе с
концом магниевой палочки, на котором находится
остаток лекарственного растительного сырья, вы¬
сушивают на водяной бане, а затем над открытым
пламенем. Сжигают, постепенно увеличивая темпе¬
ратуру, но не более чем до 600 °С. Охлаждают. При¬
бавляют 20 мл воды Р и осторожно нагревают до
кипения, перемешивая стеклянной палочкой. Филь¬
труют горячую смесь через нескладчатый фильтр в
коническую колбу. Промывают осадок на фильтре 4
раза горячей водой Р порциями по 20 мл. Фильтр и
тигель ополаскивают 50 мл горячей воды Р. Раство¬
ры объединяют и охлаждают. Нейтрализуют кисло¬
той серной разведенной Р в присутствии раствора
метилового оранжевого Р. Прибавляют 3 мл кисло¬
ты серной разведенной Р и 1 мл бромной воды Р.
Раствор становится желтым. Через 5 минут при¬
бавляют 0,6 мл раствора 50 г/л фенола Р. Раствор
становится прозрачным. Подкисляют 5 мл кислоты
фосфорной Р и прибавляют 0,2 г калия йодида Р.
Выдерживают в течение 5 мин в защищенном от
света месте. Прибавляют 1 мл раствора крахма¬
ла Р и титруют 0,01 М раствором натрия тиосуль¬
фата.
1 мл 0,01 М раствора натрия тиосульфата со¬
ответствует 0,2115 мг йода.
МАРКИРОВКА
Указывают виды присутствующих водорослей.
07/2016:1825
ХВОЩА ПОЛЕВОГО ТРАВА
Еци1зеИ ап/епз/з ЬегЬа
ЕСЦЛ8ЕП1М 8ТЕМ
ОПРЕДЕЛЕНИЕ
Собранные в течение лета и высушенные над¬
земные вегетативные побеги дикорастущего много¬
летнего травянистого растения Едшзе/ит ап/епзе 1_.
Содержание: не менее 0,3 % суммы флаво-
ноидов в пересчете на изокверцитрозид (С21Н20О12;
М.м. 464,4) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
(*») А. Внешние признаки (2.8.23). Представля¬
ет собой фрагменты бороздчатых главных стеблей,
веток с продольными острыми выступами и листьев,
расположенных по кругу и объединенных основания¬
ми в обвертку. Цвет зеленый или зеленовато-серый.
Кусочки грубые на ощупь, хрупкие и хрусткие при лом¬
ке. Главный стебель диаметром около (1—4,5) мм, по¬
лый, с утолщениями в узлах, расположенных через
интервалы в (1,5—4,5) см; на междоузлиях располо¬
жены от 4 до 14 отчетливо видимых вертикальных бо¬
роздок. Центральная полость занимает менее 50 %,
но более 25 % диаметра главного стебля. В узлах сте¬
бля расположены широко расположенные мутовки и
прямые, обычно простые, ветви, каждая из которых
толщиной около 1 мм с 3—5 продольными острыми
ребрышками; на конце каждого ребрышка находится
торчащий отчетливый колленхимный пучок, располо¬
женный под эпидермисом. Ветви не полые. Листья
мелкие, линейные, расположены мутовками в каждом
узле, сросшиеся в основании; листья образуют зубча¬
тую обвертку вокруг стебля; количество зубчиков со¬
ответствует количеству бороздок на стебле. Каждый
зубчик, зачастую коричневый, ланцетно-треугольный.
Нижнее междоузлие на каждой ветке длиннее обверт¬
ки на стебле, из которой ветка выходит.
В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет зеленовато-серый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки (рисунок
1825.-1): фрагменты эпидермиса при просматривании
с поверхности [В, С], состоящие из прямоугольных
клеток с волнистыми стенками и устьиц парацитного
типа (2.8.3), расположенных в 2—4 ряда, 2 вспомога¬
тельные клетки, находящиеся в той же плоскости, что
и эпидермис, покрывают замыкающие клетки и имеют
лучистые гребни; на поверхности вспомогательных
клеток обнаруживаются разрозненные кремниевые
диски, встречающиеся более часто у края листа и
образующие отчетливый круг, окружающий вспомога¬
тельные клетки [С]; на гребнях веток обнаруживаются
крупные прямоугольные продольно ориентированные
двухклеточные выросты [Р], на стебле они менее чет¬
ко выраженные [А]; при просматривании с поверхно¬
сти эпидермис главного стебля состоит из вытянутых
клеток [О], на эпидермисе вторичных веток видны
двухклеточные выросты, которые напоминают пары
мелких клеток, разделенные более крупными клетка¬
ми [Э]; фрагменты крупноклеточной паренхимы [Н] и
группы длинных нелигнифицированных волокон с уз¬
кими просветами; мелкие сосуды со спиральным или
круговым утолщением [Е].
Хвоща полевого трава
1333
Рисунок 1825.-1. Микроскопические признаки
травы полевого хвоща
С. Просматривают хроматограммы, получен¬
ные в испытании «Елшзе1ит ра1из1ге».
Результаты, ниже приведена последователь¬
ность зон хроматограмм раствора сравнения (Ь) и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие бледные
флуоресцирующие зоны.
Верх хроматографической пластинки
Кофейная кислота:
флуоресцирующая зона
зеленовато-голубого цвета
Две флуоресцирующих зоны
красного цвета
Две флуоресцирующих зоны
зеленовато-голубого цвета
Гиперозид:
флуоресцирующая зона
оранжевого цвета
Флуоресцирующая зона
оранжевого цвета
Две флуоресцирующих зоны
зеленовато-голубого цвета
Рутин: флуоресцирующая
зона оранжевого цвета
Раствор сравнения (Ь)
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
® Ери1зе1ит ра1из1ге. Тонкослойная хромато¬
графия (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р и
нагревают в водяной бане при температуре 60 °С в
течение 10 мин при периодическом встряхивании. Ох¬
лаждают и фильтруют.
Раствор сравнения (а). К 100,0 мг ФСО
ЕдшзеШт ра!из1ге прибавляют 10 мл метанола Р, на¬
гревают в водяной бане при температуре 60 °С в тече¬
ние 10 мин при периодическом встряхивании. Охлаж¬
дают и фильтруют.
Раствор сравнения (Ь). 1,0 мг кофейной кисло¬
ты Р, 2,5 мг гиперозида Р и 2,5 мг рутина Р раство¬
ряют в 20 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((2—10)мкм).
Подвижная фаза: кислота муравьиная безво¬
дная Р — кислота уксусная ледяная Р — вода Р —
этилацетат Р (7,5:7,5:18:67, об/об/об/об).
Наносимый объем пробы: по 5 мкл в виде полос
длиной 8 мм.
Фронт подвижной фазы: не менее 6 см от линии
старта.
Высушивание: в потоке холодного воздуха в те¬
чение 5 мин.
Проявление: пластинку нагревают при темпера¬
туре 100 °С в течение 3 мин и обрабатывают раство¬
ром 10 г/л дифенилборной кислоты аминоэтилового
эфира Р в метаноле Р и затем раствором 50 г/л ма-
крогола 400 Р в метаноле Р. Пластинку высушивают
в потоке холодного воздуха и через 10 мин просма¬
тривают в ультрафиолетовом свете при длине волны
365 нм.
Пригодность системы: на хроматограмме раст¬
вора сравнения (а) должны обнаруживаться две флу¬
оресцирующие зоны зеленоватого цвета, располо¬
женные немного выше линии нанесения пробы.
Результаты: на хроматограмме испытуемого
раствора любые флуоресцирующие зоны зеленова¬
того цвета, расположенные немного выше линии на¬
несения пробы, должны быть не интенсивнее соот¬
ветствующих зон (характерных для Е. ра!из1ге 1_.) на
хроматограмме раствора сравнения (а).
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения — не более 1 %;
другие виды хвощей — не более 4 %. Органические
примеси: не более 1 %. Минеральные примеси: не бо¬
лее 0,5 %.
Примечание. К другим видам хвощей, встречаю¬
щимся в сырье как примесь, относят:
a) Ершзе1ит зИуаНсит 1_., отличающийся от за¬
готовляемого нежестким стеблем, вторично ветвящи¬
мися вниз отклоненными тонкими ветвями. В верхней
части стебля на ребрах под лупой заметны два ряда
роговидных шипиков. Зубцы влагалища на стебле
срастаются: в сырье легко обламываются. На вер¬
хушках встречаются тупые колоски. Под микроскопом
при рассмотрении эпидермиса стебля с поверхности
в бороздках видны в один (два) ряда устьица. Ребра
гладкие, но местами по краям заметны большие со¬
сочковидные выступы: стенки клеток ребер ветвей
слабоволнистые;
b) Ершзе(ит рге{епзе 1_., отличающийся от за¬
готовляемого почти горизонтальным расположением
ветвей, книзу дуговидно отогнутых, неспаянными зуб¬
чиками влагалища и наличием в верхней части стебля
конусовидных острых сосочков, густо расположенных
по ребрышкам, очень хорошо заметных под лупой.
На верхушке стеблей могут быть тупые колоски. Под
микроскопом видно, что сосочки на эпидермисе ре¬
брышек расположены в несколько рядов. В бороздках
один, реже два ряда устьиц. Стенки клеток ребер вет¬
вей слегка волнистые;
c) Ершзе1ит ЛиV^а^^Iе отличающийся от заго¬
товляемого очень толстым стеблем, толщиной около
168. Зак. 1060.
1334
Гэсударственная фармакопея Республики Беларусь
0,5 см и высотой от 20 см до 150 см. Ветви короткие
малочисленные или отсутствуют. Влагалища с много¬
численными зубцами (от 18 до 20). На верхушках сте¬
блей встречаются тупые колоски. Под микроскопом при
рассмотрении эпидермиса стебля с поверхности вид¬
ны гладкие ребрышки, чередующиеся с широкими бо¬
роздками, несущими по 10—12 рядов устьиц в ширину;
б) Ершзе1ит ра1из(ге 1_., отличающийся от заго¬
товляемого неспаянными, снабженными широкой бе¬
лой каймой зубцами стеблевых влагалищ. Влагалища
ветвей на стебле черного цвета, а у других видов они
зеленого или темно-коричневого цвета. При отрывании
ветвей на стебле удерживаются не только влагалища,
но и первые членики в отличие от других видов хвоща.
Поверхность стеблей и ветвей поперечно-морщини¬
стая. На верхушке стеблей могут быть тупые колоски.
Под микроскопом при рассмотрении эпидермиса с по¬
верхности видны устьица, расположенные в несколько
рядов. Ребрышки стеблей и ветвей несут заостренные
зубцы. На поперечном срезе отличительными призна¬
ками являются: у ветвей — наличие центральной поло¬
сти, у стеблей — отсутствие колленхимы в бороздках.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 3,000 г измельченного сырья (2000)
(2.9.72) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не менее 12,0% и не
более 27,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.7). Не менее 3,0 % и не более 15,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
<Е> Исходный раствор. 0,800 г измельченного
сырья (355) (2.9.72) помещают в круглодонную кол¬
бу вместимостью 100 мл, прибавляют 1 мл раствора
5 г/л гексаметилентетрамина Р, 20 мл ацетона Р,
2 мл кислоты хлористоводородной Р1 и кипятят с
обратным холодильником в течение 30 мин. Извле¬
чение процеживают в колбу через ватный тампон, ко¬
торый затем присоединяют к остатку в круглодонной
колбе и еще дважды повторяют извлечение по 10 мин
каждое, прибавляя ацетон Р порциями по 20 мл. Каж¬
дое извлечение охлаждают и процеживают через ват¬
ный тампон. Объединенные ацетоновые извлечения
фильтруют через бумажный фильтр в мерную колбу
вместимостью 100 мл. Колбу и фильтр ополаскивают
ацетоном Р и доводят фильтрат до объема 100,0 мл
этим же растворителем. 20,0 мл полученного раст¬
вора помещают в делительную воронку, прибавляют
20 мл воды Р и встряхивают смесь с 15 мл этилаце-
тата Р, а затем трижды с этилацетатом Р порция¬
ми по 10 мл. Этилацетатные извлечения объединяют
в делительной воронке, дважды промывают водой Р
порциями по 50 мл и фильтруют через фильтр с 10 г
натрия сульфата безводного Р. Фильтрат доводят
этилацетатом Р до объема 50,0 мл.
Испытуемый раствор. К 10,0 мл исходного раст¬
вора прибавляют 1 мл реактива хлорида алюминия Р
и доводят раствором 5 % (об/об) кислоты уксусной
ледяной Р в метаноле Р до объема 25,0 мл.
Компенсационный раствор. 10,0 мл исходного
раствора доводят раствором 5 % (об/об) кислоты ук¬
сусной ледяной Р в метаноле Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) испытуемого раствора при 425 нм.
Содержание суммы флавоноидов в пересчете на
изокверцитрозид в процентах рассчитывают по фор¬
муле:
А- 625
т•500 ’
где:
500 — удельный показатель поглощения изо-
кверцитрозида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
07/2016:1222
ХМЕЛЯ ШИШКИ
1-ириН Лоз
НОР ЗТРОВИЕ
ОПРЕДЕЛЕНИЕ
Собранные в фазу начала созревания плодов и
высушенные шишковидные соцветия («шишки») мно¬
голетней лианы Нити1из 1ири1из \-.
ОПИСАНИЕ
Запах характерный ароматный.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
®А. Внешние признаки (2.8.23). Обычно
одиночные «шишки» длиной (2—5) см, с черешком,
яйцевидной формы, состоящие из большого количе¬
ства овальных зеленовато-желтых сидячих пленча¬
тых перекрывающихся прицветников. Внешние при¬
цветники уплощенные и симметричные. Внутренние
прицветники более длинные и асимметричные в
основании по причине изгиба, обычно окружающего
покрытый чешуйчатыми листьями плод (семянку).
Завязь или реже плод, основание прицветников и
особенно чешуйки покрыты мелкими оранжево-жел¬
тыми железками.
® В. Микроскопия (2.8.23). Исследуют из¬
мельченное сырье (355) (2.9.72). Цвет зеленовато-
желтый. Просматривают под микроскопом с исполь¬
зованием раствора хлоралгидрата Р. В порошке
обнаруживаются следующие диагностические при¬
знаки (рисунок 1222.-1): фрагменты прицветников
и прицветничков, покрытых многоугольными непра¬
вильными или извилистостенными эпидермальны¬
ми клетками р, 1_, М]; одноклеточные конические
прямые или изогнутые покровные волоски с тонки¬
ми гладкими стенками, фрагментированные [Е, О]
или присоединены к эпидермису [А]; редкие устьица
аномоцитного типа (2.8.3) [К]; железистые волоски,
обычно отдельные, с двухклеточными двухслойны¬
ми ножками и головками, состоящими из 8 мелких
клеток [Н, 14], реже волоски присоединены к эпи¬
дермису [1_а]; фрагменты мезофилла, содержащего
мелкие друзы оксалата кальция ^]; большое коли¬
чество характерных оранжево-желтых железистых
волосков с короткими двухклеточными двухслойны¬
ми ножками, несущими расширяющиеся чашевид¬
ные части диаметром (150—250) мкм, состоящие
из полусферического слоя выделительных клеток
с кутикулой, которая отделена и раздута скопле¬
нием маслянисто-смолистых выделений, видимые
при просматривании с поверхности [В] или сбоку
[С]; фрагменты вытянутых клеток семенной кожуры
с толстыми стенками, с исчерченностью и с много¬
численными порами [Р].
Чабреца блошиного трава
1335
1а
Рисунок 1222.-1. Микроскопические признаки
шишек хмеля
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г свежеизмельчен-
ного сырья (355) (2.9.12) прибавляют 10 мл смеси из
воды Р и метанола Р (3:7, об/об); встряхивают в тече¬
ние 15 мин и фильтруют.
Раствор сравнения. 1,0 мг Судана оранжево¬
го Р, 2,0 мг куркумина Р и 2,0 мг диметиламинобен-
зальдегида Р растворяют в 20 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р.
Подвижная фаза: кислота уксусная безвод¬
ная Р— этилацетат Р — циклогексан Р (2:38:60,
об/об/об).
Наносимый объем пробы: по 20 мкп в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: пластинку просматривают в уль¬
трафиолетовом свете при 254 нм.
Результаты А: на хроматограмме раствора
сравнения обнаруживаются три зоны поглощения; в
нижней четверти обнаруживается зона слабого по¬
глощения, принадлежащая куркумину, немного ниже
середины обнаруживается зона поглощения, принад¬
лежащая диметиламинобензальдегиду, и выше рас¬
положена зона поглощения, принадлежащая Судану
оранжевому. На хроматограмме испытуемого раст¬
вора обнаруживается серия зон поглощения, соответ¬
ствующих по расположению зонам на хроматограмме
раствора сравнения: около уровня зоны куркумина
обнаруживается зона слабого поглощения, принад¬
лежащая ксантагумолу, около уровня зоны димети-
ламинобензальдегида обнаруживаются зоны, при¬
надлежащие гумулонам, около уровня зоны Судана
оранжевого обнаруживаются зоны, принадлежащие
лупулонам.
Проявление В: пластинку просматривают в уль¬
трафиолетовом свете при 365 нм.
Результаты В: на хроматограмме испытуемого
раствора зоны, соответствующие лупулонам имеют
голубую флуоресценцию; зоны, соответствующие гу¬
мулонам, имеют коричневую флуоресценцию; зона,
соответствующая ксантогумолу, имеет темно-корич¬
невую флуоресценцию.
Проявление С: пластинку опрыскивают реакти¬
вом фосфорномолибденово-вольфрамовым разве¬
денным Р, выдерживают в парах аммиака и просма¬
тривают при дневном свете.
Результаты С: на хроматограмме испытуемого
раствора зоны, соответствующие гумулонам и лупу¬
лонам имеют синевато-серый цвет, зона, соответству¬
ющая ксантогумолу, имеет зеленовато-серый цвет; на
хроматограмме раствора сравнения зоны имеют цвет
от синевато-серого до коричневато-серого.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: семена — не более 4 %; осыпавшиеся
листочки — не более 25 %; другие части растения:
при машинном сборе — не более 10 %, при ручном —
не более 5 %.
® Вещества, извлекаемые 70 % (об/об) спир¬
том. Не менее 25,0 %. К 10,0 г измельченного сырья
(355) (2.9.12) прибавляют 300 мл спирта (70%, об/
об) Р и нагревают с обратным холодильником на во¬
дяной бане в течение 10 мин. Охлаждают, фильтруют,
отбрасывая первые 10 мл фильтрата. 30,0 мл филь¬
трата выпаривают досуха на водяной бане и высуши¬
вают при температуре от 100 °С до 105 °С в течение
2 ч. Масса сухого остатка должна составлять не ме¬
нее 0,250 г.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 14,0 %.
07/2016:РБ0100
ЧАБРЕЦА БЛОШИНОГО ТРАВА
ГЛут/ ри/едю/сИ ЬегЬа
1.ЕМОЫ ТНУМЕ*
ОПРЕДЕЛЕНИЕ
Собранная во время цветения, высушенная
цельная или резаная трава ТЬутиз ри/едю/дез 1_.
Содержание: не менее 1,0 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Смесь цель¬
ных или частично измельченных тонких веточек,
листьев, кусочков стеблей толщиной до 1,5 мм и
цветков. Листья обычно длиной от 4 мм до 18 мм
и шириной до 9 мм; короткочерешковые, яйцевид¬
ной или яйцевидно-эллиптической формы, цель¬
нокрайние, голые, по черешку до основания пла¬
стинки реснитчатые, с мало заметными жилками на
нижней стороне листа. При просматривании (при
увеличении 10х) по всей поверхности листа вид¬
ны многочисленные коричневатые точки (железки),
у основания листа — единичные длинные редкие
168*. Зак. 1060.
1336
Гэсударственная фармакопея Республики Беларусь
щетинистые волоски, часто обломанные. Кусочки
веточек тонкие, острочетырехгранные, опушенные
по ребрам, зеленовато-коричневого или желтовато-
коричневого цвета, часто с фиолетовым оттенком.
Цветки мелкие, одиночные или собранные по не¬
сколько штук в полумутовки. Каждый цветок состоит
из двугубой чашечки и двугубого венчика. Чашечка
колокольчатая, длиной около 4 мм, снаружи опу¬
шенная; зубцы чашечки по краю с реснитчатыми во¬
лосками. Венчик длиной (4—6) мм, тычинок четыре,
пестик с четырехраздельной верхней завязью. Цвет
листьев от зеленого до серовато-зеленого, чашеч¬
ки — лиловый; венчика — розовато-фиолетовый.
Запах ароматный.
В. Микроскопия (2.8.23). При просматривании
листа с поверхности (рисунок РБ0100.-1) видны клет¬
ки эпидермиса верхней [А] и нижней сторон листа с
извилистыми стенками [В]; на верхнем эпидермисе
иногда заметна складчатость кутикулы и четковид¬
ное утолщение стенок [А]. Устьица диацитного типа
(2.8.3) на обеих сторонах листа окружены двумя
околоустьичными клетками [А, В]. Эфиромасличные
железки крупные, состоят из шести-восьми выдели¬
тельных клеток, расположенных радиально; клетки
эпидермиса вокруг места прикрепления железки ча¬
сто образуют розетку [С]. Волоски трех типов: очень
крупные, многоклеточные бородавчатые волоски
[Э1], расположенные у основания листа (выше по
краю листа встречаются более мелкие волоски), ча¬
сто обломанные [02]; головчатые волоски с однокле¬
точной овальной головкой на короткой одноклеточ¬
ной ножке [ЭЗ]; сосочковидные выросты эпидермиса,
гладкие или слегка бородавчатые, чаще встречаются
по краю листа [04].
Рисунок РБ0100.-1. Микроскопические признаки
травы чабреца блошиного
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного сы¬
рья (355) (2.9.12), прибавляют 5 мл метиленхлорида Р,
встряхивают в течение 3 мин и фильтруют через бумаж¬
ный фильтр с 2 г натрия сульфата безводного Р.
Раствор сравнения. 5 мг тимола Р и 10 мкл кар-
вакрола Р растворяют в 10 мл метиленхлорида Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р.
Подвижная фаза: метиленхлорид Р.
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Тимол: зона поглощения
Зона поглощения
Зона поглощения (тимол)
Зоны поглощения
Раствор сравнения
Испытуемый раствор
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р, расходуя 10 мл на ква¬
дратную пластинку со стороной 200 мм, и нагревают в
течение 10 мин при температуре 105 °С.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемо¬
го раствора в нижней трети могут обнаруживаться и
другие зоны. Интенсивность зон тимола и карвакрола
зависит от испытуемого сырья (хемотип).
Верх хроматографической пластинки
Тимол: зона коричневато¬
розового цвета
Карвакрол: зона бледно¬
фиолетового цвета
Зона коричневато-розового
цвета (тимол)
Зона бледно-фиолетового
цвета (карвакрол)
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Фрагменты сырья
ТЬутиз зегруНит — не более 10 %; другие допусти¬
мые примеси — не более 3 % (в том числе ТЬутиз
VиIда^^з, ТЬутиз гуд'1з). Определение проводят из 30 г
испытуемого сырья.
Допустимые примеси могут состоять из листьев
от игольчатой до продолговато-ланцетовидной фор¬
мы, с сильно завернутыми вниз краями; на верхней
стороне листа имеются покровные волоски с боро¬
давчатыми стенками, в очертании с остроконечием;
на нижней стороне бородавчатые волоски нескольких
типов: одноклеточные, прямые или слегка изогнутые,
двух- или трехклеточные, часто коленчато изогнутые
и двух- или трехклеточные, более или менее прямые
(Путиз уи1дапз, ТЬутиз гуд/з). Стебли округло-четы¬
рехгранные, равномерно опушенные отстоящими во¬
лосками (ТЬутиз зегруНит).
Потеря в массе при высушивании (2.2.13). Не
более 13,0 %.-1,000 г измельченного сырья (2000) су¬
шат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Чабреца ползучего трава
1337
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 50,0 г ис¬
пытуемого сырья помещают в колбу вместимостью
1000 мл, прибавляют 500 мл воды Р в качестве жид¬
кости для перегонки. Перегоняют без использования
ксилола Р со скоростью (2—3) мл/мин в течение 2 ч.
07/2016:1891
ЧАБРЕЦА ПОЛЗУЧЕГО ТРАВА
8егруШ ЬегЬа (Пут/ зегруШ ЬегЬа)
УЯНО ТНУМЕ
ОПРЕДЕЛЕНИЕ
Собранная во время цветения, высушенная
цельная или резаная трава ТЬутиз зегруНит I. з./.
Содержание: не менее 1,0 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Стебель
сильно разветвлен диаметром около 1,5 мм, цилин¬
дрический или почт четырехугольный, цвет зеленый,
красноватый или красновато-фиолетовый, более ста¬
рые стебли деревянистые и коричневого цвета, мо¬
лодые стебли опушены. Листья супротивные, длиной
(3—12) мм и шириной до 4 мм, от эллиптической до
овально-ланцетовидной формы с тупой верхушкой,
короткочерешковые и клинообразные в основании;
листья цельнокрайние и заметно реснитчатые, осо¬
бенно у основания; обе поверхности более или менее
голые, однако с отчетливо видными точками. Соцве¬
тие состоит из 6—12 цветков, собранных в верхушеч¬
ные головки округлой или яйцевидной формы. Чашеч¬
ка трубчатая, двугубая с верхней губой, разделенной
на три зубчика, и нижней губой с двумя зубчиками,
окаймленными длинными волосками; внутренняя по¬
верхность сильно опушена, после отцветания волоски
образуют закрытую трубку. Венчик красновато-фио¬
летового или красного цвета, двугубый, нижняя губа
разделена на три доли, верхняя — зазубренная, вну¬
тренняя поверхность сильно опушена; 4 нелепестные
тычинки выступают из трубки венчика.
В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет серовато-зеленый
или зеленовато-коричневый. Просматривают под микро¬
скопом с использованием раствора хлоралгидрата Р. В
порошке обнаруживаются следующие диагностические
признаки (рисунок 1891.-1): фрагменты эпидермиса
листьев [А, В, Р], состоящего из клеток с извилистыми
антиклинальными стенками [Аа, Ва, Ра] с устьицами ди-
ацитного типа (2.8.3) [АЬ, ВЬ, РЬ] и с мелкоисчерченной
кутикулой; клетки эпидермиса верхней стороны ли¬
стьев [В] с извилистыми неравномерно утолщенными
антиклинальными стенками [Ва]; многочисленные
покровные волоски, расположенные на эпидермисе
обеих сторон листа и на кромке листа, небольшое
количество клеток волосков содержит очень ма¬
ленькие кристаллы оксалата кальция [А*, Са, Р6],
волоски в основном короткие, конические, однокле¬
точные, с утолщенными и бородавчатыми стенками
(при просматривании с поверхности [Вс] и при про¬
сматривании сбоку [Рс]); в небольшом количестве
169. Зак. 1060.
многоклеточные длинные заостренные покровные
волоски, состоящие из 8 клеток и слегка уширенные
в основании, с мелкоямчатыми стенками, волоски
могут обнаруживаться как вместе с эпидермисом
[Ае], так и фрагментированные [С]; многочисленные
железистые вологски, большинство из которых явля¬
ются многоклеточными слоистого типа [Ас] с однокле¬
точной ножкой и железистой головкой, состоящей из
12 малозаметных клеток, другие — с одноклеточной
ножкой и одноклеточной шаровидной или яйцевидной
головкой [Ад]; красновато-фиолетовые фрагменты
венчика, внутренний эпидермис которого состоит из
клеток с сосочковидными выростами [О], а внешний
эпидермис [Е] (с исчерченной кутикулой) состоит из
клеток с дольчатыми стенками [Еа] с одноклеточными
[ЕЬ] или многоклеточными [Ес] простыми покровны¬
ми волосками, железками с одноклеточной головкой
и одноклеточной ножкой [Еб] и железками слоистого
типа; относительно редкие зерна пыльцы диаметром
около 30 мкм с мелкоямчатой экзиной и 6 зародыше¬
выми порами [О].
Рисунок 1891.-1. Микроскопические признаки
травы чабреца ползучего
(§) С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г измельченного сы¬
рья (355) (2.9.12), прибавляют 5 мл метанола Р и об¬
рабатывают ультразвуком в течение 10 мин. Центри¬
фугируют или фильтруют; используют надосадочную
жидкость или фильтрат.
Раствор сравнения. 1 мг рутина Р и 1 мг розма¬
риновой кислоты Р растворяют в 5 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ля Р254 Р ((5—40) мкм) [или ТСХ пластинка со слоем
силикагеля Р254 Р ((2—10) мкм)].
Подвижная фаза: муравьиная кислота безвод¬
ная Р — вода Р — этилацетат Р( 1:1:15, об/об/об).
1338
Гэсударственная фармакопея Республики Беларусь
Наносимый объем пробы: по 20 мкл [или по
5 мкл] в виде полос длиной 20 мм [или 8 мм].
Фронт подвижной фазы: не менее 15 см [или
6 см] от линии старта.
Высушивание: на воздухе.
Проявление: пластинку нагревают при темпера¬
туре 100 °С в течение 3 мин; горячую пластинку обра¬
батывают раствором 5 г/л дифенилборной кислоты
аминоэтилового эфира Р в этилацетате Р, затем
раствором 50 г/л макрогола 400 Р в метиленхлори-
де Р; просматривают в ультрафиолетовом свете при
длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. Кроме того, на хроматограмме ис¬
пытуемого раствора могут обнаруживаться и другие
тусклые флуоресцирующие зоны.
Верх хроматографической пластинки
Розмариновая кислота:
флуоресцирующая зона
голубого цвета
Флуоресцирующая зона
красного цвета
Флуоресцирующая
зона голубого цвета
(розмариновая кислота)
Одна или две
флуоресцирующие зоны
голубого цвета
Рутин: флуоресцирующая
зона оранжево-желтого
цвета
Флуоресцирующая зона
желтого или оранжевого
цвета
Может обнаруживаться
флуоресцирующая зона
зеленого или голубого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
(^Допустимые примеси (2.8.2). Сумма приме¬
сей: не более 3 %. Определение проводят из 30 г ис¬
пытуемого сырья.
СЕф) ТЬутиз уи1дапз 1_. или ТЬутиз гуд/з 1_.
Фальсификация Т. уи1дапз I и Т гуд/з 1_. выявляется
путем обнаружения листьев от игольчатой до линей¬
но-ланцетовидной формы с сильно загнутым краем,
на верхней поверхности которых обнаруживаются по¬
кровные волоски, по форме напоминающие заострен¬
ные зубцы, с бородавчатыми стенками, на нижней
поверхности — волоски различных типов с бородав¬
чатыми стенками: одноклеточные, прямые или слегка
изогнутые, двух- или трехклеточные, часто коленчато
изогнутые и двух- или трехклеточные, более или ме¬
нее прямые.
Потеря в массе при высушивании (2.2.13). Не
более 13,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 50,0 г ис¬
пытуемого сырья помещают в колбу вместимостью
1000 мл, прибавляют 500 мл воды Р в качестве жид¬
кости для перегонки. Перегоняют без использования
ксилола Р со скоростью (2—3) мл/мин в течение 2 ч.
07/2016: РБ0095
ЧАГА (ЧЕРНЫЙ БЕРЕЗОВЫЙ ГРИБ)
1попо1из оЫ'щииз (Рипдиз ЬеЮНпиз)
ЗНЕ1.Г тывс/в*
ОПРЕДЕЛЕНИЕ
Собранные в течение всего года, освобожден¬
ные от остатков древесины, разрубленные на куски и
высушенные наросты бесплодной формы трутовика
косого — чаги (березового гриба) — 1попо1из оЬНдииз
(Регз.) РН.
Содержание: не менее 10% хромогенного ком¬
плекса (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Куски различной
формы размером до 10 см. Наружный слой нароста
черный, сильно растрескавшийся, внутренний — тем¬
но- или красновато-коричневый с мелкими желтыми
прожилками, число которых увеличивается к внутрен¬
ней стороне. Ткань гриба плотная, твердая.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Органические
примеси, береста, остатки древесины, в том числе от¬
деленные при анализе: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья (355)
(2.9.12) сушат при температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 14,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
10,00 г (т) измельченного сырья (1400) (2.9.12)
помещают в колбу вместимостью 500 мл, прибавляют
300 мл воды Р, выдерживают в течение 1 ч и кипятят с
обратным холодильником, поддерживая слабое кипе¬
ние в течение 2 ч. Водное извлечение фильтруют че¬
рез бумажный фильтр в мерную колбу вместимостью
500 мл, сырье переносят на фильтр и промывают те¬
плой водой. Водное извлечение в колбе охлаждают
до температуры 20 °С, доводят водой Р до объема
500,0 мл и тщательно перемешивают (раствор А).
Испытуемый раствор (а). 25,0 мл раствора А
помещают в предварительно взвешенную фарфоро¬
вую чашку, выпаривают на водяной бане досуха, су¬
шат при температуре от 100 °С до 105 °С в течение
3 ч, охлаждают в эксикаторе и быстро взвешивают,
определяя массу сухого остатка (т^).
Испытуемый раствор (Ь). 100,0 мл раствора
А помещают в лабораторный стакан вместимостью
150 мл, подкисляют кислотой хлористоводород¬
ной Р1 ((0,5—0,8) мл) до рН 1,0—2,0, перемешивают
и выдерживают в течение 30 мин. После выпадения
темно-коричневого осадка (хромогенный комплекс)
содержимое стакана фильтруют через бумажный
фильтр. 25,0 мл полученного фильтрата помещают в
предварительно взвешенную фарфоровую чашку, вы¬
паривают на водяной бане досуха, сушат при темпе¬
ратуре от 100 °С до 105 °С в течение 3 ч, охлаждают
в эксикаторе и быстро взвешивают, определяя массу
сухого остатка без хромогенного комплекса (т2).
Череды трава
1339
Содержание хромогенного комплекса в процен¬
тах рассчитывают по формуле:
(/п1 - л?2) • 2000
т
07/2016: РБ0096
ЧЕРЕДЫ ТРАВА
ВШепИз ЬегЬа
В1/Я МАШ001-0*
ОПРЕДЕЛЕНИЕ
Собранная в фазу бутонизации и начала цве¬
тения, высушенная трава однолетнего травянистого
растения Б/с/елз МрагШа 1_. и/или ВМепз {гопдоза I.
Содержание: не менее 1,0 % флавоноидов
в пересчете на лютеолин-7-глюкозид (С21Н20О1(|;
М.м. 448,4) (в пересчете на сухое сырье) и не менее
3,5 % полисахаридов (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Олиственные
стебли длиной до 20 см и их кусочки, цельные или
измельченные листья и цветочные корзинки. Листья
Б/с/елз МрагШа супротивные, на коротких сросшихся
основаниями черешках, срединные 3—5-глубокорас-
сеченные с ланцетовидными пальчатыми долями,
верхушечные — цельные, широколанцетные, дли¬
ной до 15 см. Листья Б/с/елз Iгопдоза супротивные,
на длинных черешках, тройчатосложные, реже, на
верхушке — простые, широколанцетные, длиной
до 10 см. Стебли округлоовальные, продольно-бо¬
роздчатые, толщиной до 0,5 см. Соцветия корзинки
диаметром от 0,4 см до 1,5 см. Наружные листочки
обвертки в количестве 8, зеленые, удлиненно-ланце-
видные, опушенные по краю, равные или в два раза
превышающие корзинку. Внутренние листочки об¬
вертки более короткие, удлиненноовальные, по краю
пленчатые, коричневато-желтые с многочисленными
темно-фиолетовыми жилками. Цветки мелкие, труб¬
чатые с двумя зазубренными остями вместо чашечки.
Цвет листьев зеленый или коричневато-зеленый, сте¬
блей — зеленый или зеленовато-фиолетовый, цвет¬
ков — серовато-желтый. Запах слабый.
B. Микроскопия (2.8.23). При просматривании
листа с поверхности (рисунок РБ0096.-1) видны клетки
эпидермиса верхней и нижней стороны листа и листоч¬
ков наружной обвертки с извилистыми стенками, устьи¬
ца многочисленные, аномоцитного типа (2.8.3), окруже¬
ны 3—5 клетками эпидермиса [А]. Клетки эпидермиса
стебля прямоугольные, иногда встречаются устьица [В].
У листочков внутренней обвертки клетки эпидермиса
многоугольные или округлые, у основания — прямоу¬
гольные. Клетки эпидермиса трубчатых цветков у осно¬
вания прямоугольные, в середине цветка переходят в
5—7-гранные клетки. На верхушке лепестков есть со¬
сочковидные выросты эпидермиса [С1, С2]. По жилкам
и краю листа, листочков наружной и внутренней об¬
вертки, а также по краю лепестков цветка встречаются
секреторные ходы с оранжевым содержимым [А-й].
Толстостенные простые волоски Б/с/елз МрагШа 1_.
из 2—10 клеток встречаются по краю [ОЦ и вдоль жи¬
лок листа и листочков наружной обвертки [Е1]. Тонко¬
стенные простые волоски Б/с/елз МрагШа Ь. из 4—12
клеток встречаются на листьях, листочках наружной
[Р1] и внутренней (на верхушке) обвертки [6] и цветках
[С1].Толстостенные простые волоски Б/с/елз Копдоза
Ь. из 3—11 крупных вытянутых клеток встречаются по
краю листа [02], вдоль жилок листа и листочков на¬
ружной обвертки [Е2]. Тонкостенные простые волоски
Б/с/елз Копдоза Ь. построены из 4—14 вытянутых кле¬
ток встречаются по краю и вдоль жилок листа, листоч¬
ков наружной [Р2] и внутренней обвертки [С]. Пыльца
округлая, шиповатая [Н]. Могут встречаться однокле¬
точные волоски семянок Б/с/елз МрагШа 1_. [И] и Б/с/елз
Ггопдоза Ь. [12].
Рисунок РБ0096.-1. Микроскопические признаки
травы череды
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 1 г измельченного сы¬
рья (500) (2.9.12) помещают в колбу со шлифом вме¬
стимостью 50 мл, прибавляют 10 мл спирта (70%,
об/об) Р и нагревают с обратным холодильником в
водяной бане в течение 20 мин при периодическом
перемешивании. Охлаждают и фильтруют через бу¬
мажный фильтр.
Раствор сравнения. 1 мг лютеолин-7-глю-
козида Р растворяют в 10 мл 96 % спирта Р.
Пластинка: ТСХпластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — кислота ук¬
сусная ледяная Р — вода Р(5:1:1, об/об/об).
Наносимый объем пробы: по 5 мкл в виде полос.
Фронт подвижной фазы: не менее 8 см от линии
старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
10 г/л дифенилборной кислоты аминоэтилового
эфира Р в 96% спирте Р, нагревают в сушильном
шкафу в течение (2—3) мин при температуре 105 °С и
просматривают в ультрафиолетовом свете при длине
волны 365 нм.
169*. Зак. 1060.
1340
Гэсударственная фармакопея Республики Беларусь
Результат: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Зона желтого цвета
Зона ярко-красного цвета
Лютеолин-7-глюкозид: зона
желтого цвета
Зона желтого цвета
(лютеолин-7-глюкозид)
Зона ярко-красного цвета
Зона желтого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: пожелтевшие, побуревшие и почернев¬
шие части растения — не более 8 %; семена — не
более 3 %. Органические примеси: не более 3 %. Ми¬
неральные примеси: не более 1 %.
ВМепз сегпиа 1_. Тонкослойная хроматография
(2.2.27), как указано в тесте «Подлинность (идентифи¬
кация) С». Между зоной лютеолин-7-глюкозида и ярко-
красной зоной, расположенной в верхней части пла¬
стинки, не должно быть дополнительного желтого пятна.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 14,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 3,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания флавоноидов в
пересчете на лютеолин-7-глюкозид. 2,500 г измель¬
ченного сырья (2000) (2.9.12) помещают в коническую
колбу вместимостью 100 мл, прибавляют 50 мл спир¬
та (60%, об/об) Р и нагревают с обратным холодиль¬
ником на водяной бане в течение 30 мин. Полученное
извлечение охлаждают до комнатной температуры и
фильтруют через бумажный фильтр, предварительно
смоченный спиртом (60%, об/об) Р, и доводят спир¬
том (60%, об/об) Р до объема 50,0 мл. 5,0 мл полу¬
ченного раствора доводят спиртом (60%, об/об) Р до
объема 50,0 мл (раствор А).
Испытуемый раствор. 2,5 мл раствора А, 1,5 мл
раствора 50 г/л натрия нитрита Р помещают в мер¬
ную колбу вместимостью 50 мл и оставляют на 5 мин.
Затем прибавляют 1,5 мл раствора 100 г/л алюминия
хлорида Р и снова оставляют на 5 мин. К полученному
раствору прибавляют 10,0 мл раствора 40 г/л натрия
гидроксида Р и доводят водой Р до объема 50,0 мл.
Через 10 мин измеряют оптическую плотность
испытуемого раствора при длине волны 510 нм, ис¬
пользуя воду Р в качестве коменсационного раствора.
Содержание флавоноидов в пересчете на
лютеолин-7-глюкозид в процентах рассчитывают по
формуле
А -10000
т-206 ’
где:
206 — удельный показатель поглощения
лютеолин-7-глюкозида;
А — оптическая плотность испытуемого раст¬
вора;
т — масса навески испытуемого сырья, г.
Опредление содержания полисахаридов.
10,00 г измельченного сырья (500) (2.9.12) помеща¬
ют в колбу вместимостью 500 мл, прибавляют 100 мл
воды Р и кипятят с обратным холодильником в тече¬
ние 30 мин при периодическом перемешивании. Вы¬
держивают до оседания частиц и собирают надоса-
дочную жидкость. Экстракцию повторяют еще четыре
раза с использованием воды Р порциями по 100 мл.
Водные извлечения объединяют и центрифугируют
при 5000 об/мин в течение 10 мин. Всю надосадочную
жидкость процеживают через ватный тампон и дово¬
дят водой Р до объема 500,0 мл. 25,0 мл полученного
раствора помещают в центрифужную пробирку, при¬
бавляют 75 мл 96 % спирта Р, перемешивают, подо¬
гревают в водяной бане при 60 °С в течение 5 мин.
Через 30 мин содержимое центрифугируют при 5000
об/мин в течение 30 мин. Надосадочную жидкость
фильтруют под вакуумом через высушенный при тем¬
пературе от 100 °С до 105 °С до постоянной массы
стеклянный фильтр (ПОР 16) диаметром 40 мм. Оса¬
док количественно переносят на фильтр и промывают
15 мл смеси из 96 % спирта Р и воды Р (3:1, об/об).
Фильтр с осадком высушивают сначала на воздухе,
затем при температуре от 100 °С до 105 °С до посто¬
янной массы.
Содержание полисахаридов в процентах рассчи¬
тывают по формуле:
(т2 -т^- 2000
т
где:
т1 — масса фильтра, г;
т2 — масса фильтра с осадком, г;
т — масса навески испытуемого сырья, г.
07/2016:РБ0097
ЧЕРЕМУХИ ПЛОДЫ
РасИ Iги&из
В/ЯО СНЕККУ*
ОПРЕДЕЛЕНИЕ
Высушенные зрелые плоды Радиз ау\ит МШ. и
Р. аз/аИса Кот.
Содержание: не менее 1,7 % дубильных веществ
в пересчете на танин (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Плоды — ко¬
стянки шарообразной или продолговато-яйцевидной
формы, иногда к верхушке несколько заостренные,
диаметром до 8 мм, морщинистые, без плодоножек, с
округлым белым рубцом на месте ее отпадения. Вну¬
три плода содержится одна округлая или округло-яй¬
цевидная, очень плотная, светло-коричневая косточ¬
ка диаметром до 7 мм с одним семенем. Поверхность
плодов морщинистая, косточки — поперечно-ребри¬
стая. Цвет плодов черный, матовый, реже блестящий,
иногда с беловато-серым или красноватым налетом
на складках.
B. Микроскопия (2.8.23). На поперечном срезе
плода виден эпидермис, состоящий из клеток с рав¬
номерно утолщенными стенками. Мезокарпий пред-
Черники плоды свежие
1341
ставлен рыхлой паренхимой, клетки которой запол¬
нены хромопластами разнообразной формы, изредка
встречаются проводящие пучки. Эндокарпий состоит
из двух слоев склеренхимной ткани: наружного — ка¬
менистые клетки округлой или слегка вытянутой по
радиусу формы, внутреннего — тангентально вытя¬
нутые склеренхимные волокна. В наружном слое ко¬
сточки встречаются паренхимные клетки с кристалла¬
ми оксалата кальция ромбической формы.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: пригоревшие и поврежденные насеко¬
мыми плоды — не более 3 %; недозрелые и бурые
плоды — не более 3 %; другие части растения (плодо¬
ножки, в том числе отделенные при анализе, и веточ¬
ки) — не более 3 %. Органические примеси: не более
1 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 1,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2.000 г измельченного сырья (2000) (2.9.12) поме¬
щают в коническую колбу со шлифом вместимостью
500 мл, прибавляют 250 мл кипящей воды Р и кипятят
с обратным холодильником в течение 30 мин при пе¬
риодическом перемешивании. Охлаждают до комнат¬
ной температуры, процеживают и доводят водой Р до
объема 250,0 мл.
25.0 мл полученного извлечения помещают в ко¬
ническую колбу вместимостью 750 мл, прибавляют
500 мл воды Р, 25,0 мл раствора индигосульфокисло-
ты (0,25 мг индигокармина Р растворяют в 6,5 мл кис¬
лоты серной Р, прибавляют 6,5 мл кислоты серной Р,
доводят водой Р до объема 250 мл и фильтруют). Ти¬
труют при постоянном перемешивании 0,02 М раство¬
ром калия перманганата до желтого окрашивания.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия перманганата со¬
ответствует 4,157 мг дубильных веществ в пересчете
на танин.
07/2016:1602
(в)ЧЕРНИКИ ПЛОДЫ СВЕЖИЕ
МугИШ !гис1и$ гесепз
ВИ.ВЕККУ РШЛТ, РВЕЗН
ОПРЕДЕЛЕНИЕ
Свежие или замороженные зрелые плоды
\/асстшт тугИНиз 1_.
Содержание: не менее 0,30 % антоцианов в
пересчете на цианидина З-О-глюкозида хлорид (хри-
зантемин, С^Н^О^-НС!; М.м. 484,8) (в пересчете на
сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Свежие плоды
представляют собой шаровидные черновато-синие
ягоды диаметром около 5 мм. На нижнем конце яго¬
ды — рубец или иногда фрагмент плодоножки. Верх¬
ний конец уплощенный и увенчан остатками неопа¬
дающего столбика и чашечкой, которая представляет
собой круговую складку. Мезокарпий фиолетового
цвета, представляющий собой мякоть, состоит из
4—5 гнезд, содержащих многочисленные небольшие
коричневые семена яйцевидной формы.
B. Микроскопия (2.8.23). Раздавленный свежий
плод имеет фиолетово-красный цвет. Исследуют под
микроскопом, используя раствор хлоралгидрата Р.
При исследовании наблюдаются фиолетово-розовые
склереиды эндокарпия и мезокарпия, обычно собран¬
ные группами, с толстыми бороздчатыми стенками;
красновато-коричневые фрагменты эпикарпия, со¬
стоящие из многоугольных клеток с умеренно утол¬
щенными стенками; коричневато-желтые фрагменты
внешнего слоя кожуры семян, состоящие из удлинен¬
ных клеток с 11-образными утолщенными стенками;
друзы оксалата кальция.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 5 г свежеизмельченного
сырья прибавляют 20 мл метанола Р. Перемешива¬
ют в течение 15 мин и фильтруют.
Раствор сравнения. 5 мг хризантемина Р раст¬
воряют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота муравьиная безвод¬
ная Р — вода Р — бутанол Р (16:19:65, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Хризантемин: зона
фиолетово-красного цвета
Зона фиолетово-красного
цвета
Основная зона фиолетово¬
красного цвета
Плотная группа других
основных зон:
- зона фиолетово-красного
цвета
- несколько зон фиолетово¬
синего цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Общая зола (2.4.16). Не более 0,6 %.
Потеря в массе при высушивании (2.2.32). Не
менее 80,0 % и не более 90,0 %. 5,000 г свежеизмель¬
ченного сырья сушат при температуре 105 °С.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
50 г испытуемого сырья раздавливают непосред¬
ственно перед проведением испытания. К около 5,00 г
точно взвешенного измельченного сырья прибавляют
95 мл метанола Р. Перемешивают механическим
способом в течение 30 мин и фильтруют в мерную
колбу вместимостью 100,0 мл. Ополаскивают фильтр
и доводят метанолом Р до объема 100,0 мл. Исполь¬
зуют 50-кратное разведение полученного раствора в
0,1 % (об/об) растворе кислоты хлористоводород¬
ной Р в метаноле Р.
Измеряют оптическую плотность (2.2.25) раст¬
вора при длине волны 528 нм, используя 0,1 % (об/об)
раствор кислоты хлористоводородной Р в метано¬
ле Р в качестве компенсационного раствора.
170. Зак. 1060.
1342
Гэсударственная фармакопея Республики Беларусь
Содержание антоцианов в пересчете на циани-
дина З-О-глюкозида хлорид в процентах рассчитыва¬
ют по формуле:
А•5000
718 т ’
где:
718 — удельный показатель поглощения циани-
дина З-О-глюкозида хлорида;
А — оптическая плотность раствора;
т — масса навески испытуемого сырья, г.
ХРАНЕНИЕ
В случае замороженного лекарственного расти¬
тельного сырья его хранят при температуре не выше
-18 °С.
07/2016:1588
<§> ЧЕРНИКИ ПЛОДЫ СУХИЕ
МугШН (гис!из з/ссиз
ВИВЕВКУ РРШ1Т, ОЯ/ЕО
ОПРЕДЕЛЕНИЕ
Высушенные зрелые плоды \/асстшт тугИИиз 1_.
Содержание: не менее 1,0 % дубильных веществ
в пересчете на пирогаллол (С6Н603; М.м. 126,1) (в пе¬
ресчете на сухое вещество).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Сухие плоды
представляют собой темно-синие округлые, сморщен¬
ные ягоды диаметром около 5 мм с рубцом на нижнем
конце, а на верхнем конце — неопадающей чашеч¬
кой, которая представляет собой круговую складку, и
остатками столбика. Мясистый мезокарпий темно-фи¬
олетового цвета содержит многочисленные неболь¬
шие коричневые семена яйцевидной формы.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет фиолетово-коричневый.
Просматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружива¬
ются следующие диагностические признаки: фиоле¬
тово-розовые склереиды эндокарпия и мезокарпия,
обычно собранные группами, с толстыми бороздча¬
тыми стенками; красновато-коричневые фрагменты
эпикарпия, состоящие из многоугольных клеток с уме¬
ренно утолщенными стенками; коричневато-желтые
фрагменты внешнего слоя кожуры семян, состоящие
из удлиненных клеток с 11-образными утолщенными
стенками; друзы и призмы кристаллов оксалата каль¬
ция различного размера.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 2 г измельченного сы¬
рья (355) (2.9.12) прибавляют 20 мл метанола Р.
Встряхивают в течение 15 мин и фильтруют.
Раствор сравнения. 5 мг хризантемина Р раст¬
воряют в 10 мл метанола Р.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р— вода Р— бутанол Р (16:19:65, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: просматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора.
Верх хроматографической пластинки
Хризантемин: зона
фиолетово-красного цвета
Слабая зона фиолетово¬
красного цвета
Основная зона фиолетово¬
красного цвета
Плотная группа других
основных зон:
- зона фиолетово-красного
цвета
- несколько зон фиолетово¬
синего цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ
Потеря в массе при высушивании (2.2.32). Не
более 12,0 %. 1,000 г измельченного сырья сушат при
температуре 105 °С в течение 2 ч.
Общая зола (2.4.16). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Дубильные вещества (2.8.14). Определе¬
ние проводят из 1,500 г измельченного сырья (355)
(2.9.12).
07/2016:1216
ЧЕСНОКА ПОРОШОК
АНН заШ Ьи1Ы риМз
(ЗАКИС РОИ/ОЕА?
ОПРЕДЕЛЕНИЕ
Луковицы АШит за1Ыит 1_., резаные, лиофилизи-
рованные или высушенные при температуре, не пре¬
вышающей 65 °С, и измельченные до порошкообраз¬
ного состояния.
Содержание: не менее 0,45 % аллицина
(С6Н10О32; М.м. 162,3) (в пересчете на сухое веще¬
ство).
ОПИСАНИЕ (СВОЙСТВА)
Легкий желтоватый порошок.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Исследуют под микроскопом, используя раст¬
вор хлоралгидрата Р. В порошке обнаруживаются
следующие диагностические признаки: многочислен¬
ные фрагменты паренхимы и группы спиральных или
кольцевидных ячеек с прилегающей тонкостенной па¬
ренхимой.
B. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г порошка чеснока
прибавляют 5,0 мл метанола Р, встряхивают в тече¬
ние 60 с и фильтруют.
Раствор сравнения. 5 мг аланина Р растворяют
в 10 мл воды Р и доводят метанолом Р до объема
20 мл.
Пластинка : ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: кислота уксусная ледяная
Р — пропанол Р — вода Р — этанол безводный Р
(20:20:20:40, об/об/об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 10 мкл раствора сравнения, в виде полос.
Чистотела трава
1343
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раствором
2 г/л нингидрина Р в смеси из 5 объемов кислоты ук¬
сусной ледяной Р и 95 объемов бутанола Р и нагре¬
вают при температуре от 105 °С до 110 °С в течение
(5—10) мин и просматривают в дневном свете.
Результаты: на хроматограмме раствора срав¬
нения в центральной трети обнаруживается фиоле¬
товая зона (аланин). На хроматограмме испытуемого
раствора обнаруживается фиолетовая или корич¬
невато-красная зона, соответствующая по располо¬
жению зоне на хроматограмме раствора сравнения,
соответствующей алиину. Выше и ниже этой зоны на¬
блюдаются другие в большинстве случаев более сла¬
бые фиолетовые зоны.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Крахмал. Испытуемый порошок исследуют под
микроскопом, используя водуР, и прибавляют йода
раствор Р1. Не должна появляться синяя окраска.
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 1,000 г испытуемого порошка сушат при
температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Количе¬
ственное определение проводят как можно быстрее.
Раствор внутреннего стандарта. 20,0 мг ФСО
бутилпарагидроксибензоата растворяют в 100,0 мл
смеси из равных объемов метанола Р и воды Р.
Испытуемый раствор. К 0,800 г порошка чес¬
нока прибавляют 20,0 мл воды Р и гомогенизируют
смесь в ультразвуковой бане при температуре 4 °С в
течение 5 мин. Выдерживают при комнатной темпера¬
туре в течение 30 мин и затем центрифугируют в тече¬
ние 30 мин. 10,0 мл надосадочной жидкости доводят
до объема 25,0 мл смесью из 40 объемов 1 % (об/об)
раствора кислоты муравьиной безводной Р и 60 объ¬
емов метанола Р (исходный раствор). Встряхивают
и центрифугируют в течение 5 мин. 0,50 мл раствора
внутреннего стандарта помещают в мерную колбу и
доводят до объема 10,0 мл исходным раствором.
Условия хроматографирования:
- предколонка длиной 20 мм и внутренним диа¬
метром 4 мм, заполненная силанизированным си¬
ликагелем октадецилсилильным для хроматогра¬
фии Р с размером частиц 5 мкм;
-колонка длиной 0,25м и внутренним диаме¬
тром 4 мм, заполненная силанизированным силика¬
гелем октадецилсилильным для хроматографии Р с
размером частиц 5 мкм;
- подвижная фаза: 1 % (об/об) раствор кислоты
муравьиной безводной Р— метанол Р (40:60, об/об);
- скорость подвижной фазы: 0,8 мл/мин;
-спектрофотометрический детектор, длина
волны 254 нм;
-объем вводимой пробы: петлевой инжектор,
1 мкл раствора внутреннего стандарта и 10 мкл испы¬
туемого раствора.
Процентное содержание аллицина рассчитыва¬
ют по формуле:
8, т2 22,75
$2 *Ш1 '
где:
5, — площадь пика аллицина (основной пик) на
хроматограмме испытуемого раствора;
$2 — площадь пика бутилпарагидроксибензоата
на хроматограмме испытуемого раствора;
т1 — масса испытуемого порошка, г;
т2 — масса бутилпарагидроксибензоата в
100,0 мл раствора внутреннего стандарта, г.
1 мг бутилпарагидроксибензоата соответствует
8,65 мг аллицина.
07/2016:1861
ЧИСТОТЕЛА ТРАВА
СЬеНдопН ЬегЬа
6КЕАТЕК СЕ1.АИ01ЫЕ
ОПРЕДЕЛЕНИЕ
Собранная в период цветения, высушенная,
цельная или измельченная трава СЬеНдопшт таре 1_.
Содержание: не менее 0,6 % суммы алкалои¬
дов в пересчете на хелидонин (С20Н191\Ю5; М.м. 353,4)
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
® А. Внешние признаки (2.8.23). Стебли окру¬
глые, ребристые, желтоватого или зеленовато-корич¬
невого цвета, слабоопушенные, диаметром около
(3—7) мм, полые и в основном спавшиеся. Листья
тонкие непарно-перисто-рассеченные, форма листо¬
вой пластинки от овальной до удлиненной с городча-
тым краем, конечный сегмент часто трехлопастной;
верхняя сторона листа синевато-зеленого цвета и
гладкая, нижняя сторона более бледная и опушенная,
особенно по жилкам. Цветки имеют два сильно вогну¬
товыпуклых чашелистика, которые легко отделяются,
и четыре желтых широкоовальных распластанных
лепестка длиной около (8—10) мм; тычинки многочис¬
ленны, желтого цвета; короткий столбик выходит из
верхней завязи; иногда встречаются продолговатые
стручковидные незрелые плоды.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет темный серовато-зе¬
леный или коричневато-зеленый. Просматривают под
микроскопом с использованием раствора хлоралги¬
драта Р. В порошке обнаруживаются следующие диа¬
гностические признаки (рисунок 1861 .-1): многочислен¬
ные фрагменты верхнего эпидермиса, состоящего из
клеток с извилистыми стенками при просматривании
с поверхности [В] вместе с нижерасположенной пали¬
садной паренхимой [Ва]; многочисленные фрагменты
нижнего эпидермиса при просматривании с поверхно¬
сти [А, Е] с устьицами аномоцитного типа [Аа] (2.8.3)
и основаниями покровных волосков [АЬ], иногда с ни¬
жерасположенной губчатой паренхимой [Еа]; длинные
простые многоклеточные покровные волоски, часто
поломанные, с тонкостенными, иногда спавшимися
клетками [О]; сосудистая ткань листьев и стеблей, со¬
стоящая из пористых и спиральных сосудов р]; группы
волокон [С]; членистые млечники с желтовато-коричне¬
вым содержимым [Г]; редкие фрагменты венчика [Н],
состоящего из тонкостенных клеток, содержащих блед¬
но-желтые капельки масла [На]; сферические зерна
пыльцы диаметром около (30—40) мкм с тремя порами
и мелкоямчатой экзиной [3].
170*. Зак. 1060.
1344
Гэсударственная фармакопея Республики Беларусь
Рисунок 1861.-1. Микроскопические признаки
травы чистотела
(§> С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0.4 г измельченного
сырья (710) (2.9.12) прибавляют 50 мл кислоты ук¬
сусной разведенной Р. Кипятят смесь с обратным
холодильником в водяной бане в течение 30 мин.
Охлаждают и фильтруют. К фильтрату прибавля¬
ют раствор аммиака концентрированный Р до
сильно щелочной реакции и встряхивают с 30 мл
метиленхлорида Р. Органический слой сушат с
использованием натрия сульфата безводного Р,
фильтруют и выпаривают в вакууме досуха. Оста¬
ток после выпаривания растворяют в 1,0 мл мета¬
нола Р.
Раствор сравнения. 2 мг папаверина гидро¬
хлорида Р и 2 мг метилового красного Р раство¬
ряют в 10 мл 96 % спирта Р.
Пластинка: ТСХ пластинка со слоем силика¬
геля Р.
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — пропанол Р (1:9:90, об/об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от
линии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают рас¬
твором калия йодвисмутата Р и высушивают на
воздухе. Затем опрыскивают раствором натрия
нитрита Р и снова высушивают на воздухе. Про¬
сматривают при дневном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуе¬
мого раствора могут обнаруживаться другие слабо
выраженные зоны.
Верх хроматографической пластинки
Метиловый красный: зона
красного цвета
Папаверин: зона серовато-
коричневого цвета
Зона коричневого цвета
Зона коричневого цвета
Зона серовато-коричневого
цвета
2 зоны коричневого цвета
Раствор сравнения
Испытуемый раствор
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: побуревшие и потемневшие части тра¬
вы — не более 3 %. Органические примеси: не более
1 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0%. 1,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 15,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 2,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Испытуемый раствор. К 0,750 г измель¬
ченного сырья (710) (2.9.12) прибавляют 200 мл
кислоты уксусной разведенной Р и нагревают на
водной бане в течение 30 минут при частом встря¬
хивании. Охлаждают и доводят кислотой уксусной
разведенной Р до объема 250,0 мл. Фильтруют,
отбрасывая первые 20 мл фильтрата. К 30,0 мл
фильтрата прибавляют 6,0 мл раствора аммиака
концентрированного Р и 100,0 мл метиленхлори¬
да Р. Встряхивают в течение 30 мин. Отделяют ор¬
ганический слой. 50,0 мл органического слоя поме¬
щают в круглодонную колбу вместимостью 100 мл
и выпаривают досуха в вакууме при температуре
не выше 40 °С. Остаток растворяют в (2—3) мл
96 % спирта Р, слегка нагревая. Переносят раст¬
вор в мерную колбу вместимостью 25 мл, опола¬
скивая круглодонную колбу кислотой серной раз¬
веденной Р, и доводят до объема 25,0 мл этим же
растворителем. 5,0 мл полученного раствора по¬
мещают в мерную колбу вместимостью 25 мл, при¬
бавляют 5,0 мл раствора 10 г/л натриевой соли
хромотроповой кислоты Р в кислоте серной Р,
закрывают колбу и осторожно перемешивают. По¬
лученный раствор доводят кислотой серной Р до
объема 25,0 мл и закрывают колбу.
Компенсационный раствор. Готовят анало¬
гично и одновременно с испытуемым раствором:
в мерную колбу вместимостью 25 мл помещают
5,0 мл кислоты серной разведенной Р и 5,0 мл
раствора 10 г/л натриевой соли хромотроповой
кислоты Р в кислоте серной Р, закрывают колбу и
осторожно перемешивают. Доводят кислотой сер¬
ной Р до объема 25,0 мл и закрывают колбу.
Нагревают оба раствора на водяной бане в
течение 10 мин. Охлаждают до температуры око¬
ло 20 °С и доводят, при необходимости, кислотой
серной Р до объема 25,0 мл.
Измеряют оптическую плотность (2.2.25) ис¬
пытуемого раствора при 570 нм.
Содержание суммы алкалоидов в пересчете на
хелидонин в процентах рассчитывают по формуле:
Шалфея листья
1345
А•2083
т-933 ’
где:
933 — удельный показатель поглощения хелидо-
нина;
А — оптическая плотность испытуемого раст¬
вора;
/77 — масса навески испытуемого сырья, г.
07/2016:1370
ШАЛФЕЯ ЛИСТЬЯ
За/у/'ае оШстаИз ТоНит
8А6Е1.ЕАР (ВАША ОРР1С1ЫАЫ8)
ОПРЕДЕЛЕНИЕ
Цельные или измельченные листья ЗаМа
оШтаНз 1_.
Содержание: не менее 8 мл/кг эфирного масла
(в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
®А. Внешние признаки (2.8.23). Листовая
пластинка листа шалфея имеет длину около (2—
10) см и ширину (1—2) см, форма продолговато¬
овальная, эллиптическая. Край листа от мелкого-
родчатого до ровного. Верхушка от закругленной
до подострой, основание, сморщенное у черешка,
закругленное или сердцевидное. Верхняя сторона
зеленовато-серого цвета, мелкозернистая; нижняя
сторона белая, опушенная, с плотной сеткой вы¬
ступающих жилок.
@> В. Микроскопия (2.8.23). Порошок имеет
светло-серый или коричневато-зеленый цвет. Про¬
сматривают под микроскопом с использованием
раствора хлоралгидрата Р. В порошке обнаружи¬
ваются следующие диагностические признаки (ри¬
сунок 1370.-1): очень многочисленные сочленен¬
ные и изогнутые покрывающие трихомы с узкими
продолговатыми клетками и целой [Вс] или фраг¬
ментированной базовой клеткой с очень толстыми
стенками, расположенные отдельно [С, О, Н] или
на эпидермисе (вид с поверхности [Ве], вид на по¬
перечном срезе [АЬ]); железистые трихомы ламиа-
цетного типа с одноклеточной ножкой и 8-, 12-кле¬
точной головкой, покрытой общей кутикулой, рас¬
положенные отдельно (вид с поверхности [й]) или
на эпидермисе (вид с поверхности [Ра]); неболь¬
шие железистые трихомы с одноклеточной [Аа, Вб]
или многоклеточной [РЬ] ножкой и одноклеточной
головкой, обычно расположенные на эпидермисе;
реже наблюдаются железистые трихомы (вид с по¬
верхности [ЕЬ, Ес], вид сбоку [Еб]) с одноклеточ¬
ной ножкой [Ес] и двухклеточной головкой [ЕЬ, Еб];
фрагменты верхнего эпидермиса (вид с поверхно¬
сти [Е], вид на поперечном срезе [А]) с ямчатыми
слегка полигональными клетками [Еа], покрываю¬
щие трихомы и железистые трихомы, иногда с при¬
легающими 1 или 2 слоями палисадной паренхимы
[Ас, Ее]; могут наблюдаться несколько диацитных
устьиц [2.8.3]; фрагменты нижнего эпидермиса
[В, Р] с волнистыми клетками [Ва] и многочислен¬
ными диацитными устьицами [ВЬ] (2.8.3).
листьев шалфея
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,5 г свежеизмельченно-
го сырья (355) (2.9.12) встряхивают в течение 5 мин с
5 мл этанола Р.
Раствор сравнения. 20 мкл туйона Р и 25 мкл
цинеола Р растворяют в 20 мл этанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: этилацетат Р — толуол Р
(5:95, об/об).
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
200 г/л кислоты фосфорномолибденовой Р в эта¬
ноле Р и нагревают при температуре от 100 °С до
105 °С в течение 10 мин. Просматривают при днев¬
ном свете.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться и другие зоны.
Верх хроматографической пластинки
Зона синего цвета (около
фронта растворителя)
а-туйон и р-туйон: две зоны
розовато-фиолетового цвета
Цинеол: зона синего цвета
Две зоны розовато¬
фиолетового цвета (а-туйон
и р-туйон)
Зона синего цвета (цинеол)
Зоны синего цвета
Раствор сравнения
Испытуемый раствор
171. Зак. 1060.
1346
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: кусочки стеблей и цветков — не более
13 %; почерневшие и побуревшие листья — не более
5 %. Органические примеси: не более 3 %. Минераль¬
ные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 3,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 12,0 %.
Зола, нерастворимая в хлористоводородной
кислоте (2.8.1). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 20,0 г испы¬
туемого сырья, при необходимости измельченного не¬
посредственно перед испытанием, помещают в колбу
вместимостью 500 мл, прибавляют 250 мл воды Р. В
градуированную трубку помещают 0,5 мл ксилола Р.
Перегонку проводят со скоростью (2—3) мл/мин в те¬
чение 2 ч.
07/2016:1510
ШИПОВНИКА ПЛОДЫ
Козае рзеис1о-Тгис1из
006 Я05Е
ОПРЕДЕЛЕНИЕ
Собранные в период полного созревания и вы¬
сушенные плоды кустарников различных видов Роза:
Р. та]аИз Неггт. (Р. стпатотеа I-.), Р. асюи1апз ЫпсИ.,
Р. Ьеддепапа ЗсЬгепк, Р. сал/ла I., Р. согутЬИега
ВогкЬ., Р. дачипса Ра11., Р. 1есНзсЬепкоапа Реде1,
Р. кокапюа (Реде!) Реде! ех Зит.., Р. тюгапИпа ЗтЛИ,
Р. рзатторЬИа СИгзЬап., Р. гидозаТЬипЬ., Р. №теп!оза
5тйН, Р. гапдегига Р иагоэсН. и другие виды.
Содержание:
- не менее 0,2 % аскорбиновой кислоты (СбН8Об;
М.м. 176,1) (в пересчете на сухое сырье);
- или, если сырье используют для приготовления
холосаса, каротолина и сиропов, не менее 2,6 % сво¬
бодных органических кислот в пересчете на яблочную
кислоту (С4Н605; М.м. 134,1) (в пересчете на сухое сы¬
рье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Цельные, очи¬
щенные от чашелистиков и плодоножек ложные
плоды разнообразной формы: от шаровидной, яйце¬
видной или овальной до сильно вытянутой веретено¬
видной; длина плодов от 0,7 см до 3 см, диаметр —
(0,6—1,7) см. На верхушке плода имеется небольшое
круглое отверстие или пятиугольная площадка. Пло¬
ды состоят из разросшегося мясистого, при созрева¬
нии сочного цветоложа (гипантия) и заключенных в его
полости многочисленных плодиков-орешков. Стенки
высушенных плодов твердые хрупкие, наружная по¬
верхность блестящая, реже матовая, более или ме¬
нее морщинистая. Внутри плоды обильно выстланы
длинными, очень жесткими щетинистыми волосками.
Орешки мелкие, продолговатые, со слабо выражен¬
ными гранями. Цвет плодов от оранжево-красного до
коричневато-красного, орешки светло-желтые, иногда
коричневатые.
В. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет оранжево-желтый. Вид¬
ны: обрывки наружного эпидермиса гипантия (плода)
в виде светло-желтых пластов, состоящих из много¬
угольных клеток с прямыми неодинаково утолщен¬
ными, местами четковидно-утолщенными стенками
и редкими устьицами; обрывки мякоти плода, состо¬
ящей из тонкостенных паренхимных клеток, содержа¬
щих оранжево-красные глыбки каротиноидов и много¬
численные друзы оксалата кальция; фрагменты око¬
лоплодника орешка, состоящие из групп или пластов,
реже одиночных каменистых клеток с сильно утол¬
щенными пористыми оболочками; многочисленные
крупные одноклеточные волоски двух типов (или их
обломки) — очень крупные прямые с толстой стенкой
и узкой полостью и более мелкие, слегка извилистые
с широкой полостью; обрывки проводящих пучков со
спиральными сосудами.
® С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 5 г измельченного сы¬
рья (355) (2.9.12) прибавляют 25 мл 96 % спирта Р.
Встряхивают в течение 30 мин и фильтруют.
Раствор сравнения. 10 мг аскорбиновой кисло¬
ты Р растворяют в 5,0 мл спирта (60 %, об/об) Р.
Пластинка: ТСХ пластинка со слоем силикаге-
ляР254 Р.
Подвижная фаза: кислота уксусная Р — аце¬
тон Р—метанол Р—толуол Р (5:5:20:70, об/об/об/об).
Наносимый объем пробы: 20 мкл испытуемого
раствора и 2 мкл раствора сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 254 нм.
Результаты А: на хроматограмме испытуемого
раствора обнаруживается зона поглощения, соответ¬
ствующая по расположению основной зоне на хрома¬
тограмме раствора сравнения.
Проявление В: пластинку опрыскивают раство¬
ром 0,2 г/л дихлорфенолиндофенола натриевой
соли Рв96% спирте Р. Просматривают при дневном
свете.
Результаты В: на хроматограмме испытуемого
раствора обнаруживается зона белого цвета на розо¬
вом фоне соответствующая по расположению и цвету
основной зоне на хроматограмме раствора сравне¬
ния. На хроматограмме испытуемого раствора также
обнаруживаются интенсивная зона оранжево-желтого
цвета, расположенная около фронта подвижной фазы
и зона желтого цвета, расположенная в верхней трети
хроматограммы (каротиноиды).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (кусочки веточек,
чашелистиков и плодоножек) — не более 2 %; почер¬
невшие, пригоревшие, поврежденные вредителями и
болезнями плоды — не более 1 %; частицы плодов,
в том числе орешки, проходящие сквозь сито (2800)
(2.9.12), — не более 3 %. Органические примеси: не
более 0,5 %. Минеральные примеси: не более 0,5 %.
Сырье, используемое для получения холосаса,
каротолина и сиропов, должно отвечать следую¬
щим требованиям. Несырьевые части растения:
другие части растения (кусочки веточек, чашелисти¬
ков и плодоножек) — не более 2 %; почерневшие,
Эвкалипта прутовидного листья
1347
пригоревшие, поврежденные вредителями и болез¬
нями плоды — не более 3 %; частицы плодов, в
том числе орешки, проходящие сквозь сито (2800)
(2.9.12) , — не более 3 %; недозрелые плоды (от зе¬
леного до желтого цвета) — не более 5 %. Органи¬
ческие примеси: не более 0,5 %. Минеральные при¬
меси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 15,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
@> Общая зола (2.4.16). Не более 7,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания аскорбиновой кис¬
лоты. 20,0 г измельченного сырья (2000) (2.9.12)
растирают в ступке с 5 г стеклянного порошка, по¬
степенно прибавляя 300 мл воды Р и выдерживают в
течение 10 мин, перемешивают и фильтруют через бу¬
мажный фильтр, отбрасывая первые 15 мл фильтра¬
та. 1,0 мл полученного раствора помещают в кониче¬
скую колбу, прибавляют 1 мл раствора 20 г/л кислоты
хлористоводородной Р, 13 мл воды Р и титруют из
микробюретки титрованным раствором дихлорфе-
нолиндофенола Р до появления розовой окраски, не
исчезающей в течение (30—60) с. Титрование прово¬
дят в течение не более 2 мин. В случае интенсивного
окрашивания фильтрата или высокого содержания в
нем аскорбиновой кислоты (расход титранта более
2 мл) исходное извлечение разводят водой Р в два
раза или более.
1 мл титрованного раствора дихлорфенолин-
дофенола Р соответствует 0,1 мг С6Н806.
Определение содержания свободных орга¬
нических кислот в пересчете на яблочную кис¬
лоту. 25,0 г измельченного сырья (1400) (2.9.12) по¬
мещают в колбу вместимостью 250 мл, прибавляют
200 мл воды Р и нагревают на водяной бане в тече¬
ние 2 ч. Охлаждают, количественно переносят в мер¬
ную колбу вместимостью 250 мл и доводят водой Р
до объема 250,0 мл. 10,0 мл полученного раствора
помещают в колбу вместимостью 500 мл, прибав¬
ляют (200—300) мл свежекипяченой воды Р, 1 мл
раствора фенолфталеина Р1, 1,5 мл раствора
метиленового синего Р и титруют 0,1 М раствором
натрия гидроксида до появления в пене фиолетово¬
красной окраски.
1 мл 0,1 М раствора натрия гидроксида соот¬
ветствует 6,7 мг С4Н605.
07/2016: РБ0098
ЭВКАЛИПТА ПРУТОВИДНОГО
листья
Еиса1урИ мт/паНз ТоНит
Е11СА1.УРТ118 УШЫАЫЗ /.ЕАР*
ОПРЕДЕЛЕНИЕ
Собранные поздней осенью, зимой или ранней
весной и высушенные листья Еиса1ур№з ПттаНз
1_аЬШ.
Содержание:
- цельное сырье: не менее 10 мл/кг эфирного
масла (в пересчете на сухое сырье);
- измельченное сырье: не менее 8 мл/кг эфирно¬
го масла (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Смесь двух типов
листьев: листья старых ветвей — черешковые от уз-
коланцентных до серповидно-изогнутых, остроконеч¬
ные, плотные, длиной от 4 см до 27 см, шириной от
0,5 см до 5 см; листья молодых ветвей — сидячие, с
округлым основанием или с короткими черешками, уд¬
линенно-яйцевидной формы, на верхушке заострен¬
ные, длиной от 3,5 см до 11 см, шириной от 0,7 см до
4 см. Встречаются листья, имеющие переходящую
форму от удлиненно-яйцевидной до ланцетной. Ли¬
стья голые с цельным, ровным или волнистым краем
с многочисленными точками, просвечивающимися
в проходящем ярком свете (вместилища с эфирным
маслом). Цвет листьев от светло-зеленого до серова¬
то-зеленого иногда с фиолетовым оттенком и слабым
сизоватым налетом. Запах ароматный, усиливающий¬
ся при растирании.
B. Микроскопия (2.8.23). Клетки эпидермиса ли¬
стьев как старых, так и молодых ветвей с поверхно¬
сти многоугольные, в центре их видны светло-серые
пятна (бугорки). На поперечном срезе листа — клетки
эпидермиса более или менее равносторонние с силь¬
но утолщенными наружными стенками и толстым сло¬
ем кутикулы, выступающей в виде бугорков; устьица
погружены в мезофилл листа. Листья изолатераль-
ные. В листьях молодых ветвей палисадная ткань со¬
стоит из двух, реже трех рядов клеток; губчатая ткань
и межклетники хорошо выражены. В листьях старых
ветвей палисадная ткань представлена тремя, реже
четырьмя рядами клеток, клетки губчатой ткани неяс¬
но выражены. Главная жилка листьев как старых, так
и молодых ветвей имеет кристаллоносную обкладку,
встречаются друзы оксалата кальция. Эфиромаслич¬
ные вместилища крупные, округлой или овальной
формы, погружены в мезофилл и занимают часто
более половины толщины листа; внутри их заметны
один-два слоя выделительных клеток.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 0,5 г свежеизмельченного
сырья (355) (2.9.12) прибавляют 5 мл толуола Р, встря¬
хивают в течение (2—3) мин и фильтруют через бумаж¬
ный фильтр с 2 г натрия сульфата безводного Р.
Раствор сравнения. 50 мкл цинеола Р раство¬
ряют в толуоле Р и доводят до объема 5 мл этим же
растворителем.
Пластинка: ТСХ пластинка со слоем подходяще¬
го силикагеля.
Подвижная фаза: этилацетат Р — толуол Р
(10:90, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают раствором
анисового альбегиба Р и нагревают при температуре
от 100 °С до 105 °С в течение (5—10) мин. Просма¬
тривают при дневном свете.
Результаты: на хроматограмме раствора срав¬
нения в центральной части обнаруживается зона
цинеола. На хроматограмме испытуемого раствора
обнаруживается основная зона, соответствующая по
расположению и цвету зоне цинеола на хроматограм¬
ме раствора сравнения; возле линии фронта подвиж¬
ной фазы обнаруживается интенсивная зона фиоле¬
тового цвета (углеводороды); могут обнаруживаться и
другие более слабые зоны.
171*. Зак. 1060.
1348
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: другие части растения (веточки, бутоны,
плоды) — не более 2 %; почерневшие и побуревшие
листья — не более 3 %. Органические примеси: не
более 0,5 %. Минеральные примеси: не более 0,5 %.
Потеря в массе при высушивании (2.2.32). Не
более 14,0 %. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
Общая зола (2.4.16). Не более 5,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А, В или С).
Для метода А 10,0 г испытуемого сырья измель¬
чают непосредственно перед испытанием, помещают
в круглодонную колбу вместимостью 500 мл, прибав¬
ляют 200 мл воды Р и 100 мл глицерина Р в качестве
жидкости для перегонки. В градуированную трубку
помещают 0,5 мл ксилола Р. Перегонку проводят со
скоростью (2—3) мл/мин в течение 2 ч.
Для метода В или С 10,0 г испытуемого сырья из¬
мельчают непосредственно перед испытанием (1400)
(2.9.12) , помещают в колбу вместимостью 1000 мл и
прибавляют 400 мл воды Р в качестве жидкости для
перегонки. Время перегонки 1 ч.
07/2016:1320
® ЭВКАЛИПТА ШАРОВИДНОГО
ЛИСТЬЯ
Еиса1урИ д1оЬиН ТоНит
ЕиСА/-УРП1$ 1-ЕАР
ОПРЕДЕЛЕНИЕ
Цельные или измельченные высушенные листья
старых ветвей Еиса1ур№з д1оЬи1из 1_аЬИ1.
Содержание:
-цельное сырье: не менее 20 мл/кг эфирного
масла (в пересчете на безводное вещество);
- измельченное сырье: не менее 15 мл/кг эфирно¬
го масла (в пересчете на безводное вещество).
ОПИСАНИЕ(СВОЙСТВА)
Имеет ароматный запах цинеола.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
A. Внешние признаки (2.8.23). Листья главным
образом серовато-зеленого цвета, относительно тол¬
стые, вытянутые, эллиптические и слегка серповид¬
но-изогнутые, длиной обычно до 25 см, шириной до
5 см. Черешок скрученный, сильно морщинистый,
длиной (2—3) см (реже до 5 см). Листья кожистые,
жесткие, с цельным краем, гладкие, с желтовато-зе¬
леной средней жилкой. Боковые жилки соединяются
у края листовой пластинки в сплошную линию. Край
листовой пластинки ровный, немного утолщенный.
На обеих поверхностях видны мелкие, неравномер¬
но распределенные шероховатые темно-коричневые
пятна. В проходящем ярком свете можно наблюдать
небольшие железки с эфирным маслом.
B. Микроскопия (2.8.23). Исследуют измельчен¬
ное сырье (355) (2.9.12). Цвет серовато-зеленый. Про¬
сматривают под микроскопом с использованием раст¬
вора хлоралгидрата Р. В порошке обнаруживаются
следующие диагностические признаки (1320.-1): фраг¬
менты гладкой листовой пластинки при просматрива¬
нии с поверхности [А, Ц и в поперечном разрезе [Р, Н],
с маленькими толстостенными клетками эпидермиса,
имеющими толстую кутикулу [Ра, На], многочисленны¬
ми устьицами аномоцитного типа (2.8.3) диаметром
более 80 мкм [Аа, 1_а], редкими группами коричневых
клеток пробки диаметром 300 мкм коричневато-черно¬
го цвета в центре и расположенной ниже палисадной
паренхимой [АЬ, РЬ]; фрагменты билатерального ме¬
зофилла при просматривании сбоку [С], с двумя-тремя
слоями палисадной паренхимы [6а] на каждой сторо¬
не, с несколькими слоями губчатого мезофилла [6Ь] в
центре, клетки которого вытянуты и имеют ту же ориен¬
тацию, что и палисадные клетки, и содержат призмати¬
ческие кристаллы [В, <3с1] и друзы [6с, К] оксалата каль¬
ция; крупные схизогенные масличные железки, которые
могут быть целые [Е] или чаще разрушенные, сопрово¬
ждаемые палисадной паренхимой [Еа]; фрагменты со¬
судов [)] и толстостенных и канальчатых волокон [С] с
кристаллической обкладкой [Са, ^а]ш, кристаллическая
обкладка, содержащая призмы оксалата кальция [Р].
Рисунок 1320.-1. Микроскопические признаки
листьев эвкалипта шаровидного
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 0,5 г свежеизмельченно-
го сырья (355) (2.9.12) встряхивают с 5 мл толуола
Р в течение (2—3) мин и пропускают через фильтр с
около 2 г натрия сульфата безводного Р.
Раствор сравнения. 50 мкл цинеола Р раство¬
ряют в толуоле Р и доводят этим же растворителем
до объема 5 мл.
Пластинка: ТСХ пластинка со слоем силикагеля Р.
Подвижная фаза: этилацетат Р — толуол Р
(10:90, об/об).
Наносимый объем пробы: по 10 мкл в виде полос.
Фронт подвижной фазы: не менее 15 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление: пластинку обрабатывают раство¬
ром анисового альдегида Р и нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин. Про¬
сматривают при дневном свете.
Элеутеррококка корневища и корни
1349
Результаты: на хроматограмме раствора срав¬
нения в центральной части обнаруживается зона
цинеола. На хроматограмме испытуемого раствора
обнаруживается основная зона, соответствующая по
расположению и цвету зоне цинеола на хроматограм¬
ме раствора сравнения; возле линии фронта подвиж¬
ной фазы обнаруживается интенсивная зона фиоле¬
тового цвета (углеводороды); могут обнаруживаться и
другие более слабые зоны.
ИСПЫТАНИЯ
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: черные и коричневые листья — не бо¬
лее 3 %; стебли — не более 5 %. Другие допустимые
примеси: не более 2 %. Не должны присутствовать
сердцевидные или овальные сидячие листья моло¬
дых ветвей с многочисленными железками на обеих
сторонах, видимыми в виде точек в проходящем све¬
те. Определение проводят из 30 г испытуемого сырья.
Вода (2.2.13). Не более 100 мл/кг. Определение
проводят из 20,0 г измельченного сырья (355) (2.9.12).
Общая зола (2.4.16). Не более 6,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Эфирное масло (2.8.12, метод А). 10,0 г испыту¬
емого сырья измельчают непосредственно перед испы¬
танием, помещают в круглодонную колбу вместимостью
500 мл, прибавляют 200 мл воды Р и 100 мл глицери¬
на Р в качестве жидкости для перегонки. В градуирован¬
ную трубку помещают 0,5 мл ксилола Р. Перегонку про¬
водят со скоростью (2—3) мл/мин в течение 2 ч.
07/2016:1419
ЭЛЕУТЕРРОКОККА
КОРНЕВИЩА И КОРНИ
ВеиОюгососа гЫгота е1 гасИх
Е1-ЕиТНЕ1ЮСОССи8
ОПРЕДЕЛЕНИЕ
Собранные осенью, тщательно очищенные от
земли, разрубленные на куски и высушенные корне¬
вища и корни кустарника Е1еи15егососсиз зепИсозиз
(Рирг. е1 Мах1т.) Мах1т.
Содержание:
- не менее 0,08 % суммы элеутерозида В
(М.м. 372,4) и элеутерозида Е (М.м. 742,7);
- или не менее 0,30 % суммы элеутерозидов в пе¬
ресчете на элеутерозид В (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Внешние признаки (2.8.23). Корневище уз¬
ловатое, неправильной цилиндрической формы, диа¬
метром от 1,5 см до 4,0 см; поверхность ребристая, с
продольными бороздками, цвет от серовато-коричне¬
вого до черновато-коричневого; кора толщиной около
2 мм, плотно прилегает к ксилеме; сердцевина свет¬
ло-коричневого цвета, заболонь бледно-желтого цве¬
та; излом коротковолокнистый в коре и длинноволок¬
нистый, особенно во внешней стороне ксилемы. На
нижней поверхности расположены многочисленные
цилиндрические и нитевидные корни длиной от 3,5 см
до 10 см и диаметром от 0,3 см до 1,5 см с гладкой
поверхностью от серовато-коричневого до черновато¬
коричневого цвета; кора толщиной около 0,5 мм, плот¬
но прилегает к ксилеме; излом слабоволокнистый; в
местах, где верхний слой удален, видна желтовато-ко¬
ричневая внешняя поверхность.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет желтовато-коричне¬
вый. Просматривают под микроскопом с использова¬
нием раствора хлоралгидрата Р. В порошке обна¬
руживаются следующие диагностические признаки:
многочисленные группы толстостенных лигнифици-
рованных волокон; фрагменты сетчатых и окайм¬
ленных пористых сосудов с широкими просветами;
группы секреторных каналов диаметром до 20 мкм с
коричневым содержимым; клетки паренхимы, содер¬
жащие друзы оксалата кальция диаметром от 10 мкм
до 50 мкм. Просматривают под микроскопом с ис¬
пользованием 50 % (об/об) раствора глицерина Р. В
порошке обнаруживаются мелкие зерна крахмала в
очертании от округлых до слегка угловатых, простые
или 2—3-сложные.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл спирта (50 %,
об/об) Р и кипятят с обратным холодильником в тече¬
ние 1 ч. Охлаждают и фильтруют. Фильтрат выпари¬
вают досуха на водяной бане, остаток растворяют в
2,5 мл смеси из воды Р и спирта (50 %, об/об) Р (5:20,
об/об) и фильтруют.
Раствор сравнения. 2,0 мг эскулина Р и 2,0 мг
каталпола Р растворяют в 20 мл смеси из воды Р и
спирта (50 %, об/об) Р (5:20, об/об).
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р.
Подвижная фаза: вода Р — метанол Р — мети-
ленхлорид Р (4:30:70, об/об/об).
Наносимый объем пробы: по 20 мкл в виде полос.
Фронт подвижной фазы: не менее 10 см от ли¬
нии старта.
Высушивание: на воздухе.
Проявление А: просматривают в ультрафиолето¬
вом свете при длине волны 365 нм.
Результаты А: на хроматограмме раствора
сравнения в верхней части обнаруживается флуорес¬
цирующая зона синего цвета (эскулин).
Проявление В: пластинку опрыскивают раство¬
ром анисового альдегида Р, нагревают при темпера¬
туре от 100 °С до 105 °С в течение (5—10) мин и про¬
сматривают при дневном свете.
Результаты В: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и испы¬
туемого раствора. На хроматограмме испытуемого
раствора обнаруживаются и другие слабые зоны.
Верх хроматографической пластинки
Эскулин: флуоресцирующая
зона синего цвета,
отмеченная при 365 нм
Зона коричневого цвета
(элеутерозид В)
Зона красновато-
коричневого цвета
(элеутерозид Е)
Катал пол: зона фиолетово¬
коричневого цвета
Две зоны коричневого цвета
Раствор сравнения
Испытуемый раствор
1350
Гэсударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые
части растения: почерневшие и побуревшие в из¬
ломе корневища и корни — не более 3 %; дру¬
гие части растения (остатки стеблей) — не более
1,5 %. Органические примеси: не более 1 %. Мине¬
ральные примеси: не более 1 %.
Потеря в массе при высушивании (2.2.32).
Не более 14,0 %. 2,000 г измельченного сырья
(2000) (2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 8,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение содержания суммы элеуте-
розида В и элеутерозида Е. Жидкостная хромато¬
графия (2.2.29).
Испытуемый раствор. 0,500 г измельченного
сырья (355) (2.9.12) помещают в круглодонную кол¬
бу вместимостью 100 мл, прибавляют 30 мл смеси
из 96 % спирта Р и воды Р (50:50, об/об) и нагрева¬
ют в водяной бане при температуре 60 °С в течение
30 мин. Охлаждают и фильтруют через пористый
стеклянный фильтр (2.1.2). Остаток с фильтра пе¬
реносят в ту же круглодонную колбу вместимостью
100 мл и повторяют экстракцию дважды. Фильтра¬
ты собирают в круглодонную колбу вместимостью
250 мл и упаривают при пониженном давлении до
объема около 10 мл. Полученный раствор количе¬
ственно переносят в мерную колбу вместимостью
20 мл, используя смесь из 96 % спирта Р и воды Р
(50:50, об/об), и доводят до объема 20,0 мл этой
же смесью растворителей. Фильтруют через ней¬
лоновый фильтр с размером пор 0,45 мкм.
Раствор сравнения (а). 10 мг феруловой
кислоты Р растворяют в смеси из метанола Р и
воды Р (50:50, об/об) и доводят до объема 20,0 мл
этим же растворителем.
Раствор сравнения (Ь). 10 мг кофейной кисло¬
ты Р растворяют в смеси из метанола Р и воды Р
(50:50, об/об) и доводят до объема 20,0 мл этим же
растворителем.
Раствор сравнения (с). 1 мл раствора сравне¬
ния (а) разводят смесью из метанола Р и воды Р
(50:50, об/об) до объема 25,0 мл. Фильтруют через
нейлоновый фильтр с размером пор 0,45 мкм.
Раствор сравнения (д). К 1 мл раствора срав¬
нения (а) прибавляют 1 мл раствора сравнения (Ь)
и доводят смесью из метанола Р и воды Р (50:50,
об/об) до объема 25,0 мл. Фильтруют через нейло¬
новый фильтр с размером пор 0,45 мкм.
Условия хроматографирования:
- предколонка длиной 4 мм и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- колонка длиной 0,25 м и внутренним диа¬
метром 4,6 мм, заполненная силикагелем октаде-
цилсилильным для хроматографии Р с размером
частиц 5 мкм;
- подвижная фаза:
- подвижная фаза А: кислота фосфор¬
ная Р — вода Р (0,5:99,5, об/об);
- подвижная фаза В: ацетонитрил для хро¬
матографии Р\
Рисунок 1419.-1. Спектр поглощения
элеутерозида В
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0—5
90
10
5—27
со
0
1
00
о
10 —► 20
27—30
80 —> 50
20 —* 50
30—35
50
50
- скорость подвижной фазы: 1,0 мл/мин;
- спектрофотометрический детектор, длина
волны 220 нм;
- объем вводимой пробы: по 20 мкл испытуемого
раствора и растворов сравнения (с) и (б).
Времена удерживания: элеутерозид В — около
10 мин; элеутерозид Е — около 22 мин. Отмечают пики
элеутерозида В и элеутерозида Е, используя спектры
поглощения, приведенные на рисунке 1419.-1 и рисун¬
ке 1419.-2.
Пригодность хроматографической системы:
раствор сравнения (б):
- разрешение: не менее 15 между пиками кофей¬
ной кислоты и феруловой кислоты.
Содержание суммы элеутерозида В и элеутеро¬
зида Е в процентах рассчитывают по формуле:
Эхинацеи пурпурной трава
1351
Рисунок 1419.-2. Спектр поглощения
элеутерозида Е
5В С-0,73-2 [ $Е С-1,90• 2
У А77 • т
где:
5В — площадь пика элеутерозида В на хромато¬
грамме испытуемого раствора;
5е — площадь пика элеутерозида Е на хромато¬
грамме испытуемого раствора;
— площадь пика феруловой кислоты на хро¬
матограмме раствора сравнения (с);
С — концентрация феруловой кислоты в раство¬
ре сравнения (с), мкг/мл;
т — масса навески испытуемого сырья, мг.
Определение содержания суммы элеутерози-
дов в пересчете на элеутерозид В. 1,000 г измель¬
ченного сырья (1400) (2.9.12) помещают в коническую
колбу вместимостью 100 мл и экстрагируют последо¬
вательно 20 мл спирта (70 %, об/об) Р, 20 мл 96 %
спирта Р и 20 мл смеси из хлороформа Р и 96%
спирта Р (5:1, об/об), при постоянном перемешива¬
нии при температуре не выше 50 °С в течение 1 ч.
Извлечения объединяют, фильтруют через бумажный
фильтр в круглодонную колбу и выпаривают досуха в
вакууме при температуре 40 °С. К остатку прибавляют
10 мл воды Р и 10 мл тетрахлорида углерода Р, пе¬
ремешивают и количественно переносят в делитель¬
ную воронку, используя тетрахлорид углерода Р.
Встряхивают и отбрасывают нижний органический
слой. Водный слой встряхивают дважды с тетрах¬
лоридом углерода Р порциями по 10 мл, отбрасывая
органический слой. Затем водный слой встряхивают
5 раз со смесью из хлороформа Р и 96% спирта Р
(5:1, об/об) порциями по 20 мл, 15 мл, 15 мл, 10 мл и
10 мл. Органические извлечения объединяют и филь¬
труют через бумажный фильтр с 2 г натрия сульфата
безводного Р в мерную колбу вместимостью 100 мл
и доводят смесью из хлороформа Р и 96% спирта Р
(5:1, об/об) до объема 100,0 мл. 20,0 мл полученного
раствора доводят смесью из хлороформа Р и 96%
спирта Р (5:1, об/об) до объема 50,0 мл.
Измеряют оптическую плотность (2.2.25) полу¬
ченного раствора при 278 нм, используя смесь из хло¬
роформа Р и 96% спирта Р (5:1, об/об) в качестве
компенсационного раствора.
Содержание суммы элеутерозидов в пересчете
на элеутерозид В в процентах рассчитывают по фор¬
муле:
А- 250-1,42
/77-302 ’
где:
302 — удельный показатель поглощения элеуте¬
розида В;
А — оптическая плотность раствора;
1,42 — коэффициент пересчета на сумму элеуте¬
розидов, учитывающий молекулярные массы элеуте¬
розидов В, В1? Е и соотношение их содержания;
т — масса навески испытуемого сырья, г.
07/2016:1823
ЭХИНАЦЕИ ПУРПУРНОЙ ТРАВА
ЕсМтасеае ригригеае ЬегЬа
РЦВР1.Е СОЫЕР1-<ШЕР НЕРВ
ОПРЕДЕЛЕНИЕ
Собранная в фазу цветения, цельная или реза¬
ная и высушенная трава многолетнего травянистого
растения ЕсШасеа ригригеа (Ь.) МоепсЬ.
Содержание:
-не менее 0,1% суммы кафтаровой кислоты
(С13Н1209; М.м. 312,2) и цикориевой кислоты (С22Н18012;
М.м. 474,3) (в пересчете на сухое сырье);
-или не менее 2,1 % суммы производных окси-
коричных кислот в пересчете на цикориевую кислоту
(^22Н1в^12’ М.м. 474,3) (в пересчете на сухое сырье).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, С.
Вторая идентификация: А, В, О.
® А. Внешние признаки (2.8.23). Травянистое
многолетнее растение высотой (60—150) см, реже до
180 см. Стебель от зеленого до красного цвета, прямой
1352
Гэсударственная фармакопея Республики Беларусь
и слабо разветвленный. Листья очередные, от овальной
до овально-ланцетовидной формы, неравнокрупнозуб¬
чатые, морщинистые на обеих сторонах, темно-зеленые
с выступающими светло-зелеными жилками; листовая
пластинка толстая и блестящая. Покровные прицвет¬
ники крупной головки собраны в 2—3 ряда. Плотное
цветоложе слегка выпуклое. Каждый из наружных фи¬
олетовых язычковых цветков ((4—6) см) и внутренних
фиолетово-розовых трубчатых цветков присоединен
к красноватому заостренному жесткому прицветнику,
длина которого превышает длину трубчатых цветков.
Чашечка редуцирована до очень маленького венчика,
чашелистик которого имеет длину до 1 мм.
® В. Микроскопия (2.8.23). Исследуют измель¬
ченное сырье (355) (2.9.12). Цвет зеленый. Просма¬
тривают под микроскопом с использованием раствора
хлоралгидрата Р. В порошке обнаруживаются следу¬
ющие диагностические признаки: беловато-зеленые
группы волокон длиной (150—200) мкм и диаметром
(10—15) мкм, иногда с черными отложениями; фраг¬
менты листьев при просматривании с поверхности с
устьицами аномоцитного или анизоцитного типа (2.8.3)
(диной около (25—40) мкм); простые покровные воло¬
ски или их фрагменты, состоящие в основном из 3 или
4 толстостенных клеток, конечная из которых значи¬
тельно длиннее остальных; фрагменты листьев с ро¬
зеткоподобно ориентированными клетками эпидерми¬
са вокруг основания покровных волосков; простые же¬
лезистые волоски, состоящие из клеток с очень тонки¬
ми стенками; ямчатые клетки паренхимы сердцевины
стебля, а также вытянутые ямчатые клетки мезокарпа
семянок; фрагменты паренхимы семян с капельками
масла; фрагменты эпидермиса язычковых цветков, со¬
стоящих из сосочковидных клеток от красного до фио¬
летового цвета; шаровидные зерна пыльцы диаметром
(30—40) мкм с экзиной, покрытой шипами.
@ С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. К 1,0 г измельченного
сырья (355) (2.9.12) прибавляют 10 мл метанола Р и
обрабатывают ультразвуком в течение 5 мин. Центри¬
фугируют и собирают надосадочную жидкость.
Раствор сравнения. 0,5 мг кофейной кислоты Р
и 0,5 мг хлорогеновой кислоты Р растворяют в 5,0 мл
метанола Р.
Пластинка: ТСХ пластинка со слоем силикаге¬
ля Р ((5—40) мкм) [или ТСХ пластинка со слоем сили¬
кагеля Р ((2—10) мкм)].
Подвижная фаза: кислота муравьиная безво¬
дная Р — вода Р — метилэтилкетон Р — этилаце-
тат Р (3:3:9:15, об/об/об/об).
Наносимый объем пробы. 25 мкл [или 5 мкл] ис¬
пытуемого раствора и 10 мкл [или 2 мкл] раствора
сравнения в виде полос.
Фронт подвижной фазы: не менее 15 см [или
5 см] от линии старта.
Высушивание: в потоке холодного воздуха в те¬
чение 10 мин, затем при температуре 100 °С в тече¬
ние 2 мин.
Проявление: горячую пластинку опрыскивают
раствором 5 г/л аминоэтилового эфира дифенилбор-
ной кислоты Р в этилацетате Р. Через 30 мин пла¬
стинку просматривают в ультрафиолетовом свете при
длине волны 365 нм.
Результаты: ниже приведена последователь¬
ность зон хроматограмм раствора сравнения и ис¬
пытуемого раствора. На хроматограмме испытуемого
раствора могут обнаруживаться другие слабые флуо¬
ресцирующие зоны синего цвета.
Верх хроматографической пластинки
Кофейная кислота:
флуоресцирующая зона
ярко голубого цвета
Интенсивная
флуоресцирующая зона
красного цвета
Флуоресцирующая зона
голубого цвета
Хлорогеновая кислота:
флуоресцирующая зона
голубого цвета
Флуоресцирующая зона
голубого цвета
Слабая флуоресцирующая
зона желто-оранжевого
цвета
Раствор сравнения
Испытуемый раствор
О. Просматривают хроматограммы, получен¬
ные в «Количественном определении». На хромато¬
грамме испытуемого раствора присутствует основной
пик, соответствующий цикориевой кислоте, и мень¬
ший пик, соответствующий кафтаровой кислоте. Пики
кофейной и хлорогеновой кислоты незначительны и
могут отсутствовать.
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Допустимые примеси (2.8.2). Несырьевые ча¬
сти растения: стебли — не более 55 %. Органические
примеси: не более 2,5 %. Минеральные примеси: не
более 1 %.
Потеря в массе при высушивании (2.2.32). Не
более 13,0%. 2,000 г измельченного сырья (2000)
(2.9.12) сушат при температуре 105 °С.
® Общая зола (2.4.16). Не более 12,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
® Определение содержания суммы кафтаро¬
вой кислоты и цикориевой кислоты. Жидкостная
хроматография (2.2.29).
Испытуемый раствор. 0,500 г измельченного
сырья (355) (2.9.12) помещают в мерную колбу вме¬
стимостью 100 мл, прибавляют 80 мл спирта (70 %,
об/об) Р, обрабатывают ультразвуком в течение
15 мин и доводят до объема 100,0 мл этим же раст¬
ворителем. Перемешивают и выдерживают в течение
нескольких минут для оседания видимых частиц.
Раствор сравнения. 10,0 мг ФСО хлорогено¬
вой кислоты и 10,0 мг кофейной кислоты Р раст¬
воряют в спирте (70 %, об/об) Р, обрабатывают
ультразвуком в течение 15 мин и доводят до объ¬
ема 10,0 мл этим же растворителем. 4,0 мл по¬
лученного раствора разводят спиртом (70 %,
об/об) Рдо объема 100,0 мл.
Условия хроматографирования:
- колонка длиной 0,25 м и внутренним диаме¬
тром 4,6 мм, заполненная силикагелем октадецил-
силильным для хроматографии Р с размером частиц
5 мкм;
- температура: 35 °С;
- подвижная фаза:
- подвижная фаза А: кислота фосфорная Р —
вода Р (1:999, об/об);
- подвижная фаза В: ацетонитрил Р.
Эхинацеи пурпурной трава
1353
1. Кафтаровая кислота. 2. Хлорогеновая кислота. 3. Цикориевая кислота.
Рисунок 1823.-1. Хроматограмма для количественного определения кафтаровой кислоты
и цикориевой кислоты в траве эхинацеи пурпурной
Время (мин)
Подвижная фаза А
(%, об/об)
Подвижная фаза В
(%, об/об)
0
90
10
0—13
со
о
4
"ч1
со
10—>22
13—14
78 —> 60
22 —> 40
14—20
60
40
- скорость подвижной фазы: 1,5 мл/мин;
- спектрофотометрический детектор, длина
волны 330 нм;
- объем вводимой пробы: 10 мкп.
Относительные времена удерживания (по от¬
ношению к хлорогеновой кислоте, время удержива¬
ния около 7 мин): кафтаровая кислота — около 0,8;
кофейная кислота — около 1,5; цинарин — около 1,6;
эхинакозид — около 1,7; цикориевая кислота —
около 2,3.
Пригодность хроматографической системы.
раствор сравнения:
- разрешение: не менее 5 между пиками кофей¬
ной кислоты и хлорогеновой кислоты.
Отмечают пики кофейной кислоты и хлороге¬
новой кислоты, используя хроматограмму раствора
сравнения. Отмечают пики кафтаровой кислоты и ци¬
кориевой кислоты, используя хроматограмму, приве¬
денную на рисунке 1823.-1.
Содержание кафтаровой и цикориевой кислоты
в процентах рассчитывают по формулам 1 и 2 соот¬
ветственно:
5- С2 -100*0,881 /4Ч
~— (1).
$2*0,
5Э-0,-100-0,695
32-С,
где:
51 — площадь пика кафтаровой кислоты на хро¬
матограмме испытуемого раствора;
$2 — площадь пика хлорогеновой кислоты на
хроматограмме раствора сравнения;
53 — площадь пика цикориевой кислоты на хро¬
матограмме испытуемого раствора;
С1 — концентрация испытуемого раствора, мг/мл;
С2 — концентрация хлорогеновой кислоты в рас¬
творе сравнения;
0,695 — коэффициент пересчета, учитывающий
разную чувствительность детектора;
0,881 — коэффициент пересчета между кафта¬
ровой кислотой и хлорогеновой кислотой.
Определение содержания суммы произво¬
дных оксикоричных кислот в пересчете на цико-
риевую кислоту. 1,000 г измельченного сырья (500)
(2.9.12) помещают в колбу со шлифом вместимостью
25 мл, прибавляют 0,1 г кислоты щавелевой Р, вно¬
сят магнитный стержень с полимерным покрытием
и прибавляют 10,0 мл спирта (95 %, об/об) Р. Колбу
с содержимым закрывают пробкой и взвешивают с
точностью до 0,01 г. Нагревают с обратным холодиль¬
ником при слабом кипении в течение 45 мин при по¬
стоянном перемешивании. Охлаждают, закрывают
пробкой, взвешивают и, при необходимости, доводят
спиртом (95 %, об/об) Р до первоначальной массы и
перемешивают. Содержимое колбы центрифугируют
при 3000 об/мин в течение 3 мин. Собирают надоса-
дочную жидкость.
На линию старта листа фильтровальной бумаги
размером 15^15 см наносят двумя полосами длиной
3 см по 20 мкл полученного раствора. После высыха¬
ния пятен отмечают их границы графитовым каран¬
дашом и хроматографируют восходящим способом
(2.2.26), в качестве подвижной фазы используют хло¬
роформ Р. Когда фронт подвижной фазы пройдет 5
см от линии старта, бумагу вынимают из камеры и вы¬
сушивают на воздухе до удаления запаха хлорофор¬
ма. Отмеченные участки стартовых пятен вырезают,
помещают в колбы вместимостью 25 мл, приливают
в каждую по 10,0 мл 0,1 М раствора кислоты хлори¬
стоводородной и встряхивают в течение 30 мин.
Измеряют оптическую плотность (2.2.25) полу¬
ченных растворов при 328 нм, используя 0,1 М раст¬
вор кислоты хлористоводородной в качестве ком¬
пенсационного раствора. Используют среднее значе¬
ние оптической плотности полученных растворов.
Содержание суммы производных оксикоричных
кислот в пересчете на цикориевую кислоту в процен¬
тах рассчитывают по формуле:
А•5000
л?-782’
где:
782 — удельный показатель поглощения цикори¬
евой кислоты;
А — среднее значение оптической плотности;
т — масса навески испытуемого сырья, г.
1354
Государственная фармакопея Республики Беларусь
Указатель общих фармакопейных статей
1355
УКАЗАТЕЛЬ ОБЩИХ ФАРМАКОПЕЙНЫХ СТАТЕЙ
Приведен указатель актуальных общих фармакопейных статей первого и второго томов Государственной
фармакопеи Республики Беларусь II (ГФ РБ II). Номер тома приводится после указания номера страницы.
Введение, 19-2
1. Общие сведения, 21-2
2.1.1. Каплемер, 33-1
2.1.2. Сравнительная таблица пористости
стеклянных фильтров, 33-1
2.1.3. Лампы с ультрафиолетовым излучением
для аналитических целей, 34-1
2.1.4. Сита, 34-1
2.1.5. Пробирки для сравнительных
испытаний, 35-1
2.1.6. Индикаторные трубки, 35-1
2.2.1. Определение прозрачности и степени
мутности жидкостей, 36-1
2.2.2. Определение степени окрашивания
жидкостей, 39-1
2.2.3. Потенциометрическое определение рН, 41-1
2.2.4. Зависимость между реакцией раствора,
приблизительным значением рН и цветом
индикаторов, 43-1
2.2.5. Относительная плотность, 45-1
2.2.6. Показатель преломления
(индекс рефракции), 47-1
2.2.7. Оптическое вращение, 47-1
2.2.8. Вязкость, 48-1
2.2.9. Метод капиллярной вискозиметрии, 49-1
2.2.10. Метод ротационной вискозиметрии, 51-1
2.2.11. Температурные пределы перегонки, 53-1
2.2.12. Температура кипения, 54-1
2.2.13. Определение воды методом отгонки, 54-1
2.2.14. Температура плавления — капиллярный
метод, 55-1
2.2.15. Температура плавления — открытый
капиллярный метод, 56-1
2.2.16. Температура плавления — метод
мгновенного плавления, 56-1
2.2.17. Температура каплепадения, 57-1
2.2.18. Температура затвердевания, 59-1
2.2.19. Амперометрическое титрование, 60-1
2.2.20. Потенциометрическое титрование, 33-2
2.2.21. Флуориметрия, 62-1
2.2.22. Атомно-эмиссионная спектрометрия, 62-1
2.2.23. Атомно-абсорбционная спектрометрия, 64-1
2.2.24. Абсорбционная спектрофотометрия в
инфракрасной области, 67-1
2.2.25. Абсорбционная спектрофотометрия в ультра¬
фиолетовой и видимой областях, 70-1
2.2.26. Бумажная хроматография, 76-1
2.2.27. Тонкослойная хроматография, 77-1
2.2.28. Газовая хроматография, 80-1
2.2.29. Жидкостная хроматография, 33-2
2.2.30. Эксклюзионная хроматография, 84-1
2.2.31. Электрофорез, 85-1
2.2.32. Потеря в массе при высушивании, 35-2
2.2.33. Спектрометрия ядерного магнитного
резонанса, 93-1
2.2.34. Термический анализ, 98-1
2.2.35. Осмоляльность, 101-1
2.2.36. Потенциометрическое определение
концентрации ионов с использованием
ионоселективных электродов, 103-1
2.2.37. Рентгенофлуоресцентная
спектрометрия, 104-1
2.2.38. Электропроводность, 105-1
2.2.39. Молекулярно-массовое распределение
декстранов, 106-1
2.2.40. Спектрофотометрия ближнего
инфракрасного диапазона, 109-1
2.2.41. Круговой дихроизм, 115-1
2.2.42. Плотность твердых тел, 116-1
2.2.43. Масс-спектрометрия, 117-1
2.2.44. Определение содержания общего
органического углерода в воде для
фармацевтического применения, 121-1
2.2.45. Сверхкритическая флюидная
хроматография, 123-1
2.2.46. Хроматографические методы
разделения, 123-1
2.2.47. Капиллярный электрофорез, 132-1
2.2.48. Рамановская спектрометрия, 140-1
2.2.49. Измерение вязкости на вискозиметре
с падающим шариком, 142-1
2.2.54. Изоэлектрическое фокусирование, 142-1
2.2.55. Пептидное картирование, 145-1
2.2.56. Анализ аминокислот, 150-1
2.2.57. Атомно-эмиссионная спектрометрия с
использованием индуктивно связанной
плазмы, 161-1
2.2.58. Масс-спектрометрия с использованием
индуктивно связанной плазмы, 164-1
2.2.59. Анализ гликанов в гликопротеинах, 166-1
2.2.60. Температура плавления —
инструментальный метод, 174-1
2.2.61. Определение характеристик
кристаллических твердых веществ с
помощью микрокалориметрии и калометрии
растворения, 36-2
2.2.64. Спектрометрия ядерного магнитного
резонанса для идентификации
пептидов, 39-2
2.2.65. Вольтаметрическое титрование, 40-2
2.2.66. Обнаружение и измерение
радиоактивности, 40-2
#2.2.90. Титриметрические методы анализа, 48-2
2.3.1. Реакции подлинности (идентификации) на
ионы и функциональные группы, 50-2
2.3.2. Идентификация жирных масел методом
тонкослойной хроматографии, 181-1
2.3.3. Идентификация фенотиазинов методом
тонкослойной хроматографии, 182-1
2.3.4. Определение запаха, 182-1
2.4.1. Аммония соли, 183-1
2.4.2. Мышьяк, 183-1
2.4.3. Кальций, 184-1
2.4.4. Хлориды, 185-1
2.4.5. Фториды, 185-1
2.4.6. Магний, 185-1
2.4.7. Магний и щелочноземельные
металлы, 186-1
2.4.8. Тяжелые металлы, 186-1
2.4.9. Железо, 191-1
2.4.10. Свинец в сахарах, 191-1
2.4.11. Фосфаты, 191-1
2.4.12. Калий, 191-1
2.4.13. Сульфаты, 192-1
1356
Государственная фармакопея Республики Беларусь
2.4.14. Сульфатная зола, 192-1
2.4.15. Никель в полиолах, 192-1
2.4.16. Общая зола, 192-1
2.4.17. Алюминий, 193-1
2.4.18. Свободный формальдегид, 193-1
2.4.19. Щелочные примеси в жирных маслах, 194-1
2.4.20. Определение остаточных количеств
металлических катализаторов или
металлсодержащих реактивов, 55-2
2.4.21. Посторонние масла в жирных маслах
методом тонкослойной хроматографии, 194-1
2.4.22. Посторонние жирные кислоты в маслах
методом газовой хроматографии, 195-1
2.4.23. Стерины в жирных маслах, 197-1
2.4.24. Идентификация и контроль содержания
остаточных растворителей, 200-1
2.4.25. Остаточные количества этиленоксида и
диоксана, 204-1
2.4.26. А/,Л/-Диметиланилин, 207-1
2.4.27. Тяжелые металлы в лекарственном
растительном сырье и продуктах из
лекарственного растительного сырья, 59-2
2.4.28. 2-Этилгексановая кислота, 209-1
2.4.29. Состав жирных кислот в маслах,
обогащенных омега-3 кислотами, 210-1
2.4.30. Этиленгликоль и диэтиленгликоль в
этоксилированных субстанциях, 212-1
2.4.31. Никель в гидрогенизированных
растительных маслах, 213-1
2.4.32. Общий холестерин в маслах, обогащенных
омега-3 кислотами, 213-1
2.5.1. Кислотное число, 61-2
2.5.2. Эфирное число, 215-1
2.5.3. Гидроксильное число, 215-1
2.5.4. Йодное число, 216-1
2.5.5. Перекисное (пероксидное) число, 217-1
2.5.6. Число омыления, 218-1
2.5.7. Неомыляемые вещества, 219-1
2.5.8. Определение аминного азота в
соединениях, которые содержат первичную
ароматическую аминогруппу, 219-1
2.5.9. Определение азота после минерализации
серной кислотой, 220-1
2.5.10. Метод сжигания в колбе
с кислородом, 220-1
2.5.11. Комплексометрическое титрование, 221-1
2.5.12. Вода: полумикрометод, 62-2
2.5.13. Алюминий в адсорбированных
вакцинах, 224-1
2.5.14. Кальций в адсорбированных вакцинах, 224-1
2.5.15. Фенол в иммуносыворотках и вакцинах,
224-1
2.5.16. Белок в полисахаридных вакцинах, 224-1
2.5.17. Нуклеиновые кислоты в полисахаридных
вакцинах, 225-1
2.5.18. Фосфор в полисахаридных вакцинах, 225-1
2.5.19. О-Ацетил в полисахаридных
вакцинах, 226-1
2.5.20. Гексозамины в полисахаридных
вакцинах, 226-1
2.5.21. Метилпентозы в полисахаридных
вакцинах, 227-1
2.5.22. Уроновые кислоты в полисахаридных
вакцинах, 227-1
2.5.23. Сиаловая кислота в полисахаридных
вакцинах, 228-1
2.5.24. Углерода диоксид в газах, 228-1
2.5.25. Углерода монооксид в газах, 229-1
2.5.26. Азота монооксид и азота диоксид
в газах, 230-1
2.5.27. Кислород в газах, 231-1
2.5.28. Вода в газах, 231-1
2.5.29. Серы диоксид, 231-1
2.5.30. Окисляющие вещества, 232-1
2.5.31. Рибоза в полисахаридных вакцинах, 232-1
2.5.32. Вода: микроопределение, 233-1
2.5.33. Общий белок, 233-1
2.5.34. Уксусная кислота в синтетических
пептидах, 239-1
2.5.35. Азота закись в газах, 239-1
2.5.36. Анизидиновое число, 240-1
2.5.37. Метил-, этил- и изопропилметансульфонат
в метансульфоновой кислоте, 240-1
2.5.38. Метил-, этил- и изопропилметансульфонат
в фармацевтических субстанциях, 241-1
2.5.39. Метансульфонилхлорид в
метансульфоновой кислоте, 243-1
2.5.40. Метил-, этил- и изопропилтолуолсульфонат
в фармацевтических субстанциях, 63-2
2.5.41. Метил-, этил- и изопропилбензолсульфонат
в фармацевтических субстанциях, 64-2
#2.5.50. Титрование в неводных
растворителях, 244-1
2.6.1. Стерильность, 252-1
2.6.2. Микобактерии, 257-1
2.6.7. Микоплазмы, 257-1
2.6.8. Пирогенность, 265-1
2.6.9. Аномальная токсичность, 266-1
2.6.10. Гистамин, 267-1
2.6.11. Депрессорные вещества, 268-1
2.6.12. Микробиологические испытания
нестерильной продукции: общее количество
жизнеспособных аэробов, 269-1
2.6.13. Микробиологические испытания нестерильной
продукции: испытания на наличие
специфических микроорганизмов, 276-1
2.6.14. Бактериальные эндотоксины, 288-1
2.6.15. Активатор прекалликреина, 294-1
2.6.16. Испытания на посторонние агенты в
вирусных вакцинах для медицинского
применения, 295-1
2.6.17. Испытание на антикомплементарную
активность иммуноглобулина, 298-1
2.6.18. Испытание живых вирусных вакцин на
нейровирулентность, 301-1
2.6.19. Испытание пероральной вакцины
полиомиелита на нейровирулентность, 301-1
2.6.20. Анти-А и анти-В гемагглютинины
(непрямой метод), 303-1
2.6.21. Методы амплификации нуклеиновых
кислот, 303-1
2.6.22. Активированные факторы свертывания
крови, 311-1
2.6.26. Испытание на анти-Э антитела в
иммуноглобулине человека для
внутривенного введения, 311-1
2.6.27. Микробиологический контроль клеточных
продуктов, 312-1
2.6.30. Испытание на активацию моноцитов, 314-1
2.6.31. Микробиологические испытания
лекарственных средств растительного
происхождения для внутреннего
применения и экстрактов, использующихся
для их приготовления, 65-2
2.7.1. Иммунохимические методы, 325-1
Указатель общих фармакопейных статей
1357
2.7.2.
2.7.4.
2.7.5.
2.7.6.
2.7.7.
2.7.8.
2.7.9.
2.7.10.
2.7.11.
2.7.12.
2.7.13.
2.7.14.
2.7.15.
2.7.16.
2.7.17.
2.7.18.
2.7.19.
2.7.20.
2.7.21.
2.7.22.
2.7.23.
2.7.24.
2.7.25.
2.7.27.
2.7.28.
2.7.29.
2.7.30.
2.7.31.
2.7.32.
#2.7.50,
2.8.1.
Количественное определение антибиотиков
микробиологическим методом, 327-1
Количественное определение фактора
свертывания крови VIII, 342-1
Количественное определение гепарина, 344-1
Количестенное определение адсорбирован¬
ной дифтерийной вакцины, 346-1
Количественное определение вакцины
коклюша, 353-1
Количественное определение
адсорбированной столбнячной
вакцины, 354-1
Определение функционального состояния
Рс-фрагмента иммуноглобулина, 361-1
Количественное определение фактора
свертывания крови человека VII, 363-1
Количественное определение фактора
свертывания крови человека IX, 364-1
Количественное определение гепарина
в концентратах факторов свертывания
крови, 365-1
Количественное определение анти-й-
иммуноглобулина человека, 366-1
Количественное определение антигенной
(иммуногенной) активности вакцины
гепатита А, 369-1
Количественное определение вакцины
гепатита В (рДНК), 370-1
Количественное определение вакцины
коклюша (бесклеточной), 371-1
Количественное определение
антитромбина III человека, 372-1
Количественное определение фактора
свертывания крови II человека, 373-1
Количественное определение фактора
свертывания крови X человека, 374-1
Количественное определение
инактивированной вакцины полиомиелита
т VIуо, 375-1
Количественное определение фактора
вилебранда человека, 376-1
Количественное определение фактора
свертывания крови XI человека, 378-1
Подсчет клеток СР34/С045+ в
гемопоэтических продуктах, 379-1
Проточная цитометрия, 381-1
Определение содержания ингибитора
плазмина человека, 384-1
Значение флоккуляции (/_0 токсинов
и анатоксинов дифтерии и столбняка
(проба Рамона), 385-1
Определение количества гемопоэтических
клеток-предшественников человека по
колониеобразующим клеткам, 386-1
Определение количества
и жизнеспособности ядросодержащих
клеток, 388-1
Количественное определение
человеческого белка С, 391-1
Количественное определение
человеческого белка 5, 392-1
Количественное определение ингибитора
а-1-протеиназы человека, 393-1
Определение биологической активности
инсулина, 394-1
Зола, нерастворимая в хлористоводородной
кислоте, 396-1
2.8.2. Примеси, 69-2
2.8.3. Устьичный коэффициент, 397-1
2.8.4. Коэффициент набухания, 397-1
2.8.5. Определение воды в эфирных маслах, 397-1
2.8.6. Посторонние эфиры
в эфирных маслах, 398-1
2.8.7. Жирные и минеральные масла в эфирных
маслах, 398-1
2.8.8. Запах и вкус эфирных масел, 398-1
2.8.9. Остаток после выпаривания эфирного
масла, 398-1
2.8.10. Растворимость эфирных масел
в спирте, 398-1
2.8.11. Определение 1,8-цинеола в эфирных
маслах, 399-1
2.8.12. Определение эфирного масла
в лекарственном растительном сырье, 399-1
2.8.13. Остаточное количество пестицидов, 403-1
2.8.14. Определение дубильных веществ, 405-1
2.8.15. Определение показателя горечи, 405-1
2.8.16. Сухой остаток экстрактов, 406-1
2.8.17. Потеря в массе при высушивании
экстракта, 406-1
2.8.18. Определение афлатоксина В1
в лекарственном растительном сырье, 407-1
2.8.20. Лекарственное растительное сырье: отбор
проб, 409-1
2.8.21. Испытание лекарственного
растительного сырья на наличие
аристолохиевых кислот, 69-2
2.8.22. Определение охратоксина А в
лекарственном растительном сырье, 410-1
2.8.23. Макроскопический и# микроскопический
анализ лекарственного растительного
сырья, 412-1
2.9.1. Распадаемость таблеток и капсул, 417-1
2.9.2. Распадаемость суппозиториев
и пессариев, 419-1
2.9.3. Тест «Растворение» для твердых
дозированных форм, 420-1
2.9.4. Тест «Растворение» для трансдермальных
пластырей, 428-1
2.9.5. Однородность массы
для единицы дозированного лекарственного
средства, 430-1
2.9.6. Однородность содержания действующего
вещества в единице дозированного
лекарственного средства, 431-1
2.9.7. Прочность таблеток без оболочки на
истирание, 432-1
2.9.8. Прочность таблеток на сжатие, 433-1
2.9.9. Измерение консистенции методом
пенетрометрии, 433-1
2.9.10. Содержание этанола, 71-2
2.9.11. Испытание на содержание метанола и
2-пропанола, 74-2
2.9.12. Ситовой анализ, 440-1
2.9.14. Удельная площадь поверхности, 441-1
2.9.16. Сыпучесть, 443-1
2.9.17. Определение извлекаемого объема
парентеральных лекарственных
средств, 444-1
2.9.18. Лекарственные средства для ингаляций:
аэродинамическое испытание мелких
частиц, 445-1
2.9.19. Загрязнение механическими включениями:
невидимые частицы, 459-1
1358
Гэсударственная фармакопея Республики Беларусь
2.9.20.
2.9.22.
2.9.23.
2.9.25.
2.9.26.
2.9.27.
2.9.29.
2.9.31.
2.9.32.
2.9.33.
2.9.34.
2.9.35.
2.9.36.
2.9.37.
2.9.38.
2.9.39.
2.9.40.
2.9.41.
2.9.42.
2.9.43.
2.9.44.
2.9.45.
2.9.47.
3.1.
3.1.1.
3.1.1.1.
3.1.1.2.
3.1.3.
3.1.4.
3.1.5.
Загрязнение механическими включениями:
видимые частицы, 462-1
Определение времени размягчения
липофильных суппозиториев, 469-1
Определение плотности твердых частиц
при помощи газового пикнометра, 470-1
Высвобождение действующего вещества из
лекарственных жевательных резинок, 76-2
Определение удельной площади поверхности
методом газовой адсорбции, 473-1
Однородность массы одной дозы,
высвобожденной из многодозового
контейнера, 477-1
Характеристическое растворение, 477-1
Определение размера частиц методом
дифракции лазерного излучения, 479-1
Пористость и определение размера
пор с использованием ртутной
порозиметрии, 484-1
Определение параметров кристаллических
и частично кристаллических твердых
веществ при помощи дифракции
рентгеновского излучения на порошке, 487-1
Насыпная плотность и плотность после
усадки, 494-1
Степень измельчения порошков, 497-1
Текучесть порошков, 497-1
Оптическая микроскопия, 501-1
Определение размера частиц методом
аналитического просеивания, 504-1
Взаимодействия «вода - твердое вещество»:
построение изотерм сорбции-десорбции и
определение активности воды, 80-2
Однородность дозированных единиц, 508-1
Истираемость гранул и сфероидов, 512-1
Тест «Растворение» для липофильных
твердых дозированных форм, 513-1
Наблюдаемое растворение, 515-1
Лекарственные средства для распыления:
характеристика, 515-1
Смачиваемость пористых твердых
материалов, включая порошки, 520-1
Подтверждение однородности
дозированных единиц с использованием
большого количества образцов, 84-2
Материалы, используемые для
производства контейнеров, 525-1
Материалы, используемые
для производства контейнеров
для человеческой крови и компонентов
крови, 525-1
Материалы на основе
пластифицированного поливинилхлорида,
используемые для производства
контейнеров для человеческой крови и ее
компонентов, 525-1
Материалы на основе
пластифицированного поливинилхлорида
для трубок, используемых в комплектах
для переливания крови и компонентов
крови, 530-1
Полиолефины, 533-1
Полиэтилен без добавок для контейнеров
для парентеральных и офтальмологических
лекарственных средств, 538-1
Полиэтилен с добавками для контейнеров
для парентеральных и офтальмологических
лекарственных средств, 538-1
3.1.6. Полипропилен для контейнеров
и укупорочных материалов для
парентеральных и офтальмологических
лекарственных средств, 545-1
3.1.7. Полиэтиленвинилацетат для контейнеров
и трубок для лекарственных средств для
парентерального питания, 550-1
3.1.8. Силиконовое масло, используемое в
качестве смазывающей добавки, 553-1
3.1.9. Силиконовый эластомер для укупорочных
средств и трубок, 554-1
3.1.10. Материалы на основе
непластифицированного поливинилхлорида
для контейнеров для неинъекционных
водных растворов, 556-1
3.1.11. Материалы на основе
непластифицированного поливинилхлорида
для контейнеров для твердых
лекарственных форм для перорального
применения, 559-1
3.1.13. Добавки к пластмассе, 561-1
3.1.14. Материалы на основе
пластифицированного поливинилхлорида
для контейнеров для водных растворов для
внутривенного применения, 565-1
3.1.15. Полиэтилентерефталат для контейнеров
для лекарственных средств для
непарентерального применения, 568-1
3.2. Контейнеры, 571-1
3.2.1. Стеклянные контейнеры для
фармацевтического использования, 571-1
3.2.2. Пластмассовые контейнеры и укупорочные
средства для фармацевтического
использования, 579-1
3.2.2.1. Пластмассовые контейнеры для водных
растворов для инфузий, 580-1
3.2.3. Стерильные пластмассовые контейнеры для
человеческой крови и ее компонентов, 581-1
3.2.4. Пустые стерильные контейнеры
из пластифицированного
поливинилхлорида для человеческой крови
и ее компонентов, 584-1
3.2.5. Стерильные контейнеры из
пластифицированного поливинилхлорида
для человеческой крови, содержащие
раствор антикоагулянта, 585-1
3.2.6. Комплекты для переливания крови
и компонентов крови, 586-1
3.2.8. Стерильные одноразовые пластмассовые
шприцы, 588-1
3.2.9. Резиновые укупорочные средства для
контейнеров, предназначенных для
водных лекарственных средств для
парентерального применения, порошков
и лиофилизированных порошков, 590-1
4. Реактивы, 593-1
4.1. Реактивы, эталонные растворы, буферные
растворы, 593-1
4.1.1. Реактивы, 594-1, 87-2
4.1.2. Эталонные растворы для испытаний на
предельное содержание примесей, 737-1
4.1.3. Буферные растворы, 743-1, 93-2
4.2.1. Исходные стандартные вещества для
титрованных растворов, 750-1
4.2.2. Титрованные растворы, 751-1, 93-2
5.1.1. Методы приготовления стерильных
продуктов, 761-1
Указатель общих фармакопейных статей
1359
5.1.2. Биологические индикаторы
стерилизации, 764-1
5.1.3. Эффективность антимикробных
консервантов, 766-1
5.1.4. Микробиологическая чистота
нестерильных лекарственных средств
и фармацевтических субстанций, 768-1
5.1.5. Применение 70 концепции при стерилизации
паром водных растворов, 770-1
5.1.6. Альтернативные методы контроля
микробиологической чистоты, 771-1
5.1.7. Вирусная безопасность, 789-1
5.1.8. Микробиологическая чистота
лекарственных средств для внутреннего
применения на основе лекарственного
растительного сырья, 789-1
5.1.9. Руководство по применению испытания
на стерильность, 791-1
5.1.10. Руководство по применению испытания
на бактериальные эндотоксины, 792-1
5.2.1. Общепринятая терминология в статьях
на биологические продукты, 796-1
5.2.2. Стаи кур, не имеющих конкретных
патогенов и используемых
для производства и контроля
качества вакцин, 798-1
5.2.3. Клетки-продуценты для производства
вакцин для медицинского применения, 802-1
5.2.8. Снижение риска передачи возбудителей
губчатой энцефалопатии животных при
применении медицинских лекарственных
средств, 807-1
5.3. Статистический анализ результатов
биологических испытаний и тестов, 825-1
#5.3.1. Статистический анализ результатов
химического эксперимента, 868-1
#5.3.2. Валидация аналитических методик
и испытаний, 895-1
5.4. Остаточные количества органических
растворителей, 95-2
5.5. алкоголеметрические таблицы, 918-1
5.6. Количественное определение
интерферонов, 934-1
5.7. Таблица физических характеристик
радионуклидов, упоминаемых
в фармакопее, 937-1
5.9. Полиморфизм, 943-1
5.10. Контроль примесей в субстанциях для
фармацевтического использования, 944-1
5.11. Раздел «Описание (свойства)» в частных
статьях, 949-1
5.12. Стандартные образцы, 103-2
5.14. Лекарственные средства для генной тера¬
пии для медицинского применения, 955-1
5.15. Функционально-обусловленные
характеристики вспомогательных веществ,
108-2
5.16. Кристалличность, 980-1
5.17.1. Рекомендации по проведению теста
«Растворение», 982-1
5.20. Остаточные количества металлических
катализаторов или металлсодержащих
реактивов, 110-2
#5.50. Критерии чистоты пищевых красителей, 116-2
#6. Экстемпоральные лекарственные
средства, 987-1
#6.1.1. Жидкие лекарственные средства, 987-1
#6.1.2. Твердые лекарственные средства, 1006-1
#6.1.3. Мягкие лекарственные средства, 1010-1
#6.1.4. Режимы стерилизации экстемпоральных
лекарственных средств, 1012-1
#6.2. Экспресс-анализ экстемпоральных
лекарственных средств, 1013-1
#6.3. Оценка качества экстемпоральных
лекарственных средств, 1047-1
#6.3.1. Нормы отклонений, допустимые при
изготовлении лекарственных средств (в том
числе гомеопатических) в аптеках, 1048-1
#6.3.2. Нормы отклонений, допустимые при
фасовке в аптеках лекарственных средств
промышленного производства, 1050-1
Вакцины для медицинского применения, 1053-1
Иммунные сыворотки животного происхождения для
медицинского применения, 1058-1
Лекарственное растительное сырье, 1061-1
#Лекарственное растительное сырье цельное или
измельченное фасованное, 1063-1
Лекарственные средства на основе аллергенов,
1064-1
Моноклональные антитела для медицинского
применения, 1068-1
#Настои, отвары и чаи, 1072-1
Продукты технологии рекомбинантной ДНК, 1074-1
Продукты ферментации, 1077-1
Продукты, которые могут быть переносчиками
губчатой энцефалопатии животного происхождения,
1079-1
Радиоактивные фармацевтические препараты, 147-2
Растительные жирные масла, 1088-1
Растительные чаи, 1090-1
#Сборы, 1091-1
#Регистрационные требования и правила проведения
исследований биодоступности и биоэквивалентности
генерических лекарственных средств, 1092-1
Субстанции для фармацевтического использования,
1130-1
Экстракты, 1133-1
Эфирные масла, 1136-1
Основные термины и определения, 1139-1
Глазные лекарственные средства, 1140-1
Гранулы, 1143-1
#Драже, 1145-1
Жевательные резинки лекарственные, 1145-1
Жидкие лекарственные средства для внутреннего
применения, 1146-1
Жидкие лекарственные средства для наружного
применения, 1150-1
Капсулы, 1151-1
Лекарственные средства для вагинального
применения, 1153-1
Лекарственные средства для ингаляций, 1156-1
Лекарственные средства для орошения, 1162-1
Лекарственные средства для парентерального
применения, 153-2
Лекарственные средства для ректального
применения, 1166-1
1360
Государственная фармакопея Республики Беларусь
Лекарственные средства для слизистой оболочки
полости рта, 1169-1
Лекарственные средства, находящиеся под
давлением, 1173-1
Мягкие лекарственные средства для наружного
применения, 1174-1
Назальные лекарственные средства, 1178-1
Палочки, 1180-1
Пены медицинские, 1181-1
Порошки для внутреннего применения, 1182-1
Порошки для наружного применения, 1183-1
Таблетки, 1184-1
Тампоны медицинские, 1188-1
Трансдермальные пластыри, 1188-1
Ушные лекарственные средства, 1189-1
Гомеопатические лекарственные средства, 1193-1
Методы приготовления гомеопатических базисных
препаратов и потенцирование, 1197-1
Матричные настойки для гомеопатических
лекарственных средств, 1211-1
Лекарственное растительное сырье для
гомеопатических лекарственных средств, 1213-1
Алфавитный указатель частных фармакопейных статей
1361
АЛФАВИТНЫЙ УКАЗАТЕЛЬ ЧАСТНЫХ ФАРМАКОПЕЙНЫХ СТАТЕЙ
НА СУБСТАНЦИИ ДЛЯ ФАРМАЦЕВТИЧЕСКОГО ИСПОЛЬЗОВАНИЯ
Аденозин, 157
Адипиновая кислота, 159
Адреналина тартрат, 160
Азитромицин, 162
Азота закись, 165
Азотная кислота, 166
Албендазол, 167
Аллантоин, 168
Алюминия оксид гидратированный, 169
Алюминия фосфат гидратированный, 170
Алюминия хлорид гексагидрат, 171
Амантадина гидрохлорид, 172
Амброксола гидрохлорид, 174
Амикацин, 176
Амикацина сульфат, 178
Аминалон, 182
Аминокапроновая кислота, 183
Амиодарона гидрохлорид, 184
Амитриптилина гидрохлорид, 187
Амлодипина бесилат, 189
Аммиака раствор концентрированный, 191
Аммония хлорид, 192
Амоксициллин натрия, 193
Амоксициллин тригидрат, 196
Ампициллин натрия, 199
Ампициллин тригидрат, 202
Амфотерицин В, 205
Анастрозол, 208
Анисовое масло, 210
Апельсиновое масло, 212
Аргинин, 213
Аргинина аспартат, 215
Аргинина гидрохлорид, 216
Артикаина гидрохлорид, 219
Аскорбиновая кислота, 221
Аспарагиновая кислота, 223
Аспартам, 224
Атенолол, 226
Аторвастатин кальция тригидрат, 228
Атропина сульфат, 231
Ацетилсалициловая кислота, 233
Ацетил цистеин, 235
Ацетон, 237
Ацикловир, 238
Бария сульфат, 241
Беклометазона дипропионат безводный, 242
Беклометазона дипропионат моногидрат, 245
Бендазола гидрохлорид, 247
Бензалкония хлорид, 248
Бензилбензоат, 250
Бензиловый спирт, 251
Бензилпенициллин натрия, 253
Бензойная кислота, 256
Бензокаин, 256
Бензэтония хлорид, 258
Бентонит, 259
Бетагистина дигидрохлорид, 260
Бетагистина мезилат, 261
Бетаксолола гидрохлорид, 263
Бетаметазона валериат, 265
Бетаметазона дипропионат, 267
Бисакодил, 270
Бисопролола фумарат, 272
Бифоназол, 275
Борная кислота, 277
Бромгексина гидрохлорид, 277
Бутилгидроксианизол, 279
Вазелин, 280
Валсартан, 281
Валин, 283
Ванилин, 285
Ванкомицина гидрохлорид, 286
Варфарин натрия, 288
Варфарина натрия клатрат, 290
Верапамила гидрохлорид, 292
Винная кислота, 295
Винорелбина тартрат, 296
Винпоцетин, 299
Висмута нитрат основной, тяжелый, 301
Висмута субгаллат, 302
Вода высокоочищенная, 303
Вода для инъекций, 305
Вода очищенная, 309
Водорода пероксида 3 % раствор, 311
Водорода пероксида 30 % раствор, 312
Воск пчелиный белый, 313
Воск пчелиный желтый, 313
Галактоза, 314
Галоперидол, 315
Гвайфенезин, 317
Гвоздичное масло, 319
Гексаметилентетрамин, 320
Гентамицина сульфат, 321
Гепарин натрия, 323
Гидрокортизон, 326
Гидрокортизона ацетат, 330
Гидроксикарбамид, 332
Гидроксипропилцеллюлоза, 334
Гидрохлортиазид, 336
Гиосциамина сульфат, 338
Гиосцина гидробромид,340
Гипромеллоза, 342
Гистамина дигидрохлорид, 344
Гистидин, 345
Гистидина гидрохлорид моногидрат, 347
Глибенкламид, 348
Гпикпазид, 351
Глимепирид, 353
Глицерин, 355
Глицерин 85 %, 358
Глицерина моностеарат 40-55, 360
Глицин, 361
Глутаминовая кислота, 364
Глюкоза безводная, 365
Глюкоза моногидрат, 367
Глюкозамина гидрохлорид, 369
Глюкозамина сульфат натрия хлорид, 371
Деготь березовый, 373
Дексаметазона натрия фосфат, 374
Декспантенол, 377
Декстран 40 для инъекций, 379
Декстран 60 для инъекций, 380
1362
Гэсударственная фармакопея Республики Беларусь
Декстран 70 для инъекций, 382
Декстрин, 383
Декстрометорфана гидробромид, 384
Диазепам, 386
Дигоксин, 388
Дикалия фосфат, 390
Диклофенак натрия, 391
Диметилсульфоксид, 394
Динатрия фосфат дигидрат, 395
Динатрия фосфат додекагидрат, 396
Динатрия эдетат, 396
Дифенгидрамина гидрохлорид, 398
Диэтаноламин, 399
Доксиламина гидросукцинат, 400
Доксицикпина гиклат, 401
Доксорубицина гидрохлорид, 404
Докузат натрия, 405
Допамина гидрохлорид, 407
Доцетаксел безводный, 408
Доцетаксел тригидрат, 410
Дроперидол,413
Дротаверина гидрохлорид, 415
Дутастерид, 417
Желатин, 419
Железа (II) сульфат высушенный, 424
Железа сульфат гептагидрат, 425
Железа хлорид гексагидрат, 426
Жир твердый, 427
Зопиклон, 427
Ибупрофен, 429
Идоксуридин, 432
Изолейцин, 434
Изониазид, 436
Изопропилмиристат, 437
Изопропиловый спирт, 438
Изосорбида динитрат разведенный, 440
Изосорбида мононитрат разведенный, 442
Имипенем моногидрат, 445
Индапамид, 446
Индометацин, 449
Инсулин аспарт, 451
Инсулин бычий, 453
Инсулин двухфазовый для инъекций, 456
Инсулин изофан двухфазовый для инъекций, 456
Инсулин изофан для инъекций, 457
Инсулин лизпро, 457
Инсулин растворимый для инъекций, 460
Инсулин свиной, 460
Инсулин человеческий, 463
Инсулина инъекционные лекарственные средства, 466
Инсулина цинка суспензия (аморфная)
для инъекций, 469
Инсулина цинка суспензия (кристаллическая)
для инъекций, 470
Интерферона альфа-2 концентрированный
раствор, 470
Итраконазол,474
Ихтиол, 476
Йод, 477
Какао масло, 477
Калия ацетат, 478
Калия бромид, 478
Калия гидроаспартат гем и гидрат, 480
Калия гидроксид, 481
Калия дигидрофосфат, 481
Калия йодид, 482
Калия кпавуланат, 483
Калия клавуланат разведенный, 485
Калия метабисульфит, 487
Калия перманганат, 488
Калия сорбат, 488
Калия хлорид, 489
Калия цитрат, 490
Кальция глицерофосфат, 491
Кальция глюконат, 492
Кальция глюконат безводный, 493
Кальция глюконат для инъекций, 494
Кальция карбонат, 495
Кальция лактат безводный, 496
Кальция лактат моногидрат, 497
Кальция лактат пентагидрат, 498
Кальция лактат тригидрат, 499
Кальция стеарат, 499
Кальция фолинат, 501
Кальция хлорид гексагидрат, 504
Кальция хлорид дигидрат, 505
й-Камфора, 506
Камфора рацемическая, 507
Каолин тяжелый, 508
Капсулы твердые желатиновые, 509
Каптоприл,511
Карбоплатин, 514
Карведилол, 516
Картофельный крахмал, 518
Касторовое масло нерафинированное, 519
Касторовое масло рафинированное, 520
Кетоконазол, 521
Кетопрофен, 523
Кеторолак трометамин, 526
Кетотифена гидрофумарат, 528
Кислород газообразный, 530
Кладрибин, 532
Кларитромицин, 535
Кленбутерола гидрохлорид, 537
Клиндамицина фосфат, 539
Клозапин, 542
Клонидина гидрохлорид, 543
Клопидогреля гидросульфат, 545
Клотримазол, 548
Кодеин, 550
Кодеина фосфат гемигидрат, 552
Кодеина фосфат сесквигидрат, 555
Кокосовое масло рафинированное, 557
Коповидон, 558
Кофеин, 560
Кремния диоксид коллоидный безводный, 562
Кремния диоксид коллоидный гидратированный, 563
Кроскармеллоза натрия, 564
Кросповидон, 566
Ксантановая камедь, 568
Ксилометазолина гидрохлорид, 570
Кукурузное масло рафинированное, 572
Кукурузный крахмал, 572
Лавандовое масло, 573
Лактоза безводная, 575
Лактоза моногидрат, 576
Лактулоза, 578
Алфавитный указатель частных фармакопейных статей
1363
Ламотриджин, 580
Ланолин, 583
Ланолин водный, 587
Ланолин гидрогенизированный, 588
Лансопразол, 589
Леводопа, 591
Левоментол, 592
Левотироксин натрия, 594
Лейцин, 596
Лидокаина гидрохлорид, 599
Лизина гидрохлорид, 601
Лизиноприл дигидрат, 603
Лимонная кислота безводная, 605
Лимонная кислота моногидрат, 606
Лимонное масло, 607
Линкомицина гидрохлорид, 609
Ловастатин, 611
Лоперамида гидрохлорид, 614
Лоперамида оксид моногидрат, 617
Лоратадин, 618
Магния аспартат дигидрат, 621
Магния ацетат тетрагидрат, 622
Магния гидроксид, 623
Магния карбонат основной, легкий, 624
Магния карбонат основной, тяжелый, 625
Магния оксид легкий, 626
Магния оксид тяжелый, 627
Магния стеарат, 628
Магния сульфат гептагидрат, 631
Магния хлорид 4,5-гидрат, 631
Магния хлорид гексагидрат, 632
Магния цитрат безводный, 633
Макрогола цетостеариловый эфир, 634
Макроголглицерина гидроксистеарат, 635
Макроголы, 636
Маннит (маннитол), 638
Марганца глюконат, 641
Мебендазол, 642
Меди сульфат безводный, 644
Меди сульфат пентагидрат, 644
Меклозина дигидрохлорид, 645
Мелоксикам, 647
Мельдоний дигидрат, 649
Менадион, 651
Менадиона натрия бисульфит, 651
Ментол рацемический, 653
Меропенем тригидрат, 654
Меркаптопурин, 656
Метакрезол, 656
Метакриловой кислоты
и этилакрилата сополимер (1:1), 658
Метакриловой кислоты
и этилакрилата сополимер (1:1), 30 % дисперсия, 659
Метамизол натрия моногидрат, 661
Метилпарагидроксибензоат, 663
Метилтиониния хлорид, 665
Метилцеллюлоза, 667
РЬМетионин, 669
Метионин, 670
Метокпопрамида гидрохлорид, 672
Метопролола тартрат, 674
Метотрексат, 676
Метронидазол, 679
Метронидазола бензоат, 681
Метформина гидрохлорид, 682
Миконазола нитрат, 685
Моксифлоксацина гидрохлорид, 687
Моксонидин, 689
Молочная кислота, 691
(З)-Молочная кислота, 691
Морфина гидрохлорид, 692
Морфина сульфат, 694
Мочевина, 696
Муравьиная кислота, 698
Муравьиная кислота безводная, 699
Мыло зеленое (мыло калийное), 699
Мыло хозяйственное твердое, 700
Мяты перечной масло, 702
Напроксен, 703
Натрия аминосалицилат дигидрат, 706
Натрия ацетат тригидрат, 708
Натрия бензоат, 709
Натрия бромид, 710
Натрия гиалуронат, 711
Натрия гидрокарбонат, 714
Натрия гидроксид, 715
Натрия дигидрофосфат дигидрат, 715
Натрия йодид, 716
Натрия каприлат, 717
Натрия карбонат безводный, 718
Натрия карбонат декагидрат, 718
Натрия карбонат моногидрат, 719
Натрия крахмалгликолят (тип А), 720
Натрия крахмалгликолят (тип В), 721
Натрия крахмалгликолят (тип С), 722
Натрия (5)-лактата раствор, 723
Натрия лактата раствор, 724
Натрия лаурилсульфат, 725
Натрия метабисульфит, 726
Натрия метилпарагидроксибензоат, 727
Натрия пикосульфат, 729
Натрия пропилпарагидроксибензоат, 731
Натрия салицилат, 733
Натрия сульфат безводный, 735
Натрия сульфат декагидрат, 735
Натрия сульфит безводный, 736
Натрия сульфит гептагидрат, 737
Натрия тетраборат, 738
Натрия тиосульфат, 738
Натрия фторид, 739
Натрия хлорид, 740
Натрия цетостеарилсульфат, 741
Натрия цитрат, 743
Натрия этилпарагидроксибензоат, 744
Нафазолина гидрохлорид, 745
Нафазолина нитрат, 747
Никотинамид, 749
Никотиновая кислота, 751
Нимесулид, 753
Нистатин, 755
Нитрофурал, 757
Нитрофурантоин, 758
Нифедипин, 759
Нифуроксазид, 761
Носкапина гидрохлорид, 763
Оксалиплатин, 765
Оксациллин натрия моногидрат, 768
Оксиметазолина гидрохлорид, 771
Окситоцин, 773
Окситоцина раствор концентрированный, 774
Октилдодеканол, 775
1364
Гэсударственная фармакопея Республики Беларусь
Омега-З-кислоты этиловые эфиры 60, 776
Омега-З-кислоты этиловые эфиры 90, 778
Ондансетрона гидрохлорид дигидрат, 780
Осельтамивира фосфат, 783
Офлоксацин, 785
Паклитаксел, 787
Панкреатина порошок, 791
Папаверина гидрохлорид, 794
Парафин жидкий, 796
Парафин мягкий белый, 798
Парафин мягкий желтый, 799
Парафин твердый, 799
Парацетамол, 800
Пентоксифиллин, 802
Пепсина порошок, 805
Периндоприл трет-бутиламин, 806
Пилокарпина гидрохлорид, 810
Пиперазина адипинат, 812
Пиперазин гидрат, 813
Пирантела эмбонат, 814
Пирацетам, 816
Пиридоксина гидрохлорид, 817
Плазма человеческая для фракционирования, 820
Повидон, 821
Повидон-йод, 825
Подсолнечное масло рафинированное, 826
Поливиниловый спирт, 826
Полисорбат 20, 827
Полисорбат 40, 828
Полисорбат 60, 829
Полисорбат 80, 830
Прамипексола дигидрохпорид моногидрат, 832
Преднизолон, 834
Прокаина гидрохлорид, 836
Прокаинамида гидрохлорид, 838
Пролин, 839
Прометазина гидрохлорид, 841
Пропиленгликоль, 843
Пропилпарагидроксибензоат, 844
Прополис, 846
Пропранолола гидрохлорид, 847
Протамина сульфат, 848
Псевдоэфедрина гидрохлорид, 850
Пшеничный крахмал, 852
Рамиприл, 852
Ранитидина гидрохлорид, 855
Резорцин, 857
Рибавирин, 858
Рибофлавин, 860
Рибофлавина натрия фосфат, 862
Рисовый крахмал, 864
Рисперидон, 865
Рифабутин, 867
Рифампицин, 869
Рокситромицин, 871
Ртути хлорид, 874
Рутозид тригидрат, 874
Сабаля мелкопильчатого экстракт, 876
Салициловая кислота, 879
Сальбутамола сульфат, 881
Сахарин натрий, 884
Сахароза, 886
Севофлуран, 888
Сера для наружного применения, 890
Серебра нитрат, 891
Серебра протеинат, 891
Серебро коллоидное для наружного применения, 892
Серин, 893
Серная кислота, 895
Сертаконазола нитрат, 896
Силденафила цитрат, 897
Симвастатин, 899
Соевое масло гидрогенизированное, 902
Соевое масло рафинированное, 902
Сорбиновая кислота, 903
Сорбитол (сорбит), 904
Сорбитола раствор кристаллизующийся
(сорбита раствор кристаллизующийся), 906
Сорбитола раствор некристаллизующийся
(сорбита раствор некристаллизующийся), 907
Сосны обыкновенной масло, 908
Спирамицин, 909
Спиронолактон, 912
Стеариловый спирт, 915
Стеариновая кислота, 915
Стрептомицина сульфат, 916
Суксаметония хлорид, 918
Сульпирид, 919
Сультамициллина тозилат дигидрат, 921
Сульфагуанидин, 924
Сульфаметоксазол, 926
Сульфаниламид, 928
Сульфацетамид натрия, 929
Тадалафил, 931
Тальк, 934
Таниновая кислота, 937
Таурин, 937
Тейкопланин, 939
Теобромин, 942
Теофиллин, 943
Теофиллин-этилендиамин, 945
Теофиллин-этилендиамин гидрат, 947
Теофиллин-этилендиамин для инъекций, 949
Тербинафина гидрохлорид, 951
Терпентинное масло, 953
Терпентинное масло, тип Ртиз ртаз(ег, 953
Тестостерон, 954
Тестостерона пропионат, 956
Тетракаина гидрохлорид, 958
Тетрациклин, 960
Тетризолина гидрохлорид, 961
Тиамина гидрохлорид, 963
Тимолола малеат, 965
Тирозин, 968
Титана диоксид, 970
Тозилхлорамид натрия, 971
а-Токоферилацетат, 972
а-Токоферол, 974
Я/?#-а-Токоферилацетат, 976
Толбутамид, 978
Толнафтат, 979
Трагакант, 981
Трамадола гидрохлорид, 983
Треонин, 984
Триамцинолона ацетонид, 987
Трибенозид, 990
Триметазидина дигидрохлорид, 991
Триметоприм, 993
Триптофан, 996
Троксерутин, 1000
Алфавитный указатель частных фармакопейных статей
1365
Троламин, 1002
Трометамол, 1004
Трописетрона гидрохлорид, 1005
Углерода диоксид, 1007
Уголь активированный, 1009
Уксусная кислота ледяная, 1010
Фамотидин, 1011
Феназон, 1013
Фенилаланин, 1015
Фенилэфрин, 1017
Фенилэфрина гидрохлорид, 1020
Фенирамина малеат, 1022
Фенобарбитал, 1024
Феноксиметилпенициллин, 1026
Фенол, 1028
Фенолфталеин, 1028
Фентанил, 1029
Фибриноген человеческий, 1031
Флударабина фосфат, 1032
Флуконазол, 1035
Флуоцинолона ацетонид, 1037
Флутамид, 1040
Фолиевая кислота, 1041
Формальдегида 35 % раствор, 1043
Фосфорная кислота концентрированная, 1044
Фосфорная кислота разведенная, 1045
Фрамицетина сульфат, 1045
Фруктоза, 1047
Фторурацил, 1048
Фуразолидон, 1050
Фуросемид, 1051
Химотрипсин, 1053
Хинидина сульфат, 1055
Хинина гидрохлорид, 1057
Хинина сульфат, 1058
Хлоралгидрат, 1060
Хпорамфеникол, 1061
Хлорамфеникола пальмитат, 1062
Хпорбутанол безводный, 1064
Хлорбутанол гемигидрат, 1064
Хпоргексидина диацетат, 1065
Хлоргексидина диглюконата раствор, 1067
Хлористоводородная кислота
концентрированная, 1071
Хлористоводородная кислота разведенная, 1072
Хлоркрезол, 1072
Хлороформ, 1073
Хлорталидон, 1074
Хлорфенамина малеат, 1076
Холекальциферол, 1078
Холестерин, 1079
Хондроитина натрия сульфат, 1081
Целекоксиб, 1083
Целлюлоза микрокристаллическая, 1085
Цетиловый спирт, 1088
Цетилпальмитат, 1089
Цетилпиридиния хлорид, 1090
Цетиризина дигидрохлорид, 1091
Цетостеариловый спирт, 1093
Цетостеариловый спирт (тип А) эмульсионный, 1094
Цетостеариловый спирт (тип В) эмульсионный, 1096
Цетримид, 1097
Цефазолин натрия, 1098
Цефаклор, 1101
Цефалексин моногидрат, 1103
Цефепима дигидрохлорид моногидрат, 1106
Цефоперазон натрия, 1108
Цефотаксим натрия, 1111
Цефтазидим пентагидрат, 1113
Цефтазидим пентагидрат с натрия карбонатом
для инъекций, 1116
Цефтриаксон натрия, 1119
Цианокобаламин, 1121
Циклофосфамид, 1122
Циластатин натрия, 1123
Цинка ацетат дигидрат, 1126
Цинка глюконат, 1127
Цинка оксид, 1128
Цинка сульфат гексагидрат, 1128
Цинка сульфат гептагидрат, 1129
Цинка сульфат моногидрат, 1129
Цинка хлорид, 1130
Циннаризин, 1130
Ципрофлоксацин, 1132
Ципрофлоксацина гидрохлорид, 1134
Цисплатин, 1137
Цистеина гидрохлорид моногидрат, 1139
Цитарабин, 1141
Чайного дерева масло, 1142
Шалфея мускатного масло, 1143
Шеллак, 1144
Эвкалиптовое масло, 1146
Эметина гидрохлорид пентагидрат, 1147
Эмульгатор № 1, 1148
Эналаприла малеат, 1150
Эргокальциферол, 1152
Эритритол, 1154
Эритромицин, 1156
Эритромицина эстолат, 1159
Этакридина лактат моногидрат, 1162
Этанол безводный, 1163
Этил морфина гидрохлорид, 1165
Этиловый спирт 95 %, 90 %, 80 %, 70 %,
60%, 40%, 1167
Этиловый спирт 96 %, 1167
Этилпарагидроксибензоат, 1172
Эфедрина гидрохлорид, 1173
Эфедрина гидрохлорид рацемический, 1175
Эфир, 1177
Эфир анестезирующий, 1177
1366
Государственная фармакопея Республики Беларусь
АЛФАВИТНЫЙ УКАЗАТЕЛЬ ЧАСТНЫХ ФАРМАКОПЕЙНЫХ СТАТЕЙ
НА ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ СЫРЬЕ
Аира корневища, 1179
Алтея корни, 1180
Аралии маньчжурской корни, 1180
Арники цветки, 1181
Багульника болотного побеги, 1184
Бадана корневища, 1185
Барбариса обыкновенного корни, 1185
Барбариса обыкновенного листья, 1186
Бегонии листья, 1187
Белены черной листья, 1188
Белладонны листья (красавки листья), 1189
® Березы листья, 1191
Березы почки, 1193
Бессмертника песчаного цветки, 1193
® Боярышника листья и цветки, 1194
Боярышника листья, 1196
Боярышника плоды, 1197
Боярышника цветки, 1199
Брусники листья, 1200
Бузины черной цветки, 1202
Валерианы корневища с корнями, 1204
Василька синего цветки, 1207
Вахты трехлистной листья, 1207
® Гинкго листья, 1208
® Горечавки корни, 1210
Горицвета весеннего трава, 1211
Горца змеиного корневища
(змеевика корневища), 1212
Горца перечного трава (водяного перца трава), 1213
Горца почечуйного трава, 1214
Горца птичьего трава (спорыша трава), 1214
Девясила корневища и корни, 1216
Девясила цветки, 1217
Донника трава, 1218
Дуба кора, 1220
Дурмана листья, 1221
Душицы трава, 1223
® Дягиля корни, 1224
Женьшеня корни, 1226
Жостера слабительного плоды, 1228
Зверобоя трава, 1228
Земляники лесной листья, 1230
Земляники лесной плоды, 1231
Золототысячника трава, 1231
Ивы кора, 1233
® Имбиря корневища, 1234
<®> Исландского мха слоевища, 1235
Календулы цветки (ноготков цветки), 1236
Калины кора, 1238
Каштана конского семена, 1238
Кориандра плоды, 1240
Крапивы листья, 1241
Кровохлебки корни, 1243
Крушины кора, 1244
Кукурузы столбики с рыльцами, 1246
® Лабазника вязолистного трава, 1246
Лабазника вязолистного цветки, 1248
® Лаванды цветки, 1249
Ламинарии слоевища (морская капуста), 1251
Ландыша листья, 1252
Ландыша трава, 1252
Ландыша цветки, 1253
Лапчатки белой трава, 1254
Лапчатки корневища, 1255
Левзеи сафлоровидной корневища с корнями, 1256
Левзеи сафлоровидной листья, 1257
® Лимонника плоды, 1258
Лимонника семена, 1260
Липы цветки, 1260
Льна семена, 1262
@ Любистка корни, 1262
Маклейи листья, 1264
Малины листья, 1265
Малины плоды, 1266
Марены корневища и корни, 1266
Мать-и-мачехи листья, 1268
(й) Мелиссы листья, 1269
Мелиссы трава, 1270
Многоколосника морщинистого трава
(л офанта трава), 1271
®> Можжевельника плоды, 1272
Мяты перечной листья, 1273
Наперстянки пурпурной листья, 1275
Облепихи плоды свежие, 1276
Одуванчика лекарственного корни, 1277
Ольхи серой листья, 1278
Ольхи соплодия, 1280
Ольхи черной листья, 1280
Ортосифона тычиночного листья
(почечного чая листья), 1283
Пассифлоры трава, 1284
Пастушьей сумки трава, 1285
® Первоцвета корни, 1286
Пижмы цветки, 1287
Пиона уклоняющегося
корневища и корни, 1288
Пиона уклоняющегося трава, 1289
Подорожника большого листья, 1290
® Подорожника ланцетного листья, 1291
(§•> Полыни горькой трава, 1292
Пустырника листья, 1293
Пустырника трава, 1294
Расторопши плоды, 1296
Ревеня корни, 1298
Репешка трава, 1300
Родиолы розовой
корневища и корни, 1301
Ромашки цветки, 1302
Рудбекии шершавой цветки, 1303
Рябины плоды, 1305
Алфавитный указатель частных фармакопейных статей
1367
Сабельника болотного
корневища с корнями, 1307
® Сенны листья, 1308
Сенны листья с плодами, 1309
® Сенны остролистной плоды, 1311
® Сенны узколистной плоды, 1312
Синюхи корневища с корнями, 1314
Солодки корень (лакричный корень), 1315
Сосны почки, 1317
Сушеницы топяной трава, 1317
Термопсиса ланцетного трава, 1318
® Тимьяна трава, 1319
Тмина плоды, 1321
Толокнянки листья, 1322
Тыквы семена, 1324
Тысячелистника трава, 1324
Укропа пахучего плоды, 1325
Фасоли створки, 1326
® Фенхеля горького плоды, 1327
® Фенхеля сладкого плоды, 1328
Фиалки трава, 1329
® Фукус, 1331
Хвоща полевого трава, 1332
Хмеля шишки, 1334
Чабреца блошиного трава, 1335
Чабреца ползучего трава, 1337
Чага (черный березовый гриб), 1338
Череды трава, 1339
Черемухи плоды, 1340
® Черники плоды свежие, 1341
® Черники плоды сухие, 1342
® Чеснока порошок, 1342
Чистотела трава, 1343
Шалфея листья, 1345
Шиповника плоды, 1346
Эвкалипта прутовидного листья, 1347
® Эвкалипта шаровидного листья, 1348
Элеутеррококка корневища и корни, 1349
Эхинацеи пурпурной трава, 1351
Справочное издание
ГОСУДАРСТВЕННАЯ ФАРМАКОПЕЯ
РЕСПУБЛИКИ БЕЛАРУСЬ
(ГФ РБ II)
В двух томах
Том 2
Контроль качества субстанций
для фармацевтического использования
и лекарственного растительного сырья
Редактор С. И. Марченко
Компьютерный дизайн и верстка К.А. Журавский
Корректор Т.Н. Волгина
Подписано в печать 28.03.2016. Формат 60х841/а.
Бумага офсетная. Гарнитура Апа!.
Печать офсетная. Уел. печ. л. 159,03. Уч.-изд. л. 105,1.
Тираж 1200. Заказ 1060.
Выпущено по заказу Министерства здравоохранения Республики Беларусь
Издатель и полиграфическое исполнение:
Открытое акционерное общество «Типография «Победа».
Свидетельство о государственной регистрации
издателя, изготовителя, распространителя печатных изданий
№ 1/178 от 12.02.2014, № 2/38 от 29.01.2014.
Ул. Тавлая, 11, 222310, Молодечно.